WO2010014794A1 - Benzoimidazole derivatives and glycogen synthase kinase-3 beta inhibitors containing the same - Google Patents
Benzoimidazole derivatives and glycogen synthase kinase-3 beta inhibitors containing the same Download PDFInfo
- Publication number
- WO2010014794A1 WO2010014794A1 PCT/US2009/052225 US2009052225W WO2010014794A1 WO 2010014794 A1 WO2010014794 A1 WO 2010014794A1 US 2009052225 W US2009052225 W US 2009052225W WO 2010014794 A1 WO2010014794 A1 WO 2010014794A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- benzo
- imidazole
- hydroxy
- thiophen
- group
- Prior art date
Links
- 102000019058 Glycogen Synthase Kinase 3 beta Human genes 0.000 title claims abstract description 21
- 108010051975 Glycogen Synthase Kinase 3 beta Proteins 0.000 title claims abstract description 21
- 239000003112 inhibitor Substances 0.000 title claims abstract description 10
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical class C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 title abstract description 5
- 150000001875 compounds Chemical class 0.000 claims abstract description 110
- 238000000034 method Methods 0.000 claims description 108
- -1 p-toluenesulfonylamino Chemical group 0.000 claims description 91
- ZBNZAJFNDPPMDT-UHFFFAOYSA-N 1h-imidazole-5-carboxamide Chemical compound NC(=O)C1=CNC=N1 ZBNZAJFNDPPMDT-UHFFFAOYSA-N 0.000 claims description 71
- 125000006615 aromatic heterocyclic group Chemical group 0.000 claims description 59
- 150000003839 salts Chemical class 0.000 claims description 36
- 125000000217 alkyl group Chemical group 0.000 claims description 34
- 125000003118 aryl group Chemical group 0.000 claims description 34
- 125000001424 substituent group Chemical group 0.000 claims description 30
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 29
- 239000012453 solvate Substances 0.000 claims description 28
- 125000005842 heteroatom Chemical group 0.000 claims description 24
- 229910052757 nitrogen Inorganic materials 0.000 claims description 20
- 229910052760 oxygen Inorganic materials 0.000 claims description 20
- 229910052717 sulfur Inorganic materials 0.000 claims description 20
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 19
- 150000001408 amides Chemical class 0.000 claims description 18
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 18
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 15
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 claims description 13
- 239000002253 acid Substances 0.000 claims description 13
- 125000004448 alkyl carbonyl group Chemical group 0.000 claims description 13
- 150000001412 amines Chemical class 0.000 claims description 13
- 229910052739 hydrogen Inorganic materials 0.000 claims description 13
- 239000001257 hydrogen Substances 0.000 claims description 13
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 12
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 12
- 125000005129 aryl carbonyl group Chemical group 0.000 claims description 11
- 125000004391 aryl sulfonyl group Chemical group 0.000 claims description 10
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 10
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 10
- 125000000623 heterocyclic group Chemical group 0.000 claims description 10
- 239000008194 pharmaceutical composition Substances 0.000 claims description 10
- 125000003545 alkoxy group Chemical group 0.000 claims description 9
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 8
- 201000010099 disease Diseases 0.000 claims description 8
- NKWCGTOZTHZDHB-UHFFFAOYSA-N 1h-imidazol-1-ium-4-carboxylate Chemical compound OC(=O)C1=CNC=N1 NKWCGTOZTHZDHB-UHFFFAOYSA-N 0.000 claims description 7
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 claims description 6
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 claims description 6
- 125000004656 alkyl sulfonylamino group Chemical group 0.000 claims description 6
- 229910052736 halogen Inorganic materials 0.000 claims description 6
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 6
- 208000024827 Alzheimer disease Diseases 0.000 claims description 5
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 claims description 5
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 claims description 5
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 5
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 5
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 5
- 150000002367 halogens Chemical class 0.000 claims description 5
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 5
- 125000004043 oxo group Chemical group O=* 0.000 claims description 5
- 229920006395 saturated elastomer Polymers 0.000 claims description 5
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims description 5
- 206010026749 Mania Diseases 0.000 claims description 4
- 208000019695 Migraine disease Diseases 0.000 claims description 4
- 150000001409 amidines Chemical class 0.000 claims description 4
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 4
- 206010027599 migraine Diseases 0.000 claims description 4
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 claims description 4
- BBIVHOWFUCOWKO-UHFFFAOYSA-N 7-hydroxy-n-[2-(4-nitrophenoxy)ethyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(O)=CC=C1C(=O)NCCOC1=CC=C([N+]([O-])=O)C=C1 BBIVHOWFUCOWKO-UHFFFAOYSA-N 0.000 claims description 3
- GOPDFXUMARJJEA-UHFFFAOYSA-N amino(nitro)azanide Chemical compound N[N-][N+]([O-])=O GOPDFXUMARJJEA-UHFFFAOYSA-N 0.000 claims description 3
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 claims description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 3
- 125000004674 methylcarbonyl group Chemical group CC(=O)* 0.000 claims description 3
- MVIYVSVLUFCKPM-UHFFFAOYSA-N n-(7-hydroxy-2-thiophen-2-yl-1h-benzimidazol-4-yl)-3-(1h-imidazol-5-yl)propanamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(O)=CC=C1NC(=O)CCC1=CN=CN1 MVIYVSVLUFCKPM-UHFFFAOYSA-N 0.000 claims description 3
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 claims description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 3
- MUVKYWUUGPEPCZ-INIZCTEOSA-N (2s)-2-[(7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carbonyl)amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(C=1N=2)=CC=C(O)C=1NC=2C1=CC=CS1 MUVKYWUUGPEPCZ-INIZCTEOSA-N 0.000 claims description 2
- MZHSZYCOVLPFLX-UHFFFAOYSA-N 1-(2-cyclopropyl-7-hydroxy-1h-benzimidazol-4-yl)-3-(4-hydroxyphenyl)urea Chemical compound C1=CC(O)=CC=C1NC(=O)NC1=CC=C(O)C2=C1N=C(C1CC1)N2 MZHSZYCOVLPFLX-UHFFFAOYSA-N 0.000 claims description 2
- ODQBWZNQIHEVET-UHFFFAOYSA-N 2-(furan-2-yl)-7-hydroxy-n-[2-[[5-[(4-methylphenyl)sulfonylamino]pyridin-2-yl]amino]ethyl]-1h-benzimidazole-4-carboxamide Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(C=N1)=CC=C1NCCNC(=O)C1=CC=C(O)C2=C1N=C(C=1OC=CC=1)N2 ODQBWZNQIHEVET-UHFFFAOYSA-N 0.000 claims description 2
- BSGKOICCPIZSNX-UHFFFAOYSA-N 7-hydroxy-n-[2-(pyridin-2-ylamino)ethyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(O)=CC=C1C(=O)NCCNC1=CC=CC=N1 BSGKOICCPIZSNX-UHFFFAOYSA-N 0.000 claims description 2
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 125000001153 fluoro group Chemical group F* 0.000 claims description 2
- 125000000336 imidazol-5-yl group Chemical group [H]N1C([H])=NC([H])=C1[*] 0.000 claims description 2
- DWOUPRLHBBTUHD-UHFFFAOYSA-N n-[2-hydroxy-5-[2-(1h-imidazol-5-yl)ethylcarbamoyl]phenyl]thiophene-2-carboxamide Chemical compound OC1=CC=C(C(=O)NCCC=2NC=NC=2)C=C1NC(=O)C1=CC=CS1 DWOUPRLHBBTUHD-UHFFFAOYSA-N 0.000 claims description 2
- 150000002825 nitriles Chemical class 0.000 claims description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 2
- DENPQNAWGQXKCU-UHFFFAOYSA-N thiophene-2-carboxamide Chemical compound NC(=O)C1=CC=CS1 DENPQNAWGQXKCU-UHFFFAOYSA-N 0.000 claims description 2
- 230000008878 coupling Effects 0.000 claims 7
- 238000010168 coupling process Methods 0.000 claims 7
- 238000005859 coupling reaction Methods 0.000 claims 7
- 125000004181 carboxyalkyl group Chemical group 0.000 claims 3
- 125000002490 anilino group Chemical class [H]N(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims 2
- 150000001556 benzimidazoles Chemical class 0.000 claims 2
- 230000003301 hydrolyzing effect Effects 0.000 claims 2
- RAXXELZNTBOGNW-UHFFFAOYSA-N 1H-imidazole Chemical compound C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims 1
- KIBQEHLLMWSGFX-UHFFFAOYSA-N 7-hydroxy-n-[2-(1h-imidazol-5-yl)ethyl]-1h-indole-3-carboxamide Chemical compound C=1NC=2C(O)=CC=CC=2C=1C(=O)NCCC1=CN=CN1 KIBQEHLLMWSGFX-UHFFFAOYSA-N 0.000 claims 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 claims 1
- 125000005843 halogen group Chemical group 0.000 claims 1
- VBZWSGALLODQNC-UHFFFAOYSA-N hexafluoroacetone Chemical compound FC(F)(F)C(=O)C(F)(F)F VBZWSGALLODQNC-UHFFFAOYSA-N 0.000 claims 1
- 150000002475 indoles Chemical class 0.000 claims 1
- NCFOCOFBHNWJDC-UHFFFAOYSA-N methyl 3-hydroxy-2-[(7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carbonyl)amino]propanoate Chemical compound N=1C=2C(C(=O)NC(CO)C(=O)OC)=CC=C(O)C=2NC=1C1=CC=CS1 NCFOCOFBHNWJDC-UHFFFAOYSA-N 0.000 claims 1
- DUWWHGPELOTTOE-UHFFFAOYSA-N n-(5-chloro-2,4-dimethoxyphenyl)-3-oxobutanamide Chemical compound COC1=CC(OC)=C(NC(=O)CC(C)=O)C=C1Cl DUWWHGPELOTTOE-UHFFFAOYSA-N 0.000 claims 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 claims 1
- 235000019260 propionic acid Nutrition 0.000 claims 1
- 239000000047 product Substances 0.000 description 160
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 153
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 139
- 239000007787 solid Substances 0.000 description 139
- 239000011541 reaction mixture Substances 0.000 description 131
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 110
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 90
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 89
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 80
- 238000005160 1H NMR spectroscopy Methods 0.000 description 74
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 67
- 238000004128 high performance liquid chromatography Methods 0.000 description 60
- 239000000243 solution Substances 0.000 description 51
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 46
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 45
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 44
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 41
- 235000019439 ethyl acetate Nutrition 0.000 description 34
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 33
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 33
- PZXAAFAJWASTJT-UHFFFAOYSA-N 7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxylic acid Chemical compound N=1C=2C(OC)=CC=C(C(O)=O)C=2NC=1C1=CC=CS1 PZXAAFAJWASTJT-UHFFFAOYSA-N 0.000 description 32
- 230000015572 biosynthetic process Effects 0.000 description 32
- 238000003786 synthesis reaction Methods 0.000 description 32
- 239000000377 silicon dioxide Substances 0.000 description 27
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 26
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 26
- 239000012044 organic layer Substances 0.000 description 24
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 23
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 23
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 22
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 21
- 239000007821 HATU Substances 0.000 description 19
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 18
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 18
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 18
- 229910052938 sodium sulfate Inorganic materials 0.000 description 18
- 239000007832 Na2SO4 Substances 0.000 description 17
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 14
- 239000000543 intermediate Substances 0.000 description 14
- 238000002953 preparative HPLC Methods 0.000 description 14
- 229910021529 ammonia Inorganic materials 0.000 description 13
- 238000003818 flash chromatography Methods 0.000 description 13
- 238000005342 ion exchange Methods 0.000 description 13
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 12
- 150000004677 hydrates Chemical class 0.000 description 12
- 230000026731 phosphorylation Effects 0.000 description 12
- 238000006366 phosphorylation reaction Methods 0.000 description 12
- 239000002244 precipitate Substances 0.000 description 12
- 239000000203 mixture Substances 0.000 description 11
- 229940002612 prodrug Drugs 0.000 description 11
- 239000000651 prodrug Substances 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 10
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 10
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N coumarin Chemical compound C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 10
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 230000002829 reductive effect Effects 0.000 description 9
- 239000000741 silica gel Substances 0.000 description 9
- 229910002027 silica gel Inorganic materials 0.000 description 9
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 9
- 102000002254 Glycogen Synthase Kinase 3 Human genes 0.000 description 8
- 108010014905 Glycogen Synthase Kinase 3 Proteins 0.000 description 8
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 8
- 229910019093 NaOCl Inorganic materials 0.000 description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 8
- 239000012267 brine Substances 0.000 description 8
- 238000004440 column chromatography Methods 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- 239000010410 layer Substances 0.000 description 8
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 7
- 0 Cc1ccc(*)c2c1nc(*)[n]2 Chemical compound Cc1ccc(*)c2c1nc(*)[n]2 0.000 description 7
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 7
- 229940086542 triethylamine Drugs 0.000 description 7
- QCQAZAFKSFCSIP-UHFFFAOYSA-N 2-(furan-2-yl)-7-hydroxy-1h-benzimidazole-4-carboxylic acid Chemical compound N=1C=2C(C(=O)O)=CC=C(O)C=2NC=1C1=CC=CO1 QCQAZAFKSFCSIP-UHFFFAOYSA-N 0.000 description 6
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 235000019198 oils Nutrition 0.000 description 6
- 238000010992 reflux Methods 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- VMKKFYFBTBSQCI-UHFFFAOYSA-N 2-(furan-2-yl)-7-methoxy-1h-benzimidazole-4-carboxylic acid Chemical compound N1C=2C(OC)=CC=C(C(O)=O)C=2N=C1C1=CC=CO1 VMKKFYFBTBSQCI-UHFFFAOYSA-N 0.000 description 5
- 229910015845 BBr3 Inorganic materials 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 229960000956 coumarin Drugs 0.000 description 5
- 235000001671 coumarin Nutrition 0.000 description 5
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 5
- 239000005457 ice water Substances 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- QVDWKLDUBSJEOG-UHFFFAOYSA-N methyl 3-amino-4-methoxybenzoate Chemical compound COC(=O)C1=CC=C(OC)C(N)=C1 QVDWKLDUBSJEOG-UHFFFAOYSA-N 0.000 description 5
- AOCSUUGBCMTKJH-UHFFFAOYSA-N tert-butyl n-(2-aminoethyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCN AOCSUUGBCMTKJH-UHFFFAOYSA-N 0.000 description 5
- ZXTKGGYXRGFGQZ-UHFFFAOYSA-N 2-cyclopropyl-7-methoxy-1h-benzimidazole-4-carboxylic acid Chemical compound N=1C=2C(OC)=CC=C(C(O)=O)C=2NC=1C1CC1 ZXTKGGYXRGFGQZ-UHFFFAOYSA-N 0.000 description 4
- PLIKAWJENQZMHA-UHFFFAOYSA-N 4-aminophenol Chemical compound NC1=CC=C(O)C=C1 PLIKAWJENQZMHA-UHFFFAOYSA-N 0.000 description 4
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 4
- SJZOWOQBEAFCJP-UHFFFAOYSA-N 7-hydroxy-2-phenyl-1h-benzimidazole-4-carboxylic acid Chemical compound N=1C=2C(C(=O)O)=CC=C(O)C=2NC=1C1=CC=CC=C1 SJZOWOQBEAFCJP-UHFFFAOYSA-N 0.000 description 4
- NTTTURJGCKSVGE-UHFFFAOYSA-N 7-methoxy-2-thiophen-2-yl-3h-benzimidazole-5-carboxylic acid Chemical compound N1C=2C(OC)=CC(C(O)=O)=CC=2N=C1C1=CC=CS1 NTTTURJGCKSVGE-UHFFFAOYSA-N 0.000 description 4
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- 239000005909 Kieselgur Substances 0.000 description 4
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- RUZLIIJDZBWWSA-INIZCTEOSA-N methyl 2-[[(1s)-1-(7-methyl-2-morpholin-4-yl-4-oxopyrido[1,2-a]pyrimidin-9-yl)ethyl]amino]benzoate Chemical group COC(=O)C1=CC=CC=C1N[C@@H](C)C1=CC(C)=CN2C(=O)C=C(N3CCOCC3)N=C12 RUZLIIJDZBWWSA-INIZCTEOSA-N 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- DZGWFCGJZKJUFP-UHFFFAOYSA-N tyramine Chemical compound NCCC1=CC=C(O)C=C1 DZGWFCGJZKJUFP-UHFFFAOYSA-N 0.000 description 4
- UVKHHBRUBXLKTP-QPJJXVBHSA-N (e)-3-(1h-imidazol-5-yl)-n-(7-methoxy-2-thiophen-2-yl-1h-benzimidazol-4-yl)prop-2-enamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(OC)=CC=C1NC(=O)\C=C\C1=CN=CN1 UVKHHBRUBXLKTP-QPJJXVBHSA-N 0.000 description 3
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- LXMDEJJZMNUHHS-UHFFFAOYSA-N 2-(furan-2-yl)-7-methoxy-n-(1,3-thiazol-2-yl)-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3OC=CC=3)NC=2C(OC)=CC=C1C(=O)NC1=NC=CS1 LXMDEJJZMNUHHS-UHFFFAOYSA-N 0.000 description 3
- OVWBPUJHPMWTLQ-UHFFFAOYSA-N 2-cyclopentyl-7-hydroxy-1h-benzimidazole-4-carboxylic acid Chemical compound N1C=2C(C(=O)O)=CC=C(O)C=2N=C1C1CCCC1 OVWBPUJHPMWTLQ-UHFFFAOYSA-N 0.000 description 3
- ANXCBFTZWJXBSY-UHFFFAOYSA-N 2-cyclopropyl-7-hydroxy-1h-benzimidazole-4-carboxylic acid Chemical compound N1C=2C(C(=O)O)=CC=C(O)C=2N=C1C1CC1 ANXCBFTZWJXBSY-UHFFFAOYSA-N 0.000 description 3
- HSZICKWKNHDWAD-UHFFFAOYSA-N 2-cyclopropyl-7-methoxy-1h-benzimidazol-4-amine Chemical compound N1C=2C(OC)=CC=C(N)C=2N=C1C1CC1 HSZICKWKNHDWAD-UHFFFAOYSA-N 0.000 description 3
- KMTNZCAGMPTVOX-UHFFFAOYSA-N 7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxylic acid Chemical compound N1C=2C(C(=O)O)=CC=C(O)C=2N=C1C1=CC=CS1 KMTNZCAGMPTVOX-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 229960000583 acetic acid Drugs 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 238000012512 characterization method Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 206010012601 diabetes mellitus Diseases 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 229960001340 histamine Drugs 0.000 description 3
- 150000001261 hydroxy acids Chemical class 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 238000000021 kinase assay Methods 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 150000007530 organic bases Chemical class 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- MLDSDVASYUUDLT-UHFFFAOYSA-N tert-butyl n-(3-oxopropyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCC=O MLDSDVASYUUDLT-UHFFFAOYSA-N 0.000 description 3
- CUPOOAWTRIURFT-UHFFFAOYSA-N thiophene-2-carbonitrile Chemical compound N#CC1=CC=CS1 CUPOOAWTRIURFT-UHFFFAOYSA-N 0.000 description 3
- ZUVFATUZMPMHOH-NSHDSACASA-N (2s)-2-[(7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carbonyl)amino]-3-(1h-imidazol-5-yl)propanoic acid Chemical compound C([C@@H](C(=O)O)NC(=O)C=1C=2N=C(NC=2C(O)=CC=1)C=1SC=CC=1)C1=CN=CN1 ZUVFATUZMPMHOH-NSHDSACASA-N 0.000 description 2
- RAIPHJJURHTUIC-UHFFFAOYSA-N 1,3-thiazol-2-amine Chemical compound NC1=NC=CS1 RAIPHJJURHTUIC-UHFFFAOYSA-N 0.000 description 2
- DEURIUYJZZLADZ-UHFFFAOYSA-N 2-(1h-imidazol-2-yl)ethanamine Chemical compound NCCC1=NC=CN1 DEURIUYJZZLADZ-UHFFFAOYSA-N 0.000 description 2
- CKLFJWXRWIQYOC-UHFFFAOYSA-N 2-(4-fluorophenyl)ethanamine Chemical compound NCCC1=CC=C(F)C=C1 CKLFJWXRWIQYOC-UHFFFAOYSA-N 0.000 description 2
- QIEMOKUVCNROKE-UHFFFAOYSA-N 2-(furan-2-yl)-7-hydroxy-n-[2-[(5-nitropyridin-2-yl)amino]ethyl]-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3OC=CC=3)NC=2C(O)=CC=C1C(=O)NCCNC1=CC=C([N+]([O-])=O)C=N1 QIEMOKUVCNROKE-UHFFFAOYSA-N 0.000 description 2
- GEJVLAIJXQDLML-UHFFFAOYSA-N 2-(furan-2-yl)-7-methoxy-n-[2-(1-methylpyrrol-2-yl)ethyl]-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3OC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCC1=CC=CN1C GEJVLAIJXQDLML-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- QCEWNYUNJMJBCI-UHFFFAOYSA-N 2-cyclopropyl-7-methoxy-n-[2-(4-sulfamoylphenyl)ethyl]-1h-benzimidazole-4-carboxamide Chemical compound C1=2NC(C3CC3)=NC=2C(OC)=CC=C1C(=O)NCCC1=CC=C(S(N)(=O)=O)C=C1 QCEWNYUNJMJBCI-UHFFFAOYSA-N 0.000 description 2
- RTXLXMIUTNUIGC-UHFFFAOYSA-N 2-cyclopropyl-n-[2-(4-fluorophenyl)ethyl]-7-methoxy-1h-benzimidazole-4-carboxamide Chemical compound C1=2NC(C3CC3)=NC=2C(OC)=CC=C1C(=O)NCCC1=CC=C(F)C=C1 RTXLXMIUTNUIGC-UHFFFAOYSA-N 0.000 description 2
- HQNOODJDSFSURF-UHFFFAOYSA-N 3-(1h-imidazol-2-yl)propan-1-amine Chemical compound NCCCC1=NC=CN1 HQNOODJDSFSURF-UHFFFAOYSA-N 0.000 description 2
- KXNJFDAIJDKLCZ-UHFFFAOYSA-N 3-[(2-amino-4-methoxybenzoyl)amino]piperidine-1-carboxylic acid Chemical compound NC1=CC(OC)=CC=C1C(=O)NC1CN(C(O)=O)CCC1 KXNJFDAIJDKLCZ-UHFFFAOYSA-N 0.000 description 2
- HNHTUQFIYSEUMO-UHFFFAOYSA-N 3-[1-(4-methylphenyl)sulfonylimidazol-2-yl]propan-1-ol Chemical compound C1=CC(C)=CC=C1S(=O)(=O)N1C(CCCO)=NC=C1 HNHTUQFIYSEUMO-UHFFFAOYSA-N 0.000 description 2
- LNPMZQXEPNWCMG-UHFFFAOYSA-N 4-(2-aminoethyl)aniline Chemical compound NCCC1=CC=C(N)C=C1 LNPMZQXEPNWCMG-UHFFFAOYSA-N 0.000 description 2
- FXNSVEQMUYPYJS-UHFFFAOYSA-N 4-(2-aminoethyl)benzenesulfonamide Chemical compound NCCC1=CC=C(S(N)(=O)=O)C=C1 FXNSVEQMUYPYJS-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- PEELIBUMTXGKIM-UHFFFAOYSA-N 7-hydroxy-n-[2-(3-methoxyphenyl)ethyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound COC1=CC=CC(CCNC(=O)C=2C=3N=C(NC=3C(O)=CC=2)C=2SC=CC=2)=C1 PEELIBUMTXGKIM-UHFFFAOYSA-N 0.000 description 2
- YBFGBABPODCQNN-UHFFFAOYSA-N 7-hydroxy-n-[2-(3-methylimidazol-4-yl)ethyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound CN1C=NC=C1CCNC(=O)C1=CC=C(O)C2=C1N=C(C=1SC=CC=1)N2 YBFGBABPODCQNN-UHFFFAOYSA-N 0.000 description 2
- YNPZYIACHQGCJT-UHFFFAOYSA-N 7-hydroxy-n-[2-[(5-nitropyridin-2-yl)amino]ethyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(O)=CC=C1C(=O)NCCNC1=CC=C([N+]([O-])=O)C=N1 YNPZYIACHQGCJT-UHFFFAOYSA-N 0.000 description 2
- CEWDBJZCSBURSD-UHFFFAOYSA-N 7-methoxy-2-thiophen-2-yl-1h-benzimidazol-4-amine;hydrochloride Chemical compound Cl.N1C=2C(OC)=CC=C(N)C=2N=C1C1=CC=CS1 CEWDBJZCSBURSD-UHFFFAOYSA-N 0.000 description 2
- BVOPAEYMEDOKRZ-UHFFFAOYSA-N 7-methoxy-2-thiophen-2-yl-3h-benzimidazole-5-carboxamide Chemical compound N1C=2C(OC)=CC(C(N)=O)=CC=2N=C1C1=CC=CS1 BVOPAEYMEDOKRZ-UHFFFAOYSA-N 0.000 description 2
- MBIRMGLAOUIYQA-UHFFFAOYSA-N 7-methoxy-2-thiophen-2-yl-n-[2-[5-(trifluoromethyl)pyridin-2-yl]oxyethyl]-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCOC1=CC=C(C(F)(F)F)C=N1 MBIRMGLAOUIYQA-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 208000031638 Body Weight Diseases 0.000 description 2
- 125000000041 C6-C10 aryl group Chemical group 0.000 description 2
- 125000005915 C6-C14 aryl group Chemical group 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 102100040243 Microtubule-associated protein tau Human genes 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- SORGEQQSQGNZFI-UHFFFAOYSA-N [azido(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(N=[N+]=[N-])OC1=CC=CC=C1 SORGEQQSQGNZFI-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- VPLLZMWJHRUCMF-UHFFFAOYSA-N methyl 2-cyclopentyl-7-methoxy-1h-benzimidazole-4-carboxylate Chemical compound N1C=2C(C(=O)OC)=CC=C(OC)C=2N=C1C1CCCC1 VPLLZMWJHRUCMF-UHFFFAOYSA-N 0.000 description 2
- DSXKVMFEUNHVGE-UHFFFAOYSA-N methyl 3-[1-(4-methylphenyl)sulfonylimidazol-2-yl]prop-2-enoate Chemical compound COC(=O)C=CC1=NC=CN1S(=O)(=O)C1=CC=C(C)C=C1 DSXKVMFEUNHVGE-UHFFFAOYSA-N 0.000 description 2
- UNSGMGDVIYSRCM-UHFFFAOYSA-N methyl 3-[1-(4-methylphenyl)sulfonylimidazol-2-yl]propanoate Chemical compound COC(=O)CCC1=NC=CN1S(=O)(=O)C1=CC=C(C)C=C1 UNSGMGDVIYSRCM-UHFFFAOYSA-N 0.000 description 2
- QHLFPSCMDZCUPE-UHFFFAOYSA-N methyl 3-[[amino(furan-2-yl)methylidene]amino]-4-methoxybenzoate;hydrochloride Chemical compound Cl.COC(=O)C1=CC=C(OC)C(NC(=N)C=2OC=CC=2)=C1 QHLFPSCMDZCUPE-UHFFFAOYSA-N 0.000 description 2
- XZWKHHKDPCUEOZ-UHFFFAOYSA-N methyl 4-methoxy-3-(thiophene-2-carbonylamino)benzoate Chemical compound COC(=O)C1=CC=C(OC)C(NC(=O)C=2SC=CC=2)=C1 XZWKHHKDPCUEOZ-UHFFFAOYSA-N 0.000 description 2
- OUINOQYRLVBCMS-UHFFFAOYSA-N methyl 7-fluoro-2-thiophen-2-yl-3h-benzimidazole-4-carboxylate Chemical compound N=1C=2C(C(=O)OC)=CC=C(F)C=2NC=1C1=CC=CS1 OUINOQYRLVBCMS-UHFFFAOYSA-N 0.000 description 2
- YBKCCRRVBMZAOB-UHFFFAOYSA-N methyl 7-methoxy-2-phenyl-1h-benzimidazole-4-carboxylate Chemical compound N=1C=2C(C(=O)OC)=CC=C(OC)C=2NC=1C1=CC=CC=C1 YBKCCRRVBMZAOB-UHFFFAOYSA-N 0.000 description 2
- OKWMXUZNFRSEJX-UHFFFAOYSA-N methyl 7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxylate Chemical compound N=1C=2C(C(=O)OC)=CC=C(OC)C=2NC=1C1=CC=CS1 OKWMXUZNFRSEJX-UHFFFAOYSA-N 0.000 description 2
- HZQOAXBFUCEICD-UHFFFAOYSA-N n-(2-acetamidoethyl)-7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound N=1C=2C(C(=O)NCCNC(=O)C)=CC=C(O)C=2NC=1C1=CC=CS1 HZQOAXBFUCEICD-UHFFFAOYSA-N 0.000 description 2
- COKWCLRPCPQRHW-UHFFFAOYSA-N n-(2-cyclopropyl-7-hydroxy-1h-benzimidazol-4-yl)-2-(4-hydroxyphenyl)acetamide Chemical compound C1=CC(O)=CC=C1CC(=O)NC1=CC=C(O)C2=C1N=C(C1CC1)N2 COKWCLRPCPQRHW-UHFFFAOYSA-N 0.000 description 2
- HUOMQECEWYETBW-UHFFFAOYSA-N n-(2-cyclopropyl-7-methoxy-1h-benzimidazol-4-yl)-2-(4-methoxyphenyl)acetamide Chemical compound C1=CC(OC)=CC=C1CC(=O)NC1=CC=C(OC)C2=C1N=C(C1CC1)N2 HUOMQECEWYETBW-UHFFFAOYSA-N 0.000 description 2
- BOZMVONDDOSKJB-UHFFFAOYSA-N n-(3-imidazol-1-ylpropyl)-7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCCN1C=CN=C1 BOZMVONDDOSKJB-UHFFFAOYSA-N 0.000 description 2
- MDAPGWXHICTIHL-UHFFFAOYSA-N n-[2-(3,5-dimethyl-1,2-oxazol-4-yl)ethyl]-2-(furan-2-yl)-7-methoxy-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3OC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCC=1C(C)=NOC=1C MDAPGWXHICTIHL-UHFFFAOYSA-N 0.000 description 2
- YEMDPAGCOOMZSE-UHFFFAOYSA-N n-[2-(4-hydroxyphenyl)ethyl]-7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCC1=CC=C(O)C=C1 YEMDPAGCOOMZSE-UHFFFAOYSA-N 0.000 description 2
- SXTFLHJIMWRWPZ-UHFFFAOYSA-N n-[2-(5-carbamoylpyridin-2-yl)oxyethyl]-7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCOC1=CC=C(C(N)=O)C=N1 SXTFLHJIMWRWPZ-UHFFFAOYSA-N 0.000 description 2
- PNYYRFDFEXVEPN-UHFFFAOYSA-N n-[2-(benzenesulfonamido)ethyl]-7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(O)=CC=C1C(=O)NCCNS(=O)(=O)C1=CC=CC=C1 PNYYRFDFEXVEPN-UHFFFAOYSA-N 0.000 description 2
- MLTIVPRJLZLLPK-UHFFFAOYSA-N n-[2-(dimethylamino)ethyl]-7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound N1C=2C(OC)=CC=C(C(=O)NCCN(C)C)C=2N=C1C1=CC=CS1 MLTIVPRJLZLLPK-UHFFFAOYSA-N 0.000 description 2
- YUESKMWWLICRQR-UHFFFAOYSA-N n-[2-[(5-aminopyridin-2-yl)amino]ethyl]-2-(furan-2-yl)-7-hydroxy-1h-benzimidazole-4-carboxamide;hydrochloride Chemical compound Cl.N1=CC(N)=CC=C1NCCNC(=O)C1=CC=C(O)C2=C1N=C(C=1OC=CC=1)N2 YUESKMWWLICRQR-UHFFFAOYSA-N 0.000 description 2
- YIRIHJCPYLRLBV-UHFFFAOYSA-N n-[3-(1h-imidazol-2-yl)propyl]-7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCCC1=NC=CN1 YIRIHJCPYLRLBV-UHFFFAOYSA-N 0.000 description 2
- 230000000626 neurodegenerative effect Effects 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000006408 oxalic acid Nutrition 0.000 description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- DNUTZBZXLPWRJG-UHFFFAOYSA-M piperidine-1-carboxylate Chemical compound [O-]C(=O)N1CCCCC1 DNUTZBZXLPWRJG-UHFFFAOYSA-M 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- RNSJYMNAEDRFIR-UHFFFAOYSA-N tert-butyl n-(2-cyclopropyl-7-methoxy-1h-benzimidazol-4-yl)carbamate Chemical compound N1C=2C(OC)=CC=C(NC(=O)OC(C)(C)C)C=2N=C1C1CC1 RNSJYMNAEDRFIR-UHFFFAOYSA-N 0.000 description 2
- UNXMOKLGTAULFB-UHFFFAOYSA-N tert-butyl n-(7-methoxy-2-thiophen-2-yl-1h-benzimidazol-4-yl)carbamate Chemical compound N1C=2C(OC)=CC=C(NC(=O)OC(C)(C)C)C=2N=C1C1=CC=CS1 UNXMOKLGTAULFB-UHFFFAOYSA-N 0.000 description 2
- JZSSQQFLTWLZEM-UHFFFAOYSA-N tert-butyl n-[2-(4,5-dihydro-1h-imidazol-2-yl)ethyl]carbamate Chemical compound CC(C)(C)OC(=O)NCCC1=NCCN1 JZSSQQFLTWLZEM-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- QDZZDVQGBKTLHV-UHFFFAOYSA-N (2,4-difluorophenyl)methanamine Chemical compound NCC1=CC=C(F)C=C1F QDZZDVQGBKTLHV-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- ZQISRDCJNBUVMM-RXMQYKEDSA-N (2r)-2-amino-3-(1h-imidazol-5-yl)propan-1-ol Chemical compound OC[C@H](N)CC1=CN=CN1 ZQISRDCJNBUVMM-RXMQYKEDSA-N 0.000 description 1
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- JBULSURVMXPBNA-RXMQYKEDSA-N (2s)-2-amino-3,3-dimethylbutan-1-ol Chemical compound CC(C)(C)[C@H](N)CO JBULSURVMXPBNA-RXMQYKEDSA-N 0.000 description 1
- DIVNUTGTTIRPQA-UHFFFAOYSA-N (3,4-dimethoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C=C1OC DIVNUTGTTIRPQA-UHFFFAOYSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- URCWFFJUMVIZQA-UHFFFAOYSA-N 1,3-bis(2-cyclopropyl-7-methoxy-1h-benzimidazol-4-yl)urea Chemical compound C1=2N=C(C3CC3)NC=2C(OC)=CC=C1NC(=O)NC(C=1N=2)=CC=C(OC)C=1NC=2C1CC1 URCWFFJUMVIZQA-UHFFFAOYSA-N 0.000 description 1
- CBCKQZAAMUWICA-UHFFFAOYSA-N 1,4-phenylenediamine Chemical compound NC1=CC=C(N)C=C1 CBCKQZAAMUWICA-UHFFFAOYSA-N 0.000 description 1
- SXOVYMCCZGKLGI-UHFFFAOYSA-N 1-(2-cyclopropyl-7-methoxy-1h-benzimidazol-4-yl)-3-(4-methoxyphenyl)urea Chemical compound C1=CC(OC)=CC=C1NC(=O)NC1=CC=C(OC)C2=C1N=C(C1CC1)N2 SXOVYMCCZGKLGI-UHFFFAOYSA-N 0.000 description 1
- JJDMKDXGNVJWCD-UHFFFAOYSA-N 1h-benzimidazole-4-carboxamide Chemical compound NC(=O)C1=CC=CC2=C1N=CN2 JJDMKDXGNVJWCD-UHFFFAOYSA-N 0.000 description 1
- ZMZSYUSDGRJZNT-UHFFFAOYSA-N 2-(1,3-benzothiazol-2-yl)acetonitrile Chemical compound C1=CC=C2SC(CC#N)=NC2=C1 ZMZSYUSDGRJZNT-UHFFFAOYSA-N 0.000 description 1
- CPAGZVLINCPJEH-UHFFFAOYSA-N 2-(1-methyl-1h-imidazol-5-yl)ethan-1-amine Chemical compound CN1C=NC=C1CCN CPAGZVLINCPJEH-UHFFFAOYSA-N 0.000 description 1
- ITFDYXKCBZEBDG-UHFFFAOYSA-N 2-(1-methylpyrrol-2-yl)ethanamine Chemical compound CN1C=CC=C1CCN ITFDYXKCBZEBDG-UHFFFAOYSA-N 0.000 description 1
- HOJDTRKLAOLNIT-UHFFFAOYSA-N 2-(3,5-dimethyl-1,2-oxazol-4-yl)ethanamine Chemical compound CC1=NOC(C)=C1CCN HOJDTRKLAOLNIT-UHFFFAOYSA-N 0.000 description 1
- GHKSKVKCKMGRDU-UHFFFAOYSA-N 2-(3-aminopropylamino)ethanol Chemical compound NCCCNCCO GHKSKVKCKMGRDU-UHFFFAOYSA-N 0.000 description 1
- WJBMRZAHTUFBGE-UHFFFAOYSA-N 2-(3-methoxyphenyl)ethanamine Chemical compound COC1=CC=CC(CCN)=C1 WJBMRZAHTUFBGE-UHFFFAOYSA-N 0.000 description 1
- CXJOONIFSVSFAD-UHFFFAOYSA-N 2-(4-methoxyphenyl)acetyl chloride Chemical compound COC1=CC=C(CC(Cl)=O)C=C1 CXJOONIFSVSFAD-UHFFFAOYSA-N 0.000 description 1
- CWYDJVNQLOTFNE-UHFFFAOYSA-N 2-(4-methylphenyl)benzoyl chloride Chemical compound C1=CC(C)=CC=C1C1=CC=CC=C1C(Cl)=O CWYDJVNQLOTFNE-UHFFFAOYSA-N 0.000 description 1
- WEPGUFPFXZWBCK-UHFFFAOYSA-N 2-(furan-2-yl)-7-hydroxy-n-(1,3-thiazol-2-yl)-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3OC=CC=3)NC=2C(O)=CC=C1C(=O)NC1=NC=CS1 WEPGUFPFXZWBCK-UHFFFAOYSA-N 0.000 description 1
- SBZNOMSJFLVNDA-UHFFFAOYSA-N 2-(furan-2-yl)-7-hydroxy-n-(2-phenylethyl)-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3OC=CC=3)NC=2C(O)=CC=C1C(=O)NCCC1=CC=CC=C1 SBZNOMSJFLVNDA-UHFFFAOYSA-N 0.000 description 1
- JIMLNOXLFHMJAE-UHFFFAOYSA-N 2-(furan-2-yl)-7-hydroxy-n-[2-(1-methylpyrrol-2-yl)ethyl]-1h-benzimidazole-4-carboxamide Chemical compound CN1C=CC=C1CCNC(=O)C1=CC=C(O)C2=C1N=C(C=1OC=CC=1)N2 JIMLNOXLFHMJAE-UHFFFAOYSA-N 0.000 description 1
- SKWUVECPMONQGU-UHFFFAOYSA-N 2-(furan-2-yl)-7-hydroxy-n-phenyl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3OC=CC=3)NC=2C(O)=CC=C1C(=O)NC1=CC=CC=C1 SKWUVECPMONQGU-UHFFFAOYSA-N 0.000 description 1
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 1
- HUHGPYXAVBJSJV-UHFFFAOYSA-N 2-[3,5-bis(2-hydroxyethyl)-1,3,5-triazinan-1-yl]ethanol Chemical compound OCCN1CN(CCO)CN(CCO)C1 HUHGPYXAVBJSJV-UHFFFAOYSA-N 0.000 description 1
- QQDXOCYCKJVEPP-UHFFFAOYSA-N 2-[3-[1-(4-methylphenyl)sulfonylimidazol-2-yl]propyl]isoindole-1,3-dione Chemical compound C1=CC(C)=CC=C1S(=O)(=O)N1C(CCCN2C(C3=CC=CC=C3C2=O)=O)=NC=C1 QQDXOCYCKJVEPP-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- JFZJMSDDOOAOIV-UHFFFAOYSA-N 2-chloro-5-(trifluoromethyl)pyridine Chemical compound FC(F)(F)C1=CC=C(Cl)N=C1 JFZJMSDDOOAOIV-UHFFFAOYSA-N 0.000 description 1
- CUPJXQFPTJBZOZ-UHFFFAOYSA-N 2-cyclopentyl-7-hydroxy-n-[2-(4-hydroxyphenyl)ethyl]-1h-benzimidazole-4-carboxamide Chemical compound C1=CC(O)=CC=C1CCNC(=O)C1=CC=C(O)C2=C1NC(C1CCCC1)=N2 CUPJXQFPTJBZOZ-UHFFFAOYSA-N 0.000 description 1
- HVIMPUISPMUNPS-UHFFFAOYSA-N 2-cyclopropyl-7-hydroxy-n-[2-(4-sulfamoylphenyl)ethyl]-1h-benzimidazole-4-carboxamide Chemical compound C1=CC(S(=O)(=O)N)=CC=C1CCNC(=O)C1=CC=C(O)C2=C1NC(C1CC1)=N2 HVIMPUISPMUNPS-UHFFFAOYSA-N 0.000 description 1
- MXQIXJJYQFBVJN-UHFFFAOYSA-N 2-cyclopropyl-n-(2,3-dihydroxypropyl)-7-hydroxy-1h-benzimidazole-4-carboxamide Chemical compound N1C=2C(C(=O)NCC(O)CO)=CC=C(O)C=2N=C1C1CC1 MXQIXJJYQFBVJN-UHFFFAOYSA-N 0.000 description 1
- SWMCGIYDIXUCLO-UHFFFAOYSA-N 2-cyclopropyl-n-(4-hydroxyphenyl)-7-methoxy-1h-benzimidazole-4-carboxamide Chemical compound C1=2NC(C3CC3)=NC=2C(OC)=CC=C1C(=O)NC1=CC=C(O)C=C1 SWMCGIYDIXUCLO-UHFFFAOYSA-N 0.000 description 1
- HHGDSAWNMROJTC-UHFFFAOYSA-N 2-cyclopropyl-n-[2-(4-fluorophenyl)ethyl]-7-hydroxy-1h-benzimidazole-4-carboxamide Chemical compound C1=2NC(C3CC3)=NC=2C(O)=CC=C1C(=O)NCCC1=CC=C(F)C=C1 HHGDSAWNMROJTC-UHFFFAOYSA-N 0.000 description 1
- MWOFSHRVMPYTGX-UHFFFAOYSA-N 2-cyclopropyl-n-[2-(dimethylamino)ethyl]-7-hydroxy-1h-benzimidazole-4-carboxamide Chemical compound N1C=2C(C(=O)NCCN(C)C)=CC=C(O)C=2N=C1C1CC1 MWOFSHRVMPYTGX-UHFFFAOYSA-N 0.000 description 1
- 125000006149 2-ethylbutyl sulfonyl group Chemical group 0.000 description 1
- 125000004398 2-methyl-2-butyl group Chemical group CC(C)(CC)* 0.000 description 1
- 125000004918 2-methyl-2-pentyl group Chemical group CC(C)(CCC)* 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- SDFIWGLQFXYGMQ-UHFFFAOYSA-N 2-thiophen-2-yl-1H-benzimidazole-4-carboxamide Chemical compound S1C(=CC=C1)C1=NC2=C(N1)C=CC=C2C(=O)N SDFIWGLQFXYGMQ-UHFFFAOYSA-N 0.000 description 1
- HVLUYXIJZLDNIS-UHFFFAOYSA-N 2-thiophen-2-ylethanamine Chemical compound NCCC1=CC=CS1 HVLUYXIJZLDNIS-UHFFFAOYSA-N 0.000 description 1
- GOLXRNDWAUTYKT-UHFFFAOYSA-M 3-(1H-indol-3-yl)propanoate Chemical compound C1=CC=C2C(CCC(=O)[O-])=CNC2=C1 GOLXRNDWAUTYKT-UHFFFAOYSA-M 0.000 description 1
- RMBLZFYBHYIJJL-UHFFFAOYSA-N 3-[(4-methoxy-2-nitrobenzoyl)amino]piperidine-1-carboxylic acid Chemical compound [O-][N+](=O)C1=CC(OC)=CC=C1C(=O)NC1CN(C(O)=O)CCC1 RMBLZFYBHYIJJL-UHFFFAOYSA-N 0.000 description 1
- DICPPOONERDVPQ-UHFFFAOYSA-N 3-[[amino(cyclopentyl)methylidene]amino]-4-methoxybenzoic acid;hydrochloride Chemical compound Cl.COC1=CC=C(C(O)=O)C=C1N=C(N)C1CCCC1 DICPPOONERDVPQ-UHFFFAOYSA-N 0.000 description 1
- SBTIFMGDVLXHIL-UHFFFAOYSA-N 3-[[amino(cyclopropyl)methylidene]amino]-4-methoxybenzoic acid;hydrochloride Chemical compound Cl.COC1=CC=C(C(O)=O)C=C1N=C(N)C1CC1 SBTIFMGDVLXHIL-UHFFFAOYSA-N 0.000 description 1
- IWOJQHXTUZJQKR-UHFFFAOYSA-N 3-[[amino(phenyl)methylidene]amino]-4-methoxybenzoic acid;hydrochloride Chemical compound Cl.COC1=CC=C(C(O)=O)C=C1N=C(N)C1=CC=CC=C1 IWOJQHXTUZJQKR-UHFFFAOYSA-N 0.000 description 1
- CJDKSXDQWNMKNO-UHFFFAOYSA-N 3-[[amino(thiophen-2-yl)methylidene]amino]-4-fluorobenzoic acid;hydrochloride Chemical compound Cl.C=1C=CSC=1C(N)=NC1=CC(C(O)=O)=CC=C1F CJDKSXDQWNMKNO-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- KDHWOCLBMVSZPG-UHFFFAOYSA-N 3-imidazol-1-ylpropan-1-amine Chemical compound NCCCN1C=CN=C1 KDHWOCLBMVSZPG-UHFFFAOYSA-N 0.000 description 1
- 125000004917 3-methyl-2-butyl group Chemical group CC(C(C)*)C 0.000 description 1
- 125000004919 3-methyl-2-pentyl group Chemical group CC(C(C)*)CC 0.000 description 1
- 125000004921 3-methyl-3-pentyl group Chemical group CC(CC)(CC)* 0.000 description 1
- LMLXMJPJCSUFAB-UHFFFAOYSA-N 4-(3-aminopropyl)-1,4-dihydropyrazol-5-one Chemical compound NCCCC1C=NNC1=O LMLXMJPJCSUFAB-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- CPEXPPRFYULOCJ-UHFFFAOYSA-N 4-[[amino(thiophen-2-yl)methylidene]amino]-3-methoxybenzoic acid;hydrochloride Chemical compound Cl.COC1=CC(C(O)=O)=CC=C1N=C(N)C1=CC=CS1 CPEXPPRFYULOCJ-UHFFFAOYSA-N 0.000 description 1
- ZLYBFBAHAQEEQQ-UHFFFAOYSA-N 4-chlorobenzenesulfonyl chloride Chemical compound ClC1=CC=C(S(Cl)(=O)=O)C=C1 ZLYBFBAHAQEEQQ-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- DVZBWONCSHFMMM-UHFFFAOYSA-N 4-methoxy-2-nitrobenzoic acid Chemical compound COC1=CC=C(C(O)=O)C([N+]([O-])=O)=C1 DVZBWONCSHFMMM-UHFFFAOYSA-N 0.000 description 1
- LTPVSOCPYWDIFU-UHFFFAOYSA-N 4-methoxyphenylethylamine Chemical compound COC1=CC=C(CCN)C=C1 LTPVSOCPYWDIFU-UHFFFAOYSA-N 0.000 description 1
- 125000004920 4-methyl-2-pentyl group Chemical group CC(CC(C)*)C 0.000 description 1
- ZIJAZUBWHAZHPL-UHFFFAOYSA-N 6-chloropyridine-3-carboxamide Chemical compound NC(=O)C1=CC=C(Cl)N=C1 ZIJAZUBWHAZHPL-UHFFFAOYSA-N 0.000 description 1
- HIEVUCALPWFEFA-UHFFFAOYSA-N 60814-16-6 Chemical compound NCCOC1=CC=C([N+]([O-])=O)C=C1 HIEVUCALPWFEFA-UHFFFAOYSA-N 0.000 description 1
- XVSGAPXSUQKKQJ-UHFFFAOYSA-N 7-fluoro-2-thiophen-2-yl-3h-benzimidazole-4-carboxylic acid Chemical compound N=1C=2C(C(=O)O)=CC=C(F)C=2NC=1C1=CC=CS1 XVSGAPXSUQKKQJ-UHFFFAOYSA-N 0.000 description 1
- KSMCYMNDVNDOEW-UHFFFAOYSA-N 7-fluoro-n-[2-(1h-imidazol-5-yl)ethyl]-2-thiophen-2-yl-3h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(F)=CC=C1C(=O)NCCC1=CN=CN1 KSMCYMNDVNDOEW-UHFFFAOYSA-N 0.000 description 1
- VLBUEFFYBHGVQX-UHFFFAOYSA-N 7-hydroxy-2-phenyl-n-(2-phenylethyl)-1h-benzimidazole-4-carboxamide Chemical compound C1=2NC(C=3C=CC=CC=3)=NC=2C(O)=CC=C1C(=O)NCCC1=CC=CC=C1 VLBUEFFYBHGVQX-UHFFFAOYSA-N 0.000 description 1
- JPQBWJWUGVSDBA-UHFFFAOYSA-N 7-hydroxy-2-thiophen-2-yl-3h-benzimidazole-5-carboxamide Chemical compound N=1C2=CC(C(=O)N)=CC(O)=C2NC=1C1=CC=CS1 JPQBWJWUGVSDBA-UHFFFAOYSA-N 0.000 description 1
- VDTJTBXXNGFVFO-UHFFFAOYSA-N 7-hydroxy-2-thiophen-2-yl-n-[2-[5-(trifluoromethyl)pyridin-2-yl]oxyethyl]-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(O)=CC=C1C(=O)NCCOC1=CC=C(C(F)(F)F)C=N1 VDTJTBXXNGFVFO-UHFFFAOYSA-N 0.000 description 1
- RQWPXWYHCDTLCQ-UHFFFAOYSA-N 7-hydroxy-n-(3-imidazol-1-ylpropyl)-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(O)=CC=C1C(=O)NCCCN1C=CN=C1 RQWPXWYHCDTLCQ-UHFFFAOYSA-N 0.000 description 1
- AOUOLQCMFOELTQ-CYBMUJFWSA-N 7-hydroxy-n-[(2s)-1-hydroxy-3,3-dimethylbutan-2-yl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound N=1C=2C(C(=O)N[C@H](CO)C(C)(C)C)=CC=C(O)C=2NC=1C1=CC=CS1 AOUOLQCMFOELTQ-CYBMUJFWSA-N 0.000 description 1
- ZQTVQAUSCZFRGE-AWEZNQCLSA-N 7-hydroxy-n-[(2s)-1-hydroxy-3-phenylpropan-2-yl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C([C@@H](CO)NC(=O)C=1C=2N=C(NC=2C(O)=CC=1)C=1SC=CC=1)C1=CC=CC=C1 ZQTVQAUSCZFRGE-AWEZNQCLSA-N 0.000 description 1
- KPMGOOQJFZUQKN-UHFFFAOYSA-N 7-hydroxy-n-[(4-sulfamoylphenyl)methyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=CC(S(=O)(=O)N)=CC=C1CNC(=O)C1=CC=C(O)C2=C1N=C(C=1SC=CC=1)N2 KPMGOOQJFZUQKN-UHFFFAOYSA-N 0.000 description 1
- MROBEFRBDYTTSK-UHFFFAOYSA-N 7-hydroxy-n-[2-(4-hydroxyphenyl)ethyl]-2-phenyl-1h-benzimidazole-4-carboxamide Chemical compound C1=CC(O)=CC=C1CCNC(=O)C1=CC=C(O)C2=C1NC(C=1C=CC=CC=1)=N2 MROBEFRBDYTTSK-UHFFFAOYSA-N 0.000 description 1
- PMQBYSMSFGQXEX-UHFFFAOYSA-N 7-hydroxy-n-[2-(4-hydroxyphenyl)ethyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=CC(O)=CC=C1CCNC(=O)C1=CC=C(O)C2=C1N=C(C=1SC=CC=1)N2 PMQBYSMSFGQXEX-UHFFFAOYSA-N 0.000 description 1
- FLPCFXRGUFUVHJ-UHFFFAOYSA-N 7-hydroxy-n-[2-(4-sulfamoylphenyl)ethyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=CC(S(=O)(=O)N)=CC=C1CCNC(=O)C1=CC=C(O)C2=C1N=C(C=1SC=CC=1)N2 FLPCFXRGUFUVHJ-UHFFFAOYSA-N 0.000 description 1
- YDRDTJACXJKVCD-UHFFFAOYSA-N 7-hydroxy-n-[2-[(4-methylbenzoyl)amino]ethyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=CC(C)=CC=C1C(=O)NCCNC(=O)C1=CC=C(O)C2=C1N=C(C=1SC=CC=1)N2 YDRDTJACXJKVCD-UHFFFAOYSA-N 0.000 description 1
- CTAQQUCNSORLGC-UHFFFAOYSA-N 7-hydroxy-n-[2-[4-[(4-methylphenyl)sulfonylamino]phenoxy]ethyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(C=C1)=CC=C1OCCNC(=O)C1=CC=C(O)C2=C1N=C(C=1SC=CC=1)N2 CTAQQUCNSORLGC-UHFFFAOYSA-N 0.000 description 1
- LOVNJIKQQJSSDK-UHFFFAOYSA-N 7-hydroxy-n-[3-(2-hydroxyethylamino)propyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound N=1C=2C(C(=O)NCCCNCCO)=CC=C(O)C=2NC=1C1=CC=CS1 LOVNJIKQQJSSDK-UHFFFAOYSA-N 0.000 description 1
- PZYYLNDOXCOICP-UHFFFAOYSA-N 7-hydroxy-n-[3-(5-oxo-1,4-dihydropyrazol-4-yl)propyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(O)=CC=C1C(=O)NCCCC1C=NNC1=O PZYYLNDOXCOICP-UHFFFAOYSA-N 0.000 description 1
- QJHCQFHTMOZFOW-UHFFFAOYSA-N 7-hydroxy-n-[3-(propan-2-ylamino)propyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound N=1C=2C(C(=O)NCCCNC(C)C)=CC=C(O)C=2NC=1C1=CC=CS1 QJHCQFHTMOZFOW-UHFFFAOYSA-N 0.000 description 1
- YYDDPZLBNSICAV-UHFFFAOYSA-N 7-methoxy-n-[2-(3-methylimidazol-4-yl)ethyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCC1=CN=CN1C YYDDPZLBNSICAV-UHFFFAOYSA-N 0.000 description 1
- YKYVRHJEVIGDGY-UHFFFAOYSA-N 7-methoxy-n-[2-(pyridin-4-ylsulfonylamino)ethyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCNS(=O)(=O)C1=CC=NC=C1 YKYVRHJEVIGDGY-UHFFFAOYSA-N 0.000 description 1
- XOHNBHXAPQLKBN-UHFFFAOYSA-N 7-methoxy-n-[2-[(4-methylbenzoyl)amino]ethyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCNC(=O)C1=CC=C(C)C=C1 XOHNBHXAPQLKBN-UHFFFAOYSA-N 0.000 description 1
- IQVCWYNOVLXABW-UHFFFAOYSA-N 7-methoxy-n-[3-(propan-2-ylamino)propyl]-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound N1C=2C(OC)=CC=C(C(=O)NCCCNC(C)C)C=2N=C1C1=CC=CS1 IQVCWYNOVLXABW-UHFFFAOYSA-N 0.000 description 1
- DIGZMTAFOACVBW-UHFFFAOYSA-N 7-phenylmethoxy-1h-indole Chemical compound C=1C=CC=2C=CNC=2C=1OCC1=CC=CC=C1 DIGZMTAFOACVBW-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- MRZZKPMKRVKJCS-UHFFFAOYSA-N COC(c(cc1)cc(NC(c2ccc[o]2)=N)c1OC)=O Chemical compound COC(c(cc1)cc(NC(c2ccc[o]2)=N)c1OC)=O MRZZKPMKRVKJCS-UHFFFAOYSA-N 0.000 description 1
- OFVFPQOTTLAGQX-UHFFFAOYSA-N COC(c(cc1)cc(NC(c2ccccc2)=N)c1OC)=O Chemical compound COC(c(cc1)cc(NC(c2ccccc2)=N)c1OC)=O OFVFPQOTTLAGQX-UHFFFAOYSA-N 0.000 description 1
- PTHCMJGKKRQCBF-UHFFFAOYSA-N Cellulose, microcrystalline Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC)C(CO)O1 PTHCMJGKKRQCBF-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 208000020401 Depressive disease Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 108010001483 Glycogen Synthase Proteins 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 1
- BXRMEWOQUXOLDH-LURJTMIESA-N L-Histidine methyl ester Chemical compound COC(=O)[C@@H](N)CC1=CN=CN1 BXRMEWOQUXOLDH-LURJTMIESA-N 0.000 description 1
- STVVMTBJNDTZBF-VIFPVBQESA-N L-phenylalaninol Chemical compound OC[C@@H](N)CC1=CC=CC=C1 STVVMTBJNDTZBF-VIFPVBQESA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- TYMRLRRVMHJFTF-UHFFFAOYSA-N Mafenide Chemical compound NCC1=CC=C(S(N)(=O)=O)C=C1 TYMRLRRVMHJFTF-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 101710115937 Microtubule-associated protein tau Proteins 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- BTRMVOSUMNXHFE-UHFFFAOYSA-N Oc(cc1)c2[nH]c(-c3ccc[s]3)nc2c1C(NCCc1ncc[nH]1)=O Chemical compound Oc(cc1)c2[nH]c(-c3ccc[s]3)nc2c1C(NCCc1ncc[nH]1)=O BTRMVOSUMNXHFE-UHFFFAOYSA-N 0.000 description 1
- UHNVSGAVJLZCOT-UHFFFAOYSA-N Oc1cc(C(NCCc2cnc[nH]2)=O)cc2c1[nH]c(-c1ccc[s]1)n2 Chemical compound Oc1cc(C(NCCc2cnc[nH]2)=O)cc2c1[nH]c(-c1ccc[s]1)n2 UHNVSGAVJLZCOT-UHFFFAOYSA-N 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- LSGKMZLPZFPAIN-UHFFFAOYSA-N Oxime-1H-Indole-3-carboxaldehyde Natural products C1=CC=C2C(C(=O)N)=CNC2=C1 LSGKMZLPZFPAIN-UHFFFAOYSA-N 0.000 description 1
- BHHGXPLMPWCGHP-UHFFFAOYSA-N Phenethylamine Chemical compound NCCC1=CC=CC=C1 BHHGXPLMPWCGHP-UHFFFAOYSA-N 0.000 description 1
- 108010001441 Phosphopeptides Proteins 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000009516 Protein Serine-Threonine Kinases Human genes 0.000 description 1
- 108010009341 Protein Serine-Threonine Kinases Proteins 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 239000005708 Sodium hypochlorite Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 208000034799 Tauopathies Diseases 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- ZBIKORITPGTTGI-UHFFFAOYSA-N [acetyloxy(phenyl)-$l^{3}-iodanyl] acetate Chemical compound CC(=O)OI(OC(C)=O)C1=CC=CC=C1 ZBIKORITPGTTGI-UHFFFAOYSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 125000005428 anthryl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C(*)=C([H])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 230000003190 augmentative effect Effects 0.000 description 1
- CSKNSYBAZOQPLR-UHFFFAOYSA-N benzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC=C1 CSKNSYBAZOQPLR-UHFFFAOYSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 239000000378 calcium silicate Substances 0.000 description 1
- 229910052918 calcium silicate Inorganic materials 0.000 description 1
- 235000012241 calcium silicate Nutrition 0.000 description 1
- OYACROKNLOSFPA-UHFFFAOYSA-N calcium;dioxido(oxo)silane Chemical compound [Ca+2].[O-][Si]([O-])=O OYACROKNLOSFPA-UHFFFAOYSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000011712 cell development Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- SVPZJHKVRMRREG-UHFFFAOYSA-N cyclopentanecarbonitrile Chemical compound N#CC1CCCC1 SVPZJHKVRMRREG-UHFFFAOYSA-N 0.000 description 1
- AUQDITHEDVOTCU-UHFFFAOYSA-N cyclopropyl cyanide Chemical compound N#CC1CC1 AUQDITHEDVOTCU-UHFFFAOYSA-N 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- ZCKYOWGFRHAZIQ-UHFFFAOYSA-N dihydrourocanic acid Chemical compound OC(=O)CCC1=CNC=N1 ZCKYOWGFRHAZIQ-UHFFFAOYSA-N 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 201000003104 endogenous depression Diseases 0.000 description 1
- 230000037149 energy metabolism Effects 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 125000004672 ethylcarbonyl group Chemical group [H]C([H])([H])C([H])([H])C(*)=O 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- 125000006125 ethylsulfonyl group Chemical group 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 125000001183 hydrocarbyl group Chemical group 0.000 description 1
- 150000002431 hydrogen Chemical group 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000006951 hyperphosphorylation Effects 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 125000006301 indolyl methyl group Chemical group 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 150000008040 ionic compounds Chemical class 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000011344 liquid material Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 description 1
- 229910052808 lithium carbonate Inorganic materials 0.000 description 1
- 229940071264 lithium citrate Drugs 0.000 description 1
- WJSIUCDMWSDDCE-UHFFFAOYSA-K lithium citrate (anhydrous) Chemical compound [Li+].[Li+].[Li+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O WJSIUCDMWSDDCE-UHFFFAOYSA-K 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 208000024714 major depressive disease Diseases 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229940099690 malic acid Drugs 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- KCUNTYMNJVXYKZ-JTQLQIEISA-N methyl (2s)-2-amino-3-(1h-indol-3-yl)propanoate Chemical compound C1=CC=C2C(C[C@H](N)C(=O)OC)=CNC2=C1 KCUNTYMNJVXYKZ-JTQLQIEISA-N 0.000 description 1
- NTNUDYROPUKXNA-UHFFFAOYSA-N methyl 2-(triphenyl-$l^{5}-phosphanylidene)acetate Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=CC(=O)OC)C1=CC=CC=C1 NTNUDYROPUKXNA-UHFFFAOYSA-N 0.000 description 1
- ANSUDRATXSJBLY-UHFFFAOYSA-N methyl 2-amino-3-hydroxypropanoate Chemical compound COC(=O)C(N)CO ANSUDRATXSJBLY-UHFFFAOYSA-N 0.000 description 1
- SIPPUXPIJXLNKR-UHFFFAOYSA-N methyl 3-(4-hydroxyphenyl)-2-[(7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carbonyl)amino]propanoate Chemical compound C=1C=C(O)C=2NC(C=3SC=CC=3)=NC=2C=1C(=O)NC(C(=O)OC)CC1=CC=C(O)C=C1 SIPPUXPIJXLNKR-UHFFFAOYSA-N 0.000 description 1
- VHIJRAZXPZVVMT-UHFFFAOYSA-N methyl 3-[[amino(cyclopentyl)methylidene]amino]-4-methoxybenzoate;hydrochloride Chemical compound Cl.COC(=O)C1=CC=C(OC)C(NC(=N)C2CCCC2)=C1 VHIJRAZXPZVVMT-UHFFFAOYSA-N 0.000 description 1
- ZPLDKFGQLDZAJL-UHFFFAOYSA-N methyl 3-[[amino(cyclopropyl)methylidene]amino]-4-methoxybenzoate;hydrochloride Chemical compound Cl.COC(=O)C1=CC=C(OC)C(NC(=N)C2CC2)=C1 ZPLDKFGQLDZAJL-UHFFFAOYSA-N 0.000 description 1
- AHYAHJHNDLZBNZ-UHFFFAOYSA-N methyl 3-[[amino(phenyl)methylidene]amino]-4-methoxybenzoate;hydrochloride Chemical compound Cl.COC(=O)C1=CC=C(OC)C(NC(=N)C=2C=CC=CC=2)=C1 AHYAHJHNDLZBNZ-UHFFFAOYSA-N 0.000 description 1
- DLIXZLCMFADBDS-UHFFFAOYSA-N methyl 3-[[amino(thiophen-2-yl)methylidene]amino]-4-fluorobenzoate;hydrochloride Chemical compound Cl.COC(=O)C1=CC=C(F)C(NC(=N)C=2SC=CC=2)=C1 DLIXZLCMFADBDS-UHFFFAOYSA-N 0.000 description 1
- UMDBYBUACBGSNK-UHFFFAOYSA-N methyl 3-[[amino(thiophen-2-yl)methylidene]amino]-4-methoxybenzoate Chemical compound COC(=O)C1=CC=C(OC)C(NC(=N)C=2SC=CC=2)=C1 UMDBYBUACBGSNK-UHFFFAOYSA-N 0.000 description 1
- ABELEDYNIKPYTP-UHFFFAOYSA-N methyl 3-amino-4-fluorobenzoate Chemical compound COC(=O)C1=CC=C(F)C(N)=C1 ABELEDYNIKPYTP-UHFFFAOYSA-N 0.000 description 1
- KLHIPTAXBMETKI-UHFFFAOYSA-N methyl 4-[[amino(thiophen-2-yl)methylidene]amino]-3-methoxybenzoate;hydrochloride Chemical compound Cl.COC1=CC(C(=O)OC)=CC=C1NC(=N)C1=CC=CS1 KLHIPTAXBMETKI-UHFFFAOYSA-N 0.000 description 1
- DJLFOMMCQBAMAA-UHFFFAOYSA-N methyl 4-amino-3-methoxybenzoate Chemical compound COC(=O)C1=CC=C(N)C(OC)=C1 DJLFOMMCQBAMAA-UHFFFAOYSA-N 0.000 description 1
- WUZGRQPEXDLKQN-UHFFFAOYSA-N methyl 7-methoxy-2-thiophen-2-yl-3h-benzimidazole-5-carboxylate Chemical compound N=1C2=CC(C(=O)OC)=CC(OC)=C2NC=1C1=CC=CS1 WUZGRQPEXDLKQN-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- KFDIDIIKNMZLRZ-UHFFFAOYSA-N n'-propan-2-ylpropane-1,3-diamine Chemical compound CC(C)NCCCN KFDIDIIKNMZLRZ-UHFFFAOYSA-N 0.000 description 1
- CJVFQZXNHIWPBZ-UHFFFAOYSA-N n-(2-acetamidoethyl)-7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound N1C=2C(OC)=CC=C(C(=O)NCCNC(C)=O)C=2N=C1C1=CC=CS1 CJVFQZXNHIWPBZ-UHFFFAOYSA-N 0.000 description 1
- MWEBCGQFQBAAJP-UHFFFAOYSA-N n-(2-aminoethyl)-4-chlorobenzenesulfonamide Chemical compound NCCNS(=O)(=O)C1=CC=C(Cl)C=C1 MWEBCGQFQBAAJP-UHFFFAOYSA-N 0.000 description 1
- IYTURFXZHKVZGN-UHFFFAOYSA-N n-(2-aminoethyl)-4-chlorobenzenesulfonamide;hydrochloride Chemical compound Cl.NCCNS(=O)(=O)C1=CC=C(Cl)C=C1 IYTURFXZHKVZGN-UHFFFAOYSA-N 0.000 description 1
- IISCWLATEHABQS-UHFFFAOYSA-N n-(2-aminoethyl)-4-methylbenzamide Chemical compound CC1=CC=C(C(=O)NCCN)C=C1 IISCWLATEHABQS-UHFFFAOYSA-N 0.000 description 1
- DAKZISABEDGGSV-UHFFFAOYSA-N n-(2-aminoethyl)acetamide Chemical compound CC(=O)NCCN DAKZISABEDGGSV-UHFFFAOYSA-N 0.000 description 1
- BCGXHHYBXMQECD-UHFFFAOYSA-N n-(2-aminoethyl)benzamide;hydrochloride Chemical compound Cl.NCCNC(=O)C1=CC=CC=C1 BCGXHHYBXMQECD-UHFFFAOYSA-N 0.000 description 1
- JVFJSUCEQDCCAH-UHFFFAOYSA-N n-(2-aminoethyl)benzenesulfonamide Chemical compound NCCNS(=O)(=O)C1=CC=CC=C1 JVFJSUCEQDCCAH-UHFFFAOYSA-N 0.000 description 1
- NTENADQOAWUTOH-UHFFFAOYSA-N n-(2-aminoethyl)benzenesulfonamide;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.NCCNS(=O)(=O)C1=CC=CC=C1 NTENADQOAWUTOH-UHFFFAOYSA-N 0.000 description 1
- KXBXVEGOHACSKW-UHFFFAOYSA-N n-(2-aminoethyl)pyridine-4-sulfonamide Chemical compound NCCNS(=O)(=O)C1=CC=NC=C1 KXBXVEGOHACSKW-UHFFFAOYSA-N 0.000 description 1
- AYZAQJGTNSWWBM-UHFFFAOYSA-N n-(2-aminoethyl)pyridine-4-sulfonamide;dihydrochloride Chemical compound Cl.Cl.NCCNS(=O)(=O)C1=CC=NC=C1 AYZAQJGTNSWWBM-UHFFFAOYSA-N 0.000 description 1
- LTGRYMCRXRFVCZ-UHFFFAOYSA-N n-(2-hydroxyethyl)-7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound N1C=2C(OC)=CC=C(C(=O)NCCO)C=2N=C1C1=CC=CS1 LTGRYMCRXRFVCZ-UHFFFAOYSA-N 0.000 description 1
- JHMDPDAKHZMBFU-UHFFFAOYSA-N n-(4-methoxyphenyl)carbamoyl chloride Chemical compound COC1=CC=C(NC(Cl)=O)C=C1 JHMDPDAKHZMBFU-UHFFFAOYSA-N 0.000 description 1
- KAXYSAVZDDUQAY-UHFFFAOYSA-N n-[(2,4-difluorophenyl)methyl]-7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(O)=CC=C1C(=O)NCC1=CC=C(F)C=C1F KAXYSAVZDDUQAY-UHFFFAOYSA-N 0.000 description 1
- IOOARBMSXOAWLS-UHFFFAOYSA-N n-[(3,4-dihydroxyphenyl)methyl]-7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=C(O)C(O)=CC=C1CNC(=O)C1=CC=C(O)C2=C1N=C(C=1SC=CC=1)N2 IOOARBMSXOAWLS-UHFFFAOYSA-N 0.000 description 1
- GNQXGIRJZAGFLD-UHFFFAOYSA-N n-[(3,4-dimethoxyphenyl)methyl]-7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=C(OC)C(OC)=CC=C1CNC(=O)C1=CC=C(OC)C2=C1N=C(C=1SC=CC=1)N2 GNQXGIRJZAGFLD-UHFFFAOYSA-N 0.000 description 1
- WGWOZONQSDBZKW-UHFFFAOYSA-N n-[2-(1h-imidazol-2-yl)ethyl]-7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCC1=NC=CN1 WGWOZONQSDBZKW-UHFFFAOYSA-N 0.000 description 1
- WWLAAJSXEXBHPG-UHFFFAOYSA-N n-[2-(1h-imidazol-5-yl)ethyl]-7-phenylmethoxy-1h-indole-3-carboxamide Chemical compound C=1NC2=C(OCC=3C=CC=CC=3)C=CC=C2C=1C(=O)NCCC1=CN=CN1 WWLAAJSXEXBHPG-UHFFFAOYSA-N 0.000 description 1
- NBKMAQQGBPQUAH-UHFFFAOYSA-N n-[2-(3,4-dihydroxyphenyl)ethyl]-2-(furan-2-yl)-7-hydroxy-1h-benzimidazole-4-carboxamide Chemical compound C1=C(O)C(O)=CC=C1CCNC(=O)C1=CC=C(O)C2=C1N=C(C=1OC=CC=1)N2 NBKMAQQGBPQUAH-UHFFFAOYSA-N 0.000 description 1
- PAPUOZOKUQXUCV-UHFFFAOYSA-N n-[2-(3,5-dimethyl-1,2-oxazol-4-yl)ethyl]-2-(furan-2-yl)-7-hydroxy-1h-benzimidazole-4-carboxamide Chemical compound CC1=NOC(C)=C1CCNC(=O)C1=CC=C(O)C2=C1N=C(C=1OC=CC=1)N2 PAPUOZOKUQXUCV-UHFFFAOYSA-N 0.000 description 1
- CYEZNUPYHKNUEM-UHFFFAOYSA-N n-[2-(4-aminophenyl)ethyl]-2-cyclopentyl-7-hydroxy-1h-benzimidazole-4-carboxamide Chemical compound C1=CC(N)=CC=C1CCNC(=O)C1=CC=C(O)C2=C1NC(C1CCCC1)=N2 CYEZNUPYHKNUEM-UHFFFAOYSA-N 0.000 description 1
- QSMNQEMSVXJRQS-UHFFFAOYSA-N n-[2-(4-aminophenyl)ethyl]-7-hydroxy-2-phenyl-1h-benzimidazole-4-carboxamide Chemical compound C1=CC(N)=CC=C1CCNC(=O)C1=CC=C(O)C2=C1NC(C=1C=CC=CC=1)=N2 QSMNQEMSVXJRQS-UHFFFAOYSA-N 0.000 description 1
- VDMPWEXEHQWQKM-UHFFFAOYSA-N n-[2-(4-fluorophenyl)ethyl]-7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(O)=CC=C1C(=O)NCCC1=CC=C(F)C=C1 VDMPWEXEHQWQKM-UHFFFAOYSA-N 0.000 description 1
- DCLDFGXUQVRFCZ-UHFFFAOYSA-N n-[2-(4-fluorophenyl)ethyl]-7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCC1=CC=C(F)C=C1 DCLDFGXUQVRFCZ-UHFFFAOYSA-N 0.000 description 1
- BDRBZSPAJPBMKA-UHFFFAOYSA-N n-[2-(5-carbamoylpyridin-2-yl)oxyethyl]-7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound N1=CC(C(=O)N)=CC=C1OCCNC(=O)C1=CC=C(O)C2=C1N=C(C=1SC=CC=1)N2 BDRBZSPAJPBMKA-UHFFFAOYSA-N 0.000 description 1
- UMNDDQAYXARJBV-UHFFFAOYSA-N n-[2-(benzenesulfonamido)ethyl]-7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCNS(=O)(=O)C1=CC=CC=C1 UMNDDQAYXARJBV-UHFFFAOYSA-N 0.000 description 1
- VSWOMRSVSRDHTQ-UHFFFAOYSA-N n-[2-(dimethylamino)ethyl]-7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound N=1C=2C(C(=O)NCCN(C)C)=CC=C(O)C=2NC=1C1=CC=CS1 VSWOMRSVSRDHTQ-UHFFFAOYSA-N 0.000 description 1
- JOHGYBMIJBITFP-UHFFFAOYSA-N n-[2-[(4-chlorophenyl)sulfonylamino]ethyl]-7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(O)=CC=C1C(=O)NCCNS(=O)(=O)C1=CC=C(Cl)C=C1 JOHGYBMIJBITFP-UHFFFAOYSA-N 0.000 description 1
- GETFDLMXWYOKKJ-UHFFFAOYSA-N n-[2-[(4-chlorophenyl)sulfonylamino]ethyl]-7-methoxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound C1=2N=C(C=3SC=CC=3)NC=2C(OC)=CC=C1C(=O)NCCNS(=O)(=O)C1=CC=C(Cl)C=C1 GETFDLMXWYOKKJ-UHFFFAOYSA-N 0.000 description 1
- NGQAEUFCTRTCCT-UHFFFAOYSA-N n-[2-[(5-acetamidopyridin-2-yl)amino]ethyl]-7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide Chemical compound N1=CC(NC(=O)C)=CC=C1NCCNC(=O)C1=CC=C(O)C2=C1N=C(C=1SC=CC=1)N2 NGQAEUFCTRTCCT-UHFFFAOYSA-N 0.000 description 1
- SBECMSFCBVICPN-UHFFFAOYSA-N n-[2-[(5-aminopyridin-2-yl)amino]ethyl]-7-hydroxy-2-thiophen-2-yl-1h-benzimidazole-4-carboxamide;hydrochloride Chemical compound Cl.N1=CC(N)=CC=C1NCCNC(=O)C1=CC=C(O)C2=C1N=C(C=1SC=CC=1)N2 SBECMSFCBVICPN-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000006126 n-butyl sulfonyl group Chemical group 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000005185 naphthylcarbonyl group Chemical group C1(=CC=CC2=CC=CC=C12)C(=O)* 0.000 description 1
- 125000005146 naphthylsulfonyl group Chemical group C1(=CC=CC2=CC=CC=C12)S(=O)(=O)* 0.000 description 1
- 238000011445 neoadjuvant hormone therapy Methods 0.000 description 1
- 210000003061 neural cell Anatomy 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- XKJCHHZQLQNZHY-UHFFFAOYSA-N phthalimide Chemical compound C1=CC=C2C(=O)NC(=O)C2=C1 XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- NHMOJCOXIZRTRR-UHFFFAOYSA-N pyridine-4-sulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=NC=C1 NHMOJCOXIZRTRR-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000005344 pyridylmethyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 239000011535 reaction buffer Substances 0.000 description 1
- 230000007261 regionalization Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000012056 semi-solid material Substances 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 239000012058 sterile packaged powder Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 125000006253 t-butylcarbonyl group Chemical group [H]C([H])([H])C(C(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- AKQXKEBCONUWCL-UHFFFAOYSA-N tert-butyl 3-aminopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCCC(N)C1 AKQXKEBCONUWCL-UHFFFAOYSA-N 0.000 description 1
- XDJCYKMWJCYQJM-UHFFFAOYSA-N tert-butyl n-(3-hydroxypropyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCCO XDJCYKMWJCYQJM-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000005302 thiazolylmethyl group Chemical group [H]C1=C([H])N=C(S1)C([H])([H])* 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000005301 thienylmethyl group Chemical group [H]C1=C([H])C([H])=C(S1)C([H])([H])* 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- QIQITDHWZYEEPA-UHFFFAOYSA-N thiophene-2-carbonyl chloride Chemical compound ClC(=O)C1=CC=CS1 QIQITDHWZYEEPA-UHFFFAOYSA-N 0.000 description 1
- QERYCTSHXKAMIS-UHFFFAOYSA-N thiophene-2-carboxylic acid Chemical compound OC(=O)C1=CC=CS1 QERYCTSHXKAMIS-UHFFFAOYSA-N 0.000 description 1
- LOIYMIARKYCTBW-OWOJBTEDSA-N trans-urocanic acid Chemical compound OC(=O)\C=C\C1=CNC=N1 LOIYMIARKYCTBW-OWOJBTEDSA-N 0.000 description 1
- 125000005500 uronium group Chemical group 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/18—Benzimidazoles; Hydrogenated benzimidazoles with aryl radicals directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention relates to a compound for inhibiting glycogen synthase kinase-3 (GSK3) activity, a method for the preparation thereof, and a pharmaceutical composition containing the compound as an active ingredient.
- GSK3 glycogen synthase kinase-3
- Glycogen synthase kinase-3 (GSK3) is a proline-directed serine-threonine kinase that was initially identified as a protein which inactivates glycogen synthase through phosphorylation. Two isoforms have been identified, alpha (GSK3 alpha) and beta (GSK3beta), which show a high degree of amino acid homology to each other. Previous studies have reported that the GSK3beta is involved in energy metabolism, neural cell development, and body pattern formation (Plyte SE, et al., Biochim. Biophys. Acta, 1114:147-162, 1992).
- Neurodegenerative naturopathies including Alzheimer disease, are characterized by abnormal hyperphosphorylation of the microtubule-associated protein tau at proline-directed serine/threonine phosphorylation sites (Lee VM, et al., Annu. Rev. Neurosci. 24: 1121-1159, 2001.).
- GSK3beta has been identified as a prime candidate mediating aberrant tau phosphorylation at disease-associated sites (Hanger DP, et al., Neurosci. Lett. 147: 58-62, 1992., Ishiguro K, et al., J. Biol. Chem. 267: 10897-10901, 1992., Mandelkow EM, et al., FEBS Lett.
- GSK3beta is a promising target for therapeutic intervention in neurodegenerative tauopathies including Alzheimer disease.
- Lithium carbonate, lithium citrate and lithium chloride are commonly used for the treatment of various disorders like mania, depression and migraine, and also used as an "augmenting" agent to increase the benefits of other standard drugs used for unipolar depression.
- Lithium is a GSK3beta inhibitor, and therefore, GSK3beta inhibition is a promising target for the treatment of various such disorders.
- GSK3 inhibitors are available for treatment of type 2 diabetes by reducing the activity of glucose synthase.
- GSK3beta inhibitors can be used for a broad spectrum of diseases such as Alzheimer disease, mania, depression, migraine and type 2 diabetes and there is a strong need to develop such inhibitors for the treatment and/or prevention of GSK3beta dependent diseases.
- benzoimidazole derivatives can selectively inhibit the activity of GSK3beta and are therefore useful for treatment and/or prevention of GSK3beta dependent diseases.
- ring A is (II), (III), (IV) (V), or (VI)
- X is halogen or hydroxyl
- Y is hydrogen, phenyl, thiophen-2-yl, furan-2-yl, cyclopropyl, or cyclopentyl;
- Z is a 5-10 membered heterocycle substituted carbonylamino; and Ring A is substituted by -L'-(CH 2 ) a -L 2 -M at position *;
- L 1 is -CONH-, -NHCO-, or a single bond;
- M is selected from the group consisting of hydroxyl, carboxyl, amide, Ci-C 6 alkyl, Ci-C 6 alkylcarbonyl, C 6 -Ci 4 aryl, C 6 -Ci 4 aryl Ci-C 6 alkyl, C 6 -Ci 4 arylcarbonyl, C 6 -Ci 4 arylsulfonyl, 5-14 membered saturated, unsaturated or aromatic heterocyclic group, 5-14 membered unsaturated or aromatic heterocyclic group substituted Ci-C 6 alkyl, 5-14 membered unsaturated or aromatic heterocyclic group substituted sulfonyl or -NR R ; wherein R 2 and R 3 are independently Cj-C 6 alkyl; the CpC 6 alkyl, CpC 6 alkylcarbonyl, C 6 -C 14 aryl, C 6 -Ci 4 aryl Ci-C 6 alkyl, C 6 -Cj 4 arylcarbonyl, C 6 -
- alkyl refers to a straight chain or a branched chain hydrocarbon group which does not contain any hetero atoms or unsaturated carbon-carbon bonds.
- Ci-C 6 alkyl refers to an alkyl group which has 1-6 carbon atoms.
- Ci-C 4 alkyl refers to an alkyl group which has 1-4 carbon atoms.
- Ci-C 6 alkyl examples include, but are not limited to, methyl, ethyl, 1 -propyl, 2-propyl, 2 -methyl- 1 -propyl, 2-methyl-2-propyl, 1 -butyl, 2-butyl, 1-pentyl, 2-pentyl, 3-pentyl, 2-methyl-l -butyl, 3 -methyl- 1 -butyl, 2-methyl-2-butyl, 3-methyl-2-butyl, 2,2-dimethyl-l -propyl, 1-hexyl, 2-hexyl, 3-hexyl, 2 -methyl- 1-pentyl, 3 -methyl- 1-pentyl, 4-methyl- 1-pentyl, 2-methyl-2-pentyl, 3-methyl-2-pentyl, 4-methyl-2-pentyl, 2-methy-3-pentyl, 3-methyl-3-pentyl, 2,3 -dimethyl- 1 -butyl, 3, 3
- alkoxy refers to a group represented by -OR, wherein R is alkyl.
- Ci-C 6 alkoxy refers to an alkoxy group which has 1-6 carbon atoms.
- Cj-C 4 alkoxy refers to an alkoxy group which has 1-4 carbon atoms.
- Examples OfCi-C 6 alkoxy include, but are not limited to, methoxy, ethoxy, 1-propyloxy, 2-propyloxy, 2 -methyl- 1-propyloxy, 2-methyl-2-propyloxy, and 1-butyloxy, and 2- butyloxy.
- Ci-C 6 alkylcarbonyl refers to a carbonyl group bound to the Ci-C 6 alkyl.
- C]-C 4 alkylcarbonyl refers to a carbonyl group bound to theCj-C 4 alkyl.
- Examples of “CpC 6 alkylcarbonyl” include, but are not limited to, methylcarbonyl, ethylcarbonyl, 1-propylcarbonyl, 2-propylcarbonyl, n-butylcarbonyl, s-butylcarbonyl, t-butylcarbonyl, and 2-ethylbutylcarbyl.
- amino refers to a group represented by -NH 2 in which the hydrogens are optionally replaced by a substituent.
- Cj-C 6 alkyl carbonylamino refers to an amino group bound to the
- Ci-C 6 alkylcarbonyl Ci-C 6 alkylcarbonyl.
- Cj-C 4 alkylcarbonylamino refers to an amino group bound to the CpC 4 alkylcarbonyl.
- C 1 -C 6 alkylcarbonylamino examples include, but are not limited to, methylcarbonylamino, ethylcarbonylamino, 1 -propylcarbonylamino, 2-propylcarbonylamino, n-butylcarbonylamino, s-butylcarbonylamino, t-butylcarbonylamino, and 2-ethylbutylcarbonylamino.
- sulfonyl is a group represented by -SO 2 -.
- CpC 6 alkylsulfonyl refers to a sulfonyl group bound to the CpC 6 alkyl.
- CpC 4 alkylsulfonyl refers to a sulfonyl group bound to the Cj-C 4 alkyl.
- CpC 6 alkylsulfonyl examples include, but are not limited to, methylsulfonyl, ethylsulfonyl, 1-propylsulfonyl, 2-propylsulfonyl, n-butylsulfonyl, s-butylsulfonyl, t-butylsulfonyl, and 2-ethylbutylsulfonyl.
- CpC 6 alkylsulfonylamino refers to an amino group bound to the"CpC 6 alkylsulfonyl.
- CpC 4 alkylsulfonylamino refers to an amino group bound to the"CpC 4 alkylsulfonyl.
- Cj-C 6 alkylsulfonylamino examples include, but are not limited to, methylsulfonylamino, ethylsulfonylamino, 1 -propylsulfonylamino, 2-propylsulfonylamino, n-butylsulfonylamino, s-butylsulfonylamino, t-butylsulfonylamino, and 2-ethylbutylsulfonylamino. Page: 4/97
- aryl refers to an aromatic carbon ring system.
- C 6 -Cj 4 aryl refers to a 6-14 membered aryl ring.
- C 6 -Ci 0 aryl refers to a 6-10 membered aryl ring.
- C 6 -Ci 4 aryl examples include, but are not limited to, phenyl, naphthyl, and anthryl.
- C 6 -Ci 4 aryl Cj-C 6 alkyl refers to the"Cj-C 6 alkyl" in which a hydrogen atom is substituted by the"C 6 -Cj 4 aryl.
- C 6 -Cj 0 arylC r C 4 alkyl refers to the"Cj-C 4 alkyl” in which a hydrogen atom is substituted by the"C 6 -Cjo aryl".
- C 6 -Cj 4 arylCj-C 6 alkyl examples include, but are not limited to, benzyl, phenethyl, and anthrylmethyl.
- C 6 -Cj 4 arylcarbonyl refers to a carbonyl group bound to the"C 6 -Cj 4 aryl.
- C 6 -Ci 0 arylcarbonyl refers to a carbonyl group bound to the"C 6 -C] 0 aryl.
- C 6 -Cj 4 arylcarbonyl examples include, but are not limited to, phenylcarbonyl, naphthylcarbonyl, and anthrylcarbonyl.
- C 6 -Cj 4 arylsulfonyl refers to a sulfonyl group bound to the"C 6 -Cj 4 aryl.
- C 6 -CjO arylsulfonyl refers to a sulfonyl group bound to the"C 6 -Cj 0 aryl.
- Examples of “C 6 -Ci 4 arylsulfonyl” include, but are not limited to, phenylsulfonyl, naphthylsulfonyl, and anthrylsulfonyl.
- an unsaturated or aromatic heterocyclic group refers to an unsaturated or aromatic heterocyclic group having one or more hetero atom in the ring system.
- “5-14 membered unsaturated or aromatic heterocyclic group” refers to an unsaturated or aromatic heterocyclic group in which the ring consists of 5- 14 atoms.
- “5-10 membered unsaturated or aromatic heterocyclic group” refers to a unsaturated or aromatic heterocyclic group in which the ring consists of 5-10 atoms.
- Examples of "5-14 membered unsaturated or aromatic heterocyclic group” include, but are not limited to, imidazolyl, pyrrolyl, pyridyl, thienyl, furyl, thiazolyl, pyrazolyl, pyrazolinyl, oxazolyl, isoxazolyl and indolyl.
- 5-14 membered unsaturated or aromatic heterocyclic group substituted Cj-C 6 alkyl refers to the "Cj-C 6 alkyl” in which a hydrogen atom is substituted by the"5-14 membered unsaturated or aromatic heterocyclic group.
- 5-10 membered unsaturated or aromatic heterocyclic group substituted Cj-C 4 alkyl refers to the "Cj-C 4 alkyl” in which a hydrogen atom is substituted by the"5-10 membered unsaturated or aromatic heterocyclic group”.
- Examples of “5-14 membered unsaturated or aromatic heterocyclic group substituted Cj-C 6 alkyl” include, but are not limited to, imidazolylmethyl, pyrrolylmethyl, pyridylmethyl, thienylmethyl, furylmethyl, thiazolylmethyl, pyrazolylmethyl, pyrazolinylmethyl, oxazolylmethyl, isoxazolylmethyl, and indolylmethyl.
- “5-14 membered unsaturated or aromatic heterocyclic group substituted sulfonyl” refers to a sulfonyl group bound to the 5-14 membered unsaturated or aromatic heterocyclic group”.
- 5-10 membered unsaturated or aromatic heterocyclic group substituted sulfonyl refers to a sulfonyl group bound to "5-10 membered unsaturated or aromatic heterocyclic group”.
- Examples of "5-14 membered unsaturated or aromatic heterocyclic group substituted sulfonyl” include, but are not limited to, imidazolylsulfonyl, pyrrolylsulfonyl, pyridylsulfonyl, thienylsulfonyl, furylsulfonyl, thiazolylsulfonyl, pyrazolylsulfonyl, pyrazolinylsulfonyl, oxazolylsulfonyl, isoxazolylsulfonyl, and indolylsulfonyl. Page: 5/97
- 5-10 membered unsaturated or aromatic heterocyclic group substituted carbonylamino refers to an amino group bound to a carbonyl group bound to the "5-10 membered unsaturated or aromatic heterocyclic group”.
- Examples of "5-10 membered unsaturated or aromatic heterocyclic group substituted carbonylamino” include, but are not limited to, imidazolylcarbonylamino, pyrrolylcarbonylamino, pyridylcarbonylamino, thienylcarbonylamino, furylcarbonylamino, thiazolylcarbonylamino, pyrazolylcarbonylamino, pyrazolinylcarbonylamino, oxazolylcarbonylamino, isoxazolylcarbonylamino, and indolylcarbonylamino.
- a saturated heterocyclic group refers to a saturated heterocyclic group having one or more hetero atom in the ring system.
- “5-14 membered saturated heterocyclic group” refers to a saturated heterocyclic group in which the ring consists of 5-14 atoms.
- “5-10 membered saturated heterocyclic group” refers to a saturated heterocyclic group in which the ring consists of 5-10 atoms.
- Examples of “5-14 membered saturated heterocyclic group” include, but are not limited to, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl and thiomorpholinyl.
- a salt is defined as the product formed from the neutralisation reaction of acids and bases. Salts are ionic compounds composed of cations (positively charged ions) and anions (negative ions) so that the product is electrically neutral. These component ions can be inorganic as well as organic.
- Hydrate is a term used in inorganic chemistry and organic chemistry to indicate that a substance contains water.
- Solvate refers to a molecule in a solution complexed by solvent molecules.
- Isomers are compounds with the same molecular formula but different structural formulae. More specifically, isomer includes geometric isomer, optical isomer, stereoisomer, tautomer of the compound, and mixtures thereof.
- the present invention provides a compound represented by formula (I):
- X is halogen or hydroxyl
- Y is phenyl, thiophen-2-yl, furan-2-yl, cyclopropyl, or cyclopentyl; Ring (II) is substituted by -L 1 -(CH 2 ) a -L 2 -M at position *; L 1 is -CONH- or -NHCO-; Page: 6/97
- L 2 is selected from the group consisting of -NH-, -O-, -CH(COOR 1 )-, -CH(CH 2 OH)-, and a single bond, wherein R ! is hydrogen or C 1 -C 6 alkyl;
- M is selected from the group consisting of hydroxyl, carboxyl, amide, Ci-C 6 alkyl, Ci-C 6 alkylcarbonyl, C 6 -Cj 4 aryl, C 6 -Ci 4 aryl C]-C 6 alkyl, C 6 -Ci 4 arylcarbonyl, C 6 -Ci 4 arylsulfonyl, 5-14 membered saturated, unsaturated or aromatic heterocyclic group, 5-14 membered unsaturated or aromatic heterocyclic group substituted C]-C 6 alkyl, 5-14 membered unsaturated or aromatic heterocyclic group substituted sulfonyl or -NR 2 R 3 ; wherein R 2 and R 3 are independently Ci-C 6 alkyl; the Ci-C 6 alkyl, Ci-C 6 alkylcarbonyl, C 6 -C] 4 aryl, C 6 -C] 4 aryl Ci-C 6 alkyl, C 6 -Ci 4 arylcarbonyl,
- the present invention provides compounds represented by following formula (I-II) or a salt, hydrate, solvate, or isomer thereof:
- L 1 is -CONH-;
- L 2 is a single bond;
- M is C 6 -Ci O aryl or 5-10 membered unsaturated or aromatic heterocyclic group having 1-2 hetero atom(s) selected from the group consisting of N, O, and S, which are optionally substituted by 1 or 2 substituent(s) each independently selected from the group A; and
- X, Y, and a are defined as in above embodiment represented by formula (I).
- M is selected from the group consisting of phenyl, imidazole- 1-yl, imdazole-2-yl, imidazole- 5 -yl, thiophen-2-yl, pyrole-2-yl, l,3-thiazole-2-yl, 2-pyrazoline-4-yl, and isoxazole-4-yl, which are optionally substituted by 1-2 substituent(s) each independently selected from following group B, and Y is selected from the group consisting of thiophen-2-yl, furan-2-yl, phenyl, cyclopropyl, and cyclopentyl.
- Group B consists of fluoro, hydroxyl, oxo, amino, methyl, methoxy, and sulfamoyl.
- Preferred compounds include those selected from the group consisting of: Example Nos. 8, 9, 10, 20, 21, 22, 23, 35, 37, 44, 45, 57, 62, 76, 77, 78, 79, 80, 84, 85, 86, 90, 91, 92, 93, 94, 95, 96, 101 and 102 listed in Table 1 below; and the pharmaceutically acceptable salts, prodrugs, hydrates and solvates of the forgoing compounds. Page: 7/97
- the present invention provides a compound represented by following formula (I- II) or a salt, hydrate, solvate, or isomer thereof : Page: 1 1/97
- L 2 is -NH-;
- M is Ci-C4 alkyl, Cj-C 4 alkylcarbonyl, C 6 -Ci 0 arylcarbonyl, C 6 -Ci 0 arylsulfonyl, 5-10 membered unsaturated or aromatic heterocyclic group having 1 -2 hetero atom(s) selected from the group consisting of N, O, and S or sulfonyl substituted by 5-10 membered unsaturated or aromatic heterocyclic group having 1-2 hetero atom(s) selected from the group consisting of N, O, and S, which are optionally substituted by 1 or 2 substituent(s) each independently selected form the group A; and
- X, Y, and a are defined as in the above embodiment represented by formula (I).
- M is selected from the group consisting of ethyl, isopropyl, methylcarbonyl, pyridine-2-yl, phenylcarbonyl, phenylsulfonyl, and 4-pyridilsulfonyl, which are optionally substituted by 1 -2 substituent(s) each independently selected from following group C, and Y is selected from the group consisting of thiophen-2-yl and furan-2-yl.
- Group C consists of chloro, hydroxyl, methyl, methylcarbonylamino, methylsulfonylamino, and p-toluenesulfonylamino.
- the present invention provides the compound selected from the group consisting of: Example Nos. 11, 12, 38, 39, 40, 41, 42, 43, 69, 70 and 89 listed in Table 2 below, and the pharmaceutically acceptable salts, prodrugs, hydrates and solvates of the forgoing compounds.
- the present invention provides a compound represented by following formula (I-II) or a salt thereof:
- L 1 is -CONH-
- L 2 is -CH(COOR 1 )-, wherein R 1 is hydrogen or Ci-C 4 alkyl;
- M is C 1 -C 4 alkyl, C 6 -C 10 aryl Ci-C 4 alkyl or Ci-C 4 alkyl substituted by 5-10 membered unsaturated or aromatic heterocyclic group having 1 -2 hetero atom(s) selected from the group consisting of N, O, and S , which are optionally substituted by 1 or 2 substituent(s) each independently selected from the group A; and
- X, Y, and a are defined as in the above embodiment represented by formula (I).
- M is selected from the group consisting of methyl, phenylmethyl, indole-3-ylmethyl, and imidazole-4-ylmethyl, which are optionally substituted by 1-2 hydroxyl
- Y is thiophen-2-yl.
- the present invention provides the compound selected from the group consisting of: Example Nos. 13, 14, 15, 16, 71 and 72 listed in Table 3 below, and the pharmaceutically acceptable salts, prodrugs, hydrates and solvates of the forgoing compounds.
- the present invention provides a compound represented by following formula (I- II) or a salt, hydrate, solvate, or isomer thereof:
- L 2 is - 0-
- M is C 6 -C] 0 aryl or 5-10 membered unsaturated or aromatic heterocyclic group having 1-2 hetero atom(s) selected from the group consisting of N, O, and S, which are optionally substituted by 1 or 2 substituent(s) each independently selected from the group A;
- X, Y, and a are defined in the above embodiment represented by formula (I).
- M is phenyl or pyridine-2-yl, which is optionally substituted by 1 or 2 substituent(s) each independently selected from following group D, and Y preferably consists of thiophen-2-yl.
- Group D consists of amide, nitro, trifluoromethyl, and p-toluenesulfonylamino. Page: 15/97
- the present invention provides the compound selected from the group consisting of: Example Nos. 49, 50, 73 and 74 listed in Table 4 below, and the pharmaceutically acceptable salts, prodrugs, hydrates and solvates of the forgoing compounds.
- the present invention provides the compounds represented by following formula (I-II) or a salt, hydrate, solvate, or isomer thereof:
- L 1 is -CONH-
- L 2 is - CH(CH 2 OH)-; Page: 16/97
- M is selected from the group consisting of hydroxyl, Ci-C 4 alkyl, C 6 -Ci 0 aryl Ci-C 4 alkyl, and Cj-C 4 alkyl substituted by 5-10 membered unsaturated or aromatic heterocyclic group having 1-2 hetero atom(s) selected from the group consisting of N, O, and S, the C]-C 4 alkyl, C 6 -Ci 0 aryl Ci-C 4 alkyl, and 5-10 membered unsaturated or aromatic heterocyclic group having 1-2 hetero atom(s) are optionally substituted by 1 or 2 substituent(s) each independently selected from the group A; and
- X, Y, and a are defined as in the above embodiment represented by formula (I).
- M is preferably hydroxyl, phenylmethyl, t-butyl, or imidazole-5-ylmethyl
- Y is selected from the group consisting of thiophen-2-y and cyclopropyl.
- the present invention provides the compound selected from the group consisting of: Example Nos. 17, 18, 19 and 97 listed in Table 5 below, and the pharmaceutically acceptable salts, prodrugs, hydrates and solvates of the forgoing compounds.
- the present invention provides a compound represented by following formula (I-II) or a salt, hydrate, solvate, or isomer thereof : Page: 17/97
- Y is selected from the group consisting of thiophen-2-yl and cyclopropyl.
- the present invention provides the compound selected from the group consisting of: Example Nos. 36 and 98 listed in Table 6 below, and the pharmaceutically acceptable salts, prodrugs, hydrates and solvates of the forgoing compounds.
- the present invention provides a compound represented by following formula (I-II) or a salt, hydrate, solvate, or isomer thereof:
- L 1 is -NHCO-
- M is C 6 -Ci O aryl or 5-10 membered unsaturated or aromatic heterocyclic group having 1-2 hetero atom(s) selected from the group consisting of N, O, and S, which are optionally substituted by 1 or 2 substituent(s) each independently selected from the group A; and
- X, Y, and a are defined as in the above embodiment represented by formula (I).
- M is preferably phenyl optionally having 1 or 2 hydroxyl, or imidazol-5-yl and Y is cyclopropyl or thiophen-2-yl.
- the present invention provides the compound selected from the group consisting of: Example Nos. 107, 108, 120 and 121 listed in Table 7 below, and the pharmaceutically acceptable salts, prodrugs, hydrates and solvates of the forgoing compounds.
- L is -CONH- or a single bond
- L 2 is a single bond
- M is amide or 5-10 membered unsaturated or aromatic heterocyclic group having 1 or 2 hetero atom(s) selected from the group consisting of N, O, and S optionally substituted by 1 or 2 substituent(s) each independently selected from the group A; and
- X, Y, and a are defined in the above embodiment represented by formula (I).
- Y is thiophen-2-yl.
- the present invention provides the compound selected from the group consisting of: Example Nos. 65 and 66 listed in Table 8 below, and the pharmaceutically acceptable salts, prodrugs, hydrates and solvates of the forgoing compounds.
- the present invention provides a compound represented by following formula (I-IV) or a salt, hydrate, solvate, or isomer thereof :
- L 1 is -CONH-;
- L 2 is a single bond;
- M is 5-10 membered unsaturated or aromatic heterocyclic group having 1 or 2 hetero atom(s) selected from the group consisting of N, O, and S optionally substituted by 1 or 2 substituent(s) each independently selected from the group A; and
- X, Y, and a are defined as in the embodiment represented by formula (I).
- Y is hydrogen.
- the present invention provides the compound of Example No. 110 listed in Table 9 below, and the pharmaceutically acceptable salts, prodrugs, hydrates and solvates of the forgoing compound. Page: 20/97
- the present invention provides compounds represented by following formula (I- V), (I- VI) or a salt, hydrate, solvate, or isomer thereof :
- L 1 is -CONH-;
- L 2 is a single bond;
- M is 5-10 membered saturated, unsaturated or aromatic heterocyclic group having 1 or 2 hetero atom(s) selected from the group consisting of N, O, and S optionally substituted by 1 or 2 substituent(s) each independently selected from the group A; and
- X, Z, and a are defined as in the above embodiment represented by formula (I).
- Z is preferably thiophen-2-ylcarbonylamino.
- the present invention provides the compound of Example Nos. 112 and 122 listed in Table 10 below, and the pharmaceutically acceptable salts, prodrugs, hydrates and solvates of the forgoing compound.
- the present invention provides a compound represented by formula (I- VI) or a salt, hydrate, solvate, or isomer thereof:
- Ring A is represented by the formula below;
- M is carboxyl
- X, Y, Z and a are defined as in the above embodiment represented by formula (I).
- ring A is preferably the formula (II).
- the present invention provides the compound of: Example No. 1 listed in Table 11 below, and the pharmaceutically acceptable salts, prodrugs, hydrates and solvates of the forgoing compounds.
- the compound of formula (I) of the present invention may be in the form of a pharmaceutically acceptable salt derived from an inorganic or organic acid
- representative examples of the pharmaceutically acceptable salt derived from an inorganic or organic acid include salts obtained by adding an inorganic acid such as hydrochloric acid, hydrobromic acid, phosphoric acid or sulfonic acid, or organic carboxylic acids such as acetic acid, trifluoroacetic acid, citric acid, formic acid, maleic acid, oxalic acid, succinic acid, benzoic acid, tartaric acid, fumaric acid, Page: 22/97 mandelic acid, ascorbic acid or malic acid, methanesulfonic acid, or para toluenesulfonic acid, which do not limit its scope, to the compound of formula (I).
- Such acids may be prepared by the conventional processes, and other acids, which themselves are not pharmaceutically acceptable, including oxalic acid may be employed in the preparation of the bases.
- the compound of formula (I) of the present invention may also be in the form of a pharmaceutically acceptable salt derived from an inorganic or organic base include salts obtained by adding an inorganic or organic base.
- a pharmaceutically acceptable salt derived from an inorganic or organic base include salts obtained by adding an inorganic or organic base.
- alkalis including sodium hydroxide or potassium hydroxide, or alkaline earth metal hydroxides including calcium hydroxide, magnesium hydroxide, aluminum hydroxide or ammonium hydroxide may be used for the preparation of inorganic salt of the compound.
- organic bases including triethylamine or diisopropylethylamine may also be used for the preparation of organic salt of the compound.
- p-TSA is p-toluenesulfonic acid
- HATU is
- Aniline A is reacted with a nitrile in the presence of p-toluenesulfonic acid to afford amidine B.
- Amidine B is chlorinated with sodium hypochlorite and cyclized using sodium bicarbonate to form benzimidazole C.
- Intermediate C is saponified with sodium hydroxide to afford methoxy acid D.
- Compound D is treated with boron tribromide to afford hydroxy acid E.
- Hydroxy acid E is reacted with various amines using HATU to afford compounds of formula I-II.
- Compound D is also reacted with various amines in the presence of HATU to afford amides F.
- Amides F are treated with boron tribromide to afford compounds of formula (I-III).
- the preferred inventive compound of formula (I- V) can be prepared as shown in Scheme (III).
- Aniline A is coupled with a carboxylic acid derivative to give the corresponding amide I.
- the ester and ether are cleaved with boron tribromide and the resulting acid is coupled with an amine derivative to give compounds of formula (I- V).
- Acid D is treated with diphenylphosphoryl azide, triethyl amine and r-butanol to afford intermediate J.
- the boc-group is removed by treatment with hydrogen chloride to afford the amine K.
- Amine K is treated with the requisite acid in the presence of HATU to afford amide L.
- Compound L is reacted with boron tribromide to afford the phenol M.
- Compound M is treated with hydrogen in the presence of palladium to afford compound N (Scheme IV).
- Acid O is coupled with the requisite amine to afford amide P.
- Compound P is reduced under standard hydrogenation conditions to afford aniline Q.
- the aniline is reacted with the requisite Page: 25/97 acid chloride to afford intermediate R.
- a final deprotection using boron tribromide affords compound S.
- a salt, hydrate, solvate and isomer of the inventive compound of formula (I) may be prepared by employing any of the known methods.
- the inventive compound of formula (I), a salt, hydrate, solvate or isomer thereof may be used for the treatment of GSK3beta dependent diseases such as Alzheimer disease, mania, depression, migraine and type 2 diabetes, by way of inhibiting GSK3beta activity, the inventive compound having an IC 50 value (micro M), generally in the range of 0.0001 to 100, for example 0.001 to 50, preferably 0.001 to 10, more preferably 0.001 to 5.
- the present invention includes a pharmaceutical composition which includes a therapeutically effective amount of the compound of formula (I), a salt, hydrate, solvate or isomer thereof as an active ingredient and a pharmaceutically acceptable carrier; therefore, the pharmaceutical composition of the present invention exerts superior preventive and treating effects on GSKbeta dependent diseases.
- a pharmaceutical formulation may be prepared in accordance with any of the conventional procedures.
- the active ingredient is preferably admixed or diluted with a carrier, or enclosed within a carrier, sachet or other container.
- the carrier serves as a diluent, it may be a solid, semi-solid or liquid material acting as a vehicle, excipient or medium for the active ingredient.
- the formulations may be in the form of a tablet, pill, powder, sachet, elixir, suspension, emulsion, solution, syrup, aerosol, soft and hard gelatin capsule, sterile injectable solution, sterile packaged powder and the like.
- Suitable carriers, excipients, and diluents are lactose, dextrose, sucrose, sorbitol, mannitol, calcium silicate, cellulose, methyl cellulose, microcrystalline cellulose, polyvinylpyrrolidone, water, and mineral oil.
- the formulations may additionally include fillers, antiemulsifiers, preservatives and the like.
- the compositions of the invention may be formulated so as to provide quick, sustained or delayed release of the active ingredient after their administration to a mammal by employing any of the procedures well known in the art.
- the pharmaceutical composition of the present invention can be administered via various routes including oral, transdermal, subcutaneous, intravenous and intramuscular introduction.
- the dosage and method of administration vary according to the body- weight and age of a patient and the administration method; however, one skilled in the art can routinely select a suitable method of administration. If the compound is encodable by a DNA, the DNA can be inserted into a vector for gene therapy and the vector administered to a patient to perform the therapy.
- the dosage and method of administration vary according to the body- weight, age, and symptoms of the patient; however, one skilled in the art can suitably select them.
- the dose of a compound of the present invention that regulates its activity depends on the symptoms, the dose is generally about 0.1 mg to about 100 mg per day, preferably about 1.0 mg to about 50 mg per day and more preferably about 1.0 mg to about 20 mg per day, when administered orally to a normal adult human (weight 60 kg).
- the dose is generally about 0.1 mg to about 100 mg per day, preferably about 1.0 mg to about 50 mg per day and more preferably about 1.0 mg to about 20 mg per day, when administered orally to a normal adult human (weight 60 kg).
- the appropriate dosage amount may be routinely calculated by converting to 60 kg of body-weight.
- p-Toluenesulfonic acid monohydrate (42 g, 110 mmol) was heated at 120 degrees and once the solid completely melted, it was placed under high vacuum for 1 h to remove the water. The vacuum was released, aniline (20 g, 55 mmol) and 2-thiophenecarbonitrile (24 g, 110 mmol) were added, and the reaction mixture was heated at 160 degrees for 4 h. The reaction mixture was cooled to room temperature followed by addition of satd. aq NaHCO 3 (250 mL) and ethyl acetate (250 mL).
- the combined organic layer was dried OVCr Na 2 SO 4 , concentrated, and purified by preparative HPLC (C 18 silica, 10-90% acetonitrile/water with 0.05% TFA).
- the desired product was obtained as the trifluoroacetic acid salt which was eluted through an ion-exchange column (using methanol and 7 N methanol in ammonia) to obtain the desired products.
- the desired product was treated with TFA (1-2 mL) for 1 h, concentrated and purified by preparative HPLC (Cl 8 silica, 10-90% acetonitrile/water with 0.05% TFA).
- the desired product was obtained as the trifluoroacetic acid salt which was eluted through an ion-exchange column (using methanol and 7 N methanol in ammonia) to obtain the desired products
- N-(2-(5-aminopyridin-2-ylamino)ethyl)-7-hydroxy-2-(thiophen-2-yl)-lH-benzo[d]imidazole-4- carboxamide hydrochloride (0.29 g, 0.68 mmol) in DMF (5 mL) was added DIPEA (0.44 g, 3.4 mmol) and methanesulfonyl chloride (0.085 g, 0.75 mmol) and the reaction mixture was stirred for 16 h at room temperature.
- reaction mixture was stirred at room temperature for 16 h, quenched with satd. aq NaCl (50 mL), and extracted with ethyl acetate (2x50 mL). The combined organic layers were washed with satd.
- NHTs means p-toluenesulfonamido.
- the filtrate was concentrated under reduced pressure and the crude residue was triturated in methanol and filtered.
- the filtrate was concentrated and purified by flash chromatography (silica, 0-20% methanol/dichloromethane) to afford crude product.
- the crude product was purified by preparative HPLC (C 18 silica, 10-90% acetonitrile/water with 0.05% TFA).
- Example 106 1 -(2-Cyclopropyl-7-methoxy- 1 H-benzo[d]imidazol-4-yl)-3-(4-methoxyphenyl)urea
- the resulting solids were filtered and dried to obtain the crude acid (1.0 g) as an off-white solid.
- the crude acid intermediate (0.5 g) was dissolved in DMF (5 mL) followed by the addition of HATU (0.84 g, 2.2 mmol), DIPEA (1.2 mL, 6.6 mmol), histamine (0.50 g, 4.5 mmol) and the reaction mixture was stirred at room temperature for 18 h.
- the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (3x30 mL). The combined organic layers were dried over Na 2 SO 4 , concentrated, and the residue was triturated with CH 2 Cl 2 (20 mL).
- the crude acid was dissolved in DMF (5 mL) followed by the addition of HATU (0.15 g, 0.46 mmol), DIPEA (0.20 mL, 1.1 mmol), and histamine (0.051 g, 0.46 mmol) and the reaction mixture was heated at 80 degrees for 18 h.
- the reaction mixture was cooled to room temperature, diluted with water (20 mL), and extracted with EtOAc (3x30 mL). The combined organic layers were dried over Na 2 SO 4 , concentrated, and the residue was purified by preparative HPLC (Cl 8 silica, 10-90% acetonitrile/water with 0.05% TFA).
- GSK3beta activity was measured in the presence or absence of compounds using Z'-LYTE kinase assay (Rodems SM, et al., Assay Drug Dev Technol. 1 : 9-19, 2002.) kit with SER/THR 9 peptide (Invitrogen) following the manufacturer's instruction.
- the Z'-LYTE kinase assay kit Page: 85/97 employs a fluorescence resonance energy transfer (FRET) between two fluorophores, coumarin and fluorescein, attached to each end of a substrate peptide.
- FRET fluorescence resonance energy transfer
- Test compounds were dissolved in DMSO at 12.5 mM and then serially diluted as the DMSO concentration in the assays to be 1%.
- the serially diluted compounds, 0.04 ng/mcl GSKbeta (Invitrogen) and 2 mcM SER/THR 9 peptide were reacted in a reaction buffer (50 mM HEPES pH 7.5, 0.01% Brij-35, 10 mM MgCl 2 , 1 mM EGTA, 15 mcM ATP).
- a reaction buffer 50 mM HEPES pH 7.5, 0.01% Brij-35, 10 mM MgCl 2 , 1 mM EGTA, 15 mcM ATP.
- ATP was omitted from the reaction mixture.
- SER/THR 9 phosphopeptide was used in place of the SER/THR 9 peptide.
- % phosphorylation 1 - (Co% - Cioo%) + [emission ratio x (Fioo% - Fo%)]
- coumarin emission signal (445nm) emission ratio fluorescein emission signal (520nm)
- Cioo% coumarin emission signal of the 100% phosphorylation control
- Co% coumarin emission signal of the 0% phosphorylation control
- Fioo% fluorescein emission signal of the 100% phosphorylation control
- Fo% fluorescein emission signal of the 0% phosphorylation control
- the present invention provides a novel benzoimidazole compound having GSK3beta inhibitory effect.
- the compounds of the present invention may be used for pharmaceutical composition for inhibiting GSK3-beta.
- Such pharmaceutical compositions are suitable for treating or preventing diseases involving GSK3beta.
Abstract
Description



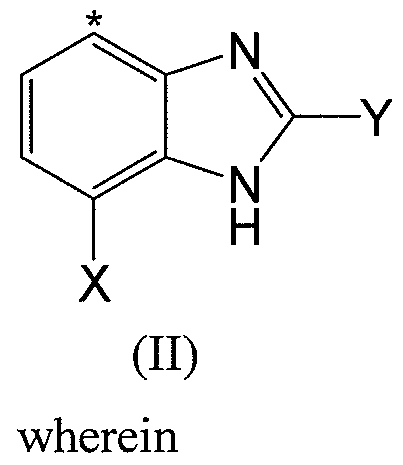










































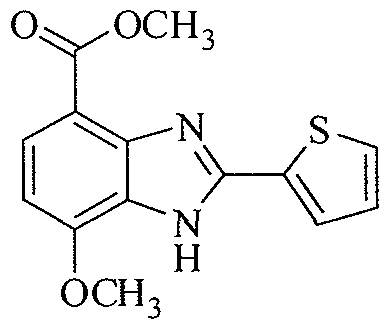

































































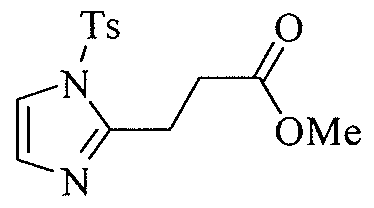

































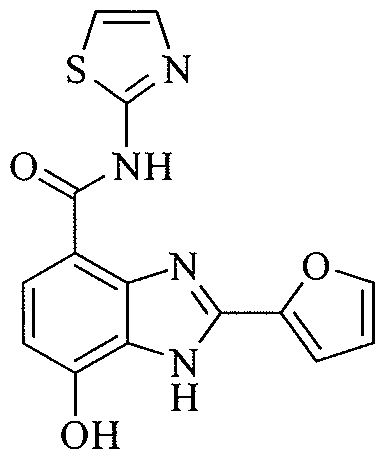








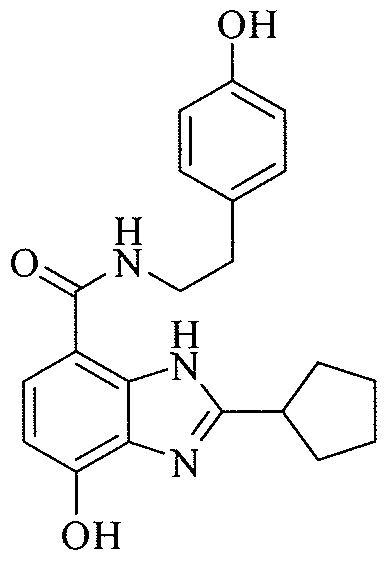






















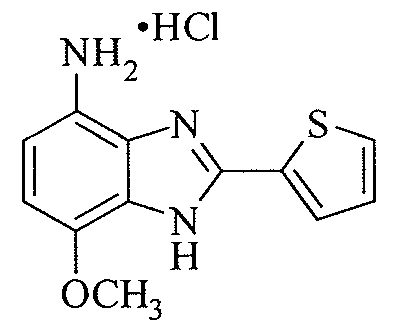










Claims






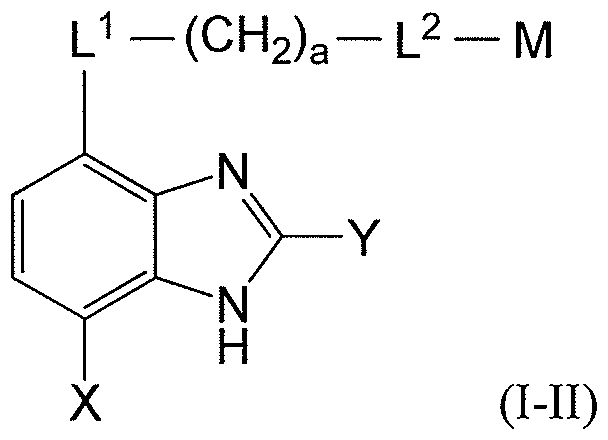






Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2009276548A AU2009276548A1 (en) | 2008-07-30 | 2009-07-30 | Benzoimidazole derivatives and glycogen synthase kinase-3 beta inhibitors containing the same |
| BRPI0916726A BRPI0916726A2 (en) | 2008-07-30 | 2009-07-30 | benzoimidazole derivatives and glycogen synthase kinase-3 beta inhibitors containing the same |
| EP09803582A EP2309855A4 (en) | 2008-07-30 | 2009-07-30 | Benzoimidazole derivatives and glycogen synthase kinase-3 beta inhibitors containing the same |
| CA2732280A CA2732280A1 (en) | 2008-07-30 | 2009-07-30 | Benzoimidazole derivatives and glycogen synthase kinase-3 beta inhibitors containing the same |
| MX2011001170A MX2011001170A (en) | 2008-07-30 | 2009-07-30 | Benzoimidazole derivatives and glycogen synthase kinase-3 beta inhibitors containing the same. |
| CN2009801387112A CN102170785A (en) | 2008-07-30 | 2009-07-30 | Benzoimidazole derivatives and glycogen synthase kinase-3 beta inhibitors containing the same |
| US13/056,591 US20110190351A1 (en) | 2008-07-30 | 2009-07-30 | Benzoimidazole Derivatives and Glycogen Synthase Kinase-3 Beta Inhibitors Containing the Same |
| JP2011521313A JP2011529903A (en) | 2008-07-30 | 2009-07-30 | Benzimidazole derivatives and glycogen synthase kinase 3β inhibitors containing the same |
| IL210863A IL210863A0 (en) | 2008-07-30 | 2011-01-25 | Benzoimidazole derivatives and glycogen synthase kinase-3 beta inhibitors containing the same |
| ZA2011/01160A ZA201101160B (en) | 2008-07-30 | 2011-02-14 | Benzoimidazole derivatives and glycogen synthase kinse-3 beta inhibitors containing the same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US8477008P | 2008-07-30 | 2008-07-30 | |
| US61/084,770 | 2008-07-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010014794A1 true WO2010014794A1 (en) | 2010-02-04 |
Family
ID=41610727
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/052225 WO2010014794A1 (en) | 2008-07-30 | 2009-07-30 | Benzoimidazole derivatives and glycogen synthase kinase-3 beta inhibitors containing the same |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US20110190351A1 (en) |
| EP (1) | EP2309855A4 (en) |
| JP (1) | JP2011529903A (en) |
| KR (1) | KR20110040958A (en) |
| CN (1) | CN102170785A (en) |
| AU (1) | AU2009276548A1 (en) |
| BR (1) | BRPI0916726A2 (en) |
| CA (1) | CA2732280A1 (en) |
| CO (1) | CO6351688A2 (en) |
| IL (1) | IL210863A0 (en) |
| MX (1) | MX2011001170A (en) |
| RU (1) | RU2011107227A (en) |
| WO (1) | WO2010014794A1 (en) |
| ZA (1) | ZA201101160B (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| US8362268B2 (en) | 2008-05-30 | 2013-01-29 | University Of Notre Dame Du Lac | Anti-bacterial agents from benzo[d]heterocyclic scaffolds for prevention and treatment of multidrug resistant bacteria |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2016133160A1 (en) * | 2015-02-19 | 2016-08-25 | 国立大学法人筑波大学 | Sulfonamide derivative or pharmaceutically acceptable acid addition salt thereof |
| WO2022079290A3 (en) * | 2020-10-16 | 2022-06-16 | Cemm - Forschungszentrum Für Molekulare Medizin Gmbh | Heterocyclic cullin ring ubiquitin ligase compounds and uses thereof |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101619058A (en) * | 2009-01-08 | 2010-01-06 | 上海交通大学 | Benzimidazole-4-acid amide type derivant |
| WO2015088000A1 (en) * | 2013-12-12 | 2015-06-18 | 国立大学法人筑波大学 | Sulfonamide derivative or pharmaceutically acceptable acid addition salt thereof |
| CN105461722B (en) * | 2014-09-04 | 2019-05-10 | 欣凯医药化工中间体(上海)有限公司 | A kind of deazapurine class compound and its derivative and its preparation method and application |
| WO2024014851A1 (en) * | 2022-07-12 | 2024-01-18 | 주식회사 넥스트젠바이오사이언스 | Novel purine derivative compounds as hif-1 protein inhibitor |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7064215B2 (en) * | 2001-07-03 | 2006-06-20 | Chiron Corporation | Indazole benzimidazole compounds |
| US7179832B2 (en) * | 2003-01-23 | 2007-02-20 | Crystalgenomics, Inc. | Glycogen synthase kinase 3β inhibitor, composition and process for the preparation thereof |
| US20070270420A1 (en) * | 2002-02-06 | 2007-11-22 | Harbeson Scott L | Heteroaryl compounds useful as inhibitors of gsk-3 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0178413A1 (en) * | 1984-08-17 | 1986-04-23 | Beecham Group Plc | Benzimidazoles |
| FR2699816B1 (en) * | 1992-12-30 | 1995-03-03 | Oreal | Dyeing compositions for keratin fibers based on paraphenylenediamines, metaphenylenediamines and benzimidazole derivatives, and dyeing process using them. |
| MX2011004414A (en) * | 2008-10-30 | 2011-06-21 | Oncotherapy Science Inc | 7-hydroxy-benzoimidazole-4-yl-methanone derivatives and pbk inhibitors containing the same. |
| CN102292083A (en) * | 2008-11-20 | 2011-12-21 | 肿瘤疗法科学股份有限公司 | Glycogen synthase kinase-3 beta inhibitors containing 7-hydroxy-benzoimidazole-4-yl-methanone derivatives |
-
2009
- 2009-07-30 MX MX2011001170A patent/MX2011001170A/en unknown
- 2009-07-30 EP EP09803582A patent/EP2309855A4/en active Pending
- 2009-07-30 CA CA2732280A patent/CA2732280A1/en not_active Abandoned
- 2009-07-30 BR BRPI0916726A patent/BRPI0916726A2/en not_active IP Right Cessation
- 2009-07-30 AU AU2009276548A patent/AU2009276548A1/en not_active Withdrawn
- 2009-07-30 KR KR1020117004602A patent/KR20110040958A/en not_active Application Discontinuation
- 2009-07-30 RU RU2011107227/13A patent/RU2011107227A/en not_active Application Discontinuation
- 2009-07-30 US US13/056,591 patent/US20110190351A1/en not_active Abandoned
- 2009-07-30 WO PCT/US2009/052225 patent/WO2010014794A1/en active Application Filing
- 2009-07-30 JP JP2011521313A patent/JP2011529903A/en not_active Withdrawn
- 2009-07-30 CN CN2009801387112A patent/CN102170785A/en not_active Withdrawn
-
2011
- 2011-01-25 IL IL210863A patent/IL210863A0/en unknown
- 2011-02-08 CO CO11014386A patent/CO6351688A2/en not_active Application Discontinuation
- 2011-02-14 ZA ZA2011/01160A patent/ZA201101160B/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7064215B2 (en) * | 2001-07-03 | 2006-06-20 | Chiron Corporation | Indazole benzimidazole compounds |
| US20070270420A1 (en) * | 2002-02-06 | 2007-11-22 | Harbeson Scott L | Heteroaryl compounds useful as inhibitors of gsk-3 |
| US7179832B2 (en) * | 2003-01-23 | 2007-02-20 | Crystalgenomics, Inc. | Glycogen synthase kinase 3β inhibitor, composition and process for the preparation thereof |
Non-Patent Citations (2)
| Title |
|---|
| MEIJER ET AL.: "Pharmacological inhibitors of glycogen synthase kinase 3", TRENDS IN PHARMACOLOGICAL SCIENCES, vol. 25, September 2004 (2004-09-01), pages 471 - 480, XP004552644 * |
| See also references of EP2309855A4 * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8362268B2 (en) | 2008-05-30 | 2013-01-29 | University Of Notre Dame Du Lac | Anti-bacterial agents from benzo[d]heterocyclic scaffolds for prevention and treatment of multidrug resistant bacteria |
| US9617249B2 (en) | 2008-05-30 | 2017-04-11 | University Of Notre Dame Du Lac | Benzoheterocyclic anti-bacterial agents |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2016133160A1 (en) * | 2015-02-19 | 2016-08-25 | 国立大学法人筑波大学 | Sulfonamide derivative or pharmaceutically acceptable acid addition salt thereof |
| WO2022079290A3 (en) * | 2020-10-16 | 2022-06-16 | Cemm - Forschungszentrum Für Molekulare Medizin Gmbh | Heterocyclic cullin ring ubiquitin ligase compounds and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2011107227A (en) | 2012-09-10 |
| IL210863A0 (en) | 2011-04-28 |
| CO6351688A2 (en) | 2011-12-20 |
| EP2309855A4 (en) | 2012-06-27 |
| ZA201101160B (en) | 2011-10-26 |
| US20110190351A1 (en) | 2011-08-04 |
| BRPI0916726A2 (en) | 2017-07-04 |
| CN102170785A (en) | 2011-08-31 |
| MX2011001170A (en) | 2011-04-05 |
| EP2309855A1 (en) | 2011-04-20 |
| KR20110040958A (en) | 2011-04-20 |
| AU2009276548A1 (en) | 2010-02-04 |
| JP2011529903A (en) | 2011-12-15 |
| CA2732280A1 (en) | 2010-02-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2010014794A1 (en) | Benzoimidazole derivatives and glycogen synthase kinase-3 beta inhibitors containing the same | |
| US9622991B2 (en) | Hematopoietic growth factor mimetic small molecule compounds and their uses | |
| JP5878859B2 (en) | Heterocyclic derivatives as histone deacetylase (HDAC) inhibitors | |
| AU781506B2 (en) | Integrin expression inhibitors | |
| CA2415010C (en) | Propane-1,3-dione derivatives useful as gnrh receptor antagonist | |
| ES2339107T3 (en) | ESPIRO HYDANTOINE COMPOUNDS USEFUL AS ANTI-INFLAMMATORY AGENTS. | |
| AU2007248341B2 (en) | Benzimidazole modulators of VR1 | |
| US20140243324A1 (en) | Use of hematopoietic growth factor mimetics | |
| JP2011529903A5 (en) | ||
| CA2525628C (en) | New benzimidazole derivatives | |
| CZ20021292A3 (en) | Derivatives of five-membered heterocycles, their preparation processes, and use as medicaments | |
| WO2007054480A1 (en) | 2-(benzimidazol-1-yl)-acetamide biaryl derivatives and their use as inhibitors of the trpv1 receptor | |
| KR20120091176A (en) | New histone deacetylase inhibitors based simultaneously on trisubstituted 1h-pyrroles and aromatic and heteroaromatic spacers | |
| JP2005501856A5 (en) | ||
| CA2577818A1 (en) | New heterocyclic amides | |
| AU734322B2 (en) | New isoquinoline derivatives | |
| JP2010533725A (en) | Aminoalkylazole compounds as histamine-3 (H3) receptors | |
| US20230000821A1 (en) | Indole carboxamide derivative and pharmaceutical composition containing same | |
| JP2009509931A (en) | Amino acid derivatives as histone deacetylase (HDAC) inhibitors | |
| JP2012509249A (en) | Glycogen synthase kinase-3 beta inhibitor comprising 7-hydroxy-benzimidazol-4-yl-methanone derivative | |
| AU2006311042A1 (en) | 2-(benzimidazol-1-yl)-acetamide biaryl derivatives and their use as inhibitors of the TRPV1 receptor | |
| BE1021253B1 (en) | NOVEL 3-INDOL SUBSTITUTED DERIVATIVES, PHARMACEUTICAL COMPOSITIONS AND METHOD OF USE |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980138711.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09803582 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011010162 Country of ref document: EG Ref document number: 12011500196 Country of ref document: PH |
|
| ENP | Entry into the national phase |
Ref document number: 2732280 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2011521313 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2011/001170 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009276548 Country of ref document: AU |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 590879 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11014386 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009803582 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1243/CHENP/2011 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2009276548 Country of ref document: AU Date of ref document: 20090730 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20117004602 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011107227 Country of ref document: RU Ref document number: A201102096 Country of ref document: UA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13056591 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0916726 Country of ref document: BR Kind code of ref document: A2 Effective date: 20110131 |