WO2010061366A1 - Manufacture of bisoprolol and intermediates therefor - Google Patents
Manufacture of bisoprolol and intermediates therefor Download PDFInfo
- Publication number
- WO2010061366A1 WO2010061366A1 PCT/IE2009/000082 IE2009000082W WO2010061366A1 WO 2010061366 A1 WO2010061366 A1 WO 2010061366A1 IE 2009000082 W IE2009000082 W IE 2009000082W WO 2010061366 A1 WO2010061366 A1 WO 2010061366A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- bisoprolol
- oxazolidinone
- oxazolidone
- benzaldehyde
- reacting
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/02—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C217/04—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C217/28—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having one amino group and at least two singly-bound oxygen atoms, with at least one being part of an etherified hydroxy group, bound to the carbon skeleton, e.g. ethers of polyhydroxy amines
- C07C217/30—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having one amino group and at least two singly-bound oxygen atoms, with at least one being part of an etherified hydroxy group, bound to the carbon skeleton, e.g. ethers of polyhydroxy amines having the oxygen atom of at least one of the etherified hydroxy groups further bound to a carbon atom of a six-membered aromatic ring
- C07C217/32—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having one amino group and at least two singly-bound oxygen atoms, with at least one being part of an etherified hydroxy group, bound to the carbon skeleton, e.g. ethers of polyhydroxy amines having the oxygen atom of at least one of the etherified hydroxy groups further bound to a carbon atom of a six-membered aromatic ring the six-membered aromatic ring or condensed ring system containing that ring being further substituted
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/08—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D263/16—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D263/18—Oxygen atoms
Definitions
- This invention relates to the manufacture of Bisoprolol fumarate and intermediates used in the process.
- DOS3205457 describes a process wherein the 2-(isopropoxy)ethoxymethyl substituent is introduced in two stages. Initially the aromatic ring of 3-isopropyl- 5-phenoxymethyl-oxazolidin-2-one is chloromethylated using HCl and paraformaldehyde followed by a Williamson ether synthesis employing metallic sodium. The resultant oxazolidine ring is cleaved by alkaline hydrolysis.
- the oxazolidone sulphonate is formed by reacting isopropylaminopropanediol with dimethylcarbonate and reacting the intermediate product thus formed with benzenesulphonylchloride.
- oxazolidinone sulphonate is not isolated prior to reaction with 4- hydroxybenzaldehyde.
- methylisobutylketone is added to the intermediate product prior to the addition of benzenesulphonylchloride.
- the reaction between the intermediate and benzenesuphonyl chloride may be performed under phase transfer conditions utilising water soluble bases such as sodium hydroxide.
- reaction between the intermediate and benzenesulphonyl chloride is performed in an organic solvent such as methylisobutylketone utilising bases soluble in organic solvents such as triethylamine.
- the process may comprise the step of purifying bisoprolol base.
- the bisoprolol base may be purified by distillation.
- the bisoprolol base may be purified by crystallisation.
- the process may comprise the step of forming bisoprolol fumarate by reacting bisoprolol base with fumaric acid.
- the process comprises converting bisoprolol fumarate to bisoprolol base.
- the invention also provides bisoprolol prepared by a process as described herein.
- the invention further provides bisoprolol fumarate prepared by a process as described herein.
- the invention also provides a process for preparing oxazolidinone benzaldehyde by reacting oxazolidinone sulphonate with 4-hydroxybenzylaldehyde.
- the process for preparing oxazolidone benzylalcohol may comprise converting oxazolidone benzaldehyde to oxazolidone benzylalcohol.
- the invention also provides oxazolidinone sulphonate having the formula:
- the invention further provides oxazolidinone benzaldehyde having the formula:
- the process described in this invention is superior to the conventional processes as the process consistently produces material of high quality suitable for use as a drug substance. Specifically product purity of greater than 99.5% is achieved with no single impurity present above a threshold of 0.10%, as described in ICH guidance Q3A(R2) for impurities in new drug substances.
- Fig. 1 is an infra-red spectrum (kBr disc) of oxazolidinone sulphonate
- Fig. 2 is a 1 H NMR spectrum (CDCl 3 , 300 MHz) of oxazolidinone benzaldehyde;
- Fig. 3 is a 13 H NMR spectrum (CDCl 3 , 75 MHz) of oxazolidinone benzaldehyde;
- Fig. 4 is an infra-red spectrum (kBr disc) of oxazolidinone benzaldehyde;
- Fig. 5 is a 1 H NMR spectrum (CDCl 3 , 300 MHz) of oxazolidinone benzyl alcohol;
- Fig. 6 is a 13 H NMR spectrum (CDCl 3 , 75 MHz) of oxazolidinone benzyl alcohol.
- Fig. 7 is an infra-red spectrum (kBr disc) of oxazolidinone benzyl alcohol.
- a reactor is charged with 392 kg of isopropylaminopropanediol, approximately 240 L of methanol, 6 to 7L of sodium methoxide (30%) and 270 L of dimethylcarbonate. The contents were heated up to allow reaction to occur. Solvent is removed by distillation.
- the reactor contents are heated and phase separation performed, removing the aqueous layer.
- Solvent is distilled and the product oxazolidone sulphonate is isolated from methyl-tert-butylether (MTBE).
- Fig. 1 illustrates the infra red spectrum of oxazolidinone sulphate.
- Oxazolidinone sulphonate is a novel intermediate and may used for the manufacture of ⁇ -blockers such as Acebutolol, Alprenolol, Atenolol, Betaxolol, Bisoprolol, Esmolol and Metoprolol.
- a reaction vessel is charged with 154.5kg of 4-hydroxybenzaldehyde, 600 L of dimethylformamide (DMF), 100 kg of potassium carbonate and 475 kg of oxazolidinone sulphonate. The mixture is agitated and heated and held until reaction completion.
- DMF dimethylformamide
- the reactor contents are then cooled down and vacuum is applied, solvent is distilled off and discarded. Water is added to the reactor to facilitate crystallisation and product isolation.
- NMR spectra of oxazolidinone benzaldehyde are illustrated in Figs. 2 and 3.
- An infra-red spectrum for the product is illustrated in Fig. 4.
- a vessel is charged with 100 g of isopropylaminopropanediol, methanol, 3-4 g of sodium methoxide 30% and 70 ml of dimethylcarbonate. The vessel contents are heated up to allow reaction to occur. Solvent is removed by distillation.
- the reactor contents are heated and phase separation performed, removing the aqueous layer.
- Solvent is distilled and the product dissolved in 380 ml dimethylforamide.
- To the reaction vessel is charged 55 g of potassium carbonate and 85 g of 4- hydroxybenzaldehyde. The mixture is agitated, heated, and held until reaction completion.
- the reactor contents are then cooled down and vacuum is applied, solvent is distilled off and discarded. Water is added to the reaction mixture to facilitate crystallisation and the product is isolated.
- Mobile phase a mixture of methanol water buffered with potassium hydrogen phosphate and triethylamine
- the key oxazolidinone benzaldehyde intermediate may also be prepared in a "telescoped" process described in this example 3, this process is more efficient as the use of MTBE for the isolation of oxazolidinone sulphonate is eliminated. This results in reduced waste disposal costs and a 50% reduction in the requirement for solids separations equipment.
- a vessel is charged with 100 g of isopropylaminopropanediol, methanol, 3-4 g of sodium methoxide 30% and 70 ml of dimethylcarbonate. The vessel contents are heated up to allow reaction to occur. Solvent is removed by distillation and ca 400 ml methylisobutylketone is then charged to the reaction. The reaction mixture is then cooled and 80g of triethylamine and 145g of benzenesulphonylchloride are added.
- reaction vessel To the reaction vessel is charged 55 g of potassium carbonate and 85 g of 4- hydroxybenzaldehyde. The mixture is agitated, heated, and held until reaction completion.
- the reactor contents are then cooled down and vacuum is applied, solvent is distilled off and discarded. Water is added to the reaction mixture to facilitate crystallisation and the product is isolated.
- Mobile phase a mixture of methanol water buffered with potassium hydrogen phosphate and triethylamine
- the key oxazolidinone benzaldehyde intermediate may also be prepared in a "telescoped" process described in this example 4.
- This process is more efficient than the process described in examples 2 and 3, as the use of triethylamine under non aqeous reaction conditions for the coupling reaction between oxazolidinone sulphonate and benzenesulphonyl chloride is more efficient. This results from a reduction in side reactions under aqueous conditions which lead to the formation of sodium benzenesulphonate as a by-product.
- the vessel contents are heated up to 100oC, cooled and ethyl acetate (100L) is charged to the vessel. Water or brine is used for washing and solvent is distilled off and the product is isolated rrom ethyl acetate.
- Mobile phase a mixture of methanol water buffered with potassium hydrogen phosphate and triethylamine
- Bisoprolol base is formed from the oxazolidinone benzyl alcohol by an acid catalysed coupling with isopropyl oxitol, followed by alkaline hydrolysis of the oxazolidinone ring.
- the resultant bisoprolol base maybe further purified either by distillation or crystallation.
- Example 7 Salt Formation The purified bisoprolol base is converted to Pharmacopoeia grade bisoprolol fumarate by addition of fumaric acid to bisoprolol base in acetone.
- Example 8 Bisoprolol Base for use as a drug substance
- bisoprolol fumarate (30Kg) in a mixture of water (301L) and sodium methyl-tert-butylether (105L), add aqueous sodium hydroxide to alkaline pH. Split the lower aqeous layer to waste and wash the product layer with water. Bisoprolol base is isolated following solvent removal by distillation.
- Bisoprolol base prepared as described in this application is particularly suited to controlled release from transdermal patch formulations.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
A process for preparing bisoprolol comprises reacting oxazolidinone sulphonate with 4-hydroxybenzylaldehyde to form oxazolidinone benzaldehyde, forming oxazolidone benzylalcohol from oxazolidone benzaldehyde, and subsequently reacting oxazolidinone benzylalcohol with isopropyl oxitol to form bisoprolol base. Oxazolidone sulphonate and oxazolidone benzaldehyde are novel intermediates.
Description
"Manufacture of Bisoprolol and intermediates therefor"
Introduction
This invention relates to the manufacture of Bisoprolol fumarate and intermediates used in the process.
The process described in DOS 2 645 710, describes a process wherein 2- (isopropoxy)ethoxymethyl-phenol is reacted with epichlorhydrin and the β-amino alcohol moiety is formed by the addition of iso-propylamine. This synthetic route suffers from several disadvantages including the handling of epichlorohydrin which is a known carcinogen and can undergo violent reactions or exothermic polymerisation on contact with amines or alkoxides. In addition, the reaction between the epoxy intermediate and isopropylamine has the potential to undergoes side reactions leading to the formation of the known impurity F (as described in the European Pharmacopeia 6.1).

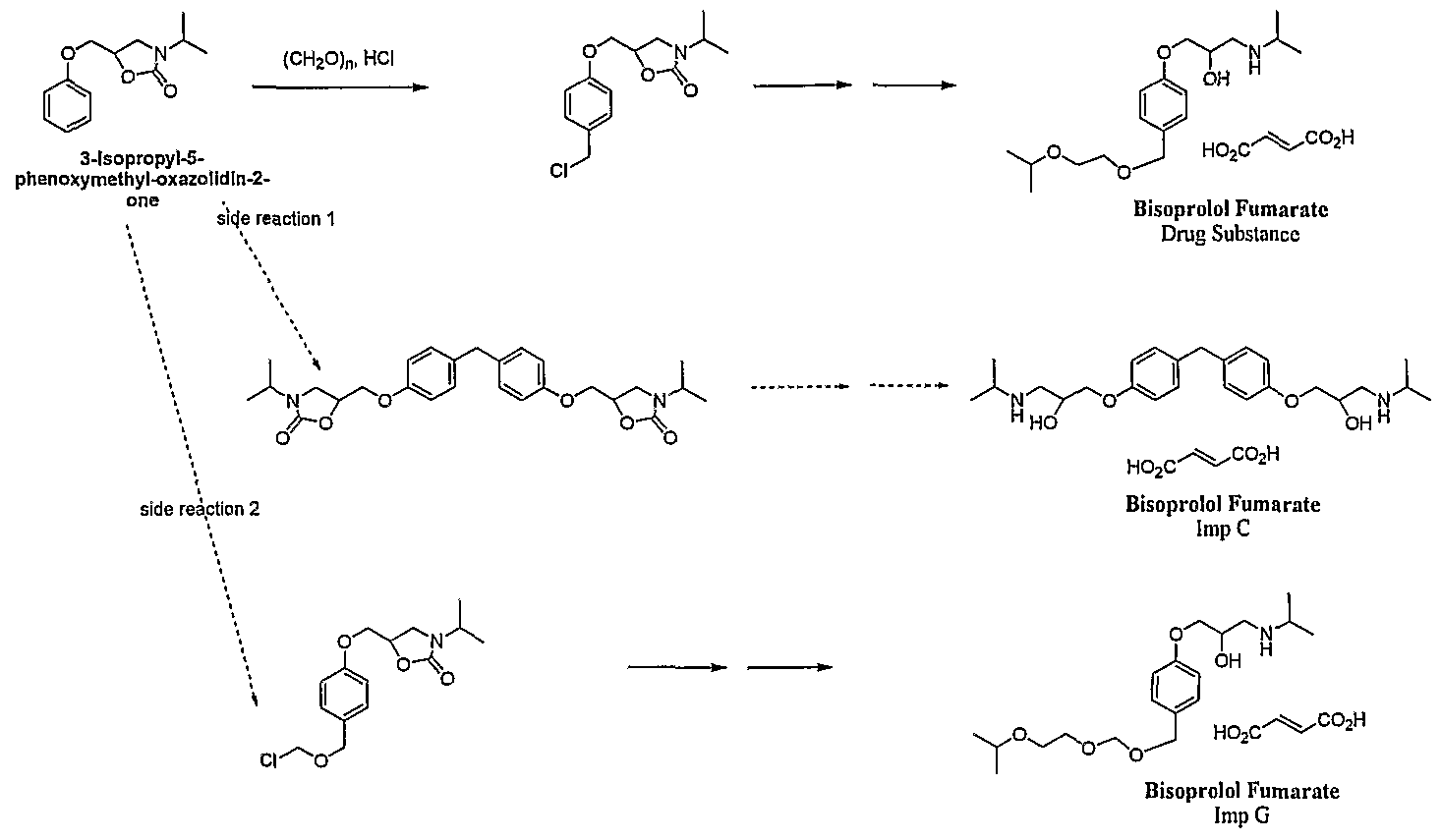
This synthetic route suffers from several disadvantages including the handling of metallic sodium which can undergo violent reactions on contact with water or alcohols. In addition the chloromethylation of 3-isopropyl-5-phenoxymethyl- oxazolidin-2-one with paraformaldehyde in the presence of HCl undergoes side reactions leading to the formation of the known impurities C and G. This process generates material of inferior quality as exemplified by a limit for impurity G of 0.5% (as described in the European Pharmacopeia 6.1).
Statements of Invention
According to the invention there is provided a process for preparing bisoprolol comprising the steps of:-
reacting oxazolidinone sulphonate with 4-hydroxybenzylaldehyde to form oxazolidinone benzaldehyde;
forming oxazolidone benzylalcohol from oxazolidone benzaldehyde; and
subsequently reacting oxazolidinone benzylalcohol with isopropyl oxitol to form bisoprolol base.
In one embodiment the oxazolidone sulphonate is formed by reacting isopropylaminopropanediol with dimethylcarbonate and reacting the intermediate product thus formed with benzenesulphonylchloride.
In one case oxazolidinone sulphonate is not isolated prior to reaction with 4- hydroxybenzaldehyde.
In one embodiment methylisobutylketone is added to the intermediate product prior to the addition of benzenesulphonylchloride. In this case the reaction between the intermediate and benzenesuphonyl chloride may be performed under phase transfer conditions utilising water soluble bases such as sodium hydroxide.
Alternatively, the reaction between the intermediate and benzenesulphonyl chloride is performed in an organic solvent such as methylisobutylketone utilising bases soluble in organic solvents such as triethylamine.
The process may comprise the step of purifying bisoprolol base. The bisoprolol base may be purified by distillation. The bisoprolol base may be purified by crystallisation.
The process may comprise the step of forming bisoprolol fumarate by reacting bisoprolol base with fumaric acid.
In one embodiment the process comprises converting bisoprolol fumarate to bisoprolol base.
The invention also provides bisoprolol prepared by a process as described herein.
The invention further provides bisoprolol fumarate prepared by a process as described herein.
The invention also provides a process for preparing oxazolidinone benzaldehyde by reacting oxazolidinone sulphonate with 4-hydroxybenzylaldehyde.
The process for preparing oxazolidone benzylalcohol may comprise converting oxazolidone benzaldehyde to oxazolidone benzylalcohol.
The invention also provides oxazolidinone sulphonate having the formula:


The process described in this invention is superior to the conventional processes as the process consistently produces material of high quality suitable for use as a drug substance. Specifically product purity of greater than 99.5% is achieved with no single impurity present above a threshold of 0.10%, as described in ICH guidance Q3A(R2) for impurities in new drug substances.
Brief Description of the Drawings
The invention will be more clearly understood from the following description of an embodiment thereof, given by way of example only, with reference to the accompanying drawings, in which: -
Fig. 1 is an infra-red spectrum (kBr disc) of oxazolidinone sulphonate;
Fig. 2 is a 1H NMR spectrum (CDCl3, 300 MHz) of oxazolidinone benzaldehyde;
Fig. 3 is a 13H NMR spectrum (CDCl3, 75 MHz) of oxazolidinone benzaldehyde;
Fig. 4 is an infra-red spectrum (kBr disc) of oxazolidinone benzaldehyde;
Fig. 5 is a 1H NMR spectrum (CDCl3, 300 MHz) of oxazolidinone benzyl alcohol;
Fig. 6 is a 13H NMR spectrum (CDCl3, 75 MHz) of oxazolidinone benzyl alcohol; and
Fig. 7 is an infra-red spectrum (kBr disc) of oxazolidinone benzyl alcohol.
Detailed Description
The process for preparing bisoprolol according to the invention can be summarised as follows:


The following details describe the manufacture of a typical batch
A reactor is charged with 392 kg of isopropylaminopropanediol, approximately 240 L of methanol, 6 to 7L of sodium methoxide (30%) and 270 L of dimethylcarbonate. The contents were heated up to allow reaction to occur. Solvent is removed by distillation.
200 L water and ca 450 L of methylisobutylketone (MIBK) are then charged to the reactor. The reactor contents are then cooled down to <25°C. Tetrabutylammoniumhydrogensulphate (ca 2.5kg) and 520 kg of benzenesulphonylchloride are then added to the vessel under cooling. 30% sodium hydroxide is added to the reactor
The reactor contents are heated and phase separation performed, removing the aqueous layer. Solvent is distilled and the product oxazolidone sulphonate is isolated from methyl-tert-butylether (MTBE).
Fig. 1 illustrates the infra red spectrum of oxazolidinone sulphate.
Purity HPLC >98%
Stationary phase: octadecylsilyl silica gel for chromatography Mobile phase: a mixture of methanol water buffered with potassium hydrogen phosphate and triethylamine
Oxazolidinone sulphonate is a novel intermediate and may used for the manufacture of β-blockers such as Acebutolol, Alprenolol, Atenolol, Betaxolol, Bisoprolol, Esmolol and Metoprolol.

A reaction vessel is charged with 154.5kg of 4-hydroxybenzaldehyde, 600 L of dimethylformamide (DMF), 100 kg of potassium carbonate and 475 kg of oxazolidinone sulphonate. The mixture is agitated and heated and held until reaction completion.
The reactor contents are then cooled down and vacuum is applied, solvent is distilled off and discarded. Water is added to the reactor to facilitate crystallisation and product isolation.
NMR spectra of oxazolidinone benzaldehyde are illustrated in Figs. 2 and 3. An infra-red spectrum for the product is illustrated in Fig. 4.
Purity HPLC >98%, <1% 4-hydroxybenzaldhyde
Stationary phase: octadecylsilyl silica gel for chromatography
Mobile phase: a mixture of methanol water buffered with potassium hydrogen phosphate and triethylamine

A vessel is charged with 100 g of isopropylaminopropanediol, methanol, 3-4 g of sodium methoxide 30% and 70 ml of dimethylcarbonate. The vessel contents are heated up to allow reaction to occur. Solvent is removed by distillation.
100 ml water and methylisobutylketone are then charged to the reaction. The reaction mixture is then cooled down to <25°C. Tetrabutylammoniumhydrogensulphate (ca 0.5g) and 145g of benzenesulphonylchloride are then added to the reaction mixture under cooling. 30% sodium hydroxide is added to the reaction mixture.
The reactor contents are heated and phase separation performed, removing the aqueous layer. Solvent is distilled and the product dissolved in 380 ml dimethylforamide.
To the reaction vessel is charged 55 g of potassium carbonate and 85 g of 4- hydroxybenzaldehyde. The mixture is agitated, heated, and held until reaction completion.
The reactor contents are then cooled down and vacuum is applied, solvent is distilled off and discarded. Water is added to the reaction mixture to facilitate crystallisation and the product is isolated.
Purity HPLC >98%, <1% 4-hydroxybenzaldhyde Stationary phase: octadecylsilyl silica gel for chromatography
Mobile phase: a mixture of methanol water buffered with potassium hydrogen phosphate and triethylamine
In the specific case of bisoprolol manufacture the key oxazolidinone benzaldehyde intermediate may also be prepared in a "telescoped" process described in this example 3, this process is more efficient as the use of MTBE for the isolation of oxazolidinone sulphonate is eliminated. This results in reduced waste disposal costs and a 50% reduction in the requirement for solids separations equipment.
]
 A vessel is charged with 100 g of isopropylaminopropanediol, methanol, 3-4 g of sodium methoxide 30% and 70 ml of dimethylcarbonate. The vessel contents are heated up to allow reaction to occur. Solvent is removed by distillation and ca 400 ml methylisobutylketone is then charged to the reaction. The reaction mixture is then cooled and 80g of triethylamine and 145g of benzenesulphonylchloride are added.
A vessel is charged with 100 g of isopropylaminopropanediol, methanol, 3-4 g of sodium methoxide 30% and 70 ml of dimethylcarbonate. The vessel contents are heated up to allow reaction to occur. Solvent is removed by distillation and ca 400 ml methylisobutylketone is then charged to the reaction. The reaction mixture is then cooled and 80g of triethylamine and 145g of benzenesulphonylchloride are added.
Water is added and the reaction mixture is heated and phase separation performed, removing the aqueous layer. Solvent is distilled and the product dissolved in 380 ml dimethylforamide.
To the reaction vessel is charged 55 g of potassium carbonate and 85 g of 4- hydroxybenzaldehyde. The mixture is agitated, heated, and held until reaction completion.
The reactor contents are then cooled down and vacuum is applied, solvent is distilled off and discarded. Water is added to the reaction mixture to facilitate crystallisation and the product is isolated.
Purity HPLC >98%, <1 % 4-hydroxybenzaldhyde
Stationary phase: octadecylsilyl silica gel for chromatography
Mobile phase: a mixture of methanol water buffered with potassium hydrogen phosphate and triethylamine
In the specific case of bisoprolol manufacture the key oxazolidinone benzaldehyde intermediate may also be prepared in a "telescoped" process described in this example 4. This process is more efficient than the process described in examples 2 and 3, as the use of triethylamine under non aqeous reaction conditions for the coupling reaction between oxazolidinone sulphonate and benzenesulphonyl chloride is more efficient. This results from a reduction in side reactions under aqueous conditions which lead to the formation of sodium benzenesulphonate as a by-product.

(0.3L), to n-butanol (300L). Water (150L) potassium carbonate and oxazolidinone benzaldehyde (271Kg) are charged to a vessel.
The vessel contents are heated up to 100ºC, cooled and ethyl acetate (100L) is charged to the vessel. Water or brine is used for washing and solvent is distilled off and the product is isolated rrom ethyl acetate.
NMR spectra of oxazolidinone benzaldehyde are illustrated in Figs. 5 and 6. An infra-red spectrum for the product is illustrated in Fig. 7.
Purity HPLC >99%
Stationary phase: octadecylsilyl silica gel for chromatography
Mobile phase: a mixture of methanol water buffered with potassium hydrogen phosphate and triethylamine
Example 6: Purification of Bisoprolol Base
Bisoprolol base is formed from the oxazolidinone benzyl alcohol by an acid catalysed coupling with isopropyl oxitol, followed by alkaline hydrolysis of the oxazolidinone ring. The resultant bisoprolol base maybe further purified either by distillation or crystallation.
Example 7: Salt Formation
The purified bisoprolol base is converted to Pharmacopoeia grade bisoprolol fumarate by addition of fumaric acid to bisoprolol base in acetone.
Example 8: Bisoprolol Base for use as a drug substance
Charge bisoprolol fumarate (30Kg) in a mixture of water (301L) and sodium methyl-tert-butylether (105L), add aqueous sodium hydroxide to alkaline pH. Split the lower aqeous layer to waste and wash the product layer with water. Bisoprolol base is isolated following solvent removal by distillation.
Patches are routinely used for the controlled release of drugs via the trans dermal route, the approach is advantageous over oral administration which can result in irregular and unpredictable blood plasma levels. Bisoprolol is normally administered in oral solid dose form as the fumarate salt, but is not suitable for controlled release from transdermal patch formulations. It has been found that
Bisoprolol base prepared as described in this application is particularly suited to controlled release from transdermal patch formulations.
The invention is not limited to the embodiment hereinbefore described, with reference to the accompanying drawings, which may be varied in and detail.
Claims
1. A process for preparing bisoprolol comprising the steps of:-
reacting oxazolidinone sulphonate with 4-hydroxybenzylaldehyde to form oxazolidinone benzaldehyde;
forming oxazolidone benzylalcohol from oxazolidone benzaldehyde; and
subsequently reacting oxazolidinone benzylalcohol with isopropyl oxitol to form bisoprolol base.
2. A process as claimed in claim 1 wherein the oxazolidone sulphonate is formed by reacting isopropylaminopropanediol with dimethylcarbonate and reacting the intermediate product thus formed with benzenesulphonylchloride.
3. A process as claimed in claim 1 or 2 wherein oxazolidinone sulphonate is not isolated prior to reaction with 4-hydroxybenzaldehyde.
4. A process as claimed in claim 2 or 3 wherein methylisobutylketone is added to the intermediate product prior to the addition of benzenesulphonylchloride.
5. A process as claimed in claim 4 wherein the reaction between the intermediate and benzenesuphonyl chloride is performed under phase transfer conditions utilising water soluble bases such as sodium hydroxide.
6. A process as claimed in claim 4 wherein the reaction between the intermediate and benzenesulphonyl chloride is performed in an organic solvent such as methylisobutylketone utilising bases soluble in organic solvents such as triethylamine.
7. A process as claimed in any of claims 1 to 6 comprising purifying bisoprolol base.
8. A process as claimed in claim 7 wherein the bisoprolol base is purified by distillation.
9. A process as claimed in claim 7 wherein the bisoprolol base is purified by crystallisation.
10. A process as claimed in any of claims 1 to 9 comprising forming bisoprolol fumarate by reacting bisoprolol base with fumaric acid.
11. A process as claimed in claim 10 comprising converting bisoprolol fumarate to bisoprolol base and isolating the bisoprolol base.
12. A process substantially as hereinbefore described.
13. Bisoprolol prepared by a process as claimed in any of claims 1 to 9.
14. Bisoprolol fumarate prepared by a process as claimed in claim 10.
15. Bisoprolol prepared by a process as claimed in claim 11.
16. A process for preparing oxazolidinone benzaldehyde by reacting oxazolidinone sulphonate with 4-hydroxybenzylaldehyde.
17. A process for preparing oxazolidone benzylalcohol comprising converting oxazolidone benzaldehyde to oxazolidone benzylalcohol.
18. Oxazolidinone sulphonate having the formula: 
19. Oxazolidinone Benzaldehyde having the formula: 
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IE2008/0949 | 2008-11-28 | ||
| IE20080949 | 2008-11-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010061366A1 true WO2010061366A1 (en) | 2010-06-03 |
Family
ID=41478803
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IE2009/000082 WO2010061366A1 (en) | 2008-11-28 | 2009-11-26 | Manufacture of bisoprolol and intermediates therefor |
Country Status (2)
| Country | Link |
|---|---|
| IE (3) | IES20090901A2 (en) |
| WO (1) | WO2010061366A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102964258A (en) * | 2012-12-11 | 2013-03-13 | 上海奥博生物医药技术有限公司 | Preparation method of related substance J of metoprolol |
| CN103664657A (en) * | 2013-11-25 | 2014-03-26 | 四川大学 | New preparation method for bisoprolol fumarate |
| CN107973761A (en) * | 2017-06-26 | 2018-05-01 | 江苏悦兴医药技术有限公司 | The synthetic method of bisoprolol fumarate's process contaminants |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0086403A2 (en) * | 1982-02-16 | 1983-08-24 | MERCK PATENT GmbH | 2-Oxazolidine-ones |
| EP0339006A1 (en) * | 1988-04-22 | 1989-10-25 | Astra Pharmaceutical Production AB | Process for preparing S-metoprolol and intermediates therefor |
| EP0605729A1 (en) * | 1992-04-30 | 1994-07-13 | Taiho Pharmaceutical Co., Ltd. | Oxazolidine derivative and pharmaceutically acceptable salt thereof |
| WO2007069266A2 (en) * | 2005-12-12 | 2007-06-21 | Unichem Laboratories Limited | A novel process for the synthesis of bisodprolol and its intermediate |
-
2009
- 2009-11-26 IE IES20090901 patent/IES20090901A2/en not_active IP Right Cessation
- 2009-11-26 IE IES20090902 patent/IES20090902A2/en not_active IP Right Cessation
- 2009-11-26 WO PCT/IE2009/000082 patent/WO2010061366A1/en active Application Filing
- 2009-11-26 IE IES20090900 patent/IES20090900A2/en not_active IP Right Cessation
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0086403A2 (en) * | 1982-02-16 | 1983-08-24 | MERCK PATENT GmbH | 2-Oxazolidine-ones |
| EP0339006A1 (en) * | 1988-04-22 | 1989-10-25 | Astra Pharmaceutical Production AB | Process for preparing S-metoprolol and intermediates therefor |
| EP0605729A1 (en) * | 1992-04-30 | 1994-07-13 | Taiho Pharmaceutical Co., Ltd. | Oxazolidine derivative and pharmaceutically acceptable salt thereof |
| WO2007069266A2 (en) * | 2005-12-12 | 2007-06-21 | Unichem Laboratories Limited | A novel process for the synthesis of bisodprolol and its intermediate |
Non-Patent Citations (3)
| Title |
|---|
| BALDWIN, JOHN J. ET AL: ".beta.1-Selective adrenoceptor antagonists: examples of the 2-[4-[3-(substituted-amino)-2-hydroxypropoxy]phenyl]imidazole class", JOURNAL OF MEDICINAL CHEMISTRY, 26(7), 950 -7 CODEN: JMCMAR; ISSN: 0022-2623, 1983, XP002562859 * |
| KASZYNSKI P ET AL: "A COMPREHENSIVE APPROACH TO (S)-(-)-2-METHYL-1-BUTANOL AS A CONVENIENT PRECURSOR FOR SYNTHESIS OF CHIRAL LIQUID CRYSTALS", MOLECULAR CRYSTALS AND LIQUID CRYSTALS, GORDON AND BREACH, LONDON, GB, vol. 174, 1 January 1989 (1989-01-01), pages 21 - 37, XP001056715, ISSN: 0026-8941 * |
| KITAORI K ET AL: "CsF in Organic Synthesis. The First and Convenient Synthesis of Enantiopure Bisoprolol by Use of Glycidyl Nosylate", TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 39, no. 20, 14 May 1998 (1998-05-14), pages 3173 - 3176, XP004116223, ISSN: 0040-4039 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102964258A (en) * | 2012-12-11 | 2013-03-13 | 上海奥博生物医药技术有限公司 | Preparation method of related substance J of metoprolol |
| CN102964258B (en) * | 2012-12-11 | 2014-01-29 | 上海奥博生物医药技术有限公司 | Preparation method of related substance J of metoprolol |
| CN103664657A (en) * | 2013-11-25 | 2014-03-26 | 四川大学 | New preparation method for bisoprolol fumarate |
| CN107973761A (en) * | 2017-06-26 | 2018-05-01 | 江苏悦兴医药技术有限公司 | The synthetic method of bisoprolol fumarate's process contaminants |
Also Published As
| Publication number | Publication date |
|---|---|
| IES20090901A2 (en) | 2010-07-07 |
| IES20090902A2 (en) | 2010-07-07 |
| IES20090900A2 (en) | 2010-07-07 |
| IE20090899A1 (en) | 2010-07-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2009203174B2 (en) | Method of synthesis of bosentan, its polymorphic forms and its salts | |
| JP2009531408A5 (en) | ||
| WO2006093281A1 (en) | METHOD FOR PRODUCING α-HYDROXY-ω-GLYCIDYL ETHER | |
| WO2010061366A1 (en) | Manufacture of bisoprolol and intermediates therefor | |
| KR101308258B1 (en) | A novel method of making Endoxifen | |
| WO2007069266A2 (en) | A novel process for the synthesis of bisodprolol and its intermediate | |
| EP2032522B1 (en) | Shortened synthesis using paraformaldehyde or trioxane | |
| US20090012328A1 (en) | Process for preparing bupropion hydrochloride | |
| WO2000068181A2 (en) | Process for preparing (1r,2s,4r) -(-)-2-[(2'- {n,n-dimethylamino} -ethoxy)] -2-[phenyl] -1,7,7-tri- [methyl] -bicyclo [2.2.1] heptane and pharmaceutically acceptable acid addition salts thereof | |
| KR101610557B1 (en) | Method for producing alkyldiol monoglycidyl ether | |
| IES85567Y1 (en) | Manufacture of beta blockers | |
| IE85808B1 (en) | Manufacture of beta blockers | |
| CN1093355A (en) | The preparation method of phenylacetic acid derivatives | |
| IES85566Y1 (en) | Manufacture of beta blockers | |
| IES85565Y1 (en) | Manufacture of beta blockers | |
| CN111592491B (en) | Preparation method of levo-hydrochloric acid demethyl phencynonate | |
| IE20090901U1 (en) | Manufacture of beta blockers | |
| US9090537B2 (en) | Process for the preparation of aliskiren | |
| CA2648329C (en) | Accelerated synthesis of substituted hydroxymethyl phenols | |
| TW201348221A (en) | Method for producing alkanediol monoglycidyl ether (meth) acrylate | |
| JP3882486B2 (en) | Method for producing ether compound having oxetane ring | |
| JP3573679B2 (en) | Polymer immobilized α-iminoester | |
| JP2008222566A (en) | Novel production method of febrifugine and isofebrifugine | |
| KR100449317B1 (en) | Process for the preparation of arbutin derivatives | |
| JP2001526253A (en) | Method for producing isopropyl-methyl- [2- (3-N-propoxyphenoxy) ethyl] amine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09764897 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09764897 Country of ref document: EP Kind code of ref document: A1 |