WO2009131687A2 - Inhibitors of protein kinases - Google Patents
Inhibitors of protein kinases Download PDFInfo
- Publication number
- WO2009131687A2 WO2009131687A2 PCT/US2009/002512 US2009002512W WO2009131687A2 WO 2009131687 A2 WO2009131687 A2 WO 2009131687A2 US 2009002512 W US2009002512 W US 2009002512W WO 2009131687 A2 WO2009131687 A2 WO 2009131687A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ylamino
- pyrrolo
- pyrimidin
- compound
- group
- Prior art date
Links
- 0 CN(***)c1nc(N(*)*)c(*(*)C(*)N2*)c2n1 Chemical compound CN(***)c1nc(N(*)*)c(*(*)C(*)N2*)c2n1 0.000 description 29
- JYTLAUZYCBKWEB-ARJAWSKDSA-N C/C=C\N=N/NC Chemical compound C/C=C\N=N/NC JYTLAUZYCBKWEB-ARJAWSKDSA-N 0.000 description 1
- WLLIHFRZHYEXQQ-UHFFFAOYSA-N CC(C(c(ccc(C)c1)c1O)[O]1(C)=CC1)C=O Chemical compound CC(C(c(ccc(C)c1)c1O)[O]1(C)=CC1)C=O WLLIHFRZHYEXQQ-UHFFFAOYSA-N 0.000 description 1
- UBEBLTRRJCIWJR-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1Nc1c(cc[nH]2)c2nc(Nc2cc(N)c(C=N)cc2)n1)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1Nc1c(cc[nH]2)c2nc(Nc2cc(N)c(C=N)cc2)n1)=O UBEBLTRRJCIWJR-UHFFFAOYSA-N 0.000 description 1
- FXGNYOJHHQDFFW-UHFFFAOYSA-N CC(N(C)c(cc1)ccc1Nc1nc(NC2CC2)c(c(C#N)c[nH]2)c2n1)=O Chemical compound CC(N(C)c(cc1)ccc1Nc1nc(NC2CC2)c(c(C#N)c[nH]2)c2n1)=O FXGNYOJHHQDFFW-UHFFFAOYSA-N 0.000 description 1
- PFQZSZKQBYZRGW-UHFFFAOYSA-N CC(N(CC1)CCN1c(cc1)ccc1Nc1nc(NC(CC2)CCC2N)c(cc[nH]2)c2n1)=O Chemical compound CC(N(CC1)CCN1c(cc1)ccc1Nc1nc(NC(CC2)CCC2N)c(cc[nH]2)c2n1)=O PFQZSZKQBYZRGW-UHFFFAOYSA-N 0.000 description 1
- ZNOASPNIGDFOQH-UHFFFAOYSA-N CC(N(CC1)CCN1c(cc1)ccc1Nc1nc(NC2CC2)c(c(-c2ccncc2)c[nH]2)c2n1)=O Chemical compound CC(N(CC1)CCN1c(cc1)ccc1Nc1nc(NC2CC2)c(c(-c2ccncc2)c[nH]2)c2n1)=O ZNOASPNIGDFOQH-UHFFFAOYSA-N 0.000 description 1
- ONLWFBZONSCPIG-UHFFFAOYSA-N CN(C(C1CC1)=O)c(cc1)ccc1Nc1nc(NC2CC2)c(cc[nH]2)c2n1 Chemical compound CN(C(C1CC1)=O)c(cc1)ccc1Nc1nc(NC2CC2)c(cc[nH]2)c2n1 ONLWFBZONSCPIG-UHFFFAOYSA-N 0.000 description 1
- WWCCTGSLIZXDTL-UHFFFAOYSA-N CN(C(CO)=O)c(cc1)ccc1Nc1nc(NC2CCC2)c(cc[nH]2)c2n1 Chemical compound CN(C(CO)=O)c(cc1)ccc1Nc1nc(NC2CCC2)c(cc[nH]2)c2n1 WWCCTGSLIZXDTL-UHFFFAOYSA-N 0.000 description 1
- PVOAHINGSUIXLS-UHFFFAOYSA-N CN1CCNCC1 Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- NSDUQINNGKHWBF-UHFFFAOYSA-N CNCCNC(CN(C(CCc1c2)=O)c1ccc2Nc1nc(NC2CCC2)c(cc[nH]2)c2n1)=O Chemical compound CNCCNC(CN(C(CCc1c2)=O)c1ccc2Nc1nc(NC2CCC2)c(cc[nH]2)c2n1)=O NSDUQINNGKHWBF-UHFFFAOYSA-N 0.000 description 1
- GSPPNZDCNBEHKN-UHFFFAOYSA-N COC(C(C(CC1)CCN1c(nc(Nc(cc1)cc(CC2)c1NC2=O)nc1)c1F)N)=O Chemical compound COC(C(C(CC1)CCN1c(nc(Nc(cc1)cc(CC2)c1NC2=O)nc1)c1F)N)=O GSPPNZDCNBEHKN-UHFFFAOYSA-N 0.000 description 1
- MCTWTZJPVLRJOU-UHFFFAOYSA-N C[n]1cncc1 Chemical compound C[n]1cncc1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 1
- JCWHYVRGACQGNO-UHFFFAOYSA-N Clc(nc1)nc(NC2CCCCC2)c1-c1ccncc1 Chemical compound Clc(nc1)nc(NC2CCCCC2)c1-c1ccncc1 JCWHYVRGACQGNO-UHFFFAOYSA-N 0.000 description 1
- BGAYHRDCCZCMCO-UHFFFAOYSA-N N#Cc1c[nH]c2nc(Cl)nc(NC3CC3)c12 Chemical compound N#Cc1c[nH]c2nc(Cl)nc(NC3CC3)c12 BGAYHRDCCZCMCO-UHFFFAOYSA-N 0.000 description 1
- DYEMRZRBHBNKAJ-CSKARUKUSA-N NC(C(CCC1)CN1/C(/N)=C(\C=CN1)/C1=N)=O Chemical compound NC(C(CCC1)CN1/C(/N)=C(\C=CN1)/C1=N)=O DYEMRZRBHBNKAJ-CSKARUKUSA-N 0.000 description 1
- KJACBIQVKKDJCK-KRWDZBQOSA-N NCCOCCNc1c(cc[nH]2)c2nc(Nc(cc2)ccc2N(CCC2)[C@@H]2C(N)=O)n1 Chemical compound NCCOCCNc1c(cc[nH]2)c2nc(Nc(cc2)ccc2N(CCC2)[C@@H]2C(N)=O)n1 KJACBIQVKKDJCK-KRWDZBQOSA-N 0.000 description 1
- JQVBZZUMWRXDSQ-BETUJISGSA-N N[C@H](CC1)CC[C@H]1NC(OCc1ccccc1)=O Chemical compound N[C@H](CC1)CC[C@H]1NC(OCc1ccccc1)=O JQVBZZUMWRXDSQ-BETUJISGSA-N 0.000 description 1
- VAVOYRCCWLRTMS-UHFFFAOYSA-N Nc(cc1)ccc1N1CCNCC1 Chemical compound Nc(cc1)ccc1N1CCNCC1 VAVOYRCCWLRTMS-UHFFFAOYSA-N 0.000 description 1
- MYGMMIDGBHQQHM-UHFFFAOYSA-N O=C(CC1)Nc(cc2)c1cc2Nc(nc1)nc(N(CCN2)CC2=O)c1F Chemical compound O=C(CC1)Nc(cc2)c1cc2Nc(nc1)nc(N(CCN2)CC2=O)c1F MYGMMIDGBHQQHM-UHFFFAOYSA-N 0.000 description 1
- XZGJAHQTUQUDGB-UHFFFAOYSA-N OC(C(CCC1)CN1c1c2nc[nH]c2nc(Nc(cc2)ccc2N2CCNCC2)n1)=O Chemical compound OC(C(CCC1)CN1c1c2nc[nH]c2nc(Nc(cc2)ccc2N2CCNCC2)n1)=O XZGJAHQTUQUDGB-UHFFFAOYSA-N 0.000 description 1
- WIQRWQXZZNVZFL-UHFFFAOYSA-N OC(CC(CC1)CCN1c1c(cc[nH]2)c2nc(Nc(cc2CC3)ccc2NC3=O)n1)=O Chemical compound OC(CC(CC1)CCN1c1c(cc[nH]2)c2nc(Nc(cc2CC3)ccc2NC3=O)n1)=O WIQRWQXZZNVZFL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/14—Vasoprotectives; Antihaemorrhoidals; Drugs for varicose therapy; Capillary stabilisers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/16—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
Definitions
- This invention is directed to pyrimidine, pyrrolopyrimidine and purine-based analogs which act as inhibitors of Spleen tyrosine kinase (syk) and/or JAK kinases.
- This invention is also directed to pharmaceutical compositions containing the pyrimidine compounds and methods of using the compounds or compositions to treat a condition characterized by undesired thrombosis.
- the invention is also directed to methods of making the compounds described herein.
- Protein kinases constitute a large family of structurally related enzymes that are responsible for the control of a variety of signal transduction processes within cells (see, e.g., Hardie and Hanks, The Protein Kinase Facts Book, I and II, Academic Press, San Diego, Calif., 1995). Protein kinases are thought to have evolved from a common ancestral gene due to the conservation of their structure and catalytic function. Almost all kinases contain a similar 250-300 amino acid catalytic domain. The kinases can be categorized into families by the substrates they phosphorylate (e.g., protein-tyrosine, protein-serine/threonine, lipids, etc.).
- TAM Immunoreceptor tyrosine activation motif
- ITAM- mediated signaling is responsible for relaying activation signals initiated at classical immune receptors such as T-cell receptors, B-cell receptors, Fc receptors in immune cells and at GPVI and Fc ⁇ RIIa in platelets to downstream intracellular molecules such as syk and ZAP-70 (Underhill, D.M and Goodridge, H. S., Trends Immunol, 28:66-73, 2007).
- the binding of a ligand to an ITAM-containing receptor triggers signaling events which allows for the recruitment of proteins from a family of nonreceptor tyrosine kinases called the Src family. These kinases phosphorylate tyrosine residues within the ITAM sequence, a region with which the tandem SH2 domains on either syk or ZAP-70 interact.
- Syk along with Zap-70, is a member of the syk family of protein tyrosine kinases.
- the interaction of syk or ZAP-70 with diphosphorylated ITAM sequences induces a conformation change in the kinases that allows for tyrosine phosphorylation of the kinase itself.
- Phosphorylated Syk family members activate a multitude of downstream signaling pathway proteins which include Src homology 2 (SH2) domain containing leukocyte-specific phosphoprotein of 76 kDa (SLP-76), Linker of Activation of T-cells (LAT) and PLC (phospholipase C) ⁇ 2.
- SH2 Src homology 2
- LAT Linker of Activation of T-cells
- PLC phospholipase C
- autoimmune diseases such as rheumatoid arthritis, systemic lupus, multiple sclerosis, hemolytic anemia, immune-thrombocytopenia purpura, and heparin-induced thrombocytopenia and arteriosclerosis.
- many of the above mentioned diseases are thought to occur through crosslinking of Fc receptors by antibodies which, via syk, activate a signaling cascade in mast, basophil and other immune cells that result in the release of cell mediators responsible for inflammatory reactions.
- Drug-induced thrombocytopenia caused by heparin- platelet factor 4 immune complexes that activate platelet Fc ⁇ RIIa, also involve syk signaling downstream of receptor engagement (Reilly, M.P., Blood, 98:2442-2447, 2001).
- Platelet agonists induce inside-out integrin signaling resulting in fibrinogen binding and platelet aggregation. This initiates outside-in signaling which produces further stimulation of platelets, syk is activated during both phases of integrin signaling, and inhibition of syk is shown to inhibit platelet adhesion to immobilized proteins (Law, D. A. et al., Blood, 93:2645-2652, 1999).
- syk inhibitors may also possess anticoagulation action.
- Arteriosclerosis is a class of diseases characterized by the thickening and hardening of the arterial walls of blood vessels. Although all blood vessels are susceptible to this serious degenerative condition, the aorta and the coronary arteries serving the heart are most often affected. Arteriosclerosis is of profound clinical importance since it can increase the risk of heart attacks, myocardial infarctions, strokes, and aneurysms.
- the traditional treatment for arteriosclerosis includes vascular recanalization procedures for less-serious blockages and coronary bypass surgery for major blockages.
- a serious shortcoming of intravascular procedures is that, in a significant number of treated individuals, some or all of the treated vessels restenose ⁇ i.e., re-narrow).
- restenosis of an atherosclerotic coronary artery after PTCA occurs in 10-50% of patients undergoing this procedure and subsequently requires either further angioplasty or a coronary artery bypass graft.
- restenosis of an atherosclerotic coronary artery after stenting occurs in 10-20% of patients undergoing this procedure and subsequently requires repeat treatments to maintain adequate blood flow through the affected artery. Restenosis generally occurs in a relatively brief time period, e.g., roughly less than six months, after treatment.
- restenosis is thought to be due in part to mechanical injury to the walls of the blood vessels caused by the balloon catheter or other intravascular device.
- the process of PTCA in addition to opening the obstructed artery, also injures resident coronary arterial smooth muscle cells (SMCs).
- SMCs resident coronary arterial smooth muscle cells
- adhering platelets, infiltrating macrophages, leukocytes, or the smooth muscle cells themselves release cell- derived growth factors such as platelet-derived growth factor (PDGF), with subsequent proliferation and migration of medial SMCs through the internal elastic lamina to the area of the vessel intima.
- PDGF platelet-derived growth factor
- syk plays a very important role in collagen-mediated signaling.
- the primary adhesive protein responsible for platelet adhesion and activation is collagen.
- Collagen is a filamentous protein contained within the fibrotic caps of atheromas which becomes exposed to blood during plaque rupture. Collagen functions initially by binding von Willebrand factor which tethers platelets through binding platelet membrane GPIb. Collagen functions secondarily by engaging the two collagen receptors on platelets, GPVI and integrin ⁇ 2 ⁇ l.
- GPVI exists in platelet membranes as a complex with FcR ⁇ , an interaction required for the expression of GPVI.
- Activation of Fc ⁇ RIIa on platelets results in platelet shape change, secretion and thrombosis.
- Signaling by the GPVI/FcR ⁇ complex is initiated by tyrosine phosphorylation of the ITAM domain of FCR ⁇ followed by the recruitment of syk.
- GPVI Activation of GPVI leads to induction of multiple platelet functions including: activation of integrins ⁇ 2 ⁇ l to achieve firm platelet adhesion, and GP Hb-IIIa which mediates platelet aggregation and thrombosis growth; platelet secretion, allowing for the delivery of inflammatory proteins such as CD40L, RANTES and TGF ⁇ to the vessel wall; and the expression of P-selectin which allows for the recruitment of leukocytes. Therefore, it is believed that syk inhibitors can inhibit thrombotic events mediated by platelet adhesion, activation and aggregation.
- Syk is important for the activation of B-cells via a B-cell antigen receptor and is involved in the phosphatidylinositol metabolism and increase in the intracellular calcium concentration caused by the antigen receptor stimulation (Hutchcroft, J E. et al., J. Biol. Chem., 267:8613-8619, 1992; and Takata, M. et al., EMBO J., 13: 1341-1349, 1994).
- syk inhibitors may be used to control the function of B-cells and are, therefore, expected to serve as therapeutic agents for antibody-related diseases.
- Syk binds to a T-cell antigen receptor, quickly undergoes tyrosine phosphorylation through crosslinking of the receptor and synergistically acts upon intracellular signals mediated by Src tyrosine kinases such as Lck (Couture, C. et al., P roc. Natl. Acad. ScL USA, 91:5301-5305, 1994; and Couture, C. et al., MoI. Cell. Biol, 14:5249-5258, 1994).
- Src tyrosine kinases such as Lck (Couture, C. et al., P roc. Natl. Acad. ScL USA, 91:5301-5305, 1994; and Couture, C. et al., MoI. Cell. Biol, 14:5249-5258, 1994).
- syk is present in mature T-cell populations, such as intraepithelial ⁇ T-cells and naive ⁇ T-cells, and has been reported to be capable of phosphorylation of multiple components of the TCR signaling cascade (Latour, S. et. al., MoI Cell Biol., 17:4434-4441, 1997).
- syk inhibitors may serve as agents for inhibiting cellular immunity mediated by T-cell antigen receptor.
- MCL Mantle Cell Lymphoma
- BCR signaling induces receptor oligomerization and phosphorylation of Ig ⁇ and ⁇ immunoreceptor tyrosine-based activated motifs by SRC family kinases. ITAM phosphorylation results in the recruitment and activation of syk that initiates downstream events and amplifies the original BCR signal.
- Syk spleen tyrosine kinase
- Leukemia is induced in mice by adoptively transferring bone marrow cells that express human TEL-Syk (Wossning, T., JEM, 2006; 203:2829-2840). Further, in mouse primary bone marrow cells, over-expression of Syk results in IL-7 independent growth in culture (Wossning, T., et.al, JEM, 2006; 203:2829- 2840).
- BCR antigen-specific B cell receptor
- the spleen tyrosine kinase (Syk) docks with and phosphorylates the IT AM, a process that enhances its kinase activity, resulting in Syk autophosphorylation and tyrosine phosphorylation of multiple downstream substrates (Rolli, Gallwitz et al. MoI Cell 10(5): 1057-69 (2002).
- This signaling pathway is active in B cells beginning at the transition from pro- to pre-B cell stage of development, when the newly formed pre-BCR is expressed. In fact, B cell development arrests at the pro-B cell stage in Syk knockout mice (Cheng, Rowley et al. 1995; Turner, Mee et al. Nature 378(6554): 303-6 (1995).
- Syk over-expression is reported in Mantle Cell Lymphoma (Rinaldi, Kwee et al. Br J Haematol 132(3): 303-16 (2006) and the TEL-Syk fusion protein (Translocated ETS Leukemia) generated by a chromosomal translocation (t(9;12)(q22;pl2)) leads to increased Syk activity and is associated with myelodysplastic syndrome (Kuno, Abe et al. Blood 97(4): 1050-5 (2001).
- TEL-Syk fusion protein Translocated ETS Leukemia
- Leukemia is induced in mice by the adoptive transfer of bone marrow cells that express human TEL-Syk (Wossning, Herzog et al. J Exp Med 203(13): 2829-40 (2006). Further, in mouse primary bone marrow cells, over-expression of Syk results in IL-7 independent growth in culture (Wossning, Herzog et al. 2006). Consistently, Syk was reported to mediate mTOR (mammalian target of Rapamycin) survival signals in follicular, mantle cell, Burkitt's, and diffuse large B-cell NHL (Leseux, Hamdi et al. Blood 108(13): 4156-62 (2006).
- mTOR mimalian target of Rapamycin
- Syk- dependant survival signals may play a role in B-cell malignancies, including DLBCL, mantle cell lymphoma and follicular lymphoma (Gururajan, Jennings et al. 2006; Irish, Czerwinski et al. J Immunol 176(10): 5715-9 (2006).
- DLBCL mantle cell lymphoma
- follicular lymphoma follicular lymphoma
- syk-dependant survival signals may play a role in B-cell malignancies, including DLBCL, mantle cell lymphoma and follicular lymphoma (see e.g., S. Linfengshen et al. Blood, Feb. 2008; 1 11 : 2230-2237; J. M. Irish et al. Blood, 2006; 108: 3135-3142; A. Renaldi et al. Brit J. Haematology, 2006; 132: 303-316; M. Guruoajan et al. J. Immunol, 2006; 176: 5715-5719; L. Laseux et al. Blood, 2006; 108: 4156- 4162.
- JAK kinases are a family of cytoplasmic protein tyrosine kinases including JAKl, JAK2, JAK3 and TYK2.
- the JAKs play a crucial role in cytokine signaling.
- Each of the JAK kinases is selective for the receptors of certain cytokines, though multiple JAK kinases can be affected by particular cytokine or signaling pathways.
- JAK3 in particular selectively binds to receptors and is part of the cytokine signaling pathway for and activated by IL-2, IL-4, TL-I, IL-9, EL- 15 and IL-21.
- JAKl interacts with, among others, the receptors for cytokines EL-2, IL-4, IL-I, IL-9 and IL- 21, while JAK2 interacts with, among others, the receptors for IL-9 and TNF- ⁇ .
- cytokines e.g., IL-2, IL-4, EL-7, IL-9, IL- 15 and IL-21
- receptor oligomerization occurs, resulting in the cytoplasmic tails of associated JAK kinases being brought into proximity and facilitating the trans-phosphorylation of tyrosine residues on the JAK kinase. This trans-phosphorylation results in the activation of the JAK kinase.
- the downstream substrates of JAK family kinases include the signal tranducer activator of transcription (STAT) proteins.
- Phosphorylated JAK kinases bind various STAT (Signal Transducer and Activator of Transcription) proteins.
- STAT proteins which are DNA binding proteins activated by phosphorylation of tyrosine residues, function both as signaling molecules and transcription factors and ultimately bind to specific DNA sequences present in the promoters of cytokine-responsive genes (Leonard et ah, (2000), J. Allergy Clin. Immunol. 105:877-888).
- JAK/STAT signaling has been implicated in the mediation of many abnormal immune responses such as allergies, asthma, autoimmune diseases such as transplant (allograft) rejection, rheumatoid arthritis, amyotrophic lateral sclerosis and multiple sclerosis, as well as in solid and hematologic malignancies such as leukemia and lymphomas.
- autoimmune diseases such as transplant (allograft) rejection
- rheumatoid arthritis such as transplant (allograft) rejection
- rheumatoid arthritis amyotrophic lateral sclerosis and multiple sclerosis
- solid and hematologic malignancies such as leukemia and lymphomas.
- JAK3 in particular has been implicated in a variety of biological processes. For example, the proliferation and survival of murine mast cells induced by IL-4 and BL-9 have been shown to be dependent on JAK3- and gamma chain-signaling (Suzuki et ah, (2000), Blood 96:2172-2180). JAK3 also plays a crucial role in IgE receptor-mediated mast cell degranulation responses (Malaviya et ah, (1999), Biochem. Biophys. Res. Commun.
- JAK3 kinases have also been implicated in the mechanism involved in early and late stages of rheumatoid arthritis (Muller-Ladner et ah, (2000), J. Immunal.
- JAKl, JAK2, and TYK2 are expressed ubiquitously, whereas JAK3 is expressed predominantly in hematopoietic cells.
- the JAK kinases, including JAK3, are abundantly expressed in primary leukemic cells from children with acute lymphoblastic leukemia, the most common form of childhood cancer, and studies have correlated STAT activation in certain cells with signals regulating apoptosis (Demoulin et al., (1996), MoI. Cell. Biol. 16:4710-6; Jurlander et al., (1997), Blood. 89:4146-52; Kaneko et al., (1997), Clin. Exp. Immun.
- JAK-3 in particular plays an essential role in the function of lymphocytes, macrophages, and mast cells.
- compounds which modulate the JAK pathway can be useful for treating diseases or conditions where the function of lymphocytes, macrophages, or mast cells is involved (Kudlacz et al., (2004) Am. J. Transplant 4:51-57; Changelian (2003) Science 302:875-878).
- Conditions in which targeting of the JAK pathway or modulation of the JAK kinases, particularly JAK3, are contemplated to be therapeutically useful include, leukemia, lymphoma, transplant rejection (e.g., pancreas islet transplant rejection, bone marrow transplant applications (e.g., graft- versus-host disease), autoimmune diseases (e.g., diabetes, rheumatoid arthritis, lupus, psoriasis), and inflammation (e.g., asthma, allergic reactions). Conditions which can benefit from JAK3 inhibition are discussed in greater detail below.
- Patents and patent applications describing substituted pyrimidinediamine compounds include: U.S. application Ser. No. 10/355,543 filed Jan. 31, 2003 (US2004/0029902A1), international application Serial No. PCT/US03/03022 filed Jan. 31, 2003 (WO 03/063794), U.S. application Ser. No. 10/631,029 filed JuI. 29, 2003, international application Serial No. PCT/US03/24087 (WO 04/014382), U.S. application Ser. No. 10/903,263 filed JuI. 30, 2004, and international application Serial No. PCT/US2004/24716 (WO 05/016893), the disclosures of which are incorporated herein by reference.
- Substituted pyrimidinediamine compounds are also described in international patent application publication numbers: WO 02/059110, WO 03/074515, WO 03/106416, WO 03/066601, WO 03/063794, WO 04/046118, WO 05/016894, WO 05/122294, WO 05/066156, WO 03/002542, WO 03/030909, WO 00/39101, WO 05/037800 and U.S. Pat. Pub. No. 2003/0149064.
- the present invention provides novel compounds having activity as inhibitors of syk activity (also referred to herein as “syk inhibitors”) and/or JAK kinase activity (also referred to herein as “JAK inhibitors”), as well as to methods for their preparation and use, and to pharmaceutical compositions containing the same.
- syk inhibitors also referred to herein as "syk inhibitors”
- JAK kinase activity also referred to herein as "JAK inhibitors”
- Such compounds have the following structure (I- II):
- the present invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of formula I- II, or a pharmaceutical acceptable salt thereof, and a pharmaceutically acceptable carrier and/or diluent.
- the compounds of the present invention have utility over a wide range of therapeutic applications, and may be used to treat a variety of conditions, mediated at least in part by syk activity, in both men and women, as well as a mammal in general (also referred to herein as a "subject").
- syk activity mediated at least in part by syk activity
- men and women as well as a mammal in general
- mammal also referred to herein as a "subject”
- such conditions include, but are not limited to, those associated with cardiovascular disease, inflammatory disease or autoimmune disease.
- the compounds of the present invention have utility for treating conditions or disorders including, but not limited to: restenosis, thrombosis, inflammation, heparin induced thrombocytopenia, dilated cardiomyopathy, sickle cell disease, atherosclerosis, myocardial infarction, vascular inflammation, unstable angina, acute coronary syndromes, allergy, asthma, rheumatoid arthritis, B-cell mediated diseases such as Non Hodgkin's lymphoma, anti-phospholipid syndrome, lupus, psoriasis, multiple sclerosis, end stage renal disease, hemolytic anemia, immune thrombocytopenic purpura, and chronic lymphocytic leukemia.
- methods are disclosed which include the administration of an effective amount of a compound of formula (I-II), typically in the form of a pharmaceutical composition, to a subject in need thereof.
- the conditions associated with cardiovascular disease is selected from the group consisting of acute coronary syndrome, myocardial infarction, unstable angina, refractory angina, occlusive coronary thrombosis occurring post-thrombolytic therapy or post-coronary angioplasty, a thrombotically mediated cerebrovascular syndrome, embolic stroke, thrombotic stroke, transient ischemic attacks, venous thrombosis, deep venous thrombosis, pulmonary embolism, coagulopathy, disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, thromboangiitis obliterans, thrombotic disease associated with heparin-induced thrombocytopenia, thrombotic complications associated with extracorporeal circulation, thrombotic complications associated with instrumentation such as cardiac or other intravascular catheterization, intra-aortic balloon pump, coronary stent or cardiac valve, and conditions requiring the fitting of prosthetic devices.
- the present invention also provides a method for inhibiting the syk activity of a blood sample comprising contacting said sample with a compound of the present invention.
- the present invention further provides compounds in purified forms, as well as chemical intermediates.
- Figure 1 shows how Syk serves as a key mediator of Fc receptor mediated signaling in cellular biology and multiple diseases.
- Figure 2 shows how gene targeting of Syk indicated that Syk serves as a key mediator in arterial platelet biology and a selective target for treating arterial thrombosis.
- Figures 3-5 show a general synthesis of compounds of the present invention.
- Figure 6 shows a graph of the effect of increasing dose of a compound of the present invention on pSTAT ⁇ formation in reponse to IL4 stimulation of B Ramos cells.
- Figure 7 provides table 1 illustrating compounds of the present invention and syk IC 50 S.
- Figure 8 provides table 2 illustrating compounds of the present invention and SYK IC 50 S.
- Figure 9 shows specificity data for selected compounds.
- Figure 10 shows the selective inhibition of selected bicyclic compounds.
- Boc t-butylcarboxy
- BOP benzotriazol-l-yloxytris(dimethylamino)-phosphoniurn hexafluorophosphate
- BPO benzoyl peroxide
- nBuOH n-butanol
- CBr4 tetrabromomethane
- mCPBA m- chloroperoxybenzoic acid
- CH2CI2 or DCM dichlorome thane
- CS2CO3 cesium carbonate
- CuCl2 copper chloride
- DIBAL diisobutylaluminum hydride
- DIEA Hunig's base or diisopropyl ethylamine
- DME dimethyl ether
- DMF dimethyl formamide
- DMSO dimethyl sulfoxide
- DPPA diphenyl phosphoryl azide
- Et3N triethylamine
- EtOAc diphenyl phosphoryl
- Alkyl by itself or as part of another substituent, means, unless otherwise stated, a straight or branched chain, fully saturated aliphatic hydrocarbon radical having the number of carbon atoms designated.
- “Ci. 8 alkyl” refers to a hydrocarbon radical straight or branched, containing from 1 to 8 carbon atoms that is derived by the removal of one hydrogen atom from a single carbon atom of a parent alkane.
- the phrase "unsubstituted alkyl” refers to alkyl groups that do not contain groups other than fully saturated aliphatic hydrocarbon radicals.
- the phrase includes straight chain alkyl groups such as methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl and the like.
- the phrase also includes branched chain isomers of straight chain alkyl groups such as isopropyl, t-butyl, isobutyl, sec-butyl, and the like.
- Representative alkyl groups include straight and branched chain alkyl groups having 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 1 1 or 12 carbon atoms.
- alkyl groups include straight and branched chain alkyl groups having 1, 2, 3, 4, 5, 6, 7 or 8 carbon atoms.
- "Alkenyl" by itself or as part of another substituent refers to a straight or branched chain, which may be mono- or polyunsaturated, having the number of carbon atoms designated.
- C 2 -C 8 alkenyl means an alkenyl radical having from 2, 3, 4, 5, 6, 7 or 8 atoms that is derived by the removal of one hydrogen atom from a single carbon atom of a parent alkane. Examples include, but are not limited to vinyl, 2-propenyl i.e.
- a "substituted" alkenyl group includes alkenyl groups in which a non-carbon or non-hydrogen atom is bonded to a carbon double bonded to another carbon and those in which one of the non- carbon or non-hydrogen atoms is bonded to a carbon not involved in a double bond to another carbon.
- Each site of unsaturation may be either cis or trans configuration about the double bond(s).
- alkynyl by itself or as part of another substituent, means a straight or branched chain hydrocarbon radical, which may be mono- or polyunsaturated, having the number of carbon atoms designated.
- C 2 -C 8 alkynyl means an alkynyl radical having from 2 to 8 carbon atoms that is derived by the removal of one hydrogen atom from a single carbon atom of a parent alkane.
- Unsubstituted alkynyl refers to straight and branched chain groups such as those described with respect to unsubstituted alkyl groups as defined above, except that at least one triple bond exists between two carbon atoms.
- a "substituted" alkynyl group includes alkynyl groups in which a non-carbon or non-hydrogen atom is bonded to a carbon triple bonded to another carbon and those in which a non-carbon or non-hydrogen atom is bonded to a carbon not involved in a triple bond to another carbon.
- Alkylene by itself or as part of another substituent means a divalent radical derived from an alkane, as exemplified by -CH 2 CH 2 CH 2 CH 2 -.
- an alkylene group will have from 1, 2, 3, 4, 5, 6, 7 or 8 carbon atoms that is derived by the removal of one hydrogen atom from a single carbon atom of a parent alkyl.
- Cycloalkyl or “carbocycle” by themselves or in combination with other terms, represent, unless otherwise stated, cyclic versions of “alkyl”, “alkenyl” and “alkynyl” in which all ring atoms are carbon.
- Cycloalkyl or “carbocycle” refers to a mono- or polycyclic group.
- polycyclic refers herein to fused and non-fused alkyl cyclic structures.
- Cycloalkyl or “carbocycle” may form a bridged ring or a spiro ring.
- the cycloalkyl group may have one or more double or triple bond(s).
- cycloalkenyl refers to a cycloalkyl group that has at least one site of alkenyl unsaturation between the ring vertices.
- cycloalkynyl refers to a cycloalkyl group that has at least one site of alkynyl unsaturation between the ring vertices.
- cycloalkyl When “cycloalkyl” is used in combination with “alkyl”, as in C 3-8 cycloalkylC 3 - 8 alkylene-, the cycloalkyl portion is meant to have the stated number of carbon atoms (e.g., from three to eight carbon atoms), while the alkylene portion has from one to eight carbon atoms. Typical cycloalkyl substituents have from 3 to 8 ring atoms. Examples of cycloalkyl include cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3-cyclohexenyl, cycloheptyl, and the like.
- Aryl by itself or as part of another substituent refers to a polyunsaturated, aromatic, hydrocarbon group containing from 6 to 14 carbon atoms, which can be a single ring or multiple rings (up to three rings) which are fused together or linked covalently.
- the phrase includes, but is not limited to, groups such as phenyl, biphenyl, anthracenyl, naphthyl by way of example.
- unsubstituted aryl groups include phenyl, 1 -naphthyl, 2-naphthyl and 4-biphenyl.
- “Substituted aryl group” includes, for example, -CH2OH (one carbon atom and one heteroatom replacing a carbon atom) and
- heteroalkylene by itself or as part of another substituent means a divalent radical derived from heteroalkyl, as exemplified by -CH2-CH2-S-CH2CH2- - and -CH2-S-CH2-CH2-NH-CH2-.
- heteroatoms can also occupy either or both of the chain termini (e.g., alkyleneoxy, alkylenedioxy, alkyleneamino, alkylenediamino, and the like). Still further, for alkylene and heteroalkylene linking groups, no orientation of the linking group is implied.
- heterocycle refers to a saturated or unsaturated non-aromatic cyclic group containing at least one heteroatom.
- heteroatom is meant to include oxygen (O), nitrogen (N), sulfur (S) and silicon (Si).
- Each heterocycle can be attached at any available ring carbon or heteroatom.
- Each heterocycle may have one or more rings. When multiple rings are present, they can be fused together or linked covalently.
- Each heterocycle typically contains 1, 2, 3, 4 or 5, independently selected heteroatoms.
- these groups contain 1, 2, 3, 4, 5, 6, 7, 8, 9 or 10 carbon atoms, 0, 1, 2, 3, 4 or 5 nitrogen atoms, 0, 1 or 2 sulfur atoms and 0, 1 or 2 oxygen atoms. More preferably, these groups contain 1, 2 or 3 nitrogen atoms, 0-1 sulfur atoms and 0-1 oxygen atoms.
- heterocycle groups include morpholin-3-one, piperazine-2-one, piperazin-1 -oxide, pyridine-2-one, piperidine, morpholine, piperazine, isoxazoline, pyrazoline, imidazoline, pyrazol-5-one, pyrrolidine-2,5- dione, imidazolidine-2,4-dione, pyrrolidine, tetrahydroquinolinyl, decahydroquinolinyl, tetrahydrobenzooxazepinyl dihydrodibenzooxepin and the like.
- Heteroaryl refers to a cyclic or polycyclic aromatic radical that contain from one to five heteroatoms selected from N, O, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized.
- a heteroaryl group can be attached to the remainder of the molecule through a heteroatom or through a carbon atom and can contain 5 to 10 carbon atoms.
- heteroaryl groups include 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 1-pyrazolyl, 3-pyrazolyl, 2-imidazolyl, 4- imidazolyl, pyrazinyl, 2-oxazolyl, 4-oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4-isoxazolyl, 5- isoxazolyl, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, 2-furyl, 3-furyl, 2-thienyl, 3-thienyl, 2- pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl and 4-pyrimidyl.
- substituted heteroaryl refers to a unsubstituted heteroaryl group as defined above in which one or more of the ring members is bonded to a non-hydrogen atom such as described above with respect to substituted alkyl groups and substituted aryl groups.
- Bicyclic heteroaryl refers to bicyclic aromatic radical that contain from one to five heteroatoms selected from N, O, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized.
- a bicyclic heteroaryl group can be attached to the remainder of the molecule through a heteroatom or through a carbon atom and can contain 5 to 10 carbon atoms.
- Non-limiting examples of bicyclic heteroaryl groups include 5-benzothiazolyl, purinyl, 2-benzimidazolyl, benzopyrazolyl, 5-indolyl, azaindole, 1-isoquinolyl, 5-isoquinolyl, 2-quinoxalinyl, 5- quinoxalinyl, 3-quinolyl and 6-quinolyl. If not specifically stated, substituents for each of the above noted aryl and heteroaryl ring systems are selected from the group of acceptable substituents described herein.
- Ci -8 designating a number of atoms e.g. "Ci -8 " is meant to include all possible embodiments that have one fewer atom.
- Non-limiting examples include Ci -7 , C 2-8 , C 2-7 , C 3-8 , C 3-7 and the like.
- each of the terms herein is meant to include both “unsubstituted” and optionally “substituted” forms of the indicated radical, unless otherwise indicated.
- each radical is substituted with 0, 1, 2 3 4 or 5 substituents, unless otherwise indicated. Examples of substituents for each type of radical are provided below.
- Substituted refers to a group as defined herein in which one or more bonds to a carbon(s) or hydrogen(s) are replaced by a bond to non-hydrogen and non-carbon atom "substituents" such as, but not limited to, a halogen atom such as F, Cl, Br, and I; an oxygen atom in groups such as hydroxyl groups, alkoxy groups, aryloxy, and acyloxy groups; a sulfur atom in groups such as thiol groups, alkyl and aryl sulfide groups, sulfone groups, sulfonyl groups, and sulfoxide groups; a nitrogen atom in groups such as amino, alkylamines, dialkylamines, arylamines, alkylarylamines, diarylamines, alkoxyamino, hydroxyamino, acylamino, sulfonylamino, N-oxides, imides, and en
- Substituents also include groups in which one or more bonds to a carbon(s) or hydrogen(s) atom is replaced by a higher-order bond (e.g., a double- or triple- bond) to a heteroatom such as oxygen in oxo, acyl, amido, alkoxycarbonyl, aminocarbonyl, carboxyl, and ester groups; nitrogen in groups such as imines, oximes, hydrazones, and nitriles. "Substituents” further include groups in which one or more bonds to a carbon(s) or hydrogen(s) atoms is replaced by a bond to a cycloalkyl, heterocyclyl, aryl, and heteroaryl groups.
- substituted alkyl groups include, among others, groups in which one or more bonds to a carbon or hydrogen atom is/are replaced by one or more bonds to fluoro, chloro, or bromo group. Another representative “substituent” is the trifluoromethyl group and other groups that contain the trifluoromethyl group. Other representative “substituents” include those in which one or more bonds to a carbon or hydrogen atom is replaced by a bond to an oxygen atom such that the substituted alkyl group contains a hydroxyl, alkoxy, or aryloxy group.
- substituted or unsubstituted alkylamine dialkylamine, arylamine, (alkyl)(aryl)amine, diarylamine, heterocyclylamine, diheterocyclylamine, (alkyl)(heterocyclyl)amine, or (aryl)(heterocyclyl)amine group.
- Still other representative "substituents” include those in which one or more bonds to a carbon(s) or hydrogen(s) atoms is replaced by a bond to an alkyl, cycloalkyl, aryl, heteroaryl, or heterocyclyl group.
- alkylamino refers to a group of the formula -NR a R b .
- R a , and R b are each independently selected from H, alkyl, alkoxy, thioalkoxy, cycloalkyl, aryl, heteroaryl, or heterocyclyl or are optionally joined together with the atom(s) to which they are attached to form a cyclic group.
- R a and R b are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 5-, 6- or 7-membered ring.
- -NR a R b is meant to include 1 -pyrrolidinyl and 4-morpholinyl.
- R c , R d , R e and R f are each independently selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, aryl, heteroaryl, heterocyclyl or alkylenearyl as defined herein.
- a particular radical will have 0, 1 , 2 or 3 substituents, with those groups having two or fewer substituents being preferred in the present invention. More preferably, a radical will be unsubstituted or monosubstituted. Most preferably, a radical will be unsubstituted.
- substituted aryl and heteroaryl groups are varied and are selected from: -halogen, -OR a , -OC(O) R a , -NR a R b , -SR a , -R a , -CN, -NO 2 , -CO 2 R a ,
- Two of the "substituents'On adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -T-C(0)-(CH 2 )q-U-, wherein T and
- U are independently -NH-, -O-, -CH 2 . or a single bond, and q is O, 1 or 2.
- two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -A-(CH 2 ) r _B-, wherein A and B are independently -CH 2 _,
- two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -(CH 2 ) S _X-(CH 2 ) ⁇ _ -, where s and t are independently integers of from O to 3, and X is -O-, -NR a -, -S- , -S(O)-, -S(O) 2 ., or -S(O) 2 NR a -.
- the substituent R a in -NR a - and -S(O) 2 NR 3 - is selected from hydrogen or unsubstituted Cj .galkyl. Otherwise, R' is as defined above.
- R c is alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, aryl, heteroaryl or heterocyclyl.
- Alkoxy refers to -OR d wherein R d is alkyl as defined herein.
- Representative examples of alkoxy groups include methoxy, ethoxy, t-butoxy, trifluoromethoxy, and the like.
- Alkoxyamino refers to the group -NH0R d where R d is alkyl.
- Alkoxyalkyleneamino refers to the group -NR a -alkylene-OR d where R d is alkyl and -NR a - is defined in amino.
- Representative alkoxycarbonyl groups include, for example, those shown below.
- Alkylheterocyclyl refers to the group -heterocyclyl-R d . where R d is alkyl.
- Alkylheterocyclyl alkylene refers to the group -alkylene-heterocyclyl-R d . where R d is alkyl.
- Alkylsulfanyl refers to the group S-R d .where R d is alkyl.
- Alkylsulfonyl groups employed in compounds of the present invention are typically C 1-6 alkylsulfinyl groups.
- Alkylsulfonyl groups employed in compounds of the present invention are typically Ci -6 alkylsulfonyl groups.
- Alkylsulfonyl groups employed in compounds of the present invention are typically Ci. 6alkylsulfonyl groups.
- Alkynyloxy refers to the group -O-alkynyl, wherein alkynyl is as defined herein. Alkynyloxy includes, by way of example, ethynyloxy, propynyloxy, and the like.
- Amino refers to a monovalent radical -NR a R b or divalent radical -NR a -.
- the term includes "alkylamino” which refers to the group -NR a R b where R a is alkyl and R b is H or alkyl.
- the term also includes "arylamino” which refers to the group -NR a R b where at least one R a or R b is aryl.
- the term also includes "(alkyl)(aryl)amino” which refers to the group - NR a R b where R a is alkyl and R b is aryl.
- dialkylamino groups the alkyl portions can be the same or different and can also be combined to form a 3-7 membered ring with the nitrogen atom to which each is attached. Accordingly, a group represented as -NR a R b is meant to include piperidinyl, pyrrolidinyl, morpholinyl, azetidinyl and the like.
- Aminoalkoxy refers to-O-alkylene-NR a R b .
- Aminoalkylene refers to -alkylene-NR a R b .
- Aminoaryl refers to-aryl-NR a R b .
- Representative aminocarbonyl groups include, for example, those shown below. These aminocarbonyl group can be further substituted as will be apparent to those having skill in the organic and medicinal chemistry arts in conjunction with the disclosure herein.
- Aminocarbonylamino refers to the group -NR a C(O)NR a R b , wherein R a is hydrogen or alkyl and R a and R b independently are selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkenyl, heteroaryl, heterocyclic, and where R a and R b are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group.
- Aminocarbonylaminoalkylene refers to the group -alkylene-NR a C(O)NR a R b , wherein R a is hydrogen or alkyl and R a and R b independently are selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkenyl, heteroaryl, heterocyclic, and where R a and R b are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group.
- Aminocarboxyalkylene refers to the group -alkylene-OC(O)NR a R b , wherein R a is hydrogen or alkyl and R a and R b independently are selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkenyl, heteroaryl, heterocyclic, and where R a and R b are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group.
- Aminosulfonyl refers to -S(O) 2 NR a R b where R is independently are selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, substituted heterocyclic and where R a and R b are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group and alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, substituted
- Aminosulfonylalkylene refers to -alkylene-S(O) 2 NR a R b where R is independently are selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, substituted heterocyclic and where R a and R b are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group and alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, substituted
- alkylaminosulfonyl refers herein to the group -S(O) 2 NR a R b where R a is alkyl and R b is H or alkyl.
- alkylarylsulfonyl refers herein to the group — S(O) 2 NR a R b where R a or R b is alkylaryl.
- Aminosulfonyloxy refers to the group -O-SO 2 NR a R b , wherein R a and R b independently are selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, cycloalkyl, cycloalkenyl, heteroaryl and heterocyclic; R a and R b are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group.
- Aminosulfonylamino refers to the group -NR a -SO 2 NR b R c , wherein R a is hydrogen or alkyl and R b and R c independently are selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R b and R c are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycl
- Aminothiocarbonyl refers to the group -C(S)NR a R b , wherein R a and R b independently are selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, heteroaryl, substituted heteroaryl, heterocyclic, and substituted heterocyclic and where R a and R b are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group, and wherein alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycl
- Aminothiocarbonylamino refers to the group -NR a C(S)NR a R b , wherein R a is hydrogen or alkyl and R b and R c are optionally joined together with the nitrogen bound thereto to form a heterocyclic or substituted heterocyclic group.
- Aryloxy refers to -OR d where R d is aryl.
- Representative examples of aryloxy groups include phenoxy, naphthoxy, and the like.
- Arylsulfanyl refers to the group S-R d .where R d is aryl.
- Arylthio refers to the group -S-aryl, wherein aryl is as defined herein.
- sulfur may be oxidized to -S(O)- or -SO 2 - moieties.
- the sulfoxide may exist as one or more stereoisomers.
- Carboxy or “carboxyl” refers to the group -CO 2 H.
- Carboxyalkylene refers to the group -alkylene-CO 2 H.
- Carboxyalkylenesulfonylamino refers to the group -NR a SO 2 -alkylene-CO 2 H.
- Carboxyl ester refers to the group -
- (Carboxyl ester)amino refers to the groups -NR a -C(O)OR c , where R a is alkyl or hydrogen.
- Cyano refers to -CN.
- Cycloalkoxy refers to -OR d where R d is cycloalkyl.
- Cycloalkylalkylene refers to a radical -R X R V wherein R x is an alkylene group and
- Ry is a cycloalkyl group as defined herein, e.g., cyclopropylmethyl, cyclohexenylpropyl, 3- cyclohexyl-2-methylpropyl, and the like.
- Cycloalkylthio refers to -S-cycloalkyl.
- sulfur may be oxidized to -S(O)- or -SO 2 - moieties.
- the sulfoxide may exist as one or more stereoisomers.
- Cycloalkenylox refers to -O-cycloalkenyl.
- Cycloalkenylthio refers to -S-cycloalkenyl.
- sulfur may be oxidized to sulfinyl or sulfonyl moieties.
- the sulfoxide may exist as one or more stereoisomers.
- Halo or "halogen” by themselves or as part of another substituent, mean, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as “haloalkyl”, are meant to include alkyl in which one or more hydrogen is substituted with halogen atoms which can be the same or different, in a number ranging from one up to the maximum number of halogens permitted e.g. for alkyl, (2m'+l), where m' is the total number of carbon atoms in the alkyl group.
- haloCj.galkyl is meant to include trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like.
- perhaloalkyl means, unless otherwise stated, alkyl substituted with (2m'+l) halogen atoms, where m' is the total number of carbon atoms in the alkyl group.
- perhaloCi _galkyl is meant to include trifluoromethyl, pentachloroethyl, l,l,l-trifluoro-2-bromo-2-chloroethyl, and the like.
- haloalkoxy refers to an alkoxy radical substituted with one or more halogen atoms.
- Heteroalkyl means an alkyl radical as defined herein with one, two or three substituents independently selected from cyano, -OR W , -NR X RY, and -S(O) n R z (where n is an integer from 0 to 2 ), with the understanding that the point of attachment of the heteroalkyl radical is through a carbon atom of the heteroalkyl radical.
- R w is hydrogen, alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, alkoxycarbonyl, aryloxycarbonyl, carboxamido, or mono- or di-alkylcarbamoyl.
- R x is hydrogen, alkyl, cycloalkyl, cycloalkyl-alkyl, aryl or araalkyl.
- Ry is hydrogen, alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, alkoxycarbonyl, aryloxycarbonyl, carboxamido, mono- or di-alkylcarbamoyl or alkylsulfonyl.
- R z is hydrogen (provided that n is 0), alkyl, cycloalkyl, cycloalkyl-alkyl, aryl, araalkyl, amino, mono- alkylamino, di-alkylamino, or hydroxyalkyl. Representative examples include, for example, 2-hydroxyethyl, 2,3-dihydroxypropyl, 2-methoxyethyl, benzyloxymethyl, 2-cyanoethyl, and
- R w , R x ,RY, and R z can be further substituted by amino, fluorine, alkylamino, di-alkylamino, OH or alkoxy.
- the prefix indicating the number of carbon atoms refers to the total number of carbon atoms in the portion of the heteroalkyl group exclusive of the cyano, -OR W , -NR X RY, or - S(O) n R 2 portions.
- Heteroarylalkenyl refers to the group -alkenyl-R c where R c is heteroaryl.
- Heteroaryloxy refers to -OR d where R d is heteroaryl.
- Heteroarylthio refers to the group — S-heteroaryl.
- sulfur may be oxidized to -S(O)- or -SO 2 - moieties.
- the sulfoxide may exist as one or more stereoisomers.
- Heterocyclylalkyl or "Cycloheteroalkyl-alkyl” means a radical -R X R V where R x is an alkylene group and R v is a heterocyclyl group as defined herein, e.g., tetrahydropyran- 2-ylmethyl, 4-(4-substituted-phenyl)piperazin-l-ylmethyl, 3-piperidinylethyl, and the like.
- Heterocyclyloxy refers to -OR d where R d is heterocyclyl.
- Heterocyclylthio refers to the group -S-heterocycyl.
- sulfur may be oxidized to -S(O)- or -SO 2 - moieties.
- the sulfoxide may exist as one or more stereoisomers.
- Hydroxyalkylene refers to the group -alkylene-OH.
- Hydroxyalkyleneamino refers to the group -NR a -alkylene-OH.
- Hydroxyalkyleneaminosulfonyl refers to the group -SO 2 NR a -alkylene-OH.
- Hydroxyamino refers to the group -NHOH.
- Niro refers to -NO 2 .
- Niroso refers to the group -NO.
- heterocyclo group optionally mono- or di- substituted with an alkyl group means that the alkyl may but need not be present, and the description includes situations where the heterocyclo group is mono- or disubstituted with an alkyl group and situations where the heterocyclo group is not substituted with the alkyl group.
- Optionally substituted means a ring which is optionally substituted independently with substituents.
- a site of a group that is unsubstituted may be substituted with hydrogen.
- Sulfanyl refers to the group -SR f where R f is as defined herein.
- Sulfonic acid refers to the group -S(O) 2 -OH.
- Sulfonyl refers to the group -S(O) 2 -R e where R e is as defined herein.
- stereoisomers Compounds that have the same molecular formula but differ in the nature or sequence of bonding of their atoms or the arrangement of their atoms in space are termed “isomers”. Isomers that differ in the arrangement of their atoms in space are termed “stereoisomers”. "Stereoisomer” and “stereoisomers” refer to compounds that exist in different stereoisomeric forms if they possess one or more asymmetric centers or a double bond with asymmetric substitution and, therefore, can be produced as individual stereoisomers or as mixtures. Stereoisomers include enantiomers and diastereomers.
- stereoisomers that are not mirror images of one another are termed “diastereomers” and those that are non-superimposable mirror images of each other are termed “enantiomers”.
- enantiomers When a compound has an asymmetric center, for example, it is bonded to four different groups, a pair of enantiomers is possible.
- An enantiomer can be characterized by the absolute configuration of its asymmetric center and is described by the R- and S-sequencing rules of Cahn and Prelog, or by the manner in which the molecule rotates the plane of polarized light and designated as dextrorotatory or levorotatory (i.e., as (+) or (-)-isomers respectively).
- a chiral compound can exist as either individual enantiomer or as a mixture thereof.
- a mixture containing equal proportions of the enantiomers is called a "racemic mixture".
- the description is intended to include individual stereoisomers as well as mixtures.
- the methods for the determination of stereochemistry and the separation of stereoisomers are well-known in the art (see discussion in Chapter 4 of ADVANCED ORGANIC CHEMISTRY, 4th edition J. March, John Wiley and Sons, New York, 1992) differ in the chirality of one or more stereocenters.
- Protecting group refers to a group of atoms that, when attached to a reactive functional group in a molecule, mask, reduce or prevent the reactivity of the functional group.
- a protecting group may be selectively removed as desired during the course of a synthesis. Examples of protecting groups can be found in Greene and Wuts, Protective Groups in Organic Chemistry, 3 rd Ed., 1999, John Wiley & Sons, NY and Harrison et al., Compendium of Synthetic Organic Methods, VoIs. 1-8, 1971-1996, John Wiley & Sons, NY.
- Representative amino protecting groups include, but are not limited to, formyl, acetyl, trifluoroacetyl, benzyl, benzyloxycarbonyl (“CBZ”), tert-butoxycarbonyl (“Boc”), trimethylsilyl (“TMS”), 2-trimethylsilyl-ethanesulfonyl (“TES”), trityl and substituted trityl groups, allyloxycarbonyl, 9-fluorenylmethyloxycarbonyl (“FMOC”), nitro- veratryloxycarbonyl (“NVOC”) and the like.
- hydroxy protecting groups include, but are not limited to, those where the hydroxy group is either acylated or alkylated such as benzyl and trityl ethers, as well as alkyl ethers, tetrahydropyranyl ethers, trialkylsilyl ethers (e.g., TMS or TIPPS groups) and allyl ethers.
- salts are meant to include salts of the active compounds which are prepared with relatively nontoxic acids or bases, depending on the particular substituents found on the compounds described herein.
- base addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired base, either neat or in a suitable inert solvent.
- salts derived from pharmaceutically-acceptable inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc and the like.
- Salts derived from pharmaceutically-acceptable organic bases include salts of primary, secondary and tertiary amines, including substituted amines, cyclic amines, naturally- occurring amines and the like, such as arginine, betaine, caffeine, choline, N,N'- dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
- acid addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired acid, either neat or in a suitable inert solvent.
- pharmaceutically acceptable acid addition salts include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from relatively nontoxic organic acids like acetic, propionic, isobutyric, malonic, benzoic, succinic, suberic, fumaric, mandelic, phthalic, benzenesulfonic, p-tolylsulfonic, citric, tartaric, methanesulfonic, and the like.
- salts of amino acids such as arginate and the like, and salts of organic acids like glucuronic or galactunoric acids and the like (see, e.g., Berge, S. M. et al., "Pharmaceutical Salts," Journal of Pharmaceutical Science, 66: 1-19, 1977).
- Certain specific compounds of the present invention contain both basic and acidic functionalities that allow the compounds to be converted into either base or acid addition salts.
- the neutral forms of the compounds may be regenerated by contacting the salt with a base or acid and isolating the parent compound in the conventional manner.
- the parent form of the compound differs from the various salt forms in certain physical properties, such as solubility in polar solvents, but otherwise the salts are equivalent to the parent form of the compound for the purposes of the present invention.
- the present invention provides compounds which are in a prodrug ester form.
- prodrug s of the compounds described herein are those compounds that readily undergo chemical changes under physiological conditions to provide the compounds of the present invention.
- prodrugs can be converted to the compounds of the present invention by chemical or biochemical methods in an ex vivo environment. For example, prodrugs can be slowly converted to the compounds of the present invention when placed in a transdermal patch reservoir with a suitable enzyme or chemical reagent. Prodrugs are frequently, but not necessarily, pharmacologically inactive until converted into the active drug.
- Prodrugs are typically obtained by masking a functional group in the drug believed to be in part required for activity with a progroup (defined below) to form a promoiety which undergoes a transformation, such as cleavage, under the specified conditions of use to release the functional group, and hence the active drug.
- the cleavage of the promoiety may proceed spontaneously, such as by way of a hydrolysis reaction, or it may be catalyzed or induced by another agent, such as by an enzyme, by light, by acid or base, or by a change of or exposure to a physical or environmental parameter, such as a change of temperature.
- the agent may be endogenous to the conditions of use, such as an enzyme present in the cells to which the prodrug is administered or the acidic conditions of the stomach, or it may be supplied exogenously.
- Progroup refers to a type of protecting group that, when used to mask a functional group within an active drug to form a promoiety, converts the drug into a prodrug.
- Progroups are typically attached to the functional group of the drug via bonds that are cleavable under specified conditions of use.
- a progroup is that portion of a promoiety that cleaves to release the functional group under the specified conditions of use.
- an amide promoiety of the formula -NH-C(O)CH 3 comprises the progroup -C(O)CH 3 .
- progroups as well as the resultant promoieties, suitable for masking functional groups in the active syk and/or JAK selective inhibitory compounds to yield prodrugs are well-known in the art.
- a hydroxyl functional group may be masked as a sulfonate, ester (such as acetate or maleate) or carbonate promoiety, which may be hydrolyzed in vivo to provide the hydroxyl group.
- An amino functional group may be masked as an amide, carbamate, imine, urea, phosphenyl, phosphoryl or sulfenyl promoiety, which may be hydrolyzed in vivo to provide the amino group.
- a carboxyl group may be masked as an ester (including methyl, ethyl, pivaloyloxymethyl, silyl esters and thioesters), amide or hydrazide promoiety, which may be hydrolyzed in vivo to provide the carboxyl group.
- the invention includes those esters and acyl groups known in the art for modifying the solubility or hydrolysis characteristics for use as sustained-release or prodrug formulations. Other specific examples of suitable progroups and their respective promoieties will be apparent to those of skill in the art. [0205] Certain compounds of the present invention can exist in unsolvated forms as well as solvated forms, including hydrated forms.
- solvent refers to a complex formed by combination of solvent molecules with molecules or ions of the solute.
- the solvent can be an organic compound, an inorganic compound, or a mixture of both.
- solvents include, but are not limited to, methanol, N,N-dimethylformamide, tetrahydrofuran, dimethylsulfoxide, and water.
- the solvated forms are equivalent to unsolvated forms and are intended to be encompassed within the scope of the present invention.
- Certain compounds of the present invention may exist in multiple crystalline or amorphous forms. In general, all physical forms are equivalent for the uses contemplated by the present invention and are intended to be within the scope of the present invention.
- Certain compounds of the present invention possess asymmetric carbon atoms (optical centers) or double bonds; the racemates, diastereomers, geometric isomers, regioisomers and individual isomers (e.g., separate enantiomers) are all intended to be encompassed within the scope of the present invention. These isomers can be resolved or asymmetrically synthesized using conventional methods to render the isomers "optically pure", i.e., substantially free of its other isomers.
- a particular enantiomer of a compound of the present invention may be prepared by asymmetric synthesis, or by derivation with a chrial auxilliary, where the resulting diastereomeric mixture is separated and the auxilliary group cleaved to provide the pure desired enantiomers.
- the molecule contains a basic functional group, such as amino, or an acidic functional group, such as carboxyl, diastereomeric salts are formed with an appropriate optically-active acid or base, followed by resolution of the diasteromers thus formed by fractional crystallization or chromatagraphic means well known in the art, and subsequent recovery of the pure enantiomers.
- the compounds of the present invention may also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds.
- the compounds may be radiolabeled with radioactive isotopes, such as for example tritium ( 3 H), iodine-125 ( 125 I) or carbon-14 ( 14 C). All isotopic variations of the compounds of the present invention, whether radioactive or not, are intended to be encompassed within the scope of the present invention.
- administering refers to oral administration, administration as a suppository, topical contact, intravenous, intraperitoneal, intramuscular, intralesional, intranasal or subcutaneous administration, or the implantation of a slow-release device e.g., a mini-osmotic pump, to a subject.
- Adminsitration is by any route, including parenteral and transmucosal (e.g., buccal, sublingual, palatal, gingival, nasal, vaginal, rectal, or transdermal).
- Parenteral administration includes, e.g., intravenous, intramuscular, intra- arteriole, intradermal, subcutaneous, intraperitoneal, intraventricular, and intracranial.
- Other modes of delivery include, but are not limited to, the use of liposomal formulations, intravenous infusion, transdermal patches, etc.
- An "agonist” or “activator” refers to an agent or molecule that binds to a receptor of the invention, stimulates, increases, opens, activates, facilitates, enhances activation or enzymatic activity, sensitizes or up regulates the activity of a receptor of the invention.
- an "antagonist” or “inhibitor” refers to an agent or molecule that inhibits or binds to, partially or totally blocks stimulation or activity, decreases, closes, prevents, delays activation or enzymatic activity, inactivates, desensitizes, or down regulates the activity of a receptor of the invention.
- antagonist also includes a reverse or inverse agonist.
- condition or disorder responsive to modulation of syk and/or JAK and related terms and phrases refer to a condition or disorder associated with inappropriate, e.g., less than or greater than normal, activity of syk and/or JAK and at least partially responsive to or affected by modulation of syk and/or JAK (e.g., syk and/or JAK antagonist or agonist results in some improvement in patient well-being in at least some patients).
- Inappropriate functional activity of syk and/or JAK might arise as the result of expression of syk and/or JAK in cells which normally do not express the receptor, greater than normal production of syk and/or JAK, or slower than normal metabolic inactivation or elimination of syk and/or JAK or its active metabolites, increased expression of syk and/or JAK or degree of intracellular activation (leading to, e.g., inflammatory and immune-related disorders and conditions) or decreased expression of syk and/or JAK.
- a condition or disorder associated with syk and/or JAK may include a " syk and/or JAK -mediated condition or disorder".
- a condition or disorder mediated at least in part by syk or JAK kinase activity refers to a condition or disorder characterized by inappropriate, e.g., greater than normal, syk and/or JAK activity. Inappropriate syk and/or JAK functional activity might arise as the result of syk and/or JAK expression in cells which normally do not express syk and/or JAK or increased syk and/or JAK expression or degree of intracellular activation (leading to, e.g., inflammatory and immune-related disorders and conditions).
- a condition or disorder mediated at least in part by syk or JAK kinase activity may be completely or partially mediated by inappropriate syk and/or JAK functional activity.
- a condition or disorder mediated at least in part by syk or JAK kinase activity is one in which modulation of syk and/or JAK results in some effect on the underlying condition or disorder (e.g., an syk and/or JAK antagonist results in some improvement in patient well-being in at least some patients).
- inflammation refers to infiltration of white blood cells (e.g., leukocytes, monocytes, etc.) into the area being treated for restenosis.
- white blood cells e.g., leukocytes, monocytes, etc.
- intervention refers to an action that produces an effect or that is intended to alter the course of a disease process.
- vascular intervention refers to the use of an intravascular procedure such as angioplasty or a stent to open an obstructed blood vessel.
- intravascular device refers to a device useful for a vascular recanalization procedure to restore blood flow through an obstructed blood vessel.
- intravascular devices include, without limitation, stents, balloon catheters, autologous venous/arterial grafts, prosthetic venous/arterial grafts, vascular catheters, and vascular shunts.
- JAK refers to a Janus kinase (RefSeq Accession No. P- 43408) or a variant thereof that is capable of mediating gene expression in vitro or in vivo.
- JAK variants include proteins substantially homologous to native JAK, i.e., proteins having one or more naturally or non-naturally occurring amino acid deletions, insertions or substitutions (e.g., JAK derivatives, homologs and fragments).
- the amino acid sequence of JAK variant preferably is at least about 80% identical to a native JAK, more preferably at least about 90% identical, and most preferably at least about 95% identical.
- leukocyte refers to any of the various blood cells that have a nucleus and cytoplasm, separate into a thin white layer when whole blood is centrifuged, and help protect the body from infection and disease.
- leukocytes include, without limitation, neutrophils, eosinophils, basophils, lymphocytes, and monocytes.
- mamal includes, without limitation, humans, domestic animals (e.g., dogs or cats), farm animals (cows, horses, or pigs), monkeys, rabbits, mice, and laboratory animals.
- modulate refers to the ability of a compound to increase or decrease the function and/or expression of syk and/or JAK, where such function may include transcription regulatory activity and/or protein-binding. Modulation may occur in vitro or in vivo. Modulation, as described herein, includes the inhibition, antagonism, partial antagonism, activation, agonism or partial agonism of a function or characteristic associated with syk and/or JAK, either directly or indirectly, and/or the upregulation or downregulation of the expression of syk and/or JAK, either directly or indirectly. In a preferred embodiment, the modulation is direct.
- Inhibitors or antagonists are compounds that, e.g., bind to, partially or totally block stimulation, decrease, prevent, inhibit, delay activation, inactivate, desensitize, or downregulate signal transduction.
- Activators or agonists are compounds that, e.g., bind to, stimulate, increase, open, activate, facilitate, enhance activation, activate, sensitize or upregulate signal transduction.
- the ability of a compound to inhibit the function of syk and/or JAK can be demonstrated in a biochemical assay, e.g., binding assay, or a cell-based assay, e.g., a transient transfection assay.
- Modulators of activity are used to refer to "ligands", “antagonists” and “agonists” identified using in vitro and in vivo assays for activity and their homologs and mimetics. Modulators include naturally occurring and synthetic ligands, antagonists, agonists, molecules and the like. Assays to identify antagonists and agonists include, e.g., applying putative modulator compounds to cells, in the presence or absence of a receptor of the invention and then determining the functional effects on a receptor of the invention activity. Samples or assays comprising a receptor of the invention that are treated with a potential activator, inhibitor, or modulator are compared to control samples without the inhibitor, activator, or modulator to examine the extent of effect.
- Control samples (untreated with modulators) are assigned a relative activity value of 100%. Inhibition is achieved when the activity value of a receptor of the invention relative to the control is about 80%, optionally 50% or 25-1%. Activation is achieved when the activity value of a receptor of the invention relative to the control is 1 10%, optionally 150%, optionally 200-500%, or 1000-3000% higher.
- "Patient” refers to human and non-human animals, especially mammals. Examples of patients include, but are not limited to, humans, cows, dogs, cats, goats, sheep, pigs and rabbits.
- the term "pharmaceutically acceptable carrier or excipient” means a carrier or excipient that is useful in preparing a pharmaceutical composition that is generally safe, non-toxic and neither biologically nor otherwise undesirable, and includes a carrier or excipient that is acceptable for veterinary use as well as human pharmaceutical use.
- a “pharmaceutically acceptable carrier or excipient” as used in the specification and claims includes both one and more than one such carrier or excipient.
- pharmaceutically effective amount refers to the amount of the subject compound that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician.
- therapeutically effective amount includes that amount of a compound that, when administered, is sufficient to prevent development of, or alleviate to some extent, one or more of the symptoms of the condition or disorder being treated. The therapeutically effective amount will vary depending on the compound, the disorder or condition and its severity and the age, weight, etc., of the mammal to be treated.
- platelet refers to a minute, nonnucleated, disklike cell found in the blood plasma of mammals that functions to promote blood clotting.
- prevent refers to a method of partially or completely delaying or precluding the onset or recurrence of a disorder or condition and/or one or more of its attendant symptoms or barring a subject from acquiring or reacquiring a disorder or condition or reducing a subject's risk of acquiring or reaquiring a disorder or condition or one or more of its attendant symptoms.
- recanalization refers to the process of restoring flow to or reuniting an interrupted channel of the body, such as a blood vessel.
- restenosis refers to a re-narrowing or blockage of an artery at the same site where treatment, such as an angioplasty or a stent procedure, has been performed.
- the phrase “selectively” or “specifically” when referring to binding to a receptor refers to a binding reaction that is determinative of the presence of the receptor, often in a heterogeneous population of receptors and other biologies. Thus, under designated conditions, the compounds bind to a particular receptor at least two times the background and more typically more than 10 to 100 times background. Specific binding of a compound under such conditions requires a compound that is selected for its specificity for a particular receptor.
- small organic molecules can be screened to obtain only those compounds that specifically or selectively bind to a selected receptor and not with other receptors or proteins.
- a variety of assay formats may be used to select compounds that are selective for a particular receptor. For example, High-throughput screening assays are routinely used to select compounds that are selective for a particular a receptor.
- the term "Sickle cell anemia” refers to an inherited disorder of the red blood cells in which both hemoglobin alleles encode the sickle hemoglobin (S) protein, i.e., the S/S genotype.
- S sickle hemoglobin
- the presence of abnormal hemoglobin results in the production of unusually shaped cells, which do not survive the usual length of time in the blood circulation. Thus, anemia results.
- Anemia refers to a decrease in the number of red blood cells and/or hemoglobin in the blood.
- sickle cell disease refers to an inherited disorder of the red blood cells in which one hemoglobin allele encodes the sickle hemoglobin (S) protein, and the other allele encodes another unusual hemoglobin protein, such as hemoglobin (S), (C), (D), (E), and ( ⁇ Thal).
- sickle cell disease genotypes include, without limitation, the S/S, S/C, S/D, S/E, and S/ ⁇ Thal genotypes.
- the most common types of sickle cell disease include sickle cell anemia, sickle-hemoglobin C disease, sickle beta-plus thalassemia, and sickle beta- zero thalassemia.
- the "subject” is defined herein to include animals such as mammals, including, but not limited to, primates (e.g., humans), cows, sheep, goats, horses, dogs, cats, rabbits, rats, mice and the like. In preferred embodiments, the subject is a human.
- syk refers to a spleen tyrosine kinase (RefSeq Accession No. P-043405) or a variant thereof that is capable of mediating a cellular response to T-cell receptors in vitro or in vivo
- syk variants include proteins substantially homologous to native syk, i.e., proteins having one or more naturally or non-naturally occurring amino acid deletions, insertions or substitutions (e.g., syk derivatives, homologs and fragments).
- the amino acid sequence of syk variant preferably is at least about 80% identical to a native syk, more preferably at least about 90% identical, and most preferably at least about 95% identical.
- syk inhibitor refers to any agent that inhibits the catalytic activity of spleen tyrosine kinase.
- thrombosis refers to the blockage or clotting of a blood vessel caused by a clumping of cells, resulting in the obstruction of blood flow.
- thrombosis refers to the clot that is formed within the blood vessel.
- treat includes partially or completely delaying, alleviating , mitigating or reducing the intensity of one or more attendant symptoms of a disorder or condition and/or alleviating, mitigating or impeding one or more causes of a disorder or condition.
- Treatments according to the invention may be applied preventively, prophylactically, pallatively or remedially.
- a blood vessel refers to any of the vessels through which blood circulates in the body.
- the lumen of a blood vessel refers to the inner open space or cavity of the blood vessel.
- the present invention provides in one embodiment, a compound having Formula (I):
- Y la is selected from the group consisting of N, CH and C;
- Z la is selected from the group consisting of a bond, -N(Ci_ 4 alkyl)-, -SO 2 -, -CO-, -NR SO 2 -, heterocyclyl, heterocyclylcarbonyl and heterocyclylsulfonyl;
- R la is selected from the group consisting of:
- Ci -8 alkyl optionally substituted with from 1 to 3 substituents selected from the group consisting of amino, hydroxy, Ci_ 8 alkoxy, heterocyclyl, aminocarbonyl, aminoCi. 8 alkoxy, aryl and heteroaryl;
- aryl optionally substituted with from 1 to 3 substituents selected from the group consisting of Ci-salkyl, Ci -8 alkoxy, Ci.galkylamino, Ci-salkylcarbonylamino; aminocarbonylCi- 8 alkoxy, aminosulfonyl, aminocarbonyl, aminoCi. 8 alkylene carbonylCi- 8 alkoxy and halo;
- heterocyclyl halogens, cyano, optionally substituted with from 1 to 3 substituents selected from the group consisting of Ci -8 alkyl, oxo and Ci_ 8 alkoxycarbonyl, cyano Ci_ 6 alkylcarbonyl, aminocarbonyl, arylCi. 4 alkoxycarbonyl, arylaminocarbonyl; and
- heteroaryl optionally substituted with from 1 to 3 substituents selected from the group consisting of Ci_ 8 alkyl, Ci_ 8 alkylsulfonyl and cyanoCi.galkylenecarbonyl;
- R 2a is H, Ci- 8 alkyl, or is taken together with R la and the nitrogen to which each is attached to form a heterocyclic or heteroaryl ring, containing 1-3 heteroatoms, including N, O or S, optionally substituted with from 1 to 2 substituents, R 2b , independently selected from the group consisting of Ci. 8 alkoxy, Ci -8 alkoxycarbonyl, Ci-salkoxycarbonylCi-salkylene, Cj- 8 alkoxycarbonylaminoCi_ 8 alkylene, amino, aminoCi -8 alkylene, aminoaryl, aminocarbonyl, aminocarbonylamino, aminocarbonylCi.
- R 3a is H, Ci.galkyl, or is taken together with the moiety R 4a -Z la and the nitrogen to which each is attached to form a heterocyclic or heteroaryl ring, optionally substituted with from 1 to 2 substituents R 2b and R 2c each of which is independently selected from the group consisting of Ci- 8 alkoxy, Ci.
- R 4a is selected from the group consisting of:
- aryl optionally substituted with from 1 to 3 substituents, R 4c , each of which is independently selected from the group consisting of Ci- 8 alkoxy, amino, Q-galkylcarbonyl and aminocarbonylCi. 8 alkoxy;
- heterocyclyl each of which is optionally substituted with from 1 to 3 substituents, R 4c , each of which is independently selected from the group consisting of Q-galkyl and oxo;
- R 4b is selected from the group consisting of H, Ci. 8 alkyl, aminoC
- gcycloalkylcarbonyl, halo, hydroxy, hydroxyCi_ 8 alkylene, aminoCi-galkyleneamino, aminoCi.salkylenecarbonyl, aminoCi.galkyleneaminocarbonylCi.galkylene, hydroxyCi.galkylenecarbonylamino, hydroxyC i -8 alkyleneamino, hydroxyC i , 8 alkyleneaminosulfonyl, hydroxyC i.salkyleneaminocarbonyl, oxo and heterocyclyl; if R 4b is heterocyclyl, it is optionally substituted with 1-3 substituents, R 4d , independently selected from the group consisting of Ci -8 alkyl, Ci_ 8 alkoxy, hydroxy, amino, halo, cyano, oxo, S, Ci- 8 alkoxycarbonyl, carboxy, aminocarbonyl, aminosulfonyl, d. 8 alkylcarbonyl and C
- R 5a is selected from the group consisting of H, Ci -8 alkyl, C 3 _ 8 cycloalkyl and Ci- 8 alkylarylsulfonyl;
- R 6a is selected from the group consisting of H, Ci_ 8 alkyl, halo, hydroxyl and oxo;
- R 7a is selected from the group consisting of H, Ci.galkyl, cyano, C 1-8 alkoxyCi_ 8 alkylene, C 2- 8 alkynyl, C 3 . 8 cycloalkyl, Q-galkylcarbonyl, hydroxy, oxo, halo and aryl, wherein each aryl and heteroaryl can be optionally substituted with halo, Ci- 8 alkyl, Ci -8 alkoxy, cyano, amino, hydroxyl, heteroaryl; and the dashed line indicates a double or single bond.
- R la is H. In another group of embodiments, R la is C
- R 2a is H. In another group of embodiments, R 2a is C]. 8 alkyl. In another group of embodiments, R 2a is taken together with R la and the nitrogen to which each is attached to form a heterocyclic ring selected from the group consisting of piperidine, piperazine, homopiperazine and pyrrolidine.
- each R is independently selected from the group consisting Of -CH 2 NH 2 , -CH 2 NHCH 3 , -CH 2 N(CH 3 ) 2 , -CH 2 CH 2 NH 2 , -C(CH 3 ) 2 NH 2 , -NH 2 , - OH, -CH 2 OH, -CH 2 CH 2 OH, -CH 2 NHC(O)NH 2 , -C(O)NH 2 and -CH 2 C(O)NH 2, - CH(NH 2 )CO 2 CH 3 , -CHNH 2 )CH 2 CO 2 Et.
- Y 1 is N. In another group of embodiments, Y la is CH. In another group of embodiments, Y la is C.
- R 3a is H. In another group of embodiments, R 3a is C
- R 4a is aryl. In another group of embodiments, R 4 is phenyl. In another group of embodiments, R 4a is heteroaryl. In another group of embodiments, R 4a is heterocyclyl.
- Z 1 is a bond. In another group of embodiments, Z 1 is -N(Ci -8 alkyl)-. In another group of embodiments, Z 1 is -SO 2 -. In another group of embodiments, Z 1 is -CO-. In another group of embodiments, Z 1 is -NR 40 SO 2 -. In another group of embodiments, Z 1 is heterocyclyl. In another group of embodiments, Z 1 is heterocyclylcarbonyl. In another group of embodiments, Z 1 is heterocyclylsulfonyl.
- R 5a is H.
- R 6a is H.
- R 7a is selected from the group consisting of H, cyano, phenyl and pyridyl.
- R 4b is selected from the group consisting of H, Ci_ 8 alkyl, Ci- 8 alkylcarbonyl, Ci_ 8 alkylcarbonylamino, Ci. 8 alkylsulfonyl, Ci. 8 alkylsulf ⁇ nyl, Ci-salkylsulfonylamino, Ci -8 alkylsulfonyl Ci -8 alkylene, Ci. 8 alkylthio, Ci-salkoxy, Ci.salkoxycarbonylamino, Ci -8 alkoxycarbonyl, Ci- 8 alkoxycarbonyl C
- the present invention provides in another embodiment, a compound, having the formula Ia:
- Z lb is heterocyclyl, -NR ⁇ SO 2 -, heterocyclylsulfonyl or a bond, R lb is selected from the group consisting of
- Ci_ 8 alkyl optionally substituted with amino, aminoCi -8 alkyl, aminocarbonyl, alkoxy, Ci- 8 alkylaminocarbonyl, aminoCi. 8 alkylene, aminoCi_ 8 alkoxy, aryl, hydroxy or heterocyclyl,
- aryl optionally substituted with alkyl, amino, aminosulfonyl, aminoCi_ 8 alkylene, Ci.salkylaminocarbonylCi-salkoxy, hydroxyl, Q-galkoxy, C )-8 alkyl aminocarbonyl, aminoCi. 8 alkylene, aminoCi-salkoxy or halo;
- heterocyclyl optionally substituted with Ci -8 alkyl, Ci -8 alkoxycarbonyl, amino, hydroxyl, C]. 8 alkoxy, Ci.salkylaminocarbonyl, aminoCi. 8 alkylene, aminoC
- heteroaryl optionally subsitituted with alkyl, amino, hydroxyl, Ci- 8 alkoxy, Ci -8 alkylaminocarbonyl, aminoCi. 8 alkylene or aminoC]. 8 alkoxy;
- R 4b is selected from the group consisting of H, hydroxy, Ci.salkylcarbonyl-, Ci.salkoxycarbonylCi-salkylene, Ci.salkoxycarbonylamino, aminocarbonylCi-galkylene, hydroxycarbonyl, hydroxycarbonylCi-salkylene, aminocarbonyl, aminoCi. 8 alkylene, hydroxy, hydroxyCi ⁇ alkylenecarbonylamino, C
- R 4d is H or C,. 8 alkyl
- R 5 is H or Ci. 8 alkyl
- R 6 is selected from the group consisting of H, Ci -8 alkyl, halo, hydroxy and oxo;
- the present invention provides in another embodiment, a compound, having the formula selected from the group consisting of:
- R lb is Ci-salkyl, optionally substituted with amino, heterocyclyl or aminoheterocyclyl, and C 3 . 8 cycloalkyl, optionally substituted with amino;
- R 7 is halo, hydroxy, Ci_ 8 alkoxyC
- R 6 is H, Ci-galkyl or halo;
- R 9 is H or C,. 8 alkyl; and
- R 10 is H, C,. 8 alkyl, C
- R Ib is Ci -8 alkyl, optionally substituted.
- R lb is C 3 . 8 cycloalkyl, optionally substituted.
- R lb is cyclohexyl, cyclopropyl or cyclobutyl, each of which is optionally substituted.
- R lb is heterocyclyl, optionally substituted.
- R lb is heterocyclyl, which is selected from the group consisting of piperidinyl, morpholino and tetrahydrothiophenyl.
- R lb is aryl.
- R lb is monocyclic heteroaryl.
- R lb is pyrazole.
- Z lb is heterocyclyl, which is selected from the group consisting of:
- R 8 is selected from the group consisting of H, Ci -8 alkyl, hydroxycarbonylCi. 8 alkylene, alkoxycarbonylCi_ 8 alkylene and C i . 8 alkylaminoC i .salkyleneaminocarbonylC i _ 8 alkylene.
- the present invention provides in another group of embodiments, a compound having the formula:
- R 2c is aminocarbonyl, aminoCi- 8 alkylene or hydroxy; and R 7 is selected from the group consisting of halo, hydroxy, Ci_ 8 alkoxyCi -8 alkylene, Q-salkylcarbonyl, cyano, phenyl and pyridinyl.
- the present invention provides in another group of embodiments, a compound having the formula:
- R Ib is Ci. 8 alkyl, C 3-8 cycloalkyl, aryl or heteroaryl; each of which is optionally substituted with hydroxy, Ci -8 alkoxy or aminoCi -8 alkoxy;
- R 4b is H or CH 3 CO-;
- R 7 is H, Ci_ 8 alkyl, Ci-salkylcarbonyl, aminocarbonyl, cyano, phenyl or pyridinyl;
- R 9 is H or C
- R 10 is H, Ci. 8 alkyl, Ci. 8 alkoxy, Ci.salkoxycarbonylCi.salkylene, aminoCi. 8 alkylene, aminocarbonylCi. 8 alkylene, carboxyCi_ 8 alkylene, C 3 _ 8 cycloalkyl and hydroxyCi. 8 alkylene.
- each R 2b is independently selected from the group consisting of amino, aminoCi -8 alkylene, aminocarbonyl, aminocarbonylCi-salkylene, carboxy, carboxyCi.galkylene, Ci. 8 alkoxycarbonyl, Ci-salkoxycarbonylCi-salkylene and hydroxy;
- R 4b is Ci -8 alkylcarbonyl
- Z lb is -heterocyclyl
- R 4c is H
- R 7b is H, cyano, pyridinyl or null; m is 0 or 1 ; and p is 0 or 1.
- R 2b is selected from the group consisting of H, aminoCi-salkylene, aminocarbonyl, aminocarbonylCi. 8 alkylene, hydroxy, hydroxycarbonylCi. 8 alkylene, Ci. 8 alkoxycarbonyl and C i -8 alkoxycarbonylC i _ 8 alkylene;
- R 4b is H or CH 3 CO-
- R 7b is H, cyano or pyridinyl; and m is 0 or 1.
- R 2b is selected from the group consisting of H, amino, aminoC 1-8 alkylene, aminocarbonyl, aminocarbonylC
- R 7a is H, C). 8 alkyl, halo, hydroxy, Ci. 8 alkoxyCi. 8 alkylene, Ci_ 8 alkylcarbonyl, cyano or pyridinyl; and m is 0 or 1.
- m is 0; and R 7a is selected from the group consisting of H, cyano or pyridinyl. In another group of embodiments, m is 1 ; and R 7a is selected from the group consisting of H, cyano or pyridinyl.
- R 4 w b i ⁇ s H or CH 3 CO-.
- R is selected from the group consisting of H, aminoC
- R 7b is H, cyano, or pyridinyl; and m is 0 or 1.
- R lc is selected from the group consisting of
- aryl each of which is optionally substituted with from 1 to 2 substituents selected from the group consisting of C
- R 2c is H or is taken together with R lc to form a heterocyclic ring, optionally cyanoCi. salkylcarbonyl, cyanoCi_ 6 alkylcarbonylamino substituted with from 1 to 2 substituents independently selected from the group consisting of aminocarbonylC
- R 3c is H or taken together with R ⁇ to form a heterocyclic or heteroaryl ring, each of which is optionally substituted with from 1 to 2 substituents independently selected from the group consisting of Q-salkyl, Ci-salkylheterocyclyl, aminoC
- R 40 is independently selected from the group consisting of
- heterocyclyl each of which is optionally substituted with from 1 to 3 substituents R 46 , R 4f and -Z lc R 4g , each of which is independently selected from the group consisting of Ci ⁇ alkyl, aminoCi_ 8 alkylene, hydroxyCi -8 alkylene, aminocarbonyl, Ci -8 alkoxycarbonyl, carboxy, amino, imino, Ci_ 8 alkylcarbonylamino, oxo, halo, aryl, heterocyclyl and heterocyclylCi-salkylene, provided that at least one of R lc and R 2c or R 3c and R 4d are combined with the nitrogen to which each is attached to form a heterocyclyl or heteroaryl ring; and
- R 7c is selected from the group consisting of H, Ci_ 8 alkyl, C 2 -salkynyl, cyano, aminocarbonyl, nitro, Ci- 8 alkoxy, halogen, , aryl, heteroaryl and C 3 . 8 cycloalkyl.
- the present invention provides in another group of embodiments, a compound having the formula Ha:
- R 3c is H.
- R 4d is phenyl. In another group of embodiments, R 4 * 1 is indazyl. In another group of embodiments, R 4d is cyclohexyl. In another group of embodiments, R 4d is dihydroindoyl.
- R 3c is is taken together with R 4d and the nitrogen to which is each is attached to form heterocyclic ring selected from the group consisting of pyridinyl, imidazolyl, tetrahydroimidazoyl, indazolyl, piperidinyl, piperazinyl, pyrrolidinyl, triazoyl and benzamidazoyl.
- R ⁇ R ⁇ N- is selected from the group consisting of: the dashed lined indicates a single or double bond; and the wavy line indicates the point of attachment to the remainder of the molecule.
- the present invention provides in another group of embodiments, a compound having the formula lib:
- R lc and R 2c are taken together to form a heterocyclic ring, optionally substituted with from 1 to 2 substituents independently selected from the group consisting of: aminocarbonylCi. 8 alkylene, Ci_ 8 alkoxycarbonyl, aminoC 1-8 alkylene, hydroxyCi_ 8 alkylene, Ci_ 8 alkoxycarbonylaminoCi. 8 alkylene, amino, aminocarbonylaminocarbonylamino, arylCi -8 alkoxycarbonylamino, aminocarbonylamino and oxo;
- Z lc is heterocyclyl, -N(C,_ 4 alkyl)-, -SO 2 - or -CO-;
- R 4g is selected from the group consisting of H, Ci_ 8 alkoxy, Ci -8 alkylcarbonyl, Ci. 8 alkylcarbonylamino, amino, aminocarbonylaminoCi. 8 alkylene, aminocarbonyl and aminosulfonyl; each R ⁇ and R 4f is independently selected from the group consisting of H-, Ci. 8 alkoxy and Ci. 8 alkylcarbonyl; or can be taken together with -Z lc -R 4g and the benzene ring to which each is attached to form a fused heterocyclic ring system, optionally substituted with from 1 to 2 oxo substituents, halogens, Ci -8 alkyl, Ci. 8 alkyl.
- the present invention provides in another group of embodiments, a compound, wherein the moiety
- R 7c is selected from the group consisting of F, Cl, Br, cyano and aminocarbonyl. In another group of embodiments, R 7c is CONH 2 or F. In another group of embodiments, R lc is C 3 - 8 cycloalkyl. In another group of embodiments, R lc is cyclohexyl. In another group of embodiments, R lc is cyclopropyl. In another group of embodiments, R lc is cyclobutyl. In another group of embodiments, R lc is aryl. In another group of embodiments, R lc is phenyl.
- R 2c is taken together with R lc and the nitrogen to which each is attached to form a pyrrolidinyl, piperidinyl or piperazinyl, diazapenyl ring.
- the present invention provides in another group of embodiments, a compound having the formula: wherein Y 2b is N, NH, C or CH; R 2d is H, Ci_ 8 alkylaminoCi. 8 alkylene, aminocarbonyl, aminocarbonylamino, aminocarbonylaminocarbonylamino, amino, oxo, aminoC
- R 7c is H, halo, aminocarbonyl, cycloalkyl, cyano or pyridinyl
- R 9 is H or Ci- 8 alkyl
- R 10 is H, Ci_ 8 alkyl, Ci- 8 alkoxy, Ci-salkoxycarbonylCi-salkylene, aminoCi. 8 alkylene, aminocarbonylCi- 8 alkylene, carboxyCi. 8 alkylene, C 3 . 8 cycloalkyl and hydroxyCi.galkylene
- m is 0 or 1
- q is 0 or 1.
- R 7c is selected from the group consisting of F, Cl, Br, cyano and aminocarbonyl. In another group of embodiments, R 7c is CONH 2 or F.
- the present invention provides in another group of embodiments, a compound having the formula: or a tautomer or a pharmaceutically acceptable salt thereof.
- the present invention provides in another embodiment, a compound selected from the group consisting of N2-(lH-indazol-6-yl)-N4-methyl-5-(pyridin-4-yl)-7H-pyrrolo[2,3- d]pyrimidine-2,4-diamine; l-(4-(4-(4-(methylamino)-5-(pyridin-4-yl)-7H-pyrrolo[2,3- d]pyrimidin-2-ylamino)phenyl)piperazin- 1 -yl)ethanone; N2-( 1 H-indazol-6-yl)-N4-methyl-5- (pyridin-3-yl)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine; l-(4-(4-(4-(4-(4-(cyclopropylamino)-5- (pyridin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-2-yla
- the present invention provides in another embodiment, a compound selected from the group consisting of l-(4-(4-(cyclobutylamino)-7H-pyrrolo[2,3-d]pyrimidin-2- ylamino)phenylsulfonyl)piperidin-4-ol; butyl 2-(4-(4-(cyclobutylamino)-7H-pyrrolo[2,3- d]pyrimidin-2-ylamino)-N-methylphenylsulfonamido)acetate; 2-(4-(4-(cyclobutylamino)-7H- pyrrolo[2,3-d]pyrimidin-2-ylamino)phenylsulfonamido)acetarnide; 2-(4-(4- (cyclobutylamino)-7H-pyrrolo[2,3-d]pyrimidin-2-ylamino)-N- methylphenylsulfonamido)acetic acid;
- the present invention provides in another embodiment, a compound of the examples.
- the present invention provides in another embodiment, a compound of any one of the tables. [0281] The present invention provides in another embodiment, a compound of any one of the figures.
- the compounds of the present invention may be prepared by known organic synthesis techniques, including the methods described in more detail in the Examples.
- the compounds of structure (I) above may be made by the following Figure 1, wherein all substituents are as defined above unless indicated otherwise.
- Compounds having formula II may be prepared according to Figure 2.
- Carboxylic acid 2.1 is converted to acid chloride 2.2 via a one-step procedure by treatment with a chlorination agent, such as thionyl chloride, and esterification with an alcohol, such as ethanol, to form compound 2.3 using conditions similar to that described below.
- Ester 2.3 is dichlorinated with a chlorinating agent, such as phosphorous oxychloride.
- a chlorinating agent such as phosphorous oxychloride.
- Benzotriazolyl ether compound 2.7 may also be prepared through a linear route. Displacement of the benzotriazolyl ether group with an appropriate amine, such as R 4 -NH.R 3 (available commercially or synthesized using methods known to those skilled in the art), gives the desired product Ha, wherein R 1 , R 2 , R 3 and R 4 are as previously defined.
- Compounds having formula II may be prepared according to Figure 3. Selective displacement of the 4-chloro group of the 2,4-dichloropyrimidine by an appropriate amine, such as R 2 -NH-R' (available commercially or synthesized using methods known to those skilled in the art), under basic conditions, such as with diisopropylamine (DIA), provides compounds of formula 3.2. Displacement of the chloro group with an appropriate amine, such as R 4 -NH-R 3 (available commercially or synthesized using methods known to those skilled in the art), gives the desired product lib, wherein R 1 , R 2 , R 3 and R 4 are as previously defined.
- an appropriate amine such as R 2 -NH-R' (available commercially or synthesized using methods known to those skilled in the art)
- DIA diisopropylamine
- the compounds of the present invention may generally be utilized as the free base.
- the compounds of this invention may be used in the form of acid addition salts as described below.
- the activity of a specified compound as an inhibitor of a JAK kinase may be assessed in vitro or in vivo.
- the activity of a specified compound can be tested in a cellular assay. Selectivity could also be ascertained in biochemical assays with isolated kinases.
- Similar types of assays can be used to assess JAK kinase inhibitory activity and to determine the degree of selectivity of the particular compound as compared to syk kinase.
- One means of assaying for such inhibition is detection of the effect of the compounds of the present invention on the upregulation of downstream gene products.
- B-cells are stimulated with the cytokine Interleukin-4 (IL-4) leading to the activation of the JAK/Stat pathway through phosphorylation of the JAK family kinases, JAKl and JAK3, which in turn phosphorylate and activate the transcription factor Stat-6.
- IL-4 cytokine Interleukin-4
- One of the genes upregulated by activated Stat-6 is the low affinity IgE receptor, CD23.
- human Ramos B-cells are stimulated with human IL-4. 10' post- stimulation, cells are subjected to intracellular flow cytometry to measure the extent of STAT-6 phosphorylation. 20 to 24 hours post-stimulation, cells are stained for upregulation of CD23 and analyzed using flow cytometry. A reduction of the amount of phospohorylated STAT-6 and/or cell surface CD23 present compared to control conditions indicates that the test compound actively inhibits the JAK kinase pathway.
- inhibitors e.g., the 2,4-substituted pyrimindinediamine compounds described herein
- IL-6 stimulation of Ramos B-cells induces JAKs 1, 2, and Tyk2, leading to Stat-3 and Erk phosphorylation.
- JAKs 1, 2, and Tyk2 leading to Stat-3 and Erk phosphorylation.
- cells are subjected to intracellular flow cytometry to measure the ability of compound to inhibit these phosphorylation events.
- JAK2 the CellSensor irfl-bla HEL cell line expressing the beta-lactamase reporter gene controlled by Stat5 will be used (Invitrogen, Carlsbad, CA).
- JAK2 inhibitory activity of compounds.
- the activity of the compounds of the invention may additionally be characterized by assaying the effect of the compounds of the present invention described herein on A549 lung epithelial cells and U937 cells.
- A549 lung epithelial cells and U937 cells up-regulate ICAM-I (CD54) surface expression in response to a variety of different stimuli. Therefore, using ICAM-I expression as readout, test compound effects on different signaling pathways can be assessed in the same cell type.
- Stimulation with IL- l ⁇ through the IL-I ⁇ receptor activates the TRAF6/NF ⁇ B pathway resulting in up-regulation of ICAM-I.
- IFN. gamma induces ICAM-I up-regulation through activation of the JAK1/JAK2 pathway.
- the up-regulation of ICAM-I can be quantified by flow cytometry across a compound dose curve and EC 50 values are calculated.
- the activity of the compounds of the invention may additionally be characterized by assaying the effect of the compounds of the present invention described herein on A549 lung epithelial cells and U937 cells.
- A549 lung epithelial cells and U937 cells up-regulate ICAM-I (CD54) surface expression in response to a variety of different stimuli. Therefore, using ICAM-I expression as readout, test compound effects on different signaling pathways can be assessed in the same cell type.
- Stimulation with IL- l ⁇ through the IL-I ⁇ receptor activates the TRAF6/NF ⁇ B pathway resulting in up-regulation of ICAM-I.
- IFN. gamma induces ICAM-I up-regulation through activation of the JAK1/JAK2 pathway.
- the up-regulation of ICAM-I can be quantified by flow cytometry across a compound dose curve and EC50 values are calculated. Exemplary assays of this type are described in greater detail in the Examples.
- Active compounds as described herein generally inhibit the JAK kinase pathway with an IC 50 in the range of about 1 mM or less, as measured in the assays described herein.
- IC 50 in the range of about 1 mM or less, as measured in the assays described herein.
- compounds which exhibit lower IC 5 oS (on the order, for example, of 100 ⁇ M, 75 ⁇ M, 50 ⁇ M, 40 ⁇ M, 30 ⁇ M, 20 ⁇ M, 15 ⁇ M, 10 ⁇ M, 5 ⁇ M, 1 ⁇ M, 500 nM, 100 nM, 10 nM, 1 nM, or even lower) can be particularly useful in therapeutic applications.
- the compound can be assayed for activity with the desired cell type and counter- screened for a lack of activity against other cell types.
- the desired degree of "inactivity" in such counter screens, or the desired ratio of activity vs. inactivity, may vary for different situations and can be selected by the user.
- the active compounds also typically inhibit IL-4 stimulated expression of CD23 in B-cells with an IC 50 in the range of about 20 ⁇ M or less, typically in the range of about 10 ⁇ M, 1 ⁇ M, 500 nM, 100 nM, 10 nM, 1 nM, or even lower.
- a suitable assay that can be used is the assay described in the Examples, "Assay for Ramos B-cell Line Stimulated with IL-4.”
- the active compounds of the present invention have an IC 50 of less than or equal to 5 ⁇ M, greater than 5 ⁇ M but less than 20 ⁇ M, greater than 20 ⁇ M, or greater than 20 ⁇ M but less than 50 ⁇ M in the assay described in the Examples.
- the active compounds also typically inhibit expression of ICAMl (CD54) induced by PEN. gamma, exposure in U937 or A549 cells with an IC 50 in the range of about 20 ⁇ M or less, typically in the range of about 10 ⁇ M, 1 ⁇ M, 500 nM, 100 nM, 10 nM, 1 nM, or even lower.
- the IC 50 against expression of ICAM (CD54) in IFN. gamma, stimulated cells can be determined in a functional cellular assay with an isolated A549 or U937 cell line.
- Suitable assays that can be used are the assays described in the Examples, "A549 Epithelial Line Stimulated with BFN ⁇ ” and “U937 EFN.gamma. ICAMl FACS Assay," respectively.
- the active compounds of the present invention have an IC 50 of less than or equal to 20 ⁇ M, greater than 20 ⁇ M, or greater than 20 ⁇ M but less than 50 ⁇ M in the assays described in the Examples.
- the present invention further provides compositions comprising one or more compounds of formula (I) or a pharmaceutically acceptable salt, ester or prodrug thereof, and a pharmaceutically acceptable carrier or diluent.
- the compounds of formula (I)) in this invention may be derivatized at functional groups to provide prodrug derivatives which are capable of conversion back to the parent compounds in vivo.
- prodrugs include the physiologically acceptable and metabolically labile ester derivatives, such as methoxymethyl esters, methylthiomethyl esters, or pivaloyloxymethyl esters derived from a hydroxyl group of the compound or a carbamoyl moiety derived from an amino group of the compound.
- any physiologically acceptable equivalents of the compounds of formula (I), similar to metabolically labile esters or carbamates, which are capable of producing the parent compounds of formula (I) in vivo are within the scope of this invention.
- salts refers to any acid or base addition salt whose counter-ions are non-toxic to the patient in pharmaceutical doses of the salts.
- a host of pharmaceutically acceptable salts are well known in the pharmaceutical field. If pharmaceutically acceptable salts of the compounds of this invention are utilized in these compositions, those salts are preferably derived from inorganic or organic acids and bases.
- acid salts include the following: acetate, adipate, alginate, aspartate, benzoate, benzene sulfonate, bisulfate, butyrate, citrate, camphorate, camphor sulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, lucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, methanesulfonate, 2- naphthalenesulfonate, nicotinate, oxalate, pamoate, pectinate, persulfate, 3-phenyl- propionate, picrate, pivalate, propionate, succinate, tartrate,
- Pharmaceutically acceptable base addition salts include, without limitation, those derived from alkali or alkaline earth metal bases or conventional organic bases, such as triethylamine, pyridine, piperidine, morpholine, N-methylmorpholine, ammonium salts, alkali metal salts, such as sodium and potassium salts, alkaline earth metal salts, such as calcium and magnesium salts, salts with organic bases, such as dicyclohexylamine salts, N-methyl-D-glucamine, and salts with amino acids such as arginine, lysine, and so forth.
- alkali or alkaline earth metal bases or conventional organic bases such as triethylamine, pyridine, piperidine, morpholine, N-methylmorpholine, ammonium salts, alkali metal salts, such as sodium and potassium salts, alkaline earth metal salts, such as calcium and magnesium salts, salts with organic bases, such as dicyclohexylamine salts, N-methyl-D
- the basic nitrogen-containing groups may be quaternized with agents like lower alkyl halides, such as methyl, ethyl, propyl and butyl chlorides, bromides and iodides; dialkyl sulfates, such as dimethyl, diethyl, dibutyl and diamyl sulfates, long chain halides, such as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides; aralkyl halides, such as benzyl and phenethyl bromides and others. Water or oil-soluble or dispersible products are thereby obtained.
- lower alkyl halides such as methyl, ethyl, propyl and butyl chlorides, bromides and iodides
- dialkyl sulfates such as dimethyl, diethyl, dibutyl and diamyl sulfates, long chain halides, such as de
- compositions and methods of this invention may also be modified by appending appropriate functionalities to enhance selective biological properties.
- modifications are known in the art and include those which increase biological penetration into a given biological system (e.g., blood, lymphatic system, central nervous system, etc.), increase oral availability, increase solubility to allow administration by injection, alter metabolism and alter rate of excretion.
- compositions of the invention can be manufactured by methods well known in the art such as conventional granulating, mixing, dissolving, encapsulating, lyophilizing, or emulsifying processes, among others.
- Compositions may be produced in various forms, including granules, precipitates, or particulates, powders, including freeze dried, rotary dried or spray dried powders, amorphous powders, tablets, capsules, syrup, suppositories, injections, emulsions, elixirs, suspensions or solutions.
- Formulations may optionally contain stabilizers, pH modifiers, surfactants, bioavailability modifiers and combinations of these.
- unit dosage form refers to physically discrete units suitable as unitary dosages for human subjects and other mammals, each unit containing a predetermined quantity of drug calculated to produce the desired onset, tolerability, and/or therapeutic effects, in association with a suitable pharmaceutical excipient (e.g., an ampoule).
- a suitable pharmaceutical excipient e.g., an ampoule
- more concentrated compositions may be prepared, from which the more dilute unit dosage compositions may then be produced.
- the more concentrated compositions thus will contain substantially more than, e.g., at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, or more times the amount of one or more syk and/or JAK inhibitors.
- compositions typically include a conventional pharmaceutical carrier or excipient and may additionally include other medicinal agents, carriers, adjuvants, diluents, tissue permeation enhancers, solubilizers, and the like.
- the composition will contain about 0.01% to about 90%, preferably about 0.1% to about 75%, more preferably about 0.1% to 50%, still more preferably about 0.1% to 10% by weight of one or more syk and/or JAK inhibitors, with the remainder consisting of suitable pharmaceutical carrier and/or excipients.
- Appropriate excipients can be tailored to the particular composition and route of administration by methods well known in the art, e.g., REMINGTON'S PHARMACEUTICAL SCIENCES, supra.
- compositions include ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances, such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose- based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene-polyoxypropylene-block polymers, polyethylene glycol and wool fat.
- buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen
- excipients include, but are not limited to, lactose, dextrose, sucrose, sorbitol, mannitol, starches, gum acacia, calcium phosphate, alginates, tragacanth, gelatin, calcium silicate, microcrystalline cellulose, polyvinylpyrrolidone, cellulose, water, saline, syrup, methylcellulose, ethylcellulose, hydroxypropylmethylcellulose, and polyacrylic acids such as Carbopols.
- compositions can additionally include lubricating agents such as talc, magnesium stearate, and mineral oil; wetting agents; emulsifying agents; suspending agents; preserving agents such as methyl-, ethyl-, and propyl-hydroxy-benzoates; pH adjusting agents such as inorganic and organic acids and bases; sweetening agents; and flavoring agents.
- lubricating agents such as talc, magnesium stearate, and mineral oil
- wetting agents such as talc, magnesium stearate, and mineral oil
- emulsifying agents such as methyl-, ethyl-, and propyl-hydroxy-benzoates
- pH adjusting agents such as inorganic and organic acids and bases
- sweetening agents such as inorganic and organic acids and bases
- flavoring agents such as talc, magnesium stearate, and mineral oil.
- Administration of a composition comprising one or more syk and/or JAK inhibitors with one or more suitable pharmaceutical excipients as advantageous can be carried out via any of the accepted modes of administration.
- administration can be, for example, oral, topical, intravenous, subcutaneous, transcutaneous, transdermal, intramuscular, intra-joint, parenteral, intra-arteriole, intradermal, intraventricular, intracranial, intraperitoneal, intralesional, intranasal, rectal, vaginal, by inhalation or via an implanted reservoir.
- parenteral includes subcutaneous, intravenous, intramuscular, intra-articular, intra-synovial, intrasternal, intrathecal, intrahepatic, intralesional and intracranial injection or infusion techniques.
- the compositions are administered orally or intravenously.
- the formulations of the invention may be designed as short-acting, fast-releasing, or long- acting.
- compounds can be administered in a local rather than systemic means, such as administration (e.g., injection) as a sustained release formulation.
- the compositions of this invention are formulated for pharmaceutical administration to a mammal, preferably a human being.
- compositions of the present invention containing one or more syk and/or JAK inhibitors can be administered repeatedly, e.g., at least 2, 3, 4, 5, 6, 7, 8, or more times, or the composition may be administered by continuous infusion.
- Suitable sites of administration include, but are not limited to, skin, bronchial, gastrointestinal, anal, vaginal, eye, and ear.
- the formulations may take the form of solid, semi-solid, lyophilized powder, or liquid dosage forms, such as, for example, tablets, pills, capsules, powders, solutions, suspensions, emulsions, suppositories, retention enemas, creams, ointments, lotions, gels, aerosols, or the like, preferably in unit dosage forms suitable for simple administration of precise dosages.
- compositions of this invention may be in any orally acceptable dosage form, including tablets, capsules, cachets, emulsions, suspensions, solutions, syrups, elixirs, sprays, boluses, lozenges, powders, granules, and sustained-release formulations.
- Suitable excipients for oral administration include pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharine, talcum, cellulose, glucose, gelatin, sucrose, magnesium carbonate, and the like.
- carriers that are commonly used include lactose and corn starch.
- Lubricating agents such as magnesium stearate, are also typically added.
- useful diluents include lactose and dried cornstarch.
- the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening, flavoring or coloring agents may also be added.
- the compositions take the form of a pill, tablet, or capsule, and thus, the composition can contain, along with one or more syk and/or JAK inhibitors, a diluent such as lactose, sucrose, dicalcium phosphate, and the like; a disintegrant such as starch or derivatives thereof; a lubricant such as magnesium stearate and the like; and/or a binder such a starch, gum acacia, polyvinylpyrrolidone, gelatin, cellulose and derivatives thereof.
- a tablet can be made by any compression or molding process known to those of skill in the art.
- Compressed tablets may be prepared by compressing in a suitable machine the syk and/or JAK inhibitors in a free-flowing form, e.g., a powder or granules, optionally mixed with accessory ingredients, e.g., binders, lubricants, diluents, disintegrants, or dispersing agents. Molded tablets can be made by molding in a suitable machine a mixture of the powdered syk and/or JAK inhibitors with any suitable carrier.
- compositions of this invention may be in the form of suppositories for rectal administration. These may be prepared by mixing the agent with a suitable non-irritating excipient which is solid at room temperature but liquid at rectal temperature and therefore will melt in the rectum to release the drug. Such materials include cocoa butter, beeswax, polyethylene glycol (PEG), hard fat, and/or hydrogenated cocoglyceride.
- a suitable non-irritating excipient which is solid at room temperature but liquid at rectal temperature and therefore will melt in the rectum to release the drug.
- Such materials include cocoa butter, beeswax, polyethylene glycol (PEG), hard fat, and/or hydrogenated cocoglyceride.
- Compositions suitable for rectal administration may also comprise a rectal enema unit containing one or more syk and/or JAK inhibitors and pharmaceutically- acceptable vehicles (e.g., 50% aqueous ethanol or an aqueous salt solution) that are physiologically compatible with the
- the rectal enema unit contains an applicator tip protected by an inert cover, preferably comprised of polyethylene, lubricated with a lubricant such as white petrolatum, and preferably protected by a one-way valve to prevent back-flow of the dispensed formula.
- the rectal enema unit is also of sufficient length, preferably two inches, to be inserted into the colon via the anus.
- Liquid compositions can be prepared by dissolving or dispersing one or more syk and/or JAK inhibitors and optionally one or more pharmaceutically acceptable adjuvants in a carrier such as, for example, aqueous saline, aqueous dextrose, glycerol, ethanol, and the like, to form a solution or suspension, e.g., for oral, topical, or intravenous administration.
- a carrier such as, for example, aqueous saline, aqueous dextrose, glycerol, ethanol, and the like
- Pharmaceutical formulations may be prepared as liquid suspensions or solutions using a sterile liquid, such as oil, water, alcohol, and combinations thereof.
- Pharmaceutically suitable surfactants, suspending agents or emulsifying agents may be added for oral or parenteral administration.
- Suspensions may include oils, such as peanut oil, sesame oil, cottonseed oil, corn oil and olive oil.
- Suspension preparation may also contain esters of fatty acids, such as ethyl oleate, isopropyl myristate, fatty acid glycerides and acetylated fatty acid glycerides.
- Suspension formulations may include alcohols, such as ethanol, isopropyl alcohol, hexadecyl alcohol, glycerol and propylene glycol.
- Ethers such as poly(ethyleneglycol), petroleum hydrocarbons, such as mineral oil and petrolatum, and water may also be used in suspension formulations.
- compositions of this invention may also be in a topical form, especially when the target of treatment includes areas or organs readily accessible by topical application, including diseases of the eye, the skin, or the lower intestinal tract. Suitable topical formulations are readily prepared for each of these areas or organs.
- the composition containing one or more syk and/or JAK inhibitors can be in the form of emulsions, lotions, gels, foams, creams, jellies, solutions, suspensions, ointments, and transdermal patches.
- Topical application for the lower intestinal tract may be effected in a rectal suppository formulation (see above) or in a suitable enema formulation. Topically- transdermal patches may also be used.
- the pharmaceutical compositions may be formulated in a suitable ointment containing the active component suspended or dissolved in one or more carriers.
- Carriers for topical administration of the compounds of this invention include, but are not limited to, mineral oil, liquid petrolatum, white petrolatum, propylene glycol, polyoxyethylene, polyoxypropylene compound, emulsifying wax and water.
- the pharmaceutical compositions may be formulated in a suitable lotion or cream containing the active components suspended or dissolved in one or more pharmaceutically acceptable carriers. Suitable carriers include mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters, wax, cetyl alcohol, 2- octyldodecanol, benzyl alcohol and water.
- compositions of this invention may also be administered by nasal aerosol or inhalation.
- the compositions can be delivered as a dry powder or in liquid form via a nebulizer.
- Such compositions are prepared according to techniques known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons and/or other conventional solubilizing or dispersing agents.
- the pharmaceutical compositions may be formulated as micronized suspensions in isotonic, pH adjusted sterile saline, or, preferably, as solutions in isotonic, pH adjusted sterile saline, either with our without a preservative, such as benzylalkonium chloride.
- the pharmaceutical compositions may be formulated in an ointment, such as petrolatum.
- compositions can be in the form of sterile injectable solutions and sterile packaged powders.
- injectable solutions are formulated at a pH of about 4.5 to about 7.5.
- Sterile injectable forms of the compositions of this invention may be aqueous or oleaginous suspension. These suspensions may be formulated according to techniques known in the art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a nontoxic parenterally acceptable diluent or solvent, for example as a solution in 1,3-butanediol.
- the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose, any bland fixed oil may be employed including synthetic mono- or di-glycerides.
- Fatty acids such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions.
- These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant, such as carboxymethyl cellulose or similar dispersing agents which are commonly used in the formulation of pharmaceutically acceptable dosage forms including emulsions and suspensions.
- a long-chain alcohol diluent or dispersant such as carboxymethyl cellulose or similar dispersing agents which are commonly used in the formulation of pharmaceutically acceptable dosage forms including emulsions and suspensions.
- Other commonly used surfactants such as Tweens, Spans and other emulsifying agents or bioavailability enhancers which are commonly used in the manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms may also be used for the purposes of formulation.
- compositions of the present invention can also be provided in a lyophilized form.
- Such compositions may include a buffer, e.g., bicarbonate, for reconstitution prior to administration, or the buffer may be included in the lyophilized composition for reconstitution with, e.g., water.
- the lyophilized composition may further comprise a suitable vasoconstrictor, e.g., epinephrine.
- the lyophilized composition can be provided in a syringe, optionally packaged in combination with the buffer for reconstitution, such that the reconstituted composition can be immediately administered to a patient.
- a therapeutically effective dose may vary depending upon the route of administration and dosage form.
- the representative compound or compounds of the invention is a formulation that exhibits a high therapeutic index.
- the therapeutic index is the dose ratio between toxic and therapeutic effects which can be expressed as the ratio between LD 5O and ED 5O .
- the LD 50 is the dose lethal to 50% of the population and the ED 50 is the dose therapeutically effective in 50% of the population.
- the LD 50 and ED 50 are determined by standard pharmaceutical procedures in animal cell cultures or experimental animals.
- compositions are generally known to those skilled in the art and are included in the invention. It should be understood that a specific dosage and treatment regimen for any particular patient will depend upon a variety of factors, including the activity of the specific compound employed, the age, body weight, general health, sex and diet of the patient, and the time of administration, rate of excretion, drug combination, judgment of the treating physician and severity of the particular disease being treated. The amount of active ingredient(s) will also depend upon the particular compound and other therapeutic agent, if present, in the composition.
- the invention provides methods of inhibiting or decreasing syk and/or JAK activity as well as treating or ameliorating a syk and/or JAK associated state, symptom, condition, disorder or disease in a patient in need thereof (e.g., human or non-human).
- a syk and/or JAK associated state, symptom, condition, disorder or disease is mediated, at least in part by syk and/or JAK kinase activity.
- the present invention provides a method for treating a condition or disorder mediated at least in part by syk and/or JAK kinase activity is cardiovascular disease, inflammatory disease or autoimmune disease.
- the invention provides methods for preventing or treating a condition in a mammal characterized by undesired thrombosis comprising the step of administering to the mammal a therapeutically effective amount of a compound of the present invention.
- Such conditions include, but are not limited, to restenosis, acute coronary syndrome, myocardial infarction, unstable angina, refractory angina, occlusive coronary thrombosis occurring post-thrombolytic therapy or post-coronary angioplasty, a thrombotically mediated cerebrovascular syndrome, embolic stroke, thrombotic stroke, transient ischemic attacks, venous thrombosis, deep venous thrombosis, pulmonary embolism, coagulopathy, disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, thromboangiitis obliterans, thrombotic disease associated with heparin-induced thrombocytopenia, thrombotic complications associated with extracorporeal circulation, thrombotic complications associated with instrumentation such as cardiac or other intravascular catheterization, intra-aortic balloon pump, coronary stent or cardiac valve, conditions requiring the fitting of prosthetic devices, and the like.
- the present invention provides a method for treating thrombosis, immune thrombocytic purura, heparin induced thrombocytopenia, dilated cardiomypathy, sickle cell disease, atherosclerosis, myocardial infarction, vacular inflammation, unstable angina or acute coronary syndromes.
- the present invention also provides a method for treating allergy, asthma, theumatoid arthritis, B Cell mediated disease such as Non-Hodgkin's Lymphoma, and phospholipids syndrome, lupus, psoriasis, multiple sclerosis, end stage renal disease or chronic lymphocytic leukemia.
- the present invention provides a method for treating hemolytic anemia or immune thrombocytopenic purpura.
- the compounds described herein are also potent and/or selective inhibitors of JAK kinases.
- the compounds can be used in a variety of in vitro, in vivo, and ex vivo contexts to regulate or inhibit JAK kinase activity, signaling cascades in which JAK kinases play a role, and the biological responses effected by such signaling cascades.
- the compounds can be used to inhibit JAK kinase, either in vitro or in vivo, in virtually any cell type expressing the JAK kinase, such as in hematopoietic cells in which, for example, JAK3 is predominantly expressed.
- JAK-dependent signal transduction cascades include, but are not limited to, the signaling cascades of cytokine receptors that involve the common gamma chain, such as, for example, the IL-4, IL-7, IL-5, IL-9, IL- 15 and IL-21, or JL-2, JL-4, IL-7, IL-9, IL-15, and IL-21 receptor signaling cascades.
- the compounds may also be used in vitro or in vivo to regulate, and in particular to inhibit, cellular or biological responses affected by such JAK-dependent signal transduction cascades.
- Such cellular or biological responses include, but are not limited to, IL-4/ramos CD23 upregulation and IL-2 mediated T-cell proliferation.
- the compounds can be used to inhibit JAK kinases in vivo as a therapeutic approach towards the treatment or prevention of diseases mediated, either wholly or in part, by a JAK kinase activity (referred to herein as "JAK kinase mediated diseases").
- Non-limiting examples of JAK kinase mediated diseases that can be treated or prevented with the compounds include, but are not limited to, the following: allergies; asthma; autoimmune diseases such as transplant rejection (e.g., kidney, heart, lung, liver, pancreas, skin, small intestine, large intestine, host versus graft reaction (HVGR), and graft versus host reaction (GVHR)), rheumatoid arthritis, and amyotrophic lateral sclerosis; T-cell mediated autoimmune diseases such as multiple sclerosis, psoraiasis, and Sjogren's syndrome; Type II inflammatory diseases such as vascular inflammation (including vasculitis, arteritis, atherosclerosis, and coronary artery disease); diseases of the central nervous system such as stroke; pulmonary diseases such as bronchitis obliteraus and primary pulmonary hypertension; solid, delayed Type IV hypersensitivity reactions; and hematologic malignancies such as leukemia and lymphomas.
- transplant rejection
- Examples of diseases that are mediated, at least in part, by JAK kinases that can be treated or prevented according to the methods include, but are not limited to, allergies, asthma, autoimmune diseases such as transplant rejection (e.g., kidney, heart, lung, liver, pancreas, skin, host versus graft reaction (HVGR), etc.), rheumatoid arthritis, and amyotrophic lateral sclerosis, multiple sclerosis, psoraiasis and Sjogren's syndrome, Type II inflammatory disease such as vascular inflammation (including vasculitis, ateritis, atherosclerosis and coronary artery disease) or other inflammatory diseases such as osteoarthritis, inflammatory bowel disease, ulcerative colitis, Crohn's disease, idiopathic inflammatory bowel disease, irritable bowel syndrome, spastic colon, low grade scarring (e.g., scleroderma, increased fibrosis, keloids, post-surgical scars, pulmonary fibro
- this invention provides a method of inhibiting an activity of a JAK kinase, comprising contacting the JAK kinase with an amount of a compound effective to inhibit an activity of the JAK kinase, wherein the compound is selected from the compounds of this invention.
- the method is carried out in vivo.
- this invention provides a method of inhibiting an activity of a JAK kinase, comprising contacting in vitro a JAK3 kinase with an amount of a compound effective to inhibit an activity of the JAK kinase, wherein the compound is selected from the compounds of this invention.
- the compounds can be used to treat and/or prevent rejection in organ and/or tissue transplant recipients (i.e., treat and/or prevent allorgraft rejection). Allografts can be rejected through either a cell-mediated or humoral immune reaction of the recipient against transplant (histocompability) antigens present on the membranes of the donor's cells. The strongest antigens are governed by a complex of genetic loci termed human leukocyte group A (HLA) antigens. Together with the ABO blood groups antigens, they are the chief transplantation antigens detectable in humans.
- HLA human leukocyte group A
- Rejection following transplantation can generally be broken into three categories: hyperacute, occurring hours to days following transplantation; acute, occurring days to months following transplantation; and chronic, occurring months to years following transplantation.
- Hyperacute rejection is caused mainly by the production of host antibodies that attack the graft tissue.
- antibodies are observed in the transplant vascular very soon after transplantation. Shortly thereafter, vascular clotting occurs, leading to ischemia, eventual necrosis and death.
- the graft infarction is unresponsive to known immunosuppressive therapies.
- pre-transplant screening is used to significantly reduce hyperacute rejection. As a consequence of this screening, hyperacute rejection is relatively uncommon today.
- Acute rejection is thought to be mediated by the accumulation of antigen specific cells in the graft tissue.
- the T-cell-mediated immune reaction against these antigens i.e., HVGR or GVHR
- HVGR or GVHR The T-cell-mediated immune reaction against these antigens
- HVGR or GVHR the principle mechanism of acute rejection. Accumulation of these cells leads to damage of the graft tissue.
- both CD4+ helper T-cells and CD8+ cytotoxic T-cells are involved in the process and that the antigen is presented by donor and host dendritic cells.
- the CD4+ helper T-cells help recruit other effector cells, such as macrophapges and eosinophils, to the graft.
- Accessing T-cell activation signal transduction cascades for example, CD28, CD40L, and CD2 cascades are also involved.
- the cell-mediated acute rejection can be reversed in many cases by intensifying immunotherapy. After successful reversal, severely damaged elements of the graft heal by fibrosis and the remainder of the graft appears normal. After resolution of acute rejection, dosages of immunosuppressive drugs can be reduced to very low levels.
- Chronic rejection which is a particular problem in renal transplants, often progresses insidiously despite increased immunosuppressive therapy. It is thought to be due, in large part, to cell-mediated Type IV hypersensitivity. The pathologic profile differs from that of acute rejection.
- the arterial endothelium is primarily involved with extensive proliferation that may gradually occlude the vessel lumen, leading to ischemia, fibrosis, a thickened intima, and atherosclerotic changes.
- Chronic rejection is mainly due to a progressive obliteration of graft vasculature and resembles a slow, vasculitic process.
- CD8 cytotoxic T-cells and CD4 helper T cells recognize either intracellular or extracellular synthesized antigen when it is complexed, respectively, with either Class I or Class II MHC molecules. Macrophages function as antigen-presenting cells and release IL-I, which promotes proliferation of helper T-cells. Helper T-cells release interferon gamma and IL-2, which together regulate delayed hyperactivity reactions mediated by macrophage activation and immunity mediated by T cells. In the case of organ transplant, the cytotoxic T-cells destroy the graft cells on contact.
- the compounds described herein can be used to treat and/or prevent many aspects of transplant rejection, and are particularly useful in the treatment and/or prevention of rejection reactions that are mediated, at least in part, by T-cells, such as HVGR or GVHR.
- the compounds can also be used to treat and/or prevent chronic rejection in transplant recipients and, in particular, in renal transplant recipients.
- the compound can also be administered to a tissue or an organ prior to transplanting the tissue or organ in the transplant recipient.
- this invention provides a method of treating a T-cell mediated autoimmune disease, comprising administering to a patient suffering from such an autoimmune disease an amount of a compound effective to treat the autoimmune disease wherein the compound is selected from the compounds of the invention.
- the autoimmune disease is multiple sclerosis (MS), psoraisis, or Sjogran's syndrome.
- MS multiple sclerosis
- psoraisis psoraisis
- Sjogran's syndrome Such autoimmune disease include, but are not limited to, those autoimmune diseases that are frequently designated as single organ or single cell-type autoimmune disorders and those autoimmune disease that are frequently designated as involving systemic autoimmune disorder.
- Non-limiting examples of diseases frequently designated as single organ or single cell-type autoimmune disorders include: Hashimoto's thyroiditis, autoimmune hemolytic anemia, autoimmune atrophic gastritis of pernicious anemia, autoimmune encephalomyelitis, autoimmune orchitis, Goodpasture's disease, autoimmune thrombocytopenia, sympathetic ophthalmia, myasthenia gravis, Graves' disease, primary biliary cirrhosis, chronic aggressive hepatitis, ulcerative colitis and membranous glomerulopathy.
- Non-limiting examples of diseases often designated as involving systemic autoimmune disorder include: systemic lupus erythematosis, rheumatoid arthritis, Sjogren's syndrome, Reiter's syndrome, polymyositis-dermatomyositis, systemic sclerosis, polyarteritis nodosa, multiple sclerosis and bullous pemphigoid.
- Additional autoimmune diseases which can be .beta.-cell (humoral) based or T-cell based, include Cogan's syndrome, ankylosing spondylitis, Wegener's granulomatosis, autoimmune alopecia, Type I or juvenile onset diabetes, and thyroiditis.
- the types of autoimmune diseases that may be treated or prevented with such prodrugs generally include those disorders involving tissue injury that occurs as a result of a humoral and/or cell-mediated response to immunogens or antigens of endogenous and/or exogenous origin. Such diseases are frequently referred to as diseases involving the nonanaphylactic (i.e., Type II, Type III and/or Type IV) hypersensitivity reactions.
- Type I hypersensitivity reactions generally result from the release of pharmacologically active substances, such as histamine, from mast and/or basophil cells following contact with a specific exogenous antigen.
- pharmacologically active substances such as histamine
- histamine pharmacologically active substances
- basophil cells following contact with a specific exogenous antigen.
- Type I reactions play a role in numerous diseases, including allergic asthma, allergic rhinitis, etc.
- Type II hypersensitivity reactions also referred to as cytotoxic, cytolytic complement-dependent or cell-stimulating hypersensitivity reactions
- cytotoxic, cytolytic complement-dependent or cell-stimulating hypersensitivity reactions result when immunoglobulins react with antigenic components of cells or tissue, or with an antigen or hapten that has become intimately coupled to cells or tissue.
- Diseases that are commonly associated with Type II hypersensitivity reactions include, but are not limited, to autoimmune hemolytic anemia, erythroblastosis fetalis and Goodpasture's disease.
- Type III hypersensitivity reactions (also referred to as toxic complex, soluble complex, or immune complex hypersensitivity reactions) result from the deposition of soluble circulating antigen-immunoglobulin complexes in vessels or in tissues, with accompanying acute inflammatory reactions at the site of immune complex deposition.
- Type III reaction diseases include the Arthus reaction, rheumatoid arthritis, serum sickness, systemic lupus erythematosis, certain types of glomerulonephritis, multiple sclerosis and bullous pemphingoid.
- Type IV hypersensitivity reactions are caused by sensitized T- lymphocytes which result from contact with a specific antigen.
- diseases cited as involving Type IV reactions are contact dermatitis and allograft rejection.
- Autoimmune diseases associated with any of the above nonanaphylactic hypersensitivity reactions may be treated or prevented with the prodrugs according to structural formulae (I) and (Ia).
- the methods may be used to treat or prevent those autoimmune diseases frequently characterized as single organ or single cell-type autoimmune disorders including, but not limited to: Hashimoto's thyroiditis, autoimmune hemolytic anemia, autoimmune atrophic gastritis of pernicious anemia, autoimmune encephalomyelitis, autoimmune orchitis, Goodpasture's disease, autoimmune thrombocytopenia, sympathetic ophthalmia, myasthenia gravis, Graves' disease, primary biliary cirrhosis, chronic aggressive hepatitis, ulcerative colitis and membranous glomerulopathy, as well as those autoimmune diseases frequently characterized as involving systemic autoimmune disorder, which include but are not limited to: systemic lupus erythematosis (SLE), rheumatoi
- SLE
- Treatment using the compounds described herein can be applied alone, or it can be applied in combination with or adjunctive to other common immunosuppressive therapies, such as, for example, the following: mercaptopurine; corticosteroids such as prednisone; methylprednisolone and prednisolone; alkylating agents such as cyclophosphamide; calcineurin inhibitors such as cyclosporine, sirolimus, and tacrolimus; inhibitors of inosine monophosphate dehydrogenase (IMPDH) such as mycophenolate, mycophenolate mofetil, and azathioprine; and agents designed to suppress cellular immunity while leaving the recipient's humoral immunologic response intact, including various antibodies (for example, antilymphocyte globulin (ALG), antithymocyte globulin (ATG), monoclonal anti-T-cell antibodies (OKT3)) and irradiation.
- ALG antilymphocyte globulin
- ATG anti
- Azathioprine is currently available from Salix Pharmaceuticals, Inc., under the brand name AZASAN; mercaptopurine is currently available from Gate Pharmaceuticals, Inc., under the brand name PURINETHOL; prednisone and prednisolone are currently available from Roxane Laboratories, Inc.; Methyl prednisolone is currently available from Pfizer; sirolimus (rapamycin) is currently available from Wyeth-Ayerst under the brand name RAPAMUNE; tacrolimus is currently available from Fujisawa under the brand name PROGRAF; cyclosporine is current available from Novartis under the brand dame SANDIMMUNE and from Abbott under the brand name GENGRAF; IMPDH inhibitors such as mycophenolate mofetil and mycophenolic acid are currently available from Roche under the brand name CELLCEPT and from Novartis under the brand name MYFORTIC; azathioprine is currently available from Glaxo Smith Kline under the brand name IMURAN; and antibodies are currently
- the compounds could be administered either in combination or adjunctively with an inhibitor of a syk kinase
- syk kinase is a tyrosine kinase known to play a critical role in Fc ⁇ receptor signaling, as well as in other signaling cascades, such as those involving B-cell receptor signaling (Turner et al., (2000), Immunology Today 21:148- 154) and integrins beta(l), beta (2), and beta (3) in neutrophils (Mocsai et al., (2002), Immunity 16:547-558).
- syk kinase plays a pivotal role in high affinity IgE receptor signaling in mast cells that leads to activation and subsequent release of multiple chemical mediators that trigger allergic attacks.
- JAK kinases which help regulate the pathways involved in delayed or cell-mediated Type IV hypersensitivity reactions
- syk kinase helps regulate the pathways involved in immediate IgE-mediated, Type I hypersensitivity reactions.
- Certain compounds that affect the syk pathway may or may not also affect the JAK pathways.
- Suitable syk inhibitory compounds are described, for example, in Ser. No. 10/355,543 filed Jan. 31, 2003 (publication no. 2004/0029902); WO 03/063794; Ser. No. 10/631,029 filed JuI. 29, 2003; WO 2004/014382; Ser. No. 10/903,263 filed JuI. 30, 2004; PCT/US2004/24716 filed JuI. 30, 2004 (WO005/016893); Ser. No. 10/903,870 filed JuI. 30, 2004; PCT/US2004/24920 filed JuI. 30, 2004; Ser. No. 60/630,808 filed Nov. 24, 2004; Ser. No. 60/645,424 filed Jan. 19, 2005; and Ser. No. 60/654,620, filed Feb. 18, 2005, the disclosures of which are incorporated herein by reference.
- the described herein and syk inhibitory compounds could be used alone or in combination with one or more conventional transplant rejection treatments, as described above.
- the compounds can be used to treat or prevent these diseases in patients that are either initially non-responsive (resistant) to or that become non- responsive to treatment with a syk inhibitory compound or one of the other current treatments for the particular disease.
- the compounds could also be used in combination with syk inhibitory compounds in patients that are syk-compound resistant or non-responsive.
- Suitable syk-inhibitory compounds with which the compounds can be administered are provided infra.
- this invention provides a method of treating a T-cell mediated autoimmune disease, comprising administering to a patient suffering from such an autoimmune disease an amount of a compound effective to treat the autoimmune disease wherein the compound is selected from the compounds of the invention, as described herein, and the compound is administered in combination with or adjunctively to a compound that inhibits syk kinase with an IC 50 in the range of at least 10 ⁇ M.
- this invention provides a method of treating or preventing allograft transplant rejection in a transplant recipient, comprising administering to the transplant recipient an amount of a compound effective to treat or prevent the rejection wherein the compound is selected from the compounds of the invention, as described herein.
- the compound is administered to a tissue or an organ prior to transplanting the tissue or organ in the transplant recipient.
- this invention provides a method of treating or preventing allograft transplant rejection in a transplant recipient, in which the rejection is acute rejection, comprising administering to the transplant recipient an amount of a compound effective to treat or prevent the rejection, wherein the compound is selected from the compounds of the invention.
- this invention provides a method of treating or preventing allograft transplant rejection in a transplant recipient, in which the rejection is chronic rejection, comprising administering to the transplant recipient an amount of a compound effective to treat or prevent the rejection, wherein the compound is selected from the compounds of the invention.
- this invention provides a method of treating or preventing allograft transplant rejection in a transplant recipient, in which the rejection is mediated by HVGR or GVHR, comprising administering to the transplant recipient an amount of a compound effective to treat or prevent the rejection, wherein the compound is selected from the compounds of this invention, as described herein.
- this invention provides a method of treating or preventing allograft transplant rejection in a transplant recipient, in which the allograft transplant is selected from a kidney, a heart, a liver, and a lung, comprising administering to the transplant recipient an amount of a compound effective to treat or prevent the rejection, wherein the compound is selected from the compounds of this invention, as described herein.
- this invention provides a method of treating or preventing allograft transplant rejection in a transplant recipient, in which the allograft transplant is selected from a kidney, a heart, a liver, and a lung, comprising administering to the transplant recipient an amount of a compound effective to treat or prevent the rejection wherein the compound is selected from the compounds of the invention, as described herein, in which the compound is administered in combination with or adjunctively to another immunosuppressant.
- this invention provides a method of treating or preventing allograft transplant rejection in a transplant recipient, in which the allograft transplant is selected from a kidney, a heart, a liver, and a lung, comprising administering to the transplant recipient an amount of a compound effective to treat or prevent the rejection, wherein the compound is selected from the compounds of the invention, as described herein, in which the compound is administered in combination with or adjunctively to another immunosuppressant, in which the immunosuppressant is selected from cyclosporine, tacrolimus, sirolimus, an inhibitor of IMPDH, mycophenolate, mycophanolate mofetil, an anti-T-Cell antibody, and OKT3.
- the compounds described herein are cytokine moderators of IL-4 signaling. As a consequence, the compounds could slow the response of Type I hypersensitivity reactions. Thus, in a specific embodiment, the compounds could be used to treat such reactions and, therefore, the diseases associated with, mediated by, or caused by such hypersensitivity reactions (for example, allergies), prophylactically. For example, an allergy sufferer could take one or more of the JAK selective compounds described herein prior to expected exposure to allergens to delay the onset or progress of, or eliminate altogether, an allergic response.
- the compounds can be administered singly, as mixtures of one or more compounds, or in mixture or combination with other agents useful for treating such diseases and/or the symptoms associated with such diseases.
- the compounds may also be administered in mixture or in combination with agents useful to treat other disorders or maladies, such as steroids, membrane stabilizers, 5-lipoxygenase (5LO) inhibitors, leukotriene synthesis and receptor inhibitors, inhibitors of IgE isotype switching or IgE synthesis, IgG isotype switching or IgG synthesis, beta.-agonists, tryptase inhibitors, aspirin, cyclooxygenase (COX) inhibitors, methotrexate, anti-TNF drugs, anti CD20 antibody, PD4 inhibitors, p38 inhibitors, PDE4 inhibitors, and antihistamines, to name a few.
- the compounds can be administered per se in the form of prodrugs or as pharmaceutical compositions, comprising an active compound or prodrug.
- this invention provides a method of treating or preventing a Type IV hypersensitivity reaction, comprising administering to a subject an amount of a compound effective to treat or prevent the hypersensitivity reaction, wherein the compound is selected from the compounds of this invention, as described herein.
- this invention provides a method of treating or preventing a Type IV hypersensitivity reaction, which is practical prophylactically, comprising administering to a subject an amount of a compound effective to treat or prevent the hypersensitivity reaction, wherein the compound is selected from the compounds of this invention, as described herein, and is administered prior to exposure to an allergen.
- this invention provides a method of inhibiting a signal transduction cascade in which JAK3 kinase plays a role, comprising contacting a cell expressing a receptor involved in such a signaling cascade with a compound wherein the compound is selected from the compounds of this invention, as described herein.
- this invention provides a method of treating or preventing a JAK kinase-mediated disease, comprising administering to a subject an amount of compound effective to treat or prevent the JAK kinase-mediated disease, wherein the compound is selected from the compounds of this invention, as described herein.
- this invention provides a method of treating or preventing a JAK kinase-mediated disease, in which the JAK-mediated disease is HVGR or GVHR, comprising administering to a subject an amount of compound effective to treat or prevent the JAK kinase-mediated disease, wherein the compound is selected from the compounds of the invention, as described herein.
- this invention provides a method of treating or preventing a JAK kinase-mediated disease, in which the JAK-mediated disease is acute allograft rejection, comprising administering to a subject an amount of compound effective to treat or prevent the JAK kinase-mediated disease, wherein the compound is selected from the compounds of the invention, as described herein.
- this invention provides a method of treating or preventing a syk and/or JAK kinase-mediated disease, in which the JAK-mediated disease is chronic allograft rejection, comprising administering to a subject an amount of compound effective to treat or prevent the JAK kinase-mediated disease, wherein the compound is selected from the compounds of the invention, as described herein.
- Active compounds of the invention typically inhibit thesyk and/or JAK/Stat pathway.
- the activity of a specified compound as an inhibitor of a syk and/or JAK kinase can be assessed in vitro or in vivo. In some embodiments, the activity of a specified compound can be tested in a cellular assay.
- Cell proliferative disorder refers to a disorder characterized by abnormal proliferation of cells.
- a proliferative disorder does not imply any limitation with respect to the rate of cell growth, but merely indicates loss of normal controls that affect growth and cell division.
- cells of a proliferative disorder can have the same cell division rates as normal cells but do not respond to signals that limit such growth.
- neoplasm or tumor which is an abnormal growth of tissue. Cancer refers to any of various malignant neoplasms characterized by the proliferation of cells that have the capability to invade surrounding tissue and/or metastasize to new colonization sites.
- cell proliferative disorders treatable with the compounds disclosed herein relate to any disorder characterized by aberrant cell proliferation. These include various tumors and cancers, benign or malignant, metastatic or non-metastatic. Specific properties of cancers, such as tissue invasiveness or metastasis, can be targeted using the methods described herein.
- Cell proliferative disorders include a variety of cancers, including, among others, ovarian cancer, renal cancer, gastrointestinal cancer, kidney cancer, bladder cancer, pancreatic cancer, lung squamous carcinoma, and adenocarcinoma.
- the cell proliferative disorder treated is a hematopoietic neoplasm, which is aberrant growth of cells of the hematopoietic system.
- Hematopoietic malignancies can have its origins in pluripotent stem cells, multipotent progenitor cells, oligopotent committed progenitor cells, precursor cells, and terminally differentiated cells involved in hematopoiesis. Some hematological malignancies are believed to arise from hematopoietic stem cells, which have the ability for self renewal. For instance, cells capable of developing specific subtypes of acute myeloid leukemia (AML) (Cynthia K. Hahn, Kenneth N. Ross, Rose M.
- AML acute myeloid leukemia
- the stem cell origin of certain hematological malignancies also finds support in the observation that specific chromosomal abnormalities associated with particular types of leukemia can be found in normal cells of hematopoietic lineage as well as leukemic blast cells.
- the reciprocal translocation t(9q34;22ql 1) associated with approximately 95% of chronic myelogenous leukemia appears to be present in cells of the myeloid, erythroid, and lymphoid lineage, suggesting that the chromosomal aberration originates in hematopoietic stem cells.
- a subgroup of cells in certain types of CML displays the cell marker phenotype of hematopoietic stem cells.
- hematopoietic neoplasms often originate from stem cells, committed progenitor cells or more terminally differentiated cells of a developmental lineage can also be the source of some leukemias.
- forced expression of the fusion protein Bcr/Abl associated with chronic myelogenous leukemia
- common myeloid progenitor or granulocyte/macrophage progenitor cells produces a leukemic-like condition.
- chromosomal aberrations associated with subtypes of leukemia are not found in the cell population with a marker phenotype of hematopoietic stem cells, but are found in a cell population displaying markers of a more differentiated state of the hematopoietic pathway (Turhan et al., 1995, Blood 85:2154-2161).
- committed progenitor cells and other differentiated cells may have only a limited potential for cell division
- leukemic cells may have acquired the ability to grow unregulated, in some instances mimicking the self-renewal characteristics of hematopoietic stem cells (Passegue et al., Proc. Natl. Acad. Sci. USA, 2003, 100: 11842-9).
- the hematopoietic neoplasm treated is a lymphoid neoplasm, where the abnormal cells are derived from and/or display the characteristic phenotype of cells of the lymphoid lineage.
- Lymphoid neoplasms can be subdivided into B-cell neoplasms, T and NK-cell neoplasms, and Hodgkin's lymphoma.
- B-cell neoplasms can be further subdivided into precursor B-cell neoplasm and mature/peripheral B-cell neoplasm.
- Exemplary B-cell neoplasms are precursor B-lymphoblastic leukemia/lymphoma (precursor B-cell acute lymphoblastic leukemia) while exemplary mature/peripheral B-cell neoplasms are B-cell chronic lymphocytic leukemia/small lymphocytic lymphoma, B-cell prolymphocytic leukemia, lymphoplasmacytic lymphoma, splenic marginal zone B-cell lymphoma, hairy cell leukemia, plasma cell myeloma/plasmacytoma, extranodal marginal zone B-cell lymphoma of MALT type, nodal marginal zone B-cell lymphoma, follicular lymphoma, mantle-cell lymphoma, diffuse large B-cell lymphoma, mediastinal large B-cell lymphoma, primary effusion lymphoma, and Burkitt's lymphoma/Burkitt cell leukemia.
- T- cell and Nk-cell neoplasms are further subdivided into precursor T-cell neoplasm and mature (peripheral) T-cell neoplasms.
- Exemplary precursor T-cell neoplasm is precursor T- lymphoblastic lymphoma/leukemia (precursor T-cell acute lymphoblastic leukemia) while exemplary mature (peripheral) T-cell neoplasms are T-cell prolymphocyte leukemia T-cell granular lymphocytic leukemia, aggressive NK-cell leukemia, adult T-cell lymphoma/leukemia (HTLV-I), extranodal NK/T-cell lymphoma, nasal type, enteropathy- type T-cell lymphoma, hepatosplenic gamma-delta T-cell lymphoma, subcutaneous panniculitis-like T-cell lymphoma, Mycosis fungoides/Sezary syndrome, Anaplastic large- cell lympho
- the third member of lymphoid neoplasms is Hodgkin's lymphoma, also referred to as Hodgkin's disease.
- Exemplary diagnosis of this class that can be treated with the compounds include, among others, nodular lymphocyte- predominant Hodgkin's lymphoma, and various classical forms of Hodgkin's disease, exemplary members of which are Nodular sclerosis Hodgkin's lymphoma (grades 1 and 2), Lymphocyte-rich classical Hodgkin's lymphoma, Mixed cellularity Hodgkin's lymphoma, and Lymphocyte depletion Hodgkin's lymphoma.
- any of the lymphoid neoplasms that are associated with aberrant JAK activity can be treated with the syk and/or JAK inhibitory compounds.
- the hematopoietic neoplasm treated is a myeloid neoplasm.
- This group comprises a large class of cell proliferative disorders involving or displaying the characteristic phenotype of the cells of the myeloid lineage.
- Myeloid neoplasms can be subdivided into myeloproliferative diseases, myelodysplastic/myeloproliferative diseases, myelodysplastic syndromes, and acute myeloid leukemias.
- Exemplary myeloproliferative diseases are chronic myelogenous leukemia (e.g., Philadelphia chromosome positive (t(9;22)(qq34;ql I)), chronic neutrophilic leukemia, chronic eosinophilic leukemia/hypereosinophilic syndrome, chronic idiopathic myelofibrosis, polycythemia vera, and essential thrombocythemia.
- Exemplary myelodysplastic/myeloproliferative diseases are chronic myelomonocytic leukemia, atypical chronic myelogenous leukemia, and juvenile myelomonocytic leukemia.
- Exemplary myelodysplastic syndromes are refractory anemia, with ringed sideroblasts and without ringed sideroblasts, refractory cytopenia (myelodysplastic syndrome) with multilineage dysplasia, refractory anemia (myelodysplastic syndrome) with excess blasts, 5q-syndrome, and myelodysplastic syndrome.
- any of the myeloid neoplasms that are associated with aberrant syk and/or JAK activity can be treated with the syk and/or JAK inhibitory compounds.
- the compounds can be used to treat Acute myeloid leukemias (AML), which represent a large class of myeloid neoplasms having its own subdivision of disorders. These subdivisions include, among others, AMLs with recurrent cytogenetic translocations, AML with multilineage dysplasia, and other AML not otherwise categorized.
- AML Acute myeloid leukemias
- Exemplary AMLs with recurrent cytogenetic translocations include, among others, AML with t(8;21)(q22;q22), AMLl(CBF-alpha)/ETO, Acute promyelocytic leukemia (AML with t(15;17)(q22;ql 1-12) and variants, PML/RAR-alpha), AML with abnormal bone marrow eosinophils (inv(16)(pl3q22) or t(16;16)(pl3;ql 1), CBFb/MYHl IX), and AML with 1 Iq23 (MLL) abnormalities.
- AML with t(8;21)(q22;q22) AMLl(CBF-alpha)/ETO
- Acute promyelocytic leukemia AML with t(15;17)(q22;ql 1-12
- PML/RAR-alpha Acute promyelocytic leukemia
- Exemplary AML with multilineage dysplasia are those that are associated with or without prior myelodysplastic syndrome.
- Other acute myeloid leukemias not classified within any definable group include, AML minimally differentiated, AML without maturation, AML with maturation, Acute myelomonocytic leukemia, Acute monocytic leukemia, Acute erythroid leukemia, Acute megakaryocy e leukemia, Acute basophilic leukemia, and Acute panmyelosis with myelofibrosis.
- Treating within the context of the invention means an alleviation of symptoms associated with a disorder or disease, or halt of further progression or worsening of those symptoms, or prevention or prophylaxis of the disease or disorder.
- mammal includes organisms which express syk and/or JAK. Examples of mammals include mice, rats, cows, sheep, pigs, goats, horses, bears, monkeys, dogs, cats and, preferably, humans. Transgenic organisms which express syk and/or JAK are also included in this definition.
- inventive methods comprise administering an effective amount of a compound or composition described herein to a mammal or non-human animal.
- "effective amount" of a compound or composition of the invention includes those amounts that antagonize or inhibit syk and/or JAK.
- An amount which antagonizes or inhibits syk and/or JAK is detectable, for example, by any assay capable of determining syk and/or JAK activity, including the one described below as an illustrative testing method.
- Effective amounts may also include those amounts which alleviate symptoms of a syk and/or JAK associated disorder treatable by inhibiting syk and/or JAK.
- antagonists of syk or “antagonists of JAK” include compounds which interact with the syk or JAK, respectively, and modulate, e.g., inhibit or decrease, the ability of a second compound, e.g., another syk or JAK ligand, to interact with the syk or JAK, respectively.
- the syk or JAK binding compounds are preferably antagonists of syk or JAK, respectively.
- syk binding compound and "JAK-binding compound” (e.g., exhibits binding affinity to the receptor) includes those compounds which interact with syk or JAK resulting in modulation of the activity of syk or JAK, respectively, syk and/or JAK binding compounds may be identified using an in vitro (e.g., cell and non-cell based) or in vivo method. A description of in vitro methods are provided below.
- compositions of this invention may further comprise another therapeutic agent.
- the second agent may be administered either as a separate dosage form or as part of a single dosage form with the compounds or compositions of this invention.
- inventive compounds can be used in an application of monotherapy to treat a disorder, disease or symptom, they also may be used in combination therapy, in which the use of an inventive compound or composition (therapeutic agent) is combined with the use of one or more other therapeutic agents for treating the same and/or other types of disorders, symptoms and diseases.
- Combination therapy includes administration of the two or more therapeutic agents concurrently or sequentially. The agents may be administered in any order. Alternatively, the multiple therapeutic agents can be combined into a single composition that can be administered to the patient.
- a single pharmaceutical composition could comprise the compound or pharmaceutically acceptable salt, ester or prodrug thereof according to the formula I, another therapeutic agent (e.g., methotrexate) or a pharmaceutically acceptable salt, ester or prodrug thereof, and a pharmaceutically acceptable excipient or carrier.
- the invention comprises a compound having the formula I, a method for making an inventive compound, a method for making a pharmaceutical composition from at least one inventive compound and at least one pharmaceutically acceptable carrier or excipient, and a method of using one or more inventive compounds to treat a variety of disorders, symptoms and diseases (e.g., inflammatory, autoimmune, neurological, neurodegenerative, oncology and cardiovascular), such as RA, osteoarthritis, irritable bowel disease IBD, asthma, chronic obstructive pulmonary disease COPD and MS.
- disorders, symptoms and diseases e.g., inflammatory, autoimmune, neurological, neurodegenerative, oncology and cardiovascular
- RA inflammatory, autoimmune, neurological, neurodegenerative, oncology and cardiovascular
- RA e.g., osteoarthritis, irritable bowel disease IBD, asthma, chronic obstructive pulmonary disease COPD and MS.
- inventive compounds and their pharmaceutically acceptable salts and/or neutral compositions may be formulated together with a pharmaceutically acceptable excipient or carrier and the resulting composition may be administered in vivo to mammals, such as men, women and animals, to treat a variety of disorders, symptoms and diseases.
- inventive compounds can be used to prepare a medicament that is useful for treating a variety of disorders, symptoms and diseases.
- All of the compounds of the present invention are either potent inhibitors of syk and/or JAK kinases, exhibiting IC 50 S in the respective assay in the range of less than 5 ⁇ M, with most being in the nanomolar, and several in the sub-nanomolar, range.
- the compounds of the present invention may be "dual" syk/JAK inhibitors in that they inhibit both syk and JAK kinase to some degree.
- the compounds of the present invention may selectively inhibit syk kinase, but not appreciably inhibit one or more JAK kinases.
- the compounds of the present invention may selectively inhibit JAK kinase, but not appreciably inhibit one or more syk kinases.
- Still another aspect of this invention is to provide a kit comprising separate containers in a single package, wherein the inventive pharmaceutical compounds, compositions and/or salts thereof are used in combination with pharmaceutically acceptable carriers to treat states, disorders, symptoms and diseases where syk and/or JAK plays a role.
- the starting materials and reagents used in preparing these compounds generally are either available from commercial suppliers, such as Aldrich Chemical Co., or are prepared by methods known to those skilled in the art following procedures set forth in references such as Fieser and Fieser's Reagents for Organic Synthesis; Wiley & Sons: New York, 1967-2004, Volumes 1-22; Rodd's Chemistry of Carbon Compounds, Elsevier Science Publishers, 1989, Volumes 1-5 and Supplementals; and Organic Reactions, Wiley & Sons: New York, 2005, Volumes 1-65.
- the starting materials and the intermediates of the synthetic reaction schemes can be isolated and purified if desired using conventional techniques, including but not limited to, filtration, distillation, crystallization, chromatography, and the like. Such materials can be characterized using conventional means, including physical constants and spectral data.
- the reactions described herein preferably are conducted under an inert atmosphere at atmospheric pressure at a reaction temperature range of from about -78°C to about 150°C, more preferably from about 0°C to about 125°C, and most preferably and conveniently at about room (or ambient) temperature, e.g., about 20°C to about 75°C.
- the compounds and/or intermediates may be characterized by high performance liquid chromatography (HPLC) using a Waters Alliance chromatography system with a 2695 Separation Module (Milford, Mass.).
- the analytical columns may be C- 18 SpeedROD RP- 18E Columns from Merck KGaA (Darmstadt, Germany).
- characterization may be performed using a Waters Unity (UPLC) system with Waters Acquity UPLC BEH C- 18 2.1 mm x 15 mm columns.
- a gradient elution may be used, typically starting with 5 % acetonitrile/95% water and progressing to 95% acetonitrile over a period of 5 minutes for the Alliance system and 1 minute for the Acquity system.
- All solvents may contain 0.1% trifluoroacetic acid (TFA).
- TFA trifluoroacetic acid
- Compounds may be detected by ultraviolet light (UV) absorption at either 220 nm or 254 nm.
- HPLC solvents may be from EMD Chemicals, Inc. (Gibbstown, NJ). In some instances, purity may be assessed by thin layer chromatography (TLC) using glass backed silica gel plates, such as, for example, EMD Silica Gel 60 2.5cm x 7.5cm plates. TLC results may be readily detected visually under ultraviolet light, or by employing well known iodine vapor and other various staining techniques.
- Mass spectrometric analysis may be performed on one of two Agilent 1100 series LCMS instruments with acetonitrile /water as the mobile phase.
- One system may use TFA as the modifier and measure in positive ion mode [reported as MH+, (M+ 1) or (M+H)+] and the other may use either formic acid or ammonium acetate and measure in both positive [reported as MH + , (M+ 1) or (M+H) + ] and negative [reported as M-, (M-I) or (M-H) ] ion modes.
- Nuclear magnetic resonance (NMR) analysis may be performed on some of the compounds with a Varian 400 MHz NMR (Palo Alto, Calif.).
- the spectral reference may be either TMS or the known chemical shift of the solvent.
- Melting points may be determined on a Laboratory Devices Mel-Temp apparatus (Holliston, Mass.).
- Preparative separations may be carried out as needed, using either an Sql ⁇ x or an SgIOOc chromatography system and prepackaged silica gel columns all purchased from Teledyne Isco, (Lincoln, NE).
- compounds and intermediates may be purified by flash column chromatography using silica gel (230-400 mesh) packing material, or by HPLC using a C- 18 reversed phase column.
- Typical solvents employed for the Isco systems and flash column chromatography may be dichloromethane, methanol, ethyl acetate, hexane, acetone, aqueous hydroxyamine and triethyl amine.
- Typical solvents employed for the reverse phase HPLC may be varying concentrations of acetonitrile and water with 0.1% trifluoroacetic acid.
- Step 1 To a stirring solution of carboxylic acid 1.1 (85g, 540mmol) in thionyl chloride (425mL) was added pyridine (8.5mL, 0.1 lmmol), slowly. The reaction was stirred at 75°C overnight at which time it was concentrated and dried under vacuum to a light yellow powder which was used immediately for the next step.
- Step 2 The yellow solid from the Step 1 was slowly diluted with 750 mL of ethanol and refluxed overnight. The next day the reaction was determined to be complete by HPLC and then cooled in an ice bath and the solid filtered and washed with diethyl ether affording the desired ethyl ester 1.3 as an off-white powder (9 Ig, 87% for two steps). MS found for C 7 H 8 N 2 O 4 as (M+H) + 185.0.
- Step 3 Ester 1.3 (22g, 120 mmol) was dissolved in phosphorous oxychloride
- Step 4 Dichloropyrimidine 1.4 (5.9g, 27 mmol) was dissolved in acetonitrile (50 mL) and treated sequentially with diisopropylamine (5.2 mL, 30 mmol) followed by cyclopropyl amine (1.9g, 27 mmol) and stirred at rt until all starting material had been consumed. The reaction mixture was then diluted with water to a total volume of 150 mL and the precipitate collected by filtration affording the desired product as a light yellow solid (6.02g, 87%).
- Step 6 Carboxylic acid 1.6 (3.15g, 15 mmol) was dissolved in N ,N- dimethylformamide (70 mL) and treated with HOBt (3.13g, 23 mmol) and EDC (4.4g, 23 mmol). After stirring ca. 25 min ammonia (0.5 M in 1,4-dioxane, 72 mL, 36 mmol) was added and the reaction stirred overnight. The following morning the reaction was diluted with water to a total volume of 500 mL and the desired product collected by filtration affording 3.62g (74%) of a light-beige solid.
- Step 7 Benzotriazolyl ether 1.7 (50 mg, 0.17 mmol), 4-(pyridyl)-aniline (0.26 mmol) and p-toluenesulfonic acid ( 30 mg, 0.17 mmol) were diluted with 1,4-dioxane (5 mL) and stirred at 12O 0 C until all starting material had been consumed. The reaction was cooled to rt, diluted with water and directly purified by preparative HPLC affording the desired product, 1, after lyophilization. MS found for Ci 9 Hi 9 N 6 O as (M) + 347.3.
- VN N- -NH
- Example 18 1 -(4-(4-(4-(4-(4-(4-(4-(aminomethyl)piperidin- 1 - yl V5-fluoropyrimidin-2- ylamino)phen yl)piperazin- 1 - vPethanone
- tert-butyl 1 -(5-fluoro-2-(4-(N-methylacetamido)phenylamino)pyrimidin-4-yl)piperidin-4- yl)methylcarbamate (65 mg). MS 473.4 (M+H); UV 205.8, 262.8 nm.
- Step 1 To a mixture of N-methyl-4-nitroaniline (2 g, 13.1 mmol) in DCM (30 mL) was added Et3N (2.75 mL, 20 mmol) and cyclopropanecarbonyl chloride (1.32 mL, 14.4 mmol) at room temperature, after stirring for 1 h at room temperature, the mixture was diluted with EtOAc, the organics was washed with Sat. NaHCO 3 , brine, dried and concentrated to give N-methyl-N-(4-nitrophenyl)cyclopropanecarboxamide as crude product (3 g).
- Step 2 To a suspension of 2-chloro-N-cyclobutyl-7H-pyrrolo[2,3- ⁇ i]pyrimidin-4- amine (67 mg, 0.3 mmol) in nBuO ⁇ (0.8 mL) was added N-(4-aminophenyl)-N- methylcyclopropanecarboxamide (85 mg, 0.45 mmol) and TMSCl (0.02 mL, 0.15 mmol).
- Step 1 To a suspension of 2-chloro-4-(methylthio)-7H-pyrrolo[2,3- d]pyrimidin-4- amine (177 mg, 0.88 mmol) in nBuO ⁇ (2 mL) was added TMSCl (0.111 mL, 0.88 mmol). After heating at 115 0 C for 24 h, the mixture was cooled and purified by preparative ⁇ PLC to give 2-(2-(lH-indazol-6-ylamino)-4-(methylthio)-7 ⁇ -pyrrolo[2,3-d]pyrimidin-4-amine (53 mg)-
- Step 2 To a suspension of 2-(2-(lH-indazol-6-ylamino)-4-(methylthio)-7H- pyrrolo[2,3-d]pyrimidin-4-amine (53 mg, 0.18 mmol) in DCM (1 mL) was added mCPBA (50 mg, 0.187 mmol). After stirring at room temperature for 2 h, the mixture was diluted with EtOAc, washed with Sat. NaHCO 3 , brine, dried and concentrated to give a mixture of corresponding sulfone and sulfoxide (100 mg total).
- Step 1 To a suspension of 2,4-dichloro-7-tosyl-7H-pyrrolo[2,3- ⁇ i]pyrirnidine (100 mg, 0.29 mmol) in nBuO ⁇ (1 mL) was added 3-aminopyrazole (26 mg, 0.32 mmol) and DIPEA (0.057 mL, 0.32 mmol). After heating at 90 °C for 3 h, the mixture was diluted with EtOAc, washed with Sat.
- Step 2 To a suspension of 2-chloro-N-(l ⁇ -pyrazol-5-yl)-7-tosyl-7H-pyrrolo[2,3- J]pyrimidine-4-amine (100 mg, 0.26 mmol) in nBuO ⁇ (1 mL) was added 6-aminoindazol (68 mg, 0.512 mmol) and TMSCl (0.016 mL, 0.128 mmol). After stirring at 1 15 0 C for 15 h, the mixture was cooled and was added MeOH (1 mL), followed by a solution of KO ⁇ (100 mg) in ⁇ 2O (0.5 mL).
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Immunology (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Diabetes (AREA)
- Heart & Thoracic Surgery (AREA)
- Pulmonology (AREA)
- Dermatology (AREA)
- Vascular Medicine (AREA)
- Rheumatology (AREA)
- Urology & Nephrology (AREA)
- Neurosurgery (AREA)
- Hospice & Palliative Care (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Oncology (AREA)
- Pain & Pain Management (AREA)
- Transplantation (AREA)
- Obesity (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description












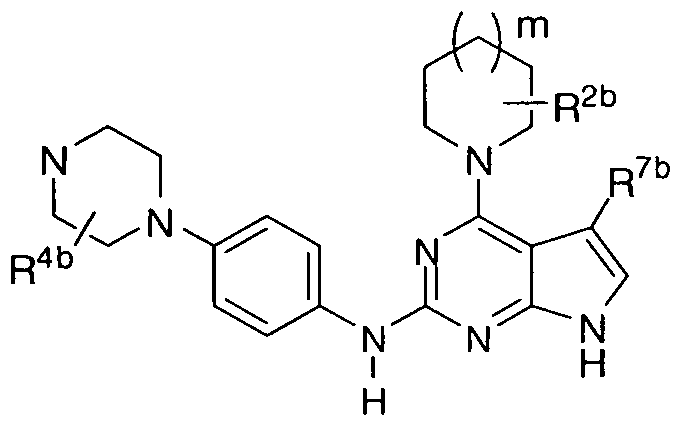
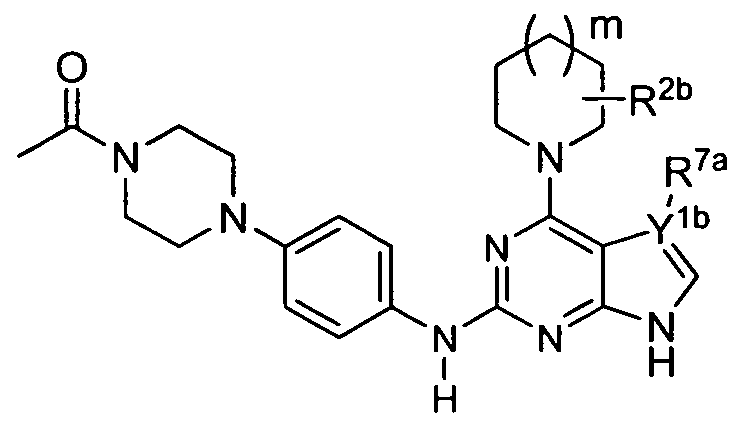








































































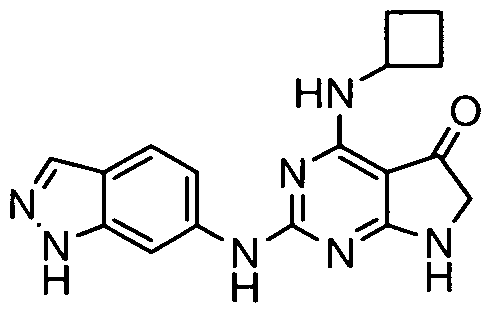



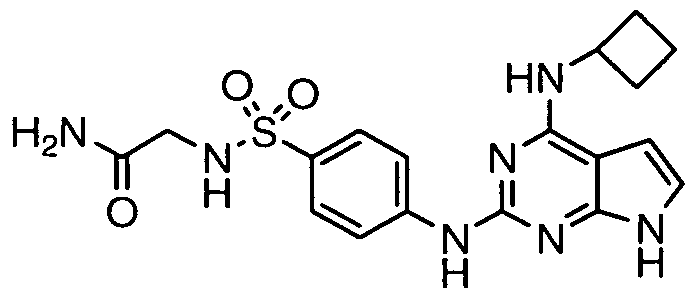































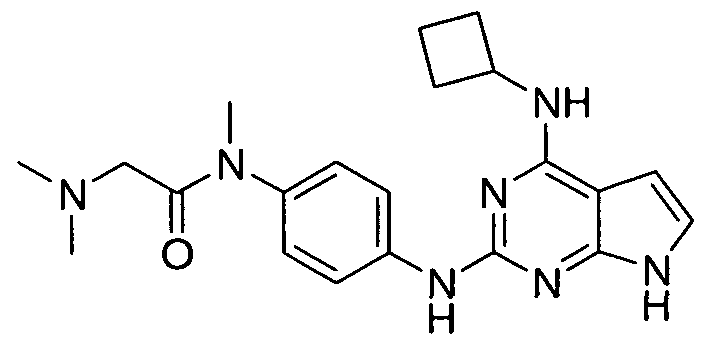

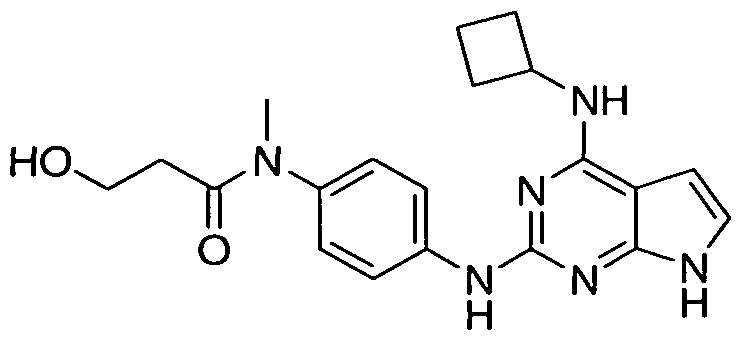









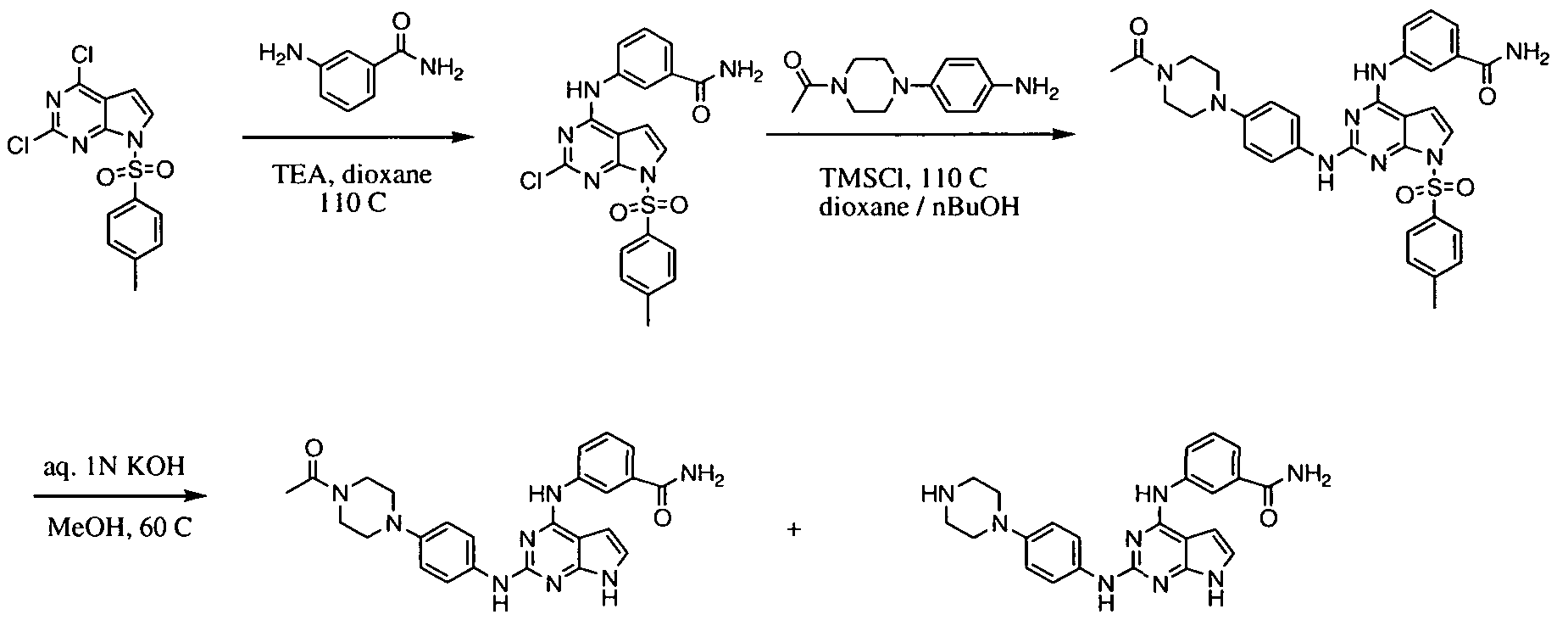





























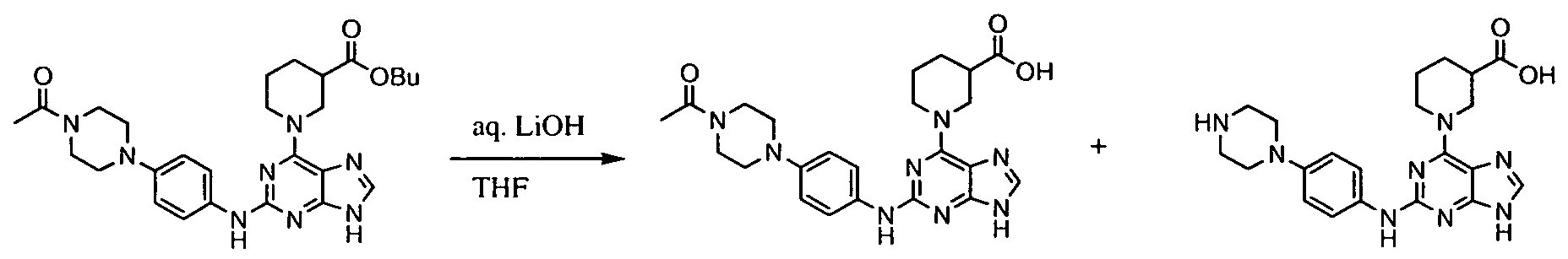















































Claims























Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| NZ588830A NZ588830A (en) | 2008-04-22 | 2009-04-22 | Inhibitors of protein kinases |
| CN2009801223945A CN102066338A (en) | 2008-04-22 | 2009-04-22 | Inhibitors of protein kinases |
| AU2009238590A AU2009238590A1 (en) | 2008-04-22 | 2009-04-22 | Inhibitors of protein kinases |
| CA2723185A CA2723185A1 (en) | 2008-04-22 | 2009-04-22 | Inhibitors of protein kinases |
| BRPI0910668A BRPI0910668A2 (en) | 2008-04-22 | 2009-04-22 | protein kinase inhibitors |
| EP09734422.0A EP2271631B1 (en) | 2008-04-22 | 2009-04-22 | Inhibitors of protein kinases |
| JP2011506293A JP2011518219A (en) | 2008-04-22 | 2009-04-22 | Inhibitors of protein kinases |
| IL208719A IL208719A0 (en) | 2008-04-22 | 2010-10-14 | Inhibitors of protein kinases |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US4707708P | 2008-04-22 | 2008-04-22 | |
| US61/047,077 | 2008-04-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009131687A2 true WO2009131687A2 (en) | 2009-10-29 |
| WO2009131687A3 WO2009131687A3 (en) | 2010-01-07 |
Family
ID=40910850
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/002512 WO2009131687A2 (en) | 2008-04-22 | 2009-04-22 | Inhibitors of protein kinases |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US8258144B2 (en) |
| EP (1) | EP2271631B1 (en) |
| JP (1) | JP2011518219A (en) |
| CN (2) | CN103224497A (en) |
| AU (1) | AU2009238590A1 (en) |
| BR (1) | BRPI0910668A2 (en) |
| CA (1) | CA2723185A1 (en) |
| IL (1) | IL208719A0 (en) |
| NZ (1) | NZ588830A (en) |
| WO (1) | WO2009131687A2 (en) |
Cited By (112)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2120573A1 (en) * | 2007-02-12 | 2009-11-25 | Merck & Co., Inc. | Piperidine derivatives |
| WO2010038081A2 (en) * | 2008-10-03 | 2010-04-08 | Astrazeneca Ab | Heterocyclic derivatives and methods of use thereof |
| WO2011143033A1 (en) | 2010-05-11 | 2011-11-17 | Amgen Inc. | Pyrimidine compounds that inhibit anaplastic lymphoma kinase |
| WO2012002577A1 (en) | 2010-06-30 | 2012-01-05 | 富士フイルム株式会社 | Novel nicotinamide derivatives or salts thereof |
| WO2012092880A1 (en) | 2011-01-07 | 2012-07-12 | Centaurus Biopharma Co., Ltd. | 2,4-DIAMINO-6,7-DIHYDRO-5H-PYRROLO[2,3]PYRIMIDINE DERIVATIVES AS FAK/Pyk2 INHIBITORS |
| EP2489663A1 (en) | 2011-02-16 | 2012-08-22 | Almirall, S.A. | Compounds as syk kinase inhibitors |
| WO2012123312A1 (en) * | 2011-03-11 | 2012-09-20 | Glaxo Group Limited | Pyrido[3,4-b]pyrazine derivatives as syk inhibitors |
| EP2554544A1 (en) | 2011-08-01 | 2013-02-06 | Almirall, S.A. | Pyridin-2(1h)-one derivatives as jak inhibitors |
| WO2013099041A1 (en) | 2011-12-28 | 2013-07-04 | 富士フイルム株式会社 | Novel nicotinamide derivative or salt thereof |
| CN103282352A (en) * | 2010-11-01 | 2013-09-04 | 波托拉医药品公司 | Benzamides and nicotinamides as syk modulators |
| CN103476776A (en) * | 2011-01-07 | 2013-12-25 | 北京赛林泰医药技术有限公司 | 2,4-diamino-6,7-dihydro-5H-pyrrolo[2,3]pyrimidine derivatives as FAK/Pyk2 inhibitors |
| US8685988B2 (en) | 2012-08-06 | 2014-04-01 | Acea Biosciences, Inc. | EGFR modulators and uses thereof |
| JP2014508808A (en) * | 2011-03-23 | 2014-04-10 | アムジエン・インコーポレーテツド | Condensed tricyclic dual inhibitors of CDK4 / 6 and FLT3 |
| AU2011218167B2 (en) * | 2010-02-17 | 2014-07-10 | Amgen Inc. | Aryl carboxamide derivatives as sodium channel inhibitors for treatment of pain |
| WO2014130693A1 (en) * | 2013-02-25 | 2014-08-28 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| WO2014113429A3 (en) * | 2013-01-16 | 2014-10-09 | Signal Pharmaceuticals, Llc | Substituted pyrrolopyrimidine compounds, compositions thereof, and methods of treatment therewith |
| US8987456B2 (en) | 2011-10-05 | 2015-03-24 | Merck Sharp & Dohme Corp. | 3-pyridyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9006444B2 (en) | 2011-10-05 | 2015-04-14 | Merck Sharp & Dohme Corp. | Phenyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9012462B2 (en) | 2008-05-21 | 2015-04-21 | Ariad Pharmaceuticals, Inc. | Phosphorous derivatives as kinase inhibitors |
| US20150119288A1 (en) * | 2009-03-23 | 2015-04-30 | Nodality, Inc. | Kits for multiparametric phospho analysis |
| WO2015061247A3 (en) * | 2013-10-21 | 2015-07-23 | Merck Patent Gmbh | Heteroaryl compounds as btk inhibitors and uses thereof |
| US9120785B2 (en) | 2011-05-10 | 2015-09-01 | Merck Sharp & Dohme Corp. | Pyridyl aminopyridines as Syk inhibitors |
| US9139534B2 (en) | 2011-04-22 | 2015-09-22 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| US9145391B2 (en) | 2011-05-10 | 2015-09-29 | Merck Sharp & Dohme Corp. | Bipyridylaminopyridines as Syk inhibitors |
| US9156845B2 (en) | 2012-06-29 | 2015-10-13 | Pfizer Inc. | 4-(substituted amino)-7H-pyrrolo[2,3-d] pyrimidines as LRRK2 inhibitors |
| WO2015164330A1 (en) | 2014-04-21 | 2015-10-29 | Millennium Pharmaceuticals, Inc. | Anti-psyk antibody molecules and use of same for syk-targeted therapy |
| US9216173B2 (en) | 2011-10-05 | 2015-12-22 | Merck Sharp & Dohme Corp. | 2-Pyridyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9242984B2 (en) | 2012-06-20 | 2016-01-26 | Merck Sharp & Dohme Corp. | Pyrazolyl derivatives as Syk inhibitors |
| US9290490B2 (en) | 2011-05-10 | 2016-03-22 | Merck Sharp & Dohme Corp. | Aminopyrimidines as Syk inhibitors |
| CN105452238A (en) * | 2013-05-24 | 2016-03-30 | 株式会社柳韩洋行 | Bicyclic derivative containing pyrimidine ring, and preparation method therefor |
| US9353066B2 (en) | 2012-08-20 | 2016-05-31 | Merck Sharp & Dohme Corp. | Substituted phenyl-Spleen Tyrosine Kinase (Syk) inhibitors |
| US9365524B2 (en) | 2014-01-30 | 2016-06-14 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US9376418B2 (en) | 2012-06-22 | 2016-06-28 | Merck Sharp & Dohme Corp. | Substituted pyridine spleen tyrosine kinase (SYK) inhibitors |
| US9382246B2 (en) | 2013-12-05 | 2016-07-05 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9416111B2 (en) | 2012-06-22 | 2016-08-16 | Merck Sharp & Dohme Corp. | Substituted diazine and triazine spleen tyrosine kinease (Syk) inhibitors |
| US9434735B2 (en) | 2014-07-14 | 2016-09-06 | Signal Pharmaceuticals, Llc | Amorphous form of 4-((4-(cyclopentyloxy)-5-(2-methylbenzo[d]oxazol-6-yl)-7h-pyrrolo[2,3-d]pyrimidin-2-yl)amino)-3-methoxy-n-methylbenzamide, compositions thereof and methods of their use |
| US9464089B2 (en) | 2012-01-13 | 2016-10-11 | Acea Biosciences Inc. | Heterocyclic compounds and uses thereof |
| US9487504B2 (en) | 2012-06-20 | 2016-11-08 | Merck Sharp & Dohme Corp. | Imidazolyl analogs as syk inhibitors |
| US9499534B2 (en) | 2013-04-26 | 2016-11-22 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminopyrimidines as spleen tyrosine kinase inhibitors |
| US9513297B2 (en) | 2014-12-16 | 2016-12-06 | Signal Pharmaceuticals, Llc | Methods for measurement of inhibition of c-Jun N-terminal kinase in skin |
| US9533984B2 (en) | 2013-04-19 | 2017-01-03 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US9580423B2 (en) | 2015-02-20 | 2017-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9586965B2 (en) | 2012-01-13 | 2017-03-07 | Acea Biosciences Inc. | Pyrrolo[2,3-d]pyrimidine compounds as inhibitors of protein kinases |
| US9586931B2 (en) | 2012-09-28 | 2017-03-07 | Merck Sharp & Dohme Corp. | Triazolyl derivatives as Syk inhibitors |
| US9586961B2 (en) | 2010-07-09 | 2017-03-07 | Leo Pharma A/S | Homopiperazine derivatives as protein tyrosine kinase inhibitors and pharmaceutical use thereof |
| US9611283B1 (en) | 2013-04-10 | 2017-04-04 | Ariad Pharmaceuticals, Inc. | Methods for inhibiting cell proliferation in ALK-driven cancers |
| US9611267B2 (en) | 2012-06-13 | 2017-04-04 | Incyte Holdings Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| WO2017059280A1 (en) * | 2015-10-02 | 2017-04-06 | The University Of North Carolina At Chapel Hill | Novel pan-tam inhibitors and mer/axl dual inhibitors |
| US9623028B2 (en) | 2014-07-14 | 2017-04-18 | Signal Pharmaceuticals, Llc | Methods of treating a cancer using substituted pyrrolopyrimidine compounds, compositions thereof |
| US9624210B2 (en) | 2012-12-12 | 2017-04-18 | Merck Sharp & Dohme Corp. | Amino-pyrimidine-containing spleen tyrosine kinase (Syk) inhibitors |
| US9670196B2 (en) | 2013-12-20 | 2017-06-06 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as Spleen Tyrosine Kinase inhibitors |
| US9695171B2 (en) | 2013-12-17 | 2017-07-04 | Pfizer Inc. | 3,4-disubstituted-1 H-pyrrolo[2,3-b]pyridines and 4,5-disubstituted-7H-pyrrolo[2,3-c]pyridazines as LRRK2 inhibitors |
| US9708318B2 (en) | 2015-02-20 | 2017-07-18 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9745295B2 (en) | 2013-04-26 | 2017-08-29 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| WO2017148995A1 (en) | 2016-03-04 | 2017-09-08 | Bayer Pharma Aktiengesellschaft | 1-(pyrimidin-2-yl)-1h-indazoles having bub1 kinase inhibiting activity |
| US9775839B2 (en) | 2014-03-13 | 2017-10-03 | Merck Sharp & Dohme Corp. | 2-pyrazine carboxamides as spleen tyrosine kinase inhibitors |
| US9783531B2 (en) | 2013-12-20 | 2017-10-10 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9796685B2 (en) | 2014-12-16 | 2017-10-24 | Signal Pharmaceuticals, Llc | Formulations of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-Methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US9815813B2 (en) | 2014-01-17 | 2017-11-14 | Novartis Ag | 1-(triazin-3-yl/pyridazin-3-yl)-piper(-azine)idine derivatives and compositions therefor for inhibiting the activity of SHP2 |
| US9822107B2 (en) | 2013-12-20 | 2017-11-21 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9834571B2 (en) | 2012-05-05 | 2017-12-05 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| US9834518B2 (en) | 2011-05-04 | 2017-12-05 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| WO2018004306A1 (en) | 2016-06-30 | 2018-01-04 | Daewoong Pharmaceutical Co., Ltd. | Pyrazolopyrimidine derivatives as kinase inhibitor |
| US9890156B2 (en) | 2015-02-20 | 2018-02-13 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| WO2018081488A1 (en) * | 2016-10-28 | 2018-05-03 | Bristol-Myers Squibb Company | Heterobicyclic compounds useful as modulators of il-12, il-23 and/or ifn alpha responses |
| WO2018117177A1 (en) | 2016-12-21 | 2018-06-28 | 小野薬品工業株式会社 | Brk INHIBITOR COMPOUND |
| US10039753B2 (en) | 2015-09-14 | 2018-08-07 | Pfizer Inc. | Imidazo[4,5-c]quinoline and imidazo[4,5-c][1,5]naphthyridine derivatives as LRRK2 inhibitors |
| US20180244676A1 (en) * | 2015-02-13 | 2018-08-30 | Dana-Farber Cancer Institute, Inc. | Lrrk2 inhibitors and methods of making and using the same |
| US10077276B2 (en) | 2014-01-17 | 2018-09-18 | Novartis Ag | N-azaspirocycloalkane substituted N-heteroaryl compounds and compositions for inhibiting the activity of SHP2 |
| US10093646B2 (en) | 2014-01-17 | 2018-10-09 | Novartis Ag | 1-pyridazin-/triazin-3-yl-piper(-azine)/idine/pyrolidine derivatives and compositions thereof for inhibiting the activity of SHP2 |
| WO2018155916A3 (en) * | 2017-02-22 | 2018-12-06 | 재단법인 대구경북첨단의료산업진흥재단 | Pyrrolo-pyrimidine derivative compound, preparation method therefor, and pharmaceutical composition comprising same compound as effective ingredient for preventing or treating protein kinase-related disease |
| EP3312182A4 (en) * | 2015-06-22 | 2018-12-12 | Ono Pharmaceutical Co., Ltd. | Brk inhibitory compound |
| US10213427B2 (en) | 2010-12-22 | 2019-02-26 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| WO2019043208A1 (en) | 2017-09-04 | 2019-03-07 | F. Hoffmann-La Roche Ag | Dihydroquinolinones |
| US10252981B2 (en) | 2015-07-24 | 2019-04-09 | Celgene Corporation | Methods of synthesis of (1R,2R,5R)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
| US10287266B2 (en) | 2015-06-19 | 2019-05-14 | Novartis Ag | Compounds and compositions for inhibiting the activity of SHP2 |
| US10308660B2 (en) | 2015-06-19 | 2019-06-04 | Novartis Ag | Compounds and compositions for inhibiting the activity of SHP2 |
| US10370371B2 (en) | 2013-08-30 | 2019-08-06 | Ptc Therapeutics, Inc. | Substituted pyrimidine Bmi-1 inhibitors |
| US10428050B2 (en) | 2012-11-21 | 2019-10-01 | Ptc Therapeutics, Inc. | Substituted reverse pyrimidine Bmi-1 inhibitors |
| US10533011B2 (en) | 2015-10-09 | 2020-01-14 | ACEA Therapeutics, Inc. | Pharmaceutical salts, physical forms, and compositions of pyrrolopyrimidine kinase inhibitors, and methods of making same |
| US10562918B2 (en) | 2013-07-11 | 2020-02-18 | ACEA Therapeutics, Inc. | Heterocyclic compounds and uses thereof |
| US10584115B2 (en) | 2013-11-21 | 2020-03-10 | Ptc Therapeutics, Inc. | Substituted pyridine and pyrazine BMI-1 inhibitors |
| US10596174B2 (en) | 2012-01-13 | 2020-03-24 | ACEA Therapeutics, Inc. | Pyrrolopyrimidine compounds as inhibitors of protein kinases |
| US10611762B2 (en) | 2017-05-26 | 2020-04-07 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| US10689351B2 (en) | 2015-01-29 | 2020-06-23 | Signal Pharmaceuticals, Llc | Isotopologues of 2-(tert butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| WO2020188015A1 (en) | 2019-03-21 | 2020-09-24 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| WO2020235902A1 (en) * | 2019-05-17 | 2020-11-26 | 주식회사 보로노이 | Heterocycle-fused pyrimidine derivative and use thereof |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10934285B2 (en) | 2016-06-14 | 2021-03-02 | Novartis Ag | Compounds and compositions for inhibiting the activity of SHP2 |
| US10975080B2 (en) | 2015-06-19 | 2021-04-13 | Novartis Ag | Compounds and compositions for inhibiting the activity of SHP2 |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| US11174257B2 (en) | 2018-05-04 | 2021-11-16 | Incyte Corporation | Salts of an FGFR inhibitor |
| US11407750B2 (en) | 2019-12-04 | 2022-08-09 | Incyte Corporation | Derivatives of an FGFR inhibitor |
| US11466004B2 (en) | 2018-05-04 | 2022-10-11 | Incyte Corporation | Solid forms of an FGFR inhibitor and processes for preparing the same |
| EP3915991A4 (en) * | 2019-01-18 | 2022-10-12 | Voronoi Co., Ltd. | Pyrrolopyrimidine derivative, and pharmaceutical composition for preventing or treating protein kinase-related disease comprising same as active ingredient |
| US11498922B2 (en) | 2017-04-07 | 2022-11-15 | ACEA Therapeutics, Inc. | Pharmaceutical composition comprising N-(3-((2-((3-fluoro-4-(4-methylpiperazin-1-yl phenyl)amino)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)phenylacrylamide |
| AU2021201532B2 (en) * | 2015-07-09 | 2022-11-24 | Merck Patent Gmbh | Pyrimidine derivatives as BTK inhibitors and uses thereof |
| US11524955B2 (en) | 2016-06-27 | 2022-12-13 | Rigel Pharmaceuticals, Inc. | 2,4-diamino-pyrimidine compounds and method for making and using the compounds |
| EP3697787A4 (en) * | 2017-10-18 | 2022-12-14 | HK inno.N Corporation | Heterocyclic compound as a protein kinase inhibitor |
| US11607416B2 (en) | 2019-10-14 | 2023-03-21 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| WO2023055731A1 (en) * | 2021-09-28 | 2023-04-06 | Sanford Burnham Prebys Medical Discovery Institute | Inhibitors of serine/threonine protein kinase stk3 or stk4 and uses thereof |
| US11628162B2 (en) | 2019-03-08 | 2023-04-18 | Incyte Corporation | Methods of treating cancer with an FGFR inhibitor |
| EP3998254A4 (en) * | 2019-07-04 | 2023-08-09 | Shenyang Pharmaceutical University | 2-aminopyrimidine compound and application thereof |
| US11897891B2 (en) | 2019-12-04 | 2024-02-13 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US11939331B2 (en) | 2021-06-09 | 2024-03-26 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US12012409B2 (en) | 2020-01-15 | 2024-06-18 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US12053470B2 (en) | 2017-01-10 | 2024-08-06 | Novartis Ag | Pharmaceutical combination comprising an ALK inhibitor and a SHP2 inhibitor |
| US12065494B2 (en) | 2021-04-12 | 2024-08-20 | Incyte Corporation | Combination therapy comprising an FGFR inhibitor and a Nectin-4 targeting agent |
| US12084453B2 (en) | 2021-12-10 | 2024-09-10 | Incyte Corporation | Bicyclic amines as CDK12 inhibitors |
| US12122767B2 (en) | 2020-09-30 | 2024-10-22 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
Families Citing this family (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2101819B1 (en) | 2006-11-20 | 2013-01-09 | President and Fellows of Harvard College | Methods, compositions, and kits for treating pain and pruritis |
| NZ589315A (en) | 2008-04-16 | 2012-11-30 | Portola Pharm Inc | 2,6-diamino-pyrimidin-5-yl-carboxamides as Spleen tryosine kinase (syk) or Janus kinase (JAK) inhibitors |
| US8138339B2 (en) | 2008-04-16 | 2012-03-20 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| US9273077B2 (en) | 2008-05-21 | 2016-03-01 | Ariad Pharmaceuticals, Inc. | Phosphorus derivatives as kinase inhibitors |
| AU2009259867A1 (en) * | 2008-06-20 | 2009-12-23 | Genentech, Inc. | Triazolopyridine JAK inhibitor compounds and methods |
| CN102131389A (en) * | 2008-06-20 | 2011-07-20 | 健泰科生物技术公司 | Triazolopyridine JAK inhibitor compounds and methods |
| CA3027255C (en) | 2009-07-10 | 2022-06-21 | The General Hospital Corporation | Permanently charged sodium and calcium channel blockers as anti-inflammatory agents |
| CN102372717B (en) * | 2010-08-20 | 2014-06-18 | 和记黄埔医药(上海)有限公司 | Pyrrolopyrimidine compound and use thereof |
| IN2014CN04065A (en) | 2011-11-23 | 2015-09-04 | Portola Pharm Inc | |
| KR101794321B1 (en) | 2012-08-14 | 2017-12-01 | 수안주 파마 코포레이션 리미티드 | Bicyclic Group Substituted Pyrimidine Compounds |
| WO2014060371A1 (en) * | 2012-10-19 | 2014-04-24 | F. Hoffmann-La Roche Ag | Inhibitors of syk |
| WO2014085154A1 (en) | 2012-11-27 | 2014-06-05 | Beth Israel Deaconess Medical Center, Inc. | Methods for treating renal disease |
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| EP2970138B1 (en) | 2013-03-13 | 2019-01-16 | Canadian Blood Services | Pyrazole derivatives and their uses thereof |
| CN104177363B (en) * | 2013-05-24 | 2018-06-05 | 江苏先声药业有限公司 | Bicyclic heterocycle amine Hedgehog signal pathway inhibitors |
| CN104311573B (en) * | 2013-09-18 | 2017-12-15 | 北京韩美药品有限公司 | Suppress the compound of BTK and/or JAK3 kinase activities |
| GB201401086D0 (en) * | 2014-01-23 | 2014-03-12 | Galapagos Nv | Novel compounds and pharmaceutical compositions thereof for the treatment of inflammatory disorders |
| JP6517319B2 (en) | 2014-03-28 | 2019-05-22 | キャリター・サイエンシーズ・リミテッド・ライアビリティ・カンパニーCalitor Sciences, Llc | Substituted heteroaryl compounds and methods of use |
| CN104086551B (en) * | 2014-06-06 | 2016-09-21 | 人福医药集团股份公司 | Compound and its production and use |
| PL3244891T3 (en) * | 2015-01-16 | 2022-12-27 | The General Hospital Corporation | Compounds for improving mrna splicing |
| CN105837577B (en) * | 2015-02-02 | 2019-01-25 | 四川大学 | 1- (pyrimidine-4-yl) 3- aminopiperidine derivatives and its preparation method and application |
| CN106188060A (en) * | 2015-04-29 | 2016-12-07 | 厦门大学 | Pyrimido azoles, its preparation method, Pharmaceutical composition and application thereof |
| WO2017011720A1 (en) * | 2015-07-16 | 2017-01-19 | Signal Pharmaceuticals, Llc | Solod forms 4-((4-(cyclopentyloxy)-5-(2-methylbenzo[d] oxazol-6-yl)17h-pyrrolo[2,3-d]pyrimidin-2-yl)amino)-3-methoxy-n-methylbenzamide, compositions thereof and methods of their use |
| JP6833811B2 (en) | 2015-08-03 | 2021-02-24 | プレジデント アンド フェローズ オブ ハーバード カレッジ | Charged ion channel blockers and how to use |
| WO2017024589A1 (en) * | 2015-08-13 | 2017-02-16 | 北京韩美药品有限公司 | Irak4 inhibitor and use thereof |
| EP3336090B1 (en) * | 2015-08-13 | 2020-07-22 | Beijing Hanmi Pharmaceutical Co., Ltd. | Irak4 inhibitor and use thereof |
| US10711002B2 (en) | 2016-04-21 | 2020-07-14 | The Royal Institution For The Advancement Of Learning/Mcgill University | Purine compounds and method for the treatment of cancer |
| CN106187899B (en) * | 2016-06-28 | 2019-07-16 | 绍兴文理学院 | A kind of synthetic method of fluoro azepine aromatic hydrocarbons |
| CN106349224A (en) * | 2016-08-03 | 2017-01-25 | 山东大学 | JAK kinase inhibitor with 4-amino-(1H)-pyrazole structure and preparation method and application thereof |
| JP2019536820A (en) * | 2016-12-01 | 2019-12-19 | アプトセ バイオサイエンシーズ インコーポレイテッド | Fused cyclic pyrimidine compounds as dual inhibitors of BRAD4 and JAK2 and methods for their use |
| SI3733673T1 (en) | 2017-12-28 | 2022-09-30 | Daewoong Pharmaceutical Co., Ltd. | Oxy-fluoropiperidine derivative as kinase inhibitor |
| EP3915985A4 (en) | 2019-01-18 | 2022-09-28 | Voronoi Co., Ltd. | Pyrrolopyridine derivative and use thereof in prevention and treatment of protein kinase-related disease |
| AU2020237474A1 (en) | 2019-03-11 | 2021-09-30 | Nocion Therapeutics, Inc. | Charged ion channel blockers and methods for use |
| CA3129089A1 (en) | 2019-03-11 | 2020-09-17 | Bridget Mccarthy Cole | Ester substituted ion channel blockers and methods for use |
| US10780083B1 (en) | 2019-03-11 | 2020-09-22 | Nocion Therapeutics, Inc. | Charged ion channel blockers and methods for use |
| US10828287B2 (en) | 2019-03-11 | 2020-11-10 | Nocion Therapeutics, Inc. | Charged ion channel blockers and methods for use |
| US11377422B2 (en) | 2019-03-11 | 2022-07-05 | Nocion Therapeutics, Inc. | Charged ion channel blockers and methods for use |
| US11591329B2 (en) | 2019-07-09 | 2023-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| WO2021076728A1 (en) | 2019-10-16 | 2021-04-22 | Incyte Corporation | Bicyclic heterocycles as fgfr inhibitors |
| US10933055B1 (en) | 2019-11-06 | 2021-03-02 | Nocion Therapeutics, Inc. | Charged ion channel blockers and methods for use |
| KR20220123381A (en) | 2019-11-06 | 2022-09-06 | 녹시온 테라퓨틱스 인코포레이티드 | Charged Ion Channel Blockers and Methods of Use |
| EP4118070A4 (en) | 2020-03-11 | 2024-04-10 | Nocion Therapeutics, Inc. | Charged ion channel blockers and methods for use |
| WO2021238908A1 (en) * | 2020-05-25 | 2021-12-02 | 上海翰森生物医药科技有限公司 | Salt and crystal form of heteroaryl derivative, and preparation method therefor |
| CN111892580B (en) * | 2020-09-29 | 2021-02-05 | 北京鑫开元医药科技有限公司 | 2-amino-4- (isoindoline-2-yl) pyrimidine-5-formamide derivative, preparation method and application |
| CN112107580A (en) * | 2020-09-29 | 2020-12-22 | 北京鑫开元医药科技有限公司 | Spleen tyrosine kinase inhibitor preparation composition and preparation method thereof |
| WO2022203466A1 (en) * | 2021-03-26 | 2022-09-29 | 주식회사 스탠다임 | Novel phenylaminopyrimidine compound having inhibitory activity against lrrk2 and use thereof |
| AU2022265032A1 (en) * | 2021-04-30 | 2023-11-16 | Wigen Biomedicine Technology (shanghai) Co., Ltd. | Fused ring compound as wee-1 inhibitor, and preparation method therefor and use thereof |
| CN118005609A (en) * | 2022-11-09 | 2024-05-10 | 沈阳药科大学 | 2-Aminopyrimidine compound and application thereof |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001009134A1 (en) * | 1999-07-30 | 2001-02-08 | Novartis Ag | Purine derivatives inhibitors of tyrosine protein kinase syk |
| WO2002051843A1 (en) * | 2000-12-26 | 2002-07-04 | Aventis Pharma S.A. | Novel purine derivatives, preparation method and use as medicines |
| WO2005097135A2 (en) * | 2004-04-05 | 2005-10-20 | Novartis Ag | Use of 9h-purine-2,6-diamine derivatives in the treatment of proliferative diseases and novel 9h-purine-2,6-diamine derivatives |
| WO2007042298A1 (en) * | 2005-10-13 | 2007-04-19 | Glaxo Group Limited | Pyrrolopyrimidine derivatives as syk inhibitors |
| US20070142402A1 (en) * | 2005-12-15 | 2007-06-21 | Rigel Pharmaceuticals, Inc. | Kinase Inhibitors And Their Uses |
| WO2007071393A2 (en) * | 2005-12-22 | 2007-06-28 | Novartis Ag | Sulphonamidoaniline derivatives being janus kinases inhibitors |
| WO2008009458A1 (en) * | 2006-07-21 | 2008-01-24 | Novartis Ag | 2, 4 -di (arylaminio) -pyrimidine-5-carboxamide compounds as jak kinases inhibitors |
Family Cites Families (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5728536A (en) * | 1993-07-29 | 1998-03-17 | St. Jude Children's Research Hospital | Jak kinases and regulation of Cytokine signal transduction |
| CN1104118C (en) * | 1995-05-19 | 2003-03-26 | 西门子公司 | Process for computer-controlled exchange of cryptographic keys between first and second computer unit |
| US6316635B1 (en) * | 1995-06-07 | 2001-11-13 | Sugen, Inc. | 2-indolinone derivatives as modulators of protein kinase activity |
| US6696448B2 (en) * | 1996-06-05 | 2004-02-24 | Sugen, Inc. | 3-(piperazinylbenzylidenyl)-2-indolinone compounds and derivatives as protein tyrosine kinase inhibitors |
| US6316429B1 (en) * | 1997-05-07 | 2001-11-13 | Sugen, Inc. | Bicyclic protein kinase inhibitors |
| US6486185B1 (en) * | 1997-05-07 | 2002-11-26 | Sugen, Inc. | 3-heteroarylidene-2-indolinone protein kinase inhibitors |
| GB9718913D0 (en) * | 1997-09-05 | 1997-11-12 | Glaxo Group Ltd | Substituted oxindole derivatives |
| US6133305A (en) * | 1997-09-26 | 2000-10-17 | Sugen, Inc. | 3-(substituted)-2-indolinones compounds and use thereof as inhibitors of protein kinase activity |
| AU4851599A (en) | 1998-06-30 | 2000-01-17 | Parker Hughes Institute | Method for inhibiting c-jun expression using jak-3 inhibitors |
| WO2000010981A1 (en) | 1998-08-21 | 2000-03-02 | Parker Hughes Institute | Quinazoline derivatives |
| GB9828511D0 (en) | 1998-12-24 | 1999-02-17 | Zeneca Ltd | Chemical compounds |
| US6841567B1 (en) | 1999-02-12 | 2005-01-11 | Cephalon, Inc. | Cyclic substituted fused pyrrolocarbazoles and isoindolones |
| US6624171B1 (en) * | 1999-03-04 | 2003-09-23 | Smithkline Beecham Corporation | Substituted aza-oxindole derivatives |
| GB9904995D0 (en) | 1999-03-04 | 1999-04-28 | Glaxo Group Ltd | Substituted aza-oxindole derivatives |
| US6080747A (en) * | 1999-03-05 | 2000-06-27 | Hughes Institute | JAK-3 inhibitors for treating allergic disorders |
| GB9905075D0 (en) * | 1999-03-06 | 1999-04-28 | Zeneca Ltd | Chemical compounds |
| US7125875B2 (en) | 1999-04-15 | 2006-10-24 | Bristol-Myers Squibb Company | Cyclic protein tyrosine kinase inhibitors |
| US7655423B2 (en) | 1999-06-14 | 2010-02-02 | Henry Ford Health System | Nitric oxide donors for inducing neurogenesis |
| CA2392554A1 (en) | 1999-11-30 | 2001-06-28 | Parker Hughes Institute | Inhibitors of thrombin induced platelet aggregation |
| EA006227B1 (en) | 1999-12-10 | 2005-10-27 | Пфайзер Продактс Инк. | PIRROLO(2,3-d)PYRIMIDINE COMPOUNDS |
| ATE299881T1 (en) * | 1999-12-21 | 2005-08-15 | Sugen Inc | 4-SUBSTITUTED 7-AZA-INDOLIN-2-ONE AND THEIR APPLICATION AS PROTEIN KINASE INHIBITORS |
| US20020065270A1 (en) | 1999-12-28 | 2002-05-30 | Moriarty Kevin Joseph | N-heterocyclic inhibitors of TNF-alpha expression |
| EP1250137B1 (en) | 2000-01-24 | 2007-08-15 | Genzyme Corporation | Jak/stat pathway inhibitors and the use thereof for the treatment of primary generalized osteoarthritis |
| IL145756A0 (en) * | 2000-02-05 | 2002-07-25 | Vertex Pharma | Pyrazole derivatives and pharmaceutical compositions containing the same |
| CN1362953A (en) | 2000-02-05 | 2002-08-07 | 沃泰克斯药物股份有限公司 | Pyrazole compositions useful as inhibitors of ERK |
| GB0004888D0 (en) * | 2000-03-01 | 2000-04-19 | Astrazeneca Uk Ltd | Chemical compounds |
| US6608048B2 (en) * | 2000-03-28 | 2003-08-19 | Wyeth Holdings | Tricyclic protein kinase inhibitors |
| AR028261A1 (en) | 2000-03-28 | 2003-04-30 | Wyeth Corp | TRICICLIC INHIBITORS OF PROTEIN QUINASA |
| CA2412560C (en) | 2000-06-26 | 2008-12-30 | Pfizer Products Inc. | Pyrrolo[2,3-d]pyrimidine compounds as immunosuppressive agents |
| WO2002043735A1 (en) | 2000-11-29 | 2002-06-06 | Parker Hughes Institute | Inhibitors of thrombin induced platelet aggregation |
| US20020115173A1 (en) * | 2000-12-11 | 2002-08-22 | Children's Medical Center Corporation | Short peptides from the 'A-region' of protein kinases which selectively modulate protein kinase activity |
| JP2004518669A (en) * | 2000-12-20 | 2004-06-24 | スージェン・インコーポレーテッド | 4-Aryl substituted indolinone |
| WO2002059110A1 (en) | 2000-12-21 | 2002-08-01 | Glaxo Group Limited | Pyrimidineamines as angiogenesis modulators |
| AUPR279101A0 (en) * | 2001-01-30 | 2001-02-22 | Cytopia Pty Ltd | Protein kinase signalling |
| CA2436487A1 (en) * | 2001-01-30 | 2002-08-08 | Cytopia Pty Ltd. | Methods of inhibiting kinases |
| KR100874791B1 (en) * | 2001-05-29 | 2008-12-18 | 바이엘 쉐링 파마 악티엔게젤샤프트 | CDV-inhibited pyrimidine, preparation method thereof and use as medicament |
| US7301023B2 (en) | 2001-05-31 | 2007-11-27 | Pfizer Inc. | Chiral salt resolution |
| DE60232510D1 (en) | 2001-06-15 | 2009-07-16 | Vertex Pharma | 5- (2-AMINOPYRIMIDIN-4-YL) BENZOXOXOLEA AS PROTEIN KINASE INHIBITOR |
| US6433018B1 (en) * | 2001-08-31 | 2002-08-13 | The Research Foundation Of State University Of New York | Method for reducing hypertrophy and ischemia |
| AU2002334355A1 (en) | 2001-09-06 | 2003-03-18 | Prochon Biotech Ltd. | Protein tyrosine kinase inhibitors |
| EP1436259A1 (en) * | 2001-09-10 | 2004-07-14 | Congxin Liang | 3-(4,5,6,7-tetrahydroindol-2-ylmethylidiene)-2-indolinone derivatives as kinase inhibitors |
| WO2003030909A1 (en) | 2001-09-25 | 2003-04-17 | Bayer Pharmaceuticals Corporation | 2- and 4-aminopyrimidines n-substtituded by a bicyclic ring for use as kinase inhibitors in the treatment of cancer |
| GT200200234A (en) | 2001-12-06 | 2003-06-27 | NEW CRYSTAL COMPOUNDS | |
| TWI329105B (en) | 2002-02-01 | 2010-08-21 | Rigel Pharmaceuticals Inc | 2,4-pyrimidinediamine compounds and their uses |
| US6998391B2 (en) * | 2002-02-07 | 2006-02-14 | Supergen.Inc. | Method for treating diseases associated with abnormal kinase activity |
| AU2003209077A1 (en) | 2002-02-08 | 2003-09-02 | Smithkline Beecham Corporation | Pyrimidine compounds |
| AU2003220970A1 (en) | 2002-03-01 | 2003-09-16 | Smithkline Beecham Corporation | Diamino-pyrimidines and their use as angiogenesis inhibitors |
| JP4681302B2 (en) | 2002-05-06 | 2011-05-11 | バーテックス ファーマシューティカルズ インコーポレイテッド | Thiadiazole or oxadiazole and their use as inhibitors of JAK protein kinase |
| WO2003095448A1 (en) | 2002-05-06 | 2003-11-20 | Bayer Pharmaceuticals Corporation | Pyridinyl amino pyrimidine derivatives useful for treating hyper-proliferative disorders |
| AU2003231880A1 (en) * | 2002-05-30 | 2003-12-19 | Vertex Pharmaceuticals Incorporated | Inhibitors of jak and cdk2 protein kinases |
| JP2005529954A (en) | 2002-06-17 | 2005-10-06 | スミスクライン ビーチャム コーポレーション | Chemical process |
| DE60330466D1 (en) | 2002-07-29 | 2010-01-21 | Rigel Pharmaceuticals Inc | METHOD FOR THE TREATMENT OR PREVENTION OF AUTOIMMUNE DISEASES WITH 2,4-PYRIMIDINE-IAMINE COMPOUNDS |
| NZ538715A (en) | 2002-08-14 | 2007-07-27 | Vertex Pharma | Protein kinase inhibitors and uses thereof |
| CA2506772A1 (en) | 2002-11-01 | 2004-05-21 | Vertex Pharmaceuticals Incorporated | Compositions useful as inhibitors of jak and other protein kinases |
| WO2004041814A1 (en) | 2002-11-04 | 2004-05-21 | Vertex Pharmaceuticals Incorporated | Heteroaryl-pyramidine derivatives as jak inhibitors |
| WO2004041810A1 (en) | 2002-11-05 | 2004-05-21 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of jak and other protein kinases |
| CL2003002353A1 (en) | 2002-11-15 | 2005-02-04 | Vertex Pharma | COMPOUNDS DERIVED FROM DIAMINOTRIAZOLS, INHIBITORS D ELA PROTEINA QUINASA; PHARMACEUTICAL COMPOSITION; PREPARATION PROCEDURE; AND ITS USE OF THE COMPOUND IN THE TREATMENT OF DISEASES OF ALLERGIC DISORDERS, PROLIFERATION, AUTOIMMUNES, CONDIC |
| CN1726192A (en) | 2002-11-21 | 2006-01-25 | 辉瑞产品公司 | 3-amino-piperadine derivatives and methods of manufacture |
| PL378246A1 (en) | 2002-11-26 | 2006-03-20 | Pfizer Products Inc. | Method of treatment of transplant rejection |
| AU2003297160A1 (en) | 2002-12-18 | 2004-07-22 | Vertex Pharmaceuticals Incorporated | Benzisoxazole derivatives useful as inhibitors of protein kinases |
| WO2004092154A1 (en) | 2003-04-03 | 2004-10-28 | Vertex Pharmaceuticals Incorporated | Compositions useful as inhibitors of protein kinases |
| CN100457728C (en) | 2003-07-24 | 2009-02-04 | 麦克公司 | Tyrosine kinase inhibitors |
| NZ545270A (en) * | 2003-07-30 | 2010-04-30 | Rigel Pharmaceuticals Inc | 2,4-Pyrimidinediamine compounds for use in the treatment or prevention of autoimmune diseases |
| WO2005016344A1 (en) | 2003-08-14 | 2005-02-24 | Pfizer Limited | Piperazine derivatives for the treatment of hiv infections |
| EP2287156B1 (en) | 2003-08-15 | 2013-05-29 | Novartis AG | 2,4-Di(phenylamino)-pyrimidines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders |
| AR045595A1 (en) | 2003-09-04 | 2005-11-02 | Vertex Pharma | USEFUL COMPOSITIONS AS INHIBITORS OF KINASE PROTEINS |
| AU2004278158B2 (en) | 2003-10-03 | 2009-08-13 | Pfizer Inc. | Imidazopyridine substituted tropane derivatives with CCR5 receptor antagonist activity for the treatment of HIV and inflammation |
| DE10349423A1 (en) | 2003-10-16 | 2005-06-16 | Schering Ag | Sulfoximine-substituted parimidines as CDK and / or VEGF inhibitors, their preparation and use as medicaments |
| PT1704145E (en) | 2004-01-12 | 2012-09-04 | Ym Biosciences Australia Pty | Selective kinase inhibitors |
| WO2005122294A1 (en) | 2004-06-14 | 2005-12-22 | Nec Corporation | Electric device packed in film |
| ES2380550T3 (en) | 2004-11-24 | 2012-05-16 | Rigel Pharmaceuticals, Inc. | Spiro-2,4-pyrimidinediamine compounds and their uses |
| EP2161275A1 (en) | 2005-01-19 | 2010-03-10 | Rigel Pharmaceuticals, Inc. | Prodrugs of 2,4-pyrimidinediamine compounds and their uses |
| US20060270694A1 (en) * | 2005-05-03 | 2006-11-30 | Rigel Pharmaceuticals, Inc. | JAK kinase inhibitors and their uses |
| KR20100015353A (en) * | 2007-04-02 | 2010-02-12 | 팔라우 파르마 에스에이 | Pyrrolopyrimidine derivatives as jak3 inhibitors |
-
2009
- 2009-04-22 JP JP2011506293A patent/JP2011518219A/en not_active Withdrawn
- 2009-04-22 NZ NZ588830A patent/NZ588830A/en not_active IP Right Cessation
- 2009-04-22 WO PCT/US2009/002512 patent/WO2009131687A2/en active Application Filing
- 2009-04-22 CA CA2723185A patent/CA2723185A1/en not_active Abandoned
- 2009-04-22 EP EP09734422.0A patent/EP2271631B1/en active Active
- 2009-04-22 US US12/386,848 patent/US8258144B2/en active Active
- 2009-04-22 CN CN2013100825419A patent/CN103224497A/en active Pending
- 2009-04-22 AU AU2009238590A patent/AU2009238590A1/en not_active Abandoned
- 2009-04-22 BR BRPI0910668A patent/BRPI0910668A2/en not_active IP Right Cessation
- 2009-04-22 CN CN2009801223945A patent/CN102066338A/en active Pending
-
2010
- 2010-10-14 IL IL208719A patent/IL208719A0/en unknown
-
2012
- 2012-06-20 US US13/528,709 patent/US9139581B2/en active Active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001009134A1 (en) * | 1999-07-30 | 2001-02-08 | Novartis Ag | Purine derivatives inhibitors of tyrosine protein kinase syk |
| WO2002051843A1 (en) * | 2000-12-26 | 2002-07-04 | Aventis Pharma S.A. | Novel purine derivatives, preparation method and use as medicines |
| WO2005097135A2 (en) * | 2004-04-05 | 2005-10-20 | Novartis Ag | Use of 9h-purine-2,6-diamine derivatives in the treatment of proliferative diseases and novel 9h-purine-2,6-diamine derivatives |
| WO2007042298A1 (en) * | 2005-10-13 | 2007-04-19 | Glaxo Group Limited | Pyrrolopyrimidine derivatives as syk inhibitors |
| US20070142402A1 (en) * | 2005-12-15 | 2007-06-21 | Rigel Pharmaceuticals, Inc. | Kinase Inhibitors And Their Uses |
| WO2007071393A2 (en) * | 2005-12-22 | 2007-06-28 | Novartis Ag | Sulphonamidoaniline derivatives being janus kinases inhibitors |
| WO2008009458A1 (en) * | 2006-07-21 | 2008-01-24 | Novartis Ag | 2, 4 -di (arylaminio) -pyrimidine-5-carboxamide compounds as jak kinases inhibitors |
Non-Patent Citations (2)
| Title |
|---|
| JOERGENSEN A ET AL: "Phosphorus pentoxide in organic synthesis. XXXVI. Synthesis of 7H-pyrrolo[2,3-d]pyrimidine-2,4-diamines and 7-diazaisoguanines from 7H-pyrrolo[2,3-d]pyrimidine-2,4-diones" CHEMICA SCRIPTA, ALMQVIST OG WIKSELL. STOCKHOLM, SE, vol. 28, no. 2, 1 January 1988 (1988-01-01), pages 201-204, XP008096228 * |
| See also references of EP2271631A2 * |
Cited By (211)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2120573A4 (en) * | 2007-02-12 | 2011-05-25 | Merck Sharp & Dohme | Piperidine derivatives |
| EP2120573A1 (en) * | 2007-02-12 | 2009-11-25 | Merck & Co., Inc. | Piperidine derivatives |
| US9012462B2 (en) | 2008-05-21 | 2015-04-21 | Ariad Pharmaceuticals, Inc. | Phosphorous derivatives as kinase inhibitors |
| WO2010038081A2 (en) * | 2008-10-03 | 2010-04-08 | Astrazeneca Ab | Heterocyclic derivatives and methods of use thereof |
| WO2010038081A3 (en) * | 2008-10-03 | 2010-05-27 | Astrazeneca Ab | 4-amino-5- (hetero) aryl-2-phenylamino-pyrimidine or pyridine and their use as dna gyrase and/or topoisomerase iv inhibitors |
| US20150119288A1 (en) * | 2009-03-23 | 2015-04-30 | Nodality, Inc. | Kits for multiparametric phospho analysis |
| AU2011218167B2 (en) * | 2010-02-17 | 2014-07-10 | Amgen Inc. | Aryl carboxamide derivatives as sodium channel inhibitors for treatment of pain |
| US9334269B2 (en) | 2010-02-17 | 2016-05-10 | Amgen Inc. | Carboxamides as inhibitors of voltage-gated sodium channels |
| JP2013527175A (en) * | 2010-05-11 | 2013-06-27 | アムジェン インコーポレイテッド | Pyrimidine compounds that inhibit anaplastic lymphoma kinase |
| WO2011143033A1 (en) | 2010-05-11 | 2011-11-17 | Amgen Inc. | Pyrimidine compounds that inhibit anaplastic lymphoma kinase |
| EP2600874A1 (en) * | 2010-05-11 | 2013-06-12 | Amgen, Inc | Pyrimidine compounds that inhibit anaplastic lymphoma kinase |
| EP2600874A4 (en) * | 2010-05-11 | 2013-12-11 | Amgen Inc | Pyrimidine compounds that inhibit anaplastic lymphoma kinase |
| WO2012002577A1 (en) | 2010-06-30 | 2012-01-05 | 富士フイルム株式会社 | Novel nicotinamide derivatives or salts thereof |
| US8895585B2 (en) | 2010-06-30 | 2014-11-25 | Fujifilm Corporation | Nicotinamide derivative or salt thereof |
| US9586961B2 (en) | 2010-07-09 | 2017-03-07 | Leo Pharma A/S | Homopiperazine derivatives as protein tyrosine kinase inhibitors and pharmaceutical use thereof |
| CN103282352B (en) * | 2010-11-01 | 2016-08-10 | 波托拉医药品公司 | Benzamides and nicotinamide as SYK regulator |
| CN103282352A (en) * | 2010-11-01 | 2013-09-04 | 波托拉医药品公司 | Benzamides and nicotinamides as syk modulators |
| US10213427B2 (en) | 2010-12-22 | 2019-02-26 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US10813930B2 (en) | 2010-12-22 | 2020-10-27 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| WO2012092880A1 (en) | 2011-01-07 | 2012-07-12 | Centaurus Biopharma Co., Ltd. | 2,4-DIAMINO-6,7-DIHYDRO-5H-PYRROLO[2,3]PYRIMIDINE DERIVATIVES AS FAK/Pyk2 INHIBITORS |
| CN103476776A (en) * | 2011-01-07 | 2013-12-25 | 北京赛林泰医药技术有限公司 | 2,4-diamino-6,7-dihydro-5H-pyrrolo[2,3]pyrimidine derivatives as FAK/Pyk2 inhibitors |
| AU2012204982B2 (en) * | 2011-01-07 | 2017-02-23 | Centaurus Biopharma Co., Ltd. | 2,4-diamino-6,7-dihydro-5H-pyrrolo[2,3]pyrimidine derivatives as FAK/Pyk2 inhibitors |
| CN103476776B (en) * | 2011-01-07 | 2016-09-28 | 北京赛林泰医药技术有限公司 | 2,4-diaminourea-6,7-dihydro-5H-pyrrolo-[2,3] pyrimidine derivatives as FAK/Pyk2 inhibitor |
| US9428508B2 (en) | 2011-01-07 | 2016-08-30 | Centaurus Biopharma Co., Ltd. | 2,4-diamino-6,7-dihydro-5H-pyrrolo[2,3]pyrimidine derivatives as FAK/Pyk2 inhibitors |
| EP2489663A1 (en) | 2011-02-16 | 2012-08-22 | Almirall, S.A. | Compounds as syk kinase inhibitors |
| WO2012123312A1 (en) * | 2011-03-11 | 2012-09-20 | Glaxo Group Limited | Pyrido[3,4-b]pyrazine derivatives as syk inhibitors |
| JP2014508808A (en) * | 2011-03-23 | 2014-04-10 | アムジエン・インコーポレーテツド | Condensed tricyclic dual inhibitors of CDK4 / 6 and FLT3 |
| US11325890B2 (en) | 2011-04-22 | 2022-05-10 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| US10040770B2 (en) | 2011-04-22 | 2018-08-07 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| US9139534B2 (en) | 2011-04-22 | 2015-09-22 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| US10919865B2 (en) | 2011-04-22 | 2021-02-16 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| US9701643B2 (en) | 2011-04-22 | 2017-07-11 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| US10266500B2 (en) | 2011-04-22 | 2019-04-23 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
| US9834518B2 (en) | 2011-05-04 | 2017-12-05 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| US9120785B2 (en) | 2011-05-10 | 2015-09-01 | Merck Sharp & Dohme Corp. | Pyridyl aminopyridines as Syk inhibitors |
| US9145391B2 (en) | 2011-05-10 | 2015-09-29 | Merck Sharp & Dohme Corp. | Bipyridylaminopyridines as Syk inhibitors |
| US9290490B2 (en) | 2011-05-10 | 2016-03-22 | Merck Sharp & Dohme Corp. | Aminopyrimidines as Syk inhibitors |
| WO2013017461A1 (en) | 2011-08-01 | 2013-02-07 | Almirall, S.A. | Pyridin-2(1h)-one derivatives as jak inhibitors |
| EP2554544A1 (en) | 2011-08-01 | 2013-02-06 | Almirall, S.A. | Pyridin-2(1h)-one derivatives as jak inhibitors |
| US9216173B2 (en) | 2011-10-05 | 2015-12-22 | Merck Sharp & Dohme Corp. | 2-Pyridyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US8987456B2 (en) | 2011-10-05 | 2015-03-24 | Merck Sharp & Dohme Corp. | 3-pyridyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9006444B2 (en) | 2011-10-05 | 2015-04-14 | Merck Sharp & Dohme Corp. | Phenyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9051310B2 (en) | 2011-12-28 | 2015-06-09 | Fujifilm Corporation | Nicotinamide derivative or salt thereof |
| WO2013099041A1 (en) | 2011-12-28 | 2013-07-04 | 富士フイルム株式会社 | Novel nicotinamide derivative or salt thereof |
| US9763949B2 (en) | 2012-01-13 | 2017-09-19 | Acea Biosciences Inc. | EGFR modulators and uses thereof |
| US9464089B2 (en) | 2012-01-13 | 2016-10-11 | Acea Biosciences Inc. | Heterocyclic compounds and uses thereof |
| US9920074B2 (en) | 2012-01-13 | 2018-03-20 | Acea Biosciences Inc. | Heterocyclic compounds and uses thereof |
| US9586965B2 (en) | 2012-01-13 | 2017-03-07 | Acea Biosciences Inc. | Pyrrolo[2,3-d]pyrimidine compounds as inhibitors of protein kinases |
| US10799504B2 (en) | 2012-01-13 | 2020-10-13 | ACEA Therapeutics, Inc. | Heterocyclic compounds and uses as anticancer agents |
| US11612602B2 (en) | 2012-01-13 | 2023-03-28 | ACEA Therapeutics, Inc. | Heterocyclic compounds and uses as anticancer agents |
| US10596174B2 (en) | 2012-01-13 | 2020-03-24 | ACEA Therapeutics, Inc. | Pyrrolopyrimidine compounds as inhibitors of protein kinases |
| US9034885B2 (en) | 2012-01-13 | 2015-05-19 | Acea Biosciences Inc. | EGFR modulators and uses thereof |
| US9834571B2 (en) | 2012-05-05 | 2017-12-05 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| US10131667B2 (en) | 2012-06-13 | 2018-11-20 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US9611267B2 (en) | 2012-06-13 | 2017-04-04 | Incyte Holdings Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US11840534B2 (en) | 2012-06-13 | 2023-12-12 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US11053246B2 (en) | 2012-06-13 | 2021-07-06 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US9487504B2 (en) | 2012-06-20 | 2016-11-08 | Merck Sharp & Dohme Corp. | Imidazolyl analogs as syk inhibitors |
| US9242984B2 (en) | 2012-06-20 | 2016-01-26 | Merck Sharp & Dohme Corp. | Pyrazolyl derivatives as Syk inhibitors |
| US9416111B2 (en) | 2012-06-22 | 2016-08-16 | Merck Sharp & Dohme Corp. | Substituted diazine and triazine spleen tyrosine kinease (Syk) inhibitors |
| US9376418B2 (en) | 2012-06-22 | 2016-06-28 | Merck Sharp & Dohme Corp. | Substituted pyridine spleen tyrosine kinase (SYK) inhibitors |
| US9156845B2 (en) | 2012-06-29 | 2015-10-13 | Pfizer Inc. | 4-(substituted amino)-7H-pyrrolo[2,3-d] pyrimidines as LRRK2 inhibitors |
| US9642855B2 (en) | 2012-06-29 | 2017-05-09 | Pfizer Inc. | Substituted pyrrolo[2,3-d]pyrimidines as LRRK2 inhibitors |
| US11007197B2 (en) | 2012-08-06 | 2021-05-18 | ACEA Therapeutics, Inc. | EGFR modulators and uses thereof |
| US10449196B2 (en) | 2012-08-06 | 2019-10-22 | ACEA Therapeutics, Inc. | EGFR modulators and uses thereof |
| US8685988B2 (en) | 2012-08-06 | 2014-04-01 | Acea Biosciences, Inc. | EGFR modulators and uses thereof |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9745311B2 (en) | 2012-08-10 | 2017-08-29 | Incyte Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9353066B2 (en) | 2012-08-20 | 2016-05-31 | Merck Sharp & Dohme Corp. | Substituted phenyl-Spleen Tyrosine Kinase (Syk) inhibitors |
| US9586931B2 (en) | 2012-09-28 | 2017-03-07 | Merck Sharp & Dohme Corp. | Triazolyl derivatives as Syk inhibitors |
| US10428050B2 (en) | 2012-11-21 | 2019-10-01 | Ptc Therapeutics, Inc. | Substituted reverse pyrimidine Bmi-1 inhibitors |
| US9624210B2 (en) | 2012-12-12 | 2017-04-18 | Merck Sharp & Dohme Corp. | Amino-pyrimidine-containing spleen tyrosine kinase (Syk) inhibitors |
| US9428509B2 (en) | 2013-01-16 | 2016-08-30 | Signal Pharmaceuticals, Llc | Substituted pyrrolopyrimidine compounds, compositions thereof, and methods of treatment therewith |
| US9346812B2 (en) | 2013-01-16 | 2016-05-24 | Signal Pharmaceuticals, Llc | Substituted pyrrolopyrimidine compounds, compositions thereof, and methods of treatment therewith |
| WO2014113429A3 (en) * | 2013-01-16 | 2014-10-09 | Signal Pharmaceuticals, Llc | Substituted pyrrolopyrimidine compounds, compositions thereof, and methods of treatment therewith |
| CN105188704A (en) * | 2013-01-16 | 2015-12-23 | 西格诺药品有限公司 | Substituted pyrrolopyrimidine compounds, compositions thereof, and methods of treatment therewith |
| EP3202403A1 (en) * | 2013-01-16 | 2017-08-09 | Signal Pharmaceuticals, LLC | Substituted pyrrolopyrimidine compounds, compositions thereof, and methods of treatment therewith |
| US9795607B2 (en) | 2013-01-16 | 2017-10-24 | Signal Pharmaceuticals, Llc | Substituted pyrrolopyrimidine compounds, compositions thereof, and methods of treatment therewith |
| CN105188704B (en) * | 2013-01-16 | 2017-09-19 | 西格诺药品有限公司 | Substituted Pyrrolopyrimidine compounds, its composition and use its treatment method |
| WO2014130693A1 (en) * | 2013-02-25 | 2014-08-28 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US9708326B2 (en) | 2013-02-25 | 2017-07-18 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| US9611283B1 (en) | 2013-04-10 | 2017-04-04 | Ariad Pharmaceuticals, Inc. | Methods for inhibiting cell proliferation in ALK-driven cancers |
| US10450313B2 (en) | 2013-04-19 | 2019-10-22 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US9533984B2 (en) | 2013-04-19 | 2017-01-03 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10040790B2 (en) | 2013-04-19 | 2018-08-07 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10947230B2 (en) | 2013-04-19 | 2021-03-16 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11530214B2 (en) | 2013-04-19 | 2022-12-20 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US9745295B2 (en) | 2013-04-26 | 2017-08-29 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9499534B2 (en) | 2013-04-26 | 2016-11-22 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminopyrimidines as spleen tyrosine kinase inhibitors |
| EP3006439A4 (en) * | 2013-05-24 | 2016-11-02 | Yuhan Corp | Bicyclic derivative containing pyrimidine ring, and preparation method therefor |
| CN105452238A (en) * | 2013-05-24 | 2016-03-30 | 株式会社柳韩洋行 | Bicyclic derivative containing pyrimidine ring, and preparation method therefor |
| US10562918B2 (en) | 2013-07-11 | 2020-02-18 | ACEA Therapeutics, Inc. | Heterocyclic compounds and uses thereof |
| US10370371B2 (en) | 2013-08-30 | 2019-08-06 | Ptc Therapeutics, Inc. | Substituted pyrimidine Bmi-1 inhibitors |
| WO2015061247A3 (en) * | 2013-10-21 | 2015-07-23 | Merck Patent Gmbh | Heteroaryl compounds as btk inhibitors and uses thereof |
| RU2703301C2 (en) * | 2013-10-21 | 2019-10-16 | Мерк Патент Гмбх | Heteroaryl compounds as btk inhibitors and use thereof |
| US10329270B2 (en) | 2013-10-21 | 2019-06-25 | Merck Patent Gmbh | Heteroaryl compounds as BTK inhibitors and uses thereof |
| US10584115B2 (en) | 2013-11-21 | 2020-03-10 | Ptc Therapeutics, Inc. | Substituted pyridine and pyrazine BMI-1 inhibitors |
| US9382246B2 (en) | 2013-12-05 | 2016-07-05 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| US9656988B2 (en) | 2013-12-05 | 2017-05-23 | Pharmacyclics Llc | Inhibitors of Bruton's tyrosine kinase |
| US9695171B2 (en) | 2013-12-17 | 2017-07-04 | Pfizer Inc. | 3,4-disubstituted-1 H-pyrrolo[2,3-b]pyridines and 4,5-disubstituted-7H-pyrrolo[2,3-c]pyridazines as LRRK2 inhibitors |
| US9822107B2 (en) | 2013-12-20 | 2017-11-21 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9783531B2 (en) | 2013-12-20 | 2017-10-10 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9670196B2 (en) | 2013-12-20 | 2017-06-06 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as Spleen Tyrosine Kinase inhibitors |
| US10077276B2 (en) | 2014-01-17 | 2018-09-18 | Novartis Ag | N-azaspirocycloalkane substituted N-heteroaryl compounds and compositions for inhibiting the activity of SHP2 |
| US11401259B2 (en) | 2014-01-17 | 2022-08-02 | Novartis Ag | 1-Pyridazin-/triazin-3-yl-piper(-azine)/idine/pyrolidine derivatives and compositions thereof for inhibiting the activity of SHP2 |
| US11952386B2 (en) | 2014-01-17 | 2024-04-09 | Novartis Ag | N-azaspirocycloalkane substituted N-heteroaryl compounds and compositions for inhibiting the activity of SHP2 |
| US10301278B2 (en) | 2014-01-17 | 2019-05-28 | Novartis Ag | 1-(triazin-3-yl/pyridazin-3-yl)-piper(-azine)idine derivatives and compositions therefor for inhibiting the activity of SHP2 |
| US10968235B2 (en) | 2014-01-17 | 2021-04-06 | Novartis Ag | N-azaspirocycloalkane substituted N-heteroaryl compounds and compositions for inhibiting the activity of SHP2 |
| US10336774B2 (en) | 2014-01-17 | 2019-07-02 | Novartis Ag | N-azaspirocycloalkane substituted N-heteroaryl compounds and compositions for inhibiting the activity of SHP2 |
| US9815813B2 (en) | 2014-01-17 | 2017-11-14 | Novartis Ag | 1-(triazin-3-yl/pyridazin-3-yl)-piper(-azine)idine derivatives and compositions therefor for inhibiting the activity of SHP2 |
| US10093646B2 (en) | 2014-01-17 | 2018-10-09 | Novartis Ag | 1-pyridazin-/triazin-3-yl-piper(-azine)/idine/pyrolidine derivatives and compositions thereof for inhibiting the activity of SHP2 |
| US10774065B2 (en) | 2014-01-17 | 2020-09-15 | Novartis Ag | 1-pyridazin-/triazin-3-yl-piper(-azine)/idine/pyrolidine derivatives and compositions thereof for inhibiting the activity of SHP2 |
| US10226461B2 (en) | 2014-01-30 | 2019-03-12 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US9814713B2 (en) | 2014-01-30 | 2017-11-14 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US11241430B2 (en) | 2014-01-30 | 2022-02-08 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US9365524B2 (en) | 2014-01-30 | 2016-06-14 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US10517873B2 (en) | 2014-01-30 | 2019-12-31 | Signal Pharmaceuticals, Llc | Solid forms of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide, compositions thereof and methods of their use |
| US9775839B2 (en) | 2014-03-13 | 2017-10-03 | Merck Sharp & Dohme Corp. | 2-pyrazine carboxamides as spleen tyrosine kinase inhibitors |
| WO2015164330A1 (en) | 2014-04-21 | 2015-10-29 | Millennium Pharmaceuticals, Inc. | Anti-psyk antibody molecules and use of same for syk-targeted therapy |
| US9434735B2 (en) | 2014-07-14 | 2016-09-06 | Signal Pharmaceuticals, Llc | Amorphous form of 4-((4-(cyclopentyloxy)-5-(2-methylbenzo[d]oxazol-6-yl)-7h-pyrrolo[2,3-d]pyrimidin-2-yl)amino)-3-methoxy-n-methylbenzamide, compositions thereof and methods of their use |
| US9623028B2 (en) | 2014-07-14 | 2017-04-18 | Signal Pharmaceuticals, Llc | Methods of treating a cancer using substituted pyrrolopyrimidine compounds, compositions thereof |
| EP3169686A4 (en) * | 2014-07-14 | 2018-01-24 | Signal Pharmaceuticals, LLC | Methods of treating a cancer using substituted pyrrolopyrimidine compounds, compositions thereof |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9513297B2 (en) | 2014-12-16 | 2016-12-06 | Signal Pharmaceuticals, Llc | Methods for measurement of inhibition of c-Jun N-terminal kinase in skin |
| US9796685B2 (en) | 2014-12-16 | 2017-10-24 | Signal Pharmaceuticals, Llc | Formulations of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-Methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US10590089B2 (en) | 2014-12-16 | 2020-03-17 | Signal Pharmaceuticals, Llc | Formulations of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US10197579B2 (en) | 2014-12-16 | 2019-02-05 | Signal Pharmaceuticals, Llc | Methods for measurement of inhibition of c-Jun N-terminal kinase in skin |
| US10131639B2 (en) | 2014-12-16 | 2018-11-20 | Signal Pharmaceuticals, Llc | Formulations of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US10975039B2 (en) | 2015-01-29 | 2021-04-13 | Signal Pharmaceuticals, Llc | Isotopologues of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US11492332B2 (en) | 2015-01-29 | 2022-11-08 | Signal Pharmaceuticals, Llc | Isotopologues of 2-(tert-butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| US10689351B2 (en) | 2015-01-29 | 2020-06-23 | Signal Pharmaceuticals, Llc | Isotopologues of 2-(tert butylamino)-4-((1R,3R,4R)-3-hydroxy-4-methylcyclohexylamino)-pyrimidine-5-carboxamide |
| EP3778605A3 (en) * | 2015-02-13 | 2021-03-10 | Dana Farber Cancer Institute, Inc. | Lrrk2 inhibitors and methods of making and using the same |
| US20180244676A1 (en) * | 2015-02-13 | 2018-08-30 | Dana-Farber Cancer Institute, Inc. | Lrrk2 inhibitors and methods of making and using the same |
| US10913744B2 (en) * | 2015-02-13 | 2021-02-09 | Dana-Farber Cancer Institute, Inc. | LRRK2 inhibitors and methods of making and using the same |
| US9580423B2 (en) | 2015-02-20 | 2017-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10016438B2 (en) | 2015-02-20 | 2018-07-10 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11667635B2 (en) | 2015-02-20 | 2023-06-06 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9890156B2 (en) | 2015-02-20 | 2018-02-13 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10632126B2 (en) | 2015-02-20 | 2020-04-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10251892B2 (en) | 2015-02-20 | 2019-04-09 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11014923B2 (en) | 2015-02-20 | 2021-05-25 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10214528B2 (en) | 2015-02-20 | 2019-02-26 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11173162B2 (en) | 2015-02-20 | 2021-11-16 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9708318B2 (en) | 2015-02-20 | 2017-07-18 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10738048B2 (en) | 2015-02-20 | 2020-08-11 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9801889B2 (en) | 2015-02-20 | 2017-10-31 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10975080B2 (en) | 2015-06-19 | 2021-04-13 | Novartis Ag | Compounds and compositions for inhibiting the activity of SHP2 |
| US10287266B2 (en) | 2015-06-19 | 2019-05-14 | Novartis Ag | Compounds and compositions for inhibiting the activity of SHP2 |
| US10308660B2 (en) | 2015-06-19 | 2019-06-04 | Novartis Ag | Compounds and compositions for inhibiting the activity of SHP2 |
| US10377759B2 (en) | 2015-06-22 | 2019-08-13 | Ono Pharmaceutical Co., Ltd. | Brk inhibitory compound |
| US11512087B2 (en) | 2015-06-22 | 2022-11-29 | Ono Pharmaceutical Co., Ltd. | BRK inhibitory compound |
| RU2719477C2 (en) * | 2015-06-22 | 2020-04-17 | Оно Фармасьютикал Ко., Лтд. | Brk inhibiting compound |
| EP3312182A4 (en) * | 2015-06-22 | 2018-12-12 | Ono Pharmaceutical Co., Ltd. | Brk inhibitory compound |
| AU2016284402B2 (en) * | 2015-06-22 | 2020-04-02 | Ono Pharmaceutical Co., Ltd. | Brk inhibitory compound |
| AU2021201532B2 (en) * | 2015-07-09 | 2022-11-24 | Merck Patent Gmbh | Pyrimidine derivatives as BTK inhibitors and uses thereof |
| US10774033B2 (en) | 2015-07-24 | 2020-09-15 | Celgene Corporation | Methods of synthesis of (1R, 2R, 5R)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
| US11780801B2 (en) | 2015-07-24 | 2023-10-10 | Celgene Corporation | Methods of synthesis of (1R,2R,5R)-5-amino-2-methyl-cyclohexanol hydrochloride and intermediates useful therein |
| US10252981B2 (en) | 2015-07-24 | 2019-04-09 | Celgene Corporation | Methods of synthesis of (1R,2R,5R)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
| US11192847B2 (en) | 2015-07-24 | 2021-12-07 | Celgene Corporation | Methods of synthesis of (1R,2R,5R)-5-amino-2-methylcyclohexanol hydrochloride and intermediates useful therein |
| US10039753B2 (en) | 2015-09-14 | 2018-08-07 | Pfizer Inc. | Imidazo[4,5-c]quinoline and imidazo[4,5-c][1,5]naphthyridine derivatives as LRRK2 inhibitors |
| US10526309B2 (en) | 2015-10-02 | 2020-01-07 | The University Of North Carolina At Chapel Hill | Pan-TAM inhibitors and Mer/Axl dual inhibitors |
| WO2017059280A1 (en) * | 2015-10-02 | 2017-04-06 | The University Of North Carolina At Chapel Hill | Novel pan-tam inhibitors and mer/axl dual inhibitors |
| US10533011B2 (en) | 2015-10-09 | 2020-01-14 | ACEA Therapeutics, Inc. | Pharmaceutical salts, physical forms, and compositions of pyrrolopyrimidine kinase inhibitors, and methods of making same |
| WO2017148995A1 (en) | 2016-03-04 | 2017-09-08 | Bayer Pharma Aktiengesellschaft | 1-(pyrimidin-2-yl)-1h-indazoles having bub1 kinase inhibiting activity |
| US10934285B2 (en) | 2016-06-14 | 2021-03-02 | Novartis Ag | Compounds and compositions for inhibiting the activity of SHP2 |
| US11905283B2 (en) | 2016-06-14 | 2024-02-20 | Novartis Ag | Compounds and compositions for inhibiting the activity of SHP2 |
| US11524955B2 (en) | 2016-06-27 | 2022-12-13 | Rigel Pharmaceuticals, Inc. | 2,4-diamino-pyrimidine compounds and method for making and using the compounds |
| RU2714206C1 (en) * | 2016-06-30 | 2020-02-13 | Даевунг Фармасьютикал Ко., Лтд. | Pyrazolopyrimidine derivatives as a kinase inhibitor |
| WO2018004306A1 (en) | 2016-06-30 | 2018-01-04 | Daewoong Pharmaceutical Co., Ltd. | Pyrazolopyrimidine derivatives as kinase inhibitor |
| US10961247B2 (en) | 2016-06-30 | 2021-03-30 | Daewoong Pharmaceutical Co., Ltd. | Pyrazolopyrimidine derivatives as kinase inhibitor |
| WO2018081488A1 (en) * | 2016-10-28 | 2018-05-03 | Bristol-Myers Squibb Company | Heterobicyclic compounds useful as modulators of il-12, il-23 and/or ifn alpha responses |
| US10781215B2 (en) | 2016-10-28 | 2020-09-22 | Bristol-Myers Squibb Company | Heterobicyclic compounds useful as modulators of IL-12, IL-23 and/or IFNα responses |
| KR102477063B1 (en) * | 2016-10-28 | 2022-12-12 | 브리스톨-마이어스 스큅 컴퍼니 | Heterobicyclic compounds useful as modulators of IL-12, IL-23 and/or IFN alpha responses |
| CN110114357A (en) * | 2016-10-28 | 2019-08-09 | 百时美施贵宝公司 | It can be used as the Heterobicyclic compounds of the regulator of IL-12, IL-23 and/or IFN α reaction |
| CN110114357B (en) * | 2016-10-28 | 2022-05-31 | 百时美施贵宝公司 | Hetero-bicyclic compounds useful as modulators of IL-12, IL-23, and/or IFN alpha response |
| KR20190068614A (en) * | 2016-10-28 | 2019-06-18 | 브리스톨-마이어스 스큅 컴퍼니 | Heterobicyclic compounds useful as modulators of IL-12, IL-23 and / or IFN alpha reactions |
| WO2018117177A1 (en) | 2016-12-21 | 2018-06-28 | 小野薬品工業株式会社 | Brk INHIBITOR COMPOUND |
| US11052091B2 (en) | 2016-12-21 | 2021-07-06 | Ono Pharmaceutical Co., Ltd. | BRK inhibitory compound |
| US12053470B2 (en) | 2017-01-10 | 2024-08-06 | Novartis Ag | Pharmaceutical combination comprising an ALK inhibitor and a SHP2 inhibitor |
| US11208412B2 (en) | 2017-02-22 | 2021-12-28 | Daegu-Gyeongbuk Medical Innovation Foundation | Pyrrolo-pyrimidine derivative compound, preparation method therefor, and pharmaceutical composition comprising same compound as effective ingredient for preventing or treating protein kinase-related disease |
| WO2018155916A3 (en) * | 2017-02-22 | 2018-12-06 | 재단법인 대구경북첨단의료산업진흥재단 | Pyrrolo-pyrimidine derivative compound, preparation method therefor, and pharmaceutical composition comprising same compound as effective ingredient for preventing or treating protein kinase-related disease |
| US11498922B2 (en) | 2017-04-07 | 2022-11-15 | ACEA Therapeutics, Inc. | Pharmaceutical composition comprising N-(3-((2-((3-fluoro-4-(4-methylpiperazin-1-yl phenyl)amino)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)phenylacrylamide |
| US11472801B2 (en) | 2017-05-26 | 2022-10-18 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| US10611762B2 (en) | 2017-05-26 | 2020-04-07 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| WO2019043208A1 (en) | 2017-09-04 | 2019-03-07 | F. Hoffmann-La Roche Ag | Dihydroquinolinones |
| US11401256B2 (en) | 2017-09-04 | 2022-08-02 | C4 Therapeutics, Inc. | Dihydroquinolinones for medical treatment |
| CN111278816B (en) * | 2017-09-04 | 2024-03-15 | C4医药公司 | Dihydro quinolinones |
| US12091397B2 (en) | 2017-09-04 | 2024-09-17 | C4 Therapeutics, Inc. | Dihydroquinolinones for medical treatment |
| CN111278816A (en) * | 2017-09-04 | 2020-06-12 | C4医药公司 | Dihydroquinolinones |
| EP3697787A4 (en) * | 2017-10-18 | 2022-12-14 | HK inno.N Corporation | Heterocyclic compound as a protein kinase inhibitor |
| US11174257B2 (en) | 2018-05-04 | 2021-11-16 | Incyte Corporation | Salts of an FGFR inhibitor |
| US12024517B2 (en) | 2018-05-04 | 2024-07-02 | Incyte Corporation | Salts of an FGFR inhibitor |
| US11466004B2 (en) | 2018-05-04 | 2022-10-11 | Incyte Corporation | Solid forms of an FGFR inhibitor and processes for preparing the same |
| EP3915991A4 (en) * | 2019-01-18 | 2022-10-12 | Voronoi Co., Ltd. | Pyrrolopyrimidine derivative, and pharmaceutical composition for preventing or treating protein kinase-related disease comprising same as active ingredient |
| US11628162B2 (en) | 2019-03-08 | 2023-04-18 | Incyte Corporation | Methods of treating cancer with an FGFR inhibitor |
| WO2020188015A1 (en) | 2019-03-21 | 2020-09-24 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| WO2020235902A1 (en) * | 2019-05-17 | 2020-11-26 | 주식회사 보로노이 | Heterocycle-fused pyrimidine derivative and use thereof |
| EP3998254A4 (en) * | 2019-07-04 | 2023-08-09 | Shenyang Pharmaceutical University | 2-aminopyrimidine compound and application thereof |
| US12083124B2 (en) | 2019-10-14 | 2024-09-10 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11607416B2 (en) | 2019-10-14 | 2023-03-21 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| US11407750B2 (en) | 2019-12-04 | 2022-08-09 | Incyte Corporation | Derivatives of an FGFR inhibitor |
| US11897891B2 (en) | 2019-12-04 | 2024-02-13 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US12012409B2 (en) | 2020-01-15 | 2024-06-18 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| US12122767B2 (en) | 2020-09-30 | 2024-10-22 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US12065494B2 (en) | 2021-04-12 | 2024-08-20 | Incyte Corporation | Combination therapy comprising an FGFR inhibitor and a Nectin-4 targeting agent |
| US11939331B2 (en) | 2021-06-09 | 2024-03-26 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| WO2023055731A1 (en) * | 2021-09-28 | 2023-04-06 | Sanford Burnham Prebys Medical Discovery Institute | Inhibitors of serine/threonine protein kinase stk3 or stk4 and uses thereof |
| US12084453B2 (en) | 2021-12-10 | 2024-09-10 | Incyte Corporation | Bicyclic amines as CDK12 inhibitors |
| US12129237B2 (en) | 2022-03-29 | 2024-10-29 | Signal Pharmaceuticals, Llc | Substituted diaminocarboxamide and diaminocarbonitrile pyrimidines, compositions thereof, and methods of treatment therewith |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2011518219A (en) | 2011-06-23 |
| EP2271631B1 (en) | 2018-07-04 |
| EP2271631A2 (en) | 2011-01-12 |
| NZ588830A (en) | 2012-11-30 |
| US20130029944A1 (en) | 2013-01-31 |
| WO2009131687A3 (en) | 2010-01-07 |
| US9139581B2 (en) | 2015-09-22 |
| BRPI0910668A2 (en) | 2019-09-24 |
| US20090298823A1 (en) | 2009-12-03 |
| US8258144B2 (en) | 2012-09-04 |
| CA2723185A1 (en) | 2009-10-29 |
| AU2009238590A1 (en) | 2009-10-29 |
| CN102066338A (en) | 2011-05-18 |
| IL208719A0 (en) | 2010-12-30 |
| CN103224497A (en) | 2013-07-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11414410B2 (en) | Inhibitors of protein kinases | |
| US9139581B2 (en) | Inhibitors of protein kinases | |
| KR101623997B1 (en) | 2,6-diamino-pyrimidin-5-yl-carboxamides as syk or jak kinases inhibitors | |
| US8952027B2 (en) | Inhibitors of syk and JAK protein kinases | |
| WO2009136995A2 (en) | Inhibitors of syk protein kinase | |
| EP2975027A1 (en) | Nicotinamides as jak kinase modulators | |
| AU2009251863B2 (en) | 2, 6-diamino-pyrimidin- 5-yl-carboxamides as syk or JAK kinases inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980122394.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09734422 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2723185 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011506293 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009734422 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 588830 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009238590 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 7300/CHENP/2010 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2009238590 Country of ref document: AU Date of ref document: 20090422 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: PI0910668 Country of ref document: BR Kind code of ref document: A2 Effective date: 20101021 |