WO2011041441A1 - Endoglin antibodies - Google Patents
Endoglin antibodies Download PDFInfo
- Publication number
- WO2011041441A1 WO2011041441A1 PCT/US2010/050759 US2010050759W WO2011041441A1 WO 2011041441 A1 WO2011041441 A1 WO 2011041441A1 US 2010050759 W US2010050759 W US 2010050759W WO 2011041441 A1 WO2011041441 A1 WO 2011041441A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antigen
- antibody
- amino acid
- substitution
- seq
- Prior art date
Links
- 108010036395 Endoglin Proteins 0.000 title claims abstract description 104
- 102000012085 Endoglin Human genes 0.000 title claims abstract description 104
- 230000027455 binding Effects 0.000 claims abstract description 344
- 108091007433 antigens Proteins 0.000 claims abstract description 267
- 102000036639 antigens Human genes 0.000 claims abstract description 267
- 239000000427 antigen Substances 0.000 claims abstract description 266
- 239000012634 fragment Substances 0.000 claims abstract description 262
- 230000033115 angiogenesis Effects 0.000 claims abstract description 83
- 230000004048 modification Effects 0.000 claims abstract description 62
- 238000012986 modification Methods 0.000 claims abstract description 62
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 56
- 239000000203 mixture Substances 0.000 claims abstract description 32
- 201000010099 disease Diseases 0.000 claims abstract description 31
- 238000001514 detection method Methods 0.000 claims abstract description 8
- 238000006467 substitution reaction Methods 0.000 claims description 281
- 238000000034 method Methods 0.000 claims description 153
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 131
- 108090000623 proteins and genes Proteins 0.000 claims description 127
- 150000001413 amino acids Chemical group 0.000 claims description 93
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 91
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 claims description 78
- 206010028980 Neoplasm Diseases 0.000 claims description 71
- 230000014509 gene expression Effects 0.000 claims description 67
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 claims description 59
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 claims description 59
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 claims description 55
- 239000004474 valine Substances 0.000 claims description 54
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 claims description 50
- 235000004279 alanine Nutrition 0.000 claims description 50
- 229920001184 polypeptide Polymers 0.000 claims description 47
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 claims description 46
- 239000004473 Threonine Substances 0.000 claims description 46
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 claims description 42
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 claims description 41
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Natural products NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 claims description 40
- 201000011510 cancer Diseases 0.000 claims description 36
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 claims description 36
- 229960000310 isoleucine Drugs 0.000 claims description 36
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 claims description 34
- 150000007523 nucleic acids Chemical class 0.000 claims description 34
- 102000039446 nucleic acids Human genes 0.000 claims description 34
- 108020004707 nucleic acids Proteins 0.000 claims description 34
- 125000000341 threoninyl group Chemical class [H]OC([H])(C([H])([H])[H])C([H])(N([H])[H])C(*)=O 0.000 claims description 32
- 150000002614 leucines Chemical class 0.000 claims description 29
- 239000004471 Glycine Substances 0.000 claims description 27
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 claims description 25
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 claims description 25
- 235000009582 asparagine Nutrition 0.000 claims description 25
- 229960001230 asparagine Drugs 0.000 claims description 25
- 239000004475 Arginine Substances 0.000 claims description 21
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 claims description 21
- 150000002333 glycines Chemical class 0.000 claims description 17
- 239000000523 sample Substances 0.000 claims description 15
- 206010012689 Diabetic retinopathy Diseases 0.000 claims description 14
- 208000002780 macular degeneration Diseases 0.000 claims description 14
- 206010029113 Neovascularisation Diseases 0.000 claims description 13
- 125000001360 methionine group Chemical class N[C@@H](CCSC)C(=O)* 0.000 claims description 13
- 230000001225 therapeutic effect Effects 0.000 claims description 13
- 239000004037 angiogenesis inhibitor Substances 0.000 claims description 12
- 229940121369 angiogenesis inhibitor Drugs 0.000 claims description 12
- 206010027476 Metastases Diseases 0.000 claims description 11
- 208000022873 Ocular disease Diseases 0.000 claims description 11
- 229930182817 methionine Natural products 0.000 claims description 11
- VEEGZPWAAPPXRB-BJMVGYQFSA-N (3e)-3-(1h-imidazol-5-ylmethylidene)-1h-indol-2-one Chemical compound O=C1NC2=CC=CC=C2\C1=C/C1=CN=CN1 VEEGZPWAAPPXRB-BJMVGYQFSA-N 0.000 claims description 9
- 239000002773 nucleotide Substances 0.000 claims description 9
- 125000003729 nucleotide group Chemical group 0.000 claims description 9
- 201000008482 osteoarthritis Diseases 0.000 claims description 9
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 9
- 208000007342 Diabetic Nephropathies Diseases 0.000 claims description 8
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 claims description 8
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 claims description 8
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims description 8
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 claims description 8
- 208000033679 diabetic kidney disease Diseases 0.000 claims description 8
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 claims description 7
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 claims description 7
- 108091008605 VEGF receptors Proteins 0.000 claims description 7
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 claims description 7
- 230000002596 correlated effect Effects 0.000 claims description 7
- -1 HIF-Ι Proteins 0.000 claims description 6
- 239000003112 inhibitor Substances 0.000 claims description 6
- 102000005962 receptors Human genes 0.000 claims description 6
- 108020003175 receptors Proteins 0.000 claims description 6
- 230000035945 sensitivity Effects 0.000 claims description 6
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 claims description 5
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 5
- 101000595923 Homo sapiens Placenta growth factor Proteins 0.000 claims description 4
- 102100035194 Placenta growth factor Human genes 0.000 claims description 4
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 claims description 4
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 claims description 4
- 201000001441 melanoma Diseases 0.000 claims description 4
- 230000009401 metastasis Effects 0.000 claims description 4
- 201000000050 myeloid neoplasm Diseases 0.000 claims description 4
- 208000026310 Breast neoplasm Diseases 0.000 claims description 3
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 3
- 239000004472 Lysine Substances 0.000 claims description 3
- 206010039491 Sarcoma Diseases 0.000 claims description 3
- 239000003153 chemical reaction reagent Substances 0.000 claims description 3
- 125000003588 lysine group Chemical class [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 claims description 3
- 239000002525 vasculotropin inhibitor Substances 0.000 claims description 3
- 206010006187 Breast cancer Diseases 0.000 claims description 2
- 206010009944 Colon cancer Diseases 0.000 claims description 2
- 206010025323 Lymphomas Diseases 0.000 claims description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 2
- 206010060862 Prostate cancer Diseases 0.000 claims description 2
- 238000002405 diagnostic procedure Methods 0.000 claims description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 2
- 208000020816 lung neoplasm Diseases 0.000 claims description 2
- 230000036210 malignancy Effects 0.000 claims description 2
- 239000013610 patient sample Substances 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 150000001294 alanine derivatives Chemical class 0.000 claims 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims 2
- 206010038389 Renal cancer Diseases 0.000 claims 2
- 208000002495 Uterine Neoplasms Diseases 0.000 claims 2
- 208000014829 head and neck neoplasm Diseases 0.000 claims 2
- 201000010982 kidney cancer Diseases 0.000 claims 2
- 208000014018 liver neoplasm Diseases 0.000 claims 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims 2
- 201000002528 pancreatic cancer Diseases 0.000 claims 2
- 208000008443 pancreatic carcinoma Diseases 0.000 claims 2
- 206010046766 uterine cancer Diseases 0.000 claims 2
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims 1
- 206010033128 Ovarian cancer Diseases 0.000 claims 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims 1
- 208000006265 Renal cell carcinoma Diseases 0.000 claims 1
- 238000002512 chemotherapy Methods 0.000 claims 1
- 208000002551 irritable bowel syndrome Diseases 0.000 claims 1
- 201000007270 liver cancer Diseases 0.000 claims 1
- 201000005202 lung cancer Diseases 0.000 claims 1
- 208000026037 malignant tumor of neck Diseases 0.000 claims 1
- 208000015347 renal cell adenocarcinoma Diseases 0.000 claims 1
- 125000000539 amino acid group Chemical group 0.000 abstract description 45
- 238000011282 treatment Methods 0.000 abstract description 27
- 230000005847 immunogenicity Effects 0.000 abstract description 15
- 238000003745 diagnosis Methods 0.000 abstract description 4
- 125000003275 alpha amino acid group Chemical group 0.000 description 236
- 235000001014 amino acid Nutrition 0.000 description 111
- 210000004027 cell Anatomy 0.000 description 104
- 229940024606 amino acid Drugs 0.000 description 86
- 102000004169 proteins and genes Human genes 0.000 description 68
- 235000018102 proteins Nutrition 0.000 description 65
- 101710117290 Aldo-keto reductase family 1 member C4 Proteins 0.000 description 63
- 210000001744 T-lymphocyte Anatomy 0.000 description 56
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 55
- 108091054438 MHC class II family Proteins 0.000 description 49
- 102000043131 MHC class II family Human genes 0.000 description 48
- 101100112922 Candida albicans CDR3 gene Proteins 0.000 description 46
- 108060003951 Immunoglobulin Proteins 0.000 description 40
- 102000018358 immunoglobulin Human genes 0.000 description 40
- 102000040430 polynucleotide Human genes 0.000 description 33
- 108091033319 polynucleotide Proteins 0.000 description 33
- 239000002157 polynucleotide Substances 0.000 description 33
- 238000003556 assay Methods 0.000 description 31
- 239000013598 vector Substances 0.000 description 31
- 230000000670 limiting effect Effects 0.000 description 30
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 27
- 210000001519 tissue Anatomy 0.000 description 26
- 208000035475 disorder Diseases 0.000 description 25
- CKLJMWTZIZZHCS-REOHCLBHSA-N aspartic acid group Chemical class N[C@@H](CC(=O)O)C(=O)O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 23
- 210000004602 germ cell Anatomy 0.000 description 23
- 108020004414 DNA Proteins 0.000 description 20
- 102000053602 DNA Human genes 0.000 description 20
- 241000699666 Mus <mouse, genus> Species 0.000 description 20
- 150000001507 asparagine derivatives Chemical class 0.000 description 20
- 235000003704 aspartic acid Nutrition 0.000 description 20
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 20
- 230000006870 function Effects 0.000 description 20
- 150000003354 serine derivatives Chemical class 0.000 description 20
- 150000003679 valine derivatives Chemical class 0.000 description 20
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 19
- 108091028043 Nucleic acid sequence Proteins 0.000 description 19
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 18
- 230000003993 interaction Effects 0.000 description 18
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 18
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 18
- 241001529936 Murinae Species 0.000 description 17
- 239000013612 plasmid Substances 0.000 description 17
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 16
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 16
- 230000001419 dependent effect Effects 0.000 description 15
- 230000002062 proliferating effect Effects 0.000 description 15
- 238000002965 ELISA Methods 0.000 description 14
- 239000002246 antineoplastic agent Substances 0.000 description 14
- 230000006907 apoptotic process Effects 0.000 description 14
- 150000002308 glutamine derivatives Chemical class 0.000 description 14
- 230000001575 pathological effect Effects 0.000 description 14
- 230000008569 process Effects 0.000 description 14
- 150000003668 tyrosines Chemical class 0.000 description 14
- 238000004458 analytical method Methods 0.000 description 13
- 230000035772 mutation Effects 0.000 description 13
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 13
- 238000012360 testing method Methods 0.000 description 13
- 230000000875 corresponding effect Effects 0.000 description 12
- 230000002401 inhibitory effect Effects 0.000 description 12
- 230000002829 reductive effect Effects 0.000 description 12
- 108700028369 Alleles Proteins 0.000 description 11
- 230000000694 effects Effects 0.000 description 11
- 230000013595 glycosylation Effects 0.000 description 11
- 238000006206 glycosylation reaction Methods 0.000 description 11
- 238000000338 in vitro Methods 0.000 description 11
- 230000001965 increasing effect Effects 0.000 description 11
- 238000004519 manufacturing process Methods 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 230000015572 biosynthetic process Effects 0.000 description 10
- 230000004663 cell proliferation Effects 0.000 description 10
- 239000003814 drug Substances 0.000 description 10
- 210000002889 endothelial cell Anatomy 0.000 description 10
- 230000012010 growth Effects 0.000 description 10
- 229940072221 immunoglobulins Drugs 0.000 description 10
- 238000001727 in vivo Methods 0.000 description 10
- 150000002519 isoleucine derivatives Chemical class 0.000 description 10
- 239000000463 material Substances 0.000 description 10
- 201000009030 Carcinoma Diseases 0.000 description 9
- 102000014914 Carrier Proteins Human genes 0.000 description 9
- 108020004705 Codon Proteins 0.000 description 9
- 241000588724 Escherichia coli Species 0.000 description 9
- 230000006044 T cell activation Effects 0.000 description 9
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 9
- 108091008324 binding proteins Proteins 0.000 description 9
- 230000008859 change Effects 0.000 description 9
- 230000003247 decreasing effect Effects 0.000 description 9
- 230000028993 immune response Effects 0.000 description 9
- 230000035755 proliferation Effects 0.000 description 9
- 238000000159 protein binding assay Methods 0.000 description 9
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 8
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 8
- 238000007792 addition Methods 0.000 description 8
- 125000003295 alanine group Chemical class N[C@@H](C)C(=O)* 0.000 description 8
- 210000004204 blood vessel Anatomy 0.000 description 8
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 8
- 208000037976 chronic inflammation Diseases 0.000 description 8
- 208000037893 chronic inflammatory disorder Diseases 0.000 description 8
- 239000013604 expression vector Substances 0.000 description 8
- 230000002163 immunogen Effects 0.000 description 8
- 239000003446 ligand Substances 0.000 description 8
- 230000004083 survival effect Effects 0.000 description 8
- 208000005590 Choroidal Neovascularization Diseases 0.000 description 7
- 206010060823 Choroidal neovascularisation Diseases 0.000 description 7
- 102000004127 Cytokines Human genes 0.000 description 7
- 108090000695 Cytokines Proteins 0.000 description 7
- 108020004511 Recombinant DNA Proteins 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 150000001875 compounds Chemical class 0.000 description 7
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 7
- 238000004520 electroporation Methods 0.000 description 7
- 238000000126 in silico method Methods 0.000 description 7
- 238000010348 incorporation Methods 0.000 description 7
- 238000003780 insertion Methods 0.000 description 7
- 230000037431 insertion Effects 0.000 description 7
- 210000004962 mammalian cell Anatomy 0.000 description 7
- 230000037361 pathway Effects 0.000 description 7
- 239000008194 pharmaceutical composition Substances 0.000 description 7
- 230000004962 physiological condition Effects 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 241000894007 species Species 0.000 description 7
- 238000001890 transfection Methods 0.000 description 7
- 210000005166 vasculature Anatomy 0.000 description 7
- 230000001594 aberrant effect Effects 0.000 description 6
- 210000004369 blood Anatomy 0.000 description 6
- 239000008280 blood Substances 0.000 description 6
- 230000000295 complement effect Effects 0.000 description 6
- 230000004540 complement-dependent cytotoxicity Effects 0.000 description 6
- 238000007796 conventional method Methods 0.000 description 6
- 231100000135 cytotoxicity Toxicity 0.000 description 6
- 230000003013 cytotoxicity Effects 0.000 description 6
- 238000012217 deletion Methods 0.000 description 6
- 230000037430 deletion Effects 0.000 description 6
- 239000000539 dimer Substances 0.000 description 6
- 239000012636 effector Substances 0.000 description 6
- 210000003527 eukaryotic cell Anatomy 0.000 description 6
- 230000001976 improved effect Effects 0.000 description 6
- 238000005259 measurement Methods 0.000 description 6
- 230000001404 mediated effect Effects 0.000 description 6
- 238000010369 molecular cloning Methods 0.000 description 6
- 210000004296 naive t lymphocyte Anatomy 0.000 description 6
- 230000004044 response Effects 0.000 description 6
- 230000002441 reversible effect Effects 0.000 description 6
- 238000012216 screening Methods 0.000 description 6
- 230000011664 signaling Effects 0.000 description 6
- SHZGCJCMOBCMKK-UHFFFAOYSA-N D-mannomethylose Natural products CC1OC(O)C(O)C(O)C1O SHZGCJCMOBCMKK-UHFFFAOYSA-N 0.000 description 5
- PNNNRSAQSRJVSB-SLPGGIOYSA-N Fucose Natural products C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C=O PNNNRSAQSRJVSB-SLPGGIOYSA-N 0.000 description 5
- SHZGCJCMOBCMKK-DHVFOXMCSA-N L-fucopyranose Chemical compound C[C@@H]1OC(O)[C@@H](O)[C@H](O)[C@@H]1O SHZGCJCMOBCMKK-DHVFOXMCSA-N 0.000 description 5
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 229930182474 N-glycoside Natural products 0.000 description 5
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 5
- 108700005078 Synthetic Genes Proteins 0.000 description 5
- 230000001640 apoptogenic effect Effects 0.000 description 5
- 238000013459 approach Methods 0.000 description 5
- 230000001580 bacterial effect Effects 0.000 description 5
- 210000004899 c-terminal region Anatomy 0.000 description 5
- 238000010367 cloning Methods 0.000 description 5
- 230000007423 decrease Effects 0.000 description 5
- 230000002950 deficient Effects 0.000 description 5
- 238000011161 development Methods 0.000 description 5
- 230000018109 developmental process Effects 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 230000002209 hydrophobic effect Effects 0.000 description 5
- 230000001900 immune effect Effects 0.000 description 5
- 230000006698 induction Effects 0.000 description 5
- 230000001939 inductive effect Effects 0.000 description 5
- 208000032839 leukemia Diseases 0.000 description 5
- 206010061289 metastatic neoplasm Diseases 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 125000001500 prolyl group Chemical class [H]N1C([H])(C(=O)[*])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 5
- 238000012552 review Methods 0.000 description 5
- 230000000638 stimulation Effects 0.000 description 5
- 235000000346 sugar Nutrition 0.000 description 5
- 238000002560 therapeutic procedure Methods 0.000 description 5
- 241000251730 Chondrichthyes Species 0.000 description 4
- 239000003155 DNA primer Substances 0.000 description 4
- 238000012286 ELISA Assay Methods 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 241000238631 Hexapoda Species 0.000 description 4
- 101000881679 Homo sapiens Endoglin Proteins 0.000 description 4
- 102100037850 Interferon gamma Human genes 0.000 description 4
- 108010074328 Interferon-gamma Proteins 0.000 description 4
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 4
- 108091034117 Oligonucleotide Proteins 0.000 description 4
- 208000002158 Proliferative Vitreoretinopathy Diseases 0.000 description 4
- 206010038934 Retinopathy proliferative Diseases 0.000 description 4
- 108091008874 T cell receptors Proteins 0.000 description 4
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 230000002491 angiogenic effect Effects 0.000 description 4
- 238000003782 apoptosis assay Methods 0.000 description 4
- 125000000637 arginyl group Chemical class N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 238000010276 construction Methods 0.000 description 4
- 238000012258 culturing Methods 0.000 description 4
- 238000013461 design Methods 0.000 description 4
- 230000004069 differentiation Effects 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 229940088598 enzyme Drugs 0.000 description 4
- 230000036541 health Effects 0.000 description 4
- 230000005745 host immune response Effects 0.000 description 4
- 102000048896 human ENG Human genes 0.000 description 4
- 238000002649 immunization Methods 0.000 description 4
- 230000003053 immunization Effects 0.000 description 4
- 210000004072 lung Anatomy 0.000 description 4
- 238000012423 maintenance Methods 0.000 description 4
- 239000003550 marker Substances 0.000 description 4
- 238000013508 migration Methods 0.000 description 4
- 238000002703 mutagenesis Methods 0.000 description 4
- 231100000350 mutagenesis Toxicity 0.000 description 4
- 208000021971 neovascular inflammatory vitreoretinopathy Diseases 0.000 description 4
- 230000003472 neutralizing effect Effects 0.000 description 4
- 208000037916 non-allergic rhinitis Diseases 0.000 description 4
- 210000001672 ovary Anatomy 0.000 description 4
- 230000035790 physiological processes and functions Effects 0.000 description 4
- 230000006785 proliferative vitreoretinopathy Effects 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 239000002904 solvent Substances 0.000 description 4
- 238000010561 standard procedure Methods 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 230000009466 transformation Effects 0.000 description 4
- 125000000430 tryptophan group Chemical class [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C2=C([H])C([H])=C([H])C([H])=C12 0.000 description 4
- 210000004881 tumor cell Anatomy 0.000 description 4
- 241000701447 unidentified baculovirus Species 0.000 description 4
- 230000004862 vasculogenesis Effects 0.000 description 4
- 108091026890 Coding region Proteins 0.000 description 3
- 101150074155 DHFR gene Proteins 0.000 description 3
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- 241000196324 Embryophyta Species 0.000 description 3
- 201000010915 Glioblastoma multiforme Diseases 0.000 description 3
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 3
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 3
- 125000000174 L-prolyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])[C@@]1([H])C(*)=O 0.000 description 3
- 101100490437 Mus musculus Acvrl1 gene Proteins 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- 239000004365 Protease Substances 0.000 description 3
- 238000012300 Sequence Analysis Methods 0.000 description 3
- 206010046865 Vaccinia virus infection Diseases 0.000 description 3
- 241000700605 Viruses Species 0.000 description 3
- 230000005875 antibody response Effects 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 210000000481 breast Anatomy 0.000 description 3
- 238000004113 cell culture Methods 0.000 description 3
- 230000010261 cell growth Effects 0.000 description 3
- 230000005754 cellular signaling Effects 0.000 description 3
- 210000001072 colon Anatomy 0.000 description 3
- 230000010339 dilation Effects 0.000 description 3
- 230000008030 elimination Effects 0.000 description 3
- 238000003379 elimination reaction Methods 0.000 description 3
- 208000005017 glioblastoma Diseases 0.000 description 3
- 210000003128 head Anatomy 0.000 description 3
- 230000028996 humoral immune response Effects 0.000 description 3
- 210000002865 immune cell Anatomy 0.000 description 3
- 229940127121 immunoconjugate Drugs 0.000 description 3
- 210000003292 kidney cell Anatomy 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 238000013507 mapping Methods 0.000 description 3
- 230000001394 metastastic effect Effects 0.000 description 3
- 229960000485 methotrexate Drugs 0.000 description 3
- 210000000822 natural killer cell Anatomy 0.000 description 3
- 210000003739 neck Anatomy 0.000 description 3
- 239000013642 negative control Substances 0.000 description 3
- 230000014399 negative regulation of angiogenesis Effects 0.000 description 3
- 230000009826 neoplastic cell growth Effects 0.000 description 3
- 230000001613 neoplastic effect Effects 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 238000003752 polymerase chain reaction Methods 0.000 description 3
- 230000005522 programmed cell death Effects 0.000 description 3
- 210000001236 prokaryotic cell Anatomy 0.000 description 3
- 210000002307 prostate Anatomy 0.000 description 3
- 230000005180 public health Effects 0.000 description 3
- 238000010188 recombinant method Methods 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 230000002207 retinal effect Effects 0.000 description 3
- 238000002864 sequence alignment Methods 0.000 description 3
- 238000012163 sequencing technique Methods 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 238000002741 site-directed mutagenesis Methods 0.000 description 3
- 210000003491 skin Anatomy 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 3
- 238000001356 surgical procedure Methods 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 239000003053 toxin Substances 0.000 description 3
- 231100000765 toxin Toxicity 0.000 description 3
- 108700012359 toxins Proteins 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- 238000011144 upstream manufacturing Methods 0.000 description 3
- 208000007089 vaccinia Diseases 0.000 description 3
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 2
- 108090000672 Annexin A5 Proteins 0.000 description 2
- 102000004121 Annexin A5 Human genes 0.000 description 2
- 101100227726 Arabidopsis thaliana FRL3 gene Proteins 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 101100120609 Caenorhabditis elegans frh-1 gene Proteins 0.000 description 2
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 2
- 241000606161 Chlamydia Species 0.000 description 2
- 241000699800 Cricetinae Species 0.000 description 2
- 238000001712 DNA sequencing Methods 0.000 description 2
- 102100024746 Dihydrofolate reductase Human genes 0.000 description 2
- 241000701959 Escherichia virus Lambda Species 0.000 description 2
- 101150065691 FRL2 gene Proteins 0.000 description 2
- 108010087819 Fc receptors Proteins 0.000 description 2
- 102000009109 Fc receptors Human genes 0.000 description 2
- 102100032789 Formin-like protein 3 Human genes 0.000 description 2
- 102000006471 Fucosyltransferases Human genes 0.000 description 2
- 108010019236 Fucosyltransferases Proteins 0.000 description 2
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 2
- 108700007698 Genetic Terminator Regions Proteins 0.000 description 2
- 208000010412 Glaucoma Diseases 0.000 description 2
- 102000003886 Glycoproteins Human genes 0.000 description 2
- 108090000288 Glycoproteins Proteins 0.000 description 2
- 101000690301 Homo sapiens Aldo-keto reductase family 1 member C4 Proteins 0.000 description 2
- 101000935587 Homo sapiens Flavin reductase (NADPH) Proteins 0.000 description 2
- 101001116548 Homo sapiens Protein CBFA2T1 Proteins 0.000 description 2
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 description 2
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 description 2
- 125000002842 L-seryl group Chemical group O=C([*])[C@](N([H])[H])([H])C([H])([H])O[H] 0.000 description 2
- 206010061309 Neoplasm progression Diseases 0.000 description 2
- 108090000526 Papain Proteins 0.000 description 2
- 102000057297 Pepsin A Human genes 0.000 description 2
- 108090000284 Pepsin A Proteins 0.000 description 2
- 241000589516 Pseudomonas Species 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 230000006052 T cell proliferation Effects 0.000 description 2
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 2
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 2
- 108010009583 Transforming Growth Factors Proteins 0.000 description 2
- 102000009618 Transforming Growth Factors Human genes 0.000 description 2
- 101150117115 V gene Proteins 0.000 description 2
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 2
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 230000003321 amplification Effects 0.000 description 2
- 210000004102 animal cell Anatomy 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- 238000004166 bioassay Methods 0.000 description 2
- 230000003115 biocidal effect Effects 0.000 description 2
- 230000008499 blood brain barrier function Effects 0.000 description 2
- 210000001218 blood-brain barrier Anatomy 0.000 description 2
- 210000001124 body fluid Anatomy 0.000 description 2
- 239000010839 body fluid Substances 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- 210000001185 bone marrow Anatomy 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 239000001110 calcium chloride Substances 0.000 description 2
- 229910001628 calcium chloride Inorganic materials 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 230000030833 cell death Effects 0.000 description 2
- 230000012292 cell migration Effects 0.000 description 2
- 238000001516 cell proliferation assay Methods 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 210000003679 cervix uteri Anatomy 0.000 description 2
- 230000002759 chromosomal effect Effects 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 208000029742 colonic neoplasm Diseases 0.000 description 2
- 239000002299 complementary DNA Substances 0.000 description 2
- 238000004590 computer program Methods 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 2
- 210000004443 dendritic cell Anatomy 0.000 description 2
- 230000001627 detrimental effect Effects 0.000 description 2
- 108020001096 dihydrofolate reductase Proteins 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 230000002124 endocrine Effects 0.000 description 2
- 230000003511 endothelial effect Effects 0.000 description 2
- 210000003989 endothelium vascular Anatomy 0.000 description 2
- 210000003238 esophagus Anatomy 0.000 description 2
- 230000001747 exhibiting effect Effects 0.000 description 2
- 230000002349 favourable effect Effects 0.000 description 2
- 238000000855 fermentation Methods 0.000 description 2
- 230000004151 fermentation Effects 0.000 description 2
- 239000012894 fetal calf serum Substances 0.000 description 2
- 101150029401 fmnl3 gene Proteins 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 238000007429 general method Methods 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 108020002326 glutamine synthetase Proteins 0.000 description 2
- 210000002443 helper t lymphocyte Anatomy 0.000 description 2
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 2
- 102000054751 human RUNX1T1 Human genes 0.000 description 2
- 210000004408 hybridoma Anatomy 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 206010022000 influenza Diseases 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 230000010354 integration Effects 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 208000037841 lung tumor Diseases 0.000 description 2
- 230000003211 malignant effect Effects 0.000 description 2
- 230000000877 morphologic effect Effects 0.000 description 2
- 210000000214 mouth Anatomy 0.000 description 2
- 201000003142 neovascular glaucoma Diseases 0.000 description 2
- 238000003199 nucleic acid amplification method Methods 0.000 description 2
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 210000000496 pancreas Anatomy 0.000 description 2
- 229940055729 papain Drugs 0.000 description 2
- 235000019834 papain Nutrition 0.000 description 2
- 230000006320 pegylation Effects 0.000 description 2
- 229940111202 pepsin Drugs 0.000 description 2
- 239000000816 peptidomimetic Substances 0.000 description 2
- 210000005259 peripheral blood Anatomy 0.000 description 2
- 239000011886 peripheral blood Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 230000008488 polyadenylation Effects 0.000 description 2
- 239000013615 primer Substances 0.000 description 2
- 230000009696 proliferative response Effects 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- 230000006798 recombination Effects 0.000 description 2
- 238000005215 recombination Methods 0.000 description 2
- 210000000664 rectum Anatomy 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000013391 scatchard analysis Methods 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 230000009870 specific binding Effects 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- PVYJZLYGTZKPJE-UHFFFAOYSA-N streptonigrin Chemical compound C=1C=C2C(=O)C(OC)=C(N)C(=O)C2=NC=1C(C=1N)=NC(C(O)=O)=C(C)C=1C1=CC=C(OC)C(OC)=C1O PVYJZLYGTZKPJE-UHFFFAOYSA-N 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 230000002381 testicular Effects 0.000 description 2
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 2
- 210000001685 thyroid gland Anatomy 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 238000010361 transduction Methods 0.000 description 2
- 230000026683 transduction Effects 0.000 description 2
- 230000009261 transgenic effect Effects 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- 230000005751 tumor progression Effects 0.000 description 2
- 241001515965 unidentified phage Species 0.000 description 2
- 241001430294 unidentified retrovirus Species 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 210000000264 venule Anatomy 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- MSTNYGQPCMXVAQ-RYUDHWBXSA-N (6S)-5,6,7,8-tetrahydrofolic acid Chemical compound C([C@H]1CNC=2N=C(NC(=O)C=2N1)N)NC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 MSTNYGQPCMXVAQ-RYUDHWBXSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- GZCWLCBFPRFLKL-UHFFFAOYSA-N 1-prop-2-ynoxypropan-2-ol Chemical compound CC(O)COCC#C GZCWLCBFPRFLKL-UHFFFAOYSA-N 0.000 description 1
- UAIUNKRWKOVEES-UHFFFAOYSA-N 3,3',5,5'-tetramethylbenzidine Chemical compound CC1=C(N)C(C)=CC(C=2C=C(C)C(N)=C(C)C=2)=C1 UAIUNKRWKOVEES-UHFFFAOYSA-N 0.000 description 1
- OSJPPGNTCRNQQC-UWTATZPHSA-N 3-phospho-D-glyceric acid Chemical compound OC(=O)[C@H](O)COP(O)(O)=O OSJPPGNTCRNQQC-UWTATZPHSA-N 0.000 description 1
- 238000010600 3H thymidine incorporation assay Methods 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- 102100038222 60 kDa heat shock protein, mitochondrial Human genes 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- WLCZTRVUXYALDD-IBGZPJMESA-N 7-[[(2s)-2,6-bis(2-methoxyethoxycarbonylamino)hexanoyl]amino]heptoxy-methylphosphinic acid Chemical compound COCCOC(=O)NCCCC[C@H](NC(=O)OCCOC)C(=O)NCCCCCCCOP(C)(O)=O WLCZTRVUXYALDD-IBGZPJMESA-N 0.000 description 1
- 102000013563 Acid Phosphatase Human genes 0.000 description 1
- 108010051457 Acid Phosphatase Proteins 0.000 description 1
- 108010032595 Antibody Binding Sites Proteins 0.000 description 1
- 206010003445 Ascites Diseases 0.000 description 1
- 206010003694 Atrophy Diseases 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- 241000193830 Bacillus <bacterium> Species 0.000 description 1
- 108010077805 Bacterial Proteins Proteins 0.000 description 1
- 201000004569 Blindness Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 208000014644 Brain disease Diseases 0.000 description 1
- 125000001433 C-terminal amino-acid group Chemical group 0.000 description 1
- 102000005701 Calcium-Binding Proteins Human genes 0.000 description 1
- 108010045403 Calcium-Binding Proteins Proteins 0.000 description 1
- 241000282836 Camelus dromedarius Species 0.000 description 1
- 239000005635 Caprylic acid (CAS 124-07-2) Substances 0.000 description 1
- 101710132601 Capsid protein Proteins 0.000 description 1
- 201000000274 Carcinosarcoma Diseases 0.000 description 1
- 108010076667 Caspases Proteins 0.000 description 1
- 102000011727 Caspases Human genes 0.000 description 1
- 206010057248 Cell death Diseases 0.000 description 1
- 108010058432 Chaperonin 60 Proteins 0.000 description 1
- 241000282552 Chlorocebus aethiops Species 0.000 description 1
- 108010077544 Chromatin Proteins 0.000 description 1
- 101710094648 Coat protein Proteins 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 102100030497 Cytochrome c Human genes 0.000 description 1
- 108010075031 Cytochromes c Proteins 0.000 description 1
- 230000004544 DNA amplification Effects 0.000 description 1
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 1
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 238000011510 Elispot assay Methods 0.000 description 1
- 101000738180 Euglena gracilis Chaperonin CPN60, mitochondrial Proteins 0.000 description 1
- 108010040476 FITC-annexin A5 Proteins 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 108700004714 Gelonium multiflorum GEL Proteins 0.000 description 1
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 1
- 102100021181 Golgi phosphoprotein 3 Human genes 0.000 description 1
- 102000006354 HLA-DR Antigens Human genes 0.000 description 1
- 108010058597 HLA-DR Antigens Proteins 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- 108091027305 Heteroduplex Proteins 0.000 description 1
- 102000018713 Histocompatibility Antigens Class II Human genes 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000693265 Homo sapiens Sphingosine 1-phosphate receptor 1 Proteins 0.000 description 1
- 102000009786 Immunoglobulin Constant Regions Human genes 0.000 description 1
- 108010009817 Immunoglobulin Constant Regions Proteins 0.000 description 1
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 101150008942 J gene Proteins 0.000 description 1
- 125000000570 L-alpha-aspartyl group Chemical group [H]OC(=O)C([H])([H])[C@]([H])(N([H])[H])C(*)=O 0.000 description 1
- 125000000510 L-tryptophano group Chemical group [H]C1=C([H])C([H])=C2N([H])C([H])=C(C([H])([H])[C@@]([H])(C(O[H])=O)N([H])[*])C2=C1[H] 0.000 description 1
- 108010001831 LDL receptors Proteins 0.000 description 1
- 102100024640 Low-density lipoprotein receptor Human genes 0.000 description 1
- 208000001344 Macular Edema Diseases 0.000 description 1
- 206010025415 Macular oedema Diseases 0.000 description 1
- 101710125418 Major capsid protein Proteins 0.000 description 1
- 102000012750 Membrane Glycoproteins Human genes 0.000 description 1
- 108010090054 Membrane Glycoproteins Proteins 0.000 description 1
- 208000034578 Multiple myelomas Diseases 0.000 description 1
- 230000004988 N-glycosylation Effects 0.000 description 1
- 125000000729 N-terminal amino-acid group Chemical group 0.000 description 1
- 108091007491 NSP3 Papain-like protease domains Proteins 0.000 description 1
- 206010028851 Necrosis Diseases 0.000 description 1
- 101710141454 Nucleoprotein Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 101150012394 PHO5 gene Proteins 0.000 description 1
- 241000237988 Patellidae Species 0.000 description 1
- 208000037273 Pathologic Processes Diseases 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 108010067902 Peptide Library Proteins 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 241001505332 Polyomavirus sp. Species 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 101710083689 Probable capsid protein Proteins 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- 108010039491 Ricin Proteins 0.000 description 1
- 108010084592 Saporins Proteins 0.000 description 1
- 102000005686 Serum Globulins Human genes 0.000 description 1
- 108010045362 Serum Globulins Proteins 0.000 description 1
- 102100025750 Sphingosine 1-phosphate receptor 1 Human genes 0.000 description 1
- 101710145796 Staphylokinase Proteins 0.000 description 1
- 108091081024 Start codon Proteins 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 241000187747 Streptomyces Species 0.000 description 1
- 238000000692 Student's t-test Methods 0.000 description 1
- 230000005867 T cell response Effects 0.000 description 1
- 108091005735 TGF-beta receptors Proteins 0.000 description 1
- 108010055044 Tetanus Toxin Proteins 0.000 description 1
- 108010022394 Threonine synthase Proteins 0.000 description 1
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 1
- 102000016715 Transforming Growth Factor beta Receptors Human genes 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 206010047513 Vision blurred Diseases 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 208000009956 adenocarcinoma Diseases 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 230000009824 affinity maturation Effects 0.000 description 1
- 229960002833 aflibercept Drugs 0.000 description 1
- 108010081667 aflibercept Proteins 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical class N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 1
- 238000012870 ammonium sulfate precipitation Methods 0.000 description 1
- 210000000648 angioblast Anatomy 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 210000001557 animal structure Anatomy 0.000 description 1
- 230000001772 anti-angiogenic effect Effects 0.000 description 1
- 210000000628 antibody-producing cell Anatomy 0.000 description 1
- 210000000612 antigen-presenting cell Anatomy 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 230000037444 atrophy Effects 0.000 description 1
- 229940120638 avastin Drugs 0.000 description 1
- 239000013602 bacteriophage vector Substances 0.000 description 1
- 210000002469 basement membrane Anatomy 0.000 description 1
- 108010079292 betaglycan Proteins 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 230000008238 biochemical pathway Effects 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 208000034158 bleeding Diseases 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 230000015624 blood vessel development Effects 0.000 description 1
- 238000004422 calculation algorithm Methods 0.000 description 1
- 229930195731 calicheamicin Natural products 0.000 description 1
- 125000000837 carbohydrate group Chemical group 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000022534 cell killing Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000009134 cell regulation Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000003822 cell turnover Effects 0.000 description 1
- 230000007541 cellular toxicity Effects 0.000 description 1
- 238000012412 chemical coupling Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 210000000038 chest Anatomy 0.000 description 1
- 210000003483 chromatin Anatomy 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 230000001447 compensatory effect Effects 0.000 description 1
- 238000012875 competitive assay Methods 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 230000000536 complexating effect Effects 0.000 description 1
- 230000002153 concerted effect Effects 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 239000012228 culture supernatant Substances 0.000 description 1
- 230000001351 cycling effect Effects 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 230000001461 cytolytic effect Effects 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 230000002074 deregulated effect Effects 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 208000018554 digestive system carcinoma Diseases 0.000 description 1
- 102000004419 dihydrofolate reductase Human genes 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 229960003983 diphtheria toxoid Drugs 0.000 description 1
- 210000001840 diploid cell Anatomy 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 230000013020 embryo development Effects 0.000 description 1
- 210000000750 endocrine system Anatomy 0.000 description 1
- 230000010595 endothelial cell migration Effects 0.000 description 1
- 238000003114 enzyme-linked immunosorbent spot assay Methods 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 231100000776 exotoxin Toxicity 0.000 description 1
- 239000002095 exotoxin Substances 0.000 description 1
- 210000001508 eye Anatomy 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- 238000013467 fragmentation Methods 0.000 description 1
- 238000006062 fragmentation reaction Methods 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 102000054766 genetic haplotypes Human genes 0.000 description 1
- 238000003205 genotyping method Methods 0.000 description 1
- 210000004907 gland Anatomy 0.000 description 1
- 230000000762 glandular Effects 0.000 description 1
- 230000002414 glycolytic effect Effects 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 210000000777 hematopoietic system Anatomy 0.000 description 1
- 108060003552 hemocyanin Proteins 0.000 description 1
- 239000000710 homodimer Substances 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 230000008348 humoral response Effects 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 206010020718 hyperplasia Diseases 0.000 description 1
- 230000002390 hyperplastic effect Effects 0.000 description 1
- 210000001822 immobilized cell Anatomy 0.000 description 1
- 230000008105 immune reaction Effects 0.000 description 1
- 230000006058 immune tolerance Effects 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000012405 in silico analysis Methods 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 238000007689 inspection Methods 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 230000010039 intracellular degradation Effects 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 125000000741 isoleucyl group Chemical group [H]N([H])C(C(C([H])([H])[H])C([H])([H])C([H])([H])[H])C(=O)O* 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 230000021633 leukocyte mediated immunity Effects 0.000 description 1
- 229940076783 lucentis Drugs 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 229940092110 macugen Drugs 0.000 description 1
- 201000010230 macular retinal edema Diseases 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 230000000116 mitigating effect Effects 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 102000035118 modified proteins Human genes 0.000 description 1
- 108091005573 modified proteins Proteins 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 230000013152 negative regulation of cell migration Effects 0.000 description 1
- 230000035407 negative regulation of cell proliferation Effects 0.000 description 1
- 230000024717 negative regulation of secretion Effects 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 230000000683 nonmetastatic effect Effects 0.000 description 1
- 239000011824 nuclear material Substances 0.000 description 1
- 238000007899 nucleic acid hybridization Methods 0.000 description 1
- 210000004940 nucleus Anatomy 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 229960002446 octanoic acid Drugs 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 231100000590 oncogenic Toxicity 0.000 description 1
- 230000002246 oncogenic effect Effects 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 230000027758 ovulation cycle Effects 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 238000005192 partition Methods 0.000 description 1
- 230000009054 pathological process Effects 0.000 description 1
- 210000003899 penis Anatomy 0.000 description 1
- 238000012510 peptide mapping method Methods 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- 210000003800 pharynx Anatomy 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 108700028325 pokeweed antiviral Proteins 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 150000004032 porphyrins Chemical class 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000035935 pregnancy Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 1
- 208000023958 prostate neoplasm Diseases 0.000 description 1
- 235000019419 proteases Nutrition 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 238000011158 quantitative evaluation Methods 0.000 description 1
- 238000003127 radioimmunoassay Methods 0.000 description 1
- 229960003876 ranibizumab Drugs 0.000 description 1
- 238000013102 re-test Methods 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 230000037390 scarring Effects 0.000 description 1
- 238000003345 scintillation counting Methods 0.000 description 1
- 239000006152 selective media Substances 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 230000037432 silent mutation Effects 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 230000003335 steric effect Effects 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 210000000106 sweat gland Anatomy 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- 239000005460 tetrahydrofolate Substances 0.000 description 1
- 238000011285 therapeutic regimen Methods 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 230000030968 tissue homeostasis Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 102000003390 tumor necrosis factor Human genes 0.000 description 1
- 231100000588 tumorigenic Toxicity 0.000 description 1
- 230000000381 tumorigenic effect Effects 0.000 description 1
- 210000003606 umbilical vein Anatomy 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- 210000002229 urogenital system Anatomy 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 238000002255 vaccination Methods 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 230000006444 vascular growth Effects 0.000 description 1
- 230000024883 vasodilation Effects 0.000 description 1
- 230000007998 vessel formation Effects 0.000 description 1
- 230000009278 visceral effect Effects 0.000 description 1
- 230000004304 visual acuity Effects 0.000 description 1
- 230000004393 visual impairment Effects 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
- 230000037314 wound repair Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/28—Compounds containing heavy metals
- A61K31/282—Platinum compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
- A61K31/4045—Indole-alkylamines; Amides thereof, e.g. serotonin, melatonin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/69—Boron compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/704—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin attached to a condensed carbocyclic ring system, e.g. sennosides, thiocolchicosides, escin, daunorubicin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/39558—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against tumor tissues, cells, antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2896—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against molecules with a "CD"-designation, not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3015—Breast
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3038—Kidney, bladder
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3046—Stomach, Intestines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3061—Blood cells
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3069—Reproductive system, e.g. ovaria, uterus, testes, prostate
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/574—Immunoassay; Biospecific binding assay; Materials therefor for cancer
- G01N33/57484—Immunoassay; Biospecific binding assay; Materials therefor for cancer involving compounds serving as markers for tumor, cancer, neoplasia, e.g. cellular determinants, receptors, heat shock/stress proteins, A-protein, oligosaccharides, metabolites
- G01N33/57492—Immunoassay; Biospecific binding assay; Materials therefor for cancer involving compounds serving as markers for tumor, cancer, neoplasia, e.g. cellular determinants, receptors, heat shock/stress proteins, A-protein, oligosaccharides, metabolites involving compounds localized on the membrane of tumor or cancer cells
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/574—Immunoassay; Biospecific binding assay; Materials therefor for cancer
- G01N33/57484—Immunoassay; Biospecific binding assay; Materials therefor for cancer involving compounds serving as markers for tumor, cancer, neoplasia, e.g. cellular determinants, receptors, heat shock/stress proteins, A-protein, oligosaccharides, metabolites
- G01N33/57496—Immunoassay; Biospecific binding assay; Materials therefor for cancer involving compounds serving as markers for tumor, cancer, neoplasia, e.g. cellular determinants, receptors, heat shock/stress proteins, A-protein, oligosaccharides, metabolites involving intracellular compounds
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6872—Intracellular protein regulatory factors and their receptors, e.g. including ion channels
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/34—Identification of a linear epitope shorter than 20 amino acid residues or of a conformational epitope defined by amino acid residues
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
- C07K2317/732—Antibody-dependent cellular cytotoxicity [ADCC]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/705—Assays involving receptors, cell surface antigens or cell surface determinants
- G01N2333/70596—Molecules with a "CD"-designation not provided for elsewhere in G01N2333/705
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/705—Assays involving receptors, cell surface antigens or cell surface determinants
- G01N2333/71—Assays involving receptors, cell surface antigens or cell surface determinants for growth factors; for growth regulators
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/16—Ophthalmology
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/16—Ophthalmology
- G01N2800/164—Retinal disorders, e.g. retinopathy
Definitions
- Endoglin also known as, inter alia, CD 105 or edg-1, is a type I homodimeric membrane glycoprotein which is expressed at high levels in proliferating vascular endothelial cells
- endoglin is primarily a
- TGF- ⁇ transforming growth factor- ⁇
- Endoglin has been targeted in antibody-based methods of reducing tumor vasculature, as EDG is a proliferation-associated antigen on endothelial and leukemia cells. Its expression is up-regulated in tumor-associated vascular endothelium, and EDG is essential for angiogenesis.
- Angiogenesis includes the formation of new capillary blood vessels leading to neovascularization as well as the maintenance of the existing vasculature. It is a complex process which includes a series of sequential steps including endothelial cell-mediated degradation of vascular basement membrane and interstitial matrices, migration of endothelial cells,
- mAb SN6 is an antibody generated from immunization of mice with glycoprotein mixtures of cell membranes of human leukemia cells (Haruta and Seon, 1986, Proc. Natl. Acad. Sci. 83:7898-7902). SN6 is a murine mAb that recognizes human endoglin.
- mAb 44G4 is an antibody generated from immunization of mice with whole cell suspensions of human pre-B leukemia cells (Gougos and Letarte, 1988, J. Immunol. 141 : 1925-1933; 1990, J. Biol. Chem.
- 44G4 is also a murine mAb that recognizes human endoglin.
- mAb MJ7/18 is an antibody generated from immunization of rats with inflamed mouse skins (Ge and Butcher, 1994, supra).
- MJ7/18 is a mAb that recognizes murine endoglin.
- mAb Tec-11 is an antibody generated from immunization of mice with human umbilical vein endothelial cells
- Tec-11 is a murine mAb with reactivity restricted to human endoglin. Antibodies against endoglin represent an important area for the development of therapies for the treatment of a variety of diseases and conditions which involve, are influenced by, or affected by angiogenesis.
- Angiogenesis is the physiological process by which new blood vessels develop from pre- existing vessels (Varner, et al, Cell Adh. Commun. 1995, 3:367-374; Blood, et al, Biochim. Biophys. Acta. 1990, 1032:89-118; Weidner, et al, J. Natl. Cancer Inst. 1992, 84: 1875-1887).
- Angiogenesis has been suggested to play a role in both normal and pathological processes. For example, angiogenic processes are involved in the development of the vascular systems of animal organs and tissues. These processes are also involved in transitory phases of
- angiogenesis for example during the menstrual cycle, in pregnancy, and in wound healing.
- a number of diseases are known to be associated with deregulated angiogenesis.
- angiogenesis is stimulated as a means to provide adequate blood and nutrient supply to the cells within affected tissue. Many of these pathological conditions involve aberrant cell proliferation and/or regulation. Therefore, inhibition of angiogenesis is a potentially useful approach to treating diseases that are characterized by new blood vessel development. For example, angiogenesis is involved in pathologic conditions including: various forms of ocular and non-ocular diseases characterized by
- angiogenesis/neovascularization e.g., macular degeneration, diabetic retinopathy), diabetic nephropathy, chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, and various forms of cancer, solid tumors, and metastases and the like.
- humanized antibodies or antigen-binding fragments thereof that bind to endoglin. Such antibodies have in vitro and in vivo purification, detection, diagnostic and therapeutic uses. Also provided herein are humanized antibodies or antigen-binding fragments thereof that bind to one or more species or variants of endoglin and inhibit angiogenesis. Further provided herein are methods of treating angiogenesis-associated diseases with humanized antibodies or antigen-binding fragments thereof that bind to endoglin.
- humanized antibodies and antigen-binding fragments which bind endoglin and are described herein can be used to treat or prevent macular degeneration, CNV, diabetic
- retinopathy or proliferative vitreoretinopathy.
- Described herein are methods of treating or preventing macular degeneration, CNV, diabetic retinopathy, or proliferative vitreoretinopathy via the administration of the antibodies and antigen-binding fragments described herein.
- the humanized antibodies and antigen-binding fragments which bind endoglin and are described herein can also shrink blood vessels, inhibit endothelial cell proliferation associated with ocular disease, clear symptoms of bleeding, treat cloudy vision, provide stasis of vision loss, and/or prevent leakage of blood vessels.
- humanized antibodies and antigen-binding fragments described herein can also be used in medicaments for the treatment of macular degeneration, CNV, diabetic retinopathy or proliferative vitreoretinopathy.
- the humanized antibodies and antigen-binding fragments described herein can also be used in medicaments for the treatment of cancer.
- antibodies, or antigen-binding fragments thereof having a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 41 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 3.
- antibodies, or antigen-binding fragments thereof that bind endoglin having comprising a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 3 and a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 41, wherein: said heavy chain variable region further comprises one or more modifications selected from the group consisting of a substitution of glycine (G) by alanine (A) at position 49; a substitution of asparagine (N) by serine (S) at position 76; a substitution of threonine (T) by arginine (R) at position 77; a substitution of leucine (L) by valine (V) at position 78; a substitution of asparagine (N) by isoleucine (I) at position 82a; a substitution of valine (V) by isoleucine (I) or leucine (L) at position 89; a substitution of threonine (T) by
- antibodies, or antigen-binding fragments thereof that bind endoglin having a heavy chain variable region and a light chain variable region, wherein said heavy chain variable region comprises:
- valine (V) a substitution of valine (V) by isoleucine (I) or leucine (L) at position 89;
- said light chain variable region comprises: (i) a CDRl of SEQ ID NO: 63, a CDR2 of SEQ ID NO: 64, and a CDR3 of SEQ ID NO:
- amino acid sequence of SEQ ID NO: 20 except for one or more substitutions selected from the group consisting of:
- amino acid sequence of SEQ ID NO: 28 except for one or more substitutions selected from the group consisting of:
- amino acid sequence of SEQ ID NO: 35 except for one or more substitutions selected from the group consisting of:
- an antibody, or antigen-binding fragment thereof comprising a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 42 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 4.
- an antibody, or antigen-binding fragment thereof, that binds endoglin comprising a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 4 and a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 42, wherein: said heavy chain variable region further comprises one or more modifications selected from the group consisting of a substitution of glycine (G) by alanine (A) at position 49; a substitution of asparagine (N) by serine (S) at position 76; a substitution of threonine (T) by arginine (R) at position 77; a substitution of leucine (L) by valine (V) at position 78; a substitution of asparagine (N) by isoleucine (I) at position 82a; a substitution of valine (V) by isoleucine (I) or leucine (L) at position 89; a substitution of arginine (R) by a substitution of glycine (G
- an antibody, or antigen-binding fragment thereof, that binds endoglin comprising a heavy chain variable region and a light chain variable region,
- said heavy chain variable region comprises:
- valine (V) a substitution of valine (V) by isoleucine (I) or leucine (L) at position 89;
- a light chain FRl having the amino acid sequence of SEQ ID NO: 6 or the amino acid sequence of SEQ ID NO: 6 except for one or more substitutions selected from the group consisting of: (a) a substitution of aspartic acid (D) by glutamine (Q) at position 1 ;
- amino acid sequence of SEQ ID NO: 20 except for one or more substitutions selected from the group consisting of:
- amino acid sequence of SEQ ID NO: 28 except for one or more substitutions selected from the group consisting of:
- amino acid sequence of SEQ ID NO: 35 except for one or more substitutions selected from the group consisting of:
- an antibody, or antigen-binding fragment thereof comprising a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 93 (VK1 AA) and a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 89 (VH1A2).
- an antibody, or antigen-binding fragment thereof that binds endoglin, comprising a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 89 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 93, wherein:
- the heavy chain variable region further comprises one or more modifications selected from the group consisting of a substitution of glycine (G) by alanine (A) or serine (S) at position 49; a substitution of alanine (A) by isoleucine (I) at position 51; a substitution of lysine (K) by arginine (R) or asparagine (Q) at position 52b; a substitution of leucine (L) by valine (V) at position 78 utilizing the Kabat numbering system; and
- the light chain variable region further comprises one or more modifications selected from the group consisting of a substitution of methionine (M) by leucine (L) at position 4; a substitution of alanine (A) by valine (V) at position 19; a substitution of threonine (T) by serine (S) at position 22; a substitution of alanine (A) by isoleucine (I) at position 48; and a substitution of threonine (T) by serine (S) at position 51 utilizing the Kabat numbering system.
- an antibody, or antigen-binding fragment thereof, of claim 2 comprising a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 88, 89, 90, 91 or 92; and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 93, 94, 95, 96, 97, 100, 102, or 103.
- the antibodies and antigen-binding fragments described herein are humanized and can be any isotype. Also encompassed herein are AVIMERs, diabodies, and heavy chain dimers (including camelids and shark heavy chain constructs).
- antibody fragment or a “functional fragment of an antibody” are used interchangeably herein to refer to one or more fragments of an antibody that retain the ability to specifically bind to an antigen.
- Non-limiting examples of antibody fragments included within such terms include, but are not limited to, (i) a Fab fragment, a monovalent fragment consisting of the V L , V H , C L and C HI domains; (ii) a F(ab') 2 fragment, a bivalent fragment containing two Fab fragments linked by a disulfide bridge at the hinge region; (iii) a Fd fragment consisting of the V H and C HI domains; (iv) a Fv fragment containing the V L and V H domains of a single arm of an antibody, (v) a dAb fragment (Ward et al, (1989) Nature 341 :544 546), which containing a V H domain; and (vi) an isolated CDR.
- an antigen-binding fragment can be any of those described herein including, but not limited to, a Fab fragment, a Fab', a F(ab') 2 fragment, an Fv fragment (including non-covalently and covalently linked Fv fragments), an scFv fragment, a single chain binding polypeptide, an Fd fragment, an Fv fragment or a dAb fragment.
- the antigen- binding fragment is a scFv which can, optionally, be further fused to a human Fc portion of an antibody.
- the antibody, or antigen-binding fragment thereof that binds endoglin comprises a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 41, 42, or 43, and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 3, 4, or 5.
- the antibody, or antigen-binding fragment thereof comprises a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 41 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 3.
- the antibody, or antigen-binding fragment thereof comprises a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO:
- the antibody, or antigen-binding fragment thereof comprises a comprising a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 41 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 5.
- the antibody, or antigen-binding fragment thereof comprises a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO:
- the antibody, or antigen-binding fragment thereof comprises a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 42 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 4.
- the antibody, or antigen-binding fragment thereof comprises a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO:
- the antibody, or antigen-binding fragment thereof comprises a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO:
- the antibody, or antigen-binding fragment thereof comprises a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 43 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 4.
- the antibody, or antigen-binding fragment thereof comprises a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 43 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 5.
- the antibody, or antigen-binding fragment thereof heavy chain variable region further comprises one or more modifications selected from the group consisting of: a substitution of asparagine (N) by serine (S) at position 76; a substitution of threonine (T) by arginine (R) at position 77; a substitution of asparagine (N) by isoleucine (I) at position 82a; a substitution of valine (V) by isoleucine (I) or leucine (L) at position 89; a substitution of threonine (T) by glycine (G) at position 94; a substitution of leucine (L) by threonine (T) at position 108; a substitution of valine (V) by leucine (L) at position 109; and a substitution of serine (S) by alanine (A) a position 113; and said light chain variable region further comprises one or more modifications selected from the group consisting of: a substitution of asparagine
- the antibodies and antigen-binding fragments described herein can be modified.
- the compound can be modified to alter a
- pharmacokinetic property of the compound such as, for example, in vivo stability, solubility, bioavailability or half-life.
- modifications include, but are not limited to, PEGylation and/or glycosylation.
- the antibodies and antigen-binding fragments described herein can be formulated for rapid or extended delivery using conventional means.
- rapid delivery is, for example, by intravenous injection.
- extended delivery is, for example, by subcutaneous deposition.
- delivery is achieved via administration by aerosol.
- compositions of the antibodies and antigen-binding fragments described herein and an acceptable carrier or excipient are provided herein.
- polynucleotides nucleic acids
- Antibodies and antigen-binding fragments thereof as described herein can be used to treat various diseases and conditions associated with angiogenesis, e.g., various forms of ocular diseases characterized by angiogenesis/neovascularization (e.g., macular degeneration, diabetic retinopathy), diabetic nephropathy, chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, and various forms of cancer, solid tumors, and metastases.
- angiogenesis/neovascularization e.g., macular degeneration, diabetic retinopathy
- diabetic nephropathy e.g., chronic inflammatory diseases (e.g., IBD)
- rheumatoid arthritis e.g., osteoarthritis
- various forms of cancer e.g., various forms of ocular diseases characterized by angiogenesis/neovascularization (e.g., macular degeneration, diabetic retinopathy), diabetic
- these antibodies and antigen-binding fragments thereof described herein can be used in the formulation of a medicament for the prophylaxis, treatment, or diagnosis of diseases and conditions associated with angiogenesis, e.g., various forms of ocular and non-ocular diseases characterized by angiogenesis/neovascularization (e.g., macular degeneration, diabetic retinopathy), diabetic nephropathy, chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, and various forms of cancer, solid tumors, and metastases.
- angiogenesis/neovascularization e.g., macular degeneration, diabetic retinopathy
- diabetic nephropathy e.g., chronic inflammatory diseases (e.g., IBD)
- rheumatoid arthritis e.g., osteoarthritis
- various forms of cancer solid tumors, and metastases.
- composition comprises a humanized anti-endoglin antibody or antigen-binding fragment thereof that induces an effective host immune response against the epitope specifically recognized by said antibody or fragment thereof.
- the host immune response can be a humoral immune response or a cell-mediated immune response. If the immune response is a humoral immune response, it can be a protective antibody response that inhibits angiogenesis, an angiogenesis-dependent disease or an angiogenesis-dependent disorder. Immune responses also include induction or blockage of cell signaling pathways (e.g. Smad signaling).
- the angiogenesis-dependent disease or disorder can be, for example, various forms of ocular and non-ocular diseases characterized by
- angiogenesis/neovascularization e.g., macular degeneration, diabetic retinopathy), diabetic nephropathy, chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, various forms of cancer (primary tumors and metastases) and the like.
- the protective antibody response inhibits angiogenesis.
- Angiogenic cells can be contacted (in vitro, in vivo or ex vivo) with an antibody or antigen-binding fragment thereof described herein in an amount sufficient to alter cell signaling pathways.
- Smad 1, 5 and/or 8 signaling is inhibited by about 1.5 fold or more in angiogenic cells.
- Smad 3 levels increase by about 1.5 fold or more, indicating that cells are returning to a quiescent state.
- angiogenesis-dependent disease or disorder can be any of the following: various forms of ocular and non-ocular diseases characterized by angiogenesis/neovascularization (e.g., macular degeneration, diabetic retinopathy), diabetic nephropathy, chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, and various forms of cancer, solid tumors and metastases.
- inhibiting angiogenesis or an angiogenesis-dependent disease or disorder alleviates symptoms associated with the disease or disorder.
- inhibiting angiogenesis or an angiogenesis-dependent disease or disorder results in decreased tumor size, prevention of tumor progression, decreased cell proliferation, increased apoptosis, or increased survival of a patient.
- Inhibiting in angiogenesis can result in decreased tumor size or prevent tumor progression.
- the method can further include surgical removal of a cancer, and/or administration of on or more additional anti-cancer agents or treatments to a patient suffering from cancer.
- a cancer/tumor to be treated includes a solid tumor; a tumor can be a primary tumor or a metastatic tumor.
- Exemplary solid tumors are of a tissue or organ selected from among skin, melanoma, lung, pancreas, breast, ovary, colon, rectum, stomach, thyroid, laryngeal, ovarian, prostate, colorectal, head, neck, eye, mouth, throat, esophagus, chest, bone, testicular, lymphoid, marrow, bone, sarcoma, renal, sweat gland, liver, kidney, brain, e.g. glioblastoma multiforme and the like tissues.
- a solid tumor is a colon tumor, a breast tumor, a kidney tumor, a lung tumor, a prostate tumor, an ovarian tumor, or metastasis of any of such tumors.
- the method can further include surgical removal of the cancer and/or administration of one or more anti-cancer agents.
- An anti-cancer agent can be administered prior to, concomitant with, or subsequent to, administration of the pharmaceutical composition.
- An anti-cancer agent can be administered within a week before the pharmaceutical composition, within a week after the pharmaceutical composition, or the anti-cancer agent can be administered on the same day as the pharmaceutical composition. If an anti-cancer agent is administered on the same day as the pharmaceutical composition, administration can be concomitant.
- Provided herein is a method for preventing or treating a cancer or a metastasis by surgical removal of the cancer/tumor and concurrent administration of an anti-cancer agent or treatment and a composition provided herein to a subject.
- angiogenesis Provided herein is a method of inhibiting angiogenesis by contacting a cell or tissue with a therapeutically effective amount of an antibody or antigen-binding fragment thereof as described herein sufficient to inhibit angiogenesis.
- an antibody or antigen-binding fragment thereof as described herein sufficient to inhibit cancer cell growth or cause apoptosis of the cancer cell.
- tissue can be a cultured tissue biopsy sample or can be present in a subject.
- a method of preventing or treating a cell proliferative (e.g., angiogenic) disorder by administering to a subject having or at risk of having a cell proliferative disorder an effective amount of a composition provided herein effective to treat the cell proliferative disorder.
- the cell proliferative disorder can be, for example a benign or malignant solid or non-solid tumor and the tumor can be metastatic or non-metastatic.
- the treatment can result in improving the subject's condition and can be assessed by determining if one or more of the following factors has occurred: decreased cell proliferation, decreased numbers of cells, increased apoptosis, or decreased survival of at least a portion of the cells comprising the cell proliferative disorder.
- the method can further include administering an anti-cancer agent or treatment to the subject.
- a method for treating diabetic retinopathy, macular degeneration, choroidal neovascularization or neovascular glaucoma in a patient by administering to the patient a therapeutically effective amount of a composition provided herein.
- the treatment can result in improving the subject's condition and can be assess by determining if one or more of the following factors has occurred: decreased macular edema, decreased areas of CNV, or increased visual acuity.
- the subject can be a human or a non-human subject.
- compositions and the anti-cancer agent or treatments provided herein can be administered once or multiple times depending on the health of the patient, the progression of the disease or condition, and the efficacy of the treatment. Adjustments to therapy and treatments can be made throughout the course of treatment. [0050] Compositions can be administered locally, regionally or systemically, such as, for example, administration by subcutaneous, subcutaneous, intravitreal, intradermal, intravenous, intra-arterial, intraperitoneal or intramuscular injection.
- humanized antibodies and antigen-binding fragments described herein can also be used in combination with known therapies and/or compounds for the treatment of macular degeneration, CNV, diabetic retinopathy or proliferative vitreoretinopathy.
- therapies and/or compounds for the treatment of macular degeneration, CNV, diabetic retinopathy or proliferative vitreoretinopathy.
- examples of such compounds include, but are not limited to, bevacizumab (AVASTIN®), ranibizumab (LUCENTIS®), aflibercept (VEGF-Trap), or Macugen.
- the humanized anti-endoglin antibodies and antigen-binding fragments can be administered via intravitreal routes.
- intravitreal modes of administration include intravitreal injection and the use of intravitreal implants.
- Another aspect is the treatment of a chronic inflammatory disease in a subject by administering a composition of an antibody or antigen-binding fragment described herein.
- chronic inflammatory diseases include IBD, Crohn's disease, and ulcerative colitis.
- Another aspect is the treatment of rheumatoid arthritis in a subject by administering a composition of an antibody or antigen-binding fragment described herein.
- Another aspect is the treatment of osteoarthritis in a subject by administering a composition of an antibody or antigen-binding fragment described herein.
- Treatment of a subject with rheumatoid arthritis and/or osteoarthritis can be assessed by various means, including improvement in the appropriate categories of ACR scores as measured according to published guidelines.
- a method of monitoring the efficacy of one or more of any of the methods provided herein is a method of monitoring the efficacy of one or more of any of the methods provided herein.
- Increased levels of soluble endoglin have been correlated with decreased survival in cancer patients.
- levels of soluble endoglin can be monitored prior to and during therapy.
- a decrease in the levels of soluble endoglin can, therefore, be one indication that a therapeutic regimen is effective in treating the patient.
- compositions of the present invention to formulate a medicament for treating a disorder of the present invention.
- Medicaments can be formulated based on the physical characteristics of the patient/subject needing treatment, and can be formulated in single or multiple formulations based on the stage of the cancerous tissue.
- Medicaments of the present invention can be packaged in a suitable pharmaceutical package with appropriate labels for the distribution to hospitals and clinics wherein the label is for the indication of treating a disorder as described herein in a subject.
- Medicaments can be packaged as a single or multiple units. Instructions for the dosage and administration of the pharmaceutical compositions of the present invention can be included with the pharmaceutical packages.
- a diagnostic method for providing a sample of cancer cells of a solid tumor or plasma from a patient to be tested, detecting in the sample the expression of at least one gene or gene product chosen from a panel of genes or gene products whose expression has been correlated with sensitivity or resistance to an angiogenesis inhibitor, wherein the at least one gene or gene product is chosen from one or more genes or gene products selected from the group consisting of VEGF, VEGF receptor, HIF- ⁇ , placental growth factor receptor, and CD 105, and comparing the level of expression of at least one gene or gene product detected in the patient sample to a level of expression of at least one gene or gene product that has been correlated with sensitivity or resistance to the angiogenesis inhibitor.
- the angiogenesis inhibitor is chosen from VEGF receptor inhibitors, VEGF inhibitors, and endoglin inhibitors.
- kits for the detection of expression levels of genes that have been correlated with sensitivity or resistance to an angiogenesis inhibitor in a sample of cancer cells or human plasma are selected from VEGF, VEGF receptor, HIF- ⁇ , placental growth factor receptor, and endoglin.
- Figure 1 provides a humanized 02-VK1-39 variable (V L ) light chain having the monoclonal murine chimeric TRC105 V L CDRs (underlined) grafted between the framework regions (FRs) 1-3 of the human sequence 02-VK1-39 and a framework region 4 from the human JK4 sequence (SEQ ID NO: 4) (all in bold). Variations that can be made to the human FRs are indicated at positions 1, 3, 4, 5, 36, 46, 47, 60, 70, 71, 100, and 106 of the sequence (sequence disclosed at SEQ ID NO: 86) utilizing the Kabat numbering system (shown in italics beneath the humanized sequence).
- Figure 2 provides a humanized VH3-15 variable (V H ) heavy chain having the monoclonal murine monoclonal murine chimeric TRC 105 V H CDRS (underlined) grafted between the framework regions (FRs) 1-3 of the human sequence VH3-15 and a framework region 4 from the human JH4 sequence (SEQ ID NO: 42) (all in bold).
- V H variable
- FRs framework regions
- SEQ ID NO: 42 all in bold.
- One or more variations that can be made to the human FRs are indicated at positions 49, 76, 77, 78, 82a, 89, 94, 108, 109, and 113 of the sequence (sequence disclosed at SEQ ID NO: 87) utilizing the Kabat numbering system (shown in italics beneath the humanized sequence).
- FIG. 3 provides diagram of the TGF-P/ALK5 signaling pathway.
- the TGF-P/ALK5 pathway (A) leads to inhibition of cell proliferation and migration.
- the TGF-p/ALKl pathway (B) induces endothelial cell proliferation and migration and requires CD 105 (endoglin) for ALK1 signaling.
- the dotted lines indicate inactive or blocked pathways.
- the bolded arrow indicates stimulation of a signaling pathway.
- Figure 4 provides an amino acid sequence alignment of exemplary mouse and humanized VK chains (SEQ ID NOS 1-5, respectively, in order of appearance) and V H chains (SEQ ID NOS 39-43, respectively, in order of appearance) produced according the invention described herein.
- Figure 5 provides an amino acid sequence alignment of exemplary mouse and super- humanized VK chains (SEQ ID NOS 1 and 69-72, respectively, in order of appearance) and V H chains (SEQ ID NOS 39 and 73-75, respectively, in order of appearance) produced according the invention described herein.
- Figure 6 provides an amino acid sequence alignment and comparison of exemplary mouse and humanized and super-humanized VK chains (SEQ ID NOS 1, 3 and 70, respectively, in order of appearance) and V H chains (SEQ ID NOS 39, 41 and 74, respectively, in order of appearance) produced according the invention described herein.
- Figure 7 illustrates binding of humanized variant constructs to endoglin in a competition ELISA assay.
- FIG. 8 Anti-CD 105 competition ELISA with humanized and humanized/deimmunized antibodies. Varying concentrations of each antibody were mixed with a fixed concentration of biotinylated reference anti-CD 105 antibody (6.25 ng/ml) and bound to CD 105 (100 ng/ml) captured on a Nunc MaxiSorp plate. Binding was detected via streptavidin-HRP and TMB substrate. Absorbance (OD) at 450nm was measured on a plate reader and this was plotted against the test antibody concentration.
- Figure 9 illustrates binding assay data for variant VK1 AAVH1 A plus VK2 containing controls.
- Figure 10 illustrates binding assay data for chimeric compared to VK1 AAVH1 A2 and VK2AAVH1A2.
- Figure 11 illustrates binding assay data for chimeric compared to VK2AAVH1Q.
- Figure 12 illustrates binding assay data for chimeric compared to VK2AAVH1R.
- Figure 13 illustrates binding assay data for chimeric compared to VK1 AAVHl A2.
- Figure 14 illustrates binding assay data for chimeric compared to VKIAAVHIQ.
- Figure 15 illustrates binding assay data for chimeric compared to VK1 AAVHIR.
- Figure 16 illustrates binding assay data for chimeric compared to VK2AAVH1 A2.
- Figure 17 illustrates the lead humanized deimmunized heavy chain variable region with CDRs in bold and underlined (sequence disclosed at SEQ ID NO: 89). Variations that can be made are indicated at the identified positions of the sequence utilizing the Kabat numbering system (sequence disclosed at SEQ ID NO: 116) (shown in italics beneath the humanized sequence). Variations may be made as a single mutation or as more than one mutation, and variations can be made with mutations in any combination.
- Figure 18 illustrates the lead humanized deimmunized light chain variable region with CDRs in bold and underlined (sequence disclosed at SEQ ID NO: 93). Variations that can be made are indicated at the identified positions of the sequence utilizing the Kabat numbering system (sequence disclosed at SEQ ID NO: 117) (shown in italics beneath the humanized sequence). Variations may be made as a single mutation or as more than one mutation, and variations can be made with mutations in any combination.
- Figure 19 illustrates Analysis of the regions of the heavy chain (SEQ ID NO: 41) using iTopeTM. Peptides spanning the entire sequence were tested as 9mer peptides in one amino acid increments. The predicted binding of each residue as the pi anchor of a core 9mer peptide to MHC class II alleles is indicated by a "O" if the binding score was 0.55-0.6 and by a "X” if the binding score was >0.6. Regions containing potentially immunogenic peptides are indicated in the "iTope” column, dark grey indicates promiscuous high affinity MHC class II binding peptides, light grey indicates promiscuous moderate affinity MHC class II binding peptides. The numbers of MHC class II alleles predicted to bind are shown in the "total” and “high affinity” columns. Potential pi anchor residues identified as germline sequences are shown in reverse type in the "Sequence” column.
- Figure 20 illustrates analysis of the variant regions of variants of the heavy chain (SEQ ID NO: 118) using iTopeTM. Peptides spanning the entire sequence were tested as 9mer peptides in one amino acid increments. The predicted binding of each residue as the pi anchor of a core 9mer peptide to MHC class II alleles is indicated by a "O” if the binding score was 0.55-0.6 and by a "X” if the binding score was >0.6. Regions containing potentially immunogenic peptides are indicated in the "iTope” column, dark grey indicates promiscuous high affinity MHC class II binding peptides, light grey indicates promiscuous moderate affinity MHC class II binding peptides.
- MHC class II alleles predicted to bind are shown in the “total” and “high affinity” columns and the difference to binding is shown.
- Potential pi anchor residues identified as germline sequences are shown in reverse type in the "Sequence” column and the amino acid differences in the variants are boxed.
- Figure 21 illustrates analysis of the regions of the light chain (SEQ ID NO: 3) using iTopeTM. Peptides spanning the entire sequence were tested as 9mer peptides in one amino acid increments. The predicted binding of each residue as the pi anchor of a core 9mer peptide to MHC class II alleles is indicated by a "O" if the binding score was 0.55-0.6 and by a "X" if the binding score was >0.6. Regions containing potentially immunogenic peptides are indicated in the "iTope” column, dark grey indicates promiscuous high affinity MHC class II binding peptides, light grey indicates promiscuous moderate affinity MHC class II binding peptides. The numbers of MHC class II alleles predicted to bind are shown in the "total” and “high affinity” columns. Potential pi anchor residues identified as germline sequences are shown in reverse type in the "Sequence” column.
- Figure 22 illustrates analysis of the variant regions of the light chain (SEQ ID NO: 101) using iTopeTM. Peptides spanning the entire sequence were tested as 9mer peptides in one amino acid increments. The predicted binding of each residue as the pi anchor of a core 9mer peptide to MHC class II alleles is indicated by a "O” if the binding score was 0.55-0.6 and by a "X” if the binding score was >0.6. Regions containing potentially immunogenic peptides are indicated in the "iTope” column, dark grey indicates promiscuous high affinity MHC class II binding peptides, light grey indicates promiscuous moderate affinity MHC class II binding peptides.
- MHC class II alleles predicted to bind are shown in the “total” and “high affinity” columns and the difference to binding is shown.
- Potential pi anchor residues identified as germline sequences are shown in reverse type in the "Sequence” column and the amino acid differences in the variants are boxed.
- Figure 23 illustrates the frequency of MHC Class II allotypes in the world population and the study population.
- FIG. 24 Chimeric anti-endoglin antibody, humanized anti-endoglin antibody VK1 VH1 and humanized/deimmunized anti-endoglin antibody VK1AAVH1 A2 were tested in EpiScreenTM time course T cell assays using PBMC from 20 donors. Bulk cultures of PBMC incubated with test antibodies were sampled on days 5, 6, 7 and 8, and pulsed with 3 H- Thymidine. Cells were harvested and incorporation of radioactivity measured by scintillation counting. Results for each triplicate sample were averaged and normalized by conversion to Stimulation Index (SI).
- SI Stimulation Index
- Murine monoclonal antibodies have been raised against endoglin which modulate endoglin activity and thereby inhibit angiogenesis and/or inhibit vasodilation of small blood vessels. These murine antibodies are described in U.S. patents 5,928,641, 6,200,566, 6,190,660, and 7,097,836 which are hereby incorporated in their entirety. Additionally, the ex vivo and in vivo efficiency of a number of these antibodies has been demonstrated; these monoclonal antibodies that bind endoglin are of interest as endoglin modulating compounds. Therapeutic use of these murine antibodies is not feasible, however, as their administration has a number of limitations, including immunogenicity in, for example, the form of human anti-mouse antibodies (HAM A). Humanized antibodies are made to address these reactions.
- HAM A human anti-mouse antibodies
- Humanized antibodies that bind endoglin are described herein that exhibit reduced immunogenicity while maintaining and/or improving their specificity. Additionally, to address problems associated with murine antibodies, humanized antibodies that bind endoglin and decrease and/or inhibit angiogenesis are described herein that exhibit reduced immunogenicity while maintaining and/or improving their specificity. These humanized endoglin antibodies are useful for the diagnosis and treatment of various conditions and diseases as well as for purification and detection of endoglin.
- endoglin can be found on cells that comprise and support existing vasculature as well as cells that are promoting the growth of, and become part of, new vasculature. These antibodies and antigen-binding fragments can bind endoglin and thereby inhibit angiogenesis, inhibit the existing vasculature or the maintenance of the existing vasculature, and/or inhibit small vessel dilation.
- antibody or “antibodies” are to be considered inclusive of any of the antigen-binding fragments described herein and the terms are to be interchangeable where applicable.
- angiogenesis is inclusive of the growth and/or development of new blood vessels (also referred to as neovascularization), dilation of the small vessels, excessive or prolonged vascular growth, and maintenance of the existing vasculature.
- Angiogenesis conditions and diseases refers to those diseases and conditions related to, caused by, or associated with angiogenesis.
- Non-limiting examples of such diseases include, for example, various forms of ocular diseases characterized by angiogenesis/neovascularization (e.g., macular degeneration, CNV, diabetic retinopathy), diabetic nephropathy, chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, and various forms of cancer (primary tumors and metastases).
- angiogenesis/neovascularization e.g., macular degeneration, CNV, diabetic retinopathy
- diabetic nephropathy e.g., chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, and various forms of cancer (primary tumors and metastases).
- antibody refers to an immunoglobulin (Ig) whether natural or partly or wholly synthetically produced.
- Ig immunoglobulin
- the term also covers any polypeptide or protein having a binding domain which is, or is homologous to, an antigen-binding domain.
- the term further includes “antigen-binding fragments” and other interchangeable terms for similar binding fragments such as described below.
- Complementarity determining region (CDR) grafted antibodies and other humanized antibodies are also contemplated by this term.
- Native antibodies and native immunoglobulins are usually heterotetrameric glycoproteins of about 150,000 Daltons, composed of two identical light (L) chains and two identical heavy (H) chains. Each light chain is typically linked to a heavy chain by one covalent disulfide bond, while the number of disulfide linkages varies among the heavy chains of different
- Each heavy and light chain also has regularly spaced intrachain disulfide bridges.
- Each heavy chain has at one end a variable domain ("V R ") followed by a number of constant domains ("C H ").
- Each light chain has a variable domain at one end (“V L ”) and a constant domain (“C L ”) at its other end; the constant domain of the light chain is aligned with the first constant domain of the heavy chain, and the light-chain variable domain is aligned with the variable domain of the heavy chain.
- Particular amino acid residues are believed to form an interface between the light- and heavy-chain variable domains.
- synthetic polynucleotide means that the corresponding polynucleotide sequence or portion thereof, or amino acid sequence or portion thereof, is derived, from a sequence that has been designed, or synthesized de novo, or modified, compared to an equivalent naturally-occurring sequence.
- Synthetic polynucleotides (antibodies or antigen binding fragments) or synthetic genes can be prepared by methods known in the art, including but not limited to, the chemical synthesis of nucleic acid or amino acid sequences.
- Synthetic genes are typically different from naturally- occurring genes, either at the amino acid, or polynucleotide level, (or both) and are typically located within the context of synthetic expression control sequences.
- synthetic gene sequences can include amino acid, or polynucleotide, sequences that have been changed, for example, by the replacement, deletion, or addition, of one or more, amino acids, or nucleotides, thereby providing an antibody amino acid sequence, or a polynucleotide coding sequence that is different from the source sequence.
- Synthetic gene polynucleotide sequences may not necessarily encode proteins with different amino acids, compared to the natural gene; for example, they can also encompass synthetic polynucleotide sequences that incorporate different codons but which encode the same amino acid (i.e., the nucleotide changes represent silent mutations at the amino acid level).
- the term "variable domain” refers to the variable domains of antibodies that are used in the binding and specificity of each particular antibody for its particular antigen. However, the variability is not evenly distributed throughout the variable domains of antibodies. Rather, it is concentrated in three segments called hypervariable regions (also known as CDRs) in both the light chain and the heavy chain variable domains.
- variable domains More highly conserved portions of variable domains are called the "framework regions" or "FRs.”
- the variable domains of unmodified heavy and light chains each contain four FRs (FR1, FR2, FR3 and FR4), largely adopting a ⁇ -sheet configuration interspersed with three CDRs which form loops connecting and, in some cases, part of the ⁇ -sheet structure.
- the CDRs in each chain are held together in close proximity by the FRs and, with the CDRs from the other chain, contribute to the formation of the antigen-binding site of antibodies (see Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health,
- hypervariable region refers to the amino acid residues of an antibody which are responsible for antigen-binding.
- the CDRs comprise amino acid residues from three sequence regions which bind in a complementary manner to an antigen and are known as CDRl, CDR2, and CDR3 for each of the V H and V L chains.
- the CDRs typically correspond to approximately residues 24-34 (CDRLl), 50- 56 (CDRL2) and 89-97 (CDRL3), and in the heavy chain variable domain the CDRs typically correspond to approximately residues 31-35 (CDRH1), 50-65 (CDRH2) and 95-102 (CDRH3) according to Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991)). It is understood that the CDRs of different antibodies may contain insertions, thus the amino acid numbering may differ.
- the Kabat numbering system accounts for such insertions with a numbering scheme that utilizes letters attached to specific residues (e.g., 27A, 27B, 27C, 27D, 27E, and 27F of CDRLl in the light chain) to reflect any insertions in the numberings between different antibodies.
- the CDRs typically correspond to
- CDRLl approximately residues 26-32
- CDRL2 50-52
- CDRL3 91-96
- CDRH1 CDRH1
- CDRH2 53-55
- CDRH3 96-101
- framework region refers to framework amino acid residues that form a part of the antigen binding pocket or groove.
- the framework residues form a loop that is a part of the antigen binding pocket or groove and the amino acids residues in the loop may or may not contact the antigen.
- Framework regions generally comprise the regions between the CDRs.
- the FRs typically correspond to approximately residues 0-23 (FRL1), 35-49 (FRL2), 57-88 (FRL3), and 98-109 and in the heavy chain variable domain the FRs typically correspond to approximately residues 0-30 (FRH1), 36-49 (FRH2), 66-94 (FRH3), and 103-133 according to Kabat et al, Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991)).
- the heavy chain too accounts for insertions in a similar manner (e.g., 35A, 35B of CDRHl in the heavy chain).
- the FRs typically correspond to approximately residues 0-25 (FRLl), 33-49 (FRL2) 53-90 (FRL3), and 97-109 (FRL4)
- the FRs typically correspond to approximately residues 0-25 (FRH1), 33-52 (FRH2), 56-95 (FRH3), and 102-113 (FRH4) according to Chothia and Lesk, J. Mol. Biol, 196: 901-917 (1987)).
- the loop amino acids of a FR can be assessed and determined by inspection of the three- dimensional structure of an antibody heavy chain and/or antibody light chain.
- the three- dimensional structure can be analyzed for solvent accessible amino acid positions as such positions are likely to form a loop and/or provide antigen contact in an antibody variable domain. Some of the solvent accessible positions can tolerate amino acid sequence diversity and others (e.g., structural positions) are, generally, less diversified.
- the three dimensional structure of the antibody variable domain can be derived from a crystal structure or protein modeling.
- Constant domains (Fc) of antibodies are not involved directly in binding an antibody to an antigen but, rather, exhibit various effector functions, such as participation of the antibody in antibody-dependent cellular toxicity via interactions with, for example, Fc receptors (FcR). Fc domains can also increase bioavailability of an antibody in circulation following administration to a patient.
- immunoglobulins can be assigned to different classes. There are five major classes of
- immunoglobulins IgA, IgD, IgE, IgG, and IgM, and several of these can be further divided into subclasses (isotypes), e.g., IgGl, IgG2, IgG3, IgG4, IgAl, and IgA2.
- the heavy-chain constant domains (Fc) that correspond to the different classes of immunoglobulins are called ⁇ , ⁇ , ⁇ , ⁇ , and ⁇ , respectively.
- the subunit structures and three-dimensional configurations of different classes of immunoglobulins are well known.
- the "light chains” of antibodies (immunoglobulins) from any vertebrate species can be assigned to one of two clearly distinct types, called kappa or (" ⁇ " or "K”) and lambda or (" ⁇ "), based on the amino acid sequences of their constant domains.
- antigen-binding portion of an antibody refers to one or more fragments of an antibody that retain the ability to specifically bind to an antigen.
- Non-limiting examples of antibody fragments included within such terms include, but are not limited to, (i) a Fab fragment, a monovalent fragment consisting of the VL, VH, CL and CHI domains; (ii) a F(ab') 2 fragment, a bivalent fragment containing two Fab fragments linked by a disulfide bridge at the hinge region; (iii) a Fd fragment consisting of the VH and CHI domains; (iv) a Fv fragment containing the VL and VH domains of a single arm of an antibody, (v) a dAb fragment (Ward et al, (1989) Nature 341 :544 546), which containing a VH domain; and (vi) an isolated CDR. Additionally included in this definition are "one-half antibodies comprising a single heavy chain and a single light chain. Other forms of single chain antibodies, such as diabodies are also encompassed herein.
- F(ab') 2 " and "Fab"' moieties can be produced by treating an Ig with a protease such as pepsin and papain, and include antibody fragments generated by digesting
- immunoglobulin near the disulfide bonds existing between the hinge regions in each of the two heavy chains.
- papain cleaves IgG upstream of the disulfide bonds existing between the hinge regions in each of the two heavy chains to generate two homologous antibody fragments in which an light chain composed of VL and CL (light chain constant region), and a heavy chain fragment composed of VH and Cu i ( ⁇ ) region in the constant region of the heavy chain) are connected at their C terminal regions through a disulfide bond.
- Fab' an antibody fragment composed of VL and CL (light chain constant region)
- Pepsin also cleaves IgG downstream of the disulfide bonds existing between the hinge regions in each of the two heavy chains to generate an antibody fragment slightly larger than the fragment in which the two above-mentioned Fab' are connected at the hinge region.
- This antibody fragment is called F(ab') 2 .
- the Fab fragment also contains the constant domain of the light chain and the first constant domain (CHI) of the heavy chain.
- Fab' fragments differ from Fab fragments by the addition of a few residues at the carboxyl terminus of the heavy chain CHI domain including one or more cysteine(s) from the antibody hinge region.
- Fab'-SH is the designation herein for Fab' in which the cysteine residue(s) of the constant domains bear a free thiol group.
- F(ab') 2 antibody fragments originally were produced as pairs of Fab' fragments which have hinge cysteines between them. Other chemical couplings of antibody fragments are also known.
- Fv refers to an antibody fragment which contains a complete antigen- recognition and antigen-binding site. This region consists of a dimer of one heavy chain and one light chain variable domain in tight, non-covalent or covalent association (disulfide linked Fv's have been described in the art, Reiter et al. (1996) Nature Biotechnology 14: 1239-1245). It is in this configuration that the three CDRs of each variable domain interact to define an antigen- binding site on the surface of the V H -V L dimer.
- a combination of one or more of the CDRs from each of the V H and V L chains confer antigen-binding specificity to the antibody.
- the CDRH3 and CDRL3 could be sufficient to confer antigen-binding specificity to an antibody when transferred to V H and V L chains of a recipient antibody or antigen-binding fragment thereof and this combination of CDRs can be tested for binding, affinity, etc. using any of the techniques described herein.
- Even a single variable domain (or half of an Fv comprising only three CDRs specific for an antigen) has the ability to recognize and bind antigen, although likely at a lower affinity than when combined with a second variable domain.
- V L and V H the two domains of a Fv fragment
- V L and V H the two domains of a Fv fragment
- they can be joined using recombinant methods by a synthetic linker that enables them to be made as a single protein chain in which the V L and V H regions pair to form monovalent molecules
- scFv single chain Fv
- Such scFvs are also intended to be encompassed within the term "antigen-binding portion" of an antibody.
- V H and V L sequences of specific scFv can be linked to an Fc region cDNA or genomic sequences, in order to generate expression vectors encoding complete Ig (e.g., IgG) molecules or other isotypes.
- V H and V L can also be used in the generation of Fab, Fv or other fragments of Igs using either protein chemistry or recombinant DNA technology.
- Single-chain Fv or “sFv” antibody fragments comprise the V H and V L domains of an antibody, wherein these domains are present in a single polypeptide chain.
- the Fv polypeptide further comprises a polypeptide linker between the V H and V L domains which enables the sFv to form the desired structure for antigen binding.
- AVIMERTM refers to a class of therapeutic proteins of human origin, which are unrelated to antibodies and antibody fragments, and are composed of several modular and reusable binding domains, referred to as A-domains (also referred to as class A module, complement type repeat, or LDL-receptor class A domain). They were developed from human extracellular receptor domains by in vitro exon shuffling and phage display (Silverman et al.,
- the resulting proteins can contain multiple independent binding domains that can exhibit improved affinity (in some cases, sub-nanomolar) and specificity compared with single-epitope binding proteins. See, for example, U.S. Patent Application Publ. Nos. 2005/0221384, 2005/0164301, 2005/0053973 and 2005/0089932, 2005/0048512, and 2004/0175756, each of which is hereby incorporated by reference herein in its entirety.
- Each of the known 217 human A-domains comprises ⁇ 35 amino acids ( ⁇ 4 kDa); and these domains are separated by linkers that average five amino acids in length.
- Native A- domains fold quickly and efficiently to a uniform, stable structure mediated primarily by calcium binding and disulfide formation.
- a conserved scaffold motif of only 12 amino acids is required for this common structure.
- the end result is a single protein chain containing multiple domains, each of which represents a separate function.
- Each domain of the proteins binds independently and the energetic contributions of each domain are additive.
- diabodies refers to small antibody fragments with two antigen-binding sites, which fragments comprise a heavy chain variable domain (V H ) connected to a light chain variable domain (V L ) in the same polypeptide chain (V H -V L ).
- V H heavy chain variable domain
- V L light chain variable domain
- the domains are forced to pair with the complementary domains of another chain and create two antigen-binding sites.
- Diabodies are described more fully in, for example, EP 404,097; WO 93/1 1161; and Hollinger et al, Proc. Natl. Acad. Sci. USA 90:6444 6448 (1993).
- Antigen-binding polypeptides also include heavy chain dimers such as, for example, antibodies from camelids and sharks.
- Camelid and shark antibodies comprise a homodimeric pair of two chains of V-like and C-like domains (neither has a light chain). Since the V H region of a heavy chain dimer IgG in a camelid does not have to make hydrophobic interactions with a light chain, the region in the heavy chain that normally contacts a light chain is changed to hydrophilic amino acid residues in a camelid. V H domains of heavy-chain dimer IgGs are called V HH domains.
- Shark Ig-NARs comprise a homodimer of one variable domain (termed a V-NAR domain) and five C-like constant domains (C-NAR domains).
- camelids the diversity of antibody repertoire is determined by the CDRs 1, 2, and 3 in the V H or V HH regions.
- the CDR3 in the camel V HH region is characterized by its relatively long length, averaging 16 amino acids (Muyldermans et al., 1994, Protein Engineering 7(9): 1129). This is in contrast to CDR3 regions of antibodies of many other species.
- the CDR3 of mouse V H has an average of 9 amino acids.
- Libraries of camelid-derived antibody variable regions which maintain the in vivo diversity of the variable regions of a camelid, can be made by, for example, the methods disclosed in U.S. Patent Application Ser. No. 20050037421.
- Humanized forms of non-human (e.g., murine) antibodies include chimeric antibodies which contain minimal sequence derived from a non-human Ig.
- humanized antibodies are human Igs (recipient antibody) in which one or more of the CDRs of the recipient are replaced by CDRs from a non-human species antibody (donor antibody) such as mouse, rat, rabbit or non-human primate having the desired specificity, affinity and binding function.
- donor antibody such as mouse, rat, rabbit or non-human primate having the desired specificity, affinity and binding function.
- one or more FR amino acid residues of the human Ig are replaced by corresponding non-human amino acid residues.
- humanized antibodies can contain residues which are not found in the recipient antibody or in the donor antibody. These modifications can be made to refine antibody performance, if needed.
- a humanized antibody can comprise substantially all of at least one and, in some cases two, variable domains, in which all or substantially all of the hypervariable regions correspond to those of a non-human
- immunoglobulin and all, or substantially all, of the FRs are those of a human immunoglobulin sequence.
- the humanized antibody optionally can also include at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin.
- Fc immunoglobulin constant region
- a humanized antibody also includes antibodies in which part, or all of the CDRs of the heavy and light chain are derived from a non-human monoclonal antibody, substantially all the remaining portions of the variable regions are derived from human variable region (both heavy and light chain), and the constant regions are derived from a human constant region.
- the CDRl, CDR2 and CDR3 regions of the heavy and light chains are derived from a non-human antibody.
- at least one CDR (e.g., a CDR3) of the heavy and light chains is derived from a non-human antibody.
- Various combinations of CDRl, CDR2, and CDR3 can be derived from a non-human antibody and are contemplated herein.
- one or more of the CDRl, CDR2 and CDR3 regions of each of the heavy and light chains are derived from a murine chimeric monoclonal antibody clone TRC105.
- the term "monoclonal antibody” as used herein refers to an antibody obtained from a population of substantially homogeneous antibodies, i.e., the individual antibodies comprising the population are identical except for possible naturally occurring mutations that may be present in minor amounts. Monoclonal antibodies are highly specific, being directed against a single antigenic site. Furthermore, in contrast to conventional (polyclonal) antibody preparations, which can include different antibodies directed against different determinants (epitopes), each monoclonal antibody is directed against a single determinant on the antigen.
- the modifier "monoclonal” indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies, and is not to be construed as requiring production of the antibody by any particular method.
- monoclonal antibodies can be made by the hybridoma method first described by Kohler et al., Nature 256:495 (1975), or can be made by recombinant DNA methods (see, e.g., U.S. Pat. No. 4,816,567).
- monoclonal antibodies can be made by the hybridoma method first described by Kohler et al., Nature 256:495 (1975), or can be made by recombinant DNA methods (see, e.g., U.S. Pat. No. 4,816,567).
- the monoclonal antibodies can be isolated from phage antibody libraries using the techniques described in Clackson et al., Nature 352:624-628 (1991) and Marks et al., J. Mol. Biol. 222:581-597 (1991), for example.
- Antibodies can be isolated and purified from the culture supernatant or ascites mentioned above by saturated ammonium sulfate precipitation, euglobulin precipitation method, caproic acid method, caprylic acid method, ion exchange chromatography (DEAE or DE52), or affinity chromatography using anti-Ig column or a protein A, G or L column such as described in more detail below.
- Exemplary antibodies for use in the compositions and methods described herein are intact immunoglobulin molecules, such as, for example, a humanized antibody or those portions of a humanized Ig molecule that contain the antigen binding site (i.e., paratope) or a single heavy chain and a single light chain, including those portions known in the art as Fab, Fab', F(ab)', F(ab') 2 , Fd, scFv, a variable heavy domain, a variable light domain, a variable NAR domain, bi-specific scFv, a bi-specific Fab 2 , a tri-specific Fab 3 and a single chain binding polypeptides and others also referred to as antigen-binding fragments.
- variable regions or portions thereof may be fused to, connected to, or otherwise joined to one or more constant regions or portions thereof to produce any of the antibodies or fragments thereof described herein. This may be accomplished in a variety of ways known in the art, including but not limited to, molecular cloning techniques or direct synthesis of the nucleic acids encoding the molecules. Exemplary non-limiting methods of constructing these molecules can also be found in the examples described herein.
- the application contemplates a single chain binding polypeptide having a heavy chain variable region, and/or a light chain variable region which binds endoglin and has, optionally, an immunoglobulin Fc region.
- the application contemplates a single chain binding polypeptide having a heavy chain variable region, and/or a light chain variable region which binds endoglin and inhibits angiogenesis and has, optionally, an immunoglobulin Fc region.
- Such a molecule is a single chain variable fragment optionally having effector function or increased half-life through the presence of the immunoglobulin Fc region.
- germline gene segments or “germline sequences” refer to the genes from the germline (the haploid gametes and those diploid cells from which they are formed).
- the germline DNA contains multiple gene segments that encode a single Ig heavy or light chain. These gene segments are carried in the germ cells but cannot be transcribed and translated into heavy and light chains until they are arranged into functional genes. During B-cell differentiation in the bone marrow, these gene segments are randomly shuffled by a dynamic genetic system capable of generating more than 10 8 specificities. Most of these gene segments are published and collected by the germline database.
- binding refers to binding agents, antibodies or fragments thereof that are specific to a sequence of amino acid residues ("binding site” or “epitope"), yet if are cross-reactive to other peptides/proteins, are not toxic at the levels at which they are formulated for administration to human use.
- binding refers to a direct association between two molecules, due to, for example, covalent, electrostatic, hydrophobic, and ionic and/or hydrogen-bond interactions under physiological conditions, and including interactions such as salt bridges and water bridges and any other conventional binding means.
- binding agent binds to the binding site with greater affinity than it binds unrelated amino acid sequences.
- affinity is at least 1-fold greater, at least 2-fold greater, at least 3-fold greater, at least 4-fold greater, at least 5-fold greater, at least 6-fold greater, at least 7-fold greater, at least 8-fold greater, at least 9-fold greater, 10-fold greater, at least 20-fold greater, at least 30-fold greater, at least 40-fold greater, at least 50-fold greater, at least 60-fold greater, at least 70-fold greater, at least 80-fold greater, at least 90-fold greater, at least 100-fold greater, or at least 1000-fold greater than the affinity of the binding agent for unrelated amino acid sequences.
- immunoreactive and
- affinity refers to the equilibrium constant for the reversible binding of two agents and is expressed as K D .
- Affinity of a binding protein to a ligand such as affinity of an antibody for an epitope can be, for example, from about 100 nanomolar (nM) to about 0.1 nM, from about 100 nM to about 1 picomolar (pM), or from about 100 nM to about 1 femtomolar (fM).
- pM nanomolar
- fM femtomolar
- the term “avidity” refers to the resistance of a complex of two or more agents to dissociation after dilution.
- Apparent affinities can be determined by methods such as an enzyme linked immunosorbent assay (ELISA) or any other technique familiar to one of skill in the art. Avidities can be determined by methods such as a Scatchard analysis or any other technique familiar to one of skill in the art.
- ELISA enzyme linked immunosorbent assay
- Epitope refers to that portion of an antigen or other macromolecule capable of forming a binding interaction with the variable region binding pocket of an antibody. Such binding interactions can be manifested as an intermolecular contact with one or more amino acid residues of one or more CDRs. Antigen binding can involve, for example, a CDR3 or a CDR3 pair or, in some cases, interactions of up to all six CDRs of the V R and V L chains.
- An epitope can be a linear peptide sequence (i.e., "continuous") or can be composed of noncontiguous amino acid sequences (i.e., "conformational” or “discontinuous").
- TRC105 is a chimeric antibody which is the same variable amino acid sequence as the murine antibody described as Y4-2F1 or SN6j in US patent numbers 5,928,641; 6,200,566; 6,190,660; and
- the term "specific” refers to a situation in which an antibody will not show any significant binding to molecules other than the antigen containing the epitope recognized by the antibody.
- the term is also applicable where for example, an antigen binding domain is specific for a particular epitope which is carried by a number of antigens, in which case the antibody or antigen-binding fragment thereof carrying the antigen binding domain will be able to bind to the various antigens carrying the epitope.
- preferentially binds or “specifically binds” mean that the antibodies or fragments thereof bind to an epitope with greater affinity than it binds unrelated amino acid sequences, and, if cross-reactive to other polypeptides containing the epitope, are not toxic at the levels at which they are formulated for administration to human use.
- such affinity is at least 1-fold greater, at least 2-fold greater, at least 3-fold greater, at least 4-fold greater, at least 5-fold greater, at least 6-fold greater, at least 7-fold greater, at least 8-fold greater, at least 9-fold greater, 10-fold greater, at least 20-fold greater, at least 30-fold greater, at least 40-fold greater, at least 50-fold greater, at least 60-fold greater, at least 70-fold greater, at least 80-fold greater, at least 90-fold greater, at least 100-fold greater, or at least 1000- fold greater than the affinity of the antibody or fragment thereof for unrelated amino acid sequences.
- immunoreactive binds
- preferentially binds and “specifically binds” are used interchangeably herein.
- binding refers to a direct association between two molecules, due to, for example, covalent, electrostatic, hydrophobic, and ionic and/or hydrogen- bond interactions under physiological conditions, and includes interactions such as salt bridges and water bridges, as well as any other conventional means of binding.
- amino acid substitution refers to grouping of amino acids on the basis of certain common properties.
- a functional way to define common properties between individual amino acids is to analyze the normalized frequencies of amino acid changes between corresponding proteins of homologous organisms (Schulz, G. E. and R. H. Schirmer, Principles of Protein Structure, Springer- Verlag).
- groups of amino acids may be defined where amino acids within a group exchange preferentially with each other, and therefore resemble each other most in their impact on the overall protein structure (Schulz, G. E. and R. H. Schirmer, Principles of Protein Structure, Springer- Verlag). Examples of amino acid groups defined in this manner include:
- a small-residue group consisting of Ser, Thr, Asp, Asn, Gly, Ala, Glu, Gin and
- each amino acid residue may form its own group, and the group formed by an individual amino acid may be referred to simply by the one and/or three letter abbreviation for that amino acid commonly used in the art as described above.
- a “conserved residue” is an amino acid that is relatively invariant across a range of similar proteins. Often conserved residues will vary only by being replaced with a similar amino acid, as described above for “conservative amino acid substitution.”
- x or xaa as used in amino acid sequences herein is intended to indicate that any of the twenty standard amino acids may be placed at this position unless specifically noted otherwise.
- an "x” or a “xaa” in an amino acid sequence may be replaced by a mimic of the amino acid present in the target sequence, or the amino acid may be replaced by a spacer of essentially any form that does not interfere with the activity of the peptidomimetic.
- Homology or “identity” or “similarity” refers to sequence similarity between two peptides or between two nucleic acid molecules. Homology and identity can each be determined by comparing a position in each sequence which may be aligned for purposes of comparison. When an equivalent position in the compared sequences is occupied by the same base or amino acid, then the molecules are identical at that position; when the equivalent site occupied by the same or a similar amino acid residue (e.g., similar in steric and/or electronic nature), then the molecules can be referred to as homologous (similar) at that position.
- Expression as a percentage of homology/similarity or identity refers to a function of the number of identical or similar amino acids at positions shared by the compared sequences.
- a sequence which is "unrelated” or “non-homologous” shares less than 40% identity, though preferably less than 25% identity with a sequence of the present invention.
- the absence of residues (amino acids or nucleic acids) or presence of extra residues also decreases the identity and homology/similarity.
- the term "homology” describes a mathematically based comparison of sequence similarities which is used to identify genes or proteins with similar functions or motifs.
- the nucleic acid (nucleotide, oligonucleotide) and amino acid (protein) sequences of the present invention may be used as a "query sequence" to perform a search against public databases to, for example, identify other family members, related sequences or homologs. Such searches can be performed using the NBLAST and XBLAST programs (version 2.0) of Altschul, et al. (1990) J. Mol. Biol. 215:403-10.
- Gapped BLAST can be utilized as described in Altschul et al, (1997) Nucleic Acids Res. 25(17):3389- 3402.
- the default parameters of the respective programs e.g., XBLAST and BLAST
- the default parameters of the respective programs e.g., XBLAST and BLAST
- identity means the percentage of identical nucleotide or amino acid residues at corresponding positions in two or more sequences when the sequences are aligned to maximize sequence matching, i.e., taking into account gaps and insertions. Identity can be readily calculated by known methods, including but not limited to those described in (Computational Molecular Biology, Lesk, A. M., ed., Oxford University Press, New York, 1988; Biocomputing: Informatics and Genome Projects, Smith, D. W., ed., Academic Press, New York, 1993; Computer Analysis of Sequence Data, Part I, Griffin, A. M., and Griffin, H.
- Computer program methods to determine identity between two sequences include, but are not limited to, the GCG program package (Devereux, J., et al, Nucleic Acids Research 12(1): 387 (1984)), BLASTP, BLASTN, and FASTA (Altschul, S. F. et al, J. Molec. Biol. 215: 403-410 (1990) and Altschul et al. Nuc. Acids Res. 25: 3389-3402 (1997)).
- the BLAST X program is publicly available from NCBI and other sources (BLAST Manual, Altschul, S., et al, NCBI NLM NIH Bethesda, Md. 20894; Altschul, S., et al, J. Mol. Biol. 215: 403-410 (1990).
- the well known Smith Waterman algorithm may also be used to determine identity.
- isolated when applied to polypeptides means a polypeptide or a portion thereof which, by virtue of its origin or manipulation: (i) is present in a host cell as the expression product of a portion of an expression vector; or (ii) is linked to a protein or other chemical moiety other than that to which it is linked in nature; or (iii) does not occur in nature, for example, a protein that is chemically manipulated by appending, or adding at least one hydrophobic moiety to the protein so that the protein is in a form not found in nature.
- isolated it is further meant a protein that is: (i) synthesized chemically; or (ii) expressed in a host cell and purified away from associated and contaminating proteins.
- the term generally means a polypeptide that has been separated from other proteins and nucleic acids with which it naturally occurs.
- the polypeptide is also separated from substances such as antibodies or gel matrices (polyacrylamide) which are used to purify it.
- angiogenesis inhibitory As used herein, the terms “angiogenesis inhibitory,” “angiogenesis inhibiting” or
- anti-angiogenic include vasculogenesis, and are intended to mean effecting a decrease in the extent, amount, or rate of neovascularization. Effecting a decrease in the extent, amount, or rate of endothelial cell proliferation or migration in the tissue is a specific example of inhibiting angiogenesis.
- angiogenesis inhibitory composition refers to a composition which inhibits angiogenesis-mediated processes such as endothelial cell migration, proliferation, tube formation and subsequently leading to the inhibition of the generation of new blood vessels from existing ones, and consequently affects angiogenesis-dependent conditions.
- angiogenesis-dependent condition is intended to mean a condition where the process of angiogenesis or vasculogenesis sustains or augments a pathological condition or beneficially influences normal physiological processes. Therefore, treatment of an angiogenesis-dependent condition in which angiogenesis sustains a pathological condition could result in mitigation of disease, while treatment of an angiogenesis-dependent condition in which angiogenesis beneficially influences normal physiological processes could result in, e.g., enhancement of a normal process.
- Angiogenesis is the formation of new blood vessels from pre-existing capillaries or post-capillary venules.
- Vasculogenesis results from the formation of new blood vessels arising from angioblasts which are endothelial cell precursors. Both processes result in new blood vessel formation and are included in the meaning of the term angiogenesis-dependent conditions.
- the term "angiogenesis” as used herein is intended to include de novo formation of vessels such as that arising from vasculogenesis as well as those arising from branching and sprouting of existing vessels, capillaries and venules.
- Angiogenesis can also be inclusive of induction of ALK1 signaling and related Smad 1/5/8 phosphorylation and/or signaling.
- CD 105 is also known to be involved in the ALK1 signaling pathway and is thus also included within the meaning of angiogenesis.
- a CD8+ IFN- ⁇ producing T cell is activated to induce a cytotoxic T lymphocyte (CTL) immune response in the patient administered the antagonist.
- CTL cytotoxic T lymphocyte
- a CD4+ IFN- ⁇ producing T cell is activated to induce a helper T cell immune response in the patient administered with the composition.
- helper T cells provide necessary immunological help (e.g., by release of cytokines) to induce and maintain not only CTL, but also a humoral immune response mediated by B cells.
- a humoral response to the antigen is activated in the patient administered with the composition.
- an adjuvant may be added to the composition to increase an immune response.
- Adjuvants are well-known in the art.
- Activation of a CD8+ and/or CD4+ T cells means causing T cells that have the ability to produce cytokines (e.g., IFN- ⁇ ) to actually produce one or more cytokine(s), or to increase their production of one or more cytokine(s).
- cytokines e.g., IFN- ⁇
- Induction of CTL response means causing potentially cytotoxic T lymphocytes to exhibit antigen specific cytotoxicity.
- Antigen specific cytotoxicity means cytotoxicity against a cell presenting an antigen that is associated with the antigen associated with the cancer that is greater than an antigen that is not associated with a cancer.
- Cytotoxicity refers to the ability of the cytotoxic T lymphocyte to kill a target cell.
- Antigen-specific cytotoxicity can be at least about 3-fold, at least about 10-fold greater, at least about 100-fold greater or more than cytotoxicity against a cell not presenting the antigen not associated with the cancer.
- Antibody dependent cell-mediated cytotoxicity also includes activation of natural killer cells ("NK cells") which mediate cell killing via antibody binding.
- NK cells natural killer cells
- the antibodies and antigen-binding fragments described herein can mediate ADCC via NK cells through the binding of endoglin.
- proliferative disorder and “proliferative condition” mean any pathological or non-pathological physiological condition characterized by aberrant or undesirable proliferation.
- cell proliferative disorder and “cell proliferative condition” mean any pathological or non-pathological physiological condition characterized by aberrant or undesirable cell proliferation, as well as including conditions characterized by undesirable or unwanted cell proliferation or cell survival (e.g., due to deficient apoptosis), conditions characterized by deficient or aberrant or deficient apoptosis, as well as conditions characterized by aberrant or undesirable or unwanted cell survival.
- differentiated means any pathological or non-pathological physiological condition characterized by aberrant or deficient differentiation.
- Proliferative or differentiative disorders amenable to treatment include diseases and non-pathological physiological conditions, benign and neoplastic, characterized by abnormal or undesirable cell numbers, cell growth or cell survival. Such disorders or conditions may therefore constitute a disease state and include all types of cancerous growths or oncogenic processes, metastatic tissues or malignantly transformed cells, tissues, or organs, or may be non- pathologic, i.e., a deviation from normal but which is not typically associated with disease.
- a specific example of a non-pathologic condition that may be treated in accordance with the invention is tissue re-growth from wound repair that results in scarring.
- Solid tumor refers to neoplasias or metastases that typically aggregate together and form a mass. Particular examples include visceral tumors such as gastric or colon cancer, hepatomas, venal carcinomas, lung and brain tumors/cancers.
- a "non- solid tumor” refers to neoplasias of the hematopoietic system, such as lymphomas, myelomas and leukemias, or neoplasias that are diffuse in nature, as they do not typically form a solid mass.
- leukemias include for example, acute and chronic lymphoblastic, myeloblastic and multiple myeloma.
- Such disorders include neoplasms or cancers, which can affect virtually any cell or tissue type, e.g., carcinoma, sarcoma, melanoma, metastatic disorders or hematopoietic neoplastic disorders.
- a metastatic tumor can arise from a multitude of primary tumor types, including but not limited to breast, lung, thyroid, head and neck, brain, lymphoid, gastrointestinal (mouth, esophagus, stomach, small intestine, colon, rectum), genito-urinary tract (uterus, ovary, cervix, bladder, testicle, penis, prostate), kidney, pancreas, liver, bone, muscle, skin, etc.
- Carcinomas refer to malignancies of epithelial or endocrine tissue, and include respiratory system carcinomas, gastrointestinal system carcinomas, genitourinary system carcinomas, testicular carcinomas, breast carcinomas, prostatic carcinomas, endocrine system carcinomas, and melanomas.
- Exemplary carcinomas include those forming from the cervix, lung, prostate, breast, head and neck, colon, liver and ovary.
- the term also includes carcinosarcomas, e.g., which include malignant tumors composed of carcinomatous and sarcomatous tissues.
- Adenocarcinoma includes a carcinoma of a glandular tissue, or in which the tumor forms a gland like structure.
- An ocular tissue to be treated is, for example, a retinal tissue of a patient with diabetic retinopathy, macular degeneration or neovascular glaucoma and the angiogenesis to be inhibited is retinal tissue angiogenesis where there is neovascularization of retinal tissue.
- a cancerous tissue to be treated is, for example, an endothelial tissue expressing an abnormal level of endoglin.
- transformed cells refers to cells that have spontaneously converted to a state of unrestrained growth, i.e., they have acquired the ability to grow through an indefinite number of divisions in culture. Transformed cells may be characterized by such terms as neoplastic, anaplastic and/or hyperplastic, with respect to their loss of growth control.
- transformed phenotype of malignant mammalian cells and “transformed phenotype” are intended to encompass, but not be limited to, any of the following phenotypic traits associated with cellular transformation of mammalian cells: immortalization, morphological or growth transformation, and tumorigenicity, as detected by prolonged growth in cell culture, growth in semi-solid media, or tumorigenic growth in immuno-incompetent or syngeneic animals.
- tumor cell antigen is defined herein as an antigen that is present in higher quantities on a tumor cell or in body fluids than unrelated tumor cells, normal cells, or in normal body fluid.
- the antigen presence may be tested by any number of assays known to those skilled in the art and include without limitation negative and/or positive selection with antibodies, such as an ELISA assay, a radioimmunoassay, or by Western Blot.
- Apoptosis refers to the physiological process by which unwanted or useless cells are eliminated during development and other normal biological processes. Apoptosis is a mode of cell death that occurs under normal physiological conditions and the cell is an active participant in its own demise ("cellular suicide”). It is most often found during normal cell turnover and tissue homeostasis, embryogenesis, induction and maintenance of immune tolerance, development of the nervous system and endocrine-dependent tissue atrophy. Cells undergoing apoptosis show characteristic morphological and biochemical features.
- apoptotic bodies membrane bound vesicles
- apoptotic bodies contain ribosomes, morphologically intact mitochondria and nuclear material.
- these apoptotic bodies are rapidly recognized and phagocytized by macrophages, dendritic cells or adjacent epithelial cells. Due to this efficient mechanism for the removal of apoptotic cells in vivo no inflammatory response is elicited.
- the apoptotic bodies as well as the remaining cell fragments ultimately swell and finally lyse.
- Apoptosis can be measured by methods known to those skilled in the art like DNA fragmentation, exposure of Annexin V, activation of caspases, release of cytochrome c, etc.
- a cell that has been induced to die is termed herein as an "apoptotic cell.”
- Apoptosis inducing agent is defined herein to induce apoptosis/programmed cell death, and include, for example, irradiation, chemotherapeutic agents or receptor ligation agents, wherein cells, for example, tumor cells are induced to undergo programmed cell death. Exemplary apoptosis inducing agents are described in more detail below.
- Apoptosis can be tested using a standard Annexin V Apoptosis Assay:
- NIH:OVCAR-3 cells are grown in 6-well plates (NUNC) and irradiated or treated with an antagonist (or in combination with another anti-cancer drug) for 4-48 hours, washed and stained with Annexin V-FITC (BD-Pharmingen) for 1 hour. Cells are analyzed by flow cytometry (Becton-Dickinson, CellQuest), counterstained with Propidium Iodide and analyzed again in the flow cytometer.
- NUNC 6-well plates
- an antagonist or in combination with another anti-cancer drug
- Annexin V-FITC BD-Pharmingen
- a chimeric monoclonal antibody has been developed that binds endoglin. This antibody is designated TR105 (also known as c-SN6j).
- TR105 also known as c-SN6j.
- the antibodies and antigen-binding fragments thereof described herein were created by humanization of the V L and V H sequences of the chimeric monoclonal TRC105 antibody (SEQ ID NOS. 1 and 39, respectively).
- Humanized immunoglobulins including humanized antibodies, have been constructed by means of genetic engineering. Most humanized immunoglobulins that have been previously described have comprised a framework that is identical to the framework of a particular human immunoglobulin chain (i.e., an acceptor or recipient), and three CDRs from a non-human (i.e., donor) immunoglobulin chain. As described herein, humanization can also include criteria by which a limited number of amino acids in the framework of a humanized immunoglobulin chain are identified and chosen to be the same as the amino acids at those positions in the donor rather than in the acceptor, in order to increase or maintain the affinity of an antibody comprising the humanized immunoglobulin chain.
- the present invention is based in part on the model that two contributing causes of the loss of affinity in prior means of producing humanized antibodies (using as examples mouse antibodies as the source of CDRs) are: (1) when the mouse CDRs are combined with a human framework, the amino acids in the frameworks close to the CDRs become human instead of mouse.
- these changed amino acids may slightly distort the CDRs (e.g., they may create different electrostatic or hydrophobic forces than in the donor mouse antibody, and the distorted-CDRs may not make as effective contacts with the antigen as the CDRs did in the donor antibody); (2) also, amino acids in the original mouse antibody that are close to, but not part of, the CDRs (i.e., still part of the framework), may make contacts with the antigen that contribute to affinity. These amino acids are lost when the antibody is humanized because, generally, all framework amino acids are made human. To circumvent these issues, and to produce humanized antibodies that have a very strong affinity for a desired antigen, humanized antibodies and antigen-binging fragments thereof can be constructed using one or more of the following principles.
- One non-limiting principle is that, for example, as acceptor, a framework is used from a particular human immunoglobulin that is unusually homologous to the donor
- immunoglobulin to be humanized or use a consensus framework from many human antibodies is used as an acceptor.
- a mouse heavy (or light) chain variable region against human heavy (or light) variable regions in a data bank (for example, the National Biomedical Research Foundation Protein Identification Resource or the protein sequence database of the National Center for Biotechnology Information - NCBI) shows that the extent of homology to different human regions can vary greatly, for example from about 40% to about 60%, about 70%>, about 80%> or higher.
- acceptor immunoglobulin one of the human heavy chain variable regions that is most homologous to the heavy chain variable region of the donor immunoglobulin, fewer amino acids will be changed in going from the donor immunoglobulin to the humanized immunoglobulin.
- the acceptor immunoglobulin By choosing as the acceptor immunoglobulin one of the human light chain variable regions that is most homologous to the light chain variable region of the donor immunoglobulin, fewer amino acids will be changed in going from the donor immunoglobulin to the humanized immunoglobulin. Generally, using such techniques, there is a reduced chance of changing an amino acid near one or more of the CDRs that distorts their conformation. Moreover, the precise overall shape of a humanized antibody comprising the humanized immunoglobulin chain may more closely resemble the shape of the donor antibody, thereby also reducing the chance of distorting the CDRs.
- the human antibody will be chosen in which the light and heavy chain variable regions sequences, taken together, are overall most homologous to the donor light and heavy chain variable region sequences. Sometimes greater weight will be given to the heavy chain sequence.
- acceptor immunoglobulin higher affinity can, in some cases, be achieved by selecting a small number of amino acids in the framework of the humanized immunoglobulin chain to be the same as the amino acids at those positions in the donor rather than in the acceptor. Methods of affinity maturation are known in the art.
- Humanized antibodies generally have at least three potential advantages over mouse or chimeric antibodies for use in human therapy. Because the effector portion of an antibody is human, it is believed to interact better with the other parts of the human immune system (e.g., destroy the target cells more efficiently by complement-dependent cytotoxicity (CDC) or antibody-dependent cellular cytotoxicity (ADCC)). Additionally, the human immune system should not recognize the framework or constant region of the humanized antibody as foreign, and therefore the antibody response against such an injected antibody should be less than against a totally foreign mouse antibody or a partially foreign chimeric antibody. Finally, mouse antibodies are known to have a half-life in the human circulation that is much shorter than the half-life of human antibodies. Humanized antibodies can, presumably, have a half-life more similar to naturally-occurring human antibodies, allowing smaller and less frequent doses to be given.
- CDC complement-dependent cytotoxicity
- ADCC antibody-dependent cellular cytotoxicity
- Another criterion that can be used for determining the relevant amino acid positions to change can be, for example, selection of framework residues that are known to be important or to contribute to CDR conformation.
- canonical framework residues are important for CDR conformation and/or structure.
- Targeting of a canonical framework residue as a relevant position to change can be used to identify a more compatible amino acid residue in context with its associated donor CDR sequence.
- the frequency of an amino acid residue at a particular framework position is another criterion which can be used for selecting relevant framework amino acid positions to change. For example, comparison of the selected framework with other framework sequences within its subfamily can reveal residues that occur at minor frequencies at a particular position or positions. Positions harboring less abundant residues are similarly applicable for selection as a position to alter in the acceptor variable region framework.
- the relevant amino acid positions to change also can be selected, for example, based on proximity to a CDR.
- FR residues can participate in CDR
- framework positions that are known or predicted to reside in a three dimensional space near the antigen-CDR interface or predicted to modulate CDR activity.
- framework residues that are known to, or predicted to, form contacts between the heavy (V H ) and light (V L ) chain variable region interface can be selected.
- Such framework positions can affect the conformation and/or affinity of a CDR by modulating the CDR binding pocket, antigen (epitope) interaction or the V H and V L interaction. Therefore, selection of these amino acid positions for constructing a diverse population for screening of binding activity can be used to identify framework changes which replace residues having detrimental effects on CDR conformation or compensate for detrimental effects of residues occurring elsewhere in the framework.
- framework residues that can be selected for alteration include amino acid positions that are inaccessible to solvent. Such residues are generally buried in the variable region and are, therefore, capable of influencing the conformation of the CDR or V H and V L interactions. Solvent accessibility can be predicted, for example, from the relative
- hydrophobicity of the environment created by the amino acid side chains of the polypeptide and/or by known three-dimensional structural data is a hydrophobicity of the environment created by the amino acid side chains of the polypeptide and/or by known three-dimensional structural data.
- amino acid changes at some or all of the selected positions can be incorporated into encoding nucleic acids for the acceptor variable region framework and donor CDRs.
- Altered framework or CDR sequences can be individually made and tested, or can be sequentially or simultaneously combined and tested.
- the variability at any or all of the altered positions can range from a few to a plurality of different amino acid residues, including all twenty naturally occurring amino acids or functional equivalents and analogues thereof. In some cases, non-naturally occurring amino acids may also be considered and are known in the art.
- a large, diverse population of altered variable regions can include all the non-identical framework region positions between the donor and acceptor framework and all single CDR amino acid position changes.
- a population of intermediate diversity can include subsets, for example, of only the proximal non-identical framework positions to be incorporated together with all single CDR amino acid position changes to, for example, increase affinity of the humanized antibodies or antigen binding fragments.
- the diversity of the above populations can be further increased by, for example, additionally including all pair-wise CDR amino acid position changes.
- populations focusing on predetermined residues or positions which incorporate variant residues at as few as one framework and/or one CDR amino acid position can similarly be constructed for screening and identification of an altered antibody variable region.
- the diversity of such focused populations can be further increased by additionally expanding the positions selected for change to include other relevant positions in either or both of the framework and CDR regions.
- Another method of humanizing antibodies includes a method termed
- Superhumanization involves the steps of obtaining a peptide sequence for a subject variable region encoded by a non-human mature antibody gene and identifying a first set of canonical CDR structure types for at least two CDRs within the non-human antibody variable region.
- Canonical CDR structure types are the structure types designated by Chothia (CITE). Chothia and coworkers found that critical portions of the CDRs of many antibodies adopt nearly identical peptide backbone conformations, despite great diversity at the level of amino acid sequence.
- Each canonical structure specifies primarily a set of peptide backbone torsion angles for a contiguous segment of amino acid residues forming a loop.
- a library of peptide sequences for human antibody variable regions for human antibodies is also obtained.
- This library contains sequences for human germline variable regions as encoded by germline nucleic acid segments, and may include mature human antibody sequences.
- the method includes identifying canonical CDR structure types (i.e., a second set of canonical CDR structure types) for at least two CDRs for each sequence within the library of human variable region sequences.
- the method uses these candidate human variable region sequences as a basis for constructing a chimeric molecule that includes at least two of the CDR sequences from the non-human variable region (e.g., of the mouse CDRs) combined with the framework regions from candidate human variable region sequences.
- the result of the construction is that the chimeric antibody contains each of the non-human CDR sequences substituted for each of the human CDR sequences at corresponding locations in the variable regions so that the framework sequences in the chimeric antibody differs from the candidate human framework sequences.
- Choosing highest homology is based on various criterion used to rank candidate human variable regions having the same canonical structure as the subject the non-human variable regions.
- the criterion for ranking members of the selected set may be by amino acid sequence identity or amino acid homology or both.
- Amino acid identity is simple a score of position by position matches of amino acid residues. Similarity by amino acid homology is position by position similarity in residue structure of character. Homology may be scored, for example, according to the tables and procedures described by Henikoff and Henikoff, (1992) Amino acid substitution matrices from protein blocks, Proc. Natl. Acad. Sci 89: 10915-10919, or by the BLOSUM series described by Henikoff and Henikoff, (1996).
- the steps are as follows: a) Determine the peptide sequences of the heavy and light chain variable domains of the subject antibody. These can be determined by any of several methods, such as DNA sequencing of the respective genes after conventional cDNA cloning; DNA sequencing of cloning products that have been amplified by the polymerase chain reaction from reverse transcripts or DNA of the subject hybridoma line; or peptide sequencing of a purified antibody protein.
- Canonical structure type 1 31, 32, 33, 34, 35.
- Canonical structure type 2 31, 32, 33, 34, 35, 35a.
- Canonical structure type 3 31, 32, 33, 34, 35, 35a, 35b.
- Canonical structure type 1 52, 53, 54, 55, 56.
- Canonical structure type 4 52, 52a, 52b, 52c, 53, 54, 55, 56.
- Canonical structure types 2 and 3 for heavy chain CDR2 have equal numbers of residues, hence must be distinguished by clues within their sequence, as discussed by Chothia et al (1992). The Kabat numbering of the segment containing these clues is: 52, 52a, 53, 54, 55.
- Canonical structure type 2 has Pro or Ser at position 52a and Gly or Ser at position 55, with no restriction at the other positions.
- Canonical structure type 3 has Gly, Ser, Asn, or Asp at position 54, with no restriction at the other positions. These criteria are sufficient to resolve the correct assignment in most cases.
- framework residue 71 is commonly Ala, Val, Leu, He, or Thr for canonical structure type 2 and commonly Arg for canonical structure type 3.
- Heavy chain CDR3 is the most diverse of all the CDRs. It is generated by genetic processes, some of a random nature, unique to lymphocytes. Consequently, canonical structures for CDR3 have been difficult to predict. In any case, human germline V gene segments do not encode any part of CDR3; because the V gene segments end at Kabat position 94, whereas positions 95 to 102 encode CDR3. For these reasons, canonical structures of CDR3 are generally not considered for choosing candidate human sequences.
- Canonical structure type 1 27, 29, 30, 31.
- Canonical structure type 2 27, 28, 29, 30, 31.
- Canonical structure type 3 27, 27a, 27b, 27c, 27d, 27e, 27f, 28, 29, 30, 31.
- Canonical structure type 4 27, 27a, 27b, 27c, 27d, 27e, 28, 29, 30, 31.
- Canonical structure type 5 27, 27a, 27b, 27c, 27d, 28, 29, 30, 31.
- Canonical structure type 6 27, 27a, 28, 29, 30, 31.
- Canonical structure type 1 91, 92, 93, 94, 95, 96, 97 (also with an obligatory Pro at position 95 and Gin, Asn, or His at position 90).
- Canonical structure type 3 91, 92, 93, 94, 95, 97.
- Canonical structure type 5 91, 92, 93, 94, 95, 96, 96a, 97.
- conformity of CDR1 and CDR2 to the mouse canonical structure types is assessed, and genes that do not conform are excluded.
- conformity of CDR1 and CDR2 of each human sequence to the canonical structure types of the subject antibody is first assessed.
- the potential of residues 89-95 of a candidate Vk gene to form a CDR3 of the same canonical structure type as the subject antibody is assessed, by positing a fusion of the gene with a J region and applying criteria for CDR3 canonical CDR structure type determination to the fused sequence, and non conforming sequences are excluded.
- variable domain of the subject antibody is of a canonical structure type not available in the human genome
- human germline V genes that have three- dimensionally similar, but not identical, canonical structure types are considered for comparison.
- Such a circumstance often occurs with kappa chain CDR1 in murine antibodies, including two of the examples described below. All 6 possible canonical structure types have been observed at this CDR in murine antibodies, whereas the human genome encodes only canonical types 2, 3, 4 and 6.
- a canonical CDR structure type having length of amino acid residues within two of the length of the amino acid residues of the subject non-human sequence may selected for the comparison.
- human Vk sequences with canonical structure type 2 are used for comparison.
- human Vk sequences with either canonical structure type 3 or 4 are be used for comparison.
- Mature, rearranged human antibody sequences can be considered for the sequence comparison. Such consideration might be warranted under a variety of circumstances, including but not limited to instances where the mature human sequence (1) is very close to germline; (2) is known not to be immunogenic in humans; or (3) contains a canonical structure type identical to that of the subject antibody, but not found in the human germline.
- residue to residue sequence identity and/or homology with the subject sequence is also evaluated to rank the candidate human sequences.
- the residues evaluated are as follows: (1) Kappa (K) light chain CDR amino acid residue positions are CDR1 (26-32), CDR2 (50-52), CDR3 (91-96); and (2) heavy chain CDR amino acid residue positions are CDR1 (31-35) and CDR2 (50-60). Additionally, heavy chain CDR3 amino acid residue positions 95 to 102 can also be considered.
- Residue-to-residue homology is first scored by the number of identical amino acid residues between the subject and the candidate human sequences.
- the human sequence used for subsequent construction of a converted antibody is chosen from among the 25 percent of candidates with the highest score.
- similarity between non-identical amino acid residues may be additionally considered as needed.
- Aliphatic-with-aliphatic, aromatic-with-aromatic, or polar- with-polar matches between subject and object residues are added to the scores.
- quantitative evaluation of sequence homology may be performed using amino acid substitution matrices such as the BLOSUM62 matrix of Henikoff and Henikoff.
- An object sequence for the framework region C-terminal to CDR3 sequence can be selected from the set of known human germline J segments.
- a J peptide sequence is selected by evaluating residue to residue homology for each J segment for sequence positions for which CDR3 and J overlap, using the scoring criteria specified for the evaluation of candidate V genes as mentioned above.
- the J gene segment peptide sequence used for subsequent construction of a converted antibody is chosen from among the 25 percent of candidates with the highest score.
- the chimeric variable chain contains at least two CDRs from a subject non-human sequence, and framework sequences from a candidate human sequence.
- chimeric light chain contains three CDRs from a subject non-human sequence and framework sequences from a candidate human sequence.
- a chimeric heavy chain contains at least two CDRs of a subject heavy chain, and framework sequence of a candidate human heavy chain, or a chimeric heavy chin contains each of the CDRs from the subject heavy chain and framework sequences of a candidate human heavy chain.
- a chimeric antibody heavy chain contains CDRs 1 and 2 from a subject non-human sequence and residues 50-60 for CDR3 and residues 61-65 of a CDR from the candidate human heavy chain, along with the framework sequences of the candidate human sequence.
- a chimeric heavy chain sequence contains each CDR from the subject non-human sequence; frameworks sequences 27-30 form the subject sequence, and the framework sequences from the candidate sequences. In all cases however, the chimeric antibody molecule contains no more than 10 amino acid residues in the framework sequence that differ from those in the framework sequence of the candidate human variable ration.
- CDRs of a converted antibody may be additionally substituted with other amino acids. Typically, no more than four amino acid residues in a CDR are changed, and most typically no more than two residues in the CDR will be changed, except for heavy chain CDR2, where as many as 10 residues may be changed. Changes in affinity can be measured by conventional methods such as those described herein (e.g., Biacore).
- Antibodies can be sequenced using conventional techniques known in the art.
- the amino acid sequences of one or more of the CDRs is inserted into a synthetic sequence of, for example, a human antibody (or antigen-binding fragment thereof) framework to create a human antibody that could limit adverse side reactions of treating a human patient with a non-human antibody.
- the amino acid sequences of one or more of the CDRs can also be inserted into a synthetic sequence of, for example, into a binding protein such as an AVIMERTM to create a construct for administration to a human patient.
- a binding protein such as an AVIMERTM
- Such techniques can be modified depending on the species of animal to be treated.
- an antibody, antigen- binding fragment or binding protein can be synthesized for administration of a non-human (e.g., a primate, a cow, a horse, etc.).
- nucleotides encoding amino acid sequences of one or more of the CDRs can inserted, for example, by recombinant techniques in restriction endonuclease sites of an existing polynucleotide that encodes an antibody, antigen-binding fragment or binding protein.
- an expression system is one which utilizes the GS system (Lonza) using a glutamine synthetase gene as the selectable marker. Briefly, a transfection is performed in CHO cells by electroporation (250V) using the GS system (Lonza) using the glutamine synthetase gene as the selectable marker. Wild type CHO cells are grown in DMEM (Sigma) containing 10% dialyzed Fetal Calf Serum (FCS) with 2 mM glutamine. 6xl0 7 CHO cells are trans fected with 300 ⁇ g of linearized DNA by electroporation.
- DMEM Sigma
- FCS dialyzed Fetal Calf Serum
- the cells are resuspended in DMEM with glutamine and plated out into 36x96-well plates (50 ⁇ /well), and incubated at 37° C. in 5% C0 2 . The following day, 150 ⁇ /well of selective medium (DMEM without glutamine) is added. After approximately 3 weeks the colonies are screened by ELISA (see below) using an irrelevant antibody as a negative control. All colonies producing >20 ⁇ g/ml are expanded into 24-well plates and then into duplicate T25 flasks.
- dhfr- dihydrofolate reductase deficient Chinese hamster ovary cells.
- the system is well known to the skilled artisan.
- the system is based upon the dehydrofolate reductase "dhfr" gene, which encodes the DHFR enzyme, which catalyzes conversion of dehydrofolate to tetrahydro folate.
- dhfr- CHO cells are transfected with an expression vector containing a functional DHFR gene, together with a gene that encodes a desired protein.
- the desired protein is recombinant antibody heavy chain and/or light chain.
- the recombinant cells develop resistance by amplifying the dhfr gene.
- the amplification unit employed is much larger than the size of the dhfr gene, and as a result the antibody heavy chain is co-amplified.
- the present application provides an isolated polynucleotide (nucleic acid) encoding an antibody or antigen-binding fragment as described herein, vectors containing such polynucleotides, and host cells and expression systems for transcribing and translating such polynucleotides into polypeptides.
- the present application also provides constructs in the form of plasmids, vectors, transcription or expression cassettes which comprise at least one polynucleotide as above.
- the present application also provides a recombinant host cell which comprises one or more constructs as above.
- a nucleic acid encoding any antibody or antigen-binding fragments thereof described herein as provided itself forms an aspect of the present application, as does a method of production of the antibody or antigen-binding fragments thereof described herein which method comprises expression from encoding nucleic acid therefrom. Expression can conveniently be achieved by culturing under appropriate conditions recombinant host cells containing the nucleic acid. Following production by expression, an antibody or antigen-binding fragment can be isolated and/or purified using any suitable technique, then used as appropriate.
- nucleic acid molecules and vectors described herein can be provided isolated and/or purified, e.g., from their natural environment, in substantially pure or homogeneous form.
- nucleic acid free or substantially free of nucleic acid or genes origin other than the sequence encoding a polypeptide with the required function.
- Nucleic acid can comprise DNA or RNA and can be wholly or partially synthetic. Methods of purification are well known in the art.
- Suitable host cells include bacteria, mammalian cells, yeast and baculovirus systems.
- Mammalian cell lines available in the art for expression of a heterologous polypeptide include Chinese hamster ovary cells, HeLa cells, baby hamster kidney cells, NSO mouse myeloma cells and many others.
- a common bacterial host is E. coli.
- Suitable vectors can be chosen or constructed, containing appropriate regulatory sequences, including promoter sequences, terminator sequences, polyadenylation sequences, enhancer sequences, marker genes and other sequences as appropriate.
- Vectors can be plasmids, viral e.g. 'phage, or phagemid, as appropriate.
- plasmids viral e.g. 'phage, or phagemid, as appropriate.
- a further aspect provides a host cell containing nucleic acid as disclosed herein.
- a still further aspect provides a method comprising introducing such nucleic acid into a host cell.
- the introduction can employ any available technique.
- suitable techniques can include, for example, calcium phosphate transfection, DEAE Dextran, electroporation, liposome -mediated transfection and transduction using retrovirus or other virus, e.g., vaccinia or, for insect cells, baculovirus.
- suitable techniques can include, for example, calcium chloride transformation, electroporation and transfection using retrovirus or other virus, e.g., vaccinia or, for insect cells, baculovirus.
- suitable techniques can include, for example, calcium chloride transformation, electroporation and transfection using
- the introduction can be followed by causing or allowing expression from the nucleic acid, e.g. by culturing host cells under conditions for expression of the gene.
- the nucleic acid is integrated into the genome (e.g.
- chromosome of the host cell. Integration can be promoted by inclusion of sequences which promote recombination with the genome, in accordance with standard techniques. Ig enhances can be initialized as needed to maximize expression.
- the present application also provides a method which comprises using a construct as stated above in an expression system in order to express the antibodies or antigen-binding fragments thereof as above.
- the present application also relates to isolated nucleic acids, such as recombinant
- the present application provides a nucleic acid which codes for an antibody or antigen-binding fragment thereof as described herein which binds endoglin.
- the full DNA sequence of the recombinant DNA molecule or cloned gene of an antibody or antigen-binding fragment described herein can be operatively linked to an expression control sequence which can be introduced into an appropriate host.
- the application accordingly extends to unicellular hosts transformed with the cloned gene or recombinant DNA molecule comprising a DNA sequence encoding the V H and/or V L , or portions thereof, of the antibody.
- DNA sequences disclosed herein can be expressed by operatively linking them to an expression control sequence in an appropriate expression vector and employing that expression vector to transform an appropriate unicellular host.
- Such operative linking of a DNA sequence to an expression control sequence includes, if not already part of the DNA sequence, the provision of an initiation codon, ATG, in the correct reading frame upstream of the DNA sequence.
- Polynucleotides and vectors can be provided in an isolated and/or a purified form
- substantially pure refers to a solution or suspension containing less than, for example, about 20% or less extraneous material, about 10% or less extraneous material, about 5% or less extraneous material, about 4% or less extraneous material, about 3% or less extraneous material, about 2% or less extraneous material, or about 1% or less extraneous material.
- a wide variety of host/expression vector combinations can be employed in expressing the DNA sequences of this invention.
- Useful expression vectors can consist of segments of chromosomal, non-chromosomal and synthetic DNA sequences.
- Suitable vectors include, but are not limited to, derivatives of SV40 and known bacterial plasmids, e.g., E.
- coli plasmids col El, Perl, Pbr322, Pmb9 and their derivatives, plasmids such as RP4; phage DNAs, e.g., the numerous derivatives of phage ⁇ , e.g., NM989, and other phage DNA, e.g., M13 and filamentous single stranded phage DNA; yeast plasmids such as the 2u plasmid or derivatives thereof; vectors useful in eukaryotic cells, such as vectors useful in insect or mammalian cells; vectors derived from combinations of plasmids and phage DNAs, such as plasmids that have been modified to employ phage DNA or other expression control sequences; and the like.
- phage DNAs e.g., the numerous derivatives of phage ⁇ , e.g., NM989, and other phage DNA, e.g., M13 and filamentous single stranded phage DNA
- a recombinant host cell which comprises one or more polynucleotide constructs.
- a polynucleotide encoding an antibody or antigen-binding fragment as provided herein forms an aspect of the present application, as does a method of production of the antibody or antigen-binding fragment which method comprises expression from the polynucleotide. Expression can be achieved, for example, by culturing under appropriate conditions recombinant host cells containing the polynucleotide. An antibody or antigen-binding fragment can then be isolated and/or purified using any suitable technique, and used as appropriate.
- any of a wide variety of expression control sequences - sequences that control the expression of a DNA sequence operatively linked to it - can be used in these vectors to express the DNA sequences.
- useful expression control sequences include, for example, the early or late promoters of SV40, CMV, vaccinia, polyoma or adenovirus, the lac system, the trp system, the TAC system, the TRC system, the LTR system, the major operator and promoter regions of phage ⁇ , the control regions of fd coat protein, the promoter for 3-phosphoglycerate kinase or other glycolytic enzymes, the promoters of acid phosphatase (e.g., Pho5), the promoters of the yeast -mating factors, and other sequences known to control the expression of genes of prokaryotic or eukaryotic cells or their viruses, and various combinations thereof.
- Suitable host cells include bacteria, mammalian cells, yeast and
- Mammalian cell lines available in the art for expression of a heterologous polypeptide include Chinese hamster ovary (CHO) cells, HeLa cells, baby hamster kidney cells, NS0 mouse myeloma cells and many others.
- a common, bacterial host can be, for example, E. coli.
- a wide variety of unicellular host cells are also useful in expressing the DNA sequences.
- These hosts include well-known eukaryotic and prokaryotic hosts, such as strains of E. coli, Pseudomonas, Bacillus, Streptomyces, fungi such as yeasts, and animal cells, such as CHO, YB/20, NS0, SP2/0, Rl.l, B-W and L-M cells, African Green Monkey kidney cells (e.g., COS 1, COS 7, BSC1, BSC40, and BMT10), insect cells (e.g., Sf9), and human cells and plant cells in tissue culture.
- eukaryotic and prokaryotic hosts such as strains of E. coli, Pseudomonas, Bacillus, Streptomyces, fungi such as yeasts, and animal cells, such as CHO, YB/20, NS0, SP2/0, Rl.l, B-W and L-M cells, African Green Mon
- the present application also provides constructs in the form of plasmids, vectors, transcription or expression cassettes as described elsewhere herein which comprise at least one polynucleotide as above.
- Suitable vectors can be chosen or constructed, containing appropriate regulatory sequences, including promoter sequences, terminator sequences, polyadenylation sequences, enhancer sequences, selectable markers and other sequences as appropriate.
- Vectors can be plasmids, viral e.g., phage, phagemid, etc., as appropriate.
- Molecular Cloning a Laboratory Manual: 2nd edition, Sambrook et al., 1989, Cold Spring Harbor Laboratory Press.
- a further aspect provides a host cell containing one or more polynucleotides as disclosed herein. Yet a further aspect provides a method of introducing such one or more polynucleotides into a host cell, any available technique.
- suitable techniques can include, for example, calcium phosphate transfection, DEAEDextran, electroporation, liposome -mediated transfection and transduction using retrovirus or other virus (e.g. vaccinia) or, for insect cells, baculovirus.
- suitable techniques can include, for example calcium chloride transformation, electroporation and transfection using bacteriophages.
- the introduction can be followed by causing or allowing expression from the one or more polynucleotides, e.g. by culturing host cells under conditions for expression of one or more polypeptides from one or more polynucleotides.
- Inducible systems can be used and expression induced by addition of an activator.
- the polynucleotides can be integrated into the genome (e.g., chromosome) of the host cell. Integration can be promoted by inclusion of sequences which promote recombination with the genome, in accordance with standard techniques.
- the nucleic acid is maintained on an episomal vector in the host cell.
- Methods are provided herein which include using a construct as stated above in an expression system in order to express a specific polypeptide.
- a polynucleotide encoding an antibody, antigen-binding fragment, or a binding protein can be prepared recombinantly/synthetically in addition to, or rather than, cloned.
- the polynucleotide can be designed with the appropriate codons for the antibody, antigen-binding fragment, or a binding protein. In general, one will select preferred codons for an intended host if the sequence will be used for expression.
- the complete polynucleotide can be assembled from overlapping oligonucleotides prepared by standard methods and assembled into a complete coding sequence. See, e.g., Edge, Nature, 292:756 (1981); Nambair et al, Science, 223: 1299 (1984); Jay et al, J. Biol. Chem., 259:6311 (1984).
- a DNA sequence encoding an antibody or antigen-binding fragment thereof can be prepared synthetically rather than cloned.
- the DNA sequence can be designed with the appropriate codons for the antibody or antigen-binding fragment amino acid sequence. In general, one will select preferred codons for the intended host if the sequence will be used for expression.
- the complete sequence is assembled from overlapping oligonucleotides prepared by standard methods and assembled into a complete coding sequence. See, e.g., Edge, Nature, 292:756 (1981); Nambair et al, Science, 223: 1299 (1984); Jay et al, J. Biol. Chem., 259:6311 (1984), each of which is which is incorporated herein by reference in its entirety.
- an antibody or an antigen binding fragment thereof described herein can be assessed for immunogenicity and, as needed, be deimmunized (i.e. the antibody is made less immuno reactive by altering one or more T cell epitopes).
- Analysis of immunogenicity and T-cell epitopes present in the humanized anti-endoglin antibodies and antigen-binding fragments described herein can be carried out via the use of software and specific databases. Exemplary software and databases include iTopeTM developed by Antitope of Cambridge, England. iTopeTM is an in silico technology for analysis of peptide binding to human MHC class II alleles.
- the iTopeTM software predicts peptide binding to human MHC class II alleles and thereby provides an initial screen for the location of such "potential T cell epitopes.”
- iTopeTM software predicts favorable interactions between amino acid side chains of a peptide and specific binding pockets within the binding grooves of 34 human MHC class II allelles. The location of key binding residues is achieved by the in silico generation of 9mer peptides that overlap by one amino acid spanning the test antibody variable region sequence. Each 9mer peptide can be tested against each of the 34 MHC class II allotypes and scored based on their potential "fit" and interactions with the MHC class II binding groove.
- Peptides that produce a high mean binding score (>0.55 in the iTopeTM scoring function) against >50% of the MHC class II alleles are considered as potential T cell epitopes.
- the core 9 amino acid sequence for peptide binding within the MHC class II groove is analyzed to determine the MHC class II pocket residues (PI, P4, P6, P7 and P9) and the possible T cell receptor (TCR) contact residues (P-l, P2, P3, P5, P8).
- amino acid residue changes, substitutions, additions, and/or deletions can be introduced to remove the identified T-cell epitope. Such changes can be made so as to preserve antibody structure and function while still removing the identified epitope. Exemplary changes can include, but are not limited to, conservative amino acid changes.
- T-cell proliferation assays examples include the use of T-cell proliferation assays to the bacterial protein staphylokinase, followed by epitope mapping using synthetic peptides to stimulate T-cell lines.
- T-cell proliferation assays using synthetic peptides of the tetanus toxin protein have resulted in definition of immunodominant epitope regions of the toxin.
- T-cell epitopes in a test protein may be determined using isolated sub-sets of human immune cells, promoting their differentiation in vitro and culture of the cells in the presence of synthetic peptides of interest and measurement of any induced proliferation in the cultured T-cells. Other techniques may also be used.
- T cell sub-sets dendritic cells, CD4+ and or CD8+ T-cells.
- the presence of T cell epitopes in an antibody may be determined by adding the antibody to isolated sub-sets of human immune cells, and assessing their differentiation in vitro and measuring any induced proliferation in the cultured T cells.
- T-cell epitope refers to an amino acid sequence which is able to bind MHC class II, able to stimulate T-cells and/or also to bind (without necessarily measurably activating) T-cells in complex with MHC class II.
- synthetic peptides or whole antibodies are tested for their ability to evoke a proliferative response in human T-cells cultured in vitro.
- the T-cells are present within a peripheral blood mononuclear cell (PBMC) layer readily obtainable by well known means from whole blood samples.
- PBMC peripheral blood mononuclear cell
- the PBMC preparation contains physiological ratios of T-cells and antigen presenting cells and is, therefore, a good source of materials with which to conduct a surrogate immune reaction in vitro.
- a stimulation index approaching or exceeding 2.0 is a useful measure of induced proliferation.
- the stimulation index may be different depending upon the antibody, or antigen-binding fragment thereof, and may be established with reference to a baseline for each antibody, or antigen-binding fragment thereof, and corresponding peptide library.
- test peptides are contacted with cells in at least two different concentrations and the concentrations would typically span a minimum two-fold concentration difference. Such a concentration range provides an off-set to the kinetic dimension to the assay and may be useful where a single time point determination, for example at day plus 7, is being conducted. In some assays, multiple time course determinations may be conducted and these too may be made using peptide immunogen provided at a minimum of two different concentrations.
- control peptides for which there is expectation that the majority of PBMC donor samples will be responsive may be included in each assay plate.
- the influenza haemagglutinin peptide 307-309, sequence PKYVKQNTLKLA (SEQ ID NO: 104); and the Chlamydia HSP 60 peptide sequence KVVDQIKKISKPVQH (SEQ ID NO: 105) are examples of control peptides to be used in such an assay.
- assays could also use a potent whole protein antigen, such as hemocyanin from Keyhole Limpet, to which all PBMC samples would be expected to exhibit an SI significantly greater than 2.0.
- Other control antigens for such use will be well-known in the art.
- the methods disclosed herein can provide an epitope map of antibodies, or antigen-binding fragments thereof, where the map has relevance to a wide spectrum of possible MHC allotypes.
- the map may be sufficiently representative to allow the design or selection of a modified protein for which the ability of the protein to evoke a T-cell driven immune response may be eliminated or at least ameliorated for the majority of patients to whom the protein is likely to be administered.
- Amelioration can refer to a reduction in an immune response (i.e., reduced immunogenicity) compared to an unmodified protein (e.g.
- antibodies, or antigen-binding fragments thereof, with reduced immunogenicity can refer to a percent reduction in its ability to elicit an immune response compared to an unmodified protein (e.g. about 1% less, about 2% less, about 3% less, about 4% less, about 5% less, about 10% less, about 20%) less, about 50%> less, about 100% less, and any range therein).
- PBMC derived T-cells from naive donors are collected from a pool of donors of sufficient immunological diversity to provide a sample of at least greater than 90% of the MHC class II repertoire (HLA-DR) extant in the human population.
- HLA-DR MHC class II repertoire
- the peptide (or antibody) in practice is contacted with PBMC preparations derived from multiple donors in isolation; the numbers of donors (or "donor pool” size), is for practical purposes not likely to be less than 20 unrelated individuals and all samples in the donor pool may be pre-selected according to their MHC class II haplotype.
- nonaive donor refers to a subject that has not been previously exposed to antibodies, or antigen-binding fragments thereof, described herein either environmentally, by vaccination, or by other means such as, for example, blood transfusions.
- T-cells can be provided from a peripheral blood sample from a multiplicity of different healthy donors but who have not been in receipt of the protein therapeutically. If needed, patient blood samples can be tested for the presence of a particular polypeptide using conventional assays such as an ELISA which uses antibodies to identify the presence or absence of one or more polypeptides.
- the assay is conducted using PBMC cultured in vitro using conventional procedures known in the art and involves contacting the PBMC with synthetic peptide species representative of the protein of interest (i.e. a library), or a whole protein, such as an antibody and following a suitable period of incubation, measurement of induced T cell activation such as cellular proliferation.
- Measurement can be by any suitable means and may, for example, be conducted using H 3 -thymidine incorporation whereby the accumulation of H 3 into cellular material is readily measured using laboratory instruments.
- the degree of cellular proliferation for each combination of PBMC sample and synthetic peptide or whole protein can be examined relative to that seen in an untreated PBMC sample. Reference may also be made to the proliferative response seen following treatment with a peptide or peptides or whole proteins for which there is an expected proliferative effect.
- it is advantageous to use a peptide or whole protein with known broad MHC restriction and especially peptide epitopes with MHC restriction to the DP or DQ isotypes although the invention is not limited to the use of such restricted peptides or proteins.
- Such peptides have been described above, for example, with respect to influenza haemagglutinin and chlamydia HSP60.
- T-cell epitopes are mapped and subsequently modified using the methods described herein.
- a library of synthetic peptides is produced. Each of the peptides is 15 amino acid residues in length and each overlapped the next peptide in the series by 12 amino acid residues; i.e. each successive peptide in the series incrementally added a further 3 amino acids to the analysis. In this way, any given adjacent pair of peptides mapped 18 amino acids of contiguous sequence.
- One method for defining a T-cell map using naive T-cell assays is illustrated in the Examples below.
- Each of the peptides identified via the method to define a T-cell map are suggested to be able to bind MHC class II and engage at least one cognate TCR with sufficient affinity to evoke a proliferative burst detectable in the assay system.
- the potential of an antibody to be processed to generate T-cell epitopes that bind MHC class II and engage at least one cognate TCR with sufficient affinity to evoke a proliferative burst detectable in the assay system is assessed.
- the molecules described herein can be prepared in any of several ways including the use of recombinant methods.
- the protein sequences and information provided herein can be used to deduce a polynucleotide (DNA) encoding an amino acid sequence. This can be achieved for example using computer software tools such as the DNAstar software suite [DNAstar Inc, Madison, Wis., USA] or similar. Any such polynucleotide encoding the polypeptides or significant homologues, variants, truncations, elongations, or further modifications thereof, are contemplated herein.
- mapping includes amino acid substitutions, deletions, or insertion made in codons of a polynucleotide encoding modified polypeptides to affect similar changes. Codons encoding amino acid residues are well known in the art. It is possible to use recombinant DNA methods to achieve directed mutagenesis of the target sequences and many such techniques are available, described herein, and known in the art such as described above. In general, the technique of site-specific mutagenesis is well known.
- a bacteriophage vector that produces a single stranded template for oligonucleotide directed PCR mutagenesis is employed.
- Phage vectors e.g. M13
- double stranded plasmids are also routinely employed in site directed mutagenesis, which eliminates the step of transferring the polynucleotide of interest from a phage to a plasmid.
- Synthetic oligonucleotide primers bearing the desired mutated sequence can be used to direct the in vitro synthesis of modified (desired mutant) DNA from this template and the heteroduplex DNA is used to transform competent E. coli for the growth selection and identification of desired clones.
- a pair of primers can be annealed to two separate strands of a double stranded vector to simultaneously synthesize both corresponding
- the Quick Change site-directed mutagenesis method using plasmid DNA templates may be employed.
- PCR amplification of the plasmid template containing the insert target gene of insert is achieved using two synthetic oligonucleotide primers containing the desired mutation.
- the oligonucleotide primers, each complementary to opposite strands of the vector, are extended during temperature cycling by mutagenesis-grade PfuTurbo DNA polymerase.
- a mutated plasmid containing staggered nicks is generated.
- Amplified un-methylated products are treated with Dpn I to digest methylated parental DNA template and select for the newly synthesized DNA containing mutations. Since DNA isolated from most E. coli strains is dam methylated, it is susceptible to Dpn I digestion, which is specific for methylated and hemimethylated DNA. The reaction products are transformed into high efficiency strains of E. coli to obtain plasmids containing the desired modifications. Additional methods for introducing amino acid
- Suitable modifications to a protein may include amino acid substitution of particular residues or combinations of residues.
- amino acid substitutions are made at appropriate points or amino acid residues within an amino acid sequence predicted to achieve reduction or elimination of the activity of the T-cell epitope.
- an appropriate point or amino acid residue will preferably equate to an amino acid residue binding within one of the pockets provided within the MHC class II binding groove.
- Such modifications may alter binding within the first pocket of the cleft at the so-called "PI" or "PI anchor” position of the peptide.
- the quality of binding interaction between the PI anchor residue of the peptide and the first pocket of the MHC class II binding groove is recognized as being a major determinant of overall binding affinity for the whole peptide.
- An appropriate substitution at this position of the amino acid sequence will generally incorporate an amino acid residue less readily accommodated within the pocket (e.g., substitution to a more hydrophilic residue).
- Amino acid residues in the peptide at positions equating to binding within other pocket regions within the MHC binding cleft are also considered and fall under the scope of the present.
- modifications can be made that alter the structure and/or reduce the biological activity of the molecule and also eliminate a T-cell epitope, thus reducing the immunogenicity of the molecule. All types of modifications are contemplated herein.
- the in silico process can test a same polypeptide sequence using >40 allotypes simultaneously. In practice this approach is able to direct the design of new sequence variants which are altered in their ability to interact with multiple MHC allotypes. As will be clear to one in the art, multiple alternative sets of substitutions could be arrived at which achieve the objective of removing undesired epitopes. The resulting sequences would however be recognized to be closely homologous with the specific compositions disclosed herein and therefore fall within the scope of the present application.
- a combined approach of using an in silico tool for the identification of MHC class II ligands and design of sequence analogues lacking MHC class II ligands, in concert with epitope mapping and re-testing optionally using biologically based assays of T-cell activation is an additional method and embodiment of the present application.
- the general method according to this embodiment comprises the following steps:
- step (ii) use of a computational scheme simulating the binding of the peptide ligand with one or more MHC allotypes to analyze the epitope regions identified in step (i) and thereby identify MHC class II ligands within the epitope region;
- naive T-cell activation assays and synthetic peptides encompassing entirely or in collection encompassing the epitope regions identified within the protein of interest and testing the sequence analogues in naive T-cell activation assay in parallel with the wild-type (parental) sequences.
- a method of making a modified antibody, or antigen-binding fragment thereof, exhibiting reduced immunogenicity compared to an unmodified antibody, or antigen-binding fragment thereof comprises identifying at least one T-cell epitope within the amino acid sequence of a antibody, or antigen-binding fragment thereof, and modifying at least one amino acid residue within at least one identified T-cell epitope.
- a modified antibody, or antigen-binding fragment thereof, exhibiting reduced immunogenicity compared to an unmodified antibody, or antigen- binding fragment thereof is produced by a process of identifying at least one T-cell epitope within the amino acid sequence of a antibody, or antigen-binding fragment thereof, and modifying at least one amino acid residue within at least one identified T-cell epitope.
- a method of selecting a modified antibody, or antigen-binding fragment thereof, that exhibits reduced immunogenicity compared to an unmodified antibody, or antigen-binding fragment thereof comprises identifying at least one T- cell epitope within the amino acid sequence of a antibody, or antigen-binding fragment thereof,, modifying at least one amino acid residue within at least one identified T-cell epitope, and selecting a modified antibody, or antigen-binding fragment thereof, that exhibits reduced immunogenicity compared to an unmodified antibody, or antigen-binding fragment thereof.
- T-cell epitopes described herein can be further characterized by the regions of the epitope. Such regions include the epitope core, the N-terminus and the C-terminus.
- epitope core refers to the core 9-mer amino acid sequences of the T-cell epitopes.
- the epitope core can further include 0, 1, 2, or 3 amino acid residues adjacent to the core 9-mer amino acid sequence on the N-terminus and/or the C-terminus.
- the epitope core in certain embodiments, can range in length from about 9 amino acids up to about 15 amino acids.
- N-terminus refers to the amino acids adjacent to the N-terminus of the epitope core and includes at least 1, 2, 3, 4, 5, 6, 7, 8 or 9 amino acids adjacent to and upstream of the N-terminus of the epitope core.
- C-terminus refers to the amino acids adjacent to the C-terminus of the epitope core and includes at least 1, 2, 3, 4, 5, 6, 7, 8, or 9 amino acids adjacent to and downstream of the C-terminus of the epitope core.
- a modified antibody, or antigen-binding fragment thereof contains one or more modifications.
- a modified antibody, or antigen-binding fragment thereof contains two modifications.
- a modified antibody, or antigen-binding fragment thereof contains three modifications.
- a modified antibody, or antigen-binding fragment thereof contains four modifications.
- a modified antibody, or antigen-binding fragment thereof contains five modifications.
- a modified antibody, or antigen-binding fragment thereof contains six modifications.
- a modified antibody, or antigen-binding fragment thereof contains seven modifications.
- a modified antibody, or antigen-binding fragment thereof contains eight modifications.
- a modified antibody, or antigen-binding fragment thereof contains nine modifications.
- a modified antibody, or antigen-binding fragment thereof contains ten modifications.
- a modified antibody, or antigen-binding fragment thereof contains up to twenty modifications.
- an antibody, or antigen-binding fragment thereof comprising a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 93—
- VK1 AA a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 89 (VH1A2).
- an antibody, or antigen-binding fragment thereof comprising a heavy chain variable region having an amino acid sequence set forth as any one of SEQ ID NOS: 88, 89, 90, 91 and 92.
- an antibody, or antigen-binding fragment thereof comprising a light chain variable region having an amino acid sequence set forth as any one of SEQ ID NOS: 93, 94, 95, 96, 97, 100, 102, and 103.
- an antibody, or antigen-binding fragment thereof, that binds endoglin comprising a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 89 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 93, wherein:
- the heavy chain variable region further comprises one or more modifications selected from the group consisting of a substitution of glycine (G) by alanine (A) or serine (S) at position 49; a substitution of alanine (A) by isoleucine (I) at position 51; a substitution of lysine (K) by arginine (R) or asparagine (Q) at position 52b; a substitution of leucine (L) by valine (V) at position 78 utilizing the Kabat numbering system; and
- the light chain variable region further comprises one or more modifications selected from the group consisting of a substitution of methionine (M) by leucine (L) at position 4; a substitution of alanine (A) by valine (V) at position 19; a substitution of threonine (T) by serine (S) at position 22; a substitution of alanine (A) by isoleucine (I) at position 48; and a substitution of threonine (T) by serine (S) at position 51 utilizing the Kabat numbering system
- an antibody, or antigen-binding fragment thereof comprising a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 88, 89, 90, 91 and 92; and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 93, 95, 96, 97, 100, 102, or 103.
- a modified antibody, or antigen-binding fragment thereof, with one or more amino acid modifications in one or more T-cell epitopes are contemplated herein.
- antibodies, or antigen-binding fragments thereof, having at least one modification in at least one T-cell epitope are provided herein.
- Additional non-limiting examples include antibodies, or antigen-binding fragments thereof, having more than one amino acid modification in more than one T-cell epitope. Any combination of the amino acid modifications in any number of the antibodies, or antigen-binding fragments thereof, T-cell epitopes described above are
- the MHC class II allotype of a subject or subjects can be easily determined by genotyping methods known in the art, and the association of T-cell epitopes with the given allotype thus easily identified, for consideration in modification of antibodies, or antigen-binding fragments thereof, tailored to that allotype. Identification of associations between T-cell epitopes and MHC class II allotypes are described in more detail in the examples below. Contemplated herein are modified antibodies, or antigen-binding fragments thereof, that have T-cell epitope modifications tailored to the MHC class II associations identified for the given epitopes.
- Simultaneous incorporation of all of the FR and/or CDR encoding nucleic acids and all of the selected amino acid position changes can be accomplished by a variety of methods known to those skilled in the art, including for example, recombinant and chemical synthesis.
- simultaneous incorporation can be accomplished by, for example, chemically synthesizing the nucleotide sequence for the acceptor variable region, fused together with the donor CDR encoding nucleic acids, and incorporating at the positions selected for harboring variable amino acid residues a plurality of corresponding amino acid codons.
- antibodies and antigen-binding fragments thereof that bind to endoglin. Also provided are antibodies and antigen-binding fragments thereof that bind endoglin and inhibit (partially or fully) or manage/treat (partially or fully)
- angiogenesis/neovascularization, dilation of small vessels, and/or diseases associated with excessive angiogenesis is also included within the meaning of inhibiting or binding endoglin.
- an antibody or antigen-binding fragment inhibits angiogenesis by binding to endoglin.
- the application also provides cell lines which can be used to produce the antibodies, methods for producing the cell lines, methods for expressing antibodies or antigen- binding fragments and purifying the same.
- antibodies and antigen-binding fragments thereof that specifically bind endoglin generated using the methods described herein can be tested using the assays provided herein or known in the art for the ability to bind to endoglin using conventional methods including, but not limited to, ELISA. Affinity of antibodies described herein can also be determined using conventional methods including, but not limited to, Biacore or surface plasmon resonance.
- the antibodies and antigen binding fragments thereof described herein were constructed by humanization of the V H and V L sequences of the TRC105 antibody. To accomplish this humanization, a 3 -dimensional model of the V H and V L chains of TRC105 was created and analyzed. The V H and V L sequences were then compared individually to a database of human germline sequences, from which human V H and V L sequences were chosen based on their homology to the V H and V L sequences of TRC105.
- the human V L sequence chosen for humanization was 02/012 (VKl-39) (SEQ ID NO. 2). 02/012 has a sequence identity with
- the human VH sequence chosen for humanization was VH3-15 (SEQ ID NO. 40).
- VH3-15 has sequence identity with TRC105 of 70% and is expressed with reasonable frequency in the human germline repertoire.
- the amino acid positions which were different between TRC105 and the human sequences were examined in the 3D model of TRC105 to determine which substitutions would be considered for modification.
- the identified and proposed substitutions for the human framework regions are incorporated into the 02 and VH3-15 human framework regions, and the CDRs of TRC105 are grafted into the corresponding 02 and VH3-15 human framework regions resulting in a multitude of humanized antibodies or antigen-binding fragments.
- the FR-4 of the light chain is derived from human J germline sequence Jk4.
- the FR-4 of the heavy chain is derived from human J germline sequence JH4.
- Described herein are humanized antibodies and antigen-binding fragments that bind endoglin. Also described herein are humanized antibodies and antigen-binding fragments that bind endoglin and inhibit angiogenesis. The antibodies and antigen-binding fragments described herein were generated as described above.
- Antibodies and antigen-binding fragments thereof can have a variable heavy (VH) chain, a variable light (VL) chain, both, or binding portions thereof.
- VH chain has an amino acid sequence set forth as any of SEQ ID NOS: 41-43, or a binding portion thereof.
- VH chains can have framework regions sequences set forth as any of SEQ ID NOS: 44-62.
- the VL chain has an amino acid sequence set forth as any of SEQ ID NOS: 3-5, or a binding portion thereof.
- Such VL chains can have framework regions sequences set forth as any of SEQ ID NOS: 6-38.
- an antibody, or antigen-binding fragment thereof comprising a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 3 and a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 41.
- an antibody, or antigen-binding fragment thereof comprising a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 3 and a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 41, wherein: the heavy chain variable region further comprises one or more modifications selected from the group consisting of a substitution of glycine (G) by alanine (A) at position 49; a substitution of asparagine (N) by serine (S) at position 76; a substitution of threonine (T) by arginine (R) at position 77; a substitution of leucine (L) by valine (V) at position 78; a substitution of asparagine (N) by isoleucine (I) at position 82a; a substitution of valine (V) by isoleucine (I) or leucine (L) at position 89; a substitution of threonine (T) by arginine (R) or
- an antibody, or antigen-binding fragment thereof that binds endoglin comprising a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 41, 42, or 43; and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 3, 4, or 5.
- An antibody, or antigen-binding fragment thereof can comprise a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 41 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 3.
- An antibody, or antigen-binding fragment thereof can comprise a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 41 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 4.
- An antibody, or antigen-binding fragment thereof can comprise a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 41 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 5.
- An antibody, or antigen-binding fragment thereof can comprise a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 42 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 3.
- An antibody, or antigen-binding fragment thereof can comprise a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 42 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 4.
- An antibody, or antigen-binding fragment thereof can comprise a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 42 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 5.
- An antibody, or antigen-binding fragment thereof can comprise a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 43 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 3.
- An antibody, or antigen-binding fragment thereof can comprise a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 43 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 4.
- An antibody, or antigen-binding fragment thereof can comprise a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 43 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 5.
- Such antibodies can bind to endoglin and inhibit angiogenesis.
- a heavy chain variable region can further comprise one ore more modifications selected from the group consisting of: a substitution of asparagine (N) by serine (S) at position 76; a substitution of threonine (T) by arginine (R) at position 77; a substitution of asparagine (N) by isoleucine (I) at position 82a; a substitution of valine (V) by isoleucine (I) or leucine (L) at position 89; a substitution of threonine (T) by glycine (G) at position 94; a substitution of leucine (L) by threonine (T) at position 108; a substitution of valine (V) by leucine (L) at position 109; and a substitution of serine (S) by alanine (A) a position 113; and the light chain variable region can further comprise one or more modifications selected from the group consisting of: a substitution of asparagine (N) by serine
- an antibody, or antigen-binding fragment thereof, that binds endoglin comprising a heavy chain variable region and a light chain variable region, wherein said heavy chain variable region comprises:
- a heavy chain FR1 having the amino acid sequence of SEQ ID NO: 44 or the amino acid sequence of SEQ ID NO: 44 except for one or more conservative substitutions
- a heavy chain FR2 having the amino acid sequence of SEQ ID NO: 45 or the amino acid sequence of SEQ ID NO: 45 except for a substitution of glycine (G) by alanine (A) at position 49 utilizing the Kabat numbering system
- G glycine
- A alanine
- valine (V) a substitution of valine (V) by isoleucine (I) or leucine (L) at position 89;
- said light chain variable region comprises:
- amino acid sequence of SEQ ID NO: 20 except for one or more substitutions selected from the group consisting of:
- amino acid sequence of SEQ ID NO: 28 except for one or more substitutions selected from the group consisting of:
- amino acid sequence of SEQ ID NO: 35 except for one or more substitutions selected from the group consisting of:
- An antibody, or antigen-binding fragment thereof, provided herein can comprise a heavy chain variable region CDR1 having an amino acid sequence as set forth in SEQ ID NO: 66, a heavy chain variable region CDR2 having an amino acid sequence as set forth in SEQ ID NO: 67, a heavy chain variable region CDR3 having an amino acid sequence as set forth in SEQ ID NO: 68, a light chain variable region CDR1 having an amino acid sequence as set forth in SEQ ID NO: 63, a light chain variable region CDR2 having an amino acid sequence as set forth in SEQ ID NO: 64, and a light chain variable region CDR3 having an amino acid sequence as set forth in SEQ ID NO: 65.
- the antibody, or antigen-binding fragment thereof binds endoglin and comprises a heavy chain variable region FR1 having an amino acid sequence as set forth in SEQ ID NO: 44; a heavy chain variable region FR2 having an amino acid sequence as set forth in SEQ ID NO: 45; a heavy chain variable region FR3 having an amino acid sequence as set forth in SEQ ID NO: 47; a heavy chain variable region FR4 having an amino acid sequence as set forth in SEQ ID NO: 56.
- the antibody, or antigen-binding fragment thereof binds endoglin and comprises a heavy chain variable region FR1 having an amino acid sequence as set forth in SEQ ID NO: 44; a heavy chain variable region FR2 having an amino acid sequence as set forth in SEQ ID NO: 46; a heavy chain variable region FR3 having an amino acid sequence as set forth in SEQ ID NO: 48; a heavy chain variable region FR4 having an amino acid sequence as set forth in SEQ ID NO: 56.
- the antibody, or antigen-binding fragment thereof comprises a light chain variable region FR1 having an amino acid sequence as set forth in SEQ ID NO: 6; a light chain variable region FR2 having an amino acid sequence as set forth in SEQ ID NO: 20; a light chain variable region FR3 having an amino acid sequence as set forth in SEQ ID NO: 28; and a light chain variable region FR4 having an amino acid sequence as set forth in SEQ ID NO: 35.
- the antibody, or antigen-binding fragment thereof binds endoglin and comprises a light chain variable region FR1 having an amino acid sequence as set forth in SEQ ID NO: 6; a light chain variable region FR2 having an amino acid sequence as set forth in SEQ ID NO: 21; a light chain variable region FR3 having an amino acid sequence as set forth in SEQ ID NO: 29; and a light chain variable region FR4 having an amino acid sequence as set forth in SEQ ID NO: 35.
- the antibody, or antigen-binding fragment thereof binds endoglin and comprises a light chain variable region FR1 having an amino acid sequence as set forth in SEQ ID NO: 7; a light chain variable region FR2 having an amino acid sequence as set forth in SEQ ID NO: 21; a light chain variable region FR3 having an amino acid sequence as set forth in SEQ ID NO: 29; and a light chain variable region FR4 having an amino acid sequence as set forth in SEQ ID NO: 35.
- an antibody, or antigen-binding fragment thereof comprising a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 42 and a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 4.
- an antibody, or antigen-binding fragment thereof, that binds endoglin comprising a light chain variable region having an amino acid sequence set forth as SEQ ID NO: 4 and a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 42, wherein: said heavy chain variable region further comprises one or more modifications selected from the group consisting of a substitution of glycine (G) by alanine (A) at position 49; a substitution of asparagine (N) by serine (S) at position 76; a substitution of threonine (T) by arginine (R) at position 77; a substitution of leucine (L) by valine (V) at position 78; a substitution of asparagine (N) by isoleucine (I) at position 82a; a substitution of valine (V) by isoleucine (I) or leucine (L) at position 89; a substitution of arginine (R) by
- an antibody, or antigen-binding fragment thereof, that binds endoglin comprising a heavy chain variable region and a light chain variable region,
- said heavy chain variable region comprises:
- a heavy chain FR1 having the amino acid sequence of SEQ ID NO: 44 or the amino acid sequence of SEQ ID NO: 44 except for one or more conservative substitutions
- a heavy chain FR2 having the amino acid sequence of SEQ ID NO: 45 or the amino acid sequence of SEQ ID NO: 45 except for a substitution of glycine (G) by alanine (A) at position 49 utilizing the Kabat numbering system
- G glycine
- A alanine
- valine (V) a substitution of valine (V) by isoleucine (I) or leucine (L) at position 89;
- said light chain variable region comprises:
- amino acid sequence of SEQ ID NO: 20 except for one or more substitutions selected from the group consisting of:
- amino acid sequence of SEQ ID NO: 28 except for one or more substitutions selected from the group consisting of:
- amino acid sequence of SEQ ID NO: 35 except for one or more substitutions selected from the group consisting of:
- a substantial portion of a variable domain will include three CDR regions, together with their intervening framework regions.
- the portion can also include at least about 50% of either or both of the first and fourth framework regions, the 50% being the C-terminal 50% of the first framework region and the N-terminal 50% of the fourth framework region. Additional residues at the N-terminal or C-terminal end of the substantial part of the variable domain may be those not normally associated with naturally occurring variable domain regions.
- construction of humanized endoglin antibodies and antigen-binding fragments described herein made by recombinant DNA techniques can result in the introduction of N- or C- terminal residues encoded by linkers introduced to facilitate cloning or other manipulation steps.
- Other manipulation steps include the introduction of linkers to join variable domains to further protein sequences including immunoglobulin heavy chains, other variable domains (for example in the production of diabodies) or protein labels as discussed in more detail below.
- Humanized endoglin CDR3 regions having amino acid sequences substantially as set out as the CDR3 regions of the antibodies described herein will be carried in a structure which allows for binding of the CDR3 regions to endoglin.
- the structure for carrying the CDR3s can be of an antibody heavy or light chain sequence or substantial portion thereof in which the CDR3 regions are located at locations corresponding to the CDR3 region of naturally-occurring V H and V L antibody variable domains encoded by rearranged immunoglobulin genes.
- variable heavy chain having a CDR3 which has an amino acid sequence set forth as SEQ ID NO: 68 and/or a variable light chain having a CDR3 which has an amino acid sequence set forth as SEQ ID NO: 65.
- variable heavy chain has an amino acid sequence set forth as SEQ ID NO: 40 except for a substitution of the CDR3 by the CDR3 amino sequence set forth as SEQ ID NO: 68.
- variable light chain has an amino acid sequence set forth as SEQ ID NO: 2 except for a substitution of the CDR3 by the CDR3 amino acid sequence set forth as SEQ ID NO: 65.
- variable regions/chains can comprise one or more FR amino acid sequences set forth as, for example, described above (or such FRs containing one or more additional modifications), where the antibodies or antigen binding fragments have 3 CDRs and 4 FRs in each of the VH and VL regions, have specific binding activity for endoglin and which are able to inhibit angiogenesis. Additionally, various antibody J segments can also be substituted within these variable regions for further variation within the variable region chains.
- variable heavy and light chains described herein can also be created by further replacing FR4 sequences.
- heavy chain FR4 sequences can be substituted for one of the following:
- light chain FR4 sequences can be substituted for one of the following:
- Such superhumanized antibodies, or antigen-binding fragments thereof can comprise a light chain variable region having an amino acid sequence set forth as SEQ ID NOS: 71 or 72 and a heavy chain variable region having an amino acid sequence set forth as SEQ ID NO: 75.
- the present application provides a humanized antibody capable of competing with a humanized anti-endoglin antibody or antigen-binding described herein under conditions in which at least 5% of an antibody having the V H and V L sequences of the antibody is blocked from binding to endoglin by competition with such an antibody in an ELISA assay.
- neutralizing antibodies or antigen-binding fragments that bind to endoglin and modulate the activity of endoglin.
- the neutralizing antibody can for example, inhibit angiogenesis by binding to endoglin.
- Percentage (%) of inhibition of angiogenesis by a humanized anti-endoglin antibody or antigen- binding fragment thereof of less than 2-fold greater than negative controls is indicative of an antibody or antigen-binding fragment thereof that does not inhibit angiogenesis.
- the neutralizing or inhibiting activity of an antibody or antigen-binding fragment can be determined using an in vitro assay and/or in vivo using art- recognized assays such as those described herein or otherwise known in the art.
- the antigen-binding fragment of any one of the humanized antibodies described above is a Fab, a Fab', a Fd, a F(ab') 2 , a Fv, a scFv, a single chain binding polypeptide (e.g., a scFv with Fc portion) or any other functional fragment thereof as described herein.
- Antibodies or antigen-binding fragments described herein are useful in detection or diagnostic applications as described in more detail below. Antibodies or antigen-binding fragments described herein are useful for binding to endoglin, which, in turn, can inhibit angiogenesis as described herein.
- Antibodies, or antigen-binding fragments thereof, described herein can be further modified to alter the specific properties of the antibody while retaining the desired functionality, if needed.
- the compound can be modified to alter a
- Antibodies, or antigen-binding fragments thereof, described herein can further comprise a therapeutic moiety, a detectable moiety, or both, for use in diagnostic and/or therapeutic applications.
- immunoconjugates refer to conjugates comprised of the humanized anti-endoglin antibodies or fragments thereof according to the present invention and at least one therapeutic label.
- Therapeutic labels include antitumor agents and angiogenesis-inhibitors.
- antitumor agents are known in the art and include, but not limited to, toxins, drugs, enzymes, cytokines, radionuclides, photodynamic agents, and angiogenesis inhibitors.
- Toxins include, but are not limited to, ricin A chain, mutant Pseudomonas exotoxins, diphtheria toxoid, streptonigrin, boamycin, saporin, gelonin, and pokeweed antiviral protein.
- Drugs include daunorubicin, methotrexate, and calicheamicins.
- Radionuclides include radiometals.
- Cytokines include, but are not limited to, transforming growth factor (TGF)-P, interleukins, interferons, and tumor necrosis factors.
- Photodynamic agents include, but are not limited to, porphyrins and their derivatives. Additional therapeutic labels will be known in the art and are also contemplated herein.
- the methods for complexing the anti-endoglin mAbs or a fragment thereof with at least one antitumor agent are well known to those skilled in the art (i.e., antibody conjugates as reviewed by Ghetie et al., 1994, Pharmacol. Ther. 63:209-34). Such methods may utilize one of several available heterobifunctional reagents used for coupling or linking molecules. Additional radionuclides are further described herein along with additional methods for linking molecules, such as therapeutic and diagnostic labels.
- Antibodies, or antigen-binding fragments thereof can be modified using techniques known in the art for various purposes such as, for example, by addition of
- PEG polyethylene glycol
- an Fc portion can be added to (e.g., recombinantly) the fragment, for example, to increase half- life of the antigen-binding fragment in circulation in blood when administered to a patient.
- Fc region of an IgG into a polypeptide of interest so as to increase its circulatory half-life, but so as not to lose its biological activity can be accomplished using conventional techniques known in the art such as, for example, described in U.S. Patent No. 6,096,871, which is hereby incorporated by reference in its entirety.
- Fc portions of antibodies can be further modified to increase half-life of the antigen-binding fragment in circulation in blood when administered to a patient. Modifications can be determined using conventional means in the art such as, for example, described in U.S. Patent No. 7,217,798, which is hereby incorporated by reference in its entirety.
- ADCC antibody dependent cell- mediated cytotoxicity
- CDC complement dependent cytotoxicity
- antibodies or antigen-binding fragments thereof that can bind endoglin can be attached at their C-terminal end to all or part of an immunoglobulin heavy chain derived from any antibody isotype, e.g., IgG, IgA, IgE, IgD and IgM and any of the isotype sub-classes, particularly IgGl, IgG2b, IgG2a, IgG3 and IgG4.
- the antibodies or antigen-binding fragments described herein can also be modified so that they are able to cross the blood-brain barrier.
- Such modification of the antibodies or antigen-binding fragments described herein allows for the treatment of brain diseases such as glioblastoma multiforme (GBM).
- GBM glioblastoma multiforme
- Exemplary modifications to allow proteins such as antibodies or antigen-binding fragments to cross the blood-brain barrier are described in US Patent Application Publication 2007/0082380 which is hereby incorporated by reference in its entirety.
- ADCC antibody dependent cell- mediated cytotoxicity
- CDC complement dependent cytotoxicity
- “defucosylated” antibodies and antigen-binding fragments may be produced through a variety of systems utilizing molecular cloning techniques known in the art, including but not limited to, transgenic animals, transgenic plants, or cell-lines that have been genetically engineered so that they no longer contain the enzymes and biochemical pathways necessary for the inclusion of a fucose in the complex N-glycoside-linked sugar chains (also known as fucosyltransferase knock- out animals, plants, or cells).
- Non-limiting examples of cells that can be engineered to be fucosyltransferase knock-out cells include CHO cells, SP2/0 cells, NSO cells, and YB2/0 cells.
- V region Glycosylation of immunoglobulins in the variable (V) region has also been observed. Sox and Hood reported that about 20% of human antibodies are glycosylated in the V region (Proc. Natl. Acad. Sci. USA 66:975 (1970)). Glycosylation of the V domain is believed to arise from fortuitous occurrences of the N-linked glycosylation signal Asn-Xaa-Ser/Thr in the V region sequence and has not been recognized in the art as playing a role in immunoglobulin function.
- Glycosylation at a variable domain framework residue can alter the binding interaction of the antibody with antigen.
- the present invention includes criteria by which a limited number of amino acids in the framework or CDRs of a humanized immunoglobulin chain are chosen to be mutated (e.g., by substitution, deletion, or addition of residues) in order to increase the affinity of an antibody.
- Affinity for binding a pre-determined polypeptide antigen can, generally, be modulated by introducing one or more mutations into the V region framework, typically in areas adjacent to one or more CDRs and/or in one or more framework regions.
- mutations involve the introduction of conservative amino acid substitutions that either destroy or create the glycosylation site sequences but do not substantially affect the hydropathic structural properties of the polypeptide.
- mutations that introduce a proline residue are avoided.
- Glycosylation of antibodies and antigen-binding fragments thereof is further described in U.S. Patent No. 6,350,861, which is incorporated by reference herein with respect to glycosylation.
- Antibodies, or antigen-binding fragments thereof can be formulated for short- term delivery or extended (long term) delivery.
- Antibodies, or antigen-binding fragments thereof, that bind to endoglin can also be used for purification of endoglin and/or to detect endoglin levels in a sample or patient to detect or diagnose a disease or disorder associated with endoglin as described in more detail below.
- Humanized antibodies, antigen-binding fragments, and binding proteins which bind endoglin generated using such methods can be tested for one or more of their binding affinity, avidity, and neutralizing capabilities.
- Useful humanized antibodies, antigen-binding fragments, and binding proteins can be used to administer a patient to prevent, inhibit, manage or treat a condition disease or disorder associated with angiogenesis.
- Antibodies and antigen-binding fragments can be evaluated for one or more of binding affinity, association rates, disassociation rates and avidity.
- antibodies can be evaluated for their ability to neutralize the activity of endoglin or a polypeptide in which the endoglin binding sequence is present.
- Measurement binding affinity, association rates, disassociation rates and avidity can be accomplished using art- recognized assays including (Surface Plasmon Resonance), but not limited to, an enzyme-linked- immunosorbent assay (ELISA), Scatchard Analysis, BIACORE analysis, etc., as well as other assays commonly used and known to those of ordinary skill in the art.
- ELISA enzyme-linked- immunosorbent assay
- Scatchard Analysis BIACORE analysis
- Measurement of binding of antibodies to endoglin and/or the ability of the antibodies and antigen-binding fragments thereof, for example, to inhibit angiogenesis can be determined using, for example, an enzyme-linked-immunosorbent assay (ELISA), a competitive binding assay, an ELISPOT assay, or any other useful assay known in the art.
- ELISA enzyme-linked-immunosorbent assay
- ELISPOT assay an enzyme-linked-immunosorbent assay
- an ELISA assay can be used to measure the binding capability of specific antibodies or antigen-binding fragments that bind to endoglin.
- Assays such as an ELISA, also can be used to identify antibodies or antigen- binding fragments thereof which exhibit increased specificity for endoglin in comparison to other antibodies or antigen-binding fragments thereof. Assays, such as an ELISA, also can be used to identify antibodies or antigen-binding fragments thereof with bind to epitopes across one or more polypeptides and across one or more species of endoglin.
- the specificity assay can be conducted by running parallel ELISAs in which a test antibodies or antigen-binding fragments thereof is screened concurrently in separate assay chambers for the ability to bind one or more epitopes on different species of the polypeptide containing the endoglin epitopes to identify antibodies or antigen-binding fragments thereof that bind to endoglin.
- Another technique for measuring apparent binding affinity familiar to those of skill in the art is a surface plasmon resonance technique (analyzed on a BIACORE 2000 system) (Liljeblad, et al., Glyco. J. 2000, 17:323-329). Standard measurements and traditional binding assays are described by Heeley, R. P., Endocr. Res. 2002, 28:217-229.
- Humanized antibodies to endoglin can also be assayed for their ability to treat various diseases and conditions associated with angiogenesis, e.g., various forms of ocular diseases characterized by angiogenesis/neovascularization (e.g., macular degeneration, diabetic retinopathy), diabetic nephropathy, chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, and various forms of cancer (primary tumors and metastases). Any suitable assay known to one of skill in the art can be used to monitor such effects. Several such techniques are described herein. In one example, the antibodies and antigen-binding fragments described herein are assayed for their ability to bind endoglin.
- angiogenesis/neovascularization e.g., macular degeneration, diabetic retinopathy
- diabetic nephropathy e.g., chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarth
- affinity constants for the antibodies and antigen-binding fragments described herein are determined by surface plasmon resonance (SPR).
- the antibodies and antigen-binding fragments described herein are assayed for their effect on the inhibition of angiogenesis.
- compositions can be used as a composition when combined with an acceptable carrier or excipient. Such compositions are useful for in vitro or in vivo analysis or for administration to a subject in vivo or ex vivo for treating a subject with the disclosed compounds.
- compositions can include, in addition to active ingredient, a pharmaceutically acceptable excipient, carrier, buffer, stabilizer or other materials well known to those skilled in the art. Such materials should be non-toxic and should not interfere with the efficacy of the active ingredient. The precise nature of the carrier or other material will depend on the route of administration.
- compositions comprising a protein of interest, e.g., an antibody or antigen-binding fragment, identified by the methods described herein can be prepared for storage by mixing the protein having the desired degree of purity with optional physiologically acceptable carriers, excipients or stabilizers (Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980)), in the form of lyophilized formulations or aqueous solutions.
- Acceptable carriers, excipients, or stabilizers are those that are non-toxic to recipients at the dosages and concentrations employed, and include buffers such as phosphate, citrate, and other organic acids; antioxidants including ascorbic acid and methionine; preservatives (such as
- octadecyldimethylbenzyl ammonium chloride hexamethonium chloride; benzalkonium chloride, benzethonium chloride; phenol, butyl or benzyl alcohol; alkyl parabens such as methyl or propyl paraben; catechol; resorcinol; cyclohexanol; 3-pentanol; and m-cresol); low molecular weight (less than about 10 residues) polypeptides; proteins, such as serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine, histidine, arginine, or lysine; monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrins; chelating agents such as EDTA; sugars such as sucrose, mannitol, trehalose or
- Acceptable carriers are physiologically acceptable to the administered patient and retain the therapeutic properties of the compounds with/in which it is administered. Acceptable carriers and their formulations are and generally described in, for example, Remington' pharmaceutical Sciences (18th Edition, ed. A. Gennaro, Mack Publishing Co., Easton, PA 1990).
- One exemplary carrier is physiological saline.
- pharmaceutically acceptable carrier means a pharmaceutically acceptable material, composition or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material, involved in carrying or transporting the subject compounds from the administration site of one organ, or portion of the body, to another organ, or portion of the body, or in an in vitro assay system. Each carrier is acceptable in the sense of being compatible with the other ingredients of the formulation and not injurious to a subject to whom it is administered. Nor should an acceptable carrier alter the specific activity of the subject compounds.
- compositions including solvents (aqueous or non-aqueous), solutions, emulsions, dispersion media, coatings, isotonic and absorption promoting or delaying agents, compatible with pharmaceutical administration.
- solvents aqueous or non-aqueous
- solutions emulsions, dispersion media, coatings, isotonic and absorption promoting or delaying agents, compatible with pharmaceutical administration.
- compositions or pharmaceutically acceptable or physiologically acceptable compositions including solvents (aqueous or non-aqueous), solutions, emulsions, dispersion media, coatings, isotonic and absorption promoting or delaying agents, compatible with pharmaceutical administration.
- formulations therefore refer to a composition suitable for pharmaceutical use in a subject.
- the pharmaceutical compositions and formulations include an amount of a compound described herein and a pharmaceutically or physiologically acceptable carrier.
- compositions can be formulated to be compatible with a particular route of administration (i.e., systemic or local).
- routes of administration i.e., systemic or local.
- compositions include carriers, diluents, or excipients suitable for administration by various routes.
- compositions can further comprise, if needed, an acceptable additive in order to improve the stability of the compounds in composition and/or to control the release rate of the composition.
- Acceptable additives do not alter the specific activity of the subject compounds.
- Exemplary acceptable additives include, but are not limited to, a sugar such as mannitol, sorbitol, glucose, xylitol, trehalose, sorbose, sucrose, galactose, dextran, dextrose, fructose, lactose and mixtures thereof.
- Acceptable additives can be combined with acceptable carriers and/or excipients such as dextrose.
- exemplary acceptable additives include, but are not limited to, a surfactant such as polysorbate 20 or polysorbate 80 to increase stability of the peptide and decrease gelling of the solution.
- the surfactant can be added to the composition in an amount of 0.01% to 5% of the solution. Addition of such acceptable additives increases the stability and half-life of the composition in storage.
- the pharmaceutical composition can be administered, for example, by injection, including, but not limited to, subcutaneous, subcutaneous, intravitreal, intradermal, intravenous, intra-arterial, intraperitoneal, or intramuscular injection.
- Excipients and carriers for use in formulation of compositions for each type of injection are contemplated herein. The following descriptions are by example only and are not meant to limit the scope of the compositions.
- compositions for injection include aqueous solutions (where water soluble) or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersion.
- suitable carriers include physiological saline, bacteriostatic water, Cremophor ELTM (BASF, Parsippany, N.J.) or phosphate buffered saline (PBS).
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, and liquid polyetheylene glycol, and the like), and suitable mixtures thereof.
- Fluidity can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersion and by the use of surfactants.
- Antibacterial and antifungal agents include, for example, parabens, chlorobutanol, phenol, ascorbic acid and thimerosal.
- Isotonic agents for example, sugars, polyalcohols such as manitol, sorbitol, and sodium chloride may be included in the composition.
- the resulting solutions can be packaged for use as is, or lyophilized; the lyophilized preparation can later be combined with a sterile solution prior to administration.
- the active ingredient will be in the form of a parenterally acceptable aqueous solution which is pyrogen- free and has suitable pH, isotonicity and stability.
- a parenterally acceptable aqueous solution which is pyrogen- free and has suitable pH, isotonicity and stability.
- Suitable solutions using, for example, isotonic vehicles such as Sodium Chloride Injection, Ringer's Injection, Lactated Ringer's Injection. Preservatives, stabilizers, buffers, antioxidants and/or other additives may be included, as needed.
- Sterile injectable solutions can be prepared by incorporating an active ingredient in the required amount in an appropriate solvent with one or a combination of ingredients enumerated above, as required, followed by filtered sterilization.
- dispersions are prepared by incorporating the active ingredient into a sterile vehicle which contains a basic dispersion medium and the required other ingredients from those enumerated above.
- a sterile vehicle which contains a basic dispersion medium and the required other ingredients from those enumerated above.
- the preferred methods of preparation are vacuum drying and freeze drying which yields a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered solution thereof.
- compositions can be conventionally administered intravitreally, sub-cutaneous, or via intravitreal implant.
- compositions can be conventionally administered intravenously, such as by injection of a unit dose, for example.
- an active ingredient can be in the form of a parenterally acceptable aqueous solution which is substantially pyrogen- free and has suitable pH, isotonicity and stability.
- suitable solutions using, for example, isotonic vehicles such as Sodium Chloride Injection, Ringer's Injection, Lactated Ringer's Injection. Preservatives, stabilizers, buffers, antioxidants and/or other additives may be included, as required.
- compositions can be administered via aerosolization.
- aerosolization (Lahn et al., Aerosolized Anti-T-cell-Receptor Antibodies Are Effective against Airway Inflammation and Hyperreactivity, Int. Arch. Allegery Immuno., 134: 49-55 (2004)).
- the composition is lyophilized, for example, to increase shelf-life in storage.
- the compositions can be substantially free of pyrogens such that the composition will not cause an inflammatory reaction or an unsafe allergic reaction when administered to a human patient.
- Testing compositions for pyrogens and preparing compositions substantially free of pyrogens are well understood to one or ordinary skill of the art and can be accomplished using commercially available kits.
- Acceptable carriers can contain a compound that stabilizes, increases or delays absorption or clearance.
- Such compounds include, for example, carbohydrates, such as glucose, sucrose, or dextrans; low molecular weight proteins; compositions that reduce the clearance or hydrolysis of peptides; or excipients or other stabilizers and/or buffers.
- Agents that delay absorption include, for example, aluminum monostearate and gelatin. Detergents can also be used to stabilize or to increase or decrease the absorption of the pharmaceutical composition, including liposomal carriers.
- the compound can be complexed with a composition to render it resistant to acidic and enzymatic hydrolysis, or the compound can be complexed in an appropriately resistant carrier such as a liposome.
- phrases "pharmaceutically acceptable” refers to molecular entities and compositions that are physiologically tolerable and do not typically produce an allergic or similar untoward reaction, such as gastric upset, dizziness and the like, when administered to a human.
- unit dose when used in reference to a therapeutic composition refers to physically discrete units suitable as unitary dosage for humans, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect in association with the required diluent; i.e., carrier, or vehicle.
- compositions can be administered in a manner compatible with the dosage formulation, and in a therapeutically effective amount.
- quantity to be administered depends on the subject to be treated, capacity of the subject's immune system to utilize the active ingredient, and degree of binding capacity desired. Precise amounts of active ingredient required to be administered depend on the judgment of the practitioner and are peculiar to each individual. Suitable regimes for initial administration and booster shots are also variable, but are typified by an initial administration followed by repeated doses at one or more hour intervals by a subsequent injection or other administration. Alternatively, continuous intravenous infusion sufficient to maintain concentrations in the blood are contemplated.
- compositions described herein to make a medicament for treating a condition, disease or disorder described herein.
- Medicaments can be formulated based on the physical characteristics of the patient/subject needing treatment, and can be formulated in single or multiple formulations based on the stage of the condition, disease or disorder.
- Medicaments can be packaged in a suitable package with appropriate labels for the distribution to hospitals and clinics wherein the label is for the indication of treating a subject having a disease described herein.
- Medicaments can be packaged as a single or multiple units. Instructions for the dosage and administration of the compositions can be included with the packages as described below.
- the invention is further directed to medicaments of a humanized anti-endoglin antibody or antigen binding fragment thereof described hereinabove and a pharmaceutically acceptable carrier.
- compositions of humanized antibodies and antigen-binding fragments thereof that bind endoglin include those such as described elsewhere herein.
- Humanized antibodies and antigen-binding fragments thereof that bind endoglin as described herein can be used for the treatment of various forms of ocular diseases characterized by angiogenesis/neovascularization (e.g., macular degeneration, diabetic retinopathy), diabetic nephropathy, chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, and various forms of cancer (primary tumors and metastases).
- angiogenesis/neovascularization e.g., macular degeneration, diabetic retinopathy
- diabetic nephropathy e.g., chronic inflammatory diseases (e.g., IBD)
- rheumatoid arthritis e.g., osteoarthritis
- various forms of cancer primary tumors and metastases.
- a composition (an antibody or an antigen-binding fragment described herein) can be administered alone or in combination with a second composition either simultaneously or sequentially dependent upon the condition to be treated.
- a second therapeutic treatment is an angiogenesis inhibitor (as described herein).
- the compositions can be administered in combination (either sequentially or simultaneously).
- a composition can be administered in a single dose or multiple doses.
- compositions are formulated to be free of pyrogens such that they are acceptable for administration to human patients. Testing compositions for pyrogens and preparing pharmaceutical compositions free of pyrogens are well understood to one of ordinary skill in the art.
- One embodiment of the present invention contemplates the use of any of the compositions of the present invention to make a medicament for treating a disorder of the present invention.
- Medicaments can be formulated based on the physical characteristics of the patient/subject needing treatment, and can be formulated in single or multiple formulations based on the disorder.
- Medicaments of the present invention can be packaged in a suitable
- Medicaments can be packaged as a single or multiple units. Instructions for the dosage and administration of the pharmaceutical compositions of the present invention can be included with the pharmaceutical packages.
- a method of inducing a response in a patient by administering to the patient a composition of an antibody or antigen-binding fragment thereof that preferentially binds to endoglin.
- the binding site to which the antibody binds can be a continuous or conformation/dis-continuous epitope.
- An effective response of the present invention is achieved when the patient experiences partial or total alleviation or reduction of signs or symptoms of illness, and specifically includes, without limitation, prolongation of survival and/or visual acuity.
- the expected progression-free survival times may be measured in months to years, depending on prognostic factors including the number of relapses, stage of disease, and other factors.
- Prolonging survival includes without limitation times of at least 1 month (mo), about at least 2 mos., about at least 3 mos., about at least 4 mos., about at least 6 mos., about at least 1 year, about at least 2 years, about at least 3 years, etc. Overall survival can be also measured in months to years. Alternatively, an effective response may be that a patient's symptoms remain static. Further indications of treatment of indications are described in more detail below.
- compositions of antibodies and antigen-binding fragments described herein can be used as non-therapeutic agents (e.g., as affinity purification agents).
- a protein of interest is immobilized on a solid phase such a Sephadex resin or filter paper, using conventional methods known in the art.
- the immobilized protein is contacted with a sample containing the target of interest (or fragment thereof) to be purified, and thereafter the support is washed with a suitable solvent that will remove substantially all the material in the sample except the target protein, which is bound to the immobilized antibody. Finally, the support is washed with another suitable solvent, such as glycine buffer, pH 5.0, which will release the target protein.
- compositions can be used for detection, diagnosis and therapy of diseases and disorders associated with endoglin and angiogenesis.
- contacting refers to adding together a solution or composition of a compound with a liquid medium bathing the polypeptides, cells, tissue or organ from an organism.
- contacting refers to mixing together a solution or composition of a compound, with a liquid such as blood, serum, or plasma derived from an organism.
- a composition can also comprise another component, such as dimethyl sulfoxide (DMSO). DMSO facilitates the uptake of the compounds or solubility of the compounds.
- DMSO dimethyl sulfoxide
- the solution comprising the test compound may be added to the medium bathing the cells, tissues, or organs, or mixed with another liquid such as blood, by utilizing a delivery apparatus, such as a pipette-based device or syringe-based device.
- a delivery apparatus such as a pipette-based device or syringe-based device.
- contacting can occur, for example, via administration of a composition to a patient by any suitable means; compositions with pharmaceutically acceptable excipients and carriers have been described in more detail above.
- a "patient” e.g., a mammal such as a human or a non-human animal such as a primate, rodent, cow, horse, pig, sheep, etc.
- a mammal e.g., a mammal such as a human or a non-human animal such as a primate, rodent, cow, horse, pig, sheep, etc.
- the patient may be asymptomatic and yet still have clinical manifestations of the disease or disorder.
- An antibody or antigen-binding fragment thereof can be conjugated to a therapeutic moiety or be a fusion protein containing a therapeutic moiety.
- An antibody or antigen-binding fragment thereof can be conjugated to a detectable moiety or be a fusion protein containing a detectable moiety. In one embodiment, the antibody or antigen-binding fragment thereof can be conjugated to both a therapeutic moiety and a detectable moiety.
- An antibody or antigen-binding fragment thereof can be conjugated to, or recombinantly engineered with, an affinity tag (e.g., a purification tag).
- affinity tag e.g., a purification tag.
- Affinity tags such as, for example, His6 tags (SEQ ID NO: 85) are conventional in the art.
- Antibodies or antigen-binding fragments thereof provided herein are such that they can be conjugated or linked to a therapeutic moiety and/or an imaging or a detectable moiety and/or an affinity tag.
- Methods for conjugating or linking polypeptides are well known in the art.
- Associations (binding) between compounds and labels include any means known in the art including, but not limited to, covalent and non-covalent interactions, chemical conjugation as well as recombinant techniques.
- Endoglin (CD 105) is expressed on the cell surface as a 180 kDa homodimeric transmembrane protein.
- the external domain binds TGF- ⁇ and -3 iso forms with high affinity (50 nM), and the transmembrane and the intracellular domains of CD 105 share a 71% sequence similarity with betaglycan.
- the human CD 105 gene is located on chromosome 9q34, identified using fluorescence in situ hybridization, and the coding region contains 14 exons, and two different iso forms (L and S) of CD 105 with capacity to bind TGF- ⁇ have been characterized.
- the L-CD105 consists of 633 amino acid residues with 47 amino acid residues in the
- cytoplasmic tail as opposed to the S-CD105, which consists of 600 amino acid residues with a 14 amino acid cytoplasmic tail.
- L-CD105 is the predominant form.
- CD 105 is
- the human CD 105 amino acid sequence contains the tripeptide arginine-glycine-aspartic acid (RGD) located in an exposed region of the extracellular domain.
- RGD arginine-glycine-aspartic acid
- the RGD peptide is a key recognition structure found on ECM proteins such as fibronectin, vitronectin, von Willebrand factor (vWF), type I collagen, and fibrinogen and is recognized by cell surface integrins.
- Integrin adhesion has been implicated in hemostasis, thrombosis, angiogenesis and inflammation, processes in which the endothelium plays a critical role. (Duff et al, FASEB J., 17:984-992 (2003)).
- CD 105 is a member of the TGF- ⁇ receptor family that is expressed by
- CD 105 proliferating endothelial cells. Normal levels of CD 105 are needed for endothelial cell proliferation. CD 105 expression is increased by cellular hypoxia through the production of hypoxia-inducible factor- 1 -a (HIF-l-a) and protects hypoxic cells from apoptosis.
- HIF-l-a hypoxia-inducible factor- 1 -a
- RI receptor I
- RII receptor II
- Binding of TGF- ⁇ to the external domains of the receptor unmasks the cytoplasmic RII kinase activity that phosphorylates the TGF- ⁇ RI, which can then interact with downstream signalers such as the Smad proteins.
- CD 105 forms part of the TGF- ⁇ receptor complex but it can exist independently on the cell surface. In many cells in vitro, CD 105 suppresses TGF- ⁇ signaling.
- CD 105 also binds other growth factors such as activin A and bone morphogenic proteins (BMP) -10, -9, -7 and -2. Binding of TGF- ⁇ or other growth factor ligands to CD 105 requires the presence of at least the receptor RII, and it cannot bind ligands by itself. CD 105 association with receptors does not alter their affinity for the ligand itself. Upon association, the cytoplasmic domain of CD 105 is phosphorylated by TGF- ⁇ RI and TGF- ⁇ RII; then TGF- ⁇ RI, but not TGF- ⁇ RII, kinase dissociates from the receptor complex.
- BMP bone morphogenic proteins
- CD 105 expression inhibits phosphorylation levels of TGF- ⁇ RII but increases that of TGF- ⁇ RI, resulting in increased phosphorylation of Smad 2 but not Smad 3. Since Smad 2 can interact with a variety of transcription factors, co-activators, and suppressors, phosphorylated Smad 2 may act as an integrator of multiple signals to modulate gene transcription. Thus, CD 105 modulates TGF- ⁇ functions via interaction with TGF- ⁇ RI and TGF- ⁇ RII and modifies the phosphorylation of downstream Smad proteins.
- CD 105 acts to modulate signaling of multiple kinase receptor complexes of the TGF- ⁇ superfamily, including TGF- ⁇ receptors (TGF ⁇ R), activin receptor- like kinases (ALK) and activin receptors.
- TGF ⁇ R TGF- ⁇ receptors
- ALK activin receptor-like kinases
- activin receptors TGF- ⁇ receptors
- TGF- ⁇ receptors TGF- ⁇ receptors
- ALK activin receptor- like kinases
- activin receptors results in phosphorylation of SMAD proteins (SMAD 2 and 3) that inhibit endothelial cell growth.
- CD 105 activation by TGF- ⁇ modulates SMAD protein phosphorylation (including the phosphorylation of SMAD 1, 5 and 8).
- the end result is release of the growth inhibitory effects of TGF- ⁇ receptor activation on endothelial cells (see Figure 3).
- prevention of CD 105 activation by anti-CD 105 antibody or antisense oligonucleotide acts synergistically with TGF- ⁇ to suppress endothelial cell growth.
- the CD 105 promoter is 2.6 kb in length but does not contain TATA or CAAT transcription initiation boxes. However, it has two GC-rich regions, consensus motifs for Spl, ets, GAT A, AP-2, NGF- ⁇ , and Mad, as well as TGF- ⁇ response elements. Nonetheless, CD 105 has a relatively restricted cellular distribution. The basal level of transcription appears to require an ets site at position -68 and the Spl sites, but the relative restriction of expression, for example, to endothelial cells, appears to involve multiple regulatory regions, in particular, one at -1294 to -932 and another very close to the transcription initiation site.
- CD105 is up-regulated by TGF- ⁇ , and this has been shown to require a Spl site at -37 to -29, also involving one or more juxtaposed upstream SBE sites binding Smads 3 and/or 4 (which are activated by TGF- ⁇ signaling).
- Hypoxia is a common feature of ischemic tissues and tumors, and is a potent stimulator for CD 105 gene expression in vascular endothelial cells (ECs). Such an effect is potentiated in combination with TGF- ⁇ 1.
- the up-regulated CD 105 can exert a self-protective role in ECs under hypoxic stress.
- Vascular EC are the major source of CD 105.
- Other cell types including vascular smooth muscle cells, fibroblasts, macrophages, leukemic cells of pre-B and myelomonocytic origin, and erythroid precursors express CD 105 to a lesser extent.
- CD 105 is involved in angiogenesis. Antisense experiments have demonstrated that suppression of CD 105 expression in HUVEC results in marked inhibition of in vitro angiogenesis in combination with TGF- ⁇ , indicating that CD 105 is a proangiogenic component in the endothelial cells. Further evidence of the important role of CD 105 in angiogenesis comes from CD 105 knockout mice.
- the CD 105 null mice exhibit multiple vascular and cardiac defects leading to death at an early embryonic stage. Severe vascular impairments observed in CD 105 null mice indicate that CD 105 is required for the formation of mature blood vessels in the extraembryonic vasculature, further confirming the direct role of endoglin in angiogenesis.
- Endoglin also known as, inter alia, CD105 or edg-1, is a type I homodimeric membrane glycoprotein which is expressed at high levels in proliferating vascular endothelial cells. Thus, endoglin is primarily a proliferation-associated marker for endothelial cells undergoing active angiogenesis. However, there may be limited expression of endoglin by the vascular endothelium of normal tissues. Human endoglin is known to specifically bind transforming growth factor- ⁇ (TGF- ⁇ ), and the deduced amino acid sequence of endoglin has strong homology to ⁇ -glycan, a type of TGF- ⁇ receptor.
- TGF- ⁇ transforming growth factor- ⁇
- Endoglin has been targeted in antibody-based methods of reducing tumor vasculature, as EDG is a proliferation-associated antigen on endothelial and leukemia cells. Its expression is up-regulated in tumor-associated vascular endothelium, and EDG is essential for angiogenesis.
- Angiogenesis includes the formation of new capillary blood vessels leading to neovascularization as well as the maintenance of the existing vasculature. It is a complex process which includes a series of sequential steps including endothelial cell-mediated degradation of vascular basement membrane and interstitial matrices, migration of endothelial cells, proliferation of endothelial cells, and formation of capillary loops by endothelial cells.
- Endoglin can be found on cells that comprise and support existing vasculature as well as cells that are promoting the growth of, and become part of, new vasculature.
- the antibodies can bind endoglin and thereby inhibit angiogenesis, inhibit the existing vasculature or the maintenance of the existing vasculature, and/or inhibit small vessel dilation.
- these antibodies are useful for purification, detection and diagnostic purposes as well as therapeutic purposes.
- the antibodies provided herein can be used for the formulation of medicaments for the treatment a variety of conditions and diseases, methods to treat said conditions and diseases and methods of detection or diagnosis.
- Murine monoclonal antibodies have been raised against endoglin which modulate endoglin activity and thereby inhibit angiogenesis and/or inhibit vasodilation of small blood vessels. These murine antibodies are described in U.S. patents 5,928,641, 6,200,566, 6,190,660, and 7,097,836, each of which is hereby incorporated in their entirety. Additionally, the ex vivo and in vivo efficiency of a number of these antibodies has been demonstrated;
- monoclonal antibodies that bind endoglin are of interest as endoglin modulating compounds.
- Therapeutic use of murine antibodies is not feasible, however, as administration of the murine antibodies has a number of limitations, including immunogenicity in, for example, the form of human anti-mouse antibodies (HAMA).
- HAMA human anti-mouse antibodies
- angiogenesis is used herein to include all aspects of blood vessel maintenance and development.
- angiogenesis includes the formation of new capillary blood vessels leading to neovascularization as well as the maintenance and control of the existing vasculature and small blood vessels.
- Angiogenesis is a complex process which includes a series of sequential steps including endothelial cell-mediated degradation of vascular basement membrane and interstitial matrices, migration of endothelial cells, proliferation of endothelial cells, and formation of capillary loops by endothelial cells.
- Angiogenesis is inclusive of the growth and/or development of new blood vessels (also referred to as neovascularization), dilation of the small vessels, excessive or prolonged vascular growth, and maintenance of the existing vasculature. Endoglin is known to be involved in the regulation of angiogenesis and is believed to be involved in multiple biochemical pathways related to the induction of angiogenesis. (Duff et al., FASEB J., 17:984-992 (2003); Bernabeu et al, J. Cell. Biochem., 102(6):1375-1388 (2007)).
- angiogenesis inhibitory As used herein, the terms "angiogenesis inhibitory,” “angiogenesis inhibiting” or
- anti-angiogenic include inhibition of vasculogenesis, and are intended to mean affecting a decrease in the extent, amount, or rate of neovascularization. Effecting a decrease in the extent, amount, or rate of endothelial cell proliferation or migration in the tissue is a specific example of inhibiting angiogenesis.
- angiogenesis inhibitory composition refers to a composition which inhibits angiogenesis-mediated processes such as endothelial cell migration, proliferation, tube formation and subsequently leading to the inhibition of the generation of new blood vessels from existing ones, and consequently affects angiogenesis-dependent conditions.
- angiogenesis-associated disease is used herein, for purposes of the specification and claims, to mean certain pathological processes in humans where angiogenesis is abnormally prolonged.
- This further includes angiogenesis conditions and diseases, such as those diseases and conditions related to, caused by, or associated with angiogenesis.
- Non- limiting examples of such diseases include various forms of ocular diseases characterized by angiogenesis/neovascularization (e.g., macular degeneration, diabetic retinopathy), diabetic nephropathy, chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, and various forms of cancers and metastasis.
- the antibodies and antigen-binding fragments thereof described herein can be used to treat an angiogenesis-associated disease by binding endoglin and inhibiting angiogenesis.
- anti-angiogenic therapy is used herein, for purposes of the
- therapy targeted to cells and/or vasculature expressing endoglin expressed at higher levels on proliferating vasculature as compared to quiescent vasculature; this further includes therapy that is directed against angiogenesis (i.e., the formation of new capillary blood vessels leading to neovascularization), therapy that is directed against existing vasculature and/or excessive vascularization or blood vessel growth, therapy directed towards the dilation of small vessels, and therapy directed to a disease or condition (e.g., vascular targeting therapy).
- exemplary diseases or conditions contemplated within the invention include, but are not limited to, various forms of ocular diseases characterized by
- angiogenesis/neovascularization e.g., macular degeneration, diabetic retinopathy), diabetic nephropathy, chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, and various forms of cancer, solid tumors, and metastases.
- Ocular disease characterized by neovascularization is used herein, for purposes of the specification and claims, to mean any ocular disease caused by, or resulting in, increased angiogenesis within any portion of the eye, including the retina, cornea, pupil, iris, vitreous humor or aqueous humor.
- diseases include for example, age-related macular degeneration, diabetic retinopathy, non-diabetic retinopathy, choroidal neovascularization (CNV) and sub- retinal neovascularization (SR or SR V) and neoplasms of the eye.
- CNV choroidal neovascularization
- SR or SR V sub- retinal neovascularization
- Humanized anti-endoglin antibodies and fragments thereof can be used for in vivo and in vitro detection, diagnostic and/or monitoring purposes. Endoglin is believed to be involved in multiple diseases and disorders as described further below. Treatment of endoglin related diseases and conditions depends, in part, upon their diagnosis, and the antibodies and antigen-binding fragments thereof described herein are useful for the diagnosis of excess endoglin or for diagnosis for diseases and conditions associated with endoglin activity.
- a method of detecting levels of endoglin in a sample or a subject comprising (i) contacting an antibody or antigen binding fragment described herein with the sample or subject, and (ii) detecting a complex of the antibody or antigen-binding fragment thereof and endoglin.
- a method of imaging or diagnosing angiogenesis or an angiogenic-dependent disease or disorder comprising contacting a composition of an antibody or antigen-binding fragment thereof as described herein with a sample.
- the sample can be, for example, blood, serum, plasma, platelets, biopsy fluid, spinal tap fluid, meninges and urine. Imaging or diagnosis method can occur in an in vitro assay.
- the angiogenesis or angiogenic-dependent disease or disorder is imaged or diagnosed in vivo.
- the antibody or antigen-binding fragment further comprises a detectable moiety. Detection can occur in vitro, in vivo or ex vivo. In vitro assays for the detection and/or determination (quantification, qualification, etc.) of endoglin with the antibodies or antigen-binding fragments thereof include but are not limited to, for example, ELISAs, RIAs and western blots. In vitro detection, diagnosis or monitoring of endoglin can occur by obtaining a sample (e.g., a blood sample) from a patient and testing the sample in, for example, a standard ELISA assay.
- a sample e.g., a blood sample
- a 96-well microtiter plate can be coated with an antibody or antigen- binding fragment thereof described herein, washed and coating with PBS-Tween/BSA to inhibit non-specific binding.
- the blood sample can be serially diluted and placed in duplicate wells compared to a serially-diluted standard curve of endoglin. After incubating and washing the wells, an anti- endoglin antibody labeled with biotin can be added, followed by addition of streptavidin-alkaline phosphatase.
- the wells can be washed and a substrate (horseradish peroxidase) added to develop the plate.
- the plate can be read using a conventional plate reader and software.
- contacting occurs via administration of the antibody or antigen binding fragment using any conventional means such as those described elsewhere herein.
- detection of endoglin in a sample or a subject can be used to diagnose a disease or disorder associated with, or correlated with the activity of endoglin such as those diseases and disorders described herein.
- a patient is administered an antibody or antigen-binding fragment that binds to endoglin, which antibody or antigen-binding fragment is bound to a detectable moiety.
- the detectable moiety can be visualized using art-recognized methods such as, but not limited to, magnetic resonance imaging (MRI), fluorescence, radioimaging, light sources supplied by endoscopes, laparoscopes, or intravascular catheter (i.e., via detection of photoactive agents), photoscanning, positron emission tomography (PET) scanning, whole body nuclear magnetic resonance (NMR), radioscintography, single photon emission computed tomography (SPECT), targeted near infrared region (NIR) scanning, X-ray, ultrasound, etc. such as described, for example, in U.S. Patent No. 6,096,289, U.S. Patent No. 7,115,716, U.S. Patent No. 7,112,412, U.S.
- MRI magnetic resonance imaging
- fluorescence fluorescence
- radioimaging light sources supplied by endoscopes, laparoscopes
- intravascular catheter i.e., via detection of photoactive agents
- photoscanning positron emission tomography (PE
- Non-limiting conditions, diseases and disorders to be considered for these methods include, but are not limited to, those associated with angiogenesis such as, for example, various forms of ocular diseases characterized by angiogenesis/neovascularization (e.g., macular degeneration, diabetic retinopathy), diabetic nephropathy, chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, and various forms of cancer (primary tumors and metastases).
- angiogenesis/neovascularization e.g., macular degeneration, diabetic retinopathy
- diabetic nephropathy chronic inflammatory diseases
- rheumatoid arthritis e.g., osteoarthritis
- various forms of cancer primary tumors and metastases.
- the subject patient is administered a composition of an antibody or antigen-binding fragment thereof described herein, which antibody or antigen-binding fragment thereof is conjugated to a detectable moiety.
- the moiety can be visualized using
- Visualization of the detectable moiety can allow for detection, diagnosis, and/or monitoring of such conditions and diseases.
- samples to be obtained from a patient include, but are not limited to, blood, tissue biopsy samples and fluid therefrom.
- the present invention provides humanized antibodies and antigen-binding fragments thereof against endoglin which are useful for detecting or diagnosing levels of endoglin associated with a disease or disorder, potentially indicating need for therapeutic treatment.
- the antibodies comprise a humanized anti-endoglin antibody described herein.
- the antibody further comprises a second agent.
- Such an agent can be a molecule or moiety such as, for example, a reporter molecule or a detectable label. Detectable labels/moieties for such detection methods are known in the art and are described in more detail below. Reporter molecules are any moiety which can be detected using an assay.
- Non-limiting examples of reporter molecules which have been conjugated to polypeptides include enzymes, radiolabels, haptens, fluorescent labels, phosphorescent molecules, chemiluminescent molecules, chromophores, luminescent molecules, photoaffinity molecules, colored particles or ligands, such as biotin.
- Detectable labels include compounds and/or elements that can be detected due to their specific functional properties, and/or chemical characteristics, the use of which allows the polypeptide to which they are attached to be detected, and/or further quantified if desired. Many appropriate detectable (imaging) agents are known in the art, as are methods for their attachment to polypeptides (see, for e.g., U.S. Pat. Nos.
- Methods of joining polypeptides such as antibodies with detectable moieties include, for example, recombinant DNA technology to form fusion proteins and conjugation (e.g., chemical conjugation). Methods for preparing fusion proteins by chemical conjugation or recombinant engineering are well-known in the art. Methods of covalently and non-covalently linking components are also known in the art. See, e.g., Williams (1995)
- linker can facilitate enhanced flexibility, and/or reduce steric hindrance between any two fragments.
- the linker can also facilitate the appropriate folding of each fragment to occur.
- the linker can be of natural origin, such as a sequence determined to exist in random coil between two domains of a protein.
- One linker sequence is the linker found between the C-terminal and N-terminal domains of the RNA polymerase a subunit.
- Other examples of naturally occurring linkers include linkers found in the 1CI and Lex A proteins.
- an amino acid sequence can be varied based on the characteristics of the linker as determined empirically or as revealed by modeling. Considerations in choosing a linker include flexibility of the linker, charge of the linker, and presence of some amino acids of the linker in the naturally-occurring subunits.
- the linker can also be designed such that residues in the linker contact deoxyribose nucleic acid (DNA), thereby influencing binding affinity or specificity, or to interact with other proteins. In some cases, such as when it is necessary to span a longer distance between subunits or when the domains must be held in a particular
- the linker can, optionally, contain an additional folded domain.
- the design of a linker can involve an arrangement of domains which requires the linker to span a relatively short distance, e.g., less than about 10 Angstroms (A). However, in certain embodiments, linkers span a distance of up to about 50 Angstroms.
- the amino acid sequence can be varied based on the characteristics of the linker as determined empirically or as revealed by modeling.
- linker Considerations in choosing a linker include flexibility of the linker, charge of the linker, and presence of some amino acids of the linker in the naturally-occurring subunits.
- the linker can also be designed such that residues in the linker contact DNA, thereby influencing binding affinity or specificity, or to interact with other proteins.
- the linker can optionally contain an additional folded domain.
- microparticle surfaces that have been modified with functional groups or coated with various antibodies or ligands such as, for example, avidin, streptavidin or biotin.
- microparticles The paramagnetic property of microparticles allows them to be separated from solution using a magnet.
- the microparticles can be easily re-suspended when removed from the magnet.
- Polypeptides can be coupled to paramagnetic polystyrene microparticles coated with a polyurethane layer in a tube.
- the hydroxy groups on the microparticle surface are activated by reaction with p-toluensulphonyl chloride (Nilsson K and Mosbach K. "p-Toluenesulfonyl chloride as an activating agent of agarose for the preparation of immobilized affinity ligands and proteins.” Eur. J. Biochem. 1980: 112: 397-402).
- paramagnetic polystyrene microparticles containing surface carboxylic acid can be activated with a carbodiimide followed by coupling to a polypeptide, resulting in a stable amide bond between a primary amino group of the polypeptide and the carboxylic acid groups on the surface of the microparticles (Nakajima N and Ikade Y, Mechanism of amide formation by carbodiimide for bioconjugation in aqueous media, Bioconjugate Chem. 1995, 6(1): 123-130; Gilles MA, Hudson AQ and Borders CL Jr, Stability of water-soluble carbodiimides in aqueous solution, Anal Biochem.
- paramagnetic polystyrene microparticles whose surfaces have been covalently linked with a monolayer of streptavidin.
- Argarana CE Kuntz ID, Birken S, Axel R, Cantor CR.
- Molecular cloning and nucleotide sequence of the streptavidin gene Nucleic Acids Res. 1986;14(4): 1871- 82; Pahler A, Hendrickson WA, Gawinowicz Kolks MA, Aragana CE, Cantor CR. Characterization and crystallization of core streptavidin. J Biol Chem 1987:262(29): 13933- 13937).
- Polypeptides can be conjugated to a wide variety of fluorescent dyes, quenchers and haptens such as fluorescein, R-phycoerythrin, and biotin. Conjugation can occur either during polypeptide synthesis or after the polypeptide has been synthesized and purified.
- Biotin is a small (244 kilodaltons) vitamin that binds with high affinity to avidin and streptavidin proteins and can be conjugated to most peptides without altering their biological activities.
- Biotin- labeled polypeptides are easily purified from unlabeled polypeptides using immobilized streptavidin and avidin affinity gels, and streptavidin or avidin-conjugated probes can be used to detect biotinylated polypeptides in, for example, ELISA, dot blot or Western blot applications.
- N- hydroxysuccinimide esters of biotin are the most commonly used type of biotinylation agent.
- N- hydroxysuccinimide-activated biotins react efficiently with primary amino groups in
- Polypeptides have primary amines at the N- terminus and can also have several primary amines in the side chain of lysine residues that are available as targets for labeling with N-hydroxysuccinimide-activated biotin reagents.
- N-hydroxysuccinimide esters of biotin are available, with varying properties and spacer arm length (Pierce, Rockford, IL).
- the sulfo-N-hydroxysuccinimide ester reagents are water soluble, enabling reactions to be performed in the absence of organic solvents.
- the mole-to-mole ratio of biotin to polypeptide can be estimated using a 2-(4'-
- biotin molecules can be conjugated to a polypeptide and each biotin molecule can bind one molecule of avidin.
- the biotin-avidin bond formation is very rapid and stable in organic solvents, extreme pH and denaturing reagents.
- a solution containing the biotinylated polypeptide is added to a mixture of 2-(4'-Hydroxyazobenzene-2-carboxylic acid) and avidin. Because biotin has a higher affinity for avidin, it displaces the 2-(4'- Hydroxyazobenzene-2-carboxylic acid) and the absorbance at 500 nanometers decreases proportionately.
- the amount of biotin in a solution can be quantitated in a single cuvette by measuring the absorbance of the 2-(4'-Hydroxyazobenzene-2-carboxylic acid)-avidin solution before and after addition of the biotin-containing peptide.
- the change in absorbance relates to the amount of biotin in the sample by the extinction coefficient of the 2-(4'-Hydroxyazobenzene- 2-carboxylic acid)-avidin complex.
- an antibody, antigen-binding fragment or binding protein can be conjugated with a fluorescent moiety
- Conjugating polypeptides with fluorescent moieties e.g., R-Phycoerythrin, fluorescein isothiocyanate (FITC), etc.
- fluorescent moieties e.g., R-Phycoerythrin, fluorescein isothiocyanate (FITC), etc.
- FITC fluorescein isothiocyanate
- an antibody antigen-binding fragment can be associated with (conjugated to) a detectable label, such as a radionuclide, iron-related compound, a dye, an imaging agent or a fluorescent agent for immunodetection of endoglin which can be used to visualize binding of the antibodies to endoglin in vitro and/or in vivo.
- a detectable label such as a radionuclide, iron-related compound, a dye, an imaging agent or a fluorescent agent for immunodetection of endoglin which can be used to visualize binding of the antibodies to endoglin in vitro and/or in vivo.
- radiolabels include, for example, P, P, K, Fe,
- Radiolabels can be attached to compounds using conventional chemistry known in the art of antibody imaging. Radiolabeled compounds are useful in in vitro diagnostics techniques and in in vivo radioimaging techniques and in
- the antibodies and antigen-binding fragments thereof can be conjugated to an imaging agent rather than a radioisotope(s), including but not limited to a magnetic resonance image enhancing agent, wherein for instance an antibody molecule is loaded with a large number of paramagnetic ions through chelating groups.
- chelating groups include EDTA, porphyrins, polyamines crown ethers and polyoximes.
- paramagnetic ions include gadolinium, iron, manganese, rhenium, europium, lanthanium, holmium and ferbium.
- Such detectable moieties also include: metals; metal chelators; lanthanides; lanthanide chelators; radiometals; radiometal chelators; positron-emitting nuclei; microbubbles (for ultrasound); liposomes; molecules microencapsulated in liposomes or nanosphere; monocrystalline iron oxide nanocompounds; magnetic resonance imaging contrast agents; light absorbing, reflecting and/or scattering agents; colloidal particles; fluorophores, such as near-infrared fluorophores.
- such secondary functionality/moiety will be relatively large, e.g., at least 25 amu in size, and in many instances can be at least 50, 100 or 250 amu in size.
- the secondary functionality is a chelate moiety for chelating a metal, e.g., a chelator for a radiometal or paramagnetic ion. In embodiments, it is a chelator for a radionuclide useful for radiotherapy or imaging procedures.
- Antagonists of the invention also can be assayed for their ability to modulate angiogenesis in a tissue. Any suitable assay known to one of skill in the art can be used to monitor such effects. Several such techniques are described herein.
- angiogenesis is an in vivo rabbit eye model and is referred to as the rabbit eye assay.
- the rabbit eye assay has been described in detail by others, and further has been used to measure both angiogenesis and neovascularization in the presence of angiogenic inhibitors such as thalidomide. See D'Amato et al. (1994) Proc. Natl. Acad. Sci. 91 :4082-4085.
- the rabbit eye assay is a well recognized assay model for in vivo angiogenesis because the neovascularization process, exemplified by rabbit blood vessels growing from the rim of the cornea into the cornea, is easily visualized through the naturally transparent cornea of the eye. Additionally, both the extent and the amount of stimulation or inhibition of
- neovascularization or regression of neovascularization can easily be monitored over time.
- the rabbit is exposed to any test reagent, and therefore the health of the rabbit is an indication of toxicity of the test reagent.
- Another assay measures angiogenesis in the chimeric mouse:human mouse model and is referred to as the chimeric mouse assay.
- the assay has been described in detail by others, and further has been described herein to measure angiogenesis, neovascularization, and regression of tumor tissues. See Yan, et al. (1993) J. Clin. Invest. 91 :986-996.
- the chimeric mouse assay is a useful assay model for in vivo angiogenesis because the transplanted skin grafts closely resemble normal human skin histologically and neovascularization of whole tissue is occurring wherein actual human blood vessels are growing from the grafted human skin into the human tumor tissue on the surface of the grafted human skin.
- the origin of the neovascularization into the human graft can be demonstrated by immunohistochemical staining of the neovasculature with human-specific endothelial cell markers.
- the chimeric mouse assay demonstrates regression of neovascularization based on both the amount and extent of regression of new vessel growth. Furthermore, it is easy to monitor effects on the growth of any tissue transplanted upon the grafted skin, such as a tumor tissue. Finally, the assay is useful because there is an internal control for toxicity in the assay system. The chimeric mouse is exposed to any test reagent, and therefore the health of the mouse is an indication of toxicity. Other animal models described herein and known in the art can also be utilized in the methods described herein.
- kits for preventing or treating one or more diseases or disorders associated with angiogenesis/neovascularization, excessive vascularization, or small vessel dilation comprising administering a composition comprising a humanized antibody or antigen-binding fragment described herein that binds to endoglin associated with the disease or disorder and prevents angiogenesis, thereby preventing, treating, ameliorating, or lessening the disease or its severity.
- kits for preventing or treating one or more diseases or disorders associated with angiogenesis/neovascularization comprising administering a composition comprising a humanized antibody or antigen-binding fragment described herein that binds to endoglin associated with the disease or disorder, decreases angiogenesis, or prevents excessive angiogenesis.
- prevention refers to prophylaxis, prevention of onset of symptoms, prevention of progression of a disease or disorder associated with angiogenesis or correlated with endoglin activity.
- inhibition refers to, for example, stasis of symptoms, prolongation of survival, partial or full amelioration of symptoms, and partial or full eradication of a condition, disease or disorder associated with angiogenesis or correlated with endoglin activity.
- compositions can be administered to a patient in a therapeutically effective amount which are effective for producing some desired therapeutic effect by inhibiting a disease or disorder such as described herein which can be associated with endoglin, at a reasonable benefit/risk ratio applicable to any medical treatment.
- a therapeutically effective amount is an amount achieves at least partially a desired therapeutic or prophylactic effect in an organ or tissue.
- the amount of a humanized anti- endoglin antibody or antigen binding fragment thereof necessary to bring about prevention and/or therapeutic treatment of a disease or disorder is not fixed per se.
- humanized anti-endoglin antibody or antigen binding fragment thereof administered may vary with the type of disease, extensiveness of the disease, and size of the mammal suffering from the disease or disorder.
- two or more humanized anti-endoglin antibodies described herein are administered to a patient in combination. Combination includes concomitant or subsequent administration of the antibodies.
- administering is defined herein as a means providing the composition to the patient in a manner that results in the composition being inside the patient's body.
- Such an administration can be by any route including, without limitation, locally, regionally or systemically by subcutaneous, intravitreal, intradermal, intravenous, intra-arterial,
- Intraperitoneal, or intramuscular administration e.g., injection
- Concurrent administration means administration within a relatively short time period from each other; such time period can be less than 2 weeks, less than 7 days, less than 1 day and could even be administered
- compositions can be varied so as to obtain an amount of the active ingredient that is effective to achieve the desired therapeutic response for a particular patient, composition, and mode of administration, without being toxic to the patient.
- the selected dosage level will depend upon a variety of factors including the activity of the particular compound employed, the route of administration, the time of administration, the rate of excretion of the particular compound being employed, the duration of the treatment, other drugs, compounds and/or materials used in combination with the particular composition employed, the age, sex, weight, condition, general health and prior medical history of the patient being treated, and like factors well known in the medical arts.
- the antibodies and antigen- binding fragments described herein can be administered to a subject in various dosing amounts and over various time frames.
- Non-limiting doses include about 0.01 mg/kg, about 0.05 mg/kg, about 0.1 mg/kg, about 0.5 mg/kg, about 1 mg/kg, about 5 mg/kg, about 10 mg/kg, about 20 mg/kg, about 30 mg/kg, about 40 mg/kg, about 50 mg/kg, about 60 mg/kg, about 70 mg/kg, about 80 mg/kg, about 90 mg/kg, about 100 mg/kg, about 125 mg/kg, about 150 mg/kg, about 175 mg/kg, about 200 mg/kg, or any integer in between.
- the dose(s) of an antibody or antigen-binding fragment can be administered twice a week, weekly, every two weeks, every three weeks, every 4 weeks, every 6 weeks, every 8 weeks, every 12 weeks, or any combination of weeks therein.
- Dosing cycles are also contemplated such as, for example, administering antibodies or antigen-binding fragments thereof one or twice a week for 4 weeks, followed by two weeks without therapy. Additional dosing cycles including, for example, different combinations of the doses and weekly cycles described herein are also contemplated within the invention.
- Contacting is defined herein as a means of bringing a composition as provided herein in physical proximity with a cell, organ, tissue or fluid as described herein. Contacting encompasses systemic or local administration of any of the compositions provided herein and includes, without limitation, in vitro, in vivo and/or ex vivo procedures and methods.
- a response is achieved when the patient experiences partial or total alleviation, or reduction of signs or symptoms of illness, and specifically includes, without limitation, prolongation of survival.
- the expected progression-free survival times can be measured in months to years, depending on prognostic factors including the number of relapses, stage of disease, and other factors.
- Prolonging survival includes without limitation times of at least 1 month (mo), about at least 2 months (mos.), about at least 3 mos., about at least 4 mos., about at least 6 mos., about at least 1 year, about at least 2 years, about at least 3 years, or more.
- Overall survival can also be measured in months to years.
- the patient's symptoms can remain static or can decrease.
- a physician or veterinarian having ordinary skill in the art can readily determine and prescribe the effective amount (ED50) of the composition required.
- the physician or veterinarian could start doses of the compounds employed in the composition at levels lower than that required in order to achieve the desired therapeutic effect and gradually increase the dosage until the desired effect is achieved.
- a dose can remain constant.
- compositions can be administered to a patient by any convenient route such as described above. Regardless of the route of administration selected, the compounds of the present invention, which can be used in a suitable hydrated form, and/or the compositions, are formulated into acceptable dosage forms such as described below or by other conventional methods known to those of skill in the art.
- Antibodies can be combined with a therapeutic moiety or to a detectable
- (imaging) moiety using methods known in the art such as, for example, chemical conjugation, covalent or non-covalent bonds or recombinant techniques to create conjugates or fusion proteins such as described in more detail below.
- methods known in the art such as, for example, chemical conjugation, covalent or non-covalent bonds or recombinant techniques to create conjugates or fusion proteins such as described in more detail below.
- antibodies and/or other agents can be combined in separate compositions for simultaneous or sequential administration.
- Toxicity and therapeutic efficacy of such ingredient can be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e.g., for
- LD 50 the dose lethal to 50% of the population
- ED 50 the dose therapeutically effective in 50% of the population.
- the dose ratio between toxic and therapeutic effects is the therapeutic index and it can be expressed as the ratio LD 5 o/ED 5 o. While compounds that exhibit toxic side effects may be used, care should be taken to design a delivery system that targets such compounds to the site of affected tissue in order to minimize potential damage to healthy cells and, thereby, reduce side effects.
- therapeutically effective dose can be estimated initially from cell culture assays.
- a dose can be formulated in animal models to achieve a circulating plasma concentration arrange that includes the IC 50 (i.e., the concentration of the test compound which achieves a half-maximal inhibition) as determined in cell culture.
- IC 50 i.e., the concentration of the test compound which achieves a half-maximal inhibition
- Levels in plasma can be measured, for example, by high performance liquid chromatography. Such information can be used to more accurately determine useful doses in humans.
- endoglin The pro-angiogenic role of endoglin has been established in many models including endothelial cell culture and knock-out mouse models. Endothelial and the associated cells are well known to express endoglin (CD 105), and the role of endoglin in angiogenesis, generally, as well as cardiac development has also been confirmed in numerous studies, culture models, and animal models. (Duff et al, FASEB J., 17:984-992 (2003); Bernabeu et al, J. Cell. Biochem., 102(6): 1375-1388 (2007); US Patent No. 7,097,836).
- methods which inhibit angiogenesis in a diseased tissue ameliorate symptoms of the disease and, depending upon the disease, can contribute to cure of the disease.
- the invention contemplates inhibition of angiogenesis in a tissue.
- the extent of angiogenesis in a tissue, and therefore the extent of inhibition achieved by the present methods, can be evaluated by a variety of methods, such as are described herein.
- the unique specificity of the antibodies which recognize (e.g., bind) an epitope on endoglin and inhibits angiogenesis provides diagnostic and therapeutic uses for diseases characterized by angiogenesis (neovascularization), small vessel dilation, and/or excessive vascularization such as described herein.
- Humanized anti-endoglin antibodies and fragments thereof can be administered to a subject such as a mammal (e.g., a human), suffering from a medical disorder, e.g., various forms of ocular diseases characterized by angiogenesis/ neovascularization (e.g., macular degeneration, diabetic retinopathy), diabetic nephropathy, chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, and various forms of cancer (primary tumors and metastases).
- a mammal e.g., a human
- a medical disorder e.g., various forms of ocular diseases characterized by angiogenesis/ neovascularization (e.g., macular degeneration, diabetic retinopathy), diabetic nephropathy, chronic inflammatory diseases (e.g., IBD), rheumatoid arthritis, osteoarthritis, and various forms of cancer (primary tumors and metasta
- a method for treating a subject having an ocular disease characterized by angiogenesis by administering a humanized antibody or fragment thereof described herein that binds endoglin and inhibits angiogenesis.
- a method for treating a subject having a chronic inflammatory disease by administering a humanized antibody or fragment thereof described herein that binds endoglin and inhibits angiogenesis. Examples of such chronic inflammatory diseases include, but are not limited to, Crohn's disease and ulcerative colitis.
- a method for treating a subject having diabetic nephropathy by administering a humanized antibody or fragment thereof described herein.
- a method for treating a subject having rheumatoid arthritis or osteoarthritis by administering a humanized antibody or fragment thereof described herein.
- anti-endoglin antibodies can be effective for treating angiogenesis, it is contemplated herein that a subject can also be treated with one or more additional angiogenesis inhibitors.
- angiogenesis inhibitor is used herein, for purposes of the specification and claims, to mean a compound or molecule including, but not limited to, peptides, proteins, enzymes, polysaccharides, oligonucleotides, DNA, RNA, recombinant vectors, and drugs which function to inhibit angiogenesis.
- Angiogenesis inhibitors are known in the art and all types are contemplated herein.
- Non-limiting examples of compounds and molecules include natural and synthetic biomolecules such as paclitaxel, O-(chloroacetyl-carbomyl) fumagillol ("TNP-470” or “AGM 1470”), thrombospondin-1, thrombospondin-2, angiostatin, human chondrocyte-derived inhibitor of angiogenesis (“hCHIAMP”), cartilage-derived angiogenic inhibitor, platelet factor-4, gro-beta, human interferon-inducible protein 10 ("IP 10"), interleukin 12, Ro 318220, tricyclodecan-9-yl xanthate (“D609”), irsogladine, 8,9-dihydroxy-7-methyl-benzo[b]
- TNP-470 O-(chloroacetyl-carbomyl) fumagillol
- thrombospondin-1 thrombospondin-2
- angiostatin human chondrocyte-derived inhibitor of angiogenesis
- quinolizinium bromide (“GPA 1734"), medroxyprogesterone, a combination of heparin and cortisone, glucosidase inhibitors, genistein, thalidomide, diamino-antraquinone, herbimycin, ursolic acid, and oleanolic acid.
- Non-limiting examples of antibodies include those directed towards molecules such as VEGF, VEGF receptor, or different epitopes of endoglin.
- VEGF receptor inhibitors include bevacizumab (AVASTIN®), ranibizumab (Lucentis), aflibercept (VEGF-Trap), sunitinib (Sutent), sorafenib (Nexavar), axitinib, pegaptanib and pazopanib.
- the VEGF receptor inhibitor is bevacizumab.
- Exemplary dosage for bevacizumab are about 7.5, about 10, or about 15 mg/kg, administered every 2 or 3 weeks.
- the VEGF receptor inhibitor is ranibizumab.
- Exemplary ocular dosages for ranibizumab include about 0.5 mg, administered intravitreally monthly.
- the VEGF receptor inhibitor is aflibercept (VEGF- Trap),.
- Exemplary dosages for VEGF -Trap include about 0.5 - about 10 mg/kg administered every 2 or 3 weeks.
- Exemplary ocular dosages for VEGF-Trap include about 0.5 - about 2.0 mg administered intravitreally monthly or quarterly.
- the VEGF receptor inhibitor is sunitinib.
- Exemplary regimens for sunitinib include about 50 mg administered for 4 weeks, followed by 2 weeks of no drug. Treatment regimens can be repeated in a cyclic or acyclic basis.
- the VEGF receptor inhibitor is sorafenib.
- Exemplary dosages for sorafenib include about 400 mg administered daily.
- the VEGF receptor inhibitor is axitinib.
- Exemplary dosages for axitinib include about 3, about 5, or about 10 mg administered twice daily.
- the VEGF receptor inhibitor is pegaptanib.
- Exemplary dosages for pegaptanib include about 0.3- about 3 mg administered intravitreally every 6 weeks.
- the VEGF receptor inhibitor is pazopanib.
- Exemplary dosages for pazopanib include about 200 - about 1000 mg administered daily.
- VEGF receptor inhibitors can be administered with the antibodies and antigen-binding fragments described herein. In one embodiment, combinations may result in the use of lower doses for the antibodies or antigen binding fragments described herein, the VEGF receptor inhibitors, or both. Such alterations in dosing may result from synergistic effects of the combinations of the antibodies and antigen-binding fragments described herein with the VEGF receptor inhibitors.
- the present invention provides a method for treating diabetic retinopathy, macular degeneration, choroidal neovascularization or neovascular glaucoma in a patient by administering to the patient a therapeutically effective amount of any of the compositions provided herein.
- Endoglin is a receptor associated with angiogenesis and the extracellular matrix.
- Macular degeneration is the loss of photoreceptors in the portion of the central retina, termed the macula, responsible for high-acuity vision. Degeneration of the macula is associated with abnormal deposition of extracellular matrix components and other debris in the membrane between the retinal pigment epithelium and the vascular choroid. This debris-like material is termed drusen. Drusen is observed with a funduscopic eye examination. Normal eyes may have maculas free of drusen, yet drusen may be abundant in the retinal periphery. The presence of soft drusen in the macula, in the absence of any loss of macular vision, is considered an early stage of AMD.
- Macular degeneration is characterized by choroidal neovascularization (CNV), the development of abnormal blood vessels beneath the retinal pigment epithelium (RPE) layer of the retina. These vessels break through the Bruch's membrane, disrupting the retinal pigmented epithelium, bleed, and eventually cause macular scarring which results in profound loss of central vision (disciform scarring).
- CNV choroidal neovascularization
- RPE retinal pigment epithelium
- Choroidal neovascularization commonly occurs in macular degeneration in addition to other ocular disorders and is associated with proliferation of choroidal endothelial cells, overproduction of extracellular matrix, and formation of a fibrovascular subretinal membrane. Retinal pigment epithelium cell proliferation and production of angiogenic factors appears to effect choroidal neovascularization.
- Diabetic retinopathy is an ocular disorder characterized by excessive angiogenesis that develops in diabetes due to thickening of capillary basement membranes, and lack of contact between pericytes and endothelial cells of the capillaries. Loss of pericytes increases leakage of the capillaries and leads to breakdown of the blood-retina barrier. Diabetic retinopathy is the result of microvascular retinal changes. Hyperglycemia-induced pericyte death and thickening of the basement membrane lead to incompetence of the vascular walls. These damages change the formation of the blood-retinal barrier and also make the retinal blood vessels become more permeable.
- Small blood vessels - such as those in the eye - are especially vulnerable to poor blood sugar (blood glucose) control.
- An over-accumulation of glucose and/or fructose damages the tiny blood vessels in the retina.
- Macular edema can also develop when the damaged blood vessels leak fluid and lipids onto the macula. These fluids make the macula swell, which blurs vision. This damage also results in a lack of oxygen at the retina.
- Fibrovascular proliferation can also cause tractional retinal detachment.
- the new blood vessels can also grow into the angle of the anterior chamber of the eye and cause neovascular glaucoma.
- Proliferative vitreoretinopathy is associated with cellular proliferation of cellular and fibrotic membranes within the vitreous membranes and on the surfaces of the retina. Retinal pigment epithelium cell proliferation and migration is common with this ocular disorder.
- the membranes associated with proliferative vitreoretinopathy contain extracellular matrix components such as collagen types I, II, and IV and fibronectin, and become progressively fibrotic.
- Age-related macular degeneration (AMD) and diabetic retinopathy are the two leading causes of blindness in the developed world.
- the recent approval of the macromolecules LUCENTIS®, AVASTIN®, and MACUGEN® have improved the treatment options available for AMD patients.
- LUCENTIS® is a Fab
- AVASTIN® is a monoclonal antibody. They both bind vascular endothelial growth factor (VEGF) and have demonstrated the most impressive results to date treating AMD; however, only a minority of treated patients experience a significant improvement in visual acuity.
- Anti-angiogenic therapy focused on a target other than VEGF may overcome some of the limitations associated with agents that target the VEGF pathway.
- the humanized antibodies and antigen-binding fragments which bind endoglin and are described herein can be used to treat or prevent macular degeneration, CNV, diabetic retinopathy, or proliferative vitreoretinopathy. Described herein are methods of treating or preventing macular degeneration, CNV, diabetic retinopathy, or proliferative vitreoretinopathy via the administration of the antibodies and antigen-binding fragments described herein.
- the humanized antibodies and antigen-binding fragments which bind endoglin and are described herein can also shrink blood vessels, inhibit endothelial cell proliferation associated with ocular disease, clear symptoms of bleeding, treat cloudy vision, provide stasis of vision loss, and/or prevent leakage of blood vessels.
- the humanized antibodies and antigen-binding fragments described herein can also be used in medicaments for the treatment of macular degeneration, CNV, diabetic retinopathy or proliferative vitreoretinopathy.
- humanized antibodies and antigen-binding fragments described herein can also be used in combination with known therapies and/or compounds for the treatment of macular degeneration, CNV, diabetic retinopathy or proliferative vitreoretinopathy.
- therapies and/or compounds for the treatment of macular degeneration, CNV, diabetic retinopathy or proliferative vitreoretinopathy.
- examples of such compounds include, but are not limited to, bevacizumab (AVASTIN®), ranibizumab (LUCENTIS®), aflibercept (VEGF-Trap), sunitinib (SUTENT®), sorafenib (NEXAVAR®), axitinib, pegaptanib, pazopanib or MACUGEN®.
- the humanized anti-endoglin antibodies and antigen-binding fragments can be administered via intravitreal routes.
- intravitreal modes of administration include intravitreal injection and the use of intravitreal implants.
- Patients can be assessed for improvement and responsiveness to treatment.
- Treatment includes, but is not limited to, decreasing the macular edema, decreased areas of CNV, and increased visual acuity. Measurements of symptoms are as known in the art and are further described in the examples below.
- tissue to be treated is an inflamed tissue and the angiogenesis to be inhibited is inflamed tissue angiogenesis where there is neovascularization of inflamed tissue.
- IBD inflammatory bowel disease
- Crohn's disease is typically characterized by inflammation of the small and large bowel, whereas ulcerative colitis is generally localized to the colon.
- Abnormal or pathological angiogenesis is central to both Crohn's disease and ulcerative colitis. Both diseases involve increased microvascular density and microvascular dysfunction, and this angiogenesis is temporally related with tissue pathology and the inflammatory cycles found in both diseases. Endoglin is known to be expressed in these tissues and to play a role in the dysregulation of angiogenesis during IBD. (Chidlow et al, Am. J. Physiol. Gastrointest. Liver Physiol, 293:5-18 (2007)).
- humanized antibodies and antigen-binding fragments which bind endoglin and are described herein can be used to treat IBD. Additionally, humanized antibodies and antigen-binding fragments which bind endoglin can be used for the treatment of Crohn's disease or ulcerative colitis.
- humanized anti-endoglin antibodies and antigen-binding fragments can also be used in combination with surgery and/or known therapies for IBD, Crohn's disease or ulcerative colitis. Examples of such known therapies include, but are not limited to,
- Aminosalicylates e.g., Mesalamine
- corticosteroids e.g., budesonide, prednisone, etc.
- antibiotics e.g., metronidizole, etc.
- immunosuppresive drugs e.g., azathioprine, 6-
- biologic drugs such as proteins and antibodies (e.g., infliximab, etc.).
- Treatment of IBD can be assessed by decreased vascularization of the inflamed tissue. Treatment can also be assessed by stasis, resolution, and/or healing of the ulcerative lesions which characterize IBD.
- Diabetic nephropathy is a major cause of morbidity and mortality in both type 1 and type 2 diabetics. It is the leading cause of end-stage renal disease world-wide. Diabetic nephropathy is characterized by glomerular microvascular injury due to the increased synthesis of pro-angiogenic factors. These pro-angiogenic factors cause increased endothelial cell proliferation and subsequent angiogenesis, and endoglin is known to be upregulated in chronic renal disease. This angiogenesis results in destruction of the glomeruli and finally renal failure. (Zent et al, Seminars in Nephrology, 27(2): 161-171 (2007); Roy-Chaudhury et al, Exp.
- the humanized antibodies and antigen-binding fragments which bind endoglin and are described herein can be used to treat or prevent diabetic nephropathy, renal failure following transplantation, and/or ischemic renal injury following transplantation.
- Described herein are methods of treating or preventing diabetic nephropathy, renal failure following transplantation, and/or ischemic renal injury following transplantation via the administration of the antibodies and antigen-binding fragments described herein.
- the humanized antibodies and antigen-binding fragments described herein can also be used in medicaments for the treatment of diabetic nephropathy, renal failure following transplantation, and/or ischemic renal injury following transplantation. Additionally, humanized antibodies and antigen-binding fragments described herein can also be used in combination with known therapies and/or compounds for the treatment of diabetic nephropathy, renal failure following transplantation, and/or ischemic renal injury following transplantation.
- Patients can be assessed with respect to the efficacy of treatment by, for example, improvement in renal function.
- Rheumatoid arthritis is characterized by excessive angiogenesis, and is well understood in this regard.
- the inflammation and destruction found in the synovial fluids is directly related to the increased angiogenesis found surrounding and in the synovial tissues.
- Numerous pro-angiogenic factors are present in the affected tissues of rheumatoid arthritis patients. (Koch and Distler, Arthritis Res. & Ther., 9(Suppl. 2): S3, 1-9 (2007).
- Osteoarthritis is a group of chronic disabling conditions that affects the synovial joints.
- Angiogenesis and inflammation are integral processes in the pathophysiology of the disease, and they contribute to joint damage through a variety of mechanisms, including but not limited to, stimulation of MMP production and endochondral ossification. Additionally, angiogenesis in osteoarthritis induces further innervation, which develops into a feedback loop where each continues to stimulate the other. (Bonnet & Walsh, Rheumatology, 44:7-16 (2005)).
- the humanized antibodies and antigen-binding fragments which bind endoglin and are described herein can be used to treat or prevent rheumatoid arthritis and osteoarthritis. Described herein are methods of treating or preventing rheumatoid arthritis and osteoarthritis via the administration of the antibodies and antigen-binding fragments described herein.
- the humanized antibodies and antigen-binding fragments described herein can also be used in medicaments for the treatment of rheumatoid arthritis and osteoarthritis.
- Paulus Criteria and The American College of Rheumatology Criteria (ACR). Paulus Criteria is defined as improvement in 4 of the following: tender and swollen joint counts, morning stiffness, patient assessment of disease activity, physician assessment of disease activity and erythrocyte sedimentation rate (ESR) rage. The level of improvement is set as a percentage improvement of each of these variables i.e. a Paulus 20 classification indicates a responder who has shown 20% improvement in 4 of the 6 parameters.
- Rheumatoid arthritis can also be assessed using American College of
- ACR Rheumatology Scoring. Briefly, ACR Classification Criteria for Determining Clinical Remission in Rheumatoid Arthritis is assessed by the presence of 5 or more of the following factors present at least two consecutive months:
- Morning stiffness ⁇ 15 minutes
- Exclusions may occur and include: clinical manifestations of active vasculitis, pericarditis, pleuritis or myositis, and unexplained recent weight loss or fever attributable to rheumatoid arthritis will prohibit a designation of complete clinical remission (Pinals RS, et.al.: Arthritis Rheum 24: 1308, 1981). Additionally, ACR Classification Criteria of Functional Status in Rheumatoid Arthritis includes classification based on the following patient abilities:
- Class I Completely able to perform usual activities of daily living (self-care, vocational, and recreational);
- Class II Able to perform usual self-care and vocational activities, but limited m benzional activities;
- Class ill Able to perform usual self-care activities, but limited in vocational and cruising activities;
- Class IV Limited ability to perform usual self-care, vocational, and cruising activities.
- Osteoarthritis can also be assessed using ACR scoring.
- Classification Criteria for Osteoarthritis of the hip is assessed utilizing patient history, physical examination and laboratory findings: a patient is assessed for pain in the hip and one of the following:
- ACR Clinical Classification Criteria for Osteoarthritis of the knee is assessed using history and physical examination utilizing the following criteria: pain in the knee in connection with three (3) of the following:
- pain in the knee can be assessed in connection with one of the following patient characteristics: (1) a patient is over 50 years of age; (2) less than 30 minutes of morning stiffness; and (3) Crepitus on active motion and osteophytes.
- patient characteristics (1) a patient is over 50 years of age; (2) less than 30 minutes of morning stiffness; and (3) Crepitus on active motion and osteophytes.
- history physical examination and laboratory findings: pain in the knee can be assessed inn connection with five (5) of the following characteristics:
- ESF is less than 40 mm/hour
- ACR Clinical Classification Criteria for Osteoarthritis of the hand can be assessed as follows: pain, aching or stiffness in the hand in connection with three (3) of the following: (1) hard tissue enlargement of two or more of the following joints (second and third distal interphalangeal, the second and third proximal interphalangeal, and the first carpometacarpal joints of both hands; (2) hard tissue enlargement of two or more distal interphalangeal joints; (3) less than three swollen MCP joints and (4) deformity of at least one of the joints listed above in (1).
- CD 105 is associated with tumor angiogenesis and is strongly up-regulated in the endothelium of various tumor tissues compared with that in normal tissues.
- CD 105 is up- regulated in a wide range of tumor endothelia including, for example, colon, breast, brain, lung, prostate, endometrial, kidney, liver, gastric, head & neck and cervical cancer. Additionally, it is known that there is stronger expression of CD 105 in tumor endothelium than corresponding normal tissues. (Duff et al, FASEB J., 17:984-992 (2003); Bernabeu et al, J. Cell. Biochem., 102(6): 1375-1388 (2007); US Patent No. 7,097,836).
- the inhibition of angiogenesis with anti-endoglin humanized antibodies represents a treatment option for cancerous tumors.
- the humanized antibodies and antigen-binding fragments which bind endoglin and are described herein can be used to treat cancerous tumors.
- the humanized antibodies and antigen-binding fragments described herein can also be used in medicaments for the treatment cancerous tumors.
- tumor is used herein to refer to a cancerous tissue expressing endoglin
- Tumors can include solid tumors and semi-solid tumors.
- Non- limiting examples of tumors include human leukemias, including non-T-cell-type (non-T) acute lymphoblastic leukemia (ALL), myelo-monocytic leukemia; and human solid and semi-solid tumors, with its surrounding vasculature expressing endoglin at moderate to high levels (as compared to expression by normal tissue of the same type) including angiosarcoma, breast carcinoma, stomach cancer, colon carcinoma, Hodgkins lymphoma, lymphoma, glioblastoma multiforme (GBM), lung carcinoma, melanoma, myeloma, lymphoma, osteosarcoma, ovarian carcinoma, parotid tumor, pharyngeal carcinoma, prostate carcinoma, hepatocellular carcinoma, renal carcinoma, and rectosigmoid carcinoma.
- ALL non-T-cell-type
- ALL acute lymphoblastic leukemia
- a cancerous tissue to be treated is, for example, an endothelial tissue expressing an abnormal level of endoglin.
- the tumor tissue In the absence of neovascularization of tumor tissue, the tumor tissue does not obtain the required nutrients, slows in growth, ceases additional growth, regresses and ultimately becomes necrotic resulting in killing of the tumor.
- the present invention provides for a method of inhibiting tumor neovascularization by inhibiting tumor angiogenesis according to the present methods. Similarly, the invention provides a method of inhibiting tumor growth by practicing the angiogenesis-inhibiting methods.
- the methods are also particularly effective against the formation of metastases because their formation requires vascularization of a primary tumor so that the metastatic cancer cells can exit the primary tumor and their establishment in a secondary site requires
- a "patient suffering from a cancer/metastasis" of the invention may express a mutant protein (tumor associated antigen) or a mutant gene and not yet be symptomatic for the disease.
- the cancer is colon cancer (which is associated with the mutant K-ras protein)
- a patient with a mutant K-ras protein in some cells of the colon is a patient according to the invention even though that patient may not yet be symptomatic for colon cancer.
- “Signs or symptoms of illness” are clinically recognized manifestations or indications of disease.
- treating a patient suffering from cancer it is meant that the patient's symptoms are partially or totally alleviated, or remain static following treatment according to the invention.
- a patient that has been treated can exhibit a partial or total alleviation of symptoms and/or tumor load. This is intended to encompass prophylaxis, therapy and cure.
- a patient suffering from a highly metastatic cancer e.g., breast cancer
- additional metastasis either do not occur, or are reduced in number as compared to a patient who does not receive treatment.
- a patient is treated where the patient's solid cancer either becomes reduced in size or does not increase in size as compared to a patient who does not receive treatment.
- the number of cancer cells in a treated patient either does not increase or is reduced as compared to the number of cancer cells in a patient who does not receive treatment.
- Improvement can also be defined, for example, as decreased cell proliferation, decreased numbers of cells, increased apoptosis, and/or increased survival of the patient being treated.
- treatment of cancer includes stasis, partial or total elimination of a cancerous growth or tumor.
- Treatment or partial elimination includes, for example, a fold reduction in growth or tumor size and/or volume such as about 2-fold, about 3- fold, about 4-fold, about 5-fold, about 10-fold, about 20-fold, about 50-fold, or any fold reduction in between.
- treatment or partial elimination can include a percent reduction in growth or tumor size and/or volume of about 1%, 2%, 3%, 4%, 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% or any percentage reduction in between.
- a tumor or cancer to be treated in the methods described herein includes, but is not limited to, a lung cancer, a gynecologic malignancy, a melanoma, a breast cancer, a brain cancer (e.g., glioblastoma multiforme, "GBM") a pancreatic cancer, an ovarian cancer, a uterine cancer, a colorectal cancer, a prostate cancer, a kidney cancer, a head cancer, a liver cancer (hepatocellular cancer), a uterine cancer, a neck cancer, a kidney cancer (renal cell cancer), a sarcoma, a myeloma, and lymphoma.
- a lung cancer e.g., glioblastoma multiforme, "GBM”
- GBM glioblastoma multiforme, a pancreatic cancer
- an ovarian cancer e.g., glioblastoma multiforme, "GBM”
- a tumor to be treated is a solid or semi-solid tumor.
- a tumor to be treated is a primary tumor.
- a tumor to be treated is a metastatic tumor.
- a tumor or cancer to be treated is of epithelial origin.
- the cancer to be treated is myeloma.
- the cancer to be treated is ovarian cancer.
- the cancer to be treated is kidney/renal cancer.
- the cancer to be treated is hepatocellular/liver cancer.
- NSCLC non-small cell lung cancer
- squamous cell carcinomas adenocarcinomas
- large cell undifferentiated carcinomas Small cell lung cancer accounts for 15-20% of lung cancers.
- Lung cancer staging is an assessment of the degree of spread of the cancer from its original source. It is an important factor affecting the prognosis and potential treatment of lung cancer.
- Non-small cell lung carcinoma is staged from IA ("one A”; best prognosis) to IV ("four"; worst prognosis).
- Small cell lung carcinoma is classified as limited stage if it is confined to one half of the chest and within the scope of a single radiotherapy field; otherwise, it is extensive stage.
- Non-small cell lung cancer may be staged using EUS (endoscopic ultrasound) or
- TX The primary tumor cannot be assessed, or there are malignant cells in the sputum or bronchoalveolar lavage but not seen on imaging or
- Tl Tumor less than 3 cm in its greatest dimension, surrounded by lung or visceral pleura and without bronchoscopic invasion into the main bronchus.
- T2 A tumor with any of: more than 3 cm in greatest dimension; extending into the main bronchus (but more than 2 cm distal to the carina), and obstructive pneumonitis (but not involving the entire lung).
- T3 A tumor with any of: invasion of the chest wall, diaphragm, mediastinal pleura, or parietal pericardium; extending into the main bronchus, within 2 cm of the carina, but not involving the carina; and obstructive pneumonitis of the entire lung.
- T4 A tumor with any of: invasion of the mediastinum, heart, great vessels, trachea, esophagus, vertebra, or carina; separate tumor nodules in the same lobe; and malignant pleural effusion.
- Lymph nodes NX: Lymph nodes cannot be assessed; NO: No lymph nodes involved; Nl : Metastasis to ipsilateral peribronchial or ipsilateral hilar lymph nodes; N2: Metastasis to ipsilateral mediastinal or subcarinal lymph nodes; and N3 : Metastasis to any of: ipsilateral supraclavicular lymph nodes; ipsilateral scalene lymph nodes; and contralateral lymph nodes.
- Distant metastasis MX: Distant metastasis cannot be assessed; MO: No distant metastasis; and Ml : Distant metastasis is present.
- Uterine cancers may refer to any of several different types of cancer which occur in the uterus, namely: uterine sarcomas (e.g., sarcomas of the myometrium, or muscular layer of the uterus, are most commonly leiomyosarcomas); endometrial cancer; and cervical cancer.
- uterine sarcomas e.g., sarcomas of the myometrium, or muscular layer of the uterus, are most commonly leiomyosarcomas
- endometrial cancer e.g., endometrial cancer
- cervical cancer e.g., uterine sarcomas of the myometrium, or muscular layer of the uterus, are most commonly leiomyosarcomas
- cervical cancer e.g., sarcomas of the myometrium, or muscular layer of the uterus, are most commonly leiomyosarcomas
- cervical cancer e.g
- Endometrial cancer is a cancer that starts in the endometrium, the inner lining of the uterus.
- Some of the examples of the cancer of uterus and endometrium include, but are not limited to, adenocarcinomas, adenoacanthomas, adenosquamous carcinomas, papillary serous
- adenocarcinomas clear cell adenocarcinomas, uterine sarcomas, stromal sarcomas, malignant mixed mesodermal tumors, and leiomyosarcomas.
- the method treats cervical cancer, preferably an
- adenocarcinoma in the cervix epithelial Two main types of this cancer exist: squamous cell carcinoma and adenocarcinomas.
- the former constitutes about 80-90% of all cervical cancers and develops where the ectocervix (portion closest to the vagina) and the endocervix (portion closest to the uterus) join. The latter develop in the mucous-producing gland cells of the endocervix.
- Some cervical cancers have characteristics of both of these and are called adenosquamous carcinomas or mixed carcinomas.
- ovarian cancer including epithelial ovarian tumors.
- Ovarian cancer is classified according to the histology of the tumor, obtained in a pathology report.
- Surface epithelial-stromal tumor also known as ovarian epithelial carcinoma, is the most typical type of ovarian cancer. It includes serous tumor, endometrioid tumor and mucinous cystadenocarcinoma.
- Sex cord-stromal tumor including estrogen-producing granulosa cell tumor and virilizing Sertoli-Leydig cell tumor or arrhenoblastoma, accounts for 8% of ovarian cancers.
- Germ cell tumor accounts for approximately 30% of ovarian tumors but only 5% of ovarian cancers because most germ cell tumors are teratomas and most teratomas are benign.
- Germ cell tumor tends to occur in young women and girls. The prognosis depends on the specific histology of germ cell tumor, but overall is favorable. Mixed tumors contain elements of more than one of the above classes of tumor histology.
- Ovarian cancer can also be a secondary cancer, the result of metastasis from a primary cancer elsewhere in the body. Common primary cancers are breast cancer and gastrointestinal cancer (in which case the ovarian cancer is a Krukenberg cancer).
- Surface epithelial-stromal tumor can originate in the peritoneum (the lining of the abdominal cavity), in which case the ovarian cancer is secondary to primary peritoneal cancer, but treatment is basically the same as for primary surface epithelial-stromal tumor involving the peritoneum.
- Ovarian cancer staging is by the FIGO staging system and uses information obtained after surgery, which can include a total abdominal hysterectomy, removal of both ovaries and fallopian tubes, the omentum, and pelvic (peritoneal) washings for cytology.
- the AJCC stage is the same as the FIGO stage.
- Stage I refers to ovarian cancer limited to one or both ovaries: IA - involves one ovary; capsule intact; no tumor on ovarian surface; no malignant cells in ascites or peritoneal washings; IB - involves both ovaries; capsule intact; no tumor on ovarian surface; negative washings; and IC - tumor limited to ovaries with any of the following: capsule ruptured, tumor on ovarian surface, positive washings.
- Stage II refers to pelvic extension or implants: IIA - extension or implants onto uterus or fallopian tube; negative washings; IIB - extension or implants onto other pelvic structures; negative washings; and IIC - pelvic extension or implants with positive peritoneal washings
- Stage III refers to microscopic peritoneal implants outside of the pelvis; or limited to the pelvis with extension to the small bowel or omentum: IIIA - microscopic peritoneal metastases beyond pelvis; IIIB - macroscopic peritoneal metastases beyond pelvis less than 2 cm in size; and IIIC - peritoneal metastases beyond pelvis > 2 cm or lymph node metastases
- Stage IV refers to distant metastases to the liver or outside the peritoneal cavity.
- the methods described herein treat an ovarian cancer selected from the following: an adenocarcinoma in the ovary and an adenocarcinoma that has migrated from the ovary into the abdominal cavity.
- a melanoma is a malignant tumor of melanocytes which are found predominantly in skin but also in the bowel and the eye (uveal melanoma). It is one of the rarer types of skin cancer but causes the majority of skin cancer related deaths. Malignant melanoma is a serious type of skin cancer caused by uncontrolled growth of pigment cells, called melanocytes. Melanomas also include, but are not limited to, a choroidea melanoma, malignant melanomas, cutaneous melanomas and intraocular melanomas.
- Melanoma may be divided into the following types: Lentigo maligna, Lentigo maligna melanoma, superficially spreading melanoma, acral lentiginous melanoma, mucosal melanoma, nodular melanoma, polypoid melanoma, desmoplastic melanoma, amelanotic melanoma, soft-tissue melanoma, and uveal melanoma.
- Melanoma stages are as follows:
- Stage I/II - invasive melanoma Tla: less than 1.00 mm primary, without ulceration, Clark Level II-III; Tib: less than 1.00 mm primary, with ulceration or Clark Level IV-V; and T2a: 1.00-2.00 mm primary, without ulceration.
- T3a 2.00-4.00 mm primary, without ulceration
- T3b 2.00-4.00 mm primary, with ulceration
- T4a 4.00 mm or greater primary without ulceration
- T4b 4.00 mm or greater primary with ulceration.
- Nl single positive lymph node
- N2 2-3 positive lymph nodes or regional skin/in-transit metastasis
- N3 4 positive lymph nodes or lymph node and regional skin/in transit metastases.
- Lung Metastasis, Normal LDH; and Mlc Other Distant Metastasis OR Any Distant Metastasis with Elevated LDH.
- the methods described herein treat a melanoma.
- Colorectal cancer also called colon cancer or large bowel cancer
- colon cancer includes cancerous growths in the colon, rectum (anus) and appendix. With 655,000 deaths worldwide per year, it is the third most common form of cancer and the second leading cause of cancer-related death in the Western world. Many colorectal cancers are thought to arise from adenomatous polyps in the colon. These mushroom-like growths are usually benign, but some may develop into cancer over time.
- Dukes classification may be used to classify colorectal cancer based on stages A-D.
- Stage A refers to colorectal cancer that is limited to mucosa (i.e., has not invaded through the bowel wall).
- Stage Bl refers to extending into muscularis intestinal, but not penetrating through it (i.e., lymph nodes have not been invaded); whereas Stage B2 cancer has penetrated through the muscularis propria, but not penetrating through it (i.e., lymph nodes have not been invaded).
- Stage CI refers to cancer that extends into the muscularis propria, but not penetrating through it (i.e., lymph nodes are involved); whereas Stage C2 refers to cancer that extends into the muscularis propria and penetrating through it (i.e., lymph nodes are involved).
- Stage D refers to distant metastatic spread.
- the TNM system may also be used to stage colorectal cancer according to conventional means known in the art.
- a lobular carcinoma in situ and a ductal carcinoma in situ are breast cancers that have developed in the lobules and ducts, respectively, but have not spread to the fatty tissue surrounding the breast or to other areas of the body.
- Infiltrating (or invasive) lobular and ductal carcinoma are cancers that have developed in the lobules and ducts, respectively, and have spread to either the breast's fatty tissue and/or other parts of the body.
- a method of treating breast cancer such as a ductal carcinoma in duct tissue in a mammary gland, a breast cancer that is Her2- and/or ER- and/or PR-.
- breast cancer such as a ductal carcinoma in duct tissue in a mammary gland
- Other cancers of the breast that would benefit from treatment by the methods are medullary carcinomas, colloid carcinomas, tubular carcinomas, and inflammatory breast cancer.
- breast cancer is staged according to the TNM system.
- Prognosis is closely linked to results of staging, and staging is also used to allocate patients to treatments both in clinical trials and clinical practice.
- TX Primary tumor cannot be assessed. TO: No evidence of tumor. Tis: Carcinoma in situ, no invasion; Tl : Tumor is 2 cm or less; T2: Tumor is more than 2 cm but not more than 5 cm; T3: Tumor is more than 5 cm; T4: Tumor of any size growing into the chest wall or skin, or inflammatory breast cancer. NX: Nearby lymph nodes cannot be assessed NO: cancer has not spread to regional lymph nodes.
- Nl cancer has spread to 1 to 3 maxillary or one internal mammary lymph node
- N2 cancer has spread to 4 to 9 maxillary lymph nodes or multiple internal mammary lymph nodes
- N3 One of the following applies: cancer has spread to 10 or more maxillary lymph nodes, or cancer has spread to the lymph nodes under the clavicle (collar bone), or cancer has spread to the lymph nodes above the clavicle, or cancer involves maxillary lymph nodes and has enlarged the internal mammary lymph nodes, or cancer involves 4 or more maxillary lymph nodes, and tiny amounts of cancer are found in internal mammary lymph nodes on sentinel lymph node biopsy.
- MX presence of distant spread (metastasis) cannot be assessed.
- M0 no distant spread.
- Ml spread to distant organs (not including the supraclavicular lymph node) has occurred.
- pancreatic cancer selected from the following: an epithelial carcinoma in the pancreatic duct tissue and an adenocarcinoma in a pancreatic duct.
- pancreatic cancer selected from the following: an epithelial carcinoma in the pancreatic duct tissue and an adenocarcinoma in a pancreatic duct.
- the most common type of pancreatic cancer is an adenocarcinoma, which occurs in the lining of the pancreatic duct.
- the methods described herein treat a pancreatic cancer.
- a method to treat prostate cancer selected from the following: an adenocarcinoma or an adenocarcinoma that has migrated to the bone.
- Prostate cancer develops in the prostate organ in men, which surrounds the first part of the urethra.
- the prostate has several cell types but 99% of tumors are adenocarcinomas that develop in the glandular cells responsible for generating seminal fluid.
- TNM system which evaluates the size of the tumor, the extent of involved lymph nodes, and any metastasis (distant spread). As with many other cancers, these are often grouped into four stages (I-IV).
- Whitmore-Jewett stage is another scheme, used less commonly, used.
- Stage I disease is cancer that is found incidentally in a small part of the sample when prostate tissue was removed for other reasons, such as benign prostatic
- Stage II more of the prostate is involved and a lump can be felt within the gland.
- Stage III the tumor has spread through the prostatic capsule and the lump can be felt on the surface of the gland.
- Stage IV disease the tumor has invaded nearby structures, or has spread to lymph nodes or other organs. Grading is based on cellular content and tissue architecture from biopsies (Gleason) which provides an estimate of the destructive potential and ultimate prognosis of the disease.
- the methods described herein treat a prostate cancer.
- Head and neck cancers refer to a group of biologically similar cancers originating from the upper aerodigestive tract, including the lip, oral cavity (mouth), nasal cavity, paranasal sinuses, pharynx, and larynx. Most head and neck cancers are squamous cell carcinomas, originating from the mucosal lining (epithelium) of these regions. Head and neck cancers often spread to the lymph nodes of the neck, and this is often the first (and sometimes only) manifestation of the disease at the time of diagnosis.
- Head and neck cancer is strongly associated with certain environmental and lifestyle risk factors, including tobacco smoking, alcohol consumption, and certain strains of the sexually transmitted human papillomavirus. Management of patients with head and neck cancers remains a daunting task. Cancers such as, hypopharyngeal cancer, laryngeal cancer, nasopharyngeal cancer, oropharyngeal cancer, may be treated using the compounds described herein.
- the methods described herein treat a head or neck cancer.
- Kidney cancer also called renal cell cancer, renal cell carcinoma, renal adenocarcinoma, and
- hypernephroma is a disease in which malignant cells are found in the lining of tubules in the kidney. Renal cell carcinoma is the most common form of kidney cancer arising from the proximal renal tubule. It is the most common type of kidney cancer in adults, responsible for approximately 80% of cases.
- the methods described herein treat a kidney cancer.
- liver cancer that begins in the liver.
- Primary liver cancer can occur in both adults and children.
- Liver cancer is characterized by the presence of malignant hepatic tumors - tumors or growths on or in the liver. They may be discovered on medical imaging (even for a different reason than the cancer itself), or may be present in patients as an abdominal mass, abdominal pain, jaundice, or some other liver dysfunction. There are several types of liver cancer.
- Hemangiomas are the most common type of benign liver tumor. They start in blood vessels. Most of these tumors do not cause symptoms, they do not need treatment. Some may bleed and need to be removed if it is mild to severe.
- Hepatic adenomas These benign epithelial liver tumors develop in the liver. They are, in most cases, located in the right hepatic lobe and are frequently seen as solitary. The size of adenomas range from 1 to 30 cm. Symptoms associated with hepatic adenomas are all associated with large lesions which can cause intense abdominal pain.
- Focal nodular hyperplasia is the second most common tumor of the liver. This tumor is the result of a congenital arteriovenous malformation hepatocyte response. This process is one in which all normal constituents of the liver are present, but the pattern by which they are presented is abnormal. Even though those conditions exist the liver still seems to perform in the normal range.
- Hepatocellular cancer is the most common cancer of the liver. It is associated with alcohol abuse and hepatitis B infection and is particularly prevalent in Asia. The majority of HCC is detected at a time when cure by surgical resection is not possible; systemic treatment of un-resectable HCC is associated with survival of less than one year.
- the methods described herein treat a liver cancer.
- Lymphoma is a type of cancer that originates in lymphocytes of the immune system. They often originate in lymph nodes, presenting as an enlargement of the node (a tumor). Lymphomas are closely related to lymphoid leukemias, which also originate in lymphocytes but typically involve only circulating blood and the bone marrow (where blood cells are generated in a process termed haematopoesis) and do not usually form tumors. There are many types of lymphomas, and in turn, lymphomas are a part of the broad group of diseases called hematological neoplasms. Some forms of lymphoma are indolent (e.g. small lymphocytic lymphoma), compatible with a long life even without treatment, whereas other forms are aggressive (e.g. Burkitt's lymphoma), causing rapid deterioration and death.
- indolent e.g. small lymphocytic lymphoma
- other forms are aggressive (e.g. Burkitt
- lymphomas by cell type (i.e., the normal cell type that most resembles the tumor) and defining phenotypic, molecular or cytogenetic characteristics. There are three large groups: the B cell, T cell, and natural killer cell tumors. Other less common groups are also recognized.
- Hodgkin's lymphoma although considered separately within the WHO (and preceding) classifications, is now recognized as being a tumor of, albeit markedly abnormal, lymphocytes of mature B cell lineage.
- the methods described herein treat a lymphoma.
- a sarcoma is a cancer of the connective tissue (bone, cartilage, fat) resulting in mesoderm proliferation.
- carcinomas which are of epithelial origin (breast, colon, pancreas, and others).
- soft tissue sarcoma is used to describe tumors of soft tissue, which includes elements that are in connective tissue, but not derived from it (such as muscles and blood vessels).
- Sarcomas are given a number of different names, based on the type of tissue from which they arise. For example, osteosarcoma arises from bone, chondrosarcoma arises from cartilage, and leiomyosarcoma arises from smooth muscle.
- chondrosarcoma and gastrointestinal stromal tumor (GIST) are more common in adults than in children.
- Most high grade bone sarcomas including Ewing's sarcoma and osteosarcoma, are much more common in children and young adults.
- the methods described herein treat a sarcoma.
- a carcinoma is any malignant cancer that arises from epithelial cells. Carcinomas invade surrounding tissues and organs and may metastasize, or spread, to lymph nodes and other sites.
- Carcinoma like all neoplasia, is classified by its histopathological appearance.
- Adenocarcinoma and squamous cell carcinoma two common descriptive terms for tumors, reflect the fact that these cells may have glandular or squamous cell appearances respectively. Severely anaplastic tumors might be so undifferentiated that they do not have a distinct histological appearance (undifferentiated carcinoma).
- a tumor is referred to by the presumptive organ of the primary (e.g., carcinoma of the prostate) or the putative cell of origin (hepatocellular carcinoma, renal cell carcinoma).
- the presumptive organ of the primary e.g., carcinoma of the prostate
- the putative cell of origin hepatocellular carcinoma, renal cell carcinoma
- Adenocarcinoma is a malignant tumor originating in the epithelial cells of glandular tissue and forming glandular structures. This is common in the lung (forming 30-40% of all lung carcinomas). It is found peripherally, arising from goblet cells or type II pneumocytes.
- Small cell carcinoma is almost certainly due to smoking. These metastasize early, and may secrete ADH (lowering patient sodium concentration).
- ADH lowering patient sodium concentration.
- Large cell undifferentiated carcinomas account for 10-15 percent of lung neoplasms. These are aggressive and difficult to recognize due to the undifferentiated nature. These are most commonly central in the lung.
- the methods described herein treat a carcinoma.
- myeloma also known as MM, myeloma, plasma cell myeloma, or as
- Kahler's disease after Otto Kahler is a cancer of plasma cells. These immune cells are formed in bone marrow, are numerous in lymphatics and produce antibodies. Myeloma is regarded as incurable, but remissions may be induced with steroids, chemotherapy, thalidomide and stem cell transplants. Myeloma is part of the broad group of diseases called hematological malignancies.
- chromosomal translocation between the immunoglobulin heavy chain gene (on the fourteenth chromosome, locus 14q32) and an oncogene (often 1 lql3, 4pl6.3, 6p21, 16q23 and 20ql 1) is frequently observed in patients with multiple myeloma.
- This mutation results in dysregulation of the oncogene which is thought to be an important initiating event in the pathogenesis of myeloma.
- the result is proliferation of a plasma cell clone and genomic instability that leads to further mutations and translocations.
- the chromosome 14 abnormality is observed in about 50% of all cases of myeloma. Deletion of (parts of) the thirteenth chromosome is also observed in about 50%) of cases.
- cytokines especially IL-6
- Angiogenesis the attraction of new blood vessels
- the methods described herein treat a myeloma.
- Stomach or gastric cancer can develop in any part of the stomach and may spread throughout the stomach and to other organs; particularly the esophagus, lungs and the liver. Stomach cancer causes about 800.000 deaths worldwide per year.
- Metastasis occurs in 80-90% of individuals with stomach cancer, with a six month survival rate of 65 % in those diagnosed in early stages and less than 15% of those diagnosed in late stages.
- Stomach cancer is often asymptomatic or causes only nonspecific symptoms in its early stages. By the time symptoms occur, the cancer has generally metastasized to other parts of the body, one of the main reasons for its poor prognosis. [00523] In one embodiment, the methods described herein treat a stomach cancer.
- Thyroid neoplasm or thyroid cancer usually refers to any of four kinds of malignant tumors of the thyroid gland: papillary, follicular, medullary or anaplastic.
- Papillary and follicular tumors are the most common. They grow slowly and may recur, but are generally not fatal in patients under 45 years of age.
- Medullary tumors have a good prognosis if restricted to the thyroid gland and a poorer prognosis if metastasis occurs.
- Anaplastic tumors are fast- growing and respond poorly to therapy.
- Thyroid cancer is usually found in a euthyroid patient, but symptoms of hyperthyroidism or hypothyroidism may be associated with a large or metastatic well- differentiated tumor. Nodules are of particular concern when they are found in those under the age of 20. The presentation of benign nodules at this age is less likely, and thus the potential for malignancy is far greater.
- Thyroid cancers can be classified according to their pathological characteristics. The following variants can be distinguished (distribution over various subtypes may show regional variation): papillary thyroid cancer (up to 75%); follicular thyroid cancer (up to 15%); medullary thyroid cancer (up to 8%); and anaplastic thyroid cancer (less than 5%). The follicular and papillary types together can be classified as "differentiated thyroid cancer". These types have a more favorable prognosis than the medullary and undifferentiated types. Thyroid adenoma is a benign neoplasm of the thyroid.
- the methods described herein treat a thyroid cancer.
- Bladder cancer refers to any of several types of malignant growths of the urinary bladder. It is a disease in which abnormal cells multiply without control in the bladder.
- the bladder is a hollow, muscular organ that stores urine; it is located in the pelvis.
- the most common type of bladder cancer begins in cells lining the inside of the bladder and is called transitional cell carcinoma (sometimes urothelial cell carcinoma).
- bladder cancers 90% are transitional cell carcinoma. The other 10% are squamous cell carcinoma, adenocarcinoma, sarcoma, small cell carcinoma and secondary deposits from cancers elsewhere in the body.
- Stage 0 Cancer cells are found only on the inner lining of the bladder.
- Stage I Cancer cells have proliferated to the layer beyond the inner lining of the urinary bladder but not to the muscles of the urinary bladder.
- Stage II Cancer cells have proliferated to the muscles in the bladder wall but not to the fatty tissue that surrounds the urinary bladder.
- Stage III Cancer cells have proliferated to the fatty tissue surrounding the urinary bladder and to the prostate gland, vagina, or uterus, but not to the lymph nodes or other organs.
- Stage IV Cancer cells have proliferated to the lymph nodes, pelvic or abdominal wall, and/or other organs.
- Recurrent Cancer has recurred in the urinary bladder or in another nearby organ after having been treated.
- Bladder TCC is staged according to the 1997 TNM system: Ta Non-invasive papillary tumor; Tl Invasive but not as far as the muscular bladder layer; T2 Invasive into the muscular layer; T3 Invasive beyond the muscle into the fat outside the bladder; and T4 Invasive into surrounding structures like the prostate, uterus or pelvic wall.
- the methods described herein treat a bladder cancer.
- the humanized endoglin antibodies or fragments thereof can be administered alone or in combination with active or inactive agents.
- the invention contemplates simultaneous or sequential administration of the humanized endoglin antibodies or antigen-binding fragments and the active or inactive agents.
- Compounds of the present invention can be, as needed, administered in combination with one or more therapeutic treatments including, but not limited to, adriamycin, cyclophosphamide, paclitaxel, pemetrexed, temozolomide, oxaliplatin, bevacizumab, erbitux, vectibix, sorafenib, sunitinib, gefitinib, erlotinib, 5-fluorouracil (5-FU) irinotecan, topotecan, leucovorin, VELCADE®, lenalidomide, thalidomide, xeloda, taxotere and many other conventional cancer therapies described herein.
- therapeutic treatments including, but not limited to, adriamycin, cyclophosphamide, paclitaxel, pemetrexed, temozolomide, oxaliplatin, bevacizumab, erbitux, vect
- radiation refers to, for example, microwaves, ultraviolet (UV), infrared (IR), or alpha-, beta- or gamma-radiation. Radiation can be “focused” or locally delivered using conventional techniques to target radiation to the site of one or more tumors without radiating the entire body.
- the cancer is ovarian cancer and the one or more therapeutic treatments is surgery, chemotherapy (e.g., doxorubicin, doxil, gemcitabine, Rubitecan, and platinum-based chemotherapeutics such as cisplatin, carboplatin and oxaliplatin), melphalan, paclitaxel, topoisomerase I inhibitors such as topotecan and irinotecan, taxane-based therapy, hormones, radiation therapy, whole body hypothermia, isoflavone derivatives such as
- cytotoxic macrolides such as Epothilones
- angiogenesis inhibitors such as bevacizumab
- signal transduction inhibitors such as trastuzumab
- gene therapy R Ai therapy
- immunotherapy monoclonal antibodies
- phosphatidylinositol-like kinase inhibitors such as rapamycin, or any combination thereof.
- the combination is a humanized anti- endoglin antibody or antigen-binding fragment thereof and doxil.
- the combination is a humanized anti-endoglin antibody or antigen-binding fragment thereof and topotecan.
- the combination is a humanized anti-endoglin antibody or antigen-binding fragment thereof and a VEGF receptor inhibitor.
- VEGF receptor inhibitors include bevacizumab (AVASTIN®), ranibizumab (LUCENTIS®), afiibercept (VEGF-Trap), sunitinib (SUTENT®), sorafenib (NEXAVAR®), axitinib, pegaptanib and pazopanib.
- the cancer is renal/kidney cancer and the one or more therapeutic treatments is surgery, chemotherapy, sunitinib, sorafenib, pazopanib, AVASTIN®, interferon-alpha, or IL-2.
- the combination is a humanized anti-endoglin antibody or antigen-binding fragment thereof and sorafenib.
- the combination is a humanized anti-endoglin antibody or antigen-binding fragment thereof and sunitinib.
- the combination is a humanized anti-endoglin antibody or antigen-binding fragment thereof and AVASTIN®.
- the combination is a humanized anti- endoglin antibody or antigen-binding fragment thereof and a VEGF receptor inhibitor.
- VEGF receptor inhibitors include bevacizumab (AVASTIN®),
- the combination therapy of the antibodies and antigen-binding fragments described herein with the kidney cancer therapies may also provide for lower doses of either therapy, or both, due to a synergistic effect from the coadministration of the therapies.
- the cancer is myeloma and the one or more therapeutic treatments is surgery, radiotherapy, VELCADE®, lenalidomide, or thalidomide.
- the combination is a humanized anti-endoglin antibody or antigen-binding fragment thereof and VELCADE®. The dosages for any of these therapies are known in the art and can be adjusted with combination therapy accordingly.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Epidemiology (AREA)
- Cell Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biomedical Technology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Microbiology (AREA)
- Oncology (AREA)
- Biotechnology (AREA)
- Physics & Mathematics (AREA)
- Food Science & Technology (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Mycology (AREA)
- Diabetes (AREA)
- Hospice & Palliative Care (AREA)
- Endocrinology (AREA)
- Cardiology (AREA)
- Wood Science & Technology (AREA)
Abstract
Description



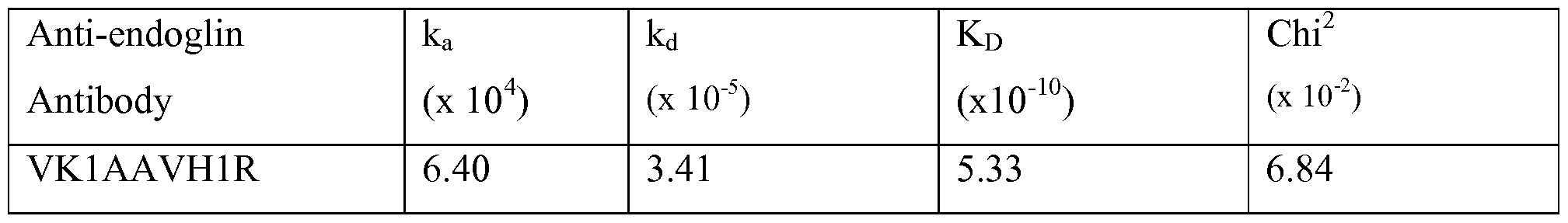



Claims
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2010300668A AU2010300668B2 (en) | 2009-09-30 | 2010-09-29 | Endoglin antibodies |
| CN201080054202.4A CN102762227B (en) | 2009-09-30 | 2010-09-29 | Endoglin antibodies |
| EA201290173A EA025174B1 (en) | 2009-09-30 | 2010-09-29 | Endoglin antibodies |
| EP10821188.9A EP2482848B1 (en) | 2009-09-30 | 2010-09-29 | Endoglin antibodies |
| BR112012007318-4A BR112012007318B1 (en) | 2009-09-30 | 2010-09-29 | Antibody or antigen binding fragment thereof, composition and use of an antibody or antigen binding fragment |
| ES10821188T ES2776977T3 (en) | 2009-09-30 | 2010-09-29 | Endoglin antibodies |
| CA2775810A CA2775810C (en) | 2009-09-30 | 2010-09-29 | Endoglin antibodies |
| JP2012532280A JP5632002B2 (en) | 2009-09-30 | 2010-09-29 | Endoglin antibody |
| KR1020127011203A KR101398707B1 (en) | 2009-09-30 | 2010-09-29 | Endoglin antibodies |
| IL218724A IL218724A0 (en) | 2009-09-30 | 2012-03-19 | Endoglin antibodies |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US24729009P | 2009-09-30 | 2009-09-30 | |
| US61/247,290 | 2009-09-30 | ||
| US12/751,907 | 2010-03-31 | ||
| US12/751,907 US8221753B2 (en) | 2009-09-30 | 2010-03-31 | Endoglin antibodies |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011041441A1 true WO2011041441A1 (en) | 2011-04-07 |
Family
ID=43780635
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/050759 WO2011041441A1 (en) | 2009-09-30 | 2010-09-29 | Endoglin antibodies |
Country Status (12)
| Country | Link |
|---|---|
| US (7) | US8221753B2 (en) |
| EP (1) | EP2482848B1 (en) |
| JP (2) | JP5632002B2 (en) |
| KR (1) | KR101398707B1 (en) |
| CN (2) | CN102762227B (en) |
| AU (1) | AU2010300668B2 (en) |
| BR (1) | BR112012007318B1 (en) |
| CA (1) | CA2775810C (en) |
| EA (1) | EA025174B1 (en) |
| ES (1) | ES2776977T3 (en) |
| IL (1) | IL218724A0 (en) |
| WO (1) | WO2011041441A1 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013074840A1 (en) * | 2011-11-15 | 2013-05-23 | Allergan, Inc. | Treatment of dry age related macular degeneration |
| WO2014039682A1 (en) * | 2012-09-05 | 2014-03-13 | Tracon Pharmaceuticals, Inc. | Antibody formulations and uses thereof |
| JP2014513089A (en) * | 2011-04-26 | 2014-05-29 | サノフイ | A composition comprising aflibercept, folinic acid, 5-fluorouracil (5-FU) and illinocetane (formyl) |
| WO2016077451A1 (en) * | 2014-11-12 | 2016-05-19 | Tracon Pharmaceuticals, Inc. | Anti-endoglin antibodies and uses thereof |
| CN105821029A (en) * | 2015-01-04 | 2016-08-03 | 彭霞 | Heterologous fusion gene modified cancer cell/dendritic cell fusion tumor vaccine and preparation method thereof |
| US9518122B2 (en) | 2009-09-30 | 2016-12-13 | Tracon Pharmaceuticals, Inc. | Endoglin antibodies |
| US9926375B2 (en) | 2014-11-12 | 2018-03-27 | Tracon Pharmaceuticals, Inc. | Anti-endoglin antibodies and uses thereof |
| US10501523B2 (en) | 2014-07-18 | 2019-12-10 | Sanofi | IL-8 level based method of predicting the outcome of colon cancer treatment |
| WO2023133595A2 (en) | 2022-01-10 | 2023-07-13 | Sana Biotechnology, Inc. | Methods of ex vivo dosing and administration of lipid particles or viral vectors and related systems and uses |
| US11883432B2 (en) | 2020-12-18 | 2024-01-30 | Century Therapeutics, Inc. | Chimeric antigen receptor system with adaptable receptor specificity |
| WO2024081820A1 (en) | 2022-10-13 | 2024-04-18 | Sana Biotechnology, Inc. | Viral particles targeting hematopoietic stem cells |
| WO2024119157A1 (en) | 2022-12-02 | 2024-06-06 | Sana Biotechnology, Inc. | Lipid particles with cofusogens and methods of producing and using the same |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100082438A1 (en) * | 2008-10-01 | 2010-04-01 | Ronnie Jack Garmon | Methods and systems for customer performance scoring |
| JP5848775B2 (en) | 2010-12-06 | 2016-01-27 | コンフルエンス・ライフ・サイエンシズ,インコーポレーテッド | Substituted pyridinone pyridinyl compounds |
| KR102011924B1 (en) | 2011-05-18 | 2019-08-21 | 메더리스 다이어비티즈, 엘엘씨 | Improved peptide pharmaceuticals for insulin resistance |
| US9468666B2 (en) * | 2011-08-01 | 2016-10-18 | Tufts Medical Center, Inc. | Treatment of heart failure and related conditions |
| KR20210133321A (en) * | 2012-11-08 | 2021-11-05 | 클리어사이드 바이오메디컬, 인코포레이드 | Methods and devices for the treatment of ocular disease in human subjects |
| CN118324870A (en) | 2012-11-20 | 2024-07-12 | 梅德瑞斯糖尿病有限责任公司 | Improved peptide drugs for insulin resistance |
| CN107880126B (en) | 2012-12-21 | 2021-03-30 | 西雅图基因公司 | anti-NTB-A antibodies and related compositions and methods |
| US10214590B2 (en) | 2013-09-20 | 2019-02-26 | Tufts Medical Center, Inc. | Inhibitors of endoglin activity for the treatment of fibrosis |
| GB201402009D0 (en) * | 2014-02-06 | 2014-03-26 | Oncomatryx Biopharma S L | Antibody-drug conjugates and immunotoxins |
| MX2016014160A (en) * | 2014-05-01 | 2017-02-16 | Genentech Inc | Anti-factor d antibody variants and uses thereof. |
| CN117582516A (en) | 2014-05-28 | 2024-02-23 | 梅德瑞斯糖尿病有限责任公司 | Improved peptide drugs against insulin resistance |
| WO2017079473A1 (en) * | 2015-11-03 | 2017-05-11 | The Brigham And Women's Hospital, Inc. | Imaging probe for angiogenic activity in pulmonary arterial hypertension |
| CN106928355B (en) * | 2015-12-30 | 2020-09-29 | 广西医科大学 | CD105 nano antibody Nb184 |
| CN106928359B (en) * | 2015-12-30 | 2020-10-13 | 广西医科大学 | CD105 nano antibody Nb59 |
| AU2017286561B2 (en) * | 2016-06-14 | 2024-08-01 | Cedars-Sinai Medical Center | Sensitization of tumors to therapies through endoglin antagonism |
| WO2017223495A1 (en) * | 2016-06-24 | 2017-12-28 | University Of South Carolina | Inhibin as targetable regulators of angiogenesis |
| WO2018067819A1 (en) * | 2016-10-06 | 2018-04-12 | Tracon Pharmaceuticals, Inc. | Compositions and methods for treatment of cancers |
| WO2018187158A1 (en) * | 2017-04-07 | 2018-10-11 | Tracon Pharmaceuticals, Inc. | Combination therapy of cancer with anti-endoglin antibodies and anti-programmed death receptor agents |
| WO2018204323A1 (en) * | 2017-05-01 | 2018-11-08 | Del Mar Pharmaceuticals (Bc) Ltd. | Use of dianhydrogalactitol or analogs and derivatives in combination with vegf inhibitors to treat cancer |
| US20200165350A1 (en) * | 2017-06-01 | 2020-05-28 | Health Research, Inc. | Strong potentiation of immune checkpoint blockade therapy of cancer by combination with endoglin-targeting therapy |
| EP3648757A4 (en) * | 2017-07-03 | 2020-11-25 | Development Center for Biotechnology | Anti-vegfr antibody and uses thereof |
| JP2017214419A (en) * | 2017-08-18 | 2017-12-07 | 学校法人関西医科大学 | Leukemia improver |
| CN111479807A (en) * | 2017-12-19 | 2020-07-31 | 百时美施贵宝公司 | Cyclohexyl acid isoxazoles azines as L PA antagonists |
| IL311775A (en) | 2018-01-03 | 2024-05-01 | Mederis Diabetes Llc | Improved peptide pharmaceuticals for treatment of nash and other disorders |
| CN108484759A (en) * | 2018-03-01 | 2018-09-04 | 东北农业大学 | Two kinds of scFv antibody, its encoding gene and its application in preparing treatment or prevention Bursal Disease preparation |
| CN108795867A (en) * | 2018-06-05 | 2018-11-13 | 华东理工大学 | The method for shifting external threedimensional model for building colon cancer cell peritonaeum |
| US20230030032A1 (en) * | 2019-12-13 | 2023-02-02 | Aggamin, LLC | Methods and systems for treating or preventing pregnancy-related hypertensive disorders |
| WO2024040194A1 (en) | 2022-08-17 | 2024-02-22 | Capstan Therapeutics, Inc. | Conditioning for in vivo immune cell engineering |
Citations (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4472509A (en) | 1982-06-07 | 1984-09-18 | Gansow Otto A | Metal chelate conjugated monoclonal antibodies |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4868103A (en) | 1986-02-19 | 1989-09-19 | Enzo Biochem, Inc. | Analyte detection by means of energy transfer |
| US4938948A (en) | 1985-10-07 | 1990-07-03 | Cetus Corporation | Method for imaging breast tumors using labeled monoclonal anti-human breast cancer antibodies |
| EP0404097A2 (en) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispecific and oligospecific, mono- and oligovalent receptors, production and applications thereof |
| US5021236A (en) | 1981-07-24 | 1991-06-04 | Schering Aktiengesellschaft | Method of enhancing NMR imaging using chelated paramagnetic ions bound to biomolecules |
| WO1993011161A1 (en) | 1991-11-25 | 1993-06-10 | Enzon, Inc. | Multivalent antigen-binding proteins |
| US5391377A (en) | 1990-10-19 | 1995-02-21 | Cortecs Limited | Biphasic release formations for lipophilic acids |
| US5631169A (en) | 1992-01-17 | 1997-05-20 | Joseph R. Lakowicz | Fluorescent energy transfer immunoassay |
| US5756097A (en) | 1985-06-28 | 1998-05-26 | Landucci; Gary R. | Lymphokine activated effector cells for antibody-dependent cellular cytotoxicity (ADCC) treatment of cancer and other diseases |
| US5928641A (en) | 1996-05-31 | 1999-07-27 | Health Research Inc. | Anti-endoglin monoclonal antibodies and their use in antiangiogenic therapy |
| US6096289A (en) | 1992-05-06 | 2000-08-01 | Immunomedics, Inc. | Intraoperative, intravascular, and endoscopic tumor and lesion detection, biopsy and therapy |
| US6096871A (en) | 1995-04-14 | 2000-08-01 | Genentech, Inc. | Polypeptides altered to contain an epitope from the Fc region of an IgG molecule for increased half-life |
| US6190660B1 (en) | 1996-05-31 | 2001-02-20 | Health Research, Inc. | Anti-endoglin monoclonal antibodies and their use in antiangiogenic therapy |
| US6350861B1 (en) | 1992-03-09 | 2002-02-26 | Protein Design Labs, Inc. | Antibodies with increased binding affinity |
| WO2002069232A2 (en) | 2001-02-19 | 2002-09-06 | Merck Patent Gmbh | Method for identification of t-cell epitopes and use for preparing molecules with reeduced immunogenicity |
| US20020136725A1 (en) * | 1996-01-17 | 2002-09-26 | Smithkline Beecham Corporation | Antithrombotic agents |
| US6610293B1 (en) | 1997-06-16 | 2003-08-26 | The Henry M. Jackson Foundation For The Advancement Of Military Medicine | Opsonic and protective monoclonal and chimeric antibodies specific for lipoteichoic acid of gram positive bacteria |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US20040175756A1 (en) | 2001-04-26 | 2004-09-09 | Avidia Research Institute | Methods for using combinatorial libraries of monomer domains |
| US20050037421A1 (en) | 2001-09-13 | 2005-02-17 | Institute For Antibodies Co., Ltd | Methods of constructing camel antibody libraries |
| US20050048512A1 (en) | 2001-04-26 | 2005-03-03 | Avidia Research Institute | Combinatorial libraries of monomer domains |
| US20050053973A1 (en) | 2001-04-26 | 2005-03-10 | Avidia Research Institute | Novel proteins with targeted binding |
| US6881557B2 (en) | 2001-07-12 | 2005-04-19 | Arrowsmith Technologies Llp | Super humanized antibodies |
| US20050089932A1 (en) | 2001-04-26 | 2005-04-28 | Avidia Research Institute | Novel proteins with targeted binding |
| US20050164301A1 (en) | 2003-10-24 | 2005-07-28 | Avidia Research Institute | LDL receptor class A and EGF domain monomers and multimers |
| US20050221384A1 (en) | 2001-04-26 | 2005-10-06 | Avidia Research Institute | Combinatorial libraries of monomer domains |
| US20050238646A1 (en) | 2001-01-17 | 2005-10-27 | Trubion Pharmaceuticals, Inc. | Binding domain-immunoglobulin fusion proteins |
| US7091321B2 (en) | 2000-02-11 | 2006-08-15 | Emd Lexigen Research Center Corp. | Enhancing the circulating half-life of antibody-based fusion proteins |
| US7097836B1 (en) | 2002-10-23 | 2006-08-29 | Health Research, Inc. | Method for increasing the efficacy of anti-tumor agents by anti-endoglin antibody |
| US7112412B1 (en) | 1996-05-06 | 2006-09-26 | Cornell Research Foundation, Inc. | Treatment and diagnosis of prostate cancer |
| US7115716B2 (en) | 2001-11-19 | 2006-10-03 | Eli Lilly And Company | Tumor specific monoclonal antibodies |
| US20060223096A1 (en) * | 2005-03-25 | 2006-10-05 | Glycart Biotechnology Ag | Antigen binding molecules directed to MCSP and having increased Fc receptor binding affinity and effector function |
| US20070082380A1 (en) | 2005-10-07 | 2007-04-12 | Pardridge William M | Nucleic acids encoding and methods of producing fusion proteins |
| US7217798B2 (en) | 2003-10-15 | 2007-05-15 | Pdl Biopharma, Inc. | Alteration of Fc-fusion protein serum half-lives by mutagenesis |
Family Cites Families (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US928641A (en) | 1908-10-09 | 1909-07-20 | Levi E Edmunds | Clasp for pilings. |
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US6407213B1 (en) | 1991-06-14 | 2002-06-18 | Genentech, Inc. | Method for making humanized antibodies |
| US5965132A (en) | 1992-03-05 | 1999-10-12 | Board Of Regents, The University Of Texas System | Methods and compositions for targeting the vasculature of solid tumors |
| US6267958B1 (en) | 1995-07-27 | 2001-07-31 | Genentech, Inc. | Protein formulation |
| US5796097A (en) | 1997-03-05 | 1998-08-18 | California Lightwave Laboratories, Inc. | Chemical sensor and method |
| AU740284B2 (en) | 1997-06-13 | 2001-11-01 | Genentech Inc. | Stabilized antibody formulation |
| EP1140840B1 (en) | 1999-01-13 | 2006-03-22 | Bayer Pharmaceuticals Corp. | -g(v)-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| US6635677B2 (en) | 1999-08-13 | 2003-10-21 | Case Western Reserve University | Methoxyamine combinations in the treatment of cancer |
| JP4340062B2 (en) | 2000-10-12 | 2009-10-07 | ジェネンテック・インコーポレーテッド | Concentrated protein preparation with reduced viscosity |
| WO2002072824A2 (en) | 2001-02-20 | 2002-09-19 | Bayer Aktiengesellschaft | Human transient receptor potential channel protein. |
| CA2444483A1 (en) | 2001-04-26 | 2002-11-07 | Board Of Regents, The University Of Texas System | Diagnostic imaging compositions, their methods of synthesis and use |
| US20030129193A1 (en) | 2001-09-27 | 2003-07-10 | Board Of Regents, The University Of Texas System And Peregrine Pharmaceuticals, Inc. | Combined methods for tumor vasculature coaguligand treatment |
| NZ534542A (en) | 2002-02-14 | 2006-08-31 | Chugai Pharmaceutical Co Ltd | Antibody-containing solutions in which the formation of degradation products and insoluble particles during storage and freeze/thaw cycles is inhibited by adding a sugar and surfactant respectively |
| AU2003243139B2 (en) | 2002-04-05 | 2007-06-21 | Amgen Inc. | Human anti-OPGL neutralizing antibodies as selective OPGL pathway inhibitors |
| US20040033228A1 (en) | 2002-08-16 | 2004-02-19 | Hans-Juergen Krause | Formulation of human antibodies for treating TNF-alpha associated disorders |
| US7754884B2 (en) | 2005-01-03 | 2010-07-13 | Vanderbilt University | Targeted, NIR imaging agents for therapy efficacy monitoring, deep tissue disease demarcation and deep tissue imaging |
| US7691374B2 (en) | 2002-10-23 | 2010-04-06 | Health Research, Inc. | Method for increasing the efficacy of anti-tumor agents by anti-endoglin antibody |
| DE10303664A1 (en) | 2003-01-23 | 2004-08-12 | Nemod Immuntherapie Ag | Detection molecules for the treatment and detection of tumors |
| US7120987B2 (en) | 2003-08-05 | 2006-10-17 | Avery Dennison Corporation | Method of making RFID device |
| JO3000B1 (en) | 2004-10-20 | 2016-09-05 | Genentech Inc | Antibody Formulations. |
| US8003108B2 (en) | 2005-05-03 | 2011-08-23 | Amgen Inc. | Sclerostin epitopes |
| US20070077310A1 (en) | 2005-10-03 | 2007-04-05 | University Of Tennessee Research Foundation | Methods of reducing the production of reactive oxygen species and methods of screening or identifying compounds and compositions that reduce the production of reactive oxygen species |
| CA2663243A1 (en) | 2006-09-28 | 2008-04-03 | Merck Serono S.A. | Junctional adhesion molecule-c (jam-c) binding compounds and methods of their use |
| AU2007307107B2 (en) | 2006-10-06 | 2011-11-03 | Amgen Inc. | Stable antibody formulations |
| WO2008051363A2 (en) | 2006-10-20 | 2008-05-02 | Amgen Inc. | Stable polypeptide formulations |
| AU2008210521A1 (en) | 2007-02-01 | 2008-08-07 | Genentech, Inc. | Combination therapy with angiogenesis inhibitors |
| ATE553771T1 (en) | 2007-06-06 | 2012-05-15 | Res Dev Foundation | RTEF-1 VARIANTS AND THEIR USE TO INHIBIT ANGIOGENESIS |
| AU2008289225A1 (en) | 2007-08-17 | 2009-02-26 | Amgen Inc. | Formulations of antibodies and Fc-fusion molecules using polycations |
| EP2212332B1 (en) | 2007-09-14 | 2012-10-24 | Bayer Intellectual Property GmbH | Substituted tricyclic compounds and methods of use thereof |
| TWI580694B (en) | 2007-11-30 | 2017-05-01 | 建南德克公司 | Anti-vegf antibodies |
| AU2009205428B2 (en) | 2008-01-14 | 2014-10-30 | Genentech, Inc. | Inhibiting angiogenesis using EGFL8 antagonists |
| CN102137846B (en) | 2008-08-29 | 2014-04-23 | 拜耳知识产权有限责任公司 | N-(2-aminophenyl)-4-[N-(pyridine-3-yl)-methoxycarbonyl-aminomethyl]-benzamide (MS-275)polymorph B |
| AU2009294414A1 (en) | 2008-09-19 | 2010-03-25 | Medimmune Llc | Antibodies directed to CD105 and uses thereof |
| US8699361B2 (en) | 2008-09-30 | 2014-04-15 | Qualcomm Incorporated | Out-of-synchronization handling method and apparatus |
| US20100082438A1 (en) | 2008-10-01 | 2010-04-01 | Ronnie Jack Garmon | Methods and systems for customer performance scoring |
| EP2196476A1 (en) | 2008-12-10 | 2010-06-16 | Novartis Ag | Antibody formulation |
| TW201039854A (en) | 2009-03-06 | 2010-11-16 | Genentech Inc | Antibody formulation |
| CN102711809B (en) | 2009-08-17 | 2015-09-30 | 特雷康制药公司 | Use anti-endoglin antibody and anti-VEGF agent therapeutic alliance cancer |
| US8221753B2 (en) | 2009-09-30 | 2012-07-17 | Tracon Pharmaceuticals, Inc. | Endoglin antibodies |
| US8754195B2 (en) | 2010-07-02 | 2014-06-17 | Medimmune, Llc | Antibody formulations |
| WO2012145539A1 (en) | 2011-04-20 | 2012-10-26 | Acceleron Pharma, Inc. | Endoglin polypeptides and uses thereof |
| US9468666B2 (en) | 2011-08-01 | 2016-10-18 | Tufts Medical Center, Inc. | Treatment of heart failure and related conditions |
| UA115789C2 (en) | 2012-09-05 | 2017-12-26 | Трейкон Фармасутікалз, Інк. | Antibody formulations and uses thereof |
| US10214590B2 (en) | 2013-09-20 | 2019-02-26 | Tufts Medical Center, Inc. | Inhibitors of endoglin activity for the treatment of fibrosis |
| US9926375B2 (en) | 2014-11-12 | 2018-03-27 | Tracon Pharmaceuticals, Inc. | Anti-endoglin antibodies and uses thereof |
| CA2966905A1 (en) | 2014-11-12 | 2016-05-19 | Tracon Pharmaceuticals, Inc. | Anti-endoglin antibodies and uses thereof |
-
2010
- 2010-03-31 US US12/751,907 patent/US8221753B2/en active Active
- 2010-09-29 WO PCT/US2010/050759 patent/WO2011041441A1/en active Application Filing
- 2010-09-29 EP EP10821188.9A patent/EP2482848B1/en active Active
- 2010-09-29 EA EA201290173A patent/EA025174B1/en not_active IP Right Cessation
- 2010-09-29 ES ES10821188T patent/ES2776977T3/en active Active
- 2010-09-29 AU AU2010300668A patent/AU2010300668B2/en not_active Ceased
- 2010-09-29 CA CA2775810A patent/CA2775810C/en not_active Expired - Fee Related
- 2010-09-29 KR KR1020127011203A patent/KR101398707B1/en active IP Right Grant
- 2010-09-29 BR BR112012007318-4A patent/BR112012007318B1/en not_active IP Right Cessation
- 2010-09-29 CN CN201080054202.4A patent/CN102762227B/en not_active Expired - Fee Related
- 2010-09-29 JP JP2012532280A patent/JP5632002B2/en not_active Expired - Fee Related
- 2010-09-29 CN CN201510115139.5A patent/CN104788562B/en not_active Expired - Fee Related
-
2012
- 2012-03-19 IL IL218724A patent/IL218724A0/en not_active IP Right Cessation
- 2012-05-31 US US13/485,702 patent/US8609094B2/en not_active Expired - Fee Related
-
2013
- 2013-10-15 US US14/054,446 patent/US9150652B2/en active Active
-
2014
- 2014-07-14 JP JP2014144256A patent/JP5960203B2/en not_active Expired - Fee Related
-
2015
- 2015-08-27 US US14/838,006 patent/US9518122B2/en not_active Expired - Fee Related
-
2016
- 2016-10-28 US US15/337,113 patent/US9944714B2/en active Active - Reinstated
-
2018
- 2018-02-28 US US15/907,505 patent/US20180194854A1/en not_active Abandoned
-
2019
- 2019-09-30 US US16/588,543 patent/US20200048360A1/en not_active Abandoned
Patent Citations (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5021236A (en) | 1981-07-24 | 1991-06-04 | Schering Aktiengesellschaft | Method of enhancing NMR imaging using chelated paramagnetic ions bound to biomolecules |
| US4472509A (en) | 1982-06-07 | 1984-09-18 | Gansow Otto A | Metal chelate conjugated monoclonal antibodies |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4683202B1 (en) | 1985-03-28 | 1990-11-27 | Cetus Corp | |
| US5756097A (en) | 1985-06-28 | 1998-05-26 | Landucci; Gary R. | Lymphokine activated effector cells for antibody-dependent cellular cytotoxicity (ADCC) treatment of cancer and other diseases |
| US4938948A (en) | 1985-10-07 | 1990-07-03 | Cetus Corporation | Method for imaging breast tumors using labeled monoclonal anti-human breast cancer antibodies |
| US4868103A (en) | 1986-02-19 | 1989-09-19 | Enzo Biochem, Inc. | Analyte detection by means of energy transfer |
| EP0404097A2 (en) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispecific and oligospecific, mono- and oligovalent receptors, production and applications thereof |
| US5391377A (en) | 1990-10-19 | 1995-02-21 | Cortecs Limited | Biphasic release formations for lipophilic acids |
| WO1993011161A1 (en) | 1991-11-25 | 1993-06-10 | Enzon, Inc. | Multivalent antigen-binding proteins |
| US5631169A (en) | 1992-01-17 | 1997-05-20 | Joseph R. Lakowicz | Fluorescent energy transfer immunoassay |
| US6350861B1 (en) | 1992-03-09 | 2002-02-26 | Protein Design Labs, Inc. | Antibodies with increased binding affinity |
| US6096289A (en) | 1992-05-06 | 2000-08-01 | Immunomedics, Inc. | Intraoperative, intravascular, and endoscopic tumor and lesion detection, biopsy and therapy |
| US6096871A (en) | 1995-04-14 | 2000-08-01 | Genentech, Inc. | Polypeptides altered to contain an epitope from the Fc region of an IgG molecule for increased half-life |
| US20020136725A1 (en) * | 1996-01-17 | 2002-09-26 | Smithkline Beecham Corporation | Antithrombotic agents |
| US7112412B1 (en) | 1996-05-06 | 2006-09-26 | Cornell Research Foundation, Inc. | Treatment and diagnosis of prostate cancer |
| US6190660B1 (en) | 1996-05-31 | 2001-02-20 | Health Research, Inc. | Anti-endoglin monoclonal antibodies and their use in antiangiogenic therapy |
| US6200566B1 (en) | 1996-05-31 | 2001-03-13 | Health Research, Inc. | Anti-endoglin monoclonal antibodies and their use in antiangiogenic therapy |
| US5928641A (en) | 1996-05-31 | 1999-07-27 | Health Research Inc. | Anti-endoglin monoclonal antibodies and their use in antiangiogenic therapy |
| US6610293B1 (en) | 1997-06-16 | 2003-08-26 | The Henry M. Jackson Foundation For The Advancement Of Military Medicine | Opsonic and protective monoclonal and chimeric antibodies specific for lipoteichoic acid of gram positive bacteria |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US7091321B2 (en) | 2000-02-11 | 2006-08-15 | Emd Lexigen Research Center Corp. | Enhancing the circulating half-life of antibody-based fusion proteins |
| US20050238646A1 (en) | 2001-01-17 | 2005-10-27 | Trubion Pharmaceuticals, Inc. | Binding domain-immunoglobulin fusion proteins |
| WO2002069232A2 (en) | 2001-02-19 | 2002-09-06 | Merck Patent Gmbh | Method for identification of t-cell epitopes and use for preparing molecules with reeduced immunogenicity |
| US20040175756A1 (en) | 2001-04-26 | 2004-09-09 | Avidia Research Institute | Methods for using combinatorial libraries of monomer domains |
| US20050089932A1 (en) | 2001-04-26 | 2005-04-28 | Avidia Research Institute | Novel proteins with targeted binding |
| US20050053973A1 (en) | 2001-04-26 | 2005-03-10 | Avidia Research Institute | Novel proteins with targeted binding |
| US20050221384A1 (en) | 2001-04-26 | 2005-10-06 | Avidia Research Institute | Combinatorial libraries of monomer domains |
| US20050048512A1 (en) | 2001-04-26 | 2005-03-03 | Avidia Research Institute | Combinatorial libraries of monomer domains |
| US6881557B2 (en) | 2001-07-12 | 2005-04-19 | Arrowsmith Technologies Llp | Super humanized antibodies |
| US20050037421A1 (en) | 2001-09-13 | 2005-02-17 | Institute For Antibodies Co., Ltd | Methods of constructing camel antibody libraries |
| US7115716B2 (en) | 2001-11-19 | 2006-10-03 | Eli Lilly And Company | Tumor specific monoclonal antibodies |
| US7097836B1 (en) | 2002-10-23 | 2006-08-29 | Health Research, Inc. | Method for increasing the efficacy of anti-tumor agents by anti-endoglin antibody |
| US7217798B2 (en) | 2003-10-15 | 2007-05-15 | Pdl Biopharma, Inc. | Alteration of Fc-fusion protein serum half-lives by mutagenesis |
| US20050164301A1 (en) | 2003-10-24 | 2005-07-28 | Avidia Research Institute | LDL receptor class A and EGF domain monomers and multimers |
| US20060223096A1 (en) * | 2005-03-25 | 2006-10-05 | Glycart Biotechnology Ag | Antigen binding molecules directed to MCSP and having increased Fc receptor binding affinity and effector function |
| US20070082380A1 (en) | 2005-10-07 | 2007-04-12 | Pardridge William M | Nucleic acids encoding and methods of producing fusion proteins |
Non-Patent Citations (130)
| Title |
|---|
| "Animal Cell Culture", 1986 |
| "Biocomputing: Informatics and Genome Projects", 1993, ACADEMIC PRESS |
| "Cell Biology: A Laboratory Handbook", vol. I-III, 1994 |
| "Computational Molecular Biology", 1988, OXFORD UNIVERSITY PRESS |
| "Computer Analysis of Sequence Data", 1994, HUMANA PRESS |
| "Current Protocols in Immunology", vol. I-III, 1994 |
| "Current Protocols in Molecular Biology", vol. I-III, 1994 |
| "Immobilized Cells And Enzymes", 1986, IRL PRESS |
| "Nucleic Acid Hybridization", 1985 |
| "Oligonucleotide Synthesis", 1984 |
| "Remington' pharmaceutical Sciences", 1990, MACK PUBLISHING CO. |
| "Remington's Pharmaceutical Sciences", 1980 |
| "Sequence Analysis Primer", 1991, M STOCKTON PRESS |
| "Short Protocols in Molecular Biology", 1992, JOHN WILEY & SONS |
| "Transcription And Translation", 1984 |
| "WHO Classification", 2001 |
| A. H. GREENBERG ET AL.: "Characteristics Of The Effector Cells Mediating Cytotoxicity Against Antibody-Coated Target Cells", I., IMMUNOLOGY, vol. 21, 1975, pages 719 |
| ALTMAN, R ET AL., ARTHRITIS RHEUM, vol. 29, 1986, pages 1039 |
| ALTMAN, R ET AL., ARTHRITIS RHEUM, vol. 34, 1991, pages 505 |
| ALTSCHUL ET AL., J. MOL. BIOL., vol. 215, 1990, pages 403 - 10 |
| ALTSCHUL ET AL., NUC. ACIDS RES., vol. 25, 1997, pages 3389 - 3402 |
| ALTSCHUL ET AL., NUCLEIC ACIDS RES., vol. 25, no. 17, 1997, pages 3389 - 3402 |
| ALTSCHUL, S. ET AL., J. MOL. BIOL., vol. 215, 1990, pages 403 - 410 |
| ALTSCHUL, S. ET AL.: "BLAST Manual", NCBI NLM NIH |
| ALTSCHUL, S. F. ET AL., J. MOLEC. BIOL., vol. 215, 1990, pages 403 - 410 |
| ARGARANA CE; KUNTZ ID; BIRKEN S; AXEL R; CANTOR CR: "Molecular cloning and nucleotide sequence of the streptavidin gene", NUCLEIC ACIDS RES., vol. 14, no. 4, 1986, pages 1871 - 82, XP002951809 |
| B. PERBAL, A PRACTICAL GUIDE TO MOLECULAR CLONING, 1984 |
| BERNABEU ET AL., J. CELL. BIOCHEM., vol. 102, no. 6, 2007, pages 1375 - 1388 |
| BIRD ET AL., SCIENCE, vol. 242, 1988, pages 423 - 426 |
| BLOOD ET AL., BIOCHIM. BIOPHYS. ACTA, vol. 1032, 1990, pages 89 - 118 |
| BONNET; WALSH, RHEUMATOLOGY, vol. 44, 2005, pages 7 - 16 |
| BROOKS ET AL., J. CLIN. INVEST, vol. 99, 1997, pages 1390 - 1398 |
| BURROWS ET AL., CLIN. CANCER RES., vol. 1, 1995, pages 1623 - 1634 |
| CARILLO, H.; LIPMAN, D., SIAM J. APPLIED MATH., vol. 48, 1988, pages 1073 |
| CHIDLOW ET AL., AM. J. PHYSIOL. GASTROINTEST. LIVER PHYSIOL., vol. 293, 2007, pages 5 - 18 |
| CHOTHIA; LESK, J. MOL. BIOL., vol. 196, 1987, pages 901 - 917 |
| CHRISTOPHER J. NOREN; SPENCER J. ANTHONY-CAHILL; MICHAEL C. GRIFFITH; PETER G. SCHULTZ, SCIENCE, vol. 244, April 1989 (1989-04-01), pages 182 - 188 |
| CLACKSON ET AL., NATURE, vol. 352, 1991, pages 624 - 628 |
| D'AMATO ET AL., PROC. NATL. ACAD. SCI. USA, vol. 91, no. 9, 1994, pages 4082 - 4085 |
| D'AMATO ET AL., PROC. NATL. ACAD. SCI., vol. 91, 1994, pages 4082 - 4085 |
| DER-BALIAN G; KAMEDA, N; ROWLEY, G., ANAL. BIOCHEM., vol. 173, 1988, pages 59 - 63 |
| DEVEREUX, J. ET AL., NUCLEIC ACIDS RESEARCH, vol. 12, no. 1, 1984, pages 387 |
| DOBELI, PROTEIN EXPR. PURIF., vol. 12, 1998, pages 404 - 414 |
| DOCHERTY ET AL., NEPHOL. DIAL. TRANSPLANT., vol. 21, 2006, pages 2106 - 2119 |
| DUFF ET AL., FASEB J., vol. 17, 2003, pages 984 - 992 |
| EDGE, NATURE, vol. 292, 1981, pages 756 |
| FIX, PHARM RES., vol. 13, 1996, pages 1760 1764 |
| FRANCIS ET AL., INTERNATIONAL JOURNAL OF HEMATOLOGY, vol. 68, 1998, pages 1 - 18 |
| FURSTENBERGER ET AL., LANCET, vol. 3, 2002, pages 298 - 302 |
| GHETIE ET AL., PHARMACOL. THER., vol. 63, 1994, pages 209 - 34 |
| GIGLI ET AL., J. IMMUNOL., vol. 100, 1986, pages 1154 - 1164 |
| GILLES MA; HUDSON AQ; BORDERS CL JR: "Stability of water-soluble carbodiimides in aqueous solution", ANAL BIOCHEM., vol. 184, no. 2, 1 February 1990 (1990-02-01), pages 244 - 248, XP024819114, DOI: doi:10.1016/0003-2697(90)90675-Y |
| GLAZER, AN; STRYER L., TRENDS BIOCHEM. SCI., vol. 9, 1984, pages 423 - 7 |
| GOUGOS; LETARTE, J. IMMUNOL., vol. 141, 1988, pages 1925 - 1933 |
| GREEN, NM.: "A spectrophotometric assay for avidin and biotin based on binding of dyes by avidin", BIOCHEM. J., vol. 94, 1965, pages 23c - 24c |
| GREEN, NM: "Avidin. In Advances in Protein Chemistry", vol. 29, 1975, ACADEMIC PRESS, pages: 85 - 133 |
| GREEN, NM: "The use of bifunctional biotinyl compounds to determine the arrangement of subunits in avidin", BIOCHEM J., vol. 125, 1971, pages 781 - 791, XP002114517 |
| HARDY RR ET AL., J. EXP. MED., vol. 159, 1984, pages 1169 - 88 |
| HARDY, RR ET AL., NATURE, vol. 306, 1983, pages 270 - 2 |
| HARUTA; SEON, PROC. NATL. ACAD. SCI., vol. 83, 1986, pages 7898 - 7902 |
| HEELEY, R. P., ENDOCR. RES., vol. 28, 2002, pages 217 - 229 |
| HENIKOFF; HENIKOFF: "Proc. Natl. Acad. Sci", vol. 89, 1992, article "Amino acid substitution matrices from protein blocks", pages: 10915 - 10919 |
| HOLLINGER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 6444 6448 |
| HUSTON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 85, 1988, pages 5879 - 5883 |
| I. C. M. MACLENNAN: "Competition For Receptors For Immunoglobulin On Cytotoxic Lymphocytes", CLIN. EXP. IMMUNOL., vol. 10, 1972, pages 275 |
| I. R. MACKAY ET AL.: "Effect On Natural Killer And Antibody-Dependent Cellular Cytotoxicity Of Adjuvant Cytotoxic Chemotherapy Including Melphalan In Breast Cancer", CANCER IMMUNOL. IMMUNOTHER., vol. 16, 1983, pages 98 - 100 |
| J. BIOL. CHEM., vol. 265, 1990, pages 8361 - 8364 |
| J. F. JONES; D. M. SEGAL: "Antibody-Dependent Cell Mediated Cytolysis (ADCC) With Antibody-Coated Effectors: New Methods For Enhancing Antibody Binding And Cytolysis", J. IMMUNOL., vol. 125, 1980, pages 926 - 33 |
| JAY ET AL., J. BIOL. CHEM., vol. 259, 1984, pages 6311 |
| JONES ET AL., NATURE, vol. 321, 1986, pages 522 - 525 |
| KABAT ET AL.: "Sequences of Proteins of Immunological Interest", 1991, PUBLIC HEALTH SERVICE, NATIONAL INSTITUTES OF HEALTH |
| KABAT ET AL.: "Sequences of Proteins of Immunological Interest", 1991, PUBLIC HEALTH SERVICE, NATIONAL INSTITUTES OF HEALTH, pages: 647 - 669 |
| KIM ET AL., BREAST CANCER RESEARCH TREATMENT, vol. 85, no. 3, 2004, pages 281 - 91 |
| KOCH; DISTLER, ARTHRITIS RES. & THER., vol. 9, no. 2, 2007, pages S3,1 - 9 |
| KOHLER ET AL., NATURE, vol. 256, 1975, pages 495 |
| KROLL, DNA CELL. BIOL., vol. 12, 1993, pages 441 - 453 |
| KRONICK, MN, J. IMMUNO. METH., vol. 92, 1986, pages 1 - 13 |
| KRONICK, MN; GROSSMAN, PD, CLIN. CHEM., vol. 29, 1983, pages 1582 - 6 |
| LAHN ET AL.: "Aerosolized Anti- T-cell-Receptor Antibodies Are Effective against Airway Inflammation and Hyperreactivity", INT. ARCH. ALLEGERY IMMUNO., vol. 134, 2004, pages 49 - 55, XP009073464, DOI: doi:10.1159/000077533 |
| LANIER, LL; LOKEN, MR, J. IMMUNOL., vol. 132, 1984, pages 151 - 156 |
| LEATHERBARROW ET AL., MOL. IMMUNOL., vol. 22, 1985, pages 407 |
| LEBRIN ET AL., J. CLIN. INVEST, vol. 99, 1997, pages 1390 - 1398 |
| LILJEBLAD ET AL., GLYCO. J., vol. 17, 2000, pages 323 - 329 |
| MARKS ET AL., J. MOL. BIOL., vol. 222, 1991, pages 581 - 597 |
| MONTESANO ET AL., J. CELL BIOL., vol. 97, 1983, pages 1648 - 1652 |
| MURAOKA; SHULMAN, J. IMMUNOL., vol. 142, 1989, pages 695 |
| MUYLDERMANS ET AL., PROTEIN ENGINEERING, vol. 7, no. 9, 1994, pages 1129 |
| NAKAJIMA N; IKADE Y: "Mechanism of amide formation by carbodiimide for bioconjugation in aqueous media", BIOCONJUGATE CHEM., vol. 6, no. 1, 1995, pages 123 - 130, XP008051282, DOI: doi:10.1021/bc00031a015 |
| NAMBAIR ET AL., SCIENCE, vol. 223, 1984, pages 1299 |
| NILSSON K; MOSBACH K: "p-Toluenesulfonyl chloride as an activating agent of agarose for the preparation of immobilized affinity ligands and proteins", EUR. J. BIOCHEM., vol. 112, 1980, pages 397 - 402, XP001011372, DOI: doi:10.1111/j.1432-1033.1980.tb07218.x |
| OSBOURN ET AL., NAT. BIOTECHNOL., vol. 16, 1998, pages 778 |
| PAHLER A; HENDRICKSON WA; GAWINOWICZ KOLKS MA; ARAGANA CE; CANTOR CR: "Characterization and crystallization of core streptavidin", J BIOL CHEM, vol. 262, no. 29, 1987, pages 13933 - 13937, XP002011788 |
| PARKS, DR ET AL., CYTOMETRY, vol. 5, 1984, pages 159 - 68 |
| PINALS RS, ARTHRITIS RHEUM, vol. 24, 1981, pages 1308 |
| PLUCKTHUN, A. BIO/TECHNOLOGY, vol. 9, 1991, pages 545 - 551 |
| PLUCKTHUN, A., BIO/TECHNOLOGY, vol. 9, 1991, pages 545 - 551 |
| PLUCKTHUN: "The Pharmacology of Monoclonal Antibodies", vol. 113, 1994, SPRINGER-VERLAG, pages: 269 - 315 |
| PRESTA, CURR. OP. STRUCT. BIOL., vol. 2, 1992, pages 593 - 596 |
| PROC. NATL. ACAD. SCI. USA, vol. 66, 1970, pages 975 |
| RAFF, M.E., CURR. OPINION BIOTECH., vol. 4, 1993, pages 573 - 576 |
| REICHMANN ET AL., NATURE, vol. 332, 1988, pages 323 - 329 |
| REITER ET AL., NATURE BIOTECHNOLOGY, vol. 14, 1996, pages 1239 - 1245 |
| ROY-CHAUDHURY ET AL., EXP. NEPHROL., vol. 5, 1997, pages 55 - 60 |
| SAMANEN, J. PHARM. PHARMACOL., vol. 48, 1996, pages 119 135 |
| SAMBROOK ET AL.: "Molecular Cloning: A Laboratory Manual", 1989 |
| SAMBROOK ET AL.: "Molecular Cloning: a Laboratory Manual", 1989, COLD SPRING HARBOR LABORATORY PRESS |
| SCHNAPER ET AL., J. CELL PHYSIOL., vol. 256, 1993, pages 235 - 246 |
| SEHGAL D; VIJAY IK: "a method for the high efficiency of water-soluble carbodiimide-mediated amidation", ANAL BIOCHEM., vol. 218, no. 1, April 1994 (1994-04-01), pages 87 - 91, XP024762985, DOI: doi:10.1006/abio.1994.1144 |
| SHE ET AL., INT. J. CANCER, vol. 108, 2004, pages 251 - 257 |
| SILVERMAN ET AL., NAT. BIOTECHNOL., vol. 23, 2005, pages 1493 - 1494 |
| SILVERMAN ET AL., NAT. BIOTECHNOL., vol. 24, 2006, pages 220 |
| SJOLANDER, S.; URBANICZKY, C., ANAL. CHEM., vol. 63, 1991, pages 2338 - 2345 |
| STEFANSSON ET AL., J. BIOL. CHEM., vol. 276, 2000, pages 8135 - 8141 |
| SU ET AL., CANCER RES., vol. 63, 2003, pages 3585 - 3592 |
| SZABO ET AL., CURR. OPIN. STRUCT. BIOL., vol. 5, 1995, pages 699 - 705 |
| SZAJANI B ET AL.: "Effects of carbodiimide structure on the immobilization of enzymes", APPL BIOCHEM BIOTECHNOL., vol. 30, no. 2, August 1991 (1991-08-01), pages 225 - 231, XP035175834, DOI: doi:10.1007/BF02921689 |
| TAO; MORRISON, J. IMMUNOL., vol. 143, 1989, pages 2595 |
| TAYLOR; WALL, MOL. CELL. BIOL., vol. 8, 1988, pages 4197 |
| TRILL J.J. ET AL., CURR. OPINION BIOTECH, vol. 6, 1995, pages 553 - 560 |
| TSUJIE ET AL., INT. J. ONCOLOGY, vol. 29, 2006, pages 1087 - 1094 |
| VARNER ET AL., CELL ADH. COMMUN., vol. 3, 1995, pages 367 - 374 |
| VOLPERT ET AL., CANCER CELL, vol. 2, no. 6, 2002, pages 473 - 83 |
| VON HEINJE, G.: "Sequence Analysis in Molecular Biology", 1987, ACADEMIC PRESS |
| WANG ET AL., INT. J. CANCER, vol. 54, 1993, pages 363 - 370 |
| WARD ET AL., NATURE, vol. 341, 1989, pages 544 - 546 |
| WARD ET AL., NATURE, vol. 341, 1989, pages 544 546 |
| WEIDNER ET AL., J. NATL. CANCER INST., vol. 84, 1992, pages 1875 - 1887 |
| WILLIAMS, BIOCHEMISTRY, vol. 34, 1995, pages 1787 1797 |
| YAN ET AL., J. CLIN. INVEST., vol. 91, 1993, pages 986 - 996 |
| ZENT ET AL., SEMINARS IN NEPHROLOGY, vol. 27, no. 2, 2007, pages 161 - 171 |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9944714B2 (en) | 2009-09-30 | 2018-04-17 | Tracon Pharmaceuticals, Inc. | Endoglin antibodies |
| US9518122B2 (en) | 2009-09-30 | 2016-12-13 | Tracon Pharmaceuticals, Inc. | Endoglin antibodies |
| US11033606B2 (en) | 2011-04-26 | 2021-06-15 | Sanofi | Composition comprising aflibercept, folinic acid, 5-fluorouracil (5-FU) and irinotecan (FOLFIRI) |
| JP2014513089A (en) * | 2011-04-26 | 2014-05-29 | サノフイ | A composition comprising aflibercept, folinic acid, 5-fluorouracil (5-FU) and illinocetane (formyl) |
| WO2013074840A1 (en) * | 2011-11-15 | 2013-05-23 | Allergan, Inc. | Treatment of dry age related macular degeneration |
| CN104684580A (en) * | 2012-09-05 | 2015-06-03 | 特雷康制药公司 | Antibody formulations and uses thereof |
| JP2015532657A (en) * | 2012-09-05 | 2015-11-12 | トラコン ファーマシューティカルズ、インコーポレイテッド | Antibody preparation and use thereof |
| US10195281B2 (en) | 2012-09-05 | 2019-02-05 | Tracon Pharmaceuticals, Inc. | Antibody formulations and uses thereof |
| EA029214B1 (en) * | 2012-09-05 | 2018-02-28 | Тракон Фармасьютикалз, Инк. | Antibody formulations and uses thereof |
| JP2019059753A (en) * | 2012-09-05 | 2019-04-18 | トラコン ファーマシューティカルズ、インコーポレイテッド | Antibody formulations and uses thereof |
| WO2014039682A1 (en) * | 2012-09-05 | 2014-03-13 | Tracon Pharmaceuticals, Inc. | Antibody formulations and uses thereof |
| AU2013312536B2 (en) * | 2012-09-05 | 2018-04-19 | Tracon Pharmaceuticals, Inc. | Antibody formulations and uses thereof |
| JP2018076350A (en) * | 2012-09-05 | 2018-05-17 | トラコン ファーマシューティカルズ、インコーポレイテッド | Antibody formulations and uses thereof |
| CN108785669A (en) * | 2012-09-05 | 2018-11-13 | 特雷康制药公司 | Antibody preparation and application thereof |
| US10501523B2 (en) | 2014-07-18 | 2019-12-10 | Sanofi | IL-8 level based method of predicting the outcome of colon cancer treatment |
| US11208461B2 (en) | 2014-07-18 | 2021-12-28 | Sanofi | Method for predicting the outcome of a treatment with aflibercept of a patient suspected to suffer from a cancer |
| US10155820B2 (en) | 2014-11-12 | 2018-12-18 | Tracon Pharmaceuticals, Inc. | Anti-endoglin antibodies and uses thereof |
| US9926375B2 (en) | 2014-11-12 | 2018-03-27 | Tracon Pharmaceuticals, Inc. | Anti-endoglin antibodies and uses thereof |
| US10336831B2 (en) | 2014-11-12 | 2019-07-02 | Tracon Pharmaceuticals, Inc. | Use of anti-endoglin antibodies for treating ocular fibrosis |
| WO2016077451A1 (en) * | 2014-11-12 | 2016-05-19 | Tracon Pharmaceuticals, Inc. | Anti-endoglin antibodies and uses thereof |
| CN105821029A (en) * | 2015-01-04 | 2016-08-03 | 彭霞 | Heterologous fusion gene modified cancer cell/dendritic cell fusion tumor vaccine and preparation method thereof |
| US11883432B2 (en) | 2020-12-18 | 2024-01-30 | Century Therapeutics, Inc. | Chimeric antigen receptor system with adaptable receptor specificity |
| WO2023133595A2 (en) | 2022-01-10 | 2023-07-13 | Sana Biotechnology, Inc. | Methods of ex vivo dosing and administration of lipid particles or viral vectors and related systems and uses |
| WO2024081820A1 (en) | 2022-10-13 | 2024-04-18 | Sana Biotechnology, Inc. | Viral particles targeting hematopoietic stem cells |
| WO2024119157A1 (en) | 2022-12-02 | 2024-06-06 | Sana Biotechnology, Inc. | Lipid particles with cofusogens and methods of producing and using the same |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9944714B2 (en) | Endoglin antibodies | |
| US20200087389A1 (en) | Combination therapy of cancer with anti-endoglin antibodies and anti-vegf agents | |
| US20100098692A1 (en) | Humanized Endoglin Antibodies | |
| US20180057602A1 (en) | Combination therapy of cancer with anti-endoglin antibodies and anti-vegf agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080054202.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10821188 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 218724 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2775810 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012532280 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010300668 Country of ref document: AU Ref document number: 2010821188 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201290173 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3643/DELNP/2012 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 20127011203 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2010300668 Country of ref document: AU Date of ref document: 20100929 Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012007318 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112012007318 Country of ref document: BR Kind code of ref document: A2 Effective date: 20120330 |