WO2011023812A1 - Microsomal prostaglandin e synthase-1 (mpges1) inhibitors - Google Patents
Microsomal prostaglandin e synthase-1 (mpges1) inhibitors Download PDFInfo
- Publication number
- WO2011023812A1 WO2011023812A1 PCT/EP2010/062596 EP2010062596W WO2011023812A1 WO 2011023812 A1 WO2011023812 A1 WO 2011023812A1 EP 2010062596 W EP2010062596 W EP 2010062596W WO 2011023812 A1 WO2011023812 A1 WO 2011023812A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- benzo
- carboxamide
- imidazol
- piperidine
- tetrahydrofuran
- Prior art date
Links
- 101710096361 Prostaglandin E synthase Proteins 0.000 title description 28
- 102100033076 Prostaglandin E synthase Human genes 0.000 title description 25
- 239000003112 inhibitor Substances 0.000 title description 6
- 150000001875 compounds Chemical class 0.000 claims abstract description 299
- 150000003839 salts Chemical class 0.000 claims abstract description 61
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 41
- 208000035475 disorder Diseases 0.000 claims abstract description 25
- 206010061218 Inflammation Diseases 0.000 claims abstract description 20
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 19
- 201000011510 cancer Diseases 0.000 claims abstract description 19
- 230000004054 inflammatory process Effects 0.000 claims abstract description 19
- 208000024827 Alzheimer disease Diseases 0.000 claims abstract description 17
- 208000023275 Autoimmune disease Diseases 0.000 claims abstract description 16
- 206010037660 Pyrexia Diseases 0.000 claims abstract description 14
- 208000022531 anorexia Diseases 0.000 claims abstract description 14
- 206010061428 decreased appetite Diseases 0.000 claims abstract description 14
- 208000024172 Cardiovascular disease Diseases 0.000 claims abstract description 13
- 208000027866 inflammatory disease Diseases 0.000 claims abstract description 13
- 230000029058 respiratory gaseous exchange Effects 0.000 claims abstract description 13
- 208000001294 Nociceptive Pain Diseases 0.000 claims abstract description 12
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 12
- DPBWFNDFMCCGGJ-UHFFFAOYSA-N 4-Piperidine carboxamide Chemical compound NC(=O)C1CCNCC1 DPBWFNDFMCCGGJ-UHFFFAOYSA-N 0.000 claims description 550
- 125000003037 imidazol-2-yl group Chemical group [H]N1C([*])=NC([H])=C1[H] 0.000 claims description 245
- 125000000217 alkyl group Chemical group 0.000 claims description 148
- 238000000034 method Methods 0.000 claims description 146
- -1 sulphonamido Chemical group 0.000 claims description 102
- 125000000623 heterocyclic group Chemical group 0.000 claims description 98
- 125000003342 alkenyl group Chemical group 0.000 claims description 63
- 125000006656 (C2-C4) alkenyl group Chemical group 0.000 claims description 59
- 125000000304 alkynyl group Chemical group 0.000 claims description 59
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 55
- 125000003118 aryl group Chemical group 0.000 claims description 48
- 125000006650 (C2-C4) alkynyl group Chemical group 0.000 claims description 45
- 208000002193 Pain Diseases 0.000 claims description 43
- 230000036407 pain Effects 0.000 claims description 42
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 40
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 37
- 229910052736 halogen Inorganic materials 0.000 claims description 37
- 150000002367 halogens Chemical class 0.000 claims description 36
- 239000000460 chlorine Substances 0.000 claims description 34
- 230000002265 prevention Effects 0.000 claims description 34
- 125000003545 alkoxy group Chemical group 0.000 claims description 31
- 239000003814 drug Substances 0.000 claims description 28
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 24
- 125000003302 alkenyloxy group Chemical group 0.000 claims description 21
- 229910052801 chlorine Inorganic materials 0.000 claims description 21
- 125000001302 tertiary amino group Chemical group 0.000 claims description 20
- 125000000392 cycloalkenyl group Chemical group 0.000 claims description 19
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 18
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 18
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 17
- 125000001072 heteroaryl group Chemical group 0.000 claims description 17
- 239000001257 hydrogen Substances 0.000 claims description 17
- 229910052739 hydrogen Inorganic materials 0.000 claims description 17
- 125000005133 alkynyloxy group Chemical group 0.000 claims description 16
- 201000010099 disease Diseases 0.000 claims description 16
- 125000003368 amide group Chemical group 0.000 claims description 15
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 15
- 125000004414 alkyl thio group Chemical group 0.000 claims description 14
- 239000003085 diluting agent Substances 0.000 claims description 14
- 125000005108 alkenylthio group Chemical group 0.000 claims description 13
- 125000005109 alkynylthio group Chemical group 0.000 claims description 13
- 239000003795 chemical substances by application Substances 0.000 claims description 13
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 13
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 claims description 12
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 12
- 201000008482 osteoarthritis Diseases 0.000 claims description 11
- 201000006107 Familial adenomatous polyposis Diseases 0.000 claims description 10
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 10
- 208000034972 Sudden Infant Death Diseases 0.000 claims description 10
- 206010042440 Sudden infant death syndrome Diseases 0.000 claims description 10
- 239000002671 adjuvant Substances 0.000 claims description 10
- 231100000504 carcinogenesis Toxicity 0.000 claims description 10
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 10
- 208000029664 classic familial adenomatous polyposis Diseases 0.000 claims description 10
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 9
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 9
- 229940124597 therapeutic agent Drugs 0.000 claims description 9
- 206010019233 Headaches Diseases 0.000 claims description 8
- 125000000000 cycloalkoxy group Chemical group 0.000 claims description 8
- 229910052731 fluorine Inorganic materials 0.000 claims description 8
- 208000024891 symptom Diseases 0.000 claims description 8
- 206010042674 Swelling Diseases 0.000 claims description 7
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 7
- 230000008961 swelling Effects 0.000 claims description 7
- 201000001320 Atherosclerosis Diseases 0.000 claims description 6
- 206010006187 Breast cancer Diseases 0.000 claims description 6
- 208000026310 Breast neoplasm Diseases 0.000 claims description 6
- 206010013935 Dysmenorrhoea Diseases 0.000 claims description 6
- 208000007571 Ovarian Epithelial Carcinoma Diseases 0.000 claims description 6
- 208000004550 Postoperative Pain Diseases 0.000 claims description 6
- 230000001413 cellular effect Effects 0.000 claims description 6
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 6
- 206010002556 Ankylosing Spondylitis Diseases 0.000 claims description 5
- 208000005623 Carcinogenesis Diseases 0.000 claims description 5
- 201000004624 Dermatitis Diseases 0.000 claims description 5
- 208000005171 Dysmenorrhea Diseases 0.000 claims description 5
- 208000001640 Fibromyalgia Diseases 0.000 claims description 5
- 201000005569 Gout Diseases 0.000 claims description 5
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims description 5
- 206010027476 Metastases Diseases 0.000 claims description 5
- 208000019695 Migraine disease Diseases 0.000 claims description 5
- 206010033128 Ovarian cancer Diseases 0.000 claims description 5
- 201000004681 Psoriasis Diseases 0.000 claims description 5
- 206010038687 Respiratory distress Diseases 0.000 claims description 5
- 125000005225 alkynyloxycarbonyl group Chemical group 0.000 claims description 5
- 208000008784 apnea Diseases 0.000 claims description 5
- 230000002917 arthritic effect Effects 0.000 claims description 5
- 208000006673 asthma Diseases 0.000 claims description 5
- 230000036952 cancer formation Effects 0.000 claims description 5
- 201000001883 cholelithiasis Diseases 0.000 claims description 5
- 239000011737 fluorine Substances 0.000 claims description 5
- 208000001130 gallstones Diseases 0.000 claims description 5
- 230000002496 gastric effect Effects 0.000 claims description 5
- 230000009401 metastasis Effects 0.000 claims description 5
- 206010061289 metastatic neoplasm Diseases 0.000 claims description 5
- GKNWKGBLYCMXTI-UHFFFAOYSA-N methyl 4-[[2-[4-(cyclopentylcarbamoyl)piperidin-1-yl]-5,6-dimethylbenzimidazol-1-yl]methyl]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1CN1C2=CC(C)=C(C)C=C2N=C1N1CCC(C(=O)NC2CCCC2)CC1 GKNWKGBLYCMXTI-UHFFFAOYSA-N 0.000 claims description 5
- 206010027599 migraine Diseases 0.000 claims description 5
- 201000006417 multiple sclerosis Diseases 0.000 claims description 5
- 208000010125 myocardial infarction Diseases 0.000 claims description 5
- 230000010309 neoplastic transformation Effects 0.000 claims description 5
- 201000003068 rheumatic fever Diseases 0.000 claims description 5
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 5
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims description 5
- 230000004614 tumor growth Effects 0.000 claims description 5
- DHNATSRXMRAFHR-UHFFFAOYSA-N 4-[[2-[4-(cyclopentylcarbamoyl)piperidin-1-yl]-5,6-dimethylbenzimidazol-1-yl]methyl]benzoic acid Chemical compound C=1C=C(C(O)=O)C=CC=1CN1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 DHNATSRXMRAFHR-UHFFFAOYSA-N 0.000 claims description 4
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 claims description 4
- 206010009944 Colon cancer Diseases 0.000 claims description 4
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 4
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 4
- 206010065390 Inflammatory pain Diseases 0.000 claims description 4
- 208000010668 atopic eczema Diseases 0.000 claims description 4
- 239000013066 combination product Substances 0.000 claims description 4
- 229940127555 combination product Drugs 0.000 claims description 4
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims description 4
- 201000001245 periodontitis Diseases 0.000 claims description 4
- 201000003651 pulmonary sarcoidosis Diseases 0.000 claims description 4
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 3
- 125000006645 (C3-C4) cycloalkyl group Chemical group 0.000 claims description 3
- 125000006374 C2-C10 alkenyl group Chemical group 0.000 claims description 3
- 125000005865 C2-C10alkynyl group Chemical group 0.000 claims description 3
- 206010012689 Diabetic retinopathy Diseases 0.000 claims description 3
- 201000009273 Endometriosis Diseases 0.000 claims description 3
- 241000124008 Mammalia Species 0.000 claims description 3
- 208000006399 Premature Obstetric Labor Diseases 0.000 claims description 3
- 206010036600 Premature labour Diseases 0.000 claims description 3
- 125000001153 fluoro group Chemical group F* 0.000 claims description 3
- 208000031209 hemophilic arthropathy Diseases 0.000 claims description 3
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 claims description 3
- 208000026440 premature labor Diseases 0.000 claims description 3
- 208000004371 toothache Diseases 0.000 claims description 3
- 230000005747 tumor angiogenesis Effects 0.000 claims description 3
- GFPWRDBDDVDCIW-HXUWFJFHSA-N 1-[1-(4-fluoro-3-methylphenyl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=C(F)C(C)=CC(N2C3=CC(C)=C(C)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 GFPWRDBDDVDCIW-HXUWFJFHSA-N 0.000 claims description 2
- YXUSPDGQUHNDDJ-UHFFFAOYSA-N 2-butyl-1-(5,6-dichloro-1-propan-2-ylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound CCCCC1CC(C(N)=O)CCN1C1=NC2=CC(Cl)=C(Cl)C=C2N1C(C)C YXUSPDGQUHNDDJ-UHFFFAOYSA-N 0.000 claims description 2
- 125000005366 cycloalkylthio group Chemical group 0.000 claims description 2
- KIEWCEJMNLXWEZ-UHFFFAOYSA-N n-butyl-1-(5,6-dimethyl-1-pentylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(C)=C(C)C=C2N(CCCCC)C=1N1CCC(C(=O)NCCCC)CC1 KIEWCEJMNLXWEZ-UHFFFAOYSA-N 0.000 claims description 2
- UTZATXBCBREYKN-UHFFFAOYSA-N n-butyl-1-[5,6-dichloro-1-[(4-fluorophenyl)methyl]benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1CC(C(=O)NCCCC)CCN1C1=NC2=CC(Cl)=C(Cl)C=C2N1CC1=CC=C(F)C=C1 UTZATXBCBREYKN-UHFFFAOYSA-N 0.000 claims description 2
- UBKAFIFYPDMZLG-UHFFFAOYSA-N n-cyclopentyl-1-[1-(4-fluoro-3-methylphenyl)-5,6-dimethylbenzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1=C(F)C(C)=CC(N2C3=CC(C)=C(C)C=C3N=C2N2CCC(CC2)C(=O)NC2CCCC2)=C1 UBKAFIFYPDMZLG-UHFFFAOYSA-N 0.000 claims description 2
- WIEFRMSSSAAMRW-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dimethyl-1-[[4-(1,2,4-triazol-1-yl)phenyl]methyl]benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C=1C=C(N2N=CN=C2)C=CC=1CN1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 WIEFRMSSSAAMRW-UHFFFAOYSA-N 0.000 claims description 2
- CGNLFNOPDHUFHT-OAQYLSRUSA-N 1-[1-(1-benzofuran-6-yl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C=C2C=COC2=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 CGNLFNOPDHUFHT-OAQYLSRUSA-N 0.000 claims 1
- LKVXOGURGIJQRA-HXUWFJFHSA-N 1-[1-(3-chloro-4-methylphenyl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=C(Cl)C(C)=CC=C1N1C2=CC(C)=C(C)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 LKVXOGURGIJQRA-HXUWFJFHSA-N 0.000 claims 1
- KUTKAKFLXPFJAI-OAQYLSRUSA-N 1-[1-(3-fluoro-5-methylphenyl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound CC1=CC(F)=CC(N2C3=CC(C)=C(C)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 KUTKAKFLXPFJAI-OAQYLSRUSA-N 0.000 claims 1
- WULYOITYRHQPEI-HSZRJFAPSA-N 1-[1-(3-tert-butylphenyl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C=CC(C(C)(C)C)=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 WULYOITYRHQPEI-HSZRJFAPSA-N 0.000 claims 1
- PBCBWAABZQXHQD-HXUWFJFHSA-N 1-[1-(4-chloro-3-methylphenyl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=C(Cl)C(C)=CC(N2C3=CC(C)=C(C)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 PBCBWAABZQXHQD-HXUWFJFHSA-N 0.000 claims 1
- SDYUCKCUEFNXCV-OAQYLSRUSA-N 1-[1-(4-fluoro-3,5-dimethylphenyl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C(C)=C(F)C(C)=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 SDYUCKCUEFNXCV-OAQYLSRUSA-N 0.000 claims 1
- QESWGSIJDHDHBS-LJQANCHMSA-N 1-[1-(5-fluoro-6-methylpyridin-2-yl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C=C(F)C(C)=NC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 QESWGSIJDHDHBS-LJQANCHMSA-N 0.000 claims 1
- PAYIWIWSLZFYTR-CQSZACIVSA-N 1-[5,6-dichloro-1-(2,2-difluoro-1,3-benzodioxol-5-yl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=C2OC(F)(F)OC2=CC=C1N(C1=CC(Cl)=C(Cl)C=C1N=1)C=1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 PAYIWIWSLZFYTR-CQSZACIVSA-N 0.000 claims 1
- RBVMWOPDEWCCQB-MRXNPFEDSA-N 1-[5,6-dichloro-1-(3-chloro-4-methylphenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=C(Cl)C(C)=CC=C1N1C2=CC(Cl)=C(Cl)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 RBVMWOPDEWCCQB-MRXNPFEDSA-N 0.000 claims 1
- WLABSQUBYIGXKN-MRXNPFEDSA-N 1-[5,6-dichloro-1-(3-chloro-5-fluorophenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound FC1=CC(Cl)=CC(N2C3=CC(Cl)=C(Cl)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 WLABSQUBYIGXKN-MRXNPFEDSA-N 0.000 claims 1
- MLYUMHAHHSBYHX-QGZVFWFLSA-N 1-[5,6-dichloro-1-(3-fluoro-5-methylphenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound CC1=CC(F)=CC(N2C3=CC(Cl)=C(Cl)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 MLYUMHAHHSBYHX-QGZVFWFLSA-N 0.000 claims 1
- UJDYLOGCORBOCM-CQSZACIVSA-N 1-[5,6-dichloro-1-(4-chloro-3-fluorophenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=C(Cl)C(F)=CC(N2C3=CC(Cl)=C(Cl)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 UJDYLOGCORBOCM-CQSZACIVSA-N 0.000 claims 1
- IQHIUGVMENYZDS-MRXNPFEDSA-N 1-[5,6-dichloro-1-(4-chloro-3-methylphenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=C(Cl)C(C)=CC(N2C3=CC(Cl)=C(Cl)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 IQHIUGVMENYZDS-MRXNPFEDSA-N 0.000 claims 1
- FCCUHWNUEXEHNH-LJQANCHMSA-N 1-[5,6-dichloro-1-(4-cyclopropylphenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C=C(C2CC2)C=CC=1N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 FCCUHWNUEXEHNH-LJQANCHMSA-N 0.000 claims 1
- FAWAHLFYOYMQGN-QGZVFWFLSA-N 1-[5,6-dichloro-1-(4-fluoro-3,5-dimethylphenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound CC1=C(F)C(C)=CC(N2C3=CC(Cl)=C(Cl)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 FAWAHLFYOYMQGN-QGZVFWFLSA-N 0.000 claims 1
- XPCACMURNVCWQN-MRXNPFEDSA-N 1-[5,6-dichloro-1-[3-(trifluoromethyl)phenyl]benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound FC(F)(F)C1=CC=CC(N2C3=CC(Cl)=C(Cl)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 XPCACMURNVCWQN-MRXNPFEDSA-N 0.000 claims 1
- WGUVGOOFSSWQIQ-OAHLLOKOSA-N 1-[5,6-dichloro-1-[4-(trifluoromethyl)pyridin-2-yl]benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound FC(F)(F)C1=CC=NC(N2C3=CC(Cl)=C(Cl)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 WGUVGOOFSSWQIQ-OAHLLOKOSA-N 0.000 claims 1
- RLORHDZEAKAILO-OAHLLOKOSA-N 1-[5,6-dichloro-1-[5-(trifluoromethyl)pyridin-2-yl]benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound N1=CC(C(F)(F)F)=CC=C1N1C2=CC(Cl)=C(Cl)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 RLORHDZEAKAILO-OAHLLOKOSA-N 0.000 claims 1
- WUZLOTOSLXQKAD-CQSZACIVSA-N 1-[5,6-dichloro-1-[6-(trifluoromethyl)pyridin-2-yl]benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound FC(F)(F)C1=CC=CC(N2C3=CC(Cl)=C(Cl)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=N1 WUZLOTOSLXQKAD-CQSZACIVSA-N 0.000 claims 1
- MZPZHTCSWWPKAY-HXUWFJFHSA-N 1-[5,6-dimethyl-1-[3-(trifluoromethyl)phenyl]benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C=CC(C(F)(F)F)=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 MZPZHTCSWWPKAY-HXUWFJFHSA-N 0.000 claims 1
- KTCVKBFKDKMNFA-HXUWFJFHSA-N 1-[5,6-dimethyl-1-[4-(trifluoromethyl)phenyl]benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C=C(C(F)(F)F)C=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 KTCVKBFKDKMNFA-HXUWFJFHSA-N 0.000 claims 1
- RHROPCJXDHCGDT-LJQANCHMSA-N 1-[5,6-dimethyl-1-[4-(trifluoromethyl)pyridin-2-yl]benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C(C(F)(F)F)=CC=NC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 RHROPCJXDHCGDT-LJQANCHMSA-N 0.000 claims 1
- GFPPBAPVOFXTRA-LJQANCHMSA-N 1-[5,6-dimethyl-1-[5-(trifluoromethyl)pyridin-2-yl]benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C=C(C(F)(F)F)C=NC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 GFPPBAPVOFXTRA-LJQANCHMSA-N 0.000 claims 1
- LFNOKOBAZJWFHB-HXUWFJFHSA-N 1-[5-chloro-1-(3,4-dimethylphenyl)-6-methylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=C(C)C(C)=CC=C1N1C2=CC(C)=C(Cl)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 LFNOKOBAZJWFHB-HXUWFJFHSA-N 0.000 claims 1
- AANHWFCQDOLJSM-HXUWFJFHSA-N 1-[5-chloro-1-(4-ethylphenyl)-6-methylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=CC(CC)=CC=C1N1C2=CC(C)=C(Cl)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 AANHWFCQDOLJSM-HXUWFJFHSA-N 0.000 claims 1
- CPUPAFFJHUHLTJ-FQEVSTJZSA-N 1-[6-chloro-1-(3,5-dimethylphenyl)-5-methylbenzimidazol-2-yl]-n-[(3s)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound CC1=CC(C)=CC(N2C3=CC(Cl)=C(C)C=C3N=C2N2CCC(CC2)C(=O)N[C@@H]2COCC2)=C1 CPUPAFFJHUHLTJ-FQEVSTJZSA-N 0.000 claims 1
- LNTOOTJDRIZRAS-HXUWFJFHSA-N 1-[6-chloro-1-(4-ethylphenyl)-5-methylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=CC(CC)=CC=C1N1C2=CC(Cl)=C(C)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 LNTOOTJDRIZRAS-HXUWFJFHSA-N 0.000 claims 1
- XTNJYEFVYIAGBD-OAQYLSRUSA-N 1-[6-chloro-5-methyl-1-(4-propan-2-ylphenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=CC(C(C)C)=CC=C1N1C2=CC(Cl)=C(C)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 XTNJYEFVYIAGBD-OAQYLSRUSA-N 0.000 claims 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 claims 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 274
- 239000011541 reaction mixture Substances 0.000 description 196
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 185
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 168
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 165
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 162
- ZMXDDKWLCZADIW-UHFFFAOYSA-N dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 151
- 229910000024 caesium carbonate Inorganic materials 0.000 description 139
- 239000007787 solid Substances 0.000 description 110
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-diisopropylethylamine Substances CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 109
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 108
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 106
- BERDEBHAJNAUOM-UHFFFAOYSA-N copper(i) oxide Chemical compound [Cu]O[Cu] BERDEBHAJNAUOM-UHFFFAOYSA-N 0.000 description 106
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 93
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 71
- 238000002360 preparation method Methods 0.000 description 69
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 64
- 229940093499 ethyl acetate Drugs 0.000 description 56
- 235000019439 ethyl acetate Nutrition 0.000 description 56
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 53
- 239000012074 organic phase Substances 0.000 description 53
- 239000000741 silica gel Substances 0.000 description 48
- 229910002027 silica gel Inorganic materials 0.000 description 48
- 239000000243 solution Substances 0.000 description 48
- 238000006243 chemical reaction Methods 0.000 description 46
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 45
- 238000004440 column chromatography Methods 0.000 description 45
- 239000000203 mixture Substances 0.000 description 44
- 229920001223 polyethylene glycol Polymers 0.000 description 44
- OYNNYIKCPGWWQH-UHFFFAOYSA-N 1-(oxolan-3-yl)piperidine-4-carboxamide Chemical compound C1CC(C(=O)N)CCN1C1COCC1 OYNNYIKCPGWWQH-UHFFFAOYSA-N 0.000 description 42
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 42
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 42
- 239000002202 Polyethylene glycol Substances 0.000 description 41
- 239000002585 base Substances 0.000 description 32
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 31
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 30
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 30
- 239000000543 intermediate Substances 0.000 description 30
- SRJOCJYGOFTFLH-UHFFFAOYSA-M piperidine-4-carboxylate Chemical compound [O-]C(=O)C1CCNCC1 SRJOCJYGOFTFLH-UHFFFAOYSA-M 0.000 description 30
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 27
- SRJOCJYGOFTFLH-UHFFFAOYSA-N isonipecotic acid Chemical class OC(=O)C1CCNCC1 SRJOCJYGOFTFLH-UHFFFAOYSA-N 0.000 description 27
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 25
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 25
- 239000005725 8-Hydroxyquinoline Substances 0.000 description 25
- 229960003540 oxyquinoline Drugs 0.000 description 25
- MCJGNVYPOGVAJF-UHFFFAOYSA-N quinolin-8-ol Chemical compound C1=CN=C2C(O)=CC=CC2=C1 MCJGNVYPOGVAJF-UHFFFAOYSA-N 0.000 description 25
- 229910052938 sodium sulfate Inorganic materials 0.000 description 25
- 235000011152 sodium sulphate Nutrition 0.000 description 25
- MOWXJLUYGFNTAL-DEOSSOPVSA-N (s)-[2-chloro-4-fluoro-5-(7-morpholin-4-ylquinazolin-4-yl)phenyl]-(6-methoxypyridazin-3-yl)methanol Chemical compound N1=NC(OC)=CC=C1[C@@H](O)C1=CC(C=2C3=CC=C(C=C3N=CN=2)N2CCOCC2)=C(F)C=C1Cl MOWXJLUYGFNTAL-DEOSSOPVSA-N 0.000 description 22
- 238000010828 elution Methods 0.000 description 22
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 22
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 21
- 150000003254 radicals Chemical class 0.000 description 21
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 20
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 17
- 239000002244 precipitate Substances 0.000 description 17
- 238000004007 reversed phase HPLC Methods 0.000 description 17
- 239000000725 suspension Substances 0.000 description 17
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 16
- 229910052799 carbon Inorganic materials 0.000 description 16
- 238000001514 detection method Methods 0.000 description 16
- 235000019253 formic acid Nutrition 0.000 description 16
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 15
- 230000000694 effects Effects 0.000 description 15
- NAMYKGVDVNBCFQ-UHFFFAOYSA-N 2-bromopropane Chemical compound CC(C)Br NAMYKGVDVNBCFQ-UHFFFAOYSA-N 0.000 description 14
- 125000004122 cyclic group Chemical group 0.000 description 14
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 13
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 13
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 13
- 238000002953 preparative HPLC Methods 0.000 description 13
- HUCLGPHNRGLENE-OAHLLOKOSA-N 1-(5,6-dimethyl-1h-benzimidazol-2-yl)-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 HUCLGPHNRGLENE-OAHLLOKOSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 12
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 12
- 125000002619 bicyclic group Chemical group 0.000 description 11
- XEYBRNLFEZDVAW-ARSRFYASSA-N dinoprostone Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEYBRNLFEZDVAW-ARSRFYASSA-N 0.000 description 11
- 239000002904 solvent Substances 0.000 description 11
- KERAHQPIDBAXAI-LLVKDONJSA-N 1-(5,6-dichloro-1h-benzimidazol-2-yl)-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 KERAHQPIDBAXAI-LLVKDONJSA-N 0.000 description 10
- 102000004226 Prostaglandin-E Synthases Human genes 0.000 description 10
- 108090000748 Prostaglandin-E Synthases Proteins 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- 239000000706 filtrate Substances 0.000 description 10
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 10
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 9
- 230000008878 coupling Effects 0.000 description 9
- 238000010168 coupling process Methods 0.000 description 9
- 238000005859 coupling reaction Methods 0.000 description 9
- 210000002683 foot Anatomy 0.000 description 9
- 238000009472 formulation Methods 0.000 description 9
- 235000019341 magnesium sulphate Nutrition 0.000 description 9
- RTJQLPHVGLWGIR-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dimethyl-1h-benzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 RTJQLPHVGLWGIR-UHFFFAOYSA-N 0.000 description 9
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 8
- 150000001408 amides Chemical class 0.000 description 8
- 206010003246 arthritis Diseases 0.000 description 8
- KXVUSQIDCZRUKF-UHFFFAOYSA-N bromocyclobutane Chemical compound BrC1CCC1 KXVUSQIDCZRUKF-UHFFFAOYSA-N 0.000 description 8
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 8
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 8
- 125000002950 monocyclic group Chemical group 0.000 description 8
- 229940002612 prodrug Drugs 0.000 description 8
- 239000000651 prodrug Substances 0.000 description 8
- XEYBRNLFEZDVAW-UHFFFAOYSA-N prostaglandin E2 Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CC=CCCCC(O)=O XEYBRNLFEZDVAW-UHFFFAOYSA-N 0.000 description 8
- 229910052701 rubidium Inorganic materials 0.000 description 8
- 229920006395 saturated elastomer Polymers 0.000 description 8
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 8
- SUQYOUJCBGMSLV-UHFFFAOYSA-N 2,5,6-trichloro-1h-benzimidazole Chemical compound ClC1=C(Cl)C=C2NC(Cl)=NC2=C1 SUQYOUJCBGMSLV-UHFFFAOYSA-N 0.000 description 7
- 229910017333 Mo(CO)6 Inorganic materials 0.000 description 7
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 7
- BXDZOYLPNAIDOC-UHFFFAOYSA-N N-[5-[(5-tert-butyl-1,3-oxazol-2-yl)methylsulfanyl]-1,3-thiazol-2-yl]-1-[2-[2-[2-[2-[2-[[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindol-4-yl]amino]ethoxy]ethoxy]ethoxy]ethylamino]-2-oxoethyl]piperidine-4-carboxamide Chemical compound CC(C)(C)c1cnc(CSc2cnc(NC(=O)C3CCN(CC(=O)NCCOCCOCCOCCNc4cccc5C(=O)N(C6CCC(=O)NC6=O)C(=O)c45)CC3)s2)o1 BXDZOYLPNAIDOC-UHFFFAOYSA-N 0.000 description 7
- 229910002666 PdCl2 Inorganic materials 0.000 description 7
- 150000001412 amines Chemical class 0.000 description 7
- 239000012267 brine Substances 0.000 description 7
- 125000004452 carbocyclyl group Chemical group 0.000 description 7
- 229960002986 dinoprostone Drugs 0.000 description 7
- 230000005764 inhibitory process Effects 0.000 description 7
- 229910052750 molybdenum Inorganic materials 0.000 description 7
- 239000011733 molybdenum Substances 0.000 description 7
- 239000003208 petroleum Substances 0.000 description 7
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 7
- BWHDROKFUHTORW-UHFFFAOYSA-O tritert-butylphosphanium Chemical compound CC(C)(C)[PH+](C(C)(C)C)C(C)(C)C BWHDROKFUHTORW-UHFFFAOYSA-O 0.000 description 7
- 239000003981 vehicle Substances 0.000 description 7
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 6
- ZDSRQBIJVZFVST-UHFFFAOYSA-N 2,5-dichloro-6-fluoro-1h-benzimidazole Chemical compound C1=C(Cl)C(F)=CC2=C1N=C(Cl)N2 ZDSRQBIJVZFVST-UHFFFAOYSA-N 0.000 description 6
- LTWREDHXPZYJOA-UHFFFAOYSA-N 2-chloro-5,6-dimethyl-1h-benzimidazole Chemical compound C1=C(C)C(C)=CC2=C1NC(Cl)=N2 LTWREDHXPZYJOA-UHFFFAOYSA-N 0.000 description 6
- BZXPLADBSZWDIH-FZSMXKCYSA-N 4-methylbenzenesulfonic acid;(3r)-oxolan-3-amine Chemical compound N[C@@H]1CCOC1.CC1=CC=C(S(O)(=O)=O)C=C1 BZXPLADBSZWDIH-FZSMXKCYSA-N 0.000 description 6
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 6
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 6
- 238000007126 N-alkylation reaction Methods 0.000 description 6
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- 239000005557 antagonist Substances 0.000 description 6
- QARVLSVVCXYDNA-UHFFFAOYSA-N bromobenzene Chemical compound BrC1=CC=CC=C1 QARVLSVVCXYDNA-UHFFFAOYSA-N 0.000 description 6
- 230000003197 catalytic effect Effects 0.000 description 6
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 6
- PQMQFEYHSVOFPG-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dichloro-1h-benzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 PQMQFEYHSVOFPG-UHFFFAOYSA-N 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 6
- ZPGVCQYKXIQWTP-UHFFFAOYSA-N 4,7-dimethoxy-1,10-phenanthroline Chemical compound C1=CC2=C(OC)C=CN=C2C2=C1C(OC)=CC=N2 ZPGVCQYKXIQWTP-UHFFFAOYSA-N 0.000 description 5
- IUJPAAPIEURDEZ-UHFFFAOYSA-N 5-(bromomethyl)-2-fluoropyridine Chemical compound FC1=CC=C(CBr)C=N1 IUJPAAPIEURDEZ-UHFFFAOYSA-N 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 5
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 238000006254 arylation reaction Methods 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 238000010549 co-Evaporation Methods 0.000 description 5
- 150000001989 diazonium salts Chemical class 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- ZXWVWQFBCSZTHB-UHFFFAOYSA-N n-cyclopentylpiperidine-4-carboxamide Chemical compound C1CNCCC1C(=O)NC1CCCC1 ZXWVWQFBCSZTHB-UHFFFAOYSA-N 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 150000003180 prostaglandins Chemical class 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 239000012312 sodium hydride Substances 0.000 description 5
- 229910000104 sodium hydride Inorganic materials 0.000 description 5
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 5
- 238000010626 work up procedure Methods 0.000 description 5
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 4
- YZWKKMVJZFACSU-UHFFFAOYSA-N 1-bromopentane Chemical compound CCCCCBr YZWKKMVJZFACSU-UHFFFAOYSA-N 0.000 description 4
- RKMGAJGJIURJSJ-UHFFFAOYSA-N 2,2,6,6-tetramethylpiperidine Chemical compound CC1(C)CCCC(C)(C)N1 RKMGAJGJIURJSJ-UHFFFAOYSA-N 0.000 description 4
- HPHAJHDXIDEYPM-UHFFFAOYSA-N 2,5-dichloro-6-methyl-1h-benzimidazole Chemical compound C1=C(Cl)C(C)=CC2=C1N=C(Cl)N2 HPHAJHDXIDEYPM-UHFFFAOYSA-N 0.000 description 4
- FORSIMRNVYOTJF-UHFFFAOYSA-N 2,5-dichloro-6-methyl-1h-benzimidazole;hydrochloride Chemical compound Cl.C1=C(Cl)C(C)=CC2=C1N=C(Cl)N2 FORSIMRNVYOTJF-UHFFFAOYSA-N 0.000 description 4
- AFABGHUZZDYHJO-UHFFFAOYSA-N 2-Methylpentane Chemical compound CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 4
- 239000000556 agonist Substances 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 229910052794 bromium Inorganic materials 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- 235000010418 carrageenan Nutrition 0.000 description 4
- 239000000679 carrageenan Substances 0.000 description 4
- 229920001525 carrageenan Polymers 0.000 description 4
- 229940113118 carrageenan Drugs 0.000 description 4
- 239000007795 chemical reaction product Substances 0.000 description 4
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 description 4
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 4
- 229910001873 dinitrogen Inorganic materials 0.000 description 4
- RUJPPJYDHHAEEK-UHFFFAOYSA-N ethyl piperidine-4-carboxylate Chemical compound CCOC(=O)C1CCNCC1 RUJPPJYDHHAEEK-UHFFFAOYSA-N 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 239000005457 ice water Substances 0.000 description 4
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- UFHSVTJSUFSCFZ-UHFFFAOYSA-N n-butyl-1-(5,6-dichloro-1h-benzimidazol-2-yl)piperidine-4-carboxamide Chemical compound C1CC(C(=O)NCCCC)CCN1C1=NC2=CC(Cl)=C(Cl)C=C2N1 UFHSVTJSUFSCFZ-UHFFFAOYSA-N 0.000 description 4
- AOCSLAYWBANWCD-UHFFFAOYSA-N n-cyclobutyl-1-(5,6-dichloro-1h-benzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCC1 AOCSLAYWBANWCD-UHFFFAOYSA-N 0.000 description 4
- 238000005016 nuclear Overhauser enhanced spectroscopy Methods 0.000 description 4
- MXQOYLRVSVOCQT-UHFFFAOYSA-N palladium;tritert-butylphosphane Chemical compound [Pd].CC(C)(C)P(C(C)(C)C)C(C)(C)C.CC(C)(C)P(C(C)(C)C)C(C)(C)C MXQOYLRVSVOCQT-UHFFFAOYSA-N 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 238000001953 recrystallisation Methods 0.000 description 4
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- UHVMMEOXYDMDKI-JKYCWFKZSA-L zinc;1-(5-cyanopyridin-2-yl)-3-[(1s,2s)-2-(6-fluoro-2-hydroxy-3-propanoylphenyl)cyclopropyl]urea;diacetate Chemical compound [Zn+2].CC([O-])=O.CC([O-])=O.CCC(=O)C1=CC=C(F)C([C@H]2[C@H](C2)NC(=O)NC=2N=CC(=CC=2)C#N)=C1O UHVMMEOXYDMDKI-JKYCWFKZSA-L 0.000 description 4
- MHOVLDXJDIEEMJ-WCCKRBBISA-N (3s)-oxolan-3-amine;hydrochloride Chemical compound Cl.N[C@H]1CCOC1 MHOVLDXJDIEEMJ-WCCKRBBISA-N 0.000 description 3
- FZWLIUBDVTWBSU-UHFFFAOYSA-N 1-(5-chloro-6-fluoro-1h-benzimidazol-2-yl)-n-cyclopentylpiperidine-4-carboxamide Chemical compound N=1C=2C=C(Cl)C(F)=CC=2NC=1N(CC1)CCC1C(=O)NC1CCCC1 FZWLIUBDVTWBSU-UHFFFAOYSA-N 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 3
- UMLFTCYAQPPZER-UHFFFAOYSA-N 4-(bromomethyl)benzonitrile Chemical compound BrCC1=CC=C(C#N)C=C1 UMLFTCYAQPPZER-UHFFFAOYSA-N 0.000 description 3
- HQBYAESCKCDTDJ-UHFFFAOYSA-N 5,6-dichloro-1,3-dihydrobenzimidazol-2-one Chemical compound ClC1=C(Cl)C=C2NC(O)=NC2=C1 HQBYAESCKCDTDJ-UHFFFAOYSA-N 0.000 description 3
- MJQDHUJPLYIJTL-UHFFFAOYSA-N 5-chloro-4-methyl-2-nitro-n-phenylaniline Chemical compound C1=C(Cl)C(C)=CC([N+]([O-])=O)=C1NC1=CC=CC=C1 MJQDHUJPLYIJTL-UHFFFAOYSA-N 0.000 description 3
- BBWHBSDZHSQECL-UHFFFAOYSA-N 5-chloro-4-methyl-2-nitroaniline Chemical compound CC1=CC([N+]([O-])=O)=C(N)C=C1Cl BBWHBSDZHSQECL-UHFFFAOYSA-N 0.000 description 3
- ZURRVGUCXLHILL-UHFFFAOYSA-N 5-chloro-6-methyl-1,3-dihydrobenzimidazol-2-one Chemical compound C1=C(Cl)C(C)=CC2=C1NC(=O)N2 ZURRVGUCXLHILL-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 229910021592 Copper(II) chloride Inorganic materials 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- 238000000297 Sandmeyer reaction Methods 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical class OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 238000005804 alkylation reaction Methods 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- 239000006286 aqueous extract Substances 0.000 description 3
- 229910052786 argon Inorganic materials 0.000 description 3
- 150000004982 aromatic amines Chemical class 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 125000001246 bromo group Chemical group Br* 0.000 description 3
- BRTFVKHPEHKBQF-UHFFFAOYSA-N bromocyclopentane Chemical compound BrC1CCCC1 BRTFVKHPEHKBQF-UHFFFAOYSA-N 0.000 description 3
- AEILLAXRDHDKDY-UHFFFAOYSA-N bromomethylcyclopropane Chemical compound BrCC1CC1 AEILLAXRDHDKDY-UHFFFAOYSA-N 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 3
- NISGSNTVMOOSJQ-UHFFFAOYSA-N cyclopentanamine Chemical compound NC1CCCC1 NISGSNTVMOOSJQ-UHFFFAOYSA-N 0.000 description 3
- 150000004985 diamines Chemical class 0.000 description 3
- 238000013265 extended release Methods 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 238000003818 flash chromatography Methods 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 125000001183 hydrocarbyl group Chemical group 0.000 description 3
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 3
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- KEELZFXRQAZCJA-UHFFFAOYSA-N n-(5-chloro-4-methyl-2-nitrophenyl)acetamide Chemical compound CC(=O)NC1=CC(Cl)=C(C)C=C1[N+]([O-])=O KEELZFXRQAZCJA-UHFFFAOYSA-N 0.000 description 3
- POKOJURNGFJIJL-UHFFFAOYSA-N n-(cyclohexylmethyl)-1-(5,6-dichloro-1h-benzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NCC1CCCCC1 POKOJURNGFJIJL-UHFFFAOYSA-N 0.000 description 3
- VAJHMIVOZYWUHA-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dichloro-1-pyridin-3-ylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound C=1C=CN=CC=1N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 VAJHMIVOZYWUHA-UHFFFAOYSA-N 0.000 description 3
- SZHIIRWFEQHSDK-UHFFFAOYSA-N n-cyclopropyl-1-(5,6-dichloro-1h-benzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CC1 SZHIIRWFEQHSDK-UHFFFAOYSA-N 0.000 description 3
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 3
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 3
- 239000012044 organic layer Substances 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- YIBNHAJFJUQSRA-YNNPMVKQSA-N prostaglandin H2 Chemical compound C1[C@@H]2OO[C@H]1[C@H](/C=C/[C@@H](O)CCCCC)[C@H]2C\C=C/CCCC(O)=O YIBNHAJFJUQSRA-YNNPMVKQSA-N 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 238000012552 review Methods 0.000 description 3
- 238000006798 ring closing metathesis reaction Methods 0.000 description 3
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 229940124530 sulfonamide Drugs 0.000 description 3
- 235000011149 sulphuric acid Nutrition 0.000 description 3
- 125000000335 thiazolyl group Chemical group 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- KWGRBVOPPLSCSI-WPRPVWTQSA-N (-)-ephedrine Chemical compound CN[C@@H](C)[C@H](O)C1=CC=CC=C1 KWGRBVOPPLSCSI-WPRPVWTQSA-N 0.000 description 2
- GJJFMKBJSRMPLA-HIFRSBDPSA-N (1R,2S)-2-(aminomethyl)-N,N-diethyl-1-phenyl-1-cyclopropanecarboxamide Chemical compound C=1C=CC=CC=1[C@@]1(C(=O)N(CC)CC)C[C@@H]1CN GJJFMKBJSRMPLA-HIFRSBDPSA-N 0.000 description 2
- AQTSNKXEMWZOGA-UHFFFAOYSA-L (2-methanidylphenyl)-bis(2-methylphenyl)phosphane;palladium(2+);diacetate Chemical compound [Pd+2].[Pd+2].CC([O-])=O.CC([O-])=O.CC1=CC=CC=C1P(C=1C(=CC=CC=1)[CH2-])C1=CC=CC=C1C.CC1=CC=CC=C1P(C=1C(=CC=CC=1)[CH2-])C1=CC=CC=C1C AQTSNKXEMWZOGA-UHFFFAOYSA-L 0.000 description 2
- TVYLLZQTGLZFBW-ZBFHGGJFSA-N (R,R)-tramadol Chemical compound COC1=CC=CC([C@]2(O)[C@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-ZBFHGGJFSA-N 0.000 description 2
- ZEUITGRIYCTCEM-KRWDZBQOSA-N (S)-duloxetine Chemical compound C1([C@@H](OC=2C3=CC=CC=C3C=CC=2)CCNC)=CC=CS1 ZEUITGRIYCTCEM-KRWDZBQOSA-N 0.000 description 2
- XNBJPITZKXMICV-UHFFFAOYSA-N 1-(5,6-dichloro-1h-benzimidazol-2-yl)piperidine-4-carboxylic acid Chemical compound C1CC(C(=O)O)CCN1C1=NC2=CC(Cl)=C(Cl)C=C2N1 XNBJPITZKXMICV-UHFFFAOYSA-N 0.000 description 2
- HLVFKOKELQSXIQ-UHFFFAOYSA-N 1-bromo-2-methylpropane Chemical compound CC(C)CBr HLVFKOKELQSXIQ-UHFFFAOYSA-N 0.000 description 2
- SNTWKPAKVQFCCF-UHFFFAOYSA-N 2,3-dihydro-1h-triazole Chemical compound N1NC=CN1 SNTWKPAKVQFCCF-UHFFFAOYSA-N 0.000 description 2
- VOHILFSOWRNVJJ-UHFFFAOYSA-N 2-(bromomethyl)oxolane Chemical compound BrCC1CCCO1 VOHILFSOWRNVJJ-UHFFFAOYSA-N 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- JWYUFVNJZUSCSM-UHFFFAOYSA-N 2-aminobenzimidazole Chemical class C1=CC=C2NC(N)=NC2=C1 JWYUFVNJZUSCSM-UHFFFAOYSA-N 0.000 description 2
- UPSXAPQYNGXVBF-UHFFFAOYSA-N 2-bromobutane Chemical compound CCC(C)Br UPSXAPQYNGXVBF-UHFFFAOYSA-N 0.000 description 2
- IMRWILPUOVGIMU-UHFFFAOYSA-N 2-bromopyridine Chemical compound BrC1=CC=CC=N1 IMRWILPUOVGIMU-UHFFFAOYSA-N 0.000 description 2
- LDJXFZUGZASGIW-UHFFFAOYSA-L 2-diphenylphosphanylethyl(diphenyl)phosphane;palladium(2+);dichloride Chemical compound Cl[Pd]Cl.C=1C=CC=CC=1P(C=1C=CC=CC=1)CCP(C=1C=CC=CC=1)C1=CC=CC=C1 LDJXFZUGZASGIW-UHFFFAOYSA-L 0.000 description 2
- JUYQSMXGDBGGSX-UHFFFAOYSA-N 2-piperidin-1-yl-1h-benzimidazole Chemical class C1CCCCN1C1=NC2=CC=CC=C2N1 JUYQSMXGDBGGSX-UHFFFAOYSA-N 0.000 description 2
- VTOQFOCYBTVOJZ-UHFFFAOYSA-N 3-bromopentane Chemical compound CCC(Br)CC VTOQFOCYBTVOJZ-UHFFFAOYSA-N 0.000 description 2
- XKTYXVDYIKIYJP-UHFFFAOYSA-N 3h-dioxole Chemical compound C1OOC=C1 XKTYXVDYIKIYJP-UHFFFAOYSA-N 0.000 description 2
- KXODOFITUJNMKA-UHFFFAOYSA-N 4-(bromomethyl)benzamide Chemical compound NC(=O)C1=CC=C(CBr)C=C1 KXODOFITUJNMKA-UHFFFAOYSA-N 0.000 description 2
- CQQSQBRPAJSTFB-UHFFFAOYSA-N 4-(bromomethyl)benzoic acid Chemical compound OC(=O)C1=CC=C(CBr)C=C1 CQQSQBRPAJSTFB-UHFFFAOYSA-N 0.000 description 2
- LMOOYAKLEOGKJR-UHFFFAOYSA-N 4-(bromomethyl)oxane Chemical compound BrCC1CCOCC1 LMOOYAKLEOGKJR-UHFFFAOYSA-N 0.000 description 2
- UIAZAEBLHYTWHA-UHFFFAOYSA-N 4-(cyclopentylcarbamoyl)piperidine-1-carboxylic acid Chemical compound C1CN(C(=O)O)CCC1C(=O)NC1CCCC1 UIAZAEBLHYTWHA-UHFFFAOYSA-N 0.000 description 2
- UAMVKOTWSHJOSY-UHFFFAOYSA-N 4-bromo-1-chloro-2-methoxybenzene Chemical compound COC1=CC(Br)=CC=C1Cl UAMVKOTWSHJOSY-UHFFFAOYSA-N 0.000 description 2
- VXKYOKPNAXNAFU-UHFFFAOYSA-N 4-bromo-1-fluoro-2-methylbenzene Chemical compound CC1=CC(Br)=CC=C1F VXKYOKPNAXNAFU-UHFFFAOYSA-N 0.000 description 2
- YPFQISHSXCFZMU-UHFFFAOYSA-N 5,6-dimethyl-1h-benzimidazol-2-amine Chemical compound C1=C(C)C(C)=CC2=C1NC(N)=N2 YPFQISHSXCFZMU-UHFFFAOYSA-N 0.000 description 2
- UDWFSJAYXTXMLM-UHFFFAOYSA-N 5-bromo-2,3-dihydro-1-benzofuran Chemical compound BrC1=CC=C2OCCC2=C1 UDWFSJAYXTXMLM-UHFFFAOYSA-N 0.000 description 2
- CEWRUTPUHGNSEY-UHFFFAOYSA-N 5-chloro-6-methyl-3-phenyl-1h-benzimidazol-2-one Chemical compound C1=2C=C(Cl)C(C)=CC=2NC(=O)N1C1=CC=CC=C1 CEWRUTPUHGNSEY-UHFFFAOYSA-N 0.000 description 2
- IFIHYLCUKYCKRH-UHFFFAOYSA-N 6-bromoquinoline Chemical compound N1=CC=CC2=CC(Br)=CC=C21 IFIHYLCUKYCKRH-UHFFFAOYSA-N 0.000 description 2
- PVFOHMXILQEIHX-UHFFFAOYSA-N 8-[(6-bromo-1,3-benzodioxol-5-yl)sulfanyl]-9-[2-(2-bromophenyl)ethyl]purin-6-amine Chemical compound C=1C=2OCOC=2C=C(Br)C=1SC1=NC=2C(N)=NC=NC=2N1CCC1=CC=CC=C1Br PVFOHMXILQEIHX-UHFFFAOYSA-N 0.000 description 2
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 2
- VOVIALXJUBGFJZ-KWVAZRHASA-N Budesonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(CCC)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O VOVIALXJUBGFJZ-KWVAZRHASA-N 0.000 description 2
- 101150071146 COX2 gene Proteins 0.000 description 2
- 101100114534 Caenorhabditis elegans ctc-2 gene Proteins 0.000 description 2
- 229910021590 Copper(II) bromide Inorganic materials 0.000 description 2
- VMQMZMRVKUZKQL-UHFFFAOYSA-N Cu+ Chemical class [Cu+] VMQMZMRVKUZKQL-UHFFFAOYSA-N 0.000 description 2
- 102100024899 Cytochrome P450 4F8 Human genes 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- 239000007821 HATU Substances 0.000 description 2
- 101000909112 Homo sapiens Cytochrome P450 4F8 Proteins 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 2
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 2
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 2
- SNIOPGDIGTZGOP-UHFFFAOYSA-N Nitroglycerin Chemical compound [O-][N+](=O)OCC(O[N+]([O-])=O)CO[N+]([O-])=O SNIOPGDIGTZGOP-UHFFFAOYSA-N 0.000 description 2
- KYYIDSXMWOZKMP-UHFFFAOYSA-N O-desmethylvenlafaxine Chemical compound C1CCCCC1(O)C(CN(C)C)C1=CC=C(O)C=C1 KYYIDSXMWOZKMP-UHFFFAOYSA-N 0.000 description 2
- 206010030113 Oedema Diseases 0.000 description 2
- 101150000187 PTGS2 gene Proteins 0.000 description 2
- 208000018737 Parkinson disease Diseases 0.000 description 2
- BYPFEZZEUUWMEJ-UHFFFAOYSA-N Pentoxifylline Chemical compound O=C1N(CCCCC(=O)C)C(=O)N(C)C2=C1N(C)C=N2 BYPFEZZEUUWMEJ-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-O Piperidinium(1+) Chemical compound C1CC[NH2+]CC1 NQRYJNQNLNOLGT-UHFFFAOYSA-O 0.000 description 2
- 102100030484 Prostaglandin E synthase 2 Human genes 0.000 description 2
- 108050003514 Prostaglandin E synthase 2 Proteins 0.000 description 2
- 241000219061 Rheum Species 0.000 description 2
- CDMGBJANTYXAIV-UHFFFAOYSA-N SB 203580 Chemical compound C1=CC(S(=O)C)=CC=C1C1=NC(C=2C=CC(F)=CC=2)=C(C=2C=CN=CC=2)N1 CDMGBJANTYXAIV-UHFFFAOYSA-N 0.000 description 2
- 229940121991 Serotonin and norepinephrine reuptake inhibitor Drugs 0.000 description 2
- 229940123445 Tricyclic antidepressant Drugs 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- 208000036142 Viral infection Diseases 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 2
- YCOXTKKNXUZSKD-UHFFFAOYSA-N as-o-xylenol Natural products CC1=CC=C(O)C=C1C YCOXTKKNXUZSKD-UHFFFAOYSA-N 0.000 description 2
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 2
- 150000001556 benzimidazoles Chemical class 0.000 description 2
- FLHFTXCMKFVKRP-UHFFFAOYSA-N bromomethylcyclobutane Chemical compound BrCC1CCC1 FLHFTXCMKFVKRP-UHFFFAOYSA-N 0.000 description 2
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 150000001733 carboxylic acid esters Chemical class 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000005660 chlorination reaction Methods 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 229940111134 coxibs Drugs 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 238000006481 deamination reaction Methods 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 239000012954 diazonium Substances 0.000 description 2
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000003596 drug target Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 229940121360 farnesoid X receptor (fxr) agonists Drugs 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 231100000869 headache Toxicity 0.000 description 2
- 150000002391 heterocyclic compounds Chemical class 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 2
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- 229940071676 hydroxypropylcellulose Drugs 0.000 description 2
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N indane Chemical compound C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- SNHMUERNLJLMHN-UHFFFAOYSA-N iodobenzene Chemical compound IC1=CC=CC=C1 SNHMUERNLJLMHN-UHFFFAOYSA-N 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- MOYKHGMNXAOIAT-JGWLITMVSA-N isosorbide dinitrate Chemical compound [O-][N+](=O)O[C@H]1CO[C@@H]2[C@H](O[N+](=O)[O-])CO[C@@H]21 MOYKHGMNXAOIAT-JGWLITMVSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 102000004311 liver X receptors Human genes 0.000 description 2
- 108090000865 liver X receptors Proteins 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- KBOPZPXVLCULAV-UHFFFAOYSA-N mesalamine Chemical compound NC1=CC=C(O)C(C(O)=O)=C1 KBOPZPXVLCULAV-UHFFFAOYSA-N 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- HNQIVZYLYMDVSB-UHFFFAOYSA-N methanesulfonimidic acid Chemical compound CS(N)(=O)=O HNQIVZYLYMDVSB-UHFFFAOYSA-N 0.000 description 2
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 2
- NQMRYBIKMRVZLB-UHFFFAOYSA-N methylamine hydrochloride Chemical compound [Cl-].[NH3+]C NQMRYBIKMRVZLB-UHFFFAOYSA-N 0.000 description 2
- 239000008108 microcrystalline cellulose Substances 0.000 description 2
- 229940016286 microcrystalline cellulose Drugs 0.000 description 2
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 2
- 230000003228 microsomal effect Effects 0.000 description 2
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 2
- SGIWFELWJPNFDH-UHFFFAOYSA-N n-(2,2,2-trifluoroethyl)-n-{4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl}benzenesulfonamide Chemical compound C1=CC(C(O)(C(F)(F)F)C(F)(F)F)=CC=C1N(CC(F)(F)F)S(=O)(=O)C1=CC=CC=C1 SGIWFELWJPNFDH-UHFFFAOYSA-N 0.000 description 2
- UGUXISYXXFZCSR-UHFFFAOYSA-N n-(oxolan-3-yl)piperidin-1-ium-4-carboxamide;2,2,2-trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F.C1C[NH2+]CCC1C(=O)NC1CCOC1 UGUXISYXXFZCSR-UHFFFAOYSA-N 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- JKBSXBTYNRZBDY-UHFFFAOYSA-N n-butyl-1-(5,6-dimethyl-1h-benzimidazol-2-yl)piperidine-4-carboxamide Chemical compound C1CC(C(=O)NCCCC)CCN1C1=NC2=CC(C)=C(C)C=C2N1 JKBSXBTYNRZBDY-UHFFFAOYSA-N 0.000 description 2
- IRGWKLGKOKVKQZ-UHFFFAOYSA-N n-butyl-1-[5,6-dichloro-1-[(6-fluoropyridin-3-yl)methyl]benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1CC(C(=O)NCCCC)CCN1C1=NC2=CC(Cl)=C(Cl)C=C2N1CC1=CC=C(F)N=C1 IRGWKLGKOKVKQZ-UHFFFAOYSA-N 0.000 description 2
- FJRVJTXZXXIUGN-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-(2-methoxyethyl)benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound N=1C2=CC(Cl)=C(Cl)C=C2N(CCOC)C=1N(CC1)CCC1C(=O)NC1CCCC1 FJRVJTXZXXIUGN-UHFFFAOYSA-N 0.000 description 2
- ZYQAOFVBPMUSRR-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-[(4-cyanophenyl)methyl]benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C=1C=C(C#N)C=CC=1CN1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 ZYQAOFVBPMUSRR-UHFFFAOYSA-N 0.000 description 2
- NHARWJZZRREPOE-UHFFFAOYSA-N n-cyclopentylpiperidin-1-ium-4-carboxamide;2,2,2-trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F.C1C[NH2+]CCC1C(=O)NC1CCCC1 NHARWJZZRREPOE-UHFFFAOYSA-N 0.000 description 2
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- WPHGSKGZRAQSGP-UHFFFAOYSA-N norcarane Chemical compound C1CCCC2CC21 WPHGSKGZRAQSGP-UHFFFAOYSA-N 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- MHOVLDXJDIEEMJ-UHFFFAOYSA-N oxolan-3-amine;hydrochloride Chemical compound Cl.NC1CCOC1 MHOVLDXJDIEEMJ-UHFFFAOYSA-N 0.000 description 2
- 102000002574 p38 Mitogen-Activated Protein Kinases Human genes 0.000 description 2
- 108010068338 p38 Mitogen-Activated Protein Kinases Proteins 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000007310 pathophysiology Effects 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 2
- UXCDUFKZSUBXGM-UHFFFAOYSA-N phosphoric tribromide Chemical compound BrP(Br)(Br)=O UXCDUFKZSUBXGM-UHFFFAOYSA-N 0.000 description 2
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 2
- FAIAAWCVCHQXDN-UHFFFAOYSA-N phosphorus trichloride Chemical compound ClP(Cl)Cl FAIAAWCVCHQXDN-UHFFFAOYSA-N 0.000 description 2
- QQJYAXDCMMXECR-UHFFFAOYSA-N piperidine-1-sulfonyl chloride Chemical class ClS(=O)(=O)N1CCCCC1 QQJYAXDCMMXECR-UHFFFAOYSA-N 0.000 description 2
- QYSPLQLAKJAUJT-UHFFFAOYSA-N piroxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=CC=CC=N1 QYSPLQLAKJAUJT-UHFFFAOYSA-N 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- FGIUAXJPYTZDNR-UHFFFAOYSA-N potassium nitrate Chemical compound [K+].[O-][N+]([O-])=O FGIUAXJPYTZDNR-UHFFFAOYSA-N 0.000 description 2
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 230000000770 proinflammatory effect Effects 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 230000000171 quenching effect Effects 0.000 description 2
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000007127 saponification reaction Methods 0.000 description 2
- 125000003548 sec-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 229940095743 selective estrogen receptor modulator Drugs 0.000 description 2
- 239000000333 selective estrogen receptor modulator Substances 0.000 description 2
- 229940124834 selective serotonin reuptake inhibitor Drugs 0.000 description 2
- 239000012896 selective serotonin reuptake inhibitor Substances 0.000 description 2
- 239000003775 serotonin noradrenalin reuptake inhibitor Substances 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 235000010288 sodium nitrite Nutrition 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 125000005017 substituted alkenyl group Chemical group 0.000 description 2
- 125000004426 substituted alkynyl group Chemical group 0.000 description 2
- 150000003456 sulfonamides Chemical class 0.000 description 2
- 239000001117 sulphuric acid Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- PYEZUOWDTVMEBF-UHFFFAOYSA-N tert-butyl 4-(1,3-thiazol-2-ylmethylcarbamoyl)piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1C(=O)NCC1=NC=CS1 PYEZUOWDTVMEBF-UHFFFAOYSA-N 0.000 description 2
- FPFRXZUFFZEQHL-UHFFFAOYSA-N tert-butyl 4-(oxolan-3-ylcarbamoyl)piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1C(=O)NC1COCC1 FPFRXZUFFZEQHL-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 2
- 230000001131 transforming effect Effects 0.000 description 2
- 239000003029 tricyclic antidepressant agent Substances 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 102000003390 tumor necrosis factor Human genes 0.000 description 2
- 230000009385 viral infection Effects 0.000 description 2
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 2
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- ZFSXKSSWYSZPGQ-TYSVMGFPSA-N (1r,2r)-2-aminocyclopentan-1-ol;hydrochloride Chemical compound Cl.N[C@@H]1CCC[C@H]1O ZFSXKSSWYSZPGQ-TYSVMGFPSA-N 0.000 description 1
- MAYZWDRUFKUGGP-VIFPVBQESA-N (3s)-1-[5-tert-butyl-3-[(1-methyltetrazol-5-yl)methyl]triazolo[4,5-d]pyrimidin-7-yl]pyrrolidin-3-ol Chemical compound CN1N=NN=C1CN1C2=NC(C(C)(C)C)=NC(N3C[C@@H](O)CC3)=C2N=N1 MAYZWDRUFKUGGP-VIFPVBQESA-N 0.000 description 1
- BVYUYDBWQMSOKM-UHFFFAOYSA-N (4-bromophenyl)methanamine;hydron;chloride Chemical compound Cl.NCC1=CC=C(Br)C=C1 BVYUYDBWQMSOKM-UHFFFAOYSA-N 0.000 description 1
- IIFVWLUQBAIPMJ-UHFFFAOYSA-N (4-fluorophenyl)methanamine Chemical compound NCC1=CC=C(F)C=C1 IIFVWLUQBAIPMJ-UHFFFAOYSA-N 0.000 description 1
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 1
- 125000006688 (C2-C4) heterocyclyl group Chemical group 0.000 description 1
- 125000006689 (C2-C5) heterocyclyl group Chemical group 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- ZGYIXVSQHOKQRZ-COIATFDQSA-N (e)-n-[4-[3-chloro-4-(pyridin-2-ylmethoxy)anilino]-3-cyano-7-[(3s)-oxolan-3-yl]oxyquinolin-6-yl]-4-(dimethylamino)but-2-enamide Chemical compound N#CC1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 ZGYIXVSQHOKQRZ-COIATFDQSA-N 0.000 description 1
- SWJPEBQEEAHIGZ-UHFFFAOYSA-N 1,4-dibromobenzene Chemical compound BrC1=CC=C(Br)C=C1 SWJPEBQEEAHIGZ-UHFFFAOYSA-N 0.000 description 1
- BDXOOYSXJZJQEI-UHFFFAOYSA-N 1-(1,3-thiazol-2-ylmethyl)piperidine-4-carboxamide Chemical compound C1CC(C(=O)N)CCN1CC1=NC=CS1 BDXOOYSXJZJQEI-UHFFFAOYSA-N 0.000 description 1
- APWRZPQBPCAXFP-UHFFFAOYSA-N 1-(1-oxo-2H-isoquinolin-5-yl)-5-(trifluoromethyl)-N-[2-(trifluoromethyl)pyridin-4-yl]pyrazole-4-carboxamide Chemical compound O=C1NC=CC2=C(C=CC=C12)N1N=CC(=C1C(F)(F)F)C(=O)NC1=CC(=NC=C1)C(F)(F)F APWRZPQBPCAXFP-UHFFFAOYSA-N 0.000 description 1
- OGFAWKRXZLGJSK-UHFFFAOYSA-N 1-(2,4-dihydroxyphenyl)-2-(4-nitrophenyl)ethanone Chemical compound OC1=CC(O)=CC=C1C(=O)CC1=CC=C([N+]([O-])=O)C=C1 OGFAWKRXZLGJSK-UHFFFAOYSA-N 0.000 description 1
- OFYVIGTWSQPCLF-UHFFFAOYSA-N 1-(4-methylphenyl)-3-azabicyclo[3.1.0]hexane Chemical compound C1=CC(C)=CC=C1C1(CNC2)C2C1 OFYVIGTWSQPCLF-UHFFFAOYSA-N 0.000 description 1
- ATPHPJIEKZUQGN-UHFFFAOYSA-N 1-(5,6-dichloro-1-propan-2-ylbenzimidazol-2-yl)-n-(2-propan-2-yloxyethyl)piperidine-4-carboxamide Chemical compound C1CC(C(=O)NCCOC(C)C)CCN1C1=NC2=CC(Cl)=C(Cl)C=C2N1C(C)C ATPHPJIEKZUQGN-UHFFFAOYSA-N 0.000 description 1
- ABJUSUMGSQXYTG-UHFFFAOYSA-N 1-(5,6-dichloro-1-propan-2-ylbenzimidazol-2-yl)-n-(oxan-4-ylmethyl)piperidine-4-carboxamide Chemical compound N=1C2=CC(Cl)=C(Cl)C=C2N(C(C)C)C=1N(CC1)CCC1C(=O)NCC1CCOCC1 ABJUSUMGSQXYTG-UHFFFAOYSA-N 0.000 description 1
- IWZWZMMMADMQCY-UHFFFAOYSA-N 1-(5,6-dichloro-1-propan-2-ylbenzimidazol-2-yl)-n-(oxolan-3-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(Cl)=C(Cl)C=C2N(C(C)C)C=1N(CC1)CCC1C(=O)NC1CCOC1 IWZWZMMMADMQCY-UHFFFAOYSA-N 0.000 description 1
- WZZCCOLETRYASZ-UHFFFAOYSA-N 1-(5,6-dichloro-1-propan-2-ylbenzimidazol-2-yl)-n-propylpiperidine-4-carboxamide Chemical compound C1CC(C(=O)NCCC)CCN1C1=NC2=CC(Cl)=C(Cl)C=C2N1C(C)C WZZCCOLETRYASZ-UHFFFAOYSA-N 0.000 description 1
- YNAURAUNEGKZLL-UHFFFAOYSA-N 1-(5,6-dichloro-1-pyridin-2-ylbenzimidazol-2-yl)-n-(oxolan-3-yl)piperidine-4-carboxamide Chemical compound C=1C=CC=NC=1N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCOC1 YNAURAUNEGKZLL-UHFFFAOYSA-N 0.000 description 1
- KXZFMVBNFFIKSK-UHFFFAOYSA-N 1-(5,6-dichloro-1-pyridin-3-ylbenzimidazol-2-yl)-n-(oxolan-3-yl)piperidine-4-carboxamide Chemical compound C=1C=CN=CC=1N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCOC1 KXZFMVBNFFIKSK-UHFFFAOYSA-N 0.000 description 1
- CPCIYGHUPLRYBW-UHFFFAOYSA-N 1-(5,6-dimethyl-1-naphthalen-2-ylbenzimidazol-2-yl)-n-(oxolan-3-yl)piperidine-4-carboxamide Chemical compound C=1C=C2C=CC=CC2=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCOC1 CPCIYGHUPLRYBW-UHFFFAOYSA-N 0.000 description 1
- NDGZOPIUBSLMBR-UHFFFAOYSA-N 1-(5,6-dimethyl-1-phenylbenzimidazol-2-yl)-n-(oxolan-3-yl)piperidine-4-carboxamide Chemical compound C=1C=CC=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCOC1 NDGZOPIUBSLMBR-UHFFFAOYSA-N 0.000 description 1
- NDGZOPIUBSLMBR-HXUWFJFHSA-N 1-(5,6-dimethyl-1-phenylbenzimidazol-2-yl)-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C=CC=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 NDGZOPIUBSLMBR-HXUWFJFHSA-N 0.000 description 1
- PBKHDVFSCKWZAK-UHFFFAOYSA-N 1-(5,6-dimethyl-1-propan-2-ylbenzimidazol-2-yl)-n-(3-propan-2-yloxypropyl)piperidine-4-carboxamide Chemical compound C1CC(C(=O)NCCCOC(C)C)CCN1C1=NC2=CC(C)=C(C)C=C2N1C(C)C PBKHDVFSCKWZAK-UHFFFAOYSA-N 0.000 description 1
- NXFNTAPSQKMOFJ-UHFFFAOYSA-N 1-(5,6-dimethyl-1-propan-2-ylbenzimidazol-2-yl)-n-[[4-(2h-tetrazol-5-yl)phenyl]methyl]piperidine-4-carboxamide Chemical compound N=1C2=CC(C)=C(C)C=C2N(C(C)C)C=1N(CC1)CCC1C(=O)NCC(C=C1)=CC=C1C=1N=NNN=1 NXFNTAPSQKMOFJ-UHFFFAOYSA-N 0.000 description 1
- FOMSSWCPKRCVAH-JOCHJYFZSA-N 1-(5,6-dimethyl-1-quinolin-6-ylbenzimidazol-2-yl)-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C=C2N=CC=CC2=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 FOMSSWCPKRCVAH-JOCHJYFZSA-N 0.000 description 1
- YJJNBQFAMSUSQV-UHFFFAOYSA-N 1-(5-chloro-1-cyclobutyl-6-fluorobenzimidazol-2-yl)-n-cyclopentylpiperidine-4-carboxamide Chemical compound C1CC(C(=O)NC2CCCC2)CCN1C1=NC=2C=C(Cl)C(F)=CC=2N1C1CCC1 YJJNBQFAMSUSQV-UHFFFAOYSA-N 0.000 description 1
- IBRAXCLPSYLNGS-UHFFFAOYSA-N 1-(6-chloro-1-cyclobutyl-5-fluorobenzimidazol-2-yl)-n-cyclopentylpiperidine-4-carboxamide Chemical compound C1CCC1N1C=2C=C(Cl)C(F)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 IBRAXCLPSYLNGS-UHFFFAOYSA-N 0.000 description 1
- KVSVNRFSKRFPIL-UHFFFAOYSA-N 1-(bromomethyl)-3,5-difluorobenzene Chemical compound FC1=CC(F)=CC(CBr)=C1 KVSVNRFSKRFPIL-UHFFFAOYSA-N 0.000 description 1
- ABDDQTDRAHXHOC-QMMMGPOBSA-N 1-[(7s)-5,7-dihydro-4h-thieno[2,3-c]pyran-7-yl]-n-methylmethanamine Chemical compound CNC[C@@H]1OCCC2=C1SC=C2 ABDDQTDRAHXHOC-QMMMGPOBSA-N 0.000 description 1
- UVYVTXXOAAUVMT-OAQYLSRUSA-N 1-[1-(2,3-dihydro-1-benzofuran-5-yl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C=C2OCCC2=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 UVYVTXXOAAUVMT-OAQYLSRUSA-N 0.000 description 1
- OVENOPZXOZOXSA-JOCHJYFZSA-N 1-[1-(3,4-dimethylphenyl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=C(C)C(C)=CC=C1N1C2=CC(C)=C(C)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 OVENOPZXOZOXSA-JOCHJYFZSA-N 0.000 description 1
- RRQCRSUZUJESRF-XMMPIXPASA-N 1-[1-(3,5-diethylphenyl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound CCC1=CC(CC)=CC(N2C3=CC(C)=C(C)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 RRQCRSUZUJESRF-XMMPIXPASA-N 0.000 description 1
- XOGHMIXYVRYMQM-HXUWFJFHSA-N 1-[1-(3-chlorophenyl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C=CC(Cl)=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 XOGHMIXYVRYMQM-HXUWFJFHSA-N 0.000 description 1
- BVELVGXABQRQAR-LJQANCHMSA-N 1-[1-(4-chloro-3-methoxyphenyl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=C(Cl)C(OC)=CC(N2C3=CC(C)=C(C)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 BVELVGXABQRQAR-LJQANCHMSA-N 0.000 description 1
- KLGJVJRUBQYIFP-JOCHJYFZSA-N 1-[1-(4-ethylphenyl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=CC(CC)=CC=C1N1C2=CC(C)=C(C)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 KLGJVJRUBQYIFP-JOCHJYFZSA-N 0.000 description 1
- CTYIGHHIUXMTBR-LJQANCHMSA-N 1-[1-(4-tert-butylphenyl)-5,6-dichlorobenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=CC(C(C)(C)C)=CC=C1N1C2=CC(Cl)=C(Cl)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 CTYIGHHIUXMTBR-LJQANCHMSA-N 0.000 description 1
- LHMFLFNAZNVMNB-HSZRJFAPSA-N 1-[1-(4-tert-butylphenyl)-5,6-dimethylbenzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C=C(C(C)(C)C)C=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 LHMFLFNAZNVMNB-HSZRJFAPSA-N 0.000 description 1
- PTRJLCTXMCQQIS-QGZVFWFLSA-N 1-[5,6-dichloro-1-(2,3-dihydro-1-benzofuran-5-yl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C=1C=C2OCCC2=CC=1N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)N[C@@H]1CCOC1 PTRJLCTXMCQQIS-QGZVFWFLSA-N 0.000 description 1
- GORFVEWHWQVUFR-MRXNPFEDSA-N 1-[5,6-dichloro-1-(3-fluorophenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound FC1=CC=CC(N2C3=CC(Cl)=C(Cl)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 GORFVEWHWQVUFR-MRXNPFEDSA-N 0.000 description 1
- XCRZESALBRMESK-UHFFFAOYSA-N 1-[5,6-dichloro-1-(3-methoxyphenyl)benzimidazol-2-yl]-n-(oxolan-3-yl)piperidine-4-carboxamide Chemical compound COC1=CC=CC(N2C3=CC(Cl)=C(Cl)C=C3N=C2N2CCC(CC2)C(=O)NC2COCC2)=C1 XCRZESALBRMESK-UHFFFAOYSA-N 0.000 description 1
- XCRZESALBRMESK-MRXNPFEDSA-N 1-[5,6-dichloro-1-(3-methoxyphenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound COC1=CC=CC(N2C3=CC(Cl)=C(Cl)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 XCRZESALBRMESK-MRXNPFEDSA-N 0.000 description 1
- LNDBPIRPTJRKGG-OAHLLOKOSA-N 1-[5,6-dichloro-1-(4-chloro-3-methoxyphenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=C(Cl)C(OC)=CC(N2C3=CC(Cl)=C(Cl)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 LNDBPIRPTJRKGG-OAHLLOKOSA-N 0.000 description 1
- DPRSVEIGCQIWJS-UHFFFAOYSA-N 1-[5,6-dichloro-1-(4-cyanophenyl)benzimidazol-2-yl]-n-(oxolan-3-yl)piperidine-4-carboxamide Chemical compound C=1C=C(C#N)C=CC=1N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCOC1 DPRSVEIGCQIWJS-UHFFFAOYSA-N 0.000 description 1
- MULVILUPCZHFFE-MRXNPFEDSA-N 1-[5,6-dichloro-1-(4-fluorophenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=CC(F)=CC=C1N1C2=CC(Cl)=C(Cl)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 MULVILUPCZHFFE-MRXNPFEDSA-N 0.000 description 1
- ANJVKPDIHJMOEN-MRXNPFEDSA-N 1-[5,6-dichloro-1-(4-methoxyphenyl)benzimidazol-2-yl]-N-[(3R)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound COc1ccc(cc1)-n1c(nc2cc(Cl)c(Cl)cc12)N1CCC(CC1)C(=O)N[C@@H]1CCOC1 ANJVKPDIHJMOEN-MRXNPFEDSA-N 0.000 description 1
- UMHKACGWDVZXPX-UHFFFAOYSA-N 1-[5,6-dichloro-1-(4-methylphenyl)benzimidazol-2-yl]-n-(oxolan-3-yl)piperidine-4-carboxamide Chemical compound C1=CC(C)=CC=C1N1C2=CC(Cl)=C(Cl)C=C2N=C1N1CCC(C(=O)NC2COCC2)CC1 UMHKACGWDVZXPX-UHFFFAOYSA-N 0.000 description 1
- IBAZOPRPKIWIIQ-UHFFFAOYSA-N 1-[5,6-dichloro-1-[(4-fluorophenyl)methyl]benzimidazol-2-yl]-n-(oxolan-3-yl)piperidine-4-carboxamide Chemical compound C1=CC(F)=CC=C1CN1C2=CC(Cl)=C(Cl)C=C2N=C1N1CCC(C(=O)NC2COCC2)CC1 IBAZOPRPKIWIIQ-UHFFFAOYSA-N 0.000 description 1
- AXXFKKHSSPOYQB-OAHLLOKOSA-N 1-[5,6-dichloro-1-[4-(trifluoromethoxy)phenyl]benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=CC(OC(F)(F)F)=CC=C1N1C2=CC(Cl)=C(Cl)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 AXXFKKHSSPOYQB-OAHLLOKOSA-N 0.000 description 1
- VHFJDSGWLUUCNE-OAQYLSRUSA-N 1-[5,6-dimethyl-1-(3-methylphenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound CC1=CC=CC(N2C3=CC(C)=C(C)C=C3N=C2N2CCC(CC2)C(=O)N[C@H]2COCC2)=C1 VHFJDSGWLUUCNE-OAQYLSRUSA-N 0.000 description 1
- ZBPICYVZXKTOSV-OAQYLSRUSA-N 1-[5,6-dimethyl-1-(4-methylphenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=CC(C)=CC=C1N1C2=CC(C)=C(C)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 ZBPICYVZXKTOSV-OAQYLSRUSA-N 0.000 description 1
- ZBPICYVZXKTOSV-NRFANRHFSA-N 1-[5,6-dimethyl-1-(4-methylphenyl)benzimidazol-2-yl]-n-[(3s)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=CC(C)=CC=C1N1C2=CC(C)=C(C)C=C2N=C1N1CCC(C(=O)N[C@@H]2COCC2)CC1 ZBPICYVZXKTOSV-NRFANRHFSA-N 0.000 description 1
- ZVVJTVWDEAMCBO-HSZRJFAPSA-N 1-[5,6-dimethyl-1-(4-propan-2-ylphenyl)benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=CC(C(C)C)=CC=C1N1C2=CC(C)=C(C)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 ZVVJTVWDEAMCBO-HSZRJFAPSA-N 0.000 description 1
- POZBTCDKJQBVTC-XMMPIXPASA-N 1-[5,6-dimethyl-1-[4-(2-methylpropyl)phenyl]benzimidazol-2-yl]-n-[(3r)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=CC(CC(C)C)=CC=C1N1C2=CC(C)=C(C)C=C2N=C1N1CCC(C(=O)N[C@H]2COCC2)CC1 POZBTCDKJQBVTC-XMMPIXPASA-N 0.000 description 1
- KGVFUGFNVKNKSC-IBGZPJMESA-N 1-[6-chloro-5-methyl-1-(4-methylphenyl)benzimidazol-2-yl]-n-[(3s)-oxolan-3-yl]piperidine-4-carboxamide Chemical compound C1=CC(C)=CC=C1N1C2=CC(Cl)=C(C)C=C2N=C1N1CCC(C(=O)N[C@@H]2COCC2)CC1 KGVFUGFNVKNKSC-IBGZPJMESA-N 0.000 description 1
- BHKKSKOHRFHHIN-MRVPVSSYSA-N 1-[[2-[(1R)-1-aminoethyl]-4-chlorophenyl]methyl]-2-sulfanylidene-5H-pyrrolo[3,2-d]pyrimidin-4-one Chemical compound N[C@H](C)C1=C(CN2C(NC(C3=C2C=CN3)=O)=S)C=CC(=C1)Cl BHKKSKOHRFHHIN-MRVPVSSYSA-N 0.000 description 1
- ZBTMRBYMKUEVEU-UHFFFAOYSA-N 1-bromo-4-methylbenzene Chemical compound CC1=CC=C(Br)C=C1 ZBTMRBYMKUEVEU-UHFFFAOYSA-N 0.000 description 1
- LSXKDWGTSHCFPP-UHFFFAOYSA-N 1-bromoheptane Chemical compound CCCCCCCBr LSXKDWGTSHCFPP-UHFFFAOYSA-N 0.000 description 1
- VMKOFRJSULQZRM-UHFFFAOYSA-N 1-bromooctane Chemical compound CCCCCCCCBr VMKOFRJSULQZRM-UHFFFAOYSA-N 0.000 description 1
- CYNYIHKIEHGYOZ-UHFFFAOYSA-N 1-bromopropane Chemical compound CCCBr CYNYIHKIEHGYOZ-UHFFFAOYSA-N 0.000 description 1
- MLRVZFYXUZQSRU-UHFFFAOYSA-N 1-chlorohexane Chemical compound CCCCCCCl MLRVZFYXUZQSRU-UHFFFAOYSA-N 0.000 description 1
- UDHAWRUAECEBHC-UHFFFAOYSA-N 1-iodo-4-methylbenzene Chemical compound CC1=CC=C(I)C=C1 UDHAWRUAECEBHC-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical group C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- HFFXLYHRNRKAPM-UHFFFAOYSA-N 2,4,5-trichloro-n-(5-methyl-1,2-oxazol-3-yl)benzenesulfonamide Chemical compound O1C(C)=CC(NS(=O)(=O)C=2C(=CC(Cl)=C(Cl)C=2)Cl)=N1 HFFXLYHRNRKAPM-UHFFFAOYSA-N 0.000 description 1
- OFPWMRMIFDHXFE-UHFFFAOYSA-N 2-(bromomethyl)pyridine Chemical compound BrCC1=CC=CC=N1 OFPWMRMIFDHXFE-UHFFFAOYSA-N 0.000 description 1
- QJYHMWJJNWZWCH-UHFFFAOYSA-N 2-(bromomethyl)pyridine;hydrochloride Chemical compound Cl.BrCC1=CC=CC=N1 QJYHMWJJNWZWCH-UHFFFAOYSA-N 0.000 description 1
- ZHAKAIBZKYHXJS-UHFFFAOYSA-N 2-(chloromethyl)-1,3-thiazole Chemical compound ClCC1=NC=CS1 ZHAKAIBZKYHXJS-UHFFFAOYSA-N 0.000 description 1
- JPMRGPPMXHGKRO-UHFFFAOYSA-N 2-(chloromethyl)pyridine hydrochloride Chemical compound Cl.ClCC1=CC=CC=N1 JPMRGPPMXHGKRO-UHFFFAOYSA-N 0.000 description 1
- KMODISUYWZPVGV-UHFFFAOYSA-N 2-bromo-6-methoxypyridine Chemical compound COC1=CC=CC(Br)=N1 KMODISUYWZPVGV-UHFFFAOYSA-N 0.000 description 1
- WRYDGMWSKBGVHS-UHFFFAOYSA-N 2-bromo-n,n-diethylethanamine Chemical compound CCN(CC)CCBr WRYDGMWSKBGVHS-UHFFFAOYSA-N 0.000 description 1
- APSMUYYLXZULMS-UHFFFAOYSA-N 2-bromonaphthalene Chemical compound C1=CC=CC2=CC(Br)=CC=C21 APSMUYYLXZULMS-UHFFFAOYSA-N 0.000 description 1
- LGAJYTCRJPCZRJ-UHFFFAOYSA-N 2-bromopentane Chemical compound CCCC(C)Br LGAJYTCRJPCZRJ-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- 125000006040 2-hexenyl group Chemical group 0.000 description 1
- ASUDFOJKTJLAIK-UHFFFAOYSA-N 2-methoxyethanamine Chemical compound COCCN ASUDFOJKTJLAIK-UHFFFAOYSA-N 0.000 description 1
- NCQSUQGHEHGLMY-UHFFFAOYSA-N 2-methyl-2-thionitrosooxypropane Chemical compound CC(C)(C)ON=S NCQSUQGHEHGLMY-UHFFFAOYSA-N 0.000 description 1
- WLAMNBDJUVNPJU-UHFFFAOYSA-N 2-methylbutyric acid Chemical compound CCC(C)C(O)=O WLAMNBDJUVNPJU-UHFFFAOYSA-N 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical compound BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- USECIYVEPXUVHT-UHFFFAOYSA-N 2-propan-2-yloxyethanamine Chemical compound CC(C)OCCN USECIYVEPXUVHT-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- GPWHFPWZAPOYNO-UHFFFAOYSA-N 3,3-dimethylbutan-1-amine Chemical compound CC(C)(C)CCN GPWHFPWZAPOYNO-UHFFFAOYSA-N 0.000 description 1
- YFEOSTXFQCDCAR-UHFFFAOYSA-N 3-(1,2,3,6-tetrahydropyridin-4-yl)-1h-benzimidazol-2-one Chemical compound O=C1NC2=CC=CC=C2N1C1=CCNCC1 YFEOSTXFQCDCAR-UHFFFAOYSA-N 0.000 description 1
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 1
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 1
- KKGUMGWNFARLSL-UHFFFAOYSA-N 3-(bromomethyl)pentane Chemical compound CCC(CC)CBr KKGUMGWNFARLSL-UHFFFAOYSA-N 0.000 description 1
- CAWYWWPWSAMGBV-UHFFFAOYSA-N 3-[(4-methylsulfonylphenyl)methylidene]pentane-2,4-dione Chemical compound CC(=O)C(C(C)=O)=CC1=CC=C(S(C)(=O)=O)C=C1 CAWYWWPWSAMGBV-UHFFFAOYSA-N 0.000 description 1
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 1
- HETQOWPCHKBEFO-UHFFFAOYSA-N 3-bromo-1-methylpyridin-1-ium Chemical compound C[N+]1=CC=CC(Br)=C1 HETQOWPCHKBEFO-UHFFFAOYSA-N 0.000 description 1
- NYPYPOZNGOXYSU-UHFFFAOYSA-N 3-bromopyridine Chemical compound BrC1=CC=CN=C1 NYPYPOZNGOXYSU-UHFFFAOYSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- 125000006041 3-hexenyl group Chemical group 0.000 description 1
- XDELKSRGBLWMBA-UHFFFAOYSA-N 3-iodopyridine Chemical compound IC1=CC=CN=C1 XDELKSRGBLWMBA-UHFFFAOYSA-N 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- VHYUNSUGCNKWSO-UHFFFAOYSA-N 3-propan-2-yloxypropan-1-amine Chemical compound CC(C)OCCCN VHYUNSUGCNKWSO-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- YGKDISJLDVGNOR-UHFFFAOYSA-N 4-(chloromethyl)-2-methyl-1,3-thiazole;hydrochloride Chemical compound Cl.CC1=NC(CCl)=CS1 YGKDISJLDVGNOR-UHFFFAOYSA-N 0.000 description 1
- LGTJOYILXBLAGP-UHFFFAOYSA-N 4-(oxolan-3-ylcarbamoyl)piperidine-1-carboxylic acid Chemical compound C1CN(C(=O)O)CCC1C(=O)NC1COCC1 LGTJOYILXBLAGP-UHFFFAOYSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- HQSCPPCMBMFJJN-UHFFFAOYSA-N 4-bromobenzonitrile Chemical compound BrC1=CC=C(C#N)C=C1 HQSCPPCMBMFJJN-UHFFFAOYSA-N 0.000 description 1
- BCJVBDBJSMFBRW-UHFFFAOYSA-N 4-diphenylphosphanylbutyl(diphenyl)phosphane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCCP(C=1C=CC=CC=1)C1=CC=CC=C1 BCJVBDBJSMFBRW-UHFFFAOYSA-N 0.000 description 1
- XOKDXPVXJWTSRM-UHFFFAOYSA-N 4-iodobenzonitrile Chemical compound IC1=CC=C(C#N)C=C1 XOKDXPVXJWTSRM-UHFFFAOYSA-N 0.000 description 1
- 125000006051 4-methyl-2-pentenyl group Chemical group 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- IRPVABHDSJVBNZ-RTHVDDQRSA-N 5-[1-(cyclopropylmethyl)-5-[(1R,5S)-3-(oxetan-3-yl)-3-azabicyclo[3.1.0]hexan-6-yl]pyrazol-3-yl]-3-(trifluoromethyl)pyridin-2-amine Chemical compound C1=C(C(F)(F)F)C(N)=NC=C1C1=NN(CC2CC2)C(C2[C@@H]3CN(C[C@@H]32)C2COC2)=C1 IRPVABHDSJVBNZ-RTHVDDQRSA-N 0.000 description 1
- MLLYPEORWDUMKB-UHFFFAOYSA-N 5-chloro-6-fluoro-1,3-dihydrobenzimidazol-2-one Chemical compound C1=C(Cl)C(F)=CC2=C1NC(=O)N2 MLLYPEORWDUMKB-UHFFFAOYSA-N 0.000 description 1
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 1
- NOYFLUFQGFNMRB-UHFFFAOYSA-N 6-bromoquinoxaline Chemical compound N1=CC=NC2=CC(Br)=CC=C21 NOYFLUFQGFNMRB-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 102000013455 Amyloid beta-Peptides Human genes 0.000 description 1
- 108010090849 Amyloid beta-Peptides Proteins 0.000 description 1
- 108010087765 Antipain Proteins 0.000 description 1
- 102100038495 Bile acid receptor Human genes 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- 229940127597 CGRP antagonist Drugs 0.000 description 1
- 101100496968 Caenorhabditis elegans ctc-1 gene Proteins 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 1
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 1
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 108010008165 Etanercept Proteins 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- 229940122957 Histamine H2 receptor antagonist Drugs 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000678026 Homo sapiens Alpha-1-antichymotrypsin Proteins 0.000 description 1
- 101000603876 Homo sapiens Bile acid receptor Proteins 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical class C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- 102000042256 MAPEG family Human genes 0.000 description 1
- 108091077604 MAPEG family Proteins 0.000 description 1
- 208000030070 Malignant epithelial tumor of ovary Diseases 0.000 description 1
- 206010064912 Malignant transformation Diseases 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 108700027649 Mitogen-Activated Protein Kinase 3 Proteins 0.000 description 1
- 102100024192 Mitogen-activated protein kinase 3 Human genes 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- PHSPJQZRQAJPPF-UHFFFAOYSA-N N-alpha-Methylhistamine Chemical compound CNCCC1=CN=CN1 PHSPJQZRQAJPPF-UHFFFAOYSA-N 0.000 description 1
- 101100221647 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) cox-1 gene Proteins 0.000 description 1
- IDRGFNPZDVBSSE-UHFFFAOYSA-N OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F Chemical compound OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F IDRGFNPZDVBSSE-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 206010061328 Ovarian epithelial cancer Diseases 0.000 description 1
- SGUKUZOVHSFKPH-UHFFFAOYSA-N PGG2 Natural products C1C2OOC1C(C=CC(OO)CCCCC)C2CC=CCCCC(O)=O SGUKUZOVHSFKPH-UHFFFAOYSA-N 0.000 description 1
- 101150062589 PTGS1 gene Proteins 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 102100038280 Prostaglandin G/H synthase 2 Human genes 0.000 description 1
- 108050003267 Prostaglandin G/H synthase 2 Proteins 0.000 description 1
- 102000004005 Prostaglandin-endoperoxide synthases Human genes 0.000 description 1
- 108090000459 Prostaglandin-endoperoxide synthases Proteins 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 206010063837 Reperfusion injury Diseases 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- GUGOEEXESWIERI-UHFFFAOYSA-N Terfenadine Chemical compound C1=CC(C(C)(C)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 GUGOEEXESWIERI-UHFFFAOYSA-N 0.000 description 1
- AUYYCJSJGJYCDS-LBPRGKRZSA-N Thyrolar Chemical class IC1=CC(C[C@H](N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-LBPRGKRZSA-N 0.000 description 1
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 1
- 102000013814 Wnt Human genes 0.000 description 1
- 108050003627 Wnt Proteins 0.000 description 1
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 1
- 229940124532 absorption promoter Drugs 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 229960002964 adalimumab Drugs 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 125000005087 alkynylcarbonyl group Chemical group 0.000 description 1
- 125000005139 alkynylsulfonyl group Chemical group 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910021502 aluminium hydroxide Inorganic materials 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 230000000954 anitussive effect Effects 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000001387 anti-histamine Effects 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- SDNYTAYICBFYFH-TUFLPTIASA-N antipain Chemical compound NC(N)=NCCC[C@@H](C=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 SDNYTAYICBFYFH-TUFLPTIASA-N 0.000 description 1
- 229940124584 antitussives Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 229940114079 arachidonic acid Drugs 0.000 description 1
- 235000021342 arachidonic acid Nutrition 0.000 description 1
- 150000001502 aryl halides Chemical class 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 210000001130 astrocyte Anatomy 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000002047 benzodioxolyl group Chemical group O1OC(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004622 benzoxazinyl group Chemical group O1NC(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 1
- OFYVIGTWSQPCLF-NWDGAFQWSA-N bicifadine Chemical compound C1=CC(C)=CC=C1[C@@]1(CNC2)[C@H]2C1 OFYVIGTWSQPCLF-NWDGAFQWSA-N 0.000 description 1
- 229950010365 bicifadine Drugs 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- GPRLTFBKWDERLU-UHFFFAOYSA-N bicyclo[2.2.2]octane Chemical compound C1CC2CCC1CC2 GPRLTFBKWDERLU-UHFFFAOYSA-N 0.000 description 1
- DCRRIOWFXXDTHV-UHFFFAOYSA-N bicyclo[4.2.0]oct-3-ene Chemical compound C1C=CCC2CCC21 DCRRIOWFXXDTHV-UHFFFAOYSA-N 0.000 description 1
- AWIIAPZLCXBIFR-UHFFFAOYSA-N bicyclo[4.3.3]dodecane Chemical compound C1CCC2CCCC1CCCC2 AWIIAPZLCXBIFR-UHFFFAOYSA-N 0.000 description 1
- AIUBUBHXJWIWOX-UHFFFAOYSA-N bicyclo[6.2.0]dec-4-yne Chemical compound C1CC2CCC#CCCC12 AIUBUBHXJWIWOX-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 150000004074 biphenyls Chemical class 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- AQNQQHJNRPDOQV-UHFFFAOYSA-N bromocyclohexane Chemical compound BrC1CCCCC1 AQNQQHJNRPDOQV-UHFFFAOYSA-N 0.000 description 1
- 229940124630 bronchodilator Drugs 0.000 description 1
- 239000000168 bronchodilator agent Substances 0.000 description 1
- 229960004436 budesonide Drugs 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- KMGBZBJJOKUPIA-UHFFFAOYSA-N butyl iodide Chemical compound CCCCI KMGBZBJJOKUPIA-UHFFFAOYSA-N 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 239000003735 calcitonin gene related peptide receptor antagonist Substances 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- FQNHWXHRAUXLFU-UHFFFAOYSA-N carbon monoxide;tungsten Chemical group [W].[O+]#[C-].[O+]#[C-].[O+]#[C-].[O+]#[C-].[O+]#[C-].[O+]#[C-] FQNHWXHRAUXLFU-UHFFFAOYSA-N 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 230000021164 cell adhesion Effects 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- 210000001612 chondrocyte Anatomy 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- WDQPAMHFFCXSNU-UHFFFAOYSA-N clofazimine Chemical compound C12=CC=CC=C2N=C2C=C(NC=3C=CC(Cl)=CC=3)C(=NC(C)C)C=C2N1C1=CC=C(Cl)C=C1 WDQPAMHFFCXSNU-UHFFFAOYSA-N 0.000 description 1
- WDQPAMHFFCXSNU-BGABXYSRSA-N clofazimine Chemical compound C12=CC=CC=C2N=C2C=C(NC=3C=CC(Cl)=CC=3)C(=N/C(C)C)/C=C2N1C1=CC=C(Cl)C=C1 WDQPAMHFFCXSNU-BGABXYSRSA-N 0.000 description 1
- 229960004287 clofazimine Drugs 0.000 description 1
- 230000008045 co-localization Effects 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 210000004922 colonic epithelial cell Anatomy 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 1
- NKNDPYCGAZPOFS-UHFFFAOYSA-M copper(i) bromide Chemical compound Br[Cu] NKNDPYCGAZPOFS-UHFFFAOYSA-M 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- KZZKOVLJUKWSKX-UHFFFAOYSA-N cyclobutanamine Chemical compound NC1CCC1 KZZKOVLJUKWSKX-UHFFFAOYSA-N 0.000 description 1
- CFBGXYDUODCMNS-UHFFFAOYSA-N cyclobutene Chemical compound C1CC=C1 CFBGXYDUODCMNS-UHFFFAOYSA-N 0.000 description 1
- MZJCFRKLOXHQIL-UHFFFAOYSA-N cyclodeca-1,3-diene Chemical compound C1CCCC=CC=CCC1 MZJCFRKLOXHQIL-UHFFFAOYSA-N 0.000 description 1
- LMGZGXSXHCMSAA-UHFFFAOYSA-N cyclodecane Chemical compound C1CCCCCCCCC1 LMGZGXSXHCMSAA-UHFFFAOYSA-N 0.000 description 1
- BGMJEAAFHCTKBY-UHFFFAOYSA-N cyclodecyne Chemical compound C1CCCCC#CCCC1 BGMJEAAFHCTKBY-UHFFFAOYSA-N 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- PAFZNILMFXTMIY-UHFFFAOYSA-N cyclohexylamine Chemical compound NC1CCCCC1 PAFZNILMFXTMIY-UHFFFAOYSA-N 0.000 description 1
- AVKNGPAMCBSNSO-UHFFFAOYSA-N cyclohexylmethanamine Chemical compound NCC1CCCCC1 AVKNGPAMCBSNSO-UHFFFAOYSA-N 0.000 description 1
- ZPWOOKQUDFIEIX-UHFFFAOYSA-N cyclooctyne Chemical compound C1CCCC#CCC1 ZPWOOKQUDFIEIX-UHFFFAOYSA-N 0.000 description 1
- MSXJLEPWWWZERM-UHFFFAOYSA-N cyclopentylmethanamine;hydrochloride Chemical compound Cl.NCC1CCCC1 MSXJLEPWWWZERM-UHFFFAOYSA-N 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- KWGRBVOPPLSCSI-UHFFFAOYSA-N d-ephedrine Natural products CNC(C)C(O)C1=CC=CC=C1 KWGRBVOPPLSCSI-UHFFFAOYSA-N 0.000 description 1
- 230000009615 deamination Effects 0.000 description 1
- 239000000850 decongestant Substances 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- 229960001623 desvenlafaxine Drugs 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- 238000006193 diazotization reaction Methods 0.000 description 1
- YSSSPARMOAYJTE-UHFFFAOYSA-N dibenzo-18-crown-6 Chemical compound O1CCOCCOC2=CC=CC=C2OCCOCCOC2=CC=CC=C21 YSSSPARMOAYJTE-UHFFFAOYSA-N 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical group OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 125000004925 dihydropyridyl group Chemical group N1(CC=CC=C1)* 0.000 description 1
- 125000005054 dihydropyrrolyl group Chemical group [H]C1=C([H])C([H])([H])C([H])([H])N1* 0.000 description 1
- XHFGWHUWQXTGAT-UHFFFAOYSA-N dimethylamine hydrochloride Natural products CNC(C)C XHFGWHUWQXTGAT-UHFFFAOYSA-N 0.000 description 1
- IQDGSYLLQPDQDV-UHFFFAOYSA-N dimethylazanium;chloride Chemical compound Cl.CNC IQDGSYLLQPDQDV-UHFFFAOYSA-N 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- AMTWCFIAVKBGOD-UHFFFAOYSA-N dioxosilane;methoxy-dimethyl-trimethylsilyloxysilane Chemical compound O=[Si]=O.CO[Si](C)(C)O[Si](C)(C)C AMTWCFIAVKBGOD-UHFFFAOYSA-N 0.000 description 1
- PCHPORCSPXIHLZ-UHFFFAOYSA-N diphenhydramine hydrochloride Chemical compound [Cl-].C=1C=CC=CC=1C(OCC[NH+](C)C)C1=CC=CC=C1 PCHPORCSPXIHLZ-UHFFFAOYSA-N 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 125000005883 dithianyl group Chemical group 0.000 description 1
- 125000005411 dithiolanyl group Chemical group S1SC(CC1)* 0.000 description 1
- 239000002934 diuretic Substances 0.000 description 1
- 230000001882 diuretic effect Effects 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 229960002866 duloxetine Drugs 0.000 description 1
- 239000012039 electrophile Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 229960002179 ephedrine Drugs 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 229960000403 etanercept Drugs 0.000 description 1
- REYMQWGKSXTIIW-UHFFFAOYSA-N ethyl 1-[6-chloro-5-methyl-1-(4-methylphenyl)benzimidazol-2-yl]piperidine-4-carboxylate Chemical compound C1CC(C(=O)OCC)CCN1C1=NC2=CC(C)=C(Cl)C=C2N1C1=CC=C(C)C=C1 REYMQWGKSXTIIW-UHFFFAOYSA-N 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- UPBDXRPQPOWRKR-UHFFFAOYSA-N furan-2,5-dione;methoxyethene Chemical compound COC=C.O=C1OC(=O)C=C1 UPBDXRPQPOWRKR-UHFFFAOYSA-N 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229940124750 glucocorticoid receptor agonist Drugs 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 229960003711 glyceryl trinitrate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000003106 haloaryl group Chemical group 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 210000000548 hind-foot Anatomy 0.000 description 1
- 239000003485 histamine H2 receptor antagonist Substances 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 229960000598 infliximab Drugs 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000002348 inosinate dehydrogenase inhibitor Substances 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 230000021995 interleukin-8 production Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 229960000201 isosorbide dinitrate Drugs 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 229960000816 magnesium hydroxide Drugs 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940057948 magnesium stearate Drugs 0.000 description 1
- 230000036212 malign transformation Effects 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 229960004963 mesalazine Drugs 0.000 description 1
- 229910001507 metal halide Inorganic materials 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- NLWBJPPMPLPZIE-UHFFFAOYSA-N methyl 4-(bromomethyl)benzoate Chemical compound COC(=O)C1=CC=C(CBr)C=C1 NLWBJPPMPLPZIE-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 229960000600 milnacipran Drugs 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 229940014456 mycophenolate Drugs 0.000 description 1
- IGLVCWPPISIXPR-UHFFFAOYSA-N n-(3-chloro-4-methylphenyl)acetamide Chemical compound CC(=O)NC1=CC=C(C)C(Cl)=C1 IGLVCWPPISIXPR-UHFFFAOYSA-N 0.000 description 1
- NTSBNRBQHMLBSH-UHFFFAOYSA-N n-(cyclohexylmethyl)-1-(5,6-dichloro-1-propan-2-ylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(Cl)=C(Cl)C=C2N(C(C)C)C=1N(CC1)CCC1C(=O)NCC1CCCCC1 NTSBNRBQHMLBSH-UHFFFAOYSA-N 0.000 description 1
- PFAGTNLQRHUFIV-UHFFFAOYSA-N n-(cyclopentylmethyl)-1-(5,6-dichloro-1-propan-2-ylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(Cl)=C(Cl)C=C2N(C(C)C)C=1N(CC1)CCC1C(=O)NCC1CCCC1 PFAGTNLQRHUFIV-UHFFFAOYSA-N 0.000 description 1
- IAUBEDFGMRRQDA-UHFFFAOYSA-N n-[(4-bromophenyl)methyl]-1-(5,6-dimethyl-1-propan-2-ylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(C)=C(C)C=C2N(C(C)C)C=1N(CC1)CCC1C(=O)NCC1=CC=C(Br)C=C1 IAUBEDFGMRRQDA-UHFFFAOYSA-N 0.000 description 1
- PEURNYLYKROBKX-UHFFFAOYSA-N n-butyl-1-(5,6-dichloro-1-pentylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(Cl)=C(Cl)C=C2N(CCCCC)C=1N1CCC(C(=O)NCCCC)CC1 PEURNYLYKROBKX-UHFFFAOYSA-N 0.000 description 1
- WGGLXXIZFXXYJM-UHFFFAOYSA-N n-butylpiperidin-1-ium-4-carboxamide;2,2,2-trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F.CCCCNC(=O)C1CC[NH2+]CC1 WGGLXXIZFXXYJM-UHFFFAOYSA-N 0.000 description 1
- GKCROKLQKJMQMJ-UHFFFAOYSA-N n-butylpiperidine-4-carboxamide Chemical compound CCCCNC(=O)C1CCNCC1 GKCROKLQKJMQMJ-UHFFFAOYSA-N 0.000 description 1
- IIECXSHLHNRLHB-UHFFFAOYSA-N n-cyclobutyl-1-(5,6-dichloro-1-propan-2-ylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(Cl)=C(Cl)C=C2N(C(C)C)C=1N(CC1)CCC1C(=O)NC1CCC1 IIECXSHLHNRLHB-UHFFFAOYSA-N 0.000 description 1
- CYGYOOROGDUQNL-UHFFFAOYSA-N n-cyclopentyl-1-(1-ethyl-5,6-dimethylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(C)=C(C)C=C2N(CC)C=1N(CC1)CCC1C(=O)NC1CCCC1 CYGYOOROGDUQNL-UHFFFAOYSA-N 0.000 description 1
- LHBXMUPBMHXCOD-UHFFFAOYSA-N n-cyclopentyl-1-(1-heptyl-5,6-dimethylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(C)=C(C)C=C2N(CCCCCCC)C=1N(CC1)CCC1C(=O)NC1CCCC1 LHBXMUPBMHXCOD-UHFFFAOYSA-N 0.000 description 1
- WJQRYYYNZPDOAS-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dichloro-1-cyclobutylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound C1CCC1N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 WJQRYYYNZPDOAS-UHFFFAOYSA-N 0.000 description 1
- VCTDQIKYXRNTRY-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dichloro-1-ethylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(Cl)=C(Cl)C=C2N(CC)C=1N(CC1)CCC1C(=O)NC1CCCC1 VCTDQIKYXRNTRY-UHFFFAOYSA-N 0.000 description 1
- CWODCRQMUZNACY-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dichloro-1-pentan-3-ylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(Cl)=C(Cl)C=C2N(C(CC)CC)C=1N(CC1)CCC1C(=O)NC1CCCC1 CWODCRQMUZNACY-UHFFFAOYSA-N 0.000 description 1
- IRRZQPYWBLONPU-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dichloro-1-phenylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound C=1C=CC=CC=1N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 IRRZQPYWBLONPU-UHFFFAOYSA-N 0.000 description 1
- ADEXTRNNZJPGDM-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dichloro-1-propan-2-ylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(Cl)=C(Cl)C=C2N(C(C)C)C=1N(CC1)CCC1C(=O)NC1CCCC1 ADEXTRNNZJPGDM-UHFFFAOYSA-N 0.000 description 1
- UZYAGLJTGBYQRU-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dimethyl-1-octylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(C)=C(C)C=C2N(CCCCCCCC)C=1N(CC1)CCC1C(=O)NC1CCCC1 UZYAGLJTGBYQRU-UHFFFAOYSA-N 0.000 description 1
- XMNXOCVDRFCZEH-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dimethyl-1-phenylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound C=1C=CC=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 XMNXOCVDRFCZEH-UHFFFAOYSA-N 0.000 description 1
- CXEBSTJMPTXJLR-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dimethyl-1-prop-2-ynylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound C#CCN1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 CXEBSTJMPTXJLR-UHFFFAOYSA-N 0.000 description 1
- FPPVJOTXZIRXTF-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dimethyl-1-propan-2-ylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(C)=C(C)C=C2N(C(C)C)C=1N(CC1)CCC1C(=O)NC1CCCC1 FPPVJOTXZIRXTF-UHFFFAOYSA-N 0.000 description 1
- MGPJDZPPQMKCGA-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dimethyl-1-propylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(C)=C(C)C=C2N(CCC)C=1N(CC1)CCC1C(=O)NC1CCCC1 MGPJDZPPQMKCGA-UHFFFAOYSA-N 0.000 description 1
- UOHXDXMCYGXIOS-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dimethyl-1-quinolin-6-ylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound C=1C=C2N=CC=CC2=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 UOHXDXMCYGXIOS-UHFFFAOYSA-N 0.000 description 1
- XPZNATSSVDNFOW-UHFFFAOYSA-N n-cyclopentyl-1-(5,6-dimethyl-1-quinoxalin-6-ylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound C=1C=C2N=CC=NC2=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 XPZNATSSVDNFOW-UHFFFAOYSA-N 0.000 description 1
- RZDLRFZAKIFDMC-UHFFFAOYSA-N n-cyclopentyl-1-[1-(2,2-dimethylpropyl)-5,6-dimethylbenzimidazol-2-yl]piperidine-4-carboxamide Chemical compound CC(C)(C)CN1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 RZDLRFZAKIFDMC-UHFFFAOYSA-N 0.000 description 1
- XAJOVLIJEPYUOX-UHFFFAOYSA-N n-cyclopentyl-1-[1-(2-ethylbutyl)-5,6-dimethylbenzimidazol-2-yl]piperidine-4-carboxamide Chemical compound N=1C2=CC(C)=C(C)C=C2N(CC(CC)CC)C=1N(CC1)CCC1C(=O)NC1CCCC1 XAJOVLIJEPYUOX-UHFFFAOYSA-N 0.000 description 1
- OSNNFRJSRLAOCH-UHFFFAOYSA-N n-cyclopentyl-1-[1-(2-methoxyethyl)-5,6-dimethylbenzimidazol-2-yl]piperidine-4-carboxamide Chemical compound N=1C2=CC(C)=C(C)C=C2N(CCOC)C=1N(CC1)CCC1C(=O)NC1CCCC1 OSNNFRJSRLAOCH-UHFFFAOYSA-N 0.000 description 1
- WNJBMNJSDYTEMC-UHFFFAOYSA-N n-cyclopentyl-1-[1-(3,4-dimethylphenyl)-5,6-dimethylbenzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1=C(C)C(C)=CC=C1N1C2=CC(C)=C(C)C=C2N=C1N1CCC(C(=O)NC2CCCC2)CC1 WNJBMNJSDYTEMC-UHFFFAOYSA-N 0.000 description 1
- LFKBLISSMQAGFE-UHFFFAOYSA-N n-cyclopentyl-1-[1-(4-ethylphenyl)-5,6-dimethylbenzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1=CC(CC)=CC=C1N1C2=CC(C)=C(C)C=C2N=C1N1CCC(C(=O)NC2CCCC2)CC1 LFKBLISSMQAGFE-UHFFFAOYSA-N 0.000 description 1
- JIHNAYSQRMTYQH-UHFFFAOYSA-N n-cyclopentyl-1-[1-(4-fluorophenyl)-5,6-dimethylbenzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C=1C=C(F)C=CC=1N1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 JIHNAYSQRMTYQH-UHFFFAOYSA-N 0.000 description 1
- WYPVDDLVGZQNHX-UHFFFAOYSA-N n-cyclopentyl-1-[1-[(3,5-difluorophenyl)methyl]-5,6-dimethylbenzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C=1C(F)=CC(F)=CC=1CN1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 WYPVDDLVGZQNHX-UHFFFAOYSA-N 0.000 description 1
- AKQZFOVMTKOKOP-UHFFFAOYSA-N n-cyclopentyl-1-[1-[[4-(dimethylcarbamoyl)phenyl]methyl]-5,6-dimethylbenzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1=CC(C(=O)N(C)C)=CC=C1CN1C2=CC(C)=C(C)C=C2N=C1N1CCC(C(=O)NC2CCCC2)CC1 AKQZFOVMTKOKOP-UHFFFAOYSA-N 0.000 description 1
- HMCQFDBOAAZVJV-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-(1-phenylethyl)benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1CC(C(=O)NC2CCCC2)CCN1C1=NC2=CC(Cl)=C(Cl)C=C2N1C(C)C1=CC=CC=C1 HMCQFDBOAAZVJV-UHFFFAOYSA-N 0.000 description 1
- IYXSCLWGDVEWPQ-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-(4-cyanophenyl)benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C=1C=C(C#N)C=CC=1N1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 IYXSCLWGDVEWPQ-UHFFFAOYSA-N 0.000 description 1
- MJOXVZGUYNQZQU-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-(4-fluorophenyl)benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1=CC(F)=CC=C1N1C2=CC(Cl)=C(Cl)C=C2N=C1N1CCC(C(=O)NC2CCCC2)CC1 MJOXVZGUYNQZQU-UHFFFAOYSA-N 0.000 description 1
- XBQDOIZXBDGSPT-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-(cyclopropylmethyl)benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1CC1CN1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 XBQDOIZXBDGSPT-UHFFFAOYSA-N 0.000 description 1
- GUOFHIWBMRNUJZ-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-(oxan-4-ylmethyl)benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1COCCC1CN1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 GUOFHIWBMRNUJZ-UHFFFAOYSA-N 0.000 description 1
- XJXHTAOMEUZOTH-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-(oxolan-2-ylmethyl)benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1CCOC1CN1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 XJXHTAOMEUZOTH-UHFFFAOYSA-N 0.000 description 1
- KPBAQBSHHWCJQO-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-(pyridin-2-ylmethyl)benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C=1C=CC=NC=1CN1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 KPBAQBSHHWCJQO-UHFFFAOYSA-N 0.000 description 1
- MKLPRYYGTGADMV-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-(pyridin-3-ylmethyl)benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C=1C=CN=CC=1CN1C=2C=C(Cl)C(Cl)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 MKLPRYYGTGADMV-UHFFFAOYSA-N 0.000 description 1
- YTLCAONEWCYPSK-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-[(4-fluorophenyl)methyl]benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1=CC(F)=CC=C1CN1C2=CC(Cl)=C(Cl)C=C2N=C1N1CCC(C(=O)NC2CCCC2)CC1 YTLCAONEWCYPSK-UHFFFAOYSA-N 0.000 description 1
- DCVZQSNNRTXLQX-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-[(6-fluoropyridin-3-yl)methyl]benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1=NC(F)=CC=C1CN1C2=CC(Cl)=C(Cl)C=C2N=C1N1CCC(C(=O)NC2CCCC2)CC1 DCVZQSNNRTXLQX-UHFFFAOYSA-N 0.000 description 1
- UNQFCNCNTPSWHN-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-[2-(diethylamino)ethyl]benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound N=1C2=CC(Cl)=C(Cl)C=C2N(CCN(CC)CC)C=1N(CC1)CCC1C(=O)NC1CCCC1 UNQFCNCNTPSWHN-UHFFFAOYSA-N 0.000 description 1
- SZWCZEBNKZEYDZ-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-[[4-(methylcarbamoyl)phenyl]methyl]benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1=CC(C(=O)NC)=CC=C1CN1C2=CC(Cl)=C(Cl)C=C2N=C1N1CCC(C(=O)NC2CCCC2)CC1 SZWCZEBNKZEYDZ-UHFFFAOYSA-N 0.000 description 1
- DOKDALCGHBLIIP-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dichloro-1-[[4-(methylsulfonylcarbamoyl)phenyl]methyl]benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1=CC(C(=O)NS(=O)(=O)C)=CC=C1CN1C2=CC(Cl)=C(Cl)C=C2N=C1N1CCC(C(=O)NC2CCCC2)CC1 DOKDALCGHBLIIP-UHFFFAOYSA-N 0.000 description 1
- FTAYCANWMJAUIE-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dimethyl-1-(2-methylpropyl)benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound N=1C2=CC(C)=C(C)C=C2N(CC(C)C)C=1N(CC1)CCC1C(=O)NC1CCCC1 FTAYCANWMJAUIE-UHFFFAOYSA-N 0.000 description 1
- BBYHZPBDYLPPIN-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dimethyl-1-(3-methylphenyl)benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound CC1=CC=CC(N2C3=CC(C)=C(C)C=C3N=C2N2CCC(CC2)C(=O)NC2CCCC2)=C1 BBYHZPBDYLPPIN-UHFFFAOYSA-N 0.000 description 1
- QSXAVJCCFXJBKZ-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dimethyl-1-(4-propan-2-ylphenyl)benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1=CC(C(C)C)=CC=C1N1C2=CC(C)=C(C)C=C2N=C1N1CCC(C(=O)NC2CCCC2)CC1 QSXAVJCCFXJBKZ-UHFFFAOYSA-N 0.000 description 1
- WYGRTFXSQADURI-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dimethyl-1-(oxolan-2-ylmethyl)benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1CCOC1CN1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 WYGRTFXSQADURI-UHFFFAOYSA-N 0.000 description 1
- OBQHJFSSCOWIRE-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dimethyl-1-[(2-methyl-1,3-thiazol-4-yl)methyl]benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound S1C(C)=NC(CN2C3=CC(C)=C(C)C=C3N=C2N2CCC(CC2)C(=O)NC2CCCC2)=C1 OBQHJFSSCOWIRE-UHFFFAOYSA-N 0.000 description 1
- ULKIJSQQQXIXAP-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dimethyl-1-[[4-(methylcarbamoyl)phenyl]methyl]benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C1=CC(C(=O)NC)=CC=C1CN1C2=CC(C)=C(C)C=C2N=C1N1CCC(C(=O)NC2CCCC2)CC1 ULKIJSQQQXIXAP-UHFFFAOYSA-N 0.000 description 1
- UVBGQNBWSLAUGU-UHFFFAOYSA-N n-cyclopentyl-1-[5,6-dimethyl-1-[[4-(methylsulfonylcarbamoyl)phenyl]methyl]benzimidazol-2-yl]piperidine-4-carboxamide Chemical compound C=1C=C(C(=O)NS(C)(=O)=O)C=CC=1CN1C=2C=C(C)C(C)=CC=2N=C1N(CC1)CCC1C(=O)NC1CCCC1 UVBGQNBWSLAUGU-UHFFFAOYSA-N 0.000 description 1
- SHVKIOBPRGCXPG-UHFFFAOYSA-N n-cyclopropyl-1-(5,6-dichloro-1-propan-2-ylbenzimidazol-2-yl)piperidine-4-carboxamide Chemical compound N=1C2=CC(Cl)=C(Cl)C=C2N(C(C)C)C=1N(CC1)CCC1C(=O)NC1CC1 SHVKIOBPRGCXPG-UHFFFAOYSA-N 0.000 description 1
- PVWOIHVRPOBWPI-UHFFFAOYSA-N n-propyl iodide Chemical compound CCCI PVWOIHVRPOBWPI-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 230000001703 neuroimmune Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 150000002826 nitrites Chemical class 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 229950009992 orazipone Drugs 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 230000003349 osteoarthritic effect Effects 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- IPBPLHNLRKRLPJ-UHFFFAOYSA-N oxan-4-ylmethanamine Chemical compound NCC1CCOCC1 IPBPLHNLRKRLPJ-UHFFFAOYSA-N 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- MIPHRQMEIYLZFZ-UHFFFAOYSA-N oxolan-3-amine Chemical compound NC1CCOC1 MIPHRQMEIYLZFZ-UHFFFAOYSA-N 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 229960001476 pentoxifylline Drugs 0.000 description 1
- 239000003444 phase transfer catalyst Substances 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- IPNPIHIZVLFAFP-UHFFFAOYSA-N phosphorus tribromide Chemical compound BrP(Br)Br IPNPIHIZVLFAFP-UHFFFAOYSA-N 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- DNUTZBZXLPWRJG-UHFFFAOYSA-M piperidine-1-carboxylate Chemical compound [O-]C(=O)N1CCCCC1 DNUTZBZXLPWRJG-UHFFFAOYSA-M 0.000 description 1
- 150000003053 piperidines Chemical class 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- 229960002702 piroxicam Drugs 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 235000010333 potassium nitrate Nutrition 0.000 description 1
- 239000004323 potassium nitrate Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 229950003700 priliximab Drugs 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000007639 printing Methods 0.000 description 1
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 1
- SGUKUZOVHSFKPH-YNNPMVKQSA-N prostaglandin G2 Chemical compound C1[C@@H]2OO[C@H]1[C@H](/C=C/[C@@H](OO)CCCCC)[C@H]2C\C=C/CCCC(O)=O SGUKUZOVHSFKPH-YNNPMVKQSA-N 0.000 description 1
- 102000017953 prostanoid receptors Human genes 0.000 description 1
- 108050007059 prostanoid receptors Proteins 0.000 description 1
- 229940126409 proton pump inhibitor Drugs 0.000 description 1
- 239000000612 proton pump inhibitor Substances 0.000 description 1
- 230000001185 psoriatic effect Effects 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 239000002510 pyrogen Substances 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- XIIOFHFUYBLOLW-UHFFFAOYSA-N selpercatinib Chemical compound OC(COC=1C=C(C=2N(C=1)N=CC=2C#N)C=1C=NC(=CC=1)N1CC2N(C(C1)C2)CC=1C=NC(=CC=1)OC)(C)C XIIOFHFUYBLOLW-UHFFFAOYSA-N 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 229940083037 simethicone Drugs 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- BTGNGJJLZOIYID-UHFFFAOYSA-N sivelestat Chemical compound C1=CC(OC(=O)C(C)(C)C)=CC=C1S(=O)(=O)NC1=CC=CC=C1C(=O)NCC(O)=O BTGNGJJLZOIYID-UHFFFAOYSA-N 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 229940125794 sodium channel blocker Drugs 0.000 description 1
- 239000003195 sodium channel blocking agent Substances 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 238000010532 solid phase synthesis reaction Methods 0.000 description 1
- 230000003381 solubilizing effect Effects 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 235000011150 stannous chloride Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 229940032147 starch Drugs 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000000859 sublimation Methods 0.000 description 1
- 230000008022 sublimation Effects 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- FDDDEECHVMSUSB-UHFFFAOYSA-N sulfanilamide Chemical compound NC1=CC=C(S(N)(=O)=O)C=C1 FDDDEECHVMSUSB-UHFFFAOYSA-N 0.000 description 1
- 150000004763 sulfides Chemical class 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- KQKPFRSPSRPDEB-UHFFFAOYSA-N sumatriptan Chemical compound CNS(=O)(=O)CC1=CC=C2NC=C(CCN(C)C)C2=C1 KQKPFRSPSRPDEB-UHFFFAOYSA-N 0.000 description 1
- 229960003708 sumatriptan Drugs 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- VQMUCYYYMQJKEV-UHFFFAOYSA-N tert-butyl 4-(cyclopentylcarbamoyl)piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1C(=O)NC1CCCC1 VQMUCYYYMQJKEV-UHFFFAOYSA-N 0.000 description 1
- IOGXOCVLYRDXLW-UHFFFAOYSA-N tert-butyl nitrite Chemical compound CC(C)(C)ON=O IOGXOCVLYRDXLW-UHFFFAOYSA-N 0.000 description 1
- 125000004192 tetrahydrofuran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000005942 tetrahydropyridyl group Chemical group 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000004495 thiazol-4-yl group Chemical group S1C=NC(=C1)* 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000002053 thietanyl group Chemical group 0.000 description 1
- 125000001730 thiiranyl group Chemical group 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 125000001166 thiolanyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 229940036555 thyroid hormone Drugs 0.000 description 1
- 239000005495 thyroid hormone Substances 0.000 description 1
- AXZWODMDQAVCJE-UHFFFAOYSA-L tin(II) chloride (anhydrous) Chemical compound [Cl-].[Cl-].[Sn+2] AXZWODMDQAVCJE-UHFFFAOYSA-L 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 229960005026 toremifene Drugs 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 229960004380 tramadol Drugs 0.000 description 1
- TVYLLZQTGLZFBW-GOEBONIOSA-N tramadol Natural products COC1=CC=CC([C@@]2(O)[C@@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-GOEBONIOSA-N 0.000 description 1
- 230000037317 transdermal delivery Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 1
- 235000019798 tripotassium phosphate Nutrition 0.000 description 1
- 125000005455 trithianyl group Chemical group 0.000 description 1
- INASOKQDNHHMRE-UHFFFAOYSA-N turofexorate isopropyl Chemical compound C1C(C)(C)C(C2=CC=CC=C2N2)=C2C(C(=O)OC(C)C)=CN1C(=O)C1=CC=C(F)C(F)=C1 INASOKQDNHHMRE-UHFFFAOYSA-N 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention relates to piperidinyl benzoimidazole derivatives and to the use thereof in disease therapy.
- the present invention relates to compounds useful in the treatment or prevention of diseases in which inhibition of the activity of microsomal prostaglandin E synthase- 1 (accession number "014684" at http://www.uniprot.org/) is desired and/or required and, more particularly, in the treatment of diseases such as inflammatory diseases, nociceptive pain, auto-immune disease, breathing disorders, fever, cancer, inflammation related anorexia, Alzheimer's disease and cardiovascular disease.
- diseases such as inflammatory diseases, nociceptive pain, auto-immune disease, breathing disorders, fever, cancer, inflammation related anorexia, Alzheimer's disease and cardiovascular disease.
- Prostaglandin (PG) E 2 is produced in a sequential action including liberation of arachidonic acid, conversion into PGG2/PGH2 by cyclooxygenase (Cox) -1 or Cox-2, and finally prostaglandin E synthase converts PGH 2 into PGE 2 .
- mPGEs-1 was regarded as the enzyme predominantly coupled with Cox-2 activity. However; later results demonstrate that mPGEs-1 can also catalyze the conversion of Cox- 1 derived PGH2 into PGE2. mPGEs-1 possesses the highest catalytic efficiency of the known PGE synthases.
- PGE2 as one of the most potent mediators of inflammation together with many in vitro reports on the presence of mPGEs-1 in different models of inflammation suggested this enzyme to be an attractive drug target for development of new anti inflammatory drugs with fewer side effects than the currently available NSAIDs and selective Cox-2 inhibitors [Samuelsson, B.;
- examples of treatable diseases of the invention include inflammatory diseases, nociceptive pain, auto-immune disease, breathing disorders, fever, cancer, inflammation related anorexia, Alzheimer's disease and cardiovascular disease. More specifically, non-limiting examples of these diseases include the treatment or prevention of rheumatoid arthritis [Murakami, M.; Nakashima, K.; Kamei, D.; Masuda, S.; Ishikawa, Y.; Ishii, T.; Ohmiya, Y.; Watanabe, K.; Kudo, I. Cellular prostaglandin E2 production by membrane- bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J. Biol.
- osteoarthritis [Kojima, F.; Naraba, H.; Miyamoto, S.; Beppu, M.; Aoki H, and Kawai S (2004) Membraneassociated prostaglandin E synthase- 1 is upregulated by proinflammatory cyto-kines in chondrocytes from patients with osteoarthritis. Arthritis Res. Ther. 2004, R355-R365; Masuko-Hongo, K.; Berenbaum, F.; Humbert, L.; Salvat, C; Goldring, M. B.; Thirion, S.
- cephalalgia headache
- PGE2 Prostaglandin E2
- Cephalalgia 2009, 29, 509-519
- pain due to gall-stones pain due to metastasis, familial adenomatous polyposis (FAP) condition
- FAP familial adenomatous polyposis
- epithelial ovarian cancer [Rask, K.; Zhu, Y.; Wang, W.; Hedin, L.; Sundfeldt, K. Ovarian epithelial cancer: a role for PGE2-synthesis and signalling in malignant transformation and progression. MoI. Cancer, 2006, 16, 62], breast cancer [Mehrotra, S.; Morimiya, A.; Agarwal, B.; Konger, R.; Badve, S. Microsomal prostaglandin E2 synthase- 1 in breast cancer: a potential target for therapy. J. Pathol. 2006, 208, 356-363], gastric
- Oshima M. Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway. Gastroenterology, 2006, 131, 1086-1095], multiple sclerosis, atherosclerosis such as myocardial infarction and stroke [Ikeda-Matsuo, Y.; Ota, A.; Fukada, T.; Uematsu, S.; Akira, S.; Sasaki, Y. Microsomal prostaglandin E synthase-1 is a critical factor of stroke-reperfusion injury. Proc. Natl. Acad. Sci. USA.
- Neutrophils CDl Ib and fibroblasts PGE(2) are elevated in Alzheimer's disease. Neurobiol. Aging, 2002, 23, 523-30], as well as the use of the said compounds in the manufacture of medicaments for the treatment of those disorders [For a reviews discussing most of the therapeutic indications mentioned above see: Samuelsson, B.; Morgenstern, R.; Jakobsson, P. -J. Membrane Prostaglandin E Synthase-1 : A Novel Therapeutic Target. Pharmacol. Rev. 2007, 5P,207-224; Murakami M, Kudo I. Prostaglandin E synthase: a novel drug target for
- mPGES-1 Other examples of medical conditions associated with mPGES-1 are pain in connection to ankylosing spondylitis, gout, or rheumatic fever, tootache, cellular neoplastic transformations and metastatic tumor growth, endometriosis, hemophilic arthropathy, Parkinson's disease, mPGES-1 -mediated proliferative disorders such as may occur in diabetic retinopathy and tumor angiogenesis, symptoms associated with viral infections, and premature labor.
- WO 2008/084218 discloses benzazole derivatives of the general formula
- One object of the present invention is to provide compounds having said desired improved properties.
- the present inventors now have surprisingly found certain piperidinyl benzoimidazoles that are capable of efficiently blocking mPGEs-1.
- R 1 is selected from branched or unbranched C 1 -C 10 alkyl, C2-C10 alkenyl or C2-C10 alkynyl; C3- Cio cycloalkyl; C4-C10 cycloalkenyl; Cs-Cio cycloalkynyl; C 1 -Cg heterocyclyl; and C 6 -CiO aryl; wherein any alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, or cycloalkynyl moiety optionally is substituted with 1, 2 or 3 groups R a ; and any heterocyclyl or aryl moiety optionally is substituted with 1, 2, 3, 4 or 5 groups R b ; R 2 is selected from branched or unbranched C 1 -Cg alkyl, C2-C9 alkenyl, or C2-C9 alkynyl; C3-C6 cycloalkyl; C 4 -C 6
- R 3 and R 4 are each independently selected from Ci -C 4 alkyl; C 2 -C 4 alkenyl; C 2 -C 4 alkynyl; C 3 -C 4 cycloalkyl; Ci-C 4 alkyloxy; C 2 -C 4 alkenyloxy; C 2 -C 4 alkynyloxy; Ci-C 4 alkylthio; C 2 -C 4 alkenylthio; C 2 -C 4 alkynylthio; halogen; Ci-C 4 alkyl, C 2 -C 4 alkenyl or C 2 -C 4 alkynyl secondary or tertiary amino; and Ci-C 4 alkyl, C 2 -C 4 alkenyl or C 2 -C 4 alkynyl carbonyl; wherein any alkyl, alkenyl, alkynyl or cycloalkyl moiety optionally is substituted with 1, 2, or 3 groups R c ; R 5 and R 6 are independently selected
- X represents a bond or branched or unbranched Ci -C 3 alkyl, C 2 -C 3 alkenyl or C 2 -C 3 alkynyl;
- Q represents carbonyl, sulfonyl or sulfmyl; m is an integer of 0, 1 or 2; each R a is independently selected from halogen; hydroxy; Ci-C 4 alkyl; C 2 -C 4 alkenyl; C 2 -C 4 alkynyl; Ci-C 4 alkyloxy; C 2 -C 4 alkenyloxy; C 2 -C 4 alkynyloxy; Ci-C 4 alkyl, C 2 -C 4 alkenyl or C 2 - C 4 alkynyl secondary or tertiary amino; Ci-C 4 alkylthio; C 2 -C 4 alkenylthio; C 2 -C 4 alkynylthio; wherein any alkyl, alkenyl or alkynyl moiety optionally is substituted with 1, 2 or 3 groups R c ; each R b is independently selected from halogen; carboxy; hydroxy; cyano; C1-C5 heteroaryl;
- Another aspect of the present invention is to provide a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, in admixture with at least one pharmaceutically acceptable adjuvant, diluent or carrier.
- Another aspect of the present invention is to provide a method of medical treatment by use of said pharmaceutical compositions.
- Another aspect of the present invention provides a compound of formula (I) or a
- Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of a disorder in which inhibition of the activity of microsomal prostaglandin E synthase- 1 is desired and/or required.
- Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of an inflammatory disease, said disease preferably being selected from rheumatoid arthritis, inflammatory bowel disease, osteoarthritis, psoriasis, eczema, swelling and periodontitis.
- Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of nociceptive pain, said pain preferably being selected from inflammatory pain, post-operative pain, fibromyalgia, dysmenorrhea, migraine, arthrotic pain, arthritic pain, pain in connection with osteoarthritis, cephalalgia, pain due to gall-stones and pain due to metastasis, ankylosing spondylitis, gout, or rheumatic fever, and tootache.
- the compound of formula (I) also may be used in preemptive treatment of surgical pain.
- Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of an autoimmune disease, said autoimmune disease preferably being selected from rheumatoid arthritis and multiple sclerosis.
- Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of a breathing disorder, said disorder preferably being selected from apnea or other respiratory distress symptoms in adults and children, sudden infant death syndrome (SIDS), asthma and other chronic obstructive lung-diseases and sarcoidosis.
- a breathing disorder preferably being selected from apnea or other respiratory distress symptoms in adults and children, sudden infant death syndrome (SIDS), asthma and other chronic obstructive lung-diseases and sarcoidosis.
- Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of cancer, said cancer preferably being selected from familial adenomatous polyposis (FAP) condition, colorectal cancer, breast cancer, gastric tumorigenesis and epithelial ovarian cancer, as well as cellular neoplastic transformations and metastatic tumor growth.
- FAP familial adenomatous polyposis
- Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of cardiovascular diseases due to atherosclerosis such as myocardial infarction and stroke.
- Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of fever- or inflammation-related anorexia.
- Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of Alzheimer's disease.
- Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of endometriosis, hemophilic arthropathy, Parkinson's disease, mPGES-1 -mediated proliferative disorders such as may occur in diabetic retinopathy and tumor angiogenesis, symptoms associated with viral infections, and premature labor.
- Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of an inflammatory disease, said disease preferably being selected from rheumatoid arthritis, inflammatory bowel disease, osteoarthritis, psoriasis, eczema, swelling and periodontitis.
- Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of nociceptive pain, said pain preferably being selected from inflammatory pain, post-operative pain, fibromyalgia, dysmenorrhea, migraine, arthrotic pain, arthritic pain, pain in connection with osteoarthritis, cephalalgia, pain due to gall-stones and pain due to metastasis, pain due to ankylosing spondylitis, pain due to gout, toothache and pain due to rheumatic fever, or for the preemptive treatment of surgical pain.
- Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of an autoimmune disease, said autoimmune disease preferably being selected from rheumatoid arthritis and multiple sclerosis.
- Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of a breathing disorder, said disorder preferably being selected from apnea or other respiratory distress symptoms in adults and children, sudden infant death syndrome (SIDS), asthma and other chronic obstructive lung- diseases and sarcoidosis.
- a breathing disorder preferably being selected from apnea or other respiratory distress symptoms in adults and children, sudden infant death syndrome (SIDS), asthma and other chronic obstructive lung- diseases and sarcoidosis.
- Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of cancer, said cancer preferably being selected from familial adenomatous polyposis (FAP) condition, colorectal cancer, breast cancer, gastric tumorigenesis and epithelial ovarian cancer, as well as cellular neoplastic transformations and metastatic tumor growth.
- FAP familial adenomatous polyposis
- Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of cardiovascular diseases due to atherosclerosis such as myocardial infarction and stroke.
- Another aspect of the present invention provides the use of a compound of formula (I), in preparation of a medicament for the treatment and/or prevention of fever- or inflammation- related anorexia.
- Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of Alzheimer's disease.
- Another aspect of the present invention provides a method of treatment of a disease in which inhibition of the activity of mPGEs-1 is desired and/or required, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient suffering from, or susceptible to, such a condition.
- Another aspect of the present invention provides a method of treatment of an inflammatory disease, said disease preferably being selected from rheumatoid arthritis, inflammatory bowel disease, osteoarthritis, psoriasis, eczema, swelling and periodontitis, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
- Another aspect of the present invention provides a method of treatment of nociceptive pain, said pain preferably being selected from inflammatory pain, post-operative pain, fibromyalgia, dysmenorrhea, migraine, arthrotic pain, arthritic pain, pain in connection with osteoarthritis, cephalalgia, pain due to gall-stones and pain due to metastasis, pain due to ankylosing spondylitis, gout, toothache and pain due to rheumatic fever, as well as preemptive treatment of surgical pain, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
- a compound of formula (I) or a pharmaceutically-acceptable salt thereof
- Another aspect of the present invention provides a method of treatment of an autoimmune disease, said autoimmune disease preferably being selected from rheumatoid arthritis and multiple sclerosis, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
- Another aspect of the present invention provides a method of treatment of a breathing disorder, said disorder preferably being selected from apnea or other respiratory distress symptoms in adults and children, sudden infant death syndrome (SIDS), asthma and other chronic obstructive lung-diseases and sarcoidosis, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
- SIDS sudden infant death syndrome
- asthma chronic obstructive lung-diseases and sarcoidosis
- Another aspect of the present invention provides a method of treatment of cancer, said cancer preferably being selected from familial adenomatous polyposis (FAP) condition, colorectal cancer, breast cancer, gastric tumorigenesis and epithelial ovarian cancer, as well as cellular neoplastic transformations and metastatic tumor growth, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically- acceptable salt thereof, to a patient in the need of such treatment.
- FAP familial adenomatous polyposis
- Another aspect of the present invention provides a method of treatment of cardiovascular diseases due to atherosclerosis such as myocardial infarction and stroke, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
- Another aspect of the present invention provides a method of treatment of fever- or
- inflammation-related anorexia which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
- Another aspect of the present invention provides a method of treatment of Alzheimer's disease, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
- Figure 1 is a bar diagram showing results of a carrageenan- induced paw edema assay in Wistar rats using the compound synthesised in Example 41.
- the compound at 10, 30 and 100 mg/kg, as well as vehicle was administered intraperitoneally immediately before carrageenan challenge.
- Hind paw edema a measure of inflammation, was recorded 6 hours after carrageenan administration.
- Volume increase is paw volume at 6 hours after carrageenan administration minus paw volume before carrageenan administration.
- Figure 2 is a bar diagram showing inhibition of CFA- induced paw swelling by the compound synthesised in Example 41 compared to vehicle control.
- any alkyl, alkenyl or alkynyl group as referred to herein may be branched or unbranched. This also applies to said groups when present in moieties such as alkoxy groups, and the like.
- alkyl refers to an acyclic straight or branched chain radical, unless otherwise specified containing 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 or 12 carbons in the normal chain, which includes methyl, ethyl, /? -propyl, /? -butyl, n-pentyl, n-hexyl, n-heptyl and n-octyl.
- Alkyl also includes a straight or branched alkyl group that contains or is interrupted by a carbocyclyl group, exemplified by cyclopropane, as exemplified below: (CH ⁇ -J- ⁇ ( CH ⁇ )z—
- alkyl portions can be attached at any variable point of attachment to the carbocyclyl, including the same ring carbon, as exemplified below:
- substituted alkyl when substituted alkyl is present, this refers to a straight or branched alkyl group, substituted with 1, 2, 3 groups of R a , which groups may be the same or different at any available point, as defined with respect to each variable.
- alkenyl refers to a straight or branched chain radical, unless otherwise specified containing 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 or 12, containing at least one carbon to carbon double bond, which includes in the normal chain vinyl, 2-propenyl, 3-butenyl, 2-butenyl, 4-pentenyl, 3-pentenyl, 2-hexenyl, 3-hexenyl, 2-heptenyl, 3- heptenyl, 4-heptenyl, 3-octenyl, and the like.
- branched chain radicals examples include 2-buten-2-yl, 4-methyl-2-pentenyl, 6-methylene- 2-octenyl, and the like. All possible (E)- and (Z)-isomers is contemplated within the scope of the invention.
- the straight or branched portion of the alkenyl group may be optionally substituted when a substituted alkenyl group is provided, and the chain may be interrupted by a carbocyclyl group.
- substituted alkenyl when substituted alkenyl is present, this refers to a straight or branched alkyl group, substituted with 1, 2, 3 groups of R a , which groups may be the same or different at any available point, as defined with respect to each variable.
- alkynyl refers to a straight or branched chain radical, unless otherwise specified containing 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 or 12 carbons, which contains at least one carbon to carbon triple bond, which in the normal chain such as 2-propynyl, 3-butynyl, 2-butynyl, 4-pentynyl, 3-pentynyl, 2-hexynyl, 3-hexynyl, 2- heptynyl, 3-heptynyl, 4-heptynyl, 3-octynyl, and the like.
- Examples of branched chain radicals are 3-methyl-l-butynyl, 2-methyl-4- pentynyl, 6-methyl-4-octynyl, 6-methyl-3-propyl-4-octynyl, and the like.
- the alkynyl can in addition to carbon to carbon triple bonds also include a to carbon double bond.
- Examples of such radicals, not excuding any of the possible not mentioned, are 3-buten-l-ynyl, l-buten-3-ynyl, 6- methylene-2-octynyl, 3-allyl-6-methyl-l-octen-4-ynyl, and the like.
- the straight or branched portion of the alkynyl group may be optionally substituted when a substituted alkynyl group is provided, and the chain may be interrupted by a carbocyclyl group.
- substituted alkynyl this refers to a straight or branched alkyl group, substituted with 1, 2, 3 groups of R a , which groups may be the same or different at any available point, as defined with respect to each variable.
- carbocyclyl refers to a cyclic moiety containing only carbon atoms.
- bicyclic refers to a cyclic moiety containing two rings.
- a bicyclic group may, for example, be fused or bridged.
- heterocyclyl mean a mono- or bicyclic, saturated or unsaturated cyclic group, possibly aromatic, containing one or more heteroatom(s) preferably selected from N, O and S, such as, but not limited to, aziridinyl, azetidinyl, dihydropyranyl, dihydropyridyl, dihydropyrrolyl, dioxolanyl, dioxanyl, dithianyl, dithiolanyl, imidazolidinyl, imidazolinyl, morpholinyl, oxetanyl, oxiranyl, pyrrolidinyl, pyrrolidinonyl, piperidyl, piperazinyl, piperidinyl, pyrazolidinyl, quinuclidinyl, sulfalonyl, 3-sulfolenyl, tetrahydrofuranyl tetrahydropyranyl,
- benzobenzoxadiazolyl benzoxazinyl, benzoxazolyl, benzo morpholinyl, benzoselenadiazolyl, benzothienyl, purinyl, pteridinyl, and the like.
- halogen refers to fluorine, chlorine, bromine and iodine, where the preferred halogen radicals are fluoro and chloro.
- aryl means an aromatic group, monocyclic or bicyclic such as, but not limited to phenyl or naphthyl, and the like. The aryl group is preferably a monocyclic C 6 aryl (phenyl).
- cycloalkyl as employed herein alone or as part of another group includes saturated cyclic hydrocarbyl groups, mono- or bicyclic rings(s), having a total of 3, 4, 5, 6, 7, 8, 9 or 10 carbons forming the ring(s), which includes cyclopropyl, cyclobutyl, cyclohexyl, cycloheptyl, cyclodecane, bicyclo[4.1.0]heptane, bicyclo[4.3.3]dodecane, cyclopentyl, decahydronaphthalene, bicyclo[2.2.2]octane, and the like, optionally substituted with 1, 2 or 3 groups of R a .
- cycloalkenyl as employed herein alone or as part of another group includes unsaturated cyclic rings(s), mono- or bicyclic, having a total of 4, 5, 6, 7, 8, 9 or 10 carbons forming the ring(s), which includes cyclohexene, cyclobutene, bicyclo[4.2.0]-3-octene, 1,3- cyclodecadiene and the like, optionally substituted with 1, 2 or 3 groups of R a .
- cycloalkynyl as employed herein alone or as part of another group includes mono- or bicyclic, rings having one carbon to carbon triple bond, and having a total of 8, 9 or 10 carbons forming the ring, which includes cyclooctyne, cyclodecyne, bicyclo[6.2.0]-4-decyne and the like, optionally substituted with 1, 2 or 3 groups of R a .
- the cycloalkynyl can in addition to carbon to carbon triple bonds also contain a carbon to carbon double bond, which includes l-cyclodecen-5- yne and the like.
- Carbocyclyl as employed herein alone or as part of another group includes saturated cyclic hydrocarbyl groups or unsaturated (at least one carbon to carbon double bond) cyclic hydrocarbyl groups, containing one ring and a total of 3, 4, 5, 6, 7, 8, 9 or 10 carbons forming the ring, which includes cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, phenyl, cycloheptyl, and the like.
- the cyclic hydrocarbyl may be mono- or bicyclic.
- tern ⁇ 'unsaturated when referring to a bicyclic system, is meant a ring system
- both rings may be unsaturated or only one ring may be unsaturated, the other one being saturated.
- unsaturated bicyclic also is intended to refer to a non- aromatic bicyclic system comprising a ring that is either unsaturated or saturated fused to a ring that by itself would be aromatic, such as in indane or 4,5-dihydro-l -indole.
- aromatic refers to an unsaturated cyclic moiety that has an aromatic character
- non-aromatic refers to a cyclic moiety, that may be unsaturated, but that does not have an aromatic character
- the rings may be both saturated or both unsaturated, e.g. both aromatic.
- the rings may also be of different degrees of saturation, and one ring may be aromatic whereas the other is non-aromatic.
- the rings also may comprise different numbers of atoms, e.g. one ring being 5-membered and the other one being 6-membered.
- a bicyclic heterocyclyl as referred to herein, one or both of the rings may contain one or several heteroatoms.
- hydroxy (i), carboxy (ii) and aminocarbonyl (iii), refer to radicals of the type:
- alkyloxy, alkenyloxy, alkynyloxy and cycloalkyloxy refer to a radical of the type:
- R is an alkyl, alkenyl, alkynyl or cycloalkyl moiety.
- alkylthio, alkenylthio, and alkynylthio refer to a radical of the type:
- R is an alkyl, alkenyl or alkynyl moiety.
- alkyl, alkenyl and alkynyl secondary amino refer to a radical of the type:
- R is an alkyl, alkenyl or alkynyl moiety.
- alkyl, alkenyl and alkynyl tertiary amino refer to a radical of the type:
- R and R' are each an independently selected alkyl, alkenyl or alkynyl moiety.
- alkyl, alkenyl and alkynyl secondary amido refer to a radical of the type:
- R is an alkyl, alkenyl or alkynyl moiety.
- alkyl, alkenyl and alkynyl tertiary amido refer to a radical of the type:
- R and R' are each an independently selected alkyl, alkenyl or alkynyl moiety.
- alkyl, alkenyl and alkynyl carbonyl refer to a radical of the type:
- R is an alkyl, alkenyl or alkynyl moiety.
- alkyl, alkenyl and alkynyl sulfonyl refer to a radical of the type:
- R is an alkyl, alkenyl or alkynyl moiety.
- alkyl, alkenyl and alkynyl secondary sulphonamido refer to a radical of the type:
- R is an alkyl, alkenyl or alkynyl moiety.
- alkyl, alkenyl and alkynyl tertiary sulphonamido refer to a radical of the type:
- R and R' are each independently selected from alkyl, alkenyl or alkynyl moiety.
- alkyl, alkenyl and alkynyl secondary acylsulphonamido refer to a radical of the type:
- R is an alkyl, alkenyl or alkynyl moiety.
- alkyl, alkenyl and alkynyl tertiary acylsulphonamido refer to a radical of the type:
- each R is independently selected from an alkyl, alkenyl or alkynyl moiety.
- alkyl, alkenyl and alkynyl alkynyloxy carbonyl refer to a radical of the type: o
- R is an alkyl, alkenyl or alkynyl moiety.
- cyclic refers to an atom that is a member of at least one ring in a carbocycle or heterocycle.
- a cyclyl according to the invention may comprise 1-10 carbons, it being understood that in case the cyclyl comprises less than 3 carbons, it must necessarily also comprise at least one heteroatom. Unless otherwise indicated or apparent from the context, in a bicyclic moiety according to the invention, each constituent monocycle may independently be selected from aromatic or non- aromatic carbo- and heterocycles.
- R 1 in formula (I) is selected from branched or unbranched C 1 -C 10 alkyl, C 2 -C 10 alkenyl or C2-C10 alkynyl; C3-C10 cycloalkyl; C4-C10 cycloalkenyl; Cs-Cio cycloalkynyl; C1-C9 heterocyclyl; and C 6 - Cio aryl; wherein any alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, or cycloalkynyl moiety optionally is substituted with 1, 2 or 3 groups R a ; and any heterocyclyl or aryl moiety optionally is substituted with 1, 2, 3, 4 or 5 groups R b , as defined herein.
- R 1 in formula (I) is selected from branched or unbranched C 1 -C 10 alkyl; C3- Cio cycloalkyl; C1-C9 heterocyclyl; and C 6 -CiO aryl; said alkyl and cycloalkyl optionally being substituted with 1 , 2 or 3 groups R a ; and said heterocyclyl and aryl optionally being substituted with 1, 2, 3, 4 or 5 groups R b .
- R 1 in formula (I) is optionally substituted branched or unbranched C 1 -C 10 alkyl, e.g. Ci-Cs alkyl, i.e. methyl, ethyl and branched or unbranched propyl, butyl, pentyl, hexyl, heptyl, and octyl, such as methyl, ethyl, n-propyl, ⁇ o-propyl, n-butyl, iso- butyl, sec-butyl, n-pentyl, pentan-2-yl, pentan-3-yl, n-hexyl, n-heptyl, and n-octyl.
- Ci-Cs alkyl i.e. methyl, ethyl and branched or unbranched propyl, butyl, pentyl, hexyl, heptyl, and octyl
- R 1 in formula (I) may be selected from optionally substituted branched or unbranched Ci-C 6 alkyl, e.g. methyl, ethyl, n-propyl, ⁇ o-propyl, n-butyl, ⁇ o-butyl, sec-butyl, n -pentyl, pentan-2-yl, pentan-3-yl, n-hexyl, or from optionally substituted branched or unbranched Ci -C 4 alkyl.
- R 1 may be optinally substituted iso- propyl.
- R 1 in formula (I) when R 1 in formula (I) is selected from optionally substituted branched or unbranched Ci -C 10 alkyl, m is 0. In another embodiment, R 1 in formula (I) is optionally substituted C 3 -C 10 cycloalkyl, in particular C 3 -C 6 cycloalkyl, or C3-C5 cycloalkyl.
- R 1 in formula (I) also may be optionally substituted C1-C9 heterocyclyl, in particular C1-C5 heterocyclyl, such as a 6-membered C1-C5 heterocyclyl, C2-C5 heterocyclyl, C3-C5 heterocyclyl, C4-C5 heterocyclyl or C5 heterocyclyl, e.g.
- R 1 in formula (I) also may be optionally substituted C 6 -CiO aryl, preferably phenyl.
- R 1 is alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, it optionally is substituted with 1, 2 or 3 groups, e.g. 1 or 2 independently selected groups R a , e.g. 1 group R a , as defined herein, i.e.
- halogen selected from halogen; hydroxy; C1-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; C1-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amino; C1-C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; wherein any alkyl, alkenyl or alkynyl moiety optionally is substituted with 1, 2 or 3 groups R c .
- each R a is indpendently selected from hydroxy, C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; and C1-C4 alkyl, C2-C4 alkenyl or C 2 -C 4 alkynyl secondary or tertiary amino, in particular from hydroxy, C 1 -C 4 alkoxy, such as methoxy, or C 1 -C 4 alkyl secondary or tertiary amino, e.g. such as ethyl secondary or tertiary amino, e.g. diethylamino.
- R 1 is not substituted with any R a .
- R 1 is heterocyclyl or aryl, it optionally is substituted with 1, 2, 3, 4 or 5 independently selected groups R b , as defined herein, i.e. selected from halogen; carboxy; hydroxy; cyano; C1-C5 heteroaryl; C1-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; Ci-C 4 alkylthio; C 2 -C 4 alkenylthio; C 2 -C 4 alkynylthio; Ci-C 4 alkyl, C 2 -C 4 alkenyl or C 2 -C 4 alkynyl secondary or tertiary amino; amino carbonyl; Ci-C 4 alkyl, C 2 -C 4 alkenyl or C 2 -C 4 alkynyl secondary or tertiary amido; Ci-C 4 alky
- R b is selected from halogen; carboxy; cyano; Ci-C 5 heteroaryl; Ci-C 4 alkyl; C 2 -C 4 alkenyl; C 2 -C 4 alkynyl; amino carbonyl; Ci-C 4 alkyl, C 2 -C 4 alkenyl or C 2 -C 4 alkynyl secondary or tertiary amido; Ci-C 4 alkyl, C 2 -C 4 alkenyl or C 2 -C 4 alkynyl secondary or tertiary acylsulphonamido; Ci-C 4 alkyloxy, C 2 -C 4 alkenyloxy, or C 2 -C 4 alkynyloxy carbonyl; and C 1 -C 5 heterocyclyl; wherein any alkyl, alkenyl and alkynyl moiety optionally is substituted with 1, 2 or 3 groups Rc.
- R b is selected from halogen, i.e. F, Cl and Br, in particular F; carboxy; cyano; 5- or 6-membered C1-C5 heteroaryl, in particular 5- membered Ci-C 4 heteroaryl, such as triazole; Ci-C 4 alkyl, such as methyl; amino carbonyl; Ci-C 4 alkyl secondary or tertiary amido, such as methyl secondary or tertiary amido; Ci-C 4 alkyl secondary or tertiary acylsulphonamido, such as methyl secondary or tertiary acylsulphonamido; C1-C4 alkyloxy carbonyl, such as methoxy carbonyl; and C1-C5 heterocyclyl, such as 5- or 6- membered heterocyclyl, e.g. 5-membered C1-C4 heterocyclyl, e.g. tetrazole.
- R 1 is substituted with 1-3 groups R b as defined herein, e.g. with 1 or 2 groups, in particular with 1 group R b . In another embodiment, R 1 is not substituted with any R b .
- R 1 when R 1 is a 6-membered heterocyclyl or aryl, it is substituted with at least one group R b in para -position relative to the linking bond or chain attaching R 1 to the benzimidazol moiety.
- R 2 in formula (I) is selected from branched or unbranched C1-C9 alkyl, C2-C9 alkenyl or C2-C9 alkynyl; C3-C6 cycloalkyl, C 4 -C 6 cycloalkenyl; Cs-Cio cycloalkynyl; C 1-C9 heterocyclyl; and C 6 - Cio aryl; said alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl optionally being substituted with 1 , 2 or 3 groups R a ; and said heterocyclyl or aryl optionally being substituted with 1, 2, 3, 4 or 5 groups R b .
- R 2 is optionally substituted branched or unbranched C1-C9 alkyl, C2-C9 alkenyl or C2-C9 alkynyl.
- R 2 more particularly may be Ci-C 6 alkyl, C2-C6 alkenyl or C 2 -C 6 alkynyl, e.g. C 1 -C 4 alkyl, C 2 -C 4 alkenyl or C 2 -C 4 alkynyl.
- R 2 may be branched or unbranched C1-C9 alkyl, or Ci-C 6 alkyl, e.g. C1-C4 alkyl, e.g. C2-C4 alkyl.
- R 2 when R 2 is optionally substituted branched or unbranched C1-C9 alkyl, C2- C 9 alkenyl or C 2 -C 9 alkynyl, X is a bond.
- R 2 is optionally substituted C 3 -C 6 cycloalkyl or C 4 -C 6 cycloalkenyl, and then more particularly may be C 3 -C 6 cycloalkyl, in particular pentyl.
- R 2 is optionally substituted C 1 -C 9 heterocyclyl.
- R 2 may in particular be C1-C5 heterocyclyl, e.g. 5-membered C1-C4 heterocyclyl or 6-membered C1-C5 heterocyclyl, in particular 5-membered C1-C4 heterocyclyl, e.g. 5-membered C3-C4 heterocyclyl.
- R 2 may be selected from tetrahydrofuryl and thiazolyl.
- R 2 is optionally substituted C 6 -CiO aryl, and then preferably is phenyl.
- R 2 is alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, or cycloalkynyl, it optionally is substituted with 1, 2 or 3 groups, e.g. 1 or 2 groups, R a , as defined herein, i.e.
- halogen selected from halogen; hydroxy; C1-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; C1-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amino; C 1 - C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; wherein any alkyl, alkenyl or alkynyl moiety optionally is substituted with 1 , 2 or 3 groups R c .
- each R a is independently selected from C 1 - C4 alkyloxy; C2-C4 alkenyloxy; and C2-C4 alkynyloxy, in particular C1-C4 alkyloxy, such as Ci- C3 alkyloxy, more particularly methoxy and iso-propoxy.
- R 2 is substituted with 1 group R a and in another embodiment, R 2 is not substituted with any group R a .
- R 2 when R 2 is cycloalkyl, cycloalkenyl, or cycloalkynyl, it is not substituted with any group R a .
- R 2 When R 2 is heterocyclyl or aryl, it optionally is substituted with 1, 2, 3, 4 or 5 groups R b , as defined herein above. In one embodiment, R 2 is substituted with 1, 2, 3, 4 or 5 groups R b each independently selected from halogen, e.g. F, Cl and Br, in particular F or Br; cyano; and C1-C5 heterocyclyl, e.g. f ⁇ ve-membered C 1-C4 heterocyclyl.
- halogen e.g. F, Cl and Br, in particular F or Br
- cyano cyano
- C1-C5 heterocyclyl e.g. f ⁇ ve-membered C 1-C4 heterocyclyl.
- R 2 may be 6-membered heterocyclyl or aryl, such as phenyl, substituted with at least one R b in para -position in relation to the bond attaching R 2 to X or - in case X is a bond - in para -position in relation to the bond X.
- R 2 is substituted with 1-3 groups R b , e.g. 1 or 2 groups R b , in particular 1 group R b .
- R 3 and R 4 are each independently selected from C1-C4 alkyl; C2-C4 alkenyl; C 2 -C 4 alkynyl; C3-C4 cycloalkyl; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; Ci-C 4 alkylthio; C 2 -C 4 alkenylthio; C 2 -C 4 alkynylthio; halogen; Ci-C 4 alkyl, C 2 -C 4 alkenyl or C 2 - C 4 alkynyl secondary or tertiary amino; and Ci-C 4 alkyl, C 2 -C 4 alkenyl or C 2 -C 4 alkynyl carbonyl; wherein any alkyl, alkenyl, alkynyl or cycloalkyl moiety optionally is substituted with 1, 2, or 3 groups R c .
- R 3 and R 4 are each independently selected from Ci-C 4 alkyl and
- R 3 and R 4 may be both halogen or both Ci-C 4 alkyl, e.g. both are Cl or both are methyl, or one may be halogen, e.g. Cl, and the other one Ci-C 4 alkyl, e.g. methyl.
- R 3 is selected from C 1 -C 4 alkyl, e.g. methyl
- R 4 is selected from halogen and C 1 -C 4 alkyl, e.g. Cl and methyl.
- R 3 is selected from halogen, e.g. Cl
- R 4 is selected from halogen and C1-C4 alkyl, e.g. Cl and methyl.
- R 3 is selected from halogen and C 1 -C 4 alkyl, e.g. Cl and methyl, and R 4 is selected from C 1 -C 4 alkyl, e.g. methyl.
- R 3 is selected from halogen and C1-C4 alkyl, e.g. Cl and methyl, and R 4 is selected from halogen, e.g. Cl.
- R 5 and R 6 in formula (I) are independently selected from hydrogen, C1-C4 alkyl, C2-C4 alkenyl, C 2 -C 4 alkynyl and halogen; whereby any alkyl; alkenyl; or alkynyl is optionally substituted with 1, 2, or 3 groups R c .
- R 5 and R 6 are independently selected from hydrogen and C 1 -C 4 alkyl, more particularly from H and methyl, for example, R 5 and R 6 may both be H; or R 5 may be H and R 6 methyl.
- X represents a bond (i.e. a single, covalent bond) or branched or unbranched Ci- C3 alkyl, C2-C3 alkenyl or C2-C3 alkynyl.
- X represents a bond or C1-C3 alkyl, more preferably a bond or methylene (-CH2-).
- Q in formula (I) is selected from carbonyl, sulfonyl and sulf ⁇ nyl and preferably is carbonyl.
- m is an integer of 0, 1 or 2, preferably 0 or 1. In one particular embodiment, m is 0. In another embodiment, m is 1.
- Each R a is independently selected from halogen; hydroxy; C1-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; C1-C4 alkyl, C2-C4 alkenyl or C2- C4 alkynyl secondary or tertiary amino; C1-C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; wherein any alkyl, alkenyl or alkynyl moiety optionally is substituted with 1, 2 or 3 groups R c .
- each R a is independently selected from hydroxy; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; and C1-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amino; wherein any alkyl, alkenyl and alkynyl moiety optionally is substituted with 1, 2 or 3 groups R c .
- each R a is independently selected from hydroxy; C 1 -C 4 alkyloxy, e.g. methoxy and ⁇ o-propoxy; and C1-C4 alkyl secondary or tertiary amino, e.g.diethylamino.
- Each R b in formula (I) is independently selected from halogen; carboxy; hydroxy; cyano; C 1 -C 5 heteroaryl; Ci-C 4 alkyl; C 2 -C 4 alkenyl; C 2 -C 4 alkynyl; C 3 -C 5 cycloalkyl; Ci-C 4 alkyloxy; C 2 -C 4 alkenyloxy; C 2 -C 4 alkynyloxy; C3-C5 cycloalkyloxy; Ci-C 4 alkylthio; C 2 -C 4 alkenylthio; C 2 -C 4 alkynylthio; C3-C5 cycloalkylthio; Ci-C 4 alkyl, C 2 -C 4 alkenyl or C 2 -C 4 alkynyl secondary or tertiary amino; amino carbonyl; Ci-C 4 alkyl, C 2 -C 4 alkenyl or C 2 -C 4 alkynyl secondary
- acylsulphonamido Ci-C 4 alkyloxy, C 2 -C 4 alkenyloxy, or C 2 -C 4 alkynyloxy carbonyl; C1-C5 heterocyclyl; wherein any alkyl, alkenyl, alkynyl or cycloalkyl moiety optionally is substituted with 1 , 2 or 3 groups R c as defined herein.
- each R b is independently selected from halogen; carboxy; hydroxy; cyano; Ci -C 5 heteroaryl; Ci-C 4 alkyl; C 3 -C 5 cycloalkyl; Ci-C 4 alkyloxy; C 3 -C 5 cycloalkyloxy; Ci-C 4 alkylthio; Ci-C 4 alkyl secondary or tertiary amino; amino carbonyl; Ci-C 4 alkyl secondary or tertiary amido; Ci-C 4 alkyl carbonyl; Ci-C 4 alkyl sulfonyl; Ci-C 4 alkyl secondary or tertiary sulphonamido; Ci-C 4 alkyl secondary or tertiary acylsulphonamido; Ci-C 4 alkyloxy carbonyl; Ci-C 5 heterocyclyl; wherein any alkyl or cycloalkyl moiety optionally is substituted with 1, 2 or 3 groups R c as defined herein.
- each R b is independently selected from halogen; carboxy, cyano; Ci-C 5 heteroaryl; Ci-C 4 alkyl; C 2 -C 4 alkenyl; C 3 -C 5 cycloalkyl; Ci-C 4 alkyloxy; C 3 -C 5 cycloalkyloxy; C 2 -C 4 alkynyl; amino carbonyl; Ci-C 4 alkyl, C 2 -C 4 alkenyl or C 2 -C 4 alkynyl secondary or tertiary amido; Ci-C 4 alkyl, C 2 -C 4 alkenyl or C 2 -C 4 alkynyl secondary or tertiary acylsulphonamido; C 1 - C 4 alkyloxy carbonyl; and Ci-C 5 heterocyclyl; wherein any alkyl, alkenyl and alkynyl moiety optionally is substituted with 1 , 2 or 3 groups R c as defined herein.
- each R b is independently selected from halogen; carboxy; cyano; Ci-C 5 heteroaryl; Ci-C 4 alkyl; C 3 -C 5 cycloalkyl; Ci-C 4 alkyloxy; C 3 -C 5 cycloalkyloxy;
- Ci-C 4 alkyl secondary or tertiary amido Ci-C 4 alkyl secondary or tertiary acylsulphonamido
- Ci-C 4 alkyloxy carbonyl Ci-C 5 heterocyclyl; wherein any alkyl, moiety optionally is substituted with 1, 2 or 3 groups R c as defined herein.
- each R b is independently selected from halogen; carboxy, cyano; C 1-C5 heteroaryl; C1-C4 alkyl; amino carbonyl; C1-C4 alkyl secondary or tertiary amido; C1-C4 alkyl secondary or tertiary acylsulphonamido; C1-C4 alkyloxy carbonyl; and C1-C5 heterocyclyl; wherein any alkyl, moiety optionally is substituted with 1, 2 or 3 groups R c as defined herein.
- R b When R b is C1-C5 heterocyclyl, it may be C1-C4 heterocyclyl, and preferably is 6-membered Ci- C5 heterocyclyl or 5-membered C1-C4 heterocyclyl, more preferably 5-membered C1-C4 heterocyclyl.
- R b When R b is C1-C5 heteroaryl, it may be C1-C4 heteroaryl, and prefereably is 6- membered C1-C5 heteroaryl or 5-membered C1-C4 heteroaryl.
- R c in formula (I) is selected from fluorine and chlorine. In one embodiment, R c is absent. In another embodiment, R c is fluorine.
- X represents methylene or a bond and R 2 represents C3-C 6 cycloalkyl or heterocyclyl.
- X represents a bond and R 2 represents cyclopentyl.
- R 1 is selected from branched or unbranched C 1 -C 10 alkyl; C3-C10 cycloalkyl; C1-C9 heterocyclyl; and C 6 -CiO aryl; wherein any alkyl or cycloalkyl moiety optionally is substituted with 1, 2 or 3 groups R a ; and any heterocyclyl or aryl moiety optionally is substituted with 1, 2, 3, 4 or 5 groups R b ;
- R 2 is selected from branched or unbranched C1-C9 alkyl; C 3 -C 6 cycloalkyl; C1-C9 heterocyclyl; and C 6 -CiO aryl; wherein any alkyl or cycloalkyl moiety optionally is substituted with 1, 2 or 3 groups R a ; and any heterocyclyl or aryl moiety optionally is substituted with 1, 2, 3, 4 or 5 groups R b ;
- R 3 and R 4 are each independently selected from C 1 -C 4 alkyl and halogen;
- R 5 and R 6 are each selected from H and methyl; m is 0 or 1 ;
- Q is carbonyl;
- X represents a bond or methylene;
- each R a is independently selected from hydroxy; C1-C4 alkyloxy; C1-C4 alkyl secondary or tertiary amino;
- each R b is independently selected from halogen; cyano; C1-C5 heteroaryl, carboxy, C1-C4 alkyl; C 1 -C 4 alkyl secondary or tertiary amido; C 1 -C 4 alkyl carbonyl; C 1 -C 4 alkyloxy carbonyl; C 1 -C 4 alkyl secondary or tertiary acylsulphonamido; and C1-C5 heterocyclyl; and
- R c is absent.
- R 1 is selected from branched or unbranched C 1 -C 10 alkyl; C3-C6 cycloalkyl; 5- or 6-membered C 3 -C 5 heterocyclyl; and phenyl; wherein any alkyl or cycloalkyl . moiety optionally is substituted with 1 or 2 groups R a ; and any heterocyclyl or phenyl moiety optionally is substituted with 1 or 2 groups R b ;
- R 2 is selected from branched or unbranched Ci-C 6 alkyl; C3-C6 cycloalkyl; 5- or 6-membered C3- C 5 heterocyclyl; and phenyl; wherein any alkyl or cycloalkyl moiety optionally is substituted with 1 or 2 groups R a ; and any heterocyclyl or phenyl moiety optionally is substituted with 1 or 2 groups R b ;
- R 3 and R 4 are each independently selected from methyl and chloro
- R 5 and R 6 are each selected from H.
- R 1 represents C 1 -C 10 alkyl, e.g. Ci-Cs alkyl, or C 1 - C 6 alkyl
- R 2 represents C4-C6 cycloalkyl or C4-C5 heterocyclyl, e.g. C4-C6 cycloalkyl or 5- or 6- membered C4-C5 heterocyclyl
- R 3 and R 4 are independently selected from methyl and chlorine
- R 5 and R 6 are hydrogen
- X represents a bond
- Q represents carbonyl.
- the compound of formula (I) may be represented by the formula (Ia):
- intej ger p is 0-11, e.g. 0-9, or 0-7.
- R 1 represents C3-C6 cycloalkyl
- R 2 represents C 4 -C 6 cycloalkyl or C4-C5 heterocyclyl
- R 3 and R 4 are independently selected from methyl and chlorine
- X represents a bond
- Q represents carbonyl
- R 1 represents phenyl or C 4 -C 5 heterocyclyl
- R 2 represents C 4 -C 6 cycloalkyl or C4-C5 heterocyclyl
- R 3 and R 4 are independently selected from methyl and chlorine
- X represents a bond
- Q represents carbonyl
- R 1 represents phenyl or C4-C5 heterocyclyl, e.g. 5- or 6-membered C4-C5 heterocyclyl, optionally substituted with one or more R b ;
- R 2 represents cyclopentyl;
- R 3 and R 4 are independently selected from methyl and chlorine; the integer m is equal to 1 or 0 (zero);
- R 5 and R 6 are hydrogen;
- X represents a bond; and Q represents carbonyl.
- the compound of formula (I) may be represented by the formula (Ib):
- R 1 represents C 1 -C 10 alkyl, e.g. Ci-Cs alkyl, or Ci- C 6 alkyl
- R 2 represents Ci-C 9 alkyl, e.g. Ci-C 6 alkyl, or Ci-C 4 alkyl
- R 3 and R 4 are independently selected from methyl and chlorine, R 5 and R 6 are hydrogen
- Q represents carbonyl.
- the compound of formula (I) may be represented by the formula (Ic):
- R 1 represents C4-C5 saturated heterocyclyl, e.g. 5- or 6-membered C4-C5 heterocyclyl
- R 2 represents cyclopentyl
- R 3 and R 4 are independently selected from methyl and chlorine
- the integer m is equal to 1
- R 5 and R 6 are hydrogen
- X represents a bond
- Q represents carbonyl.
- the compound of formula (I) may be represented by the formula (Id):
- R 1 represents C 3 -C 5 cycloalkyl
- R 2 represents cyclopentyl or tetrahydrofuranyl
- R 3 and R 4 are independently selected from methyl and chlorine
- the integer m is equal to either 0 (zero) or 1
- R 5 and R 6 are hydrogen
- X represents a bond
- Q represents carbonyl
- the compounds of formula (I) of the invention can be prepared according to the synthetic routes outlined herein and by following the methods described herein below and illustrated in the Examples. Alternative synthetic routes to these compounds can easily be visualized by any person skilled in the art and the present synthetic routes are not limiting for the invention. With respect to the reaction schemes below, although the various R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , Q and X moieties sometimes are specifically defined, unless otherwise indicated, it is to be understood that R 1 , R 2 , R 3 , R 4 and X may be any of the groups encompassed thereby.
- a compound of formula (I) as defined herein may be formed in a three step procedure wherein, first, a suitable substituted 2-aminobenzoimidazol (i) is diazotizated and subjected to the Sandmeyer reaction, or any suitable modification of this method, the formed chloro intermediate (ii) is coupled with a piperidine-4-carboxamide derivative (iii) to form the product (iv), which subsequently is JV-alkylated or arylated by Y- (CR 5 R 6 ) m -R 1 (v) to yield the final compound of formula (I).
- the entire synthetic route is depicted below in Reaction Scheme 1.
- diazonium compounds from aromatic or heteroaromatic amines has been known for more than 150 years and involves an amine that reacts with a nitrite salt such as sodium nitrite to form a diazonium salt at low temperature, a procedure that should be well known for the one skilled in the art.
- a nitrite salt such as sodium nitrite
- diazonium compounds are not isolated, but used immediately in further reactions as described below in the Sandmeyer reaction.
- Diazonium compounds can, however, also be isolated as tetrafluoroborate salts.
- Conditions suitable for the Sandmeyer reaction are well known to the person skilled in the art and comprise an diazonium salt of an aromatic or heterocyclic kept at low temperature, which is decomposed in the presence of copper(I) salts, such as copper(I) chloride or copper(I) bromide, to form the desired chloro or bromo derivative.
- copper(I) salts such as copper(I) chloride or copper(I) bromide
- the chloro intermediate (ii) is reacted with the piperidine-4-carboxamide derivative (iii) to form the coupled product (iv) in the presence of a base, such as as N, ⁇ /-diisopropylethylamine (Hunig's base, DIEA, CAS Registry Number: 7087-68-5), in a suitable solvent, such as 1,4-dioxane.
- a base such as as N, ⁇ /-diisopropylethylamine (Hunig's base, DIEA, CAS Registry Number: 7087-68-5)
- a suitable solvent such as 1,4-dioxane.
- TMP 2,2,6,6- tetramethylpiperidine
- JV-alkylation or JV-arylation by Y-R 1 is employed.
- JV-alkylation is treated with a chloro-, bromo or iodoalkyl electrophile in the presence of a base such as caesium carbonate in a solvent such as acetonitrile and/or N, ⁇ /-dimethylformamide.
- a palladium catalyst such as, but not limited to [1,1'- bis(diphenylphosphino)ferrocene] dichloropalladium(II) (PdCl2(dppf), CAS Registry Number: 72287-26-4) can also be employed in this reaction.
- (iv) is treated with a haloaryl in the presence of a base such as caesium carbonate, and an agent as 1,10-phenanthroline, 4,7-dimethoxy-l,10- phenanthroline or 8-hydroxyquinoline, polyethylene glycol and copper(I) oxide in a solvent such as dimethylsulfoxide.
- a base such as caesium carbonate
- an agent such as 1,10-phenanthroline, 4,7-dimethoxy-l,10- phenanthroline or 8-hydroxyquinoline, polyethylene glycol and copper(I) oxide
- solvent such as dimethylsulfoxide
- Alternative reagents include phosphorus pentachloride (CAS Registry Number: 10026-13-8) or phosphorus trichloride (CAS Registry Number: 7719-12-2), the former also, alone or as mixtures in various ratios, or any other chlorination agent, all that exists in abundance in the scientific literature, for the purpose of transforming a hydroxyl group to a chlorine group.
- the corresponding bromo derivative of (ii) also can be useful as an intermediate in the reaction schemes, leading to the target compounds of the invention, and could be prepared by the action of phosphoryl bromide (CAS Registry Number: 7789-59-5) phosphorus tribromide (CAS
- the carboxylic acid ester (xi) is saponified with a base such as aqueous sodium hydroxide and a solvent such as ethanol. Acidification of the completed reaction mixture is followed by standard work-up and crystallization or chromatography, to yield the carboxylic acid (xii).
- a base such as aqueous sodium hydroxide
- a solvent such as ethanol.
- Acidification of the completed reaction mixture is followed by standard work-up and crystallization or chromatography, to yield the carboxylic acid (xii).
- a wide variety of other protecting groups for the carboxylic acid can be employed, and their usage is known to those skilled in the art (references describing protecting group strategy include, for example, "Protecting Groups in Organic Chemistry", J. F. W. McOmie, Plenum Press, London, New York, 1973, and "Protective Groups in Organic Synthesis", T. W. Greene, Wiley, New York, 1984).
- a suitable amide coupling reagent such as 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (HATU; CAS Registry Number: 148893-10-1) and N,N- diisopropylethylamine in a suitable solvent, such as dichloromethane, or by reacting the acid derivative in the presence of e.g. triethylamine in a suitable solvent, such as chloroform or in the presence of N,7V-diisopropylethylamine in a suitable solvent, such as dichloromethane.
- a suitable amide coupling reagent such as 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (HATU; CAS Registry Number: 148893-10-1) and N,N- diisopropylethy
- the present invention also involve the use of microwave irridation for heating purposes, which today is well known for the one skilled in the art, as witnessed by a large number of book chapters and published books [eg. Strauss, C. R., Application of microwaves for environmentally benign organic chemistry, In Handbook of Green Chemistry and Technology, Clark, J.;
- reaction mixtures are purified by standard purification procedures as extraction, washing, recrystallization, and column chromatography employing silica gel, aluminia or reverse phase and the like, employing the appropriate eluents for the purification of the Examples described herein.
- the compounds of the invention can also be prepared by the application of combinatorical and/or parallell chemistry techniques eg. methods of making libraries of compounds, techniques well known for the one skilled in the art, which can include solution phase synthesis, and solid phase synthesis including design of resins, linkers and the like.
- combinatorical and/or parallell chemistry techniques eg. methods of making libraries of compounds, techniques well known for the one skilled in the art, which can include solution phase synthesis, and solid phase synthesis including design of resins, linkers and the like.
- (x) can be saponif ⁇ cated to give the intermediate (xiv) that can serve as a suitable scaffold for further manipulation.
- JV-alkylation with (v), followed by amide coupling with (xiii) can be conducted in one pot (in the same reaction vessel) if the two reactions are selective in terms of regioselectivity (a term that is well known for the one skilled in the art), which also includes the reverse reaction order eg. amide coupling followed by N-alkylation, or alternatively both reactions acting at the same time.
- regioselectivity a term that is well known for the one skilled in the art
- Outlined in Reaction Scheme 7 is the one of the synthetic strategies of the invention that involves tautomeric forms of the intermediate compounds. Accordingly, the tautomeric mixture of (iia) and (iib) is treated with a nucleophilic agent, as exemplified by (iii), but not restricted to, then yet two tautomeric compounds (iva) and (ivb) are formed. In the final step an N-alkylating agent (v) is employed and a regioisomeric mixture of end products (I) is formed, which might be tested directly in biological assays.
- a nucleophilic agent as exemplified by (iii)
- iva tautomeric compounds
- ivb N-alkylating agent
- the regioisomers of the mixture can be separated before biological testing, using for instance column chromatography, recrystallization, sublimation or any other methods available for the one skilled in the art of chemical separation.
- the final purification can only take palace after the final synthetic step, since all previous intermediates are tautomers.
- N-alkylation or N-arylation can be introduced at an earlier stage in the synthetic procedure, as exemplified shown in Reaction Scheme 7.
- This will provide the compounds (xia) and (xib), which already at this stage can be separated using any of the techniques mentioned above.
- this strategy will ultimately lead to the end products (I).
- This synthetic strategy can, for example, be considered when the final Examples are difficult to separate, or because of any other advantageous reason.
- the invention at hand also comprises a synthetic method that is strictly
- N-alkylation when N-alkylation is mentioned in the text or in the Reaction Schemes, then the N-arylation of the available -NH- or -NH 2 group is not excluded, but an important aspect of the present invention. Also, when only N-arylation is mentioned, then should neither N-alkylation be excluded.
- the present invention also comprises stereoisomers that, for example, are prepared with the available metods described above.
- a large number of available single enantiomers are available commercially or in the literature.
- the single enantiomers can be amines that are used in the amide coupling of present invention, such as, but not limited to, (R)-(+)-tetrahydrofuran-3 -amine 4-methylbenzenesulfonate (CAS Registry Number: 111769-27-8), (S)-(-)-tetrahydrofuran-3- amine hydrochloride (CAS Registry Number: 204512-95-8), (lS,2S)-trans-2- aminocyclopentanol hydrochloride (CAS Registry Number: 68327-04-8), (lR,2R)-trans-2- aminocyclopentanol hydrochloride (CAS Registry Number: 68327-11-7).
- (xxii), which includes a protection group PG, is coupled with an amine (xiii) to give the sulphonamide (xxiii), that can be coupled, after de-protection with a suitable halobenzimidazole (ii), and finally N-alkylated or N-arylated with (v) to the end products (I).
- a suitable halobenzimidazole ii
- v N-alkylated or N-arylated with (v)
- H a and H d correlates as a pair, as H b and H c .
- the compounds according to formula (I) will be useful for treating various diseases such as inflammatory diseases, nociceptive pain, auto-immune disease, breathing disorders, fever, cancer, inflammation related anorexia, Alzheimer's disease and cardiovascular disease.
- the treatment may be preventive, palliative or curative.
- pharmaceutically acceptable addition salts for use in the pharmaceutical compositions of the present invention include those derived from mineral acids, such as hydrochloric, hydrobromic, phosphoric, metaphosphoric, nitric and sulphuric acids, and organic acids, such as tartaric, acetic, citric, malic, lactic, fumaric, benzoic, glycolic, gluconic, succinic, and arylsulphonic acids.
- the pharmaceutically acceptable excipients described herein for example, vehicles, adjuvants, carriers or diluents, are well-known to those who are skilled in the art and are readily available to the public.
- the pharmaceutically acceptable carrier may be one that is chemically inert to the active compounds and that has no detrimental side effects or toxicity under the conditions of use. Pharmaceutical formulations are found e.g. in Remington: The Science and Practice of Pharmacy, 19th ed., Mack Printing Company, Easton, Pennsylvania (1995).
- All stereoisomers of the compounds of the instant invention are contemplated, either in admixture or in pure or substantially pure form.
- the compounds of the present invention can have asymmetric centers at any of the carbon atoms including any one of the R substitutents. Consequently, compounds of formula (I) can exist in enantiomeric or diasteromeric forms or in mixtures thereof.
- the processes for preparation can utilize racemates, enantiomers or diasteromers as starting materials. When diastereomeric or enantiomeric products are prepared, they can be separated by conventional methods, which for example is chromatographic or fractional crystallization.
- Prodrugs of the compounds of formula (I) may be prepared by modifying functional groups present on the compound in such a way that the modifications are cleaved, in vivo when such prodrug is administered to a mammalian subject. The modifications typically are achieved by synthesizing the parent compound with a prodrug substituent.
- Prodrugs include compounds of formula (I) wherein a hydroxy, amino, sulfhydryl, carboxy or carbonyl group in a compound of formula (I) is bonded to any group that may be cleaved in vivo to regenerate the free hydroxyl, amino, or sulfhydryl group, respectively.
- prodrugs include, but are not limited to, esters and carbamates of hydroxy functional groups, esters groups of carboxyl functional groups, N-acyl derivatives, JV-Mannich bases.
- General information on prodrugs may be found in the scientific literature [eg. Bundegaard, H. "Design of Prodrugs” p 1-92, Elesevier, New York- Oxford, 1985; The Practice of Medicinal Chemistry, Camille G. Wermuth et al., chapter 31, Academic Press, 1996; A Textbook of Drug Design and Development, P. Krogsgaard-Larson and H. Bundgaard, eds. chapter 5, pgs 113 - 191, Harwood Academic Publishers, 1991). Said references are incorporated herein by reference.
- the compounds of the formula (I) can be administered for any of the uses described herein by any suitable means, for example, orally, such as in the form of tablets, aqueous or oily suspensions or solutions, elexirs, syrups, capsules, granules or powders; sublingually; buccally; ocular; parenterally, such as by subcutaneous, intravenous, intramuscular, or intrasternal injection or infusion techniques (e.g., as sterile injectable aqueous or non-aqueous solutions or suspensions). Also, the compounds of the formula (I) may be applied as gargles and mouth washes.
- a parenterally acceptable aqueous or oily suspension, emulsion or solution is employed, which is pyrogen free and has requisite pH, isotonicity and stability.
- a parenterally acceptable aqueous or oily suspension, emulsion or solution is employed, which is pyrogen free and has requisite pH, isotonicity and stability.
- the compounds of the formula (I) can be administered nasally, including administration to the nasal membranes, such as by inhalation spray; topically, such as in the form of a gel, cream or ointment; or rectally such as in the form of suppositories; in dosage unit formulations containing non-toxic, pharmaceutically acceptable vehicles or diluents.
- the present compounds can, for example, be administered in a form suitable for immediate release or extended release. Immediate release or extended release can be achieved by the use of suitable pharmaceutical compositions comprising the present compounds, or, particularly in the case of extended release, by the use of devices such as subcutaneous implants or osmotic pumps.
- the present compounds can also be administered liposomally.
- the precise nature of the carrier or other material will depend on the route of administration and those skilled in the art are well able to prepare suitable solutions and numerous methods are described in the literature.
- compositions for oral administration include suspensions which can contain, for example, microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners or flavoring agents such as those known in the art; and immediate release tablets which can contain, for example, microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and/or lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants such as those known in the art.
- the compounds of formula (I) can also be delivered through the oral cavity by sublingual and/or buccal administration.
- Molded tablets, compressed tablets or freeze-dried tablets are exemplary forms which may be used.
- Exemplary compositions include those formulating the present compound(s) with fast dissolving diluents such as mannitol, lactose, sucrose and/or cyclodextrins. Also included in such formulations may be high molecular weight excipients such as celluloses (avicel) or polyethylene glycols (PEG).
- Such formulations can also include an excipient to aid mucosal adhesion such as hydroxy propyl cellulose (HPC), hydroxy propyl methyl cellulose (HPMC), sodium carboxy methyl cellulose (SCMC), maleic anhydride copolymer (e.g., Gantrez), and agents to control release such as polyacrylic copolymer (e.g. Carbopol 934).
- HPC hydroxy propyl cellulose
- HPMC hydroxy propyl methyl cellulose
- SCMC sodium carboxy methyl cellulose
- maleic anhydride copolymer e.g., Gantrez
- agents to control release such as polyacrylic copolymer (e.g. Carbopol 934).
- Lubricants, glidants, flavors, coloring agents and stabilizers may also be added for ease of fabrication and use.
- compositions for nasal aerosol or inhalation administration include solutions in saline which can contain, for example, benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, and/or other solubilizing or dispersing agents such as those known in the art.
- compositions for parenteral administration include injectable solutions, emulsions or suspensions which can contain, for example, suitable non-toxic, parenterally acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, oil or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid, or Cremaphor.
- suitable non-toxic, parenterally acceptable diluents or solvents such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, oil or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid, or Cremaphor.
- compositions for rectal administration include suppositories which can contain, for example, a suitable non-irritating excipient, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures, but liquify and/or dissolve in the rectal cavity to release the drug.
- a suitable non-irritating excipient such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures, but liquify and/or dissolve in the rectal cavity to release the drug.
- compositions for topical administration include a topical carrier such as Plastibase (mineral oil gelled with polyethylene).
- a topical carrier such as Plastibase (mineral oil gelled with polyethylene).
- the dose administered to a mammal, particularly a human, in the context of the present invention should be sufficient to effect a therapeutic response in the mammal over a reasonable time frame.
- dosage will depend upon a variety of factors including the potency of the specific compound, the age, condition and body weight of the patient, the nature and extent of the condition being treated, recommendations of the treating physician, and the therapeutics or combination of therapeutics selected for administration, as well as the stage and severity of the disease.
- the dose will also be determined by the route (administration form), timing and frequency of administration.
- Oral dosages of the present invention when used for the indicated effects, will range between about 0.01 mg per kg of body weight per day (mg/kg/day) to about 100 mg/kg/day, preferably 0.01 mg per kg of body weight per day (mg/kg/day) to 20 mg/kg/day, and most preferably 0.1 to 10 mg/kg/day, for adult humans.
- the compositions are preferably provided in the form of tablets or other forms of presentation provided in discrete units containing 0.5 to 1000 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated, for example 0.5, 1.0, 2.5, 5.0, 10.0, 15.0, 25.0, 50.0, 100, 200, 400, 500, 600 and 800 mg..
- the most preferred doses will range from about 0.001 to about 10 mg/kg/hour during a constant rate infusion.
- compounds of the present invention may be administered in a single daily dose, or the total daily dosage may be
- a nasal, sublingual or buccal administration typically contains from about 0.01 mg to about 500 mg of the active ingredient, preferably from about 1 mg to about 100 mg of active ingredient, per dose.
- the compounds of the present invention may be also be used or administered in combination with one or more additional drugs useful in the treatment of inflammatory diseases, nociceptive pain, auto-immune diseases, breathing disorders, fever, cancer, inflammation related anorexia, Alzheimer's disease and cardiovascular diseases.
- the components may be in the same formulation or in separate formulations for administration simultaneously or sequentially.
- the compounds of the present invention may also be used or administered in combination with other treatment such as irradiation for the treatment of cancer.
- (B) is formulated in admixture with a pharmaceutically-acceptable adjuvant, diluent or carrier.
- a pharmaceutically-acceptable adjuvant, diluent or carrier Such combination products provide for the administration of a compound of the invention in conjunction with the other therapeutic agent, and may thus be presented either as separate formulations, wherein at least one of those formulations comprises a compound of the invention, and at least one comprises the other therapeutic agent, or may be presented (i.e. formulated) as a combined preparation (i.e. presented as a single formulation including a compound of the invention and the other therapeutic agent).
- a pharmaceutical formulation including a compound of the invention, as hereinbefore defined, another therapeutic agent, and a pharmaceutically-acceptable adjuvant, diluent or carrier;
- cyclooxygenase-2 inhibitors and conventional NSAIDs in preparations wherein they are presently co-administered with other agents or ingredients.
- agents for the administration in combination with compounds of the invention include suitable anti-inflammatory, anti-pain, anti-autoimmune, anti-fever, anti-cancer and anti- anorexia (inflammatoriy) agents, and agents for the treatment or prevention of breathing disorders, cardiovascular diseases and Alzheimer's disease, a potentiator including caffeine, an H2 antagonist, aluminium or magnesium hydroxide, simethicone, a decongestant, an antitussive, an antihistamine, a diuretic, a proton pump inhibitor, a bradykinin-1 antagonist, a sodium channel blocker, a 5 -HT agonist or a CGRP antagonist.
- compounds for use in combination with the compounds of the present invention include, but is not limited to, prednisone (CAS Registry Number: 53-03-2);
- dexamethasone (CAS Registry Number: 50-02-2); any of the selective glucocorticoid receptor agonists (SEGRAs) exemplified by All 6515 (Lin, C. et al. MoL Pharm., 2002, 62, 297-303), and ZK209614 (Schacke, H. et al, Proc. Natl. Acad. Sci.
- budesonide (CAS Registry Number: 51333-22-3); clofazimine (CAS Registry Number: 2030-63-9); selective thyroid hormone agonists such as GC-I (Johansson, L. et al,Proc. N ⁇ tl. Ac ⁇ d. Sci. USA. 2005, 102, 10297-10302), KB2115 (Berkenstam, A. et al., Proc. N ⁇ tl Ac ⁇ d. Sci. USA. 2008, 105, 663-667); KB-141 (Graver, G. J. et al., Proc. N ⁇ tl. Ac ⁇ d. Sci. USA.
- FXRs farnesoid X receptor agonists
- WAY-362450 farnesoid X receptor agonists
- CD4 antagonists such as priliximab (CAS Registry Number: 147191-91-1); p38 mitogen-activated protein kinase inhibitors such as SB203580 (CAS Registry Number: 152121-47-6); mesalazine (CAS Registry Number: 89-57-6); azathioprine (CAS Registry Number: 446-86-6); sumatriptan (CAS Registry Number: 103628- 46-2); paracetamol (CAS Registry Number: 103-90-2); fast-acting bronchodilators such as salbutamol (CAS Registry Number: 18559-94-9) and ephedrine (CAS Registry Number: 299-42- 3); rituximab; NO-releasing drugs such as nitroglycerine (CAS Registry Number: 55-63-0) and isosorbide dinitrate (CAS Registry Number: 87-33-2); Selective Estrogen Receptor Modulators (SERMs) such as tamoxifen (CAS Registry Number: 147191-91-1);
- antidepressant drugs and pain-relivers such as tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) such as desvenlafaxine (CAS Registry Number: 93413-62-8); duloxetine (CAS Registry Number:
- treating encompasses not only treating a patient to relieve the patient of the signs and symptoms of the disease or condition but also prophylactically treating an asymptomatic patient to prevent the onset or progression of the disease or condition.
- patients include mammalian (including human) patients.
- the term "effective amount” refers to an amount of a compound, which confers a therapeutic effect on the treated patient.
- the effect may be objective (i.e. measurable by some test or marker) or subjective (i.e. the subject gives an indication of or feels an effect).
- the expression "purified on reverse- phase hplc" will mean utilizing a preparative hplc-system comprising the following conditions and equipment: Shimadzu LC8A, C 18 column (22 x 250 mm, lO ⁇ m), acetonitrile/water gradient (0.1% trifluoroacetic acid), flowrate 15 mL/min.
- N-cyclopentyl- 1 -(5,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (17.0 mg, 0.05 mmol), 1-iodopropane (17.0 mg, 0.10 mmol), caesium carbonate (48.9 mg, 0.15 mmol) and acetonitrile (2 mL) were mixed and the reaction mixture heated at 100 0 C over night.
- N-cyclopentyl-l-(l-heptyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide was prepared from ⁇ /-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (17.0 mg, 0.05 mmol), 1-bromo heptane (17.9 mg, 0.10 mmol), caesium carbonate (48.9 mg, 0.15 mmol) and acetonitrile (2 mL), using the method described in Example 2 to give 12.2 mg (56 % yield) of the title compound.
- N-cyclopentyl- 1 -(5,6-dimethyl- 1 -octyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide was prepared from N-cyclopentyl- 1 -(5,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (17.0 mg, 0.05 mmol), 1-bromooctane (19.3 mg, 0.1 mmol), caesium carbonate (48.9 mg, 0.15 mmol) and acetonitrile (2 mL), using the method described in Example 2. This gave 8.4 mg (37% yield) of the title compound.
- the reaction mixture was concentrated in vacuo and purified on preparative hplc (performed on a Gilson- Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 ⁇ m, 21.2 x 150 mm) column, using acetonitrile/water (0.05 % formic acid) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection). This gave 0.7 mg (3 % yield) of JV-cyclopentyl-1- (5,6-dimethyl-l-(pyridin-2-ylmethyl)-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC- MS (m/z) 432.3 (M+ 1).
- N-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-3-ylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide was prepared from sodium hydride (72 mg, 0.30 mmol) in N, ⁇ /-dimethylformamide (2 mL), ⁇ /-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (80 mg, 0.20 mmol) in N, ⁇ /-dimethylformamide (2 mL) and 3 -bromo methyl pyridinium
- N-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-2-ylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide was prepared from sodium hydride (72 mg, 0.30 mmol) in N, ⁇ /-dimethylformamide (2 mL), ⁇ /-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (80 mg, 0.20 mmol) in N, ⁇ /-dimethylformamide (2 mL) and 2-bromomethyl pyridinium
- the resulting reaction mixture was subjected to microwave irradiation for 45 min at 12O 0 C.
- the reaction mixture was concentrated in vacuo, the solid residue filtered off, washed with ethyl acetate and the filtrate extracted with ethyl acetate (2 x 15 mL).
- the combined organic phases were washed with water (3 x 15 mL), dried over anhydrous sodium sulfate and concentrated in vacuo.
- the reaction mixture was filtered, cooncentrated in vacuo and subjected to preparative hplc (performed on a Gilson-Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 ⁇ m, 21.2 x 150 mm) column, using acetonitrile/water (0.05 % formaldehyde) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 1.9 mg (18 %) of the title compound.
- N-cyclopentyl- 1 -(5,6-dichloro- 1 -ethyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide was prepared from ⁇ /-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (10 mg, 0.026 mmol), caesium carbonate (25.6 mg, 0.079 mmol), acetonitrile (2 mL), ⁇ /, ⁇ /-dimethylformamide (0.5 mL), iodoethane (6.1 mg, 0.039 mmol) using the method described in Example 18 to give 5.4 mg (50 % yield) of the title compound.
- N-cyclopentyl- 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide was prepared from ⁇ /-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (10 mg, 0.026 mmol), caesium carbonate (25.6 mg, 0.079 mmol), acetonitrile (2 mL), ⁇ /, ⁇ /-dimethylformamide (0.5 mL), 2-bromopropane (9.7 mg, 0.079 mmol) using the method described in Example 18 to give 5.0 mg (45 % yield) of the title compound.
- N-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-fluorobenzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide was prepared from ⁇ /-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (10 mg, 0.026 mmol), caesium carbonate (25.6 mg, 0.079 mmol), acetonitrile (2 mL), N, ⁇ /-dimethylformamide (0.5 mL), l-(bromomethyl)-4-fluorobenzene (4.96 mg, 0.026 mmol) using the method described in Example 18 to give 4.2 mg (33 % yield) of the title compound.
- Example 23 methyl 4-((2-(4-(cyclopentylcarbamoyl)piperidin-l-yl)-5,6-dimethyl-lH- benzo[d]imidazol-l-yl)methyl)benzoate
- N-cyclopropyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (0.09 g, 0.25 mmol) was dissolved in N, ⁇ /-dimethylformamide (1 mL).
- Caesium carbonate (332 mg, 1 mmol) and isopropyl bromide (56 mg, 0.45 mmol) were added, and the reaction mixture irradiated under microwave conditions at 120 0 C for 60 minutes.
- the reaction mixture was quenched with water (2 mL) and extracted with ethyl acetate (2 x25 mL).
- N-cyclobutyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide was prepared from 1 -(5,6-dichloro- lH-benzoimidazol-2-yl)-piperidine-4-carboxylic acid (0.05 g, 0.16 mmol), ⁇ /, ⁇ /-dimethylformamide (2 mL), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate ( ⁇ ATU, 91 mg, 0.24 mmol), ⁇ /, ⁇ /-diisopropylethylamine ( ⁇ unig's base,
- N-cyclo butyl- 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide was prepared from N-cyclobutyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.28g, 0.76mmol), N, ⁇ /-dimethylformamide (3 mL), caesium carbonate (0.99 g, 3 mmol) and isopropyl bromide (168 mg, 1.3 mmol) using the method described in Example 26(d) to give 61 mg (20 % yield) of the title compound as a light yellow solid.
- l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-propylpiperidine-4-carboxamide was prepared from l-(5,6-dichloro-lH-benzoimidazol-2-yl)-piperidine-4-carboxylic acid (0.07 g, 0.22 mmol), N, ⁇ /-dimethylformamide (4 mL), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate ( ⁇ ATU, 127 mg, 0.33 mmol), JV,JV-diisopropylethylamine ( ⁇ unig's base, DIEA, 0.052 mL, 0.45 mmol) and propan-1 -amine (0.27 mmol), using the method described in Example 26(c) to give 10 mg (13 % yield) of l-(5,6-d
- N-(cyclohexylmethyl)- 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide was prepared from N-(cyclohexylmethyl)-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.15 g, 0.36 mmol), N, ⁇ /-dimethylformamide (3 mL), caesium carbonate (0.716 g, 2.2 mmol) and isopropyl bromide (0.09 g, 0.72 mmol) using the method described in Example 29(b) to give 20 mg (12 % yield) of the title compound.
- the title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.29 mmol), N, ⁇ /-dimethylformamide (2 mL), caesium carbonate (0.566 g, 1.7 mmol) and 4-(bromomethyl)benzonitrile (0.103 g, 0.52 mmol), using the method described in Example 36 to give 0.08 g (62 % yield) of l-(l-(4-cyanobenzyl)-5,6- dimethyl-lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as a white solid.
- the title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.29 mmol), N, ⁇ /-dimethylformamide (2 mL), caesium carbonate (0.566 g, 1.7 mmol) and cyclobutylbromide (0.07 g, 0.527 mmol), using the method described in Example 36, to give 9 mg (8 % yield) of l-(l-cyclobutyl-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as an off-white solid.
- a reaction mixture consisting of l-(l-(4-bromobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)-N-cyclopentylpiperidine-4-carboxamide (0.1 g, 0.19 mmol), £ra/?s-di( ⁇ -acetato)-bis[o-(di-o- tolylphosphino)-benzyl]dipalladium(II) (Hermann palladacycle, 0.089 g, 0.095 mmol), N 5 N- diisopropylethylamine (Hunig's base, DIEA, 0.07 mL, 0.39 mmol), 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU, 0.19 mmol), tri tertiarybutyl phosphonium
- the title compound was prepared from l-(l-(4-bromobenzyl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide (0.1 g, 0.19 mmol), trans-di( ⁇ - acetato)-bis[o-(di-o-tolylphosphino)-benzyl]dipalladium(II) (Hermann palladacycle, 0.089 g, 0.095 mmol), N,N-diisopropylethylamine (Hunig's base, 0.07 mL, 0.39 mmol), 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU, 0.028 mL, 0.19 mmol), tri tertiarybutyl phosphonium hexafluoborate ([(t-Bu) 3 PH]BF 4 , 0.0055 g,
- Example 46 7V-cyclopentyl-l-(5,6-dimethyl-l-(4-(methylsulfonylcarbamoyl)benzyl)-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide l-(l-(4-bromobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4- carboxamide (O.lg, 0.19mmol), £rans-di( ⁇ -acetato)-bis[o-(di-o-tolylphosphino)- benzyl]dipalladium(II) (Hermann palladacycle, 0.089 g, 0.095 mmol), N 5 N- diisopropylethylamine (Hunig's base, DIEA, 0.07 niL, 0.39 mmol), 1,8- diazabicyclo
- reaction mixture was allowed to warm to room temperature and was stirred for 10 hours.
- the reaction mixture was washed with aqueous hydrochloric acid (3 x15 mL, 2N), saturated aqueous sodium bicarbonate (2 x15 mL), dried over magnesium sulfate, filtered and the filtrate concentrated in vacuo.
- the residue was recrystallized from dichloro methane/pet ether to give 1.15 g (88%) of tert-butyl 4-(tetrahydrofuran-3-ylcarbamoyl)piperidine-l-carboxylate.
- Example 49 l-(5-chloro-l-cyclobutyl-6-methyl-lH-benzo[d]imidazol-2-yl)-7V-cyclopentyl- piperidine-4-carboxamide and l-(6-chloro-l-cyclobutyl-5-methyl-lH-benzo[d]imidazol-2- yl)-7V-cyclopentylpiperidine-4-carboxamide
- aqueous extract was acidified to pH 5 via addition of aqueous hydrochloric acid (1.57V) and the resulting brown precipitate was filtered, washed with water (3 ⁇ 20mL) and dried under high vacuum to obtain 0.28 g (80 % yield) of 5-chloro-6-methyl-lH- benzo[d]imidazol-2(3H)-one.
- N-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-cyanobenzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide was prepared from ⁇ /-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (50 mg, 0.13 mmol), N, ⁇ /-dimethylformamide (0.5 mL), caesium carbonate (171 mg, 0.52 mmol) and 4-(bromomethyl)benzonitrile (31 mg, 0.15 mmol), using the method described in Example 52 to give 20 mg (15 % yield) of the title compound.
- N-cyclopentyl- 1 -(5,6-dichloro- 1 -(cyclopropylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide was prepared from ⁇ /-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (100 mg, 0.26 mmol), N, ⁇ /-dimethylformamide (0.5 mL), caesium carbonate (512 mg, 1.5 mmol) and (bromomethyl)cyclopropane (43 mg, 0.32 mmol), using the method described in Example 52 to give 19 mg (17 % yield) of the title compound.
- N-cyclopentyl- 1 -(5,6-dichloro- 1 -((tetrahydro-2H-pyran-4-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide was prepared from N-cyclopentyl- 1 -(5 ,6-dichloro- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide (100 mg, 0.26 mmol), N,N-dimethylformamide (0.5 mL), caesium carbonate (512 mg, 1.5 mmol), and 4-(bromomethyl)tetrahydro-2H-pyran (80 mg, 0.47 mmol), using the method described in Example 55 to give 70 mg (57 % yield) of the title compound.
- a reaction mixture consisting of l-(l-(4-bromobenzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)- N-cyclopentylpiperidine-4-carboxamide (Example 51, 100 mg, 0.182 mmol), trans-di( ⁇ - acetato)-bis[o-(di-o-tolylphosphino)-benzyl]dipalladium(II) (Hermann palladacycle, 9 mg, 0.009 mmol), ⁇ /, ⁇ /-diisopropylethylamine (Hunig's base, DIEA, 0.06 mL, 0.36 mmol), 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU, 0.028 mL, 1.82 mmol), tri tertiarybutyl phosphonium hexafluoborate ([(t-Bu)sPH]BF4,
- the reaction mixture was filtered, concentrated in vacuo and subjected to preparative hplc (performed on a Gilson-Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 ⁇ m, 21.2 x 150 mm) column, using acetonitrile/water (0.05% HCOOH) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 3.6 mg (15 %) of the title compound.
- N-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-cyanobenzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (Example 53, 50 mg, 0.1 mmol), sodium azide (78 mg, 1.2 mmol), ammonium chloride (64 mg, 1.2 mmol), lithium chloride (catalytic amount) was refluxed in N 5 N- dimethylformamide (3 mL) for 2 hours.
- the title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.1 g, 0.26 mmol), N, ⁇ /-dimethylformamide (2 mL), caesium carbonate (0.508 g, 1.56 mmol) and 1-bromopentane (0.033 mL, 0.31mmol) using the method described in Example 64(b), to give 17 mg (14 % yield) of l-(5,6-dichloro-l-pentyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid.
- the title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.1 g, 0.26 mmol), N, ⁇ /-dimethylformamide (2 mL), caesium carbonate (0.508 g, 1.56 mmol) and 4-(bromomethyl)benzonitrile (0.062 g, 0.31 mmol) using the method described in Example 64(b), to give 13 mg (10 % yield) of l-(5,6- dichloro- 1 -(4-cyanobenzyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a white solid.
- Example 72 l-(5,6-dichloro-l-((6-fluoropyridin-3-yl)methyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide
- the title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.1 g, 0.26 mmol), N, ⁇ /-dimethylformamide (2 mL), caesium carbonate (0.508 g, 1.56 mmol) and l-bromo-4-(bromomethyl)benzene (0.078 g, 0.31 mmol) using the method described in Example 74, to give 22 mg (15 % yield) of l-(l-(4- bromobenzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a white solid.
- the title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.30 g, 0.829 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.316 mmol), l-bromo-4-iodobenzene (3.316 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.132 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.044 mmol) using the method described in Example 76.
- the title compound was prepared from N-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.293 mmol), N, ⁇ /-dimethylformamide (3 mL), caesium carbonate (1.172 mmol), (bromomethyl)cyclo butane (1.172 mmol) and dichlorobis(triphenyl- phosphine)palladium (II) (Pd(PPhS) 2 Cl 2 (0.0293 mmol) using the method described in Example 14.
- the title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.293 mmol), N, ⁇ /-dimethylformamide (3 mL), caesium carbonate (1.172 mmol), 3-bromoprop-l-yne (1.172 mmol) and dichlorobis(triphenyl- phosphine)palladium (II) (Pd(PPlIs) 2 Cl 2 (0.0293 mmol) using the method described in Example 14.
- the title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.293 mmol), N, ⁇ /-dimethylformamide (3 mL), caesium carbonate (1.172 mmol), 3-(bromomethyl)pentane (1.172 mmol) and dichlorobis(triphenyl- phosphine)palladium (II) (Pd(PPh 3 ) 2 Cl2 (0.0293 mmol) using the method described in Example 14.
- the title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.293 mmol), N, ⁇ /-dimethylformamide (3 mL), caesium carbonate (1.172 mmol), l-bromo-2,2-dimethylpropane (1.172 mmol) and dichlorobis(triphenyl- phosphine)palladium (II) (Pd(PPh 3 ) 2 Cl2 (0.0293 mmol) using the method described in Example 14.
- the title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.293 mmol), N, ⁇ /-dimethylformamide (3 mL), caesium carbonate ( 1.172 mmo 1), bromocyclohexane ( 1.172 mmo 1) and dichlorobis(triphenyl- phosphine)palladium (II) (Pd(PPh 3 ) 2 Cl2 (0.0293 mmol) using the method described in Example 14.
- the title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.293 mmol), N, ⁇ /-dimethylformamide (3 mL), caesium carbonate (1.172 mmol), 2-bromobutane (1.172 mmol) and dichlorobis(triphenylphosphine)- palladium (II) (Pd(PPlIs) 2 Cl 2 (0.0293 mmol) using the method described in Example 14.
- N-cyclopentyl- 1 -(5 ,6-dichloro- 1 -(2-methoxyethyl)- lH-benzo [d]imidazol-2-yl)piperidine-4- carboxamide was dissolved in dichloromethane and stirred under nitrogen at O 0 C for 15 minutes, treated with tribromoborane via drop-wise addition and gradually warmed to room temperature.
- the reaction mixture was stirred at room temperature for 1 hour, quenched with aqueous sodium hydrogencarbonate (10%) to ashive neutral p ⁇ and extracted with dichloromethane (2x 50 mL).
- reaction mixture was filtered, concentrated in vacuo and purified on preparative hplc (performed on a Gilson- Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 ⁇ m, 21.2 x 150 mm) column, using acetonitrile/water (0.05 % formic acid) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 8.3 mg (19 % yield) of l-(l-benzyl-5,6- dimethyl- lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide.
Abstract
A compound of formula (I): as well as pharmaceutically acceptable salts thereof, and a pharmaceutical composition comprising the compound. The compound is useful for the treatment of disorder selected from inflammatory diseases, nociceptive pain, auto-immune disease, breathing disorders, fever, cancer, inflammation related anorexia, Alzheimer's disease and cardiovascular diseases.
Description
MICROSOMAL PROSTAGLANDIN E SYNTHASE-1 (MPGES1) INHIBITORS
FIELD OF THE INVENTION
The present invention relates to piperidinyl benzoimidazole derivatives and to the use thereof in disease therapy. Particularly, the present invention relates to compounds useful in the treatment or prevention of diseases in which inhibition of the activity of microsomal prostaglandin E synthase- 1 (accession number "014684" at http://www.uniprot.org/) is desired and/or required and, more particularly, in the treatment of diseases such as inflammatory diseases, nociceptive pain, auto-immune disease, breathing disorders, fever, cancer, inflammation related anorexia, Alzheimer's disease and cardiovascular disease.
BACKGROUND OF THE INVENTION
The following is provided as background information only and represents the current state-of-art in the scientific literature. Furthermore, this background information should not be taken as an admission that any subject matter discussed or that any reference mentioned is prior art to the instant invention.
Prostaglandin (PG) E2 is produced in a sequential action including liberation of arachidonic acid, conversion into PGG2/PGH2 by cyclooxygenase (Cox) -1 or Cox-2, and finally prostaglandin E synthase converts PGH2 into PGE2. There exist three known enzymes that catalyze the latter reaction, i.e. microsomal prostaglandin E synthase-1 (mPGEs-1), cytosolic prostaglandin E synthase (cPGEs), and microsomal prostaglandin E synthase-2 (mPGEs-2). The latter two enzymes are constitutively expressed whereas mPGEs-1 is inducible. Initially, mPGEs-1 was regarded as the enzyme predominantly coupled with Cox-2 activity. However; later results demonstrate that mPGEs-1 can also catalyze the conversion of Cox- 1 derived PGH2 into PGE2. mPGEs-1 possesses the highest catalytic efficiency of the known PGE synthases. The role of PGE2 as one of the most potent mediators of inflammation together with many in vitro reports on the presence of mPGEs-1 in different models of inflammation suggested this enzyme to be an attractive drug target for development of new anti inflammatory drugs with fewer side effects than the currently available NSAIDs and selective Cox-2 inhibitors [Samuelsson, B.;
Morgenstern, R.; Jakobsson, P. -J. Membrane Prostaglandin E Synthase-1 : A Novel Therapeutic Target. Pharmacol. Rev. 2007, 5P,207-224; Jachak, S. M. PGE synthase inhibitors as an alternative to COX-2 inhibitors. Curr. Opin. Investig. Drugs, 2007, 8, 411-415]. The rationale is that mPGEs-1 is predominantly expressed during inflammation and that other enzymes exist as mediating "house keeping functions".
Furthermore, mPGEs-1 -derived PGE2 has been implicated in a range of diseases, not confined to inflammation, and it is suggested that inhibitors of mPGEs-1 are effective for the treatment or prevention of these diseases also. Consequently, examples of treatable diseases of the invention include inflammatory diseases, nociceptive pain, auto-immune disease, breathing disorders, fever, cancer, inflammation related anorexia, Alzheimer's disease and cardiovascular disease. More specifically, non-limiting examples of these diseases include the treatment or prevention of rheumatoid arthritis [Murakami, M.; Nakashima, K.; Kamei, D.; Masuda, S.; Ishikawa, Y.; Ishii, T.; Ohmiya, Y.; Watanabe, K.; Kudo, I. Cellular prostaglandin E2 production by membrane- bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J. Biol. Chem. 2003, 278, 37937-37947; Westman, M.; Korotkova, M.; af Klint, E.; Stark, A.; Audoly, L. P.; Klareskog, L.; Ulfgren, A. K.; Jakobsson, P. J. Expression of microsomal prostaglandin E synthase 1 in rheumatoid arthritis synovium. Arthritis Rheum. 2004, 50, 1774-1780] or other inflammatory diseases related to arthritis [Kojima, F.; Kato, S.; Kawai, S. Prostaglandin E synthase in the pathophysiology of arthritis. Fundam. Clin. Pharmacol. 2005, 19, 255-261; Fahmi, H. mPGEs-1 as a novel target for arthritis. Curr. Opin. Rheumatol. 2004, 16, 623-627] inflammatory bowel disease [Dey, L; Giembycz, M. A.; Chadee, K. Prostaglandin E(2) couples through EP(4) prostanoid receptors to induce IL-8 production in human colonic epithelial cell lines. Br. J. Pharmacol. 2009, 156, 475-85], osteoarthritis [Kojima, F.; Naraba, H.; Miyamoto, S.; Beppu, M.; Aoki H, and Kawai S (2004) Membraneassociated prostaglandin E synthase- 1 is upregulated by proinflammatory cyto-kines in chondrocytes from patients with osteoarthritis. Arthritis Res. Ther. 2004, R355-R365; Masuko-Hongo, K.; Berenbaum, F.; Humbert, L.; Salvat, C; Goldring, M. B.; Thirion, S. Up-regulation of microsomal prostaglandin E synthase 1 in osteoarthritic human cartilage: critical roles of the ERK-1/2 and p38 signaling pathways. Arthritis Rheum. 2004, 50, 2829-2838], dermatitis including psoriasis [Stark, K.; Tόrma, H.; Oliw, E. H. Co- localization of COX-2, CYP4F8, and mPGEs-1 in epidermis with prominent expression of CYP4F8 mRNA in psoriatic lesions. Prostaglandins Other Lipid Mediat. 2006, 79, 114-125] and eczema, swelling, inflammatory pain [Trebino, C. E.; Stock, J. L.; Gibbons, C. P.; Naiman, B. M.; Wachtmann, T. S.; Umland, J. P.; Pandher, K.; Lapointe, J. M.; Saha, S.; Roach, M. L, Carter, D.; Thomas, N. A.; Durtschi, B. A.; McNeish, J. D.; Hambor, J. E.; Jakobsson, P. J.; Carty, T. J.; Perez, J. R.; Audoly, L. P. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc. Natl. Acad. Sci. USA, 2004, 100, 9044- 9049], post-operative pain [Shavit, Y.; Fridel, K.; Beilin, B. Postoperative pain management and proinflammatory cytokines: animal and human studies. J. Neuroimmune Pharmacol. 2006, 443- 451], fibromyalgia, dysmenorrhea [Proctor, M.; Farquhar, C. Dysmenorrhoea. Clin. Evid., 2002,
7,1639-1653], migraine, arthrotic and arthritic pain [Schaible, H. G.; Ebersberger, A.; von Banchet, G. S. Mechanisms of pain in arthritis. Ann. N Y Acad. Sci. 2002, 966, 343-354], pain in connection with osteoarthritis, cephalalgia (headache) [Wienecke, T.; Olesen, J.; Oturai, P. S.; Ashina, M. Prostaglandin E2 (PGE2) induces headache in healthy subjects. Cephalalgia, 2009, 29, 509-519], pain due to gall-stones, pain due to metastasis, familial adenomatous polyposis (FAP) condition [Takeda, H.; Sonoshita, M.; Oshima, H.; Sugihara, K.; Chulada, P. C;
Langenbach, R.; Oshima, M.; Taketo, M. M. Cooperation of cyclooxygenase 1 and
cyclooxygenase 2 in intestinal polyposis. Cancer Res. 2003, 63, 4872-4877], colorectal cancer [Kamei, D.; Murakami, M.; Nakatani, Y.; Ishikawa, Y.; Ishii, T.; Kudo, I. Potential role of microsomal prostaglandin E synthase- 1 in tumorigenesis. J. Biol. Chem., 2003, 278, 19396-
19405; Yoshimatsu, K.; Golijanin, D.; Paty, P. B.; Soslow, R. A.; Jakobsson, P. -J.; DeLellis, R. A.; Subbaramaiah, K.; Dannenberg, A. J. Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin. Cancer Res. 2001, 7, 3971-3976;
Elander, N.; Ungerback, J.; Olsson, H.; Uematsu, S.; Akira, S.; Sόderkvist, P. Genetic deletion of mPGEs-1 accelerates intestinal tumorigenesis in APC(Min/+) mice. Biochem. Biophys. Res.
Commun. 2008, 372, 249-253], epithelial ovarian cancer [Rask, K.; Zhu, Y.; Wang, W.; Hedin, L.; Sundfeldt, K. Ovarian epithelial cancer: a role for PGE2-synthesis and signalling in malignant transformation and progression. MoI. Cancer, 2006, 16, 62], breast cancer [Mehrotra, S.; Morimiya, A.; Agarwal, B.; Konger, R.; Badve, S. Microsomal prostaglandin E2 synthase- 1 in breast cancer: a potential target for therapy. J. Pathol. 2006, 208, 356-363], gastric
tumorigenesis [Oshima, H.; Matsunaga, A.; Fujimura, T.; Tsukamoto, T.; Taketo, M. M.;
Oshima, M. Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway. Gastroenterology, 2006, 131, 1086-1095], multiple sclerosis, atherosclerosis such as myocardial infarction and stroke [Ikeda-Matsuo, Y.; Ota, A.; Fukada, T.; Uematsu, S.; Akira, S.; Sasaki, Y. Microsomal prostaglandin E synthase-1 is a critical factor of stroke-reperfusion injury. Proc. Natl. Acad. Sci. USA. 2006, 103, 11790-11795], apnea or other respiratory distress symptoms in adults and children [Hofstetter, A. O.; Saha, S.; Siljehav, V.; Jakobsson, P. J.; Herlenius, E. The induced prostaglandin E2 pathway is a key regulator of the respiratory response to infection and hypoxia in neonates. Proc. Natl. Acad. Sci. USA. 2007, 104, 9894-9899], sudden infant death syndrome (SIDS), asthma, fever [Engblom, D.; Saha, S.;
Engstrόm, L.; Westman, M.; Audoly, L. P.; Jakobsson, P. J.; Blomqvist, A. Microsomal prostaglandin E synthase-1 is the central switch during immune-induced pyresis. Nat. Neurosci. 2003, 6, 1137-1138; Saha, S.; Engstrόm, L.; Mackerlova, L.; Jakobsson, P. J.; Blomqvist, A. Impaired febrile responses to immune challenge in mice deficient in microsomal prostaglandin E synthase-1. Am. J. Physiol. 2005, 288, Rl 100-Rl 107], inflammation related anorexia
[Samuelsson, B.; Morgenstern, R.; Jakobsson, P. -J. Membrane Prostaglandin E Synthase-1 : A Novel Therapeutic Target. Pharmacol. Rev. 2007, 5P,207-224], Alzheimer's disease [Satoh, K.; Nagano, Y.; Shimomura, C; Suzuki, N.; Saeki, Y.; Yokota, H. Expression of prostaglandin E synthase mRNA is induced in beta-amyloid treated rat astrocytes. Neurosci. Lett. 2000, 283, 221-223; Licastro, F.; Davis, L. J.; Pedrini, S.; Galasko, D.; Masliah, E. Prostaglandin E2 induced polymerization of human alpha- 1-antichymotrypsin and suppressed its protease inhibitory activity: implications for Alzheimer's disease. Biochem. Biophys. Res. Commun. 1998, 249, 182- 186; Chaudhry, U. A.; Zhuang, H.; Crain, B. J.; Dore, S. Elevated microsomal prostaglandin-E synthase-1 in Alzheimer's disease. Akheimers Dement. 2008, 4, 6-13; Hoozemans, J. J.;
Veerhuis, R.; Janssen, L; van Elk, E. J.; Rozemuller, A. J.; Eikelenboom, P. The role of cyclo- oxygenase 1 and 2 activity in prostaglandin E(2) secretion by cultured human adult microglia: implications for Alzheimer's disease. Brain Res. 2002, 951, 218-226; Scali, C; Prosperi, C; Bracco, L.; Piccini, C; Baronti, R.; Ginestroni, A.; Sorbi, S.; Pepeu, G.; Casamenti, F.
Neutrophils CDl Ib and fibroblasts PGE(2) are elevated in Alzheimer's disease. Neurobiol. Aging, 2002, 23, 523-30], as well as the use of the said compounds in the manufacture of medicaments for the treatment of those disorders [For a reviews discussing most of the therapeutic indications mentioned above see: Samuelsson, B.; Morgenstern, R.; Jakobsson, P. -J. Membrane Prostaglandin E Synthase-1 : A Novel Therapeutic Target. Pharmacol. Rev. 2007, 5P,207-224; Murakami M, Kudo I. Prostaglandin E synthase: a novel drug target for
inflammation and cancer. Curr Pharm Des. 2006, 12, 943-954; Kojima, F.; Kato, S.; Kawai, S. Prostaglandin E synthase in the pathophysiology of arthritis. Fundam. Clin. Pharmacol. 2005, 19, 255-261].
Other examples of medical conditions associated with mPGES-1 are pain in connection to ankylosing spondylitis, gout, or rheumatic fever, tootache, cellular neoplastic transformations and metastatic tumor growth, endometriosis, hemophilic arthropathy, Parkinson's disease, mPGES-1 -mediated proliferative disorders such as may occur in diabetic retinopathy and tumor angiogenesis, symptoms associated with viral infections, and premature labor. WO 2008/084218 discloses benzazole derivatives of the general formula

as useful in the treatment of diseases in which it is desired and/or required to inhibit the activity of a member of the MAPEG family, e.g. microsomal prostaglandin £ synthase- 1 (mPGEs- 1). SUMMARY OF THE INVENTION
There still is a large unmet need for drugs that are as effective as NSAID inhibitors but having significantly improved side effect profile, for the treatment of conditions such as inflammatory diseases, nociceptive pain, auto-immune diseases, breathing disorders, fever, cancer, inflammation related anorexia, Alzheimer's disease and cardiovascular diseases. One object of the present invention is to provide compounds having said desired improved properties. The present inventors now have surprisingly found certain piperidinyl benzoimidazoles that are capable of efficiently blocking mPGEs-1.
Consequently, in one aspect the present invention provides a compound of formula (I)

(I)
wherein:
R1 is selected from branched or unbranched C1-C10 alkyl, C2-C10 alkenyl or C2-C10 alkynyl; C3- Cio cycloalkyl; C4-C10 cycloalkenyl; Cs-Cio cycloalkynyl; C1-Cg heterocyclyl; and C6-CiO aryl; wherein any alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, or cycloalkynyl moiety optionally is substituted with 1, 2 or 3 groups Ra; and any heterocyclyl or aryl moiety optionally is substituted with 1, 2, 3, 4 or 5 groups Rb; R2 is selected from branched or unbranched C1-Cg alkyl, C2-C9 alkenyl, or C2-C9 alkynyl; C3-C6 cycloalkyl; C4-C6 cycloalkenyl; Cs-Cio cycloalkynyl; C 1-C9 heterocyclyl; and C6-CiO aryl; wherein any alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl or cycloalkynyl moiety optionally
is substituted with 1 , 2 or 3 groups Ra; and any heterocyclyl or aryl moeiety optionally is substituted with 1, 2, 3, 4 or 5 groups Rb;
R3 and R4 are each independently selected from Ci -C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C3-C4 cycloalkyl; Ci-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; Ci-C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; halogen; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amino; and Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl carbonyl; wherein any alkyl, alkenyl, alkynyl or cycloalkyl moiety optionally is substituted with 1, 2, or 3 groups Rc; R5 and R6 are independently selected from hydrogen; Ci-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; and halogen; wherein any alkyl; alkenyl or alkynyl moiety optionally is substituted with 1, 2, or 3 groups Rc;
X represents a bond or branched or unbranched Ci -C3 alkyl, C2-C3 alkenyl or C2-C3 alkynyl;
Q represents carbonyl, sulfonyl or sulfmyl; m is an integer of 0, 1 or 2; each Rais independently selected from halogen; hydroxy; Ci-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; Ci-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; Ci-C4 alkyl, C2-C4 alkenyl or C2- C4 alkynyl secondary or tertiary amino; Ci-C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; wherein any alkyl, alkenyl or alkynyl moiety optionally is substituted with 1, 2 or 3 groups Rc; each Rb is independently selected from halogen; carboxy; hydroxy; cyano; C1-C5 heteroaryl; C1- C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C3-C5 cycloalkyl; Ci-C4 alkyloxy; C2-C4 alkenyloxy; C2- C4 alkynyloxy; C3-C5 cycloalkyloxy; Ci-C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; C1- C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amino; amino carbonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amido; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl carbonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl sulfonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary sulphonamido; Ci-C4 alkyl, C2-C4 alkenyl or C2- C4 alkynyl secondary or tertiary acylsulphonamido; Ci-C4 alkyloxy, C2-C4 alkenyloxy, or C2-C4 alkynyloxy carbonyl; Ci-C5 heterocyclyl; wherein any alkyl, alkenyl, alkynyl or cycloalkyl moiety optionally is substituted with 1, 2 or 3 groups Rc;
each Rc is independently selected from fluorine and chlorine; and pharmaceutically acceptable salts thereof. Another aspect of the present invention is to provide a compound of formula (I) for use as a medicament.
Another aspect of the present invention is to provide a pharmaceutical composition comprising a therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, in admixture with at least one pharmaceutically acceptable adjuvant, diluent or carrier.
Another aspect of the present invention is to provide a method of medical treatment by use of said pharmaceutical compositions.
Another aspect of the present invention provides a compound of formula (I) or a
pharmaceutically acceptable salt thereof, as defined herein above, for use in the treatment of a disorder in which inhibition of the activity of microsomal prostaglandin E synthase- 1 is desired and/or required.
Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of a disorder in which inhibition of the activity of microsomal prostaglandin E synthase- 1 is desired and/or required. Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of an inflammatory disease, said disease preferably being selected from rheumatoid arthritis, inflammatory bowel disease, osteoarthritis, psoriasis, eczema, swelling and periodontitis. Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of nociceptive pain, said pain preferably being selected from inflammatory pain, post-operative pain, fibromyalgia, dysmenorrhea, migraine, arthrotic pain, arthritic pain, pain in connection with osteoarthritis, cephalalgia, pain due to gall-stones and pain due to metastasis, ankylosing spondylitis, gout, or rheumatic fever, and tootache. The compound of formula (I) also may be used in preemptive treatment of surgical pain.
Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of an autoimmune disease, said autoimmune disease preferably being selected from rheumatoid arthritis and multiple sclerosis.
Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of a breathing disorder, said disorder preferably being selected from apnea or other respiratory distress symptoms in adults and children, sudden infant death syndrome (SIDS), asthma and other chronic obstructive lung-diseases and sarcoidosis.
Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of cancer, said cancer preferably being selected from familial adenomatous polyposis (FAP) condition, colorectal cancer, breast cancer, gastric tumorigenesis and epithelial ovarian cancer, as well as cellular neoplastic transformations and metastatic tumor growth.
Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of cardiovascular diseases due to atherosclerosis such as myocardial infarction and stroke.
Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of fever- or inflammation-related anorexia.
Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of Alzheimer's disease.
Another aspect of the present invention provides a compound of formula (I), for use in the treatment and/or prevention of endometriosis, hemophilic arthropathy, Parkinson's disease, mPGES-1 -mediated proliferative disorders such as may occur in diabetic retinopathy and tumor angiogenesis, symptoms associated with viral infections, and premature labor.
Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of an inflammatory disease, said disease preferably being selected from rheumatoid arthritis, inflammatory bowel disease, osteoarthritis, psoriasis, eczema, swelling and periodontitis.
Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of nociceptive pain, said pain preferably being selected from inflammatory pain, post-operative pain, fibromyalgia, dysmenorrhea, migraine, arthrotic pain, arthritic pain, pain in connection with osteoarthritis, cephalalgia, pain due to gall-stones and pain due to metastasis, pain due to ankylosing spondylitis, pain due to gout, toothache and pain due to rheumatic fever, or for the preemptive treatment of surgical pain. Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of an autoimmune disease, said autoimmune disease preferably being selected from rheumatoid arthritis and multiple sclerosis.
Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of a breathing disorder, said disorder preferably being selected from apnea or other respiratory distress symptoms in adults and children, sudden infant death syndrome (SIDS), asthma and other chronic obstructive lung- diseases and sarcoidosis.
Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of cancer, said cancer preferably being selected from familial adenomatous polyposis (FAP) condition, colorectal cancer, breast cancer, gastric tumorigenesis and epithelial ovarian cancer, as well as cellular neoplastic transformations and metastatic tumor growth.
Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of cardiovascular diseases due to atherosclerosis such as myocardial infarction and stroke.
Another aspect of the present invention provides the use of a compound of formula (I), in preparation of a medicament for the treatment and/or prevention of fever- or inflammation- related anorexia.
Another aspect of the present invention provides the use of a compound of formula (I), in the preparation of a medicament for the treatment and/or prevention of Alzheimer's disease.
Another aspect of the present invention provides a method of treatment of a disease in which inhibition of the activity of mPGEs-1 is desired and/or required, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient suffering from, or susceptible to, such a condition.
Another aspect of the present invention provides a method of treatment of an inflammatory disease, said disease preferably being selected from rheumatoid arthritis, inflammatory bowel disease, osteoarthritis, psoriasis, eczema, swelling and periodontitis, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
Another aspect of the present invention provides a method of treatment of nociceptive pain, said pain preferably being selected from inflammatory pain, post-operative pain, fibromyalgia, dysmenorrhea, migraine, arthrotic pain, arthritic pain, pain in connection with osteoarthritis, cephalalgia, pain due to gall-stones and pain due to metastasis, pain due to ankylosing spondylitis, gout, toothache and pain due to rheumatic fever, as well as preemptive treatment of surgical pain, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
Another aspect of the present invention provides a method of treatment of an autoimmune disease, said autoimmune disease preferably being selected from rheumatoid arthritis and multiple sclerosis, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
Another aspect of the present invention provides a method of treatment of a breathing disorder, said disorder preferably being selected from apnea or other respiratory distress symptoms in adults and children, sudden infant death syndrome (SIDS), asthma and other chronic obstructive lung-diseases and sarcoidosis, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
Another aspect of the present invention provides a method of treatment of cancer, said cancer preferably being selected from familial adenomatous polyposis (FAP) condition, colorectal cancer, breast cancer, gastric tumorigenesis and epithelial ovarian cancer, as well as cellular neoplastic transformations and metastatic tumor growth, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically- acceptable salt thereof, to a patient in the need of such treatment.
Another aspect of the present invention provides a method of treatment of cardiovascular diseases due to atherosclerosis such as myocardial infarction and stroke, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
Another aspect of the present invention provides a method of treatment of fever- or
inflammation-related anorexia, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
Another aspect of the present invention provides a method of treatment of Alzheimer's disease, which method comprises administration of a therapeutically effective amount of a compound of formula (I), or a pharmaceutically-acceptable salt thereof, to a patient in the need of such treatment.
In another aspect of the invention, methods for preparing a compound of formula (I) are provided. BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 is a bar diagram showing results of a carrageenan- induced paw edema assay in Wistar rats using the compound synthesised in Example 41. The compound at 10, 30 and 100 mg/kg, as well as vehicle was administered intraperitoneally immediately before carrageenan challenge. Hind paw edema, a measure of inflammation, was recorded 6 hours after carrageenan administration. Volume increase is paw volume at 6 hours after carrageenan administration minus paw volume before carrageenan administration. The bars represent average ±standard error of the mean (n=8).
Figure 2 is a bar diagram showing inhibition of CFA- induced paw swelling by the compound synthesised in Example 41 compared to vehicle control. Data represent average inhibition of paw swelling ±standard error of the mean for acute (A) or delayed (B) phase (n=5). Inhibition of paw swelling = (paw volume (vehicle) - paw volume (treatment))/paw volume (vehicle).
DETAILED DESCRIPTION OF THE INVENTION
Unless otherwise indicated or apparent from the context, any alkyl, alkenyl or alkynyl group as referred to herein may be branched or unbranched. This also applies to said groups when present in moieties such as alkoxy groups, and the like.
The term "alkyl" as employed herein, alone or as part of another group, refers to an acyclic straight or branched chain radical, unless otherwise specified containing 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 or 12 carbons in the normal chain, which includes methyl, ethyl, /? -propyl, /? -butyl, n-pentyl, n-hexyl, n-heptyl and n-octyl. Examples of branched chain radicals, not excluding any of the possible isomers not mentioned, are ώo-propyl, sec-butyl, ώo-pentyl, 3-methylpentyl, 2,3- dimethylhexyl, 3-ethylhexyl, and the like. Alkyl also includes a straight or branched alkyl group that contains or is interrupted by a carbocyclyl group, exemplified by cyclopropane, as exemplified below: (CH \ -J- \ (CH∑)z—
wherein, w and z are 1 to 8, the sum of w and z is not more than 9. The alkyl portions can be attached at any variable point of attachment to the carbocyclyl, including the same ring carbon, as exemplified below:

(CH2)W
When substituted alkyl is present, this refers to a straight or branched alkyl group, substituted with 1, 2, 3 groups of Ra, which groups may be the same or different at any available point, as defined with respect to each variable.
The term "alkenyl" as used herein, alone or as part of another group, refers to a straight or branched chain radical, unless otherwise specified containing 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 or 12, containing at least one carbon to carbon double bond, which includes in the normal chain vinyl, 2-propenyl, 3-butenyl, 2-butenyl, 4-pentenyl, 3-pentenyl, 2-hexenyl, 3-hexenyl, 2-heptenyl, 3- heptenyl, 4-heptenyl, 3-octenyl, and the like. Examples of branched chain radicals, not excluding
any of the possible isomers not mentioned, are 2-buten-2-yl, 4-methyl-2-pentenyl, 6-methylene- 2-octenyl, and the like. All possible (E)- and (Z)-isomers is contemplated within the scope of the invention. As described above with respect to the "alkyl", the straight or branched portion of the alkenyl group may be optionally substituted when a substituted alkenyl group is provided, and the chain may be interrupted by a carbocyclyl group. When substituted alkenyl is present, this refers to a straight or branched alkyl group, substituted with 1, 2, 3 groups of Ra, which groups may be the same or different at any available point, as defined with respect to each variable.
The term "alkynyl" as used herein by itself or as part of another group refers to a straight or branched chain radical, unless otherwise specified containing 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 or 12 carbons, which contains at least one carbon to carbon triple bond, which in the normal chain such as 2-propynyl, 3-butynyl, 2-butynyl, 4-pentynyl, 3-pentynyl, 2-hexynyl, 3-hexynyl, 2- heptynyl, 3-heptynyl, 4-heptynyl, 3-octynyl, and the like. Examples of branched chain radicals, not excluding any of the possible isomers not mentioned, are 3-methyl-l-butynyl, 2-methyl-4- pentynyl, 6-methyl-4-octynyl, 6-methyl-3-propyl-4-octynyl, and the like. The alkynyl can in addition to carbon to carbon triple bonds also include a to carbon double bond. Examples of such radicals, not excuding any of the possible not mentioned, are 3-buten-l-ynyl, l-buten-3-ynyl, 6- methylene-2-octynyl, 3-allyl-6-methyl-l-octen-4-ynyl, and the like. As described above with respect to the "alkyl", the straight or branched portion of the alkynyl group may be optionally substituted when a substituted alkynyl group is provided, and the chain may be interrupted by a carbocyclyl group. When substituted alkynyl is present, this refers to a straight or branched alkyl group, substituted with 1, 2, 3 groups of Ra, which groups may be the same or different at any available point, as defined with respect to each variable. As used herein, the term "carbocyclyl" refers to a cyclic moiety containing only carbon atoms.
As used herein with respect to any carbocyclyl, heterocyclyl, cycloalkyl, cycloalkenyl, cycloalkynyl the term "monocyclic" refers to a cyclic moiety containing only one ring. The term "bicyclic" refers to a cyclic moiety containing two rings. A bicyclic group may, for example, be fused or bridged.
The concept of fused and bridged rings are well known to the one skilled in the art, and for example is explained in "Fused Ring and Bridged Fused Ring Nomenclature, FR-I from IUPAC Recommendations 1998"
As used herein, the terms "(E)- and (Z)-isomers" refer to the terms "Entgegen" and "Zusammen", respectively. Whether a molecular configuration is designated as Entgegen or Zusammen is determined by the "Cahn-Ingold-Prelog" priority rules, which is well known for the one skilled in the art.
As used herein, the terms "heterocyclyl" mean a mono- or bicyclic, saturated or unsaturated cyclic group, possibly aromatic, containing one or more heteroatom(s) preferably selected from N, O and S, such as, but not limited to, aziridinyl, azetidinyl, dihydropyranyl, dihydropyridyl, dihydropyrrolyl, dioxolanyl, dioxanyl, dithianyl, dithiolanyl, imidazolidinyl, imidazolinyl, morpholinyl, oxetanyl, oxiranyl, pyrrolidinyl, pyrrolidinonyl, piperidyl, piperazinyl, piperidinyl, pyrazolidinyl, quinuclidinyl, sulfalonyl, 3-sulfolenyl, tetrahydrofuranyl tetrahydropyranyl, tetrahydropyridyl, thietanyl, thiiranyl, thiolanyl, thiomorpholinyl, trithianyl, tropanyl, IH- indazolyl, monosaccharide, pyridyl, quinolinyl, furanyl, thienyl, oxadiazolyl, thiadiazolyl, thiazolyl, oxazolyl, pyrazolyl, triazolyl, tetrazolyl, isoxazolyl, isothiazolyl, isoquinolinyl, naphthyridinyl, imidazolyl, phenazinyl, phenothiazinyl, phthalazinyl, indolyl, pyridazinyl, quinazolinyl, quinolizinyl, quinoxalinyl, tetrahydroisoquinolinyl, pyrazinyl, indazolyl, indolinyl, pyrimidinyl, thiophenetyl, pyranyl, carbazolyl, chromanyl, cinnolinyl, acridinyl, benzimidazolyl, benzodioxanyl, benzodioxepinyl, benzodioxolyl, benzo furanyl, benzothiazolyl,
benzobenzoxadiazolyl, benzoxazinyl, benzoxazolyl, benzo morpholinyl, benzoselenadiazolyl, benzothienyl, purinyl, pteridinyl, and the like.
The term "halogen" refers to fluorine, chlorine, bromine and iodine, where the preferred halogen radicals are fluoro and chloro. As used herein, the term "aryl" means an aromatic group, monocyclic or bicyclic such as, but not limited to phenyl or naphthyl, and the like. The aryl group is preferably a monocyclic C6 aryl (phenyl).
The term "cycloalkyl" as employed herein alone or as part of another group includes saturated cyclic hydrocarbyl groups, mono- or bicyclic rings(s), having a total of 3, 4, 5, 6, 7, 8, 9 or 10 carbons forming the ring(s), which includes cyclopropyl, cyclobutyl, cyclohexyl, cycloheptyl, cyclodecane, bicyclo[4.1.0]heptane, bicyclo[4.3.3]dodecane, cyclopentyl, decahydronaphthalene, bicyclo[2.2.2]octane, and the like, optionally substituted with 1, 2 or 3 groups of Ra.
The term "cycloalkenyl" as employed herein alone or as part of another group includes unsaturated cyclic rings(s), mono- or bicyclic, having a total of 4, 5, 6, 7, 8, 9 or 10 carbons forming the ring(s), which includes cyclohexene, cyclobutene, bicyclo[4.2.0]-3-octene, 1,3- cyclodecadiene and the like, optionally substituted with 1, 2 or 3 groups of Ra.
The term "cycloalkynyl" as employed herein alone or as part of another group includes mono- or bicyclic, rings having one carbon to carbon triple bond, and having a total of 8, 9 or 10 carbons forming the ring, which includes cyclooctyne, cyclodecyne, bicyclo[6.2.0]-4-decyne and the like, optionally substituted with 1, 2 or 3 groups of Ra. The cycloalkynyl can in addition to carbon to carbon triple bonds also contain a carbon to carbon double bond, which includes l-cyclodecen-5- yne and the like.
The term "carbocyclyl" as employed herein alone or as part of another group includes saturated cyclic hydrocarbyl groups or unsaturated (at least one carbon to carbon double bond) cyclic hydrocarbyl groups, containing one ring and a total of 3, 4, 5, 6, 7, 8, 9 or 10 carbons forming the ring, which includes cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, phenyl, cycloheptyl, and the like. The cyclic hydrocarbyl may be mono- or bicyclic.
By the ternϊ'unsaturated", when referring to a bicyclic system, is meant a ring system
comprising at least one double or triple bond in at least one ring. Thus, it is contemplated that both rings may be unsaturated or only one ring may be unsaturated, the other one being saturated. Furthermore, the term "unsaturated bicyclic" also is intended to refer to a non- aromatic bicyclic system comprising a ring that is either unsaturated or saturated fused to a ring that by itself would be aromatic, such as in indane or 4,5-dihydro-l -indole.
The term "aromatic", as used herein, refers to an unsaturated cyclic moiety that has an aromatic character, while the term "non-aromatic", as used herein, refers to a cyclic moiety, that may be unsaturated, but that does not have an aromatic character. In a bicyclic ring system, as referred to herein, the rings may be both saturated or both unsaturated, e.g. both aromatic. The rings may also be of different degrees of saturation, and one ring may be aromatic whereas the other is non-aromatic. The rings also may comprise different numbers of atoms, e.g. one ring being 5-membered and the other one being 6-membered.
In a bicyclic heterocyclyl, as referred to herein, one or both of the rings may contain one or several heteroatoms.
The terms hydroxy (i), carboxy (ii) and aminocarbonyl (iii), refer to radicals of the type:

(i) (ϋ) (iϋ)
The terms alkyloxy, alkenyloxy, alkynyloxy and cycloalkyloxy refer to a radical of the type:
RCr ^
wherein R is an alkyl, alkenyl, alkynyl or cycloalkyl moiety.
The terms alkylthio, alkenylthio, and alkynylthio refer to a radical of the type:
RS^
wherein R is an alkyl, alkenyl or alkynyl moiety. The terms alkyl, alkenyl and alkynyl secondary amino refer to a radical of the type:

wherein R is an alkyl, alkenyl or alkynyl moiety.
The terms alkyl, alkenyl and alkynyl tertiary amino refer to a radical of the type:
Rγv
R
wherein R and R' are each an independently selected alkyl, alkenyl or alkynyl moiety.
The terms alkyl, alkenyl and alkynyl secondary amido refer to a radical of the type:

wherein R is an alkyl, alkenyl or alkynyl moiety.
The terms alkyl, alkenyl and alkynyl tertiary amido refer to a radical of the type:
R'
N \'
O
wherein R and R' are each an independently selected alkyl, alkenyl or alkynyl moiety.
The terms alkyl, alkenyl and alkynyl carbonyl refer to a radical of the type:
wherein R is an alkyl, alkenyl or alkynyl moiety.
The terms alkyl, alkenyl and alkynyl sulfonyl refer to a radical of the type:
1VV
ό'h
wherein R is an alkyl, alkenyl or alkynyl moiety.
The terms alkyl, alkenyl and alkynyl secondary sulphonamido refer to a radical of the type:

wherein R is an alkyl, alkenyl or alkynyl moiety.
The terms alkyl, alkenyl and alkynyl tertiary sulphonamido refer to a radical of the type:

wherein R and R' are each independently selected from alkyl, alkenyl or alkynyl moiety.
The terms alkyl, alkenyl and alkynyl secondary acylsulphonamido refer to a radical of the type:

wherein R is an alkyl, alkenyl or alkynyl moiety.
The terms alkyl, alkenyl and alkynyl tertiary acylsulphonamido refer to a radical of the type:

wherein each R is independently selected from an alkyl, alkenyl or alkynyl moiety.
The terms alkyl, alkenyl and alkynyl alkynyloxy carbonyl refer to a radical of the type:
o
wherein R is an alkyl, alkenyl or alkynyl moiety.
The terms carbonyl (i), sulfonyl (ii) and sulfmyl (iii), when used to define the scope of Q, refer to di-radicals of the type:

(i) (ϋ) (iϋ)
The term "cyclic", as used herein in respect of an atom, refers to an atom that is a member of at least one ring in a carbocycle or heterocycle.
A cyclyl according to the invention may comprise 1-10 carbons, it being understood that in case the cyclyl comprises less than 3 carbons, it must necessarily also comprise at least one heteroatom. Unless otherwise indicated or apparent from the context, in a bicyclic moiety according to the invention, each constituent monocycle may independently be selected from aromatic or non- aromatic carbo- and heterocycles.
In a first aspect of the invention, there is provided a compound of formula (I):

(I)
as defined herein above.
R1 in formula (I) is selected from branched or unbranched C1-C10 alkyl, C2-C 10 alkenyl or C2-C10 alkynyl; C3-C10 cycloalkyl; C4-C10 cycloalkenyl; Cs-Cio cycloalkynyl; C1-C9 heterocyclyl; and C6- Cio aryl; wherein any alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, or cycloalkynyl moiety optionally is substituted with 1, 2 or 3 groups Ra; and any heterocyclyl or aryl moiety optionally is substituted with 1, 2, 3, 4 or 5 groups Rb, as defined herein.
In one embodiment, R1 in formula (I) is selected from branched or unbranched C1-C10 alkyl; C3- Cio cycloalkyl; C1-C9 heterocyclyl; and C6-CiO aryl; said alkyl and cycloalkyl optionally being substituted with 1 , 2 or 3 groups Ra; and said heterocyclyl and aryl optionally being substituted with 1, 2, 3, 4 or 5 groups Rb.
In one embodiment of the invention, R1 in formula (I) is optionally substituted branched or unbranched C1-C10 alkyl, e.g. Ci-Cs alkyl, i.e. methyl, ethyl and branched or unbranched propyl, butyl, pentyl, hexyl, heptyl, and octyl, such as methyl, ethyl, n-propyl, ώo-propyl, n-butyl, iso- butyl, sec-butyl, n-pentyl, pentan-2-yl, pentan-3-yl, n-hexyl, n-heptyl, and n-octyl. More specifically, in this embodiment, R1 in formula (I) may be selected from optionally substituted branched or unbranched Ci-C6 alkyl, e.g. methyl, ethyl, n-propyl, ώo-propyl, n-butyl, ώo-butyl, sec-butyl, n -pentyl, pentan-2-yl, pentan-3-yl, n-hexyl, or from optionally substituted branched or unbranched Ci -C4 alkyl. For example, in this embodiment, R1 may be optinally substituted iso- propyl.
In one embodiment of the invention, when R1 in formula (I) is selected from optionally substituted branched or unbranched Ci -C 10 alkyl, m is 0. In another embodiment, R1 in formula (I) is optionally substituted C3-C10 cycloalkyl, in particular C3-C6 cycloalkyl, or C3-C5 cycloalkyl.
R1 in formula (I) also may be optionally substituted C1-C9 heterocyclyl, in particular C1-C5 heterocyclyl, such as a 6-membered C1-C5 heterocyclyl, C2-C5 heterocyclyl, C3-C5 heterocyclyl, C4-C5 heterocyclyl or C5 heterocyclyl, e.g. tetrahydropyryl or pyridyl, such as pyridin-2-yl, pyridin-3-yl, pyridin-4-yl, or tetrahydropyran-4-yl; or 5-membered Ci -C4 heterocyclyl, C2-C4 heterocyclyl, or C3-C4 heterocyclyl, e.g. thiazolyl or tetrahydrofuryl, such as thiazol-4-yl and tetrahydrofuran-2-yl. R1 in formula (I) also may be optionally substituted C6-CiO aryl, preferably phenyl.
When R1 is alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, it optionally is substituted with 1, 2 or 3 groups, e.g. 1 or 2 independently selected groups Ra, e.g. 1 group Ra, as defined herein, i.e. selected from halogen; hydroxy; C1-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; C1-C4 alkyl, C2-C4 alkenyl or C2-C4
alkynyl secondary or tertiary amino; C1-C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; wherein any alkyl, alkenyl or alkynyl moiety optionally is substituted with 1, 2 or 3 groups Rc.
In one embodiment, when R1 is substituted with Ra, each Ra is indpendently selected from hydroxy, C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; and C1-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amino, in particular from hydroxy, C1-C4 alkoxy, such as methoxy, or C1-C4 alkyl secondary or tertiary amino, e.g. such as ethyl secondary or tertiary amino, e.g. diethylamino. In one embodiment, R1 is not substituted with any Ra.
When R1 is heterocyclyl or aryl, it optionally is substituted with 1, 2, 3, 4 or 5 independently selected groups Rb, as defined herein, i.e. selected from halogen; carboxy; hydroxy; cyano; C1-C5 heteroaryl; C1-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; Ci-C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amino; amino carbonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amido; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl carbonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl sulfonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary sulphonamido; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary acylsulphonamido; Ci-C4 alkyloxy, C2-C4 alkenyloxy, or C2-C4 alkynyloxy carbonyl; Ci- C5 heterocyclyl; wherein any alkyl, alkenyl or alkynyl moiety optionally is substituted with 1, 2 or 3 groups Rc.
In one embodiment, when R1 is substituted with Rb, Rb is selected from halogen; carboxy; cyano; Ci-C5 heteroaryl; Ci-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; amino carbonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amido; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary acylsulphonamido; Ci-C4 alkyloxy, C2-C4 alkenyloxy, or C2-C4 alkynyloxy carbonyl; and C1-C5 heterocyclyl; wherein any alkyl, alkenyl and alkynyl moiety optionally is substituted with 1, 2 or 3 groups Rc.
In one particular embodiment, when R1 is substituted with Rb, Rb is selected from halogen, i.e. F, Cl and Br, in particular F; carboxy; cyano; 5- or 6-membered C1-C5 heteroaryl, in particular 5- membered Ci-C4 heteroaryl, such as triazole; Ci-C4 alkyl, such as methyl; amino carbonyl; Ci-C4 alkyl secondary or tertiary amido, such as methyl secondary or tertiary amido; Ci-C4 alkyl secondary or tertiary acylsulphonamido, such as methyl secondary or tertiary acylsulphonamido;
C1-C4 alkyloxy carbonyl, such as methoxy carbonyl; and C1-C5 heterocyclyl, such as 5- or 6- membered heterocyclyl, e.g. 5-membered C1-C4 heterocyclyl, e.g. tetrazole.
In one embodiment, R1 is substituted with 1-3 groups Rb as defined herein, e.g. with 1 or 2 groups, in particular with 1 group Rb. In another embodiment, R1 is not substituted with any Rb.
In one embodiment, when R1 is a 6-membered heterocyclyl or aryl, it is substituted with at least one group Rb in para -position relative to the linking bond or chain attaching R1 to the benzimidazol moiety.
R2 in formula (I) is selected from branched or unbranched C1-C9 alkyl, C2-C9 alkenyl or C2-C9 alkynyl; C3-C6 cycloalkyl, C4-C6 cycloalkenyl; Cs-Cio cycloalkynyl; C 1-C9 heterocyclyl; and C6- Cio aryl; said alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl optionally being substituted with 1 , 2 or 3 groups Ra; and said heterocyclyl or aryl optionally being substituted with 1, 2, 3, 4 or 5 groups Rb.
In one embodiment, R2 is optionally substituted branched or unbranched C1-C9 alkyl, C2-C9 alkenyl or C2-C9 alkynyl. In this embodiment, R2 more particularly may be Ci-C6 alkyl, C2-C6 alkenyl or C2-C6 alkynyl, e.g. C1-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl. In particular, R2 may be branched or unbranched C1-C9 alkyl, or Ci-C6 alkyl, e.g. C1-C4 alkyl, e.g. C2-C4 alkyl.
In one embodiment, when R2 is optionally substituted branched or unbranched C1-C9 alkyl, C2- C9 alkenyl or C2-C9 alkynyl, X is a bond. In another embodiment, R2 is optionally substituted C3-C6 cycloalkyl or C4-C6 cycloalkenyl, and then more particularly may be C3-C6 cycloalkyl, in particular pentyl.
In still another embodiment, R2 is optionally substituted C 1-C9 heterocyclyl. In this embodiment, R2 may in particular be C1-C5 heterocyclyl, e.g. 5-membered C1-C4 heterocyclyl or 6-membered C1-C5 heterocyclyl, in particular 5-membered C1-C4 heterocyclyl, e.g. 5-membered C3-C4 heterocyclyl. For example, R2 may be selected from tetrahydrofuryl and thiazolyl.
In another embodiment, R2 is optionally substituted C6-CiO aryl, and then preferably is phenyl.
When R2 is alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, or cycloalkynyl, it optionally is substituted with 1, 2 or 3 groups, e.g. 1 or 2 groups, Ra, as defined herein, i.e. selected from halogen; hydroxy; C1-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; C1-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amino; C1- C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; wherein any alkyl, alkenyl or alkynyl moiety optionally is substituted with 1 , 2 or 3 groups Rc.
In one embodiment, when R2 is substituted with Ra, each Ra is independently selected from C1- C4 alkyloxy; C2-C4 alkenyloxy; and C2-C4 alkynyloxy, in particular C1-C4 alkyloxy, such as Ci- C3 alkyloxy, more particularly methoxy and iso-propoxy. In one embodiment, R2 is substituted with 1 group Ra and in another embodiment, R2 is not substituted with any group Ra. In one embodiment, when R2 is cycloalkyl, cycloalkenyl, or cycloalkynyl, it is not substituted with any group Ra. When R2 is heterocyclyl or aryl, it optionally is substituted with 1, 2, 3, 4 or 5 groups Rb, as defined herein above. In one embodiment, R2 is substituted with 1, 2, 3, 4 or 5 groups Rb each independently selected from halogen, e.g. F, Cl and Br, in particular F or Br; cyano; and C1-C5 heterocyclyl, e.g. fϊve-membered C 1-C4 heterocyclyl. For example, R2 may be 6-membered heterocyclyl or aryl, such as phenyl, substituted with at least one Rb in para -position in relation to the bond attaching R2 to X or - in case X is a bond - in para -position in relation to the bond X. In one embodiment, R2 is substituted with 1-3 groups Rb, e.g. 1 or 2 groups Rb, in particular 1 group Rb.
In a compound of formula (I), R3 and R4 are each independently selected from C1-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C3-C4 cycloalkyl; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; Ci-C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; halogen; Ci-C4 alkyl, C2-C4 alkenyl or C2- C4 alkynyl secondary or tertiary amino; and Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl carbonyl; wherein any alkyl, alkenyl, alkynyl or cycloalkyl moiety optionally is substituted with 1, 2, or 3 groups Rc.
In one embodiment, R3 and R4 are each independently selected from Ci-C4 alkyl and
halogen, in particular from methyl and chloro. For example, R3 and R4 may be both halogen or both Ci-C4 alkyl, e.g. both are Cl or both are methyl, or one may be halogen, e.g. Cl, and the other one Ci-C4 alkyl, e.g. methyl.
In one embodiment, R3 is selected from C1-C4 alkyl, e.g. methyl, and R4 is selected from halogen and C1-C4 alkyl, e.g. Cl and methyl. In another embodiment, R3 is selected from halogen, e.g. Cl, and R4 is selected from halogen and C1-C4 alkyl, e.g. Cl and methyl. In still another embodiment, R3 is selected from halogen and C1-C4 alkyl, e.g. Cl and methyl, and R4 is selected from C1-C4 alkyl, e.g. methyl. In still another embodiment, R3 is selected from halogen and C1-C4 alkyl, e.g. Cl and methyl, and R4 is selected from halogen, e.g. Cl.
R5 and R6 in formula (I) are independently selected from hydrogen, C1-C4 alkyl, C2-C4 alkenyl, C2-C4 alkynyl and halogen; whereby any alkyl; alkenyl; or alkynyl is optionally substituted with 1, 2, or 3 groups Rc.
In one embodiment, R5 and R6 are independently selected from hydrogen and C1-C4 alkyl, more particularly from H and methyl, for example, R5 and R6 may both be H; or R5 may be H and R6 methyl.
In formula (I), X represents a bond (i.e. a single, covalent bond) or branched or unbranched Ci- C3 alkyl, C2-C3 alkenyl or C2-C3 alkynyl. Preferably X represents a bond or C1-C3 alkyl, more preferably a bond or methylene (-CH2-).
Q in formula (I) is selected from carbonyl, sulfonyl and sulfϊnyl and preferably is carbonyl.
In formula (I), m is an integer of 0, 1 or 2, preferably 0 or 1. In one particular embodiment, m is 0. In another embodiment, m is 1.
Each Rais independently selected from halogen; hydroxy; C1-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; C1-C4 alkyl, C2-C4 alkenyl or C2- C4 alkynyl secondary or tertiary amino; C1-C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; wherein any alkyl, alkenyl or alkynyl moiety optionally is substituted with 1, 2 or 3 groups Rc.
In one embodiment, each Ra is independently selected from hydroxy; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; and C1-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amino; wherein any alkyl, alkenyl and alkynyl moiety optionally is substituted with 1, 2 or 3 groups Rc.
In another embodiment, each Ra is independently selected from hydroxy; C1-C4 alkyloxy, e.g. methoxy and ώo-propoxy; and C1-C4 alkyl secondary or tertiary amino, e.g.diethylamino.
Each Rb in formula (I) is independently selected from halogen; carboxy; hydroxy; cyano; C1-C5 heteroaryl; Ci-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C3-C5 cycloalkyl; Ci-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; C3-C5 cycloalkyloxy; Ci-C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; C3-C5 cycloalkylthio; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amino; amino carbonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amido; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl carbonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl sulfonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary sulphonamido; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary
acylsulphonamido; Ci-C4 alkyloxy, C2-C4 alkenyloxy, or C2-C4 alkynyloxy carbonyl; C1-C5 heterocyclyl; wherein any alkyl, alkenyl, alkynyl or cycloalkyl moiety optionally is substituted with 1 , 2 or 3 groups Rc as defined herein.
In one embodiment, each Rb is independently selected from halogen; carboxy; hydroxy; cyano; Ci -C5 heteroaryl; Ci-C4 alkyl; C3-C5 cycloalkyl; Ci-C4 alkyloxy; C3-C5 cycloalkyloxy; Ci-C4 alkylthio; Ci-C4 alkyl secondary or tertiary amino; amino carbonyl; Ci-C4 alkyl secondary or tertiary amido; Ci-C4 alkyl carbonyl; Ci-C4 alkyl sulfonyl; Ci-C4 alkyl secondary or tertiary sulphonamido; Ci-C4 alkyl secondary or tertiary acylsulphonamido; Ci-C4 alkyloxy carbonyl; Ci-C5 heterocyclyl; wherein any alkyl or cycloalkyl moiety optionally is substituted with 1, 2 or 3 groups Rc as defined herein.
In another embodiment, each Rb is independently selected from halogen; carboxy, cyano; Ci-C5 heteroaryl; Ci-C4 alkyl; C2-C4 alkenyl; C3-C5 cycloalkyl; Ci-C4 alkyloxy; C3-C5 cycloalkyloxy; C2-C4 alkynyl; amino carbonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amido; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary acylsulphonamido; C1- C4 alkyloxy carbonyl; and Ci-C5 heterocyclyl; wherein any alkyl, alkenyl and alkynyl moiety optionally is substituted with 1 , 2 or 3 groups Rc as defined herein.
In still another embodiment, each Rb is independently selected from halogen; carboxy; cyano; Ci-C5 heteroaryl; Ci-C4 alkyl; C3-C5 cycloalkyl; Ci-C4 alkyloxy; C3-C5 cycloalkyloxy;
aminocarbonyl; Ci-C4 alkyl secondary or tertiary amido; Ci-C4 alkyl secondary or tertiary acylsulphonamido; Ci-C4 alkyloxy carbonyl; and Ci-C5 heterocyclyl; wherein any alkyl, moiety optionally is substituted with 1, 2 or 3 groups Rc as defined herein.
In still another embodiment, each Rb is independently selected from halogen; carboxy, cyano; C 1-C5 heteroaryl; C1-C4 alkyl; amino carbonyl; C1-C4 alkyl secondary or tertiary amido; C1-C4 alkyl secondary or tertiary acylsulphonamido; C1-C4 alkyloxy carbonyl; and C1-C5 heterocyclyl; wherein any alkyl, moiety optionally is substituted with 1, 2 or 3 groups Rc as defined herein.
When Rb is C1-C5 heterocyclyl, it may be C1-C4 heterocyclyl, and preferably is 6-membered Ci- C5 heterocyclyl or 5-membered C1-C4 heterocyclyl, more preferably 5-membered C1-C4 heterocyclyl. When Rb is C1-C5 heteroaryl, it may be C1-C4 heteroaryl, and prefereably is 6- membered C1-C5 heteroaryl or 5-membered C1-C4 heteroaryl.
Rc in formula (I) is selected from fluorine and chlorine. In one embodiment, Rc is absent. In another embodiment, Rc is fluorine.
In a particular embodiment of the invention, X represents methylene or a bond and R2 represents C3-C 6 cycloalkyl or heterocyclyl.
In another embodiment of the invention, X represents a bond and R2 represents cyclopentyl.
In one embodiment of the invention, in a compound of formula (I)
R1 is selected from branched or unbranched C1-C10 alkyl; C3-C10 cycloalkyl; C1-C9 heterocyclyl; and C6-CiO aryl; wherein any alkyl or cycloalkyl moiety optionally is substituted with 1, 2 or 3 groups Ra; and any heterocyclyl or aryl moiety optionally is substituted with 1, 2, 3, 4 or 5 groups Rb;
R2 is selected from branched or unbranched C1-C9 alkyl; C3-C6 cycloalkyl; C1-C9 heterocyclyl; and C6-CiO aryl; wherein any alkyl or cycloalkyl moiety optionally is substituted with 1, 2 or 3 groups Ra; and any heterocyclyl or aryl moiety optionally is substituted with 1, 2, 3, 4 or 5 groups Rb;
R3 and R4 are each independently selected from C1-C4 alkyl and halogen;
R5 and R6 are each selected from H and methyl;
m is 0 or 1 ;
Q is carbonyl; X represents a bond or methylene; each Rais independently selected from hydroxy; C1-C4 alkyloxy; C1-C4 alkyl secondary or tertiary amino; each Rb is independently selected from halogen; cyano; C1-C5 heteroaryl, carboxy, C1-C4 alkyl; C1-C4 alkyl secondary or tertiary amido; C1-C4 alkyl carbonyl; C1-C4 alkyloxy carbonyl; C1-C4 alkyl secondary or tertiary acylsulphonamido; and C1-C5 heterocyclyl; and
Rc is absent. In one embodiment of the invention, in a compound of formula (I)
R1 is selected from branched or unbranched C1-C10 alkyl; C3-C6 cycloalkyl; 5- or 6-membered C3-C5 heterocyclyl; and phenyl; wherein any alkyl or cycloalkyl. moiety optionally is substituted with 1 or 2 groups Ra; and any heterocyclyl or phenyl moiety optionally is substituted with 1 or 2 groups Rb;
R2 is selected from branched or unbranched Ci-C6 alkyl; C3-C6 cycloalkyl; 5- or 6-membered C3- C5 heterocyclyl; and phenyl; wherein any alkyl or cycloalkyl moiety optionally is substituted with 1 or 2 groups Ra; and any heterocyclyl or phenyl moiety optionally is substituted with 1 or 2 groups Rb;
R3 and R4 are each independently selected from methyl and chloro; and
R5 and R6 are each selected from H. In yet another embodiment of the invention, R1 represents C1-C10 alkyl, e.g. Ci-Cs alkyl, or C1- C6 alkyl; R2 represents C4-C6 cycloalkyl or C4-C5 heterocyclyl, e.g. C4-C6 cycloalkyl or 5- or 6- membered C4-C5 heterocyclyl; R3 and R4 are independently selected from methyl and chlorine; R5 and R6 are hydrogen; X represents a bond; and Q represents carbonyl. Thus, in this embodiment, the compound of formula (I) may be represented by the formula (Ia):

wherein the intej ger p is 0-11, e.g. 0-9, or 0-7.
In yet another embodiment of the invention, R1 represents C3-C6 cycloalkyl; R2 represents C4-C6 cycloalkyl or C4-C5 heterocyclyl; R3 and R4 are independently selected from methyl and chlorine; X represents a bond; and Q represents carbonyl.
In yet another embodiment of the invention, R1 represents phenyl or C4-C5 heterocyclyl; R2 represents C4-C6 cycloalkyl or C4-C5 heterocyclyl; R3 and R4 are independently selected from methyl and chlorine; X represents a bond; and Q represents carbonyl.
In yet another embodiment of the invention, R1 represents phenyl or C4-C5 heterocyclyl, e.g. 5- or 6-membered C4-C5 heterocyclyl, optionally substituted with one or more Rb; R2 represents cyclopentyl; R3 and R4 are independently selected from methyl and chlorine; the integer m is equal to 1 or 0 (zero); R5 and R6 are hydrogen; X represents a bond; and Q represents carbonyl. Thus, in this embodiment, the compound of formula (I) may be represented by the formula (Ib):

In yet another embodiment of the invention, R1 represents C1-C10 alkyl, e.g. Ci-Cs alkyl, or Ci- C6 alkyl; R2 represents Ci-C9 alkyl, e.g. Ci-C6 alkyl, or Ci-C4 alkyl; R3 and R4 are independently selected from methyl and chlorine, R5 and R6 are hydrogen; and Q represents carbonyl. Thus, in this embodiment, the compound of formula (I) may be represented by the formula (Ic):

wherein the integer p is 0-11, e.g. 0-9 , or 0-7; and the integer r is 0-12, e.g. 0-10, or 0-8.
In yet another embodiment of the invention, R1 represents C4-C5 saturated heterocyclyl, e.g. 5- or 6-membered C4-C5 heterocyclyl; R2 represents cyclopentyl; R3 and R4 are independently selected from methyl and chlorine; the integer m is equal to 1 ; R5 and R6 are hydrogen; X represents a bond; and Q represents carbonyl. Thus, in this embodiment, the compound of formula (I) may be represented by the formula (Id):

In yet another embodiment of the invention, R1 represents C3-C5 cycloalkyl; R2 represents cyclopentyl or tetrahydrofuranyl; R3 and R4 are independently selected from methyl and chlorine; the integer m is equal to either 0 (zero) or 1; R5 and R6 are hydrogen; X represents a bond and Q represents carbonyl. Thus, in this embodiment, the compound of formula (I) may be represented by the formula (Ie):

wherein the integer t is 0, 1 or 2, and W is oxygen or methylene.
Unless otherwise specified, or apparent from the context, in preferred embodiments of the compounds of formulae (Ia), (Ib), (Ic), (Id), and (Ie), the identity of R1, R2, R3, R4, R5, R6, m, X,
Q, Ra, Rb, and Rc, is as defined herein above in relation to the inventive compound of the general formula (I).
In another embodiment of the invention, there is provided a compound selected from:
Λ/-cyclopentyl-l-(l-ethyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -propyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide; l-(l-butyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4-carboxamide; Λ/-cyclopentyl-l-(l-heptyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -octyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -pentyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl-l-(l-hexyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -(pyridin-2-ylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -( 1 -isopropyl-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(2-(diethylamino)ethyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-
4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-4-ylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-3-ylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-2-ylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
l-(l-sec-butyl-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4-carboxamide; Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(pentan-3-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5 ,6-dichloro- 1 -cyclobutyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l -(5,6-dichloro- l-(l-phenylethyl)-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -methyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -ethyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5 ,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-fluorobenzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(3,5-difluorobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
methyl 4-((2-(4-(cyclopentylcarbamoyl)piperidin- 1 -yl)-5,6-dimethyl- lH-benzo[d]imidazol- 1 - yl)methyl)benzoate;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -((2-methylthiazol-4-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -phenyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopropyl- 1 -(5 ,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclo butyl- 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-propylpiperidine-4-carboxamide; l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-(2-methoxyethyl)piperidine-4- carboxamide;
l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-(isopropoxymethyl)piperidine-4- carboxamide;
l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
Λ/-(cyclohexylmethyl)-l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-((tetrahydro-2H-pyran-4- yl)methyl)piperidine-4-carboxamide;
Λ/-butyl- 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-(cyclopentylmethyl)- 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -( 1 -((6-fluoropyridin-3-yl)methyl)-5 ,6-dimethyl- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
l-(l-(4-cyanobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(cyclopropylmethyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -((tetrahydrofuran-2-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
1 -( 1 -(4-( IH- 1 ,2,4-triazol- 1 -yl)benzyl)-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide;
1 -( 1 -cyclobutyl-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
l-(l-(4-bromobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -(4-(methylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl-l-(l-(4-(dimethylcarbamoyl)benzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
l-(l-(4-carbamoylbenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5 ,6-dimethyl- 1 -(4-(methylsulfonylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3-yl)piperidine-4- carboxamide;
1 -( 1 -(4-fluorobenzyl)-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)-JV-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
1 -(5-chloro- 1 -cyclobutyl-6-methyl- lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentyl-piperidine-4- carboxamide;
1 -(6-chloro- 1 -cyclobutyl-5 -methyl- lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -cyclopentyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
l-(l-(4-bromobenzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -((6-fluoropyridin-3-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-cyanobenzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(cyclopropylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -((tetrahydrofuran-2-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -((tetrahydro-2H-pyran-4-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5 ,6-dichloro- 1 -(4-(methylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-(dimethylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
l-(l-(4-carbamoylbenzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-cyclopentyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4- carboxamide;
l-(l-(4-chlorobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
l-(l-(4-(lH-tetrazol-5-yl)benzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5 ,6-dichloro- 1 -(4-(methylsulfonylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
TV-butyl- 1 -(5,6-dichloro- 1 -cy clo butyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-butyl- 1 -(5 ,6-dichloro- 1 -pentyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
N-butyl- 1 -(5 ,6-dichloro- 1 -(4-fluorobenzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
N-butyl- 1 -(5,6-dichloro- 1 -((6-fluoropyridin-3-yl)methyl)- lH-benzo[d]imidazol-2-yl)piperidine- 4-carboxamide;
1 -(5,6-dichloro- 1-cyclo butyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
1 -(5,6-dichloro- l-pentyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
1 -(5,6-dichloro- l-(4-cyanobenzyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(5,6-dichloro- l-(cyclopropylmethyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(5,6-dichloro- l-((6-fluoropyridin-3-yl)methyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -((tetrahydro-2H-pyran-4-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
1 -(5,6-dichloro- l-cyclopentyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
l-(l-(4-bromobenzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide; l-(l-(4-(lH-tetrazol-5-yl)benzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide;
1 -(5,6-dichloro- 1 -(4-fluorobenzyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1.( 1.(4-( IH- 1 ,2,4-triazol- 1 -yl)benzyl)-5 ,6-dichloro- lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide;
N-cyclopentyl- 1 -(5,6-dichloro- 1 -(2-methoxyethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(4-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
l-(l-(cyclobutylmethyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -(prop-2-ynyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(2-ethylbutyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5 ,6-dimethyl- 1 -neopentyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
l-(l-cyclohexyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
l-(l-sec-butyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentyl-piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(2-hydroxyethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -( 1 ,5 ,6-trimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl-l-(l-isobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide; l-(l-benzyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4-carboxamide; Λ/-cyclopentyl-l-(l-(2-methoxyethyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(4-fluorobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
1 -(5-chloro- 1 -cyclobutyl-6-fluoro- lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4- carboxamide;
1 -(6-chloro- 1 -cyclobutyl-5-fluoro- lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -(pentan-2-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-butyl- 1 -( 1 -cyclobutyl-5 ,6-dimethyl- lH-benzo [d]imidazo l-2-yl)piperidine-4-carboxamide;
Λ/-butyl-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
N-butyl- 1 -(5,6-dimethyl- 1 -pentyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-butyl- 1 -( 1 -((6-fluoropyridin-3 -yl)methyl)-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)piperidine-
4-carboxamide;
Λ/-butyl- 1 -( 1 -(4-fluorobenzyl)-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-benzyl-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-(4-fluorobenzyl)- 1 -( 1 -isopropyl-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)piperidine-4- carboxamide;
Λ/-(3-isopropoxypropyl)-l -(I -isopropyl-5, 6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-(3,3-dimethylbutyl)-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
JV-(4-bromobenzyl)- 1 -(I -isopropyl-5, 6-dimethyl- 1 H-benzo [d]imidazol-2-yl)piperidine-4- carboxamide;
JV-cyclohexyl- 1 -( 1 -isopropyl-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-(4-cyanobenzyl)- 1 -( 1 -isopropyl-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)piperidine-4- carboxamide;
(S)- 1 -(5, 6-dimethyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
4-((2-(4-(cyclopentylcarbamoyl)piperidin- 1 -yl)-5, 6-dimethyl- lH-benzo[d]imidazol- 1 - yl)methyl)benzoic acid
1 -(I -isopropyl-5, 6-dimethyl- lH-benzo[d]imidazol-2-yl)-Λ/-(thiazol-2-ylmethyl)piperidine-4- carboxamide;
l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-(thiazol-2-ylmethyl)piperidine-4- carboxamide;
Λ/-(4-(lH-tetrazol-5-yl)benzyl)-l -(I -isopropyl-5, 6-dimethyl- lH-benzo[d]imidazo 1-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5, 6-dimethyl- 1 -(pentan-3-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
1 -(5, 6-dimethyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-fluorophenyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
l-(l-(4-bromophenyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(I -(4-fluorophenyl)-5, 6-dimethyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
l-(l-(3-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
l-(l-(3,5-difluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
l-(l-(4-chlorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(5,6-dimethyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-JV-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
l-(l-(3,5-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(5,6-dimethyl- 1 -(naphthalen-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -( 1 -(benzo[d] [1,3] dioxo 1-5 -yl)-5 ,6-dimethyl- lH-benzo [d]imidazo l-2-yl)-JV-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
l-(5,6-dichloro-l-(3-methoxyphenyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
l-(l-(benzo[d][l,3]dioxol-5-yl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
l-(5,6-dichloro-l-(4-cyanophenyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -m-tolyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -( 1 -(3 ,4-dimethylphenyl)-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(4-fluoro-3-methylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl-l-(l-(4-ethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(4-isopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -(quinolin-6-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -(quinoxalin-6-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
l-(5,6-dichloro-l-(4-(methylcarbamoyl)phenyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran- 3-yl)piperidine-4-carboxamide;
(R)-l-(5,6-dichloro-l-(3-methoxyphenyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-3-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-3-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(6-methoxypyridin-2-yl)- lH-benzo[d]imidazol-2-yl)piperidine-
4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-cyanophenyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-fluorophenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3 -fluorophenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-methoxyphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-tert-butylphenyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-(trifluoromethoxy)phenyl)- lH-benzo[d]imidazol-2-yl)-JV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide;
1 -(5,6-dichloro- l-phenyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
1 -(5,6-dichloro- l-(pyridin-2-yl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-
4-carboxamide;
1 -(5,6-dichloro- l-(pyridin-3-yl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-
4-carboxamide;
1 -(5,6-dichloro- l-p-tolyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
(R)-l-(l-(4-chloro-3-methoxyphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/- (tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-chloro-3-methoxyphenyl)- lH-benzo[d]imidazol-2-yl)-JV-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;.
(R)-l-(l-(2,3-dihydrobenzofuran-5-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(2,3-dihydrobenzofuran-5-yl)- lH-benzo[d]imidazol-2-yl)-JV-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(3,5-diethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(5,6-dichloro- l-(cyclobutylmethyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-fluoro-3-methylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(4-ethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3,4-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -(quinolin-6-yl)- lH-benzo[d]imidazol-2-yl)-JV-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -( 1 -(4-isopropylphenyl)-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)-JV-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-tert-butylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3-chlorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(6-chloro-5 -methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
(R)- 1 -(5-chloro-6-methyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(6-chloro-5-methyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide;
l-(l-(4-carbamoylphenyl)-6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)-Λ/- cyclopentylpiperidine-4-carboxamide;
(R)-l-(l-(4-carbamoylphenyl)-6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(S-chloro-β-methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]-imidazol-2-yl)-/V-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
1 -(S-chloro-β-methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-/V- cyclopentylpiperidine-4-carboxamide;
1 -(S-chloro-β-methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-/V-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(β-chloro-S-methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]-imidazol-2-yl)-/V-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
1 -(6-chloro-5 -methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-/V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide;
1 -(6-chloro-5 -methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-/V- cyclopentylpiperidine-4-carboxamide;:
(R)- 1 -(6-chloro-5-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-/V-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(S)- l-(6-chloro-5-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-/V-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
/V-butyl- 1 -(5-chloro-6-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide; TV- butyl- 1 -(6-chloro-5-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
1 -(5-chloro-6-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine- 4-carboxamide;
1 -(6-chloro-5 -methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-
4-carboxamide;
1 -(5-chloro- 1 -cyclobutyl-6-methyl- lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(6-chloro- 1 -cyclobutyl-5 -methyl- lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(5-chloro- 1 -(cyclobutylmethyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-7V-cyclopentylpiperidine-
4-carboxamide;
1 -(6-chloro- 1 -(eye Io butylmethyl)-5 -methyl- lH-benzo[d]imidazol-2-yl)-7V-cyclopentylpiperidine- 4-carboxamide;
1 -(5-chloro-6-methyl- 1 -pentyl- lH-benzo[d]imidazol-2-yl)-7V-cyclopentylpiperidine-4- carboxamide;
1 -(6-chloro-5 -methyl- 1 -pentyl- lH-benzo[d]imidazol-2-yl)-7V-cyclopentylpiperidine-4- carboxamide;
1 -(5-chloro- 1 -(4-fluorophenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
1 -(6-chloro- 1 -(4-fluorophenyl)-5 -methyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
1 -(S-chloro-β-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
1 -(6-chloro-5 -methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
Λ/-butyl- 1 -(5-chloro- l-cyclobutyl-6-methyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-butyl- 1 -(6-chloro- 1 -eye Io butyl-5 -methyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -(3-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -(5-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(3-ethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3-isopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(6-chloro- l-(4-isopropylphenyl)-5 -methyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-chlorophenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-fluoro-3-methylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5 ,6-dichloro- 1 -(3 ,5-difluorophenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-cyanophenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(5-methylpyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(6-methoxypyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-isopropylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-methylpyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(6-methylpyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-(difluoromethoxy)phenyl)- lH-benzo[d]imidazol-2-yl)-JV-
(tetrahydro furan-3 -yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-(trifluoromethoxy)phenyl)- lH-benzo[d]imidazol-2-yl)-JV-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-isobutylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5 ,6-dichloro- 1 -(3 ,4-dimethylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-chlorophenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(5-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(5-fluoropyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -( 1 -(4-isobutylphenyl)-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)-N-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
(R)-l-(6-chloro-l-(3,5-dimethylphenyl)-5-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(4-chloro-3-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-chloro-3 -fluorophenyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3,5-diethylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3-tert-butylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3-tert-butylphenyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-ethylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -m-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydro furan-3 -yl)piperidine-4- carboxamide;
(R)- 1 -(5 ,6-dichloro- 1 -(3 ,5-dimethylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-ethylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(6-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydro furan-3 -yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-ethoxyphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-isopropoxyphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-propoxyphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -( 1 -(4-fluoro-3 ,5-dimethylphenyl)-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-chloro-3-methylphenyl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-chloro-4-methylphenyl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5 ,6-dimethyl- 1 -phenyl- 1 H-benzo [d]imidazol-2-yl)-N-(tetrahydro furan-3 -yl)piperidine-4- carboxamide;
(R)- 1 -(5 ,6-dimethyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
(R)- 1 -(5 ,6-dimethyl- 1 -m-tolyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
(R)-l-(l-(3-cyanophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -(naphthalen-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-chloro-3-methylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(3-chloro-4-methylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(benzo[d]thiazol-6-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(benzo[d]thiazol-5-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(R)-l-(5-chloro-l-(3,4-dimethylphenyl)-6-methyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(6-chloro-l-(3,4-dimethylphenyl)-5-methyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5-chloro- l-(4-isopropylphenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(6-chloro- l-(4-isopropylphenyl)-5 -methyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran- 3-yl)piperidine-4-carboxamide;
(S)- l-(5-chloro-6-methyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(S)- l-(6-chloro-5-methyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(S)-l-(6-chloro-l-(3,5-dimethylphenyl)-5-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3-yl)piperidine-4- carboxamide;
l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-((15f,25)-2- hydroxy cyclopentyl)piperidine-4-carboxamide;
l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-((lR,2R)-2- hydroxycyclopentyl)piperidine-4-carboxamide;
(R)-l-(l-(benzofuran-5-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-fluoro-3,5-dimethylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(3-fluoro-5-methylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-fluoro-5-methylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-cyclopropylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-cyclopropylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -( 1 -(benzo furan-6-yl)-5 ,6-dimethyl- lH-benzo [d]imidazo l-2-yl)-JV-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
(R)- 1 -(5-chloro- l-(4-ethylphenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(6-chloro- l-(4-ethylphenyl)-5 -methyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5-chloro- l-(4-ethylphenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-tert-butylphenyl)-6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide;
(R)-l-(l-(4-cyclopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3-cyclopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-(cyclopentyloxy)phenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-cyclobutoxyphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
l-(5-chloro-6-fluoro-l-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
l-(6-chloro-5-fluoro-l-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
1 -(5-chloro- 1 -ethyl-6-fluoro- lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
1 -(6-chloro- 1 -ethyl-5-fluoro- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
l-(5-chloro-6-fluoro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
l-(6-chloro-5-fluoro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
l-(l-butyl-5-chloro-6-fluoro-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
l-(l-butyl-6-chloro-5-fluoro-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
1 -(5 ,6-dimethyl- 1 -p-tolyl- 1 H-benzo [d]imidazo l-2-yl)-JV-(tetrahydrofuran-3 -yl)piperidine-4- carboxamide;
(R)- 1 -(6-chloro- l-(4-fluorophenyl)-5 -methyl- lH-benzo[d]imidazol-2-yl)-JV-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5-chloro- l-(4-fluorophenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-JV-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(6-chloro-5-methyl- 1 -m-tolyl- lH-benzo[d]imidazol-2-yl)-JV-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5-chloro-6-methyl- 1 -m-tolyl- lH-benzo[d]imidazol-2-yl)-JV-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5-chloro-6-methyl- 1 -(5-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-JV-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(6-chloro-5-methyl- 1 -(5-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-((2S)-2- methoxycyclopentyl)piperidine-4-carboxamide;
l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-((2S)-2- ethoxycyclopentyl)piperidine-4-carboxamide;
l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-((2S)-2- propoxycyclopentyl)piperidine-4-carboxamide;
l-(5,6-dichloro-l-(4-(dimethylcarbamoyl)phenyl)-lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)-N-((lS,2S)-2- hydroxycyclopentyl)piperidine-4-carboxamide;
l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)-N-((lS,2S)-2- methoxycyclopentyl)piperidine-4-carboxamide;
l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)-N-((lS,2S)-2- ethoxycyclopentyl)piperidine-4-carboxamide;
1 -( 1 -cyclobutyl-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)-Λ/-(furan-2-ylmethyl)piperidine-4- carboxamide;
1 -( 1 -cyclobutyl-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)-Λ/-(thiophen-2-ylmethyl)piperidine-4- carboxamide;
(R)- 1 -( 1 -(5 -bromopyridin-2-yl)-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)-JV-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
(R)-l-(l-(5-ethylpyridin-2-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3-chloro-5-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3 -chloro-5 -fluorophenyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(2,2-difluorobenzo[d] [ 1 ,3]dioxol-5-yl)- lH-benzo[d]imidazol-2-yl)-Λ/- (tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(S)- 1 -( 1 -(3 ,5-dimethylphenyl)-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(5-chloropyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(5-fluoro-6-methylpyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydro furan-3 -yl)piperidine-4-carboxamide;
(R)-l-(l-(3-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- l-(l-(3,5-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydro furan-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3,5-difluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-chlorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
(R)-l-(l-(5-chloropyridin-2-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -(4-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -( 1 -(5 -fluoro-6-methylpyridin-2-yl)-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-ethoxyphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-methoxy-3-methylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide; and
(R)- 1 -( 1 -cyclobutyl-5 ,6-dimethyl- 1 H-benzo [d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
or a pharmaceutically acceptable salt thereof.
The compounds of formula (I) of the invention can be prepared according to the synthetic routes outlined herein and by following the methods described herein below and illustrated in the Examples. Alternative synthetic routes to these compounds can easily be visualized by any person skilled in the art and the present synthetic routes are not limiting for the invention. With respect to the reaction schemes below, although the various R1, R2, R3, R4, R5, R6, Q and X moieties sometimes are specifically defined, unless otherwise indicated, it is to be understood that R1, R2, R3, R4 and X may be any of the groups encompassed thereby.
As a non- limiting example, a compound of formula (I) as defined herein may be formed in a three step procedure wherein, first, a suitable substituted 2-aminobenzoimidazol (i) is diazotizated and subjected to the Sandmeyer reaction, or any suitable modification of this method, the formed chloro intermediate (ii) is coupled with a piperidine-4-carboxamide derivative (iii) to form the product (iv), which subsequently is JV-alkylated or arylated by Y-
(CR5R6)m-R1 (v) to yield the final compound of formula (I). The entire synthetic route is depicted below in Reaction Scheme 1.
Reaction Scheme 1
1 ) diazotization
2) Sandmeyer



The process of forming diazonium compounds from aromatic or heteroaromatic amines has been known for more than 150 years and involves an amine that reacts with a nitrite salt such as sodium nitrite to form a diazonium salt at low temperature, a procedure that should be well known for the one skilled in the art. Typically diazonium compounds are not isolated, but used immediately in further reactions as described below in the Sandmeyer reaction. Diazonium compounds can, however, also be isolated as tetrafluoroborate salts. Conditions suitable for the Sandmeyer reaction are well known to the person skilled in the art and comprise an diazonium salt of an aromatic or heterocyclic kept at low temperature, which is decomposed in the presence of copper(I) salts, such as copper(I) chloride or copper(I) bromide, to form the desired chloro or bromo derivative. Several modifications to the standard procedures above have been made and are exemplified by the use of t-butyl nitrite and anhydrous copper(II) chloride or copper(II) bromide at elevated temperatures [Doyle, M. P.; Siegfried, B.; Dellaria Jr, J. F. Alkyl nitrite- metal halide deamination reactions. 2. Substitutive deamination of arylamines by alkyl nitrites and copper(II) halides. A direct and remarkably efficient conversion of arylamines to aryl halides. J. Org. Chem. 1977, 42, 2426-2431], t-butyl thionitrite and copper(II) chloride or copper(II) bromide employed at room temperature [Oae, A.; Shinhama, K.; Kim, Y. H. Direct conversion of arylamines to the corresponding halides, biphenyls and sulfides with t-buty thionitrate. Chem. Lett., 1979, 939-942] and mixtures of Cu(I) and Cu(II) chloro and bromo salts, used in catalytic amounts in the presence of the bidentate ligand phenanthroline and phase-
transfer catalyst dibenzo-18-crown-6 [Beletskaya, I, P.; Sigeev, A. S.; Peregudov, A. S.;
Petrovskii, P. V. Catalytic Sandmeyer Bromination. Synthesis, 2007, 2534-2538]. In this context, it should be contemplated that a wide variety of methods exists for the transformation of (i) into (ii) and several exhaustive review articles on the topic can be found in the literature.
Conditions suitable for the coupling of aromatic and heteroaromatic halides, such as chlorides, with aliphatic amine chains or secondary amine aliphatic ring structures such as piperidine derivatives as (iii) are well known for the one skilled in the art, and there exists a wealth of different methods in the scientific literature, summarized in for example: "Advanced Organic Chemistry", 4th edition, Jerry March, Wiley-Interscience publication, 1992, p 656-657, including the references cited therein. In the precent invention the chloro intermediate (ii) is reacted with the piperidine-4-carboxamide derivative (iii) to form the coupled product (iv) in the presence of a base, such as as N,Λ/-diisopropylethylamine (Hunig's base, DIEA, CAS Registry Number: 7087-68-5), in a suitable solvent, such as 1,4-dioxane. Obviously other alternative good bases that are poor nucleophiles can be employed, such as, but not limited to 2,2,6,6- tetramethylpiperidine (TMP, CAS Registry Number: 768-66-1).
In the transformation of (iv) into the final compound of formula (I), JV-alkylation or JV-arylation by Y-R1 (v) is employed. For the JV-alkylation, (iv) is treated with a chloro-, bromo or iodoalkyl electrophile in the presence of a base such as caesium carbonate in a solvent such as acetonitrile and/or N,Λ/-dimethylformamide. A palladium catalyst, such as, but not limited to [1,1'- bis(diphenylphosphino)ferrocene] dichloropalladium(II) (PdCl2(dppf), CAS Registry Number: 72287-26-4) can also be employed in this reaction. Possible use of alternative bidentate palladium-catalysts for a successful N-alkylation might involve [l,2-bis(diphenylphosphino)- ethane]dichloro-palladium(II) (PdCl2(dppe), CAS Registry Number: 19978-61-1), [1,4- bis(diphenylphosphino)-butane] dichloropalladium(II), PdCl2dppb), and the like, all well known for the one skilled in the art. For the JV-arylation, (iv) is treated with a haloaryl in the presence of a base such as caesium carbonate, and an agent as 1,10-phenanthroline, 4,7-dimethoxy-l,10- phenanthroline or 8-hydroxyquinoline, polyethylene glycol and copper(I) oxide in a solvent such as dimethylsulfoxide. Yet other combinations of bases, catalysts, solvents and agents can be appropriately employed and are well known for those skilled in the art [eg. Preston, P. N.
Synthesis, reactions, and spectroscopic properties of benzimidazoles. Chem. Rev., 1974, 74, 279- 314], and are well within the scope of the invention.
An alternative preparation of (ii) is depicted in Reaction Scheme 2 and involves the treatment of a suitable substituted diamine (vi) with excess stoichiometric amounts of the carbonyl source
N,N-carbonyl dimidazole (CDI, CAS Registry Number: 530-62-1) in a solvent such as tetrahydrofuran, in order to achieve ring-closure to the corresponding tautomeric most stable benzoimidazol-2-one (vii). The ring-closure of aromatic or heteroaromatic diamines is well- known in the literature and might involve the use of alternative of carbonyl sources exemplified by the usage of carbon monoxide in the presence of catalytic amounts of tungsten hexacarbonyl (CAS Registry Number: 14040-11-0). Subsequently, the tautomeric -ol form (viii) can be transformed to the intermediary (ii) via treatment with phosphoryl trichloride (CAS Registry Number: 10025-87-3), as exemplified in the invention. Alternative reagents include phosphorus pentachloride (CAS Registry Number: 10026-13-8) or phosphorus trichloride (CAS Registry Number: 7719-12-2), the former also, alone or as mixtures in various ratios, or any other chlorination agent, all that exists in abundance in the scientific literature, for the purpose of transforming a hydroxyl group to a chlorine group. In this context, it should be contemplated that the corresponding bromo derivative of (ii) also can be useful as an intermediate in the reaction schemes, leading to the target compounds of the invention, and could be prepared by the action of phosphoryl bromide (CAS Registry Number: 7789-59-5) phosphorus tribromide (CAS
Registry Number: 7789-60-8) and/or phosphorus pentabromide (CAS Registry Number: 7789- 69-7), or any other brominating agent, all that exists in abundance in the scientific literature, for the purpose of transforming a hydroxyl group to a bromine group.
Reaction Scheme 2
(ii)

Alternative synthetic strategies for the preparation of a compound of formula (I) are shown in Scheme 3. For example, the chloro intermediate (ii), can be coupled with piperidine-4-carboxylic acid derivative (ix) to form the coupled product (x), which subsequently is N-alkylated by Y- (CR5R6)m-R1 (v), the ethyl ester saponificated to give the carboxylic acid derivative, which can be subjected to amide coupling with H2N-X-R2, to yield the final compound of formula (I). This method is foremost useful when the Reposition needs to be varied, while the R'-substituent is kept constant during the preparation.
Reaction Scheme 3


The carboxylic acid ester (xi) is saponified with a base such as aqueous sodium hydroxide and a solvent such as ethanol. Acidification of the completed reaction mixture is followed by standard work-up and crystallization or chromatography, to yield the carboxylic acid (xii). A wide variety of other protecting groups for the carboxylic acid can be employed, and their usage is known to those skilled in the art (references describing protecting group strategy include, for example, "Protecting Groups in Organic Chemistry", J. F. W. McOmie, Plenum Press, London, New York, 1973, and "Protective Groups in Organic Synthesis", T. W. Greene, Wiley, New York, 1984).
Conditions suitable for amide coupling, as exemplified herein by the acid derivative (xii) and the amine H2N-X-R2 (xiii), are well-known to the person of ordinary skill in the art, and may be eg. obtained by performing the reaction of acid and amine in the presence of a suitable amide coupling reagent, such as 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (HATU; CAS Registry Number: 148893-10-1) and N,N- diisopropylethylamine in a suitable solvent, such as dichloromethane, or by reacting the acid derivative in the presence of e.g. triethylamine in a suitable solvent, such as chloroform or in the presence of N,7V-diisopropylethylamine in a suitable solvent, such as dichloromethane.
A number of other chemical transformations may also be used within the present invention, all which readily could be modified and improved by the one skilled in the art.
Apart from traditional heating, such as the use of heating blocks, heat transfer via oil baths or the like, the present invention also involve the use of microwave irridation for heating purposes, which today is well known for the one skilled in the art, as witnessed by a large number of book chapters and published books [eg. Strauss, C. R., Application of microwaves for environmentally benign organic chemistry, In Handbook of Green Chemistry and Technology, Clark, J.;
Macquarrie, D., Eds. Blackwell Science Ltd: Oxford, 2002, pp 297-415; Kappe, C. O.; Stadler, A. Microwaves in Organic and Medicinal Chemistry, Wiley- VCH, Weinheim, 2005; Watley, B.; Tierney, J.; Lidstrόm, P.; Westman, J. The impact of microwave assisted organic chemistry on drug discovery. Drug Discov. Today, 2002, 7, 373-380].
All reaction mixtures are purified by standard purification procedures as extraction, washing, recrystallization, and column chromatography employing silica gel, aluminia or reverse phase and the like, employing the appropriate eluents for the purification of the Examples described herein.
The compounds of the invention can also be prepared by the application of combinatorical and/or parallell chemistry techniques eg. methods of making libraries of compounds, techniques well known for the one skilled in the art, which can include solution phase synthesis, and solid phase synthesis including design of resins, linkers and the like. For example (Scheme 4), (x) can be saponifϊcated to give the intermediate (xiv) that can serve as a suitable scaffold for further manipulation. It can then be contemplated that JV-alkylation with (v), followed by amide coupling with (xiii) can be conducted in one pot (in the same reaction vessel) if the two reactions are selective in terms of regioselectivity (a term that is well known for the one skilled in the art), which also includes the reverse reaction order eg. amide coupling followed by N-alkylation, or alternatively both reactions acting at the same time. This can lead to compounds of formula (I) of the invention in a shorther time than traditional consecutive synthesis, albeit an approach not excluded from the present invention.
Reaction Scheme 4
N-alkylation
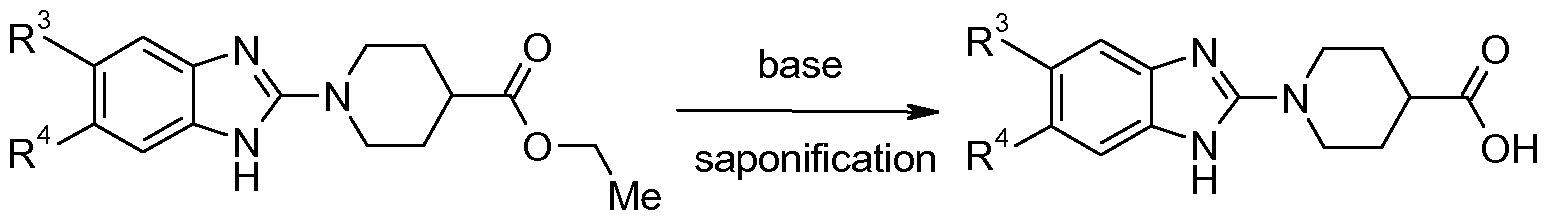
(X) (xiv)
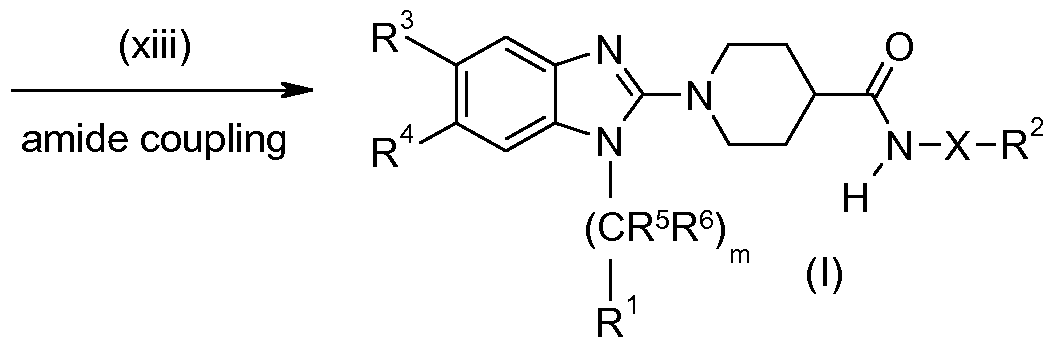
All above is provided when R3 and R4 is the same atom or group. On the other hand, when R3 and R4 is dissimilar, for instance when R3 is methyl and R4 is chlorine, then two tautomeric forms of (ii) will exist as shown in Reaction Scheme 6 (iia and iib). The knowledge of tautomers is well known for the one skilled in the art, and numerous scientific book chapters, reviews and papers have examined various aspects of this phenomenon, [eg. The Chemistry of Heterocyclic Compounds, Benzimidazoles and Cogeneric Tricyclic Compounds (Chemistry of Heterocyclic Compounds: A Series Of Monographs), Preston, P. N. Smith, D. M. and Tennant, G. (Eds), John Wiley & Sons Inc, 1981]. Reaction Scheme 5

(iia) (iib)
Outlined in Reaction Scheme 7 is the one of the synthetic strategies of the invention that involves tautomeric forms of the intermediate compounds. Accordingly, the tautomeric mixture of (iia) and (iib) is treated with a nucleophilic agent, as exemplified by (iii), but not restricted to, then yet two tautomeric compounds (iva) and (ivb) are formed. In the final step an N-alkylating agent (v) is employed and a regioisomeric mixture of end products (I) is formed, which might be tested directly in biological assays. Alternatively, the regioisomers of the mixture can be separated before biological testing, using for instance column chromatography, recrystallization, sublimation or any other methods available for the one skilled in the art of chemical separation. In the present method the final purification can only take palace after the final synthetic step, since all previous intermediates are tautomers.
Reaction Scheme 6

(I) (I)
R 1 R '
Alternatively, and also within the scope of the invention, N-alkylation or N-arylation can be introduced at an earlier stage in the synthetic procedure, as exemplified shown in Reaction Scheme 7. This will provide the compounds (xia) and (xib), which already at this stage can be separated using any of the techniques mentioned above. As exemplified in Reaction Scheme 7, this strategy will ultimately lead to the end products (I). This synthetic strategy can, for example, be considered when the final Examples are difficult to separate, or because of any other advantageous reason. Reaction Scheme 7

(xia) (xib)
R1 R'
1 ) saponification ^ saponification
2) amide coupling 2) amide coupling

(I) (I)
R' R'
Furthermore, the invention at hand also comprises a synthetic method that is strictly
regioselective when R4 and R5 are dissimilar. This is outlined in Reaction Scheme 8 and the synthetic strategy involves the introduction of the N-arylating agent (v) at an early stage in the synthesis in order to obtain control over regioselectivity. The ortho-nitroanline (xiv) is arylated at the at the amine to give (xv), the nitro group reduced to the diamine (xvi), ring-closed to the tautomers (xvii) and (xviii) and finally chlorinated to give (xix). The resulting compound (xix) can then be transformed to the appropriate end-product by any of the methods described above.
Reaction Scheme 8
Reduction Ring-closure
 R6)m R6),
R6)m R6),


(CR5R6), (CR5R6), (xix) (CRSR<%
(xvii) (xviii)
R1
In this context it should be understood that when N-alkylation is mentioned in the text or in the Reaction Schemes, then the N-arylation of the available -NH- or -NH2 group is not excluded, but an important aspect of the present invention. Also, when only N-arylation is mentioned, then should neither N-alkylation be excluded.
The present invention also comprises stereoisomers that, for example, are prepared with the available metods described above. A large number of available single enantiomers are available commercially or in the literature. The single enantiomers can be amines that are used in the amide coupling of present invention, such as, but not limited to, (R)-(+)-tetrahydrofuran-3 -amine 4-methylbenzenesulfonate (CAS Registry Number: 111769-27-8), (S)-(-)-tetrahydrofuran-3- amine hydrochloride (CAS Registry Number: 204512-95-8), (lS,2S)-trans-2- aminocyclopentanol hydrochloride (CAS Registry Number: 68327-04-8), (lR,2R)-trans-2- aminocyclopentanol hydrochloride (CAS Registry Number: 68327-11-7).
When Q is sulfonyl, this results in compounds of the invention that are sulfonamides, which can be prepared employing reaction conditions that are very well known from the scientific literature (see for example: "Advanced Organic Chemistry", 4th edition, Jerry March, Wiley-Interscience publication, 1992, p 499). For example, if a suitable substituted piperidine sulfonyl chloride (xv) is reacted with a suitable substituted amine derivative (xiii), a sulfonamide derivative of formula (I) can be formed as shown in Reaction Scheme 9. Reaction Scheme 9

Compounds wherein Q is sulfϊnyl can be prepared using an analogous method as described in Reaction Scheme 9, as well as treatment of sulfonamides with reductive agents, procedures well known for the one skilled in the art.
Alternatively, and within the scope of the invention, (xxii), which includes a protection group PG, is coupled with an amine (xiii) to give the sulphonamide (xxiii), that can be coupled, after de-protection with a suitable halobenzimidazole (ii), and finally N-alkylated or N-arylated with (v) to the end products (I). The synthetic procedure is shown in Reaction Scheme 10.
Reaction Scheme 10

The above methods, as outlined in Schemes 1, 2, 3, 4, 5, 6, 7, 8, 9 and 10 are only exemplary and it is contemplated that the skilled person will be able to modify them in view of the particular compound according to formula (I) that is to be prepared, or even to identify other equally suitable methods.
It should be contemplated that several spectroscopic methods are available for the structural determination of regio isomers. For example, when R3 is dissimilar to R4, the Nuclear Overhauser effect (NOE) can be utlilized to determine the exact structure of the Example, since the NOE is observed through space, and not through bonds. Consequently, all atoms that are in proximity to each other give a NOE. For example, and utilized in the present invention, the NMR tequnique NOESY (Nuclear Overhauser Effect Spectroscopy) is used for determination of the nature of the
R3 and R4substituents. For example, A NOE effect can be seen between the Ha, Hb and Hc protons in (xxv), and reveals that Hb correlates with both Ha and Hc in the 2-dimensional plot. On the other hand, in (xxvi) Ha and Hd correlates as a pair, as Hb and Hc.
Reaction Scheme 11

The compounds according to formula (I) will be useful for treating various diseases such as inflammatory diseases, nociceptive pain, auto-immune disease, breathing disorders, fever, cancer, inflammation related anorexia, Alzheimer's disease and cardiovascular disease. The treatment may be preventive, palliative or curative. Examples of pharmaceutically acceptable addition salts for use in the pharmaceutical compositions of the present invention include those derived from mineral acids, such as hydrochloric, hydrobromic, phosphoric, metaphosphoric, nitric and sulphuric acids, and organic acids, such as tartaric, acetic, citric, malic, lactic, fumaric, benzoic, glycolic, gluconic, succinic, and arylsulphonic acids. The pharmaceutically acceptable excipients described herein, for example, vehicles, adjuvants, carriers or diluents, are well-known to those who are skilled in the art and are readily available to the public. The pharmaceutically acceptable carrier may be one that is chemically inert to the active compounds and that has no detrimental side effects or toxicity under the conditions of use. Pharmaceutical formulations are found e.g. in Remington: The Science and Practice of Pharmacy, 19th ed., Mack Printing Company, Easton, Pennsylvania (1995).
All stereoisomers of the compounds of the instant invention are contemplated, either in admixture or in pure or substantially pure form. The compounds of the present invention can have asymmetric centers at any of the carbon atoms including any one of the R substitutents. Consequently, compounds of formula (I) can exist in enantiomeric or diasteromeric forms or in mixtures thereof. The processes for preparation can utilize racemates, enantiomers or diasteromers as starting materials. When diastereomeric or enantiomeric products are prepared,
they can be separated by conventional methods, which for example is chromatographic or fractional crystallization.
Prodrugs of the compounds of formula (I) may be prepared by modifying functional groups present on the compound in such a way that the modifications are cleaved, in vivo when such prodrug is administered to a mammalian subject. The modifications typically are achieved by synthesizing the parent compound with a prodrug substituent. Prodrugs include compounds of formula (I) wherein a hydroxy, amino, sulfhydryl, carboxy or carbonyl group in a compound of formula (I) is bonded to any group that may be cleaved in vivo to regenerate the free hydroxyl, amino, or sulfhydryl group, respectively. Examples of prodrugs include, but are not limited to, esters and carbamates of hydroxy functional groups, esters groups of carboxyl functional groups, N-acyl derivatives, JV-Mannich bases. General information on prodrugs may be found in the scientific literature [eg. Bundegaard, H. "Design of Prodrugs" p 1-92, Elesevier, New York- Oxford, 1985; The Practice of Medicinal Chemistry, Camille G. Wermuth et al., chapter 31, Academic Press, 1996; A Textbook of Drug Design and Development, P. Krogsgaard-Larson and H. Bundgaard, eds. chapter 5, pgs 113 - 191, Harwood Academic Publishers, 1991). Said references are incorporated herein by reference. All prodrugs of the compounds of the invention are included within the scope of the invention. The compounds of the formula (I) can be administered for any of the uses described herein by any suitable means, for example, orally, such as in the form of tablets, aqueous or oily suspensions or solutions, elexirs, syrups, capsules, granules or powders; sublingually; buccally; ocular; parenterally, such as by subcutaneous, intravenous, intramuscular, or intrasternal injection or infusion techniques (e.g., as sterile injectable aqueous or non-aqueous solutions or suspensions). Also, the compounds of the formula (I) may be applied as gargles and mouth washes. For parenteral administration, a parenterally acceptable aqueous or oily suspension, emulsion or solution is employed, which is pyrogen free and has requisite pH, isotonicity and stability. Those skilled in the art are well able to prepare suitable formulations and numerous methods are described in the literature. A brief review of methods of drug delivery is also found in the scientific literature [eg. Langer, Science 249:1527-1533 (1990)]. Furthermore, the compounds of the formula (I) can be administered nasally, including administration to the nasal membranes, such as by inhalation spray; topically, such as in the form of a gel, cream or ointment; or rectally such as in the form of suppositories; in dosage unit formulations containing non-toxic, pharmaceutically acceptable vehicles or diluents. The present compounds can, for example, be administered in a form suitable for immediate release or extended release.
Immediate release or extended release can be achieved by the use of suitable pharmaceutical compositions comprising the present compounds, or, particularly in the case of extended release, by the use of devices such as subcutaneous implants or osmotic pumps. The present compounds can also be administered liposomally. The precise nature of the carrier or other material will depend on the route of administration and those skilled in the art are well able to prepare suitable solutions and numerous methods are described in the literature.
Exemplary compositions for oral administration include suspensions which can contain, for example, microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners or flavoring agents such as those known in the art; and immediate release tablets which can contain, for example, microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and/or lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants such as those known in the art. The compounds of formula (I) can also be delivered through the oral cavity by sublingual and/or buccal administration. Molded tablets, compressed tablets or freeze-dried tablets are exemplary forms which may be used. Exemplary compositions include those formulating the present compound(s) with fast dissolving diluents such as mannitol, lactose, sucrose and/or cyclodextrins. Also included in such formulations may be high molecular weight excipients such as celluloses (avicel) or polyethylene glycols (PEG). Such formulations can also include an excipient to aid mucosal adhesion such as hydroxy propyl cellulose (HPC), hydroxy propyl methyl cellulose (HPMC), sodium carboxy methyl cellulose (SCMC), maleic anhydride copolymer (e.g., Gantrez), and agents to control release such as polyacrylic copolymer (e.g. Carbopol 934). Lubricants, glidants, flavors, coloring agents and stabilizers may also be added for ease of fabrication and use.
Exemplary compositions for nasal aerosol or inhalation administration include solutions in saline which can contain, for example, benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, and/or other solubilizing or dispersing agents such as those known in the art.
Exemplary compositions for parenteral administration include injectable solutions, emulsions or suspensions which can contain, for example, suitable non-toxic, parenterally acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, oil or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid, or Cremaphor.
Exemplary compositions for rectal administration include suppositories which can contain, for example, a suitable non-irritating excipient, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures, but liquify and/or dissolve in the rectal cavity to release the drug.
Exemplary compositions for topical administration include a topical carrier such as Plastibase (mineral oil gelled with polyethylene). The dose administered to a mammal, particularly a human, in the context of the present invention should be sufficient to effect a therapeutic response in the mammal over a reasonable time frame. One skilled in the art will recognize that dosage will depend upon a variety of factors including the potency of the specific compound, the age, condition and body weight of the patient, the nature and extent of the condition being treated, recommendations of the treating physician, and the therapeutics or combination of therapeutics selected for administration, as well as the stage and severity of the disease. The dose will also be determined by the route (administration form), timing and frequency of administration. Oral dosages of the present invention, when used for the indicated effects, will range between about 0.01 mg per kg of body weight per day (mg/kg/day) to about 100 mg/kg/day, preferably 0.01 mg per kg of body weight per day (mg/kg/day) to 20 mg/kg/day, and most preferably 0.1 to 10 mg/kg/day, for adult humans. For oral administration, the compositions are preferably provided in the form of tablets or other forms of presentation provided in discrete units containing 0.5 to 1000 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated, for example 0.5, 1.0, 2.5, 5.0, 10.0, 15.0, 25.0, 50.0, 100, 200, 400, 500, 600 and 800 mg..
Parenterally, especially intravenously, the most preferred doses will range from about 0.001 to about 10 mg/kg/hour during a constant rate infusion. Advantageously, compounds of the present invention may be administered in a single daily dose, or the total daily dosage may be
administered in divided doses of two, three or four times daily. Furthermore, preferred compounds for the present invention can be administered in intranasal form via topical use of suitable intranasal vehicles, or via transdermal routes, using those forms of transdermal skin patches well known to those of ordinary skill in the art. To be administered in the form of a transdermal delivery system, the dosage administration will, of course, be continuous rather than intermittent throughout the dosage regimen. A nasal, sublingual or buccal administration
typically contains from about 0.01 mg to about 500 mg of the active ingredient, preferably from about 1 mg to about 100 mg of active ingredient, per dose.
The compounds of the present invention may be also be used or administered in combination with one or more additional drugs useful in the treatment of inflammatory diseases, nociceptive pain, auto-immune diseases, breathing disorders, fever, cancer, inflammation related anorexia, Alzheimer's disease and cardiovascular diseases. The components may be in the same formulation or in separate formulations for administration simultaneously or sequentially. The compounds of the present invention may also be used or administered in combination with other treatment such as irradiation for the treatment of cancer.
Accordingly, in a further aspect of the invention, there is provided a combination product comprising:
(A) a compound of the invention, as hereinbefore defined; and
(B) another therapeutic agent that is useful in the treatment of inflammatory diseases, nociceptive pain, auto-immune disease, breathing disorders, fever, cancer, inflammation related anorexia, Alzheimer's disease and cardiovascular diseases., wherein each of components (A) and
(B) is formulated in admixture with a pharmaceutically-acceptable adjuvant, diluent or carrier. Such combination products provide for the administration of a compound of the invention in conjunction with the other therapeutic agent, and may thus be presented either as separate formulations, wherein at least one of those formulations comprises a compound of the invention, and at least one comprises the other therapeutic agent, or may be presented (i.e. formulated) as a combined preparation (i.e. presented as a single formulation including a compound of the invention and the other therapeutic agent).
Thus, there is further provided:
(1) a pharmaceutical formulation including a compound of the invention, as hereinbefore defined, another therapeutic agent, and a pharmaceutically-acceptable adjuvant, diluent or carrier; and
(2) a kit of parts comprising components:
(a) a pharmaceutical formulation including a compound of the invention, as hereinbefore defined, in admixture with a pharmaceutically-acceptable adjuvant, diluent or carrier; and
(b) a pharmaceutical formulation including another therapeutic agent in admixture with a pharmaceutically-acceptable adjuvant, diluent or carrier, which components (a) and (b) are each provided in a form that is suitable for administration in conjunction with the other. Compounds of the formula (I) will be useful as partial or complete substitute for
cyclooxygenase-2 inhibitors and conventional NSAIDs in preparations wherein they are presently co-administered with other agents or ingredients.
Examples of such agents for the administration in combination with compounds of the invention, include suitable anti-inflammatory, anti-pain, anti-autoimmune, anti-fever, anti-cancer and anti- anorexia (inflammatoriy) agents, and agents for the treatment or prevention of breathing disorders, cardiovascular diseases and Alzheimer's disease, a potentiator including caffeine, an H2 antagonist, aluminium or magnesium hydroxide, simethicone, a decongestant, an antitussive, an antihistamine, a diuretic, a proton pump inhibitor, a bradykinin-1 antagonist, a sodium channel blocker, a 5 -HT agonist or a CGRP antagonist.
More specifically compounds for use in combination with the compounds of the present invention include, but is not limited to, prednisone (CAS Registry Number: 53-03-2);
dexamethasone (CAS Registry Number: 50-02-2); any of the selective glucocorticoid receptor agonists (SEGRAs) exemplified by All 6515 (Lin, C. et al. MoL Pharm., 2002, 62, 297-303), and ZK209614 (Schacke, H. et al, Proc. Natl. Acad. Sci. USA., 2004, 101, 227-232); aspirin (CAS Registry Number: 50-78-2); indomethacin (CAS Registry Number: 53-86-1) particularly dosed as local treatment with gel or spray; ibuprofen (CAS Registry Number: 15687-27-1); piroxicam (CAS Registry Number: 36322-90-4,; CTLA4-Ig agonists/antagonists (Kremer, J. M. et al, N EnglJ Med. 2003, 349, 1907-1915); Inosine-5 '-monophosphate dehydrogenase inhibitors (Whitby, F. G. et al., Biochemistry, 1997, 36, 10666-10674), such as mycophenolate (CAS Registry Number: 24280-93-1); tumor necrosis factor (TNF) antagonists as infliximab (CAS
Registry Number: 170277-31-3); adalimumab (CAS Registry Number: 331731-18-1); etanercept (CAS Registry Number: 185243-69-0) and pentoxifylline (CAS Registry Number: 6493-05-6); orazipone (OR- 1384); integrin antagonists; cell adhesion inhibitors interferon gamma
antagonists; budesonide (CAS Registry Number: 51333-22-3); clofazimine (CAS Registry Number: 2030-63-9); selective thyroid hormone agonists such as GC-I (Johansson, L. et al,Proc. Nαtl. Acαd. Sci. USA. 2005, 102, 10297-10302), KB2115 (Berkenstam, A. et al., Proc. Nαtl Acαd. Sci. USA. 2008, 105, 663-667); KB-141 (Graver, G. J. et al., Proc. Nαtl. Acαd. Sci. USA. 2003, 100, 10067-10072); and MB07811 (Erion, MD et al., Proc. Nαtl. Acαd. Sci. USA. 2007, 104, 15490-15495); selective farnesoid X receptor agonists (FXRs) such as WAY-362450 (Flatt, B. et al., J. Med. Chem. 2009, 52, 904-907); CD4 antagonists such as priliximab (CAS
Registry Number: 147191-91-1); p38 mitogen-activated protein kinase inhibitors such as SB203580 (CAS Registry Number: 152121-47-6); mesalazine (CAS Registry Number: 89-57-6); azathioprine (CAS Registry Number: 446-86-6); sumatriptan (CAS Registry Number: 103628- 46-2); paracetamol (CAS Registry Number: 103-90-2); fast-acting bronchodilators such as salbutamol (CAS Registry Number: 18559-94-9) and ephedrine (CAS Registry Number: 299-42- 3); rituximab; NO-releasing drugs such as nitroglycerine (CAS Registry Number: 55-63-0) and isosorbide dinitrate (CAS Registry Number: 87-33-2); Selective Estrogen Receptor Modulators (SERMs) such as tamoxifen (CAS Registry Number: 10540-29-1); raloxifene (CAS Registry Number: 84449-90-1) and toremifene (CAS Registry Number: 89778-26-7); selective Liver X receptor (LXR) agonists such as T0901317 (CAS Registry Number: 293754-55-9, Repa, J. J. et al, Science, 2000, 289, 1524-1529) and GW3965 (CAS Registry Number: 405911-17-3); antidepressant drugs and pain-relivers, such as tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) such as desvenlafaxine (CAS Registry Number: 93413-62-8); duloxetine (CAS Registry Number:
116539-59-4); milnacipran (CAS Registry Number: 92623-85-3); tramadol (CAS Registry Number: 27203-92-5) and bicifadine (CAS Registry Number: 71195-57-8).
The term "treating" encompasses not only treating a patient to relieve the patient of the signs and symptoms of the disease or condition but also prophylactically treating an asymptomatic patient to prevent the onset or progression of the disease or condition.
The term "patients" include mammalian (including human) patients.
The term "effective amount" refers to an amount of a compound, which confers a therapeutic effect on the treated patient. The effect may be objective (i.e. measurable by some test or marker) or subjective (i.e. the subject gives an indication of or feels an effect).
The invention is illustrated by the following non-limiting Examples. In connection to these Examples, it should be noted that unless otherwise indicated, the expression "purified on reverse- phase hplc" will mean utilizing a preparative hplc-system comprising the following conditions and equipment: Shimadzu LC8A, C18 column (22 x 250 mm, lOμm), acetonitrile/water gradient (0.1% trifluoroacetic acid), flowrate 15 mL/min.
EXAMPLES
Example 1: 7V-cyclopentyl-l-(l-ethyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide

(a) Preparation of the intermediary compound 2-chloro-5,6-dimethyl-lH-benzo[d]imidazole:

To a solution of sodium nitrite (3.77 g, 54.6 mmol) in water (220 mL) was added an aqueous solution of copper(II) chloride (12.24 g, 91.06 mmol, in 160 mL of water) The mixture was stirred at room temperature for 5 minutes and 5,6-dimethyl-lH-benzo[d]imidazol-2-amine (2.936 g, 18.21 mmol) was added to the mixture in portions for 5 minutes. The reaction mixture was stirred at room temperature for 1 hour, heated at 1000C for 1 hour and cooled down. The resulting reaction mixture was extracted with ethyl acetate (3 x300 mL), the combined organic phases washed with brine (200 mL) and dried over anhydrous magnesium sulphate. After concentration in vacuo, the residue was subjected to column chromathography (silica gel, methanol/dichloromethane gradient elution consisting of 1-5% methanol), to give 1.054 g (29 % yield, 90 % purity) of 2-chloro-5,6-dimethyl-lH-benzo[d]imidazole that was used directly in the next step.
(b) Preparation of the intermediary compound iV-cyclopentyl-l-(5,6-dimethyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide:
 2-Chloro-5,6-dimethyl-lH-benzo[d]imidazole (3.59 mmol), iV-cyclopentylpiperidine-4- carboxamide (3.95mmol), JV,JV-diisopropylethylamine (Ηunig's base, DIEA, 927.3 mg, 7.18 mmol) and 1,4-dioxane (2 mL) were mixed in a microwave vial and sealed. The reaction mixture was heated at 18O0C in a microwave oven for 1 hour. After cooling down the reaction mixture, the resulting slurry was filtered, the precipitate washed with 1 ,4-dioxane (5 mL) and dried under
vacuum over night. This gave 796 mg (65% yield) of N-cyclopentyl-1 -(5,6-dimethyl- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 341.2 (M+l).
2-Chloro-5,6-dimethyl-lH-benzo[d]imidazole (3.59 mmol), iV-cyclopentylpiperidine-4- carboxamide (3.95mmol), JV,JV-diisopropylethylamine (Ηunig's base, DIEA, 927.3 mg, 7.18 mmol) and 1,4-dioxane (2 mL) were mixed in a microwave vial and sealed. The reaction mixture was heated at 18O0C in a microwave oven for 1 hour. After cooling down the reaction mixture, the resulting slurry was filtered, the precipitate washed with 1 ,4-dioxane (5 mL) and dried under
vacuum over night. This gave 796 mg (65% yield) of N-cyclopentyl-1 -(5,6-dimethyl- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 341.2 (M+l).
(c) Λ/-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (17.0 mg, 0.05 mmol), iodoethane (9.4 mg, 0.06 mmol), caesium carbonate (32.6 mg, O.lmmol) and acetonitrile (2 mL) were mixed and heated at 1000C. After 2 hours additional iodoethane was added and the reaction mixture was continued to be stirred over night at 1000C. After concentration in vacuo, the residue was subjected to column chromatography (Silica gel, methanol/dichloromethane gradient elution 0-10% methanol) to afford 5.4 mg (29% yield) of the title N-cyclopentyl- 1 -( 1 -ethyl-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide. LC-MS (m/z) 369.3 (M+l).
Example 2: 7V-cyclopentyl-l-(5,6-dimethyl-l-propyl-lH-benzo[d]imidazol-2-yl)piperidine- 4-carboxamide

N-cyclopentyl- 1 -(5,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (17.0 mg, 0.05 mmol), 1-iodopropane (17.0 mg, 0.10 mmol), caesium carbonate (48.9 mg, 0.15 mmol) and acetonitrile (2 mL) were mixed and the reaction mixture heated at 1000C over night. After concentration in vacuo, the residue was subjected to column chromathography (Silica gel, methanol/dichloromethane gradient elution 0-10% methanol) to give lO.lmg (53% yield) of N- cyclopentyl- 1 -(5,6-dimethyl- 1 -propyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC- MS (m/z) 383.3 (M+l).
Example 3: l-(l-butyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V-cyclopentylpiperidine-4- carboxamide

Example 4: 7V-cyclopentyl-l-(l-heptyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide

N-cyclopentyl-l-(l-heptyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide was prepared fromΛ/-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (17.0 mg, 0.05 mmol), 1-bromo heptane (17.9 mg, 0.10 mmol), caesium carbonate (48.9 mg, 0.15 mmol) and acetonitrile (2 mL), using the method described in Example 2 to give 12.2 mg (56 % yield) of the title compound. LC-MS (m/z) 439.4 (M+l).
Example 5: 7V-cyclopentyl-l-(5,6-dimethyl-l-octyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide


Example 7: 7V-cyclopentyl-l-(l-hexyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide

Example 8: 7V-cyclopentyl-l-(5,6-dimethyl-l-(pyridin-2-ylmethyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

Example 9: 7V-cyclopentyl-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

Λ/-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (356 mg, 1.046 mmol), caesium carbonate (1022 mg, 3.137 mmol), acetonitrile (30 mL), N,N- dimethylformamide (3 mL) and 2-bromopropane (386 mg, 3.14 mmol) were mixed and heated at 6O0C over night. The reaction mixture was filtered, concentrated in vacuo and subjected to column chromatography (silica gel, methanol/dichloromethane, gradient elution using 1-5 % methanol) to give 314 mg (78 %) of N-cyclopentyl-l-(l-isopropyl-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 383.2 (M+ 1).
Example 10: 7V-cyclopentyl-l-(l-(2-(diethylamino)ethyl)-5,6-dichloro-lH-benzo[d]imidazol- 2-yl)piperidine-4-carboxamide

(a) Preparation of the intermediary compound 5,6-dichloro-lH-benzo[d]imidazol-2(3H)-one:

To a stirred solution of 4,5-dichlorobenzene-l,2-diamine (1.0 g, 5.6 mmol) in tetrahydrofuran (10 rnL) was added N,N-carbonyl diimidazole (CDI, 1.36g, 8.4 mmol). The mixture was stirred at room temperature for 8 hours under Argon. The reaction mixture was concentrated in vacuo, the residue partitioned between diethyl ether (60 mL) and aqueous sodium hydroxide (60 mL, 1.0 N). The aqueous extract was acidified to reach a pH of approximately 5 by careful addition of aqueous hydrochloric acid (1.5 N). The resulting off- white solid was filtered off, washed with water (3 χ20 mL) and dried under high vacuum to give 0.68 mg (60 % yield) 5,6-dichloro-lH- benzo[d]imidazol-2(3H)-one. (b) Preparation of the intermediary compound 2,5,6-trichloro-l -benzo[d]imidazole:

To 5,6-dichloro-lH-benzo[d]imidazol-2(3H)-one (1.0 g, 4.5 mmol) was added phosphoryl trichloride (4 mL) and the reaction mixture was heated in a screw-cap pressure tube at 12O0C for 2 hours. The reaction mixture was concentrated in vacuo, the residue was diluted with water (30 mL) and the resulting off-white solid was filtered off. The precipitate was washed with water (3x20 mL) and dried under high vacuum to afford 0.77 g (71 % yield) of 2,5,6-trichloro-lH- benzo[d]imidazole.
(c) Preparation of the intermediary compound N-cyclopentyl-l-(5,6-dichloro-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide:

To a suspension of 2,5,6-trichloro-lH-benzo[d]imidazole (Ig, 4.5mmol) in 1,4-dioxane (5 mL) was added iV,jV-diisopropylethylamine (Ηunig's base, DIEA, 0.094 mL, 5.4 mmol) followed by N-cyclopentylpiperidine-4-carboxamide. The mixture was heated in a screw cap pressure tube at 18O0C for 2 hours. The reaction mixture was concentrated in vacuo, the residue washed with water and filtered off to afford 1.1 g (65 % yield) of N-cyclopentyl-l-(5,6-dichloro-lH-
benzo[d]imidazol-2-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 381.3 (M+l).
(d) To a suspension of sodium hydride (0.021 g, 0.52 mmol) in N,Λ/-dimethylformamide (0.5 rnL) was added N-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (0.10 g, 0.26 mmol) in N,Λ/-dimethylformamide (0.5 mL) drop-wise at O0C under Nitrogen. The reaction mixture was stirred at room temperature for 30 min and 2-bromo-N,N- diethylethylamine (0.101 g, 0.39 mmol) was added drop-wise. The reaction mixture was stirred at room temperature for 12 hours. The reaction mixture was quenched with ice-water and extracted with ethyl acetate (2 x 15 mL). The combined organic phases were washed with water (3 x 15 mL), dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by column chromatography (silica gel) to obtain 12 mg (9.6 % yield) of N-cyclopentyl- l-(l-(2-(diethylamino)ethyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 480.3 (M+l).
Example 11: 7V-cyclopentyl-l-(5,6-dichloro-l-(pyridin-4-ylmethyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

To a suspension of sodium hydride (0.021 g, 0.52 mmol) in N,Λ/-dimethylformamide (0.5 mL) was added Λ/-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (0.10 g, 0.26mmol) in Λ/,iV-dimethylformamide (0.5 mL) drop-wise at O0C under nitrogen gas. The reaction mixture was stirred at room temperature for 30 minutes and 4-
(bromomethyl)pyridine hydrochloride (110 mg, 0.46 mmol) was added. The reaction mixture was stirred at room temperature for 12 hours, quenched with ice and extracted with ethyl acetate (2 x15 mL). The combined organic phases were washed with water (3χ 15 mL), dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was subjected to preparative reverse-phase hplc to afford 81 mg (74 %) of Λ/-cyclopentyl-l-(5,6-dichloro-l-(pyridin-4- ylmethyl)-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 472.1 (M+l).
Example 12: 7V-cyclopentyl-l-(5,6-dichloro-l-(pyridin-3-ylmethyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

N-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-3-ylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide was prepared from sodium hydride (72 mg, 0.30 mmol) in N,Λ/-dimethylformamide (2 mL), Λ/-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (80 mg, 0.20 mmol) in N,Λ/-dimethylformamide (2 mL) and 3 -bromo methyl pyridinium
hydrobromide (0.063g, 0.25mmol), using the method described in Example 11 to give 5 mg (5 % yield) of the title compound. LC-MS (m/z) 472.3 (M+l).
Example 13: 7V-cyclopentyl-l-(5,6-dichloro-l-(pyridin-2-ylmethyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

N-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-2-ylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide was prepared from sodium hydride (72 mg, 0.30 mmol) in N,Λ/-dimethylformamide (2 mL), Λ/-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (80 mg, 0.20 mmol) in N,Λ/-dimethylformamide (2 mL) and 2-bromomethyl pyridinium
hydrobromide (0.063g, 0.25mmol), using the method described in Example 11 to give 5 mg (5 % yield) of the title compound. LC-MS (m/z) 472.2 (M+l).
Example 14: l-(l-sec-butyl-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide


To a solution of Λ/-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (0.10 g, 0.26 mmol) in N,Λ/-dimethylformamide (0.5 mL) caesium carbonate (0.33g, 1 mmol) and [l,l'-bis(diphenylphosphino)ferrocene] dichloropalladium(II) (PdCl2(dppf), 1.0 mg, 0.0026 mmol) were added, followed by 3-bromopentane (59 mg, 0.39 mmol). The reaction mixture was heated at 12O0C for 12 hours, concentrated in vacuo and the residue extracted with ethyl acetate (2 x15 mL). The combined organic phases were washed with water (3 x 15 mL), dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was subjected to column chromatography (silica gel) to give 10 mg (17%) of N-cyclopentyl-l-(5,6- dichloro- 1 -(pentan-3-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 451.3 (M+l).
Example 16: 7V-cyclopentyl-l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

Example 17: 7V-cyclopentyl-l-(5,6-dichloro-l-(l-phenylethyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

To a suspension of sodium hydride (0.01 Ig, 0.46 mmol) in N,Λ/-dimethylformamide (0.5 mL) was added Λ/-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (100 mg, 0.26 mmol) in N,Λ/-dimethylformamide (0.5 mL) drop-wise at O0C under nitrogen gas. The reaction mixture was stirred at room temperature for 30 min, 2-bromoethylbenzene (57 mg, 0.31 mmol) was added in portions and stirred at room temperature for 16 hours. The reaction mixture was quenched with ice water and extracted with ethyl acetate (2xl5mL). The collected organic phases were washed with water (3χ 15mL), dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by column chromatography (silica gel) to obtain 8 mg (6 % yield) of N-cyclopentyl-l-(5,6-dichloro-l-(l-phenylethyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide. LC-MS (m/z) 485.3 (M+l).
Example 18 : 7V-cyclopentyl- l-(5,6-dichloro- 1-methyl- lH-benzo [d] imidazol-2-yl)piperidine- 4-carboxamide

Example 19: 7V-cyclopentyl-l-(5,6-dichloro-l-ethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide

N-cyclopentyl- 1 -(5,6-dichloro- 1 -ethyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide was prepared from Λ/-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (10 mg, 0.026 mmol), caesium carbonate (25.6 mg, 0.079 mmol), acetonitrile (2 mL), Λ/,Λ/-dimethylformamide (0.5 mL), iodoethane (6.1 mg, 0.039 mmol) using the method described in Example 18 to give 5.4 mg (50 % yield) of the title compound. LC-MS (m/z) 409.2 (M+l).
Example 20: 7V-cyclopentyl-l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

N-cyclopentyl- 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide was prepared from Λ/-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (10 mg, 0.026 mmol), caesium carbonate (25.6 mg, 0.079 mmol), acetonitrile (2 mL), Λ/,Λ/-dimethylformamide (0.5 mL), 2-bromopropane (9.7 mg, 0.079 mmol) using the method described in Example 18 to give 5.0 mg (45 % yield) of the title compound. LC-MS (m/z) 423.2 (M+l).
Example 21: 7V-cyclopentyl-l-(5,6-dichloro-l-(4-fluorobenzyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

N-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-fluorobenzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide was prepared from Λ/-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (10 mg, 0.026 mmol), caesium carbonate (25.6 mg, 0.079 mmol), acetonitrile (2 mL), N,Λ/-dimethylformamide (0.5 mL), l-(bromomethyl)-4-fluorobenzene (4.96 mg, 0.026 mmol) using the method described in Example 18 to give 4.2 mg (33 % yield) of the title compound. LC-MS (m/z) 489.2 (M+ 1). Example 22: 7V-cyclopentyl-l-(l-(3,5-difluorobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

Λ/-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (20 mg, 0.059 mmol), caesium carbonate (57.4 mg, 0.176 mmol), N,Λ/-dimethylformamide (4 mL), 1- (bromomethyl)-3,5-difluorobenzene (15.2 mg, 0.073 mmol) was stirred at room temperature for 3-4 hours. The reaction mixture was filtered, the filtrate concentrated in vacuo and subjected to column chromatography (Silica gel, methanol/dichloromethane, gradient elution 1-15 % methanol) to give 14.2 mg (52 % yield) of N-cyclopentyl-l-(l-(3,5-difluorobenzyl)-5,6- dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 467.3 (M+l). Example 23: methyl 4-((2-(4-(cyclopentylcarbamoyl)piperidin-l-yl)-5,6-dimethyl-lH- benzo[d]imidazol-l-yl)methyl)benzoate

Methyl 4-((2-(4-(cyclopentylcarbamoyl)piperidin- 1 -yl)-5,6-dimethyl- lH-benzo[d]imidazol- 1 - yl)methyl)benzoate was prepared from Λ/-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (20 mg, 0.059 mmol), caesium carbonate (57.4 mg, 0.176 mmol), Λ/,Λ/-dimethylfbrmamide (4 mL), methyl 4-(bromomethyl)benzoate (16.8 mg, 0.073 mmol) using the method described in Example 22 to give 20.4 mg (71 % yield) of the title compound. LC-MS (m/z) 489.3 (M+l).
Example 24: 7V-cyclopentyl-l-(5,6-dimethyl-l-((2-methylthiazol-4-yl)methyl)- IH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

Λ/-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (20 mg, 0.059 mmol), caesium carbonate (76.6 mg, 0.235 mmol), N,Λ/-dimethylformamide (2 mL) and 4- (chloromethyl)-2-methylthiazole hydrochloride (16.2 mg, 0.088 mmol) were mixed in a microwave vial and irridated in a microwave oven at 6O0C for 1 hour and then at 1000C over night. The reaction mixture was filtered, concentrated in vacuo and subjected to preparative hplc (performed on a Gilson-Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using acetonitrile/water (0.05% ΗCOOΗ) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 3.1 mg (12 % yield) of N-cyclopentyl- 1 -(5,6-dimethyl- 1 -((2-methylthiazol-4-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide. LC-MS (m/z) 452.3 (M+l).
Example 25: 7V-cyclopentyl-l-(5,6-dichloro-l-phenyl-lH-benzo[d]imidazol-2-yl)piperidine- 4-carboxamide

To a solution of Λ/-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (0.1 g, 0.26 mmol) in dimethylsulfoxide (1 niL) was added caesium carbonate (119 mg, 0.36 mmol), followed by bromobenzene (0.033 mL, 0.31 mmol), 1,10-phenanthroline (9 mg, 0.39 mmol), polyethylene glycol (294 mg) and copper(I) oxide (1.8 mg, 0.013 mmol). The reaction mixture was refluxed at 1100C for 24 hours, quenched with water and extracted with ethyl acetate (2 x15 mL). The combined organic phases were washed with water (3 x15 mL), dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was subjected to column chromatography (silica gel) to give 5 mg (4 % yield) of N-cyclopentyl-l-(5,6-dichloro-l- phenyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 457.3 (M+ 1).
Example 26: 7V-cyclopropyl-l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

(a) Preparation of the intermediary compound ethyl l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylate

To a suspension of 2,5,6-trichloro-lH-benzo[d]imidazole (2.8 g, 12.6 mmol) in 1,4-dioxane (15 mL) was added Λ/,Λ/-diisopropylethylamine (Ηunig's base, DIEA, 6.5 mL, 38 mmol) followed by ethyl piperidine-4-carboxylate (2.38 g, 15 mmol), and the reaction mixture was heated in a screw cap pressure tube at 18O0C for 2 hours. The reaction mixture was concentrated in vacuo, the residue formed washed with water and filtered off to give 2.5 g (58 % yield) of ethyl l-(5,6- dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate as an off-white solid.
(b) Preparation of the intermediary compound l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylic acid

Ethyl l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (2.5 g), aqueous sodium hydroxide (10 mL, IN) and ethanol (20 mL) was stirred for 5 hours at room temperature. The reaction mixture was concentrated in vacuo, the residue partitioned between water and ethyl acetate and the aqueous layer acidified with aqueous hydrochloric acid (1.5 N) to neutral pΗ. The precipitate was filtered off and dried in air to give 2.3 g (99 % yield) of l-(5,6-dichloro-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid as a brown solid. (c) Preparation of the intermediary compound N-cyclopropyl- 1 -(5 ,6-dichloro- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide:

To a solution of l-(5,6-dichloro-lH-benzoimidazol-2-yl)-piperidine-4-carboxylic acid (0.05 g, 0.16 mmol) in N,Λ/-dimethylformamide (2 mL) was added 2-(7-Aza-lH-benzotriazole-l-yl)- 1,1,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 91 mg, 0.24 mmol) followed by N,N- diisopropylethylamine (Ηunig's base, DIEA, 0.04 mL, 0.32 mmol) and cyclopropylamine (0.13 mL, 0.19 mmol). The reaction mixture was stirred at room temperature for 5 hours, concentrated in vacuo and extracted with ethyl acetate (2 x20 mL). The combined organic phases were dried over anhydrous sodium sulphate and finally recrystallized from dichloromethane/n-hexane to give 10 mg (18 % yield) of N-cyclopropyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine- 4-carboxamide. LC-MS (m/z) 353.3 (M+l).
(d) N-cyclopropyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (0.09 g, 0.25 mmol) was dissolved in N,Λ/-dimethylformamide (1 mL). Caesium carbonate (332 mg, 1 mmol) and isopropyl bromide (56 mg, 0.45 mmol) were added, and the reaction mixture irradiated under microwave conditions at 1200C for 60 minutes. The reaction mixture was quenched with water (2 mL) and extracted with ethyl acetate (2 x25 mL). The combined organic phases were washed with water (5 x25 mL), once with brine and dried over anhydrous sodium sulfate. The organic phase was concentrated in vacuo and the residue subjected to column
chromatography (silica gel, chloroform/methanol 99:1), to give 25 mg (12 % yield) of N- cyclopropyl- 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a light yellow solid. LC-MS (m/z) 395.3 (M+l).
Example 27: 7V-cyclobutyl-l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

(a) Preparation of the intermediary compound N-cyclo butyl- 1 -(5,6-dichloro- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide:
 N-cyclobutyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide was prepared from 1 -(5,6-dichloro- lH-benzoimidazol-2-yl)-piperidine-4-carboxylic acid (0.05 g, 0.16 mmol), Λ/,Λ/-dimethylformamide (2 mL), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 91 mg, 0.24 mmol), Λ/,Λ/-diisopropylethylamine (Ηunig's base, DIEA, 0.04 mL, 0.32 mmol) and cyclobutylamine (0.16 mL, 0.19 mmol), using the method described in Example 26(c) to give 43 mg of N-cyclobutyl-1 -(5,6-dichloro- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 367.3 (M+l).
N-cyclobutyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide was prepared from 1 -(5,6-dichloro- lH-benzoimidazol-2-yl)-piperidine-4-carboxylic acid (0.05 g, 0.16 mmol), Λ/,Λ/-dimethylformamide (2 mL), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 91 mg, 0.24 mmol), Λ/,Λ/-diisopropylethylamine (Ηunig's base, DIEA, 0.04 mL, 0.32 mmol) and cyclobutylamine (0.16 mL, 0.19 mmol), using the method described in Example 26(c) to give 43 mg of N-cyclobutyl-1 -(5,6-dichloro- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 367.3 (M+l).
(b) N-cyclo butyl- 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide was prepared from N-cyclobutyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.28g, 0.76mmol), N,Λ/-dimethylformamide (3 mL), caesium carbonate (0.99 g, 3 mmol) and isopropyl bromide (168 mg, 1.3 mmol) using the method described in Example 26(d) to give 61 mg (20 % yield) of the title compound as a light yellow solid. LC-MS (m/z) 409.5 (M+l).
Example 28: l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-7V-propylpiperidine-4- carboxamide

(a) Preparation of the intermediary compound l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- propylpiperidine-4-carboxamide :

(b) 1 -(5 ,6-Dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)-N-propylpiperidine-4-carboxamide was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-propylpiperidine-4- carboxamide (0.05 g, 0.14 mmol), N,Λ/-dimethylformamide (1 mL), caesium carbonate (0.19 g, 0.58 mmol) and isopropyl bromide (26 mg, 0.21 mmol), using the method described in Example 26(d) to give 10 mg (17 % yield) of the title compound as a light yellow solid. LC-MS (m/z) 397.2 (M+l).
Example 29: l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-7V-(2- methoxyethyl)piperidine-4-carboxamide

(a) Preparation of the intermediary compound l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-(2- methoxyethyl)piperidine-4-carboxamide:

To a solution of l-(5,6-dichloro-lH-benzoimidazol-2-yl)-piperidine-4-carboxylic acid (0.07 g, 0.22 mmol) in N,N-dimethylformamide (2 niL) was added 2-(7-Aza-lH-benzotriazole-l-yl)- 1,1,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 0.127 g, 0.33 mmol) followed by N5N- diisopropylethylamine (Ηunig's base, DIEA, 0.052 mL, 0.44 mmol) and 2-methoxy-ethylamine (0.0198g, 0.24 mmol). The reaction mixture was stirred at room temperature for 5 hours, concentrated in vacuo and extracted with ethyl acetate (2 x50 mL). The combined organic phases were dried over anhydrous sodium sulphate and recrystallized from ethyl acetate/n-hexane to give 20 mg (24 % yield) of l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-(2- methoxyethyl)piperidine-4-carboxamide . LC-MS (m/z) 371.2 (M+ 1 ) .
(b) l-(5,6-Dichloro-lH-benzo[d]imidazol-2-yl)-N-(2-methoxyethyl)piperidine-4-carboxamide (0.1 g, 0.27 mmol) was dissolved in N,N-dimethylformamide (4 mL). Caesium carbonate (0.527 mg, 1.6 mmol) and isopropyl bromide (66 mg, 0.54 mmol) were added, and the reaction mixture irradiated under microwave conditions at 1200C for 60 minutes. The reaction mixture was quenched with water (2 mL) and extracted with ethyl acetate (2 x25 mL). The combined organic phases were washed with water (5 x25 mL), once with brine and dried over anhydrous sodium sulfate. The organic phase was concentrated in vacuo and the residue recrystallized from ethyl acetate and n-hexane to give 20 mg (18 % yield) of 1 -(5, 6-dichloro-l -isopropyl- IH- benzo[d]imidazol-2-yl)-Ν-(2-methoxyethyl)piperidine-4-carboxamide. LC-MS (m/z) 413.3 (M+l).
Example 30: l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-JV- (isopropoxymethyl)piperidine-4-carboxamide

(a) Preparation of the intermediary compound l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-(2- isopropoxyethyl)piperidine-4-carboxamide:
 l-(5,6-Dichloro-lH-benzo[d]imidazol-2-yl)-N-(2-isopropoxyethyl)piperidine-4-carboxamide was prepared from l-(5,6-dichloro-lH-benzoimidazol-2-yl)-piperidine-4-carboxylic acid (0.3 g, 0.95 mmol) in N,N-dimethylformamide (5 mL), 2-(7-aza-lH-benzotriazole-l-yl)-l, 1,3,3- tetramethyluronium hexafluorophosphate (ΗATU, 0.544 g, 1.4 mmol), N5N- diisopropylethylamine (Ηunig's base, DIEA, 0.33 mL, 1.9 mmol) and 2-isopropoxyethanamine (0.118 g, 1.1 mmol), using the method described for Example 29(a) to give 0.35 g (92% yield) of l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-(2-isopropoxyethyl)piperidine-4-carboxamide. LC-MS (m/z) 399.2 (M+l). (b) 1 -(5 ,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)-N-(2-isopropoxyethyl)piperidine-4- carboxamide was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-(2- isopropoxyethyl)piperidine-4-carboxamide (0.1 g, 0.25 mmol), N,N-dimethylformamide (1 mL), caesium carbonate (0.5 g, 1.5 mmol) and isopropyl bromide (55 mg, 0.45 mmol) using the method described in Example 29(b) to give 60 mg (18 % yield) of the title compound as a light yellow solid. LC-MS (m/z) 441.3 (M+l). 1H ΝMR (300 MHz, DMSO-d6) δ 7.84 (s, IH), 7.66 (s, IH), 4.54 (m, IH), 3.53 (m, IH), 3.47-3.28 (m, 4H), 3.18 (dd, 2H), 2.89 (dd, 2H), 2.36 (m, IH), 1.79 (m, 4H), 1.51 (d, 6H), 1.09 (d, 6H).
l-(5,6-Dichloro-lH-benzo[d]imidazol-2-yl)-N-(2-isopropoxyethyl)piperidine-4-carboxamide was prepared from l-(5,6-dichloro-lH-benzoimidazol-2-yl)-piperidine-4-carboxylic acid (0.3 g, 0.95 mmol) in N,N-dimethylformamide (5 mL), 2-(7-aza-lH-benzotriazole-l-yl)-l, 1,3,3- tetramethyluronium hexafluorophosphate (ΗATU, 0.544 g, 1.4 mmol), N5N- diisopropylethylamine (Ηunig's base, DIEA, 0.33 mL, 1.9 mmol) and 2-isopropoxyethanamine (0.118 g, 1.1 mmol), using the method described for Example 29(a) to give 0.35 g (92% yield) of l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-(2-isopropoxyethyl)piperidine-4-carboxamide. LC-MS (m/z) 399.2 (M+l). (b) 1 -(5 ,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)-N-(2-isopropoxyethyl)piperidine-4- carboxamide was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-(2- isopropoxyethyl)piperidine-4-carboxamide (0.1 g, 0.25 mmol), N,N-dimethylformamide (1 mL), caesium carbonate (0.5 g, 1.5 mmol) and isopropyl bromide (55 mg, 0.45 mmol) using the method described in Example 29(b) to give 60 mg (18 % yield) of the title compound as a light yellow solid. LC-MS (m/z) 441.3 (M+l). 1H ΝMR (300 MHz, DMSO-d6) δ 7.84 (s, IH), 7.66 (s, IH), 4.54 (m, IH), 3.53 (m, IH), 3.47-3.28 (m, 4H), 3.18 (dd, 2H), 2.89 (dd, 2H), 2.36 (m, IH), 1.79 (m, 4H), 1.51 (d, 6H), 1.09 (d, 6H).
Example 31: l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-iV-(tetrahydrofuran-3- yl)piperidine-4-carboxamide

(a) Preparation of the intermediary compound l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide
 To a solution of l-(5,6-dichloro-lH-benzoimidazol-2-yl)-piperidine-4-carboxylic acid (0.3 g, 0.95 mmol) in N,Λ/-dimethylformamide (5 niL) was added 2-(7-aza-lH-benzotriazole-l-yl)- 1,1,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 0.544 g, 1.4 mmol) followed by N,N- diisopropylethylamine (Ηunig's base, DIEA, 0.33 mL, 1.9 mmol) and tetrahydrofuran-3 -amine (0.1 g, 1.1 mmol). The reaction mixture was stirred at room temperature for 5 hours,
To a solution of l-(5,6-dichloro-lH-benzoimidazol-2-yl)-piperidine-4-carboxylic acid (0.3 g, 0.95 mmol) in N,Λ/-dimethylformamide (5 niL) was added 2-(7-aza-lH-benzotriazole-l-yl)- 1,1,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 0.544 g, 1.4 mmol) followed by N,N- diisopropylethylamine (Ηunig's base, DIEA, 0.33 mL, 1.9 mmol) and tetrahydrofuran-3 -amine (0.1 g, 1.1 mmol). The reaction mixture was stirred at room temperature for 5 hours,
concentrated in vacuo and extracted with ethyl acetate (2 x50 mL). The combined organic phases were dried over anhydrous sodium sulphate and subjected to column chromatography
(chloroform/methanol 95:5) to give 0.135 g (37 % yield) of l-(5,6-dichloro-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 383.2 (M+l).
(b) l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide (0.1 g, 0.25 mmol) was dissolved in N,Λ/-dimethylformamide (1 mL). Caesium carbonate (0.5 g, 1.5 mmol) and isopropyl bromide (60 mg, 0.46 mmol) were added, and the reaction mixture irradiated under microwave conditions at 1200C for 60 minutes. The reaction mixture was quenched with water (2 mL) and extracted with ethyl acetate (2 x25 mL). The combined organic phases were washed with water (5 x25 mL), once with brine and dried over anhydrous sodium sulfate. The organic phase was concentrated in vacuo to give 75 mg (68 % yield) of 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide. LC-MS (m/z) 425.3 (M+l).
Example 32 : TV-(cyclohexylmethyl)- l-(5,6-dichloro- 1-isopropyl- lH-benzo [d] imidazol-2- yl)piperidine-4-carboxamide

(a) Preparation of the intermediary compound N-(cyclohexylmethyl)- 1 -(5 ,6-dichloro- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide:

(a) To a solution of l-(5,6-dichloro-lH-benzoimidazol-2-yl)-piperidine-4-carboxylic acid (0.07 g, 0.22 mmol) in N,Λ/-dimethylformamide (2 niL) was added 2-(7-aza-lH-benzotriazole-l-yl)- 1,1,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 0.127 g, 0.33 mmol) followed by N,N- diisopropylethylamine (Ηunig's base, DIEA, 0.05 mL, 0.44 mmol) and cyclohexylmethanamine (0.049 g, 0.44 mmol). The reaction mixture was stirred at room temperature for 5 hours, quenced with water to give a precipitate that was filtered off, washed with water and dried in air, to give 0.1 g (76 %) yield of N-(cyclohexylmethyl)-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 409.3 (M+l). (b) N-(cyclohexylmethyl)- 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide was prepared from N-(cyclohexylmethyl)-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.15 g, 0.36 mmol), N,Λ/-dimethylformamide (3 mL), caesium carbonate (0.716 g, 2.2 mmol) and isopropyl bromide (0.09 g, 0.72 mmol) using the method described in Example 29(b) to give 20 mg (12 % yield) of the title compound. LC-MS (m/z) 451.4 (M+l).
Example 33: l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-7V-((tetrahydro-2H- pyran-4-yl)methyl)piperidine-4-carboxamide

(a) Preparation of the intermediary compound l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- ((tetrahydro-2H-pyran-4-yl)methyl)piperidine-4-carboxamide.

(b) 1 -(5 ,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)-N-((tetrahydro-2H-pyran-4- yl)methyl)piperidine-4-carboxamide was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)-N-((tetrahydro-2H-pyran-4-yl)methyl)piperidine-4-carboxamide (0.09 g, 0.19 mmol), N,N- dimethylformamide (1 mL), caesium carbonate (0.38 g, 1.1 mmol) and isopropyl bromide (43 mg, 0.35 mmol) using the method described in Example 26(d) to give 20 mg (7 % yield) of the title compound. LC-MS (m/z) 453.3 (M+l).
Example 34: 7V-butyl-l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide

(b) l-(5,6-Dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (0.05 g, 0.16 mmol) was treated with 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 0.96 g, 0.25 mmol) in N,Λ/-dimethylformamide (Im L) followed by Λ/,Λ/-diisopropylethylamine (Ηunig's base, DIEA, 0.05 mL, 0.32 mmol), and butan-1-amine (0.18 mL, 0.19 mmol). The reaction mixture was stirred at room temperature for 4 hours, quenched with with water (1 mL), extracted with ethyl acetate (2 x20 mL), washed once with brine (1OmL) and dried over anhydrous sodium sulfate. The organic phase was filtered, the filtrate concentrated in vacuo and subjected to column chromatography (silica gel,
chloroform/methanol 98:2) to give 20 mg (29 % yield) of iV-butyl-l-(5,6-dichloro-l-isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 411.4 (M+l).
Example 35 : 7V-(cy clopentylm ethyl)- l-(5,6-dichloro- 1-isopropyl- lH-benzo [d] imidazol-2- yl)piperidine-4-carboxamide

cyclopentylmethanamine hydrochloride (0.069 mL, 0.5 mmol). The reaction mixture was stirred at room temperature for 12 hours, quenched with water (1 mL), extracted with ethyl acetate (2 x20 mL), washed with brine (10 mL) and dried over anhydrous sodium sulfate. The organic phase was concentrated in vacuo, recrystallized with petroleum ether to give to give 0.02 g (11 % yield) of N-(cyclopentylmethyl)-l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 437.3 (M+l).
Example 36: 7V-cyclopentyl-l-(l-((6-fluoropyridin-3-yl)methyl)-5,6-dimethyl-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

Example 37: l-(l-(4-cyanobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.29 mmol), N,Λ/-dimethylformamide (2 mL), caesium carbonate (0.566 g, 1.7 mmol) and 4-(bromomethyl)benzonitrile (0.103 g, 0.52 mmol), using the method described in Example 36 to give 0.08 g (62 % yield) of l-(l-(4-cyanobenzyl)-5,6- dimethyl-lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as a white solid. LC-MS (m/z) 456.2 (M+l). Example 38: 7V-cyclopentyl-l-(l-(cyclopropylmethyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

To a solution of N-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (0.08 g, 0.23 mmol) in N,Λ/-dimethylformamide (3 mL) cesium carbonate (0.459 g, 1.4 mmol) was added, followed by (bromomethyl)cyclopropane (0.037 g, 0.276 mmol). The reaction mixture was irradiated under microwave conditions at 12O0C for 45 minutes. The reaction mixture was concentrated in vacuo and the residue was extracted with ethyl acetate (2 x 15 mL). The combined organic phases were washed with water (3><15 mL), dried over anhydrous sodium sulfate and concentrated. The residue was recrystallized to give 0.05 g (54 %)
ofN-cyclopentyl-l-(l-(cyclopropylmethyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine- 4-carboxamide as an orange solid. LC-MS (m/z) 395.3 (M+l).
Example 39: 7V-cyclopentyl-l-(5,6-dimethyl-l-((tetrahydrofuran-2-yl)methyl)-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.29 mmol), Λ/,Λ/-dimethylformamide (2 mL), caesium carbonate (0.566 g, 1.7 mmol) and 2-(bromomethyl)tetrahydrofuran (0.08 g, 0.52 mmol), using the method described in Example 36, to give 11 mg (9 % yield) of N-cyclopentyl-l-(5,6- dimethyl- 1 -((tetrahydrofuran-2-yl)methyl)- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 425.2 (M+l).
Example 40: l-(l-(4-(lH-l,2,4-triazol-l-yl)benzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)- 7V-cyclopentylpiperidine-4-carboxamide

Example 41: l-(l-cyclobutyl-5,6-dimethyl-l//-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.29 mmol), N,Λ/-dimethylformamide (2 mL), caesium carbonate (0.566 g, 1.7 mmol) and cyclobutylbromide (0.07 g, 0.527 mmol), using the method described in Example 36, to give 9 mg (8 % yield) of l-(l-cyclobutyl-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 395.2 (M+l).
Example 42: l-(l-(4-bromobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.9 g, 2.6 mmol), N,Λ/-dimethylformamide (5 mL), caesium carbonate (5 g, 15.6 mmol) and l-bromo-4-(bromomethyl)benzene (1.18 g, 4.7 mmol), using the method described in Example 36, to give 0.5 g (38 % yield) of l-(l-(4-bromobenzyl)-5,6- dimethyl- lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 509.6 (M+l).
Example 43: 7V-cyclopentyl-l-(5,6-dimethyl-l-(4-(methylcarbamoyl)benzyl)-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

A reaction mixture consisting of l-(l-(4-bromobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)-N-cyclopentylpiperidine-4-carboxamide (0.1 g, 0.19 mmol), £ra/?s-di(μ-acetato)-bis[o-(di-o- tolylphosphino)-benzyl]dipalladium(II) (Hermann palladacycle, 0.089 g, 0.095 mmol), N5N- diisopropylethylamine (Hunig's base, DIEA, 0.07 mL, 0.39 mmol), 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU, 0.19 mmol), tri tertiarybutyl phosphonium
hexafluoborate ([(t-Bu)sPH]BF4, 5.5 mg, 0.019 mmol), molybdenum hexacarbonyl (Mo(CO)6, 0.025 g, 0.095 mmol) and methyl amine hydrochloride (0.1 mg, 1.52 mmol) in 1,4-dioxane (3 mL), was irradiated under microwave conditions at 1500C for 30 minutes. The reaction mixture was quenched with water (5 mL), extracted with dichloro methane (2 x50 mL) and the combined organic phases dried over anhydrous sodium sulphate. After concentration in vacuo, the residue was subjected to column chromatography (basic aluminia, chloroform/methanol 99:1) to give 45 mg (50 % yield) of N-cyclopentyl-l-(5,6-dimethyl-l-(4-(methylcarbamoyl)benzyl)-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 488.1 (M+l). Example 44: 7V-cyclopentyl-l-(l-(4-(dimethylcarbamoyl)benzyl)-5,6-dimethyl-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

The title compound was prepared from l-(l-(4-bromobenzyl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide (0.1 g, 0.19 mmol), trans-di(μ- acetato)-bis[o-(di-o-tolylphosphino)-benzyl]dipalladium(II) (Hermann palladacycle, 0.089 g, 0.095 mmol), N,N-diisopropylethylamine (Hunig's base, 0.07 mL, 0.39 mmol), 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU, 0.028 mL, 0.19 mmol), tri tertiarybutyl phosphonium hexafluoborate ([(t-Bu)3PH]BF4, 0.0055 g, 0.019 mmol), molybdenum hexacarbonyl (Mo(CO)6,
0.025 g, 0.095 mmol), dimethyl amine hydrochloride (0.1 g, 1.23 mmol) and 1,4-dioxane (3mL), using the method described in Example 43, to give 5 mg (5.5 % yield) of N-cyclopentyl-l-(l-(4- (dimethylcarbamoyl)benzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 502.1 (M+ 1).
Example 45: l-(l-(4-carbamoylbenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide

To a solution of l-(l-(4-bromobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide (0.1 g, 0.29 mmol) in N,Λ/-dimethylformamide (0.5 mL) was added caesium carbonate (0.566 g, 1.7 mmol), followed by 4-(bromomethyl)benzamide (0.112 g, 0.52 mmol, which was prepared from 4-(bromomethyl)benzoic acid by reaction with thionyl chloride (SOCl2) and quenching the formed acid chloride with aqueous ammonia (28 %)). The resulting mixture was irradiated under microwave conditions at 1200C for 45 min. The reaction mixture was concentrated in vacuo, the residue extracted with ethyl acetate (2 x15 mL), the combined organic phases washed with water (3χ15mL) and dried over anhydrous sodium sulphate. This gave 3.6 g (2.6 % yield) of l-(l-(4-carbamoylbenzyl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as a white solid. LC-MS (m/z) 474.1 (M+l). Example 46: 7V-cyclopentyl-l-(5,6-dimethyl-l-(4-(methylsulfonylcarbamoyl)benzyl)-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide
 l-(l-(4-bromobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4- carboxamide (O.lg, 0.19mmol), £rans-di(μ-acetato)-bis[o-(di-o-tolylphosphino)- benzyl]dipalladium(II) (Hermann palladacycle, 0.089 g, 0.095 mmol), N5N- diisopropylethylamine (Hunig's base, DIEA, 0.07 niL, 0.39 mmol), 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU, 0.028 mL, 0.19 mmol) tri tertiarybutyl phosphonium hexafluoborate ([(t-Bu)3PH]BF4, 5.5 mg, 0.019 mmol), molybdenum hexacarbonyl (Mo(CO)6, 0.025 g, 0.095 mmol), methane sulfonamide (0.037 g, 0.39 mmol) and 1,4-dioxane (3 mL) was irradiated under microwave conditions at 1500C for 30 minutes. The reaction mixture was quenched with water (5 mL) and extracted with dichloro methane (2 x50 mL), the combined organic phases dried over anhydrous sodium sulfate and subjected to column chromatography (basic alumina, chloroform/methanol 99:1) to give 36 mg (33 % yield) of N-cyclopentyl-l-(5,6- dimethyl- 1 -(4-(methylsulfonylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide as a white solid. LC-MS (m/z) 552.0 (M+ 1).
l-(l-(4-bromobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4- carboxamide (O.lg, 0.19mmol), £rans-di(μ-acetato)-bis[o-(di-o-tolylphosphino)- benzyl]dipalladium(II) (Hermann palladacycle, 0.089 g, 0.095 mmol), N5N- diisopropylethylamine (Hunig's base, DIEA, 0.07 niL, 0.39 mmol), 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU, 0.028 mL, 0.19 mmol) tri tertiarybutyl phosphonium hexafluoborate ([(t-Bu)3PH]BF4, 5.5 mg, 0.019 mmol), molybdenum hexacarbonyl (Mo(CO)6, 0.025 g, 0.095 mmol), methane sulfonamide (0.037 g, 0.39 mmol) and 1,4-dioxane (3 mL) was irradiated under microwave conditions at 1500C for 30 minutes. The reaction mixture was quenched with water (5 mL) and extracted with dichloro methane (2 x50 mL), the combined organic phases dried over anhydrous sodium sulfate and subjected to column chromatography (basic alumina, chloroform/methanol 99:1) to give 36 mg (33 % yield) of N-cyclopentyl-l-(5,6- dimethyl- 1 -(4-(methylsulfonylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide as a white solid. LC-MS (m/z) 552.0 (M+ 1).
Example 47: l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-iV-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide

(a) Preparation of the intermediary compound tert-butyi 4-(tetrahydrofuran-3- ylcarbamoyl)piperidine- 1 -carboxylate.

To a solution of l-(tert-butoxycarbonyl)piperidine-4-carboxylic acid (1.0 g, 4.37 mmol) in dry dichloromethane (10 rnL), cooled to 0 0C, was added JV,JV-diisopropylethylamine (Hunig's base, DIEA, 8.7 mmol), Λ/-(3-Dimethylaminopropyl)-Λf'-ethylcarbodiimide hydrochloride (EDCΗC1, CAS Registry Number: 25952-53-8, 8.7 mmol), 1-hydroxybenzotriazole (HOBt, CAS Registry Number: 2592-95-2, 2.2 mmol) and tetrahydrofuran-3 -amine hydrochloride (5.2 mmol). The reaction mixture was allowed to warm to room temperature and was stirred for 10 hours. The reaction mixture was washed with aqueous hydrochloric acid (3 x15 mL, 2N), saturated aqueous sodium bicarbonate (2 x15 mL), dried over magnesium sulfate, filtered and the filtrate concentrated in vacuo. The residue was recrystallized from dichloro methane/pet ether to give 1.15 g (88%) of tert-butyl 4-(tetrahydrofuran-3-ylcarbamoyl)piperidine-l-carboxylate.
(b) Preparation of the intermediary compound 4-(tetrahydrofuran-3-ylcarbamoyl)piperidinium 2,2,2-trifluoroacetate:

(c) Preparation of the intermediary compound l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-JV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide.
 To a suspension of 2-chloro-5,6-dimethyl-lH-benzo[d]imidazole (0.3 g, 1.6 mmol) in 1,4- dioxane (5 mL), 4-(tetrahydrofuran-3-ylcarbamoyl)piperidinium 2,2,2-trifluoroacetate (0.38 g, 1.9 mmol) in 1,4-dioxane (3 mL) was added. Λ/,Λ/-diisopropylethylamine (Ηunig's base, DIEA 0.89 mL, 5.0 mmol) was added and the reaction mixture was heated in a sealed tube at 18O0C for 3 hours. The reaction mixture was quenched with water, extracted with ethyl acetate, dried over anhydrous sodium sulphate and subjected to column chromatography (silica gel,
To a suspension of 2-chloro-5,6-dimethyl-lH-benzo[d]imidazole (0.3 g, 1.6 mmol) in 1,4- dioxane (5 mL), 4-(tetrahydrofuran-3-ylcarbamoyl)piperidinium 2,2,2-trifluoroacetate (0.38 g, 1.9 mmol) in 1,4-dioxane (3 mL) was added. Λ/,Λ/-diisopropylethylamine (Ηunig's base, DIEA 0.89 mL, 5.0 mmol) was added and the reaction mixture was heated in a sealed tube at 18O0C for 3 hours. The reaction mixture was quenched with water, extracted with ethyl acetate, dried over anhydrous sodium sulphate and subjected to column chromatography (silica gel,
chloroform/methanol 95:5), to give 0.12 g (18 % yield) of l-(5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid.
(d) To a solution of l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide (0.05 g, 0.14 mmol) in N,Λ/-dimethylformamide (2 mL) was added caesium carbonate (0.285 g, 0.87 mmol), followed by cyclobutylbromide (0.02 mL, 0.21 mmol).
The reaction mixture was irradiated under microwave conditions at 12O0C for 1 hour, concentrated in vacuo and extracted with ethyl acetate (2><15mL). The combined organic phases were washed with water (3 ><15 mL), dried over anhydrous sodium sulfate and subjected to column chromatography (silica gel, chloroform/methanol 97.5:2.5) to give 6 mg (10 % yield) of l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a brown solid. LC-MS (m/z) 397.2 (M+ 1).
Example 48: l-(l-(4-fluorobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

To a solution of l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide (0.05 g, 0.14 mmol) in N,Λ/-dimethylformamide (2 mL) was added caesium carbonate (0.285 g, 0.87 mmol), followed by l-(bromomethyl)-4-fluorobenzene (0.032 mL, 0.17 mmol). The reaction mixture was irradiated under microwave conditions at 12O0C for 1 hour, concentrated in vacuo and extracted with ethyl acetate (2><15mL). The combined organic phases were washed with water (3 ><15 mL), dried over anhydrous sodium sulfate and recrystallized with dichloromethane and n-hexane to give 0.01 g (15 % yield) of l-(l-(4- fluorobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a yellow solid. LC-MS (m/z) 451.1 (M+l).
Example 49: l-(5-chloro-l-cyclobutyl-6-methyl-lH-benzo[d]imidazol-2-yl)-7V-cyclopentyl- piperidine-4-carboxamide and l-(6-chloro-l-cyclobutyl-5-methyl-lH-benzo[d]imidazol-2- yl)-7V-cyclopentylpiperidine-4-carboxamide


To a stirred solution of 4-chloro-5-methylbenzene-l,2-diamine (0.3 g, 1.9 mmol) in
tetrahydrofuran (5 mL) was added JV,iV-carbonyl dimidazole (CDI, 0.465 g, 2.8 mmol). The reaction mixture was stirred at room temperature for 3 hours under Argon gas, concentrated in vacuo and the residue partitioned between ethyl acetate (50 mL) and aqueous sodium hydroxide (50 mL, IN). The aqueous extract was acidified to pH 5 via addition of aqueous hydrochloric acid (1.57V) and the resulting brown precipitate was filtered, washed with water (3χ20mL) and dried under high vacuum to obtain 0.28 g (80 % yield) of 5-chloro-6-methyl-lH- benzo[d]imidazol-2(3H)-one.
(b) Preparation of the intermediate compound 2,5-dichloro-6-methyl-lH-benzo[d]imidazole:

To 5-chloro-6-methyl-lH-benzo[d]imidazol-2(3H)-one (0.25 g), phosphoryl trichloride (POCI3, 4 mL) was added, the reaction mixture was heated in a screw cap pressure tube at 12O0C for 2 hours and concentrated in vacuo. The residue was neutralized with sodium hydroxide, the resulting off-pale yellow solid was filtered, washed with water (3χ50 mL) and dried under high vacuum to give 0.27 g (98 % yield) of 2,5-dichloro-6-methyl-lH-benzo[d]imidazole.
(c) Preparation of the intermediary compound tert-butyl 4-(cyclopentylcarbamoyl)piperidine-l- carboxylate.
M

To a mixture consisting of l-(tert-butoxycarbonyl)piperidine-4-carboxylic acid (6.4 g, 27.9 mmol), dry dichloromethane (66 mL), triethylamine (4.24 g, 41.9 mmol) was carefully added thionyl chloride (3.99 g, 33.5 mmol) in 16 mL dry dichloromethane. The reaction mixture was stirred for 20 minutes at room temperature, cyclopentanamine (5.94 g, 69.8 mmol) was added and the reaction mixture was heated at reflux over night under nitrogen gas. The reaction mixture was washed with aqueous hydrochloric acid (3 x60 mL, 2N), saturated aqueous sodium bicarbonate (2 x60 mL), dried over magnesium sulphate, filtered and the filtrate concentrated in vacuo. The residue was dried under vacuum to give 5.859 g of tert-butyl 4- (cyclopentylcarbamoyl)piperidine-l-carboxylate. LC-MS (m/z) 297.3 (M+l).
(d) Preparation of the intermediary compound 4-(cyclopentylcarbamoyl)piperidine:

(e) Preparation of the intermediate compound l-(6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)- JV-cyclopentylpiperidine-4-carboxamide:
 To a suspension of 2,5-dichloro-6-methyl-lH-benzo[d]imidazole (0.1 g, 0.49 mmol) in 1,4- dioxane (4 niL), 4-(cyclopentylcarbamoyl)piperidine (0.185 g) in 1,4-dioxane (2 niL) was added N,Λ/-diisopropylethylamine (Ηunig's base, DIEA, 0.25 mL, 1.4 mmol) and heated at 18O0C for 3 hours. The reaction mixture was concentrated in vacuo, partitioned between ethyl acetate (15 mL) and water (15 mL). The aqueous phase was extracted with ethyl acetate, the combined organic phases dried over anhydrous sodium sulphate and subjected to chromatography (silica gel, chloroform/methanol 98:2) to give 46 mg (26 % yield) of l-(6-chloro-5-methyl-lH- benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4-carboxamide. LC-MS (m/z) 361.2 (M+l).
To a suspension of 2,5-dichloro-6-methyl-lH-benzo[d]imidazole (0.1 g, 0.49 mmol) in 1,4- dioxane (4 niL), 4-(cyclopentylcarbamoyl)piperidine (0.185 g) in 1,4-dioxane (2 niL) was added N,Λ/-diisopropylethylamine (Ηunig's base, DIEA, 0.25 mL, 1.4 mmol) and heated at 18O0C for 3 hours. The reaction mixture was concentrated in vacuo, partitioned between ethyl acetate (15 mL) and water (15 mL). The aqueous phase was extracted with ethyl acetate, the combined organic phases dried over anhydrous sodium sulphate and subjected to chromatography (silica gel, chloroform/methanol 98:2) to give 46 mg (26 % yield) of l-(6-chloro-5-methyl-lH- benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4-carboxamide. LC-MS (m/z) 361.2 (M+l).
(f) To a solution of l-(6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine- 4-carboxamide (0.1 g, 0.27 mmol) in N,Λ/-dimethylformamide (2 mL), caesium carbonate (0.541 g, 1.6 mmol) was added, followed by cyclobutylbromide (0.032 mL, 0.33 mmol) and [1,1'- bis(diphenylphosphino)ferrocene] dichloropalladium(II) (PdCl2(dppf) in catalytic amounts. The reaction mixture was irradiated under microwave conditions at 12O0C for 1 hour, concentrated in vacuo and the residue extracted with ethyl acetate (2x15mL). The combined organic phases were washed with water (3χ 15mL), dried over anhydrous sodium sulphate, concentrated in vacuo and subjected to column chromatography (silica gel, chloroform/methanol 98:2) to give 13 mg (11 %) of a 1 :1 mixture of l-(5-chloro-l -cyclobutyl-6-methyl- lH-benzo[d]imidazol-2-yl)-JV- cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro- 1 -cyclobutyl-5 -methyl- IH- benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4-carboxamide in a mixture as a yellow solid. LC-MS (m/z) 415.3 (M+l).
Example 50 : 7V-cyclopentyl- l-(5,6-dichloro- 1-cyclopentyl- lH-benzo [d] imidazol-2- yl)piperidine-4-carboxamide

To a solution of Λ/-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (100 mg, 0.26 mmol) in N,Λ/-dimethylformamide (0.5 mL) was added caesium carbonate (500 mg, 1.53 mmol), followed by cyclopentylbromide (70 mg, 0.46 mmol). The reaction mixture was irradiated under microwave conditions at 1200C for 60 minutes, concentrated in vacuo and the residue extracted with ethyl acetate (2x15 mL). The combined organic phases were washed with water (3><15 mL), dried over anhydrous sodium sulfate
concentrated in vacuo. The residue was subjected to column chromathography (silica gel) to give 75 mg (64 % yield) Λ/-cyclopentyl-l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 449.3 (M+ 1).
Example 51: l-(l-(4-bromobenzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide

Example 52: 7V-cyclopentyl-l-(5,6-dichloro-l-((6-fluoropyridin-3-yl)methyl)-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

To a solution of l-(5,6-dichloro-lH-benzoimidazole-2-yl)-piperdine-4-carboxylic acid cyclopentylamide (60 mg, 0.16 mmol) in N,Λ/-dimethylformamide (0.5 mL) was added caesium carbonate (300 mg, 0.94 mmol), followed by 2-fluoro-5-bromomethylpyridine (36 mg, 0.19 mmol). The reaction mixture was irradiated under microwave conditions at 1200C for 45 minutes, concentrated in vacuo and the residue extracted with ethyl acetate (2 x15 mL). The combined organic phases were washed with water (3 x15 mL), dried over anhydrous sodium sulphate and concentrated in vacuo. The residue was subjected to column chromatography (silica
gel) to give 20 mg (26 %) N-cyclopentyl-l-(5,6-dichloro-l-((6-fluoropyridin-3-yl)methyl)-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 490.3 (M+l).
Example 53: 7V-cyclopentyl-l-(5,6-dichloro-l-(4-cyanobenzyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

N-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-cyanobenzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide was prepared from Λ/-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (50 mg, 0.13 mmol), N,Λ/-dimethylformamide (0.5 mL), caesium carbonate (171 mg, 0.52 mmol) and 4-(bromomethyl)benzonitrile (31 mg, 0.15 mmol), using the method described in Example 52 to give 20 mg (15 % yield) of the title compound. LC-MS (m/z) 496.2 (M+l).
Example 54: 7V-cyclopentyl-l-(5,6-dichloro-l-(cyclopropylmethyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

Example 55: 7V-cyclopentyl-l-(5,6-dichloro-l-((tetrahydrofuran-2-yl)methyl)-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

To a solution of Λ/-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (100 mg, 0.26mmol) in N,N-dimethylformamide (0.5 niL) was added cesium carbonate (512 mg, 1.5 mmol), followed by 2-(bromomethyl)tetrahydrofuran (77 mg, 0.47 mmol). The reaction mixture was irradiated under microwave conditions at 12O0C for 45 min, concentrated in vacuo and the residue extracted with ethyl acetate (2 x15 mL). The combined organic phases were washed with water (3 x15 mL), dried over anhydrous sodium sulphate and concentrated in vacuo. The residue was washed with n-hexane to afford 50 mg (41 % yield) of N-cyclopentyl- 1 -(5,6-dichloro- 1 -((tetrahydrofuran-2-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide. LC-MS (m/z) 465.2 (M+ 1).
Example 56: 7V-cyclopentyl-l-(5,6-dichloro-l-((tetrahydro-2H-pyran-4-yl)methyl)-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

N-cyclopentyl- 1 -(5,6-dichloro- 1 -((tetrahydro-2H-pyran-4-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide was prepared from N-cyclopentyl- 1 -(5 ,6-dichloro- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide (100 mg, 0.26 mmol), N,N-dimethylformamide (0.5 mL), caesium carbonate (512 mg, 1.5 mmol), and 4-(bromomethyl)tetrahydro-2H-pyran (80 mg, 0.47 mmol), using the method described in Example 55 to give 70 mg (57 % yield) of the title compound. LC-MS (m/z) 479.3 (M+l). Example 57: 7V-cyclopentyl-l-(5,6-dichloro-l-(4-(methylcarbamoyl)benzyl)-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

A reaction mixture consisting of l-(l-(4-bromobenzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)- N-cyclopentylpiperidine-4-carboxamide (Example 51, 100 mg, 0.182 mmol), trans-di(μ- acetato)-bis[o-(di-o-tolylphosphino)-benzyl]dipalladium(II) (Hermann palladacycle, 9 mg, 0.009 mmol), Λ/,Λ/-diisopropylethylamine (Hunig's base, DIEA, 0.06 mL, 0.36 mmol), 1,8- diazabicyclo[5.4.0]undec-7-ene (DBU, 0.028 mL, 1.82 mmol), tri tertiarybutyl phosphonium hexafluoborate ([(t-Bu)sPH]BF4, 5 mg, 0.018 mmol), molybdenum hexacarbonyl (Mo(CO)6, 0.024 g, 0.09 mmol) and methyl amine (25 mg, 0.36 mmol) in 1,4-dioxane, was irradiated under microwave conditions at 15O0C for 30 minutes. The reaction mixture was quenched with water (5 mL), extracted with dichloro methane (2 x50 mL) and the combined organic phases dried over anhydrous sodium sulphate. After concentration in vacuo, the residue was subjected to column chromatography (basic aluminia, chloroform/methanol 96:4) to give 12 mg (12 % yield) of N- cyclopentyl- 1 -(5 ,6-dichloro- 1 -(4-(methylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 528.4 (M+l). Example 58: 7V-cyclopentyl-l-(5,6-dichloro-l-(4-(dimethylcarbamoyl)benzyl)-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide


To a solution of l-(5,6-dichloro-lH-benzoimidazole-2-yl)-piperdine-4-carboxylic acid cyclopentylamide (50 mg, 0.13 mmol) in N,Λ/-dimethylformamide (0.5 mL) was added caesium carbonate (260 mg, 0.28 mmol), followed by 4-(bromomethyl)benzamide (34 mg, 0.15 mmol, that was prepared from 4-(bromomethyl)benzoic acid by reacting with thionyl chloride (SOCl2) and quenching the resulting acid chloride with aqueous ammonia (28 %)). The reaction mixture was irradiated under microwave conditions at 12O0C for 45 minutes and concentrated in vacuo. The residue was extracted with ethyl acetate (2 x15 mL), the collected organic phases washed with water (3 x15 mL), dried over anhydrous sodium sulphate and concentrated in vacuo to afford 20 mg (30 % yield) of l-(l-(4-carbamoylbenzyl)-5,6-dichloro-lH-benzo[d]imidazol-2- yl)-N-cyclopentylpiperidine-4-carboxamide as a white solid. LC-MS (m/z) 514.6 (M+l).
Example 60: 7V-cyclopentyl-l-(l-cyclopentyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

Λ/-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (20 mg, 0.059 mmol), caesium carbonate (76.6 mg, 0.235 mmol), N,Λ/-dimethylformamide (4 mL) and bromocyclopentane (4.96 mg, 0.026 mmol) were mixed in a microwave vial and irridated in a
microwave oven at 600C over night. The reaction mixture was filtered, concentrated in vacuo and subjected to preparative hplc (performed on a Gilson-Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using acetonitrile/water (0.05% HCOOH) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 3.6 mg (15 %) of the title compound. LC-MS (m/z) 409.3 (M+ 1).
Example 61: l-(l-(4-chlorobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-iV- cyclopentylpiperidine-4-carboxamide


N-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-cyanobenzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (Example 53, 50 mg, 0.1 mmol), sodium azide (78 mg, 1.2 mmol), ammonium chloride (64 mg, 1.2 mmol), lithium chloride (catalytic amount) was refluxed in N5N- dimethylformamide (3 mL) for 2 hours. The reaction mixture was quenched with water and extracted with ethyl acetate (2 x25 mL), the organic phase was dried over anhydrous sodium
sulphate, filtered and concentrated in vacuo, to give 15 mg (28 % yield) of l-(l-(4-(lH-tetrazol- 5-yl)benzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as a white solid. LC-MS (m/z) 539.3 (M+l).
Example 63: 7V-cyclopentyl-l-(5,6-dichloro-l-(4-(methylsulfonylcarbamoyl)benzyl)-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

(l-(l-(4-Bromobenzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4- carboxamide) (Example 51, 0.1 g, 0.182 mmol), £rαns-di(μ-acetato)-bis[o-(di-o-tolylphosphino)- benzyl]dipalladium(II) (Hermann palladacycle, 9 mg, 0.009 mmol), Λ/,Λ/-diisopropylethylamine (Hύnig's base, DIEA, 0.06 mL, 0.36 mmol), l,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 0.028 mL, 1.82 mmol), tri tertiarybutyl phosphonium hexafluoborate [(t-Bu)sPH]BF4 (5 mg, 0.018 mmol), molybdenum hexacarbonyl (Mo(CO)6, 24 mg, 0.09 mmol), methane sulfonamide (34 mg, 0.36 mmol) and 1,4-dioxane was irradiated under microwave conditions at 15O0C for 30 minutes. The reaction mixture was quenched with water (5 mL), extracted with dichloromethane (2 x50 mL), the combined organic phases dried over anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by column chromatography (basic aluminia,
chloroform/methanol 96:4) to give 80 mg (74 % yield) of N-cyclopentyl-l-(5,6-dichloro-l-(4- (methylsulfonylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 592.4 (M+l). Example 64: 7V-butyl-l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide


To a suspension of 2,5,6-trichloro-lH-benzo[d]imidazole (0.3 g, 1.35 mmol) in 1,4-dioxane (6 mL) was added Λ/,Λ/-diisopropylethylamine (Ηunig's base, DIEA, 0.67 mL, 4.0 mmol) followed by N-butylpiperidine-4-carboxamide (0.3 g, 1.6 mmol). The reaction mixture was heated in a sealed tube at 18O0C for 3 hours, concentrated in vacuo, the residue washed with water and filtered off to give 0.3 g (60 %) of N-butyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 369.5 (M+l).
(b) To a solution of Λ/-butyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (0.3 g, 0.81 mmol) in N,Λ/-dimethylformamide (2 mL) was added caesium carbonate (0.79 g, 2.4 mmol), followed by cyclobutylbromide (0.164 mL, 1.2 mmol). The reaction mixture was irradiated under microwave conditions at 12O0C for 45 minutes, concentrated in vacuo and the residue extracted with ethyl acetate (2 x15 mL). The combined organic phases were washed with water (3 x15 mL), dried over anhydrous sodium sulphate and concentrated in vacuo. The residue was subjected to column chromatography (silica gel, chloroform/methanol 98:2) to give 0.09 g (26 % yield) of iV-butyl-l-(5,6-dichloro-l-cyclobutyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 423.3 (M+l).
Example 65: 7V-butyl-l-(5,6-dichloro-l-pentyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide


The title compound was prepared fromN-butyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.27 mmol), N,Λ/-dimethylformamide (2 mL), caesium carbonate (0.527 g, 1.6 mmol) and l-(bromomethyl)-4-fluorobenzene (0.164 mL, 1.2 mmol) using the method described in Example 64(b), to give 0.02 g (16 % yield) of iV-butyl-l-(5,6- dichloro-l-(4-fluorobenzyl)-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a off-white solid. LC-MS (m/z) 411.2 (M+l).
Example 67: 7V-butyl-l-(5,6-dichloro-l-((6-fluoropyridin-3-yl)methyl)- IH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

The title compound was prepared fromN-butyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.27 mmol), N,Λ/-dimethylformamide (2 mL), caesium
carbonate (0.527 g, 1.6 mmol) and 5-(bromomethyl)-2-fluoropyridine (0.092 g, 0.48 mmol) using the method described in Example 64(b), to give 2 mg (1.5 % yield) of N-butyl-l-(5,6- dichloro- 1 -((6-fluoropyridin-3-yl)methyl)- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a off-white solid. LC-MS (m/z) 478.3 (M+ 1).
Example 68: l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3- yl)piperidine-4-carboxamide

(a) Preparation of the intermediary compound l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide:

To a suspension of 2,5,6-trichloro-lH-benzo[d]imidazole (0.3 g, 1.36 mmol) in 1,4-dioxane (6 mL) was added Λ/,Λ/-diisopropylethylamine (Ηunig's base, DIEA, 0.36 mL, 2.0 mmol) followed by Λ/-(tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.636 g, 2.0 mmol) and the reaction mixture was heated in a sealed tube at 18O0C for 3 hours. The reaction mixture was concentrated in vacuo, the residue washed with water and filtered off, to give 0.14 g (26 % yield) of l-(5,6- dichloro- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 383.1 (M+l).
(b) The title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-JV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.05 g, 0.13 mmol), N,Λ/-dimethylformamide (2 mL), caesium carbonate (0.254 g, 0.78 mmol) and cyclobutylbromide(0.025 mL, 0.26 mmol) using the method described in Example 64(b), to give 0.011 g (19 % yield) of l-(5,6-dichloro-l- cyclo butyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 437.4 (M+l).
Example 69: l-(5,6-dichloro-l-pentyl-lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3- yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.1 g, 0.26 mmol), N,Λ/-dimethylformamide (2 mL), caesium carbonate (0.508 g, 1.56 mmol) and 1-bromopentane (0.033 mL, 0.31mmol) using the method described in Example 64(b), to give 17 mg (14 % yield) of l-(5,6-dichloro-l-pentyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 453.3 (M+ 1). Example 70: l-(5,6-dichloro-l-(4-cyanobenzyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.1 g, 0.26 mmol), N,Λ/-dimethylformamide (2 mL), caesium carbonate (0.508 g, 1.56 mmol) and 4-(bromomethyl)benzonitrile (0.062 g, 0.31 mmol) using the method described in Example 64(b), to give 13 mg (10 % yield) of l-(5,6- dichloro- 1 -(4-cyanobenzyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a white solid. LC-MS (m/z) 498.1 (M+l).
Example 71: l-(5,6-dichloro-l-(cyclopropylmethyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-JV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.1 g, 0.26 mmol), N,Λ/-dimethylformamide (2 mL), caesium carbonate (0.508 g, 1.56 mmol) and (bromomethyl)cyclopropane (0.031 mL, 0.31 mmol) using the method described in Example 64(b), to give 14 mg (11 % yield) of l-(5,6- dichloro- 1 -(cyclopropylmethyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine- 4-carboxamide as an off-white solid. LC-MS (m/z) 437.2 (M+ 1).
Example 72: l-(5,6-dichloro-l-((6-fluoropyridin-3-yl)methyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

To a solution of N-butyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (0.1 g, 0.26 mmol) in N,Λ/-dimethylformamide (2 mL) was added caesium carbonate (0.508 g, 1.56 mmol), followed by 5-(bromomethyl)-2-fluoropyridine (0.059 mL, 0.31 mmol). The reaction mixture was irradiated under microwave conditions at 12O0C for 45 minutes, concentrated in vacuo and the residue extracted with ethyl acetate (2 x15 mL). The combined organic phases were washed with water (3 x15 mL), dried over anhydrous sodium sulphate and concentrated in vacuo. The residue was subjected to column chromatography (silica gel, chloroform/methanol 97:3) to give 25 mg (19 % yield) of l-(5,6-dichloro-l-((6-fluoropyridin-3- yl)methyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 492.5 (M+l).
Example 73: 7V-cyclopentyl-l-(5,6-dimethyl-l-((tetrahydro-2H-pyran-4-yl)methyl)-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

To a solution of N-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (0.1 g, 0.29 mmol) in N,Λ/-dimethylformamide (2 niL) was added caesium carbonate (0.566 g, 1.7 mmol), followed by 4-(bromomethyl)tetrahydro-2H-pyran (0.08 g, 0.52 mmol). The reaction mixture was irradiated under microwave conditions at 12O0C for 1 hour, concentrated in vacuo and the residue extracted with ethyl acetate (2 x15 mL). The combined organic phases were washed with water (3 x15 mL), dried over anhydrous sodium sulphate and concentrated in vacuo. The residue was subjected to column chromatography (silica gel, chloroform/methanol 99:1) to give 0.043 g (33.5 % yield) ofN-cyclopentyl-l-(5,6-dimethyl-l- ((tetrahydro-2H-pyran-4-yl)methyl)-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 439.2 (M+l).
Example 74: l-(5,6-dichloro-l-cyclopentyl-lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide


The title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.1 g, 0.26 mmol), N,Λ/-dimethylformamide (2 mL), caesium carbonate (0.508 g, 1.56 mmol) and l-bromo-4-(bromomethyl)benzene (0.078 g, 0.31 mmol) using the method described in Example 74, to give 22 mg (15 % yield) of l-(l-(4- bromobenzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a white solid. LC-MS (m/z) 552.5 (M+l). 1U NMR (300.1 MHz, DMSO-d6) δ 8.04 (s, IH), 7.67 (s, IH), 7.54 (d, 2H), 7.53 (s, IH), 5.29 (s, 2H), 4.21 (m, IH), 3.70 (m, 2H), 3.53 (m, 2H), 2.91 (m, 2H), 2.05 (m, IH), 1.70 (m).
Example 76: 7V-cyclopentyl-l-(5,6-dimethyl-l-phenyl-lH-benzo[d]imidazol-2-yl)piperidine- 4-carboxamide

I l l
Example 77: l-(l-(4-(lH-tetrazol-5-yl)benzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide

Example 78: l-(5,6-dichloro-l-(4-fluorobenzyl)-lH-benzo[d]imidazol-2-yl)-JV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

(tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.1 g, 0.26 mmol), N,N-dimethylformamide (2 mL), caesium carbonate (0.508 g, 1.56 mmol) and l-(bromomethyl)-4-fluorobenzene (0.058 mL, 0.31 mmol) using the method described in Example 74, to give 27 mg (21 % yield) of l-(5,6- dichloro- 1 -(4-fluorobenzyl)- lH-benzo[d]imidazol-2-yl)-Ν-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a brown solid.
Example 79: l-(l-(4-(lH-l,2,4-triazol-l-yl)benzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)- JV-cyclopentylpiperidine-4-carboxamide

To a solution of N-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (0.1 g, 0.26mmol) in N,Λ/-dimethylformamide (0.5 niL) was added cesium carbonate (0.512 g, 1.5 mmol), followed by l-(4-(bromomethyl)phenyl)-lH-l,2,4-triazole (0.112 g, 0.47 mmol). The reaction mixture was irradiated under microwave conditions at 12O0C for 45 minutes, concentrated in vacuo and the residue extracted with ethyl acetate (2 x15 mL). The organic layer was washed with water (3 x15 mL), dried over anhydrous sodium sulphate, concentrated in vacuo and subjected to column chromatography (silica gel, chloroform/methanol 99:1), to give 0.04 g (28 % yield) of l-(l-(4-(lH-l,2,4-triazol-l-yl)benzyl)-5,6-dichloro-lH- benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4-carboxamide as a white solid. LC-MS (m/z) 538.3 (M+l).
Example 80: 7V-cyclopentyl-l-(5,6-dichloro-l-(2-methoxyethyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide


The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.30 g, 0.829 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.316 mmol), l-bromo-4-iodobenzene (3.316 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.132 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.044 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 20 mg (5.2 % yield) of N-cyclopentyl- 1 -( 1 -(4-fluorophenyl)-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide as an off- white solid. LC-MS (m/z) 435.1 (M+ 1).
Example 82: l-(l-(cyclobutylmethyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide

Example 83: 7V-cyclopentyl-l-(5,6-dimethyl-l-(prop-2-ynyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.293 mmol), N,Λ/-dimethylformamide (3 mL), caesium carbonate (1.172 mmol), 3-bromoprop-l-yne (1.172 mmol) and dichlorobis(triphenyl- phosphine)palladium (II) (Pd(PPlIs)2Cl2 (0.0293 mmol) using the method described in Example 14. The reaction mixture was heated at 12O0C for 1 hour under microwave conditions and purified with column chromathography, to give 15 mg (13 % yield) of N-cyclopentyl-l-(5,6- dimethyl-l-(prop-2-ynyl)-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 379.0 (M+ 1). Example 84: 7V-cyclopentyl-l-(l-(2-ethylbutyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.293 mmol), N,Λ/-dimethylformamide (3 mL), caesium carbonate (1.172 mmol), 3-(bromomethyl)pentane (1.172 mmol) and dichlorobis(triphenyl- phosphine)palladium (II) (Pd(PPh3)2Cl2 (0.0293 mmol) using the method described in Example 14. The reaction mixture was heated at 12O0C for 1 hour under microwave conditions and purified with column chromathography, to give 26 mg (22 % yield) of N-cyclopentyl-l-(l-(2- ethylbutyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 425.1 (M+l).
Example 85: 7V-cyclopentyl-l-(5,6-dimethyl-l-neopentyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.293 mmol), N,Λ/-dimethylformamide (3 mL), caesium carbonate (1.172 mmol), l-bromo-2,2-dimethylpropane (1.172 mmol) and dichlorobis(triphenyl- phosphine)palladium (II) (Pd(PPh3)2Cl2 (0.0293 mmol) using the method described in Example 14. The reaction mixture was heated at 12O0C for 1 hour under microwave conditions and purified with column chromathography, to give 55 mg (23 % yield) of N-cyclopentyl-l-(5,6- dimethyl-l-neopentyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as an off- white solid. LC-MS (m/z) 411.0 (M+ 1). Example 86: l-(l-cyclohexyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.293 mmol), N,Λ/-dimethylformamide (3 mL), caesium carbonate ( 1.172 mmo 1), bromocyclohexane ( 1.172 mmo 1) and dichlorobis(triphenyl- phosphine)palladium (II) (Pd(PPh3)2Cl2 (0.0293 mmol) using the method described in Example 14. The reaction mixture was heated at 12O0C for 1 hour under microwave conditions and purified with column chromathography, to give 8 mg (3.2 % yield) of l-(l-cyclohexyl-5,6- dimethyl-lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as an off- white solid. LC-MS (m/z) 423.1 (M+l).
Example 87: l-(l-sec-butyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V-cyclopentyl- piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.293 mmol), N,Λ/-dimethylformamide (3 mL), caesium carbonate (1.172 mmol), 2-bromobutane (1.172 mmol) and dichlorobis(triphenylphosphine)- palladium (II) (Pd(PPlIs)2Cl2 (0.0293 mmol) using the method described in Example 14. The reaction mixture was heated at 12O0C for 1 hour under microwave conditions and purified with column chromathography, to give 20 mg (8.6 % yield) of l-(l-sec-butyl-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-cyclopentyl-piperidine-4-carboxamide as a white solid. LC-MS (m/z) 397.0 (M+l). Example 88: 7V-cyclopentyl-l-(5,6-dichloro-l-(2-hydroxyethyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

N-cyclopentyl- 1 -(5 ,6-dichloro- 1 -(2-methoxyethyl)- lH-benzo [d]imidazol-2-yl)piperidine-4- carboxamide was dissolved in dichloromethane and stirred under nitrogen at O0C for 15 minutes, treated with tribromoborane via drop-wise addition and gradually warmed to room temperature. The reaction mixture was stirred at room temperature for 1 hour, quenched with aqueous sodium hydrogencarbonate (10%) to ashive neutral pΗ and extracted with dichloromethane (2x 50 mL). The combined organic phases were dried over anhydrous sodium sulfate and concentrated in vacuo to give 0.02 g (42 % yield) of Λ/-cyclopentyl-l-(5,6-dichloro-l-(2-hydroxyethyl)-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 425.3 (M+l).
Example 89: 7V-cyclopentyl-l-(l,5,6-trimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide

Example 90: 7V-cyclopentyl-l-(l-isobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

Λ/-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (6.8 mg, 0.02 mmol), l-bromo-2-methylpropane (3.3 mg, 2.6 mL, 0.024 mmol), caesium carbonate (13 mg, 0.04 mmol) and acetonitrile (1.0 mL) and N,N-dimethylformamide (5 drops) were mixed and stirred at 5O0C. After 75 minutes, additional l-bromo-2-methylpropane (3.3 mg, 2.6 mL, 0.024 mmol) was added and the reaction mixture was stirred at 6O0C over night, filtered and concentrated in vacuo. The residue was and purified on preparative hplc (performed on a Gilson- Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using acetonitrile/water (0.05 % formic acid) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 1.7 mg (22 % yield) of N-cyclopentyl-l-(l- isobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 397.2 (M+ 1).
Example 91: l-(l-benzyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V-cyclopentylpiperidine- 4-carboxamide


Λ/-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (34 mg, 0.10 mmol), l-bromo-2-methoxyethane (16.7 mg, 11 mL, 0.120 mmol), caesium carbonate (65.2 mg, 0.20 mmol) and acetonitrile (2 mL) were mixed and stirred at 1000C over night, filtered, concentrated in vacuo and subjected to column chromathography (silica gel, methanol/ dichloromethane gradient elution 3-20 % methanol) to give 4.7 mg (12 %) of N-cyclopentyl-1- (l-(2-methoxyethyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 399.2 (M+l). Example 93: 7V-cyclopentyl-l-(l-(4-fluorobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-JV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (34 mg, 0.10 mmol), acetonitrile (2 mL),
caesium carbonate (65.2 mg, 0.20 mmol) and l-(bromomethyl)-4-fluorobenzene (16.7 mg, 0.12 mmol) using the method described in Example 92, to give 4.7 mg (12 % yield) of iV-cyclopentyl- l-(l-(4-fluorobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC- MS (m/z) 449.2 (M+ 1). Example 94: l-(5-chloro-l-cyclobutyl-6-fluoro-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide and l-(6-chloro-l-cyclobutyl-5-fluoro-lH- benzo [d] imidazol-2-yl)-7V-cyclopentylpiperidine-4-carboxamide

(a) Preparation of the intermediate compound 5-chloro-6-fluoro-lH-benzo[d]imidazol-2(3Η)- one:

To a stirred solution of 4-chloro-5-fluorobenzene-l,2-diamine (1 g, 6.22 mmol) in
tetrahydrofuran (10 mL) was added JV,iV-carbonyl dimidazole (CDI, 9.33 mmol). The reaction mixture was stirred at room temperature for 3 hours under Argon gas, concentrated in vacuo and the residue partitioned between ethyl acetate and aqueous sodium hydroxide. The aqueous extract was acidified to pH 5 via addition of aqueous hydrochloric acid and the resulting brown precipitate was filtered, washed with water and dried to obtain 0.8 g (68 % yield) of 5-chloro-6- fluoro-lH-benzo[d]imidazol-2(3H)-one after recrystallization from petroleum ether.
(b) Preparation of the intermediate compounds 2,5-dichloro-6-fluoro-lH-benzo[d]imidazole and 2,6-dichloro-5-fluoro-lH-benzo[d]imidazole.

To 5-chloro-6-fluoro-lH-benzo[d]imidazol-2(3Η)-one (1 g, 5.3 mmol), phosphoryl trichloride (POCI3, 132 mmol)) was added, the reaction mixture was heated at 12O0C for 2 hours and then
concentrated in vacuo. The residue was neutralized with sodium hydroxide, the resulting was filtered off, washed with water and dried to give 0.85 g (77 % yield) of 2,5-dichloro-6-fluoro- lH-benzo[d]imidazole and 2,6-dichloro-5-fluoro-lH-benzo[d]imidazole after recrystallization after petroleum ether. No effort was made to determine the ratios of the tautomers. (c) Preparation of the intermediary compound 4-(cyclopentylcarbamoyl)piperidinium 2,2,2- trifluoroacetate.

To a solution of tert-EvXy\ 4-(cyclopentylcarbamoyl)piperidine-l-carboxylate (1.0 g, 3.37 mmol) in dichloromethane (10 mL), cooled to 0 0C, was added 2,2,2-trifluoroacetic acid (8.4 mmol). The reaction was allowed to warm to room temperature and stirred for 16 hours. The mixture was concentrated in vacuo (repeated co-evaporation with toluene), and dried under vacuum to give 4-(cyclopentylcarbamoyl)piperidinium 2,2,2-trifluoroacetate in quantitative yield.
(d) Preparation of the intermediary compounds l-(5-chloro-6-fluoro-lH-benzo[d]imidazol-2-yl)- N-cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro-5-fluoro- lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide.

To a suspension of the tautomeric compounds 2,5-dichloro-6-fluoro-lH-benzo[d]imidazole and 2,6-dichloro-5-fluoro-lH-benzo[d]imidazole (1 g, 4.87 mmol), 1,4-dioxane (10 mL) and 4- (cyclopentylcarbamoyl)piperidinium 2,2,2-trifluoroacetate (7.3 mmol) was added NJSf- diisopropylethylamine (Ηunig's base, DIEA, 14.6 mmol) and heated at 18O0C for 4 hours. After the appropriate work-up, the residue was recrystallized from a mixture of dichloromethane, petroleum ether and diethyl ether, to give 0.8 g (45 % yield) of l-(5-chloro-6-fluoro-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro-5-fluoro- IH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide. (e) A tautomeric mixture of l-(5-chloro-6-fluoro-lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro-5-fluoro- lH-benzo[d]imidazol-2-yl)-N-
cyclopentylpiperidine-4-carboxamide (0.1 g, 0.274 mmol), bromocyclo butane (1.096 mmol), caesium carbonate (1.096 mmol), [l,r-bis(diphenyl-phosphino)ferrocene] dichloropalladium(II) (PdCl2(dppf), 0.0274 mmol) and N,Λ/-dimethyl-formamide (3 mL) was subjected to microwave conditions at 12O0C for one hour in a sealed tube and purified with column chromathography, to give 5 mg (4.4 % yield) of l-(5-chloro-l-cyclobutyl-6-fluoro-lH-benzo[d]imidazol-2-yl)-N- cyclopentyl-piperidine-4-carboxamide and 1 -(6-chloro- 1 -cyclobutyl-5-fluoro- IH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as an off-white solid. No effort was made to determine the ratio of the regioisomers. LC-MS (m/z) 419.1 (M+l).
Example 95: 7V-cyclopentyl-l-(5,6-dimethyl-l-(pentan-2-yl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

Λ/-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (20 mg, 0.059 mmol), 2-bromopentane (17.8 mg, 0.117 mmol), caesium carbonate (57.4 mg, 0.176 mmol), Λ/,Λ/-dimethylformamide (3.0 mL) was heated at 6O0C over night. The reaction mixture was filtered, concentrated in vacuo and purified on preparative hplc (performed on a Gilson- Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 μm, 21.2 xl50 mm) column, using acetonitrile/water (0.05 % formic acid) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection). This gave 7.2 mg (30 % yield) of JV-cyclopentyl- 1 -(5 ,6-dimethyl- 1 -(pentan-2-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 411.4 (M+l).
Example 96: 7V-butyl-l-(l-cyclobutyl-5,6-dimethyl-l//-benzo[d]imidazol-2-yl)piperidine-4- carboxamide

(a) Preparation of the intermediary compound tert-butyl 4-(/?-butylcarbamoyl)piperidine-l- carboxylate.

To a solution of l-(tert-butoxycarbonyl)piperidine-4-carboxylic acid (1.0 g, 4.37 mmol) in dry dichloromethane (10 rnL), cooled to 0 0C, was added JV,JV-diisopropylethylamine (Hunig's base, DIEA, 8.7 mmol), Λ/-(3-dimethylaminopropyl)-Λf'-ethylcarbodiimide hydrochloride (EDCΗC1, , 8.7 mmol, CAS Registry Number: 25952-53-8), 1-hydroxybenzotriazole (HOBt, CAS Registry Number: 2592-95-2, 2.2 mmol) and n-butylamine (5.2 mmol). The reaction mixture was allowed to warm to room temperature and was stirred for 8 hours. The reaction mixture was washed with aqueous hydrochloric acid (3 x15 mL, 2 N), saturated aqueous sodium bicarbonate (2 x15 mL), dried over magnesium sulfate, filtered and the filtrate concentrated in vacuo. The residue was recrystallized from dichloro methane/pet ether to give 1.1 g (92%) of tert-butyl A-(n- butylcarbamoyl)piperidine- 1 -carboxylate.
(b) Preparation of the intermediary compound 4-(/?-butylcarbamoyl)piperidinium 2,2,2- trifluoroacetate :


(d) To a solution of ΛMxityl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide (0.1 g, 0.3 mmol) in N,Λ/-dimethylformamide (2 mL), caesium carbonate (0.6 g, 1.8 mmol) was added cyclobutylbromide (0.074 g, 0.54 mmol). The reaction mixture was irradiated under microwave conditions at 12O0C for 1 hour, concentrated in vacuo and the residue extracted with ethyl acetate (2 x15 mL). The combined organic phases were washed with water (3 x15 mL), dried over anhydrous sodium sulphate, filtered and concentrated in vacuo. The residue was subjected to column chromatography (silica gel, chloroform/methanol 99:8) to give 23 mg (20 % yield) ofN-butyl-l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide as a white solid. LC-MS (m/z) 383.3 (M+l).
Example 97: 7V-butyl-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide

To a solution of Λ/-butyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide (0.1 g, 0.3 mmol) in N,7V-dimethylformamide (2 mL), was added caesium carbonate (0.6 g, 1.8 mmol), followed by 2-bromopropane (67 mg, 0.54 mmol). The reaction mixture was irradiated under microwave conditions at 12O0C for 1 hour, concentrated in vacuo, the residue extracted with ethyl acetate (2 x15 mL). The combined organic phases were washed with water (3 x15 mL), dried over anhydrous sodium sulphate, filtered and concentrated in vacuo. The residue was subjected to column chromatography (silica gel, chloroform/methanol 99: 1) to give 49 mg (44 % yield) ofN-butyl-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamid. LC-MS (m/z) 371.3 (M+l).
Example 98: 7V-butyl-l-(5,6-dimethyl-l-pentyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide

The title compound was prepared fromΛMxityl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.3 mmol), N,Λ/-dimethylformamide (2 mL), caesium carbonate (0.6 g, 1.8 mmol) and 1-bromopentane (0.082 g, 0.54 mmol) using the method described in Example 97, to give 25 mg (21 % yield) of ΛMxityl-l-(5,6-dimethyl-l-pentyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 399.4 (M+l).
Example 99: TV-butyl-Ηl-ftό-fluoropyridin-S-yOmethyO-S^-dimethyl- IH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide


The title compound was prepared fromΛMxityl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.3 mmol), N,Λ/-dimethylformamide (2 mL), caesium
carbonate (0.6 g, 1.8 mmol) and l-(bromomethyl)-4-fluorobenzene (0.1 g, 0.54 mmol) using the method described in Example 97, to give 28 mg (28 % yield) of JV-butyl-l-(l-(4-fluorobenzyl)- 5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide. LC-MS (m/z) 437.1 (M+l).
Example 101: 7V-benzyl-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine- 4-carboxamide

(a) Preparation of the intermediate compound ethyl l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylate.

(b) Preparation of the intermediate compound ethyl l-(l-isopropyl-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylate.

Ethyl l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (280 mg, 0.929 mmol) was dissolved in acetonitrile (25 mL) and Λ/,Λ/-dimethylformamide (5 mL). 2-
Bromopropane (342.8 mg, 2.79 mmol) and potassium carbonate (385.2 mg, 2.79 mmol) was added and the reaction mixture was heated at 6O0C over night. 2-Bromopropane (342.8 mg, 2.79
mmol) and potassium carbonate (128.4 mg, 0.929 mmol) was added, the reaction mixture was heated at 6O0C for 7 hours, filtered and the filtrate concentrated in vacuo. The residue was subjected to column chromatography (silica gel, dichloromethane/methanol, gradient elution 2- 10 % methanol) to give 143 mg (45 % yield, 90 % pure) of ethyl l-(l-isopropyl-5,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate. LC-MS (m/z) 344.3 (M+ 1).
(c) Preparation of the intermediate compound l-(l-isopropyl-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid.

A mixture of ethyl l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxylate (143 mg, 0.416 mmol), sodium hydroxide (83.3 mg, 2.08 mmol) and water (1 mL) was heated at reflux for 50 minutes. Aqueous hydrochloric acid (1 mL, 2N) was added and the reaction mixture concentrated in vacuo. LC-MS (m/z) 344.3 (M+l). This gave l-(l-isopropyl- 5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid in a mixture with sodium chloride. LC-MS (m/z) 316.2 (M+l). (d) l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (10 mg, 0.0317 mmol) was dissolved in N,Λ/-dimethylformamide (2 mL). 2-(7-Aza-lH-benzotriazo Ie-I- yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (13.3 mg, 0.0349 mmol), N,N- diisopropylethylamine (8.2 mg, 0.0634 mmol) and phenylmethanamine (4.08 mg, 0.0380 mmol) was added and the reaction mixture was stirred at room temperature for 1 hour, concentrated in vacuo and filtered through a pad of silica gel (dichloromethane/methanol 95:5). The pooled fractions containg the title compound was concentrated in vacuo and subjected to purification on hplc (performed on a Gilson-Finnigan ThermoQuest AQA system equipped with a Zorbax SB- C8 (5 μm, 21.2 x 150 mm) column with UV (214 or 254 nm) and MS (ESI) detection), using acetonitrile/water (containing 0.05 % formic acid) gradient elution 10-50 %), to give 7.3 mg (57 % yield) of 7V-benzyl-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide. LC-MS (m/z) 405.3 (M+l).
Example 102: 7V-(4-fluorobenzyl)-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared from l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylic acid (10 mg, 0.0317 mmol), N,N-dimethylformamide (2 mL), 2-(7- aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (13.3 mg, 0.0349 mmol), N,N-diisopropylethylamine (8.2 mg, 0.0634 mmol) and (4-fluorophenyl)methanamine (4.76 mg, 0.0380 mmol) using the method described in Example 101, to give 8.5 mg (63 % yield) ofN-(4-fluorobenzyl)-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine- 4-carboxamide. LC-MS (m/z) 423.3 (M+l).
Example 103: 7V-(3-isopropoxypropyl)-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

0.0380 mmol) was added and the reaction mixture was stirred at room temperature for 1 hour, and concentrated in vacuo. The residue was subjected to purification on hplc (performed on a Gilson-Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 μm, 21.2 xl50 mm) column with UV (214 or 254 nm) and MS (ESI) detection, using acetonitrile/water (containing 0.05 % formic acid) gradient elution 10-50 %), to give 9.2 mg (70 % yield) of N-(3- isopropoxypropyl)-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide. LC-MS (m/z) 415.3 (M+l).
Example 104: 7V-(3,3-dimethylbutyl)-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared from l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylic acid (10 mg, 0.0317 mmol), N,N-dimethylformamide (1 mL), 2-(7- Aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (13.3 mg, 0.0349 mmol), N,N-diisopropylethylamine (8.2 mg, 0.0634 mmol) and 3,3-dimethylbutan-l -amine (3.85 mg, 0.0380 mmol) using the method described in Example 103, to give 6.1 mg (48 % yield) of Λ/-(3,3-dimethylbutyl)-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide. LC-MS (m/z) 399.4 (M+l).
Example 105: 7V-(4-bromobenzyl)-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

Example 106: 7V-cyclohexyl-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

Example 107: 7V-(4-cyanobenzyl)-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

JV-(4-Bromobenzyl)- 1 -( 1 -isopropyl-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)piperidine-4- carboxamide (20 mg, 0.0414 mmol), dicyanozinc (ZnCN2, 4.9 mg, 0.0414 mmol), bis(tri-tert- butylphosphine)palladium (Pd[P(t-bu)3]2, 2.1 mg, 0.00414 mmol) and N,Λ/-dimethylformamide (1 mL) was irridated in a microwave oven at 15O0C for 30 minutes. The reaction mixture was filtred, concentrated in vacuo, subjected to column chromatography (silica gel), to give 8.8 mg (50 % yield) of Λ/-(4-cyanobenzyl)-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide. LC-MS (m/z) 430.3 (M+l).
Example 108: (S)- l-(5,6-dimethyl- 1-p-tolyl- lH-benzo [d] imidazol-2-yl)-7V-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide

(a) Preparation of the intermediate compound ethyl l-(5,6-dimethyl-l-p-to IyI- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxylate:

(b) Preparation of the intermediate compound l-(5,6-dimethyl-l-p-tolyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylic acid:

To ethyl l-(5,6-dimethyl-l-p-tolyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (40 mg, 0.10 mmol) was added ethanol (2 mL) and aqueous sodium hydroxide (1 mL, 1 N) and the
reaction mixture was stirred at 50 0C for one hour. The reaction mixtures were neutralized with aqueous hydrochloric acid (1 N), concentrated in vacuo to give l-(5,6-dimethyl-l-p-to IyI- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid in quantitative yield. LC-MS (m/z) 364.0 (M+l). (c) l-(5,6-Dimethyl-l-p-tolyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (37 mg, 0.10 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 58 mg, 0.153 mmol), (S)-tetrahydrofuran-3 -amine hydrochloride (18.9 mg, 0.153 mmol) and iV,jV-diisopropyl-ethylamine (Ηunig's base, DIEA, 170 μL, 1.02 mmol) were dissolved in N,N-dimethylformamide (1 mL). The reaction mixture was stirred overnight at ambient temperature, filtered and subjected to preparative hplc (preparative RP-LC was performed on a Gilson system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using methano I/water (0.05% formic acid) gradients at a flow rate of 4 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 33 mg (75 % yield) of (S)-l-(5,6-dimethyl-l-p-tolyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 433.0. (M+l).
Example 109: 4-((2-(4-(cyclopentylcarbamoyl)piperidin-l-yl)-5,6-dimethyl-lH- benzo[d]imidazol-l-yl)methyl)benzoic acid

Methyl 4-((2-(4-(cyclopentylcarbamoyl)piperidin- 1 -yl)-5,6-dimethyl- lH-benzo[d]imidazol- 1 - yl)methyl)benzoate (17 mg, 0.035 mmol) was dissolved in methanol (1 mL). Sodium hydroxide (13.9 mg, 0.348 mmol) and water (0.17 mL) was added and the reaction mixture was stirred at room temperature for 2 hours. Aqueous hydrochloride (0.085 mL, 2N) was added, the reaction mixture concentrated in vacuo and subjected to preparative hplc (performed on a Gilson- Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 μm, 21.2 xl50 mm) column, using acetonitrile/water (0.05 % formic acid) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 8.1 mg (49 % yield) of 4-((2-(4- (cyclopentylcarbamoyl)-piperidin- 1 -yl)-5,6-dimethyl- lH-benzo[d]imidazol- 1 -yl)methyl)benzoic acid. LC-MS (m/z) 475.3 (M+l).
Example 110: l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V-(thiazol-2- ylmethyl)piperidine-4-carboxamide

(a) Preparation of the intermediate compound tert-butyl 4-(thiazol-2-ylmethylcarbamoyl)- piperidine- 1 -carboxylate :

To a mixture of l-(tert-butoxycarbonyl)piperidine-4-carboxylic acid (500 mg, 2.18 mmol), triethylamine (220.7 mg, 2.18 mmol) and dichloromethane (10 mL) was carefully added thionyl chloride (311.3 mg, 2.62 mmol) and the reaction mixture was stirred at room temperature (reaction mixture 1). In a separate vial was mixed 2-(chloromethyl)thiazole, triethylamine (882.7 mg, 8.723 mmol) and dichloromethane (10 mL) that was stirred and cooled on ice-batch and reaction micture 1 was added over 5 minutes while stirring. The reaction mixture was allowed to reach room temperature, washed with aqueous hydrochloric acid (2 x20 mL, 2 N), aqueous saturated sodium bicarbonate (2 x20 mL), dried over magnesium sulphate, filtered and concentrated in vacuo. The residue was collected and the acidic and basic washes were combined and extracted with dichloromethane (3 x30 mL), the organic phase dried over magnesium sulphate and concentrated. The two residues were combined to give 474 mg (67 % yield) of tert- butyl 4-(thiazol-2-ylmethylcarbamoyl)-piperidine-l -carboxylate. LC-MS (m/z) 326.2 (M+ 1).
(b) Preparation of the intermediate compound 2-((4-piperidiniumcarboxamido)methyl)-3- thiazolium dichloride:

(c) Preparation of the intermediate compound l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-JV- (thiazo l-2-ylmethyl)piperidine-4-carboxamide :

2-Chloro-5,6-dimethyl-lH-benzo[d]imidazole (54.2 mg, 0.30 mmol), 2-((4-piperidinium- carboxamido)methyl)-3-thiazolium dichloride (113.4 mg, 0.39 mmol, 90 % purity), N,N- diisopropylethylamine (116.3 mg, 0.9 mmol) and 1,4-dioxane (2 mL) was irridated in a microwave oven at 17O0C for 1 hour. The reaction mixture was concentrated in vacuo and the residue subjected to column chromatography (silica gel, dichloromethane/methanol, gradient elution using 5-20 % methanol) to give 83 mg (75 % yield) of l-(5,6-dichloro-lH- benzo[d]imidazol-2-yl)-Λ/-(thiazol-2-ylmethyl)piperidine-4-carboxamide. LC-MS (m/z) 370.3 (M+ 1). (d) The title compound was prepared from l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-JV-
(thiazol-2-ylmethyl)piperidine-4-carboxamide (11.1 mg, 0.03 mmol), N,Λ/-dimethylformamide (1 mL), acetonitrile (3 mL), caesium carbonate (29.3 mg, 0.09 mmol) and 2-bromopropane (11.1 mg, 0.09 mmol) using the method described in Example 111, to give 2.7 mg (22 % yield) of 1- (l-Isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(thiazol-2-ylmethyl)piperidine-4- carboxamide. LC-MS (m/z) 412.3 (M+l).
Example 111: l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-7V-(thiazol-2- ylmethyl)piperidine-4-carboxamide

(a) Preparation of the intermediate compound l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-JV- (thiazo l-2-ylmethyl)piperidine-4-carboxamide :

(b) l-(5,6-Dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-(thiazol-2-ylmethyl)piperidine-4-carboxamide (12.3 mg, 0.03 mmol), 2-bromopropane (11.1 mg, 0.09 mmol), caesium carbonate (29.3 mg, 0.09 mmol), acetonitrile (3 mL) and Λ/,Λ/-dimethylformamide (1 mL) was mixed and heated at 6O0C over night. The reaction mixture concentrated in vacuo and subjected to preparative hplc (performed on a Gilson-Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 μm, 21.2 Xx 150 mm) column, using acetonitrile/water (0.05 % formic acid) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 1.6 mg (12 % yield) of l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-(thiazol-2- ylmethyl)piperidine-4-carboxamide. LC-MS (m/z) 452.2 (M+ 1).
Example 112: 7V-(4-(lH-tetrazol-5-yl)benzyl)-l-(l-isopropyl-5,6-dimethyl-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

JV-(4-Bromobenzyl)- 1 -( 1 -isopropyl-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)piperidine-4- carboxamide (20 mg, 0.0414 mmol), dicyanozinc (ZnCN2, 4.9 mg, 0.0414 mmol), bis(tri-tert- butylphosphine)palladium (Pd[P(t-bu)3]2, 2.1 mg, 0.00414 mmol) and Λ/,Λ/-dimethylformamide (1 mL) were irridated in a microwave oven at 14O0C for 10 minutes. Sodium azide (NaNi, 32.3 mg, 0.496 mmol) and ammonium chloride (26.6 mg, 0.496 mmol) was added to the reaction mixture, which was heated at 15O0C for 40 minutes. The reaction mixture was filtered, the filtrate concentrated in vacuo and the residue subjected to preparative hplc (performed on a Gilson-Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 μm, 21.2 xl50 mm) column, using acetonitrile/water (0.05 % formic acid) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 3.5 mg (18 % yield) of N-(4-(lH- tetrazol-5-yl)benzyl)-l -(I -isopropyl-5, 6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide. LC-MS (m/z) 473.3 (M+ 1).
Example 113: 7V-cyclopentyl-l-(5,6-dimethyl-l-(pentan-3-yl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.1 g, 0.293 mmol), N,Λ/-dimethylformamide (3 mL), caesium carbonate (1.172 mmol), 3-bromopentane (1.172 mmol) and dichlorobis(triphenylphosphine)- palladium (II) (Pd(PPhS)2Cl2 (0.0293 mmol) using the method described in Example 14. The reaction mixture was heated at 12O0C for 1 hour under microwave conditions and purified with column chromathography, to give 27 mg (11.2 % yield) ofN-cyclopentyl-l-(5,6-dimethyl-l- (pentan-3-yl)-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 411.1 (M+l).
Example 114: l-(5,6-dimethyl-l-phenyl-lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3- yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.30 g, 0.877 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.51 mmol), iodobenzene (3.51 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.132 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0438 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 2.9 mg (0.8 % yield) of 1 -(5,6-dimethyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a brown solid. LC-MS (m/z) 419.2 (M+ 1).
Example 115: 7V-cyclopentyl-l-(5,6-dichloro-l-(4-fluorophenyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

(tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.3 g, 0.78 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.092 mmol), l-fluoro-4-iodobenzene (0.936 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.117 mmol), polyethylene glycol (0.039 mmol) and copper(I) oxide (0.039 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 18 hours in a sealed tube and purified with column chromathography, to give 21 mg (6 % yield) of N-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-fluorophenyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide as a white solid. LC-MS (m/z) 475.5 (M+ 1).
Example 116: l-(l-(4-bromophenyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.3 g, 0.78 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.092 mmol), 1 ,4-dibromobenzene (0.936 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.117 mmol), polyethylene glycol (0.039 mmol) and copper(I) oxide (0.039 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 18 hours in a sealed tube and purified with column chromathography, to give 6 mg (1.4 % yield) of l-(l-(4-bromophenyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as a white solid. Example 117: l-(l-(4-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

(a) Preparation of the intermediate compound l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylic acid:

Ethyl l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (1.00 g, 3.31 mmol) was dissolved in ethanol (50 mL) and aqueous sodium hydroxide (1 N, 17.0 mL) was added. The reaction mixture was stirred at room temperature 1 hour, concentrated in vacuo and pΗ adjusted to 4-5 with addition of aqueous hydrochloric acid (6 N). The formed precipitate was filtered off, the aqueous mother liquid extracted three times with dichloromethane (50 mL) and the combined organic layers were dried with MgSO4 and concentrated in vacuo. The precipitate and the residue
from the extraction were combined and rechrystallized in petroleum ether to give 0.538 g (59%) of l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid.
(b) Preparation of the intermediate compound l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-iV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide
 l-(5,6-Dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (1.0 g, 3.7 mmol), tetrahydrofuran-3 -amine hydrochloride (0.54 g, 4.4 mmol), 2-(7-aza-lH-benzotriazole-l-yl)- 1,1,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 2.8 g, 7.4 mmol), N,N- diisopropylethylamine (Ηunig's base, DIEA, 0.96 g, 7.4 mmol) and N,N-dimethylformamide (25 mL) were stirred at room temperature for 1 hour. After an extractive work-up, the crude was chrystallized from dichloromethane/petroleum ether to give 1.0 g (83% yield) of l-(5,6- dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide.
l-(5,6-Dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (1.0 g, 3.7 mmol), tetrahydrofuran-3 -amine hydrochloride (0.54 g, 4.4 mmol), 2-(7-aza-lH-benzotriazole-l-yl)- 1,1,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 2.8 g, 7.4 mmol), N,N- diisopropylethylamine (Ηunig's base, DIEA, 0.96 g, 7.4 mmol) and N,N-dimethylformamide (25 mL) were stirred at room temperature for 1 hour. After an extractive work-up, the crude was chrystallized from dichloromethane/petroleum ether to give 1.0 g (83% yield) of l-(5,6- dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide.
(c) The title compound was prepared from l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-JV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.30 g, 0.87 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.48 mmol), l-fluoro-4-iodobenzene (3.48 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.130 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0435 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 9 mg (2.3 % yield) of l-(l-(4-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as a pale green solid. LC-MS (m/z) 437.2 (M+l).
Example 118: l-(l-(3-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.30 g, 0.87 mmol), dimethylsulfoxide (6 mL),
caesium carbonate (3.48 mmol), l-bromo-3-fluorobenzene (3.48 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.130 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0435 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 22 mg (6 % yield) of l-(l-(3-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as a pale green solid. LC-MS (m/z) 437.1 (M+l).
Example 119: l-(l-(3,5-difluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

(tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.30 g, 0.87 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.48 mmol), l-bromo-3,5-difluorobenzene (3.48 mmol), 4,7-dimethoxy- 1,10-phenanthroline (0.130 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0435 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 32 mg (8 % yield) of l-(l-(3,5-difluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as a off-white solid. LC-MS (m/z) 455.2 (M+l).
Example 120: l-(l-(4-chlorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-JV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.30 g, 0.87 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.48 mmol), l-bromo-4-chlorobenzene (3.48 mmol), 4,7-dimethoxy-l,10-
phenanthroline (0.130 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0435 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 61 mg (15 % yield) of l-(l-(4-chlorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as a off-white solid. LC-MS (m/z) 453.3 (M+l).
Example 121: l-CS^-dimethyl-l-C^CtrifluoromethyOpheny^-lH-benzoIdlimidazol-l-yO-TV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-iV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.30 g, 0.87 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.48 mmol), l-bromo-4-(trifluoromethyl)benzene (3.48 mmol), 4,7- dimethoxy-l,10-phenanthroline (0.130 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0435 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 54 mg (12 % yield) of l-(5,6-dimethyl-l-(4-(trifluoromethyl)phenyl)-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale green solid. LC-MS (m/z) 487.2 (M+l).
Example 122: l-(l-(3,5-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-JV-(tetra- hydrofuran-3-yl)piperidine-4-carboxamide (0.30 g, 0.87 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.48 mmol), l-bromo-3,5-dimethylbenzene (3.48 mmol), 4,7-dimethoxy-
1,10-phenanthroline (0.130 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0435 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 24 mg (6.1 % yield) of l-(l-(3,5-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydro- furan-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 447.2 (M+l).
Example 123: l-(5,6-dimethyl-l-(naphthalen-2-yl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-iV-(tetra- hydrofuran-3-yl)piperidine-4-carboxamide (0.30 g, 0.87 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.48 mmol), 2-bromonaphthalene (3.48 mmol), 4, 7-dimethoxy- 1,10- phenanthroline (0.130 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0435 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 5 mg (1.2 % yield) of 1 -(5,6-dimethyl- 1 -(naphthalen-2-yl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)- piperidine-4-carboxamide as a pale green solid. LC-MS (m/z) 469.1 (M+l).
Example 124: l-(l-(benzo[d] [l,3]dioxol-5-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide


The title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.3 g, 0.78 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.092 mmol), l-bromo-3-methoxybenzene (0.936 mmol), 4,7-dimethoxy-
1,10-phenanthroline (0.117 mmol), polyethylene glycol (0.039 mmol) and copper(I) oxide (0.039 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 18 hours in a sealed tube and purified with column chromathography, to give 52 mg (13.6 % yield) of 1 -(5,6-dichloro- 1 -(3-methoxyphenyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 489.4 (M+ 1).
Example 126: l-(l-(benzo[d] [l,3]dioxol-5-yl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.3 g, 0.78 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.092 mmol), 5-bromobenzo[d][l,3]dioxole (0.936 mmol), 4,7-dimethoxy- 1,10-phenanthroline (0.117 mmol), polyethylene glycol (0.039 mmol) and copper(I) oxide (0.039 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for
18 hours in a sealed tube and purified with column chromathography, to give 41 mg (10.5 % yield) of l-(l-(benzo[d][l,3]dioxol-5-yl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 503.4 (M+l). Example 127: l-(5,6-dichloro-l-(4-cyanophenyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.3 g, 0.78 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.092 mmol), 4-bromobenzonitrile (0.936 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.117 mmol), polyethylene glycol (0.039 mmol) and copper(I) oxide (0.039 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 18 hours in a sealed tube and purified on preperative reverse-phase hplc, to give 13 mg (3.4 % yield) of 1 -(5,6-dichloro- 1 -(4-cyanophenyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 484.4 (M+l).
Example 128: 7V-cyclopentyl-l-(5,6-dimethyl-l-m-tolyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.30 g, 0.87 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.48 mmol), l-bromo-3-methylbenzene (3.48 mmol), 4,7-dimethoxy-l,10-phenanthroline (0.130 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.044 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube
and purified with column chromathography, to give 12 mg (3.2 % yield) of N-cyclopentyl-1- (5,6-dimethyl-l-m-tolyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 431.2 (M+ 1).
Example 129: 7V-cyclopentyl-l-(l-(3,4-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol- 2-yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.30 g, 0.829 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.316 mmol), 4-bromo-l,2-dimethylbenzene (3.316 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.132 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.044 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 2 mg (0.52 % yield) of N-cyclopentyl- 1 -( 1 -(3 ,4-dimethylphenyl)-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide as a white solid. LC-MS (m/z) 445.2 (M+l). Example 130: 7V-cyclopentyl-l-(l-(4-fluoro-3-methylphenyl)-5,6-dimethyl-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.30 g, 0.829 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.316 mmol), 4-bromo-l-fluoro-2-methylbenzene (3.316 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.132 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.044 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 5 mg (1.3 % yield) of
N-cyclopentyl-l-(l-(4-fluoro-3-methylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 449.1 (M+ 1).
Example 131: 7V-cyclopentyl-l-(l-(4-ethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.30 g, 0.829 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.316 mmol), l-bromo-4-ethylbenzene (3.316 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.132 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.044 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 10 mg (2.5 % yield) of N-cyclopentyl-l-(l-(4-ethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide as an off-white solid. LC-MS (m/z) 445.2 (M+ 1).
Example 132: 7V-cyclopentyl-l-(l-(4-isopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.30 g, 0.829 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.316 mmol), l-bromo-4-isopropylbenzene (3.316 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.132 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.044 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 6 mg (1.5 % yield) of
N-cyclopentyl-l-(l-(4-isopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide as an off-white solid. LC-MS (m/z) 459.2 (M+ 1).
Example 133: 7V-cyclopentyl-l-(5,6-dimethyl-l-(quinolin-6-yl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.30 g, 0.829 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.316 mmol), 6-bromoquinoline (3.316 mmol), 4,7-dimethoxy-l,10-phenanthroline (0.132 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.044 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 16 mg (3.9 % yield) of N-cyclopentyl-1- (5,6-dimethyl-l-(quinolin-6-yl)-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 468.2 (M+l).
Example 134: 7V-cyclopentyl-l-(5,6-dimethyl-l-(quinoxalin-6-yl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.30 g, 0.829 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.316 mmol), 6-bromoquinoxaline (3.316 mmol), 4,7-dimethoxy-l,10-phenanthroline (0.132 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.044 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 15 mg (3.6 % yield) of N-cyclopentyl-1-
(5 ,6-dimethyl- 1 -(quinoxalin-6-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a yellow solid. LC-MS (m/z) 469.2 (M+l).
Example 135: l-(5,6-dichloro-l-(4-(methylcarbamoyl)phenyl)-lH-benzo[d]imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

A reaction mixture consisting of l-(l-(4-bromophenyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)- N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.19 mmol), trans -di(μ-acQtato)- bis[o-(di-o-tolylphosphino)-benzyl]dipalladium(II) (Hermann palladacycle, 0.0095 mmol), N,N- diisopropylethylamine (Hunig's base, DIEA, 0.38 mmol), l,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 0.19 mmol), tri tertiarybutyl phosphonium tetrafluoborate ([(t-Bu)3PH]BF4, 0.019 mmol), molybdenum hexacarbonyl (Mo(CO)6, 0.095 mmol) and methyl amine hydrochloride (0.38 mmol) in 1,4-dioxane (3 mL), was irradiated under microwave conditions at 15O0C for 30 minutes. The reaction mixture was quenched with water (5 mL), extracted with dichloromethane (2 x50 mL) and the combined organic phases dried over anhydrous sodium sulphate. After concentration in vacuo, the residue was subjected to column chromatography to give 10 mg (10 % yield) of l-(5,6-dichloro-l-(4-(methylcarbamoyl)phenyl)-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide.
Example 136: (R)-l-(5,6-dichloro-l-(3-methoxyphenyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

(a) Preparation of the intermediate compound ethyl l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylate

2,5,6-trichloro-lH-benzo[d]imidazole (1 g, 5.0 mmol) was mixed with 1,4-dioxane (9 mL) and ethyl piperidine-4-carboxylate (0.85 g, 5.0 mmol) was added, followed by JV,iV-diisopropyl- ethylamine (Ηunig's base, DIEA, 1.034 g, 8 mmol). The reaction mixture was subjected to microwave conditions for one hour at 18O0C, purified on column (silica gel, hexane/ethyl acetate, gradient elution, 20-70 % ethyl acetate) to give 1.33 g (87 % yield) of ethyl l-(5,6- dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate as a brown solid. LC-MS (m/z) 342.0 (M+l).
(b) Preparation of the intermediate compound l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylic acid

Ethyl l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (0.60 g, 2.0 mmol) was dissolved in ethanol (150 mL) and aqueous sodium hydroxide (1 N, 17.5 mL) was added. The reaction mixture was stirred at room temperature for two days, concentrated in vacuo and pΗ adjusted to 4-5 with addition of aqueous hydrochloric acid (1 N). The formed precipitate was filtered off, the aqueous mother liquid extracted three times with dichloromethane (100 mL) and the combined organic phases concentrated in vacuo. The crude carboxylic acid (precipitate and the residue from the extraction) was purified on column (silica gel, ethyl acetate/methanol/formic acid, gradient elution 10-15 % methanol, 0.1 % formic acid) to give 0.53 g (90% purity) of 1- (5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid that was used directly in the next step.
(c) Preparation of the intermediate compound (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide
 l-(5,6-Dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (628 mg, 2.0 mmol), (R)-(+)-tetrahydrofuran-3-amine 4-methylbenzenesulfonate (622 mg, 2.4 mmol), 2-(7-aza-lH-
benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (HATU, 912 mg, 2.4 mmol), JV,JV-diisopropylethylamine (Hunig's base, DIEA, 775 mg, 6.0 mmol) and N5N- dimethylformamide (25 niL) was stirred at room temperature for 100 minutes. The reaction mixure was concentrated in vacuo, the residue purified on column (silica gel, flash
l-(5,6-Dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (628 mg, 2.0 mmol), (R)-(+)-tetrahydrofuran-3-amine 4-methylbenzenesulfonate (622 mg, 2.4 mmol), 2-(7-aza-lH-
benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (HATU, 912 mg, 2.4 mmol), JV,JV-diisopropylethylamine (Hunig's base, DIEA, 775 mg, 6.0 mmol) and N5N- dimethylformamide (25 niL) was stirred at room temperature for 100 minutes. The reaction mixure was concentrated in vacuo, the residue purified on column (silica gel, flash
chromatography, dichloromethane/methanol, gradient elution 4-20 % methanol) and finally precipitated from chloroform to give 378 mg (49 % yield) of (R)-l-(5,6-dichloro-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid. LC- MS (m/z) 382.9 (M+ 1).
(d) A mixture of (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide (57.5 mg, 0.15 mmol), dimethylsulfoxide (0.7 mL), caesium carbonate (68.4 mg, 0.21 mmol), l-iodo-3-methoxybenzene (70.2 mg, 0.3 mmol), 8- hydroxyquinoline (4.4 mg, 0.03 mmol), polyethylene glycol 400 (60 mg, 0.15 mmol) and copper(I) oxide (2.1 mg, 0.015 mmol) was subjected to microwave conditions for one hour at 15O0C. The reaction mixture was filtered and subjected to preparative hplc (performed on a Gilson-Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using methano I/water (0.05 % formic acid) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 21.7 mg (30 % yield) of (R)- 1 -(5,6- dichloro-l-(3-methoxyphenyl)-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide. LC-MS (m/z) 488.9 (M+ 1). Example 137: 7V-cyclopentyl-l-(5,6-dichloro-l-(pyridin-3-yl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (38 mg, 0.0997 mmol), dimethylsulfoxide (0.7 mL), caesium carbonate (32 mg, 0.0997 mmol), 3-iodopyridine (61.3 mg, 0.299 mmol), 8-hydroxyquinoline (3 mg, 0.0199 mmol), polyethylene glycol 400 (20 mg, 0.0498 mmol) and copper(I) oxide (1.4 mg, 0.00997 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for one hour at 15O0C and subsequently purified on reverse-phase hplc,
to give 12 mg (26 % yield) of N-cyclopentyl-l-(5,6-dichloro-l-(pyridin-3-yl)-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a brown solid. LC-MS (m/z) 458.0 (M+l).
Example 138: 7V-cyclopentyl-l-(5,6-dichloro-l-(pyridin-3-yl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (38 mg, 0.0997 mmol), dimethylsulfoxide (0.7 mL), caesium carbonate (32 mg, 0.0997 mmol), 2-bromopyridine (47.2 mg, 0.299 mmol), 8-hydroxyquinoline (3 mg, 0.0199 mmol), polyethylene glycol 400 (20 mg, 0.0498 mmol) and copper(I) oxide (1.4 mg, 0.00997 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for one hour at 15O0C and subsequently purified on reverse- phase hplc, to give 13 mg (28 % yield) of N-cyclopentyl-l-(5,6-dichloro-l-(pyridin-3-yl)-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a brownish solid. LC-MS (m/z) 458.0 (M+l). Example 139: 7V-cyclopentyl-l-(5,6-dichloro-l-(6-methoxypyridin-2-yl)-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

The title compound was prepared from N-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (38 mg, 0.0997 mmol), dimethylsulfoxide (0.5 mL), caesium carbonate (32 mg, 0.0997 mmol), 2-bromo-6-methoxypyridine (56.2 mg, 0.299 mmol), 8- hydroxyquinoline (3 mg, 0.0199 mmol), polyethylene glycol 400 (20 mg, 0.0498 mmol) and copper(I) oxide (1.4 mg, 0.00997 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for one hour at 15O0C and subsequently purified on reverse-phase hplc, to give 22 mg (45 % yield) of N-cyclopentyl-l-(5,6-dichloro-l-
(pyridin-3-yl)-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a beige solid. LC-MS (m/z) 488.0 (M+ 1).
Example 140: 7V-cyclopentyl-l-(5,6-dichloro-l-(4-cyanophenyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

The title compound was prepared fromN-cyclopentyl-l-(5,6-dichloro-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (38 mg, 0.0997 mmol), dimethylsulfoxide (0.7 mL), caesium carbonate (45.5 mg, 0.14 mmol), 4-iodobenzonitrile (68.5 mg, 0.299 mmol), 8-hydroxyquinoline (3 mg, 0.0199 mmol), polyethylene glycol 400 (20 mg, 0.0498 mmol) and copper(I) oxide (1.4 mg, 0.00997 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for one hour at 15O0C and subsequently purified on reverse- phase hplc, to give 10.5 mg (22 % yield) of N-cyclopentyl-l-(5,6-dichloro-l-(4-cyanophenyl)- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a brownish solid. LC-MS (m/z) 482.0 (M+l). Example 141: (R)-l-(5,6-dichloro-l-(4-fluorophenyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (38.3 mg, 0.10 mmol), dimethylsulfoxide (0.7 mL), caesium carbonate (45.6 mg, 0.14 mmol), l-fluoro-4-iodobenzene (44.4 mg, 0.20 mmol), 8-hydroxyquinoline (2.9 mg, 0.02 mmol), polyethylene glycol 400 (40 mg, 0.1 mmol) and copper(I) oxide (1.4 mg, 0.01 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for one hour at 15O0C and subsequently purified
on reverse-phase hplc, to give 14 mg (29 % yield) of (R)-l-(5,6-dichloro-l-(4-fluorophenyl)-lH- benzo[d]-imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 476.9 (M+l).
Example 142: (R)-l-(5,6-dichloro-l-(3-fluorophenyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (38.3 mg, 0.10 mmol), dimethylsulfoxide (0.7 mL), caesium carbonate (45.6 mg, 0.14 mmol), l-fluoro-3-iodobenzene (44.4 mg, 0.20 mmol), 8-hydroxyquinoline (2.9 mg, 0.02 mmol), polyethylene glycol 400 (40 mg, 0.1 mmol) and copper(I) oxide (1.4 mg, 0.01 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for one hour at 15O0C and subsequently purified on reverse-phase hplc, to give 6 mg (13 % yield) of (R)-l-(5,6-dichloro-l-(3-fluorophenyl)-lH- benzo[d]-imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 477.0 (M+l).
Example 143: (R)-l-(5,6-dichloro-l-(4-methoxyphenyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (38.3 mg, 0.10 mmol), dimethylsulfoxide (0.7 mL), caesium carbonate (45.6 mg, 0.14 mmol), l-iodo-4-methoxybenzene (46.8 mg, 0.20 mmol), 8-hydroxyquinoline (2.9 mg, 0.02 mmol), polyethylene glycol 400 (40 mg, 0.1 mmol) and copper(I) oxide (1.4 mg, 0.01 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for one hour at 15O0C and subsequently
purified on reverse-phase hplc, to give 9.5 mg (49 % yield) of (R)-l-(5,6-dichloro-l-(4- methoxyphenyl)- lH-benzo [d]imidazo l-2-yl)-N-(tetrahydrofuran-3 -yl)piperidine-4-carboxamide . LC-MS (m/z) 489.0 (M+l).
Example 144: (R)-l-(l-(4-tert-butylphenyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (38.3 mg, 0.10 mmol), dimethylsulfoxide (0.7 mL), caesium carbonate (45.6 mg, 0.14 mmol), l-bromo-4-tert-butylbenzene (42.6 mg, 0.20 mmol), 8-hydroxyquinoline (5.8 mg, 0.04 mmol), polyethylene glycol 400 (40 mg, 0.1 mmol) and copper(I) oxide (2.9 mg, 0.02 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for one hour at 15O0C and subsequently purified on reverse-phase hplc, to give 8.2 mg (16 % yield) of (R)-l-(l-(4-tert-butylphenyl)-5,6- dichloro-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 515.0 (M+l).
Example 145: (R)-l-(5,6-dichloro-l-(4-(trifluoromethoxy)phenyl)-lH-benzo[d]imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (38.3 mg, 0.10 mmol), dimethylsulfoxide (0.7 mL), caesium carbonate (45.6 mg, 0.14 mmol), l-iodo-4-(trifluoromethoxy)benzene (57.6 mg, 0.20 mmol), 8-hydroxyquinoline (2.9 mg, 0.02 mmol), polyethylene glycol 400 (40 mg, 0.1
mmol) and copper(I) oxide (1.4 mg, 0.01 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 15O0C, filtered and subsequently purified on reverse-phase hplc, to give 15.2 mg (28 % yield) of (R)-I -(5,6- dichloro- 1 -(4-(trifluoromethoxy)phenyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide. LC-MS (m/z) 543.0 (M+ 1).
Example 146: l-(5,6-dichloro-l-phenyl-lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3- yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.3 g, 0.78 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.092 mmol), iodobenzene (0.936 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.117 mmol), polyethylene glycol (0.039 mmol) and copper(I) oxide (0.039 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 18 hours in a sealed tube and purified with column chromathography, to give 10 mg (2.8 % yield) of l-(5,6-dichloro-l-phenyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as a brown solid. LC-MS (m/z) 459.1 (M+ 1).
Example 147: l-(5,6-dichloro-l-(pyridin-2-yl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

(tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.3 g, 0.78 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.092 mmol), 2-bromopyridine (0.936 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.117 mmol), polyethylene glycol (0.039 mmol) and copper(I) oxide (0.039 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 18 hours in a sealed tube and purified with column chromathography, to give 26 mg (7.2 %
yield) of 1 -(5,6-dichloro- 1 -(pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 460.6 (M+l).
Example 148: l-(5,6-dichloro-l-(pyridin-3-yl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.3 g, 0.78 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.092 mmol), 3-bromopyridine (0.936 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.117 mmol), polyethylene glycol (0.039 mmol) and copper(I) oxide (0.039 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 18 hours in a sealed tube and purified with column chromathography, to give 5 mg (1.4 % yield) of 1 -(5,6-dichloro- l-(pyridin-3-yl)-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 460.5 (M+l).
Example 149: l-(5,6-dichloro-l-p-tolyl-lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3- yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.3 g, 0.78 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.092 mmol), 4-bromotoluene (0.936 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.117 mmol), polyethylene glycol (0.039 mmol) and copper(I) oxide (0.039 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 18 hours in a sealed tube and purified with column chromathography, to give 27 mg (7.3 % yield) of 1 -(5,6-dichloro- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 473.4 (M+l).
Example 150: (R)- l-(l-(4-chloro-3-methoxyphenyl)-5,6-dimethyl- lH-benzo [d] imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

(a) Preparation of the intermediate compound (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)- N-(tetrahydrofuran-3 -yl)piperidine-4-carboxamide :
 l-(5,6-Dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (1.0 g, 3.6 mmol), (R)- (+)-tetrahydrofuran-3 -amine 4-methylbenzenesulfonate (4.3 mmol), 2-(7-aza-lH-benzotriazole- l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 7.2 mmol), N5N- diisopropylethylamine (Ηunig's base, DIEA, 7.2 mmol) and N,N-dimethylformamide (25 mL) was stirred at room temperature for 1 hour. After the appropriate work-up, the residue was recrystallized from dichloromethane/petroleum ether to give 0.96 g (77 % yield) of (R)-l-(5,6- dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide.
l-(5,6-Dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (1.0 g, 3.6 mmol), (R)- (+)-tetrahydrofuran-3 -amine 4-methylbenzenesulfonate (4.3 mmol), 2-(7-aza-lH-benzotriazole- l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 7.2 mmol), N5N- diisopropylethylamine (Ηunig's base, DIEA, 7.2 mmol) and N,N-dimethylformamide (25 mL) was stirred at room temperature for 1 hour. After the appropriate work-up, the residue was recrystallized from dichloromethane/petroleum ether to give 0.96 g (77 % yield) of (R)-l-(5,6- dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide.
(b) The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (100 mg, 0.292 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.584 mmol), 4-bromo-l-chloro-2-methoxybenzene (0.876 mmol), 8- hydroxyquinoline (0.117 mmol), polyethylene glycol (0.146 mmol) and copper(I) oxide (0.0584 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase hplc, to give 15 mg (10 % yield) of (R)-l-(l-(4-chloro-3-methoxyphenyl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a yellow solid. LC-MS (m/z) 483.3 (M+l).
Example 151: (R)- l-(5,6-dichloro- l-(4-chloro-3-methoxyphenyl)- lH-benzo [d] imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 4-bromo-l-chloro-2-methoxybenzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase hplc, to give 23 mg (17 % yield) of (R)-l-(5,6-dichloro-l-(4-chloro-3-methoxyphenyl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a yellow solid. LC-MS (m/z) 523.5 (M+l).
Example 152: (R)-l-(l-(2,3-dihydrobenzofuran-5-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (100 mg, 0.292 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.584 mmol), 5-bromo-2,3-dihydrobenzofuran (0.876 mmol), 8- hydroxyquinoline (0.117 mmol), polyethylene glycol (0.146 mmol) and copper(I) oxide (0.0584 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase hplc, to give 5 mg (3.7 % yield) of (R)-l-(l-(2,3-dihydrobenzofuran-5-yl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 461.2 (M+l).
Example 153: (R)-l-(5,6-dichloro-l-(2,3-dihydrobenzofuran-5-yl)-lH-benzo[d]imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 5-bromo-2,3-dihydrobenzofuran (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase hplc, to give 7 mg (5.3 % yield) of (R)-l-(5,6-dichloro-l-(2,3-dihydrobenzofuran-5-yl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a yellow solid. LC-MS (m/z) 501.6 (M+l).
Example 154: (RJ-l-Cl-CS^-diethylpheny^-S^-dimethyl-lH-benzoIdlimidazoU-yO-TV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (200 mg, 0.584 mmol) dimethylsulfoxide (2 mL), caesium carbonate (1.17 mmol), l-bromo-3,5-diethylbenzene (1.75 mmol), 8- hydroxyquinoline (0.234 mmol), polyethylene glycol (0.292 mmol) and copper(I) oxide (0.117 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase hplc, to give 4 mg (1.5 % yield) of (R)-l-(l-(3,5-diethylphenyl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a brown solid. LC- MS (m/z) 475.2 (M+l).
Example 155: l-(5,6-dichloro-l-(cyclobutylmethyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.1 g, 0.26 mmol), (bromomethyl)cyclobutane (1.04 mmol), caesium carbonate (1.04 mmol), [l,r-bis(diphenylphosphino)ferrocene] dichloropalladium(II) (PdCl2(dppf), 0.026 mmol) and N,Λ/-dimethylformamide (8 mL) using the method described in Example 16. The reaction mixture was heated at 13O0C for two hours in a sealed tube and purified with column chromathography, to give 15 mg (12.8 % yield) of l-(5,6- dichloro-l-(cyclobutylmethyl)-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a pale yellow solid. LC-MS (m/z) 449.3 (M+l). Example 156: (R)-l-(l-(4-fluoro-3-methylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

(c) The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (120 mg, 0.35 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.4 mmol), 4-bromo-l-fluoro-2-methylbenzene (1.4 mmol), 8- hydroxyquinoline (0.07 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0175 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on column chromatography, to give 30.5 mg (30 % yield) of (R)-l-(l-(4-fluoro-3-methylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol- 2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 451.1 (M+l).
Example 157: (R)-l-(l-(4-ethylphenyl)-5,6-dimethyl-lΗ-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (120 mg, 0.35 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.4 mmol), l-bromo-4-ethylbenzene (1.4 mmol), 8-hydroxyquinoline (0.07 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0175 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on column chromatography (silica gel), to give 19 mg (12 % yield) of (R)-l-(l-(4-ethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 447.2 (M+l). 1H NMR (400 MHz, DMSO-d6): (for the aromatic region and -NH-)
δ 8.03 (IH, NH), 7.44 (4H), 7.21 (IH), 6.82 (IH).
Example 158: (RJ-l-Cl-CS^-dimethylphenylJ-S^-dimethyl-lH-benzoIdlimidazol-l-yl)-^- (tetrahydrofuran-3-yl)piperidine-4-carboxamide


The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (120 mg, 0.35 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.4 mmol), 6-bromoquinoline (1.4 mmol), 8-hydroxyquinoline (0.07 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0175 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on column chromatography, to give 15 mg (9 % yield) of (R)- 1 -(5,6-dimethyl- 1 -(quinolin-6-yl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 470.1 (M+ 1).
Example 160: (R)-l-(l-(4-isopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide


The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (120 mg, 0.35 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.4 mmol), l-bromo-4-tert-butylbenzene (1.4 mmol), 8-hydroxy- quinoline (0.07 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0175 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on column chromatography, to give 27 mg (16 % yield) of (R)-l-(l-(4-tert-butylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 475.2 (M+l).
Example 162: (R)-l-(l-(3-chlorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (120 mg, 0.35 mmol), dimethylsulfoxide (6 mL), caesium carbonate (1.4 mmol), l-bromo-3-chlorobenzene (1.4 mmol), 8-hydroxy-quinoline (0.07 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0175 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on column chromatography, to give 54 mg (34 % yield) of (R)-l-(l-(3-chlorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 453.2 (M+l).
Example 163: l-(6-chloro-5-methyl-l-phenyl-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide

(a) Preparation of the intermediary compound N-(5-chloro-4-methyl-2-nitrophenyl)acetamide:

Potassium nitrate (10.11 g, 0.10 mol) in concentrated sulphuric acid (21.3 mL) was added drop- wise to N-(3-chloro-4-methylphenyl)acetamide (18.36 g, 0.10 mol) in concentrated sulphuric acid (101.3 mL) at 5°C. After 5 hours stirring below 5°C, the reaction mixture was carefully poured out on ice water (500 mL), the formed precipitate filtered off, and washed with water (50 mL), ώo-propanol (10 mL) and ώo-hexane (30 mL). The precipitate was recrystallized from ethanol (160 mL, 95 %) to give 10.3 g (45 % yield) of N-(5-chloro-4-methyl-2-nitrophenyl)- acetamide.
(b) Preparation of the intermediary compound 5-chloro-4-methyl-2-nitroaniline:

Sodium ethoxide (8.27 g, 45.9 mmol) in ethanol (10 mL) was added to (N-(5-chloro-4-methyl-2- nitrophenyl)acetamide (7.0 g, 30.6 mmol) in ethanol (10 mL) while stirring at room temperature. After one hour the reaction mixture was poured out in water (100 mL), the precipitate filtered off, and washed with a mixture of ώo-propanol and ώo-hexane (1:3, 10 mL) followed by iso- hexane (10 mL). This gave 5.71 g (85 % yield) of 5-chloro-4-methyl-2-nitroaniline. LC-MS (m/z) 187.0 (M+l).
(c) Preparation of the intermediary compound 5-chloro-4-methyl-2-nitro-N-phenylaniline

5-Chloro-4-methyl-2-nitroaniline (560 mg, 3.0 mmol), bromobenzene (3.0 g, 0.019 mmol), tripotassium phosphate (1.27 g, 6 mmol), bis(tri-tert-butylphosphine)palladium(0) (Pd[P(t- Bu)3]2, 0.05, 0.0978 mmol) and toluene (5 mL) was heated at 1200C under nitrogen gas over night. The reaction mixture was purified on column (silica gel, flash chromatography, gradient elution iso-hexane/ethyl acetate 100-90 % : 0-10 %) to give 503 mg (64 %) of 5-chloro-4- methyl-2-nitro-N-phenylaniline. LC-MS (m/z) 262.9 (M+l).
(d) Preparation of the intermediary compound 5-chloro-4-methyl-N1-phenylbenzene-l,2-diamine

(e) Preparation of the intermediary compound 6-chloro-5-methyl-l -phenyl- IH- benzo[d]imidazol-2(3Η)-one.

(f) 6-Chloro-5-methyl-l -phenyl- lH-benzo[d]imidazol-2(3H)-one (42.4 mg, 0.164 mmol) was stirred in phosphoryl trichloride (POCI3, 5 g) at reflux for 6 hours and concentrated in vacuo (co- evaporation with toluene). Water (10 ml) was added to the residue, followed by an aqueous saturated sodium bicarbonate solution (10 mL). The aqueous layer was extracted twice with dichloromethane (20 mL), the combined organic layer dried over magnesium sulfate and concentrated in vacuo. The residue, N-cyclopentylpiperidine-4-carboxamide (29. mg, 0.136 mmol) and 1 ,2-dimethoxyethane (DME, 0.8 mL) was subjected to microwave conditions for one hour at 18O0C. After work-up, the residue was subjected to preparative hplc (performed on a Gilson-Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using methano I/water (0.05 % formic acid) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 2.8 mg of l-(6-chloro-5-methyl-l- phenyl- lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide. LC-MS (m/z) 437.1 (M+l).
Example 164 : (R)- l-(5-chloro-6-methyl- 1-p-tolyl- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

hydrochloride :

To 5-chloro-6-methyl-lH-benzo[d]imidazol-2(3H)-one (0.627 g, 3.43 mmol), phosphoryl trichloride (POCI3, 21.1 g, 0.137 mol) was added, the reaction mixture was heated at 1000C for 6 hours and subsequently cooled down to room temperature. The formed precipitate was filtered off and washed with toluene. The filtrate was concentrated in vacuo and collected. This gave 674 mg (83 % yield) of 2,5-dichloro-6-methyl-lH-benzo[d]imidazole hydrochloride and 2,6- dichloro-5-methyl-lH-benzo[d]imidazole hydrochloride. LC-MS (m/z) 200.9 (M+ 1).
(b) Preparation of the intermediary tautomeric compounds ethyl l-(5-chloro-6-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylate and ethyl l-(6-chloro-5-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylate :

2,5-dichloro-6-methyl-lH-benzo[d]imidazole hydrochloride and 2,6-dichloro-5-methyl-lH- benzo[d]imidazole hydrochloride (0.60 g, 2.526 mmol), 1,4-dioxane (10 mL), N,N- diisopropylethylamine (Ηunig's base, DIEA, 0.979 g, 7.578 mmol) and ethyl piperidine-4- carboxylate (0.477 g, 3.03 mmol) was subjected to microwave conditions for one hour at 19O0C. The reaction mixture was concentrated in vacuo and purified on column (silica gel,
dichloromethane/methanol 98:2) to give 0.498 g (61 % yield) of ethyl l-(5-chloro-6-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylate and ethyl l-(6-chloro-5-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylate. No effort was done to to determine the ratio of the tautomers. LC-MS (m/z) 321.9 (M+ 1 )
(c) Preparation of the intermediary intermediate compound ethyl l-(5-chloro-6-methyl-l-p-tolyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate:

Example 165: (R)- l-(6-chloro-5-methyl- 1-p-tolyl- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

Ethyl 1 -(β-chloro-S-methyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (4.5 mg, eluting second after (R)- l-(5-chloro-6-methyl- 1-p-tolyl- lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide as described in example 164d), ethanol (1 mL) and aqueous sodium hydroxide (1 mL, 1 N) was stirred at room temperature for two hours. Aqueous hydrochloric acid (0.5 mL, 2 N) was added and the reaction mixture concentrated in vacuo. The residue was mixed with N,N-dimethylformamide (3 mL), JV,iV-diisopropylethyl- amine (Ηunig's base, DIEA, 0.02 mL), 2-(7-aza-lH-benzotriazole-l-yl)-l, 1,3,3- tetramethyluronium hexafluorophosphate (ΗATU, 10 mg) and (R)-(+)-tetrahydro-3-furylamine p-toluenesulfonate salt (6.5 mg, CAS Registry Number: 111769-27-8). The reaction mixture was stirred at room temperature for one hour, concentrated in vacuo and purified on column (silica gel, dichloromethane/methanol, gradient elution using 2.5-5.0 % methanol) to give 3.3 mg of : (R)- 1 -(5-chloro-6-methyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide after further purification on preparative reverse-phase hplc
(performed on a Gilson-Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using methano I/water (0.05 % formic acid) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection). LC-MS (m/z) 452.9 (M+ 1).
Example 166: (R)- l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl- lH-benzo [d] imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide


A tautomeric mixture of ethyl l-(5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxylate and ethyl l-(6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (80.4 mg, 0.25 mmol), dimethylsulfoxide (0.5 mL), caesium carbonate (114 mg, 0.35 mmol), 4- iodobenzonitrile (115.5 mg, 0.5 mmol), 8-hydroxy-quinoline (7.3 mg, 0.05 mmol), polyethylene glycol 400 (100 mg, 0.25 mmol) and copper(I) oxide (3.6 mg, 0.025 mmol) was subjected to microwave conditions for one hour at 15O0C. The reaction mixture was poured out on water (5 mL) and loaded on a short column reverse-phase material (Silica gel 60, C18). The short column was washed with a mixture of acetonitrile and water (approximately 10 mL, 1 :3), and then eluted with acetonitrile. The elute was concentrated in vacuo and further purified on column (silica gel, iso-hexane/ethyl acetate, gradient elution using 25-40 % ethyl acetate), to give 23.2 mg (22 % yield ) of ethyl l-(5-chloro-l-(4-cyanophenyl)-6-methyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxylate, eluting ahead of ethyl l-(6-chloro-l-(4-cyanophenyl)-5-methyl-lH-benzo[d]- imidazol-2-yl)piperidine-4-carboxylate (22.1 mg, 21 % yield) on the column. LC-MS (m/z) 422.9 (M+l).
(b) Preparation of the intermediate compound l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid:

Ethyl 1 -(5-chloro- 1 -(4-cyanophenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxylate (23.2 mg), ethanol (2 mL) and aqueous sodium hydroxide (1 mL, 1 N) was stirred at 5O0C for one hour. The reaction mixture was neutralized with aqueous hydrochloric acid and concentrated in vacuo to give l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl-lH-
benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid in quantitative yield. LC-MS (m/z) 412.9 (M+l).
(c) l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxylic acid (11.6 mg, 0.0281 mmol), N,N-dimethylformamide (3.0 niL), JV,iV-diisopropyl- ethylamine (Ηunig's base, DIEA, 10.9 mg, 0.0843 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-
1,1,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 12.8 mmol, 0.034 mmol) and (R)-(+)- tetrahydro-3-furylamine p-toluenesulfonate salt (8.7 mg, 0.0337 mmol, CAS Registry Number: 111769-27-8). The reaction mixture was stirred at room temperature over night, concentrated in vacuo and purified on column (silica gel, dichloromethane/methanol, gradient elution using 5.0- 10 % methanol) to give 10.3 mg (76 % yield) of (R)-l-(l-(4-carbamoylphenyl)-5-chloro-6- methyl- 1 H-benzo [d]imidazo l-2-yl)-N-(tetrahydrofuran-3 -yl)piperidine-4-carboxamide . LC-MS (m/z) 481.9 (M+l).
Example 167: l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide

The title compound was prepared from l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (11.6 mg, 0.0281 mmol), N ,N- dimethylformamide (3.0 mL), JV,jV-diisopropyl-ethylamine (Ηunig's base, DIEA, 10.9 mg, 0.0843 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluoro- phosphate (ΗATU, 12.8 mmol, 0.034 mmol) and cyclopentanamine (2.9 mg, 0.0337 mmol) using the method described in Example 166 c). The reaction mixture was purified on column (silica gel, dichloromethane/methanol, gradient elution using 2.5-7.0 % methanol), to give 10.6 mg (79 % yield) of l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide. LC-MS (m/z) 480.0 (M+l). Example 168: l-(l-(4-carbamoylphenyl)-6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide

(a) Preparation of the intermediate compound l-(l-(4-carbamoylphenyl)-6-chloro-5-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid:

(b) The title compound was prepared from l-(l-(4-carbamoylphenyl)-6-chloro-5-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (11 mg, 0.0266 mmol), N5N- dimethylformamide (3.0 mL), JV,jV-diisopropyl-ethylamine (Ηunig's base, DIEA, 10.3 mg, 0.0799 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluoro- phosphate (ΗATU, 12.2 mg, 0.033 mmol) and cyclopentanamine (2.7 mg, 0.032 mmol) using the method described in Example 166 c). The reaction mixture was purified on column (silica gel, dichloromethane/methanol, gradient elution using 2.5-7.0 % methanol), to give 10.0 mg (78 % yield) of l-(l-(4-carbamoylphenyl)-6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide. LC-MS (m/z) 480.0 (M+l).
Example 169: (R)- l-(l-(4-carbamoylphenyl)-6-chloro-5-methyl- lH-benzo [d] imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(l-(4-carbamoylphenyl)-6-chloro-5-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (11 mg, 0.0266 mmol), N5N- dimethylformamide (3.0 mL), JV,JV-diisopropylethylamine (Ηunig's base, DIEA, 10.3 mg, 0.0799 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate
(ΗATU, 12.2 mmol, 0.033 mmol) and (R)-(+)-tetrahydro-3-furylamine p-toluenesulfonate salt (8.3 mg, 0.032 mmol, CAS Registry Number: 111769-27-8) using the method described in Example 166 c). The reaction mixture was purified on column chromatography (silica gel, dichloromethane/methanol, gradient elution using 5-10 % methanol), to give 10.9 mg (85 % yield) of (R)- 1 -( 1 -(4-carbamoylphenyl)-6-chloro-5-methyl- lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 481.9 (M+ 1).
Example 170 : (R)- l-(5-chloro-6-methyl- l-(4-(trifluoromethyl)phenyl)- lH-benzo [d] - imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide


This compound was prepared from a tautomeric mixture of ethyl l-(5-chloro-6-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylate and ethyl l-(6-chloro-5-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylate (80.4 mg, 0.25 mmol), dimethylsulfoxide (0.7
niL), caesium carbonate (114 mg, 0.35 mmol), l-iodo-4-(trifluoromethyl)benzene (136 mg, 0.5 mmol), 8-hydroxyquinoline (7.3 mg, 0.05 mmol), polyethylene glycol 400 (100 mg, 0.25 mmol) and copper(I) oxide (3.6 mg, 0.025 mmol) using the method described in Example 166. This gave, after column chromathography (silica gel), 30.4 mg (26 % yield) of ethyl l-(5-chloro-6- methyl- 1 -(4-(trifluoromethyl)-phenyl)- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (26.4 mg, 23 % yield) eluting ahead of ethyl l-(6-chloro-5-methyl-l-(4-(trifluoromethyl)phenyl)-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylate on the column. LC-MS (m/z) 465.9 (M+l). b) Preparation of the intermediate compound l-(5-chloro-6-methyl-l-(4-(trifluoromethyl)- phenyl)- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid:

Ethyl 1 -(5-chloro-6-methyl- 1 -(4-(trifluoromethyl)-phenyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylate (30.4 mg), ethanol (2 mL) and aqueous sodium hydroxide (0.5 mL, 1 N) was stirred at room temperature for one hour. The reaction mixture was neutralized with aqueous hydrochloric acid and concentrated in vacuo to give l-(l-(4-carbamoylphenyl)-5- chloro-6-methyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid in quantitative yield. LC-MS (m/z) 437.8 (M+l). c) The title compound was prepared from l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (9.4 mg, 0.021 mmol), N ,N- dimethylformamide (3.0 mL), JV,jV-diisopropylethylamine (Ηunig's base, DIEA, 8.3 mg, 0.064 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate
(ΗATU, 9.8 mg, 0.026 mmol) and (R)-(+)-tetrahydro-3-furylamine p-toluenesulfonate salt (6.7 mg, 0.026 mmol, CAS Registry Number: 111769-27-8) using the method described in Example 166 c). The reaction mixture was purified on column chromatography (silica gel,
dichloromethane/methanol, gradient elution using 2-10 % methanol), to give (R)-l-(5-chloro-6- methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]-imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide in quantitative yield. LC-MS (m/z) 506.9 (M+l).
Example 171: l-(5-chloro-6-methyl-l-(4-(trifluoromethyl)phenyl)-lH-benzo[d]imidazol-2- yl)-7V-cyclopentylpiperidine-4-carboxamide

The title compound was prepared from l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (9.4 mg, 0.0215 mmol), N5N- dimethylformamide (3.0 mL), JV,jV-diisopropyl-ethylamine (Ηunig's base, DIEA, 8.3 mg, 0.0644 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluoro-phosphate
(ΗATU, 9.79 mg, 0.026 mmol) and cyclopentanamine (2.2 mg, 0.0258 mmol) using the method described in Example 166 c). The reaction mixture was purified on column (silica gel, dichloromethane/methanol, gradient elution using 2-7 % methanol), to give l-(5-chloro-6- methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4- carboxamide in quantitative yield. LC-MS (m/z) 504.9 (M+ 1).
Example 172: l-(5-chloro-6-methyl-l-(4-(trifluoromethyl)phenyl)-lH-benzo[d]imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (9.4 mg, 0.021 mmol), N5N- dimethylformamide (3.0 mL), N,Λ/-diisopropylethylamine (Ηunig's base, DIEA, 8.3 mg, 0.064 mmol), 2-(7-aza- lH-benzotriazole- 1 -yl)- 1 , 1 ,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 9.8 mg, 0.026 mmol) and cyclopentanamine hydrochloride (3.2 mg, 0.026 mmol) using the method described in Example 166. The reaction mixture was purified on column
chromatography (silica gel, dichloromethane/methanol, gradient elution using 2 to 8 % methanol), to give l-(5-chloro-6-methyl-l-(4-(trifluoromethyl)phenyl)-lH-benzo[d]imidazol-2- yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide in quantitative yield. LC-MS (m/z) 506.9 (M+ 1).
Example 173 : (R)- l-(6-chloro-5-methyl- l-(4-(trifluoromethyl)phenyl)- lH-benzo [d] - imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide


Ethyl 1 -(6-chloro-5-methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)piperidine- 4-carboxylate (26.4 mg, eluting after of ethyl l-(5-chloro-6-methyl-l-(4-(trifluoromethyl)- phenyl)-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate as described in Example 170), ethanol (2 mL) and aqueous sodium hydroxide (0.5 mL, 1 N) was stirred at room temperature for one hour. The reaction mixture was neutralized with aqueous hydrochloric acid and concentrated in vacuo to give l-(6-chloro-5-methyl-l-(4-(trifluoromethyl)-phenyl)-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylic acid in quantitative yield. LC-MS (m/z) 437.8 (M+l).
(b) The title compound was prepared from l-(6-chloro-5 -methyl- l-(4-(trifluoromethyl)-phenyl)- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (8.3 mg, 0.019 mmol), N5N- dimethylformamide (3.0 mL), JV,JV-diisopropylethylamine (Ηunig's base, DIEA, 7.4 mg, 0.0569 mmol), 2-(7-aza- lH-benzotriazole- 1 -yl)- 1 , 1 ,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 8.6 mg, 0.023 mmol) and (R)-(+)-tetrahydro-3-furylamine p-toluenesulfonate salt (5.9 mg, 0.023 mmol, CAS Registry Number: 111769-27-8) using the method described in Example 166 c). The reaction mixture was purified on column chromatography (silica gel,
dichloromethane/methanol, gradient elution using 2-8 % methanol), to give (R)-l-(l-(4- carbamoylphenyl)-6-chloro-5 -methyl- 1 H-benzo [d]imidazo l-2-yl)-N-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide in quantitative yield. LC-MS (m/z) 506.9 (M+l).
Example 174: l-(6-chloro-5-methyl-l-(4-(trifluoromethyl)phenyl)-lH-benzo[d]imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(6-chloro-5-methyl-l-(4-(trifluoromethyl)-phenyl)-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (8.5 mg, 0.019 mmol), N5N- dimethylformamide (3.0 mL), N57V-diisopropylethylamine (Ηunig's base, DIEA, 7.5 mg, 0.058 mmol), 2-(7-aza- lH-benzotriazole- 1 -yl)- 1 , 1 ,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 8.8 mg, 0.023 mmol) and tetrahydrofuran-3 -amine hydrochloride (2.9 mg, 0.023 mmol, CAS Registry Number: 204512-94-7) using the method described in Example 166 c). The reaction mixture was purified on column chromatography (silica gel, dichloromethane/methanol, gradient elution using 2-8 % methanol), to give l-(6-chloro-5-methyl-l-(4- (trifluoromethyl)phenyl)-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide in quantitative yield. LC-MS (m/z) 506.9 (M+ 1).
Example 175: l-(6-chloro-5-methyl-l-(4-(trifluoromethyl)phenyl)-lH-benzo[d]imidazol-2- yl)-7V-cyclopentylpiperidine-4-carboxamide:

The title compound was prepared from l-(6-chloro-5-methyl-l-(4-(trifluoromethyl)-phenyl)-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (8.3 mg, 0.019 mmol), N5N- dimethylformamide (3.0 mL), N,7V-diisopropyl-ethylamine (Ηunig's base, DIEA, 7.4 mg, 0.057 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluoro-phosphate
(ΗATU, 8.6 mg, 0.023 mmol) and cyclopentanamine (1.9 mg, 0.0227 mmol) using the method described in Example 166. The reaction mixture was purified on column (silica gel,
dichloromethane/methanol, gradient elution using from 2 to 8 % methanol), to give l-(6-chloro-
5-methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4- carboxamide in quantitative yield. LC-MS (m/z) 504.9 (M+ 1).
Example 176 : (R)- l-(6-chloro-5-methyl- 1-phenyl- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

(a) Preparation of the intermediate compound 2,6-dichloro-5-methyl- 1-phenyl- IH- benzo[d]imidazole hydrochloride

6-Chloro-5-methyl-l -phenyl- lH-benzo[d]imidazol-2(3Η)-one (100 mg, 0.387 mmol) was stirred in phosphoryl trichloride (POCI3, 5.926 g) over night at relux, and concentrated in vacuo (co- evaporation with toluene), to give 2,6-dichloro-5-methyl-l -phenyl- lH-benzo[d]imidazo Ie hydrochloride in quantitative yield.
(b) Preparation of the intermediary compound ethyl l-(6-chloro-5-methyl- 1-phenyl- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxylate:

2,6-dichloro-5-methyl-l -phenyl- lH-benzo[d]imidazole hydrochloride (121.2 mg, 0.387 mmol), ethyl piperidine-4-carboxylate (303.8 mg, 0.2 mmol), toluene (2 mL) and N,N-dimethyl- formamide (0.5 mL) was subjected to microwave conditions at 2000C. After 30 minutes the reaction mixture was concentrated in vacuo, the residue purified on column (silica gel, iso- hexane/ethyl acetate, gradient elution from 10 to 100 % ethyl acetate) to give 27.5 mg (18 %
yield) of ethyl l-(6-chloro-5-methyl-l -phenyl- lH-benzo[d]-imidazol-2-yl)piperidine-4- carboxylate. LC-MS (m/z) 398.0 (M+l).
(c) Preparation of the intermediary compound l-(6-chloro-5-methyl-l -phenyl- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid:

A mixture of ethyl l-(6-chloro-5-methyl-l -phenyl- lH-benzo[d]-imidazol-2-yl)piperidine-4- carboxylate (17.5 mg, 0.044 mmol), sodium hydroxide (8.8 mg in 1 mL water), ethanol (1 mL) was stirred at 4OC for two hours. The reaction mixture was neutralized with hydrochloric acid (2 N) and concentrated in vacuo, to give a quantitative yield of l-(6-chloro-5-methyl-l -phenyl- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid. LC-MS (m/z) 369.9 (M+l).
(d) The title compound was prepared from l-(6-chloro-5 -methyl- 1 -phenyl- lH-benzo[d]imidazol- 2-yl)piperidine-4-carboxylic acid (9.3 mg, 0.025 mmol), N,N-dimethylformamide (3.0 mL), Λ/,Λ/-diisopropyl-ethylamine (Ηunig's base, DIEA, 6.5 mg, 0.050 mmol), 2-(7-aza-lH- benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluoro-phosphate (ΗATU, 9.6 mg, 0.025 mmol) and (R)-(+)-tetrahydro-3-furylamine p-toluenesulfonate salt (6.5 mg, 0.025 mmol, CAS Registry Number: 111769-27-8) using the method described in Example 166. The reaction mixture was purified on column (silica gel, dichloromethane/methanol, gradient elution using 0 to 8 % methanol), to give 7.3 mg (66 % yield) of (R)-I -(6-chloro-5-methyl-l -phenyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 439.0 (M+l). On measure on optical rotation this compound gave (+)-rotation. 1H NMR (400 MHz, CD3OD): δ 7.67-7.60 (m, 2H), 7.55-7.48 (m, 3H), 7.34 (s, IH), 7.01 (s, IH), 4.34-4.28 (m, IH), 3.91-3.72 (m, 3H), 3.65-3.52 (m, 3H), 2.90-2.81 (m, 2H), 2.40 (s, 3H), 2.36-2.27 (m, IH), 2.23- 2.13 (m, IH), 1.83-1.74 (m, IH), 1.70-1.60 (m, 4H).
Example 177: (S)-l-(6-chloro-5-methyl-l-phenyl-l//-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from l-(6-chloro-5 -methyl- 1 -phenyl- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylic acid (16.3 mg, 0.044 mmol), N,N-dimethylformamide (4.0 mL), N5N- diisopropyl-ethylamine (Ηunig's base, DIEA, 17.1 mg, 0.132 mmol), 2-(7-aza-lH-benzotriazole- l-yl)-l,l,3,3-tetramethyluronium hexafluoro -phosphate (ΗATU, 20.1 mg, 0.053 mmol) and (S)- tetrahydrofuran-3 -amine hydrochloride (6.5 mg, 0.0529 mmol) using the method described in Example 166 c). The reaction mixture was purified on column (silica gel,
dichloromethane/methanol, gradient elution using from 2 to 8 % methanol), to give 15.5 mg (81 % yield) of (S)- l-(6-chloro-5 -methyl- 1 -phenyl- lH-benzo[d]imidazo l-2-yl)-N-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide. On measure on optical rotation this compound gave (-)-rotation. LC-MS (mix) 438.9 (M+ 1). 1H NMR (400 MHz, CD3OD): δ 7.67-7.60 (m, 2H), 7.55-7.48 (m, 3H), 7.34 (s, IH), 7.01 (s, IH), 4.34-4.28 (m, IH), 3.91-3.72 (m, 3H), 3.65-3.52 (m, 3H), 2.90- 2.81 (m, 2H), 2.40 (s, 3H), 2.36-2.27 (m, IH), 2.23-2.13 (m, IH), 1.83-1.74 (m, IH), 1.70-1.60 (m, 4H). Example 178: 7V-butyl-l-(5-chloro-6-methyl-l-phenyl-lH-benzo[d]imidazol-2-yl)piperidine- 4-carboxamide and 7V-butyl-l-(6-chloro-5-methyl-l-phenyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide

(a) Preparation of the intermediary tautomeric compounds 2,5-dichloro-6-methyl-lH- benzo[d]imidazole and 2,6-dichloro-5-methyl- lH-benzo[d]imidazole:

To 5-chloro-6-methyl-lH-benzo[d]imidazol-2(3H)-one (1.0 g, 5.48 mmol), phosphoryl trichloride (POCl3, 0.137 mol) was added, the reaction mixture was heated at 12O0C for 3 hours
and subsequently cooled down to room temperature. The reaction mixture was quenched with ice (without evaporating POCI3) and neutralized with aqueous sodium hydroxide (10%) to pH 7. The solid thus obtained was filtered off and the residue was recrystallized from petroleum ether to give 850 mg (86 % yield) of 2,5-dichloro-6-methyl-lH-benzo[d]imidazole and 2,6-dichloro-5- methyl- 1 H-benzo [d]imidazo Ie.
(b) Preparation of the intermediary tautomeric compounds N-butyl- l-(5-chloro-6-methyl- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide and N-butyl- 1 -(6-chloro-5-methyl- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide:

(c) The title compounds was prepared from N-butyl- l-(5-chloro-6-methyl-lH-benzo[d]imidazol- 2-yl)piperidine-4-carboxamide and N-butyl- 1 -(6-chloro-5-methyl- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide (0.30 g, 0.87 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.48 mmol), iodobenzene (3.48 mmol), 4,7-dimethoxy-l,10-phenanthroline (0.130 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0435 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 10 mg (2.7 % yield) of N-butyl- l-(5-chloro-6-methyl-l- phenyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide and N-butyl- 1 -(6-chloro-5-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as an off- white solid. LC-MS (m/z) 425.3 (M+l).
Example 179: l-(5-chloro-6-methyl-l-phenyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide and l-(6-chloro-5-methyl-l-phenyl-lH- benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide


2,5-dichloro-6-methyl- lH-benzo[d]imidazole and 2,6-dichloro-5-methyl- lH-benzo[d]imidazole (1 g, 4.98 mmol), 1,4-dioxane (10 mL), JV,JV-diisopropylethylamine (Ηunig's base, DIEA, 14.9 mmol) and 4-(tetrahydrofuran-3-ylcarbamoyl)piperidinium 2,2,2-trifluoroacetate (7.47 mmol) was heated at 18O0C for 4 hours. The reaction mixture was concentrated in vacuo and
recrystallized from petrolium ether to give 1.1 g (65 % yield) l-(5-chloro-6-methyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide and 1 -(6-chloro-5- methyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. No effort was done in order to determine the ratios of the tautomers.
(b) The title compounds was prepared from l-(5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide and 1 -(6-chloro-5-methyl- lH-benzo[d]imidazol- 2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.30 g, 0.83 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.32 mmol), iodobenzene (3.32 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.124 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0415 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 22 mg (6 % yield) of 1 -(5-chloro-6-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine- 4-carboxamide and l-(6-chloro-5-methyl-l -phenyl- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid. No effort was done in order to determine the ratios of the compounds. LC-MS (m/z) 439.1 (M+ 1).
Example 180: l-(5-chloro-l-cyclobutyl-6-methyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide compound and l-(6-chloro-l-cyclobutyl- 5-methyl- lH-benzo [d] imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

(PdCl2(dppf), 0.028 mmol) and N,Λ/-dimethylformamide (3 mL) was subjected to microwave conditions at 12O0C for one hour. The reaction mixture was purified with column
chromathography, to give 12 mg (10 % yield) of l-^-chloro-l-cyclobutyl-β-methyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide and l-(6-chloro-l- cyclobutyl-5-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a brown solid. No effort was done in order to determine the ratios of the compounds. LC-MS (m/z) All .1 (M+l).
Example 181: l-(5-chloro- l-(cyclobutylmethyl)-6-methyl- lH-benzo [d] imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide and l-(6-chloro-l-(cyclobutylmethyl)-5-methyl-lH- benzo [d] imidazol-2-yl)-7V-cyclopentylpiperidine-4-carboxamide


(b) The title compounds were prepared from a tautomeric mixture of l-(5-chloro-6-methyl-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide and l-(6-chloro-5-methyl-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide (0.1 g, 0.28 mmol),
bromocyclobutane (1.12 mmol), caesium carbonate (1.12 mmol), [l,l'-bis(diphenylphosphino)- ferrocene] dichloropalladium(II) (PdCl2(dppf), 0.028 mmol) and N,Λ/-dimethylformamide (3 mL) using the method described in Example 180. This gave 20 mg (17 % yield) of l-(5-chloro-l- (cyclobutylmethyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4- carboxamide and 1 -(6-chloro- 1 -(eye Io butylmethyl)-5 -methyl- 1 H-benzo [d]imidazo l-2-yl)-N- cyclopentylpiperidine-4-carboxamide as a brown solid. No effort was done in order to determine the regioisomeric ratio of the compounds. LC-MS (m/z) 429.1 (M+l).
Example 182: l-(5-chloro-6-methyl-l-pentyl-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide and l-(6-chloro-5-methyl-l-pentyl-lH- benzo [d] imidazol-2-yl)-7V-cyclopentylpiperidine-4-carboxamide

The title compounds were prepared from 1 -(5 -chloro-6-methyl-l H-benzo [d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro-5 -methyl- 1 H-benzo [d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide (0.1 g, 0.28 mmol), 1-bromopentane (1.12 mmol), caesium carbonate (1.12 mmol), [l,l'-bis(diphenylphosphino)ferrocene] dichloropalladium(II)
(PdCl2(dppf), 0.028 mmol) and N,Λ/-dimethylformamide (3 mL) using the method described in Example 180. This gave 20 mg (17 % yield) of l-(5-chloro-6-methyl-l-pentyl-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro-5-methyl- 1 -
pentyl-lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as an off-white solid. No effort was done in order to determine the ratios of the compounds. LC-MS (m/z) 431.3 (M+l).
Example 183 : l-(5-chloro- l-(4-fluorophenyl)-6-methyl- lH-benzo [d] imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide and l-(6-chloro-l-(4-fluorophenyl)-5-methyl-lH- benzo [d] imidazol-2-yl)-7V-cyclopentylpiperidine-4-carboxamide

The title compounds was prepared from l-(5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro-5 -methyl- lH-benzo [d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide (0.30 g, 0.83 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.32 mmol), l-fluoro-4-iodobenzene (3.32 mmol), 4,7-dimethoxy-l,10- phenanthroline (0.124 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0415 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 11 mg (2.9 % yield) of 1 -(5-chloro- 1 -(4-fluorophenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4- carboxamide and 1 -(6-chloro- 1 -(4-fluorophenyl)-5 -methyl- lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide as a brown solid. No effort was done in order to determine the regioisomeric ratio of the compounds.
Example 184: l-(5-chloro-6-methyl-l-phenyl-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide and l-(6-chloro-5-methyl-l-phenyl-lH- benzo [d] imidazol-2-yl)-7V-cyclopentylpiperidine-4-carboxamide

The title compounds was prepared from l-(5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro-5 -methyl- lH-benzo [d]imidazol-2-yl)-N-
cyclopentylpiperidine-4-carboxamide (0.30 g, 0.83 mmol), dimethylsulfoxide (6 niL), caesium carbonate (3.32 mmol), iodobenzene (3.32 mmol), 4,7-dimethoxy-l,10-phenanthroline (0.124 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0415 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 5 mg (1.4 % yield) of l-(5-chloro-6-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro-5- methyl-1 -phenyl- lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as a white solid. No effort was done in order to determine the ratios of the compounds. LC-MS (m/z) 437.7 (M+l). Example 185: 7V-butyl-l-(5-chloro-l-cyclobutyl-6-methyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide and 7V-butyl-l-(6-chloro-l-cyclobutyl-5-methyl-lH- benzo [d] imidazol-2-yl)piperidine-4-carboxamide

The title compounds were prepared from N-butyl- 1 -(5 -chloro-6-methyl-lH-benzo[d]imidazo 1-2- yl)piperidine-4-carboxamide and N-butyl- 1 -(6-chloro-5 -methyl- lH-benzo [d]imidazo 1-2- yl)piperidine-4-carboxamide (0.1 g, 0.28 mmol), bromocyclobutane (1.12 mmol), caesium carbonate (1.12 mmol), [l,l'-bis(diphenylphosphino)ferrocene] dichloropalladium(II)
(PdCl2(dppf), 0.028 mmol) and N,Λ/-dimethylformamide (3 mL) using the method described in Example 180. This gave 15 mg (13 % yield) of N-butyl-l-(5-chloro-l-cyclobutyl-6-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxamide and N-butyl- 1 -(6-chloro- 1 -cyclobutyl-5- methyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide as a white solid. No effort was done in order to determine the ratios of the compounds. LC-MS (m/z) 403.3 (M+l).
Example 186: (R)- l-(5,6-dimethyl- l-(3-(trifluoromethyl)phenyl)- lH-benzo [d] imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide


The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (150 mg, 0.438 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.657 mmol), 5-bromo-2,2-difluorobenzo[d][l,3]dioxole (1.31 mmol), 8-hydroxyquinoline (0.175 mmol), polyethylene glycol (0.219 mmol) and copper(I) oxide (0.0876 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 15O0C and subsequently purified on reverse-phase hplc, to give 10 mg (4.6 % yield) of (R)-l-(l-(2,2-difiuorobenzo[d][l,3]dioxol-5-yl)-5,6- dimethyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 499.2 (M+l).
Example 188: (R)- l-(5,6-dimethyl- l-(5-(trifluoromethyl)pyridin-2-yl)- IH- benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (150 mg, 0.438 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.657 mmol), 2-bromo-5-(trifluoromethyl)pyridine (1.31 mmol), 8- hydroxyquinoline (0.175 mmol), polyethylene glycol (0.219 mmol) and copper(I) oxide (0.0876 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 15O0C and subsequently purified on reverse-phase hplc, to give 37 mg (17 % yield) of (R)-l-(5,6-dimethyl-l-(5-(trifluoromethyl)pyridin-2-yl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 488.1 (M+l).
Example 189: (R)-l-(l-(3-ethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (150 mg, 0.438 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.657 mmol), l-bromo-3-ethylbenzene (1.31 mmol), 8- hydroxyquinoline (0.175 mmol), polyethylene glycol (0.219 mmol) and copper(I) oxide (0.0876 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on reverse-phase hplc, to give 15 mg (7.6 % yield) of (R)-l-(l-(3-ethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale green solid. LC-MS (m/z) 447.2 (M+l).
Example 190: (R)-l-(l-(3-isopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (150 mg, 0.438 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.657 mmol), l-bromo-3-isopropylbenzene (1.31 mmol), 8- hydroxyquinoline (0.175 mmol), polyethylene glycol (0.219 mmol) and copper(I) oxide (0.0876 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on reverse-phase hplc, to give 5 mg (2.5 % yield) of (R)-l-(l-(3-isopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol- 2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 461.4 (M+l).
Example 191: (R)- l-(6-chloro- l-(4-isopropylphenyl)-5-methyl- lH-benzo [d] imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-4-isopropylbenzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on reverse-phase hplc, to give 6 mg (5 % yield) of (R)-l-(l-(4-isopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 501.5 (M+l).
Example 192 : (R)- l-(5,6-dichloro- l-(3-chlorophenyl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-3-chlorobenzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 2 mg (2 % yield) of (R)-l-(5,6-dichloro-l-(3-chlorophenyl)-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 493.1 (M+l).
Example 193 : (R)- l-(5,6-dichloro- l-(4-fluoro-3-methylphenyl)- lH-benzo [d] imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 4-bromo-l-fluoro-2-methylbenzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on reverse-phase hplc, to give 15 mg (12 % yield) of (R)-l-(5,6-dichloro-l-(4-fiuoro-3-methylphenyl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale green solid. LC-MS (m/z) 491.1 (M+l).
Example 194 : (R)- l-(5,6-dichloro- l-(3,5-difluorophenyl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-3,5-difluorobenzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 3 mg (2.3 % yield) of (R)-l-(5,6-dichloro-l-(3,5-difluorophenyl)-lH-benzo[d]imidazol-2- yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 495.4 (M+l).
Example 195: (R)- l-(5,6-dichloro- l-(3-cyanophenyl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 3-bromobenzonitrile (0.783 mmol), 8 -hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 5 mg (3.9 % yield) of (R)- 1 -(5,6-dichloro- 1 -(3-cyanophenyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 484.1 (M+l).
Example 196: (R)- l-(5,6-dichloro- l-(pyridin-2-yl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 2-iodopyridine (0.783 mmol), 8-hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 30 mg (25 % yield) of (R)- 1 -(5,6-dichloro- 1 -(pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 460.3 (M+l). Example 197: (RJ-l-CS^-dichloro-l-CS-methylpyridin-l-yO-lH-benzoIdlimidazol-l-yO-TV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 2-bromo-5-methylpyridine (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 13 mg (11 % yield) of (R)-l-(5,6-dichloro-l-(5-methylpyridin-2-yl)-lH-benzo[d]imidazol- 2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 474.4 (M+l).
Example 198: (R)- l-(5,6-dichloro- l-(6-methoxypyridin-2-yl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 2-bromo-6-methoxypyridine (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 3 mg (2.3 % yield) of (R)-l-(5,6-dichloro-l-(6-methoxypyridin-2-yl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 490.4 (M+ 1).
Example 199: (R)- l-(5,6-dichloro- l-(3-isopropylphenyl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-3-isopropylbenzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 5 mg (3.8 % yield) of (R)-l-(5,6-dichloro-l-(3-isopropylphenyl)-lH-benzo[d]imidazol-2- yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off- white solid. LC-MS (m/z) 501.2 (M+l).
Example 200 : (R)- l-(5,6-dichloro- l-(4-methylpyridin-2-yl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 2-bromo-4-methylpyridine (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 5 mg (3.9 % yield) of (R)-l-(5,6-dichloro-l-(4-methylpyridin-2-yl)-lH-benzo[d]imidazol- 2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 474.2 (M+l).
Example 201: (R)- l-(5,6-dichloro- l-(6-methylpyridin-2-yl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 2-bromo-6-methylpyridine (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 25 mg (20 % yield) of (R)-l-(5,6-dichloro-l-(6-methylpyridin-2-yl)-lH-benzo[d]imidazol- 2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 474.2 (M+l).
Example 202 : (R)- l-(5,6-dichloro- l-(3-(difluoromethoxy)phenyl)- lH-benzo [d] imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-3-(difluoromethoxy)benzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 5 mg (3.6 % yield) of (R)-l-(5,6-dichloro-l-(3-(difluoromethoxy)phenyl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a gray solid. LC- MS (m/z) 525.2 (M+ 1).
Example 203 : (R)- l-(5,6-dichloro- l-(3-(trifluoromethoxy)phenyl)- lH-benzo [d] imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-3-(trifluoromethoxy)benzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 9 mg (6.4 % yield) of (R)-l-(5,6-dichloro-l-(3-(trifiuoromethoxy)phenyl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 543.0 (M+l).
Example 204: (R)-l-(5,6-dichloro-l-(4-isobutylphenyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-4-isobutylbenzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase hplc, to give 10 mg (7.4 % yield) of (R)-l-(5,6-dichloro-l-(4-isobutylphenyl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 515.5 (M+l).
Example 205 : (R)- l-(5,6-dichloro- l-(3,4-dimethylphenyl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (120 mg, 0.313 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.470 mmol), l-bromo-3-(cyclopentyloxy)benzene (0.939 mmol), 8- hydroxyquinoline (0.125 mmol), polyethylene glycol (0.157 mmol) and copper(I) oxide (0.0626 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 5 mg (3.2 % yield) of (R)-l-(5,6-dichloro-l-(3,4-dimethylphenyl)-lH-benzo[d]imidazol-2- yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale green solid. LC-MS (m/z) 487.2 (M+l).
Example 206 : (R)- l-(5,6-dichloro- l-(4-chlorophenyl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (200 mg, 0.522 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.783 mmol), l-chloro-4-iodobenzene (1.565 mmol), 8- hydroxyquinoline (0.209 mmol), polyethylene glycol (0.261 mmol) and copper(I) oxide (0.104 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 9 mg (3.6 % yield) of (R)-l-(5,6-dichloro-l-(4-chlorophenyl)-lH-benzo[d]imidazol-2-yl)- N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 493.5 (M+l).
Example 207: (R)-l-(5,6-dichloro-l-(5-(trifluoromethyl)pyridin-2-yl)-lH-benzo[d]imidazol- 2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (150 mg, 0.391 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.587 mmol), 2-bromo-5-(trifluoromethyl)pyridine (1.17 mmol), 8- hydroxyquinoline (0.157 mmol), polyethylene glycol (0.196 mmol) and copper(I) oxide (0.0783 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 5 mg (2.5 % yield) of (R)-l-(5,6-dichloro-l-(5-(trifluoromethyl)pyridin-2-yl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale green solid. LC-MS (m/z) 528.3 (M+l).
Example 208 : (R)- l-(5,6-dichloro- l-(5-fluoropyridin-2-yl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (150 mg, 0.391 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.587 mmol), 2-bromo-5-fluoropyridine (1.17 mmol), 8-hydroxy- quinoline (0.157 mmol), polyethylene glycol (0.196 mmol) and copper(I) oxide (0.0783 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 35 mg (19 % yield) of (R)-l-(5,6-dichloro-l-(5-fluoropyridin-2-yl)-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 478.2 (M+l).
Example 209: (R)-l-(l-(4-isobutylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide



A tautomeric mixture of ethyl l-(5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxylate and ethyl l-(6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (80 mg, 0.249 mmol), dimethylsulfoxide (0.5 mL), caesium carbonate (113 mg, 0.348 mmol), 1- bromo-3,5-dimethylbenzene (138 mg, 0.746 mmol), 8-hydroxy-quinoline (14.4 mg, 0.099 mmol), polyethylene glycol 400 (50.0 mg, 0.124 mmol) and copper(I) oxide (7.1 mg, 0.05 mmol) was heated at 15O0C for 4 hours. The reaction mixture was filtered, washed with N ,N- dimethylformamide, the filtrated concentrated in vacuo (co-evaporation with N5N- dimethylformamide) and purified on column (silica gel, iso-hexane/ethyl acetate, gradient elution from 20 to 30 % ethyl acetate). This gave 16.3 mg (15 % yield) of ethyl l-(6-chloro-l- (3,5-dimethylphenyl)-5-methyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (LC-MS (m/z) 426.0 (M+ 1)) that eluted as the second peak after ethyl l-(5-chloro-l-(3,5- dimethylphenyl)-6-methyl-lH-benzo[d]-imidazol-2-yl)piperidine-4-carboxylate (18.0 mg, 17 % yield). (b) To ethyl l-(6-chloro-l-(3,5-dimethylphenyl)-5-methyl-lH-benzo[d]imidazol-2-yl)piperidine- 4-carboxylate (8.6 mg) was added ethanol (2 mL) and aqueous sodium hydroxide (1 mL, 2 N). The reaction mixture was stirred at 4O0C for two hours, neutralized with aqueous hydrochloric acid (2 N) and concentrated in vacuo. The residue was mixed (R)-(+)-tetrahydrofuran-3 -amine A- methylbenzenesulfonate (6.3 mg, 0.024 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l, 1,3,3-
tetramethyluronium hexafluorophosphate (HATU, 9.2 mg, 0.024 mmol), 7V,iV-diisopropyl- ethylamine (Hunig's base, DIEA, 7.8 mg, 0.060 mmol) and N,N-dimethylformamide (5 mL) was stirred at room temperature over the weekend. The reaction mixure was concentrated in vacuo, the residue subjected to preperative hplc (Preparative RP-LC was performed on a Gilson system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using methano I/water (0.05% formic acid) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 7.7 mg (82 % yield) (R)-l-(6-chloro-l-(3,5-dimethylphenyl)-5-methyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. MS (m/z) 467.0 (M+l). Example 211: (R)-l-(l-(4-chloro-3-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (120 mg, 0.350 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.701 mmol), 4-bromo-l-chloro-2-fluorobenzene (1.051 mmol), 8- hydroxyquinoline (0.140 mmol), polyethylene glycol (0.175 mmol) and copper(I) oxide (0.0701 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase hplc, to give 3 mg (1.8 % yield) of (R)-l-(l-(3-fluoro-5-methylphenyl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 471.2 (M+l).
Example 212: (R)-l-(5,6-dichloro-l-(4-chloro-3-fluorophenyl)-lH-benzo[d]imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide


The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (200 mg, 0.522 mmol) dimethylsulfoxide (2 mL), caesium carbonate (1.04 mmol), l-bromo-3,5-diethylbenzene (1.57 mmol), 8- hydroxyquinoline (0.209 mmol), polyethylene glycol (0.261 mmol) and copper(I) oxide (0.104 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase hplc, to give 5 mg (1.9 % yield) of (R)-l-(5,6-dichloro-l-(3,5-diethylphenyl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid. LC- MS (m/z) 515.5 (M+l).
Example 214: (R)-l-(l-(3-tert-butylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide


The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-3-tert-butylbenzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase hplc, to give 5 mg (3.7 % yield) of (R)-I -(I -(3-tert-butylphenyl)-5,6-dichloro- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off- white solid. LC-MS (m/z) 515.5 (M+l).
Example 216: (R)- l-(5,6-dichloro- 1-phenyl- lH-benzo [d] imidazol-2-yl)-7V-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide

Example 217: (R)- l-(5,6-dichloro- l-(4-ethylphenyl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-4-ethylbenzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 34 mg (27 % yield) of (R)-l-(5,6-dichloro-l-(4-ethylphenyl)-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale green solid. LC-MS (m/z) 487.1 (M+l).
Example 218: (R)- l-(5,6-dichloro- 1-m-tolyl- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide


The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-3,5-dimethylbenzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 5 mg (4 % yield) of (R)-l-(5,6-dichloro-l-(3,5-dimethylphenyl)-lH-benzo[d]imidazol-2- yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a yellow solid. LC-MS (m/z) 487.1 (M+l).
Example 220: (R)-l-(5,6-dichloro-l-(3-ethylphenyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

Example 221 : (R)- l-(5,6-dichloro- l-(6-(trifluoromethyl)pyridin-2-yl)- lH-benzo [d] imidazol- 2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 2-bromo-6-(trifluoromethyl)pyridine (0.783 mmol), 8- hydroxy-quinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 25 mg (18 % yield) of (R)-l-(5,6-dichloro-l-(6-(trifluoromethyl)pyridin-2-yl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid. LC- MS (m/z) 528.4 (M+l). 1H NMR (400 MHz, CDCl3): δ 8.11-8.06 (m, IH), 7.98-7.93 (m,
1H),7.74 (s, IH), 7.71-7.68 (m, IH), 7.61 (s, IH), 5.77 (br s, IH), 4.48-4.40 (m, IH), 3.90-3.82 (m, IH), 3.77-3.68 (m, 2H), 3.65-3.55 (m, 3H), 3.07-2.91 (m, 2H), 2.26-2.16 (m, 2H), 1.86-1.77 (m, 6H).
Example 222: (R)-l-(5,6-dichloro-l-(3-ethoxyphenyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-3-ethoxybenzene (0.783 mmol, CAS Registry Number: 2655-84-7), 8-hydroxy-quinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 5 mg (3.8 % yield) of (R)-l-(5,6-dichloro-l-(3-ethoxyphenyl)-lH- benzo[d]-imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 503.2 (M+ 1).
Example 223 : (R)- l-(5,6-dichloro- l-(3-isopropoxyphenyl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-3-isopropoxybenzene (0.783 mmol, CAS Registry Number: 131738-73-3), 8-hydroxy-quinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 2 mg (1.2 % yield) of (R)-l-(5,6-dichloro-l-(3- isopropoxyphenyl)-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a pale green solid. LC-MS (m/z) 517.2 (M+l).
Example 224 : (R)- l-(5,6-dichloro- l-(3-propoxyphenyl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-3-propoxybenzene (0.783 mmol, CAS Registry Number: 149557-17-5), 8-hydroxy-quinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 14 mg (10 % yield) of (R)-l-(5,6-dichloro-l-(3-propoxyphenyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 517.5 (M+l).
Example 225: (R)-l-(l-(4-fluoro-3,5-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (100 mg, 0.292 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.584 mmol), 5-bromo-2-fluoro-l,3-dimethylbenzene (0.876 mmol), 8- hydroxyquinoline (0.117 mmol), polyethylene glycol (0.146 mmol) and copper(I) oxide (0.0584 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase hplc, to give 17 mg (13 % yield) of (R)-I -(I -(4-fiuoro-3,5-dimethylphenyl)-5,6-dimethyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 465.2 (M+l).
Example 226: (R)-l-(5,6-dichloro-l-(4-chloro-3-methylphenyl)-lH-benzo[d]imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 4-bromo-l-chloro-2-methylbenzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc (Shimadzu LC8A, C18 column (22 x 250 mm, lOμm), acetonitrile/water gradient (0.1% trifluoroacetic acid), flowrate 15 mL/min) , to give 15 mg (11 % yield) of (R)-l-(5,6-dichloro-l- (4-chloro-3-methylphenyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a pale brown solid. LC-MS (m/z) 507.8 (M+ 1).
Example 227: (R)-l-(5,6-dichloro-l-(3-chloro-4-methylphenyl)-lH-benzo[d]imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide


The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (100 mg, 0.292 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.438 mmol), iodobenzene (0.876 mmol), 8-hydroxyquinoline (0.117 mmol), polyethylene glycol (0.146 mmol) and copper(I) oxide (0.0584 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on reverse-phase hplc, to give 34 mg (28 % yield) of (R)- 1 -(5 ,6-dimethyl- 1 -phenyl- 1 H-benzo [d]imidazol-2-yl)-N-(tetrahydrofuran-3 -yl)piperidine-4- carboxamide as a pale yellow solid. LC-MS (m/z) 419.1 (M+ 1).
Example 229 : (R)- l-(5,6-dimethyl- 1-p-tolyl- lH-benzo [d] imidazol-2-yl)-7V-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide


The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (100 mg, 0.292 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.438 mmol), l-bromo-3-methylbenzene (0.876 mmol), 8- hydroxyquinoline (0.117 mmol), polyethylene glycol (0.146 mmol) and copper(I) oxide (0.0584 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on reverse-phase hplc, to give 4 mg (2.6 % yield) of (R)-l-(5,6-dimethyl-l-m-tolyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 433.3 (M+l).
Example 231: (R)-l-(l-(3-cyanophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (100 mg, 0.292 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.438 mmol), 3-bromobenzonitrile (0.876 mmol), 8-hydroxyquinoline (0.117 mmol), polyethylene glycol (0.146 mmol) and copper(I) oxide (0.0584 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on reverse-phase hplc, to give 8 mg (4.6 % yield) of (R)- 1 -( 1 -(3 -cyanophenyl)-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)-N-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide as a pale green solid. LC-MS (m/z) 444.1 (M+l).
Example 232 : (R)- l-(5,6-dimethyl- l-(naphthalen-2-yl)- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (100 mg, 0.292 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.438 mmol), 2-bromonaphthalene (0.876 mmol), 8-hydroxyquinoline (0.117 mmol), polyethylene glycol (0.146 mmol) and copper(I) oxide (0.0584 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on reverse-phase hplc, to give 3 mg (2.2 % yield) of (R)- 1 -(5,6-dimethyl- 1 -(naphthalen-2-yl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 469.1 (M+l).
Example 233 : (R)- l-(l-(4-chloro-3-methylphenyl)-5,6-dimethyl- lH-benzo [d] imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (100 mg, 0.292 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.438 mmol), 4-bromo-l-chloro-2-methylbenzene (0.876 mmol), 8- hydroxyquinoline (0.117 mmol), polyethylene glycol (0.146 mmol) and copper(I) oxide (0.0584 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on reverse-phase hplc, to give 35 mg (26 % yield) of (R)-I -(I -(4-chloro-3-methylphenyl)-5,6-dimethyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 467.2 (M+l).
Example 234 : (R)- l-(l-(3-chloro-4-methylphenyl)-5,6-dimethyl- lH-benzo [d] imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (100 mg, 0.292 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.438 mmol), 4-bromo-2-chloro-l-methylbenzene (0.876 mmol), 8- hydroxyquinoline (0.117 mmol), polyethylene glycol (0.146 mmol) and copper(I) oxide (0.0584 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on reverse-phase hplc, to give 20 mg (15 % yield) of (R)-l-(l-(3-chloro-4-methylphenyl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale green solid. LC-MS (m/z) 467.2 (M+l).
Example 235 : (R)- l-(l-(benzo [d] thiazol-6-yl)-5,6-dimethyl- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (120 mg, 0.350 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.526 mmol), 6-bromobenzo[d]thiazole (1.051 mmol), 8- hydroxyquinoline (0.140 mmol), polyethylene glycol (0.175 mmol) and copper(I) oxide (0.0701 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on reverse-phase hplc, to give 3 mg (1.8 % yield) of (R)-l-(l-(benzo[d]thiazol-6-yl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 476.2 (M+l).
Example 236 : (R)- l-(l-(benzo [d] thiazol-5-yl)-5,6-dimethyl- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (120 mg, 0.350 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.526 mmol), 5-bromobenzo[d]thiazole (1.051 mmol), 8- hydroxyquinoline (0.140 mmol), polyethylene glycol (0.175 mmol) and copper(I) oxide (0.0701 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 12O0C and subsequently purified on reverse-phase hplc, to give 5 mg (3 % yield) of (R)-l-(l-(benzo[d]thiazol-5-yl)-5,6-dimethyl-lH-benzo[d]imidazol- 2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale green solid. LC-MS (m/z) 476.1 (M+l).
Example 237: (R)-l-(5-chloro-l-(3,4-dimethylphenyl)-6-methyl-lH-benzo[d]imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

(a) Preparation of the intermediate tautomeric mixture l-(5-chloro-6-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid and l-(6-chloro-5-methyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid

temperature two hours, concentrated in vacuo and pΗ adjusted to 4-5 with addition of aqueous hydrochloric acid (6 N). The formed precipitate was filtered off, the aqueous mother liquid extracted three times with dichloro methane (50 mL) and the combined organic layers were dried with MgSO4 and concentrated in vacuo. The precipitate and the residue from the extraction were combined and rechrystallized in petroleum ether to give 0.82 g (90%) of a tautomeric mixture of l-(5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid and l-(6-chloro-5- methyl- 1 H-benzo [d]imidazo l-2-yl)piperidine-4-carboxylic.
(b) Preparation of the intermediate tautomeric mixture of (R)-I -(5-chloro-6-methyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide and (R)- 1 -(6-chloro- 5-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide:

A tautomeric mixture of 1 -(5 -chloro-6-methyl-l H-benzo [d]imidazol-2-yl)piperidine-4- carboxylic acid and l-(6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (1.0 g, 3.4 mmol), (R)-(+)-tetrahydrofuran-3 -amine 4-methylbenzenesulfonate (4.1 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 6.8 mmol), Λ/,Λ/-diisopropylethylamine (Ηunig's base, DIEA, 6.8 mmol) and N5N- dimethylformamide (25 mL) was stirred at room temperature for 1 hour. After the appropriate work-up, the residue was recrystallized from dichloromethane/petroleum ether to give 1.0 g (83 % yield) of a tautomeric mixture of (R)-I -(5-chloro-6-methyl- lH-benzo[d]imidazo l-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide and (R)-l-(6-chloro-5-methyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide.
(c) The title compound was prepared from a tautomeric mixture of (R)-I -(5-chloro-6-methyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide and (R)- 1 -(6-chloro- 5-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide (500 mg, 1.38 mmol) dimethylsulfoxide (2 mL), caesium carbonate (2.07 mmol), 4-bromo-l,2- dimethylbenzene (4.13 mmol), 8-hydroxyquinoline (0.551 mmol), polyethylene glycol (0.689
mmol) and copper(I) oxide (0.276 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 15 mg (2.3 % yield) of (R)-l-(5-chloro-l-(3,4- dimethylphenyl)-6-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as an off-white solid, eluting after (R)-l-(6-chloro-l-(3,4-dimethylphenyl)-5- methyl- lH-benzo[d]-imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide on the column. LC-MS (m/z) 467.2 (M+l).
Example 238: (RJ-l-Cό-chloro-l-CS^-dimethylphenylJ-S-methyl-lH-benzoIdlimidazol-l-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was obtained as described in Example 237 eluting ahead of (R)-l-(5-chloro- l-(3,4-dimethylphenyl)-6-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide on the column. This gave 16 mg (2.5 % yield) of (R)-I -(6-chloro-l- (3,4-dimethylphenyl)-5-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine- 4-carboxamide as an off-white solid. MS (m/z) 467.2 (M+l).
Example 239: (R)-l-(5-chloro-l-(4-isopropylphenyl)-6-methyl-lH-benzo[d]imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from a tautomeric mixture of (R)-I -(5-chloro-6-methyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide and (R)-l-(6-chloro- 5-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide (500 mg, 1.38 mmol) dimethylsulfoxide (2 mL), caesium carbonate (2.07 mmol), l-bromo-4- isopropylbenzene (4.13 mmol), 8-hydroxyquinoline (0.551 mmol), polyethylene glycol (0.689
mmol) and copper(I) oxide (0.276 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 27 mg (4.3 % yield) of (R)-l-(5-chloro-l-(4- isopropylphenyl)-6-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a white solid, eluting after (R)-l-(6-chloro-l-(4-isopropylphenyl)-5-methyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide on the column. LC- MS (m/z) 481.2 (M+l).
Example 240 : (R)- l-(6-chloro- l-(4-isopropylphenyl)-5-methyl- lH-benzo [d] imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was obtained as described in Example 239 eluting ahead of (R)-l-(5-chloro- l-(4-isopropylphenyl)-6-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine- 4-carboxamide on the column. This gave 4 mg (6 % yield) of (R)-I -(6-chloro-l -(4-isopropyl- phenyl)-5 -methyl- 1 H-benzo [d]imidazo l-2-yl)-N-(tetrahydrofuran-3 -yl)piperidine-4-carboxamide as an off-white solid. MS (m/z) 481.3 (M+ 1).
Example 241: (S)-l-(5-chloro-6-methyl-l-p-tolyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

A tautomeric mixture of ethyl l-(6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxylate and ethyl l-(5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (80.5 mg, 0.25 mmol) dimethylsulfoxide (0.5 mL), caesium carbonate (114 mg, 0.35 mmol), 1- iodo-4-methylbenzene (109 mg, 0.50 mmol), 8-hydroxyquinoline (7.3 mg, 0.05 mmol), polyethylene glycol 400 (60.4 mg, 0.515 mmol) and copper(I) oxide (3.6 mg, 0.025 mmol) was
heated at 15O0C for 4 hours. The temperature was lowered to 9O0C and water (0.004 mL) was added to the reaction mixture, which was stirred for one hour. Aqueous sodium hydroxide (0.5 mL, 1 N) and ethanol (1 mL) was added and the reaction mixture stirred over night. Aqueous hydrochloric acid (0.25 mL, 2 N) was added and the reaction mixture concentrated in vacuo. The residue was mixed with N,N-dimethylformamide (5 mL), JV,jV-diisopropylethylamine (Hunig's base, DIEA, 97 mg, 0.75 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluoro-phosphate (ΗATU, 114 mg, 0.3 mmol) and (S)-tetrahydrofuran-3 -amine
hydrochloride (31 mg, 0.25 mmol). The reaction mixture was stirred at room temperature for 90 minutes, concentrated in vacuo and purified on column (silica gel, dichloromethane/methanol 96:4) to give 4.6 mg (4 % yield) of (S)-l-(5-chloro-6-methyl-l-p-tolyl-lH-benzo[d]imidazol-2- yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 452.9 (M+l). Also eluting from the column was the other regioisomer (S)-l-(6-chloro-5-methyl-l-p-tolyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide.
Example 242: (S)-l-(6-chloro-5-methyl-l-p-tolyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was obtained as described in Example 241 to give 5.8 mg (5 % yield) of (S)- 1 -(6-chloro-5 -methyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine- 4-carboxamide. LC-MS (m/z) 452.9 (M+l).
Example 243: (S)-l-(6-chloro-l-(3,5-dimethylphenyl)-5-methyl-lH-benzo[d]imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

chromathography, the appropriate combined fractions concentrated in vacuo and dissolved in ethanol. Aqueous sodium hydroxide (0.5 mL, 1 N) was added and the reaction mixture was stirred at room temperature over night. Aqueous hydrochloric acid (0.25 mL, 2 N) was added and the reaction mixture concentrated in vacuo. The residue was mixed with N ,N- dimethylformamide (5 mL), JV,jV-diisopropylethylamine (Ηunig's base, DIEA, 73 mg, 0.563 mmol), 2-(7-aza- lH-benzotriazole- 1 -yl)- 1 , 1 ,3,3-tetramethyluronium hexafluoro-phosphate (ΗATU, 85.7 mg, 0.225 mmol) and (S)-tetrahydrofuran-3 -amine hydrochloride (23 mg, 0.188 mmol). The reaction mixture was stirred at room temperature for 105 minutes, concentrated in vacuo and purified on column (silica gel, dichloromethane/methanol, gradient elution using from 3 to 7 % methanol) and further on hplc (preparative RP-LC was performed on a Gilson system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using methano I/water (0.05% formic acid) gradients at a flow rate of 15 niL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 1.9 mg of (S)-l-(6-chloro-l-(3,5-dimethylphenyl)-5-methyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 466.9 (M+l).
Example 244 : (R)- l-(5,6-dichloro- 1-p-tolyl- lH-benzo [d] imidazol-2-yl)-7V-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide

1,1,3,3-tetramethyluronium hexafluoro-phosphate (ΗATU, 14.1 mg, 0.037 mmol) and (R)-(+)- tetrahydrofuran-3 -amine 4-methylbenzenesulfonate (9.6 mg, 0.0371 mmol). The reaction mixture was stirred at room temperature for 2 hours, concentrated in vacuo and purified on hplc (preparative RP-LC was performed on a Gilson system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using methano I/water (0.05% formic acid) gradients at a flow rate of 4 niL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 7.7 mg of (R)- 1 -(5,6- dichloro- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide. LC-MS (m/z) 473.1 (M+ 1).
Example 245: l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V-((lS,25)-2- hydroxycyclopentyl)piperidine-4-carboxamide

(a) Preparation of the intermediate compound ethyl l-(l-cyclobutyl-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylate:

chromathography, to give 510 mg (43 % yield) of ethyl l-(l-cyclobutyl-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylate.
(b) Preparation of the intermediate compound l-(l-cyclobutyl-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid:

Ethyl l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (1 g, 2.79 mmol), aqueous sodium hydroxide (14 mmol), ethanol and water (50 mL) was stirred at room temperature for 30 minutes. After the appropriate work-up, the target compound was recrystallized from petroleum ether to give 500 mg (54 % yield) of l-(l-cyclobutyl-5,6- dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid.
(c) The title compound was prepared from l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol- 2-yl)piperidine-4-carboxylic acid (0.1 g, 0.3 mmol), N,N-dimethylformamide (3 mL), N,N- diisopropylethylamine (Ηϋnig's base, DIEA, 0.6 mmol), 2-(7-aza-lH-benzotriazole-l-yl)- 1,1,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 0.6 mmol) and (lS,2S)-2- aminocyclopentanol (0.36 mmol) using the method described in Example 166. The reaction mixture was stirred at room temperature for one hour and purified with recrystallization from petroleum ether, to give 47 mg (37 % yield) of l-(l-cyclobutyl-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-((15',25)-2-hydroxycyclopentyl)piperidine-4-carboxamide as an off- white solid. LC-MS (m/z) 410.9 (M+l).
Example 246: l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V-((lR,2R)-2- hydroxycyclopentyl)piperidine-4-carboxamide

The title compound was prepared from l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylic acid (0.1 g, 0.3 mmol), N,N-dimethylformamide (3 mL), N,N- diisopropylethylamine (Ηϋnig's base, DIEA, 0.6 mmol), 2-(7-aza-lH-benzotriazole-l-yl)- 1,1,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 0.6 mmol) and (lR,2R)-2- aminocyclopentanol (0.36 mmol) using the method described in Example 166. The reaction
mixture was stirred at room temperature for one hour and purified with recrystallization from petroleum ether, to give 10 mg (8 % yield) of l-(l-cyclobutyl-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-((lR,2R)-2-hydroxycyclopentyl)piperidine-4-carboxamidee as an off- white solid. LC-MS (m/z) 410.9 (M+l). Example 247: (R)-l-(l-(benzofuran-5-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (120 mg, 0.350 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.701 mmol), 5-bromobenzofuran (1.051 mmol), 8-hydroxyquinoline (0.140 mmol), polyethylene glycol (0.175 mmol) and copper(I) oxide (0.0701 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on preperative hplc (Shimadzu LC8A, C18 column (22 x 250 mm, lOμm), acetonitrile/water gradient (0.1% trifluoroacetic acid), flowrate 15 mL/min), to give 5 mg (3.1 % yield) of (R)-l-(l-(benzofuran-5-yl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 459.2 (M+l).
Example 248: (R)-l-(5,6-dichloro-l-(4-fluoro-3,5-dimethylphenyl)-lH-benzo[d]imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 5-bromo-2-fluoro-l,3-dimethylbenzene (0.783 mmol), 8-
hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on preparative hplc (Shimadzu LC8A, C18 column (22 x 250 mm, lOμm), acetonitrile/water gradient (0.1% trifluoroacetic acid), flowrate 15 niL/min), to give 10 mg (7.6 % yield) of (R)-l-(5,6-dichloro-l- (4-fluoro-3,5-dimethylphenyl)-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a pale yellow, solid. LC-MS (m/z) 505.4 (M+ 1).
Example 249 : (R)- l-(l-(3-fluoro-5-methylphenyl)-5,6-dimethyl- lH-benzo [d] imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (120 mg, 0.350 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.701 mmol), l-bromo-3-fluoro-5-methylbenzene (1.051 mmol), 8- hydroxyquinoline (0.140 mmol), polyethylene glycol (0.175 mmol) and copper(I) oxide (0.0701 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 3 mg (1.9 % yield) of (R)-l-(l-(3-fluoro-5-methylphenyl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid. LC- MS (m/z) 451.1 (M+ 1). Example 250: (R)-l-(5,6-dichloro-l-(3-fluoro-5-methylphenyl)-lH-benzo[d]imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (120 mg, 0.313 mmol) dimethylsulfoxide (2
niL), caesium carbonate (0.470 mmol), l-bromo-3-fluoro-5-methylbenzene (0.939 mmol), 8- hydroxyquinoline (0.125 mmol), polyethylene glycol (0.157 mmol) and copper(I) oxide (0.0626 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 14 mg (9.1 % yield) of (R)-l-(5,6-dichloro-l-(3-fiuoro-5-methylphenyl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 491.5 (M+l).
Example 251: (R)-l-(5,6-dichloro-l-(4-cyclopropylphenyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (120 mg, 0.313 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.470 mmol), l-bromo-4-cyclopropylbenzene (0.939 mmol), 8- hydroxyquinoline (0.125 mmol), polyethylene glycol (0.157 mmol) and copper(I) oxide (0.0626 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 5 mg (3.2 % yield) of (R)-l-(5,6-dichloro-l-(4-cyclopropylphenyl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 499.3 (M+l). Example 252: (R)-l-(5,6-dichloro-l-(3-cyclopropylphenyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

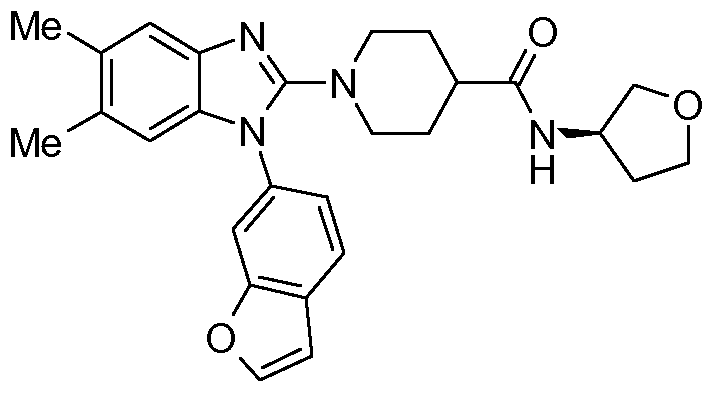
The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (120 mg, 0.350 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.701 mmol), 6-bromobenzofuran (1.051 mmol), 8-hydroxyquinoline (0.140 mmol), polyethylene glycol (0.175 mmol) and copper(I) oxide (0.0701 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 20 mg (12 % yield) of (R)-I -(l-(benzofuran-6-yl)-5,6-dimethyl- lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 459.1 (M+l).
Example 254: (R)-l-(5-chloro-l-(4-ethylphenyl)-6-methyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from a tautomeric misture of (R)-I -(5-chloro-6-methyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide and (R)- 1 -(6-chloro- 5-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide (500 mg, 1.38 mmol) dimethylsulfoxide (2 mL), caesium carbonate (2.76 mmol), l-bromo-4- ethylbenzene (4.13 mmol), 8-hydroxyquinoline (0.551 mmol), polyethylene glycol (0.689 mmol) and copper(I) oxide (0.276 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase reverse-phase hplc, to give 20 mg (3.3 % yield) of (R)-l-(5-chloro-l- (4-ethylphenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as an off-white solid solid, eluting after (R)-l-(6-chloro-l-(4-ethylphenyl)-5- methyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide on the column. LC-MS (m/z) 467.2 (M+l).
Example 255: (R)-l-(6-chloro-l-(4-ethylphenyl)-5-methyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was obtained as described in Example 254 as eluting before (R)-l-(5-chloro- 1 -(4-ethylphenyl)-6-methyl- lH-benzo [d]imidazo l-2-yl)-N-(tetrahydrofuran-3 -yl)piperidine-4- carboxamide on the column. This gave 24 mg (3.7 % yield) of (R)-l-(6-chloro-l-(4- ethylphenyl)-5 -methyl- 1 H-benzo [d]imidazo l-2-yl)-N-(tetrahydrofuran-3 -yl)piperidine-4- carboxamide as an off-white solid. MS (m/z) 467.2 (M+l).
Example 256: (R)-l-(5-chloro-l-(4-ethylphenyl)-6-methyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from a tautomeric mixture of (R)-I -(5-chloro-6-methyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide and (R)- 1 -(6-chloro- 5-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide (500 mg, 1.38 mmol) dimethylsulfoxide (2 mL), caesium carbonate (2.76 mmol), l-bromo-4-tert- butylbenzene (4.13 mmol), 8-hydroxyquinoline (0.551 mmol), polyethylene glycol (0.689 mmol) and copper(I) oxide (0.276 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase reverse-phase hplc, to give 69 mg (10 % yield) of (R)-l-(l-(4-tert- butylphenyl)-5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine- 4-carboxamide as a white solid, eluting after (R)-l-(l-(4-tert-butylphenyl)-6-chloro-5-methyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide on the column. LC-MS (m/z) 495.3 (M+ 1).
Example 257: (R)-l-(l-(4-tert-butylphenyl)-6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was obtained as described in Example 256 eluting before (R)-l-(l-(4-tert- butylphenyl)-5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine- 4-carboxamide on the column. This gave 63 mg (9 % yield) of (R)-I -(I -(4-tert-butylphenyl)-6- chloro-5 -methyl- 1 H-benzo [d]imidazo l-2-yl)-N-(tetrahydrofuran-3 -yl)piperidine-4-carboxamide as a white solid. MS (m/z) 495.3 (M+l).
Example 258: (R)-l-(l-(4-cyclopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (120 mg, 0.350 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.701 mmol), l-bromo-4-cyclopropylbenzene (1.051 mmol), 8- hydroxyquinoline (0.140 mmol), polyethylene glycol (0.175 mmol) and copper(I) oxide (0.0701 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hours at 14O0C and subsequently purified on reverse-phase hplc, to give 13 mg (8.1 % yield) of (R)-I -(I -(4-cyclopropylphenyl)-5,6-dimethyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a pale yellow. solid. LC-MS (m/z) 459.1 (M+ 1).
Example 259: (R)-l-(l-(3-cyclopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (120 mg, 0.350 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.701 mmol), l-bromo-3-cyclopropylbenzene (1.051 mmol), 8- hydroxyquinoline (0.140 mmol), polyethylene glycol (0.175 mmol) and copper(I) oxide (0.0701 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 5 mg (3.1 % yield) of (R)-I -(I -(3-cyclopropylphenyl)-5,6-dimethyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 459.2 (M+l).
Example 260 : (R)- l-(5,6-dichloro- l-(3-(cyclopentyloxy)phenyl)- lH-benzo [d] imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-3-(cyclopentyloxy)benzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 2 mg (1.4 % yield) of (R)-l-(5,6-dichloro-l-(3-(cyclopentyloxy)phenyl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid. LC- MS (m/z) 543.2 (M+ 1).
Example 261: (R)-l-(5,6-dichloro-l-(3-cyclobutoxyphenyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), l-bromo-3-cyclobutoxybenzene (0.783 mmol, prepared by alkylation of 3-bromophenol using bromocyclobutane and potassium carbonate in N ,N- dimethylformamide), 8-hydroxy-quinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for two hour at 12O0C and subsequently purified on reverse-phase hplc, to give 2 mg (1.5 % yield) of (R)-l-(5,6-dichloro-l-(3-
cyclobutoxyphenyl)-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a pale yellow solid. LC-MS (m/z) 529.4 (M+ 1).
Example 262: l-(5-chloro-6-fluoro-l-methyl-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide and l-(6-chloro-5-fluoro-l-methyl-lH- benzo [d] imidazol-2-yl)-7V-cyclopentylpiperidine-4-carboxamide

The title compound was prepared from a tautomeric mixture of l-(5-chloro-6-fluoro-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro-5-fluoro- IH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide (0.1 g, 0.27 mmol), methyl iodide (1.096 mmol), caesium carbonate (1.096 mmol), [l,l'-bis(diphenyl-phosphino)ferrocene] dichloropalladium(II) (PdCl2(dppf), 0.0274 mmol) and N,Λ/-dimethyl-formamide (3 mL) using the method described in Example 94. This gave 30 mg (29 % yield) of l-(5-chloro-6-fluoro-l- methyl- lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro-5- fluoro-l-methyl-lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as an off- white solid. No effort was made to determine the ratio of the regioisomers. LC-MS (m/z) 379.1 (M+l).
Example 263: l-(5-chloro-l-ethyl-6-fluoro-l//-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide and l-(6-chloro-l-ethyl-5-fluoro-lH- benzo [d] imidazol-2-yl)-7V-cyclopentylpiperidine-4-carboxamide

The title compound was prepared from a tautomeric mixture of l-(5-chloro-6-fluoro-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide and l-(6-chloro-5-fluoro-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide (0.1 g, 0.27 mmol), ethyl iodide (1.096 mmol), caesium carbonate (1.096 mmol), [l,l'-bis(diphenyl-phosphino)ferrocene] dichloropalladium(II) (PdCl2(dppf), 0.0274 mmol) and N,Λ/-dimethyl-formamide (3 mL) using the method described in Example 94. This gave 35 mg (33 % yield) of l-(5-chloro-l-ethyl-6- fluoro-lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide and l-(6-chloro-l-
ethyl-5-fluoro-lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as an off- white solid. No effort was made to determine the ratio of the regioisomers. LC-MS (m/z) 393.0 (M+l).
Example 264: l-(5-chloro-6-fluoro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide and l-(6-chloro-5-fluoro-l-isopropyl-lH- benzo [d] imidazol-2-yl)-7V-cyclopentylpiperidine-4-carboxamide

The title compound was prepared from a tautomeric mixture of l-(5-chloro-6-fluoro-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro-5-fluoro- IH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide (0.1 g, 0.27 mmol), isopropyl iodide (1.096 mmol), caesium carbonate (1.096 mmol), [l,l'-bis(diphenyl-phosphino)ferrocene] dichloropalladium(II) (PdCl2(dppf), 0.0274 mmol) and N,Λ/-dimethyl-formamide (3 mL) using the method described in Example 94. This gave 50 mg (45 % yield) of l-(5-chloro-6-fluoro-l- isopropyl- lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro- 5-fluoro-l -isopropyl- lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide as an off-white solid. No effort was made to determine the ratio of the regioisomers. LC-MS (m/z) 407.1 (M+l).
Example 265: l-(l-butyl-5-chloro-6-fluoro-l//-benzo[d]imidazol-2-yl)-7V- cyclopentylpiperidine-4-carboxamide and l-(l-butyl-6-chloro-5-fluoro-l//- benzo [d] imidazol-2-yl)-7V-cyclopentylpiperidine-4-carboxamide

The title compound was prepared from a tautomeric mixture of l-(5-chloro-6-fluoro-lH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide and 1 -(6-chloro-5-fluoro- IH- benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4-carboxamide (0.1 g, 0.27 mmol), 1- bromobutane (1.096 mmol), caesium carbonate (1.096 mmol), [l,l'-bis(diphenyl- phosphino)ferrocene] dichloropalladium(II) (PdCl2(dppf), 0.0274 mmol) and N,7V-dimethyl-
formamide (3 niL) using the method described in Example 94. This gave 8 mg (6.8 % yield) of l-(l-butyl-5-chloro-6-fluoro-lH-benzo[d]imidazol-2-yl)-N-cyclopentylpiperidine-4- carboxamide and l-(l-butyl-6-chloro-5-fluoro-lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide as an off-white solid. No effort was made to determine the ratio of the regioisomers. LC-MS (m/z) 421.1 (M+ 1).
Example 266: l-(5,6-dimethyl-l-p-tolyl-lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3- yl)piperidine-4-carboxamide

The title compound was prepared from l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-iV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (0.30 g, 0.877 mmol), dimethylsulfoxide (6 mL), caesium carbonate (3.51 mmol), l-iodo-4-methylbenzene (3.51 mmol), 4,7-dimethoxy- 1,10-phenanthroline (0.132 mmol), polyethylene glycol (0.89 g) and copper(I) oxide (0.0438 mmol) using the method described in Example 76. The reaction mixture was heated at 12O0C for 12 hours in a sealed tube and purified with column chromathography, to give 43 mg (11.3 % yield) of l-(5,6-dimethyl-l-p-tolyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)- piperidine-4-carboxamide as a pale green solid. LC-MS (m/z) 432.2 (M+ 1).
Example 267: (R)-l-(6-chloro-l-(4-fluorophenyl)-5-methyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

Example 268 : (R)- l-(5-chloro- l-(4-fluorophenyl)-6-methyl- lH-benzo [d] imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was obtained as described in Example 267 eluting after (R)-l-(6-chloro-l- (4-fluorophenyl)-5-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide on the column. This gave 47 mg (7.4 % yield) of (R)-l-(5-chloro-l-(4- fluorophenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as an off-white solid. LC-MS (m/z) 457.1 (M+l).
Example 269: (R)-l-(6-chloro-5-methyl-l-m-tolyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from a tautomeric misture of (R)-I -(5-chloro-6-methyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide and (R)- 1 -(6-chloro- 5-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide (500
mg, 1.38 mmol) dimethylsulfoxide (2 niL), caesium carbonate (2.76 mmol), l-bromo-3- methylbenzene (4.13 mmol), 8-hydroxyquinoline (0.551 mmol), polyethylene glycol (0.689 mmol) and copper(I) oxide (0.276 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase reverse-phase hplc, to give 24 mg (3.8 % yield) of (R)-I - (6-chloro-5-methyl-l-m-tolyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4- carboxamide as a pale yellow solid, eluting ahead of (R)-I -(5-chloro-6-methyl- 1-m-to IyI- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide on the column. LC- MS (m/z) 453.2 (M+l). Example 270: (R)-l-(5-chloro-6-methyl-l-m-tolyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was obtained as described in Example 269 eluting after (R)-l-(6-chloro-5- methyl- 1 -m-tolyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide on the column. This gave 23 mg (3.7 % yield) of (R)-I -(5-chloro-6-methyl- 1-m-to IyI- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as an off-white solid. LC-MS (mix) 453.3 (M+l).
Example 271: (R)-l-(5-chloro-6-methyl-l-(5-(trifluoromethyl)pyridin-2-yl)-lH- benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from a tautomeric misture of (R)-I -(5-chloro-6-methyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide and (R)- 1 -(6-chloro- 5-methyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide (500
mg, 1.38 mmol) dimethylsulfoxide (2 niL), caesium carbonate (2.76 mmol), 2-bromo-5- (trifluoromethyl)pyridine (4.13 mmol), 8-hydroxyquinoline (0.551 mmol), polyethylene glycol (0.689 mmol) and copper(I) oxide (0.276 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 12O0C and subsequently purified on reverse-phase reverse-phase hplc, to give 12 mg (1.7 % yield) of (R)-I- (5-chloro-6-methyl- 1 -(5-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide as a yellow solid, eluting ahead of (R)-I -(6- chloro-5-methyl- 1 -(5-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide on the column. LC-MS (m/z) 508.3 (M+l). Example 272: (R)-l-(6-chloro-5-methyl-l-(5-(trifluoromethyl)pyridin-2-yl)-lH- benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was obtained as described in Example 271 eluting after (R)-l-(5-chloro-6- methyl- 1 -(5-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide on the column. This gave 16 mg (2.3 % yield) of (R)-I -(6-chloro-5- methyl- 1 -(5-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as an yellow solid. LC-MS (m/z) 508.2 (M+l).
Example 273: l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V-((2S)-2- methoxycyclopentyl)piperidine-4-carboxamide
A solution of l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-((15f,25)-2- hydroxycyclopentyl)piperidine-4-carboxamid (0.10 g, 0.24 mmol) in N,N-dimethylformamide (DMF, 2 mL) was cooled to 0 0C. Sodium hydride (0.26 mmol) was added followed by methyl iodide (0.24 mmol) and the reaction was stirred at 0 0C for 3 h. The reaction mixture was poured
onto ice and the precipitate was collected by filtration. The crude was rechrystallized from petroleum ether to give 17 mg (17%) of l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)-N-((2S)-2-methoxycyclopentyl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 425.2 (M+l). Example 274: l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V-((2S)-2- ethoxycyclopentyl)piperidine-4-carboxamide

A solution of l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-((lS,25)-2- hydroxycyclopentyl)piperidine-4-carboxamid (0.10 g, 0.24 mmol) in N,N-dimethylformamide (DMF, 2 mL) was cooled to 0 0C. Sodium hydride (0.48 mmol) was added followed by ethyl iodide (0.48 mmol) and the reaction was stirred at 0 0C for 3 h. The reaction mixture was poured onto ice and the precipitate was collected by filtration. The crude was rechrystallized from petroleum ether to give 42 mg (39%) of l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)-N-((2S)-2-ethoxycyclopentyl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 439.2 (M+l).
Example 275: l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V-((2S)-2- propoxycyclopentyl)piperidine-4-carboxamide

A solution of l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-((lS,25)-2- hydroxy cyclopentyl)piperidine-4-carboxamid (0.10 g, 0.24 mmol) in N,N-dimethylformamide (DMF, 2 mL) was cooled to 0 0C. Sodium hydride (0.48 mmol) was added followed by 1- iodopropane (0.48 mmol) and the reaction was stirred at 0 0C for 3 hours. The reaction mixture was concentrated under reduced pressure and the crude purified by silica chromatography
(chloroform/methanol gradient) to give 51 mg (46%) of l-(l-cyclobutyl-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-((2S)-2-propoxycyclopentyl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 453.2 (M+ 1).
Example 276: l-(5,6-dichloro-l-(4-(dimethylcarbamoyl)phenyl)-lH-benzo[d]imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

A reaction mixture consisting of l-(l-(4-bromophenyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)- N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.19 mmol), trans-di(μ-acetato)- bis[o-(di-o-tolylphosphino)-benzyl]dipalladium(II) (Hermann palladacycle, 0.0095 mmol), N5N- diisopropylethylamine (Hunig's base, DIEA, 0.38 mmol), l,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 0.19 mmol), tri tertiarybutyl phosphonium tetrafluoborate ([(t-Bu)3PH]BF4, 0.019 mmol), molybdenum hexacarbonyl (Mo(CO)6, 0.095 mmol) and N, N-dimethyl amine hydrochloride (0.38 mmol) in 1,4-dioxane (3 mL), was irradiated under microwave conditions at 15O0C for 30 minutes. The reaction mixture was quenched with water (5 mL), extracted with dichloromethane (2 x50 mL) and the combined organic phases dried over anhydrous sodium sulphate. After concentration in vacuo, the residue was subjected to column chromatography to give 5 mg (5 % yield) of 1 -(5,6-dichloro- 1 -(4-(dimethylcarbamoyl)phenyl)- lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 530.5 (M+l).
Example 277: l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)-iV-((lS,2S)-2- hydroxycyclopentyl)piperidine-4-carboxamide

(a) Preparation of the intermediate compound ethyl 1 -(5,6-dichloro- 1-cyclo butyl- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxylate:

Ethyl 1 -(5,6-dichloro- 1 -cyclobutyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate was prepared from ethyl l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (1.0 g, 2.9 mmol), (bromomethyl)cyclo butane (11.6 mmol), caesium carbonate (11.6 mmol), [1,1'- bis(diphenylphosphino)ferrocene] dichloropalladium(II) (PdCl2(dppf), 0.29 mmol) and N,N- dimethylformamide (8 mL) using the method described in Example 16. The reaction mixture was heated at 1300C for two hours in a sealed tube and purified with column chromathography, to give 480 mg (42 % yield) of ethyl l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxylate as a pale yellow solid. LC-MS (m/z) 451.3 (M+l). (b) Preparation of the intermediate compound 1 -(5,6-dichloro- 1 -cyclobutyl- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid:

Ethyl l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylate (1.0 g, 2.5 mmol), aqueous sodium hydroxide (12.5) and ethano I/water (4:1) was stirred for 30 minutes at room temperature. After the appropriate work-up, the residue was recrystallized from petroleum ether, to give 0.70 g (83 % yield) of 1 -(5,6-dichloro- 1 -cyclobutyl- IH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid.
(c) l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (1.0 mg, 2.7 mmol), N,N-dimethylformamide (10 mL), N,7V-diisopropyl-ethylamine (Ηunig's base, DIEA, 5.4 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium
hexafluorophosphate (ΗATU, 5.4 mmol) and (lS,2S)-2-aminocyclopentanol (3.24 mmol) was stirred at room temperature for one hour. After the appropriate work-up, the residue was recrystallized from petroleum ether, to give 1.0 g (83 % yield) of 1 -(5,6-dichloro- 1-cyclobutyl- lH-benzo[d]imidazol-2-yl)-N-((lS,2S)-2-hydroxycyclopentyl)piperidine-4-carboxamide.
Example 278: l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)-7V-((lS,2S)-2- methoxycyclopentyl)piperidine-4-carboxamide

A solution of l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)-N-((lS,2S)-2- hydroxy cyclopentyl)piperidine-4-carboxamide (0.10 g, 0.22 mmol) in N,N-dimethylformamide (DMF, 3 mL) was cooled to 0 0C. Sodium hydride (0.24 mmol) was added followed by methyl iodide (0.22 mmol) and the reaction was stirred at 0 0C for 30 minutes. The reaction mixture was poured onto ice and the precipitate was collected by filtration. The crude was recrystallized from petroleum ether to give 5 mg (5%) of l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)- N-((lS,2S)-2-methoxycyclopentyl)piperidine-4-carboxamide as a yellow solid. LC-MS (m/z) 465.4 (M+l).
Example 279: l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)-7V-((lS,2S)-2- ethoxycyclopentyl)piperidine-4-carboxamide

Example 280: l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V-(furan-2- ylmethyl)piperidine-4-carboxamide
 l-(l-Cyclobutyl-5,6-dimethyl-lH-benzoimidazol-2-yl)-piperidine-4-carboxylic acid (30 mg, 0.092 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 42 mg, 0.11 mmol), 2-aminomethylfϊιrane (25 μL, 0.28 mmol) and JV,iV-diisopropyl- 5 ethylamine (Ηunig's base, DIEA, 46 μL, 0.28 mmol) were dissolved in N, N-dimethylformamide (DMF, 1 mL). The reaction mixture was stirred overnight at ambient temperature. The reaction mixture was filtered and the crude product was purified on preparative hplc (preparative RP-LC was performed on a Gilson system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using methano I/water (0.05% formic acid) gradients at a flow rate of 4 mL/min with UV 10 (214 or 254 nm) and MS (ESI) detection) to give 18.2 mg (72 % yield) of l-(l-cyclobutyl-5,6- dimethyl-lH-benzo[d]imidazol-2-yl)-N-(furan-2-ylmethyl)piperidine-4-carboxamide as an off- white solid. LC-MS (m/z) 407.0 (M+l).
l-(l-Cyclobutyl-5,6-dimethyl-lH-benzoimidazol-2-yl)-piperidine-4-carboxylic acid (30 mg, 0.092 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 42 mg, 0.11 mmol), 2-aminomethylfϊιrane (25 μL, 0.28 mmol) and JV,iV-diisopropyl- 5 ethylamine (Ηunig's base, DIEA, 46 μL, 0.28 mmol) were dissolved in N, N-dimethylformamide (DMF, 1 mL). The reaction mixture was stirred overnight at ambient temperature. The reaction mixture was filtered and the crude product was purified on preparative hplc (preparative RP-LC was performed on a Gilson system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using methano I/water (0.05% formic acid) gradients at a flow rate of 4 mL/min with UV 10 (214 or 254 nm) and MS (ESI) detection) to give 18.2 mg (72 % yield) of l-(l-cyclobutyl-5,6- dimethyl-lH-benzo[d]imidazol-2-yl)-N-(furan-2-ylmethyl)piperidine-4-carboxamide as an off- white solid. LC-MS (m/z) 407.0 (M+l).
Example 281: l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V-(thiophen-2- ylmethyl)piperidine-4-carboxamide

temperature. The reaction mixture was filtered and the crude product was purified on preparative hplc (preparative RP-LC was performed on a Gilson system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using methano I/water (0.05% formic acid) gradients at a flow rate of 4 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 2.5 mg (6 % yield) of 1- 5 (1 -cyclobutyl-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)-N-(thiophen-2-ylmethyl)piperidine-4- carboxamide as an off-white solid. LC-MS (m/z) 423.0 (M+l).
Example 282: (R)-l-(l-(5-bromopyridin-2-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (290 mg, 0.757 mmol) dimethylsulfoxide (1 mL), caesium carbonate (345 mg, 1.06 mmol), 2,5-dibromopyridine (0.72 g, 3.0 mmol), 8- hydroxyquinoline (0.15 mmol), polyethylene glycol 400 (0.38 mmol) and copper(I) oxide (0.076 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 60 minutes at 15O0C. Most of the DMSO was removed under reduced pressure and the residue purified by silica chromatography (0-6% MeOH in dichloromethane) to give 97 mg (37 % yield) of (R)-l-(l-(5-bromopyridin-2-yl)-5,6-dimethyl-lH-benzo[d]imidazol- 2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a brown solid. LC-MS (m/z) 540.2 (M+l).
Example 283: (R)-l-(l-(5-ethylpyridin-2-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

To a solution of (R)-l-(l-(5-bromopyridin-2-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (20 mg, 0.037 mmol) and bis(tri-tert- butylphosphine)palladium(O) (CAS Registry Number: 53199-31-8, 1.2 mg, 0.0024 mmol) in dry tetrahydrofurane under nitrogen was added diethylzinc (1 N in hexane, 0.11 mL, 0.11 mmol) and the mixture microwave heated at 120 0C for 1 hour. The reaction mixture was purified by preparative reversed phase ΗPLC to give 7.7 mg (43 % yield) of (R)-l-(l-(5-ethylpyridin-2-yl)- 5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide as a white solid. LC-MS (m/z) 488.2 (M+l).
Example 284: (R)-l-(l-(3-chloro-5-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (100 mg, 0.292 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.584 mmol), 3-chloro-5-fluoro-bromobenzene (0.876 mmol), 8- hydroxyquinoline (0.117 mmol), polyethylene glycol (0.146 mmol) and copper(I) oxide (0.0584 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 10 mg (7 % yield) of (R)-l-(l-(3-chloro-5-fiuorophenyl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 471.2 (M+l).
Example 285: (R)-l-(5,6-dichloro-l-(3-chloro-5-fluorophenyl)-lH-benzo[d]imidazol-2-yl)- 7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol), dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 3-chloro-5-fluoro-bromobenzene (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 9 mg (7 % yield) of (R)-l-(5,6-dichloro-l-(3-chloro-5-fluoro-phenyl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 511.3 (M+l).
Example 286: (R)-l-(5,6-dichloro-l-(3-(trifluoromethyl)phenyl)-lH-benzo[d]imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol), dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 3-bromo-benzotrifluoride (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 12 mg (9 % yield) of (R)-l-(5,6-dichloro-l-(3-trifluoromethylphenyl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 521 A (M+l).
Example 287: (R)-l-(5,6-dichloro-l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl)-lH- benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol), dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 5-bromo-2,2-difluorobenzo[d][l,3]dioxol (0.783 mmol), 8-hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 11 mg (8 % yield) of (R)-l-(5,6-dichloro-l-(2,2-difiuorobenzo[d][l,3]dioxol-5-yl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 539.3 (M+l).
Example 288 (SJ-l-Cl-CS^-dimethylpheny^-S^-dimethyl-lH-benzoIdlimidazoH-yO-TV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

(a) Preparation of the intermediate compound l-(l-(3,5-dimethylphenyl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid:

Ethyl l-(l-(3,5-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxylate (80.5 mg, 0.267 mmol), l-bromo-3,5-dimethylbenzene (73 μL, 0.534 mmol), 8- hydroxyquinoline (7.8 mg, 0.053 mmol), copper(I) oxide (3.8 mg, 0.027 mmol), polyethylene glycol 400 (107 mg, 0.267 mmol), caesium carbonate (122 mg, 0.374 mmol) and dimethyl- sulfoxide (0.5 mL) were mixed in a sealed reaction tube. The reaction mixture was heated at 1500C for two hour in a heating block and then diluted with ethylacetate and water. The water phase was extracted with ethyl acetate, the combined organic phases was dried over magnesium sulphate and concentrated in vacuo. To the residue was added ethanol (2 mL) and aquesous sodium hydroxide (1 mL, 1 N). The reaction mixture was stirred at 50 0C for one hour, neutralized with aqueous hydrochloric acid (1 N) and concentrated in vacuo. This gave 90 mg of l-(l-(3,5-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid. LC-MS (m/z) 378.0 (M+l).
(b) l-(l-(3,5-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxylic acid (90 mg, 0.24 mmol), 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (ΗATU, 136 mg, 0.358 mmol), (S)-(-)-tetrahydrofuran-3 -amine
hydrochloride (29.5 mg, 0.238 mmol) and JV,JV-diisopropyl-ethylamine (Ηunig's base, DIEA, 994 μL, 5.96 mmol) were dissolved in N,N-dimethylformamide (1 mL). The reaction mixture was stirred overnight at ambient temperature, filtered and subjected to preparative hplc
(preparative RP-LC was performed on a Gilson system equipped with a Zorbax SB-C8 (5 μm,
21.2 x 150 mm) column, using methano I/water (0.05% formic acid) gradients at a flow rate of 4 niL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 2 mg (2 % yield) of (S)-I -(I - (3,5-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide as an off-white solid. LC-MS (m/z) 447.0 (M+l). Example 289: (R)-l-(5,6-dichloro-l-(4-(trifluoromethyl)pyridin-2-yl)-lH- benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol), dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 2-bromo-4-trifluoromethylpyridine (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 41 mg (30 % yield) of (R)-l-(5,6-dichloro-l-(4-(trifluoromethyl)pyridin-2-yl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 528.2 (M+l).
Example 290: (R)-l-(5,6-dichloro-l-(5-chloropyridin-2-yl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

(tetrahydrofuran-3-yl)piperidine-4-carboxamide (100 mg, 0.261 mmol), dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 2-bromo-4-chloropyridine (0.783 mmol), 8- hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522
mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 20 mg (17 % yield) of (R)-l-(5,6-dichloro-l-(5-chloropyridin-2-yl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 494.3 (M+l).
Example 291: (R)-l-(5,6-dichloro-l-(5-fluoro-6-methylpyridin-2-yl)-lH-benzo[d]imidazol- 2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (120 mg, 0.313 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.470 mmol), 2-bromo-5-fluoro-6-methylpyridine (0.939 mmol), 8- hydroxyquinoline (0.125 mmol), polyethylene glycol (0.157 mmol) and copper(I) oxide (0.0626 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 5 mg (3.2 % yield) of (R)-l-(5,6-dichloro-l-(5-fiuoro-6-methylpyridin-2-yl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamid. LC-MS (m/z) 492.3 (M+l).
Example 292: (R)-l-(l-(3-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (100 mg, 0.292 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.584 mmol), 3-fluoro-bromobenzene (0.876 mmol), 8-
hydroxyquinoline (0.117 mmol), polyethylene glycol (0.146 mmol) and copper(I) oxide (0.0584 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 7 mg (6 % yield) of (R)-l-(l-(3-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol- 2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 437.1 (M+ 1).
Example 293: (R)-l-(l-(3,5-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

hexafluorophosphate (ΗATU, 136 mg, 0.358 mmol), (R)-(+)-tetrahydrofuran-3 -amine 4- methylbenzenesulfonate (61.8 mg, 0.238 mmol) and iV,jV-diisopropyl-ethylamine (Ηunig's base, DIEA, 994 μL, 5.96 mmol) were dissolved in N,N-dimethylformamide (1 mL). The reaction mixture was stirred overnight at ambient temperature, filtered and subjected to preparative hplc (preparative RP-LC was performed on a Gilson system equipped with a Zorbax SB-C8 (5 μm, 21.2 x 150 mm) column, using methano I/water (0.05% formic acid) gradients at a flow rate of 4 niL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 11 mg (10 % yield) of (R)-I- (l-(3,5-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamid as an off-white solid. LC-MS (m/z) 447.0 (M+l). Example 294: (R)-l-(l-(4-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (100 mg, 0.292 mmol) dimethylsulfoxide (2
niL), caesium carbonate (0.584 mmol), 4-fluoro-bromobenzene (0.876 mmol), 8- hydroxyquinoline (0.117 mmol), polyethylene glycol (0.146 mmol) and copper(I) oxide (0.0584 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 25 mg (22 % yield) of (R)-I -(I -(4-fiuorophenyl)-5,6-dimethyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 437.1 (M+l).
Example 295: (RJ-l-Cl-CS^-difluoropheny^-S^-dimethyl-lH-benzoIdlimidazoU-yO-TV- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (120 mg, 0.350 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.701 mmol), 3,5-difluoro-bromobenzene (1.05 mmol), 8- hydroxyquinoline (0.140 mmol), polyethylene glycol (0.175 mmol) and copper(I) oxide (0.0701 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 31 mg (19 % yield) of (R)-l-(l-(3,5-difiuorophenyl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 455.1 (M+l). Example 296: (R)-l-(l-(4-chlorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide


The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (120 mg, 0.350 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.701 mmol), 4-bromo-benzotrifluoride (1.05 mmol), 8- hydroxyquinoline (0.140 mmol), polyethylene glycol (0.175 mmol) and copper(I) oxide (0.0701 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 3 mg (1.8 % yield) of (R)-I -(I -(4-trifluoromethyl-phenyl)-5,6-dimethyl- IH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 487.1 (M+l).
Example 298: (R)-l-(5,6-dimethyl-l-p-tolyl-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide


The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (120 mg, 0.350 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.701 mmol), 2-bromo-5-chloropyridine (1.05 mmol), 8- hydroxyquinoline (0.140 mmol), polyethylene glycol (0.175 mmol) and copper(I) oxide (0.0701 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 18 mg (12 % yield) of (R)-l-(l-(5-chloropyridin-2-yl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 454.2 (M+l).
Example 300: (R)-l-(5,6-dimethyl-l-(4-(trifluoromethyl)pyridin-2-yl)-lH- benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide


The title compound was prepared from (R)-l-(5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide, (120 mg, 0.350 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.701 mmol), 2-bromo-5-fluoro-6-methylpyridine (1.05 mmol), 8- hydroxyquinoline (0.140 mmol), polyethylene glycol (0.175 mmol) and copper(I) oxide (0.0701 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 11 mg (7 % yield) of (R)-l-(l-(5-fiuoro-6-methylpyridin-2-yl)-5,6-dimethyl-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 452.1 (M+l).
Example 302: (R)-l-(5,6-dichloro-l-(4-ethoxyphenyl)-lH-benzo[d]imidazol-2-yl)-7V- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

Example 303: (R)-l-(5,6-dichloro-l-(4-methoxy-3-methylphenyl)-lH-benzo[d]imidazol-2- yl)-7V-(tetrahydrofuran-3-yl)piperidine-4-carboxamide

The title compound was prepared from (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide (120 mg, 0.313 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.470 mmol), 4-methoxy-3-methylbenzene (0.939 mmol), 8- hydroxyquinoline (0.125 mmol), polyethylene glycol (0.157 mmol) and copper(I) oxide (0.0626 mmol) using the method described in Example 136. The reaction mixture was subjected to microwave conditions for 150 minutes at 14O0C and subsequently purified on reverse-phase hplc, to give 15 mg (10 % yield) of (R)-l-(5,6-dichloro-l-(4-methoxy-3-methylphenyl)-lH- benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)piperidine-4-carboxamide. LC-MS (m/z) 503.4 (M+l).
Example 304: (RJ-l-Cl-cyclobutyl-S^-dimethyl-lH-benzoIdlimidazoU-yl)-^- (tetrahydrofuran-3-yl)piperidine-4-carboxamide

BIOLOGICAL TESTS
Prostaglandin detection kits were purchased from Cayman Chemicals and used according to the instruction of the manufacturer.
Thiobarbituric acid assay (TBA-MDA assay or Malondialdehyde assay):
Malondialdehyde is a product of lipid peroxidation and reacts with thiobarbituric acid forming a red product that absorbs at 535 nm (W.G. Niehaus, Jr and B. Samuelsson, Eur. J. Biochem 6, 126 (1968) and is also fluorescent. The extinction coefficient of the TBA-MDA conjugate is 1.56 x 105 M"1 cm"1 (E.D. Wills. Biochem. J. 113, 315 (1969). The method used for detection of inhibition of mPGEs-1 is based on the detection of the amount of remaining PGH2. This method was described more than 20 years ago by Basevich et al (Bioorg. Khim. 1983, 9(5), 658-665).
The assay used was a modified variant and used citric acid instead of the TCA-TBA-HCl reagent described in the original assay. In this assay recombinant, membrane-bound human or rat mPGEs-1 was incubated with PGH2. The reaction was stopped by adding citric acid with a final pH of 3 and a large excess of iron(II) chloride (20 mM) to convert any remaining PGH2 into MDA and 12-HHT. TBA reagent was finally added (0.67 %) and the samples were heated at 80 0C for 30 min. The fluorescence of the conjugate was measured at 485/545 nm.
The product of mPGEs-1 (PGE2) was not measured directly in this assay, but rather the remaining substrate (PGH2) indirectly by adding FeCl2 that converts PGH2 into MDA and 12- HHT. As a positive control a known mPGEs-1 inhibitor, MK-886, was used and the new inhibitors were compared with the inhibition of MK-886.
Selected results from the human and rat mPGEs-1 TBA-MDA assay (IC50 in μM) are shown in Table 1.
A549 cell-assay
A549 lung carcinoma cells, seeded at a density of 10, 000 cells/well, were grown in 96 well tissue culture plates. TNFalfa (5 ng/mL) and IL-I beta (5 ng/mL) were added and the cells were incubated for 16 hours. Cells were washed in PBS and test compounds at the appropriate concentration in HBSS/0.1% BSA were added. After 30 minutes incubation with test compounds, 10 μM arachidonic acid was added and cells were further incubated for 30 minutes. Supernatant was collected and analyzed for PGE2 content by EIA according to manufacturer's instructions. Selected results from the A549 cell-assay are shown in Table 1.
Table 1. Inhibition of human and rat mPGEs-1 and cellular PGE2 synthesis.
















The human mPGEs-1 IC50 values of the compounds of Examples 284-287, 289-292 and 294-303 are within the range of 10-250 nM (i.e. 0.01-0.25 iM).
Carrageenan-induced paw edema
Method
Chemicals
λ-Carrageenan (Fluka, Switzerland), Carboxymethyl cellulose (CMC) (Sigma, USA), Dimethyl sulfoxide (DMSO) (Merck, Germany), Pyrogen free saline (Sintong, R. O. C.) and Tween 80 (Sigma, USA).
Equipment
Animal cage (Allentown, USA), Glass syringes (1 mL, 2 mL, Mitsuba, Japan), Hypodermic needle 27G x 1" (Top Corporation, Japan), Paw/Tail stimulator analgesia meter (IITC Model- 336G, IITC, USA), Plethysmometer (PV-Ol, Paw Volume Apparatus, SINGA Technology Co., R. O .C), Rat oral needle (Natsume, Japan) and Rat scale (1 g ~ 1000 g, TANITA, Japan).
Animals
Male Wistar rats weighing 165 ± 15 g were provided by BioLasco Taiwan (under Charles River Laboratories Technology Licensee). Space allocation for 5 animals was 45 x 23 x 21 cm.
Animals were housed in animal cages and maintained in a controlled temperature (21 - 230C) and humidity (50% - 70%) environment with 12 hours light/dark cycles for at least three days at test site prior to use. The rats were divided into groups of eight, each group to receive either
vehicle (1% Tween 80/0.5% CMC), or 10, 30 or 100 mg/kg bodyweight of the compound of EXAMPLE 41 (in the following referred to as "EXAMPLE 41"). The rats were fasted overnight prior to use. EXAMPLE 41 was suspended in vehicle and administered intraperitoneally immediately before the right hind paw received intraplantar injection of carrageenan (0.1 mL of 1% suspension). Hind paw edema, a measure of inflammation, was recorded 6 hours after carrageenan administration using a plethysmometer.
Results
Intraperitoneal administration of EXAMPLE 41 at 10, 30 and 100 mg/kg bodyweight to rats resulted in a decrease in paw swelling of 22%, 34% and 39%, respectively, compared to vehicle- treated rats (figure qzl).
Conclusion
EXAMPLE 41 is capable of reducing carrageenan- induced paw swelling, a symptom of inflammation, in rats.
Complete Freund's Adjuvant-induced arthritis
EXAMPLE 41, was evaluated for anti- inflammatory activity in Complete Freund's Adjuvant (CFA)-induced arthritis in rats. EXAMPLE 41 at 3, 10 and 30 mg/kg was administered orally once daily for 5 consecutive days; the first dose given one hour before CFA was injected into the right hind paw on day 1. The right hind paw volume was measured on day 0 (before CFA), 4 hours after the single CFA injection (denoted day 1) and on days 5, 8, 11, 14, 18 and 21 (acute phase), while the left hind paw (without CFA) volume was measured on days 0 (before CFA), 14, 18 and 21 (delayed phase).
Method
Test Substance and Dosing Pattern
EXAMPLE 41 at 3, 10 and 30 mg/kg was administered orally once daily for 5 consecutive days, with the first dose given one hour before CFA (Complete Freund's Adjuvant) challenge. A dosing volume of 2 mL/kg was used.
Animals
Female Lewis rats weighing 210 ± 10 g were obtained from Charles River USA Inc. Space allocation for 5 animals was 45 x 23 x 21 cm. All animals were maintained in a controlled temperature (21-230C) and humidity (50% - 70%) environment with 12 hours light/dark cycles
for at least three days at test site prior to use. Free access to standard lab chow for rats and water was granted.
EXAMPLE 41 at 3, 10 and 30 mg/kg was administered orally (PO) on day 1 beginning one hour before injection of CFA; once daily dosing was performed for 5 consecutive days. A well-ground suspension of killed Mycobacterium butyricum (0.3 mg in 0.1 mL of light mineral oil; CFA, was administered in a single dose into the subplantar region of the right hind paw 1 hour following the first dose of test substance (denoted day 1). Right hind paw volume was measured by plethysmometry on day 0 (before CFA treatment), day 1 (4 hr after CFA treatment) and days 5, 8, 11, 14, 18 and 21 after CFA treatment.The left paw volume measured days 0 (before CFA treatment), 14, 18 and 21 after administration of CFA in the right paw. For CFA-injected rats, the increase in paw volume (net swelling) on days 1 5, 8, 11, 14, 18 and 21 was obtained by subtracting the volume at day 0 (acute phase of inflammation); and the left paw volume on days 14, 18 and 21 was obtained by subtracting the volume at day 0 (delayed phase of inflammation). The net swelling of each treatment group was then compared to vehicle-treated group and expressed as percentage of inhibition. Anti- inflammatory activity in this model was denoted by values calculated during the acute phase as well as the delayed phase.
Equipment
Animal cage (Allentown, USA), Glass syringe (1 mL, Mitsuba, Japan), Hypodermic needle (27G x 1", Top Corporation, Japan), Stainless forceps (Klappenecker, Germany), Stainless scissors (Klappenecker, Germany) and TERUMO Syringe with the needle (1 mL, TERUMO CO., Philippines). Plethysmometer (PV-Ol Paw volume apparatus, SINGA Technology Co. R. O. C), Rat scale (1 ~ 1000 g, TANITA, Japan) and Rat oral needle (Natsume, Japan), Centrifuge 5810R (Eppendrof, Germany), Eppendorf tube (2 mL, Safe-lock, Eppendorf, Germany), Micropipettes (100 P & 200 P, Gilson, France)
Results
Maximum inhibition of paw swelling in EXAMPLE 41 -treated animals compared to vehicle- treated animals was recorded on day 1 and 14 for acute and delayed phase, respectively (figure qz2).
Conlusion
Oral administration of EXAMPLE 41 inhibits CF A- induced paw swelling in rats.
Claims
1. A compound of formula (I)

(I)
wherein
R1 is selected from branched or unbranched C1-C10 alkyl, C2-C10 alkenyl or C2-C10 alkynyl; C3- Cio cycloalkyl; C4-C10 cycloalkenyl; Cs-Cio cycloalkynyl; C1-Cg heterocyclyl; and C6-CiO aryl; wherein any alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, or cycloalkynyl moiety optionally is substituted with 1, 2 or 3 groups Ra; and any heterocyclyl or aryl moiety optionally is substituted with 1, 2, 3, 4 or 5 groups Rb;
R2 is selected from branched or unbranched C1-Cg alkyl, C2-C9 alkenyl, or C2-C9 alkynyl; C3-C6 cycloalkyl; C4-C6 cycloalkenyl; C8-C10 cycloalkynyl; C1-Cg heterocyclyl; and C6-CiO aryl;
wherein any alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl or cycloalkynyl moiety optionally is substituted with 1 , 2 or 3 groups Ra; and any heterocyclyl or aryl moeiety optionally is substituted with 1, 2, 3, 4 or 5 groups Rb;
R3 and R4 are each independently selected from Ci -C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C3-C4 cycloalkyl; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; Ci-C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; halogen; C1-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amino; and C1-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl carbonyl; wherein any alkyl, alkenyl, alkynyl or cycloalkyl moiety optionally is substituted with 1, 2, or 3 groups Rc; R5 and R6 are independently selected from hydrogen; C1-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; and halogen; wherein any alkyl; alkenyl or alkynyl moiety optionally is substituted with 1, 2, or 3 groups Rc;
X represents a bond or branched or unbranched C1-C3 alkyl, C2-C3 alkenyl or C2-C3 alkynyl;
Q represents carbonyl, sulfonyl or sulfmyl;
m is an integer of 0, 1 or 2; each Rais independently selected from halogen; hydroxy; C1-C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C1-C4 alkyloxy; C2-C4 alkenyloxy; C2-C4 alkynyloxy; C1-C4 alkyl, C2-C4 alkenyl or C2- C4 alkynyl secondary or tertiary amino; C1-C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; wherein any alkyl, alkenyl or alkynyl moiety optionally is substituted with 1, 2 or 3 groups Rc; each Rb is independently selected from halogen; carboxy; hydroxy; cyano; C1-C5 heteroaryl; C1- C4 alkyl; C2-C4 alkenyl; C2-C4 alkynyl; C3-C5 cycloalkyl; Ci-C4 alkyloxy; C2-C4 alkenyloxy; C2- C4 alkynyloxy; C3-C5 cycloalkyloxy; Ci-C4 alkylthio; C2-C4 alkenylthio; C2-C4 alkynylthio; C3- C5 cycloalkylthio; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amino; aminocarbonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary amido; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl carbonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl sulfonyl; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary sulphonamido; Ci-C4 alkyl, C2-C4 alkenyl or C2-C4 alkynyl secondary or tertiary acylsulphonamido; Ci-C4 alkyloxy, C2-C4 alkenyloxy, or C2-C4 alkynyloxy carbonyl; C1-C5 heterocyclyl; wherein any alkyl, alkenyl or alkynyl moiety optionally is substituted with 1, 2 or 3 groups Rc; each Rc is independently selected from fluorine and chlorine; or a pharmaceutically acceptable salt thereof.
2. A compound according to claim 1, wherein
R1 is selected from branched or unbranched C1-C10 alkyl, C3-C10 cycloalkyl; C1-Cg heterocyclyl; and C6-CiO aryl; wherein any alkyl or cycloalkyl moiety optionally is substituted with 1, 2 or 3 groups Ra; and any heterocyclyl or aryl moiety optionally is substituted with 1, 2, 3, 4 or 5 groups Rb;
R2 is selected from branched or unbranched C1-Cg alkyl; C3-C6 cycloalkyl; C1-Cg heterocyclyl; and C6-CiO aryl; wherein any alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl or cycloalkynyl moiety optionally is substituted with 1, 2 or 3 groups Ra; and any heterocyclyl or aryl moeiety optionally is substituted with 1, 2, 3, 4 or 5 groups Rb;
R3 and R4 are each independently selected from Ci -C4 alkyl and halogen, wherein any alkyl moiety optionally is substituted with 1, 2 or 3 groups Rc;
R5 and R6 are independently selected from hydrogen and Ci-C4 alkyl, wherein any alkyl moiety optionally is substituted with 1 , 2 or 3 groups Rc;
X represents a bond or branched or unbranched C1-C3 alkyl;
Q represents carbonyl; m is 0 or 1; each Rais independently selected from hydroxy; C1-C4 alkyloxy; C1-C4 alkyl secondary or tertiary amino; wherein any alkyl moiety optionally is substituted with 1, 2 or 3 groups Rc; each Rb is independently selected from halogen; carboxy; cyano; C1-C5 heteroaryl, C1-C4 alkyl; C3-C5 cycloalkyl; C1-C4 alkyloxy; C3-C5 cycloalkyloxy; aminocarbonyl; C1-C4 alkyl secondary or tertiary amido; C1-C4 alkyl carbonyl; C1-C4 alkyloxy carbonyl; C1-C4 alkyl secondary or tertiary acylsulphonamido; and C1-C5 heterocyclyl; wherein any alkyl, alkenyl or alkynyl moiety optionally is substituted with 1 , 2 or 3 groups Rc; each Rc is fluoro or is absent; or a pharmaceutically acceptable salt thereof.
3. A compound according to claim 1 or claim 2, wherein
R1 is selected from branched or unbranched C1-C10 alkyl; C3-C6 cycloalkyl; 5- or 6-membered C3-C5 heterocyclyl; and phenyl; wherein any alkyl or cycloalkyl moiety optionally is substituted with 1 or 2 groups Ra; and any heterocyclyl or phenyl moiety optionally is substituted with 1 or 2 groups Rb;
or a pharmaceutically acceptable salt thereof.
4. A compound according to claim 3, wherein R1 is selected from branched or unbranched C1- Cio alkyl, optionally substituted with 1 or 2 groups Ra;
or a pharmaceutically acceptable salt thereof.
5. A compound according to claim 3, wherein R1 is selected from C3-C6 cycloalkyl, optionally substituted with 1 or 2 groups Ra;
or a pharmaceutically acceptable salt thereof.
6. A compound according to claim 3, wherein R1 is selected from 5- or 6-membered C3-C5 heterocyclyl optionally substituted with 1 or 2 groups Rb;
or a pharmaceutically acceptable salt thereof.
7. A compound according to any one of the claims 1-6, wherein R2 is selected from branched or unbranched Ci-C6 alkyl; C3-C6 cycloalkyl; 5- or 6-membered C3-C5 heterocyclyl; and phenyl; wherein any alkyl or cycloalkyl moiety optionally is substituted with 1 or 2 groups Ra; and any heterocyclyl or phenyl moiety optionally is substituted with 1 or 2 groups Rb;
or a pharmaceutically acceptable salt thereof.
8. A compound according to claim 7, wherein R2 is selected from C3-C6 cycloalkyl, 5-membered C3-C4 heterocyclyl and 6-membered C3-C5 heterocyclyl, wherein any cycloalkyl moiety optionally is substituted with 1 or 2 groups Ra; and any heterocyclyl moiety optionally is substituted with 1 or 2 groups Rb;
or a pharmaceutically acceptable salt thereof.
9. A compound according to claim 7, wherein R2 is selected from C4-C6 cycloalkyl and 5- or 6- membered C4-C5 heterocyclyl, wherein any cycloalkyl moiety optionally is substituted with 1 or 2 groups Ra; and any heterocyclyl moiety optionally is substituted with 1 or 2 groups Rb; or a pharmaceutically acceptable salt thereof.
10. A compound according to claim 7, wherein R2 is selected from C4-C6 cycloalkyl and 5- membered C3-C4 heterocyclyl, wherein any cycloalkyl moiety optionally is substituted with 1 or 2 groups Ra; and any heterocyclyl moiety optionally is substituted with 1 or 2 groups Rb;
or a pharmaceutically acceptable salt thereof.
11. A compound according to claim 7, wherein R2 is selected from pentyl, optionally substituted with 1 or 2 groups Ra; and tetrahydrofuryl, optionally substituted with 1 or 2 groups Rb;
or a pharmaceutically acceptable salt thereof.
12. A compound according to claim 7, wherein R2 is selected from branched or unbranched C1-
C6 alkyl;
or a pharmaceutically acceptable salt thereof.
13. A compound according to any one of the claims 1-12, wherein R3 and R4 are each independently selected from methyl and chloro, wherein any methyl optionally is substituted with 1 , 2 or 3 groups Rc;
or a pharmaceutically acceptable salt thereof.
14. A compound according to any one of the claims 1-13, wherein R5 and R6 are each independently selected from hydrogen and methyl, wherein any methyl optionally is substituted with 1 , 2 or 3 groups Rc;
or a pharmaceutically acceptable salt thereof.
15. A compound according to claim 14, wherein R5 and R6 are each hydrogen;
or a pharmaceutically acceptable salt thereof.
16. A compound according to claim 14, wherein R5 is hydrogen and R6 is methyl;
or a pharmaceutically acceptable salt thereof.
17. A compound according to any one of the claims 1-16, wherein X represents a bond or methylene;
or a pharmaceutically acceptable salt thereof.
18. A compound according to claim 17, wherein X represents a bond;
or a pharmaceutically acceptable salt thereof.
19. A compound according to any one of the claims 1 to 18, wherein m is 0;
or a pharmaceutically acceptable salt thereof.
20. A compound according to any one of the claims 1 to 19, wherein Rc is absent;
or a pharmaceutically acceptable salt thereof.
21. A compound according to claim 1, wherein R1 represents C1-C10 alkyl; R2 represents C4-C6 cycloalkyl or C4-C5 heterocyclyl; R3 and R4 are independently selected from methyl and chlorine; R5 is hydrogen; X represents a bond; and Q represents carbonyl;
or a pharmaceutically acceptable salt thereof.
22. A compound according to claim 1, wherein R1 represents C3-C6 cycloalkyl; R2 represents C4- C6 cycloalkyl or C4-C5 heterocyclyl; R3 and R4 are independently selected from methyl and chlorine; X represents a bond and Q represents carbonyl;
or a pharmaceutically acceptable salt thereof.
23. A compound according to claim 1, wherein R1 represents phenyl or C4-C5 heterocyclyl; R2 represents C4-C6 cycloalkyl or C4-C5 heterocyclyl; R3 and R4 are independently selected from methyl or chlorine; X represents a bond; and Q represents carbonyl;
or a pharmaceutically acceptable salt thereof.
24. A compound according to claim 1, wherein R1 represents phenyl or C4-C5 heterocyclyl, optionally substituted with one or more Rb; R2 represents cyclopentyl; R3 and R4 are
independently selected from methyl and chlorine; the integer m is equal to 1 or 0 (zero); R5 and R6 are hydrogen; X represents a bond; and Q represents carbonyl;
or a pharmaceutically acceptable salt thereof.
25. A compound according to claim 1, wherein R1 represents Ci -C 10 alkyl; R2 represents C1-C9 alkyl; R3 and R4 are independently selected from methyl and chlorine; R5 and R6 are hydrogen; and Q represents carbonyl;
or a pharmaceutically acceptable salt thereof.
26. A compound according to claim 1, wherein R1 represents C4-C5 saturated heterocyclyl; R2 represents cyclopentyl; R3 and R4 are independently selected from methyl and chlorine; the iinntteeggeerr mm iiss eeqquuaall ttoo 11 ;; RR55 aanndd RR66 aarree hhyyddrrooggξen; X represents a bond; and Q represents carbonyl; or a pharmaceutically acceptable salt thereof.
27. A compound according to claim 1, wherein R1 represents C3-C5 cycloalkyl; R2 represents cyclopentyl or tetrahydrofuranyl; R3 and R4 are independently selected from methyl and chlorine; the integer m is equal to either 0 (zero) or 1 ; R5 and R6 are hydrogen; X represents a bond; and Q represents carbonyl;
or a pharmaceutically acceptable salt thereof.
28. A compound according to claim 1, selected from
Λ/-cyclopentyl-l-(l-ethyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide; Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -propyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide; l-(l-butyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4-carboxamide;
Λ/-cyclopentyl-l-(l-heptyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -octyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -pentyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide; Λ/-cyclopentyl- 1 -( 1 -hexyl-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -(pyridin-2-ylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -( 1 -isopropyl-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(2-(diethylamino)ethyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)piperidine-
4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-4-ylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-3-ylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-2-ylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
1 -( 1 -sec-butyl-5 ,6-dichloro- lH-benzo [d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(pentan-3-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5 ,6-dichloro- 1 -cyclobutyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l -(5,6-dichloro- l-(l-phenylethyl)-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -methyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -ethyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5 ,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-fluorobenzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(3,5-difluorobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
methyl 4-((2-(4-(cyclopentylcarbamoyl)piperidin- 1 -yl)-5,6-dimethyl- lH-benzo[d]imidazol- 1 - yl)methyl)benzoate;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -((2-methylthiazol-4-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -phenyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
JV-cyclopropyl- 1 -(5 ,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclo butyl- 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
1 -(5,6-dichloro- l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-propylpiperidine-4-carboxamide;
1 -(5,6-dichloro- l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-(2-methoxyethyl)piperidine-4- carboxamide;
1 -(5,6-dichloro- l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-(isopropoxymethyl)piperidine-4- carboxamide;
1 -(5,6-dichloro- l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
Λ/-(cyclohexylmethyl)-l -(5,6-dichloro- l-isopropyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
1 -(5,6-dichloro- l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-((tetrahydro-2H-pyran-4- yl)methyl)piperidine-4-carboxamide;
Λ/-butyl- 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-(cyclopentylmethyl)- 1 -(5,6-dichloro- 1 -isopropyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-((6-fluoropyridin-3-yl)methyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
l-(l-(4-cyanobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(cyclopropylmethyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -((tetrahydrofuran-2-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
1 -( 1 -(4-( IH- 1 ,2,4-triazol- 1 -yl)benzyl)-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide;
1 -( 1 -cyclobutyl-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
l-(l-(4-bromobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -(4-(methylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl-l-(l-(4-(dimethylcarbamoyl)benzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
l-(l-(4-carbamoylbenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5 ,6-dimethyl- 1 -(4-(methylsulfonylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3-yl)piperidine-4- carboxamide;
1 -( 1 -(4-fluorobenzyl)-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)-JV-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
1 -(5-chloro- 1 -cyclobutyl-6-methyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentyl-piperidine-4- carboxamide;
1 -(6-chloro- 1 -cyclobutyl-5 -methyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -cyclopentyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
l-(l-(4-bromobenzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -((6-fluoropyridin-3-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-cyanobenzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(cyclopropylmethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -((tetrahydrofuran-2-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -((tetrahydro-2H-pyran-4-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5 ,6-dichloro- 1 -(4-(methylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-(dimethylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
l-(l-(4-carbamoylbenzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-cyclopentyl-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4- carboxamide;
l-(l-(4-chlorobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
l-(l-(4-(lH-tetrazol-5-yl)benzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5 ,6-dichloro- 1 -(4-(methylsulfonylcarbamoyl)benzyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
N-butyl- 1 -(5,6-dichloro- 1 -cy clo butyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-butyl- 1 -(5 ,6-dichloro- 1 -pentyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
N-butyl- 1 -(5 ,6-dichloro- 1 -(4-fluorobenzyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-butyl- 1 -(5,6-dichloro- 1 -((6-fluoropyridin-3-yl)methyl)- lH-benzo[d]imidazol-2-yl)piperidine- 4-carboxamide;
1 -(5,6-dichloro- 1-cyclo butyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
1 -(5,6-dichloro- l-pentyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
1 -(5,6-dichloro- l-(4-cyanobenzyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(5,6-dichloro- l-(cyclopropylmethyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(5,6-dichloro- l-((6-fluoropyridin-3-yl)methyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -((tetrahydro-2H-pyran-4-yl)methyl)- lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
1 -(5,6-dichloro- l-cyclopentyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
l-(l-(4-bromobenzyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide; l-(l-(4-(lH-tetrazol-5-yl)benzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- cyclopentylpiperidine-4-carboxamide;
l-(5,6-dichloro-l-(4-fluorobenzyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
1.( 1.(4-( IH- 1 ,2,4-triazol- 1 -yl)benzyl)-5 ,6-dichloro- lH-benzo[d]imidazol-2-yl)-JV- cyclopentylpiperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(2-methoxyethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(4-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
l-(l-(cyclobutylmethyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
JV-cyclopentyl- 1 -(5,6-dimethyl- 1 -(prop-2-ynyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(2-ethylbutyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5 ,6-dimethyl- 1 -neopentyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
l-(l-cyclohexyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
l-(l-sec-butyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentyl-piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(2-hydroxyethyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -( 1 ,5 ,6-trimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl-l-(l-isobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide; l-(l-benzyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4-carboxamide;
Λ/-cyclopentyl-l-(l-(2-methoxyethyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(4-fluorobenzyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
1 -(5-chloro- 1 -cyclobutyl-6-fluoro- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
1 -(6-chloro- 1 -cyclobutyl-5-fluoro- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -(pentan-2-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-butyl-l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-butyl-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
N-butyl- 1 -(5,6-dimethyl- 1 -pentyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
TV-butyl- 1 -( 1 -((6-fluoropyridin-3 -yl)methyl)-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)piperidine-
4-carboxamide;
Λ/-butyl- 1 -( 1 -(4-fluorobenzyl)-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-benzyl-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide; JV-(4-fluorobenzyl)- 1 -(I -isopropyl-5,6-dimethyl- 1 H-benzo [d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-(3-isopropoxypropyl)-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-(3,3-dimethylbutyl)-l-(l-isopropyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-(4-bromobenzyl)- 1 -( 1 -isopropyl-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)piperidine-4- carboxamide;
Λ/-cyclohexyl- 1 -( 1 -isopropyl-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-(4-cyanobenzyl)- 1 -( 1 -isopropyl-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)piperidine-4- carboxamide;
(S)- 1 -(5,6-dimethyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3-yl)piperidine-4- carboxamide;
4-((2-(4-(cyclopentylcarbamoyl)piperidin- 1 -yl)-5,6-dimethyl- lH-benzo[d]imidazol- 1 - yl)methyl)benzoic acid
1 -(I -isopropyl-5, 6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(thiazol-2-ylmethyl)piperidine-4- carboxamide;
l-(5,6-dichloro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-(thiazol-2-ylmethyl)piperidine-4- carboxamide;
Λ/-(4-(lH-tetrazol-5-yl)benzyl)-l -(I -isopropyl-5, 6-dimethyl-lH-benzo[d]imidazo 1-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -(pentan-3-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
1 -(5,6-dimethyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-fluorophenyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
l-(l-(4-bromophenyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
l-(l-(4-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
l-(l-(3-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
l-(l-(3,5-difluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
l-(l-(4-chlorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
1 -(5,6-dimethyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
l-(l-(3,5-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
1 -(5,6-dimethyl- 1 -(naphthalen-2-yl)- lH-benzo[d]imidazol-2-yl)-JV-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -( 1 -(benzo[d] [1,3] dioxo 1-5 -yl)-5 ,6-dimethyl- lH-benzo [d]imidazo l-2-yl)-JV-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
1 -(5,6-dichloro- l-(3-methoxyphenyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
l-(l-(benzo[d][l,3]dioxol-5-yl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
1 -(5,6-dichloro- l-(4-cyanophenyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -m-tolyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -( 1 -(3 ,4-dimethylphenyl)-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)piperidine-4- carboxamide;
N-cyclopentyl-l-(l-(4-fluoro-3-methylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl-l-(l-(4-ethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl-l-(l-(4-isopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -(quinolin-6-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dimethyl- 1 -(quinoxalin-6-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
l-(5,6-dichloro-l-(4-(methylcarbamoyl)phenyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide;
(R)-l-(5,6-dichloro-l-(3-methoxyphenyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-3-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
JV-cyclopentyl- 1 -(5,6-dichloro- 1 -(pyridin-3-yl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(6-methoxypyridin-2-yl)- lH-benzo[d]imidazol-2-yl)piperidine-
4-carboxamide;
Λ/-cyclopentyl- 1 -(5,6-dichloro- 1 -(4-cyanophenyl)- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-fluorophenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3 -fluorophenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-methoxyphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-tert-butylphenyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-(trifluoromethoxy)phenyl)- lH-benzo[d]imidazol-2-yl)-Λ/- (tetrahydrofuran-3-yl)piperidine-4-carboxamide;
1 -(5,6-dichloro- l-phenyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
1 -(5,6-dichloro- l-(pyridin-2-yl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-
4-carboxamide;
l-(5,6-dichloro-l-(pyridin-3-yl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-
4-carboxamide;
l-(5,6-dichloro-l-p-tolyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
(R)-l-(l-(4-chloro-3-methoxyphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-chloro-3-methoxyphenyl)- lH-benzo[d]imidazol-2-yl)-JV-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;.
(R)-l-(l-(2,3-dihydrobenzoflιran-5-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(2,3-dihydrobenzofuran-5-yl)- lH-benzo[d]imidazol-2-yl)-JV-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(3,5-diethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(5,6-dichloro- l-(cyclobutylmethyl)-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-fluoro-3-methylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(4-ethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3,4-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -(quinolin-6-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-isopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-tert-butylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3-chlorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(6-chloro-5 -methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
(R)- 1 -(5-chloro-6-methyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(β-chloro-S-methyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-/V-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
l-(l-(4-carbamoylphenyl)-5-chloro-6-methyl-lH-benzo[d]imidazol-2-yl)-Λ/- cyclopentylpiperidine-4-carboxamide;
l-(l-(4-carbamoylphenyl)-6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)-Λ/- cyclopentylpiperidine-4-carboxamide;
(R)-l-(l-(4-carbamoylphenyl)-6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)-Λ/- (tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5-chloro-6-methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]-imidazol-2-yl)-/V-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
1 -(5-chloro-6-methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-/V- cyclopentylpiperidine-4-carboxamide;
1 -(5-chloro-6-methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-/V-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(6-chloro-5-methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]-imidazol-2-yl)-/V-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
1 -(6-chloro-5 -methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-Λ/- (tetrahydrofuran-3-yl)piperidine-4-carboxamide;
1 -(6-chloro-5 -methyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-Λ/- cyclopentylpiperidine-4-carboxamide;:
(R)- 1 -(6-chloro-5-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(S)- l-(6-chloro-5-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
Λ/-butyl- 1 -(5-chloro-6-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide; TV- butyl- 1 -(6-chloro-5-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)piperidine-4-carboxamide;
1 -(5-chloro-6-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine- 4-carboxamide;
1 -(6-chloro-5 -methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3-yl)piperidine-
4-carboxamide;
1 -(5-chloro- 1 -cyclobutyl-6-methyl- lH-benzo[d]imidazol-2-yl)-7V-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(6-chloro- 1 -cyclobutyl-5 -methyl- lH-benzo[d]imidazol-2-yl)-JV-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
1 -(5-chloro- 1 -(cyclobutylmethyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-
4-carboxamide;
1 -(6-chloro- 1 -(eye Io butylmethyl)-5 -methyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-
4-carboxamide;
1 -(5-chloro-6-methyl- 1 -pentyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
1 -(6-chloro-5 -methyl- 1 -pentyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
1 -(5-chloro- 1 -(4-fluorophenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
1 -(6-chloro- 1 -(4-fluorophenyl)-5 -methyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
1 -(5-chloro-6-methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
1 -(6-chloro-5 -methyl- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
Λ/-butyl- 1 -(5-chloro- l-cyclobutyl-6-methyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
Λ/-butyl- 1 -(6-chloro- 1 -cyclobutyl-5 -methyl- lH-benzo[d]imidazol-2-yl)piperidine-4- carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -(3-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -(5-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(3-ethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3-isopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(6-chloro- l-(4-isopropylphenyl)-5 -methyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-chlorophenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-fluoro-3-methylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5 ,6-dichloro- 1 -(3 ,5-difluorophenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-cyanophenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(5-methylpyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(6-methoxypyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-isopropylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-methylpyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(6-methylpyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-(difluoromethoxy)phenyl)- lH-benzo[d]imidazol-2-yl)-JV-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-(trifluoromethoxy)phenyl)- lH-benzo[d]imidazol-2-yl)-JV-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-isobutylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5 ,6-dichloro- 1 -(3 ,4-dimethylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-chlorophenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(5-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(5-fluoropyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -( 1 -(4-isobutylphenyl)-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)-JV-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
(R)-l-(6-chloro-l-(3,5-dimethylphenyl)-5-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(4-chloro-3-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-
3 -yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-chloro-3 -fluorophenyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3,5-diethylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -( 1 -(3 -tert-butylphenyl)-5 ,6-dimethyl- 1 H-benzo [d]imidazol-2-yl)-N-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
(R)-l-(l-(3-tert-butylphenyl)-5,6-dichloro-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -phenyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-ethylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -m-to IyI- 1 H-benzo [d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3 -yl)piperidine-4- carboxamide;
(R)- 1 -(5 ,6-dichloro- 1 -(3 ,5-dimethylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-ethylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(6-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-ethoxyphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-isopropoxyphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-propoxyphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -( 1 -(4-fluoro-3 ,5-dimethylphenyl)-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-chloro-3-methylphenyl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-chloro-4-methylphenyl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5 ,6-dimethyl- 1 -phenyl- 1 H-benzo [d]imidazol-2-yl)-JV-(tetrahydrofuran-3 -yl)piperidine-4- carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3-yl)piperidine-4- carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -m-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3-yl)piperidine-4- carboxamide;
(R)-l-(l-(3-cyanophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -(naphthalen-2-yl)- lH-benzo[d]imidazol-2-yl)-JV-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-chloro-3-methylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(3-chloro-4-methylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(benzo[d]thiazol-6-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(benzo[d]thiazol-5-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(R)-l-(5-chloro-l-(3,4-dimethylphenyl)-6-methyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(6-chloro-l-(3,4-dimethylphenyl)-5-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5-chloro- l-(4-isopropylphenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(6-chloro- l-(4-isopropylphenyl)-5 -methyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran- 3-yl)piperidine-4-carboxamide;
(S)- l-(5-chloro-6-methyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(S)- l-(6-chloro-5-methyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydroflιran-3- yl)piperidine-4-carboxamide;
(S)-l-(6-chloro-l-(3,5-dimethylphenyl)-5-methyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-((15f,25)-2- hydroxycyclopentyl)piperidine-4-carboxamide;
l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-((lR,2R)-2- hydroxycyclopentyl)piperidine-4-carboxamide;
(R)- 1 -( 1 -(benzo furan-5 -yl)-5 ,6-dimethyl- lH-benzo [d]imidazo l-2-yl)-JV-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-fluoro-3,5-dimethylphenyl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(3-fluoro-5-methylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-fluoro-5-methylphenyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-cyclopropylphenyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-cyclopropylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -( 1 -(benzo furan-6-yl)-5 ,6-dimethyl- lH-benzo [d]imidazo l-2-yl)-N-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
(R)- 1 -(5-chloro- l-(4-ethylphenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(6-chloro- l-(4-ethylphenyl)-5 -methyl- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5-chloro- l-(4-ethylphenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-tert-butylphenyl)-6-chloro-5-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran- 3-yl)piperidine-4-carboxamide;
(R)-l-(l-(4-cyclopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3-cyclopropylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-(cyclopentyloxy)phenyl)- lH-benzo[d]imidazol-2-yl)-JV-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-cyclobutoxyphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
l-(5-chloro-6-fluoro-l-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
l-(6-chloro-5-fluoro-l-methyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
1 -(5-chloro- 1 -ethyl-6-fluoro- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
1 -(6-chloro- 1 -ethyl-5-fluoro- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
l-(5-chloro-6-fluoro-l-isopropyl-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
1 -(6-chloro-5-fluoro- 1-isopropyl- lH-benzo[d]imidazol-2-yl)-JV-cyclopentylpiperidine-4- carboxamide;
l-(l-butyl-5-chloro-6-fluoro-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
l-(l-butyl-6-chloro-5-fluoro-lH-benzo[d]imidazol-2-yl)-Λ/-cyclopentylpiperidine-4- carboxamide;
1 -(5 ,6-dimethyl- 1 -p-tolyl- 1 H-benzo [d]imidazo l-2-yl)-JV-(tetrahydrofuran-3 -yl)piperidine-4- carboxamide;
(R)- 1 -(6-chloro- l-(4-fluorophenyl)-5 -methyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5-chloro- l-(4-fluorophenyl)-6-methyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(6-chloro-5-methyl- 1 -m-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5-chloro-6-methyl- 1 -m-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5-chloro-6-methyl- 1 -(5-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(6-chloro-5-methyl- 1 -(5-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-JV-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-((2S)-2- methoxycyclopentyl)piperidine-4-carboxamide;
l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-((2S)-2- ethoxycyclopentyl)piperidine-4-carboxamide;
l-(l-cyclobutyl-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-N-((2S)-2- propoxycyclopentyl)piperidine-4-carboxamide;
l-(5,6-dichloro-l-(4-(dimethylcarbamoyl)phenyl)-lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)-N-((lS,2S)-2- hydroxycyclopentyl)piperidine-4-carboxamide;
l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)-N-((lS,2S)-2- methoxycyclopentyl)piperidine-4-carboxamide;
l-(5,6-dichloro-l-cyclobutyl-lH-benzo[d]imidazol-2-yl)-N-((lS,2S)-2- ethoxycyclopentyl)piperidine-4-carboxamide;
1 -( 1 -cyclobutyl-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)-Λ/-(furan-2-ylmethyl)piperidine-4- carboxamide;
1 -( 1 -cyclobutyl-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)-Λ/-(thiophen-2-ylmethyl)piperidine-4- carboxamide;
(R)- 1 -( 1 -(5 -bromopyridin-2-yl)-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)-Λ/-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
(R)-l-(l-(5-ethylpyridin-2-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3-chloro-5-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3 -chloro-5 -fluorophenyl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-
3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(3-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(2,2-difluorobenzo[d] [ 1 ,3]dioxol-5-yl)- lH-benzo[d]imidazol-2-yl)-N- (tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(S)- 1 -( 1 -(3 ,5-dimethylphenyl)-5 ,6-dimethyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(5-chloropyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(5-fluoro-6-methylpyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-JV-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)-l-(l-(3-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3,5-dimethylphenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-fluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(3,5-difluorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)-l-(l-(4-chlorophenyl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -(4-(trifluoromethyl)phenyl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -p-tolyl- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3-yl)piperidine-4- carboxamide;
(R)-l-(l-(5-chloropyridin-2-yl)-5,6-dimethyl-lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dimethyl- 1 -(4-(trifluoromethyl)pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -( 1 -(5 -fluoro-6-methylpyridin-2-yl)-5 ,6-dimethyl- 1 H-benzo [d]imidazo l-2-yl)-N-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-ethoxyphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3- yl)piperidine-4-carboxamide;
(R)- 1 -(5,6-dichloro- 1 -(4-methoxy-3-methylphenyl)- lH-benzo[d]imidazol-2-yl)-Λ/-
(tetrahydrofuran-3-yl)piperidine-4-carboxamide; and
(R)- 1 -( 1 -cyclobutyl-5 ,6-dimethyl- 1 H-benzo [d]imidazol-2-yl)-Λ/-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide;
or a pharmaceutically acceptable salt thereof.
29. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use as a medicament.
30. A pharmaceutical composition comprising a therapeutically effective amount of a compound according to any of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, in admixture with at least one pharmaceutically acceptable adjuvant, diluent or carrier.
31. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prevention of an inflammatory disease, said disease preferably being selected from rheumatoid arthritis, inflammatory bowel disease, osteoarthritis, psoriasis, eczema, swelling and periodontitis.
32. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prevention of nociceptive pain, said pain preferably being selected from inflammatory pain, post-operative pain, fibromyalgia, dysmenorrhea, migraine, arthrotic pain, arthritic pain, pain in connection with osteoarthritis, cephalalgia, pain due to gall-stones, pain due to metastasis, pain due to ankylosing spondylitis, pain due to gout, toothache and pain due to rheumatic fever, or for use in the preemptive treatment of surgical pain.
33. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prevention of an autoimmune disease, said autoimmune disease preferably being selected from rheumatoid arthritis and multiple sclerosis.
34. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prevention of a breathing disorder, said disorder preferably being selected from apnea or other respiratory distress symptoms in adults and children, sudden infant death syndrome (SIDS), asthma and other chronic obstructive lung- diseases and sarcoidosis.
35. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prevention of cancer, said cancer preferably being selected from familial adenomatous polyposis (FAP) condition, colorectal cancer, breast cancer, gastric tumorigenesis and epithelial ovarian cancer, or cellular neoplastic transformations and metastatic tumor growth.
36. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prevention of a cardiovascular disease due to atherosclerosis, such as myocardial infarction and stroke.
37. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prevention of fever-related or inflammation-related anorexia.
38. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prevention of Alzheimer's disease.
39. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prevention of endometriosis.
40. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prevention of hemophilic arthropathy.
41. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prevention of diabetic retinopathy.
42. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prevention of and tumor angiogenesis.
43. A compound according to any one of the claims 1 to 28, or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prevention of premature labor.
44. A combination product comprising: (A) a compound according to any one of the claims 1 to 28, or a pharmaceutically-acceptable salt thereof, and (B) another therapeutic agent, wherein each of components (A) and (B) is formulated in admixture with a pharmaceutically-acceptable adjuvant, diluent or carrier.
45. A combination product as claimed in claim 44, wherein the other therapeutic agent is an agent useful in the treatment of inflammation.
46. A combination product as claimed in claim 44 or claim 45, which comprises a kit of parts comprising components: (A) a pharmaceutical formulation including a compound as defined above, or a pharmaceutically-acceptable salt thereof, in admixture with a pharmaceutically- acceptable adjuvant, diluent or carrier; and (B) a pharmaceutical formulation including another therapeutic agent in admixture with a pharmaceutically acceptable adjuvant, diluent or carrier, which components (A) and (B) are each provided in a form that is suitable for administration in conjunction with the other.
47. A method of treatment of a disorder selected from an inflammatory disease, nociceptive pain, an autoimmune disease, a breathing disorder, cancer, a cardiovascular disease due to
atherosclerosis, fever or inflammation-related anorexia, or Alzheimer's disease, which method comprises administration of a therapeutically effective amount of a compound according to any one of the claims 1 to 28, or a pharmaceutically-acceptable salt thereof, to a mammal subject suffering from, or susceptible to, such a disorder.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP09168830.9 | 2009-08-27 | ||
| EP09168830 | 2009-08-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011023812A1 true WO2011023812A1 (en) | 2011-03-03 |
Family
ID=41181063
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2010/062596 WO2011023812A1 (en) | 2009-08-27 | 2010-08-27 | Microsomal prostaglandin e synthase-1 (mpges1) inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011023812A1 (en) |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2495244A1 (en) * | 2011-03-02 | 2012-09-05 | NovaSaid AB | Piperidinyl benzoimidazole derivatives as mPGEs-1 inhibitors |
| US20130210844A1 (en) * | 2012-02-09 | 2013-08-15 | Glenmark Pharmaceuticals S.A. | BICYCLIC COMPOUNDS AS mPGES-1 INHIBITORS |
| WO2013146970A1 (en) * | 2012-03-29 | 2013-10-03 | 第一三共株式会社 | Novel quinoline derivative |
| WO2013146969A1 (en) * | 2012-03-29 | 2013-10-03 | 第一三共株式会社 | Novel disubstituted cyclohexane derivative |
| WO2014204371A1 (en) * | 2013-06-20 | 2014-12-24 | Cadila Pharmaceuticals Ltd. | Piperidinyl benzoimidazole derivatives as mpge-1 inhibitors |
| WO2014204370A1 (en) * | 2013-06-20 | 2014-12-24 | Cadila Pharmaceuticals Ltd. | Novel piperidinyl benzoimidazole derivatives as mpges-1 inhibitors |
| CN105968112A (en) * | 2016-05-16 | 2016-09-28 | 青岛云天生物技术有限公司 | Method for preparing linagliptin intermediate for treating II-type diabetis |
| KR20170055025A (en) * | 2014-10-29 | 2017-05-18 | 일라이 릴리 앤드 캄파니 | Novel methyl-piperidine compounds useful for inhibiting microsomal prostaglandin e2 synthase-1 |
| JP2019510079A (en) * | 2016-02-25 | 2019-04-11 | ジェシンタ・ファーマ・アーベー | Methods of treating diseases characterized by vasoconstriction |
| WO2019101826A1 (en) | 2017-11-22 | 2019-05-31 | Khondrion Ip B.V. | Compounds as mpges-1 inhibitors |
| US10501421B1 (en) | 2017-01-27 | 2019-12-10 | Vanderbilt University | Substituted benzimidazoles as modulators of Ras signaling |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| WO2021076908A1 (en) | 2019-10-18 | 2021-04-22 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| WO2021087064A1 (en) | 2019-10-31 | 2021-05-06 | Forty Seven, Inc. | Anti-cd47 and anti-cd20 based treatment of blood cancer |
| WO2021096860A1 (en) | 2019-11-12 | 2021-05-20 | Gilead Sciences, Inc. | Mcl1 inhibitors |
| WO2021130638A1 (en) | 2019-12-24 | 2021-07-01 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| WO2021132472A1 (en) | 2019-12-25 | 2021-07-01 | 日本新薬株式会社 | Prophylactic and/or therapeutic agent for chronic prostatitis/chronic pelvic pain syndrome |
| WO2021163064A2 (en) | 2020-02-14 | 2021-08-19 | Jounce Therapeutics, Inc. | Antibodies and fusion proteins that bind to ccr8 and uses thereof |
| WO2021222522A1 (en) | 2020-05-01 | 2021-11-04 | Gilead Sciences, Inc. | Cd73 inhibiting 2,4-dioxopyrimidine compounds |
| US11332459B2 (en) | 2017-10-19 | 2022-05-17 | Teijin Pharma Limited | Benzimidazole derivatives and their uses |
| WO2022221304A1 (en) | 2021-04-14 | 2022-10-20 | Gilead Sciences, Inc. | CO-INHIBITION OF CD47/SIRPα BINDING AND NEDD8-ACTIVATING ENZYME E1 REGULATORY SUBUNIT FOR THE TREATMENT OF CANCER |
| WO2022245671A1 (en) | 2021-05-18 | 2022-11-24 | Gilead Sciences, Inc. | Methods of using flt3l-fc fusion proteins |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2023076983A1 (en) | 2021-10-28 | 2023-05-04 | Gilead Sciences, Inc. | Pyridizin-3(2h)-one derivatives |
| WO2023122615A1 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023122581A2 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023147418A1 (en) | 2022-01-28 | 2023-08-03 | Gilead Sciences, Inc. | Parp7 inhibitors |
| EP4245756A1 (en) | 2022-03-17 | 2023-09-20 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023183817A1 (en) | 2022-03-24 | 2023-09-28 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| WO2023196784A1 (en) | 2022-04-05 | 2023-10-12 | Gilead Sciences, Inc. | Combinations of antibody therapies for treating colorectal cancer |
| WO2023205719A1 (en) | 2022-04-21 | 2023-10-26 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2024006929A1 (en) | 2022-07-01 | 2024-01-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2024064668A1 (en) | 2022-09-21 | 2024-03-28 | Gilead Sciences, Inc. | FOCAL IONIZING RADIATION AND CD47/SIRPα DISRUPTION ANTICANCER COMBINATION THERAPY |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008084218A1 (en) | 2007-01-12 | 2008-07-17 | Boehringer Ingelheim International Gmbh | Benzazole derivatives for the treatment of inflammations |
-
2010
- 2010-08-27 WO PCT/EP2010/062596 patent/WO2011023812A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008084218A1 (en) | 2007-01-12 | 2008-07-17 | Boehringer Ingelheim International Gmbh | Benzazole derivatives for the treatment of inflammations |
Non-Patent Citations (64)
| Title |
|---|
| "A Textbook ofdrug Design and Development", 1991, HARWOOD ACADEMIC PUBLISHERS, pages: 113 - 191 |
| "Remington: The Science and Practice of Pharmacy,19th ed", 1995, MACK PRINTING COMPANY |
| BASEVICH ET AL., BIOORG. KHIM., vol. 9, no. 5, 1983, pages 658 - 665 |
| BELETSKAYA, I, P.; SIGEEV, A. S.; PEREGUDOV, A. S.; PETROVSKII, P. V.: "Catalytic Sandmeyer Bromination", SYNTHESIS, 2007, pages 2534 - 2538 |
| BERKENSTAM, A. ET AL., PROC. NATL. ACAD. SCI. USA., vol. 105, 2008, pages 663 - 667 |
| BUNDEGAARD, H.: "Design of Prodrugs", 1985, ELESEVIER, pages: 1 - 92 |
| CAMILLE G. WERMUTH ET AL.: "The Practice ofmedicinal Chemistry", 1996, ACADEMIC PRESS |
| CHAUDHRY, U. A.; ZHUANG, H.; CRAIN, B. J.; DORE, S.: "Elevated microsomal prostaglandin-E synthase-1 in Alzheimer's disease", ALZHEIMERS DEMENT., vol. 4, 2008, pages 6 - 13 |
| DEY, 1.; GIEMBYCZ, M. A.; CHADEE, K.: "Prostaglandin E(2) couples through EP(4) prostanoid receptors to induce IL-8 production in human colonic epithelial cell lines", BR. J. PHARMACOL., vol. 156, 2009, pages 475 - 85 |
| DOYLE, M. P.; SIEGFRIED, B.; DELLARIA JR, J. F.: "Alkyl nitrite- metal halide deamination reactions. 2. Substitutive deamination of arylamines by alkyl nitrites and copper(II) halides. A direct and remarkably efficient conversion of arylamines to aryl halides", J. ORG. CHEM., vol. 42, 1977, pages 2426 - 2431 |
| E.D. WILLS., BIOCHEM. J., vol. 113, 1969, pages 315 |
| ELANDER, N.; UNGERBÄCK, J.; OLSSON, H.; UEMATSU, S.; AKIRA, S.; S6DERKVIST, P.: "Genetic deletion of mPGEs-1 accelerates intestinal tumorigenesis in APC(Min/+) mice", BIOCHEM. BIOPHYS. RES. COMMUN., vol. 372, 2008, pages 249 - 253 |
| ENGBLOM, D.; SAHA, S.; ENGSTR6M, L.; WESTMAN, M.; AUDOLY, L. P.; JAKOBSSON, P. J.; BLOMQVIST, A.: "Microsomal prostaglandin E synthase-1 is the central switch during immune-induced pyresis", NAT. NEUROSCI., vol. 6, 2003, pages 1137 - 1138 |
| ERION, MD ET AL., PROC. NATL. ACAD. SCI. USA., vol. 104, 2007, pages 15490 - 15495 |
| FAHMI, H.: "mPGEs-1 as a novel target for arthritis", CURR. OPIN. RHEUMATOL., vol. 16, 2004, pages 623 - 627 |
| FLATT, B. ET AL., J. MED. CHEM., vol. 52, 2009, pages 904 - 907 |
| GROVER, G. J. ET AL., PROC. NATL. ACAD. SCI. USA., vol. 100, 2003, pages 10067 - 10072 |
| HOFSTETTER, A. 0.; SAHA, S.; SILJEHAV, V.; JAKOBSSON, P. J.; HERLENIUS, E.: "The induced prostaglandin E2 pathway is a key regulator of the respiratory response to infection and hypoxia in neonates", PROC. NATL. ACAD. SCI. USA., vol. 104, 2007, pages 9894 - 9899 |
| HOOZEMANS, J. J.; VEERHUIS, R.; JANSSEN, I.; VAN ELK, E. J.; ROZEMULLER, A. J.; EIKELENBOOM, P.: "The role of cyclooxygenase 1 and 2 activity in prostaglandin E(2) secretion by cultured human adult microglia: implications for Alzheimer's disease", BRAIN RES., vol. 951, 2002, pages 218 - 226 |
| IKEDA-MATSUO, Y.; OTA, A.; FUKADA, T.; UEMATSU, S.; AKIRA, S.; SASAKI, Y.: "Microsomal prostaglandin E synthase-1 is a critical factor of stroke-reperfusion injury", PROC. NATL. ACAD. SCI. USA., vol. 103, 2006, pages 11790 - 11795 |
| J. F. W. MCOMIE: "Protecting Groups in Organic Chemistry", 1973, PLENUM PRESS |
| JACHAK S M: "PGE synthase inhibitors as an alternative to COX-2 inhibitors", CURRENT OPINION IN INVESTIGATIONAL DRUGS, PHARMAPRESS, US, vol. 8, no. 5, 1 May 2007 (2007-05-01), pages 411 - 415, XP009124702, ISSN: 1472-4472 * |
| JACHAK, S. M.: "PGE synthase inhibitors as an alternative to COX-2 inhibitors", CURR. OPIN. INVESTIG. DRUGS, vol. 8, 2007, pages 411 - 415 |
| JERRY MARCH: "Advanced Organic Chemistry, 4th edition", 1992, WILEY-INTERSCIENCE PUBLICATION, pages: 499 |
| JERRY MARCH: "Advanced Organic Chemistry,4th edition,", 1992, WILEY-INTERSCIENCE PUBLICATION, pages: 656 - 657 |
| JOHANSSON, L. ET AL., PROC. NATL. ACAD. SCI. USA., vol. 102, 2005, pages 10297 - 10302 |
| KAMEI, D.; MURAKAMI, M.; NAKATANI, Y.; ISHIKAWA, Y.; ISHII, T.; KUDO, I.: "Potential role of microsomal prostaglandin E synthase-1 in tumorigenesis", J. BIOL. CHEM., vol. 278, 2003, pages 19396 - 19405 |
| KAPPE, C. 0.; STADLER, A.: "Microwaves in Organic and Medicinal Chemistry", 2005, WILEY-VCH |
| KOJIMA, F.; KATO, S.; KAWAI, S.: "Prostaglandin E synthase in the pathophysiology of arthritis", FUNDAM. CLIN. PHARMACOL., vol. 19, 2005, pages 255 - 261 |
| KOJIMA, F.; NARABA, H.; MIYAMOTO, S.; BEPPU, M.; AOKI H; KAWAI S: "Membraneassociated prostaglandin E synthase-1 is upregulated by proinflammatory cyto-kines in chondrocytes from patients with osteoarthritis", ARTHRITIS RES. THER., 2004, pages R355 - R365 |
| KREMER, J. M. ET AL., N ENGL J MED., vol. 349, 2003, pages 1907 - 1915 |
| LANGER, SCIENCE, vol. 249, 1990, pages 1527 - 1533 |
| LICASTRO, F.; DAVIS, L.J.; PEDRINI, S.; GALASKO, D.; MASLIAH, E.: "Prostaglandin E2 induced polymerization of human alpha-1-antichymotrypsin and suppressed its protease inhibitory activity: implications for Alzheimer's disease", BIOCHEM. BIOPHYS. RES. COMMUN., vol. 249, 1998, pages 182 - 186 |
| LIN, C. ET AL., MOL. PHARM., vol. 62, 2002, pages 297 - 303 |
| MASUKO-HONGO, K.; BERENBAUM, F.; HUMBERT, L.; SALVAT, C.; GOLDRING, M. B.; THIRION, S.: "Up-regulation of microsomal prostaglandin E synthase 1 in osteoarthritic human cartilage: critical roles of the ERK-1/2 and p38 signaling pathways", ARTHRITIS RHEUM., vol. 50, 2004, pages 2829 - 2838 |
| MEHROTRA, S.; MORIMIYA, A.; AGARWAL, B.; KONGER, R.; BADVE, S.: "Microsomal prostaglandin E2 synthase-1 in breast cancer: a potential target for therapy", J. PATHOL., vol. 208, 2006, pages 356 - 363 |
| MURAKAMI M; KUDO 1.: "Prostaglandin E synthase: a novel drug target for inflammation and cancer", CURR PHARM DES., vol. 12, 2006, pages 943 - 954 |
| MURAKAMI, M.; NAKASHIMA, K.; KAMEI, D.; MASUDA, S.; ISHIKAWA, Y.; ISHII, T.; OHMIYA, Y.; WATANABE, K.; KUDO, I.: "Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2", J. BIOL. CHEM., vol. 278, 2003, pages 37937 - 37947 |
| OAE, A.; SHINHAMA, K.; KIM, Y. H.: "Direct conversion of arylamines to the corresponding halides, biphenyls and sulfides with t-buty thionitrate", CHEM. LETT., 1979, pages 939 - 942 |
| OSHIMA, H.; MATSUNAGA, A.; FUJIMURA, T.; TSUKAMOTO, T.; TAKETO, M. M.; OSHIMA, M.: "Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway", GASTROENTEROLOGY, vol. 131, 2006, pages 1086 - 1095 |
| PRESTON, P. N. SMITH, D. M. AND TENNANT, G.: "The Chemistry of Heterocyclic Compounds, Benzimidazoles and Cogeneric Tricyclic Compounds", 1981, JOHN WILEY & SONS INC |
| PRESTON, P. N.: "Synthesis, reactions, and spectroscopic properties of benzimidazoles", CHEM. REV., vol. 74, 1974, pages 279 - 314 |
| PROCTOR, M.; FARQUHAR, C.: "Dysmenorrhoea", CLIN. EVID., vol. 7, 2002, pages 1639 - 1653 |
| RASK, K.; ZHU, Y.; WANG, W.; HEDIN, L.; SUNDFELDT, K.: "Ovarian epithelial cancer: a role for PGE2-synthesis and signalling in malignant transformation and progression", MOL. CANCER, vol. 16, 2006, pages 62 |
| REPA, J. J. ET AL., SCIENCE, vol. 289, 2000, pages 1524 - 1529 |
| SAHA, S.; ENGSTROM, L.; MACKERLOVA, L.; JAKOBSSON, P. J.; BLOMQVIST, A.: "Impaired febrile responses to immune challenge in mice deficient in microsomal prostaglandin E synthase-1", AM. J. PHYSIOL., vol. 288, 2005, pages R1 100 - R1 107 |
| SAMUELSSON, B.; MORGENSTERN, R.; JAKOBSSON, P.-J.: "Membrane Prostaglandin E Synthase-1: A Novel Therapeutic Target", PHARMACOL. REV., vol. 59, 2007, pages 207 - 224 |
| SAMUELSSON, B.; MORGENSTERN, R.; JAKOBSSON, P.-J.: "Membrane Prostaglandin E Synthase-1: A Novel Therapeutic Target.", PHARMACOL. REV., vol. 59, 2007, pages 207 - 224 |
| SATOH, K.; NAGANO, Y.; SHIMOMURA, C.; SUZUKI, N.; SAEKI, Y.; YOKOTA, H.: "Expression of prostaglandin E synthase mRNA is induced in beta-amyloid treated rat astrocytes", NEUROSCI. LETT., vol. 283, 2000, pages 221 - 223 |
| SCALI, C.; PROSPERI, C.; BRACCO, L.; PICCINI, C.; BARONTI, R.; GINESTRONI, A.; SORBI, S.; PEPEU, G.; CASAMENTI, F.: "Neutrophils CD Ib and fibroblasts PGE(2) are elevated in Alzheimer's disease", NEUROBIOL. AGING, vol. 23, 2002, pages 523 - 30 |
| SCHÄCKE, H. ET AL., PROC. NATL. ACAD. SCI. USA., vol. 101, 2004, pages 227 - 232 |
| SCHAIBLE, H. G.; EBERSBERGER, A.; VON BANCHET, G. S.: "Mechanisms of pain in arthritis", ANN. N YACAD. SCI., vol. 966, 2002, pages 343 - 354 |
| SHAVIT, Y.; FRIDEL, K.; BEILIN, B.: "Postoperative pain management and proinflammatory cytokines: animal and human studies", J. NEUROIMMUNE PHARMACOL., 2006, pages 443 - 451 |
| STARK, K.; TORMA, H.; OLIW, E. H.: "Co- localization of COX-2, CYP4F8, and mPGEs-1 in epidermis with prominent expression of CYP4F8 mRNA in psoriatic lesions", PROSTAGLANDINS OTHER LIPID MEDIAT., vol. 79, 2006, pages 114 - 125 |
| STRAUSS, C. R.: "Handbook of Green Chemistry and Technology", 2002, BLACKWELL SCIENCE LTD, article "Application of microwaves for environmentally benign organic chemistry", pages: 297 - 415 |
| T. W. GREENE: "Protective Groups in Organic Synthesis", 1984, WILEY |
| TAKEDA, H.; SONOSHITA, M.; OSHIMA, H.; SUGIHARA, K.; CHULADA, P. C.; LANGENBACH, R.; OSHIMA, M.; TAKETO, M. M.: "Cooperation of cyclooxygenase 1 and cyclooxygenase 2 in intestinal polyposis", CANCER RES., vol. 63, 2003, pages 4872 - 4877 |
| TREBINO, C. E.; STOCK, J. L.; GIBBONS, C. P.; NAIMAN, B. M.; WACHTMANN, T. S.; UMLAND, J. P.; PANDHER, K.; LAPOINTE, J. M.; SAHA,: "Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase", PROC. NATL. ACAD. SCI. USA, vol. 100, 2004, pages 9044 - 9049 |
| W.G. NIEHAUS, JR; B. SAMUELSSON, EUR. J. BIOCHEM, vol. 6, 1968, pages 126 |
| WATLEY, B.; TIERNEY, J.; LIDSTR6M, P.; WESTMAN, J.: "The impact of microwave assisted organic chemistry on drug discovery", DRUG DISCOV. TODAY, vol. 7, 2002, pages 373 - 380 |
| WESTMAN, M.; KOROTKOVA, M.; AF KLINT, E.; STARK, A.; AUDOLY, L. P.; KLARESKOG, L.; ULFGREN, A. K.; JAKOBSSON, P. J.: "Expression of microsomal prostaglandin E synthase 1 in rheumatoid arthritis synovium", ARTHRITIS RHEUM., vol. 50, 2004, pages 1774 - 1780 |
| WHITBY, F. G. ET AL., BIOCHEMISTRY, vol. 36, 1997, pages 10666 - 10674 |
| WIENECKE, T.; OLESEN, J.; OTURAI, P. S.; ASHINA, M.: "Prostaglandin E2 (PGE2) induces headache in healthy subjects", CEPHALALGIA, vol. 29, 2009, pages 509 - 519 |
| YOSHIMATSU, K.; GOLIJANIN, D.; PATY, P. B.; SOSLOW, R. A.; JAKOBSSON, P.-J.; DELELLIS, R. A.; SUBBARAMAIAH, K.; DANNENBERG, A. J.: "Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer", CLIN. CANCER RES., vol. 7, 2001, pages 3971 - 3976 |
Cited By (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2495244A1 (en) * | 2011-03-02 | 2012-09-05 | NovaSaid AB | Piperidinyl benzoimidazole derivatives as mPGEs-1 inhibitors |
| WO2012117062A1 (en) * | 2011-03-02 | 2012-09-07 | Novasaid Ab | Piperidinyl benzoimidazole derivatives as mpges-1 inihibitors |
| US20130210844A1 (en) * | 2012-02-09 | 2013-08-15 | Glenmark Pharmaceuticals S.A. | BICYCLIC COMPOUNDS AS mPGES-1 INHIBITORS |
| US9006257B2 (en) * | 2012-02-09 | 2015-04-14 | Glenmark Pharmaceuticals S.A. | Bicyclic compounds as mPGES-1 inhibitors |
| WO2013146970A1 (en) * | 2012-03-29 | 2013-10-03 | 第一三共株式会社 | Novel quinoline derivative |
| WO2013146969A1 (en) * | 2012-03-29 | 2013-10-03 | 第一三共株式会社 | Novel disubstituted cyclohexane derivative |
| WO2014204371A1 (en) * | 2013-06-20 | 2014-12-24 | Cadila Pharmaceuticals Ltd. | Piperidinyl benzoimidazole derivatives as mpge-1 inhibitors |
| WO2014204370A1 (en) * | 2013-06-20 | 2014-12-24 | Cadila Pharmaceuticals Ltd. | Novel piperidinyl benzoimidazole derivatives as mpges-1 inhibitors |
| KR101898829B1 (en) | 2014-10-29 | 2018-09-13 | 일라이 릴리 앤드 캄파니 | Novel methyl-piperidine compounds useful for inhibiting microsomal prostaglandin e2 synthase-1 |
| KR20170055025A (en) * | 2014-10-29 | 2017-05-18 | 일라이 릴리 앤드 캄파니 | Novel methyl-piperidine compounds useful for inhibiting microsomal prostaglandin e2 synthase-1 |
| JP2019510079A (en) * | 2016-02-25 | 2019-04-11 | ジェシンタ・ファーマ・アーベー | Methods of treating diseases characterized by vasoconstriction |
| CN105968112A (en) * | 2016-05-16 | 2016-09-28 | 青岛云天生物技术有限公司 | Method for preparing linagliptin intermediate for treating II-type diabetis |
| US10501421B1 (en) | 2017-01-27 | 2019-12-10 | Vanderbilt University | Substituted benzimidazoles as modulators of Ras signaling |
| US11332459B2 (en) | 2017-10-19 | 2022-05-17 | Teijin Pharma Limited | Benzimidazole derivatives and their uses |
| WO2019101826A1 (en) | 2017-11-22 | 2019-05-31 | Khondrion Ip B.V. | Compounds as mpges-1 inhibitors |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| WO2021076908A1 (en) | 2019-10-18 | 2021-04-22 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| EP4349413A2 (en) | 2019-10-18 | 2024-04-10 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| WO2021087064A1 (en) | 2019-10-31 | 2021-05-06 | Forty Seven, Inc. | Anti-cd47 and anti-cd20 based treatment of blood cancer |
| WO2021096860A1 (en) | 2019-11-12 | 2021-05-20 | Gilead Sciences, Inc. | Mcl1 inhibitors |
| WO2021130638A1 (en) | 2019-12-24 | 2021-07-01 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| WO2021132472A1 (en) | 2019-12-25 | 2021-07-01 | 日本新薬株式会社 | Prophylactic and/or therapeutic agent for chronic prostatitis/chronic pelvic pain syndrome |
| KR20220120646A (en) | 2019-12-25 | 2022-08-30 | 니뽄 신야쿠 가부시키가이샤 | Prevention and/or treatment of chronic prostatitis/chronic pelvic pain syndrome |
| WO2021163064A2 (en) | 2020-02-14 | 2021-08-19 | Jounce Therapeutics, Inc. | Antibodies and fusion proteins that bind to ccr8 and uses thereof |
| US11692038B2 (en) | 2020-02-14 | 2023-07-04 | Gilead Sciences, Inc. | Antibodies that bind chemokine (C-C motif) receptor 8 (CCR8) |
| WO2021222522A1 (en) | 2020-05-01 | 2021-11-04 | Gilead Sciences, Inc. | Cd73 inhibiting 2,4-dioxopyrimidine compounds |
| WO2022221304A1 (en) | 2021-04-14 | 2022-10-20 | Gilead Sciences, Inc. | CO-INHIBITION OF CD47/SIRPα BINDING AND NEDD8-ACTIVATING ENZYME E1 REGULATORY SUBUNIT FOR THE TREATMENT OF CANCER |
| WO2022245671A1 (en) | 2021-05-18 | 2022-11-24 | Gilead Sciences, Inc. | Methods of using flt3l-fc fusion proteins |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2023076983A1 (en) | 2021-10-28 | 2023-05-04 | Gilead Sciences, Inc. | Pyridizin-3(2h)-one derivatives |
| WO2023122581A2 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023122615A1 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023147418A1 (en) | 2022-01-28 | 2023-08-03 | Gilead Sciences, Inc. | Parp7 inhibitors |
| EP4245756A1 (en) | 2022-03-17 | 2023-09-20 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023178181A1 (en) | 2022-03-17 | 2023-09-21 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023183817A1 (en) | 2022-03-24 | 2023-09-28 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| WO2023196784A1 (en) | 2022-04-05 | 2023-10-12 | Gilead Sciences, Inc. | Combinations of antibody therapies for treating colorectal cancer |
| WO2023205719A1 (en) | 2022-04-21 | 2023-10-26 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2024006929A1 (en) | 2022-07-01 | 2024-01-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2024064668A1 (en) | 2022-09-21 | 2024-03-28 | Gilead Sciences, Inc. | FOCAL IONIZING RADIATION AND CD47/SIRPα DISRUPTION ANTICANCER COMBINATION THERAPY |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2011023812A1 (en) | Microsomal prostaglandin e synthase-1 (mpges1) inhibitors | |
| JP5324437B2 (en) | Indole as a 5-HT6 modulator | |
| US6506747B1 (en) | Substituted 1-(4-aminophenyl)pyrazoles and their use as anti-inflammatory agents | |
| US7851474B2 (en) | Dipiperazinyl ketones and related analogues | |
| EP1300398B1 (en) | Propane-1,3-dione derivatives | |
| WO2000044743A1 (en) | Amide derivatives and drug compositions | |
| EA002907B1 (en) | 6-phenylpyridyl-2-amine derivatives useful as non inhibitors | |
| JP2000509719A (en) | Substituted azabicyclo compounds and their use as inhibitors of TNF and cyclic AMP phosphodiesterase production | |
| KR20010070946A (en) | Heterocyclic compounds and methods to treat cardiac failure and other disorders | |
| AU2006218125A1 (en) | Pyrrolidine and piperidine acetylene derivatives for use as mGluR5 antagonists | |
| TW201204361A (en) | Nitrogen-containing heterocyclic compound having kynurenine production inhibitory activity | |
| EP1996196A2 (en) | Somatostatin agonists | |
| JP5069119B2 (en) | Nicotinamide pyridine urea as a vascular endothelial growth factor (VEGF) receptor kinase inhibitor | |
| WO2006089076A9 (en) | Thiazole amides, imidazole amides and related analogues | |
| AU2007307105A1 (en) | N-substituted-azacyclylamines as histamine-3 antagonists | |
| EP2454237B1 (en) | 3-oxo-2,3,-dihydro-1h-isoindole-4-carboxamides with selective parp-1 inhibition | |
| CN110234640B (en) | Benzimidazole derivatives as anticancer agents | |
| EP1303508A1 (en) | Substituted 1-(4-aminophenyl)indoles and their use as anti-inflammatory agents | |
| MX2014004701A (en) | Bicyclic heterocyclic compound. | |
| JP2012211085A (en) | Hedgehog signal inhibitor | |
| KR20160018686A (en) | Bicyclic nitrogen-containing aromatic heterocyclic amide compound | |
| WO2012117062A1 (en) | Piperidinyl benzoimidazole derivatives as mpges-1 inihibitors | |
| JP2013533844A (en) | Azaindole derivatives | |
| CN107466294B (en) | Indole derivatives | |
| JP5694958B2 (en) | Substituted 3-benzofuranyl-indol-2-one-3-acetamidopiperazine derivatives, their preparation, and their therapeutic use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10747047 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10747047 Country of ref document: EP Kind code of ref document: A1 |