WO2010048477A2 - Improved process for preparation of coupled products from 4-amino-3-cyanoquinolines using stabilized intermediates - Google Patents
Improved process for preparation of coupled products from 4-amino-3-cyanoquinolines using stabilized intermediates Download PDFInfo
- Publication number
- WO2010048477A2 WO2010048477A2 PCT/US2009/061787 US2009061787W WO2010048477A2 WO 2010048477 A2 WO2010048477 A2 WO 2010048477A2 US 2009061787 W US2009061787 W US 2009061787W WO 2010048477 A2 WO2010048477 A2 WO 2010048477A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino
- chloride
- butenoyl
- acid
- group
- Prior art date
Links
- WRGKROVGVSWJMI-UHFFFAOYSA-N CCOc(c(N)cc1c2Nc(cc3Cl)ccc3OCc3ccccn3)cc1ncc2C#N Chemical compound CCOc(c(N)cc1c2Nc(cc3Cl)ccc3OCc3ccccn3)cc1ncc2C#N WRGKROVGVSWJMI-UHFFFAOYSA-N 0.000 description 1
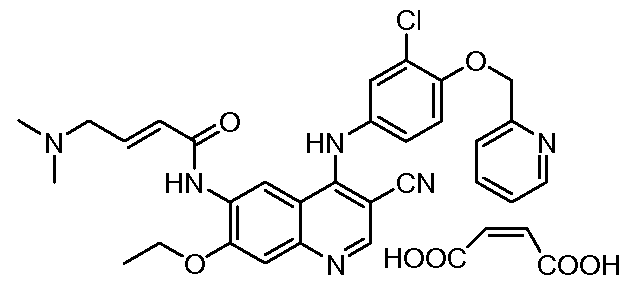
- JWNPDZNEKVCWMY-VQHVLOKHSA-N CCOc(c(NC(/C=C/CN(C)C)=O)cc1c2Nc(cc3)cc(Cl)c3OCc3ncccc3)cc1ncc2C#N Chemical compound CCOc(c(NC(/C=C/CN(C)C)=O)cc1c2Nc(cc3)cc(Cl)c3OCc3ncccc3)cc1ncc2C#N JWNPDZNEKVCWMY-VQHVLOKHSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the acid chloride (Step 5) is generated from 4- dimethylaminocrotonic acid hydrochloride by reacting it with oxalyl chloride in THF as a solvent and small amount of DMF as a catalyst. The acid chloride is then coupled with an appropriate substituted aniline to afford the free base form of HKI-272 (Step 6).
- the present invention provides a method for coupling a 4-(amino)-2-butenoyl group to an amino group at the 6- or 7-position of a 4-amino-3-quinolinecarbonitrile, said method comprising: at between about 5° C and 25° C, generating 4-(amino)-2-butenoyl chloride by reacting dimethylaminocrotonic acid with oxalyl chloride in isopropyl acetate as a reaction solvent, forming 4-(amino)-2-butenoyl chloride as a hydrochloride salt in the form of a solid or a slurry, and reacting said 4-(amino)-2-butenoyl chloride hydrochloride salt with said 4-amino-3- quinolinecarbonitrile.
- the present invention also provides a method for coupling a 4-(amino)-2-butenoyl group to an amino group (-NH 2 ) at the 6- or 7-position of a 4-amino-3-quinolinecarbonitrile, said method comprising: generating an 4-(amino)-2-butenoyl chloride at very low or lowered temperatures using phosphorous oxychloride (POCI 3 ) and dimethylacetamide (DMAc) as a reaction solvent.
- POCI 3 phosphorous oxychloride
- DMAc dimethylacetamide
- the reaction solvent is believed to act as a catalyst in generating the 4-(amino)-2-butenoyl chloride from a corresponding acid.
- Figure 4 summarizes formation and stability of 4-dimethylaminocrotonoyl chloride in iPAc at 15 °C.
- Figure 5 summarizes formation and stability of 4-dimethylaminocrotonoyl chloride in THF at 20 0 C.
- Figure 6 summarizes LC-MS data for degradation products via cyclic intermediates, typically observed in existing methods.
- Figure 9 summarizes reaction conditions for preparing the coupled product, 4-amino-3- quinolinecarbonitriles having a 4-(amino)-2-butenamido group at the 6- or 7-position, using A- (amino)-2-butenoyl chloride derivatives using process 2.
- the present invention provides inventive solutions to address shortcomings of the existing manufacturing methods for preparing 3-cyanoquinolines, including HKI-272, by providing an improved method for coupling a 4-(amino)-2-butenoyl group to an amino group (- NH 2 ) at the 6- or 7-position of a 4-amino-3-quinolinecarbonitrile. More specifically the present addresses the known limitation of 4-(amino)-2-butenoyl chloride instability by providing new and inventive methods for stabilizing such acid chlorides. Exemplary 4-(amino)-2-butenoyl chlorides usefully employed in accordance with the present invention are disclosed in U.S. Pat. No. 7,126,025. Scheme 2 summarizes the stabilizing methods for 4-(amino)-2-butenoyl chlorides in connection with one embodiment of the invention.
- the present invention provides a method for coupling a A- (amino)-2-butenoyl group to an amino group at the 6- or 7-position of a 4-amino-3- quinolinecarbonitrile, said method comprising: at between 5° and 25° C, generating a 4-(amino)- 2-butenoyl chloride by reacting dimethylaminocrotonic acid with oxalyl chloride in iPAc as a solvent, forming the 4-(dimethylamino)-2-butenoyl chloride hydrochloride at low temperature in solution, stabilizing the 4-(amino)-2-butenoyl chloride with a reaction solvent, isolating the A- (amino)-2-butenoyl chloride as a solid or as a slurry, and reacting the 4-(amino)-2-butenoyl chloride with the 4-amino-3-quinolinecarbonitrile.
- the 4-(amino)-2-butenoyl chloride is generated using reactants described in an existing method, however, the acid chloride is stabilized by forcing it to precipitate out of solution, at preferably 5° to 25° C. This is achieved by using an appropriate inert reaction solvent in which the acid chloride hydrochloride is not soluble, for example isopropyl acetate (iPAc), ethyl acetate and like solvents.
- iPAc isopropyl acetate
- the solid 4-(amino)-2-butenoyl chloride can be isolated by filtration or the slurry of the 4-(amino)-2-butenoyl chloride can be used directly in the next step.
- the optimal temperature is 15° C.
- optimal and acceptable reaction conditions, reagents and inert solvents are as follows: Reagent: oxalyl chloride; amount 0.85-1.00 equivalents. (0.90- 0.95 optimal). Catalyst: DMF, DMAc; amount 5-25 mol%, optimal 5-10 mol%. Inert solvent: iPAc, MeAc or EtAc. Temperature range: 10-20 0 C, optimal 13-17°C. Reaction time: 2 to 6 hrs, optimal 2-3 hrs.
- Advantages of the homogeneous method for coupling a 4-(amino)-2-butenoyl group to an amino group at the 6- or 7-position of a 4-amino-3-quinolinecarbonitrile, including HKI-272, include minimizing effects of mass transfer, minimizing sample variability, and providing a narrow manufacturing window, related to product stability.
- the acid chloride (4-(dimethylamino)-2-butenoyl chloride) is generated at very low or lowered temperatures (-5 to -25 0 C) using phosphorous oxychloride (POCI 3 ) and dimethylacetamide (DMAc) as a reaction solvent.
- POCI 3 phosphorous oxychloride
- DMAc dimethylacetamide
- reaction solvent also acts as a catalyst in converting a corresponding acid to the acid chloride.
- One advantage of the new invented processes include greater tolerability towards impurities in reactants and solvents (trace amounts of residual amines and alcohols result in generation of impurities in the product) as compared to existing processes. These impurities are tolerated in the inventive process 2.
- process 3 parameter ranges are as follows: Acid chloride formation.
- Solvent DMAc (optimal), NMP, other R 1 CONR 2 R 3 may work to some extent but results are likely to be substantially inferior).
- Reagent SOCI 2 amount 0.95 to 1.2 equivalents, relative to crotonic acid. Temperature -13 to -18 0 C optimal, -5 to -25 0 C may still work well enough to get some product out. Reaction time 3-6 hr optimal, 2-12 hours will work also.
- the conversion was calculated as a ratio of peak areas corresponding to the starting acid and its ethyl ester formed by reaction of the acid chloride with ethanol. After conversion reached the maximum (usually 80 to 90% after 2 to 3 hours), the mixture was cooled to 0-5 0 C. A solution of an appropriate substituted aniline (12.9 kg, 28.9 mol) in NMP (173 kg) was added at 0-5 0 C to the slurry of acid chloride and the mixture was stirred at this temperature for 15 hr. Reaction progress was monitored by HPLC. The thick suspension of the hydrochloride salt of the coupled product was quenched with water (106 kg) at 0 to 10 0 C resulting in complete dissolution of the solids.
- the iPAc layer was separated, extracted with water (28 kg) and discarded.
- the aqueous phase combined with the aqueous extract was warmed to 34-40 0 C and its pH was adjusted to 10-1 1 with a 16.9% aqueous solution of NaOH (approx. 44 kg).
- the product crystallized out as a free base dihydrate.
- the suspension was further diluted with water (80 kg) and was filtered on a polypropylene cloth.
Abstract
This invention discloses improved methods for coupling a 4-(amino)-2-butenoyl group to an amino group at the 6- or 7-position of a 4-amino-3-quinolinecarbonitrile by generating a stabilized 4-(amino)-2-butenoyl chloride hydrochloride.
Description
IMPROVED PROCESS FOR PREPARATION OF COUPLED PRODUCTS FROM 4- AMINO-3-CYANOQUINOLINES USING STABILIZED INTERMEDIATES
BACKGROUND OF THE INVENTION
Protein kinases are enzymes that catalyze the transfer of a phosphate group from ATP to an amino acid residue, such as tyrosine, serine, threonine, or histidine on a protein. Regulation of these protein kinases is essential for the control of a wide variety of cellular events including proliferation and migration. Specific protein kinases have been implicated in adverse conditions including cancer [Traxler, P. M., Exp. Opin. Ther. Patents, 8, 1599 (1998); Bridges, A. J., Emerging Drugs, 3, 279 (1998)], restenosis [Mattsson, E., Trends Cardiovas. Med. 5, 200 (1995); Shaw, Trends Pharmacol. Sci. 16, 401 (1995) ], atherosclerosis [Raines, E. W., Bioessays, 18, 271 (1996)], angiogenesis [Shawver, L. K., Drug Discovery Today, 2, 50 (1997); Folkman, J., Nature Medicine, 1 , 27 (1995)] and osteoporosis [Boyce, J. Clin. Invest., 90,1622 (1992)]. Compounds derived from this invention are for example, useful in the treatment of cancers, including but not limited to for example, non-small cell lung cancer (NSLC), breast cancer polycystic kidney disease, colonic polyps, and stroke in mammals.
U.S. Pat. Nos. 6,002,008, 6,288,082, 6,297,258, 6,384,051 and 7,399,865 describe certain substituted 3-cyanoquinolines and 4-amino-3-cyanoquinolines, methods of making them and their biological activity. In particular, methods for preparing HKI-272, (.=)-Λ/-(4-(3-chloro-4- (pyridin-2-ylmethoxy)phenylamino)-3-cyano-7-ethoxyquinolin-6-yl)-4-(dimethylamino)but-2- enamide have been described by H. R. Tsou, et al in J. Med. Chem., Vol. 48, 1 107 (2005). Several known processes for preparing HKI-272 and related compounds including but not limited to for example EKB-569, while useful for the laboratory scale production, suffer a number of limitations if employed in the commercial scale production, namely non-optimal yields, degradation products and undesirable crystalline and amorphous forms of such 3- cyanoquinolines. In one process, disclosed in U.S. Pat. No. 7,126,025, an acid chloride is generated from 4-dimethylaminocrotonic acid hydrochloride by reacting it with oxalyl chloride in THF as a solvent and small amount of DMF as a catalyst, and then coupled with an appropriate substituted aniline to afford the free base of HKI-272. This process has two major limitations: 1 ) the intermediate acid chloride is unstable and degrades considerably during commercial, large scale (kg) preparations, and 2) the process is very sensitive towards minor impurities in the reactants and solvents which results in formation of impurities in the coupled product. More efficient methods of synthesis, particularly for the commercial scale synthesis, would be highly
desirable. Major process improvements have been discovered in the present invention and have been developed to overcome the commercial manufacturing limitations.
U.S. Pat. No. 7,126,025 discloses certain novel 4-amino-2-butenoyl chlorides, processes for their preparation and their use as intermediates in the synthesis of pharmaceutically active protein kinase inhibitors, including but not limited to for example HKI-272 and EKB-569.
The sequence illustrated below and summarized in Scheme 1 describes one existing process for preparing HKI-272, (E)-Λ/-(4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-3- cyano-7-ethoxyquinolin-6-yl)-4-(dimethylamino)but-2-enamide in the form of the maleate salt, also known as Neratinib™.

O
^
Step 5 OH 16 h HCI

In one existing process, the acid chloride (Step 5) is generated from 4- dimethylaminocrotonic acid hydrochloride by reacting it with oxalyl chloride in THF as a solvent and small amount of DMF as a catalyst. The acid chloride is then coupled with an appropriate substituted aniline to afford the free base form of HKI-272 (Step 6).
This process has two major drawbacks: 1 ) the intermediate acid chloride is unstable and degrades considerably during commercial, large scale (kg quantities) preparations, and 2) the process is very sensitive towards minor impurities in the reactants and solvents which results in
formation of impurities in the coupled product. Manufacturing improvements have been discovered and disclosed in the present invention and developed to address the commercial scale limitations.
Figure 1 summarizes problems associated with the 4-(amino)-2-butenoyl chloride. The stability of the acid chloride in THF solution at 30° C is likely responsible for degradation products and tar formation for example. Figures 2 and 3 summarize impurities and degradation products associated with the existing process and related small scale processes for preparing
HKI-272.
SUMMARY OF THE INVENTION
This invention provides methods for manufacturing certain 4-amino-3- quinolinecarbonitrile derivatives, including but not limited to those disclosed in U.S. Pat. Nos. 6,002,008, 6,288,082, 6,297,258, 6,384,051 and 7,399,865, using stabilized 4-(amino)-2- butenoyl chloride intermediates (for example, 4-(dimethylamino)-2-butenoyl chloride) for coupling a 4-(amino)-2-butenoyl group to an amino group (-NH2) at the 6- or 7-position of a 4- amino-3-quinolinecarbonitrile. The present invention is further directed to an improved process for the preparation of HKI-272, (E)-Λ/-(4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-3-cyano- 7-ethoxyquinolin-6-yl)-4-(dimethylamino)but-2-enamide free base and maleate (Neratinib™). Such substituted quinolines, as well as the pharmaceutically acceptable salts thereof, inhibit the action of certain protein kinases (PK) thereby inhibiting the abnormal growth of certain cell types.
The present invention provides a method for coupling a 4-(amino)-2-butenoyl group to an amino group at the 6- or 7-position of a 4-amino-3-quinolinecarbonitrile, said method comprising: at between about 5° C and 25° C, generating 4-(amino)-2-butenoyl chloride by reacting dimethylaminocrotonic acid with oxalyl chloride in isopropyl acetate as a reaction solvent, forming 4-(amino)-2-butenoyl chloride as a hydrochloride salt in the form of a solid or a slurry, and reacting said 4-(amino)-2-butenoyl chloride hydrochloride salt with said 4-amino-3- quinolinecarbonitrile.
The present invention also provides a method for coupling a 4-(amino)-2-butenoyl group to an amino group (-NH2) at the 6- or 7-position of a 4-amino-3-quinolinecarbonitrile, said method comprising: generating an 4-(amino)-2-butenoyl chloride at very low or lowered temperatures using phosphorous oxychloride (POCI3) and dimethylacetamide (DMAc) as a reaction solvent. In certain embodiments, the reaction solvent is believed to act as a catalyst in generating the 4-(amino)-2-butenoyl chloride from a corresponding acid.
The following experimental details are set forth to aid in an understanding of the invention, and are not intended, and should not be construed, to limit in any way the invention set forth in the claims that follow thereafter.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 summarizes limitations of existing methods of amidation and isolation for preparing coupled products having a 4-(amino)-2-butenamido group at the 6- or 7-position of a 4-amino-3-quinolinecarbonitrile.
Figure 2 summarizes the impurity profile for 4-amino-3-quinolinecarbonitriles having a A- (amino)-2-butenamido group at the 6- or 7-position, in accordance with existing methods of amidation and isolation for preparing coupled products.
Figure 3 summarizes NMR spectra for degradation products of a 4-(amino)-2-butenoyl chloride derivative, 4-dimethylaminocrotonoyl chloride, typically observed in existing methods.
Figure 4 summarizes formation and stability of 4-dimethylaminocrotonoyl chloride in iPAc at 15 °C.
Figure 5 summarizes formation and stability of 4-dimethylaminocrotonoyl chloride in THF at 20 0C.
Figure 6 summarizes LC-MS data for degradation products via cyclic intermediates, typically observed in existing methods.
Figure 7 summarizes optimal reaction conditions for preparing the coupled product, A- amino-3-quinolinecarbonitriles having a 4-(amino)-2-butenamido group at the 6- or 7-position, using 4-(amino)-2-butenoyl chloride derivatives.
Figure 8 summarizes reaction conditions for preparing the coupled product, 4-amino-3- quinolinecarbonitriles having a 4-(amino)-2-butenamido group at the 6- or 7-position, using A- (amino)-2-butenoyl chloride derivatives using process 1.
Figure 9 summarizes reaction conditions for preparing the coupled product, 4-amino-3- quinolinecarbonitriles having a 4-(amino)-2-butenamido group at the 6- or 7-position, using A- (amino)-2-butenoyl chloride derivatives using process 2.
Figure 10 summarizes reaction conditions for preparing the coupled product, 4-amino-3- quinolinecarbonitriles having a 4-(amino)-2-butenamido group at the 6- or 7-position, using A- (amino)-2-butenoyl chloride derivatives using process 3.
Figure 1 1 summarizes a comparison of methods using oxalyl chloride and phosphorus oxychloride for preparing the coupled product, 4-amino-3-quinolinecarbonitriles having a 4- (amino)-2-butenamido group at the 6- or 7-position, using 4-(amino)-2-butenoyl chloride derivatives.
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides inventive solutions to address shortcomings of the existing manufacturing methods for preparing 3-cyanoquinolines, including HKI-272, by providing an improved method for coupling a 4-(amino)-2-butenoyl group to an amino group (- NH2) at the 6- or 7-position of a 4-amino-3-quinolinecarbonitrile. More specifically the present addresses the known limitation of 4-(amino)-2-butenoyl chloride instability by providing new and inventive methods for stabilizing such acid chlorides. Exemplary 4-(amino)-2-butenoyl chlorides usefully employed in accordance with the present invention are disclosed in U.S. Pat. No. 7,126,025. Scheme 2 summarizes the stabilizing methods for 4-(amino)-2-butenoyl chlorides in connection with one embodiment of the invention.
Scheme 2

In one embodiment, the 4-(amino)-2-butenoyl chloride is generated using reactants described in an existing method, however, the acid chloride is stabilized by forcing it to precipitate out of solution, at preferably 5° to 25° C. This is achieved by using an appropriate inert reaction solvent in which the acid chloride hydrochloride is not soluble, for example isopropyl acetate (iPAc), ethyl acetate and like solvents. The solid 4-(amino)-2-butenoyl chloride can be isolated by filtration or the slurry of the 4-(amino)-2-butenoyl chloride can be used directly in the next step. In one embodiment, the optimal temperature is 15° C. If isolated, solid acid chloride hydrochloride salt can be stored for weeks in the absence of air and moisture. As a slurry in iPAc, the acid chloride can be kept at 25 0C or lower for several days. The Figures 4 and 5 summarize and illustrate relative stability of the acid chloride in THF and iPAc. The inventive process is reliable, reproducible and scalable (16 kg), increasing yields of the crude coupled product (HKI-272 free base) from 60-75% to 85-98% and the purity of the HKI-272 product from 85-90% to 96-98%, as compared to existing methods.
According to one embodiment, optimal and acceptable reaction conditions, reagents and inert solvents are as follows: Reagent: oxalyl chloride; amount 0.85-1.00 equivalents. (0.90- 0.95 optimal). Catalyst: DMF, DMAc; amount 5-25 mol%, optimal 5-10 mol%. Inert solvent: iPAc, MeAc or EtAc. Temperature range: 10-200C, optimal 13-17°C. Reaction time: 2 to 6 hrs, optimal 2-3 hrs.
Advantages of the homogeneous method for coupling a 4-(amino)-2-butenoyl group to an amino group at the 6- or 7-position of a 4-amino-3-quinolinecarbonitrile, including HKI-272, include minimizing effects of mass transfer, minimizing sample variability, and providing a narrow manufacturing window, related to product stability.
The present invention also provides a method for coupling a 4-(amino)-2-butenoyl group to an amino group (-NH2) at the 6- or 7-position of a 4-amino-3-quinolinecarbonitrile, said
method comprising: at between -5° to -25° C, preferably between -10° to -20° C, generating a 4-(amino)-2-butenoyl chloride hydrochloride by reacting dimethylaminocrotonic acid with phosphorous oxychloride (POCI3) and dimethylacetamide (DMAc) as a reaction solvent. In certain embodiments, the reaction solvent is believed to act as a catalyst in generating the A- (amino)-2-butenoyl chloride from a corresponding acid.
The acid chloride (4-(dimethylamino)-2-butenoyl chloride) is generated at very low or lowered temperatures (-5 to -25 0C) using phosphorous oxychloride (POCI3) and dimethylacetamide (DMAc) as a reaction solvent. In one embodiment, it is believed that reaction solvent also acts as a catalyst in converting a corresponding acid to the acid chloride. The very low or lowered operation temperature (as compared to the temperature of the previous embodiment using iPAc as a reaction solvent) slows down degradation of the acid chloride resulting in a more robust and efficient process for converting 4-(amino)-2-butenoyl chlorides to coupled products, with the inventive selection of solvents and other reaction conditions, allowing the reaction to proceed despite the low temperatures.
One advantage of the new invented processes include greater tolerability towards impurities in reactants and solvents (trace amounts of residual amines and alcohols result in generation of impurities in the product) as compared to existing processes. These impurities are tolerated in the inventive process 2.
According to one embodiment, optimal and acceptable reaction conditions, reagents and inert solvents are as follows: Reagent: POCI3; amount 0.9 to 1.0 equivalents, (optimal 0.98). Inert solvents: DMAc, NMP, DMF, other R1CONR2 2 where R's are alkyls, aryls or mixtures thereof or with other inert solvents; preferred DMAc, other solvents produce impurities. Temperature range: -5 to -25 0C (optimal -14 to -18 0C). Reaction time 1 to 10 hrs (optimal 3 to 5 hrs).
According to a separate embodiment, the invention provides a thionyl chloride coupling method, referred to as process 3. It was found that when 4-dimethylaminocrotonic acid hydrochloride is treated with thionyl chloride in DMAc at -15 0C it converts rapidly to the acid chloride HCI salt, which, when treated further with a solution of an appropriate substituted aniline in DMAc at the same temperature, provide a high yield of HKI-272 (Scheme 3).
However, when during isolation of the product, the reaction mixture is quenched with water and pH is adjusted from acidic to basic with aqueous base we observe formation of variable amounts of an impurity (Scheme 4 and Figure 10). It forms via a Michael addition of the bisulfite anion to the side chain of the HKI-272 according to Scheme 2 and the rate of
formation was found to be pH-dependent. The formation is fastest at neutral pH where concentration of the bisulfite anion is fastest. Because during pH adjustment we have to go from acidic pH to basic through neutral, the amount of the impurity will depend on the rate of pH adjustment. On smaller scale, the adjustment can be carried out quickly and the amount of the impurity can be kept to the minimum (below 1 %), however, on multikilogram scale this has been known to limit efficiency of the process.
Scheme 3. Formation of acid chloride with SOCI2 in DMAc and coupling with a substituted aniline.
SOCl2
/ Nv^-^'C02H HCI DMAc HCI


According to one embodiment process 3 parameter ranges are as follows: Acid chloride formation. Solvent: DMAc (optimal), NMP, other R1CONR2R3 may work to some extent but results are likely to be substantially inferior). Reagent SOCI2, amount 0.95 to 1.2 equivalents, relative to crotonic acid. Temperature -13 to -18 0C optimal, -5 to -25 0C may still work well enough to get some product out. Reaction time 3-6 hr optimal, 2-12 hours will work also.
This invention is not intended to be limited in any manner by the permissible substituents of organic compounds.
As used herein the solvent is the liquid or homogeneous mixture of liquids in which extractant(s) and possible modifier(s) may be dissolved to form the solvent phase, a solution.
The present invention is described by the following examples, which are exemplary and are not intended to be limiting.
Example 1 : Process 1
4-Dimethylaminocrotonoyl chloride hydrochloride and its coupling with 6-amino- 4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-7-ethoxyquinoline-3-carbonitrile (procedure with oxalyl chloride and iPAc).
A suspension of 4-dimethylaminocrotonic acid hydrochloride (1 1.5 kg, 69.6 mol) in a mixture of iPAc (1 12 kg) and DMF (0.407 kg, 5.57 mol, 8 mol%) was cooled to 0-10 0C. Oxalyl chloride (7.68 kg, 60.5 mol) was added to the suspension, maintaining the batch temperature between 1 and 6 0C. The resulting mixture was warmed to 10-15 0C and stirred in this temperature range for 5 hr. Conversion of the acid to acid chloride was monitored by HPLC: an aliquot of the reaction mixture was quenched into anhydrous ethanol and the solution was analyzed by HPLC using SIELC Primesep™ 200 column capable of retaining polar compounds. The conversion was calculated as a ratio of peak areas corresponding to the starting acid and its ethyl ester formed by reaction of the acid chloride with ethanol. After conversion reached the maximum (usually 80 to 90% after 2 to 3 hours), the mixture was cooled to 0-5 0C. A solution of an appropriate substituted aniline (12.9 kg, 28.9 mol) in NMP (173 kg) was added at 0-5 0C to the slurry of acid chloride and the mixture was stirred at this temperature for 15 hr. Reaction progress was monitored by HPLC. The thick suspension of the hydrochloride salt of the coupled product was quenched with water (106 kg) at 0 to 10 0C resulting in complete dissolution of the solids. The iPAc layer was separated, extracted with water (28 kg) and discarded. The aqueous phase combined with the aqueous extract was warmed to 34-40 0C and its pH was adjusted to 10-1 1 with a 16.9% aqueous solution of NaOH (approx. 44 kg). The product crystallized out as a free base dihydrate. The suspension was further diluted with water (80 kg) and was filtered on a polypropylene cloth. The cake was washed with water until neural pH of the washes, dried first by suction on the filter for 3 hr and then in a vacuum oven at 40 to 50 0C to afford crude (E)-N- (4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-3-cyano-7-ethoxyquinolin-6-yl)-4- (dimethylamino)but-2-enamide 13.8 kg (85%) as a bright-yellow crystalline solid.
Example 2: Process 2
4-Dimethylaminocrotonoyl chloride hydrochloride and its coupling with 6-amino-
4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-7-ethoxyquinoline-3-carbonitrile (procedure with phosphorus oxychloride and DMAc).
A suspension of 4-dimethylaminocrotonic acid (6.70 kg, 40.4 mol) in DMAc (64.3 kg) was cooled to -14 to -19 0C. Neat POCI3 (6.15 kg, 40.1 mol) was added to the slurry at a rate to maintain the temperature in the reactor in the above range (moderate exotherm). The reaction mixture was held at -15 0C for 2-3 hrs. Conversion of the acid to acid chloride was monitored by HPLC: an aliquot of the reaction mixture was quenched into anhydrous ethanol and the solution was analyzed by HPLC using SIELC Primesep 200 column capable of retaining polar compounds. The conversion was calculated as a ratio of peak areas corresponding to the starting acid and its ethyl ester formed by reaction of the acid chloride with ethanol. After conversion reached the maximum (usually 85 to 95% after 2 to 3 hours), a solution of the aminoquinoline (12.6 kg, 28.3 mol) in DMAc (99.4 kg) was added to the reactor maintaining the temperature in the -14 to -19 0C range. Resulting mixture was held for 5 hr at approximately - 15 0C. At this point HPLC analysis showed residual aniline level at or below 0.5%. The thick suspension of the hydrochloride salt of the coupled product was quenched with water (87.1 kg) maintaining the batch temperature between -7 to -16 0C. The resulting clear solution in was warmed to 37-42 0C and its pH was adjusted to 10-1 1 with a 25% aqueous solution of KOH (approx. 53 kg of the solution was added). The suspension was further diluted with water (95 kg) and the solids were filtered on a polypropylene cloth filter. The cake was washed with water until neutral pH of the washes and dried first in the nitrogen flow on the filter and then on trays in vacuum at 45 to 50 0C to afford crude (E)-Λ/-(4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)- 3-cyano-7-ethoxyquinolin6-yl)-4-(dimethylamino)but-2-enamide (15.75 kg, 94%) as a bright- yellow crystalline solid.
Example 3:
Comparison of formation and stability of 4-dimethylaminocrotonoyl chloride THF and iPAc.
In THF: 4-Dimethylaminocrotonic acid hydrochloride (662 mg, 4.00 mmol) was suspended in a mixture of THF (6.6 mL) and DMF (16 μL, 0.20 mmol). The mixture was chilled to 0 0C in an ice bath. Oxalyl chloride (343 μL, 3.92 mmol) was added slowly to the reaction mixture via a syringe keeping the temperature below 1 0C. The reaction mixture was warmed up to 20 0C and kept at that temperature for the duration of the experiment. To analyze the composition of the reaction mixture, an 0.2-mL aliquot was drawn, the solvent was removed in vacuum, and the residue was dissolved in deuterated acetonitrile and the solution was promptly analyzed by 1H NMR. The ratios of the mixture components (acid chloride, starting acid, cyclic degradant) was determined by integration of the corresponding signals in the NMR spectra.
In iPAc: Charged 4-dimethylaminocrotonic acid hydrochloride (1.00 g, 6.04 mmol), isopropyl acetate (8 ml_) and dimethylformamide (10 mol%, 0.042 g, 0.0005796 mol) to a reactor tube and stirred. Cooled the mixture to 1 O0C and added oxalyl chloride (0.699 g, 0.00551 mol) over a period of 10 min. The temperature of the reaction mixture was adjusted to 150C and maintained at this temperature for the duration of the experiment. To analyze the composition of the reaction mixture, an 0.2-mL aliquot was drawn, the solvent was removed in vacuum, and the residue was dissolved in deuterated acetonitrile and the solution was promptly analyzed by 1H NMR. The ratios of the mixture components (acid chloride, starting acid, cyclic degradant) was determined by integration of the corresponding signals in the NMR spectra.
Example 4: Process 3
4-Dimethylaminocrotonoyl chloride hydrochloride and its coupling with 6-amino- 4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-7-ethoxyquinoline-3-carbonitrile (procedure with thionyl chloride and DMAc).
A suspension of 4-dimethylaminocrotonic acid (17.0 g, 97.5 mmol) in DMAc (170 ml_) was cooled to -15 0C under nitrogen atmosphere. Neat thionyl chloride (12.8 g, 7.83 mmol) was added to the slurry at a rate to maintain the temperature in the reactor in the range of -18 to -14 0C (moderate exotherm). The reaction mixture was held at -17 to -15 0C for 4 hrs. A solution of the aminoquinoline (36.2 g, 81.3 mmol) in DMAc (440 ml_) was added to the reactor maintaining the temperature in the -14 to -19 0C range. The resulting mixture was held for 18 hr at approximately -15 0C. At this point HPLC analysis showed residual aniline level at 2.5%. The thick suspension of the hydrochloride salt of the coupled product was quenched with water (200 ml_) maintaining the batch temperature between -5 and -16 0C. The pH of the resulting clear solution was adjusted to 1 1 with a 13% aqueous solution of NaOH (approx. 210 ml_ of the solution was added). The suspension was further diluted with water (350 ml_) and the solids were filtered on a polypropylene cloth filter. The cake was washed with water until neutral pH of the washes and dried first in the nitrogen flow on the filter and then on a tray in vacuum at 45 to 50 0C to afford crude (.=)-/\/-(4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-3-cyano-7- ethoxyquinolin-6-yl)-4-(dimethylamino)but-2-enamide (42.0 g, 91 %) as a bright-yellow crystalline solid.
Claims
1. A method for coupling a 4-(amino)-2-butenoyl group to an amino group at the 6- or 7- position of a 4-amino-3-quinolinecarbonitrile, said method comprising: at between about 5° C and 25° C, generating 4-(amino)-2-butenoyl chloride hydrochloride by reacting an aminocrotonic acid with oxalyl chloride in isopropyl acetate, forming said 4-(amino)-2- butenoyl chloride hydrochloride as a solid or as a slurry, and reacting said 4-(amino)-2- butenoyl chloride with said 4-amino-3-quinolinecarbonitrile.
2. The method of claim 1 comprising generating 4-(dimethylamino)-2-butenoyl chloride hydrochloride by reacting an dimethylaminocrotonic acid with oxalyl chloride.
3. The method of claim 1 wherein said method is performed at between about 10° C and
200 C.
4. The method of claim 1 wherein the isolated slurry is stored at 5° C or less for up to 30 hours as a hydrochloride salt.
5. The method of claim 1 wherein the solid is isolated by filtering and stored as a hydrochloride chloride at 25° C or less.
6. The method of claims 1-5 wherein the coupled product is (E)-Λ/-(4-(3-chloro-4-(pyridin-2- ylmethoxy)phenylamino)-3-cyano-7-ethoxyquinolin-6-yl)-4-(dimethylamino)but-2- enamide or a pharmaceutically acceptable salt.
7. A method for preparing a method for coupling a 4-(amino)-2-butenoyl group to an amino group at the 6- or 7-position of a 4-amino-3-quinolinecarbonitrile, said method comprising: at between about -5° C and -25° C, generating 4-(amino)-2-butenoyl chloride by reacting an aminocrotonic acid with phosphorous oxychloride (POCI3) in dimethylacetamide (DMAc), and reacting said 4-(amino)-2-butenoyl chloride with said 4- amino-3-quinolinecarbonitrile.
8. The method of claim 1 comprising generating 4-(dimethylamino)-2-butenoyl chloride by reacting dimethylaminocrotonic acid with phosphorous oxychloride.
9. The method of claim 7 wherein the coupled product is (E)-Λ/-(4-(3-chloro-4-(pyridin-2- ylmethoxy)phenylamino)-3-cyano-7-ethoxyquinolin-6-yl)-4-(dimethylamino)but-2- enamide or a pharmaceutically acceptable salt.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10825408P | 2008-10-24 | 2008-10-24 | |
| US61/108,254 | 2008-10-24 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2010048477A2 true WO2010048477A2 (en) | 2010-04-29 |
| WO2010048477A3 WO2010048477A3 (en) | 2011-02-24 |
Family
ID=41590915
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/061787 WO2010048477A2 (en) | 2008-10-24 | 2009-10-23 | Improved process for preparation of coupled products from 4-amino-3-cyanoquinolines using stabilized intermediates |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2010048477A2 (en) |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102838539A (en) * | 2011-06-21 | 2012-12-26 | 苏州迈泰生物技术有限公司 | Quinolinylenamide derivative and its application in preparation of anti-malignant tumor drugs |
| WO2015101554A1 (en) * | 2013-12-31 | 2015-07-09 | Boehringer Ingelheim International Trading (Shanghai) Co., Ltd: | Process for the manufacture of (e)-4-n,n-dialkylamino crotonic acid in hx salt form and use thereof for synthesis of egfr tyrosine kinase inhibitors |
| US9139558B2 (en) | 2007-10-17 | 2015-09-22 | Wyeth Llc | Maleate salts of (E)-N-{4-[3-Chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide and crystalline forms thereof |
| CN105085485A (en) * | 2015-08-21 | 2015-11-25 | 哈尔滨珍宝制药有限公司 | Preparation method of neratinib |
| US9211291B2 (en) | 2009-04-06 | 2015-12-15 | Wyeth Llc | Treatment regimen utilizing neratinib for breast cancer |
| US9211264B2 (en) | 2009-11-09 | 2015-12-15 | Wyeth Llc | Coated drug spheroids and uses thereof for eliminating or reducing conditions such as emesis and diarrhea |
| US9265784B2 (en) | 2008-08-04 | 2016-02-23 | Wyeth Llc | Antineoplastic combinations of 4-anilino-3-cyanoquinolines and capecitabine |
| CN105461689A (en) * | 2015-05-19 | 2016-04-06 | 上海麦步医药科技有限公司 | Novel preparation method of epidermal growth factor receptor (EGFR) inhibitor neratinib |
| CN105949176A (en) * | 2016-06-24 | 2016-09-21 | 浙江海正药业股份有限公司 | Method for purifying neratinib |
| US9511063B2 (en) | 2008-06-17 | 2016-12-06 | Wyeth Llc | Antineoplastic combinations containing HKI-272 and vinorelbine |
| US9539258B2 (en) | 2005-11-11 | 2017-01-10 | Boehringer Ingelheim International Gmbh | Quinazoline derivatives for the treatment of cancer diseases |
| WO2017186140A1 (en) * | 2016-04-28 | 2017-11-02 | 江苏恒瑞医药股份有限公司 | Method for preparing tyrosine kinase inhibitor and derivative thereof |
| WO2018005418A1 (en) * | 2016-06-27 | 2018-01-04 | Pliva Hrvatska D.O.O. | Solid state forms of neratinib and salts thereof |
| US10004743B2 (en) | 2009-07-06 | 2018-06-26 | Boehringer Ingelheim International Gmbh | Process for drying of BIBW2992, of its salts and of solid pharmaceutical formulations comprising this active ingredient |
| CN110357854A (en) * | 2018-03-26 | 2019-10-22 | 江苏创诺制药有限公司 | A kind of preparation method of linatinib |
| CN110357855A (en) * | 2018-03-26 | 2019-10-22 | 江苏创诺制药有限公司 | A kind of linatinib hydrochloride Form and preparation method thereof |
| US10596162B2 (en) | 2005-02-03 | 2020-03-24 | Wyeth Llc | Method for treating gefitinib resistant cancer |
| US10729672B2 (en) | 2005-11-04 | 2020-08-04 | Wyeth Llc | Antineoplastic combinations with mTOR inhibitor, trastuzumab and/or HKI-272 |
| CN111943933A (en) * | 2020-09-02 | 2020-11-17 | 重庆医科大学 | Preparation method of neratinib impurity D |
| CN111995618A (en) * | 2020-09-02 | 2020-11-27 | 重庆医科大学 | Preparation method of neratinib impurity G |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004066919A2 (en) * | 2003-01-21 | 2004-08-12 | Wyeth | Synthesis of 4amino-2-butenoyl chlorides and their use in the preparation of 3-cyano quinolines |
-
2009
- 2009-10-23 WO PCT/US2009/061787 patent/WO2010048477A2/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004066919A2 (en) * | 2003-01-21 | 2004-08-12 | Wyeth | Synthesis of 4amino-2-butenoyl chlorides and their use in the preparation of 3-cyano quinolines |
Non-Patent Citations (1)
| Title |
|---|
| TSOU H-R ET AL: "Optimization of 6,7-Disubstituted-4-(arylamino)quinoline-3 -carbonitr iles as Orally Active, Irreverible Inhibitors of HEGFR-2 Kinase Activity" JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, WASHINGTON, US, vol. 48, 27 January 2005 (2005-01-27), pages 1107-1131, XP002414228 ISSN: 0022-2623 cited in the application * |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10603314B2 (en) | 2005-02-03 | 2020-03-31 | The General Hospital Corporation | Method for treating gefitinib resistant cancer |
| US10596162B2 (en) | 2005-02-03 | 2020-03-24 | Wyeth Llc | Method for treating gefitinib resistant cancer |
| US10729672B2 (en) | 2005-11-04 | 2020-08-04 | Wyeth Llc | Antineoplastic combinations with mTOR inhibitor, trastuzumab and/or HKI-272 |
| US9539258B2 (en) | 2005-11-11 | 2017-01-10 | Boehringer Ingelheim International Gmbh | Quinazoline derivatives for the treatment of cancer diseases |
| US9139558B2 (en) | 2007-10-17 | 2015-09-22 | Wyeth Llc | Maleate salts of (E)-N-{4-[3-Chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide and crystalline forms thereof |
| US10035788B2 (en) | 2007-10-17 | 2018-07-31 | Wyeth Llc | Maleate salts of (E)-N-{4[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide and crystalline forms thereof |
| US9630946B2 (en) | 2007-10-17 | 2017-04-25 | Wyeth Llc | Maleate salts of (E)-N-{4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide and crystalline forms thereof |
| US10111868B2 (en) | 2008-06-17 | 2018-10-30 | Wyeth Llc | Antineoplastic combinations containing HKI-272 and vinorelbine |
| US9511063B2 (en) | 2008-06-17 | 2016-12-06 | Wyeth Llc | Antineoplastic combinations containing HKI-272 and vinorelbine |
| US9265784B2 (en) | 2008-08-04 | 2016-02-23 | Wyeth Llc | Antineoplastic combinations of 4-anilino-3-cyanoquinolines and capecitabine |
| US9211291B2 (en) | 2009-04-06 | 2015-12-15 | Wyeth Llc | Treatment regimen utilizing neratinib for breast cancer |
| US10004743B2 (en) | 2009-07-06 | 2018-06-26 | Boehringer Ingelheim International Gmbh | Process for drying of BIBW2992, of its salts and of solid pharmaceutical formulations comprising this active ingredient |
| US9211264B2 (en) | 2009-11-09 | 2015-12-15 | Wyeth Llc | Coated drug spheroids and uses thereof for eliminating or reducing conditions such as emesis and diarrhea |
| CN102838539A (en) * | 2011-06-21 | 2012-12-26 | 苏州迈泰生物技术有限公司 | Quinolinylenamide derivative and its application in preparation of anti-malignant tumor drugs |
| WO2015101554A1 (en) * | 2013-12-31 | 2015-07-09 | Boehringer Ingelheim International Trading (Shanghai) Co., Ltd: | Process for the manufacture of (e)-4-n,n-dialkylamino crotonic acid in hx salt form and use thereof for synthesis of egfr tyrosine kinase inhibitors |
| JP2017503020A (en) * | 2013-12-31 | 2017-01-26 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Process for the preparation of (E) -4-N, N-dialkylaminocrotonic acid in the form of an HX salt and its use for the synthesis of EGFR tyrosine kinase inhibitors |
| US9242965B2 (en) | 2013-12-31 | 2016-01-26 | Boehringer Ingelheim International Gmbh | Process for the manufacture of (E)-4-N,N-dialkylamino crotonic acid in HX salt form and use thereof for synthesis of EGFR tyrosine kinase inhibitors |
| CN105461689A (en) * | 2015-05-19 | 2016-04-06 | 上海麦步医药科技有限公司 | Novel preparation method of epidermal growth factor receptor (EGFR) inhibitor neratinib |
| CN105085485A (en) * | 2015-08-21 | 2015-11-25 | 哈尔滨珍宝制药有限公司 | Preparation method of neratinib |
| CN108290867A (en) * | 2016-04-28 | 2018-07-17 | 江苏恒瑞医药股份有限公司 | A method of preparing tyrosine kinase inhibitor and its derivative |
| JP2019520305A (en) * | 2016-04-28 | 2019-07-18 | ジエンス ヘンルイ メデイシンカンパニー リミテッドJiangsu Hengrui Medicine Co.,Ltd. | Method for producing tyrosine kinase inhibitor and derivative thereof |
| CN108290867B (en) * | 2016-04-28 | 2022-02-08 | 江苏恒瑞医药股份有限公司 | Method for preparing tyrosine kinase inhibitor and derivative thereof |
| WO2017186140A1 (en) * | 2016-04-28 | 2017-11-02 | 江苏恒瑞医药股份有限公司 | Method for preparing tyrosine kinase inhibitor and derivative thereof |
| TWI739825B (en) * | 2016-04-28 | 2021-09-21 | 大陸商江蘇恆瑞醫藥股份有限公司 | Method for preparing tyrosine kinase inhibitor and derivatives thereof |
| US10793548B2 (en) | 2016-04-28 | 2020-10-06 | Jiangsu Hengrui Medicine Co., Ltd. | Method for preparing tyrosine kinase inhibitor and derivative thereof |
| US11198683B2 (en) | 2016-04-28 | 2021-12-14 | Jiangsu Hengrui Medicine Co., Ltd. | Method for preparing tyrosine kinase inhibitor and derivative thereof |
| CN105949176A (en) * | 2016-06-24 | 2016-09-21 | 浙江海正药业股份有限公司 | Method for purifying neratinib |
| WO2018005418A1 (en) * | 2016-06-27 | 2018-01-04 | Pliva Hrvatska D.O.O. | Solid state forms of neratinib and salts thereof |
| CN110357855A (en) * | 2018-03-26 | 2019-10-22 | 江苏创诺制药有限公司 | A kind of linatinib hydrochloride Form and preparation method thereof |
| CN110357854A (en) * | 2018-03-26 | 2019-10-22 | 江苏创诺制药有限公司 | A kind of preparation method of linatinib |
| CN111943933B (en) * | 2020-09-02 | 2021-05-28 | 重庆医科大学 | Preparation method of neratinib impurity D |
| CN111995618B (en) * | 2020-09-02 | 2021-06-11 | 重庆医科大学 | Preparation method of neratinib impurity G |
| CN111995618A (en) * | 2020-09-02 | 2020-11-27 | 重庆医科大学 | Preparation method of neratinib impurity G |
| CN111943933A (en) * | 2020-09-02 | 2020-11-17 | 重庆医科大学 | Preparation method of neratinib impurity D |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2010048477A3 (en) | 2011-02-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2010048477A2 (en) | Improved process for preparation of coupled products from 4-amino-3-cyanoquinolines using stabilized intermediates | |
| EP1730113B1 (en) | Process for the synthesis of a cxcr4 antagonist | |
| US10040778B2 (en) | Anhydrous lenalidomide form-I | |
| US9199970B2 (en) | 4-[5-(pyridin-4-yl)-1H-1,2,4-triazol-3-yl]pyridine-2-carbonitrile crystalline polymorph and production method therefor | |
| CN102985416B (en) | Process of preparing a thrombin specific inhibitor | |
| US20040010151A1 (en) | Lansoprazole polymorphs and processes for preparation thereof | |
| JP2011513497A (en) | Preparation of lenalidomide | |
| EP2755959B1 (en) | Rilpivirine hydrochloride | |
| EP2794610B1 (en) | Processes and intermediates for preparing pralatrexate | |
| WO2014086326A1 (en) | A method for the preparation and purification of new and known polymorphs and solvates of dasatinib | |
| WO2016150340A1 (en) | Salts of quinazoline derivative and method for preparing same | |
| US20190241511A1 (en) | Polymorphic forms of belinostat and processes for preparation thereof | |
| EP1789412B1 (en) | Crystalline alfuzosin base | |
| WO2012090221A1 (en) | Novel salts of imatinib | |
| JP2020050632A (en) | Novel crystal form of production intermediate of alogliptin benzoate | |
| US6306886B1 (en) | Crystalline roxifiban | |
| US20140155607A1 (en) | Novel Salts of Raltegravir | |
| JP2014504281A (en) | Separation of triazine derivative enantiomers with tartaric acid. | |
| KR20130134407A (en) | Process for preparing gefitinib and an intermediate used for preparing thereof | |
| US20090137844A1 (en) | Crystallization process | |
| CN109705118B (en) | Preparation method of tricyclic EGFR kinase inhibitor | |
| CN109153652A (en) | The preparation process of 1- (aryl methyl) quinazoline -2,4 (1H, 3H)-diketone | |
| ES2909302T3 (en) | Process for the preparation of Raltegravir | |
| JP2011195500A (en) | Method for producing (s)-4-[4-[(4-chlorophenyl)(2-pyridyl)methoxy]piperidino]butanoic acid/benzenesulfonic acid salt | |
| US20170022220A1 (en) | High-purity crystals of active blood coagulation factor x (fxa) inhibitor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09740618 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09740618 Country of ref document: EP Kind code of ref document: A2 |