WO2010039922A1 - Calcilytic compounds - Google Patents
Calcilytic compounds Download PDFInfo
- Publication number
- WO2010039922A1 WO2010039922A1 PCT/US2009/059175 US2009059175W WO2010039922A1 WO 2010039922 A1 WO2010039922 A1 WO 2010039922A1 US 2009059175 W US2009059175 W US 2009059175W WO 2010039922 A1 WO2010039922 A1 WO 2010039922A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- alkyl
- pyridinyl
- fluoro
- pyrimidinone
- Prior art date
Links
- 0 *c(cccc1C(N(*)C2=O)=NC(*)=C2Br)c1OCc1ccccc1 Chemical compound *c(cccc1C(N(*)C2=O)=NC(*)=C2Br)c1OCc1ccccc1 0.000 description 3
- GHUFWTFUFIAIQJ-UHFFFAOYSA-N Cc([s]c(C1=C(C)N=C(c2cccc(F)c2O)N(CCc2ccccc2)C1=O)c1)c1-c1ncccc1C Chemical compound Cc([s]c(C1=C(C)N=C(c2cccc(F)c2O)N(CCc2ccccc2)C1=O)c1)c1-c1ncccc1C GHUFWTFUFIAIQJ-UHFFFAOYSA-N 0.000 description 1
- JRBHUQWUJHBNQV-UHFFFAOYSA-N Cc(nc1)ccc1-c1ccc(C2=C(C)N=C(c3cccc(F)c3O)N(CCc3ccccc3)C2=O)[s]1 Chemical compound Cc(nc1)ccc1-c1ccc(C2=C(C)N=C(c3cccc(F)c3O)N(CCc3ccccc3)C2=O)[s]1 JRBHUQWUJHBNQV-UHFFFAOYSA-N 0.000 description 1
- HWGQBUXKBICFRT-UHFFFAOYSA-N Cc1cccc(C(N2CCc3ccccc3)=NC(C)=C(c3ccc(-c4ccccn4)[s]3)C2=O)c1O Chemical compound Cc1cccc(C(N2CCc3ccccc3)=NC(C)=C(c3ccc(-c4ccccn4)[s]3)C2=O)c1O HWGQBUXKBICFRT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention relates to novel calcilytic compounds, pharmaceutical compositions containing these compounds, processes for their preparation and their use as calcium receptor antagonists.
- extracellular Ca ⁇ + In mammals, extracellular Ca ⁇ + is under rigid homeostatic control and regulates various processes such as blood clotting, nerve and muscle excitability, and proper bone formation.
- Extracellular Ca ⁇ + inhibits the secretion of parathyroid hormone ("PTH") from parathyroid cells, inhibits bone resorption by osteoclasts, and stimulates secretion of calcitonin from C-cells.
- PTH parathyroid hormone
- Calcium receptor proteins enable certain specialized cells to respond to changes in extracellular Ca ⁇ + concentration.
- PTH is the principal endocrine factor regulating Ca ⁇ + homeostasis in the blood and extracellular fluids.
- PTH by acting on bone and kidney cells, increases the level Of Ca ⁇ + in the blood. This increase in extracellular Ca ⁇ + then acts as a negative feedback signal, depressing PTH secretion.
- the reciprocal relationship between extracellular Ca ⁇ + and PTH secretion forms an important mechanism maintaining bodily Ca ⁇ + homeostasis.
- Extracellular Ca ⁇ + acts directly on parathyroid cells to regulate PTH secretion.
- parathyroid cell surface protein which detects changes in extracellular Ca 2+ has been confirmed. See Brown et al., Nature 366:574, 1993.
- this protein the calcium receptor, acts as a receptor for extracellular Ca 2+ , detects changes in the ion concentration of extracellular Ca ⁇ +, and initiates a functional cellular response,
- Calcilytics are compounds able to inhibit calcium receptor activity, thereby causing a decrease in one or more calcium receptor activities evoked by extracellular Ca ⁇ +.
- Calcilytics are useful as lead molecules in the discovery, development, design, modification and/or construction of useful calcium modulators, which are active at Ca ⁇ + receptors.
- Such calcilytics are useful in the treatment of various disease states characterized by abnormal levels of one or more components, e.g., polypeptides such as hormones, enzymes or growth factors, the expression and/or secretion of which is regulated or affected by activity at one or more Ca ⁇ + receptors.
- Target diseases or disorders for calcilytic compounds include diseases involving abnormal bone and mineral homeostasis.
- Abnormal calcium homeostasis is characterized by one or more of the following activities: an abnormal increase or decrease in serum calcium; an abnormal increase or decrease in urinary excretion of calcium; an abnormal increase or decrease in bone calcium levels (for example, as assessed by bone mineral density measurements); an abnormal absorption of dietary calcium; an abnormal increase or decrease in the production and/or release of messengers which affect serum calcium levels such as PTH and calcitonin; and an abnormal change in the response elicited by messengers which affect serum calcium levels.
- calcium receptor antagonists offer a unique approach towards the pharmacotherapy of diseases associated with abnormal bone or mineral homeostasis, such as hypoparathyroidism, osteosarcoma, periodontal disease, bone fracture, osteoarthritis, rheumatoid arthritis, Paget's disease, humoral hypercalcemia associated with malignancy and bone fracture, osteopenia, and osteoporosis.
- the present invention involves novel compounds according to Formula (I):
- R 1 is a 6-membered heteroaryl group, containing 1-2 nitrogen atoms, optionally substituted by -O-(Ci-C2)alkyl-O-, or one to three times, independently, by halogen, (Ci-C 4 )alkyl, -CF 3 , amino, (Ci-C 4 )alkylamino, (Ci-C 4 )alkyl(Ci-C 4 )alkylamino, (Ci-C 4 )alkoxy, hydroxy(Ci-C 4 )alkyl, or (Ci-C 4 )alkoxy(Ci-C 4 )alkyl;
- R 2 is hydrogen or (Ci-C 4 )alkyl; wherein R 1 is located at either the 4- or 5 -position of the thiophene ring and when R 1 is located at the 4-position, R 2 is located at the 5 -position, and when R 1 is located at the 5 -position, R 2 is located at the 4-position;
- R 3 is selected from the group consisting of (Ci-C 4 )alkyl, phenyl, and heteroaryl, optionally substituted, one to three times, independently, by halogen, (Ci-C 4 )alkyl, -CF 3 , or (C 1 -C 4 )alkoxy;
- R 4 is selected from the group consisting of (C 5 -C 6 )cycloalkyl(Ci-C 4 )alkyl, heterocycloalkyl(Ci-C 4 )alkyl, aryl(Ci-C 4 )alkyl, and heteroaryl(Ci-C 4 )alkyl, wherein any cycloalkyl, heterocycloalkyl, aryl, or heteroaryl group is optionally substituted by -O-(Ci-C 2 )alkyl-O-, or one to three times, independently, by halogen, (Ci-C 4 )alkyl, -CF 3 , amino, (Ci-C 4 )alkylamino, (Ci-C 4 )alkyl(Ci-C 4 )alkylamino, or (Ci-C 4 )alkoxy;
- R 5 is hydrogen or fluorine; or a salt thereof.
- the present invention is also directed to formulations comprising compounds of Formula (I), or a salt thereof, and their use as calcium receptor antagonists in the treatment of a variety of diseases associated with abnormal bone or mineral homeostasis, including but not limited to hypoparathyroidism, osteosarcoma, periodontal disease, bone fracture, osteoarthritis, rheumatoid arthritis, Paget' s disease, humoral hypercalcemia associated with malignancy and bone fracture, osteopenia, and osteoporosis.
- diseases associated with abnormal bone or mineral homeostasis including but not limited to hypoparathyroidism, osteosarcoma, periodontal disease, bone fracture, osteoarthritis, rheumatoid arthritis, Paget' s disease, humoral hypercalcemia associated with malignancy and bone fracture, osteopenia, and osteoporosis.
- the present invention further provides a method for antagonizing calcium receptors in a mammal, including a human, which comprises administering to a mammal in need thereof an effective amount of a compound of Formula (I), or a salt thereof.
- the present invention further provides a method for increasing serum parathyroid levels in a mammal, including a human, which comprises administering to a mammal in need thereof an effective amount of a compound of Formula (I), or a salt thereof.
- alkyl refers to a straight- or branched-chain hydrocarbon radical having the specified number of carbon atoms.
- (Ci-C 4 )alkyl refers to an alkyl group having at least 1 and up to 4 carbon atoms. Examples of such branched or straight-chained alkyl groups useful in the present invention include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, s-butyl, and t-butyl.
- cycloalkyl refers to a non-aromatic, saturated, cyclic hydrocarbon ring containing the specified number of carbon atoms.
- Cs-C 6 cycloalkyl refers to a non-aromatic cyclic hydrocarbon ring having from five to six carbon atoms.
- Exemplary "Cs-C 6 cycloalkyl” groups useful in the present invention include cyclopentyl and cyclohexyl.
- Alkoxy means an alkyl radical containing the specified number of carbon atoms attached through an oxygen linking atom.
- (Ci-C 4 )alkoxy refers to a straight- or branched-chain hydrocarbon radical having at least 1 and up to 4 carbon atoms attached through an oxygen linking atom.
- Exemplary "(Ci-C 4 )alkoxy” groups useful in the present invention include, but are not limited to, methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, 5-butoxy, and t-butoxy.
- Heterocycloalkyl means a non-aromatic heterocyclic ring containing 5-6 ring atoms, being saturated or having one or more degrees of unsaturation, and containing one or more heteroatom substitutions selected from O, S, and/or N.
- heterocycloalkyl moieties include, but are not limited to, tetrahydrofuranyl, dihydropyranyl, tetrahydropyranyl, 1 ,4-dioxanyl, 1,3-dioxanyl, piperidinyl, piperazinyl, 2,4-piperazinedionyl, pyrrolidinyl, pyrrolinyl, imidazolidinyl, pyrazolidinyl, pyrazolinyl, morpholinyl, thiomorpholinyl, tetrahydrothiopyranyl, tetrahydrothienyl, and the like.
- Aryl refers to optionally substituted monocyclic or fused polycarbocyclic groups having 6 to 14 carbon atoms and having at least one aromatic ring that complies with H ⁇ ckel's Rule.
- aryl groups are phenyl, naphthyl, anthracenyl, phenanthrenyl, and the like.
- Preferably aryl refers to optionally substituted phenyl.
- Heteroaryl means an optionally substituted aromatic monocyclic ring containing 5-6 ring atoms that complies with H ⁇ ckel's Rule and contains at least one and up to three heteroatoms independently selected from N, O, and/or S.
- 5-membered “heteroaryl” groups include furanyl, thienyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, thiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiadiazolyl, and isothiazolyl.
- 6-membered "heteroaryl” groups include oxo-pyridyl, pyridinyl, pyridazinyl, pyrazinyl, and pyrimidinyl.
- any group or moiety such as alkyl, aryl, cycloalkyl, heterocycloalkyl, or heteroaryl, is defined herein as being “optionally substituted, one to two (or three) times, independently, by" the recited substituents, it is to be understood that the group or moiety is unsubstituted or is substituted by one to two (or three) substituents, wherein each substituent is independently selected from the recited substituents.
- R 1 is selected from the group consisting of pyridinyl, pyrazinyl, and pyrimidinyl, optionally substituted, one to three times, independently, by (Ci-C 4 )alkyl, -CF 3 , amino, (Ci-C 4 )alkylamino, (Ci-C 4 )alkyl(Ci-C 4 )alkylamino, (Ci-C 4 )alkoxy, hydroxy(Ci-C 4 )alkyl, or (Ci-C 4 )alkoxy(Ci-C 4 )alkyl.
- R 1 is pyridinyl, optionally substituted, one to two times, independently, by (Ci-C 4 )alkyl, -CF 3 , (Ci-C 4 )alkoxy, hydroxy(Ci-C 4 )alkyl, or (Ci-C 4 )alkoxy(Ci-C 4 )alkyl.
- R 1 is 2-pyridinyl, 3-methyl-2-pyridinyl, 6-methyl-2- pyridinyl, 6-[(methyloxy)methyl]-2 -pyridinyl, 3-pyridinyl, 6-methyl-3 -pyridinyl, 6- trifluoromethyl-3 -pyridinyl, 6-hydroxymethyl-3 -pyridinyl, 4-pyridinyl, 2-pyrimidinyl, 5- pyrimidinyl, or 2-pyrazinyl.
- R 1 is 2-pyridinyl.
- R 2 is hydrogen or methyl.
- R 3 is (Ci-C 4 )alkyl. In a specific embodiment, R 3 is methyl. In a yet another embodiment, R 4 is a phenyl(Ci-C 2 )alkyl group, optionally substituted by -O-(Ci-C2)alkyl-O-, or one to two times, independently, by F, Cl, (Ci-C 4 )alkyl, (Ci-C 4 )alkoxy, or -CF 3 .
- R 4 is phenethyl, optionally substituted by -O-(Ci-C2)alkyl-O-, or one to two times, independently, by F, Cl, (Ci-C 4 )alkyl, (Ci-C 4 )alkoxy, or -CF 3 .
- R 4 is phenethyl.
- R 5 is fluorine.
- One particular embodiment of the invention is a compound of Formula (I) wherein:
- R 1 is selected from the group consisting of pyridinyl, pyrazinyl, and pyrimidinyl, optionally substituted, one to three times, independently, by (Ci-C 4 )alkyl, -CF 3 , amino, (Ci-C 4 )alkylamino, (Ci-C 4 )alkyl(Ci-C 4 )alkylamino, (Ci-C 4 )alkoxy, hydroxy(Ci-C 4 )alkyl, or (Ci-C 4 )alkoxy(Ci-C 4 )alkyl;
- R 2 is hydrogen or (Ci-C 4 )alkyl
- R 3 is (Ci-C 4 )alkyl
- R 4 is phenethyl, optionally substituted by -O-(Ci-C2)alkyl-O-, or one to two times, independently, by F, Cl, (Ci-C 4 )alkyl, (Ci-C 4 )alkoxy, or -CF 3 ;
- R 5 is hydrogen or fluorine; or a salt thereof.
- Another particular embodiment of the invention is a compound of Formula (I) wherein:
- R 1 is pyridinyl, optionally substituted, one to two times, independently, by (Ci-C 4 )alkyl, -CF 3 , (Ci-C 4 )alkoxy, hydroxy(Ci-C 4 )alkyl, or (Ci-C 4 )alkoxy(Ci-C 4 )alkyl;
- R 2 is hydrogen or methyl; R is methyl;
- R 4 is phenethyl
- R 5 is fluorine; or a salt thereof.
- R 1 , R 2 , R 3 , R 4 and R 5 are as defined hereinabove.
- R 1 , R 2 , R 3 , R 4 and R 5 are as defined hereinabove.
- Specific compounds exemplified herein are:
- solvate refers to a complex of variable stoichiometry formed by a solute and a solvent. Such solvents for the purpose of the invention may not interfere with the biological activity of the solute.
- suitable solvents include, but are not limited to, water, MeOH, EtOH and AcOH.
- the solvent used is a pharmaceutically acceptable solvent.
- suitable pharmaceutically acceptable solvents include, without limitation, water, EtOH and AcOH.
- Solvates wherein water is the solvent molecule are typically referred to as "hydrates". Hydrates include compositions containing stoichiometric amounts of water, as well as compositions containing variable amounts of water. Solvates, particularly hydrates, of the compounds of Formula (I), (Ia), and (Ib), and salts thereof, are within the scope of the invention.
- the compound or salt including solvates (particularly, hydrates) thereof, may exist in crystalline forms, non-crystalline forms or a mixture thereof.
- the compound or salt, or solvates (particularly, hydrates) thereof may also exhibit polymorphism (i.e. the capacity to occur in different crystalline forms). These different crystalline forms are typically known as "polymorphs.”
- polymorphs typically known as “polymorphs.”
- the disclosed compound, or solvates (particularly, hydrates) thereof also include all polymorphs thereof. Polymorphs have the same chemical composition but differ in packing, geometrical arrangement, and other descriptive properties of the crystalline solid state.
- Polymorphs therefore, may have different physical properties such as shape, density, hardness, deformability, stability, and dissolution properties. Polymorphs typically exhibit different melting points, IR spectra, and X-ray powder diffraction patterns, which may be used for identification. One of ordinary skill in the art will appreciate that different polymorphs may be produced, for example, by changing or adjusting the conditions used in crystallizing/recrystallizing the compound.
- the invention also includes various isomers of the compounds of Formula (I), (Ia), and (Ib), and mixtures thereof.
- “Isomer” refers to compounds that have the same composition and molecular weight but differ in physical and/or chemical properties. The structural difference may be in constitution (geometric isomers) or in the ability to rotate the plane of polarized light (stereoisomers). With regard to stereoisomers, the present compounds may have one or more asymmetric carbon atom and may occur as racemates, racemic mixtures, and as individual enantiomers or diastereomers. All such isomeric forms are included within the present invention, including mixtures thereof.
- salts of the compounds of Formula (I) are preferably pharmaceutically acceptable.
- Suitable pharmaceutically acceptable salts can include acid or base addition salts.
- salts and solvates e.g. hydrates and hydrates of salts
- the counterion or associated solvent is pharmaceutically acceptable.
- salts and solvates having non-pharmaceutically acceptable counterions or associated solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of the invention and their pharmaceutically acceptable salts and solvates.
- a pharmaceutically acceptable acid addition salt can be formed by reaction of a compound of Formula (I) containing a basic moiety with a suitable inorganic or organic acid (such as acetic, aspartic, benzenesulfonic, benzoic, bicarbonic, camphorsulfonic, carbonic, citric, dodecyl sulfonic, 1 ,2-ethanedisulfonic, ethanesulfonic, formic, fumaric, (3i?,45 * ,5i?,6i?)-2,3,4,5,6,7-hexahydroxyheptanoic acid, galacturonic, gluconic, glutamic, hexanoic, hydrobromide, hydrochloride, 2-hydroxyethanesulfonic, hydroxynaphthoic, lactic, lactobionic, malic, maleic, mandelic, methanesulfonic, mucic, naphthalene-2- sulfonic, nitric, pamoi
- Pharmaceutically acceptable acid addition salts of a compound of Formula (I) include acetate, aspartate, benzenesulfonate, benzoate, bicarbonate, bitartrate, bromide, calcium edetate, camsylate, carbonate, chloride, citrate, dihydrochloride, edetate, edisylate, estolate, esylate, formate, fumarate, galacturonate, gluceptate, gluconate, glutamate, glycollylarsanilate, hexanoate, hydrobromide, hydrochloride, hydroxynaphthoate, iodide, isethionate, lactate, lactobionate, malate, maleate, mandelate, mesylate, methylsulfate, mucate, napsylate, nitrate, pamoate, pantothenate, phosphate/diphosphate, polygalacturonate, propionate, salicylate, ste
- a pharmaceutically acceptable base addition salt can be formed by reaction of a compound of Formula (I) containing an acidic moiety with a suitable inorganic or organic base (e.g. triethylamine, ethanolamine, triethanolamine, choline, arginine, lysine, or histidine), optionally in a suitable solvent such as an organic solvent, to give the base addition salt which is usually isolated for example by crystallization and filtration.
- suitable pharmaceutically acceptable salts include pharmaceutically acceptable metal salts, for example pharmaceutically acceptable alkali-metal or alkaline- earth-metal salts such as sodium, potassium, calcium, or magnesium salts; in particular pharmaceutically acceptable metal salts of one or more carboxylic acid moieties that may be present in the compound of Formula (I).
- Other non-pharmaceutically acceptable salts e.g. trifluoroacetate, may be used, for example in the isolation of compounds of the invention, and are included within the scope of this invention.
- the invention includes within its scope all possible stoichiometric and non- stoichiometric forms of the salts of the compounds of Formula (I).
- a compound of Formula (I) or a salt thereof for the treatment of humans and other mammals, it is normally formulated in accordance with standard pharmaceutical practice as a pharmaceutical composition.
- the calcilytic compounds can be administered by different routes including intravenous, intraperitoneal, subcutaneous, intramuscular, oral, topical (transdermal), or transmucosal administration.
- oral administration is preferred.
- the compounds can be formulated into conventional oral dosage forms such as capsules, tablets, and liquid preparations such as syrups, elixirs, and concentrated drops.
- the calcilytic compounds can be administered by injection (parenteral administration), e.g., for intramuscular, intravenous, intraperitoneal, and subcutaneous administration.
- parenteral administration e.g., for intramuscular, intravenous, intraperitoneal, and subcutaneous administration.
- the compounds of the invention are formulated in liquid solutions, preferably, in physiologically compatible buffers or solutions, such as saline solution, Hank's solution, or Ringer's solution.
- the compounds may be formulated in solid form and redissolved or suspended immediately prior to use. Lyophilized forms can also be produced.
- Systemic administration can also be by transmucosal or transdermal means. For transmucosal or transdermal administration, penetrants appropriate to the barrier to be permeated are used in the formulation.
- penetrants are generally known in the art, and include, for example, for transmucosal administration, bile salts and fusidic acid derivatives.
- detergents may be used to facilitate permeation.
- Transmucosal administration for example, may be through nasal sprays, rectal suppositories, or vaginal suppositories.
- the compounds of the invention can be formulated into ointments, salves, gels, or creams, as is generally known in the art.
- compositions which comprise a compound of Formula (I) and salts, solvates, and the like, and one or more pharmaceutically acceptable carriers, diluents, or excipients.
- the compounds of Formula (I) and salts, solvates, etc. are as described above.
- the carrier(s), diluent(s), or excipient(s) must be acceptable in the sense of being compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- a process for the preparation of a pharmaceutical formulation including admixing a compound of Formula (I), or salts, solvates, etc., with one or more pharmaceutically acceptable carriers, diluents or excipients.
- pro-drugs examples include Drugs of Today, Volume 19, Number 9, 1983, pp 499 - 538 and in Topics in Chemistry, Chapter 31 , pp 306 - 316 and in "Design of Prodrugs" by H. Bundgaard, Elsevier, 1985, Chapter 1 (the disclosures in which documents are incorporated herein by reference). It will further be appreciated by those skilled in the art, that certain moieties, known to those skilled in the art as “pro-moieties”, for example as described by H. Bundgaard in "Design of Prodrugs” (the disclosure in which document is incorporated herein by reference) may be placed on appropriate functionalities when such functionalities are present within compounds of the invention.
- Preferred "pro-moieties" for compounds of the invention include ester, carbonate ester, hemi-ester, phosphate ester, nitro ester, sulfate ester, sulfoxide, amide, carbamate, azo-, phosphamide, glycoside, ether, acetal, and ketal derivatives of the compounds of Formula (I).
- the amounts of various calcilytic compounds to be administered can be determined by standard procedures taking into account factors such as the compound IC 50 , EC50, the biological half-life of the compound, the age, size, and weight of the patient, and the disease or disorder associated with the patient. The importance of these and other factors to be considered are known to those of ordinary skill in the art. Amounts administered also depend on the routes of administration and the degree of oral bioavailability. For example, for compounds with low oral bioavailability, relatively higher doses will have to be administered.
- the composition is in unit dosage form.
- a tablet, or capsule may be administered, for nasal application, a metered aerosol dose may be administered, for transdermal application, a topical formulation or patch may be administered and for transmucosal delivery, a buccal patch may be administered.
- dosing is such that the patient may administer a single dose.
- Each dosage unit for oral administration contains suitably from 0.01 to 500 mg/kg, and preferably from 0.1 to 50 mg/kg, of a compound of Formula (I) or a salt thereof, calculated as the free base.
- the daily dosage for parenteral, nasal, oral inhalation, transmucosal or transdermal routes contains suitably from 0.01 mg to 100 mg/kg, of a compound of Formula (I).
- a topical formulation contains suitably 0.01 to 5.0% of a compound of Formula (I).
- the active ingredient may be administered, for example, from 1 to 6 times per day, preferably once, sufficient to exhibit the desired activity, as is readily apparent to one skilled in the art.
- treatment includes, but is not limited to prevention, retardation, and prophylaxis of the disease.
- Diseases and disorders which might be treated or prevented, based upon the affected cells include bone and mineral-related diseases or disorders; hypoparathyroidism; those of the central nervous system such as seizures, stroke, head trauma, spinal cord injury, hypoxia-induced nerve cell damage, such as occurs in cardiac arrest or neonatal distress, epilepsy, neurodegenerative diseases such as Alzheimer's disease, Huntington's disease and Parkinson's disease, dementia, muscle tension, depression, anxiety, panic disorder, obsessive-compulsive disorder, post-traumatic stress disorder, schizophrenia, neuroleptic malignant syndrome, and Tourette's syndrome; diseases involving excess water reabsorption by the kidney, such as syndrome of inappropriate ADH secretion (SIADH), cirrhosis, congestive heart failure, and nephrosis; hypertension; preventing and/or decreasing renal toxicity from cationic antibiotics (e.g., aminoglycoside antibiotics); gut motility disorders such as diarrhea and spastic colon; GI ulcer diseases; GI diseases with excessive calcium absorption such as s
- the present compounds are used to increase serum parathyroid hormone ("PTH") levels.
- PTH serum parathyroid hormone
- Increasing serum PTH levels can be helpful in treating diseases such as hypoparathyroidism, osteosarcoma, periodontal disease, fracture, osteoarthritis, rheumatoid arthritis, Paget's disease, humoral hypercalcemia malignancy, osteopenia, and osteoporosis.
- Another aspect of the present invention describes a method of treating a human comprising administering to said human a therapeutically effective amount of a compound of Formula (I), (Ia), or (Ib) to increase the serum PTH level.
- the method is carried out by administering an amount of the compound effective to cause an increase in duration and/or quantity of serum PTH level sufficient to have a therapeutic effect.
- the compound administered to a patient causes an increase in serum PTH for a period of time of up to one hour, about one to about twenty- four hours, about one to about twelve hours, about one to about six hours, about one to about five hours, about one to about four hours, about two to about five hours, about two to about four hours, or about three to about six hours.
- the compound administered to a patient causes an increase in serum PTH for a period of more than about twenty- four hours provided that it is co-administered with an anti resorptive agent.
- the compound administered to a patient causes an increase in serum PTH of up to two fold, two to five fold, five to ten fold, and at least 10 fold, greater than peak serum PTH in the patient.
- the peak serum level is measured with respect to a patient not undergoing treatment.
- compounds of Formula (I) are co-administered with an anti-resorptive agent.
- Suitable anti-resorptive agents for co- administration include, but are not limited to, estrogens, l ⁇ ,25 -(O H) 2 D 3 , Ia-(OH)D 3 , calcitonin, denosumab, selective estrogen receptor modulators, vitronectin receptor antagonists, V-H+-ATPase inhibitors, src SH2 antagonists, bisphosphonates and cathepsin K inhibitors.
- selective estrogen receptor modulators which can be used in combination with compounds of Formula (I) include, but are not limited to, lasofoxifene (Oporia ® /Fablyn ® ), raloxifene (Evista ® ), arzoxifene, apeledoxifene, ospemifene, Chiesi's CHF-4227, and Prostrakan's PSK-3471.
- bisphosphonates which can be used in combination with compounds of Formula (I) include, but are not limited to, tiludronate (Skelid ® ), clondronate (Bonefos ® ), etidronate (Didronel ® ), alendronate (Fosamax ® ), risedronate (Actonel ® ), ibandronate (Boniva ® ), zoledronate (Zometa ® ), minodronate (Onobis ® ), neridronate, and pamidronate.
- estrogens which can be used in combination with compounds of Formula (I) include, but are not limited to, estradiol, conjugated equine estrogens (Premarin ® ), or other estrogens.
- cathepsin K inhibitors which can be used in combination with compounds of Formula (I) include, but are not limited to, Novartis's AAE-581, balicatib, GlaxoSmithKline's SB-462795 and odanacatib.
- the calcitonin that is used in combination with compounds of Formula (I) may be used as an injectable or intranasal formulation, such as Miacalcin ® , Miacalcic ® , Calcitonia ® , Fortical ® , or Elcitonin ® , or as an oral formulation, such as Novartis' SMC-021, Bone Medical's BN-002 (Capsitonin ® ), or Nobex's NCT-025 (Oratonin ® ).
- an injectable or intranasal formulation such as Miacalcin ® , Miacalcic ® , Calcitonia ® , Fortical ® , or Elcitonin ®
- an oral formulation such as Novartis' SMC-021, Bone Medical's BN-002 (Capsitonin ® ), or Nobex's NCT-025 (Oratonin ® ).
- Composition of Formula (I) and their pharmaceutically acceptable salts, which are active when given orally, can be formulated as syrups, tablets, capsules and lozenges.
- a syrup formulation will generally consist of a suspension or solution of the compound or salt in a liquid carrier for example, ethanol, peanut oil, olive oil, glycerine or water with a flavoring or coloring agent.
- a liquid carrier for example, ethanol, peanut oil, olive oil, glycerine or water with a flavoring or coloring agent.
- any pharmaceutical carrier routinely used for preparing solid formulations may be used. Examples of such carriers include magnesium stearate, terra alba, talc, gelatin, acacia, stearic acid, starch, lactose and sucrose.
- composition is in the form of a capsule
- any routine encapsulation is suitable, for example using the aforementioned carriers in a hard gelatin capsule shell.
- composition is in the form of a soft gelatin shell capsule
- any pharmaceutical carrier routinely used for preparing dispersions or suspensions may be considered, for example aqueous gums, celluloses, silicates or oils, and are incorporated in a soft gelatin capsule shell.
- Typical parenteral compositions consist of a solution or suspension of a compound or salt in a sterile aqueous or non-aqueous carrier optionally containing a parenterally acceptable oil, for example polyethylene glycol, polyvinylpyrrolidone, lecithin, arachis oil or sesame oil.
- a parenterally acceptable oil for example polyethylene glycol, polyvinylpyrrolidone, lecithin, arachis oil or sesame oil.
- compositions for inhalation are in the form of a solution, suspension or emulsion that may be administered as a dry powder or in the form of an aerosol using a conventional propellant such as dichlorodifluoromethane or trichlorofluoromethane.
- a typical suppository formulation comprises a compound of Formula (I) or a salt thereof which is active when administered in this way, with a binding and/or lubricating agent, for example polymeric glycols, gelatins, cocoa-butter or other low melting vegetable waxes or fats or their synthetic analogs.
- a binding and/or lubricating agent for example polymeric glycols, gelatins, cocoa-butter or other low melting vegetable waxes or fats or their synthetic analogs.
- Typical dermal and transdermal formulations comprise a conventional aqueous or non-aqueous vehicle, for example a cream, ointment, lotion or paste or are in the form of a medicated plaster, patch or membrane.
- the composition is in unit dosage form, for example a tablet, capsule or metered aerosol dose, so that the patient may administer a single dose.
- the invention includes the use of compounds of the invention for the preparation of a composition for treating or ameliorating bone and mineral-related diseases or disorders in a subject in need thereof, wherein the composition comprises a mixture of one or more of the compounds of the invention and an optional pharmaceutically acceptable carrier.
- the invention further includes the use of compounds of the invention as an active therapeutic substance, in particular in the treatment of bone and mineral-related diseases or disorders.
- the invention includes the use of compounds of the invention in the treatment of hypoparathyroidism, osteosarcoma, periodontal disease, fracture, osteoarthritis, rheumatoid arthritis, Paget's disease, humoral hypercalcemia malignancy, osteopenia, and osteoporosis.
- the invention includes the use of compounds of the invention in the manufacture of a medicament for use in the treatment of the above disorders. No unacceptable toxo logical effects are expected when compounds of the present invention are administered in accordance with the present invention.
- Calcilytic activity was measured by determining the IC 50 of the test compound for blocking increases of intracellular Ca ⁇ + elicited by extracellular Ca ⁇ + in HEK 293 4.0-7 cells stably expressing the human calcium receptor.
- HEK 293 4.0-7 cells were constructed as described by Rogers et al, J. Bone Miner. Res. 10 Suppl. 1 :S483, 1995 (hereby incorporated by reference herein).
- Intracellular Ca ⁇ + increases were elicited by increasing extracellular Ca ⁇ + from 1 to 1.75 mM.
- Intracellular Ca ⁇ + was measured using fluo-3, a fluorescent calcium indicator.
- the medium was decanted and the cell monolayer was washed twice with phosphate-buffered saline (PBS) kept at 37 0 C. After the second wash, 6 mL of 0.02% EDTA in PBS was added and incubated for 4 min. at 37 0 C. Following the incubation, cells were dispersed by gentle agitation.
- PBS phosphate-buffered saline
- Sulfate- and phosphate-free parathyroid cell buffer contains 20 mM Na-Hepes, pH 7.4, 126 mM NaCl, 5 mM KCl, and 1 mM MgC ⁇ .
- SPF-PCB was made up and stored at 4 0 C. On the day of use, SPF-PCB was supplemented with 1 mg/mL of D-glucose and 1 mM CaCl2 and then split into two fractions. To one fraction, bovine serum albumin (BSA; fraction V, ICN) was added at 5 mg/mL (SPF-PCB+). This buffer was used for washing, loading and maintaining the cells. The BSA-free fraction was used for diluting the cells in the cuvette for measurements of fluorescence.
- BSA bovine serum albumin
- the pellet was resuspended in 10 mL of SPF-PCB+ containing 2.2 ⁇ M fluo-3 (Molecular Probes) and incubated at room temperature for 35 min..
- test compound or vehicle as a control
- Calcilytic compounds were detected by their ability to block, in a concentration-dependent manner, increases in the concentration of intracellular Ca ⁇ + elicited by extracellular Ca ⁇ +.
- Method B 48 hours prior to running the CaSR assay, frozen HEK293 CaRec4.0-cl7 cells are thawed, counted, and diluted to 3e5/mL (15K/50 ⁇ L).
- the cell medium used to dilute the cells consists of DMEM/F12 (HAM'S) 1 :1 with L-Glutamine, 15mM HEPES, phenol red, 10% Fetal Bovine Serum, and 1% Penicillin-Streptomycin solution.
- Cell solution is seeded at 15K cells/50 ⁇ L/well in Greiner Poly-D-Lysine coated 384well, black, clear bottom, tissue culture plates and left at room temperature for one hour to reduce edge effect. After the first hour at room temperature, cell plates are then placed into a 37 0 C, 5% CO 2 incubator for 48 hours.
- Dye load buffer consists of Hank's Buffered Saline Solution with .75 mM Calcium, without Magnesium, without Sodium Bicarbonate, with 20 mM Hepes, Probenecid (2.5 mM final concentration), Fluo4 (2 ⁇ M final concentration), and Brilliant Black (500 ⁇ M final concentration).
- Compound dilution buffer consists of Hank's Buffered Saline Solution without Calcium, without Magnesium (without Sodium Bicarbonate) and CHAPS (0.01% final concentration).
- a ligand curve plate is also prepared fresh.
- a 16 pt curve is dispensed into a Greiner 384 well polypropylene plate.
- the top concentration of the ligand, CaCl is 2.875 mM (final concentration) and the lowest concentration is .375 mM (final concentration).
- An ECgo value is generated from the curve data.
- the CaSR assay begins when the cell media is aspirated from the cell plate using a Tecan Plate Washer, leaving nothing but the cell monolayer.
- Dye Load Buffer is added to the cell plate at 20 ⁇ L/well using a Multidrop and the loaded plate incubates for 45 min. at 37 0 C, 5% CO 2 .
- Compound plates are received with 1 ⁇ L of compound stamped at 5 mM- top concentration (25 ⁇ M final cone, in cell plate).
- Compound Dilution Buffer is added to columns 1-24 in the compound plate at 65 ⁇ L/well using a Multidrop.
- Column 6 is pre- stamped with 1 ⁇ L DMSO to represent the high control, and column 18 receives 65 ⁇ L of buffer as the low control.
- the compound addition takes place on a Cybi Well dispenser when 10 ⁇ L of diluted compound is added to the dye loaded cell plate. The cell plate with compound is then incubated at room temperature for 5 min..
- the Antagonist addition takes place on the FLIPR when 10 ⁇ L of ECso challenge is added to the cell plate and fluorescence imagining proceeds for 65 sec.
- Column 18 of the ECso challenge plate contains only buffer to represent a low control or tool antagonist.
- HEK 293 4.0-7 cells stably transfected with the Human Parathyroid Calcium Receptor (“HuPCaR”) were scaled up in T 180 tissue culture flasks.
- Plasma membrane is obtained by polytron homogenization or glass douncing in buffer (50 mM Tris-HCl pH 7.4, 1 mM EDTA, 3 mM MgCl2) in the presence of a protease inhibitor cocktail containing 1 ⁇ M Leupeptin, 0.04 ⁇ M Pepstatin, and 1 mM PMSF. Aliquoted membrane was snap frozen and stored at -80 0 C. 3 H labeled compound was radiolabeled to a radiospecific activity of 44 Ci/mmole and was aliquoted and stored in liquid nitrogen for radiochemical stability.
- a typical reaction mixture contains 2 nM -1H compound ((R,R)-N-4'-Methoxy-t-3- 3'-methyl-r-ethylphenyl-l-(l-naphthyl)ethylamine), or 3 H compound (R)-N- [2-Hy droxy- 3-(3-chloro-2-cyanophenoxy)propyl]- 1 , 1 -dimethyl-2-(4-methoxyphenyl)ethylamine 4-10 ug membrane in homogenization buffer containing 0.1% gelatin and 10% EtOH in a reaction volume of 0.5 mL. Incubation is performed in 12 x 75 polyethylene tubes in an ice water bath.
- Method B Ligand binding assays were performed in a 96-well filtration plate assembly
- membrane -bound radio labeled ligand was separated from the unbound by filtration using a Millipore filtration manifold, followed by washing with ice-cold buffer (2 X 0.1 mL). Bound radioactivity remaining on the filters was determined in 1 mL Bio-Safe (Research Products international Corp. Order No. 111195) in a liquid scintillation counter. The data was analyzed with GraphPad PRISM software.
- Examples 1-20 were tested according to the above assay conditions and each demonstrated an IC50 of ⁇ 1 ⁇ M.
- the compound of example 8 demonstrated an IC50 in a ligand binding assay of approximately 0.009 ⁇ M.
- the compounds of this invention may be made by a variety of methods, including standard chemistry. Any previously defined variable will continue to have the previously defined meaning unless otherwise indicated. Illustrative general synthetic methods are set out below and then specific compounds of the invention as prepared are given in the examples.
- Nuclear magnetic resonance spectra were recorded at either 300 or 400 MHz using, respectively, a Bruker ARX 300 or Bruker AVANCE 400 spectrometer.
- DMSO- ⁇ f ⁇ is hexadeuteriodimethylsulfoxide and CHLOROFORM -d is deuteriochloroform. Chemical shifts are reported in parts per million ( ⁇ ) downfield from the internal standard tetramethylsilane.
- FTIR Fourier transform infrared
- FTIR spectra were recorded on a Nicolet 510 infrared spectrometer.
- FTIR spectra were recorded in transmission mode, and band positions are reported in inverse wavenumbers (cm'l).
- Mass spectra were taken on either a SCIEX5 or Micromass instruments, using electrospray (ES) ionization techniques. Elemental analyses were obtained using a Perkin-Elmer 240C elemental analyzer. Melting points were taken on a Thomas-Hoover melting point apparatus and are uncorrected. All temperatures are reported in degrees Celsius.
- ODS refers to an octadecylsilyl derivatized silica gel chromatographic support. 5 ⁇ Apex-ODS indicates an octadecylsilyl derivatized silica gel chromatographic support having a nominal particle size of 5 ⁇ , made by Jones Chromatography, Littleton, Colorado.
- YMC ODS-AQ ® is an ODS chromatographic support and is a registered trademark of YMC Co. Ltd., Kyoto, Japan.
- PRP- 1 ® is a polymeric (styrene-divinylbenzene) chromatographic support, and is a registered trademark of Hamilton Co., Reno, Nevada.
- Celite ® is a filter aid composed of acid- washed diatomaceous silica, and is a registered trademark of Manville Corp., Denver, Colorado.
- the solution was degassed with N 2 for 10 min., at which time 2-(tributylstannyl)pyridine (3.10 g, 7.58 mmol) was added to the tube.
- the vessel was sealed and heated tol 10 0 C for 16 h.
- the reaction mixture was allowed to cool to rt, diluted with 10% aq. KF (50 mL), and stirred for 30 min, at which time the suspension was filtered through a pad of Celite ® which was then rinsed with EtOAc and water.
- the layers were separated and the aqueous layer was back-extracted with EtOAc (2 x 25 mL).
- the combined organic layers were washed with Brine (1 x 25 mL).
- the vessel was heated with a heat gun until just before reflux and stirred for 30 min. as white crystals crashed out.
- the crystals were filtered, rinsed with EtOAc, and dried with airflow for 30 min. to provide the title compound (1.51 g, 60.2%).
- Tablet Formulation An exemplary tablet formulation is formed by tableting the following mixture:
- a pharmaceutical composition for parenteral administration is prepared by dissolving an appropriate amount of a compound of Formula (I) in polyethylene glycol with heating. This solution is then diluted with water for injections (to 100 mL). The solution is then rendered sterile by filtration through a 0.22 micron membrane filter and sealed in sterile containers.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Novel calcilytic compounds, pharmaceutical compositions, methods of synthesis and methods of using them are provided.
Description
CALCILYTIC COMPOUNDS FIELD OF THE INVENTION
The present invention relates to novel calcilytic compounds, pharmaceutical compositions containing these compounds, processes for their preparation and their use as calcium receptor antagonists.
BACKGROUND OF THE INVENTION
In mammals, extracellular Ca^+ is under rigid homeostatic control and regulates various processes such as blood clotting, nerve and muscle excitability, and proper bone formation. Extracellular Ca^+ inhibits the secretion of parathyroid hormone ("PTH") from parathyroid cells, inhibits bone resorption by osteoclasts, and stimulates secretion of calcitonin from C-cells. Calcium receptor proteins enable certain specialized cells to respond to changes in extracellular Ca^+ concentration.
PTH is the principal endocrine factor regulating Ca^+ homeostasis in the blood and extracellular fluids. PTH, by acting on bone and kidney cells, increases the level Of Ca^+ in the blood. This increase in extracellular Ca^+ then acts as a negative feedback signal, depressing PTH secretion. The reciprocal relationship between extracellular Ca^+ and PTH secretion forms an important mechanism maintaining bodily Ca^+ homeostasis. Extracellular Ca^+ acts directly on parathyroid cells to regulate PTH secretion.
The existence of a parathyroid cell surface protein which detects changes in extracellular Ca 2+ has been confirmed. See Brown et al., Nature 366:574, 1993. In parathyroid cells, this protein, the calcium receptor, acts as a receptor for extracellular Ca 2+ , detects changes in the ion concentration of extracellular Ca^+, and initiates a functional cellular response,
PTH secretion.
Extracellular Ca^+ influences various cell functions, reviewed in Nemeth et al.,
Cell Calcium 11 :319, 1990. For example, extracellular Ca^+ plays a role in parafollicular
(C-cells) and parathyroid cells. See Nemeth, Cell Calcium 11 :323, 1990. The role of extracellular Ca^+ on bone osteoclasts has also been studied. See Zaidi, Bioscience
Reports 10:493, 1990.
Various compounds are known to mimic the effects of extra-cellular Ca^+ on a calcium receptor molecule. Calcilytics are compounds able to inhibit calcium receptor activity, thereby causing a decrease in one or more calcium receptor activities evoked by extracellular Ca^+. Calcilytics are useful as lead molecules in the discovery, development, design, modification and/or construction of useful calcium modulators, which are active at Ca^+ receptors. Such calcilytics are useful in the treatment of various disease states characterized by abnormal levels of one or more components, e.g., polypeptides such as hormones, enzymes or growth factors, the expression and/or secretion of which is regulated or affected by activity at one or more Ca^+ receptors. Target diseases or disorders for calcilytic compounds include diseases involving abnormal bone and mineral homeostasis.
Abnormal calcium homeostasis is characterized by one or more of the following activities: an abnormal increase or decrease in serum calcium; an abnormal increase or decrease in urinary excretion of calcium; an abnormal increase or decrease in bone calcium levels (for example, as assessed by bone mineral density measurements); an abnormal absorption of dietary calcium; an abnormal increase or decrease in the production and/or release of messengers which affect serum calcium levels such as PTH and calcitonin; and an abnormal change in the response elicited by messengers which affect serum calcium levels. Thus, calcium receptor antagonists offer a unique approach towards the pharmacotherapy of diseases associated with abnormal bone or mineral homeostasis, such as hypoparathyroidism, osteosarcoma, periodontal disease, bone fracture, osteoarthritis, rheumatoid arthritis, Paget's disease, humoral hypercalcemia associated with malignancy and bone fracture, osteopenia, and osteoporosis.
SUMMARY OF THE INVENTION
The present invention involves novel compounds according to Formula (I):

R1 is a 6-membered heteroaryl group, containing 1-2 nitrogen atoms, optionally substituted by -O-(Ci-C2)alkyl-O-, or one to three times, independently, by halogen, (Ci-C4)alkyl, -CF3, amino, (Ci-C4)alkylamino, (Ci-C4)alkyl(Ci-C4)alkylamino, (Ci-C4)alkoxy, hydroxy(Ci-C4)alkyl, or (Ci-C4)alkoxy(Ci-C4)alkyl; R2 is hydrogen or (Ci-C4)alkyl; wherein R1 is located at either the 4- or 5 -position of the thiophene ring and when R1 is located at the 4-position, R2 is located at the 5 -position, and when R1 is located at the 5 -position, R2 is located at the 4-position;
R3 is selected from the group consisting of (Ci-C4)alkyl, phenyl, and heteroaryl, optionally substituted, one to three times, independently, by halogen, (Ci-C4)alkyl, -CF3, or (C1-C4)alkoxy;
R4 is selected from the group consisting of (C5-C6)cycloalkyl(Ci-C4)alkyl, heterocycloalkyl(Ci-C4)alkyl, aryl(Ci-C4)alkyl, and heteroaryl(Ci-C4)alkyl, wherein any cycloalkyl, heterocycloalkyl, aryl, or heteroaryl group is optionally substituted by -O-(Ci-C2)alkyl-O-, or one to three times, independently, by halogen, (Ci-C4)alkyl, -CF3, amino, (Ci-C4)alkylamino, (Ci-C4)alkyl(Ci-C4)alkylamino, or (Ci-C4)alkoxy;
R5 is hydrogen or fluorine; or a salt thereof.
The present invention is also directed to formulations comprising compounds of Formula (I), or a salt thereof, and their use as calcium receptor antagonists in the treatment of a variety of diseases associated with abnormal bone or mineral homeostasis, including but not limited to hypoparathyroidism, osteosarcoma, periodontal disease, bone fracture, osteoarthritis, rheumatoid arthritis, Paget' s disease, humoral hypercalcemia associated with malignancy and bone fracture, osteopenia, and osteoporosis.
The present invention further provides a method for antagonizing calcium receptors in a mammal, including a human, which comprises administering to a mammal in need thereof an effective amount of a compound of Formula (I), or a salt thereof. The present invention further provides a method for increasing serum parathyroid levels in a mammal, including a human, which comprises administering to a mammal in need thereof an effective amount of a compound of Formula (I), or a salt thereof.
DETAILED DESCRIPTION OF THE INVENTION
As used herein, the term "alkyl" refers to a straight- or branched-chain hydrocarbon radical having the specified number of carbon atoms. As used herein, the term
"(Ci-C4)alkyl" refers to an alkyl group having at least 1 and up to 4 carbon atoms. Examples of such branched or straight-chained alkyl groups useful in the present invention include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, s-butyl, and t-butyl. As used herein, the term "cycloalkyl" refers to a non-aromatic, saturated, cyclic hydrocarbon ring containing the specified number of carbon atoms. The term "Cs-C6 cycloalkyl" refers to a non-aromatic cyclic hydrocarbon ring having from five to six carbon atoms. Exemplary "Cs-C6 cycloalkyl" groups useful in the present invention include cyclopentyl and cyclohexyl. "Alkoxy" means an alkyl radical containing the specified number of carbon atoms attached through an oxygen linking atom. The term "(Ci-C4)alkoxy" refers to a straight- or branched-chain hydrocarbon radical having at least 1 and up to 4 carbon atoms attached through an oxygen linking atom. Exemplary "(Ci-C4)alkoxy" groups useful in the present invention include, but are not limited to, methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, 5-butoxy, and t-butoxy.
"Heterocycloalkyl" means a non-aromatic heterocyclic ring containing 5-6 ring atoms, being saturated or having one or more degrees of unsaturation, and containing one or more heteroatom substitutions selected from O, S, and/or N. Examples of "heterocycloalkyl" moieties include, but are not limited to, tetrahydrofuranyl, dihydropyranyl, tetrahydropyranyl, 1 ,4-dioxanyl, 1,3-dioxanyl, piperidinyl, piperazinyl, 2,4-piperazinedionyl, pyrrolidinyl, pyrrolinyl, imidazolidinyl, pyrazolidinyl, pyrazolinyl, morpholinyl, thiomorpholinyl, tetrahydrothiopyranyl, tetrahydrothienyl, and the like.
"Aryl" refers to optionally substituted monocyclic or fused polycarbocyclic groups having 6 to 14 carbon atoms and having at least one aromatic ring that complies with Hϋckel's Rule. Examples of "aryl" groups are phenyl, naphthyl, anthracenyl, phenanthrenyl, and the like. Preferably aryl refers to optionally substituted phenyl.
"Heteroaryl" means an optionally substituted aromatic monocyclic ring containing 5-6 ring atoms that complies with Hϋckel's Rule and contains at least one and up to three heteroatoms independently selected from N, O, and/or S. Examples of 5-membered "heteroaryl" groups include furanyl, thienyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, thiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiadiazolyl, and isothiazolyl. Examples of 6-membered "heteroaryl" groups include oxo-pyridyl, pyridinyl, pyridazinyl, pyrazinyl, and pyrimidinyl.
When any group or moiety, such as alkyl, aryl, cycloalkyl, heterocycloalkyl, or heteroaryl, is defined herein as being "optionally substituted, one to two (or three) times, independently, by" the recited substituents, it is to be understood that the group or moiety is unsubstituted or is substituted by one to two (or three) substituents, wherein each substituent is independently selected from the recited substituents.
As used herein, "halogen" or "halo" refers to F, Cl, Br, or I. In one embodiment of this invention, R1 is selected from the group consisting of pyridinyl, pyrazinyl, and pyrimidinyl, optionally substituted, one to three times, independently, by (Ci-C4)alkyl, -CF3, amino, (Ci-C4)alkylamino, (Ci-C4)alkyl(Ci-C4)alkylamino, (Ci-C4)alkoxy, hydroxy(Ci-C4)alkyl, or (Ci-C4)alkoxy(Ci-C4)alkyl.
In another embodiment, R1 is pyridinyl, optionally substituted, one to two times, independently, by (Ci-C4)alkyl, -CF3, (Ci-C4)alkoxy, hydroxy(Ci-C4)alkyl, or (Ci-C4)alkoxy(Ci-C4)alkyl.
In a specific embodiment, R1 is 2-pyridinyl, 3-methyl-2-pyridinyl, 6-methyl-2- pyridinyl, 6-[(methyloxy)methyl]-2 -pyridinyl, 3-pyridinyl, 6-methyl-3 -pyridinyl, 6- trifluoromethyl-3 -pyridinyl, 6-hydroxymethyl-3 -pyridinyl, 4-pyridinyl, 2-pyrimidinyl, 5- pyrimidinyl, or 2-pyrazinyl. In a selected embodiment, R1 is 2-pyridinyl.
In a further embodiment, R2 is hydrogen or methyl.
In another embodiment, R3 is (Ci-C4)alkyl. In a specific embodiment, R3 is methyl. In a yet another embodiment, R4 is a phenyl(Ci-C2)alkyl group, optionally substituted by -O-(Ci-C2)alkyl-O-, or one to two times, independently, by F, Cl, (Ci-C4)alkyl, (Ci-C4)alkoxy, or -CF3. Specifically, R4 is phenethyl, optionally substituted by -O-(Ci-C2)alkyl-O-, or one to two times, independently, by F, Cl, (Ci-C4)alkyl, (Ci-C4)alkoxy, or -CF3. In a selected embodiment, R4 is phenethyl. In a specific embodiment, R5 is fluorine.
One particular embodiment of the invention is a compound of Formula (I) wherein:
R1 is selected from the group consisting of pyridinyl, pyrazinyl, and pyrimidinyl, optionally substituted, one to three times, independently, by (Ci-C4)alkyl, -CF3, amino, (Ci-C4)alkylamino, (Ci-C4)alkyl(Ci-C4)alkylamino, (Ci-C4)alkoxy, hydroxy(Ci-C4)alkyl, or (Ci-C4)alkoxy(Ci-C4)alkyl;
R2 is hydrogen or (Ci-C4)alkyl;
R3 is (Ci-C4)alkyl;
R4 is phenethyl, optionally substituted by -O-(Ci-C2)alkyl-O-, or one to two times, independently, by F, Cl, (Ci-C4)alkyl, (Ci-C4)alkoxy, or -CF3; R5 is hydrogen or fluorine; or a salt thereof.
Another particular embodiment of the invention is a compound of Formula (I) wherein:
R1 is pyridinyl, optionally substituted, one to two times, independently, by (Ci-C4)alkyl, -CF3, (Ci-C4)alkoxy, hydroxy(Ci-C4)alkyl, or (Ci-C4)alkoxy(Ci-C4)alkyl;
R2 is hydrogen or methyl;
R is methyl;
R4 is phenethyl;
R5 is fluorine; or a salt thereof.
It is to be understood that the phrase "when R1 is located at the 4-position, R2 is located at the 5-position" means that the intended structure is that of a compound of Formula (Ia):

It is also to be understood that the phrase "when R1 is located at the 5-position, R2 is located at the 4-position" means that the intended structure is that of a compound of Formula (Ib):

2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[5-(2-pyridinyl)-2- thienyl] -4(3H)-pyrimidinone;
2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-[4-methyl-5-(2-pyridinyl)-2-thienyl]-3- (2-phenylethyl)-4(3H)-pyrimidinone;
2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[4-(2-pyridinyl)-2- thienyl] -4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 - [5 -methyl-4-(2-pyridinyl)-2-thienyl] -3 - (2-phenylethyl)-4(3H)-pyrimidinone; 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-[5-(3-methyl-2-pyridinyl)-2-thienyl]-3-
(2-phenylethyl)-4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 - [4-methyl-5 -(3 -methyl-2-pyridinyl)-2- thienyl]-3-(2-phenylethyl)-4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 - [5 -methyl-4-(3 -methyl-2-pyridinyl)-2- thienyl]-3-(2-phenylethyl)-4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 - [5 -(6-methyl-2-pyridinyl)-2-thienyl]-3 - (2-phenylethyl)-4(3H)-pyrimidinone;
2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[5-(3-pyridinyl)-2- thienyl] -4(3H)-pyrimidinone; 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-[5-(6-methyl-3-pyridinyl)-2-thienyl]-3-
(2-phenylethyl)-4(3H)-pyrimidinone;
2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-{5-[6- (trifluoromethyl)-3-pyridinyl]-2-thienyl}-4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-5 - {5 - [6-(hydroxymethyl)-3 -pyridinyl] -2-thienyl} -6- methyl-3 -(2-phenylethyl)-4(3H)-pyrimidinone;
2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[5-(4-pyridinyl)-2- thienyl] -4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-3 -(2-phenylethyl)-5 - [5 -(2-pyrimidinyl)-2- thienyl] -4(3H)-pyrimidinone; 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[4-(2-pyrimidinyl)-2- thienyl] -4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-3 -(2-phenylethyl)-5 - [5 -(5 -pyrimidinyl)-2- thienyl] -4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-3 -(2-phenylethyl)-5 - [4-(5 -pyrimidinyl)-2- thienyl] -4(3H)-pyrimidinone;
2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[5-(2-pyrazinyl)-2- thienyl] -4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 -(5 -methyl-4- {6- [(methyloxy)methyl] -2- pyridinyl} -2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone; and 2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 -(4-methyl-5 - {6- [(methyloxy)methyl] -2- pyridinyl}-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone.
The term "solvate" refers to a complex of variable stoichiometry formed by a solute and a solvent. Such solvents for the purpose of the invention may not interfere with the biological activity of the solute. Examples of suitable solvents include, but are not limited to, water, MeOH, EtOH and AcOH. Preferably, the solvent used is a pharmaceutically acceptable solvent. Examples of suitable pharmaceutically acceptable solvents include, without limitation, water, EtOH and AcOH. Solvates wherein water is the solvent molecule are typically referred to as "hydrates". Hydrates include compositions containing stoichiometric amounts of water, as well as compositions containing variable amounts of water. Solvates, particularly hydrates, of the compounds of Formula (I), (Ia), and (Ib), and salts thereof, are within the scope of the invention.
When a disclosed compound or its salt is named or depicted by structure, it is to be understood that the compound or salt, including solvates (particularly, hydrates) thereof, may exist in crystalline forms, non-crystalline forms or a mixture thereof. The compound or salt, or solvates (particularly, hydrates) thereof, may also exhibit polymorphism (i.e. the capacity to occur in different crystalline forms). These different crystalline forms are typically known as "polymorphs." It is to be understood that when named or depicted by structure, the disclosed compound, or solvates (particularly, hydrates) thereof, also include all polymorphs thereof. Polymorphs have the same chemical composition but differ in packing, geometrical arrangement, and other descriptive properties of the crystalline solid state. Polymorphs, therefore, may have different physical properties such as shape, density, hardness, deformability, stability, and dissolution properties. Polymorphs typically exhibit different melting points, IR spectra, and X-ray powder diffraction patterns, which may be used for identification. One of ordinary skill in the art will
appreciate that different polymorphs may be produced, for example, by changing or adjusting the conditions used in crystallizing/recrystallizing the compound.
The invention also includes various isomers of the compounds of Formula (I), (Ia), and (Ib), and mixtures thereof. "Isomer" refers to compounds that have the same composition and molecular weight but differ in physical and/or chemical properties. The structural difference may be in constitution (geometric isomers) or in the ability to rotate the plane of polarized light (stereoisomers). With regard to stereoisomers, the present compounds may have one or more asymmetric carbon atom and may occur as racemates, racemic mixtures, and as individual enantiomers or diastereomers. All such isomeric forms are included within the present invention, including mixtures thereof.
Because of their potential use in medicine, the salts of the compounds of Formula (I) are preferably pharmaceutically acceptable. Suitable pharmaceutically acceptable salts can include acid or base addition salts.
As used herein, the term "pharmaceutically acceptable" means a compound which is suitable for pharmaceutical use. Salts and solvates (e.g. hydrates and hydrates of salts) of the compounds of the invention which are suitable for use in medicine are those wherein the counterion or associated solvent is pharmaceutically acceptable. However, salts and solvates having non-pharmaceutically acceptable counterions or associated solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of the invention and their pharmaceutically acceptable salts and solvates.
A pharmaceutically acceptable acid addition salt can be formed by reaction of a compound of Formula (I) containing a basic moiety with a suitable inorganic or organic acid (such as acetic, aspartic, benzenesulfonic, benzoic, bicarbonic, camphorsulfonic, carbonic, citric, dodecyl sulfonic, 1 ,2-ethanedisulfonic, ethanesulfonic, formic, fumaric, (3i?,45*,5i?,6i?)-2,3,4,5,6,7-hexahydroxyheptanoic acid, galacturonic, gluconic, glutamic, hexanoic, hydrobromide, hydrochloride, 2-hydroxyethanesulfonic, hydroxynaphthoic, lactic, lactobionic, malic, maleic, mandelic, methanesulfonic, mucic, naphthalene-2- sulfonic, nitric, pamoic, pantoic, phosphoric/diphosphoric, polygalacturonic, propionic, salicylic, stearic, succinic, sulfonic, tannic, tartaric, or/?-toluenesulfonic acid), optionally in a suitable solvent such as an organic solvent, to give the salt which is usually isolated
for example by crystallization and filtration. Pharmaceutically acceptable acid addition salts of a compound of Formula (I) include acetate, aspartate, benzenesulfonate, benzoate, bicarbonate, bitartrate, bromide, calcium edetate, camsylate, carbonate, chloride, citrate, dihydrochloride, edetate, edisylate, estolate, esylate, formate, fumarate, galacturonate, gluceptate, gluconate, glutamate, glycollylarsanilate, hexanoate, hydrobromide, hydrochloride, hydroxynaphthoate, iodide, isethionate, lactate, lactobionate, malate, maleate, mandelate, mesylate, methylsulfate, mucate, napsylate, nitrate, pamoate, pantothenate, phosphate/diphosphate, polygalacturonate, propionate, salicylate, stearate, subacetate, succinate, sulfate, tannate, tartrate, teoclate, or tosylate salts. A pharmaceutically acceptable base addition salt can be formed by reaction of a compound of Formula (I) containing an acidic moiety with a suitable inorganic or organic base (e.g. triethylamine, ethanolamine, triethanolamine, choline, arginine, lysine, or histidine), optionally in a suitable solvent such as an organic solvent, to give the base addition salt which is usually isolated for example by crystallization and filtration. Other suitable pharmaceutically acceptable salts include pharmaceutically acceptable metal salts, for example pharmaceutically acceptable alkali-metal or alkaline- earth-metal salts such as sodium, potassium, calcium, or magnesium salts; in particular pharmaceutically acceptable metal salts of one or more carboxylic acid moieties that may be present in the compound of Formula (I). Other non-pharmaceutically acceptable salts, e.g. trifluoroacetate, may be used, for example in the isolation of compounds of the invention, and are included within the scope of this invention.
The invention includes within its scope all possible stoichiometric and non- stoichiometric forms of the salts of the compounds of Formula (I). In order to use a compound of Formula (I) or a salt thereof for the treatment of humans and other mammals, it is normally formulated in accordance with standard pharmaceutical practice as a pharmaceutical composition.
The calcilytic compounds can be administered by different routes including intravenous, intraperitoneal, subcutaneous, intramuscular, oral, topical (transdermal), or transmucosal administration. For systemic administration, oral administration is preferred. For oral administration, for example, the compounds can be formulated into conventional
oral dosage forms such as capsules, tablets, and liquid preparations such as syrups, elixirs, and concentrated drops.
Alternatively, the calcilytic compounds can be administered by injection (parenteral administration), e.g., for intramuscular, intravenous, intraperitoneal, and subcutaneous administration. For injection, the compounds of the invention are formulated in liquid solutions, preferably, in physiologically compatible buffers or solutions, such as saline solution, Hank's solution, or Ringer's solution. In addition, the compounds may be formulated in solid form and redissolved or suspended immediately prior to use. Lyophilized forms can also be produced. Systemic administration can also be by transmucosal or transdermal means. For transmucosal or transdermal administration, penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants are generally known in the art, and include, for example, for transmucosal administration, bile salts and fusidic acid derivatives. In addition, detergents may be used to facilitate permeation. Transmucosal administration, for example, may be through nasal sprays, rectal suppositories, or vaginal suppositories.
For topical administration, the compounds of the invention can be formulated into ointments, salves, gels, or creams, as is generally known in the art.
While it is possible that, for use in therapy, a compound of Formula (I), as well as salts, solvates and the like, may be administered as a neat preparation, i.e. no additional carrier, the more usual practice is to present the active ingredient confected with a carrier or diluent. Accordingly, the invention further provides pharmaceutical compositions, which comprise a compound of Formula (I) and salts, solvates, and the like, and one or more pharmaceutically acceptable carriers, diluents, or excipients. The compounds of Formula (I) and salts, solvates, etc., are as described above. The carrier(s), diluent(s), or excipient(s) must be acceptable in the sense of being compatible with the other ingredients of the formulation and not deleterious to the recipient thereof. In accordance with another aspect of the invention there is also provided a process for the preparation of a pharmaceutical formulation including admixing a compound of Formula (I), or salts, solvates, etc., with one or more pharmaceutically acceptable carriers, diluents or excipients.
It will be appreciated by those skilled in the art that certain protected derivatives of compounds of Formula (I), which may be made prior to a final deprotection stage, may not possess pharmacological activity as such, but may, in certain instances, be administered orally or parenterally and thereafter metabolized in the body to form compounds of the invention which are pharmacologically active. Such derivatives may therefore be described as "prodrugs". Further, certain compounds of the invention may act as prodrugs of other compounds of the invention. All protected derivatives and prodrugs of compounds of the invention are included within the scope of the invention. Examples of suitable pro-drugs for the compounds of the present invention are described in Drugs of Today, Volume 19, Number 9, 1983, pp 499 - 538 and in Topics in Chemistry, Chapter 31 , pp 306 - 316 and in "Design of Prodrugs" by H. Bundgaard, Elsevier, 1985, Chapter 1 (the disclosures in which documents are incorporated herein by reference). It will further be appreciated by those skilled in the art, that certain moieties, known to those skilled in the art as "pro-moieties", for example as described by H. Bundgaard in "Design of Prodrugs" (the disclosure in which document is incorporated herein by reference) may be placed on appropriate functionalities when such functionalities are present within compounds of the invention. Preferred "pro-moieties" for compounds of the invention include ester, carbonate ester, hemi-ester, phosphate ester, nitro ester, sulfate ester, sulfoxide, amide, carbamate, azo-, phosphamide, glycoside, ether, acetal, and ketal derivatives of the compounds of Formula (I). The amounts of various calcilytic compounds to be administered can be determined by standard procedures taking into account factors such as the compound IC50, EC50, the biological half-life of the compound, the age, size, and weight of the patient, and the disease or disorder associated with the patient. The importance of these and other factors to be considered are known to those of ordinary skill in the art. Amounts administered also depend on the routes of administration and the degree of oral bioavailability. For example, for compounds with low oral bioavailability, relatively higher doses will have to be administered.
Preferably, the composition is in unit dosage form. For oral application, for example, a tablet, or capsule may be administered, for nasal application, a metered aerosol dose may be administered, for transdermal application, a topical formulation or patch may
be administered and for transmucosal delivery, a buccal patch may be administered. In each case, dosing is such that the patient may administer a single dose.
Each dosage unit for oral administration contains suitably from 0.01 to 500 mg/kg, and preferably from 0.1 to 50 mg/kg, of a compound of Formula (I) or a salt thereof, calculated as the free base. The daily dosage for parenteral, nasal, oral inhalation, transmucosal or transdermal routes contains suitably from 0.01 mg to 100 mg/kg, of a compound of Formula (I). A topical formulation contains suitably 0.01 to 5.0% of a compound of Formula (I). The active ingredient may be administered, for example, from 1 to 6 times per day, preferably once, sufficient to exhibit the desired activity, as is readily apparent to one skilled in the art.
As used herein, "treatment" of a disease includes, but is not limited to prevention, retardation, and prophylaxis of the disease.
Diseases and disorders which might be treated or prevented, based upon the affected cells, include bone and mineral-related diseases or disorders; hypoparathyroidism; those of the central nervous system such as seizures, stroke, head trauma, spinal cord injury, hypoxia-induced nerve cell damage, such as occurs in cardiac arrest or neonatal distress, epilepsy, neurodegenerative diseases such as Alzheimer's disease, Huntington's disease and Parkinson's disease, dementia, muscle tension, depression, anxiety, panic disorder, obsessive-compulsive disorder, post-traumatic stress disorder, schizophrenia, neuroleptic malignant syndrome, and Tourette's syndrome; diseases involving excess water reabsorption by the kidney, such as syndrome of inappropriate ADH secretion (SIADH), cirrhosis, congestive heart failure, and nephrosis; hypertension; preventing and/or decreasing renal toxicity from cationic antibiotics (e.g., aminoglycoside antibiotics); gut motility disorders such as diarrhea and spastic colon; GI ulcer diseases; GI diseases with excessive calcium absorption such as sarcoidosis; autoimmune diseases and organ transplant rejection; squamous cell carcinoma; and pancreatitis.
In a preferred embodiment of the present invention, the present compounds are used to increase serum parathyroid hormone ("PTH") levels. Increasing serum PTH levels can be helpful in treating diseases such as hypoparathyroidism, osteosarcoma, periodontal disease, fracture, osteoarthritis, rheumatoid arthritis, Paget's disease, humoral hypercalcemia malignancy, osteopenia, and osteoporosis.
Another aspect of the present invention describes a method of treating a human comprising administering to said human a therapeutically effective amount of a compound of Formula (I), (Ia), or (Ib) to increase the serum PTH level. Preferably, the method is carried out by administering an amount of the compound effective to cause an increase in duration and/or quantity of serum PTH level sufficient to have a therapeutic effect.
In various embodiments, the compound administered to a patient causes an increase in serum PTH for a period of time of up to one hour, about one to about twenty- four hours, about one to about twelve hours, about one to about six hours, about one to about five hours, about one to about four hours, about two to about five hours, about two to about four hours, or about three to about six hours.
In an alternative embodiment of the present invention, the compound administered to a patient causes an increase in serum PTH for a period of more than about twenty- four hours provided that it is co-administered with an anti resorptive agent.
In additional different embodiments, the compound administered to a patient causes an increase in serum PTH of up to two fold, two to five fold, five to ten fold, and at least 10 fold, greater than peak serum PTH in the patient. The peak serum level is measured with respect to a patient not undergoing treatment.
In a selected embodiment of the present invention, compounds of Formula (I) are co-administered with an anti-resorptive agent. Suitable anti-resorptive agents for co- administration include, but are not limited to, estrogens, lα,25 -(O H)2D3, Ia-(OH)D3, calcitonin, denosumab, selective estrogen receptor modulators, vitronectin receptor antagonists, V-H+-ATPase inhibitors, src SH2 antagonists, bisphosphonates and cathepsin K inhibitors.
Examples of selective estrogen receptor modulators which can be used in combination with compounds of Formula (I) include, but are not limited to, lasofoxifene (Oporia®/Fablyn®), raloxifene (Evista®), arzoxifene, bazedoxifene, ospemifene, Chiesi's CHF-4227, and Prostrakan's PSK-3471. Examples of bisphosphonates which can be used in combination with compounds of Formula (I) include, but are not limited to, tiludronate (Skelid®), clondronate (Bonefos®), etidronate (Didronel®), alendronate (Fosamax®), risedronate (Actonel®), ibandronate (Boniva®), zoledronate (Zometa®), minodronate (Onobis®), neridronate, and pamidronate. Examples of estrogens which can be used in
combination with compounds of Formula (I) include, but are not limited to, estradiol, conjugated equine estrogens (Premarin®), or other estrogens. Examples of cathepsin K inhibitors which can be used in combination with compounds of Formula (I) include, but are not limited to, Novartis's AAE-581, balicatib, GlaxoSmithKline's SB-462795 and odanacatib. The calcitonin that is used in combination with compounds of Formula (I) may be used as an injectable or intranasal formulation, such as Miacalcin®, Miacalcic®, Calcitonia®, Fortical®, or Elcitonin®, or as an oral formulation, such as Novartis' SMC-021, Bone Medical's BN-002 (Capsitonin®), or Nobex's NCT-025 (Oratonin®).
Composition of Formula (I) and their pharmaceutically acceptable salts, which are active when given orally, can be formulated as syrups, tablets, capsules and lozenges. A syrup formulation will generally consist of a suspension or solution of the compound or salt in a liquid carrier for example, ethanol, peanut oil, olive oil, glycerine or water with a flavoring or coloring agent. Where the composition is in the form of a tablet, any pharmaceutical carrier routinely used for preparing solid formulations may be used. Examples of such carriers include magnesium stearate, terra alba, talc, gelatin, acacia, stearic acid, starch, lactose and sucrose. Where the composition is in the form of a capsule, any routine encapsulation is suitable, for example using the aforementioned carriers in a hard gelatin capsule shell. Where the composition is in the form of a soft gelatin shell capsule, any pharmaceutical carrier routinely used for preparing dispersions or suspensions may be considered, for example aqueous gums, celluloses, silicates or oils, and are incorporated in a soft gelatin capsule shell.
Typical parenteral compositions consist of a solution or suspension of a compound or salt in a sterile aqueous or non-aqueous carrier optionally containing a parenterally acceptable oil, for example polyethylene glycol, polyvinylpyrrolidone, lecithin, arachis oil or sesame oil.
Typical compositions for inhalation are in the form of a solution, suspension or emulsion that may be administered as a dry powder or in the form of an aerosol using a conventional propellant such as dichlorodifluoromethane or trichlorofluoromethane.
A typical suppository formulation comprises a compound of Formula (I) or a salt thereof which is active when administered in this way, with a binding and/or lubricating
agent, for example polymeric glycols, gelatins, cocoa-butter or other low melting vegetable waxes or fats or their synthetic analogs.
Typical dermal and transdermal formulations comprise a conventional aqueous or non-aqueous vehicle, for example a cream, ointment, lotion or paste or are in the form of a medicated plaster, patch or membrane.
Preferably the composition is in unit dosage form, for example a tablet, capsule or metered aerosol dose, so that the patient may administer a single dose.
The invention includes the use of compounds of the invention for the preparation of a composition for treating or ameliorating bone and mineral-related diseases or disorders in a subject in need thereof, wherein the composition comprises a mixture of one or more of the compounds of the invention and an optional pharmaceutically acceptable carrier.
The invention further includes the use of compounds of the invention as an active therapeutic substance, in particular in the treatment of bone and mineral-related diseases or disorders. In particular, the invention includes the use of compounds of the invention in the treatment of hypoparathyroidism, osteosarcoma, periodontal disease, fracture, osteoarthritis, rheumatoid arthritis, Paget's disease, humoral hypercalcemia malignancy, osteopenia, and osteoporosis.
In another aspect, the invention includes the use of compounds of the invention in the manufacture of a medicament for use in the treatment of the above disorders. No unacceptable toxo logical effects are expected when compounds of the present invention are administered in accordance with the present invention.
The biological activity of the compounds of Formula (I) is demonstrated by the following tests:
(D Calcium Receptor Inhibitor Assay: Method A:
Calcilytic activity was measured by determining the IC50 of the test compound for blocking increases of intracellular Ca^+ elicited by extracellular Ca^+ in HEK 293 4.0-7 cells stably expressing the human calcium receptor. HEK 293 4.0-7 cells were constructed as described by Rogers et al, J. Bone Miner. Res. 10 Suppl. 1 :S483, 1995 (hereby incorporated by reference herein). Intracellular Ca^+ increases were elicited by increasing
extracellular Ca^+ from 1 to 1.75 mM. Intracellular Ca^+ was measured using fluo-3, a fluorescent calcium indicator.
The procedure was as follows:
1. Cells were maintained in T- 150 flasks in selection media (DMEM supplemented with 10% fetal bovine serum and 200 ug/mL hygromycin B), under 5% Cθ2:95% air at 37
0C and were grown up to 90% confluency.
2. The medium was decanted and the cell monolayer was washed twice with phosphate-buffered saline (PBS) kept at 37 0C. After the second wash, 6 mL of 0.02% EDTA in PBS was added and incubated for 4 min. at 37 0C. Following the incubation, cells were dispersed by gentle agitation.
3. Cells from 2 or 3 flasks were pooled and pelleted (100 x g). The cellular pellet was resuspended in 10-15 mL of SPF-PCB+ and pelleted again by centrifugation. This washing was done twice.
Sulfate- and phosphate-free parathyroid cell buffer (SPF-PCB) contains 20 mM Na-Hepes, pH 7.4, 126 mM NaCl, 5 mM KCl, and 1 mM MgC^. SPF-PCB was made up and stored at 4 0C. On the day of use, SPF-PCB was supplemented with 1 mg/mL of D-glucose and 1 mM CaCl2 and then split into two fractions. To one fraction, bovine serum albumin (BSA; fraction V, ICN) was added at 5 mg/mL (SPF-PCB+). This buffer was used for washing, loading and maintaining the cells. The BSA-free fraction was used for diluting the cells in the cuvette for measurements of fluorescence.
4. The pellet was resuspended in 10 mL of SPF-PCB+ containing 2.2 μM fluo-3 (Molecular Probes) and incubated at room temperature for 35 min..
5. Following the incubation period, the cells were pelleted by centrifugation. The resulting pellet was washed with SPF-PCB+. After this washing, cells were resuspended in SPF-PCB+ at a density of 1 -2 x 106 cells/mL.
6. For recording fluorescent signals, 300 μL of cell suspension were diluted in 1.2 mL of SPF buffer containing 1 mM CaCl2 and 1 mg/mL of D-glucose. Measurements of fluorescence were performed at 37 0C with constant stirring using a spectra fluorimeter. Excitation and emission wavelengths were measured at 485 and 535 nm, respectively. To calibrate fluorescence signals, digitonin (5 mg/mL in EtOH) was added to obtain Fmax, and the apparent Fmin was determined by adding Tris-EGTA (2.5 M Tris-Base, 0.3 M
EGTA). The concentration of intracellular calcium was calculated using the following equation:
Intracellular calcium = (F-Fmm/Fmax) x K^; where K^ = 400 nM. 7. To determine the potential calcilytic activity of test compounds, cells were incubated with test compound (or vehicle as a control) for 90 seconds before increasing the concentration of extracellular Ca^+ from 1 to 2mM. Calcilytic compounds were detected by their ability to block, in a concentration-dependent manner, increases in the concentration of intracellular Ca^+ elicited by extracellular Ca^+. Method B: 48 hours prior to running the CaSR assay, frozen HEK293 CaRec4.0-cl7 cells are thawed, counted, and diluted to 3e5/mL (15K/50 μL). The cell medium used to dilute the cells consists of DMEM/F12 (HAM'S) 1 :1 with L-Glutamine, 15mM HEPES, phenol red, 10% Fetal Bovine Serum, and 1% Penicillin-Streptomycin solution. Cell solution is seeded at 15K cells/50 μL/well in Greiner Poly-D-Lysine coated 384well, black, clear bottom, tissue culture plates and left at room temperature for one hour to reduce edge effect. After the first hour at room temperature, cell plates are then placed into a 37 0C, 5% CO2 incubator for 48 hours.
On the day of the experiment, plate confluency is first checked via microscope. Wells should be -100% confluent in an even cell monolayer. Assay reagents are prepared fresh. Dye load buffer consists of Hank's Buffered Saline Solution with .75 mM Calcium, without Magnesium, without Sodium Bicarbonate, with 20 mM Hepes, Probenecid (2.5 mM final concentration), Fluo4 (2 μM final concentration), and Brilliant Black (500 μM final concentration). Compound dilution buffer consists of Hank's Buffered Saline Solution without Calcium, without Magnesium (without Sodium Bicarbonate) and CHAPS (0.01% final concentration). A ligand curve plate is also prepared fresh. A 16 pt curve is dispensed into a Greiner 384 well polypropylene plate. The top concentration of the ligand, CaCl is 2.875 mM (final concentration) and the lowest concentration is .375 mM (final concentration). An ECgo value is generated from the curve data.
The CaSR assay begins when the cell media is aspirated from the cell plate using a Tecan Plate Washer, leaving nothing but the cell monolayer. Dye Load Buffer is added to the cell plate at 20 μL/well using a Multidrop and the loaded plate incubates for 45 min. at
37 0C, 5% CO2 . Compound plates are received with 1 μL of compound stamped at 5 mM- top concentration (25 μM final cone, in cell plate). Compound Dilution Buffer is added to columns 1-24 in the compound plate at 65 μL/well using a Multidrop. Column 6 is pre- stamped with 1 μL DMSO to represent the high control, and column 18 receives 65 μL of buffer as the low control. The compound addition takes place on a Cybi Well dispenser when 10 μL of diluted compound is added to the dye loaded cell plate. The cell plate with compound is then incubated at room temperature for 5 min.. The Antagonist addition takes place on the FLIPR when 10 μL of ECso challenge is added to the cell plate and fluorescence imagining proceeds for 65 sec. Column 18 of the ECso challenge plate contains only buffer to represent a low control or tool antagonist.
The compounds of Examples 1-20 were tested according to the above assay conditions and each demonstrated an IC50 of < 1 μM.
(II) Calcium Receptor Binding Assay Method A:
HEK 293 4.0-7 cells stably transfected with the Human Parathyroid Calcium Receptor ("HuPCaR") were scaled up in T 180 tissue culture flasks. Plasma membrane is obtained by polytron homogenization or glass douncing in buffer (50 mM Tris-HCl pH 7.4, 1 mM EDTA, 3 mM MgCl2) in the presence of a protease inhibitor cocktail containing 1 μM Leupeptin, 0.04 μM Pepstatin, and 1 mM PMSF. Aliquoted membrane was snap frozen and stored at -80 0C. 3H labeled compound was radiolabeled to a radiospecific activity of 44 Ci/mmole and was aliquoted and stored in liquid nitrogen for radiochemical stability.
A typical reaction mixture contains 2 nM -1H compound ((R,R)-N-4'-Methoxy-t-3- 3'-methyl-r-ethylphenyl-l-(l-naphthyl)ethylamine), or 3H compound (R)-N- [2-Hy droxy- 3-(3-chloro-2-cyanophenoxy)propyl]- 1 , 1 -dimethyl-2-(4-methoxyphenyl)ethylamine 4-10 ug membrane in homogenization buffer containing 0.1% gelatin and 10% EtOH in a reaction volume of 0.5 mL. Incubation is performed in 12 x 75 polyethylene tubes in an ice water bath. To each tube 25 μL of test sample in 100% EtOH is added, followed by 400 μL of cold incubation buffer, and 25 μL of 40 nM 3H-compound in 100% EtOH for a final concentration of 2 nM. The binding reaction is initiated by the addition of 50 μL of
80-200 ug/mL HEK 293 4.0-7 membrane diluted in incubation buffer, and allowed to incubate at 4 0C for 30 min. Wash buffer is 50 mM Tris-HCl containing 0.1% PEL Nonspecific binding is determined by the addition of 100-fold excess of unlabeled homologous ligand, and is generally 20% of total binding. The binding reaction is terminated by rapid filtration onto 1% PEI pretreated GF/C filters using a Brandel Harvester. Filters are placed in scintillation fluid and radioactivity assessed by liquid scintillation counting.
Method B: Ligand binding assays were performed in a 96-well filtration plate assembly
(MADVN65, Millipore Corporation). The wells were pre -blocked with 1% PEI in 50 mM Tris/HCl, pH 7.4. Membranes prepared from HEK293 expressing calcium receptors were incubated with [3H] compound (2-(2-hydroxyphenyl)-6-methyl-5-(2-methylpropyl)-3-(2- phenylethyl)-4(3H)-pyrimidinone) in the absence or presence of various concentrations of unlabeled ligand or compound in 50 mM Tris/HCl, pH 7.4, 3 mM MgCl2, 1 mM EDTA containing 0.1% PEI for 1 hour at 4 0C. After the incubation, membrane -bound radio labeled ligand was separated from the unbound by filtration using a Millipore filtration manifold, followed by washing with ice-cold buffer (2 X 0.1 mL). Bound radioactivity remaining on the filters was determined in 1 mL Bio-Safe (Research Products international Corp. Order No. 111195) in a liquid scintillation counter. The data was analyzed with GraphPad PRISM software.
The compounds of Examples 1-20 were tested according to the above assay conditions and each demonstrated an IC50 of < 1 μM. For instance, the compound of example 8 demonstrated an IC50 in a ligand binding assay of approximately 0.009 μM.
Definitions: d - day(s), h - hour(s), min. - minute(s), sat. - saturated, aq. - aqueous, rt - room temperature, g - gram(s), mg - milligram(s), mL - milliliter(s), μl - microliter(s), mmol - millimole(s),
M - molar,
N2 - nitrogen gas, BnBr - benzyl bromide,
MeI - methyl iodide,
K2CO3 - potassium carbonate,
Na2CO3 - sodium carbonate,
NaHCO3 - sodium bicarbonate, Na2SO4 - sodium sulfate,
CsF - cesium fluoride,
KF - potassium fluoride,
H2O2 - hydrogen peroxide,
BBr3 - boron tribromide, n-BuLi - n-butyllithium,
NaH - sodium hydride
NaOAc - sodium acetate,
AcOH - acetic acid,
HBr - hydrobromic acid, HCl - hydrochloric acid,
NaOH - sodium hydroxide,
NH4Cl - ammonium chloride
Sn2Me6 - hexamethylditin,
Pd[POBu)3J2 - føχtri-t-butylphosphine)palladium(O),
 DMF - Λf,Λ/-dimethylformamide,
DMF - Λf,Λ/-dimethylformamide,
CH2Cl2 - dichloromethane,
MeOH - methanol,
EtOH - ethanol,
Et2O - diethyl ether, EtOAc - ethyl acetate,
THF - tetrahydrofuran.
Chemical Background:
The compounds of this invention may be made by a variety of methods, including standard chemistry. Any previously defined variable will continue to have the previously defined meaning unless otherwise indicated. Illustrative general synthetic methods are set out below and then specific compounds of the invention as prepared are given in the examples.
Compounds of general Formula (I) may be prepared by methods known in the art of organic synthesis as set forth in part by the following synthetic scheme. In the scheme described below, it is well understood that protecting groups for sensitive or reactive groups are employed where necessary in accordance with general principles of chemistry.
Protecting groups are manipulated according to standard methods of organic synthesis (T.
W. Green and P. G. M. Wuts (1991) Protecting Groups in Organic Synthesis, John Wiley
& Sons). These groups are removed at a convenient stage of the compound synthesis using methods that are readily apparent to those skilled in the art. The selection of processes, as well as the reaction conditions and order of their execution, shall be consistent with the preparation of compounds of Formula (I). Those skilled in the art will recognize if a stereocenter exists in compounds of Formula (I). Accordingly, the present invention includes both possible stereoisomers and includes not only racemic compounds but the individual enantiomers as well. When a compound is desired as a single enantiomer, it may be obtained by stereospecific synthesis or by resolution of the final product or any
convenient intermediate. Resolution of the final product, an intermediate, or a starting material may be effected by any suitable method known in the art. See, for example, Stereochemistry of Organic Compounds by E. L. Eliel, S. H. Wilen, and L. N. Mander (Wiley-Interscience, 1994). The compounds described herein may be made from commercially available starting materials or synthesized using known organic, inorganic, and/or enzymatic processes.
Illustrated Methods of preparation Schemes
Included in the present invention is a process according to Schemes 1 and 2 for the synthesis of the compounds:
Scheme 1

Scheme 2

Examples
Nuclear magnetic resonance spectra were recorded at either 300 or 400 MHz using, respectively, a Bruker ARX 300 or Bruker AVANCE 400 spectrometer. DMSO-ύfø is hexadeuteriodimethylsulfoxide and CHLOROFORM -d is deuteriochloroform. Chemical shifts are reported in parts per million (δ) downfield from the internal standard tetramethylsilane. Abbreviations for NMR data are as follows: s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, dd=doublet of doublets, dt=doublet of triplets, app=apparent, br=broad. J indicates the NMR coupling constant measured in Hertz. Fourier transform infrared (FTIR) spectra were recorded on a Nicolet 510 infrared spectrometer. FTIR spectra were recorded in transmission mode, and band positions are reported in inverse wavenumbers (cm'l). Mass spectra were taken on either a SCIEX5 or Micromass instruments, using electrospray (ES) ionization techniques. Elemental analyses were obtained using a Perkin-Elmer 240C elemental analyzer. Melting points were taken
on a Thomas-Hoover melting point apparatus and are uncorrected. All temperatures are reported in degrees Celsius.
Analtech Silica Gel GF and E. Merck Silica Gel 60 F-254 thin layer plates were used for thin layer chromatography. Both flash and gravity chromatography were carried out on E. Merck Kieselgel 60 (230-400 mesh) silica gel. Analytical and preparative HPLC were carried out on Rainin or Beckman chromatographs. ODS refers to an octadecylsilyl derivatized silica gel chromatographic support. 5 μ Apex-ODS indicates an octadecylsilyl derivatized silica gel chromatographic support having a nominal particle size of 5 μ, made by Jones Chromatography, Littleton, Colorado. YMC ODS-AQ® is an ODS chromatographic support and is a registered trademark of YMC Co. Ltd., Kyoto, Japan. PRP- 1® is a polymeric (styrene-divinylbenzene) chromatographic support, and is a registered trademark of Hamilton Co., Reno, Nevada. Celite® is a filter aid composed of acid- washed diatomaceous silica, and is a registered trademark of Manville Corp., Denver, Colorado.
The following examples are intended to be illustrative only and not limiting in any way:
Example 1

2-(3-fluoro-2-hvdroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-r5-(2-pyridinyl)-2-thienyll- 4(3H)-pyrimidinone a) 2- (3-fluoro-2-r(phenylmethyr)oxy|phenvU -6-methyl-3-(2-phenylethylV5-r5-(2- pyridinyl)-2-thienyll-4(3H)-pyrimidinone
To a 50 mL round bottom flask containing 5-bromo-2-{3-fluoro-2- [(phenylmethyl)oxy]phenyl} -6-methyl-3-(2-phenylethyl)-4(3H)-pyrimidinone (618 mg, 1.25 mmol) (prepared by the method described in WO 2007/062370) was added 2-(5-
bromo-2-thienyl)pyridine (300 mg, 1.25 mmol), Pd[POBu)3J2 (30 mg, 0.058 mmol), Sn2Me6 (0.273 niL, 1.32 mmol), and 1,4-dioxane (15 mL). The solution was degassed. The reaction mixture was heated for 2 d at 100 0C. The reaction mixture was diluted with 10% aq. KF, filtered through a plug of Celite®, and then extracted with EtOAc. The organic layer was dried over Na2SO4, evaporated, and concentrated in vacuo. Purification by flash column chromatography (30% EtOAc/hexanes) provided the title compound (350 mg, 48%). LC/MS (m/z): 574 [M+H]+. b) 2-(3-fluoro-2-hvdroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-r5-(2-pyridinyl)-2- thienyl] -4(3H)-pyrimidinone
A solution of 2- {3-fluoro-2-[(phenylmethyl)oxy]phenyl} -6-methyl-3-(2- phenylethyl)-5-[5-(2-pyridinyl)-2-thienyl]-4(3H)-pyrimidinone (200 mg, 0.348 mmol) in CH2Cl2 (5 mL) at -78 0C was degassed by bubbling N2. BBr3 (0.047 mL, 0.522 mmol) was added and the reaction stirred for 1 h at this temperature. The reaction mixture was warmed to 0 0C, poured onto ice, and then extracted with CH2Cl2. The organic layer was washed with sat. aq. NaHCO3 and then concentrated in vacuo. Purification by flash column chromatography provided the title compound (60 mg, 36%). 1H NMR (400 MHz, CDCl3) δ ppm 2.51 (s, 3 H), 2.98 (t, 2 H), 4.29 (t, 2 H), 6.92-6.93 (m, 3 H), 6.99-7.13 (m, 1 H), 7.14-7.26 (m, 5 H), 7.27-7.28 (m, 1 H), 7.60-7.67 (m, 3 H), 8.57 (d, 1 H), 8.58 (br. s., 1 H). LC/MS (m/z): 484 [M+H]+.
Example 2

2-(3-fluoro-2-hvdroxyphenvπ-6-methyl-5-r4-methyl-5-(2-pyridinvπ-2-thienyl1-3-(2- phenylethyl)-4(3H)-pyrimidinone
a) 2- {3-fluoro-2-[(phenylmethyl)oxylphenyl| -6-methyl-5-(4-methyl-2-thienyl)-3- (2-phenylethyl)-4(3H)-pyrimidinone
A 100 niL round bottom flask was charged with 5-bromo-2-{3-fluoro-2- [(phenylmethyl)oxy]phenyl}-6-methyl-3-(2-phenylethyl)-4(3H)-pyrimidinone (3.0 g, 6.08 mmol) (prepared by the method described in WO 2007/062370), deoxygenated toluene (15 mL), EtOH (10 mL), water (5 mL), K2CO3 (1.67 g, 12.08 mmol), 4-methyl thiophene-2- boronic acid (1.71 g, 12.04 mmol), and Pd[P(^-Bu)3J2 (0.30 g, 0.587 mmol). The flask was sealed and stirred at 110 0C for 18 h. The reaction was allowed to cool to rt and the crude reaction mixture was filtered through Celite® and concentrated in vacuo. Purification by flash column chromatography (2-30% EtOAc/hexanes) provided the title compound (2.8 g, 90%). b) 5-(5-bromo-4-methyl-2-thienyl)-2- {3-fluoro-2-r(phenylmethyl)oxylphenyl| -6- methyl-3-(2-phenylethyl)-4(3H)-pyrimidinone
To a solution of 2-{3-fluoro-2-[(phenylmethyl)oxy]phenyl}-6-methyl-5-(4-methyl- 2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone (2.8 g, 5.48 mmol) in Et2O (13 mL) was added 48% aq. HBr (0.874 mL, 7.71 mmol). The reaction was cooled to -25 0C and 30% aq. H2O2 (0.18 mL, 1.76 mmol) was added dropwise and allowed to warm to 10 0C. The reaction was diluted with EtOAc and water. The organic layer was separated, washed with sat. aq. NaHCO3, dried over Na2SO4, and concentrated in vacuo to provide the title compound (2.5 g, 77%). c) 2-{3-fluoro-2-[(phenylmethyl)oxylphenyl|-6-methyl-5-[4-methyl-5-(2- pyridinvD-2-thienvπ -3 -(2-phenylethyl)-4(3H)-pyrimidinone
To a mixture of 5-(5-bromo-4-methyl-2-thienyl)-2-{3-fluoro-2- [(phenylmethyl)oxy]phenyl} -6-methyl-3-(2-phenylethyl)-4(3H)-pyrimidinone (0.60 g, 1.02 mmol), Pd(PPh3)4 (0.06 g, 0.052 mmol), and 2-(tributylstannyl)pyridine (0.44 g, 1.20 mmol) was added toluene (10 mL). The reaction mixture was degassed for 30 min. by bubbling with argon and then refluxed for 16 h. The reaction was allowed to cool to rt and toluene was removed under reduced pressure. The resulting residue was dissolved in EtOAc, washed with water, dried over Na2SO4, and concentrated in vacuo. Purification by flash column chromatography (30% EtOAc/hexanes) provided the title compound (0.45 g, 75%).
d) 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-[4-methyl-5-(2-pyridinyl)-2-thienyll- 3-(2-phenylethyl)-4(3H)-pyrimidinone
2- {3 -fluoro-2- [(phenylmethyl)oxy]phenyl} -6-methyl-5 - [4-methyl-5 -(2-pyridinyl)- 2-thienyl]-3-(2-phenylethyl)-4(3H)-pyrimidinone (0.45 g, 0.766 mmol) was dissolved in 33% HBr in AcOH (15 mL) and stirred at rt for 4 h. The reaction mixture was evaporated under reduced pressure and the crude residue was washed with EtOAc (2 x 25 mL). To the resulting solid were added sat. aq. NaHCO3 and CH2Cl2 until neutral pH. The organic layer was separated, dried over Na2SO4, and concentrated in vacuo. Purification by flash column chromatography (60% EtOAc/hexanes) provided the title compound (0.22 g, 58%). 1H NMR (400MHz, CDCl3) δ ppm 2.52 (s, 3 H), 2.60 (s, 3 H), 2.99 (t, 2 H), 4.30 (t, 2 H), 6.91-7.30 (m, 11 H), 7.53-7.55 (m, 2 H), 8.47 (s, 1 H). LC/MS (m/z): 498 [M+H]+.
Example 3

To a solution of 2-(3-thienyl)pyridine (3.00 g, 18.61 mmol) and NaOAc (6.12 g, 74.6 mmol) in AcOH (52 mL) was added dropwise a solution of bromine (2.8 mL, 54.4 mmol) in AcOH (35 mL) over 15 min. The reaction was heated at reflux for 2.5 h. The reaction was allowed to cool to rt, poured into water, and neutralized to pΗ ~6-7 with Na2COβ (solid). The aqueous layer was then extracted twice with EtOAc. The combined organic layers were washed with sat. aq. Na2COβ and brine, dried over Na2SOφ filtered, and concentrated in vacuo to provide the title compound (4.90 g, 83%). LC/MS (m/z): 320 [M+Η]+. b) 2-(5-bromo-3-thienyl)pyridine
A solution of 2-(2,5-dibromo-3-thienyl)pyridine (4.90 g, 15.36 mmol) in THF (96 niL) under N2 was cooled to -75 0C. n-BuLi (8.0 mL, 16.00 mmol) was added dropwise and the reaction stirred at -75 0C for 30 min. The reaction mixture was poured into water (~ 200 mL). The aqueous layer was extracted twice with Et2O. The combined organic layers were washed with brine, dried over Na2SOφ filtered, and concentrated in vacuo. Purification via flash column chromatography (0-50% EtOAc/hexanes) provided the title compound (2.81 g, 76%). LC/MS (m/z): 240/242 [M+H]+. c) 2- {3-fluoro-2-r(phenylmethyl)oxylphenyl| -6-methyl-3-(2-phenylethyl)-5-r4-(2- pyridinyl)-2-thienyll-4(3H)-pyrimidinone A solution of 2-(5-bromo-3-thienyl)pyridine (2.81 g, 11.70 mmol) in 1,4-dioxane
(100 mL) was deoxygenated for 30 min. with N2. Sn2Me6 (5.0 mL, 24.11 mmol) and Pd[P(t-Bu)3]2 (1.789 g, 3.50 mmol) were added and the reaction stirred at reflux for 1.5 h. The reaction was cooled to rt and filtered through a Celite®-plugged filter frit and rinsed with EtOAc. The filtrate was concentrated and carried to the next step without further purification or analysis. The crude 2-[5-(trimethylstannanyl)-3-thienyl]pyridine (3.79 g, 11.70 mmol) was dissolved in 1,4-dioxane (105 mL) under N2. CsF (3.26 g, 21.46 mmol), Pd[P(J-Bu)3J2 (1.366 g, 2.67 mmol), and 5-bromo-2-{3-fluoro-2- [(phenylmethyl)oxy]phenyl} -6-methyl-3-(2-phenylethyl)-4(3H)-pyrimidinone (5.27 g, 10.68 mmol) (prepared by the method described in WO 2007/062370) were added and the reaction vessel was heated at 105 0C for 17 h. The reaction was cooled to rt, filtered through a Celite®-plugged filter frit, rinsed with MeOH and CΗ2CI2, and concentrated in vacuo. Purification via flash column chromatography (0-80% EtOAc/hexanes) provided the title compound (3.97 g, 65%). LC/MS (m/z): 574 [M+H]+. d) 2-(3-fluoro-2-hvdroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-r4-(2-pyridinyl)-2- thienyll-4(3H)-pyrimidinone hydrochloride
HBr (33% in AcOH, 12.0 mL, 68.5 mmol) was added to a solution of 2-{3-fluoro- 2-[(phenylmethyl)oxy]phenyl}-6-methyl-3-(2-phenylethyl)-5-[4-(2-pyridinyl)-2-thienyl]- 4(3H)-pyrimidinone (3.93 g, 6.85 mmol) in CH2Cl2 (30 mL) and stirred at rt for 21 h. The reaction mixture was diluted with EtOAc and washed with sat. aq. Na2CO3. The aqueous layer was back-extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SOφ filtered, and concentrated in vacuo. The residue was purified
via flash column chromatography (0-80% EtOAc/hexanes). The resulting solid was suspended in MeOH and HCl (1.25 M solution in MeOH) was added. The solution was stirred for ~15 min. before being concentrated in vacuo to provide the title compound (3.44 g, 97%). 1H NMR (400 MHz, DMSO-J6) δ ppm 2.51 (s, 3H), 2.74 - 2.88 (m, 2 H), 3.89 - 4.03 (m, 2 H), 6.76 - 6.86 (m, 2 H), 6.86 - 7.03 (m, 2 H), 7.12 - 7.27 (m, 3 H), 7.27 - 7.46 (m, 2 H), 7.84 - 8.01 (m, 3 H), 8.36 (s, 1 H), 8.63 (d, J=4.29 Hz, 1 H), 10.50 (s, 1 H). LC/MS (m/z): 484 [M+H]+.
Example 4

2-(3-fluoro-2-hvdroxyphenyl)-6-methyl-5-r5-methyl-4-(2-pyridinyl)-2-thienyll-3-(2- phenylethyl)-4(3H)-pyrimidinone hydrochloride a) 2- {3-fluoro-2-r(phenylmethyl)oxylphenyl| -6-methyl-5-(5-methyl-2-thienyl)-3- (2-phenylethyl)-4(3H)-pyrimidinone To a 500 mL round bottom flask was added 2-(3-fluoro-2-hydroxyphenyl)-6- methyl-5-(5-methyl-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone (11.0 g, 26.2 mmol) (prepared by the method described in WO 2007/062370), K2CO3 (3.98 g, 28.8 mmol), and BnBr (3.49 mL, 28.8 mmol) in DMF (100 mL). The reaction mixture was stirred overnight at rt, diluted with water and Et2O, and the layers were separated. The aqueous layer was extracted twice with Et2O. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified via flash column chromatography (0-30% EtOAc/hexanes) to afford the title compound (13.45 g, 99%). 1H NMR (400 MHz, CHLOROFORM-^) δ ppm 2.42 (s, 3 H), 2.57 (s, 3 H), 2.79 (m, 1 H), 2.87 - 3.01 (m, 1 H), 3.71 (m, 1 H), 4.98 (d, J=I 1.37 Hz, 1 H), 5.22 (d, J=I 1.37 Hz, 1 H), 6.81 (dd, J=3.54, 1.01 Hz, 1 H), 6.84 - 6.92 (m, 2 H), 7.03 (d, J=3.54 Hz, 1 H), 7.09 - 7.23 (m, 9 H), 7.23 - 7.33 (m, 3 H). LC/MS (m/z): 511 [M+H]+.
b) 5-(4-bromo-5-methyl-2-thienyl)-2- {3-fluoro-2-[(phenylmethyl)oxy]phenyU -6- methyl-3-(2-phenylethyl)-4(3H)-pyrimidinone
To a solution of 2-{3-fluoro-2-[(phenylmethyl)oxy]phenyl}-6-methyl-5-(5-methyl- 2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone (11.40 g, 21.88 mmol) in Et2O (40 mL) at 0 0C was added 48% aq. HBr (5.45 mL, 48.1 mmol) followed by 30% aq. H2O2 (2.235 mL, 21.88 mmol). The cooling bath was removed and the solution was allowed to warm to rt and stirred overnight. The reaction mixture was diluted with Et2O, washed with brine, and the layers were separated. The aqueous phase was extracted twice with Et2O. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified via flash column chromatography (0-20% EtOAc/hexanes) to afford the title compound (9.09 g, 70.5%). 1H NMR (400 MHz, CHLOROFORM-J) δ ppm 2.43 (s, 3 H), 2.45 (s, 3 H), 2.72 - 2.84 (m, 1 H), 2.87 - 3.00 (m, 1 H), 3.63 - 3.77 (m, 1 H), 4.28 - 4.41 (m, I H), 4.97 (d, J=I 1.37 Hz, 1 H), 5.25 (d, J=I 1.37 Hz, 1 H), 6.80 - 6.91 (m, 3 H), 7.05 (s, 1 H), 7.08 - 7.34 (m, 10 H). LC/MS (m/z): 589/591 [M+H]+. c) 2-(3-fluoro-2-r(phenylmethvπoxy1phenyl|-6-methyl-5-r5-methyl-4-(2- pyridinvD-2-thienvπ -3 -(2-phenylethyl)-4(3H)-pyrimidinone
In a 150 mL sealed tube was 5-(4-bromo-5-methyl-2-thienyl)-2-{3-fluoro-2- [(phenylmethyl)oxy]phenyl} -6-methyl-3-(2-phenylethyl)-4(3H)-pyrimidinone (4.06 g, 6.89 mmol), CsF (2.09 g, 13.77 mmol), and Pd[P(?-Bu)3]2 (0.352 g, 0.689 mmol) in 1,4- dioxane (45.9 mL) to give a brown solution. The solution was degassed with N2 for 10 min., at which time 2-(tributylstannyl)pyridine (3.10 g, 7.58 mmol) was added to the tube. The vessel was sealed and heated tol 10 0C for 16 h. The reaction mixture was allowed to cool to rt, diluted with 10% aq. KF (50 mL), and stirred for 30 min, at which time the suspension was filtered through a pad of Celite® which was then rinsed with EtOAc and water. The layers were separated and the aqueous layer was back-extracted with EtOAc (2 x 25 mL). The combined organic layers were washed with Brine (1 x 25 mL). The organic phase was dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified via flash column chromatography (0-30% EtOAc/hexanes) to provide the title compound (2.76 g, 67.5%) as a pale yellow oil which expanded to a yellow foam under high vacuum. 1U NMR (400 MHz, CHLOROFORM-^) δ ppm 2.49 (s, 3 H), 2.72 (s, 3 H), 2.78 - 2.88 (m, 1 H), 2.88 - 3.00 (m, 1 H), 3.72 - 3.86 (m, 1 H), 4.23 - 4.37 (m, 1 H), 4.98
(d, J=I 1.37 Hz, I H), 5.25 (d, J=I 1.37 Hz, 1 H), 6.81 - 6.92 (m, 3 H), 7.07 - 7.35 (m, 11 H), 7.46 - 7.58 (m, 2 H), 7.75 (td, J=Ul, 1.77 Hz, 1 H), 8.65 - 8.75 (m, 1 H). LC/MS (m/z): 588 [M+H]+. d) 2-(3-fluoro-2-hvdroxyphenyl)-6-methyl-5-r5-methyl-4-(2-pyridinyl)-2-thienyll- 3-(2-phenylethyl)-4(3H)-pyrimidinone hydrochloride
To a solution of 2-{3-fluoro-2-[(phenylmethyl)oxy]phenyl}-6-methyl-5-[5-methyl- 4-(2-pyridinyl)-2-thienyl]-3-(2-phenylethyl)-4(3H)-pyrimidinone (2.76 g, 4.70 mmol) in CH2Cl2 (47.0 niL) was added HBr in AcOH (8.34 M, 5.63 mL, 47.0 mmol). The reaction mixture was stirred overnight (16 h) at rt, followed by quenching with sat. aq. Na2CO3 (50 mL) and stirring for 45 min. until a homogenous biphasic mixture resulted (all solid was in solution). The layers were separated and the aqueous phase was extracted with CH2Cl2 (2 x 25 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified via flash column chromatography (0-80% EtOAc/hexanes). The resulting yellow foam was taken up in 64 mL of EtOAc and HCl (1 M solution in Et2O, 16.2 mL) was added. The solution was stirred for 15 min. and then volatiles were removed. The resulting white residue was taken up in 49 mL of CH2Cl2 and MeOH (~5 mL) was added to ensure complete dissolution. While stirring, EtOAc was added just until the solution became cloudy (~33 mL). The vessel was heated with a heat gun until just before reflux and stirred for 30 min. as white crystals crashed out. The crystals were filtered, rinsed with EtOAc, and dried with airflow for 30 min. to provide the title compound (1.51 g, 60.2%). 1H NMR (400 MHz, DMSO- dβ) δ ppm 2.53 (s, 3 H), 2.71 (s, 3 H), 2.75 - 2.87 (m, 2 H), 3.87 - 4.05 (m, 2 H), 6.82 (dd, J=7.71, 1.64 Hz, 2 H), 6.87 - 7.08 (m, 2 H), 7.09 - 7.32 (m, 3 H), 7.41 (ddd, J=I 1.12, 7.83, 2.02 Hz, 1 H), 7.70 (d, J=3.03 Hz, 1 H), 7.75 (br. s., IH), 8.07 (br. s., 1 H), 8.39 (br. s., 1 H), 8.83 (d, J=5.31 Hz, 1 H), 10.53 (br. s., 1 H). LC/MS (m/z): 498 [M+H]+.
Example 5

2-(3-fluoro-2-hvdroxyphenvπ-6-methyl-5-r5-(3-methyl-2-pyridinvπ-2-thienyl1-3-(2- phenylethyl)-4(3H)-pyrimidinone a) 2-(tributylstannyl)-3-methylpyridine
To a solution of 2-bromo-3 -methyl pyridine (3 g, 17.44 mmol) in TΗF (40 mL) was added n-BuLi (13 mL, 20.93 mmol) at -78 0C under N2. The reaction mixture was maintained at -78 0C for 30 min. and then tributyltin chloride (5.6 mL, 20.93 mmol) was added dropwise. The resultant mixture was maintained at -78 0C for Ih and then allowed to slowly warm to rt. The reaction mixture was diluted with EtOAc (40 mL) and washed with 10% aq. KF (3 x 30 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo to afford 6 g of the title compound, which was used for next step without further purification. LC/MS (m/z): 384 ([M+Η]+). b) 2- {3-fluoro-2-[(phenylmethyl)oxylphenyl| -6-methyl-5-|"5-(3-methyl-2- pyridinyl)-2-thienyll -3 -(2-phenylethyl)-4(3H)-pyrimidinone
A solution of 5-bromo-2- {3-fluoro-2-[(phenylmethyl)oxy]phenyl} -6-methyl-3-(2- phenylethyl)-4(3H)-pyrimidinone (600 mg, 1.043 mmol) (prepared by the method described in WO 2007/062370), 2-(tributylstannyl)-3-methylpyridine (600 mg, 1.565 mmol), Pd(PPh3 )4 (54 mg, 0.0469 mmol) in toluene (15 mL) was deoxygenated with argon for 20 min. and then heated to reflux overnight. The reaction mixture was filtered through Celite® and concentrated in vacuo. Purification via flash column chromatography (0-20% EtO Ac/petroleum ether) provided the title compound (330 mg, 54%) as a pale yellow viscous oil. LC/MS (m/z): 588 ([M+Η]+). c) 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-[5-(3-methyl-2-pyridinyl)-2-thienyll- 3-(2-phenylethyl)-4(3H)-pyrimidinone
A solution of 33% HBr in AcOH (20 mL) was added to 2-{3-fluoro-2- [(phenylmethyl)oxy]phenyl}-6-methyl-5-[5-(3-methyl-2-pyridinyl)-2-thienyl]-3-(2-
phenylethyl)-4(3H)-pyrimidinone (330 mg, 0.561 mmol) and resultant reaction mixture was stirred for 2 h at rt. Concentration in vacuo was followed by neutralization with sat. aq. NaHCO3 and extraction with chloroform (2 x 25 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Purification via flash column chromatography (0-40% EtO Ac/petroleum ether) provided the title compound as a pale yellow solid. 1H NMR (400MHz, CDCl3) δ ppm 2.52 (s, 3 H), 2.60 (s, 3 H), 2.99 (t, 2 H), 4.30 (t, 2 H), 6.91-7.30 (m, 11 H), 7.53-7.55 (m, 2 H), 8.47 (s, 1 H). LC/MS (m/z): 498 [M+H]+.
Example 6

2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-[4-methyl-5-(3-methyl-2-pyridinyl)-2-thienyll- 3-(2-phenylethyl)-4(3H)-pyrimidinone
The title compound was prepared following the general procedures of Example 2 except substituting 2-(tributylstannanyl)-3-methylpyridine for 2-(tributylstannanyl)- pyridine. LC/MS (m/z): 512 [M+Η]+.
Example 7

2-(3-fluoro-2-hvdroxyphenyl)-6-methyl-5-r5-methyl-4-(3-methyl-2-pyridinyl)-2-thienyll- 3-(2-phenylethyl)-4(3H)-pyrimidinone
The title compound was prepared following the general procedures of Example 4 except substituting 2-(tributylstannanyl)-3-methylpyridine for 2-(tributylstannanyl)- pyridine. LC/MS (m/z): 512 [M+H]+.
Example 8

2-(3-fluoro-2-hvdroxyphenvπ-6-methyl-5-r5-(6-methyl-2-pyridinvπ-2-thienyl1-3-(2- phenylethyl)-4(3H)-pyrimidinone a) 2-(tributylstannyl)-6-methylpyridine To a solution of 2-bromo-6-methyl pyridine (3 g, 17.44 mmol) in TΗF (40 mL) was added n-BuLi (13 mL, 20.93 mmol) at -78 0C under N2. The reaction mixture was maintained at -78 0C for 30 min. and then tributyltin chloride (5.6 mL, 20.93 mmol) was added dropwise. The resultant mixture was maintained at -78 0C for Ih and then allowed to slowly warm to rt. The reaction mixture was diluted with EtOAc (40 mL) and washed with 10% aq. KF (3 x 30 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo to afford 6 g of the title compound, which was used for next step without further purification. 1H NMR (400MΗz, CDCl3) δ ppm 0.88-1.59 (m, 27 H), 2.56 (s, 3 H), 6.95(d, 1 H), 7.20 (d, 1 H), 7.40 (d, 1 H). LC/MS (m/z): 384 [M+H]+. b) 2- (3-fluoro-2-IYphenylmethv0oxylphenvU -6-methyl-5-r5-(6-methyl-2- pyridinyl)-2-thienyll -3 -(2-phenylethyl)-4(3H)-pyrimidinone
A solution of 5-bromo-2- {3-fluoro-2-[(phenylmethyl)oxy]phenyl} -6-methyl-3-(2- phenylethyl)-4(3H)-pyrimidinone (3.5 g, 6.08 mmol) (prepared by the method described in WO 2007/062370), 2-(tributylstannyl)-6-methylpyridine (2.9 mL, 9.13 mmol), Pd(PPh3)4 (210 mg, 0.18 mmol) in toluene (25 mL) was deoxygenated with argon for 20 min. and then heated to reflux overnight. The reaction mixture was filtered through Celite® and concentrated in vacuo. Purification via flash column chromatography (0-30% EtO Ac/petroleum ether) provided the title compound (2.5 g, 71%) as a pale yellow viscous
oil. 1H NMR (400MHz, CDCl3) δ ppm 2.45 (s, 3 H), 2.60 (s, 3 H), 2.82 (m, 1 H), 2.95 (m, 1 H), 3.71 (m, 1 H), 4.3 l(m, 1 H), 4.96 (d, 1 H), 5.22 (d, 1 H), 6.84-6.88 (m, 3 H); 7.01- 7.65 (m, 15 H). LC/MS (m/z): 588 ([M+H]+). c) 2-(3-fluoro-2-hvdroxyphenyl)-6-methyl-5-r5-(6-methyl-2-pyridinyl)-2-thienyll- 3-(2-phenylethyl)-4(3H)-pyrimidinone
A solution of 33% HBr in AcOH (20 mL) was added to 2-{3-fluoro-2- [(phenylmethyl)oxy]phenyl}-6-methyl-5-[5-(6-methyl-2-pyridinyl)-2-thienyl]-3-(2- phenylethyl)-4(3H)-pyrimidinone (2.5 g, 4.25 mmol) and resultant reaction mixture was stirred for 4 h at rt. Concentration in vacuo was followed by neutralization with sat. aq. NaHCO3 and extraction with chloroform (2 x 40 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Trituration with Et2O and n-pentane provided the title compound (1.4 g, 66% yield) as a pale yellow solid. 1H NMR (400MHz, CDCl3) δ ppm 2.51 (s, 3 H), 2.57 (s, 3 H), 2.99 (t, 2 H), 4.29 (t, 2 H), 6.88-7.59 (m, 13 H), 8.78 (s, 1 H). LC/MS (m/z): 498 [M+H]+.

2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[5-(3-pyridinyl)-2-thienyll-
4(3H)-pyrimidinone a) 2-{3-fluoro-2-r(phenylmethyl)oxy1phenyl}-6-methyl-3-(2-phenylethyl)-5-(2- thienyl)-4(3H)-pyrimidinone
A solution of 5-bromo-2- {3-fluoro-2-[(phenylmethyl)oxy]phenyl} -6-methyl-3-(2- phenylethyl)-4(3H)-pyrimidinone (1O g, 17.4 mmol) (prepared by the method described in
WO 2007/062370), 2-tributylstannylthiophene (5.7 mL, 17.9 mmol), Pd(PPh3)4 (1.5 g, 1.29 mmol) in toluene (25 mL) was deoxygenated with N2 for 20 min. and then heated to reflux for 48 h. The reaction mixture was allowed to cool to rt and then filtered through Celite®, diluted with water, and extracted with EtOAc. The organic layer was dried over Na2SO4,
filtered, and concentrated in vacuo. Purification via flash column chromatography provided the title compound (8 g, 93%). b) 5-(5-bromo-2-thienyl)-2-{3-fluoro-2-[(phenylmethyl)oxylphenyl|-6-methyl-3- (2-phenylethyl)-4(3H)-pyrimidinone To a solution of 2- {3-fluoro-2-[(phenylmethyl)oxy]phenyl} -6-methyl-3-(2- phenylethyl)-5-(2-thienyl)-4(3H)-pyrimidinone (1.2 g, 2.42 mmol) in Et2O (10 mL) was added 48% aq. HBr (5.45 mL, 48.1 mmol). The reaction mixture was cooled to -25 0C and 30% aq. H2O2 (0.28 mL, 2.74 mmol) was added dropwise, allowing the temperature to rise to 10 0C. The solution was diluted with Et2O and washed with water and sat. aq. NaHCO3, dried over Na2SO4, filtered, and concentrated in vacuo to afford the title compound (1.1 g, 79%). c) 2- {3-fluoro-2-r(phenylmethvDoxy1phenyl} -6-methyl-3-(2-phenylethyl)-5-r5-(3- pyridinyl)-2-thienyll-4(3H)-pyrimidinone
A solution of 5-(5-bromo-2-thienyl)-2- {3-fluoro-2-[(phenylmethyl)oxy]phenyl} -6- methyl-3-(2-phenylethyl)-4(3H)-pyrimidinone (0.7 g, 1.22 mmol), Pd(PPh3)4 (0.07 g, 0.06 mmol), and 3-(tributylstannyl)pyridine (0.55 g, 1.49 mmol) in toluene (5 mL) was deoxygenated with argon for 15 min. and then heated to reflux overnight. The reaction mixture was allowed to cool to rt, concentrated in vacuo, diluted with EtOAc, washed with water, dried over Na2SO4, filtered, and concentrated in vacuo. Purification via flash column chromatography (0-30% EtOAc/hexanes) provided the title compound (0.55 g, 79%). d) 2-(3-fluoro-2-hvdroxyphenvn-6-methyl-3-(2-phenylethvn-5-r5-(3-pyridinvn-2- thienyl] -4(3H)-pyrimidinone
2-{3-fluoro-2-[(phenylmethyl)oxy]phenyl}-6-methyl-3-(2-phenylethyl)-5-[5-(3- pyridinyl)-2-thienyl]-4(3H)-pyrimidinone (0.55 g, 0.96 mmol) was dissolved in HBr in AcOH (15 mL) and stirred at rt for 4 h. The reaction mixture was concentrated in vacuo, diluted with CH2Cl2 (50 mL) and neutralized with sat. aq. NaHCO3. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo to afford the title compound. 1H NMR (400 MHz, CDCl3) δ ppm 2.48 (s, 3 H), 2.78 - 2.82 (m, 2 H), 3.93 - 3.97 (m, 2 H), 6.80 - 6.82 (m, 2 H), 6.92 - 6.99 (m, 2 H), 7.16 - 7.22 (m, 3 H), 7.36 - 7.47 (m, 3 H), 7.67
(d, 1 H), 8.07 (d, 3 H), 8.51 (d, 1 H), 8.95 (s, 1 H), 10.45 (s, 1 H). LC/MS (m/z): 484.2 [M+H]+.

2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-[5-(6-methyl-3-pyridinyl)-2-thienyll-3-(2- phenylethvD-4(3H)-pyrimidinone a) 2-{3-fluoro-2-[(phenylmethyl)oxylphenyl|-6-methyl-5-[5-(6-methyl-3- pyridinyl)-2-thienyll -3 -(2-phenylethyl)-4(3H)-pyrimidinone A solution of 5-bromo-2- {3-fluoro-2-[(phenylmethyl)oxy]phenyl} -6-methyl-3-(2- phenylethyl)-4(3H)-pyrimidinone (1 g, 1.74 mmol) (prepared by the method described in WO 2007/062370), 2-methylpyridine-5-boronic acid (285 mg, 2.08 mmol), K2CO3 (480 mg, 3.48 mmol), Pd(PPh3)4 (100 mg, 0.08 mmol), EtOΗ:water (2:1, 2 mL), in toluene (15 mL) was deoxygenated with argon for 20 min. and then heated to reflux overnight. The reaction mixture was allowed to cool to rt, filtered through Celite®, and washed with water (2 x 20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. Purification via flash column chromatography (0-70% EtOAc/hexanes) provided the title compound (650 mg, 64%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ ppm 2.50 (s, 3 H), 2.60 (s, 3 H), 2.81 (m, 1 H), 2.95 (m, 1 H), 3.78 (m, 1 H), 4.36(m, 1 H), 5.01 (d, 1 H), 5.22 (d, 1 H), 6.82-6.89 (m, 3 H), 7.06-7.32 (m, 12 H), 7.36 (d, 1 H), 7.83 (d, 1 H), 8.82 (s, 1 H). LC/MS (m/z): 588 [M+H]+. b) 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-[5-(6-methyl-3-pyridinyl)-2-thienyll- 3-(2-phenylethyl)-4(3H)-pyrimidinone
A solution of 33% HBr in AcOH (20 mL) was added to 2-{3-fluoro-2- [(phenylmethyl)oxy]phenyl}-6-methyl-5-[5-(6-methyl-3-pyridinyl)-2-thienyl]-3-(2- phenylethyl)-4(3H)-pyrimidinone (650 mg, 1.107 mmol) and resultant reaction mixture was stirred for 3 h at rt. Concentration in vacuo was followed by neutralization with sat.
aq. NaHCO3 and extraction with chloroform (2 x 25 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Purification via flash column chromatography (0-70% EtOAc/hexanes) provided the title compound (480 mg, 87%). 1H NMR (400MHz, CDCl3) δ ppm 2.52 (s, 3 H), 2.55 (s, 3 H), 3.01 (t, 2 H), 4.30 (t, 2 H), 6.91-7.35 (m, 11 H), 7.80 (d, 1 H), 8.80 (s, 1 H), 9.20 (br. s., 1 H). LC/MS (mix): 498 [M+H]+.

A solution of 5-bromo-2- {3-fluoro-2-[(phenylmethyl)oxy]phenyl} -6-methyl-3-(2- phenylethyl)-4(3H)-pyrimidinone (1 g, 2.02 mmol) (prepared by the method described in WO 2007/062370), 2-trifluoromethylpyridine-5-boronic acid (463 mg, 2.42 mmol), Pd(PPh3)4 (117 mg, 0.101 mmol), K2CO3 (557 mg, 4.04 mmol), EtOΗ:water (2:1, 2 mL), in dimethoxy ethane (15 mL) was deoxygenated with argon for 20 min. and then heated to reflux overnight. The reaction mixture was allowed to cool to rt, filtered through Celite®, and washed with water (2 x 20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. Purification via flash column chromatography (0-70% EtO Ac/petroleum ether) provided the title compound (1.1 g, 85%) as a yellow solid. LC/MS (m/z): 642 [M+H]+. b) 2-(3-fluoro-2-hvdroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-{5-r6- (trifluoromethyl)-3-pyridinyll-2-thienvU-4(3H)-pyrimidinone
A solution of 33% HBr in AcOH (20 mL) was added to 2-{3-fluoro-2- [(phenylmethyl)oxy]phenyl}-6-methyl-3-(2-phenylethyl)-5-{5-[6-(trifluoromethyl)-3-
pyridinyl]-2-thienyl}-4(3H)-pyrimidinone (1.1 g, 1.713 mmol) and resultant reaction mixture was stirred for 3 h at rt. Concentration in vacuo was followed by neutralization with sat. aq. NaHCO3 and extraction with chloroform (2 x 25 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Purification via flash column chromatography (0-70% EtOAc/hexanes) provided the title compound (0.75 g, 79%). 1H NMR (400MHz, CDCl3) δ ppm 2.60 (s, 3 H), 2.99 (t, 2 H), 4.30 (t, 2 H), 6.91- 7.30 (m, 11 H), 7.53-7.55 (m, 2 H), 8.47 (s, 1 H). LC/MS (m/z): 552 [M+H]+.
Example 12

2-(3 -fluoro-2-hvdroxyphenyl)-5 - (5 - r6-(hvdroxymethyl)-3 -pyridinyll -2-thienyll -6-methyl-
3-(2-phenylethyl)-4(3H)-pyrimidinone a) 2- {3-fluoro-2-r(phenylmethyl)oxylphenyl| -5- {5-r6-(hvdroxymethyl)-3- pyridinyll-2-thienyl|-6-methyl-3-(2-phenylethyl)-4(3H)-pyrimidinone A solution of 5-bromo-2- {3-fluoro-2-[(phenylmethyl)oxy]phenyl} -6-methyl-3-(2- phenylethyl)-4(3H)-pyrimidinone (1.2 g, 2.15 mmol) (prepared by the method described in WO 2007/062370), 2-hydroxymethyl pyridine-5-boronic acid (394 mg, 2.58 mmol), Pd(PPh3)4 (123 mg, 0.10 mmol), K2CO3 (594 mg, 4.3 mmol), EtOΗ:water (5:2 7 mL), in dimethoxyethane (20 mL) was deoxygenated with argon for 20 min. and then heated to reflux overnight. The reaction mixture was allowed to cool to rt, filtered through Celite®, and washed with water (2 x 20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. Purification via flash column chromatography (0-70% EtO Ac/petroleum ether) provided the title compound (500 mg, 39%) as a yellow solid. 1H NMR (400MHz, CDCl3) δ ppm 2.48 (s, 3 H), 2.81 (m, 1 H), 2.95 (m, 1 H), 3.52 (br. s., 1 H), 3.78 (m, 1 H), 4.36 (m, 1 H), 4.80 (s, 2 H), 5.01 (d, 1 H), 5.25 (d, 1 H), 6.82-6.89 (m, 3 H), 7.06-7.40 (m, 13 H), 7.96 (d, 1 H), 8.90 (s, 1 H). LC/MS (m/z): 604 [M+H]+.
b) (5-{5-[2-(3-fluoro-2-hydroxyphenyl)-4-methyl-6-oxo-l-(2-phenylethyl)-l,6- dihvdro-5-pyrimidinyll-2-thienvU -2-pyridinyl)methyl acetate
A solution of 33% HBr in AcOH (20 mL) was added to 2-{3-fluoro-2- [(phenylmethyl)oxy]phenyl} -5- {5-[6-(hydroxymethyl)-3-pyridinyl]-2-thienyl} -6-methyl- 3-(2-phenylethyl)-4(3H)-pyrimidinone (400 mg, 0.66 mmol) and resultant reaction mixture was stirred for 3 h at rt. Concentration in vacuo was followed by neutralization with sat. aq. NaHCO3 and extraction with chloroform (2 x 25 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. LCMS analysis of the crude compound shows the formation of O-acetylated product. The crude compound was taken into the next step without further purification. LC/MS (m/z): 542 [M+H]+. c) 2-(3-fluoro-2-hydroxyphenyl)-5-{5-[6-(hydroxymethyl)-3-pyridinyll-2-thienyl|- 6-methyl-3-(2-phenylethyl)-4(3H)-pyrimidinone
To a solution of (5-{5-[2-(3-fluoro-2-hydroxyphenyl)-4-methyl-6-oxo-l-(2- phenylethyl)-l,6-dihydro-5-pyrimidinyl]-2-thienyl}-2-pyridinyl)methyl acetate (320 mg, 0.574 mmol) in TΗF was added 10% aq. NaOH (12 mL) and the reaction mixture was stirred for 2 h at rt. TΗF was removed from reaction mixture under reduced pressure and the aqueous layer was extracted with Et2O. The aqueous layer was neutralized with 2 M aq. HCl and extracted with EtOAc (2 x 15 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Purification via flash column chromatography (0-3% MeOΗ/chloroform) followed by preparative ΗPLC afforded the title compound (160 mg, 54%) as a pale yellow solid. 1H NMR (400MHz, CDCl3) δ ppm 2.50 (s, 3 H), 2.95 (t, 2 H), 4.30 (t, 2 H), 4.78 (s, 2 H), 6.82-6.98 (m, 4 H), 7.15-7.39 (m, 10 H),7.90 (d, 1 H), 8.89 (s, 1 H). LC/MS (m/z): 514 [M+H]+.
Example 13

The title compound was prepared following the general procedures of Example 9 except substituting 4-(tributylstannanyl)pyridine for 3-(tributylstannanyl)pyridine. LC/MS (m/z): 484 [M+Η]+.
Example 14

2-(3-fluoro-2-hvdroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-r5-(2-pyrimidinyl)-2-thienyll- 4(3H)-pyrimidinone
The title compound was prepared following the general procedures of Example 9 except substituting 2-(tributylstannanyl)pyrimidine for 3-(tributylstannanyl)pyridine. LC/MS (m/z): 485 [M+Η]+.
Example 15

2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[4-(2-pyrimidinyl)-2-thienyll- 4(3H)-pyrimidinone
The title compound was prepared following the general procedures of Example 17 except substituting 2-(tributylstannanyl)pyrimidine for 5-(tributylstannanyl)pyrimidine. 1H
NMR (400 MHz, DMSO-J6) δ ppm 2.52 (s, 3 H), 2.71 - 2.92 (m, 2 H), 3.84 - 4.08 (m, 2
H), 6.83 (dd, J=7.71, 1.64 Hz, 2 H), 6.87 - 7.12 (m, 2 H), 7.12 - 7.30 (m, 3 H), 7.30 - 7.48
(m, 2 H), 8.01 (d, J=I.26 Hz, 1 H), 8.52 (d, J=I.52 Hz, 1 H), 8.87 (d, J=4.80 Hz, 2 H), 10.51 (br. s., 1 H). LC/MS (m/z): 485 [M+H]+.

2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[5-(5-pyrimidinyl)-2-thienyll- 4(3H)-pyrimidinone
The title compound was prepared following the general procedures of Example 9 except substituting 5-(tributylstannanyl)pyrimidine for 3-(tributylstannanyl)pyridine. LC/MS (m/z): 485 [M+Η]+.
Example 17

2-(3-fluoro-2-hvdroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-r4-(5-pyrimidinyl)-2-thienyll- 4(3H)-pyrimidinone a) 5-(4-bromo-2-thienvP)-2- {3-fluoro-24(phenylmethyl)oxy1phenyl| -β-methyl-3- (2-phenylethyl)-4(3H)-pyrimidinone
An oven dried 500 mL round bottom flask was charged with argon, 5-bromo-2-{3- fluoro-2-[(phenylmethyl)oxy]phenyl}-6-methyl-3-(2-phenylethyl)-4(3Η)-pyrimidinone (20 g, 40.5 mmol) and THF (40 mL). A suspension of Rieke Zinc (55 mL, 42.0 mmol) in THF was then added via syringe. The reaction was placed in a 50 0C heating mantle to ensure continued heating. After 2 h an additional portion of Rieke Zinc (20 mL, 15.28 mmol) was
added, followed another 2 h later by a third portion of Rieke Zinc (20 mL, 15.28 mmol), and finally a fourth batch of Rieke Zinc (20 mL, 15.28 mmol) another 2 h later. The reaction was continued for 2.5 h, at which point it was allowed to cool to rt and settle without stirring for 30 min. 2,4-Dibromothiophene (4.58 mL, 40.5 mmol) was dissolved in THF (30 mL) in an oven dried 50OmL flask. Pd(PPh3)4 (5 g, 4.33 mmol) was added to the yellow solution followed by the organozinc reagent solution (transferred carefully to leave precipitate in the flask, 20 mL at a time by syringe over approximately 15 min.). The reaction was then stirred at rt. After 14 h the reaction was quenched with sat. aq. NH4Cl and stirred until the color had changed to bright yellow, extracted with EtOAc, washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. Repeated flash column chromatography (10-50% and 5-30% EtOAc/Hexanes) afforded the title compound (12.9 g, 47%) as a yellow oil. LC/MS (m/z): 575 [M+H]+. b) 2- {3-fluoro-2-[(phenylmethyl)oxylphenyl| -6-methyl-3-(2-phenylethyl)-5-[4-(5- pyrimidinyl)-2-thienvH-4(3H)-pyrimidinone 5-(4-bromo-2-thienyl)-2- {3-fluoro-2-[(phenylmethyl)oxy]phenyl} -6-methyl-3-(2- phenylethyl)-4(3H)-pyrimidinone (58 mg, 0.10 mmol), 5-(tributylstannanyl)pyrimidine (74.4 mg, 0.20 mmol) and CsF (46 mg, 0.30 mmol) was degassed by bubbling with N2 for 20 min. followed by the addition of Pd[P(?-Bu)3]2 (7.7 mg, 0.015 mmol). The solution was heated to 150 0C in a microwave synthesizer for 20 min. The reaction mixture was allowed to cool to rt, filtered through Celite®, and concentrated in vacuo. Purification via flash column chromatography (0-50% EtOAc/hexanes) provided the title compound (35 mg, 60%). c) 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[4-(5-pyrimidinyl)- 2-thienyll -4(3H)-pyrimidinone 2- {3-fluoro-2-[(phenylmethyl)oxy]phenyl} -6-methyl-3-(2-phenylethyl)-5-[4-(5- pyrimidinyl)-2-thienyl]-4(3H)-pyrimidinone (38 mg, 0.066 mmol) was dissolved in CH2Cl2 (1 mL) and then 30% HBr in AcOH (0.23 mL, 1.32 mmol) was added slowly. After 16 h, sat. aq. NaHCOβ was added until the solution was neutralized. The product was extracted thrice with CH2Cl2 and the combined organic extracts were concentrated in vacuo and purified by flash column chromatography (0-80% EtOAc/hexanes) to afford the title compound (0.022 g, 68%) as a yellow solid. 1U NMR (400 MHz, DMSO-J6) δ ppm
2.52 (s, 3 H), 2.73 - 2.90 (m, 2 H), 3.84 - 4.09 (m, 2 H), 6.71 - 6.87 (m, 2 H), 6.87 - 7.12 (m, 2 H), 7.12 - 7.30 (m, 3 H), 7.39 (d, J=9.09 Hz, 1 H), 7.91 (d, J=I.52 Hz, 1 H), 8.35 (d, J=I.26 Hz, 1 H), 9.13 (s, 1 H), 9.25 - 9.27 (m, 2 H), 10.51 (br. s., 1 H). LC/MS (m/z): 485 [M+H]+.
Example 18

2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[5-(2-pyrazinyl)-2-thienyll- 4(3H)-pyrimidinone The title compound was prepared following the general procedures of Example 9 except substituting 2-(tributylstannanyl)pyrazine for 3-(tributylstannanyl)pyridine. LC/MS (m/z): 485 [M+Η]+.
Example 19

2-(3 -fluoro-2-hvdroxyphenyl)-6-methyl-5 -(5 -methyl-4- (6- r(methyloxy)methyll -2- PyridinvU-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone hydrochloride
a) 2-bromo-6- [(methyloxy)methyl]pyridine
To a solution of (6-bromo-2-pyridinyl)methanol (1.100 g, 5.85 mmol) in DMF (29.3 mL) at 0 0C was added NaH (0.257 g, 6.44 mmol) and the reaction mixture was stirred at 0 0C for 5 min, placed in a rt water bath for 20 min, and then cooled back to 0 0C for 5 min. MeI (0.402 mL, 6.44 mmol) was added dropwise and the cooling bath was removed. After stirring overnight at rt, the reaction was quenched with sat. aq. NH4Cl and extracted three times with Et2O. The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification via flash column chromatography (10% EtOAc/hexanes) provided the title compound (530 mg, 45%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ ppm 3.49 (s, 3 H), 4.57 (s, 2 H), 7.40 (t, 2 H), 7.57 (t, 1 H). LC/MS (m/z): 203 [M+H]+. b) 2- {3-fluoro-2-r(phenylmethyl)oxylphenyl| -6-methyl-5-(5-methyl-4- (6- [(methyloxy)methyll-2-pyridinyU-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone
A solution of 5-(4-bromo-5-methyl-2-thienyl)-2- {3-fluoro-2-[(phenylmethyl)oxy]- phenyl}-6-methyl-3-(2-phenylethyl)-4(3H)-pyrimidinone (500 mg, 0.848 mmol) and
Pd[P(t-Bu)3]2 (43.3 mg, 0.085 mmol) in 1,4-dioxane (5.65 mL) was degassed with N2 for 15 min. Sn2Me6 (0.185 mL, 0.891 mmol) was added and the reaction vessel was heated at 110 0C for 2 h. Upon cooling, CsF (234 mg, 1.542 mmol), Pd[P(?-Bu)3]2 (39.4 mg, 0.077 mmol), and 2-bromo-6-[(methyloxy)methyl]pyridine (156 mg, 0.771 mmol) were added and the reaction vessel was heated at 110 0C overnight. Upon cooling, the reaction was filtered through Celite® and concentrated in vacuo. Purification via flash column chromatography (0-25% EtOAc/hexanes) provided the title compound (45.4 mg, 9.3%) as a yellow oil. LC/MS (m/z): 632 [M+Η]+. c) 2-(3-fluoro-2-hvdroxyphenyl)-6-methyl-5-(5-methyl-4-{6-r(methyloxy)methyll- 2-pyridinyU-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone
To a solution of 2-{3-fluoro-2-[(phenylmethyl)oxy]phenyl}-6-methyl-5-(5-methyl- 4-{6-[(methyloxy)methyl]-2-pyridinyl}-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone (45.4 mg, 0.072 mmol) in CH2Cl2 (0.72 mL) was added HBr (48% solution in AcOH, 0.086 mL, 0.719 mmol). The reaction mixture was for 24 h at rt, followed by quenching with 1 M aq. NaOH. The layers were separated and the aqueous phase was extracted twice with CH2Cl2. The combined organic layers were passed though a plug OfMgSO4 and
concentrated in vacuo. The residue was purified via flash column chromatography (0-25% EtOAc/hexanes) to afford the title compound (22.6 mg, 45%). LC/MS (m/z): 542 [M+H]+. d) 2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 -(5 -methyl-4- (6- [(methyloxy)methyl] - 2-pyridinvU-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone hydrochloride To a solution of 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-(5-methyl-4-{6-
[(methyloxy)methyl]-2-pyridinyl}-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone (60.8 mg, 0.112 mmol) in EtOAc (2.25 mL) was added HCl (1 M solution in Et2O, 0.561 mL, 0.561 mmol). After stirring for 5 min at rt, the resulting precipitate was collected by filtration and rinsed with EtOAc. Purification via flash column chromatography (0-5% acetone/CΗ2Cl2) provided the title compound (27 mg, 42%) as a yellow semisolid. 1H NMR (400 MHz, CDCl3) δ ppm 2.54 (s, 3 H), 2.71 (s, 3 H), 3.00 (t, 2 H), 3.52 (s, 3 H), 4.30 (t, 2 H), 4.66 (s, 2 H), 6.91-6.96 (m, 3 H), 7.02 (d, 1 H), 7.17-7.26 (m, 5 H), 7.34-7.45 (m, 3 H), 7.75 (t, 1 H), 8.72 (br s, 1 H). LC/MS (m/z): 542 [M+H]+.
Example 20

2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 -(4-methyl-5 - (6- [(methyloxy)methyl] -2- pyridinvU-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone hydrochloride a) 2- {3-fluoro-2-|Yphenylmethyl)oxy]phenyU -6-methyl-5 -(5 -methyl-4- (6- r(methyloxy)methyll-2-pyridinvU-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone
A solution of 5-(5-bromo-4-methyl-2-thienyl)-2- {3-fluoro-2-[(phenylmethyl)oxy]- phenyl}-6-methyl-3-(2-phenylethyl)-4(3Η)-pyrimidinone (500 mg, 0.848 mmol) and Pd[P(?-Bu)3]2 (43.3 mg, 0.085 mmol) in 1,4-dioxane (5.65 mL) was degassed with N2 for 15 min. Sn2Me6 (0.185 mL, 0.891 mmol) was added and the reaction vessel was heated at 110 0C for 2 h. Upon cooling, CsF (234 mg, 1.542 mmol), Pd[P(t-Bu)3]2 (39.4 mg, 0.077 mmol), and 2-bromo-6-[(methyloxy)methyl]pyridine (156 mg, 0.771 mmol) were added
and the reaction vessel was heated at 110 0C overnight. Upon cooling, the reaction was filtered through Celite® and concentrated in vacuo. Purification via flash column chromatography (0-25% EtOAc/hexanes) provided the title compound (127 mg, 26%). LC/MS (m/z): 632 [M+H]+. b) 2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 -(4-methyl-5 - (6- [(methyloxy)methyl] -
2-pyridinvU-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone
To a solution of 2-{3-fluoro-2-[(phenylmethyl)oxy]phenyl}-6-methyl-5-(4-methyl- 5-{6-[(methyloxy)methyl]-2-pyridinyl}-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone (127 mg, 0.201 mmol) in CH2Cl2 (2.01 mL) was added HBr (48% solution in AcOH, 0.241 mL, 2.010 mmol). The reaction mixture was for 24 h at rt, followed by quenching with 1 M aq. NaOH. The layers were separated and the aqueous phase was extracted twice with CH2Cl2. The combined organic layers were passed though a plug Of MgSO4 and concentrated in vacuo. The residue was combined with the crude reaction mixture from another run (performed on 0.037 mmol scale) and purified via flash column chromatography (0-25% EtOAc/hexanes) to afford the title compound (59.2 mg, 44% combined yield) as a yellow oil. LC/MS (m/z): 542 [M+H]+. c) 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-(4-methyl-5-{6-[(methyloxy)methyll- 2-pyridinvU-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone hydrochloride
To a solution of 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-(4-methyl-5-{6- [(methyloxy)methyl]-2-pyridinyl}-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone (59.2 mg, 0.109 mmol) in EtOAc (2.18 mL) was added HCl (1 M solution in Et2O, 0.545 mL, 0.545 mmol). After stirring for 15 min at rt, the resulting precipitate was collected by filtration, rinsed with EtOAc, and dried under high vacuum for 4 h to afford the title compound (36 mg, 54%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ ppm 2.48 (s, 3 H), 2.53 (s, 3 H), 2.81 (t, 2 H), 3.41 (s, 3 H), 3.95 (t, 2 H), 4.55 (s, 2 H), 6.82 (d, 2 H), 6.92-6.97 (m, 1 H), 6.99 (d, 1 H), 7.17-7.25 (m, 4 H), 7.33 (d, 1 H), 7.38-7.43 (m, 1 H), 7.63 (d, 1 H), 7.91 (t, 1 H), 10.51 (br s, 1 H). LC/MS (m/z): 542 [M+H]+.
Example 21 Pharmaceutical formulations:
Tablet Formulation An exemplary tablet formulation is formed by tableting the following mixture:
Ingredient Wt%
Compound* 50.8
Povidone K30 2.2
Microcrystalline cellulose 40.7
Croscarmellose sodium 5.3
Magnesium Stearate 1.0
* Compound is a compound of this invention
Parenteral formulation
A pharmaceutical composition for parenteral administration is prepared by dissolving an appropriate amount of a compound of Formula (I) in polyethylene glycol with heating. This solution is then diluted with water for injections (to 100 mL). The solution is then rendered sterile by filtration through a 0.22 micron membrane filter and sealed in sterile containers.
All publications, including but not limited to patents and patent applications cited in this specification are herein incorporated by reference as if each individual publication were specifically and individually indicated to be incorporated by reference as though fully set forth.
Claims
1. A compound according to Formula (I):

R1 is a 6-membered heteroaryl group, containing 1-2 nitrogen atoms, optionally substituted by -O-(Ci-C2)alkyl-O-, or one to three times, independently, by halogen, (Ci-C4)alkyl, -CF3, amino, (Ci-C4)alkylamino, (Ci-C4)alkyl(Ci-C4)alkylamino, (Ci-C4)alkoxy, hydroxy(Ci-C4)alkyl, or (Ci-C4)alkoxy(Ci-C4)alkyl; R2 is hydrogen or (Ci-C4)alkyl; wherein R1 is located at either the 4- or 5 -position of the thiophene ring and when R1 is located at the 4-position, R2 is located at the 5 -position, and when R1 is located at the 5 -position, R2 is located at the 4-position;
R3 is selected from the group consisting of (Ci-C4)alkyl, phenyl, and heteroaryl, optionally substituted, one to three times, independently, by halogen, (Ci-C4)alkyl, -CF3, or (Ci-C4)alkoxy;
R4 is selected from the group consisting of (C5-C6)cycloalkyl(Ci-C4)alkyl, heterocycloalkyl(Ci-C4)alkyl, aryl(Ci-C4)alkyl, and heteroaryl(Ci-C4)alkyl, wherein any cycloalkyl, heterocycloalkyl, aryl, or heteroaryl group is optionally substituted by -O-(Ci-C2)alkyl-O-, or one to three times, independently, by halogen, (Ci-C4)alkyl, -CF3, amino, (Ci-C4)alkylamino, (Ci-C4)alkyl(Ci-C4)alkylamino, or (Ci-C4)alkoxy; R5 is hydrogen or fluorine; or a salt thereof.
2. A compound according to Formula (Ia):
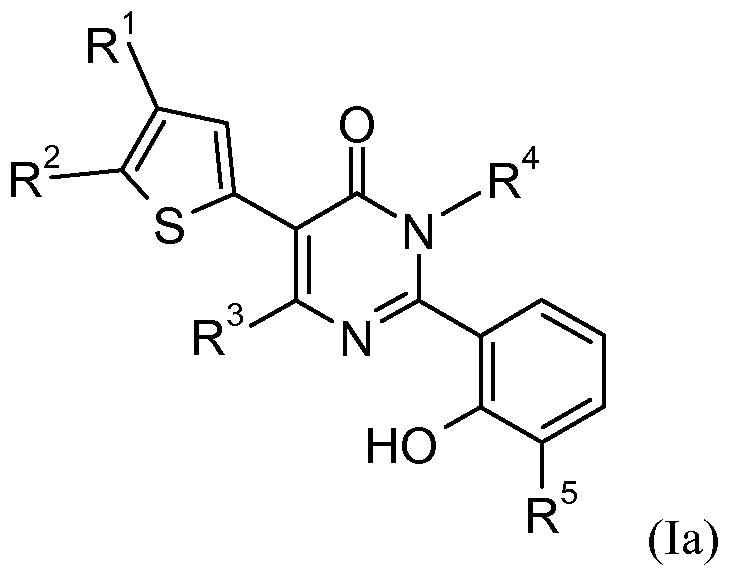
R1 is a 6-membered heteroaryl group, containing 1-2 nitrogen atoms, optionally substituted by -O-(Ci-C2)alkyl-O-, or one to three times, independently, by halogen, (Ci-C4)alkyl, -CF3, amino, (d-C4)alkylamino, (Ci-C4)alkyl(Ci-C4)alkylamino, (Ci-C4)alkoxy, hydroxy(Ci-C4)alkyl, or (Ci-C4)alkoxy(Ci-C4)alkyl;
R2 is hydrogen or (Ci-C4)alkyl;
R3 is selected from the group consisting of (Ci-C4)alkyl, phenyl, and heteroaryl, optionally substituted, one to three times, independently, by halogen, (Ci-C4)alkyl, -CF3, or (Ci-C4)alkoxy;
R4 is selected from the group consisting of (C5-C6)cycloalkyl(Ci-C4)alkyl, heterocycloalkyl(Ci-C4)alkyl, aryl(Ci-C4)alkyl, and heteroaryl(Ci-C4)alkyl, wherein any cycloalkyl, heterocycloalkyl, aryl, or heteroaryl group is optionally substituted by -O-(Ci-C2)alkyl-O-, or one to three times, independently, by halogen, (Ci-C4)alkyl, -CF3, amino, (Ci-C4)alkylamino, (Ci-C4)alkyl(Ci-C4)alkylamino, or (Ci-C4)alkoxy;
R5 is hydrogen or fluorine; or a salt thereof.
3. A compound according to Formula (Ib):

R1 is a 6-membered heteroaryl group, containing 1-2 nitrogen atoms, optionally substituted by -O-(Ci-C2)alkyl-O-, or one to three times, independently, by halogen, (Ci-C4)alkyl, -CF3, amino, (d-C4)alkylamino, (Ci-C4)alkyl(Ci-C4)alkylamino, (Ci-C4)alkoxy, hydroxy(Ci-C4)alkyl, or (Ci-C4)alkoxy(Ci-C4)alkyl;
R2 is hydrogen or (Ci-C4)alkyl;
R3 is selected from the group consisting of (Ci-C4)alkyl, phenyl, and heteroaryl, optionally substituted, one to three times, independently, by halogen, (Ci-C4)alkyl, -CF3, or (Ci-C4)alkoxy;
R4 is selected from the group consisting of (C5-C6)cycloalkyl(Ci-C4)alkyl, heterocycloalkyl(Ci-C4)alkyl, aryl(Ci-C4)alkyl, and heteroaryl(Ci-C4)alkyl, wherein any cycloalkyl, heterocycloalkyl, aryl, or heteroaryl group is optionally substituted by -O-(Ci-C2)alkyl-O-, or one to three times, independently, by halogen, (Ci-C4)alkyl, -CF3, amino, (Ci-C4)alkylamino, (Ci-C4)alkyl(Ci-C4)alkylamino, or (Ci-C4)alkoxy;
R5 is hydrogen or fluorine; or a salt thereof.
4. The compound or salt according to any one of Claims 1-3, wherein R4 is phenethyl, optionally substituted by -O-(Ci-C2)alkyl-O-, or one to two times, independently, by F, Cl, (Ci-C4)alkyl, (CrC4)alkoxy, or -CF3.
5. The compound or salt according to any one of Claims 1-3, wherein:
R1 is selected from the group consisting of pyridinyl, pyrazinyl, and pyrimidinyl, optionally substituted, one to three times, independently, by (Ci-C4)alkyl, -CF3, amino, (Ci-C4)alkylamino, (Ci-C4)alkyl(Ci-C4)alkylamino, (Ci-C4)alkoxy, hydroxy(Ci-C4)alkyl, or (Ci-C4)alkoxy(Ci-C4)alkyl;
R2 is hydrogen or (Ci-C4)alkyl; R3 is (Ci-C4)alkyl;
R4 is phenethyl, optionally substituted by -O-(Ci-C2)alkyl-O-, or one to two times, independently, by F, Cl, (Ci-C4)alkyl, (Ci-C4)alkoxy, or -CF3; and R5 is hydrogen or fluorine.
6. The compound or salt according to any one of Claims 1-3, wherein:
R1 is pyridinyl, optionally substituted, one to two times, independently, by (Ci-C4)alkyl, -CF3, (Ci-C4)alkoxy, hydroxy(Ci-C4)alkyl, or (Ci-C4)alkoxy(Ci-C4)alkyl; R2 is hydrogen or methyl;
R3 is methyl; R4 is phenethyl; and R5 is fluorine.
7. The compound or salt according to any one of Claims 1-6, wherein R1 is
2-pyridinyl, 3-methyl-2-pyridinyl, 6-methyl-2 -pyridinyl, 6-[(methyloxy)methyl]-2- pyridinyl, 3 -pyridinyl, 6-methyl-3 -pyridinyl, 6-trifluoromethyl-3 -pyridinyl, 6-hydroxymethyl-3 -pyridinyl, 4-pyridinyl, 2-pyrimidinyl, 5 -pyrimidinyl, or 2-pyrazinyl.
8. A compound which is:
2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[5-(2-pyridinyl)-2- thienyl] -4(3H)-pyrimidinone;
2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-[4-methyl-5-(2-pyridinyl)-2-thienyl]-3- (2-phenylethyl)-4(3H)-pyrimidinone; 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[4-(2-pyridinyl)-2- thienyl] -4(3H)-pyrimidinone; 2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 - [5 -methyl-4-(2-pyridinyl)-2-thienyl] -3 - (2-phenylethyl)-4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 - [5 -(3 -methyl-2-pyridinyl)-2-thienyl]-3 - (2-phenylethyl)-4(3H)-pyrimidinone; 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-[4-methyl-5-(3-methyl-2-pyridinyl)-2- thienyl]-3-(2-phenylethyl)-4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 - [5 -methyl-4-(3 -methyl-2-pyridinyl)-2- thienyl]-3-(2-phenylethyl)-4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 - [5 -(6-methyl-2-pyridinyl)-2-thienyl]-3 - (2-phenylethyl)-4(3H)-pyrimidinone;
2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[5-(3-pyridinyl)-2- thienyl] -4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 - [5 -(6-methyl-3 -pyridinyl)-2-thienyl]-3 - (2-phenylethyl)-4(3H)-pyrimidinone; 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5- {5-[6-
(trifluoromethyl)-3-pyridinyl]-2-thienyl}-4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-5 - {5 - [6-(hydroxymethyl)-3 -pyridinyl] -2-thienyl} -6- methyl-3-(2-phenylethyl)-4(3H)-pyrimidinone;
2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[5-(4-pyridinyl)-2- thienyl]-4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-3 -(2-phenylethyl)-5 - [5 -(2-pyrimidinyl)-2- thienyl] -4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-3 -(2-phenylethyl)-5 - [4-(2-pyrimidinyl)-2- thienyl] -4(3H)-pyrimidinone; 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[5-(5-pyrimidinyl)-2- thienyl] -4(3H)-pyrimidinone;
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-3 -(2-phenylethyl)-5 - [4-(5 -pyrimidinyl)-2- thienyl] -4(3H)-pyrimidinone;
2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5-[5-(2-pyrazinyl)-2- thienyl] -4(3H)-pyrimidinone; 2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 -(5 -methyl-4- {6- [(methyloxy)methyl] -2- pyridinyl} -2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone; or
2-(3 -fluoro-2-hydroxyphenyl)-6-methyl-5 -(4-methyl-5 - {6- [(methyloxy)methyl] -2- pyridinyl}-2-thienyl)-3-(2-phenylethyl)-4(3H)-pyrimidinone; or a salt thereof.
9. A compound which is 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5- [5-(2-pyridinyl)-2-thienyl]-4(3H)-pyrimidinone.
10. A compound which is 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-[5-methyl-4-(2- pyridinyl)-2-thienyl]-3-(2-phenylethyl)-4(3H)-pyrimidinone.
11. A compound which is 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-[5-methyl-4-(2- pyridinyl)-2-thienyl]-3-(2-phenylethyl)-4(3H)-pyrimidinone hydrochloride.
12. A compound which is 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-3-(2-phenylethyl)-5- [5-(2-pyridinyl)-2-thienyl]-4(3H)-pyrimidinone or a pharmaceutically acceptable salt thereof.
13. A compound which is 2-(3-fluoro-2-hydroxyphenyl)-6-methyl-5-[5-methyl-4-(2- pyridinyl)-2-thienyl]-3-(2-phenylethyl)-4(3H)-pyrimidinone or a pharmaceutically acceptable salt thereof.
14. A pharmaceutical composition which comprises the compound or salt according to any one of Claims 1-13 and a pharmaceutically acceptable diluent, carrier and/or excipient.
15. A process for preparing the composition as defined in Claim 14, the process comprising mixing the compound or salt as defined in Claims 1-13 with a pharmaceutically acceptable diluent, carrier, and/or excipient.
16. A method of antagonizing a calcium receptor, which comprises administering to a human in need thereof, an effective amount of the compound or salt according to any one of Claims 1-13.
17. A method of treating a disease or disorder characterized by an abnormal bone or mineral homeostasis, which comprises administering to a human in need thereof, an effective amount of the compound or salt according to any one of Claims 1-13.
18. A method according to Claim 17 wherein the abnormal bone or mineral homeostasis disease or disorder is selected from the group consisting of osteosarcoma, periodontal disease, bone fracture, osteoarthritis, rheumatoid arthritis, Paget's disease, humoral hypercalcemia, malignancy, osteopenia, and osteoporosis.
19. A method according to Claim 18 wherein the bone or mineral disease or disorder is osteoporosis.
20. A method of increasing serum parathyroid levels which comprises administering to a human in need thereof, an effective amount of the compound or salt according to any one of Claims 1-13.
21. A method according to Claim 18 wherein the compound according to Formula (I) is co-administered with an anti-resorptive agent.
22. A method according to Claim 21 wherein the anti-resorptive agent is selected from the group consisting of estrogens, 101,25-(OH)2Ds, lα-(0H)U3, calcitonin, denosumab, selective estrogen receptor modulators, vitronectin receptor antagonists, V-H+-ATPase inhibitors, src SH2 antagonists, bisphosphonates and cathepsin K inhibitors.
23. A method according to Claim 22 wherein the selective estrogen receptor modulator is selected from the group consisting of lasofoxifene, raloxifene, arzoxifene, bazedoxifene, and ospemifene.
24. A method according to Claim 22 wherein the bisphosphonate is selected from the group consisting of tiludronate, clondronate, etidronate, alendronate, risedronate, ibandronate, zoledronate, minodronate, neridronate, and pamidronate.
INTERNATIONAL SEARCH REPORT International application No PCT/US 09/59175
Box No. 11 Observations where certain claims were found unsearchable (Continuation of item 2 of first sheet)
This international search report has not been established in respect of certain claims under Article 17(2)(a) for the following reasons
1 I I Claims Nos because they relate to subject matter not required to be searched by this Authority, namely
Claims Nos because they relate to parts of the international application that do not comply with the prescribed requirements to such an extent that no meaningful international search can be earned out, specifically
IAI Claims Nos 7 and 14-24 because they are dependent claims and are not drafted in accordance with the second and third sentences of Rule 6 4(a)
Box No. Ill Observations where unity of invention is lacking (Continuation of item 3 of first sheet)
This International Searching Authority found multiple inventions in this international application, as follows
1 I I As all required additional search fees were timely paid by the applicant, this international search report covers all searchable claims
2 I I As all searchable claims could be searched without effort justifying additional fees, this Authority did not invite payment of additional fees
3 I I As only some of the required additional search fees were timely paid by the applicant, this international search report covers only those claims for which fees were paid, specifically claims Nos
No required additional search fees were timely paid by the applicant Consequently, this international search report is restπcted to the invention first mentioned in the claims, it is covered by claims Nos
Remark on Protest | | The additional search fees were accompanied by the applicant's protest and, where applicable, the payment of a protest fee
I I The additional search fees were accompanied by the applicant's protest but the applicable protest
^_^ fee was not paid within the time limit specified in the invitation
I I No protest accompanied the payment of additional search fees
Form PCT/ISA/210 (continuation of first sheet (2)) (April 2007)
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10270808P | 2008-10-03 | 2008-10-03 | |
| US61/102,708 | 2008-10-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010039922A1 true WO2010039922A1 (en) | 2010-04-08 |
Family
ID=42073878
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/059175 WO2010039922A1 (en) | 2008-10-03 | 2009-10-01 | Calcilytic compounds |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2010039922A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013098588A1 (en) * | 2011-12-27 | 2013-07-04 | Ubaldo Armato | Use of calcilytic drugs as a pharmacological approach to the treatment and prevention of alzheimer's disease, alzheimer's disease-related disorders, and down's syndrome neuropathies |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007062370A2 (en) * | 2005-11-22 | 2007-05-31 | Smithkline Beecham Corporation | Calcilytic compounds |
| US20070232628A1 (en) * | 2004-05-06 | 2007-10-04 | Luengo Juan I | Calcilytic Compounds |
-
2009
- 2009-10-01 WO PCT/US2009/059175 patent/WO2010039922A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070232628A1 (en) * | 2004-05-06 | 2007-10-04 | Luengo Juan I | Calcilytic Compounds |
| WO2007062370A2 (en) * | 2005-11-22 | 2007-05-31 | Smithkline Beecham Corporation | Calcilytic compounds |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013098588A1 (en) * | 2011-12-27 | 2013-07-04 | Ubaldo Armato | Use of calcilytic drugs as a pharmacological approach to the treatment and prevention of alzheimer's disease, alzheimer's disease-related disorders, and down's syndrome neuropathies |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4504375B2 (en) | 2,4-di (hetero) -arylaminopyrimidine derivatives as ZAP-70 and / or SYK inhibitors | |
| JP2021508686A (en) | Triazole N-linked carbamoylcyclohexyl as an LPA antagonist | |
| TWI767410B (en) | Triazole carbamate pyridyl sulfonamides as lpa receptor antagonists and uses thereof | |
| KR20200100719A (en) | Cyclohexyl acid triazole azine as an LPA antagonist | |
| JP4944286B1 (en) | Cyclopropane compound | |
| NO314503B1 (en) | N-Heter Garyl pyridine sulfonamide derivatives, their use and preparation, and pharmaceutical composition | |
| WO2014079136A1 (en) | Compounds, pharmaceutical compositions and uses as glutaminase inhibitors for treating cancers thereof | |
| CZ302359B6 (en) | Triazole compounds and their use | |
| TW201927778A (en) | Cyclohexyl acid triazole azoles as LPA antagonists | |
| WO2011143426A1 (en) | Compounds useful as inhibitors of atr kinase | |
| US8198285B2 (en) | Pyrazine derivatives | |
| JPWO2010038465A1 (en) | 8-substituted isoquinoline derivatives and uses thereof | |
| JP6559699B2 (en) | Substituted nitrogen-containing heterocyclic derivatives, pharmaceutical compositions containing them, and their antitumor applications | |
| CA2830706A1 (en) | Quinolinone derivatives | |
| JP5753625B2 (en) | 5- (Phenyl / pyridinyl-ethynyl) -2-pyridine / 2-pyrimidine-carboxamide as mGluR5 modulator | |
| JP6883045B2 (en) | Halo-substituted piperidine as an orexin receptor regulator | |
| US20090093494A1 (en) | 4-Pyrimidineamine Compounds And Uses As Anti-Proliferative Agents | |
| JP5326108B2 (en) | Substituted sulfoximines and salts thereof as Tie2 inhibitors, pharmaceutical compositions containing them, methods of preparing them and uses thereof | |
| EA015574B1 (en) | Triazolyl aminopyrimidine compounds | |
| JP2010513553A (en) | Imidazolidinylaminopyrimidine compounds for cancer treatment | |
| WO2010039922A1 (en) | Calcilytic compounds | |
| JPWO2016098793A1 (en) | Thiazole derivatives having a cyclic guanidyl group | |
| WO2021117759A1 (en) | Histone deacetylase inhibitor having nitrogen-containing aromatic heterocyclic group | |
| US20050222228A1 (en) | Novel diazine derivatives | |
| WO2010039913A1 (en) | Calcilytic compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09818494 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009818494 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09818494 Country of ref document: EP Kind code of ref document: A1 |