WO2009144548A1 - Imidazo [2,1-b] purine derivatives as trpa1 modulators - Google Patents
Imidazo [2,1-b] purine derivatives as trpa1 modulators Download PDFInfo
- Publication number
- WO2009144548A1 WO2009144548A1 PCT/IB2009/005530 IB2009005530W WO2009144548A1 WO 2009144548 A1 WO2009144548 A1 WO 2009144548A1 IB 2009005530 W IB2009005530 W IB 2009005530W WO 2009144548 A1 WO2009144548 A1 WO 2009144548A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acetamide
- methyl
- imidazo
- oxo
- purin
- Prior art date
Links
- 0 CC(*1)=NC=C1c(cc1N)cc(N)c1O Chemical compound CC(*1)=NC=C1c(cc1N)cc(N)c1O 0.000 description 1
- UWDLUBUGDCCBFZ-UHFFFAOYSA-N CN(C1=NCCN1c1c2[n](CC(Nc3nc(-c(cc4)ccc4Br)c[s]3)=O)cn1)C2=O Chemical compound CN(C1=NCCN1c1c2[n](CC(Nc3nc(-c(cc4)ccc4Br)c[s]3)=O)cn1)C2=O UWDLUBUGDCCBFZ-UHFFFAOYSA-N 0.000 description 1
- AUCIAJVLLMGCOS-UHFFFAOYSA-O N#Cc(cc1)ccc1-c1c[s]c(NC(C[n]2c(C([NH2+]C3=NCCN33)=O)c3nc2)=O)n1 Chemical compound N#Cc(cc1)ccc1-c1c[s]c(NC(C[n]2c(C([NH2+]C3=NCCN33)=O)c3nc2)=O)n1 AUCIAJVLLMGCOS-UHFFFAOYSA-O 0.000 description 1
- MJXISBQXEHLYLE-UHFFFAOYSA-N OC(C[n]1c(C(NC2=NCCN22)=O)c2nc1)NC1SC=C(c(cc2Cl)cc(Cl)c2OCc2c(C(F)(F)F)cccc2)N1 Chemical compound OC(C[n]1c(C(NC2=NCCN22)=O)c2nc1)NC1SC=C(c(cc2Cl)cc(Cl)c2OCc2c(C(F)(F)F)cccc2)N1 MJXISBQXEHLYLE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/18—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 one oxygen and one nitrogen atom, e.g. guanine
Definitions
- the present invention relates to novel TRPAl (Transient Receptor Potential subfamily A, member 1) modulators, in particular TRPAl antagonists and uses thereof for treating diseases, conditions and/or disorders modulated by TRPAl.
- TRPAl Transient Receptor Potential subfamily A, member 1
- TRP Transient Receptor Potential
- TRPC canonical
- TRPV vanilloid
- TRPM melastatin
- TRPP polycystin
- TRPML mucolipin
- TRPA ankyrin, ANKTMl
- TRPN TRPN
- TRPV5 and TRPV6 are more closely related to each other than to TRPVl, TRPV2, TRPV3, or TRPV4.
- TRPAl is most closely related to TRPV3, and is more closely related to TRPVl and TRPV2 than to TRPV5 and TRPV6.
- the TRPM family has 8 members.
- Constituents include the following: the founding member TRPMl (melastatin or LTRPCl), TRPM3 (KIAAl 616 or LTRPC3), TRPM7 (TRP-PLIK, ChaK(l), LTRPC7), TRPM6 (ChaK2), TRPM2 (TRPC7 or LTRPC2), TRPM8 (TRP-p8 or CMRl), TRPM5 (MTRl or LTRPC5), and TRPM4 (FLJ20041 or LTRPC4).
- TRPMl melastatin or LTRPCl
- TRPM3 KAAl 616 or LTRPC3
- TRPM7 TRP-PLIK, ChaK(l), LTRPC7
- TRPM6 ChoK2
- TRPM2 TRPC7 or LTRPC2
- TRPM8 TRP-p8 or CMRl
- TRPM5 MMRl or LTRPC5
- TRPM4 FLJ20041 or LTRPC4
- TRPP family consists of two groups of channels: those predicted to have six transmembrane domains and those that have eleven.
- TRPP2 PPD2
- TRPP3 PPD2L1
- TRPP5 PPD2L2
- TRPPl PPDl, PCl
- PKD-REJ PKD-REJ
- PKD-ILl The sole mammalian member of the TRPA family is ANKTMl. It is believed TRPAl is expressed in nociceptive neurons. The nociceptive neurons of the nervous system sense the peripheral damage and transmit a pain signal.
- TRPAl is activated by a variety of noxious stimuli, including cold temperatures (activated at 17°C), pungent natural compounds (e.g., mustard, cinnamon and garlic), and environmental irritants (MacPherson LJ et al, Nature, (2007), 445; 541-545). Noxious compounds activate TRPAl ion channels through covalent modification of cysteines. TRPAl is membrane bound and most likely acts as a heterodimeric voltage gated channel. It is believed to have a particular secondary structure: its N-terminus is lined with a large number of ankyrin repeats which are believed to form a spring-like edifice.
- TRPAl responds to a variety of stimuli it works through different system. It forms covalently linked adducts with electrophilic compounds. The difference with other TRP receptors is that TRPAl ligand binding persists for hours. The physiological response (e.g., pain) is greatly prolonged. Hence to dissociate the electrophile an effective antagonist is required.
- WO 2009/002933, WO 2008/0949099, WO 2007/073505, WO 2004/055054, and WO 2005/089206 describe the TRP channels as the targets for the treatment of pain and related conditions.
- R 1 , R 2 and R 3 independently represents hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted heterocyclic ring or substituted or unsubstituted heterocyclylalkyl;
- R 4 and R 5 independently represents hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkylalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted heterocyclic ring or substituted or unsubstituted heterocyclylalkyl;
- the compounds of formula I may involve one or more embodiments. It is to be understood that the embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified.
- R 1 is alkyl. In this embodiment, preferably R 1 is methyl.
- R 5 is substituted or unsubstituted aryl, preferably phenyl.
- R 5 is substituted or unsubstituted heteroaryl, preferably thiazole.
- R 5 is substituted or unsubstituted alkylaryl, preferably ethylphenyl and the substituent is halogen.
- R 6 represents hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkylalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted heterocyclic ring and substituted or unsubstituted heterocyclylalkyl;
- R 7 independently represents hydrogen or alkyl
- R 6 and R 7 together with the carbon atoms which they are attached to, form a optionally substituted 3 to 7 saturated, unsaturated or partially saturated cyclic ring, which may optionally include one or more heteroatoms selected from O, N or S;
- R 7 is alkyl preferably methyl.
- R 6 is substituted or unsubstituted aryl, preferably phenyl, wherein one or more substituent is independently selected from halogen (for eg., F, Cl or Br), cyano, alkyl (for eg., methyl, ethyl, n-pentyl, n-hexyl, or zso-butyl), cycloalkyl (for eg., cyclohexyl), haloalkyl (for eg., trifluoromethyl), fully or partially substituted haloalkyloxy (for eg., trifluoromethyloxy, OCHF 2 , OCH 2 CF 3 , OCH 2 CH 2 CF 3 , or OCH 2 CH 2 CF 2 CF 3 ), alkoxy (for eg., wo-pentyloxy or weo-pentyloxy), cycloalkyl
- alkylaryl for eg., CH 3 Ph or (CH 3 ) 3 C-Ph
- alkylalkynyl for eg., (CH 3 )3C-C ⁇ C-
- heteroaryl for eg., thiophene
- halophenyl chlorophenyl
- haloalkylphenylalkoxy for eg., trifluoroalkyl benzyloxy
- R 6 is aryl, wherein aryl is fully or partially aromatic, preferably partially aromatic.
- R 6 is tetrahydronaphthalene.
- substituents on carbocyclic ring is alkyl for eg., tert- butyl.
- R a is independently selected from hydrogen, halogen, cyano, haloalkyl, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkyloxy, substituted or unsubstituted cycloalkylalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted arylalkoxy, substituted or unsubstituted heteroaryl, substituted or un
- R a is selected from one or more halogen and/or haloalkyl (for eg. trifluoromethyl).
- R a is selected from substituted or unsubstitued aryloxy (for eg. phenyloxy) and substituted or unsubstituted aryl (for eg. phenyl), and the substitutent is alkyl (for eg. methyl or tert.-buty ⁇ ) or cyclopentyloxy.
- R a is selected from substituted or unsubstitued aryloxy (for eg. phenyloxy) and substituted or unsubstituted aryl (for eg. phenyl), and the substitutent is alkyl (for eg. methyl or tert.-buty ⁇ ) or cyclopentyloxy.
- the present patent application also provides a pharmaceutical composition that includes at least one compound described herein and at least one pharmaceutically acceptable excipient (such as a pharmaceutically acceptable carrier or diluent).
- the pharmaceutical composition comprises a therapeutically effective amount of at least one compound described herein.
- the compounds described in the present patent application may be associated with a pharmaceutically acceptable excipient (such as a carrier or a diluents) or be diluted by a carrier, or enclosed within a carrier which can be in the form of a capsule, sachet, paper or other container.
- the compounds and pharmaceutical compositions of the present invention are useful for modulating TRPAl receptors, which modulation is believed to be related to a variety of disease states.
- the present patent application further provides a method of inhibiting TRPAl receptors in a subject in need thereof by administering to the subject one or more compounds described herein in an amount effective to cause inhibition of such receptor.
- alkyl refers to a straight or branched hydrocarbon chain radical consisting solely of carbon and hydrogen atoms, containing no unsaturation, having from one to eight carbon atoms, and which is attached to the rest of the molecule by a single bond, e.g., methyl, ethyl, n-propyl, 1-methylethyl (isopropyl), n-butyl, n-pentyl, and 1,1- dimethylethyl (tert-buty ⁇ ).
- Ci -6 alkyl refers to an alkyl chain having 1 to 6 carbon atoms.
- alkenyl refers to an aliphatic hydrocarbon group containing a carbon- carbon double bond and which may be a straight or branched chain having 2 to about 10 carbon atoms, e.g., ethenyl, 1-propenyl, 2-propenyl (allyl), iso-propenyl, 2-methyl-l- propenyl, 1-butenyl, and 2-butenyl.
- alkynyl refers to a straight or branched chain hydrocarbyl radical having at least one carbon-carbon triple bond, and having 2 to about 12 carbon atoms (with radicals having 2 to about 10 carbon atoms being preferred), e.g., ethynyl, propynyl, and butynyl.
- alkoxy denotes an alkyl group attached via an oxygen linkage to the rest of the molecule. Representative examples of such groups are -OCH 3 and -OC 2 Hs.
- halogen or halo includes fluorine, chlorine, bromine, or iodine.
- haloalkyl is used to denote a group comprised of an alkyl group substituted with halogen atom, where alkyl group is as defined above and halogen is used to denote fluorine, chlorine, bromine or iodine, an example of such group is trifiuoromethyl, difluoromethyl.
- cycloalkyl denotes a non-aromatic mono or multicyclic ring system of 3 to about 12 carbon atoms, such as cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- multicyclic cycloalkyl groups include, but are not limited to, perhydronapththyl, adamantyl and norbornyl groups, bridged cyclic groups or sprirobicyclic groups, e.g., sprio (4,4) non-2-yl.
- cycloalkylalkyl refers to a cyclic ring-containing radical having 3 to about 8 carbon atoms directly attached to an alkyl group.
- the cycloalkylalkyl group may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure.
- Non-limiting examples of such groups include cyclopropylmethyl, cyclobutylethyl, and cyclopentylethyl.
- cycloalkenyl refers to a cyclic ring-containing radical having 3 to about 8 carbon atoms with at least one carbon-carbon double bond, such as cyclopropenyl, cyclobutenyl, and cyclopentenyl.
- aryl refers to an aromatic radical having 6 to 14 carbon atoms such as phenyl, naphthalenyl, thienyl, tetrahydronapthenyl, tetrahydrobenzothiazolyl indanyl, and biphenyl.
- arylalkyl refers to an aryl group as defined above directly bonded to an alkyl group as defined above, e.g., -CH 2 C 6 Hs or -C 2 H 5 C 6 H 5 .
- heterocyclic ring refers to a stable 3- to 15-membered ring radical which consists of carbon atoms and from one to five heteroatoms selected from nitrogen, phosphorus, oxygen and sulfur.
- the heterocyclic ring radical may be a monocyclic, bicyclic or tricyclic ring system, which may include fused, bridged or spiro ring systems, and the nitrogen, phosphorus, carbon, oxygen or sulfur atoms in the heterocyclic ring radical may be optionally oxidized to various oxidation states.
- the nitrogen atom may be optionally quaternized; and the ring radical may be partially or fully saturated (i.e., heterocyclic or heteroaryl).
- heterocyclic ring radicals include, but are not limited to, azetidinyl, acridinyl, benzodioxolyl, benzodioxanyl, benzofurnyl, carbazolyl, cinnolinyl, dioxolanyl, indolizinyl, naphthyridinyl, perhydroazepinyl, phenazinyl, phenothiazinyl, phenoxazinyl, phthalazinyl, pyridyl, pteridinyl, purinyl, quinazolinyl, quinoxalinyl, quinolinyl, isoquinolinyl, tetrazoyl, imidazolyl, tetrahydroisouinolyl, piperidinyl, piperazinyl, 2- oxopiperazinyl, 2-oxopiperidinyl, 2-oxopyrrolidin
- heterocyclyl refers to a heterocyclic ring radical as defined above.
- the heterocyclyl ring radical may be attached to the main structure at any heteroatom or carbon atom that results in the creation of a stable structure.
- heterocyclylalkyl refers to a heterocyclic ring radical directly bonded to an alkyl group.
- the heterocyclylalkyl radical may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure.
- heteroaryl refers to an aromatic heterocyclic ring radical.
- the heteroaryl ring radical may be attached to the main structure at any heteroatom or carbon atom that results in the creation of a stable structure.
- heteroarylalkyl refers to a heteroaryl ring radical directly bonded to an alkyl group.
- the heteroarylalkyl radical may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure.
- the substituents in the aforementioned "substituted” groups cannot be further substituted.
- the substituent on “substituted alkyl” is "substituted aryl”
- the substituent on “substituted aryl” cannot be “substituted alkenyl”.
- protecting group refers to a substituent that is employed to block or protect a particular functionality while other functional groups on the compound may remain reactive.
- an "amino-protecting group” is a substituent attached to an amino group that blocks or protects the amino functionality in the compound. Suitable amino-protecting groups include, but are not limited to, acetyl, benzyl, trifluoroacetyl, t-butoxycarbonyl (BOC), benzyloxycarbonyl (CBz) and 9- fluorenylmethylenoxycarbonyl (Fmoc).
- a "hydroxy-protecting group” refers to a substituent of a hydroxy group that blocks or protects the hydroxy functionality.
- Suitable hydroxy-protecting groups include, but are not limited to, acetyl, benzyl, tetrahydropyranyl and silyl.
- a "carboxy-protecting group” refers to a substituent of the carboxy group that blocks or protects the carboxy functionality.
- Suitable carboxy- protecting groups include, but are not limited to, -CH 2 CH 2 SO 2 Ph, cyanoethyl, 2- (trimethylsilyl)ethyl, 2-(trimethylsilyl)ethoxymethyl, 2-(p-toluenesulfonyl)ethyl, 2-(p- nitrophenylsulfenyl)ethyl, 2-(diphenylphosphino)-ethyl, and nitroethyl.
- protecting .groups and their use T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
- treating or “treatment” of a state, disorder or condition includes:
- the benefit to a subject to be treated is either statistically significant or at least perceptible to the subject or to the physician.
- a “therapeutically effective amount” means the amount of a compound that, when administered to a subject for treating a state, disorder or condition, is sufficient to effect such treatment.
- the “therapeutically effective amount” will vary depending on the compound, the disease and its severity and the age, weight, physical condition and responsiveness of the subject to be treated.
- the compound described in the present patent application may form salts.
- Non- limiting examples of pharmaceutically acceptable salts forming part of this patent application include salts derived from inorganic bases salts of organic bases salts of chiral bases, salts of natural amino acids and salts of non-natural amino acids.
- Certain compounds of present patent application are capable of existing in stereoisomeric forms (e.g. diastereomers and enantiomers). With respect to the overall compounds described by the Formula (I), the present patent application extends to these stereoisomeric forms and to mixtures thereof.
- the pharmaceutical composition provided in the present patent application includes at least one compound described herein and at least one pharmaceutically acceptable excipient (such as a pharmaceutically acceptable carrier or diluent).
- the contemplated pharmaceutical compositions include the compound(s) described herein in an amount sufficient to inhibit TRPAl receptor in a subject.
- the inhibitory activity of compounds falling within the formula (I) may be measured by an assay provided herein below.
- the subjects contemplated include, for example, a living cell and a mammal, including human mammal.
- the compound of the present patent application may be associated with a pharmaceutically acceptable excipient (such as a carrier or a diluent) or be diluted by a carrier, or enclosed within a carrier which can be in the form of a capsule, sachet, paper or other container.
- suitable carriers include, but are not limited to, water, salt solutions, alcohols, polyethylene glycols, polyhydroxyethoxylated castor oil, peanut oil, olive oil, gelatin, lactose, terra alba, sucrose, dextrin, magnesium carbonate, sugar, cyclodextrin, amylose, magnesium stearate, talc, gelatin, agar, pectin, acacia, stearic acid or lower alkyl ethers of cellulose, silicic acid, fatty acids, fatty acid amines, fatty acid monoglycerides and diglycerides, pentaerythritol fatty acid esters, polyoxyethylene, hydroxymethyl- cellulose and polyvinylpyrrolidone.
- the carrier or diluent may include a sustained release material, such as glyceryl monostearate or glyceryl distearate, alone or mixed with a wax.
- the pharmaceutical composition may also include one or more pharmaceutically acceptable auxiliary agents, wetting agents, emulsifying agents, suspending agents, preserving agents, salts for influencing osmetic pressure, buffers, sweetening agents, flavoring agents, colorants, or any combination of the foregoing.
- the pharmaceutical composition may be formulated so as to provide quick, sustained, or delayed release of the active ingredient after administration to the subject by employing procedures known in the art.
- compositions may be prepared by conventional techniques known in the art (Remington: The Science and Practice of Pharmacy, 20 th Ed., 2003, Lippincott Williams & Wilkins).
- the active compound can be mixed with a carrier, or diluted by a carrier, or enclosed within a carrier, which may be in the form of an ampoule, capsule, sachet, paper, or other container.
- the carrier serves as a diluent, it may be a solid, semi-solid, or liquid material that acts as a vehicle, excipient, or medium for the active compound.
- the active compound can be adsorbed on a granular solid container, for example, in a sachet.
- compositions may be in conventional forms, for example, capsules, tablets, aerosols, solutions, suspensions or products for topical application.
- the route of administration may be any route which effectively transports the active compound of the invention to the appropriate or desired site of action.
- Suitable routes of administration include, but are not limited to, oral, nasal, pulmonary, buccal, subdermal, intradermal, transdermal, parenteral, rectal, depot, subcutaneous, intravenous, intraurethral, intramuscular, intranasal, ophthalmic (such as with an ophthalmic solution) or topical (such as with a topical ointment).
- the oral route. is preferred.
- Solid oral formulations include, but are not limited to, tablets, capsules (soft or hard gelatin), dragees (containing the active ingredient in powder or pellet form), troches and lozenges. Tablets, dragees, or capsules having talc and/or a carbohydrate carrier or binder or the like are particularly suitable for oral application. Preferable carriers for tablets, dragees, or capsules include lactose, cornstarch, and/or potato starch. A syrup or elixir can be used in cases where a sweetened vehicle can be employed.
- Liquid formulations include, but are not limited to, syrups, emulsions, soft gelatin and sterile injectable liquids, such as aqueous or non-aqueous liquid suspensions or solutions.
- sterile injectable liquids such as aqueous or non-aqueous liquid suspensions or solutions.
- injectable solutions or suspensions preferably aqueous solutions with the active compound dissolved in polyhydroxylated castor oil.
- the present patent application provides compounds and pharmaceutical formulations thereof that are useful in the treatment of diseases, conditions and/or disorders modulated by TRPAl antagonists.
- the compounds described herein and pharmaceutical formulations thereof have TRPAl activity and are believed to be of potential use for the treatment or prophylaxis of certain diseases or disorders mediated or associated with the activity of TRPAl receptor, including disorders such as pain, chronic pain, complex regional pain syndrome, neuropathic pain, postoperative pain, rheumatoid arthritic pain, osteoarthritic pain, back pain, visceral pain, cancer pain, algesia, neuralgia, migraine, neuropathies, diabetic neuropathy, sciatica, HIV-related neuropathy, postherpetic neuralgia, fibromyalgia, nerve injury, ischaemia, neurodegeneration, stroke, post stroke pain, multiple sclerosis, respiratory diseases, asthma, cough, COPD, inflammatory disorders, oesophagitis, gastroeosophagal reflux disorder (GERD), irritable bowel syndrome,
- TRPAl The connection between therapeutic effect and inhibition of TRPAl is illustrated, for example, in Story GM et al, Cell, (2003), JJ2, 819-829; McMahon SB and Wood JN, Cell, (2006), 124, 1123-1125; Voorhoeve PM et al, Cell, (2006), 124, 1169-1181; Horbach U, Niemeyer BA and Flockerzi V, Biology of the Cell, (2004), 96, 47-54; Dayne YO, Albert YH & Michael X, Expert Opinion on Therapeutic Targets (2007), 11(3), 391-401 and the references cited therein.
- Diamines of formula (2) are converted to compounds of formula (3) by reductive amination with benazaldehyde and sodium cyanoborohydride followed by cyclization with triethyl orthoformate.
- Compound of formula (3) is converted to a compound of a formula (5) by reaction with phosphorous oxychloride followed by addition of suitable amino ethanol of the formula (4) (wherein R 2 and R 3 are hydrogen).
- the 2-haloacetamides of formula (7a) (Scheme 2) (wherein R b is as described above in description) required for the synthesis of compounds of the present invention can be prepared according to methods known to one skilled in the art (Carroll, L. et al. J. Am. Chem. Soc. (1950), 72, 3722-3725; Ohkubo, M. et al; Chem. Pharm. Bull (1995), 43 (9), 1497-1504).
- acylation of an amine of formula (12) with bromoacetyl bromide in the presence of a suitable base such as triethylamine or pyridine gives N- substituted bromoacetamide of the general formula (7a) (Scheme 2).
- this transformation can be carried out using excess thionyl chloride.
- the acid chloride of formula (9) was converted to corresponding Weinreb amide of formula (10) by treating with N,O-dimethyl hydroxylamine hydrochloride in the presence of a suitable base such as triethylamine. Addition of methyl magnesium iodide to Weinreb amide of formula (10) gives acetophenone derivative of the formula (11).
- the aryl alkyl ketone of the formula (11) is converted to 2-aminothiazole of the formula (12) in one step by its reaction with thiourea in the presence of iodine in ethanol.
- This conversion is similar to the one described by Carroll, K. et al. J. Am. Chem. Soc. (1950), 3722; and ⁇ aik, S., J.; Halkar, U. P. ARKIVOC (2005) xiii, 141-149.
- 2-aminothiazoles of the formula (12) can be prepared by the reaction of compounds of formula (11) with bromine in acetic acid to give the alpha halo intermediate, which on reaction with thiourea in THF at reflux condition give compounds of the formula (12).
- the compound of the formula (12) is transferred to (2-bromo-N-l,3- thiazol-2-yl)acetamide of the formula (7a) by acylation with bromoacetyl bromide in the presence of a suitable base such as pyridine or triethylamine as base in a suitable solvent such as THF.
- 2-Bromoacetamide derivatives of general formula (7a) used in the coupling reactions are prepared by acylation of appropriate amines with bromoacetyl bromide as shown in 'General Methods of preparation'. Detailed experimental procedure and characterization data for selected 2-bromoacetamides is given below.
- Step 1 intermediate (18 g, 105.803 mmol) in 12.5 % aqueous NH 4 OH (500 ml) was prepared by heating at 70 0 C.
- Sodium hydrosulphite 36.84 g, 211.602 mmol was added in small portions with vigorous stirring over a period of 20 min. The volume of the solution was reduced under reduced pressure until the product started to crystallize out.
- Step 2 intermediate To a suspension of Step 2 intermediate (8.16 g, 55.155 mmol) in water (80 ml) was added acetic acid (0.8 ml) followed by benzaldehyde (8.32 g, 78.411 mmol) over 15 min at room temperature.
- Step 3 intermediate 8 g, 32.487 mmol
- triethyl orthoformate 40 ml
- the reaction mixture was cooled to room temperature, solid formed was filtered, washed with diethyl ether (40 ml), and dried to give 7.65 g (91%) of the product as a pale yellow solid; 3108, 2877, 1708, 1675, 1413, 1214, 1009, 718 cm- 1 ; 1 H NMR (300 MHz, DMSO-J 6 ) ⁇ 3.14 (s, 3H), 5.42 (s, 2H), 7.29 (s, 5H), 8.13 (s, IH), 11.84 (br s, IH).
- Step 5 7-Benzyl-2-chloro-l-methyl-l,7-dihydro-6H-purin-6-one
- Step 4 intermediate (7.6 g, 29.658 mmol) and POCl 3 (200 ml) was heated to reflux overnight. Excess POCl 3 was removed under reduced pressure and the residue was partitioned between ethyl acetate (200 ml) and aqueous saturated NaHCO 3 (100 ml) and the organic layer was washed with brine (100 ml), dried (Na 2 SO 4 ) and concentrated under reduced pressure to afford 7.1 g (86%) of the product as an off- white solid; IR (KBr) 3098, 1694, 1566, 1392, 1226, 1051, 697 cm “1 ; 1 HNMR (300 MHz, DMSO-J 6 ) ⁇ 3.58 (s, 3H), 5.54 (s, 2H), 7.30 (s, 5H), 8.44 (s, IH); ESI-MS (m/z) 275.35 (M+H) + .
- Step_6 3-Benzyl-5-methyl-7,8
- Step 5 intermediate (280 mg, 1.021 mmol) in CH 3 CN (10 ml) was added triethylamine (213 ⁇ l, 1.531 mmol) and ethanolamine (218 mg, 3.571 mmol) at room temperature. The resulting solution was heated to reflux overnight. After this time, the reaction mixture was concentrated under reduced pressure and the residue was partitioned between ethyl acetate (100 ml) and water (50 ml).
- Step 6 intermediate 160 mg, 0.571 mmol
- 20% palladium hydroxide on carbon 176 mg, 1.254 mmol
- MeOH 20 ml
- Step 1 A mixture of appropriate aryl alkyl ketone (1.0 equiv.), thiourea (2.0 eqiv.) and iodine (1.0 equiv.) in dry ethanol (5 volumes) was refluxed for 24h. The reaction mixture was diluted with ethyl acetate and washed with saturated solution of sodium thiosulphate.
- the organic layer was treated with IN HCl and the precipitated salt collected by filtration.
- the salt was then treated with saturated solution OfNaHCO 3 and extracted with dichloromethane, washed with brine, dried over sodium sulfate and the solvent was evaporated to afford the 2-aminothiazole derivative.
- Step 2 To a stirred and cooled (O 0 C) solution of appropriate amine (1.0 equiv.) and pyridine (1.2 equiv.) in dichloromethane (5 volume) was added bromoacetyl bromide (1.2 eq.) over 5 min and the resulting mixture was allowed to warm to room temperature and then further stirred at room temperature for 2 h. The reaction mixture was diluted with dichloromethane (50 ml) and water (50 ml). The layers were separated.
- the aqueous layer was extracted with dichloromethane (2 x 50 ml) and the combined organic layers were washed with water (2 x 50 ml) followed by brine (50 ml), dried (Na 2 SO 4 ) and filtered. The filtrate was concentrated under reduced pressure. The residue obtained after the evaporation of the solvent was purified by silica gel column chromatography using 5-10% ethyl acetate in petroleum ether to obtain the desired product as an off-white solid.
- Examples 1-57 were prepared by coupling Intermediate 1 with appropriate N- substituted bromoacetamides of the formula (7a) (Scheme 2) in the presence of a suitable base using DMF as solvent.
- the illustrative examples of the present invention are screened for TRPAl activity according to a modified procedure described in (a) T ⁇ th, A., Kedei, N., Wang, Y. and Blumberg, P. M. Life Sciences (2003), 73, 487-498. (b) McNamara C, R. et al. (2007) PNAS 104, 13525-13530.
- the screening of the compounds can be carried out by other methods and procedures known to persons skilled in the art.
- TRPAl receptor activation was followed as inhibition of allyl isothiocyanate (AITC) induced cellular uptake of radioactive calcium.
- Test compounds were dissolved in DMSO to prepare 10 mM stock solution and then diluted using plain medium with 0.1% BSA and 1.8 mM CaCl 2 to get desired concentration. Final concentration of DMSO in the reaction was 0.5% (v/v).
- Human TRPAl expressing CHO cells were grown in F- 12 DMEM medium with 10% FBS, 1% penicillin-streptomycin solution, 400 ⁇ g / ml of G-418. Cells were seeded 24 h prior to the assay in 96 well plates so as to get ⁇ 50,000 cells per well on the day of experiment.
- IC 50 (nM) values of the compounds are set forth in Table 2 wherein "A” refers to an IC 50 value of less than 500 nM, “B” refers to IC 50 value in range of 500.1 to 1000.0 nM and “C” refers to an IC 50 value of more than 1000 nM
Abstract
The present invention provides TRPA (Transient Receptor Potential subfamily A) modulators. In particular, compounds described herein are useful for treating or preventing diseases, conditions and/or disorders modulated by TRPAl (Transient Receptor Potential subfamily A, member 1) modulators. Also provided herein are processes for preparing compounds described herein, intermediates used in their synthesis, pharmaceutical compositions thereof, and methods for treating or preventing diseases, conditions and/or disorders modulated by TRPAl.
Description
IMIDAZO[2,1-£]PURINE DERIVATIVES AS TRPAl MODULATORS
Related applications
This application claims the benefit of Indian Patent Application Nos. 1138/MUM/2008 filed on May 28, 2008; 2511/MUM/2008 filed on Dec 1, 2008 and US Provisional Application Nos 61/074,115 filed on Jun 19, 2008 and 61/138,431 filed on Dec 17, 2008 all of which are hereby incorporated by reference.
Technical Field
The present invention relates to novel TRPAl (Transient Receptor Potential subfamily A, member 1) modulators, in particular TRPAl antagonists and uses thereof for treating diseases, conditions and/or disorders modulated by TRPAl.
Background
The Transient Receptor Potential (TRP) channels or receptors are pain receptors. They have been classified into seven subfamilies: TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin), TRPA (ankyrin, ANKTMl) and TRPN (NOMPC) families. The TRPC family can be divided into 4 subfamilies (i) TRPCl (ii) TRPC2 (iii) TRPC3, TRPC6, TRPC7 and (iv) TRPC4, TRPC5 based on sequence functional similarities. Currently the TRPV family has 6 members. TRPV5 and TRPV6 are more closely related to each other than to TRPVl, TRPV2, TRPV3, or TRPV4. TRPAl is most closely related to TRPV3, and is more closely related to TRPVl and TRPV2 than to TRPV5 and TRPV6. The TRPM family has 8 members. Constituents include the following: the founding member TRPMl (melastatin or LTRPCl), TRPM3 (KIAAl 616 or LTRPC3), TRPM7 (TRP-PLIK, ChaK(l), LTRPC7), TRPM6 (ChaK2), TRPM2 (TRPC7 or LTRPC2), TRPM8 (TRP-p8 or CMRl), TRPM5 (MTRl or LTRPC5), and TRPM4 (FLJ20041 or LTRPC4). The TRPML family consists of the mucolipins, which include TRPMLl (mucolipin 1), TRPML2 (mucolipin 2), and TRPML3 (mucolipin 3). The TRPP family consists of two groups of channels: those predicted to have six transmembrane domains and those that have eleven. TRPP2 (PKD2), TRPP3 (PKD2L1), TRPP5 (PKD2L2) are all predicted to have six transmembrane domains. TRPPl (PKDl, PCl), PKD-REJ and PKD-ILl are all thought to have eleven transmembrane domains. The sole mammalian member of the TRPA family is ANKTMl.
It is believed TRPAl is expressed in nociceptive neurons. The nociceptive neurons of the nervous system sense the peripheral damage and transmit a pain signal. TRPAl is activated by a variety of noxious stimuli, including cold temperatures (activated at 17°C), pungent natural compounds (e.g., mustard, cinnamon and garlic), and environmental irritants (MacPherson LJ et al, Nature, (2007), 445; 541-545). Noxious compounds activate TRPAl ion channels through covalent modification of cysteines. TRPAl is membrane bound and most likely acts as a heterodimeric voltage gated channel. It is believed to have a particular secondary structure: its N-terminus is lined with a large number of ankyrin repeats which are believed to form a spring-like edifice. Most receptors have intricate pockets which are specific to certain kind of ligand, and the slightest alteration of either the pocket or the ligand has drastic effects. Since TRPAl responds to a variety of stimuli it works through different system. It forms covalently linked adducts with electrophilic compounds. The difference with other TRP receptors is that TRPAl ligand binding persists for hours. The physiological response (e.g., pain) is greatly prolonged. Hence to dissociate the electrophile an effective antagonist is required.
WO 2009/002933, WO 2008/0949099, WO 2007/073505, WO 2004/055054, and WO 2005/089206 describe the TRP channels as the targets for the treatment of pain and related conditions.
In efforts to discover better analgesics for the treatment of both acute and chronic pain and to develop treatments for various neuropathic and nociceptive pain states, there exists a need for a more effective and safe therapeutic treatment of diseases, conditions and/or disorders modulated by TRPAl.
Summary The present patent application provides TRPAl modulators of formula (I)

R1, R2 and R3 independently represents hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted
or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted heterocyclic ring or substituted or unsubstituted heterocyclylalkyl;
R4 and R5 independently represents hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkylalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted heterocyclic ring or substituted or unsubstituted heterocyclylalkyl;
The compounds of formula I may involve one or more embodiments. It is to be understood that the embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified.
According to one embodiment, specifically provided are compounds of the formula (I) in which, R1 is alkyl. In this embodiment, preferably R1 is methyl.
According to one embodiment, specifically provided are compounds of the formula (I) in which, R2 and R3 is hydrogen.
According to one embodiment, specifically provided are compounds of the formula (I) in which, R4 is hydrogen.
According to one embodiment, specifically provided are compounds of the formula (I) in which, R5 is substituted or unsubstituted aryl, preferably phenyl.
According to one embodiment, specifically provided are compounds of the formula (I) in which, R5 is substituted or unsubstituted heteroaryl, preferably thiazole.
According to one embodiment, specifically provided are compounds of the formula (I) in which, R5 is substituted or unsubstituted alkylaryl, preferably ethylphenyl and the substituent is halogen.
According to one embodiment, specifically provided are compounds of the formula (Ia)

R6 represents hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkylalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted heterocyclic ring and substituted or unsubstituted heterocyclylalkyl;
R7 independently represents hydrogen or alkyl; and
R6 and R7 together with the carbon atoms which they are attached to, form a optionally substituted 3 to 7 saturated, unsaturated or partially saturated cyclic ring, which may optionally include one or more heteroatoms selected from O, N or S;
According to one embodiment, specifically provided are compounds of the formula (Ia) in which, R7 is hydrogen.
According to one embodiment, specifically provided are compounds of the formula (Ia) in which, R7 is alkyl preferably methyl.
According to one embodiment, specifically provided are compounds of the formula (Ia) in which, R6 is substituted or unsubstituted aryl, preferably phenyl, wherein one or more substituent is independently selected from halogen (for eg., F, Cl or Br), cyano, alkyl (for eg., methyl, ethyl, n-pentyl, n-hexyl, or zso-butyl), cycloalkyl (for eg., cyclohexyl), haloalkyl (for eg., trifluoromethyl), fully or partially substituted haloalkyloxy (for eg., trifluoromethyloxy, OCHF2, OCH2CF3, OCH2CH2CF3, or OCH2CH2CF2CF3), alkoxy (for eg., wo-pentyloxy or weo-pentyloxy), cycloalkyl, cycloalkyloxy (for eg., cyclopentyloxy), cycloalkylalkoxy (for eg. cyclobutylmethoxy or cyclohexylmethoxy), alkylaryl (for eg., CH3Ph or (CH3)3C-Ph), alkylalkynyl (for eg., (CH3)3C-C≡C-), heteroaryl (for eg., thiophene), halophenyl (chlorophenyl) or haloalkylphenylalkoxy (for eg., trifluoroalkyl benzyloxy).
According to one embodiment, specifically provided are compounds of the formula (Ia) in which, R6 is aryl, wherein aryl is fully or partially aromatic, preferably partially aromatic. In particular variant of this embodiment R6 is tetrahydronaphthalene.
According to one embodiment, specifically provided are compounds of the formula (Ia) in which, R6 and R7 together form a substituted or unsubstituted carbocyclic ring, wherein the carbocyclic ring is cyclohexane which includes two carbon atoms in thiazole ring. In this embodiment substituents on carbocyclic ring is alkyl for eg., tert- butyl.
According to one embodiment, specifically provided are compounds of the formula (Ia) in which, R6 is substituted or unsubstituted heteroaryl, preferably thienyl, wherein one or more substituents are independently selected from halogen (F, Cl or Br) or alkyl (for eg., methyl).
According to one embodiment, specifically provided are compounds of the formula (Ib)

According to one embodiment, specifically provided are compounds of the formula (Ib) in which, Ra is selected from one or more halogen and/or haloalkyl (for eg. trifluoromethyl).
According to one embodiment, specifically provided are compounds of the formula (Ib) in which, Ra is selected from substituted or unsubstitued aryloxy (for eg. phenyloxy) and substituted or unsubstituted aryl (for eg. phenyl), and the substitutent is alkyl (for eg. methyl or tert.-butyϊ) or cyclopentyloxy.
Representative examples of compounds of the present invention are provided below. These compounds are illustrative in nature only and do not limit the scope of the invention.
2-(5-Methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-Z»]purin-3-yl)-N-(4- phenoxyphenyl)acetamide;
N-[2-(4-Chlorophenyl)ethyl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -δ]purin-3-yl)acetamide;
N-[4-(4-Bromophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2,l-έ]purin-3-yl)acetamide;
N-[4-(4-Methylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2, 1 -έ]purin-3-yl)acetamide;
N-[4-(4-Chlorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2,l-ό]purin-3-yl)acetamide;
N-[4-Bromo-3-(trifluoromethyl)phenyl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2, 1 -Z>]purin-3-yl)acetamide;
N-(4-Bromo-2-chlorophenyl)-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -έ]purin-3-yl)acetamide;
N-[4-(4-Brόmophenyl)-5-methyl-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide;
2-(5-Methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[25l-δ]purin-3-yl)-N-{4-[4- (trifluoromethyl)phenyl]-l,3-thiazol-2-yl}acetamide;
N-[2-(4-Bromophenyl)ethyl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -έ]purin-3-yl)acetamide;
N-[4-(4-Cyanophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -Z>]purin-3-yl)acetamide;
N-μ-CS^-DichlorophenyO-l^-thiazol^-ylj^-CS-methyl^-oxo^^J^- tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide;
2-(5-Methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)-N-[4-(4-tert- butylphenyl)-l,3-thiazol-2-yl]acetamide;
Nl-{4-[4-(rert-Butyl)phenoxy]phenyl}-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -b]purin-3-yl)acetamide;
M-{4-[4-(4-Methylphenyl]phenyl]-l,3-thiazol-2-yl}2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -b]purin-3-yl)acetamide;
M-{4-[4-(ϊert-Butyl)phenyl]phenyl}-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2,l-έ]puiϊn-3-yl)acetamide;,
M-{4-[4-(3,3-Dimethyl-l-butynyl)phenyl]-l,3-thiazol-2-yl}2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-b]purin-3-yl)acetamide;
N-[4-(4-Ethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -έ]purin-3-yl)acetamide;
N-^-Cl^^^-Tetrahydro-β-naphthalenyO-l^-thiazol^-yll^-CS-methyl^-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide;
N-[5-(tert-Butyl)-4,5,6,7-tetrahydrobenzo[cO[l,3]thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide;
M -(4- {4-[4-(tert-Butyl)phenyl]phenyl} - 1 ,3 -thiazol-2-yl)-2-(5 -methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide;
Nl -[4-(4-Cyclopentyloxyphenyl)- 1 ,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -6]purin-3-yl)acetamide;
Nl-[4-(4-Isopentyloxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -Z>]purin-3-yl)acetamide;
Nl-[4-(3-Isopentyloxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide;,
Nl-[4-(4-Cyclopentyloxyphenoxy)phenyl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2,l-έ]purin-3-yl)acetamide;
Nl-[4-(3-Fluoro-4-trifluoromethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide;
Nl-[4-(4-Pentylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2,l-δ]purin-3-yl)acetamide;,
Nl-[4-(4-Cyclohexylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -ό]purin-3-yl)acetamide;
Nl-[4-(4-Isobutylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2, 1 -δ]purin-3 -yl)acetamide;
Nl-[4-(3-Trifluoromethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -6]purin-3-yl)acetamide;
Nl-[4-(2,5-Dimethyl-3-thienyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -ό]purin-3-yl)acetamide;
Nl-[4-(4-Trifluoromethoxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -ό]purin-3-yl)acetamide;
M-{4-[4-(4-Chlorophenyl)phenyl]-l,3-thiazol-2-yl}2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2,l-b]purin-3-yl)acetamide;
M-{4-[4-(3-Thienyl)phenyl]-l,3-thiazol-2-yl}-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide;
N-[4-(3-Bromo-4-fluorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide;
Nl-[4-(2,5-Dichloro-3-thienyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -6]purin-3-yl)acetamide;
Nl-[4-(2-Fluoro-4-trifluoromethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2, 1 -δ]purin-3-yl)acetamide;
Nl-{4-[3-Methyl-4-(3-trifluoromethylbenzyloxy)phenyl]-l,3-thiazol-2-yl}-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide;
Nl-μ^-ΗexylphenyO-l^-thiazol^-y^^-CS-methyl^-oxo^^^^-tetrahydro- 3H-imidazo[2, 1 -ό]purin-3-yl)acetamide;
Nl-[4-(4-Chloro-3-trifluoromethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5, 7, 8-tetrahydro-3H-imidazo [2, 1 -Z>]purin-3 -yl)acetamide;
Nl-[4-(3-Chloro-4-isopentyloxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2, 1 -6]purin-3-yl)acetamide;,
Nl-[4-(3-Chloro-4-difluoromethoxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2, 1 -δ]purin-3-yl)acetamide;
Nl-[4-(3,5-Dichloro-4-difluoromethoxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4- oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-δ]purin-3-yl)acetamide;
Nl-[4-(4-Difluoromethoxy-3,5-difluorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4- oxo-4,5,7,8-tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide;
Nl-{4-[3,5-dichloro-4-(3-methylbutoxy)]phenyl}-l,3-thiazol-2-yl]-2-(5-methyl-4- oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide;
Nl-{4-[3,5-dichloro-4-(3,3,3-trifluoropropoxy)]phenyl}-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide;
Nl:{4-[3,5-bis(trifluoromethyl)phenyl]-l,3-thiazol-2-yl}-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-δ]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-4-(2,2-dimethylpropoxy)phenyl)-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-Z>]purin-3-yl)acetamide;
Nl-[4-(4-Cyclobutylmethoxy)-3,5-dichlorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl- 4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-δ]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-4-[4-(triflυoromethyl)benzyloxy]phenyl)-l,3-thiazol-2-yl]-2-
(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-ό]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-4-[4-(trifluoromethyl)benzyloxy]phenyl)-l,3-thiazol-2-yl]-2- (5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-δ]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-[4-(3,4-difluorobenzyloxy)]phenyl)-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-6]purin-3-yl)acetamide;
Nl-^-CS^-dichloro-^^S^^^^-pentafluorobutoxy^phenyO-l^-thiazol^-yl]^- (5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-ό]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-[4-(cyclohexylmethoxy)]phenyl)-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l~£]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-{4-[2-(trifluoromethyl)benzyloxy]}phenyl)-l,3-thiazol-2-yl]- 2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-{4-[3-(trifluoromethyl)benzyloxy]}phenyl)-l,3-thiazol-2-yl]- 2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin~3-yl)acetamide;
Nl -[4-(3 ,5-difluoro-4-(3 ,3 ,3-trifluof oethoxy)phenyl)- 1 ,3 -thiazol-2-yl]-2-(5 - methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2, 1 -ό]purin-3-yl)acetamide; and
Nl-[4-(2,3,4-Trichlorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -ό]purin-3-yl)acetamide; or a pharmaceutically acceptable salt thereof, an ester thereof, a tautomer thereof, regioisomer thereof or a stereoisomer thereof,
The present patent application also provides a pharmaceutical composition that includes at least one compound described herein and at least one pharmaceutically acceptable excipient (such as a pharmaceutically acceptable carrier or diluent). Preferably, the pharmaceutical composition comprises a therapeutically effective amount of at least one compound described herein. The compounds described in the present patent application may be associated with a pharmaceutically acceptable excipient (such as a carrier or a diluents) or be diluted by a carrier, or enclosed within a carrier which can be in the form of a capsule, sachet, paper or other container.
The compounds and pharmaceutical compositions of the present invention are useful for modulating TRPAl receptors, which modulation is believed to be related to a variety of disease states.
The present patent application further provides a method of inhibiting TRPAl receptors in a subject in need thereof by administering to the subject one or more compounds described herein in an amount effective to cause inhibition of such receptor.
Detailed Description of the Invention
Definitions
The term "alkyl" refers to a straight or branched hydrocarbon chain radical consisting solely of carbon and hydrogen atoms, containing no unsaturation, having from one to eight carbon atoms, and which is attached to the rest of the molecule by a single bond, e.g., methyl, ethyl, n-propyl, 1-methylethyl (isopropyl), n-butyl, n-pentyl, and 1,1- dimethylethyl (tert-butyϊ). The term "Ci-6 alkyl" refers to an alkyl chain having 1 to 6 carbon atoms.
The term "alkenyl" refers to an aliphatic hydrocarbon group containing a carbon- carbon double bond and which may be a straight or branched chain having 2 to about 10 carbon atoms, e.g., ethenyl, 1-propenyl, 2-propenyl (allyl), iso-propenyl, 2-methyl-l- propenyl, 1-butenyl, and 2-butenyl.
The term "alkynyl" refers to a straight or branched chain hydrocarbyl radical having at least one carbon-carbon triple bond, and having 2 to about 12 carbon atoms (with radicals having 2 to about 10 carbon atoms being preferred), e.g., ethynyl, propynyl, and butynyl.
The term "alkoxy" denotes an alkyl group attached via an oxygen linkage to the rest of the molecule. Representative examples of such groups are -OCH3 and -OC2Hs.
The terms "halogen" or "halo" includes fluorine, chlorine, bromine, or iodine.
The term "haloalkyl" is used to denote a group comprised of an alkyl group substituted with halogen atom, where alkyl group is as defined above and halogen is used to denote fluorine, chlorine, bromine or iodine, an example of such group is trifiuoromethyl, difluoromethyl.
The term "cycloalkyl" denotes a non-aromatic mono or multicyclic ring system of 3 to about 12 carbon atoms, such as cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl. Examples of multicyclic cycloalkyl groups include, but are not limited to, perhydronapththyl, adamantyl and norbornyl groups, bridged cyclic groups or sprirobicyclic groups, e.g., sprio (4,4) non-2-yl.
The term "cycloalkylalkyl" refers to a cyclic ring-containing radical having 3 to about 8 carbon atoms directly attached to an alkyl group. The cycloalkylalkyl group may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure. Non-limiting examples of such groups include cyclopropylmethyl, cyclobutylethyl, and cyclopentylethyl.
The term "cycloalkenyl" refers to a cyclic ring-containing radical having 3 to about 8 carbon atoms with at least one carbon-carbon double bond, such as cyclopropenyl, cyclobutenyl, and cyclopentenyl.
The term "aryl" refers to an aromatic radical having 6 to 14 carbon atoms such as phenyl, naphthalenyl, thienyl, tetrahydronapthenyl, tetrahydrobenzothiazolyl indanyl, and biphenyl.
The term "arylalkyl" refers to an aryl group as defined above directly bonded to an alkyl group as defined above, e.g., -CH2C6Hs or -C2H5C6H5.
The term "heterocyclic ring" refers to a stable 3- to 15-membered ring radical which consists of carbon atoms and from one to five heteroatoms selected from nitrogen, phosphorus, oxygen and sulfur. For purposes of this invention, the heterocyclic ring radical may be a monocyclic, bicyclic or tricyclic ring system, which may include fused, bridged or spiro ring systems, and the nitrogen, phosphorus, carbon, oxygen or sulfur atoms in the heterocyclic ring radical may be optionally oxidized to various oxidation states. In addition, the nitrogen atom may be optionally quaternized; and the ring radical may be partially or fully saturated (i.e., heterocyclic or heteroaryl). Examples of such heterocyclic ring radicals include, but are not limited to, azetidinyl, acridinyl, benzodioxolyl, benzodioxanyl, benzofurnyl, carbazolyl, cinnolinyl, dioxolanyl, indolizinyl, naphthyridinyl, perhydroazepinyl, phenazinyl, phenothiazinyl, phenoxazinyl, phthalazinyl, pyridyl, pteridinyl, purinyl, quinazolinyl, quinoxalinyl, quinolinyl, isoquinolinyl, tetrazoyl, imidazolyl, tetrahydroisouinolyl, piperidinyl, piperazinyl, 2- oxopiperazinyl, 2-oxopiperidinyl, 2-oxopyrrolidinyl, 2-oxoazepinyl, azepinyl, pyrrolyl, 4- piperidonyl, pyrrolidinyl, pyrazinyl, pyrimidinyl, pyridazinyl, oxazolyl, oxazolinyl, oxasolidinyl, triazolyl, indanyl, isoxazolyl, isoxasolidinyl, morpholinyl, thiazolyl, thiazolinyl, thiazolidinyl, isothiazolyl, quinuclidinyl, isothiazolidinyl, indolyl, isoindolyl, indolinyl, isoindolinyl, octahydroindolyl, octahydroisoindolyl, quinolyl, isoquinolyl, decahydroisoquinolyl, benzimidazolyl, thiadiazolyl, benzopyranyl, benzothiazolyl, benzooxazolyl, furyl, tetrahydrofurtyl, tetrahydropyranyl, thienyl, benzothienyl, thiamorpholinyl, thiamorpholinyl sulfoxide, thiamorpholinyl sulfone, dioxaphospholanyl, oxadiazolyl, chromanyl, and isochromanyl. The heterocyclic ring radical may be attached to the main structure at any heteroatom or carbon atom that results in the creation of a stable structure.
The term "heterocyclyl" refers to a heterocyclic ring radical as defined above. The
heterocyclyl ring radical may be attached to the main structure at any heteroatom or carbon atom that results in the creation of a stable structure.
The term "heterocyclylalkyl" refers to a heterocyclic ring radical directly bonded to an alkyl group. The heterocyclylalkyl radical may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure.
The term "heteroaryl" refers to an aromatic heterocyclic ring radical. The heteroaryl ring radical may be attached to the main structure at any heteroatom or carbon atom that results in the creation of a stable structure.
The term "heteroarylalkyl" refers to a heteroaryl ring radical directly bonded to an alkyl group. The heteroarylalkyl radical may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure.
Unless otherwise specified, the term "substituted" as used herein refers to substitution with any one or any combination of the following substituents: hydroxy, halogen, carboxyl, cyano, nitro, oxo (=0), thio (=S), substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkenylalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted amino, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocyclylalkyl ring, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted heterocyclic ring, substituted or unsubstiuted guanidine, -COORX, -C(O)RX, -C(S)RX, -C(O)NRxRy, -C(O)ONRxRy, -NRxC0NRyRz, - N(Rx)S0Ry, -N(Rx)SO2Ry, -(=N-N(Rx)Ry), -NRxC(O)ORy, -NRxRy, -NRxC(O)Ry, - NRxC(S)Ry, -NRxC(S)NRyRz, -SONRxRy, -SO2NRxRy, -ORX, -ORxC(O)NRyRz, - ORxC(O)ORy, -OC(O)RX, -OC(O)NRxRy, -RxNRyC(O)Rz, -RxORy, -RxC(O)ORy, - RxC(0)NRyRz, -RxC(O)Ry, -RxOC(O)Ry, -SRX, -SORX, -SO2RX, and -ONO2, wherein Rx, Ry and Rz are independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted amino, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted heterocyclylalkyl ring, substituted or unsubstituted heteroarylalkyl, or substituted or unsubstituted heterocyclic ring. According to one embodiment, the substituents in the aforementioned "substituted"
groups cannot be further substituted. For example, when the substituent on "substituted alkyl" is "substituted aryl", the substituent on "substituted aryl" cannot be "substituted alkenyl".
The term "protecting group" or "PG" refers to a substituent that is employed to block or protect a particular functionality while other functional groups on the compound may remain reactive. For example, an "amino-protecting group" is a substituent attached to an amino group that blocks or protects the amino functionality in the compound. Suitable amino-protecting groups include, but are not limited to, acetyl, benzyl, trifluoroacetyl, t-butoxycarbonyl (BOC), benzyloxycarbonyl (CBz) and 9- fluorenylmethylenoxycarbonyl (Fmoc). Similarly, a "hydroxy-protecting group" refers to a substituent of a hydroxy group that blocks or protects the hydroxy functionality. Suitable hydroxy-protecting groups include, but are not limited to, acetyl, benzyl, tetrahydropyranyl and silyl. A "carboxy-protecting group" refers to a substituent of the carboxy group that blocks or protects the carboxy functionality. Suitable carboxy- protecting groups include, but are not limited to, -CH2CH2SO2Ph, cyanoethyl, 2- (trimethylsilyl)ethyl, 2-(trimethylsilyl)ethoxymethyl, 2-(p-toluenesulfonyl)ethyl, 2-(p- nitrophenylsulfenyl)ethyl, 2-(diphenylphosphino)-ethyl, and nitroethyl. For a general description of protecting .groups and their use, T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
The term "treating" or "treatment" of a state, disorder or condition includes:
(a) preventing or delaying the appearance of clinical symptoms of the state, disorder or condition developing in a subject that may be afflicted with or predisposed to the state, disorder or condition but does not yet experience or display clinical or subclinical symptoms of the state, disorder or condition; (b) inhibiting the state, disorder or condition, i.e., arresting or reducing the development of the disease or at least one clinical or subclinical symptom thereof; or (c) relieving the disease, i.e., causing regression of the state, disorder or condition or at least one of its clinical or subclinical symptoms.
The benefit to a subject to be treated is either statistically significant or at least perceptible to the subject or to the physician.
The term "subject" includes mammals (especially humans) and other animals, such as domestic animals (e.g., household pets including cats and dogs) and non-domestic animals (such as wildlife).
A "therapeutically effective amount" means the amount of a compound that, when administered to a subject for treating a state, disorder or condition, is sufficient to effect such treatment. The "therapeutically effective amount" will vary depending on the compound, the disease and its severity and the age, weight, physical condition and responsiveness of the subject to be treated.
The compound described in the present patent application may form salts. Non- limiting examples of pharmaceutically acceptable salts forming part of this patent application include salts derived from inorganic bases salts of organic bases salts of chiral bases, salts of natural amino acids and salts of non-natural amino acids. Certain compounds of present patent application are capable of existing in stereoisomeric forms (e.g. diastereomers and enantiomers). With respect to the overall compounds described by the Formula (I), the present patent application extends to these stereoisomeric forms and to mixtures thereof. To the extent prior art teaches synthesis or separation of particular stereoisomers, the different stereoisomeric forms of the present patent application may be separated from one another by the method known in the art, or a given isomer may be obtained by stereospecific or asymmetric synthesis. Tautomeric forms and mixtures of compounds described herein are also contemplated.
Pharmaceutical Compositions
The pharmaceutical composition provided in the present patent application includes at least one compound described herein and at least one pharmaceutically acceptable excipient (such as a pharmaceutically acceptable carrier or diluent). Preferably, the contemplated pharmaceutical compositions include the compound(s) described herein in an amount sufficient to inhibit TRPAl receptor in a subject. The inhibitory activity of compounds falling within the formula (I) may be measured by an assay provided herein below.
The subjects contemplated include, for example, a living cell and a mammal, including human mammal. The compound of the present patent application may be associated with a pharmaceutically acceptable excipient (such as a carrier or a diluent) or be diluted by a carrier, or enclosed within a carrier which can be in the form of a capsule, sachet, paper or other container.
Examples of suitable carriers include, but are not limited to, water, salt solutions, alcohols, polyethylene glycols, polyhydroxyethoxylated castor oil, peanut oil, olive oil, gelatin, lactose, terra alba, sucrose, dextrin, magnesium carbonate, sugar, cyclodextrin,
amylose, magnesium stearate, talc, gelatin, agar, pectin, acacia, stearic acid or lower alkyl ethers of cellulose, silicic acid, fatty acids, fatty acid amines, fatty acid monoglycerides and diglycerides, pentaerythritol fatty acid esters, polyoxyethylene, hydroxymethyl- cellulose and polyvinylpyrrolidone. The carrier or diluent may include a sustained release material, such as glyceryl monostearate or glyceryl distearate, alone or mixed with a wax.
The pharmaceutical composition may also include one or more pharmaceutically acceptable auxiliary agents, wetting agents, emulsifying agents, suspending agents, preserving agents, salts for influencing osmetic pressure, buffers, sweetening agents, flavoring agents, colorants, or any combination of the foregoing. The pharmaceutical composition may be formulated so as to provide quick, sustained, or delayed release of the active ingredient after administration to the subject by employing procedures known in the art.
The pharmaceutical compositions may be prepared by conventional techniques known in the art (Remington: The Science and Practice of Pharmacy, 20th Ed., 2003, Lippincott Williams & Wilkins). For example, the active compound can be mixed with a carrier, or diluted by a carrier, or enclosed within a carrier, which may be in the form of an ampoule, capsule, sachet, paper, or other container. When the carrier serves as a diluent, it may be a solid, semi-solid, or liquid material that acts as a vehicle, excipient, or medium for the active compound. The active compound can be adsorbed on a granular solid container, for example, in a sachet.
The pharmaceutical compositions may be in conventional forms, for example, capsules, tablets, aerosols, solutions, suspensions or products for topical application.
The route of administration may be any route which effectively transports the active compound of the invention to the appropriate or desired site of action. Suitable routes of administration include, but are not limited to, oral, nasal, pulmonary, buccal, subdermal, intradermal, transdermal, parenteral, rectal, depot, subcutaneous, intravenous, intraurethral, intramuscular, intranasal, ophthalmic (such as with an ophthalmic solution) or topical (such as with a topical ointment). The oral route.is preferred.
Solid oral formulations include, but are not limited to, tablets, capsules (soft or hard gelatin), dragees (containing the active ingredient in powder or pellet form), troches and lozenges. Tablets, dragees, or capsules having talc and/or a carbohydrate carrier or binder or the like are particularly suitable for oral application. Preferable carriers for
tablets, dragees, or capsules include lactose, cornstarch, and/or potato starch. A syrup or elixir can be used in cases where a sweetened vehicle can be employed.
Liquid formulations include, but are not limited to, syrups, emulsions, soft gelatin and sterile injectable liquids, such as aqueous or non-aqueous liquid suspensions or solutions. For parenteral application, particularly suitable are injectable solutions or suspensions, preferably aqueous solutions with the active compound dissolved in polyhydroxylated castor oil.
Methods of Treatment
The present patent application provides compounds and pharmaceutical formulations thereof that are useful in the treatment of diseases, conditions and/or disorders modulated by TRPAl antagonists. The compounds described herein and pharmaceutical formulations thereof have TRPAl activity and are believed to be of potential use for the treatment or prophylaxis of certain diseases or disorders mediated or associated with the activity of TRPAl receptor, including disorders such as pain, chronic pain, complex regional pain syndrome, neuropathic pain, postoperative pain, rheumatoid arthritic pain, osteoarthritic pain, back pain, visceral pain, cancer pain, algesia, neuralgia, migraine, neuropathies, diabetic neuropathy, sciatica, HIV-related neuropathy, postherpetic neuralgia, fibromyalgia, nerve injury, ischaemia, neurodegeneration, stroke, post stroke pain, multiple sclerosis, respiratory diseases, asthma, cough, COPD, inflammatory disorders, oesophagitis, gastroeosophagal reflux disorder (GERD), irritable bowel syndrome, inflammatory bowel disease, pelvic hypersensitivity, urinary incontinence, cystitis, burns, psoriasis, eczema, emesis, stomach duodenal ulcer and pruritus. The connection between therapeutic effect and inhibition of TRPAl is illustrated, for example, in Story GM et al, Cell, (2003), JJ2, 819-829; McMahon SB and Wood JN, Cell, (2006), 124, 1123-1125; Voorhoeve PM et al, Cell, (2006), 124, 1169-1181; Wissenbach U, Niemeyer BA and Flockerzi V, Biology of the Cell, (2004), 96, 47-54; Dayne YO, Albert YH & Michael X, Expert Opinion on Therapeutic Targets (2007), 11(3), 391-401 and the references cited therein.
General method of preparation
The compounds described herein, including compounds of general formula I and specific examples are prepared using techniques known to one skilled in the art through the reaction sequences depicted in Scheme 1-2. Furthermore, in the following schemes, where specific acids, bases, reagents, coupling agents, solvents, etc. are mentioned, it is
understood that other suitable acids, bases, reagents, coupling agents, etc., may be used and are included within the scope of the present invention. The compounds obtained by using the general reaction sequences may be of insufficient purity. These compounds can be purified by using any of the methods for purification of organic compounds known to persons skilled in the art, for example, crystallization or silica gel or alumina column chromatography using different solvents in suitable ratios. All possible stereoisomers are envisioned within the scope of this invention.
A general approach for the synthesis of the compounds of formula I is described in Scheme 1. The required 6-aminouracil derivatives (1) (wherein R1 is alkyl, for example methyl) are commercially available or can be prepared according to methods known to one skilled in the art (Wright, G. E. et al. J. Heterocyclic Chem. (1976), 13., 539-544; Muller, C. E. et al. Tetrahedron Lett. (1991), 32(45). 6539-6540). Diamine of general formula (2) is prepared by nitrosation of intermediate (1) followed by reduction of nitroso compound using sodium hydrosulphite or by catalytic hydrogenation using catalytic amounts of palladium on carbon (Muller, C. E. et al. Synthesis (1993), 125-128; Muller C. E. et al; Synthesis, (1995), 1295-1299 and references cited there in). Diamines of formula (2) are converted to compounds of formula (3) by reductive amination with benazaldehyde and sodium cyanoborohydride followed by cyclization with triethyl orthoformate. Compound of formula (3) is converted to a compound of a formula (5) by reaction with phosphorous oxychloride followed by addition of suitable amino ethanol of the formula (4) (wherein R2 and R3 are hydrogen). Compound of formula (5) is converted to tricyclic compound of the formula (6) by one-pot halogenation and cyclization with thionyl chloride followed by debenzylation using hydrogen in the presence of catalytic amounts of Pd(OH)2 on carbon (Suzuki, H. et al; Chem. Pharrn. Bull (2002), 50(9), 1163-1168). Alkylation of tricyclic compound of formula (6) using an appropriate 2- bromoacetamide derivative (7) (wherein R4 and R5 are as defined above in description) in the presence of a suitable base such as potassium carbonate or sodium hydride in a suitable solvent such as DMF or THF affords compounds of the general formula I.
Scheme 1

The 2-haloacetamides of formula (7a) (Scheme 2) (wherein Rb is as described above in description) required for the synthesis of compounds of the present invention can be prepared according to methods known to one skilled in the art (Carroll, L. et al. J. Am. Chem. Soc. (1950), 72, 3722-3725; Ohkubo, M. et al; Chem. Pharm. Bull (1995), 43 (9), 1497-1504). Thus, acylation of an amine of formula (12) with bromoacetyl bromide in the presence of a suitable base such as triethylamine or pyridine gives N- substituted bromoacetamide of the general formula (7a) (Scheme 2).
A few of aniline derivatives, arylalkylamines and 2-amino-4-arylthiazoles were commercially available. Many of the disubstituted and trisubstituted arylaminothiazoles were prepared from appropriate aryl alkyl ketones. Commercially unavailable aryl alkyl ketones were prepared from the corresponding benzoic acids as shown in Scheme 2. Substituted benzoic acid of the formula (8) was converted to the corresponding acetophenone in three steps as shown in Scheme 2. Thus, acid of formula (8) was converted to the corresponding acid chloride of formula (9) using oxalyl chloride in the presence of catalytic amounts of DMF in dry dichloromethane. Alternatively, this transformation can be carried out using excess thionyl chloride. The acid chloride of formula (9) was converted to corresponding Weinreb amide of formula (10) by treating with N,O-dimethyl hydroxylamine hydrochloride in the presence of a suitable base such as triethylamine. Addition of methyl magnesium iodide to Weinreb amide of formula (10) gives acetophenone derivative of the formula (11).
The aryl alkyl ketone of the formula (11) is converted to 2-aminothiazole of the formula (12) in one step by its reaction with thiourea in the presence of iodine in ethanol. This conversion is similar to the one described by Carroll, K. et al. J. Am. Chem. Soc. (1950), 3722; and Νaik, S., J.; Halkar, U. P. ARKIVOC (2005) xiii, 141-149.
Alternatively, 2-aminothiazoles of the formula (12) can be prepared by the reaction of compounds of formula (11) with bromine in acetic acid to give the alpha halo intermediate, which on reaction with thiourea in THF at reflux condition give compounds of the formula (12). The compound of the formula (12) is transferred to (2-bromo-N-l,3- thiazol-2-yl)acetamide of the formula (7a) by acylation with bromoacetyl bromide in the presence of a suitable base such as pyridine or triethylamine as base in a suitable solvent such as THF.
Scheme 2

The intermediates and examples described in the present invention are prepared using the procedure described below. However, it is understood that these intermediates and examples can be prepared by alternate approaches which are within the scope of the present invention.
. EXPERIMENTAL Preparation of Intermediates
5-Methyl-7,8-dihydro-3H-imidazo[2,l-Z>]purin-4(5H)-one (Intermediate 1), required for the synthesis of compound of the present invention, is prepared in 7 steps as described in the 'General Methods of Preparation' starting from commercially available 6-amino-3-methyl pyrimidine-2,4(lH,3H)-dione. Stepwise experimental procedure and characterization data for each intermediate is given below.
2-Bromoacetamide derivatives of general formula (7a) used in the coupling reactions are prepared by acylation of appropriate amines with bromoacetyl bromide as shown in 'General Methods of preparation'. Detailed experimental procedure and characterization data for selected 2-bromoacetamides is given below.
Intermediate 1 5-Methyl-7,8-dihydro-3H-imidazo[2,l-δ]purin-4(5H)-one
Step 1: 6-Amino-3-methyl-5-nitrosopyrimidine-2,4(lH,3H)-dione
A solution of 6-amino-3-methylpyrimidine-2,4(lH,3H)-dione (16.5 g, 116.913 mmol) in DMF (150 ml) and water (50 ml) was prepared by heating at 90 0C. The solution was brought to room temperature and NaNO2 (16.13 g, 233.826 mmol) dissolved in water (25 ml) was added. A few drops of concentrated HCl were added until, upon further addition, no further deepening of the color occurred. The mixture was stirred for Ih. Then, the solvent was removed under reduced pressure, the residue obtained was suspended in water (200 ml), brought to pΗ 4 by treatment with concentrated HCl, cooled and the dark violet precipitate was collected by filtration and washed with water to get 18.2 g (90%) of the product as a violet solid; IR (KBr) 3027, 1726, 1686, 1531, 1444, 1092 "cm"1; 1H NMR (300 MHz, DMSO-J6) δ 3.18 (s, 3H), 8.09 (br s, 2H), 11.21 (br s, IH); ESI-MS (m/z) 171.33 (M+H)+. Step 2: 5,6-Diamino-3-methylpyrimidine-2,4(lH,3H)-dione
A solution of Step 1 intermediate (18 g, 105.803 mmol) in 12.5 % aqueous NH4OH (500 ml) was prepared by heating at 70 0C. Sodium hydrosulphite (36.84 g, 211.602 mmol) was added in small portions with vigorous stirring over a period of 20 min. The volume of the solution was reduced under reduced pressure until the product started to crystallize out. The mixture was cooled and the precipitate was collected by filtration and washed with a small amount of cold water to get 9.75 g (59%) of the product as an off-white solid; IR (KBr) 3193, 1682, 1662, 1548, 1460, 1108, 723 cm'1; 1H NMR (300 MHz, DMSO-J6) δ 3.05 (s, 3H), 5.54 (s, 2H), 6.09 (s, IH), 8.76 (s, 2H); ESI- MS (m/z) 157.30 (M+H)+. Step 3: 6-Amino-5-(benzylamino)-3-methylpyrimidine-2,4(lH,3H)-dione
To a suspension of Step 2 intermediate (8.16 g, 55.155 mmol) in water (80 ml) was added acetic acid (0.8 ml) followed by benzaldehyde (8.32 g, 78.411 mmol) over 15 min at room temperature. The solution was cooled and the precipitate was filtered, washed with cold water followed by acetonitrile to give 11.6 g (91%) of 6-amino-3- methyl-5-{[(l£)-phenylmethylene]amino}pyrimidine-2,4(lH,3H)-dione as a light yellow solid; IR (KBr) 3209, 1712, 1628, 1444, 1171, 752 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 3.10 (s, 3H), 6.62 (br s, 2H), 7.36 (d, J= 7.2 Hz, 3H), 7.83 (d, J= 6.9 Hz, 2H), 9.65 (s, IH), 10.76 (br s, IH); ESI-MS (m/z) 245.20 (M)+.
To a suspension of 6-amino-3~methyl-5-{[(li^-phenylmethylene]amino} pyrimidine-2,4(lH,3H)-dione (11.5 g, 47.082 mmol) in dichloromethane (200 ml) and MeOH (200 ml) was added acetic acid (2.82 g, 47.011 mmol) followed by NaBH3CN (2.95 g, 46.944 mmol). The reaction mixture was stirred at room temperature for 2 h. After this time, acetic acid (0.28 g, 4.701 mmol) and NaBH3CN (0.29 g, 4.694 mmol) were added and reaction mixture was further stirred for 30 min. The reaction mixture was concentrated under reduced pressure, chilled and precipitate was filtered, washed with cold methanol (50 ml). This residue was stirred in boiling methanol (50 ml) for 15 min, chilled, filtered, and dried to get 8.01 g (70%) of the product as an off-white solid; IR (KBr) 3189, 1703, 1575, 1456, 1087, 727 cm-1; 1HNMR (300 MHz, DMSO-J6) δ 3.32 (s, 3H), 3.79 (s, 2H), 5.71 (s, 2H), 7.19-7.24 (m, 3H), 7.35 (d, J= 6.9 Hz, 2H), 10.23 (br s, IH); ESI-MS (m/z) 247.07 (M+H)+. Step 4: 7-Benzyl-l-methyl-3,7-dihydro-lH-purine-2,6-dione
A suspension of Step 3 intermediate (8 g, 32.487 mmol) in triethyl orthoformate (40 ml) was heated to reflux for 5 h. After this time, the reaction mixture was cooled to room temperature, solid formed was filtered, washed with diethyl ether (40 ml), and dried to give 7.65 g (91%) of the product as a pale yellow solid; 3108, 2877, 1708, 1675, 1413, 1214, 1009, 718 cm-1; 1H NMR (300 MHz, DMSO-J6) δ 3.14 (s, 3H), 5.42 (s, 2H), 7.29 (s, 5H), 8.13 (s, IH), 11.84 (br s, IH). Step 5: 7-Benzyl-2-chloro-l-methyl-l,7-dihydro-6H-purin-6-one
A suspension of Step 4 intermediate (7.6 g, 29.658 mmol) and POCl3 (200 ml) was heated to reflux overnight. Excess POCl3 was removed under reduced pressure and the residue was partitioned between ethyl acetate (200 ml) and aqueous saturated NaHCO3 (100 ml) and the organic layer was washed with brine (100 ml), dried (Na2SO4) and concentrated under reduced pressure to afford 7.1 g (86%) of the product as an off- white solid; IR (KBr) 3098, 1694, 1566, 1392, 1226, 1051, 697 cm"1; 1HNMR (300 MHz, DMSO-J6) δ 3.58 (s, 3H), 5.54 (s, 2H), 7.30 (s, 5H), 8.44 (s, IH); ESI-MS (m/z) 275.35 (M+H)+. Step_6: 3-Benzyl-5-methyl-7,8-dihydro-3H-imidazo[2,l-έ]purin-4(5H)-one
To a stirred solution of Step 5 intermediate (280 mg, 1.021 mmol) in CH3CN (10 ml) was added triethylamine (213 μl, 1.531 mmol) and ethanolamine (218 mg, 3.571 mmol) at room temperature. The resulting solution was heated to reflux overnight. After this time, the reaction mixture was concentrated under reduced pressure and the residue was partitioned between ethyl acetate (100 ml) and water (50 ml). The organic layer was
washed with brine, dried (Na2SO4) and evaporation under reduced pressure afforded 221 mg (72%) of 7-benzyl-2-[(2-hydroxyethyl)amino]-l-methyl-l,7-dihydro-6H-purin-6-one as an off-white solid; IR (Neat) 3266, 2941, 1676, 1559, 1383, 1215, 756 cm'1; 1H NMR (300 MHz, DMSO-J6) δ 3.30 (s, 3H), 3.32-3.38 (m, 2H), 3.54 (d, J= 5.4 Hz, 2H), 4.73 (s, IH), 5.42 (s, 2H), 6.73 (br s, IH), 7.26 (s, 5H), 8.09 (s, IH); ESI-MS (m/z) 300.35 (M+H)+.
To a solution of 7-benzyl-2-[(2-hydroxyethyl)amino]-l-methyl-l,7-dihydro-6H- purin-6-one (500 mg, 1.671 mmol) in dichloromethane (15 ml) was added SOCl2 (596 mg, 5.011 mmol) at 0 0C. The reaction mixture was stirred for 12 h at room temperature. After this time, the reaction mixture was adjusted to pΗ 7-8 with 4M NaOH solution. The organic layer was washed with brine, dried (Na2SO4) and concentrated under reduced pressure to afford 351 mg (74%) of the product as an off-white solid; IR (KBr) 2953, 1679, 1634, 1544, 1472, 1295 cm"1; 1HNMR (300 MHz, CDCl3) δ 3.35 (s, 3H), 3.99 (d, J = 8.4 Hz, 2H), 4.08 (d, J = 7.8 Hz, 2H), 5.42 (s, 2H), 7.29-7.41 (m, 6H); ESI-MS (m/z) 282.41 (M+H)+. Step 7: 5-Methyl-7,8-dihydro-3H-imidazo[2, 1 -Z>]purin-4(5H)-one
A mixture of Step 6 intermediate (160 mg, 0.571 mmol) and 20% palladium hydroxide on carbon (176 mg, 1.254 mmol) in MeOH (20 ml) was shaken under hydrogen atmosphere (50 psi) for 24 h. Then the catalyst was filtered, the filtrate was concentrated under reduced pressure and the residue recrystallized form ethyl acetate and methanol to give 121 mg (50%) of the product as an off-white solid; IR (KBr) 3019, 1717, 1666, 1437, 1215, 756 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 3.30 (s, 3H), 3.92 (t, J= 9.3 Hz, 2H), 4.24 (t, J = 8.4 Hz, 2H), 8.07 (s, IH), 10.18 (br s, IH); ESI-MS (m/z) 192.61 (M+H)+.
General procedure for the preparation of 2-bromo-N-thiazolyl acetamide derivatives (Intermediate 7a, Scheme 2):
Some of the (2-amino-4-aryl)thiazoles used for the synthesis of 2-bromo-N- thiazolyl acetamides of general formula (7a) were not commercially available and were prepared from appropriately substituted acetophenones and thiourea as described below.

Step 1: A mixture of appropriate aryl alkyl ketone (1.0 equiv.), thiourea (2.0 eqiv.) and iodine (1.0 equiv.) in dry ethanol (5 volumes) was refluxed for 24h. The reaction mixture
was diluted with ethyl acetate and washed with saturated solution of sodium thiosulphate.
The organic layer was treated with IN HCl and the precipitated salt collected by filtration. The salt was then treated with saturated solution OfNaHCO3 and extracted with dichloromethane, washed with brine, dried over sodium sulfate and the solvent was evaporated to afford the 2-aminothiazole derivative.
Step 2: To a stirred and cooled (O 0C) solution of appropriate amine (1.0 equiv.) and pyridine (1.2 equiv.) in dichloromethane (5 volume) was added bromoacetyl bromide (1.2 eq.) over 5 min and the resulting mixture was allowed to warm to room temperature and then further stirred at room temperature for 2 h. The reaction mixture was diluted with dichloromethane (50 ml) and water (50 ml). The layers were separated. The aqueous layer was extracted with dichloromethane (2 x 50 ml) and the combined organic layers were washed with water (2 x 50 ml) followed by brine (50 ml), dried (Na2SO4) and filtered. The filtrate was concentrated under reduced pressure. The residue obtained after the evaporation of the solvent was purified by silica gel column chromatography using 5-10% ethyl acetate in petroleum ether to obtain the desired product as an off-white solid.
Table 1: Structure and characterization data of 2-bromoacetamide derivatives







Examples 1-57 were prepared by coupling Intermediate 1 with appropriate N- substituted bromoacetamides of the formula (7a) (Scheme 2) in the presence of a suitable base using DMF as solvent.
Examples 1
2-(5-Methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-Z?]purin-3-yl)-N-(4-phenoxy- phenyl)acetamide

To a mixture of Intermediate 1, 5-Methyl-7,8-dihydro-3H-imidazo[2,l-έ]purin-4(5H)-one (46 mg, 0.241 mmol) and anhydrous K2CO3 (50 mg, 0.362 mmol) in DMF (3 ml) was added 2-bromo-N-(4-phenoxyphenyl)acetamide (110 mg, 0.362 mmol) and the reaction mixture was stirred at room temperature overnight. After this time, the reaction mixture was concentrated under reduced pressure and the residue was partitioned between EtOAc (50 ml) and water (10 ml). The organic layer was washed with brine, dried (Na2SO4) and evaporation under reduced pressure afforded 23 mg of 2-(5-methyl-4-oxo-4,5,7,8-
tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)-N-(4-phenoxyphenyl)acetamide as an off-white soild; IR (Neat) 2925, 1681, 1634, 1448, 1228, 757 cm'1; 1H NMR (300 MHz, DMSO-J6) δ 3.15 (s, 3H), 3.81 (t, J= 8.4 Hz, 2H), 3.99 (t, J= 8.4 Hz, 2H), 5.10 (s, 2H), 6.92-6.99 (m, 4H)5 7.07 (t, J= 7.2 Hz5 IH), 7.34 (t, J= 7.2 Hz, 2H), 7.55 (d, J= 9.3 Hz, 2H), 7.86 (s, IH), 10.38 (br s, IH); ESI-MS (m/z) 417.35 (M+H)+.
Example 2
N-[2-(4-Chlorophenyl)ethyl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l- έ]purin-3 -yl)acetamide

The title compound was prepared from Intermediate 1 (45 mg, 0.136 mmol) and 2- bromo-N-[2-(4-chlorophenyl)ethyl]acetamide (0.021 ml, 0.138 mmol) using anhydrous K2CO3 (50 mg, 0.362 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 1 to give 25 mg of the product as a white solid; IR (KBr) 3288, 2937, 1679, 1637, 1474, 1088 CnT1; 1H ΝMR (300 MHz, DMSO-J6) δ 2.69 (t, J= 6.9 Hz, 2H), 3.16 (s, 3H), 3.27 (d, J= 6.6 Hz, 2H), 3.80 (t, J= 9.0 Hz, 2H), 3.97 (t, J= 7.8 Hz, 2H), 4.84 (s, 2H), 7.20-7.31 (m, 4H), 7.79 (s, IH), 8.21 (br s, IH); ESI-MS (m/z) 387.44 (M+H)+.
Example 3
N-[4-(4-Bromophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetra-hydro-3H- imidazo[2, 1 -6]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (45 mg, 0.136 mmol) and 2- bromo-N-[4-(4-bromophenyl)-l,3-thiazol-2-yl]acetamide (0.021 ml, 0.138 mmol) using anhydrous K2CO3 (50 mg, 0.362 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 1 to give 35 mg of the product as a white solid; IR (KBr) 3019, 2927, 1662, 1638, 1427, 1215, 756 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 3.13 (s, 3H), 3.82 (d, J= 9.0 Hz, 2H), 3.98 (t, J= 8.1 Hz, 2H), 5.25 (s, 2H), 7.61 (d, J= 8.1 Hz, 2H), 7.72 (s, IH), 7.81-7.88 (m, 3H), 12.72 (br s, IH); ESI-MS (m/z) 484.31 (M-HT.
Example 4 i\r-[4-(4-Chlorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetra-hydro-3H- imidazo[2, 1 -ό]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (45 mg, 0.136 mmol) and 2- bromo-N-[4-(4-chlorophenyl)-l,3-thiazol-2-yl]acetamide (0.021 ml, 0.138 mmol) using anhydrous K2CO3 (50 mg, 0.362 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 1 to give 30 mg of the product as a white solid; IR (KBr) 3436, 1638, 1560, 1232, 744 cm"1; 1H ΝMR (300 MHz, OMSO-d6) δ 3.14 (s, 3H), 3.83 (d, J= 8.1 Hz, 2H), 4.00 (t, J= 6.3 Hz, 2H), 5.26 (s, 2H), 7.48 (d, J= 6.9 Hz, 2H), 7.70 (s, IH), 7.90 (s, IH), 12.79 (br s, IH); ESI-MS (m/z) 440.33 (M-H)".
Example 5
N-[4-Bromo-3-(trifluoromethyl)phenyl]-2-(5-methyl-4-oxo-4,5,7,8-tetra-hydro-3H- imidazo[2, 1 -ό]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (45 mg, 0.136 mmol) and 2- bromo-N-[4-bromo-3-(trifluoromethyl)phenyl]acetamide (0.021 ml, 0.138 mmol) using anhydrous K2CO3 (50 mg, 0.362 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 1 to give 40 mg of the product as a white solid; IR (KBr) 3116, 2927, 1678, 1638, 1547, 1322, 756 cm4; 1H NMR (300 MHz, DMSO-O δ 3.14 (s, 3H), 3.82 (d, J= 6.9 Hz, 2H), 3.98 (d, J= 6.3 Hz, 2H), 5.13 (s, 2H), 7.68 (d, J= 6.3 Hz, IH), 7.80-7.87 (m, 2H), 8.13 (s, IH), 10.81 (s, IH); ESI-MS (m/z) 471.56 (M+H)+.
Example 6
N-(4-Bromo-2-chlorophenyl)-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l- έ]purin-3 -yl)acetamide

The title compound was prepared from Intermediate 1 (40 mg, 0.209 mmol) and 2- bromo-N-(4-bromo-2-chlorophenyl)acetamide (82 mg, 0.251 mmol) using anhydrous
K2CO3 (50 mg, 0.362 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 1 to give 16 mg of the product as a white solid; IR (Neat) 3019, 2928, 1637, 1547, 1215, 764 cm"1; 1H NMR (300 MHz, DMSO-^6) δ 3.16 (s, 3H), 3.80 (t, J= 8.7 Hz, 2H), 3.98 (t, J= 7.8 Hz, 2H), 5.19 (s, 2H), 7.51 (d, J= 9.3 Hz, IH), 7.67 (d, J = 8.1 Hz, IH), 7.78 (s, IH), 7.87 (s, IH), 10.03 (s, IH); ESI-MS (m/z) 439.15 (M+H)+.
Example 7 iV-[4-(4-Bromophenyl)-5-methyl-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7 ,8- tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (40 mg, 0.209 mmol) and 2- bromo-N-[4-(4-bromophenyl)-5-methyl-l,3-thiazol-2-yl]acetamide (98 mg, 0.251 mmol) using anhydrous K2CO3 (50 mg, 0.362 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 1 to give 70 mg of the product as a white solid; IR (KBr) 3095, 2947, 1697, 1628, 1575, 1220, 1007 cm"1; 1H ΝMR (300 MHz, DMSOO δ 2.45 (s, 3H), 3.14 (s, 3H), 3.81 (t, J= 9.0 Hz, 2H), 4.00 (t, J= 8.7 Hz, 2H), 5.22 (s, 2H), 7.61 (dd, J= 8.4, 7.2 Hz, 4H), 7.88 (s, IH), 12.57 (s, IH); ESI-MS {m/z) 498.65 (M-H)'.
Example 8
2-(5-Methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)-N-{4-[4- (trifluoromethyl)phenyl]-l,3-thiazol-2-yl}acetamide

The title compound was prepared from Intermediate 1 (33 mg, 0.173 mmol) and 2- bromo-N-{4-[4-(trifluoromethyl)phenyl]-l,3-thiazol-2-yl}acetamide using anhydrous K2CO3 (11 mg, 0.259 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 1 to give 30 mg of the product as a white solid; IR (Neat) 3136, 2923, 1697, 1638, 1559, 1109, 847 cm"1; 1HNMR (300 MHz, CDCl3) δ 3.14 (s, 3H), 3.82 (t, J= 8.4 Hz, 2H), 4.00 (t, J= 9.3 Hz, 2H), 5.26 (s, 2H), 7.79 (d, J= 8.4 Hz, 2H), 7.89 (s, 2H), 8.10 (d, J= 8.1 Hz, 2H), 12.81 (br s, IH); ESI-MS (m/z) 476.22 (M+H)+.
Example 9
N-[2-(4-Bromophenyl)ethyl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l- ό]purin-3 -yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.157 mmol) and 2- bromo-N-[2-(4-bromophenyl)ethyl]acetamide (60 mg, 0.188 mmol) using anhydrous K2CO3 (10 mg, 0.259 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 1 to give 12 mg of the product as a white solid; IR (Neat) 3109, 2925, 1668, 1634, 1463, 1377, 1050 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 2.67 (br s, 2H), 3.17 (s, 3H), 3.81 (t, J= 9.3 Hz, 2H), 3.99 (t, J= 8.4 Hz, 2H), 4.85 (s, 2H), 7.16 (d, J = 7.8 Hz, 2H), 7.43 (d, J= 6.3 Hz, 2H), 7.80 (s, IH), 8.23 (s, IH); ESI-MS (m/z) 433.96 (M+H)+.
Example 10
N-[4-(4-Cyanophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetra-hydro-3H- imidazo[2,l-έ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.157 mmol) and 2- bromo-N-[4-(4-cyanophenyl)-l,3-thiazol-2-yl]acetamide (60 mg, 0.188 mmol) using anhydrous K2CO3 (10 mg, 0.259 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 1 to give 48 mg of the product as a white solid; IR (KBr) 3138, 2988, 2232, 1661, 1635, 1560, 1257, 752 cm"1; 1H ΝMR (300 MHz, DMSO-^6) δ 3.14 (s, 3H), 3.82 (t, J= 8.7 Hz, 2H), 4.00 (t, J= 9.9 Hz, 2H), 5.26 (s, 2H), 7.88-7.95 (m, 4H), 8.06 (d, J= 8.4 Hz, 2H), 12.80 (br s, IH); ESI-MS (m/z) 433.23 (M+H)+.
Example 11
N-[4-(3,4-Dichlorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -Z>]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (26 mg, 0.136 mmol) and 2- bromo-N-[4-(3,4-dichlorophenyl)-l,3-thiazol-2-yl]acetamide (60 mg, 0.163 mmol) using anhydrous K2CO3 (8 mg, 0.204 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 1 to give 40 mg of the product as a white solid; IR (KBr)
3143, 2988, 1661, 1636, 1552, 1258, 749 cm"1; 1H NMR (300 MHz, DMSO-cfe) δ 3.14 (s,
3H), 3.82 (t, J= 8.1 Hz, 2H), 3.99 (t, J= 7.8 Hz, 2H), 5.26 (s, 2H), 7.69 (d, J= 9.0 Hz, 2H), 7.90 (d, J = 8.4 Hz, 2H), 8.12 (s, IH), 12.78 (br s, IH); ESI-MS (m/z) 476.49 (M+H)+.
Example 12
2-(5-Methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)-N-[4-(4-tert- butylphenyl)-l,3-thiazol-2-yl]acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.157 mmol) and 2- bromo-N-[4-(4-tert-butylphenyl)-l,3-thiazol-2-yl]acetamide (67 mg, 0.188 mmol) using anhydrous K2CO3 (8 mg, 0.204 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 1 to give 40 mg of the product as a white solid; IR (KBr) 3132, 2961, 1657, 1639, 1557, 1264, 1016 cm'1; 1H ΝMR (300 MHz, DMSO-^6) δ 3.14 (s, 3H), 3.82 (d, J= 6.9 Hz, 2H), 3.98 (d, J= 6.3 Hz, 2H), 5.13 (s, 2H), 7.68 (d, J= 6.3 Hz, IH), 7.80-7.87 (m, 2H), 8.13 (s, IH), 10.81 (s, IH); ESI-MS (m/z) 464.26 (M+H)+.
Example 13
Nl-{4-[4-(tert-Butyl)phenoxy]phenyl}-2-(5-methyl-4-oxo-4,5,7,8-tetra-hydro-3H- imidazo[2, 1 -b]purin-3-yl)acetamide

To a stirred mixture of Intermediate 1 (25 mg, 0.131 mmol) and ΝaΗ (60 wt. % dispersion in mineral oil ) (8.0 mg, 0.196 mmol) in anhydrous DMF (3 ml) was added 2- bromo-N-[4-(4-ter?-butylphenoxy)phenyl]acetamide (57 mg, 0.157 mmol) and the reaction mixture was stirred at 80 0C overnight. After this time, the reaction mixture was cooled and concentrated under reduced pressure and the residue was partitioned between ethyl acetate (25 ml) and water (10 ml). The organic layer was washed with brine, dried (Na2SO4) and evaporation under reduced pressure afforded 18 mg of the product as an off-white solid; IR (Neat) 2927, 1636, 1501, 1462, 1215, 760 cm"1; 1H NMR (300 MHz, DMSO-Cf6) δ 1.26 (s, 9H), 3.15 (s, 3H), 3.81 (t, J= 8.47Hz, 2H), 3.99 (t, J= 9.0 Hz, 2H),
5.10 (s, 2H), 6.87 (d, J= 9.0 Hz, 2H), 6.5 (d, J= 9.0 Hz, 2H), 7.35 (d, J= 8.7 Hz, 2H),
7.54 (d, J= 8.7 Hz, 2H), 7.87 (s, IH), 10.37 (br s, IH); ESI-MS (m/z) 473.68 (M+H)+.
Example 14
M-{4-[4-(4-Methylphenyl]phenyl]-l,3-thiazol-2-yl}2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -b]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (27 mg, 0.141 mmol) and 2- bromo-N-[4-(4'-methylbiphenyl-4-yl)-l,3-thiazol-2-yl]acetamide (65 mg, 0.169 mmol) using NaH ( 9.0 mg, 0.212 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 21 mg of the product as a white solid; IR (Neat) 3020, 1638, 1424, 1215, 757 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 2.34 (s, 3H), 3.14 (s, 3H), 3.82 (t, J= 8.7 Hz, 2H), 4.00 (t, J= 8.4 Hz, 2H), 5.26 (s, 2H), 7.26 (d, J= 8.1 Hz, 2H), 7.60 (d, J= 8.4 Hz, 2H), 7.68-7.25 (m, 3H), 7.89 (s, IH), 7.96 (d, J= 8.4 Hz, 2H), 12.74 (br s, IH); ESI-MS {m/z) 496.31 (M-H)".
Example 15
Nl-{4-[4-(tert-Butyl)phenyl]phenyl}-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2,l-έ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.157 mmol) and 2- bromo-N-(4'-tert-butylbiphenyl-4-yl)acetamide (65 mg, 0.188 mmol) using NaH (9.0 mg, 0.212 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 26 mg of the product as a white solid; IR (Neat) 2924, 1635, 1463, 1158, 822 cm4; 1H NMR (300 MHz, DMS(W6) δ 1.30 (s, 9H), 3.15 (s, 3H), 3.81 (t, J = 9.3 Hz, 2H), 4.00 (t, J= 8.1 Hz, 2H), 5.13 (s, 2H), 7.30-7.43 (m, 3H), 7.49-7.64 (m, 5H), 7.88 (s, IH), 10.44 (br s, IH); ESI-MS (m/z) 457.46 (M+H)+.
Example 16
Nl-{4-[4-(3,3-Dimethyl-l-butynyl)phenyl]-l,3-thiazol-2-yl}2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-irnidazo[2,l-b]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.157 mmol) and 2- bromo-N- {4-[4-(3 ,3-dimethylbut- 1 -yn- 1 -yl)pheny I]- 1 ,3 -thiazol-2-yl} acetamide (71 mg, 0.188 mmol) using NaH (9.0 mg, 0.212 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 56 mg of the product as a white solid; IR (Neat) 3019, 2937, 1638, 1637, 1215, 756 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 1.30 (s, 9H), 3.14 (s, IH), 3.84 (t, J= 8.4 Hz, 2H), 4.00 (t, J= 8.1 Hz, 2H), 5.25 (s, 2H), 7.39 (d, J = 7.8 Hz, 2H), 7.70 (s, IH), 7.83-7.88 (m, 3H), 12.73 (br s, IH); ESI-MS (m/z) 486.37 (M-H)".
Example 17
N-[4-(4-Ethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetra-hydro-3H- imidazo[2,l-&]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.157 mmol) and 2- bromo-N-[4-(4-ethylphenyl)-l,3-thiazol-2-yl]acetamide (61 mg, 0.188 mmol) using ΝaΗ (9.0 mg, 0.212 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 39 mg of the product as a white solid; IR (Neat) 2922, 1638, 1462, 1082 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 1.20 (t, J= 7.8 Hz, 3H), 2.62 (q, J= 7.2 Hz, 2H), 3.14 (s, 3H), 3.82 (t, J= 9.0 Hz, 2H), 4.00 (t, J= 8.1 Hz, 2H), 5.25 (s, 2H), 7.25 (d, J = 8.4 Hz, 2H), 7.57 (s, IH), 7.25 (d, J= 7.8 Hz, 2H), 7.89 (s, IH), 12.70 (br s, IH); ESI- MS (m/z) 436.28 (M+H)+.
Example 18
N-[4-(l,2,3,4-Tetrahydro-6-naphthalenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -6]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.157 mmol) and 2- bromo-N-[4-(5,6,7,8-tetrahydronaphthalen-2-yl)-l,3-thiazol-2-yl]acetamide (66 mg, 0.188 mmol) using ΝaΗ (9.0 mg, 0.212 mmol) in DMF (3 ml) as solvent according to the
procedure described in Example 13 to give 45 mg of the product as a white solid; IR
(KBr) 3178, 2930, 1698, 1638, 1471, 1258, 1019, 745 cm"1; 1H NMR (300 MHz, DMSO- d6) δ 1.73-1.77 (m, 4H), 2.70-2.77 (m, 4H), 3.13 (s, 3H), 3.81 (t, J= 8.7 Hz, 2H), 3.99 (t, J= 9.3 Hz, 2H), 5.24 (s, 2H), 7.07 (d, J= 8.1 Hz, IH), 7.52-7.58 (m, 3H), 7.88 (s, IH), 12.68 (br s, IH); ESI-MS (m/z) 462.35 (M+H)+.
Example 19 N-[5-(tert-Butyl)-4,5,6,7-tetrahydrobenzo[β(l[l,3]thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -δ]purin-3 -yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.156 mmol) and 2- bromo-N-(6-tert-butyl-4,5,6,7-tetrahydro-l,3-benzothiazol-2-yl)acetamide (62 mg, 0.314 mmol) using NaH (9.4 mg, 0.208 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 25 mg of the product as a white solid; 1H NMR (300 MHz, DMSCW6) δ 0.91 (s, 9H), 1.08 (t, J = 6.9 Hz, IH), 1.30-1.48 (m, 2H), 1.89-1.98 (m, 2H), 2.34-2.42 (m, 2H), 2.64-2.72 (m, 2H), 3.16 (s, 3H), 3.82 (t, J= 8.1 Hz, 2H), 4.00 (t, J= 8.1 Hz, 2H), 5.18 (s, 2H), 7.87 (s, IH), 12.23 (br s, IH); ESI-MS (m/z) 442.35 (M+H)+.
Example 20
Nl-(4-{4-[4-(ført-Butyl)phenyl]phenyl}-l,3-thiazol-2-yl)-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -&]purin-3 -yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.1156 mmol) and 2- bromo-N-[4-(4'-/ert-butylbiphenyl-4-yl)-l,3-thiazol-2-yl]acetamide (80 mg, 0.188 mmol) using ΝaΗ (9.0 mg, 0.212 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 25 mg of the product as a white solid; IR (Neat) 3429, 2923, 1658, 1638, 1258, 1019, 720 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 1.32 (s, 9H), 3.14 (s, 3H), 3.82 (t, J= 8.1 Hz, 2H), 4.00 (t, J= 8.1 Hz, 2H), 5.26 (s, 2H), 7.46 (d, J = 8.1 Hz, 2H), 7.62 (d, J= 8.4 Hz, 2H), 7.68-7.73 (m, 3H), 7.89 (s, IH), 7.95 (d, J= 8.4 Hz, 2H), 12.73 (br s, IH); ESI-MS (m/z) 540.18 (M+H)+.
Example 21
Nl-[4-(4-Cyclopentyloxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2, 1 -έ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.156 mmol) and 2- bromo-N-{4-[4-(cyclopentyloxy)phenyl]-l,3-thiazol-2-yl}acetamide (71.79 mg, 0.188 mmol) using ΝaΗ ( 9.3 mg, 0.234 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 19 mg of the product as an off-white solid; IR (Neat) 3428, 1638, 1454, 1325, 1017, 771 cm"1; 1H NMR (300 MHz, DMSCM6) δ 1.58- 1.70 (m, 5H), 1.91-1-93 (m, 3H), 3.14 (s, 3H), 3.81 (t, J= 7.8 Hz, 2H), 3.99 (t, J = 8.4 Hz, 2H), 4.82-4.84 (m, IH), 5.24 (s, 2H)5 6.92 (d, J= 8.1 Hz, IH)5 7.44 (s, IH)5 7.77 (d, J = 7.8 Hz, 2H), 7.88 (s, IH)5 12.65 (br s, IH); ESI-MS (m/z) 490.26 (M-H)".
Example 22
M-[4-(4-Isopentyloxyphenyl)-l53-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2, 1 -Z>]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (40 mg, 0.209 mmol) and 2- bromo-N-{4-[4-(3-methylbutoxy)phenyl]-l53-thiazol-2-yl}acetamide (82 mg, 0.251 mmol) using ΝaΗ (13 mg, 0.314 mmol) in DMF as solvent (4 ml) according to the procedure described in Example 13 to give 16 mg of the product as a white solid; IR (KBr) 3390, 2873, 1640, 1563, 1272, 745 cm"1; 1H ΝMR (300 MHz5 DMSCW6) δ 0.93 (d, J = 6.3 Hz, 6H), 1.61-1.63 (m, 2H), 1.76-1.81 (m, IH), 3.13 (s, 3H), 3.81 (t, J= 8.4 Hz, 2H), 3.97-4.01(111, 4H)5 5.24 (s, 2H), 6.96 (d, J= 8.7 Hz, 2H), 7.46 (s, IH), 7.78 (d J = 8.4 Hz, 2H), 7.88 (s, IH), 12.65 (br s, IH); ESI-MS (m/z) 492.28 (M-H)"
Example 23
Nl-[4-(3-Isopentyloxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2,l-έ]purin-3-yl)acetamide
V-J
The title compound was prepared from Intermediate 1 (40 mg, 0.209 mmol) and 2- bromo-N-{4-[3-(3-methylbutoxy)phenyl]-l,3-thiazol-2-yl}acetamide (96 mg, 0.251 mmol) using NaH (13 mg, 0.314 mmol) in DMF as solvent (4 ml) according to the procedure described in Example 13 to give 40 mg of the product as a white solid; IR (Neat) 3020, 1637, 1522, 1215, 770 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 0.94 (d, J = 6.3 Hz, 6H), 1.63 (q, J= 6.3 Hz, 2H), 1.77-1.81 (m, IH), 3.16 (s, 3H), 3.85 (t, J= 8.4 Hz, 2H), 4.01-4.09 (m, 4H), 5.27 (s, 2H), 6.88 (d, J= 7.8 Hz, IH), 7.31 (t, J = 7.8 Hz, IH), 132-1 Al (m, 2H), 7.68 (s, IH), 7.94 (s, IH), 12.71 (br s, IH); ESI-MS (m/z) 494.46 (M+H)+.
Example 24 M-[4-(4-Cyclopentyloxyphenoxy)phenyl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -δ]purin-3 -yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.156 mmol) and 2- bromo-N-{4-[4-(cyclopentyloxy)phenoxy]phenyl}acetamide (73 mg, 0.187 mmol) using ΝaΗ (9.4 mg, 0.235 mmol) in DMF as solvent (3 ml) according to the procedure described in Example 13 to give 25 mg of the product as an off-white solid; IR (KBr) 3289, 2958, 1682, 1633, 1497, 1217, 831 cm"1; 1H ΝMR (300 MHz, DMSO-^6) δ 1.55- 1.70 (m, 6H), 1.86-1.88 (m, 2H), 3.15 (s, 3H), 3.81 (t, J= 8.7 Hz, 2H), 3.99 (t, J= 9.0 Hz5 2H), 4.73-4.75 (m, IH), 5.08 (s, 2H), 6.86-6.89 (m, 6H), 7.50 (d, J= 9.3Hz, 2H), 7.86 (s, IH), 10.32 (s, IH); ESI-MS (m/z) 501.43 (M+H)+.
Example 25
Nl-[4-(3-Fluoro-4-trifluoromethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (45 mg, 0.235 mmol) and 2- bromo-N-{4-[3-fluoro-4-(trifluoromethyl)phenyl]-l,3-thiazol-2-yl}acetamide (108 mg,
0.282 mmol) using NaH (14.12 mg, 0.358 mmol) in DMF as solvent (4 ml) according to the procedure described in Example 13 to give 35 mg of the product as an off-white solid; 1H NMR (300 MHz, DMSO-*) δ 3.14 (s, 3H), 3.82 (t, J = 8.1 Hz, 2H), 4.00 (t, J= 7.2 Hz, 2H), 5.26 (s, 2H), 7.84-7.98 (m, 4H), 8.00 (s, IH), 12.83 (br s, IH); ESI-MS (m/z) 476.22 (M+H)+.
Example 26
Nl-[4-(4-Pentylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazoβ, 1 -έ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (40 mg, 0.209 mmol) and 2- bromo-N-[4-(4-pentylphenyl)-l,3-thiazol-2-yl]acetamide (92 mg, 0.251 mmol) using NaH (14.12 mg, 0.358 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 13 to give 30 mg of the product as an off-white solid; IR (KBr) 3173, 2924, 1659, 1634, 1463, 1557, 1258, 1064, 751 cm"1; 1H NMR (300 MHz, DMSO- d6) δ 0.86 (t, J= 6.3 Hz, 3H), 1.28-1.30 (m, 4H)5 1.57-1.59 (m, 2H), 2.58 (t, J= 7.5 Hz5 2H)5 3.14 (s, 3H), 3.81 (t, J= 8.4 Hz, 2H), 3.99 (t, J= 8.4 Hz, 2H), 5.25 (s, 2H)5 7.22 (d, J = 7.8 Hz5 2H)5 7.56 (s, IH)5 7.78 (d, J= 7.8 Hz, 2H), 7.88 (s, IH)5 12.68 (br s5 IH); ESI- MS (m/z) 478.58 (M+H)+.
Example 27
M-[4-(4-Cyclohexylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,758-tetrahydro-3H- imidazo[2,l-έ]purin-3-yl)acetamide

2H), 7.88 (s, IH), 12.69 (br s, IH); ESI-MS (m/z) 490.36 (M+H)+.
Example 28 iVl-[4-(4-Isobutylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -δ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (40 mg, 0.209 mmol) and 2- bromo-N-[4-(4-isobutylphenyl)-l,3-thiazol-2-yl]acetamide (88 mg, 0.249 mmol) using ΝaΗ (12 mg, 0.312 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 13 to give 30 mg of the product as a white solid; IR (KBr) 3428, 2927, 1698, 1638, 1557, 1258, 752 cm4; 1H ΝMR (300 MHz, DMSO-J6) δ 0.87-0.89 (m, 6H), 1.85- 1.87 (m, IH), 3.14 (s, 3H), 3.82 (t, J= 8.1 Hz, 2H), 4.00 (t, J= 7.8 Hz, 2H), 5.25 (s, 2H), 7.20 (d, J= 8.7 Hz, 2H), 7.57 (s, IH), 7.78 (d, J= 8.7 Hz, 2H), 7.89 (s, IH), 12.69 (br s, IH); ESI-MS (m/z) 464.56 (M+H)+.
Example 29
Nl-[4-(3-Trifluoromethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2, 1 -ό]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (40 mg, 0.209 mmol) and 2- bromo-N-{4-[3-(trifluoromethyl)phenyl]-l,3-thiazol-2-yl}acetamide (91 mg, 0.255 mmol) using NaH (12 mg, 0.312 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 13 to give 25 mg of the product as an off-white solid; IR (KBr) 3235, 2962, 1697, 1664, 1635, 1557, 1342, 1106, 1018, 750 cm"1; 1H NMR (300 MHz, DMSCW6) δ 3.14 (s, 3H), 3.82 (t, J= 7.8 Hz, 2H), 4.01 (t, J= 8.7 Hz, 2H)5 5.26 (s, 2H), 7.68 (s, 2H), 7.09 (s, 2H), 8.21-8.23 (m, 2H), 12.80 (br s, IH); ESI-MS (m/z) 474.33 (M-H)-.
Example 30
Nl-[4-(2,5-Dimethyl-3-thienyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,758-tetrahydro- 3H-imidazo[2, 1 -6]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (40 mg, 0.209 mmol) and 2- bromo-N-[4-(2,5-dimethyl-3-thienyl)-l,3-thiazol-2-yl]acetamide (83 mg, 0.250 mmol) using NaH (12 mg, 0.312 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 13 to give 18 mg of the product as an off-white solid; 1H NMR (300 MHz, DMSO-J6) δ 2.38 (s, 3H), 2.57 (s, 3H), 3.14 (s, 3H), 3.82 (t, J= 7.8 Hz, 2H), 4.00 (t, J= 8.7 Hz, 2H), 5.24 (s, 2H), 7.0 (s, IH), 7.22 (s, IH), 7.88 (s, IH), 12.59 (br s, IH); ESI-MS (m/z) 442.17 (M+H)+.
Example 31
Nl-[4-(4-Trifluoromethoxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -Z>]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (40 mg, 0.209 mmol) and 2- bromo-N-{4-[4-(trifluoromethoxy)phenyl]-l,3-thiazol-2-yl}acetamide (96 mg, 0.251 mmol) using NaH (12 mg, 0.312 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 13 to give 42 mg of the product as a white solid; IR (Neat) 3166, 2958, 1663, 1638, 1560, 1173, 752 cm"1; 1H NMR (300 MHz, DMSO-^6) δ 3.14 (s, 3H), 3.82 (d, J= 9.3 Hz, 2H), 4.00 (t, J= 8.1 Hz, 2H), 5.26 (s, 2H), 7.42 (d, J = 7.8 Hz, 2H), 7.73 (s, IH), 7.89 (s, IH), 8.00 (d, J= 7.8 Hz, 2H), 12.76 (br s, IH); ESI-MS (m/z) 492.31 (M+H)+.
Example 32
Nl-{4-[4-(4-Chlorophenyl)phenyl]-l,3-thiazol-2-yl}2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -b]purin-3 -yl)acetamide

The title compound was prepared from Intermediate 1 (40 mg, 0.209 mmol) and 2- bromo-N-[4-(4'-chlorobiphenyl-4-yl)-l,3-thiazol-2-yl]acetamide (102 mg, 0.251 mmol) using NaH (13 mg, 0.314) in DMF (4 ml) as solvent according to the procedure described
in Example 13 to give 45 mg of the product as a white solid; IR (KBr) 3198, 2933, 1662,
1635, 1476, 1286, 1064 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 3.14 (s, 3H), 3.82 (t, J = 8.1 Hz, 2H), 4.00 (t, J= 8.7 Hz, 2H), 5.26 (s, 2H), 7.50 (d, J= 9.0 Hz, 2H), 1.12-1.16 (m, 4H), 7.89 (s, IH), 7.95-8.00 (m, 3H), 12.73 (br s, IH); ESI-MS (m/z) 519.06 (M+H)+.
Example 33
M-{4-[4-(3-Thienyl)phenyl]-l,3-thiazol-2-yl}-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -ό]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (50 mg, 0.262 mmol) and 2- bromo-N-{4-[4-(3-thienyl)phenyl]-l,3-thiazol-2-yl}acetamide (119 mg, 0.314 mmol) using NaH (16 mg, 0.392 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 13 to give 65 mg of the product as a white solid; IR (KBr) 3421, 2927, 1698, 1644, 1557, 1478, 1255, 1064, 748 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 3.15 (s, 3H), 3.83 (t, J= 6.9 Hz, 2H), 4.02 (t, J= 7.8 Hz, 2H), 5.27 (s, 2H), 7.59-7.70 (m, 3H), 7.78 (d, J= 7.2 Hz, 2H), 7.90-7.92 (m, 4H), 12.74 (br s, IH); ESI-MS (m/z) 490.59 (M+H)+.
Example 34
N-[4-(3-Bromo-4-fluorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2, 1 -Z>]purin-3-yl)acetamide

The title compound was prepare Νdi fΝrom Int 'ermediate 1 (40 mg, 0.209 mmol) and 2- bromo-N-[4-(3-bromo-4-fluorophenyl)-l,3-thiazol-2-yl]acetamide (98 mg, 0.248 mmol) using ΝaΗ (12 mg, 0.291 mmol) in DMF (4 ml) as solvent according to the procedure described in Example 13 to give 20 mg of the product as an off-white solid; IR (KBr) 3409, 2926, 1710, 1651, 1580, 1478, 1216, 1071 cm"1; 1HNMR (300 MHz, DMSO-J6) δ 3.14 (s, 3H), 3.98 (t, J= 7.8 Hz, 2H), 4.35 (t, J= 7.2 Hz, 2H), 5.36 (s, 2H), 7.44-7.46 (m, IH), 7.78 (s, IH), 7.91-7.93 (m, IH), 8.18-8.20 (m, 2H), 12.85 (br s, IH); ESI-MS (m/z) 506.03 (M+H)+.
Example 35
Nl-[4-(2,5-Dichloro-3-thienyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-
3H-imidazo[2, 1 -έ]purin-3-yl)acetamide

Example 36
Nl-[4-(2-Fluoro-4-trifluoromethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -έ]purin-3 -yl)acetamide

The title compound was prepared from Intermediate 1 (50 mg, 0.261 mmol) and 2- bromo-N-{4-[2-fluoro-4-(trifluoromethyl)phenyl]-l,3-thiazol-2-yl}acetamide (120 mg, 0.313 mmol) using ΝaΗ (15 mg, 0.392 mmol) in DMF (5 ml) as solvent according to the procedure described in Example 13 to give 25 mg of the product as a white solid; IR (KBr) 3204, 2952, 1663, 1634, 1542, 1336, 1124, 1077 cm"1; 1H ΝMR (300 MHz, DMSO-J6) δ 3.14 (s, 3H)5 3.82 (t, J= 9.0 Hz, 2H), 4.00 (t, J= 8.4 Hz, 2H), 5.27 (s, 2H), 7.69-7.84 (m, 2H), 7.89 (s, IH), 8.24-8.25 (m, IH), 12.84 (br s, IH); ESI-MS (m/z) 494.44 (M+H)+.
Example 37
Nl-{4-[3-Methyl-4-(3-trifluoromethylbenzyloxy)phenyl]-l,3-thiazol-2-yl}-2-(5-methyl- 4-0X0-4, 5,7, 8-tetrahydro-3H-imidazo [2, 1 -έ]purin-3 -y l)acetamide
 The title compound was prepared from Intermediate 1 (55 mg, 0.287 mmol) and 2- bromo-N-[4-(3-methyl-4-{[3-(trifluoromethyl)benzyl]oxy}phenyl)-l,3-thiazol-2- yl]acetamide (167 mg, 0.344 mmol) using NaH (17 mg, 0.416 mmol) in DMF (5 ml) as solvent according to the procedure described in Example 13 to give 40 mg of the product as an off-white solid; IR (KBr) 3194, 2949, 1658, 1637, 1557, 1331, 1123, 1073 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 2.26 (s, 3H), 3.15 (s, 3H), 3.82 (t, J= 8.7 Hz, 2H), 4.03 (t, J= 7.8 Hz, 2H), 5.26 (s, 2H), 7.06 (d, J = 7.2 Hz, IH), 7.46 (s, IH), 7.67-7.78 (m, 4H), 7.79-7.82 (m, 2H), 7.91 (s, IH), 12.68 (br s, IH); ESI-MS (m/z) 496.24 (M+H)+.
The title compound was prepared from Intermediate 1 (55 mg, 0.287 mmol) and 2- bromo-N-[4-(3-methyl-4-{[3-(trifluoromethyl)benzyl]oxy}phenyl)-l,3-thiazol-2- yl]acetamide (167 mg, 0.344 mmol) using NaH (17 mg, 0.416 mmol) in DMF (5 ml) as solvent according to the procedure described in Example 13 to give 40 mg of the product as an off-white solid; IR (KBr) 3194, 2949, 1658, 1637, 1557, 1331, 1123, 1073 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 2.26 (s, 3H), 3.15 (s, 3H), 3.82 (t, J= 8.7 Hz, 2H), 4.03 (t, J= 7.8 Hz, 2H), 5.26 (s, 2H), 7.06 (d, J = 7.2 Hz, IH), 7.46 (s, IH), 7.67-7.78 (m, 4H), 7.79-7.82 (m, 2H), 7.91 (s, IH), 12.68 (br s, IH); ESI-MS (m/z) 496.24 (M+H)+.
Example 38
M-[4-(4-Hexylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2,l-έ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (50 mg, 0.261 mmol) and 2- bromo-N-[4-(4-hexylphenyl)-l,3-thiazol-2-yl]acetamide (119 mg, 0.312 mmol) using ΝaΗ (15 mg, 0.375 mmol) in DMF (5 ml) as solvent according to the procedure described in Example 13 to give 35 mg of the product as an off-white solid; IR (KBr) 3173, 2925, 1660, 1638, 1559, 1259, 1018 Cm-1; 1H NMR (300 MHz, DMSO-^6) δ 0.84-0.85 (m, 3H), 1.27-1.29 (m, 5H), 1.56-1.58 (m, 2H), 2.57-2.59 (m, 3H), 3.14 (s, 3H), 3.82 (t, J= 8.4 Hz, 2H), 4.00 (t, J= 8.4 Hz, 2H), 5.25 (s, 2H), 7.23 (d, J= 6.9 Hz, 2H), 7.56 (s, IH), 7.78 (d, J= 6.9 Hz, 2H), 7.88 (s, IH), 12.69 (br s, IH); ESI-MS (m/z) 490.43 (M-H)".
Example 39
Nl-[4-(4-Chloro-3-trifluoromethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -ό]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.261 mmol) and 2- bromo-N-{4-[4-chloro-3-(trifluoromethyl)phenyl]-l,3-thiazol-2-yl}acetamide (125 mg,
0.313 mmol) using NaH (10 mg, 0.375 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 35 mg of the product as an off-white solid; IR (KBr) 3140, 2988, 1663, 1634, 1557, 1470, 1256, 1136, 746 cm"1; 1H NMR (300 MHz5 DMSO-J6) δ 3.13 (s, 3H), 3.82 (t, J= 6.9 Hz, 2H), 4.00 (t, J= 6.3 Hz, 2H), 5.26 (s, 2H), 7.80-7.96 (m, 3H), 8.19 (d, J= 7.5 Hz, IH), 8.32 (s, IH), 12.83 (br s, IH); ESI-MS (m/z) 510.89 (M+H)+.
Example 40
Nl-[4-(3-Chloro-4-isopentyloxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5s7,8- tetrahydro~3H-imidazo[2, 1 ~6]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (50 mg, 0.261 mmol) and 2- bromo-N-{4-[3-chloro-4-(3-methylbutoxy)phenyl]-l,3-thiazol-2-yl}acetamide (131 mg, 0.313 mmol) using ΝaΗ (15 mg, 0.375 mmol) in DMF (5 ml) as solvent according to the procedure described in Example 13 to give 30 mg of the product as a white solid; IR (KBr) 3137, 2954, 1659, 1637, 1556, 1472, 1262, 1013 cm"1; 1H ΝMR (300 MHz, DMSO-J6) δ 0.95 (d, J = 6.9 Hz, 6H), 1.65 (q, J = 6.3 Hz, 2H), 1.79-1.83 (m, IH), 3.19 (s, 3H), 3.88 (t, J= 8.7 Hz, 2H), 4.11 (t, J= 6.6 Hz, 2H), 5.29 (s, 2H), 7.21 (d, J= 9.0 Hz, IH), 7.62 (s, IH), 7.62-7.82 (m, IH), 7.93 (s, IH), 8.00 (s, IH), 12.74 (br s, IH); ESI-MS (m/z) 528.45 (M+H)+.
Example 41
Nl-[4-(3-Chloro-4-difluoromethoxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -δ]purin-3-yl)acetamide
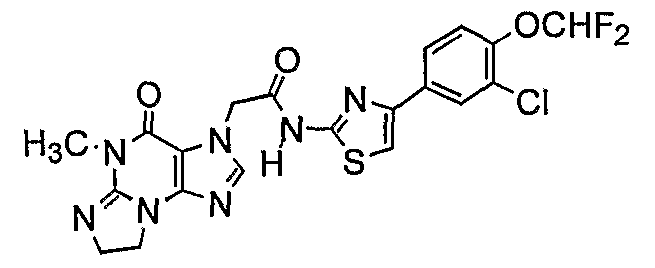
The title compound was prepared from Intermediate 1 (50 mg, 0.261 mmol) and 2- bromo-N-{4-[3-chloro-4-(difluoromethoxy)phenyl]-l,3-thiazol-2-yl}acetamide (124 mg, 0.313 mmol) using NaH (15 mg, 0.375 mmol) in DMF (5 ml) as solvent according to the procedure described in Example 13 to give 25 mg of the product as a white solid; IR (KBr) 3171, 2993, 1658, 1634, 1558, 1471, 1255, 1046, 748 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 3.14 (s, 3H), 3.82 (t, J= 8.1 Hz, 2H), 4.00 (t, J= 8.4 Hz, 2H), 5.25 (s, 2H),
7.31 (t, J= 72.9 Hz, IH), 7.42 (d, J= 8.4 Hz, IH), 7.80 (s, IH), 7.87-7.92 (m, 2H), 8.08
(s, IH), 12.76 (br s, IH); ESI-MS (m/z) 508.49 (M+H)+.
Example 42
Nl-[4-(3,5-Dichloro-4-difluoromethoxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (43 mg, 0.224 mmol) and 2- bromo-N-{4-[3,5-dichloro-4-(difluoromethoxy)phenyl]-l,3-thiazol-2-yl}acetamide (116 mg, 0.269 mmol) using ΝaΗ (13 mg, 0.337 mmol) in DMF (5 ml) as solvent according to the procedure described in Example 13 to give 30 mg of the product as a white solid; IR (KBr) 3176, 2924, 1698, 1669, 1636, 1572, 1387, 1143, 758 cm"1; 1H ΝMR (300 MHz, DMSO-J6) δ 3.13 (s, 3H), 3.81 (t, J= 8.2 Hz, 2H), 3.99 (t, J= 8.1 Hz, 2H), 5.25 (s, 2H), 7.17 (t, J= 72.9 Hz, IH), 7.88 (s, IH), 7.96 (s, IH), 8.09 (s, IH), 12.77 (br s, IH); ESI-MS (m/z) 542.64 (M+H)+.
Example 43
Nl-[4-(4-Difluoromethoxy-3,5-difluorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-δ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (43 mg, 0.224 mmol) and 2- bromo-N-{4-[4-(difluoromethoxy)-3,5-difluorophenyl]-l,3-thiazol-2-yl}acetamide (107 mg, 0.268 mmol) using NaH (13 mg, 0.337 mmol) in DMF (5 ml) as solvent according to the procedure described in Example 13 to give 30 mg of the product as a white solid; IR (KBr) 3137, 2993, 1700, 1661, 1637, 1432, 1232, 1061, 749 cm4; 1H NMR (300 MHz, DMSO-J6) δ 3.13 (s, 3H), 3.81 (t, J= 9.3 Hz, 2H), 4.00 (t, J= 9.3 Hz, 2H), 5.26 (s, 2H), 7.26 (t, J= 72.3 Hz, IH), 7.77 (d, J= 9.3 Hz, 2H), 7.89 (d, J= 6.0 Hz, 2H), 12.78 (br s, IH); ESI-MS (m/z) 510.51 (M+H)+.
Example 44
M-{4-[3,5-dichloro-4-(3-methylbutoxy)]phenyl}-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2, 1 -/b]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.156 mmol) and 2- bromo-N-{4-[3,5-dichloro-4-(3-methylbutoxy)phenyl]-l,3-thiazol-2-yl}acetamide (85 mg, 0.187 mmol) using NaH (9 mg, 0.375 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 45 mg of the product as an off-white solid; IR (KBr) 2957, 1634, 1563, 1471, 1257, 804 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 0.96 (d, J = 6.3 Hz, 6H), 1.85-1.92 (m, IH), 3.17 (s, 3H), 3.82-3.87 (m, 2H), 3.98-4.11 (m, 6H), 5.27 (s, 2H), 7.87 (s, IH), 7.90 (s, IH), 8.00 (s, 2H), 12.37 (br s, IH); ESI-MS (m/z) 562.86 (M)+.
Example 45
Nl-{4-[3,5-dichloro-4-(3,3,3-trifluoropropoxy)]phenyl}-l,3-thiazol-2-yl]-2-(5-methyl-4- oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-Z>]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (32 mg, 0.167 mmol) and 2- bromo-N- {4- [3 ,5 -dichloro-4-(3 ,3 , 3 -trifluoropropoxy)phenyl] - 1 ,3 -thiazol-2-yl } acetamide (96 mg, 0.201 mmol) using ΝaΗ (10.0 mg, 0.416 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 40 mg of the product as a white solid; IR (KBr) 3132, 1634, 1330, 1244, 806 cm"1; 1H ΝMR (300 MHz, DMSO-J6) δ 2.80-2.89 (m, 2H), 3.13 (s, 3H), 3.81 (t, J= 8.4 Hz, 2H), 3.99 (t, J= 8.7 Hz, 2H), 4.19 (t, J = 6.0 Hz, 2H), 5.25 (s, 2H), 7.87 (s, 2H), 8.00 (s, 2H), 12.74 (br s, IH); ESI-MS (m/z) 589.43 (M+H)+.
Example 46
Nl-{4-[3,5-bis(trifluoromethyl)phenyl]-l,3-thiazol-2-yl}-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -ό]purin-3-yl)acetamide

[3,5-bis(trifluoromethyl)phenyl]-l,3-thiazol-2-yl}-2-bromoacetamide (68 mg, 0.157 mmol) using NaH (9 mg, 0.208 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 25 mg of the product as a white solid; IR (KBr) 2962, 1637, 1550, 1287, 898 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 3.17 (s, 3H), 3.81-3.86 (m, 2H), 3.99-4.06 (m, 2H), 5.29 (s, 2H), 7.92 (s, IH), 8.08 (s, IH), 8.22 (s, IH), 8.57 (s, 2H), 12.89 (br s, IH); ESI-MS (m/z) 544.40 (M+H)+.
Example 47
Nl-[4-(3,5-dichloro-4-(2,2-dimethylpropoxy)phenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4- oxo-4,5,7,8-tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.157 mmol) and 2- bromo-N- {4-[3 , 5 -dichloro-4-(2,2-dimethylpropoxy)phenyl] -1,3 -thiazol-2-yl } acetamide (85 mg, 0.188 mmol) using ΝaΗ (9 mg, 0.375 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 30 mg of the product as a white solid; IR (KBr) 2871, 1634, 1470, 1038, 804 cm"1; 1H ΝMR (300 MHz, DMSO-J6) δ 1.80 (s, 9H), 3.15 (s, 3H), 3.67 (s, 2H), 3.80-3.86 (m, 2H), 3.98-4.04 (m, 2H), 5.27 (s, 2H), 7.87 (s, IH), 7.91 (s, IH), 8.00 (s, 2H), 12.78 (br s, IH); ESI-MS (m/z) 562.79 (M+H)+.
Example 48
Nl-[4-(4-Cyclobutylmethoxy)-3,5-dichlorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-δ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.157 mmol) and 2- bromo-N-{4-[3,5-dichloro-4-(cyclobutylmethoxy)phenyl]-l ,3-thiazol-2-yl} acetamide (84 mg, 0.188 mmol) using NaH (9.0 mg, 0.375 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 35 mg of the product as a white solid; IR (KBr) 2954, 1635, 1470, 1216, 753 cm4; 1H NMR (300 MHz, DMSO-J6) δ 1.88-1.96 (m, 4H), 2.02-2.12 (m, 2H), 2.72-2.79 (m, IH), 3.14 (s, 3H), 3.78-3.86 (m, 2H), 3.95-4.03
(s, 4H), 5.27 (s, 2H), 7.87 (s, IH), 7.91 (s, IH), 8.00 (s, 2H), 12.79 (br s, IH); ESI-MS
(m/z) 560.21 (M+H)+.
Example 49 Nl-[4-(3,5-dichloro-4-[4-(trifluoromethyl)benzyloxy]phenyl)-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.157 mmol) and 2- bromo-N-[4-(3,5-dichloro-4-{[4-(trifluoromethyl)benzyl]oxy}phenyl)-l,3-thiazol-2- yl]acetamide (101 mg, 0.187 mmol) using NaH (9.0 mg, 0.375 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 36 mg of the product as a white solid; IR (KBr) 3070, 1634, 1334, 1121, 1068, 723 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 3.15 (s, 3H), 3.81-3.88 (m, 2H), 3.99-4.05 (m, 2H), 5.18 (s, 2H), 5.28 (s, 2H), 7.75-7.84 (m, 4H), 7.92 (s, 2H), 8.06 (s, 2H), 12.81 (br s, IH); ESI-MS {m/z) 650.16 (M)+.
Example 50
M-[4-(3,5-difluoro-4-[2-(trifluoromethyl)benzyloxy]phenyl)-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-Z>]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (30 mg, 0.157 mmol) and 2- bromo-N-[4-(3,5-difluoro-4-{[2-(trifluoromethyl)benzyl]oxy}phenyl)-l,3-thiazol-2- yl]acetamide (95 mg, 0.187 mmol) using NaH (9 mg, 0.375 mmol) in DMF (3 ml) as solvent according to the procedure described in Example 13 to give 36 mg of the product as a white solid; IR (KBr) 2991, 1637, 1313, 1127, 1034, 751 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 3.15 (s, 3H), 3.79-3.87 (m, 2H), 3.96-4.05 (m, 2H), 5.27 (s, 2H), 5.35 (s, 2H), 7.64-7.70 (m, 3H), 7.76-7.84 (m, 4H), 7.91 (s, IH), 12.79 (br s, IH); APCI-MS (m/z) 618.26 (M+H)+.
Example 51
Nl-[4-(3,5-dichloro-[4-(3,4-difluorobenzyloxy)]phenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4- oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-ib]purin-3-yl)acetamide
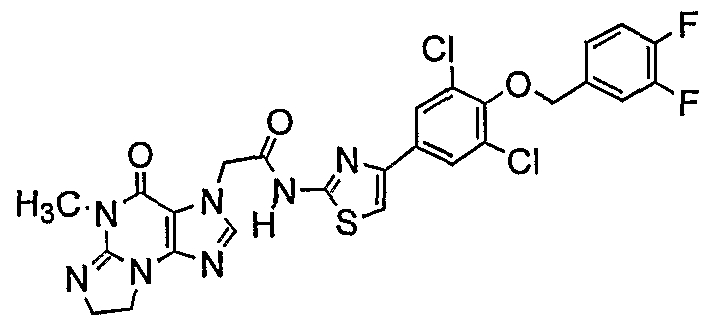
The title compound was prepared from Intermediate 1 (50 mg, 0.261 mmol) and 2- bromo-N-(4-{3,5-dichloro-4-[(3,4-difluorobenzyl)oxy]phenyl}-l,3-thiazol-2- yl)acetamide (159 mg, 0.312 mmol) using ΝaΗ (15 mg, 0.625 mmol) in DMF (5 ml) as solvent according to the procedure described in Example 13 to give 40 mg of the product as a white solid; IR (KBr) 3130, 1661, 1634, 1518, 1256, 754 cm"1; 1H ΝMR (300 MHz, DMSO-J6) δ 3.15 (s, 3H), 3.83 (t, J= 8.4 Hz, 2H), 4.01 (t, J= 8.7 Hz, 2H), 5.07 (s, 2H), 5.27 (s, 2H), 7.36-7.42 (m, IH), 7.47 (d, J= 8.1 Hz, IH), 7.57-7.65 (m, IH), 7.91 (s, 2H), 8.04 (s, 2H), 12.79 (br s, IH); APCI-MS (m/z) 618.44 (M)+.
Example 52
Nl-[4-(3,5-dichloro-[4-(3,3,4,4,4-pentafluorobutoxy)]phenyl)-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-6]purin-3~yl)acetamide
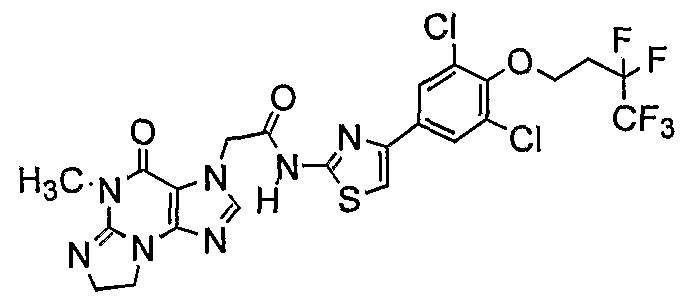
The title compound was prepared from Intermediate 1 (50 mg, 0.261 mmol) and 2- bromo-N-{4-[3,5-dichloro-4-(3,3,4,4,4-pentafluorobutoxy)phenyl]-l,3-thiazol-2- yl}acetamide (165 mg, 0.313 mmol) using ΝaΗ (15 mg, 0.625 mmol) in DMF (5 ml) as solvent according to the procedure described in Example 13 to give 41 mg of the product as an off-white solid; IR (KBr) 3188, 1634, 1452, 1193, 806 cm4; 1H ΝMR (300 MHz, CDCl3) δ 2.75-2.89 (m, 2H), 3.15 (s, 3H), 3.83 (t, J= 8.7 Hz, 2H), 4.01 (t, J = 8.7 Hz, 2H), 4.25-4.32 (m, 2H), 5.27 (s, 2H), 7.91 (s, 2H), 8.03 (s, 2H), 12.78 (br s, IH); ESI-MS (m/z) 638.26 (M)+.
Example 53
Nl-[4-(3,5-dichloro-[4-(cyclohexylmethoxy)]phenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4- oxo-4,5,7,8-tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (50 mg, 0.261 mmol) and 2- bromo-N-{4-[3,5-dichloro-4-(cyclohexylmethoxy)phenyl]-l,3-thiazol-2-yl}acetamide (145 mg, 0.313 mmol) using NaH (15 mg, 0.625 mmol) in DMF (5 ml) as solvent according to the procedure described in Example 13 to give 71 mg of the product as a white solid; IR (KBr) 2926, 1634, 1564, 1450, 1255, 804 cnT1; 1H NMR (300 MHz, DMSO-J6) δ 1.11-1.30 (m, 5H), 1.70-1.76 (m, 3H), 1.84-1.90 (m, 3H), 3.15 (s, 3H), 3.81- 3.87 (m, 4H), 3.98-4.05 (m, 2H), 5.27 (s, 2H), 7.84 (s, IH), 7.90 (s, IH), 7.99 (s, 2H), 12.73 (br s, IH); APCI-MS (m/z) 588.17 (M)+.
Example 54
Nl-[4-(3,5-dichloro-{4-[2-(trifluoromethyl)benzyloxy]}phenyl)-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-&]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (50 mg, 0.261 mmol) and 2- bromo-N-[4-(3 , 5 -dichloro-4- { [2-(trifluoromethyl)benzyl] oxy } phenyl)- 1 ,3 -thiazol-2- yl]acetamide (169 mg, 0.313 mmol) using ΝaΗ (15 mg, 0.625 mmol) in DMF (5 ml) as solvent according to the procedure described in Example 13 to give 71 mg of the product as a white solid; IR (KBr) 3073, 1634, 1548, 1214, 754 cm"1; 1H ΝMR (300 MHz, DMSO-J6) δ 3.15 (s, 3H), 3.83 (t, J= 8.1 Hz, 2H)5 4.01 (t, J= 8.7 Hz, 2H), 5.24 (s, 2H), 5.28 (s, 2H), 7.63 (d, J= 7.2 Hz, IH), 7.75-7.83 (m, 2H), 7.90-7.97 (m, 3H), 8.06 (s, 2H), 12.79 (br s, IH); ESI-MS (m/z) 650.08 (M)+.
Example 55
Nl-[4-(3,5-dichloro-{4-[3-(trifluoromethyl)benzyloxy]}phenyl)-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (50 mg, 0.261 mmol) and 2- bromo-N-[4-(3,5-dichloro-4-{[3-(trifluoromethyl)benzyl]oxy}phenyl)-l,3-thiazol-2-
yl]acetamide (169 mg, 0.312 mmol) using NaH (15 mg, 0.375 mmol) in DMF (5ml) as solvent according to the procedure described in Example 13 to give 72 mg of the product as an off-white solid; IR (KBr) 3182, 2949, 1666, 1638, 1469, 1332, 1111, 800 cm-'; 1H NMR (300 MHz, DMSO-J6) δ 3.15 (s, 3H), 3.78-3.86 (m, 2H), 3.97-4.02 (m, 2H), 5.19 (s, 2H), 5.27 (s, 2H), 7.69 (d, J= 6.9 Hz, IH), 7.77 (d, J = 6.9 Hz, IH), 7.87-7.93 (m, 4H), 8.05 (s, 2H), 12.79 (br s, IH); ESI-MS (m/z) 650.12 (M)+.
Example 56
5-Methyl-4-oxo-N-[4-(2,3,4-trichlorophenyl)-l,3-thiazol-2-yl]-4,5,7,8-tetrahydro-3H- imidazo[2, 1 ~έ]purine-3-carboxamide

The title compound was prepared from Intermediate 1 (50 mg, 0.262 mmol) and 2- bromo-N-[4-(2,3,4-trichlorophenyl)-l,3-thiazol-2-yl]acetamide (125 mg, 0.314 mmol) using NaH (16 mg, 0.392 mmol) in DMF (5 ml) as solvent according to the procedure described in Example 13 to give 85 mg of the product as a white solid; IR (KLBr) 2961, 1641, 1564, 1261, 1089, 758 cm"1; 1HNMR (300 MHz, DMSO-J6) δ 3.15 (s, 3H), 3.83 (t, J= 8.4 Hz, 2H), 4.01 (t, J= 8.7 Hz, 2H), 5.27 (s, 2H), 7.72 (s, IH), 7.77 (s, 2H), 7.91 (s, IH), 12.82 (br s, IH); APCI-MS (m/z) 510.24 (M+H)+
Example 57
Nl-[4-(3,5-difluoro-4-(3,3,3-trifluoroethoxy)phenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-ό]purin-3-yl)acetamide

The title compound was prepared from Intermediate 1 (50 mg, 0.262 mmol) and 2- bromo-N-{4-[3,5-difluoro-4-(2,2,2-trifluoroethoxy)phenyl]-l,3-thiazol-2-yl}acetamide (135 mg, 0.314 mmol) using NaH (16 mg, 0.392 mmol) in DMF (5, ml) as solvent according to the procedure described in Example 13 to give 70 mg of the product as an off-white solid; IR (KBr) 2989, 1638, 1560, 1349, 1292, 1034, 753 cm"1; 1H NMR (300 MHz, DMSO-J6) δ 3.14 (s, 3H), 3.83 (t, J= 8.4 Hz, 2H), 4.01 (t, J= 8.7 Hz, 2H), 4.86 (q, J= 8.7 Hz, 2H), 5.27 (s, 2H), 7.70 (d, J= 9.3 Hz, 2H), 7.85 (s, IH), 7.91 (s, IH), 12.80 (br s, IH); APCI-MS (m/z) 542.30 (M+H)+.
Pharmacological activity
The illustrative examples of the present invention are screened for TRPAl activity according to a modified procedure described in (a) Tόth, A., Kedei, N., Wang, Y. and Blumberg, P. M. Life Sciences (2003), 73, 487-498. (b) McNamara C, R. et al. (2007) PNAS 104, 13525-13530. The screening of the compounds can be carried out by other methods and procedures known to persons skilled in the art.
Screening for TRPAl antagonist using the 45Calcium uptake assay
The inhibition of TRPAl receptor activation was followed as inhibition of allyl isothiocyanate (AITC) induced cellular uptake of radioactive calcium. Test compounds were dissolved in DMSO to prepare 10 mM stock solution and then diluted using plain medium with 0.1% BSA and 1.8 mM CaCl2 to get desired concentration. Final concentration of DMSO in the reaction was 0.5% (v/v). Human TRPAl expressing CHO cells were grown in F- 12 DMEM medium with 10% FBS, 1% penicillin-streptomycin solution, 400 μg / ml of G-418. Cells were seeded 24 h prior to the assay in 96 well plates so as to get ~ 50,000 cells per well on the day of experiment. Cells were treated with test compounds for 10 min followed by addition of AITC at a final concentration of 30 μM and 5 μCi/ml 45Ca+2 for 3 min. Cells were washed and lysed using buffer containing 1% Triton X-IOO5 0.1 % deoxycholate and 0.1% SDS. Radioactivity in the lysate was measured in Packardt Top count after addition of liquid scintillant. Concentration response curves were plotted as a % of maximal response obtained in the absence of test antagonist. IC50 value was calculated from concentration response curve by nonlinear regression analysis using GraphPad PRISM software.
The compounds prepared were tested using the above assay procedure and the results obtained are given in Table 2, Percentage inhibition at concentrations of 1.0 μM and 10.0 μM are given in the table along with IC50 (nM) details for selected examples.
The IC50 (nM) values of the compounds are set forth in Table 2 wherein "A" refers to an IC50 value of less than 500 nM, "B" refers to IC50 value in range of 500.1 to 1000.0 nM and "C" refers to an IC50 value of more than 1000 nM
Table 2: In-vitro screening results of compounds of invention



Claims
1. A compound of Formula (I)

R1, R2 and R3 independently represents hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted heterocyclic ring or substituted or unsubstituted heterocyclylalkyl;
R4 and R5 independently represents hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkylalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted heterocyclic ring or substituted or unsubstituted heterocyclylalkyl;
2. A compound according to claim 1, wherein R1 is methyl.
3. A compound according to any of claim 1 or 2, wherein R2 is hydrogen.
4. A compound according to any of claims 1 to 3, wherein R3 is hydrogen.
5. A compound according to any of claims 1 to 4, wherein R4 is hydrogen.
6. A compound according to any of claims 1 to 5, wherein R5 is substituted or unsubstituted thiazole.
7. A compound according to any of claims 1 to 5, wherein R5 is substituted or unsubstituted phenyl.
8. The compound of claim 1, wherein, the compound is selected from N-[2-(4-Chlorophenyl)ethyl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo
[2, 1 -ό]purin-3-yl)acetamide; N-[2-(4-Bromophenyl)ethyl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo
[2, 1 -δ]purin-3-yl)acetamide;
N-[5-(ter?-Butyl)-4,5,6,7-tetrahydrobenzo[cri[l,3]thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2, 1 -ό]purin-3-yl)acetamide;
Nl-[4-(4-Chloro-3-trifluoromethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5, 7, 8-tetrahydro-3H-imidazo [2, 1 -Z?]purin-3 -yl)acetamide;
Nl-{4-[3,5-dichloro-4-(3-methylbutoxy)]phenyl}-l,3-thiazol-2-yl]-2-(5-methyl-4- oxo-4,5,7,8-tetrahydro-3Η-imidazo[2,l-b]purin-3-yl)acetamide;
Nl-{4-[3,5-dichloro-4-(3,3,3-trifluoropropoxy)]ρhenyl}-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-b]purin-3-yl)acetamide; iVi-[4-(4-Cyclobutylmethoxy)-3,5-dichlorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl- 4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-b]purin-3-yl)acetamide; and
Nl-[4-(3,5-dichloro-4-[4-(trifluoromethyl)benzyloxy]phenyl)-l,3-thiazol-2-yl]-2- (5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-b]purin-3-yl)acetamide; or a pharmaceutically acceptable salt thereof, an ester thereof, a tautomer thereof, regioisomer thereof or a stereoisomer thereof,
9. A compound of formula (Ia)

R6 represents hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkylalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted heterocyclic ring and substituted or unsubstituted heterocyclylalkyl;
R7 independently represents hydrogen or alkyl; and R and R together with the carbon atoms which they are attached to, tbrm a optionally substituted 3 to 7 saturated, unsaturated or partially saturated cyclic ring, which may optionally include one or more heteroatoms selected from O, Nor S;
10. A compound according to claim 9, wherein R6 is substituted of unsubstituted phyenyl.
11. A compound according to claim 9, wherein R6 is substituted of unsubstituted thienyl.
12. A compound according to any of claims 9 to 11, wherein R7 is hydrogen.
13. A compound according to any of claims 9 to 11, wherein R7 is methyl.
14. The compound of claim 9, wherein, the compound is selected from
N-[4-(4-Bromophenyl)-l,3~thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2,l-έ]purin-3-yl)acetamide;
N-[4-(4-Methylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2, 1 -ό]purin-3-yl)acetamide;
N-[4-(4-Chlorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2, 1 -έ]purin-3-yl)acetamide;
N-[4-(4-Bromophenyl)-5 -methyl- 1 ,3 -thiazol-2-yl]-2-(5-methyl-4-oxo-4, 5,7,8- tetrahydro-3H-imidazo[2, 1 -έ]purin-3 -yl)acetamide;
2-(5-Methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-δ]purin-3-yl)-N-{4-[4- (trifluoromethy l)phenyl] - 1 ,3 -thiazol-2-yl } acetamide ;
N-[4-(4-Cyanophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -έ]purin-3-yl)acetamide;
N-[4-(3 ,4-Dichlorophenyl)- 1 ,3 -thiazol-2-yl]-2-(5 -methyl-4-oxo-4,5 ,7, 8-tetra- hydro-3H~imidazo[2, 1 -δ]purin-3-yl)acetamide;
2-(5-Methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)-N-[4-(4-tert- butylphenyl)-l,3-thiazol-2-yl]acetamide;
Nl-{4-[4-(4-Methylphenyl]phenyl]-l,3-thiazol-2-yl}2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -b]purin-3-yl)acetamide;
Nl-{4-[4-(3,3-Dimethyl-l-butynyl)phenyl]-l,3-thiazol-2-yl}2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-b]purin-3-yl)acetamide;
N-[4-(4-Ethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -δ]purin-3-yl)acetamide;
N-[4-(l,2,3,4-Tetrahydro-6-naphthalenyl)-l,3-thiazol-2-yi]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide; Nl -(4- {4~[4-(/ert-Butyl)phenyl]phenyl} - 1 ,3 -thiazol-2-yl)-2-(5 -methyl-4-oxo-
4,5,7,8-tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide;
Nl-[4-(4-Cyclopentyloxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide;
Nl-[4-(4-Isopentyloxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5s7,8-tetra- hydro-3H-imidazo[2,l-ό]purin-3-yl)acetamide;
M-^-CS-Isopentyloxypheny^-l^-thiazol^-yy^-CS-methyl^-oxo^.S^.δ-tetra- hydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide;,
Nl-[4-(3-Fluoro-4-trifluoromethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-6]purin-3-yl)acetamide;
Nl-[4-(4-Pentylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2, 1 -Z>]purin-3-yl)acetamide;,
Nl-[4-(4-Cyclohexylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -&]purin-3-yl)acetamide;
Nl-[4-(4-Isobutylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2,l-Z>]purin-3-yl)acetamide;
Nl-[4-(3-Trifluoromethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide;
Nl-[4-(2,5-Dimethyl-3-thienyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetra- hydro-3H-imidazo[2, 1 -&]purin-3-yl)acetamide;
Nl-[4-(4-Trifluoromethoxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -Z>]purin-3-yl)acetamide;
Nl-{4-[4-(4-Chlorophenyl)phenyl]-l,3-thiazol-2-yl}2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -b]purin-3-yl)acetamide;
Nl-{4-[4-(3-Thienyl)phenyl]-l,3-thiazol-2-yl}-2-(5-methyl-4-oxo-4,5,7,8-tetra- hydro-3H-imidazo[2, 1 -&]purin-3-yl)acetamide;
N-[4-(3-Bromo-4-fluorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetra- hydro-3H-imidazo[2,l-Z>]purin-3-yl)acetamide;
Nl-[4-(2,5-Dichloro-3-thienyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetra- hydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide;
Nl-[4-(2-Fluoro-4-trifluoromethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-δ]purin-3-yl)acetamide;
Nl-{4-[3-Methyl-4-(3-trifluoromethylbenzyloxy)phenyl]-l,3-thiazol-2-yl}-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)acetamide; M-[4-(4-Hexylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-
3H-imidazo[2,l-Z>]purin-3-yl)acetamide;
Nl-[4-(4-Chloro-3-trifluoromethylphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin~3-yl)acetamide;
Nl-[4-(3-Chloro-4-isopentyloxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5, 7,8-tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide;,
Nl-[4-(3-Chloro-4-difluoromethoxyphenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo- 4,5,7,8-tetrahydro-3H~imidazo[2,l-ά]purin-3-yl)acetamide;
Nl-^-CS^-Dichloro^-difluoromethoxypheny^-l^-thiazol^-yll^-CS-methyl^- oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-ό]purin-3-yl)acetamide;
Nl-[4-(4-Difluoromethoxy-3,5-difluorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4- oxo-4,5,7,8-tetrahydro-3H-imidazo[2, 1 -έ]purin-3-yl)acetamide;
Nl -{4-[3,5-dichloro-4-(3-methylbutoxy)]phenyl} - 1 ,3-thiazol-2-yl]-2-(5-methyl-4- oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-δ]purin-3-yl)acetamide;
Nl-{4-[3,5-dichloro-4-(3,3,3-trifluoropropoxy)]phenyl}-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-δ]purin-3-yl)acetamide;
Nl-{4-[3,5-bis(trifluoromethyl)phenyl]-l,3-thiazol-2-yl}-2-(5-methyl-4-oxo-4,5, 7,8-tetrahydro-3H-imidazo[2,l-δ]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-4-(2,2-dimethylpropoxy)phenyl)-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-Z>]purin-3-yl)acetamide;
Nl-[4-(4-Cyclobutylmethoxy)-3,5-dichlorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl- 4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-ό]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-4-[4-(trifluoromethyl)benzyloxy]phenyl)-l,3-thiazol-2-yl]-2- (5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-ό]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-4-[4-(trifluoromethyl)benzyloxy]phenyl)-l,3-thiazol-2-yl]-2- (5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-6]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-[4-(3,4-difluorobenzyloxy)]phenyl)-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-ό]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-[4-(3,3,4,4,4-pentafluorobutoxy)]phenyl)-l,3-thiazol-2-yl]-2- (5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-Z>]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-[4-(cyclohexylmethoxy)]phenyl)-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-δ]purin-3-yl)acetamide;
Nl-[4-(3,5-dichloro-{4-[2-(trifluoromethyl)benzyloxy]}phenyl)-l,3-thiazol-2-yl]- 2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-&]purin-3-yl)acetamide; Nl-[4-(3,5-dichloro-{4-[3-(trifluoromethyl)benzyloxy]}phenyl)-l,3-thiazol-2-yl]-
2~(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-%urin-3-yl)acetamide;
Nl-[4-(3,5-difluoro-4-(3,3,3-trifluoroethoxy)phenyl)-l,3-thiazol-2-yl]-2-(5- methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-ό]purin-3-yl)acetamide; and
Nl-[4-(2,3,4-Trichlorophenyl)-l,3-thiazol-2-yl]-2-(5-methyl-4-oxo-4,5,7,8- tetrahydro-3H-imidazo[2, 1 -δ]purin-3-yl)acetamide; or a pharmaceutically acceptable salt thereof, an ester thereof, a tautomer thereof, regioisomer thereof or a stereoisomer thereof,
15. A compound of formula (Ib)

16. The compound of claim 15, wherein, the compound is selected from
2-(5-Methyl-4-oxo-4,5,7,8-tetrahydro-3H-imidazo[2,l-έ]purin-3-yl)-N-(4- phenoxyphenyl)acetamide;
N-[4-Bromo-3-(trifluoromethyl)phenyl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2, 1 -ό]purin-3-yl)acetamide; N-(4-Bromo-2-chlorophenyl)-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -&]purin-3-yl)acetamide;
Nl-{4-[4-(te^-Butyl)phenoxy]phenyl}-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2, 1 -b]purin-3 -yl)acetamide;
Nl-{4-[4-(tert-Butyl)phenyl]phenyl}-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro-3H- imidazo[2,l-6]purin-3-yl)acetamide; and
Nl-[4-(4-Cyclopentyloxyphenoxy)phenyl]-2-(5-methyl-4-oxo-4,5,7,8-tetrahydro- 3H-imidazo[2,l-Z>]purin-3-yl)acetamide; or a pharmaceutically acceptable salt thereof, an ester thereof, a tautomer thereof, regioisomer thereof or a stereoisomer thereof,
17. A pharmaceutical composition comprising one or more compounds selected from the compounds of any one of claims 1 to 16, and one or more pharmaceutically acceptable excipients, carriers, diluents or mixture thereof.
18. A method for treating disease or condition associated with TRPAl function in a subject in need thereof comprising administering to the subject an effective amount of a compound according to any of claims 1 to 16.
19. A method according to claim 18, wherein the symptoms of a disease or condition associated with TRPAl function is selected from pain, chronic pain, complex regional pain syndrome, neuropathic pain, postoperative pain, rheumatoid arthritic pain, osteoarthritic pain, back pain, visceral pain, cancer pain, algesia, neuralgia, migraine, neuropathies, diabetic neuropathy, sciatica, ΗIV-related neuropathy, post-herpetic neuralgia, fibromyalgia, nerve injury, ischaemia, neurodegeneration, stroke, post stroke pain, multiple sclerosis, respiratory diseases, asthma, cough, COPD, inflammatory disorders, oesophagitis, gastroeosophagal reflux disorder (GERD), irritable bowel syndrome, inflammatory bowel disease, pelvic hypersensitivity, urinary incontinence, cystitis, burns, psoriasis, eczema, emesis, stomach duodenal ulcer and pruritus.
20. A method according to claim 19, wherein the symptoms of a disease or condition is associated with chronic pain.
21. A method according to claim 19, wherein the symptoms of a disease or condition is associated with neuropathic pain.
22. A method according to claim 19, wherein the symptoms of a disease or condition is associated with rheumatoid arthritic pain or osteoarthritic pain.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1138/MUM/2008 | 2008-05-28 | ||
| IN1138MU2008 | 2008-05-28 | ||
| US7411508P | 2008-06-19 | 2008-06-19 | |
| US61/074,115 | 2008-06-19 | ||
| US13843108P | 2008-12-17 | 2008-12-17 | |
| US61/138,431 | 2008-12-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009144548A1 true WO2009144548A1 (en) | 2009-12-03 |
Family
ID=41376642
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2009/005530 WO2009144548A1 (en) | 2008-05-28 | 2009-05-08 | Imidazo [2,1-b] purine derivatives as trpa1 modulators |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009144548A1 (en) |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011114184A1 (en) * | 2010-03-15 | 2011-09-22 | Glenmark Pharmaceuticals S.A. | Amides of heterocyclic compounds as trpa1 inhibitors |
| WO2012085662A1 (en) | 2010-12-20 | 2012-06-28 | Glenmark Pharmaceuticals S.A. | 2-amino-4-arylthiazole compounds as trpa1 antagonists |
| EP2520566A1 (en) | 2011-05-06 | 2012-11-07 | Orion Corporation | New Pharmaceutical Compounds |
| WO2012172475A1 (en) | 2011-06-13 | 2012-12-20 | Glenmark Pharmaceuticals S.A. | Treatment of respiratory disorders using trpa1 antagonists |
| WO2012176143A1 (en) | 2011-06-22 | 2012-12-27 | Glenmark Pharmaceuticals Sa | Pharmaceutical composition comprising a trpa1 antagonist and a beta-2 agonist |
| WO2012176105A1 (en) | 2011-06-22 | 2012-12-27 | Glenmark Pharmaceuticals Sa | Pharmaceutical composition comprising a trpa1 antagonist and a leukotriene receptor antagonist |
| WO2013014597A1 (en) | 2011-07-25 | 2013-01-31 | Glenmark Pharmaceuticals Sa | Pharmaceutical composition comprising a trpa1 antagonist and a steroid |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013084153A1 (en) | 2011-12-05 | 2013-06-13 | Glenmark Pharmaceuticals S.A. | Pharmaceutical composition comprising a trpa1 antagonist and an anticholinergic agent |
| JP2014534271A (en) * | 2011-11-29 | 2014-12-18 | タイヴェックス・セラピューティクス・コーポレイションTaivex Therapeutics Corporation | Improved modulators for HEC1 activity and methods thereof |
| WO2015056094A2 (en) | 2013-10-15 | 2015-04-23 | Glenmark Pharmaceuticals S.A. | Pharmaceutical composition comprising a trpa1 antagonist and an analgesic agent |
| US9193729B2 (en) | 2011-08-09 | 2015-11-24 | Cubist Pharmaceuticals, Inc. | Inhibiting transient receptor potential ion channel TRPA1 |
| WO2016042501A1 (en) | 2014-09-16 | 2016-03-24 | Glenmark Pharmaceuticals S.A. | Trpa1 antagonist for the treatment of pain associated to diabetic neuropathic pain |
| US9394308B2 (en) | 2013-01-18 | 2016-07-19 | Merck Sharp & Dohme Corp. | Inhibiting the transient receptor potential A1 ion channel |
| US9533952B2 (en) | 2012-10-01 | 2017-01-03 | Orion Corporation | N-prop-2-ynyl carboxamide derivatives and their use as TRPA1 antagonists |
| WO2017060488A1 (en) | 2015-10-09 | 2017-04-13 | Almirall, S.A. | New trpa1 antagonists |
| WO2017064068A1 (en) | 2015-10-14 | 2017-04-20 | Almirall, S.A. | New trpa1 antagonists |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994019351A1 (en) * | 1993-02-26 | 1994-09-01 | Schering Corporation | 2-benzyl-polycyclic guanine derivatives and process for preparing them |
| US20070219222A1 (en) * | 2005-12-22 | 2007-09-20 | Hydra Biosciences | Methods and compositions for treating pain |
| WO2007150026A2 (en) * | 2006-06-23 | 2007-12-27 | Incyte Corporation | Purinone derivatives as hm74a agonists |
-
2009
- 2009-05-08 WO PCT/IB2009/005530 patent/WO2009144548A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994019351A1 (en) * | 1993-02-26 | 1994-09-01 | Schering Corporation | 2-benzyl-polycyclic guanine derivatives and process for preparing them |
| US20070219222A1 (en) * | 2005-12-22 | 2007-09-20 | Hydra Biosciences | Methods and compositions for treating pain |
| WO2007150026A2 (en) * | 2006-06-23 | 2007-12-27 | Incyte Corporation | Purinone derivatives as hm74a agonists |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011114184A1 (en) * | 2010-03-15 | 2011-09-22 | Glenmark Pharmaceuticals S.A. | Amides of heterocyclic compounds as trpa1 inhibitors |
| US9073955B2 (en) | 2010-12-20 | 2015-07-07 | Glenmark Pharmaceuticals, S.A. | 2-amino-4-arylthiazole compounds as TRPA1 antagonists |
| EA023141B1 (en) * | 2010-12-20 | 2016-04-29 | Гленмарк Фармасьютикалс С.А. | 2-amino-4-arylthiazole compounds as trpa1 antagonists |
| WO2012085662A1 (en) | 2010-12-20 | 2012-06-28 | Glenmark Pharmaceuticals S.A. | 2-amino-4-arylthiazole compounds as trpa1 antagonists |
| KR101591153B1 (en) | 2010-12-20 | 2016-02-02 | 그렌마크 파머수티칼스 에스. 아. | 2-amino-4-arylthiazole compounds as trpa1 antagonists |
| CN103261201B (en) * | 2010-12-20 | 2015-04-22 | 格兰马克药品股份有限公司 | 2-amino-4-arylthiazole compounds as TRPA1 antagonists |
| CN103261201A (en) * | 2010-12-20 | 2013-08-21 | 格兰马克药品股份有限公司 | 2-amino-4-arylthiazole compounds as TRPA1 antagonists |
| US8592398B2 (en) | 2010-12-20 | 2013-11-26 | Glenmark Pharmaceuticals, S.A. | 2-amino-4-arylthiazole compounds as TRPA1 antagonists |
| US8889862B2 (en) | 2010-12-20 | 2014-11-18 | Glenmark Pharmaceuticals, S.A. | 2-amino-4-arylthiazole compounds as TRPA1 antagonists |
| EP2520566A1 (en) | 2011-05-06 | 2012-11-07 | Orion Corporation | New Pharmaceutical Compounds |
| WO2012152983A1 (en) | 2011-05-06 | 2012-11-15 | Orion Corporation | Phenyl- sulfonyl derivatives as mediators of trpa1 receptor activity for the treatment of pain |
| WO2012172475A1 (en) | 2011-06-13 | 2012-12-20 | Glenmark Pharmaceuticals S.A. | Treatment of respiratory disorders using trpa1 antagonists |
| US9186360B2 (en) | 2011-06-13 | 2015-11-17 | Glenmark Pharmaceuticals S.A. | Treatment of respiratory disorders using TRPA1 antagonists |
| WO2012176105A1 (en) | 2011-06-22 | 2012-12-27 | Glenmark Pharmaceuticals Sa | Pharmaceutical composition comprising a trpa1 antagonist and a leukotriene receptor antagonist |
| WO2012176143A1 (en) | 2011-06-22 | 2012-12-27 | Glenmark Pharmaceuticals Sa | Pharmaceutical composition comprising a trpa1 antagonist and a beta-2 agonist |
| WO2013014597A1 (en) | 2011-07-25 | 2013-01-31 | Glenmark Pharmaceuticals Sa | Pharmaceutical composition comprising a trpa1 antagonist and a steroid |
| US9193729B2 (en) | 2011-08-09 | 2015-11-24 | Cubist Pharmaceuticals, Inc. | Inhibiting transient receptor potential ion channel TRPA1 |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| JP2014534271A (en) * | 2011-11-29 | 2014-12-18 | タイヴェックス・セラピューティクス・コーポレイションTaivex Therapeutics Corporation | Improved modulators for HEC1 activity and methods thereof |
| WO2013084153A1 (en) | 2011-12-05 | 2013-06-13 | Glenmark Pharmaceuticals S.A. | Pharmaceutical composition comprising a trpa1 antagonist and an anticholinergic agent |
| US9533952B2 (en) | 2012-10-01 | 2017-01-03 | Orion Corporation | N-prop-2-ynyl carboxamide derivatives and their use as TRPA1 antagonists |
| US9394308B2 (en) | 2013-01-18 | 2016-07-19 | Merck Sharp & Dohme Corp. | Inhibiting the transient receptor potential A1 ion channel |
| WO2015056094A2 (en) | 2013-10-15 | 2015-04-23 | Glenmark Pharmaceuticals S.A. | Pharmaceutical composition comprising a trpa1 antagonist and an analgesic agent |
| US9532988B2 (en) | 2013-10-15 | 2017-01-03 | Glenmark Pharmaceuticals S.A. | Pharmaceutical composition comprising a TRPA1 antagonist and an analgesic agent |
| WO2016042501A1 (en) | 2014-09-16 | 2016-03-24 | Glenmark Pharmaceuticals S.A. | Trpa1 antagonist for the treatment of pain associated to diabetic neuropathic pain |
| WO2017060488A1 (en) | 2015-10-09 | 2017-04-13 | Almirall, S.A. | New trpa1 antagonists |
| WO2017064068A1 (en) | 2015-10-14 | 2017-04-20 | Almirall, S.A. | New trpa1 antagonists |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009144548A1 (en) | Imidazo [2,1-b] purine derivatives as trpa1 modulators | |
| EP2708538B1 (en) | Process for preparing fused pyrimidine-dione derivatives, useful as as TRPA1 modulators | |
| EP2411395B1 (en) | Furopyrimidinedione derivatives as trpa1 modulators | |
| WO2009118596A2 (en) | Phthalimide derivatives as trpa1 modulators | |
| EP2411397B1 (en) | Isothiazolo-pyrimidinedione derivatives as trpa1 modulators | |
| US9073955B2 (en) | 2-amino-4-arylthiazole compounds as TRPA1 antagonists | |
| US8623880B2 (en) | Fused pyrimidine-dione derivatives as TRPA1 modulators | |
| WO2011114184A1 (en) | Amides of heterocyclic compounds as trpa1 inhibitors | |
| WO2011132017A1 (en) | Pyrido[3,4-d]pyrimidinyl acetamide derivatives as trpa1 modulators | |
| WO2010004390A1 (en) | Quinazoline dione derivatives as trpa1 modulators | |
| WO2010010435A2 (en) | Fused oxazole and thiazole derivatives as trpms modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09754176 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09754176 Country of ref document: EP Kind code of ref document: A1 |