WO2009134624A1 - Hcv ns3 protease inhibitors - Google Patents
Hcv ns3 protease inhibitors Download PDFInfo
- Publication number
- WO2009134624A1 WO2009134624A1 PCT/US2009/040815 US2009040815W WO2009134624A1 WO 2009134624 A1 WO2009134624 A1 WO 2009134624A1 US 2009040815 W US2009040815 W US 2009040815W WO 2009134624 A1 WO2009134624 A1 WO 2009134624A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- alkyl
- compound according
- hcv
- mmol
- Prior art date
Links
- 0 O*1(N[C@]1C1*CC*C1)O Chemical compound O*1(N[C@]1C1*CC*C1)O 0.000 description 3
- JUIZBSJNCXXRBR-UHFFFAOYSA-N C=CCC(C(N(C=CC=C1)C1=N1)=O)=C1O Chemical compound C=CCC(C(N(C=CC=C1)C1=N1)=O)=C1O JUIZBSJNCXXRBR-UHFFFAOYSA-N 0.000 description 1
- ABZCXRWPERHSHQ-UHFFFAOYSA-N C=CCCCC(COC1)C1O Chemical compound C=CCCCC(COC1)C1O ABZCXRWPERHSHQ-UHFFFAOYSA-N 0.000 description 1
- WJHBUQJLLBQXAC-BBRMVZONSA-N CC(C)(C)OC(N(CC[C@@H]1OC(N2CC=C)=CC=CC2=O)[C@@H]1C(OC)=O)=O Chemical compound CC(C)(C)OC(N(CC[C@@H]1OC(N2CC=C)=CC=CC2=O)[C@@H]1C(OC)=O)=O WJHBUQJLLBQXAC-BBRMVZONSA-N 0.000 description 1
- UJRVHKVJCIECLA-UHFFFAOYSA-N CC(Nc1cc(Br)ccc1C(NCC=C)=O)=O Chemical compound CC(Nc1cc(Br)ccc1C(NCC=C)=O)=O UJRVHKVJCIECLA-UHFFFAOYSA-N 0.000 description 1
- NCMGSRKXWZSLRF-UHFFFAOYSA-N COC(C=CN12)=CC1=NC(O)=C(CC=C)C2=O Chemical compound COC(C=CN12)=CC1=NC(O)=C(CC=C)C2=O NCMGSRKXWZSLRF-UHFFFAOYSA-N 0.000 description 1
- LIEMDDNGLLSHET-PKNPKLBSSA-N COC([C@H](C[C@H](C1)OC(N2CC(C3)C3CCCC(CCC3)[C@@H]3OC(N[C@H]3C4CCCC4)=O)=Cc(cccc4)c4C2=O)N1C3=O)=O Chemical compound COC([C@H](C[C@H](C1)OC(N2CC(C3)C3CCCC(CCC3)[C@@H]3OC(N[C@H]3C4CCCC4)=O)=Cc(cccc4)c4C2=O)N1C3=O)=O LIEMDDNGLLSHET-PKNPKLBSSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/12—Cyclic peptides with only normal peptide bonds in the ring
- C07K5/123—Tripeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/18—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0812—Tripeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Definitions
- the present invention relates to macrocyclic compounds that are useful as inhibitors of the hepatitis C virus (HCV) NS3 protease, the synthesis of such compounds, and the use of such compounds for treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection.
- HCV hepatitis C virus
- HCV infection is a major health problem that leads to chronic liver disease, such as cirrhosis and hepatocellular carcinoma, in a substantial number of infected individuals, estimated to be 2-15% of the world's population.
- HCV human immunodeficiency virus
- WHO World Health Organization
- NS3 protease is located in the N-terminal domain of the NS 3 protein. Because it is responsible for an intramolecular cleavage at the NS 3/4 A site and for downstream intermolecular processing at the NS4A/4B, NS4B/5A and NS5 A/5B junctions, the NS3 protease is considered a prime drug target.
- the present invention relates to macrocyclic compounds of formula (I) and pharmaceutically acceptable salts thereof. These compounds are useful in the inhibition of HCV (hepatitis C virus) NS3 (non-structural 3) protease, the prevention or treatment of one or more of the symptoms of HCV infection, either as compounds or their pharmaceutically acceptable salts, or as pharmaceutical composition ingredients.
- these compounds and salts may be the primary active therapeutic agent, and, when appropriate, may be combined with other therapeutic agents including but not limited to other HCV antivirals, anti- infectives, immunomodulators, antibiotics or vaccines. More particularly, the present invention relates to a compound of formula (I) and a pharmaceutically acceptable salt thereof:
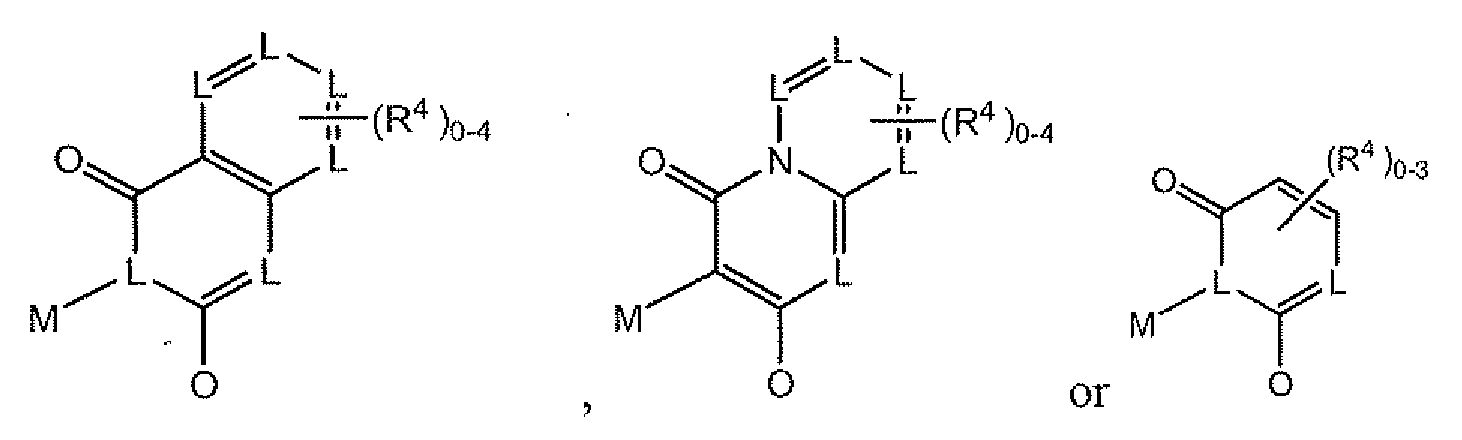
- each L is independently selected from the group consisting of N and CH, provided that the total number of L that are N is from 1 to 4;
- R 1 is selected from the group consisting of -CO 2 H, and -CONHSO 2 (cyclopropyl);
- R 2 is selected from the group consisting of ethyl and ethenyl;
- R 3 is selected from the group consisting of cyclopentyl, cyclohexyl, and
- each R 4 is independently selected from the group consisting of halogen atoms, C 1 -C 4 alkoxy, C 1 -C 7 alkyl, C 2 -C 7 alkenyl, -CN, -CF 3 , -OCF 3 , SCH 3 , -SO 2 (CH 3 ), C 3 -C 8 cycloalkyl, C 3 -C 8 cycloalkoxy, C 1 -C 6 haloalkyl, phenyl, naphthyl and heteroaryl groups, wherein each said R 4 heteroaryl is selected from the group consisting of 5- and
- Y is selected from the group consisting of -C(O)-, -C(O)O- and -C(O)NH-;
- M is selected from the group consisting of C 4 -C 7 alkylene and C 4 -C 7 alkenylene, wherein said M is substituted with O to 3 substituents independently selected C 1 -C 8 alkyl, provided that two adjacent substituents can together form a 3 to 6 membered ring.
- the present invention also includes pharmaceutical compositions containing a compound of the present invention and methods of preparing such pharmaceutical compositions.
- the present invention further includes methods of treating or reducing the likelihood or severity of one or more symptoms of HCV infection.
- the present invention includes compounds of formula (I), and pharmaceutically acceptable salts thereof.
- HCV protease inhibitors e.g., HCV NS3 protease inhibitors
- Preferred compounds are those with high activity (e.g., Ki of 5 nM or less, 1 nM or less, 0.5 nM or less, or 0.1 raM or less) against HCV NS3 genotype Ib Rl 55K.
- Rl 55K is a HCV Ib mutation that occurs in nature and which provides resistance against some NS3 protease inhibitors.
- Example 85 infra illustrates the ability of different compounds to provide high activity against such a mutation.
- Reference to formula I compounds throughout the present application includes reference to compounds within formula I including different embodiment and subgeneric formula (formula Ia and Ib).
- O or 1 R 4 is present and, if present, is selected from the group consisting of -Br, -Cl, -CN, phenyl, -O-phenyl, -OCF 3 , -OCH 3 , -C(O)OH 3 -CH 3 and -C(O)CH 3 .
- R 1 is -CO 2 H, and the other substituents are as provided in formula I above or in the above first embodiment.
- R 1 is -C(0)NHS ⁇ 2 Cyclopropyl, and the other substituents are as provided in formula I above or in the first embodiment.
- R 2 is -CH 2 CH 3 , and the other substituents are as provided in formula I above or in the first to third embodiments.
- R 3 is cyclopentyl, and the other substituents are as provided in formula I above or the first through fifth embodiments.
- R 3 is cyclohexyl, and the other substituents are as provided in formula I above or in the first through fifth embodiments.
- R 3 is -C(CH 3 ) 3 , and the other substituents are as provided in formula I above or the first through fifth embodiments.
- M is selected from the group consisting of
- the formula I compound has the following structure:
- R 4 if present is selected from the group consisting of -Br, -Cl, -CN, phenyl, -O-phenyl, -OCF 3 , -OCH 3 , -C(O)OH, -CH 3 and -C(O)CH 3 .
- R 4 is present and is selected from the group consisting of
- R 4 is not present.
- the formula I compound has the following structure:
- R 4 if present is selected from the group consisting of -Br, -Cl, -CN, phenyl, -O-phenyl, -OCF 3 , -OCH 3 , -C(O)OH, -CH 3 and -C(O)CH 3 .
- R 4 if present is selected from the group consisting of -Br, -Cl, -CN, phenyl, -O-phenyl, -OCF 3 , -OCH 3 , -C(O)OH, -CH 3 and -C(O)CH 3 .
- R 4 is present and is selected from the group consisting of -Br, -Cl, -CN, -OCF 3 , -OCH 3 , -C(O)OH, -CH 3 and -C(O)CH 3 .
- R 4 is not present.
- R 4 is as provided in the general formula Ia above, or in the first or second embodiments,
- the compound of the invention is a compound provided in Examples 1 through 84 shown below or a pharmaceutically acceptable salt thereof.
- a pharmaceutical composition comprising an effective amount of a compound of formula (I) and a pharmaceutically acceptable carrier.
- HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NS 5 B polymerase inhibitors.
- a pharmaceutical combination which is (i) a compound of formula (I) and (ii) a second therapeutic agent selected from the group consisting of HCV antiviral agents, immunomodulators, and anti-infective agents; wherein the compound of formula (I) and the second therapeutic agent are each employed in an amount that renders the combination effective for inhibiting HCV NS 3 protease, or for treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection.
- HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NS5B polymerase inhibitors.
- HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NS5B polymerase inhibitors.
- t (j) A method of inhibiting HCV NS3 protease in a subject in need thereof which comprises administering to the subject the pharmaceutical composition of (a), (b), or (c) or the combination of (d) or (e).
- k A method of treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection in a subject in need thereof which comprises administering to the subject the pharmaceutical composition of (a), (b), or (c) or the combination of (d) or (e).
- the present invention also includes a compound of the present invention for use (i) in, (ii) as a medicament for, or (iii) in the preparation of a medicament for: (a) inhibiting HCV NS 3 protease, or (b) treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection.
- the compounds of the present invention can optionally be employed in combination with one or more second therapeutic agents selected from HCV antiviral agents, anti -infective agents, and immunomodulators.
- Additional embodiments of the invention include the pharmaceutical compositions, combinations and methods set forth in (a)-(k) above and the uses set forth in the preceding paragraph, wherein the compound of the present invention employed therein is a compound of one of the embodiments, aspects, classes, sub-classes, or features of the compounds described above. In all of these embodiments, the compound may optionally be used in the form of a pharmaceutically acceptable salt.
- alkyl refers to any linear or branched chain alkyl group having a number of carbon atoms in the specified range.
- C ⁇ e alkyl (or “C 1 -Ce alkyl”) refers to all of the hexyl alkyl and pentyl alkyl isomers as well as n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl.
- C 1 ⁇ alkyl refers to n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl.
- Alkyl groups may be substituted as indicated.
- halogenated refers to a group or molecule in which a hydrogen atom has been replaced by a halogen.
- haloalkyl refers to a halogenated alkyl group.
- halogen or “halo” refers to atoms of fluorine, chlorine, bromine and iodine (alternatively referred to as fluoro, chloro, bromo, and iodo)
- alkoxy refers to an "alkyl-O-" group. Alkoxy groups may be substituted as indicated.
- alkenylene refers to any linear or branched chain alkenylene group containing a double and having a number of carbon atoms in the specified range.
- -C 2-6 alkenylene - refers to any of the C 2 to C 6 linear or branched alkenylene.
- Alkenylene groups may be substituted as indicated.
- alkylene refers to any linear or branched chain alkylene group (or alternatively “alkanediyl”) having a number of carbon atoms in the specified range.
- -C 1-6 alkylene- refers to any of the C 1 to C 6 linear or branched alkylenes.
- alkylenes include -(CH 2 ) 1-6 -, -(CH 2 ) 1-4 -, -(CH 2 ) 1-3 -, -(CH 2 ) 1-2 -, -CH 2 - and -CH(CH 3 )-.
- Alkylene groups may be substituted as indicated.
- cycloalkyl refers to any cyclic ring of an alkane or alkene having a number of carbon atoms in the specified range.
- C 3-8 cycloalkyl refers to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyc ⁇ oheptyl, and cyclooctyl.
- cycloalkoxy refers to a "cycloalkyl-O-" group. Cycloalkyl groups may be substituted as indicated.
- Carbocycle (and variations thereof such as “carbocyclic” or “carbocyclyl”) as used herein, unless otherwise indicated, refers to (i) a C 3 to C 8 monocyclic, saturated or unsaturated ring or (ii) a C 7 to C 12 bicyclic saturated or unsaturated ring system. Each ring in (ii) is either independent of, or fused to, the other ring, and each ring is saturated or unsaturated. Carbocycle groups maybe substituted as indicated, for example with C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, aryl, halogen, -NH2 or -OH.
- the carbocycle may be attached to the rest of the molecule at any carbon atom which results in a stable compound.
- the fused bicyclic carbocycles are a subset of the carbocycles; i.e., the term "fused bicyclic carbocycle” generally refers to a C 7 to C 10 bicyclic ring system in which each ring is saturated or unsaturated and two adjacent carbon atoms are shared by each of the rings in the ring system.
- a fused bicyclic carbocycle in which both rings are saturated is a saturated bicyclic ring system.
- Saturated carbocyclic rings are also referred to as cycloalkyl rings, e.g., cyclopropyl, cyclobutyl, etc.
- a fused bicyclic carbocycle in which one or both rings are unsaturated is an unsaturated bicyclic ring system.
- a subset of the fused bicyclic unsaturated carbocycles are those bicyclic carbocycles in which one ring is a benzene ring and the other ring is saturated or unsaturated, with attachment via any carbon atom that results in a stable compound.
- Depicted ring systems include, where appropriate, an indication of the variable to
- variable R 4 is shown as a floating variable which can be attached to any ring atom, provided that such attachment results in formation of a stable ring.
- aryl refers to aromatic mono- and poly-carbocyclic ring systems, also referred to as “arenes,” wherein the individual carbocyclic rings in the polyring systems are fused or attached to each other via a single bond. Suitable aryl groups include phenyl, naphthyl, and biphenyleny ⁇ . Aryl groups may be substituted as indicated.
- heteroaryl and “heteroaromatic ring” refer to a stable 5- or
- 6-membered monocyclic aromatic ring a stable 7- to 12-membered bicyclic ring system, or a stable 11- to 15-mernbered tricyclic ring system, which consists of carbon atoms and one or more heteroatoms selected from N, O and S.
- substituted heteraromatic rings containing at least one nitrogen atom e.g., pyridine
- substitutions can be those resulting in N-oxide formation.
- heteroaromatic rings include pyridyl, pyrrolyl, pyrazinyl, pyrimidinyl, pyridazinyl, thienyl (or thiophenyl), thiazolyl, furanyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isooxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, and thiadiazolyl.
- a particular group such as alkyl, cycloalkyl, aryl and heteroaryl groups are unsubstituted.
- the alkyl, cycloalkyl, aryl and heteroaryl groups are substituted with one to three substitutents selected from the group consisting of: halo, C 1 -C 20 alkyl, -CF 3 , -NH 2 , -N(C 1 -C 6 alkyl) 2 , -NO 2 , oxo, -CN, -N 3 , -OH, -0(C 1 -C 6 alkyl), C 3 -C] 0 cycloalkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, (C ⁇ -C 6 alkyl) S(O) 0 ⁇ 2 -, aiyl- S(O) 0-2 -, (C 0 -C 6 alkyl)S(0)o.2(C 0 -C 6 alkyl)-, (C 0 -C 6 alkyl)C(O)NH-, H 2
- heteroaryl ring described as containing from “1 to 3 heteroatoms” means the ring can contain 1, 2, or 3 heteroatoms. It is also to be understood that any range cited herein includes within its scope all of the sub-ranges within that range. The oxidized forms of the heteroatoms N and S are also included within the scope of the present invention.
- variable e.g. L
- its definition on each occurrence is independent of its definition at every other occurrence. Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- a “stable” compound is a compound which can be prepared and isolated and whose structure and properties remain or can be caused to remain essentially unchanged for a period of time sufficient to allow use of the compound for the purposes described herein (e.g., therapeutic or prophylactic administration to a subject).
- certain of the compounds of the present invention can have asymmetric centers and can occur as mixtures of stereoisomers, or as individual diastereomers, or enantiomers. All isomeric forms of these compounds, whether isolated or in mixtures, are within the scope of the present invention.
- Reference to a compound also includes stable complexes of the compound such as a stable hydrate.
- certain of the compounds of the present invention can exist as tautomers.
- a reference to a compound of formula (I) is a reference to the compound per se, or to any one of its tautomers per se, or to mixtures of two or more tautomers.
- the compounds of the present inventions are useful in the inhibition of HCV protease (e.g., HCV NS3 protease) and the treatment of HCV infection and/or reduction of the likelihood or severity of symptoms of HCV infection.
- the compounds of this invention are useful in treating infection by HCV after suspected past exposure to HCV by such means as blood transfusion, exchange of body fluids, bites, accidental needle stick, or exposure to patient blood during surgery.
- the compounds of this invention are useful in the preparation and execution of screening assays for antiviral compounds.
- the compounds of this invention are useful for isolating enzyme mutants, which are excellent screening tools for more powerful antiviral compounds.
- the compounds of this invention are useful in establishing or determining the binding site of other antivirals to HCV protease, e.g., by competitive inhibition.
- the compounds of this invention may be commercial products to be sold for these purposes.
- the compounds of the present invention may be administered in the form of pharmaceutically acceptable salts.
- pharmaceutically acceptable salt refers to a salt which possesses the effectiveness of the parent compound and which is not biologically or otherwise undesirable (e.g., is neither toxic nor otherwise deleterious to the recipient thereof).
- Suitable salts include acid addition salts which may, for example, be formed by mixing a solution of the compound of the present invention with a solution of a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, acetic acid, trifluoroacetic acid, or benzoic acid.
- suitable pharmaceutically acceptable salts thereof can include alkali metal salts (e.g., sodium or potassium salts), alkaline earth metal salts (e.g., calcium or magnesium salts), and salts formed with suitable organic ligands such as quaternary ammonium salts.
- suitable pharmaceutically acceptable esters can be employed to modify the solubility or hydrolysis characteristics of the compound.
- administration and variants thereof (e.g., “administering” a compound) in reference to a compound of the invention mean providing the compound or a prodrug of the compound to the individual in need of treatment.
- administration and its variants are each understood to include concurrent and sequential provision of the compound or salt and other agents.
- prodrug is intended to encompass an inactive drug form or compound that is converted into an active drag form or compound by the action of enzymes, chemicals or metabolic processes in the body of an individual to whom it is administered.
- composition is intended to encompass a product comprising the specified ingredients, as well as any product which results, directly or indirectly, from combining the specified ingredients,
- pharmaceutically acceptable is meant that the ingredients of the pharmaceutical composition must be compatible with each other and not deleterious to the recipient thereof.
- subject refers to an animal, preferably a mammal, most preferably a human, who has been the object of treatment, observation or experiment.
- the term "effective amount” as used herein means that amount of active compound or pharmaceutical agent that elicits the biological or medicinal response in a tissue, system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician.
- the effective amount is a "therapeutically effective amount” for the alleviation of one or more symptoms of the disease or condition being treated.
- the effective amount is a "prophylactically effective amount” for reduction of the severity or likelihood of one or more symptoms of the disease or condition.
- the term also includes herein the amount of active compound sufficient to inhibit HCV NS 3 protease and thereby elicit the response being sought (i.e., an "inhibition effective amount").
- the compounds of the present invention can be administered by means that produces contact of the active agent with the agent's site of action. They can be administered by conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. They can be administered alone, but typically are administered with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice.
- the compounds of the invention can, for example, be administered by one or more of the following routes: orally, parenterally (including subcutaneous injections, intravenous, intramuscular, intrasternal injection or infusion techniques), by inhalation (such as in a spray form), or rectally, in the form of a unit dosage of a pharmaceutical composition containing an effective amount of the compound and conventional non-toxic pharmaceutically-acceptable carriers, adjuvants and vehicles.
- routes e.g., parenterally (including subcutaneous injections, intravenous, intramuscular, intrasternal injection or infusion techniques), by inhalation (such as in a spray form), or rectally, in the form of a unit dosage of a pharmaceutical composition containing an effective amount of the compound and conventional non-toxic pharmaceutically-acceptable carriers, adjuvants and vehicles.
- Liquid preparations suitable for oral administration e.g., suspensions, syrups, elixirs and the like
- Solid preparations suitable for oral administration can be prepared according to techniques known in the art and can employ such solid excipients as starches, sugars, kaolin, lubricants, binders, disintegrating agents and the like.
- Parenteral compositions can be prepared according to techniques known in the art and typically employ sterile water as a carrier and optionally other ingredients, such as solubility aids.
- injectable solutions can be prepared according to methods known in the art wherein the carrier comprises a saline solution, a glucose solution or a solution containing a mixture of saline and glucose. Further guidance for methods suitable for use in preparing pharmaceutical compositions of the present invention and of ingredients suitable for use in said compositions is provided in Remington's Pharmaceutical Sciences, 20 th edition (ed. A. R. Gennaro, Mack Publishing Co., 2000).
- the compounds of this invention can be administered orally in a dosage range of 0.001 to 1000 mg/kg of mammal (e.g., human) body weight per day in a single dose or in divided doses.
- mammal e.g., human
- One dosage range is 0.01 to 500 mg/kg body weight per day orally in a single dose or in divided doses.
- Another dosage range is 0.1 to 100 mg/kg body weight per day orally in single or divided doses.
- the compositions can be provided in the form of tablets or capsules containing 1.0 to 500 mg of the active ingredient, particularly 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, and 500 mg of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- the specific dose level and frequency of dosage for any particular patient may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the host undergoing therapy.
- the present invention also relates to a method of inhibiting HCV NS3 protease, inhibiting HCV replication, or preventing or treating HCV infection with a compound of the present invention in combination with one or more therapeutic agents and a pharmaceutical composition comprising a compound of the present invention and one or more therapeutic agents selected from the group consisting of a HCV antiviral agent, an immunomodulator, and an anti-infective agent.
- Such therapeutic agents active against HCV include ribavirin, levovirin, viramidine, thymosin alpha-1, interferon- ⁇ , interferon- ⁇ , pegylated interferon- ⁇ (peginterferon- ⁇ ), a combination of interferon- ⁇ and ribavirin, a combination of peginterferon- ⁇ and ribavirin, a combination of interferon- ⁇ and levovirin, and a combination of peginterferon- ⁇ and levovirin.
- Interferon- ⁇ includes recombinant interferon- ⁇ 2a (such as ROFERON interferon available from Hoffmann-LaRoche, Nutley, NJ), pegylated interferon- ⁇ 2a (PEGASYS), interferon- ⁇ 2b (such as INTRON-A interferon available from Schering Corp., Kenilworth, NJ), pegylated interferon- ⁇ 2b (PEGINTRON), a recombinant consensus interferon (such as interferon alphacon-1), and a purified interferon- ⁇ product.
- Amgen's recombinant consensus interferon has the brand name INFERGEN.
- Levovirin is the L-enantiomer of ribavirin which has shown immunomodulatory activity similar to ribavirin
- Viramidine represents an analog of ribavirin disclosed in WO 01/60379.
- the individual components of the combination can be administered separately at different times during the course of therapy or concurrently in divided or single combination forms.
- the compounds of the present invention may also be administered in combination with an agent that is an inhibitor of HCV NS3 serine protease.
- HCV NS3 serine protease is an essential viral enzyme and has been described to be an excellent target for inhibition of HCV replication.
- Both substrate and non-substrate based inhibitors of HCV NS3 protease inhibitors are disclosed in International Patent Application Publications WO 98/22496, WO 98/46630, WO 99/07733, WO 99/07734, WO 99/38888, WO 99/50230, WO 99/64442, WO 00/09543, WO 00/59929, WO 02/48116 and WO 02/48172, British Patent No.
- Ribavirin, levovirin, and viramidine may exert their anti-HCV effects by modulating intracellular pools of guanine nucleotides via inhibition of the intracellular enzyme inosine monophosphate dehydrogenase (IMPDH).
- IMPDH inosine monophosphate dehydrogenase
- DVIPDH is the rate-limiting enzyme on the biosynthetic route in de novo guanine nucleotide biosynthesis.
- Ribavirin is readily phosphorylated intracellularly and the monophosphate derivative is an inhibitor of IMPDH.
- inhibition of EVIPDH represents another useful target for the discovery of inhibitors of HCV replication.
- the compounds of the present invention may also be administered in combination with an inhibitor of IMPDH, such as VX-497, which is disclosed in International Patent Application Publications WO 97/41211 and WO 01/00622; another IMPDH inhibitor, such as that disclosed in WO 00/25780; or mycophenolate mofetil. See A.C. Allison and E.M. Eugui, 44 (Suppl.) Agents Action 165 (1993).
- an inhibitor of IMPDH such as VX-497, which is disclosed in International Patent Application Publications WO 97/41211 and WO 01/00622
- another IMPDH inhibitor such as that disclosed in WO 00/25780
- mycophenolate mofetil See A.C. Allison and E.M. Eugui, 44 (Suppl.) Agents Action 165 (1993).
- the compounds of the present invention may also be administered in combination with the antiviral agent amantadine (1 -aminoadamantane).
- amantadine 1 -aminoadamantane
- the compounds of the present invention may also be administered in combination with the antiviral agent polymerase inhibitor R7128 (Roche).
- the compounds of the present invention may also be combined for the treatment of HCV infection with antiviral 2'-C-branched ribonucleosides disclosed in R. E. Harry-O'Kuru et al., 62 J. Ore. Chem. 1754-59 (1997); M. S. Wolfe et al, 36 Tet. Lett. 7611-14 (1995); U.S. Patent No. 3,480,613; and International Patent Application Publications WO 01/90121, WO 01/92282, WO 02/32920, WO 04/002999, WO 04/003000 and WO 04/002422; the contents of each of which are incorporated by reference in their entirety.
- Such 2'-C-branched ribonucleosides include, but are not limited to, 2'-C-methyl-cytidine, 2'-C-methyl-uridine, 2'-C- methyl- adenosine, 2'-C-methyl-guanosine, and 9-(2-C-methyl- ⁇ -D-ribofuranosyl)-2,6- diaminopurine, and the corresponding amino acid ester of the ribose C-2', C-3', and C-5' hydroxyls and the corresponding optionally substituted cyclic 1,3 -propanediol esters of the 5'- phosphate derivatives.
- the compounds of the present invention may also be combined for the treatment of HCV infection with other nucleosides having anti-HCV properties, such as those disclosed in International Patent Application Publications WO 02/51425, assigned to Mitsubishi Pharma Corp.; WO 01/79246, WO 02/32920, WO 02/48165 and WO2005/003147 (including Rl 656, (2'/?)-2'-deoxy-2'-iluoro-2'-C-methylcytidine, shown as compounds 3 ⁇ 6 on page 77); WO 01/68663; WO 99/43691; WO 02/18404 and WO2006/021341, and U.S.
- Patent Application Publication US 2005/0038240 including 4'-azido nucleosides such as Rl 626, 4'-azidocytidine; U.S. Patent Application Publications US 2002/0019363, US 2003/0236216, US 2004/0006007 and US 2004/0063658; and International Patent Application Publications WO 02/100415, WO 03/026589, WO 03/026675, WO 03/093290, WO 04/011478, WO 04/013300 and WO 04/028481 ; the content of each is incorporated herein by reference in its entirety.
- the compounds of the present invention may also be administered in combination with an agent that is an inhibitor of HCV NS5B polymerase.
- HCV NS5B polymerase inhibitors that may be used as combination therapy include, but are not limited to, those disclosed in International Patent Application Publications WO 02/057287, WO 02/057425, WO 03/068244, WO 2004/000858, WO 04/003138 and WO 2004/007512; U.S. Patent No. 6,777,392 and U.S. Patent Application Publication US2004/0067901 ; the content of each is incorporated herein by reference in its entirety.
- nucleoside HCV NS5B polymerase inhibitors that are used in combination with the present HCV NS3 protease inhibitors are selected from the following compounds: 4-amino-7-(2-C-methyl- ⁇ -D-arabinofuranosyl)-7H-pyrrolo[2,3-d]pyriniidine; 4- amino-7-(2-C-methyl- ⁇ -D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidine; 4-methylamino-7-(2-C- methyl- ⁇ -D-riboruranosyl)-7H-pyrrolo[2,3-d]pyrimidine; 4-dimethylamino-7-(2-C-methyl- ⁇ -D- ribofuranosyl)-7H-pyrrol
- the compounds of the present invention may also be combined for the treatment of ⁇ CV infection with non-nucleoside inhibitors of ⁇ CV polymerase such as those disclosed in International Patent Application Publications WO 01/77091 ; WO 01/47883 ; WO 02/04425 ; WO 02/06246; WO 02/20497; WO 2005/016927 (in particular JTK003); the content of each is incorporated herein by reference in its entirety; and ⁇ CV-796 (Viropharma Inc.).
- non-nucleoside inhibitors of ⁇ CV polymerase such as those disclosed in International Patent Application Publications WO 01/77091 ; WO 01/47883 ; WO 02/04425 ; WO 02/06246; WO 02/20497; WO 2005/016927 (in particular JTK003); the content of each is incorporated herein by reference in its entirety; and ⁇ CV-796 (Viropharma Inc.).
- non-nucleoside HCV NS5B polymerase inhibitors that are used in combination with the present HCV NS3 protease inhibitors are selected from the following compounds: 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-7-oxo-5,6,7,8- tetrahydroindolo[2,1- ⁇ ][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-6-(2-morpholin- 4-ylethyl)-5,6,7,8-tetrahydroindolo[2,1- ⁇ ][2,5]benzodiazocine-l 1-carboxylic acid; 14- cyclohexyl-6-[2-(dimethylammo)ethyl]-3-methoxy"5,6,7,8-tetrahydroindolo[2,1- ⁇ ] [2,5]benzodiazocine-l 1-carboxylic acid;
- the ⁇ CV NS3 protease inhibitory activity of the present compounds may be tested using assays known in the art.
- One such assay is ⁇ CV NS 3 protease time-resolved fluorescence (TRF) assay as described below and in International Patent Application Publication WO 2006/102087.
- TRF time-resolved fluorescence
- Other examples of such assays are described in e.g., International Patent Application Publication WO 2005/046712.
- a NS3 protease assay can be performed, for example, in a final volume of 100 ⁇ l assay buffer containing 50 niM HEPES, pH 7.5, 150 mM NaCl, 15 % glycerol, 0.15 % TRITON X- 100, 10 mM DTT, and 0.1 % PEG 8000.
- NS3 and NS4A protease is pre-incubated with various concentrations of inhibitors in DMSO for 30 minutes. The reaction is initiated by adding the TRF peptide substrate (final concentration 100 nM).
- NS3 mediated hydrolysis of the substrate is quenched after 1 hour at room temperature with 100 ⁇ l of 500 mM MES, pH 5.5.
- Product fluorescence is detected using either a VICTOR V2 or FUSION fluorophotometer (Perkin Elmer Life and Analytical Sciences) with excitation at 340 nm and emission at 615 nm with a 400 ⁇ s delay. Testing concentrations of different enzyme forms are selected to result in a signal to background ratio (S/B) of 10-30.
- S/B signal to background ratio
- IC 50 values are derived using a standard four- parameter fit to the data.
- K i values are derived from IC 50 values using the following formula,
- the present invention also includes processes for making compounds of formula (I).
- the compounds of the present invention can be readily prepared according to the following reaction schemes and examples, or modifications thereof, using readily available starting materials, reagents and conventional synthesis procedures. In these reactions, it is also possible to make use of variants which are themselves known to those of ordinary skill in this art, but are not mentioned in greater detail Furthermore, other methods for preparing compounds of the invention will be readily apparent to the person of ordinary skill in the art in light of the following reaction schemes and examples. Unless otherwise indicated, all variables are as defined above. The following reaction schemes and examples serve only to illustrate the invention and its practice.
- Olefin metathesis catalysts include the following Ruthenium based species: F. Miller et al., 118 J. AM. CHEM. Soc. 9606 (1996); G. Kingsbury et al., 121 J. Am. Chem. Soc. 791 (1999); H. Scholl et al., 1 ORG. LETT. 953 (1999); U.S. Patent Application Publication US2002/0107138; K. Furstner et al., 64 J. ORG. CHEM. 8275 (1999).
- the utility of these catalysts in ring closing metathesis is well known in the literature (e.g. Trnka and Grubbs, 34 ACC. CHEM. RES. 18 (2001).
- LiOH-H 2 O Lithium hydroxide monohydrate
- Step 1 t-Butyl (Y 1 R2R)- 1 - ([ (cyclopropylsulfonyl)aminolcarbonyl ⁇ -2- ethyl c yclopropyl)carb amate
- a hydrogenation vessel was charged with a MeOH (1000 mL) slurry of ⁇ -butyl (( IR,2S)- 1 - ⁇ [(cyclopropylsulfonyljamino] carbonyl ⁇ -2»vinylcyclopropyl)carbamate ( 164 g, 0.50 mol) (US 6,995,174) and 5% Ru/C (dry, 7.5 wt%, 12.4 g) and stirred.
- the vessel was placed under N 2 (20 psi) and vented to atmospheric pressure (3x) to remove residual oxygen.
- the vessel was then placed under H 2 (50 psi). After 20 hours, the vessel was vented to atmospheric pressure.
- the reaction slurry was then transferred out of the reaction vessel and filtered through SOLKA FLOK (34 g, wetted with 100 mL MeOH) to yield a clear, light brown solution.
- the SOLKA FLOK was rinsed with MeOH (200 mL x 2).
- the combined MeOH solutions were concentrated under reduced pressure to yield crude product as a white solid (153 g).
- the crude product was slurried in EtOAc (800 mL), warmed to 40°C and aged 30 minutes.
- the solution was then seeded, aged 30 minutes, and heptane (500 mL) was added via addition funnel over 30 minutes.
- the partially crystallized solid was cooled to RT and aged overnight, after which additional heptane (500 mL) was added.
- AMANO LIPASE PS (7.0 g, 64.7 mmol) was added to a solution of ⁇ r ⁇ r ⁇ -2-pent-
- Step 2 Methyl (2 i 5 f )-cvclopentyl[(i ⁇ ' ⁇ i?.2/gV2- ⁇ ent-4-en-1-vlcvclot)entyl]oxylcarbonvl) amino] acetate
- Step 3 (2S)- Cyclop entyl [ ( ⁇ [ ( 1 R2R)-2 -p ent-4-en- 1 -yl cyclopent ylj oxy ⁇ carbonyl)amino] ac.eti c acid
- Step 1 Methyl (2S)-[( ⁇ [ " f ljf?,2jy)-2-allylcyclopentyl]oxy ⁇ carbonyl)amino](cyclohexyl)acetate
- NjN'-disuccininiidyl carbonate (8.28 g, 32.3 mmol) and triethylamine (4.51 ml, 32.3 mmol) were added to a solution of Intermediate Al 1 (3.4 g, 26.9 mmol) in acetonitrile (70 ml) and left stir under a N 2 atmosphere for 6 hours.
- Methyl (2S)-amino(cyclohexyl)acetate hydrochloride (8.39 g, 40.4 mmol) and triethylamine (7.51 ml, 53.9 mmol) were added to the reaction mixture and left stir under a N 2 atmosphere for 12 hours.
- Step 2 (2S)- ⁇ ( ⁇ ( lJ?,2 1 S f )-2-allylcvclopentyl]oxy ⁇ carbonyl)amino](cyclohexyl)acetic acid
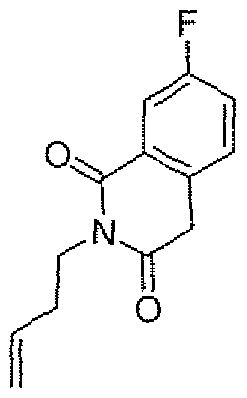
- Step 2 2-But-3-en-l -yl-6-methoxyisoquinoline-l ,3(2H,4H)-dione
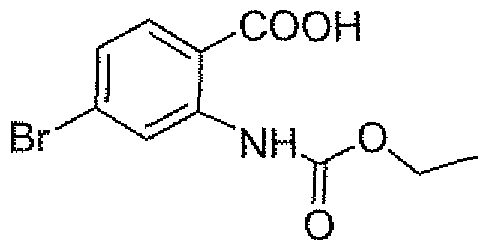
- the title compound was prepared according to the procedure for Intermediate CI, using 2-(carboxymethyl)-4-methoxybenzoic acid in place of homophthalic anhydride.
- LCMS (ES+) m/z 246.04 (M+H) + .
- Intermediate C3 2-but-3-en- 1 -yl- ⁇ -chloroisoquinoline- 1 ,3(2H,4H)-dio:ne
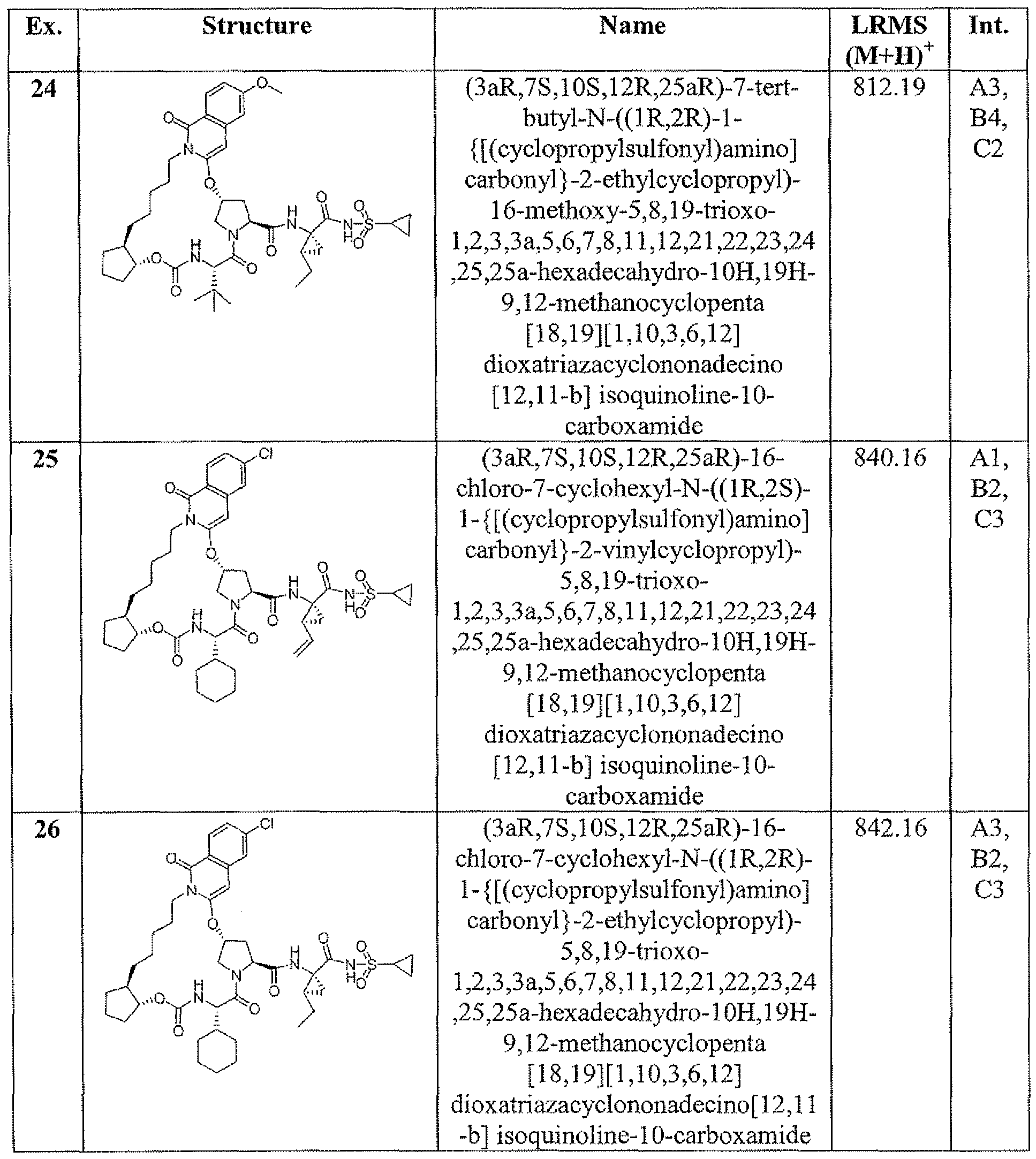
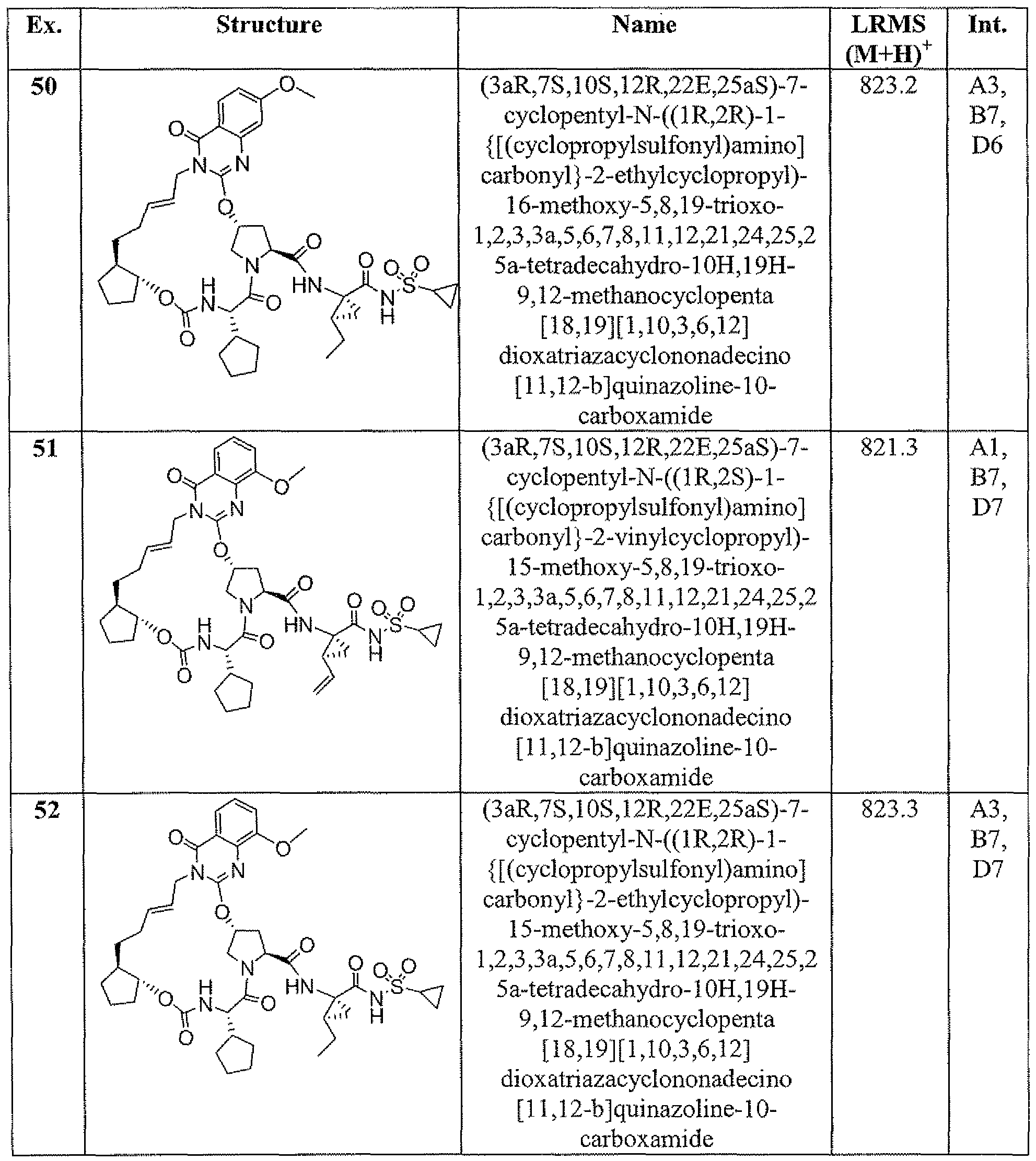
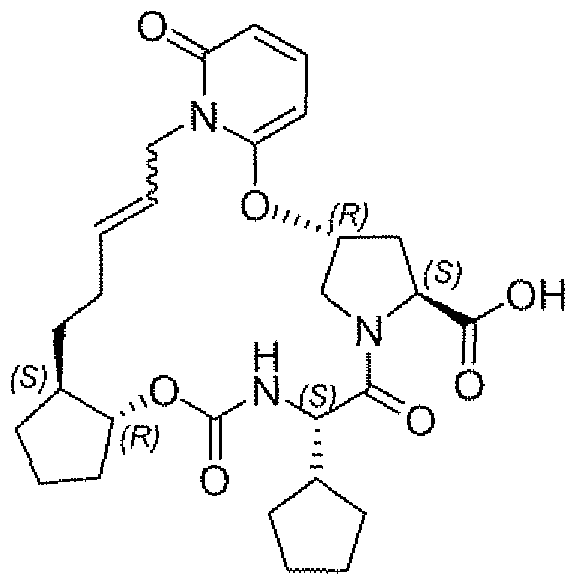
- Example 1 (3a ⁇ J ⁇ lQ ⁇ 12-R,23g,25a ⁇ -7-cvclopentyl-N-(fli?,2 ⁇ -1- ⁇ [(cyclo ⁇ rgpylsulfonyl)amino3carbonyl ⁇ -2-vinylcyclopropyl)-5,8J9-trioxo- L2,3,3a,5 > 6J,8.11J2,21.22,25,25a-tetradecahvdro40H.19H-9J2- methanocyclopentaf 18, 19][ ⁇ , 10,3,6, ⁇ 2]dipxatriazacyclononadecinoj ' 12 ,11 -bjisoquinoline- 10- carboxamide
- Step 1 1 -tert-Butyl 2-methvl f2ff.4R)-4-
- Step 3 Methyl C4i?)-1- ⁇ (26 f )-2-Ffl ⁇ lJ?,2 ⁇ -2-allvIcvclopentvl]oxy)carborjyl)amino1-2- cyclopentylacetyl)-4-[(2-but-3-en-1-yl-1-oxo-L2-dihydrpisoq ⁇ iinolin-3-yl)oxyl-L-prolinate
- Step 4 Methyl (SaRJS, 1 OS ⁇ 2&23&25a5V7-cvclopentyl-5,8.19-trioxo- 1 ,2,3 ,3a,5,6.7,8, 1 U 2,21 ,22,25,25a-tetradecahvdro- 1 OH, 19H-9 J 2- me thanpcvclopentaf 18 J 911- 1 ,10 ,3 ,6 J 2]dioxatriazacyclononadecino [ 1241 -b ⁇ soquinoline- 10- carboxylate
- Step 5 OaRJS ⁇ 0S ⁇ 2R23E25aS)-7-c ⁇ c ⁇ OQnt ⁇ l-5$ ⁇ 9-t ⁇ oxo- 1.2.3.3a.5.6.7,8,l l,12.21 ⁇ 2,25.25a-tetradecahvdiO-10H,19H-9.12- methanocyclopentaf 18 , 19] [ 1 , 10,3 ,6, 12] dioxatriazacyclononadecino[ 12,11 -b]isoquinoline- 10- carboxylic acid
- Example 13 (3aRJS ⁇ QS ⁇ 2R25&RY7-c ⁇ d ⁇ Omiyl- ⁇ N-((lR2S)A- ⁇ [(cydopropylsulfonyl)amino] carbonyl I -2- vinylcyclopropyl)-5 ,8 , 19-trioxo- 1,2,3,3a,5,6,7,8,l U2,2U2,23,24,25,25a-hexadecahvdro-10H.19H-9.12- methanocyclopentaf 18 , 19] [ IJ 0,3 ,6 J 2] dioxatriazacyclononadecino [ 12, 11 -blisoquinoline- 1 Q- carboxaroide
- Step 2 (3aRJS ⁇ 0S ⁇ 2R25aR)-l-cydovQ ⁇ Ayl-5.8 ⁇ 9-t ⁇ oxo- l ,2,3,3a,5,6,7,8J lJ2,21,22,23,24,25,25a-hexadecahvdro-10HJ9H-9J2- methanocyclo ⁇ enta[ 18, 19] [ 1 ,10,3 ,6, 12]dioxatriazacyclononadecino[ 12,11 -b]isoquinoline- 10- carboxylic acid
- Step 3 (3aRJS ⁇ 0S ⁇ 2R25aR)-7-oychy&ntvl- ' N'( ⁇ lR2 ⁇ -l-
- Step 1 Methyl (3&RJS ⁇ QS ⁇ 2R,22E26aR)-7-cvdoyentv ⁇ 5$ ⁇ 9 ⁇ tnoxo- 2.3.3a.5.6J,8.11.12 ⁇ 1.24.25.26,26a-tetradecahvdro-1H.10H,19H-9,12- methanocyclopenta[ 19,20] [ 1 J0,3,6,12]dioxatnazacycloicosmo[l 2,1 l-b]isoquinolme-10- carboxylate
- Step 2- Methyl (3aRJS ⁇ 0S ⁇ 2R25aR)-7-c ⁇ dopentv ⁇ -5£ ⁇ 9-tnoxo- U ⁇ 3a,5A7,8 ⁇ U22Ula,22,22a,2324.25.25a-octadecahydro40HJ9H-9J2- methanocyclopentaf 19,20] cyclopropa[ 14, 151 f l , 10,3 ,6 , 121 dioxat ⁇ azacycloicosino [12,11- bi isoq umohne- 10-carbox ylate
- PdOAc 2 (6.67 mg, 0 030 mmol) was added to a mixture of methyl (3aR,lS, ⁇ 0S,l2R22E,26aR)-7-cydopmty ⁇ -5 ⁇ ,l 9-tnoxo-2,3,3a,5.6,7,8,l 1,12,21 ,24,25,26,26a- tetradecahydro-1H,l OH, 19H-9.12-methanocyclopenta[ 19,20][ 1 , 10,3 ,6,12] dioxatnazacycloicosino[12,l l-b]isoqumohne-10-carboxylate (90 mg, 0 149 mmol) m diethyl ether/THF (2 ml), and the mixture allowed to stir for 5 mm To this mixture was added dropwise a solution of diazomethane (0.97 mmol, 10 eq) in diethyl ether (2 mL) The reaction was stirred for
- Step 3 (3aRJS ⁇ 0S ⁇ 2R25aR)-7-c ⁇ c ⁇ opQnt ⁇ h53 ⁇ 9-t ⁇ oxo- 123 ,3a ⁇ 6 ,7,8 , 1 U 2,21 ,21 a.22,22a.23.24.25.25a ⁇ ctadecahvdro- 1 QH, 19H-9, 12- methanoc yclopenta[ 19,20] cyclopropaf 14, 15] [ IJ 0,3 ,6, 121 dioxatriazacycloicosino[ 12,11- blisoqumoline-10-carbo ⁇ ylic acid
- Step 4 (3aR.lS ⁇ 0S ⁇ 2R25aRY7-c ⁇ c ⁇ o ⁇ entyl- ' N-(aR2S) ⁇ ' ⁇ ffcyclopropylsulfonyl)aminolcarbonvU-Z-vinylcyclopropylVS ⁇ ⁇ ⁇ -trioxo- 1,23.3a3,6.7.8.11.12,2L21a,22,22a,23.24,25.25a-octadecahvdro-lQH,19H-9J2- methanocvclopenta[19,20]cyclopropa[14J5][l J0,3,6,121dioxatriazacycloicosino[12J l- biisoquinoline-1 O-carboxamide
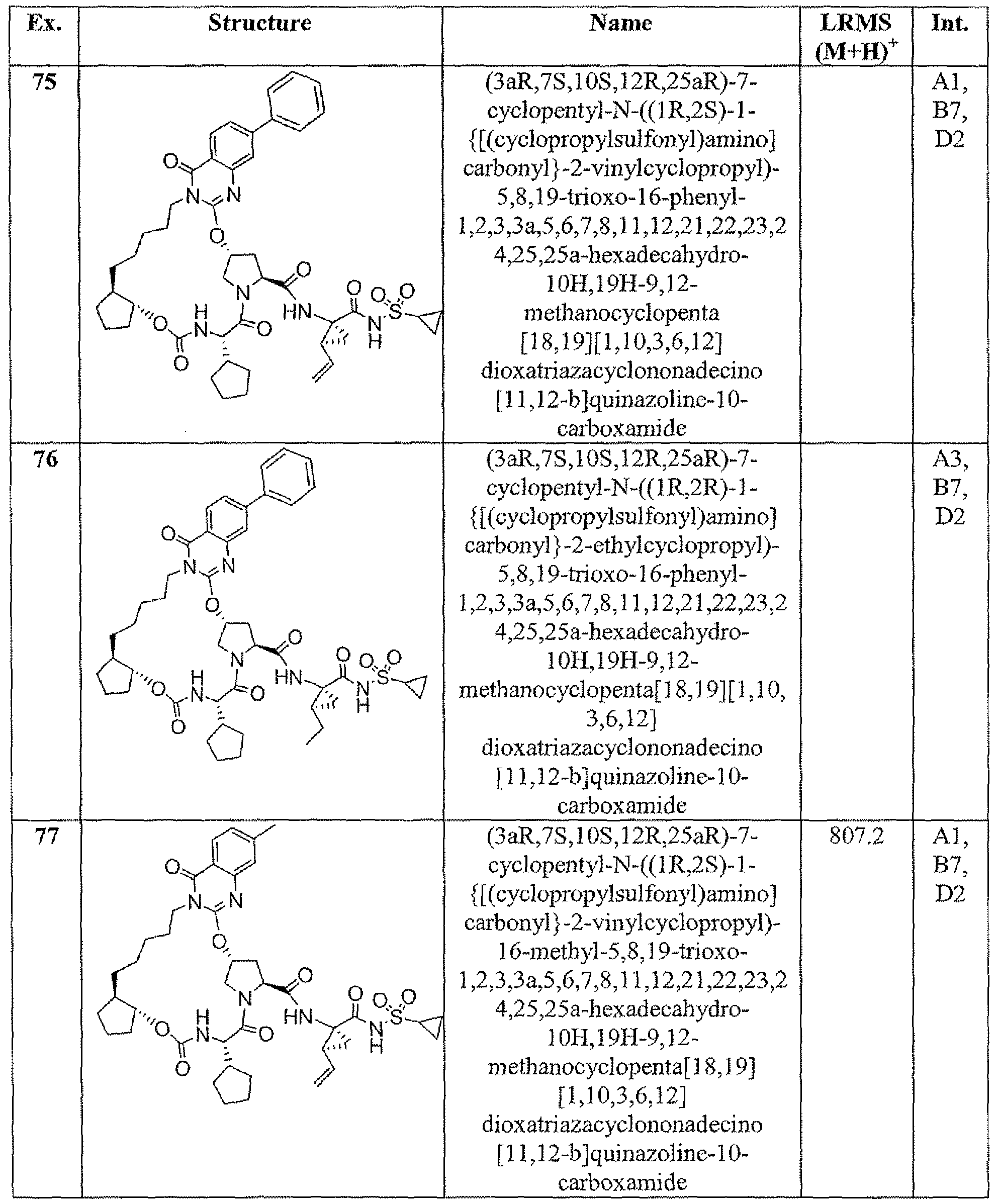
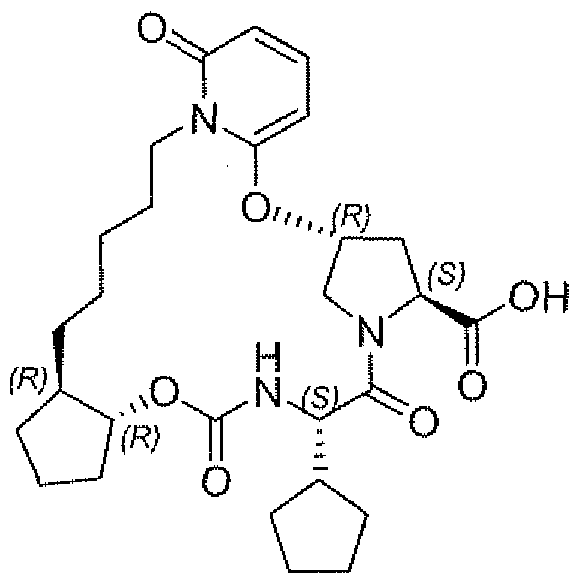
- Example 38 r3ai;,75.1Q>S,12i?,24aS r )-16-chloro-7-cvclo ⁇ entyl-N-fflj;,2y>4- ⁇ [fcyclopropylsulfonyl)amino]carbonyl ⁇ -2-vinylc ⁇ clopropyl)-5,8,19-trioxo- 2,3.3a.5,6J,8.11.12.21.22,22a t 23,23a ⁇ 4.24a-hexadecahvdro-1H,10H,19H-9,12- methanocyclopenta[l 8,191cyclopropa[l 5,16][1 ,10,3i6,121dioxatriazacyclononadecin ⁇ [12,l 1 - blisoquinoline-10-carboxamide
- Example 39 (3aRJSA0SA2R22E25aSh7-cvclOO&Ayl-N- ⁇ aR2S)-l- ⁇ [ (cyclopropyisulfonyl)amino] carbonyl ) -2- vinylcyclopropyl)-5 ,8,19-trioxo- 1.2.3.3a.5,6J.8.11.12.21 , 24,25,25a-tetradecahydro-I0HJ9H-9,12- methanocyclopentaf 18 , 19] F IJ 0,3 ,6, 12] dioxatriazacyclononadecinof 11 ,12-blq ⁇ j.inazoline- 10- carboxamide
- Step 1 1-fert-butvl 2-methyl (2»y.4J? ' )-4-rf3-allyl-4-oxo-3.4-dihydroq ⁇ inazolin-2- yl)oxy1pyrrolidine- 1 ,2-dicarboxylate
- Step 2 Methyl (4J?)-4-f(3-allyl-4-oxo-3,4-dihydroquinazolin-2-yl ' )oxy1-L-prolinate
- Step 3 Methyl (4J ⁇ M4f3-allyl-4-oxo-3,4-dihvdroq ⁇ i p azolin-2-vnoxyl-lWf25V2-r(f [(1R.2SV2- but-3-en-1-ylcyclopentyl1oxy)carbonyl)amino1-2-cyclo ⁇ >entylacetvU-L-prolinate
- Step 4 Methyl f3aRJ-?,105,12f1,22g,25a ⁇ -7-cyclopentyl-5 ⁇ 8,19-trioxo- 123,3a,5,6.7,8J U2,21,24.25,25a4etradecahydro-10HJ9H-9J2- methanocvclopentaf 18, 19][ 1 ,10,3,6 J2]dioxatriazacyclpnonadecino[ 11,12-b]quinazoline-l O- carboxylate
- Step 5 (3&RJSA0SA2R22E25aS)-7'Cvc ⁇ opQnt ⁇ -S$A9-t ⁇ oxo- 1.233a,5,6j,8,l lJ2.2L24.25,25a4etradecahvdro-10H,19H-9,12- methanocyciopenta[l 8 ,19-[[I A 0,3,6,12Jdioxatriazacvclononadeeino[ 1 l J2-b]quinazoline-l 0- carboxylic acid
- Step 6 OaRJS. I OS, 12i?,22£,25a£)-7-cyclopentvl-N-( (IR2SY- 1 - ( [(cyclopropylsulfonyl)aroinoi carbon yl i -2- vinylcyclopropyl)- 5 , 8.19-trioxo- 1.2.3,3a.S.6.7.8.11.1221,24.25.25a-tetradecahvdro-10HJ9H-9.12- methanocyclopentaf 18 , 19] [ 1 , 10,3 ,6 J 2]dioxatri azac yclononadecino [ 11 , 12-b] quinazo ⁇ ine- 10- carboxamide
- Example 59 (3aR.7.S',10 ⁇ 12/?,22g,25a5)-7-cvclopentyl-N-ffli;,25)-1- ⁇ [fcyclopropyls ⁇ dfonyl)amino]ca ⁇ bonyl ⁇ -2-vinylcvclopropy ⁇ -5,8,19-trioxo-16-phenyl- 1,233a,5.6.7,8J L122K24,25,25a-tetradecahvdro-10HJ9H-9.12- methanocyclopenta[l 8, 19] [ 1 ,10,3,6, 12]dioxatriazacyclononadecino[ 11 J2-b-]quinazoline-l 0- carboxamide
- Step 1 Methyl Gai?J5U05a2i?,22£25a5yi6-bromo-7-cyclopentyl-5,8a94rioxo-
- Step 2 Methyl (3siRJSJ0S ⁇ 2R22E25aS)-7-cyclopent ⁇ l-5S ⁇ 9-t ⁇ oxo-16-phen ⁇ l ⁇
- Step 3 ( 3 aRJS ⁇ 0S ⁇ 2 R22E25aS)-7- cyclopentyl-N-fd R2S)- ⁇ -
- Example 60 (3aR.7S,10SJ2R.22E,25aSV7-cvclopeatyl-N-f(lR,2RVl-
- Example 61 (3d ⁇ RJS ⁇ 0S ⁇ 2R22E25aS)-7-c ⁇ chOQnt ⁇ - ⁇ -(( ⁇ R2S)-l- ⁇
- Step 1 MethvIOa ⁇ J ⁇ lOS.U ⁇ lE ⁇ Sayy-y-cvclopentyl-l ⁇ -methyl-S ⁇ jg-trioxo- 1,2,3,3a.5,6J,8,11.12.21.24,25,25a-tetradecahvdro-10H.19H-9,12- methanocvclopenta[18J91f l J0,3,6J2]dioxatriazacyclononadecino[m2-blq ⁇ tma2oline-10- carboxylate
- Tetramethyltin (0.041 ml, 0.298 mmol), lithium chloride (50.5 mg, 1.191 ⁇ raiol), triphenylphosphine (15.62 mg, 0.060 mmol) and bis(triphenylphosphine)palladium(II) chloride (15.68 mg, 0.022 mmol) were added to a nitrogen purged solution of methyl (3ai?/7S,l 05,12/ ⁇ 22£,25aS> 16-bromo-7-cyclopentyl-5.8, 19-trioxo- 1,2,3,38,5,6,7,8,11,12,21,24,25,258-16 ⁇ 3(1608117 ⁇ 0-1011,1913-9,12- methanocyclopentaf 18 , 19] [ 1 , 10 , 3 ,6 , 12] di ox atri azacycl ononadecino [ 11 , 12 -b ] quinazoline- 10- carboxylate, Example 59 Step 1 (100
- Step 2 (3aJ?.7 ⁇ 105,12i;,22£,25a5 f )-7-cvclopentyl-N- ⁇ i ⁇ .2 ⁇ )-1-
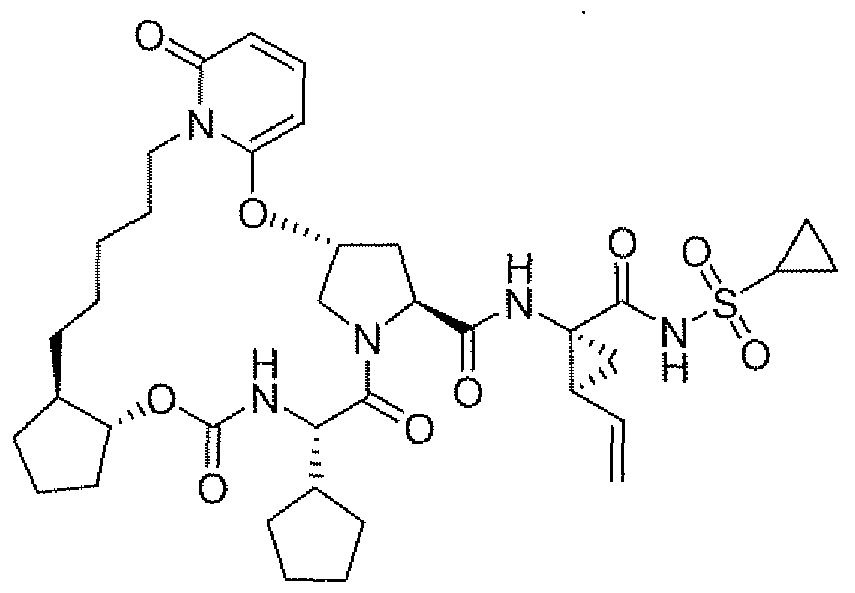
- Example 62 ( ⁇ R ⁇ 4E ⁇ 8 ⁇ .22 J?,26 ⁇ 29 ⁇ )-26-cvclopentyl-iV-(( li?,2i?)-l - ⁇ j-fcvclopropylsulfonyl)amino1carbon ⁇ l)-2-ethylcvclopropyl)-7-methyl-l K24,27-trioxo-2,23- dioxa-4J225,28-tetraaza ⁇ entacvclo[ ' 26.2, 1.0 3 ' 12 ,0 5;1() .0 l8 ' 22 ]hentriaconta-3,5,7,9.14-pentaene-29- carboxamide
- Example 63 (3&RJS ⁇ 0S ⁇ 2R22E25aS)-l6-cvax ⁇ o-7-c ⁇ clopentyl-N-( ⁇ R2S) ⁇ l- ⁇ [(cyclopropylsulfonyl)aminol carbonyl I -2-vinylcyclopropyl)-5 ,8,19-trioxo- 1.2.3,3a,5,6J.8.11.12.21 ,24.25.25a-tetradecahvdro-10H.19H-9.12- methanocyclopenta[ 18 , 19] [ IJ 0,3 ,6, 12]dioxatriazacyclononadecino[ ' 11,12-b]quina2oline- 10- carboxamide
- Step 1 Methyl (3ai?JSJ0£J2i ⁇ 2£.2Sa5)-16-cyano-7-cvclopentyl-5,8J94rioxo-
- Zinc cyanide (5.20 ⁇ l, 0.082 mniol) was added to a nitrogen-purged solution of methyl (3aR,7S,l OS, 12R,22E,25s£)- 16-bromo-7-cyclo ⁇ entyl-5,8, 19-trioxo- 1,2,3 ; 3a ; 5,6,7,8J l,12,21,24,25,25a-tetradecahydro-10HJ9H-9J2- methanocyclopenta[l 8 J 9] [ 1 ,10,3,6 J2]dioxatriazacyclononadecino[ 11 J 2-b]quinazoline-l 0- carboxylate,
- Example 59 Step 1 (50 mg, 0.074 mmol) in DMF (1 ml), followed by Pd(Ph 3 P) 4 (8.60 mg, 7.45 ⁇ mol), and the mixture was heated at 100°C for 3 hrs. The reaction was filtered and purified by reverse phase chromatography to
- Step 2 (3aRJS ⁇ 0S ⁇ 2R22E25aS)-l6-c ⁇ ano-7-C ⁇ c ⁇ opent ⁇ l-N-((W2S)-l-
- Example 64 (3aRJS ⁇ 0S ⁇ 2R22E25aS)-l 6-cvano-7-cvclopentyl-N-((lJ;,2J;)-1- (rCcvclopropylsulfonyl)amino]carbonyl ⁇ -2-ethylcvclopropyl)-5,8,19-trioxo- 1,2,3,3 ⁇ 5,6,7,8,1 U2,21,24,2S.25a-tetradecahvdro-10H.19H-9,12- methanocvclopentafl8J9][U0,3,6 ⁇ ,12]dioxatria2acvclononadecino[l l, ⁇ 12-b]quinazolirse-10- carboxamjde
- Example 65 (38 ⁇ ,75.10 ⁇ 12J?,25aR)-7-cvclope ⁇ tvi-N-f(lJ?,25>-1-
- Step 1 Methyl (3eJtJS ⁇ OS ⁇ 2R22E25aS)-7 ⁇ vclop&ityl-5,8 ⁇ 9-t ⁇ oxo-
- Step 2 (3aRJSJ0-?,12jR.22Jg,25aS)-7-cvclopentv ⁇ -5 ⁇ 8J9-trioxo- 1.2.3.3a.5,6,7,8.11 J2.21.24,25.25a-tetradecahvdro-10H,19H-9J2- methanocyclopentafl8J91
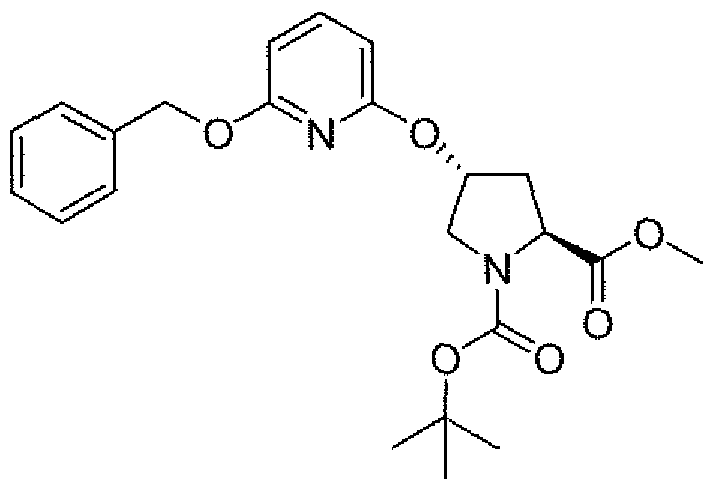
- Step 2 (4J?V4- ⁇ j " 6-fBenzyloxy) ⁇ yridin-2-ylloxyl-1-ffert-butoxycarbonyl)-L- ⁇ roline
- Step 4 1 -/erf-Butyl 2-methyl f2-? ⁇ 4J?)-4-[f6-hvdro ⁇ ypyridin-2-yl)oxy1 ⁇ vrrolidine-l 2- dicarboxvlate
- Step 6 1-tert-But ⁇ l 2-methy ⁇ (2-?,4/gV4-rf l-aIlyl-6-oxo-l ,6-dihydropvridin-2- ypoxyjp yrrolidme- 1 ,2-di carboxylate
- Step 7 (25 r ,4i?)-4-[(l -Allyl-6-oxo-l ,6-dihydropyridin-2-vDo ⁇ 1-2-(methoxycarbonyl)- pyrrolidinium chloride
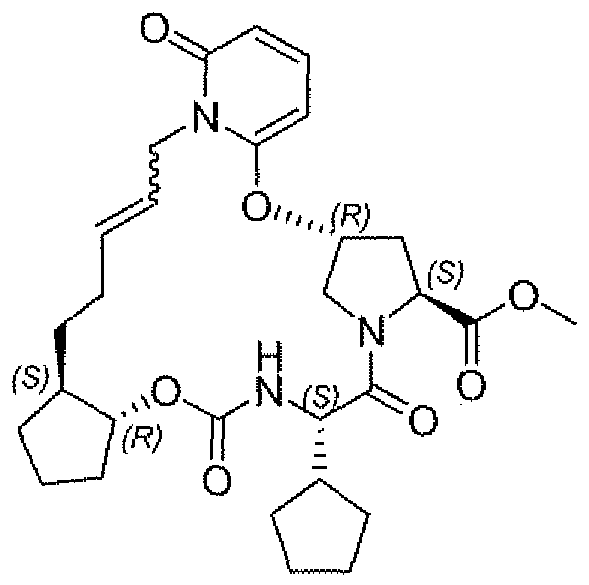
- Step 8 Methyl (4JgM-ffl-allyl-6-oxo-L6-dihvdropwidifl-2- ⁇ 1)oxv]-l -If 25)-2-[( ⁇ [f lJg,2S)-2- but-3-en- 1 -ylcyclopentyljoxyl carbon ⁇ l)amino]-2-cyclopentylacetyl) -L-prolinate
- Step 9 Methyl (IRAOE, and 10Z,14>S F J8ig,225',255)-22-cyclopentyl-7,20,234rioxo-2J9-dioxa- 8,21.24-triazatetracyclor22.2.1.0 3 ' 8 .0 14 ' 18 lhe ⁇ tacosa-3,5.10-triene-25-carboxylate O
- Step 10 (IR ⁇ OE, and 10ZJ4JU8i?22£25 ⁇ -22-CvclopentyI-7,20.23-trioxo-2.19-dioxa- 8.21.24-triazatetracvclof22.2.1 ,0 3 ' 8 .0 14 ' l8 ]heptacosa-3,5,l O-triene-25-carboxylic acid
- Step 11 (IR ⁇ OE, and 10Z.145,18J?.22.S',2S5)-22-Cvclopentyl-.V-ffl J R ⁇ J Sf)-1-Ufcvclo- propylsulfonyl)aminoicarbonyl ⁇ -2-vinylcyclopropyl)-7,20,23-trioxo-2,l 9-dioxa-8,21 ,24- triazatetracvcloF22.2.1.0 3 ' 8 , ⁇ ' 4 ⁇ 8 lheptacosa-3,5JO-triene-25-carboxamide
- Step 1 Methyl (lJ?.14 ⁇ ,18ig,225,2S.?)-22-cvclopentyl-7.2023-trioxo-2,19-dioxa-8 ⁇ 2 ] .24- triazatetracyclor22.2.1.0 3 ' S . ⁇ ' 4 ' iS 1heptacosa--3,5-diene-25-carboxvlate
- Step 2 (lJ?J4 J RJ8i?,22 ⁇ 255 r )-22-Cvclopentyl-7,20,23-trioxo-2.19-dioxa-8,21,24- triazatetracvclo[22.2.1.0 3 ' S .Q' 4 ' 18 lheptacosa-3,5-diene-25-carboxylic acid
- Step 3 d ⁇ J4 ⁇ J8 ⁇ ,22 ⁇ 25 ⁇ -22-cvclopentyl-JV-f(lJ ! ?,25 r )-1- ⁇ r(cvclopropylsulfonyl)- aminol carbonyl I -2-vinyl cvclo ⁇ ro ⁇ yl)-7 ,20,23 -trioxo-2 J 9-dioxa- 8.21 ,24- triazatetracvcloi-22.2.1.0 3 ' s .0 l4 ' ls 1heptaco$a-3,5-diene-25-carboxamide
- Example 83 (IR ⁇ 4R ⁇ SR22S25,SV 6- bromo-22 -cyclopent yl-iV-Cf 1 R2S)- 1 - ([(cyclopropylsulfonyl)aminolcarbonyl ⁇ -2-vinylcyclopropyl')-7,20,23-trioxc)-2,19-dioxa-
- Step 1 Methyl fli?J4i?J8 ⁇ ,225,255 r )-6-bromo-22-cvclopentyl-7.20.23-trioxo-2J9-dioxa- 8,2L24-triazatetracvclor22.2.1.Q 3 ' 8 ,0 14 ' ls ]heptacosa-3.5-diene-25-carboxylate
- Step 2 flJ?,14j?J8i;,225',25y>-6-bromo-22-cvclopentyl-7,20,23-trioxo-2.19-dioxa-8,21.24- triazatetracvclo[22.2.1.0 3 ' 8 ,0 14 ' 18 lheptacosa-3,5-dieae-25-carboxvlic acid
- Step 3 (l ⁇ a4 ⁇ J 8 ⁇ 22 ⁇ 25 i $ f )-6-bromo-22-cyclopentvl-N-r ⁇ J?,25)-1- ⁇ [fcyclopropylsijlfonyl)amino]carbonyl)-2-vinylcvclo ⁇ ro ⁇ yl)-7,20,23-trioxo-2,19-dioxa- 8,21,24-triazatetracvclof 22,2, 1.0 3 ' 8 .0 14 ' ls lheptacosa-3,5-diene-25-carboxamide
- Example 84 aR ⁇ 4R ⁇ R22S25S) ⁇ 22-cvc ⁇ Oent ⁇ -N-((lR2S)- ⁇
- Step 1 Methyl flJ?,147?J8J?.22 ⁇ .25.y>-22-cyclopentyl-7.2Q,23-trioxo-6-t>henvl-2J9-dioxa- 8,21 ,24-triazatetracyclor22.2.1.Q 3 ' 8 .0 14>ls 1heptacosa-3 ,5-diene-25-carboxylate
- Step 2 ⁇ J?J4J?J8/?.22 ⁇ 25 ⁇ -22-cvclopentyl-7,20,23-trioxo-6-phenyl-2J9-dioxa-8,2L24- triazatetracvclof22.2,1.0 3 ' s .0 ⁇ 4 ' ls 1heptacosa-3.5-diene-25-carbox:ylic acid
- Step 3 (lJ ⁇ ,14J?.18i;,22 J ?,25 ⁇ )-22-cvdopentyl- ⁇ -f (1 ⁇ .25)-!-
- the HCV NS3 protease inhibitory activity was measured using the protease time- resolved fluorescence (TRF) assay as described below and in International Patent Application Publication WO 2006/102087.
- TRF protease time- resolved fluorescence
- the assay was performed with HCV genotype Ib (BK) NS3 modified with a R 155K mutation .
- the assay was performed in a final volume of 100 ⁇ l in assay buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 15 % glycerol, 0.15 % TRITON X-100, 10 mM DTT, and 0.1 % PEG 8000.
- NS3 and NS4A protease is pre-incubated with various concentrations of inhibitors in DMSO for 30 minutes. The reaction is initiated by adding the TRF peptide substrate (final concentration 100 nM).
- NS3 mediated hydrolysis of the substrate is quenched after 1 hour at room temperature with 100 ⁇ l of 500 mM MES 3 pH 5.5.
- Product fluorescence is detected using either a VICTOR V2 or FUSION fluorophotometer (Perkin Elmer Life and Analytical Sciences) with excitation at 340 nm and emission at 615 nm with a 400 ⁇ s delay. Testing concentrations of the enzyme containing the R155K mutation were selected to result in a signal to background ratio (S/B) of 10-30.
- S/B signal to background ratio
- IC 50 values are derived using a standard four- parameter fit to the data. K; values are derived from IC 50 values using the following formula,
- IC 50 K 1 (1 + [S] / KM), Eqn (I), where [S] is the concentration of substrate peptide in the reaction and K M is the Michaelis constant. See P. Gallinari et al., 38 BiOCHEM. 5620-32(1999); P. Gallinari et al, 72 J. ViROL. 6758-69 (1998); M. Taliani et al, 240 ANAL. BIOCHEM. 60-67 (1996).
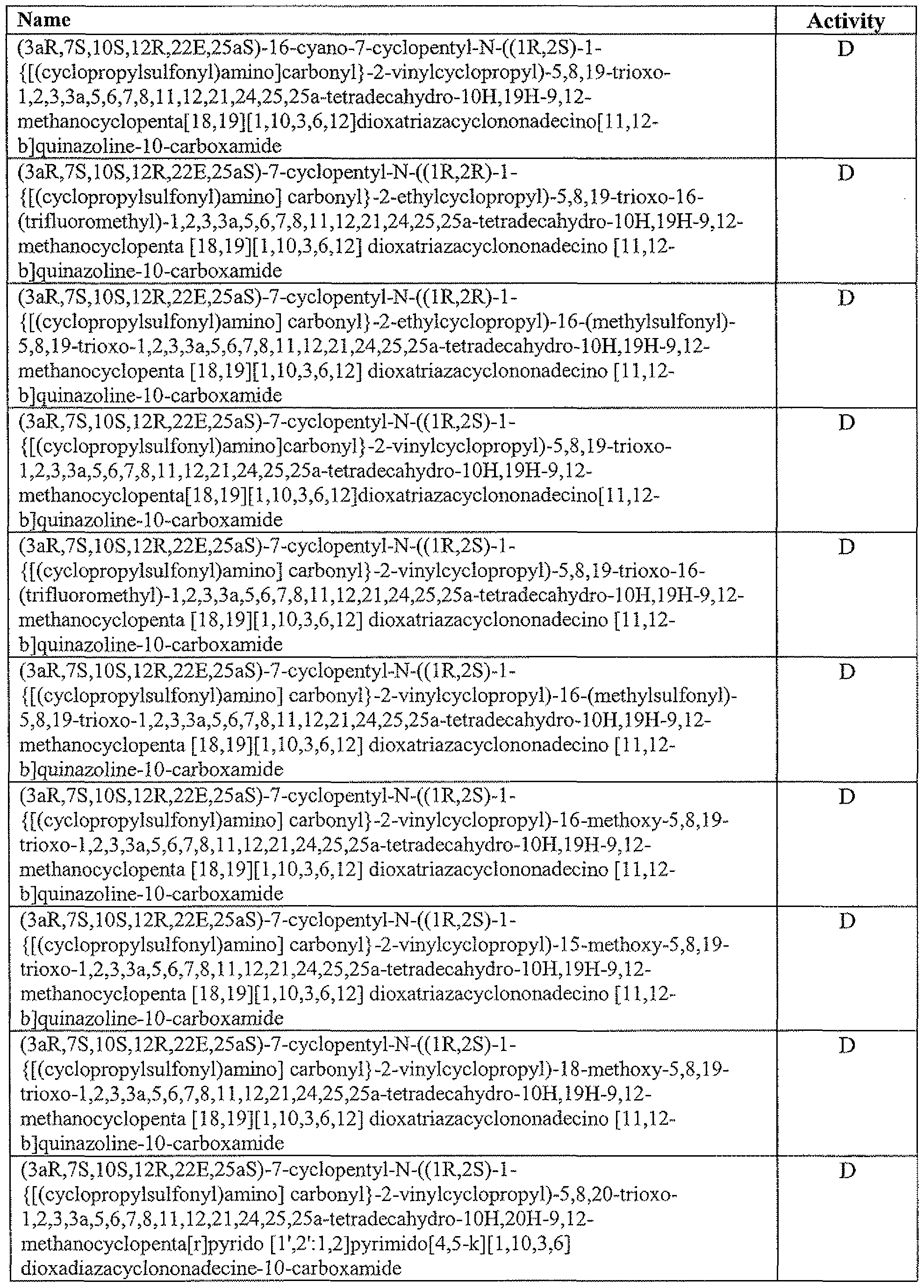
- the activity table provided below illustrates the observed activity, where compound activity fell within the following ranges: A: Ki 1 nM to 5 nM B: K, 0.5 nM to 1 nM C: K, 0.1 nM to 0.5 nM D: K 1 ⁇ 0.1 nM Activity Table
Abstract
Description





































































































































Claims









Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/989,672 US8461107B2 (en) | 2008-04-28 | 2009-04-16 | HCV NS3 protease inhibitors |
| EP20090739426 EP2271345B1 (en) | 2008-04-28 | 2009-04-16 | Hcv ns3 protease inhibitors |
| JP2011507531A JP2011518882A (en) | 2008-04-28 | 2009-04-16 | HCV NS3 protease inhibitor |
| AU2009241445A AU2009241445A1 (en) | 2008-04-28 | 2009-04-16 | HCV NS3 protease inhibitors |
| CN200980115230XA CN102014911A (en) | 2008-04-28 | 2009-04-16 | HCV NS3 protease inhibitors |
| CA2720850A CA2720850A1 (en) | 2008-04-28 | 2009-04-16 | Hcv ns3 protease inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12568808P | 2008-04-28 | 2008-04-28 | |
| US61/125,688 | 2008-04-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009134624A1 true WO2009134624A1 (en) | 2009-11-05 |
Family
ID=41255357
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/040815 WO2009134624A1 (en) | 2008-04-28 | 2009-04-16 | Hcv ns3 protease inhibitors |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8461107B2 (en) |
| EP (1) | EP2271345B1 (en) |
| JP (1) | JP2011518882A (en) |
| CN (1) | CN102014911A (en) |
| AU (1) | AU2009241445A1 (en) |
| CA (1) | CA2720850A1 (en) |
| WO (1) | WO2009134624A1 (en) |
Cited By (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011091757A1 (en) | 2010-01-27 | 2011-08-04 | AB Pharma Ltd. | Polyheterocyclic compounds highly potent as hcv inhibitors |
| JP2011528713A (en) * | 2008-07-22 | 2011-11-24 | メルク・シャープ・エンド・ドーム・コーポレイション | Macrocyclic quinoxaline compounds as HCV NS3 protease inhibitors |
| US8372802B2 (en) | 2008-03-20 | 2013-02-12 | Enanta Pharmaceuticals, Inc. | Fluorinated macrocyclic compounds as hepatitis C virus inhibitors |
| WO2013028470A1 (en) | 2011-08-19 | 2013-02-28 | Merck Sharp & Dohme Corp. | Process and intermediates for preparing macrolactams |
| WO2013074386A2 (en) | 2011-11-15 | 2013-05-23 | Merck Sharp & Dohme Corp. | Hcv ns3 protease inhibitors |
| EP2618665A1 (en) * | 2010-09-21 | 2013-07-31 | Merck Sharp & Dohme Corp. | Hcv ns3 protease inhibitors |
| WO2014009509A1 (en) * | 2012-07-13 | 2014-01-16 | Janssen R&D Ireland | Macrocyclic purines for the treatment of viral infections |
| US8648037B2 (en) | 2010-09-21 | 2014-02-11 | Enanta Pharmaceuticals, Inc. | Macrocyclic proline derived HCV serine protease inhibitors |
| US8828930B2 (en) | 2009-07-30 | 2014-09-09 | Merck Sharp & Dohme Corp. | Hepatitis C virus NS3 protease inhibitors |
| US8936781B2 (en) | 2009-05-13 | 2015-01-20 | Enanta Pharmaceuticals, Inc. | Macrocyclic compounds as hepatitis C virus inhibitors |
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8962810B2 (en) | 2011-06-16 | 2015-02-24 | AB Pharma Ltd. | Macrocyclic heterocyclic compound for inhibiting hepatitis C virus and preparation and use thereof |
| US9120818B2 (en) | 2010-12-14 | 2015-09-01 | Merck Sharp & Dohme Corp. | Process and intermediates for preparing macrolactams |
| US9193740B2 (en) | 2009-10-19 | 2015-11-24 | Enanta Pharmaceuticals, Inc. | Bismacrocyclic compounds as hepatitis C virus inhibitors |
| US9284304B2 (en) | 2012-08-10 | 2016-03-15 | Janssen Sciences Ireland Uc | Substituted pyrimidines as toll-like receptor modulators |
| US9296782B2 (en) | 2012-07-03 | 2016-03-29 | Gilead Sciences, Inc. | Inhibitors of hepatitis C virus |
| US9334279B2 (en) | 2012-11-02 | 2016-05-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9409943B2 (en) | 2012-11-05 | 2016-08-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9499550B2 (en) | 2012-10-19 | 2016-11-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9580463B2 (en) | 2013-03-07 | 2017-02-28 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9598433B2 (en) | 2012-11-02 | 2017-03-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9617310B2 (en) | 2013-03-15 | 2017-04-11 | Gilead Sciences, Inc. | Inhibitors of hepatitis C virus |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9873707B2 (en) | 2013-10-18 | 2018-01-23 | Merck Sharp & Dohme Corp. | Methods and intermediates for preparing macrolactams |
| US10253003B2 (en) | 2012-11-16 | 2019-04-09 | Janssen Sciences Ireland Uc | Heterocyclic substituted 2-amino quinazoline derivatives for the treatment of viral infections |
| US10259814B2 (en) | 2012-10-10 | 2019-04-16 | Janssen Sciences Ireland Uc | Pyrrolo[3,2-d]pyrimidine derivatives for the treatment of viral infections and other diseases |
| US10259793B2 (en) | 2013-02-21 | 2019-04-16 | Janssen Sciences Ireland Uc | 2-aminopyrimidine derivatives for the treatment of viral infections |
| US10266543B2 (en) | 2013-03-29 | 2019-04-23 | Janssen Sciences Ireland Uc | Macrocyclic deaza-purinones for the treatment of viral infections |
| US10272085B2 (en) | 2011-04-08 | 2019-04-30 | Janssen Sciences Ireland Uc | Pyrimidine derivatives for the treatment of viral infections |
| US10280167B2 (en) | 2011-11-09 | 2019-05-07 | Janssen Sciences Ireland Uc | Purine derivatives for the treatment of viral infections |
| US10316043B2 (en) | 2013-07-30 | 2019-06-11 | Janssen Sciences Ireland Unlimited Company | Thieno[3,2-d]pyrimidines derivatives for the treatment of viral infections |
| US10377738B2 (en) | 2013-05-24 | 2019-08-13 | Janssen Sciences Ireland Unlimited Company | Pyridone derivatives for the treatment of viral infections and further diseases |
| US10385054B2 (en) | 2013-06-27 | 2019-08-20 | Janssen Sciences Ireland Unlimited Company | Pyrrolo[3,2-d]pyrimidine derivatives for the treatment of viral infections and other diseases |
| US10968184B2 (en) | 2016-09-29 | 2021-04-06 | Janssen Sciences Ireland Unlimited Company | Pyrimidine prodrugs for the treatment of viral infections and further diseases |
| US11053256B2 (en) | 2016-07-01 | 2021-07-06 | Janssen Sciences Ireland Unlimited Company | Dihydropyranopyrimidines for the treatment of viral infections |
| US11484534B2 (en) | 2013-03-14 | 2022-11-01 | Abbvie Inc. | Methods for treating HCV |
| US11597704B2 (en) | 2018-03-01 | 2023-03-07 | Janssen Sciences Ireland Unlimited Company | 2,4-diaminoquinazoline derivatives and medical uses thereof |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2994471B1 (en) * | 2013-05-06 | 2017-05-17 | Merck Patent GmbH | Macrocycles as kinase inhibitors |
| WO2017197055A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| CN109562113A (en) | 2016-05-10 | 2019-04-02 | C4医药公司 | Loop coil degron body for target protein degradation |
| WO2017197046A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | C3-carbon linked glutarimide degronimers for target protein degradation |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040229818A1 (en) | 2003-03-05 | 2004-11-18 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compound |
| US20040229776A1 (en) | 2003-04-02 | 2004-11-18 | Boehringer Ingelheim International Gmbh | Pharmaceutical compositions for hepatitis C viral protease inhibitors |
| US20050020503A1 (en) | 2003-05-21 | 2005-01-27 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compounds |
| US20060257980A1 (en) * | 2003-04-16 | 2006-11-16 | Wenying Li | Macrocyclic isoquinoline peptide inhibitors of hepatitis C virus |
| US20070027071A1 (en) * | 2005-07-20 | 2007-02-01 | Holloway M K | HCV NS3 protease inhibitors |
| WO2007016441A1 (en) | 2005-08-01 | 2007-02-08 | Merck & Co., Inc. | Macrocyclic peptides as hcv ns3 protease inhibitors |
Family Cites Families (96)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3480613A (en) * | 1967-07-03 | 1969-11-25 | Merck & Co Inc | 2-c or 3-c-alkylribofuranosyl - 1-substituted compounds and the nucleosides thereof |
| US6128582A (en) | 1996-04-30 | 2000-10-03 | Vertex Pharmaceuticals Incorporated | Molecules comprising an IMPDH-like binding pocket and encoded data storage medium capable of graphically displaying them |
| GB9623908D0 (en) | 1996-11-18 | 1997-01-08 | Hoffmann La Roche | Amino acid derivatives |
| GB9707659D0 (en) | 1997-04-16 | 1997-06-04 | Peptide Therapeutics Ltd | Hepatitis C NS3 Protease inhibitors |
| AU757783B2 (en) | 1997-08-11 | 2003-03-06 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis C inhibitor peptides |
| EP1012180B1 (en) | 1997-08-11 | 2004-12-01 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor peptide analogues |
| IT1299134B1 (en) | 1998-02-02 | 2000-02-29 | Angeletti P Ist Richerche Bio | PROCEDURE FOR THE PRODUCTION OF PEPTIDES WITH PROTEAS INHIBITING THE NS3 PROTEASIS OF THE HCV VIRUS, PEPTIDES SO OBTAINABLE AND PEPTIDES |
| EP1058686B1 (en) | 1998-02-25 | 2006-11-02 | Emory University | 2'-fluoronucleosides |
| GB9806815D0 (en) | 1998-03-30 | 1998-05-27 | Hoffmann La Roche | Amino acid derivatives |
| WO1999050230A1 (en) | 1998-03-31 | 1999-10-07 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hepatitis c virus ns3 protease |
| GB9812523D0 (en) | 1998-06-10 | 1998-08-05 | Angeletti P Ist Richerche Bio | Peptide inhibitors of hepatitis c virus ns3 protease |
| WO2000009546A1 (en) | 1998-08-10 | 2000-02-24 | Hokkaido Electric Power Company, Incorporated | Process for the preparation of regular glycopeptides |
| US6323180B1 (en) * | 1998-08-10 | 2001-11-27 | Boehringer Ingelheim (Canada) Ltd | Hepatitis C inhibitor tri-peptides |
| AU764479B2 (en) | 1998-10-29 | 2003-08-21 | Bristol-Myers Squibb Company | Compounds derived from an amine nucleus that are inhibitors of IMPDH enzyme |
| US6608027B1 (en) * | 1999-04-06 | 2003-08-19 | Boehringer Ingelheim (Canada) Ltd | Macrocyclic peptides active against the hepatitis C virus |
| UA74546C2 (en) | 1999-04-06 | 2006-01-16 | Boehringer Ingelheim Ca Ltd | Macrocyclic peptides having activity relative to hepatitis c virus, a pharmaceutical composition and use of the pharmaceutical composition |
| MXPA02000294A (en) | 1999-06-25 | 2002-06-21 | Vertex Pharma | Prodrugs of carbamate inhibitors of impdh. |
| ID30204A (en) | 1999-12-27 | 2001-11-15 | Japan Tobacco Inc | COMPOUNDS OF DIFFUSED RING AND ITS USE AS A MEDICINE |
| US6455508B1 (en) | 2000-02-15 | 2002-09-24 | Kanda S. Ramasamy | Methods for treating diseases with tirazole and pyrrolo-pyrimidine ribofuranosyl nucleosides |
| US6495677B1 (en) | 2000-02-15 | 2002-12-17 | Kanda S. Ramasamy | Nucleoside compounds |
| EA200200778A1 (en) * | 2000-02-18 | 2003-06-26 | Шайре Байокем Инк. | METHOD OF TREATMENT OR PREVENTION OF HEPATITIS C INFECTION IN THE ORGANISM ORGANISM, PHARMACEUTICAL COMPOSITION AGAINST FLAVIVIRUS, CONNECTION OF FORMULA Ib - AN ACTIVE AGENT FOR THE TREATMENT OR PREVENTION OF EFFECTECH EFFECTURES Ib - AN ACTIVE AGENT FOR THE TREATMENT OR PREVENTION INEKEKHEKUSA Ib - AN ACTIVE AGENT FOR THE TREATMENT OR PROTECTION |
| AU2001253206A1 (en) | 2000-04-05 | 2001-10-23 | Tularik, Inc. | Ns5b hcv polymerase inhibitors |
| EP1268525B1 (en) | 2000-04-05 | 2008-12-31 | Schering Corporation | Macrocyclic ns3-serine protease inhibitors of hepatitis c virus comprising n-cyclic p2 moieties |
| BR0110023A (en) | 2000-04-13 | 2003-12-30 | Pharmasset Ltd | Substituted 3'-or-2 'nucleoside derivatives for treatment of hepatitis virus infections |
| MY164523A (en) | 2000-05-23 | 2017-12-29 | Univ Degli Studi Cagliari | Methods and compositions for treating hepatitis c virus |
| EP1294735A2 (en) | 2000-05-26 | 2003-03-26 | Novirio Pharmaceuticals Limited | Methods and compositions for treating flaviviruses and pestiviruses |
| US6448281B1 (en) | 2000-07-06 | 2002-09-10 | Boehringer Ingelheim (Canada) Ltd. | Viral polymerase inhibitors |
| GB0017676D0 (en) | 2000-07-19 | 2000-09-06 | Angeletti P Ist Richerche Bio | Inhibitors of viral polymerase |
| JP3943015B2 (en) * | 2000-08-10 | 2007-07-11 | トラスティーズ オブ ボストン カレッジ | Recyclable metathesis catalyst |
| US6955174B2 (en) * | 2000-08-18 | 2005-10-18 | Uryovascular Systems, Inc. | Cryotherapy method for detecting and treating vulnerable plaque |
| US20030008841A1 (en) | 2000-08-30 | 2003-01-09 | Rene Devos | Anti-HCV nucleoside derivatives |
| WO2002020497A1 (en) | 2000-09-01 | 2002-03-14 | Shionogi & Co., Ltd. | Compounds having anti-hepatitis c virus effect |
| KR101201552B1 (en) | 2000-10-18 | 2012-11-15 | 파마셋 인코포레이티드 | Modified nucleosides for treatment of viral infections and abnormal cellular proliferation |
| WO2002048172A2 (en) | 2000-12-12 | 2002-06-20 | Schering Corporation | Diaryl peptides as ns3-serine protease inhibitors of hepatits c virus |
| AU2002230763A1 (en) | 2000-12-13 | 2008-01-03 | Bristol-Myers Squibb Pharma Company | Inhibitors of hepatitis c virus ns3 protease |
| AU2002232660A1 (en) | 2000-12-15 | 2002-06-24 | Pharmasset Ltd. | Antiviral agents for treatment of flaviviridae infections |
| US20040063651A1 (en) | 2000-12-26 | 2004-04-01 | Masahiko Morioka | Remedies for hepatitis c |
| RS50236B (en) | 2001-01-22 | 2009-07-15 | Merck & Co.Inc., | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| GB0114286D0 (en) * | 2001-06-12 | 2001-08-01 | Hoffmann La Roche | Nucleoside Derivatives |
| KR20040037062A (en) | 2001-08-14 | 2004-05-04 | 텔 아비브 유니버시티 퓨쳐 테크날러지 디벨로프멘트 엘피 | Lipidated Glycosaminoglycan Particles and Their Use in Drug and Gene Delivery for Diagnosis and Therapy |
| EP1435974A4 (en) | 2001-09-28 | 2006-09-06 | Idenix Cayman Ltd | Methods and compositions for treating hepatitis c virus using 4'-modified nucleosides |
| US20040006002A1 (en) | 2001-09-28 | 2004-01-08 | Jean-Pierre Sommadossi | Methods and compositions for treating flaviviruses and pestiviruses using 4'-modified nucleoside |
| EP1465862A1 (en) | 2002-01-17 | 2004-10-13 | SmithKline Beecham Corporation | Cycloalkyl ketoamides derivatives useful as cathepsin k inhibitors |
| GB0201179D0 (en) | 2002-01-18 | 2002-03-06 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| CA2369711A1 (en) | 2002-01-30 | 2003-07-30 | Boehringer Ingelheim (Canada) Ltd. | Macrocyclic peptides active against the hepatitis c virus |
| US7323453B2 (en) | 2002-02-13 | 2008-01-29 | Merck & Co., Inc. | Methods of inhibiting orthopoxvirus replication with nucleoside compounds |
| MXPA04010983A (en) | 2002-05-06 | 2005-02-14 | Genelabs Tech Inc | Nucleoside derivatives for treating hepatitis c virus infection. |
| MY140680A (en) | 2002-05-20 | 2010-01-15 | Bristol Myers Squibb Co | Hepatitis c virus inhibitors |
| AU2003269890A1 (en) | 2002-06-21 | 2004-01-06 | Isis Pharmaceuticals, Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| US20060234962A1 (en) | 2002-06-27 | 2006-10-19 | Olsen David B | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| MXPA04012779A (en) | 2002-06-28 | 2005-08-19 | Idenix Cayman Ltd | 2' and 3'-nucleoside prodrugs for treating flaviviridae infections. |
| KR20050035194A (en) | 2002-06-28 | 2005-04-15 | 이데닉스 (케이만) 리미티드 | 2'-c-methyl-3'-o-l-valine ester ribofuranosyl cytidine for treatment of flaviviridae infections |
| RS114004A (en) | 2002-06-28 | 2007-02-05 | Idenix (Cayman) Limited, | Modified 2' and 3'-nucleoside produgs for treating flaviridae infections |
| JP2005533108A (en) | 2002-07-16 | 2005-11-04 | メルク エンド カムパニー インコーポレーテッド | Nucleoside derivatives as inhibitors of RNA-dependent RNA viral polymerase |
| CN1671727A (en) | 2002-07-25 | 2005-09-21 | 麦克洛基克斯生物技术有限公司 | Anti-viral 7-deaza D-nucleosides and uses thereof |
| JP2006507235A (en) | 2002-08-01 | 2006-03-02 | フアーマセツト・インコーポレイテツド | Compounds having bicyclo [4.2.1] nonane system for the treatment of Flaviviridae virus infection |
| NZ538457A (en) | 2002-09-30 | 2008-04-30 | Genelabs Tech Inc | Nucleoside derivatives for treating hepatitis C virus infection |
| CA2504344A1 (en) | 2002-11-01 | 2004-05-21 | Viropharma Incorporated | Benzofuran compounds, compositions and methods for treatment and prophylaxis of hepatitis c viral infections and associated diseases |
| US20040254159A1 (en) * | 2003-02-27 | 2004-12-16 | Hasvold Lisa A. | Heterocyclic kinase inhibitors |
| GB0307891D0 (en) | 2003-04-04 | 2003-05-14 | Angeletti P Ist Richerche Bio | Chemical compounds,compositions and uses |
| US7176208B2 (en) * | 2003-04-18 | 2007-02-13 | Enanta Pharmaceuticals, Inc. | Quinoxalinyl macrocyclic hepatitis C serine protease inhibitors |
| EP2345661A1 (en) | 2003-05-30 | 2011-07-20 | Pharmasset, Inc. | Modified fluorinated nucleoside analogues |
| GB0313250D0 (en) | 2003-06-09 | 2003-07-16 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| DE602004003389T2 (en) * | 2003-06-19 | 2007-09-13 | F. Hoffmann-La Roche Ag | PROCESS FOR THE PREPARATION OF 4'-AZIDONUCLEOSIDE DERIVATIVES |
| TW200510425A (en) | 2003-08-13 | 2005-03-16 | Japan Tobacco Inc | Nitrogen-containing fused ring compound and use thereof as HIV integrase inhibitor |
| GB0321003D0 (en) | 2003-09-09 | 2003-10-08 | Angeletti P Ist Richerche Bio | Compounds, compositions and uses |
| GB0323845D0 (en) | 2003-10-10 | 2003-11-12 | Angeletti P Ist Richerche Bio | Chemical compounds,compositions and uses |
| US7132504B2 (en) | 2003-11-12 | 2006-11-07 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| ES2358333T3 (en) | 2004-01-21 | 2011-05-09 | Boehringer Ingelheim International Gmbh | MACROCYCLIC PEPTIDES WITH ACTION AGAINST THE VIRUS OF HEPATITIS C. |
| DE602005013922D1 (en) | 2004-02-24 | 2009-05-28 | Japan Tobacco Inc | CONDENSED HETEROTETRACYCLIC COMPOUNDS AND THEIR USE AS HCV POLYMERASE INHIBITOR |
| GB0413087D0 (en) | 2004-06-11 | 2004-07-14 | Angeletti P Ist Richerche Bio | Therapeutic compounds |
| GB0416396D0 (en) | 2004-07-22 | 2004-08-25 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| US7153848B2 (en) | 2004-08-09 | 2006-12-26 | Bristol-Myers Squibb Company | Inhibitors of HCV replication |
| EP1794172B1 (en) | 2004-08-23 | 2009-07-15 | F.Hoffmann-La Roche Ag | Antiviral 4'-azido-nucleosides |
| GB0419850D0 (en) | 2004-09-07 | 2004-10-13 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| JP2008517987A (en) | 2004-10-26 | 2008-05-29 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー | Tetracyclic indole derivatives as antiviral agents |
| AU2006242475B2 (en) | 2005-05-02 | 2011-07-07 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| GB0509326D0 (en) | 2005-05-09 | 2005-06-15 | Angeletti P Ist Richerche Bio | Therapeutic compounds |
| WO2007028789A1 (en) | 2005-09-07 | 2007-03-15 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Quinazoline derivatives as antiviral agents |
| GB0518390D0 (en) | 2005-09-09 | 2005-10-19 | Angeletti P Ist Richerche Bio | Therapeutic compounds |
| GB0519797D0 (en) | 2005-09-29 | 2005-11-09 | Istituto Di Ricerche D Biolog | Therapeutic agents |
| GB0609492D0 (en) | 2006-05-15 | 2006-06-21 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| US20090203008A1 (en) | 2006-06-08 | 2009-08-13 | Ludmerer Steven W | Rapid method to determine inhibitor sensitivity of NS3/4A protease sequences cloned from clinical samples |
| GB0612423D0 (en) | 2006-06-23 | 2006-08-02 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| JP2010507656A (en) | 2006-10-24 | 2010-03-11 | メルク エンド カムパニー インコーポレーテッド | HCV NS3 protease inhibitor |
| US8309540B2 (en) | 2006-10-24 | 2012-11-13 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US8138164B2 (en) | 2006-10-24 | 2012-03-20 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| KR101615500B1 (en) | 2006-10-27 | 2016-04-27 | 머크 샤프 앤드 돔 코포레이션 | HCV NS3 protease inhibitors |
| ES2444575T3 (en) | 2006-10-27 | 2014-02-25 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| EP2079568B1 (en) | 2006-11-09 | 2014-12-24 | Twinblade Technologies Holding Sweden AB | Hub device |
| WO2008112108A1 (en) | 2007-03-09 | 2008-09-18 | Merck & Co., Inc. | In vivo hcv resistance to anti-viral inhibitors |
| TWI434849B (en) | 2007-06-29 | 2014-04-21 | Gilead Sciences Inc | Modulators of toll-like receptor 7 |
| WO2009010804A1 (en) | 2007-07-19 | 2009-01-22 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Macrocyclic compounds as antiviral agents |
| CL2008003384A1 (en) | 2007-11-14 | 2009-12-11 | Enanta Pharm Inc | Macrocyclic quinoxaline derived compounds, serine protease inhibitors; pharmaceutical composition comprising them; and its use in the treatment of hepatitis c. |
| WO2009064955A1 (en) | 2007-11-14 | 2009-05-22 | Enanta Pharmaceuticals, Inc. | Macrocyclic tetrazolyl hepatitis c serine protease inhibitors |
| RS53420B (en) * | 2008-07-22 | 2014-12-31 | Msd Italia S.R.L. | Combinations of a macrocyclic quinoxaline compound which is an hcv ns3 protease inhibitor with other hcv agents |
-
2009
- 2009-04-16 AU AU2009241445A patent/AU2009241445A1/en not_active Abandoned
- 2009-04-16 CN CN200980115230XA patent/CN102014911A/en active Pending
- 2009-04-16 CA CA2720850A patent/CA2720850A1/en not_active Abandoned
- 2009-04-16 WO PCT/US2009/040815 patent/WO2009134624A1/en active Application Filing
- 2009-04-16 JP JP2011507531A patent/JP2011518882A/en active Pending
- 2009-04-16 EP EP20090739426 patent/EP2271345B1/en active Active
- 2009-04-16 US US12/989,672 patent/US8461107B2/en active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040229818A1 (en) | 2003-03-05 | 2004-11-18 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compound |
| US20040229776A1 (en) | 2003-04-02 | 2004-11-18 | Boehringer Ingelheim International Gmbh | Pharmaceutical compositions for hepatitis C viral protease inhibitors |
| US20060257980A1 (en) * | 2003-04-16 | 2006-11-16 | Wenying Li | Macrocyclic isoquinoline peptide inhibitors of hepatitis C virus |
| US20050020503A1 (en) | 2003-05-21 | 2005-01-27 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compounds |
| US20070027071A1 (en) * | 2005-07-20 | 2007-02-01 | Holloway M K | HCV NS3 protease inhibitors |
| WO2007016441A1 (en) | 2005-08-01 | 2007-02-08 | Merck & Co., Inc. | Macrocyclic peptides as hcv ns3 protease inhibitors |
Non-Patent Citations (5)
| Title |
|---|
| "Specific Antiviral Therapy", INTERVIROLOGY, vol. 40, 1997, pages 378 - 393 |
| B.W. DYMOCK: "Emerging therapies for hepatitis C virus infection", EMERGING DRUGS, vol. 6, 2001, pages 13 - 42 |
| C. CRABB: "Hard-Won Advances Spark Excitement about Hepatitis C", SCIENCE, 2001, pages 506 - 507 |
| G.M. LAUER; B.D. WALKER: "Hepatitis C Virus Infection", N. ENGL. J. MED., vol. 345, 2001, pages 41 - 52 |
| See also references of EP2271345A4 * |
Cited By (64)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8372802B2 (en) | 2008-03-20 | 2013-02-12 | Enanta Pharmaceuticals, Inc. | Fluorinated macrocyclic compounds as hepatitis C virus inhibitors |
| JP2011528713A (en) * | 2008-07-22 | 2011-11-24 | メルク・シャープ・エンド・ドーム・コーポレイション | Macrocyclic quinoxaline compounds as HCV NS3 protease inhibitors |
| US8936781B2 (en) | 2009-05-13 | 2015-01-20 | Enanta Pharmaceuticals, Inc. | Macrocyclic compounds as hepatitis C virus inhibitors |
| US8828930B2 (en) | 2009-07-30 | 2014-09-09 | Merck Sharp & Dohme Corp. | Hepatitis C virus NS3 protease inhibitors |
| US9193740B2 (en) | 2009-10-19 | 2015-11-24 | Enanta Pharmaceuticals, Inc. | Bismacrocyclic compounds as hepatitis C virus inhibitors |
| WO2011091757A1 (en) | 2010-01-27 | 2011-08-04 | AB Pharma Ltd. | Polyheterocyclic compounds highly potent as hcv inhibitors |
| US9220748B2 (en) | 2010-09-21 | 2015-12-29 | Enanta Pharmaceuticals, Inc. | Macrocyclic proline derived HCV serine protease inhibitors |
| EP2618665A1 (en) * | 2010-09-21 | 2013-07-31 | Merck Sharp & Dohme Corp. | Hcv ns3 protease inhibitors |
| US8648037B2 (en) | 2010-09-21 | 2014-02-11 | Enanta Pharmaceuticals, Inc. | Macrocyclic proline derived HCV serine protease inhibitors |
| EP2618665A4 (en) * | 2010-09-21 | 2014-08-20 | Merck Sharp & Dohme | Hcv ns3 protease inhibitors |
| US9120818B2 (en) | 2010-12-14 | 2015-09-01 | Merck Sharp & Dohme Corp. | Process and intermediates for preparing macrolactams |
| US10780089B2 (en) | 2011-04-08 | 2020-09-22 | Janssen Sciences Ireland Uc | Pyrimidine derivatives for the treatment of viral infections |
| US10272085B2 (en) | 2011-04-08 | 2019-04-30 | Janssen Sciences Ireland Uc | Pyrimidine derivatives for the treatment of viral infections |
| US10420767B2 (en) | 2011-04-08 | 2019-09-24 | Janssen Sciences Ireland Uc | Pyrimidine derivatives for the treatment of viral infections |
| US11541050B2 (en) | 2011-04-08 | 2023-01-03 | Janssen Sciences Ireland Uc | Pyrimidine derivatives for the treatment of viral infections |
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9527885B2 (en) | 2011-05-05 | 2016-12-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8962810B2 (en) | 2011-06-16 | 2015-02-24 | AB Pharma Ltd. | Macrocyclic heterocyclic compound for inhibiting hepatitis C virus and preparation and use thereof |
| US9073825B2 (en) | 2011-08-19 | 2015-07-07 | Merck Sharp & Dohme Limited | Methods and intermediates for preparing macrolactams |
| WO2013028470A1 (en) | 2011-08-19 | 2013-02-28 | Merck Sharp & Dohme Corp. | Process and intermediates for preparing macrolactams |
| US9238604B2 (en) | 2011-08-19 | 2016-01-19 | Merck Sharp & Dohme Corp. | Process and intermediates for preparing macrolactams |
| US9242917B2 (en) | 2011-08-19 | 2016-01-26 | Merck Sharp & Dohme Limited | Crystal forms of a HCV protease inhibitor |
| US10280167B2 (en) | 2011-11-09 | 2019-05-07 | Janssen Sciences Ireland Uc | Purine derivatives for the treatment of viral infections |
| US11104678B2 (en) | 2011-11-09 | 2021-08-31 | Janssen Sciences Ireland Unlimited Company | Purine derivatives for the treatment of viral infections |
| EP2780026A4 (en) * | 2011-11-15 | 2016-06-08 | Merck Sharp & Dohme | Hcv ns3 protease inhibitors |
| US9328138B2 (en) | 2011-11-15 | 2016-05-03 | Msd Italia S.R.L. | HCV NS3 protease inhibitors |
| WO2013074386A2 (en) | 2011-11-15 | 2013-05-23 | Merck Sharp & Dohme Corp. | Hcv ns3 protease inhibitors |
| US10603318B2 (en) | 2012-07-03 | 2020-03-31 | Gilead Pharmasset Llc | Inhibitors of hepatitis C virus |
| US9296782B2 (en) | 2012-07-03 | 2016-03-29 | Gilead Sciences, Inc. | Inhibitors of hepatitis C virus |
| US10335409B2 (en) | 2012-07-03 | 2019-07-02 | Gilead Pharmasset Llc | Inhibitors of hepatitis C virus |
| EA035790B1 (en) * | 2012-07-13 | 2020-08-11 | Янссен Сайенсиз Айрлэнд Юси | Macrocyclic purines for the treatment of viral infections |
| US10822349B2 (en) | 2012-07-13 | 2020-11-03 | Janssen Sciences Ireland Unlimited Company | Macrocyclic purines for the treatment of viral infections |
| CN104703990B (en) * | 2012-07-13 | 2017-10-13 | 爱尔兰詹森科学公司 | Big ring purine for treating viral infection |
| WO2014009509A1 (en) * | 2012-07-13 | 2014-01-16 | Janssen R&D Ireland | Macrocyclic purines for the treatment of viral infections |
| CN104703990A (en) * | 2012-07-13 | 2015-06-10 | 爱尔兰詹森研发公司 | Macrocyclic purines for the treatment of viral infections |
| US10280180B2 (en) | 2012-07-13 | 2019-05-07 | Janssen Sciences Ireland Uc | Macrocyclic purines for the treatment of viral infections |
| US9284304B2 (en) | 2012-08-10 | 2016-03-15 | Janssen Sciences Ireland Uc | Substituted pyrimidines as toll-like receptor modulators |
| US11220504B2 (en) | 2012-10-10 | 2022-01-11 | Janssen Sciences Ireland Unlimited Company | Pyrrolo[3,2-d] pyrimidine derivatives for the treatment of viral infections and other diseases |
| US10259814B2 (en) | 2012-10-10 | 2019-04-16 | Janssen Sciences Ireland Uc | Pyrrolo[3,2-d]pyrimidine derivatives for the treatment of viral infections and other diseases |
| US9499550B2 (en) | 2012-10-19 | 2016-11-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9598433B2 (en) | 2012-11-02 | 2017-03-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9334279B2 (en) | 2012-11-02 | 2016-05-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9409943B2 (en) | 2012-11-05 | 2016-08-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US10253003B2 (en) | 2012-11-16 | 2019-04-09 | Janssen Sciences Ireland Uc | Heterocyclic substituted 2-amino quinazoline derivatives for the treatment of viral infections |
| US10723707B2 (en) | 2012-11-16 | 2020-07-28 | Janssen Sciences Ireland Unlimited Company | Heterocyclic substituted 2-amino quinazoline derivatives for the treatment of viral infections |
| US10259793B2 (en) | 2013-02-21 | 2019-04-16 | Janssen Sciences Ireland Uc | 2-aminopyrimidine derivatives for the treatment of viral infections |
| US10647684B2 (en) | 2013-02-21 | 2020-05-12 | Janssen Sciences Ireland Unlimited Company | 2-aminopyrimidine derivatives for the treatment of viral infections |
| US9580463B2 (en) | 2013-03-07 | 2017-02-28 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US11484534B2 (en) | 2013-03-14 | 2022-11-01 | Abbvie Inc. | Methods for treating HCV |
| US9617310B2 (en) | 2013-03-15 | 2017-04-11 | Gilead Sciences, Inc. | Inhibitors of hepatitis C virus |
| US11702426B2 (en) | 2013-03-29 | 2023-07-18 | Janssen Sciences Ireland Unlimited Company | Macrocyclic deaza-purinones for the treatment of viral infections |
| US10266543B2 (en) | 2013-03-29 | 2019-04-23 | Janssen Sciences Ireland Uc | Macrocyclic deaza-purinones for the treatment of viral infections |
| US10829494B2 (en) | 2013-03-29 | 2020-11-10 | Janssen Sciences Ireland Unlimited Company | Macrocyclic deaza-purinones for the treatment of viral infections |
| US10377738B2 (en) | 2013-05-24 | 2019-08-13 | Janssen Sciences Ireland Unlimited Company | Pyridone derivatives for the treatment of viral infections and further diseases |
| US10865193B2 (en) | 2013-05-24 | 2020-12-15 | Janssen Sciences Ireland Unlimited Company | Pyridone derivatives for the treatment of viral infections and further diseases |
| US10781216B2 (en) | 2013-06-27 | 2020-09-22 | Janssen Sciences Ireland Unlimited Company | Pyrrolo [3,2-d]pyrimidine derivatives for the treatment of viral infections and other diseases |
| US10385054B2 (en) | 2013-06-27 | 2019-08-20 | Janssen Sciences Ireland Unlimited Company | Pyrrolo[3,2-d]pyrimidine derivatives for the treatment of viral infections and other diseases |
| US10822347B2 (en) | 2013-07-30 | 2020-11-03 | Janssen Sciences Ireland Unlimited Company | Thieno[3,2-d]pyrimidines derivatives for the treatment of viral infections |
| US10316043B2 (en) | 2013-07-30 | 2019-06-11 | Janssen Sciences Ireland Unlimited Company | Thieno[3,2-d]pyrimidines derivatives for the treatment of viral infections |
| US9873707B2 (en) | 2013-10-18 | 2018-01-23 | Merck Sharp & Dohme Corp. | Methods and intermediates for preparing macrolactams |
| US11053256B2 (en) | 2016-07-01 | 2021-07-06 | Janssen Sciences Ireland Unlimited Company | Dihydropyranopyrimidines for the treatment of viral infections |
| US10968184B2 (en) | 2016-09-29 | 2021-04-06 | Janssen Sciences Ireland Unlimited Company | Pyrimidine prodrugs for the treatment of viral infections and further diseases |
| US11597704B2 (en) | 2018-03-01 | 2023-03-07 | Janssen Sciences Ireland Unlimited Company | 2,4-diaminoquinazoline derivatives and medical uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2011518882A (en) | 2011-06-30 |
| CA2720850A1 (en) | 2009-11-05 |
| EP2271345B1 (en) | 2015-05-20 |
| CN102014911A (en) | 2011-04-13 |
| EP2271345A1 (en) | 2011-01-12 |
| EP2271345A4 (en) | 2011-08-31 |
| US20110046161A1 (en) | 2011-02-24 |
| AU2009241445A1 (en) | 2009-11-05 |
| US8461107B2 (en) | 2013-06-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2271345A1 (en) | Hcv ns3 protease inhibitors | |
| EP2086982B1 (en) | Hcv ns3 protease inhibitors | |
| AU2007318164B2 (en) | HCV NS3 protease inhibitors | |
| EP1910404B9 (en) | Hcv ns3 protease inhibitors | |
| CA2615896C (en) | Macrocyclic peptides as hcv ns3 protease inhibitors | |
| CA2667266C (en) | Hcv ns3 protease inhibitors | |
| EP1879607A2 (en) | Hcv ns3 protease inhibitors | |
| WO2012040040A1 (en) | Hcv ns3 protease inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980115230.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09739426 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 5935/CHENP/2010 Country of ref document: IN Ref document number: 2009739426 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009241445 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2720850 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2009241445 Country of ref document: AU Date of ref document: 20090416 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12989672 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011507531 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |