WO2010093419A1 - Novel amino azaheterocyclic carboxamides - Google Patents
Novel amino azaheterocyclic carboxamides Download PDFInfo
- Publication number
- WO2010093419A1 WO2010093419A1 PCT/US2010/000313 US2010000313W WO2010093419A1 WO 2010093419 A1 WO2010093419 A1 WO 2010093419A1 US 2010000313 W US2010000313 W US 2010000313W WO 2010093419 A1 WO2010093419 A1 WO 2010093419A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- quinazoline
- carboxylic acid
- acid amide
- phenyl
- amino
- Prior art date
Links
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 title claims abstract description 30
- 150000003857 carboxamides Chemical class 0.000 title description 7
- -1 carboxamide compounds Chemical class 0.000 claims abstract description 250
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 29
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 28
- 201000010099 disease Diseases 0.000 claims abstract description 25
- 201000011510 cancer Diseases 0.000 claims abstract description 22
- 230000003463 hyperproliferative effect Effects 0.000 claims abstract description 10
- 238000004519 manufacturing process Methods 0.000 claims abstract description 9
- 150000001875 compounds Chemical class 0.000 claims description 488
- 238000000034 method Methods 0.000 claims description 353
- 238000002360 preparation method Methods 0.000 claims description 155
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 claims description 53
- 150000003839 salts Chemical class 0.000 claims description 48
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 35
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 35
- 229910052757 nitrogen Inorganic materials 0.000 claims description 33
- 239000000651 prodrug Substances 0.000 claims description 32
- 229940002612 prodrug Drugs 0.000 claims description 32
- 239000012453 solvate Substances 0.000 claims description 31
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 24
- GYBHRTQLUQOYID-UHFFFAOYSA-N quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1 GYBHRTQLUQOYID-UHFFFAOYSA-N 0.000 claims description 22
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 claims description 16
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 15
- 229920006395 saturated elastomer Polymers 0.000 claims description 14
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 claims description 13
- 229910052727 yttrium Inorganic materials 0.000 claims description 13
- DXASQZJWWGZNSF-UHFFFAOYSA-N n,n-dimethylmethanamine;sulfur trioxide Chemical group CN(C)C.O=S(=O)=O DXASQZJWWGZNSF-UHFFFAOYSA-N 0.000 claims description 12
- 239000004480 active ingredient Substances 0.000 claims description 11
- 150000002148 esters Chemical class 0.000 claims description 11
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 11
- 239000008194 pharmaceutical composition Substances 0.000 claims description 11
- 125000006367 bivalent amino carbonyl group Chemical group [H]N([*:1])C([*:2])=O 0.000 claims description 9
- 239000003814 drug Substances 0.000 claims description 9
- 229910052760 oxygen Inorganic materials 0.000 claims description 9
- 230000008569 process Effects 0.000 claims description 9
- 125000004434 sulfur atom Chemical group 0.000 claims description 9
- 229910052801 chlorine Inorganic materials 0.000 claims description 8
- 229910052731 fluorine Inorganic materials 0.000 claims description 8
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 claims description 8
- 229910052794 bromium Inorganic materials 0.000 claims description 7
- 125000004432 carbon atom Chemical group C* 0.000 claims description 7
- 206010061218 Inflammation Diseases 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 6
- 230000004054 inflammatory process Effects 0.000 claims description 6
- 125000000217 alkyl group Chemical group 0.000 claims description 5
- 230000033115 angiogenesis Effects 0.000 claims description 5
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 5
- 206010012601 diabetes mellitus Diseases 0.000 claims description 5
- 210000002307 prostate Anatomy 0.000 claims description 5
- 125000004076 pyridyl group Chemical group 0.000 claims description 5
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 claims description 4
- 201000004624 Dermatitis Diseases 0.000 claims description 4
- 206010012689 Diabetic retinopathy Diseases 0.000 claims description 4
- 208000032612 Glial tumor Diseases 0.000 claims description 4
- 206010018338 Glioma Diseases 0.000 claims description 4
- 208000007766 Kaposi sarcoma Diseases 0.000 claims description 4
- 201000004681 Psoriasis Diseases 0.000 claims description 4
- 206010038933 Retinopathy of prematurity Diseases 0.000 claims description 4
- 206010064930 age-related macular degeneration Diseases 0.000 claims description 4
- 208000010668 atopic eczema Diseases 0.000 claims description 4
- 125000004744 butyloxycarbonyl group Chemical group 0.000 claims description 4
- 208000002780 macular degeneration Diseases 0.000 claims description 4
- 201000001441 melanoma Diseases 0.000 claims description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 4
- 125000003386 piperidinyl group Chemical group 0.000 claims description 4
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 4
- 208000017520 skin disease Diseases 0.000 claims description 4
- 125000000335 thiazolyl group Chemical group 0.000 claims description 4
- 230000005747 tumor angiogenesis Effects 0.000 claims description 4
- 230000004862 vasculogenesis Effects 0.000 claims description 4
- 150000001408 amides Chemical class 0.000 claims description 3
- 125000000623 heterocyclic group Chemical group 0.000 claims description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 3
- 125000001424 substituent group Chemical group 0.000 claims description 3
- HITVHYHCWXALKZ-MRXNPFEDSA-N 4-[[(1s)-2-(ethylamino)-1-phenylethyl]amino]quinazoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)CNCC)=CC=CC=C1 HITVHYHCWXALKZ-MRXNPFEDSA-N 0.000 claims description 2
- UFMCGKYLIYMLOY-CQSZACIVSA-N 4-[[(1s)-2-amino-1-phenylethyl]amino]quinazoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)CN)=CC=CC=C1 UFMCGKYLIYMLOY-CQSZACIVSA-N 0.000 claims description 2
- NOOJGWFQMPPHRX-UHFFFAOYSA-N 4-[[1-[3-[(2,4-difluorobenzoyl)amino]phenyl]-3-methoxypropyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CCOC)C(C=1)=CC=CC=1NC(=O)C1=CC=C(F)C=C1F NOOJGWFQMPPHRX-UHFFFAOYSA-N 0.000 claims description 2
- GYEINYAKLFRDRF-UHFFFAOYSA-N 4-[[1-[3-[(2-fluoro-4-methoxybenzoyl)amino]phenyl]-2-(methylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C(C=1)=CC=CC=1NC(=O)C1=CC=C(OC)C=C1F GYEINYAKLFRDRF-UHFFFAOYSA-N 0.000 claims description 2
- UVOCNINDPZNPIC-UHFFFAOYSA-N 6-methoxy-4-[[2-(methylamino)-1-phenylethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=C(OC)C=C2C=1NC(CNC)C1=CC=CC=C1 UVOCNINDPZNPIC-UHFFFAOYSA-N 0.000 claims description 2
- 125000000815 N-oxide group Chemical group 0.000 claims description 2
- 229910006074 SO2NH2 Inorganic materials 0.000 claims description 2
- 125000002947 alkylene group Chemical group 0.000 claims description 2
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 claims description 2
- 125000004429 atom Chemical group 0.000 claims description 2
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 claims description 2
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 claims description 2
- 229910052740 iodine Inorganic materials 0.000 claims description 2
- 125000002950 monocyclic group Chemical group 0.000 claims description 2
- 125000006413 ring segment Chemical group 0.000 claims description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 claims description 2
- 208000002193 Pain Diseases 0.000 claims 3
- 206010033645 Pancreatitis Diseases 0.000 claims 3
- 201000011066 hemangioma Diseases 0.000 claims 3
- 206010020718 hyperplasia Diseases 0.000 claims 3
- 208000017169 kidney disease Diseases 0.000 claims 3
- 125000000896 monocarboxylic acid group Chemical group 0.000 claims 3
- 208000037803 restenosis Diseases 0.000 claims 3
- SGZFLJKVODZGJM-UHFFFAOYSA-N 4-[[2-amino-1-(3,4-dimethoxyphenyl)ethyl]amino]quinazoline-8-carboxamide Chemical compound C1=C(OC)C(OC)=CC=C1C(CN)NC1=NC=NC2=C(C(N)=O)C=CC=C12 SGZFLJKVODZGJM-UHFFFAOYSA-N 0.000 claims 2
- PRHCRPUDJXUXCO-UHFFFAOYSA-N 4-[[2-amino-1-(4-methoxyphenyl)ethyl]amino]quinazoline-8-carboxamide Chemical compound C1=CC(OC)=CC=C1C(CN)NC1=NC=NC2=C(C(N)=O)C=CC=C12 PRHCRPUDJXUXCO-UHFFFAOYSA-N 0.000 claims 2
- QVHGKNYBLQTWEC-UHFFFAOYSA-N 4-[[2-amino-1-(4-methylphenyl)ethyl]amino]quinazoline-8-carboxamide Chemical compound C1=CC(C)=CC=C1C(CN)NC1=NC=NC2=C(C(N)=O)C=CC=C12 QVHGKNYBLQTWEC-UHFFFAOYSA-N 0.000 claims 2
- XMZNZUXMEXIQDA-UHFFFAOYSA-N 4-[(3,4-dichlorophenyl)methylamino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NCC1=CC=C(Cl)C(Cl)=C1 XMZNZUXMEXIQDA-UHFFFAOYSA-N 0.000 claims 1
- CKSGWNWLYXUENQ-UHFFFAOYSA-N 4-[(3,4-dimethylphenyl)methylamino]quinazoline-8-carboxamide Chemical compound C1=C(C)C(C)=CC=C1CNC1=NC=NC2=C(C(N)=O)C=CC=C12 CKSGWNWLYXUENQ-UHFFFAOYSA-N 0.000 claims 1
- YNPQIFHMRSVLHW-UHFFFAOYSA-N 4-[1-[3-(1,3-benzodioxole-5-carbonylamino)phenyl]ethylamino]quinazoline-8-carboxamide Chemical compound C1=C2OCOC2=CC(C(=O)NC=2C=CC=C(C=2)C(NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)C)=C1 YNPQIFHMRSVLHW-UHFFFAOYSA-N 0.000 claims 1
- BZNBBOLMYRXASK-UHFFFAOYSA-N 4-[1-[3-[(3-fluoro-4-methylbenzoyl)amino]phenyl]ethylamino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(C)C(C=1)=CC=CC=1NC(=O)C1=CC=C(C)C(F)=C1 BZNBBOLMYRXASK-UHFFFAOYSA-N 0.000 claims 1
- QBWMPYZCKOEWSU-UHFFFAOYSA-N 4-[1-[3-[(4-bromobenzoyl)amino]phenyl]ethylamino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(C)C(C=1)=CC=CC=1NC(=O)C1=CC=C(Br)C=C1 QBWMPYZCKOEWSU-UHFFFAOYSA-N 0.000 claims 1
- SICPNVSSMIOAEC-UHFFFAOYSA-N 4-[1-[3-[(4-fluoro-3-hydroxybenzoyl)amino]phenyl]ethylamino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(C)C(C=1)=CC=CC=1NC(=O)C1=CC=C(F)C(O)=C1 SICPNVSSMIOAEC-UHFFFAOYSA-N 0.000 claims 1
- POBKWGYBFDTZBY-UHFFFAOYSA-N 4-[1-[3-[(6-methoxypyridine-3-carbonyl)amino]phenyl]propylamino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CC)C(C=1)=CC=CC=1NC(=O)C1=CC=C(OC)N=C1 POBKWGYBFDTZBY-UHFFFAOYSA-N 0.000 claims 1
- YNPQIFHMRSVLHW-CQSZACIVSA-N 4-[[(1r)-1-[3-(1,3-benzodioxole-5-carbonylamino)phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound C1=C2OCOC2=CC(C(=O)NC=2C=CC=C(C=2)[C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)C)=C1 YNPQIFHMRSVLHW-CQSZACIVSA-N 0.000 claims 1
- VQTZMSIOEYJERT-ZVDHGWRTSA-N 4-[[(1r)-1-[3-[(2,2-difluorocyclopropanecarbonyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound C=1C([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)C)=CC=CC=1NC(=O)C1CC1(F)F VQTZMSIOEYJERT-ZVDHGWRTSA-N 0.000 claims 1
- NXUJCUODKLXIPE-CQSZACIVSA-N 4-[[(1r)-1-[3-[(2-methoxypyridine-4-carbonyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound C1=NC(OC)=CC(C(=O)NC=2C=C(C=CC=2)[C@@H](C)NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 NXUJCUODKLXIPE-CQSZACIVSA-N 0.000 claims 1
- KWBXLLQBOZJIAV-QGZVFWFLSA-N 4-[[(1r)-1-[3-[(3,4-dimethylbenzoyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound C=1C([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)C)=CC=CC=1NC(=O)C1=CC=C(C)C(C)=C1 KWBXLLQBOZJIAV-QGZVFWFLSA-N 0.000 claims 1
- CVCLGNCOASWGPB-OAHLLOKOSA-N 4-[[(1r)-1-[3-[(4-chloro-3-methylbenzoyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound C=1C([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)C)=CC=CC=1NC(=O)C1=CC=C(Cl)C(C)=C1 CVCLGNCOASWGPB-OAHLLOKOSA-N 0.000 claims 1
- ZSTHXAKLQBVNDX-CQSZACIVSA-N 4-[[(1r)-1-[3-[(5,6-dimethoxypyridine-3-carbonyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound N1=C(OC)C(OC)=CC(C(=O)NC=2C=C(C=CC=2)[C@@H](C)NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 ZSTHXAKLQBVNDX-CQSZACIVSA-N 0.000 claims 1
- SHFIJZVQMKWQNN-CYBMUJFWSA-N 4-[[(1r)-1-[3-[(5-chloro-6-methoxypyridine-3-carbonyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound C1=C(Cl)C(OC)=NC=C1C(=O)NC1=CC=CC([C@@H](C)NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 SHFIJZVQMKWQNN-CYBMUJFWSA-N 0.000 claims 1
- GLMIBJBCEWSYSZ-CQSZACIVSA-N 4-[[(1r)-1-[3-[(6-cyanopyridine-3-carbonyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound C=1C([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)C)=CC=CC=1NC(=O)C1=CC=C(C#N)N=C1 GLMIBJBCEWSYSZ-CQSZACIVSA-N 0.000 claims 1
- GTUAZZUFAUMXII-CQSZACIVSA-N 4-[[(1r)-1-[3-[(6-methoxypyridine-3-carbonyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound C1=NC(OC)=CC=C1C(=O)NC1=CC=CC([C@@H](C)NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 GTUAZZUFAUMXII-CQSZACIVSA-N 0.000 claims 1
- JMHMGIHHTONCMK-OAHLLOKOSA-N 4-[[(1r)-1-[3-[(6-methylpyridine-3-carbonyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound C=1C([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)C)=CC=CC=1NC(=O)C1=CC=C(C)N=C1 JMHMGIHHTONCMK-OAHLLOKOSA-N 0.000 claims 1
- RWBMPHFTXFAKIZ-CYBMUJFWSA-N 4-[[(1r)-1-[3-[[2-fluoro-4-(trifluoromethyl)benzoyl]amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound C=1C([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)C)=CC=CC=1NC(=O)C1=CC=C(C(F)(F)F)C=C1F RWBMPHFTXFAKIZ-CYBMUJFWSA-N 0.000 claims 1
- PELODSQJHQJMMD-CYBMUJFWSA-N 4-[[(1r)-1-[3-[[2-fluoro-5-(trifluoromethyl)benzoyl]amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound C=1C([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)C)=CC=CC=1NC(=O)C1=CC(C(F)(F)F)=CC=C1F PELODSQJHQJMMD-CYBMUJFWSA-N 0.000 claims 1
- KFUGVCOVSKGHMS-HNNXBMFYSA-N 4-[[(1r)-2-(methylamino)-1-phenylethyl]amino]quinazoline-8-carboxamide Chemical compound C1([C@@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)CNC)=CC=CC=C1 KFUGVCOVSKGHMS-HNNXBMFYSA-N 0.000 claims 1
- KTEVQEUEMLYYMP-MRXNPFEDSA-N 4-[[(1s)-1-(3-chlorophenyl)-2-(methylamino)ethyl]amino]-6-methoxyquinazoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC(OC)=CC(=C3N=CN=2)C(N)=O)CNC)=CC=CC(Cl)=C1 KTEVQEUEMLYYMP-MRXNPFEDSA-N 0.000 claims 1
- XQZHZCUJORPEOS-MRXNPFEDSA-N 4-[[(1s)-1-(3-fluorophenyl)-2-(methylamino)ethyl]amino]-6-methoxyquinazoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC(OC)=CC(=C3N=CN=2)C(N)=O)CNC)=CC=CC(F)=C1 XQZHZCUJORPEOS-MRXNPFEDSA-N 0.000 claims 1
- RBMOLIOJAWRTIV-OAHLLOKOSA-N 4-[[(1s)-1-(3-fluorophenyl)-2-(methylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)CNC)=CC=CC(F)=C1 RBMOLIOJAWRTIV-OAHLLOKOSA-N 0.000 claims 1
- BFLHCYRERXMTKZ-QGZVFWFLSA-N 4-[[(1s)-2-(dimethylamino)-1-(3-fluorophenyl)ethyl]amino]-6-methoxyquinazoline-8-carboxamide Chemical compound C1([C@@H](CN(C)C)NC2=NC=NC3=C(C(N)=O)C=C(C=C32)OC)=CC=CC(F)=C1 BFLHCYRERXMTKZ-QGZVFWFLSA-N 0.000 claims 1
- AAEAMMIUQZAASJ-MRXNPFEDSA-N 4-[[(1s)-2-(dimethylamino)-1-phenylethyl]amino]quinazoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)CN(C)C)=CC=CC=C1 AAEAMMIUQZAASJ-MRXNPFEDSA-N 0.000 claims 1
- GEECDZLHNUPFCU-MRXNPFEDSA-N 4-[[(1s)-2-(ethylamino)-1-(3-fluorophenyl)ethyl]amino]quinazoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)CNCC)=CC=CC(F)=C1 GEECDZLHNUPFCU-MRXNPFEDSA-N 0.000 claims 1
- XAVPWAIYBBCDNE-QGZVFWFLSA-N 4-[[(1s)-2-(ethylamino)-1-phenylethyl]amino]-6-methoxyquinazoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC(OC)=CC(=C3N=CN=2)C(N)=O)CNCC)=CC=CC=C1 XAVPWAIYBBCDNE-QGZVFWFLSA-N 0.000 claims 1
- MUTXAXOXPDZSGG-MRXNPFEDSA-N 4-[[(1s)-2-amino-1-phenylethyl]amino]quinoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC=CC(=C3N=CC=2)C(N)=O)CN)=CC=CC=C1 MUTXAXOXPDZSGG-MRXNPFEDSA-N 0.000 claims 1
- CHKCVVJJVFZWOP-KXCGHJCSSA-N 4-[[(3r,4e)-1-amino-4-ethenylhepta-4,6-dien-3-yl]amino]quinazoline-8-carboxamide Chemical compound C1=CC=C2C(N[C@H](CCN)C(\C=C)=C\C=C)=NC=NC2=C1C(N)=O CHKCVVJJVFZWOP-KXCGHJCSSA-N 0.000 claims 1
- AGDQZNITYJKXQO-UHFFFAOYSA-N 4-[[1-(3,4-dichlorophenyl)-2-(dimethylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C)C1=CC=C(Cl)C(Cl)=C1 AGDQZNITYJKXQO-UHFFFAOYSA-N 0.000 claims 1
- BYUUFRNHCFTPTI-UHFFFAOYSA-N 4-[[1-(3,4-dichlorophenyl)-2-(methylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C1=CC=C(Cl)C(Cl)=C1 BYUUFRNHCFTPTI-UHFFFAOYSA-N 0.000 claims 1
- QZMSENZZQWAYFQ-UHFFFAOYSA-N 4-[[1-(3-bromophenyl)-2-(dimethylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C)C1=CC=CC(Br)=C1 QZMSENZZQWAYFQ-UHFFFAOYSA-N 0.000 claims 1
- ODIBHBBABRKJIO-UHFFFAOYSA-N 4-[[1-(3-chlorophenyl)-2-(dimethylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C)C1=CC=CC(Cl)=C1 ODIBHBBABRKJIO-UHFFFAOYSA-N 0.000 claims 1
- KTEVQEUEMLYYMP-UHFFFAOYSA-N 4-[[1-(3-chlorophenyl)-2-(methylamino)ethyl]amino]-6-methoxyquinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=C(OC)C=C2C=1NC(CNC)C1=CC=CC(Cl)=C1 KTEVQEUEMLYYMP-UHFFFAOYSA-N 0.000 claims 1
- OPYPJZSJHHSMJL-UHFFFAOYSA-N 4-[[1-(3-chlorophenyl)-2-(methylamino)ethyl]amino]-6-phenylmethoxyquinazoline-8-carboxamide Chemical compound C=1C=CC(Cl)=CC=1C(CNC)NC(C1=C2)=NC=NC1=C(C(N)=O)C=C2OCC1=CC=CC=C1 OPYPJZSJHHSMJL-UHFFFAOYSA-N 0.000 claims 1
- LAJUPICIRDRINN-UHFFFAOYSA-N 4-[[1-(3-chlorophenyl)-2-(methylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C1=CC=CC(Cl)=C1 LAJUPICIRDRINN-UHFFFAOYSA-N 0.000 claims 1
- XQZHZCUJORPEOS-UHFFFAOYSA-N 4-[[1-(3-fluorophenyl)-2-(methylamino)ethyl]amino]-6-methoxyquinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=C(OC)C=C2C=1NC(CNC)C1=CC=CC(F)=C1 XQZHZCUJORPEOS-UHFFFAOYSA-N 0.000 claims 1
- RBMOLIOJAWRTIV-UHFFFAOYSA-N 4-[[1-(3-fluorophenyl)-2-(methylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C1=CC=CC(F)=C1 RBMOLIOJAWRTIV-UHFFFAOYSA-N 0.000 claims 1
- ZJKRQNYRWHKMJV-UHFFFAOYSA-N 4-[[1-(4-methoxyphenyl)-2-(methylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C1=CC=C(OC)C=C1 ZJKRQNYRWHKMJV-UHFFFAOYSA-N 0.000 claims 1
- MFRRIEBIBJQMAF-UHFFFAOYSA-N 4-[[1-[3-[(2,4-difluorobenzoyl)amino]phenyl]-3-(dimethylamino)propyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CCN(C)C)C(C=1)=CC=CC=1NC(=O)C1=CC=C(F)C=C1F MFRRIEBIBJQMAF-UHFFFAOYSA-N 0.000 claims 1
- JKUFJTIKVQADGJ-UHFFFAOYSA-N 4-[[1-[3-[(2,6-difluorobenzoyl)amino]phenyl]-2-(methylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C(C=1)=CC=CC=1NC(=O)C1=C(F)C=CC=C1F JKUFJTIKVQADGJ-UHFFFAOYSA-N 0.000 claims 1
- FFJNCCISXWZBSC-UHFFFAOYSA-N 4-[[1-[3-[(2,6-difluorobenzoyl)amino]phenyl]-3-methoxypropyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CCOC)C(C=1)=CC=CC=1NC(=O)C1=C(F)C=CC=C1F FFJNCCISXWZBSC-UHFFFAOYSA-N 0.000 claims 1
- NERGHRXOVVUXNP-UHFFFAOYSA-N 4-[[1-[3-[(2-chloropyridine-4-carbonyl)amino]phenyl]-2-(dimethylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C)C(C=1)=CC=CC=1NC(=O)C1=CC=NC(Cl)=C1 NERGHRXOVVUXNP-UHFFFAOYSA-N 0.000 claims 1
- FIZSPRJMYGKSMB-UHFFFAOYSA-N 4-[[1-[3-[(3,4-difluorobenzoyl)amino]phenyl]-2-(methylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C(C=1)=CC=CC=1NC(=O)C1=CC=C(F)C(F)=C1 FIZSPRJMYGKSMB-UHFFFAOYSA-N 0.000 claims 1
- YRQSEFHVBKRZFV-UHFFFAOYSA-N 4-[[1-[3-[(4-bromobenzoyl)amino]phenyl]-2-(dimethylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C)C(C=1)=CC=CC=1NC(=O)C1=CC=C(Br)C=C1 YRQSEFHVBKRZFV-UHFFFAOYSA-N 0.000 claims 1
- RHWHWWRJINHZQN-UHFFFAOYSA-N 4-[[1-[3-[(4-bromobenzoyl)amino]phenyl]-2-(methylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C(C=1)=CC=CC=1NC(=O)C1=CC=C(Br)C=C1 RHWHWWRJINHZQN-UHFFFAOYSA-N 0.000 claims 1
- YGQJBWJOHRVWHU-UHFFFAOYSA-N 4-[[1-[3-[(4-bromobenzoyl)amino]phenyl]-3-(dimethylamino)propyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CCN(C)C)C(C=1)=CC=CC=1NC(=O)C1=CC=C(Br)C=C1 YGQJBWJOHRVWHU-UHFFFAOYSA-N 0.000 claims 1
- CNFJGGPOIJUVKL-UHFFFAOYSA-N 4-[[1-[3-[(4-methoxybenzoyl)amino]phenyl]-2-(methylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C(C=1)=CC=CC=1NC(=O)C1=CC=C(OC)C=C1 CNFJGGPOIJUVKL-UHFFFAOYSA-N 0.000 claims 1
- JHGDIFKUZFGJGH-UHFFFAOYSA-N 4-[[1-[3-[[2-[3-(diethylamino)pyrrolidin-1-yl]pyridine-4-carbonyl]amino]phenyl]-2-(dimethylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound C1C(N(CC)CC)CCN1C1=CC(C(=O)NC=2C=C(C=CC=2)C(CN(C)C)NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=CC=N1 JHGDIFKUZFGJGH-UHFFFAOYSA-N 0.000 claims 1
- GWUYLDMVDQHMJN-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-(3-fluorophenyl)ethyl]amino]-6-ethoxyquinazoline-8-carboxamide Chemical compound C12=CC(OCC)=CC(C(N)=O)=C2N=CN=C1NC(CN(C)C)C1=CC=CC(F)=C1 GWUYLDMVDQHMJN-UHFFFAOYSA-N 0.000 claims 1
- ALLGNFRRMBCRHD-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-(3-methoxyphenyl)ethyl]amino]quinazoline-8-carboxamide Chemical compound COC1=CC=CC(C(CN(C)C)NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 ALLGNFRRMBCRHD-UHFFFAOYSA-N 0.000 claims 1
- PPACVGVOVMKIML-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-(3-nitrophenyl)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C)C1=CC=CC([N+]([O-])=O)=C1 PPACVGVOVMKIML-UHFFFAOYSA-N 0.000 claims 1
- POQNJRCZSQACTE-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-[3-[(2-piperidin-1-ylpyridine-4-carbonyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C)C(C=1)=CC=CC=1NC(=O)C(C=1)=CC=NC=1N1CCCCC1 POQNJRCZSQACTE-UHFFFAOYSA-N 0.000 claims 1
- VWYGEDAFWDXUMQ-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-[3-[(2-pyrrolidin-1-ylpyridine-4-carbonyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C)C(C=1)=CC=CC=1NC(=O)C(C=1)=CC=NC=1N1CCCC1 VWYGEDAFWDXUMQ-UHFFFAOYSA-N 0.000 claims 1
- LYOMZOFITVVBNX-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-[3-[(3-fluoro-4-methoxybenzoyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound C1=C(F)C(OC)=CC=C1C(=O)NC1=CC=CC(C(CN(C)C)NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 LYOMZOFITVVBNX-UHFFFAOYSA-N 0.000 claims 1
- IUEUJHZZCUYXKU-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-[3-[(4-methoxybenzoyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound C1=CC(OC)=CC=C1C(=O)NC1=CC=CC(C(CN(C)C)NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 IUEUJHZZCUYXKU-UHFFFAOYSA-N 0.000 claims 1
- DCRRXSUQGXVDLQ-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-[3-[(5-pyrrolidin-1-ylpyridine-3-carbonyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C)C(C=1)=CC=CC=1NC(=O)C(C=1)=CN=CC=1N1CCCC1 DCRRXSUQGXVDLQ-UHFFFAOYSA-N 0.000 claims 1
- IVOSBLQIUULOJZ-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-[3-[[2-(2-methylpyrrolidin-1-yl)pyridine-4-carbonyl]amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound CC1CCCN1C1=CC(C(=O)NC=2C=C(C=CC=2)C(CN(C)C)NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=CC=N1 IVOSBLQIUULOJZ-UHFFFAOYSA-N 0.000 claims 1
- YFTUDCDZHZYZAB-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-[3-[[2-(dimethylamino)pyridine-4-carbonyl]amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C)C(C=1)=CC=CC=1NC(=O)C1=CC=NC(N(C)C)=C1 YFTUDCDZHZYZAB-UHFFFAOYSA-N 0.000 claims 1
- DHEDUDJQNPBXHX-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-[3-[[2-fluoro-4-(trifluoromethyl)benzoyl]amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C)C(C=1)=CC=CC=1NC(=O)C1=CC=C(C(F)(F)F)C=C1F DHEDUDJQNPBXHX-UHFFFAOYSA-N 0.000 claims 1
- RXAZPQCDGXHCAO-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-[3-[[4-(trifluoromethyl)benzoyl]amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C)C(C=1)=CC=CC=1NC(=O)C1=CC=C(C(F)(F)F)C=C1 RXAZPQCDGXHCAO-UHFFFAOYSA-N 0.000 claims 1
- ZJDZJPZEQDNRHD-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-[4-(trifluoromethyl)phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C)C1=CC=C(C(F)(F)F)C=C1 ZJDZJPZEQDNRHD-UHFFFAOYSA-N 0.000 claims 1
- ZHHSRQFICBOYCE-UHFFFAOYSA-N 4-[[2-(methylamino)-1-[3-[[4-(trifluoromethoxy)benzoyl]amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C(C=1)=CC=CC=1NC(=O)C1=CC=C(OC(F)(F)F)C=C1 ZHHSRQFICBOYCE-UHFFFAOYSA-N 0.000 claims 1
- JNZCFUTUPRDOSS-UHFFFAOYSA-N 4-[[2-(methylamino)-1-[3-[[4-(trifluoromethyl)benzoyl]amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C(C=1)=CC=CC=1NC(=O)C1=CC=C(C(F)(F)F)C=C1 JNZCFUTUPRDOSS-UHFFFAOYSA-N 0.000 claims 1
- KFUGVCOVSKGHMS-UHFFFAOYSA-N 4-[[2-(methylamino)-1-phenylethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C1=CC=CC=C1 KFUGVCOVSKGHMS-UHFFFAOYSA-N 0.000 claims 1
- UALHJBCHHRMERK-UHFFFAOYSA-N 4-[[2-amino-1-(3,4-dichlorophenyl)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN)C1=CC=C(Cl)C(Cl)=C1 UALHJBCHHRMERK-UHFFFAOYSA-N 0.000 claims 1
- IKONXWFEIGCAGN-UHFFFAOYSA-N 4-[[2-amino-1-(3-fluorophenyl)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN)C1=CC=CC(F)=C1 IKONXWFEIGCAGN-UHFFFAOYSA-N 0.000 claims 1
- YCUXZFDOLJQXOU-UHFFFAOYSA-N 4-[[2-amino-1-(3-methoxyphenyl)ethyl]amino]quinazoline-8-carboxamide Chemical compound COC1=CC=CC(C(CN)NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 YCUXZFDOLJQXOU-UHFFFAOYSA-N 0.000 claims 1
- UUKGDBLVFLHJLM-UHFFFAOYSA-N 4-[[2-amino-1-[3-[(4-fluorobenzoyl)amino]phenyl]ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN)C(C=1)=CC=CC=1NC(=O)C1=CC=C(F)C=C1 UUKGDBLVFLHJLM-UHFFFAOYSA-N 0.000 claims 1
- BMYPGRZADREWCU-UHFFFAOYSA-N 4-[[3-(dimethylamino)-1-[3-[[4-(trifluoromethyl)benzoyl]amino]phenyl]propyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CCN(C)C)C(C=1)=CC=CC=1NC(=O)C1=CC=C(C(F)(F)F)C=C1 BMYPGRZADREWCU-UHFFFAOYSA-N 0.000 claims 1
- LJMKRAKDJVPMIO-UHFFFAOYSA-N 4-[[3-[(2,4-dichlorobenzoyl)amino]phenyl]methylamino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NCC(C=1)=CC=CC=1NC(=O)C1=CC=C(Cl)C=C1Cl LJMKRAKDJVPMIO-UHFFFAOYSA-N 0.000 claims 1
- DHNWCWGJFSTRBB-UHFFFAOYSA-N 4-[[3-[(2,4-difluorobenzoyl)amino]phenyl]methylamino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NCC(C=1)=CC=CC=1NC(=O)C1=CC=C(F)C=C1F DHNWCWGJFSTRBB-UHFFFAOYSA-N 0.000 claims 1
- SKTCDUXPKLCJIP-UHFFFAOYSA-N 4-[[3-[(5-pyrrolidin-1-ylpyridine-3-carbonyl)amino]phenyl]methylamino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NCC(C=1)=CC=CC=1NC(=O)C(C=1)=CN=CC=1N1CCCC1 SKTCDUXPKLCJIP-UHFFFAOYSA-N 0.000 claims 1
- FTLGJGCPUAMLIH-UHFFFAOYSA-N 4-[[3-[[2-[2-hydroxyethyl(methyl)amino]pyridine-4-carbonyl]amino]phenyl]methylamino]quinazoline-8-carboxamide Chemical compound C1=NC(N(CCO)C)=CC(C(=O)NC=2C=C(CNC=3C4=CC=CC(=C4N=CN=3)C(N)=O)C=CC=2)=C1 FTLGJGCPUAMLIH-UHFFFAOYSA-N 0.000 claims 1
- YZSOMODRJHNWOR-UHFFFAOYSA-N 4-[[4-(trifluoromethyl)phenyl]methylamino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NCC1=CC=C(C(F)(F)F)C=C1 YZSOMODRJHNWOR-UHFFFAOYSA-N 0.000 claims 1
- BXSXPCFESWIUHR-UHFFFAOYSA-N 4-[[4-chloro-3-(trifluoromethyl)phenyl]methylamino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NCC1=CC=C(Cl)C(C(F)(F)F)=C1 BXSXPCFESWIUHR-UHFFFAOYSA-N 0.000 claims 1
- MDBLXTCBPCQULU-UHFFFAOYSA-N 6-(cyclopropylmethoxy)-4-[[2-(dimethylamino)-1-(3-fluorophenyl)ethyl]amino]quinazoline-8-carboxamide Chemical compound C=1C=CC(F)=CC=1C(CN(C)C)NC(C1=C2)=NC=NC1=C(C(N)=O)C=C2OCC1CC1 MDBLXTCBPCQULU-UHFFFAOYSA-N 0.000 claims 1
- NKFAPWMJVXBXHU-OAHLLOKOSA-N 6-chloro-4-[[(1s)-1-(3-fluorophenyl)-2-(methylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC(Cl)=CC(=C3N=CN=2)C(N)=O)CNC)=CC=CC(F)=C1 NKFAPWMJVXBXHU-OAHLLOKOSA-N 0.000 claims 1
- UVOCNINDPZNPIC-MRXNPFEDSA-N 6-methoxy-4-[[(1s)-2-(methylamino)-1-phenylethyl]amino]quinazoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC(OC)=CC(=C3N=CN=2)C(N)=O)CNC)=CC=CC=C1 UVOCNINDPZNPIC-MRXNPFEDSA-N 0.000 claims 1
- FTCVWEWECUNCTD-UHFFFAOYSA-N 6-methoxy-4-[[4-(trifluoromethyl)phenyl]methylamino]quinazoline-8-carboxamide Chemical compound C12=CC(OC)=CC(C(N)=O)=C2N=CN=C1NCC1=CC=C(C(F)(F)F)C=C1 FTCVWEWECUNCTD-UHFFFAOYSA-N 0.000 claims 1
- 125000000565 sulfonamide group Chemical group 0.000 claims 1
- DLYKSKLOFRPDPD-UHFFFAOYSA-N tert-butyl n-[2-(3-benzamidophenyl)-2-[(8-carbamoylquinazolin-4-yl)amino]ethyl]-n-methylcarbamate Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C(=O)OC(C)(C)C)C(C=1)=CC=CC=1NC(=O)C1=CC=CC=C1 DLYKSKLOFRPDPD-UHFFFAOYSA-N 0.000 claims 1
- UTOWQUDTUIJADK-UHFFFAOYSA-N tert-butyl n-[2-[(8-carbamoylquinazolin-4-yl)amino]-2-(3-nitrophenyl)ethyl]-n-methylcarbamate Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CN(C)C(=O)OC(C)(C)C)C1=CC=CC([N+]([O-])=O)=C1 UTOWQUDTUIJADK-UHFFFAOYSA-N 0.000 claims 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 273
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 269
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 241
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 216
- 239000007787 solid Substances 0.000 description 211
- 239000000243 solution Substances 0.000 description 154
- 239000000047 product Substances 0.000 description 153
- 239000000203 mixture Substances 0.000 description 134
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 130
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 120
- 235000019439 ethyl acetate Nutrition 0.000 description 107
- 238000006243 chemical reaction Methods 0.000 description 104
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 102
- 238000005160 1H NMR spectroscopy Methods 0.000 description 80
- 239000011541 reaction mixture Substances 0.000 description 71
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 69
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 68
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 68
- 238000003756 stirring Methods 0.000 description 66
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 60
- 239000002244 precipitate Substances 0.000 description 48
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 45
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 44
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical class CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 42
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 42
- 229910052938 sodium sulfate Inorganic materials 0.000 description 42
- XMIIGOLPHOKFCH-UHFFFAOYSA-N 3-phenylpropionic acid Chemical compound OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 41
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 41
- 239000002904 solvent Substances 0.000 description 40
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 39
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 36
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 34
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 32
- 239000000463 material Substances 0.000 description 31
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 30
- 239000007832 Na2SO4 Substances 0.000 description 30
- 238000004128 high performance liquid chromatography Methods 0.000 description 28
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 26
- 238000002953 preparative HPLC Methods 0.000 description 24
- 238000000746 purification Methods 0.000 description 24
- 230000015572 biosynthetic process Effects 0.000 description 23
- 239000003921 oil Substances 0.000 description 23
- 235000019198 oils Nutrition 0.000 description 23
- 239000000758 substrate Substances 0.000 description 23
- 238000003786 synthesis reaction Methods 0.000 description 23
- 230000000694 effects Effects 0.000 description 22
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 22
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 21
- 239000012267 brine Substances 0.000 description 21
- 230000002829 reductive effect Effects 0.000 description 21
- 239000000725 suspension Substances 0.000 description 21
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 20
- 239000000543 intermediate Substances 0.000 description 20
- 239000012071 phase Substances 0.000 description 19
- 102000001253 Protein Kinase Human genes 0.000 description 18
- 238000004440 column chromatography Methods 0.000 description 18
- 239000003112 inhibitor Substances 0.000 description 18
- 108060006633 protein kinase Proteins 0.000 description 18
- 108091000080 Phosphotransferase Proteins 0.000 description 17
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 17
- 210000004027 cell Anatomy 0.000 description 17
- 238000001914 filtration Methods 0.000 description 17
- 102000020233 phosphotransferase Human genes 0.000 description 17
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 15
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 15
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 15
- 241000124008 Mammalia Species 0.000 description 14
- 239000012043 crude product Substances 0.000 description 14
- 229910052739 hydrogen Inorganic materials 0.000 description 14
- CAZALFALOKHBET-UHFFFAOYSA-N methyl 4-chloroquinazoline-8-carboxylate Chemical compound N1=CN=C2C(C(=O)OC)=CC=CC2=C1Cl CAZALFALOKHBET-UHFFFAOYSA-N 0.000 description 14
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 14
- MEMUNXSUKDSSRN-UHFFFAOYSA-N 4-[(3-aminophenyl)methylamino]quinazoline-8-carboxamide;hydrochloride Chemical compound Cl.N1=CN=C2C(C(=O)N)=CC=CC2=C1NCC1=CC=CC(N)=C1 MEMUNXSUKDSSRN-UHFFFAOYSA-N 0.000 description 13
- 238000001816 cooling Methods 0.000 description 13
- 239000000706 filtrate Substances 0.000 description 13
- 102000004190 Enzymes Human genes 0.000 description 12
- 108090000790 Enzymes Proteins 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 12
- 235000011152 sodium sulphate Nutrition 0.000 description 12
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 11
- 102000003989 Aurora kinases Human genes 0.000 description 11
- 108090000433 Aurora kinases Proteins 0.000 description 11
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 11
- 230000005764 inhibitory process Effects 0.000 description 11
- 239000012299 nitrogen atmosphere Substances 0.000 description 11
- 239000012044 organic layer Substances 0.000 description 11
- 230000026731 phosphorylation Effects 0.000 description 11
- 238000006366 phosphorylation reaction Methods 0.000 description 11
- 108090000765 processed proteins & peptides Proteins 0.000 description 11
- ZEYHEAKUIGZSGI-UHFFFAOYSA-N 4-methoxybenzoic acid Chemical compound COC1=CC=C(C(O)=O)C=C1 ZEYHEAKUIGZSGI-UHFFFAOYSA-N 0.000 description 10
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 10
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 10
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 10
- 108091007960 PI3Ks Proteins 0.000 description 10
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 10
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 10
- 108091008611 Protein Kinase B Proteins 0.000 description 10
- 239000002253 acid Substances 0.000 description 10
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 10
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 10
- 239000000741 silica gel Substances 0.000 description 10
- 229910002027 silica gel Inorganic materials 0.000 description 10
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- 239000005441 aurora Substances 0.000 description 9
- 230000010261 cell growth Effects 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- 230000002159 abnormal effect Effects 0.000 description 8
- 239000000908 ammonium hydroxide Substances 0.000 description 8
- 229910000024 caesium carbonate Inorganic materials 0.000 description 8
- 239000000460 chlorine Substances 0.000 description 8
- 238000004587 chromatography analysis Methods 0.000 description 8
- 238000003818 flash chromatography Methods 0.000 description 8
- 230000006870 function Effects 0.000 description 8
- 230000002401 inhibitory effect Effects 0.000 description 8
- 239000010410 layer Substances 0.000 description 8
- FOIQBHSPCYEVGH-UHFFFAOYSA-N methyl 4-oxo-1h-quinazoline-8-carboxylate Chemical compound N1C=NC(=O)C2=C1C(C(=O)OC)=CC=C2 FOIQBHSPCYEVGH-UHFFFAOYSA-N 0.000 description 8
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 8
- 108090000623 proteins and genes Proteins 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 8
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 8
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 7
- 102100033810 RAC-alpha serine/threonine-protein kinase Human genes 0.000 description 7
- 150000001412 amines Chemical class 0.000 description 7
- 239000008346 aqueous phase Substances 0.000 description 7
- 235000019441 ethanol Nutrition 0.000 description 7
- 239000001257 hydrogen Substances 0.000 description 7
- 230000001965 increasing effect Effects 0.000 description 7
- 229960004592 isopropanol Drugs 0.000 description 7
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 7
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 7
- 239000007858 starting material Substances 0.000 description 7
- 238000005406 washing Methods 0.000 description 7
- 238000010626 work up procedure Methods 0.000 description 7
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 6
- 150000007513 acids Chemical class 0.000 description 6
- 230000003321 amplification Effects 0.000 description 6
- 230000006907 apoptotic process Effects 0.000 description 6
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 6
- 230000022131 cell cycle Effects 0.000 description 6
- 230000004663 cell proliferation Effects 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- WQWSKFSXFWLOHG-UHFFFAOYSA-N methyl 4-chloro-6-iodoquinazoline-8-carboxylate Chemical compound N1=CN=C2C(C(=O)OC)=CC(I)=CC2=C1Cl WQWSKFSXFWLOHG-UHFFFAOYSA-N 0.000 description 6
- 238000003199 nucleic acid amplification method Methods 0.000 description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 description 6
- 230000019491 signal transduction Effects 0.000 description 6
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 6
- MCCOWFUKWQGOSO-UHFFFAOYSA-N 4-[[1-(3-aminophenyl)-3-pyrrolidin-1-ylpropyl]amino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NC(C=1C=C(N)C=CC=1)CCN1CCCC1 MCCOWFUKWQGOSO-UHFFFAOYSA-N 0.000 description 5
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 5
- 208000026310 Breast neoplasm Diseases 0.000 description 5
- 102100039872 Inner centromere protein Human genes 0.000 description 5
- 101710162819 Inner centromere protein Proteins 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 5
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 description 5
- 102000013530 TOR Serine-Threonine Kinases Human genes 0.000 description 5
- 229960000583 acetic acid Drugs 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 239000002246 antineoplastic agent Substances 0.000 description 5
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 5
- 230000000875 corresponding effect Effects 0.000 description 5
- 230000012010 growth Effects 0.000 description 5
- 208000013403 hyperactivity Diseases 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- RMXSAKHTLYDYIP-UHFFFAOYSA-N methyl 6-iodo-4-oxo-1h-quinazoline-8-carboxylate Chemical compound N1C=NC(=O)C2=C1C(C(=O)OC)=CC(I)=C2 RMXSAKHTLYDYIP-UHFFFAOYSA-N 0.000 description 5
- 230000035772 mutation Effects 0.000 description 5
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 5
- 229910052763 palladium Inorganic materials 0.000 description 5
- 238000001556 precipitation Methods 0.000 description 5
- 230000035755 proliferation Effects 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 239000000377 silicon dioxide Substances 0.000 description 5
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 5
- SSNBDGOCMUSQKH-UHFFFAOYSA-N tert-butyl n-[3-[[[8-carbamoyl-6-(1,2-dihydroxyethyl)quinazolin-4-yl]amino]methyl]phenyl]carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=CC(CNC=2C3=CC(=CC(=C3N=CN=2)C(N)=O)C(O)CO)=C1 SSNBDGOCMUSQKH-UHFFFAOYSA-N 0.000 description 5
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 4
- KDKUVYLMPJIGKA-UHFFFAOYSA-N 4-[[5-amino-1-[(2,6-difluorophenyl)-oxomethyl]-1,2,4-triazol-3-yl]amino]benzenesulfonamide Chemical compound N=1N(C(=O)C=2C(=CC=CC=2F)F)C(N)=NC=1NC1=CC=C(S(N)(=O)=O)C=C1 KDKUVYLMPJIGKA-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 4
- 239000007821 HATU Substances 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 4
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 4
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 4
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 4
- 102000002278 Ribosomal Proteins Human genes 0.000 description 4
- 108010000605 Ribosomal Proteins Proteins 0.000 description 4
- 102100024908 Ribosomal protein S6 kinase beta-1 Human genes 0.000 description 4
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- 101710200273 Targeting protein for Xklp2 Proteins 0.000 description 4
- 102100024813 Targeting protein for Xklp2 Human genes 0.000 description 4
- ARVVGPPODCHFPK-UHFFFAOYSA-N [3-[(2-methylpropan-2-yl)oxycarbonylamino]-3-(3-nitrophenyl)propyl] 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OCCC(NC(=O)OC(C)(C)C)C1=CC=CC([N+]([O-])=O)=C1 ARVVGPPODCHFPK-UHFFFAOYSA-N 0.000 description 4
- 235000011054 acetic acid Nutrition 0.000 description 4
- 235000001014 amino acid Nutrition 0.000 description 4
- CBHOOMGKXCMKIR-UHFFFAOYSA-N azane;methanol Chemical compound N.OC CBHOOMGKXCMKIR-UHFFFAOYSA-N 0.000 description 4
- CELPHAGZKFMOMR-UHFFFAOYSA-N azanium;dichloromethane;methanol;hydroxide Chemical compound [NH4+].[OH-].OC.ClCCl CELPHAGZKFMOMR-UHFFFAOYSA-N 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- HTZCNXWZYVXIMZ-UHFFFAOYSA-M benzyl(triethyl)azanium;chloride Chemical compound [Cl-].CC[N+](CC)(CC)CC1=CC=CC=C1 HTZCNXWZYVXIMZ-UHFFFAOYSA-M 0.000 description 4
- 230000033228 biological regulation Effects 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 239000003480 eluent Substances 0.000 description 4
- 230000014509 gene expression Effects 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 4
- 208000030159 metabolic disease Diseases 0.000 description 4
- ATIONWGROLCVEA-UHFFFAOYSA-N methyl 6-ethenyl-4-[[3-[(2-methylpropan-2-yl)oxycarbonylamino]phenyl]methylamino]quinazoline-8-carboxylate Chemical compound N1=CN=C2C(C(=O)OC)=CC(C=C)=CC2=C1NCC1=CC=CC(NC(=O)OC(C)(C)C)=C1 ATIONWGROLCVEA-UHFFFAOYSA-N 0.000 description 4
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 4
- 230000037361 pathway Effects 0.000 description 4
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 230000001105 regulatory effect Effects 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- 229960002930 sirolimus Drugs 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 230000004083 survival effect Effects 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- 230000014616 translation Effects 0.000 description 4
- 239000003643 water by type Substances 0.000 description 4
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 3
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 3
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 3
- NJXPYZHXZZCTNI-UHFFFAOYSA-N 3-aminobenzonitrile Chemical compound NC1=CC=CC(C#N)=C1 NJXPYZHXZZCTNI-UHFFFAOYSA-N 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- 125000001999 4-Methoxybenzoyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C(*)=O 0.000 description 3
- HAWMGYVPJGGBFN-UHFFFAOYSA-N 4-[(3-aminophenyl)methylamino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NCC1=CC=CC(N)=C1 HAWMGYVPJGGBFN-UHFFFAOYSA-N 0.000 description 3
- NPSTWKVVOHABJN-UHFFFAOYSA-N 4-[(4-aminophenyl)methylamino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NCC1=CC=C(N)C=C1 NPSTWKVVOHABJN-UHFFFAOYSA-N 0.000 description 3
- WEGBYDBCGCQGMC-UHFFFAOYSA-N 4-[[3-[(4-methoxybenzoyl)-methylamino]phenyl]methylamino]quinazoline-8-carboxamide Chemical compound C1=CC(OC)=CC=C1C(=O)N(C)C1=CC=CC(CNC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 WEGBYDBCGCQGMC-UHFFFAOYSA-N 0.000 description 3
- YMZRVSIMAIQEJB-UHFFFAOYSA-N 4-oxo-1h-quinazoline-8-carboxylic acid Chemical compound N1=CNC(=O)C2=C1C(C(=O)O)=CC=C2 YMZRVSIMAIQEJB-UHFFFAOYSA-N 0.000 description 3
- 206010006187 Breast cancer Diseases 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- 108091007914 CDKs Proteins 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 3
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 239000007995 HEPES buffer Substances 0.000 description 3
- 101001051706 Homo sapiens Ribosomal protein S6 kinase beta-1 Proteins 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 206010034764 Peutz-Jeghers syndrome Diseases 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 230000018199 S phase Effects 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 230000002547 anomalous effect Effects 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 239000013058 crude material Substances 0.000 description 3
- 230000021953 cytokinesis Effects 0.000 description 3
- 230000004069 differentiation Effects 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 238000001952 enzyme assay Methods 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 239000012458 free base Substances 0.000 description 3
- 239000003102 growth factor Substances 0.000 description 3
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 208000032839 leukemia Diseases 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 108020004999 messenger RNA Proteins 0.000 description 3
- 230000002503 metabolic effect Effects 0.000 description 3
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 3
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 3
- 150000004702 methyl esters Chemical class 0.000 description 3
- JTSLALYXYSRPGW-UHFFFAOYSA-N n-[5-(4-cyanophenyl)-1h-pyrrolo[2,3-b]pyridin-3-yl]pyridine-3-carboxamide Chemical compound C=1C=CN=CC=1C(=O)NC(C1=C2)=CNC1=NC=C2C1=CC=C(C#N)C=C1 JTSLALYXYSRPGW-UHFFFAOYSA-N 0.000 description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 3
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 3
- DGTNSSLYPYDJGL-UHFFFAOYSA-N phenyl isocyanate Chemical compound O=C=NC1=CC=CC=C1 DGTNSSLYPYDJGL-UHFFFAOYSA-N 0.000 description 3
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 230000005855 radiation Effects 0.000 description 3
- 238000001959 radiotherapy Methods 0.000 description 3
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 3
- 150000003384 small molecules Chemical class 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- HABQSOAEHLQKKG-UHFFFAOYSA-N tert-butyl n-[3-hydroxy-1-(3-nitrophenyl)propyl]carbamate Chemical compound CC(C)(C)OC(=O)NC(CCO)C1=CC=CC([N+]([O-])=O)=C1 HABQSOAEHLQKKG-UHFFFAOYSA-N 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 3
- 238000013519 translation Methods 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- 238000011144 upstream manufacturing Methods 0.000 description 3
- KRRWAASSBQOAHT-MRVPVSSYSA-N (1s)-2-azido-1-phenylethanamine Chemical compound [N-]=[N+]=NC[C@@H](N)C1=CC=CC=C1 KRRWAASSBQOAHT-MRVPVSSYSA-N 0.000 description 2
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 2
- DPXXYMQUWQIWCM-UHFFFAOYSA-N 1-(3-nitrophenyl)-3-pyrrolidin-1-ylpropan-1-amine Chemical compound C=1C=CC([N+]([O-])=O)=CC=1C(N)CCN1CCCC1 DPXXYMQUWQIWCM-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- YLYPIBBGWLKELC-RMKNXTFCSA-N 2-[2-[(e)-2-[4-(dimethylamino)phenyl]ethenyl]-6-methylpyran-4-ylidene]propanedinitrile Chemical compound C1=CC(N(C)C)=CC=C1\C=C\C1=CC(=C(C#N)C#N)C=C(C)O1 YLYPIBBGWLKELC-RMKNXTFCSA-N 0.000 description 2
- 125000006275 3-bromophenyl group Chemical group [H]C1=C([H])C(Br)=C([H])C(*)=C1[H] 0.000 description 2
- 125000006284 3-fluorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C(F)=C1[H])C([H])([H])* 0.000 description 2
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 2
- SLZGHLVKUFSFBL-UHFFFAOYSA-N 4-(benzylamino)quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NCC1=CC=CC=C1 SLZGHLVKUFSFBL-UHFFFAOYSA-N 0.000 description 2
- RATSANVPHHXDCT-UHFFFAOYSA-N 4-(trifluoromethoxy)benzoic acid Chemical compound OC(=O)C1=CC=C(OC(F)(F)F)C=C1 RATSANVPHHXDCT-UHFFFAOYSA-N 0.000 description 2
- UOYTTXGBPMYPLE-UHFFFAOYSA-N 4-[1-(3-aminophenyl)ethylamino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(C)C1=CC=CC(N)=C1 UOYTTXGBPMYPLE-UHFFFAOYSA-N 0.000 description 2
- YFCIFWOJYYFDQP-PTWZRHHISA-N 4-[3-amino-6-[(1S,3S,4S)-3-fluoro-4-hydroxycyclohexyl]pyrazin-2-yl]-N-[(1S)-1-(3-bromo-5-fluorophenyl)-2-(methylamino)ethyl]-2-fluorobenzamide Chemical compound CNC[C@@H](NC(=O)c1ccc(cc1F)-c1nc(cnc1N)[C@H]1CC[C@H](O)[C@@H](F)C1)c1cc(F)cc(Br)c1 YFCIFWOJYYFDQP-PTWZRHHISA-N 0.000 description 2
- MFPCXWBJHINRKG-QGZVFWFLSA-N 4-[[(1s)-2-hydroxy-1-phenylethyl]amino]quinoline-8-carbonitrile Chemical compound C1([C@H](NC=2C3=CC=CC(=C3N=CC=2)C#N)CO)=CC=CC=C1 MFPCXWBJHINRKG-QGZVFWFLSA-N 0.000 description 2
- DAJCKHCRJIXKHC-UHFFFAOYSA-N 4-[[1-(3-aminophenyl)-3-methoxypropyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CCOC)C1=CC=CC(N)=C1 DAJCKHCRJIXKHC-UHFFFAOYSA-N 0.000 description 2
- YCNIACHCKKBPGW-UHFFFAOYSA-N 4-[[1-(3-nitrophenyl)-3-pyrrolidin-1-ylpropyl]amino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NC(C=1C=C(C=CC=1)[N+]([O-])=O)CCN1CCCC1 YCNIACHCKKBPGW-UHFFFAOYSA-N 0.000 description 2
- FRCWIKMEVRFKBJ-UHFFFAOYSA-N 4-[[2-(methylamino)-1-phenylethyl]amino]-6-phenylmethoxyquinazoline-8-carboxamide Chemical compound C=1C=CC=CC=1C(CNC)NC(C1=C2)=NC=NC1=C(C(N)=O)C=C2OCC1=CC=CC=C1 FRCWIKMEVRFKBJ-UHFFFAOYSA-N 0.000 description 2
- MGAKKWGHIRKPMT-UHFFFAOYSA-N 4-[[3-methoxy-1-(3-nitrophenyl)propyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CCOC)C1=CC=CC([N+]([O-])=O)=C1 MGAKKWGHIRKPMT-UHFFFAOYSA-N 0.000 description 2
- NLSGLAZSZPWMLF-UHFFFAOYSA-N 4-amino-4-(3-nitrophenyl)butanoic acid Chemical compound OC(=O)CCC(N)C1=CC=CC([N+]([O-])=O)=C1 NLSGLAZSZPWMLF-UHFFFAOYSA-N 0.000 description 2
- CZKLEJHVLCMVQR-UHFFFAOYSA-N 4-fluorobenzoyl chloride Chemical compound FC1=CC=C(C(Cl)=O)C=C1 CZKLEJHVLCMVQR-UHFFFAOYSA-N 0.000 description 2
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 2
- WGGOYMCUJRCWRJ-UHFFFAOYSA-N 6-amino-4-[[4-chloro-3-(trifluoromethyl)phenyl]methylamino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC(N)=CC2=C1NCC1=CC=C(Cl)C(C(F)(F)F)=C1 WGGOYMCUJRCWRJ-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 102000004228 Aurora kinase B Human genes 0.000 description 2
- 108090000749 Aurora kinase B Proteins 0.000 description 2
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 2
- 229940123587 Cell cycle inhibitor Drugs 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- 102100032857 Cyclin-dependent kinase 1 Human genes 0.000 description 2
- 101710106279 Cyclin-dependent kinase 1 Proteins 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 2
- PNKUSGQVOMIXLU-UHFFFAOYSA-N Formamidine Chemical compound NC=N PNKUSGQVOMIXLU-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 102000006947 Histones Human genes 0.000 description 2
- 108010033040 Histones Proteins 0.000 description 2
- 206010022489 Insulin Resistance Diseases 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- JLTDJTHDQAWBAV-UHFFFAOYSA-N N,N-dimethylaniline Chemical compound CN(C)C1=CC=CC=C1 JLTDJTHDQAWBAV-UHFFFAOYSA-N 0.000 description 2
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 2
- LFTLOKWAGJYHHR-UHFFFAOYSA-N N-methylmorpholine N-oxide Chemical compound CN1(=O)CCOCC1 LFTLOKWAGJYHHR-UHFFFAOYSA-N 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- 208000008589 Obesity Diseases 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 102100037414 Rac GTPase-activating protein 1 Human genes 0.000 description 2
- 101710196218 Rac GTPase-activating protein 1 Proteins 0.000 description 2
- 201000000582 Retinoblastoma Diseases 0.000 description 2
- 101710108924 Ribosomal protein S6 kinase beta-1 Proteins 0.000 description 2
- 102100026715 Serine/threonine-protein kinase STK11 Human genes 0.000 description 2
- 101710181599 Serine/threonine-protein kinase STK11 Proteins 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- 102000000763 Survivin Human genes 0.000 description 2
- 108010002687 Survivin Proteins 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 2
- 102000016663 Vascular Endothelial Growth Factor Receptor-3 Human genes 0.000 description 2
- YKNZTUQUXUXTLE-UHFFFAOYSA-N [3-(trifluoromethyl)phenyl]methanamine Chemical compound NCC1=CC=CC(C(F)(F)F)=C1 YKNZTUQUXUXTLE-UHFFFAOYSA-N 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000004037 angiogenesis inhibitor Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 239000004305 biphenyl Substances 0.000 description 2
- 235000010290 biphenyl Nutrition 0.000 description 2
- 210000000481 breast Anatomy 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 230000000973 chemotherapeutic effect Effects 0.000 description 2
- 238000004296 chiral HPLC Methods 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 238000006911 enzymatic reaction Methods 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 239000002198 insoluble material Substances 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- TWBYWOBDOCUKOW-UHFFFAOYSA-N isonicotinic acid Chemical compound OC(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-N 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 239000004310 lactic acid Substances 0.000 description 2
- 235000014655 lactic acid Nutrition 0.000 description 2
- 125000005647 linker group Chemical group 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 239000001630 malic acid Substances 0.000 description 2
- 235000011090 malic acid Nutrition 0.000 description 2
- 230000035800 maturation Effects 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- WCYSVIMUYDWOPM-UHFFFAOYSA-N methyl 3-amino-3-(3-nitrophenyl)propanoate Chemical compound COC(=O)CC(N)C1=CC=CC([N+]([O-])=O)=C1 WCYSVIMUYDWOPM-UHFFFAOYSA-N 0.000 description 2
- MXAFBADEGVDPJL-OAHLLOKOSA-N methyl 4-[[(1s)-2-azido-1-phenylethyl]amino]quinazoline-8-carboxylate Chemical compound C1([C@@H](CN=[N+]=[N-])NC2=C3C=CC=C(C3=NC=N2)C(=O)OC)=CC=CC=C1 MXAFBADEGVDPJL-OAHLLOKOSA-N 0.000 description 2
- QNVPUSHEFBLTBI-UHFFFAOYSA-N methyl 4-[[1-(3-nitrophenyl)-3-pyrrolidin-1-ylpropyl]amino]quinazoline-8-carboxylate Chemical compound N1=CN=C2C(C(=O)OC)=CC=CC2=C1NC(C=1C=C(C=CC=1)[N+]([O-])=O)CCN1CCCC1 QNVPUSHEFBLTBI-UHFFFAOYSA-N 0.000 description 2
- NJKAZVLZWRJRCF-UHFFFAOYSA-N methyl 4-[[3-methoxy-1-(3-nitrophenyl)propyl]amino]quinazoline-8-carboxylate Chemical compound N=1C=NC2=C(C(=O)OC)C=CC=C2C=1NC(CCOC)C1=CC=CC([N+]([O-])=O)=C1 NJKAZVLZWRJRCF-UHFFFAOYSA-N 0.000 description 2
- SFTIODPDUDDGCZ-UHFFFAOYSA-N methyl 4-amino-4-(3-nitrophenyl)butanoate Chemical compound COC(=O)CCC(N)C1=CC=CC([N+]([O-])=O)=C1 SFTIODPDUDDGCZ-UHFFFAOYSA-N 0.000 description 2
- XPSLQEXRZDNOFZ-UHFFFAOYSA-N methyl 6-iodo-4-[[3-[(2-methylpropan-2-yl)oxycarbonylamino]phenyl]methylamino]quinazoline-2-carboxylate Chemical compound C=12C=C(I)C=CC2=NC(C(=O)OC)=NC=1NCC1=CC=CC(NC(=O)OC(C)(C)C)=C1 XPSLQEXRZDNOFZ-UHFFFAOYSA-N 0.000 description 2
- LEGKDPRUXDCBCA-UHFFFAOYSA-N methyl 6-iodo-4-[[3-[(2-methylpropan-2-yl)oxycarbonylamino]phenyl]methylamino]quinazoline-8-carboxylate Chemical compound N1=CN=C2C(C(=O)OC)=CC(I)=CC2=C1NCC1=CC=CC(NC(=O)OC(C)(C)C)=C1 LEGKDPRUXDCBCA-UHFFFAOYSA-N 0.000 description 2
- 125000004458 methylaminocarbonyl group Chemical group [H]N(C(*)=O)C([H])([H])[H] 0.000 description 2
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 230000011278 mitosis Effects 0.000 description 2
- 230000000394 mitotic effect Effects 0.000 description 2
- IKCLWFBBSBDUHC-UHFFFAOYSA-N n-[3-(aminomethyl)phenyl]-4-methoxy-n-methylbenzamide Chemical compound C1=CC(OC)=CC=C1C(=O)N(C)C1=CC=CC(CN)=C1 IKCLWFBBSBDUHC-UHFFFAOYSA-N 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 235000020824 obesity Nutrition 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 229910000489 osmium tetroxide Inorganic materials 0.000 description 2
- 239000012285 osmium tetroxide Substances 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- SGNPIGAGPRLGEJ-UHFFFAOYSA-N oxathiazolidine-3-carboxylic acid Chemical compound OC(=O)N1CCOS1 SGNPIGAGPRLGEJ-UHFFFAOYSA-N 0.000 description 2
- BHAAPTBBJKJZER-UHFFFAOYSA-N p-anisidine Chemical compound COC1=CC=C(N)C=C1 BHAAPTBBJKJZER-UHFFFAOYSA-N 0.000 description 2
- 201000002528 pancreatic cancer Diseases 0.000 description 2
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 2
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 2
- WLJVNTCWHIRURA-UHFFFAOYSA-N pimelic acid Chemical compound OC(=O)CCCCCC(O)=O WLJVNTCWHIRURA-UHFFFAOYSA-N 0.000 description 2
- DNUTZBZXLPWRJG-UHFFFAOYSA-M piperidine-1-carboxylate Chemical compound [O-]C(=O)N1CCCCC1 DNUTZBZXLPWRJG-UHFFFAOYSA-M 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 230000002685 pulmonary effect Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 230000001235 sensitizing effect Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 239000012258 stirred mixture Substances 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- 238000003419 tautomerization reaction Methods 0.000 description 2
- LSOZALWRNWQPLK-UHFFFAOYSA-N tert-butyl n-[(3-aminophenyl)methyl]carbamate Chemical compound CC(C)(C)OC(=O)NCC1=CC=CC(N)=C1 LSOZALWRNWQPLK-UHFFFAOYSA-N 0.000 description 2
- IUBFAZSFGODJPA-UHFFFAOYSA-N tert-butyl n-[3-(1-aminoethyl)phenyl]carbamate Chemical compound CC(N)C1=CC=CC(NC(=O)OC(C)(C)C)=C1 IUBFAZSFGODJPA-UHFFFAOYSA-N 0.000 description 2
- NXQNEOIMZVWHQW-UHFFFAOYSA-N tert-butyl n-[3-(aminomethyl)phenyl]carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=CC(CN)=C1 NXQNEOIMZVWHQW-UHFFFAOYSA-N 0.000 description 2
- JSSSBSWQIXUCLQ-UHFFFAOYSA-N tert-butyl n-[3-[1-[(8-carbamoylquinazolin-4-yl)amino]ethyl]phenyl]carbamate Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(C)C1=CC=CC(NC(=O)OC(C)(C)C)=C1 JSSSBSWQIXUCLQ-UHFFFAOYSA-N 0.000 description 2
- NQPCKXMZXDBUNY-UHFFFAOYSA-N tert-butyl n-[3-[[(8-carbamoyl-6-formylquinazolin-4-yl)amino]methyl]phenyl]carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=CC(CNC=2C3=CC(C=O)=CC(=C3N=CN=2)C(N)=O)=C1 NQPCKXMZXDBUNY-UHFFFAOYSA-N 0.000 description 2
- BMXVHUJLRNUTPB-UHFFFAOYSA-N tert-butyl n-[3-[[[8-carbamoyl-6-(hydroxymethyl)quinazolin-4-yl]amino]methyl]phenyl]carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=CC(CNC=2C3=CC(CO)=CC(=C3N=CN=2)C(N)=O)=C1 BMXVHUJLRNUTPB-UHFFFAOYSA-N 0.000 description 2
- OJHNWARLLWFOFI-UHFFFAOYSA-N tert-butyl n-[3-methoxy-1-(3-nitrophenyl)propyl]carbamate Chemical compound CC(C)(C)OC(=O)NC(CCOC)C1=CC=CC([N+]([O-])=O)=C1 OJHNWARLLWFOFI-UHFFFAOYSA-N 0.000 description 2
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- QIWRFOJWQSSRJZ-UHFFFAOYSA-N tributyl(ethenyl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C=C QIWRFOJWQSSRJZ-UHFFFAOYSA-N 0.000 description 2
- 238000001665 trituration Methods 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 238000000825 ultraviolet detection Methods 0.000 description 2
- IKBMZMKEBCIDPD-FYZOBXCZSA-N (1r)-1-(3-nitrophenyl)ethanamine;hydrochloride Chemical compound Cl.C[C@@H](N)C1=CC=CC([N+]([O-])=O)=C1 IKBMZMKEBCIDPD-FYZOBXCZSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- DNISEZBAYYIQFB-PHDIDXHHSA-N (2r,3r)-2,3-diacetyloxybutanedioic acid Chemical compound CC(=O)O[C@@H](C(O)=O)[C@H](C(O)=O)OC(C)=O DNISEZBAYYIQFB-PHDIDXHHSA-N 0.000 description 1
- JUSWZYFYLXTMLJ-JTQLQIEISA-N (2s)-1-(benzenesulfonyl)pyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1S(=O)(=O)C1=CC=CC=C1 JUSWZYFYLXTMLJ-JTQLQIEISA-N 0.000 description 1
- RQYKQWFHJOBBAO-JTQLQIEISA-N (2s)-1-benzoylpyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1C(=O)C1=CC=CC=C1 RQYKQWFHJOBBAO-JTQLQIEISA-N 0.000 description 1
- IJXJGQCXFSSHNL-MRVPVSSYSA-N (2s)-2-amino-2-phenylethanol Chemical compound OC[C@@H](N)C1=CC=CC=C1 IJXJGQCXFSSHNL-MRVPVSSYSA-N 0.000 description 1
- HGRWHBQLRXWSLV-DEOSSOPVSA-N (4s)-3'-(3,6-dihydro-2h-pyran-5-yl)-1'-fluoro-7'-(3-fluoropyridin-2-yl)spiro[5h-1,3-oxazole-4,5'-chromeno[2,3-c]pyridine]-2-amine Chemical compound C1OC(N)=N[C@]21C1=CC(C=3COCCC=3)=NC(F)=C1OC1=CC=C(C=3C(=CC=CN=3)F)C=C12 HGRWHBQLRXWSLV-DEOSSOPVSA-N 0.000 description 1
- SPEUIVXLLWOEMJ-UHFFFAOYSA-N 1,1-dimethoxyethane Chemical compound COC(C)OC SPEUIVXLLWOEMJ-UHFFFAOYSA-N 0.000 description 1
- 125000005919 1,2,2-trimethylpropyl group Chemical group 0.000 description 1
- 125000004509 1,3,4-oxadiazol-2-yl group Chemical group O1C(=NN=C1)* 0.000 description 1
- 125000005940 1,4-dioxanyl group Chemical group 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- YZUPZGFPHUVJKC-UHFFFAOYSA-N 1-bromo-2-methoxyethane Chemical compound COCCBr YZUPZGFPHUVJKC-UHFFFAOYSA-N 0.000 description 1
- FBQJSTRRHBTMMQ-UHFFFAOYSA-N 1-chloro-7-methylisoquinoline Chemical compound C1=CN=C(Cl)C2=CC(C)=CC=C21 FBQJSTRRHBTMMQ-UHFFFAOYSA-N 0.000 description 1
- 125000006218 1-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000006219 1-ethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000005808 2,4,6-trimethoxyphenyl group Chemical group [H][#6]-1=[#6](-[#8]C([H])([H])[H])-[#6](-*)=[#6](-[#8]C([H])([H])[H])-[#6]([H])=[#6]-1-[#8]C([H])([H])[H] 0.000 description 1
- JIZVMILRYBKPFN-UHFFFAOYSA-N 2,6-dibromo-4-methoxyaniline Chemical compound COC1=CC(Br)=C(N)C(Br)=C1 JIZVMILRYBKPFN-UHFFFAOYSA-N 0.000 description 1
- HNUJOYMRHWMPOM-UHFFFAOYSA-N 2-(3-fluorophenyl)-2-hydroxyacetic acid Chemical compound OC(=O)C(O)C1=CC=CC(F)=C1 HNUJOYMRHWMPOM-UHFFFAOYSA-N 0.000 description 1
- YCWRFIYBUQBHJI-UHFFFAOYSA-N 2-(4-aminophenyl)acetonitrile Chemical group NC1=CC=C(CC#N)C=C1 YCWRFIYBUQBHJI-UHFFFAOYSA-N 0.000 description 1
- CTBARTMXUVSCRR-UHFFFAOYSA-N 2-(6-bromopyridin-3-yl)acetonitrile Chemical compound BrC1=CC=C(CC#N)C=N1 CTBARTMXUVSCRR-UHFFFAOYSA-N 0.000 description 1
- MOPFCAZSSMNDDE-UHFFFAOYSA-N 2-(dimethylamino)-1-(3-methoxyphenyl)ethanone Chemical compound COC1=CC=CC(C(=O)CN(C)C)=C1 MOPFCAZSSMNDDE-UHFFFAOYSA-N 0.000 description 1
- UZYQSNQJLWTICD-UHFFFAOYSA-N 2-(n-benzoylanilino)-2,2-dinitroacetic acid Chemical compound C=1C=CC=CC=1N(C(C(=O)O)([N+]([O-])=O)[N+]([O-])=O)C(=O)C1=CC=CC=C1 UZYQSNQJLWTICD-UHFFFAOYSA-N 0.000 description 1
- OXQGTIUCKGYOAA-UHFFFAOYSA-N 2-Ethylbutanoic acid Chemical compound CCC(CC)C(O)=O OXQGTIUCKGYOAA-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- KMONSDYJWOGBBU-UHFFFAOYSA-N 2-[3-[1-[(8-carbamoylquinazolin-4-yl)amino]ethyl]anilino]-1,3-oxazole-5-carboxylic acid Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(C)C(C=1)=CC=CC=1NC1=NC=C(C(O)=O)O1 KMONSDYJWOGBBU-UHFFFAOYSA-N 0.000 description 1
- YSLPTGOAVRWPDS-UHFFFAOYSA-N 2-amino-2-(3-nitrophenyl)ethanol Chemical compound OCC(N)C1=CC=CC([N+]([O-])=O)=C1 YSLPTGOAVRWPDS-UHFFFAOYSA-N 0.000 description 1
- KQOFWDBENVYNSA-UHFFFAOYSA-N 2-amino-5-methoxybenzene-1,3-dicarbonitrile Chemical compound COC1=CC(C#N)=C(N)C(C#N)=C1 KQOFWDBENVYNSA-UHFFFAOYSA-N 0.000 description 1
- AGCMUJTYJYULPT-UHFFFAOYSA-N 2-amino-5-methoxybenzene-1,3-dicarboxylic acid Chemical compound COC1=CC(C(O)=O)=C(N)C(C(O)=O)=C1 AGCMUJTYJYULPT-UHFFFAOYSA-N 0.000 description 1
- DYZDIWNRWSNVPT-UHFFFAOYSA-N 2-amino-6-methoxybenzoic acid Chemical compound COC1=CC=CC(N)=C1C(O)=O DYZDIWNRWSNVPT-UHFFFAOYSA-N 0.000 description 1
- LDOMKUVUXZRECL-UHFFFAOYSA-N 2-aminobenzene-1,3-dicarboxylic acid Chemical compound NC1=C(C(O)=O)C=CC=C1C(O)=O LDOMKUVUXZRECL-UHFFFAOYSA-N 0.000 description 1
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 description 1
- KMGUEILFFWDGFV-UHFFFAOYSA-N 2-benzoyl-2-benzoyloxy-3-hydroxybutanedioic acid Chemical compound C=1C=CC=CC=1C(=O)C(C(C(O)=O)O)(C(O)=O)OC(=O)C1=CC=CC=C1 KMGUEILFFWDGFV-UHFFFAOYSA-N 0.000 description 1
- IOOHBIFQNQQUFI-UHFFFAOYSA-N 2-bromo-1-(3-methoxyphenyl)ethanone Chemical compound COC1=CC=CC(C(=O)CBr)=C1 IOOHBIFQNQQUFI-UHFFFAOYSA-N 0.000 description 1
- LSZMVESSGLHDJE-UHFFFAOYSA-N 2-bromo-4-methylpyridine Chemical compound CC1=CC=NC(Br)=C1 LSZMVESSGLHDJE-UHFFFAOYSA-N 0.000 description 1
- 125000006276 2-bromophenyl group Chemical group [H]C1=C([H])C(Br)=C(*)C([H])=C1[H] 0.000 description 1
- IMRWILPUOVGIMU-UHFFFAOYSA-N 2-bromopyridine Chemical compound BrC1=CC=CC=N1 IMRWILPUOVGIMU-UHFFFAOYSA-N 0.000 description 1
- BSQLQMLFTHJVKS-UHFFFAOYSA-N 2-chloro-1,3-benzothiazole Chemical compound C1=CC=C2SC(Cl)=NC2=C1 BSQLQMLFTHJVKS-UHFFFAOYSA-N 0.000 description 1
- SSXYKBHZZRYLLY-UHFFFAOYSA-N 2-chloro-1,3-oxazole Chemical compound ClC1=NC=CO1 SSXYKBHZZRYLLY-UHFFFAOYSA-N 0.000 description 1
- GBNPVXZNWBWNEN-UHFFFAOYSA-N 2-chloro-4-(trifluoromethyl)pyridine Chemical compound FC(F)(F)C1=CC=NC(Cl)=C1 GBNPVXZNWBWNEN-UHFFFAOYSA-N 0.000 description 1
- CZQXWSPZVZKNRM-UHFFFAOYSA-N 2-chloro-6-(trifluoromethyl)-1h-benzimidazole Chemical compound FC(F)(F)C1=CC=C2N=C(Cl)NC2=C1 CZQXWSPZVZKNRM-UHFFFAOYSA-N 0.000 description 1
- FVUFTABOJFRHSU-UHFFFAOYSA-N 2-chloro-6-methoxy-1,3-benzothiazole Chemical compound COC1=CC=C2N=C(Cl)SC2=C1 FVUFTABOJFRHSU-UHFFFAOYSA-N 0.000 description 1
- QORVXCUHOOQAJL-UHFFFAOYSA-N 2-chloro-n-[3-[2-(dimethylamino)-1-(1,3-dioxoisoindol-2-yl)ethyl]phenyl]pyridine-4-carboxamide Chemical compound O=C1C2=CC=CC=C2C(=O)N1C(CN(C)C)C(C=1)=CC=CC=1NC(=O)C1=CC=NC(Cl)=C1 QORVXCUHOOQAJL-UHFFFAOYSA-N 0.000 description 1
- 125000006282 2-chlorobenzyl group Chemical group [H]C1=C([H])C(Cl)=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000004182 2-chlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(*)C([H])=C1[H] 0.000 description 1
- OKDGRDCXVWSXDC-UHFFFAOYSA-N 2-chloropyridine Chemical compound ClC1=CC=CC=N1 OKDGRDCXVWSXDC-UHFFFAOYSA-N 0.000 description 1
- QRXBTPFMCTXCRD-UHFFFAOYSA-N 2-chloropyridine-4-carbonitrile Chemical compound ClC1=CC(C#N)=CC=N1 QRXBTPFMCTXCRD-UHFFFAOYSA-N 0.000 description 1
- UNCQVRBWJWWJBF-UHFFFAOYSA-N 2-chloropyrimidine Chemical compound ClC1=NC=CC=N1 UNCQVRBWJWWJBF-UHFFFAOYSA-N 0.000 description 1
- OFUFXTHGZWIDDB-UHFFFAOYSA-N 2-chloroquinoline Chemical compound C1=CC=CC2=NC(Cl)=CC=C21 OFUFXTHGZWIDDB-UHFFFAOYSA-N 0.000 description 1
- LQLJZSJKRYTKTP-UHFFFAOYSA-N 2-dimethylaminoethyl chloride hydrochloride Chemical compound Cl.CN(C)CCCl LQLJZSJKRYTKTP-UHFFFAOYSA-N 0.000 description 1
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 1
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004198 2-fluorophenyl group Chemical group [H]C1=C([H])C(F)=C(*)C([H])=C1[H] 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- ONLRUXGFBWJVJE-UHFFFAOYSA-N 2-methoxy-1-(3-nitrophenyl)ethanamine Chemical compound COCC(N)C1=CC=CC([N+]([O-])=O)=C1 ONLRUXGFBWJVJE-UHFFFAOYSA-N 0.000 description 1
- 125000006179 2-methyl benzyl group Chemical group [H]C1=C([H])C(=C(C([H])=C1[H])C([H])([H])*)C([H])([H])[H] 0.000 description 1
- HDECRAPHCDXMIJ-UHFFFAOYSA-N 2-methylbenzenesulfonyl chloride Chemical compound CC1=CC=CC=C1S(Cl)(=O)=O HDECRAPHCDXMIJ-UHFFFAOYSA-N 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- CESUXLKAADQNTB-SSDOTTSWSA-N 2-methylpropane-2-sulfinamide Chemical compound CC(C)(C)[S@](N)=O CESUXLKAADQNTB-SSDOTTSWSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004485 2-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004362 3,4,5-trichlorophenyl group Chemical group [H]C1=C(Cl)C(Cl)=C(Cl)C([H])=C1* 0.000 description 1
- 125000003762 3,4-dimethoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C(OC([H])([H])[H])C([H])=C1* 0.000 description 1
- 125000004211 3,5-difluorophenyl group Chemical group [H]C1=C(F)C([H])=C(*)C([H])=C1F 0.000 description 1
- GNFTZDOKVXKIBK-UHFFFAOYSA-N 3-(2-methoxyethoxy)benzohydrazide Chemical compound COCCOC1=CC=CC(C(=O)NN)=C1 GNFTZDOKVXKIBK-UHFFFAOYSA-N 0.000 description 1
- ZDBWYUOUYNQZBM-UHFFFAOYSA-N 3-(aminomethyl)aniline Chemical compound NCC1=CC=CC(N)=C1 ZDBWYUOUYNQZBM-UHFFFAOYSA-N 0.000 description 1
- KLDLRDSRCMJKGM-UHFFFAOYSA-N 3-[chloro-(2-oxo-1,3-oxazolidin-3-yl)phosphoryl]-1,3-oxazolidin-2-one Chemical compound C1COC(=O)N1P(=O)(Cl)N1CCOC1=O KLDLRDSRCMJKGM-UHFFFAOYSA-N 0.000 description 1
- BFGCKEHSFRPNRZ-UHFFFAOYSA-N 3-amino-2-fluorobenzonitrile Chemical compound NC1=CC=CC(C#N)=C1F BFGCKEHSFRPNRZ-UHFFFAOYSA-N 0.000 description 1
- SJBFILRQMRECCK-UHFFFAOYSA-N 3-amino-3-(3-nitrophenyl)propanoic acid Chemical compound OC(=O)CC(N)C1=CC=CC([N+]([O-])=O)=C1 SJBFILRQMRECCK-UHFFFAOYSA-N 0.000 description 1
- 125000004179 3-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(Cl)=C1[H] 0.000 description 1
- HYNNNQDQEORWEU-UHFFFAOYSA-N 3-fluoro-4-methoxybenzoic acid Chemical compound COC1=CC=C(C(O)=O)C=C1F HYNNNQDQEORWEU-UHFFFAOYSA-N 0.000 description 1
- LOSBAHQDKBWMBY-UHFFFAOYSA-N 3-fluoro-4-methoxybenzoyl chloride Chemical compound COC1=CC=C(C(Cl)=O)C=C1F LOSBAHQDKBWMBY-UHFFFAOYSA-N 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- FAKWCPNASXXVJG-UHFFFAOYSA-N 3-methoxy-1-(3-nitrophenyl)propan-1-amine Chemical compound COCCC(N)C1=CC=CC([N+]([O-])=O)=C1 FAKWCPNASXXVJG-UHFFFAOYSA-N 0.000 description 1
- 125000006497 3-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C(OC([H])([H])[H])=C1[H])C([H])([H])* 0.000 description 1
- 125000006180 3-methyl benzyl group Chemical group [H]C1=C([H])C(=C([H])C(=C1[H])C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- OIRRIMBUPKXMSS-UHFFFAOYSA-N 4-(2,3-dihydro-1h-inden-1-ylamino)quinazoline-8-carboxamide Chemical compound C1CC2=CC=CC=C2C1NC1=C2C=CC=C(C(=O)N)C2=NC=N1 OIRRIMBUPKXMSS-UHFFFAOYSA-N 0.000 description 1
- NBJHDLKSWUDGJG-UHFFFAOYSA-N 4-(2-chloroethyl)morpholin-4-ium;chloride Chemical compound Cl.ClCCN1CCOCC1 NBJHDLKSWUDGJG-UHFFFAOYSA-N 0.000 description 1
- VABBMMIPDSWANM-UHFFFAOYSA-N 4-(3-chloro-4-fluoroanilino)quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NC1=CC=C(F)C(Cl)=C1 VABBMMIPDSWANM-UHFFFAOYSA-N 0.000 description 1
- LWKVORKGASLSCM-UHFFFAOYSA-N 4-(benzylamino)-5-methoxyquinazoline-8-carbonitrile Chemical compound C=12C(OC)=CC=C(C#N)C2=NC=NC=1NCC1=CC=CC=C1 LWKVORKGASLSCM-UHFFFAOYSA-N 0.000 description 1
- PMFORDWFGARVAB-UHFFFAOYSA-N 4-(benzylamino)-5-methoxyquinazoline-8-carboxamide Chemical compound C=12C(OC)=CC=C(C(N)=O)C2=NC=NC=1NCC1=CC=CC=C1 PMFORDWFGARVAB-UHFFFAOYSA-N 0.000 description 1
- WKMQQUXWGSZFEY-UHFFFAOYSA-N 4-[(2-aminophenyl)methylamino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NCC1=CC=CC=C1N WKMQQUXWGSZFEY-UHFFFAOYSA-N 0.000 description 1
- IHULTJPTZMOSNS-UHFFFAOYSA-N 4-[(2-fluorophenyl)methylamino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NCC1=CC=CC=C1F IHULTJPTZMOSNS-UHFFFAOYSA-N 0.000 description 1
- QRWNIJRYSPRJQI-VZYDHVRKSA-N 4-[(3-amino-1-phenylpropyl)amino]quinazoline-8-carboxamide benzyl N-[(3R)-3-[(8-carbamoylquinazolin-4-yl)amino]-3-phenylpropyl]carbamate Chemical compound C(C1=CC=CC=C1)OC(NCC[C@H](C1=CC=CC=C1)NC1=NC=NC2=C(C=CC=C12)C(N)=O)=O.NCCC(C1=CC=CC=C1)NC1=NC=NC2=C(C=CC=C12)C(=O)N QRWNIJRYSPRJQI-VZYDHVRKSA-N 0.000 description 1
- XVGWNXRXZBSCHZ-UHFFFAOYSA-N 4-[(3-aminophenyl)methylamino]-6-(hydroxymethyl)quinazoline-8-carboxamide;hydrochloride Chemical compound Cl.N1=CN=C2C(C(=O)N)=CC(CO)=CC2=C1NCC1=CC=CC(N)=C1 XVGWNXRXZBSCHZ-UHFFFAOYSA-N 0.000 description 1
- VJPPLCNBDLZIFG-ZDUSSCGKSA-N 4-[(3S)-3-(but-2-ynoylamino)piperidin-1-yl]-5-fluoro-2,3-dimethyl-1H-indole-7-carboxamide Chemical compound C(C#CC)(=O)N[C@@H]1CN(CCC1)C1=C2C(=C(NC2=C(C=C1F)C(=O)N)C)C VJPPLCNBDLZIFG-ZDUSSCGKSA-N 0.000 description 1
- ZRKPLPCYBYJEHV-UHFFFAOYSA-N 4-[(5-amino-5-piperidin-1-ylcyclohexa-1,3-dien-1-yl)-propylamino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1N(CCC)C(C1)=CC=CC1(N)N1CCCCC1 ZRKPLPCYBYJEHV-UHFFFAOYSA-N 0.000 description 1
- UZEGEPCLJOWZGN-UHFFFAOYSA-N 4-[1-[3-[(1-methylpyrrole-2-carbonyl)amino]phenyl]ethylamino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(C)C(C=1)=CC=CC=1NC(=O)C1=CC=CN1C UZEGEPCLJOWZGN-UHFFFAOYSA-N 0.000 description 1
- UOYTTXGBPMYPLE-SNVBAGLBSA-N 4-[[(1r)-1-(3-aminophenyl)ethyl]amino]quinazoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)C)=CC=CC(N)=C1 UOYTTXGBPMYPLE-SNVBAGLBSA-N 0.000 description 1
- FQJHFJNZUIRWLD-SNVBAGLBSA-N 4-[[(1r)-1-(3-nitrophenyl)ethyl]amino]quinazoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)C)=CC=CC([N+]([O-])=O)=C1 FQJHFJNZUIRWLD-SNVBAGLBSA-N 0.000 description 1
- QVKPUHCBABRGLM-JOCHJYFZSA-N 4-[[(1s)-2-[ethyl-(4-nitrophenyl)sulfonylamino]-1-phenylethyl]amino]quinazoline-8-carboxamide Chemical compound C1([C@H](NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)CN(CC)S(=O)(=O)C=2C=CC(=CC=2)[N+]([O-])=O)=CC=CC=C1 QVKPUHCBABRGLM-JOCHJYFZSA-N 0.000 description 1
- ZNBNDHNWIUYYAW-CQSZACIVSA-N 4-[[(1s)-2-azido-1-phenylethyl]amino]quinazoline-8-carboxamide Chemical compound C1([C@@H](CN=[N+]=[N-])NC2=C3C=CC=C(C3=NC=N2)C(=O)N)=CC=CC=C1 ZNBNDHNWIUYYAW-CQSZACIVSA-N 0.000 description 1
- LEZXTADOUHXXBO-QGZVFWFLSA-N 4-[[(1s)-2-azido-1-phenylethyl]amino]quinoline-8-carbonitrile Chemical compound C1([C@H](NC=2C3=CC=CC(=C3N=CC=2)C#N)CN=[N+]=[N-])=CC=CC=C1 LEZXTADOUHXXBO-QGZVFWFLSA-N 0.000 description 1
- ZRKPLPCYBYJEHV-QHCPKHFHSA-N 4-[[(5r)-5-amino-5-piperidin-1-ylcyclohexa-1,3-dien-1-yl]-propylamino]quinazoline-8-carboxamide Chemical compound N1([C@@]2(N)C=CC=C(C2)N(CCC)C=2C3=CC=CC(=C3N=CN=2)C(N)=O)CCCCC1 ZRKPLPCYBYJEHV-QHCPKHFHSA-N 0.000 description 1
- WKUKPCDTSWRVPY-QFIPXVFZSA-N 4-[[(5r)-5-amino-5-pyrrolidin-1-ylcyclohexa-1,3-dien-1-yl]-propylamino]quinazoline-8-carboxamide Chemical compound N1([C@@]2(N)C=CC=C(C2)N(CCC)C=2C3=CC=CC(=C3N=CN=2)C(N)=O)CCCC1 WKUKPCDTSWRVPY-QFIPXVFZSA-N 0.000 description 1
- FGCXCLHDTHADTE-UHFFFAOYSA-N 4-[[1-(3-aminophenyl)-2-hydroxyethyl]amino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NC(CO)C1=CC=CC(N)=C1 FGCXCLHDTHADTE-UHFFFAOYSA-N 0.000 description 1
- HZCSYARAUKXOFF-UHFFFAOYSA-N 4-[[1-(3-benzamidophenyl)-2-(methylamino)ethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C(C=1)=CC=CC=1NC(=O)C1=CC=CC=C1 HZCSYARAUKXOFF-UHFFFAOYSA-N 0.000 description 1
- AOBJHUQGPIRXMJ-UHFFFAOYSA-N 4-[[1-(3-benzamidophenyl)-3-pyrrolidin-1-ylpropyl]amino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NC(C=1C=C(NC(=O)C=2C=CC=CC=2)C=CC=1)CCN1CCCC1 AOBJHUQGPIRXMJ-UHFFFAOYSA-N 0.000 description 1
- VLEPKMISMIOUBK-UHFFFAOYSA-N 4-[[1-(3-chlorophenyl)-2-(dimethylamino)ethyl]amino]-6-phenylmethoxyquinazoline-8-carboxamide Chemical compound C=1C=CC(Cl)=CC=1C(CN(C)C)NC(C1=C2)=NC=NC1=C(C(N)=O)C=C2OCC1=CC=CC=C1 VLEPKMISMIOUBK-UHFFFAOYSA-N 0.000 description 1
- SZCJQZJURBSEEN-UHFFFAOYSA-N 4-[[1-[3-[(4-bromobenzoyl)amino]phenyl]-3-methoxypropyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CCOC)C(C=1)=CC=CC=1NC(=O)C1=CC=C(Br)C=C1 SZCJQZJURBSEEN-UHFFFAOYSA-N 0.000 description 1
- VKFMVGVEVCJSRM-UHFFFAOYSA-N 4-[[1-[3-[(5-cyclopropyl-1h-pyrazole-3-carbonyl)amino]phenyl]-3-oxopropyl]amino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NC(CC=O)C(C=1)=CC=CC=1NC(=O)C(NN=1)=CC=1C1CC1 VKFMVGVEVCJSRM-UHFFFAOYSA-N 0.000 description 1
- CXIHDFKIPQCBNU-UHFFFAOYSA-N 4-[[2-(dimethylamino)-1-(3-fluorophenyl)ethyl]amino]-6-phenylmethoxyquinazoline-8-carboxamide Chemical compound C=1C=CC(F)=CC=1C(CN(C)C)NC(C1=C2)=NC=NC1=C(C(N)=O)C=C2OCC1=CC=CC=C1 CXIHDFKIPQCBNU-UHFFFAOYSA-N 0.000 description 1
- YOBZFADVBDPXMF-UHFFFAOYSA-N 4-[[2-(methylamino)-1-phenylethyl]amino]quinazoline-8-carboxamide;hydrochloride Chemical compound Cl.N=1C=NC2=C(C(N)=O)C=CC=C2C=1NC(CNC)C1=CC=CC=C1 YOBZFADVBDPXMF-UHFFFAOYSA-N 0.000 description 1
- QWMDXMFYSJOKEB-UHFFFAOYSA-N 4-[[2-fluoro-5-[(4-methoxybenzoyl)amino]phenyl]methylamino]quinazoline-8-carboxamide Chemical compound C1=CC(OC)=CC=C1C(=O)NC1=CC=C(F)C(CNC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 QWMDXMFYSJOKEB-UHFFFAOYSA-N 0.000 description 1
- VSDTUEPFDSPDEE-UHFFFAOYSA-N 4-[[3-(dimethylamino)-1-[3-[(2-fluoro-4-methoxybenzoyl)amino]phenyl]propyl]amino]quinazoline-8-carboxamide Chemical compound FC1=CC(OC)=CC=C1C(=O)NC1=CC=CC(C(CCN(C)C)NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 VSDTUEPFDSPDEE-UHFFFAOYSA-N 0.000 description 1
- SGOPVPGKQMBONR-UHFFFAOYSA-N 4-[[3-(trifluoromethyl)phenyl]methylamino]quinazoline-8-carboxylic acid Chemical compound N1=CN=C2C(C(=O)O)=CC=CC2=C1NCC1=CC=CC(C(F)(F)F)=C1 SGOPVPGKQMBONR-UHFFFAOYSA-N 0.000 description 1
- NCLXOZZMSLYBND-UHFFFAOYSA-N 4-[[3-(trifluoromethyl)phenyl]methylamino]quinoline-8-carbonitrile Chemical compound FC(F)(F)C1=CC=CC(CNC=2C3=CC=CC(=C3N=CC=2)C#N)=C1 NCLXOZZMSLYBND-UHFFFAOYSA-N 0.000 description 1
- CGRQRIYZFRKJTH-UHFFFAOYSA-N 4-[[3-[(3-hydroxybenzoyl)amino]phenyl]methylamino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1NCC(C=1)=CC=CC=1NC(=O)C1=CC=CC(O)=C1 CGRQRIYZFRKJTH-UHFFFAOYSA-N 0.000 description 1
- IHJKNMFUUHSUNR-UHFFFAOYSA-N 4-[[3-[[5-[(1,3-dioxoisoindol-2-yl)methyl]-1,3-thiazol-2-yl]amino]phenyl]methylamino]quinazoline-8-carboxamide;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.O=C1C2=CC=CC=C2C(=O)N1CC(S1)=CN=C1NC1=CC(CNC2=C3C=CC=C(C3=NC=N2)C(=O)N)=CC=C1 IHJKNMFUUHSUNR-UHFFFAOYSA-N 0.000 description 1
- NWCVETNDCNXQME-UHFFFAOYSA-N 4-[[4-chloro-3-(trifluoromethyl)phenyl]methylamino]-6-nitroquinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC([N+]([O-])=O)=CC2=C1NCC1=CC=C(Cl)C(C(F)(F)F)=C1 NWCVETNDCNXQME-UHFFFAOYSA-N 0.000 description 1
- HLHJMHGNKSIYSU-UHFFFAOYSA-N 4-[[4-chloro-3-(trifluoromethyl)phenyl]methylamino]-6-nitroquinazoline-8-carboxylic acid Chemical compound N1=CN=C2C(C(=O)O)=CC([N+]([O-])=O)=CC2=C1NCC1=CC=C(Cl)C(C(F)(F)F)=C1 HLHJMHGNKSIYSU-UHFFFAOYSA-N 0.000 description 1
- CKSFNWDGZCXQON-UHFFFAOYSA-N 4-[[5-[(4-methoxybenzoyl)amino]-2-methylphenyl]methylamino]quinazoline-8-carboxamide Chemical compound C1=CC(OC)=CC=C1C(=O)NC1=CC=C(C)C(CNC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 CKSFNWDGZCXQON-UHFFFAOYSA-N 0.000 description 1
- 125000004042 4-aminobutyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])N([H])[H] 0.000 description 1
- 125000004800 4-bromophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Br 0.000 description 1
- MQKWVLSWSZHDGR-UHFFFAOYSA-N 4-bromoquinoline-8-carbonitrile Chemical compound C1=CC=C2C(Br)=CC=NC2=C1C#N MQKWVLSWSZHDGR-UHFFFAOYSA-N 0.000 description 1
- AFBRUNQKLYJRCA-UHFFFAOYSA-N 4-chloro-1-methylimidazo[4,5-c]pyridine Chemical compound N1=CC=C2N(C)C=NC2=C1Cl AFBRUNQKLYJRCA-UHFFFAOYSA-N 0.000 description 1
- GVRRXASZZAKBMN-UHFFFAOYSA-N 4-chloroquinazoline Chemical compound C1=CC=C2C(Cl)=NC=NC2=C1 GVRRXASZZAKBMN-UHFFFAOYSA-N 0.000 description 1
- OXKQMBHOOISBLS-UHFFFAOYSA-N 4-chloroquinoline-8-carbonitrile Chemical compound C1=CC=C2C(Cl)=CC=NC2=C1C#N OXKQMBHOOISBLS-UHFFFAOYSA-N 0.000 description 1
- 125000004176 4-fluorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1F)C([H])([H])* 0.000 description 1
- 125000006306 4-iodophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1I 0.000 description 1
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- QRAOZQGIUIDZQZ-UHFFFAOYSA-N 4-methyl-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3-dihydro-1,4-benzoxazine Chemical compound C=1C=C2N(C)CCOC2=CC=1B1OC(C)(C)C(C)(C)O1 QRAOZQGIUIDZQZ-UHFFFAOYSA-N 0.000 description 1
- GCHMJLGIJQKFDV-UHFFFAOYSA-N 4-morpholin-4-ylquinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC=CC2=C1N1CCOCC1 GCHMJLGIJQKFDV-UHFFFAOYSA-N 0.000 description 1
- JXRGUPLJCCDGKG-UHFFFAOYSA-N 4-nitrobenzenesulfonyl chloride Chemical compound [O-][N+](=O)C1=CC=C(S(Cl)(=O)=O)C=C1 JXRGUPLJCCDGKG-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical compound [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 1
- 102100036009 5'-AMP-activated protein kinase catalytic subunit alpha-2 Human genes 0.000 description 1
- HHTRAISBAAXRKZ-UHFFFAOYSA-N 5-amino-2-fluorobenzonitrile Chemical compound NC1=CC=C(F)C(C#N)=C1 HHTRAISBAAXRKZ-UHFFFAOYSA-N 0.000 description 1
- YDZVQWCVKXYGIU-UHFFFAOYSA-N 5-amino-2-methylbenzonitrile Chemical compound CC1=CC=C(N)C=C1C#N YDZVQWCVKXYGIU-UHFFFAOYSA-N 0.000 description 1
- 125000004539 5-benzimidazolyl group Chemical group N1=CNC2=C1C=CC(=C2)* 0.000 description 1
- GNWMHLBUCXEXPE-UHFFFAOYSA-N 5-cyclopropyl-1h-pyrazole-3-carboxylic acid Chemical compound N1N=C(C(=O)O)C=C1C1CC1 GNWMHLBUCXEXPE-UHFFFAOYSA-N 0.000 description 1
- YJEVNITWKNWUKX-UHFFFAOYSA-N 5-morpholin-4-ylpyridine-3-carboxylic acid Chemical compound OC(=O)C1=CN=CC(N2CCOCC2)=C1 YJEVNITWKNWUKX-UHFFFAOYSA-N 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- OMMPPDGBYMQCGF-UHFFFAOYSA-N 6-(2-morpholin-4-ylethoxy)-4-[[4-(trifluoromethyl)phenyl]methylamino]quinazoline-8-carboxamide Chemical compound C=1C2=C(NCC=3C=CC(=CC=3)C(F)(F)F)N=CN=C2C(C(=O)N)=CC=1OCCN1CCOCC1 OMMPPDGBYMQCGF-UHFFFAOYSA-N 0.000 description 1
- SWYUPOALZWKXQY-UHFFFAOYSA-N 6-(hydroxymethyl)-4-[[3-[(4-methoxybenzoyl)amino]phenyl]methylamino]quinazoline-8-carboxamide Chemical compound C1=CC(OC)=CC=C1C(=O)NC1=CC=CC(CNC=2C3=CC(CO)=CC(=C3N=CN=2)C(N)=O)=C1 SWYUPOALZWKXQY-UHFFFAOYSA-N 0.000 description 1
- ZKBQDFAWXLTYKS-UHFFFAOYSA-N 6-Chloro-1H-purine Chemical compound ClC1=NC=NC2=C1NC=N2 ZKBQDFAWXLTYKS-UHFFFAOYSA-N 0.000 description 1
- NVDJVEQITUWZDT-UHFFFAOYSA-N 6-Methoxy-pyridine-3-carboxylic acid Chemical compound COC1=CC=C(C(O)=O)C=N1 NVDJVEQITUWZDT-UHFFFAOYSA-N 0.000 description 1
- ILQICKVTZSTVOE-UHFFFAOYSA-N 6-[3-(dimethylamino)propoxy]-4-[[2-(methylamino)-1-phenylethyl]amino]quinazoline-8-carboxamide Chemical compound N=1C=NC2=C(C(N)=O)C=C(OCCCN(C)C)C=C2C=1NC(CNC)C1=CC=CC=C1 ILQICKVTZSTVOE-UHFFFAOYSA-N 0.000 description 1
- SLXKOJJOQWFEFD-UHFFFAOYSA-N 6-aminohexanoic acid Chemical compound NCCCCCC(O)=O SLXKOJJOQWFEFD-UHFFFAOYSA-N 0.000 description 1
- ORMLOSONIQIDBI-UHFFFAOYSA-N 6-hydroxy-4-[[4-(trifluoromethyl)phenyl]methylamino]quinazoline-8-carboxamide Chemical compound N1=CN=C2C(C(=O)N)=CC(O)=CC2=C1NCC1=CC=C(C(F)(F)F)C=C1 ORMLOSONIQIDBI-UHFFFAOYSA-N 0.000 description 1
- ZAUBVRZBSPZYHI-UHFFFAOYSA-N 6-methoxy-4-oxo-1h-quinazoline-8-carboxylic acid Chemical compound N1=CNC(=O)C2=CC(OC)=CC(C(O)=O)=C21 ZAUBVRZBSPZYHI-UHFFFAOYSA-N 0.000 description 1
- LFBFLVREWCTKLY-UHFFFAOYSA-N 6-nitro-4-oxo-1h-quinazoline-8-carboxylic acid Chemical compound N1=CNC(=O)C2=C1C(C(=O)O)=CC([N+]([O-])=O)=C2 LFBFLVREWCTKLY-UHFFFAOYSA-N 0.000 description 1
- BBRDYPDWYYISDZ-UHFFFAOYSA-N 6-phenylmethoxy-4-[[4-(trifluoromethyl)phenyl]methylamino]quinazoline-8-carbonitrile Chemical compound C1=CC(C(F)(F)F)=CC=C1CNC(C1=C2)=NC=NC1=C(C#N)C=C2OCC1=CC=CC=C1 BBRDYPDWYYISDZ-UHFFFAOYSA-N 0.000 description 1
- ZVRDIELKVSLWLQ-UHFFFAOYSA-N 6-phenylmethoxy-4-[[4-(trifluoromethyl)phenyl]methylamino]quinazoline-8-carboxamide Chemical compound C=1C2=C(NCC=3C=CC(=CC=3)C(F)(F)F)N=CN=C2C(C(=O)N)=CC=1OCC1=CC=CC=C1 ZVRDIELKVSLWLQ-UHFFFAOYSA-N 0.000 description 1
- UNQYAAAWKOOBFQ-UHFFFAOYSA-N 7-[(4-chlorophenyl)methyl]-8-[4-chloro-3-(trifluoromethoxy)phenoxy]-1-(3-hydroxypropyl)-3-methylpurine-2,6-dione Chemical compound C=1C=C(Cl)C=CC=1CN1C=2C(=O)N(CCCO)C(=O)N(C)C=2N=C1OC1=CC=C(Cl)C(OC(F)(F)F)=C1 UNQYAAAWKOOBFQ-UHFFFAOYSA-N 0.000 description 1
- 108010013238 70-kDa Ribosomal Protein S6 Kinases Proteins 0.000 description 1
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 208000010507 Adenocarcinoma of Lung Diseases 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- 102100033972 Amyloid protein-binding protein 2 Human genes 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 102000042871 Aurora family Human genes 0.000 description 1
- 108091082291 Aurora family Proteins 0.000 description 1
- 108700020463 BRCA1 Proteins 0.000 description 1
- 102000036365 BRCA1 Human genes 0.000 description 1
- 101150072950 BRCA1 gene Proteins 0.000 description 1
- 108700020462 BRCA2 Proteins 0.000 description 1
- 102000052609 BRCA2 Human genes 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- 101150008921 Brca2 gene Proteins 0.000 description 1
- FGUUSXIOTUKUDN-IBGZPJMESA-N C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 Chemical compound C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 FGUUSXIOTUKUDN-IBGZPJMESA-N 0.000 description 1
- 101100356682 Caenorhabditis elegans rho-1 gene Proteins 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 229920002284 Cellulose triacetate Polymers 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- 102100034484 DNA repair protein RAD51 homolog 3 Human genes 0.000 description 1
- 102100036912 Desmin Human genes 0.000 description 1
- 108010044052 Desmin Proteins 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 101000785279 Dictyostelium discoideum Calcium-transporting ATPase PAT1 Proteins 0.000 description 1
- 241000255925 Diptera Species 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 102100021238 Dynamin-2 Human genes 0.000 description 1
- 241000854350 Enicospilus group Species 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 241000975394 Evechinus chloroticus Species 0.000 description 1
- 101150025764 FGFR3 gene Proteins 0.000 description 1
- 101150009958 FLT4 gene Proteins 0.000 description 1
- GISRWBROCYNDME-PELMWDNLSA-N F[C@H]1[C@H]([C@H](NC1=O)COC1=NC=CC2=CC(=C(C=C12)OC)C(=O)N)C Chemical compound F[C@H]1[C@H]([C@H](NC1=O)COC1=NC=CC2=CC(=C(C=C12)OC)C(=O)N)C GISRWBROCYNDME-PELMWDNLSA-N 0.000 description 1
- 102100037813 Focal adhesion kinase 1 Human genes 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 102000053171 Glial Fibrillary Acidic Human genes 0.000 description 1
- 101710193519 Glial fibrillary acidic protein Proteins 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000783681 Homo sapiens 5'-AMP-activated protein kinase catalytic subunit alpha-2 Proteins 0.000 description 1
- 101000779309 Homo sapiens Amyloid protein-binding protein 2 Proteins 0.000 description 1
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 1
- 101001132271 Homo sapiens DNA repair protein RAD51 homolog 3 Proteins 0.000 description 1
- 101000817607 Homo sapiens Dynamin-2 Proteins 0.000 description 1
- 101000878536 Homo sapiens Focal adhesion kinase 1 Proteins 0.000 description 1
- 101000599951 Homo sapiens Insulin-like growth factor I Proteins 0.000 description 1
- 101000628949 Homo sapiens Mitogen-activated protein kinase 10 Proteins 0.000 description 1
- 101000692455 Homo sapiens Platelet-derived growth factor receptor beta Proteins 0.000 description 1
- 101000713296 Homo sapiens Proton-coupled amino acid transporter 1 Proteins 0.000 description 1
- 101001106406 Homo sapiens Rho GTPase-activating protein 1 Proteins 0.000 description 1
- 101000944909 Homo sapiens Ribosomal protein S6 kinase alpha-1 Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101000864800 Homo sapiens Serine/threonine-protein kinase Sgk1 Proteins 0.000 description 1
- 101000864342 Homo sapiens Tyrosine-protein kinase BTK Proteins 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 102100021854 Inhibitor of nuclear factor kappa-B kinase subunit beta Human genes 0.000 description 1
- 101710205525 Inhibitor of nuclear factor kappa-B kinase subunit beta Proteins 0.000 description 1
- 102100037852 Insulin-like growth factor I Human genes 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- 101150009057 JAK2 gene Proteins 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- 101150081525 LIMK1 gene Proteins 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 1
- 208000001145 Metabolic Syndrome Diseases 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 102000029749 Microtubule Human genes 0.000 description 1
- 108091022875 Microtubule Proteins 0.000 description 1
- 102100026931 Mitogen-activated protein kinase 10 Human genes 0.000 description 1
- 102100026888 Mitogen-activated protein kinase kinase kinase 7 Human genes 0.000 description 1
- 208000034578 Multiple myelomas Diseases 0.000 description 1
- 101100335081 Mus musculus Flt3 gene Proteins 0.000 description 1
- 101100268066 Mus musculus Zap70 gene Proteins 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- 102000003505 Myosin Human genes 0.000 description 1
- 108060008487 Myosin Proteins 0.000 description 1
- FEYNFHSRETUBEM-UHFFFAOYSA-N N-[3-(1,1-difluoroethyl)phenyl]-1-(4-methoxyphenyl)-3-methyl-5-oxo-4H-pyrazole-4-carboxamide Chemical compound COc1ccc(cc1)N1N=C(C)C(C(=O)Nc2cccc(c2)C(C)(F)F)C1=O FEYNFHSRETUBEM-UHFFFAOYSA-N 0.000 description 1
- MVFWIFOFNJEDPL-UHFFFAOYSA-N N-[3-[3-(dimethylamino)-1-(quinazolin-4-ylamino)propyl]phenyl]-2,6-difluorobenzamide Chemical compound CN(CCC(C1=CC(=CC=C1)NC(C1=C(C=CC=C1F)F)=O)NC1=NC=NC2=CC=CC=C12)C MVFWIFOFNJEDPL-UHFFFAOYSA-N 0.000 description 1
- 229910017912 NH2OH Inorganic materials 0.000 description 1
- 101150111783 NTRK1 gene Proteins 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 1
- 206010029719 Nonspecific reaction Diseases 0.000 description 1
- 101150073900 PTEN gene Proteins 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 108090000315 Protein Kinase C Proteins 0.000 description 1
- 102000003923 Protein Kinase C Human genes 0.000 description 1
- 102000005569 Protein Phosphatase 1 Human genes 0.000 description 1
- 108010059000 Protein Phosphatase 1 Proteins 0.000 description 1
- 102000009516 Protein Serine-Threonine Kinases Human genes 0.000 description 1
- 108010009341 Protein Serine-Threonine Kinases Proteins 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 101150111584 RHOA gene Proteins 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 108050002653 Retinoblastoma protein Proteins 0.000 description 1
- 102100021433 Rho GTPase-activating protein 1 Human genes 0.000 description 1
- 108010034782 Ribosomal Protein S6 Kinases Proteins 0.000 description 1
- 102000009738 Ribosomal Protein S6 Kinases Human genes 0.000 description 1
- 102100033536 Ribosomal protein S6 kinase alpha-1 Human genes 0.000 description 1
- 101150001535 SRC gene Proteins 0.000 description 1
- 101001092180 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) RHO GTPase-activating protein RGD1 Proteins 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- 102100030070 Serine/threonine-protein kinase Sgk1 Human genes 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 102000001742 Tumor Suppressor Proteins Human genes 0.000 description 1
- 108010040002 Tumor Suppressor Proteins Proteins 0.000 description 1
- 102100029823 Tyrosine-protein kinase BTK Human genes 0.000 description 1
- 102000016549 Vascular Endothelial Growth Factor Receptor-2 Human genes 0.000 description 1
- 108010053100 Vascular Endothelial Growth Factor Receptor-3 Proteins 0.000 description 1
- 102000013127 Vimentin Human genes 0.000 description 1
- 108010065472 Vimentin Proteins 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- NNLVGZFZQQXQNW-ADJNRHBOSA-N [(2r,3r,4s,5r,6s)-4,5-diacetyloxy-3-[(2s,3r,4s,5r,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6s)-4,5,6-triacetyloxy-2-(acetyloxymethyl)oxan-3-yl]oxyoxan-2-yl]methyl acetate Chemical compound O([C@@H]1O[C@@H]([C@H]([C@H](OC(C)=O)[C@H]1OC(C)=O)O[C@H]1[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O1)OC(C)=O)COC(=O)C)[C@@H]1[C@@H](COC(C)=O)O[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O NNLVGZFZQQXQNW-ADJNRHBOSA-N 0.000 description 1
- QMCZMSKKAHWYBJ-GFCCVEGCSA-N [(2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-2-phenylethyl] methanesulfonate Chemical compound CC(C)(C)OC(=O)N[C@H](COS(C)(=O)=O)C1=CC=CC=C1 QMCZMSKKAHWYBJ-GFCCVEGCSA-N 0.000 description 1
- PRDBLLIPPDOICK-UHFFFAOYSA-N [4-(trifluoromethyl)phenyl]methanamine Chemical compound NCC1=CC=C(C(F)(F)F)C=C1 PRDBLLIPPDOICK-UHFFFAOYSA-N 0.000 description 1
- SRHQOQMNBVOXCF-UHFFFAOYSA-N [4-chloro-3-(trifluoromethyl)phenyl]methanamine Chemical compound NCC1=CC=C(Cl)C(C(F)(F)F)=C1 SRHQOQMNBVOXCF-UHFFFAOYSA-N 0.000 description 1
- DGYIJVNZSDYBOE-UHFFFAOYSA-N [CH2]C1=CC=NC=C1 Chemical group [CH2]C1=CC=NC=C1 DGYIJVNZSDYBOE-UHFFFAOYSA-N 0.000 description 1
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- IKHGUXGNUITLKF-XPULMUKRSA-N acetaldehyde Chemical compound [14CH]([14CH3])=O IKHGUXGNUITLKF-XPULMUKRSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 238000010640 amide synthesis reaction Methods 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229960002684 aminocaproic acid Drugs 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- 125000003705 anilinocarbonyl group Chemical group O=C([*])N([H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- MXMOTZIXVICDSD-UHFFFAOYSA-N anisoyl chloride Chemical compound COC1=CC=C(C(Cl)=O)C=C1 MXMOTZIXVICDSD-UHFFFAOYSA-N 0.000 description 1
- 230000002280 anti-androgenic effect Effects 0.000 description 1
- 230000003388 anti-hormonal effect Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000000051 antiandrogen Substances 0.000 description 1
- 229940030495 antiandrogen sex hormone and modulator of the genital system Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 125000000732 arylene group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 238000002820 assay format Methods 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 230000015097 attachment of spindle microtubules to kinetochore Effects 0.000 description 1
- 230000035578 autophosphorylation Effects 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- CSKNSYBAZOQPLR-UHFFFAOYSA-N benzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC=C1 CSKNSYBAZOQPLR-UHFFFAOYSA-N 0.000 description 1
- IDIPWEYIBKUDNY-UHFFFAOYSA-N benzenesulfonyl fluoride Chemical compound FS(=O)(=O)C1=CC=CC=C1 IDIPWEYIBKUDNY-UHFFFAOYSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- RDHPKYGYEGBMSE-UHFFFAOYSA-N bromoethane Chemical compound CCBr RDHPKYGYEGBMSE-UHFFFAOYSA-N 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical class C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 239000003560 cancer drug Substances 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 230000006369 cell cycle progression Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 210000003793 centrosome Anatomy 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 150000001793 charged compounds Chemical class 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- DGLFSNZWRYADFC-UHFFFAOYSA-N chembl2334586 Chemical compound C1CCC2=CN=C(N)N=C2C2=C1NC1=CC=C(C#CC(C)(O)C)C=C12 DGLFSNZWRYADFC-UHFFFAOYSA-N 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 239000007958 cherry flavor Substances 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 208000037976 chronic inflammation Diseases 0.000 description 1
- 208000037893 chronic inflammatory disorder Diseases 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 201000011024 colonic benign neoplasm Diseases 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 230000005757 colony formation Effects 0.000 description 1
- 238000005056 compaction Methods 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 108010057108 condensin complexes Proteins 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000004966 cyanoalkyl group Chemical group 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- UKJLNMAFNRKWGR-UHFFFAOYSA-N cyclohexatrienamine Chemical group NC1=CC=C=C[CH]1 UKJLNMAFNRKWGR-UHFFFAOYSA-N 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 210000000172 cytosol Anatomy 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 229960001251 denosumab Drugs 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000030609 dephosphorylation Effects 0.000 description 1
- 238000006209 dephosphorylation reaction Methods 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 210000005045 desmin Anatomy 0.000 description 1
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- CDHICTNQMQYRSM-UHFFFAOYSA-N di(propan-2-yl)alumane Chemical compound CC(C)[AlH]C(C)C CDHICTNQMQYRSM-UHFFFAOYSA-N 0.000 description 1
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- WBKFWQBXFREOFH-UHFFFAOYSA-N dichloromethane;ethyl acetate Chemical compound ClCCl.CCOC(C)=O WBKFWQBXFREOFH-UHFFFAOYSA-N 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- 230000007783 downstream signaling Effects 0.000 description 1
- 229940125436 dual inhibitor Drugs 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 229960001776 edrecolomab Drugs 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 150000002085 enols Chemical group 0.000 description 1
- HKSZLNNOFSGOKW-UHFFFAOYSA-N ent-staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 HKSZLNNOFSGOKW-UHFFFAOYSA-N 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 230000014392 establishment of spindle localization Effects 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- ASCRXOPBUNVSNJ-UHFFFAOYSA-N ethyl 2-[3-[1-[(8-carbamoylquinazolin-4-yl)amino]ethyl]anilino]-1,3-oxazole-5-carboxylate Chemical compound O1C(C(=O)OCC)=CN=C1NC1=CC=CC(C(C)NC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 ASCRXOPBUNVSNJ-UHFFFAOYSA-N 0.000 description 1
- 239000002024 ethyl acetate extract Substances 0.000 description 1
- DNJIEGIFACGWOD-UHFFFAOYSA-N ethyl mercaptane Natural products CCS DNJIEGIFACGWOD-UHFFFAOYSA-N 0.000 description 1
- SFNALCNOMXIBKG-UHFFFAOYSA-N ethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCO SFNALCNOMXIBKG-UHFFFAOYSA-N 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 238000001125 extrusion Methods 0.000 description 1
- 239000010685 fatty oil Substances 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 210000005046 glial fibrillary acidic protein Anatomy 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- HHLFWLYXYJOTON-UHFFFAOYSA-N glyoxylic acid Chemical compound OC(=O)C=O HHLFWLYXYJOTON-UHFFFAOYSA-N 0.000 description 1
- 239000003979 granulating agent Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 239000008241 heterogeneous mixture Substances 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 235000014304 histidine Nutrition 0.000 description 1
- 150000002411 histidines Chemical class 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 238000013415 human tumor xenograft model Methods 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 229940071676 hydroxypropylcellulose Drugs 0.000 description 1
- 201000001421 hyperglycemia Diseases 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 239000000367 immunologic factor Substances 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 229940060367 inert ingredients Drugs 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 239000012444 intercalating antibiotic Substances 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 238000000021 kinase assay Methods 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 1
- 229950002950 lintuzumab Drugs 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 125000000040 m-tolyl group Chemical group [H]C1=C([H])C(*)=C([H])C(=C1[H])C([H])([H])[H] 0.000 description 1
- UEGPKNKPLBYCNK-UHFFFAOYSA-L magnesium acetate Chemical compound [Mg+2].CC([O-])=O.CC([O-])=O UEGPKNKPLBYCNK-UHFFFAOYSA-L 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 230000021121 meiosis Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 230000031864 metaphase Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- DYRRGNKJTXKVCX-UHFFFAOYSA-N methyl 3-[(2-methylpropan-2-yl)oxycarbonylamino]-3-(3-nitrophenyl)propanoate Chemical compound CC(C)(C)OC(=O)NC(CC(=O)OC)C1=CC=CC([N+]([O-])=O)=C1 DYRRGNKJTXKVCX-UHFFFAOYSA-N 0.000 description 1
- JLITVWGQYVGHRZ-UHFFFAOYSA-N methyl 4,6-dichloroquinazoline-8-carboxylate Chemical compound N1=CN=C2C(C(=O)OC)=CC(Cl)=CC2=C1Cl JLITVWGQYVGHRZ-UHFFFAOYSA-N 0.000 description 1
- RKFUKUMWXUPZBB-UHFFFAOYSA-N methyl 4-[(2-methylpropan-2-yl)oxycarbonylamino]-4-(3-nitrophenyl)butanoate Chemical compound COC(=O)CCC(NC(=O)OC(C)(C)C)C1=CC=CC([N+]([O-])=O)=C1 RKFUKUMWXUPZBB-UHFFFAOYSA-N 0.000 description 1
- CNVFYELKJXIOFN-LLVKDONJSA-N methyl 4-[[(1r)-1-(3-nitrophenyl)ethyl]amino]quinazoline-8-carboxylate Chemical compound C1([C@@H](C)NC2=C3C=CC=C(C3=NC=N2)C(=O)OC)=CC=CC([N+]([O-])=O)=C1 CNVFYELKJXIOFN-LLVKDONJSA-N 0.000 description 1
- PWRZFYWVXZJIRI-UHFFFAOYSA-N methyl 4-[[1-[3-[(2-chloropyridine-4-carbonyl)amino]phenyl]-2-(dimethylamino)ethyl]amino]quinazoline-8-carboxylate Chemical compound N1=CN=C2C(C(=O)OC)=CC=CC2=C1NC(CN(C)C)C(C=1)=CC=CC=1NC(=O)C1=CC=NC(Cl)=C1 PWRZFYWVXZJIRI-UHFFFAOYSA-N 0.000 description 1
- GEWKUMNCVZQXJP-UHFFFAOYSA-N methyl 4-[[3-(trifluoromethyl)phenyl]methylamino]quinazoline-8-carboxylate Chemical compound N1=CN=C2C(C(=O)OC)=CC=CC2=C1NCC1=CC=CC(C(F)(F)F)=C1 GEWKUMNCVZQXJP-UHFFFAOYSA-N 0.000 description 1
- KUAFHKVSEUQECY-UHFFFAOYSA-N methyl 4-[[3-[(4-methoxybenzoyl)-methylamino]phenyl]methylamino]quinazoline-8-carboxylate Chemical compound N1=CN=C2C(C(=O)OC)=CC=CC2=C1NCC(C=1)=CC=CC=1N(C)C(=O)C1=CC=C(OC)C=C1 KUAFHKVSEUQECY-UHFFFAOYSA-N 0.000 description 1
- LXIPCHFJMNLFMF-UHFFFAOYSA-N methyl 4-chloro-6-methoxyquinazoline-8-carboxylate Chemical compound N1=CN=C2C(C(=O)OC)=CC(OC)=CC2=C1Cl LXIPCHFJMNLFMF-UHFFFAOYSA-N 0.000 description 1
- NTQOSXFAJKWLCU-UHFFFAOYSA-N methyl 4-chloro-6-phenylmethoxyquinazoline-8-carboxylate Chemical compound C=1C2=C(Cl)N=CN=C2C(C(=O)OC)=CC=1OCC1=CC=CC=C1 NTQOSXFAJKWLCU-UHFFFAOYSA-N 0.000 description 1
- AAJZXPWBILCHAW-UHFFFAOYSA-N methyl 5-bromopyridine-3-carboxylate Chemical compound COC(=O)C1=CN=CC(Br)=C1 AAJZXPWBILCHAW-UHFFFAOYSA-N 0.000 description 1
- JKKSUVYKLLZBOJ-UHFFFAOYSA-N methyl 5-morpholin-4-ylpyridine-3-carboxylate Chemical compound COC(=O)C1=CN=CC(N2CCOCC2)=C1 JKKSUVYKLLZBOJ-UHFFFAOYSA-N 0.000 description 1
- FHKUIGCQUJXPSI-UHFFFAOYSA-N methyl 6-chloro-4-[1-[3-[(2-methylpropan-2-yl)oxycarbonylamino]phenyl]ethylamino]quinazoline-8-carboxylate Chemical compound N1=CN=C2C(C(=O)OC)=CC(Cl)=CC2=C1NC(C)C1=CC=CC(NC(=O)OC(C)(C)C)=C1 FHKUIGCQUJXPSI-UHFFFAOYSA-N 0.000 description 1
- KTBYZONFSIWERM-ZDUSSCGKSA-N methyl 6-iodo-4-[[(3s)-1-[(2-methylpropan-2-yl)oxycarbonyl]piperidin-3-yl]amino]quinazoline-8-carboxylate Chemical compound N1=CN=C2C(C(=O)OC)=CC(I)=CC2=C1N[C@H]1CCCN(C(=O)OC(C)(C)C)C1 KTBYZONFSIWERM-ZDUSSCGKSA-N 0.000 description 1
- ZVJNIISINPNCPE-UHFFFAOYSA-N methyl 6-nitro-4-oxo-1h-quinazoline-8-carboxylate Chemical compound N1=CNC(=O)C2=C1C(C(=O)OC)=CC([N+]([O-])=O)=C2 ZVJNIISINPNCPE-UHFFFAOYSA-N 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- 239000003226 mitogen Substances 0.000 description 1
- 230000008747 mitogenic response Effects 0.000 description 1
- 125000004312 morpholin-2-yl group Chemical group [H]N1C([H])([H])C([H])([H])OC([H])(*)C1([H])[H] 0.000 description 1
- 125000004572 morpholin-3-yl group Chemical group N1C(COCC1)* 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 230000004899 motility Effects 0.000 description 1
- 210000000066 myeloid cell Anatomy 0.000 description 1
- JFPYVWYSQXLONF-UHFFFAOYSA-N n-(3-cyanophenyl)-4-methoxy-n-methylbenzamide Chemical compound C1=CC(OC)=CC=C1C(=O)N(C)C1=CC=CC(C#N)=C1 JFPYVWYSQXLONF-UHFFFAOYSA-N 0.000 description 1
- HXDBEVLWHUIGRV-UHFFFAOYSA-N n-(3-cyanophenyl)-4-methoxybenzamide Chemical compound C1=CC(OC)=CC=C1C(=O)NC1=CC=CC(C#N)=C1 HXDBEVLWHUIGRV-UHFFFAOYSA-N 0.000 description 1
- FMASTMURQSHELY-UHFFFAOYSA-N n-(4-fluoro-2-methylphenyl)-3-methyl-n-[(2-methyl-1h-indol-4-yl)methyl]pyridine-4-carboxamide Chemical compound C1=CC=C2NC(C)=CC2=C1CN(C=1C(=CC(F)=CC=1)C)C(=O)C1=CC=NC=C1C FMASTMURQSHELY-UHFFFAOYSA-N 0.000 description 1
- HIFQSGLCAZLODB-UHFFFAOYSA-N n-(benzenesulfonyl)-n-(5-bromopyridin-3-yl)benzenesulfonamide Chemical compound BrC1=CN=CC(N(S(=O)(=O)C=2C=CC=CC=2)S(=O)(=O)C=2C=CC=CC=2)=C1 HIFQSGLCAZLODB-UHFFFAOYSA-N 0.000 description 1
- LRJILWRMLJFZEX-MRXNPFEDSA-N n-[(2s)-2-amino-2-phenylethyl]-n-ethyl-4-nitrobenzenesulfonamide Chemical compound C1([C@H](N)CN(CC)S(=O)(=O)C=2C=CC(=CC=2)[N+]([O-])=O)=CC=CC=C1 LRJILWRMLJFZEX-MRXNPFEDSA-N 0.000 description 1
- JQAQYRGSABEZBJ-UHFFFAOYSA-N n-[2-(dimethylamino)-1-(3-methoxyphenyl)ethylidene]hydroxylamine Chemical compound COC1=CC=CC(C(CN(C)C)=NO)=C1 JQAQYRGSABEZBJ-UHFFFAOYSA-N 0.000 description 1
- JMHLMDXDEGJECY-UHFFFAOYSA-N n-[3-(aminomethyl)phenyl]-6-methoxypyridine-3-carboxamide Chemical compound C1=NC(OC)=CC=C1C(=O)NC1=CC=CC(CN)=C1 JMHLMDXDEGJECY-UHFFFAOYSA-N 0.000 description 1
- SQLDPDURRFUEHG-UHFFFAOYSA-N n-[3-[1-amino-2-(dimethylamino)ethyl]phenyl]-2-chloropyridine-4-carboxamide Chemical compound CN(C)CC(N)C1=CC=CC(NC(=O)C=2C=C(Cl)N=CC=2)=C1 SQLDPDURRFUEHG-UHFFFAOYSA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- DOWVMJFBDGWVML-UHFFFAOYSA-N n-cyclohexyl-n-methyl-4-(1-oxidopyridin-1-ium-3-yl)imidazole-1-carboxamide Chemical compound C1=NC(C=2C=[N+]([O-])C=CC=2)=CN1C(=O)N(C)C1CCCCC1 DOWVMJFBDGWVML-UHFFFAOYSA-N 0.000 description 1
- GTUAWMBSCRFHKR-UHFFFAOYSA-N n-ethyl-4-nitrobenzenesulfonamide Chemical compound CCNS(=O)(=O)C1=CC=C([N+]([O-])=O)C=C1 GTUAWMBSCRFHKR-UHFFFAOYSA-N 0.000 description 1
- YZMHQCWXYHARLS-UHFFFAOYSA-N naphthalene-1,2-disulfonic acid Chemical class C1=CC=CC2=C(S(O)(=O)=O)C(S(=O)(=O)O)=CC=C21 YZMHQCWXYHARLS-UHFFFAOYSA-N 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000009826 neoplastic cell growth Effects 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 125000003261 o-tolyl group Chemical group [H]C1=C([H])C(*)=C(C([H])=C1[H])C([H])([H])[H] 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 210000000287 oocyte Anatomy 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 239000007968 orange flavor Substances 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 1
- 125000000636 p-nitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)[N+]([O-])=O 0.000 description 1
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 150000003906 phosphoinositides Chemical class 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 210000004508 polar body Anatomy 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 238000010837 poor prognosis Methods 0.000 description 1
- LZMJNVRJMFMYQS-UHFFFAOYSA-N poseltinib Chemical compound C1CN(C)CCN1C(C=C1)=CC=C1NC1=NC(OC=2C=C(NC(=O)C=C)C=CC=2)=C(OC=C2)C2=N1 LZMJNVRJMFMYQS-UHFFFAOYSA-N 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical group CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 239000003909 protein kinase inhibitor Substances 0.000 description 1
- 230000009822 protein phosphorylation Effects 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 230000004850 protein–protein interaction Effects 0.000 description 1
- 125000002206 pyridazin-3-yl group Chemical group [H]C1=C([H])C([H])=C(*)N=N1 0.000 description 1
- 125000004940 pyridazin-4-yl group Chemical group N1=NC=C(C=C1)* 0.000 description 1
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 1
- 125000005551 pyridylene group Chemical group 0.000 description 1
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 1
- 125000004943 pyrimidin-6-yl group Chemical group N1=CN=CC=C1* 0.000 description 1
- 125000004547 quinazolin-6-yl group Chemical group N1=CN=CC2=CC(=CC=C12)* 0.000 description 1
- UPGZAERNNNZSPM-UHFFFAOYSA-N quinazoline-8-carboxylic acid Chemical compound N1=CN=C2C(C(=O)O)=CC=CC2=C1 UPGZAERNNNZSPM-UHFFFAOYSA-N 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- HPQRQAOVNXWEEQ-UHFFFAOYSA-N quinoline-8-carboxamide Chemical compound C1=CN=C2C(C(=O)N)=CC=CC2=C1 HPQRQAOVNXWEEQ-UHFFFAOYSA-N 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 230000022983 regulation of cell cycle Effects 0.000 description 1
- 230000024122 regulation of cell motility Effects 0.000 description 1
- 102000037983 regulatory factors Human genes 0.000 description 1
- 108091008025 regulatory factors Proteins 0.000 description 1
- 230000008844 regulatory mechanism Effects 0.000 description 1
- 201000010174 renal carcinoma Diseases 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 125000003548 sec-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 230000004747 sister chromatid biorientation Effects 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical compound COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 1
- 230000020347 spindle assembly Effects 0.000 description 1
- 230000024355 spindle assembly checkpoint Effects 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical compound C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 1
- CGPUWJWCVCFERF-UHFFFAOYSA-N staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(OC)O1 CGPUWJWCVCFERF-UHFFFAOYSA-N 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000000967 suction filtration Methods 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- IIACRCGMVDHOTQ-UHFFFAOYSA-N sulfamic acid Chemical compound NS(O)(=O)=O IIACRCGMVDHOTQ-UHFFFAOYSA-N 0.000 description 1
- 125000001010 sulfinic acid amide group Chemical group 0.000 description 1
- 239000001117 sulphuric acid Substances 0.000 description 1
- 235000011149 sulphuric acid Nutrition 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000004654 survival pathway Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229960000235 temsirolimus Drugs 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- QZEQMEOSQLWVRN-SFHVURJKSA-N tert-butyl (3s)-3-[(8-carbamoyl-6-phenylquinazolin-4-yl)amino]piperidine-1-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCC[C@@H]1NC1=NC=NC2=C(C(N)=O)C=C(C=3C=CC=CC=3)C=C12 QZEQMEOSQLWVRN-SFHVURJKSA-N 0.000 description 1
- AKQXKEBCONUWCL-QMMMGPOBSA-N tert-butyl (3s)-3-aminopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC[C@H](N)C1 AKQXKEBCONUWCL-QMMMGPOBSA-N 0.000 description 1
- ZKMVGXBRWLICLF-UHFFFAOYSA-N tert-butyl 3-[[3-[[(8-carbamoylquinazolin-4-yl)amino]methyl]phenyl]carbamoyl]-6,7-dihydro-4h-pyrazolo[1,5-a]pyrazine-5-carboxylate Chemical compound C1=CC=C2C(NCC=3C=CC=C(C=3)NC(=O)C=3C=NN4CCN(CC4=3)C(=O)OC(C)(C)C)=NC=NC2=C1C(N)=O ZKMVGXBRWLICLF-UHFFFAOYSA-N 0.000 description 1
- PXLWDYOUWTYABH-LLVKDONJSA-N tert-butyl n-[(1s)-1-(3-fluorophenyl)-2-hydroxyethyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@H](CO)C1=CC=CC(F)=C1 PXLWDYOUWTYABH-LLVKDONJSA-N 0.000 description 1
- UGKAWIQUIOZIMD-UHFFFAOYSA-N tert-butyl n-[1-(3-aminophenyl)-3-hydroxypropyl]carbamate Chemical compound CC(C)(C)OC(=O)NC(CCO)C1=CC=CC(N)=C1 UGKAWIQUIOZIMD-UHFFFAOYSA-N 0.000 description 1
- FPUJDBMWNXZNAV-UHFFFAOYSA-N tert-butyl n-[1-(3-nitrophenyl)-3-pyrrolidin-1-ylpropyl]carbamate Chemical compound C=1C=CC([N+]([O-])=O)=CC=1C(NC(=O)OC(C)(C)C)CCN1CCCC1 FPUJDBMWNXZNAV-UHFFFAOYSA-N 0.000 description 1
- QCQLTRAUGWYUFV-UHFFFAOYSA-N tert-butyl n-[2-[(8-carbamoyl-6-hydroxyquinazolin-4-yl)amino]-2-phenylethyl]-n-methylcarbamate Chemical compound N=1C=NC2=C(C(N)=O)C=C(O)C=C2C=1NC(CN(C)C(=O)OC(C)(C)C)C1=CC=CC=C1 QCQLTRAUGWYUFV-UHFFFAOYSA-N 0.000 description 1
- FYEHHEDXJKWPIB-UHFFFAOYSA-N tert-butyl n-[2-[(8-carbamoyl-6-methoxyquinazolin-4-yl)amino]-2-phenylethyl]-n-methylcarbamate Chemical compound C12=CC(OC)=CC(C(N)=O)=C2N=CN=C1NC(CN(C)C(=O)OC(C)(C)C)C1=CC=CC=C1 FYEHHEDXJKWPIB-UHFFFAOYSA-N 0.000 description 1
- IIPQFFCDLPLTDA-UHFFFAOYSA-N tert-butyl n-[2-[(8-carbamoyl-6-phenylmethoxyquinazolin-4-yl)amino]-2-(3-chlorophenyl)ethyl]-n-methylcarbamate Chemical compound C=1C=CC(Cl)=CC=1C(CN(C)C(=O)OC(C)(C)C)NC(C1=C2)=NC=NC1=C(C(N)=O)C=C2OCC1=CC=CC=C1 IIPQFFCDLPLTDA-UHFFFAOYSA-N 0.000 description 1
- BZMFKCZBICAGJX-UHFFFAOYSA-N tert-butyl n-[2-[(8-carbamoyl-6-phenylmethoxyquinazolin-4-yl)amino]-2-phenylethyl]-n-methylcarbamate Chemical compound C=1C=CC=CC=1C(CN(C)C(=O)OC(C)(C)C)NC(C1=C2)=NC=NC1=C(C(N)=O)C=C2OCC1=CC=CC=C1 BZMFKCZBICAGJX-UHFFFAOYSA-N 0.000 description 1
- CNAHTFLEMVTQQC-UHFFFAOYSA-N tert-butyl n-[2-amino-2-(3-nitrophenyl)ethyl]-n-methylcarbamate Chemical compound CC(C)(C)OC(=O)N(C)CC(N)C1=CC=CC([N+]([O-])=O)=C1 CNAHTFLEMVTQQC-UHFFFAOYSA-N 0.000 description 1
- FUPDUVSANAQCMB-UHFFFAOYSA-N tert-butyl n-[3-[1-[(8-carbamoyl-6-chloroquinazolin-4-yl)amino]ethyl]phenyl]carbamate Chemical compound N=1C=NC2=C(C(N)=O)C=C(Cl)C=C2C=1NC(C)C1=CC=CC(NC(=O)OC(C)(C)C)=C1 FUPDUVSANAQCMB-UHFFFAOYSA-N 0.000 description 1
- AXWHGWQLVTVMQA-UHFFFAOYSA-N tert-butyl n-[3-[[(8-carbamoylquinazolin-4-yl)amino]methyl]phenyl]carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=CC(CNC=2C3=CC=CC(=C3N=CN=2)C(N)=O)=C1 AXWHGWQLVTVMQA-UHFFFAOYSA-N 0.000 description 1
- QXMOQSGCRNWKDY-UHFFFAOYSA-N tert-butyl n-[3-cyano-1-(3-nitrophenyl)propyl]carbamate Chemical compound CC(C)(C)OC(=O)NC(CCC#N)C1=CC=CC([N+]([O-])=O)=C1 QXMOQSGCRNWKDY-UHFFFAOYSA-N 0.000 description 1
- SLJMTERFCITLKA-UHFFFAOYSA-N tert-butyl n-[4-[[(8-carbamoylquinazolin-4-yl)amino]methyl]phenyl]carbamate Chemical compound C1=CC(NC(=O)OC(C)(C)C)=CC=C1CNC1=NC=NC2=C(C(N)=O)C=CC=C12 SLJMTERFCITLKA-UHFFFAOYSA-N 0.000 description 1
- SFPKTHKAZYMGLV-UHFFFAOYSA-N tert-butyl n-[4-[[3-[[(8-carbamoylquinazolin-4-yl)amino]methyl]phenyl]carbamoyl]-1,3-thiazol-2-yl]carbamate Chemical compound S1C(NC(=O)OC(C)(C)C)=NC(C(=O)NC=2C=C(CNC=3C4=CC=CC(=C4N=CN=3)C(N)=O)C=CC=2)=C1 SFPKTHKAZYMGLV-UHFFFAOYSA-N 0.000 description 1
- RGAODFTWIJGPHT-UHFFFAOYSA-N tert-butyl n-[[3-[(5-morpholin-4-ylpyridine-3-carbonyl)amino]phenyl]methyl]carbamate Chemical compound CC(C)(C)OC(=O)NCC1=CC=CC(NC(=O)C=2C=C(C=NC=2)N2CCOCC2)=C1 RGAODFTWIJGPHT-UHFFFAOYSA-N 0.000 description 1
- ZNMNFIZDYPXSGQ-UHFFFAOYSA-N tert-butyl n-[[3-[(6-methoxypyridine-3-carbonyl)amino]phenyl]methyl]carbamate Chemical compound C1=NC(OC)=CC=C1C(=O)NC1=CC=CC(CNC(=O)OC(C)(C)C)=C1 ZNMNFIZDYPXSGQ-UHFFFAOYSA-N 0.000 description 1
- 230000002381 testicular Effects 0.000 description 1
- 108091008743 testicular receptors 4 Proteins 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 230000017423 tissue regeneration Effects 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 210000005048 vimentin Anatomy 0.000 description 1
- QDLHCMPXEPAAMD-QAIWCSMKSA-N wortmannin Chemical compound C1([C@]2(C)C3=C(C4=O)OC=C3C(=O)O[C@@H]2COC)=C4[C@@H]2CCC(=O)[C@@]2(C)C[C@H]1OC(C)=O QDLHCMPXEPAAMD-QAIWCSMKSA-N 0.000 description 1
- QDLHCMPXEPAAMD-UHFFFAOYSA-N wortmannin Natural products COCC1OC(=O)C2=COC(C3=O)=C2C1(C)C1=C3C2CCC(=O)C2(C)CC1OC(C)=O QDLHCMPXEPAAMD-UHFFFAOYSA-N 0.000 description 1
- 229950009002 zanolimumab Drugs 0.000 description 1
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/04—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to the ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/04—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to the ring carbon atoms
- C07D215/06—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to the ring carbon atoms having only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, attached to the ring nitrogen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/94—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the invention relates to a series of substituted amino azaheterocyclic carboxamide compounds that are useful in the treatment of hyperproliferative diseases, such as cancer, in mammals. Also encompassed by the present invention is the use of such compounds in the treatment of hyperproliferative diseases in mammals, especially humans, and pharmaceutical compositions containing such compounds.
- Protein kinases constitute a large family of structurally related enzymes that are responsible for the control of a wide variety of signal transduction processes within the cell (Hardie, G. and Hanks, S. (1995) The Protein Kinase Facts Book. I and II, Academic Press, San Diego, CA).
- the kinases may be categorized into families by the substrates they phosphorylate (e.g., protein-tyrosine, protein-serine/threonine, lipids, etc.).
- kinases regulate many different cell processes including, but not limited to, proliferation, differentiation, apoptosis, motility, transcription, translation and other signalling processes, by adding phosphate groups to target proteins.
- phosphorylation events act as molecular on/off switches that can modulate or regulate the target protein biological function. Phosphorylation of target proteins occurs in response to a variety of extracellular signals (hormones, neurotransmitters, growth and differentiation factors, etc.), cell cycle events, environmental or nutritional stresses, etc.
- the appropriate protein kinase functions in signalling pathways to activate or inactivate (either directly or indirectly), for example, a metabolic enzyme, regulatory protein, receptor, cytoskeletal protein, ion channel or pump, or transcription factor.
- Uncontrolled signalling due to defective control of protein phosphorylation has been implicated in a number of diseases, including, for example, inflammation, cancer, allergy/asthma, diseases and conditions of the immune system, diseases and conditions of the central nervous system, and angiogenesis.
- the enzyme PI3K is activated by a range of growth and survival factors e.g. EGF, PDGF and through the generation of polyphosphatidylinositols, initiates the activation of the downstream signalling events including the activity of the kinases PDK1 and protein kinase B (PKB) also known as Akt.
- Protein kinase 70S6K the 70 kDa ribosomal protein kinase p70S6K (also known as SK6, p70/p85 S6 kinase, p70/p85 ribosomal S6 kinase and pp70S6K), is a member of the AGC subfamily of protein kinases.
- p70S6K is a serine-threonine kinase that is a component of the phosphatidylinositol 3 kinase (PI3K)/AKT pathway.
- p70S6K is downstream of PI3K, and activation occurs through phosphorylation at a number of sites in response to numerous mitogens, hormones and growth factors. p70S6K activity is also under the control of a mTOR-containing complex (TORC1) since rapamycin acts to inhibit p70S6K activity. p70S6K is regulated by PI3K downstream targets AKT and PKC ⁇ . Akt directly phosphorylates and inactivates TSC2, thereby activating mTOR. In addition, studies with mutant alleles of p70S6K that inhibited by Wortmannin but not by rapamycin suggest that the PI3K pathway can exhibit effects on p70S6K independent of the regulation of mTOR activity.
- TORC1 mTOR-containing complex
- the enzyme p70S6K modulates protein synthesis by phosphorylation of the S6 ribosomal protein.
- S6 phosphorylation correlates with increased translation of mRNAs encoding components of the translational apparatus, including ribosomal proteins and translational elongation factors whose increased expression is essential for cell growth and proliferation.
- mRNAs contain an oligopyrimidime tract at their 5' transcriptional start (termed 5 1 TOP) 1 which has been shown to be essential for their regulation at the translational level.
- p70S6K activation has also been implicated in cell cycle control, neuronal cell differentiation, regulation of cell motility and a cellular response that is important in tumor metastases, the immune response and tissue repair.
- Antibodies to p70S6K abolish the mitogenic response driven entry of rat fibroblasts into S phase, indication that p70S6K function is essential for the progression from G1 to S phase in the cell cycle.
- inhibition of cell cycle proliferation at the G1 to S phase of the cell cycle by rapamycin has been identified as a consequence of inhibition of the production of the hyperphosphorylated, activated form of p70S6K.
- Chromosome 17q23 is amplified in up to 20% of primary breast tumors, in 87% of breast tumors containing BRCA2 mutations and in 50% of tumors containing BRCA1 mutations, as well as other cancer types such as pancreatic, bladder and neuroblastoma (see M. Barlund, O. Monni, J. Kononen, R. Cornelison, J. Torhorst, G. Sauter, O.-P. Kallioniemi and Kallioniemi A., Cancer Res., 2000, 60:5340-5346). It has been shown that 17q23 amplifications in breast cancer involve the PAT1 , RAD51C, PS6K, and SIGMA1 B genes (Cancer Res. (2000): 60, pp. 5371-5375). The p70S6K gene has been identified as a target of amplification and overexpression in this region, and statistically significant association between amplification and poor prognosis has been observed.
- p70S6K A role for p70S6K in metabolic diseases and disorders such as obesity, diabetes, metabolic syndrome, insulin resistance, hyperglycemia, hyperaminoacidemia, and hyperlipidmia is supported based upon the findings.
- Compounds described as suitable for p70S6K inhibition are disclosed in WO 03/064397, WO 04/092154, WO 05/054237, WO 05/056014, WO 05/033086, WO 05/117909, WO 05/039506, WO 06/120573, WO 06/136821 , WO 06/071819, WO 06/131835 and WO 08/140947.
- TPX2 XKLP 2
- Aurora A also functions in meiosis promoting oocyte maturation, polar-body extrusion, spindle positioning and exit from metaphase I. Regulation of Aurora A occurs through phosphorylation/dephosphorylation and degradation. Protein phosphatase 1 negatively regulates Aurora and this interaction is modulated by TPX2.
- Aurora B is a chromosomal-passenger protein with multiple functions in mitosis.
- Aurora B is essential for completion of cytokinesis.
- Myosin Il regulatory chain, vimentin, desmin and glial fibrillary acidic protein are among its cleavage furrow substrates.
- Aurora B phosphorylates MgcRacGAP, transforming it into an activator of RhoA in the contractile ring (Minoshima Y, Kawashima T, Hirose K 1 Tonozuka Y, Kawajiri A, Bao Y, Deng X, Tatsuka M, Narumiya S 1 May W Phosphorylation by aurora B converts MgcRacGAP to a RhoGAP during cytokinesis. Dev. Cell , (2003) 4:549-560. Much less is known about Aurora C kinase, other than that it seems to be preferentially expressed in meiotic cells. During the cell cycle, Aurora kinases travel to their subcellular targets aided by their binding partner-substrates, INCENP, survivin and TPX2. This provides an additional level of regulation that might be essential for the choreography of mitotic events.
- Aurora A and B kinases are frequently elevated in human cancers making them attractive targets for therapeutic intervention.
- Small molecule inhibitors of Aurora kinases have recently been reported, but their effect on cytokinesis has yet to be investigated in detail.
- a high selective and potent small-molecule inhibitor of Aurora kinases, VX-680 blocks cell-cycle progression and induces apoptosis in a diverse range of human tumor types.
- This compound causes profound inhibition of tumor growth in a variety of in vivo xenograft models, leading to regression of leukemia, colon and pancreatic tumors at well-tolerated doses (Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T. Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JM, Miller KM VX-680, a potent and selective small- molecule inhibitor of the aurora kinases, suppresses tumor growth in vivo, Nat. Med., (2004) 10: 262-267.
- JNJ-7706621 inhibited cell growth independent of p53, retinoblastoma, or P-glycoprotein status; activated apoptosis; and reduced colony formation. At low concentrations, JNJ- 7706621 slowed the growth of cells and at higher concentrations induced cytotoxicity. Inhibition of CDK1 kinase activity, altered CDK1 phosphorylation status, and interference with downstream substrates such as retinoblastoma were also shown in human tumor cells following drug treatment.
- JNJ-7706621 delayed progression through G1 and arrested the cell cycle at the G2-M phase (Emanuel S, Rugg CA, Gruninger RH, Lin R, Fuentes-Pesquera A, Connolly PJ, Wetter SK, Hollister B, Kruger WW, Napier C, JoINfTe L, Middleton SA, The in vitro and in vivo effects of JNJ-7706621 : A dual inhibitor of cyclin-dependent kinases and aurora kinases, Cancer Res., (2005) 65:9038-9046). Additional cellular effects due to inhibition of Aurora kinases included endoreduplication and inhibition of histone H3 phosphorylation. In a human tumor xenograft model, several intermittent dosing schedules were identified that produced significant antitumor activity.
- PDK1 phosphoinositide-dependent kinase 1
- AGC protein kinase family comprising PKB, SGK, S6K and PKC isoforms. These kinases are involved in the PI3K signal transduction pathway and control basic cellular functions, such as survival, growth and differentiation. PDK1 is thus an important regulator of diverse metabolic, proliferative and life-sustaining effects.
- Intramalous activity relates either to: (1) the expression in cells which do not usually express these protein kinases; (2) increased kinase expression which results in undesired cell proliferation, such as cancer; (3) increased kinase activity which results in undesired cell proliferation, such as cancer, and/or in hyperactivity of the corresponding protein kinases.
- Hyperactivity relates either to amplification of the gene which encodes a certain protein kinase or the generation of an activity level which can be correlated with a cell proliferation disease (i.e.
- the bioavailability of a protein kinase can also be influenced by the presence or absence of a set of binding proteins of this kinase.
- anomalous activity of the substrates PKB and S6K of this kinase has been observed in a large number of types of cancer which exhibit point mutation of the PTEN gene, which results in uncontrolled proliferation and an increased survival rate.
- Inhibitors of PDK1 should therefore prove advantageous in the treatment of cancer cells with constitutively activated AGC kinases.
- this invention provides novel, substituted azaheterocyclic carboxamide compounds and pharmaceutically acceptable salts, solvates or prodrugs thereof, that are kina e inhibitors and useful in the treatment of the above mentioned diseases.
- the compounds are defined by Formula (I):
- X is N or C-R 3 ,
- Y is NH, O or absent
- R 1 is L 1 -R 4 -L 2 -R 5 -L 3 -R 6 , L 1 -R 4 -L 2 -R 5 or L 1 -R 4 ,
- NHA NHA, NH-L 1 -Ar, NHCOA, NHCO-L 1 -Ar, NHSO 2 A, NHSO 2 -L 1 -Ar, NHCONHA or NHCONH-L 1 -Ar, L 1 -Ar, O- L 1 -Ar, L 1 -R 4 ,
- L 1 , L 3 each, independently of one another, are a single bond, u ⁇ bra ⁇ ched or branched alkylene having 1, 2, 3, 4 or 5 C atoms, which may be unsubstltuted or mono- or dlsubstltuted with Hal, OH 1 CN, NH 2 , NH(LA) 1 N(LA)a, NO 2 , COOH, N 3 , ethenyl or ethynyl, and/or monosubstituted with R 4 , and In which one or two CH 2 groups may be replaced by an O or S atom or by an -NH-, -N(LA)-, -CONH-, -N(LA)COO-, -SO r or -NHCO- group,
- R 3 is H, A, HaI 1 OH, COOH, SH, NH 2 , NO 2 or CN,
- Ar is a mono- or bicyclic aromatic homo- or heterocycle having O, 1 , 2, 3 or 4 N 1 O and/or S atoms and 5, 6, 7, 8, 9, or 10 skeleton atoms, which may be unsubstituted or, independently of one another, mono-, di- or trisubsti tuted by Hal, A, OH, SH, OA, NH 2 , NHA, NA 2 , NO 2 , CN, OCN, SCN, COOH, COOA, CONH 2 , CONHA, CONA 2 , NHCOA, NHCONHA, NHCONH 2 , NHSO 2 A, CHO, COA, SO 2 NH 2 , SO 2 A and/or SO 2 HaI, and in which a ring N-atom may be substituted by an O-atom to form an N- oxide group, and in which in the case of a bicyclic aromatic cycle on of the two rings may be partly saturated,
- A is unbranched or branched linear or cyclic alkyl having 1 , 2, 3, 4, 5, 6, 7 or 8 C atoms, in which one or two CH 2 groups may be replaced by an O or S atom and/or by an -NH-, -CO-, -NHCOO-, -NHCONH-.
- LA is unbranched or branched, linear alkyl having 1 , 2, 3 or 4 C atoms,
- Hal is F, Cl, Br or I.
- the invention relates, in particular, to the compounds of the Formula (I) in which at least one of the said residues has one of the preferred meanings indicated below.
- Hal denotes fluorine, chlorine, bromine or iodine, in particular fluorine or chlorine.
- one or two CH 3 groups is replaced by OH, SH, NH 2 , N(LA)H, N(LA) 2 or CN, such as, for example, N,N'-dimethylaminoalkyl, 2-aminoethyl, 3-amino- propyl, 4-aminobutyl, 5-aminopentyl, 3-aminomethylcyclobutyl or cyanoalkyl.
- Cyclic A preferably denotes cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cycloheptyl.
- LA denotes unbranched or branched, linear alkyl having 1 , 2, 3 or 4 C atoms, i.e. methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl or tert-butyl.
- Ar denotes, for example, unsubstituted phenyl, naphthyl or biphenyl, furthermore preferably, for example, phenyl, naphthyl or biphenyl, each of which is mono-, di- or trisubstituted by A, fluorine, chlorine, bromine, iodine, hydroxyl, methoxy, ethoxy, propoxy, butoxy, pentyloxy, hexyloxy, nitro, cyano, formyl, acetyl, propionyl, trifluoromethyl, amino, methylamino, ethylamino, dimethylamino, diethylamino, benzyloxy, sulfonamido, methylsulfonamido, ethylsulfonamido, propylsulfonamido, butylsulfonamido, dimethylsulfonamido, phenyls
- Ar furthermore denotes phenyl, o-, m- or p-tolyl, o-, m- or p-ethylphenyl, o-, m- or p-propylphenyl, o-, m- or p-isopropylphenyl, o-, m- or p-tert-butylphenyl, o-, m- or p-hydroxyphenyl, o-, m- or p-nitrophenyl, o-, m- or p-aminophenyl, o-, m- or p-(N-methyl- amino)phenyl, o-, m- or p-(N-methylaminocarbonyl)phenyl, o-, m- or p-acetamidophenyl, o-, m- or p-methoxyphenyl, o-, m- or p-e
- “Ar” furthermore preferably denotes 2-, 3- or 4-phenyl, 2-, 3- or 4-phenylmethyl, 2-, 3- or 4-phenylethyl, 2- or 3-furyl, 2- or 3-thienyl, 1-, 2- or 3-pyrrolyl, 1-, 2, 4- or 5-imidazolyl, 1-, 3-, 4- or 5-pyrazolyl, 2-, 4- or 5-oxazolyl, 3-, 4- or 5-isoxazolyl, 2-, 4- or 5-thiazolyl, 3-, A- or 5-isothiazolyl, 2-, 3- or 4-pyridyl, 2-, 3- or 4-pyridylmethyl, 2-, 3- or 4-pyridylethyl, 2-, A-, 5- or 6-pyrimidinyl, 2-, 3-, 5-, or 6-pyrazin-1- or 4-yl, furthermore preferably 1 ,2,3-tria- zol-1-, -4- or -5-yl, 1 ,2,4-triazoM-, -3- or 5-yl, 1-
- heterocyclic "Ar" residues may also be partially or fully hydrogenated and also denote, for example, 2,3-dihydro-2-, -3-, -A- or -5-furyl, 2,5-dihydro-2-, -3-, -A- or -5-furyl, tetrahydro-2- or -3-furyl, 1 ,3-dioxolan-4-yl, tetrahydro-2- or -3-thienyl, 2,3-dihydro-1-, -2-, -3-, -4- or -5-pyrrolyl, 2,5-dihydro-1-, -2-, -3-, -4- or -5-pyrrolyl, 1-, 2- or 3-pyrrolidinyl, tetrahydro-1-, -2- or -4-imidazolyl, 2,3-dihydro-1-, -2-, -3-, -4- or -5-pyrazolyl, tetra
- residue R 4 obviously has a bridging function, and is substituted by linkers L 1 and L 2 , independently of any further substitutions it may have.
- residue R 5 in those cases where R 1 is L 1 -R 4 -L 2 -R 5 -L 3 -R 6 .
- substituted preferably relates to the substitution by the above-mentioned substituents, where a plurality of different degrees of substitution are possible, unless indicated otherwise.
- the compounds of the Formula (I) may have one or more centres of chirality. They may accordingly occur in various enantiomeric forms and be in racemic or optically active form.
- the invention therefore also relates to the optically active forms (stereoisomers), the enantiomers, the racemates, the diastereomers and hydrates and solvates of these compounds.
- the pharmaceutical activity of the racemates or stereoisomers of the compounds according to the invention may differ, it may be desirable to use the enantiomers.
- the end product or even the intermediates can be separated into enantiomeric compounds by chemical or physical measures known to the person skilled in the art or even employed as such in the synthesis.
- diastereomers are formed from the mixture by reaction with an optically active resolving agent.
- optically active acids such as the R and S forms of tartaric acid, diacetyltartaric acid, dibenzoyltartaric acid, mandelic acid, malic acid, lactic acid, suitably N-protected amino acids (for example N-benzoylproline or N-benzenesulfonylproline), or the various optically active camphorsulfonic acids.
- chromatographic enantio- mer resolution with the aid of an optically active resolving agent (for example dinitrobenzoylphenylglycine, cellulose triacetate or other derivatives of carbohydrates or chirally derivatised methacrylate polymers immobilised on silica gel).
- optically active resolving agent for example dinitrobenzoylphenylglycine, cellulose triacetate or other derivatives of carbohydrates or chirally derivatised methacrylate polymers immobilised on silica gel.
- Suitable eluents for this purpose are aqueous or alcoholic solvent mixtures, such as, for example, hexane/isopropanol/ acetonitrile, for example in the ratio 82:15:3.
- An elegant method for the resolution of racemates containing ester groups is the use of enzymes, in particular esterases.
- a preferred group of compounds of Formula (I) conform to Formulae (II) or (III) in which R 2 has the meaning indicated for R 2 , R 2 of Formula (I) 1 and R 1 , X and Y have the meaning indicated for Formula (I). Particularly preferred are the compounds according to Formula (II).
- R 3 is H
- Subformula 4 X is N 1 Y is NH 1 in Subformula 5 X is N,
- Y is NH
- Y is NH
- L 1 is methylene
- Y is NH
- L 1 is methylene
- R 2 is H, methoxy, ethoxy or amino
- L 1 is methylene which is unsubstituted or substituted with methyl, aminomethyl, methoxymethyl, azidomethyl or triazolylmethyl
- R 2 is H, methoxy, ethoxy or amino
- Y is NH
- L 1 is methylene which is substituted with aminomethyl
- L 1 is methylene which is substituted with aminomethyl
- R 2 is H, methoxy, ethoxy or amino
- Y is NH
- R 1 is L 1 -R 4
- L 1 is methylene which is substituted with aminomethyl
- R 2 is H, methoxy, ethoxy or amino
- R 1 is L 1 -R 4 -L 2 -R 5 or L 1 -R 4 ,
- L 1 is methylene which is unsubstituted or substituted with aminomethyl
- R 2 is H, methoxy, ethoxy or amino
- Y is NH
- R 1 is L 1 -R 4 _L 2 -R 5 or L 1 -R 4 ,
- L 1 is methylene
- R 2 is H, methoxy, ethoxy or amino
- Y is NH
- R 1 is L 1 -R 4 -L 2 -R 5 or L 1 -R 4
- L 1 is methylene
- R 2 is H, methoxy or amino
- R 1 is L 1 -R 4 ,
- L 1 is methylene
- R 2 is H, methoxy or amino
- Y is NH
- R 1 is L 1 -R 4 ,
- L 1 is methylene
- R 4 is phenyl which is unsubstituted or monosubstituted with Hal or CF 3 , or disubstituted with Hal,
- R 2 is H, methoxy or amino
- Y is NH
- R 1 is L 1 -R 4 ,
- L 1 is methylene
- R 4 is phenyl which is unsubstituted or monosubstituted with Hal or CF 3 , or disubstituted with Hal,
- R 2 is H
- Y is NH
- R 1 is L 1 -R 4 -L 2 -R 5 ,
- L 1 is methylene
- R 4 is phenyl
- L 2 is NHCO or NHCONH 1
- R 2 is H or methoxy
- Y is NH
- R 1 is L 1 -R 4 -L 2 -R 5 ,
- L 1 is methylene
- R 4 is phenyl
- L 2 is NHCO or NHCONH
- R 5 is phenyl which is unsubstituted or mono- or disubstituted with Hal
- R 2 is H or methoxy
- Y is NH
- R 1 is L 1 -R 4 -L 2 -R 5 ,
- L 1 is methylene
- R 4 is phenyl
- L 2 is NHCO
- R 5 is phenyl which is unsubstituted or mono- or disubstituted with Hal
- R 2 is H or methoxy
- Y is NH
- R 1 is L 1 -R 4 -L 2 -R 5 ,
- L 1 is methylene
- R 4 is phenyl
- L 2 is NHCO or NHCONH
- R 5 is phenyl which is unsubstituted, or mono- or disubstituted with Hal,
- R 2 is H
- Subformula 23 X is N, R 1 is L 1 -R 4 -L 2 -R 5 , R 4 is phenyl, R 5 benzo-1,3-dioxolyl,
- L 1 is methylene which is unsubstituted or substituted with aminomethyl, (methyl- amino)methyl, (dimethyl-amino)methyl, methyl, ethyl, 2-hydroxyethyl, methoxymethyl, 2- (dimethyl-amino)ethyl, (ethyl-amino)methyl, 2-(methoxy)ethyl, 2-(allyl-methyl- amino)ethyl, ((tert.
- L 1 is methylene which is unsubstituted or substituted with (methyl-amino)methyl, (dimethyl-amino)methyl, methyl or 2-(dimethyl-amino)ethyl,
- Y is NH
- R 1 is L 1 -R 4 -L 2 -R 5 ,
- R 4 is phenyl
- L 2 is -NHCO- , -NH-, -NHCH 2 -, NHCOOCH 2 - or -NHCONH-,
- Y is NH
- R 1 is L 1 -R 4 -L 2 -R 5 ,
- R 4 is phenyl
- L 2 is -NHCO- , -NH-, -NHCH 2 -, NHCOOCH 2 - or -NHCONH-,
- R 5 is Ar which is unsubstituted or substituted as defined for Ar in Claim 1 ,
- X is N
- Y is NH 1
- R 1 is L 1 -R 4 -L 2 -R 5
- R 4 is phenyl
- L 2 is -NHCO-, -NH-, -NHCH 2 -, NHCOOCH 2 - or -NHCONH-
- R 5 is phenyl, pyridyl, benzo-1 ,3-dioxolyl, pyrazolyl or thiazolyl, all of which are unsubstituted or substituted as defined for Ar in Claim 1 ,
- Y is NH
- L 1 is methylene which is unsubstituted or substituted with aminomethyl, (methyl- amino)methyl, (dimethyl-amino)methyl, methyl, ethyl, 2-hydroxyethyl, methoxymethyl, 2- (dimethyl-amino)ethyl, (ethyl-amino)methyl, 2-(methoxy)ethyl, 2-(allyl-methyl- amino)ethyl, ((tert.
- Y is NH
- L 1 is methylene which is unsubstituted or substituted with (methyl-amino)methyl, (dimethyl-amino)methyl, methyl or 2-(dimethyl-amino)ethyl, R 2 is H or methoxy,
- Y is NH
- R 1 is L 1 -R 4 -L 2 -R 5 , R 4 is phenyl,
- L 2 is -NHCO-, -NH-, -NHCH 2 -, NHCOOCH 2 - or -NHCONH-,
- R 2 is H or methoxy
- R 1 is L 1 -R 4 -L 2 -R 5 ,
- R 4 is phenyl
- L 2 is -NHCO-, -NH-, -NHCH 2 -, NHCOOCH 2 - or -NHCONH-,
- R 5 is Ar which is unsubstituted or substituted as defined for Ar in Claim 1 ,
- R 2 is H or methoxy,
- R 1 is L 1 -R 4 -L 2 -R 5 , R 4 is phenyl,
- L 2 is -NHCO-, -NH-, -NHCH 2 -, NHCOOCH 2 - or -NHCONH-
- R 5 is phenyl, pyridyl, benzo-1 ,3-dioxolyl, pyrazolyl or thiazolyl, all of which are unsubstituted or substituted as defined for Ar in Claim 1
- R 2 is H or methoxy
- X is N
- Y is NH
- R 1 is L 1 -R 4 -L 2 -R 5 ,
- L 1 is methylene which is unsubstituted or substituted with (methyl-amino)methyl
- R 4 is phenyl, L 2 is -NHCO-, -NH-, -NHCH 2 -, NHCOOCH 2 - or -NHCONH-,
- R 5 is Ar which is unsubstituted or substituted as defined for Ar in Claim 1 ,
- R 2 is H or methoxy
- Y is NH
- R 1 is L 1 -R 4
- R 2 is L 1 -Ar
- Y is NH
- R 1 is L 1 -R 4
- R 4 is piperidinyl
- Y is NH
- R 1 is L 1 -R 4 ,
- R 4 is piperidinyl, R 2 is L 1 -Ar, L 1 is a bond,
- Y is NH
- R 1 is L 1 -R 4 ,
- L 1 is methylene which is substituted with aminomethyl, (methyl-amino)methyl, (dimethyl-amino)methyl or 2-aminoprop-2-yl,
- R 4 is phenyl which is unsubstituted or substituted as defined for Ar in Claim 1 ,
- R 2 is H, methoxy, methyl, ethyl, hydroxymethyl, methoxymethyl or cyano
- R 4 is meta-phenylene.
- Especially preferred compounds according to Formula (I), (II) and/or Formula (III) include those listed in Tables 1 , 2 and 3 below, or their pharmaceutically acceptable salts, solvates or prodrugs. Table 1
- the compounds of the present invention can be in the form of a prodrug compound.
- Prodrug compound means a derivative that is converted into a biologically active compound according to the present invention under physiological conditions in the living body, e.g., by oxidation, reduction, hydrolysis or the like, each of which is carried out enzymatically, or without enzyme involvement.
- prodrugs are compounds, wherein the amino group in a compound of the present invention is acylated, alkylated or phosphorylated, e.g., eicosanoylamino, alanylamino, pivaloyloxymethylamino or wherein the hydroxyl group is acylated, alkylated, phosphorylated or converted into the borate, e.g. acetyloxy, palmitoyloxy, pivaloyloxy, succinyloxy, fumaryloxy, alanyloxy or wherein the carboxyl group is esterified or amidated, or wherein a sulfhydryl group forms a disulfide bridge with a carrier molecule, e.g.
- a peptide that delivers the drug selectively to a target and/or to the cytosol of a cell.
- prodrugs are compounds, wherein the carboxylate in a compound of the present invention is for example converted into an alkyl-, aryl-, choline-, amino, acyloxymethylester, linolenoyl-ester. Metabolites of compounds of the present invention are also within the scope of the present invention.
- tautomerism e.g., keto-enol tautomerism
- the individual forms e.g., the keto or the enol form
- stereoisomers e.g., enantiomers, cis/trans isomers, conformers and the like.
- isomers can be separated by methods well known in the art, e.g. by liquid chromatography. The same applies for enantiomers, e.g., by using chiral stationary phases. Additionally, enantiomers may be isolated by converting them into diastereomers, i.e., coupling with an enantiomerically pure auxiliary compound, subsequent separation of the resulting diastereomers and cleavage of the auxiliary residue. Alternatively, any enantiomer of a compound of the present invention may be obtained from stereoselective synthesis using optically pure starting materials
- the compounds of the present invention can be in the form of a pharmaceutically acceptable salt or a solvate, or a solvate of a salt.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids, including inorganic bases or acids and organic bases or acids.
- the invention also comprises their corresponding pharmaceutically or toxicologically acceptable salts, in particular their pharmaceutically utilizable salts.
- the compounds of the present invention which contain acidic groups can be present in salt form, and can be used according to the invention, for example, as alkali metal salts, alkaline earth metal salts or as ammonium salts.
- salts include sodium salts, potassium salts, calcium salts, magnesium salts or salts with ammonia or organic amines such as, for example, ethylamine, ethanolamine, triethanolamine or amino acids.
- Compounds of the present invention which contain one or more basic groups, i.e. groups which can be protonated, can be present in salt form, and can be used according to the invention in the form of their addition salts with inorganic or organic acids.
- acids include hydrogen chloride, hydrogen bromide, phosphoric acid, sulfuric acid, nitric acid, methanesulfonic acid, p- toluenesulfonic acid, naphthalenedisulfonic acids, oxalic acid, acetic acid, tartaric acid, lactic acid, salicylic acid, benzoic acid, formic acid, propionic acid, pivalic acid, diethylacetic acid, malonic acid, succinic acid, pimelic acid, fumaric acid, maleic acid, malic acid, sulfaminic acid, phenylpropionic acid, gluconic acid, ascorbic acid, isonicotinic acid, citric acid, adipic acid, and other acids known to the person skilled in the art.
- the invention also includes, in addition to the salt forms mentioned, inner salts or betaines (zwitterions).
- inner salts or betaines can be obtained by customary methods which are known to a person skilled in the art, for example by contacting these with an organic or inorganic acid or base in a solvent or dispersant, or by anion exchange or cation exchange with other salts.
- the present invention also includes all salts of the compounds of the present invention which, owing to low physiological compatibility, are not directly suitable for use in pharmaceuticals but which can be used, for example, as intermediates for chemical reactions or for the preparation of pharmaceutically acceptable salts.
- the present invention relates to pharmaceutical compositions comprising a compound of the present invention, or a prodrug compound thereof, or a pharmaceutically acceptable salt or solvate thereof as an active ingredient together with a pharmaceutically acceptable carrier.
- “Pharmaceutical composition” means one or more active ingredients, and one or more inert ingredients that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients. Accordingly, the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the present invention and a pharmaceutically acceptable carrier.
- a pharmaceutical composition of the present invention may additionally comprise one or more other compounds as active ingredients, such as one or more additional compounds of the present invention, or a prodrug compound or other p70S6K inhibitors.
- said compounds and pharmaceutical composition are for the treatment of cancer such as brain, lung, colon, epidermoid, squamous cell, bladder, gastric, pancreatic, breast, head, neck, renal, kidney, liver, ovarian, prostate, colorectal, uterine, rectal, oesophageal, testicular, gynecological, thyroid cancer, melanoma, hematologic malignancies such as acute myelogenous leukemia, multiple myeloma, chronic myelogneous leukemia, myeloid cell leukemia, glioma, Kaposi's sarcoma, or any other type of solid or liquid tumors.
- the cancer to be treated is chosen from breast, colorectal, lung, prostate or pancreatic cancer or glioblastoma.
- the invention also relates to the use of compounds according to the invention for the preparation of a medicament for the treatment of hyperproliferative diseases related to the hyperactivity of p70S6K as well as diseases modulated by the p70S6K cascade in mammals, or disorders mediated by aberrant proliferation, such as cancer and inflammation.
- the invention also relates to a compound or pharmaceutical composition for treating a disease related to vasculogenesis or angiogenesis in a mammal which comprises a therapeutically effective amount of a compound of the present invention, or a pharmaceutically acceptable salt, prodrug or hydrate thereof, and a pharmaceutically acceptable carrier.
- said compound or pharmaceutical composition is for treating a disease selected from the group consisting of tumor angiogenesis, chronic inflammatory disease such as rheumatoid arthritis, inflammatory bowel disease, atherosclerosis, skin diseases such as psoriasis, eczema, and sclerodema, diabetes, diabetic retinopathy, retinopathy of prematurity and age-related macular degeneration.
- a disease selected from the group consisting of tumor angiogenesis, chronic inflammatory disease such as rheumatoid arthritis, inflammatory bowel disease, atherosclerosis, skin diseases such as psoriasis, eczema, and sclerodema, diabetes, diabetic retinopathy, retinopathy of prematurity and age-related macular degeneration.
- This invention also relates to a compound or pharmaceutical composition for inhibiting abnormal cell growth in a mammal which comprises an amount of a compound of the present invention, or a pharmaceutically acceptable salt or solvate or prodrug thereof, in combination with an amount of another anti-cancer therapeutic, wherein the amounts of the compound, salt, solvate, or prodrug, and of the chemotherapeutic are together effective in inhibiting abnormal cell growth.
- a compound or pharmaceutical composition for inhibiting abnormal cell growth in a mammal which comprises an amount of a compound of the present invention, or a pharmaceutically acceptable salt or solvate or prodrug thereof, in combination with an amount of another anti-cancer therapeutic, wherein the amounts of the compound, salt, solvate, or prodrug, and of the chemotherapeutic are together effective in inhibiting abnormal cell growth.
- Many anti-cancer therapeutics are presently known in the art.
- the anti-cancer therapeutic is a chemotherapeutic selected from the group consisting of mitotic inhibitors, alkylating agents, anti- metabolites, intercalating antibiotics, growth factor inhibitors, cell cycle inhibitors, enzymes, topoisomerase inhibitors, biological response modifiers, anti-hormones, angiogenesis inhibitors, and anti-androgens.
- the anti-cancer therapeutic is an antibody selected from the group consisting of bevacizumab, CD40- specific antibodies, chTNT-1/B, denosumab, zanolimumab, IGF1 R-specific antibodies, lintuzumab, edrecolomab, WX G250, rituximab, ticilimumab, trastuzumab and cetuximab.
- the anti-cancer therapeutic is an inhibitor of another protein kinase, such as Akt, AxI, Aurora A, Aurora B, dyrk2, epha2, fgfr3, igflr, IKK2, JNK3, Vegfri , Vegfr2, Vegfr3 (also known as Flt-4), KDR, MEK, MET, PIkI 1 RSK1 , Src, TrkA, Zap70, cKit, bRaf, EGFR, Jak2, PI3K, NPM-AIk, c-Abl, BTK, FAK, PDGFR, TAK1 , LimK, Flt-3, PDK1 and Erk.
- another protein kinase such as Akt, AxI, Aurora A, Aurora B, dyrk2, epha2, fgfr3, igflr, IKK2, JNK3, Vegfri , Vegfr2, Vegfr3 (
- This invention further relates to a method for inhibiting abnormal cell growth in a mammal or treating a hyperproliferative disorder that comprises administering to the mammal an amount of a compound of the present invention, or a pharmaceutically acceptable salt or solvate or prodrug thereof, in combination with radiation therapy, wherein the amounts of the compound, salt, solvate, or prodrug, is in combination with the radiation therapy effective in inhibiting abnormal cell growth or treating the hyperproliferative disorder in the mammal.
- Techniques for administering radiation therapy are known in the art, and these techniques can be used in the combination therapy described herein.
- the administration of a compound of the invention in this combination therapy can be determined as described herein. It is believed that the compounds of the present invention can render abnormal cells more sensitive to treatment with radiation for purposes of killing and/or inhibiting the growth of such cells.
- this invention further relates to a method for sensitizing abnormal cells in a mammal to treatment with radiation which comprises administering to the mammal an amount of a compound of the present invention or pharmaceutically acceptable salt or solvate or prodrug thereof, which amount is effective is sensitizing abnormal cells to treatment with radiation.
- the amount of the compound, salt, or solvate in this method can be determined according to the means for ascertaining effective amounts of such compounds described herein.
- the invention also relates to a method for inhibiting abnormal cell growth in a mammal that comprises an amount of a compound of the present invention, or a pharmaceutically acceptable salt or solvate thereof, a prodrug thereof, or an isotopically-labeled derivative thereof, and an amount of one or more substances selected from anti-angiogenesis agents, signal transduction inhibitors, and antiproliferative agents.
- the compounds of the present invention can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques.
- the carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g., oral or parenteral (including intravenous).
- any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents and the like.
- any of the usual pharmaceutical media may be employed, such as, for example, suspensions, elixirs and solutions; or carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like.
- the composition may take forms such as, for example, powders, hard and soft capsules and tablets, with the solid oral preparations being preferred over the liquid preparations.
- tablets and capsules represent the most advantageous oral dosage unit form in which case solid pharmaceutical carriers are obviously employed. If desired, tablets may be coated by standard aqueous or nonaqueous techniques. Such compositions and preparations should contain at least 0.1 percent of active compound. The percentage of active compound in these compositions may, of course, be varied and may conveniently be between about 2 percent to about 60 percent of the weight of the unit. The amount of active compound in such therapeutically useful compositions is such that an effective dosage will be obtained.
- the active compounds can also be administered intranasally as, for example, liquid drops or spray.
- the tablets, pills, capsules, and the like may also contain a binder such as gum tragacanth, acacia, corn starch or gelatin; excipients such as dicalcium phosphate; a disintegrating agent such as corn starch, potato starch, alginic acid; a lubricant such as magnesium stearate; and a sweetening agent such as sucrose, lactose or saccharin.
- a dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier such as a fatty oil.
- tablets may be coated with shellac, sugar or both.
- a syrup or elixir may contain, in addition to the active ingredient, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and a flavoring such as cherry or orange flavor.
- Compounds of the present invention may also be administered parenterally. Solutions or suspensions of these active compounds can be prepared in water suitably mixed with a surfactant such as hydroxy-propylcellulose. Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
- the pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions.
- the form must be sterile and must be fluid to the extent that easy syringability exists. It must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g., glycerol, propylene glycol and liquid polyethylene glycol), suitable mixtures thereof, and vegetable oils.
- Any suitable route of administration may be employed for providing a mammal, especially a human, with an effective dose of a compound of the present invention.
- oral, rectal, topical, parenteral, ocular, pulmonary, nasal, and the like may be employed.
- Dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols, and the like.
- compounds of the present invention are administered orally.
- the effective dosage of active ingredient employed may vary depending on the particular compound employed, the mode of administration, the condition being treated and the severity of the condition being treated. Such dosage may be ascertained readily by a person skilled in the art.
- the compounds of the present invention are administered at a daily dosage of from about 0.01 milligram to about 100 milligram per kilogram of animal body weight, preferably given as a single daily dose.
- the total daily dosage is from about 0.1 milligrams to about 1000 milligrams, preferably from about 0.2 milligram to about 50 milligrams.
- the total daily dose will generally be from about 0.2 milligrams to about 200 milligrams. This dosage regimen may be adjusted to provide the optimal therapeutic response.
- the invention also relates to a set (kit) consisting of separate packs of a) an effective amount of a compound according to the invention or a physiologically acceptable salt, solvate or prodrug thereof, and b) an effective amount of a further medicament active ingredient.
- the set comprises suitable containers, such as boxes, individual bottles, bags or ampoules.
- the set may, for example, comprise separate ampoules, each containing an effective amount of a compound according to the invention and/or pharmaceutically usable derivatives, solvates and stereoisomers thereof, including mixtures thereof in all ratios, and an effective amount of a further medicament active ingredient in dissolved or lyophilised form.
- the compounds of the present invention can be prepared according to the procedures of the following Schemes and Examples, using appropriate materials and are further exemplified by the following specific examples. Moreover, by utilizing the procedures described herein, in conjunction with ordinary skills in the art, additional compounds of the present invention claimed herein can be readily prepared.
- the compounds illustrated in the examples are not, however, to be construed as forming the only genus that is considered as the invention.
- the examples further illustrate details for the preparation of the compounds of the present invention. Those skilled in the art will readily understand that known variations of the conditions and processes of the following preparative procedures can be used to prepare these compounds.
- the instant compounds are generally isolated in the form of their pharmaceutically acceptable salts, such as those described above.
- the amine-free bases corresponding to the isolated salts can be generated by neutralization with a suitable base, such as aqueous sodium hydrogencarbonate, sodium carbonate, sodium hydroxide and potassium hydroxide, and extraction of the liberated amine-free base into an organic solvent, followed by evaporation.
- a suitable base such as aqueous sodium hydrogencarbonate, sodium carbonate, sodium hydroxide and potassium hydroxide
- the amine-free base, isolated in this manner can be further converted into another pharmaceutically acceptable salt by dissolution in an organic solvent, followed by addition of the appropriate acid and subsequent evaporation, precipitation or crystallization.
- the present invention relates to a process for the manufacture of compounds of Formula (I) 1 wherein X is N and Y is NH, and all other substituents have the meaning as defined for Formula (I) in Claim 1 , wherein a carboxylic acid compound of Formula (l-lll)
- Scheme 1 illustrates the general route used for the synthesis of Examples 1 - 49 according to the Subformula (Ia) of Formula (I):
- R 1 has the meaning as defined for Formula (I) above.
- the present invention relates to a process for the manufacture of compounds of Formula (Ia) 1 wherein an ester of Formula (Id) is reacted with H 2 N-R 1 to an amine compound of Formula (Ic), which is then saponified to a carboxylic acid of Formula (Ib) 1 which is finally converted to a carboxamide of Formula (Ia).
- Scheme 2
- the present invention furthermore relates to a process for the manufacture of compounds of Formula (If), wherein a compound of Formula (Ih) is reacted with H 2 N- R 1 to an amine compound of Formula (Ig) 1 which is then converted to a carboxamide of Formula (If).
- R is H, A, L 1 -Ar, COA, CO-L 1 -Ar, SO 2 A, SO 2 -L 1 -Ar, CONHA or CONH-L 1 -Ar, and A, L 1 , Ar and R 1 have the meaning as defined for Formula (I) above.
- the present invention furthermore relates to a process for the manufacture of compounds of Formula (Ii), wherein an ester of Formula (lo) is reacted with H 2 N-R 1 to an amine compound of Formula (In), which is then saponified to a carboxylic acid of Formula (Im), which is further converted to a carboxamide of Formula (Ik), which is then reduced to an compound of Formula (Ij), which is finally converted to a compound of Formula (Ii).
- Scheme 4 illustrates the general route used for the synthesis of examples 255, 275, 281 , 286, 300, 319, 322, 333, 338, 353, 366, 370, 379, 403, 405, 462, 486, 510, 529 and 712 according to the Formula (I): HCI
- Scheme 5 illustrates the general route used for the synthesis of Examples 189, 196, 208, 209, 212, 215, 219, 223, 228, 233, 249, 252, 254, 265, 273, 287, 296, 313, 314, 332, 335, 360, 361 , 363, 365, 391 , 392, 399, 418, 422, 437, 450, 458, 490, 493, 495, 500, 524, 527, 664, 695, 697, 698, 700, 703, 704 according to Formula (I):
- Scheme 6 illustrates the general route used for the synthesis of examples 240, 244, 246, 247, 250, 261 , 266, 272, 280, 291 , 292, 294, 299, 301 , 308, 309, 321 , 323, 331 , 334, 339, 358, 359, 371 , 383, 385, 386, 390, 394, 395, 402, 404, 414, 421 , 425, 426, 429, 430, 434, 440, 442, 446, 452, 456, 461 , 463, 464, 471 , 472, 475, 476, 496, 497, 498, 501 , 506, 507, 512, 525, 543, 544, 546, 551 , 552, 553, 554, 557,
- Scheme 7 illustrates the general route used for the synthesis of examples 564, 577, 590, 612, 626, 638 according to Formula (I):
- Methyl 4-oxo-3,4-dihydroquinazoline-8-carboxylate (5.00 g; 24.49 mmol; 1.00 eq.) was dissolved in sulfuric acid (50.00 ml; 938.01 mmol; 38.31 eq.) while cooling with water bath.
- N-iododsuccinamide (44.07 g; 195.90 mmol; 8.00 eq.) was then added. The mixture was stirred at RT for 21 hours, then heated to 40°C and stirred at same temperature for 8 days. Poured the reaction mixture into a cooled solution of 2N NaOH. 50ml 5% -NaS 2 SO 3 solution was added and stirred for 1h at RT. Filtered the product methyl 6-iodo-4-oxo-3,4-dihydroquinazoline-8-carboxylate to get a white solid (3.5g, 43.5%).
- reaction mixture was stirred for 20 min at 90°C, poured into 2N NaOH soltuion (22ml) containing crushed ice. Filtered, washed with water and collected 850 mg of the 4-Chloro-6-iodo-quinazoline-8-carboxylic acid methyl ester in 80% yield.
- Scheme 8 illustrates the general route used for the synthesis of examples 550, 618, 674, 743 according to Formula (I): step c
- Steps (a) to (e) are carried out as described in Example 743.
- Scheme 9 illustrates the general route used for the synthesis of example 539.
- Scheme 10 illustrates the general route used for the synthesis of examples 477, 526, 549,569, 574, 594, 603, 611 , 616, 621 628 and 642.
- Scheme 11 illustrates the general route used for the synthesis of examples 427, 540, 581 , 595, 602, 615, 619, 630 and 684
- Scheme 12 illustrates the general route used for the synthesis of examples 541 , 545 and 548.
- Scheme 13 illustrates the general route used for the synthesis of examples 398, 609, 614, 634, 635 and 673
- Scheme 14 illustrates the general route used for the synthesis of examples 538 and 559
- Scheme 15 illustrates the general route used for the synthesis of examples 555, 556, 562, 578, 579, 597, and 744.
- Method A A Discovery C 18 , 5 ⁇ m, 3 x 30 mm column was used at a flow rate of 400 ⁇ L/min, sample loop 5 ⁇ l_, mobile phase: (A) water with 0.1% formic acid, mobile phase, (B) methanol with 0.1% formic acid; retention times are given in minutes.
- Method B A Waters Symmetry C 18 , 3.5 ⁇ m, 4.6 x 75 mm column at a flow rate of 1 ml_ /min, sample loop 10 ⁇ l_, mobile phase (A) is water with 0.05% TFA, mobile phase (B) is ACN with 0.05% TFA; retention times are given in minutes.
- Method C Gradient: 4.2 min/ Flow: 2 ml/min 99:01 - 0:100 Water + 0.1%(Vol.) TFA; Acetonitril + 0.1%(Vol.) TFA; 0.0 to 0.2 min: 99:01 ; 0.2 to 3.8 min: 99:01 ⁇ 0:100; 3.8 to 4.2 min: 0:100; Column: Chromolith Performance RP18e; 100 mm long, 3 mm diameter; Wavelength: 220nm.
- Analytical chiral HPLC was performed using a ChiralPak AD-H column (250 X 4.6 mm) from Daicel Chemical Industries, Ltd. on an Agilent 1100 Series system. The method used a 5.0 ⁇ L injection volume, with a flow rate of 1 ml_/min of 100% methanol for 15 min at 25 °C, and UV-detection at 254 and 280 nm.
- Preparative HPLC was performed using either a Waters Atlantis dC 18 OBD TM 10 ⁇ M (30 X 250 mm) column or a Waters Sunfire Prep C 18 OBD 10 ⁇ M (30 X 250 mm) column. The columns were used at a flow rate of 60 ml_/min on a Waters Prep LC 4000 System equipped with a sample loop (10 mL) and an ISCO UA-6 UV/Vis detector. The mobile phase was drawn from two solvent reservoirs containing (A) water and (B) HPLC-grade acetonitrile. A typical preparative run used a linear gradient (e.g., 0-60 % solvent B over 60 min).
- 4-oxo-3,4-dihydroquinazoline-8-carboxylic acid (5.0 g, 26.3 mmol) was treated with a solution of sulfuric acid ((1.2 equivalents) in anhydrous MeOH (100 mL) under refluxing for 2 days. After cooling to rt, 2N NaOH solution was added to the reaction mixture to adjust pH ⁇ 8. After removal of MeOH, methyl ester was collected by filtration, and washing with water and ethyl acetate as pale yellow solid in 94% yield.
- Phenyl isocyanate (240 ⁇ l_, 0.5 M in anhydrous DCM, 0.11 mmol, 1.1 equiv.) was added to a solution of 4-[(4-aminobenzyl)amino]quinazoline-8-carboxamide (32 mg, 0.1 mmol, 1.0 equiv.) and triehtylamine (40 ⁇ l_, 0.3 mmol, 3.0 equiv.) in anhydrous DCM (2ml_). The resulting mixture was stirred at rt overnight. The title compound was obtained by pre- HPLC in 27% yield. Mass: M+H + : 413.
- Phenyl isocyanate (240 ⁇ L, 0.5 M in anhydrous DCM, 0.11 mmol, 1.1 equiv.) was added to a solution of 4-[(3-aminobenzyl)amino]quinazoline-8-carboxamide (32 mg, 0.1 mmol, 1.0 equiv.) and triehtylamine (40 ⁇ L, 0.3 mmol, 3.0 equiv.) in anhydrous DCM (2mL). The resulting mixture was stirred at rt overnight. The title compound was obtained by pre- HPLC in 34% yield. Mass: M+H + : 413.
- Methyl ⁇ -chloro-3- ⁇ rifluoromethyObenzyllaminoJ- ⁇ -nitroquinazoline-8-carboxylate (190 mg, 0.43 mmol) was treated with a mixture of 650 ⁇ l_ of THF and 650 ⁇ l_ of 2N LiOH (1.3 mmol, 3 equiv.)) at rt for 2h. Water was added and pH was adjusted to ⁇ 4 with 2N HCI. The precipitate was collected as the desired acid by filtration, and washing with water in 97% yield.
- Zinc powder (115 mg, 1.76 mmol, 5.0 equiv.) was added to a suspension of 4- ⁇ [4-chloro- 3-(trifluoromethyl)benzyl]amino ⁇ -6-nitroquinazoline-8-carboxamide (150 mg, 0.35 mmol, 1.0 equiv.) in acetic acid (12 mL). The resulting mixture was stirred at 80°C for 10 min. The reaction mixture was filtered. The filtrate was concentrated and purified by pre- HPLC to yield the title compound as yellow solid in 68% yield. Mass: M+H + : 396. 63. 6-(acetylamino)-4-(r4-chloro-3-(trifluoromethyl)benzyllamino)quinazoline-8- carboxamide (compound No. 173)
- Phenyl isocyanate (70 ⁇ L, 0.5 M in anhydrous DCM, 0.03 mmol, 1.1 equiv.) was added to a solution of TFA salt of 6-amino-4- ⁇ [4-chloro-3-
- Step 1 A 1 L round bottom flask was charged with 2-aminoisophthalic acid (25.0 g, 138.1 mmol) and formaldehyde (125 mL) and heated to 130 °C for 4 hours. The crude mixture was cooled to room temperature, poured on to ice water and the filtrate collected, rinsed with water and heptanes and dried (A). The aqueous solution was then concentrated and poured in to acetone and more precipitate was collected, washed with acetone and heptanes and dried (B). Both isomers were carried on in a similar fashion through the methylation step.
- Step 2 A 1 L round bottom flask was charged with 4-hydroxyquinazoline-8-carboxylic acid (A), MeOH (300 ml_), and H 2 SO 4 (10 mL). The reaction was heated to 50 °C for 2 days. Upon cooling to room temperature, the reaction mixture was neutralized with NaHCO 3 and diluted with water. The methanol was removed in vacuo, and the precipitate was collected, washed with water and heptanes, then dried to give methyl 4- hydroxyquinazoline-8-carboxylate (18.3 g, 65%, over 2 steps combining both isomers from A and B). Step 3.
- A 4-hydroxyquinazoline-8-carboxylic acid
- MeOH 300 ml_
- H 2 SO 4 10 mL
- Step 4 To a solution of the above chloroester in THF was added diisopropylethylamine at 25 °C. After 5 minutes the desired amine was added and heated to 50 °C. After the reaction was complete the reaction was concentrated in vacuo to dryness. The residue was re-dissolved in methylene chloride and washed with aqueous brine solution. The organic layer was separated and dried with sodium sulfate, concentrated and purified by ISCO Companion system. Step 5. Formation of the carboxamide was accomplished by one of the following methods:
- Step 1 A 1000 mL round bottom flask equipped with a stir bar and nitrogen inlet was charged with anisidine, (25 g, 200 mmol) and THF, (400 mL) . To this solution at RT was added 4.0M HCI in dioxane, (100 mL, 400 mmol). A ppt formed almost immediately. The suspension was allowed 30 min and then was filtered. The ppt was washed with Et 2 O and dried under vacuum. A 1000 mL round bottom flask equipped with a stir bar and nitrogen inlet was charged with anisidine hydrochloride, (32 g, 200 mmol) and AcOH, (400 mL).
- Step 2 A 250 mL round bottom flask equipped with a stir bar, Vigreux column and nitrogen inlet was charged with 2,6-dibromo-4-methoxy-phenylamine, (14 g, 50 mmol) and NMP, (80 mL). Copper cyanide, (18 g, 200 mmol) was then added at ambient temperature and the reaction was heated at 140 °C oil bath temperature. The reaction was allowed left to stir for 24 h. The reaction was cooled, diluted with EtOAc, (1000 mL) and poured into 1000 mL of 10% ethylene diamine solution. The mixture was stirred vigorously for 2 h.
- Step 4 A 100 mL round bottom flask equipped with a stir bar, Vigreux column and nitrogen inlet was charged with 2-amino-5-methoxy-isophthalic acid, (3.8 g, 18 mmol) and formamide, (36 mL). The mixture was stirred and heated at 140 °C overnight. The reaction was poured into rapidly stirred ice water (100 mL) causing a precipitate to form and the pH was adjusted to approx. pH 4 using 1 M HCI. The material was collected by suction filtration and was washed with Et 2 O. Amount obtained: 2.2 g, 10 mmol, 56% yield. The material was used as is in the next step. Step 5.
- Step 1 To a solution of 3-fluoromandelic acid (4.0 g, 23.5 mmol) in 30.0 mL of N 1 N- dimethylformamide (DMF) was added CS 2 CO 3 (11.49 g, 35.3 mmol) and the suspension was stirred at room temperature until gas evolution ceased. To the stirring suspension was added at room temperature ethyl iodide (2.28 mL, 28.2 mmol) dropwise. The reaction was stirred at room temperature for 16 hours and then saturated aqueous sodium chloride ( ⁇ 50 mL), water ( ⁇ 50 mL) and ethyl acetate ( ⁇ 50 mL) were added.
- DMF N 1 N- dimethylformamide
- Step 2 A 2M solution of oxalyl chloride in dichloromethane (17.63 mL, 35.2 mmol) was diluted further with 20 mL of additional dichloromethane and cooled to -78 °C.
- Step 3 The glyoxylate from Step 2 was dissolved in 75 mL of THF and (R)-2-methyl-2- propanesulfinamide (3.09 g, 24.8 mmol) was added followed by Ti(OEt) 4 (21.1 mL, 99.2 mmol).
- Step 4 A 500 mL round-bottom flask containing a solution of the sulfinimine (5.72 g,
- Step 5 To a solution of the sulfinamide reactant (3.62 g, 12.0 mmol) in ethanol (80 mL) was added at room temperature 4N HCI in dioxane (15.0 mL, 60 mmol). The reaction was stirred for 4 hours and then concentrated in vacuo to an off-white solid (2.3 g, 100%).
- Step 6 The HCI amine salt (2.3 g, 12.0 mmol) was suspended in dioxane (40 mL) and 1N NaOH (80 mL) was added at room temperature. To the vigorously stirring yellow solution was then added di-tert-butyl dicarbonate (3.27 g, 15 mmol) and the reaction was stirred at room temperature for 16 hours. At this time water (150 mL) and ethyl acetate (100 mL) were added and the mixture was extracted with ethyl acetate three times.
- N-Ethyl-4-nitro-benzenesulfonamide (Ref.: Ragactives, S. L. Patent: EP1813618 (2007)).
- Ethylamine solution (0.91 g, 20 mmol, 1.3 mL, 70% w/v in water, 4.5 eq.) and methanol (5 mL).
- the reaction vial was cooled to 0 °C.
- the Nosyl chloride (1.0 g, 4.5 mmol, 1 eq.) was added portion-wise keeping the temperature between 0 - 5 °C and stirring continued x 15 min. after addition was complete. Water (10 mL) was then added.
- Step 1 To a 100-mL round bottom flask with magnetic stir bar at 25 °C was added the powdered KOH (0.37 g, 6.55 mmol, 2 eq.) and acetonitrile (10 mL).
- Example F (0.83 g, 3.6 mmol, 1.1 eq.) was then added and stirring continued x 10 minutes.
- Example C (0.98 g, 3.3 mmol, 1 eq.) was dissolved in acetonitrile (16 ml.) and added to the reaction flask. Stirring was continued x 4 hours at 25 °C. The reaction was quenched by addition of an approximately equal volume of 0.5N aq. HCI solution ( ⁇ 25 ml_).
- Step 2 To a 100-mL round bottom flask with magnetic stir bar at 25 °C was added ((S)- 2-Ethyl-(4-nitro-benzenesulfonyl)-amino]-1-phenyl-ethyl ⁇ -carbamic acid tert-butyl ester (1.06 g, 2.4 mmol, 1 eq.) and THF (20 mL). The reaction flask was cooled to 0 °C and the HCI in Dioxane (30 mL, 4M, excess) added with stirring. The reaction was stirred vigorously x 16 hours (0 °C - 25 °C). The solvent was then evaporated in vacuo.
- Examples A - G were also used for the synthesis of other scaffolds and buiilding blocks that were commonly used for the compounds according to Formula (I).
- reaction mixture was stirred at RT for 30 min, filtered, got the tert-butyl [3-( ⁇ [8-(aminocarbonyl)-6- formylquinazolin-4-yl]amino ⁇ methyl)phenyl]carbamate as a solid, which was used directly for the next reaction.
- Step 1 To a 40-mL vial with magnetic stir bar at 25 °C under a nitrogen atmosphere was added 4-Chloro-quinazoline-8-carboxylic acid methyl ester (0.46 g, 2.08 mmol, 1 eq.) and anhydrous THF (15 mL). The Diisopropylethylamine (0.81 g, 1.09 ml_, 6.25 mmol, 3 eq.) was then added followed by the amine (0.6 g, 2.3 mmol, 1.1 eq.). The resulting mixture was heated in a capped vial at 50 °C overnight with stirring. The solvent was evaporated in vacuo and the resulting residue re-dissolved in EtOAc (50 mL).
- Step 2 To a 40-mL vial with magnetic stirbar at r.t. was added the 4- ⁇ 3-[(2-Chloro- pyridine ⁇ -carbonyl)-aminoj-benzylamino ⁇ quinazoline-8-carboxylic acid methyl ester (0.6 g, 1.34 mmol, 1 eq.) and THF (6 mL) and 2-propanol (6 mL). An approx. equal volume (i.e.; 6 mL) of concentrated aqueous ammonium hydroxide solution (28-30% soln.) was then added and stirring continued overnight. Water (15 mL) was added to the reaction mixture and a white precipitate immediately began to form. The precipitate was collected and dried thoroughly in vacuo and afforded 314 mg (55% yield). Material was carried on to the next synthetic step without further purification. LCMS (ESI) 433 (M+H)
- 6-(2-Morpholin-4-yl-ethoxy)-4-(4-trifluoromethyl-benzylamino)-quinazoline-8-carboxylic acid amide To a solution of 6-Hydroxy-4-(4-trifluoromethyl-benzylamino)-quinazoline-8-carboxylic acid amide (85 mg, 0.2 mmol) in N,N-dimethylformamide (3.0 mL) was added cesium carbonate (229 mg, 0.6 mmol).
- Example 184 6-methoxy-4-(2-methylamino-1 -phenyl-ethylamino)-quinazoline-8-carboxylic acid amide
- a scintillation vial equipped with a stir bar was charged with [2-(8-carbamoyl-6-methoxy- quinazolin-4-ylamino)-2-phenyl-ethyl]-methyl-carbamic acid tert-butyl ester (150 mg, 0.33 mmol) and THF, (5 mL).
- 4 M HCI in dioxane, (5 mL) was added at RT and the mixture was stirred overnight. After 18 h, a white precipitate had formed and LCMS indicated consumption of SM.
- Example 201 The title compound was synthesized according to the procedure described for the preparation of Example 462. LCMS [520.8 (M+2)]. Example 201
- Step 2 To a 40-mL vial with magnetic stir bar at 25 °C under a nitrogen atmosphere was added 4-Chloro-quinazoline-8-carboxylic acid methyl ester (0.25 g, 1.14 mmol) and anhydrous THF (15 mL). DIEA (0.6 mL, 3.4 mmol) was then added followed with N-[3- (1-Amino-2-dimethylamino-ethyl)-phenyl]-2-chloro-isonicotinamide (0.4 g, 1.25 mmol). The resulting mixture was heated in a capped vial at 50 - 55 °C x 16 hours with stirring.
- Step 3 To a 40-mL vial with magnetic stirbar at 25 °C was added 4-(1- ⁇ 3-[(2-Chloro- pyridine-4-carbonyl)-amino]-phenyl ⁇ -2-dimethylamino-ethylamino)-quinazoline-8- carboxylic acid methyl ester (0.25 g, 0.495 mmol, 1 eq.) and THF (5 mL) and iPrOH (5 ml_). An approx. equal volume of concentrated aq. NH 4 OH solution (28-30% soln.) was then added and stirring continued overnight. The reaction mixture was heated at 50 °C x 4 hours in order to drive the reaction to completion. H 2 O (25 mL) was added to the reaction mixture and a precipitate immediately began to form. The precipitate was collected and discarded, and the aqueous layer was evaporated under nitrogen affording the desired product, 0.23 g (96% yield).
- Example 209 4-(1-[3-(2.6-Difluoro-benzoylamino)-phenyl1-2-methylamino-ethylamino)-quinazoline--8- carboxylic acid amide
- Example 228 4-(1-[3-(2-fluoro-4-methoxy-benzoylamino)-phenyl]-2-methylamino-ethylamino ⁇ - quinazoline--8-carboxylic acid amide
- Example 246 4-(1-[3-(3-Fluoro-4-methyl-benzoylamino)-phenyl1-ethylamino)-quinazoline-8-carboxylic acid amide.
- Example 250 The title compound was synthesized according to the procedure described for the preparation of Example 462. LCMS [505 (M+1)]. Example 250
- Example 266 4-((R)-1-(3-[(6-Cvano-pyridine-3-carbonyl)-aminol-phenyl ⁇ -ethylamino)-quinazoline-8- carboxylic acid amide
- Step 2 To a solution of 2-Dimethylamino-1-(3-methoxy-phenyl)-ethanone (1.0g, 5.64 mmol) in pyridine (10.0 mL) was added NH 2 OH. HCI (1.9g, 28.2 mmol) and the reaction mixture was stirred at 25 °C for 16h. The mixture was diluted with water (100 mL) and was extracted with DCM (x3), dried (Na 2 SO 4 ) and the solvent was evaporated under reduced pressure to give crude product, (0.75g, 64%). The material was used as is in the next step.
- Step 3 To a solution of 2-Dimethylamino-1-(3-methoxy-phenyl)-ethanone oxime (0.75g, 3.6 mmol) in THF (8.0 mL) was added LAH (4.5 mL of 2.0M THF solution, 9.01 mmol) at 0 °C. After the addition is over, the reaction was refluxed for 3h. The reaction was carefully quenched with water (5.0 mL) followed by 2N NaOH (10.0 ml). Additional 20 mL of THF was added and the organic layer was separated from the white solid and concentrated.
- LAH 4.5 mL of 2.0M THF solution, 9.01 mmol
- a Wheaton vial equipped with a stir bar was charged with 4-[2-dimethylamino-1-(3- fluoro-phenyl)-ethylaminol-6-hydroxy-quinazoline-8-carboxylic acid amide, (37 mg, 0.1 mmol) Cs 2 CO 3 , (100 mg, 0.3 mmol) and dry DMF, (1 ml_).
- the mixture was heated at 60 °C for 1 h. It was then cooled to RT and ethylbromide (11 mg, 0.1 mmol) was added a solution in DMF (0.5 ml_). The mixture was left to stir overnight. After 18 h, LCMS indicated consumption of SM.
- Step 1 To a 250-mL round bottom flask with magnetic stir bar at 25 °C under a nitrogen atmosphere and fitted with a Vigreux column was added 4-Chloro-quinazoline-8- carboxylic acid methyl ester (0.5 g, 2.25 mmol, 1 eq.) and anhydrous THF (40 mL). DIEA (0.87 g, 1.17 mL, 6.7 mmol, 3 eq.) was then added followed by Example G (0.86 g, 2.5 mmol, 1.1 eq.). The resulting mixture was heated at 70- 75 °C x 16 hours with stirring. The solvent was evaporated in vacuo and the resulting residue re-dissolved in EtOAc (50 mL).
- Step 2 To a 40-mL vial with magnetic stirbar at 25 °C was added the 4- ⁇ (S)-2-[Ethyl-(4- nitro-benzenesulfonyl)-amino]-1-phenyl-ethylaminoJ-quinazoline-8-carboxylic acid methyl ester (0.69 g, 1.29 mmol, 1 eq.) and THF (10 mL) and 2-propanol (10 mL). An approx. equal volume (i.e.; 10 mL) of concentrated aqueous ammonium hydroxide solution (28- 30% soln.) was then added and stirring continued over the weekend (x 96 hours).
- Step 3 To a 40-mL vial with magnetic stirbar at 25 °C under a nitrogen atmosphere was added 4- ⁇ (S)-2-[Ethyl-(4-nitro-benzenesulfonyl)-amino]-1-phenyl-ethylamino ⁇ - quinazoline-8-carboxylic acid amide (0.45 g, 0.86 mmol, 1 eq.) and anhydrous acetonitrile (25 mL). The Cesium carbonate (0.84 g, 2.6 mmol, 3 eq.) was added followed by the thiophenol (0.14 g, 1.3 mmol, 0.13 mL, 1.5 eq.). Stirring was continued at 25 °C x 16 hours.
- Example 294 4-(1-[3-(4-Cvano-3-fluoro-benzoylamino)-phenyll-ethylamino)-quinazoline-8-carboxylic acid amide.
- Example 301 4-(1-(3-[(6-Chloro-pyridine-3-carbonyl)-amino1-phenyl)-ethylamino)-quinazoline-8- carboxylic acid amide.
- 6-hvdroxy-4-(2-methylamino-1 -phenyl-ethylamino)-quinazoline-8-carboxylic acid amide A 250 mL round bottom flask equipped with a stir bar was evacuated and flushed with nitrogen. To this flask was added Pd/C (5%), (6 mg) and EtOH, (30 mL). Then 6- benzyloxy-4-(2-methylamino-1 -phenyl-ethylamino)-quinazoline-8-carboxylic acid amide, (64 mg, 0.15 mmol) was added as a solid. The solution was evacuated and flushed three times with nitrogen and then evacuated and flushed three times with hydrogen. The mixture was left to stir over the weekend.
- Example 322 4- ( 3-Methoxy-1-[3- ( 2.5-difluoro-benzoylamino)-phenyll-Dropylamino)-quinazoline--8- carboxylic acid amide
- Example 333 The title compound was synthesized according to the procedure described for the preparation of Example 462. LCMS [559.1 (M), 561.0 (M+2H)]. Example 333
- Step 1 To a 40-mL vial with magnetic stir bar at 25 °C under a nitrogen atmosphere was added Methyl 5-Bromo-nicotinate (0.5 g, 2.3 mmol, 1 eq.), morpholine (0.3 g, 0.3 mL, 3.5 mmol, 1.5 eq.), and toluene (5 mL). Cesium carbonate (2.26 g, 6.9 mmol, 3 eq.), Palladium (II) acetate (0.052 g, 0.23 mmol, 0.1 eq.), and BINAP (0.29 g, 0.46 mmol, 0.2 eq.) were then added and the reaction vial heated with stirring at 80° C x 16 hours.
- Methyl 5-Bromo-nicotinate 0.5 g, 2.3 mmol, 1 eq.
- morpholine 0.3 g, 0.3 mL, 3.5 mmol, 1.5 eq.
- Step 2 To a 40-mL vial with magnetic stir bar at 25 °C was added Methyl 5-Morpholin-4- yl-nicotinate (0.42 g, 1.89 mmol, 1 eq.) and methanol (10 mL). Aqueous sodium hydroxide solution (0.94 mL, 10M, 9.45 mmol, 5 eq.) was then added and the reaction vial heated with stirring at 65 °C x 16 hours. The reaction mixture was concentrated in vacuo and the resulting residue dissolved in a minimal volume of H 2 O (2 - 3 mL). The mixture was acidified with glacial acetic acid (AcOH) to pH 3.
- AcOH glacial acetic acid
- Step 3 To a 40-mL vial with magnetic stir bar at 25 °C under a nitrogen atmosphere was added (3-Amino-benzyl)-carbamic acid tert-butyl ester (0.23 g, 1.05 mmol, 1 eq.) and anhydrous DMF (10 mL).
- the 5-Morpholin-4-yl-nicotinic acid (0.24 g, 1.15 mmol, 1.1 eq.) was added followed by the Diisopropylethylamine (0.68 g, 0.91 mL, 5.2 mmol, 5 eq.) and the HATU (0.48 g, 1.26 mmol, 1.2 eq.).
- the reaction mixture was stirred overnight at 25 °C.
- the reaction mixture was then taken up in EtOAc (50 mL) and washed with H 2 O (20 mL), saturated aq. LiCI solution (20 mL), brine (20 mL), dried (e.g., Na 2 SO 4 ), filtered and concentrated.
- the resulting residue was purified by column chromatography (ISCO CombiFlash) using a 0-100% gradient (EtOAc/Heptane) to afford 250 mg (57% yield).
- Step 4 To a 40-mL vial with magnetic stir bar at 25 °C was added ⁇ 3-[(5-Morpholin-4-yl- pyridine-3-carbonyl)-amino]-benzyl ⁇ -carbamic acid tert-butyl ester (0.25 g, 0.61 mmol, 1 eq.) and anhydrous DCM (3 mL). The reaction vial was cooled to 0 °C and the HCI in 1 ,4-Dioxane (0.75 mL, 4M, 3 mmol, 5 eq.) was added drop-wise with vigorous stirring. Stirring was continued x 16 hours and was allowed to equilibrate to 25 °C.
- Step 5 To a 40-mL vial with magnetic stir bar at 25 °C under a nitrogen atmosphere was added 4-Chloro-quinazoline-8-carboxylic acid methyl ester (0.14 g, 0.61 mmol, 1 eq.) and anhydrous THF (10 mL).
- the Diisopropylethylamine (0.24 g, 0.32 mL, 1.8 mmol, 3 eq.) was then added followed by the amine (0.21 g, 0.67 mmol, 1.1 eq.).
- the resulting mixture was heated in a capped vial at 50 °C x 16 hours with stirring.
- the solvent was evaporated in vacuo and the resulting residue re-dissolved in EtOAc (30 mL).
- the mixture was washed with saturated aqueous NaHCO 3 solution (20 mL), H 2 O (20 mL), brine (20 mL), and dried (e.g., Na 2 SO 4 ), filtered and concentrated.
- Example 353 4-(3-Methoxy-1-[3-(3,4-difluoro-benzoylamino)-phenvn-propylamino)-quinazoline--8- carboxylic acid amide
- Example 358 4-f 1 -P-Benzoylamino-phenyl)-ethylaminoi-quinazoline-8-carboxylic acid Amide.
- the title compound was synthesized according to the procedure described for the preparation of Example 425. MS (M+1) 412
- Example 359 4-(1-(3-[(2-Methyl-furan-3-carbonyl)-amino1-phenyl)-ethylamino)-quinazoline-8-carboxylic acid amide.
- Example 367 4-f2-(ethyl-methyl-amino)-1-phenyl-ethylaminoi-quinazoline-8-carboxylic acid amide
- a 20 mL scintillation vial equipped with a stir bar was charged with 4-(2-methylamino-1- phenyl-ethylamino)-quinazoline-8-carboxylic acid amide hydrochloride salt, (32 mg, 0.1 mmol), EtOH, (5 mL) and Et 3 N, (0.03 mL, 0.2 mmol). The mixture was stirred until the amine had dissolved.
- Example 418 4-(1-[3-(2.4-Difluoro-benzoylamino)-phenyll-3-pyrrolidin-1-yl-propylamino)-quinazoline-- 8-carboxylic acid amide
- the di-tert-butyl dicarbonate (9.8 g, 45 mmol, 1.1 eq.) was dissolved in anhydrous DCM (15 mL) and added rapidly drop-wise to the reaction vessel. The reaction was then stirred overnight and allowed to equilibrate to room temperature. The mixture was washed with saturated aqueous NaHC ⁇ 3 solution (50 mL), brine (50 mL), and dried (e.g., Na 2 SO 4 ), filtered and concentrated to afford 10.8 g Step 2.
- Step 3 To a 40-mL vial with magnetic stir bar at 25 °C was added ⁇ 3-[(6-Methoxy- pyridine-3-carbonyl)-amino]-benzyl ⁇ -carbamic acid tert-butyl ester (1.66g, 4.7 mmol, 1 eq.) and anhydrous DCM (20 mL). The reaction vial was cooled to 0 °C and the HCI in 1 ,4-Dioxane (5.9 mL, 4M, 23.8 mmol, 5 eq.) was added drop-wise with vigorous stirring. Stirring was continued overnight and was allowed to equilibrate to 25 °C.
- Step 5 To a 40-mL vial with magnetic stirbar at 25 °C was added 4- ⁇ 3-[(6-Methoxy- pyridine-3-carbonyl)-aminol-benzylaminoJ-quinazoline-8-carboxylic acid methyl ester (0.18 g, 0.4 mmol, 1 eq.) and THF (2 mL) and 2-propanol (2 mL). An approx. equal volume (i.e.; 2 mL) of concentrated aqueous ammonium hydroxide solution (28-30% soln.) was then added and stirring continued overnight. Water (15 ml_) was added to the reaction mixture and a precipitate immediately began to form. The precipitate was collected and purified further via preparative HPLC afforded the desired compound as a white solid (13.9 mg) LCMS (ESI) 429.2 (M+H).
- Product is the hydrochloride salt.
- Example 442 4-(1- ⁇ 3-[(Furan-3-carbonyl)-amino1-phenyl)-ethylamino)-quinazoline-8-carboxylic acid amide.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Diabetes (AREA)
- Pain & Pain Management (AREA)
- Dermatology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Obesity (AREA)
- Ophthalmology & Optometry (AREA)
- Heart & Thoracic Surgery (AREA)
- Biomedical Technology (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Rheumatology (AREA)
- Emergency Medicine (AREA)
- Urology & Nephrology (AREA)
- Cardiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description
































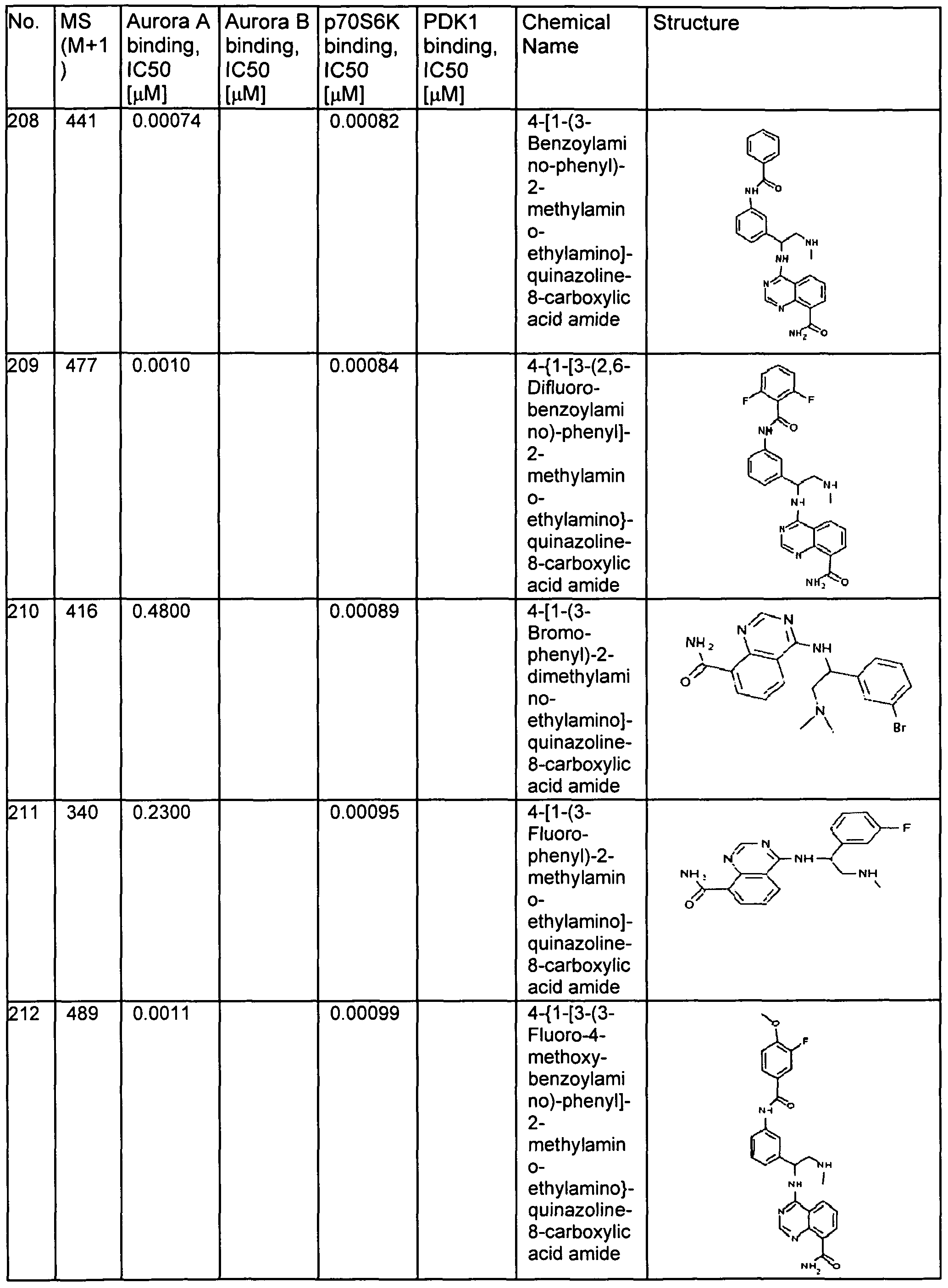














































































































































Claims






Priority Applications (22)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MX2011008395A MX338354B (en) | 2009-02-11 | 2010-02-04 | Novel amino azaheterocyclic carboxamides. |
| CA2751886A CA2751886C (en) | 2009-02-11 | 2010-02-04 | Novel amino azaheterocyclic carboxamides |
| SG2011055001A SG173473A1 (en) | 2009-02-11 | 2010-02-04 | Novel amino azaheterocyclic carboxamides |
| BRPI1008325-1A BRPI1008325A2 (en) | 2009-02-11 | 2010-02-04 | amino azaeterocyclic carboxamides |
| AU2010214095A AU2010214095B2 (en) | 2009-02-11 | 2010-02-04 | Novel amino azaheterocyclic carboxamides |
| KR1020117021335A KR101699991B1 (en) | 2009-02-11 | 2010-02-04 | Novel amino azaheterocyclic carboxamides |
| US13/148,903 US8637532B2 (en) | 2009-02-11 | 2010-02-04 | Amino azaheterocyclic carboxamides |
| JP2011550122A JP5642714B2 (en) | 2009-02-11 | 2010-02-04 | A novel aminoazaheterocyclic carboxamide |
| PL10705453T PL2396307T3 (en) | 2009-02-11 | 2010-02-04 | Novel amino azaheterocyclic carboxamides |
| NZ595087A NZ595087A (en) | 2009-02-11 | 2010-02-04 | Novel amino azaheterocyclic carboxamides |
| CN201080007165.1A CN102317269B (en) | 2009-02-11 | 2010-02-04 | Novel amino azaheterocyclic carboxamides |
| DK10705453.8T DK2396307T3 (en) | 2009-02-11 | 2010-02-04 | NOVEL AMINO-azaheterocyclic carboxamides |
| EA201101182A EA020731B1 (en) | 2009-02-11 | 2010-02-04 | Amino azaheterocyclic carboxamides |
| UAA201110411A UA107188C2 (en) | 2009-02-11 | 2010-02-04 | CARBOXAMID DERIVATIVES OF QUINASOLINES OR QUINOLINES AND PHARMACEUTICAL COMPOSITION THEREOF |
| SI201030849T SI2396307T1 (en) | 2009-02-11 | 2010-02-04 | Novel amino azaheterocyclic carboxamides |
| ES10705453.8T ES2527940T3 (en) | 2009-02-11 | 2010-02-04 | New amino-azaheterocyclic carboxamides |
| EP10705453.8A EP2396307B1 (en) | 2009-02-11 | 2010-02-04 | Novel amino azaheterocyclic carboxamides |
| IL214412A IL214412A0 (en) | 2009-02-11 | 2011-08-02 | Novel amino azaheterocyclic carboxamides |
| ZA2011/06624A ZA201106624B (en) | 2009-02-11 | 2011-09-09 | Novel amino azaheterocyclic carboxamides |
| HK12106745.1A HK1166070A1 (en) | 2009-02-11 | 2012-07-10 | Novel amino azaheterocyclic carboxamides |
| US14/105,974 US9040560B2 (en) | 2009-02-11 | 2013-12-13 | Amino azaheterocyclic carboxamides |
| HRP20141239AT HRP20141239T1 (en) | 2009-02-11 | 2014-12-19 | Novel amino azaheterocyclic carboxamides |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US20735409P | 2009-02-11 | 2009-02-11 | |
| US61/207,354 | 2009-02-11 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/148,903 A-371-Of-International US8637532B2 (en) | 2009-02-11 | 2010-02-04 | Amino azaheterocyclic carboxamides |
| US14/105,974 Division US9040560B2 (en) | 2009-02-11 | 2013-12-13 | Amino azaheterocyclic carboxamides |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010093419A1 true WO2010093419A1 (en) | 2010-08-19 |
Family
ID=42199208
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/000313 WO2010093419A1 (en) | 2009-02-11 | 2010-02-04 | Novel amino azaheterocyclic carboxamides |
Country Status (26)
| Country | Link |
|---|---|
| US (2) | US8637532B2 (en) |
| EP (1) | EP2396307B1 (en) |
| JP (1) | JP5642714B2 (en) |
| KR (1) | KR101699991B1 (en) |
| CN (1) | CN102317269B (en) |
| AR (1) | AR075396A1 (en) |
| AU (1) | AU2010214095B2 (en) |
| BR (1) | BRPI1008325A2 (en) |
| CA (1) | CA2751886C (en) |
| CL (1) | CL2011001967A1 (en) |
| DK (1) | DK2396307T3 (en) |
| EA (1) | EA020731B1 (en) |
| ES (1) | ES2527940T3 (en) |
| HK (1) | HK1166070A1 (en) |
| HR (1) | HRP20141239T1 (en) |
| IL (1) | IL214412A0 (en) |
| MX (1) | MX338354B (en) |
| MY (1) | MY160468A (en) |
| NZ (1) | NZ595087A (en) |
| PL (1) | PL2396307T3 (en) |
| PT (1) | PT2396307E (en) |
| SG (1) | SG173473A1 (en) |
| SI (1) | SI2396307T1 (en) |
| UA (1) | UA107188C2 (en) |
| WO (1) | WO2010093419A1 (en) |
| ZA (1) | ZA201106624B (en) |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012013282A1 (en) * | 2010-07-29 | 2012-02-02 | Merck Patent Gmbh | Bicyclic azaheterocyclic carboxamides as inhibitors of the kinase p70s6k |
| WO2012016001A1 (en) * | 2010-07-29 | 2012-02-02 | Merck Patent Gmbh | Cyclic amine azaheterocyclic carboxamides |
| WO2012069146A1 (en) | 2010-11-24 | 2012-05-31 | Merck Patent Gmbh | Quinazoline carboxamide azetidines |
| WO2013040059A1 (en) | 2011-09-12 | 2013-03-21 | Merck Patent Gmbh | Novel imidazole amines as modulators of kinase activity |
| WO2013040044A1 (en) | 2011-09-12 | 2013-03-21 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| WO2013096194A1 (en) * | 2011-12-22 | 2013-06-27 | Merck Patent Gmbh | Novel heterocyclic carboxamides as modulators of kinase activity |
| WO2014078637A1 (en) | 2012-11-16 | 2014-05-22 | Merck Patent Gmbh | Novel heterocyclic derivatives as modulators of kinase activity |
| WO2014078634A1 (en) | 2012-11-16 | 2014-05-22 | Merck Patent Gmbh | Novel imidazol-piperidinyl derivatives as modulators of kinase activity |
| WO2014085528A1 (en) | 2012-11-29 | 2014-06-05 | Merck Patent Gmbh | Azaquinazoline carboxamide derivatives |
| WO2014143612A1 (en) | 2013-03-11 | 2014-09-18 | Merck Patent Gmbh | 6-[4-(1 h-imidazol-2-yl)piperidin-1 -yl]pyrimidin-4-amine derivatives as modulators of kinase activity |
| WO2015149909A1 (en) | 2014-04-03 | 2015-10-08 | Merck Patent Gmbh | Combinations of cancer therapeutics |
| US9701664B2 (en) | 2013-10-04 | 2017-07-11 | Cancer Research Technology Limited | Fused 1,4-dihydrodioxin derivatives as inhibitors of heat shock transcription factor 1 |
| EP3209299A4 (en) * | 2014-10-22 | 2018-04-04 | The Board of Regents of The University of Texas System | Small-molecule inhibitors targeting discoidin domain receptor 1 and uses thereof |
| WO2018215557A1 (en) * | 2017-05-23 | 2018-11-29 | Centre National De La Recherche Scientifique | Ion channel inhibitor compounds for cancer treatment |
| WO2019231271A1 (en) | 2018-05-31 | 2019-12-05 | C&C Research Laboratories | Heterocyclic derivatives and use thereof |
| US10647678B2 (en) | 2015-04-01 | 2020-05-12 | Cancer Research Technology Limited | Quinoline derivatives as inhibitors of heat shock factor 1 pathway activity |
| WO2021083936A1 (en) | 2019-11-01 | 2021-05-06 | Syngenta Crop Protection Ag | Pesticidally active fused bicyclic heteroaromatic compounds |
| WO2022101459A1 (en) | 2020-11-16 | 2022-05-19 | Merck Patent Gmbh | Kinase inhibitor combinations for cancer treatment |
| WO2022268648A1 (en) | 2021-06-24 | 2022-12-29 | Syngenta Crop Protection Ag | 2-[3-[1 [(quinazolin-4-yl)amino]ethyl]pyrazin-2-yl]thiazole-5-carbonitrile derivatives and similar compounds as pesticides |
| WO2023201185A1 (en) * | 2022-04-11 | 2023-10-19 | Merck Patent Gmbh | Pyrido[3,2-d]pyrimidines as hpk1 inhibitors |
| US11814370B2 (en) | 2016-10-07 | 2023-11-14 | Cancer Research Technology Limited | Deuterated N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-((4-ethylpiperazin-1-yl)methyl)quinoline-6-carboxamide |
| WO2023247360A1 (en) | 2022-06-21 | 2023-12-28 | Syngenta Crop Protection Ag | Pesticidally active fused bicyclic heteroaromatic compounds |
| US11993613B2 (en) | 2022-03-31 | 2024-05-28 | Abbvie Inc. | Thiazolo[5,4-b]pyridine MALT-1 inhibitors |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104968200B (en) | 2013-02-01 | 2018-03-06 | 维尔斯达医疗公司 | Amine compounds having anti-inflammatory, antifungal, antiparasitic and anticancer activity |
| CN104292170B (en) * | 2014-09-22 | 2016-06-29 | 广西师范大学 | There is quinazoline-Arylurea derivatives and the application thereof of antitumor action |
| US11254654B2 (en) * | 2017-06-30 | 2022-02-22 | The University Of North Carolina At Chapel Hill | Heterochromatin gene repression inhibitors |
| SI3762368T1 (en) | 2018-03-08 | 2022-06-30 | Incyte Corporation | Aminopyrazine diol compounds as pi3k-y inhibitors |
| CN109369634B (en) * | 2018-12-19 | 2021-07-27 | 陕西国际商贸学院 | Preparation method and application of 2-methoxynicotinamide derivative with antitumor activity |
| RU2723481C1 (en) * | 2019-10-03 | 2020-06-11 | ФЕДЕРАЛЬНОЕ ГОСУДАРСТВЕННОЕ УНИТАРНОЕ ПРЕДПРИЯТИЕ "ИНСТИТУТ ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ ХИМИЧЕСКИХ ВЕЩЕСТВ НАЦИОНАЛЬНОГО ИССЛЕДОВАТЕЛЬСКОГО ЦЕНТРА "КУРЧАТОВСКИЙ ИНСТИТУТ" (НИЦ "Курчатовский институт - ИРЕА) | 4-[methyl 4-(aminomethyl)cyclohexane carboxylate]quinazoline and method for production thereof |
| CN112778217B (en) * | 2019-11-08 | 2024-01-26 | 沈阳化工研究院有限公司 | Quinazoline compound and application thereof |
| CN115521474B (en) * | 2022-09-30 | 2023-08-22 | 陕西科技大学 | Organic framework material based on five-fold interpenetrating hydrogen bonds and preparation method and application thereof |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999009024A1 (en) * | 1997-08-14 | 1999-02-25 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives as hfgan72 antagonists |
| WO2001021596A1 (en) * | 1999-09-21 | 2001-03-29 | Astrazeneca Ab | Quinazoline derivatives and their use as pharmaceuticals |
| WO2002000649A1 (en) * | 2000-06-28 | 2002-01-03 | Astrazeneca Ab | Substituted quinazoline derivatives and their use as inhibitors |
| US20020151544A1 (en) * | 2000-04-27 | 2002-10-17 | Masahiko Hayakawa | Fused heteroaryl derivatives |
| WO2003055491A1 (en) * | 2001-12-24 | 2003-07-10 | Astrazeneca Ab | Substituted quinazoline derivatives as inhibitors of aurora kinases |
| WO2008049047A2 (en) * | 2006-10-18 | 2008-04-24 | Wyeth | Quinoline compounds |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA805652B (en) * | 1979-09-14 | 1981-09-30 | New Zealand Dev Finance | Compounds having antitumour properties |
| GB8804445D0 (en) * | 1988-02-25 | 1988-03-23 | Smithkline Beckman Intercredit | Compounds |
| ES2360933T3 (en) * | 2000-04-27 | 2011-06-10 | Astellas Pharma Inc. | CONDENSED HETEROARILO DERIVATIVES. |
| TW200306819A (en) | 2002-01-25 | 2003-12-01 | Vertex Pharma | Indazole compounds useful as protein kinase inhibitors |
| CA2522595A1 (en) | 2003-04-03 | 2004-10-28 | Vertex Pharmaceuticals Incorporated | Compositions useful as inhibitors of protein kinases |
| WO2005033086A1 (en) | 2003-09-30 | 2005-04-14 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| CA2541989C (en) | 2003-10-24 | 2013-10-01 | Exelixis, Inc. | P70s6 kinase modulators and method of use |
| KR20060117329A (en) | 2003-11-21 | 2006-11-16 | 노파르티스 아게 | 1h-imidazoquinoline derivatives as protein kinase inhibitors |
| TR200808208T1 (en) | 2003-12-09 | 2008-12-22 | The Government Of The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Suppression of an immune response or treatment of a proliferative disease |
| CA2563699C (en) | 2004-04-23 | 2014-03-25 | Exelixis, Inc. | Kinase modulators and method of use |
| JP5274842B2 (en) | 2004-12-28 | 2013-08-28 | エグゼリクシス, インコーポレイテッド | [1H-piperazo [3,4-d] pyrimidin-4-yl] -piperazine as a serine-threonine kinase modulator (p70S6K, Akt-1 and Akt-2) for the treatment of immune, inflammatory and proliferative disorders Or [1H-piperazo [3,4-d] pyrimidin-4-yl] -piperazine compounds |
| GB0501999D0 (en) | 2005-02-01 | 2005-03-09 | Sentinel Oncology Ltd | Pharmaceutical compounds |
| JP2008546751A (en) | 2005-06-22 | 2008-12-25 | アステックス・セラピューティクス・リミテッド | Pharmaceutical composition |
| WO2007015569A1 (en) * | 2005-08-01 | 2007-02-08 | Eisai R & D Management Co., Ltd. | Method for prediction of the efficacy of vascularization inhibitor |
| US7776857B2 (en) * | 2007-04-05 | 2010-08-17 | Amgen Inc. | Aurora kinase modulators and method of use |
| UA99284C2 (en) | 2007-05-11 | 2012-08-10 | Елі Ліллі Енд Компані | P70 s6 kinase inhibitors |
| US7829574B2 (en) * | 2008-05-09 | 2010-11-09 | Hutchison Medipharma Enterprises Limited | Substituted quinazoline compounds and their use in treating angiogenesis-related diseases |
-
2010
- 2010-02-04 BR BRPI1008325-1A patent/BRPI1008325A2/en not_active IP Right Cessation
- 2010-02-04 CN CN201080007165.1A patent/CN102317269B/en active Active
- 2010-02-04 PT PT107054538T patent/PT2396307E/en unknown
- 2010-02-04 MY MYPI2011003710A patent/MY160468A/en unknown
- 2010-02-04 ES ES10705453.8T patent/ES2527940T3/en active Active
- 2010-02-04 KR KR1020117021335A patent/KR101699991B1/en active IP Right Grant
- 2010-02-04 AU AU2010214095A patent/AU2010214095B2/en active Active
- 2010-02-04 JP JP2011550122A patent/JP5642714B2/en active Active
- 2010-02-04 SG SG2011055001A patent/SG173473A1/en unknown
- 2010-02-04 SI SI201030849T patent/SI2396307T1/en unknown
- 2010-02-04 DK DK10705453.8T patent/DK2396307T3/en active
- 2010-02-04 MX MX2011008395A patent/MX338354B/en active IP Right Grant
- 2010-02-04 EP EP10705453.8A patent/EP2396307B1/en active Active
- 2010-02-04 NZ NZ595087A patent/NZ595087A/en unknown
- 2010-02-04 US US13/148,903 patent/US8637532B2/en active Active
- 2010-02-04 CA CA2751886A patent/CA2751886C/en active Active
- 2010-02-04 UA UAA201110411A patent/UA107188C2/en unknown
- 2010-02-04 EA EA201101182A patent/EA020731B1/en not_active IP Right Cessation
- 2010-02-04 WO PCT/US2010/000313 patent/WO2010093419A1/en active Application Filing
- 2010-02-04 PL PL10705453T patent/PL2396307T3/en unknown
- 2010-02-11 AR ARP100100376A patent/AR075396A1/en not_active Application Discontinuation
-
2011
- 2011-08-02 IL IL214412A patent/IL214412A0/en active IP Right Grant
- 2011-08-11 CL CL2011001967A patent/CL2011001967A1/en unknown
- 2011-09-09 ZA ZA2011/06624A patent/ZA201106624B/en unknown
-
2012
- 2012-07-10 HK HK12106745.1A patent/HK1166070A1/en unknown
-
2013
- 2013-12-13 US US14/105,974 patent/US9040560B2/en active Active
-
2014
- 2014-12-19 HR HRP20141239AT patent/HRP20141239T1/en unknown
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999009024A1 (en) * | 1997-08-14 | 1999-02-25 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives as hfgan72 antagonists |
| WO2001021596A1 (en) * | 1999-09-21 | 2001-03-29 | Astrazeneca Ab | Quinazoline derivatives and their use as pharmaceuticals |
| US20020151544A1 (en) * | 2000-04-27 | 2002-10-17 | Masahiko Hayakawa | Fused heteroaryl derivatives |
| WO2002000649A1 (en) * | 2000-06-28 | 2002-01-03 | Astrazeneca Ab | Substituted quinazoline derivatives and their use as inhibitors |
| WO2003055491A1 (en) * | 2001-12-24 | 2003-07-10 | Astrazeneca Ab | Substituted quinazoline derivatives as inhibitors of aurora kinases |
| WO2008049047A2 (en) * | 2006-10-18 | 2008-04-24 | Wyeth | Quinoline compounds |
Cited By (72)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8710044B2 (en) | 2010-07-29 | 2014-04-29 | Merck Patent Gmbh | Bicyclic azaheterocyclic carboxamides |
| WO2012016001A1 (en) * | 2010-07-29 | 2012-02-02 | Merck Patent Gmbh | Cyclic amine azaheterocyclic carboxamides |
| EA023132B1 (en) * | 2010-07-29 | 2016-04-29 | Мерк Патент Гмбх | Bicyclic azaheterocyclic carboxamides |
| AU2011282684B2 (en) * | 2010-07-29 | 2015-05-21 | Merck Patent Gmbh | Cyclic amine azaheterocyclic carboxamides |
| AU2011285247B2 (en) * | 2010-07-29 | 2016-08-04 | Merck Patent Gmbh | Bicyclic azaheterocyclic carboxamides as inhibitors of the kinase p70S6K |
| US10087166B2 (en) | 2010-07-29 | 2018-10-02 | Merck Patent Gmbh | Cyclic amine azaheterocyclic carboxamides |
| US9586938B2 (en) | 2010-07-29 | 2017-03-07 | Merck Patent Gmbh | Cyclic amine azaheterocyclic carboxamides |
| JP2013535473A (en) * | 2010-07-29 | 2013-09-12 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Cyclic amine azaheterocyclic carboxamide |
| WO2012013282A1 (en) * | 2010-07-29 | 2012-02-02 | Merck Patent Gmbh | Bicyclic azaheterocyclic carboxamides as inhibitors of the kinase p70s6k |
| EA028012B1 (en) * | 2010-07-29 | 2017-09-29 | Мерк Патент Гмбх | Cyclic amine-azaheterocyclic carboxamide |
| KR101894116B1 (en) * | 2010-11-24 | 2018-08-31 | 메르크 파텐트 게엠베하 | Quinazoline carboxamide azetidines |
| AU2011334173B2 (en) * | 2010-11-24 | 2015-07-16 | Merck Patent Gmbh | Quinazoline carboxamide azetidines |
| WO2012069146A1 (en) | 2010-11-24 | 2012-05-31 | Merck Patent Gmbh | Quinazoline carboxamide azetidines |
| JP2013544257A (en) * | 2010-11-24 | 2013-12-12 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Quinazoline carboxamidoazetidine |
| KR20130130753A (en) * | 2010-11-24 | 2013-12-02 | 메르크 파텐트 게엠베하 | Quinazoline carboxamide azetidines |
| EA023821B1 (en) * | 2010-11-24 | 2016-07-29 | Мерк Патент Гмбх | Quinazoline carboxamide azetidines, use thereof for treating hyperproliferative diseases, and compositions comprising the same |
| CN103228649A (en) * | 2010-11-24 | 2013-07-31 | 默克专利股份公司 | Quinazoline carboxamide azetidines |
| US8946247B2 (en) | 2010-11-24 | 2015-02-03 | Merck Patent Gmbh | Quinazoline carboxamide azetidines |
| WO2013040044A1 (en) | 2011-09-12 | 2013-03-21 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| WO2013040059A1 (en) | 2011-09-12 | 2013-03-21 | Merck Patent Gmbh | Novel imidazole amines as modulators of kinase activity |
| US10080750B2 (en) | 2011-09-12 | 2018-09-25 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| US9662330B2 (en) | 2011-09-12 | 2017-05-30 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| US9145392B2 (en) | 2011-09-12 | 2015-09-29 | Merck Patent Gmbh | Imidazole amines as modulators of kinase activity |
| US9321760B2 (en) | 2011-09-12 | 2016-04-26 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| AU2012355494B2 (en) * | 2011-12-22 | 2017-07-13 | Merck Patent Gmbh | Novel heterocyclic carboxamides as modulators of kinase activity |
| US10080753B2 (en) | 2011-12-22 | 2018-09-25 | Merck Patent Gmbh | Heterocyclic carboxamides as modulators of kinase activity |
| KR102098358B1 (en) * | 2011-12-22 | 2020-04-08 | 메르크 파텐트 게엠베하 | Novel heterocyclic carboxamides as modulatrs of kinase activity |
| US9139568B2 (en) | 2011-12-22 | 2015-09-22 | Merck Patent Gmbh | Heterocyclic carboxamides as modulators of kinase activity |
| JP2015503521A (en) * | 2011-12-22 | 2015-02-02 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツングMerck Patent Gesellschaft mit beschraenkter Haftung | Novel heterocyclic carboxamides as modulators of kinase activity |
| JP2017197587A (en) * | 2011-12-22 | 2017-11-02 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツングMerck Patent Gesellschaft mit beschraenkter Haftung | Novel heterocyclic carboxamides as modulators of kinase activity |
| WO2013096194A1 (en) * | 2011-12-22 | 2013-06-27 | Merck Patent Gmbh | Novel heterocyclic carboxamides as modulators of kinase activity |
| EA027968B1 (en) * | 2011-12-22 | 2017-09-29 | Мерк Патент Гмбх | Heterocyclic carboxamides as modulators of kinase activity |
| CN104169258A (en) * | 2011-12-22 | 2014-11-26 | 默克专利有限公司 | Novel heterocyclic carboxamides as modulators of kinase activity |
| KR20140107317A (en) * | 2011-12-22 | 2014-09-04 | 메르크 파텐트 게엠베하 | Novel heterocyclic carboxamides as modulatrs of kinase activity |
| US10287300B2 (en) | 2012-11-16 | 2019-05-14 | Merck Patent Gmbh | Heterocyclic derivatives as modulators of kinase activity |
| WO2014078637A1 (en) | 2012-11-16 | 2014-05-22 | Merck Patent Gmbh | Novel heterocyclic derivatives as modulators of kinase activity |
| US9315517B2 (en) | 2012-11-16 | 2016-04-19 | Merck Patent Gmbh | Imidazol-piperidinyl derivatives as modulators of kinase activity |
| US9580443B2 (en) | 2012-11-16 | 2017-02-28 | Merck Patent Gmbh | Heterocyclic derivatives as modulators of kinase activity |
| EP3299362A1 (en) | 2012-11-16 | 2018-03-28 | Merck Patent GmbH | Imidazol-piperidinyl derivatives as modulators of kinase activity |
| US9850255B2 (en) | 2012-11-16 | 2017-12-26 | Merck Patent Gmbh | Heterocyclic derivatives as modulators of kinase activity |
| WO2014078634A1 (en) | 2012-11-16 | 2014-05-22 | Merck Patent Gmbh | Novel imidazol-piperidinyl derivatives as modulators of kinase activity |
| WO2014085528A1 (en) | 2012-11-29 | 2014-06-05 | Merck Patent Gmbh | Azaquinazoline carboxamide derivatives |
| US10233160B2 (en) | 2012-11-29 | 2019-03-19 | Merck Patent Gmbh | Substituted pyrido[3,4-d]pyrimidines and pyrido[4,3-d]pyrimidines as p70S6K inhibitors |
| EA029144B1 (en) * | 2012-11-29 | 2018-02-28 | Мерк Патент Гмбх | Azaquinazoline carboxamide derivatives |
| AU2013352261B2 (en) * | 2012-11-29 | 2018-05-10 | Merck Patent Gmbh | Azaquinazoline carboxamide derivatives |
| US9981925B2 (en) | 2012-11-29 | 2018-05-29 | Merck Patent Gmbh | Substituted benzo[d][1,2,3]triazines as p70S6K inhibitors |
| EA033655B1 (en) * | 2012-11-29 | 2019-11-13 | Merck Patent Gmbh | Azaquinazoline carboxamide derivatives |
| US9440968B2 (en) | 2012-11-29 | 2016-09-13 | Merck Patent Gmbh | Substituted pyrido[3,2-d]pyrimidines for treating cancer |
| US9867826B2 (en) | 2013-03-11 | 2018-01-16 | Merck Patent Gmbh | Heterocycles as modulators of kinase activity |
| WO2014143612A1 (en) | 2013-03-11 | 2014-09-18 | Merck Patent Gmbh | 6-[4-(1 h-imidazol-2-yl)piperidin-1 -yl]pyrimidin-4-amine derivatives as modulators of kinase activity |
| US9458134B2 (en) | 2013-03-11 | 2016-10-04 | Merck Patent Gmbh | Heterocycles as modulators of kinase activity |
| US9701664B2 (en) | 2013-10-04 | 2017-07-11 | Cancer Research Technology Limited | Fused 1,4-dihydrodioxin derivatives as inhibitors of heat shock transcription factor 1 |
| US10189821B2 (en) | 2013-10-04 | 2019-01-29 | Cancer Research Technology Limited | Fused 1,4-dihydrodioxin derivatives as inhibitors of heat shock transcription factor I |
| US11787786B2 (en) | 2013-10-04 | 2023-10-17 | Cancer Research Technology Limited | Fused 1,4-dihydrodioxin derivatives as inhibitors of heat shock transcription factor 1 |
| US11124501B2 (en) | 2013-10-04 | 2021-09-21 | Cancer Research Technology Limited | Fused 1,4-dihydrodioxin derivatives as inhibitors of heat shock transcription factor I |
| WO2015149909A1 (en) | 2014-04-03 | 2015-10-08 | Merck Patent Gmbh | Combinations of cancer therapeutics |
| US10370360B2 (en) | 2014-10-22 | 2019-08-06 | The Board Of Regents Of The University Of Texas System | Small-molecule inhibitors targeting discoidin domain receptor 1 and uses thereof |
| EP3209299A4 (en) * | 2014-10-22 | 2018-04-04 | The Board of Regents of The University of Texas System | Small-molecule inhibitors targeting discoidin domain receptor 1 and uses thereof |
| US10647678B2 (en) | 2015-04-01 | 2020-05-12 | Cancer Research Technology Limited | Quinoline derivatives as inhibitors of heat shock factor 1 pathway activity |
| US11814370B2 (en) | 2016-10-07 | 2023-11-14 | Cancer Research Technology Limited | Deuterated N-(5-(2,3-dihydrobenzo[b][1,4]dioxine-6-carboxamido)-2-fluorophenyl)-2-((4-ethylpiperazin-1-yl)methyl)quinoline-6-carboxamide |
| WO2018215557A1 (en) * | 2017-05-23 | 2018-11-29 | Centre National De La Recherche Scientifique | Ion channel inhibitor compounds for cancer treatment |
| US11932638B2 (en) | 2017-05-23 | 2024-03-19 | Centre National De La Recherche Scientifque | Ion channel inhibitor compounds for cancer treatment |
| WO2019231271A1 (en) | 2018-05-31 | 2019-12-05 | C&C Research Laboratories | Heterocyclic derivatives and use thereof |
| EP3802495A4 (en) * | 2018-05-31 | 2022-04-13 | C&C Research Laboratories | Heterocyclic derivatives and use thereof |
| CN112204010B (en) * | 2018-05-31 | 2024-03-19 | C&C新药研究所 | Heterocyclic derivatives and uses thereof |
| CN112204010A (en) * | 2018-05-31 | 2021-01-08 | C&C新药研究所 | Heterocyclic derivatives and use thereof |
| WO2021083936A1 (en) | 2019-11-01 | 2021-05-06 | Syngenta Crop Protection Ag | Pesticidally active fused bicyclic heteroaromatic compounds |
| WO2022101459A1 (en) | 2020-11-16 | 2022-05-19 | Merck Patent Gmbh | Kinase inhibitor combinations for cancer treatment |
| WO2022268648A1 (en) | 2021-06-24 | 2022-12-29 | Syngenta Crop Protection Ag | 2-[3-[1 [(quinazolin-4-yl)amino]ethyl]pyrazin-2-yl]thiazole-5-carbonitrile derivatives and similar compounds as pesticides |
| US11993613B2 (en) | 2022-03-31 | 2024-05-28 | Abbvie Inc. | Thiazolo[5,4-b]pyridine MALT-1 inhibitors |
| WO2023201185A1 (en) * | 2022-04-11 | 2023-10-19 | Merck Patent Gmbh | Pyrido[3,2-d]pyrimidines as hpk1 inhibitors |
| WO2023247360A1 (en) | 2022-06-21 | 2023-12-28 | Syngenta Crop Protection Ag | Pesticidally active fused bicyclic heteroaromatic compounds |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DK2396307T3 (en) | NOVEL AMINO-azaheterocyclic carboxamides | |
| AU2011285247B2 (en) | Bicyclic azaheterocyclic carboxamides as inhibitors of the kinase p70S6K | |
| CA2770155C (en) | Novel azaheterocyclic compounds | |
| KR20120055608A (en) | Novel bicyclic urea compounds | |
| AU2017239625B2 (en) | Novel heterocyclic carboxamides as modulators of kinase activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080007165.1 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10705453 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010705453 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 214412 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2751886 Country of ref document: CA Ref document number: MX/A/2011/008395 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011001967 Country of ref document: CL Ref document number: 2011550122 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: a201110411 Country of ref document: UA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010214095 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 595087 Country of ref document: NZ Ref document number: 3725/KOLNP/2011 Country of ref document: IN Ref document number: 201101182 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 20117021335 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2010214095 Country of ref document: AU Date of ref document: 20100204 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13148903 Country of ref document: US |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: PI1008325 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: PI1008325 Country of ref document: BR Kind code of ref document: A2 Effective date: 20110810 |