WO2010004343A1 - Piperidinyl gpcr agonists - Google Patents
Piperidinyl gpcr agonists Download PDFInfo
- Publication number
- WO2010004343A1 WO2010004343A1 PCT/GB2009/050825 GB2009050825W WO2010004343A1 WO 2010004343 A1 WO2010004343 A1 WO 2010004343A1 GB 2009050825 W GB2009050825 W GB 2009050825W WO 2010004343 A1 WO2010004343 A1 WO 2010004343A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pharmaceutically acceptable
- acceptable salt
- compound according
- preparation
- mmol
- Prior art date
Links
- 125000003386 piperidinyl group Chemical group 0.000 title claims 2
- 229940125633 GPCR agonist Drugs 0.000 title abstract description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 235
- 150000003839 salts Chemical class 0.000 claims abstract description 65
- 238000011282 treatment Methods 0.000 claims abstract description 41
- 208000008589 Obesity Diseases 0.000 claims abstract description 20
- 235000020824 obesity Nutrition 0.000 claims abstract description 20
- 206010012601 diabetes mellitus Diseases 0.000 claims abstract description 14
- 238000000034 method Methods 0.000 claims description 143
- 125000000217 alkyl group Chemical group 0.000 claims description 27
- -1 3-azetidinyl Chemical group 0.000 claims description 25
- 239000001257 hydrogen Substances 0.000 claims description 22
- 229910052739 hydrogen Inorganic materials 0.000 claims description 22
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 claims description 20
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 19
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 16
- 239000008194 pharmaceutical composition Substances 0.000 claims description 15
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 14
- 201000010099 disease Diseases 0.000 claims description 13
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 9
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 8
- 239000003937 drug carrier Substances 0.000 claims description 8
- 125000003709 fluoroalkyl group Chemical group 0.000 claims description 8
- 208000001145 Metabolic Syndrome Diseases 0.000 claims description 7
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 claims description 7
- 239000003814 drug Substances 0.000 claims description 7
- 125000001153 fluoro group Chemical group F* 0.000 claims description 7
- 206010020772 Hypertension Diseases 0.000 claims description 6
- 125000000623 heterocyclic group Chemical group 0.000 claims description 6
- 238000004519 manufacturing process Methods 0.000 claims description 6
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 5
- 208000002705 Glucose Intolerance Diseases 0.000 claims description 5
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 5
- 230000036186 satiety Effects 0.000 claims description 5
- 235000019627 satiety Nutrition 0.000 claims description 5
- 125000003545 alkoxy group Chemical group 0.000 claims description 4
- 201000009104 prediabetes syndrome Diseases 0.000 claims description 4
- 208000011580 syndromic disease Diseases 0.000 claims description 4
- 208000035150 Hypercholesterolemia Diseases 0.000 claims description 3
- 208000031226 Hyperlipidaemia Diseases 0.000 claims description 3
- 125000002393 azetidinyl group Chemical group 0.000 claims description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 3
- 208000006575 hypertriglyceridemia Diseases 0.000 claims description 3
- 230000002265 prevention Effects 0.000 claims description 3
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 claims description 2
- 230000033228 biological regulation Effects 0.000 claims description 2
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 claims description 2
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 2
- VUCWMAJEUOWLEY-UHFFFAOYSA-N 1-$l^{1}-azanylpiperidine Chemical compound [N]N1CCCCC1 VUCWMAJEUOWLEY-UHFFFAOYSA-N 0.000 claims 2
- 238000002360 preparation method Methods 0.000 description 142
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 89
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 81
- 239000000243 solution Substances 0.000 description 81
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 68
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 64
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 54
- 239000000203 mixture Substances 0.000 description 50
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 45
- 239000011541 reaction mixture Substances 0.000 description 45
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 41
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 40
- 239000002904 solvent Substances 0.000 description 35
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 34
- 239000002253 acid Substances 0.000 description 31
- 239000012267 brine Substances 0.000 description 30
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 30
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 29
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 26
- 238000006243 chemical reaction Methods 0.000 description 26
- 238000004440 column chromatography Methods 0.000 description 23
- 238000000746 purification Methods 0.000 description 22
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 21
- 210000004027 cell Anatomy 0.000 description 19
- 239000000284 extract Substances 0.000 description 18
- 238000001914 filtration Methods 0.000 description 18
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 17
- 230000002829 reductive effect Effects 0.000 description 17
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 16
- 239000000883 anti-obesity agent Substances 0.000 description 14
- 239000000556 agonist Substances 0.000 description 13
- 229940125710 antiobesity agent Drugs 0.000 description 13
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 13
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 239000007788 liquid Substances 0.000 description 11
- 239000012074 organic phase Substances 0.000 description 11
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 11
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 10
- 102000004877 Insulin Human genes 0.000 description 10
- 108090001061 Insulin Proteins 0.000 description 10
- IVOMOUWHDPKRLL-UHFFFAOYSA-N UNPD107823 Natural products O1C2COP(O)(=O)OC2C(O)C1N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-UHFFFAOYSA-N 0.000 description 10
- 210000004369 blood Anatomy 0.000 description 10
- 239000008280 blood Substances 0.000 description 10
- 229940095074 cyclic amp Drugs 0.000 description 10
- 229940125396 insulin Drugs 0.000 description 10
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 10
- 239000007787 solid Substances 0.000 description 10
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 10
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 9
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 9
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 9
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 9
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 9
- 239000004480 active ingredient Substances 0.000 description 9
- 238000003556 assay Methods 0.000 description 9
- 125000004494 ethyl ester group Chemical group 0.000 description 9
- 239000003112 inhibitor Substances 0.000 description 9
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 8
- 150000001412 amines Chemical group 0.000 description 8
- 229910052786 argon Inorganic materials 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 8
- 102000005962 receptors Human genes 0.000 description 8
- 108020003175 receptors Proteins 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- 210000005253 yeast cell Anatomy 0.000 description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 7
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 7
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 7
- 241000700159 Rattus Species 0.000 description 7
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 7
- 150000001408 amides Chemical class 0.000 description 7
- 239000008346 aqueous phase Substances 0.000 description 7
- JXHYCCGOZUGBFD-UHFFFAOYSA-N benzoic acid;hydrochloride Chemical compound Cl.OC(=O)C1=CC=CC=C1 JXHYCCGOZUGBFD-UHFFFAOYSA-N 0.000 description 7
- 239000000969 carrier Substances 0.000 description 7
- 239000008103 glucose Substances 0.000 description 7
- 238000005984 hydrogenation reaction Methods 0.000 description 7
- 239000012044 organic layer Substances 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- 125000006239 protecting group Chemical group 0.000 description 7
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- 239000005557 antagonist Substances 0.000 description 6
- 230000037396 body weight Effects 0.000 description 6
- 239000000872 buffer Substances 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- FCZCIXQGZOUIDN-UHFFFAOYSA-N ethyl 2-diethoxyphosphinothioyloxyacetate Chemical compound CCOC(=O)COP(=S)(OCC)OCC FCZCIXQGZOUIDN-UHFFFAOYSA-N 0.000 description 6
- 230000003914 insulin secretion Effects 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- 238000005259 measurement Methods 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 238000000935 solvent evaporation Methods 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 5
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 5
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 5
- 229910002092 carbon dioxide Inorganic materials 0.000 description 5
- 235000013305 food Nutrition 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 150000002431 hydrogen Chemical group 0.000 description 5
- 239000004615 ingredient Substances 0.000 description 5
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium;hydroxide;hydrate Chemical compound [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 238000004007 reversed phase HPLC Methods 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- FQFILJKFZCVHNH-UHFFFAOYSA-N tert-butyl n-[3-[(5-bromo-2-chloropyrimidin-4-yl)amino]propyl]carbamate Chemical compound CC(C)(C)OC(=O)NCCCNC1=NC(Cl)=NC=C1Br FQFILJKFZCVHNH-UHFFFAOYSA-N 0.000 description 5
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 5
- CEJAHXLRNZJPQH-UHFFFAOYSA-N 2,5-dichloropyrimidine Chemical compound ClC1=CN=C(Cl)N=C1 CEJAHXLRNZJPQH-UHFFFAOYSA-N 0.000 description 4
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- 241000699666 Mus <mouse, genus> Species 0.000 description 4
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 4
- 210000000577 adipose tissue Anatomy 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 235000005911 diet Nutrition 0.000 description 4
- 230000037213 diet Effects 0.000 description 4
- WQOXQRCZOLPYPM-UHFFFAOYSA-N dimethyl disulfide Chemical compound CSSC WQOXQRCZOLPYPM-UHFFFAOYSA-N 0.000 description 4
- 238000006073 displacement reaction Methods 0.000 description 4
- 235000019441 ethanol Nutrition 0.000 description 4
- 239000003925 fat Substances 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- RXXUFTBOBMUSNM-UHFFFAOYSA-N n-(2-hydroxyethyl)-2-methyl-4-(3-piperidin-4-ylpropoxy)benzamide;hydrochloride Chemical compound Cl.C1=C(C(=O)NCCO)C(C)=CC(OCCCC2CCNCC2)=C1 RXXUFTBOBMUSNM-UHFFFAOYSA-N 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 231100000252 nontoxic Toxicity 0.000 description 4
- 230000003000 nontoxic effect Effects 0.000 description 4
- 230000037361 pathway Effects 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 4
- 229940044601 receptor agonist Drugs 0.000 description 4
- 239000000018 receptor agonist Substances 0.000 description 4
- 238000001953 recrystallisation Methods 0.000 description 4
- 238000012799 strong cation exchange Methods 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 238000000825 ultraviolet detection Methods 0.000 description 4
- RSJKGSCJYJTIGS-UHFFFAOYSA-N undecane Chemical compound CCCCCCCCCCC RSJKGSCJYJTIGS-UHFFFAOYSA-N 0.000 description 4
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 3
- ZTOBILYWTYHOJB-WBCGDKOGSA-N 3',6'-bis[[(2s,3r,4s,5r,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy]spiro[2-benzofuran-3,9'-xanthene]-1-one Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1OC1=CC=C2C3(C4=CC=CC=C4C(=O)O3)C3=CC=C(O[C@H]4[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O4)O)C=C3OC2=C1 ZTOBILYWTYHOJB-WBCGDKOGSA-N 0.000 description 3
- WMUWDPLTTLJNPE-UHFFFAOYSA-N 4-bromo-3,5-dimethylphenol Chemical compound CC1=CC(O)=CC(C)=C1Br WMUWDPLTTLJNPE-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 102100026189 Beta-galactosidase Human genes 0.000 description 3
- 206010065687 Bone loss Diseases 0.000 description 3
- 239000007821 HATU Substances 0.000 description 3
- 239000007995 HEPES buffer Substances 0.000 description 3
- 229910013596 LiOH—H2O Inorganic materials 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 108010005774 beta-Galactosidase Proteins 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 3
- 238000013262 cAMP assay Methods 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000006071 cream Substances 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- WGLUMOCWFMKWIL-UHFFFAOYSA-N dichloromethane;methanol Chemical compound OC.ClCCl WGLUMOCWFMKWIL-UHFFFAOYSA-N 0.000 description 3
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 125000001072 heteroaryl group Chemical group 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- XIXADJRWDQXREU-UHFFFAOYSA-M lithium acetate Chemical compound [Li+].CC([O-])=O XIXADJRWDQXREU-UHFFFAOYSA-M 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- JABYJIQOLGWMQW-UHFFFAOYSA-N undec-4-ene Chemical compound CCCCCCC=CCCC JABYJIQOLGWMQW-UHFFFAOYSA-N 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- UKSZBOKPHAQOMP-SVLSSHOZSA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 UKSZBOKPHAQOMP-SVLSSHOZSA-N 0.000 description 2
- ZGYIXVSQHOKQRZ-COIATFDQSA-N (e)-n-[4-[3-chloro-4-(pyridin-2-ylmethoxy)anilino]-3-cyano-7-[(3s)-oxolan-3-yl]oxyquinolin-6-yl]-4-(dimethylamino)but-2-enamide Chemical compound N#CC1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 ZGYIXVSQHOKQRZ-COIATFDQSA-N 0.000 description 2
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 2
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- XFAMUOYNXFXQTC-UHFFFAOYSA-N 2,5-difluoropyridine Chemical compound FC1=CC=C(F)N=C1 XFAMUOYNXFXQTC-UHFFFAOYSA-N 0.000 description 2
- ZFEMFPXEHOVVFA-UHFFFAOYSA-N 2,6-dimethyl-4-(3-piperidin-4-ylpropoxy)benzoic acid;hydrochloride Chemical compound Cl.CC1=C(C(O)=O)C(C)=CC(OCCCC2CCNCC2)=C1 ZFEMFPXEHOVVFA-UHFFFAOYSA-N 0.000 description 2
- KBUNTTPQADGDOL-UHFFFAOYSA-N 2,6-dimethyl-4-[3-[1-(5-propan-2-ylpyridin-2-yl)piperidin-4-yl]propoxy]benzoic acid Chemical compound N1=CC(C(C)C)=CC=C1N1CCC(CCCOC=2C=C(C)C(C(O)=O)=C(C)C=2)CC1 KBUNTTPQADGDOL-UHFFFAOYSA-N 0.000 description 2
- BGLLZQRUXJGTAD-UHFFFAOYSA-N 2-chloro-5-ethylpyrimidine Chemical compound CCC1=CN=C(Cl)N=C1 BGLLZQRUXJGTAD-UHFFFAOYSA-N 0.000 description 2
- JREOGLNRKHWRQK-UHFFFAOYSA-N 2-methyl-4-[3-[1-[(2-methylpropan-2-yl)oxycarbonyl]piperidin-4-yl]propoxy]benzoic acid Chemical compound C1=C(C(O)=O)C(C)=CC(OCCCC2CCN(CC2)C(=O)OC(C)(C)C)=C1 JREOGLNRKHWRQK-UHFFFAOYSA-N 0.000 description 2
- JNOGUUKHGBYFKP-UHFFFAOYSA-N 2-methyl-6-[3-[1-[(2-methylpropan-2-yl)oxycarbonyl]piperidin-4-yl]propoxy]pyridine-3-carboxylic acid Chemical compound C1=C(C(O)=O)C(C)=NC(OCCCC2CCN(CC2)C(=O)OC(C)(C)C)=C1 JNOGUUKHGBYFKP-UHFFFAOYSA-N 0.000 description 2
- XKTYXVDYIKIYJP-UHFFFAOYSA-N 3h-dioxole Chemical compound C1OOC=C1 XKTYXVDYIKIYJP-UHFFFAOYSA-N 0.000 description 2
- SXCRMBVRFRJOBL-UHFFFAOYSA-N 4-[3-[1-(5-ethylpyridin-2-yl)piperidin-4-yl]propoxy]-2-methylbenzoic acid Chemical compound N1=CC(CC)=CC=C1N1CCC(CCCOC=2C=C(C)C(C(O)=O)=CC=2)CC1 SXCRMBVRFRJOBL-UHFFFAOYSA-N 0.000 description 2
- KECCFSZFXLAGJS-UHFFFAOYSA-N 4-methylsulfonylphenol Chemical compound CS(=O)(=O)C1=CC=C(O)C=C1 KECCFSZFXLAGJS-UHFFFAOYSA-N 0.000 description 2
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 229920000858 Cyclodextrin Polymers 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- 208000032928 Dyslipidaemia Diseases 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 102000016267 Leptin Human genes 0.000 description 2
- 108010092277 Leptin Proteins 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N Methyl benzoate Natural products COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 2
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 108050006759 Pancreatic lipases Proteins 0.000 description 2
- 102000019280 Pancreatic lipases Human genes 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- 239000004743 Polypropylene Substances 0.000 description 2
- 239000004793 Polystyrene Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 101001032756 Rattus norvegicus Granzyme-like protein 1 Proteins 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 230000003187 abdominal effect Effects 0.000 description 2
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 238000010976 amide bond formation reaction Methods 0.000 description 2
- 230000003579 anti-obesity Effects 0.000 description 2
- 229940125708 antidiabetic agent Drugs 0.000 description 2
- 239000003472 antidiabetic agent Substances 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- 150000001559 benzoic acids Chemical class 0.000 description 2
- 229910000085 borane Inorganic materials 0.000 description 2
- 235000019577 caloric intake Nutrition 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000004296 chiral HPLC Methods 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- WBKFWQBXFREOFH-UHFFFAOYSA-N dichloromethane;ethyl acetate Chemical compound ClCCl.CCOC(C)=O WBKFWQBXFREOFH-UHFFFAOYSA-N 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 230000003291 dopaminomimetic effect Effects 0.000 description 2
- 238000000132 electrospray ionisation Methods 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- UEZSEPUDNLUVNJ-UHFFFAOYSA-N ethyl 2-methyl-6-oxo-1h-pyridine-3-carboxylate Chemical compound CCOC(=O)C=1C=CC(=O)NC=1C UEZSEPUDNLUVNJ-UHFFFAOYSA-N 0.000 description 2
- BGSUEZSKWUKYHT-UHFFFAOYSA-N ethyl 6-[3-[1-(5-chloropyrimidin-2-yl)piperidin-4-yl]propoxy]-2-methylpyridine-3-carboxylate Chemical compound N1=C(C)C(C(=O)OCC)=CC=C1OCCCC1CCN(C=2N=CC(Cl)=CN=2)CC1 BGSUEZSKWUKYHT-UHFFFAOYSA-N 0.000 description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N ethyl acetate Substances CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 235000019197 fats Nutrition 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 229940039781 leptin Drugs 0.000 description 2
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- FINKSGWSBJRISB-UHFFFAOYSA-N methyl 4-methoxy-2-methylbenzoate Natural products COC(=O)C1=CC=C(O)C=C1C FINKSGWSBJRISB-UHFFFAOYSA-N 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 2
- SJYNFBVQFBRSIB-UHFFFAOYSA-N norbornadiene Chemical compound C1=CC2C=CC1C2 SJYNFBVQFBRSIB-UHFFFAOYSA-N 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 229940116369 pancreatic lipase Drugs 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- 229920002223 polystyrene Polymers 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 230000000171 quenching effect Effects 0.000 description 2
- JZCPYUJPEARBJL-UHFFFAOYSA-N rimonabant Chemical compound CC=1C(C(=O)NN2CCCCC2)=NN(C=2C(=CC(Cl)=CC=2)Cl)C=1C1=CC=C(Cl)C=C1 JZCPYUJPEARBJL-UHFFFAOYSA-N 0.000 description 2
- 229960003015 rimonabant Drugs 0.000 description 2
- 230000000862 serotonergic effect Effects 0.000 description 2
- 239000012265 solid product Substances 0.000 description 2
- 238000013222 sprague-dawley male rat Methods 0.000 description 2
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- WWSVGVQAKIRKTK-OAHLLOKOSA-N tert-butyl 4-[(2r)-4-(4-bromo-3,5-dimethylphenoxy)butan-2-yl]piperidine-1-carboxylate Chemical compound C([C@@H](C)C1CCN(CC1)C(=O)OC(C)(C)C)COC1=CC(C)=C(Br)C(C)=C1 WWSVGVQAKIRKTK-OAHLLOKOSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- BKMMTJMQCTUHRP-GSVOUGTGSA-N (2r)-2-aminopropan-1-ol Chemical compound C[C@@H](N)CO BKMMTJMQCTUHRP-GSVOUGTGSA-N 0.000 description 1
- ZVYWTYGEMCKQAA-SNVBAGLBSA-N (3r)-3-[1-[(2-methylpropan-2-yl)oxycarbonyl]piperidin-4-yl]butanoic acid Chemical compound OC(=O)C[C@@H](C)C1CCN(C(=O)OC(C)(C)C)CC1 ZVYWTYGEMCKQAA-SNVBAGLBSA-N 0.000 description 1
- AXJQVVLKUYCICH-OAQYLSRUSA-N (4s)-5-(4-chlorophenyl)-n-(4-chlorophenyl)sulfonyl-n'-methyl-4-phenyl-3,4-dihydropyrazole-2-carboximidamide Chemical compound C=1C=C(Cl)C=CC=1C([C@H](C1)C=2C=CC=CC=2)=NN1C(=NC)NS(=O)(=O)C1=CC=C(Cl)C=C1 AXJQVVLKUYCICH-OAQYLSRUSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- XHAFSILEIAKMSU-UHFFFAOYSA-N 2,6-dimethyl-4-[3-[1-[(2-methylpropan-2-yl)oxycarbonyl]piperidin-4-yl]propoxy]benzoic acid Chemical compound CC1=C(C(O)=O)C(C)=CC(OCCCC2CCN(CC2)C(=O)OC(C)(C)C)=C1 XHAFSILEIAKMSU-UHFFFAOYSA-N 0.000 description 1
- OYUNTGBISCIYPW-UHFFFAOYSA-N 2-chloroprop-2-enenitrile Chemical compound ClC(=C)C#N OYUNTGBISCIYPW-UHFFFAOYSA-N 0.000 description 1
- HJDJKAUQCRWFLC-UHFFFAOYSA-N 2-methyl-3-methylsulfonyl-6-(3-piperidin-4-ylpropoxy)pyridine;hydrochloride Chemical compound Cl.C1=C(S(C)(=O)=O)C(C)=NC(OCCCC2CCNCC2)=C1 HJDJKAUQCRWFLC-UHFFFAOYSA-N 0.000 description 1
- MJIGJEHKYMOQOT-UHFFFAOYSA-N 2-methyl-4-(3-piperidin-4-ylpropoxy)benzoic acid;hydrochloride Chemical compound Cl.C1=C(C(O)=O)C(C)=CC(OCCCC2CCNCC2)=C1 MJIGJEHKYMOQOT-UHFFFAOYSA-N 0.000 description 1
- ZPUCOTJTNSFQPD-UHFFFAOYSA-N 2-methyl-4-[3-[1-(5-propan-2-ylpyridin-2-yl)piperidin-4-yl]propoxy]benzoic acid Chemical compound N1=CC(C(C)C)=CC=C1N1CCC(CCCOC=2C=C(C)C(C(O)=O)=CC=2)CC1 ZPUCOTJTNSFQPD-UHFFFAOYSA-N 0.000 description 1
- MXKVFEDVBUNNFR-UHFFFAOYSA-N 2-methyl-6-(3-piperidin-4-ylpropoxy)pyridine-3-carboxylic acid;hydrochloride Chemical compound Cl.C1=C(C(O)=O)C(C)=NC(OCCCC2CCNCC2)=C1 MXKVFEDVBUNNFR-UHFFFAOYSA-N 0.000 description 1
- LTNUSYNQZJZUSY-UHFFFAOYSA-N 3,3-dimethylbutanal Chemical compound CC(C)(C)CC=O LTNUSYNQZJZUSY-UHFFFAOYSA-N 0.000 description 1
- GUSWJGOYDXFJSI-UHFFFAOYSA-N 3,6-dichloropyridazine Chemical compound ClC1=CC=C(Cl)N=N1 GUSWJGOYDXFJSI-UHFFFAOYSA-N 0.000 description 1
- ZLPSLHMURAMHQW-UHFFFAOYSA-N 3-[1-(5-chloropyrimidin-2-yl)piperidin-4-yl]propan-1-ol Chemical compound C1CC(CCCO)CCN1C1=NC=C(Cl)C=N1 ZLPSLHMURAMHQW-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- OENDTQFJHJVHQK-UHFFFAOYSA-N 3-piperidin-4-ylpropan-1-ol;hydrochloride Chemical compound Cl.OCCCC1CCNCC1 OENDTQFJHJVHQK-UHFFFAOYSA-N 0.000 description 1
- ROLMZTIHUMKEAI-UHFFFAOYSA-N 4,5-difluoro-2-hydroxybenzonitrile Chemical compound OC1=CC(F)=C(F)C=C1C#N ROLMZTIHUMKEAI-UHFFFAOYSA-N 0.000 description 1
- WFHPXSHLCFHEIA-UHFFFAOYSA-N 4,6,11-tris(2-methylpropyl)-1,4,6,11-tetraza-5-phosphabicyclo[3.3.3]undecane Chemical compound C1CN(CC(C)C)P2N(CC(C)C)CCN1CCN2CC(C)C WFHPXSHLCFHEIA-UHFFFAOYSA-N 0.000 description 1
- MQXUITMEZRMWSB-UHFFFAOYSA-N 4-(3-hydroxypropyl)piperidine-1-carboxylic acid Chemical compound OCCCC1CCN(C(O)=O)CC1 MQXUITMEZRMWSB-UHFFFAOYSA-N 0.000 description 1
- FXSRMDPUALJNGV-SECBINFHSA-N 4-[(2R)-4-methylsulfonyloxybutan-2-yl]piperidine-1-carboxylic acid Chemical compound CS(=O)(=O)OCC[C@@H](C)C1CCN(CC1)C(=O)O FXSRMDPUALJNGV-SECBINFHSA-N 0.000 description 1
- VAIIPBRDQQISIN-BTQNPOSSSA-N 4-[(2r)-4-(4-methylsulfonylphenoxy)butan-2-yl]piperidine;hydrochloride Chemical compound Cl.C([C@@H](C)C1CCNCC1)COC1=CC=C(S(C)(=O)=O)C=C1 VAIIPBRDQQISIN-BTQNPOSSSA-N 0.000 description 1
- LMCBBEPSOHBHPU-UHFFFAOYSA-N 4-[3-(4-carboxy-3,5-dimethylphenoxy)propyl]piperidine-1-carboxylic acid Chemical compound CC1=C(C(O)=O)C(C)=CC(OCCCC2CCN(CC2)C(O)=O)=C1 LMCBBEPSOHBHPU-UHFFFAOYSA-N 0.000 description 1
- IZDMEWABANLQMD-UHFFFAOYSA-N 4-[3-(4-methoxycarbonyl-3-methylphenoxy)propyl]piperidine-1-carboxylic acid Chemical compound C1=C(C)C(C(=O)OC)=CC=C1OCCCC1CCN(C(O)=O)CC1 IZDMEWABANLQMD-UHFFFAOYSA-N 0.000 description 1
- NWYWEUXZMIAOAB-UHFFFAOYSA-N 4-[3-[1-(5-chloropyrimidin-2-yl)piperidin-4-yl]propoxy]-n-(2-hydroxyethyl)-2-methylbenzamide Chemical compound C1=C(C(=O)NCCO)C(C)=CC(OCCCC2CCN(CC2)C=2N=CC(Cl)=CN=2)=C1 NWYWEUXZMIAOAB-UHFFFAOYSA-N 0.000 description 1
- ZULRQGBHWBQPFE-UHFFFAOYSA-N 5-chloro-2-fluoropyridine Chemical compound FC1=CC=C(Cl)C=N1 ZULRQGBHWBQPFE-UHFFFAOYSA-N 0.000 description 1
- WJRKNLONLOMALV-UHFFFAOYSA-N 5-chloropyridine Chemical compound ClC1=C=NC=C[CH]1 WJRKNLONLOMALV-UHFFFAOYSA-N 0.000 description 1
- LEEJCKGABMHXDD-UHFFFAOYSA-N 5-ethyl-2-fluoropyridine Chemical compound CCC1=CC=C(F)N=C1 LEEJCKGABMHXDD-UHFFFAOYSA-N 0.000 description 1
- MJTSPTRANGPNRJ-UHFFFAOYSA-N 5-ethylpyrimidine Chemical compound CCC1=CN=CN=C1 MJTSPTRANGPNRJ-UHFFFAOYSA-N 0.000 description 1
- ULUJLZZYBGEJHW-UHFFFAOYSA-N 5-fluoro-2-[4-[3-(6-methyl-5-methylsulfonylpyridin-2-yl)oxypropyl]piperidin-1-yl]pyrimidine Chemical compound C1=C(S(C)(=O)=O)C(C)=NC(OCCCC2CCN(CC2)C=2N=CC(F)=CN=2)=C1 ULUJLZZYBGEJHW-UHFFFAOYSA-N 0.000 description 1
- ODQJQEROUCVZNG-UHFFFAOYSA-N 5-fluoropyridine Chemical compound FC1=C=NC=C[CH]1 ODQJQEROUCVZNG-UHFFFAOYSA-N 0.000 description 1
- DMLWFRXQEOZFTC-UHFFFAOYSA-N 6-[3-[1-(5-chloropyrimidin-2-yl)piperidin-4-yl]propoxy]-2-methylpyridine-3-carboxylic acid Chemical compound C1=C(C(O)=O)C(C)=NC(OCCCC2CCN(CC2)C=2N=CC(Cl)=CN=2)=C1 DMLWFRXQEOZFTC-UHFFFAOYSA-N 0.000 description 1
- OQARSAOXPZPVJQ-UHFFFAOYSA-N 6-[3-[1-(5-fluoropyridin-2-yl)piperidin-4-yl]propoxy]-2-methyl-3-methylsulfonylpyridine Chemical group C1=C(S(C)(=O)=O)C(C)=NC(OCCCC2CCN(CC2)C=2N=CC(F)=CC=2)=C1 OQARSAOXPZPVJQ-UHFFFAOYSA-N 0.000 description 1
- IPAGEVKSCYHZQF-UHFFFAOYSA-N 6-fluoro-2-methyl-3-methylsulfonylpyridine Chemical compound CC1=NC(F)=CC=C1S(C)(=O)=O IPAGEVKSCYHZQF-UHFFFAOYSA-N 0.000 description 1
- 208000004611 Abdominal Obesity Diseases 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 229940077274 Alpha glucosidase inhibitor Drugs 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 238000003276 AlphaScreen cAMP assay kit Methods 0.000 description 1
- 208000002679 Alveolar Bone Loss Diseases 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 210000002237 B-cell of pancreatic islet Anatomy 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 229940123208 Biguanide Drugs 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 0 CC(C)(C)OC(N1CCC(*CCOS(=C)=O)CC1)=O Chemical compound CC(C)(C)OC(N1CCC(*CCOS(=C)=O)CC1)=O 0.000 description 1
- FKSZOBZKWJWQHK-UHFFFAOYSA-N CC(C)c1cnc(N2CCC(CCCOc3ccc(C(NC(CO)CO)=O)c(C)n3)CC2)nc1 Chemical compound CC(C)c1cnc(N2CCC(CCCOc3ccc(C(NC(CO)CO)=O)c(C)n3)CC2)nc1 FKSZOBZKWJWQHK-UHFFFAOYSA-N 0.000 description 1
- ZEOVKUVEPGIQPI-LLVKDONJSA-N C[C@H](CCO)C(CC1)CCN1C(OC(C)(C)C)=O Chemical compound C[C@H](CCO)C(CC1)CCN1C(OC(C)(C)C)=O ZEOVKUVEPGIQPI-LLVKDONJSA-N 0.000 description 1
- LVWOONRGULOBJE-XMSQKQJNSA-N C[C@H]([C@@H](CO)O)NC(c(c(C)c1)c(C)cc1OCCCC(CC1)CCN1c(nc1)ncc1Cl)=O Chemical compound C[C@H]([C@@H](CO)O)NC(c(c(C)c1)c(C)cc1OCCCC(CC1)CCN1c(nc1)ncc1Cl)=O LVWOONRGULOBJE-XMSQKQJNSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 208000002177 Cataract Diseases 0.000 description 1
- YCXVVFALGLBRNK-UHFFFAOYSA-N Cc1c(C(O)=O)c(C)cc(OCCCC(CC2)CCN2c(nc2)ncc2Cl)c1 Chemical compound Cc1c(C(O)=O)c(C)cc(OCCCC(CC2)CCN2c(nc2)ncc2Cl)c1 YCXVVFALGLBRNK-UHFFFAOYSA-N 0.000 description 1
- 206010065941 Central obesity Diseases 0.000 description 1
- 102000004859 Cholecystokinin Receptors Human genes 0.000 description 1
- 108090001085 Cholecystokinin Receptors Proteins 0.000 description 1
- 102100032165 Corticotropin-releasing factor-binding protein Human genes 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- 208000002249 Diabetes Complications Diseases 0.000 description 1
- 206010012655 Diabetic complications Diseases 0.000 description 1
- 229940124213 Dipeptidyl peptidase 4 (DPP IV) inhibitor Drugs 0.000 description 1
- 208000030814 Eating disease Diseases 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- JNCMHMUGTWEVOZ-UHFFFAOYSA-N F[CH]F Chemical compound F[CH]F JNCMHMUGTWEVOZ-UHFFFAOYSA-N 0.000 description 1
- 208000019454 Feeding and Eating disease Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical compound OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 102000030782 GTP binding Human genes 0.000 description 1
- 108091000058 GTP-Binding Proteins 0.000 description 1
- 102100031416 Gastric triacylglycerol lipase Human genes 0.000 description 1
- 208000018522 Gastrointestinal disease Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 102400000321 Glucagon Human genes 0.000 description 1
- 108060003199 Glucagon Proteins 0.000 description 1
- 102000030595 Glucokinase Human genes 0.000 description 1
- 108010021582 Glucokinase Proteins 0.000 description 1
- 229940121931 Gluconeogenesis inhibitor Drugs 0.000 description 1
- 206010018429 Glucose tolerance impaired Diseases 0.000 description 1
- 102000007390 Glycogen Phosphorylase Human genes 0.000 description 1
- 108010046163 Glycogen Phosphorylase Proteins 0.000 description 1
- 206010018910 Haemolysis Diseases 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 208000015580 Increased body weight Diseases 0.000 description 1
- 206010022489 Insulin Resistance Diseases 0.000 description 1
- 108010041872 Islet Amyloid Polypeptide Proteins 0.000 description 1
- 102000036770 Islet Amyloid Polypeptide Human genes 0.000 description 1
- 239000007836 KH2PO4 Substances 0.000 description 1
- 108010009384 L-Iditol 2-Dehydrogenase Proteins 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 229910009891 LiAc Inorganic materials 0.000 description 1
- 238000005684 Liebig rearrangement reaction Methods 0.000 description 1
- 229940127470 Lipase Inhibitors Drugs 0.000 description 1
- 239000000867 Lipoxygenase Inhibitor Substances 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- 108010008364 Melanocortins Proteins 0.000 description 1
- 101710151321 Melanostatin Proteins 0.000 description 1
- 208000019022 Mood disease Diseases 0.000 description 1
- 208000023178 Musculoskeletal disease Diseases 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 102400000064 Neuropeptide Y Human genes 0.000 description 1
- 108090000189 Neuropeptides Proteins 0.000 description 1
- 102000003797 Neuropeptides Human genes 0.000 description 1
- WMWAWDQNBZDLPX-UHFFFAOYSA-N OCC(CO)NC(=O)C=1C=CC(=NC1C)OCCCC1CCN(CC1)C(=O)O Chemical compound OCC(CO)NC(=O)C=1C=CC(=NC1C)OCCCC1CCN(CC1)C(=O)O WMWAWDQNBZDLPX-UHFFFAOYSA-N 0.000 description 1
- 208000010191 Osteitis Deformans Diseases 0.000 description 1
- 208000003076 Osteolysis Diseases 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 206010033307 Overweight Diseases 0.000 description 1
- 108010016731 PPAR gamma Proteins 0.000 description 1
- 229940124754 PPAR-alpha/gamma agonist Drugs 0.000 description 1
- 208000027868 Paget disease Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 102100038825 Peroxisome proliferator-activated receptor gamma Human genes 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 102100026974 Sorbitol dehydrogenase Human genes 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 229940100389 Sulfonylurea Drugs 0.000 description 1
- 239000007984 Tris EDTA buffer Substances 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- 230000001800 adrenalinergic effect Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 239000003288 aldose reductase inhibitor Substances 0.000 description 1
- 229940090865 aldose reductase inhibitors used in diabetes Drugs 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 239000003888 alpha glucosidase inhibitor Substances 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 238000012440 amplified luminescent proximity homogeneous assay Methods 0.000 description 1
- 229940125709 anorectic agent Drugs 0.000 description 1
- 230000003178 anti-diabetic effect Effects 0.000 description 1
- 239000003524 antilipemic agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 235000019789 appetite Nutrition 0.000 description 1
- 230000036528 appetite Effects 0.000 description 1
- 239000002830 appetite depressant Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229940125388 beta agonist Drugs 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 235000013877 carbamide Nutrition 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 229940082638 cardiac stimulant phosphodiesterase inhibitors Drugs 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 239000013553 cell monolayer Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 230000001713 cholinergic effect Effects 0.000 description 1
- 238000010549 co-Evaporation Methods 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229910052681 coesite Inorganic materials 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004737 colorimetric analysis Methods 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 208000029078 coronary artery disease Diseases 0.000 description 1
- 108010083720 corticotropin releasing factor-binding protein Proteins 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 229910052906 cristobalite Inorganic materials 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- AFABGHUZZDYHJO-UHFFFAOYSA-N dimethyl butane Natural products CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 239000003603 dipeptidyl peptidase IV inhibitor Substances 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- BVTBRVFYZUCAKH-UHFFFAOYSA-L disodium selenite Chemical compound [Na+].[Na+].[O-][Se]([O-])=O BVTBRVFYZUCAKH-UHFFFAOYSA-L 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 235000014632 disordered eating Nutrition 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 238000010410 dusting Methods 0.000 description 1
- 201000006549 dyspepsia Diseases 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 238000001952 enzyme assay Methods 0.000 description 1
- 238000006345 epimerization reaction Methods 0.000 description 1
- XIRRHMRGKFDBIA-UHFFFAOYSA-N ethyl 2,4-dimethyl-6-oxo-1h-pyridine-3-carboxylate Chemical compound CCOC(=O)C=1C(C)=CC(=O)NC=1C XIRRHMRGKFDBIA-UHFFFAOYSA-N 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 239000013604 expression vector Substances 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000012631 food intake Nutrition 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 208000020694 gallbladder disease Diseases 0.000 description 1
- 230000030136 gastric emptying Effects 0.000 description 1
- 108010091264 gastric triacylglycerol lipase Proteins 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 1
- 229960004666 glucagon Drugs 0.000 description 1
- 239000003850 glucocorticoid receptor antagonist Substances 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000003979 granulating agent Substances 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 230000008588 hemolysis Effects 0.000 description 1
- 238000001511 high performance liquid chromatography nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- 230000000742 histaminergic effect Effects 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 201000001421 hyperglycemia Diseases 0.000 description 1
- 229940126904 hypoglycaemic agent Drugs 0.000 description 1
- 230000002267 hypothalamic effect Effects 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 238000012528 insulin ELISA Methods 0.000 description 1
- 230000037041 intracellular level Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 229940125425 inverse agonist Drugs 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 235000014666 liquid concentrate Nutrition 0.000 description 1
- 208000028755 loss of height Diseases 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 208000029791 lytic metastatic bone lesion Diseases 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 208000027202 mammary Paget disease Diseases 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000002865 melanocortin Substances 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 208000037819 metastatic cancer Diseases 0.000 description 1
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 1
- MGNBKHCWNYBOAN-UHFFFAOYSA-N methyl 2-methyl-4-(3-piperidin-4-ylpropoxy)benzoate Chemical compound C1=C(C)C(C(=O)OC)=CC=C1OCCCC1CCNCC1 MGNBKHCWNYBOAN-UHFFFAOYSA-N 0.000 description 1
- LDOQVZIBZVWMLR-UHFFFAOYSA-N methyl 2-methyl-4-[3-[1-(5-propan-2-ylpyrimidin-2-yl)piperidin-4-yl]propoxy]benzoate Chemical compound C1=C(C)C(C(=O)OC)=CC=C1OCCCC1CCN(C=2N=CC(=CN=2)C(C)C)CC1 LDOQVZIBZVWMLR-UHFFFAOYSA-N 0.000 description 1
- MQWVQAWQGGYVJT-UHFFFAOYSA-N methyl 4-[3-[1-(5-ethylpyridin-2-yl)piperidin-4-yl]propoxy]-2-methylbenzoate Chemical compound N1=CC(CC)=CC=C1N1CCC(CCCOC=2C=C(C)C(C(=O)OC)=CC=2)CC1 MQWVQAWQGGYVJT-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 230000014925 multi-organism signaling Effects 0.000 description 1
- 230000003387 muscular Effects 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- IHPOWKBAUMFSHB-UHFFFAOYSA-N n-(1,3-dihydroxypropan-2-yl)-2-methyl-6-(3-piperidin-4-ylpropoxy)pyridine-3-carboxamide;hydrochloride Chemical compound Cl.C1=C(C(=O)NC(CO)CO)C(C)=NC(OCCCC2CCNCC2)=C1 IHPOWKBAUMFSHB-UHFFFAOYSA-N 0.000 description 1
- RLRHPCKWSXWKBG-UHFFFAOYSA-N n-(2-azaniumylethyl)carbamate Chemical compound NCCNC(O)=O RLRHPCKWSXWKBG-UHFFFAOYSA-N 0.000 description 1
- UQWSWYKGKNLOEQ-HNNXBMFYSA-N n-[(2s)-2,3-dihydroxypropyl]-2-methyl-6-(3-piperidin-4-ylpropoxy)pyridine-3-carboxamide Chemical compound C1=C(C(=O)NC[C@H](O)CO)C(C)=NC(OCCCC2CCNCC2)=C1 UQWSWYKGKNLOEQ-HNNXBMFYSA-N 0.000 description 1
- 201000001119 neuropathy Diseases 0.000 description 1
- 230000007823 neuropathy Effects 0.000 description 1
- 235000005152 nicotinamide Nutrition 0.000 description 1
- 239000011570 nicotinamide Substances 0.000 description 1
- 229960003966 nicotinamide Drugs 0.000 description 1
- URPYMXQQVHTUDU-OFGSCBOVSA-N nucleopeptide y Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 URPYMXQQVHTUDU-OFGSCBOVSA-N 0.000 description 1
- 125000003261 o-tolyl group Chemical group [H]C1=C([H])C(*)=C(C([H])=C1[H])C([H])([H])[H] 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 230000000406 opioidergic effect Effects 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 238000007410 oral glucose tolerance test Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 201000008482 osteoarthritis Diseases 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000013618 particulate matter Substances 0.000 description 1
- 230000007310 pathophysiology Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 208000028169 periodontal disease Diseases 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 208000019899 phobic disease Diseases 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- 239000002571 phosphodiesterase inhibitor Substances 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 1
- 235000014483 powder concentrate Nutrition 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 238000003672 processing method Methods 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 230000006340 racemization Effects 0.000 description 1
- 230000009892 regulation of energy homeostasis Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- UNAANXDKBXWMLN-UHFFFAOYSA-N sibutramine Chemical compound C=1C=C(Cl)C=CC=1C1(C(N(C)C)CC(C)C)CCC1 UNAANXDKBXWMLN-UHFFFAOYSA-N 0.000 description 1
- 229960004425 sibutramine Drugs 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 235000020374 simple syrup Nutrition 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000011781 sodium selenite Substances 0.000 description 1
- 229960001471 sodium selenite Drugs 0.000 description 1
- 235000015921 sodium selenite Nutrition 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 229910052682 stishovite Inorganic materials 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000020679 tap water Nutrition 0.000 description 1
- 239000008399 tap water Substances 0.000 description 1
- LZXLJOLNLRQANR-UHFFFAOYSA-N tert-butyl 4-[3-(4-bromo-3,5-dimethylphenoxy)propyl]piperidine-1-carboxylate Chemical compound CC1=C(Br)C(C)=CC(OCCCC2CCN(CC2)C(=O)OC(C)(C)C)=C1 LZXLJOLNLRQANR-UHFFFAOYSA-N 0.000 description 1
- KARHCQNPTHLRLD-UHFFFAOYSA-N tert-butyl 4-[3-[4-(2-hydroxyethylcarbamoyl)-3-methylphenoxy]propyl]piperidine-1-carboxylate Chemical compound C1=C(C(=O)NCCO)C(C)=CC(OCCCC2CCN(CC2)C(=O)OC(C)(C)C)=C1 KARHCQNPTHLRLD-UHFFFAOYSA-N 0.000 description 1
- LFKDJXLFVYVEFG-UHFFFAOYSA-N tert-butyl carbamate Chemical compound CC(C)(C)OC(N)=O LFKDJXLFVYVEFG-UHFFFAOYSA-N 0.000 description 1
- LZNOKWJGNPKUSE-UHFFFAOYSA-N tert-butyl n-(3-amino-2-hydroxypropyl)carbamate Chemical compound CC(C)(C)OC(=O)NCC(O)CN LZNOKWJGNPKUSE-UHFFFAOYSA-N 0.000 description 1
- SYTNTKVUQUYHLM-UHFFFAOYSA-N tert-butyl n-[2-[[4-[3-[1-(5-chloropyrimidin-2-yl)piperidin-4-yl]propoxy]-2-methylbenzoyl]amino]ethyl]carbamate Chemical compound C1=C(C(=O)NCCNC(=O)OC(C)(C)C)C(C)=CC(OCCCC2CCN(CC2)C=2N=CC(Cl)=CN=2)=C1 SYTNTKVUQUYHLM-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- NQRYJNQNLNOLGT-UHFFFAOYSA-N tetrahydropyridine hydrochloride Natural products C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 230000000929 thyromimetic effect Effects 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 229910052905 tridymite Inorganic materials 0.000 description 1
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 239000012137 tryptone Substances 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 239000013585 weight reducing agent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
Definitions
- the present invention is directed to G-protein coupled receptor (GPCR) agonists.
- GPCR G-protein coupled receptor
- the present invention is directed to agonists of GPRl 19 that are useful for the treatment of obesity, e g as regulators of satiety, metabolic syndrome and for the treatment of diabetes.
- Obesity is characterized by an excessive adipose tissue mass relative to body size.
- body fat mass is estimated by the body mass index (BMI; weight(kg)/height(m) 2 ), or waist circumference
- BMI body mass index
- Individuals are considered obese when the BMI is greater than 30 and there are established medical consequences of being overweight. It has been an accepted medical view for some time that an increased body weight, especially as a result of abdominal body fat, is associated with an increased risk for diabetes, hypertension, heart disease, and numerous other health complications, such as arthritis, stroke, gallbladder disease, muscular and respiratory problems, back pam and even certain cancers
- Hypothalamic centres are also able to sense peripheral hormones involved in the maintenance of body weight and degree of adiposity, such as insulin and leptin, and fat tissue derived peptides
- Drugs aimed at the pathophysiology associated with insulin dependent Type I diabetes and non-insulin dependent Type II diabetes have many potential side effects and do not adequately address the dyslipidaemia and hyperglycaemia in a high proportion of patients. Treatment is often focused at individual patient needs using diet, exercise, hypoglycaemic agents and insulin, but there is a continuing need for novel antidiabetic agents, particularly ones that may be better tolerated with fewer adverse effects.
- metabolic syndrome places people at high risk of coronary artery disease, and is characterized by a cluster of risk factors including central obesity (excessive fat tissue in the abdominal region), glucose intolerance, high triglycerides and low HDL cholesterol, and high blood pressure.
- central obesity excessive fat tissue in the abdominal region
- glucose intolerance high triglycerides
- low HDL cholesterol high blood pressure
- Myocardial ischemia and microvascular disease is an established morbidity associated with untreated or poorly controlled metabolic syndrome.
- GPRl 19 (previously referred to as GPRl 16) is a GPCR identified as SNORF25 in WO00/50562 which discloses both the human and rat receptors, US 6,468,756 also discloses the mouse receptor (accession numbers. AAN95194 (human), AAN95195 (rat) and ANN95196
- GPRl 19 is expressed in the pancreas, small intestine, colon and adipose tissue.
- the expression profile of the human GPRl 19 receptor indicates its potential utility as a target for the treatment of obesity and diabetes.
- International patent applications WO2005/061489, WO2006/070208 and WO2006/067532 disclose heterocyclic derivatives as GPRl 19 receptor agonists
- International patent applications WO2006/067531, WO2007/003960, WO2007/003961, WO2007/003962 and WO2007/003964, WO2007/116230 and WO2007/116229 disclose GPRl 19 receptor agonists
- the present invention relates to agonists of GPRl 19 which are useful for the treatment of diabetes and as peripheral regulators of satiety, e g for the treatment of obesity and metabolic syndrome
- (I) or pharmaceutically acceptable salts thereof are agonists of GPRl 19 and are useful for the prophylactic or therapeutic treatment of diabetes and obesity.
- the present invention is directed to a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- R 1 is -SO 2 Me or -CONHR 6 ;
- R 2 , R 3 and R 4 are independently selected from hydrogen and methyl, n is 0, 1 or 2,
- R 5 is independently Ci 4 alkyl, C M alkoxy, fluoro, chloro, Ci 3 fluoroalkyl or benzyl
- R 6 is hydrogen, 3-azetidinyl, 3-pyrrolidinyl, 3-pipendinyl or 4-piperidinyl, wherein the azetidinyl, pyrrolidinyl and pipe ⁇ dinyl rings may be optionally substituted with OH, CH 2 OH or CH 3 , Ci 3 alkyl, C 2 A alkyl substituted by -N(R 7 ) 2 and/or one or two hydroxy groups, or alkyl substituted by a 4- to 6-membered nitrogen-containing heterocyclic ring; and
- R 7 is independently hydrogen or methyl In one embodiment of the invention, Q is CH In another Q is N. Q is preferably CH
- R 1 is -SO 2 Me In another, R 1 is -CONHR 6
- R 1 is preferably -CONHR 6
- R 2 and R 3 are preferably methyl, for example R 2 is methyl and R 3 is hydrogen.
- R 4 is hydrogen and m another R 4 is methyl
- R 4 is preferably hydrogen.
- the stereocenter produced preferably has the (R)- configuration.
- n is preferably 1.
- R 5 is preferably meta or para, more preferably para, to the point of attachment to the piperidmyl nitrogen.
- R 5 is preferably Ci 3 alkyl, fluoro, chloro or Ci 3 fluoroalkyl, more preferably C 2 3 alkyl, chloro or Ci 3 fluoroalkyl, especially chloro, ethyl or isopropyl, more especially chloro.
- W and X are preferably CH, for example W, X and Y may be CH Y is preferably N
- R 6 is preferably hydrogen or C 2 3 alkyl substituted by -N(R 7 ) 2 or one or two hydroxy groups, for example hydrogen or C 2 3 alkyl substituted by one or two hydroxy groups.
- R 6 is more preferably C 2 3 alkyl substituted by one or two hydroxy groups, e g. 2-hydroxyethyl, 2- hydroxy-1-methylethyl, 2,3-dihydroxypropyl or 2-hydroxy-l-hydroxymethylethyl.
- R 6 is C 2 ⁇ alkyl substituted a 4- to 6-membered nitrogen-containing heterocyclic ring
- suitable 4- to 6-membered nitrogen-containing heterocyclic rings include pyrrolidmyl and azetidinyl
- R 7 is preferably hydrogen
- Q is N; one of W, X and Y is N or CH and the others are CH where the H may be replaced by
- R 1 is -SO 2 Me or -CONHR 6 ;
- R 2 , R 3 and R 4 are independently selected from hydrogen and methyl, n is 0, 1 or 2, R 5 is Ci 4 alkyl, Ci 4 alkoxy, fluoro, chloro, Ci 3 fluoroalkyl or benzyl; and
- R 6 is hydrogen, 3-azetidinyl, 3-pyrrolidinyl, Ci 3 alkyl, or C 2 3 alkyl substituted by amino or one or two hydroxy groups; and pharmaceutically acceptable salts thereof
- a further group of compounds which may be mentioned are those where: Q is CH; one of W, X and Y is N or CH and the others are CH where the H may be replaced by R 5 when present;
- R 1 is -SO 2 Me or -CONHR 6 ; when R 1 is -SO 2 Me, R 2 and R 3 are hydrogen, and when R 1 is -CONHR 6 , R 2 and R 3 are independently selected from hydrogen and methyl,
- R 4 is hydrogen or methyl; n is 0, 1 or 2,
- R 5 is independently Ci 4 alkyl, CM alkoxy, fluoro, chloro or Ci 3 fluoroalkyl
- R 6 is hydrogen, 3-azetidinyl, 3-pyrrolidinyl, 4-pipendinyl, Ci 3 alkyl, or C 2 ⁇ alkyl substituted by amino and/or one or two hydroxy groups, or by a 4- or 5-membered nitrogen- containing heterocyclic ring.
- R 5 is hydrogen, 3-azetidinyl, 3-pyrrolidinyl, Ci 3 alkyl, or C 2 3 alkyl substituted by amino or one or two hydroxy groups
- preferred compounds of this invention include those in which several or each variable in formula (I) is selected from the preferred, more preferred or particularly listed groups for each variable Therefore, this invention is intended to include all combinations of preferred, more preferred and particularly listed groups
- alkyl means carbon chains which may be linear or branched or combinations thereof. Examples of alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, sec- and fert-butyl.
- Fluoroalkyl means alkyl groups substituted by one or more fluoro atoms, e g. CHF 2 and CF 3 .
- Compounds described herein may contain one or more asymmetric centers and may thus give rise to diastereomers and optical isomers.
- the present invention includes all such possible diastereomers as well as their raceinic mixtures, their substantially pure resolved enantiomers, all possible geometric isomers, and pharmaceutically acceptable salts thereof
- the above formula (I) is shown without a definitive stereochemistry at certain positions.
- the present invention includes all stereoisomers of formula (I) and pharmaceutically acceptable salts thereof Further, mixtures of stereoisomers as well as isolated specific stereoisomers are also included During the course of the synthetic procedures used to prepare such compounds, or in using racemization or epimerization procedures known to those skilled in the art, the products of such procedures can be a mixture of stereoisomers.
- the present invention includes any possible solvates and polymorphic forms.
- a type of a solvent that forms the solvate is not particularly limited so long as the solvent is pharmacologically acceptable.
- water, ethanol, propanol, acetone or the like can be used
- salts derived from bases include those derived from bases such as, for example, potassium and sodium salts and the like.
- Salts derived from pharmaceutically acceptable non-toxic acids include those derived from inorganic and organic acids such as, for example, hydrochloric, methanesulfonic, sulfuric, p-toluenesulfonic acid and the like. Since the compounds of formula (I) are intended for pharmaceutical use they are preferably provided in substantially pure form, for example at least 60% pure, more suitably at least 75% pure, especially at least 98% pure (% are on a weight for weight basis).
- the compounds of formula (I) can be prepared as described below, wherein R 1 , R 2 , R 3 , R 4 , R 5 , Q, W, X and Y are defined as above, PG represents a protecting group, DG represents a displaceable group e g, F, Cl, Br, MeSC> 2 , and Ak is Ci 2 alkyl.
- Unichiral building blocks for compounds of formula (I), where R 4 is Me can be readily prepared from known compounds (Scheme 1).
- Scheme 1 the ethyl ester of compound (II) where PG is Boc has been previously reported (US Patent 6,518,423) Saponification and hydrogenation, under standard conditions, will yield the racemic compound of formula (III) Chiral reduction of the alkenoic acid (II) under suitable conditions, such as a hydrogenation in the presence of a chiral catalyst, yields compounds of formula (III) in high enantiomeric excess.
- a suitable catalyst is [Rh(norbornadiene) 2 ]BF 4 and (S)-l-[(R)-2-(di-tert- butylphosphmo)ferrocenyl]ethylbis(2-methylphenyl)phosphine
- compounds of formula (IV) can then be obtained by reduction of the carboxylic acids of formula (III) under standard conditions, for example employing borane in a suitable solvent such as THF.
- Compounds of formula (I) where Q is CH and R 1 is -SO 2 Me can also be prepared from compounds of formula (VI), by reaction with the appropriate 4-methanesulfonylphenol under Mitsunobu conditions (for example using a suitable solvent such as THF, at between O 0 C and room temperature, followed by the addition of triphenylphosphme and diisopropylazo- dicarboxylate) followed by removal of the protecting group under conditions well known to those with skill in the art.
- Mitsunobu conditions for example using a suitable solvent such as THF, at between O 0 C and room temperature, followed by the addition of triphenylphosphme and diisopropylazo- dicarboxylate
- the compounds of formula (I) may be prepared singly or as compound libraries comprising at least 2, for example 5 to 1,000, compounds and more preferably 10 to 100 compounds of formula (I)
- Compound libraries may be prepared by a combinatorial "split and mix” approach or by multiple parallel synthesis using either solution or solid phase chemistry, using procedures known to those skilled in the art.
- labile functional groups in the intermediate compounds e.g. hydroxy, carboxy and amino groups
- the protecting groups may be removed at any stage in the synthesis of the compounds of formula (I) or may be present on the final compound of formula (I).
- a comprehensive discussion of the ways in which various labile functional groups may be protected and methods for cleaving the resulting protected derivatives is given in, for example, Protective Groups in Organic Chemistry, T.W. Greene and P.G M. Wuts, (1991) Wiley-Interscience, New York, 2 nd edition.
- the compounds of formula (I) are useful as GPRl 19 agonists, e.g. for the treatment and/or prophylaxis of obesity and diabetes.
- the compounds of formula (I) will generally be administered in the form of a pharmaceutical composition.
- the invention also provides a compound of formula (I), or a pharmaceutically acceptable salt thereof, for use as a pharmaceutical.
- the invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I), in combination with a pharmaceutically acceptable carrier.
- composition is comprised of a pharmaceutically acceptable carrier and a non-toxic therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- the invention also provides a pharmaceutical composition for the treatment of disease by modulating GPRl 19, resulting in the prophylactic or therapeutic treatment of obesity, e.g. by regulating satiety, or for the treatment of diabetes, comprising a pharmaceutically acceptable carrier and a non-toxic therapeutically effective amount of compound of formula (I), or a pharmaceutically acceptable salt thereof
- compositions may optionally comprise other therapeutic ingredients or adjuvants.
- the compositions include compositions suitable for oral, rectal, topical, and parenteral (including subcutaneous, intramuscular, and intravenous) administration, although the most suitable route m any given case will depend on the particular host, and nature and severity of the conditions for which the active ingredient is being administered.
- the pharmaceutical compositions may be conveniently presented in unit dosage form and prepared by any of the methods well known in the art of pharmacy
- the compounds of formula (I), or pharmaceutically acceptable salts thereof can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques
- a pharmaceutical carrier may take a wide variety of forms depending on the form of preparation desired for administration, e g oral or parenteral (including intravenous).
- compositions can be presented as discrete units suitable for oral administration such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient. Further, the compositions can be presented as a powder, as granules, as a solution, as a suspension m an aqueous liquid, as a non-aqueous liquid, as an oil-m-water emulsion, or as a water-m-oil liquid emulsion.
- the compound of formula (I), or a pharmaceutically acceptable salt thereof may also be administered by controlled release means and/or delivery devices
- the compositions may be prepared by any of the methods of pharmacy In general, such methods include a step of bringing into association the active ingredient with the carrier that constitutes one or more necessary ingredients In general, the compositions are prepared by uniformly and intimately admixing the active ingredient with liquid carriers or finely divided solid carriers or both. The product can then be conveniently shaped into the desired presentation
- the compounds of formula (I), or pharmaceutically acceptable salts thereof, can also be included in pharmaceutical compositions in combination with one or more other therapeutically active compounds.
- the pharmaceutical carrier employed can be, for example, a solid, liquid, or gas.
- solid carriers include lactose, terra alba, sucrose, talc, gelatin, agar, pectm, acacia, magnesium stearate, and stearic acid
- liquid carriers are sugar syrup, peanut oil, olive oil, and water.
- gaseous carriers include carbon dioxide and nitrogen
- any convenient pharmaceutical media may be employed.
- oral liquid preparations such as suspensions, elixirs and solutions
- carriers such as starches, sugars, microcrystallme cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents, and the like may be used to form oral solid preparations such as powders, capsules and tablets Because of their ease of administration, tablets and capsules are the preferred oral dosage units whereby solid pharmaceutical carriers are employed.
- tablets may be coated by standard aqueous or nonaqueous techniques
- a tablet containing the composition of this invention may be prepared by compression or molding, optionally with one or more accessory ingredients or adjuvants
- Compressed tablets may be prepared by compressing, in a suitable machine, the active ingredient in a free -flowing form such as powder or granules, optionally mixed with a binder, lubricant, inert diluent, surface active or dispersing agent.
- Molded tablets may be made by molding in a suitable machine, a mixture of the powdered compound moistened with an inert liquid diluent
- Each tablet preferably contains from about 0 05mg to about 5g of the active ingredient and each cachet or capsule preferably containing from about 0.05mg to about 5g of the active ingredient
- a formulation intended for the oral administration to humans may contain from about 0 5mg to about 5g of active agent, compounded with an appropriate and convenient amount of carrier material which may vary from about 5 to about 95 percent of the total composition.
- Unit dosage forms will generally contain between from about lmg to about 2g of the active ingredient, typically 25mg, 50mg, lOOmg, 200mg, 300mg, 400mg, 500mg, 600mg, 800mg, or lOOOmg
- Pharmaceutical compositions of the present invention suitable for parenteral administration may be prepared as solutions or suspensions of the active compounds in water.
- a suitable surfactant can be included such as, for example, hydroxypropylcellulose.
- Dispersions can also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof in oils. Further, a preservative can be included to prevent the detrimental growth of microorganisms.
- Pharmaceutical compositions of the present invention suitable for injectable use include sterile aqueous solutions or dispersions Furthermore, the compositions can be in the form of sterile powders for the extemporaneous preparation of such sterile injectable solutions or dispersions. In all cases, the final injectable form must be sterile and must be effectively fluid for easy syringability.
- the pharmaceutical compositions must be stable under the conditions of manufacture and storage; thus, preferably should be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g. glycerol, propylene glycol and liquid polyethylene glycol), vegetable oils, and suitable mixtures thereof.
- compositions of the present invention can be in a form suitable for topical use such as, for example, an aerosol, cream, ointment, lotion, dusting powder, or the like.
- compositions can be in a form suitable for use in transdermal devices.
- These formulations may be prepared, using a compound of formula (I), or a pharmaceutically acceptable salt thereof, via conventional processing methods
- a cream or ointment is prepared by admixing hydrophilic material and water, together with about 5wt% to about 10wt% of the compound, to produce a cream or ointment having a desired consistency
- compositions of this invention can be in a form suitable for rectal administration wherein the carrier is a solid It is preferable that the mixture forms unit dose suppositories.
- suitable carriers include cocoa butter and other materials commonly used in the art.
- the suppositories may be conveniently formed by first admixing the composition with the softened or melted carrier(s) followed by chilling and shaping in molds.
- the pharmaceutical formulations described above may include, as appropriate, one or more additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface -active agents, thickeners, lubricants, preservatives (including anti-oxidants) and the like.
- additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface -active agents, thickeners, lubricants, preservatives (including anti-oxidants) and the like.
- additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface -active agents, thickeners, lubricants, preservatives (including anti-oxidants) and the like.
- additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface -active agents, thickeners, lubricants, preservatives (including anti-oxidants) and the like.
- other adjuvants can be included to render the formulation isotonic with the blood
- dosage levels on the order of 0.01mg/kg to about 150mg/kg of body weight per day are useful m the treatment of the above-indicated conditions, or alternatively about
- obesity may be effectively treated by the administration of from about 0.01 to 50mg of the compound per kilogram of body weight per day, or alternatively about 0.5mg to about 3.5g per patient per day.
- the compounds of formula (I) may be used in the treatment of diseases or conditions in which GPRl 19 plays a role
- the invention also provides a method for the treatment of a disease or condition in which GPRl 19 plays a role comprising a step of administering to a subject in need thereof an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof
- Diseases or conditions in which GPRl 19 plays a role include obesity and diabetes.
- the treatment of obesity is intended to encompass the treatment of diseases or conditions such as obesity and other eating disorders associated with excessive food intake e.g by reduction of appetite and body weight, maintenance of weight reduction and prevention of rebound and diabetes (including Type 1 and Type 2 diabetes, impaired glucose tolerance, insulin resistance and diabetic complications such as neuropathy, nephropathy, retinopathy, cataracts, cardiovascular complications and dyslipidaemia). And the treatment of patients who have an abnormal sensitivity to ingested fats leading to functional dyspepsia
- the compounds of the invention may also be used for treating metabolic diseases such as metabolic syndrome (syndrome X), impaired glucose tolerance, hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels and hypertension.
- the compounds of the invention may offer advantages over compounds acting via different mechanisms for the treatment of the above mentioned disorders in that they may offer beta-cell protection, increased cAMP and insulin secretion and also slow gastric emptying.
- the compounds of the invention may also be used for treating conditions characterised by low bone mass such asosteopenia, osteoporosis, rheumatoid arthritis, osteoarthritis, periodontal disease, alveolar bone loss, osteotomy bone loss, childhood idiopathic bone loss, Paget's disease, bone loss due to metastatic cancer, osteolytic lesions, curvature of the spine and loss of height.
- low bone mass such asosteopenia, osteoporosis, rheumatoid arthritis, osteoarthritis, periodontal disease, alveolar bone loss, osteotomy bone loss, childhood idiopathic bone loss, Paget's disease, bone loss due to metastatic cancer, osteolytic lesions, curvature of the spine and loss of height.
- the invention also provides a method for the regulation of satiety comprising a step of administering to a subject in need thereof an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- the invention also provides a method for the treatment of obesity comprising a step of administering to a subject in need thereof an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- the invention also provides a method for the treatment of diabetes, including Type 1 and Type 2 diabetes, particularly type 2 diabetes, comprising a step of administering to a patient m need thereof an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof
- the invention also provides a method for the treatment of metabolic syndrome (syndrome X), impaired glucose tolerance, hyper lipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels or hypertension comprising a step of administering to a patient in need thereof an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- metabolic syndrome sekunder X
- impaired glucose tolerance hyper lipidemia
- hypertriglyceridemia hypercholesterolemia
- low HDL levels or hypertension comprising a step of administering to a patient in need thereof an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- the invention also provides a compound of formula (I), or a pharmaceutically acceptable salt thereof, for use in the treatment of a condition as defined above.
- the invention also provides the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of a condition as defined above
- treatment includes both therapeutic and prophylactic treatment
- the compounds of formula (I) may exhibit advantageous properties compared to known
- the compounds may exhibit improved potency or half-life, or solubility or metabolic profiles, or improved anti-obesity effects, or other advantageous properties for compounds to be used as pharmaceuticals.
- the compounds of formula (I), or pharmaceutically acceptable salts thereof, may be administered alone or in combination with one or more other therapeutically active compounds.
- the other therapeutically active compounds may be for the treatment of the same disease or condition as the compounds of formula (I) or a different disease or condition.
- the therapeutically active compounds may be administered simultaneously, sequentially or separately.
- the compounds of formula (I) may be administered with other active compounds for the treatment of obesity and/or diabetes, for example insulin and insulin analogs, gastric lipase inhibitors, pancreatic lipase inhibitors, sulfonyl ureas and analogs, biguanides, ⁇ 2 agonists, glitazones, PPAR- ⁇ agonists, mixed PPAR- ⁇ / ⁇ agonists, RXR agonists, fatty acid oxidation inhibitors, ⁇ -glucosidase inhibitors, dipeptidyl peptidase IV inhibitors, GLP-I agonists e g GLP-I analogues and mimetics, ⁇ -agonists, phosphodiesterase inhibitors, lipid lowering agents, glycogen phosphorylase inhibitors, antio
- pancreatic lipase inhibitors MCH-I antagonists and CB-I antagonists (or inverse agonists), amylin antagonists, lipoxygenase inhibitors, somostatin analogs, glucokinase activators, glucagon antagonists, insulin signalling agonists, PTPlB inhibitors, gluconeogenesis inhibitors, antilypolitic agents, GSK inhibitors, galanm receptor agonists, anorectic agents, CCK receptor agonists, leptin, serotonergic/dopaminergic antiobesity drugs, reuptake inhibitors e g.
- sibutramine CRF antagonists, CRF binding proteins, thyromimetic compounds, aldose reductase inhibitors, glucocorticoid receptor antagonists, NHE-I inhibitors or sorbitol dehydrogenase inhibitors.
- Combination therapy comprising the administration of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and at least one other antiobesity agent represents a further aspect of the invention.
- the present invention also provides a method for the treatment of obesity in a mammal, such as a human, which method comprises administering an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and another antiobesity agent, to a mammal in need thereof
- the invention also provides the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and another antiobesity agent for the treatment of obesity.
- the invention also provides the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for use in combination with another antiobesity agent, for the treatment of obesity.
- the compound of formula (I), or a pharmaceutically acceptable salt thereof, and the other antiobesity agent(s) may be co-administered or administered sequentially or separately.
- Co-administration includes administration of a formulation which includes both the compound of formula (I), or a pharmaceutically acceptable salt thereof, and the other antiobesity agent(s), or the simultaneous or separate administration of different formulations of each agent
- the invention also provides the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and another antiobesity agent in the manufacture of a medicament for the treatment of obesity.
- the invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I), or a pharmaceutically acceptable salt thereof, and another antiobesity agent, and a pharmaceutically acceptable carrier.
- the invention also encompasses the use of such compositions in the methods described above.
- GPRl 19 agonists are of particular use m combination with centrally acting antiobesity agents
- the other antiobesity agent for use in the combination therapies according to this aspect of the invention is preferably a CB-I modulator, e g. a CB-I antagonist or inverse agonist.
- CB-I modulators include SR141716 (rimonabant) and SLV-319 ((45)-(-)-3-(4- chlorophenyl)- ⁇ -methyl-]V-[(4-chlorophenyl)sulfonyl]-4-phenyl-4,5-dihydro-lH-pyrazole-l- carboxamide); as well as those compounds disclosed m EP576357, EP656354, WO 03/018060, WO 03/020217, WO 03/020314, WO 03/026647, WO 03/026648, WO 03/027076, WO
- GPRl 19 has been suggested to play a role
- diseases or conditions in which GPRl 19 has been suggested to play a role include those described in WO 00/50562 and US 6,468,756, for example cardiovascular disorders, hypertension, respiratory disorders, gestational abnormalities, gastrointestinal disorders, immune disorders, musculoskeletal disorders, depression, phobias, anxiety, mood disorders and
- HOBt H 2 O (77.0 mg, 660 ⁇ mol) was added to a stirred solution of 4-[3-(4-carboxy-3- methylphenoxy)propyl]piperidine-l-carboxylic acid tert-butyl ester (Preparation 7, 200 mg, 530 ⁇ mol) and EDCI (126 mg, 660 ⁇ mol) in THF (10 mL) After 1 h, 2-aminoethanol (64 ⁇ L,
- NEt 3 (13.0 mL, 93.4 mmol) was added to a stirred solution of terf-butyl 4-((R)-3- hydroxy-l-methylpropyl)piperidme-l-carboxylate (Preparation 3, 20.0 g, 77.9 mmol) and methanesulfonyl chloride (9 80 g, 85 6 mmol) m DCM (200 mL). The reaction mixture was warmed to ambient temperature and stirred for 3 h.
- HOBt H 2 O (771 mg, 5.71 mmol) was added to a stirred solution of 6-[3-(l-tert- butoxycarbonylpiperidin-4-yl)propoxy]-2-methylnicotinic acid (Preparation 49, 1.80 g, 4.76 mmol), EDCI (1.10 g, 5.71 mmol) and DIPEA (2.50 niL, 14.3 mmol) in THF (50 mL). After 15 min, 2-aminopropane-l,3-diol (650 mg, 7.13 mmol) was added and the resulting mixture was stirred at ambient temperature for 16 h.
- 2,5-Dichloropyrimidine (17.4 g, 117 mmol) was added portionwise, then the reaction was heated to 110 0 C for 4 h After cooling to 2O 0 C, the reaction was poured into H 2 O (200 mL) and extracted with EtOAc (3 x 500 mL) The combined organic extracts were washed with IM HCl (2 x 200 mL), before being dried (MgSO 4 ) and concentrated.
- Method 2 Products were purified by column chromotography (DCM-MeOH, 98 2).
- HOBt H 2 O 50 0 mg, 370 ⁇ mol
- EDCI 70 5 mg, 370 ⁇ mol
- THF 7 inL
- 2-aminoethanol 68 ⁇ L, 1.13 mmol
- HOBt H 2 O 100 mg, 735 ⁇ mol
- 4- ⁇ 3-[l-(5-chloro- pyrimidin-2-yl)pipendin-4-yl]propoxy ⁇ -2-methylbenzoic acid Preparation 12, 220 mg, 565 ⁇ mol
- EDCI 141 mg, 735 ⁇ mol
- THF 12 mL
- DMF 600 ⁇ L
- (2- aminoethyl)carbamic acid terf-butyl ester 268 ⁇ L, 1.694 mmol
- Examples 110 and 111 N-(( ⁇ )-3-Amino-2-hydroxypropyl)-4- ⁇ 3-[l-(5-chloropyrimidin-2-yl)- piperidin-4-yl]propoxy ⁇ -2,6-dimethylbenzamide and N-((S)-3-Amino-2-hydroxypropyl)-4- ⁇ 3- [l-(5-chloropyrimidin-2-yl)piperidin-4-yl]propoxy ⁇ -2,6-dimethylbenzamide
- amides listed in Table 7 were obtained in enantiomerically pure form employing procedures similar to that outlined for Examples 110 and 111, with the exception that the individual enantiomers of the Boc-protected intermediates were separated by preparative chiral HPLC using a Daicel Chiralpak IA column (250 x 20mm, 5 ⁇ m), with an eluent of IHAPrOH (7:3) at a flow rate of 15 mL/min, and UV detection at 250 nm.
- Example 128 4-[3-(5'-Chloro-3,4,5,6-tetrahydro-2H-[l,2']bipyridinyl-4-yl)propoxy]-N-(2- hydroxyethyl)-2-methylbenzamide
- HATU (82.0 m , 220 ⁇ mol) was added to a stirr ted sxolution of 4-[3-(5'-chloro- 3,4,5,6-tetrahydro-2H-[l,2']bipyridinyl-4-yl)propoxy]-2-methylbenzoic acid (Preparation 27, 118 mg, 180 ⁇ mol), ethanolamine (22.0 ⁇ L, 360 ⁇ mol) and DIPEA (63.0 ⁇ L, 360 ⁇ L) in THF (7 mL), and the resulting solution was stirred at ambient temperature for 16 h.
- Example 141 4- ⁇ 3-[ 1 -(5-Fluoropyrimidin-2-yl)pipendm-4-yl]propoxy ⁇ -N-(2-hydroxyethyl)-2- methylbenzamide
- Example 142 4- ⁇ 3-[l-(5-Chloropyrimidin-2-yl)pipendin-4-yl]propoxy ⁇ -N-((li?,2S)-2,3- dihydroxy-l-methylpropyl)-2,6-dimethylbenzamide
- Example 146 N-(2-Hydroxy- 1 -hydroxymethylethyl)-6- ⁇ 3 -[ 1 -(5-isopropylpyrimidin-2-yl)- pipe ⁇ din-4-yl] propoxy ⁇ -2-methylnicotmamide
- Example 148 6-[3-(5'-Chloro-3,4,5,6-tetrahydro-2H-[l,2']bipyridmyl-4-yl)propoxy]-N-(2- hydroxy- 1 -hydroxymethylethyl)-2-methylnicotinamide
- Example 160 N-((5)-2,3-Dihydroxypropyl)-6- ⁇ 3-[l-(5-ethylpyrimidin-2-yl)piperidin-4-yl]- propoxy ⁇ -2-methylnicotinamide
- Example 161 5'-Fluoro-4-[3-(5-methanesulfonyl-6-methylpyridin-2-yloxy)propyl]-3,4,5,6- tetrahydro-2H-[ 1 ,2']bipyridinyl
- 2,5-Difluoropyridine (65.0 mg, 490 ⁇ mol) and DBU (72.0 ⁇ L, 490 ⁇ mol) were added to a solution of 3-methanesulfonyl-2-methyl-6-(3-piperidin-4-yl-propoxy)pyridine hydrochloride (Preparation 54, 125 mg, 330 ⁇ mol) m DMSO (500 ⁇ L) and the resulting solution stirred at 50 0 C for 72 h.
- the biological activity of the compounds of the invention may be tested in the following assay systems-
- yeast cell-based reporter assays have previously been described in the literature (e.g. see Miret J. J. et al, 2002, J. Biol. Chem., 277:6881-6887, Campbell R.M. et al, 1999, Bioorg. Med. Chem. Lett., 9:2413-2418; King K. et al, 1990, Science, 250: 121-123); WO 99/14344; WO 00/12704, and US 6,100,042). Briefly, yeast cells have been engineered such that the endogenous yeast G-alpha (GPAl) has been deleted and replaced with G-protein chimeras constructed using multiple techniques.
- G-alpha G-alpha
- yeast GPCR Ste3 has been deleted to allow for heterologous expression of a mammalian GPCR of choice
- elements of the pheromone signaling transduction pathway which are conserved in eukaryotic cells (for example, the mitogen-activated protein kinase pathway), drive the expression of Fusl.
- ⁇ -galactosidase LacZ
- Fuslp Fusl promoter
- Yeast cells were transformed by an adaptation of the lithium acetate method described by Agatep et al, (Agatep, R. et al, 1998, Transformation of Saccharomyces cerevisiae by the lithium acetate/single-stranded carrier DNA/polyethylene glycol (LiAc/ss-DNA/PEG) protocol.
- yeast cells were grown overnight on yeast tryptone plates (YT) Carrier smgle-stranded DNA (lO ⁇ g), 2 ⁇ g of each of two Fuslp- LacZ reporter plasmids (one with URA selection marker and one with TRP), 2 ⁇ g of GPRl 19 (human or mouse receptor) in yeast expression vector (2 ⁇ g origin of replication) and a lithium acetate/ polyethylene glycol/ TE buffer was pipetted into an Eppendorf tube The yeast expression plasrmd containing the receptor/ no receptor control has a LEU marker. Yeast cells were inoculated into this mixture and the reaction proceeds at 30 0 C for 60min.
- the yeast cells were then heat-shocked at 42 0 C for 15min The cells were then washed and spread on selection plates
- the selection plates are synthetic defined yeast media minus LEU, URA and TRP (SD- LUT). After incubating at 30 0 C for 2-3 days, colonies that grow on the selection plates were then tested in the LacZ assay.
- yeast cells carrying the human or mouse GPRl 19 receptor were grown overnight in liquid SD-LUT medium to an unsaturated concentration (i.e. the cells were still dividing and had not yet reached stationary phase) They were diluted m fresh medium to an optimal assay concentration and 90 ⁇ l of yeast cells added to 96-well black polystyrene plates (Costar) Compounds, dissolved in DMSO and diluted in a 10% DMSO solution to 1OX concentration, were added to the plates and the plates placed at 30 0 C for 4h.
- Fluorescein di ⁇ -D-galactopyranoside
- FDG Fluorescein di ( ⁇ -D-galactopyranoside)
- a stable cell line expressing recombinant human GPRl 19 was established and this cell line may be used to investigate the effect of compounds of the invention on intracellular levels of cyclic AMP (cAMP).
- cAMP cyclic AMP
- the cell monolayers are washed with phosphate buffered saline and stimulated at 37 0 C for 30mm with various concentrations of compound in stimulation buffer plus 1 % DMSO. Cells are then lysed and cAMP content determined using the Perkin Elmer
- AlphaScreenTM Analogously fluorescent Luminescent Proximity Homogeneous Assay
- Buffers and assay conditions are as described in the manufacturer's protocol.
- Animals are maintained on a reverse phase light-dark cycle (lights off for 8 h from 09.30-17.30 h) during which time the room was illuminated by red light Animals have free access to a standard powdered rat diet and tap water during a two week acclimatization period
- the diet is contained in glass feeding jars with aluminum lids Each lid had a 3-4 cm hole in it to allow access to the food.
- Animals, feeding jars and water bottles are weighed (to the nearest 0.1 g) at the onset of the dark period. The feeding jars and water bottles are subsequently measured 1 , 2, 4, 6 and 24 h after animals are dosed with a compound of the invention and any significant differences between the treatment groups at baseline compared to vehicle -treated controls.
- HIT-T15 cells (passage 60) were obtained from ATCC, and were cultured in RPMI1640 medium supplemented with 10% fetal calf serum and 3OnM sodium selenite All experiments were done with cells at less than passage 70, in accordance with the literature, which describes altered properties of this cell line at passage numbers above 81 (Zhang HJ, Walseth TF, Robertson RP. Insulin secretion and cAMP metabolism in HIT cells. Reciprocal and serial passage-dependent relationships Diabetes 1989 Jan;38(l) 44-8)
- HIT-T15 cells were plated in standard culture medium in 96-well plates at 100,000 cells/ 0 1ml/ well and cultured for 24 hr and the medium was then discarded Cells were incubated for 15min at room temperature with lOO ⁇ l stimulation buffer (Hanks buffered salt solution, 5mM HEPES, 0.5mM IBMX, 0.1% BSA, pH 7 4).
- lOO ⁇ l stimulation buffer Hormoned salt solution, 5mM HEPES, 0.5mM IBMX, 0.1% BSA, pH 7 4).
- HIT-T 15 cells were plated in standard culture medium m 12-well plates at 106 cells/ 1 ml/ well and cultured for 3 days and the medium was then discarded. Cells were washed x 2 with supplemented Krebs-Ringer buffer (KRB) containing 119 mM NaCl, 4.74 mM KCl, 2.54 inM CaCl 2 , 1.19 mM MgSO 4 , 1.19 mM KH2PO4, 25 mM NaHCO 3 , 1OmM HEPES at pH 7.4 and 0 1% bovine serum albumin Cells were incubated with ImI KRB at 37°C for 30 min which was then discarded.
- KRB Krebs-Ringer buffer
- Representative compounds of the invention were found to increase insulin secretion at an EC 50 of less than 10 ⁇ M Compounds showing an EC 50 of less than 1 ⁇ M in the insulin secretion assay may be preferred
- Blood samples (20 ⁇ L) are then taken 25, 50, 80, 120, and 180 min after GIc administration.
- the 20 ⁇ L blood samples for measurement of GIc levels are taken from the cut tip of the tail into disposable micro-pipettes (Dade Diagnostics Inc., Puerto Rico) and the sample added to 480 ⁇ L of haemolysis reagent.
- Duplicate 20 ⁇ L aliquots of the diluted haemolysed blood are then added to 180 ⁇ L of Trinders glucose reagent (Sigma enzymatic (Trinder) colorimetric method) in a 96-well assay plate. After mixing, the samples are left at rt for 30 min before being read against GIc standards (Sigma glucose/urea nitrogen combined standard set).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Diabetes (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Child & Adolescent Psychology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description






























































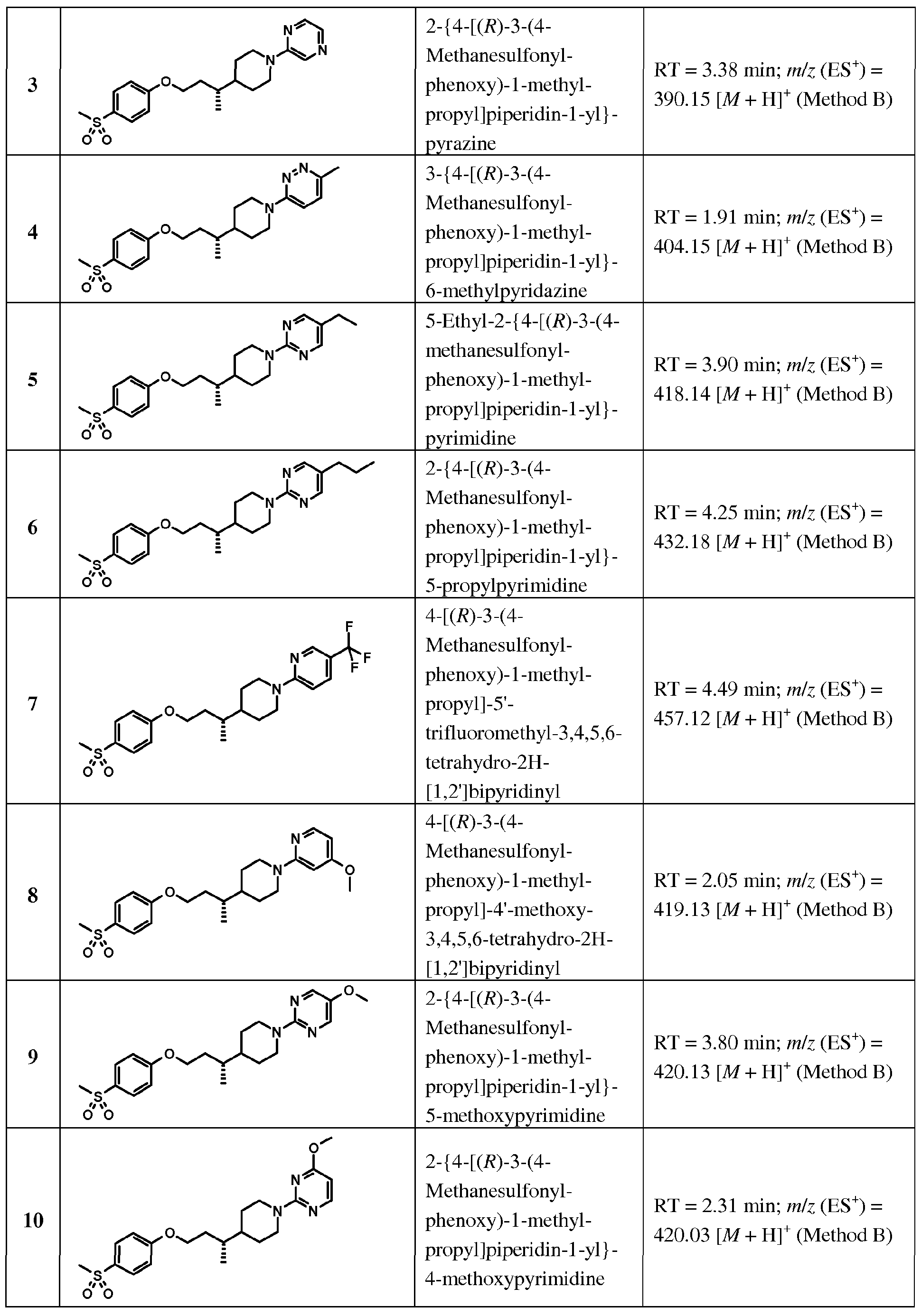




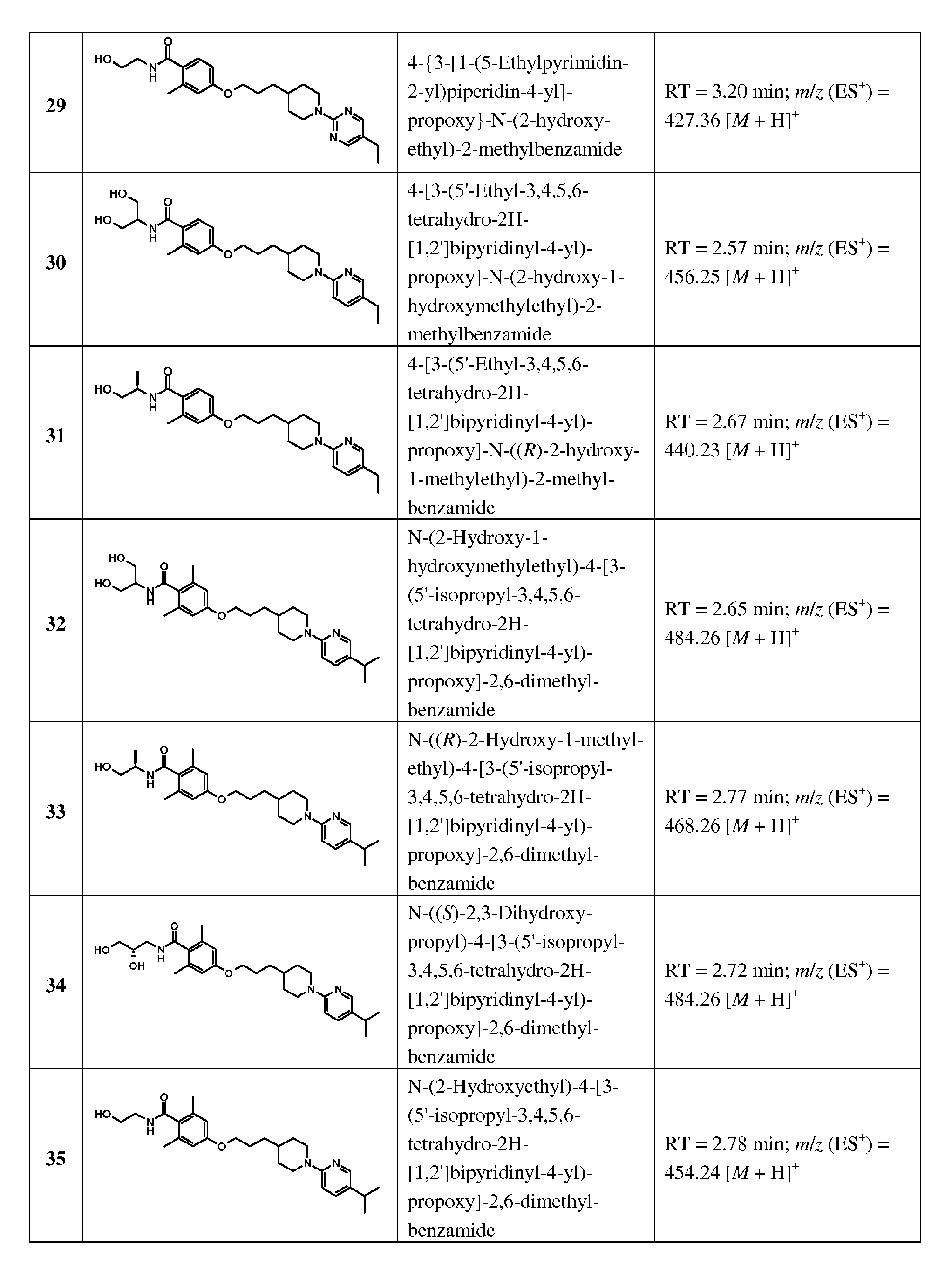
















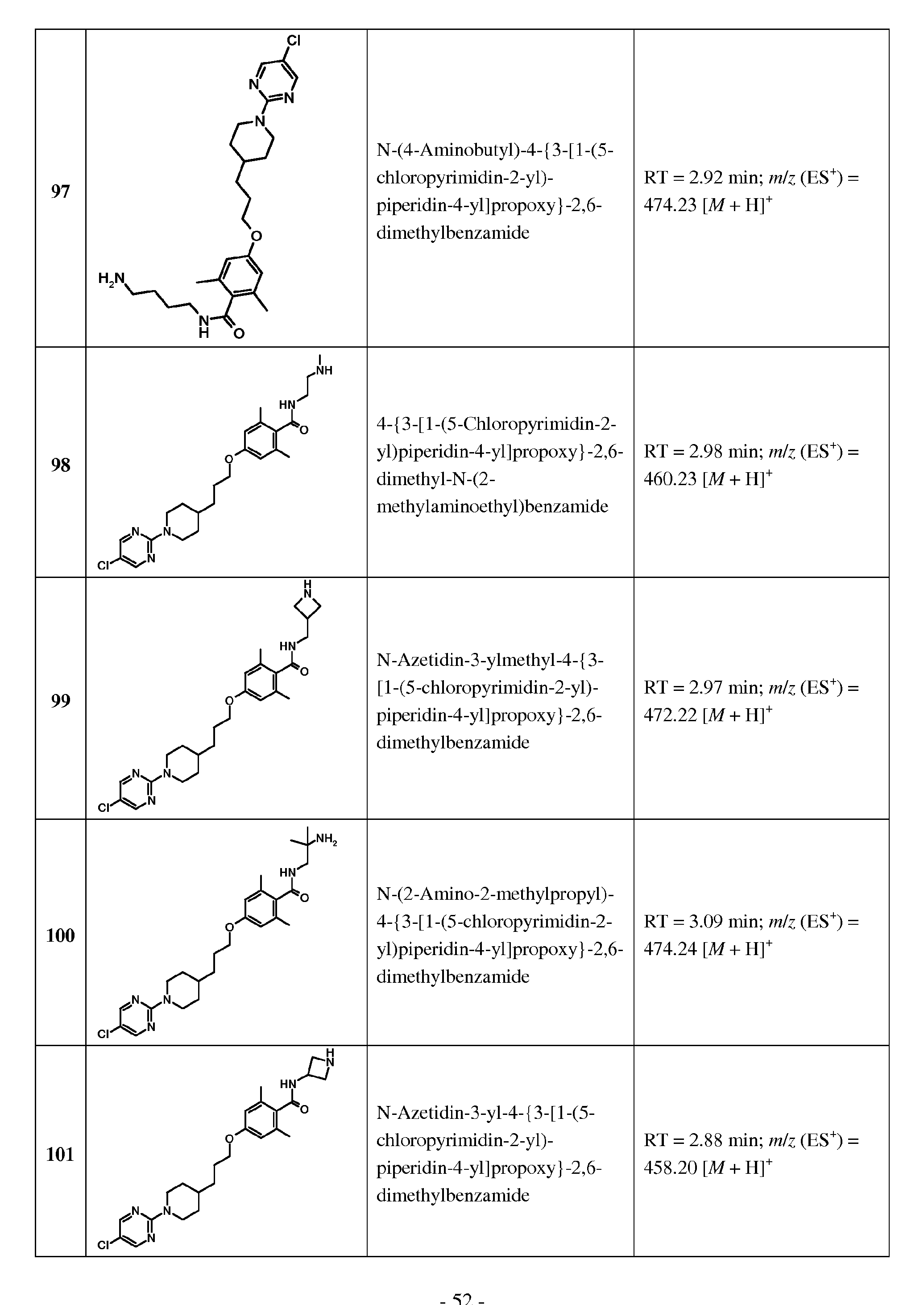




















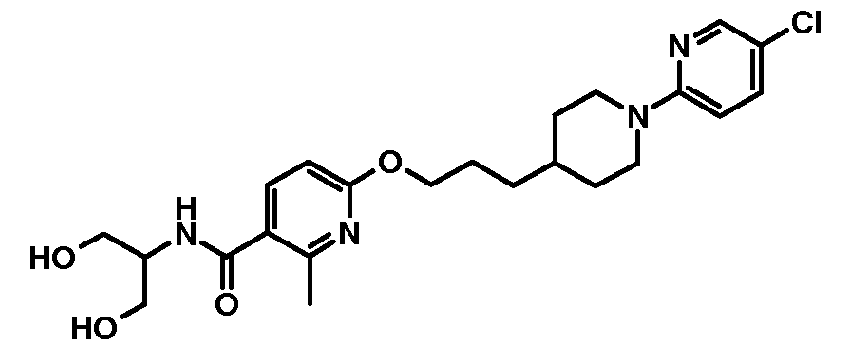








Claims

Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011517245A JP2011527331A (en) | 2008-07-10 | 2009-07-10 | Piperidinyl GPCR agonist |
| EA201170151A EA201170151A1 (en) | 2008-07-10 | 2009-07-10 | ПИПЕРИДИНИЛОВЫЕ АГОНИСТЫ GPCR |
| EP09785303A EP2321297A1 (en) | 2008-07-10 | 2009-07-10 | Piperidinyl gpcr agonists |
| CN2009801263533A CN102083813A (en) | 2008-07-10 | 2009-07-10 | Piperidinyl gpcr agonists |
| CA2728042A CA2728042A1 (en) | 2008-07-10 | 2009-07-10 | Piperidinyl gpcr agonists |
| AU2009269772A AU2009269772A1 (en) | 2008-07-10 | 2009-07-10 | Piperidinyl GPCR agonists |
| US13/003,546 US20110269734A1 (en) | 2008-07-10 | 2009-07-10 | Piperidinyl gpcr agonists |
| MX2011000060A MX2011000060A (en) | 2008-07-10 | 2009-07-10 | Piperidinyl gpcr agonists. |
| IL209978A IL209978A0 (en) | 2008-07-10 | 2010-12-13 | Piperidinyl gpcr agonists |
| ZA2010/08955A ZA201008955B (en) | 2008-07-10 | 2010-12-13 | Piperidinyl gpcr agonists |
| MA33508A MA32467B1 (en) | 2008-07-10 | 2011-01-10 | GPCR AGONISTS PIPERIDINYL DERIVATIVES |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0812646A GB0812646D0 (en) | 2008-07-10 | 2008-07-10 | Compounds |
| GB0812622.9 | 2008-07-10 | ||
| GB0812646.8 | 2008-07-10 | ||
| GB0812622A GB0812622D0 (en) | 2008-07-10 | 2008-07-10 | Compounds |
| GB0902391A GB0902391D0 (en) | 2009-02-13 | 2009-02-13 | Compounds |
| GB0902391.2 | 2009-02-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010004343A1 true WO2010004343A1 (en) | 2010-01-14 |
Family
ID=41070447
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2009/050825 WO2010004343A1 (en) | 2008-07-10 | 2009-07-10 | Piperidinyl gpcr agonists |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US20110269734A1 (en) |
| EP (1) | EP2321297A1 (en) |
| JP (1) | JP2011527331A (en) |
| KR (1) | KR20110036609A (en) |
| CN (1) | CN102083813A (en) |
| AU (1) | AU2009269772A1 (en) |
| CA (1) | CA2728042A1 (en) |
| CL (1) | CL2011000051A1 (en) |
| EA (1) | EA201170151A1 (en) |
| IL (1) | IL209978A0 (en) |
| MA (1) | MA32467B1 (en) |
| MX (1) | MX2011000060A (en) |
| PE (1) | PE20110329A1 (en) |
| WO (1) | WO2010004343A1 (en) |
| ZA (1) | ZA201008955B (en) |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010103335A1 (en) | 2009-03-12 | 2010-09-16 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| WO2010103333A1 (en) | 2009-03-12 | 2010-09-16 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| WO2010103334A1 (en) | 2009-03-12 | 2010-09-16 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| WO2011113947A1 (en) | 2010-03-18 | 2011-09-22 | Boehringer Ingelheim International Gmbh | Combination of a gpr119 agonist and the dpp-iv inhibitor linagliptin for use in the treatment of diabetes and related conditions |
| WO2011128395A1 (en) | 2010-04-14 | 2011-10-20 | Prosidion Limited | N- substituted 3-amino 4 - ( pyrrolidine - 1 - carbonyl) pyrrolidine and its derivatives for use in the treatment of metabolic disorders |
| WO2011128394A1 (en) | 2010-04-14 | 2011-10-20 | Prosidion Limited | 3-substituted 5-(pyrrolidine-1-carbonyl) pyrrolidine and its derivatives for use in the treatment of metabolic disorders |
| WO2011147951A1 (en) | 2010-05-28 | 2011-12-01 | Prosidion Limited | Cycloamino derivatives as gpr119 antagonists |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012066077A1 (en) | 2010-11-18 | 2012-05-24 | Prosidion Limited | 1,4 di substituted pyrrolidine - 3 - yl -amine derivatives and their use for the treatment of metabolic disorders |
| US8293729B2 (en) | 2009-06-24 | 2012-10-23 | Boehringer Ingelheim International Gmbh | Compounds, pharmaceutical composition and methods relating thereto |
| WO2013026587A1 (en) | 2011-08-22 | 2013-02-28 | Prosidion Limited | 1,4 disubstituted pyrrolidine - 3 - yl -amine derivatives and their use for the treatment of metabolic disorders |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8481731B2 (en) | 2009-06-24 | 2013-07-09 | Boehringer Ingelheim International Gmbh | Compounds, pharmaceutical composition and methods relating thereto |
| US8822430B2 (en) | 2009-05-13 | 2014-09-02 | Gilead Pharmasset Llc | Antiviral compounds |
| US8957062B2 (en) | 2011-04-08 | 2015-02-17 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment |
| US9006228B2 (en) | 2011-06-16 | 2015-04-14 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds, and methods of treatment |
| US9018200B2 (en) | 2011-10-24 | 2015-04-28 | Merck Sharp & Dohme Corp. | Substituted piperidinyl compounds useful as GPR119 agonists |
| US9018224B2 (en) | 2011-11-15 | 2015-04-28 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds useful as GPR119 agonists |
| US9206217B2 (en) | 2009-05-20 | 2015-12-08 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US9284342B2 (en) | 2009-05-20 | 2016-03-15 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US9393256B2 (en) | 2011-09-16 | 2016-07-19 | Gilead Pharmasset Llc | Methods for treating HCV |
| US9422266B2 (en) | 2011-09-30 | 2016-08-23 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment |
| WO2018068295A1 (en) * | 2016-10-14 | 2018-04-19 | Merck Sharp & Dohme Corp. | ARYL AND HETEROARYL ETHER DERIVATIVES AS LIVER X RECEPTOR β AGONISTS, COMPOSITIONS, AND THEIR USE |
| US10039779B2 (en) | 2013-01-31 | 2018-08-07 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US10086011B2 (en) | 2013-08-27 | 2018-10-02 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US12037340B2 (en) | 2021-05-21 | 2024-07-16 | Gilead Sciences, Inc. | Pentacyclic derivatives as Zika virus inhibitors |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20110053949A1 (en) * | 2008-04-17 | 2011-03-03 | Pfizer Inc. | 4-[3-(aryloxy)benzylidene]-3-methyl piperidine aryl carboxamide compounds useful as faah inhibitors |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007003962A2 (en) * | 2005-06-30 | 2007-01-11 | Prosidion Limited | Gpcr agonists |
-
2009
- 2009-07-10 PE PE2011000013A patent/PE20110329A1/en not_active Application Discontinuation
- 2009-07-10 EA EA201170151A patent/EA201170151A1/en unknown
- 2009-07-10 JP JP2011517245A patent/JP2011527331A/en not_active Withdrawn
- 2009-07-10 MX MX2011000060A patent/MX2011000060A/en not_active Application Discontinuation
- 2009-07-10 CN CN2009801263533A patent/CN102083813A/en active Pending
- 2009-07-10 EP EP09785303A patent/EP2321297A1/en not_active Withdrawn
- 2009-07-10 WO PCT/GB2009/050825 patent/WO2010004343A1/en active Application Filing
- 2009-07-10 CA CA2728042A patent/CA2728042A1/en not_active Abandoned
- 2009-07-10 KR KR1020117002904A patent/KR20110036609A/en not_active Application Discontinuation
- 2009-07-10 AU AU2009269772A patent/AU2009269772A1/en not_active Abandoned
- 2009-07-10 US US13/003,546 patent/US20110269734A1/en not_active Abandoned
-
2010
- 2010-12-13 ZA ZA2010/08955A patent/ZA201008955B/en unknown
- 2010-12-13 IL IL209978A patent/IL209978A0/en unknown
-
2011
- 2011-01-10 MA MA33508A patent/MA32467B1/en unknown
- 2011-01-10 CL CL2011000051A patent/CL2011000051A1/en unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007003962A2 (en) * | 2005-06-30 | 2007-01-11 | Prosidion Limited | Gpcr agonists |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010103333A1 (en) | 2009-03-12 | 2010-09-16 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| WO2010103334A1 (en) | 2009-03-12 | 2010-09-16 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| WO2010103335A1 (en) | 2009-03-12 | 2010-09-16 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| US8822430B2 (en) | 2009-05-13 | 2014-09-02 | Gilead Pharmasset Llc | Antiviral compounds |
| US9511056B2 (en) | 2009-05-13 | 2016-12-06 | Gilead Pharmasset Llc | Antiviral compounds |
| US9981955B2 (en) | 2009-05-13 | 2018-05-29 | Gilead Pharmasset Llc | Antiviral compounds |
| US9206217B2 (en) | 2009-05-20 | 2015-12-08 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US9284342B2 (en) | 2009-05-20 | 2016-03-15 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8293729B2 (en) | 2009-06-24 | 2012-10-23 | Boehringer Ingelheim International Gmbh | Compounds, pharmaceutical composition and methods relating thereto |
| US8481731B2 (en) | 2009-06-24 | 2013-07-09 | Boehringer Ingelheim International Gmbh | Compounds, pharmaceutical composition and methods relating thereto |
| WO2011113947A1 (en) | 2010-03-18 | 2011-09-22 | Boehringer Ingelheim International Gmbh | Combination of a gpr119 agonist and the dpp-iv inhibitor linagliptin for use in the treatment of diabetes and related conditions |
| WO2011128395A1 (en) | 2010-04-14 | 2011-10-20 | Prosidion Limited | N- substituted 3-amino 4 - ( pyrrolidine - 1 - carbonyl) pyrrolidine and its derivatives for use in the treatment of metabolic disorders |
| WO2011128394A1 (en) | 2010-04-14 | 2011-10-20 | Prosidion Limited | 3-substituted 5-(pyrrolidine-1-carbonyl) pyrrolidine and its derivatives for use in the treatment of metabolic disorders |
| WO2011147951A1 (en) | 2010-05-28 | 2011-12-01 | Prosidion Limited | Cycloamino derivatives as gpr119 antagonists |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012066077A1 (en) | 2010-11-18 | 2012-05-24 | Prosidion Limited | 1,4 di substituted pyrrolidine - 3 - yl -amine derivatives and their use for the treatment of metabolic disorders |
| US8957062B2 (en) | 2011-04-08 | 2015-02-17 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment |
| US9006228B2 (en) | 2011-06-16 | 2015-04-14 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds, and methods of treatment |
| WO2013026587A1 (en) | 2011-08-22 | 2013-02-28 | Prosidion Limited | 1,4 disubstituted pyrrolidine - 3 - yl -amine derivatives and their use for the treatment of metabolic disorders |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US9393256B2 (en) | 2011-09-16 | 2016-07-19 | Gilead Pharmasset Llc | Methods for treating HCV |
| US10456414B2 (en) | 2011-09-16 | 2019-10-29 | Gilead Pharmasset Llc | Methods for treating HCV |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US9422266B2 (en) | 2011-09-30 | 2016-08-23 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment |
| US9018200B2 (en) | 2011-10-24 | 2015-04-28 | Merck Sharp & Dohme Corp. | Substituted piperidinyl compounds useful as GPR119 agonists |
| US9018224B2 (en) | 2011-11-15 | 2015-04-28 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds useful as GPR119 agonists |
| US10039779B2 (en) | 2013-01-31 | 2018-08-07 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US10086011B2 (en) | 2013-08-27 | 2018-10-02 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US11116783B2 (en) | 2013-08-27 | 2021-09-14 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US11707479B2 (en) | 2013-08-27 | 2023-07-25 | Gilead Sciences, Inc. | Combination formulation of two antiviral compounds |
| WO2018068295A1 (en) * | 2016-10-14 | 2018-04-19 | Merck Sharp & Dohme Corp. | ARYL AND HETEROARYL ETHER DERIVATIVES AS LIVER X RECEPTOR β AGONISTS, COMPOSITIONS, AND THEIR USE |
| US11655216B2 (en) | 2016-10-14 | 2023-05-23 | Merck Sharp & Dohme Llc | Aryl and heteroaryl ether derivatives as liver X receptor beta agonists, compositions, and their use |
| US12037340B2 (en) | 2021-05-21 | 2024-07-16 | Gilead Sciences, Inc. | Pentacyclic derivatives as Zika virus inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102083813A (en) | 2011-06-01 |
| IL209978A0 (en) | 2011-02-28 |
| CL2011000051A1 (en) | 2012-03-16 |
| JP2011527331A (en) | 2011-10-27 |
| EP2321297A1 (en) | 2011-05-18 |
| CA2728042A1 (en) | 2010-01-14 |
| EA201170151A1 (en) | 2011-08-30 |
| US20110269734A1 (en) | 2011-11-03 |
| AU2009269772A1 (en) | 2010-01-14 |
| MA32467B1 (en) | 2011-07-03 |
| MX2011000060A (en) | 2011-02-22 |
| PE20110329A1 (en) | 2011-06-03 |
| ZA201008955B (en) | 2011-08-31 |
| KR20110036609A (en) | 2011-04-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2321297A1 (en) | Piperidinyl gpcr agonists | |
| EP2114935B1 (en) | Piperidine gpcr agonists | |
| EP2321308B1 (en) | Piperidine gpcr agonists | |
| EP2114931B1 (en) | Piperidine gpcr agonists | |
| EP2114933B1 (en) | Piperidine gpcr agonists | |
| WO2010004347A1 (en) | Heterocyclic gpcr agonists | |
| US8173807B2 (en) | Pyridine, pyrimidine and pyrazine derivatives as GPCR agonists | |
| EP2318399B1 (en) | Piperidinyl gpcr agonists | |
| WO2010004345A1 (en) | Piperidinyl gpcr agonists | |
| EP2321305A1 (en) | Heterocyclic gpcr agonists | |
| EP2114932A1 (en) | Piperidine gpcr agonists | |
| EP2215078A1 (en) | Azetidinyl g-protein coupled receptor agonists | |
| WO2009050522A1 (en) | Azetidinyl g-protein coupled receptor agonists | |
| CA2674360A1 (en) | Piperidine gpcr agonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980126353.3 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09785303 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 209978 Country of ref document: IL Ref document number: 2728042 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 8224/CHENP/2010 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2011/000060 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011010039 Country of ref document: EG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 590354 Country of ref document: NZ Ref document number: 000013-2011 Country of ref document: PE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011000051 Country of ref document: CL Ref document number: 2009269772 Country of ref document: AU Ref document number: 12011500059 Country of ref document: PH |
|
| ENP | Entry into the national phase |
Ref document number: 2011517245 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009785303 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2009269772 Country of ref document: AU Date of ref document: 20090710 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20117002904 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201170151 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201101486 Country of ref document: UA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13003546 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0915661 Country of ref document: BR Kind code of ref document: A2 Effective date: 20110106 |