WO2011068898A1 - Protein kinase c inhibitors and uses thereof - Google Patents
Protein kinase c inhibitors and uses thereof Download PDFInfo
- Publication number
- WO2011068898A1 WO2011068898A1 PCT/US2010/058597 US2010058597W WO2011068898A1 WO 2011068898 A1 WO2011068898 A1 WO 2011068898A1 US 2010058597 W US2010058597 W US 2010058597W WO 2011068898 A1 WO2011068898 A1 WO 2011068898A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- fluoro
- ylamino
- methyl
- tetrazol
- phenyl
- Prior art date
Links
- 0 *c1c(*)c([N+]([O-])=O)c(*)c(N)c1* Chemical compound *c1c(*)c([N+]([O-])=O)c(*)c(N)c1* 0.000 description 8
- TZWFNWLLLUISAG-UHFFFAOYSA-N [O-][N+](c1cc(F)c(C2CC2)c(N=C=O)c1)=O Chemical compound [O-][N+](c1cc(F)c(C2CC2)c(N=C=O)c1)=O TZWFNWLLLUISAG-UHFFFAOYSA-N 0.000 description 2
- BFIBCXLPSFGLAF-UHFFFAOYSA-N CC(C)(C(F)(F)F)Oc(c(N1N=NN(C)C1=O)c1)ccc1Nc(nc1)nc(NC2CC(C)(C)N(C)C(C)(C)C2)c1F Chemical compound CC(C)(C(F)(F)F)Oc(c(N1N=NN(C)C1=O)c1)ccc1Nc(nc1)nc(NC2CC(C)(C)N(C)C(C)(C)C2)c1F BFIBCXLPSFGLAF-UHFFFAOYSA-N 0.000 description 1
- XBEXCZLWOWTTAU-UHFFFAOYSA-N CC(C)(C1)N(C)C(C)(C)CC1Nc1nc(Nc(cc2)cc(N3N=NN(C)C3=O)c2OC2COC2)ncc1F Chemical compound CC(C)(C1)N(C)C(C)(C)CC1Nc1nc(Nc(cc2)cc(N3N=NN(C)C3=O)c2OC2COC2)ncc1F XBEXCZLWOWTTAU-UHFFFAOYSA-N 0.000 description 1
- OSXHISIFYQFLOS-UHFFFAOYSA-N CC(C)(C1)N(C)C(C)(C)CC1Nc1nc(Nc2cc(N3N=NN(C)C3=O)c(C)c(C([N]C)=O)c2)ncc1F Chemical compound CC(C)(C1)N(C)C(C)(C)CC1Nc1nc(Nc2cc(N3N=NN(C)C3=O)c(C)c(C([N]C)=O)c2)ncc1F OSXHISIFYQFLOS-UHFFFAOYSA-N 0.000 description 1
- CIORCHOFGHDWCX-UHFFFAOYSA-N CC(C)(C1)N(C)C(C)(C)CC1Nc1nc(Nc2cc(N3N=NN(C)C3=O)c(C3CC3)c(C(F)(F)F)c2)ncc1F Chemical compound CC(C)(C1)N(C)C(C)(C)CC1Nc1nc(Nc2cc(N3N=NN(C)C3=O)c(C3CC3)c(C(F)(F)F)c2)ncc1F CIORCHOFGHDWCX-UHFFFAOYSA-N 0.000 description 1
- HLYVGISNDNHMRA-UHFFFAOYSA-N CC(C)(C1)N(C)C(C)(C)CC1Nc1nc(Nc2cc(N3N=NN(C)C3=O)cc(OC3COC3)c2)ncc1F Chemical compound CC(C)(C1)N(C)C(C)(C)CC1Nc1nc(Nc2cc(N3N=NN(C)C3=O)cc(OC3COC3)c2)ncc1F HLYVGISNDNHMRA-UHFFFAOYSA-N 0.000 description 1
- JWQXDHKFVICOAN-UHFFFAOYSA-N CC(C)(C1)NC(C)(C)CC1Nc1nc(Cl)ncc1F Chemical compound CC(C)(C1)NC(C)(C)CC1Nc1nc(Cl)ncc1F JWQXDHKFVICOAN-UHFFFAOYSA-N 0.000 description 1
- WXGOHMWDDPBVDZ-UHFFFAOYSA-N CC(C)(C1)NC(C)(C)CC1Nc1nc(Nc2cc(N3N=NN(C)C3=O)cc(C(F)(F)F)c2)ncc1F Chemical compound CC(C)(C1)NC(C)(C)CC1Nc1nc(Nc2cc(N3N=NN(C)C3=O)cc(C(F)(F)F)c2)ncc1F WXGOHMWDDPBVDZ-UHFFFAOYSA-N 0.000 description 1
- GITSRSYOHJKAJD-UHFFFAOYSA-N CC(C)N(C1=O)N=NN1c(cc1N)c(C2CC2)cc1F Chemical compound CC(C)N(C1=O)N=NN1c(cc1N)c(C2CC2)cc1F GITSRSYOHJKAJD-UHFFFAOYSA-N 0.000 description 1
- VCZGDUKBJLQXMM-UHFFFAOYSA-N CN(C1=O)N=NN1c(cc(c(F)c1)[N+]([O-])=O)c1Cl Chemical compound CN(C1=O)N=NN1c(cc(c(F)c1)[N+]([O-])=O)c1Cl VCZGDUKBJLQXMM-UHFFFAOYSA-N 0.000 description 1
- NJJMISVHZLOPCA-UHFFFAOYSA-N CN(C1=O)N=NN1c(cc(cc1)N)c1F Chemical compound CN(C1=O)N=NN1c(cc(cc1)N)c1F NJJMISVHZLOPCA-UHFFFAOYSA-N 0.000 description 1
- LVTKOGDYYOCSDQ-UHFFFAOYSA-N CN(C1=O)N=NN1c(cc(cc1)N)c1OC1CCOCC1 Chemical compound CN(C1=O)N=NN1c(cc(cc1)N)c1OC1CCOCC1 LVTKOGDYYOCSDQ-UHFFFAOYSA-N 0.000 description 1
- LMUDBRIHFQQGTH-UHFFFAOYSA-N CN(C1=O)N=NN1c(cc(cc1)N)c1OC1COC1 Chemical compound CN(C1=O)N=NN1c(cc(cc1)N)c1OC1COC1 LMUDBRIHFQQGTH-UHFFFAOYSA-N 0.000 description 1
- LZDURIBLDSFEPZ-UHFFFAOYSA-N CN(C1=O)N=NN1c(cc(cc1)[N+]([O-])=O)c1Cl Chemical compound CN(C1=O)N=NN1c(cc(cc1)[N+]([O-])=O)c1Cl LZDURIBLDSFEPZ-UHFFFAOYSA-N 0.000 description 1
- LPSVTZNCXLQWGV-UHFFFAOYSA-N CN(C1=O)N=NN1c(cc(cc1C(F)(F)F)N)c1Cl Chemical compound CN(C1=O)N=NN1c(cc(cc1C(F)(F)F)N)c1Cl LPSVTZNCXLQWGV-UHFFFAOYSA-N 0.000 description 1
- ASEYWVJGUFTUQB-UHFFFAOYSA-N CN(C1=O)N=NN1c(cc(cc1C(F)(F)F)[N+]([O-])=O)c1Cl Chemical compound CN(C1=O)N=NN1c(cc(cc1C(F)(F)F)[N+]([O-])=O)c1Cl ASEYWVJGUFTUQB-UHFFFAOYSA-N 0.000 description 1
- WZFPTGWVOQDPAW-UHFFFAOYSA-N CN(C1=O)N=NN1c(ccc(F)c1)c1Cl Chemical compound CN(C1=O)N=NN1c(ccc(F)c1)c1Cl WZFPTGWVOQDPAW-UHFFFAOYSA-N 0.000 description 1
- ZMXIAYZYZQNCRI-UHFFFAOYSA-N CN(C1=O)N=NN1c1c(C2CC2)ccc([N+]([O-])=O)c1 Chemical compound CN(C1=O)N=NN1c1c(C2CC2)ccc([N+]([O-])=O)c1 ZMXIAYZYZQNCRI-UHFFFAOYSA-N 0.000 description 1
- BPPNMOJYUOWJDI-UHFFFAOYSA-N CN(C1=O)N=NN1c1cc(Cl)cc(N)c1 Chemical compound CN(C1=O)N=NN1c1cc(Cl)cc(N)c1 BPPNMOJYUOWJDI-UHFFFAOYSA-N 0.000 description 1
- BAKFDCOLBFBSJA-UHFFFAOYSA-N CN(C1=O)N=NN1c1cc(OC)cc(N)c1 Chemical compound CN(C1=O)N=NN1c1cc(OC)cc(N)c1 BAKFDCOLBFBSJA-UHFFFAOYSA-N 0.000 description 1
- WYYVXCIHYUOEDW-UHFFFAOYSA-N CN(C1=O)N=NN1c1cc(OC2COC2)cc(N)c1 Chemical compound CN(C1=O)N=NN1c1cc(OC2COC2)cc(N)c1 WYYVXCIHYUOEDW-UHFFFAOYSA-N 0.000 description 1
- JJMOPKRXGVDDST-UHFFFAOYSA-N CN(C1=O)N=NN1c1cccc(N)c1 Chemical compound CN(C1=O)N=NN1c1cccc(N)c1 JJMOPKRXGVDDST-UHFFFAOYSA-N 0.000 description 1
- NDHKNGVBDLFYBP-UHFFFAOYSA-N Cc(c(C(F)(F)F)cc([N+]([O-])=O)c1)c1N1N=NNC1=O Chemical compound Cc(c(C(F)(F)F)cc([N+]([O-])=O)c1)c1N1N=NNC1=O NDHKNGVBDLFYBP-UHFFFAOYSA-N 0.000 description 1
- GVYLZBJHCDJAMS-UHFFFAOYSA-N Cc(c(N=C=O)c1)c(C(F)(F)F)cc1[N+]([O-])=O Chemical compound Cc(c(N=C=O)c1)c(C(F)(F)F)cc1[N+]([O-])=O GVYLZBJHCDJAMS-UHFFFAOYSA-N 0.000 description 1
- BMKGNOGDUIRULI-UHFFFAOYSA-N Cc1cc(N)cc(N2N=NN(C)C2=O)c1C Chemical compound Cc1cc(N)cc(N2N=NN(C)C2=O)c1C BMKGNOGDUIRULI-UHFFFAOYSA-N 0.000 description 1
- ZGDLGVNNATVVSI-UHFFFAOYSA-N Cc1cc([N+]([O-])=O)cc(N2N=NN(C)C2=O)c1C Chemical compound Cc1cc([N+]([O-])=O)cc(N2N=NN(C)C2=O)c1C ZGDLGVNNATVVSI-UHFFFAOYSA-N 0.000 description 1
- WHPFEQUEHBULBW-UHFFFAOYSA-N Fc(cnc(Cl)n1)c1Cl Chemical compound Fc(cnc(Cl)n1)c1Cl WHPFEQUEHBULBW-UHFFFAOYSA-N 0.000 description 1
- DSHRRUJSFSGICH-UHFFFAOYSA-N Nc1cc(C(F)(F)F)c(C2CC2)c([N+]([O-])=O)c1 Chemical compound Nc1cc(C(F)(F)F)c(C2CC2)c([N+]([O-])=O)c1 DSHRRUJSFSGICH-UHFFFAOYSA-N 0.000 description 1
- SGKPJAWEHPLNMV-UHFFFAOYSA-N Nc1cc(N(C(c2c3cccc2)=O)C3=O)cc(C(F)(F)F)c1C1CC1 Chemical compound Nc1cc(N(C(c2c3cccc2)=O)C3=O)cc(C(F)(F)F)c1C1CC1 SGKPJAWEHPLNMV-UHFFFAOYSA-N 0.000 description 1
- ZZOZQNMXRYAAEM-UHFFFAOYSA-N Nc1cc([N+]([O-])=O)cc(F)c1C1CC1 Chemical compound Nc1cc([N+]([O-])=O)cc(F)c1C1CC1 ZZOZQNMXRYAAEM-UHFFFAOYSA-N 0.000 description 1
- ZQLDCUVRANMURJ-UHFFFAOYSA-N [O-][N+](c(cc1)cc(N2N=NN(CCF)C2=O)c1F)=O Chemical compound [O-][N+](c(cc1)cc(N2N=NN(CCF)C2=O)c1F)=O ZQLDCUVRANMURJ-UHFFFAOYSA-N 0.000 description 1
- ROUXETNGZAENHK-UHFFFAOYSA-N [O-][N+](c(cc1)cc(N2N=NNC2=O)c1Br)=O Chemical compound [O-][N+](c(cc1)cc(N2N=NNC2=O)c1Br)=O ROUXETNGZAENHK-UHFFFAOYSA-N 0.000 description 1
- UVAHMYAJJBHFRX-UHFFFAOYSA-N [O-][N+](c(cc1)cc(N2N=NNC2=O)c1Cl)=O Chemical compound [O-][N+](c(cc1)cc(N2N=NNC2=O)c1Cl)=O UVAHMYAJJBHFRX-UHFFFAOYSA-N 0.000 description 1
- FQFOOISQGKXZMY-UHFFFAOYSA-N [O-][N+](c(cc1)cc(N2N=NNC2=O)c1F)=O Chemical compound [O-][N+](c(cc1)cc(N2N=NNC2=O)c1F)=O FQFOOISQGKXZMY-UHFFFAOYSA-N 0.000 description 1
- XCDYDLFUPMSLGZ-UHFFFAOYSA-N [O-][N+](c(cc1)cc(N=C=O)c1F)=O Chemical compound [O-][N+](c(cc1)cc(N=C=O)c1F)=O XCDYDLFUPMSLGZ-UHFFFAOYSA-N 0.000 description 1
- AVVRMLYMVUFHIH-UHFFFAOYSA-N [O-][N+](c(cc1N=C=O)ccc1Br)=O Chemical compound [O-][N+](c(cc1N=C=O)ccc1Br)=O AVVRMLYMVUFHIH-UHFFFAOYSA-N 0.000 description 1
- OELTYULIZKSSFC-UHFFFAOYSA-N [O-][N+](c1cc(C(F)(F)F)c(C2CC2)c([N+]([O-])=O)c1)=O Chemical compound [O-][N+](c1cc(C(F)(F)F)c(C2CC2)c([N+]([O-])=O)c1)=O OELTYULIZKSSFC-UHFFFAOYSA-N 0.000 description 1
- QGCSAJPKKLBJBM-UHFFFAOYSA-N [O-][N+](c1cc(N(C(c2c3cccc2)=O)C3=O)cc(C(F)(F)F)c1C1CC1)=O Chemical compound [O-][N+](c1cc(N(C(c2c3cccc2)=O)C3=O)cc(C(F)(F)F)c1C1CC1)=O QGCSAJPKKLBJBM-UHFFFAOYSA-N 0.000 description 1
- YHGJVJJKJULLEB-UHFFFAOYSA-N [O-][N+](c1cc(N2N=NNC2=O)c(C2CC2)c(F)c1)=O Chemical compound [O-][N+](c1cc(N2N=NNC2=O)c(C2CC2)c(F)c1)=O YHGJVJJKJULLEB-UHFFFAOYSA-N 0.000 description 1
- KCPAMBMOIAPSQR-UHFFFAOYSA-N [O-][N+](c1cc(N2N=NNC2=O)cc(C2CC2)c1)=O Chemical compound [O-][N+](c1cc(N2N=NNC2=O)cc(C2CC2)c1)=O KCPAMBMOIAPSQR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
- A61P5/16—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4 for decreasing, blocking or antagonising the activity of the thyroid hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
Definitions
- PKC Protein kinase C
- the PKC family of isozymes includes at least 11 different protein kinases that can be divided into at least three subfamilies based on their homology and sensitivity to activators. Each isozyme includes a number of homologous (“conserved” or “C”) domains interspersed with isozyme-unique (“variable” or “V”) domains.
- PKC ⁇ , ⁇ , ⁇ ⁇ and ⁇ contain four homologous domains (CI, C2, C3 and C4) and require calcium, phosphatidylserine, and diacylglycerol or phorbol esters for activation.
- CI, C2, C3 and C4 homologous domains
- nPKC nPKC subfamily
- PKC ⁇ and ⁇ /i lack both the C2 and one-half of the CI homologous domains and are insensitive to diacylglycerol, phorbol esters and calcium.
- This disclosure concerns compounds which are useful as inhibitors of protein kinase C (PKC) and are thus useful for treating a variety of diseases and disorders that are mediated or sustained through the activity of PKC.
- PKC protein kinase C
- This disclosure also relates to pharmaceutical compositions comprising these compounds, methods of using these compounds in the treatment of various diseases and disorders, processes for preparing these compounds and intermediates useful in these processes.
- R 5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
- Y 1 and Y 2 are independently selected from hydrogen, alkyl, and acyl;
- R is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
- R a and R b are independently selected from hydrogen and alkyl
- R c and R d are independently selected from hydrogen and alkyl
- Q is selected from N and CR 7b ;
- R 6a , R 6b , R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O- alk-A;
- alk is a bond, alkylene or substituted alkylene
- A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
- a ring can be substituted or unsubstituted
- R 7y is selected from hydrogen, alkyl, cycloalkyl, and substituted alkyl
- This disclosure concerns compounds which are useful as inhibitors of protein kinase C (PKC) and are thus useful for treating a variety of diseases and disorders that are mediated or sustained through the activity of PKC.
- PKC protein kinase C
- This disclosure also relates to pharmaceutical compositions comprising these compounds, methods of using these compounds in the treatment of various diseases and disorders, processes for preparing these compounds and intermediates useful in these processes.
- Alkyl refers to monovalent saturated aliphatic hydrocarbyl groups having from 1 to 10 carbon atoms and preferably 1 to 6 carbon atoms. This term includes, by way of example, linear and branched hydrocarbyl groups such as methyl (CH 3 -), ethyl (CH 3 CH 2 -), n-propyl (CH 3 CH 2 CH 2 -), isopropyl ((CH 3 ) 2 CH-), n-butyl (CH 3 CH 2 CH 2 CH 2 -), isobutyl ((CH 3 ) 2 CHCH 2 -), sec-butyl ((CH 3 )(CH 3 CH 2 )CH-), t-butyl ((CH 3 ) 3 C-), n-pentyl
- substituted alkyl refers to an alkyl group as defined herein wherein one or more carbon atoms in the alkyl chain have been optionally replaced with a heteroatom such as -0-, -N-, -S-, -S(0) n - (where n is 0 to 2), -NR- (where R is hydrogen or alkyl) and having from 1 to 5 substituents selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, oxo, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheter
- Alkylene refers to divalent aliphatic hydrocarbyl groups preferably having from 1 to 6 and more preferably 1 to 3 carbon atoms that are either straight-chained or branched, and which are optionally interrupted with one or more groups selected from -0-, - NR 10 -, -NR 10 C(O , -C(0)NR 10 - and the like.
- This term includes, by way of example, methylene (-CH 2 -), ethylene (-CH 2 CH 2 -), n-propylene (-CH 2 CH 2 CH 2 -), iso-propylene (-CH 2 CH(CH 3 )-), (-C(CH 3 ) 2 CH 2 CH 2 -), (-C(CH 3 ) 2 CH 2 C(0)-), (-C(CH 3 ) 2 CH 2 C(0)NH-), (-CH(CH 3 )CH 2 -), and the like.
- Substituted alkylene refers to an alkylene group having from 1 to 3 hydrogens replaced with substituents as described for carbons in the definition of "substituted” below.
- alkane refers to alkyl group and alkylene group, as defined herein.
- alkylaminoalkyl refers to the groups RNHR - where R is alkyl group as defined herein and R is alkylene, alkenylene or alkynylene group as defined herein.
- alkaryl or “aralkyl” refers to the groups -alkylene-aryl and
- alkylene, substituted alkylene and aryl are defined herein.
- Alkoxy refers to the group -O-alkyl, wherein alkyl is as defined herein.
- Alkoxy includes, by way of example, methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, t- butoxy, sec-butoxy, n-pentoxy, and the like.
- alkoxy also refers to the groups alkenyl-O-, cycloalkyl-O-, cycloalkenyl-O-, and alkynyl-O-, where alkenyl, cycloalkyl, cycloalkenyl, and alkynyl are as defined herein.
- substituted alkoxy refers to the groups substituted alkyl-O-, substituted alkenyl-O-, substituted cycloalkyl-O-, substituted cycloalkenyl-O-, and substituted alkynyl-O- where substituted alkyl, substituted alkenyl, substituted cycloalkyl, substituted cycloalkenyl and substituted alkynyl are as defined herein.
- alkoxyamino refers to the group -NH-alkoxy, wherein alkoxy is defined herein.
- haloalkoxy refers to the groups alkyl-O- wherein one or more hydrogen atoms on the alkyl group have been substituted with a halo group and include, by way of examples, groups such as trifluoromethoxy, and the like.
- haloalkyl refers to a substituted alkyl group as described above, wherein one or more hydrogen atoms on the alkyl group have been substituted with a halo group.
- groups include, without limitation, fluoroalkyl groups, such as trifluoromethyl, difluoromethyl, trifluoroethyl and the like.
- alkylalkoxy refers to the groups -alkylene-O-alkyl, alkylene-O- substituted alkyl, substituted alkylene-O-alkyl, and substituted alkylene-O-substituted alkyl wherein alkyl, substituted alkyl, alkylene and substituted alkylene are as defined herein.
- alkylthioalkoxy refers to the group -alkylene-S -alkyl, alkylene-S- substituted alkyl, substituted alkylene-S-alkyl and substituted alkylene-S-substituted alkyl wherein alkyl, substituted alkyl, alkylene and substituted alkylene are as defined herein.
- Alkenyl refers to straight chain or branched hydrocarbyl groups having from 2 to 6 carbon atoms and preferably 2 to 4 carbon atoms and having at least 1 and preferably from 1 to 2 sites of double bond unsaturation. This term includes, by way of example, bi-vinyl, allyl, and but-3-en-l-yl. Included within this term are the cis and trans isomers or mixtures of these isomers.
- substituted alkenyl refers to an alkenyl group as defined herein having from 1 to 5 substituents, or from 1 to 3 substituents, selected from alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, oxo, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, hydroxyamino, alkoxy
- Alkynyl refers to straight or branched monovalent hydrocarbyl groups having from 2 to 6 carbon atoms and preferably 2 to 3 carbon atoms and having at least 1 and preferably from 1 to 2 sites of triple bond unsaturation. Examples of such alkynyl groups include acetylenyl (-C ⁇ CH), and propargyl (-CH 2 C ⁇ CH).
- substituted alkynyl refers to an alkynyl group as defined herein having from 1 to 5 substituents, or from 1 to 3 substituents, selected from alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, oxo, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, hydroxyamino, al
- Alkynyloxy refers to the group -O-alkynyl, wherein alkynyl is as defined herein. Alkynyloxy includes, by way of example, ethynyloxy, propynyloxy, and the like.
- Acyl refers to the groups H-C(O)-, alkyl-C(O)-, substituted alkyl-C(O)-, alkenyl-C(O)-, substituted alkenyl-C(O)-, alkynyl-C(O)-, substituted alkynyl-C(O)-, cycloalkyl-C(O)-, substituted cycloalkyl-C(O)-, cycloalkenyl-C(O)-, substituted
- acyl includes the "acetyl" group CH 3 C(0)-
- Acylamino refers to the groups -NR 20 C(O)alkyl, -NR 20 C(O)substituted alkyl, N R 20 C(O)cycloalkyl, -NR 20 C(O)substituted cycloalkyl, -NR 20 C(O)cycloalkenyl,
- R 20 is hydrogen or alkyl and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, substituted cycloalkenyl, wherein R 20 is hydrogen or alkyl and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, substituted cycloalkenyl, substituted cycloalkenyl, wherein R 20 is hydrogen or alkyl and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycl
- aminocarbonyl or the term “aminoacyl”_refers to the group -C(0)NR 21 R 22 ,
- R and R independently are selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R 21
- R are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic are as defined herein.
- alkoxycarbonylamino refers to the group -NRC(0)OR where each R is independently hydrogen, alkyl, substituted alkyl, aryl, heteroaryl, or heterocyclyl wherein alkyl, substituted alkyl, aryl, heteroaryl, and heterocyclyl are as defined herein.
- acyloxy refers to the groups alkyl-C(0)0-, substituted alkyl-C(0)0-, cycloalkyl-C(0)0-, substituted cycloalkyl-C(0)0-, aryl-C(0)0-, heteroaryl-C(0)0-, and heterocyclyl-C(0)0- wherein alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, aryl, heteroaryl, and heterocyclyl are as defined herein.
- Aminosulfonyl refers to the group -S0 2 NR 21 R 22 , wherein R 21 and R 22 independently are selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted
- heteroaryl heteroaryl, heterocyclic, substituted heterocyclic and where R and R are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group and alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic and substituted heterocyclic are as defined herein.
- Sulfonylamino refers to the group -NR 1 S0 2 R , wherein R and R independently are selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R 21 and R 22 are optionally joined together with the atoms bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, ary
- Aryl or “Ar” refers to a monovalent aromatic carbocyclic group of from 6 to 14 carbon atoms having a single ring (e.g., phenyl) or multiple condensed rings (e.g., naphthyl or anthryl) which condensed rings may or may not be aromatic, provided that the point of attachment is through an atom of the aromatic aryl group.
- This term includes, by way of example, phenyl and naphthyl.
- such aryl groups can optionally be substituted with from 1 to 5 substituents, or from 1 to 3 substituents, selected from acyloxy, hydroxy, thiol, acyl, alkyl, alkoxy, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, substituted alkyl, substituted alkoxy, substituted alkenyl, substituted alkynyl, substituted cycloalkyl, substituted cycloalkenyl, amino, substituted amino, aminoacyl, acylamino, alkaryl, aryl, aryloxy, azido, carboxyl, carboxylalkyl, cyano, halogen, nitro, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, aminoacyloxy, oxyacylamino, thioalkoxy, substituted thioalkoxy, thioaryloxy, thi
- Aryloxy refers to the group -O-aryl, wherein aryl is as defined herein, including, by way of example, phenoxy, naphthoxy, and the like, including optionally substituted aryl groups as also defined herein.
- Amino refers to the group -NH 2 .
- substituted amino refers to the group -NRR where each R is independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, alkenyl, substituted alkenyl, cycloalkenyl, substituted cycloalkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, and heterocyclyl provided that at least one R is not hydrogen.
- Carboxylalkyl refers to the groups -C(0)0-alkyl, -C(0)0-substituted alkyl,
- alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic are as defined herein.
- (Carboxyl ester)oxy refers to the groups -0-C(0)0-alkyl, -0-C(0)0-substituted alkyl, -0-C(0)0-alkenyl, -0-C(0)0-substituted alkenyl, -0-C(0)0- alkynyl, -0-C(0)0-substituted alkynyl, -0-C(0)0-aryl, -0-C(0)0-substituted aryl, -O- C(0)0-cycloalkyl, -0-C(0)0-substituted cycloalkyl, -0-C(0)0-cycloalkenyl, -0-C(0)0- substituted cycloalkenyl, -0-C(0)0-heteroaryl, -0-C(0)0-substituted heteroaryl, -O- C(0)0-heterocyclic, and -0-C(0)0-substituted heterocyclic, wherein alkyl, substituted alkyl, substituted alkyl, substitute
- Cycloalkyl refers to cyclic alkyl groups of from 3 to 10 carbon atoms having single or multiple cyclic rings including fused, bridged, and spiro ring systems.
- suitable cycloalkyl groups include, for instance, adamantyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclooctyl and the like.
- Such cycloalkyl groups include, by way of example, single ring structures such as cyclopropyl, cyclobutyl, cyclopentyl, cyclooctyl, and the like, or multiple ring structures such as adamantanyl, and the like.
- substituted cycloalkyl refers to cycloalkyl groups having from 1 to 5 substituents, or from 1 to 3 substituents, selected from alkyl, substituted alkyl, alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy,
- oxyaminoacyl azido, cyano, halogen, hydroxyl, oxo, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, -SO-alkyl, -SO-substituted alkyl, -SO-aryl, -SO-heteroaryl, -S0 2 -alkyl, - S0 2 -substituted alkyl, -S0 2 -aryl and -S0 2 -heteroaryl.
- Cycloalkenyl refers to non-aromatic cyclic alkyl groups of from 3 to 10 carbon atoms having single or multiple rings and having at least one double bond and preferably from 1 to 2 double bonds.
- substituted cycloalkenyl refers to cycloalkenyl groups having from 1 to 5 substituents, or from 1 to 3 substituents, selected from alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, hydroxyamino, alkoxyamin
- Cycloalkynyl refers to non-aromatic cycloalkyl groups of from 5 to 10 carbon atoms having single or multiple rings and having at least one triple bond.
- Cycloalkoxy refers to -O-cycloalkyl
- Cycloalkenyloxy refers to -O-cycloalkenyl.
- Halo or "halogen” refers to fluoro, chloro, bromo, and iodo.
- Heteroaryl refers to an aromatic group of from 1 to 10 carbon atoms and 1 to 4 heteroatoms selected from the group consisting of oxygen, nitrogen, and sulfur within the ring.
- Such heteroaryl groups can have a single ring (e.g., pyridinyl, imidazolyl or furyl) or multiple condensed rings (e.g., indolizinyl, quinolinyl, benzimidazolyl or benzothienyl), wherein the condensed rings may or may not be aromatic and/or contain a heteroatom, provided that the point of attachment is through an atom of the aromatic heteroaryl group.
- the nitrogen and/or sulfur ring atom(s) of the heteroaryl group are optionally oxidized to provide for the N-oxide (N ⁇ 0), sulfinyl, or sulfonyl moieties.
- This term includes, by way of example, pyridinyl, pyrrolyl, indolyl, thiophenyl, and furanyl.
- heteroaryl groups can be optionally substituted with 1 to 5 substituents, or from 1 to 3 substituents, selected from acyloxy, hydroxy, thiol, acyl, alkyl, alkoxy, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, substituted alkyl, substituted alkoxy, substituted alkenyl, substituted alkynyl, substituted cycloalkyl, substituted cycloalkenyl, amino, substituted amino, aminoacyl, acylamino, alkaryl, aryl, aryloxy, azido, carboxyl, carboxylalkyl, cyano, halogen, nitro, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, aminoacyloxy, oxyacylamino, thioalkoxy, substituted thioalkoxy, thioaryloxy, thio
- heteroaryl refers to the groups -alkylene-heteroaryl where alkylene and heteroaryl are defined herein. This term includes, by way of example, pyridylmethyl, pyridylethyl, indolylmethyl, and the like.
- Heteroaryloxy refers to -O-heteroaryl.
- Heterocycle refers to a saturated or unsaturated group having a single ring or multiple condensed rings, including fused bridged and spiro ring systems, and having from 3 to 15 ring atoms, including 1 to 4 hetero atoms.
- These ring atoms are selected from the group consisting of nitrogen, sulfur, or oxygen, wherein, in fused ring systems, one or more of the rings can be cycloalkyl, aryl, or heteroaryl, provided that the point of attachment is through the non-aromatic ring.
- the nitrogen and/or sulfur atom(s) of the heterocyclic group are optionally oxidized to provide for the N-oxide, -S(O)-, or -S0 2 - moieties.
- heterocycles and heteroaryls include, but are not limited to, azetidine, pyrrole, imidazole, pyrazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, dihydroindole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, phenanthroline, isothiazole, phenazine, isoxazole, phenoxazine, phenothiazine, imidazolidine, imidazoline, piperidine, piperazine, indoline, phthalimide, 1,2,3,4-tetrahydroisoquinoline,
- heterocyclic groups can be optionally substituted with 1 to 5, or from 1 to 3 substituents, selected from alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, oxo, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, hydroxyamino, alkoxyamino,
- Heterocyclyloxy refers to the group -O-heterocyclyl.
- heterocyclylthio refers to the group heterocyclic-S-.
- heterocyclene refers to the diradical group formed from a heterocycle, as defined herein.
- hydroxyamino refers to the group -NHOH.
- Neitro refers to the group -N0 2 .
- Sulfonyl refers to the group S0 2 -alkyl, S0 2 -substituted alkyl, S0 2 -alkenyl, S0 2 -substituted alkenyl, S0 2 -cycloalkyl, S0 2 -substituted cylcoalkyl, S0 2 -cycloalkenyl, S0 2 -substituted cylcoalkenyl, S0 2 -aryl, S0 2 -substituted aryl, S0 2 -heteroaryl, SO 2 - substituted heteroaryl, S0 2 -heterocyclic, and S0 2 -substituted heterocyclic, wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalken
- Sulfonyloxy refers to the group -OS0 2 -alkyl, OS0 2 -substituted alkyl, OS0 2 - alkenyl, OS0 2 -substituted alkenyl, OS0 2 -cycloalkyl, OS0 2 -substituted cylcoalkyl, OSO 2 - cycloalkenyl, OS0 2 -substituted cylcoalkenyl, OS0 2 -aryl, OS0 2 -substituted aryl, OSO 2 - heteroaryl, OS0 2 -substituted heteroaryl, OS0 2 -heterocyclic, and OSO 2 substituted heterocyclic, wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkyl, cycl
- cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic are as defined herein.
- aminocarbonyloxy refers to the group -OC(0)NRR where each R is independently hydrogen, alkyl, substituted alkyl, aryl, heteroaryl, or heterocyclic wherein alkyl, substituted alkyl, aryl, heteroaryl and heterocyclic are as defined herein.
- Thiol refers to the group -SH.
- Alkylthio or the term “thioalkoxy” refers to the group -S-alkyl, wherein alkyl is as defined herein.
- sulfur may be oxidized to -S(O)-.
- the sulfoxide may exist as one or more stereoisomers.
- substituted thioalkoxy refers to the group -S-substituted alkyl.
- thioaryloxy refers to the group aryl-S- wherein the aryl group is as defined herein including optionally substituted aryl groups also defined herein.
- heteroaryloxy refers to the group heteroaryl-S- wherein the heteroaryl group is as defined herein including optionally substituted aryl groups as also defined herein.
- heterocyclooxy refers to the group heterocyclyl-S- wherein the heterocyclyl group is as defined herein including optionally substituted heterocyclyl groups as also defined herein.
- substituted when used to modify a specified group or radical, can also mean that one or more hydrogen atoms of the specified group or radical are each, independently of one another, replaced with the same or different substituent groups as defined below.
- Each M + may independently be, for example, an alkali ion, such as K + , Na + , Li + ; an ammonium ion, such as + N(R 60 )4; or an alkaline earth ion, such as [Ca 2+ ]o . s, [Mg 2+ ]o . s, or [Ba 2+ ]o.5 ("subscript 0.5 means e.g.
- one of the counter ions for such divalent alkali earth ions can be an ionized form of a compound of the invention and the other a typical counter ion such as chloride, or two ionized compounds of the invention can serve as counter ions for such divalent alkali earth ions, or a doubly ionized compound of the invention can serve as the counter ion for such divalent alkali earth ions).
- -NR 80 R 80 is meant to include -NH 2 , -NH-alkyl, N-pyrrolidinyl, N-piperazinyl, 4N-methyl-piperazin-l- yl and N-morpholinyl.
- substituent groups for hydrogens on unsaturated carbon atoms in "substituted" alkene, alkyne, aryl and heteroaryl groups are, unless otherwise specified, -R 60 , halo, -0 " M + , -OR 70 , -SR 70 , -S ⁇ M + , -NR 80 R 80 , trihalomethyl, -CF 3 , -CN, -OCN, -SCN, -NO, -N0 2 , -N 3 , -S0 2 RTM, -S0 3 ⁇ M + , -SO 3 RTM, -OSO 2 RTM, -OS0 3 M + , -OSO 3 R 70 , -P0 3 ⁇ 2 (M + ) 2 , -P(O)(OR 70 )O ⁇ M + , -P(O)(OR 70 ) 2 ,
- -OC(S)OR 70 -NR 70 C(O)R 70 , -NR 70 C(S)R 70 , -NR 70 CO 2 M + , -NR 70 CO 2 R 70 , -NR 70 C(S)OR 70 , -NR 70 C(O)NR 80 R 80 , -NR 70 C(NR 70 )R 70 and -NR 70 C(NR 70 )NR 80 R 80 , where R 60 , R 70 , R 80 and M + are as previously defined, provided that in case of substituted alkene or alkyne, the substituents are not -0 " M + , -OR 70 , -SR 70 , or -STVT ⁇
- substituent groups for hydrogens on nitrogen atoms in "substituted" heteroalkyl and cycloheteroalkyl groups are, unless otherwise specified, -R 60 , -0 " M + , -OR 70 , -SR 70 , -S " M + , -NR 80 R 80 , trihalomethyl, -CF 3 , -CN, -NO, -N0 2 , -S(0) 2 R 70 , -S(0) 2 0 " M + , -S(0) 2 OR 70 , -OS(0) 2 R 70 , -OS(0) 2 0 " M + , -OS(0) 2 OR 70 ,
- -OC(0)OR 70 -OC(S)OR 70 , -NR 70 C(O)R 70 , -NR 70 C(S)R 70 , -NR 70 C(O)OR 70 , -NR 70 C(S)OR 70 , -NR 70 C(O)NR 80 R 80 , -NR 70 C(NR 70 )R 70 and -NR 70 C(NR 70 )NR 80 R 80 , where R 60 , R 70 , R 80 and M + are as previously defined.
- a group that is substituted has 1, 2, 3, or 4 substituents, 1, 2, or 3 substituents, 1 or 2 substituents, or 1 substituent.
- any of the groups disclosed herein which contain one or more substituents it is understood, of course, that such groups do not contain any substitution or substitution patterns which are sterically impractical and/or synthetically non-feasible.
- the subject compounds include all stereochemical isomers arising from the substitution of these compounds.
- pharmaceutically acceptable salt means a salt which is acceptable for administration to a patient, such as a mammal (e.g., salts having acceptable mammalian safety for a given dosage regime).
- Such salts can be derived from pharmaceutically acceptable inorganic or organic bases and from pharmaceutically acceptable inorganic or organic acids.
- “Pharmaceutically acceptable salt” refers to pharmaceutically acceptable salts of a compound, which salts are derived from a variety of organic and inorganic counter ions well known in the art and include, by way of example only, sodium, potassium, calcium, magnesium, ammonium, tetraalkylammonium, and the like; and when the molecule contains a basic functionality, salts of organic or inorganic acids, such as hydrochloride,
- hydrobromide formate, tartrate, besylate, mesylate, acetate, maleate, oxalate, and the like.
- salt thereof means a compound formed when the hydrogen of an acid is replaced by a cation, such as a metal cation or an organic cation and the like. Where applicable, the salt is a pharmaceutically acceptable salt, although this is not required for salts of compounds that are not intended for administration to a patient.
- salts of the present compounds include those wherein the compound is protonated by an inorganic or organic acid to form a cation, with the conjugate base of the inorganic or organic acid as the anionic component of the salt.
- solvent refers to a complex formed by combination of solvent molecules with molecules or ions of the solute.
- the solvent can be an organic compound, an inorganic compound, or a mixture of both.
- Some examples of solvents include, but are not limited to, methanol, ⁇ , ⁇ -dimethylformamide, tetrahydrofuran, dimethylsulfoxide, and water. When the solvent is water, the solvate formed is a hydrate.
- Stereoisomers refer to compounds that have same atomic connectivity but different atomic arrangement in space. Stereoisomers include cis-trans isomers, E and Z isomers, enantiomers, and diastereomers.
- pyrazoles imidazoles, benzimidazoles, triazoles, and tetrazoles.
- “Pharmaceutically effective amount” and “therapeutically effective amount” refer to an amount of a compound sufficient to treat a specified disorder or disease or one or more of its symptoms and/or to prevent the occurrence of the disease or disorder.
- a pharmaceutically or therapeutically effective amount comprises an amount sufficient to, among other things, cause the tumor to shrink or decrease the growth rate of the tumor.
- Patient refers to human and non-human animals, especially mammals.
- treating means the treating or treatment of a disease or medical condition in a patient, such as a mammal (particularly a human) that includes: (a) preventing the disease or medical condition from occurring, i.e., prophylactic treatment of a patient; (b) ameliorating the disease or medical condition, i.e., eliminating or causing regression of the disease or medical condition in a patient; (c) suppressing the disease or medical condition, i.e., slowing or arresting the development of the disease or medical condition in a patient; or (d) alleviating the symptoms of the disease or medical condition in a patient.
- compositions of the present disclosure include compounds of formula I, shown below.
- Pharmaceutical compositions and methods of the present disclosure also contemplate compounds of formula I.
- the present embodiments provide a compound of formula (I):
- R is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
- Y 1 and Y 2 are independently selected from hydrogen, alkyl, and acyl;
- R 1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
- R a and R b are independently selected from hydrogen and alkyl
- R c and R d are independently selected from hydrogen and alkyl
- Q is selected from N and CR 7b ;
- R 6a , R 6b , R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O- alk-A;
- alk is a bond, alkylene or substituted alkylene
- A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
- a ring can be substituted or unsubstituted
- R 7y is selected from hydrogen, alkyl, cycloalkyl, and substituted alkyl
- R 5 can be selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl.
- R 5 is cyano, halogen, acyl, aminoacyl, or nitro.
- R 5 is halogen.
- R 5 is fluoro.
- R 5 is cyano.
- R 5 is fluoro, cyano, or aminoacyl.
- R 5 is cyano or aminoacyl.
- Y 1 and Y 2 can be independently selected from hydrogen, alkyl, and acyl.
- Y 1 is hydrogen.
- Y 1 is alkyl.
- Y 1 is acyl.
- Y 2 is hydrogen.
- Y 2 is alkyl.
- Y 2 is acyl.
- R 1 can be selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical. In certain instances, R 1 is hydrogen or alkyl. In certain instances, R 1 is hydrogen. In certain instances, R 1 is alkyl. In certain instances, R 1 is methyl. In certain instances, R 1 is hydrogen, alkyl, substituted alkyl, or oxy radical. In certain instances, R 1 is hydrogen, alkyl, substituted alkyl, acyl, or cycloalkyl.
- R a and R b can be independently selected from hydrogen and alkyl. In certain instances, R a and R b are both alkyl. In certain instances, R a and R b are both methyl. In certain instances, at least one of R a and R b is alkyl.
- R c and R d can be independently selected from hydrogen and alkyl. In certain instances, R c and R d are both alkyl. In certain instances, R c and R d are both methyl. In certain instances, at least one of R c and R d is alkyl.
- Q can be selected from N and CR 7b . In certain instances, Q is CR 7b . In certain instances, Q is N.
- R 6a , R 6b , R 7b and R 8 can be independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A.
- R 6a , R 6b , R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, cycloalkyl, substituted cycloalkyl, and - O-alk-A.
- R 6a , R 6b , R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, acyl, carboxyl, carboxyl ester, aminoacyl, and -O-alk-A.
- at least one of R 6a , R 6b , R 7b and R 8 is alkyl or substituted alkyl.
- at least one of R 6a , R 6b , R 7b and R 8 is halogen.
- At least one of R 6a , R 6b , R 7b and R 8 is alkoxy, substituted alkoxy, or -O-alk-A. In certain instances, at least one of R 6a , R 6b , R 7b and R 8 is cyano, acyl, carboxyl, carboxyl ester, or aminoacyl. In certain instances, at least one of R 6a , R 6b , R 7b and R 8 is cycloalkyl or substituted cycloalkyl.
- R 6a , R 6b , R 7b and R 8 are independently selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
- one of R 6a and R 6b is fluoro and the other is hydrogen.
- R 6a is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain instances, R 6a is selected from hydrogen, alkyl, substituted alkyl, and halogen. In certain instances, R 6a is hydrogen. In certain instances, R 6a is selected from alkyl and substituted alkyl. In certain instances, R 6a is halogen. In certain instances, R 6a is fluoro.
- R 6b is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain instances, R 6b is selected from hydrogen, alkyl, substituted alkyl, and halogen. In certain instances, R 6b is hydrogen. In certain instances, R 6b is selected from alkyl and substituted alkyl. In certain instances, R 6b is halogen. In certain instances, R 6b is fluoro.
- R 7b is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain instances, R is selected from hydrogen, alkyl, and substituted alkyl. In certain instances, R 7b is hydrogen. In certain instances, R 7b is selected from alkyl and substituted alkyl.
- R 7b is selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, acyl, carboxyl, carboxyl ester, aminoacyl, and -O-alk-A.
- R 8 is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl.
- R 8 is hydrogen.
- R 8 is alkyl or substituted alkyl.
- R 8 is methyl.
- R 8 is halogen.
- R 8 is fluoro.
- R 8 is alkoxy or substituted alkoxy.
- R 8 is fluoro.
- R 8 is cycloalkyl or substituted cycloalkyl.
- R 8 is cyclopropyl.
- any of R 7b or R 8 is selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, acyl, carboxyl, carboxyl ester, aminoacyl, and -O-alk-A.
- at least one of R 7b or R 8 is cycloalkyl, or substituted cycloalkyl.
- at least one of R 7b or R 8 is
- at least one of R' u or R° is alkyl, substituted alkyl, or halogen.
- alk is a bond, alkylene or substituted alkylene. In certain instances, alk is a bond. In certain instances, alk is alkylene. In certain instances, alk is ethylene or propylene. In certain instances, alk is substituted alkylene. In certain instances, alk is substituted ethylene or substituted propylene.
- alk is a bond or alk is ethylene, substituted ethylene, propylene, or -0( ⁇ 3 ⁇ 4) 2 ⁇ 3 ⁇ 4-.
- alk is a bond or alk is substituted propylene, -C(CH 3 ) 2 CH 2 CH 2 -, or
- A can be selected from aryl, cycloalkyl, heteroaryl, and
- heterocyclyl wherein the A ring can be substituted or unsubstituted.
- A is aryl or substituted aryl.
- A is cycloalkyl or substituted cycloalkyl.
- A is heteroaryl or substituted heteroaryl.
- A is heterocyclyl or substituted heterocyclyl.
- A is heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl.
- A is selected from azetidine, imidazole, pyrazole, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, dihydroindole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, pteridine, carbazole, carboline, isothiazole, phenazine, isoxazole, imidazolidine, imidazoline, oxazole, oxazolidine, piperidine, piperazine, indoline, phthalimide, 1,2,3,4-tetrahydroisoquinoline, tetrazole, triazole, thiazole, thiazolidine, thiophene, thiomorpholinyl, 1,1-dioxo
- A is selected from 1-triazole, 3-pyrrolidine, 4- piperidine, and 1-imidazolidine; wherein the A ring can be substituted or unsubstituted.
- A is selected from piperidine,
- tetrahydropyranyl tetrahydrothiopyranyl, azetidinyl, azepanyl, and furanyl; wherein the A ring can be substituted or unsubstituted.
- R 7y is selected from hydrogen, alkyl, cycloalkyl, and substituted alkyl. In certain instances, R 7y is hydrogen. In certain instances, R 7y is alkyl. In certain instances, R 7y is methyl. In certain instances, R 7y is isopropyl. In certain instances, R 7y is cycloalkyl. In certain instances, R 7y is substituted alkyl.
- R 5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
- Y 1 and Y 2 are independently selected from hydrogen, alkyl, and acyl;
- R 1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
- R a and R b are independently selected from hydrogen and alkyl
- R c and R d are independently selected from hydrogen and alkyl
- R a , R , R and R are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O- alk-A;
- alk is a bond, alkylene or substituted alkylene
- A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
- a ring can be substituted or unsubstituted
- R 5 can be selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl.
- R 5 is cyano, halogen, acyl, aminoacyl, or nitro.
- R 5 is halogen.
- R 5 is fluoro.
- R 5 is fluoro, cyano, or aminoacyl.
- R 5 is cyano or aminoacyl.
- Y 1 and Y 2 can be independently selected from hydrogen, alkyl, and acyl.
- Y 1 is hydrogen.
- Y 1 is alkyl.
- Y 1 is acyl.
- Y 2 is hydrogen.
- Y 2 is alkyl.
- Y 2 is acyl.
- R 1 can be selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical. In certain instances, R 1 is hydrogen or alkyl. In certain instances, R 1 is hydrogen. In certain instances, R 1 is alkyl. In certain instances, R 1 is methyl. In certain instances, R 1 is hydrogen, alkyl, substituted alkyl, or oxy radical. In certain instances, R 1 is hydrogen, alkyl, substituted alkyl, acyl, or cycloalkyl.
- R a and R b can be independently selected from hydrogen and alkyl. In certain instances, R a and R b are both alkyl. In certain instances, R a and R b are both methyl. In certain instances, at least one of R a and R b is alkyl.
- R c and R d can be independently selected from hydrogen and alkyl. In certain instances, R c and R d are both alkyl. In certain instances, R c and R d are both methyl. In certain instances, at least one of R c and R d is alkyl. [00130] In formula II, Q can be selected from N and CR . In certain instances, Q is CR 7b . In certain instances, Q is N.
- R 7b and R 8 can be independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino,
- aminocarbonylamino acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A.
- R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, and -O-alk-A.
- R 7b and R 8 are independently selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
- R 7b is selected from hydrogen, alkyl, and substituted alkyl. In certain instances, R 7b is hydrogen. In certain instances, R 7b is selected from alkyl and substituted alkyl.
- R 8 is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, and substituted alkoxy. In certain instances, R 8 is hydrogen. In certain instances, R 8 is alkyl or substituted alkyl. In certain instances, R 8 is methyl. In certain instances, R 8 is halogen. In certain instances, R 8 is fluoro. In certain instances, R 8 is alkoxy or substituted alkoxy.
- alk can be present or not present and is alkyl or substituted alkyl. In certain instances, alk is not present. In certain instances, alk is present and is alkyl. In certain instances, alk is present and is ethylene or propylene. In certain instances, alk is present and is substituted alkyl. In certain instances, alk is present and is substituted ethylene or substituted propylene.
- alk is not present or alk is present and is ethylene, substituted ethylene, propylene, or -C(CH 3 ) 2 CH 2 -.
- alk is not present or alk is present and is substituted propylene, -C(CH 3 ) 2 CH 2 CH 2 -, or -C(CH 3 ) 2 CH 2 C(0)-.
- A can be selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein the A ring can be substituted or unsubstituted.
- A is aryl or substituted aryl.
- A is cycloalkyl or substituted cycloalkyl.
- A is heteroaryl or substituted heteroaryl.
- A is heterocyclyl or substituted heterocyclyl.
- A is heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl.
- A is selected from azetidine, imidazole, pyrazole, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, dihydroindole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, pteridine, carbazole, carboline, isothiazole, phenazine, isoxazole, imidazolidine, imidazoline, oxazole, oxazolidine, piperidine, piperazine, indoline, phthalimide, 1,2,3,4-tetrahydroisoquinoline, tetrazole, triazole, thiazole, thiazolidine, thiophene, thiomorpholinyl, 1,1-dioxo
- A is selected from 1-triazole, 3-pyrrolidine, 4- piperidine, and 1 -imidazolidine; wherein the A ring can be substituted or unsubstituted.
- A is selected from piperidine,
- tetrahydropyranyl tetrahydrothiopyranyl, azetidinyl, azepanyl, and furanyl; wherein the A ring can be substituted or unsubstituted.
- the present embodiments provide a compound of formula (III):
- Y 1 and Y 2 are independently selected from hydrogen, alkyl, and acyl;
- R 1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
- R a and R b are independently selected from hydrogen and alkyl
- R c and R d are independently selected from hydrogen and alkyl
- Q is selected from N and CR 7b ;
- R 6a , R 6b , R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O- alk-A;
- alk is a bond, alkylene or substituted alkylene
- A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
- a ring can be substituted or unsubstituted
- R 7y is selected from hydrogen, alkyl, and substituted alkyl
- Y 1 and Y 2 can be independently selected from hydrogen, alkyl, and acyl.
- Y 1 is hydrogen.
- Y 1 is alkyl.
- Y 1 is acyl.
- Y 2 is hydrogen.
- Y 2 is alkyl.
- Y 2 is acyl.
- R 1 can be selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical. In certain instances, R 1 is hydrogen or alkyl. In certain instances, R 1 is hydrogen. In certain instances, R 1 is alkyl. In certain instances, R 1 is methyl. In certain instances, R 1 is hydrogen, alkyl, substituted alkyl, or oxy radical. In certain instances, R 1 is hydrogen, alkyl, substituted alkyl, acyl, or cycloalkyl.
- R a and R b can be independently selected from hydrogen and alkyl. In certain instances, R a and R b are both alkyl. In certain instances, R a and R b are both methyl. In certain instances, at least one of R a and R b is alkyl.
- R c and R d can be independently selected from hydrogen and alkyl. In certain instances, R c and R d are both alkyl. In certain instances, R c and R d are both methyl. In certain instances, at least one of R c and R d is alkyl.
- Q can be selected from N and CR 7b . In certain instances, Q is CR 7b . In certain instances, Q is N.
- R 7b and R 8 can be independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A.
- R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, and -O-alk-A.
- R 7b and R 8 are independently selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
- R 7b is selected from hydrogen, alkyl, and substituted alkyl. In certain instances, R 7b is hydrogen. In certain instances, R 7b is selected from alkyl and substituted alkyl.
- R 8 is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, and substituted alkoxy. In certain instances, R 8 is hydrogen. In certain instances, R 8 is alkyl or substituted alkyl. In certain instances, R 8 is methyl. In certain instances, R 8 is halogen. In certain instances, R 8 is fluoro. In certain instances, R 8 is alkoxy or substituted alkoxy.
- alk can be present or not present and is alkyl or substituted alkyl. In certain instances, alk is not present. In certain instances, alk is present and is alkyl. In certain instances, alk is present and is ethylene or propylene. In certain instances, alk is present and is substituted alkyl. In certain instances, alk is present and is substituted ethylene or substituted propylene.
- alk is not present or alk is present and is ethylene, substituted ethylene, propylene, or -0( ⁇ 3 ⁇ 4) 2 ⁇ 3 ⁇ 4-.
- formula I for "-O-alk-A,” alk is not present or alk is present and is substituted propylene, -C(CH 3 ) 2 CH 2 CH 2 -, or -C(CH 3 ) 2 CH 2 C(0)-.
- A can be selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein the A ring can be substituted or unsubstituted.
- A is aryl or substituted aryl.
- A is cycloalkyl or substituted cycloalkyl.
- A is heteroaryl or substituted heteroaryl.
- A is heterocyclyl or substituted heterocyclyl.
- A is heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl.
- A is selected from azetidine, imidazole, pyrazole, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, dihydroindole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, pteridine, carbazole, carboline, isothiazole, phenazine, isoxazole, imidazolidine, imidazoline, oxazole, oxazolidine, piperidine, piperazine, indoline, phthalimide, 1,2,3,4-tetrahydroisoquinoline, tetrazole, triazole, thiazole, thiazolidine, thiophene, thiomorpholinyl, 1,1-dioxo
- A is selected from 1-triazole, 3-pyrrolidine, 4- piperidine, and 1 -imidazolidine; wherein the A ring can be substituted or unsubstituted.
- A is selected from piperidine,
- tetrahydropyranyl tetrahydrothiopyranyl, azetidinyl, azepanyl, and furanyl; wherein the A ring can be substituted or unsubstituted.
- R 7y is selected from hydrogen, alkyl, and substituted alkyl. In certain instances, R 7y is hydrogen. In certain instances, R 7y is alkyl. In certain instances, R 7y is methyl. In certain instances, R 7y is isopropyl. In certain instances, R 7y is substituted alkyl.
- R 5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
- Y 1 and Y 2 are independently selected from hydrogen, alkyl, and acyl;
- R 1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
- R a and R b are independently selected from hydrogen and alkyl
- R c and R d are independently selected from hydrogen and alkyl
- Q is selected from N and CR 7b ;
- R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A;
- alk is a bond, alkylene or substituted alkylene
- A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
- a ring can be substituted or unsubstituted
- R 7y is selected from hydrogen, alkyl, and substituted alkyl
- R 5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
- Y 1 and Y 2 are independently selected from hydrogen, alkyl, and acyl;
- R 1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
- R a and R b are independently selected from hydrogen and alkyl
- R c and R d are independently selected from hydrogen and alkyl
- Q is selected from N and CR 7b ;
- R 6a , R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A;
- alk is a bond, alkylene or substituted alkylene
- A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein the A ring can be substituted or unsubstituted;
- R 7y is selected from hydrogen, alkyl, and substituted alkyl
- Q can be selected from N and CR 7b . In certain instances, Q is CR 7b . In certain instances, Q is N.
- R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, and -O-alk-A.
- R 7b and R 8 are independently selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
- R 7b is selected from hydrogen, alkyl, and substituted alkyl. In certain instances, R 7b is hydrogen. In certain instances, R 7b is selected from alkyl and substituted alkyl.
- R 8 is selected from cycloalkyl, substituted cycloalkyl, heterocyclyl, substituted heterocyclyl, and -O-alk-A. In certain instances, R 8 is cycloalkyl or substituted cycloalkyl. In certain instances, R 8 is cycloalkyl. In certain instances, R 8 is substituted cycloalkyl. In certain instances, R 8 is heterocyclyl or substituted heterocyclyl. In certain instances, R 8 is heterocyclyl. In certain instances, R 8 is substituted heterocyclyl. In certain instances, R 8 is -O-alk-A.
- R 8 is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, and substituted alkoxy. In certain instances, R 8 is hydrogen. In certain instances, R 8 is alkyl or substituted alkyl. In certain instances, R 8 is methyl. In certain instances, R 8 is halogen. In certain instances, R 8 is fluoro. In certain instances, R 8 is alkoxy or substituted alkoxy.
- alk can be present or not present and is alkyl or substituted alkyl. In certain instances, alk is not present. In certain instances, alk is present and is alkyl. In certain instances, alk is present and is ethylene or propylene. In certain instances, alk is present and is substituted alkyl. In certain instances, alk is present and is substituted ethylene or substituted propylene.
- alk is not present or alk is present and is ethylene, substituted ethylene, propylene, or -C(CH 3 ) 2 CH 2 -.
- formula I for "-O-alk-A,” alk is not present or alk is present and is substituted propylene, -C(CH 3 ) 2 CH 2 CH 2 -, or -C(CH 3 ) 2 CH 2 C(0)-.
- A can be selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein the A ring can be substituted or unsubstituted.
- A is aryl or substituted aryl.
- A is cycloalkyl or substituted cycloalkyl.
- A is heteroaryl or substituted heteroaryl.
- A is heterocyclyl or substituted heterocyclyl.
- A is heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl.
- R 5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
- Y 1 and Y 2 are independently selected from hydrogen, alkyl, and acyl;
- R 1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
- R a and R b are independently selected from hydrogen and alkyl
- R c and R d are independently selected from hydrogen and alkyl
- Q is selected from N and CR 7b ;
- R 6a , R 6b , and R 7b are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A;
- alk is a bond, alkylene or substituted alkylene
- A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein the A ring can be substituted or unsubstituted;
- R 7y is selected from hydrogen, alkyl, and substituted alkyl
- At least one of R 6a and R 6b is selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
- one of R 6a and R 6b is fluoro and the other is hydrogen.
- R 6a is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain instances, R 6a is selected from hydrogen, alkyl, substituted alkyl, and halogen. In certain instances, R 6a is hydrogen. In certain instances, R 6a is selected from alkyl and substituted alkyl. In certain instances, R 6a is halogen. In certain instances, R 6a is fluoro.
- R 6b is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain instances, R 6b is selected from hydrogen, alkyl, substituted alkyl, and halogen. In certain instances, R 6b is hydrogen. In certain instances, R 6b is selected from alkyl and substituted alkyl. In certain instances, R 6b is halogen. In certain instances, R 6b is fluoro.
- alk is a bond, alkylene or substituted alkylene. In certain instances, alk is a bond. In certain instances, alk is alkylene. In certain instances, alk is ethylene or propylene. In certain instances, alk is substituted alkylene. In certain instances, alk is substituted ethylene or substituted propylene.
- alk is a bond or alk is ethylene, substituted ethylene, propylene, or -0( ⁇ 3 ⁇ 4) 2 ⁇ 3 ⁇ 4-.
- alk is a bond or alk is substituted propylene, -C(CH 3 ) 2 CH 2 CH 2 -, or -C(CH 3 ) 2 CH 2 C(0)-.
- A can be selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein the A ring can be substituted or unsubstituted.
- A is aryl or substituted aryl.
- A is cycloalkyl or substituted cycloalkyl.
- A is heteroaryl or substituted heteroaryl.
- A is heterocyclyl or substituted heterocyclyl.
- A is heterocyclyl.
- A is substituted heterocyclyl.
- A is heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl.
- the present embodiments provide a compound of formula (VII):
- R is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
- Y 1 and Y 2 are independently selected from hydrogen, alkyl, and acyl;
- R 1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
- R a and R b are independently selected from hydrogen and alkyl
- R c and R d are independently selected from hydrogen and alkyl
- Q is selected from N and CR 7b ;
- R 6a , R 6b , and R 7b are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A;
- alk is a bond, alkylene or substituted alkylene
- A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
- a ring can be substituted or unsubstituted
- R 7y is selected from hydrogen, alkyl, and substituted alkyl
- At least one of R a and R is selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
- R 6a and R 6b are fluoro and the other is hydrogen.
- R a is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl.
- R 6a is selected from hydrogen, alkyl, substituted alkyl, and halogen.
- R 6a is hydrogen.
- R 6a is selected from alkyl and substituted alkyl.
- R 6a is halogen.
- R 6a is fluoro.
- R 6b is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain instances, R 6b is selected from hydrogen, alkyl, substituted alkyl, and halogen. In certain instances, R 6b is hydrogen. In certain instances, R 6b is selected from alkyl and substituted alkyl. In certain instances, R 6b is halogen. In certain instances, R 6b is fluoro.
- R 5 can be selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl.
- R 5 is cyano, halogen, acyl, aminoacyl, or nitro.
- R 5 is halogen.
- R 5 is fluoro.
- R 5 is fluoro, cyano, or aminoacyl.
- R 5 is cyano or aminoacyl.
- Y 1 and Y 2 can be independently selected from hydrogen, alkyl, and acyl.
- Y 1 is hydrogen.
- Y 1 is alkyl.
- Y 1 is acyl.
- Y 2 is hydrogen.
- Y 2 is alkyl.
- Y 2 is acyl.
- R 1 can be selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical.
- R 1 is hydrogen or alkyl.
- R 1 is hydrogen.
- R 1 is alkyl.
- R 1 is methyl.
- R 1 is hydrogen, alkyl, substituted alkyl, or oxy radical.
- R 1 is hydrogen, alkyl, substituted alkyl, acyl, or cycloalkyl.
- R a and R b can be independently selected from hydrogen and alkyl. In certain instances, R a and R b are both alkyl. In certain instances, R a and R b are both methyl. In certain instances, at least one of R a and R b is alkyl.
- R c and R d can be independently selected from hydrogen and alkyl. In certain instances, R c and R d are both alkyl. In certain instances, R c and R d are both methyl. In certain instances, at least one of R c and R d is alkyl.
- Q can be selected from N and CR 7b . In certain instances, Q is CR 7b . In certain instances, Q is N.
- R 5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
- Y is selected from hydrogen and alkyl
- R 1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
- R a and R b are independently selected from hydrogen and alkyl
- R c and R d are independently selected from hydrogen and alkyl
- Q is selected from N and CR 7b ;
- R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteraryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A;
- alk if present, is alkyl or substituted alkyl
- A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
- a ring can be substituted or unsubstituted
- R 7y is selected from hydrogen, alkyl, and substituted alkyl
- R 5 can be selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl.
- R 5 is cyano, halogen, acyl, aminoacyl, or nitro.
- R 5 is halogen.
- R 5 is fluoro.
- R 5 is fluoro, cyano, or aminoacyl.
- R 5 is cyano or aminoacyl.
- Y can be selected from hydrogen and alkyl. In certain instances, Y is hydrogen. In certain instances, Y is alkyl.
- R 1 can be selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical. In certain instances, R 1 is hydrogen or alkyl. In certain instances, R 1 is hydrogen. In certain instances, R 1 is alkyl. In certain instances, R 1 is methyl. In certain instances, R 1 is hydrogen, alkyl, substituted alkyl, or oxy radical. In certain instances, R 1 is hydrogen, alkyl, substituted alkyl, acyl, or cycloalkyl.
- R a and R b can be independently selected from hydrogen and alkyl. In certain instances, R a and R b are both alkyl. In certain instances, R a and R b are both methyl. In certain instances, at least one of R a and R b is alkyl.
- R c and R d can be independently selected from hydrogen and alkyl. In certain instances, R c and R d are both alkyl. In certain instances, R c and R d are both methyl. In certain instances, at least one of R c and R d is alkyl.
- Q can be selected from N and CR 7b . In certain instances, Q is CR 7b . In certain instances, Q is N.
- R 7b and R 8 can be independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino,
- aminocarbonylamino acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A.
- R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, and -O-alk-A.
- R 7b and R 8 are independently selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
- R 7b is selected from hydrogen, alkyl, and substituted alkyl. In certain instances, R 7b is hydrogen. In certain instances, R 7b is selected from alkyl and substituted alkyl. [00200] In certain instances, R is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, and substituted alkoxy. In certain instances, R 8 is hydrogen. In certain instances, R 8 is alkyl or substituted alkyl. In certain instances, R 8 is methyl. In certain instances, R 8 is halogen. In certain instances, R 8 is fluoro. In certain instances, R 8 is alkoxy or substituted alkoxy.
- alk can be present or not present and is alkyl or substituted alkyl. In certain instances, alk is not present. In certain instances, alk is present and is alkyl. In certain instances, alk is present and is ethylene or propylene. In certain instances, alk is present and is substituted alkyl. In certain instances, alk is present and is substituted ethylene or substituted propylene.
- alk is not present or alk is present and is ethylene, substituted ethylene, propylene, or -0( ⁇ 3 ⁇ 4) 2 ⁇ 3 ⁇ 4-.
- formula I for "-O-alk-A,” alk is not present or alk is present and is substituted propylene, -C(CH 3 ) 2 CH 2 CH 2 -, or -C(CH 3 ) 2 CH 2 C(0)-.
- A can be selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein the A ring can be substituted or unsubstituted.
- A is aryl or substituted aryl.
- A is cycloalkyl or substituted cycloalkyl.
- A is heteroaryl or substituted heteroaryl.
- A is heterocyclyl or substituted heterocyclyl.
- A is heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl.
- A is selected from azetidine, imidazole, pyrazole, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, dihydroindole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, pteridine, carbazole, carboline, isothiazole, phenazine, isoxazole, imidazolidine, imidazoline, oxazole, oxazolidine, piperidine, piperazine, indoline, phthalimide, 1,2,3,4-tetrahydroisoquinoline, tetrazole, triazole, thiazole, thiazolidine, thiophene, thiomorpholinyl, 1,1-dioxo
- A is selected from 1-triazole, 3-pyrrolidine, 4-piperidine, and 1 -imidazolidine; wherein the A ring can be substituted or unsubstituted.
- A is selected from piperidine,
- R y is selected from hydrogen, alkyl, and substituted alkyl.
- R 7y is hydrogen.
- R 7y is alkyl.
- R 7y is methyl.
- R 7y is isopropyl.
- R 7y is substituted alkyl.
- 1-34 l-(2-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one; 1-35: l-(3-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
- Particular compounds of interest, and salts or solvates or stereoisomers thereof, include:
- Particular compounds of interest, and salts or solvates or stereoisomers thereof, include:
- Particular compounds of interest, and salts or solvates or stereoisomers thereof include: 1-70: l-(4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-((lS,2S)-2-(trifluoromethyl)cyclopropyl)phenyl)-4-methyl-lH-tetrazol-5(4H)- one;
- the compounds described also include isotopically labeled compounds where one or more atoms have an atomic mass different from the atomic mass conventionally found in nature.
- isotopes that may be incorporated into the compounds disclosed herein include, but are not limited to, 2 H, 3 H, n C, 13 C, 14 C, 15 N, 18 0, 17 0, etc.
- the disclosed compounds may be enriched in one or more of these isotopes relative to the natural abundance of such isotope.
- deuterium ( 2 H) has a natural abundance of about 0.015%. Accordingly, for approximately every 6,500 hydrogen atoms occurring in nature, there is one deuterium atom.
- Specifically contemplated herein are compounds enriched in deuterium at one or more positions.
- deuterium containing compounds of the disclosure have deuterium at one or more positions (as the case may be) in an abundance of greater than 0.015%.
- compositions comprising a pharmaceutically acceptable carrier and a therapeutically effective amount of a compound of formula I- VIII or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
- a disclosed compound can be administered alone, as the sole active
- the therapeutic agents can be formulated as separate compositions that are administered simultaneously or at different times, or the therapeutic agents can be administered together as a single composition combining two or more therapeutic agents.
- compositions disclosed herein containing a compound of formula I- VIII optionally contain other therapeutic agents. Accordingly, certain embodiments are directed to such pharmaceutical compositions, wherein the composition further comprises a therapeutically effective amount of an agent selected as is known to those of skill in the art.
- the subject compounds can inhibit a protein kinase C activity. Accordingly, the compounds are useful for treating a disease or disorder that is mediated through the activity of a PKC activity in a subject. Also, the compounds are useful for treating a disease or disorder that is associated with the activation of T-cells in a subject.
- the present disclosure provides a method of treating an inflammatory disease in a subject, the method comprising administering to the subject with a compound of formula I- VIII or a salt or solvate or stereoisomer thereof.
- the present disclosure also provides a method of treating an autoimmune disease in a subject, the method comprising administering to the subject with a compound of formula I- VIII or a salt or solvate or stereoisomer thereof.
- the present disclosure also provides a method of treating an ocular disease or disorder involving inflammatory and/or neovascular events.
- the present disclosure also provides a method of treating diseases or conditions of interest including, but are not limited to, atherosclerosis, vascular occlusion due to vascular injury, angioplasty, restenosis, obesity, syndrome X, impaired glucose tolerance, polycystic ovary syndrome, hypertension, heart failure, chronic obstructive pulmonary disease, CNS diseases, Alzheimer disease, amyotrophic lateral sclerosis, bipolar disease, cancer, infectious disease, AIDS, septic shock, adult respiratory distress syndrome, ischemia/reperfusion injury, myocardial infarction, stroke, gut ischemia, renal failure, hemorrhage shock, and traumatic shock, and traumatic brain injury.
- diseases or conditions of interest including, but are not limited to, atherosclerosis, vascular occlusion due to vascular injury, angioplasty, restenosis, obesity, syndrome X, impaired glucose tolerance, polycystic ovary syndrome, hypertension, heart failure, chronic obstructive pulmonary disease, CNS diseases, Alzheimer disease,
- the present disclosure also provides a method of treating diseases or conditions of interest including, but are not limited to, T-cell mediated acute or chronic inflammatory diseases or disorders or autoimmune diseases, rheumatoid arthritis, osteoarthritis, systemic lupus erythematosus, Hashimoto's thyroiditis, multiple sclerosis, myasthenia gravis, diabetes type I or II and the disorders associated therewith, transplant rejection, graft versus host disease, respiratory diseases, asthma, inflammatory lung injury, inflammatory liver injury, inflammatory glomerular injury, cutaneous manifestations of immunologically-mediated disorders or illnesses, inflammatory and hyperproliferative skin diseases, psoriasis, atopic dermatitis, allergic contact dermatitis, irritant contact dermatitis and further eczematous dermatitises, seborrhoeic dermatitis, inflammatory eye diseases, Sjoegren's syndrome, keratoconjunctivitis, uveitis, inflammatory T-cell
- the subject compounds can be used for treating a cell proliferative disorder.
- the present disclosure also provides a method of treating diseases or conditions of interest including, but are not limited to hematopoietic neoplasm, lymphoid neoplasm, T cell neoplasm, T lymphoblastic leukemia, B cell neoplasm, B-lymphoblastic leukemia, Burkitt's lymphoma, myeloid neoplasm, myeloproferative disease, chronic myelogenous leukemia (CML), myelodysplastic disease, chronic myelomonocytic leukemia, myelodysplastic syndrome, and acute myeloid leukemia.
- CML chronic myelogenous leukemia
- myelodysplastic disease chronic myelomonocytic leukemia
- myelodysplastic syndrome and acute myeloid leukemia.
- the disclosure also provides for a method for using a compound of formula I- VIII or a salt or solvate or stereoisomer thereof as a research tool for studying a biological system or sample, or for discovering new chemical compounds having PKC inhibitory properties.
- the embodiments are also directed to processes and novel intermediates useful for preparing compounds of formula I- VIII or a salt or solvate or stereoisomer thereof.
- the above process further comprises the step of forming a salt of a compound of formula I- VIII.
- Embodiments are directed to the other processes described herein; and to the product prepared by any of the processes described herein.
- the embodiments are also directed to a compound of formula I- VIII or a salt or solvate or stereoisomer thereof, for use in therapy or as a medicament.
- the embodiments are directed to the use of a compound of formula I- VIII or a salt or solvate or stereoisomer thereof, for the manufacture of a medicament; especially for the manufacture of a medicament for the inhibition of protein kinase C (PKC) activity.
- PKC protein kinase C
- the embodiments are also directed to the use of a compound of formula I- VIII or a salt or solvate or stereoisomer thereof for the manufacture of a medicament for the treatment of a disease or disorder mediated or sustained through the activity of PKC activity.
- the embodiments are also directed to the use of a compound of formula I- VIII or a salt or solvate or stereoisomer thereof for the manufacture of a medicament for the treatment of a disease or disorder associated with the activation of T-cells.
- Diseases or conditions of interest include, but are not limited to, an inflammatory disease, an immunological disorder, an autoimmune disease, an ocular disease or disorder involving inflammatory and/or neovascular events, organ and bone marrow transplant rejection, acute or chronic inflammation, allergies, contact dermatitis, psoriasis, rheumatoid arthritis, multiple sclerosis, type I diabetes, type II diabetes, inflammatory bowel disease, Guillain-Barre syndrome, Crohn's disease, ulcerative colitis, graft versus host disease, and lupus erythematosus.
- the embodiments are also directed to the use of a compound of formula I- VIII or a salt or solvate or stereoisomer thereof for the manufacture of a medicament for the treatment of a cell proliferative disorder.
- Diseases or conditions of interest include, but are not limited to, hematopoietic neoplasm, lymphoid neoplasm, T cell neoplasm, T
- lymphoblastic leukemia B cell neoplasm, B -lymphoblastic leukemia, Burkitt's lymphoma, myeloid neoplasm, myeloproferative disease, chronic myelogenous leukemia (CML), myelodysplastic disease, chronic myelomonocytic leukemia, myelodysplastic syndrome, acute myeloid leukemia.
- CML chronic myelogenous leukemia
- myelodysplastic disease chronic myelomonocytic leukemia
- myelodysplastic syndrome acute myeloid leukemia.
- any suitable stationary phase can be used, including normal and reversed phases as well as ionic resins.
- Most typically the disclosed compounds are purified via silica gel and/or alumina chromatography. See, e.g., Introduction to Modern Liquid Chromatography, 2nd Edition, ed. L. R. Snyder and J. J. Kirkland, John Wiley and Sons, 1979; and Thin Layer
- the subject compounds can be synthesized via a variety of different synthetic routes using commercially available starting materials and/or starting materials prepared by conventional synthetic methods. Suitable exemplary methods that can be routinely adapted to synthesize the 2,4-pyrimidinediamine compounds and prodrugs of the invention are found in U.S. Patent No. 5,958,935, the disclosure of which is incorporated herein by reference. Specific examples describing the synthesis of numerous 2,4-pyrimidinediamine compounds and prodrugs, as well as intermediates therefore, are described in the U.S. publication No. US2004/0029902A1, the contents of which are incorporated herein by reference.
- Suitable exemplary methods that can be routinely used and/or adapted to synthesize active 2,4- pyrimidinediamine compounds can also be found in WO 03/063794, U.S. application Serial No. 10/631,029 filed July 29, 2003, WO2004/014382, U.S. publication No. 2005-0234049 Al, and WO005/016893, the disclosures of which are incorporated herein by reference. All of the compounds described herein (including prodrugs) can be prepared by routine adaptation of these methods.
- the compounds can be synthesized from substituted or unsubstituted uracils as illustrated in Scheme 1, below: Scheme 1
- uracil A-l is dihalogenated at the 2- and 4-positions using a standard dehydrating-halogenating agent such as POCI 3 (phosphorus oxychloride) (or other standard halogenating agent) under standard conditions to yield 2,4
- a standard dehydrating-halogenating agent such as POCI 3 (phosphorus oxychloride) (or other standard halogenating agent) under standard conditions to yield 2,4
- dichloropyrimidine A-2 Depending upon the substituents in pyrimidinediamine A-2, the chloride at the C4 position is more reactive towards nucleophiles than the chloride at the C2 position. This differential reactivity can be exploited by first reacting 2,4
- the C4 halide is more reactive towards nucleophiles, as illustrated in the scheme.
- the identity of the substituent may alter this reactivity.
- the substituent is trifluoromethyl, a 50:50 mixture of 4N-substituted-4-pyrimidineamine A-4 and the corresponding 2N- substituted-2-pyrimidineamine is obtained.
- the regioselectivity of the reaction can also be controlled by adjusting the solvent and other synthetic conditions (such as temperature), as is well-known in the art.
- nucleophilic aromatic substitution can be used to couple compounds with an electrophilic leaving group, such as halides or pseudohalides, and compounds with an amino group.
- a halogen substituent on Compound A-2 and the amino group on Compound A-3 can react.
- a halogen substituent on Compound A-4 and the amino group on Compound A-5 can react.
- Conditions for nucleophilic aromatic substitution include the compounds reacting in a polar aprotic solvent or polar protic solvent. Suitable solvents include alcohols (such as isopropanol, methanol, ethanol), formic acid, dimethylsulfoxide, dimethylformamide, dioxane, and tetrahydrofuran. The reaction can be run at room temperature or can be heated.
- a coupling reaction such as a Buchwald coupling reaction
- the Buchwald coupling reaction involves palladium-catalyzed synthesis of aryl amines.
- Starting materials are aryl halides or pseudohalides (for example, triflates) and primary or secondary amines.
- Such reaction can be performed using a variety of methods well known in the art and specific examples can be had by reference to the Examples hereunder described.
- Asymmetric 2N,4N-disubstituted-5-fluoro-2,4-pyrimidinediamine A- 10 can be obtained by reacting 2,4-dichloro-5-fluoropyrimidine A-8 with one equivalent of amine A-3 (to yield 2-chloro-N4-substituted-5-fluoro-4-pyrimidineamine A-9) followed by one or more equivalents of amine A-5.
- Asymmetric 2N,4N-disubstituted-5-cyano-2,4-pyrimidinediamine A- 15 can be obtained by reacting 2,4-dichloro-5-carbamoylpyrimidine A-12 with one equivalent of amine A-3 (to yield 2-chloro-N4-substituted-5-carbamoyl-4-pyrimidineamine A-13).
- the amide group of Compound A-13 is converted to a cyano group to yield Compound A-14, followed by reaction with one or more equivalents of amine A-5. Conversion of the amide group to the cyano group can be accomplished with dehydration, such as with use of Burgess reagent or trifluoroacetic anhydride.
- aniline A-5 may also be reacted with intermediate A-13, and the resultant N2,N4-disubstituted diaminopyrimidine-5-carbamoylpyrimidine can be dehydrated to yield the corresponding 5-cyano compound A-15.
- the uracil A-l, A-7, and A-ll starting materials can be purchased from commercial sources or prepared using standard techniques of organic chemistry.
- uracils that can be used as starting materials in the schemes disclosed herein include, by way of example and not limitation, uracil (Aldrich #13,078-8;
- Amines, such as A-3 and A-5 can be purchased from commercial sources or, alternatively, can be synthesized utilizing standard techniques. For example, suitable amines can be synthesized from nitro precursors using standard chemistry. See also Vogel, 1989, Practical Organic Chemistry, Addison Wesley Longman, Ltd. and John Wiley & Sons, Inc.
- Compound A-5 was reacted to form tetrazolone Compound A-17 by treatment with phosgene and azidotrimethylsilane. The reaction is general to any appropriate aminophenyl compound. Compound A-17 was reacted to reduce the nitro group to form Compound A-5.
- Compound A-5 can also be prepared according to the procedures provided by Satoh et al., Tetrahedron Lett, 1995, 36, 1749; Gupta et al. Tetrahedron Lett, 2004, 45, 4113; Su et al. Eur. J. Org. Chem., 2006, 2723; and Potewar et al., Tetrahedron Lett, 2007, 48, 172.
- substitution of the ring with substituents can be performed with standard chemistry.
- substitution of the ring with substituents can be performed with nucleophilic aromatic substitution.
- a halogen substituent can be replaced with another substituent with nucleophilic aromatic substitution.
- substitution of the ring with substituents can be performed with a metal catalyzed coupling reaction.
- a halogen substituent can be replaced with another substituent with utilization of a metal catalyst. Suitable metal catalyzed reactions to place appropriate substituents include Suzuki coupling, Stille coupling, and Buchwald coupling.
- the nitro group of Compound A-17 was converted to an amino group to produce Compound A-5.
- the conversion of the nitro group to an amino group can be accomplished by various methods.
- a suitable method for reduction of nitro group is catalytic
- hydrogenation which uses hydrogen and a catalyst, such as, but not limited to, palladium on carbon, platinum oxide, Raney nickel, and samarium diiodide.
- a catalyst such as, but not limited to, palladium on carbon, platinum oxide, Raney nickel, and samarium diiodide.
- Compound A- 16 can be purchased from commercial sources or prepared using standard techniques of organic chemistry.
- Compound A- 16 can be prepared from the corresponding amine with standard techniques of organic chemistry.
- Compound A-16 can be prepared from the corresponding dinitro compound in which one of the nitro groups is reduced to an amino group.
- Myriad textbook references teaching suitable synthetic methods are provided infra.
- Prodrugs as described herein can be prepared by routine modification of the above-described methods.
- such prodrugs can be prepared by reacting a suitably protected 2,4-pyrimidinediamine with a suitable progroup. Conditions for carrying out such reactions and for deprotecting the product to yield prodrugs as described herein are well-known.
- the disclosed compounds are useful, at least, for the inhibition of PKC activity and the treatment of a disease or disorder that is mediated through the activity of a PKC activity. Accordingly, pharmaceutical compositions comprising at least one disclosed compound are also described herein.
- a pharmaceutical composition comprising a subject compound may be administered to a patient alone, or in combination with other supplementary active agents.
- the pharmaceutical compositions may be manufactured using any of a variety of processes, including, without limitation, conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping, and lyophilizing.
- the pharmaceutical composition can take any of a variety of forms including, without limitation, a sterile solution, suspension, emulsion, lyophilisate, tablet, pill, pellet, capsule, powder, syrup, elixir or any other dosage form suitable for administration.
- a subject compound may be administered to the host using any convenient means capable of resulting in the desired reduction in disease condition or symptom.
- a subject compound can be incorporated into a variety of formulations for therapeutic administration. More particularly, a subject compound can be formulated into
- compositions by combination with appropriate pharmaceutically acceptable carriers or diluents, and may be formulated into preparations in solid, semi-solid, liquid or gaseous forms, such as tablets, capsules, powders, granules, ointments, solutions, suppositories, injections, inhalants and aerosols.
- Formulations for pharmaceutical compositions are well known in the art. For example, Remington's Pharmaceutical Sciences, by E. W. Martin, Mack Publishing Co., Easton, Pa., 19th Edition, 1995, describes exemplary formulations (and components thereof) suitable for pharmaceutical delivery of disclosed compounds.
- Pharmaceutical compositions comprising at least one of the subject compounds can be formulated for use in human or veterinary medicine. Particular formulations of a disclosed pharmaceutical composition may depend, for example, on the mode of administration and/or on the location of the infection to be treated.
- formulations include a pharmaceutically acceptable carrier in addition to at least one active ingredient, such as a subject compound.
- other medicinal or pharmaceutical agents for example, with similar, related or complementary effects on the affliction being treated can also be included as active ingredients in a pharmaceutical composition.
- compositions e.g., powder, pill, tablet, or capsule forms
- conventional non-toxic solid carriers can include, for example, pharmaceutical grades of mannitol, lactose, starch, or magnesium stearate.
- compositions to be administered can optionally contain minor amounts of non-toxic auxiliary substances (e.g., excipients), such as wetting or emulsifying agents, preservatives, and pH buffering agents and the like; for example, sodium acetate or sorbitan monolaurate.
- excipients include, nonionic solubilizers, such as cremophor, or proteins, such as human serum albumin or plasma preparations.
- Some examples of materials which can serve as pharmaceutically- acceptable carriers include: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as corn starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients, such as cocoa butter and suppository waxes; (9) oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol, and polyethylene glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14) buffering agents, such as magnesium hydrox
- compositions may be formulated as a
- Pharmaceutically acceptable salts are non-toxic salts of a free base form of a compound that possesses the desired pharmacological activity of the free base. These salts may be derived from inorganic or organic acids. Non-limiting examples of suitable inorganic acids are hydrochloric acid, nitric acid, hydrobromic acid, sulfuric acid, hydroiodic acid, and phosphoric acid.
- Non- limiting examples of suitable organic acids are acetic acid, propionic acid, glycolic acid, lactic acid, pyruvic acid, malonic acid, succinic acid, malic acid, maleic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, methyl sulfonic acid, salicylic acid, formic acid, trichloroacetic acid, trifluoroacetic acid, gluconic acid, asparagic acid, aspartic acid, benzenesulfonic acid, para- toluenesulfonic acid, naphthalenesulfonic acid, and the like.
- suitable pharmaceutically acceptable salts are found in Remington's Pharmaceutical Sciences, 17th Edition, Mack Publishing Company, Easton, Pa., 1985.
- a pharmaceutically acceptable salt may also serve to adjust the osmotic pressure of the composition.
- a subject compound can be used alone or in combination with appropriate additives to make tablets, powders, granules or capsules, for example, with conventional additives, such as lactose, mannitol, corn starch or potato starch; with binders, such as crystalline cellulose, cellulose derivatives, acacia, corn starch or gelatins; with disintegrators, such as corn starch, potato starch or sodium carboxymethylcellulose; with lubricants, such as talc or magnesium stearate; and if desired, with diluents, buffering agents, moistening agents, preservatives and flavoring agents.
- Such preparations can be used for oral administration.
- a subject compound can be formulated into preparations for injection by dissolving, suspending or emulsifying them in an aqueous or nonaqueous solvent, such as vegetable or other similar oils, synthetic aliphatic acid glycerides, esters of higher aliphatic acids or propylene glycol; and if desired, with conventional additives such as solubilizers, isotonic agents, suspending agents, emulsifying agents, stabilizers and preservatives.
- the preparation may also be emulsified or the active ingredient encapsulated in liposome vehicles.
- Formulations suitable for injection can be administered by an intravitreal, intraocular, intramuscular, subcutaneous, sublingual, or other route of administration, e.g., injection into the gum tissue or other oral tissue. Such formulations are also suitable for topical administration.
- a subject compound can be delivered by a continuous delivery system.
- continuous delivery system is used interchangeably herein with “controlled delivery system” and encompasses continuous (e.g., controlled) delivery devices (e.g., pumps) in combination with catheters, injection devices, and the like, a wide variety of which are known in the art.
- a subject compound can be utilized in aerosol formulation to be administered via inhalation.
- a subject compound can be formulated into pressurized acceptable propellants such as dichlorodifluoromethane, propane, nitrogen and the like.
- a subject compound can be made into suppositories by mixing with a variety of bases such as emulsifying bases or water-soluble bases.
- bases such as emulsifying bases or water-soluble bases.
- a subject compound can be administered rectally via a suppository.
- the suppository can include vehicles such as cocoa butter, carbowaxes and polyethylene glycols, which melt at body temperature, yet are solidified at room temperature.
- unit dosage form refers to physically discrete units suitable as unitary dosages for human and animal subjects, each unit containing a predetermined quantity of a subject compound calculated in an amount sufficient to produce the desired effect in association with a pharmaceutically acceptable diluent, carrier or vehicle.
- the specifications for a subject compound depend on the particular compound employed and the effect to be achieved, and the pharmacodynamics associated with each compound in the host.
- the dosage form of a disclosed pharmaceutical composition will be determined by the mode of administration chosen.
- topical or oral dosage forms may be employed.
- Topical preparations may include eye drops, ointments, sprays and the like.
- Oral formulations may be liquid (e.g., syrups, solutions or suspensions), or solid (e.g., powders, pills, tablets, or capsules). Methods of preparing such dosage forms are known, or will be apparent, to those skilled in the art.
- compositions comprising a subject compound may be formulated in unit dosage form suitable for individual administration of precise dosages.
- amount of active ingredient administered will depend on the subject being treated, the severity of the affliction, and the manner of administration, and is known to those skilled in the art.
- the formulation to be administered will contain a quantity of the extracts or compounds disclosed herein in an amount effective to achieve the desired effect in the subject being treated.
- Each therapeutic compound can independently be in any dosage form, such as those described herein, and can also be administered in various ways, as described herein.
- the compounds may be formulated together, in a single dosage unit (that is, combined together in one form such as capsule, tablet, powder, or liquid, etc.) as a combination product.
- an individual subject compound may be administered at the same time as another therapeutic compound or sequentially, in any order thereof.
- the subject compounds can inhibit a protein kinase C activity. Accordingly, the subject compounds are useful for treating a disease or disorder that is mediated through the activity of a PKC activity in a subject. Accordingly, the subject compounds are useful for treating a disease or disorder that is associated with the activation of T-cells in a subject.
- the route of administration will be selected according to a variety of factors including, but not necessarily limited to, the condition to be treated, the formulation and/or device used, the patient to be treated, and the like.
- Routes of administration useful in the disclosed methods include but are not limited to oral and parenteral routes, such as intravenous (iv), intraperitoneal (ip), rectal, topical, ophthalmic, nasal, and transdermal. Formulations for these dosage forms are described herein.
- an effective amount of a subject compound will depend, at least, on the particular method of use, the subject being treated, the severity of the affliction, and the manner of administration of the therapeutic composition.
- a "therapeutically effective amount" of a composition is a quantity of a specified compound sufficient to achieve a desired effect in a subject (host) being treated. For example, this may be the amount of a subject compound necessary to prevent, inhibit, reduce or relieve a disease or disorder that is mediated through the activity of a PKC activity in a subject.
- a therapeutically effective amount of a compound is an amount sufficient to prevent, inhibit, reduce or relieve a disease or disorder that is mediated through the activity of a PKC activity in a subject without causing a substantial cytotoxic effect on host cells.
- Therapeutically effective doses (or growth inhibitory amounts) of a subject compound or pharmaceutical composition can be determined by one of skill in the art, with a goal of achieving local (e.g., tissue) concentrations that are at least as high as the ICso of an applicable compound disclosed herein.
- An example of a dosage range is from about 0.1 to about 200 mg/kg body weight orally in single or divided doses.
- a dosage range is from about 1.0 to about 100 mg/kg body weight orally in single or divided doses, including from about 1.0 to about 50 mg/kg body weight, from about 1.0 to about 25 mg/kg body weight, from about 1.0 to about 10 mg/kg body weight (assuming an average body weight of approximately 70 kg; values adjusted accordingly for persons weighing more or less than average).
- the compositions are, for example, provided in the form of a tablet containing from about 50 to about 1000 mg of the active ingredient, particularly about 75 mg, about 100 mg, about 200 mg, about 400 mg, about 500 mg, about 600 mg, about 750 mg, or about 1000 mg of the active ingredient for the symptomatic adjustment of the dosage to the subject being treated.
- a tablet containing from about 500 mg to about 1000 mg active ingredient is administered once (e.g., a loading dose) followed by administration of 1/2 dosage tablets (e.g., from about 250 to about 500 mg) each 6 to 24 hours for at least 3 days.
- the specific dose level and frequency of dosage for any particular subject may be varied and will depend upon a variety of factors, including the activity of the subject compound, the metabolic stability and length of action of that compound, the age, body weight, general health, sex and diet of the subject, mode and time of administration, rate of excretion, drug combination, and severity of the condition of the host undergoing therapy.
- the present disclosure also contemplates combinations of one or more disclosed compounds with one or more other agents or therapies useful in the treatment of a disease or disorder.
- the disease or disorder is mediated through the activity of a PKC activity in a subject.
- the disease or disorder is cell proliferative disorder.
- one or more disclosed compounds may be administered in combination with effective doses of other medicinal and pharmaceutical agents, or in combination other non- medicinal therapies, such as hormone or radiation therapy.
- administration in combination with refers to both concurrent and sequential administration of the active agents.
- PKC is a family of enzymes that function as serine/threonine kinases.
- the isoenzymes of PKC differ in their tissue distribution, enzymatic selectivity, requirement for Ca 2+ , and regulation. PKCs play an important role in cell-cell signaling, gene expression and in the control of cell differentiation and growth.
- the subject compound can be a selective inhibitor of PKC, e.g. an inhibitor selective for PKC over one or more other protein kinases, e.g. over one or more tyrosine kinases, for instance, over one or more non- receptor or receptor tyrosine kinases, e.g. over one or more of PKA, PKB, Abl Met, Src, Ins- R, Flt-3, JAK-2, KDR and/or Ret proteins.
- the selective PKC inhibitors may optionally be selective over one or more serine/threonine kinases, e.g. one or more serine/threonine kinases which do not belong to the CDK family.
- the subject compounds can exhibit a selectivity of at least 10 fold, or 20 fold, or 100 fold for the PKC over one or more other protein kinases, e.g. over one or more tyrosine kinases, e.g. over Flt-3, JAK-2, KDR and/or Ret proteins, or over one or more serine/threonine kinases which do not belong to the CDK family.
- other protein kinases e.g. over one or more tyrosine kinases, e.g. over Flt-3, JAK-2, KDR and/or Ret proteins, or over one or more serine/threonine kinases which do not belong to the CDK family.
- the selectivity of a selective inhibitor of PKC over other protein kinases may be calculated as the ratio of the IC 50 measured for PKC in an assay described herein over the IC 50 determined for another kinase.
- a PKC inhibitor for which the ratio of the IC 50 value as determined in an Allogeneic Mixed Lymphocyte Reaction (MLR) assay to the IC 50 value as determined in a BM assay is higher than 5, 10, 20, or 30.
- MLR and BM assays can be done according to known methods, e.g. mouse or human MLR and BM assays, such as disclosed herein.
- the disclosure provides an inhibitor of PKC, which can be an isozyme-selective PKC inhibitor, wherein the subject compound possesses selectivity for the isoforms ⁇ and a of PKC over one or more of the other PKC isoforms.
- the subject compound possesses selectivity for the isoform ⁇ of PKC over one or more of the other PKC isoforms.
- the subject compound possesses selectivity for the isoform a of PKC over one or more of the other PKC isoforms.
- the disclosed compounds exhibit selectivity for PKC ⁇ and PKC a over at least one PKC isoform.
- a subject compound can show a selectivity of at least 10 fold, or 20 fold, or 100 fold for the isoforms ⁇ or a of PKC over one or more of the other PKC isoforms.
- Selectivity for the isoforms ⁇ or a of PKC over one or more of the other PKC isoforms can be measured by comparing the IC 50 of the subject compound for the isoforms ⁇ or a of PKC to the IC 50 of the subject compound for the other PKC isoforms.
- the selectivity can be determined by calculating the ratio of IC 50 of the subject compound for the other isoforms of PKC to the IC 50 of the subject compound for ⁇ or a isoforms of PKC.
- subject compounds exhibit a selectivity for PKC ⁇ , a or both over another PKC isoform of at least about 2-fold, such as from about 3-fold to about 300-fold, from about 10-fold to about 100-fold or from about 5-fold to 50-fold.
- IC 50 values are obtained, for example, according to PKC assays described herein.
- the subject compounds can show an IC 50 value for the isoforms ⁇ or a of PKC of 1 ⁇ or less, such as less than about 300 nM, such as from about 1 nM to about 250 nM, less than 100 nM or even less than 10 nM in the assays disclosed herein.
- the subject compounds can show a selectivity of the isoforms ⁇ or ⁇ of PKC over other isoforms of PKC, as well as a selectivity over one or more of the other protein kinases, e.g. over one or more tyrosine kinases, or over one or more serine/threonine kinases which do not belong to the CDK-family, e.g. over one or more of PKA, PKB, Abl, Met, Src, Ins- it, Flt-3, JAK-2, KDR and Ret proteins, e.g. over one or more of Fit- 3, JAK-2, KDR and Ret proteins.
- Certain isozymes of PKC have been implicated in the mechanisms of various disease states, including, but not necessarily limited to, the following: cancer (PKC ⁇ , ⁇ , ⁇ , and ⁇ ); cardiac hypertrophy and heart failure (PKC ⁇ and PKC ⁇ ) nociception (PKC ⁇ and ⁇ ); ischemia including myocardial infarction (PKC ⁇ and 5); immune response, particularly T-cell mediated (PKC ⁇ and a); and fibroblast growth and memory (PKC ⁇ and ⁇ ).
- PKC inhibitors can also be used for treating an ocular disease or disorder involving inflammatory and/or neovascular events.
- the subject compounds can be used in the treatment of mammalian (especially human) disease states characterized by aberrant, elevated activity of a PKC isozyme in a tissue as compared to non-disease tissue of the same origin.
- PKC isozymes and disease states and/or biological functions amenable to therapy by inhibition of activity of the PKC isozyme include, but are not necessarily limited to: PKC a (hyperproliferative cellular diseases, such as cancer); PKC ⁇ and PKC ⁇ (cardiac hypertrophy and heart failure); PKC ⁇ (pain management); PKC ⁇ (ischemia, hypoxia (e.g,.
- PKC ⁇ pain management, myocardial dysfunction
- PKC ⁇ immune system diseases, particularly those involving T-cell mediated responses
- PKC ⁇ memory and fibroblast growth
- PKC ⁇ is expressed predominantly in lymphoid tissue and skeletal muscle.
- PKC ⁇ is selectively expressed in T-cells and plays a role in mature T-cell activation. It has been shown that PKC ⁇ is involved in T-cell receptor (TCR) -mediated T-cell activation but inessential during TCR-dependent thymocyte development. PKC ⁇ , but not other PKC isoforms, translocates to the site of cell contact between antigen-specific T-cells and antigen presenting cells (APC), where it localizes with the TCR in the central core of the T-cell activation.
- TCR T-cell receptor
- APC antigen presenting cells
- PKC ⁇ can selectively activate a FasL promoter-reporter gene and upregulate the mRNA or cell surface expression of endogenous FasL.
- PKC ⁇ and ⁇ can promote T-cell survival by protecting the cells from Fas-induced apoptosis, and this protective effect was mediated by promoting p90Rsk- dependent phosphorylation of BCL-2 family member BAD.
- PKC ⁇ appears to play a dual regulatory role in T-cell apoptosis.
- PKC ⁇ inhibitors can find use in the treatment or prevention of disorders or diseases mediated by T lymphocytes, for example, autoimmune disease such as rheumatoid arthritis, psoriasis and lupus erythematosus, and inflammatory disease such as asthma and inflammatory bowel diseases.
- autoimmune disease such as rheumatoid arthritis, psoriasis and lupus erythematosus
- inflammatory disease such as asthma and inflammatory bowel diseases.
- PKC ⁇ is a drug target for immunosuppression in transplantation and autoimmune diseases (Isakov et al. (2002) Annual Review of Immunology, 20, 761-794).
- PKC ⁇ As a target for treatment of transplant rejection and multiple sclerosis. PKC ⁇ also plays a role in inflammatory bowel disease (The Journal of Pharmacology and Experimental Therapeutics (2005), 313 (3), 962-982), asthma (WO 2005062918), and lupus (Current Drug Targets: Inflammation & Allergy (2005), 4 (3), 295-298).
- PKC ⁇ is highly expressed in gastrointestinal stromal tumors (Blay, P. et al. (2004) Clinical Cancer Research, 10, 12, Pt. 1), it has been suggested that PKC ⁇ is a molecular target for treatment of gastrointestinal cancer (Wiedmann, M. et al. (2005) Current Cancer Drug Targets 5(3), 171).
- the subject compounds are useful for treating a disease or disorder that is mediated through, or exacerbated by, the activity of a PKC in a subject in need of treatment. Also, the compounds are useful for treating a disease or disorder that is associated with aberrant or otherwise undesirable T cell activation in a subject.
- the present disclosure provides methods of treating an inflammatory disease in a subject by administering an effective amount of a subject compound, including a salt or solvate or stereoisomer thereof, so as to treat inflammation.
- Inflammatory diseases contemplated for therapy include acute and chronic inflammation mediated or exacerbated by PKC activity
- the present disclosure also provides methods of treating an autoimmune disease in a subject by administering to the subject an effective amount of a subject compound, including a salt or solvate or stereoisomer thereof, so as to treat the autoimmune disease.
- the present disclosure also provides methods of treating an ocular disease or disorder involving inflammatory and/or neovascular events by administration of a subject compound, including a salt or solvate or stereoisomer thereof, in an effective amount.
- Diseases or conditions of interest for treatment according to the present disclosure include, but are not limited to, atherosclerosis, vascular occlusion due to vascular injury such as angioplasty, restenosis, obesity, syndrome X, impaired glucose tolerance, polycystic ovary syndrome, hypertension, heart failure, chronic obstructive pulmonary disease, CNS diseases such as Alzheimer disease or amyotrophic lateral sclerosis, cancer, infectious diseases such as: AIDS, septic shock or adult respiratory distress syndrome, ischemia/reperfusion injury, e.g.: myocardial infarction, stroke, gut ischemia, renal failure or hemorrhage shock, and traumatic shock, e.g. traumatic brain injury.
- vascular occlusion due to vascular injury such as angioplasty, restenosis, obesity, syndrome X, impaired glucose tolerance, polycystic ovary syndrome, hypertension, heart failure, chronic obstructive pulmonary disease, CNS diseases such as Alzheimer disease or amyotrophic lateral
- Further diseases or conditions of interest for treatment according to the present disclosure include, but are not limited to, T-cell mediated acute or chronic inflammatory diseases or disorders or autoimmune diseases, rheumatoid arthritis, osteoarthritis, systemic lupus erythematosus, Hashimoto's thyroiditis, multiple sclerosis, myasthenia gravis, diabetes type I or II and the disorders associated therewith, transplant rejection, graft versus host disease, respiratory diseases, asthma, inflammatory lung injury, inflammatory liver injury, inflammatory glomerular injury, cutaneous manifestations of immunologically-mediated disorders or illnesses, inflammatory and hyperproliferative skin diseases (such as psoriasis, atopic dermatitis, allergic contact dermatitis, irritant contact dermatitis and further eczematous dermatitises, seborrhoeic dermatitis), inflammatory eye diseases (such as Sjoegren's syndrome, keratoconjunctivitis, uveitis)
- Ocular diseases or disorders involving inflammation and/or neovascularization include, but are not limited to, macular degeneration (AMD), diabetic ocular diseases or disorders, uveitis, optic neuritis, ocular edema, ocular angiogenesis, ischemic retinopathy, anterior ischemic optic neuropathy, optic neuropathy and neuritis, macular edema, cystoid macular edema (CME), retinal disease or disorder, such as retinal detachment, retinitis pigmentosa (RP), Stargart's disease, Best's vitelliform retinal degeneration, Leber's congenital amaurosis and other hereditary retinal degenerations, Sorsby's fundus dystrophy, pathologic myopia, retinopathy of prematurity (ROP), Leber's
- cell proliferative disorders treatable with the subject compound disclosed herein relate to any disorder characterized by aberrant cell proliferation. These include various tumors and cancers, benign or malignant, metastatic or non-metastatic. Specific properties of cancers, such as tissue invasiveness or metastasis, can be targeted using the methods described herein.
- Cell proliferative disorders include a variety of cancers, including, among others, breast cancer, ovarian cancer, renal cancer, gastrointestinal cancer, kidney cancer, bladder cancer, pancreatic cancer, lung squamous carcinoma, and adenocarcinoma.
- the cell proliferative disorder treated is a hematopoietic neoplasm, which is aberrant growth of cells of the hematopoietic system.
- Hematopoietic malignancies can have its origins in pluripotent stem cells, multipotent progenitor cells, oligopotent committed progenitor cells, precursor cells, and terminally differentiated cells involved in hematopoiesis. Some hematological malignancies are believed to arise from hematopoietic stem cells, which have the ability for self renewal.
- cells capable of developing specific subtypes of acute myeloid leukemia (AML) upon transplantation display the cell surface markers of hematopoietic stem cells, implicating hematopoietic stem cells as the source of leukemic cells.
- Blast cells that do not have a cell marker characteristic of hematopoietic stem cells appear to be incapable of establishing tumors upon AML.
- hematopoietic neoplasms often originate from stem cells, committed progenitor cells or more terminally differentiated cells of a developmental lineage can also be the source of some leukemias.
- forced expression of the fusion protein Bcr/Abl associated with chronic myelogenous leukemia
- common myeloid progenitor or granulocyte/macrophage progenitor cells produces a leukemic-like condition.
- chromosomal aberrations associated with subtypes of leukemia are not found in the cell population with a marker phenotype of hematopoietic stem cells, but are found in a cell population displaying markers of a more differentiated state of the hematopoietic pathway (Turhan et al., 1995, Blood 85:2154-2161).
- committed progenitor cells and other differentiated cells may have only a limited potential for cell division
- leukemic cells may have acquired the ability to grow unregulated, in some instances mimicking the self -renewal characteristics of hematopoietic stem cells (Passegue et al., Proc. Natl. Acad. Sci. USA, 2003, 100: 11842-9).
- the hematopoietic neoplasm treated is a lymphoid neoplasm, where the abnormal cells are derived from and/or display the characteristic phenotype of cells of the lymphoid lineage.
- Lymphoid neoplasms can be subdivided into B- cell neoplasms, T and NK-cell neoplasms, and Hodgkin's lymphoma.
- B-cell neoplasms can be further subdivided into precursor B-cell neoplasm and mature/peripheral B-cell neoplasm.
- Exemplary B-cell neoplasms are precursor B-lymphoblastic leukemia/lymphoma (precursor B-cell acute lymphoblastic leukemia) while exemplary mature/peripheral B-cell neoplasms are B-cell chronic lymphocytic leukemia/small lymphocytic lymphoma, B-cell
- prolymphocytic leukemia lymphoplasmacytic lymphoma, splenic marginal zone B-cell lymphoma, hairy cell leukemia, plasma cell myeloma/plasmacytoma, extranodal marginal zone B-cell lymphoma of MALT type, nodal marginal zone B-cell lymphoma, follicular lymphoma, mantle-cell lymphoma, diffuse large B-cell lymphoma, mediastinal large B-cell lymphoma, primary effusion lymphoma, and Burkitt's lymphoma/Burkitt cell leukemia.
- T- cell and Nk-cell neoplasms are further subdivided into precursor T-cell neoplasm and mature (peripheral) T-cell neoplasms.
- Exemplary precursor T-cell neoplasm is precursor T- lymphoblastic lymphoma/leukemia (precursor T-cell acute lymphoblastic leukemia) while exemplary mature (peripheral) T-cell neoplasms are T-cell prolymphocytic leukemia T-cell granular lymphocytic leukemia, aggressive NK-cell leukemia, adult T-cell
- lymphoma/leukemia HTLV-1
- extranodal NK T-cell lymphoma nasal type
- enteropathy- type T-cell lymphoma hepatosplenic gamma-delta T-cell lymphoma
- subcutaneous panniculitis-like T-cell lymphoma Mycosis fungoides/Sezary syndrome
- Anaplastic large- cell lymphoma T/null cell
- primary cutaneous type Peripheral T-cell lymphoma, not otherwise characterized
- Angioimmunoblastic T-cell lymphoma Anaplastic large-cell lymphoma, T/null cell, primary systemic type.
- the third member of lymphoid neoplasms is Hodgkin's lymphoma, also referred to as Hodgkin's disease.
- Hodgkin's lymphoma also referred to as Hodgkin's disease.
- Exemplary diagnosis of this class that can be treated with the compounds include, among others, nodular lymphocyte- predominant Hodgkin's lymphoma, and various classical forms of Hodgkin's disease, exemplary members of which are Nodular sclerosis Hodgkin's lymphoma (grades 1 and 2), Lymphocyte-rich classical Hodgkin's lymphoma, Mixed cellularity Hodgkin's lymphoma, and Lymphocyte depletion Hodgkin's lymphoma.
- the hematopoietic neoplasm treated is a myeloid neoplasm.
- This group comprises a large class of cell proliferative disorders involving or displaying the characteristic phenotype of the cells of the myeloid lineage.
- Myeloid neoplasms can be subdivided into myeloproliferative diseases,
- myelodysplastic/myeloproliferative diseases myelodysplastic/myeloproliferative diseases, myelodysplastic syndromes, and acute myeloid leukemias.
- myeloproliferative diseases are chronic myelogenous leukemia (e.g., Philadelphia chromosome positive (t(9;22)(qq34;ql l)), chronic neutrophilic leukemia, chronic eosinophilic leukemialhypereosinophilic syndrome, chronic idiopathic
- myelofibrosis myelofibrosis, polycythemia vera, and essential thrombocythemia.
- myelodysplastic/myeloproliferative diseases are chronic myelomonocytic leukemia, atypical chronic myelogenous leukemia, and juvenile myelomonocytic leukemia.
- Exemplary myelodysplastic syndromes are refractory anemia, with ringed sideroblasts and without ringed sideroblasts, refractory cytopenia (myelodysplastic syndrome) with multilineage dysplasia, refractory anemia (myelodysplastic syndrome) with excess blasts, 5q-syndrome, and myelodysplastic syndrome with t(9;12)(q22;pl2) (TEL-Syk fusion; see, e.g., Kuno et al., 2001, Blood 97: 1050).
- the composition can be used to treat acute myeloid leukemias (AML), which represent a large class of myeloid neoplasms having its own subdivision of disorders. These subdivisions include, among others, AMLs with recurrent cytogenetic translocations, AML with multilineage dysplasia, and other AML not otherwise categorized.
- AML acute myeloid leukemias
- Exemplary AMLs with recurrent cytogenetic translocations include, among others, AML with t(8;21)(q22;q22), AML 1 (CB F- alpha)/ETO , Acute promyelocytic leukemia (AML with t(15;17)(q22;ql l-12) and variants, PML/RAR-alpha), AML with abnormal bone marrow eosinophils (inv(16)(pl3q22) or t(16;16)(pl3;ql l),
- AML with multilineage dysplasia are those that are associated with or without prior myelodysplastic syndrome.
- Other acute myeloid leukemias not classified within any definable group include, AML minimally differentiated, AML without maturation, AML with maturation, Acute myelomonocytic leukemia, Acute monocytic leukemia, Acute erythroid leukemia, Acute megakaryocytic leukemia, Acute basophilic leukemia, and Acute panmyelosis with myelofibrosis.
- cell proliferative disorders comprise virally mediated tumors. These can arise from infection of cells by an oncogenic virus that has the capability of transforming a normal cell into a tumor cell. Because rates of viral infection far exceed the number of actual incidence of cell transformation, viral mediated transformation generally act together with other cellular factors to generate a transformed tumor cell. Thus, a virally mediated tumor does not require the virus to be the sole causative agent of the cell proliferative disorder, but rather that the viral infection or persistent presence of virus is associated with the generation of the tumor.
- tumors where the causative agent is a virus typically has continual expression of a limited number of viral genes and that viral these oncogenes, expressed as part of the viral infection or through persistence of the virus, disrupts the normal cellular gene expression and signal transduction pathways.
- viral oncogenes involved in cell transformation appear to disrupt four main cellular processes: cell surface receptors that interact with growth factors and extracellular matrix, transmembrane signaling networks, cytosolic elements such as soluble proteins and second messengers, and nuclear proteins including DNA binding proteins and factors which function directly and indirectly in gene regulation and replication.
- the inhibition of PKC activity is measured by monitoring the production of phosphorylated peptide by fluorescence polarization at different concentrations of the inhibitor. Reactions are carried out in 96-well plate format with a total volume of 20 ⁇ L containing 20 mM HEPES, pH 7.4, 5 mM MgCl 2 , 0.2 mM CaCl 2 , 1 mM DTT, 0.02% Brij- 35, 0.1 mg/mL phosphatidylserine, 0.02 mg/mL dioleoyl-sn-glycerol and 5 ⁇ each of ATP and the peptide substrate.
- Human primary T cell isolation and culture Human primary T cells were prepared as follows. Fresh PBMC's from All Cells (Cat # PB002) were re-suspended in RPMI (RPMI-1640 with L-Glutamine; Mediatech, Inc., Herndon VA, cat. #10-040-CM) with 10% FBS and seeded into flasks and incubated at 37°C for 2 hours to allow the monocytes to adhere.
- RPMI RPMI-1640 with L-Glutamine; Mediatech, Inc., Herndon VA, cat. #10-040-CM
- the non-adherent cells were then centrifuged and re-suspended in RPMI medium containing 40 U/ml IL2 and seeded into a flask pre-coated with 1 ⁇ g/ml aCD3 and 5 ug/ml aCD28 (Anti-Human CD3, BD Pharmingen Catalog #555336, Anti- Human CD28, Beckman Coulter Catalog #IM1376).
- the cells were stimulated for 3-4 days, then transferred to a fresh flask and maintained in RPMI (RPMI-1640 with L-Glutamine; Mediatech, Inc., Herndon VA, cat. #10-040-CM) with 10% FBS and 40 U/mL IL-2.
- the subject compounds can be tested for activity on different PKC isoforms according to the following method. Assay is performed in a white with clear bottom 384- well microtiterplate with non-binding surface.
- the reaction mixture (25 ⁇ ) contains 1.5 ⁇ of a tridecapeptide acceptor substrate that mimics the pseudo substrate sequence of PKC a with the Ala ⁇ Ser replacement, 10 ⁇ 33 P-ATP, 10 mM Mg(N0 3 ) 2 , 0.2 mM CaCl 2 , PKG at a protein concentration varying from 25 to 400 ng/ml (depending on the isotype used), lipid vesicles (containing 30 mol % phosphatidylserine, 5 mol % DAG and 65 mol %
- phosphatidylcholine at a final lipid concentration of 0.5 mM, in 20 mM Tris-HCl buffer pH 7.4+0.1% BSA. Incubation is performed for 60 minutes at room temperature. Reaction is stopped by adding 50 ⁇ of stop mix (100 mM EDTA, 200 ⁇ ATP, 0.1% Triton X-100, 0.375 mg/well streptavidin-coated SPA beads in phosphate buffered saline w/o Ca, Mg. After 10 minutes incubation at room temperature, the suspension is spun down for 10 minutes at 300 g. Incorporated radioactivity is measured in a Trilux counter for 1 minute. IC 50 measurement is performed on a routine basis by incubating a serial dilution of inhibitor at concentrations ranging between 1-1000 ⁇ . ICso values are calculated from the graph by curve fitting with XL Fit® software.
- the assay is performed with Jurkat cells transfected with a human interleukin-2 promoter/reporter gene construct as described by Baumann G et al. in Transplant. Proc. 1992; 24:43-8, the ⁇ -galactosidase reporter gene being replaced by the luciferase gene (de Wet J., et al., Mol. Cell. Biol. 1987, 7(2), 725-737).
- Cells are stimulated by solid phase- coupled antibodies or phorbol myristate acetate (PMA) and the Ca ++ ionophore ionomycin as follows.
- PMA phorbol myristate acetate
- Microlite TM1 micro titer plates (Dynatech) are coated with 3 ⁇ g/ml goat anti-mouse IgG Fc antibodies (Jackson) in 55 ⁇ phosphate- buffered saline (PBS) per well for three hours at room temperature. Plates are blocked after removing the antibodies by incubation with 2% bovine serum albumin (BSA) in PBS (300 ⁇ per well) for 2 hours at room temperature.
- BSA bovine serum albumin
- test compounds in duplicates in assay medium RPMI 1640/10% fetal calf serum (FCS) containing 50 ⁇ 2-mercaptoethanol, 100 units/ml penicillin and 100 ⁇ g/ml streptomycin
- FCS fetal calf serum
- 100 ⁇ of this mixture containing 1 x 10 5 cells are then transferred to the antibody- coated assay plates.
- 100 ⁇ are incubated with 40 ng/ml PMA and 2 ⁇ ionomycin.
- the level of luciferase is determined by bioluminescence measurement.
- the plates are centrifuged for 10 minutes at 500 g and the supernatant is removed by flicking.
- Lysis buffer containing 25 mM Tris- phosphate, pH 7.8, 2 mM DTT, 2 mM l,2-diaminocyclohexane-N,N,N',N-tetraacetic acid, 10% (v/v) glycerol and 1% (v/v) Triton X-100 is added (20 ⁇ per well). The plates are incubated at room temperature for 10 minutes under constant shaking.
- Luciferase activity is assessed with a bioluminescence reader (Labsystem, Helsinki, Finland) after automatic addition of 50 ⁇ per well luciferase reaction buffer containing 20 mM Tricine, 1.07 mM (MgC0 3 ) 4 Mg(OH) 2 5H 2 0, 2.67 mM MgS0 4 , 0.1 mM EDTA, 33.3 mM DTT, 270 ⁇ coenzyme A, 470 ⁇ luciferin (Chemie Brunschwig AG), 530 ⁇ ATP, pH 7.8.
- Lag time is 0.5 seconds, total measuring time is 1 or 2 seconds.
- Low control values are light units from anti-T cell receptor- or PMA- stimulated cells, high controls are from anti-T cell receptor/anti-CD28- or PMA/ionomycin-stimulated cells without any test sample. Low controls are subtracted from all values.
- the inhibition obtained in the presence of a test compound is calculated as percent inhibition of the high control.
- the concentration of test compounds resulting in 50% inhibition (IC 50 ) is determined from the dose-response curves. 11.
- BM Bone Marrow Proliferation
- Bone marrow cells from CBA mice are incubated in 100 ⁇ RPMI medium containing 10% FCS, 100 U/ml penicillin, 100 ⁇ g/ml streptomycin (Gibco BRL, Basel, Switzerland), 50 UM 2- mercaptoethanol (Fluke, Buchs, Switzerland), WEHI-3 conditioned medium (7.5% v/v) and L929 conditioned medium (3% v/v) as a source of growth factors and serially diluted compounds. Seven three-fold dilution steps in duplicates per test compound are performed. After four days of incubation 1 ⁇ 3 H-thymidine is added.
- WEHI-3 cells 1 (ATCC TIB68) and L929 cells (ATCC CCL 1) are grown in RPMI medium until confluence for 4 days and one week, respectively.
- Cells are harvested, resuspended in the same culture flasks in medium C containing 1% FCS (Schreier and Tees 1981) for WEHI-3 cells and RPMI medium for L929 cells and incubated for 2 days (WEHI-3) or one week (L929). The supernatant is collected, filtered through 0.2 ⁇ and stored in aliquots at -80°C.
- the two-way MLR is performed according to standard procedures (J. Immunol. Methods, 1973, 2, 279 and Meo T. et al., Immunological Methods, New York, Academic Press, 1979, 227-39). Briefly, spleen cells from CBA and BALB/c mice (1.6xl0 5 cells from each strain per well in flat bottom tissue culture micro titer plates, 3.2 l0 5 in total) are incubated in RPMI medium containing 10% FCS, 100 U/ml penicillin, 100 ⁇ g/ml streptomycin (Gibco BRL, Basel, Switzerland), 50 ⁇ 2-mercaptoethanol (Fluka, Buchs, Switzerland) and serially diluted compounds.
- the strain combination used Male Lewis (RT 1 haplotype) and BN (RT 1 haplotype).
- the animals are anaesthetised using inhalational isofluorane. Following heparinisation of the donor rat through the abdominal inferior vena cava with simultaneous exsanguination via the aorta, the chest is opened and the heart rapidly cooled. The aorta is ligated and divided distal to the first branch and the brachiocephalic trunk is divided at the first bifurcation. The left pulmonary artery is ligated and divided and the right side divided but left open. All other vessels are dissected free, ligated and divided and the donor heart is removed into iced saline.
- the recipient is prepared by dissection and cross-clamping of the infra-renal abdominal aorta and vena cava.
- the graft is implanted with end-to-side anastomoses, using 1010 monofilament suture, between the donor brachiocephalic trunk and the recipient aorta and the donor right pulmonary artery to the recipient vena cava.
- the clamps are removed, the graft tethered retroabdominally, the abdominal contents washed with warm saline and the animal is closed and allowed to recover under a heating lamp.
- Graft survival is monitored by daily palpation of the beating donor heart through the abdominal wall. Rejection is considered to be complete when-heart beat stops. Graft survival is monitored in animals treated with compounds.
- Spleen cells (2xl0 7 ) from Wistar/F rats are injected subcutaneously into the right hind footpad of (Wistar/F x Fischer 344)Fi hybrid rats.
- the left footpad is left untreated.
- the animals are treated with the test compounds on 4 consecutive days (0-3).
- the popliteal lymph nodes are removed on day 7, and the weight differences between two corresponding lymph nodes are determined.
- the results are expressed as the inhibition of lymph node enlargement (given in percent) comparing the lymph node weight differences in the experimental groups to the weight difference between the corresponding lymph nodes from a group of animals left untreated with a test compound.
- the test compound is a selective PKC inhibitor.
- disclosed compounds that are particularly useful for treating graft versus host disease and related disorders are selective PKC a and ⁇ inhibitors.
- RA Rheumatoid arthritis
- IgG-containing IC are abundant in the synovial tissue of patients with RA. While it is still debated what role these complexes play in the etiology and pathology of the disease, IC communicate with the hematopoetic cells via the FcyR.
- CIA is a widely accepted animal model of RA that results in chronic
- Syngeneic LOU rats are immunized with native type II collagen on Day 0, and efficacy of a test compound is evaluated in a prevention regimen and a treatment regimen.
- a prevention protocol either vehicle or various doses of a test compound are administered via oral gavage starting on day of immunization (Day 0).
- treatment protocol after clinical signs of arthritis develop on Day 10, treatment with a test compound is initiated (e.g., 300 mg/kg by oral gavage, qd) and continued until sacrifice on Day 28.
- clinical scores are obtained daily, and body weights are measured twice weekly.
- radiographic scores are obtained, and serum levels of collagen II antibody are measured by ELISA.
- rats can develop clinical CIA, as determined by an increase in their arthritis scores.
- the mean arthritic score gradually increases in the rats treated with vehicle alone after Day 10, and by Day 28 the mean clinical score can reach about 6.75.
- Mean clinical scores in animals treated from the day of immunization (Day 0) with a test compound can be significantly reduced on Days 10-28 compared with vehicle controls. In the rats treated with a test compound at disease onset, there can be a
- Blinded radiographic scores (scale 0-6) can be obtained on Day 28 of CIA and compared between the animals in the vehicle group, animals in the prevention group, and animals in the treatment group.
- the groups administered with a test compound can preclude the development of erosions and reduced soft tissue swelling.
- the groups administered with a test compound can result in reduction of serum anti-collagen II antibody.
- EAE experimental autoimmune encephalomyelitis
- EAE is a useful model for multiple sclerosis (MS), an autoimmune disease of the CNS that is caused by immune-cell infiltration of the CNS white matter. Inflammation and subsequent destruction of myelin cause progressive paralysis.
- MS multiple sclerosis
- EAE is associated with peripheral activation of T cells autoreactive with myelin proteins, such as myelin basic protein (MBP), proteolipid protein (PLP), or myelin oligodendrocyte protein (MOG).
- MBP myelin basic protein
- PGP proteolipid protein
- MOG myelin oligodendrocyte protein
- Activated neuroantigen-specific T cells pass the blood-brain barrier, leading to focal mononuclear cell infiltration and demyelination.
- EAE can be induced in susceptible mouse strains by immunization with myelin- specific proteins in combination with adjuvant.
- mice hind limb and tail paralysis is apparent by Day 10 after immunization, the peak of disease severity can be observed between Days 10 and 14, and a cycle of partial spontaneous remission followed by relapse can be observed up to Day 35.
- the results can demonstrate the potential of the test compound to suppress disease severity and prevent relapse of disease symptoms that may be the result of FcyR-mediated cytokine release from immune cells.
- each mouse is sensitized with PLP/CFA. (150 ⁇ g PLP139-151 with 200 ⁇ g CFA in 0.05 ml of homogenate on four sites of hind flank for a total of 0.2 ml emulsion is used to induce EAE).
- a suppression protocol either vehicle or various doses of a test compound are administered via oral gavage starting on the day of immunization (Day 0).
- a treatment protocol at onset of disease, animals are separated to achieve groups with a similar mean clinical score at onset and administered vehicle or various dose frequencies of test compounds via oral gavage. In both protocols, clinical scores are monitored daily, and body weights are measured twice weekly.
- SJL mice treated with a test compound at disease onset can show a significant decrease in CDI. Further, there can be a decrease in the number of relapses in animals treated with a test compound compared with the number of relapses in animals treated with vehicle.
- subject compounds can inhibit a PKC activity, such compounds are also useful as research tools.
- the present disclosure also provides a method for using subject compounds as a research tool for studying a biological system or sample, or for discovering new chemical compounds that can inhibit a PKC activity.
- the disclosure provides for a method of studying a biological system or sample known to comprise PKC, the method comprising: (a) contacting the biological sample with a compound of formula I- VIII or a salt or solvate or stereoisomer thereof; and (b) determining the inhibiting effects caused by the compound on the biological sample.
- Any suitable biological sample having PKC can be employed in such studies which can be conducted either in vitro or in vivo.
- Representative biological samples suitable for such studies include, but are not limited to, cells, cellular extracts, plasma membranes, tissue samples, isolated organs, mammals (such as mice, rats, guinea pigs, rabbits, dogs, pigs, humans, and so forth), and the like, with mammals being of particular interest.
- a biological sample comprising PKC is typically contacted with a PKC activity-inhibiting amount of a subject compound.
- Exposure encompasses contacting the biological sample with the compound or administering the compound to a subject.
- the determining step can involve measuring a response (a quantitative analysis) or can involve making an observation (a qualitative analysis).
- Measuring a response involves, for example, determining the effects of the compound on the biological sample using conventional procedures and equipment, such as radioligand binding assays and measuring ligand-mediated changes in functional assays.
- the assay results can be used to determine the activity level as well as the amount of compound necessary to achieve the desired result, that is, a PKC activity-inhibiting amount.
- subject compounds can be used as research tools for evaluating other chemical compounds, and thus are also useful in screening assays to discover, for example, new compounds having a PKC inhibiting activity.
- a subject compound can be used as a standard in an assay to allow comparison of the results obtained with a test compound and with the subject compounds to identify those test compounds that have about equal or superior activity, if any.
- IC 50 data for a test compound or a group of test compounds is compared to the IC 50 data for a subject compound to identify those test compounds that have the desired properties, for example, test compounds having an IC 50 about equal or superior to a subject compound, if any.
- a test compound can be evaluated in a biological assay, by a method comprising the steps of: (a) conducting a biological assay with a test compound to provide a first assay value; (b) conducting the biological assay with a subject compound to provide a second assay value; wherein step (a) is conducted either before, after or concurrently with step (b); and (c) comparing the first assay value from step (a) with the second assay value from step (b).
- the assays that can be used for generation of comparison data are disclosed herein, such as the PKC assays.
- Trifluoroacetic anhydride (9.4 mL; 67.3 mmol) was added dropwise over 30-45 minutes to a stirred solution of 5-carboxyamide-2-chloro-N4-(2,2,6,6-tetramethylpiperidin-4- yl)-4-pyrimidineamine (2.1 g, 6.7 mmol) and Et 3 N (11.3 mL; 80.8 mmol) in THF (40 mL) at -78 °C under nitrogen. After complete addition, the mixture was stirred at -78 °C for a further 60 minutes, then a saturated solution of NaHCC>3 (30 mL) was added dropwise keeping the internal temperature below -30 °C.
- Trifluoroacetic anhydride (9.35 mL; 67.3 mmol, 10 eq) was added dropwise over 30-45 minutes to a stirred solution of 5-carboxyamide-2-chloro-N4-(l,2,2,6,6- pentamethylpiperidin-4-yl)-4-pyrimidineamine hydrochloride (2.19 g, 6.73 mmol, 1 eq) and Et 3 N (11.26 mL; 80.76 mmol, 12 eq) in THF (45 mL) at -78 °C under nitrogen.
- the combined organic extracts were washed with brine (1 x 50 mL), dried (Na 2 S0 4 ), filtered and the solvent removed under vacuum to leave a crude solid with TFAA and Et 3 N residues.
- the crude solid was dissolved in 100 mL of EtOAc and partitioned with 1 N aqueous NaOH (50 mL). The ethyl acetate layer was extracted with 2 x 50 mL aqueous IN NaOH.
- the combined organic extracts were washed with brine (1 x 50 mL), dried (Na 2 S0 4 ), filtered and the solvent removed under vacuum to give light yellow solid (1.80 g, 87 %).
- Step 1 Preparation of l-(2-Bromo-4-fluorophenyl)-lH-tetrazol-5(4H)-one [00388] A mixture of 2-bromo-4-fluoro-l-isocyanatobenzene (20g, 92.6 mmol;
- Step 2 l-(2-Bromo-4-fluorophenyl)-4-methyl-lH-tetrazol-5(4H)-one
- K 2 CO 3 (26.8g, 194 mmol) and Mel (9.7 mL, 155 mmol) were added at -78 °C to a solution of l-(2-bromo-4-fluorophenyl)-lH-tetrazol-5(4H)-one (20.1 g, 77.6 mmol) in DMF (150 mL) under N 2 .
- the mixture was stirred from -78 °C to room temperature over 2 days.
- the mixture was poured in to EtOAc (300 mL) and 3 ⁇ 40 (500 mL) and the aqueous and organic layers were partitioned.
- Step 3 l-(2-Bromo-4-fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one
- Step 1 Preparatio -(2-Bromo-4-fluorophenyl)-lH-tetrazol-5(4H)-one:
- Trimethylsilyl azide (65 mL, 494 mmol; ca. 6 equiv.) was added to a stirred mixture of 2-bromo-4-fluorobenzoyl chloride (20.4 g, 86 mmol; Apollo Scientific Ltd) under N 2 at 0 to 10 °C. The mixture was then heated slowly to 50-70 °C (gas evolution starts at approximately 50-60 °C and becomes vigorous after ca. 65 °C). The mixture was briefly removed from the heat until nitrogen evolution was more controlled, then the mixture returned to the heat. The mixture was then heated to 90 °C under nitrogen overnight.
- a safety notice for the procedure The reaction was performed behind a blast shield in a 250 mL round-bottom flask. A nitrogen balloon and vent (needle vent) was used in the set-up, especially the first part of the reaction in which nitrogen gas is evolved.
- the mixture was purified by filtration through a 4-to-5 inch plug of silica (the residue was dry-loaded on to silica) eluting with EtOAc / hexane (3:7 to 4:6; fractions collected in conical flasks) to give the product (11.0 g, 86%) as a solid.
- Step 2 l-(2-CYCLOPROPYL-4-FLUORO-5-NITROPHENYL)-lH-TETRAZOL-5(4H)-ONE
- Step 3 l-(2-CYCLOPROPYL-4-FLUORO-5-NITROPHENYL)-4-METHYL-lH-TETRAZOL- 5(4H)-ONE
- K 2 C0 3 (13.4 g, 97.1 mmol) and Mel (4.8 mL, 77.7 mmol) were added sequentially to a stirred solution of l-(2-cyclopropyl-4-fluoro-5-nitrophenyl)-lH-tetrazol- 5(4H)-one (10.3 g, 38.8 mmol) in DMF (100 mL) at -78 °C under N 2 . The mixture was allowed to warm to room temperature over 3 hours. TLC indicated complete reaction, so poured mixture into H 2 0 (1 L) and EtOAc (500 mL).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Neurology (AREA)
- Cardiology (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Pulmonology (AREA)
- Hematology (AREA)
- Heart & Thoracic Surgery (AREA)
- Neurosurgery (AREA)
- Endocrinology (AREA)
- Obesity (AREA)
- Biomedical Technology (AREA)
- Physical Education & Sports Medicine (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pain & Pain Management (AREA)
- Virology (AREA)
- Reproductive Health (AREA)
- Hospice & Palliative Care (AREA)
- Urology & Nephrology (AREA)
- Rheumatology (AREA)
- Dermatology (AREA)
- Emergency Medicine (AREA)
- Psychiatry (AREA)
Abstract
This disclosure concerns compounds which are useful as inhibitors of protein kinase C (PKC) and are thus useful for treating a variety of diseThis disclosure concerns compounds which are useful as inhibitors of protein kinase C (PKC) and are thus useful for treating a variety of diseases and disorders that are mediated or sustained through the activity of PKC. This disclosure also relates to pharmaceutical compositions comprising these compounds, methods of using these compounds in the treatment of various diseases and disorders, processes for preparing these compounds and intermediates useful in these processes.ases and disorders that are mediated or sustained through the activity of PKC. This disclosure also relates to pharmaceutical compositions comprising these compounds, methods of using these compounds in the treatment of various diseases and disorders, processes for preparing these compounds and intermediates useful in these processes.
Description
PROTEIN KINASE C INHIBITORS AND USES THEREOF
BACKGROUND
[0001] Protein kinase C ("PKC") is a key enzyme in signal transduction involved in a variety of cellular functions, including cell growth, regulation of gene expression, and ion channel activity. The PKC family of isozymes includes at least 11 different protein kinases that can be divided into at least three subfamilies based on their homology and sensitivity to activators. Each isozyme includes a number of homologous ("conserved" or "C") domains interspersed with isozyme-unique ("variable" or "V") domains. Members of the "classical" or "cPKC" subfamily, PKC α, βι, βϋ and γ, contain four homologous domains (CI, C2, C3 and C4) and require calcium, phosphatidylserine, and diacylglycerol or phorbol esters for activation. Members of the "novel" or "nPKC" subfamily, PKC δ, ε, η and Θ, lack the C2 homologous domain and do not require calcium for activation. Finally, members of the "atypical" or "aPKC" subfamily, PKC ζ and λ/i, lack both the C2 and one-half of the CI homologous domains and are insensitive to diacylglycerol, phorbol esters and calcium.
SUMMARY
[0002] This disclosure concerns compounds which are useful as inhibitors of protein kinase C (PKC) and are thus useful for treating a variety of diseases and disorders that are mediated or sustained through the activity of PKC. This disclosure also relates to pharmaceutical compositions comprising these compounds, methods of using these compounds in the treatment of various diseases and disorders, processes for preparing these compounds and intermediates useful in these processes.
[0003] Exemplary chemical structures are provided throughout the disclosure. By way of example, such compounds are represented by the following formula:

(I)
wherein
R5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
Y1 and Y2 are independently selected from hydrogen, alkyl, and acyl;
R is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
Ra and Rb are independently selected from hydrogen and alkyl;
Rc and Rd are independently selected from hydrogen and alkyl;
Q is selected from N and CR7b;
R6a, R6b, R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O- alk-A;
alk is a bond, alkylene or substituted alkylene;
A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
wherein the A ring can be substituted or unsubstituted;
R7y is selected from hydrogen, alkyl, cycloalkyl, and substituted alkyl;
or a salt or stereoisomer thereof.
DETAILED DESCRIPTION
[0004] This disclosure concerns compounds which are useful as inhibitors of protein kinase C (PKC) and are thus useful for treating a variety of diseases and disorders that are mediated or sustained through the activity of PKC. This disclosure also relates to pharmaceutical compositions comprising these compounds, methods of using these compounds in the treatment of various diseases and disorders, processes for preparing these compounds and intermediates useful in these processes.
[0005] Before the present invention is further described, it is to be understood that this invention is not limited to particular embodiments described, as such may, of course, vary. It is also to be understood that the terminology used herein is for the purpose of describing particular embodiments only, and is not intended to be limiting, since the scope of the present invention will be limited only by the appended claims.
[0006] It must be noted that as used herein and in the appended claims, the singular forms "a," "an," and "the" include plural referents unless the context clearly dictates otherwise. It is further noted that the claims may be drafted to exclude any optional element. As such, this statement is intended to serve as antecedent basis for use of such exclusive
terminology as "solely," "only" and the like in connection with the recitation of claim elements, or use of a "negative" limitation.
[0007] Where a range of values is provided, it is understood that each intervening value, to the tenth of the unit of the lower limit unless the context clearly dictates otherwise, between the upper and lower limit of that range and any other stated or intervening value in that stated range, is specifically contemplated. The upper and lower limits of these smaller ranges may independently be included in the smaller ranges, and are also encompassed within the invention, subject to any specifically excluded limit in the stated range. Where the stated range includes one or both of the limits, ranges excluding either or both of those included limits are also included in the invention.
[0008] The publications discussed herein are provided solely for their disclosure prior to the filing date of the present application. Nothing herein is to be construed as an admission that the present invention is not entitled to antedate such publication by virtue of prior invention. Further, the dates of publication provided may be different from the actual publication dates which may need to be independently confirmed.
[0009] Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. Although any methods and materials similar or equivalent to those described herein can also be used in the practice or testing of the present invention, the preferred methods and materials are now described. All publications mentioned herein are incorporated herein by reference to disclose and describe the methods and/or materials in connection with which the publications are cited.
[0010] Except as otherwise noted, the methods and techniques of the present embodiments are generally performed according to conventional methods well known in the art and as described in various general and more specific references that are cited and discussed throughout the present specification. See, e.g., Loudon, Organic Chemistry, Fourth Edition, New York: Oxford University Press, 2002, pp. 360-361, 1084-1085; Smith and March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Fifth Edition, Wiley-Interscience, 2001; or Vogel, A Textbook of Practical Organic Chemistry, Including Qualitative Organic Analysis, Fourth Edition, New York: Longman, 1978.
[0011] The nomenclature used herein to name the subject compounds is illustrated in the Examples herein. This nomenclature has generally been derived using the commercially- available AutoNom software (MDL, San Leandro, Calif.).
TERMS
[0012] The following terms have the following meanings unless otherwise indicated. Any undefined terms have their art recognized meanings.
[0013] "Alkyl" refers to monovalent saturated aliphatic hydrocarbyl groups having from 1 to 10 carbon atoms and preferably 1 to 6 carbon atoms. This term includes, by way of example, linear and branched hydrocarbyl groups such as methyl (CH3-), ethyl (CH3CH2-), n-propyl (CH3CH2CH2-), isopropyl ((CH3)2CH-), n-butyl (CH3CH2CH2CH2-), isobutyl ((CH3)2CHCH2-), sec-butyl ((CH3)(CH3CH2)CH-), t-butyl ((CH3)3C-), n-pentyl
(CH3CH2CH2CH2CH2-), and neopentyl ((CH3)3CCH2-).
[0014] The term "substituted alkyl" refers to an alkyl group as defined herein wherein one or more carbon atoms in the alkyl chain have been optionally replaced with a heteroatom such as -0-, -N-, -S-, -S(0)n- (where n is 0 to 2), -NR- (where R is hydrogen or alkyl) and having from 1 to 5 substituents selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, oxo, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, -SO-alkyl, -SO-aryl, -SO-heteroaryl, -S02-alkyl, -S02-aryl, -S02-heteroaryl, and -NRaRb, wherein R and R may be the same or different and are chosen from hydrogen, optionally substituted alkyl, cycloalkyl, alkenyl, cycloalkenyl, alkynyl, aryl, heteroaryl and heterocyclic.
[0015] "Alkylene" refers to divalent aliphatic hydrocarbyl groups preferably having from 1 to 6 and more preferably 1 to 3 carbon atoms that are either straight-chained or branched, and which are optionally interrupted with one or more groups selected from -0-, - NR10-, -NR10C(O , -C(0)NR10- and the like. This term includes, by way of example, methylene (-CH2-), ethylene (-CH2CH2-), n-propylene (-CH2CH2CH2-), iso-propylene (-CH2CH(CH3)-), (-C(CH3)2CH2CH2-), (-C(CH3)2CH2C(0)-), (-C(CH3)2CH2C(0)NH-), (-CH(CH3)CH2-), and the like.
[0016] "Substituted alkylene" refers to an alkylene group having from 1 to 3 hydrogens replaced with substituents as described for carbons in the definition of "substituted" below.
[0017] The term "alkane" refers to alkyl group and alkylene group, as defined herein.
[0018] The term "alkylaminoalkyl", "alkylaminoalkenyl" and "alkylaminoalkynyl" refers to the groups RNHR - where R is alkyl group as defined herein and R is alkylene, alkenylene or alkynylene group as defined herein.
[0019] The term "alkaryl" or "aralkyl" refers to the groups -alkylene-aryl and
-substituted alkylene-aryl where alkylene, substituted alkylene and aryl are defined herein.
[0020] "Alkoxy" refers to the group -O-alkyl, wherein alkyl is as defined herein.
Alkoxy includes, by way of example, methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, t- butoxy, sec-butoxy, n-pentoxy, and the like. The term "alkoxy" also refers to the groups alkenyl-O-, cycloalkyl-O-, cycloalkenyl-O-, and alkynyl-O-, where alkenyl, cycloalkyl, cycloalkenyl, and alkynyl are as defined herein.
[0021] The term "substituted alkoxy" refers to the groups substituted alkyl-O-, substituted alkenyl-O-, substituted cycloalkyl-O-, substituted cycloalkenyl-O-, and substituted alkynyl-O- where substituted alkyl, substituted alkenyl, substituted cycloalkyl, substituted cycloalkenyl and substituted alkynyl are as defined herein.
[0022] The term "alkoxyamino" refers to the group -NH-alkoxy, wherein alkoxy is defined herein.
[0023] The term "haloalkoxy" refers to the groups alkyl-O- wherein one or more hydrogen atoms on the alkyl group have been substituted with a halo group and include, by way of examples, groups such as trifluoromethoxy, and the like.
[0024] The term "haloalkyl" refers to a substituted alkyl group as described above, wherein one or more hydrogen atoms on the alkyl group have been substituted with a halo group. Examples of such groups include, without limitation, fluoroalkyl groups, such as trifluoromethyl, difluoromethyl, trifluoroethyl and the like.
[0025] The term "alkylalkoxy" refers to the groups -alkylene-O-alkyl, alkylene-O- substituted alkyl, substituted alkylene-O-alkyl, and substituted alkylene-O-substituted alkyl wherein alkyl, substituted alkyl, alkylene and substituted alkylene are as defined herein.
[0026] The term "alkylthioalkoxy" refers to the group -alkylene-S -alkyl, alkylene-S- substituted alkyl, substituted alkylene-S-alkyl and substituted alkylene-S-substituted alkyl wherein alkyl, substituted alkyl, alkylene and substituted alkylene are as defined herein.
[0027] "Alkenyl" refers to straight chain or branched hydrocarbyl groups having from 2 to 6 carbon atoms and preferably 2 to 4 carbon atoms and having at least 1 and preferably from 1 to 2 sites of double bond unsaturation. This term includes, by way of example, bi-vinyl, allyl, and but-3-en-l-yl. Included within this term are the cis and trans isomers or mixtures of these isomers.
[0028] The term "substituted alkenyl" refers to an alkenyl group as defined herein having from 1 to 5 substituents, or from 1 to 3 substituents, selected from alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl,
acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, oxo, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, -SO-alkyl, -SO-substituted alkyl, -SO-aryl, -SO-heteroaryl, -S02-alkyl, - S02-substituted alkyl, -S02-aryl and -S02-heteroaryl.
[0029] "Alkynyl" refers to straight or branched monovalent hydrocarbyl groups having from 2 to 6 carbon atoms and preferably 2 to 3 carbon atoms and having at least 1 and preferably from 1 to 2 sites of triple bond unsaturation. Examples of such alkynyl groups include acetylenyl (-C≡CH), and propargyl (-CH2C≡CH).
[0030] The term "substituted alkynyl" refers to an alkynyl group as defined herein having from 1 to 5 substituents, or from 1 to 3 substituents, selected from alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, oxo, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, -SO-alkyl, -SO-substituted alkyl, -SO-aryl, -SO-heteroaryl, -S02-alkyl, -S02-substituted alkyl, -S02-aryl, and -S02-heteroaryl.
[0031] "Alkynyloxy" refers to the group -O-alkynyl, wherein alkynyl is as defined herein. Alkynyloxy includes, by way of example, ethynyloxy, propynyloxy, and the like.
[0032] "Acyl" refers to the groups H-C(O)-, alkyl-C(O)-, substituted alkyl-C(O)-, alkenyl-C(O)-, substituted alkenyl-C(O)-, alkynyl-C(O)-, substituted alkynyl-C(O)-, cycloalkyl-C(O)-, substituted cycloalkyl-C(O)-, cycloalkenyl-C(O)-, substituted
cycloalkenyl-C(O)-, aryl-C(O)-, substituted aryl-C(O)-, heteroaryl-C(O)-, substituted heteroaryl-C(O)-, heterocyclyl-C(O)-, and substituted heterocyclyl-C(O)-, wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic are as defined herein. For example, acyl includes the "acetyl" group CH3C(0)-
[0033] "Acylamino" refers to the groups -NR20C(O)alkyl, -NR20C(O)substituted alkyl, N R20C(O)cycloalkyl, -NR20C(O)substituted cycloalkyl, -NR20C(O)cycloalkenyl,
-NR20C(O)substituted cycloalkenyl, -NR20C(O)alkenyl, -NR20C(O)substituted alkenyl, -NR20C(O)alkynyl, -NR20C(O)substituted alkynyl, -NR20C(O)aryl, -NR20C(O)substituted
aryl, -NR2UC(0)heteroaryl, -NR2UC(0)substituted heteroaryl, -NR2UC(0)heterocyclic, and -NR20C(O)substituted heterocyclic, wherein R20 is hydrogen or alkyl and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic are as defined herein.
[0034] "Aminocarbonyl" or the term "aminoacyl"_refers to the group -C(0)NR21R22,
21 22
wherein R and R independently are selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R21
22
and R are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic are as defined herein.
[0035] The term "alkoxycarbonylamino" refers to the group -NRC(0)OR where each R is independently hydrogen, alkyl, substituted alkyl, aryl, heteroaryl, or heterocyclyl wherein alkyl, substituted alkyl, aryl, heteroaryl, and heterocyclyl are as defined herein.
[0036] The term "acyloxy" refers to the groups alkyl-C(0)0-, substituted alkyl-C(0)0-, cycloalkyl-C(0)0-, substituted cycloalkyl-C(0)0-, aryl-C(0)0-, heteroaryl-C(0)0-, and heterocyclyl-C(0)0- wherein alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, aryl, heteroaryl, and heterocyclyl are as defined herein.
[0037] "Aminosulfonyl" refers to the group -S02NR21R22, wherein R21 and R22 independently are selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted
21 22
heteroaryl, heterocyclic, substituted heterocyclic and where R and R are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group and alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic and substituted heterocyclic are as defined herein.
[0038] "Sulfonylamino" refers to the group -NR 1S02R , wherein R and R independently are selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R21 and R22 are optionally joined together with the atoms bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic are as defined herein.
[0039] "Aryl" or "Ar" refers to a monovalent aromatic carbocyclic group of from 6 to 14 carbon atoms having a single ring (e.g., phenyl) or multiple condensed rings (e.g., naphthyl or anthryl) which condensed rings may or may not be aromatic, provided that the point of attachment is through an atom of the aromatic aryl group. This term includes, by way of example, phenyl and naphthyl. Unless otherwise constrained by the definition for the aryl substituent, such aryl groups can optionally be substituted with from 1 to 5 substituents, or from 1 to 3 substituents, selected from acyloxy, hydroxy, thiol, acyl, alkyl, alkoxy, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, substituted alkyl, substituted alkoxy, substituted alkenyl, substituted alkynyl, substituted cycloalkyl, substituted cycloalkenyl, amino, substituted amino, aminoacyl, acylamino, alkaryl, aryl, aryloxy, azido, carboxyl, carboxylalkyl, cyano, halogen, nitro, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, aminoacyloxy, oxyacylamino, thioalkoxy, substituted thioalkoxy, thioaryloxy, thioheteroaryloxy, -SO-alkyl, -SO-substituted alkyl, -SO-aryl, -SO-heteroaryl, -S02-alkyl, -S02-substituted alkyl, -S02- aryl, -S02-heteroaryl and trihalomethyl.
[0040] "Aryloxy" refers to the group -O-aryl, wherein aryl is as defined herein, including, by way of example, phenoxy, naphthoxy, and the like, including optionally substituted aryl groups as also defined herein.
[0041] "Amino" refers to the group -NH2.
[0042] The term "substituted amino" refers to the group -NRR where each R is independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, alkenyl, substituted alkenyl, cycloalkenyl, substituted cycloalkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, and heterocyclyl provided that at least one R is not hydrogen.
[0043] The term "azido" refers to the group -N3.
[0044] "Carboxyl," "carboxy" or "carboxylate" refers to -C02H or salts thereof.
[0045] "Carboxyl ester" or "carboxy ester" or the terms "carboxyalkyl" or
"carboxylalkyl" refers to the groups -C(0)0-alkyl, -C(0)0-substituted alkyl,
-C(0)0-alkenyl, -C(0)0-substituted alkenyl, -C(0)0-alkynyl, -C(0)0-substituted alkynyl, -C(0)0-aryl, -C(0)0-substituted aryl, -C(0)0-cycloalkyl, -C(0)0-substituted cycloalkyl, -C(0)0-cycloalkenyl, -C(0)0-substituted cycloalkenyl, -C(0)0-heteroaryl,
-C(0)0-substituted heteroaryl, -C(0)0-heterocyclic, and -C(0)0-substituted heterocyclic, wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic are as defined herein.
[0046] "(Carboxyl ester)oxy" or "carbonate" refers to the groups -0-C(0)0-alkyl, -0-C(0)0-substituted alkyl, -0-C(0)0-alkenyl, -0-C(0)0-substituted alkenyl, -0-C(0)0- alkynyl, -0-C(0)0-substituted alkynyl, -0-C(0)0-aryl, -0-C(0)0-substituted aryl, -O- C(0)0-cycloalkyl, -0-C(0)0-substituted cycloalkyl, -0-C(0)0-cycloalkenyl, -0-C(0)0- substituted cycloalkenyl, -0-C(0)0-heteroaryl, -0-C(0)0-substituted heteroaryl, -O- C(0)0-heterocyclic, and -0-C(0)0-substituted heterocyclic, wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic are as defined herein.
[0047] "Cyano" or "nitrile" refers to the group -CN.
[0048] "Cycloalkyl" refers to cyclic alkyl groups of from 3 to 10 carbon atoms having single or multiple cyclic rings including fused, bridged, and spiro ring systems. Examples of suitable cycloalkyl groups include, for instance, adamantyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclooctyl and the like. Such cycloalkyl groups include, by way of example, single ring structures such as cyclopropyl, cyclobutyl, cyclopentyl, cyclooctyl, and the like, or multiple ring structures such as adamantanyl, and the like.
[0049] The term "substituted cycloalkyl" refers to cycloalkyl groups having from 1 to 5 substituents, or from 1 to 3 substituents, selected from alkyl, substituted alkyl, alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy,
oxyaminoacyl, azido, cyano, halogen, hydroxyl, oxo, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, hydroxyamino,
alkoxyamino, nitro, -SO-alkyl, -SO-substituted alkyl, -SO-aryl, -SO-heteroaryl, -S02-alkyl, - S02-substituted alkyl, -S02-aryl and -S02-heteroaryl.
[0050] "Cycloalkenyl" refers to non-aromatic cyclic alkyl groups of from 3 to 10 carbon atoms having single or multiple rings and having at least one double bond and preferably from 1 to 2 double bonds.
[0051] The term "substituted cycloalkenyl" refers to cycloalkenyl groups having from 1 to 5 substituents, or from 1 to 3 substituents, selected from alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, -SO-alkyl, -SO-substituted alkyl, -SO-aryl, -SO-heteroaryl, -S02-alkyl, -S02-substituted alkyl, -S02- aryl and -S02-heteroaryl.
[0052] "Cycloalkynyl" refers to non-aromatic cycloalkyl groups of from 5 to 10 carbon atoms having single or multiple rings and having at least one triple bond.
[0053] "Cycloalkoxy" refers to -O-cycloalkyl.
[0054] "Cycloalkenyloxy" refers to -O-cycloalkenyl.
[0055] "Halo" or "halogen" refers to fluoro, chloro, bromo, and iodo.
[0056] "Hydroxy" or "hydroxyl" refers to the group -OH.
[0057] "Heteroaryl" refers to an aromatic group of from 1 to 10 carbon atoms and 1 to 4 heteroatoms selected from the group consisting of oxygen, nitrogen, and sulfur within the ring. Such heteroaryl groups can have a single ring (e.g., pyridinyl, imidazolyl or furyl) or multiple condensed rings (e.g., indolizinyl, quinolinyl, benzimidazolyl or benzothienyl), wherein the condensed rings may or may not be aromatic and/or contain a heteroatom, provided that the point of attachment is through an atom of the aromatic heteroaryl group. In certain embodiments, the nitrogen and/or sulfur ring atom(s) of the heteroaryl group are optionally oxidized to provide for the N-oxide (N→0), sulfinyl, or sulfonyl moieties. This term includes, by way of example, pyridinyl, pyrrolyl, indolyl, thiophenyl, and furanyl. Unless otherwise constrained by the definition for the heteroaryl substituent, such heteroaryl groups can be optionally substituted with 1 to 5 substituents, or from 1 to 3 substituents, selected from acyloxy, hydroxy, thiol, acyl, alkyl, alkoxy, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, substituted alkyl, substituted alkoxy, substituted alkenyl, substituted alkynyl, substituted cycloalkyl, substituted cycloalkenyl, amino, substituted amino, aminoacyl,
acylamino, alkaryl, aryl, aryloxy, azido, carboxyl, carboxylalkyl, cyano, halogen, nitro, heteroaryl, heteroaryloxy, heterocyclyl, heterocyclooxy, aminoacyloxy, oxyacylamino, thioalkoxy, substituted thioalkoxy, thioaryloxy, thioheteroaryloxy, -SO-alkyl, -SO- substituted alkyl, -SO-aryl, -SO-heteroaryl, -S02-alkyl, -S02-substituted alkyl, -S02-aryl and -S02-heteroaryl, and trihalomethyl.
[0058] The term "heteroaralkyl" refers to the groups -alkylene-heteroaryl where alkylene and heteroaryl are defined herein. This term includes, by way of example, pyridylmethyl, pyridylethyl, indolylmethyl, and the like.
[0059] "Heteroaryloxy" refers to -O-heteroaryl.
[0060] "Heterocycle," "heterocyclic," "heterocycloalkyl," and "heterocyclyl" refer to a saturated or unsaturated group having a single ring or multiple condensed rings, including fused bridged and spiro ring systems, and having from 3 to 15 ring atoms, including 1 to 4 hetero atoms. These ring atoms are selected from the group consisting of nitrogen, sulfur, or oxygen, wherein, in fused ring systems, one or more of the rings can be cycloalkyl, aryl, or heteroaryl, provided that the point of attachment is through the non-aromatic ring. In certain embodiments, the nitrogen and/or sulfur atom(s) of the heterocyclic group are optionally oxidized to provide for the N-oxide, -S(O)-, or -S02- moieties.
[0061] Examples of heterocycles and heteroaryls include, but are not limited to, azetidine, pyrrole, imidazole, pyrazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, dihydroindole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, phenanthroline, isothiazole, phenazine, isoxazole, phenoxazine, phenothiazine, imidazolidine, imidazoline, piperidine, piperazine, indoline, phthalimide, 1,2,3,4-tetrahydroisoquinoline, 4,5,6,7-tetrahydrobenzo[b]thiophene, thiazole, thiazolidine, thiophene, benzo[b]thiophene, morpholinyl, thiomorpholinyl (also referred to as thiamorpholinyl), 1,1-dioxothiomorpholinyl, piperidinyl, pyrrolidine, tetrahydrofuranyl, and the like.
[0062] Unless otherwise constrained by the definition for the heterocyclic substituent, such heterocyclic groups can be optionally substituted with 1 to 5, or from 1 to 3 substituents, selected from alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, substituted amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, oxo, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclyl,
heterocyclooxy, hydroxyamino, alkoxyamino, nitro, -SO-alkyl, -SO-substituted alkyl, -SO- aryl, -SO-heteroaryl, -S02-alkyl, -S02-substituted alkyl, -S02-aryl, -S02-heteroaryl, and fused heterocycle.
[0063] "Heterocyclyloxy" refers to the group -O-heterocyclyl.
[0064] The term "heterocyclylthio" refers to the group heterocyclic-S-.
[0065] The term "heterocyclene" refers to the diradical group formed from a heterocycle, as defined herein.
[0066] The term "hydroxyamino" refers to the group -NHOH.
[0067] "Nitro" refers to the group -N02.
[0068] "Oxo" refers to the atom (=0).
[0069] "Sulfonyl" refers to the group S02-alkyl, S02-substituted alkyl, S02-alkenyl, S02-substituted alkenyl, S02-cycloalkyl, S02-substituted cylcoalkyl, S02-cycloalkenyl, S02-substituted cylcoalkenyl, S02-aryl, S02-substituted aryl, S02-heteroaryl, SO2- substituted heteroaryl, S02-heterocyclic, and S02-substituted heterocyclic, wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic are as defined herein. Sulfonyl includes, by way of example, methyl-S02-, phenyl-S02-, and 4- methylphenyl- S O2- .
[0070] "Sulfonyloxy" refers to the group -OS02-alkyl, OS02-substituted alkyl, OS02- alkenyl, OS02-substituted alkenyl, OS02-cycloalkyl, OS02-substituted cylcoalkyl, OSO2- cycloalkenyl, OS02-substituted cylcoalkenyl, OS02-aryl, OS02-substituted aryl, OSO2- heteroaryl, OS02-substituted heteroaryl, OS02-heterocyclic, and OSO2 substituted heterocyclic, wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted
cycloalkenyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic are as defined herein.
[0071] The term "aminocarbonyloxy" refers to the group -OC(0)NRR where each R is independently hydrogen, alkyl, substituted alkyl, aryl, heteroaryl, or heterocyclic wherein alkyl, substituted alkyl, aryl, heteroaryl and heterocyclic are as defined herein.
[0072] "Thiol" refers to the group -SH.
[0073] "Thioxo" or the term "thioketo" refers to the atom (=S).
[0074] "Alkylthio" or the term "thioalkoxy" refers to the group -S-alkyl, wherein alkyl is as defined herein. In certain embodiments, sulfur may be oxidized to -S(O)-. The sulfoxide may exist as one or more stereoisomers.
[0075] The term "substituted thioalkoxy" refers to the group -S-substituted alkyl.
[0076] The term "thioaryloxy" refers to the group aryl-S- wherein the aryl group is as defined herein including optionally substituted aryl groups also defined herein.
[0077] The term "thioheteroaryloxy" refers to the group heteroaryl-S- wherein the heteroaryl group is as defined herein including optionally substituted aryl groups as also defined herein.
[0078] The term "thioheterocyclooxy" refers to the group heterocyclyl-S- wherein the heterocyclyl group is as defined herein including optionally substituted heterocyclyl groups as also defined herein.
[0079] In addition to the disclosure herein, the term "substituted," when used to modify a specified group or radical, can also mean that one or more hydrogen atoms of the specified group or radical are each, independently of one another, replaced with the same or different substituent groups as defined below.
[0080] In addition to the groups disclosed with respect to the individual terms herein, substituent groups for substituting for one or more hydrogens (any two hydrogens on a single carbon can be replaced with =0, =NR70, =N-OR70, =N2 or =S) on saturated carbon atoms in the specified group or radical are, unless otherwise specified, -R60, halo, =0, -OR70, -SR70, -NR80R80, trihalomethyl, -CN, -OCN, -SCN, -NO, -N02, =N2, -N3, -S02R™, -S020 M+, -S02OR70, -OS02R70, -OS020 M+, -OS02OR70, -P(0)(0~)2(M+)2, -P(O)(OR70)O~M+, -P(O)(OR70) 2, -C(0)R70, -C(S)R70, -C(NR70)R70, -C(0)O M+, -C(0)OR70, -C(S)OR70, -C(O)NR80R80, -C(NR70)NR80R80, -OC(0)R70, -OC(S)R70, -OC(0)0"M+, -OC(0)OR70, -OC(S)OR70, -NR70C(O)R70, -NR70C(S)R70, -NR70CO2 M+, -NR70CO2R70, -NR70C(S)OR70, -NR70C(O)NR80R80, -NR70C(NR70)R70 and -NR70C(NR70)NR80R80, where R60 is selected from the group consisting of optionally substituted alkyl, cycloalkyl, heteroalkyl, heterocycloalkylalkyl, cycloalkylalkyl, aryl, arylalkyl, heteroaryl and heteroarylalkyl, each R70 is independently hydrogen or R60; each R80 is independently R70 or alternatively, two R80s, taken together with the nitrogen atom to which they are bonded, form a 5-, 6- or 7- membered heterocycloalkyl which may optionally include from 1 to 4 of the same or different additional heteroatoms selected from the group consisting of O, N and S, of which N may have -H or C1-C3 alkyl substitution; and each M+ is a counter ion with a net single positive charge. Each M+ may independently be, for example, an alkali ion, such as K+, Na+,
Li+; an ammonium ion, such as +N(R60)4; or an alkaline earth ion, such as [Ca2+]o.s, [Mg2+]o.s, or [Ba2+]o.5 ("subscript 0.5 means e.g. that one of the counter ions for such divalent alkali earth ions can be an ionized form of a compound of the invention and the other a typical counter ion such as chloride, or two ionized compounds of the invention can serve as counter ions for such divalent alkali earth ions, or a doubly ionized compound of the invention can serve as the counter ion for such divalent alkali earth ions). As specific examples, -NR80R80 is meant to include -NH2, -NH-alkyl, N-pyrrolidinyl, N-piperazinyl, 4N-methyl-piperazin-l- yl and N-morpholinyl.
[0081] In addition to the groups disclosed with respect to the individual terms herein, substituent groups for hydrogens on unsaturated carbon atoms in "substituted" alkene, alkyne, aryl and heteroaryl groups are, unless otherwise specified, -R60, halo, -0"M+, -OR70, -SR70, -S~M+, -NR80R80, trihalomethyl, -CF3, -CN, -OCN, -SCN, -NO, -N02, -N3, -S02R™, -S03 ~M+, -SO3R™, -OSO2R™, -OS03 M+, -OSO3R70, -P03 ~2(M+)2, -P(O)(OR70)O~M+, -P(O)(OR70)2, -C(0)R70, -C(S)R70, -C(NR70)R70, -C02 ~M+, -C02R70, -C(S)OR70,
-C(O)NR80R80, -C(NR70)NR80R80, -OC(0)R70, -OC(S)R70, -OC02 M+, -OC02R70,
-OC(S)OR70, -NR70C(O)R70, -NR70C(S)R70, -NR70CO2 M+, -NR70CO2R70, -NR70C(S)OR70, -NR70C(O)NR80R80, -NR70C(NR70)R70 and -NR70C(NR70)NR80R80, where R60, R70, R80 and M+ are as previously defined, provided that in case of substituted alkene or alkyne, the substituents are not -0"M+, -OR70, -SR70, or -STVT\
[0082] In addition to the disclosure herein, substituent groups for hydrogens on nitrogen atoms in "substituted" heteroalkyl and cycloheteroalkyl groups are, unless otherwise specified, -R60, -0"M+, -OR70, -SR70, -S"M+, -NR80R80, trihalomethyl, -CF3, -CN, -NO, -N02, -S(0)2R70, -S(0)20"M+, -S(0)2OR70, -OS(0)2R70, -OS(0)20"M+, -OS(0)2OR70,
-P(0)(0")2(M+)2, -P(O)(OR70)O"M+, -P(O)(OR70)(OR70), -C(0)R70, -C(S)R70, -C(NR70)R70, -C(0)OR70, -C(S)OR70, -C(O)NR80R80, -C(NR70)NR80R80, -OC(0)R70, -OC(S)R70,
-OC(0)OR70, -OC(S)OR70, -NR70C(O)R70, -NR70C(S)R70, -NR70C(O)OR70, -NR70C(S)OR70, -NR70C(O)NR80R80, -NR70C(NR70)R70 and -NR70C(NR70)NR80R80, where R60, R70, R80 and M+ are as previously defined.
[0083] In addition to the disclosure herein, in a certain embodiment, a group that is substituted has 1, 2, 3, or 4 substituents, 1, 2, or 3 substituents, 1 or 2 substituents, or 1 substituent.
[0084] It is understood that in all substituted groups defined above, polymers arrived at by defining substituents with further substituents to themselves (e.g., substituted aryl having a substituted aryl group as a substituent which is itself substituted with a substituted aryl
group, which is further substituted by a substituted aryl group, etc.) are not intended for inclusion herein. In such cases, the maximum number of such substitutions is three. For example, serial substitutions of substituted aryl groups are limited to substituted aryl- (substituted aryl)-substituted aryl.
[0085] Unless indicated otherwise, the nomenclature of substituents that are not explicitly defined herein are arrived at by naming the terminal portion of the functionality followed by the adjacent functionality toward the point of attachment. For example, the substituent "arylalkyloxycarbonyl" refers to the group (aryl)-(alkyl)-0-C(0)-.
[0086] As to any of the groups disclosed herein which contain one or more substituents, it is understood, of course, that such groups do not contain any substitution or substitution patterns which are sterically impractical and/or synthetically non-feasible. In addition, the subject compounds include all stereochemical isomers arising from the substitution of these compounds.
[0087] The term "pharmaceutically acceptable salt" means a salt which is acceptable for administration to a patient, such as a mammal (e.g., salts having acceptable mammalian safety for a given dosage regime). Such salts can be derived from pharmaceutically acceptable inorganic or organic bases and from pharmaceutically acceptable inorganic or organic acids. "Pharmaceutically acceptable salt" refers to pharmaceutically acceptable salts of a compound, which salts are derived from a variety of organic and inorganic counter ions well known in the art and include, by way of example only, sodium, potassium, calcium, magnesium, ammonium, tetraalkylammonium, and the like; and when the molecule contains a basic functionality, salts of organic or inorganic acids, such as hydrochloride,
hydrobromide, formate, tartrate, besylate, mesylate, acetate, maleate, oxalate, and the like.
[0088] The term "salt thereof" means a compound formed when the hydrogen of an acid is replaced by a cation, such as a metal cation or an organic cation and the like. Where applicable, the salt is a pharmaceutically acceptable salt, although this is not required for salts of compounds that are not intended for administration to a patient. By way of example, salts of the present compounds include those wherein the compound is protonated by an inorganic or organic acid to form a cation, with the conjugate base of the inorganic or organic acid as the anionic component of the salt.
[0089] "Solvate" refers to a complex formed by combination of solvent molecules with molecules or ions of the solute. The solvent can be an organic compound, an inorganic compound, or a mixture of both. Some examples of solvents include, but are not limited to,
methanol, Ν,Ν-dimethylformamide, tetrahydrofuran, dimethylsulfoxide, and water. When the solvent is water, the solvate formed is a hydrate.
[0090] "Stereoisomer" and "stereoisomers" refer to compounds that have same atomic connectivity but different atomic arrangement in space. Stereoisomers include cis-trans isomers, E and Z isomers, enantiomers, and diastereomers.
[0091] "Tautomer" refers to alternate forms of a molecule that differ only in electronic bonding of atoms and/or in the position of a proton, such as enol-keto and imine-enamine tautomers, or the tautomeric forms of heteroaryl groups containing a -N=C(H)-NH- ring atom arrangement, such as pyrazoles, imidazoles, benzimidazoles, triazoles, and tetrazoles. A person of ordinary skill in the art would recognize that other tautomeric ring atom arrangements are possible.
[0092] It will be appreciated that the term "or a salt or solvate or stereoisomer thereof" is intended to include all permutations of salts, solvates and stereoisomers, such as a solvate of a pharmaceutically acceptable salt of a stereoisomer of subject compound.
[0093] "Pharmaceutically effective amount" and "therapeutically effective amount" refer to an amount of a compound sufficient to treat a specified disorder or disease or one or more of its symptoms and/or to prevent the occurrence of the disease or disorder. In reference to tumorigenic proliferative disorders, a pharmaceutically or therapeutically effective amount comprises an amount sufficient to, among other things, cause the tumor to shrink or decrease the growth rate of the tumor.
[0094] "Patient" refers to human and non-human animals, especially mammals.
[0095] The term "treating" or "treatment" as used herein means the treating or treatment of a disease or medical condition in a patient, such as a mammal (particularly a human) that includes: (a) preventing the disease or medical condition from occurring, i.e., prophylactic treatment of a patient; (b) ameliorating the disease or medical condition, i.e., eliminating or causing regression of the disease or medical condition in a patient; (c) suppressing the disease or medical condition, i.e., slowing or arresting the development of the disease or medical condition in a patient; or (d) alleviating the symptoms of the disease or medical condition in a patient.
Representative Embodiments
[0096] The following substituents and values are intended to provide representative examples of various aspects and embodiments. These representative values are intended to further define and illustrate such aspects and embodiments and are not intended to exclude other embodiments or to limit the scope of this invention. In this regard, the representation
that a particular value or substituent is preferred is not intended in any way to exclude other values or substituents from this invention unless specifically indicated.
[0097] These compounds may contain one or more chiral centers and therefore, the embodiments are directed to racemic mixtures; pure stereoisomers (i.e., enantiomers or diastereomers); stereoisomer-enriched mixtures and the like unless otherwise indicated. When a particular stereoisomer is shown or named herein, it will be understood by those skilled in the art that minor amounts of other stereoisomers may be present in the compositions unless otherwise indicated, provided that the desired utility of the composition as a whole is not eliminated by the presence of such other isomers.
[0098] The compositions of the present disclosure include compounds of formula I, shown below. Pharmaceutical compositions and methods of the present disclosure also contemplate compounds of formula I.
Formula I
[0099] In one of its composition aspects, the present embodiments provide a compound of formula (I):

wherein
R is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
Y1 and Y2 are independently selected from hydrogen, alkyl, and acyl;
R1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
Ra and Rb are independently selected from hydrogen and alkyl;
Rc and Rd are independently selected from hydrogen and alkyl;
Q is selected from N and CR7b;
R6a, R6b, R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl,
substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O- alk-A;
alk is a bond, alkylene or substituted alkylene;
A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
wherein the A ring can be substituted or unsubstituted;
R7y is selected from hydrogen, alkyl, cycloalkyl, and substituted alkyl;
or a salt or stereoisomer thereof.
[00100] In formula I, R5 can be selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl. In certain instances, R5 is cyano, halogen, acyl, aminoacyl, or nitro. In certain instances, R5 is halogen. In certain instances, R5 is fluoro. In certain instances, R5 is cyano. In certain instances, R5 is fluoro, cyano, or aminoacyl. In certain instances, R5 is cyano or aminoacyl.
[00101] In formula I, Y1 and Y2 can be independently selected from hydrogen, alkyl, and acyl. In certain instances, Y1 is hydrogen. In certain instances, Y1 is alkyl. In certain instances, Y1 is acyl. In certain instances, Y2 is hydrogen. In certain instances, Y2 is alkyl. In certain instances, Y2 is acyl.
[00102] In formula I, R1 can be selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical. In certain instances, R1 is hydrogen or alkyl. In certain instances, R1 is hydrogen. In certain instances, R1 is alkyl. In certain instances, R1 is methyl. In certain instances, R1 is hydrogen, alkyl, substituted alkyl, or oxy radical. In certain instances, R1 is hydrogen, alkyl, substituted alkyl, acyl, or cycloalkyl.
[00103] In formula I, Ra and Rb can be independently selected from hydrogen and alkyl. In certain instances, Ra and Rb are both alkyl. In certain instances, Ra and Rb are both methyl. In certain instances, at least one of Ra and Rb is alkyl.
[00104] In formula I, Rc and Rd can be independently selected from hydrogen and alkyl. In certain instances, Rc and Rd are both alkyl. In certain instances, Rc and Rd are both methyl. In certain instances, at least one of Rc and Rd is alkyl.
[00105] In formula I, Q can be selected from N and CR7b. In certain instances, Q is CR7b. In certain instances, Q is N.
[00106] In formula I, R6a, R6b, R7b and R8 can be independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino,
substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A.
[00107] In certain instances, in formula I, R6a, R6b, R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, cycloalkyl, substituted cycloalkyl, and - O-alk-A.
[00108] In certain instances, in formula I, R6a, R6b, R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, acyl, carboxyl, carboxyl ester, aminoacyl, and -O-alk-A. In certain instances, at least one of R6a, R6b, R7b and R8 is alkyl or substituted alkyl. In certain instances, at least one of R6a, R6b, R7b and R8 is halogen. In certain instances, at least one of R6a, R6b, R7b and R8 is alkoxy, substituted alkoxy, or -O-alk-A. In certain instances, at least one of R6a, R6b, R7b and R8 is cyano, acyl, carboxyl, carboxyl ester, or aminoacyl. In certain instances, at least one of R6a, R6b, R7b and R8 is cycloalkyl or substituted cycloalkyl.
[00109] In certain instances, at least one of R6a, R6b, R7b and R8 are independently selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
[00110] In certain instances, one of R6a and R6b is fluoro and the other is hydrogen.
[00111] In certain instances, R6a is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain instances, R6a is selected from hydrogen, alkyl, substituted alkyl, and halogen. In certain instances, R6a is hydrogen. In certain instances, R6a is selected from alkyl and substituted alkyl. In certain instances, R6a is halogen. In certain instances, R6a is fluoro.
[00112] In certain instances, R6b is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain instances, R6b is selected from hydrogen, alkyl, substituted alkyl, and halogen. In certain instances, R6b is hydrogen. In certain instances, R6b is selected from alkyl and substituted alkyl. In certain instances, R6b is halogen. In certain instances, R6b is fluoro.
[00113] In certain instances, R7b is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain
instances, R is selected from hydrogen, alkyl, and substituted alkyl. In certain instances, R7b is hydrogen. In certain instances, R7b is selected from alkyl and substituted alkyl.
[00114] In certain instances, R7b is selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, acyl, carboxyl, carboxyl ester, aminoacyl, and -O-alk-A.
[00115] In certain instances, R8 is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain instances, R8 is hydrogen. In certain instances, R8 is alkyl or substituted alkyl. In certain instances, R8 is methyl. In certain instances, R8 is halogen. In certain instances, R8 is fluoro. In certain instances, R8 is alkoxy or substituted alkoxy. In certain instances, R8 is fluoro. In certain instances, R8 is cycloalkyl or substituted cycloalkyl. In certain instances, R8 is cyclopropyl.
[00116] In certain instances, any of R7b or R8 is selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, acyl, carboxyl, carboxyl ester, aminoacyl, and -O-alk-A. In certain instances, at least one of R7b or R8 is cycloalkyl, or substituted cycloalkyl. In certain instances, at least one of R7b or R8 is
7b 8 alkoxy, substituted alkoxy, or -O-alk-A. In certain instances, at least one of R'u or R° is alkyl, substituted alkyl, or halogen.
[00117] In formula I, for "-O-alk-A," alk is a bond, alkylene or substituted alkylene. In certain instances, alk is a bond. In certain instances, alk is alkylene. In certain instances, alk is ethylene or propylene. In certain instances, alk is substituted alkylene. In certain instances, alk is substituted ethylene or substituted propylene.
[00118] In certain instances, in formula I, for "-O-alk-A," alk is a bond or alk is ethylene, substituted ethylene, propylene, or -0(Ο¾)2θ¾-. In certain instances, in formula I, for "- O-alk-A," alk is a bond or alk is substituted propylene, -C(CH3)2CH2CH2-, or
-C(CH3)2CH2C(0)-.
[00119] In formula I, A can be selected from aryl, cycloalkyl, heteroaryl, and
heterocyclyl; wherein the A ring can be substituted or unsubstituted. In certain instances, A is aryl or substituted aryl. In certain instances, A is cycloalkyl or substituted cycloalkyl. In certain instances, A is heteroaryl or substituted heteroaryl. In certain instances, A is heterocyclyl or substituted heterocyclyl. In certain instances, A is heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl.
[00120] In certain instances, in formula I, A is selected from azetidine, imidazole, pyrazole, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, dihydroindole,
indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, pteridine, carbazole, carboline, isothiazole, phenazine, isoxazole, imidazolidine, imidazoline, oxazole, oxazolidine, piperidine, piperazine, indoline, phthalimide, 1,2,3,4-tetrahydroisoquinoline, tetrazole, triazole, thiazole, thiazolidine, thiophene, thiomorpholinyl, 1,1-dioxothiomorpholinyl, piperidinyl, 3-pyrrolidine; wherein the A ring can be substituted or unsubstituted.
[00121] In certain instances, in formula I, A is selected from 1-triazole, 3-pyrrolidine, 4- piperidine, and 1-imidazolidine; wherein the A ring can be substituted or unsubstituted.
[00122] In certain instances, in formula I, A is selected from piperidine,
tetrahydropyranyl, tetrahydrothiopyranyl, azetidinyl, azepanyl, and furanyl; wherein the A ring can be substituted or unsubstituted.
[00123] In formula I, R7y is selected from hydrogen, alkyl, cycloalkyl, and substituted alkyl. In certain instances, R7y is hydrogen. In certain instances, R7y is alkyl. In certain instances, R7y is methyl. In certain instances, R7y is isopropyl. In certain instances, R7y is cycloalkyl. In certain instances, R7y is substituted alkyl.
Formula II
[00124] In one of its composition aspects, the present embodiments provide a compound of formula (II):

R5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
Y1 and Y2 are independently selected from hydrogen, alkyl, and acyl;
R1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
Ra and Rb are independently selected from hydrogen and alkyl;
Rc and Rd are independently selected from hydrogen and alkyl;
Q is selected from N and CR7b;
R a, R , R and R are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O- alk-A;
alk is a bond, alkylene or substituted alkylene;
A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
wherein the A ring can be substituted or unsubstituted;
or a salt or stereoisomer thereof.
[00125] In formula II, R5 can be selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl. In certain instances, R5 is cyano, halogen, acyl, aminoacyl, or nitro. In certain instances, R5 is halogen. In certain instances, R5 is fluoro. In certain instances, R5 is fluoro, cyano, or aminoacyl. In certain instances, R5 is cyano or aminoacyl.
[00126] In formula II, Y1 and Y2 can be independently selected from hydrogen, alkyl, and acyl. In certain instances, Y1 is hydrogen. In certain instances, Y1 is alkyl. In certain instances, Y1 is acyl. In certain instances, Y2 is hydrogen. In certain instances, Y2 is alkyl. In certain instances, Y2 is acyl.
[00127] In formula II, R1 can be selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical. In certain instances, R1 is hydrogen or alkyl. In certain instances, R1 is hydrogen. In certain instances, R1 is alkyl. In certain instances, R1 is methyl. In certain instances, R1 is hydrogen, alkyl, substituted alkyl, or oxy radical. In certain instances, R1 is hydrogen, alkyl, substituted alkyl, acyl, or cycloalkyl.
[00128] In formula II, Ra and Rb can be independently selected from hydrogen and alkyl. In certain instances, Ra and Rb are both alkyl. In certain instances, Ra and Rb are both methyl. In certain instances, at least one of Ra and Rb is alkyl.
[00129] In formula II, Rc and Rd can be independently selected from hydrogen and alkyl. In certain instances, Rc and Rd are both alkyl. In certain instances, Rc and Rd are both methyl. In certain instances, at least one of Rc and Rd is alkyl.
[00130] In formula II, Q can be selected from N and CR . In certain instances, Q is CR7b. In certain instances, Q is N.
[00131] In formula II, R7b and R8 can be independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino,
aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A.
[00132] In certain instances, in formula II, R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, and -O-alk-A.
[00133] In certain instances, R7b and R8 are independently selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
[00134] In certain instances, R7b is selected from hydrogen, alkyl, and substituted alkyl. In certain instances, R7b is hydrogen. In certain instances, R7b is selected from alkyl and substituted alkyl.
[00135] In certain instances, R8 is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, and substituted alkoxy. In certain instances, R8 is hydrogen. In certain instances, R8 is alkyl or substituted alkyl. In certain instances, R8 is methyl. In certain instances, R8 is halogen. In certain instances, R8 is fluoro. In certain instances, R8 is alkoxy or substituted alkoxy.
[00136] In formula II, for "-O-alk-A," alk can be present or not present and is alkyl or substituted alkyl. In certain instances, alk is not present. In certain instances, alk is present and is alkyl. In certain instances, alk is present and is ethylene or propylene. In certain instances, alk is present and is substituted alkyl. In certain instances, alk is present and is substituted ethylene or substituted propylene.
[00137] In certain instances, in formula II, for "-O-alk-A," alk is not present or alk is present and is ethylene, substituted ethylene, propylene, or -C(CH3)2CH2-. In certain instances, in formula I, for "-O-alk-A," alk is not present or alk is present and is substituted propylene, -C(CH3)2CH2CH2-, or -C(CH3)2CH2C(0)-.
[00138] In formula II, A can be selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein the A ring can be substituted or unsubstituted. In certain instances, A
is aryl or substituted aryl. In certain instances, A is cycloalkyl or substituted cycloalkyl. In certain instances, A is heteroaryl or substituted heteroaryl. In certain instances, A is heterocyclyl or substituted heterocyclyl. In certain instances, A is heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl.
[00139] In certain instances, in formula II, A is selected from azetidine, imidazole, pyrazole, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, dihydroindole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, pteridine, carbazole, carboline, isothiazole, phenazine, isoxazole, imidazolidine, imidazoline, oxazole, oxazolidine, piperidine, piperazine, indoline, phthalimide, 1,2,3,4-tetrahydroisoquinoline, tetrazole, triazole, thiazole, thiazolidine, thiophene, thiomorpholinyl, 1,1-dioxothiomorpholinyl, piperidinyl, 3-pyrrolidine; wherein the A ring can be substituted or unsubstituted.
[00140] In certain instances, in formula II, A is selected from 1-triazole, 3-pyrrolidine, 4- piperidine, and 1 -imidazolidine; wherein the A ring can be substituted or unsubstituted.
[00141] In certain instances, in formula II, A is selected from piperidine,
tetrahydropyranyl, tetrahydrothiopyranyl, azetidinyl, azepanyl, and furanyl; wherein the A ring can be substituted or unsubstituted.
Formula III
[00142] In one of its composition aspects, the present embodiments provide a compound of formula (III):

(III)
wherein
Y1 and Y2 are independently selected from hydrogen, alkyl, and acyl;
R1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
Ra and Rb are independently selected from hydrogen and alkyl;
Rc and Rd are independently selected from hydrogen and alkyl;
Q is selected from N and CR7b;
R6a, R6b, R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino,
acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O- alk-A;
alk is a bond, alkylene or substituted alkylene;
A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
wherein the A ring can be substituted or unsubstituted;
R7y is selected from hydrogen, alkyl, and substituted alkyl;
or a salt or stereoisomer thereof.
[00143] In formula III, Y1 and Y2 can be independently selected from hydrogen, alkyl, and acyl. In certain instances, Y1 is hydrogen. In certain instances, Y1 is alkyl. In certain instances, Y1 is acyl. In certain instances, Y2 is hydrogen. In certain instances, Y2 is alkyl. In certain instances, Y2 is acyl.
[00144] In formula III, R1 can be selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical. In certain instances, R1 is hydrogen or alkyl. In certain instances, R1 is hydrogen. In certain instances, R1 is alkyl. In certain instances, R1 is methyl. In certain instances, R1 is hydrogen, alkyl, substituted alkyl, or oxy radical. In certain instances, R1 is hydrogen, alkyl, substituted alkyl, acyl, or cycloalkyl.
[00145] In formula III, Ra and Rb can be independently selected from hydrogen and alkyl. In certain instances, Ra and Rb are both alkyl. In certain instances, Ra and Rb are both methyl. In certain instances, at least one of Ra and Rb is alkyl.
[00146] In formula III, Rc and Rd can be independently selected from hydrogen and alkyl. In certain instances, Rc and Rd are both alkyl. In certain instances, Rc and Rd are both methyl. In certain instances, at least one of Rc and Rd is alkyl.
[00147] In formula III, Q can be selected from N and CR7b. In certain instances, Q is CR7b. In certain instances, Q is N.
[00148] In formula III, R7b and R8 can be independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino,
aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A.
[00149] In certain instances, in formula III, R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, and -O-alk-A.
[00150] In certain instances, R7b and R8 are independently selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
[00151] In certain instances, R7b is selected from hydrogen, alkyl, and substituted alkyl. In certain instances, R7b is hydrogen. In certain instances, R7b is selected from alkyl and substituted alkyl.
[00152] In certain instances, R8 is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, and substituted alkoxy. In certain instances, R8 is hydrogen. In certain instances, R8 is alkyl or substituted alkyl. In certain instances, R8 is methyl. In certain instances, R8 is halogen. In certain instances, R8 is fluoro. In certain instances, R8 is alkoxy or substituted alkoxy.
[00153] In formula III, for "-O-alk-A," alk can be present or not present and is alkyl or substituted alkyl. In certain instances, alk is not present. In certain instances, alk is present and is alkyl. In certain instances, alk is present and is ethylene or propylene. In certain instances, alk is present and is substituted alkyl. In certain instances, alk is present and is substituted ethylene or substituted propylene.
[00154] In certain instances, in formula III, for "-O-alk-A," alk is not present or alk is present and is ethylene, substituted ethylene, propylene, or -0(Ο¾)2θ¾-. In certain instances, in formula I, for "-O-alk-A," alk is not present or alk is present and is substituted propylene, -C(CH3)2CH2CH2-, or -C(CH3)2CH2C(0)-.
[00155] In formula III, A can be selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein the A ring can be substituted or unsubstituted. In certain instances, A is aryl or substituted aryl. In certain instances, A is cycloalkyl or substituted cycloalkyl. In certain instances, A is heteroaryl or substituted heteroaryl. In certain instances, A is heterocyclyl or substituted heterocyclyl. In certain instances, A is heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl.
[00156] In certain instances, in formula III, A is selected from azetidine, imidazole, pyrazole, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, dihydroindole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine,
quinoxaline, quinazoline, pteridine, carbazole, carboline, isothiazole, phenazine, isoxazole, imidazolidine, imidazoline, oxazole, oxazolidine, piperidine, piperazine, indoline, phthalimide, 1,2,3,4-tetrahydroisoquinoline, tetrazole, triazole, thiazole, thiazolidine, thiophene, thiomorpholinyl, 1,1-dioxothiomorpholinyl, piperidinyl, 3-pyrrolidine; wherein the A ring can be substituted or unsubstituted.
[00157] In certain instances, in formula III, A is selected from 1-triazole, 3-pyrrolidine, 4- piperidine, and 1 -imidazolidine; wherein the A ring can be substituted or unsubstituted.
[00158] In certain instances, in formula III, A is selected from piperidine,
tetrahydropyranyl, tetrahydrothiopyranyl, azetidinyl, azepanyl, and furanyl; wherein the A ring can be substituted or unsubstituted.
[00159] In formula III, R7y is selected from hydrogen, alkyl, and substituted alkyl. In certain instances, R7y is hydrogen. In certain instances, R7y is alkyl. In certain instances, R7y is methyl. In certain instances, R7y is isopropyl. In certain instances, R7y is substituted alkyl.
Formula IV
[00160] In one of its composition aspects, the present embodiments provide a compound of formula (IV):

wherein
R5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
Y1 and Y2 are independently selected from hydrogen, alkyl, and acyl;
R1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
Ra and Rb are independently selected from hydrogen and alkyl;
Rc and Rd are independently selected from hydrogen and alkyl;
Q is selected from N and CR7b;
R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl,
alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A;
alk is a bond, alkylene or substituted alkylene;
A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
wherein the A ring can be substituted or unsubstituted;
R7y is selected from hydrogen, alkyl, and substituted alkyl;
or a salt or stereoisomer thereof.
Formula V
[00161] In one of its composition aspects, the present embodiments provide a compound of formula (V):

(V)
wherein
R5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
Y1 and Y2 are independently selected from hydrogen, alkyl, and acyl;
R1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
Ra and Rb are independently selected from hydrogen and alkyl;
Rc and Rd are independently selected from hydrogen and alkyl;
Q is selected from N and CR7b;
R6a, R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A;
alk is a bond, alkylene or substituted alkylene;
A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
wherein the A ring can be substituted or unsubstituted;
R7y is selected from hydrogen, alkyl, and substituted alkyl;
or a salt or stereoisomer thereof.
[00162] In formula V, Q can be selected from N and CR7b. In certain instances, Q is CR7b. In certain instances, Q is N.
[00163] In certain instances, in formula V, R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, and -O-alk-A.
[00164] In certain instances, R7b and R8 are independently selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
[00165] In certain instances, R7b is selected from hydrogen, alkyl, and substituted alkyl. In certain instances, R7b is hydrogen. In certain instances, R7b is selected from alkyl and substituted alkyl.
[00166] In certain instances, R8 is selected from cycloalkyl, substituted cycloalkyl, heterocyclyl, substituted heterocyclyl, and -O-alk-A. In certain instances, R8 is cycloalkyl or substituted cycloalkyl. In certain instances, R8 is cycloalkyl. In certain instances, R8 is substituted cycloalkyl. In certain instances, R8 is heterocyclyl or substituted heterocyclyl. In certain instances, R8 is heterocyclyl. In certain instances, R8 is substituted heterocyclyl. In certain instances, R8 is -O-alk-A.
[00167] In certain instances, R8 is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, and substituted alkoxy. In certain instances, R8 is hydrogen. In certain instances, R8 is alkyl or substituted alkyl. In certain instances, R8 is methyl. In certain instances, R8 is halogen. In certain instances, R8 is fluoro. In certain instances, R8 is alkoxy or substituted alkoxy.
[00168] In formula V, for "-O-alk-A," alk can be present or not present and is alkyl or substituted alkyl. In certain instances, alk is not present. In certain instances, alk is present and is alkyl. In certain instances, alk is present and is ethylene or propylene. In certain instances, alk is present and is substituted alkyl. In certain instances, alk is present and is substituted ethylene or substituted propylene.
[00169] In certain instances, in formula V, for "-O-alk-A," alk is not present or alk is present and is ethylene, substituted ethylene, propylene, or -C(CH3)2CH2-. In certain
instances, in formula I, for "-O-alk-A," alk is not present or alk is present and is substituted propylene, -C(CH3)2CH2CH2-, or -C(CH3)2CH2C(0)-.
In formula V, A can be selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein the A ring can be substituted or unsubstituted. In certain instances, A is aryl or substituted aryl. In certain instances, A is cycloalkyl or substituted cycloalkyl. In certain instances, A is heteroaryl or substituted heteroaryl. In certain instances, A is heterocyclyl or substituted heterocyclyl. In certain instances, A is heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl.
Formula VI
[00170] In one of its composition aspects, the present embodiments provide a compound of formula (VI):

wherein
R5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
Y1 and Y2 are independently selected from hydrogen, alkyl, and acyl;
R1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
Ra and Rb are independently selected from hydrogen and alkyl;
Rc and Rd are independently selected from hydrogen and alkyl;
Q is selected from N and CR7b;
R6a, R6b, and R7b are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A;
alk is a bond, alkylene or substituted alkylene;
A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
wherein the A ring can be substituted or unsubstituted;
R7y is selected from hydrogen, alkyl, and substituted alkyl;
or a salt or stereoisomer thereof.
[00171] In certain instances, in formula VI, at least one of R6a and R6b is selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
[00172] In certain instances, one of R6a and R6b is fluoro and the other is hydrogen.
[00173] In certain instances, in formula VI, R6a is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain instances, R6a is selected from hydrogen, alkyl, substituted alkyl, and halogen. In certain instances, R6a is hydrogen. In certain instances, R6a is selected from alkyl and substituted alkyl. In certain instances, R6a is halogen. In certain instances, R6a is fluoro.
[00174] In certain instances, in formula VI, R6b is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain instances, R6b is selected from hydrogen, alkyl, substituted alkyl, and halogen. In certain instances, R6b is hydrogen. In certain instances, R6b is selected from alkyl and substituted alkyl. In certain instances, R6b is halogen. In certain instances, R6b is fluoro.
[00175] In formula VI, for "-O-alk-A," alk is a bond, alkylene or substituted alkylene. In certain instances, alk is a bond. In certain instances, alk is alkylene. In certain instances, alk is ethylene or propylene. In certain instances, alk is substituted alkylene. In certain instances, alk is substituted ethylene or substituted propylene.
[00176] In certain instances, in formula VI, for "-O-alk-A," alk is a bond or alk is ethylene, substituted ethylene, propylene, or -0(Ο¾)2θ¾-. In certain instances, in formula I, for "-O-alk-A," alk is a bond or alk is substituted propylene, -C(CH3)2CH2CH2-, or -C(CH3)2CH2C(0)-.
[00177] In formula VI, A can be selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein the A ring can be substituted or unsubstituted. In certain instances, A is aryl or substituted aryl. In certain instances, A is cycloalkyl or substituted cycloalkyl. In certain instances, A is heteroaryl or substituted heteroaryl. In certain instances, A is heterocyclyl or substituted heterocyclyl. In certain instances, A is heterocyclyl. In certain instances, A is substituted heterocyclyl. In certain instances, A is heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl.
Formula VII
[00178] In one of its composition aspects, the present embodiments provide a compound of formula (VII):

wherein
R is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
Y1 and Y2 are independently selected from hydrogen, alkyl, and acyl;
R1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
Ra and Rb are independently selected from hydrogen and alkyl;
Rc and Rd are independently selected from hydrogen and alkyl;
Q is selected from N and CR7b;
R6a, R6b, and R7b are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A;
alk is a bond, alkylene or substituted alkylene;
A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
wherein the A ring can be substituted or unsubstituted;
R7y is selected from hydrogen, alkyl, and substituted alkyl;
or a salt or stereoisomer thereof.
[00179] In certain instances, in formula VII, at least one of R a and R is selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
[00180] In certain instances, one of R6a and R6b is fluoro and the other is hydrogen.
[00181] In certain instances, in formula VII, R a is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain instances, R6a is selected from hydrogen, alkyl, substituted alkyl, and halogen. In certain instances, R6a is hydrogen. In certain instances, R6a is selected from alkyl and substituted alkyl. In certain instances, R6a is halogen. In certain instances, R6a is fluoro.
[00182] In certain instances, in formula VII, R6b is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl. In certain instances, R6b is selected from hydrogen, alkyl, substituted alkyl, and halogen. In certain instances, R6b is hydrogen. In certain instances, R6b is selected from alkyl and substituted alkyl. In certain instances, R6b is halogen. In certain instances, R6b is fluoro.
Formulae IV-VII
[00183] In formulae IV-VII, R5 can be selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl. In certain instances, R5 is cyano, halogen, acyl, aminoacyl, or nitro. In certain instances, R5 is halogen. In certain instances, R5 is fluoro. In certain instances, R5 is fluoro, cyano, or aminoacyl. In certain instances, R5 is cyano or aminoacyl.
[00184] In formulae IV-VII, Y1 and Y2 can be independently selected from hydrogen, alkyl, and acyl. In certain instances, Y1 is hydrogen. In certain instances, Y1 is alkyl. In certain instances, Y1 is acyl. In certain instances, Y2 is hydrogen. In certain instances, Y2 is alkyl. In certain instances, Y2 is acyl.
[00185] In formulae IV-VII, R1 can be selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical. In certain instances, R1 is hydrogen or alkyl. In certain instances, R1 is hydrogen. In certain instances, R1 is alkyl. In certain instances, R1 is methyl. In certain instances, R1 is hydrogen, alkyl, substituted alkyl, or oxy radical. In certain instances, R1 is hydrogen, alkyl, substituted alkyl, acyl, or cycloalkyl.
[00186] In formulae IV-VII, Ra and Rb can be independently selected from hydrogen and alkyl. In certain instances, Ra and Rb are both alkyl. In certain instances, Ra and Rb are both methyl. In certain instances, at least one of Ra and Rb is alkyl.
[00187] In formulae IV-VII, Rc and Rd can be independently selected from hydrogen and alkyl. In certain instances, Rc and Rd are both alkyl. In certain instances, Rc and Rd are both methyl. In certain instances, at least one of Rc and Rd is alkyl.
[00188] In formulae IV-VII, Q can be selected from N and CR7b. In certain instances, Q is CR7b. In certain instances, Q is N.
Formula VIII
[00189] In one of its composition aspects, the present embodiments provide a compound of formula (V

wherein
R5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
Y is selected from hydrogen and alkyl;
R1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
Ra and Rb are independently selected from hydrogen and alkyl;
Rc and Rd are independently selected from hydrogen and alkyl;
Q is selected from N and CR7b;
R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteraryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A;
alk, if present, is alkyl or substituted alkyl;
A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
wherein the A ring can be substituted or unsubstituted;
R7y is selected from hydrogen, alkyl, and substituted alkyl;
or a salt or stereoisomer thereof.
[00190] In formula VIII, R5 can be selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted
alkynyl. In certain instances, R5 is cyano, halogen, acyl, aminoacyl, or nitro. In certain instances, R5 is halogen. In certain instances, R5 is fluoro. In certain instances, R5 is fluoro, cyano, or aminoacyl. In certain instances, R5 is cyano or aminoacyl.
[00191] In formula VIII, Y can be selected from hydrogen and alkyl. In certain instances, Y is hydrogen. In certain instances, Y is alkyl.
[00192] In formula VIII, R1 can be selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical. In certain instances, R1 is hydrogen or alkyl. In certain instances, R1 is hydrogen. In certain instances, R1 is alkyl. In certain instances, R1 is methyl. In certain instances, R1 is hydrogen, alkyl, substituted alkyl, or oxy radical. In certain instances, R1 is hydrogen, alkyl, substituted alkyl, acyl, or cycloalkyl.
[00193] In formula VIII, Ra and Rb can be independently selected from hydrogen and alkyl. In certain instances, Ra and Rb are both alkyl. In certain instances, Ra and Rb are both methyl. In certain instances, at least one of Ra and Rb is alkyl.
[00194] In formula VIII, Rc and Rd can be independently selected from hydrogen and alkyl. In certain instances, Rc and Rd are both alkyl. In certain instances, Rc and Rd are both methyl. In certain instances, at least one of Rc and Rd is alkyl.
[00195] In formula VIII, Q can be selected from N and CR7b. In certain instances, Q is CR7b. In certain instances, Q is N.
[00196] In formula VIII, R7b and R8 can be independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino,
aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A.
[00197] In certain instances, in formula VIII, R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, and -O-alk-A.
[00198] In certain instances, R7b and R8 are independently selected from fluoro, trifluoromethyl, difluoromethoxy, hydroxyl, and isopropoxy.
[00199] In certain instances, R7b is selected from hydrogen, alkyl, and substituted alkyl. In certain instances, R7b is hydrogen. In certain instances, R7b is selected from alkyl and substituted alkyl.
[00200] In certain instances, R is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, and substituted alkoxy. In certain instances, R8 is hydrogen. In certain instances, R8 is alkyl or substituted alkyl. In certain instances, R8 is methyl. In certain instances, R8 is halogen. In certain instances, R8 is fluoro. In certain instances, R8 is alkoxy or substituted alkoxy.
[00201] In formula VIII, for "-O-alk-A," alk can be present or not present and is alkyl or substituted alkyl. In certain instances, alk is not present. In certain instances, alk is present and is alkyl. In certain instances, alk is present and is ethylene or propylene. In certain instances, alk is present and is substituted alkyl. In certain instances, alk is present and is substituted ethylene or substituted propylene.
[00202] In certain instances, in formula VIII, for "-O-alk-A," alk is not present or alk is present and is ethylene, substituted ethylene, propylene, or -0(Ο¾)2θ¾-. In certain instances, in formula I, for "-O-alk-A," alk is not present or alk is present and is substituted propylene, -C(CH3)2CH2CH2-, or -C(CH3)2CH2C(0)-.
[00203] In formula VIII, A can be selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl; wherein the A ring can be substituted or unsubstituted. In certain instances, A is aryl or substituted aryl. In certain instances, A is cycloalkyl or substituted cycloalkyl. In certain instances, A is heteroaryl or substituted heteroaryl. In certain instances, A is heterocyclyl or substituted heterocyclyl. In certain instances, A is heteroaryl, substituted heteroaryl, heterocyclyl, or substituted heterocyclyl.
[00204] In certain instances, in formula VIII, A is selected from azetidine, imidazole, pyrazole, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, dihydroindole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, pteridine, carbazole, carboline, isothiazole, phenazine, isoxazole, imidazolidine, imidazoline, oxazole, oxazolidine, piperidine, piperazine, indoline, phthalimide, 1,2,3,4-tetrahydroisoquinoline, tetrazole, triazole, thiazole, thiazolidine, thiophene, thiomorpholinyl, 1,1-dioxothiomorpholinyl, piperidinyl, 3-pyrrolidine; wherein the A ring can be substituted or unsubstituted.
[00205] In certain instances, in formula VIII, A is selected from 1-triazole, 3-pyrrolidine, 4-piperidine, and 1 -imidazolidine; wherein the A ring can be substituted or unsubstituted.
[00206] In certain instances, in formula VIII, A is selected from piperidine,
tetrahydropyranyl, tetrahydrothiopyranyl, azetidinyl, azepanyl, and furanyl; wherein the A ring can be substituted or unsubstituted.
[00207] In formula VIII, R y is selected from hydrogen, alkyl, and substituted alkyl. In certain instances, R7y is hydrogen. In certain instances, R7y is alkyl. In certain instances, R7y is methyl. In certain instances, R7y is isopropyl. In certain instances, R7y is substituted alkyl.
[00208] Particular compounds of interest are shown illustrated the following table.
Table 1


cmpd R1 R5 R6a R6b R8 R7y salt
1-21 -H -F -H -H -H -CH3
1-22 -H -F -H -H -F -H -CH3
1-23 -CH3 -F -H -H -F -H -CH3
1-24 -H -F -H -H -H -CH3
1-25 -CH3 -F -H -H -H -CH3
1-26 -CH3 -F -H -H -H -CH3
1-27 -H -F -H -H -H -CH3
1-28 -H -F -H -H -H -CH3
1-29 -CH3 -F -H -H -H -Cl -CH3
1-30 -H -F -H -H -H -CI -CH3
1-31 -CH3 -F -H -H -OCH3 -H -CH3
1-32 -H -F -H -H -OCH3 -H -CH3
1-33 -H -F -H -H -H -\ -CH3
1-34 -CH3 -F -H -H -H -hd -CH3
1-35 -H -F -H -H -W -H -CH3
1-36 -CH3 -F -H -H -\< -H -CH3
1-37 -H -F -H -H -CI -H -CH3
1-38 -H -F -H -H -CF3 -Cl -CH3
1-39 -H -F -H -H -CF3 -\ -CH3
1-40 -CH3 -F -H -H -CF3 -\ -CH3
1-41 -H -CN -H -H -CF3 -\ -CH3
1-42 -CH3 -CN -H -H -CF3 -\ -CH3
1-43 -H -F -H -H -F -CH3

cmpd R1 R5 R6a R6b R8 R7y salt
1-44 -H -CN -H -H -F -CH3
1-45 -CH3 -F -H -H -CH3 -CH3 -CH3
1-46 -H -F -H -H -CH3 -CH3 -CH3
1-47 -CH3 -F -H -H -CF3 -H -CH3
1-48 -H -F -H -H -CF3 -H -CH3
1-49 -CH3 -F -H -H -CN -H -CH3
1-50 -H -F -H -H -CN -H -CH3
1-51 -H -F -H -H -CF3 -0-CH(CH3)2 -CH3
1-52 -CH3 -F -H -H -CF3 -0-CH(CH3)2 -CH3
1-53 -H -F -H -H -CF3 -CH3 -CH3
1-54 -CH3 -F -H -H -CF3 -CH3 -CH3
1-55 -H -F -H -H -CI -CH3 -CH3
1-56 -CH3 -F -H -H -CI -CH3 -CH3
1-57 -H -H -H -F
CONH2 ~\ -CH3
1-58 -H -F -H -H -H -CN -CH3
1-59 -H -F -H -H -F -\ -CH3
1-60 -H -F -H -H -COOCH3 -CH3 -CH3
1-61 -H -F -H -H -COOH -CH3 -CH3
1-62 -H -CN -H -H -F -\ -CH3
1-63 -CH3 -F -H -H -COOCH3 -CH3 -CH3
1-64 -CH3 -F -H -H -CH3 -CH3
CONHCH3
1-65 -CH3 -F -H -H -COOH -CH3 -CH3
1-66 -CH3 -F -F -H -H -F -CH3
1-67 -H -F -F -H -H -F -CH3
1-68 -H -F -H -F -H -CI -CH3
1-69 -CH3 -F -H -F -H -CI -CH3
1-70 -H -F -H -F -H -CH3
¾F3

cmpd R1 R5 R6a R6b R8 R7y salt
1-71 -H -F -H -F -H -H -CH3
1-72 -H -F -H -F -H - -CD3
1-73 -H -F -H -F -H - K
1-74 -H -F -H -F -H - -H formate salt
1-75 -H -F -H -F -H -Kl -H citrate salt
1-76 -H -F -H -F -H -K -H maleate salt umarate salt (2: 1
1-77 -H -F -H -F -H -Kl f
-H
ratio)
1-78 -H -F -H -F -H -Kl -H L-tartarate salt
1-79 -H -F -H -F -H -Kl -H HSO4 " salt
1-80 -H -F -H -F -H -K -H HC1 salt
1-81 -H -F -H -F -H -Kl -H benzoate salt
1-82 -H -F -H -F -H -K -H Tosylate salt
1-83 -H -F -H -F -H -K -H Besylate salt
1-84 -H -F -H -F -H -Kl -H Mesylate salt
1-85 -H -F -H -F -H -KJ -H Acetate salt
1-86 -H -F -H -F -H -OCH3 -CH3
1-87 -CH3 -F -H -F -H -OCH3 -CH3
[00209] Particular compounds of interest, and salts or solvates or stereoisomers thereof, include:
1-1: 5-Fluoro-N2-{4-fluoro-[3-(4-H)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-2: 5-Fluoro-N2-{4-fluoro-[3-(4-H)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-3: 5-Huoro-N2-{4-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-4: 5-Fluoro-N2-{4-fluoro-[3-(4-isopropyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-5: 5-Fluoro-N2-{4-fluoro-[3-(4-isopropyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-6: 5-Fluoro-N2-{4-fluoro-3-[4-(2-fluoroethyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-7: 5-Fluoro-N2-{4-fluoro-3-[4-(2-fluoroethyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-8: 5-Fluoro-N2-{4-methyl-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-9: 5-Fluoro-N2-{4-methyl-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (2,2,6, 6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
I- 10 : 5 -Fluoro-N2- { 4-isopropoxy- [3 -(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 -yl] } phenyl- N4-(l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
I- 11 : 5 -Fluoro-N2- { 4-isopropoxy- [3 -(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 -yl] } phenyl- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-12: 5-Fluoro-N2-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl]phenyl-N4-(l,2,2,6,6- pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-13: 5-Fluoro-N2-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl]phenyl-N4-(2,2,6,6- tetramethylpiperidin-4- yl) -2 ,4-pyrimidinediamine ;
1-14: l-(2-cyclopropyl-4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-15: 2-(4-cyclopropyl-2-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carbonitrile;
1-16: l-{2-Cyclopropyl-4-fluoro-5-[5-fluoro-4-(l, 2,2,6, 6-pentamethyl-piperidin-4- ylamino)-pyrimidin-2-ylamino]-phenyl}-4-methyl-l,4-dihydro-tetrazol-5-one;
1-17: 2-[4-Cyclopropyl-2-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-tetrazol-l-yl)- phenylamino]-4-(l,2,2,6,6-pentamethyl-piperidin-4-ylamino)-pyrimidine-5-carbonitrile;
1-18: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(l,l,l-trifluoro-2-methylpropan-2-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)- one;
1-19: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(l ,1 ,l-trifluoro-2-methylpropan-2-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)- one;
1-20: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-21: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-22: l-(3-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-23: l-(3-fluoro-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-24: l-(3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-25: l-(3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-26: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(tetrahydro-2H-pyran-4-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-27: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(tetrahydro-2H-pyran-4-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-28: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-((3-methyloxetan-3-yl)methoxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-29: l-(2-chloro-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-30: l-(2-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-31: l-(3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-methoxyphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-32: l-(3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-methoxyphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-33: l-(2-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-34: l-(2-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-35: l-(3-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-36: l-(3-cyclopropyl-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-37: l-(3-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-38: l-(2-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino) - 3 - (trifluoromethyl)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-39: l-(2-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-40: l-(2-cyclopropyl-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-41: 2-(4-cyclopropyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)-5- (trifluoromethyl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5- carbonitrile;
1-42: 2-(4-cyclopropyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)-5- (trifluoromethyl)phenylamino)-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidine-5- carbonitrile;
1-43: l-(3-cyclopropyl-2-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-44: 2-(3-cyclopropyl-4-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carbonitrile;
1-45: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2,3-dimethylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-46: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2,3-dimethylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-47: l-(3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino) -5 - (trifluoromethyl)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-48: l-(3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino) -5 - (trifluoromethyl)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-49: 3-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzonitrile;
1-50: 3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2-ylamino)-5- (4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzonitrile;
1-51: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-isopropoxy-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-52: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-isopropoxy-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-53: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-54: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-55: l-(3-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino)-2-methylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-56: l-(3-chloro-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)-2-methylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-57: 2-(4-cyclopropyl-3-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carboxamide;
1-58: 4-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2-ylamino)-2- (4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzonitrile;
1-59: l-(2-cyclopropyl-3-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-60: methyl 5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoate;
1-61: 5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2-ylamino)-2- methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoic acid;
1-62: 2-(4-cyclopropyl-3-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carbonitrile;
1-63 : methyl 5-(5-fluoro-4-(l ,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoate;
1-64: 5-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-N,2-dimethyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzamide;
1-65: 5-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoic acid;
1-66: l-(2,6-difluoro-3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-67: l-(2,6-difluoro-3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-68: l-(2-chloro-4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-69: l-(2-chloro-4-fluoro-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-70: l-(4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-((lS,2S)-2-(trifluoromethyl)cyclopropyl)phenyl)-4-methyl-lH-tetrazol-5(4H)- one;
1-71: l-(4-fluoro-3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-72: l-(2-cyclopropyl-4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-(trideuteromethyl)-lH-tetrazol-5(4H)-one;
1-73: N2-{4-Cyclopropyl-6-fluoro-[3-(4-isopropyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-74: N2-{4-Cyclopropyl-6-fluoro-3-(l,2,3,4-tetrazol-5-one-l-yl)}phenyl-5-fluoro- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, formate salt;
1-75: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, citrate salt;
1-76: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, maleate salt;
1-77 : N2- { 4-Cyclopropyl-6-fluoro- [3-(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 - yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, fumarate salt;
1-78: N2- { 4-Cyclopropyl-6-fluoro- [3-(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 - yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, L- tartarate salt;
1-79: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, hydrogen sulfate salt;
1-80: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, hydrogen chloride salt;
1-81 : N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediami benzoate salt;
1-82: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, tosylate salt;
1-83: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, besylate salt;
1-84: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, mesylate salt;
1-85: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, acetate salt;
1-86: N2-(4-fluoro-2-methoxy-3-(4-methyl-4,5-dihydro-5-oxo-lH-tetrazol-l- yl)phenyl)-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)pyrimidine-2,4-diamine; and
1-87: N2-(4-fluoro-2-methoxy-3-(4-methyl-4,5-dihydro-5-oxo-lH-tetrazol-l- yl)phenyl)-5-fluoro-N4-(l,2,2,6,6-pentamethylpiperidin-4-yl)pyrimidine-2,4-diamine.
[00210] Particular compounds of interest, and salts or solvates or stereoisomers thereof, include:
1-1 : 5-Fluoro-N2-{4-fluoro-[3-(4-H)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-2: 5-Fluoro-N2-{4-fluoro-[3-(4-H)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (2,2,6, 6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-3: 5-Fluoro-N2-{4-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-4: 5-Fluoro-N2-{4-fluoro-[3-(4-isopropyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (2,2,6, 6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-5: 5-Fluoro-N2-{4-fluoro-[3-(4-isopropyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-6: 5-Fluoro-N2-{4-fluoro-3-[4-(2-fluoroethyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-7: 5-Fluoro-N2-{4-fluoro-3-[4-(2-fluoroethyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-8: 5-Fluoro-N2-{4-methyl-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-9: 5-Fluoro-N2-{4-methyl-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (2,2,6, 6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
I- 10 : 5 -Fluoro-N2- { 4-isopropoxy- [3 -(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 -yl] } phenyl- N4-(l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
I- 11 : 5 -Fluoro-N2- { 4-isopropoxy- [3 -(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 -yl] } phenyl- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-12: 5-Fluoro-N2-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl]phenyl-N4-(l,2,2,6,6- pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine; and
1-13: 5-Fluoro-N2-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl]phenyl-N4-(2,2,6,6- tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine.
[00211] Particular compounds of interest, and salts or solvates or stereoisomers thereof, include:
1-14: l-(2-cyclopropyl-4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-15: 2-(4-cyclopropyl-2-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carbonitrile;
1-16: l-{2-Cyclopropyl-4-fluoro-5-[5-fluoro-4-(l, 2,2,6, 6-pentamethyl-piperidin-4- ylamino)-pyrimidin-2-ylamino]-phenyl}-4-methyl-l,4-dihydro-tetrazol-5-one; and
1-17: 2-[4-Cyclopropyl-2-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-tetrazol-l-yl)- phenylamino]-4-(l,2,2,6,6-pentamethyl-piperidin-4-ylamino)-pyrimidine-5-carbonitrile.
[00212] Particular compounds of interest, and salts or solvates or stereoisomers thereof, include:
1-18: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(l,l,l-trifluoro-2-methylpropan-2-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)- one;
1-19: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(l,l,l-trifluoro-2-methylpropan-2-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)- one;
1-20: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-21: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-22: l-(3-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-23: l-(3-fluoro-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-24: l-(3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-25: l-(3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-26: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(tetrahydro-2H-pyran-4-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-27: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(tetrahydro-2H-pyran-4-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-28: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-((3-methyloxetan-3-yl)methoxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-29: l-(2-chloro-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-30: l-(2-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-31: l-(3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-methoxyphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-32: l-(3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-methoxyphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-33: l-(2-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-34: l-(2-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-35: l-(3-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-36: l-(3-cyclopropyl-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-37: l-(3-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-38: l-(2-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino) - 3 - (trifluoromethyl)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-39: l-(2-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-40: l-(2-cyclopropyl-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-41: 2-(4-cyclopropyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)-5- (trifluoromethyl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5- carbonitrile;
1-42: 2-(4-cyclopropyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)-5- (trifluoromethyl)phenylamino)-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidine-5- carbonitrile;
1-43: l-(3-cyclopropyl-2-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-44: 2-(3-cyclopropyl-4-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carbonitrile;
1-45: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2,3-dimethylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-46: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2,3-dimethylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-47: l-(3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino) -5 - (trifluoromethyl)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-48: l-(3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino) -5 - (trifluoromethyl)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-49: 3-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzonitrile;
1-50: 3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2-ylamino)-5- (4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzonitrile;
1-51 : l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-isopropoxy-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-52: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-isopropoxy-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-53: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-54: l-(5-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-memyl-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-55: l-(3-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino)-2-methylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-56: l-(3-chloro-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)-2-methylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-57: 2-(4-cyclopropyl-3-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carboxamide;
1-58: 4-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2-ylamino)-2- (4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzonitrile;
1-59: l-(2-cyclopropyl-3-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-60: methyl 5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoate;
1-61 : 5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2-ylamino)-2- methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoic acid;
1-62: 2-(4-cyclopropyl-3-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carbonitrile;
1-63 : methyl 5-(5-fluoro-4-(l ,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoate;
1-64: 5-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-N,2-dimethyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzamide;
1-65: 5-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoic acid;
1-66: l-(2,6-difluoro-3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-67: l-(2,6-difluoro-3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-68: l-(2-chloro-4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one; and
1-69: l-(2-chloro-4-fluoro-5-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one.
[00213] Particular compounds of interest, and salts or solvates or stereoisomers thereof, include:
1-70: l-(4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-((lS,2S)-2-(trifluoromethyl)cyclopropyl)phenyl)-4-methyl-lH-tetrazol-5(4H)- one;
1-71: l-(4-fluoro-3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-72: l-(2-cyclopropyl-4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-(trideuteromethyl)-lH-tetrazol-5(4H)-one;
1-73: N2-{4-Cyclopropyl-6-fluoro-[3-(4-isopropyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-74: N2-{4-Cyclopropyl-6-fluoro-3-(l,2,3,4-tetrazol-5-one-l-yl)}phenyl-5-fluoro- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, formate salt;
1-75: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, citrate salt;
1-76: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, maleate salt;
1-77 : N2- { 4-Cyclopropyl-6-fluoro- [3-(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 - yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, fumarate salt;
1-78: N2- { 4-Cyclopropyl-6-fluoro- [3-(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 - yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, L- tartarate salt;
1-79: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, hydrogen sulfate salt;
1-80: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, hydrogen chloride salt;
1-81: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, benzoate salt;
1-82: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediam tosylate salt;
1-83: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediami besylate salt;
1-84: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediami mesylate salt;
1-85: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, acetate salt;
1-86: N2-(4-fluoro-2-methoxy-3-(4-methyl-4,5-dihydro-5-oxo-lH-tetrazol-l- yl)phenyl)-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)pyrimidine-2,4-diamine; and
1-87: N2-(4-fluoro-2-methoxy-3-(4-methyl-4,5-dihydro-5-oxo-lH-tetrazol-l- yl)phenyl)-5-fluoro-N4-(l,2,2,6,6-pentamethylpiperidin-4-yl)pyrimidine-2,4-diamine.
[00214] The compounds described also include isotopically labeled compounds where one or more atoms have an atomic mass different from the atomic mass conventionally found in nature. Examples of isotopes that may be incorporated into the compounds disclosed herein include, but are not limited to, 2H, 3H, nC, 13C, 14C, 15N, 180, 170, etc. Thus, the disclosed compounds may be enriched in one or more of these isotopes relative to the natural abundance of such isotope. By way of example, deuterium (2H) has a natural abundance of about 0.015%. Accordingly, for approximately every 6,500 hydrogen atoms occurring in nature, there is one deuterium atom. Specifically contemplated herein are compounds enriched in deuterium at one or more positions. Thus, deuterium containing compounds of the disclosure have deuterium at one or more positions (as the case may be) in an abundance of greater than 0.015%.
[00215] The present disclosure also provides pharmaceutical compositions comprising a pharmaceutically acceptable carrier and a therapeutically effective amount of a compound of formula I- VIII or a pharmaceutically acceptable salt or solvate or stereoisomer thereof.
[00216] A disclosed compound can be administered alone, as the sole active
pharmaceutical agent, or in combination with one or more additional compounds of formula I- VIII or in conjunction with other agents. When administered as a combination, the therapeutic agents can be formulated as separate compositions that are administered
simultaneously or at different times, or the therapeutic agents can be administered together as a single composition combining two or more therapeutic agents. Thus, the
pharmaceutical compositions disclosed herein containing a compound of formula I- VIII optionally contain other therapeutic agents. Accordingly, certain embodiments are directed to such pharmaceutical compositions, wherein the composition further comprises a therapeutically effective amount of an agent selected as is known to those of skill in the art.
[00217] The subject compounds can inhibit a protein kinase C activity. Accordingly, the compounds are useful for treating a disease or disorder that is mediated through the activity of a PKC activity in a subject. Also, the compounds are useful for treating a disease or disorder that is associated with the activation of T-cells in a subject.
[00218] The present disclosure provides a method of treating an inflammatory disease in a subject, the method comprising administering to the subject with a compound of formula I- VIII or a salt or solvate or stereoisomer thereof.
[00219] The present disclosure also provides a method of treating an autoimmune disease in a subject, the method comprising administering to the subject with a compound of formula I- VIII or a salt or solvate or stereoisomer thereof.
[00220] The present disclosure also provides a method of treating an ocular disease or disorder involving inflammatory and/or neovascular events.
[00221] The present disclosure also provides a method of treating diseases or conditions of interest including, but are not limited to, atherosclerosis, vascular occlusion due to vascular injury, angioplasty, restenosis, obesity, syndrome X, impaired glucose tolerance, polycystic ovary syndrome, hypertension, heart failure, chronic obstructive pulmonary disease, CNS diseases, Alzheimer disease, amyotrophic lateral sclerosis, bipolar disease, cancer, infectious disease, AIDS, septic shock, adult respiratory distress syndrome, ischemia/reperfusion injury, myocardial infarction, stroke, gut ischemia, renal failure, hemorrhage shock, and traumatic shock, and traumatic brain injury.
[00222] The present disclosure also provides a method of treating diseases or conditions of interest including, but are not limited to, T-cell mediated acute or chronic inflammatory diseases or disorders or autoimmune diseases, rheumatoid arthritis, osteoarthritis, systemic lupus erythematosus, Hashimoto's thyroiditis, multiple sclerosis, myasthenia gravis, diabetes type I or II and the disorders associated therewith, transplant rejection, graft versus host disease, respiratory diseases, asthma, inflammatory lung injury, inflammatory liver injury, inflammatory glomerular injury, cutaneous manifestations of immunologically-mediated disorders or illnesses, inflammatory and hyperproliferative skin diseases, psoriasis, atopic
dermatitis, allergic contact dermatitis, irritant contact dermatitis and further eczematous dermatitises, seborrhoeic dermatitis, inflammatory eye diseases, Sjoegren's syndrome, keratoconjunctivitis, uveitis, inflammatory bowel disease, Crohn's disease or ulcerative colitis, Guillain-Barre syndrome, and allergies.
[00223] The subject compounds can be used for treating a cell proliferative disorder. The present disclosure also provides a method of treating diseases or conditions of interest including, but are not limited to hematopoietic neoplasm, lymphoid neoplasm, T cell neoplasm, T lymphoblastic leukemia, B cell neoplasm, B-lymphoblastic leukemia, Burkitt's lymphoma, myeloid neoplasm, myeloproferative disease, chronic myelogenous leukemia (CML), myelodysplastic disease, chronic myelomonocytic leukemia, myelodysplastic syndrome, and acute myeloid leukemia.
[00224] Since subject compounds possess PKC inhibitory properties, such compounds are also useful as research tools. Accordingly, the disclosure also provides for a method for using a compound of formula I- VIII or a salt or solvate or stereoisomer thereof as a research tool for studying a biological system or sample, or for discovering new chemical compounds having PKC inhibitory properties.
[00225] The embodiments are also directed to processes and novel intermediates useful for preparing compounds of formula I- VIII or a salt or solvate or stereoisomer thereof.
[00226] In one embodiment, the above process further comprises the step of forming a salt of a compound of formula I- VIII. Embodiments are directed to the other processes described herein; and to the product prepared by any of the processes described herein.
[00227] The embodiments are also directed to a compound of formula I- VIII or a salt or solvate or stereoisomer thereof, for use in therapy or as a medicament.
[00228] Additionally, the embodiments are directed to the use of a compound of formula I- VIII or a salt or solvate or stereoisomer thereof, for the manufacture of a medicament; especially for the manufacture of a medicament for the inhibition of protein kinase C (PKC) activity. The embodiments are also directed to the use of a compound of formula I- VIII or a salt or solvate or stereoisomer thereof for the manufacture of a medicament for the treatment of a disease or disorder mediated or sustained through the activity of PKC activity. The embodiments are also directed to the use of a compound of formula I- VIII or a salt or solvate or stereoisomer thereof for the manufacture of a medicament for the treatment of a disease or disorder associated with the activation of T-cells. Diseases or conditions of interest include, but are not limited to, an inflammatory disease, an immunological disorder, an autoimmune disease, an ocular disease or disorder involving inflammatory and/or neovascular events,
organ and bone marrow transplant rejection, acute or chronic inflammation, allergies, contact dermatitis, psoriasis, rheumatoid arthritis, multiple sclerosis, type I diabetes, type II diabetes, inflammatory bowel disease, Guillain-Barre syndrome, Crohn's disease, ulcerative colitis, graft versus host disease, and lupus erythematosus.
[00229] The embodiments are also directed to the use of a compound of formula I- VIII or a salt or solvate or stereoisomer thereof for the manufacture of a medicament for the treatment of a cell proliferative disorder. Diseases or conditions of interest include, but are not limited to, hematopoietic neoplasm, lymphoid neoplasm, T cell neoplasm, T
lymphoblastic leukemia, B cell neoplasm, B -lymphoblastic leukemia, Burkitt's lymphoma, myeloid neoplasm, myeloproferative disease, chronic myelogenous leukemia (CML), myelodysplastic disease, chronic myelomonocytic leukemia, myelodysplastic syndrome, acute myeloid leukemia.
General Synthetic Procedures
[00230] Many general references providing commonly known chemical synthetic schemes and conditions useful for synthesizing the disclosed compounds are available (see, e.g., Smith and March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Fifth Edition, Wiley-Interscience, 2001; or Vogel, A Textbook of Practical Organic Chemistry, Including Qualitative Organic Analysis, Fourth Edition, New York: Longman, 1978).
[00231] Compounds as described herein can be purified by any of the means known in the art, including chromatographic means, such as HPLC, preparative thin layer
chromatography, flash column chromatography and ion exchange chromatography. Any suitable stationary phase can be used, including normal and reversed phases as well as ionic resins. Most typically the disclosed compounds are purified via silica gel and/or alumina chromatography. See, e.g., Introduction to Modern Liquid Chromatography, 2nd Edition, ed. L. R. Snyder and J. J. Kirkland, John Wiley and Sons, 1979; and Thin Layer
Chromatography, ed E. Stahl, Springer- Verlag, New York, 1969.
[00232] During any of the processes for preparation of the subject compounds, it may be necessary and/or desirable to protect sensitive or reactive groups on any of the molecules concerned. This may be achieved by means of conventional protecting groups as described in standard works, such as J. F. W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York 1973, in T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", Third edition, Wiley, New York 1999, in "The Peptides"; Volume 3 (editors: E. Gross and J. Meienhofer), Academic Press, London and New York
1981, in "Methoden der organischen Chemie", Houben-Weyl, 4 edition, Vol. 15/1, Georg Thieme Verlag, Stuttgart 1974, in H.-D. Jakubke and H. Jescheit, "Aminosauren, Peptide, Proteine", Verlag Chemie, Weinheim, Deerfield Beach, and Basel 1982, and/or in Jochen Lehmann, "Chemie der Kohlenhydrate: Monosaccharide and Derivate", Georg Thieme Verlag, Stuttgart 1974. The protecting groups may be removed at a convenient subsequent stage using methods known from the art.
[00233] The subject compounds can be synthesized via a variety of different synthetic routes using commercially available starting materials and/or starting materials prepared by conventional synthetic methods. Suitable exemplary methods that can be routinely adapted to synthesize the 2,4-pyrimidinediamine compounds and prodrugs of the invention are found in U.S. Patent No. 5,958,935, the disclosure of which is incorporated herein by reference. Specific examples describing the synthesis of numerous 2,4-pyrimidinediamine compounds and prodrugs, as well as intermediates therefore, are described in the U.S. publication No. US2004/0029902A1, the contents of which are incorporated herein by reference. Suitable exemplary methods that can be routinely used and/or adapted to synthesize active 2,4- pyrimidinediamine compounds can also be found in WO 03/063794, U.S. application Serial No. 10/631,029 filed July 29, 2003, WO2004/014382, U.S. publication No. 2005-0234049 Al, and WO005/016893, the disclosures of which are incorporated herein by reference. All of the compounds described herein (including prodrugs) can be prepared by routine adaptation of these methods.
[00234] Exemplary synthetic methods for the 2,4-substituted pyrimidinediamines described herein are described below. Those of skill in the art will also be able to readily adapt these methods for the synthesis of specific 2,4-substituted pyrimidinediamines as described herein.
[00235] A variety of exemplary synthetic routes that can be used to synthesize the 2,4- pyrimidinediamine compounds of the invention are described in scheme below. These methods can be routinely adapted to synthesize the 2,4-pyrimidinediamine compounds and prodrugs described herein.
Synthesis of Compounds
[00236] In a certain embodiment, the compounds can be synthesized from substituted or unsubstituted uracils as illustrated in Scheme 1, below:
Scheme 1

[00237] In Scheme 1, R1, Ra, Rb, Rc, Rd, R5, R6a, R6b, R7b, R7y, R8are as set forth hereinbefore.
[00238] According to Scheme 1, uracil A-l is dihalogenated at the 2- and 4-positions using a standard dehydrating-halogenating agent such as POCI3 (phosphorus oxychloride) (or other standard halogenating agent) under standard conditions to yield 2,4
dichloropyrimidine A-2. Depending upon the substituents in pyrimidinediamine A-2, the chloride at the C4 position is more reactive towards nucleophiles than the chloride at the C2 position. This differential reactivity can be exploited by first reacting 2,4
dichloropyrimidine A-2 with one equivalent of amine A-3, yielding 4N-substituted-2-chloro- 4-pyrimidineamine A-4, followed by amine A-5 to yield a 2,4-pyrimidinediamine derivative A-6.
[00239] Typically, the C4 halide is more reactive towards nucleophiles, as illustrated in the scheme. However, as will be recognized by skilled artisans, the identity of the substituent may alter this reactivity. For example, when the substituent is trifluoromethyl, a 50:50 mixture of 4N-substituted-4-pyrimidineamine A-4 and the corresponding 2N- substituted-2-pyrimidineamine is obtained. The regioselectivity of the reaction can also be controlled by adjusting the solvent and other synthetic conditions (such as temperature), as is well-known in the art.
[00240] In a certain embodiment, to couple compounds with an electrophilic leaving group, such as halides or pseudohalides, and compounds with an amino group, nucleophilic aromatic substitution can be used. For example, a halogen substituent on Compound A-2 and the amino group on Compound A-3 can react. Also for example, a halogen substituent on Compound A-4 and the amino group on Compound A-5 can react. Conditions for nucleophilic aromatic substitution include the compounds reacting in a polar aprotic solvent or polar protic solvent. Suitable solvents include alcohols (such as isopropanol, methanol, ethanol), formic acid, dimethylsulfoxide, dimethylformamide, dioxane, and tetrahydrofuran. The reaction can be run at room temperature or can be heated.
[00241] In a certain embodiment, to couple compounds with an electrophilic leaving group, such as halides or pseudohalides, and aryl compounds with an amino group, a coupling reaction, such as a Buchwald coupling reaction, can be used. The Buchwald coupling reaction involves palladium-catalyzed synthesis of aryl amines. Starting materials are aryl halides or pseudohalides (for example, triflates) and primary or secondary amines. Such reaction can be performed using a variety of methods well known in the art and specific examples can be had by reference to the Examples hereunder described.
[00242] The reactions depicted in Scheme 1 may proceed more quickly when the reaction mixtures are heated via microwave. When heating in this fashion, the following conditions can be used: heat to 175 °C in ethanol for 5-20 minutes in a Smith Reactor (Personal Chemistry, Uppsala, Sweden) in a sealed tube (at 20 bar pressure).
[00243] A specific embodiment of Scheme 1 utilizing 5-fluorouracil (Aldrich #32,937-1) as a starting material is illustrated in Scheme 2, below.
heme 2

[00244] In Scheme 2, R1, Ra, Rb, Rc, Rd, R6a, R6b, R7b, R7y, R8are as set forth hereinbefore.
[00245] Asymmetric 2N,4N-disubstituted-5-fluoro-2,4-pyrimidinediamine A- 10 can be obtained by reacting 2,4-dichloro-5-fluoropyrimidine A-8 with one equivalent of amine A-3 (to yield 2-chloro-N4-substituted-5-fluoro-4-pyrimidineamine A-9) followed by one or more equivalents of amine A-5.
[00246] A specific embodiment of Scheme 1 to form cyano derivatives is illustrated in Scheme 3, below.
heme 3

A-5
[00247] In Scheme 3, R1, Ra, Rb, Rc, Rd, R6a, R6b, R7b, R7y, R8are as set forth hereinbefore.
[00248] Asymmetric 2N,4N-disubstituted-5-cyano-2,4-pyrimidinediamine A- 15 can be obtained by reacting 2,4-dichloro-5-carbamoylpyrimidine A-12 with one equivalent of amine A-3 (to yield 2-chloro-N4-substituted-5-carbamoyl-4-pyrimidineamine A-13). The amide group of Compound A-13 is converted to a cyano group to yield Compound A-14, followed by reaction with one or more equivalents of amine A-5. Conversion of the amide group to the cyano group can be accomplished with dehydration, such as with use of Burgess reagent or trifluoroacetic anhydride. As will be recognized by those of skill in the art and exemplified herein, aniline A-5 may also be reacted with intermediate A-13, and the resultant N2,N4-disubstituted diaminopyrimidine-5-carbamoylpyrimidine can be dehydrated to yield the corresponding 5-cyano compound A-15.
Uracil Starting Materials and Intermediates
[00249] The uracil A-l, A-7, and A-ll starting materials can be purchased from commercial sources or prepared using standard techniques of organic chemistry.
Commercially available uracils that can be used as starting materials in the schemes disclosed herein include, by way of example and not limitation, uracil (Aldrich #13,078-8;
CAS Registry 66-22-8); 5 bromouracil (Aldrich #85,247-3; CAS Registry 51-20-7; 5 fluorouracil (Aldrich #85,847-1; CAS Registry 51-21-8); 5 iodouracil (Aldrich #85,785-8;
CAS Registry 696-07-1); 5 nitrouracil (Aldrich #85,276-7; CAS Registry 611-08-5); 5
(trifluoromethyl)-uracil (Aldrich #22,327-1 ; CAS Registry 54-20-6). Additional 5- substituted uracils are available from General Intermediates of Canada, Inc., Edmonton, CA and/or Interchim, Cedex, France, or can be prepared using standard techniques. Myriad textbook references teaching suitable synthetic methods are provided infra.
Amino Starting Materials and Intermediates
[00250] Amines, such as A-3 and A-5 can be purchased from commercial sources or, alternatively, can be synthesized utilizing standard techniques. For example, suitable amines can be synthesized from nitro precursors using standard chemistry. See also Vogel, 1989, Practical Organic Chemistry, Addison Wesley Longman, Ltd. and John Wiley & Sons, Inc.
Tetrazole Intermediates
[00251] Compound A-5 with an N-linked tetrazole in Schemes 1-3 was prepared as illustrated in Scheme 4 and may be incorporated into the present compounds according to the procedure illustrated in Scheme 4.
Scheme 4

A-17 A-5
A-16
[00252] In Scheme 4, R , R , R b, R , and R y are as previously defined.
[00253] To prepare Compound A-5, Compound A-16 was reacted to form tetrazolone Compound A-17 by treatment with phosgene and azidotrimethylsilane. The reaction is general to any appropriate aminophenyl compound. Compound A-17 was reacted to reduce the nitro group to form Compound A-5. Compound A-5 can also be prepared according to the procedures provided by Satoh et al., Tetrahedron Lett, 1995, 36, 1749; Gupta et al. Tetrahedron Lett, 2004, 45, 4113; Su et al. Eur. J. Org. Chem., 2006, 2723; and Potewar et al., Tetrahedron Lett, 2007, 48, 172.
[00254] Substitution of the ring with substituents can be performed with standard chemistry. In certain embodiment, substitution of the ring with substituents can be performed with nucleophilic aromatic substitution. For example, a halogen substituent can be replaced with another substituent with nucleophilic aromatic substitution. In certain embodiment, substitution of the ring with substituents can be performed with a metal catalyzed coupling reaction. For example, a halogen substituent can be replaced with another substituent with utilization of a metal catalyst. Suitable metal catalyzed reactions to
place appropriate substituents include Suzuki coupling, Stille coupling, and Buchwald coupling.
[00255] The nitro group of Compound A-17 was converted to an amino group to produce Compound A-5. The conversion of the nitro group to an amino group can be accomplished by various methods. A suitable method for reduction of nitro group is catalytic
hydrogenation which uses hydrogen and a catalyst, such as, but not limited to, palladium on carbon, platinum oxide, Raney nickel, and samarium diiodide.
[00256] Compound A- 16 can be purchased from commercial sources or prepared using standard techniques of organic chemistry. For example, Compound A- 16 can be prepared from the corresponding amine with standard techniques of organic chemistry. In certain embodiment, Compound A-16 can be prepared from the corresponding dinitro compound in which one of the nitro groups is reduced to an amino group. Myriad textbook references teaching suitable synthetic methods are provided infra.
[00257] Although many of the synthetic schemes discussed above do not illustrate the use of protecting groups, skilled artisans will recognize that in some instances certain substituents may include functional groups requiring protection. The exact identity of the protecting group used will depend upon, among other things, the identity of the functional group being protected and the reaction conditions used in the particular synthetic scheme, and will be apparent to those of skill in the art. Guidance for selecting protecting groups, their attachment and removal suitable for a particular application can be found, for example, in Greene & Wuts, supra.
[00258] Prodrugs as described herein can be prepared by routine modification of the above-described methods. Alternatively, such prodrugs can be prepared by reacting a suitably protected 2,4-pyrimidinediamine with a suitable progroup. Conditions for carrying out such reactions and for deprotecting the product to yield prodrugs as described herein are well-known.
[00259] Myriad references teaching methods useful for synthesizing pyrimidines generally, as well as starting materials described in Schemes (I)-(VII), are known in the art. For specific guidance, the reader is referred to Brown, D. J., "The Pyrimidines", in The Chemistry of Heterocyclic Compounds, Volume 16 (Weissberger, A., Ed.), 1962,
Interscience Publishers, (A Division of John Wiley & Sons), New York ("Brown I"); Brown, D. J., "The Pyrimidines", in The Chemistry of Heterocyclic Compounds, Volume 16, Supplement I (Weissberger, A. and Taylor, E. C, Ed.), 1970, Wiley-Interscience, (A
Division of John Wiley & Sons), New York (Brown Π"); Brown, D. J., "The Pyrimidines",
in The Chemistry of Heterocyclic Compounds, Volume 16, Supplement II (Weissberger, A. and Taylor, E. C, Ed.), 1985, An Interscience Publication (John Wiley & Sons), New York ("Brown III"); Brown, D. J., "The Pyrimidines" in The Chemistry of Heterocyclic
Compounds, Volume 52 (Weissberger, A. and Taylor, E. C, Ed.), 1994, John Wiley & Sons, Inc., New York, pp. 1-1509 (Brown IV"); Kenner, G. W. and Todd, A., in Heterocyclic Compounds, Volume 6, (Elderfield, R. C, Ed.), 1957, John Wiley, New York, Chapter 7 (pyrimidines); Paquette, L. A., Principles of Modern Heterocyclic Chemistry, 1968, W. A. Benjamin, Inc., New York, pp. 1 - 401 (uracil synthesis pp. 313, 315; pyrimidinediamine synthesis pp. 313-316; amino pyrimidinediamine synthesis pp. 315); Joule, J. A., Mills, K. and Smith, G. F., Heterocyclic Chemistry, 3rd Edition, 1995, Chapman and Hall, London, UK, pp. 1 - 516; Vorbriiggen, H. and Ruh-Pohlenz, C, Handbook of Nucleoside Synthesis, John Wiley & Sons, New York, 2001, pp. 1-631 (protection of pyrimidines by acylation pp. 90-91; silylation of pyrimidines pp. 91-93); Joule, J. A., Mills, K. and Smith, G. F.,
Heterocyclic Chemistry, 4th Edition, 2000, Blackwell Science, Ltd, Oxford, UK, pp. 1 - 589; and Comprehensive Organic Synthesis, Volumes 1-9 (Trost, B. M. and Fleming, I., Ed.), 1991, Pergamon Press, Oxford, UK.
Pharmaceutical Compositions
[00260] The disclosed compounds are useful, at least, for the inhibition of PKC activity and the treatment of a disease or disorder that is mediated through the activity of a PKC activity. Accordingly, pharmaceutical compositions comprising at least one disclosed compound are also described herein.
[00261] A pharmaceutical composition comprising a subject compound may be administered to a patient alone, or in combination with other supplementary active agents. The pharmaceutical compositions may be manufactured using any of a variety of processes, including, without limitation, conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping, and lyophilizing. The pharmaceutical composition can take any of a variety of forms including, without limitation, a sterile solution, suspension, emulsion, lyophilisate, tablet, pill, pellet, capsule, powder, syrup, elixir or any other dosage form suitable for administration.
[00262] A subject compound may be administered to the host using any convenient means capable of resulting in the desired reduction in disease condition or symptom. Thus, a subject compound can be incorporated into a variety of formulations for therapeutic administration. More particularly, a subject compound can be formulated into
pharmaceutical compositions by combination with appropriate pharmaceutically acceptable
carriers or diluents, and may be formulated into preparations in solid, semi-solid, liquid or gaseous forms, such as tablets, capsules, powders, granules, ointments, solutions, suppositories, injections, inhalants and aerosols.
[00263] Formulations for pharmaceutical compositions are well known in the art. For example, Remington's Pharmaceutical Sciences, by E. W. Martin, Mack Publishing Co., Easton, Pa., 19th Edition, 1995, describes exemplary formulations (and components thereof) suitable for pharmaceutical delivery of disclosed compounds. Pharmaceutical compositions comprising at least one of the subject compounds can be formulated for use in human or veterinary medicine. Particular formulations of a disclosed pharmaceutical composition may depend, for example, on the mode of administration and/or on the location of the infection to be treated. In some embodiments, formulations include a pharmaceutically acceptable carrier in addition to at least one active ingredient, such as a subject compound. In other embodiments, other medicinal or pharmaceutical agents, for example, with similar, related or complementary effects on the affliction being treated can also be included as active ingredients in a pharmaceutical composition.
[00264] Pharmaceutically acceptable carriers useful for the disclosed methods and compositions are conventional in the art. The nature of a pharmaceutical carrier will depend on the particular mode of administration being employed. For example, parenteral formulations usually comprise injectable fluids that include pharmaceutically and physiologically acceptable fluids such as water, physiological saline, balanced salt solutions, aqueous dextrose, glycerol or the like as a vehicle. For solid compositions (e.g., powder, pill, tablet, or capsule forms), conventional non-toxic solid carriers can include, for example, pharmaceutical grades of mannitol, lactose, starch, or magnesium stearate. In addition to biologically neutral carriers, pharmaceutical compositions to be administered can optionally contain minor amounts of non-toxic auxiliary substances (e.g., excipients), such as wetting or emulsifying agents, preservatives, and pH buffering agents and the like; for example, sodium acetate or sorbitan monolaurate. Other non- limiting excipients include, nonionic solubilizers, such as cremophor, or proteins, such as human serum albumin or plasma preparations.
[00265] Some examples of materials which can serve as pharmaceutically- acceptable carriers include: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as corn starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients, such as cocoa butter and suppository waxes; (9) oils, such as
peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol, and polyethylene glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14) buffering agents, such as magnesium hydroxide and aluminum hydroxide; (15) alginic acid; (16) pyrogen-free water; (17) isotonic saline; (18) Ringer's solution; (19) ethyl alcohol; (20) pH buffered solutions; (21) polyesters, polycarbonates and/or poly anhydrides; and (22) other non-toxic compatible substances employed in pharmaceutical formulations.
[00266] The disclosed pharmaceutical compositions may be formulated as a
pharmaceutically acceptable salt of a disclosed compound. Pharmaceutically acceptable salts are non-toxic salts of a free base form of a compound that possesses the desired pharmacological activity of the free base. These salts may be derived from inorganic or organic acids. Non-limiting examples of suitable inorganic acids are hydrochloric acid, nitric acid, hydrobromic acid, sulfuric acid, hydroiodic acid, and phosphoric acid. Non- limiting examples of suitable organic acids are acetic acid, propionic acid, glycolic acid, lactic acid, pyruvic acid, malonic acid, succinic acid, malic acid, maleic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, methyl sulfonic acid, salicylic acid, formic acid, trichloroacetic acid, trifluoroacetic acid, gluconic acid, asparagic acid, aspartic acid, benzenesulfonic acid, para- toluenesulfonic acid, naphthalenesulfonic acid, and the like. Lists of other suitable pharmaceutically acceptable salts are found in Remington's Pharmaceutical Sciences, 17th Edition, Mack Publishing Company, Easton, Pa., 1985. A pharmaceutically acceptable salt may also serve to adjust the osmotic pressure of the composition.
[00267] A subject compound can be used alone or in combination with appropriate additives to make tablets, powders, granules or capsules, for example, with conventional additives, such as lactose, mannitol, corn starch or potato starch; with binders, such as crystalline cellulose, cellulose derivatives, acacia, corn starch or gelatins; with disintegrators, such as corn starch, potato starch or sodium carboxymethylcellulose; with lubricants, such as talc or magnesium stearate; and if desired, with diluents, buffering agents, moistening agents, preservatives and flavoring agents. Such preparations can be used for oral administration.
[00268] A subject compound can be formulated into preparations for injection by dissolving, suspending or emulsifying them in an aqueous or nonaqueous solvent, such as vegetable or other similar oils, synthetic aliphatic acid glycerides, esters of higher aliphatic acids or propylene glycol; and if desired, with conventional additives such as solubilizers,
isotonic agents, suspending agents, emulsifying agents, stabilizers and preservatives. The preparation may also be emulsified or the active ingredient encapsulated in liposome vehicles. Formulations suitable for injection can be administered by an intravitreal, intraocular, intramuscular, subcutaneous, sublingual, or other route of administration, e.g., injection into the gum tissue or other oral tissue. Such formulations are also suitable for topical administration.
[00269] In some embodiments, a subject compound can be delivered by a continuous delivery system. The term "continuous delivery system" is used interchangeably herein with "controlled delivery system" and encompasses continuous (e.g., controlled) delivery devices (e.g., pumps) in combination with catheters, injection devices, and the like, a wide variety of which are known in the art.
[00270] A subject compound can be utilized in aerosol formulation to be administered via inhalation. A subject compound can be formulated into pressurized acceptable propellants such as dichlorodifluoromethane, propane, nitrogen and the like.
[00271] Furthermore, a subject compound can be made into suppositories by mixing with a variety of bases such as emulsifying bases or water-soluble bases. A subject compound can be administered rectally via a suppository. The suppository can include vehicles such as cocoa butter, carbowaxes and polyethylene glycols, which melt at body temperature, yet are solidified at room temperature.
[00272] The term "unit dosage form," as used herein, refers to physically discrete units suitable as unitary dosages for human and animal subjects, each unit containing a predetermined quantity of a subject compound calculated in an amount sufficient to produce the desired effect in association with a pharmaceutically acceptable diluent, carrier or vehicle. The specifications for a subject compound depend on the particular compound employed and the effect to be achieved, and the pharmacodynamics associated with each compound in the host.
[00273] The dosage form of a disclosed pharmaceutical composition will be determined by the mode of administration chosen. For example, in addition to injectable fluids, topical or oral dosage forms may be employed. Topical preparations may include eye drops, ointments, sprays and the like. Oral formulations may be liquid (e.g., syrups, solutions or suspensions), or solid (e.g., powders, pills, tablets, or capsules). Methods of preparing such dosage forms are known, or will be apparent, to those skilled in the art.
[00274] Certain embodiments of the pharmaceutical compositions comprising a subject compound may be formulated in unit dosage form suitable for individual administration of
precise dosages. The amount of active ingredient administered will depend on the subject being treated, the severity of the affliction, and the manner of administration, and is known to those skilled in the art. Within these bounds, the formulation to be administered will contain a quantity of the extracts or compounds disclosed herein in an amount effective to achieve the desired effect in the subject being treated.
[00275] Each therapeutic compound can independently be in any dosage form, such as those described herein, and can also be administered in various ways, as described herein. For example, the compounds may be formulated together, in a single dosage unit (that is, combined together in one form such as capsule, tablet, powder, or liquid, etc.) as a combination product. Alternatively, when not formulated together in a single dosage unit, an individual subject compound may be administered at the same time as another therapeutic compound or sequentially, in any order thereof.
Methods of Administration
[00276] The subject compounds can inhibit a protein kinase C activity. Accordingly, the subject compounds are useful for treating a disease or disorder that is mediated through the activity of a PKC activity in a subject. Accordingly, the subject compounds are useful for treating a disease or disorder that is associated with the activation of T-cells in a subject.
[00277] The route of administration will be selected according to a variety of factors including, but not necessarily limited to, the condition to be treated, the formulation and/or device used, the patient to be treated, and the like. Routes of administration useful in the disclosed methods include but are not limited to oral and parenteral routes, such as intravenous (iv), intraperitoneal (ip), rectal, topical, ophthalmic, nasal, and transdermal. Formulations for these dosage forms are described herein.
[00278] An effective amount of a subject compound will depend, at least, on the particular method of use, the subject being treated, the severity of the affliction, and the manner of administration of the therapeutic composition. A "therapeutically effective amount" of a composition is a quantity of a specified compound sufficient to achieve a desired effect in a subject (host) being treated. For example, this may be the amount of a subject compound necessary to prevent, inhibit, reduce or relieve a disease or disorder that is mediated through the activity of a PKC activity in a subject. Ideally, a therapeutically effective amount of a compound is an amount sufficient to prevent, inhibit, reduce or relieve a disease or disorder that is mediated through the activity of a PKC activity in a subject without causing a substantial cytotoxic effect on host cells.
[00279] Therapeutically effective doses (or growth inhibitory amounts) of a subject compound or pharmaceutical composition can be determined by one of skill in the art, with a goal of achieving local (e.g., tissue) concentrations that are at least as high as the ICso of an applicable compound disclosed herein.
[00280] An example of a dosage range is from about 0.1 to about 200 mg/kg body weight orally in single or divided doses. In particular examples, a dosage range is from about 1.0 to about 100 mg/kg body weight orally in single or divided doses, including from about 1.0 to about 50 mg/kg body weight, from about 1.0 to about 25 mg/kg body weight, from about 1.0 to about 10 mg/kg body weight (assuming an average body weight of approximately 70 kg; values adjusted accordingly for persons weighing more or less than average). For oral administration, the compositions are, for example, provided in the form of a tablet containing from about 50 to about 1000 mg of the active ingredient, particularly about 75 mg, about 100 mg, about 200 mg, about 400 mg, about 500 mg, about 600 mg, about 750 mg, or about 1000 mg of the active ingredient for the symptomatic adjustment of the dosage to the subject being treated. In one exemplary oral dosage regimen, a tablet containing from about 500 mg to about 1000 mg active ingredient is administered once (e.g., a loading dose) followed by administration of 1/2 dosage tablets (e.g., from about 250 to about 500 mg) each 6 to 24 hours for at least 3 days.
[00281] The specific dose level and frequency of dosage for any particular subject may be varied and will depend upon a variety of factors, including the activity of the subject compound, the metabolic stability and length of action of that compound, the age, body weight, general health, sex and diet of the subject, mode and time of administration, rate of excretion, drug combination, and severity of the condition of the host undergoing therapy.
[00282] The present disclosure also contemplates combinations of one or more disclosed compounds with one or more other agents or therapies useful in the treatment of a disease or disorder. In certain instances, the disease or disorder is mediated through the activity of a PKC activity in a subject. In certain instances, the disease or disorder is cell proliferative disorder. For example, one or more disclosed compounds may be administered in combination with effective doses of other medicinal and pharmaceutical agents, or in combination other non- medicinal therapies, such as hormone or radiation therapy. The term "administration in combination with" refers to both concurrent and sequential administration of the active agents.
Protein Kinase C
Protein Kinase C
[00283] PKC is a family of enzymes that function as serine/threonine kinases. The isoenzymes of PKC differ in their tissue distribution, enzymatic selectivity, requirement for Ca2+, and regulation. PKCs play an important role in cell-cell signaling, gene expression and in the control of cell differentiation and growth.
[00284] The subject compound can be a selective inhibitor of PKC, e.g. an inhibitor selective for PKC over one or more other protein kinases, e.g. over one or more tyrosine kinases, for instance, over one or more non- receptor or receptor tyrosine kinases, e.g. over one or more of PKA, PKB, Abl Met, Src, Ins- R, Flt-3, JAK-2, KDR and/or Ret proteins. The selective PKC inhibitors may optionally be selective over one or more serine/threonine kinases, e.g. one or more serine/threonine kinases which do not belong to the CDK family. The subject compounds can exhibit a selectivity of at least 10 fold, or 20 fold, or 100 fold for the PKC over one or more other protein kinases, e.g. over one or more tyrosine kinases, e.g. over Flt-3, JAK-2, KDR and/or Ret proteins, or over one or more serine/threonine kinases which do not belong to the CDK family.
[00285] The selectivity of a selective inhibitor of PKC over other protein kinases may be calculated as the ratio of the IC50 measured for PKC in an assay described herein over the IC50 determined for another kinase. In a certain instance, there is provided a PKC inhibitor for which the ratio of the IC50 value as determined in an Allogeneic Mixed Lymphocyte Reaction (MLR) assay to the IC50 value as determined in a BM assay is higher than 5, 10, 20, or 30. MLR and BM assays can be done according to known methods, e.g. mouse or human MLR and BM assays, such as disclosed herein.
[00286] The disclosure provides an inhibitor of PKC, which can be an isozyme-selective PKC inhibitor, wherein the subject compound possesses selectivity for the isoforms Θ and a of PKC over one or more of the other PKC isoforms. In a certain instance, the subject compound possesses selectivity for the isoform Θ of PKC over one or more of the other PKC isoforms. In a certain instance, the subject compound possesses selectivity for the isoform a of PKC over one or more of the other PKC isoforms. In one embodiment, the disclosed compounds exhibit selectivity for PKC Θ and PKC a over at least one PKC isoform.
[00287] A subject compound can show a selectivity of at least 10 fold, or 20 fold, or 100 fold for the isoforms Θ or a of PKC over one or more of the other PKC isoforms. Selectivity for the isoforms Θ or a of PKC over one or more of the other PKC isoforms can be measured by comparing the IC50 of the subject compound for the isoforms Θ or a of PKC to the IC50 of
the subject compound for the other PKC isoforms. In a certain instance, the selectivity can be determined by calculating the ratio of IC50 of the subject compound for the other isoforms of PKC to the IC50 of the subject compound for Θ or a isoforms of PKC. In certain examples subject compounds exhibit a selectivity for PKC θ, a or both over another PKC isoform of at least about 2-fold, such as from about 3-fold to about 300-fold, from about 10-fold to about 100-fold or from about 5-fold to 50-fold. IC50 values are obtained, for example, according to PKC assays described herein. The subject compounds can show an IC50 value for the isoforms Θ or a of PKC of 1 μΜ or less, such as less than about 300 nM, such as from about 1 nM to about 250 nM, less than 100 nM or even less than 10 nM in the assays disclosed herein.
[00288] The subject compounds can show a selectivity of the isoforms Θ or μ of PKC over other isoforms of PKC, as well as a selectivity over one or more of the other protein kinases, e.g. over one or more tyrosine kinases, or over one or more serine/threonine kinases which do not belong to the CDK-family, e.g. over one or more of PKA, PKB, Abl, Met, Src, Ins- it, Flt-3, JAK-2, KDR and Ret proteins, e.g. over one or more of Fit- 3, JAK-2, KDR and Ret proteins.
[00289] Certain isozymes of PKC have been implicated in the mechanisms of various disease states, including, but not necessarily limited to, the following: cancer (PKC α, βΐ, βΙΙ, and δ); cardiac hypertrophy and heart failure (PKC βΐ and PKC βΙΙ) nociception (PKC γ and ε); ischemia including myocardial infarction (PKC ε and 5); immune response, particularly T-cell mediated (PKC Θ and a); and fibroblast growth and memory (PKC δ and ζ). The role of PKC ε is also implicated in pain perception. PKC inhibitors can also be used for treating an ocular disease or disorder involving inflammatory and/or neovascular events.
[00290] The subject compounds can be used in the treatment of mammalian (especially human) disease states characterized by aberrant, elevated activity of a PKC isozyme in a tissue as compared to non-disease tissue of the same origin. PKC isozymes and disease states and/or biological functions amenable to therapy by inhibition of activity of the PKC isozyme include, but are not necessarily limited to: PKC a (hyperproliferative cellular diseases, such as cancer); PKC βΐ and PKC βΙΙ (cardiac hypertrophy and heart failure); PKC γ (pain management); PKC δ (ischemia, hypoxia (e.g,. such as in myocardial infarction and in stroke); apoptosis induced by UV irradiation; and aberrant fibroblast growth (e.g., as may occur in wound healing)); PKC ε (pain management, myocardial dysfunction); PKC Θ (immune system diseases, particularly those involving T-cell mediated responses); and PKC ζ (memory and fibroblast growth).
PKC theta
[00291] PKC Θ is expressed predominantly in lymphoid tissue and skeletal muscle.
PKC Θ is selectively expressed in T-cells and plays a role in mature T-cell activation. It has been shown that PKC Θ is involved in T-cell receptor (TCR) -mediated T-cell activation but inessential during TCR-dependent thymocyte development. PKC Θ, but not other PKC isoforms, translocates to the site of cell contact between antigen-specific T-cells and antigen presenting cells (APC), where it localizes with the TCR in the central core of the T-cell activation. PKC Θ, but not the α, ε, or ζ isoenzymes, can selectively activate a FasL promoter-reporter gene and upregulate the mRNA or cell surface expression of endogenous FasL. On the other hand, PKC Θ and ε can promote T-cell survival by protecting the cells from Fas-induced apoptosis, and this protective effect was mediated by promoting p90Rsk- dependent phosphorylation of BCL-2 family member BAD. Thus, PKC Θ appears to play a dual regulatory role in T-cell apoptosis.
[00292] PKC Θ inhibitors can find use in the treatment or prevention of disorders or diseases mediated by T lymphocytes, for example, autoimmune disease such as rheumatoid arthritis, psoriasis and lupus erythematosus, and inflammatory disease such as asthma and inflammatory bowel diseases.
[00293] PKC Θ is a drug target for immunosuppression in transplantation and autoimmune diseases (Isakov et al. (2002) Annual Review of Immunology, 20, 761-794). PCT
Publication WO2004/043386 identifies PKC Θ as a target for treatment of transplant rejection and multiple sclerosis. PKC Θ also plays a role in inflammatory bowel disease (The Journal of Pharmacology and Experimental Therapeutics (2005), 313 (3), 962-982), asthma (WO 2005062918), and lupus (Current Drug Targets: Inflammation & Allergy (2005), 4 (3), 295-298).
[00294] In addition, PKC Θ is highly expressed in gastrointestinal stromal tumors (Blay, P. et al. (2004) Clinical Cancer Research, 10, 12, Pt. 1), it has been suggested that PKC Θ is a molecular target for treatment of gastrointestinal cancer (Wiedmann, M. et al. (2005) Current Cancer Drug Targets 5(3), 171).
[00295] Experiments induced in PKC Θ knock-out mice led to the conclusion that PKC Θ inactivation prevented fat-induced defects in insulin signalling and glucose transport in skeletal muscle (Kim J. et al, 2004, The J. of Clinical Investigation 114 (6), 823). This data indicates PKC Θ is a therapeutic target for the treatment of type 2 diabetes, and hence PKC Θ inhibitors can be useful for treating such disease.
Therapeutic Applications
[00296] The subject compounds are useful for treating a disease or disorder that is mediated through, or exacerbated by, the activity of a PKC in a subject in need of treatment. Also, the compounds are useful for treating a disease or disorder that is associated with aberrant or otherwise undesirable T cell activation in a subject.
[00297] Accordingly, the present disclosure provides methods of treating an inflammatory disease in a subject by administering an effective amount of a subject compound, including a salt or solvate or stereoisomer thereof, so as to treat inflammation. Inflammatory diseases contemplated for therapy include acute and chronic inflammation mediated or exacerbated by PKC activity
[00298] The present disclosure also provides methods of treating an autoimmune disease in a subject by administering to the subject an effective amount of a subject compound, including a salt or solvate or stereoisomer thereof, so as to treat the autoimmune disease.
[00299] The present disclosure also provides methods of treating an ocular disease or disorder involving inflammatory and/or neovascular events by administration of a subject compound, including a salt or solvate or stereoisomer thereof, in an effective amount.
[00300] Diseases or conditions of interest for treatment according to the present disclosure include, but are not limited to, atherosclerosis, vascular occlusion due to vascular injury such as angioplasty, restenosis, obesity, syndrome X, impaired glucose tolerance, polycystic ovary syndrome, hypertension, heart failure, chronic obstructive pulmonary disease, CNS diseases such as Alzheimer disease or amyotrophic lateral sclerosis, cancer, infectious diseases such as: AIDS, septic shock or adult respiratory distress syndrome, ischemia/reperfusion injury, e.g.: myocardial infarction, stroke, gut ischemia, renal failure or hemorrhage shock, and traumatic shock, e.g. traumatic brain injury.
[00301] Further diseases or conditions of interest for treatment according to the present disclosure include, but are not limited to, T-cell mediated acute or chronic inflammatory diseases or disorders or autoimmune diseases, rheumatoid arthritis, osteoarthritis, systemic lupus erythematosus, Hashimoto's thyroiditis, multiple sclerosis, myasthenia gravis, diabetes type I or II and the disorders associated therewith, transplant rejection, graft versus host disease, respiratory diseases, asthma, inflammatory lung injury, inflammatory liver injury, inflammatory glomerular injury, cutaneous manifestations of immunologically-mediated disorders or illnesses, inflammatory and hyperproliferative skin diseases (such as psoriasis, atopic dermatitis, allergic contact dermatitis, irritant contact dermatitis and further eczematous dermatitises, seborrhoeic dermatitis), inflammatory eye diseases (such as
Sjoegren's syndrome, keratoconjunctivitis, uveitis) inflammatory bowel disease, Crohn's disease or ulcerative colitis, Guillain-Barre syndrome, and allergies.
[00302] The subject compounds can also be used for preventing or treating or delaying ocular diseases and disorders involving inflammation and/or neovascularization. Ocular diseases or disorders involving inflammatory and/or neovascular events include, but are not limited to, macular degeneration (AMD), diabetic ocular diseases or disorders, uveitis, optic neuritis, ocular edema, ocular angiogenesis, ischemic retinopathy, anterior ischemic optic neuropathy, optic neuropathy and neuritis, macular edema, cystoid macular edema (CME), retinal disease or disorder, such as retinal detachment, retinitis pigmentosa (RP), Stargart's disease, Best's vitelliform retinal degeneration, Leber's congenital amaurosis and other hereditary retinal degenerations, Sorsby's fundus dystrophy, pathologic myopia, retinopathy of prematurity (ROP), Leber's hereditary optic neuropathy, corneal transplantation or refractive corneal surgery, keratoconjunctivitis, or dry eye.
[00303] Generally, cell proliferative disorders treatable with the subject compound disclosed herein relate to any disorder characterized by aberrant cell proliferation. These include various tumors and cancers, benign or malignant, metastatic or non-metastatic. Specific properties of cancers, such as tissue invasiveness or metastasis, can be targeted using the methods described herein. Cell proliferative disorders include a variety of cancers, including, among others, breast cancer, ovarian cancer, renal cancer, gastrointestinal cancer, kidney cancer, bladder cancer, pancreatic cancer, lung squamous carcinoma, and adenocarcinoma.
[00304] In some embodiments, the cell proliferative disorder treated is a hematopoietic neoplasm, which is aberrant growth of cells of the hematopoietic system. Hematopoietic malignancies can have its origins in pluripotent stem cells, multipotent progenitor cells, oligopotent committed progenitor cells, precursor cells, and terminally differentiated cells involved in hematopoiesis. Some hematological malignancies are believed to arise from hematopoietic stem cells, which have the ability for self renewal. For instance, cells capable of developing specific subtypes of acute myeloid leukemia (AML) upon transplantation display the cell surface markers of hematopoietic stem cells, implicating hematopoietic stem cells as the source of leukemic cells. Blast cells that do not have a cell marker characteristic of hematopoietic stem cells appear to be incapable of establishing tumors upon
transplantation (Blaire et al., 1997, Blood 89:3104-3112). The stem cell origin of certain hematological malignancies also finds support in the observation that specific chromosomal abnormalities associated with particular types of leukemia can be found in normal cells of
hematopoietic lineage as well as leukemic blast cells. For instance, the reciprocal translocation t(9q34;22ql l) associated with approximately 95% of chronic myelogenous leukemia appears to be present in cells of the myeloid, erythroid, and lymphoid lineage, suggesting that the chromosomal aberration originates in hematopoietic stem cells. A subgroup of cells in certain types of CML displays the cell marker phenotype of
hematopoietic stem cells.
[00305] Although hematopoietic neoplasms often originate from stem cells, committed progenitor cells or more terminally differentiated cells of a developmental lineage can also be the source of some leukemias. For example, forced expression of the fusion protein Bcr/Abl (associated with chronic myelogenous leukemia) in common myeloid progenitor or granulocyte/macrophage progenitor cells produces a leukemic-like condition. Moreover, some chromosomal aberrations associated with subtypes of leukemia are not found in the cell population with a marker phenotype of hematopoietic stem cells, but are found in a cell population displaying markers of a more differentiated state of the hematopoietic pathway (Turhan et al., 1995, Blood 85:2154-2161). Thus, while committed progenitor cells and other differentiated cells may have only a limited potential for cell division, leukemic cells may have acquired the ability to grow unregulated, in some instances mimicking the self -renewal characteristics of hematopoietic stem cells (Passegue et al., Proc. Natl. Acad. Sci. USA, 2003, 100: 11842-9).
[00306] In some embodiments, the hematopoietic neoplasm treated is a lymphoid neoplasm, where the abnormal cells are derived from and/or display the characteristic phenotype of cells of the lymphoid lineage. Lymphoid neoplasms can be subdivided into B- cell neoplasms, T and NK-cell neoplasms, and Hodgkin's lymphoma. B-cell neoplasms can be further subdivided into precursor B-cell neoplasm and mature/peripheral B-cell neoplasm. Exemplary B-cell neoplasms are precursor B-lymphoblastic leukemia/lymphoma (precursor B-cell acute lymphoblastic leukemia) while exemplary mature/peripheral B-cell neoplasms are B-cell chronic lymphocytic leukemia/small lymphocytic lymphoma, B-cell
prolymphocytic leukemia, lymphoplasmacytic lymphoma, splenic marginal zone B-cell lymphoma, hairy cell leukemia, plasma cell myeloma/plasmacytoma, extranodal marginal zone B-cell lymphoma of MALT type, nodal marginal zone B-cell lymphoma, follicular lymphoma, mantle-cell lymphoma, diffuse large B-cell lymphoma, mediastinal large B-cell lymphoma, primary effusion lymphoma, and Burkitt's lymphoma/Burkitt cell leukemia. T- cell and Nk-cell neoplasms are further subdivided into precursor T-cell neoplasm and mature (peripheral) T-cell neoplasms. Exemplary precursor T-cell neoplasm is precursor T-
lymphoblastic lymphoma/leukemia (precursor T-cell acute lymphoblastic leukemia) while exemplary mature (peripheral) T-cell neoplasms are T-cell prolymphocytic leukemia T-cell granular lymphocytic leukemia, aggressive NK-cell leukemia, adult T-cell
lymphoma/leukemia (HTLV-1), extranodal NK T-cell lymphoma, nasal type, enteropathy- type T-cell lymphoma, hepatosplenic gamma-delta T-cell lymphoma, subcutaneous panniculitis-like T-cell lymphoma, Mycosis fungoides/Sezary syndrome, Anaplastic large- cell lymphoma, T/null cell, primary cutaneous type, Peripheral T-cell lymphoma, not otherwise characterized, Angioimmunoblastic T-cell lymphoma, Anaplastic large-cell lymphoma, T/null cell, primary systemic type. The third member of lymphoid neoplasms is Hodgkin's lymphoma, also referred to as Hodgkin's disease. Exemplary diagnosis of this class that can be treated with the compounds include, among others, nodular lymphocyte- predominant Hodgkin's lymphoma, and various classical forms of Hodgkin's disease, exemplary members of which are Nodular sclerosis Hodgkin's lymphoma (grades 1 and 2), Lymphocyte-rich classical Hodgkin's lymphoma, Mixed cellularity Hodgkin's lymphoma, and Lymphocyte depletion Hodgkin's lymphoma.
[00307] In some embodiments, the hematopoietic neoplasm treated is a myeloid neoplasm. This group comprises a large class of cell proliferative disorders involving or displaying the characteristic phenotype of the cells of the myeloid lineage. Myeloid neoplasms can be subdivided into myeloproliferative diseases,
myelodysplastic/myeloproliferative diseases, myelodysplastic syndromes, and acute myeloid leukemias. Exemplary myeloproliferative diseases are chronic myelogenous leukemia (e.g., Philadelphia chromosome positive (t(9;22)(qq34;ql l)), chronic neutrophilic leukemia, chronic eosinophilic leukemialhypereosinophilic syndrome, chronic idiopathic
myelofibrosis, polycythemia vera, and essential thrombocythemia. Exemplary
myelodysplastic/myeloproliferative diseases are chronic myelomonocytic leukemia, atypical chronic myelogenous leukemia, and juvenile myelomonocytic leukemia. Exemplary myelodysplastic syndromes are refractory anemia, with ringed sideroblasts and without ringed sideroblasts, refractory cytopenia (myelodysplastic syndrome) with multilineage dysplasia, refractory anemia (myelodysplastic syndrome) with excess blasts, 5q-syndrome, and myelodysplastic syndrome with t(9;12)(q22;pl2) (TEL-Syk fusion; see, e.g., Kuno et al., 2001, Blood 97: 1050).
[00308] In some embodiments, the composition can be used to treat acute myeloid leukemias (AML), which represent a large class of myeloid neoplasms having its own subdivision of disorders. These subdivisions include, among others, AMLs with recurrent
cytogenetic translocations, AML with multilineage dysplasia, and other AML not otherwise categorized. Exemplary AMLs with recurrent cytogenetic translocations include, among others, AML with t(8;21)(q22;q22), AML 1 (CB F- alpha)/ETO , Acute promyelocytic leukemia (AML with t(15;17)(q22;ql l-12) and variants, PML/RAR-alpha), AML with abnormal bone marrow eosinophils (inv(16)(pl3q22) or t(16;16)(pl3;ql l),
CBFb/MYHl IX), and AML with l lq23 (MLL) abnormalities. Exemplary AML with multilineage dysplasia are those that are associated with or without prior myelodysplastic syndrome. Other acute myeloid leukemias not classified within any definable group include, AML minimally differentiated, AML without maturation, AML with maturation, Acute myelomonocytic leukemia, Acute monocytic leukemia, Acute erythroid leukemia, Acute megakaryocytic leukemia, Acute basophilic leukemia, and Acute panmyelosis with myelofibrosis.
[00309] In other aspects, cell proliferative disorders comprise virally mediated tumors. These can arise from infection of cells by an oncogenic virus that has the capability of transforming a normal cell into a tumor cell. Because rates of viral infection far exceed the number of actual incidence of cell transformation, viral mediated transformation generally act together with other cellular factors to generate a transformed tumor cell. Thus, a virally mediated tumor does not require the virus to be the sole causative agent of the cell proliferative disorder, but rather that the viral infection or persistent presence of virus is associated with the generation of the tumor. Generally, tumors where the causative agent is a virus typically has continual expression of a limited number of viral genes and that viral these oncogenes, expressed as part of the viral infection or through persistence of the virus, disrupts the normal cellular gene expression and signal transduction pathways. Without being bound by theory, viral oncogenes involved in cell transformation appear to disrupt four main cellular processes: cell surface receptors that interact with growth factors and extracellular matrix, transmembrane signaling networks, cytosolic elements such as soluble proteins and second messengers, and nuclear proteins including DNA binding proteins and factors which function directly and indirectly in gene regulation and replication.
Characterization of Functional Properties
[00310] The following are exemplary assays useful in characterizing activities of a compound of interest.
A. In Vitro
1. Protein Kinase C assay
[00311] The inhibition of PKC activity is measured by monitoring the production of phosphorylated peptide by fluorescence polarization at different concentrations of the inhibitor. Reactions are carried out in 96-well plate format with a total volume of 20 \L containing 20 mM HEPES, pH 7.4, 5 mM MgCl2, 0.2 mM CaCl2, 1 mM DTT, 0.02% Brij- 35, 0.1 mg/mL phosphatidylserine, 0.02 mg/mL dioleoyl-sn-glycerol and 5 μΜ each of ATP and the peptide substrate. Compounds are first diluted serially in DMSO and then transferred to a solution containing the above concentrations of HEPES, MgCl2, CaCl2, DTT, and Brij-35 to yield 5x compound solutions in 2% DMSO, which is then added to the reaction solution. Reactions are initiated by the addition of PKC at a typical concentration as described in the table below, and then allowed to incubate at room temperature for 20 minutes. At the end of this time, a combination of quench (EDTA) and detection (peptide tracer and antibody) reagents is added using the protocol of Invitrogen P2748 (Carlsbad, CA), a Protein Kinase C Fluorescence polarization Assay Kit. After a 30 minute period of incubation, the amount of phosphorylated peptide generated is measured by fluorescence polarization (Ex = 485 nm, Em = 535 nm) using a Tecan Polarian instrument (Switzerland).
Table 2

2. IL-2 ELISA, Human primary T cell, anti-CD3+CD28+ Assays
[00312] Human primary T cell isolation and culture: Human primary T cells were prepared as follows. Fresh PBMC's from All Cells (Cat # PB002) were re-suspended in RPMI (RPMI-1640 with L-Glutamine; Mediatech, Inc., Herndon VA, cat. #10-040-CM) with 10% FBS and seeded into flasks and incubated at 37°C for 2 hours to allow the monocytes to adhere. The non-adherent cells were then centrifuged and re-suspended in RPMI medium containing 40 U/ml IL2 and seeded into a flask pre-coated with 1 μg/ml
aCD3 and 5 ug/ml aCD28 (Anti-Human CD3, BD Pharmingen Catalog #555336, Anti- Human CD28, Beckman Coulter Catalog #IM1376). The cells were stimulated for 3-4 days, then transferred to a fresh flask and maintained in RPMI (RPMI-1640 with L-Glutamine; Mediatech, Inc., Herndon VA, cat. #10-040-CM) with 10% FBS and 40 U/mL IL-2.
[00313] Primary T cell stimulation and IL2 ELISA: Human primary T cells (100,000 cells per well) were pre-incubated with or without test compound in RPMI-1640 with L- Glutamine and 10% FBS for 1 hour at 37 °C. Cells were then stimulated by transferring them to round-bottom 96-well plates pre-coated with 1 μg/ml aCD3 and 5 μg/ml aCD28. For counter assay, cells were instead stimulated by adding 8X stock solutions of PMA and ionomycin in RPMI-1640 with L-Glutamine and 10% FBS (for final concentrations of 0.5ng/ml PMA and 0.1 μΜ ionomycin, both from Calbiochem). Cells were incubated at 37 °C for 24 hours before 100 μΐ^ supernatants were harvested for quantification of IL-2 by ELISA using Human IL-2 Duoset ELISA Kit from R and D Systems, Cat. # DY202E.
3. Protein Kinase C assay
[00314] The subject compounds can be tested for activity on different PKC isoforms according to the following method. Assay is performed in a white with clear bottom 384- well microtiterplate with non-binding surface. The reaction mixture (25 μΐ) contains 1.5 μΜ of a tridecapeptide acceptor substrate that mimics the pseudo substrate sequence of PKC a with the Ala→Ser replacement, 10 μΜ 33P-ATP, 10 mM Mg(N03)2, 0.2 mM CaCl2, PKG at a protein concentration varying from 25 to 400 ng/ml (depending on the isotype used), lipid vesicles (containing 30 mol % phosphatidylserine, 5 mol % DAG and 65 mol %
phosphatidylcholine) at a final lipid concentration of 0.5 mM, in 20 mM Tris-HCl buffer pH 7.4+0.1% BSA. Incubation is performed for 60 minutes at room temperature. Reaction is stopped by adding 50 μΐ of stop mix (100 mM EDTA, 200 μΜ ATP, 0.1% Triton X-100, 0.375 mg/well streptavidin-coated SPA beads in phosphate buffered saline w/o Ca, Mg. After 10 minutes incubation at room temperature, the suspension is spun down for 10 minutes at 300 g. Incorporated radioactivity is measured in a Trilux counter for 1 minute. IC50 measurement is performed on a routine basis by incubating a serial dilution of inhibitor at concentrations ranging between 1-1000 μΜ. ICso values are calculated from the graph by curve fitting with XL Fit® software.
4. Protein Kinase C a Assay
[00315] Human recombinant PKC a is obtained from Oxford Biomedical Research and is used under the assay conditions as described under Section A. l above.
5. Protein Kinase C βΐ Assay
[00316] Human recombinant PKC βΐ is obtained from Oxford Biomedical Research and is used under the assay conditions as described under Section A. l above.
6. Protein Kinase C δ Assay
[00317] Human recombinant PKC δ is obtained from Oxford Biomedical Research and is used under the assay conditions as described under Section A. l above.
7. Protein Kinase C ε Assay
[00318] Human recombinant PKC ε is obtained from Oxford Biomedical Research and is used under the assay conditions as described under Section A. l above.
8. Protein Kinase C η Assay
[00319] Human recombinant PKC η is obtained from Pan Vera and is used under the assay conditions as described under Section A.1 above.
9. Protein Kinase C Θ Assay
[00320] Human recombinant PKC Θ is used under the assay conditions as described above.
10. CD28 Costimulation Assay
[00321] The assay is performed with Jurkat cells transfected with a human interleukin-2 promoter/reporter gene construct as described by Baumann G et al. in Transplant. Proc. 1992; 24:43-8, the β-galactosidase reporter gene being replaced by the luciferase gene (de Wet J., et al., Mol. Cell. Biol. 1987, 7(2), 725-737). Cells are stimulated by solid phase- coupled antibodies or phorbol myristate acetate (PMA) and the Ca++ ionophore ionomycin as follows. For antibody-mediated stimulation Microlite TM1 micro titer plates (Dynatech) are coated with 3 μg/ml goat anti-mouse IgG Fc antibodies (Jackson) in 55 μΐ phosphate- buffered saline (PBS) per well for three hours at room temperature. Plates are blocked after removing the antibodies by incubation with 2% bovine serum albumin (BSA) in PBS (300 μΐ per well) for 2 hours at room temperature. After washing three times with 300 μΐ PBS per well, 10 ng/ml anti-T cell receptor antibodies (WT31, Becton & Dickinson) and 300 ng/ml anti-CD28 antibodies (15E8) in 50 μΐ 2% BSA/PBS are added as stimulating antibodies and incubated overnight at 4°C. Finally the plates are washed three times with 300 μΐ PBS per well. Seven three-fold serial dilutions of test compounds in duplicates in assay medium (RPMI 1640/10% fetal calf serum (FCS) containing 50 μΜ 2-mercaptoethanol, 100 units/ml penicillin and 100 μg/ml streptomycin) are prepared in separate plates, mixed with transfected Jurkat cells (clone K22 290_H23) and incubated for 30 minutes at 37°C in 5% CO2 100 μΐ of this mixture containing 1 x 105 cells are then transferred to the antibody-
coated assay plates. In parallel 100 μΐ are incubated with 40 ng/ml PMA and 2 μΜ ionomycin. After incubation for 5.5 hours at 37°C in 5% CO2, the level of luciferase is determined by bioluminescence measurement. The plates are centrifuged for 10 minutes at 500 g and the supernatant is removed by flicking. Lysis buffer containing 25 mM Tris- phosphate, pH 7.8, 2 mM DTT, 2 mM l,2-diaminocyclohexane-N,N,N',N-tetraacetic acid, 10% (v/v) glycerol and 1% (v/v) Triton X-100 is added (20 μΐ per well). The plates are incubated at room temperature for 10 minutes under constant shaking. Luciferase activity is assessed with a bioluminescence reader (Labsystem, Helsinki, Finland) after automatic addition of 50 μΐ per well luciferase reaction buffer containing 20 mM Tricine, 1.07 mM (MgC03)4Mg(OH)2 5H20, 2.67 mM MgS04, 0.1 mM EDTA, 33.3 mM DTT, 270 μΜ coenzyme A, 470 μΜ luciferin (Chemie Brunschwig AG), 530 μΜ ATP, pH 7.8. Lag time is 0.5 seconds, total measuring time is 1 or 2 seconds. Low control values are light units from anti-T cell receptor- or PMA- stimulated cells, high controls are from anti-T cell receptor/anti-CD28- or PMA/ionomycin-stimulated cells without any test sample. Low controls are subtracted from all values. The inhibition obtained in the presence of a test compound is calculated as percent inhibition of the high control. The concentration of test compounds resulting in 50% inhibition (IC50) is determined from the dose-response curves. 11. Bone Marrow Proliferation (BM) Assay
[00322] Bone marrow cells from CBA mice (2.5 x 104 cells per well in flat bottom tissue culture microtiter plates) are incubated in 100 μΐ RPMI medium containing 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin (Gibco BRL, Basel, Switzerland), 50 UM 2- mercaptoethanol (Fluke, Buchs, Switzerland), WEHI-3 conditioned medium (7.5% v/v) and L929 conditioned medium (3% v/v) as a source of growth factors and serially diluted compounds. Seven three-fold dilution steps in duplicates per test compound are performed. After four days of incubation 1 μθ 3H-thymidine is added. Cells are harvested after an additional five-hour incubation period, and incorporated 3H-thymidine is determined according to standard procedures. Conditioned media are prepared as follows. WEHI-3 cells 1 (ATCC TIB68) and L929 cells (ATCC CCL 1) are grown in RPMI medium until confluence for 4 days and one week, respectively. Cells are harvested, resuspended in the same culture flasks in medium C containing 1% FCS (Schreier and Tees 1981) for WEHI-3 cells and RPMI medium for L929 cells and incubated for 2 days (WEHI-3) or one week (L929). The supernatant is collected, filtered through 0.2 μιη and stored in aliquots at -80°C. Cultures without test compounds and without WEHI-3 and L929 supernatants are used as low control values. Low control values are subtracted from all values. High controls
without any sample are taken as 100% proliferation. Percent inhibition by the samples is calculated and the concentrations required for 50% inhibition (IC50 values) are determined. 12. Allogeneic Mixed Lymphocyte Reaction (MLR)
[00323] The two-way MLR is performed according to standard procedures (J. Immunol. Methods, 1973, 2, 279 and Meo T. et al., Immunological Methods, New York, Academic Press, 1979, 227-39). Briefly, spleen cells from CBA and BALB/c mice (1.6xl05 cells from each strain per well in flat bottom tissue culture micro titer plates, 3.2 l05 in total) are incubated in RPMI medium containing 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin (Gibco BRL, Basel, Switzerland), 50 μΜ 2-mercaptoethanol (Fluka, Buchs, Switzerland) and serially diluted compounds. Seven three-fold dilution steps in duplicates per test compound are performed. After four days of incubation 1 μθ 3H-thymidine is added. Cells are harvested after an additional five-hour incubation period, and incorporated 3H-thymidine is determined according to standard procedures. Background values (low control) of the MLR are the proliferation of BALB/c cells alone. Low controls are subtracted from all values. High controls without any sample are taken as 100%
proliferation. Percent inhibition by the samples is calculated, and the concentrations required for 50% inhibition (IC50 values) are determined.
B. In vivo
Heart Transplantation Model
[00324] The strain combination used: Male Lewis (RT1 haplotype) and BN (RT1 haplotype). The animals are anaesthetised using inhalational isofluorane. Following heparinisation of the donor rat through the abdominal inferior vena cava with simultaneous exsanguination via the aorta, the chest is opened and the heart rapidly cooled. The aorta is ligated and divided distal to the first branch and the brachiocephalic trunk is divided at the first bifurcation. The left pulmonary artery is ligated and divided and the right side divided but left open. All other vessels are dissected free, ligated and divided and the donor heart is removed into iced saline.
[00325] The recipient is prepared by dissection and cross-clamping of the infra-renal abdominal aorta and vena cava. The graft is implanted with end-to-side anastomoses, using 1010 monofilament suture, between the donor brachiocephalic trunk and the recipient aorta and the donor right pulmonary artery to the recipient vena cava. The clamps are removed, the graft tethered retroabdominally, the abdominal contents washed with warm saline and the animal is closed and allowed to recover under a heating lamp. Graft survival is monitored by daily palpation of the beating donor heart through the abdominal wall. Rejection is
considered to be complete when-heart beat stops. Graft survival is monitored in animals treated with compounds.
Graft v. Host Model
[00326] Spleen cells (2xl07) from Wistar/F rats are injected subcutaneously into the right hind footpad of (Wistar/F x Fischer 344)Fi hybrid rats. The left footpad is left untreated. The animals are treated with the test compounds on 4 consecutive days (0-3). The popliteal lymph nodes are removed on day 7, and the weight differences between two corresponding lymph nodes are determined. The results are expressed as the inhibition of lymph node enlargement (given in percent) comparing the lymph node weight differences in the experimental groups to the weight difference between the corresponding lymph nodes from a group of animals left untreated with a test compound. In certain instances the test compound is a selective PKC inhibitor. For example, disclosed compounds that are particularly useful for treating graft versus host disease and related disorders are selective PKC a and Θ inhibitors.
Rat Collagen-Induced Arthritis Model
[00327] Rheumatoid arthritis (RA) is characterized by chronic joint inflammation eventually leading to irreversible cartilage destruction. IgG-containing IC are abundant in the synovial tissue of patients with RA. While it is still debated what role these complexes play in the etiology and pathology of the disease, IC communicate with the hematopoetic cells via the FcyR.
[00328] CIA is a widely accepted animal model of RA that results in chronic
inflammatory synovitis characterized by pannus formation and joint degradation. In this model, intradermal immunization with native type II collagen, emulsified with incomplete Freund's adjuvant, results in an inflammatory polyarthritis within 10 or 11 days and subsequent joint destruction in 3 to 4 weeks.
Study Protocol
[00329] Syngeneic LOU rats are immunized with native type II collagen on Day 0, and efficacy of a test compound is evaluated in a prevention regimen and a treatment regimen. In the prevention protocol, either vehicle or various doses of a test compound are administered via oral gavage starting on day of immunization (Day 0). In the treatment protocol, after clinical signs of arthritis develop on Day 10, treatment with a test compound is initiated (e.g., 300 mg/kg by oral gavage, qd) and continued until sacrifice on Day 28. In both protocols, clinical scores are obtained daily, and body weights are measured twice
weekly. At Day 28, radiographic scores are obtained, and serum levels of collagen II antibody are measured by ELISA.
Determination of Results
[00330] By 10 days after immunization, rats can develop clinical CIA, as determined by an increase in their arthritis scores. The mean arthritic score gradually increases in the rats treated with vehicle alone after Day 10, and by Day 28 the mean clinical score can reach about 6.75. Mean clinical scores in animals treated from the day of immunization (Day 0) with a test compound can be significantly reduced on Days 10-28 compared with vehicle controls. In the rats treated with a test compound at disease onset, there can be a
significantly lower arthritis score beginning around Day 16, and this difference can be observed until the end of the study on Day 28.
[00331] Blinded radiographic scores (scale 0-6) can be obtained on Day 28 of CIA and compared between the animals in the vehicle group, animals in the prevention group, and animals in the treatment group.
[00332] The groups administered with a test compound, either prophylactically (at immunization) or after disease onset can preclude the development of erosions and reduced soft tissue swelling. Similarly, the groups administered with a test compound can result in reduction of serum anti-collagen II antibody.
Mouse Experimental Autoimmune Encephalomyelitis
[00333] The in vivo efficacy of a test compound towards autoimmune diseases can be demonstrated in a mouse model of experimental autoimmune encephalomyelitis (EAE).
Model Description
[00334] EAE is a useful model for multiple sclerosis (MS), an autoimmune disease of the CNS that is caused by immune-cell infiltration of the CNS white matter. Inflammation and subsequent destruction of myelin cause progressive paralysis. Like the human disease, EAE is associated with peripheral activation of T cells autoreactive with myelin proteins, such as myelin basic protein (MBP), proteolipid protein (PLP), or myelin oligodendrocyte protein (MOG). Activated neuroantigen-specific T cells pass the blood-brain barrier, leading to focal mononuclear cell infiltration and demyelination. EAE can be induced in susceptible mouse strains by immunization with myelin- specific proteins in combination with adjuvant. In the SJL mouse model used in these studies, hind limb and tail paralysis is apparent by Day 10 after immunization, the peak of disease severity can be observed between Days 10 and 14, and a cycle of partial spontaneous remission followed by relapse can be observed up to Day 35. The results can demonstrate the potential of the test compound to suppress disease
severity and prevent relapse of disease symptoms that may be the result of FcyR-mediated cytokine release from immune cells.
Study Protocol
[00335] In the SJL murine model of EAE, each mouse is sensitized with PLP/CFA. (150 μg PLP139-151 with 200 μg CFA in 0.05 ml of homogenate on four sites of hind flank for a total of 0.2 ml emulsion is used to induce EAE). In a suppression protocol, either vehicle or various doses of a test compound are administered via oral gavage starting on the day of immunization (Day 0). In a treatment protocol, at onset of disease, animals are separated to achieve groups with a similar mean clinical score at onset and administered vehicle or various dose frequencies of test compounds via oral gavage. In both protocols, clinical scores are monitored daily, and body weights are measured twice weekly.
Determination of Results
[00336] By 10 days after PLP immunization, SJL mice can develope clinical EAE, as evidenced by an increase in their mean clinical scores. The paralytic score can gradually increase in the animals treated with vehicle only from the day of immunization (Day 0), and by Day 14 the mean score can reach a peak of about 5.1. At disease peak (e.g., Day 14), the mean clinical score in animals treated with either daily or twice daily can be significantly reduced. By Day 16, animals can exhibit a partial remission of mean clinical severity, which is a characteristic of the SJL model. The lower clinical scores in animals treated twice daily with a test compound can remain significant throughout the experiment until the animals are sacrificed on Day 30. These lower scores throughout the treatment period are reflected in the significantly lower cumulative disease index (CDI) and increase in cumulative weight index (CWI).
[00337] SJL mice treated with a test compound at disease onset (e.g., Day 11) can show a significant decrease in CDI. Further, there can be a decrease in the number of relapses in animals treated with a test compound compared with the number of relapses in animals treated with vehicle.
Research Applications
[00338] Since subject compounds can inhibit a PKC activity, such compounds are also useful as research tools. The present disclosure also provides a method for using subject compounds as a research tool for studying a biological system or sample, or for discovering new chemical compounds that can inhibit a PKC activity.
[00339] The disclosure provides for a method of studying a biological system or sample known to comprise PKC, the method comprising: (a) contacting the biological sample with a
compound of formula I- VIII or a salt or solvate or stereoisomer thereof; and (b) determining the inhibiting effects caused by the compound on the biological sample.
[00340] Any suitable biological sample having PKC can be employed in such studies which can be conducted either in vitro or in vivo. Representative biological samples suitable for such studies include, but are not limited to, cells, cellular extracts, plasma membranes, tissue samples, isolated organs, mammals (such as mice, rats, guinea pigs, rabbits, dogs, pigs, humans, and so forth), and the like, with mammals being of particular interest.
[00341] When used as a research tool, a biological sample comprising PKC is typically contacted with a PKC activity-inhibiting amount of a subject compound. After the biological sample is exposed to the compound, the effects of inhibition of a PKC activity are determined using conventional procedures and equipment, such as the assays disclosed herein. Exposure encompasses contacting the biological sample with the compound or administering the compound to a subject. The determining step can involve measuring a response (a quantitative analysis) or can involve making an observation (a qualitative analysis). Measuring a response involves, for example, determining the effects of the compound on the biological sample using conventional procedures and equipment, such as radioligand binding assays and measuring ligand-mediated changes in functional assays. The assay results can be used to determine the activity level as well as the amount of compound necessary to achieve the desired result, that is, a PKC activity-inhibiting amount.
[00342] Additionally, subject compounds can be used as research tools for evaluating other chemical compounds, and thus are also useful in screening assays to discover, for example, new compounds having a PKC inhibiting activity. In this manner, a subject compound can be used as a standard in an assay to allow comparison of the results obtained with a test compound and with the subject compounds to identify those test compounds that have about equal or superior activity, if any. For example, IC50 data for a test compound or a group of test compounds is compared to the IC50 data for a subject compound to identify those test compounds that have the desired properties, for example, test compounds having an IC50 about equal or superior to a subject compound, if any.
[00343] This aspect includes, as separate embodiments, both the generation of comparison data (using the appropriate assays) and the analysis of test data to identify test compounds of interest. Thus, a test compound can be evaluated in a biological assay, by a method comprising the steps of: (a) conducting a biological assay with a test compound to provide a first assay value; (b) conducting the biological assay with a subject compound to provide a second assay value; wherein step (a) is conducted either before, after or
concurrently with step (b); and (c) comparing the first assay value from step (a) with the second assay value from step (b). The assays that can be used for generation of comparison data are disclosed herein, such as the PKC assays.
EXAMPLES
[00344] The following examples are put forth so as to provide those of ordinary skill in the art with a complete disclosure and description of how to make and use the embodiments, and are not intended to limit the scope of what the inventors regard as their invention nor are they intended to represent that the experiments below are all or the only experiments performed. Efforts have been made to ensure accuracy with respect to numbers used (e.g. amounts, temperature, etc.) but some experimental errors and deviations should be accounted for. As will be understood, by those of skill in the art of organic synthesis and medicinal chemistry the specific conditions set forth below are exemplary and can be varied or adapted to other reagents and products in routine fashion. Unless indicated otherwise, parts are parts by weight, molecular weight is weight average molecular weight, temperature is in degrees Celsius, and pressure is at or near atmospheric. Standard abbreviations may be used.
EXAMPLE 1:
5-FLUORO-N2-[4-(4-METHYLPIPERAZINO)-3-TRIFLUOROMETHYL]PHENYL-N4-(1,2,2,6,6- PENTAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00345] 2-Fluoro-5-nitrobenzotrifluoride (2 g) and 1-methylpiperazine (2 mL) were dissolved in methanol (5 mL). The yellow solution was stirred at room temperature overnight. The reaction mixture was diluted with water (100 mL) and extracted with ethyl acetate (2 x 100 mL). The organic solutions were evaporated to give 2-(4- methylpiperazino)-5-nitrobenzotrifluoride.
[00346] 2-(4-Methylpiperazino)-5-nitrobenzotrifluoride was dissolved in methanol (100 mL) and to the solution was added 10% Pd-C. The reaction mixture was reacted under hydrogen atmosphere (~ 40 psi) for 1 hour. The catalyst was filtered off over cellite and washed with methanol. The filtrate was evaporated to give [4-(4-methylpiperazino)-3- trifluoromethyl] aniline (2.25 g, 91% in two steps). JH NMR (DMSO-d6): δ 2.19 (s, 3H), 2.38 (br, 4H), 2.70 (t, J= 4.5 Hz, 4H), 5.31 (br, 2H), 6.73 (dd, J= 2.4, 8.7 Hz, 1H), 6.78 (d, J= 2.7 Hz, 1H), 7.20 (d, J= 8.1 Hz, 1H).
[00347] 4- Amino- 1, 2,2,6, 6-pentamethylpiperidine (1 g) and 2,6-dichloro-5- fluoropyrimidine (1.5 g) were dissolved in methanol (10 mL). The reaction solution was stirred at room temperature overnight. The reaction solution was evaporated and crystallized
from ethyl acetate and hexanes to give 2-chloro-5-fluoro-N4-( 1,2,2, 6,6- pentamethylpiperidin-4-yl)-4-pyrimidineamine HC1 salt (1.65 g, 93%). JH NMR (DMSO- d6): δ 1.38 (s, 6H), 1.48 (s, 6H), 2.02 (m, 4H), 2.68 (d, J= 4.8 Hz, 3H), 4.33 (br, 1H), 8.10 (d, J= 3.3 Hz, 1H), 8.32 (d, J= 6.9 Hz, 1H), 9.66 (br, 1H).
[00348] 2-Chloro-5-fluoro-N4-(l,2,2,6,6-pentamethylpiperidin-4-yl)-4-pyrimidineamine (300 mg) and [4-(4-methylpiperazino)-3-trifluoromethyl]aniline (300 mg) were suspended in isopropanol (1 mL) and TFA (5 drops). The solution was heated at 100 °C overnight, then cooled to room temperature. The solution was evaporated and purified by flash column chromatography (2.0 M NH3/MeOH in dichloromethane = 2, 4, 6, 10%) to give 5-fluoro-N2- [4-(4-methylpiperazino)-3-trifluoromethyl]phenyl-N4-(l , 2,2,6, 6-pentamethylpiperidin-4-yl)- 2,4-pyrimidinediamine (440 mg, 84%). JH NMR (DMSO-d6): δ 1.04 (s, 6H), 1.07 (s, 6H), 1.44 (t, J= 11.7 Hz, 2H), 1.68 (d, J= 9.9 Hz, 2H), 2.18 (s, 3H), 2.20 (s, 3H), 2.41 (br, 4H), 2.76 (t, J= 4.2 Hz, 4H), 4.29 (br, 1H), 7.16 (d, J= 8.4 Hz, 1H), 7.32 (d, J= 8.4 Hz, 1H), 7.75 (d, J= 2.1 Hz, 1H), 7.84 (d, J= 3.6 Hz, 1H), 8.07 (d, J= 8.7 Hz, 1H), 9.13 (s, 1H); 19F NMR (282 MHz, DMSO-d6): δ - 165.87, - 59.89; LCMS : purity: 100%; MS (m/e): 524.43 (MH+).
[00349] The following compounds were made in a similar fashion to the Example 1 or by methods described herein or known to skilled artisans.
EXAMPLE 2:
1-1: 5-FLUORO-N2-{4-FLUORO-[3-(4-H)-1,2,3,4-TETRAZOL-5-ONE-1-YL]}PHENYL- N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00350] JH NMR (DMSO d6, 300MHz): δ 9.21 (s, 1H), 7.88 (m, 2H), 7.63 (bs, 1H), 7.35 (d, / = 8.1Hz, 1H), 7.19 (t, / = 9.6Hz, 1H), 4.48 (bs, 1H), 1.86 (d, / = 11.4Hz, 2H), 1.65 (bs, 2H), 1.22 (s, 6H), 1.15 (s 6H); LCMS (m/z): 460.07 (MH+).
EXAMPLE 3:
1-2: 5-FLUORO-N2-{4-FLUORO-[3-(4-H)-1,2,3,4-TETRAZOL-5-ONE-1-YL]}PHENYL- N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00351] JH NMR (DMSO d6, 300MHz): δ 9.19 (s, 1H), 7.90 (m, 2H), 7.48 - 7.34 (m, 2H), 7.15 (t, / = 9.9Hz, 1H), 4.44 (s, 1H), 1.90 (d, / = 10.5Hz, 2H), 1.51 (t, / = 12Hz, 2H), 1.29 (d, / = 7.5Hz, 12H); LCMS (m/z): 446.10 (MH+).
EXAMPLE 4:
1-3: 5-FLUORO-N2-{4-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00352] Ή NMR (DMSO d6, 300MHz): δ 9.37 (s, IH), 9.09 (s, IH), 8.02 - 7.71 (m, 3H), 7.65 (d, / = 7.5Hz, IH), 7.36 (t, / = 9.9Hz, IH), 4.38 (bs, IH), 3.62 (s, 3H), 2.68 (d, / = 3.9Hz, 3H), 2.05 - 2.00 (m, 2H), 1.87 (m, 2H), 1.41 (s, 6H), 1.31 (s, 6H); LCMS (m/z):
474.11 (MH+).
EXAMPLE 5:
1-4: 5-FLUORO-N2-{4-FLUORO-[3-(4-ISOPROPYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00353] JH NMR (DMSO d6, 300MHz): δ 9.37 (s, IH), 8.02 (d, / = 6.6Hz, IH), 7.87 (d, / = 3.6Hz, IH), 7.77 (m, IH), 7.32 (t, / = 9.6Hz, IH), 7.23 (d, / = 7.8Hz, 4.45 (bs, IH), 3.6 (s, 3H), 1.65 (d, / = 9Hz, 2H), 1.14 (t, / = 11.4Hz, 2H), 1.04 (s, 6H), 0.99 (s, 6H); LCMS (m/z): 460.15 (MH+).
EXAMPLE 6:
1-5: 5-FLUORO-N2-{4-FLUORO-[3-(4-ISOPROPYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00354] JH NMR (DMSO d6, 300MHz): δ 9.34 (s, IH), 7.98 (m, IH), 7.86 (m, 2H), 7.32 (m, 2H), 4.44 (m, IH), 4.29 (bs, IH), 2.14 (bs, 3H), 1.67 (m, 2H), 1.46 (s, 6H), 1.43 (s, 6H), 1.52 (m, 2H), 1.05 (m, 12H); LCMS (m/z): 502.19 (MH+).
EXAMPLE 7:
1-6: 5-FLUORO-N2-{4-FLUORO-3-[4-(2-FLUOROETHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00355] JH NMR (DMSO d6, 300MHz): δ 9.34 (s, IH), 7.98 (m, IH), 7.88 (d, / = 3.6Hz, IH), 7.8 (m, IH), 7.29 (m, 2H), 4.44 (m, IH), 4.29 (bs, IH), 1.67 (m, 2H), 1.46 (s, 6H), 1.43 (s, 6H), 1.52 (m, 2H), 1.05 (m, 12H); LCMS (m/z): 488.28 (MH+).
EXAMPLE 8:
1-7: 5-FLUORO-N2-{4-FLUORO-3-[4-(2-FLUOROETHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00356] JH NMR (DMSO d6, 300MHz): δ 9.35 (s, IH), 8.25 (s, IH), 7.91 (d, / = 6.9Hz, IH), 7.88 (m, IH), 7.34 (m, 2H), 4.87 (m, IH), 4.71 (m, IH), 4.36 (m, 2H), 4.28 (m, IH), 1.74 (d, / = 11.4Hz, 2H), 1.26 (t, / = 12.9Hz, 2H), 1.15 (s, 6H), 1.09 (s, 6H); LCMS (m/z):
492.12 (MH+).
EXAMPLE 9:
1-8: 5-FLUORO-N2-{4-METHYL-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00357] JH NMR (DMSO d6, 300MHz): δ 9.25 (s, 1H), 8.15 (s, 1H), 7.86 (d, / = 3.9Hz, 1H), 7.78 - 7.74 (m, 2H), 7.22 (m, 2H), 4.26 (s, 1H), 3.60 (s, 3H), 2.19 (s, 3H), 2.05 (s, 3H), 1.68 (d, / = 11.4Hz, 2H), 1.45 (t, / = 12Hz, 2H), 1.07 (s, 6H), 0.96 (s, 6H); LCMS (m/z): 470.14 (MH+).
EXAMPLE 10:
1-9: 5-FLUORO-N2-{4-METHYL-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00358] JH NMR (DMSO d6, 300MHz): δ 9.26 (s, 1H), 8.29 (s, 1H), 7.88 (d, / = 3.7Hz, 1H), 7.74 (s, 1H), 7.35 (d, / = 7.8Hz, 2H), 7.22 (d, / = 9.3Hz, 1H), 4.39 (s, 1H), 3.60 (s, 3H), 2.05 (s, 3H), 1.77 (d, / = 12.9Hz, 2H), 1.33 (t, / = 12.9Hz, 2H), 1.17 (m, 12H); LCMS (m/z): 456.16 (MH+).
EXAMPLE 11:
I- 10: 5-FLUORO-N2-{4-ISOPROPOXY-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00359] JH NMR (DMSO d6, 300MHz): δ 9.10 (s, 1H), 8.16 (s, 3H), 7.83 (d, / = 3.6Hz, 1H), 7.79 (s, 1H), 7.71 (d, / = 9Hz, 1H), 7.17 (d, / =8.1Hz 1H), 7.1 (d, 9Hz, 1H), 4.46 (m, 1H), 4.25 (s, 1H), 3.59 (s, 3H), 2.18 (s, 3H), 1.66 (d, / = 10.2Hz, 2H), 1.45 (t, / = 12.1Hz, 9H), 1.12 (d, / = 6Hz, 6H), 1.06 (s, 6H), 0.93 (s, 6H); LCMS (m/z): 514.17 (MH+).
EXAMPLE 12:
1-11: 5-FLUORO-N2-{4-ISOPROPOXY-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00360] JH NMR (DMSO d6, 300MHz): δ 9.12 (s, 1H), 8.30 (s, 1H), 7.85 (d, / = 3.6Hz, 1H), 7.8 (s, 1H), 7.69 (d, / = 9.0Hz, 1H), 7.28 (d, / = 8. lHz, 1H), 7.12 (d, / = 9Hz, 1H), 4.46 (m, 1H), 4.35 (s, 1H), 3.58 (s, 3H), 1.74 (d, / = 12Hz, 10H), 1.31 (t, / =12.3Hz, 2H), 1.12 (m, 12H); LCMS (m/z): 500.16 (MH+).
EXAMPLE 13:
1-12: 5-FLUORO-N2-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1-YL]PHENYL-N4- (1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00361] JH NMR (DMSO d6, 300MHz): δ 9.33 (s, 1H), 8.08 (s, 1H), 7.84 (dd, / = 3.9Hz, 1H), 7.8 (d, / = 7.5Hz, 1H), 7.29 (m, 3H), 4.33 (m, 1H), 3.59 (s, 3H), 2.22 (s, 3H), 1.72 (d, /
= 11.7Hz, 2H), 1.47 (t, / = 12.0Hz, 2H), 1.08 (s, 6H), 0.99 (s, 6H); LCMS (m/z): 456.17 (MH+).
EXAMPLE 14:
1-13: 5-FLUORO-N2-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1-YL]PHENYL-N4- (2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE
[00362] JH NMR (DMSO d6, 300MHz): δ 9.38 (s, 1H), 8.72 (s, 1H), 8.07 (s, 1H), 7.86 (dd, / = 3.6Hz, 1H), 7.77 (d, / = 7.8Hz, 1H), 7.55 (bs, 1H), 7.39 - 7.3 (m, 2H), 4.43 (s, 1H), 3.59 (s, 3H), 1.93 (bs, 2H), 1.53 (bs, 2H), 1.33 (s, 12H); LCMS (m/z): 442.17 (MH+).
EXAMPLE 15
SYNTHESIS OF 2-CHLORO-5-FLUORO-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)-4- PYRIMIDINEAMI

[00363] 4-Amino-l, 2,2,6, 6-pentamethylpiperidine (1 g) and 2,6-dichloro-5- fluoropyrimidine (1.5 g) were dissolved in methanol (10 mL). The reaction solution was stirred at room temperature overnight. The reaction solution was evaporated and crystallized from ethyl acetate and hexanes to give 2-chloro-5-fluoro-N4-(l, 2,2,6,6- pentamethylpiperidin-4-yl)-4-pyrimidineamine HCl salt (1.65 g, 93%). See also Example 1 for this synthesis.
[00364] JH NMR (DMSO-d6): δ 9.66 (br. s, 1H), 8.32 (d, / = 6.9 Hz, 1H), 8.10 (d, / = 3.3 Hz, 1H), 4.33 (br. s, 1H), 2.68 (d, / = 4.8 Hz, 3H), 2.02 (m, 4H), 1.48 (s, 6H), 1.38 (s, 6H). EXAMPLE 16
SYNTHESIS OF 2-CHLORO-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-4- PYRIMIDINEAMI

[00365] 2,4-Dichloro-5-fluoropyrimidine (21.7 g) was dissolved in methanol (400 mL) and cooled to 0 °C. 4-Amino-2,2,6,6-tetramethylpiperidine (19.2 mL) was added dropwise, The resulting mixture was slowly warmed to room temperature and stirred overnight. The
reaction solution was evaporated and triturated with ethyl to 2-chloro-5-fluoro-N-(2,2,6,6- tetramethylpiperidin-4-yl)-4-pyrimidineamine, HC1 salt (36.2 g, 93%).
[00366] JH NMR (DMSO-d6): δ 8.24 (d, 1H), 8.16 (d, 1H), 4.38 (m, 1H), 1.92 (d, 2H),
1.63 (t, 2H), 1.39 (d, 12H); mJz = 287 (M+H)+.
EXAMPLE 17
SYNTHESIS OF 5-CARBOXYAMIDE-2,4-DICHLOROPYRIMIDINE

[00367] To a 2L round bottom flask equipped with water condenser and a CaCl2 drying tube, 2,4-dihydroxypyrimidine (25g, 0.16mole) was added to PC15 (117g, 0.56mole), and POCI3 (250ml, 2.6mole). The mixture was heated at 115 °C overnight to give a clear, slightly light yellow solution. The mixture was cooled to room temperature, and was concentrated under reduced pressure to give pale yellowish oil.
[00368] To this oil, anhydrous 1,4-dioxane (300ml) was added and the mixture was cooled to 0°C in an ice/water bath. 35ml of N¾ in water (28%) was added dropwise to the mixture with stirring, temperature was kept below 5 °C. The mixture changed from clear to white with precipitate forming, and was stirred for 1 hour at 0 °C, reaction was followed by TLC (1 : 1 Hexanes: Ethyl Acetate). Ethyl acetate (700ml) and water (500ml) were added to the mixture, the 2 layers were separated. The organic layer was dried with Na2S04, and filtered. The solution was concentrated under reduced pressure to give a light yellow solid. This light yellow solid was sonicated with methylene chloride (200ml), and filtered to give a pale yellow solid (16g). This pale yellow solid was dissolved into ethyl acetate (1.5L) and washed with saturated NaHCC>3 (500ml). The organic layer was dried with Na2S04, filtered, and concentrated under reduced pressure to give 13.1 g of product as a white solid (44% yield).
[00369] JH NMR (DMSO-d6, 300MHz): δ 8.86 (s, 1H), 8.14 (bs, 1H), 8.02 (bs, 1H).
EXAMPLE 18
SYNTHESIS OF 5-CARBOXYAMIDE-2,4-DICHLOROPYRIMIDINE

[00370] Concentrated ammonium hydroxide solution in ¾0 (assumed to be 8.5M; 14.1 mL; 120 mmol) was added over 15-20 minutes to a stirred solution of 2,4- dichloropyrimidine-5-carbonyl chloride (12.5 g; 60 mmol; Manchester Organics, Sutton Weaver, England) in CH2C12 (300 mL) at -15 to -20 °C (internal temperature) [n.b.: a precipitate is formed during the addition]. After complete addition, the mixture was filtered (the filter cake comprises desired product and an impurity - for purification see below). H2O (50 mL) was added to the filtrate, which was partitioned. The organic layer was dried (NaS04), filtered and the solvent removed under vacuum to give the title compound (l. lg) as a solid. The filter cake from above was triturated with hot (ca. 50 °C) EtOAc (300 mL) and the mixture filtered - this was repeated another 2 times. The combined filtrates from the trituration were concentrated under vacuum to give another 9.1g of the title compound. The total yield from the reaction is 10.2g (88 ). Data identical to those of Example 17.
EXAMPLE 19
SYNTHESIS OF 5-CARBOXYAMIDE-2-CHLORO-JV4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4- YL)-4-PYRIMIDINEAMINE, HCL SALT

[00371] 5-Carboxyamide-2,4-dichloropyrimidine (7.5g, 0.04mole) was dissolved into MeOH (300ml)/ ¾0 (30ml). The solution was cooled to 0 °C in a ice/water bath, 4-amino- 1,2,2,6,6-pentamethylpiperidine (6.65g, 0.04mole) was added dropwise. The mixture was stirred at 0 °C and let warmed up to room temperature over 2 days. Solution was concentrated under reduced pressure to give a light yellow slush. Ethyl acetate (250ml x 2) was added and then concentrated under reduced pressure to remove the remaining traces of methanol and water to give a light yellowish solid. This solid was then sonicated with
methylene chloride (100ml), and filtered using a Buchner funnel, to give 9.5g of pale yellow solid (75% yield) of the title compound as a HC1 salt.
[00372] JH NMR (DMSO d6, 300MHz): δ 9.74 (s, 1H), 9.23 (s, 1H), 8.6 (bs, 1H), 8.39 (bs, 1H), 7.76 (s, 1H), 4.36 (bs, 1H), 2.68 (s, 3H), 2.14 (d, 2H), 1.88 (t, 2H), 1.48 (s, 6H), 1.39 (s, 6H).
EXAMPLE 20
SYNTHESIS OF 5-CARBOXYAMIDE-2-CHLORO-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4- YL)-4-PYRIMIDINEAMINE FREE BASE

[00373] 5-Carboxyamide-2,4-dichloropyrimidine (7.5g, 0.04 mol) was dissolved into MeOH (300ml) / ¾0 (30ml). The solution was cooled to 0 °C in a ice/water bath, 4-amino- 2,2,6, 6-tetramethylpiperidine (6.8ml, 0.04mole) was added dropwise. The mixture was stirred at 0 °C and let warmed up to room temperature over 2 days. Solution was concentrated under reduced pressure to give a light yellow slush. Ethyl acetate (250ml x 2) was added and then concentrated under reduced pressure to remove the remaining traces of methanol and water to give a light yellowish solid. This solid was then sonicated with methylene chloride (100ml), and filtered using a Buchner funnel, to give a pale yellow solid.
[00374] This solid was treated with ethyl acetate (2L), and saturated NaHCOs, the 2 layers were separated, and the organic layer was dried with Na2S04. The drying agent was filtered off and the solution was concentrated under reduced pressure to give a white solid (5g, 41% yield). Additional product can be retrieved from the aqueous layer by back extracting it with additional ethyl acetate.
[00375] JH NMR (DMSO-if6, 300MHz): δ 9.14 (d, 1H), 8.54 (s, 1H), 8.18 (bs, 1H), 7.68 (s, 1H), 4.30 (bs, 1H), 1.79 (d, 2H), 1.15 (s, 6H), 1.02 (s, 6H); mJz = 312.2 (M+H)+.
EXAMPLE 21
SYNTHESIS OF 5-CYANO-2-CHLORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDLN-4-YL)-4- PYRIMIDINEAMINE

[00376] Burgess reagent - methyl (N-triethylammoniumsulfonyl)carbamate - (238 mg; 1.0 mmol) was added in one portion to a stirred solution of 5-carboxamide-2-chloro-N4- (2,2,6,6-tetramethylpiperidin-4-yl)-4-pyrimidineamine (156 mg; 0.5 mmol) in 1,2- dichloroethane (3 mL) at room temperature. The mixture was heated to 70 °C and stirred for 2 hours. After allowing to cool to room temperature the mixture was diluted with further 1 ,2-dichloroethane (20 mL) and ¾0 (30 mL). The aqueous and organic layers were partitioned and the organic layer washed with saturated NaHCC>3 then dried (Na2S04), filtered and the solvent removed under vacuum to leave a crude viscous oil (NMR shows this to be product and unreacted Burgess reagent). The crude oil was purified by column chromatography on silica gel using EtOAc:MeOH (9: 1) then EtO Ac : MeOH : Et3N (90:8:2) as eluent to give the title compound (75 mg, 51%) as a foam solid. This solid was suitable for use without further purification.
EXAMPLE 22
SYNTHESIS OF 5-CYANO-2-CHLORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDLN-4-YL)-4- PYRIMIDINEAMINE

[00377] Trifluoroacetic anhydride (9.4 mL; 67.3 mmol) was added dropwise over 30-45 minutes to a stirred solution of 5-carboxyamide-2-chloro-N4-(2,2,6,6-tetramethylpiperidin-4- yl)-4-pyrimidineamine (2.1 g, 6.7 mmol) and Et3N (11.3 mL; 80.8 mmol) in THF (40 mL) at -78 °C under nitrogen. After complete addition, the mixture was stirred at -78 °C for a further 60 minutes, then a saturated solution of NaHCC>3 (30 mL) was added dropwise keeping the internal temperature below -30 °C. After complete addition of the NaHCC>3, EtOAc (150 mL) and ¾0 (100 mL) was added and the mixture was stirred for 10 minutes. Further ¾0 (200 mL) was added and the organic and aqueous layers were partitioned. The aqueous layer was extracted with EtO Ac (4 x 150 mL) - until substantially all precipitated
material had gone in to solution. The combined organic extracts were washed with brine (1 x 50 mL), dried (Na2S04), filtered and the solvent removed under vacuum to leave a crude solid with TFAA and Et3N residues. The solid was triturated with Et20 (50 mL) and filtered to give the product (2.1g) as a TFA salt.
EXAMPLE 23
FORMATION OF FREE BASE OF 5-CYANO-2-CHLORO-JV4-(2,2,6,6- TETRAMETHYLPIPERIDIN-4-YL)-4-PYRIMIDINEAMINE
[00378] 5-Cyano-2-chloro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-4-pyrimidineamine TFA salt (2.1 g) was partitioned between EtOAc (100 mL) and 0.2 M NaOH (50 mL). The organic layer was washed with brine (1 x 50 mL), dried (Na2S04), filtered and the solvent removed under vacuum to leave the product (1.35 g, 68%) as a solid.
[00379] JH NMR (DMSO-if6, 300MHz): δ 8.51 (s, 1H), 8.34 (br. S, 1H), 4.42 (t, 1H), 1.61 (br. d, 2H), 1.23 (t, 2H), 1.14 (s, 6H), 1.02 (s, 6H); m/z = 294.1 (M+H)+ for 35C1.
EXAMPLE 24
SYNTHESIS OF 5-CYANO-2-CHLORO-JV4-(1, 2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)-4- PYRIMIDI

[00380] Trifluoroacetic anhydride (9.35 mL; 67.3 mmol, 10 eq) was added dropwise over 30-45 minutes to a stirred solution of 5-carboxyamide-2-chloro-N4-(l,2,2,6,6- pentamethylpiperidin-4-yl)-4-pyrimidineamine hydrochloride (2.19 g, 6.73 mmol, 1 eq) and Et3N (11.26 mL; 80.76 mmol, 12 eq) in THF (45 mL) at -78 °C under nitrogen. After complete addition, the mixture was stirred at -78 °C for a further 60 minutes, then a saturated solution of NaHC03 (30 mL) was added dropwise keeping the internal temperature below - 30 °C. After complete addition of the NaHC03, EtOAc (100 mL) and H20 (100 mL) was added and the mixture was stirred for 10 minutes. Further ¾0 (100 mL) was added and the organic and aqueous layers were partitioned. The aqueous layer was extracted with EtOAc (4 x 100 mL) - until all precipitated material had gone in to solution. The combined organic extracts were washed with brine (1 x 50 mL), dried (Na2S04), filtered and the solvent removed under vacuum to leave a crude solid with TFAA and Et3N residues. The crude solid was dissolved in 100 mL of EtOAc and partitioned with 1 N aqueous NaOH (50 mL). The ethyl acetate layer was extracted with 2 x 50 mL aqueous IN NaOH. The combined
organic extracts were washed with brine (1 x 50 mL), dried (Na2S04), filtered and the solvent removed under vacuum to give light yellow solid (1.80 g, 87 %).
[00381] JH NMR (DMSO-if6, 300MHz): δ 8.51 (s, 1H), 8.37 (d, 1H), 4.31 (bm, 1H), 2.15
(s, 3H), 1.47-1.66 (m, 4H), 1.06 (s, 6H), 1.00 (s, 6H); m/z = 309 (M+H)+.
EXAMPLE 25
SYNTHESIS OF 2-BROMO-4-FLUORO-5-NITROANILINE

[00382] 2-Bromo-4-fluoroaniline (47.5g, 250 mmol) was added to a solution of concentrated ]¾SC)4 (300 mL) keeping the internal temperature below 30 °C. The mixture was aged for ca. 30-60 minutes then cooled to -20 °C. 90% HNO3 (35 g) was added dropwise over ca. 60 minutes keeping the internal temperature between -15 to -20 °C. TLC indicated a slight amount of starting material, so a further aliquot of 90% HNO3 (3g) was added over 5 minutes at -15 to -20 °C. The cold mixture was then poured on to ice ¾0 (ca. 1L ice + 500 mL H20) and EtOAc (1 L). The aqueous and organic layers were partitioned and the organic layer was washed with saturated NaHCC>3 (2 x 500 mL), dried (Na2S04), filtered and the solvent removed under vacuum to leave a dark solid (35g, 60%).
[00383] JH NMR (DMSO-if6, 300MHz): δ 8.27 (br. S, 2H), 7.70 (d 1H), 7.47 (d, 1H); m/z = 275.9 (M+MeCN+H)+ for 79Br.
EXAMPLE 26
SYNTHESIS OF l-(2-BROMO-4-FLU -5-NiTROPHENYL)-lH-TETRAZOL-5(4H)-ONE

[00384] 2-Bromo-4-fluoro-5-nitrobenzenamine (2.4g, 10.2 mmol) was added to phosgene (20% weight in toluene, 25 mL, excess) in a 100 mL single neck round bottom flask equipped with a water condenser. The mixture was heated to 80 °C and stirred for 2.5 hours under N2. The mixture was cooled to room temperature and was concentrated under reduced pressure to give a dark residue. To this residue, azidotrimethylsilane (20 mL, excess) was added and the mixture was heated at 80 °C overnight under N2. The mixture was cooled to room temperature, then concentrated under reduced pressure. Ethyl acetate was added to the residue and the product was extracted with saturated NaHCC>3 (x 2 - until substantially all product had gone in to the aqueous layer). The aqueous layers were combined and washed
with a small amount of ethyl acetate, then the aqueous layer was acidified with 2N HCl. The emerging precipitate from the acidic aqueous layer was extracted with ethyl acetate, and the organic layer was washed with brine and dried (Na2S04), filtered and concentrated under vacuum to give the title compound (1.35 g, 44%) as a light brown solid.
[00385] JH NMR (300 MHz, DMSO) δ 8.66 (d, / = 7.5 Hz, 1H), 8.37 (d, / = 10.8 Hz, lH); m/z = 303.9 (M-H)+.
EXAMPLE 27
SYNTHESIS OF l-(2-BROMO-4-FLUORO-5-NiTROPHENYL)-4-METHYL-lH-TETRAZOL-
5(4H)-ONE

[00386] l-(2-Bromo-4-fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (1.4g, 4.6 mmol) was added to anhydrous DMF (45 mL) in a single neck round bottom flask followed by K2CO3 (1.8g, 13.8 mmol) and chilled to -40 °C in an acetone/dry ice bath. Iodomethane (0.35mL, 6.9 mmol) was added, and the mixture was further chilled to -78 °C. The mixture was stirred under N2 and allowed to warm up to room temperature overnight. Ethyl acetate (300 mL) was added and the mixture was washed with brine (2 x 200 mL). The organic and aqueous layers were separated and the organic layer was dried (Na2S04), filtered and concentrated under vacuum. Dichloromethane (300 mL) was added to the residue and the organic layer was washed with brine (x2) dried (Na2S04), filtered and the solvent concentrated under vacuum. To the residue, EtOAc was added, and the mixture was sonicated and filtered. The mother liquor was concentrated under reduced pressure to give the title compound (1.4g) as a yellowish brown solid.
[00387] JH NMR (300 MHz, DMSO) δ 8.64 (d, / = 7.2 Hz, 1H), 8.38 (d, / = 10.8 Hz, 1H), 3.63 (s, 3H); m/z = 317.95 (M+H)+.
EXAMPLE 28
ALTERNATIVE SYNTHESIS OF l-(2-BROMO-4-FLUORO-5-NiTROPHENYL)-lH-TETRAZOL-
5( -ONE

Step 1: Preparation of l-(2-Bromo-4-fluorophenyl)-lH-tetrazol-5(4H)-one
[00388] A mixture of 2-bromo-4-fluoro-l-isocyanatobenzene (20g, 92.6 mmol;
UkrOrgSynthesis, Kiev, Ukraine) and trimethylsilyazide (50 mL) was heated to reflux under N2 and stirred overnight. After allowing to cool to room temperature the mixture was concentrated under vacuum and the residue partitioned between EtOAc (300 mL) and saturated NaHCC>3 (300 mL). The organic layer was then extracted with saturated NaHCC>3 (200 mL portions) until TLC indicated all the product had been removed from the organic layer {ca. 5 extractions). EtOAc (500 mL) was added to the combined aqueous layers and the mixture was acidified using 6N HC1 (to pH < 3). The aqueous and organic layers were partitioned and the organic layer was washed with brine (300 mL), dried (Na2S04), filtered and the solvent removed under vacuum to leave the product (20. lg, 84%) as a solid.
[00389] JH NMR (DMSO-if6, 300MHz): δ 7.91 (dt, 1H), 7.79-7.74 (m, 1H), 7.52-7.45 (m, 1H); 19F NMR (DMSO-if6, 282 MHz): 108.0 (dd); m/z = 258.9 (M+H)+ for 79Br.
Step 2: l-(2-Bromo-4-fluorophenyl)-4-methyl-lH-tetrazol-5(4H)-one
[00390] K2CO3 (26.8g, 194 mmol) and Mel (9.7 mL, 155 mmol) were added at -78 °C to a solution of l-(2-bromo-4-fluorophenyl)-lH-tetrazol-5(4H)-one (20.1 g, 77.6 mmol) in DMF (150 mL) under N2. The mixture was stirred from -78 °C to room temperature over 2 days. The mixture was poured in to EtOAc (300 mL) and ¾0 (500 mL) and the aqueous and organic layers were partitioned. The organic layer was washed with ¾0 (3 x 500 mL) then brine (300 mL), dried (Na2S04), filtered and the solvent removed under vacuum to leave the product (20.3g, 96%) as a solid. This methylation reaction can be conducted at room temperature with similar results.
[00391] JH NMR (DMSO-if6, 300MHz): δ 7.93 (dd, 1H), 7.77 (dd, 1H), 7.51 (dt, 1H); 19F NMR (DMSO- , 282 MHz): 107.7 (dd); m/z = 274.9 (M+H)+ for 81Br.
Step 3: l-(2-Bromo-4-fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one
[00392] ΗΝΟ3 (25g) was added dropwise over ca. 30 minutes to a solution of l-(2- bromo-4-fluorophenyl)-4-methyl-lH-tetrazol-5(4H)-one (23 g, 84.2 mmol) in H2S04 (250 mL) at 0 to -10°C. After complete addition of the HNO3, TLC indicated some unreacted starting material so a further aliquot of HNO3 (5g) was added dropwise over ca. 10 minutes TLC indicated complete reaction so the mixture was poured in to a mixture of ice / H20 (750g ice : 500 mL H20) and EtOAc (500 mL). The aqueous ad organic layers were partitioned and the aqueous layer was washed with saturated NaHC03 (300 mL; with care: acid residues present). The organic layer was dried (Na2S04), filtered and the solvent removed under vacuum to leave a crude residue. The crude residue was triturated with EtOAc and filtered. The filtrate was concentrated under vacuum and the trituration
procedure repeated. The solid collected in the triturations above were combined to give the title compound (20. lg, 75%) as a solid [note: the filtrate still contains some desired product which can be purified by column chromatography on silica gel if need be]. Data for this product identical to l-(2-Bromo-4-fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one produced above.
EXAMPLE 29
SYNTHESIS OF 1-(5-AMINO-2-CYCLOPROPYL-4-FLUOROPHENYL)-4-METHYL-1H- TETRAZOL-5(4H)-ONE
,

Step 1: Preparatio -(2-Bromo-4-fluorophenyl)-lH-tetrazol-5(4H)-one:

[00393] Trimethylsilyl azide (65 mL, 494 mmol; ca. 6 equiv.) was added to a stirred mixture of 2-bromo-4-fluorobenzoyl chloride (20.4 g, 86 mmol; Apollo Scientific Ltd) under N2 at 0 to 10 °C. The mixture was then heated slowly to 50-70 °C (gas evolution starts at approximately 50-60 °C and becomes vigorous after ca. 65 °C). The mixture was briefly removed from the heat until nitrogen evolution was more controlled, then the mixture returned to the heat. The mixture was then heated to 90 °C under nitrogen overnight.
[00394] A safety notice for the procedure: The reaction was performed behind a blast shield in a 250 mL round-bottom flask. A nitrogen balloon and vent (needle vent) was used in the set-up, especially the first part of the reaction in which nitrogen gas is evolved.
[00395] After allowing to cool to room temperature, the mixture was concentrated under vacuum and the residue partitioned between EtOAc (200 mL) and saturated NaHCC>3 (100 mL). The organic layer was then extracted with saturated NaHCC>3 (150 mL portions) until
TLC indicated the desired product had been removed from the organic layer (ca. 5 extractions). EtOAc (300 mL) was added to the combined aqueous layers and the mixture was acidified using 6N HC1 (to pH < 3). The aqueous and organic layers were partitioned and the aqueous layer ws extracted with EtOAc (1 x 150 mL). The combined organic extracts were dried (MgS04), filtered and the solvent removed under vacuum to leave the product (19.9g, 89%) as a solid.
[00396] JH NMR (DMSO-if6, 300MHz): δ 7.91 (dt, 1H), 7.79-7.74 (m, 1H), 7.52-7.45 (m, 1H) 19F NMR (DMSO-if6, 282 MHz): -107.6 (dd). m/z = 301.95 (M+MeCN+H)+ for 81Br.
[00397] The other steps in the scheme of Example 29 are disclosed the examples herein. EXAMPLE 30
SYNTHESIS OF l-(2-CYCLOPROPYL-4-FLUORO-5-NiTROPHENYL)-4-METHYL-lH-
TETRAZOL-5(4H)-ONE

[00398] l-(2-Bromo-4-fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (350mg, 1.1 mmol) was added to toluene / water (4.6mL:0.9mL), in a 60ml round bottom flask fitted with a water condenser. To this solution was added tricyclohexylphosphine (92mg, 0.3 mmol), CS2CO3 (2.14g, 6.6 mmol) and cyclopropylboronic acid MIDA ester (303mg, 1.5 mmol). This mixture was degassed by bubbling N2 into the solution for 15 minutes.
Pd(OAc)2 (37mg, 0.2 mmol) was added, and the mixture was heated to 100° C under N2 and stirred overnight. After allowing to cool to room temperature, EtOAc and saturated K2CO3 were added, and the organic and aqueous layers were partitioned. The organic layer was dried (Na2S04), filtered and the solvent was concentrated under vacuum to leave a crude residue. The residue was purified by column chromatography on silica gel using EtOAc / hexanes (1:3 to 1:2.5) as an eluent to give the title compound as a yellow solid (200mg).
[00399] JH NMR (300 MHz, DMSO) δ 8.37 (d, / = 7.5Hz, 1H), 7.32 (d, / = 12.6Hz, 1H), 3.61 (s, 3H), 1.87 (m, 1H), 1.08-1.04 (m, 2H), 0.97-0.93 (m, 2H); m/z = 279.95 (M+H)+. ALTERNATIVE SUZUKI CONDITIONS FOR SYNTHESIS OF 1-(2-CYCLOPROPYL-4-FLUORO-5- NITROPHENYL)-4-METHYL-LH-TETRAZOL-5(4H)-ONE
[00400] Toluene (140 mL) then H20 (50 mL) was added to a mixture of l-(2-bromo-4- fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (14.5 g, 45.6 mmol), potassium
cyclopropyltrifluoroborate (7.42 g, 50.1 mmol), palladium(II) acetate (205 mg, 0.91 mmol), tricyclohexylphosphine (511 mg, 1.82 mmol) and K2CO3 (12.6 g, 91.1 mmol) under N2. The mixture was sparged with N2 for 10 mins then heated to 90 °C and stirred overnight. TLC did not indicate complete reaction, so more palladium(II) acetate (103 mg, 0.45 mmol), tricyclohexylphosphine (255 mg, 0.91 mmol) and potassium cyclopropyltrifluoroborate (3.7 g, 25.5 mmol) were added. The mixture was sparged with N2 once again and heated to 90 °C under N2 overnight. TLC only indicated a little further reaction, so after allowing to cool to room temperature, the mixture was filtered through a small plug of celite and the filter cake washed with EtOAc (5 x 50 mL). The filtrate was partitioned and the organic layer was dried (MgS04), filtered and the solvent removed under vacuum to leave a solid residue. To the solid residue was added potassium cyclopropyltrifluoroborate (3.7 g, 25.5 mmol), palladium(II) acetate (103 mg, 0.45 mmol), tricyclohexylphosphine (255 mg, 0.91 mmol) and K2CO3 (6.3 g, 91.1 mmol). The mixture was placed under N2 and toluene (140 mL) and H2O (50 mL) were added. The mixture was sparged with N2 for 10 min then the mixture heated to 90 °C under N2 overnight. TLC indicated complete reaction. After allowing to cool to room temperature, the mixture was partitioned and the organic layer dried (MgS04), filtered and the solvent removed under vacuum. The mixture was purified by filtration through a 4-to-5 inch plug of silica (the residue was dry-loaded on to silica) eluting with EtOAc / hexane (3:7 to 4:6; fractions collected in conical flasks) to give the product (11.0 g, 86%) as a solid.
EXAMPLE 31
ALTERNATIVE SYNTHESIS OF 1-(2-CYCLOPROPYL-4-FLUORO-5-NITROPHENYL)-4- M -LH-TETRAZOL-5(4H)-ONE

Step 1: 2-CYCLOPROPYL-4-FLUORO-5-NITRO ANILINE
[00401] A mixture of 2-bromo-4-fluoro-5-nitroaniline (12 g, 51 mmol),
cyclopropylboronic acid MIDA ester (Aldrich; 20.1 g, 102 mmol), Pd(OAc)2 (1.72 g, 7.7 mmol), Cy3P (4.3 g, 15.3 mmol) and Cs2C03 (98.8 g, 306 mmol) in toluene (120 mL) and Η2Ο (40 mL) was de-gassed with N2 for 15 minutes. The mixture was then heated at 100 °C (oil bath temperature) overnight (the reaction mixture can also be heated to reflux). After allowing to cool to room temperature, the mixture was diluted with EtOAc (200 mL) and Η2Ο (100 mL) and the mixture filtered through Celite. The filter cake was washed with
EtOAc (2 x 100 mL) and the filtrate partitioned. The organic layer was dried (Na2S04), filtered and the solvent removed under vacuum to leave a crude residue. The residue was purified by column chromatography on silica gel (residue dry-loaded on to silica gel) using EtOAc / hexanes (1 :4 to 3:7) as eluent to give the product (8.1 g, 81%) as a dark solid.
[00402] JH NMR (DMSO-if6, 300MHz): δ 7.27 (d 1H), 6.84 (d, 1H), 5.52 (br. s, 2H), 1.74-1.83 (m, 1H), 0.92-0.98 (m, 2H), 0.62-0.73 (m, 2H); m/z = 238.0 (M+MeCN+H)+.
Alternative Step 1: 2-CYCLOPROPYL-4-FLUORO-5-NITROANILINE USING POTASSIUM CYCLOPROPYLTRIFLUOROBORATE
[00403] A mixture of 2-bromo-4-fluoro-5-nitroaniline (13.1 g, 56 mmol), potassium cyclopropyltrifluoroborate (16.5 g, 112 mmol), Pd(OAc)2 (1.89 g, 8.4 mmol), CysP (4.7 g, 16.8 mmol) and Cs2C03 (109.5 g, 336 mmol) in toluene (150 mL) and H20 (60 mL) was degassed with N2 for 15 minutes. The mixture was then heated at reflux overnight (120°C oil bath temperature). After allowing to cool to room temperature, the mixture was diluted with EtOAc (200 mL) and ¾0 (200 mL) and the mixture filtered through Celite. The filter cake was washed with EtOAc (3 x 100 mL) and the filtrate transferred to a separating funnel. Brine (200 mL) was added and the aqueous and organic layers partitioned. The organic layer was dried (Na2S04), filtered and the solvent removed under vacuum to leave a crude residue. The residue was purified by column chromatography on silica gel (residue dry- loaded on to silica gel) using EtOAc / hexanes (1:9 to 1 :4) as eluent to give the product (8.3 g, 76%) as a dark solid. Data same as above.
Step 2: l-(2-CYCLOPROPYL-4-FLUORO-5-NITROPHENYL)-lH-TETRAZOL-5(4H)-ONE
[00404] 2-Cyclopropyl-4-fluoro-5-nitroaniline (10.2 g, 52 mmol) was added over 5 minutes to a stirred solution of phosgene in toluene (20% wt. / vol; 100 mL) at ca. -10 °C under N2. Residual aniline from the flask was washed in to the mixture by rinsing with toluene (10 mL). The mixture was stirred at -10 °C for 10 minutes then heated to 80 °C and stirred for 3 hours. After allowing to cool to room temperature, the mixture was concentrated under vacuum to leave a crude residue. The residue was suspended in EtOAc (150 mL) and then concentrated under vacuum. The residue was placed under N2 and trimethylsilylazide (50g) was added. A reflux condenser was added to the apparatus and the mixture was heated to 80 °C under N2 with stirring overnight (note: the mixture does not reflux, but condenser used to minimize any loss of trimethylsilylazide). After allowing to cool to room
temperature, the mixture was concentrated under vacuum and the residue partitioned between EtOAc (200 mL) and H20 (60 mL). The organic layer was washed with H20 (60
mL) and then extracted with saturated NaHCC>3 (7 x 100 mL - until TLC indicated all product had been removed from the EtOAc layer). EtOAc (500 mL) was added to the combined aqueous extracts, and the pH was adjusted to <3 using 2N HCl. The aqueous and organic layers were partitioned and the aqueous layer was extracted with EtOAc (2 x 200 mL). The combined organic layers were dried (Na2S04), filtered and the solvent removed under vacuum to leave the product (10.3 g, 75%) as a solid.
[00405] JH NMR (300 MHz, DMSO) δ 8.38 (d, 1H), 7.3 (d, 1H), 1.82-1.78 (m, 1H), 1.09- 1.04 (m, 2H), 0.92-0.91 (m, 2H); 19F NMR (DMSO- , 282 MHz): 115.7 (dd).
Step 3: l-(2-CYCLOPROPYL-4-FLUORO-5-NITROPHENYL)-4-METHYL-lH-TETRAZOL- 5(4H)-ONE
[00406] K2C03 (13.4 g, 97.1 mmol) and Mel (4.8 mL, 77.7 mmol) were added sequentially to a stirred solution of l-(2-cyclopropyl-4-fluoro-5-nitrophenyl)-lH-tetrazol- 5(4H)-one (10.3 g, 38.8 mmol) in DMF (100 mL) at -78 °C under N2. The mixture was allowed to warm to room temperature over 3 hours. TLC indicated complete reaction, so poured mixture into H20 (1 L) and EtOAc (500 mL). The aqueous and organic layers were partitioned and the organic layer was washed with H20 (2 x 300 mL), brine (300 mL), then dried (Na2S04), filtered and the solvent removed under vacuum to leave the product (10.2 g, 94%) as a solid. Data identical to preparation of l-(2-cyclopropyl-4-fluoro-5-nitrophenyl)-4- methyl-lH-tetrazol-5(4H)-one described above.
EXAMPLE 32
SYNTHESIS OF l-(5-AMiNO-2-CYCLOPROPYL-4-FLUOROPHENYL)-4-METHYL-lH-
TETRAZOL-5(4H)-ONE

[00407] l-(2-Cyclopropyl-4-fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (170mg, 0.6mmol) was added to a Radley's Carousel reactor tube, ethanol (18ml) was added to the solid, followed by SnCl2.2H20 (460mg, 2.4mmol) and concentrated HCl (0.6ml). The mixture was heated at 80° C, reaction followed by thin layer chromatography. Once the reaction was determined to be completed, the reaction mixture was allowed to cool down to room temperature and concentrated under reduced pressure. Ethyl acetate was added to the residue, the solution was washed with saturated K2CO3. The layers were separated, and the organic layer was washed with brine, dried with Na2S04, and the solid was filtered off. The
mother liquor was then concentrated under reduced pressure, and the title compound was obtained as light brown oil (125mg).
[00408] JH NMR (300 MHz, DMSO) δ 6.79 (d, /=12.3Hz, 1H), 6.72 (d, /=8.4Hz, 1H), 5.36 (s, 2H), 3.59 (s, 3H), 1.56 (m, 1H), 0.68 (d, /=8.7Hz, 2H), 0.46 (d, /=5.1Hz, 2H); m/z = 250.09 (M+H)+.
EXAMPLE 33
ALTERNATIVE SYNTHESIS OF 1-(5-AMINO-2-CYCLOPROPYL-4-FLUOROPHENYL)-4- METHYL-LH-TETRAZOL-5(4H)-ON

[00409] Palladium on carbon (2.0 g, 20% by wt.) was added to a suspension of l-(2- cyclopropyl-4-fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (10.2 g, 36.5 mmol) in EtOH (150 mL) and AcOH (5.1 mL) under N2 in a 1L Parr hydrogenation flask. The mixture was evacuated then filled with ¾ on a Parr hydrogenation apparatus. The mixture was hydrogenated at 25-30 psi for ca. 5 hours until LC/MS indicated completion of the reaction (note: the hydrogen pressure decreases rapidly in the first 15 minutes and is replenished to 25-30 psi). After completion of the reaction the mixture is filtered through a small layer of Celite and the filter cake is washed with EtOH (4 x 50 mL). The filtrate was concentrated under vacuum to leave a crude solid that was dissolved in EtOAc (250 mL) and washed with saturated NaHC03 (150 mL), then dried (Na2S04), filtered and the solvent removed under vacuum to leave a crude solid. The solid was purified by column chromatography on silica gel (dry-loaded solid on silica gel) using EtOAc / hexanes (3:7 to 6:4) as an eluent to give the product (8.6 g, 86%) as a solid. Data identical to preparation of l-(5-amino-2- cyclopropyl-4-fluorophenyl)-4-methyl-lH-tetrazol-5(4H)-one described above. Also collected from the column were some less pure fractions (0.3 g) that were kept separate from the 8.6g lot of product. The less pure fractions contained ca. 93% of the title compound and ca. 4% of l-(5-amino-2-isopropyl-4-fluorophenyl)-4-methyl-lH-tetrazol-5(4H)-one, together with ca. 3% of other impurities by LC/MS analysis.
EXAMPLE 34
SYNTHESIS OF A^2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE- 1-YL]}PHENYL-5-FLUORO-A^4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE (1-1

[00410] 2-Chloro-5-fluoro-N-(2,2,6,6-tetramethylpiperidin-4-yl)pyrimidin-4-amine hydrochloride (72mg, 0.3 mmol) and l-(5-amino-2-cyclopropyl-4-fluorophenyl)-4-methyl- lH-tetrazol-5(4H)-one (62.5mg, 0.3 mmol) were added to a Radley's Carousel reactor tube. Isopropyl alcohol (2 mL) was added, followed by para-toluenesulfonic acid monohydrate (48mg, 0.3 mmol). The mixture was heated at 100° C under Ν2 overnight and the progress of the reaction was followed by LCMS. Upon completion of the reaction, the mixture was cooled, then neutralized by adding 2Ν ΝΗ3 in MeOH. The mixture was concentrated under vacuum and the residue was purified by column chromatography on silica gel using EtOAc : 2Ν ΝΗ3 in MeOH (100:0 to 90: 10) as an eluent to give the title compound as an off-white solid.
[00411] Ή NMR (300 MHz, DMSO) δ 8.47 (s, 1H), 7.82-7.76 (m, 2H), 7.17 (d, / = 8.1 Hz, 1H), 6.95 (d, / = 12.0 Hz, 1H), 4.22 (bs, 1H), 3.59 (s, 3H), 1.64-1.55 (m, 3H), 1.07 (t, / = 12.0 Hz, 2H), 0.97 (s, 12H), 1.56 (m, 1H), 0.79 (d, / = 8.1 Hz, 2H), 0.58 (d, / = 5.1 Hz, 2H); m/z = 500.20 (M+H)+.
EXAMPLE 35
SYNTHESIS OF 5-CYANO-N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4- TETRAZOL-5-ONE-1-YL]}PHENYL-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE (1-1

[00412] 5-Carboxyamide-2-chloro-N-(2,2,6,6-tetramethylpiperidin-4-yl)pyrimidin-4- amine hydrochloride (78mg, 0.3 mmol) and l-(5-amino-2-cyclopropyl-4-fluorophenyl)-4- methyl-lH-tetrazol-5(4H)-one (62.5mg, 0.3 mmol) were added to a Radley's Carousel reactor tube. Isopropyl alcohol (1.25 mL) was added, followed by para-toluenesulfonic acid monohydrate (48mg, 0.3 mmol). The mixture was heated at 100° C under Ν2 overnight and
the progress of the reaction was followed by LCMS. Upon completion of the reaction, the mixture was cooled, then neutralized by adding 2N N¾ in MeOH. The mixture was concentrated under vacuum to leave a crude residue. Anhydrous THF (2 mL) was added to the residue, and the mixture cooled to 0° C in an ice/water bath. Et3N (0.07 mL, 0.6 mmol) was added, followed by the addition of TFAA (0.05 mL, 0.45 mmol) dropwise. The progress of the reaction was followed by LCMS, additional Et3N (2 x 0.07mL) and TFAA (2 x 0.05 mL) being added to push the reaction to completion. The mixture was concentrated under vacuum and the residue was purified by column chromatography on silica gel using EtOAc : 2N NH3 in MeOH (100:0 to 90: 10) as an eluent to give the title compound as an off-white solid.
[00413] Ή NMR (300 MHz, DMSO) δ 9.56 (bs, 1H), 8.52 (bs, 1H), 8.33 (s, 1H), 7.74 (m, 2H), 7.48 (s, 1H), 7.03 (d, / = 11.4 Hz, 1H), 4.27 (bs, 1H), 3.59 (s, 3H), 1.73-1.69 (m, 3H), 1.52 (m, 2H), 1.27 (bs, 6H), 1.08 (bs, 6H), 0.86 (d, / = 8.1 Hz, 2H), 0.64 (bs, 2H); m/z = 506.58 (M+H)+.
EXAMPLE 36
SYNTHESIS OF 7Y2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE- 1-YL]}PHENYL-5-FLUORO-/Y4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE (1-1

[00414] A mixture of l-(5-amino-2-cyclopropyl-4-fluorophenyl)-4-methyl-lH-tetrazol- 5(4H)-one (200 mg, 0.8 mmol), 2-chloro-5-fluoro-N4-( 1,2,2, 6,6-pentamethylpiperidin-4-yl)- 4-pyrimidineamine, HC1 salt (246 mg, 0.73 mmol) and /?ara-toluenesulfonic acid monohydrate (139 mg, 0.73 mmol) in isopropanol (2 mL) was heated at 110 °C in a sealed vial and stirred for 2 days. After 2 days, the mixture was cooled and 3-amino benzoic acid (excess) was added. The mixture was re -heated to 110 °C and stirred for 1 day (to ensure reaction of all 2-chloro-5-fluoro-N4-(l,2,2,6,6-pentamethylpiperidin-4-yl)-4- pyrimidineamine). After allowing to cool the solvent was concentrated under vacuum to leave a crude residue. The residue was partitioned between EtOAc (50 L) and IN NaOH (50 mL). The organic layer was washed with IN NaOH (20 mL) and brine (20 mL), then dried (Na2S04), filtered and the solvent removed under vacuum to leave a crude solid. The solid was triturated with Et20 and filtered. The filter cake was re-triturated with Et20 and filtered
to give the title compound (30 mg) as a solid [note: the filtrates from the triturations were also kept and contain desired product].
[00415] JH NMR (300 MHz, _fc-DMSO) δ 8.53 (s, 1H), 7.84 (d, 1H), 7.80 (d, 1H), 7.24 (d, 1H), 7.00 (d, 1H), 4.21-4.07 (m, 1H), 3.62 (s, 3H), 2.14 (s, 3H), 1.66-1.58 (m, 3H), 1.38 (t, 2H), 1.03 (s, 6H), 0.84 (br. s, 8H), 0.62-0.60 (m, 2H); m/z = 514.45 (M+H)+.
EXAMPLE 37
SYNTHESIS OF 5-CYANO-N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4- TETRAZOL-5-ONE-1-YL]}PHENYL-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)-2,4-

Step 1:
[00416] A mixture of 5-carboxamide-2-chloro-N4-( 1,2,2, 6,6, -pentamethylpiperidin-4-yl)- 4-pyrimidineamine, HC1 salt (326 mg, 1.0 mmol), l-(5-amino-2-cyclopropyl-4- fluorophenyl)-4-methyl-lH-tetrazol-5(4H)-one (250 mg, 1.0 mmol) and para- toluenesulfonic acid monohydrate (190 mg, 1.0 mmol) in isopropanol (5 mL) was heated in a sealed vial at 100 °C overnight. After allowing to cool to room temperature the mixture was concentrated under vacuum. The residue was partitioned between IN NaOH (50 mL) and EtOAc (50 mL) and the organic layer was washed with IN NaOH (25 mL), brine (25 mL), then dried (Na2S04), filtered and the solvent removed under vacuum to leave a crude solid. The solid was triturated with Et20 and filtered to give the product (285 mg, 53%) that was used directly in the next step.
Step 2:
[00417] TFAA (1.4 mL, 5.0 mmol) was added over 10 minutes to a stirred suspension of the amide product from step 1 above (270 mg, 0.5 mmol) and Et3N (1.67 mL, 6.0 mmol) in THF (5 mL) at -50 °C under N2. The mixture was stirred at -50 °C for 20 minutes then allowed to warm to 0 °C over 1 hour and stirred for another 2 hours at 0 °C (LC/MS indicated complete reaction). The mixture was quenched by pouring in to saturated NaHC03 (50 mL) and EtOAc (100 mL). The aqueous and organic layers were partitioned and the organic layer was washed with brine (50 mL), dried (Na2S04), filtered and the solvent removed under vacuum to leave a crude solid. The solid was triturated with Et20 and filtered and the filter cake was then recrystallized from EtOAc / MeOH. JH NMR indicated the
recrystallized product to be a trifluoroacetate salt, so partitioned recrystallized solid between 0.5N NaOH (30 mL) and EtOAc (50 mL). The organic layer was washed with brine (30 mL), dried (Na2S04), filtered and the solvent removed under vacuum to leave a solid (65 mg, 25%) as a solid [note: much product in Et20 filtrate and EtOAc / MeOH filtrate].
[00418] JH NMR (300 MHz, _fc-DMSO) δ 9.44 (s, 1H), 8.28 (s, 1H), 7.49 (d, 1H), 7.39 (d, 1H), 6.99 (d, 1H), 4.16 (m, 1H), 3.59 (s, 3H), 2.08 (s, 3H), 1.69 (m, 1H), 1.43-1.33 (m, 4H), 0.98 (s, 6H), 0.88-0.83 (m, 2H), 0.73 (s, 6H), 0.62-0.60 (m, 2H); m/z = 521.37 (M+H)+. EXAMPLE 38:
STEPI: PREPARATION OF 1-(2-(1,1,1,-TRIFLUORO-2-METHYLPROPAN-2-YLOXY)-5- N

[00419] ΚΗ (35% wt. in mineral oil; 126 mg, 1.1 mmol of ΚΗ) was washed with hexanes and the solvent removed with a pipette. The solid ΚΗ remaining (about 44 mg, 1.1 mmol) was cooled to 0 °C under an atmosphere of nitrogen and THF (2 mL) was added. A solution of 2-trifluoromethyl-2-propanol (116 mg, 1.0 mmol) in THF (1 mL) was slowly added (after complete addition, the vial containing 2-trifluoromethyl-2-propanol was rinsed with an additional 0.5 mL of THF). The mixture was stirred at 0 °C for 20 minutes then l-(2-fluoro- 5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (120 mg, 0.5 mmol) was added in one portion. The mixture was stirred at 0 °C for 5 minutes then allowed to warm to room temperature and stirred for 2 hours. The solvent was removed under vacuum and the residue was partitioned between CH2CI2 (30 mL) and ¾0 (20 mL). The aqueous layer was extracted with CH2CI2 (10 mL) and the combined organic extracts were dried (Na2S04), filtered and the solvent removed under vacuum to leave a residue (150 mg) that crystallized on standing.
[00420] JH NMR (300 MHz; _fc-DMSO) δ 8.60 (m, 1H), 8.40 (m, 1H), 7.75 (d, 1H), 3.60 (s, 3H), 1.5 (s, 6H); 19F NMR (282 MHz; _¼-DMSO) δ -83.0 ; m/z = 348.2 (M+H)+.
STEP 2: PREPARATION OF 1-(2-(1,1,1,-TRIFLUORO-2-METHYLPROPAN-2-YLOXY)-5- AMINOPHENYL)-4-METHYL-LH-TETRAZOL-5(4H)-ONE
[00421] Pd(C), 10%w/w Pd, wet Degussa grade E101 (30 mg) was added to a stirred mixture of l-(2-(l,l,l,-trifluoro-2-methylpropan-2-yloxy)-5-nitrophenyl)-4-methyl-lH- tetrazol-5(4H)-one (150 mg) in MeOH (10 mL) under nitrogen. The mixture was evacuated
and placed under an atmosphere of hydrogen (balloon of ¾ used). The evacuation and hydrogen- fill procedure was repeated twice, then the mixture was placed under an atmosphere of ¾ and stirred for 1 hour. After completion of the reaction, the mixture was filtered through Celite and the filter cake was washed with MeOH (3 x 10 mL). The solvent was removed under vacuum to leave a crude residue (112 mg, 70% over 2 steps) that crystallized on standing. mJz = 318.2 (M+H)+.
EXAMPLE 39:
PREPARATION OF 1-(2-(1,1,1-TRIFLUORO-2-METHYLPROPAN-2-YLOXY)-5-(4-(1,2,2,6,6- PENTAMETHYLPIPERIDIN-4-YLAMINO)-5-FLUOROPYRIMIDIN-2-YLAMINO)PHENYL)-4- METHYL- LH-TETRAZOL-5(4H)-ONE (I- 18)

[00422] A mixture of 2-chloro-5-fluoro-N4-(l ,2,2,6,6-pentamethylpiperidin-4-yl)-4- pyrimidineamine (56 mg, 0.19 mmol), l-(2-(l,l,l,-trifluoro-2-methylpropan-2-yloxy)-5- aminophenyl)-4-methyl-lH-tetrazol-5(4H)-one (56 mg, 0.18 mmol) and para- toluenesulfonic acid monohydrate (17 mg, 0.09 mmol) in isopropanol (2 mL) was heated to reflux and stirred overnight. After allowing to cool to room temperature, the mixture was dry- loaded on to silica gel and then purified by column chromatography on silica gel using CH2C12 / 2N NH3 in MeOH (1:0 too 95:5) to give a solid. The solid was triturated with Et20 and filtered - the filter cake comprised the desired product and /?-TsOH. The filter cake was suspended in CH2CI2 and washed with 0.5 N NaOH. The organic layer was dried (Na2S04), filtered and the solvent removed under vacuum to leave the product as a solid.
[00423] JH NMR (300 MHz; _fc-DMSO) δ 9.44 (s, 1H), 7.87-7.78 (m, 3H), 7.24-7.19 (m, 2H), 4.28-4.25 (m, 1H), 3.58 (s, 3H), 2.14 (s, 3H), 1.66-1.63 (m, 2H), 1.43 (app. t, 2H), 1.25 (s, 6H), 1.04 (s, 6H), 0.91 (s, 6H); m/z = 580.3 (M-H)+.
EXAMPLE 40:
PREPARATION OF 1-(2-(1,1,1-TRIFLUORO-2-METHYLPROPAN-2-YLOXY)-5-(4-(2,2,6,6- TETRAMETHYLPIPERIDIN-4-YLAMINO)-5-FLUOROPYRIMIDIN-2-YLAMINO)PHENYL)-4- METHYL-L -TETRAZOL-5(4H)-ONE (1-19)

[00424] Reaction performed in a manner similar to that described for Example 39, to give the product as a solid.
[00425] JH NMR (300 MHz; _fc-DMSO) δ 9.35 (s, 1H), 7.79-7.87 (m, 3H), 7.19-7.22 (m, 2H), 4.37-4.35 (m, 1H), 3.57 (s, 3H), 1.62-1.66 (m, 2H), 1.24 (s, 6H), 1.14 (app. t, 2H), 1.05 (s, 6H), 1.00 (s, 6H); m/z = 568.4 (M+H)+.
EXAMPLE 41:
STEP 1: PREPARATION OF l-METHYL-4-(5-NiTRO-2-(oxETAN-3-YLOXY)PHENYL)-lH-
TETRAZOL-5-(4H)-ONE

[00426] tert-BuOK (123 mg, 1.1 mmol) was added to a stirred solution of oxetan-3-ol (74 mg, 1.0 mmol) in THF (3 mL) at 0 °C under nitrogen. The mixture was stirred at 0 °C for 20 minutes then l-(2-fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (120 mg, 9.5 mmol) was added in one portion. The mixture was stirred at 0 °C for 10 minutes, then allowed to warm to room temperature and stirred for 2 hours. The solvent was removed under vacuum and the residue was partitioned between CH2CI2 (30 mL) and ¾0 (20 mL). The aqueous layer was extracted with CH2CI2 (1 x 20 mL) and the combined organic extracts were dried (Na2S04), filtered and the solvent removed under vacuum to leave a residue (150 mg; theoretical = 147 mg) that crystallized on standing.
[00427] JH NMR (300 MHz; _fc-DMSO) δ 8.51-8.50 (m, 1H), 8.41-8.36 (m, 1H), 7.18- 7.15 (m, 1H), 5.58-5.41 (m, 1H), 4.92 (t, 2H), 4.47 (t, 2H), 3.63 (s, 3H).
STEP 2: PREPARATION OF l-(5-AMiNO-2-(oxETAN-3-YLOXY)PHENYL)-4-METHYL-lH-
TETRAZOL-5-(4H)-ONE
[00428] Pd(C), 10%w/w Pd, wet Degussa grade E101 (30 mg) was added to a stirred mixture of l-methyl-4-(5-nitro-2-(oxetan-3-yloxy)phenyl)-lH-tetrazol-5-(4H)-one (147 mg, 0.5 mmol) in MeOH (10 mL) under nitrogen. The mixture was evacuated and placed under an atmosphere of hydrogen (balloon of ¾ used). The evacuation and hydrogen-fill procedure was repeated twice, then the mixture was placed under an atmosphere of ¾ and stirred for 1 hour. After completion of the reaction, the mixture was filtered through Celite and the filter cake was washed with MeOH (3 x 10 mL). The solvent was removed under vacuum to leave a crude residue that crystallized on standing (119 mg, 90% over 2 steps). The compound was used directly in the next step.
EXAMPLE 42:
PREPARATION OF 1-(5-(4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YLAMINO)-5- FLUOROPYRIMIDIN-2-YLAMINO)-2-(OXETAN-3-YLOXY)PHENYL)-4-METHYL-LH- TETRAZOL- -ONE (1-20)

[00429] A mixture of 2-chloro-5-fluoro-N4-(l ,2,2,6,6-pentamethylpiperidin-4-yl)-4- pyrimidineamine (72 mg, 0.24 mmol), l-(5-amino-2-(oxetan-3-yloxy)phenyl)-4-methyl-lH- tetrazol-5-(4H)-one (60 mg, 0.23 mmol), Pd(OAc)2 (5.1 mg, 0.02 mmol), BINAP (28.4 mg, 0.05 mmol) and CS2CO3 (223 mg, 0. 68 mmol) were combined in a 10 mL CEM microwave vial. 1,4-Dioxane (3 mL) was added and the mixture was degassed with nitrogen for 5-10 minutes. The mixture was sealed and then heated to 120 °C under microwave irradiation for 60 minutes. After completion of the first microwave heating cycle, the mixture was then heated under microwave irradiation for a further 60 minutes. After allowing to cool to room temperature, the mixture was filtered through Celite and the filter cake was washed with 1,4- dioxane (3 x 5 mL). The filtrate was concentrated under vacuum and purified by column chromatography on silica gel using CH2CI2 : 2N N¾ in MeOH (1:0 to 95:5) to give a solid. The solid was triturated with Et20 and filtered to give the title compound (50 mg, 42%) as a solid.
[00430] JH NMR (300 MHz; _fc-DMSO) δ 9.15 (s, 1H), 7.85 (m, 2H), 7.67 (d, 1H), 7.18 (br. d, 1H), 6.77 (d, 1H), 5.26 (m, 1H), 4.81 (t, 2H), 4.36 (t, 2H), 3.61 (s, 3H), 2.17 (s, 3H), 1.67-1.63 (m, 2H), 1.44 (app. t, 2H), 1.05 (s, 6H), 0.92 (s, 6H); m/z = 528.6 (M+H)+.
I l l
EXAMPLE 43:
PREPARATION OF 1-(5-(4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YLAMINO)-5- FLUOROPYRIMIDIN-2-YLAMINO)-2-(OXETAN-3-YLOXY)PHENYL)-4-METHYL-LH- TETRAZOL- -ONE (1-21)

[00431] Reaction performed in a manner similar to that described for Example 42, to give the product (60 mg, 56%) as a solid.
[00432] JH NMR (300 MHz; _fc-DMSO) δ 9.15 (s, 1H), 7.90 (d, 1H), 7.65-7.68 (m, 1H), 7.14 (br. d, 1H), 6.75 (d, 1H), 5.22-5.27 (m, 1H), 4.80 (t, 2H), 4.40 (t, 2H), 4.26-4.40 (m, 1H), 3.60 (s, 3H), 1.61-1.64 (m, 2H), 1.07-1.24 (m, 2H), 1.03 (s, 6H), 0.99 (s, 6H); mJz = 514.3 (M+H)+.
EXAMPLE 44:
STEP 1: PREPARATION OF l-(3-FLUORO-5-NiTROPHENYL)-lH-TETRAZOL-5(4H)-ONE

[00433] A mixture of 5-fluoro-3-nitroaniline (1.56 g, 10.0 mmol) and phosgene in toluene (20% wt. / vol; 25 mL) was heated to reflux and stirred for 3 hours. After allowing to cool to room temperature, the solvent was removed under vacuum and the residue was suspended in trimethylsilyl azide (25 mL) and the contents transferred to a sealed tube. The mixture was heated at 105 °C overnight. After allowing to cool to room temperature, the mixture was concentrated under vacuum and the residue partitioned between EtOAc (40 mL) and saturated NaHCC>3 (40 mL). The organic layer was extracted with further saturated NaHCC>3 (3 x 40 mL - TLC being used to monitor the removal of the tetrazolone product from the EtOAc layer). The combined aqueous layers were acidified using 6N HCl and the emerging solid was extracted with EtOAc (4 x 40 mL). The combined organic extracts were dried (Na2S04), filtered and the solvent removed under vacuum to leave the product (1.16 g, 52%) as a solid.
[00434] JH NMR (300 MHz; _fc-DMSO) δ 8.59-8.58 (m, 1H), 8.20-8.16 (m, 2H); m/z = 224.1 (M-H)+.
STEP 2: PREPARATION OF l-(3-FLUORO-5-NiTROPHENYL)-4-METHYL-lH-TETRAZOL-
5(4H)-ONE
[00435] A mixture of l-(3-fluoro-5-nitrophenyl)-lH-tetrazol-5(4H)-one (1.12 g, 5.0 mmol), K2C03 (1.73 g, 12.5 mmol) and iodomethane (1.42 g, 10.0 mmol) in DMF (20 mL) was stirred at room temperature overnight. I¾0 (100 mL) and EtOAc (50 mL) were then added, and the aqueous and organic layers were partitioned. The organic layer was washed with Η2Ο (3 x 75 mL), brine (1 x 50 mL), then dried (Na2S04), filtered and the solvent removed under vacuum to leave the product (1.18 g, 98%) as a solid.
[00436] JH NMR (300 MHz; _fc-DMSO) δ 8.58 (s, 1H), 8.22-8.18 (m, 2H), 3.63 (s, 3H). STEP 3: PREPARATION OF l-(3-AMiNO-5-FLUOROPHENYL)-4-METHYL-lH-TETRAZOL-
5(4H)-ONE
[00437] Pd(C), 10%w/w Pd, wet Degussa grade E101 (90 mg) was added to a stirred suspension of l-(3-fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (500 mg, 2.1 mmol) in MeOH (20 mL) under nitrogen. The mixture was evacuated and placed under an atmosphere of hydrogen (balloon of ¾ used). The evacuation and hydrogen-fill procedure was repeated twice, then the mixture was placed under an atmosphere of ¾ and stirred for 3 hours. After completion of the reaction, the mixture was filtered through Celite and the filter cake was washed with MeOH (4 x 10 mL). The solvent was removed under vacuum. The crude residue was purified by column chromatography on silica gel using EtOAc / hexane (4:6) as eluent to give the product (396 mg, 91%) as a solid.
[00438] JH NMR (300 MHz; _fc-DMSO) δ 6.95 (t, 1H), 6.79 (dt, 1H), 6.31 (dt, 1H), 5.85 (br. s, 2H), 3.57 (s, 3H); m/z = 210.2 (M-H)+.
EXAMPLE 45:
PREPARATION OF 1-(3-(4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YLAMINO)-5-
FLUOROPYRIMIDIN-2-YLAMINO)-5-FLUOROPHENYL)-4-METHYL-LH-TETRAZOL-5(4H)-ONE
(1-22)

[00439] A mixture of 2-chloro-5-fluoro-N4-(l ,2,2,6,6-pentamethylpiperidin-4-yl)-4- pyrimidineamine hydrochloride (202 mg, 0.6 mmol), l-(3-amino-5-fluorophenyl)-4-methyl- lH-tetrazol-5(4H)-one (190 mg, 0.9 mmol) and TFA (186 \lL, 2.4 mmol) in isopropanol (6 mL) was heated at 100 °C overnight in a sealed vial. Additional TFA (186 \lL, 2.4 mmol)
was added and the mixture was heated at 100 °C over 2 days. After allowing to cool to room temperature, the solvent was removed under vacuum and the residue purified by column chromatography on silica gel using CH2CI2 / 2N N¾ in MeOH (1:0 to 95:5) as eluent to give the product as a solid.
[00440] JH NMR (300 MHz; _fc-DMSO) δ 9.58 (s, 1H), 7.90-7.92 (m, 1H), 7.85 (br. s, 1H), 7.30 (d, 1H), 7.16-7.19 (m, 1H), 4.31-4.34 (m, 1H), 3.59 (s, 3H), 2.15 (s, 3H), 1.66- 1.69 (m, 2H), 1.44 (t, 2H), 1.05 (s, 6H), 0.99 (s, 6H); 19F NMR (282 MHz; _fc-DMSO) δ - 110.6 (t), -165.3 (s); m/z = 474.3 (M+H)+; m/z = 472.3 (M-H)+.
EXAMPLE 46:
PREPARATION OF 1-(3-(4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YLAMINO)-5-
FLUOROPYRIMIDIN-2-YLAMINO)-5-FLUOROPHENYL)-4-METHYL-LH-TETRAZOL-5(4H)-ONE
(1-23)

[00441] Reaction performed in a manner similar to that described for Example 45, to give the product as a solid.
[00442] JH NMR (300 MHz; i¾-DMSO) δ 9.58 (s, 1H), 7.87 (d, 1H), 7.85 (d, 1H), 7.29 (br. d, 1H), 7.15-7.18 (m, 1H), 4.39-4.42 (m, 1H), 3.58 (s, 3H), 1.66-1.69 (m, 2H), 1.19-1.12 (m, 2H), 1.12 (s, 6H), 1.00 (s, 6H); 19F NMR (282 MHz; i¾-DMSO) δ -110.6 (t), -165.2 (s); m/z = 460.4 (M+H)+.
EXAMPLE 47:
STEP 1: PREPARATION OF l-METHYL-4-(3-NiTRO-5-(oxETAN-3-YLOXY)PHENYL)-lH-
TETRA -5(4H)-ONE

[00443] tert-BuOK (123 mg, 1.1 mmol) was added to a stirred solution of oxetan-3-ol (74 mg, 1.0 mmol) in THF (3 mL) at 0 °C under nitrogen. The mixture was stirred at 0 °C for 30 minutes then l-(3-fluoro-5-nitrophenyl)-lH-tetrazol-5(4H)-one (120 mg, 9.5 mmol) was added in THF (1 mL). The mixture was allowed to warm to room temperature and then
stirred overnight. The solvent was removed under vacuum and the residue was partitioned between EtOAc (20 mL) and ¾0 / brine (40 mL). The aqueous layer was extracted with EtOAc (2 x 20 mL) and the combined organic extracts were washed with brine (1 x 20 mL), dried (Na2S04), filtered and the solvent removed under vacuum to leave a crude residue. The residue was purified by column chromatography on silica gel using EtOAc / hexane (3:7 to 1:1) as eluent to give the product (70 mg, 48%) as a solid.
[00444] JH NMR (300 MHz; CDC13) δ 8.52 (m, 1H), 7.84 (m, 1H), 7.53 (m, 1H), 5.36 (app. q, 1H), 5.06 (dd, 2H), 4.81 (dd, 2H), 3.74 (s, 3H).
[00445] Note: the reaction was repeated starting with 500mg of l-(3-fluoro-5- nitrophenyl)-lH-tetrazol-5(4H)-one to give the product (249 mg, 41%).
STEP 2: PREPARATION OF l-(3-AMiNO-5-(oxETAN-3-YLOXY)PHENYL)-4-METHYL-lH-
TETRAZOL-5(4H)-ONE
[00446] Pd(C), 10%w/w Pd, wet Degussa grade E101 (50 mg) was added to a stirred suspension of l-methyl-4-(3-nitro-5-(oxetan-3-yloxy)phenyl)-lH-tetrazol-5(4H)-one (320 mg, 1.1 mmol) in MeOH (25 mL) under nitrogen. The mixture was evacuated and placed under an atmosphere of hydrogen (balloon of ¾ used). The evacuation and hydrogen-fill procedure was repeated twice, then the mixture was placed under an atmosphere of ¾ and stirred for 6 hours. Additional Pd(C) (40 mg) was added under an atmosphere of nitrogen, and the mixture was hydrogenated for a further 3 hours. After completion of the reaction, the mixture was filtered through Celite and the filter cake was washed with MeOH (3 x 15 mL). The solvent was removed under vacuum and the crude residue was purified by column chromatography on silica gel using EtOAc / hexane (3:7 to 6:4) as eluent to give the product (225 mg, 79%) as a solid.
[00447] JH NMR (300 MHz; CDC13) δ 6.98 (t, 1H), 6.70 (t, 1H), 6.05 (t, 1H), 5.21 (app. q, 1H), 4.97 (t, 2H), 4.76 (t, 2H), 3.89 (br. s, 2H), 3.69 (s, 3H).
EXAMPLE 48:
PREPARATION OF 1-(3-(4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YLAMINO)-5- FLUOROPYRIMIDIN-2-YLAMINO)-5-(OXETAN-3-YLOXY)PHENYL)-4-METHYL-LH- TETRAZOL- -ONE (1-24)

[00449] JH NMR (300 MHz; _¼-DMSO) δ 9.32 (s, IH), 7.89 (d, IH), 7.72 (s, IH), 7.40 (s, IH), 7.24 (br. d, IH), 6.68 (s, IH), 5.22-5.25 (m, IH), 4.89 (t, 2H), 4.53 (dd, 2H), 4.30 (m, IH), 3.58 (s, 3H), 2.14 (s, 3H), 1.64-1.68 (m, 2H), 1.42 (t, 2H), 1.04 (s, 6H), 0.98 (s, 6H); m/z = 528.4 (M+H)+.
EXAMPLE 49:
PREPARATION OF 1-(3-(4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YLAMINO)-5- FLUOROPYRIMIDIN-2-YLAMINO)-5-(OXETAN-3-YLOXY)PHENYL)-4-METHYL-LH- TETRAZOL- -ONE (1-25)

[00450] Reaction performed in a manner similar to that described for Example 42, except a temperature of 160 °C for 1 hour was used for the reaction. Purification by column chromatography on silica gel using CH2CI2 / 2N N¾ in MeOH (1:0 to 95:5) gave the product (30 mg, 27%) as a solid.
[00451] JH NMR (300 MHz; _¾-DMSO) δ 9.31 (s, IH), 7.88-7.89 (m, IH), 7.72 (m, IH), 7.41 (m, IH), 7.22 (br. d, IH), 6.70 (m, IH), 5.21-5.23 (m, IH), 4.88 (t, 2H), 4.52 (t, 2H), 4.41 (m, IH), 3.56 (s, 3H), 1.65-1.68 (m, 4H), 1.26-1.11 (m, 4H), 1.11 (s, 6H), 1.00 (s, 6H); m/z = 514.5 (M+H)+.
EXAMPLE 50:
L-(2-FLUORO-5-NITROPHENYL)-LH-TETRAZOL-5(4H)-ONE

[00453] JH NMR (300 MHz, DMSO) δ 8.66 (m, 1H), 8.46 (m, 1H), 7.82 (t, /=9.6Hz, 1H).
EXAMPLE 51:
L-(5-AMINO-2-F -LH-TETRAZOL-5(4H)-ONE
[00454] l-(2-Fluoro-5-nitrophenyl lH-tetrazol-5(4H)-one (240mg, 1. lmmol) was dissolved into MeOH (5ml), Pd/C (36mg, 10% weight) was added to the solution. The mixture was degassed and stirred under 1 atmosphere of ¾ via a balloon at room
temperature overnight. After filtering off Pd/C first through fluted filter paper and then again through a bed of Celite, the solution was concentrated under reduced pressure to give the title compound (150mg, 72% yield) as an off-colored solid.
[00455] JH NMR (300 MHz, DMSO) δ 7.10 (m, 1H), 6.67 (m, 2H), 5.38 (bs, 2H); LCMS (m/z): 196.01 (MH+).
EXAMPLE 52:
L-(2-FLUORO-5-NI -4-METHYL-LH-TETRAZOL-5(4H)-ONE

[00456] l-(2-Fluoro-5-nitrophenyl)-lH-tetrazol-5(4H)-one (500mg, 2.2 mmol) was dissolved into DMF (5ml), K2CO3 (lg, 7.2 mmol) was added to this solution and followed by Mel (0.15ml, 2.4 mmol). The mixture was stirred at room temperature overnight. Ethyl acetate (15ml) and water (15ml) was added to this mixture, the 2 layers were separated. Aqueous layer was extracted with ethyl acetate (15ml), the organic layers were combined and concentrated under reduced pressure. DCM (40ml) was added to the residue and washed twice with brine (20ml). The layers were separated and the organic layer was dried with Na2S04. After filtering off the solid, the solution was concentrated under reduced pressure
and the residue was purified by flash column chromatography (1:1 Ethyl Acetate : Hexanes) to give the title compound as a white solid (400mg, 75% yield).
[00457] JH NMR (300 MHz, DMSO) δ 8.64 (m, 1H), 8.48 (m, 1H), 7.85 (t, /=9.3Hz, 1H), 3.63 (s, 3H); LCMS (m/z): 239.99 (MH+).
EXAMPLE 53:
L-(5-AMINO-2-FLUOROPHENYL)-4- -LH-TETRAZOL-5(4H)-ONE

[00458] l-(2-Fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (360mg, 1.5mmol) was suspended in MeOH (8ml) and ethyl acetate (3ml) mixture, Pd/C (50mg, 10% weight) was added to the solution. The mixture was degassed and stirred under 1 atmosphere of ¾ via a balloon at room temperature overnight. After filtering off Pd/C first through fluted filter paper and then again through a bed of Celite, the solution was concentrated under reduced pressure to give the title compound (200mg, 63% yield) as an off-colored solid.
[00459] JH NMR (300 MHz, DMSO) δ 7.11 (t, /=9.9Hz, 1H), 6.68 (m, 2H), 5.38 (s, 2H), 3.58 (s, 3H); LCMS (m/z): 210.02 (MH+).
EXAMPLE 54:
L-(2-FLUORO-5-NITROPHENYL)-4- -LH-TETRAZOL-5(4H)-ONE

[00460] l-(2-Fluoro-5-nitrophenyl)-lH-tetrazol-5(4H)-one (500mg, 2.2 mmol) was dissolved into DMF (5ml), K2CO3 (lg, 7.2 mmol) was added to this solution and followed by 2-iodopropane (0.25ml, 2.4 mmol). The mixture was stirred at room temperature overnight. Ethyl acetate (15ml) and water (15ml) was added to this mixture, the 2 layers were separated. Aqueous layer was extracted with ethyl acetate (15ml), the organic layers were combined and concentrated under reduced pressure. DCM (40ml) was added to the residue and washed twice with brine (20ml). The layers were separated and the organic layer was dried with Na2S04. After filtering off the solid, the solution was concentrated under reduced pressure and the residue was purified by flash column chromatography (1:2 Ethyl acetate : Hexanes) to give the title compound as a white solid (200mg, 75% pure).
[00461] JH NMR (300 MHz, DMSO) δ 8.68 (m, 1H), 8.48 (m, 1H), 7.84 (t, /=9.6Hz, 1H), 4.47 (m, 1H), 1.45 (d, /=3.3Hz, 6H); LCMS (m/z): 268.01 (MH+).
EXAMPLE 55:
l-(5-AMINO-2-FLUOROPHENYL)- -ISOPROPYL-lH-TETRAZOL-5(4H)-ONE

[00462] l-(2-Fluoro-5-nitrophenyl)-4-isopropyl-lH-tetrazol-5(4H)-one (200mg, 0.75mmol) was suspended in MeOH (10ml), Pd/C (20mg, 10% weight) was added to the solution. The mixture was degassed and stirred under 1 atmosphere of ¾ via a balloon at room temperature overnight. Pd/C was removed by using 2 layers of fluted filter paper and the mother liquor was concentrated under reduced pressure. Dichloromethane (20ml) was added and the solution was filtered through another 2 layers of fluted filter paper, thereafter the mother liquor was concentrated under reduced pressure to give the title compound (130mg, 73% yield) as an off-white solid.
[00463] JH NMR (300 MHz, DMSO) δ 7.12 (t, /=9.3Hz, 1H), 6.69 (m, 2H), 5.38 (s, 2H), 3.57 (s, 3H), 4.43 (m, 1H), 1.43 (d, /=3.4Hz, 6H); LCMS (m/z): 238.05 (MH+).
EXAMPLE 56:
L-(2-FLUORO-5-NITROPHENYL)- -(2-FLUOROETHYL)-LH-TETRAZOL-5(4H)-ONE

[00464] l-(2-Fluoro-5-nitrophenyl)-lH-tetrazol-5(4H)-one (500mg, 2.2 mmol) was dissolved into DMF (5ml), K2CO3 (lg, 7.2 mmol) was added to this solution and followed by l-bromo-2-fluoroethane (0.31g, 2.4 mmol). The mixture was stirred at room temperature overnight. Ethyl acetate (15ml) and water (15ml) was added to this mixture, the 2 layers were separated. Aqueous layer was extracted with ethyl acetate (15ml), the organic layers were combined and concentrated under reduced pressure. DCM (40ml) was added to the residue and washed twice with brine (20ml). The layers were separated and the organic layer was dried with Na2S04. After filtering off the solid, the solution was concentrated under reduced pressure and the residue was purified by flash column chromatography (1:2 Ethyl acetate : Hexanes) to give the title compound as a white solid (70mg, 11.6% yield).
[00465] Ή NMR (300 MHz, DMSO) δ 8.69-8.67 (m, 1H), 8.51-8.47 (m, 1H), 7.86 (t, /=9.3Hz, 1H), 4.87 (t, /=4.5Hz, 1H), 4.72 (t, /=4.2Hz, 1H), 4.39 (t, /=4.5Hz, 1H), 4.3 (t, /=4.8Hz, 1H); LCMS (m/z): 271.05 (MH+).
EXAMPLE 57:
L-(5-AMINO-2-FLUOROPHENYL)- -(2-FLUOROETHYL)-LH-TETRAZOL-5(4H)-ONE

[00466] l-(2-Fluoro-5-nitrophenyl)-4-(2-fluoroethyl)- lH-tetrazol-5(4H)-one (70mg, 0.2mmol) was dissolved into MeOH (1ml), Pd/C (lOmg, 10% weight) was added to the solution. The mixture was degassed and stirred under 1 atmosphere of ¾ via a balloon at room temperature overnight. Pd/C was removed by using 2 layers of fluted filter paper and the mother liquor was concentrated under reduced pressure. Dichloromethane (10ml) was added and the solution was filtered through another 2 layers of fluted filter paper, thereafter the mother liquor was concentrated under reduced pressure to give the title compound (36mg, 58% yield).
[00467] JH NMR (300 MHz, DMSO) δ 7.13 (t, /=9.3Hz, 1H), 6.69 (m, 2H), 5.38 (s, 2H), 4.85 (t, /=4.8Hz, 1H), 4.7 (t, /=4.5Hz, 1H), 4.35 (t, /=4.8Hz, 1H), 4.26 (t, /=4.8Hz, 1H); LCMS (m/z): 242.02 (MH+).
EXAMPLE 58:
L-(2-METHYL-5-NITROPHENYL)-L -TETRAZOL-5(4H)-ONE

[00468] To 2-methyl-5-nitrobenzoisocyanate (2g, 11.2mmol), azidotrimethylsilane (3.7ml, 2.5eq) was added and the mixture was heated at 100° C overnight. The mixture was cooled to room temperature, concentrated under reduced pressure. Traces of
azidotrimethylsilane were removed by co-evaporating with ethyl acetate under reduced pressure. To this residue, dichloromethane was added and the mixture was sonicated, the title compound (1.9g, 80% yield) was obtained as a fluffy white solid after filtering through the Buchner funnel fitted with filter paper.
[00469] Ή NMR (300 MHz, DMSO) δ 8.38 (s, 1H), 8.3 (d, /=8.4Hz, 1H), 7.74 (d, /=8.7Hz, 1H), 2.35 (s, 3H); LCMS (m/z): 211.01 (MH+).
EXAMPLE 59:
l-(2-METHYL-5-NITROPHENYL)-4- -lH-TETRAZOL-5(4H)-ONE

[00470] l-(2-Methyl-5-nitrophenyl)-lH-tetrazol-5(4H)-one (900mg, 4.3mmol) was dissolved into DMF (13ml), K2CO3 (1.95g, 14.1mmol) was added to this solution and followed by iodomethane (0.32ml, 5.1mmol). The mixture was stirred at room temperature overnight. Ethyl acetate (20ml) and water (20ml) was added to this mixture, the 2 layers were separated and the organic layer was dried with Na2S04. After filtering off the solid, the solution was concentrated under reduced pressure and the residue was sonicated with ether and then filtered off mother liquor to give the title compound as a white solid (700mg, 73% yield).
[00471] JH NMR (300 MHz, DMSO) δ 8.36 (s, 1H), 8.31 (d, 7=8. lHz, 1H), 7.75 (d, /=8.7Hz, 1H), 3.62 (s, 3H), 2.36 (s, 3H); LCMS (m/z): 236.05 (MH+).
EXAMPLE 60:
L-(5-AMINO-2-METHYLPHENYL)-4- -LH-TETRAZOL-5(4H)-ONE

[00472] l-(2-Methyl-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (300mg, 0.75mmol) was suspended in MeOH (10ml), Pd/C (30mg, 10% weight) was added to the solution. The mixture was degassed and stirred under 1 atmosphere of ¾ via a balloon at room temperature for 4 hours. Pd/C was removed by using 2 layers of fluted filter paper and the mother liquor was concentrated under reduced pressure to give the title compound (140mg, 54% yield) as brown oil.
[00473] JH NMR (300 MHz, DMSO) δ 7.02 (d, /=8.1Hz, 1H), 6.63 (d, /=8.4Hz, 1H), 6.52 (s, 1H), 5.26 (s, 2H), 3.58 (s, 3H), 1.96 (s, 3H); LCMS (m/z): 206.07 (MH+).
EXAMPLE 61:
L-(2-ISOPROPOX

[00475] JH NMR (300 MHz, DMSO) δ 8.45-8.39 (m, 2H), 7.52 (d, /=9.3Hz, 1H), 4.93- 4.89 (m, 1H), 3.60 (s, 3H), 1.24 (d, /=6.0Hz, 6H); LCMS (m/z): 280.05 (MH+).
EXAMPLE 62:
L-(5-AMINO-2-ISOPROPOXYPHENY -4-METHYL-LH-TETRAZOL-5(4H)-ONE

[00476] l-(2-Isopropxy-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (356mg, 1.27mmol) was suspended in MeOH (10ml), Pd/C (40mg, 10% weight) was added to the solution. The mixture was degassed and stirred under 1 atmosphere of ¾ via a balloon at room temperature for 6 hours. Another 15mg of Pd/C was added to the solution and stirred under 1 atmosphere of hydrogen until TLC (1:1 Hexanes : Ethyl Acetate) indicated the completion of reaction. Pd/C was removed by using 2 layers of fluted filter paper and the mother liquor was concentrated under reduced pressure. Dichloromethane (5ml) was added to the residue and it was filtered through another double layers of fluted filter paper again. The mother liquor was concentrated under reduced pressure to give the title compound (200mg, 63% yield) as an off white solid.
[00477] JH NMR (300 MHz, DMSO) δ 6.95 (d, /=8.7Hz, 1H), 6.69 (d, /=8.7Hz, 1H), 6.55 (s, 1H), 5.03 (s, 2H), 4.25 (m, 1H), 3.57 (s, 3H), 1.07 (d, /=6.0Hz, 6H); LCMS (m/z): 250.08 (MH+).
EXAMPLE 63:
l-(3-NlTROPHENYL)-lH-TETRAZO -5(4H)-ONE

[00478] To 3-nitrobenzoisocyanate (2g, 12.2mmol), azidotrimethylsilane (3.2ml, 24.4mmol) was added and the mixture was heated at 100° C overnight. The mixture was cooled to room temperature, concentrated under reduced pressure. Residue was sonicated with hexanes (100ml), and filtered to give a pale yellow solid (2.3g). This solid was further purified by flash column chromatography (2: 1 Hexanes : Ethyl Acetate to 4: 1 Ethyl Acetate: Hexanes) to give 1.8g of the title compound (75% pure). This solid was dissolved into ethyl acetate (100ml) and washed with saturated NaHCC>3, the layers were separated. The aqueous layer was acidified with IN HC1, and extracted with ethyl acetate (100ml). The organic layer was dried with Na2S04, the solid was filtered off and mother liquor was concentrated under reduced pressure to give the title compound (1.6g, 64% yield).
[00479] JH NMR (300 MHz, DMSO) δ 8.73 (s, 1H), 8.3-8.23 (m, 2H), 7.85 (t, 7=8. lHz, 1H); LCMS (m/z): 208.13 (MH+).
EXAMPLE 64:
l-(3-NlTROPHENYL)-4-METHYL-l -TETRAZOL-5(4H)-ONE

[00480] l-(3-Nitrophenyl)-lH-tetrazol-5(4H)-one (500mg, 2.4mmol) was dissolved into DMF (5ml), K2CO3 (l. lg, 6.0mmol) was added to this solution and followed by
iodomethane (0.17ml, 2.7mmol). The mixture was stirred at room temperature over 2 days. Ethyl acetate (20ml) was added to this mixture, after filtering off solid, the solution was concentrated under reduced pressure. Dichloromethane was added and the solution was washed with saturated NaHCC>3, brine (4x50ml) and dried with Na2S04. The solid was filtered off and mother liquor was concentrated under reduced pressure to give the title compound as a white solid (360mg, 67% yield).
[00481] Ή NMR (300 MHz, DMSO) δ 8.71 (s, 1H), 8.32-8.25 (m, 2H), 7.87 (t, /=8.1Hz, 1H), 3.62 (s, 3H); LCMS (m/z): 222.16 (MH+).
EXAMPLE 65:
l-(3-AMINOPHENYL)-4-METHYL-l -TETRAZOL-5(4H)-ONE

[00482] l-(3-Nitrophenyl)-4-methyl- lH-tetrazol-5(4H)-one (360mg, 1.6mmol) was suspended in MeOH (10ml), Pd/C (40mg, 10% weight) was added to the solution. The mixture was degassed and stirred under 1 atmosphere of ¾ via a balloon at room temperature for 2 hours. Reaction was monitored by TLC (2: 1 Ethyl Acetate : Hexanes) and an addition of 25 mg of Pd/C was added to the mixture and left stirred at room temperature overnight. Pd/C was removed by using 2 layers of fluted filter paper and the mother liquor was concentrated under reduced pressure. To this residue, dichloromethane was added and the solution was filtered again through double layer of fluted filter paper. Mother liquor was concentrated under reduced pressure to give the title compound (200mg, 64% yield) as light grey solid.
[00483] Ή NMR (300 MHz, DMSO) δ 7.13 (t, /=8.1Hz, 1H), 7.04 (s, 1H), 6.94 (d, /=7.8Hz, 1H), 6.56 (d, /=8.1Hz, 1H), 5.48 (s, 2H), 3.57 (s, 3H); LCMS (m/z): 192.19
(MH+).
EXAMPLE 66:
1- [5-NLTRO-2-(4-TETRAHYDROPYRAN-4- YLOXY) PHENYL] -4-METHYL- 1H-TETRAZOL-
5(4H)-ONE

[00484] Oxan-4-ol (0.28ml, 3mmol) was dissolved into DMF (5ml), the solution was chilled to 0° C in a ice/water bath. NaH (90mg, 3.7mmol) was added and the mixture was stirred at 0° C for 10 minutes. l-(2-Fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (600mg, 2.5mmol) in DMF (5ml) was added into the mixture dropwise, and the solution was allowed to warm up to room temperature, progress of reaction followed by thin layer chromatography. The mixture was first warmed to 45° C, then increased to 65° C and left at that temperature overnight. Ethyl acetate (20ml) and water (20ml) were added to the mixture, the 2 layers were separated, and the organic layer was dried with Na2S04. The organic layer was concentrated under reduced pressure and purified by flash column
chromatrography (2: 1 Hexanes : Ethyl acetate) to give the title compound (600mg, 75% yield).
[00485] JH NMR (300 MHz, DMSO) δ 8.47 (s, IH), 8.4 (d, /=9.3Hz, IH), 7.59 (d, /=9.3Hz, IH), 4.37 (1, IH), 3.68 (bs, 2H), 3.62 (s, 3H), 3.47 (bs, 2H), 1.9 (bs, 2H), 1.58 (bs, 2H); LCMS (m/z): 322.12 (MH+).
EXAMPLE 67:
1- [5-AMINO-2-(4-TETRAHYDROPYRAN-4- YLOXY) PHENYL] ^-METHYL- 1H-TETRAZOL-
5(4H)-ONE

[00486] l-[5-Nitro-2-(4-tetrahydropyran-4-yloxy)phenyl]-4-methyl- lH-tetrazol-5(4H)- one (260mg, 0.8mmol) was suspended in MeOH (5ml), Pd/C (30mg, 10% weight) was added to the solution. The mixture was degassed and stirred under 1 atmosphere of ¾ via a balloon at room temperature overnight. Pd/C was removed by using 2 layers of fluted filter paper and the mother liquor was concentrated under reduced pressure. To this residue, dichloromethane was added and the solution was filtered again through double layer of fluted filter paper. Mother liquor was concentrated under reduced pressure to give the title compound (lOOmg, 43% yield) as light grey solid.
[00487] JH NMR (300 MHz, DMSO) δ 6.99 (d, /=9.0Hz, IH), 6.7 (d, /=9.0Hz, IH), 6.57 (s, IH), 5.06 (s, 2H), 4.3 (m, IH), 3.6 (m, 2H), 3.58 (s, 3H), 3.35 (m, 2H), 1.78 (m, 2H), 1.45 (m, 2H); LCMS (m/z): 292.28 (MH+).
EXAMPLE 68:
N2-{[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1-YL]-4-(TETRAHYDROPYRAN-4- YLOXY)}PHENYL-5-FLUORO-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE (1-2

EXAMPLE 69:
N2-{4-(TETRAHYDROPYRAN-4-YLOXY)-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE (1-2

[00489] Ή NMR (300 MHz, DMSO) δ 9.13 (s, IH), 7.89 (d, /=3Hz, IH), 7.83 (d, /=3.6Hz, IH), 7.67 (d, /=9Hz, IH), 7.17 (m, 2H), 4.51 (m, IH), 4.33 (m, IH), 3.66-3.59 (m, 5H), 3.36 (m, 2H), 1.78 (m, 2H), 1.64 (d, /=9.6Hz, 2H), 1.44 (m, 2H), 1.15 (t, /=11.7Hz, 2H), 1.03 (s, 6H), 1.0 (s, 6H); LCMS (m/z): 542.58 (MH+).
EXAMPLE 70:
L-{2-[(3-METHYLOXETAN-3-YL)METHOXY]-5-NITROPHENYL}-4-METHYL-LH-TETRAZOL-
5(4H)-ONE

[00490] (3-Methyloxetan-3-yl)methanol (0.3ml, 3mmol) was dissolved into DMF (4ml), the solution was chilled to 0° C in a ice/water bath. NaH (90mg, 3.7mmol) was added and the mixture was stirred at 0° C for 10 minutes. l-(2-Fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol- 5(4H)-one (600mg, 2.5mmol) in DMF (5ml) was added into the mixture dropwise, and the solution was allowed to warm up to room temperature, progress of reaction followed by thin layer chromatography. The mixture was first warmed to 45° C, then increased to 60° C and left at that temperature overnight. Additional 0.3ml of (3-methyloxetan-3-yl)methanol and lOOmg of NaH were added to the mixture and left for another day. Ethyl acetate (20ml) and water (20ml) were added to the mixture, the 2 layers were separated, and the organic layer
was dried with Na2S04. The organic layer was concentrated under reduced pressure and purified by flash column chromatrography (2: 1 Hexanes : Ethyl acetate) to give the title compound (466mg, 58% yield).
[00491] JH NMR (300 MHz, DMSO) δ 8.51-8.45 (m, 2H), 7.53 (d, J=9.3Hz, 1H), 4.36 (d, /=5.7Hz, 2H), 4.25 (s, 2H), 4.19 (d, /=5.7Hz, 2H), 3.58 (s, 3H), 1.23 (s, 3H); LCMS (m/z): 322.11 (MH+).
EXAMPLE 71:
1-{5-AMIN0-2-[(3-METHYL0XETAN-3-YL)METH0XY]- PHENYL}-4-METHYL- 1H-TETRAZOL- 5(4H)-ONE

[00492] 1 - { 2- [(3-Methyloxetan-3 -yl)methoxy] -5 -nitrophenyl } -4-methyl- lH-tetrazol- 5(4H)-one (466mg, 1.45mmol) was suspended in MeOH (8ml), Pd/C (50mg, 10% weight) was added to the solution. The mixture was degassed and stirred under 1 atmosphere of ¾ via a balloon at room temperature overnight. Pd/C was removed by using 2 layers of fluted filter paper and the mother liquor was concentrated under reduced pressure. To this residue, dichloromethane was added and the solution was filtered again through double layer of fluted filter paper. Mother liquor was concentrated under reduced pressure to give the title compound (300mg, 71 % yield) as light grey solid.
[00493] JH NMR (300 MHz, DMSO) δ 7.0 (d, /=9Hz, 1H), 6.72 (d, /=8.7Hz, 1H), 6.62 (d, /=2.7Hz, 1H), 5.05 (s, 2H), 4.30 (d, /=5.7Hz, 2H), 4.14 (d, /=5.4Hz, 2H), 3.87 (s, 2H), 3.55 (s, 3H), 1.16 (s, 3H); LCMS (m/z): 292.24 (MH+).
EXAMPLE 72:
5-FLUORO-N2-{4-(3-METHYLOXETAN-3-YL)METHOXY-3-[(4-METHYL)-1,2,3,4-TETRAZOL- 5-ONE-1-YL] }PHENYL-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE (1-

EXAMPLE 73:
L-(2-CHLORO-5-NITROPHENYL)-L -TETRAZOL-5(4H)-ONE

[00495] To 2-chloro-5-nitrobenzoisocyanate (2g, lO.lmmol), azidotrimethylsilane (3.3ml, 25.2mmol) was added and the mixture was heated at 100° C overnight. The mixture was cooled to room temperature, concentrated under reduced pressure. Residue was dissolved into ethyl acetate (100ml) and washed with saturated NaHCC>3 (100ml), the layers were separated. The aqueous layer was acidified with 2N HC1, and extracted with ethyl acetate (100ml). The organic layer was dried with Na2S04, the solid was filtered off and mother liquor was concentrated under reduced pressure to give the title compound (l.Og, 41% yield).
[00496] Ή NMR (300 MHz, DMSO) δ 8.67 (s, IH), 8.43 (d, /=9.0Hz, IH), 8.05 (d, /=8.7Hz, IH); LCMS (m/z): 241.99 (MH+).
EXAMPLE 74:
L-(2-CHLORO-5-NITROPHENYL)-4- -LH-TETRAZOL-5(4H)-ONE

[00497] l-(2-Chloro-5-nitrophenyl)-lH-tetrazol-5(4H)-one (lg, 4.1mmol) was dissolved into DMF (20ml), K2C03 (1.9g, 10.25mmol) was added to this solution and followed by iodomethane (0.32ml, 4.92mmol). The mixture was stirred at room temperature over 2 days. Ethyl acetate (20ml) was added to this mixture, after filtering off solid, the solution was concentrated under reduced pressure. Dichloromethane was added and the solution was washed with saturated NaHCC>3, brine (4x50ml) and dried with Na2S04. The solid was filtered off and mother liquor was concentrated under reduced pressure to give the title compound as a white solid (950mg, 89.6% yield).
[00498] Ή NMR (300 MHz, DMSO) δ 8.65 (s, IH), 8.44 (d, /=9Hz, IH), 8.07 (d, /=9Hz, IH), 3.63 (s, 3H); LCMS (m/z): 256.00 (MH+).
EXAMPLE 75:
l-(5-AMINO-2-CHLOROPHENYL)-4-METHYL-lH-TETRAZOL-5(4H)-ONE

[00499] l-(2-Chloro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (500mg, 1.96mmol) was added to EtOH (15ml), followed by SnCl2.2H20 (l.lg, 5.88mmol) and heated at 75° C for 4.5 hours. The solution was allowed to cool to room temperature and concentrated under reduced pressure. To this residue, ethyl acetate (50ml) was added and washed with aqueous K2C(¾ (50ml). The 2 layers were separated, the organic layer was washed with brine (50ml) and dried with Na2S04. The solid was filtered off and the solution was concentrated under reduced pressure to give the title compound (300mg, 68% yield).
[00500] JH NMR (300 MHz, DMSO) δ 7.27 (d, /=8.4Hz, 1H), 6.75-6.7 (m, 2H), 5.69 (s, 2H), 3.59 (s, 3H); LCMS (m/z): 226.08 (MH+).
EXAMPLE 76:
N2-{4-CHLORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1-YL]}PHENYL-5-FLUORO-N4- (2,2,6,6-TETRAMETHYLPI -4-YL)-2,4-PYRIMIDINEDIAMINE (1-30)

[00501] Ή NMR (300 MHz, DMSO) δ 9.53 (s, 1H), 8.05 (s, 1H), 7.85 (m, 2H), 7.49 (d, /=8.7Hz, 1H), 7.29 (d, /=7.2Hz, 1H), 4.33 (m, 1H), 3.61 (s, 3H), 1.65 (d, /=10.2Hz, 2H), 1.15 (m, 2H), 1.06 (s, 6H), 0.99 (s, 6H); LCMS (m/z): 476.32 (MH+).
EXAMPLE 77:
N2-{4-CHLORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1-YL]}PHENYL-5-FLUORO-N4- (1,2,2,6,6-PENTAMETHYL -4-YL)-2,4-PYRIMIDINEDIAMINE (1-29)

[00502] Ή NMR (300 MHz, DMSO) δ 9.53 (s, 1H), 8.03 (s, 1H), 7.88 (m, 2H), 7.49 (d, /=8.7Hz, 1H), 7.3 (d, /=8.1Hz, 1H), 4.24 (m, 1H), 3.62 (s, 3H), 2.15 (s, 3H), 1.65 (d,
7=11.1Hz, 2H), 1.43 (t, J=12.3Hz, 2H), 1.05 (s, 6H), 0.93 (s, 6H); LCMS (m/z): 490.40
(MH+).
EXAMPLE 78:
L-(3- -5-NITROPHENYL)-LH-TETRAZOL-5(4H)-ONE

[00503] 3-Methoxy-5-nitroaniline (2g, 11.9mmol) was added to 32ml of phosgene (20% in toluene), the mixture was heated at 100° C. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. To this residue, azidotrimethylsilane (6ml, 47.6mmol) was added and the solution was heated at 100° C overnight. After cooled to room temperature, TMSN3 was removed under reduced pressure. Ethyl Acetate (50ml) was added to this residue and treated with saturated NaHCC>3 (50ml). The aqueous layer was separated and acidified with 2N HC1, extracted with ethyl acetate (150ml). The organic layer was washed with brine, dried with Na2S04, filtered off solid, and concentrated under reduced pressure to give the title compound (2g, 71.2% yield).
[00504] JH NMR (300 MHz, DMSO) δ 8.35 (s, 1H), 7.87 (s, 1H), 7.78 (s, 1H), 3.96 (s, 3H); LCMS (m/z): 237.00 (MH+).
EXAMPLE 79:
L-(3-METHOXY-5-NITROPHENYL)-4-METHYL-LH-TETRAZOL-5(4H)-ONE

[00505] l-(3-Methoxy-5-nitrophenyl)-lH-tetrazol-5(4H)-one (lg, 4.2mmol) was dissolved into DMF (20ml), K2CO3 (1.9g, 10.25mmol) was added to this solution and followed by iodomethane (0.4ml, 6.3mmol). The mixture was stirred at room temperature overnight. Dichloromethane (100ml) and water (100ml) were added to the mixture, and the layers were separated. The organic layer was washed with brine (3x50ml) and dried with Na2S04. The solid was filtered off and mother liquor was concentrated under reduced pressure to give the title compound as a white solid (800mg, 74% yield).
[00506] Ή NMR (300 MHz, DMSO) δ 8.32 (s, 1H), 7.84 (s, 1H), 7.78 (s, 1H), 3.95 (s, 3H), 3.63 (s, 3H); LCMS (m/z): 256.00 (MH+).
EXAMPLE 80:
l-(5-AMINO-3-METHOXYPHENYL)- -METHYL-lH-TETRAZOL-5(4H)-ONE

[00507] l-(3-Methoxy-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (600mg,
2.3mmol) was suspended in MeOH (10ml), Pd/C (80mg, 10% weight) was added to the solution. The mixture was degassed and stirred under 1 atmosphere of ¾ via a balloon at room temperature overnight. Pd/C was removed by using 2 layers of fluted filter paper and the mother liquor was concentrated under reduced pressure. To this residue, dichloromethane was added and the solution was filtered again through double layers of fluted filter paper. Mother liquor was concentrated under reduced pressure to give the title compound (450mg, 86% yield) as light grey solid.
[00508] JH NMR (300 MHz, DMSO) δ 6.7 (s, 1H), 6.58 (s, 1H), 6.14 (s, 1H), 5.52 (s, 2H), 3.68 (s, 3H), 3.57 (s, 3H); LCMS (m/z): 222.19 (MH+).
EXAMPLE 81:
N2-{5-METHOXY-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1-YL] }PHENYL-5-FLUORO-N4- (2,2,6,6-TETRAMETHYLPI -4-YL)-2,4-PYRIMIDINEDIAMINE (1-32)

[00509] Ή NMR (300 MHz, DMSO) δ 9.23 (s, 1H), 7.87 (d, /=3.6Hz, 1H), 7.78 (s, 1H), 7.37 (s, 1H), 7.18 (d, /=7.2Hz, 1H), 6.89 (s, 1H), 4.38 (m, 1H), 3.72 (s, 3H), 3.57 (s, 3H), 1.66 (d, /=9.9Hz, 2H), 1.15 (m, 2H), 1.06 (s, 6H), 0.99 (s, 6H); LCMS (m/z): 472.45 (MH+). EXAMPLE 82:
N2-{5-METHOXY-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1-YL] }PHENYL-5-FLUORO-N4- (1,2,2,6,6-PENTAMETHYL -4-YL)-2,4-PYRIMIDINEDIAMINE (1-31)

EXAMPLE 83:
L-(2-CYCLOP

[00511] l-(2-Chloro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (500mg, 1.96mmol) was added to toluene (10ml), followed by cyclopropyl boronic acid (250mg, 2.9mmol), tricyclohexylphosphine (55mg, 0.2mmol), cesium carbonate (1.9g, 5.9mmol). The mixture was degassed by bubbling N2 into it for 15 minutes, and Pd(OAc)2 (22mg, O.lmmol) was added. The mixture was heated at 100° C for 4 hours, followed by thin layer chromatography (1:1 Hexanes : Ethyl Acetate). Additional cyclopropyl boronic acid (125mg, 1.45mmol), tricyclohexylphosphine (25mg, 0.09mmol), cesium carbonate (lg, 3.1mmol) and Pd(OAc)2 (lOmg, 0.045mmol) were added to the mixture and left heated at 100° C overnight. The solution was cooled to room temperature, ethyl acetate (50ml) and water (50ml) were added to the mixture. The 2 layers were separated and the organic layer was dried with Na2S04, filtered and concentrated under reduced pressure. The title compound (230mg, 45% yield) was obtained after purifying residue using flash column chromatography (2: 1 Hexanes : Ethyl Acetate).
[00512] JH NMR (300 MHz, DMSO) δ 8.37 (s, IH), 8.28 (d, /=9Hz, IH), 7.34 (d, /=8.7Hz, IH), 3.62 (s, 3H), 1.91 (m, IH), 1.05 (m, 2H), 0.87 (m, 2H); LCMS (m/z): 262.26 (MH+).
EXAMPLE 84:
L-(5-AMINO-2-C -4-METHYL-LH-TETRAZOL-5(4H)-ONE

[00514] JH NMR (300 MHz, DMSO) δ 6.84 (d, /=8.4Hz, 1H), 6.63 (d, /=8.7Hz, 1H), 6.5 (s, 1H), 5.32 (s, 2H), 3.59 (s, 3H), 1.56 (m, 1H), 0.64 (d, /=8.7Hz, 2H), 0.38 (d, /=3.9Hz, 2H); LCMS (m/z): 232.21 (MH+).
EXAMPLE 85:
N2-{4-CYCLOPROPYL-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1-YL] }PHENYL-5- FLUORO-N4-(2,2,6,6-TET -4-YL)-2,4-PYRIMIDINEDIAMINE (1-33)

[00515] Ή NMR (300 MHz, DMSO) δ 9.25 (s, 1H), 7.85 (d, /=3.9Hz, 1H), 7.78 (s, 1H), 7.72 (d, /=8.4Hz, 1H), 7.2 (d, /=8.4Hz, 1H), 6.98 (d, /=8.4Hz, 1H), 4.34 (m, 1H), 3.6 (s, 3H), 1.63 (m, 3H), 1.14 (t, /=12Hz, 2H), 1.05 (s, 6H), 0.99 (s, 6H), 0.72 (d, /=8.7Hz, 2H), 0.46 (d, /=4.8Hz, 2H); LCMS (m/z): 482.47 (MH+).
EXAMPLE 86:
N2-{4-CYCLOPROPYL-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1-YL] }PHENYL-5- FLUORO-N4-(1,2,2,6,6-PE -4-YL)-2,4-PYRIMIDINEDIAMINE (1-34)

[00516] Ή NMR (300 MHz, DMSO) δ 9.25 (s, 1H), 7.85 (d, /=3.9Hz, 1H), 7.75-7.71 (m, 2H), 7.2 (d, /=8.1Hz, 1H), 6.98 (d, /=8.4Hz, 1H), 4.26 (m, 1H), 3.61 (s, 3H), 2.15 (s, 3H), 1.62 (m, 3H), 1.42 (t, /=12Hz, 2H), 1.04 (s, 6H), 0.92 (s, 6H), 0.73 (d, J=7.2Hz, 2H), 0.46 (d, /=4.8Hz, 2H); LCMS (m/z): 496.35 (MH+).
EXAMPLE 87:
1-CHLORO-3,5-DINITROBENZENE

[00517] 3,5-Dinitroaniline (3g, 16.4mmol), was added to trifluoroacetic acid (60ml, excess), followed by NaNC>2 (2.26g, 32.8mmol), and the mixture was cooled to 0° C. Cu(I)Cl
(6g, 60mmol) in concentrated HCI (60ml) was added dropwise to the mixture and stirred at
0° C for 1 hour. It was poured onto ice/water and extracted with ethyl acetate, the 2 layers were separated and the organic layer was washed with saturated NaHCC>3, followed with brine and dried with Na2S04. The solid was filtered off and concentrated under reduced pressure, and purified by flash column chromatography (3: 1 Hexanes : Ethyl Acetate) to give the title compound (2.5g, 76% yield) as a light yellow solid.
[00518] JH NMR (300 MHz, DMSO) δ 8.75 (s, 3H), JH NMR (300 MHz, CDC13) δ 8.97 (s, 1H), 8.57 (s, 2H); LCMS (m/z): 201.90 (MH+).
EXAMPLE 88:
3-CHLORO-5-NITRO

[00519] l-Chloro-3,5-dinitrobenzene (512mg, 2.5mmol) was dissolved into EtOH (8ml) with the aid of sonication. (NH4)2S (3ml, 20% in ¾0) was added to the solution and heated at 80° C for 1 hour, followed by thin layer chromatography (3: 1 Hexanes : Ethyl Acetate). After the completion of reaction, the solution was allowed to cool to room temperature, ethyl acetate (75ml) was added and washed with brine (30ml). The 2 layers were separated, the organic layer was dried with Na2S04 and concentrated under reduced pressure after filtering off solid. The title compound (225mg, 53% yield) was obtained by purifying the residue with flash column chromatography (3: 1 Hexanes : Ethyl Acetate) as an orange solid.
[00520] JH NMR (300 MHz, DMSO) δ 7.32 (s, 1H), 7.24 (s, 1H), 6.94 (s, 1H), 6.14 (s, 2H); LCMS (m/z): 173.04 (MH+).
EXAMPLE 89:
l-(3-CHLORO-5-NITROPHENYL)-lH-TETRAZOL-5(4H)-ONE

[00521] To 3-chloro-5-nitroaniline (138 mg, 0.8mmol), was added to 6ml of phosgene (20% in toluene), the mixture was heated at 100° C. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. To this residue,
azidotrimethylsilane (5ml, excess) was added and the solution was heated at 100° C overnight. Cooled to room temperature, TMSN3 was removed under reduced pressure. Ethyl acetate (10ml) was added to this residue and treated with saturated NaHCC>3 (10ml). The aqueous layer was separated and acidified with 2N HCl, extracted with ethyl acetate (50ml). The organic layer was washed with brine, dried with Na2S04, filtered off solid, and concentrated under reduced pressure to give the title compound (118 mg, 61% yield).
[00522] JH NMR (300 MHz, DMSO) δ 8.66 (s, 1H), 8.36 (s, 1H), 8.32 (s, 1H), 3.95 (s, 3H), 3.63 (s, 3H); LCMS (m/z): 240.07 (MH-).
EXAMPLE 90:
L-(3-CHLORO-5-NITROPHENYL)-4-METHYL-LH-TETRAZOL-5(4H)-ONE

[00523] l-(3-Chloro-5-nitrophenyl)-lH-tetrazol-5(4H)-one (118mg, 0.49mmol) was dissolved into DMF (1.5ml), K2CO3 (224mg, 1.6mmol) was added to this solution and followed by iodomethane (0.05ml, 0.73mmol). The mixture was stirred at room temperature overnight. Dichloromethane (10ml) and water (10ml) were added to the mixture, and the layers were separated. The organic layer was washed with brine (3x5ml) and dried with Na2S04. The solid was filtered off and mother liquor was concentrated under reduced pressure to give the title compound (86mg, 69% yield).
[00524] Ή NMR (300 MHz, DMSO) δ 8.64 (s, 1H), 8.36 (s, 2H), 3.63 (s, 3H); LCMS (m/z): 255.13 (MH+).
EXAMPLE 91:
l-(5-AMINO-3-CHLOROPHENYL)-4-METHYL-lH-TETRAZOL-5(4H)-ONE

[00525] l-(3-Chloro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (86mg, 0.34mmol) was added to EtOH (5.1ml), followed by SnCl2.2H20 (228mg, lmmol), and heated at 70° C for 4.5 hours. The solution was allowed to cool to room temperature and concentrated under reduced pressure. To this residue, ethyl acetate (50ml) was added and washed with aqueous K2CO3 (50ml). The 2 layers were separated, the organic layer was washed with brine (50ml) and dried with Na2S04. The solid was filtered off and the solution was concentrated under reduced pressure to give the title compound (42mg, 72% in purity).
[00526] JH NMR (300 MHz, DMSO) δ 7.04 (s, IH), 7.00 (s, IH), 6.58 (s, IH), 5.87 (s, 2H), 3.58 (s, 3H); LCMS (m/z): 226.11 (MH+).
EXAMPLE 92:
N2-{5-CHLORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1-YL]}PHENYL-5-FLUORO-N4- (2,2,6,6-TETRAMETHYLPI -4-YL)-2,4-PYRIMIDINEDIAMINE (1-37)

[00527] Ή NMR (300 MHz, DMSO) δ 9.54 (s, IH), 8.05 (s, IH), 7.99 (s, IH), 7.93 (bs, IH), 7.65 (bs, IH), 7.42 (s, IH), 4.43 (m, IH), 3.59 (s, 3H), 1.93 (m, 2H), 1.56 (m, 2H), 1.36 (bs, 12H); LCMS (m/z): 476.30 (MH+).
EXAMPLE 93:
1-CYCLOPROPYL-3,5-DINITROBENZE

[00528] 3,5-Dinitrobromobenzene (600mg, 2.4mmol) was added to toluene / water (10:2ml), in a 60ml round bottom flask fitted with a water condenser. To this solution, tricyclohexylphosphine (200mg, 0.7mmol), cesium carbonate (4.74g, 14.5mmol), cyclopropylboronic acid MIDA ester (670mg, 3.4mmol) were added subsequently. This
mixture was degassed by bubbling N2 into the solution for 15 mintues, and finally palladium acetate (82mg, 0.36mmol) was added to the mixture and was heated at 100° C with stirring under N2 overnight. The solution was cooled to room temperature. Ethyl acetate and saturated K2CO3 was added to the mixture, and the two layers were separated. Organic layer was dried with Na2S04> after filtering off the solid, the mother liquor was concentrated under reduced pressure. The residue was purified with chromatography using ethyl acetate and hexanes (1:4) as an eluent to give the title compound as a yellow solid (350mg, 69% yield).
[00529] Ή NMR (300 MHz, DMSO) δ 8.57 (s, 1H), 8.31 (s, 2H), 2.33 (m, 1H), 1.14 (d, /=6Hz, 2H), 0.95 (m, 2H).
EXAMPLE 94:
3-CYCLOPROPYL-5-NITROANILINE

[00530] l-Chloro-3,5-dinitrobenzene (570mg, 2.7mmol) was dissolved into EtOH (14ml) with the aid of sonication. (NH4)2S (3.6ml, 20% in H20) was added to the solution and heated at 80° C for 1 hour. Upon completion of reaction, the solution was cooled to room temperature and concentrated under reduced pressure. Hexanes was added to the residue, sonicated and filtered to give ~600mg of dark red solid. The title compound (270mg, 55% yield) was obtained by purifying the filtrate with flash column chromatography (4:1 Hexanes : Ethyl Acetate) as an orange solid.
[00531] JH NMR (300 MHz, DMSO) δ 7.13 (s, 1H), 7.01 (s, 1H), 6.63 (s, 1H), 5.68 (s, 2H), 1.89 (m, 1H), 0.94 (d, /=6.6Hz, 2H), 0.64 (d, /=4.8Hz, 2H).
EXAMPLE 95:
L-(3-CYCLOPROPYL-5-NITROPHEN -LH-TETRAZOL-5(4H)-ONE

[00532] To 3-cyclopropyl-5-nitroaniline (270 mg, 2.25mmol), was added 6ml of phosgene (20% in toluene), the mixture was heated at 100° C for 2 hours. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. To this residue, azidotrimethylsilane (11ml, excess) was added and the solution was heated at 100° C
overnight. Cooled to room temperature, TMSN3 was removed under reduced pressure. Ethyl Acetate (20ml) was added to this residue and treated with saturated NaHCC>3 (20ml). The aqueous layer was separated and acidified with 2N HCl, extracted with ethyl acetate (50ml). The organic layer was washed with brine, dried with Na2S04, filtered off solid, and concentrated under reduced pressure to give the title compound (200mg, 54% yield).
[00533] JH NMR (300 MHz, DMSO) δ 8.47 (s, 1H), 7.97 (s, 1H), 7.93 (s, 1H), 2.24 (m, 1H), 1.1 (d, /=8.4Hz, 2H), 0.84 (d, /=4.5Hz, 2H); LCMS (m/z): 246.12 (MH-).
EXAMPLE 96:
L-(3-CYCLOPROPYL-5-NITROPHEN -4-METHYL-LH-TETRAZOL-5(4H)-ONE

[00534] l-(3-Cyclopropyl-5-nitrophenyl)-lH-tetrazol-5(4H)-one (200 mg, 0.8mmol) was dissolved into DMF (3ml), K2CO3 (370mg, 2.64mmol) was added to this solution and followed by iodomethane (0.78ml, 1.2mmol). The mixture was stirred at room temperature overnight. Dichloromethane (50ml) and water (2x25ml) were added to the mixture, and the layers were separated. The organic layer was washed with brine (2x50ml) and dried with Na2S04. The solid was filtered off and mother liquor was concentrated under reduced pressure to give the title compound as a white solid (150mg, 71% yield).
[00535] JH NMR (300 MHz, DMSO) δ 8.46 (s, 1H), 7.97 (s, 1H), 7.95 (s, 1H), 3.62 (s, 3H), 2.45 (m, 1H), 1.11 (d, /=8.4Hz, 2H), 0.85 (m, 2H); LCMS (m/z): 262.21 (MH-).
EXAMPLE 97:
L-(5-AMINO-3-CYCLOPROPYLPHEN -4-METHYL-LH-TETRAZOL-5(4H)-ONE

[00536] l-(3-Cyclopropyl-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (150mg, 0.57mmol) was added to EtOH (16ml), followed by SnCl2.2H20 (436mg, 2.3mmol), cone. HCl (0.6ml) and heated at 80° C for 1 hours. The solution was allowed to cool to room temperature and concentrated under reduced pressure. To this residue, ethyl acetate (20ml) was added and washed with aqueous K2CO3 (20ml). The 2 layers were separated, the
organic layer was washed with brine (20ml) and dried with Na2S04. The solid was filtered off and the solution was concentrated under reduced pressure to give the title compound (140mg, 100% yield).
[00537] JH NMR (300 MHz, DMSO) δ 6.83 (s, IH), 6.68 (s, IH), 6.28 (s, IH), 5.38 (s, 2H), 3.57 (s, 3H), 1.8 (m, IH), 0.89 (d, /=7.8Hz, 2H), 0.57 (d, /=6.3Hz, 2H); LCMS (m/z): 232.13 (MH+).
EXAMPLE 98:
N2-{5-CYCLOPROPYL-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1-YL]}PHENYL-5- FLUORO-N4-(2,2,6,6-TET -4-YL)-2,4-PYRIMIDINEDIAMINE (1-35)

[00538] Ή NMR (300 MHz, DMSO) δ 9.11 (s, IH), 8.09 (s, IH), 7.86 (d, J=3.6Hz, IH), 7.22 (s, IH), 7.14 (d, /=8.1Hz, IH), 6.94 (s, IH), 4.3 (m, IH), 3.56 (s, 3H), 1.9 (m, IH), 1.65 (d, /=9.9Hz, 2H), 1.1 (t, /=12Hz, 2H), 0.97 (m, 14H), 0.64 (d, J=6.3Hz, 2H); LCMS (m/z): 482.43 (MH+).
EXAMPLE 99:
N2-{5-CYCLOPROPYL-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1-YL]}PHENYL-5- FLUORO-N4-(1,2,2,6,6-PE -4-YL)-2,4-PYRIMIDINEDIAMINE (1-36)

[00539] Ή NMR (300 MHz, DMSO) δ 9.11 (s, IH), 8.08 (s, IH), 7.86 (d, J=3.9Hz, IH), 7.24 (s, IH), 7.15 (d, /=8.4Hz, IH), 6.95 (s, IH), 4.23 (m, IH), 3.57 (s, 3H), 2.11 (s, 3H), 1.9 (m, IH), 1.65 (d, 7=11. lHz, 2H), 1.38 (t, /=12Hz, 2H), 1.01 (s, 6H), 0.95 (d, /=6Hz, 2H), 0.86 (s, 6H), 0.64 (d, J=6.3Hz, 2H); LCMS (m/z): 496.37 (MH+).
EXAMPLE 100:
L-(2-CHLORO-5-NITRO-3-TRIFLUO -LH-TETRAZOL-5(4H)-ONE

[00541] Ή NMR (300 MHz, DMSO) δ 9.01 (d, /=2.7Hz, 1H), 8.7 (d, /=2.7Hz, 1H); LCMS (m/z): 308.07 (MH-).
EXAMPLE 101:
L-(2-CHLORO-5-NITRO-3-TRIFLUOROMETHYLPHENYL)-4-METHYL-LH-TETRAZOL-5(4H)-
ONE

[00542] l-(2-Chloro-5-nitro-3-trifluoromethyl-phenyl)-lH-tetrazol-5(4H)-one (42 mg, 0.13mmol) was dissolved into DMF (1ml), K2CO3 (62mg, 0.43mmol) was added to this solution and followed by iodomethane (0.013ml, 0.195mmol). The mixture was stirred at room temperature overnight. Ethyl acetate (10ml), was added to the mixture and was washed water (2x25ml), with brine (2x50ml) and dried with Na2S04. The solid was filtered off and mother liquor was concentrated under reduced pressure to give the title compound as a white solid (33mg, 75% yield).
[00543] JH NMR (300 MHz, DMSO) δ 8.97 (d, /=2.7Hz, 1H), 8.71 (d, /=2.4Hz, 1H), 3.65 (s, 3H).
EXAMPLE 102:
L-(5-AMINO-2-CHLORO-3-TRIFLUOROMETHYLPHENYL)-4-METHYL-LH-TETRAZOL-5(4H)-
ONE

[00544] l-(2-Chloro-3-trifluoro-5-nitrophenyl)-4-methyl- lH-tetrazol-5(4H)-one (33mg, O.lmmol) was added to MeOH (1ml), followed by Fe (33mg, 0.6mmol), acetic acid (2 drops) and stirred at room temperature overnight. Fe was filtered off, and ethyl acetate (5ml) was added to the mother liquor. The solution was concentrated under reduced pressure to give the title compound (26.9mg, 75% in purity).
[00545] JH NMR (300 MHz, DMSO) δ 7.29 (s, 1H), 7.20 (s, 1H), 6.22 (s, 2H), 3.62 (s, 3H); LCMS (m/z): 293.13 (MH+).
EXAMPLE 103:
N2-{4-CHLORO-5-TRIFLUOROMETHYL-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE (1-3

[00546] Ή NMR (300 MHz, DMSO) δ 9.77 (s, 1H), 8.47 (s, 1H), 8.26 (s, 1H), 7.94 (d, /=3.6Hz, 1H), 7.4 (bs, 1H), 4.34 (m, 1H), 3.63 (s, 3H), 1.66 (d, /=12.6Hz, 1H), 1.21 (m, 2H), 1.06 (s, 6H), 1.02 (s, 6H); LCMS (m/z): 544.37 (MH+).
EXAMPLE 104:
2-CYCLOPROPYL-1-TRIFLUOROMET -3,5-DINITROBENZENE

[00547] 2-Chloro-l-(trifluoromethyl)-3,5-dinitrobenzene (5g, 18.5mmol) was added to toluene (91.6ml) and water (18ml), followed by cyclopropylboronic acid MID A ester (5.1g, 25.9mmol), tricyclohexylphosphine (1.55g, 5.5mmol), cesium carbonate (36g, O.l lmol). The mixture was degassed by bubbling N2 into it for 30 minutes, and Pd(OAc)2 (620mg,
2.76mmol) was added. The mixture was heated at 100° C overnight, followed by thin layer chromatography (9.75 :0.25 Hexanes : Ethyl Acetate). The solution was cooled to room temperature, ethyl acetate (250ml) and saturated NaHCC>3 (250ml) were added to the mixture. The 2 layers were separated, the organic layer washed with brine (250ml) and dried with Na2S04, filtered and concentrated under reduced pressure. The title compound (3g, 59% yield) was obtained after purifying residue using flash column chromatography (9.75 :0.25 Hexanes : Ethyl Acetate).
[00548] JH NMR (300 MHz, DMSO) δ 8.94 (d, /=2.4Hz, 1H), 8.62 (d, /=2.1Hz, 1H), 2.25 (m, 1H), 1.05 (m, 2H), 0.61 (m, 2H); LCMS (m/z): 277.14 (MH+).
EXAMPLE 105:
4-CYCLOPROPYL- -TRIFLUOROMETHYL-5-NITRO ANILINE

[00549] Na2S.9H20 (2.5g, 10.4mmol) and NaHC03 (0.88g, 10.4mmol) was mixed together in MeOH (30ml) and water (20ml). This mixture, was added to 2-cyclopropyl- l- trifluoromethyl-3,5-dinitrobenzene (1.44g, 5.2mmol) in MeOH (20ml) and it was heated at 70° C for 0.5 hour. The reaction was followed by thin layer chromatography (4: 1 Hexanes : Ethyl Acetate), upon completion, it was cooled to room temperature. The solution was concentrated under reduced pressure. Dichloromethane (125ml) was added to the residue and washed twice with water (150ml). The 2 layers were separated, organic layer was dried with Na2S04, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography (4: 1 Hexanes : Ethyl Acetate) to give the title compound (640mg, 50% yield).
[00550] JH NMR (300 MHz, DMSO) δ 7.1 (s, 1H), 7.01 (s, 1H), 6.08 (s, 2H), 1.87 (m, 1H), 0.815 (m, 2H), 0.34 (d, J=5.1Hz, 2H); LCMS (m/z): 247.15 (MH+).
EXAMPLE 106:
2-(4-CYCLOPROPYL-3-TRIFLUOROMETHYL-5-NITROPHENYL)ISOINDOLE-1,3-DIONE

[00552] JH NMR (300 MHz, DMSO) δ 8.23 (s, 1H), 8.2 (s, 1H), 8.0-7.92 (m, 4H), 2.2 (m, 1H), 1.0 (m, 2H), 0.61 (d, /=5.4Hz, 2H); LCMS (m/z): 377.11 (MH+).
EXAMPLE 107:
2-(3-AMINO- -CYCLOPROPYL-5-TRIFLUOROMETHYLPHENYL)ISOINDOLE-1,3-DIONE

[00553] 2-(4-Cyclopropyl-3-trifluoromethyl-5-nitrophenyl)isoindole- l,3-dione (1 lOmg, 0.3mmol) was added to EtOH (15ml), followed by SnCl2.2H20 (264mg, 1.2mmol), cone. HCl (0.5ml) and heated at 80° C for 1 hour. The solution was allowed to cool to room temperature and concentrated under reduced pressure. To this residue, ethyl acetate (10ml) was added and washed with aqueous K2CO3 (10ml). The 2 layers were separated, the organic layer was washed with brine (20ml) and dried with Na2S04. The solid was filtered off and the solution was concentrated under reduced pressure. The residue was purified by flash column chromatography (4: 1 Hexanes : Ethyl Acetate) to give the title compound (40mg, 40% yield).
[00554] JH NMR (300 MHz, DMSO) δ 7.95-7.86 (m, 4H), 6.92 (s, 2H), 5.63 (s, 2H), 1.59 (m, 1H), 1.06 (m, 2H), 0.57 (d, /=5.1Hz, 2H); LCMS (m/z): 347.14 (MH+).
EXAMPLE 108:
2-[4-CYCLOPROPYL-3-TRIFLUOROMETHYL-5-(4,5-DIHYDRO-4-METHYL-5-OXOTETRAZOL- 1-YL)PHENYL]ISOINDOLE-1,3-DI

[00555] 2-(3-Amino-4-cyclopropyl-5-trifluoromethylphenyl)isoindole-l,3-dione (1.06g, 3.1mmol), was added to 22.8ml of phosgene (20% in toluene), the mixture was heated at 100° C for 1.5 hours. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. To this residue, azidotrimethylsilane (42ml, excess) was added and the solution was heated at 100° C overnight. Cooled to room temperature, TMSN3 was removed under reduced pressure. Ethyl acetate (10ml) was added to this residue and concentrated under reduced pressure to give 2-[4-cyclopropyl-3-trifluoromethyl-5-(4,5- dihydro-4-H-5-oxotetrazol-l-yl)phenyl]isoindole-l,3-dione (1.28g, 50% in purity) as a white solid. This solid was added to DMF (18ml), followed by K2CO3 (1.24g), and iodomethane (0.1ml). The mixture was stirred under N2, at room temperature overnight. Ethyl acetate (100ml) and water (100ml) was added to the mixture, the 2 layers were separated, the organic layer was washed with brine, dried with Na2S04. The solid was filtered off, and the mother liquor was concentrated under reduced pressure. The title compound (500mg, 38%) was obtained after purified through flash column chromatography (2: 1 Hexanes : Ethyl Acetate).
[00556] JH NMR (300 MHz, DMSO) δ 8.12 (s, 2H), 8.0-7.9 (m, 4H), 3.65 (s, 3H), 2.01 (m, 1H), 0.86 (m, 2H), 0.35 (d, /=5.4Hz, 2H); LCMS (m/z): 430.22 (MH+).
EXAMPLE 109:
1-(5-AMINO-2-CYCLOPROPYL-3-TRIFLUOROMETHYLPHENYL)-4-METHYL-1H-TETRAZOL- 5(4H)-ONE

[00557] 2-[4-Cyclopropyl-3-trifluoromethyl-5-(4,5-dihydro-4-methyl-5-oxotetrazol-l- yl)phenyl]isoindole-l,3-dione (500mg, 1.16mmol) was suspended in EtOH (10ml),
N2H4.2H20 was added and the mixture was heated at 80° C. Solution turned from turbid to
clear, then white solid crashed out, additional EtOH (1ml) was added, and the solution was heated at 80° C for 15 minutes. The solution was concentrated under reduced pressure and was sonicated with ethyl acetate (10ml). The title compound (265mg, 76% yield) as a white solid was obtained by filtering solution through a Buchner funnel fitted with a filter paper.
[00558] JH NMR (300 MHz, DMSO) δ 7.05 (s, 1H), 7.78 (s, 1H), 5.89 (s, 2H), 3.61 (s, 3H), 1.7 (m, 1H), 0.65 (m, 2H), 0.12 (d, /=5.1Hz, 2H); LCMS (m/z): 300.23 (MH+).
EXAMPLE 110:
N2-{4-CYCLOPROPYL-5-TRIFLUOROMETHYL-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL] }PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE (1-3

[00559] Ή NMR (300 MHz, DMSO) δ 9.54 (s, 1H), 8.26 (s, 1H), 8.04 (s, 1H), 7.91 (d, /=3.6Hz, 1H), 7.29 (d, /=9.0Hz, 1H), 4.25 (m, 1H), 3.63 (s, 3H), 1.81 (m, 1H), 1.64 (d, /=9.6Hz, 2H), 1.24 (m, 2H), 1.02 (s, 6H), 0.99 (s, 6H), 0.72 (d, /=8.1Hz, 2H), 0.16 (d, /=4.8Hz, 2H); LCMS (m/z): 550.45 (MH+).
EXAMPLE 111:
N2-{4-CYCLOPROPYL-5-TRIFLUOROMETHYL-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL] }PHENYL-5-FLUORO-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE (1-4

[00560] Ή NMR (300 MHz, DMSO) δ 9.54 (s, 1H), 8.25 (s, 1H), 8.05 (s, 1H), 7.91 (d, /=3.6Hz, 1H), 7.29 (d, /=8.1Hz, 1H), 4.21 (m, 1H), 3.64 (s, 3H), 2.13 (s, 3H), 1.8 (m, 1H), 1.64 (d, /=8.7Hz, 2H), 1.4 (t, /=12.3Hz, 2H), 1.03 (s, 6H), 0.88 (s, 6H), 0.72 (d, /=7.8Hz, 2H), 0.17 (d, /=5.4Hz, 2H); LCMS (m/z): 564.59 (MH+).
EXAMPLE 112:
5-CYANO-N2-{4-CYCLOPROPYL-5-TRIFLOUROMETHYL-[3-(4-METHYL)-1,2,3,4-TETRAZOL- 5-ONE-1-YL] }PHENYL-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE (1-4

[00561] Ή NMR (300 MHz, DMSO) δ 8.57 (bs, IH), 8.44 (s, IH), 8.1 (bs, 2H), 7.85- 7.75 (m, 2H), 4.42 (m, IH), 3.64 (s, 3H), 1.87 (d, /=10.8Hz, 2H), 1.6 (m, 2H), 1.32 (m, 12H), 0.77 (d, /=8.4Hz, 2H), 0.18 (s, 2H); LCMS (m/z): 557.48 (MH+).
EXAMPLE 113:
5-CYANO-N2-{4-CYCLOPROPYL-5-TRIFLOUROMETHYL-[3-(4-METHYL)-1,2,3,4-TETRAZOL- 5-ONE-1-YL] }PHENYL-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE (1-4

[00562] Ή NMR (300 MHz, DMSO) δ 8.54 (bs, IH), 8.44 (s, IH), 8.07 (bs, 2H), 7.87 (d, /=7.8Hz, IH), 4.42 (m, IH), 3.65 (s, 3H), 2.69 (s, 3H), 2.05 (m, IH), 1.98 (bs, 2H), 1.82 (m, 2H), 1.36 (m, 12H), 0.77 (d, /=8.4Hz, 2H), 0.19 (s, 2H); LCMS (m/z): 571.47 (MH+).
EXAMPLE 114:
1-(3-BROMO-2-FLUORO-5-NITROPHENYL)-1H-TETRAZOL-5(4H)-ONE

[00563] To 3-bromo-2-fluoro-5-nitroaniline (400mg, 1.7mmol), was added to 4.23ml of phosgene (20% in toluene), the mixture was heated at 100° C for 1 hour. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. To this residue, azidotrimethylsilane (16ml, excess) was added and the solution was heated at 100° C overnight. Cooled to room temperature, TMSN3 was removed under reduced pressure. Ethyl Acetate (50ml) was added to this residue and treated with saturated NaHCC>3 (50ml). The aqueous layer was separated and acidified with 2N HC1, extracted with ethyl acetate (70ml).
The organic layer was washed with brine, dried with Na2S04, filtered off solid, and concentrated under reduced pressure to give the title compound (210mg, 42% yield).
[00564] JH NMR (300 MHz, DMSO) δ 8.77-8.75 (m, 1H), 8.67-8.64 (m, 1H); LCMS (m/z): 303.88 (MH-).
EXAMPLE 115:
1-(3-BROMO-2-FLUORO-5-NITROPHENYL)-4-METHYL-1H-TETRAZOL-5(4H)-ONE

[00565] l-(3-Bromo-2-fluoro-5-nitrophenyl)-lH-tetrazol-5(4H)-one (210mg, 0.7mmol) was dissolved into DMF (3.5ml), K2CO3 (286mg, 2.1mmol) was added to this solution and followed by iodomethane (0.064ml, 1.03mmol). The mixture was stirred at room
temperature overnight. Ethyl acetate (10ml) and brine (10ml) were added to the mixture, and the layers were separated. The aqueous layer was extracted with ethyl acetate (2x10ml), the organic layers were combined and dried with Na2S04. The solid was filtered off and mother liquor was concentrated under reduced pressure. This residue was purified by flash column chromatography (3 : 1 Hexanes : Ethyl Acetate) to give the title compound as a white solid (120mg, 55% yield).
[00566] JH NMR (300 MHz, DMSO) δ 8.8-8.77 (m, 1H), 8.64-8.61 (m, 1H), 3.65 (s, 3H); LCMS (m/z): 315.90 (MH-).
EXAMPLE 116:
1-(3-CYCLOPROPYL-2-FLUORO-5- -4-METHYL-1H-TETRAZOL-5(4H)-ONE

[00567] l-(3-Bromo-2-fluoro-5-nitrophenyl)-4-methyl- lH-tetrazol-5(4H)-one (120mg, 0.38mmol) was added to toluene / water (1.5ml:0.3ml), in a carousel reaction tube. To this solution, tricyclohexylphosphine (35mg, O. l lmmol), cesium carbonate (750mg, 2.3mmol), cyclopropylboronic acid MIDA ester (104mg, 0.53mmol) were added subsequently. This mixture was degassed by bubbling N2 into the solution for 15 minutes, and finally palladium acetate (13mg, 0.057mmol) was added to the mixture and was heated at 100° C with stirring under N2 overnight. The solution was cooled to room temperature. Ethyl acetate and
saturated K2CO3 was added to the mixture, and the two layers were separated. Organic layer was dried with Na2S04> after filtering off the solid, the mother liquor was concentrated under reduced pressure. The residue was purified with chromatography using ethyl acetate and hexanes (1 :4) as an eluent to give the title compound (80mg, 76% yield).
[00568] JH NMR (300 MHz, DMSO) δ 8.4-8.38 (m, 1H), 7.99-7.96 (m, 1H), 3.62 (s, 3H),
2.21 (m, 1H), 1.12 (m, 2H), 0.96 (m, 2H).
EXAMPLE 117:
1-(5-AMINO-3-CYCLOPROPYL-2-FL -4-METHYL-1H-TETRAZOL-5(4H)-ONE

[00569] l-(3-Cyclopropyl-2-fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (80mg, 0.29mmol) was added to EtOH (2ml), followed by SnCl2.2H20 (217mg, 1.16mmol), concentrated HCl (0.6ml) and heated at 80° C for 0.5 hour. The solution was allowed to cool to room temperature and concentrated under reduced pressure. To this residue, ethyl acetate (5ml) was added and washed with aqueous K2CO3 (5ml). The 2 layers were separated and the aqueous layer was extracted with ethyl acetate (5ml). The combined organic layers were dried with Na2S04. The solid was filtered off and the solution was concentrated under reduced pressure to give the title compound (60mg, 84% yield).
[00570] JH NMR (300 MHz, DMSO) δ 6.48-6.45 (m, 1H), 6.29-6.26 (m, 1H), 5.23 (s, 2H), 3.59 (s, 3H), 1.97 (m, 1H), 0.96 (m, 2H), 0.64 (m, 2H); LCMS (m/z): 250.18 (MH+). EXAMPLE 118:
N2-{5-CYCLOPROPYL-4-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE (1-4

[00571] Ή NMR (300 MHz, DMSO) δ 9.1 (s, 1H), 8.07 (d, /=6.0Hz, 1H), 7.85 (d, /=3.9Hz, 1H), 7.2 (d, /=8.7Hz, 1H), 7.11 (d, /=3.6Hz, 1H), 4.28 (m, 1H), 3.6 (s, 3H), 2.45
(m, 1H), 1.62 (d, /=9.9Hz, 2H), 1.12 (m, 4H), 0.98 (m, 12H), 0.7 (m, 2H); LCMS (m/z): 500.47 (MH+).
EXAMPLE 119:
5-CYANO-N2-{5-CYCLOPROPYL-4-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-N4-(2,2,6,6- -4-YL)-2,4-PYRIMIDINEDIAMINE (1-44)

[00572] Ή NMR (300 MHz, DMSO) δ 9.84 (bs, 1H), 8.33 (s, 1H), 7.82 (bs, 1H), 7.45 (bs, 1H), 7.14 (bs, 1H), 4.26 (m, 1H), 3.59 (s, 3H), 2.05 (m, 1H), 1.58 (bs, 2H), 1.26 (m, 4H), 1.01 (m, 12H), 0.73 (m, 2H); LCMS (m/z): 507.41 (MH+).
EXAMPLE 120:
2,3-DLMETH -5-NITROPHENYLISOCYANATE

[00573] A solution of 2,3-dimethyl-5-nitroaniline (1.0 g, 6.02 mmol, 1 equiv) in 20% of phosgene in toluene (9.5 mL, 18.06 mmol, 3 equiv) was refluxed for 5 hours. The reaction mixture was then concentrated and the resulting residue was directly used in the next step.
[00574] JH NMR (CDC13, 300 MHz): δ 7.88 (s, 1H), 7.83 (s, 1H), 2.39 (s, 3H), 2.33 (s, 3H) .
EXAMPLE 121:
1-(2,3-DIM -5-NITROPHENYL)-4,5-DIHYDRO-5H-TETRAZOL-5-ONE

[00575] To the above residue of Example 120, 3.2 mL of TMSN3 (24.08 mmol, 4 equiv) was added and then the resulting mixture was heated at 100° C overnight. After being cooled to room temperature, the reaction mixture was concentrated. The residue was diluted with EtOAc (50 mL) and saturated aqueous NaHCC>3 (50 mL). The aqueous layer was separated, neutralized with 2 N HC1 to pH 4-5 and extracted with EtOAc (2x50 mL). The organic
layers were combined, dried over MgS04 and concentrated to give a beige solid (1.0 g, 71% two steps) which was directly used in the next step.
[00576] JH NMR (DMSO-d6, 300 MHz): δ 8.26 (s, 1H), 8.22 (s, 1H), 2.45 (s, 3H), 2.17 (s, 3H); m/z = 236 (M)+ .
EXAMPLE 122:
1-(2,3-DIM -5-NITROPHENYL)-4-METHYL-5H-TETRA -5-ONE
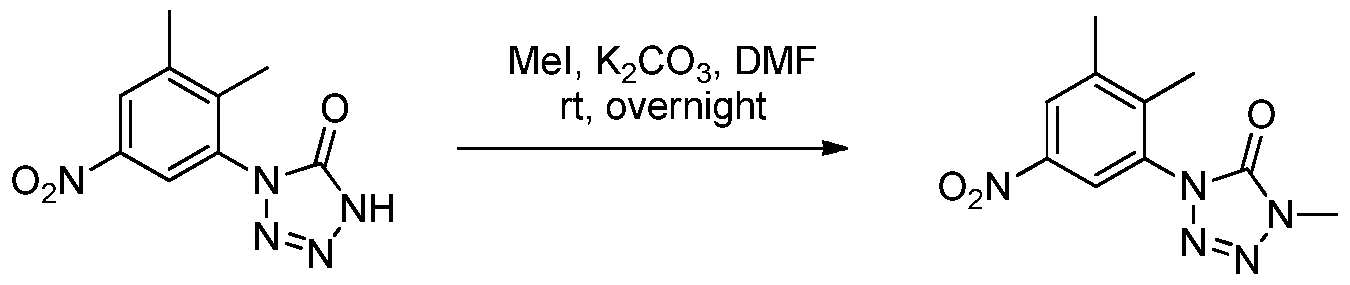
[00577] To a mixture of l-(2,3-dimethyl-5-nitrophenyl)-4,5-dihydr-5H-tetrazol-5-one g, 4.26 mmol, 1 equiv) and K2CO3 (1.76 g, 12.76 mmol, 3 equiv) in 8 mL of DMF was added Mel (0.8 mL, 12.76 mmol, 3 equiv) and the resulting mixture was stirred at room temperature overnight. A pale yellow solid crashed out, filtered, washed with water and dried to give 1.0 g of product (94%). m/z = 250 (M)+ .
EXAMPLE 123:
l-(5-Amin -2,3-dimethylphenyl)-4-methyl-5H-tetrazol-5-one

[00578] A solution of l-(2,3-dimethyl-5-nitrophenyl)-4-methyl-5H-tetrazol-5-one (1.0 g, 4.01 mmol, 1 equiv) in 20 mL of MeOH in the presence of 50 mg of 10% Pd/C was hydrogenated at room temperature for 3 hours. The reaction mixture was then filtered over a pad of Celite and washed with MeOH. The solvent was concentrated to give the product in quantitative yield.
[00579] JH NMR (CDC13, 300 MHz): δ 6.91 (s, 1H), 6.85 (s, 1H), 6.75 (br. s, 1H), 5.22 (br. s, 1H), 2.31 (s, 3H), 2.05 (s, 3H); m/z = 220 (M+H)+ .
EXAMPLE 124:
N2-(4,5-DIMETHYL-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)PHENYL)-5- FLUORO-N4-(1,2,2,6,6-PE E-2,4-DIAMINE (1-45)

EXAMPLE 125:
N2-(4,5-DIMETHYL-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)PHENYL)-5- FLUORO-N4-(2,2,6,6-TET -4-YL)PYRIMIDINE-2,4-DIAMINE (1-46)

[00581] Ή NMR (DMSO-d6, 300 MHz): 59.11 (s, 1H), 7.85 (d, / = 3.9 Hz, 1H), 7.78 (s, 1H), 7.44 (s, 1H), 7.14 (d, / = 8.7 Hz, 1H), 4.35-4.25 (m, 1H), 3.59 (s, 3H), 2.24 (s, 3H), 1.88 (s, 3H), 1.62 (dm, / = 10.8 Hz, 2H), 1.14-1.08 (m, 2H), 0.99 (s, 6H), 0.98 (s, 6H); m/z = 470 (M+H)+.
EXAMPLE 126:
5-NITRO-3-T

[00582] A solution of 3-amino-5-nitrobenzotrifluoride (3.3 g, 16.01 mmol, 1 equiv) in 20% of phosgene in toluene (16.85 mL, 32.02 mmol, 2 equiv) was refluxed for 5 hours. The reaction mixture was then concentrated and the resulting residue was directly used in the next step.
EXAMPLE 127:
1-(5-NITR -3-TRIFLUOROMETHYLPHENYL)-4,5-DIHYDRO-5H-TETRAZOL-5-ONE

[00583] To the above residue 4.3 mL of TMSN3 (32.02 mmol, 2 equiv) was added and then the resulting mixture was heated at 100° C overnight. After being cooled to room temperature, the reaction mixture was concentrated. The residue was diluted with EtOAc (100 mL) and saturated aqueous NaHCC>3 (50 mL). The organic layer was separated and
concentrated. The residue was purified by column chromatography on silica gel using EtOAc / hexanes / HO Ac (6:4:0.05) as eluent to give 1.42 g of product (32% two steps). m/z = 276 (M+H)+ .
EXAMPLE 128:
1-(5-NITR -3-TRIFLUOROMETHYLPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE
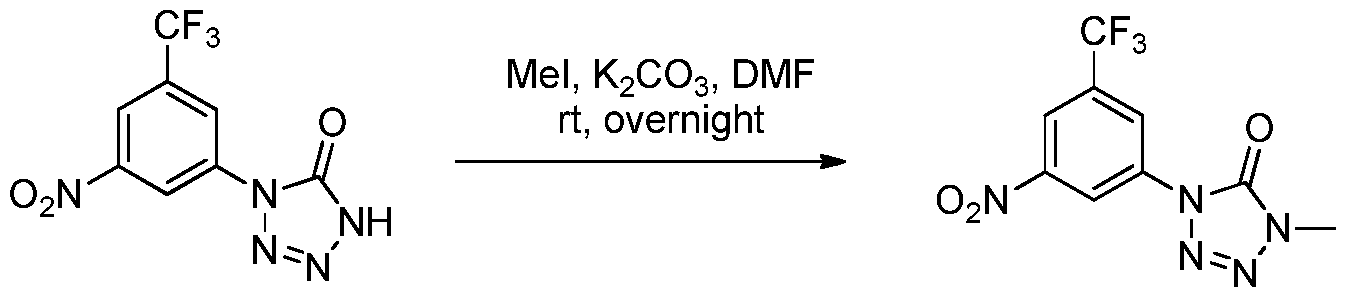
[00584] To a mixture of l-(5-nitro-3-trifluoromethylphenyl)-4,5-dihydro-5H-tetrazol-5- one (1.42 g, 5.16 mmol, 1 equiv) and K2CO3 (2.14 g, 15.49 mmol, 3 equiv) in 25 mL of DMF was added Mel (1.0 mL, 15.49 mmol, 3 equiv) and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was separated and concentrated. The residue was triturated in water to give 1.41 g of product (95%) as a yellow solid, m/z = 289 (M)+ .
EXAMPLE 129:
1-(5-AMIN -3-TRIFLUOROMETHYLPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE
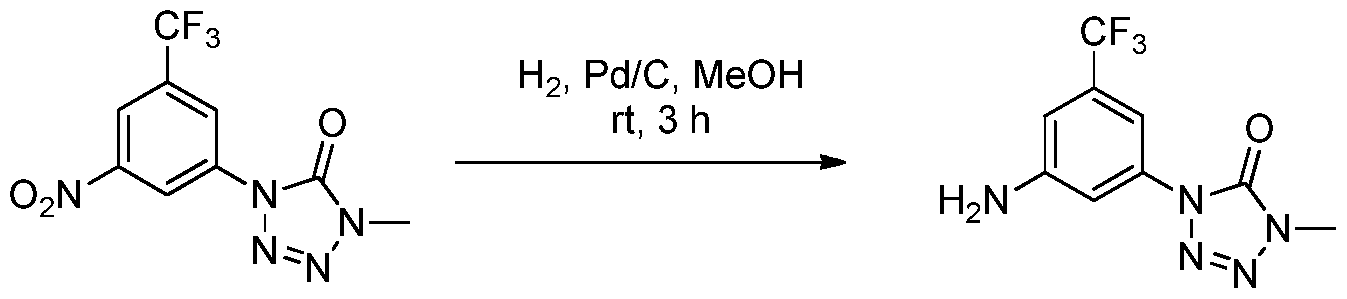
[00585] A solution of l-(5-nitro-3-trifluoromethylphenyl)-4-methyl-5H-tetrazol-5-one (0.7 g, 2.42 mmol, 1 equiv) in 10 mL of MeOH in the presence of 50 mg of 10% Pd/C was hydrogenated at room temperature for 3 hours. The reaction mixture was then filtered over a pad of Celite and washed with MeOH. The solvent was concentrated to give the product in quantitative yield, m/z = 259 (M)+ .
EXAMPLE 130:
N2-(3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)-5- TRIFLUOROMETHYLPHENYL)-5-FLUORO-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4- YL)PYRIMIDINE-2,4-DIAM

EXAMPLE 131:
N2-(3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)-5- TRIFLUOROMETHYLPHENYL)-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4- YL)PYRIMIDINE-2,4-DIAM -48)

[00587] Ή NMR (DMSO-d6, 300 MHz): δ 9.58 (s, 1H), 8.56 (s, 1H), 8.03 (s, 1H), 7.94
(d, / = 3.9 Hz, 1H), 7.64 (s, 1H), 7.45 (s, 1H), 7.30 (d, / = 7.2 Hz, 1H), 4.45-4.35 (m, 1H),
3.59 (s, 3H), 1.69 (dm, / = 10.8 Hz, 2H), 1.15-1.10(m, 2H), 1.08 (s, 6H), 1.02 (s, 6H); m/z =
510 (M+H)+.
EXAMPLE 132:
3-BROMO-5-NITROANILINE

[00588] To a solution of l-bromo-3,5-dinitrobenzene (3.0 g, 12 mmol, 1 equiv) in EtOH (15 mL) at room temperature was added 20% of (NH4)2S in water (9.0 mL, 26 mmol, 2.2 equiv). The mixture was then refluxed for 2 hours. After being cooled to room temperature, the reaction mixture was diluted with EtOAc and water. The organic layer was separated, washed with brine and concentrated. The residue was purified by column chromatography on silica gel using EtOAc / hexanes (1 :4) as eluent to give 2.2 g of 3-bromo-5-nitroaniline (84%) as an orange solid.
[00589] Ή NMR (CDC13, 300 MHz): δ 7.70 (s, 1H), 7.41 (d, / = 1.8 Hz, 1H), 7.08 (d, / = 1.5 Hz, 1H), 4.07 (br. s, 2H); m/z = 217 (M)+ .
EXAMPLE 133:
3-BROMO-5-NITROPHENYLISOCYANATE

[00590] A solution of 3-bromo-5-nitroaniline (500 mg, 2.3 mmol, 1 equiv) in 20% of phosgene in toluene (2.5 mL, 4.6 mmol, 2 equiv) was refluxed for 5 hours. The reaction mixture was then concentrated and the resulting residue was directly used in the next step.
[00591] JH NMR (CDC13, 300 MHz): δ 8.21 (s, 1H), 7.90 (s, 1H), 7.58 (s, 1H).
EXAMPLE 134:
1-(3-BROMO-5-NITROPHENYL)-4,5-DIHYDRO-5H-TETRAZOL-5-ONE

[00592] To the above residue 1.2 mL of TMSN3 (9.2 mmol, 4 equiv) was added and then the resulting mixture was heated at 100° C overnight. After being cooled to room
temperature, the reaction mixture was concentrated. The residue was diluted with EtOAc (20 mL) and saturated aqueous NaHCC>3 (20 mL). The aqueous layer was separated, neutralized with 2 N HC1 to pH 4-5 and extracted with EtOAc (2x30 mL). The organic layers were combined, dried over MgS04 and concentrated to give the product (370 mg, 56% two steps) which was directly used in the next step. . m/z = 283.97 (M-H)+ for 79Br.
EXAMPLE 135:
1-(3-BROMO-5-NITROPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

[00593] To a mixture of l-(3-bromo-5-nitrophenyl)-4,5-dihydro-5H-tetrazol-5-one (200 mg, 0.7 mmol, 1 equiv) and K2CO3 (290 mg, 2.1 mmol, 3 equiv) in 5 mL of DMF was added Mel (0.13 mL, 2.1 mmol, 3 equiv) and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was separated and concentrated. The residue was triturated in water to give 150 mg of product (74%) as a pale yellow solid.
[00594] Ή NMR (DMSO-d6, 300 MHz): δ 8.66 (d, / = 0.9 Hz, 1H), 8.47 (d, /
1H), 8.44 (t, / = 2.1 , 1.5 Hz, 1H), 3.62 (s, 3H); m/z = 300.0 (M+H)+ for 79Br.
EXAMPLE 136:
3-(4,5-DIHYDRO-4-METHYL-5-OXOTETRAZOL-1-YL)-5-NITROBENZONITRILE

[00595] A mixture of l-(3-bromo-5-nitrophenyl)-4-methyl-5H-tetrazol-5-one (100 mg, 0.34 mmol, 1 equiv), K4[Fe(CN)6].3H20 (215 mg, 0.51 mmol, 1.5 equiv), Pd(OAc)2 (8 mg, 0.034 mmol, 0.1 equiv) and Na2C03 (54 mg, 0.51 mmol, 1.5 equiv) in 3 mL of DMA was degassed under N2 for 1 minute and then irridated under MW at 120° C for 90 minutes. The reaction mixture was directly purified by column chromatography on silica gel using EtOAc / hexanes (3:7) as eluent to give 47 mg of product (56%).
[00596] JH NMR (CDC13, 300 MHz): δ 9.15 (dd, / = 1.8, 1.2 Hz, 1H), 8.76 (dd, / = 1.5, 1.2 Hz, 1H), 8.48 (dd, / = 1.5, 1.2 Hz, 1H), 3.76 (s, 3H);
EXAMPLE 137:
ALTERNATIVE PREPARATION OF 3-(4,5-DIHYDRO-4-METHYL-5-OXOTETRAZOL-1-YL)-5- NITROBENZONITRILE

[00597] A mixture of 3,5-dinitrobenzonitrile (386 mg, 2.0 mmol), 1 -methyl- lH-tetrazol- 5(4H)-one (400 mg, 4.0 mmol) and K2C03 (552 mg, 4.0 mmol) in NMP (6 mL) was heated to 110 °C and stirred overnight. After allowing to cool, the mixture was poured in to H20 (75 mL) and EtOAc (40 mL). The aqueous and organic layers were partitioned and the aqueous layer was extracted with EtOAc (2 x 30 mL). The combined organic extracts were washed with brine (1 x 20 mL), dried (Na2S04), filtered and the solvent removed under vacuum to leave a crude residue. The residue was purified by column chromatography on silica gel using EtOAc / hexane (3:7 to 4:6) as eluent to give the product (182 mg, 37%) as a solid.
[00598] Data identical to previously synthesized material. (Example 136)
[00599] Note: l-methyl-lH-tetrazol-5(4H)-one was prepared according to procedure detailed in EP643049 (1995); Preparation of l-substituted-5(4H)-tetrazolinones by desulfurization of tetrazolinethiones, which is hereby incorporated by reference.
EXAMPLE 138:
5-AMINO-3-(4,5-DIHYDRO-4-METHYL-5-OXOTETRAZOL-1-YL)BENZONITRILE

[00600] A solution of l-(3-cyano-5-nitrophenyl)-4-methyl-5H-tetrazol-5-one (200 mg, 0.81 mmol, 1 equiv) in 10 mL of MeOH in the presence of 20 mg of 10% Pd/C was hydrogenated at room temperature for 1 hour. The reaction mixture was then filtered over a pad of Celite and washed with MeOH. The solvent was concentrated to give the product in quantitative yield. . m/z = 217.2 (M+H)+ .
EXAMPLE 139:
N2-(5-CYANO-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)PHENYL)-5- FLUORO-N4-(1,2,2,6,6-PE -4-YL)PYRIMIDINE-2,4-DIAMINE (1-49)

[00601] Ή NMR (DMSO-d6, 300 MHz): δ 9.66 (s, 1H), 8.40 (s, 1H), 8.20 (s, 1H), 7.94 (d, / = 3.6 Hz, 1H), 7.72 (s, 1H), 7.34 (d, / = 7.2 Hz, 1H), 4.35-4.25 (m, 1H), 3.60 (s, 3H), 1.97 (s, 3H), 1.66 (dm, / = 10.8 Hz, 2H), 1.41 (tm, / = 12.3, 12.0 Hz, 2H), 1.04 (s, 6H), 0.97 (s, 6H); m/z = 481.4 (M+H)+.
EXAMPLE 140:
N2-(5-CYANO-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)PHENYL)-5- FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)PYRIMIDINE-2,4-DIAMINE (1-50)

[00602] Ή NMR (DMSO-d6, 300 MHz): δ 9.66 (s, 1H), 8.41 (s, 1H), 8.19 (s, 1H), 7.94 (d, / = 3.6 Hz, 1H), 7.72 (s, 1H), 7.34 (d, / = 8.1 Hz, 1H), 4.40-4.30 (m, 1H), 3.59 (s, 3H),
1.68 (dm, 7 = 9.6 Hz, 2H), 1.14 (tm, / = 12.0 Hz, 2H), 1.11 (s, 6H), 1.00 (s, 6H); m/z = 467.4
(M+H)+.
EXAMPLE 141:
3,5-DLNITRO- -ISOPROPOXYBENZOTRIFLUORIDE

[00603] iPrOH (4.3 mL, 55.44 mmol, 3 equiv) was added to a suspension of NaH (1.1 lg, 27.72 mmol, 1.5 equiv, 60%) in 20 mL of THF at 0° C and stirred for 30 minutes. A solution of 2-chloro-3,5-dinitrobenzotrifluoride (5.0 g, 18.48 mmol, 1 equiv) in 10 mL of THF was then added dropwise. The reaction was left to reach room temperature overnight. THF was evaporated and water was added to the residue. The aqueous phase was extracted with a mixture of hexanes / EtOAc (4/1, 2x100 mL). The organic layers were combined and concentrated. The residue was purified by column chromatography on silica gel using EtOAc / hexanes (1 :9) as eluent to give 3.3 g of product (60%) as a dark yellow oil.
[00604] JH NMR (CDC13, 300 MHz): δ 8.82 (s, 1H), 8.70 (s, 1H), 4.59 (sept, / = 6.0 Hz, 1H), 1.36 (d, 7 = 6.0 Hz, 6H).
EXAMPLE 142:
3-HYDROX

[00605] A mixture of 2.2 g of 3,5-dinitro-2-isopropoxybenzotrifluoride (7.5 mmol, 1 equiv) and 5.0 g of 8ηθ2.2¾0 (22.5 mmol, 3 equiv) in 50 mL of EtOH was stirred at room temperature for 3 hours. The solvent was evaporated and the residue was diluted with 2N NaOH and EtOAc. The organic layer was separated and concentrated. The residue was purified by column chromatography on silica gel using EtOAc / hexanes (1:4) as eluent to give 1.1 g of product (52%).
[00606] JH NMR (CDC13, 300 MHz): δ 8.30 (d, / = 2.1 Hz, 1H), 8.08 (d, / = 2.4 Hz, 1H), 7.19 (br. s, 1H), 5.56 (br. s, 1H), 4.67 (sept, / = 6.3 Hz, 1H), 1.31 (d, / = 6.0 Hz, 6H). m/z = 279.2 (M-H)+.
EXAMPLE 143:
3-AMINO-2- -5-NITROBENZOTRIFLUORIDE

[00607] To a solution of 3-hydroxyamino-2-isopropoxy-5-nitrobenzotrifluoride (32 mg, 0.11 mmol, 1 equiv) in EtOH (1 iriL) at room temperature was added 20% of (NH4)2S in water (0.04 mL, 0.11 mmol, 1.0 equiv). The mixture was then refluxed for 2 hours. After being cooled to rt, the reaction mixture was diluted with EtOAc and water. The organic layer was separated, washed with brine and concentrated. The residue was purified by column chromatography on silica gel using EtOAc / hexanes (1 :4) as eluent to give 16 mg of product (55%) as a yellow solid. JH NMR (CDC13, 300 MHz): δ 7.86 (s, 1H), 7.73 (s, 1H), 4.66 (sept, / = 6.0 Hz, 1H), 1.34 (d, / = 6.3 Hz, 6H). m/z = 263.1 (M-H)+.
EXAMPLE 144:
2-ISOPROPO -5-NITRO-3-TRIFLUOROMETHYLPHENYLISOCYANATE

[00608] A solution of 3-amino-2-isopropoxy-5-nitrobenzotrifluoride (520 mg, 1.97 mmol, 1 equiv) in 20% of phosgene in toluene (6.0 mL, 11.82 mmol, 6 equiv) was refluxed for 5 hours. The reaction mixture was then concentrated and the resulting residue was directly used in the next step.
[00609] JH NMR (CDC13, 300 MHz): δ 8.34 (s, 1H), 8.12 (s, 1H), 4.89 (m, 1H), 1.40 (d, 7 = 6.3 Hz, 6H) .
EXAMPLE 145:
1-(2-ISOPROPOXY-5-NITRO-3-TRIFLUOROMETHYLPHENYL)-4,5-DIHYDRO-5H-TETRAZOL- 5-ONE

[00610] To the above residue 1.1 mL of TMSN3 (7.88 mmol, 4 equiv) was added and then the resulting mixture was heated at 100° C overnight. After being cooled to room
temperature, the reaction mixture was concentrated. The residue was diluted with EtOAc (20 mL) and saturated aqueous NaHCC>3 (20 mL). The aqueous layer was separated, neutralized with 2 N HC1 to pH 4-5 and extracted with EtOAc (2x20 mL). The organic layers were combined, dried over MgS04 and concentrated to give a yellow solid (270 mg, 41% two steps) which was directly used in the next step. m/z = 332.2 (M)+ .
EXAMPLE 146:
1-(2-ISOPROPOXY-5-NITRO-3-TRIFLUOROMETHYLPHENYL)-4-METHYL-5H-TETRAZOL-5-
ONE

[00611] To a mixture of l-(2-isopropoxy-5-nitro-3-trifluoromethylphenyl)-4,5-dihydro- 5H-tetrazol-5-one (270 mg, 0.81 mmol, 1 equiv) and K2CO3 (335 mg, 2.43 mmol, 3 equiv) in 5 mL of DMF was added Mel (0.15 mL, 2.43 mmol, 3 equiv) and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was separated and concentrated. The residue was triturated in water to give 200 mg of product (71 ). m/z = 347 (M)+ .
EXAMPLE 147:
1-(5-AMINO-2-ISOPROPOXY-3-TRIFLUOROMETHYLPHENYL)-4-METHYL-5H-TETRAZOL-5-
ONE
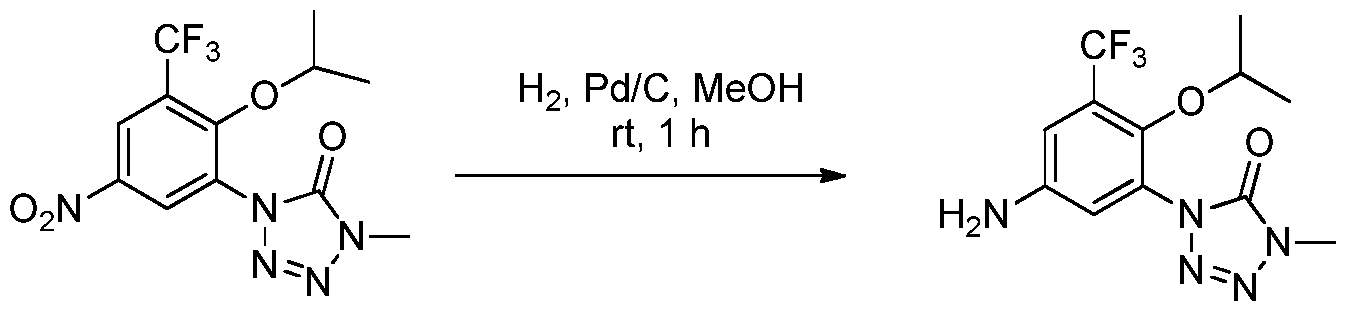
[00612] A solution of l-(2-isopropoxy-5-nitro-3-trifluoromethylphenyl)-4-methyl-5H- tetrazol-5-one (200 mg, 0.58 mmol, 1 equiv) in 10 mL of MeOH in the presence of 20 mg of 10% Pd/C was hydrogenated at room temperature for 1 hour. The reaction mixture was then filtered over a pad of Celite and washed with MeOH. The solvent was concentrated to give the product in quantitative yield, m/z = 318.3 (M+H)+ .
EXAMPLE 148:
N2-(4-ISOPROPOXY-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)-5- TRIFLUOROMETHYLPHENYL)-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4- YL)PYRIMIDINE-2,4-DIAM

[00613] Ή NMR (DMSO-d6, 300 MHz): δ 9.42 (s, 1H), 8.17 (s, 1H), 8.02 (s, 1H), 7.89 (d, / = 3.6 Hz, 1H), 7.25 (d, / = 9.3 Hz, 1H), 4.30-4.20 (m, 1H), 3.82 (sept. / = 6.9 Hz, 1 H), 3.62 (s, 3H), 1.63 (dm, / = 11.7 Hz, 2H), 1.13 (dm, / = 10.8 Hz, 2H), 1.01 (s, 6H), 0.99 (d, / = 7.8 Hz, 6H), 0.98 (s, 6H); m/z = 568 (M+H)+.
EXAMPLE 149:
N2-(4-ISOPROPOXY-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)-5- TRIFLUOROMETHYLPHENYL)-5-FLUORO-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4- YL)PYRIMIDINE-2,4-DIAM

[00614] Ή NMR (DMSO-d6, 300 MHz): δ 9.42 (s, 1H), 8.15 (s, 1H), 8.03 (s, 1H), 7.89 (d, / = 3.6 Hz, 1H), 7.26 (d, / = 7.8 Hz, 1H), 4.25-4.15 (m, 1H), 3.82 (sept. / = 6.9 Hz, 1 H), 3.62 (s, 3H), 2.13 (s, 3H), 1.64 (dm, / = 10.8 Hz, 2H), 1.40 (dm, / = 11.4 Hz, 2H), 1.03 (s, 6H), 0.99 (d, / = 6.3 Hz, 6H), 0.89 (s, 6H); m/z = 582.4 (M+H)+.
EXAMPLE 150:
3-AMINO-2-MET -5-NITROBENZOTRIFLUORIDE

[00615] 70% HNO3 (1.96 mL, 32.6 mmol, 1.1 equiv) was added dropwise to a mixture of 3-amino-2-methylbenzotrifluoride (5.0 g, 28.55 mmol, 1 equiv) in 30 mL of fuming H2SO4 at 0° C. Then the mixture was left to reach room temperature and stirred for 1 hour. Poured into ice and the aqueous phase was extracted with EtOAc. The organic layer was separated, washed with saturated NaHCC>3 and concentrated. The residue was purified by column
chromatography on silica gel using EtOAc / hexanes (3:7) as eluent to give 1.6 g of product (25%) as a yellow solid.
[00616] JH NMR (CDC13, 300 MHz): δ 7.91 (s, 1H), 7.67 (s, 1H), 4.13 (br. s, 2H), 2.31 (s, 3H). m/z = 221.1 (M+H)+.
EXAMPLE 151:
2-METHYL- -NITRO-3-TRIFLUOROMETHYLPHENYLISOCYANATE

[00617] A solution of 3-amino-2-methyl-5-nitrobenzotrifluoride (700 mg, 3.2 mmol, 1 equiv) in 20% of phosgene in toluene (3.4 mL, 6.4 mmol, 2 equiv) was refluxed for 5 hours. The reaction mixture was then concentrated and the resulting residue was directly used in the next step.
[00618] JH NMR (CDC13, 300 MHz): δ 8.36 (s, 1H), 8.17 (s, 1H), 2.55 (s, 3H).
EXAMPLE 152:
1-(2-METHYL-5-NITRO-3-TRIFLUOROMETHYLPHENYL)-4,5-DIHYDRO-5H-TETRAZOL-5-
ONE

[00619] To the above residue of Example 151, 1.7 mL of TMSN3 (12.8 mmol, 4 equiv) was added and then the resulting mixture was heated at 100° C overnight. After being cooled to room temperature, the reaction mixture was concentrated. The residue was diluted with EtOAc (30 mL) and saturated aqueous NaHC03 (30 mL). The aqueous layer was separated, neutralized with 2 N HC1 to pH 4-5 and extracted with EtOAc (2x20 mL). The organic layers were combined, dried over MgS04 and concentrated to give a yellow solid (560 mg, 60% two steps) which was directly used in the next step. mJz = 289 (M)+ .
EXAMPLE 153:
1-(2-METHYL-5-NITRO-3-TRIFLUOROMETHYLPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

EXAMPLE 154:
1-(5-AMIN -2-METHYL-3-TRIFLUOROMETHYLPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

[00621] A solution of l-(2-methyl-5-nitro-3-trifluoromethylphenyl)-4-methyl-5H- tetrazol-5-one (520 mg, 1.72 mmol, 1 equiv) in 10 mL of MeOH in the presence of 30 mg of 10% Pd/C was hydrogenated at room temperature for 1 hour. The reaction mixture was then filtered over a pad of Celite and washed with MeOH. The solvent was concentrated to give the product in quantitative yield, m/z = 274.2 (M+H)+ .
EXAMPLE 155:
N2-(4-METHYL-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)-5- TRIFLUOROMETHYLPHENYL)-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4- YL)PYRIMIDINE-2,4-DIAM -53)

[00622] Ή NMR (DMSO-d6, 300 MHz): δ 9.50 (s, 1H), 8.21 (s, 1H), 8.09 (s, 1H), 7.90 (d, 7 = 3.6 Hz, 1H), 7.27 (d, 7 = 7.5 Hz, 1H), 4.35-4.25 (m, 1H), 3.61 (s, 3 H), 2.11 (s, 3H), 1.64 (dm, / = 9.3 Hz, 2H), 1.13 (dm, / = 9.3 Hz, 2H), 1.03 (s, 6H), 0.99 (s, 6H); m/z = 524 (M+H)+.
EXAMPLE 156:
N2-(4-METHYL-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)-5- TRIFLUOROMETHYLPHENYL)-5-FLUORO-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4- YL)PYRIMIDINE-2,4-DIAM

[00623] JH NMR (DMSO-d6, 300 MHz): δ 9.51 (s, 1H), 8.20 (s, 1H), 8.10 (s, 1H), 7.90 (d, / = 3.6 Hz, 1H), 7.28 (d, / = 8.7 Hz, 1H), 4.25-4.15 (m, 1H), 3.62 (s, 3 H), 2.14 (s, 3H), 2.11 (s, 3H), 1.64 (dm, / = 9.9 Hz, 2H), 1.41 (dm, / = 9.3 Hz, 2H), 1.03 (s, 6H), 0.89 (s, 6H); m/z = 538.3 (M+H)+.
EXAMPLE 157:
3-CHLORO-2-METHYL-5-NITROPHENYLISOCYANATE

[00624] A solution of 3-chloro-2-methyl-5-nitroaniline (1.0 g, 5.36 mmol, 1 equiv) in 20% of phosgene in toluene (17.0 mL, 32.16 mmol, 6 equiv) was refluxed for 5 hours. The reaction mixture was then concentrated and the resulting residue was directly used in the next step.
[00625] JH NMR (CDC13, 300 MHz): δ 8.10 (d, / = 1.8 Hz, 1H), 7.89 (d, / = 2.4 Hz, 1H), 2.48 (s, 3H).
EXAMPLE 158:
1-(3-CHLORO-2-METHYL-5-NITROPHENYL)-4,5-DIHYDRO-5H-TETRAZOL-5-ONE

[00626] To the above residue 2.8 mL of TMSN3 (21.44 mmol, 4 equiv) was added and then the resulting mixture was heated at 100° C overnight. After being cooled to room temperature, the reaction mixture was concentrated. The residue was diluted with EtOAc (30 mL) and saturated aqueous NaHCC>3 (30 mL). The aqueous layer was separated, neutralized with 2 N HC1 to pH 4-5 and extracted with EtOAc (2x20 mL). The organic layers were
combined, dried over MgS04 and concentrated to give a yellow solid (320 mg, 24% two steps) which was directly used in the next step.
EXAMPLE 159:
l-(3-Chloro-2-methyl-5-nitrophenyl)-4-methyl-5H-tetrazol-5-one

[00627] To a mixture of l-(3-chloro-2-methyl-5-nitrophenyl)-4,5-dihydro-5H-tetrazol-5- one (320 mg, 1.3 mmol, 1 equiv) and K2CO3 (538 mg, 3.9 mmol, 3 equiv) in 6 mL of DMF was added Mel (0.24 mL, 3.9 mmol, 3 equiv) and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was separated and concentrated. The residue was triturated in water to give 280 mg of product (80%) as a light yellow solid.
EXAMPLE 160:
1-(5-AMINO-3-CHLORO-2-METHYLPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

[00628] A mixture of 280 mg of l-(3-chloro-2-methyl-5-nitrophenyl)-4-methyl-5H- tetrazol-5-one (1.04 mmol, 1 equiv) and 704 mg of 8ηθ2.2¾0 (3.12 mmol, 3 equiv) in 10 mL of EtOH was refluxed for 16 hours. After being cooled to room temperature, the reaction mixture was diluted with 2N NaOH and EtOAc. The organic layer was separated, dried over MgS04 and concentrated to give the product as a yellow solid in quantitative yield.
[00629] Ή NMR (CDC13, 300 MHz): δ 6.84 (s, 1H), 6.54 (d, / = 1.8 Hz, 1H), 3.79 (br. s, 2H), 3.70 (s, 3H), 2.13 (s, 3H). mJz = 240.2 (M+H)+ .
EXAMPLE 161:
N2-(5-CHLORO-4-METHYL-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1- YL)PHENYL)-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)PYRIMIDINE-2,4-
DIAMINE (1-55)

[00630] Ή NMR (DMSO-dg, 300 MHz): δ 9.39 (s, 1H), 7.98 (s, 1H), 7.89 (d, / = 3.6 Hz, 1H), 7.78 (d, / = 1.5 Hz, 1H), 7.26 (d, / = 7.2 Hz, 1H), 4.35-4.25 (m, 1H), 3.60 (s, 3 H), 2.05 (s, 3H), 1.64 (dm, / = 8.7 Hz, 2H), 1.19-1.13 (m, 2H), 1.06(s, 6H), 0.99 (s, 6H); m/z = 490.3 (M+H)+.
EXAMPLE 162:
N2-(5-CHLORO-4-METHYL-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1- YL)PHENYL)-5-FLUORO-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)PYRIMIDINE-2,4-
DIAMINE (1-56)

[00631] Ή NMR (DMSO-dg, 300 MHz): δ 9.39 (s, 1H), 7.98 (s, 1H), 7.89 (d, / = 3.3 Hz, 1H), 7.75 (s, 1H), 7.27 (d, / = 8.4 Hz, 1H), 4.25-4.15 (m, 1H), 3.61 (s, 3 H), 2.14 (s, 3H), 2.05 (s, 3H), 1.64 (dm, / = 9.9 Hz, 2H), 1.42 (tm, / = 12.6 Hz, 2H), 1.04 (s, 6H), 0.93 (s, 6H); m/z = 504.4 (M+H)+.
EXAMPLE 163:
4,6-DLNITRO-2-FLUOROPHENOL

[00632] To 2-fluorophenol (10 mL, 12.1 g, 108 mmol, 1 equiv) in anhydrous DCM at 0° C was added 90% HNO3 (12.6 mL, 17.0 g, 270 mmol, 2.5 equiv) dropwise. The mixture was warmed to room temperature and stirred for 2 hours, then cooled to 0° C again and quenched with 2N NaOH solution to pH 5 (ca. 80 mL). The mixture was concentrated, diluted with water and extracted with EtOAc (3x150 mL). The combined organic layers was dried over
MgS04, filtered and concentrated. The residue was triturated in hexanes to give the product (18 g, 82%) as a yellow solid.
[00633] JH NMR (CDC13, 300 MHz): δ 10.97 (br. s, 1H), 8.92-8.90 (m, 1H), 8.32 (dm, / = 9.3 Hz, lH); m/z = 201.1 (M-H)+ .
EXAMPLE 164:
2-BROMO-1,5-DINITRO-3-FLUOROBENZENE

[00634] To a solution of 4,6-dinitro-2-fluorophenol (8 g, 39.60 mmol, 1 equiv) in DMF (24 mL) and toluene (160 mL), PBr3 (5.6 mL, 59.40 mmol, 1.5 equiv) was added at room temperature. Then the reaction mixture was heated at 110° C for 1 hour. After allowing to cool to room temperature, the upper colorless layer was poured into a separate funnel and diluted with hexanes. The organic layer was washed with water, dried over MgS04 and evaporated to dryness to give the product (10.3g, 98%) as a yellow solid.
[00635] JH NMR (CDC13, 300 MHz): δ 8.54 (d, / = 1.2 Hz, 1H), 8.22 (dd, / = 7.5, 0.9 Hz, y, m/z = 263.0 (M)+ .
EXAMPLE 165:
2- AND 4-BROMO-3-FLUORO-5-NIT

[00636] A mixture of 2-bromo-l,5-dinitro-3-fluorobenzene (100 mg, 0.38 mmol, 1 equiv) and iron powder (64 mg, 1.14 mmol, 3 equiv) in 3 mL of HOAc was stirred at room temperature for 90 minutes. The reaction mixture was diluted with EtOAc (20 mL) and saturated NaHCC>3 (to ca. pH 7-8). The organic layer was separated and evaporated under vacuum. The crude residue was purified by column chromatography on silica gel using EtOAc / hexanes (1:4) as eluent to give 47 mg of 2-bromo-3-fluoro-5-nitroaniline (52%).
[00637] JH NMR (CDC13, 300 MHz): δ 7.43-7.32 (m, 2H), 4.63 (br. s, 2H), 1.40-1.35 (m, 1H), 1.25-1.20 (m, 2H), 0.85-0.80 (m, 2H); m/z = 237.0 (M+H)+ for 81Br.
[00638] A later fraction gave 28 mg of 4-Bromo-3-fluoro-5-nitroaniline (31%).
[00639] JH NMR (CDC13, 300 MHz): δ 6.94 (d, / = 2.7 Hz, 1H), 6.62 (dd, / = 9.6, 2.7 Hz, 1H), 4.15 (br. s, 2H), 1.40-1.35 (m, 1H), 1.28-1.23 (m, 2H), 0.88-0.85 (m, 2H); m/z = 237.0
(M+H)+ for 81Br.
EXAMPLE 166:
2-CYCLOPROPYL-3-FLUORO-5-NITRO ANILINE

[00640] A mixture of 2-bromo-3-fluoro-5-nitroaniline (1.6 g, 6.81 mmol, 1 equiv), cyclopropylboronic acid MID A ester (Aldrich; 4.0 g, 20.43 mmol, 3 equiv), Pd(OAc)2 (238 mg, 1.06 mmol, 0.15 equiv), Cy3P (578 mg, 2.06 mmol, 0.3 equiv) and Cs2C03 (13.26 g, 40.8 mmol, 6 equiv) in toluene (70 mL) and ¾0 (14 mL) was de-gassed with N2 for 5 minutes. The mixture was then heated at 100 °C (oil bath temperature) overnight. After allowing to cool to room temperature, the mixture was diluted with EtOAc (100 mL) and H2O (50 mL) and the mixture filtered through Celite. The filter cake was washed with EtOAc (2 x 50 mL) and the filtrate partitioned. The organic layer was evaporated under vacuum to leave a crude residue which was purified by column chromatography on silica gel using EtOAc / hexanes (1:4) as eluent to give the product (1.2 g, 90%) as a dark yellow solid.
[00641] Ή NMR (CDCI3, 300MHz): δ 7.29-7.21 (m,2H), 4.44 (br. s, 2H), 1.52-1.42 (m, 1H), 1.11-1.05 (m, 2H), 0.73-0.67 (m, 2H); m/z = 197.2 (M+H)+
EXAMPLE 167:
2-CYCLOPR -3-FLUORO-5-NITROPHENYLISOCYANATE

[00642] A solution of 2-cyclopropyl-3-fluoro-5-nitroaniline (413 mg, 2.1 mmol, 1 equiv) in 20% of phosgene in toluene (2.1 mL, 4.2 mmol, 2 equiv) was refluxed for 5 hours. The reaction mixture was then concentrated and the resulting residue was directly used in the next step.
EXAMPLE 168:
1-(2-CYCL -3-FLUORO-5-NITROPHENYL)-4,5-DIHYDRO-5H-TETRAZOL-5-ONE

EXAMPLE 169:
1-(2-CYCL -3-FLUORO-5-NITROPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

[00644] To a mixture of l-(2-cyclopropyl-3-fluoro-5-nitrophenyl)-4,5-dihydro-5H- tetrazol-5-one (300 mg, 1.13 mmol, 1 equiv) and K2CO3 (470 mg, 3.4 mmol, 3 equiv) in 6 mL of DMF was added Mel (0.22 mL, 3.4 mmol, 3 equiv) and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was separated and concentrated. The residue was triturated in water to give 270 mg of product (86%) as a pale yellow solid, m/z = 280.2 (M+H)+ .
EXAMPLE 170:
1-(5-AMIN -2-CYCLOPROPYL-3-FLUOROPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

[00645] A mixture of 270 mg of l-(3-chloro-2-methyl-5-nitrophenyl)-4-methyl-5H- tetrazol-5-one (0.97 mmol, 1 equiv) and 655 mg of SnCl2.2H20 (2.9 mmol, 3 equiv) in 8 mL of EtOH was refluxed for 2 hours. After being cooled to room temperature, the reaction mixture was diluted with 2N NaOH and EtOAc. The organic layer was separated, dried over MgS04 and concentrated to give 230 mg of product (95%). m/z = 250.2 (M+H)+ .
EXAMPLE 171:
N2-(4-CYCLOPROPYL-5-FLUORO-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1- YL)PHENYL)-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)PYRIMIDINE-2,4-DIAMINO-5- CARBOXYAMIDE (1-57)

[00646] *H NMR (DMSO-d6, 300 MHz): δ 9.91 (s, IH), 9.25 (br. s, IH), 8.53 (s, IH), 7.87 (d, / = 12.9 Hz, IH), 7.85 (br. s, IH), 7.57 (s, IH), 7.22 (br. s, IH), 4.40-4.30 (m, IH), 3.62 (s, 3 H), 1.80-1.70 (m, 2H), 1.60-1.56 (m, IH), 1.20-1.10 (m, 2H), 1.07 (s, 6H), 1.00 (s, 6H), 0.71-0.68 (m, 2H), 0.26-0.24 (m, 2H); mJz = 525.5 (M+H)+.
EXAMPLE 172:
N2-(4-CYCLOPROPYL-5-FLUORO-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1- YL)PHENYL)-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)PYRIMIDINE-2,4-
DIAMINE (1-59)

[00647] *H NMR (DMSO-d6, 300 MHz): δ 9.50 (s, IH), 7.89 (d, / = 3.6 Hz, IH), 7.81 (d, / = 13.5 Hz, IH), 7.52 (s, IH), 7.30 (d, / = 7.8 Hz, IH), 4.40-4.30 (m, IH), 3.62 (s, 3 H), 1.67-1.63 (m, 2H), 1.60-1.50 (m, IH), 1.20-1.10 (m, 2H), 1.09 (s, 6H), 1.00 (s, 6H), 0.69- 0.67 (m, 2H), 0.26-0.24 (m, 2H); m/z = 500.3 (M+H)+.
EXAMPLE 173:
N2-(4-CYCLOPROPYL-5-FLUORO-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1- YL)PHENYL)-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)PYRIMIDINE-2,4-DIAMINO-5- CARBONITRILE (1-62)

[00648] JH NMR (DMSO-d6, 300 MHz): δ 10.16 (s, IH), 8.36 (s, IH), 7.84 (d, / = 13.2 Hz, IH), 7.52 (d, / = 9.0 Hz, IH), 7.48 (s, IH), 4.50-4.40 (m, IH), 3.62 (s, 3 H), 1.61-1.57
(m, 2H), 1.28-1.19 (m, 2H), 1.16-1.13 (m, 1H), 1.05 (s, 6H), 0.99 (s, 6H), 0.72-0.70 (m, 2H), 0.27-0.25 (m, 2H); m/z = 507.4 (M+H)+.
EXAMPLE 174:
2-BROMO-5-NITROPHENYLISOCYANATE
Phosgene, toluene

[00649] A solution of 2-bromo-5-nitroaniline (1.0 g, 4.61 mmol, 1 equiv) in 20% of phosgene in toluene (4.85 mL, 9.22 mmol, 2 equiv) was refluxed for 5 hours. The reaction mixture was then concentrated and the resulting residue was directly used in the next step. EXAMPLE 175:
1-(2-BROMO-5-NITROPHENYL)-4,5-DIHYDRO-5H-TETRAZOL-5-ONE

[00650] To the above residue 2.4 mL of TMSN3 (18.44 mmol, 4 equiv) was added and then the resulting mixture was heated at 100° C overnight. After being cooled to room temperature, the reaction mixture was concentrated. The residue was diluted with EtOAc (50 mL) and saturated aqueous NaHCC>3 (50 mL). The aqueous layer was separated, neutralized with 2 N HC1 to pH 4-5 and extracted with EtOAc (2x50 mL). The organic layers were combined, dried over MgS04 and concentrated to give a yellow solid (500 mg, 38% two
+ 81
steps) which was directly used in the next step, m/z = 288.0 (M+H) for Br.
EXAMPLE 176:
1-(2-BROM -5-NITROPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

[00651] To a mixture of l-(2-bromo-5-nitrophenyl)-4,5-dihydro-5H-tetrazol-5-one (500 mg, 1.75 mmol, 1 equiv) and K2CO3 (724 g, 5.25 mmol, 3 equiv) in 8 mL of DMF was added Mel (0.33 mL, 5.25 mmol, 3 equiv) and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was separated and concentrated. The residue was triturated in water to give 460 mg of product (88%) as a pale yellow solid, m/z = 302.0 (M+H)+ for 81Br.
EXAMPLE 177:
2-(4,5-DIHYDRO-4-METHYL-5-OXOTETRAZOL-1-YL)-4-NITROBENZONITRILE

[00652] A mixture of l-(2-bromo-5-nitrophenyl)-4-methyl-5H-tetrazol-5-one (100 mg, 0.34 mmol, 1 equiv), K4[Fe(CN)6].3H20 (215 mg, 0.51 mmol, 1.5 equiv), Pd(OAc)2 (8 mg, 0.034 mmol, 0.1 equiv) and Na2C(¾ (54 mg, 0.51 mmol, 1.5 equiv) in 3 mL of DMA was degassed under N2 for 1 minute and then irridated under MW at 120° C for 90 minutes. The reaction mixture was directly purified by column chromatography on silica gel using EtOAc / hexanes (3:7) as eluent to give 58 mg of product (70%). m/z = 247.2 (M+H)+.
EXAMPLE 178:
4-AMINO- -(4,5-DIHYDRO-4-METHYL-5-OXOTETRAZOL-1-YL)BENZONITRILE

[00653] A solution of 2-(4,5-dihydro-4-methyl-5-oxotetrazol-l-yl)-4-nitrobenzonitrile (58 mg, 0.24 mmol, 1 equiv) in 5 mL of MeOH in the presence of 5 mg of 10% Pd/C was hydrogenated at room temperature for 3 hours. The reaction mixture was then filtered over a pad of Celite and washed with MeOH. The solvent was concentrated to give the product in quantitative yield, m/z = 217.2 (M+H)+ .
EXAMPLE 179:
N2-(4-CYANO-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)PHENYL)-5- FLUORO-N4-(2,2,6,6-TET -4-YL)PYRIMIDINE-2,4-DIAMINE (1-58)

[00654] Ή NMR (DMSO-d6, 300 MHz): δ 9.99 (s, 1H), 8.15 (s, 1H), 7.95 (d, 7 = 3.6 Hz, 1H), 7.94 (s, 1H), 7.81 (d, / = 8.1 Hz, 1H), 7.43 (d, 7 = 7.8 Hz, 1 H), 4.28-4.18 (m, 1H), 3.62 (s, 3H), 1.66 (dm, / = 9.6 Hz, 2H), 1.14 (tm, / = 12.0 Hz, 2H), 1.06 (s, 6H), 1.03 (s, 6H); m/z = 467.3 (M+H)+.
EXAMPLE 180:
METHYL 3-ISOCYANATO-2-METHYL-5-NITROBENZOATE

[00655] A solution of methyl 3-amino-2-methyl-5-nitrobenzoate (2.0 g, 9.52 mmol, 1 equiv) in 20% of phosgene in toluene (10.0 mL, 19.03 mmol, 2 equiv) was refluxed for 5 hours. The reaction mixture was then concentrated and the resulting residue was directly used in the next step.
[00656] JH NMR (CDC13, 300 MHz): δ 8.54 (s, 1H), 8.10 (s, 1H), 3.96 (s, 3H), 2.66 (s, 3H).
EXAMPLE 181:
METHYL 3- -DIHYDRO-5-OXOTETRAZOL-1-YL)-2-METHYL-5-NITROBENZOATE

[00657] To the above residue 5.0 mL of TMSN3 (38.06 mmol, 4 equiv) was added and then the resulting mixture was heated at 100° C overnight. After being cooled to room temperature, the reaction mixture was concentrated. The residue was diluted with EtOAc (50 mL) and saturated aqueous NaHCC>3 (50 mL). The aqueous layer was separated, neutralized with 2 N HC1 to pH 4-5 and extracted with EtOAc (2x50 mL). The organic layers were combined, dried over MgS04 and concentrated to give a yellow solid (430 mg, 16% two steps) which was directly used in the next step.
EXAMPLE 182:
METHYL 3-(4,5-DIHYDRO-4-METHYL-5-OXOTETRAZOL-1-YL)-2-METHYL-5- NITROBEN

[00658] To a mixture of methyl 3-(4,5-dihydro-5-oxotetrazol-l-yl)-2-methyl-5- nitrobenzoate (430 mg, 1.54 mmol, 1 equiv) and K2CO3 (640 g, 4.62 mmol, 3 equiv) in 8 mL of DMF was added Mel (0.29 mL, 4.62 mmol, 3 equiv) and the resulting mixture was stirred
at room temperature overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was separated and concentrated. The residue was triturated in water to give 400 mg of product (89%) as a pale yellow solid. m/z = 294.2 (M+H)+ .
EXAMPLE 183:
METHYL 5- AMINO-3-(4,5-DIHYDRO-4-METHYL-5-OXOTETRAZOL-1-YL)-2- METHYLBE

[00659] A solution of methyl 3-(4,5-dihydro-4-methyl-5-oxotetrazol-l-yl)-2-methyl-5- nitrobenzoate (400 mg, 1.37 mmol, 1 equiv) in 10 mL of MeOH in the presence of 40 mg of 10% Pd/C was hydrogenated at room temperature for 3 hours. The reaction mixture was then filtered over a pad of Celite and washed with MeOH. The solvent was concentrated to give the product in quantitative yield, m/z = 264.2 (M+H)+ .
EXAMPLE 184:
METHYL 5-((5-FLUORO-4-((2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)AMINO)-2- PYRIMIDINYL)AMINO)-2-METHYL-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1- YL)BENZOATE (1-60)

[00660] Ή NMR (DMSO-d6, 300 MHz): δ 9.38 (s, 1H), 8.13 (s, 1H), 8.08 (d, / = 2.7 Hz, 1H), 7.88 (d, / = 3.0 Hz, 1H), 7.21 (d, / = 7.8 Hz, 1 H), 4.30-4.20 (m, 1H), 3.83 (s, 3H), 3.60 (s, 3H), 2.12 (s, 3H), 1.64 (dm, / = 11.7 Hz, 2H), 1.15-1.11 (m, 2H), 1.02 (s, 6H), 0.98 (s, 6H); m/z = 514.3 (M+H)+.
EXAMPLE 185:
METHYL 5-((5-FLUORO-4-((1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)AMINO)-2- PYRIMIDINYL)AMINO)-2-METHYL-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1- YL)BENZOATE (1-63)

[00661] Ή NMR (DMSO-d6, 300 MHz): δ 9.35 (s, 1H), 8.13 (s, 1H), 8.05 (s, 1H), 7.96 (d, / = 3.0 Hz, 1H), 7.62 (d, / = 7.8 Hz, 1 H), 4.40-4.30 (m, 1H), 3.85 (s, 3H), 3.63 (s, 3H), 2.71 (s, 3H), 2.16 (s, 3H), 2.05 (dm, / = 13.2 Hz, 2H), 1.78 (tm, / = 13.2 Hz, 2H), 1.39 (s, 6H), 1.32 (s, 6H); m/z = 528.5 (M+H)+.
EXAMPLE 186:
5-((5-FLUORO-4-((2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)AMINO)-2-PYRIMIDINYL)AMINO)- 2-METHYL-3-(4-METHYL- -DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)BENZOIC ACID (1-61)

[00662] A mixture of 80 mg of methyl 5-((5-fluoro-4-((2,2,6,6-tetramethylpiperidin-4- yl)amino)-2-pyrimidinyl)amino)-2-methyl-3-(4-methyl-4,5-dihydro-5-oxo-lH-tetrazol-l- yl)benzoate (0.16 mmol, 1 equiv) in THF / ¾0 (1.5 mL / 1.5 mL) in the presence of LiOH (37 mg, 1.6 mmol, 10 equiv) was stirred at room temperature for 5 hours. Then the reaction mixture was neutralized with 2N HC1 to pH 3. The product crashed out as a white solid which was filtered and dried (77 mg, 90%).
[00663] Ή NMR (DMSO-d6, 300 MHz): δ 9.37 (s, 1H), 8.72 (br. s, 1H), 8.13 (s, 1H), 7.95 (d, / = 4.2 Hz, 1H), 7.78 (br. s, 1H), 7.57 (d, / = 7.2 Hz, 1 H), 4.40-4.30 (m, 1H), 3.62 (s, 3H), 2.15 (s, 3H), 1.95 (dm, / = 11.7 Hz, 2H), 1.60-1.50 (m, 2H), 1.34 (s, 6H), 1.32 (s, 6H); m/z = 500.3 (M+H)+.
EXAMPLE 187:
5-((5-FLUORO-4-((1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)AMINO)-2- PYRIMIDINYL)AMINO)-2-METHYL-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1- YL)BENZOIC ACID (1-65)

[00664] A mixture of 120 mg of methyl 5-((5-fluoro-4-((l,2,2,6,6-pentamethylpiperidin- 4-yl)amino)-2-pyrimidinyl)amino)-2-methyl-3-(4-methyl-4,5-dihydro-5-oxo-lH-tetrazol-l- yl)benzoate (0.23 mmol, 1 equiv) in THF / ¾0 (2.0 mL / 2.0 mL) in the presence of LiOH (55 mg, 2.3 mmol, 10 equiv) was stirred at room temperature for 5 hours. Then the reaction mixture was neutralized with 2N HC1 to pH 3. The product crashed out as a white solid which was filtered and dried (55 mg, 47%).
[00665] JH NMR (DMSO-d6, 300 MHz): δ 9.35 (s, 1H), 8.51 (br. s, 1H), 8.15 (s, 1H), 7.95 (d, / = 1.5 Hz, 1H), 7.63 (d, / = 8.1 Hz, 1 H), 4.40-4.30 (m, 1H), 3.62 (s, 3H), 2.70 (s, 3H), 2.17 (s, 3H), 2.05 (dm, / = 12.9 Hz, 2H), 1.74 (tm, / = 13.2 Hz, 2H), 1.36 (s, 6H), 1.30 (s, 6H); m/z = 514.4 (M+H)+.
EXAMPLE 188:
N-METHYL 5-((5-FLUORO-4-((1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)AMINO)-2- PYRIMIDINYL)AMINO)-2-METHYL-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1- YL)BENZENECARBOXYAMIDE (1-67)

[00666] A mixture of 120 mg of methyl 5-((5-fluoro-4-((l,2,2,6,6-pentamethylpiperidin- 4-yl)amino)-2-pyrimidinyl)amino)-2-methyl-3-(4-methyl-4,5-dihydro-5-oxo-lH-tetrazol-l- yl)benzoate (0.23 mmol, 1 equiv) in THF / ¾0 (2.0 mL / 2.0 mL) in the presence of LiOH (55 mg, 2.3 mmol, 10 equiv) was stirred at room temperature for 5 hours. Then the reaction mixture was neutralized with 2N HC1 to pH 3. The product crashed out as a white solid which was filtered and dried (55 mg, 47%).
[00667] Ή NMR (DMSO-d6, 300 MHz): δ 9.27 (s, 1H), 8.35-8.30 (m, 1H), 7.96 (s, 1H), 7.87 (d, / = 3.9 Hz, 1H), 7.64 (s, 1H), 7.20 (d, / = 8.1 Hz, 1 H), 4.30-4.20 (m, 1H), 3.61 (s,
3H), 2.73 (d, / = 4.8 Hz, 3H), 2.13 (s, 3H), 1.96 (s, 3H), 1.62 (dm, /
(tm, / = 12.3 Hz, 2H), 1.36 (s, 6H), 1.30 (s, 6H); m/z = 527.5 (M+H)
EXAMPLE 189:
1-(2,6-DIFLUOROPHENYL)-4,5-DIHYDRO-5H-TETRAZOL-5-ONE

[00668] A mixture of 2,6-difluorophenylisocyanate (6.0 g, 38.68 mmol, 1 equiv) in 10.32 mL of TMSN3 (77.36 mmol, 2 equiv) was heated at 100° C overnight. After being cooled to room temperature, the reaction mixture was concentrated. The residue was diluted with EtOAc (100 mL) and saturated aqueous NaHCC>3 (100 mL). The aqueous layer was separated, neutralized with 2 N HCl to pH 4-5 and extracted with EtOAc (2x100 mL). The organic layers were combined, dried over MgS04 and concentrated to give a white solid (7.1 g, 93%).
[00669] Ή NMR (CDC13, 300MHz): δ 7.59-7.49 (m, 1H), 7.15 (t, / = 9.0, 8.4 Hz, 2H); m/z = 199.3 (M+H)+ .
EXAMPLE 190:
1-(2,6-DIFLUO -4-METHYL-5H-TETRAZOL-5-ONE

[00670] To a mixture of l-(2,6-difluorophenyl)-4,5-dihydro-5H-tetrazol-5-one (3.55 g, 17.92 mmol, 1 equiv) and K2C03 (7.42 g, 53.75 mmol, 3 equiv) in 60 mL of DMF was added Mel (3.35 mL, 53.75 mmol, 3 equiv) and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was separated and concentrated. The residue was triturated in water to give 3.45 g of product (91 ) as a white solid.
[00671] JH NMR (CDC13, 300MHz): δ 7.54-7.46 (m, 1H), 7.12 (t, / = 8.4, 7.5 Hz, 2H), 3.73 (s, 3H); m/z = 213.4 (M+H)+ .
EXAMPLE 191:
1-(2,6-DIFLUORO-5-NITROPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE
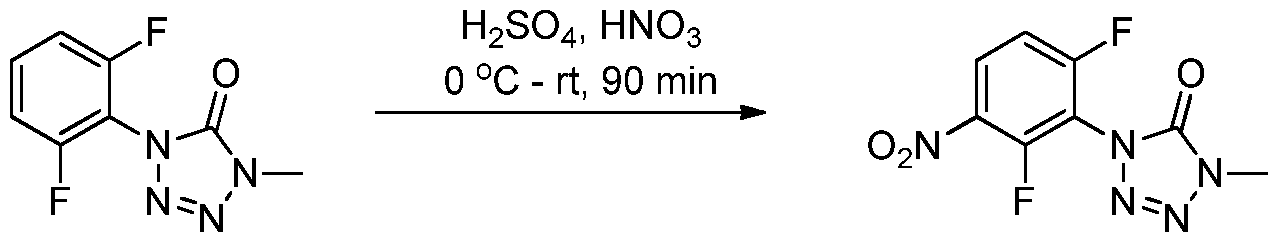
[00672] 90% HNO3 (0.37 mL, 8.3 mmol, 1.1 equiv) was added dropwise to a mixture of l-(2,6-difluorophenyl)-4-methyl-5H-tetrazol-5-one (1.6 g, 7.55 mmol, 1 equiv) in 6 mL of fuming H2SO4 at 0° C. Then the mixture was left to room temperature and stirred for 1 hour. Poured into ice and the aqueous phase was extracted with EtOAc. The organic layer was separated, washed with saturated NaHCC>3 and concentrated to give 1.98 g of product (quantitative) as a yellow solid.
[00673] JH NMR (CDC13, 300 MHz): δ 8.40-8.33 (m, 1H), 7.29 (td, / = 8.1, 1.2 Hz, 1H), 3.75 (s, 3H); m/z = 258.4 (M+H)+.
EXAMPLE 192:
1-(5-AMI -2,6-DIFLUOROPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

[00674] A mixture of l-(2,6-difluoro-5-nitrophenyl)-4-methyl-5H-tetrazol-5-one (500 mg, 1.95 mmol, 1 equiv), iron powder (543 mg, 9.73 mmol, 5 equiv) and ammonium chloride (520 mg, 9.73 mmol, 5 equiv) in iPrOH (15 mL) and water (3 mL) was refluxed for 3 hours. After cooling to room temperature, the reaction mixture was filtered through a pad of Celite, and the pad of Celite was rinsed with EtOAc (50 mL). The filtrate was washed with water. The organic layer was separated, dried over MgS04 and evaporated to give 420 mg of product (95%) as a dark oil. m/z = 228.4 (M+H)+.
EXAMPLE 193:
N2-(2,4-DIFLUORO-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)PHENYL)-5- FLUORO-N4-(1,2,2,6,6-PE -4-YL)PYRIMIDINE-2,4-DIAMINE (1-66)

[00675] Ή NMR (DMSO-d6, 300 MHz): δ 9.01 (s, 1H), 8.53 (br. s, 1H), 7.93 (d, / = 3.6 Hz, 1H), 7.75 (d, / = 6.9 Hz, 1H), 7.36 (t, / = 9.0 Hz, 1 H), 4.25-4.15 (m, 1H), 3.64 (s, 3H),
2.67 (d, / = 5.1 Hz, 3H), 1.98 (dm, / = 12.6 Hz, 2H), 1.71 (tm, / = 13.2, 12.6 Hz, 2H), 1.35 (s, 6H), 1.23 (s, 6H); m/z = 492.4 (M+H)+.
EXAMPLE 194:
N2-(2,4-DIFLUORO-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1-YL)PHENYL)-5- FLUORO-N4-(2,2,6,6-TET -4-YL)PYRIMIDINE-2,4-DIAMINE (1-67)
[00676] Ή NMR (DMSO-d6, 300 MHz): δ 8.99 (s, 1H), 8.57 (br. s, 1H), 7.92 (d, / = 3.6 Hz, 1H), 7.71 (d, / = 6.9 Hz, 1H), 7.37 (t, / = 9.3 Hz, 1 H), 4.40-4.30 (m, 1H), 3.64 (s, 3H), 1.88 (dm, / = 12.6 Hz, 2H), 1.48 (tm, / = 13.2, 12.6 Hz, 2H), 1.32 (s, 6H), 1.26 (s, 6H); m/z = 478.4 (M+H)+.
EXAMPLE 195:
2-CHLORO-4-FLUOROPHENYLISOCYANATE
Phosgene, toluene

[00677] A solution of 2-chloro-4-fluoroaniline (5.0 g, 34.35 mmol, 1 equiv) in 20% of phosgene in toluene (36 mL, 68.7- mmol, 2 equiv) was refluxed for 5 hrs. The reaction mixture was then concentrated and the resulting residue was directly used in the next step. EXAMPLE 196:
1-(2-CHLORO-4-FLUOROPHENYL)-4,5-DIHYDRO-5H-TETRAZOL-5-ONE

[00678] To the above residue of Example 195, 9.12 mL of TMSN3 (68.7 mmol, 2 equiv) was added and then the resulting mixture was heated at 100° C overnight. After being cooled to room temperature, the reaction mixture was concentrated. The residue was diluted with EtOAc (100 mL) and saturated aqueous NaHC(¾ (100 mL). The aqueous layer was separated, neutralized with 2 N HCl to pH 4-5 and extracted with EtOAc (2x100 mL). The organic layers were combined, dried over MgS04 and concentrated to give a white solid (1.8 g, 24% two steps) which was directly used in the next step, m/z = 215.3 (M+H)+ .
EXAMPLE 197:
1-(2-CHLOR -4-FLUOROPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

[00679] To a mixture of l-(2-chloro-4-fluorophenyl)-4,5-dihydro-5H-tetrazol-5-one (1.8 g, 8.4 mmol, 1 equiv) and K2C03 (3.48 g, 25.23 mmol, 3 equiv) in 25 mL of DMF was added Mel (1.57 mL, 25.23 mmol, 3 equiv) and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was separated and concentrated. The residue was triturated in water to give 1.68 g of product (88%) as a white solid, m/z = 229 A (M+H)+ .
EXAMPLE 198:
1-(2-CHLORO-4-FLUORO-5-NITROPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

[00680] 90% ΗΝΟ3 (0.19 mL, 3.86 mmol, 1.1 equiv) was added dropwise to a mixture of l-(2-chloro-4-fluorophenyl)-4-methyl-5H-tetrazol-5-one (800 mg, 3.51 mmol, 1 equiv) in 3 mL of fuming H2S04 at 0° C. Then the mixture was left to room temperature and stirred for 2 hours. Poured into ice and the aqueous phase was extracted with EtOAc. The organic layer was separated, washed with saturated NaHCC>3 and concentrated to give 980 mg of product (quantitative) as a yellow solid.
[00681] JH NMR (CDC13, 300 MHz): δ 8.31 (d, / = 7.5 Hz, 1H), 7.61 (d, / = 10.2 Hz, 1H), 3.75 (s, 3H); m/z = 274.4 (M+H)+.
EXAMPLE 199:
1-(5-AMI -2-CHLORO-4-FLUOROPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

[00682] A mixture of l-(2-chloro-4-fluoro-5-nitrophenyl)-4-methyl-5H-tetazol-5-one (600 mg, 2.2 mmol, 1 equiv), iron powder (614 mg, 10.99 mmol, 5 equiv) and ammonium chloride (588 mg, 10.99 mmol, 5 equiv) in iPrOH (10 mL) and water (2 mL) was refluxed for 3 hours. After cooling to room temperature, the reaction mixture was filtered through a
pad of Celite, and the pad of Celite was rinsed with EtOAc (50 mL). The filtrate was washed with water. The organic layer was separated, dried over MgS04 and evaporated to give the product quantitatively. m/z = 244.4 (M+H)+.
EXAMPLE 200:
N2-(2-CHLORO-4-FLUORO-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1- YL)PHENYL)-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)PYRIMIDINE-2,4-
DIAMINE (1-68)

[00683] Ή NMR (DMSO-d6, 300 MHz): δ 8.75 (br. s, 1H), 8.15 (d, / = 7.8 Hz, 1H), 7.87 (d, / = 3.6 Hz, 1H), 7.76 (d, / = 10.5 Hz, 1H), 7.29 (br. s, 1H), 4.30-4.20 (m, 1H), 3.61 (s, 3H), 1.70-1.60 (m, 2H), 1.40-1.50 (m, 2H), 1.02 (s, 12H); m/z = 494.3 (M+H)+.
EXAMPLE 201:
N2-(2-CHLORO-4-FLUORO-3-(4-METHYL-4,5-DIHYDRO-5-OXO-1H-TETRAZOL-1- YL)PHENYL)-5-FLUORO-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)PYRIMIDINE-2,4-
DIAMINE (1-69)

[00684] Ή NMR (DMSO-d6, 300 MHz): δ 8.73 (br. s, 1H), 8.15 (d, / = 6.9 Hz, 1H), 7.86 (br. s, 1H), 7.74 (d, / = 10.8 Hz, 1H), 7.28 (s, 1H), 4.20-4.10 (m, 1H), 3.62 (s, 3H), 2.12 (s, 3H), 1.64-1.58 (m, 2H), 1.37 (tm, / = 11.7 Hz, 2H), 1.02 (s, 6H), 0.84 (s, 6H); m/z = 508.3 (M+H)+.
SYNTHESIS OF 1-(4-FLUORO-5-(5-FLUORO-4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4- YLAMINO)PYRIMIDIN-2-YLAMINO)-2-((LS,2S)-2-
(TRIFLUOROMETHYL)CYCLOPROPYL)PHENYL)-4-METHYL-1H-TETRAZOL-5(4H)-ONE (I- 70)

Compound 1-70
EXAMPLE 202:
PREPARATION OF raAA¾-2-(TRiFLUOROMETHYL)cYCLOPROPYLBORONic ACID MIDA
ESTER

vinylboronic acid Trans- somer MIDA ester (racemic)
Step 1: Preparation of trifluoromethyl diazomethane
[00685] Sodium nitrite (4.6 g, 66 mmol) in water (10 mL) was added in one portion to a stirred solution of 2,2,2-trifluoroethylamine hydrochloride (8.1 g, 60 mmol) in water (25 mL) and ether (45 mL) at 0 °C. The reaction vessel was sealed with a teflon stopper and the mixture stirred from 0 °C to room temperature and stirred at room temperature for approximately 3 hours. The mixture was then partitioned in a separating funnel and the ether layer containing the product was used directly in the next step without further purification. The yield of the trifluoromethyl diazomethane product was assumed to be approximately 50% based on literature citation herein (= 3.32 g).
[00686] A safety notice for the procedure: Diazo compounds are potentially explosive. The reaction was performed behind a blast shield in glassware free from cracks or prominent scratches and glassware was inspected prior to use.
[00687] Reference for the procedure is made to /. Am. Chem. Soc. 1943, 65, 1458, which is hereby incorporated by reference in its entirety.
Step 2: Preparation of trans-2-(trifluoromethyl)cyclopropylboronic acid MIDA ester
[00688] A mixture of trifluoromethyl diazomethane (3.32 g, 30 mmol) in Et20 (45 mL) was added dropwise to a stirred suspension of vinylboronic acid MIDA ester (Sigma- Aldrich, St. Louis, MO; 1.65 g, 9.0 mmol) and Pd(OAc)2 (50 mg) in Et20 at room temperature. After adding for 10 minutes (about a quarter of the trifluoromethyl diazomethane had been added at this stage), more Pd(OAc)2 (50 mg) and Et20 (100 mL) was added, and trifluoromethyl diazomethane was added dropwise for another 20 minutes (approximately three quarters added after this time). EtOAc (50 mL) and Pd(OAc)2 (50 mg) were added at this point and the remaining trifloromethyl diazomethane was added dropwise over 10 minutes. After complete addition of the trifloromethyl diazomethane the mixture was analysed by TLC which indicated complete reaction. The solvent was removed under vacuum and the residue was dry- loaded on to silica gel and purified by column
chromatography on silica gel using EtOAc as eluent to give the product (1.45 g, 61%) as a solid. A sample was recrystallised from EtOAc, and then a small sample recrystallized again from 1 ,2-dichloroethane, to give crystals suitable for analysis by x-ray crystallography. X- ray studies confirmed the material to be the fraws-isomer.
[00689] Reference for the procedure is made to Tetrahedron Letters 2010, 51, 1009-1011, which is hereby incorporated by reference in its entirety. Reference for the procedure and procedures below is made to U.S. Provisional Patent Application Serial No. 61/418,654 (Attorney Docket No. RIGL-071PRV), entitled "Cyclopropyl MIDA Boronate," filed concurrently herewith, which is hereby incorporated by reference in its entirety.
[00690] JH NMR (DMSO-if6, 300MHz): δ 3.99-3.72 (m, 4H), 2.70 (s, 3H), 1.28 (m, 1H), 0.53 (m, 1H), 0.31 (m, 1H), 0.00 (m, 1H). 19F NMR (DMSO- , 282 MHz): -65.4
EXAMPLE 203:
PREPARATION OF raA^vs-l-(4-FLUORO-2-(2-(TRiFLUOROMETHYL)cYCLOPROPYL)-5-
NITROPHENYL)-4-METHYL-lH-TETRAZOL-5(4H)-ONE
Pd(OAc)2, Cy3P,
Cs2C03, toluene, Η20

Trans-isomer
(racemic)
[00691] A mixture of l-(2-bromo-4-fluoro-5-nitrophenyl)-4-methyl- lH-tetrazol-5(4H)- one (145 mg, 0.46 mmol), £ra«s-2-(trifluoromethyl)cyclopropylboronic acid MIDA ester (240 mg, 0.91 mmol), Pd(OAc)2 (20 mg, 0.09 mmol), Cy3P (50 mg, 0.18 mmol) and Cs2C03 (593 mg, 1.82 mmol) in toluene (6 mL) and H20 (2 mL) was de-gassed with N2 for 10 minutes, then placed under a nitrogen atmosphere and heated to 80 °C overnight (a reflux condenser was used in the apparatus). After completion of the reaction (Note: TLC showed product and starting material to be very close), the mixture was cooled and partitioned between EtOAc (50 mL) and H20 (50 mL). The aqueous and organic layers were partitioned and the organic layer was washed with brine (1 x 20 mL), dried (MgS04), filtered and the solvent removed under vacuum to leave a crude residue. The residue was dry-loaded on to silica gel and purified by column chromatography on silica gel using EtOAc / hexane (3 :7 to 4:6) as eluent to give the product (107 mg, 67%).
[00692] The above reaction was also undertaken starting with 106mg of l-(2-bromo-4- fluoro-5-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one to give the product (55 mg, 47%).
[00693] JH NMR (CDC13, 300MHz): δ 8.19 (dd, 1H), 7.22 (d, 1H), 3.74 (s, 3H), 2.69- 2.62 (m, 1H), 1.78-1.69 (m, 1H), 1.50- 1.43 (m, 1H), 1.29- 1.22 (m, 1H). 19F NMR (CDC13, 282 MHz): -67.6, - 112.9. m/z = 389.2 (M+MeCN+H)+
EXAMPLE 204:
PREPARATION OF 77fAA¾-l-(5-AMiNO-4-FLUORO-2-(2-
(TRIFLUOROMETHYL)CYCLOPROPYL)PHENYL)-4-METHYL-lH-TETRAZOL-5(4H)-ONE

[00694] Palladium on charcoal, wet (Degussa grade E101 ; 29 mg) was added to a mixture of ira«5- l-(4-fluoro-2-(2-(trifluoromethyl)cyclopropyl)-5-nitrophenyl)-4-methyl-lH- tetrazol-5(4H)-one (145 mg, 0.46 mmol), EtOH (10 mL) and AcOH (75 μν> under nitrogen. The mixture was evacuated and filled with hydrogen - this procedure was repeated another two times. The mixture was hydrogenated at 30 psi for 7 days (topping-off the hydrogen if necessary). More Pd on charcoal, wet (Degussa grade E101 ; 15 mg) was added and the mixture hydrogenated at 30 psi for another 10 days (LC/MS was used to monitor the progression of the reaction over the 2 week experiment). After complete reaction, the mixture was filtered through a small plug of Celite and the filter cake washed with EtOH (3 x 10 mL). The filtrate was concentrated under vacuum (dry-loaded on to silica) and purified by column chromatography on silica gel using EtOAc / hexane (1 :4 to 2:3) as eluent to give the product (117 mg, 89%) as a solid.
[00695] Ή NMR ( -DMSO, 300MHz): δ 6.99 (d, 1H), 6.78 (dd, 1H), 5.53 (br. s 3.58 (s, 3H), 2.18-2.11 (m, 1H), 1.92- 1.87 (m, 1H), 1.23- 1.09 (m, 2H). 19F NMR (d6
DMSO, 282 MHz): -65.5, -132.0. m/z = 318.1 (M+H)+
EXAMPLE 205:
PREPARATION OF raA^vs-l-(5-(4-(2,2,6,6-TETRAMETHYLPiPERiDiN-4-YLAMiNo)-5-
FLUOROPYRIMIDIN-2-YLAMINO)-4-FLUORO-2(2-
(TRIFLUOROMETHYL)CYCLOPROPYL)PHENYL)-4-METHYL-lH-TETRAZOL-5(4H)-ONE (I-

[00696] A mixture of tra«s-l-(5-arnino-4-fluoro-2-(2-
(trifluoromethyl)cyclopropyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one (110 mg, 0.35 mmol), 2-chloro-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-4-pyrimidineamine hydrochloride (112 mg, 0.35 mmol) and /?ara-toluenesulfonic acid monohydrate (66 mg, 0.35 mmol) in IPA (7.5 mL) was heated to reflux and stirred for 7 days. After allowing to cool, 3- aminobenzoic acid (100 mg) added and the mixture stirred at reflux overnight. After cooling, the mixture was concentrated under vacuum and the residue portioned between EtOAc (30 mL) and IN NaOH (30 mL). The aqueous and organic layers were partitioned and the organic layer was dried (MgS04), filtered and the solvent removed under vacuum to leave a residue (LC/MS indicates this to be product and unreacted aniline). The residue was triturated with Et20 and the emerging precipitate was filtered and the filter cake washed with Et20 to give the product (48 mg, 24%) as a solid. [Note: there is still a lot of product in the filtrate].
[00697] JH NMR ( -DMSO, 300MHz): δ 8.56 (br. s, 1H), 7.92 (d, 1H), 7.84 (d, 1H), 7.25-7.19 (m, 2H), 4.22 (m, 1H), 3.58 (s, 3H), 2.23 (m, 1H), 2.02 (m, 1H), 1.57 (m, 2H), 1.26 (m, 2H), 1.14-1.07 (m, 2H), 0.97 (s, 12H). 19F NMR ( -DMSO, 282 MHz): -65.6, - 120.1, -165.8. mJz = 568.7 (M+H)+
SYNTHESIS OF 1-(4-FLUORO-3-(5-FLUORO-4-(2,2,6,6-TETRAMETHYLPIPERIDLN-4- -2-YLAMINO)PHENYL)-4-METHYL-1H-TETRAZOL-5(4H)-ONE

EXAMPLE 206:
PREPARATION -(4-FLUORO-3-NiTROPHENYL)-lH-TETRAZOL-5(4H)-ONE

[00698] A mixture of 4-fluoro-3-nitro-benzoyl chloride (2.04 g, 10 mmol) and azidotrimethylsilane (7.9 mL, 60 mmol) was slowly heated to 80-90 °C and the mixture was stirred overnight. The reaction was performed behind a blast shield. After allowing to cool, the solvent was removed under vacuum and the residue partitioned between EtOAc (50 mL) and ¾0 (50 mL). The organic layer was extracted with saturated NaHCC>3 (3 x 50 mL - until TLC indicated all desired product removed from organic layer). The aqueous extracts were combined and EtOAc (100 mL) was added. The mixture was acidified to pH < 2 using 6M HC1. The aqueous and organic layers were partitioned and the organic layer was dried (MgS04), filtered and the solvent removed under vacuum to leave the product (1.96 g, 78%) as a solid.
[00699] JH NMR ( -DMSO, 300MHz): δ 8.64-8.62 (m, 1H), 8.28-8.22 (m, 1H), 7.83- 7.77 (m, 1H). 19F NMR (if6-DMSO, 282 MHz): -119.7. m/z = 224.0 (M-H)+
EXAMPLE 207:
PREPARATION OF l-(4-FLUORO-3-NiTROPHENYL)-4-METHYL-lH-TETRAZOL-5(4H)-ONE

[00700] K2CO3 (2.82 g, 20.4 mmol) was added in one portion to a stirred mixture of l-(4- fluoro-3-nitrophenyl)-lH-tetrazol-5(4H)-one (1.84 g, 8.2 mmol) and Mel (2.32 g, 16.3 mmol) in DMF (17.5 mL) under nitrogen at -78 °C. The mixture was allowed to warm to room temperature overnight. Analysis by TLC indicated complete reaction, so the mixture was poured in to EtOAc (50 mL) and ¾0 (75 mL). The aqueous and organic layers were partitioned and the organic layer was washed with ¾0 (2 x 75 mL). The combined aqueous layers were back-extracted with EtOAc (1 x 50 mL) and the combined organic extracts were dried (MgS04), filtered and the solvent removed under vacuum to leave a crude residue. The residue (dry- loaded on to silica) was purified by column chromatography on silica gel using EtOAc / hexane (3:7 to 4:6) as eluent to give the product (1.68 g, 86%) as a solid.
[00701] Ή NMR ( -DMSO, 300ΜΗζ): δ 8.62-8.58 (m, 1H), 8.28-8.22 (m, 1H), 7.84- 7.77 (m, 1H), 3.62 (s, 3H). 19F NMR ( -DMSO, 282 MHz): -119.2. m/z = 281.1
(M+MeCN+H)+
EXAMPLE 208:
PREPARATION OF l-(3-AMiNO-4-FLUOROPHENYL)-4-METHYL-lH-TETRAZOL-5(4H)-ONE

[00702] Palladium on charcoal, wet (Degussa grade E101; 25 mg) was added to a mixture of l-(4-fluoro-3-nitrophenyl)-4-methyl-lH-tetrazol-5(4H)-one (120 mg, 0.5 mmol), EtOH (15 mL) and AcOH (120 \L) under nitrogen. The mixture was evacuated and filled with hydrogen - this procedure was repeated another two times. The mixture was hydrogenated at 30 psi overnight (LC/MS was used to monitor the progression of the reaction). After complete reaction, the mixture was filtered through a small plug of Celite and the filter cake washed with EtOH. The filtrate was concentrated under vacuum to give the product (106 mg, 100%) as a solid.
[00703] Ή NMR (_fc-DMSO, 300MHz): δ 7.27 (m, IH), 7.16-7.10 (m, IH), 6.97-6.92 (m, IH), 5.53 (br. s, 2H), 3.58 (s, 3H). 19F NMR (_fc-DMSO, 282 MHz): -135.6. m/z =
210.0 (M+H)+
EXAMPLE 209:
PREPARATION OF 1-(3-(4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YLAMINO)-5-
FLUOROPYRIMIDIN-2-YLAMINO)-4-FLUOROPHENYL)-4-METHYL-LH-TETRAZOL-5(4H)-ONE
(1-

[00704] A mixture of l-(3-amino-4-fluorophenyl)-4-methyl-lH-tetrazol-5(4H)-one (96 mg, 0.46 mmol), 2-chloro-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-4- pyrimidineamine hydrochloride (141 mg, 0.44 mmol) and /?ara-toluenesulfonic acid monohydrate (84 mg, 0.44 mmol) suspended in IPA (7.5 mL) was heated to reflux and stirred for 3 days. The mixture was then cooled, and 3-aminobenzoic acid (100 mg) was added and the mixture stirred at reflux overnight. After allowing to cool, the mixture was concentrated under vacuum and the residue partitioned between saturated NaHCC>3 (30 mL) and EtOAc (30 mL). The aqueous and organic layers were partitioned and the organic layer was washed with saturated NaHCC>3 (1 x 30 mL), dried (MgS04), filtered and the solvent removed under vacuum to leave a crude residue (ca. 180 mg). The residue was triturated with Et20 (solid emerges) and the Et20 mixture cooled in a -18 °C freezer for 15 minutes before filtering. The filter cake was washed with Et20 (2 x 10 mL) to give the product (78 mg, 39%) as a solid.
[00705] Ή NMR (_fc-DMSO, 300MHz): δ 8.62 (br. s, IH), 8.29 (m, IH), 7.85 (d, IH), 7.42-7.32 (m, 2H), 7.17 (d, IH), 4.21 (m, IH), 3.57 (s, 3H), 1.57 (m, 2H), 1.10-1.02 (m, 2H), 0.95 (s, 6H), 0.90 (s, 6H). 19F NMR (_½-DMSO, 282 MHz): -124.2, -165.8. m/z = 460.4
(M+H) +
SYNTHESIS OF 1-(2-CYCLOPROPYL-4-FLUORO-5-(5-FLUORO-4-(2,2,6,6- TETRAMETHYLPIPERIDIN-4-YLAMINO)PYRIMIDIN-2-YLAMINO)PHENYL)-4-
(DEUTERIOMETHYL)-1H-TETRAZOL-5(4H)-ONE (1-72)

ompound I-'
EXAMPLE 210:
PREPARATION OF 1-(2-CYCLOPROPYL-4-FLUORO-5-NITROPHENYL)-4-DEUTERIOMETHYL- lH-TETRAZO -5(4H)-ONE

[00706] K2CO3 (276 mg, 2.0 mmol) was added in one portion to a stirred mixture of l-(2- cyclopropyl-4-fluoro-5-nitrophenyl)-lH-tetrazol-5(4H)-one (265 mg, 1.0 mmol) and d3- iodomethane (Aldrich; 187 ^L, 3.0 mmol) in DMF (3 mL) under nitrogen at -78 °C (mixture solidified after K2CO3 addition). The mixture was allowed to warm to room temperature overnight. The mixture was poured in to EtOAc (50 mL) and ¾0 (50 mL). The aqueous and organic layers were partitioned and the organic layer was washed with ¾0 (2 x 30 mL), then brine (1 x 30 mL). The organic layer was dried (MgS04), filtered and the solvent removed under vacuum to leave the product (257 mg, 91%) as a solid.
[00707] Ή NMR ( -DMSO, 300ΜΗζ): δ 8.38 (d, 1H), 7.29 (d, 1H), 1.88-1.82 (m, 1H), 1.10-1.04 (m, 2H), 0.96-0.91 (m, 2H) [Note: no Me peak in JH NMR as CD3 analogue]. 19F NMR (ife-DMSO, 282 MHz): -115.3. m/z = 283.1 (M+H)+
EXAMPLE 211:
PREPARATION OF 1-(5-AMINO-2-CYCLOPROPYL-4-FLUOROPHENYL)-4-DEUTERIOMETHYL- lH-TETRAZOL-5(4H)-ONE

[00708] Palladium on charcoal, wet (Degussa grade E101; 50 mg) was added to a mixture of l-(2-cyclopropyl-4-fluoro-5-nitrophenyl)-4-deuteriomethyl-lH-tetrazol-5(4H)-one (250 mg, 0.89 mmol) in EtOH (15 mL) under nitrogen. The mixture was evacuated and filled with hydrogen - this procedure was repeated another two times. The mixture was hydrogenated at 30 psi for 3-4 hours (LC/MS was used to monitor the progression of the reaction). After complete reaction, the mixture was filtered through a small plug of Celite and the filter cake washed with EtOH (3 x 20 mL). The filtrate was concentrated under vacuum to give the product, which was used directly in the next step (yield assumed quantitative = 222 mg).
[00709] mJz = 253.1 (M+H)+
EXAMPLE 212:
PREPARATION OF A^2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-DEUTERIOMETHYL)-1,2,3,4- TETRAZOL-5-ONE-1-YL]}PHENYL-5-FLUORO-A^4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-

[00710] A mixture of l-(5-amino-2-cyclopropyl-4-fluorophenyl)-4-deuteriomethyl-lH- tetrazol-5(4H)-one (222 mg, 0.89 mmol), 2-chloro-5-fluoro-N4-(2,2,6,6- tetramethylpiperidin-4-yl)-4-pyrimidineamine hydrochloride (270 mg, 0.83 mmol) and para- toluenesulfonic acid monohydrate (159 mg, 0.83 mmol) suspended in IPA (5 mL) was heated to reflux and stirred for 2 days. The mixture was then cooled, and 3-aminobenzoic acid (100 mg) was added and the mixture stirred at reflux overnight. After allowing to cool, the mixture was concentrated under vacuum and the residue partitioned between IN NaOH
(50 mL) and EtOAc (50 mL). The aqueous and organic layers were partitioned and the organic layer was washed with brine (1 x 30 mL), dried (MgS04), filtered and the solvent removed under vacuum to leave a crude residue. The residue was triturated with MeCN and filtered to give the product (140 mg, 34%) as a solid.
[00711] JH NMR (_fc-DMSO, 300MHz): δ 8.49 (br. s, 1H), 7.81-7.76 (m, 2H), 7.19 (d, 1H), 6.94 (d, 1H), 4.21 (m, 1H), 1.65-1.56 (m, 3H), 1.30-1.05 (m, 2H), 0.98-0.96 (s, 12H), 0.81-0.78 (m, 2H), 0.59-0.57 (m, 2H) [Note: no Me peak in JH NMR as CD3 analogue]. 19F NMR (ife-DMSO, 282 MHz): -120.1, -166.8. m/z = 503.4 (M+H)+
EXAMPLE 213:
L-(2-CYCLOPROPYL-4-FLUORO-5- -4-ISOPROPYL-LH-TETRAZOL-5(4H)-ONE

[00712] l-(2-Cyclopropyl-4-fluoro-5-nitrophenyl)-lH-tetrazol-5(4H)-one (500mg, 1.9 mmol) was dissolved into DMF (9.4ml), the solution was chilled to -70 °C. K2CO3 (780mg, 5.7 mmol) was added to this solution and followed by 2-iodopropane (0.23ml, 2.28 mmol). The mixture was allowed to warm up to room temperature overnight with stirring. Ethyl acetate and brine were added to this mixture, the 2 layers were separated. The organic layer was dried with Na2S04, filtered off solid and concentrated under reduced pressure to give the product (208mg, 75% pure).
[00713] JH NMR (300 MHz, DMSO- ) δ 8.43 (d, /=6.9Hz, 1H), 7.33 (d, /=12.6Hz, 1H), 4.45 (m, 1H), 1.82 (m, 1H), 1.47 (s, 3H), 1.45 (s, 3H), 1.05 (m, 2H), 0.95 (m, 2H); LCMS (m/z): 307.11 (MH+).
EXAMPLE 214:
L-(5-AMINO-2-CYCLOPROPYL-4-FLUOROPHENYL)-4-ISOPROPYL-LH-TETRAZOL-5(4H)-
ONE

[00714] l-(2-Cyclopropyl-4-fluoro-5-nitrophenyl)-4-isopropyl-lH-tetrazol-5(4H)-one (208mg, OJmmol) was added to ethyl alcohol (4.8ml), SnCl2.2H20 (501mg, 2.7mmol) and cone. HC1 (0.6ml) were added to the solution. The mixture was heated at 80 °C for 1 hour.
The solution was allowed to cool to room temperature, and concentrated under reduced pressure. Ethyl acetate was added and washed with saturated K2CO3, the 2 layers were separated. The organic layer was washed with brine and dried with Na2S04. Solid was filtered off and mother liquor was concentrated under reduced pressure to give the product (140 mg) as a white solid.
[00715] JH NMR (300 MHz, DMSO- ) δ 6.83 (d, /=12.3Hz, IH), 6.75 (d, /=8.7Hz, IH), 5.36 (s, 2H), 4.43 (m, IH), 1.54 (m, IH), 1.44 (s, 3H), 1.42 (s, 3H), 0.65 (m, 2H), 0.43 (m, 2H); LCMS (m/z): 278.11 (MH+).
EXAMPLE 215:
N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-ISOPROPYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE (1-73)

[00716] Ή NMR (DMSO-i¾, 300MHz): δ 8.49 (s, IH), 7.81 (bs, IH), 7.76 (d, / = 7.2Hz, IH), 7.19 (bs, IH), 6.99 (d, /=11.7Hz, IH), 4.43 (m, IH), 4.25 (bs, IH), 1.6 (m, 2H), 1.45 (s, 3H), 1.43 (s, 3H), 1.51 (m, 2H), 0.97 (m, 12H), 0.77 (m, 2H), 0.53 (m, 2H); LCMS (m/z): 528.40 (MH+).
EXAMPLE 216:
L-(5-AMINO-2-CYCLOPROPYL-4-FL -LH-TETRAZOL-5(4H)-ONE

[00717] l-(2-Cyclopropyl-4-fluoro-5-nitrophenyl)-4-H-lH-tetrazol-5(4H)-one (145mg, 0.5mmol) was added to ethyl alcohol (4.0ml), SnCl22H20 (409mg, 2.2mmol) and concentrated HCl (0.47ml) were added to the solution. The mixture was heated at 80 °C for 1 hour. The solution was allowed to cool to room temperature, and concentrated under reduced pressure. Ethyl acetate was added and washed first with water (2x10ml), and then brine (10ml). The 2 layers were separated and the organic layer was dried with Na2S04. Solid was filtered off and mother liquor was concentrated under reduced pressure to give the product (95.1mg, 74% yield) as a white solid.
[00718] Ή NMR (300 MHz, DMSO- ) δ 6.8 (d, /=12.6Hz, 1H), 6.72 (d, /=8.4Hz, 1H), 5.33 (s, 2H), 1.56 (m, 1H), 0.67 (d, /=7.8Hz, 2H), 0.45 (d, /=4.5Hz, 2H); LCMS (m/z): 236.05 (MH+).
EXAMPLE 217:
N2-{4-CYCLOPROPYL-6-FLUORO-3-(1,2,3,4-TETRAZOL-5-ONE-1-YL)}PHENYL-5-FLUORO- N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4-PYRIMIDINEDIAMINE, FORMATE SALT (I- 74)

[00719] Ή NMR (DMSO-i¾, 300MHz): δ 8.88 (bs, 1H), 8.55 (d, /=12.3Hz, 1H), 7.95 (d, J=3.3Hz, 1H), 7.69 (d, / = 7.5Hz, 1H), 7.45 (d, /=7.5Hz, 1H), 7.09 (d, /=7.5Hz, 1H), 7.03 (d, /=11.7Hz, 1H), 4.26 (m, 1H), 2.71 (bs, 1H), 1.84 (d, /=11.7Hz, 2H), 1.66 (m, 1H), 1.49 (t, /=12.9Hz, 2H), 1.32 (s, 6H), 1.21 (s, 6H), 0.85 (m, 2H), 0.6 (m, 2H); LCMS (m/z): 486.13 (MH+).
EXAMPLE 218:
N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE CITRATE SALT (1-75)
[00720] N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl- 5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine (50mg) was dissolved into isopropyl alcohol (4ml) in a 20ml scintillation vial, citric acid monohydrate (21.1mg, 1.0 eq.) was added to the solution, solid formed immediately. The turbid solution was heated to 60 °C in an oil bath until the solution turned clear, turned off the heat and the vial was left in the oil bath until the temperature returned to room temperature (25 °C). The vial was removed from the bath and left in the cabinet for two days. The product was collected by filtering through a Buchner funnel fitted with filter paper, and pumped under reduced pressure overnight to remove residual solvent, gave 40 mg of white solid.
[00721] JH NMR (300 MHz, DMSO- ) δ 8.61 (s, 1H), 7.88 (s, 1H), 7.72 (d, /=7.2Hz, 1H), 7.49 (d, /=7.8Hz, 1H), 6.99 (d, /=12.3Hz, 1H), 4.31 (bs, 1H), 3.6 (s, 3H), 1.84 (d, /=13.5Hz, 2H), 1.66 (m, 1H), 1.45 (t, /=12.6Hz, 2H), 1.29 (s, 6H), 1.21 (s, 6H), 0.83 (m, 2H), 0.61 (m, 2H); LCMS (m/z): 500.20 (MH+)
EXAMPLE 219:
N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL] }PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE MALEATE SALT (1-76)
[00722] N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l ,2,3,4-tetrazol-5-one- l-yl] }phenyl- 5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4^yrimidinediamine (lOOmg) was suspended in water (4ml), maleic acid (23.3mg, l .Oeq.) was added to the suspension, a clear solution formed. A few minutes later tiny crystals can be seen forming at the bottom of vial. The vial was left inside the cabinet for 1 week. Product was collected by filtering off water, and dried under reduced pressure overnight, gave 85 mg of white solid.
[00723] JH NMR (300 MHz, DMSO- ) δ 8.62 (s, 1H), 8.48 (d, /=13.2Hz, 1H), 7.88 (d, /=3.9Hz, 1H), 7.71 (d, /=6.9Hz, 1H), 7.65 (bs, 1H), 7.51 (d, /=7.5Hz, 1H), 6.99 (d, /=12.3Hz, 1H), 5.99(s, 2H), 4.31 (bs, 1H), 3.6 (s, 3H), 1.86 (d, /=12Hz, 2H), 1.65 (m, 1H), 1.49 (t, /=12.3Hz, 2H), 1.31 (s, 6H), 1.22 (s, 6H), 0.83 (m, 2H), 0.6 (m, 2H); LCMS (m/z): 500.20 (MH+)
EXAMPLE 220:
N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL] }PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE FUMARATE SALT (2:1 RATIO) (1-77)
[00724] N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l ,2,3,4-tetrazol-5-one- l-yl] }phenyl- 5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine (50 mg) was suspended in 2 ml of ¾0 in a 20ml scintillation vial, fumaric acid (11.65 mg, 1.0 eq.) was added, a clear solution was obtained. The vial was left for 1 week at room temperature inside the cabinet, white stringy solid formed. The product was collected by filtering through a Buchner funnel fitted with filter paper and then pumped overnight under reduced pressure to dry, gave around 15 mg of white solid.
[00725] JH NMR (300 MHz, DMSO- ) δ 8.52 (s, 1H), 7.84 (bs, 1H), 7.75 (d, /=7.5Hz, 1H), 7.34 (bs, 1H), 6.97 (d, /=12Hz, 1H), 6.38 (s, 1H), 4.24 (bs, 1H), 3.6 (s, 3H), 1.67 (m, 3H), 1.29 (t, /=11.4Hz, 2H), 1.14 (s, 6H), 1.09 (s, 6H), 0.83 (m, 2H), 0.6 (m, 2H); LCMS (m/z): 500.20 (MH+)
EXAMPLE 221:
N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE L-TARTARATE SALT (1-78)
[00726] N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl- 5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine (50mg) in a 20 ml scintillation vial was dissolved into isopropyl alcohol (3ml) at 55 °C in an oil bath. l.Oeq of L-tartaric acid (15.3 mg, 1.0 eq.) was added, without stirring, temperature increased to 75 °C (salt precipitates at about 55 °C) to minimize quick salt formation. Heating was turned off after 10 minutess at 75 °C and vial was left inside the oil bath to cool down overnight. White solid was obtained after filtering off the solvent and was dried under reduced pressure for 3 days, and then overnight at 60 °C under high vacuum to give 30 mg of white solid.
[00727] JH NMR (300 MHz, DMSO- ) δ 8.53 (s, 1H), 7.85 (d, /=3.6Hz, 1H), 7.75 (d, /=7.8Hz, 1H), 7.35 (bs, 1H), 6.98 (d, /=12Hz, 1H), 4.25 (bs, 1H), 3.73 (s, 2H), 3.6 (s, 3H), 1.69 (m, 3H), 1.28 (m, 2H), 1.14 (s, 6H), 1.09 (s, 6H), 0.81 (m, 2H), 059 (m, 2H); LCMS (m/z): 500.20 (MH+)
EXAMPLE 222:
N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE HYDROGEN SULFATE SALT (1-79)
[00728] N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl- 5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine (100 mg) was suspended in water (2ml), IN H2SO4 (0.2 ml, 1.0 eq.) was added, a clear solution was formed. The hydrogen sulfate salt was obtained by lyophilizing for 2 days and confirmed by 1H NMR. This salt (50mg) was added to a 20ml scintillation vial and dissolved in 10 ml of MeOH, the solution was filtered through filter paper to ensure clear solution were obtained. The vial was put into a glass jar filled with THF, kept in the cabinet with the lid closed. After 3 days, with no signs of crystal formation, THF was poured out and replaced with diethyl ether. The jar was again kept with the lid closed and inside the cabinet for 9 days, long needle shaped crystals can be seen forming at the bottom and the wall of the vial. The solution from the vial was carefully poured out onto a funnel fitted with filter paper. The solid (15mg) collected was washed with ether, and dried under reduced pressure for 48 hours.
[00729] Ή NMR (300 MHz, DMSO- ) δ 8.56 (s, IH), 7.86 (d, /=3.6Hz, IH), 7.74 (d, /=7.8Hz, IH), 7.41 (bs, IH), 6.99 (d, /=12.3Hz, IH), 4.28 (bs, IH), 3.6 (s, 3H), 1.74 (m, 2H), 1.65 (m, IH), 1.14 (m, 14H), 0.82 (m, 2H), 0.59 (m, 2H); LCMS (m/z): 500.20 (MH+) EXAMPLE 223:
N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL] }PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE HYDROGEN CHLORIDE SALT (1-80)
[00730] N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l ,2,3,4-tetrazol-5-one- l-yl] }phenyl- 5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4^yrimidinediamine (100 mg) suspended in water in a 20 ml scintillation vial, 1 N HCl (0.2 ml, 1.0 eq.) was added and a clear solution was obtained. Water was removed by lyophilizing over two days to give white solid. Salt formation was confirmed by 1H NMR, 50mg of this salt was dissolved completely into ethyl alcohol (6 ml) in a 20 ml scintillation vial and it was placed into a glass TLC chamber filled with EtOAc. After 3 days with no signs of crystal formation, EtOAc was removed from the TLC chamber, and replaced with ether. The TLC chamber was closed with a glass top, left inside the cabinet. Crystals started to form at the bottom of the vial after sitting inside the cabinet for two days and it was left there for around 9 days. The vial was carefully removed from the chamber and the solid was collected by filtering through a funnel fitted with filter paper. The solid (30 mg) was dried under reduced pressure for 48 hours.
[00731] JH NMR (300 MHz, DMSO-i¾ δ 8.9 (bs, IH), 8.61 (s, IH), 7.88 (d, /=3.6Hz, IH), 7.72 (d, /=7.8Hz, IH), 7.52 (d, /=7.2Hz, IH), 6.99 (d, /=12Hz, IH), 4.33 (bs, IH), 3.6 (s, 3H), 1.83 (d, /=11.7Hz, 2H)1.66 (m, IH), 1.5 (t, /=13.2Hz, 2H), 1.34 (s, 6H), 1.23 (s, 6H), 0.81 (m, 2H), 0.6 (m, 2H); LCMS (m/z): 500.20 (MH+)
EXAMPLE 224:
N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL] }PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE BENZOATE SALT (1-81)
[00732] N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l ,2,3,4-tetrazol-5-one- l-yl] }phenyl- 5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine (lOOmg) was suspended in water, benzoic acid (24.5 mg, 1.0 eq) was added. Solution remained milky even after sonication, left in cabinet for 1 week. Filtered off solid to get a clear solution, small sparkling crystal started to form in mother liqior while sitting inside the cabinet. It was left undisturbed for 2 more weeks, filtered to get around 25 mg of solid.
[00733] Ή NMR (300 MHz, DMSO- ) δ 8.49 (s, IH), 7.9 (d, /=6.9Hz, 2H), 7.83 (d, /=3.6Hz, IH), 7.77 (d, /=8.1Hz, IH), 7.53 (m, IH), 7.43 (m, 2H), 7.24 (d, /=7.8Hz, IH), 6.97 (d, J=12.3Hz, IH), 4.26 (bs, IH), 3.6 (s, 3H), 1.63 (m, 3H), 1.17 (t, /=12Hz, 2H), 1.05 (s, 6H), 1.02 (s, 6H), 0.81 (m, 2H), 0.59 (m, 2H); LCMS (m/z): 500.20 (MH+)
EXAMPLE 225:
N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE TOSYLATE SALT (1-82)
[00734] N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl- 5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4^yrimidinediamine (50mg) in a 20 ml scintillation vial was dissolved into anhydrous THF (lml), para-TSA (19.1 mg, 1.0 eq.) was added, a clear solution was obtained, small crystal started to form at the bottom of vial. It was left inside the cabinet for 3 days, removed THF by filtering through Buchner funnel fitted with filter paper. White solid (40mg) collected was dried under reduced pressure for 2 days.
[00735] JH NMR (300 MHz, DMSO- ) δ 8.62 (s, IH), 8.49 (d, /=12.6Hz, IH), 7.89 (d, /=3.6Hz, IH), 7.72 (d, /=7.8Hz, IH), 7.51 (d, J=8.1Hz, IH), 7.46 (d, J=7.8Hz, 2H), 7.09 (d, /=8.4Hz, 2H), 7.0 (d, /=12Hz, IH), 4.3 (bs, IH), 3.6 (s, 3H), 2.27 (s, 3H), 1.86 (d, /=12Hz, 2H), 1.67 (m, IH), 1.48 (t, /=11.7Hz, 2H), 1.31 (s, 6H), 1.22 (s, 6H), 0.83 (m, 2H), 0.60 (m, 2H); LCMS (m/z): 500.20 (MH+)
EXAMPLE 226:
N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL]}PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)-2,4- PYRIMIDINEDIAMINE BESYLATE SALT (1-83)
[00736] N2- { 4-Cyclopropyl-6-fluoro- [3 -(4-methyl)- 1,2,3 ,4-tetrazol-5 -one- 1 -yl] Jphenyl- 5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine (50mg) in a 20 ml scintillation vial was dissolved into anhydrous THF (1 ml), benzenesulfonic acid (15.8 mg, 1.0 eq.) was added, a clear solution was obtained, no crystal formed right away. It was left inside the cabinet for 3 days, crystals formed at the bottom of the vial. It was collected after THF was removed by filtering through Buchner funnel fitted with filter paper. White solid (25 mg) collected was pumped under reduced pressure for 2 days.
[00737] JH NMR (300 MHz, DMSO-i¾) δ 8.62 (s, IH), 8.49 (d, /=12.6Hz, IH), 7.89 (d, /=3.6Hz, IH), 7.72 (d, /=7.5Hz, IH), 7.59 (m, 3H), 7.3 (m, 2H), 7.0 (d, /=12Hz, IH), 4.31
(bs, IH), 3.6 (s, 3H), 1.86 (d, /=13.8Hz, 2H), 1.66 (b,s, IH), 1.48 (t, /=12.3Hz, 2H), 1.31 (s, 6H), 1.22 (s, 6H), 0.82 (m, 2H), 0.6 (m, 2H); LCMS (m/z): 500.20 (MH+)
EXAMPLE 227:
N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL] }PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDLN-4-YL)-2,4- PYRIMIDINEDIAMINE MESYLATE SALT (1-84)
[00738] N2- {4-Cyclopropyl-6-fluoro-[3-(4-methyl)- l,2,3,4-tetrazol-5-one-l-yl] }phenyl- 5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine (50 mg) in a 20 ml scintillation vial dissolved into 1ml of anhydrous THF, methanesulfonic acid (6.5 ul, 1.0 eq) was added, a milky solution formed, heated at 70 °C until clear solution obtained, cooled to room temperature in oil bath. Filtered off liquid the next day and dried under reduced pressure for 3 days, gave around 38 mg of white solid.
[00739] JH NMR (300 MHz, DMSO- ) δ 9.07 (s, IH), 8.54 (d, /=10.5Hz, IH), 8.08 (s, IH), 8.01 (s, IH), 7.73 (s IH), 7.68 (d, /=7.8Hz, IH), 7.05 (d, /=11.7Hz, IH), 4.28 (bs, IH), 3.6 (s, 3H), 2.29 (s, 3H), 1.83 (d, /=12.6Hz, 2H), 1.68 (m, IH), 1.50 (t, /=12.6Hz, 2H), 1.32 (s, 6H), 1.19 (s, 6H), 0.85 (m, 2H), 0.63 (m, 2H); LCMS (m/z): 500.20 (MH+)
EXAMPLE 228:
N2-{4-CYCLOPROPYL-6-FLUORO-[3-(4-METHYL)-1,2,3,4-TETRAZOL-5-ONE-1- YL] }PHENYL-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDLN-4-YL)-2,4- PYRIMIDINEDIAMINE ACETATE SALT (1-85)
[00740] N2- {4-Cyclopropyl-6-fluoro-[3-(4-methyl)- l,2,3,4-tetrazol-5-one-l-yl] }phenyl- 5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine (50mg) was dissolved into anhydrous THF in a 20 ml scintillation vial. Acetic acid (5.7 ul, 1.0 eq.) was added to the solution, a clear solution formed, this solution was put inside the refrigerator for 4 days. Solid formed at the bottom of the vial was collected by filtering off THF through filter paper to obtain white solids (25 mg). It was dried under reduced pressure for 4 days.
[00741] JH NMR (300 MHz, DMSO- ) δ 8.46 (s, IH), 7.81 (m, 2H), 7.17 (d, /=8.4Hz, IH), 6.96 (d, /=12Hz, IH), 4.23 (bs, IH), 3.6 (s, 2H), 1.88 (s, 3H), 1.60 (m, 3H), 1.09 (t, /=12Hz, 2H), 0.98 (s, 12H), 0.81 (m, 2H), 0.59 (m, 2H); LCMS (m/z): 500.20 (MH+)
EXAMPLE 229:
4-FLUORO-2-M
 [00742] A solution of 5-fluoro-2-nitroanisole (5.0 g, 29.22 mmol, 1 equiv) in the presence of 250 mg of Pd/C (5%) in MeOH (60 mL) was hydrogenated at room temperature for 6 hours. The reaction mixture was filtered over a pad of Celite and then concentrated to give a brown oil (4.06 g, 98%) which was directly used in the next step. mJz = 142 (M+H)+.
[00742] A solution of 5-fluoro-2-nitroanisole (5.0 g, 29.22 mmol, 1 equiv) in the presence of 250 mg of Pd/C (5%) in MeOH (60 mL) was hydrogenated at room temperature for 6 hours. The reaction mixture was filtered over a pad of Celite and then concentrated to give a brown oil (4.06 g, 98%) which was directly used in the next step. mJz = 142 (M+H)+.
EXAMPLE 230:
4-FLUORO-2-METHOXYPHENYLISOCYANATE
Phosgene, toluene

[00743] A solution of 4-fluoro-2-methoxyaniline (2.0 g, 14.2 mmol, 1 equiv) in 20% of phosgene in toluene (15 mL, 28.4 mmol, 2 equiv) was refluxed for 5 hours. The reaction mixture was then concentrated and the resulting residue was directly used in the next step. EXAMPLE 231:
1-(4-FLUORO-2-METHOXYPHENYL)-4,5-DIHYDRO-5H-TETRAZOL-5-ONE

[00744] To the above residue of Example 230, 3.77 mL of TMSN3 (28.4 mmol, 2 equiv) was added and then the resulting mixture was heated at 100 °C overnight. After being cooled to room temperature, the reaction mixture was concentrated. The residue was diluted with EtOAc (50 mL) and saturated aqueous NaHCC>3 (50 mL). The aqueous layer was separated, neutralized with 2 N HC1 to pH 4-5 and extracted with EtOAc (2x50 mL). The organic layers were combined, dried over MgS04 and concentrated to give a white solid (450 mg, 15% two steps) which was directly used in the next step, m/z = 211 (M+H)+ . EXAMPLE 232:
1-(4-FLUORO- -METHOXYPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

[00745] To a mixture of l-(4-fluoro-2-methoxyphenyl)-4,5-dihydro-5H-tetrazol-5-one (450 mg, 2.14 mmol, 1 equiv) and K2C03 (887 mg, 6.43 mmol, 3 equiv) in 6 mL of DMF was added Mel (0.4 mL, 6.43 mmol, 3 equiv) and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with
EtOAc. The organic layer was separated and concentrated. The residue was triturated in water to give 410 mg of product (85%) as an off-white solid. m/z = 270 (M+H)+ .
EXAMPLE 233:
1-(4-FLUORO-2-METHOXY-5-NITROPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

[00746] 90% HNO3 (0.056 mL, 1.13 mmol, 1.1 equiv) was added dropwise to a mixture of l-(4-fluoro-2-methoxyphenyl)-4-methyl-5H-tetrazol-5-one (230 mg, 1.03 mmol, 1 equiv) in 2 mL of fuming H2SO4 at 0 °C. Then the mixture was left to room temperature and stirred for 2 hours. Poured into ice and the aqueous phase was extracted with EtOAc. The organic layer was separated, washed with saturated NaHCC>3 and concentrated to give 277 mg of product (quantitative) as a yellow solid.
[00747] JH NMR (CDC13, 300 MHz): δ 8.28 (d, / = 7.8 Hz, 1H), 6.94 (d, / = 12.0 Hz, 1H), 3.98 (s, 3H), 3.72 (s, 3H); m/z = 270 (M+H)+.
EXAMPLE 234:
1-(5-AMI -4-FLUORO-2-METHOXYPHENYL)-4-METHYL-5H-TETRAZOL-5-ONE

[00748] A mixture of l-(4-fluoro-2-methoxy-5-nitrophenyl)-4-methyl-5H-tetazol-5-one (320 mg, 1.2 mmol, 1 equiv), iron powder (332 mg, 6.0 mmol, 5 equiv) and ammonium chloride (318 mg, 6.0 mmol, 5 equiv) in iPrOH (6 mL) and water (1.2 mL) was refluxed for 3 hours. After cooling to room temperature, the reaction mixture was filtered through a pad of Celite, and the pad of Celite was rinsed with EtOAc (50 mL). The filtrate was washed with water. The organic layer was separated, dried over MgS04 and evaporated to give the product quantitatively, m/z = 240 (M+H)+.
EXAMPLE 235:
N2-(4-FLUORO-2-METHOXY-3-(4-METHYL-4,5-DIHYDRO-5-OXO-LH-TETRAZOL-L- YL)PHENYL)-5-FLUORO-N4-(2,2,6,6-TETRAMETHYLPIPERIDIN-4-YL)PYRIMIDINE-2,4-
DIAMINE (1-88)

[00749] Ή NMR (DMSO-i¾, 300 MHz): δ 8.23 (br. s, 1H), 7.76 (d, / = 3.6 Hz, 1H), 7.72 (d, 7 = 8.1 Hz, 1H), 7.20 (d, 7 = 11.1 Hz, 1H), 7.02 (br. s, 1H), 4.25-4.15 (m, 1H), 3.75 (s, 3H), 3.57 (s, 3H), 1.61-1.27 (m, 2H), 1.16-1.03 (m, 2H), 0.98 (s, 12H); mJz = 490 (M+H)+. EXAMPLE 236:
N2-(4-FLUORO-2-METHOXY-3-(4-METHYL-4,5-DIHYDRO-5-OXO-LH-TETRAZOL-L- YL)PHENYL)-5-FLUORO-N4-(1,2,2,6,6-PENTAMETHYLPIPERIDIN-4-YL)PYRIMIDINE-2,4-
DIAMINE (1-87)

[00750] Ή NMR (DMSO-i¾, 300 MHz): δ 8.50 (br. s, 1H), 7.85 (d, / = 3.9 Hz, 1H), 7.62 (d, / = 8.4 Hz, 1H), 7.47 (d, 7 = 7.5 Hz, 1H), 7.26 (d, / = 12.0 Hz, 1H), 4.28-4.18 (m, 1H), 3.77 (s, 3H), 3.58 (s, 3H), 2.66 (s, 3H), 2.00-1.90 (m, 2H), 1.73-1.65 (m, 2H), 1.35 (s, 6H), 1.18 (s, 6H); m/z = 504 (M+H)+.
BIOLOGICAL EXAMPLES EXAMPLE 237: PKC ASSAY
[00751] The inhibition of PKC-alpha, PKC-beta, PKC-delta, PKC epsilon and PKC-theta activity is determined via ELISA as follows: NUNC MAXISORP (#436110) or Costar High
Binding (#3922) plates are coated with 0.01 mg/mL Neutravidin (Pierce #PI-31000) in lx
PBS (100
 for 18-24 hours at 4 °C. When ready to be used, plates are washed with lx PBST and then blocked with 2% BSA in lx PBST (100 μΕΛ βΠ) for a minimum of 1 hour at room temperature. The reactions are conducted in a volume of 60 μΕΛ βΙΙ. When ready to begin, the plates are washed with lx PBST to remove the 2% BSA blocking solution. Reaction solution containing the necessary buffer components as well as the appropriate concentrations of ATP and peptide substrate is then added to each well (see
Table 3). Appropriate concentrations of test compound is then added - with the volume added should taking into consideration the DMSO tolerance of the kinases being about 0.2%. The reaction is then initiated by the addition of kinase - the approximate final concentration of which is listed in Table 3 ( note that this will vary depending on the batch to batch variability in the activity of enzymes). After allowing the reaction to stand at room temperature for 20 minutes, the plates are then washed with lx PBST.
for 18-24 hours at 4 °C. When ready to be used, plates are washed with lx PBST and then blocked with 2% BSA in lx PBST (100 μΕΛ βΠ) for a minimum of 1 hour at room temperature. The reactions are conducted in a volume of 60 μΕΛ βΙΙ. When ready to begin, the plates are washed with lx PBST to remove the 2% BSA blocking solution. Reaction solution containing the necessary buffer components as well as the appropriate concentrations of ATP and peptide substrate is then added to each well (see
Table 3). Appropriate concentrations of test compound is then added - with the volume added should taking into consideration the DMSO tolerance of the kinases being about 0.2%. The reaction is then initiated by the addition of kinase - the approximate final concentration of which is listed in Table 3 ( note that this will vary depending on the batch to batch variability in the activity of enzymes). After allowing the reaction to stand at room temperature for 20 minutes, the plates are then washed with lx PBST.
Table 3

[00752] After removal of the reaction mixture from the plate and washing with lx PBST, an antibody developing solution containing a 1: 10,000 dilution of the appropriate primary and secondary antibodies (Table 3) in a 0.1% BSA solution in lx PBST is then added to each well (100 μΕΛ εΠ). This is then allowed to stand at room temperature for a minimum of 1 hour. After this time, the plates are once again washed with lx PBST. The SuperSignal ELISA Pico Chemiluminescent substrate (Pierce #PI-37069) is then added (100 μΕ/well) and the plate is read on a luminescence plate reader.
EXAMPLE 238: PKC ASSAY
[00753] Alternatively, the inhibition of PKC activity is measured by monitoring the production of phosphorylated peptide by fluorescence polarization at different
concentrations of the inhibitor. Reactions are carried out in 96-well plate format with a total volume of 20 [lL containing 20 mM HEPES, pH 7.4, 5 mM MgCl2, 0.2 mM CaCl2, 1 mM DTT, 0.02% Brij-35, 0.1 mg/mL phosphatidylserine, 0.02 mg/mL dioleoyl-sn-glycerol and 5 μΜ each of ATP and the peptide substrate. Compounds are first diluted serially in DMSO and then transferred to a solution containing the above concentrations of HEPES, MgCl2, CaCl2, DTT, and Brij-35 to yield 5x compound solutions in 2% DMSO, which is then added to the reaction solution. Reactions are initiated by the addition of PKC at a typical concentration as described in Table 4, and then allowed to incubate at room temperature for
20 minutes. At the end of this time, a combination of quench (EDTA) and detection (peptide tracer and antibody) reagents is added using the protocol of Invitrogen P2748. After a 30 minutes period of incubation, the amount of phosphorylated peptide generated is measured by fluorescence polarization (Ex = 485 nm, Em = 535 nm) using a Tecan Polarian instrument.
Table 4

EXAMPLE 239: CALCIUM INFLUX
[00754] HEK-FLPTREX cells are stably transfected with pcDNA5/FRT/TO+hTRPV4a, rat TRPV1-HA or rTRPAl-HA are grown in Dulbecco's Modified Eagle's Medium
(DMEM) containing 10% tetracycline-free fetal bovine serum, hygromycin (50 μg/ml) and blasticidin (10 μg/ml). Cells are treated with tetracycline (0.1 μg/ml, 20 h) to induce TRP expression. DRG from thoracic and lumbar spinal cord of rats or mice are minced in cold Hank's Balanced Salt Solution (HBSS) and incubated for 60 at 37 °C in DMEM containing 1 mg/ml of collagenase type IA and 0.1 mg/ml of DNAse type IV, pelleted and incubated with 0.25% trypsin for 30 minutes. Neurons are pelleted, suspended in DMEM containing 10% fetal bovine serum, 10% horse serum, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 2 mM glutamine, dissociated by gentle trituration until the solution appears cloudy and
homogeneous and plated on glass coverslips coated with PolyOnitine/laminin. Neurons are cultured for 3-4 days before the experiment.
[00755] Cells grown on coverslips or on a 96 multiwell plate are incubated in HBSS (pH 7.4) containing Ca2+ and Mg2+, 20 mM HEPES buffer, 0.1% BSA, 100 U/ml penicillin, 100 μg/ml streptomycin, with 2.5-5 μΜ Fura-2AM (Invitrogen) for 20-45 minutes at 37 °C. Cells are washed and fluorescence is measured at 340 nm and 380 nm excitation and 510 nm emission in a F-2500 spectrophotometer, or in a Flexstation 3 Microplate Reader III (for the measurement of the calcium in the cell population) or using a Zeiss Axiovert microscope, an
ICCD video camera and a video microscopy acquisition program (for the measurement of the calcium influx in the single neurons). Substances are injected directly into the chamber (20 ml into 2 ml, for the spectrophotometer; 20 ml in 200 ml for the Flexstation, 50 ml in 350 ml in the chamber for the single cells).
EXAMPLE 240: IN VIVO HYPERPLASIA
[00756] Mechanical pain is quantified as the number of times the hind paw is withdrawn in response to 5 applications of a 0.173 mN von Frey hair. Responses are expressed as a percentage (e.g. 3 withdrawals out of 5 are recorded as 60%) and mechanical hyperalgesia defined as increase in the percentage of withdrawal compared to basal measurement. 2) Mechanical pain is quantified using the 'up-down paradigm', determining the 50% response threshold to the von Frey filaments applied to the mid-plantar surface for 5 s or until a withdrawal response occurred. Von Frey filaments are in this range of intensities: 1.65, 2.44, 2.83, 3.22, 3.61, 3.84, 4.08.
[00757] Thermal hyperalgesia is assessed in mice using a plantar test apparatus and quantified as the latency of paw withdrawal to a radiant heat. Thermal hyperalgesia is defined as a decrease in the withdrawal latency compared to the basal measurement. After measuring basal level mice, under light halothane anesthesia (5%), are injected with testing compound into the left or right paws (5-10 μΐ intraplantar injection) and paw withdrawal measurements repeated at different time point. To assess PAR2 TRPVl, TRPV4 and TRPAl mediated hyperalgesia and potentiation of TRPV-mediated responses, mice are treated with PAR2-AP for 15 minutes followed by capsaicin, 4ocPDD or HNE. To assess the role of protein kinases, the antagonists or the corresponding vehicles are injected 20-30 minutes before the challenge with agonists. The effects induced by the different treatments are evaluated within the same rat comparing the responses recorded in the right paw (receiving for example saline, or vehicle) with the responses obtained in the left paw (receiving for example PAR2-AP or 4aPDD).
[00758] Formalin induced hyperalgeisa is assessed using 5% solution of formalin administered by intradermal injection into the dorsal surface of the mouse or rat forepaw to induce a painful behavior. Pain is accessed on a four-level scale related to posture: 0, normal posture; 1, with the injected paw remaining on the ground but not supporting the animal; 2, with the injected paw clearly raised; and 3, with the injected paw being licked, nibbled, or shaken. Animals are observed and scored for behavior at 3 minutes after the injection (defined as initial phase that results from the direct stimulation of nociceptors), and then at 30-60 minutes after the injection (defined as second phase that involves a period of
sensitization during which inflammatory phenomena occur). The nociceptive behavioral score for each 3-minutes interval is calculated as the weighted average of the number of seconds spent in each behavior. 2.5% solution of formalin is administered by intraplantar injection and thermal and mechanical pain measured as described above after 30-60 minutes. To assess the role of protein kinases, antagonists or their vehicles (control) are injected into the right paws 20-30 minutes before formalin. Nociceptive behavior will be scored for each rats and compared to control.
EXAMPLE 241: IL-2 ELISA, HUMAN PRIMARY T CELL, ANTI CD3+CD28+
[00759] Human primary T cell isolation and culture: Human primary T cells were prepared as follows. Whole blood was obtained from a healthy volunteer, mixed 1: 1 with PBS, layered on to Ficoll Hypaque (Amersham Pharmacia Biotech, Piscataway, NJ, Catalog #17-1440-03) in 2: 1 blood/PBS :ficoll ratio and centrifuged for 30 minutes at 4 °C at 1750 rpm. The cells at the serum: ficoll interface were recovered and washed twice with 5 volumes of PBS. These freshly isolated human peripheral blood mononuclear cells were cultured in Yssel's medium containing 40 U/mL IL2 in a flask pre-coated with 1 μg/mL aCD3 and 5 μg/mL aCD28 (Anti-Human CD3, BD Pharmingen Catalog #555336, Anti- Human CD28, Beckman Coulter Catalog #IM1376). The cells were stimulated for 3-4 days, then transferred to a fresh flask and maintained in RPMI (RPMI-1640 with L-Glutamine; Mediatech, Inc., Herndon VA, cat. #10-040-CM) with 10% FBS and 40 U/mL IL-2. The primary T-cells were then washed twice with PBS to remove the IL-2.
[00760] Primary T cell stimulation and IL2 ELISA: Human primary T cells (100,000 cells per well) were pre-incubated with or without test compound in Yssel's medium for 1 hour at 37 °C. Cells were then stimulated by transferring them to round-bottom 96-well plates pre-coated with 1 μg/ml ocCD3 and 5 μg/ml ocCD28. For counter assay, cells were instead stimulated by adding 8X stock solutions of PMA and ionomycin in Yssels (for final concentrations of 0.5ng/ml PMA and O.luM ionomycin, both from Calbiochem). Cells were incubated at 37 °C for 24 hours before 100 μΐ^ supernatants were harvested for quantification of IL-2 by ELISA using Human IL-2 Duoset ELISA Kit from R and D Systems, Cat. # DY202E.
[00761] Table 5 shows the IC50 values for compounds tested in the assays described in this example. For ELISA data in Table 5, "E" indicates an IC50 in the indicated assay of less than 0.5 μΜ; "F" is 0.51-2.99 μΜ; "G" is 3-25 μΜ; and "H" is from greater than 25 μΜ.
Table 5

1-36 A
1-37 B
1-38 A
1-39 A
1-40 A
1-41 A
1-42 A
1-43 A
1-44 A
1-45 A
1-46 A
1-47 A
1-48 A
1-49 A
1-50 A
1-51 A
1-52 A
1-53 A
1-54 A
1-55 A
1-56 A
1-57 A
1-58 A
1-59 A
1-60 A
1-61 C
1-62 A
1-63 A
1-64 A
1-65 C
1-66 C
1-67 c
1-68 A
1-69 A
[00762] While the present invention has been described with reference to the specific embodiments thereof, it should be understood by those skilled in the art that various changes may be made and equivalents may be substituted without departing from the true spirit and scope of the invention. In addition, many modifications may be made to adapt a particular situation, material, composition of matter, process, process step or steps, to the objective, spirit and scope of the present invention. All such modifications are intended to be within the scope of the claims appended hereto.
Claims
1. A c

wherein
R5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
Y1 and Y2 are independently selected from hydrogen, alkyl, and acyl;
R1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
Ra and Rb are independently selected from hydrogen and alkyl;
Rc and Rd are independently selected from hydrogen and alkyl;
Q is selected from N and CR7b;
R6a, R6b, R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O- alk-A;
alk is a bond, alkylene or substituted alkylene;
A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
wherein the A ring can be substituted or unsubstituted;
R7y is selected from hydrogen, alkyl, cycloalkyl, and substituted alkyl;
or a salt or stereoisomer thereof.
2.
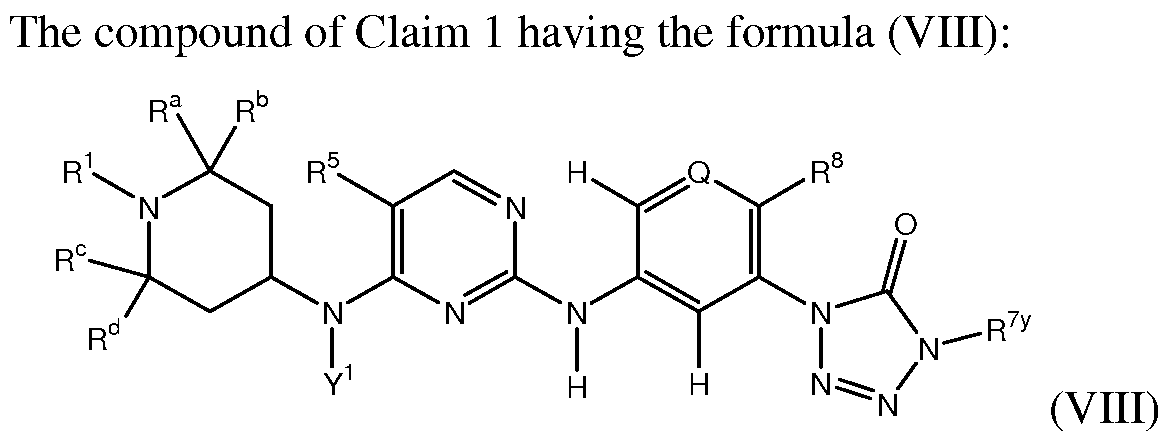
R5 is selected from alkyl, substituted alkyl, hydroxy, alkoxy, substituted alkoxy, amino, substituted amino, carboxyl, carboxyl ester, cyano, halogen, acyl, aminoacyl, nitro, alkenyl, substituted alkenyl, alkynyl, and substituted alkynyl;
Y1 is selected from hydrogen and alkyl;
R1 is selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, acyl, and oxy radical;
Ra and Rb are independently selected from hydrogen and alkyl;
Rc and Rd are independently selected from hydrogen and alkyl;
Q is selected from N and CR7b;
R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, alkoxycarbonylamino, aminocarbonylamino, acyl, carboxyl, carboxyl ester, aminoacyl, sulfonyl, sulfonylamino, aminosulfonyl, and -O-alk-A;
alk, if present, is alkyl or substituted alkyl;
A is selected from aryl, cycloalkyl, heteroaryl, and heterocyclyl;
wherein the A ring can be substituted or unsubstituted;
R7y is selected from hydrogen, alkyl, and substituted alkyl;
or a salt or stereoisomer thereof.
3. The compound of Claim 1 or 2, wherein R5 is cyano, halogen, acyl, aminoacyl, or nitro.
4. The compound of Claim 1 or 2, wherein R5 is fluoro.
5. The compound of Claim 1 or 2, wherein R5 is cyano.
6. The compound of Claim 1 or 2, wherein Y1 is hydrogen.
7. The compound of Claim 1, wherein Y2 is hydrogen.
8. The compound of Claim 1 or 2, wherein R is hydrogen.
9. The compound of Claim 1 or 2, wherein R1 is alkyl.
10. The compound of Claim 1 or 2, wherein Ra and Rb are both alkyl.
11. The compound of Claim 1 or 2, wherein Rc and Rd are both alkyl.
12. The compound of Claim 1 or 2, wherein Q is CR7b.
13. The compound of Claim 1 or 2, wherein Q is N.
14. The compound of Claim 1, wherein R6a, R6b, R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, cycloalkyl, substituted cycloalkyl, and -O-alk-A.
15. The compound of Claim 1 or 2, wherein R 7b and R 8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, hydroxyl, alkoxy, substituted alkoxy, amino, substituted amino, acylamino, aminocarbonyloxy, heteroaryl, substituted heteroaryl, heterocyclyl, substituted heterocyclyl, cycloalkyl, substituted cycloalkyl, and -O-alk-A.
16. The compound of Claims 1 or 2, wherein R6a, R6b, R7b and R8 are independently selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, acyl, carboxyl, carboxyl ester, aminoacyl, and -O-alk-A.
17. The compound of Claims 1 or 2, wherein at least one of R6a, R6b, R7b and Rs is alkyl or substituted alkyl.
18. The compound of Claims 1 or 2, wherein at least one of R6a, R6b, R7b and Rs is halogen.
19. The compound of Claims 1 or 2, wherein at least one of R6a, R6b, R7b and Rs is alkoxy, substituted alkoxy, or -O-alk-A.
20. The compound of Claims 1 or 2, wherein at least one of R6a, R6b, R7b and Rs is cyano, acyl, carboxyl, carboxyl ester, or aminoacyl.
21. The compound of Claims 1 or 2, wherein at least one of R6a, R6b, R7b and Rs is cycloalkyl or substituted cycloalkyl.
22. The compound of Claim 1, wherein R6a is selected from hydrogen, alkyl, substituted alkyl, and halogen.
23. The compound of Claim 1, wherein R6b is selected from hydrogen, alkyl, substituted alkyl, and halogen.
24. The compound of Claim 1 or 2, wherein R is selected from hydrogen, alkyl, and substituted alkyl.
25. The compound of Claim 1 or 2, wherein R7b is selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, acyl, carboxyl, carboxyl ester, aminoacyl, and -O-alk-A.
26. The compound of Claim 1 or 2, wherein R7b is hydrogen.
27. The compound of Claim 1 or 2, wherein R8 is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy, cycloalkyl, and substituted cycloalkyl.
28. The compound of Claim 1 or 2, wherein R8 is selected from hydrogen, alkyl, substituted alkyl, halogen, alkoxy, substituted alkoxy.
29. The compound of Claim 1 or 2, wherein R8 is halogen.
30. The compound of Claim 1 or 2, wherein any of R 7b or R 8 is selected from hydrogen, alkyl, substituted alkyl, halogen, cyano, alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, acyl, carboxyl, carboxyl ester, aminoacyl, and -O-alk-A.
31. The compound of Claim 1 or 2, wherein at least one of R7b or R8 is cycloalkyl, or substituted cycloalkyl.
32. The compound of Claim 1 or 2, wherein at least one of R 7b or R 8 is alkoxy, substituted alkoxy, or -O-alk-A.
33. The compound of Claim 1 or 2, wherein at least one of R7b or R8 is alkyl, substituted alkyl, or halogen.
34. The compound of Claim 1 or 2, wherein R7y is hydrogen.
35. The compound of Claim 1 or 2, wherein R7y is alkyl.
36. The compound of Claim 1 or 2, wherein R7y is methyl.
37. The compound of Claim 1 or 2, wherein R7y is substituted alkyl.
38. The compound of Claim 1, wherein the compound selected from:
1-1: 5-Fluoro-N2-{4-fluoro-[3-(4-H)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-2: 5-Fluoro-N2-{4-fluoro-[3-(4-H)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-3: 5-Fluoro-N2-{4-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-4: 5-Fluoro-N2-{4-fluoro-[3-(4-isopropyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (2,2,6, 6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine; 1-5: 5-Fluoro-N2-{4-fluoro-[3-(4-isopropyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-6: 5-Fluoro-N2-{4-fluoro-3-[4-(2-fluoroethyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-7: 5-Fluoro-N2-{4-fluoro-3-[4-(2-fluoroethyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-8: 5-Fluoro-N2-{4-methyl-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-9: 5-Fluoro-N2-{4-methyl-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (2,2,6, 6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
I- 10 : 5 -Fluoro-N2- { 4-isopropoxy- [3 -(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 -yl] } phenyl- N4-(l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
I- 11 : 5 -Fluoro-N2- { 4-isopropoxy- [3 -(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 -yl] } phenyl- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-12: 5-Fluoro-N2-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl]phenyl-N4-(l,2,2,6,6- pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-13: 5-Fluoro-N2-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl]phenyl-N4-(2,2,6,6- tetramethylpiperidin-4- yl) -2 ,4-pyrimidinediamine ;
1-14: l-(2-cyclopropyl-4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-15: 2-(4-cyclopropyl-2-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carbonitrile;
1-16: l-{2-Cyclopropyl-4-fluoro-5-[5-fluoro-4-(l, 2,2,6, 6-pentamethyl-piperidin-4- ylamino)-pyrimidin-2-ylamino]-phenyl}-4-methyl-l,4-dihydro-tetrazol-5-one;
1-17: 2-[4-Cyclopropyl-2-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-tetrazol-l-yl)- phenylamino]-4-(l,2,2,6,6-pentamethyl-piperidin-4-ylamino)-pyrimidine-5-carbonitrile;
1-18: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(l,l,l-trifluoro-2-methylpropan-2-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)- one;
1-19: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(l,l,l-trifluoro-2-methylpropan-2-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)- one;
1-20: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one; 1-21: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-22: l-(3-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-23: l-(3-fluoro-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-24: l-(3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-25: l-(3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-26: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(tetrahydro-2H-pyran-4-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-27: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(tetrahydro-2H-pyran-4-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-28: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-((3-methyloxetan-3-yl)methoxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-29: l-(2-chloro-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-30: l-(2-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-31: l-(3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-methoxyphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-32: l-(3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-methoxyphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-33: l-(2-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-34: l-(2-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5s(4H)-one;
1-35: l-(3-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-36: l-(3-cyclopropyl-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-37: l-(3-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one; 1-38: l-(2-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino) - 3 - (trifluoromethyl)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-39: l-(2-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-40: l-(2-cyclopropyl-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-41: 2-(4-cyclopropyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)-5- (trifluoromethyl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5- carbonitrile;
1-42: 2-(4-cyclopropyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)-5- (trifluoromethyl)phenylamino)-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidine-5- carbonitrile;
1-43: l-(3-cyclopropyl-2-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-44: 2-(3-cyclopropyl-4-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carbonitrile;
1-45: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2,3-dimethylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-46: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2,3-dimethylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-47: l-(3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino) -5 - (trifluoromethyl)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-48: l-(3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino) -5 - (trifluoromethyl)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-49: 3-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzonitrile;
1-50: 3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2-ylamino)-5- (4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzonitrile;
1-51 : l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-isopropoxy-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-52: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-isopropoxy-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-53: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one; 1-54: l-(5-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-55: l-(3-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino)-2-methylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-56: l-(3-chloro-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)-2-methylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-57: 2-(4-cyclopropyl-3-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carboxamide;
1-58: 4-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2-ylamino)-2- (4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzonitrile;
1-59: l-(2-cyclopropyl-3-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-60: methyl 5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoate;
1-61: 5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2-ylamino)-2- methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoic acid;
1-62: 2-(4-cyclopropyl-3-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carbonitrile;
1-63 : methyl 5-(5-fluoro-4-(l ,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoate;
1-64: 5-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-N,2-dimethyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzamide;
1-65: 5-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoic acid;
1-66: l-(2,6-difluoro-3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-67: l-(2,6-difluoro-3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-68: l-(2-chloro-4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-69: l-(2-chloro-4-fluoro-5-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one; 1-70: l-(4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-((lS,2S)-2-(trifluoromethyl)cyclopropyl)phenyl)-4-methyl-lH-tetrazol-5(4H)- one;
1-71: l-(4-fluoro-3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-72: l-(2-cyclopropyl-4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-(trideuteromethyl)-lH-tetrazol-5(4H)-one;
1-73: N2-{4-Cyclopropyl-6-fluoro-[3-(4-isopropyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-74: N2-{4-Cyclopropyl-6-fluoro-3-(l,2,3,4-tetrazol-5-one-l-yl)}phenyl-5-fluoro- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, formate salt;
1-75: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, citrate salt;
1-76: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, maleate salt;
1-77 : N2- { 4-Cyclopropyl-6-fluoro- [3-(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 - yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, fumarate salt;
1-78: N2- { 4-Cyclopropyl-6-fluoro- [3-(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 - yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, L- tartarate salt;
1-79: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, hydrogen sulfate salt;
1-80: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, hydrogen chloride salt;
1-81: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, benzoate salt; 1-82: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediami tosylate salt;
1-83: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, besylate salt;
1-84: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, mesylate salt;
1-85: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, acetate salt;
1-86: N2-(4-fluoro-2-methoxy-3-(4-methyl-4,5-dihydro-5-oxo-lH-tetrazol-l- yl)phenyl)-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)pyrimidine-2,4-diamine; and
1-87: N2-(4-fluoro-2-methoxy-3-(4-methyl-4,5-dihydro-5-oxo-lH-tetrazol-l- yl)phenyl)-5-fluoro-N4-(l,2,2,6,6-pentamethylpiperidin-4-yl)pyrimidine-2,4-diamine;
or a solvate, prodrug, or a pharmaceutically acceptable salt thereof.
39. The compound of Claim 1, wherein the compound selected from:
1-1: 5-Fluoro-N2-{4-fluoro-[3-(4-H)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-2: 5-Fluoro-N2-{4-fluoro-[3-(4-H)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (2,2,6, 6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-3: 5-Fluoro-N2-{4-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-4: 5-Fluoro-N2-{4-fluoro-[3-(4-isopropyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (2,2,6, 6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-5: 5-Fluoro-N2-{4-fluoro-[3-(4-isopropyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-6: 5-Fluoro-N2-{4-fluoro-3-[4-(2-fluoroethyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-7: 5-Fluoro-N2-{4-fluoro-3-[4-(2-fluoroethyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine; 1-8: 5-Fluoro-N2-{4-methyl-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-9: 5-Fluoro-N2-{4-methyl-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl] }phenyl-N4- (2,2,6, 6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
I- 10 : 5 -Fluoro-N2- { 4-isopropoxy- [3 -(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 -yl] } phenyl- N4-(l,2,2,6,6-pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine;
I- 11 : 5 -Fluoro-N2- { 4-isopropoxy- [3 -(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 -yl] } phenyl- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-12: 5-Fluoro-N2-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl]phenyl-N4-(l,2,2,6,6- pentamethylpiperidin-4-yl)-2,4-pyrimidinediamine; and
1-13: 5-Fluoro-N2-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l-yl]phenyl-N4-(2,2,6,6- tetramethylpiperidin-4- yl) -2 ,4-pyrimidinediamine ;
or a solvate, prodrug, or a pharmaceutically acceptable salt thereof.
40. The compound of Claim 1, wherein the compound selected from:
1-14: l-(2-cyclopropyl-4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-15: 2-(4-cyclopropyl-2-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carbonitrile;
1-16: l-{2-Cyclopropyl-4-fluoro-5-[5-fluoro-4-(l, 2,2,6, 6-pentamethyl-piperidin-4- ylamino)-pyrimidin-2-ylamino]-phenyl}-4-methyl-l,4-dihydro-tetrazol-5-one; and
1-17: 2-[4-Cyclopropyl-2-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-tetrazol-l-yl)- phenylamino]-4-(l,2,2,6,6-pentamethyl-piperidin-4-ylamino)-pyrimidine-5-carbonitrile; or a solvate, prodrug, or a pharmaceutically acceptable salt thereof.
41. The compound of Claim 1, wherein the compound is l-(2-cyclopropyl-4- fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2-ylamino)phenyl)- 4-methyl-lH-tetrazol-5(4H)-one (1-14).
42. The compound of Claim 1, wherein the compound selected from:
1-18: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(l,l,l-trifluoro-2-methylpropan-2-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)- one; 1-19: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(l ,1 ,l-trifluoro-2-methylpropan-2-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)- one;
1-20: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-21: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-22: l-(3-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-23: l-(3-fluoro-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-24: l-(3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-25: l-(3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-(oxetan-3-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-26: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(tetrahydro-2H-pyran-4-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-27: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-(tetrahydro-2H-pyran-4-yloxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-28: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-((3-methyloxetan-3-yl)methoxy)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-29: l-(2-chloro-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-30: l-(2-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-31: l-(3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-methoxyphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-32: l-(3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-methoxyphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-33: l-(2-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-34: l-(2-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one; 1-35: l-(3-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-36: l-(3-cyclopropyl-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-37: l-(3-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-38: l-(2-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino) - 3 - (trifluoromethyl)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-39: l-(2-cyclopropyl-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-40: l-(2-cyclopropyl-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-41: 2-(4-cyclopropyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)-5- (trifluoromethyl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5- carbonitrile;
1-42: 2-(4-cyclopropyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)-5- (trifluoromethyl)phenylamino)-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidine-5- carbonitrile;
1-43: l-(3-cyclopropyl-2-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-44: 2-(3-cyclopropyl-4-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carbonitrile;
1-45: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2,3-dimethylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-46: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2,3-dimethylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-47: l-(3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino) -5 - (trifluoromethyl)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-48: l-(3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino) -5 - (trifluoromethyl)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-49: 3-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzonitrile;
1-50: 3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2-ylamino)-5- (4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzonitrile; 1-51 : l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-isopropoxy-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-52: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-isopropoxy-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-53: l-(5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-54: l-(5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(trifluoromethyl)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-55: l-(3-chloro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin- 2-ylamino)-2-methylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-56: l-(3-chloro-5-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)-2-methylphenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-57: 2-(4-cyclopropyl-3-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carboxamide;
1-58: 4-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2-ylamino)-2- (4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzonitrile;
1-59: l-(2-cyclopropyl-3-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-60: methyl 5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoate;
1-61: 5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2-ylamino)-2- methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoic acid;
1-62: 2-(4-cyclopropyl-3-fluoro-5-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l- yl)phenylamino)-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidine-5-carbonitrile;
1-63 : methyl 5-(5-fluoro-4-(l ,2,2,6,6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoate;
1-64: 5-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-N,2-dimethyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzamide;
1-65: 5-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-methyl-3-(4-methyl-5-oxo-4,5-dihydro-lH-tetrazol-l-yl)benzoic acid;
1-66: l-(2,6-difluoro-3-(5-fluoro-4-(l,2,2,6,6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
1-67: l-(2,6-difluoro-3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one; 1-68: l-(2-chloro-4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one; and
1-69: l-(2-chloro-4-fluoro-5-(5-fluoro-4-(l, 2,2,6, 6-pentamethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-methyl-lH-tetrazol-5(4H)-one;
or a solvate, prodrug, or a pharmaceutically acceptable salt thereof.
43. The compound of Claim 1, wherein the compound selected from:
1-70: l-(4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)-2-((lS,2S)-2-(trifluoromethyl)cyclopropyl)phenyl)-4-methyl-lH-tetrazol-5(4H)- one;
1-71: l-(4-fluoro-3-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4-ylamino)pyrimidin-2- ylamino)phenyl) -4-methyl- 1 H-tetrazol- 5 (4H) -one ;
1-72: l-(2-cyclopropyl-4-fluoro-5-(5-fluoro-4-(2,2,6,6-tetramethylpiperidin-4- ylamino)pyrimidin-2-ylamino)phenyl)-4-(trideuteromethyl)-lH-tetrazol-5(4H)-one;
1-73: N2-{4-Cyclopropyl-6-fluoro-[3-(4-isopropyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine;
1-74: N2-{4-Cyclopropyl-6-fluoro-3-(l,2,3,4-tetrazol-5-one-l-yl)}phenyl-5-fluoro- N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, formate salt;
1-75: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, citrate salt;
1-76: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, maleate salt;
1-77 : N2- { 4-Cyclopropyl-6-fluoro- [3-(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 - yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, fumarate salt;
1-78: N2- { 4-Cyclopropyl-6-fluoro- [3-(4-methyl)- 1 ,2,3 ,4-tetrazol-5 -one- 1 - yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, L- tartarate salt;
1-79: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, hydrogen sulfate salt; 1-80: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediami hydrogen chloride salt;
1-81: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, benzoate salt;
1-82: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, tosylate salt;
1-83: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, besylate salt;
1-84: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, mesylate salt;
1-85: N2-{4-Cyclopropyl-6-fluoro-[3-(4-methyl)-l,2,3,4-tetrazol-5-one-l- yl] }phenyl-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)-2,4-pyrimidinediamine, acetate salt;
1-86: N2-(4-fluoro-2-methoxy-3-(4-methyl-4,5-dihydro-5-oxo-lH-tetrazol-l- yl)phenyl)-5-fluoro-N4-(2,2,6,6-tetramethylpiperidin-4-yl)pyrimidine-2,4-diamine; and
1-87: N2-(4-fluoro-2-methoxy-3-(4-methyl-4,5-dihydro-5-oxo-lH-tetrazol-l- yl)phenyl)-5-fluoro-N4-(l,2,2,6,6-pentamethylpiperidin-4-yl)pyrimidine-2,4-diamine.
44. A pharmaceutical composition comprising a compound of any of Claims 1 to 43 and a pharmaceutically acceptable carrier.
45. A method of inhibiting a protein kinase C (PKC) activity in a biological sample or a patient, which method comprises contacting the biological sample or administering to the patient a compound of any of Claims 1 to 43.
46. The method of Claim 45, wherein the inhibition of PKC results in treatment of a disease or disorder that is mediated or sustained through the activity of a PKC activity.
47. The method of Claim 46, wherein the disease or disorder is associated with activation of T cells.
48. The method of Claim 46, wherein the disease or disorder is an inflammatory disease.
49. The method of Claim 46, wherein the disease or disorder is an autoimmune disease.
50. The method of Claim 46, wherein the disease or disorder is an ocular disease or disorder involving inflammatory and/or neovascular events.
51. The method of Claim 46, wherein the disease or disorder is selected from atherosclerosis, vascular occlusion due to vascular injury, angioplasty, restenosis, obesity, syndrome X, impaired glucose tolerance, polycystic ovary syndrome, hypertension, heart failure, chronic obstructive pulmonary disease, CNS diseases, Alzheimer disease, amyotrophic lateral sclerosis, bipolar disease, cancer, infectious disease, AIDS, septic shock, adult respiratory distress syndrome, ischemia/reperfusion injury, myocardial infarction, stroke, gut ischemia, renal failure, hemorrhage shock, and traumatic shock, and traumatic brain injury.
52. The method of Claim 46, wherein the disease or disorder is selected from T- cell mediated acute or chronic inflammatory diseases or disorders or autoimmune diseases, rheumatoid arthritis, osteoarthritis, systemic lupus erythematosus, Hashimoto's thyroiditis, multiple sclerosis, myasthenia gravis, diabetes type I or II and the disorders associated therewith, transplant rejection, graft versus host disease, respiratory diseases, asthma, inflammatory lung injury, inflammatory liver injury, inflammatory glomerular injury, cutaneous manifestations of immunologically-mediated disorders or illnesses, inflammatory and hyperproliferative skin diseases, psoriasis, atopic dermatitis, allergic contact dermatitis, irritant contact dermatitis and further eczematous dermatitises, seborrhoeic dermatitis, inflammatory eye diseases, Sjoegren's syndrome, keratoconjunctivitis, uveitis, inflammatory bowel disease, Crohn's disease or ulcerative colitis, Guillain-Barre syndrome, and allergies.
53. A method of studying a biological sample known to comprise PKC, the method comprising:
(a) contacting the biological sample with a compound of any of Claims 1 to 43; and
(b) determining the PKC activity inhibiting effects caused by the compound on the biologic sample.
54. The method of Claim 53, wherein the determination step is performed using an assay of inhibition of PKC activity.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10796518.8A EP2507227B1 (en) | 2009-12-01 | 2010-12-01 | Tetrazolones as protein kinase c inhibitors and uses thereof |
| CA2780759A CA2780759A1 (en) | 2009-12-01 | 2010-12-01 | Protein kinase c inhibitors and uses thereof |
| JP2012542165A JP5802677B2 (en) | 2009-12-01 | 2010-12-01 | Protein kinase C inhibitor and use thereof |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US26564809P | 2009-12-01 | 2009-12-01 | |
| US61/265,648 | 2009-12-01 | ||
| US36646910P | 2010-07-21 | 2010-07-21 | |
| US61/366,469 | 2010-07-21 | ||
| US40596810P | 2010-10-22 | 2010-10-22 | |
| US61/405,968 | 2010-10-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011068898A1 true WO2011068898A1 (en) | 2011-06-09 |
Family
ID=43902714
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/058597 WO2011068898A1 (en) | 2009-12-01 | 2010-12-01 | Protein kinase c inhibitors and uses thereof |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US20110130415A1 (en) |
| EP (1) | EP2507227B1 (en) |
| JP (1) | JP5802677B2 (en) |
| CA (1) | CA2780759A1 (en) |
| WO (1) | WO2011068898A1 (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012012619A1 (en) | 2010-07-21 | 2012-01-26 | Rigel Pharmaceuticals, Inc. | Protein kinase c inhibitors and uses thereof |
| WO2014089112A1 (en) | 2012-12-04 | 2014-06-12 | Rigel Pharmaceuticals, Inc. | Protein kinase c inhibitors and uses thereof |
| WO2014151900A1 (en) | 2013-03-14 | 2014-09-25 | Rigel Pharmaceuticals, Inc. | Protein kinase c inhibitors and uses thereof |
| JP2015519307A (en) * | 2012-04-04 | 2015-07-09 | ライジェル ファーマシューティカルズ, インコーポレイテッド | Protein kinase C inhibitor and use thereof |
| WO2016020864A1 (en) * | 2014-08-06 | 2016-02-11 | Novartis Ag | Protein kinase c inhibitors and methods of their use |
| CN105683169A (en) * | 2013-10-28 | 2016-06-15 | 住友化学株式会社 | Tetrazolinone compound and application for same |
| WO2019213545A1 (en) * | 2018-05-04 | 2019-11-07 | Portola Pharmaceuticals, Inc. | Synthesis of cerdulatinib |
| US10736895B2 (en) | 2015-12-04 | 2020-08-11 | Portola Pharmaceuticals, Inc. | Cerdulatinib for treating hematological cancers |
| US10865198B2 (en) | 2018-05-04 | 2020-12-15 | Alexion Pharmaceuticals, Inc. | Solid forms of cerdulatinib |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1663242B1 (en) * | 2003-08-07 | 2011-04-27 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds and uses as anti-proliferative agents |
| US8722916B2 (en) | 2010-12-01 | 2014-05-13 | Rigel Pharmaceuticals, Inc. | Cyclopropyl MIDA boronate |
| CN111499580A (en) | 2011-04-22 | 2020-08-07 | 西格诺药品有限公司 | Substituted diaminocarboxamides and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| WO2012174412A2 (en) * | 2011-06-16 | 2012-12-20 | La Jolla Institute For Allergy And Immunology | Compositions targeting pkc-theta and uses and methods of treating pkc-theta pathologies, adverse immune responses and diseases |
| GB201200555D0 (en) * | 2012-01-13 | 2012-02-29 | Univ Birmingham | Peptide |
| US9839671B2 (en) | 2012-01-13 | 2017-12-12 | The University Of Birmingham | Peptide and uses therefor |
| US8754251B2 (en) | 2012-06-06 | 2014-06-17 | Rigel Pharmaceuticals, Inc. | Cyclopropyl PIDA boronate |
| EP3010893B1 (en) | 2013-06-20 | 2019-10-02 | Bayer CropScience Aktiengesellschaft | Arylsulfide and arylsulfoxide derivatives as acaricides and insecticides |
| NZ715903A (en) | 2014-01-30 | 2017-06-30 | Signal Pharm Llc | Solid forms of 2-(tert-butylamino)-4-((1r,3r,4r)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| CN106132939B (en) * | 2014-03-28 | 2018-11-13 | 住友化学株式会社 | The manufacturing method of Terazololine-one compound |
| JP6517319B2 (en) | 2014-03-28 | 2019-05-22 | キャリター・サイエンシーズ・リミテッド・ライアビリティ・カンパニーCalitor Sciences, Llc | Substituted heteroaryl compounds and methods of use |
| US10059689B2 (en) | 2014-10-14 | 2018-08-28 | Calitor Sciences, Llc | Substituted heteroaryl compounds and methods of use |
| WO2016100308A1 (en) | 2014-12-16 | 2016-06-23 | Signal Pharmaceuticals, Llc | Methods for measurement of inhibition of c-jun n-terminal kinase in skin |
| EP3233809B1 (en) | 2014-12-16 | 2021-04-21 | Signal Pharmaceuticals, LLC | Formulations of 2-(tert-butylamino)-4-((1r,3r,4r)-3-hydroxy-4-methycyclohexylamino)-pyrimidine-5-carboxamide |
| EP3250565B1 (en) | 2015-01-26 | 2019-07-03 | Rigel Pharmaceuticals, Inc. | Tetrazolones as carboxylic acid bioisosteres |
| EP3250557A4 (en) | 2015-01-29 | 2018-06-20 | Signal Pharmaceuticals, LLC | Isotopologues of 2-(tert-butylamino)-4-((1r,3r,4r)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US10252981B2 (en) | 2015-07-24 | 2019-04-09 | Celgene Corporation | Methods of synthesis of (1R,2R,5R)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
| WO2017044434A1 (en) | 2015-09-11 | 2017-03-16 | Sunshine Lake Pharma Co., Ltd. | Substituted heteroaryl compounds and methods of use |
| CN106316864A (en) * | 2016-08-22 | 2017-01-11 | 苏州天马精细化学品股份有限公司 | Preparation method of 4-methyl-3-trifluoromethyl phenylamine |
| WO2018097318A1 (en) * | 2016-11-28 | 2018-05-31 | Sumitomo Chemical Company, Limited | Tetrazolinone compounds and its use as pest control agents |
| EP3710006A4 (en) | 2017-11-19 | 2021-09-01 | Sunshine Lake Pharma Co., Ltd. | Substituted heteroaryl compounds and methods of use |
| EP3560913A1 (en) | 2018-04-25 | 2019-10-30 | Dynamit Nobel GmbH Explosivstoff- und Systemtechnik | Process for the production of tetrazolinones |
| EP3616724A1 (en) | 2018-08-27 | 2020-03-04 | SmartDyeLivery GmbH | Pkc inhibitors for the treatment of septic cholestasis with ctm targeting |
| EP3616725A1 (en) | 2018-08-27 | 2020-03-04 | SmartDyeLivery GmbH | Pkc inhibitors for the treatment of septic cholestasis with polymethine dye targeting |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5136868A (en) * | 1983-11-04 | 1992-08-11 | Fmc Corporation | Herbicidal 1-aryl-4-substituted-1,4-dihydro-5h-tetrazol-5-ones and sulfur analogs thereof |
| EP0643049A1 (en) | 1993-09-14 | 1995-03-15 | Nihon Bayer Agrochem K.K. | A process for the preparation of 1-substituted-5(4H)-tetrazolinones |
| WO1997019065A1 (en) * | 1995-11-20 | 1997-05-29 | Celltech Therapeutics Limited | Substituted 2-anilinopyrimidines useful as protein kinase inhibitors |
| WO2003032997A1 (en) * | 2001-10-17 | 2003-04-24 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Pyrimidine derivatives, pharmaceutical agent containing said compounds, use and method for making same |
| WO2003063794A2 (en) | 2002-02-01 | 2003-08-07 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds and their uses |
| WO2004014382A1 (en) | 2002-07-29 | 2004-02-19 | Rigel Pharmaceuticals | Methods of treating or preventing autoimmune diseases with 2,4-pyrimidinediamine compounds |
| WO2004043386A2 (en) | 2002-11-08 | 2004-05-27 | Tolerrx, Inc. | Molecules preferentially associated with effector t cells and methods of their use |
| WO2005013996A2 (en) * | 2003-08-07 | 2005-02-17 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds and uses as anti-proliferative agents |
| WO2005016893A2 (en) | 2003-07-30 | 2005-02-24 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds for use in the treatment or prevention of autoimmune diseases |
| WO2005062918A2 (en) | 2003-12-24 | 2005-07-14 | Wyeth | Methods of treating asthma |
| US20060270694A1 (en) * | 2005-05-03 | 2006-11-30 | Rigel Pharmaceuticals, Inc. | JAK kinase inhibitors and their uses |
| WO2007146981A2 (en) * | 2006-06-15 | 2007-12-21 | Boehringer Ingelheim International Gmbh | 2-anilino-4-(heterocyclic)amino-pyrimidines as inhibitors of protein kinase c-alpha |
| WO2009012421A1 (en) * | 2007-07-17 | 2009-01-22 | Rigel Pharmaceuticals, Inc. | Cyclic amine substituted pyrimidinediamines as pkc inhibitors |
| WO2009158571A1 (en) * | 2008-06-27 | 2009-12-30 | Avila Therapeutics And Uses Thereof | Heteroaryl compounds and uses thereof |
| WO2010090875A1 (en) * | 2009-01-21 | 2010-08-12 | Rigel Pharmaceuticals, Inc. | Derivatives of n2-(3-pyridil or phenyl)-n4-(4-piperidyl)-2,4-pyrimidinediamine useful in the treatment of inflammatory, autoimmune or proliferative diseases |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2355374A1 (en) * | 1999-01-13 | 2000-07-20 | Warner-Lambert Company | 1-heterocycle substituted diarylamines |
| WO2005020921A2 (en) | 2003-08-29 | 2005-03-10 | Exelixis, Inc. | C-kit modulators and methods of use |
-
2010
- 2010-12-01 EP EP10796518.8A patent/EP2507227B1/en not_active Not-in-force
- 2010-12-01 US US12/958,284 patent/US20110130415A1/en not_active Abandoned
- 2010-12-01 CA CA2780759A patent/CA2780759A1/en not_active Abandoned
- 2010-12-01 WO PCT/US2010/058597 patent/WO2011068898A1/en active Application Filing
- 2010-12-01 JP JP2012542165A patent/JP5802677B2/en not_active Expired - Fee Related
-
2016
- 2016-09-09 US US15/261,294 patent/US9732070B2/en active Active
Patent Citations (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5136868A (en) * | 1983-11-04 | 1992-08-11 | Fmc Corporation | Herbicidal 1-aryl-4-substituted-1,4-dihydro-5h-tetrazol-5-ones and sulfur analogs thereof |
| EP0643049A1 (en) | 1993-09-14 | 1995-03-15 | Nihon Bayer Agrochem K.K. | A process for the preparation of 1-substituted-5(4H)-tetrazolinones |
| WO1997019065A1 (en) * | 1995-11-20 | 1997-05-29 | Celltech Therapeutics Limited | Substituted 2-anilinopyrimidines useful as protein kinase inhibitors |
| US5958935A (en) | 1995-11-20 | 1999-09-28 | Celltech Therapeutics Limited | Substituted 2-anilinopyrimidines useful as protein kinase inhibitors |
| US6235746B1 (en) * | 1995-11-20 | 2001-05-22 | Celltech Therapeutics, Limited | Substituted 2-anilinopyrimidines useful as protein kinase inhibitors |
| WO2003032997A1 (en) * | 2001-10-17 | 2003-04-24 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Pyrimidine derivatives, pharmaceutical agent containing said compounds, use and method for making same |
| WO2003063794A2 (en) | 2002-02-01 | 2003-08-07 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds and their uses |
| US20040029902A1 (en) | 2002-02-01 | 2004-02-12 | Rajinder Singh | 2,4-Pyrimidinediamine compounds and their uses |
| WO2004014382A1 (en) | 2002-07-29 | 2004-02-19 | Rigel Pharmaceuticals | Methods of treating or preventing autoimmune diseases with 2,4-pyrimidinediamine compounds |
| WO2004043386A2 (en) | 2002-11-08 | 2004-05-27 | Tolerrx, Inc. | Molecules preferentially associated with effector t cells and methods of their use |
| WO2005016893A2 (en) | 2003-07-30 | 2005-02-24 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds for use in the treatment or prevention of autoimmune diseases |
| US20050234049A1 (en) | 2003-07-30 | 2005-10-20 | Rajinder Singh | Methods of treating or preventing autoimmune diseases with 2, 4-pyrimidinediamine compounds |
| WO2005013996A2 (en) * | 2003-08-07 | 2005-02-17 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds and uses as anti-proliferative agents |
| WO2005062918A2 (en) | 2003-12-24 | 2005-07-14 | Wyeth | Methods of treating asthma |
| US20060270694A1 (en) * | 2005-05-03 | 2006-11-30 | Rigel Pharmaceuticals, Inc. | JAK kinase inhibitors and their uses |
| WO2007146981A2 (en) * | 2006-06-15 | 2007-12-21 | Boehringer Ingelheim International Gmbh | 2-anilino-4-(heterocyclic)amino-pyrimidines as inhibitors of protein kinase c-alpha |
| WO2009012421A1 (en) * | 2007-07-17 | 2009-01-22 | Rigel Pharmaceuticals, Inc. | Cyclic amine substituted pyrimidinediamines as pkc inhibitors |
| WO2009158571A1 (en) * | 2008-06-27 | 2009-12-30 | Avila Therapeutics And Uses Thereof | Heteroaryl compounds and uses thereof |
| WO2010090875A1 (en) * | 2009-01-21 | 2010-08-12 | Rigel Pharmaceuticals, Inc. | Derivatives of n2-(3-pyridil or phenyl)-n4-(4-piperidyl)-2,4-pyrimidinediamine useful in the treatment of inflammatory, autoimmune or proliferative diseases |
Non-Patent Citations (44)
| Title |
|---|
| "Comprehensive Organic Synthesis", vol. 1-9, 1991, PERGAMON PRESS |
| "Introduction to Modem Liquid Chromatography", 1979, JOHN WILEY AND SONS |
| "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING COMPANY |
| "The Peptides", vol. 3, ACADEMIC PRESS |
| "Thin Layer Chromatography", 1969, SPRINGER-VERLAG |
| BAUMANN G ET AL., TRANSPLANT. PROC., vol. 24, 1992, pages 43 - 8 |
| BLAIRE ET AL., BLOOD, vol. 89, 1997, pages 3104 - 3112 |
| BLAY, P. ET AL., CLINICAL CANCER RESEARCH, vol. 10, 2004, pages 12 |
| BROWN, D. J.: "The Chemistry of Heterocyclic Compounds", vol. 16, 1962, INTERSCIENCE PUBLISHERS, article "The Pyrimidines" |
| BROWN, D. J.: "The Chemistry of Heterocyclic Compounds", vol. 16, 1970, WILEY-INTERSCIENCE, article "The Pyrimidines" |
| BROWN, D. J.; BROWN III: "The Chemistry of Heterocyclic Compounds", vol. 16, 1985, AN INTERSCIENCE PUBLICATION, article "The Pyrimidines" |
| BROWN, D. J.; BROWN IV: "The Chemistry of Heterocyclic Compounds", vol. 52, 1994, JOHN WILEY & SONS, INC., article "The Pyrimidines", pages: 1 - 1509 |
| CURRENT DRUG TARGETS: INFLAMMATION & ALLERGY, vol. 4, no. 3, 2005, pages 295 - 298 |
| DE WET J. ET AL., MOL. CELL. BIOL., vol. 7, no. 2, 1987, pages 725 - 737 |
| E. W. MARTIN: "Remington's Pharmaceutical Sciences", 1995, MACK PUBLISHING CO. |
| GUPTA ET AL., TETRAHEDRON LETT, vol. 45, 2004, pages 4113 |
| H.-D. JAKUBKE; H. JESCHEIT: "Aminosauren, Peptide, Proteine", 1982, VERLAG CHEMIE, WEINHEIM, DEERFIELD BEACH, AND BASEL |
| HOUBEN-WEYL: "Methoden der organischen Chemie", vol. 15, 1974, GEORG THIEME VERLAG |
| ISAKOV ET AL., ANNUAL REVIEW OF IMMUNOLOGY, vol. 20, 2002, pages 761 - 794 |
| J. F. W. MCOMIE: "Protective Groups in Organic Chemistry", 1973, PLENUM PRESS |
| J. IMMUNOL. METHODS, vol. 2, 1973, pages 279 |
| JOCHEN LEHMANN: "Chemie der Kohlenhydrate: Monosaccharide and Derivate", 1974, GEORG THIEME VERLAG |
| JOULE, J. A.; MILLS, K.; SMITH, G. F.: "Heterocyclic Chemistry", 1995, CHAPMAN AND HALL, LONDON, pages: 1 - 516 |
| JOULE, J. A.; MILLS, K.; SMITH, G. F.: "Heterocyclic Chemistry", 2000, BLACKWELL SCIENCE, LTD, pages: 1 - 589 |
| KENNER, G. W.; TODD, A.: "Heterocyclic Compounds", vol. 6, 1957, JOHN WILEY |
| KIM J. ET AL., THE J. OF CLINICAL INVESTIGATION, vol. 114, no. 6, 2004, pages 823 |
| KUNO ET AL., BLOOD, vol. 97, 2001, pages 1050 |
| LOUDON: "Organic Chemistry", 2002, OXFORD UNIVERSITY PRESS, pages: 360 - 361,1084 |
| MEO T. ET AL.: "Immunological Methods", 1979, ACADEMIC PRESS, pages: 227 - 39 |
| PAQUETTE, L. A.: "Principles of Modern Heterocyclic Chemistry", 1968, W. A. BENJAMIN, INC., pages: 1 - 401 |
| PASSEGUE ET AL., PROC. NATL. ACAD. SCI. USA, vol. 100, 2003, pages 11842 - 9 |
| POTEWAR ET AL., TETRAHEDRON LETT, vol. 48, 2007, pages 172 |
| SATOH ET AL., TETRAHEDRON LETT, vol. 36, 1995, pages 1749 |
| SMITH; MARCH: "March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure", 2001, WILEY-INTERSCIENCE |
| SU ET AL., EUR. J. ORG. CHEM., 2006, pages 2723 |
| T. W. GREENE; P. G. M. WUTS: "Protective Groups in Organic Synthesis", 1999, WILEY |
| TETRAHEDRON LETTERS, vol. 51, 2010, pages 1009 - 1011 |
| THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS, vol. 313, no. 3, 2005, pages 962 - 982 |
| TURHAN ET AL., BLOOD, vol. 85, 1995, pages 2154 - 2161 |
| V ORBRÜGGEN, H.; RUH-POHLENZ, C.: "Handbook of Nucleoside Synthesis", 2001, JOHN WILEY & SONS, pages: 1 - 631 |
| VOGEL: "A Textbook of Practical Organic Chemistry, Including Qualitative Organic Analysis", 1978, I DNGMAN |
| VOGEL: "A Textbook of Practical Organic Chemistry, Including Qualitative Organic Analysis", 1978, LONGMAN |
| VOGEL: "Practical Organic Chemistry", 1989, ADDISON WESLEY LONGMAN, LTD. AND JOHN WILEY & SONS, INC |
| WIEDMANN, M. ET AL., CURRENT CANCER DRUG TARGETS, vol. 5, no. 3, 2005, pages 171 |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8710223B2 (en) | 2010-07-21 | 2014-04-29 | Rigel Pharmaceuticals, Inc. | Protein kinase C inhibitors and uses thereof |
| WO2012012619A1 (en) | 2010-07-21 | 2012-01-26 | Rigel Pharmaceuticals, Inc. | Protein kinase c inhibitors and uses thereof |
| US9181222B2 (en) | 2010-07-21 | 2015-11-10 | Rigel Pharmaceuticals, Inc. | Protein kinase C inhibitors and uses thereof |
| US9682976B2 (en) | 2012-04-04 | 2017-06-20 | Rigel Pharmaceuticals, Inc. | Protein kinase C inhibitors and uses thereof |
| JP2015519307A (en) * | 2012-04-04 | 2015-07-09 | ライジェル ファーマシューティカルズ, インコーポレイテッド | Protein kinase C inhibitor and use thereof |
| US10005777B2 (en) | 2012-04-04 | 2018-06-26 | Rigel Pharmaceuticals, Inc. | Protein kinase C inhibitors and uses thereof |
| US9321763B2 (en) | 2012-04-04 | 2016-04-26 | Rigel Pharmaceuticals, Inc. | Protein kinase C inhibitors and uses thereof |
| WO2014089112A1 (en) | 2012-12-04 | 2014-06-12 | Rigel Pharmaceuticals, Inc. | Protein kinase c inhibitors and uses thereof |
| US9913843B2 (en) | 2012-12-04 | 2018-03-13 | Rigel Pharmaceuticals, Inc. | Protein kinase C inhibitors and uses thereof |
| US9062068B2 (en) | 2012-12-04 | 2015-06-23 | Rigel Pharmaceuticals, Inc. | Protein kinase C inhibitors and uses thereof |
| US10792282B2 (en) | 2013-03-14 | 2020-10-06 | Rigel Pharmaceuticals, Inc. | Protein kinase C inhibitors and uses thereof |
| JP2016519657A (en) * | 2013-03-14 | 2016-07-07 | ライジェル ファーマシューティカルズ, インコーポレイテッド | Protein kinase C inhibitor and use thereof |
| US9566278B2 (en) | 2013-03-14 | 2017-02-14 | Rigel Pharmaceuticals, Inc. | Protein kinase C inhibitors and uses thereof |
| US9169249B2 (en) | 2013-03-14 | 2015-10-27 | Rigel Pharmaceuticals, Inc. | Protein kinase C inhibitors and uses thereof |
| WO2014151900A1 (en) | 2013-03-14 | 2014-09-25 | Rigel Pharmaceuticals, Inc. | Protein kinase c inhibitors and uses thereof |
| CN105683169B (en) * | 2013-10-28 | 2018-11-06 | 住友化学株式会社 | Terazololine-one compound and application thereof |
| CN105683169A (en) * | 2013-10-28 | 2016-06-15 | 住友化学株式会社 | Tetrazolinone compound and application for same |
| AU2015298364B2 (en) * | 2014-08-06 | 2018-08-16 | Novartis Ag | Protein kinase C inhibitors and methods of their use |
| US9452998B2 (en) | 2014-08-06 | 2016-09-27 | Novartis Ag | Protein kinase C inhibitors and methods of their use |
| EP3514151A1 (en) * | 2014-08-06 | 2019-07-24 | Novartis AG | Protein kinase c inhibitors and methods of their use |
| EA033361B1 (en) * | 2014-08-06 | 2019-10-31 | Novartis Ag | Protein kinase c inhibitors and methods of their use |
| US9845309B2 (en) | 2014-08-06 | 2017-12-19 | Novartis Ag | Protein kinase C inhibitors and methods of their use |
| US10508101B2 (en) | 2014-08-06 | 2019-12-17 | Novartis Ag | Protein kinase C inhibitors and methods of their use |
| WO2016020864A1 (en) * | 2014-08-06 | 2016-02-11 | Novartis Ag | Protein kinase c inhibitors and methods of their use |
| KR102605181B1 (en) | 2014-08-06 | 2023-11-27 | 노파르티스 아게 | Protein kinase c inhibitors and methods of their use |
| KR20230051605A (en) * | 2014-08-06 | 2023-04-18 | 노파르티스 아게 | Protein kinase c inhibitors and methods of their use |
| US11059804B2 (en) | 2014-08-06 | 2021-07-13 | Novartis Ag | Protein kinase C inhibitors and methods of their use |
| US11505541B2 (en) | 2014-08-06 | 2022-11-22 | Novartis Ag | Protein kinase C inhibitors and methods of their use |
| US11446300B2 (en) | 2015-12-04 | 2022-09-20 | Alexion Pharmaceuticals, Inc. | Cerdulatinib for treating hematological cancers |
| US10736895B2 (en) | 2015-12-04 | 2020-08-11 | Portola Pharmaceuticals, Inc. | Cerdulatinib for treating hematological cancers |
| WO2019213545A1 (en) * | 2018-05-04 | 2019-11-07 | Portola Pharmaceuticals, Inc. | Synthesis of cerdulatinib |
| US11339145B2 (en) | 2018-05-04 | 2022-05-24 | Alexion Pharmaceuticals, Inc. | Synthesis of cerdulatinib |
| US10865198B2 (en) | 2018-05-04 | 2020-12-15 | Alexion Pharmaceuticals, Inc. | Solid forms of cerdulatinib |
| TWI805751B (en) * | 2018-05-04 | 2023-06-21 | 美商普托拉製藥有限公司 | Synthesis of cerdulatinib |
| US11713309B2 (en) | 2018-05-04 | 2023-08-01 | Alexion Pharmaceuticals, Inc. | Solid forms of Cerdulatinib |
| US10851087B2 (en) | 2018-05-04 | 2020-12-01 | Portola Pharmaceuticals, Inc. | Synthesis of cerdulatinib |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2780759A1 (en) | 2011-06-09 |
| EP2507227B1 (en) | 2014-10-08 |
| US20110130415A1 (en) | 2011-06-02 |
| JP2013512281A (en) | 2013-04-11 |
| JP5802677B2 (en) | 2015-10-28 |
| US20170152246A1 (en) | 2017-06-01 |
| US9732070B2 (en) | 2017-08-15 |
| EP2507227A1 (en) | 2012-10-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9732070B2 (en) | Protein kinase C inhibitors and uses thereof | |
| CA2801781C (en) | Protein kinase c inhibitors and uses thereof | |
| EP2389373B1 (en) | Derivatives of n2-(3-pyridyl or phenyl)-n4-(4-piperidyl)-2,4-pyrimidinediamine useful in the treatment of inflammatory, autoimmune or proliferative diseases | |
| CA2867760C (en) | Indolizinyl derivatives as protein kinase c inhibitors and uses thereof | |
| EP2928891B1 (en) | Protein kinase c inhibitors and uses thereof | |
| EP2970275B1 (en) | Protein kinase c inhibitors and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10796518 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2780759 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012542165 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010796518 Country of ref document: EP |