WO2010037688A1 - Process for the synthesis of halogenated cyclic compounds - Google Patents
Process for the synthesis of halogenated cyclic compounds Download PDFInfo
- Publication number
- WO2010037688A1 WO2010037688A1 PCT/EP2009/062462 EP2009062462W WO2010037688A1 WO 2010037688 A1 WO2010037688 A1 WO 2010037688A1 EP 2009062462 W EP2009062462 W EP 2009062462W WO 2010037688 A1 WO2010037688 A1 WO 2010037688A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- process according
- compound
- alkyl group
- group
- Prior art date
Links
- 0 *C(CC(*)=O)Cl Chemical compound *C(CC(*)=O)Cl 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
- C07D213/80—Acids; Esters in position 3
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
- C07D213/803—Processes of preparation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/34—One oxygen atom
- C07D239/36—One oxygen atom as doubly bound oxygen atom or as unsubstituted hydroxy radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to a process for the synthesis of halogenated cyclic compounds.
- Halogenated cyclic compounds are useful for example as intermediates for the preparation of various herbicides, insecticides, miticides, pesticides etc.
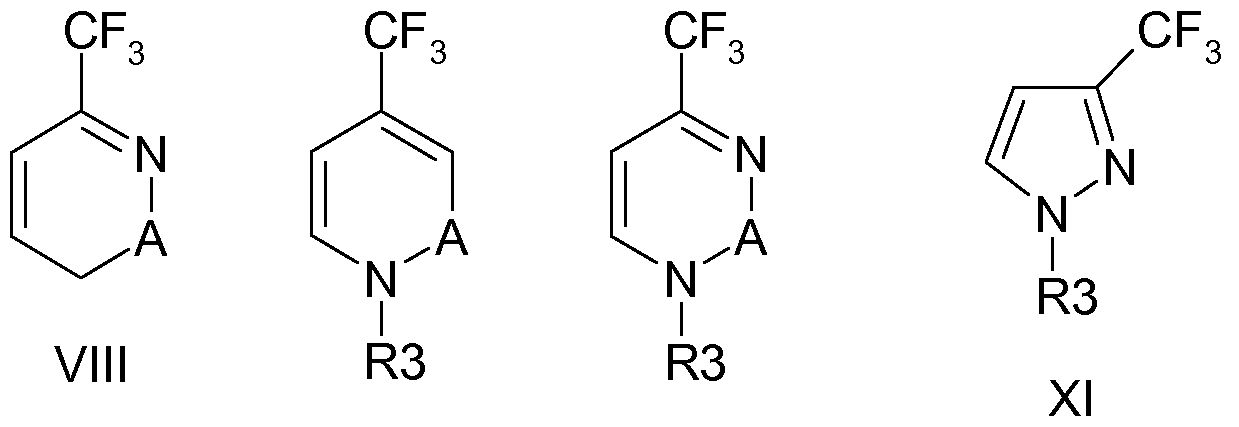
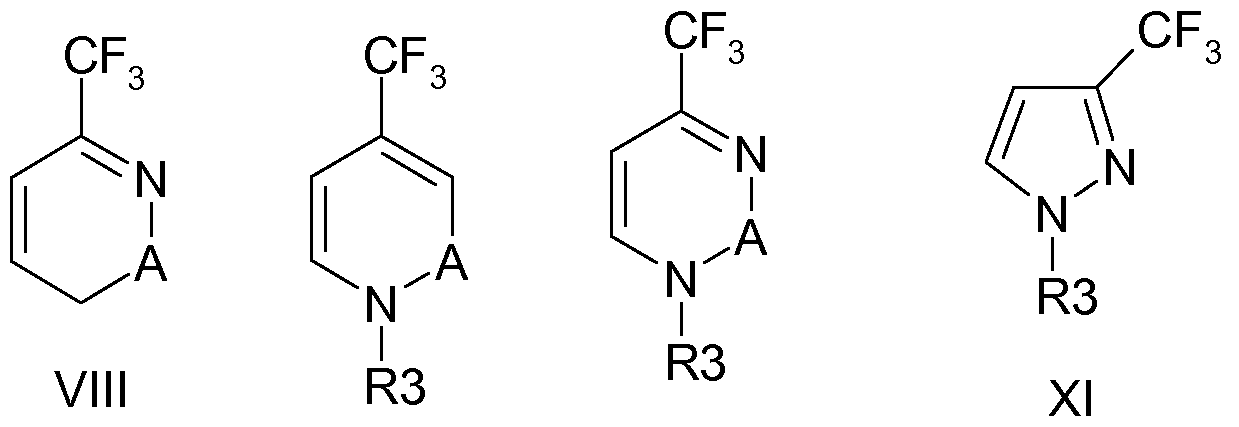
- U.S. patent application US 2006/0128702 Al describes the synthesis of 3-trifluoromethyl-lH-Pyrazole (TFPZO) by reacting 4-ethoxy- 1,1,1 -trifluoro- 3-buten-2-one with hydrazine dihydrochloride.
- Atherton and Fields, J. Chem. Soc. (C), 1968, p.1507-1513 describe the synthesis of 3-trifluoromethyl-lH-Pyrazole from 2,2,2-trifluorodiazoethane.
- the TFPZO compound is used as intermediate in chemical synthesis.
- European patent application EP-A- 163280 discloses the manufacture of 2-hydroxy-4- trifluoromethyl pyrimidine (TFPMO) from the reaction product of TFAH with EVE.
- TFPMO 2-hydroxy-4- trifluoromethyl pyrimidine
- the TFPMO compound is used as intermediate in chemical synthesis, to produce pyrimidinylphosphates as pesticides. It is an object of the present invention to provide an efficient process for manufacturing halogenated cyclic compounds, in particular halogenated nitrogen containing heterocycles which allows for good yields and product purities, starting from readily available starting materials.
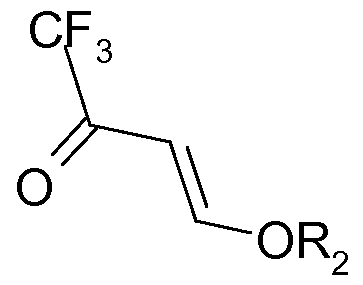
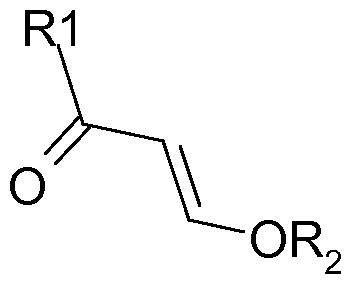
- the invention concerns in consequence a process for the manufacture of a cyclic compound of formula (I) :
- Ri is a halogenated alkyl group Z and Y designate independently carbon or a heteroatom
- the addition product of an acid halide in particular acid chloride with a vinyl ether as specified above is particularly suitable as starting material for producing cyclic compounds as detailed above.
- Overall yield starting from acid halide and vinyl ether is good.
- a purification of the addition product may be unnecessary before further reaction.
- the addition product, in particular if hydrogen halide is present, may increase the reaction rate in the reaction with the compound of formula (III) and allow for easier separation of the cyclic compound produced from the reaction mixture, in particular when a nitrogen-containing heterocycle is produced.
- Rl preferably contains at least 1, more preferably 2 fluorine atoms.
- Rl is a perfluoroalkyl group.
- Rl is often a Cl to C3 halogenated alkyl group, preferably a Cl to C3 fluorinated alkyl group, in particular as described here before.
- Rl is more preferably a Cl fluorinated alkyl group selected in particular from CF3, CF 2 H and CF 2 Cl.
- a CF 3 group is more particularly preferred.
- Rl preferably contains at least 1, more preferably 2 fluorine atoms and at least 1 other halogen atom.
- Rl is more preferably a fluorinated Cl alkyl group selected in particular from CF 2 Br and CF 2 Cl.
- X is often selected from Cl and Br and is more preferably Cl.
- Acid halides used in the present invention can be obtained, for example, by photo xidation of halogenated precursor alkanes, in particular as described in US 5,569,782 the contents of which is incorporated by reference in the present application.
- trifluoro acetyl chloride which is a particularly preferred starting material in the present invention, can be obtained by photooxidation of 1,1,1 -Trifluoro-2,2-dichloroethane (HCFC-123).
- R 2 is often selected from a Cl to C4 alkyl group, preferably a methyl, an ethyl, an isopropyl or a n-butyl group and is more preferably an ethyl group.
- the acid halide is trifluoro acetyl chloride and the vinyl ether is ethyl vinyl ether or methyl vinyl ether, more preferably ethyl vinyl ether.
- Z and Y can be independently selected from oxygen, sulphur, nitrogen and carbon. The process according to the invention applies in particularly advantageous manner when at least one of Z and Y is nitrogen.
- Z and/or Y are selected from -NH2, -NHR3, wherein R3 is an alkyl aryl or aralkyl residue, preferably a Cl to C6 alkyl, or hydrogen halide salts thereof, -OH, -SH and donor carbon atoms for example a CH2 group in ⁇ -position to a carbonyl function such as a keto or ester group.
- A is a linking group between Z and Y.
- Such linking group can simply be a covalent bond, in which case A contains 0 atoms.
- the linking group A can also contain 1, 2 or 3 atoms, which form part of the ring (annular atoms) when A forms part of the structure of compound (I) and which are catenary (i.e. in the chain linking Z and Y) when A forms part of the structure of compound (III).
- A comprises 1 or 2 catenary/annular atoms, in particular, optionally substituted carbon atoms.
- compound of formula (III) are selected from hydrazine or its hydrate or hydrochloride, urea, thiourea, malonic acid monoamide (for example methyl or ethyl ester of malonic acid monoamide), and malomononitrile (2-cyano-acetic acid alkyl ester, for example methyl or ethyl ester).
- step (b) comprises 1 catenary/annular atoms, optionally substituted carbon atoms.
- the reaction of step (b) is generally carried out at a temperature in the range from -15° C to +80 0 C, preferably from 0° C to +20 0 C.
- the reaction of step (b) can be carried out in the presence of a non-hindered amine.
- Non-hindered amines refers to chemical compounds containing an amine functional group bonded to non- sterically hindering groups.
- Typical examples of non-sterically hindering groups are short linear aliphatic groups such as methyl, ethyl, propyl and n-butyl
- Typical examples of non-hindered amines are for example methylamine, diethylamine, triethylamine and tri-n-butylamine. Triethylamine is most preferred non- hindered amine.
- the molar ratio of the non- hindered amine to the compound of formula (III) is advantageously from 0.5:1 to 1.7:1, preferably from 0.6:1 to 1.5:1, and more preferably from 0.8:1 to 1.2:1. Most preferably, the molar ratio is about 1.
- the reaction of step (b) can optionally be carried out in the presence of an additive. Such additive generally increases the polarity of the reaction medium. Ionic liquids can be suitably used as additives.
- additives are for instance 1,3-dialkylimidazolium or 1,3-dialkyl piridinium salts in particular l-ethyl-3- methylimidazolium trifluoromethanesulfonate (EMIMOtf).
- Amines such as 4-Dimethylaminopyridine (DMAP), Diazabicyclo [5.4.0] undec-7-ene (DBU) and Diazabicyclononan (DBN) as such or under salt form, for example as trifluoro acetic acid salt can also be suitably used as additive.
- DMAP 4-Dimethylaminopyridine
- DBU Diazabicyclo [5.4.0] undec-7-ene
- DBN Diazabicyclononan
- the term "ionic liquid” refers to a homogeneous composition consisting of a single salt (one cationic species and one anionic species) or it may refer to a heterogeneous composition containing more than one species of cation and/or more than one species of anion.
- the reaction of step (b) is often carried out in an organic solvent, in particular a polar organic solvent.
- Alcohols such as methanol or ethanol give good results in the reaction of step (b). Methanol is particularly preferred.
- the process according to the invention suitably further comprises isolating the compound of formula (I) from the reaction medium of step (b) by solid/liquid separation, for example filtration or by distillation.
- the addition product comprises a compound of formula (IV) :
- the addition product comprises a compound of formula (V) :
- R 2 is as defined above.
- the addition product comprises a compound of formula (IV) or (V)
- its content is generally at least 0.1 wt. % relative to the total weight of addition product. Often this content is at least 0.5 %. In some embodiments this content does not exceed 10 wt. %.
- the addition product can also consist essentially of compound of formula (IV) or (V). In this case its content is generally from 90-99.9, often from 95 to 99.0 wt. % relative to the total weight of addition product.
- the addition product comprises a compound of formula (VI) :
- the addition product comprises a compound of formula (VII) :
- R 2 is as defined above.
- the compounds of formulae (VI) and (VII) can be obtained preferably by a process comprising (a) adding, as described above, acid halide to vinyl ether to obtain a reaction product comprising compound of formula (IV) or (V) and (b) eliminating hydrogen halide to produce a compound of formula (VI) or (VII) respectively.
- Such reaction can be carried out in the presence of base such as described in US patent 5,708,174 or, preferably, in the absence of base such as described in US patent 7,405,328, the entire contents of said two US patents being incorporated by reference into the present application.
- the addition product comprises a compound of formula (VI) or (VII)
- its content is generally at least 0.1 wt. % relative to the total weight of addition product. Often this content is at least 0.5 %. In some embodiments this content does not exceed 10 wt. %.
- the addition product can also consist essentially of compound of formula (VI) or (VII). In this case its content is generally from 90-99.9, often from 95 to 99.0 wt. % relative to the total weight of addition product.
- the addition product can be a mixture of compounds of formula (IV) and (VI) or (V) and (VII) respectively.
- the molar ratio between compounds (IV) and (V) on the one hand and (VI ) and (VII) on the other hand is generally from 0.01 to 100, preferably from O.l to lO.
- HX, in particular HCl is fed to step (b).
- HX, in particular HCl, produced in step (a) may suitably be fed to step (b).
- at least one of Z or Y is nitrogen, it has been found that it is possible to precipitate the heterocycle from the reaction medium of step (b) by addition of HX in particular HCl and to isolate it for example by filtration.
- the HX addition can be carried out, for example during reaction or during work-up.
- the invention also concerns a process for the synthesis of a cyclic compound of formula (I) :
- Ri is a halogenated alkyl group Z and Y designate independently carbon or a heteroatom
- Example 1 Manufacture of 6-(trifluoromethyl)pyrimidin-2( 1 H)-on To a solution of 8.74 moles of urea in 2.2L of methanol in a 3-necked flask, equipped with a mechanical stirrer, a reflux condenser and a dropping funnel was added dropwise over about 3 hours under N 2 atmosphere an equimolar amount of 4-ethoxy- 1,1,1 -trifluoro-3-buten-2-one (ETFBO) which had been manufactured by addition of trifluoroacetyl chloride to ethyl vinyl ether. The temperature of the reaction mixture was kept below 15 0 C.
- ETFBO 4-ethoxy- 1,1,1 -trifluoro-3-buten-2-one
- CETFBO could be obtained quantitatively without any elimination of HCl.
- 6-(trifluoromethyl)pyrimidin-2( lH)-on was obtained as pale beige crystals which were filtered off and washed with water. After drying on a rotary evaporator under reduced pressure, 6-(trifluoromethyl)pyrimidin-2(lH)-on was obtained in 82 % yield.
- Example 4 6-(trifluoromethyl)pyrimidin-2( lH)-on was obtained as pale beige crystals which were filtered off and washed with water. After drying on a rotary evaporator under reduced pressure, 6-(trifluoromethyl)pyrimidin-2(lH)-on was obtained in 82 % yield.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pyridine Compounds (AREA)
Abstract
Description





Claims





Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2009801384364A CN102164898A (en) | 2008-09-30 | 2009-09-25 | Process for the synthesis of halogenated cyclic compounds |
| US13/120,505 US8431710B2 (en) | 2008-09-30 | 2009-09-25 | Process for the synthesis of halogenated cyclic compounds |
| KR1020197018768A KR20190080972A (en) | 2008-09-30 | 2009-09-25 | Process for the synthesis of halogenated cyclic compounds |
| JP2011529509A JP6180705B2 (en) | 2008-09-30 | 2009-09-25 | Method for the synthesis of halogenated cyclic compounds |
| KR1020167031136A KR20160131129A (en) | 2008-09-30 | 2009-09-25 | Process for the synthesis of halogenated cyclic compounds |
| CA2735921A CA2735921C (en) | 2008-09-30 | 2009-09-25 | Process for the synthesis of halogenated cyclic compounds |
| EP09783436.0A EP2334645B1 (en) | 2008-09-30 | 2009-09-25 | Process for the synthesis of fluorinated cyclic compounds |
| US13/872,860 US8981115B2 (en) | 2008-09-30 | 2013-04-29 | Process for the synthesis of halogenated cyclic compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08165547 | 2008-09-30 | ||
| EP08165547.4 | 2008-09-30 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/120,505 A-371-Of-International US8431710B2 (en) | 2008-09-30 | 2009-09-25 | Process for the synthesis of halogenated cyclic compounds |
| US13/872,860 Continuation US8981115B2 (en) | 2008-09-30 | 2013-04-29 | Process for the synthesis of halogenated cyclic compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010037688A1 true WO2010037688A1 (en) | 2010-04-08 |
Family
ID=40351699
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/062462 WO2010037688A1 (en) | 2008-09-30 | 2009-09-25 | Process for the synthesis of halogenated cyclic compounds |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US8431710B2 (en) |
| EP (1) | EP2334645B1 (en) |
| JP (5) | JP6180705B2 (en) |
| KR (3) | KR20110066201A (en) |
| CN (2) | CN106083710B (en) |
| CA (1) | CA2735921C (en) |
| WO (1) | WO2010037688A1 (en) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8426650B2 (en) | 2009-07-06 | 2013-04-23 | Solvay Sa | Process for the manufacture of halogenated precursors of alkenones in the presence of a solvent |
| US8431710B2 (en) | 2008-09-30 | 2013-04-30 | Solvay Sa | Process for the synthesis of halogenated cyclic compounds |
| US8519195B2 (en) | 2008-07-04 | 2013-08-27 | Solvay Sa | Process for the manufacture of alkenones |
| EP2662359A1 (en) | 2012-05-08 | 2013-11-13 | Solvay Sa | Ionic Liquids, Method for manufacturing thereof, and Electrochemical Devices Comprising the Same |
| WO2014029786A1 (en) * | 2012-08-22 | 2014-02-27 | Solvay Sa | Process for the manufacture of alkenones |
| WO2014096284A1 (en) | 2012-12-20 | 2014-06-26 | Solvay Sa | Salts of n-containing heterocyclic anions as components in electrolytes |
| WO2014202599A1 (en) * | 2013-06-20 | 2014-12-24 | Basf Se | Process for preparing pyridylpyrazole compounds and derivatives thereof from pyridylhydrazine |
| US8957254B2 (en) | 2009-07-06 | 2015-02-17 | Solvay Sa | Process for chemical synthesis from an alkenone made from a halogenated precursor |
| EP2987782A1 (en) | 2014-08-22 | 2016-02-24 | Solvay SA | Distillation process comprising at least two distillation steps to obtain purified halogenated carboxylic acid halide, and use of the purified halogenated carboxylic acid halide |
| WO2016079122A1 (en) | 2014-11-17 | 2016-05-26 | Solvay Sa | A method for producing a chemical compound and apparatus therefor |
| WO2016079126A1 (en) | 2014-11-17 | 2016-05-26 | Solvay Sa | Distillation process comprising at least two distillation steps to obtain purified halogenated carboxylic acid halide, and use of the purified halogenated carboxylic acid halide |
| EP3031791A1 (en) | 2014-12-10 | 2016-06-15 | Solvay SA | Halogenation process of 1,1-dihaloethene |
| EP3199522A1 (en) | 2016-02-01 | 2017-08-02 | Solvay SA | Aminothioate derivatives, production thereof and use |
| WO2019043238A1 (en) | 2017-09-04 | 2019-03-07 | Solvay Sa | Process and intermediate for the manufacture of difluoroacetyl chloride |
| EP3650443A1 (en) | 2018-11-07 | 2020-05-13 | Fujian Yongjing Technology Co., Ltd. | Continuous flow synthesis of fluorinated or non-fluorinated pyrazoles |
| WO2021152055A1 (en) | 2020-01-31 | 2021-08-05 | Solvay Sa | Process for the manufacture of haloalkyl substituted pyridine compounds |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0903749D0 (en) * | 2009-03-04 | 2009-04-15 | Syngenta Participations Ag | Chemical process |
| US11390587B2 (en) * | 2018-12-27 | 2022-07-19 | Corteva Agriscience Llc | Preparation of sulfonamide herbicide process intermediates |
| JP2021079146A (en) * | 2021-02-22 | 2021-05-27 | 株式会社三洋物産 | Game machine |
| JP7315071B2 (en) * | 2021-03-03 | 2023-07-26 | 株式会社三洋物産 | game machine |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0163280A1 (en) * | 1984-05-30 | 1985-12-04 | BASF Aktiengesellschaft | Pyrimidylphosphoric acid esters, process for their preparation and their use for pest control |
Family Cites Families (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0502740B1 (en) * | 1991-03-07 | 1998-05-27 | E.I. Du Pont De Nemours And Company | Herbicidal pyridine sulfonamide |
| US5235060A (en) * | 1991-07-11 | 1993-08-10 | E. I. Du Pont De Nemours And Company | Certain three component ionic substituted pyridine compounds as intermediates for preparation as herbicides |
| DE4128828A1 (en) | 1991-08-30 | 1993-03-04 | Basf Ag | AMMONIUM OR UREA-CONTAINED DISPENSERS AND METHOD FOR THEIR PRODUCTION |
| WO1994008994A1 (en) | 1992-10-13 | 1994-04-28 | Smithkline Beecham Plc | Heterocyclic -esters or -amides used as 5-ht4 receptor antagonists |
| EP0659729B1 (en) * | 1993-12-23 | 1998-06-24 | Solvay Fluor und Derivate GmbH | Process for preparing chlorides of polyfluorochloro- and perfluoro carboxylic acids in the presence of chlorine |
| JP3843152B2 (en) * | 1995-08-08 | 2006-11-08 | 石原産業株式会社 | Process for producing 4-alkoxy-1,1,1-trifluoro-3-buten-2-one |
| GB2305174A (en) * | 1995-09-15 | 1997-04-02 | Zeneca Ltd | Chemical process |
| GB9711114D0 (en) * | 1997-05-29 | 1997-07-23 | Merck Sharp & Dohme | Therapeutic agents |
| DE19821088A1 (en) | 1998-05-12 | 1999-11-18 | Basf Ag | New polyaspartic acids modified with pyrazole compounds, useful as stable nitrification inhibitors for addition to mineral fertilizers |
| WO2002053518A2 (en) | 2000-12-29 | 2002-07-11 | Honeywell International, Inc. | HALOGENATED-α,β-UNSATURATED-β-(SUBSTITUTED-AMINO) CARBOXYLATE ESTERS |
| JP4673293B2 (en) | 2003-03-07 | 2011-04-20 | シンジェンタ パーティシペーションズ アクチェンゲゼルシャフト | Process for the production of substituted nicotinic acid esters |
| DE10325715A1 (en) | 2003-06-06 | 2004-12-23 | Solvay Fluor Und Derivate Gmbh | Simplified production of alkenones |
| US20060128702A1 (en) | 2004-11-23 | 2006-06-15 | Manojit Pal | Heterocyclic and bicyclic compounds, compositions and methods |
| DE102004061593A1 (en) | 2004-12-21 | 2006-06-22 | Abbott Gmbh & Co. Kg | Substituted N-heterocyclic compounds and their therapeutic use |
| EP1940842B1 (en) | 2005-09-29 | 2012-05-30 | Merck Sharp & Dohme Corp. | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| WO2007088876A1 (en) * | 2006-02-02 | 2007-08-09 | Kumiai Chemical Industry Co., Ltd. | Pyridone derivative and herbicide |
| EP2422790A1 (en) * | 2007-02-09 | 2012-02-29 | Emory University | Paramyxovirus family inhibitors and methods of use thereof |
| EP1987717A1 (en) * | 2007-04-30 | 2008-11-05 | Bayer CropScience AG | Pyridon carboxamides, agents containing these but not impacting useful plants and method for their manufacture and application |
| EP2008996A1 (en) | 2007-06-27 | 2008-12-31 | Syngeta Participations AG | Process for the production of pyrazoles |
| WO2010002577A1 (en) * | 2008-07-01 | 2010-01-07 | Dow Agrosciences Llc | Improved process for the preparation of 2-trifluoromethyl-5-(1-substituted)alkylpyridines |
| US8519195B2 (en) | 2008-07-04 | 2013-08-27 | Solvay Sa | Process for the manufacture of alkenones |
| EP2154167A1 (en) * | 2008-07-30 | 2010-02-17 | Bayer MaterialScience AG | Electromechanical converter with a polymer element on a polyisocyanate basis |
| KR20110066201A (en) * | 2008-09-30 | 2011-06-16 | 솔베이(소시에떼아노님) | Process for the synthesis of halogenated cyclic compounds |
| CN102471202B (en) | 2009-07-06 | 2015-10-07 | 索尔维公司 | Manufacture the method for the halogenated precursors of ketenes in the presence of solvent |
| CN102471201A (en) | 2009-07-06 | 2012-05-23 | 索尔维公司 | Process for the manufacture of halogenated precursors of alkenones under specific conditions |
| EP2451764B1 (en) | 2009-07-06 | 2016-01-13 | Solvay Sa | Process for the manufacture of alkenones |
| JP6194245B2 (en) * | 2013-12-27 | 2017-09-06 | 株式会社Subaru | Traffic light recognition device |
-
2009
- 2009-09-25 KR KR1020117009786A patent/KR20110066201A/en active Search and Examination
- 2009-09-25 US US13/120,505 patent/US8431710B2/en not_active Expired - Fee Related
- 2009-09-25 KR KR1020167031136A patent/KR20160131129A/en active Search and Examination
- 2009-09-25 WO PCT/EP2009/062462 patent/WO2010037688A1/en active Application Filing
- 2009-09-25 KR KR1020197018768A patent/KR20190080972A/en not_active Application Discontinuation
- 2009-09-25 CA CA2735921A patent/CA2735921C/en not_active Expired - Fee Related
- 2009-09-25 CN CN201610380671.4A patent/CN106083710B/en active Active
- 2009-09-25 CN CN2009801384364A patent/CN102164898A/en active Pending
- 2009-09-25 JP JP2011529509A patent/JP6180705B2/en active Active
- 2009-09-25 EP EP09783436.0A patent/EP2334645B1/en active Active
-
2013
- 2013-04-29 US US13/872,860 patent/US8981115B2/en not_active Expired - Fee Related
-
2015
- 2015-06-23 JP JP2015125708A patent/JP6511346B2/en active Active
-
2017
- 2017-11-24 JP JP2017225725A patent/JP6647268B2/en active Active
-
2019
- 2019-10-10 JP JP2019186868A patent/JP2020023533A/en active Pending
-
2021
- 2021-11-12 JP JP2021184880A patent/JP2022028792A/en active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0163280A1 (en) * | 1984-05-30 | 1985-12-04 | BASF Aktiengesellschaft | Pyrimidylphosphoric acid esters, process for their preparation and their use for pest control |
Non-Patent Citations (4)
| Title |
|---|
| AGENOR COLLA ET AL: "Trihaloacetylated enol ethers - General synthetic procedure and heterocyclic ring closure reactions with hydroxylamine", SYNTHESIS, GEORG THIEME VERLAG, STUTTGART, DE, 1991, pages 483 - 486, XP000196317, ISSN: 0039-7881 * |
| FRANZ EFFENBERGER ET AL: "Die Acylierung von Enolethern mit reaktiven Carbonsäurechloriden", CHEMISCHE BERICHTE, VERLAG CHEMIE GMBH. WEINHEIM, DE, vol. 115, 1982, pages 2766 - 2782, XP002295515, ISSN: 0009-2940 * |
| MANFRED SCHLOSSER ET AL: "Switchable reactivity: the site-selective functionalization of trifluoromethyl-substituted pyrazoles", EUROPEAN JOURNAL OF ORGANIC CHEMISTRY, WILEY-VCH VERLAG, WEINHEIM, DE, no. 17, 2002, pages 2913 - 2920, XP002462801, ISSN: 1434-193X * |
| MARINE G. GORBUNOVA ET AL: "Synthesis and properties of ß-ethoxyvinyl polyfluoroalkyl ketones", SYNTHESIS, GEORG THIEME VERLAG, STUTTGART, DE, no. 5, 2000, pages 738 - 742, XP002295516, ISSN: 0039-7881 * |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8519195B2 (en) | 2008-07-04 | 2013-08-27 | Solvay Sa | Process for the manufacture of alkenones |
| US8431710B2 (en) | 2008-09-30 | 2013-04-30 | Solvay Sa | Process for the synthesis of halogenated cyclic compounds |
| US8981115B2 (en) | 2008-09-30 | 2015-03-17 | Solvay Sa | Process for the synthesis of halogenated cyclic compounds |
| US8957254B2 (en) | 2009-07-06 | 2015-02-17 | Solvay Sa | Process for chemical synthesis from an alkenone made from a halogenated precursor |
| US8426650B2 (en) | 2009-07-06 | 2013-04-23 | Solvay Sa | Process for the manufacture of halogenated precursors of alkenones in the presence of a solvent |
| EP2662359A1 (en) | 2012-05-08 | 2013-11-13 | Solvay Sa | Ionic Liquids, Method for manufacturing thereof, and Electrochemical Devices Comprising the Same |
| WO2013167586A1 (en) | 2012-05-08 | 2013-11-14 | Solvay Sa | Ionic liquids, method for manufacturing thereof, and electrochemical devices comprising the same |
| US9365480B2 (en) | 2012-08-22 | 2016-06-14 | Solvay Sa | Process for the manufacture of alkenones |
| WO2014029786A1 (en) * | 2012-08-22 | 2014-02-27 | Solvay Sa | Process for the manufacture of alkenones |
| WO2014096284A1 (en) | 2012-12-20 | 2014-06-26 | Solvay Sa | Salts of n-containing heterocyclic anions as components in electrolytes |
| WO2014202599A1 (en) * | 2013-06-20 | 2014-12-24 | Basf Se | Process for preparing pyridylpyrazole compounds and derivatives thereof from pyridylhydrazine |
| EP2987782A1 (en) | 2014-08-22 | 2016-02-24 | Solvay SA | Distillation process comprising at least two distillation steps to obtain purified halogenated carboxylic acid halide, and use of the purified halogenated carboxylic acid halide |
| US10017452B2 (en) | 2014-08-22 | 2018-07-10 | Solvay Sa | Distillation process comprising at least two distillation steps to obtain purified halogenated carboxylic acid halide, and use of the purified halogenated carboxylic acid halide |
| WO2016079126A1 (en) | 2014-11-17 | 2016-05-26 | Solvay Sa | Distillation process comprising at least two distillation steps to obtain purified halogenated carboxylic acid halide, and use of the purified halogenated carboxylic acid halide |
| WO2016079122A1 (en) | 2014-11-17 | 2016-05-26 | Solvay Sa | A method for producing a chemical compound and apparatus therefor |
| EP3031791A1 (en) | 2014-12-10 | 2016-06-15 | Solvay SA | Halogenation process of 1,1-dihaloethene |
| EP3199522A1 (en) | 2016-02-01 | 2017-08-02 | Solvay SA | Aminothioate derivatives, production thereof and use |
| WO2019043238A1 (en) | 2017-09-04 | 2019-03-07 | Solvay Sa | Process and intermediate for the manufacture of difluoroacetyl chloride |
| EP3650443A1 (en) | 2018-11-07 | 2020-05-13 | Fujian Yongjing Technology Co., Ltd. | Continuous flow synthesis of fluorinated or non-fluorinated pyrazoles |
| US11299463B2 (en) | 2018-11-07 | 2022-04-12 | Fujian Yongjing Technology Co., Ltd. | Process for the manufacture of pyrazoles or pyrimidones |
| WO2021152055A1 (en) | 2020-01-31 | 2021-08-05 | Solvay Sa | Process for the manufacture of haloalkyl substituted pyridine compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| US20130237710A1 (en) | 2013-09-12 |
| KR20190080972A (en) | 2019-07-08 |
| JP6511346B2 (en) | 2019-05-15 |
| JP2018058869A (en) | 2018-04-12 |
| JP2020023533A (en) | 2020-02-13 |
| EP2334645A1 (en) | 2011-06-22 |
| KR20160131129A (en) | 2016-11-15 |
| KR20110066201A (en) | 2011-06-16 |
| CA2735921C (en) | 2017-03-21 |
| CN106083710B (en) | 2021-03-12 |
| CN102164898A (en) | 2011-08-24 |
| EP2334645B1 (en) | 2019-11-06 |
| US8981115B2 (en) | 2015-03-17 |
| CN106083710A (en) | 2016-11-09 |
| JP2022028792A (en) | 2022-02-16 |
| US8431710B2 (en) | 2013-04-30 |
| US20110178297A1 (en) | 2011-07-21 |
| CA2735921A1 (en) | 2010-04-08 |
| JP2015227339A (en) | 2015-12-17 |
| JP2012504153A (en) | 2012-02-16 |
| JP6647268B2 (en) | 2020-02-14 |
| JP6180705B2 (en) | 2017-08-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2735921C (en) | Process for the synthesis of halogenated cyclic compounds | |
| CA2607934C (en) | Method for preparation of optionally 2-substituted 1,6-dihydro-6-oxo-4-pyrimidinecarboxylic acids | |
| EP1723156A1 (en) | Process for the preparation of pyridine derivatives | |
| US7847117B2 (en) | Process for preparing alkyl(methoxymethyl)trimethylsilanylmethylamines | |
| US5925764A (en) | Process and intermediated for the manufacture of pyridine-2, 3-dicarboxylate compounds | |
| US7772395B2 (en) | Process for the preparation of phenyl 2-pyrimidinyl ketones and their novel intermediates | |
| US6320053B1 (en) | Preparation of heteroarylcarboxamides | |
| US6080867A (en) | Process and intermediates for the manufacture of pyridine-2,3-dicarboxylate compounds | |
| US5663365A (en) | Process for the preparation of pyrazolones | |
| JPH11130752A (en) | Production of heteroaryl carboxylic amide and ester | |
| JP2991832B2 (en) | Method for producing pyrimidine derivative | |
| US6462195B1 (en) | Methods for highly selectively o-alkylating amide compounds with the use of copper salts | |
| JPH02101064A (en) | Production of pyrazole carboxylic acid amides | |
| JPH0478632B2 (en) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980138436.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09783436 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009783436 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2735921 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13120505 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011529509 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2737/CHENP/2011 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 20117009786 Country of ref document: KR Kind code of ref document: A |