WO2010141550A2 - 11-beta hsd1 inhibitors - Google Patents
11-beta hsd1 inhibitors Download PDFInfo
- Publication number
- WO2010141550A2 WO2010141550A2 PCT/US2010/037024 US2010037024W WO2010141550A2 WO 2010141550 A2 WO2010141550 A2 WO 2010141550A2 US 2010037024 W US2010037024 W US 2010037024W WO 2010141550 A2 WO2010141550 A2 WO 2010141550A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- sulfonyl
- phenyl
- trifluoromethyl
- trifluoro
- methylpiperazin
- Prior art date
Links
- 0 C[C@](*)(CN(CC1)c2c(*)cc(*)cc2)N1S(C1C(*C(*)(*)*)CCCC1*)(=O)=O Chemical compound C[C@](*)(CN(CC1)c2c(*)cc(*)cc2)N1S(C1C(*C(*)(*)*)CCCC1*)(=O)=O 0.000 description 5
- NILCNOBWWYMTTF-SSJGJQFISA-N C[C@H](CN(CC1)c(c(OC)c2)ccc2F)N1S(c1ccc(C(C(F)(F)F)(C(N)=O)O)[s]1)(=O)=O Chemical compound C[C@H](CN(CC1)c(c(OC)c2)ccc2F)N1S(c1ccc(C(C(F)(F)F)(C(N)=O)O)[s]1)(=O)=O NILCNOBWWYMTTF-SSJGJQFISA-N 0.000 description 1
- GJCQDQQQMSLZAR-SNVBAGLBSA-N C[C@H](CN(CC1)c(cc2)c(C(F)(F)F)cc2F)N1S(c([s]1)ccc1Br)(=O)=O Chemical compound C[C@H](CN(CC1)c(cc2)c(C(F)(F)F)cc2F)N1S(c([s]1)ccc1Br)(=O)=O GJCQDQQQMSLZAR-SNVBAGLBSA-N 0.000 description 1
- LDNXCAVPYKBBSI-BLVKFPJESA-N C[C@H](CN(CC1)c(cc2)c(C(F)(F)F)cc2F)N1S(c1cc([C@@](C(F)(F)F)(C(N)=O)O)ccc1)(=O)=O Chemical compound C[C@H](CN(CC1)c(cc2)c(C(F)(F)F)cc2F)N1S(c1cc([C@@](C(F)(F)F)(C(N)=O)O)ccc1)(=O)=O LDNXCAVPYKBBSI-BLVKFPJESA-N 0.000 description 1
- WCYKILLGEZYXPX-CPONVNJPSA-N C[C@H](CNc(cc1)c(C(F)(F)F)cc1F)NS(C1=CCC(C)(C(C)(C(F)(F)F)O)S1)(=O)=O Chemical compound C[C@H](CNc(cc1)c(C(F)(F)F)cc1F)NS(C1=CCC(C)(C(C)(C(F)(F)F)O)S1)(=O)=O WCYKILLGEZYXPX-CPONVNJPSA-N 0.000 description 1
- NUHLQHCMQWGQFN-YQDUUYOCSA-N C[C@H](CNc(cc1)c(C(F)(F)F)cc1F)NS(c1ccc(C(C)(C(F)(F)F)NC(C)=O)[s]1)(=O)=O Chemical compound C[C@H](CNc(cc1)c(C(F)(F)F)cc1F)NS(c1ccc(C(C)(C(F)(F)F)NC(C)=O)[s]1)(=O)=O NUHLQHCMQWGQFN-YQDUUYOCSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/04—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- This invention features compounds that inhibit 11 -beta HSDl.
- Diabetes is generally characterized by relatively high levels of plasma glucose (hyperglycemia) in the fasting state.
- Patients having type 2 diabetes non-insulin dependent diabetes mellitus (NIDDM)) produce insulin (and even exhibit hyperinsulinemia), whilst demonstrating hyperglycemia.
- NIDDM non-insulin dependent diabetes mellitus
- Type 2 diabetics can often develop insulin resistance, in which the effect of insulin in stimulating glucose and lipid metabolism is diminished. Further, patients having insulin resistance, but have not developed type 2 diabetes, are also at risk of developing Syndrome X (metabolic syndrome). Syndrome X is characterized by insulin resistance, along with obesity (e.g., abdominal obesity), hyperinsulinemia, high blood pressure, relatively low HDL and relatively high VLDL.
- obesity e.g., abdominal obesity
- hyperinsulinemia e.g., high blood pressure
- relatively low HDL e.g., high HDL
- VLDL relatively high VLDL
- Glucocorticoids are counter regulatory hormones that oppose the action of insulin. It is established that glucocorticoid activity is controlled at the tissue level by intracellular interconversion of active Cortisol and inactive cortisone by the 11 -beta hydroxysteroid dehydrogenases, ll ⁇ HSDl, which activates cortisone and ll ⁇ HSD2, which inactivates Cortisol. Excess levels of glucocorticoids (e.g., Cortisol) can cause metabolic complications. For example, excess Cortisol is associated with disorders including NIDDM, obesity, dyslipidemia, insulin resistance, and hypertension.
- 1 l ⁇ HSDl can reduce the effects of excessive amounts of 1 l ⁇ -hydroxysteroids, e.g., Cortisol, and therefore can be useful for the treatment and control of diseases mediated by abnormally high levels of Cortisol and other 1 l ⁇ -hydroxysteroids, e.g., NIDDM, obesity, dyslipidemia, and hypertension.
- this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof).
- a chemical entity E which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof).
- Formula I is provided below:
- R 1 is halo, -CH 3 , -CH 2 X, -CHX 2 , -CF 3 , -OCH 3 or -OCF 3 ;
- R 2 is halo, -CN, -C(O)-NH 2 , -C(O)-NH-(C L3 alkyl), -C(0)-N(d_ 3 alkyl) 2 , or 5- to 6-membered heteroaryl containing 1-4 ring heteroatoms independently selected from O, N, N(R 2sl ) and S, wherein R 2sl is -H, -CH 3 or ethyl, and wherein said heteroaryl is unsubstituted or substituted with one or two substituents R 2s2 independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 ;
- R 3 is -H, halo, -CH 3 , ethyl, -OH or -OCH 3 ; one of R 4 and R 5 is H, and the other is -CH 3 , -CH 2 X, -CHX 2 , -CF 3 or ethyl;
- A is phenyl, or 5- to 6-membered heteroaryl containing 1-3 ring heteroatoms independently selected from O, N, N(R al ) and S, and in which the atom bonded to the sulfonyl sulfur is carbon, wherein R al is -H, -CH 3 or ethyl; R 9 is -H, halo, -CH 3 , -CH 2 X, -CHX 2 , -CF 3 , ethyl, -OCH 3 or -OCF 3 ; provided that when A is 5- membered heteroaryl containing 3 ring heteroatoms, then R 9 is absent;
- R 6 is -CH 3 , ethyl, -OH or -OCH 3 ;
- R 7 is -CH 3 , -CH 2 X, -CHX 2 , -CF 3 or phenyl, wherein said phenyl is unsubstituted or substituted with one or two substituents R 7sl independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 ;
- R 8 is -C(O)-NH 2 , -C(O)-NH-(CL 3 alkyl), -C(O)-N(CL 3 alkyl) 2 , -NH-C(O)-(CL 3 alkyl), -N(C L3 alkyl)-C(O)-(C 1-3 alkyl), or 5- to 6-membered heteroaryl containing 1-4 ring heteroatoms independently selected from O, N, N(R 8sl ) and S, wherein R 8sl is -H, -CH 3 or ethyl, and wherein said heteroaryl
- this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof), in which: R 6 is -OH, -CH 3 , or ethyl;
- R 7 is -CF 3 , -CH 3 , or phenyl, wherein said phenyl is unsubstituted or substituted with one or two substituents R 7sl independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 ; provided that when A is phenyl, then R 6 and R 7 cannot both be -CH 3 ; and R 1 , R 2 , R 3 , R 4 , R 5 , R 8 , and R 9 can be as defined anywhere herein.
- this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof), in which:
- R 6 is -OH
- R 7 is -CF 3 ;
- R 1 , R 2 , R 3 , R 4 , R 5 , R 8 , and R 9 can be as defined anywhere herein.
- this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof), in which: R 6 is -OH;
- R 7 is -CF 3 ;
- R 8 is -C(O)-NH 2 , -C(O)-NH-(C L3 alkyl), or -C(O)-N(C L3 alkyl) 2 (e.g., -C(O)-NH 2 );
- R 1 , R 2 , R 3 , R 4 , R 5 , and R 9 can be as defined anywhere herein.
- this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof), in which: R 8 is -C(O)-NH 2 , -C(O)-NH-(C L3 alkyl), or -C(O)-N(C L3 alkyl) 2 (e.g.,
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , and R 9 can be as defined anywhere herein.
- this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof), in which:
- A is 5- to 6-membered heteroaryl containing 1-3 ring heteroatoms independently selected from O, N, N(R al ) and S, and in which the atom bonded to the sulfonyl sulfur is carbon, wherein R al is -H, -CH 3 or ethyl; and
- this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof), in which:
- A is a 5-membered heteroaryl, which has formula (A-I):
- a 1 is doubly bonded to A 4
- a 3 is singly bonded to A 4 ;
- a 3 is O, S, or NR al ; and one of A 1 , A 2 , and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 1 , A 2 , and A 4 is C-R 9 , and one of A 1 , A 2 , and A 4 is N or C-H; and R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , and R 9 can be as defined anywhere herein.
- the chemical entity E can be a compound of formula (I) (including any subgenus of formula (I) or specific compound of formula (I)).
- the chemical entity E can be a salt (e.g., a pharmaceutically acceptable salt) of a compound of formula (I) (including any subgenus of formula (I) or specific compound of formula (I)).
- the chemical entity E can be an N-oxide of a compound of formula (I) (including any subgenus of formula (I) or specific compound of formula (I)).
- the chemical entity E can be a salt (e.g., a pharmaceutically acceptable salt) of an N-oxide of a compound of formula (I) (including any subgenus of formula (I) or specific compound of formula (I)).
- this invention features a composition (e.g., a pharmaceutical composition), which includes a chemical entity E as defined anywhere herein and a pharmaceutically acceptable carrier.
- the composition can include an effective amount of a chemical entity E as defined anywhere herein.
- the composition can further include an additional therapeutic agent.
- this invention features a dosage form, which includes from about
- the dosage form can further include a pharmaceutically acceptable carrier and/or an additional therapeutic agent.
- the invention also relates generally to inhibiting 11 -beta HSDl with a chemical entity E as defined anywhere herein.
- the methods can include, e.g., contacting an 11- ⁇ HSDl in a sample (e.g., a tissue) with a chemical entity E as defined anywhere herein.
- the methods can include administering a chemical entity E as defined anywhere herein to a subject (e.g., a mammal, such as a human).
- this invention includes methods of screening for compounds that inhibit 11 - ⁇ HSD 1.
- this invention also features methods for treating (e.g., controlling, relieving, ameliorating, alleviating, or slowing the progression of) or methods for preventing (e.g., delaying the onset of or reducing the risk of developing) a disease or condition that is mediated by excess (e.g., abnormally high) levels or uncontrolled (e.g., resulting in abnormally high) levels of Cortisol and/or other corticosteroids (e.g., 1 l ⁇ - hydroxysteroids), which include administering to a subject in need thereof an effective amount of a chemical entity E as defined anywhere herein.
- a chemical entity E as defined anywhere herein.
- diseases or conditions include, but are not limited to diabetes (e.g., type 1 or type 2 diabetes), Syndrome X, hyperglycemia, low glucose tolerance, insulin resistance, obesity, lipid disorders, dyslipidemia, hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels, high LDL levels, atherosclerosis and its sequelae, vascular restenosis, pancreatitis, abdominal obesity, neurodegenerative disease, retinopathy, nephropathy, neuropathy, hypertension, coronary heart disease, stroke, peripheral vascular disease, Cushing's syndrome, glaucoma, osteoperosis, hyperinsulinemia, tuberculosis, psoriasis, cognitive disorders and dementia (e.g., impairment associated with aging and of neuronal dysfunction, e.g., Alzheimer's disease), depression, viral diseases, inflammatory disorders, immune disorders); or promoting wound healing.
- diabetes e.g., type 1 or type 2 diabetes
- Syndrome X hyperglycemia, low
- this invention features methods for treating or preventing diabetes (e.g., type I diabetes, type 2 diabetes), which includes administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
- diabetes e.g., type I diabetes, type 2 diabetes
- this invention features methods for treating or preventing Syndrome X, which includes administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
- this invention features methods for treating or preventing hyperglycemia, which includes administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
- this invention features methods for treating or preventing hyperglycemia, which includes administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
- this invention features methods for treating or preventing obesity, which include administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
- this invention features methods for treating or preventing a lipid disorder selected from the group consisting of dyslipidemia, hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL and high LDL, which include administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
- this invention features methods for treating or preventing atherosclerosis, which include administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
- this invention features methods for treating or preventing a cognitive disorder (e.g., Alzheimer's disease), which include administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
- a cognitive disorder e.g., Alzheimer's disease
- this invention features methods for promoting wound healing, which include administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
- the subject can be a subject in need thereof (e.g., a subject identified as being in need of such treatment, such as a subject having, or at risk of having, one or more of the diseases or conditions described herein). Identifying a subject in need of such treatment can be in the judgment of a subject or a health care professional and can be subjective (e.g. opinion) or objective (e.g. measurable by a test or diagnostic method).
- the subject can be a mammal. In certain embodiments, the subject can be a human.
- this invention also relates to methods of making compounds described herein.
- the methods include taking any one of the intermediate compounds described herein and reacting it with one or more chemical reagents in one or more steps to produce a chemical entity E as defined anywhere herein.
- this invention relates to a packaged product.
- the packaged product includes a container, one of the aforementioned compounds in the container, and a legend (e.g., a label or an insert) associated with the container and indicating administration of the compound for treatment and control of the diseases or disorders described herein.
- any chemical entity, composition, or method described herein can also include any one or more of the other features delineated in the detailed description and/or in the claims.
- mammal includes organisms, which include mice, rats, cows, sheep, pigs, rabbits, goats, horses, monkeys, dogs, cats, and humans.
- an effective amount refers to an amount of a compound that confers a therapeutic effect (e.g., treats, e.g., controls, relieves, ameliorates, alleviates, or slows the progression of; or prevents, e.g., delays the onset of or reduces the risk of developing, a disease, disorder, or condition or symptoms thereof) on the treated subject.
- the therapeutic effect may be objective (i.e., measurable by some test or marker) or subjective (i.e., subject gives an indication of or feels an effect).
- An effective amount of the compound described above may range from about 0.01 mg/kg to about 1000 mg/kg, (e.g., from about 0.1 mg/kg to about 100 mg/kg, from about 1 mg/kg to about 100 mg/kg). Effective doses will also vary depending on route of administration, as well as the possibility of co-usage with other agents.
- halo or halogen refers to any radical of fluorine, chlorine, bromine or iodine.
- substituent (radical) prefix names are derived from the parent hydride by either (i) replacing the "ane” in the parent hydride with the suffix "yl;” or (ii) replacing the "e” in the parent hydride with the suffix "yl;” (here the atom(s) with the free valence, when specified, is (are) given numbers as low as is consistent with any established numbering of the parent hydride).
- Accepted contracted names e.g., furyl, pyridyl, and piperidyl, and trivial names, e.g., phenyl and thienyl are also used herein throughout. Conventional numbering/lettering systems are also adhered to for substituent numbering.
- the term "Ci_3 alkyl” refers to methyl, ethyl, n-propyl, and ⁇ opropyl.
- heteroaryl refers to an aromatic monocyclic or bicyclic hydrocarbon groups having one or more (e.g., 1-3 or 1-4) heteroatom ring atoms independently selected from O, N, or S (and mono and dioxides thereof, e.g., N ⁇ O ⁇ , S(O), SO 2 ). Any atom can be optionally substituted by one or more substituents.
- Heteroaryl groups include pyridyl, thienyl, furyl (furanyl), imidazolyl, and pyrrolyl.
- This invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof).
- a chemical entity E which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof).
- Formula I is provided below:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , and A can be as defined anywhere herein.
- R 1 can be -CH3, -CH 2 X, -CHX 2 , or -CF3. In some embodiments, R 1 can be -CH 2 X, -CHX 2 , or -CF3. In some embodiments, R 1 can be -CF 3 .
- R 1 can be -CH 2 X, Or-CHX 2 . In some embodiments, R 1 can be -CH3.
- each instance of X can be, independently, -F or -Cl, e.g., -F.
- R 1 can be halo (e.g., -F or -Cl).
- R 1 can be -OCH 3 or -OCF 3 .
- R 1 can be -CF 3 , -F, -Cl, -CH 3 , or -OCH 3 .
- R 2 can be: (i) halo
- R 2 can be:
- halo or (iv) 5- to 6-membered heteroaryl containing 1-4 ring heteroatoms independently selected from O, N, N(R 2sl ) and S, in which R 2sl is -H, -CH 3 or ethyl, and in which said heteroaryl is unsubstituted or substituted with one or two substituents R 2s2 independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 .
- R 2 can be halo (e.g., -F).
- R 2 can be 5- to 6-membered heteroaryl containing 1-4 (e.g., 2-3, 2-4, 3-4, 1-3, 1-2, 1, 2, 3, or 4) ring heteroatoms independently selected from O, N, N(R 2sl ) and S, in which R 2sl is -H, -CH 3 or ethyl, and in which said heteroaryl is unsubstituted or substituted with one or two substituents R 2s2 independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 .
- each instance of R 2sl can be, independently, -H or -CH 3 (e.g., -H).
- each instance of R 2s2 can be, independently, -CH 3 , ethyl, or -CF 3 (e.g., -CH 3 or ethyl; e.g., -CH 3 ).
- the 5- to 6-membered heteroaryl can contain 1-3 (e.g., 2-
- the 5- to 6-membered heteroaryl can contain 2-4 (e.g., 2- 3, 3-4, 2, 3, or 4) ring heteroatoms.
- the 5- to 6-membered heteroaryl can contain 3 or 4 ring heteroatoms.
- the 3 or 4 ring heteroatoms can be independently selected from O, N, and N(R 2sl ).
- each of the 3 or 4 ring heteroatoms can be independently selected from N and N(R 2sl ).
- one of the 3 or 4 ring heteroatoms can be O or N(R 2sl ), and the others can each be N.
- the 5- to 6-membered heteroaryl is bonded to the phenyl ring via a ring carbon atom and not via one of the aforementioned ring heteroatoms.
- R 2 can be 5- membered heteroaryl containing 1-4 (e.g., 2-3, 2-4, 3-4, 1-3, 1-2, 1, 2, 3, or 4) ring heteroatoms independently selected from O, N, N(R 2sl ) and S, in which R 2sl can be as defined anywhere herein; and in which said heteroaryl can be unsubstituted or substituted with one or two substituents R 2s2 , in which each instance of R 2s2 can be as defined anywhere herein.
- 1-4 e.g., 2-3, 2-4, 3-4, 1-3, 1-2, 1, 2, 3, or 4
- R 2sl can be as defined anywhere herein
- said heteroaryl can be unsubstituted or substituted with one or two substituents R 2s2 , in which each instance of R 2s2 can be as defined anywhere herein.
- Embodiments can include one or more of the following features.
- R 2sl can be -H.
- R 2s2 can be -CH 3 or ethyl; e.g., -CH 3 .
- the 5- membered heteroaryl can contain 3 ring heteroatoms.
- each of the 3 ring heteroatoms can be independently selected from O, N, and N(R 2sl ).
- each of the 3 ring heteroatoms can be independently selected from N and N(R 2sl ).
- one of the 3 ring heteroatoms can be O, and the others can be N.
- the 5- membered heteroaryl can be l,2,4-oxadiazol-5-yl (unsubstituted or substituted with, e.g., -CH3) or l,3,4-oxadiazol-2-yl (unsubstituted or substituted with, e.g., -CH 3 ).
- one of the 3 ring heteroatoms can be N(R 2sl ), and the others can be N.
- the 5- membered heteroaryl can be l,2,4-triazol-3-yl (unsubstituted or substituted with, e.g., -CH 3 ).
- the 5- membered heteroaryl can contain 4 ring heteroatoms.
- the 5- membered heteroaryl can be tetrazol-5-yl.
- R 2 can be -C(O)-NH 2 . In some embodiments, R 2 can be -CN.
- R 2 can be -F; -CN; -C(O)-NH 2 ; l,2,4-oxadiazol-5-yl (unsubstituted or substituted with, e.g., -CH 3 ); l,3,4-oxadiazol-2-yl (unsubstituted or substituted with, e.g., -CH 3 ); l,2,4-triazol-3-yl (unsubstituted or substituted with, e.g., - CH 3 ); or tetrazol-5-yl.
- R 1 is -CH 2 X, -CHX 2 , or -CF 3 ;
- R 2 is:
- halo e.g., -F
- ⁇ CN
- Embodiments can include one or more of the following features.
- R 1 can be -CF 3 .
- R 2 can be -F.
- R 1 is -CH 2 X, -CHX 2 , or -CF 3 ;
- R 2 is:
- halo e.g., -F
- 5- to 6-membered heteroaryl containing 1-4 ring heteroatoms independently selected from O, N, N(R 2sl ) and S, in which R 2sl is -H, -CH 3 or ethyl, and in which said heteroaryl is unsubstituted or substituted with one or two substituents R 2s2 independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 .
- Embodiments can include one or more of the following features.
- R 1 can be -CF 3 .
- X; the ring heteroatom content of the 5- to 6-membered heteroaryl; R 2sl ; and R 2s2 can each be, independently, as defined anywhere herein.
- R 2 can -F.
- R 1 is -CH 2 X, -CHX 2 , or -CF 3 ;
- R 2 is halo (e.g., -F).
- Embodiments can include one or more of the following features.
- R 1 can be -CF 3 .
- R 2 can -F.
- R 1 is -CF 3
- R 2 is -F
- R 3 can be -H.
- Variables R 4 and R 5 are -H.
- the carbon atom attached to R 4 and R 5 can have the R- configuration (such compounds are referred to herein as having "the i?-CR 4 R 5 configuration"). In some embodiments, the carbon atom attached to R 4 and R 5 can have the ⁇ -configuration (such compounds are referred to herein as compounds having "the S- CR 4 R 5 configuration").
- one of R 4 and R 5 can be H, and the other can be -CH3. In certain embodiments, R 4 can be H, and R 5 can be -CH3. In other embodiments, R 4 can be H, and R 5 can be -CH 3 .
- A can be 5- to 6-membered heteroaryl containing 1-3 (e.g., 1-2, 2-3, 1, 2, or 3) ring heteroatoms independently selected from O, N, N(R al ) and S, and in which the atom bonded to the sulfonyl sulfur is carbon.
- R al can be -H or -CH 3 .
- the 5- to 6-membered heteroaryl can contain 1 or 2 ring heteroatoms.
- the ring heteroatoms can be independently selected from
- A can be 5-membered heteroaryl containing 1-3 (e.g., 1-2, 2-3, 1, 2, or 3) ring heteroatoms independently selected from O, N, N(R al ) and S, and in which the atom bonded to the sulfonyl sulfur is carbon, and in which R al can be as defined anywhere herein.
- the 5-membered heteroaryl can contain 1 ring heteroatom (e.g., S).
- the 5-membered heteroaryl can contain 2 ring heteroatoms.
- the 2 ring heteroatoms can be independently selected from N, N(R al ) and S.
- one of the 2 ring heteroatoms can be N, and the other can be S.
- one of the 2 ring heteroatoms can be N, and the other can be S.
- the 5- membered heteroaryl can contain 3 ring heteroatoms.
- one of the 3 ring heteroatoms can be N(R al ) or S, (e.g., N(R a1 )), and the others can each be N.
- A can be a 5-membered heteroaryl, which has formula
- a 1 is singly bonded to A 4
- a 3 is doubly bonded to A 4 ;
- a 1 is doubly bonded to A 4
- a 3 is singly bonded to A 4 ;
- a 3 is O, S, or NR al ; and one of A 1 , A 2 , and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 1 , A 2 , and A 4 is C-R 9 , and one of A 1 , A 2 , and A 4 is N or C-H.
- the definitions set forth under (i) ⁇ supra) apply, i.e.: A 1 is singly bonded to A 4 , and A 3 is doubly bonded to A 4 ; A 1 Is O, S, or NR al ; and one of A 2 , A 3 ,and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 2 , A 3 and A 4 is C- R 9 , and one of A 2 , A 3 , and A 4 is C-H or N.
- A is 5- membered heteroaryl, which has formula (A-2):
- a 1 is O, S, or NR al ; and one of A 2 , A 3 ,and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 2 , A 3 and A 4 is C-R 9 , and one of A 2 , A 3 , and A 4 is C-H or N.
- Embodiments of formula (A-2) can include one or more of the following features.
- a 1 can be S.
- a 4 can be C-C(R 6 )(R 7 )(R 8 ).
- a 2 canbe C-H.
- a 2 can be N.
- a 3 canbe C-R 9 .
- R 9 can be -H.
- a 1 canbe S
- a 4 canbe C-C(R 6 )(R 7 )(R 8 )
- a 2 canbe C-H orN.
- a 1 canbe S
- a 3 canbe C-C(R 6 )(R 7 )(R 8 )
- a 2 canbe C-H orN.
- a 1 canbe S,A 4 canbe C-C(R 6 )(R 7 )(R 8 ),A 2 can be C-H, andA 3 canbe C-R 9 .
- R 9 can be -H.
- a 1 canbe S,A 4 canbe C-C(R 6 )(R 7 )(R 8 ),A 2 can beN, andA 3 canbe C-R 9 .
- R 9 can be -H.
- the definitions set forth under (ii) ⁇ supra) apply, i.e.: A 1 is doubly bonded to A 4 , and A 3 is singly bonded to A 4 ; A 3 is O, S, or NR al ; and one of A 1 , A 2 , and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 1 , A 2 , and A 4 is C-R 9 , and one of A 1 , A 2 , and A 4 is N or C-H.
- A is 5- membered heteroaryl, which has formula (A-3):
- a 3 is O, S, or NR al ; and one of A 1 , A 2 , and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 1 , A 2 , and A 4 is C-R 9 , and one of A 1 , A 2 , and A 4 is N or C-H.
- Embodiments of formula (A-3) can include one or more of the following features.
- a 3 can be NR al .
- R al can be -CH 3 .
- a 2 and A 4 can be C-C(R 6 )(R 7 )(R 8 ), and one of A 2 and A 4 can be C-R 9 .
- a 1 can be N or C-H (e.g., N).
- a 3 can be NR al ; one of A 2 and A 4 can be C-C(R 6 )(R 7 )(R 8 ), and one of A 2 and A 4 can be C-R 9 ; and A 1 can be N.
- A can be phenyl.
- -C(R 6 )(R 7 )(R 8 ) can be attached to the phenyl ring carbon that is meta or para with respect to the phenyl carbon that is attached to the sulfonyl sulfur in Formula I.
- R 6 can be -OH or -OCH 3 . In certain embodiments, R 6 can be -OH.
- R 6 can be CH 3 or ethyl. In certain embodiments, R 6 can be -CH 3 .
- R 6 can be -OH or -CH 3 .
- R 7 can be -CH 3 , -CH 2 X, -CHX 2 , or -CF 3 .
- R 7 can be -CH 2 X, -CHX 2 , or -CF 3 . In some embodiments, R 7 can be -CF 3 .
- R 7 can be -CH 2 X, Or-CHX 2 .
- R 7 can be -CH 3 .
- each instance of X can be, independently, -F or -Cl, e.g., -F.
- R 7 can be phenyl, wherein said phenyl is unsubstituted or substituted with one or two substituents R 7sl independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 .
- R 7 can be unsubstituted phenyl.
- R 8 can be -C(O)-NH 2 , -C(O)-NH-(d_ 3 alkyl), or -C(O)-N(C L3 alkyl) 2 .
- R 8 can be -C(O)-NH 2 .
- R 8 can be -C(O)-NH-(Ci_ 3 alkyl), or -C(O)-N(Ci_ 3 alkyl) 2 , e.g., -C(O)-NH-(CH 3 ), or -C(O)-N(CH 3 ) 2 .
- R 8 can be -NH-C(O)-(Ci_ 3 alkyl) or
- R 8 can be -NH-C(O)-(C L3 alkyl), e.g., -NH-C(O)-(CH 3 ).
- R 8 can be 5- to 6-membered heteroaryl containing 1-4 (e.g., 2-3, 2-4, 3-4, 1-3, 1-2, 1, 2, 3, or 4) ring heteroatoms independently selected from O, N, N(R 2sl ) and S, in which R 8sl is -H, -CH 3 or ethyl, and in which said heteroaryl is unsubstituted or substituted with one or two substituents R 8s2 independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 .
- 1-4 e.g., 2-3, 2-4, 3-4, 1-3, 1-2, 1, 2, 3, or 4
- R 8sl is -H, -CH 3 or ethyl
- said heteroaryl is unsubstituted or substituted with one or two substituents R 8s2 independently selected from halo, -CH 3 , ethyl, -CF 3 ,
- each instance of R 8sl can be, independently, -H or -CH 3 (e.g., -H).
- each instance of R 8s2 can be, independently, -CH 3 , ethyl, or -CF 3 (e.g., -CH 3 or ethyl; e.g., -CH 3 ).
- the 5- to 6-membered heteroaryl can contain 1-3 (e.g., 2- 3, 1, 2, or 3) ring heteroatoms.
- the 5- to 6-membered heteroaryl can contain 2-4 (e.g., 2- 3, 3-4, 2, 3, or 4) ring heteroatoms.
- the 5- to 6-membered heteroaryl can contain 3 or 4 ring heteroatoms.
- the 3 or 4 ring heteroatoms can be independently selected from O, N, and N(R 8sl ).
- each of the 3 or 4 ring heteroatoms can be independently selected from N and N(R 8sl ).
- one of the 3 or 4 ring heteroatoms can be O or N(R 8sl ), and the others can each be N.
- the 5- to 6-membered heteroaryl is bonded to the quaternary carbon via a ring carbon atom and not via one of the aforementioned ring heteroatoms.
- R 8 can be 5- membered heteroaryl containing 1-4 (e.g., 2-3, 2-4, 3-4, 1-3, 1-2, 1, 2, 3, or 4) ring heteroatoms independently selected from O, N, N(R 8sl ) and S, in which R 8sl can be as defined anywhere herein; and in which said heteroaryl can be unsubstituted or substituted with one or two substituents R 8s2 , in which each instance of R 8s2 can be as defined anywhere herein.
- 1-4 e.g., 2-3, 2-4, 3-4, 1-3, 1-2, 1, 2, 3, or 4
- R 8sl can be as defined anywhere herein
- said heteroaryl can be unsubstituted or substituted with one or two substituents R 8s2 , in which each instance of R 8s2 can be as defined anywhere herein.
- Embodiments can include one or more of the following features.
- R 8sl can be -H.
- R 8s2 can be -CH 3 or ethyl; e.g., -CH 3 .
- the 5- membered heteroaryl can contain 1-3 (e.g., 2-3, 1, 2, or 3) ring heteroatoms.
- the 5- membered heteroaryl can contain 3 ring heteroatoms.
- each of the 3 ring heteroatoms can be independently selected from O, N, and N(R 8sl ).
- each of the 3 ring heteroatoms can be independently selected from N and N(R 8sl ).
- one of the 3 ring heteroatoms can be O, and the others can be N.
- the 5- membered heteroaryl can be l,2,4-oxadiazol-5-yl (unsubstituted or substituted with, e.g., -CH3) or l,3,4-oxadiazol-2-yl (unsubstituted or substituted with, e.g., -CH 3 ).
- one of the 3 ring heteroatoms can be N(R 8sl ), and the others can be N.
- the 5- membered heteroaryl can be l,2,4-triazol-3-yl (unsubstituted or substituted with, e.g., -CH 3 ).
- the 5- membered heteroaryl can contain 4 ring heteroatoms.
- the 5- membered heteroaryl can be tetrazol-5-yl.
- R 8 can be -C(O)-NH 2 ; -C(O)-NH-(CH 3 ); -C(O)-N(CH 3 ) 2 ; -NH-C(O)-(CH 3 ); or 5-membered heteroaryl containing 1-4 (e.g., 1-3) ring heteroatoms independently selected from O, N, N(R 8sl ) and S, wherein said heteroaryl is unsubstituted or substituted with one or two substituents R 8s2 independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 (e.g., l,2,4-triazol-3-yl or 1,2,4- oxadiazol-3-yl, each of which is unsubstituted or substituted with one or two substituents R 8s2 independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH
- the carbon atom attached to R , R , and R can have the R- configuration (referred to herein as "the i?-CR 6 R 7 R 8 configuration"). In some embodiments, the carbon atom attached to R , R , and R can have the ⁇ -configuration (referred to herein as "the ,S-CR 6 R 7 R 8 configuration").
- R 9 can be -H, halo (e.g., chloro or bromo), or -CH 3 .
- R 9 can be -H.
- R 9 can be absent. [1] In some embodiments:
- R 6 is -OH, -CH 3 , or ethyl
- R 7 is -CF 3 , -CH3, or phenyl, wherein said phenyl is unsubstituted or substituted with one or two substituents R 7sl independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 .
- R 6 is -OH, -CH 3 , or ethyl
- R 7 is -CF 3 , -CH 3 , or phenyl that is unsubstituted or substituted with one or two substituents R 7sl independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3
- R 6 is -OH, -CH 3 , or ethyl
- R 7 is -CF 3 , -CH 3 , or phenyl that is unsubstituted or substituted with one or two substituents R 7sl independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3
- halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 can also include any one or more of the features described herein, including (but not limited) to those delineated
- R 6 can be -OH.
- R 7 can be -CF 3 .
- A can be 5- to 6-membered heteroaryl containing 1-3 (e.g., 1-2, 2-3, 1, 2, or 3) ring heteroatoms independently selected from O, N, N(R al ) and S, and in which the atom bonded to the sulfonyl sulfur is carbon.
- R al can be -H or -CH 3 .
- the 5- to 6-membered heteroaryl can contain 1 or 2 ring heteroatoms.
- the ring heteroatoms can be independently selected from N, N(R al ) and S.
- A can be 5-membered heteroaryl containing 1-3 (e.g., 1 or 2) ring heteroatoms independently selected from O, N, N(R al ) and S, and in which the atom bonded to the sulfonyl sulfur is carbon, and in which R al can be as defined anywhere herein.
- A can have formula (A-I):
- (A-I) in which the carbon atom shown as C in formula (A-I) represents the carbon that is bonded to the sulfonyl sulfur in Formula I; and in which A 1 , A 2 , A 3 , A 4 , and the dotted lines between A 1 and A 4 and A 3 and A 4 are all defined according to the definitions under (i) below or are all defined according to the definitions under (ii) below: (i): A 1 is singly bonded to A 4 , and A 3 is doubly bonded to A 4 ;
- a 1 is doubly bonded to A 4
- a 3 is singly bonded to A 4 ;
- a 3 is O, S, or NR al ; and one of A 1 , A 2 , and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 1 , A 2 , and A 4 is C-R 9 , and one of A 1 , A 2 , and A 4 is N or C-H.
- A can have formula (A-2):
- a 1 is O, S, or NR al ; one of A 2 , A 3 , and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 2 , A 3 and A 4 is C-R 9 , and one of A 2 , A 3 , and A 4 is C-H or N.
- a 1 , A 2 , A 3 , and A 4 can be as defined anywhere herein in (e.g., A 1 can be S, A 4 can be C-C(R 6 )(R 7 )(R 8 ), and A 2 can be C-H or N).
- A can be 5-membered heteroaryl having formula (A-3) as described herein.
- A can be phenyl.
- -C(R 6 )(R 7 )(R 8 ) can be attached to the phenyl ring carbon that is meta ox para with respect to the phenyl carbon that is attached to the sulfonyl sulfur in Formula I.
- R 8 can be -C(O)-NH 2 , -C(O)-NH-(CH 3 ), -C(O)-N(CH 3 ) 2 , -NH-C(O)-(CH 3 ), or
- R 8 can be -C(O)-NH 2 .
- R 8 can be l,2,4-triazol-3-yl or l,2,4-oxadiazol-3-yl, each of which is unsubstituted or substituted with one or two substituents R 8s2 independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 (e.g., -CH 3 ).
- the carbon atom attached to R 4 and R 5 can have the ⁇ -configuration.
- the carbon atom attached to R , R , and R can have the ⁇ -configuration.
- R 9 can be 5 --HH oorr aabbsseenntt..
- R and R is H, and the other is -CH 3 (e.g., R can be H, and R can be -CH 3 ).
- R 1 and R 2 can be, independently, as defined as defined anywhere herein.
- R 1 can be -CF 3 .
- R 2 can be -F.
- R 1 can be -CF 3 , and R 2 can be -F.
- R 3 can be -H.
- R 1 can be -CF 3
- R 2 can be -F
- R 3 can be -H.
- R 6 is -OH; and R 7 is -CF 3 .
- Embodiments in which R 6 is -OH, and R 7 is -CF 3 can also include any one or more of the features described herein, including (but not limited to) those delineated below.
- A can be 5- to 6-membered heteroaryl containing 1-3 (e.g., 1-2, 2-3, 1, 2, or 3) ring heteroatoms independently selected from O, N, N(R al ) and S, and in which the atom bonded to the sulfonyl sulfur is carbon.
- R al can be -H or -CH 3 .
- the 5- to 6-membered heteroaryl can contain 1 or 2 ring heteroatoms.
- the ring heteroatoms can be independently selected from N, N(R al ) and S.
- A can be 5-membered heteroaryl containing 1-3 (e.g., 1 or 2) ring heteroatoms independently selected from O, N, N(R al ) and S, and in which the atom bonded to the sulfonyl sulfur is carbon, and in which R al can be as defined anywhere herein.
- A can have formula (A-I):
- a 1 is singly bonded to A 4 , and A 3 is doubly bonded to A 4 ; AMs O 5 S 5 Or NR" 1 ; and one of A 2 , A 3 ,and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 2 , A 3 and A 4 is C-R 9 , and one of A 2 , A 3 , and A 4 is C-H or N; or
- a 3 is O, S, or NR al ; and one of A 1 , A 2 , and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 1 , A 2 , and A 4 is C-R 9 , and one of A 1 , A 2 , and A 4 is N or C-H.
- A can have formula (A-2):
- a 1 is O, S, or NR al ; and one of A 2 , A 3 ,and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 2 , A 3 and A 4 is C-R 9 , and one of A 2 , A 3 , and A 4 is C-H or N.
- a 1 , A 2 , A 3 , and A 4 can be as defined anywhere herein in (e.g., A 1 can be S, A 4 can be C-C(R 6 XR 7 XR 8 ), and A 2 can be C-H or N).
- A can be 5-membered heteroaryl having formula (A-3) as described herein.
- A can be phenyl.
- -C(R 6 )(R 7 )(R 8 ) can be attached to the phenyl ring carbon that is meta ox para with respect to the phenyl carbon that is attached to the sulfonyl sulfur in Formula I.
- R 8 can be -C(O)-NH 2 , -C(O)-NH-(CH 3 ), -C(O)-N(CH 3 ) 2 , -NH-C(O)-(CH 3 ), or
- N, N(R , 8s s l ) and S wherein said heteroaryl is unsubstituted or substituted with one or two substituents R ,8s2 independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 .
- R 8 can be -C(O)-NH 2 .
- R 8 can be l,2,4-triazol-3-yl or l,2,4-oxadiazol-3-yl, each of which is unsubstituted or substituted with one or two substituents R 8s2 independently selected from halo, -CH 3 , ethyl, -CF 3 , -OH and -OCH 3 (e.g., -CH 3 ).
- the carbon atom attached to R 4 and R 5 can have the ⁇ -configuration.
- the carbon atom attached to R 6 , R 7 , and R 8 can have the ⁇ -configuration.
- R 9 can be -H or absent.
- R 4 and R 5 are H, and the other is -CH 3 (e.g., R 4 can be H, and R 5 can be -CH 3 ).
- R 1 and R 2 can be, independently, as defined as defined anywhere herein.
- R 1 can be -CF 3 .
- R 2 can be -F.
- R 1 can be -CF 3
- R 2 can be -F.
- R 3 can be -H.
- R 1 canbe -CF 3
- R 2 canbe -F
- R 3 can be -H.
- R 6 is -OH
- R 7 is -CF 3 ;
- R 8 is -C(O)-NH 2 , -C(O)-NH-(CL 3 alkyl), or -C(O)-N(CL 3 alkyl) 2 (e.g., R 8 is -C(O)-NH 2 ).
- R 6 is -OH
- R 7 is -CF 3
- R 8 is -C(O)-NH 2 , -C(O)-NH-(C L3 alkyl), or -C(O)-N(C L3 alkyl) 2 (e.g., R 8 is -C(O)-NH 2 )
- R 8 is -C(O)-NH 2
- R 8 is -C(O)-NH 2
- A can be 5- to 6-membered heteroaryl containing 1-3 (e.g., 1-2, 2-3, 1, 2, or 3) ring heteroatoms independently selected from O, N, N(R al ) and S, and in which the atom bonded to the sulfonyl sulfur is carbon.
- R al can be -H or -CH 3 .
- the 5- to 6-membered heteroaryl can contain 1 or 2 ring heteroatoms.
- the ring heteroatoms can be independently selected from N, N(R al ) and S.
- A can be 5-membered heteroaryl containing 1-3 (e.g., 1 or 2) ring heteroatoms independently selected from O, N, N(R al ) and S, and in which the atom bonded to the sulfonyl sulfur is carbon, and in which R al can be as defined anywhere herein.
- A can have formula (A-I):
- a 1 is singly bonded to A 4 , and A 3 is doubly bonded to A 4 ; AMs O 5 S 5 Or NR" 1 ; and one of A 2 , A 3 ,and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 2 , A 3 and A 4 is C-R 9 , and one of A 2 , A 3 , and A 4 is C-H or N; or
- a 3 is O, S, or NR al ; and one of A 1 , A 2 , and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 1 , A 2 , and A 4 is C-R 9 , and one of A 1 , A 2 , and A 4 is N or C-H.
- A can have formula (A-2):
- a 1 is O, S, or NR al ; and one of A 2 , A 3 ,and A 4 is C-C(R 6 )(R 7 )(R 8 ), one of A 2 , A 3 and A 4 is C-R 9 , and one of A 2 , A 3 , and A 4 is C-H or N.
- a 1 , A 2 , A 3 , and A 4 can be as defined anywhere herein in (e.g., A 1 can be S, A 4 can be C-C(R 6 )(R 7 )(R 8 ), and A 2 can be C-H or N).
- A can be 5-membered heteroaryl having formula (A-3) as described herein.
- A can be phenyl.
- -C(R 6 )(R 7 )(R 8 ) can be attached to the phenyl ring carbon that is meta ox para with respect to the phenyl carbon that is attached to the sulfonyl sulfur in Formula I.
- the carbon atom attached to R 4 and R 5 can have the ⁇ -configuration.
- the carbon atom attached to R 6 , R 7 , and R 8 can have the ⁇ -configuration.
- R 9 can be -H or absent.
- R 4 and R 5 are H, and the other is -CH 3 (e.g., R 4 can be H, and R 5 can be -CH 3 ).
- R 1 and R 2 can be, independently, as defined as defined anywhere herein.
- R 1 can be -CF 3 .
- R can be -F.
- R 1 can be -CF 3
- R 2 can be -F.
- R 3 can be -H.
- R 1 can be -CF 3
- R 2 can be -F
- R 3 can be -H.
- R 6 is -OH;
- R 7 is -CF 3 ; and one, more than one, or all of the following features is(are) present:
- R 8 is -C(O)-NH 2 .
- A is a 5-membered heteroaryl, which has formula (A-2):
- a 1 , A 2 , A 3 , and A 4 can be as defined anywhere herein in (e.g., A 1 can be S, A 4 can be C- C(R 6 )(R 7 )(R 8 ), and A 2 can be C-H or N; e.g., A 1 can be S, A 4 can be C- C(R 6 )(R 7 )(R 8 ), and A 2 can be C-H).
- R 9 is -H or absent.
- R 4 is H
- R 5 is -CH 3 .
- R 1 is -CF 3 .
- the chemical entity E can have formula II (i.e., in which A in formula I is 2-thienyl):
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , and R 9 can be as defined anywhere herein.
- Embodiments of formula II can include any one or more of the features described herein, including but not limited to those delineated below.
- R 6 can be -OH.
- R 7 can be -CF 3 .
- R 8 can be -C(O)-NH 2 .
- R 6 can be -OH, and R 7 can be -CF 3 .
- R 6 can be -OH
- R 7 can be -CF 3
- R 8 can be -C(O)-NH 2 .
- the carbon atom attached to R 4 and R 5 can have the ⁇ -configuration.
- the carbon atom attached to R , R , and R can have the ⁇ -configuration.
- R 9 can be -H.
- R 4 can be H, and R 5 can be -CH 3 .
- R 1 and R 2 can be, independently, as defined as defined anywhere herein.
- R 1 can be -CF 3 .
- R 2 can be -F.
- R 3 can be -H.
- R 1 can be -CF 3
- R 2 can be -F.
- R 1 can be -CF 3
- R 2 can be -F
- R 3 can be -H.
- the chemical entity E can be (2R)-3,3,3-trifluoro-2-[5-( ⁇ (2R)-4-[4-fluoro-2- (trifluoromethyl)phenyl]-2-methylpiperazin- 1 -yl ⁇ sulfonyl)thiophen-2-yl]-2- hydroxypropanamide or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof; (e.g., or a pharmaceutically acceptable salt thereof).
- a salt e.g., a pharmaceutically acceptable salt
- N-oxide thereof e.g., or a pharmaceutically acceptable salt thereof
- the actual structure can instead be some hybrid or weighted average of two or more canonical forms, known collectively as resonance forms or structures.
- Resonance structures are not discrete chemical entities and exist only on paper. They differ from one another only in the placement or "localization" of the bonding and nonbonding electrons for a particular chemical entity. It can be possible for one resonance structure to contribute to a greater extent to the hybrid than the others.
- the written and graphical descriptions of the embodiments of the present invention are made in terms of what the art recognizes as the predominant resonance form for a particular species.
- the compounds described herein can be synthesized according to methods described herein (or variations thereof) and/or conventional, organic chemical synthesis methods from commercially available starting materials and reagents or from starting materials and reagents that can be prepared according to conventional organic chemical synthesis methods. It is also possible to make use of variants of any of the aforementioned process steps. As can be appreciated by the skilled artisan, further methods of synthesizing the compounds of the formulae herein will be evident to those skilled in the art. Additionally, the various synthetic steps may be performed in an alternate sequence or order to give the desired compounds.
- Synthetic chemistry transformations and protecting group methodologies useful in synthesizing the compounds described herein are known in the art and include, for example, those such as described in R. C. Larock, Comprehensive Organic Transformations, 2d.ed., Wiley- VCH Publishers (1999); P. G. M. Wuts and T. W. Greene, Protective Groups in Organic Synthesis, 4th Ed., John Wiley and Sons (2007); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994); and L. Paquette, ed., Encyclopedia of Reagents or Organic Synthesis, John Wiley and Sons (1995), and subsequent editions thereof.
- the compounds described herein can be separated from a reaction mixture and further purified by a method such as column chromatography, high- performance liquid chromatography (HPLC), or recrystallization.
- the compounds of this invention can be readily prepared according to the following schemes from commercially available starting materials or starting materials which can be prepared using literature procedures.
- the schemes show the preparation of representative compounds of this invention. It is also possible to make use of variants of these process steps, which in themselves are known to and well within the preparatory skill of the skilled artisan.
- R 1 to R 9 , and A are selected from groups defined above (unless otherwise indicated).
- piperazine 1 e.g., (R)-2-methyl-piperazine, which can be obtained commercially from Aldrich Chemical Company or synthesized according the procedure described in Xiang, J. et al, Journal Medicinal Chemistry, 2008, 57(14), 4068- 4071
- halobenzene 2 Hal 1 can be Br or I, typically Br
- a transition metal catalyst typically a palladium- based catalyst, e.g., Pd 2 (dba) 3
- sulfonyl halide 4 Reaction of intermediate 3 with sulfonyl halide 4 (Hal 2 is typically chloro; Z is halo, e.g., bromo; or Z is hydrogen) results in the formation sulfonamide 5.
- sulfonyl halide 4 can be synthesized by reacting the corresponding thiol with a halogenating reagent, e.g., SO 2 CI 2 /KNO 3 , in a polar aprotic solvent such as acetonitrile.
- the process of converting 3 to 5 can also include one or more steps needed to introduce certain R 2 substituents (e.g., when R 2 is 5-membered heteroaryl).
- Metallation of 5 e.g., by metal-halogen exchange when Z is halo; or by deprotonation with a strong base, e.g., LDA or n-BuLi, when Z is hydrogen
- a carbon electrophile e.g., a ketone, such as 3,3,3-trifluoropyruvate
- E is a substituent precursor (e.g., a tertiary alcohol) to -CR 6 R 7 R 8 in the final compound 7.
- the process of converting 6 to 7 can also include one or more steps needed to introduce certain R 2 substituents (e.g., when R 2 is -C(O)NH 2 or 5- membered heteroaryl).
- the process of converting 6 to 7 in Scheme I can also include one or more steps needed to introduce certain R 2 substituents.
- compounds in which R 2 is -CN can be partially hydrolyzed to provide amide 13 or can undergo an intermolecular dipolar addition reaction to produce tetrazole 14 (see Scheme III).
- compounds in which R 8 is -NH-C(O)-(Ci_3 alkyl) can be prepared according to Scheme IV, which shows for illustrative purposes only the synthesis of compounds in which R 8 is -NH-C(O)-(CHs).
- compounds in which R 8 is 5-membered heteroaryl can be prepared according to Scheme V, which shows for illustrative purposes only the synthesis of compounds in which R 6 is -OH, R 7 is CF 3 , and R 8 is 5-membered heteroaryl from compounds in which R 6 is -OH, R 7 is CF 3 , and R 8 is CONH 2 .
- Scheme V shows for illustrative purposes only the synthesis of compounds in which R 6 is -OH, R 7 is CF 3 , and R 8 is 5-membered heteroaryl from compounds in which R 6 is -OH, R 7 is CF 3 , and R 8 is CONH 2 .
- compound 9 (c/v Scheme I) can be converted to compound 18 using DMF dimethyl acetal. Exposure of compound 18 to hydrazine results in the formation of triazine 19, while reaction of compound 18 with hydroxylamine affords oxadiazole 20.
- the tertiary hydroxyl group in 9 can be protected, e.g., as an ether, e.g., as a silyl ether.
- nitrile 21 (Hal is typically -Br) can be hydrolyzed under basic conditions (e.g., 3N NaOH in methanol and water) to carboxylic acid 22, which in turn can be converted to the corresponding hydrazide 23 under conventional conditions.
- Formation of the oxadiazole ring in 24 can be achieved upon reacting hydrozide 23 with an ortho formate, such as triethylorthoformate, in the presence of a catalytic amount of acid, e.g., PSA.
- Compounds 25 and 26 can be obtained using the chemistries described in the preceding schemes.
- compound 5 (cf. Scheme I) can be coupled with silyl enol ether 27 to form ester 28, which is hydrolyzed to the carboxylic acid 29. Conversion of 29 to the amide 30 can be achieved, e.g., using CDI and NH3.
- the compounds of this invention may contain one or more asymmetric centers and thus occur as racemates and racemic mixtures, single enantiomers, individual diastereomers and diastereomeric mixtures. All such isomeric forms of these compounds are expressly included in the present invention.
- a chemical entity E can have the i?-CR 4 CR 5 configuration and the i?-CR 6 R 7 R 8 configuration ("a i?-CR 4 CR 5 , i?-CR 6 R 7 R 8 stereoisomer"). In other embodiments, a chemical entity E can have the i?-CR 4 CR 5 configuration and the S- CR 6 R 7 R 8 configuration ("a i?-CR 4 CR 5 , ,S-CR 6 R 7 R 8 stereoisomer").
- a i?-CR 4 CR 5 , i?-CR 6 R 7 R 8 stereoisomer can be present as a mixture with its corresponding i?-CR 4 CR 5 , S-CR 6 R 7 R 8 stereoisomer.
- the mixture can contain greater than about 50% of the i?-CR 4 CR 5 , i?-CR 6 R 7 R 8 stereoisomer (e.g., about 60%, about 70%, about 80%, about 90%, about 95%, about 99%).
- the mixture can further include one or more other substances (e.g., one or more pharmaceutically acceptable carriers, biological fluids, cellular culture, or any combination thereof).
- the i?-CR 4 CR 5 , i?-CR 6 R 7 R 8 stereoisomer can be substantially free of its i?-CR 4 CR 5 , ,S-CR 6 R 7 R 8 stereoisomer.
- the i?-CR 4 CR 5 , i?-CR 6 R 7 R 8 stereoisomer can be in substantially pure form.
- the compounds of this invention may also contain linkages (e.g., carbon-carbon bonds, carbon-nitrogen bonds such as amide bonds) wherein bond rotation is restricted about that particular linkage, e.g. restriction resulting from the presence of a ring or double bond. Accordingly, all cis/trans and E/Z isomers and rotational isomers are expressly included in the present invention.
- the compounds of this invention may also be represented in multiple tautomeric forms, in such instances, the invention expressly includes all tautomeric forms of the compounds described herein, even though only a single tautomeric form may be represented (e.g., alkylation of a ring system may result in alkylation at multiple sites, the invention expressly includes all such reaction products). All such isomeric forms of such compounds are expressly included in the present invention.
- the compounds of this invention include the compounds themselves, as well as their salts and their prodrugs, if applicable.
- a salt for example, can be formed between an anion and a positively charged substituent (e.g., amino) on a compound described herein. Suitable anions include chloride, bromide, iodide, sulfate, nitrate, phosphate, citrate, methanesulfonate, trifluoroacetate, and acetate.
- a salt can also be formed between a cation and a negatively charged substituent (e.g., carboxylate) on a compound described herein.
- Suitable cations include sodium ion, potassium ion, magnesium ion, calcium ion, and an ammonium cation such as tetramethylammonium ion.
- prodrugs include Ci_6 alkyl esters of carboxylic acid groups, which, upon administration to a subject, are capable of providing active compounds.
- Pharmaceutically acceptable salts of the compounds of this invention include those derived from pharmaceutically acceptable inorganic and organic acids and bases.
- suitable acid salts include acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptanoate, glycolate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2- hydroxyethanesulfonate, lactate, maleate, malonate, methanesulfonate, 2- naphthalenesulfonate, nicotinate, nitrate, palmoate, pectinate, persulfate, 3- phenylpropionate, phosphate,
- Salts derived from appropriate bases include alkali metal (e.g., sodium), alkaline earth metal (e.g., magnesium), ammonium and N-(alkyl) 4 salts.
- alkali metal e.g., sodium
- alkaline earth metal e.g., magnesium
- ammonium e.g., ammonium
- N-(alkyl) 4 salts e.g., ammonium
- This invention also envisions the quaternization of any basic nitrogen-containing groups of the compounds disclosed herein. Water or oil-soluble or dispersible products may be obtained by such quaternization.
- Salt forms of the compounds of any of the formulae herein can be amino acid salts of carboxy groups (e.g. L-arginine, -lysine, -histidine salts).
- pharmaceutically acceptable carrier refers to a carrier that may be administered to a subject (e.g., a patient), together with a chemical entity E as defined anywhere herein, and which does not destroy the pharmacological activity thereof and is nontoxic when administered in doses sufficient to deliver a therapeutic amount of the chemical entity E.
- compositions described herein can include any one or more of the following: ion exchangers, alumina, aluminum stearate, lecithin, self-emulsifying drug delivery systems (SEDDS) such as d- ⁇ -tocopherol polyethyleneglycol 1000 succinate, surfactants used in pharmaceutical dosage forms such as Tweens or other similar polymeric delivery matrices, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts, or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene-polyoxypropylene-block polymers, polyethylene glycol
- Cyclodextrins such as ⁇ -, ⁇ -, and ⁇ -cyclodextrin, or chemically modified derivatives such as hydroxyalkylcyclodextrins, including 2- and 3-hydroxypropyl- ⁇ -cyclodextrins, or other solubilized derivatives may also be advantageously used to enhance delivery of compounds of the formulae described herein.
- the chemical entities described herein can be used for treating (e.g., controlling, relieving, ameliorating, alleviating, or slowing the progression of) or methods for preventing (e.g., delaying the onset of or reducing the risk of developing) a disease or condition that is mediated by excess (e.g., abnormally high) levels or uncontrolled (e.g., resulting in abnormally high) levels of Cortisol and/or other corticosteroids (e.g., 11 ⁇ - hydroxy steroids.
- excess e.g., abnormally high
- uncontrolled e.g., resulting in abnormally high
- Cortisol and/or other corticosteroids e.g., 11 ⁇ - hydroxy steroids.
- the diseases, disorders, conditions or symptoms mediated by excess or uncontrolled amounts of Cortisol and/or other corticosteroids can include diabetes (e.g., type 1 or type 2 diabetes), Syndrome X, hyperglycemia, low glucose tolerance, insulin resistance, obesity, lipid disorders, dyslipidemia, hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels, high LDL levels, atherosclerosis and its sequelae, vascular restenosis, pancreatitis, abdominal obesity, neurodegenerative disease, retinopathy, nephropathy, neuropathy, hypertension, coronary heart disease, stroke, peripheral vascular disease, Cushing's syndrome, glaucoma, osteoperosis, hyperinsulinemia, tuberculosis, psoriasis,cognitive disorders and dementia (e.g., impairment associated with aging and of neuronal dysfunction, e.g., Alzheimer's disease), depression, viral diseases, inflammatory disorders, immune disorders.
- the diseases, disorders conditions e
- the chemical entities described herein can be coadministered with one or more other threapeutic agents.
- the additional agents may be administered separately, as part of a multiple dose regimen, from the chemical entities described herein (e.g., sequentially, e.g., on different overlapping schedules with the administration of one or more compounds of formula (I)).
- these agents may be part of a single dosage form, mixed together with the chemical entities described herein in a single composition.
- these agents can be given as a separate dose that is administered at about the same time that one or more chemical entities described herein are administered (e.g., simultaneously with the administration of one or more chemical entities described herein).
- compositions described herein include a combination of a chemical entity E as defined anywhere herein and one or more additional therapeutic or prophylactic agents
- both the chemical entity and the additional agent should be present at dosage levels of between about 1 to 100%, and more preferably between about 5 to 95% of the dosage normally administered in a monotherapy regimen.
- Other therapeutic agents can include DP-IV inhibitors; insulin sensitizers (e.g., (i) PPAR agonists and (ii) biguanides); insulin and insulin analogues and mimetics; sulfonylureas and other insulin secretagogues; prandial glucose regulators, alpha.- glucosidase inhibitors; glucagon receptor antagonists; GLP-I, GLP-I mimetics, and GLP-I receptor agonists; GIP 5 GIP mimetics, and GIP receptor agonists; PACAP,
- PACAP mimetics and PACAP receptor 3 agonists; cholesterol lowering agents (e.g., (i) HMG-CoA reductase inhibitors, (ii) sequestrants, (iii) nicotinyl alcohol, nicotinic acid and salts thereof, (iv) PPAR. alpha, agonists, (v) PPAR. alpha./, gamma, dual agonists, (vi) inhibitors of cholesterol absorption, (vii) acyl CoAxholesterol acyltransferase inhibitors, and (viii) anti-oxidants; PPAR.delta.
- cholesterol lowering agents e.g., (i) HMG-CoA reductase inhibitors, (ii) sequestrants, (iii) nicotinyl alcohol, nicotinic acid and salts thereof, (iv) PPAR. alpha, agonists, (v) PPAR. alpha./, gam
- antiobesity compounds e.g., sibutramine and orlisat
- an ileal bile acid transporter inhibitor e.g., an ileal bile acid transporter inhibitor
- anti-inflammatory agents excluding glucocorticoids (e.g., aspirin); protein tyrosine phosphatase- IB (PTP-IB) inhibitors; agents that suppress hepatic glucose output (e.g., metformin); agents designed to reduce the absorption of glusoce from the intestine (e.g., acarbose); agents designed to treat the complications of prolonged hyperglycemia (e.g., aldose reductase inhibitors); antidiabetic agents (e.g., glusoce phosphatase inhibitors, glucose -6-phosphatase inhibitors, glucagon receptor antagonists, glucose kinase activators, glycogen phosphorylase inhibitors, fructose 1,6 bisphosphatase inhibitors
- the chemical entities and compositions described herein can, for example, be administered orally, parenterally (e.g., subcutaneously, intracutaneously, intravenously, intramuscularly, intraarticularly, intraarterially, intrasynovially, intrasternally, intrathecally, intralesionally and by intracranial injection or infusion techniques), by inhalation spray, topically, rectally, nasally, buccally, vaginally, via an implanted reservoir, by injection, subdermally, intraperitoneally, transmucosally, or in an ophthalmic preparation, with a dosage ranging from about 0.01 mg/Kg to about 1000 mg/Kg, (e.g., from about 0.01 to about 100 mg/kg, from about 0.1 to about 100 mg/Kg, from about 1 to about 100 mg/Kg, from about 1 to about 10 mg/kg) every 4 to 120 hours, or according to the requirements of the particular drug.
- parenterally e.g., subcutaneously, intracutaneously, intra
- compositions are administered by oral administration or administration by injection.
- the methods herein contemplate administration of an effective amount of compound or compound composition to achieve the desired or stated effect.
- the pharmaceutical compositions of this invention will be administered from about 1 to about 6 times per day or alternatively, as a continuous infusion. Such administration can be used as a chronic or acute therapy.
- the amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration.
- a typical preparation will contain from about 5% to about 95% active compound (w/w).
- such preparations contain from about 20% to about 80% active compound.
- a maintenance dose of a compound, composition or combination of this invention may be administered, if necessary.
- compositions of this invention may contain any conventional non-toxic pharmaceutically-acceptable carriers, adjuvants or vehicles.
- pH of the formulation may be adjusted with pharmaceutically acceptable acids, bases or buffers to enhance the stability of the formulated compound or its delivery form.
- compositions may be in the form of a sterile injectable preparation, for example, as a sterile injectable aqueous or oleaginous suspension.
- This suspension may be formulated according to techniques known in the art using suitable dispersing or wetting agents (such as, for example, Tween 80) and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3- butanediol.
- suitable vehicles and solvents that may be employed are mannitol, water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- Fatty acids, such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions.
- These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant, or carboxymethyl cellulose or similar dispersing agents which are commonly used in the formulation of pharmaceutically acceptable dosage forms such as emulsions and or suspensions.
- surfactants such as Tweens or Spans and/or other similar emulsifying agents or bioavailability enhancers which are commonly used in the manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms may also be used for the purposes of formulation.
- compositions of this invention may be orally administered in any orally acceptable dosage form including, but not limited to, capsules, tablets, emulsions and aqueous suspensions, dispersions and solutions.
- carriers which are commonly used include lactose and corn starch.
- Lubricating agents such as magnesium stearate, are also typically added.
- useful diluents include lactose and dried corn starch.
- compositions of this invention may also be administered in the form of suppositories for rectal administration.
- These compositions can be prepared by mixing a compound of this invention with a suitable non-irritating excipient which is solid at room temperature but liquid at the rectal temperature and therefore will melt in the rectum to release the active components.
- suitable non-irritating excipient include, but are not limited to, cocoa butter, beeswax and polyethylene glycols.
- topical administration of the compounds and compositions described herein may be presented in the form of an aerosol, a semi-solid pharmaceutical composition, a powder, or a solution.
- a semi-solid composition is meant an ointment, cream, salve, jelly, or other pharmaceutical composition of substantially similar consistency suitable for application to the skin. Examples of semi-solid compositions are given in Chapter 17 of The Theory and Practice of Industrial Pharmacy, Lachman, Lieberman and Kanig, published by Lea and Febiger (1970) and in Remington's Pharmaceutical Sciences, 21st Edition (2005) published by Mack Publishing Company, which is incorporated herein by reference in its entirety.
- Topical administration of the compositions of this invention is useful when the desired treatment involves areas or organs readily accessible by topical application.
- the composition should be formulated with a suitable ointment containing the active components suspended or dissolved in a carrier.
- Carriers for topical administration of the compounds of this invention include, but are not limited to, mineral oil, liquid petroleum, white petroleum, propylene glycol, polyoxyethylene polyoxypropylene compound, emulsifying wax and water.
- the composition can be formulated with a suitable lotion or cream containing the active compound suspended or dissolved in a carrier with suitable emulsifying agents.
- Suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water.
- the compositions of this invention may also be topically applied to the lower intestinal tract by rectal suppository formulation or in a suitable enema formulation.
- Topically-transdermal patches are also included in this invention. Also within the invention is a patch to deliver active chemotherapeutic combinations herein.
- a patch includes a material layer (e.g., polymeric, cloth, gauze, bandage) and the compound of the formulae herein as delineated herein. One side of the material layer can have a protective layer adhered to it to resist passage of the compounds or compositions.
- the patch can additionally include an adhesive to hold the patch in place on a subject.
- An adhesive is a composition, including those of either natural or synthetic origin, that when contacted with the skin of a subject, temporarily adheres to the skin. It can be water resistant. The adhesive can be placed on the patch to hold it in contact with the skin of the subject for an extended period of time.
- the adhesive can be made of a tackiness, or adhesive strength, such that it holds the device in place subject to incidental contact, however, upon an affirmative act (e.g., ripping, peeling, or other intentional removal) the adhesive gives way to the external pressure placed on the device or the adhesive itself, and allows for breaking of the adhesion contact.
- the adhesive can be pressure sensitive, that is, it can allow for positioning of the adhesive (and the device to be adhered to the skin) against the skin by the application of pressure (e.g., pushing, rubbing,) on the adhesive or device.
- compositions of this invention may be administered by nasal aerosol or inhalation.
- Such compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art.
- a composition having a chemical entity as defined anywhere herein and an additional agent e.g., a therapeutic agent
- an additional agent e.g., a therapeutic agent
- a composition having the compound of the formulae herein and an additional agent can be administered using an implantable device.
- Implantable devices and related technology are known in the art and are useful as delivery systems where a continuous, or timed- release delivery of compounds or compositions delineated herein is desired. Additionally, the implantable device delivery system is useful for targeting specific points of compound or composition delivery (e.g., localized sites, organs). Negrin et al, Biomaterials, 22(6): 563 (2001). Timed-release technology involving alternate delivery methods can also be used in this invention. For example, timed-release formulations based on polymer technologies, sustained-release techniques and encapsulation techniques (e.g., polymeric, liposomal) can also be used for delivery of the compounds and compositions delineated herein. The invention will be further described in the following examples. It should be understood that these examples are for illustrative purposes only and are not to be construed as limiting this invention in any manner.
- Step IB To a solution of 5-bromothiophene-2-sulfonyl chloride (26.2 g, 100 mmol) and (3R)-l-(4-fluoro-2-(trifluoromethyl)phenyl)-3-methylpiperazine (27.6 g, lOOmmol) in DCM (200 ml) was added Et 3 N (41.8 ml, 300 mmol) at room temp. The reaction mixture was stirred at room temperature until completion of the reaction (about 6 hours) and then washed with aq. NaHCO 3 . The basic washes were back extracted with dichloromethane (DCM). The combined organic layers were washed with brine and dried over Na 2 SO 4 .
- DCM dichloromethane
- Step 1C To a solution of (R)-l-(5-bromothiophen-2-ylsulfonyl)-4-(4-fluoro-2- (trifluoromethyl)phenyl)-2-methylpiperazine (28.1 g, 57.7 mmol) in anhydrous THF (200 ml) was added Butyllithium (28.8 ml, 57.7 mmol) at -78 0 C. The reaction mixture was Stirred under N 2 for 15 min. and then a solution of methyl 3,3,3-trifluoropyruvate (6.07 ml, 57.7 mmol) in THF (20 mL) was added via a cannula.
- reaction mixture was stirred at -78 0 C for 2 h. and then quenched with a 10 mL of 10% aq. HCl.
- the reaction mixture was dried over MgSO 4 and CombiFlashed with DCM/hexane (15 - 100%) to provide methyl 3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2-(trifluoromethyl)phenyl)-2- methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2-hydroxypropanoate as a sticky, light yellow solid (22 g, 39.0 mmol, 67.6 % yield).
- Step ID Method 1: To a solution of methyl 3,3,3-trifluoro-2-(5-((R)-4-(4-fiuoro- 2-(trifluoromethyl)phenyl)-2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2- hydroxypropanoate (21.5 g, 38.1 mmol) in MeOH (200 ml) was added aq. NH 3 (-28-
- the title compound was isolated from the crude reaction mixture that was obtained after performing step D in Example 1.1.
- the title compound was separated using a chiral column (Chiralpak ADH) in SFC Analytical Instrument. Mobile Phase was 90% CO2 /10%Methanol. (900 mg, 3.76% yield).
- TLC thin layer chromatography
- Step 2B To a stirred solution of (3i?)-l-[4-fluoro-2-(trifluoromethyl)phenyl]-3- methylpiperazine (100 mg, 0.38 mmol) and diisopropylethylamine (0.13 mL, 0.76 mmol) in anhydrous dichloromethane (2 mL) was added thiazole-2-sulfonyl chloride (70 mg, 0.38 mmol) at O 0 C. The reaction mixture was stirred at O 0 C for 15 min, then stirred at room temperature for 3 hrs, after which time the reaction was judged complete by TLC.
- Example 2.1 was separated with a chiral column (Chiralpak ADH) in SFC Analytical Instrument. Mobile Phase was 90% CO2 /10%MethanoL. The title compounds of Example 2.2 and 2.3 were isolated in good yield.
- Step 3 A To a stirred solution of (3i?)-l-[4-fluoro-2-(trifluoromethyl)phenyl]-3- methylpiperazine (1.57 g, 5.97 mmol) and diisopropylethylamine (3.1 rnL, 17.91 mmol) in anhydrous dichloromethane (20 rnL) was added 2H-[l,2,4]triazole-3-sulfonyl chloride (1.0 g, 5.97 mmol) at O 0 C. The mixture was stirred at O 0 C for 30 min, then stirred at room temperature for o/n. The reaction mixture was washed with H 2 O and extracted with DCM. The organic layer was dried over Na 2 SO 4 and concentrated.
- Step 3B To a stirred solution of (2i?)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methyl-l-(lH-l,2,4-triazol-5-ylsulfonyl)piperazine (695 mg, 1.77 mmol) in 10 mL of anhydrous T ⁇ F was added NaH (77.9 mg, 1.947 mmol) at room temperature. The reaction mixture was heated to reflux for 6 hrs. The reaction mixture was cooled to room temperature, and methyl iodide (MeI) (0.12 mL, 1.947 mmol) was added. The reaction mixture was heated to reflux again for lhr, after which time the reaction was determined to be complete by TLC.
- MeI methyl iodide
- reaction mixture was cooled to room temperature and stirred o/n.
- the solvent was evaporated to afford a residue that was diluted with DCM, then washed with H2O and sat. brine, dried over MgSO4, and concentrated to afford an orange gum, which was purified via flash column chromatography, eluting with 40-80% EtOAc/Hexane to yield (2i?)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methyl-l-[(l- methyl-lH-l,2,4-triazol-5-yl)sulfonyl]piperazine as a light yellow solid (522 mg, 72.4% yield).
- Step 3C To a solution of (2i?)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methyl-l- [(l-methyl-lH-l,2,4-triazol-5-yl)sulfonyl]piperazine (484 mg, 1.19 mmol) in anhydrous T ⁇ F (7 ml) was added n-Butyllithium (0.52 ml, 1.31 mmol) at -78 0 C. The reaction mixture was stirred under N 2 for 30 min., then methyl 3,3,3-trifluoropyruvate (0.18 ml, 1.785 mmol) was added.
- Step 3D To methyl 3,3,3-trifluoro-2-[3-( ⁇ (2i?)-4-[4-fluoro-2-
- Step 4A To a stirred solution of (3i?)-l-[4-fluoro-2-(trifluoromethyl)phenyl]-3- methylpiperazine (1.038 g, 3.96 mmol) and diisopropylethylamine (1.38 rnL, 7.92 mmol) in anhydrous dichloromethane (15 rnL) was added 1 -methyl- l ⁇ -imidazole-4-sulfonyl chloride (0.715 g, 3.96 mmol) at O 0 C. The mixture was stirred at O 0 C for 30 min, then stirred at room temperature for o/n. The reaction mixture was washed with H2O, extracted with DCM.
- Step 4B To a solution of (2i?)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methyl-l- [(I -methyl- lH-imidazol-4-yl)sulfonyl]piperazine (552 mg, 1.36 mmol) in anhydrous
- Step 5 A A mixture of (R)-2-methyl-piperazine (6.0 g, 59.9 mmol), 4-chloro-3- trifluoromethyl-benzonitrile (11.2 g, 54.5 mmol), tris(dibenzylidineacetone)dipalldium (0) (0.499 g, 0.545 mmol), rac-2,2'-bis(diphenylphosphino)-l,l '-binaphthyl (1.02 g, 1.635 mmol)and sodium tert-butoxide (6.55 g, 68.12 mmol) was mixed and purged with N 2 .
- Step 5B To a stirred solution of 4-[(3i?)-3-methylpiperazin-l-yl]-3- (trifluoromethyl)benzonitrile (2.69 g, 10 mmol) and TEA (4.18 rnL, 30 mmol) in anhydrous dichloromethane (20 mL) was added thiophene-2-sulfonyl chloride (1.92 g, 10 mmol) at O 0 C. The mixture was stirred at O 0 C for 15 min, then stirred at room temperature for o/n, after which time the reaction was determined to be complete by
- Step 5C To a solution of 5-chloro-N- ⁇ 4-[(6-cyano-2- ⁇ 3-[4- (dimethylamino)piperidin-l-yl]phenyl ⁇ pyrazolo[l,5-a]pyrimidin-7-yl)amino]-2- methoxyphenyl ⁇ -l-benzofuran-2-carboxamide (4.15 g, 10 mmol) in anhydrous THF (60 ml) was added freshly made LDA (12 mmol) at -78 0 C. The reaction mixture was stirred under N 2 for 20 min., and then methyl 3,3,3-trifluoropyruvate (2.1 mL, 20 mmol) was added.
- Step 5D To methyl 2-[5-( ⁇ (2i?)-4-[4-cyano-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl ⁇ sulfonyl)-2-thienyl] -3 ,3 ,3 -trifluoro-2-hydroxypropanoate (120 mg, 0.21) was added NH 3 in EtOH (2.0M, 5 mL). The resultant mixture was stirred at room temperature for 2 days.
- reaction mixture was then concentrated and purified on a SiO 2 gel column, eluting with MeOH in DCM to give 2-[5-( ⁇ (2i?)-4-[4-cyano-2- (trifluoromethyl)phenyl]-2-methylpiperazin-l-yl ⁇ sulfonyl)-2-thienyl]-3,3,3-trifluoro-2- hydroxypropanamide as a white solid (58 mg, 50% yield).
- Step 5E To a solution of 2-[5-( ⁇ (2i?)-4-[4-cyano-2-(trifluoromethyl)phenyl]-2- methylpiperazin-l-yl ⁇ sulfonyl)-2-thienyl]-3,3,3-trifluoro-2-hydroxypropanamide (200 mg, 0.36 mmol) in t-BuOH (2 niL) was added powdered KOH (100 mg, 1.8 mmol) at room temperature. The reaction mixture was heated to 8O 0 C for 15 min., after which the Reaction was determined to be complete by LC/MS. The reaction mixture was diluted with EtOAc, washed with H 2 O three times, then washed with sat. brine.
- Step 6A The starting material, 4-[(3i?)-3-methylpiperazin-l-yl]-3- (trifluoromethyl)benzonitrile, was prepared according to a procedure similar to that described in Example 5.1, step 5 A.
- 4-[(3i?)-3-methylpiperazin-l- yl]-3-(trifluoromethyl)benzonitrile (3.24 g, 12.03 mmol) and diisopropylethylamine (4.19 mL, 24.06 mmol) in anhydrous dichloromethane (30 mL) was added 5-bromo-thiophene- 2-sulfonyl chloride (3.15 g, 12.03 mmol) at O 0 C.
- Step 6C To a 50 mL flask containing 4- ⁇ (3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3- methylpiperazin-l-yl ⁇ -3-(trifluoromethyl)benzamide was added N,N-dimethylacetamide dimethylacetal (6 mL, 37 mmol). The reaction mixture was stirred at 85 0 C for 20 min., after which time the reaction was determined to be complete by LC/MS. The reaction mixture was cooled to room temperature, and the solvent was removed under reduced pressure.
- Step 6D To a solution of hydroxy lamine hydrochloride (93.2 mg, 1.34 mmol) in a mixture of 6N NaOH solution (223 ⁇ L, 1.34 mmol) and 7 mL of 70% aqueous acetic acid was added 4- ⁇ (3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3-methylpiperazin-l-yl ⁇ - ⁇ /- [(lZ)-l-(dimethylamino) ethylidene]-3-(trifluoromethyl)benzamide (650 mg, 1.12 mmol). The reaction mixture was stirred at room temperature for 1.5 hrs., after which time the reaction was determined to be complete by LC/MS.
- Step 6E To a solution of (2i?)-l-[(5-bromo-2-thienyl)sulfonyl]-2-methyl-4-[4-(3- methyl-l,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenyl]piperazine (500 mg, 0.91 mmol) in anhydrous THF (10 ml) was added n-butyllithium (0.36 ml, 2.5 M in hexane, 0.91 mmol) at -78 0 C. The reaction mixture was stirred under N 2 for 5 min. and then methyl 3,3,3-trifluoropyruvate (0.20 ml, 1.36 mmol) was added.
- Step 6F To a solution of 3,3,3-trifluoro-2-hydroxy-2-[5-( ⁇ (2i?)-2-methyl-4-[4-(3- methyl-l,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenyl]piperazin-l- yl ⁇ sulfonyl)thiophen-2-yl]propanoate (200 mg, 0.31 mmol) in 20 mL of MeOH was bubbled NH3 (g) for 30 seconds at -78 0 C. The reaction mixture was warmed up to room temperature and stirred under empty balloon for o/n., after which the reaction was determined to be complete by LC/MS.
- Example 7.1 The title compound of Example 7.1 was prepared as shown in Scheme 7 below. Detailed synthesis procedures are provided below.
- Step 7A The starting material, 4- ⁇ (3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3- methylpiperazin-l-yl ⁇ -3-(trifluoromethyl)benzonitrile, was prepared according to a procedure similar to that described in Example 6.1, step 6 A.
- Step 7B To a solution of 4- ⁇ (3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3- methylpiperazin-l-yl ⁇ -3-(trifluoromethyl)benzoic acid (700 mg, 1.36 mmol) in 7 mL of anhydrous 1 ,2-dichloroethane was added SOCl 2 (0.6 mL, 8.18 mmol). The reaction mixture was stirred at room temperature for 2 hours, heated at reflux for 5hrs., and then cooled to room temperature and stirred o/n. The solvent was evaporated to afford a residue, which was taken up in 7 mL of DCM.
- the DCM mixture was cooled to O 0 C, and NH 2 NH 2 (0.26 mL, 8.18 mmol) was added. Stirring was continued at O 0 C for 30 min., after which the reaction was determined to be complete by LC/MS. The solvent was removed under reduced pressure. To the resulting residue was added H2O, and the aqueous mixture was extracted with DCM (3x). The combined organic layers were washed with sat.
- Step 7C To a solution of 4- ⁇ (3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3- methylpiperazin-l-yl ⁇ -3-(trifluoromethyl)benzohydrazide (548 mg, 1.04 mmol) in 3 mL of EtOH were added triethylortho formate (5 mL) and a few mg of PSA. The reaction mixture was heated to 8O 0 C for 3 hrs., after which the reaction was determined to be complete by LC/MS. The reaction mixture was cooled to room temperature, and the solvent removed under reduced pressure.
- Step 7D The penultimate compound in Scheme 7 was prepared according to a procedure similar to that described in Example 6.1, step 6E.
- Step 7E The final compound in Scheme 7 was prepared according to a procedure similar to that described in Example 6.1, step 6F (50 mg, 83.4% yield).
- HRMS calcd for C 2 IHi 9 F 6 N 5 O 5 S 2 + H+, 600.08045; found (ESI, [M+H]+),
- Example 8.1 The title compound of Example 8.1 was prepared as shown in Scheme 8 below. Detailed synthesis procedures are provided below.
- Step 8A The starting material, 2-[5-( ⁇ (2i?)-4-[4-cyano-2-
- Example 9.1 The title compound of Example 9.1 was prepared as shown in Scheme 9 below. Detailed synthesis procedures are provided below.
- Step 9A The starting material, 4- ⁇ (3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3- methylpiperazin-l-yl ⁇ - ⁇ /-[(lZ)-l-(dimethylamino) ethylidene]-3-
- Step 9B The penultimate compound in Scheme 9 was prepared according to a procedure similar to that described in Example 6.1, step 6E.
- Step 9C The final compound was prepared according to a procedure similar to that described in Example 6.1, step 6F (58 mg, 94.7% yield).
- Example 10.1 The title compound of Example 10.1 was prepared as shown in Scheme 10 below. Detailed synthesis procedures are provided below.
- Step 1OA The starting material, 3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2- (trifluoromethyl)phenyl)-2-methylpiperazin- 1 -ylsulfonyl)thiophen-2-yl)-2- hydroxypropanamide, was prepared according to a procedure similar to that described in Example 1.1, step IA to ID.
- Step 1OB To a solution of hydrazine (8 uL, 0.28 mmol) in 3 mL of acetic acid was added N-dimethylaminomethylene-3,3,3-trifluoro-2- ⁇ 5-[4-(4-fluoro-2- trifluoromethyl-phenyl)-2-methyl-piperazine- 1 -sulfonyl]-thiophen-2-yl ⁇ -2-hydroxy- propionamide (96 mg, 0.14 mmol) at room temperature. The reaction mixture was then stirred at room temperature for 1 hour., after which time the reaction was determined to be complete by LC/MS. The solvent was evaporated, and the resultant residue diluted with EtOAc.
- Step 1 IA The starting material, 3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2- (trifluoromethyl)phenyl)-2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2- hydroxypropanamide, was prepared according to a procedure similar to that described in Example 1.1, step IA to ID.
- Step 12A-12B The starting material was prepared according to a procedure similar to that described in Example 5.1, steps 5 A to 5D.
- Step 12C The title compound was prepared according to a procedure similar to that described in Example 5.1, step 5E (80 mg, 72%yield).
- Step 13 A The starting material, 2- ⁇ 5-[4-(4-Cyano-2-trifluoromethyl-phenyl)-2- methyl-piperazine-l-sulfonyl]-thiophen-2-yl ⁇ -N-dimethylaminomethylene-3,3,3- trifluoro-2-hydroxy-propionamide, was prepared according to a procedure similar to that described in Example 12.1, step 12A.
- Step 13B To a solution of hydroxylamine hydrochloride (40 mg, 0.61 mmol) in a mixture of 3N NaOH solution (100 ⁇ L, 0.61 mmol) and 6 mL of 70% aqueous acetic acid was added 2- ⁇ 5-[4-(4-Cyano-2-trifluoromethyl-phenyl)-2-methyl-piperazine- 1 -sulfonyl]- thiophen-2-yl ⁇ -N-dimethylaminomethylene-3,3,3-trifluoro-2-hydroxy-propionamide (373.1 mg, 0.61 mmol). The reaction mixture was stirred at room temperature for 10 min., after which time the reaction was determined to be complete by LC/MS.
- the reaction mixture was then diluted with 3 mL Of H 2 O and extracted with DCM. The organic layer was washed with sat.brine, dried over Na 2 SO 4 , and concentrated.
- the crude product (without any further purification) was treated with a mixture of acetic acid (3 mL) and 1,4-dioxane (3 mL). The reaction mixture was stirred at 9O 0 C for 2 hours, after which time the reaction was determined to be complete by LC/MS.
- the reaction mixture was cooled to room temperature, diluted with H 2 O, and extracted with DCM (3x). The organic layer was washed with sat. brine, dried over Na 2 SO 4 , and concentrated.
- Example 14.1 The title compound of Example 14.1 was prepared as shown in Scheme 14 below. Detailed synthesis procedures are provided below.
- Step 14A The starting material, (R)-l-(5-bromothiophen-2-ylsulfonyl)-4-(4- fluoro-2-(trifluoromethyl)phenyl)-2-methylpiperazine, was prepared according to procedure similar to that described in Example 1.1, steps IA- IB.
- Step 14B To a solution of l,l,l-trifluoro-2-[5-( ⁇ (2i?)-4-[4-fluoro-2- (trifluoromethyl) phenyl] -2-methylpiperazin- 1 -yl ⁇ sulfonyl)-2-thienyl]propan-2-ol (100 mg, 0.192mmol) in MeCN (6 mL) was added sulfuric acid (0.4 rnL, 7.5 mmol) dropwise. The reaction mixture was stirred at room temperature o/n and then diluted with EtOAc, washed with H 2 O, and sat. brine.
- Step 15 A The starting material, (3i?)-l-[4-fluoro-2-(trifluoromethyl)phenyl]-3- methylpiperazine, was prepared according to a procedure similar to that described in Example 1.1, step IA.
- Step 15B To a solution of (2i?)-l-[(3-bromophenyl)sulfonyl]-4-[4-fluoro-2- (trifluoromethyl)phenyl]-2-methylpiperazine (1.0 g, 2.1 mmol) in anhydrous toluene (5 ml) was added Butyllithium (0.84 ml, 2.1 mmol) at -78 0 C. Stirring was continued under N 2 for 15 min. and then methyl 3,3,3-trifluoropyruvate (0.4 ml, 2.5 mmol) was added. The reaction mixture was stirred at -78 0 C for 2 hours and then was quenched with a few mL of 10% aq.
- Step 15C To a solution of 3,3,3-trifiuoro-2-[3-( ⁇ (2i?)-4-[4-fiuoro-2- (trifluoromethyl) phenyl]-2-methylpiperazin- 1 -yl ⁇ sulfonyl)phenyl]-2-hydroxypropanoate (250 mg, 0.45 mmol) in 10 mL of MeOH was bubbled NH3 (g) for 30 seconds at -78 0 C. The reaction mixture was warmed up room temperature and stirred under empty balloon for o/n., after which time the reaction was determined to be complete by LC/MS. The solvent was evaporated, and the resultant residue washed with aq.
- Example 15.1 steps 15A-15C (200 mg, 82%yield), but using using 4-bromobenzene sulfonylchloride instead of 3-bromobenzene sulfonylchloride.
- Step 17A To a solution of (2i?)-2-[2-bromo-5-( ⁇ (2i?)-4-[4-fluoro-2- (trifluoromethyl)phenyl]-2-methylpiperazin-l-yl ⁇ sulfonyl)-3-thienyl]-3,3,3-trifluoro-2- hydroxypropanamide (300 mg, 0.477 mmol) in MeOH (10 mL) was added palladium 10% dry on carbon powder (30 mg, 0.282 mmol) under N 2 . Then the reaction mixture was stirred under 1 atm H 2 at room temperature for over night., after which time the reaction was determined to be complete by LC/MS.
- Example 20.1 The title compound of Example 20.1 was prepared using a procedure analogous to that described in Example 1.1, steps IA - ID, but using l-bromo-2-chloro-4- fluorobenzene instead of 2-bromo-5-fluorobenzotrifluoride in step IA.
- Example 20.2 was prepared using a procedure analogous to that described in Example 1.1, steps IA - ID, but using l-bromo-4-fluoro-2- methylbenzene instead of 2-bromo-5-fluorobenzotrifluoride in step IA.
- the title compound of Example 20.3 was prepared using a procedure analogous to that described in Example 1.1, steps IA - ID, but using l-bromo-4-fluoro-2- methoxybenzene instead of 2-bromo-5-fluorobenzotrifluoride in step IA.
- Example 20.4 The title compound of Example 20.4 was prepared using a procedure analogous to that described in Example 1.1, steps IA - ID, but using l-bromo-2,4-difluorobenzene instead of 2-bromo-5-fluorobenzotrifluoride in step IA.
- Example 21.1 The title compound of Example 21.1 was prepared using a procedure analogous to that described in Example 1.1, steps IA - ID, but using methylamine instead OfNH 3 in step ID.
- Example 21.2 The title compound of Example 21.2 was prepared using a procedure analogous to that described in Example 1.1, steps IA - ID, but using dimethylamine instead of NH 3 in step ID.
- Chinese Hamster Ovary (CHO) cell line expressing human 1 Ib-HSDl An exemplary procedure is described below.
- Cells are plated at 20,000 cells/well in 384 well plates and incubated overnight (12-16 hrs) at 37 °C/5% CO 2 . Cells are treated with different concentration of compound in 90 microliter serum-free media and incubated for 30 minutes at 37 °C/5%CO2. 10 microliter of 5 uM cortisone (final concentration 500 nM) is then added to the cells and the plate is incubated at 37 °C/5%CO2 for 120 minutes. 15 microliter of media is withdrawn and amount of Cortisol in the media is measured using the DiscoverX HitHunter Cortisol Assay (DiscoverX corp, CA), following manufacturer's instructions. Briefly, 15 microliter media is transferred to white 384 well assay plate.
- DiscoverX HitHunter Cortisol Assay DiscoverX corp, CA
Abstract
This invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof). Formula I is provided below: formula (I) R1, R2, R3, R4, R5, R6, R7, R8, R9, and A can be as defined anywhere herein.
Description
11 -beta HSDl Inhibitors
TECHNICAL FIELD
This invention features compounds that inhibit 11 -beta HSDl.
BACKGROUND
Diabetes is generally characterized by relatively high levels of plasma glucose (hyperglycemia) in the fasting state. Patients having type 2 diabetes (non-insulin dependent diabetes mellitus (NIDDM)) produce insulin (and even exhibit hyperinsulinemia), whilst demonstrating hyperglycemia.
Type 2 diabetics can often develop insulin resistance, in which the effect of insulin in stimulating glucose and lipid metabolism is diminished. Further, patients having insulin resistance, but have not developed type 2 diabetes, are also at risk of developing Syndrome X (metabolic syndrome). Syndrome X is characterized by insulin resistance, along with obesity (e.g., abdominal obesity), hyperinsulinemia, high blood pressure, relatively low HDL and relatively high VLDL.
Glucocorticoids (e.g., Cortisol in humans, corticosterone in rodents) are counter regulatory hormones that oppose the action of insulin. It is established that glucocorticoid activity is controlled at the tissue level by intracellular interconversion of active Cortisol and inactive cortisone by the 11 -beta hydroxysteroid dehydrogenases, llβHSDl, which activates cortisone and llβHSD2, which inactivates Cortisol. Excess levels of glucocorticoids (e.g., Cortisol) can cause metabolic complications. For example, excess Cortisol is associated with disorders including NIDDM, obesity, dyslipidemia, insulin resistance, and hypertension.
It is believed that inhibition of 1 lβHSDl can reduce the effects of excessive amounts of 1 lβ-hydroxysteroids, e.g., Cortisol, and therefore can be useful for the treatment and control of diseases mediated by abnormally high levels of Cortisol and other 1 lβ-hydroxysteroids, e.g., NIDDM, obesity, dyslipidemia, and hypertension.
SUMMARY
In one aspect, this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof). Formula I is provided below:

R1 is halo, -CH3, -CH2X, -CHX2, -CF3, -OCH3 or -OCF3;
R2 is halo, -CN, -C(O)-NH2, -C(O)-NH-(CL3 alkyl), -C(0)-N(d_3 alkyl)2, or 5- to 6-membered heteroaryl containing 1-4 ring heteroatoms independently selected from O, N, N(R2sl) and S, wherein R2sl is -H, -CH3 or ethyl, and wherein said heteroaryl is unsubstituted or substituted with one or two substituents R2s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3;
R3 is -H, halo, -CH3, ethyl, -OH or -OCH3; one of R4 and R5 is H, and the other is -CH3, -CH2X, -CHX2, -CF3 or ethyl;
A is phenyl, or 5- to 6-membered heteroaryl containing 1-3 ring heteroatoms independently selected from O, N, N(Ral) and S, and in which the atom bonded to the sulfonyl sulfur is carbon, wherein Ral is -H, -CH3 or ethyl; R9 is -H, halo, -CH3, -CH2X, -CHX2, -CF3, ethyl, -OCH3 or -OCF3; provided that when A is 5- membered heteroaryl containing 3 ring heteroatoms, then R9 is absent;
R6 is -CH3, ethyl, -OH or -OCH3;
R7 is -CH3, -CH2X, -CHX2, -CF3 or phenyl, wherein said phenyl is unsubstituted or substituted with one or two substituents R7sl independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3;
R8 is -C(O)-NH2, -C(O)-NH-(CL3 alkyl), -C(O)-N(CL3 alkyl)2, -NH-C(O)-(CL3 alkyl), -N(CL3 alkyl)-C(O)-(C1-3 alkyl), or 5- to 6-membered heteroaryl containing 1-4 ring heteroatoms independently selected from O, N, N(R8sl) and S, wherein R8sl is -H, -CH3 or ethyl, and wherein said heteroaryl is unsubstituted or substituted with one or two substituents R8s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3; and each instance of X is independently -F, -Cl or -Br; provided that when A is phenyl, then R6 and R7 cannot both be -CH3.
In another aspect, this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof), in which: R6 is -OH, -CH3, or ethyl;
R7 is -CF3, -CH3, or phenyl, wherein said phenyl is unsubstituted or substituted with one or two substituents R7sl independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3; provided that when A is phenyl, then R6 and R7 cannot both be -CH3; and R1, R2, R3, R4, R5, R8, and R9 can be as defined anywhere herein.
In another aspect, this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof), in which:
R6 is -OH;
R7 is -CF3; and
R1, R2, R3, R4, R5, R8, and R9 can be as defined anywhere herein.
In a further aspect, this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof), in which: R6 is -OH;
R7 is -CF3;
R8 is -C(O)-NH2, -C(O)-NH-(CL3 alkyl), or -C(O)-N(CL3 alkyl)2 (e.g., -C(O)-NH2); and
R1, R2, R3, R4, R5, and R9 can be as defined anywhere herein.
In another aspect, this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof), in which: R8 is -C(O)-NH2, -C(O)-NH-(CL3 alkyl), or -C(O)-N(CL3 alkyl)2 (e.g.,
-C(O)-NH2); provided that when A is phenyl, then R6 and R7 cannot both be -CH3; and
R1, R2, R3, R4, R5, R6, R7, and R9 can be as defined anywhere herein.
In a further aspect, this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof), in which:
A is 5- to 6-membered heteroaryl containing 1-3 ring heteroatoms independently selected from O, N, N(Ral) and S, and in which the atom bonded to the sulfonyl sulfur is carbon, wherein Ral is -H, -CH3 or ethyl; and
R1, R2, R3, R4, R5, R6, R7, R8, and R9 can be as defined anywhere herein.
In another aspect, this invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof), in which:
A is a 5-membered heteroaryl, which has formula (A-I):

(i): A1 is singly bonded to A4, and A3 is doubly bonded to A4;
AMs O5 S5 Or NR"1; and one of A2, A3,and A4 is C-C(R6)(R7)(R8), one of A2, A3 and A4 is C-R9, and one of A2, A3, and A4 is C-H or N; or (ii):
A1 is doubly bonded to A4, and A3 is singly bonded to A4;
A3 is O, S, or NRal; and one of A1, A2, and A4 is C-C(R6)(R7)(R8), one of A1, A2, and A4 is C-R9, and one of A1, A2, and A4 is N or C-H; and R1, R2, R3, R4, R5, R6, R7, R8, and R9 can be as defined anywhere herein.
In some embodiments, the chemical entity E can be a compound of formula (I) (including any subgenus of formula (I) or specific compound of formula (I)).
In some embodiments, the chemical entity E can be a salt (e.g., a pharmaceutically acceptable salt) of a compound of formula (I) (including any subgenus of formula (I) or specific compound of formula (I)).
In some embodiments, the chemical entity E can be an N-oxide of a compound of formula (I) (including any subgenus of formula (I) or specific compound of formula (I)).
In some embodiments, the chemical entity E can be a salt (e.g., a pharmaceutically acceptable salt) of an N-oxide of a compound of formula (I) (including any subgenus of formula (I) or specific compound of formula (I)).
In one aspect, this invention features a composition (e.g., a pharmaceutical composition), which includes a chemical entity E as defined anywhere herein and a pharmaceutically acceptable carrier. In some embodiments, the composition can include an effective amount of a chemical entity E as defined anywhere herein. In some embodiments, the composition can further include an additional therapeutic agent. In one aspect, this invention features a dosage form, which includes from about
0.05 milligrams to about 2,000 milligrams (e.g., from about 0.1 milligrams to about 1,000 milligrams, from about 0.1 milligrams to about 500 milligrams, from about 0.1 milligrams to about 250 milligrams, from about 0.1 milligrams to about 100 milligrams, from about 0.1 milligrams to about 50 milligrams, or from about 0.1 milligrams to about 25 milligrams) of a chemical entity E as defined anywhere herein. The dosage form can further include a pharmaceutically acceptable carrier and/or an additional therapeutic agent.
The invention also relates generally to inhibiting 11 -beta HSDl with a chemical entity E as defined anywhere herein. In some embodiments, the methods can include, e.g., contacting an 11-βHSDl in a sample (e.g., a tissue) with a chemical entity E as defined anywhere herein. In other embodiments, the methods can include administering a chemical entity E as defined anywhere herein to a subject (e.g., a mammal, such as a human). Accordingly, in yet another aspect, this invention includes methods of screening for compounds that inhibit 11 -βHSD 1.
In one aspect, this invention also features methods for treating (e.g., controlling, relieving, ameliorating, alleviating, or slowing the progression of) or methods for preventing (e.g., delaying the onset of or reducing the risk of developing) a disease or condition that is mediated by excess (e.g., abnormally high) levels or uncontrolled (e.g., resulting in abnormally high) levels of Cortisol and/or other corticosteroids (e.g., 1 lβ- hydroxysteroids), which include administering to a subject in need thereof an effective amount of a chemical entity E as defined anywhere herein.
Examples of such diseases or conditions include, but are not limited to diabetes (e.g., type 1 or type 2 diabetes), Syndrome X, hyperglycemia, low glucose tolerance, insulin resistance, obesity, lipid disorders, dyslipidemia, hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels, high LDL levels, atherosclerosis and its sequelae, vascular restenosis, pancreatitis, abdominal obesity, neurodegenerative disease, retinopathy, nephropathy, neuropathy, hypertension, coronary heart disease, stroke, peripheral vascular disease, Cushing's syndrome, glaucoma, osteoperosis, hyperinsulinemia, tuberculosis, psoriasis, cognitive disorders and dementia (e.g., impairment associated with aging and of neuronal dysfunction, e.g., Alzheimer's disease), depression, viral diseases, inflammatory disorders, immune disorders); or promoting wound healing.
In another aspect, this invention features methods for treating or preventing diabetes (e.g., type I diabetes, type 2 diabetes), which includes administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
In a further aspect, this invention features methods for treating or preventing Syndrome X, which includes administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
In another aspect, this invention features methods for treating or preventing hyperglycemia, which includes administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
In a further aspect, this invention features methods for treating or preventing hyperglycemia, which includes administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
In another aspect, this invention features methods for treating or preventing obesity, which include administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
In a further aspect, this invention features methods for treating or preventing a lipid disorder selected from the group consisting of dyslipidemia, hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL and high LDL, which include administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
In another aspect, this invention features methods for treating or preventing atherosclerosis, which include administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
In a further aspect, this invention features methods for treating or preventing a cognitive disorder (e.g., Alzheimer's disease), which include administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
In another aspect, this invention features methods for promoting wound healing, which include administering to a subject (e.g., a subject in need thereof) an effective amount of a chemical entity E as defined anywhere herein.
In some embodiments, the subject can be a subject in need thereof (e.g., a subject identified as being in need of such treatment, such as a subject having, or at risk of having, one or more of the diseases or conditions described herein). Identifying a subject in need of such treatment can be in the judgment of a subject or a health care professional and can be subjective (e.g. opinion) or objective (e.g. measurable by a test or diagnostic method). In some embodiments, the subject can be a mammal. In certain embodiments, the subject can be a human.
In a further aspect, this invention also relates to methods of making compounds described herein. In embodiments, the methods include taking any one of the intermediate compounds described herein and reacting it with one or more chemical reagents in one or more steps to produce a chemical entity E as defined anywhere herein.
In one aspect, this invention relates to a packaged product. The packaged product includes a container, one of the aforementioned compounds in the container, and a legend (e.g., a label or an insert) associated with the container and indicating administration of the compound for treatment and control of the diseases or disorders described herein.
In embodiments, any chemical entity, composition, or method described herein can also include any one or more of the other features delineated in the detailed description and/or in the claims.
The term "mammal" includes organisms, which include mice, rats, cows, sheep, pigs, rabbits, goats, horses, monkeys, dogs, cats, and humans.
"An effective amount" refers to an amount of a compound that confers a therapeutic effect (e.g., treats, e.g., controls, relieves, ameliorates, alleviates, or slows the progression of; or prevents, e.g., delays the onset of or reduces the risk of developing, a disease, disorder, or condition or symptoms thereof) on the treated subject. The therapeutic effect may be objective (i.e., measurable by some test or marker) or subjective (i.e., subject gives an indication of or feels an effect). An effective amount of the compound described above may range from about 0.01 mg/kg to about 1000 mg/kg, (e.g., from about 0.1 mg/kg to about 100 mg/kg, from about 1 mg/kg to about 100 mg/kg). Effective doses will also vary depending on route of administration, as well as the possibility of co-usage with other agents.
The term "halo" or "halogen" refers to any radical of fluorine, chlorine, bromine or iodine.
In general, and unless otherwise indicated, substituent (radical) prefix names are derived from the parent hydride by either (i) replacing the "ane" in the parent hydride with the suffix "yl;" or (ii) replacing the "e" in the parent hydride with the suffix "yl;" (here the atom(s) with the free valence, when specified, is (are) given numbers as low as is consistent with any established numbering of the parent hydride). Accepted contracted names, e.g., furyl, pyridyl, and piperidyl, and trivial names, e.g., phenyl and thienyl are also used herein throughout. Conventional numbering/lettering systems are also adhered to for substituent numbering.
The term "Ci_3 alkyl" refers to methyl, ethyl, n-propyl, and ώopropyl.
The term "heteroaryl" refers to an aromatic monocyclic or bicyclic hydrocarbon groups having one or more (e.g., 1-3 or 1-4) heteroatom ring atoms independently selected from O, N, or S (and mono and dioxides thereof, e.g., N→O~, S(O), SO2). Any atom can be optionally substituted by one or more substituents. Heteroaryl groups include pyridyl, thienyl, furyl (furanyl), imidazolyl, and pyrrolyl.
The details of one or more embodiments of the invention are set forth in the description below. Other features and advantages of the invention will be apparent from the description and from the claims.
DETAILED DESCRIPTION
This invention features a chemical entity E, which is a compound of Formula I, or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof (e.g., a compound of Formula I, or a pharmaceutically acceptable salt thereof). Formula I is provided below:

Here and throughout this specification, R1, R2, R3, R4, R5, R6, R7, R8, R9, and A can be as defined anywhere herein.
For ease of exposition, it is also understood that where in this specification (including the claims), a group is defined by "as defined anywhere herein" (or the like), the definitions for that particular group include the first occurring and broadest generic definition as well as any sub-generic and specific definitions delineated anywhere in this specification.
Variables R1, R2, and R3
In some embodiments, R1 can be -CH3, -CH2X, -CHX2, or -CF3. In some embodiments, R1 can be -CH2X, -CHX2, or -CF3. In some embodiments, R1 can be -CF3.
In some embodiments, R1 can be -CH2X, Or-CHX2. In some embodiments, R1 can be -CH3.
In embodiments, each instance of X can be, independently, -F or -Cl, e.g., -F. In some embodiments, R1 can be halo (e.g., -F or -Cl). In some embodiments, R1 can be -OCH3 or -OCF3.
In certain embodiments, R1 can be -CF3, -F, -Cl, -CH3, or -OCH3.
In some embodiments, R2 can be: (i) halo;
(ϋ)-CN;
(iii) -C(O)-NH2; or
(iv) 5- to 6-membered heteroaryl containing 1-4 ring heteroatoms independently selected from O, N, N(R2sl) and S, in which R2sl is -H, -CH3 or ethyl, and in which said heteroaryl is unsubstituted or substituted with one or two substituents R2s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3.
In some embodiments, R2 can be:
(i) halo; or (iv) 5- to 6-membered heteroaryl containing 1-4 ring heteroatoms independently selected from O, N, N(R2sl) and S, in which R2sl is -H, -CH3 or ethyl, and in which said heteroaryl is unsubstituted or substituted with one or two substituents R2s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3.
In some embodiments, R2 can be halo (e.g., -F).
In some embodiments, R2 can be 5- to 6-membered heteroaryl containing 1-4 (e.g., 2-3, 2-4, 3-4, 1-3, 1-2, 1, 2, 3, or 4) ring heteroatoms independently selected from O, N, N(R2sl) and S, in which R2sl is -H, -CH3 or ethyl, and in which said heteroaryl is unsubstituted or substituted with one or two substituents R2s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3.
In certain embodiments, each instance of R2sl can be, independently, -H or -CH3 (e.g., -H).
In certain embodiments, each instance of R2s2 can be, independently, -CH3, ethyl, or -CF3 (e.g., -CH3 or ethyl; e.g., -CH3). In certain embodiments, the 5- to 6-membered heteroaryl can contain 1-3 (e.g., 2-
3, 1, 2, or 3) ring heteroatoms.
In certain embodiments, the 5- to 6-membered heteroaryl can contain 2-4 (e.g., 2- 3, 3-4, 2, 3, or 4) ring heteroatoms.
In certain embodiments, the 5- to 6-membered heteroaryl can contain 3 or 4 ring heteroatoms. In embodiments, the 3 or 4 ring heteroatoms can be independently selected from O, N, and N(R2sl). In other embodiments, each of the 3 or 4 ring heteroatoms can be independently selected from N and N(R2sl). For example, one of the 3 or 4 ring heteroatoms can be O or N(R2sl), and the others can each be N.
In embodiments, the 5- to 6-membered heteroaryl is bonded to the phenyl ring via a ring carbon atom and not via one of the aforementioned ring heteroatoms.
In certain embodiments, R2 can be 5- membered heteroaryl containing 1-4 (e.g., 2-3, 2-4, 3-4, 1-3, 1-2, 1, 2, 3, or 4) ring heteroatoms independently selected from O, N, N(R2sl) and S, in which R2sl can be as defined anywhere herein; and in which said heteroaryl can be unsubstituted or substituted with one or two substituents R2s2, in which each instance of R2s2 can be as defined anywhere herein.
Embodiments can include one or more of the following features. R2sl can be -H.
R2s2 can be -CH3 or ethyl; e.g., -CH3. The 5- membered heteroaryl can contain 3 ring heteroatoms. In certain embodiments, each of the 3 ring heteroatoms can be independently selected from O, N,
and N(R2sl). In certain embodiments, each of the 3 ring heteroatoms can be independently selected from N and N(R2sl).
In embodiments, one of the 3 ring heteroatoms can be O, and the others can be N. For example, the 5- membered heteroaryl can be l,2,4-oxadiazol-5-yl (unsubstituted or substituted with, e.g., -CH3) or l,3,4-oxadiazol-2-yl (unsubstituted or substituted with, e.g., -CH3).
In embodiments, one of the 3 ring heteroatoms can be N(R2sl), and the others can be N. For example, the 5- membered heteroaryl can be l,2,4-triazol-3-yl (unsubstituted or substituted with, e.g., -CH3). The 5- membered heteroaryl can contain 4 ring heteroatoms. For example, the 5- membered heteroaryl can be tetrazol-5-yl.
In some embodiments, R2 can be -C(O)-NH2. In some embodiments, R2 can be -CN.
In certain embodiments, R2 can be -F; -CN; -C(O)-NH2; l,2,4-oxadiazol-5-yl (unsubstituted or substituted with, e.g., -CH3); l,3,4-oxadiazol-2-yl (unsubstituted or substituted with, e.g., -CH3); l,2,4-triazol-3-yl (unsubstituted or substituted with, e.g., - CH3); or tetrazol-5-yl.
In some embodiments:
R1 is -CH2X, -CHX2, or -CF3; and
R2 is:
(i) halo (e.g., -F); (ϋ)-CN;
(iii) -C(O)-NH2; or
(iv) 5- to 6-membered heteroaryl containing 1-4 ring heteroatoms independently selected from O, N, N(R2sl) and S, in which R2sl is -H, -CH3 or ethyl, and in which said heteroaryl is unsubstituted or substituted with one or two substituents R2s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3.
Embodiments can include one or more of the following features.
R1 can be -CF3.
X; the ring heteroatom content of the 5- to 6-membered heteroaryl; R2sl; and R2s2 can each be, independently, as defined anywhere herein. R2 can be -F.
In some embodiments:
R1 is -CH2X, -CHX2, or -CF3; and
R2 is:
(i) halo (e.g., -F); or (iv) 5- to 6-membered heteroaryl containing 1-4 ring heteroatoms independently selected from O, N, N(R2sl) and S, in which R2sl is -H, -CH3 or ethyl, and in which said heteroaryl is unsubstituted or substituted with one or two substituents R2s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3.
Embodiments can include one or more of the following features. R1 can be -CF3.
X; the ring heteroatom content of the 5- to 6-membered heteroaryl; R2sl; and R2s2 can each be, independently, as defined anywhere herein.
R2 can -F.
In some embodiments:
R1 is -CH2X, -CHX2, or -CF3; and
R2 is halo (e.g., -F).
Embodiments can include one or more of the following features.
R1 can be -CF3. R2 can -F.
In some embodiments, R1 is -CF3, and R2 is -F.
In some embodiments, R3 can be -H.
Variables R4 and R5
In some embodiments, the carbon atom attached to R4 and R5 can have the R- configuration (such compounds are referred to herein as having "the i?-CR4R5 configuration"). In some embodiments, the carbon atom attached to R4 and R5 can have the ^-configuration (such compounds are referred to herein as compounds having "the S- CR4R5 configuration").
In some embodiments, one of R4 and R5 can be H, and the other can be -CH3. In certain embodiments, R4 can be H, and R5 can be -CH3. In other embodiments, R4 can be H, and R5 can be -CH3.
Variables A, R6, R7, R8, and R9
Variable A
In some embodiments, A can be 5- to 6-membered heteroaryl containing 1-3 (e.g., 1-2, 2-3, 1, 2, or 3) ring heteroatoms independently selected from O, N, N(Ral) and S, and in which the atom bonded to the sulfonyl sulfur is carbon. In certain embodiments, Ral can be -H or -CH3.
In certain embodiments, the 5- to 6-membered heteroaryl can contain 1 or 2 ring heteroatoms. In certain embodiments, the ring heteroatoms can be independently selected from
N, N(Ral) and S.
In some embodiments, A can be 5-membered heteroaryl containing 1-3 (e.g., 1-2, 2-3, 1, 2, or 3) ring heteroatoms independently selected from O, N, N(Ral) and S, and in which the atom bonded to the sulfonyl sulfur is carbon, and in which Ral can be as defined anywhere herein.
In certain embodiments, the 5-membered heteroaryl can contain 1 ring heteroatom (e.g., S).
In certain embodiments, the 5-membered heteroaryl can contain 2 ring heteroatoms. In embodiments, the 2 ring heteroatoms can be independently selected from N, N(Ral) and S. For example, one of the 2 ring heteroatoms can be N, and the other can
be S. As another example, one of the 2 ring heteroatoms can be N, and the other can be
N(Ral). In certain embodiments, the 5- membered heteroaryl can contain 3 ring heteroatoms. For example, one of the 3 ring heteroatoms can be N(Ral) or S, (e.g., N(Ra1)), and the others can each be N.
In certain embodiments, A can be a 5-membered heteroaryl, which has formula
(A-I):

(i):
A1 is singly bonded to A4, and A3 is doubly bonded to A4;
AMs O5 S5 Or NR"1; and one of A2, A3,and A4 is C-C(R6)(R7)(R8), one of A2, A3 and A4 is C-R9, and one of A2, A3, and A4 is C-H or N; or
(ii):
A1 is doubly bonded to A4, and A3 is singly bonded to A4;
A3 is O, S, or NRal; and one of A1, A2, and A4 is C-C(R6)(R7)(R8), one of A1, A2, and A4 is C-R9, and one of A1, A2, and A4 is N or C-H.
In certain embodiments, the definitions set forth under (i) {supra) apply, i.e.: A1 is singly bonded to A4, and A3 is doubly bonded to A4; A1 Is O, S, or NRal; and one of A2, A3,and A4 is C-C(R6)(R7)(R8), one of A2, A3 and A4 is C- R9, and one of A2, A3, and A4 is C-H or N.
When the definitions set forth under (i) {supra) apply, it is understood that A is 5- membered heteroaryl, which has formula (A-2):

A1 is O, S, or NRal; and one of A2, A3,and A4 is C-C(R6)(R7)(R8), one of A2, A3 and A4 is C-R9, and one of A2, A3, and A4 is C-H or N.
Embodiments of formula (A-2) can include one or more of the following features.
A1 can be S.
A4 can be C-C(R6)(R7)(R8).
A2 canbe C-H. A2 can be N. A3 canbe C-R9. R9 can be -H.
A1 canbe S,A4 canbe C-C(R6)(R7)(R8), andA2 canbe C-H orN.
A1 canbe S,A3 canbe C-C(R6)(R7)(R8), andA2 canbe C-H orN.
A1 canbe S,A4 canbe C-C(R6)(R7)(R8),A2 can be C-H, andA3 canbe C-R9. R9 can be -H. A1 canbe S,A4 canbe C-C(R6)(R7)(R8),A2 can beN, andA3 canbe C-R9. R9 can be -H.
In certain embodiments, the definitions set forth under (ii) {supra) apply, i.e.: A1 is doubly bonded to A4, and A3 is singly bonded to A4; A3 is O, S, or NRal; and one of A1, A2, and A4 is C-C(R6)(R7)(R8), one of A1, A2, and A4 is C-R9, and one of A1, A2, and A4 is N or C-H.
When the definitions set forth under (ii) {supra) apply, it is understood that A is 5- membered heteroaryl, which has formula (A-3):

A3 is O, S, or NRal; and one of A1, A2, and A4 is C-C(R6)(R7)(R8), one of A1, A2, and A4 is C-R9, and one of A1, A2, and A4 is N or C-H.
Embodiments of formula (A-3) can include one or more of the following features.
A3 can be NRal. Ral can be -CH3.
One of A2 and A4 can be C-C(R6)(R7)(R8), and one of A2 and A4 can be C-R9. A1 can be N or C-H (e.g., N).
A3 can be NRal; one of A2 and A4 can be C-C(R6)(R7)(R8), and one of A2 and A4 can be C-R9; and A1 can be N.
In some embodiments, A can be phenyl. In certain embodiments, -C(R6)(R7)(R8) can be attached to the phenyl ring carbon that is meta or para with respect to the phenyl carbon that is attached to the sulfonyl sulfur in Formula I.
Variables R6. R7. and R8
In some embodiments, R6 can be -OH or -OCH3. In certain embodiments, R6 can be -OH.
In some embodiments, R6 can be CH3 or ethyl. In certain embodiments, R6 can be -CH3.
In certain embodiments, R6 can be -OH or -CH3.
In some embodiments, R7 can be -CH3, -CH2X, -CHX2, or -CF3.
In some embodiments, R7 can be -CH2X, -CHX2, or -CF3. In some embodiments, R7 can be -CF3.
In some embodiments, R7 can be -CH2X, Or-CHX2.
In some embodiments, R7 can be -CH3.
In certain embodiments, each instance of X can be, independently, -F or -Cl, e.g., -F. In some embodiments, R7 can be phenyl, wherein said phenyl is unsubstituted or substituted with one or two substituents R7sl independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3. In certain embodiments, R7 can be unsubstituted phenyl.
In some embodiments, R8 can be -C(O)-NH2, -C(O)-NH-(d_3 alkyl), or -C(O)-N(CL3 alkyl)2.
In some embodiments, R8 can be -C(O)-NH2.
In some embodiments, R8 can be -C(O)-NH-(Ci_3 alkyl), or -C(O)-N(Ci_3 alkyl)2, e.g., -C(O)-NH-(CH3), or -C(O)-N(CH3)2.
In some embodiments, R8 can be -NH-C(O)-(Ci_3 alkyl) or
-N(CL3 alkyl)-C(O)-(Ci_3 alkyl). In certain embodiments, R8 can be -NH-C(O)-(CL3 alkyl), e.g., -NH-C(O)-(CH3).
In some embodiments, R8 can be 5- to 6-membered heteroaryl containing 1-4 (e.g., 2-3, 2-4, 3-4, 1-3, 1-2, 1, 2, 3, or 4) ring heteroatoms independently selected from O, N, N(R2sl) and S, in which R8sl is -H, -CH3 or ethyl, and in which said heteroaryl is
unsubstituted or substituted with one or two substituents R8s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3.
In certain embodiments, each instance of R8sl can be, independently, -H or -CH3 (e.g., -H). In certain embodiments, each instance of R8s2 can be, independently, -CH3, ethyl, or -CF3 (e.g., -CH3 or ethyl; e.g., -CH3).
In certain embodiments, the 5- to 6-membered heteroaryl can contain 1-3 (e.g., 2- 3, 1, 2, or 3) ring heteroatoms.
In certain embodiments, the 5- to 6-membered heteroaryl can contain 2-4 (e.g., 2- 3, 3-4, 2, 3, or 4) ring heteroatoms.
In certain embodiments, the 5- to 6-membered heteroaryl can contain 3 or 4 ring heteroatoms. In embodiments, the 3 or 4 ring heteroatoms can be independently selected from O, N, and N(R8sl). In other embodiments, each of the 3 or 4 ring heteroatoms can be independently selected from N and N(R8sl). For example, one of the 3 or 4 ring heteroatoms can be O or N(R8sl), and the others can each be N.
In embodiments, the 5- to 6-membered heteroaryl is bonded to the quaternary carbon via a ring carbon atom and not via one of the aforementioned ring heteroatoms.
In certain embodiments, R8 can be 5- membered heteroaryl containing 1-4 (e.g., 2-3, 2-4, 3-4, 1-3, 1-2, 1, 2, 3, or 4) ring heteroatoms independently selected from O, N, N(R8sl) and S, in which R8sl can be as defined anywhere herein; and in which said heteroaryl can be unsubstituted or substituted with one or two substituents R8s2, in which each instance of R8s2 can be as defined anywhere herein.
Embodiments can include one or more of the following features. R8sl can be -H.
R8s2 can be -CH3 or ethyl; e.g., -CH3.
In certain embodiments, the 5- membered heteroaryl can contain 1-3 (e.g., 2-3, 1, 2, or 3) ring heteroatoms.
The 5- membered heteroaryl can contain 3 ring heteroatoms. In certain embodiments, each of the 3 ring heteroatoms can be independently selected from O, N,
and N(R8sl). In certain embodiments, each of the 3 ring heteroatoms can be independently selected from N and N(R8sl).
In embodiments, one of the 3 ring heteroatoms can be O, and the others can be N. For example, the 5- membered heteroaryl can be l,2,4-oxadiazol-5-yl (unsubstituted or substituted with, e.g., -CH3) or l,3,4-oxadiazol-2-yl (unsubstituted or substituted with, e.g., -CH3).
In embodiments, one of the 3 ring heteroatoms can be N(R8sl), and the others can be N. For example, the 5- membered heteroaryl can be l,2,4-triazol-3-yl (unsubstituted or substituted with, e.g., -CH3). The 5- membered heteroaryl can contain 4 ring heteroatoms. For example, the 5- membered heteroaryl can be tetrazol-5-yl.
In certain embodiments, R8 can be -C(O)-NH2; -C(O)-NH-(CH3); -C(O)-N(CH3)2; -NH-C(O)-(CH3); or 5-membered heteroaryl containing 1-4 (e.g., 1-3) ring heteroatoms independently selected from O, N, N(R8sl) and S, wherein said heteroaryl is unsubstituted or substituted with one or two substituents R8s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3 (e.g., l,2,4-triazol-3-yl or 1,2,4- oxadiazol-3-yl, each of which is unsubstituted or substituted with one or two substituents R8s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3).
In some embodiments, the carbon atom attached to R , R , and R can have the R- configuration (referred to herein as "the i?-CR6R7R8 configuration"). In some embodiments, the carbon atom attached to R , R , and R can have the ^-configuration (referred to herein as "the ,S-CR6R7R8 configuration").
Variable R9
In some embodiments, R9 can be -H, halo (e.g., chloro or bromo), or -CH3.
In some embodiments, R9 can be -H.
In some embodiments, R9 can be absent.
[1] In some embodiments:
R6 is -OH, -CH3, or ethyl; and
R7 is -CF3, -CH3, or phenyl, wherein said phenyl is unsubstituted or substituted with one or two substituents R7sl independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3.
Embodiments in which R6 is -OH, -CH3, or ethyl; and R7 is -CF3, -CH3, or phenyl that is unsubstituted or substituted with one or two substituents R7sl independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3 can also include any one or more of the features described herein, including (but not limited) to those delineated below.
R6 can be -OH.
R7 can be -CF3.
A can be 5- to 6-membered heteroaryl containing 1-3 (e.g., 1-2, 2-3, 1, 2, or 3) ring heteroatoms independently selected from O, N, N(Ral) and S, and in which the atom bonded to the sulfonyl sulfur is carbon. Ral can be -H or -CH3. The 5- to 6-membered heteroaryl can contain 1 or 2 ring heteroatoms. The ring heteroatoms can be independently selected from N, N(Ral) and S.
A can be 5-membered heteroaryl containing 1-3 (e.g., 1 or 2) ring heteroatoms independently selected from O, N, N(Ral) and S, and in which the atom bonded to the sulfonyl sulfur is carbon, and in which Ral can be as defined anywhere herein. E.g., A can have formula (A-I):

(A-I) in which the carbon atom shown as C in formula (A-I) represents the carbon that is bonded to the sulfonyl sulfur in Formula I; and in which A1, A2, A3, A4, and the dotted lines between A1 and A4 and A3 and A4 are all defined according to the definitions under (i) below or are all defined according to the definitions under (ii) below: (i):
A1 is singly bonded to A4, and A3 is doubly bonded to A4;
AMs O5 S5 Or NR"1; and one of A2, A3,and A4 is C-C(R6)(R7)(R8), one of A2, A3 and A4 is C-R9, and one of A2, A3, and A4 is C-H or N; or
(ii):
A1 is doubly bonded to A4, and A3 is singly bonded to A4;
A3 is O, S, or NRal; and one of A1, A2, and A4 is C-C(R6)(R7)(R8), one of A1, A2, and A4 is C-R9, and one of A1, A2, and A4 is N or C-H.
In certain embodiments, the definitions set forth under (i) {supra) apply, and A can have formula (A-2):

A can be 5-membered heteroaryl having formula (A-3) as described herein. A can be phenyl. In certain embodiments, -C(R6)(R7)(R8) can be attached to the phenyl ring carbon that is meta ox para with respect to the phenyl carbon that is attached to the sulfonyl sulfur in Formula I. R8 can be -C(O)-NH2, -C(O)-NH-(CH3), -C(O)-N(CH3)2, -NH-C(O)-(CH3), or
5-membered heteroaryl containing 1-3 ring heteroatoms independently selected from O, N, N(R8sl) and S, wherein said heteroaryl is unsubstituted or substituted with one or two substituents R8s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3. In certain embodiments, R8 can be -C(O)-NH2.
In other embodiments, R8 can be l,2,4-triazol-3-yl or l,2,4-oxadiazol-3-yl, each of which is unsubstituted or substituted with one or two substituents R8s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3 (e.g., -CH3).
The carbon atom attached to R4 and R5 can have the ^-configuration.
The carbon atom attached to R , R , and R can have the ^-configuration.
R9 can be 5 --HH oorr aabbsseenntt..
One of R and R is H, and the other is -CH3 (e.g., R can be H, and R can be -CH3).
R1 and R2 can be, independently, as defined as defined anywhere herein. R1 can be -CF3.
R2 can be -F.
R1 can be -CF3, and R2 can be -F. R3 can be -H. R1 can be -CF3, R2 can be -F, and R3 can be -H.
[2] In some embodiments: R6 is -OH; and R7 is -CF3.
Embodiments in which R6 is -OH, and R7 is -CF3 can also include any one or more of the features described herein, including (but not limited to) those delineated below.
A can be 5- to 6-membered heteroaryl containing 1-3 (e.g., 1-2, 2-3, 1, 2, or 3) ring heteroatoms independently selected from O, N, N(Ral) and S, and in which the atom bonded to the sulfonyl sulfur is carbon. Ral can be -H or -CH3. The 5- to 6-membered heteroaryl can contain 1 or 2 ring heteroatoms. The ring heteroatoms can be independently selected from N, N(Ral) and S.
A can be 5-membered heteroaryl containing 1-3 (e.g., 1 or 2) ring heteroatoms independently selected from O, N, N(Ral) and S, and in which the atom bonded to the sulfonyl sulfur is carbon, and in which Ral can be as defined anywhere herein. E.g., A can have formula (A-I):

(A-I) in which the carbon atom shown as C in formula (A-I) represents the carbon that is bonded to the sulfonyl sulfur in Formula I; and in which A1, A2, A3, A4, and the dotted lines between A1 and A4 and A3 and A4 are all defined according to the definitions under (i) below or are all defined according to the definitions under (ii) below:
(i):
A1 is singly bonded to A4, and A3 is doubly bonded to A4; AMs O5 S5 Or NR"1; and one of A2, A3,and A4 is C-C(R6)(R7)(R8), one of A2, A3 and A4 is C-R9, and one of A2, A3, and A4 is C-H or N; or
(ii): A1 is doubly bonded to A4, and A3 is singly bonded to A4;
A3 is O, S, or NRal; and one of A1, A2, and A4 is C-C(R6)(R7)(R8), one of A1, A2, and A4 is C-R9, and one of A1, A2, and A4 is N or C-H.
In certain embodiments, the definitions set forth under (i) {supra) apply, and A can have formula (A-2):

A can be 5-membered heteroaryl having formula (A-3) as described herein.
A can be phenyl. In certain embodiments, -C(R6)(R7)(R8) can be attached to the phenyl ring carbon that is meta ox para with respect to the phenyl carbon that is attached to the sulfonyl sulfur in Formula I. R8 can be -C(O)-NH2, -C(O)-NH-(CH3), -C(O)-N(CH3)2, -NH-C(O)-(CH3), or
5-membered heteroaryl containing 1-3 ring heteroatoms independently selected from O,
N, N(R , 8ssl ) and S, wherein said heteroaryl is unsubstituted or substituted with one or two substituents R ,8s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3.
In certain embodiments, R8 can be -C(O)-NH2. In other embodiments, R8 can be l,2,4-triazol-3-yl or l,2,4-oxadiazol-3-yl, each of which is unsubstituted or substituted with one or two substituents R8s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3 (e.g., -CH3).
The carbon atom attached to R4 and R5 can have the ^-configuration. The carbon atom attached to R6, R7, and R8 can have the ^-configuration. R9 can be -H or absent.
One of R4 and R5 is H, and the other is -CH3 (e.g., R4 can be H, and R5 can be -CH3).
R1 and R2 can be, independently, as defined as defined anywhere herein. R1 can be -CF3. R2 can be -F.
R1 can be -CF3, and R2 can be -F.
R3 can be -H.
R1 canbe -CF3, R2 canbe -F, and R3 can be -H.
[3] In some embodiments: R6 is -OH; R7 is -CF3; and
R8 is -C(O)-NH2, -C(O)-NH-(CL3 alkyl), or -C(O)-N(CL3 alkyl)2 (e.g., R8 is -C(O)-NH2).
Embodiments in which R6 is -OH, R7 is -CF3, and R8 is -C(O)-NH2, -C(O)-NH-(CL3 alkyl), or -C(O)-N(CL3 alkyl)2 (e.g., R8 is -C(O)-NH2) can also include any one or more of the features described herein, including (but not limited to) those delineated below.
A can be 5- to 6-membered heteroaryl containing 1-3 (e.g., 1-2, 2-3, 1, 2, or 3) ring heteroatoms independently selected from O, N, N(Ral) and S, and in which the atom bonded to the sulfonyl sulfur is carbon. Ral can be -H or -CH3. The 5- to 6-membered heteroaryl can contain 1 or 2 ring heteroatoms. The ring heteroatoms can be independently selected from N, N(Ral) and S.
A can be 5-membered heteroaryl containing 1-3 (e.g., 1 or 2) ring heteroatoms independently selected from O, N, N(Ral) and S, and in which the atom bonded to the sulfonyl sulfur is carbon, and in which Ral can be as defined anywhere herein. E.g., A can have formula (A-I):

(A-I) in which the carbon atom shown as C in formula (A-I) represents the carbon that is bonded to the sulfonyl sulfur in Formula I; and in which A1, A2, A3, A4, and the dotted lines between A1 and A4 and A3 and A4 are all defined according to the definitions under (i) below or are all defined according to the definitions under (ii) below:
(i):
A1 is singly bonded to A4, and A3 is doubly bonded to A4; AMs O5 S5 Or NR"1; and
one of A2, A3,and A4 is C-C(R6)(R7)(R8), one of A2, A3 and A4 is C-R9, and one of A2, A3, and A4 is C-H or N; or
(ii): A1 is doubly bonded to A4, and A3 is singly bonded to A4;
A3 is O, S, or NRal; and one of A1, A2, and A4 is C-C(R6)(R7)(R8), one of A1, A2, and A4 is C-R9, and one of A1, A2, and A4 is N or C-H.
In certain embodiments, the definitions set forth under (i) {supra) apply, and A can have formula (A-2):

A can be 5-membered heteroaryl having formula (A-3) as described herein. A can be phenyl. In certain embodiments, -C(R6)(R7)(R8) can be attached to the phenyl ring carbon that is meta ox para with respect to the phenyl carbon that is attached to the sulfonyl sulfur in Formula I.
The carbon atom attached to R4 and R5 can have the ^-configuration.
The carbon atom attached to R6, R7, and R8 can have the ^-configuration. R9 can be -H or absent.
One of R4 and R5 is H, and the other is -CH3 (e.g., R4 can be H, and R5 can be -CH3).
R1 and R2 can be, independently, as defined as defined anywhere herein.
R1 can be -CF3.
R can be -F.
R1 can be -CF3, and R2 can be -F.
R3 can be -H.
R1 can be -CF3, R2 can be -F, and R3 can be -H.
[4] In some embodiments:
R6 is -OH; R7 is -CF3; and one, more than one, or all of the following features is(are) present:
• R8 is -C(O)-NH2.
• A is a 5-membered heteroaryl, which has formula (A-2):

• in which the carbon atom shown as C in formula (A-2) represents the carbon that is bonded to the sulfonyl sulfur in Formula I; A1 is O, S, or NRal; and one of A2, A3,and A4 is C-C(R6)(R7)(R8), one of A2, A3 and A4 is C-R9, and one of A2, A3, and A4 is C-H or N. Each of A1, A2, A3, and A4 can be as defined anywhere herein in (e.g., A1 can be S, A4 can be C- C(R6)(R7)(R8), and A2 can be C-H or N; e.g., A1 can be S, A4 can be C- C(R6)(R7)(R8), and A2 can be C-H).
• The carbon atom attached to R4 and R5 has the ^-configuration. • The carbon atom attached to R , R , and R has the ^-configuration.
• R9 is -H or absent.
• R4 is H, and R5 is -CH3.
• R1 is -CF3.
• R2 is -F. • R3 is -H.
In some embodiments, the chemical entity E can have formula II (i.e., in which A in formula I is 2-thienyl):

Embodiments of formula II can include any one or more of the features described herein, including but not limited to those delineated below.
R6 can be -OH.
R7 can be -CF3.
R8 can be -C(O)-NH2.
R6 can be -OH, and R7 can be -CF3.
R6 can be -OH, R7 can be -CF3, and R8 can be -C(O)-NH2.
-CR6R7R8 can be attached to C5 of the thienyl ring.
The carbon atom attached to R4 and R5 can have the ^-configuration.
The carbon atom attached to R , R , and R can have the ^-configuration.
R9 can be -H.
R4 can be H, and R5 can be -CH3.
R1 and R2 can be, independently, as defined as defined anywhere herein.
R1 can be -CF3.
R2 can be -F.
R3 can be -H.
R1 can be -CF3, and R2 can be -F.
R1 can be -CF3, and R2 can be -F, and R3 can be -H.
The chemical entity E can be (2R)-3,3,3-trifluoro-2-[5-({(2R)-4-[4-fluoro-2- (trifluoromethyl)phenyl]-2-methylpiperazin- 1 -yl} sulfonyl)thiophen-2-yl]-2-
hydroxypropanamide or a salt (e.g., a pharmaceutically acceptable salt), or an N-oxide thereof; (e.g., or a pharmaceutically acceptable salt thereof).
It is understood that the actual electronic structure of some chemical entities cannot be adequately represented by only one canonical form (i.e. Lewis structure).
While not wishing to be bound by theory, the actual structure can instead be some hybrid or weighted average of two or more canonical forms, known collectively as resonance forms or structures. Resonance structures are not discrete chemical entities and exist only on paper. They differ from one another only in the placement or "localization" of the bonding and nonbonding electrons for a particular chemical entity. It can be possible for one resonance structure to contribute to a greater extent to the hybrid than the others. Thus, the written and graphical descriptions of the embodiments of the present invention are made in terms of what the art recognizes as the predominant resonance form for a particular species. The compounds described herein can be synthesized according to methods described herein (or variations thereof) and/or conventional, organic chemical synthesis methods from commercially available starting materials and reagents or from starting materials and reagents that can be prepared according to conventional organic chemical synthesis methods. It is also possible to make use of variants of any of the aforementioned process steps. As can be appreciated by the skilled artisan, further methods of synthesizing the compounds of the formulae herein will be evident to those skilled in the art. Additionally, the various synthetic steps may be performed in an alternate sequence or order to give the desired compounds.
Synthetic chemistry transformations and protecting group methodologies (protection and deprotection) useful in synthesizing the compounds described herein are known in the art and include, for example, those such as described in R. C. Larock, Comprehensive Organic Transformations, 2d.ed., Wiley- VCH Publishers (1999); P. G. M. Wuts and T. W. Greene, Protective Groups in Organic Synthesis, 4th Ed., John Wiley and Sons (2007); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994); and L. Paquette, ed., Encyclopedia of Reagents or Organic Synthesis, John Wiley and Sons (1995), and subsequent editions thereof.
The compounds described herein can be separated from a reaction mixture and further purified by a method such as column chromatography, high- performance liquid chromatography (HPLC), or recrystallization.
The compounds of this invention can be readily prepared according to the following schemes from commercially available starting materials or starting materials which can be prepared using literature procedures. The schemes show the preparation of representative compounds of this invention. It is also possible to make use of variants of these process steps, which in themselves are known to and well within the preparatory skill of the skilled artisan. In the following reaction schemes, R1 to R9, and A are selected from groups defined above (unless otherwise indicated).
In general, the compounds described herein can be prepared according to Scheme
I below.
Scheme I


steps

 Referring to Scheme I, piperazine 1 (e.g., (R)-2-methyl-piperazine, which can be obtained commercially from Aldrich Chemical Company or synthesized according the procedure described in Xiang, J. et al, Journal Medicinal Chemistry, 2008, 57(14), 4068- 4071) can be coupled to halobenzene 2 (Hal1 can be Br or I, typically Br) at elevated temperatures and in the presence of a transition metal catalyst (typically a palladium- based catalyst, e.g., Pd2(dba)3) to provide N-phenyl piperazine 3. Reaction of intermediate 3 with sulfonyl halide 4 (Hal2 is typically chloro; Z is halo, e.g., bromo; or Z is hydrogen) results in the formation sulfonamide 5. In some embodiments, sulfonyl halide 4 can be synthesized by reacting the corresponding thiol with a halogenating reagent, e.g., SO2CI2/KNO3, in a polar aprotic solvent such as acetonitrile. In some embodiments, the process of converting 3 to 5 can also include one or more steps needed to introduce certain R2 substituents (e.g., when R2 is 5-membered heteroaryl). Metallation of 5 (e.g., by metal-halogen exchange when Z is halo; or by deprotonation with a strong base, e.g., LDA or n-BuLi, when Z is hydrogen) followed by addition of a carbon electrophile (e.g., a ketone, such as 3,3,3-trifluoropyruvate) provides 6, in which E is a substituent precursor (e.g., a tertiary alcohol) to -CR6R7R8 in the final compound 7. In some embodiments, the process of converting 6 to 7 can also include one or more steps needed to introduce certain R2 substituents (e.g., when R2 is -C(O)NH2 or 5- membered heteroaryl).
Referring to Scheme I, piperazine 1 (e.g., (R)-2-methyl-piperazine, which can be obtained commercially from Aldrich Chemical Company or synthesized according the procedure described in Xiang, J. et al, Journal Medicinal Chemistry, 2008, 57(14), 4068- 4071) can be coupled to halobenzene 2 (Hal1 can be Br or I, typically Br) at elevated temperatures and in the presence of a transition metal catalyst (typically a palladium- based catalyst, e.g., Pd2(dba)3) to provide N-phenyl piperazine 3. Reaction of intermediate 3 with sulfonyl halide 4 (Hal2 is typically chloro; Z is halo, e.g., bromo; or Z is hydrogen) results in the formation sulfonamide 5. In some embodiments, sulfonyl halide 4 can be synthesized by reacting the corresponding thiol with a halogenating reagent, e.g., SO2CI2/KNO3, in a polar aprotic solvent such as acetonitrile. In some embodiments, the process of converting 3 to 5 can also include one or more steps needed to introduce certain R2 substituents (e.g., when R2 is 5-membered heteroaryl). Metallation of 5 (e.g., by metal-halogen exchange when Z is halo; or by deprotonation with a strong base, e.g., LDA or n-BuLi, when Z is hydrogen) followed by addition of a carbon electrophile (e.g., a ketone, such as 3,3,3-trifluoropyruvate) provides 6, in which E is a substituent precursor (e.g., a tertiary alcohol) to -CR6R7R8 in the final compound 7. In some embodiments, the process of converting 6 to 7 can also include one or more steps needed to introduce certain R2 substituents (e.g., when R2 is -C(O)NH2 or 5- membered heteroaryl).
In some embodiments, compounds in which R6 is -OH, R7 is CF3, and R8 is CONH2, can be prepared according to Scheme II. Scheme II

chiral column separation


11
Referring to Scheme II above, metallation of 5 (cf: Scheme I), followed by addition of 3,3,3-trifluoropyruvate results in the formation of methyl ester 8, which in turn can be transformed into the corresponding amide 9 using, e.g., NH3 (g). The aforementioned sequence of reactions typically provides a mixture of diastereoisomers 10 and 11, which can be separated using, e.g., chiral column chromatography.
As mentioned above, the process of converting 6 to 7 in Scheme I can also include one or more steps needed to introduce certain R2 substituents. By way of example, compounds in which R2 is -CN can be partially hydrolyzed to provide amide 13 or can undergo an intermolecular dipolar addition reaction to produce tetrazole 14 (see Scheme III).
Scheme III

14
In some embodiments, compounds in which R8 is -NH-C(O)-(Ci_3 alkyl) can be prepared according to Scheme IV, which shows for illustrative purposes only the synthesis of compounds in which R8 is -NH-C(O)-(CHs).
Scheme IV

15
Referring to Scheme IV, metallation of 5 (cf: Scheme I), followed by addition of a ketone provides a tertiary alcohol intermediate (not shown), which upon exposure to acetonitrile and aqueous sulfuric acid results in the conversion of the hydroxyl group in the tertiary alcohol intermediate to a -NH-C(O)-(CHs) group (see compound 15 in Scheme IV).
In some embodiments, compounds in which R8 is 5-membered heteroaryl can be prepared according to Scheme V, which shows for illustrative purposes only the synthesis of compounds in which R6 is -OH, R7 is CF3, and R8 is 5-membered heteroaryl from compounds in which R6 is -OH, R7 is CF3, and R8 is CONH2.
Scheme V

NH2OHHCI, HOAc, dioxane

20
Referring to Scheme V, compound 9 (c/v Scheme I) can be converted to compound 18 using DMF dimethyl acetal. Exposure of compound 18 to hydrazine results in the formation of triazine 19, while reaction of compound 18 with hydroxylamine affords oxadiazole 20. In certain embodiments, the tertiary hydroxyl group in 9 can be protected, e.g., as an ether, e.g., as a silyl ether.
Similar chemistries to those shown in Scheme V can be used to synthesize compounds in which R2 is triazinyl or oxadiazolyl. In certain embodiments, compounds in which R2 is oxadiazolyl can be prepared according to Scheme VI.
Scheme VI

21 22
1 SOCI2, DCE
2 NH2NH2

23
24 n-BuLι CF3COCO2Me

25 26
Referring to Scheme VI, nitrile 21 (Hal is typically -Br) can be hydrolyzed under basic conditions (e.g., 3N NaOH in methanol and water) to carboxylic acid 22, which in turn can be converted to the corresponding hydrazide 23 under conventional conditions. Formation of the oxadiazole ring in 24 can be achieved upon reacting hydrozide 23 with an ortho formate, such as triethylorthoformate, in the presence of a catalytic amount of acid, e.g., PSA. Compounds 25 and 26 can be obtained using the chemistries described in the preceding schemes.
In some embodiments, compounds in which R6 is -CH3, R7 is -CH3, and R8 is - CONH2 can be prepared according to Scheme VII.
Scheme VII

LiOH, THF/H2O

30 29
Referring to Scheme VII above, compound 5 (cf. Scheme I) can be coupled with silyl enol ether 27 to form ester 28, which is hydrolyzed to the carboxylic acid 29. Conversion of 29 to the amide 30 can be achieved, e.g., using CDI and NH3.
The compounds of this invention may contain one or more asymmetric centers and thus occur as racemates and racemic mixtures, single enantiomers, individual diastereomers and diastereomeric mixtures. All such isomeric forms of these compounds are expressly included in the present invention.
In some embodiments, a chemical entity E can have the i?-CR4CR5 configuration and the i?-CR6R7R8 configuration ("a i?-CR4CR5, i?-CR6R7R8 stereoisomer"). In other embodiments, a chemical entity E can have the i?-CR4CR5 configuration and the S- CR6R7R8 configuration ("a i?-CR4CR5, ,S-CR6R7R8 stereoisomer").
In certain embodiments, a i?-CR4CR5, i?-CR6R7R8 stereoisomer can be present as a mixture with its corresponding i?-CR4CR5, S-CR6R7R8 stereoisomer. In embodiments, the mixture can contain greater than about 50% of the i?-CR4CR5, i?-CR6R7R8 stereoisomer (e.g., about 60%, about 70%, about 80%, about 90%, about 95%, about 99%). In these embodiments, the mixture can further include one or more other
substances (e.g., one or more pharmaceutically acceptable carriers, biological fluids, cellular culture, or any combination thereof). In still other embodiments, the i?-CR4CR5, i?-CR6R7R8 stereoisomer can be substantially free of its i?-CR4CR5, ,S-CR6R7R8 stereoisomer. The i?-CR4CR5, i?-CR6R7R8 stereoisomer can be in substantially pure form.
The compounds of this invention may also contain linkages (e.g., carbon-carbon bonds, carbon-nitrogen bonds such as amide bonds) wherein bond rotation is restricted about that particular linkage, e.g. restriction resulting from the presence of a ring or double bond. Accordingly, all cis/trans and E/Z isomers and rotational isomers are expressly included in the present invention. The compounds of this invention may also be represented in multiple tautomeric forms, in such instances, the invention expressly includes all tautomeric forms of the compounds described herein, even though only a single tautomeric form may be represented (e.g., alkylation of a ring system may result in alkylation at multiple sites, the invention expressly includes all such reaction products). All such isomeric forms of such compounds are expressly included in the present invention.
The compounds of this invention include the compounds themselves, as well as their salts and their prodrugs, if applicable. A salt, for example, can be formed between an anion and a positively charged substituent (e.g., amino) on a compound described herein. Suitable anions include chloride, bromide, iodide, sulfate, nitrate, phosphate, citrate, methanesulfonate, trifluoroacetate, and acetate. Likewise, a salt can also be formed between a cation and a negatively charged substituent (e.g., carboxylate) on a compound described herein. Suitable cations include sodium ion, potassium ion, magnesium ion, calcium ion, and an ammonium cation such as tetramethylammonium ion. Examples of prodrugs include Ci_6 alkyl esters of carboxylic acid groups, which, upon administration to a subject, are capable of providing active compounds.
Pharmaceutically acceptable salts of the compounds of this invention include those derived from pharmaceutically acceptable inorganic and organic acids and bases. Examples of suitable acid salts include acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, digluconate,
dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptanoate, glycolate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2- hydroxyethanesulfonate, lactate, maleate, malonate, methanesulfonate, 2- naphthalenesulfonate, nicotinate, nitrate, palmoate, pectinate, persulfate, 3- phenylpropionate, phosphate, picrate, pivalate, propionate, salicylate, succinate, sulfate, tartrate, thiocyanate, tosylate and undecanoate. Other acids, such as oxalic, while not in themselves pharmaceutically acceptable, may be employed in the preparation of salts useful as intermediates in obtaining the compounds of the invention and their pharmaceutically acceptable acid addition salts. Salts derived from appropriate bases include alkali metal (e.g., sodium), alkaline earth metal (e.g., magnesium), ammonium and N-(alkyl)4 salts. This invention also envisions the quaternization of any basic nitrogen-containing groups of the compounds disclosed herein. Water or oil-soluble or dispersible products may be obtained by such quaternization. Salt forms of the compounds of any of the formulae herein can be amino acid salts of carboxy groups (e.g. L-arginine, -lysine, -histidine salts).
The term "pharmaceutically acceptable carrier" refers to a carrier that may be administered to a subject (e.g., a patient), together with a chemical entity E as defined anywhere herein, and which does not destroy the pharmacological activity thereof and is nontoxic when administered in doses sufficient to deliver a therapeutic amount of the chemical entity E.
Pharmaceutical compositions described herein can include any one or more of the following: ion exchangers, alumina, aluminum stearate, lecithin, self-emulsifying drug delivery systems (SEDDS) such as d-α-tocopherol polyethyleneglycol 1000 succinate, surfactants used in pharmaceutical dosage forms such as Tweens or other similar polymeric delivery matrices, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts, or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene-polyoxypropylene-block polymers, polyethylene glycol and wool fat.
Cyclodextrins such as α-, β-, and γ-cyclodextrin, or chemically modified derivatives such as hydroxyalkylcyclodextrins, including 2- and 3-hydroxypropyl-β-cyclodextrins, or other solubilized derivatives may also be advantageously used to enhance delivery of compounds of the formulae described herein. In general, the chemical entities described herein can be used for treating (e.g., controlling, relieving, ameliorating, alleviating, or slowing the progression of) or methods for preventing (e.g., delaying the onset of or reducing the risk of developing) a disease or condition that is mediated by excess (e.g., abnormally high) levels or uncontrolled (e.g., resulting in abnormally high) levels of Cortisol and/or other corticosteroids (e.g., 11 β- hydroxy steroids. While not wishing to be bound by any theory, it is believed that the chemical entities described herein can reduce the levels of Cortisol and other corticosteroids (e.g., llβ-hydroxysteroids) by inhibiting the reductase activity of llβ- HSDl. The diseases, disorders, conditions or symptoms mediated by excess or uncontrolled amounts of Cortisol and/or other corticosteroids can include diabetes (e.g., type 1 or type 2 diabetes), Syndrome X, hyperglycemia, low glucose tolerance, insulin resistance, obesity, lipid disorders, dyslipidemia, hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels, high LDL levels, atherosclerosis and its sequelae, vascular restenosis, pancreatitis, abdominal obesity, neurodegenerative disease, retinopathy, nephropathy, neuropathy, hypertension, coronary heart disease, stroke, peripheral vascular disease, Cushing's syndrome, glaucoma, osteoperosis, hyperinsulinemia, tuberculosis, psoriasis,cognitive disorders and dementia (e.g., impairment associated with aging and of neuronal dysfunction, e.g., Alzheimer's disease), depression, viral diseases, inflammatory disorders, immune disorders. In some embodiments, the diseases, disorders conditions or symptoms can further include those where insulin resistance is a component. In other embodiments, the chemical entities described herein can be used for promoting wound healing.
In some embodiments, the chemical entities described herein can be coadministered with one or more other threapeutic agents. In certain embodiments, the additional agents may be administered separately, as part of a multiple dose regimen, from the chemical entities described herein (e.g., sequentially, e.g., on different overlapping schedules with the administration of one or more compounds of formula (I)).
Alternatively, these agents may be part of a single dosage form, mixed together with the chemical entities described herein in a single composition. In still another embodiment, these agents can be given as a separate dose that is administered at about the same time that one or more chemical entities described herein are administered (e.g., simultaneously with the administration of one or more chemical entities described herein). When the compositions described herein include a combination of a chemical entity E as defined anywhere herein and one or more additional therapeutic or prophylactic agents, both the chemical entity and the additional agent should be present at dosage levels of between about 1 to 100%, and more preferably between about 5 to 95% of the dosage normally administered in a monotherapy regimen.
Other therapeutic agents can include DP-IV inhibitors; insulin sensitizers (e.g., (i) PPAR agonists and (ii) biguanides); insulin and insulin analogues and mimetics; sulfonylureas and other insulin secretagogues; prandial glucose regulators, alpha.- glucosidase inhibitors; glucagon receptor antagonists; GLP-I, GLP-I mimetics, and GLP-I receptor agonists; GIP5GIP mimetics, and GIP receptor agonists; PACAP,
PACAP mimetics, and PACAP receptor 3 agonists; cholesterol lowering agents (e.g., (i) HMG-CoA reductase inhibitors, (ii) sequestrants, (iii) nicotinyl alcohol, nicotinic acid and salts thereof, (iv) PPAR. alpha, agonists, (v) PPAR. alpha./, gamma, dual agonists, (vi) inhibitors of cholesterol absorption, (vii) acyl CoAxholesterol acyltransferase inhibitors, and (viii) anti-oxidants; PPAR.delta. agonists); antiobesity compounds (e.g., sibutramine and orlisat); an ileal bile acid transporter inhibitor; anti-inflammatory agents excluding glucocorticoids (e.g., aspirin); protein tyrosine phosphatase- IB (PTP-IB) inhibitors; agents that suppress hepatic glucose output (e.g., metformin); agents designed to reduce the absorption of glusoce from the intestine (e.g., acarbose); agents designed to treat the complications of prolonged hyperglycemia (e.g., aldose reductase inhibitors); antidiabetic agents (e.g., glusoce phosphatase inhibitors, glucose -6-phosphatase inhibitors, glucagon receptor antagonists, glucose kinase activators, glycogen phosphorylase inhibitors, fructose 1,6 bisphosphatase inhibitors, glutamine:fructose-6-phosphate amidotransferase inhibitors); antihypertensive agents (e.g., β blockers (e.g., atenolol, inderal), ACE inhibitors (e.g., lisinopril), calcium agonists (e.g., nifedipine), angiotensin receptor antagonists (e.g., candesartan), a agonists and diuretic agents (e.g., furosemide,
benzthiazide)); and haemostasis modulators (e.g., antithrombotics, activators of fibrinolysis and antiplatelet agents (e.g., clopidogrel, aspirin), thrombin antogonists, factor Xa inhibitors, factor Vila inhibitors, anticoagulants (e.g., heparin and low molecular weight analogues, hirudin), warfarin). The chemical entities and compositions described herein can, for example, be administered orally, parenterally (e.g., subcutaneously, intracutaneously, intravenously, intramuscularly, intraarticularly, intraarterially, intrasynovially, intrasternally, intrathecally, intralesionally and by intracranial injection or infusion techniques), by inhalation spray, topically, rectally, nasally, buccally, vaginally, via an implanted reservoir, by injection, subdermally, intraperitoneally, transmucosally, or in an ophthalmic preparation, with a dosage ranging from about 0.01 mg/Kg to about 1000 mg/Kg, (e.g., from about 0.01 to about 100 mg/kg, from about 0.1 to about 100 mg/Kg, from about 1 to about 100 mg/Kg, from about 1 to about 10 mg/kg) every 4 to 120 hours, or according to the requirements of the particular drug. The interrelationship of dosages for animals and humans (based on milligrams per meter squared of body surface) is described by Freireich et al., Cancer Chemother. Rep. 50, 219 (1966). Body surface area may be approximately determined from height and weight of the patient. See, e.g., Scientific Tables, Geigy Pharmaceuticals, Ardsley, New York, 537 (1970). In certain embodiments, the compositions are administered by oral administration or administration by injection. The methods herein contemplate administration of an effective amount of compound or compound composition to achieve the desired or stated effect. Typically, the pharmaceutical compositions of this invention will be administered from about 1 to about 6 times per day or alternatively, as a continuous infusion. Such administration can be used as a chronic or acute therapy. The amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration. A typical preparation will contain from about 5% to about 95% active compound (w/w). Alternatively, such preparations contain from about 20% to about 80% active compound.
Lower or higher doses than those recited above may be required. Specific dosage and treatment regimens for any particular patient will depend upon a variety of factors, including the activity of the specific compound employed, the age, body weight, general
health status, sex, diet, time of administration, rate of excretion, drug combination, the severity and course of the disease, condition or symptoms, the patient's disposition to the disease, condition or symptoms, and the judgment of the treating physician.
Upon improvement of a patient's condition, a maintenance dose of a compound, composition or combination of this invention may be administered, if necessary.
Subsequently, the dosage or frequency of administration, or both, may be reduced, as a function of the symptoms, to a level at which the improved condition is retained when the symptoms have been alleviated to the desired level. Patients may, however, require intermittent treatment on a long-term basis upon any recurrence of disease symptoms. The compositions of this invention may contain any conventional non-toxic pharmaceutically-acceptable carriers, adjuvants or vehicles. In some cases, the pH of the formulation may be adjusted with pharmaceutically acceptable acids, bases or buffers to enhance the stability of the formulated compound or its delivery form.
The compositions may be in the form of a sterile injectable preparation, for example, as a sterile injectable aqueous or oleaginous suspension. This suspension may be formulated according to techniques known in the art using suitable dispersing or wetting agents (such as, for example, Tween 80) and suspending agents. The sterile injectable preparation may also be a sterile injectable solution or suspension in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3- butanediol. Among the acceptable vehicles and solvents that may be employed are mannitol, water, Ringer's solution and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose, any bland fixed oil may be employed including synthetic mono- or diglycerides. Fatty acids, such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions. These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant, or carboxymethyl cellulose or similar dispersing agents which are commonly used in the formulation of pharmaceutically acceptable dosage forms such as emulsions and or suspensions. Other commonly used surfactants such as Tweens or Spans and/or other similar emulsifying agents or bioavailability enhancers which are commonly used in the
manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms may also be used for the purposes of formulation.
The compositions of this invention may be orally administered in any orally acceptable dosage form including, but not limited to, capsules, tablets, emulsions and aqueous suspensions, dispersions and solutions. In the case of tablets for oral use, carriers which are commonly used include lactose and corn starch. Lubricating agents, such as magnesium stearate, are also typically added. For oral administration in a capsule form, useful diluents include lactose and dried corn starch. When aqueous suspensions and/or emulsions are administered orally, the active ingredient may be suspended or dissolved in an oily phase is combined with emulsifying and/or suspending agents. If desired, certain sweetening and/or flavoring and/or coloring agents may be added.
The compositions of this invention may also be administered in the form of suppositories for rectal administration. These compositions can be prepared by mixing a compound of this invention with a suitable non-irritating excipient which is solid at room temperature but liquid at the rectal temperature and therefore will melt in the rectum to release the active components. Such materials include, but are not limited to, cocoa butter, beeswax and polyethylene glycols.
In some embodiments, topical administration of the compounds and compositions described herein may be presented in the form of an aerosol, a semi-solid pharmaceutical composition, a powder, or a solution. By the term "a semi-solid composition" is meant an ointment, cream, salve, jelly, or other pharmaceutical composition of substantially similar consistency suitable for application to the skin. Examples of semi-solid compositions are given in Chapter 17 of The Theory and Practice of Industrial Pharmacy, Lachman, Lieberman and Kanig, published by Lea and Febiger (1970) and in Remington's Pharmaceutical Sciences, 21st Edition (2005) published by Mack Publishing Company, which is incorporated herein by reference in its entirety.
Topical administration of the compositions of this invention is useful when the desired treatment involves areas or organs readily accessible by topical application. For application topically to the skin, the composition should be formulated with a suitable ointment containing the active components suspended or dissolved in a carrier. Carriers for topical administration of the compounds of this invention include, but are not limited
to, mineral oil, liquid petroleum, white petroleum, propylene glycol, polyoxyethylene polyoxypropylene compound, emulsifying wax and water. Alternatively, the composition can be formulated with a suitable lotion or cream containing the active compound suspended or dissolved in a carrier with suitable emulsifying agents. Suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water. The compositions of this invention may also be topically applied to the lower intestinal tract by rectal suppository formulation or in a suitable enema formulation.
Topically-transdermal patches are also included in this invention. Also within the invention is a patch to deliver active chemotherapeutic combinations herein. A patch includes a material layer (e.g., polymeric, cloth, gauze, bandage) and the compound of the formulae herein as delineated herein. One side of the material layer can have a protective layer adhered to it to resist passage of the compounds or compositions. The patch can additionally include an adhesive to hold the patch in place on a subject. An adhesive is a composition, including those of either natural or synthetic origin, that when contacted with the skin of a subject, temporarily adheres to the skin. It can be water resistant. The adhesive can be placed on the patch to hold it in contact with the skin of the subject for an extended period of time. The adhesive can be made of a tackiness, or adhesive strength, such that it holds the device in place subject to incidental contact, however, upon an affirmative act (e.g., ripping, peeling, or other intentional removal) the adhesive gives way to the external pressure placed on the device or the adhesive itself, and allows for breaking of the adhesion contact. The adhesive can be pressure sensitive, that is, it can allow for positioning of the adhesive (and the device to be adhered to the skin) against the skin by the application of pressure (e.g., pushing, rubbing,) on the adhesive or device.
The compositions of this invention may be administered by nasal aerosol or inhalation. Such compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art.
A composition having a chemical entity as defined anywhere herein and an additional agent (e.g., a therapeutic agent) can be administered using any of the routes of administration described herein. In some embodiments, a composition having the compound of the formulae herein and an additional agent (e.g., a therapeutic agent) can be administered using an implantable device. Implantable devices and related technology are known in the art and are useful as delivery systems where a continuous, or timed- release delivery of compounds or compositions delineated herein is desired. Additionally, the implantable device delivery system is useful for targeting specific points of compound or composition delivery (e.g., localized sites, organs). Negrin et al, Biomaterials, 22(6): 563 (2001). Timed-release technology involving alternate delivery methods can also be used in this invention. For example, timed-release formulations based on polymer technologies, sustained-release techniques and encapsulation techniques (e.g., polymeric, liposomal) can also be used for delivery of the compounds and compositions delineated herein. The invention will be further described in the following examples. It should be understood that these examples are for illustrative purposes only and are not to be construed as limiting this invention in any manner.
EXAMPLES The title compounds of Examples 1.1, 1.2, and 1.3 were prepared as shown in
Scheme 1 below. Detailed synthesis procedures are provided below.
Scheme 1

Example 1.1

3,3,3-trifluoro-2-r5-({(2R)-4-r4-fluoro-2-(trifluoromethyl)phenyll-2-methylpiperazin- l-yl}sulfonyl)-2-thienyll-2-hvdroxypropanamide Step IA: A mixture of (R)-2-methyl-piperazine (25.0 g, 250 mmol), 2-bromo 5- fluoro benzotrifluoride (55.1 g, 227 mmol), tris(dibenzylidineacetone)dipalldium (0) (2.08g, 2.27 mmol), rac-2,2'-bis(diphenylphosphino)-l,r-binaphthyl (4.24 g, 6.81 mmol) and sodium tert-butoxide (27.3 g, 280 mmol) was mixed and purged with N2. Anhydrous toluene (500 mL) was added and purged with N2 again. The resulting mixture was heated in an oil bath at 105 0C under N2 for 3.5 hours. After cooling, the reaction mixture was concentrated and then filtered through a pad of Celite, washed with Et2O. The organic layer was concentrated, diluted with Et2O (500 mL), filtered through a pad of Celite again, and washed with IN aq. HCl (2 x 150 mL). The aqueous layer was basified with NaOH at 0 0C (pH = -10) and then was extracted with Et2O (3 x 200 mL). The combined organic layer was dried over MgSO4 and concentrated under vacuum to give (3i?)-l-[4-
fluoro-2-(trifluoromethyl)phenyl]-3-methylpiperazine as a brown oil (58.5 g, 98%), which was used without further purification.
Step IB: To a solution of 5-bromothiophene-2-sulfonyl chloride (26.2 g, 100 mmol) and (3R)-l-(4-fluoro-2-(trifluoromethyl)phenyl)-3-methylpiperazine (27.6 g, lOOmmol) in DCM (200 ml) was added Et3N (41.8 ml, 300 mmol) at room temp. The reaction mixture was stirred at room temperature until completion of the reaction (about 6 hours) and then washed with aq. NaHCO3. The basic washes were back extracted with dichloromethane (DCM). The combined organic layers were washed with brine and dried over Na2SO4. The crude product was purified on a SiO2 column using hexanes/DCM as the eluent to give (R)-l-(5-bromothiophen-2-ylsulfonyl)-4-(4-fluoro-2- (trifluoromethyl)phenyl)-2-methylpiperazine as a white solid (38 g, 78 mmol, 78 % yield).
Step 1C: To a solution of (R)-l-(5-bromothiophen-2-ylsulfonyl)-4-(4-fluoro-2- (trifluoromethyl)phenyl)-2-methylpiperazine (28.1 g, 57.7 mmol) in anhydrous THF (200 ml) was added Butyllithium (28.8 ml, 57.7 mmol) at -780C. The reaction mixture was Stirred under N2 for 15 min. and then a solution of methyl 3,3,3-trifluoropyruvate (6.07 ml, 57.7 mmol) in THF (20 mL) was added via a cannula. The reaction mixture was stirred at -780C for 2 h. and then quenched with a 10 mL of 10% aq. HCl. The reaction mixture was dried over MgSO4 and CombiFlashed with DCM/hexane (15 - 100%) to provide methyl 3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2-(trifluoromethyl)phenyl)-2- methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2-hydroxypropanoate as a sticky, light yellow solid (22 g, 39.0 mmol, 67.6 % yield).
Step ID, Method 1: To a solution of methyl 3,3,3-trifluoro-2-(5-((R)-4-(4-fiuoro- 2-(trifluoromethyl)phenyl)-2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2- hydroxypropanoate (21.5 g, 38.1 mmol) in MeOH (200 ml) was added aq. NH3 (-28-
30%, 50 mL). The reaction mixture was stirred at room temperature o/n and then diluted with ice water (700 mL). The resultant white ppt was collected by filtration, washed with water, and dried in an oven at 60 0C to give the desired product 3,3,3-trifluoro-2-(5-((R)- 4-(4-fluoro-2-(trifluoromethyl)phenyl)-2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2- hydroxypropanamide (15 g, 27.3 mmol, 71.7 % yield). The aqueous layer was extracted with DCM (4 x 100 mL), and the combined organic layers were concentrated.
Purification of the concentrate by column chromatography with EA/DCM (0-40%) gave an additional 1.5 g of product.
Method 2: To a solution of methyl 3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2-
(trifluoromethyl)phenyl)-2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2- hydroxypropanoate (200 mg) in MeOH (20 ml) at -780C was bubbled NH3 gas. The resultant mixture was stirred at room temperature overnight, concentrated, and dissolved in fresh DCM. The organic layer was washed with aq. NaHCO3 and dried to give 3,3,3- trifluoro-2-(5 -((R)-4-(4-fluoro-2-(trifluoromethyl)phenyl)-2-methylpiperazin- 1 - ylsulfonyl)thiophen-2-yl)-2-hydroxypropanamide as a white solid (150 mg). It was found that competing hydrolysis of the ester group to the corresponding acid occurred to a greater extent when using Method 1. Thus, in some instances, it may be preferable to use Method 2 when performing step D.
HRMS: calcd for Ci9Hi8F7N3O4S2 + H+, 550.06997; found (ESI-FTMS,
[M+H]1+), 550.07165. Example 1.2

13.5 grams of 3,3,3-trifluoro-2-(5-((R)-4-(4-fiuoro-2-(trifluoromethyl)phenyl)-2- methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2-hydroxypropanamide (prepared according to a procedure similar to that described in Example 1.1) was separated was separated with a chiral column (Chiralpak ADH) in SFC Analytical Instrument; Mobile Phase was 90% CO2 /10%Methanol at flow rate 5mL/min. Early fraction (Retention 4.4min) was collected to give the title compound (5.7g); late fraction was collected to give the diastereomer described in Example 1.3 (6g, retention time 6. lmin).
HRMS: calcd for Ci9Hi8F7N3O4S2 + H+, 550.06997; found (ESI, [M+H]+), 550.0697.
Example 1.3

HRMS: calcd for Ci9Hi8F7N3O4S2 + H+, 550.06997; found (ESI, [M+H]+), 550.0701.
Example 1.4

(2R)-2- [2-bromo-5-({(2R)-4- r4-fluoro-2-(trifluoromethyl)phenyll -2-methylpiperazin- l-yl}sulfonyl)-3-thienyll-3,3i3-trifluoro-2-hvdroxypropanamide
The title compound was isolated from the crude reaction mixture that was obtained after performing step D in Example 1.1. The title compound was separated using a chiral column (Chiralpak ADH) in SFC Analytical Instrument. Mobile Phase was 90% CO2 /10%Methanol. (900 mg, 3.76% yield).
HRMS: calcd for Ci9HnBrF7N3O4S2 + H+, 627.98048; found (ESI, [M+H]+), 627.9805.
Example 1.5

The title compound was prepared using a procedure analogous to that described in Example 1.1 (235 mg, 54.1% yield), but using 2-oxo-propionic acid ethyl ester instead of methyl 3,3,3-trifluoropyruvate in Step 1C.
HRMS: calcd for Ci9H2IF4N3O4S2 + H+, 496.09824; found (ESI, [M+H]+), 496.0997.

The title compound was prepared using a procedure analogous to that described in Example 1.1 (85 mg, 27.3% yield), but using oxo-phenyl-acetic acid methyl ester instead of methyl 3,3,3-trifluoropyruvate in Step 1C.
Example 2.1
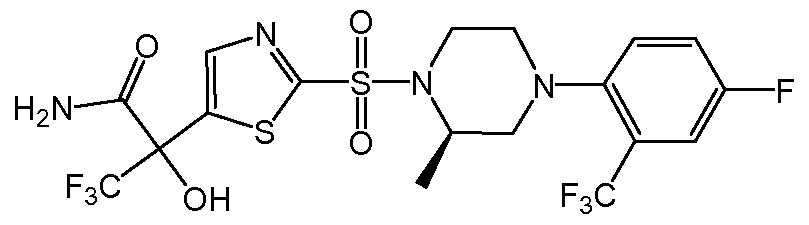
3,3,3-trifluoro-2-r2-α(2R)-4-r4-fluoro-2-(trifluoromethyl)phenyll-2-methylpiperazin- 1-yl} sulfonyl)- 1 ,3-thiazol-5-yll -2-hydroxypropanamide The title compound of Example 2.1 was prepared as shown in Scheme 2 below.
Detailed synthesis procedures are provided below.
Scheme 2

Step 2A: To a suspension of 2-mercaptothiazole (1.0 g, 8.53 mmol) with KNO3 (2.15 g, 21.3 mmol) in 17 mL of anhydrous MeCN was added SO2Cl2 (1.73 mL, 21.3 mmol) at O0C under N2. The suspension was stirred at O0C for lhr, after which time the reaction was determined to be complete by thin layer chromatography (TLC). The reaction mixture was diluted with ether and neutralized with sat. NaHCO3 solution until a pH=6-7 was attained. The reaction mixture was extracted first with ether, then with EtOAc. The combined organic layers were washed with sat. brine, dried over Na2SO4, concentrated down to give thiazole-2-sulfonyl chloride as a brown-yellow color liquid (350 mg), which was used in next step without purification.
Step 2B: To a stirred solution of (3i?)-l-[4-fluoro-2-(trifluoromethyl)phenyl]-3- methylpiperazine (100 mg, 0.38 mmol) and diisopropylethylamine (0.13 mL, 0.76 mmol) in anhydrous dichloromethane (2 mL) was added thiazole-2-sulfonyl chloride (70 mg, 0.38 mmol) at O0C. The reaction mixture was stirred at O0C for 15 min, then stirred at room temperature for 3 hrs, after which time the reaction was judged complete by TLC. Another batch was prepared using (400 mg) of (3i?)-l-[4-fluoro-2- (trifluoromethyl)phenyl]-3-methylpiperazine. The reaction mixtures were combined, washed with H2O, extracted with DCM. The organic layers were dried over Na2SO4 and concentrated to afford a brown oil. The crude product was purified via flash column chromatography, eluting with 20-40% EtOAc/Hexane to yield (2i?)-4-[4-fluoro-2- (trifluoromethyl)phenyl]-2-methyl-l-(l,3-thiazol-2-ylsulfonyl)piperazine in 64% yield (497 mg) as a light yellow solid. Step 2C: To a solution of (2i?)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methyl-l-
(l,3-thiazol-2-ylsulfonyl)piperazine (258 mg, 0.63 mmol) in anhydrous THF (4 ml) was added lithium diisopropyl amide (0.38 ml, 0.76 mmol) at -780C. The reaction mixture
was stirred under N2 for 20 min, the methyl 3,3,3-trifluoropyruvate (0.096 ml, 0.945 mmol) was added. The resultant mixture was stirred at -780C for 15 min, warmed to room temperature, quenched with H2O, and extracted with DCM (3x). The combined organic layers were dried over MgSO4. The crude product was purified by chromatography (CombiFlash) with 30-50% EtOAc/Hexane to give methyl 3,3,3- trifluoro-2-[2-({(2i?)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methylpiperazin-l- yl}sulfonyl)-l,3-thiazol-5-yl]-2-hydroxypropanoate as a yellow solid (229.5 mg, 64.4 % yield).
Step 2D: To a solution of 3,3,3-trifiuoro-2-[2-({(2i?)-4-[4-fluoro-2- (trifluoromethyl)phenyl]-2-methylpiperazin- 1 -yl} sulfonyl)-l ,3-thiazol-5-yl]-2- hydroxypropanoate (213 mg, 0.38 mmol) in MeOH (2 ml) was added aq. NH3 (-28-30%, 1 mL). The reaction mixture was stirred at room temperature for 5 hrs, after which the reaction was determined to be complete by LC/MS. Most of the solvent was removed under reduced pressure. The concentrate was then neutralized with IN HCl solution until a pH=6 was attained. The concentrate was extracted with EtOAc first, then extracted with DCM. The combined organic layers were concentrated and purified via flash column chromatography, eluting with 3-5% MeOH/DCM to yield (25)-3,3,3-trifluoro-2- [5-( {(2i?)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methylpiperazin- 1 - yl}sulfonyl)thiophen-2-yl]-2-hydroxypropanamide as an off-white solid (112 mg, 53.5% yield).
HRMS: calcd for Ci8Hi7F7N4O4S2 + H+, 551.06522; found (ESI, [M+H]+), 551.0639.
Example 2.1 was separated with a chiral column (Chiralpak ADH) in SFC Analytical Instrument. Mobile Phase was 90% CO2 /10%MethanoL. The title compounds of Example 2.2 and 2.3 were isolated in good yield.
Example 2.2

HRMS: calcd for Ci8Hi7F7N4O4S2 + H+, 551.06522; found (ESI, [M+H]+), 551.0650.
Example 2.3

αS)-3,3,3-trifluoro-2-r2-ααR)-4-r4-fluoro-2-(trifluoromethyl)phenyll-2- methylpiperazin- 1-vU sulfonyl)- 1.,3-thiazol-5-yH -2-hvdroxypropanamide
HRMS: calcd for Ci8Hi7F7N4O4S2 H+, 551.06522; found (ESI, [M+H]+), 551.0651.
Example 3.1

3-,3.,3-trifluoro-2-[3-f{f2R)-4-[4-fluoro-2-ftrifluoromethyl)phenyll-2-methylpiperazin- 1-yll sulfonyl)- 1-methyl- IH- 1 ,2,4-triazol-5-yll -2-hydroxypropanamide
The title compound of Example 3.1 was prepared as shown in Scheme 3 below. Detailed synthesis procedures are provided below. Scheme 3
THF



Step 3C: To a solution of (2i?)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methyl-l- [(l-methyl-lH-l,2,4-triazol-5-yl)sulfonyl]piperazine (484 mg, 1.19 mmol) in anhydrous TΗF (7 ml) was added n-Butyllithium (0.52 ml, 1.31 mmol) at -780C. The reaction mixture was stirred under N2 for 30 min., then methyl 3,3,3-trifluoropyruvate (0.18 ml, 1.785 mmol) was added. Stirring at -780C was continued for 1.5 hrs, then the reaction mixture was warmed to room temperature. The reaction mixture was quenched with H2O and extracted with DCM (3x). The combined organic layers were dried over MgSO4. The crude product was purified by chromatography (CombiFlash) with 35-70%
EtOAc/Hexane to give methyl 3,3,3-trifluoro-2-[3-({(2i?)-4-[4-fluoro-2-
(trifluoromethyl)phenyl]-2-methylpiperazin-l-yl}sulfonyl)-l-methyl-lH-l,2,4-triazol-5- yl]-2-hydroxypropanoate as a colorless gum (230 mg, 34.3 % yield).
Step 3D: To methyl 3,3,3-trifluoro-2-[3-({(2i?)-4-[4-fluoro-2-
(trifluoromethyl)phenyl]-2-methylpiperazin-l-yl}sulfonyl)-l-methyl-lH-l,2,4-triazol-5- yl]-2-hydroxypropanoate (213.5 mg, 0.379 mmol) was added 3 mL OfNH3 in MeOH (2.0 M in MeOH, 1.5 mmol). The reaction mixture was stirred at room temperature o/n, after which time the reaction was determined to be complete by TLC. Evaporation of the solvent and purification via flash column chromatography (eluting with 30-50% EtOAc/Hexane) afforded 3,3,3-trifluoio-2-[3-({(2i?)-4-[4-fluoio-2- (trifluoromethyl)phenyl]-2-methylpiperazin-l-yl}sulfonyl)-l-methyl-lH-l,2,4-triazol-5- yl]-2-hydroxypropanamide as a white powder (75.6 mg, 36.4% yield).
ΗRMS: calcd for Ci8Hi9F7N6O4S + H+, 549.11495; found (ESI, [M+H]+), 549.1146.
The title compounds of Examples 4.1 and 4.2 were prepared as shown in Scheme 4 below. Detailed synthesis procedures are provided below.
Scheme 4

Example 4.1
 αR)-3,3,3-trifluoro-2-r4-ααR)-4-r4-fluoro-2-qrifluoromethyl)phenyll-2- methylpiperazin-l-yl}sulfonyl)-l-methyl-lH-imidazol-5-yll-2-hvdroxypropanamide
αR)-3,3,3-trifluoro-2-r4-ααR)-4-r4-fluoro-2-qrifluoromethyl)phenyll-2- methylpiperazin-l-yl}sulfonyl)-l-methyl-lH-imidazol-5-yll-2-hvdroxypropanamide
Step 4A: To a stirred solution of (3i?)-l-[4-fluoro-2-(trifluoromethyl)phenyl]-3- methylpiperazine (1.038 g, 3.96 mmol) and diisopropylethylamine (1.38 rnL, 7.92 mmol) in anhydrous dichloromethane (15 rnL) was added 1 -methyl- lΗ-imidazole-4-sulfonyl chloride (0.715 g, 3.96 mmol) at O0C. The mixture was stirred at O0C for 30 min, then stirred at room temperature for o/n. The reaction mixture was washed with H2O, extracted with DCM. The organic layer was dried over Na2SO4 and concentrated. The crude product was purified via flash column chromatography, eluting with 2-4% MeOH/DCM to yield (2i?)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methyl-l-[(l- methyl-lH-imidazol-4-yl)sulfonyl]piperazine as a light yellow solid (1.22 g, 75.8% yield).
Step 4B: To a solution of (2i?)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methyl-l- [(I -methyl- lH-imidazol-4-yl)sulfonyl]piperazine (552 mg, 1.36 mmol) in anhydrous
TΗF (7 ml) was added n-Butyllithium (0.60 ml, 1.49 mmol) at -780C. The reaction was stirred at this tempeature under N2 for 40 min., then methyl 3,3,3-trifluoropyruvate (0.21 ml, 2.04 mmol) was added. The reaction mixture was stirred at -780C for 1 hr, then warmed to room temperature and Quenched with a Η2O. The quenched reaction mixture was extracted with DCM (3x). The combined organic layers were dried over MgSO4. Evaporation of the solvent and purification by chromatography (CombiFlash) with 40- 80% EtOAc/Hexane afforded methyl 3,3,3-trifluoro-2-[4-({(2i?)-4-[4-fluoro-2- (trifluoromethyl)phenyl]-2-methylpiperazin-l-yl}sulfonyl)-l-methyl-lH-imidazol-5-yl]- 2-hydroxypropanoate as a white solid (330 mg, 43.1 % yield). Step 4C: To a solution of methyl 3,3,3-trifluoro-2-[4-({(2i?)-4-[4-fluoro-2-
(trifluoromethyl)phenyl]-2-methylpiperazin-l-yl}sulfonyl)-l-methyl-lH-imidazol-5-yl]- 2-hydroxypropanoate (230 mg, 0.41 mmol) in 2 mL of MeOH was added aq. NΗ3 (~28-
30%, 2 mL). The reaction mixture was stirred at room temperature for o/n, after which
time the reaction was determined to be complete as by TLC. Evaporation of the solvent and purification via preparative HPLC under neutral condition yielded (2i?)-3,3,3- trifluoro-2-[4-({(2i?)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methylpiperazin-l- yl}sulfonyl)-l-methyl-lH-imidazol-5-yl]-2-hydroxypropanamide as a white solid (1.7 mg) and (2lS)-3,3,3-trifluoro-2-[4-({(2i?)-4-[4-fluoro-2-(trifluoromethyl) phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)- 1 -methyl- lH-imidazol-5-yl]-2-hydroxypropanamide as a white solid (12.3 mg). The chiral column and conditions described elsewhere were used to effect separation of the above-described isomers.
ΗRMS: calcd for Ci9H20F7N5O4S + H+, 548.11970; found (ESI, [M+H]+), 548.1195.
Example 4.2

Example 4.1.
ΗRMS: calcd for Ci9H20F7N5O4S + H+, 548.11970; found (ESI, [M+H]+), 548.1197.
The title compounds of Examples 5.1 and 5.2 were prepared as shown in Scheme
5 below. Detailed synthesis procedures are provided below.


Example 5.1

4-r(3R)-4-{r5-α-carbamoyl-2,2,2-trifluoro-l-hvdroxyethyl)-2-thienyllsulfonyl}-3- methylpiperazin-l-yll-3-ftrifluoromethyl)benzamide
Step 5 A: A mixture of (R)-2-methyl-piperazine (6.0 g, 59.9 mmol), 4-chloro-3- trifluoromethyl-benzonitrile (11.2 g, 54.5 mmol), tris(dibenzylidineacetone)dipalldium (0) (0.499 g, 0.545 mmol), rac-2,2'-bis(diphenylphosphino)-l,l '-binaphthyl (1.02 g, 1.635 mmol)and sodium tert-butoxide (6.55 g, 68.12 mmol) was mixed and purged with N2. Anhydrous toluene (75 mL) was added and the resultant mixture was purged with N2 again. The resultant mixture was heated in an oil bath at 107 0C under N2 for 3 hours. After cooling, the reaction mixture was diluted with DCM (200 mL), washed with H2O (50 mL), and washed with saturated brine (50 mL). The combined organic layers were dried over Na2SO4, concentrated under reduced pressure to give a dark brown liquid. The crude product was purified using a SiO2 gel column, eluting with 5-7% MeOH in DCM to give 4-[(3i?)-3-methylpiperazin-l-yl]-3-(trifluoromethyl)benzonitrile as a brownish red color oil (l 1.2 g, 76.2%).
Step 5B: To a stirred solution of 4-[(3i?)-3-methylpiperazin-l-yl]-3- (trifluoromethyl)benzonitrile (2.69 g, 10 mmol) and TEA (4.18 rnL, 30 mmol) in anhydrous dichloromethane (20 mL) was added thiophene-2-sulfonyl chloride (1.92 g, 10 mmol) at O0C. The mixture was stirred at O0C for 15 min, then stirred at room temperature for o/n, after which time the reaction was determined to be complete by
TLC. The reaction mixture was washed with aq. NaHCO3, then extracted with DCM. The organic layer was dried over Na2SO4 and concentrated. The crude product was purified via flash column chromatography to yield 5-chloro-Λ/-{4-[(6-cyano-2-{3-[4- (dimethylamino)piperidin-l-yl]phenyl}pyrazolo[l,5-a]pyrimidin-7-yl)amino]-2- methoxyphenyl}-l-benzofuran-2-carboxamide in 96% yield (3.98 g) as a white solid.
Step 5C: To a solution of 5-chloro-N-{4-[(6-cyano-2-{3-[4- (dimethylamino)piperidin-l-yl]phenyl}pyrazolo[l,5-a]pyrimidin-7-yl)amino]-2- methoxyphenyl}-l-benzofuran-2-carboxamide (4.15 g, 10 mmol) in anhydrous THF (60 ml) was added freshly made LDA (12 mmol) at -780C. The reaction mixture was stirred under N2 for 20 min., and then methyl 3,3,3-trifluoropyruvate (2.1 mL, 20 mmol) was added. Stirring at -780C was maintained for 30 min. The reaction mixture was then warmed to room temperature and quenched with a H2O. The quenched reaction mixture was extracted with DCM (3x). The combined organic layers were dried over MgSO4 and concentrated. The crude product was purified by chromatography (CombiFlash) with EtOAc/Hexane to give methyl 2-[5-( {(2i?)-4-[4-cyano-2-(trifluoromethyl)phenyl]-2- methylpiperazin-l-yl}sulfonyl)-2-thienyl]-3,3,3-trifluoro-2-hydroxypropanoate as a light yellow solid (4.3 g, 78 % yield).
Step 5D: To methyl 2-[5-({(2i?)-4-[4-cyano-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)-2-thienyl] -3 ,3 ,3 -trifluoro-2-hydroxypropanoate (120 mg, 0.21) was added NH3 in EtOH (2.0M, 5 mL). The resultant mixture was stirred at room temperature for 2 days. The reaction mixture was then concentrated and purified on a SiO2 gel column, eluting with MeOH in DCM to give 2-[5-({(2i?)-4-[4-cyano-2- (trifluoromethyl)phenyl]-2-methylpiperazin-l-yl}sulfonyl)-2-thienyl]-3,3,3-trifluoro-2- hydroxypropanamide as a white solid (58 mg, 50% yield). Step 5E: To a solution of 2-[5-({(2i?)-4-[4-cyano-2-(trifluoromethyl)phenyl]-2- methylpiperazin-l-yl}sulfonyl)-2-thienyl]-3,3,3-trifluoro-2-hydroxypropanamide (200
mg, 0.36 mmol) in t-BuOH (2 niL) was added powdered KOH (100 mg, 1.8 mmol) at room temperature. The reaction mixture was heated to 8O0C for 15 min., after which the Reaction was determined to be complete by LC/MS. The reaction mixture was diluted with EtOAc, washed with H2O three times, then washed with sat. brine. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure to give 4-[(3i?)-4-{[5-(l-carbamoyl-2,2,2-trifluoro-l-hydroxyethyl)-2-thienyl]sulfonyl}-3- methylpiperazin-l-yl]-3-(trifluoromethyl)benzamide as a white solid (160 mg, 77.4% yield).
HRMS: calcd for C20H20F6N4O5S2 + H+, 575.08521; found (ESI, [M+H]+), 575.0852.
Example 5.2

2-[5-f{f2/?)-4-[4-cvano-2-ftrifluoromethyl)phenyll-2-methylpiperazin-l-yl}sulfonyl)- 2-thienyll-3,3i3-trifluoro-2-hvdroxypropanamide
The title compound was prepared using a procedure similar to that described in Example 5.1, steps 5A-5D (58 mg, 50% yield).
HRMS: calcd for C20Hi8F6N4O4S2 + H+, 557.07464; found (ESI, [M+H]+), 557.0749.
Example 6.1

3,3,3-trifluoro-2-hvdroxy-2-r5-({(2R)-2-methyl-4-r4-(3-methyl-l,2,4-oxadiazol-5-yl)- 2-ftrifluoromethyl)phenyllpiperazin-l-yl}sulfonyl)thiophen-2-yllpropanamide The title compound of Example 6.1 was prepared as shown in Scheme 6 below.
Detailed synthesis procedures are provided below.
Scheme 6

Step 6A: The starting material, 4-[(3i?)-3-methylpiperazin-l-yl]-3- (trifluoromethyl)benzonitrile, was prepared according to a procedure similar to that described in Example 5.1, step 5 A. To a stirred solution of 4-[(3i?)-3-methylpiperazin-l- yl]-3-(trifluoromethyl)benzonitrile (3.24 g, 12.03 mmol) and diisopropylethylamine (4.19 mL, 24.06 mmol) in anhydrous dichloromethane (30 mL) was added 5-bromo-thiophene- 2-sulfonyl chloride (3.15 g, 12.03 mmol) at O0C. The mixture was stirred at O0C for 30 min, then stirred at room temperature for o/n. The reaction mixture was washed with H2O and extracted with DCM. The organic layer was dried over Na2SO4 and concentrated. The crude product was purified via flash column chromatography, eluting with 30-50% EtOAc/Hexane to yield 4-{(3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3- methylpiperazin-l-yl}-3-(trifluoromethyl)benzonitrile in 74.5% yield (4.432 g) as a yellow solid.
Step 6B: To 4-{(3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3-methylpiperazin-l-yl}-3- (trifluoromethyl)benzonitrile (2.51 g, 5.08 mmol) was added a mixture of TFA/H2SO4 (15 mL, TFA//H2SO4 = 2/1, v/v) at room temperature. The reaction mixture was stirred at room temperature for over night and then carefully poured onto ice. The aqueous mixture was neutralized with 6 N NaOH solution until pH =5-6 was attained and then extracted with DCM (3x). The combined organic layers were washed with sat. brine,
dried over Na2SO4, and concentrated to give a tan solid. This solid was re-dissolved in DCM, then hexane was added to form a slurry. The slurry was filtered, washed with hexane, to give 4-{(3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3-methylpiperazin-l-yl}-3- (trifluoromethyl)benzamide as a tan color solid (2.20 g, 84.8%). Step 6C: To a 50 mL flask containing 4-{(3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3- methylpiperazin-l-yl}-3-(trifluoromethyl)benzamide was added N,N-dimethylacetamide dimethylacetal (6 mL, 37 mmol). The reaction mixture was stirred at 850C for 20 min., after which time the reaction was determined to be complete by LC/MS. The reaction mixture was cooled to room temperature, and the solvent was removed under reduced pressure. The crude product was purified via flash column chromatography, eluting with 1-3% MeOH/DCM to yield 4-{(3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3-methylpiperazin- l-yl}-Λ/-[(lZ)-l-(dimethylamino) ethylidene]-3-(trifluoromethyl)benzamide as a light yellow solid (1.36 g, 100% yield).
Step 6D: To a solution of hydroxy lamine hydrochloride (93.2 mg, 1.34 mmol) in a mixture of 6N NaOH solution (223 μL, 1.34 mmol) and 7 mL of 70% aqueous acetic acid was added 4-{(3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3-methylpiperazin-l-yl}-Λ/- [(lZ)-l-(dimethylamino) ethylidene]-3-(trifluoromethyl)benzamide (650 mg, 1.12 mmol). The reaction mixture was stirred at room temperature for 1.5 hrs., after which time the reaction was determined to be complete by LC/MS. The reaction mixture was diluted with H2O, which resulted in the formation of a slurry. Filtration of the slurry gave (2R)- 1 - [(5 -bromo-2-thienyl)sulfonyl]-2-methyl-4-[4-(3 -methyl- 1 ,2,4-oxadiazol-5-yl)-2- (trifluoromethyl)phenyl]piperazine as an off- white solid (532 mg, 86.1% yield).
Step 6E: To a solution of (2i?)-l-[(5-bromo-2-thienyl)sulfonyl]-2-methyl-4-[4-(3- methyl-l,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenyl]piperazine (500 mg, 0.91 mmol) in anhydrous THF (10 ml) was added n-butyllithium (0.36 ml, 2.5 M in hexane, 0.91 mmol) at -780C. The reaction mixture was stirred under N2 for 5 min. and then methyl 3,3,3-trifluoropyruvate (0.20 ml, 1.36 mmol) was added. Stirring at -780C was maintained for 1 hr, then the reaction mixture was warmed to room temperature and diluted with EtOAc. The EtOAc layer was washed with aq. NaHCO3 solution (3x) and dried over Na2SO4. Purification by chromatography (CombiFlash) with 10-80%
EtOAc/Hexane afforded methyl 3,3,3-trifluoro-2-hydroxy-2-[5-({(2i?)-2-methyl-4-[4-(3-
methyl-l,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenyl]piperazin-l- yl}sulfonyl)thiophen-2-yl]propanoate as a yellow solid (200 mg, 35% yield).
Step 6F: To a solution of 3,3,3-trifluoro-2-hydroxy-2-[5-({(2i?)-2-methyl-4-[4-(3- methyl-l,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenyl]piperazin-l- yl}sulfonyl)thiophen-2-yl]propanoate (200 mg, 0.31 mmol) in 20 mL of MeOH was bubbled NH3 (g) for 30 seconds at -780C. The reaction mixture was warmed up to room temperature and stirred under empty balloon for o/n., after which the reaction was determined to be complete by LC/MS. The solvent was evaporated, and the residue was washed with aq. NaHCO3 solution and extracted with EtOAc. The organic layer was dried over Na2SO4 and concentrated to yield 3,3,3-trifluoro-2-hydroxy-2-[5-({(2i?)-2- methyl-4-[4-(3 -methyl- 1 ,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenyl] piperazin- 1 - yl}sulfonyl)thiophen-2-yl]propanamide as a pale yellow powder (150 mg, 79% yield ).
HRMS: calcd for C22H2IF6N5O5S2 + H+, 614.09610; found (ESI, [M+H]+), 614.0957.

150 mg of 3,3,3-trifluoro-2-hydroxy-2-[5-({(2i?)-2-methyl-4-[4-(3-methyl-l,2,4- oxadiazol-5-yl)-2-(trifluoromethyl)phenyl]piperazin- 1 -yl} sulfonyl)thiophen-2- yl]propanamide (prepared according to a procedure similar to that described in Example 6.1, step 6A-6F) separated with a chiral column (Chiralpak ADH) in SFC Analytical Instrument. Mobile Phase was 90% CO2 /10%Methanol. Early fraction was collected to give the title compound (60 mg); late fraction was collected to give the diastereomer described in Example 6.3 (53 mg).
HRMS: calcd for C22H2IF6N5O5S2 + H+, 614.09610; found (ESI, [M+H]+), 614.0963.
Example 6.3

HRMS: calcd for C22H2IF6N5O5S2 + H+, 614.09610; found (ESI, [M+H]+), 614.0958.
Example 7.1

3,3,3-trifluoro-2-hvdroxy-2-r5-ααR)-2-methyl-4-r4-q,3,4-oxadiazol-2-yl)-2- ftrifluoromethyl)phenyllpiperazin-l-yl}sulfonyl)thiophen-2-yllpropanamide
The title compound of Example 7.1 was prepared as shown in Scheme 7 below. Detailed synthesis procedures are provided below.
Scheme 7

Step 7A: The starting material, 4-{(3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3- methylpiperazin-l-yl}-3-(trifluoromethyl)benzonitrile, was prepared according to a procedure similar to that described in Example 6.1, step 6 A.
To a solution of 4-{(3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3-methylpiperazin-l- yl}-3-(trifluoromethyl)benzonitrile (1.908 g, 3.86 mmol) in a mixture of MeOH (10 mL) and H2O (10 mL) was added 3N NaOH (7.72 mL, 23.16 mmol) at room temperature. The reaction mixture was heated to reflux for over night., after which time the reaction was determined to be complete by TLC. The reaction mixture was cooled to room temperature, acidified with 6 N HCl solution until pH =5, and extracted with EtOAc three times. The combined organic layers were washed with sat. brine and dried over Na2SO4. Removal of the solvent under reduced pressure afforded 4-{(3i?)-4-[(5-bromo-2- thienyl)sulfonyl]-3-methylpiperazin- 1 -yl} -3-(trifluoromethyl)benzoic acid as a yellow solid (1.737 g, 87.7% yield).
Step 7B: To a solution of 4-{(3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3- methylpiperazin-l-yl}-3-(trifluoromethyl)benzoic acid (700 mg, 1.36 mmol) in 7 mL of anhydrous 1 ,2-dichloroethane was added SOCl2 (0.6 mL, 8.18 mmol). The reaction mixture was stirred at room temperature for 2 hours, heated at reflux for 5hrs., and then cooled to room temperature and stirred o/n. The solvent was evaporated to afford a residue, which was taken up in 7 mL of DCM. The DCM mixture was cooled to O0C, and NH2NH2 (0.26 mL, 8.18 mmol) was added. Stirring was continued at O0C for 30 min., after which the reaction was determined to be complete by LC/MS. The solvent was
removed under reduced pressure. To the resulting residue was added H2O, and the aqueous mixture was extracted with DCM (3x). The combined organic layers were washed with sat. brine, dried over Na2SO4, concentrated, and purified via flash column chromatography, eluting with 2-4% MeOH/DCM to yield 4-{(3i?)-4-[(5-bromo-2- thienyl)sulfonyl]-3-methylpiperazin- 1 -yl} -3-(trifluoromethyl)benzohydrazide as a white solid (548 mg, 76% yield).
Step 7C: To a solution of 4-{(3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3- methylpiperazin-l-yl}-3-(trifluoromethyl)benzohydrazide (548 mg, 1.04 mmol) in 3 mL of EtOH were added triethylortho formate (5 mL) and a few mg of PSA. The reaction mixture was heated to 8O0C for 3 hrs., after which the reaction was determined to be complete by LC/MS. The reaction mixture was cooled to room temperature, and the solvent removed under reduced pressure. The crude product was purified via flash column chromatography, eluting with 30-50% EtOAc/Hexane to yield (2i?)-l-[(5-bromo- 2-thienyl)sulfonyl]-2-methyl-4-[4-(l,3,4-oxadiazol-2-yl)-2- (trifluoromethyl)phenyl]piperazine as a white solid (500 mg, 89.5% yield).
Step 7D: The penultimate compound in Scheme 7 was prepared according to a procedure similar to that described in Example 6.1, step 6E.
Step 7E: The final compound in Scheme 7 was prepared according to a procedure similar to that described in Example 6.1, step 6F (50 mg, 83.4% yield). HRMS: calcd for C2IHi9F6N5O5S2 + H+, 600.08045; found (ESI, [M+H]+),
600.0804.
Example 8.1

3,3,3-trifluoro-2-hvdroxy-2-r5-ααR)-2-methyl-4-r4-qH-tetrazol-5-yl)-2- (ϊrifluoromethyl)phenyllpiperazin-l-yl}sulfonyl)-2-thienyllpropanamide
The title compound of Example 8.1 was prepared as shown in Scheme 8 below. Detailed synthesis procedures are provided below.

Step 8A: The starting material, 2-[5-({(2i?)-4-[4-cyano-2-
(trifluoromethyl)phenyl]-2-methylpiperazin-l-yl}sulfonyl)-2-thienyl]-3,3,3-trifluoro-2- hydroxypropanamide, was prepared according to a procedure similar to that described in Example 5.1, steps 5 A to 5D.
In a microwave vial with 2-[5-({(2i?)-4-[4-cyano-2-(trifluoromethyl)phenyl]-2- methylpiperazin-l-yl}sulfonyl)-2-thienyl]-3,3,3-trifluoro-2-hydroxypropanamide (55 mg, 0.1 mmol) was added a mixture of isopropanol (0.3 mL) and H2O (0.1 mL). ZnBr2 (45 mg, 0.2 mmol) and NaN3 (33 mg, 0.5 mmol) were then added. The resultant mixture was heated to 8O0C in oil bath for o/n., after which time the reaction was determined to be complete as by LC/MS. The resultant mixture was cooled to room temperature and concentrated down to remove the isopropanol. The crude product was triturated with cold H2O to give 3,3,3-trifluoro-2-hydroxy-2-[5-({(2i?)-2-methyl-4-[4-(lH-tetrazol-5-yl)-2- (trifluoromethyl)phenyl]piperazin-l-yl}sulfonyl)-2-thienyl]propanamide as a white powder (68 mg, 100% yield).
ΗRMS: calcd for C20Hi9F6N7O4S2 + H+, 600.09169; found (ESI, [M+H]+), 600.0908.
Example 9.1

3,3,3-trifluoro-2-hvdroxy-2-r5-ααR)-2-methyl-4-r4-(4H-l,2,4-triazol-3-yl)-2- ftrifluoromethyl)phenyllpiperazin-l-yl}sulfonyl)-2-thienyllpropanamide
The title compound of Example 9.1 was prepared as shown in Scheme 9 below. Detailed synthesis procedures are provided below.

Step 9A: The starting material, 4-{(3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3- methylpiperazin-l-yl}-Λ/-[(lZ)-l-(dimethylamino) ethylidene]-3-
(trifluoromethyl)benzamide, was prepared according to a procedure similar to that described in Example 6.1, steps 6A to 6C.
To a solution of hydrazine (0.16 mL, 5.17 mmol) in 25 mL of acetic acid was added 4- {(3i?)-4-[(5-bromo-2-thienyl)sulfonyl]-3-methylpiperazin- 1 -yl} -N-[(IZ)- 1 - (dimethylamino) ethylidene]-3-(trifluoromethyl)benzamide (1.0 g, 1.72 mmol) at room temperature. The reaction mixture was then stirred at room temperature for 10 min., after which time the reaction was determined to be complete by LC/MS. The reaction mixture was concentrated to remove most of the HOAc. The resulting residue was poured into H2O, which resulted in formation of a solid. The solid was filtered, washed with H2O, and air dried under suction, to provide (2i?)-l-[(5-bromo-2-thienyl)sulfonyl]-2-methyl-4- [4-(5-methyl-4H-l,2,4-triazol-3-yl)-2-(trifluoromethyl)phenyl]piperazine as a white solid (1.0 g, 94.7% Yield).
Step 9B: The penultimate compound in Scheme 9 was prepared according to a procedure similar to that described in Example 6.1, step 6E. Step 9C: The final compound was prepared according to a procedure similar to that described in Example 6.1, step 6F (58 mg, 94.7% yield).
ΗRMS: calcd for C2IH20F6N6O4S2 + H+, 599.09644; found (ESI, [M+H]+), 599.0962.
Example 9.2

3,3,3-trifluoro-2-hvdroxy-2-r5-ααR)-2-methyl-4-r4-(5-methyl-4H-l,2,4-triazol-3-yl)- 2-ftrifluoromethyl)phenyllpiperazin-l-yl}sulfonyl)-2-thienyllpropanamide
The title compound was prepared using a procedure analogous to that described in Example 9.1, steps 9A-9C (60 mg, 100% yield), but using 4-{(3R)-4-[(5-bromo-2- thienyl)sulfonyl]-3-methylpiperazin- 1 -yl} -N-[(l E)- (dimethylamino)methylene]-3- (trifluoromethyl)benzamide instead of 4- {(3R)-4-[(5-bromo-2-thienyl)sulfonyl]-3- methylpiperazin- 1 -yl} -N-[( 1 Z)- 1 - (dimethylamino)ethylidene] -3 - (trifluoromethyl)benzamide .
ΗRMS: calcd for C22H22F6N6O4S2 + H+, 613.11209; found (ESI, [M+H]+), 613.1121.

2,2,2-trifluoro-l-r5-({(2R)-4-r4-fluoro-2-(trifluoromethyl)phenyll-2- methylpiperazin-l-yl}sulfonyl)-2-thienyll-l-(4H-l,2,4-triazol-3-yl)ethanol
The title compound of Example 10.1 was prepared as shown in Scheme 10 below. Detailed synthesis procedures are provided below.
Scheme 10

Step 1OA: The starting material, 3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2- (trifluoromethyl)phenyl)-2-methylpiperazin- 1 -ylsulfonyl)thiophen-2-yl)-2- hydroxypropanamide, was prepared according to a procedure similar to that described in Example 1.1, step IA to ID.
To a 50 mL flask containing 3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2- (trifluoromethyl)phenyl)-2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2- hydroxypropanamide (76.9 mg, 0.14 mmol) was added N,N-dimethylformamide dimethylacetal (1 mL). The reaction mixture was stirred at room temperature for 20 min., after which time the reaction was determined to be complete by LC/MS. The reaction mixture was concentrated to give N-dimethylaminomethylene-3,3,3-trifluoro-2-{5-[4-(4- fluoro-2-trifluoromethyl-phenyl)-2-methyl-piperazine- 1 -sulfonyl]-thiophen-2-yl} -2- hydroxy-propionamide as a crude oil (96 mg, 100% Yield), which was used in next step directly without purification.
Step 1OB: To a solution of hydrazine (8 uL, 0.28 mmol) in 3 mL of acetic acid was added N-dimethylaminomethylene-3,3,3-trifluoro-2-{5-[4-(4-fluoro-2- trifluoromethyl-phenyl)-2-methyl-piperazine- 1 -sulfonyl]-thiophen-2-yl} -2-hydroxy- propionamide (96 mg, 0.14 mmol) at room temperature. The reaction mixture was then stirred at room temperature for 1 hour., after which time the reaction was determined to be complete by LC/MS. The solvent was evaporated, and the resultant residue diluted with EtOAc. The EtO Ac-diluted residue was washed with aq. NaHCO3 solution and extracted with EtOAc (3x). The combined organic layers were dried over Na2SO4 and concentrated. The crude product was purified on SiO2 gel column, eluting with 0-8%
MeOH in DCM to give 2,2,2-trifluoro-l-[5-({(2i?)-4-[4-fluoro-2- (trifluoromethyl)phenyl]-2-methylpiperazin-l -yl} sulfonyl)-2-thienyl]- 1 -(AH- 1 ,2,4- triazol-3-yl)ethanol as a light yellow solid (60 mg, 75% Yield).
HRMS: calcd for C20Hi8F7N5O3S2 + H+, 574.08120; found (ESI, [M+H]+), 574.0808.
Example 11.1

2,2,2-trifluoro-l-[5-f{f2R)-4-[4-fluoro-2-ftrifluoromethyl)phenyll-2-methylpiperazin- l-yllsulfonyl)-2-thienyll-l-(5-methyl-4H-l,2,4-triazol-3-yl)ethanol
The title compound of Example 11.1 was prepared as shown in Scheme 11 below. Detailed synthesis procedures are provided below. Scheme 11

Step 1 IA: The starting material, 3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2- (trifluoromethyl)phenyl)-2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2- hydroxypropanamide, was prepared according to a procedure similar to that described in Example 1.1, step IA to ID.
To a solution of 3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2-(trifluoromethyl)phenyl)- 2-methylpiperazin-l-ylsulfonyl)thiophen-2-yl)-2-hydroxypropanamide (2 g, 3.6 mmol) in
DCM (40 niL) was added Et3N( 0.61 mL, 4.4 mmol). The reaction mixture was cooled to O0C and TBDPSCl (0.93 mL, 3.6 mmol) was added under N2. The reaction mixture was stirred at room temperature for 3 days., after which time the reaction was determined to be complete by LC/MS. The reaction mixture was washed with aq. NaHCO3 solution, and the organic layer was dried over Na2SO4 and concentrated. The crude product was purified on SiO2 gel column, eluting with 5-40% EtOAc in Hexane to give 2-(tert-butyl- diphenyl-silanyloxy)-3,3,3-trifluoro-2-{5-[4-(4-fluoro-2-trifluoromethyl-phenyl)-2- methyl-piperazine-l-sulfonyl]-thiophen-2-yl}-propionamide as a white solid (750 mg, 27% yield). Step 1 IB: To 2-(tert-butyl-diphenyl-silanyloxy)-3,3,3-trifiuoro-2-{5-[4-(4-fluoro-
2-trifluoromethyl-phenyl)-2-methyl-piperazine- 1 -sulfonyl]-thiophen-2-yl} -propionamide (500 mg, 0.6 mmol) was added 10 mL of DMA-DMA. The reaction mixture was stirred at room temperature for 30 min., after which time the reaction was determined to be complete by LC/MS. The reaction mixture was concentrated, and the resultant residue dissolved in minimum volume of HOAc. This HO Ac-diluted residue was then treated with a solution of hydrazine (0.05 mL, 1.5 mmol) in 2.5 mL of acetic acid and stirred at room temperature for 1 hr., after which time the reaction was determined to be complete by LC/MS. Addition of H2O resulted in the formation of a precipitate, which was filtered to yield 2,2,2-trifluoro-l-[5-({(2i?)-4-[4-fiuoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin-l-yl}sulfonyl)-2-thienyl]-l-(5-methyl-4H-l,2,4-triazol-3-yl)ethanol as a light yellow solid (70 mg, 20% yield).
The title compounds of Examples 12.1 and 12.2 were prepared as shown in Scheme 12 below. Detailed synthesis procedures are provided below.
Scheme 12

Example 12 .1

4-r(3R)-3-methyl-4-α5-r2,2,2-trifluoro-l-hvdroxy-l-(4H-l,2,4-triazol-3-yl)ethyll-2- thienyl}sulfonyl)piperazin-l-yll-3-ftrifluoromethyl)benzamide
Step 12A-12B: The starting material was prepared according to a procedure similar to that described in Example 5.1, steps 5 A to 5D.
4-[(3R)-3-methyl-4-( {5-[2,2,2-trifiuoro- 1 -hydroxy- 1 -(4H-1 ,2,4-triazol-3- yl)ethyl]-2-thienyl} sulfonyl)piperazin- 1 -yl]-3-(trifluoromethyl)benzonitrile was prepared according to a procedure similar to that described in Example 10.1, steps 1OA to 1OB.
Step 12C: The title compound was prepared according to a procedure similar to that described in Example 5.1, step 5E (80 mg, 72%yield).
ΗRMS: calcd for C2IH20F6N6O4S2 + H+, 599.09644; found (ESI, [M+H]+), 599.0964.
Example 12.2

4-r(3R)-3-methyl-4-α5-r2,2,2-trifluoro-l-hvdroxy-l-(4H-l,2,4-triazol-3-yl)ethyll-2- thienyl}sulfonyl)piperazin-l-yll-3-(ϊrifluoromethyl)benzonitrile
The title compound was prepared according to a procedure similar to that described in Example 12.1, steps 12A-12B (110 mg, 31%yield).
ΗRMS: calcd for C2IHi8F6N6O3S2 + H+, 581.08587; found (ESI, [M+H]+), 581.0857.
Example 12.3

4-r(3R)-3-methyl-4-α5-r2,2,2-trifluoro-l-hvdroxy-l-(5-methyl-4H-l,2,4-triazol-3- yl)ethyll-2-thienyl}sulfonyl)piperazin-l-yll-3-ftrifluoromethyl)benzonitrile
The title compound was prepared using a procedure analogous to that described in Example 12.1, steps 12A-12B (19 mg, 5%yield), but using N,N-dimethylacetamide dimethyl acetal instead of N,N-dimethylformamide dimethyl acetal.
Example 13.1

The title compound of Example 13.1 was prepared as shown in Scheme 13 below. Detailed synthesis procedures are provided below.
Scheme 13
NH2OHHCl, HOAc dioxane


Step 13 A: The starting material, 2-{5-[4-(4-Cyano-2-trifluoromethyl-phenyl)-2- methyl-piperazine-l-sulfonyl]-thiophen-2-yl}-N-dimethylaminomethylene-3,3,3- trifluoro-2-hydroxy-propionamide, was prepared according to a procedure similar to that described in Example 12.1, step 12A.
Step 13B: To a solution of hydroxylamine hydrochloride (40 mg, 0.61 mmol) in a mixture of 3N NaOH solution (100 μL, 0.61 mmol) and 6 mL of 70% aqueous acetic acid was added 2- {5-[4-(4-Cyano-2-trifluoromethyl-phenyl)-2-methyl-piperazine- 1 -sulfonyl]- thiophen-2-yl}-N-dimethylaminomethylene-3,3,3-trifluoro-2-hydroxy-propionamide (373.1 mg, 0.61 mmol). The reaction mixture was stirred at room temperature for 10 min., after which time the reaction was determined to be complete by LC/MS. The reaction mixture was then diluted with 3 mL Of H2O and extracted with DCM. The organic layer was washed with sat.brine, dried over Na2SO4, and concentrated. The crude product (without any further purification) was treated with a mixture of acetic acid (3 mL) and 1,4-dioxane (3 mL). The reaction mixture was stirred at 9O0C for 2 hours, after which time the reaction was determined to be complete by LC/MS. The reaction mixture was cooled to room temperature, diluted with H2O, and extracted with DCM (3x). The organic layer was washed with sat. brine, dried over Na2SO4, and concentrated. The crude product was purified on SiO2 gel column to give 4-[(3i?)-3-methyl-4-({5- [2,2,2-trifluoro- 1 -hydroxy- 1 -( 1 ,2,4-oxadiazol-5-yl)ethyl]-2-thienyl} sulfonyl)piperazin- 1 - yl]-3-(trifluoromethyl)benzonitrile_as a white solid (2 mg, 0.56% Yield).
HRMS: calcd for C2IHi7F6N5O4S2 + H+, 582.06989; found (ESI, [M+H]+), 582.0696.
Example 14.1

7V-{2,2,2-trifluoro-l-r5-ααR)-4-r4-fluoro-2-(trifluoromethyl)phenyll-2- methylpiperazin-l-yl}sulfonyl)-2-thienyll-l-methylethyl}acetamide
The title compound of Example 14.1 was prepared as shown in Scheme 14 below. Detailed synthesis procedures are provided below.
Scheme 14 H2O

Step 14A: The starting material, (R)-l-(5-bromothiophen-2-ylsulfonyl)-4-(4- fluoro-2-(trifluoromethyl)phenyl)-2-methylpiperazine, was prepared according to procedure similar to that described in Example 1.1, steps IA- IB.
To a solution of (R)-l-(5-bromothiophen-2-ylsulfonyl)-4-(4-fluoro-2- (trifluoromethyl)phenyl)-2-methylpiperazine (4.0 g, 8.23 mmol) in anhydrous THF (30 ml) was added n-Butyllithium (5.4 mL, 8.64 mmol, 1.6 M in Hexane) at -780C. The reaction mixture was stirred under N2 for 15 min. and then trifluoromethyl ketone (2.7 mL, 30 mmol) was added. Stirring at -780C was continued for 30 min. and then the reaction mixture was warmed to room temperature, quenched with a H2O, and extracted with DCM (3x). The combined organic layers were dried over MgSO4 and purified by chromatography (CombiFlash) with DCM/Hexane to give l,l,l-trifluoro-2-[5-({(2i?)-4- [4-fluoro-2-(trifluoromethyl)phenyl]-2-methylpiperazin-l-yl}sulfonyl)-2-thienyl]propan- 2-ol as a white solid (3.89 g, 91 % yield).
Step 14B: To a solution of l,l,l-trifluoro-2-[5-({(2i?)-4-[4-fluoro-2- (trifluoromethyl) phenyl] -2-methylpiperazin- 1 -yl} sulfonyl)-2-thienyl]propan-2-ol (100 mg, 0.192mmol) in MeCN (6 mL) was added sulfuric acid (0.4 rnL, 7.5 mmol) dropwise. The reaction mixture was stirred at room temperature o/n and then diluted with EtOAc, washed with H2O, and sat. brine. Purification by column chromatography (DCM/EtOAc) afforded N-{2,2,2-trifiuoro-l-[5-({(2i?)-4-[4-fiuoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin-l-yl}sulfonyl)-2-thienyl]-l-methylethyl}acetamide as light yellow solid (28 mg, 26% yield).
HRMS: calcd for C2IH22F7N3O3S2 + H+, 562.10635; found (ESI, [M+H]+), 562.1065.
The title compounds of Examples 15.1 - 15.6 were prepared as shown in Scheme 15 below. Detailed synthesis procedures are provided below.
Scheme 15

chiral column separation

Example 15.1

Step 15 A: The starting material, (3i?)-l-[4-fluoro-2-(trifluoromethyl)phenyl]-3- methylpiperazine, was prepared according to a procedure similar to that described in Example 1.1, step IA.
To a stirred solution of (3i?)-l-[4-fluoro-2-(trifluoromethyl)phenyl]-3- methylpiperazine (480 mg, 1.83 mmol) and diisopropylethylamine (0.64 mL, 3.66 mmol) in anhydrous dichloromethane (5 mL) was added 3-bromo benzenesulfonyl chloride (467.6 mg, 1.83 mmol). The reaction mixture was stirred at room temperature over night., after which time the reaction was determined to be complete as by TLC.
Purification of the reaction mixture via flash chromatography afforded (2i?)-l-[(3- bromophenyl)sulfonyl] -4- [4-fluoro-2-(trifluoromethyl)phenyl] -2-methylpiperazine in 59.4% yield (524 mg) as a light yellow solid.
Step 15B: To a solution of (2i?)-l-[(3-bromophenyl)sulfonyl]-4-[4-fluoro-2- (trifluoromethyl)phenyl]-2-methylpiperazine (1.0 g, 2.1 mmol) in anhydrous toluene (5 ml) was added Butyllithium (0.84 ml, 2.1 mmol) at -780C. Stirring was continued under N2 for 15 min. and then methyl 3,3,3-trifluoropyruvate (0.4 ml, 2.5 mmol) was added. The reaction mixture was stirred at -780C for 2 hours and then was quenched with a few mL of 10% aq. HCl and dried over MgSO4. Purification of the reaction mixture via chromatography (CombiFlash, 10-80% EtOAc/Hexane) afforded methyl 3,3,3-trifluoro- 2-[3-( {(2i?)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methylpiperazin- 1 - yl}sulfonyl)phenyl]-2-hydroxypropanoate as a colorless oil (250 mg, 21.3 % yield).
Step 15C: To a solution of 3,3,3-trifiuoro-2-[3-({(2i?)-4-[4-fiuoro-2- (trifluoromethyl) phenyl]-2-methylpiperazin- 1 -yl} sulfonyl)phenyl]-2-hydroxypropanoate (250 mg, 0.45 mmol) in 10 mL of MeOH was bubbled NH3 (g) for 30 seconds at -780C. The reaction mixture was warmed up room temperature and stirred under empty balloon for o/n., after which time the reaction was determined to be complete by LC/MS. The solvent was evaporated, and the resultant residue washed with aq. NaHCO3 solution and extracted with EtOAc. The organic layer was dried over Na2SO4 and concentrated to yield 3,3,3-trifiuoro-2-[3-({(2i?)-4-[4-fiuoro-2-(trifiuoromethyl) phenyl]-2-
methylpiperazin-l-yl}sulfonyl)phenyl]-2-hydroxypropanamide as a white solid (200 mg, 82% yield ).
HRMS: calcd for C2IH20F7N3O4S + H+, 544.11355; found (ESI, [M+H]+), 544.1137.
Example 15.2

HRMS: calcd for C2IH20F7N3O4S + H+, 544.11355; found (ESI, [M+H]+), 544.1136.
Example 15.3

The title compound was obtained as the late fraction using the separation method described in Example 15.2.
HRMS: calcd for C2IH20F7N3O4S + H+, 544.11355; found (ESI, [M+H]+), 544.1135.
Example 15.4

3,3i3-trifluoro-2-[4-f{f2/?)-4-[4-fluoro-2-ftrifluoromethyl)phenyll-2-methylpiperazin- l-yl}sulfonyl)phenyll-2-hvdroxypropanamide The title compound was prepared using a procedure analogous to that described in
Example 15.1, steps 15A-15C (200 mg, 82%yield), but using using 4-bromobenzene sulfonylchloride instead of 3-bromobenzene sulfonylchloride.
HRMS: calcd for C2IH20F7N3O4S + H+, 544.11355; found (ESI, [M+H]+), 544.1142.
Example 15.5

Example 15.6

The title compound was obtained as the late fraction using the separation method described in Example 15.5. HRMS: calcd for C2IH20F7N3O4S + H+, 544.11355; found (ESI, [M+H]+),
544.1137.
Example 17.1

The title compound of Example 17.1 was prepared as shown in Scheme 17 below. Detailed synthesis procedures are provided below. Scheme 17

Step 17A: To a solution of (2i?)-2-[2-bromo-5-( {(2i?)-4-[4-fluoro-2- (trifluoromethyl)phenyl]-2-methylpiperazin-l-yl}sulfonyl)-3-thienyl]-3,3,3-trifluoro-2- hydroxypropanamide (300 mg, 0.477 mmol) in MeOH (10 mL) was added palladium 10% dry on carbon powder (30 mg, 0.282 mmol) under N2. Then the reaction mixture was stirred under 1 atm H2 at room temperature for over night., after which time the reaction was determined to be complete by LC/MS. The reaction mixture was filtered through a pad of celite, rinsed with EtOAc, and concentrated under reduced pressure. The crude oil was purified via prep HPLC to yield (2i?)-3,3,3-trifluoro-2-[5-({(2i?)-4-[4- fluoro-2-(trifluoromethyl)phenyl]-2-methylpiperazin-l-yl}sulfonyl)thiophen-3-yl]-2- hydroxypropanamide in 71.7% yield (188 mg) as a white solid.
HRMS: calcd for Ci9Hi8F7N3O4S2 + H+, 550.06997; found (ESI, [M+H]+), 550.0694.
The title compound of Example 20.1 was prepared using a procedure analogous to that described in Example 1.1, steps IA - ID, but using l-bromo-2-chloro-4- fluorobenzene instead of 2-bromo-5-fluorobenzotrifluoride in step IA.
The title compound of Example 20.2 was prepared using a procedure analogous to that described in Example 1.1, steps IA - ID, but using l-bromo-4-fluoro-2- methylbenzene instead of 2-bromo-5-fluorobenzotrifluoride in step IA. The title compound of Example 20.3 was prepared using a procedure analogous to that described in Example 1.1, steps IA - ID, but using l-bromo-4-fluoro-2- methoxybenzene instead of 2-bromo-5-fluorobenzotrifluoride in step IA.
The title compound of Example 20.4 was prepared using a procedure analogous to that described in Example 1.1, steps IA - ID, but using l-bromo-2,4-difluorobenzene instead of 2-bromo-5-fluorobenzotrifluoride in step IA.
The title compound of Example 21.1 was prepared using a procedure analogous to that described in Example 1.1, steps IA - ID, but using methylamine instead OfNH3 in step ID.
The title compound of Example 21.2 was prepared using a procedure analogous to that described in Example 1.1, steps IA - ID, but using dimethylamine instead of NH3 in step ID.
Example 20.1

Example 20.2

3,3,3-trifluoro-2-(5-{[(2R)-4-(4-fluoro-2-methylphenyl)-2-methylpiperazin-l- yllsulfonyl}thiophen-2-yl)-2-hvdroxypropanamide
MS (LC-ESIMS) m/z 496.1; MS (LC-ESIMS) m/z 494.1.
Example 20.3

3,3,3-trifluoro-2-(5-{[(2R)-4-(4-fluoro-2-methoxyphenyl)-2-methylpiperazin-l- yllsulfonyl}thiophen-2-yl)-2-hvdroxypropanamide
MS (LC-ESIMS) m/z 512.1; MS (LC-ESIMS) m/z 510.
Example 20.4

3-,3i3-trifluoro-2-hvdroxypropanamide
MS (LC-ESIMS) m/z 500; MS (LC-ESIMS) m/z 498.1.
Example 21.1

3-,3.,3-trifluoro-2-[5-f{f2R)-4-[4-fluoro-2-ftrifluoromethyl)phenyll-2-methylpiperazin- l-yl}sulfonyl)thiophen-2-yll-2-hvdroxy-7V-methylpropanamide
MS (LC-ESIMS) m/z 563.3; MS (LC-ESIMS) m/z 561.5.
Example 21.2

3-,3.,3-trifluoro-2-[5-f{f2R)-4-[4-fluoro-2-ftrifluoromethyl)phenyll-2-methylpiperazin- l-yl}sulfonyl)thiophen-2-yll-2-hvdroxy-7VJV-dimethylpropanamide
HRMS: calcd for C2IH22F7N3O4S2 + H+, 578.10127; found (ESI, [M+H]+ Obs'd), 578.1017.
The chemical names and structures of the title compounds of Examples 1.1-21.2 are set forth in the table below.










Example 22
Determination of ICsn against human 1 Ib-HSDl Compounds described herein can be tested in a cell-based assay using a stable
Chinese Hamster Ovary (CHO) cell line expressing human 1 Ib-HSDl. An exemplary procedure is described below.
Cells are plated at 20,000 cells/well in 384 well plates and incubated overnight (12-16 hrs) at 37 °C/5% CO2. Cells are treated with different concentration of compound in 90 microliter serum-free media and incubated for 30 minutes at 37 °C/5%CO2. 10 microliter of 5 uM cortisone (final concentration 500 nM) is then added to the cells and the plate is incubated at 37 °C/5%CO2 for 120 minutes. 15 microliter of media is withdrawn and amount of Cortisol in the media is measured using the DiscoverX HitHunter Cortisol Assay (DiscoverX corp, CA), following manufacturer's instructions. Briefly, 15 microliter media is transferred to white 384 well assay plate. 5 microliter of anti-cortisol antibody and 10 microliter of ED complex is added to each well of the assay plate. The assay plate is incubated at room temperature for 60 minutes with gentle shaking. Galacton Star, Emerald Green, and chemiluminescence substrate buffer are mixed 1 :5:19. The mixture is then combined in equal parts with EA complex. 20 microliter of the mix is added to each well of the assay plate and the assay plate is incubated at room temperature for another 60 minutes. The plate is read in Envision multilabel plate reader (Perkin Elmer, MA) in the enhanced luminescence mode.
Background subtracted data is fitted to y = a + (b/(l+x / IC50)) and IC50 values are calculated using XLFit (IDBS, MA).
The IC50 values that were obtained for the title compounds of Examples 1.1 - 21.2 using the above-described assay are shown in Table 1.
Table 1


A number of embodiments of the invention have been described. Nevertheless, it will be understood that various modifications may be made without departing from the spirit and scope of the invention. Accordingly, other embodiments are within the scope of the following claims.
Claims
1. A chemical entity E, which is a compound of Formula I, or a pharmaceutically acceptable salt thereof:

R2 is halo, -CN, -C(O)-NH2, -C(O)-NH-(CL3 alkyl), -C(0)-N(d_3 alkyl)2, or 5- to 6-membered heteroaryl containing 1-4 ring heteroatoms independently selected from O, N, N(R2sl) and S, wherein R2sl is -H, -CH3 or ethyl, and wherein said heteroaryl is unsubstituted or substituted with one or two substituents R2s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3;
R3 is -H, halo, -CH3, ethyl, -OH or -OCH3; one of R4 and R5 is H, and the other is -CH3, -CH2X, -CHX2, -CF3 or ethyl;
A is phenyl, or 5- to 6-membered heteroaryl containing 1-3 ring heteroatoms independently selected from O, N, N(Ral) and S, and in which the atom bonded to the sulfonyl sulfur is carbon, wherein Ral is -H, -CH3 or ethyl;
R9 is -H, halo, -CH3, -CH2X, -CHX2, -CF3, ethyl, -OCH3 or -OCF3; provided that when A is 5- membered heteroaryl containing 3 ring heteroatoms, then R9 is absent; R6 is -CH3, ethyl, -OH or -OCH3;
R7 is -CH3, -CH2X, -CHX2, -CF3 or phenyl, wherein said phenyl is unsubstituted or substituted with one or two substituents R7sl independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3; R8 -C(O)-NH2, -C(O)-NH-(CL3 alkyl), -C(O)-N(CL3 alkyl)2, -NH-C(O)-(CL3 alkyl), -N(CL3 alkyl)-C(O)-(C1-3 alkyl), or 5- to 6-membered heteroaryl containing 1-4 ring heteroatoms independently selected from O, N, N(R8sl) and S, wherein R8sl is -H, -CH3 or ethyl, and wherein said heteroaryl is unsubstituted or substituted with one or two substituents R8s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3; and each instance of X is independently -F, -Cl or -Br; provided that when A is phenyl, then R6 and R7 cannot both be -CH3.
2. The chemical entity E of claim 1, wherein A is 5- to 6-membered heteroaryl containing 1-3 ring heteroatoms independently selected from O, N, N(Ral) and S, and in which the atom bonded to the sulfonyl sulfur is carbon.
3. The chemical entity E of claim 2, wherein A is a 5-membered heteroaryl, which has formula (A-I):

A1 is singly bonded to A4, and A3 is doubly bonded to A4; AMs O5 S, or NRal; and one of A2, A3,and A4 is C-C(R6)(R7)(R8), one of A2, A3 and A4 is C-R9, and one of A2, A3, and A4 is C-H or N; or (ii):
A1 is doubly bonded to A4, and A3 is singly bonded to A4;
A3 is O, S, or NRal; and one of A1, A2, and A4 is C-C(R6)(R7)(R8), one of A1, A2, and A4 is C-R9, and one of A1, A2, and A4 is N or C-H.
4. The chemical entity E of claim 3, wherein:
A1 is singly bonded to A4, and A3 is doubly bonded to A4; AMs O5 S5 Or NR"1; and one of A2, A3,and A4 is C-C(R6)(R7)(R8), one of A2, A3 and A4 is C-R9, and one of
A2, A3, and A4 is C-H or N.
5. The chemical entity E of claim 4, wherein A1 is S.
6. The chemical entity E of claim 5, wherein A4 is C-C(R6)(R7)(R8).
7. The chemical entity E of claim 6, wherein A2 is C-H or N.
8. The chemical entity E of claim 3, wherein: A1 is doubly bonded to A4, and A3 is singly bonded to A4;
A3 is O, S, or NRal; and one of A1, A2, and A4 is C-C(R6)(R7)(R8), one of A1, A2, and A4 is C-R9, and one of A1, A2, and A4 is N or C-H.
9. The chemical entity E of claim 8, wherein A3 is NRal .
10. The chemical entity E of claim 1, wherein
R6 is -OH, -OCH3, CH3 or ethyl; X is, independently, -F or -Cl; R1 is -CF3; R2 is halo; R3 is -H; R7 is -CF3, -CH3 or phenyl, wherein said phenyl is unsubstituted or substituted with one or two substituents R7sl independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3; R8 is -C(O)-NH2, -C(O)-NH-(CL3 alkyl), or -C(0)-N(d_3 alkyl)2, -NH-C(O)-(Ci-3 alkyl) or -N(Ci-3 alkyl)-C(O)-(Ci_3 alkyl), 5- to 6-membered heteroaryl containing 1-3 ring heteroatoms independently selected from O, N, N(R8sl) and S, wherein said heteroaryl is unsubstituted or substituted with one or two substituents R8s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3; R9 is -H or halo; one of R4 and R5 is H, and the other is -CH3; wherein the carbon atom attached to R4 and R5 has the ^-configuration.
11. The chemical entity E of claim 10, wherein R8 is l,2,4-triazol-3-yl or l,2,4-oxadiazol-3-yl, each of which is unsubstituted or substituted with one or two substituents R8s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3; R6 is -OH or -CH3; R4 is H, and R5 is -CH3; R2 is -F.
12. The chemical entity E of claim 1, wherein: R6 is -OH, -CH3, or ethyl; and
R7 is -CF3, -CH3, or phenyl, wherein said phenyl is unsubstituted or substituted with one or two substituents R7sl independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3.
13. The chemical entity E of claim 12, wherein R6 is -OH; R7 is -CF3;
A is a 5-membered heteroaryl, which has formula (A-I):

(i):
A1 is singly bonded to A4, and A3 is doubly bonded to A4; AMs O5 S, or NRal; and one of A2, A3,and A4 is C-C(R6)(R7)(R8), one of A2, A3 and A4 is C-R9, and one of A2, A3, and A4 is C-H or N; or
(ii): A1 is doubly bonded to A4, and A3 is singly bonded to A4;
A3 is O, S, or NRal; and one of A1, A2, and A4 is C-C(R6)(R7)(R8), one of A1, A2, and A4 is C-R9, and one of A1, A2, and A4 is N or C-H.
14. The chemical entity E of claim 13, wherein:
A1 is singly bonded to A4, and A3 is doubly bonded to A4;
A1 Is O, S, or NRal; and one of A2, A3,and A4 is C-C(R6)(R7)(R8), one of A2, A3 and A4 is C-R9, and one of A2, A3, and A4 is C-H or N.
15. The chemical entity E of claim 14, wherein A1 is S, A4 is C-C(R6)(R7)(R8), and A2 is C-H or N.
16. The chemical entity E of claim 12, wherein R8 is -C(O)-NH2, -C(O)-NH-(CH3), -C(O)-N(CHs)2, -NH-C(O)-(CH3), or 5-membered heteroaryl containing 1-3 ring heteroatoms independently selected from O, N, N(R8sl) and S, wherein said heteroaryl is unsubstituted or substituted with one or two substituents R8s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3; R9 is -H; one of R4 and R5 is H, and the other is -CH3; R1 is -CF3; R2 is -F; and R3 is -H.
17. The chemical entity E of claim 16, wherein R8 is l,2,4-triazol-3-yl or l,2,4-oxadiazol-3-yl, each of which is unsubstituted or substituted with one or two substituents R8s2 independently selected from halo, -CH3, ethyl, -CF3, -OH and -OCH3; R4 is H, and R5 is -CH3.
18. The chemical entity E of claim 1, wherein: R6 is -OH; and R7 is -CF3.
A is a 5-membered heteroaryl, which has formula (A-2):

A1 is singly bonded to A4, and A3 is doubly bonded to A4; AMs O5 S, or NRal; and one of A2, A3,and A4 is C-C(R6)(R7)(R8), one of A2, A3 and A4 is C-R9, and one of A2, A3, and A4 is C-H or N;
R8 is -C(O)-NH2; R9 is -H; R1 is -CF3; R2 is -F; R3 is -H.; R4 is H; and R5 is -CH3.
19. The chemical entity E of claim 18, wherein A1 is S, A4 is C-C(R6)(R7)(R8), and A2 is C-H or N.
20. The chemical entity E of claim 19, wherein A2 is C-H.
21. The chemical entity E of claim 1 , wherein the chemical entity E is selected from:
3,3,3-trifluoro-2-[5-({(2R)-4-[4-fiuoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin-l-yl}sulfonyl)-2-thienyl]-2-hydroxypropanamide;
(2R)-3,3,3-trifluoro-2-[5-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)thiophen-2-yl]-2-hydroxypropanamide;
(2S)-3,3,3-trifiuoro-2-[5-({(2R)-4-[4-fiuoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)thiophen-2-yl]-2-hydroxypropanamide;
(2R)-2-[2-bromo-5-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin-l-yl}sulfonyl)-3-thienyl]-3,3,3-trifluoro-2-hydroxypropanamide; 2-[5-( {(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methylpiperazin- 1 - yl}sulfonyl)thiophen-2-yl]-2-hydroxypropanamide;
2-[5-( {(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methylpiperazin- 1 - yl}sulfonyl)thiophen-2-yl]-2-hydroxy-2-phenylacetamide; 3,3,3-trifluoro-2-[2-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)- 1 ,3-thiazol-5-yl]-2-hydroxypropanamide;
(2R)-3,3,3-trifluoro-2-[2-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)- 1 ,3-thiazol-5-yl]-2-hydroxypropanamide;
(2S)-3,3,3-trifiuoro-2-[2-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)- 1 ,3-thiazol-5-yl]-2-hydroxypropanamide;
3,3,3-trifluoro-2-[3-({(2R)-4-[4-fiuoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)- 1 -methyl- IH-1 ,2,4-triazol-5-yl]-2-hydroxypropanamide;
(2R)-3,3,3-trifluoro-2-[4-({(2R)-4-[4-fluoro-2-(trifiuoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)- 1 -methyl- 1 H-imidazol-5 -yl] -2-hydroxypropanamide; (2R)-3,3,3-trifluoro-2-[4-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)- 1 -methyl- 1 H-imidazol-5 -yl] -2-hydroxypropanamide;
4-[(3R)-4-{[5-(l-carbamoyl-2,2,2-trifluoro-l-hydroxyethyl)-2-thienyl]sulfonyl}- 3 -methylpiperazin- 1 -yl] -3 -(trifluoromethyl)benzamide;
2-[5-( {(2R)-4-[4-cyano-2-(trifluoromethyl)phenyl]-2-methylpiperazin- 1 - yl}sulfonyl)-2-thienyl]-3,3,3-trifluoro-2-hydroxypropanamide;
3,3,3-trifluoro-2-hydroxy-2-[5-({(2R)-2-methyl-4-[4-(3-methyl-l,2,4-oxadiazol- 5-yl)-2-(trifluoromethyl)phenyl]piperazin- 1 -yl} sulfonyl)thiophen-2-yl]propanamide;
(2S)-3,3,3-trifluoro-2-hydroxy-2-[5-({(2R)-2-methyl-4-[4-(3-methyl-l,2,4- oxadiazol-5-yl)-2-(trifluoromethyl)phenyl]piperazin-l-yl}sulfonyl)-2- thienyl]propanamide;
(2R)-3,3,3-trifiuoro-2-hydroxy-2-[5-({(2R)-2-methyl-4-[4-(3-methyl-l,2,4- oxadiazol-5-yl)-2-(trifluoromethyl)phenyl]piperazin-l-yl}sulfonyl)-2- thienyl]propanamide;
3,3,3-trifluoro-2-hydroxy-2-[5-({(2R)-2-methyl-4-[4-(l,3,4-oxadiazol-2-yl)-2- (trifluoromethyl)phenyl]piperazin- 1 -yl} sulfonyl)thiophen-2-yl]propanamide; 3,3,3-trifluoro-2-hydroxy-2-[5-({(2R)-2-methyl-4-[4-(lH-tetrazol-5-yl)-2- (trifluoromethyl)phenyl]piperazin- 1 -yl} sulfonyl)-2-thienyl]propanamide;
3,3,3-trifluoro-2-hydroxy-2-[5-({(2R)-2-methyl-4-[4-(4H-l,2,4-triazol-3-yl)-2- (trifluoromethyl)phenyl]piperazin- 1 -yl} sulfonyl)-2-thienyl]propanamide; 3,3,3-trifluoro-2-hydroxy-2-[5-({(2R)-2-methyl-4-[4-(5-methyl-4H-l,2,4-triazol-
3-yl)-2-(trifluoromethyl)phenyl]piperazin- 1 -yl} sulfonyl)-2-thienyl]propanamide;
2,2,2-trifluoro-l-[5-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin-l-yl}sulfonyl)-2-thienyl]-l-(4H-l,2,4-triazol-3-yl)ethanol;
2,2,2-trifluoro-l-[5-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin-l-yl}sulfonyl)-2-thienyl]-l-(5-methyl-4H-l,2,4-triazol-3-yl)ethanol;
4-[(3R)-3-methyl-4-({5-[2,2,2-trifluoro-l-hydroxy-l-(4H-l,2,4-triazol-3- yl)ethyl]-2-thienyl}sulfonyl)piperazin-l-yl]-3-(trifluoromethyl)benzamide;
4-[(3R)-3-methyl-4-({5-[2,2,2-trifluoro-l-hydroxy-l-(4H-l,2,4-triazol-3- yl)ethyl]-2-thienyl} sulfonyl)piperazin- 1 -yl]-3-(trifluoromethyl)benzonitrile; 4-[(3R)-3-methyl-4-({5-[2,2,2-trifluoro-l-hydroxy-l-(5-methyl-4H-l,2,4-triazol-
3-yl)ethyl]-2-thienyl} sulfonyl)piperazin- 1 -yl]-3-(trifluoromethyl)benzonitrile;
4-[(3R)-3-methyl-4-({5-[2,2,2-trifluoro-l-hydroxy-l-(l,2,4-oxadiazol-5-yl)ethyl]- 2-thienyl} sulfonyl)piperazin- 1 -yl]-3-(trifluoromethyl)benzonitrile;
N-{2,2,2-trifluoro-l-[5-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)-2-thienyl]- 1 -methylethyl} acetamide;
3,3,3-trifluoro-2-[3-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)phenyl]-2-hydroxypropanamide;
(2S)-3,3,3-trifluoro-2-[3-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)phenyl]-2-hydroxypropanamide; (2R)-3,3,3-trifluoro-2-[3-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)phenyl]-2-hydroxypropanamide;
3,3,3-trifluoro-2-[4-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)phenyl]-2-hydroxypropanamide;
(2S)-3,3,3-trifluoro-2-[4-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)phenyl]-2-hydroxypropanamide; (2R)-3,3,3-trifluoro-2-[4-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)phenyl]-2-hydroxypropanamide;
(2R)-3,3,3-trifluoro-2-[5-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)thiophen-3-yl]-2-hydroxypropanamide; 2-(5-{[(2R)-4-(2-chloro-4-fluorophenyl)-2-methylpiperazin-l- yl]sulfonyl}thiophen-2-yl)-3,3,3-trifluoro-2-hydroxypropanamide;
3 ,3 ,3 -trifluoro-2-(5 - { [(2R)-4-(4-fluoro-2-methylphenyl)-2-methylpiperazin- 1 - yl] sulfonyl} thiophen-2-yl)-2-hydroxypropanamide;
3 ,3 ,3 -trifluoro-2-(5 - { [(2R)-4-(4-fluoro-2-methoxyphenyl)-2-methylpiperazin- 1 - yl] sulfonyl} thiophen-2-yl)-2-hydroxypropanamide;
2-(5-{[(2R)-4-(2,4-difluorophenyl)-2-methylpiperazin-l-yl]sulfonyl}thiophen-2- yl)-3,3,3-trifluoro-2-hydroxypropanamide;
3,3,3-trifluoro-2-[5-({(2R)-4-[4-fiuoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)thiophen-2-yl]-2-hydroxy-N-methylpropanamide; and 3,3,3-trifluoro-2-[5-({(2R)-4-[4-fiuoro-2-(trifluoromethyl)phenyl]-2- methylpiperazin- 1 -yl} sulfonyl)thiophen-2-yl]-2-hydroxy-N,N-dimethylpropanamide; or a pharmaceutically acceptable salt thereof.
22. The chemical entity E of claim 1, wherein the chemical entity E is (2R)- 3,3,3-trifluoro-2-[5-({(2R)-4-[4-fluoro-2-(trifluoromethyl)phenyl]-2-methylpiperazin-l- yl}sulfonyl)thiophen-2-yl]-2-hydroxypropanamide; or a pharmaceutically acceptable salt thereof.
23. A composition comprising a chemical entity E as claimed in claim 1 and a pharmaceutically acceptable carrier.
24. A method for treating a disease or condition mediated by excess or uncontrolled amounts of Cortisol and/or other corticosteroids, the method comprising administering to a subject in need thereof an effective amount of a chemical entity E as claimed in claim 1.
25. A method for treating type 2 diabetes, the method comprising administering to a subject in need thereof an effective amount of a chemical entity E as claimed in claim 1.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US18412009P | 2009-06-04 | 2009-06-04 | |
| US61/184,120 | 2009-06-04 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2010141550A2 true WO2010141550A2 (en) | 2010-12-09 |
| WO2010141550A3 WO2010141550A3 (en) | 2011-09-01 |
Family
ID=42790974
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/037024 WO2010141550A2 (en) | 2009-06-04 | 2010-06-02 | 11-beta hsd1 inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2010141550A2 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8524894B2 (en) | 2009-06-04 | 2013-09-03 | Laboratorios Salvat, S.A. | Inhibitor compounds of 11-beta-hydroxysteroid dehydrogenase type 1 |
| WO2016040449A1 (en) * | 2014-09-10 | 2016-03-17 | Raze Therapeutics, Inc. | 3-phosphoglycerate dehydrogenase inhibitors and uses thereof |
| WO2020048826A1 (en) | 2018-09-03 | 2020-03-12 | Bayer Aktiengesellschaft | 5-substituted 1-oxa-3,9-diazaspiro[5.5]undecan-2-one compounds |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007515490A (en) * | 2003-12-22 | 2007-06-14 | アムジェン インコーポレーティッド | Arylsulfonamide compounds and related uses |
| PA8713501A1 (en) * | 2006-02-07 | 2009-09-17 | Wyeth Corp | 11-BETA HYDROXIESTEROID DEHYDROGENASA INHIBITORS - 11ßHSD1 |
-
2010
- 2010-06-02 WO PCT/US2010/037024 patent/WO2010141550A2/en active Application Filing
Non-Patent Citations (3)
| Title |
|---|
| FREIREICH ET AL., CANCER CHEMOTHER. REP., vol. 50, 1966, pages 219 |
| NEGRIN ET AL., BIOMATERIALS, vol. 22, no. 6, 2001, pages 563 |
| XIANG, J. ET AL., JOURNAL MEDICINAL CHEMISTRY, vol. 51, no. 14, 2008, pages 4068 - 4071 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8524894B2 (en) | 2009-06-04 | 2013-09-03 | Laboratorios Salvat, S.A. | Inhibitor compounds of 11-beta-hydroxysteroid dehydrogenase type 1 |
| US8822452B2 (en) | 2009-06-04 | 2014-09-02 | Laboratorios Salvat, S.A. | Inhibitor compounds of 11-beta-hydroxysteroid dehydrogenase type 1 |
| WO2016040449A1 (en) * | 2014-09-10 | 2016-03-17 | Raze Therapeutics, Inc. | 3-phosphoglycerate dehydrogenase inhibitors and uses thereof |
| WO2020048826A1 (en) | 2018-09-03 | 2020-03-12 | Bayer Aktiengesellschaft | 5-substituted 1-oxa-3,9-diazaspiro[5.5]undecan-2-one compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2010141550A3 (en) | 2011-09-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5180099B2 (en) | Substituted imidazole derivatives, compositions and methods of use as PTPase inhibitors | |
| TWI404715B (en) | Biaryl ether urea compounds | |
| US20100311748A1 (en) | Heterocyclic amides useful for the treatment of cancer and psoriasis | |
| JP4894517B2 (en) | 2-Phenylpyridine derivative | |
| US11447453B2 (en) | Metabotropic glutamate receptor negative allosteric modulators (NAMs) and uses thereof | |
| TWI429642B (en) | Compounds useful for treating disorders associated with acyl coa-diacylglycerol acyl transferase 1 activity, pharmaceutical composition comprising the same and use thereof | |
| CN101365696A (en) | Compounds and compositions as modulators of steroid hormone nuclear receptors | |
| AU2008345681A1 (en) | Imidazo [1,2-a] pyridine compounds | |
| US20080004325A1 (en) | PTP1B inhibitors | |
| CN101600712A (en) | Novel oxadiazole derivative and thiadiazoles derivative with neovascularization inhibiting activity | |
| PT885212E (en) | QUINOXALINODIONAS | |
| JPH10195063A (en) | Ethynylthiazole derivative | |
| AU2008345687A1 (en) | Pyrazolo [1,5-a] pyrimidine compounds | |
| JP6574705B2 (en) | Novel dihydroquinolin-2-one derivatives as inhibitors of aldosterone synthase (CYP11B2 or CYP11B1) | |
| WO2009020683A2 (en) | Quinazoline compounds | |
| CA2072775A1 (en) | Substituted triazolinones | |
| WO2010141550A2 (en) | 11-beta hsd1 inhibitors | |
| JP6117948B2 (en) | Dihydroquinolin-2-one derivatives for use as aldosterone synthase inhibitors | |
| TW201422596A (en) | Novel oxazolidinone compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10740041 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10740041 Country of ref document: EP Kind code of ref document: A2 |