WO2009118753A2 - Process for preparation of naratriptan hydrochloride - Google Patents
Process for preparation of naratriptan hydrochloride Download PDFInfo
- Publication number
- WO2009118753A2 WO2009118753A2 PCT/IN2009/000165 IN2009000165W WO2009118753A2 WO 2009118753 A2 WO2009118753 A2 WO 2009118753A2 IN 2009000165 W IN2009000165 W IN 2009000165W WO 2009118753 A2 WO2009118753 A2 WO 2009118753A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- naratriptan
- methyl
- indole
- preparation
- Prior art date
Links
- 0 C*S(CCc(cc1)ccc1NN=C(C)*)(=*)=O Chemical compound C*S(CCc(cc1)ccc1NN=C(C)*)(=*)=O 0.000 description 4
- GUWAGLRUTZOGPY-UHFFFAOYSA-N CCCC(CCC)c1c[nH]c2c1C=C(CCS(NC)(=O)=O)CC2 Chemical compound CCCC(CCC)c1c[nH]c2c1C=C(CCS(NC)(=O)=O)CC2 GUWAGLRUTZOGPY-UHFFFAOYSA-N 0.000 description 1
- CJTDQTJKWVZIAZ-UHFFFAOYSA-N CN(CC1)CCC1c1c[nH]c2c1cc(CCS(N)(=O)=O)cc2 Chemical compound CN(CC1)CCC1c1c[nH]c2c1cc(CCS(N)(=O)=O)cc2 CJTDQTJKWVZIAZ-UHFFFAOYSA-N 0.000 description 1
- NFIXDHOWGMUDCT-UHFFFAOYSA-N CNC(CCCc1ccc2[nH]ccc2c1)=O Chemical compound CNC(CCCc1ccc2[nH]ccc2c1)=O NFIXDHOWGMUDCT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Definitions
- the present invention relates to an improved process for the preparation of N-methyl-3-
- N-methyl-3-( 1 -methyl-4-piperidinyl)- 1 H-indole-5-ethanesulfonamide hydrochloride is also known by the name "Naratriptan hydrochloride.”
- Naratriptan hydrochloride exhibits selective vasoconstrictor activity used as the active ingredient in pharmaceutical products and being sold by Glaxo with trademark AMERGETM for treating migraine.
- Naratriptan hydrochloride is an indole derivative disclosed in US 4997841 and US 5066660.
- Process A A solution of 4-hydrazino-N-methyl-benzenethanesulfonamide (II) with 1- methyl-4-piperidineacetaldehyde (A) in a mixture of water and 2N hydrochloric acid followed by stirring for 2 days at room temperature to yield the hydrazone compound which is treated with polyphosphate ester in chloroform to get crude Naratriptan base (IX).
- the crude base (IX) was purified by column chromatography on silica gel using mixture of dichloromethane, ethanol and ammonia.
- the main disadvantage of this process is use of the aldehyde intermediate which is commercially not available and there are no synthetically feasible routes available for its synthesis in the literature.
- the reaction is represented by scheme below. ester
- Process B A mixture of 5-bromo indole (B), N-methyl-4-piperidone and potassium hydroxide in ethanol is refluxed to obtain compound of formula (C).
- the compound (C) is treated with ethanolic HCl followed by hydrogenation in presence of Platinum oxide to obtain 5-bromo-3-(l-methyl-4-piperidinyl)-lH-indole (D).
- the purified compound (E) is hydrogenated in presence of 10% palladium on carbon in ethanolic hydrochloric acid and DMF to get N-methyl-3-(l-methyl-4-piperidinyl)-lH- indole-5-ethanesulfonamide (IX).
- the obtained compound (IX) is treated with ethanolic hydrochloric acid to get Naratriptan hydrochloride (I).
- the reaction is represented by scheme below.
- Process C A mixture of N-methyl-lH-indole-5-ethanesulfonamide (VII), N-methyl-4- piperidone and potassium hydroxide in methanol is refiuxed to produce compound (VIII).
- the compound (VIII) is hydrogenated in the presence of 5% Pd/C to obtain N-Methyl-3- (4-piperidinyl)-lH-indole-5-ethanesulfonamide (IX).
- the compound (IX) is purified by column chromatography on silica gel using a mixture of dichloromethane, ethanol and ammonia. The reaction is represented by the below scheme.
- the purified compound (F) is hydrogenated in presence of 10% palladium oxide on charcoal in mixture of ethyl acetate and methanol to get N-methyl-lH-indole-5-ethanesulfonamide (VII).
- the reported yield is 22-23%.
- the reaction is represented by the below scheme.
- the compound (G) is hydrogenated in presence of 10% palladium oxide on charcoal in a mixture of dimethylformamide, water and 2N hydrochloric acid to get crude Naratriptan hydrochloride, which is crystallized with hot IMS(Industrial methylated spirit)/water to get pure Naratriptan hydrochloride (I).
- the main disadvantage of this process is the use of high quantity of catalyst. The reaction is represented by the scheme below.
- WO2006010079 describes process for preparation of Naratriptan hydrochloride which comprises reaction of diazonium salt of N-Methyl-2-(4-aminophenyl)ethane sulfonamide with ⁇ -ketoester compound , which further upon cyclization, quaternization, reduction (twice), saponification and decarboxylation gives Naratriptan hydrochloride (I). This process is time consuming and economically not viable as it involves many steps.
- the object of the present invention is to provide a simple and cost effective process for the preparation of Naratriptan hydrochloride i.e. N-methyl-3-(l-methyl-4-piperidinyl)-l
- Another object of the invention is to provide novel amorphous Naratriptan hydrochloride and process for preparation thereof.
- Another object of the invention is to provide desmethyl Naratriptan (IA) as a reference standard to quantify the amount of impurity in a sample of Naratriptan or salts thereof.
- IA desmethyl Naratriptan
- Another object of the invention is to provide pharmaceutical compositions comprising
- Naratriptan or pharmaceutically acceptable salts thereof along with at least one pharmaceutically acceptable excipient is provided.
- a process for preparation of N-methyl-2-[3-(l-methyl-4-piperidinyl)- 1 H-indole-5-ethanesulfonamide salts, particularly hydrochloride of formula (I) which comprises the following steps; a) combining compound (VII) with N-Methyl-4-piperidone to produce compound (VIII); b) combining compound (VIII) in suitable solvent with catalyst under reduced condition and adding acid to the obtained reaction mixture to get Naratriptan salt; a) optionally purifying the obtained salt to get pure compound with purity more than 90%.
- process for preparation of key intermediate indole sulfonamide (VII) used for the preparation of Naratriptan hydrochloride which comprises the steps of; a) combining 5-bromo indole (B) with aryl vinyl sulfonate wherein aryl is substituted or unsubstituted phenyl compound to get aryl lH-indole-5-ethenesulfonate (X). b) hydrogenating the compound (X) to get lH-indole-5-aryl ethanesulfonamide (XI); c) combining compound (XI) with methyamine to obtain desired compound (VII).
- the present invention provides process for preparation of key intermediate indole sulfonamide (VII) comprising a) combining compound (X) with methylamine to obtain N-methyl- 1 H- indole-5- ethenesulfonamide (F); b) hydrogenating the compound (X) to get lH-indole-5-aryl ethanesulfonamide (XI) c) treating the obtained compound (XI) with methylamine to get compound (VII).
- a process for preparation of key intermediate indole sulfonamide (VII) used for the preparation of Naratriptan hydrochloride which comprises the steps of; a) combining isethionic acid or salts thereof with chlorinating agent to get chloroethane sulfonyl chloride; b) combining obtained compound of step a) with methylamine in suitable solvent to get N-methyl vinyl sulfonamide;
- the present invention provides process for preparation of Naratriptan hydrochloride comprising the steps of; a) combining hydrazino derivative (II) with a pyruvic acid ester compound (III) in ethanol and methane sulphonic acid at 25- 100 0 C for 2 to 4 hours to obtain hydrazone compound (IV);
- R an alkyl group having 1 to 5 carbon atoms
- R an alkyl group having 1 to 5 carbon atoms (V) c) refluxing a mixture of carboxylic ester (V) and base for 2-3 hours to get carboxylic acid compound (VI);
- the present invention provides process for preparation of desmethyl Naratriptan (IA).
- Another aspect of the invention is to provide desmethyl Naratriptan (IA) as a reference standard to quantify the amount of impurity in a sample of Naratriptan or salts thereof.
- IA desmethyl Naratriptan
- the present invention provides amorphous Naratriptan hydrochloride and process for preparation thereof.
- compositions comprising Naratriptan or pharmaceutically acceptable salts thereof.
- Fig.1 is a characteristic X-ray Powder diffraction pattern of Naratriptan base.
- Fig.2 is a characteristic X-ray Powder diffraction pattern of Naratriptan hydrochloride
- Fig.3 is a characteristic X-ray Powder diffraction pattern of pure Naratriptan hydrochloride.
- Fig. 4 is a characteristic X-ray Powder diffraction pattern of Naratriptan hydrochloride
- Fig. 5 is a characteristic X-ray Powder diffraction pattern of Amorphous Naratriptan hydrochloride Description of the invention:
- the present invention describes process for preparation of Naratriptan hydrochloride (I) having less than 0.15% area by HPLC of 3-(l-methyl-4-piperidinyl)-lH-indole-5- ethanesulfonamide (IA) or desmethyl Naratriptan.
- Another embodiment of the present invention provides process for preparation of Naratriptan salts comprising the steps of: c) combining compound (VII) with N-Methyl-4-piperidone to produce compound (VIII); d) combining compound (VIII) in suitable solvent under reducing condition with catalyst and adding acid to the obtained reaction mixture to get Naratriptan salt; e) optionally purifying the obtained salt to get pure compound with purity more than 90%.
- Naratriptan hydrochloride is prepared by condensing compound (VII) with N-methyl-4-piperidone at temperature of about 60 to 100 0 C for about 8 to 14 hours, preferably 8 to 10 hours in presence of base in suitable solvent to obtain compound (VIII).
- the obtained compound (VIII) is optionally further dissolved in suitable solvent and refluxed for 1-2 hours and filtered.
- the solvent is partially distilled out and the reaction mass is cooled at 10-15 0 C to get pure compound (VIII).
- the compound (VIII) dissolved in suitable solvent is reduced in presence of catalyst and hydrogen gas at temperature of about 25 to 80 0 C, preferably 50 to 60 0 C for about 5 to 12 hours, preferably 6 to 8 hours.
- the compound (VIII) may be reduced using catalytic transfer hydrogenation.
- the reaction is carried out in a solvent or in a mixture of solvent and acid.
- the reaction mixture is filtered and distilled to get an oil, which is stripped with suitable solvent followed by addition of acid and refluxing the obtained reaction mixture for 1-2 hours.
- the precipitated solid is stirred at 5-2O 0 C.
- the solid thus obtained is washed with solvent and dried to get Naratriptan salt with high yield.
- the process of producing Naratriptan salt according to the present invention i.e. without isolating Naratriptan base is economically and industrially feasible.
- Naratriptan hydrochloride obtained according to the present invention is optionally purified by dissolving in suitable solvent at 70-80 0 C for 30 minutes to 1 hour. The solution is filtered and the filtrate is stirred for 30 minutes at 5-20 0 C to get solid of pure Naratriptan hydrochloride having purity more than 90% by HPLC.
- the above reaction is represented by the scheme below,
- the present invention provides process for preparation of N-methyl-lH-indole-5-ethanesulfonamide (VII) used for the preparation of Naratriptan hydrochloride which comprises the steps of; a) combining 5-bromo indole (B) with aryl vinyl sulfonate compound wherein aryl is substituted or unsubstituted phenyl to get lH-indole-5-aryl ethenesulfonamide (X). b) hydrogenating the compound (X) to get lH-indole-5-aryl ethanesulfonamide
- 5-bromo indole (B) is treated with aryl vinyl sulfonate wherein wherein aryl is substituted or unsubstituted phenyl compound in the presence of Pd(OAc)2, tri-o- tolyphosphine and triethylamine in suitable solvent at temperature of about 80 to 120 0 C, preferably about 80 0 C for a period of about 2 to 4 hours to get compound (X).
- the compound (X) is reduced to compound (XI) in the presence of hydrogen gas and catalyst at temperature about 25 to 80 0 C, preferably 40 to 50 0 C for about 8 to 24 hours, preferably 14 to 16 hour.
- the reduction can be carried out in solvent or mixtures of solvents, preferably mixture of methanol and terahydrofuran.
- the compound (XI) is further converted into indole sulfonamide compound (VII) using methylamine in water or organic solvent or mixtures thereof at temperature of about 25 to 80 0 C preferably about 40 to 50 0 C.
- the reaction is carried out for 2 to 6 hours preferably about 3 to 4 hours.
- the above reaction is represented in the scheme below,
- the present invention provides process for preparation of N-methyl-lH-indole-5-ethanesulfonamide (VII) which comprises the steps of; a) combining compound (B) with N-methyl vinyl sulfonamide in suitable solvent to get compound (F). b) reducing obtained compound (F) in the presence of catalyst at temperature about 25 to 80 0 C for about 8 to 24 hours to get compound (VII).
- a mixture of palladium acetate and tri-o-tolyl phosphine in suitable solvent is stirred under nitrogen atmosphere and a mixture of compound (B), N-methyl vinyl sulfonamide and base is added to the reaction mixture followed by heating for 8 to 10 hrs.
- the present invention provides process for preparation of N-methyl-lH-indole-5-ethanesulfonamide (VII) comprises the steps of; a) combining lH-indole-5-aryl ethenesulfonamide (X) with methylamine to obtain N-methyl- 1 H-indole-5-ethenesulfonamide (F); b) reducing obtained compound (F) in the presence of catalyst at temperature about 25 to 80 0 C for about 8 to 24 hours to get compound (VII).
- lH-indole-5-aryl ethenesulfonamide (X) with methylamine in water or suitable solvent or mixtures thereof, preferably mixture of methanol and tetrahydrofuran at temperature of about 25 to 80 0 C preferably about 40 to 50 0 C and high pressure to get compound (F).
- the reaction is carried out for 2 to 6 hours preferably about 3 to 4 hours.
- the obtained compound (F) is further reduced in the presence of hydrogen gas using a catalyst at temperature about 25 to 80 0 C, preferably 40 to 50 0 C for about 8 to 24 hours, preferably 14 to 16 hours to get compound (VII).
- the reduction can be carried out in solvent or mixtures of solvents.
- the above reaction is represented in the scheme below,
- compound (F) is purified by refluxing in suitable solvent to get clear solution.
- the solvent is partially distilled out and reaction mass is cooled at 1O 0 C with stirring to get pure compound (F).
- the obtained compound (F) is optionally further refluxed in a suitable solvent to obtain pure compound (F) in high yield.
- the suitable solvent used is selected from ethanol, methanol, isopropanol, hexane, ethylacetate and acetone or mixtures thereof, preferably mixture of hexane: ethyl acetate.
- N-methyl vinyl sulfonamide is an unstable compound, expensive and not easily available. Also it is difficult to produce N-methyl vinyl sulfonamide at a commercial scale due to its unstability.
- the present invention provides simple process for preparation of N-methyl vinyl sulfonamide which comprises the steps of; a) combining isethionic acid salt with chlorinating agent to get chloroethane sulfonyl chloride; b) optionally distilling the compound obtained in step a) under vacuum; c) treating obtained pure with methylamine to get N- methyl vinyl sulfonamide and d) optionally distilling N-methyl vinyl sulfonamide under high vacuum or short path distillation to get pure N- methyl vinyl sulfonamide.
- thionyl chloride is added dropwise to a mixture of isethionic acid salt in DMF and dichloromethae at 20-30°C.
- the reaction mixture is heated slowly from 30-60°C. After completion of the reaction chilled water is added.
- the reaction mixture is extracted with dichloromethane.
- the solvent is distilled out from the reaction mixture to get an oil which is distilled under vacuum to get pure chloroethane sulfonyl chloride with yield of 90%.
- a mixture of chloroethane sulfonyl chloride in suitable solvent is stirred under nitrogen atmosphere.
- the reaction mixture is cooled to -50 to -60°C followed by addition of hydroquinone in the catalytic amount and purging N- methyl amine gas (aqueous methylamine was added in potassium hydroxide) in the reaction mixture maintaining temperature - 40 to - 60°C.
- the reaction reaction mixture is filtered and filtrate is evaporated under vacuum not more than 50 0 C to get thick oil which is stored under nitrogen atmosphere and distilled using short path distillation or high vacuum distillation to get pure N-methyl vinyl sulfonamide.
- N-methyl vinyl sulfonamide thus obtained has less than about 0.5% of vinyl sulfonamide.
- process for preparation of aryl vinyl sulfonate which comprises the steps of; a) combining isethionic acid or salt thereof with chlorinating agent to get chloroethane sulphonyl chloride; b) optionally distilling the obtained compound of step a) under vacuum; c) adding aryl-OH, wherein aryl is substituted or unsubstituted phenyl to the obtained pure chloroethane sulphonyl chloride to get aryl vinyl sulfonate; d) optionally distilling aryl vinyl sulphonate under high vacuum or short path distillation to get pure aryl vinyl sulfonate.
- thionyl chloride is added to a mixture of isethionic acid salt in DMF and dichloromethane at 20-30°C.
- the obtained reaction mixture is heated slowly at temperature range from 30-60°C.
- After completion of the reaction chilled water is added to the reaction mixture and the reaction mixture is extracted with dichloromethane.
- the solvent is distilled out from the reaction mixture to get oil.
- the obtained oil is distilled under vacuum to get pure chloroethane sulfonyl chloride.
- a mixture of chloroethane sulfonyl chloride and dichloromethane is stirred under nitrogen atmosphere and hydroquinone is added in the catalytic amount.
- the reaction mixture is chilled at -15 to -20 0 C and a mixture of phenol and triethylamine in dichloromethane is added to it maintaining the same temperature followed by stirring at 25 to 30°C for one hour. After completion of reaction dichloromethane is added.
- the separated organic layer is treated with hydrochloric acid followed by aqueous sodium hydroxide and water.
- the organic layer is distilled out to get an oil which is distilled using short path distillation or high vacuum distillation to get pure desired compound.
- the reaction is performed in presence of solvent selected from the group consisting of dimethylformamide, dichloromethane, ethylacetate, tetrahydrofuran.
- the isethionic acid salt is isethionic acid sodium salt.
- the high vacuum used is in the range of 0.1mm to 0.5mm preferably 0.1mm to 0.2 mm optionally using glass bids.
- the methylamine used herein is in the form of solution or gas.
- the treatment of chloroethane sulfonyl chloride with methylamine is performed in presence of catalytic amount of hydroquinone.
- the present invention provides process for preparation of key intermediate N-methyl- lH-indole-5-ethanesulfonamide (VII) comprising the steps of; a) reacting hydrazino derivative (II) with a pyruvic acid ester compound (III) to obtain hydrazone compound (FV); ft
- reaction of the present invention is represented by the following scheme; wherein R represents an alkyl group having 1 to 5 carbon atoms
- the hydrazino derivative (III) is condensed with pyruvic acid ester (II), maintaining the temperature at about 25 to 100 0 C, preferably about 80 0 C for a period of about 2 to 4 hours to obtain hydrazone compound (IV).
- the compound (IV) can be prepared by reacting the compound (II) with compound (III) in presence or absence of catalytic amount of acid in suitable solvent, preferably methanesulphonic acid in ethanol is used.
- the hydrazone compound (IV) is cyclized using an acid in presence or absence of solvent, preferably methane sulphonic acid in acetic acid, for 2-6 hours at temperature 100-120 0 C. After the completion of reaction the obtained solid is neutralized with base to get indole derivative (V).
- the obtained indole derivative (V) is hydrolyzed by refluxing a mixture of compound (V) and suitable base in water or water-solvent mixture for 2-3 hours.
- the obtained reaction mixture is acidified with cone, or dil hydrochloric acid to precipitate out the compound
- the compound (VI) is decarboxylated in the presence of copper catalyst such as cuprous oxide, copper oxide, copper bronze in the presence of quinoline at temperature in the range of about 140 to 220 0 C for 2 to 6 hours to obtain compound (VII).
- copper catalyst such as cuprous oxide, copper oxide, copper bronze
- Another embodiment of the present invention provides a process for preparation of Naratriptan or salts thereof comprising the steps of: a) condensing indole sulfonamide (VII) with N-methyl-4-piperidone at 60 to 100 0 C in presence of base to obtain compound (VIII);
- compound (VII) is further condensed with N-methyl-4-piperidone at temperature of about 60 to 100 0 C for about 8 to 14 hours, preferably 8 to 10 hours in presence of base in suitable solvent to obtain compound (VIII).
- the obtained compound (VIII) is optionally further dissolved in suitable solvent and refluxed for 1-2 hours and filtered.
- the solvent is partially distilled out and the reaction mass is cooled at 10-15 0 C to get pure compound (VIII).
- the obtained compound (VIII) is further reduced using catalyst at temperature of about 25 to 80 0 C, preferably 40 to 50 0 C for about 5 to 12 hours, preferably 8 to 12 hours.
- the reaction mixture is then filtered and filtrate is concentrated.
- Naratriptan base (IX) is characterized by XRPD having the following characteristic peaks at 2 ⁇ angle positions ⁇ 0.2 degrees represented in below table (Fig. 1):
- Naratriptan base (IX) thus obtained is further converted into its acid addition salt (I).
- Naratriptan base (IX) is dissolved in suitable solvent at refluxed temperature for 1-2 hours and cooled at room temperature. An acid is added to the cooled solution and the mixture is refluxed. A mixture of suitable solvent is added to it and refluxed further for 1 hour. The hot solution is filtered and the filtrate is stirred for 1 hour at 5-1O 0 C to get pure desired salt of Naratriptan.
- Naratriptan base (IX) is dissolved in suitable solvent at refluxed temperature for 30 minutes and solution is filtered. The filtrate is cooled to 30-35 0 C and acid is added slowly to it followed by cooling at 10-15 0 C to get crude desired salt of Naratriptan.
- the crude salt of Naratriptan is further dissolved in suitable solvent at refluxed temperature of about 25 to 100 ° C, preferably 60 to 80 ° C and the solution is filtered followed by cooling and stirring at 10-15 0 C for 1 hour to get pure Naratriptan salt with high yield of more than 85% and purity more than 90%, more preferably more than 99.8%.
- Naratriptan hydrochloride obtained according to the present invention has purity of more than 99.8% by HPLC and characterized by XRPD having the following characteristic peaks at 2 ⁇ angle positions ⁇ 0.2 degrees represented in below table (Fig. 2):
- Naratriptan hydrochloride thus obtained may be further purified and characterized by XRPD having the following characteristic peaks at 2 ⁇ angle positions ⁇ 0.2 degrees (Fig. 3):
- Naratriptan hydrochloride is listed in US pharmacopoeia.
- the USP reference standard shows XRPD having the following characteristic peaks at 2 ⁇ angle positions ⁇ 0.2 degrees (Fig. 4).
- Naratriptan HCl obtained by the process of the present invention has particle size cUo less than about 250 ⁇ preferably less than about lOO ⁇ more preferably less than about 80 ⁇ which may be further micronized using conventional micronization techniques to get desired less particle size of Naratriptan or salts thereof.
- the process for preparation of desmethyl Naratriptan comprises the steps of; a) treating chloroethane sulfonyl chloride with ammonia in suitable solvent to get vinyl sulfonamide;
- Step a) A mixture of chloroethane sulfonyl chloride and tetrahydrofuran is stirred under nitrogen atmosphere with cooling to -30 to -60°C followed by purging ammonia gas slowly to the reaction mixture at same temperature. The reaction is monitored on GC. After completion of reaction, the temperature of the reaction is maintained at 25 °C and filtered through hyflow. The solvent is distilled out from the obtained filtrate to get thick oil which is treated with treated with suitable solvent to get vinyl sulfonamide 98% pure on GC in high yield.
- Step c) A mixture of lH-indole-5-ethenesulfonamide (IXa) is hydrogenated using 5-60 % of catalyst (50% wet palladium on charcoal) in a mixture of suitable solvent preferably methanol: ethyl acetate for 2 to 8h at 25°-30° C under hydrogen pressure to get 2-(1H- indol-5yl)-ethane sulfonamide (IXb).
- suitable solvent preferably methanol: ethyl acetate for 2 to 8h at 25°-30° C under hydrogen pressure to get 2-(1H- indol-5yl)-ethane sulfonamide (IXb).
- the reaction mixture is cooled to 25- 40°C and quenched in water at temperature belowlO°C to precipitate out solid of 2-[3-(l, 2,3,6- tetrahydro-l-methyl-4-pyridinyl)-lH-indol-5-yl]ethanesulfonamide (IXc) or extracting the reaction mixture with 1-butanol.
- Step e) A mixture of compound (IXc) is reduced using platinum oxide in a mixture of 50% acetic acid in methanol for 8h at .70 psi at 50 0 C. After completion of reaction, the reaction mixture is filtered and filtrate is concentrated, basified with aqueous sodium carbonate solution and extracted with solvent. The organic layer is separated, washed with EDTA, water and concentrated to get the 2[3-(l,2,3,6-tetrahydro-l-methyl-4- piperidinyl)- 1 H-indol-5-yl]ethanesulfonamide (IA).
- the invention provides desmethyl Naratriptan (IA) as a reference standard to quantify the amount of impurity in a sample of Naratriptan or salts thereof.
- IA desmethyl Naratriptan
- the acid used is selected from the group consisting of methane sulfonic acid, acetic acid, phosphoric acid, hydrochloric acid, sulfuric acid, para-toluenesulphonic acid and lewis acid such as boron trifluoride and the like optionally in presence or absence of solvent.
- the suitable solvent used is alcohol such as methanol, ethanol, propanol, isopropanol, chlorinated solvent such as dichloromethane, chloroform, ethers such as 1,4-dioxane, tetrahydrofuran, esters such as ethyl acetate, isopropyl acetate, butyl acetate, hydrocarbons such as pentane, hexane, heptane, amide such as dimethylformamide, water, acetonitrile or mixtures thereof.
- alcohol such as methanol, ethanol, propanol, isopropanol
- chlorinated solvent such as dichloromethane, chloroform
- ethers such as 1,4-dioxane, tetrahydrofuran
- esters such as ethyl acetate, isopropyl acetate, butyl acetate
- hydrocarbons such as pentane, hexane,
- the base used is selected from the group consisting of sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate potassium carbonate, triethylamine, diisopropylethylamine and the like.
- the catalyst used is selected from the group consisting of palladium oxide/charcoal, palladium/charcoal, platinum oxide/charcoal, platinum/charcoal, rhodium/charcoal in presence of hydrogen gas and used in an amount less than about 60%, preferably 20% more preferably 10%.
- the said methylamine used in above processes is in the form of solution or gas optionally in presence of catalytic amount of hydroquinone.
- the coupling catalyst used is palladium acetate and others which are conventionally known.
- the ligand used is tri-o-tolylphosphine or tri phenyl phosphine.
- the present invention provides amorphous Naratriptan hydrochloride.
- the XRPD of amorphous Naratriptan hydrochloride is shown in Fig.5.
- Another embodiment of the present invention provides process for preparation of amorphous Naratriptan hydrochloride which comprises the steps of ; a) dissolving Naratriptan HCl in a suitable solvent to get clear solution or suspending Naratriptan in suitable solvent and adding conc.HCl to the suspension and b) lyophilizing the resultant solution of step a) to obtain the amorphous Naratriptan hydrochloride.
- the solvent used in step a) is water.
- the concentration of Naratriptan in solution used for lyophilization is 4-10% preferably 4-8 %.
- the solution of Naratriptan is lyophilized for 24 hrs to get an amorphous Form.
- Another embodiment of the present invention provides process for preparation of amorphous Naratriptan hydrochloride which comprises the steps of ; a) dissolving Naratriptan HCl in a suitable solvent to get clear solution or suspending Naratriptan in suitable solvent and adding conc.HCl to the suspension Naratriptan HCl and b) spray drying the resultant clear solution to get the amorphous form.
- the solvent used in step a) is polar protic solvent such as water or Ci -Ci alcohols or mixture thereof, preferably water.
- the alcohols used is selected from methanol and ethanol preferably methanol.
- the concentration of Naratriptan in solution used for spray drying is 4-10% preferably 4-8 %.
- the solution of Naratriptan is spray dried at inlet temperature of 40 to 170 0 C and outlet temperature of 35-85 0 C.
- Another embodiment of the present invention provides process for preparation of amorphous Naratriptan hydrochloride which comprises the steps of ; a) dissolving Naratriptan HCl in a suitable solvent to get clear solution or suspending Naratriptan in suitable solvent and adding conc.HCl to the suspension to get Naratriptan HCl. b) evaporating the solvent from the solution of step a) under vacuum to get amorphous Naratriptan HCl.
- the dissolving solvent used is polar protic solvent such as water or Ci-C 4 alcohols or mixtures thereof, preferably water.
- the alcohols used is methanol, ethanol preferably methanol.
- the solvent from the solution of Naratriptan is evaporated at 65 0 C, under vacuum to get the amorphous Naratriptan HCl.
- compositions comprising an active ingredient and at least one pharmaceutical excipient.
- the pharmaceutical compositions are normally employed as tablets, capsules, powders, syrups, solutions, suspensions and the like, optionally containing flavoring agents, sweeteners, etc., in suitable solid or liquid carriers or diluents; and in suitable sterile media to form injectable solutions or suspensions.
- the pharmaceutical composition preferably a tablet, comprises Naratriptan or pharmaceutically acceptable salts thereof in the range of 0.5-2 % by weight of the composition and suitable pharmaceutically acceptable excipients such as fillers, disintegrants, glidants, lubricants and others known in the pharmaceutical field.
- suitable pharmaceutically acceptable excipients such as fillers, disintegrants, glidants, lubricants and others known in the pharmaceutical field.
- the filler and disintegrants may include Lactose, Microcrystalline cellulose, dibasic calcium phosphate, monobasic calcium phosphate, Mannitol and the like.
- the glidants may include croscarmellose sodium, sodium starch glycolate and the like.
- the lubricants may include stearic acid, calcium stearate and the like.
- the amount of fillers may be in the range of about 40-80 % based on the total weight of the formulation.
- the amount of disintegrants may be in the range of about 20-40 % based on the total weight of the formulation.
- the amount of glidants may be in the range of about 1-10 % based on the total weight of the formulation.
- the amount of lubricants may be in the range of about 0.1-2.5 % based on the total weight of the formulation.
- a method of formulating the composition into tablets may be selected from any conventional methods including wet granulation method, dry granulation method and direct compression method.
- the tablet formed may be optionally film coated. The coating can be performed according to a conventional coating method. Naratriptan hydrochloride prepared according to the present invention is used in the manufacture of pharmaceutical composition for the treatment of migraine.
- N-Methyl-3-(l-methyl-4-piperidinyl)-l H-indole-5- ethanesulfonamide hydrochloride used in the pharmaceutical compositions can be prepared by any processes known in the art and the present invention is not dependent on a particular synthetic method described herein. Further, any polymorphic form of the Naratriptan is suitable for use to prepare Naratriptan hydrochloride of this invention.
- the crystallization of the polymorphic form of this invention can be performed as a final step in a synthesis of the compound, and it is not always necessary to isolate a product having another polymorphic form before performing the crystallization.
- the present invention provides a method of treating a patient suffering from migraine administering to the patient a pharmaceutical compositions comprising Naratriptan or pharmaceutically acceptable salts thereof or its amorphous form prepared according to the present invention.
- Method 2 A mixture of the compound (VII) (51 g, 21.42mmol), N-Methyl-4- pipperidone (60.61g, 53.56mmol) and potassium hydroxide (30g, 53.57mmol) in ethanol
- the compound (VIII) (38g, 11.14mmol) was reduced using platinum oxide (7.6g) in a mixture of 50% acetic acid in methanol (600 ml) for 8h under hydrogen pressure at 40 psi for 8 hours at temperature 40 0 C. After completion of reaction, the reaction mixture was filtered through hyflow. The filtrate was concentrated, basified with aqueous sodium carbonate solution and extracted with ethyl acetate (1 lit). The ethyl acetate layer was separated, washed with EDTA, water and concentrated to get the solid. [ Yield 24.54g,
- the obtained compound (I) (50g, 1347mmol) was further dissolved in 400 ml of ethanol and 100 ml of water at 78 to 80°C and 2g of activated charcoal was added. The reaction mixture was refluxed for half an hour. The obtained solution was filtered through hyflow and washed with 100 ml of ethanol. The filtrate was stirred for half an hour at 5 to 20°C to precipitates out the solid which was filtered, washed with chilled ethanol and dried.
- PART B A mixture of chloroethane sulfonyl chloride (10Og, 613 mmole) and dichloromethane (0.4L) was stirred under nitrogen atmosphere. To the obtained reaction mixture hydroquinone was added in the catalytic amount and reaction mixture was chilled to -15 to -20°C. A mixture of phenol (57.6gm, 613mmol) and triethylamine (0.171 L, 813mmol) in dichloromethane (0.4L) was added to the reaction mixture maintaining the same temperature. The mixture was then stirred at 25 to 30°C for one hour and monitored on GC. After completion of reaction dichloromethane ( 0.2 L) was added.
- the separated organic layer was treated with hydrochloric acid (2%, 1.5L) followed by aqueous sodium hydroxide ( IL, 5%) and water (2xlL). Then the organic layer was distilled out to get an oil (86gm, 76.78%) which was distilled under high vacuum or shortpath distillation to get pure compound which gets solidified after cooling. [Yield 64gm, 74.41%].
- Method I A mixture of N-methyl- lH-indole- 5 -ethenesulfonamide (4Og, 169mmol) was hydrogenated using 50 % wet 10% palladium on charcoal (40 g) in a mixture of
- Step IV Preparation of 2-[3-(l,2,3,6-tetrahydro-l-methyl-4-pyridinyl)-lH-indol-5- yl]ethanesulfonamide (IXc)
- Step V Preparation of 2[3-(l,2,3,6-tetrahydro-l-methyl-4-piperidinyl)-lH-indol-5- yl]ethanesulfonamide (IA)
- Naratriptan 1Og Naratriptan was suspended in 250 ml water at 40-50 0 C. 2.6 ml cone. HCl was added to the obtained suspension. The hot solution was filtered to remove insoluble matter. The clear solution was lyophilized for 24hr to get amorphous Naratriptan HCl
- Naratriptan HCl 2Og was dissolved in 400 ml water at 50-60 0 C. The solution was filtered to get clear solution. The clear solution was spray dried. Spray drying was carried out at the inlet temperature 120 0 C and outlet temperature 65 0 C to get amorphous Naratriptan HCl.
- Naratriptan HCl 2Og was dissolved in 400 ml water at 50-60 0 C. 5.2 ml cone. HCl was added to the obtained suspension. The solution was filtered to get clear solution. The clear solution was spray dried. Spray drying was carried out at the inlet temperature 120 0 C and outlet temperature 65 0 C to get amorphous Naratriptan HCl.
- Naratriptan HCl 5g was dissolved in 150 ml water at a temperature range of 50-60 0 C. The clear solution was concentrated under vacuum at 65 0 C to get amorphous Naratriptan HCl.
- Naratriptan was suspended in 250 ml water at 40-50 0 C. 2.6 ml cone. HCl was added to the obtained suspension. The hot solution was filtered to remove insoluble matter. The clear solution was concentrated under vacuum at 65 0 C to get amorphous Naratriptan. HCl.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Pain & Pain Management (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Indole Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
The present invention relates to an improved process for the preparation of N-methyl-3- (1-methyl-4-piperidinyl)-1H-indole-5-ethanesulfonamide hydrochloride of formula (I) having less than 0.15 % area by HPLC of 3-(1-methyl-4-piperidinyl)-1H-indole-5- ethanesulfonamide (1A) and intermediates thereof.
Description
Related application:
This application claims the benefit of Indian Provisional Application No.
00464/MUM/2008 filed on 7th March 2008.
Technical field:
The present invention relates to an improved process for the preparation of N-methyl-3-
(l-methyl-4-piperidinyl)-lH-indole-5-ethanesulfonamide hydrochloride of formula (I) having less than 0.15% area by HPLC of 3-(l-methyl-4-piperidinyl)-lH-indole-5- ethanesulfonamide (IA) and intermediates thereof.

(I) Background of invention:
The compound N-methyl-3-( 1 -methyl-4-piperidinyl)- 1 H-indole-5-ethanesulfonamide hydrochloride is also known by the name "Naratriptan hydrochloride." Naratriptan hydrochloride exhibits selective vasoconstrictor activity used as the active ingredient in pharmaceutical products and being sold by Glaxo with trademark AMERGE™ for treating migraine. Naratriptan hydrochloride is an indole derivative disclosed in US 4997841 and US 5066660.
US 4997841 describes three different processes for preparation of Naratriptan: Process A: A solution of 4-hydrazino-N-methyl-benzenethanesulfonamide (II) with 1- methyl-4-piperidineacetaldehyde (A) in a mixture of water and 2N hydrochloric acid followed by stirring for 2 days at room temperature to yield the hydrazone compound which is treated with polyphosphate ester in chloroform to get crude Naratriptan base (IX). The crude base (IX) was purified by column chromatography on silica gel using mixture of dichloromethane, ethanol and ammonia. The main disadvantage of this process is use of the aldehyde intermediate which is commercially not available and there are no synthetically feasible routes available for its synthesis in the literature. The reaction is represented by scheme below.
ester


(D) (A) (K)
Scheme 1
Process B: A mixture of 5-bromo indole (B), N-methyl-4-piperidone and potassium hydroxide in ethanol is refluxed to obtain compound of formula (C). The compound (C) is treated with ethanolic HCl followed by hydrogenation in presence of Platinum oxide to obtain 5-bromo-3-(l-methyl-4-piperidinyl)-lH-indole (D). The mixture of obtained compound (D), N-methyl vinyl sulfonamide, Pd(OAc)2 and tri-o-tolylphosphine and triethyl amine in acetonitrile is heated to get crude N-Methyl-2-[3-(l-methyl-4- piperidinyl)-lH-indole-5-yl]ethenesulfonamide (E) which is purified by column chromatography on silica gel using a mixture of dichloromethane, ethanol and ammonia. The purified compound (E) is hydrogenated in presence of 10% palladium on carbon in ethanolic hydrochloric acid and DMF to get N-methyl-3-(l-methyl-4-piperidinyl)-lH- indole-5-ethanesulfonamide (IX). The obtained compound (IX) is treated with ethanolic hydrochloric acid to get Naratriptan hydrochloride (I). The reaction is represented by scheme below.

(E) (IX) Scheme 2
The disadvantage of this process is that cumbersome operations like Heck reaction is involved. Other reagents such as Pd(OAc)2 and tri-o-tolylphosphine are very expensive. Besides the above process using these expensive intermediates and reagents yield only 5- 7% of the final product.
Process C: A mixture of N-methyl-lH-indole-5-ethanesulfonamide (VII), N-methyl-4- piperidone and potassium hydroxide in methanol is refiuxed to produce compound (VIII). The compound (VIII) is hydrogenated in the presence of 5% Pd/C to obtain N-Methyl-3- (4-piperidinyl)-lH-indole-5-ethanesulfonamide (IX). The compound (IX) is purified by column chromatography on silica gel using a mixture of dichloromethane, ethanol and ammonia. The reaction is represented by the below scheme.

C IX)
Scheme 3
The main disadvantage of the processes as described in Process A, B and C is the use of column chromatography for purification of Naratriptan base thus these processes are time consuming, impractical at industrial scale and economically not viable. US 4994483 describes process for preparation of N-methyl-lH-indole-5- ethanesulfonamide (VII) which comprises heating the mixture of 5-bromo indole (B), N- methyl vinyl sulfonamide, Pd(OAc)2, tri-o-tolylphosphine, and triethylamine in acetonitrile to get crude 2-(lH-indol-5-yl)-N-methyl ethane sulfonamide (F) which is purified by column chromatography and resulted in poor yield. The purified compound (F) is hydrogenated in presence of 10% palladium oxide on charcoal in mixture of ethyl acetate and methanol to get N-methyl-lH-indole-5-ethanesulfonamide (VII). The reported yield is 22-23%. The reaction is represented by the below scheme.
"  US 5659040 describes process for preparation of Naratriptan hydrochloride comprising heating the mixture of compound (C), N-methyl vinyl sulfonamide, Pd(OAc)2, tri-o- tolyphosphine and triethylamine in dimethylformamide to get (E)-N-Methyl-2-[3- ( 1 ,2,3,6-tetrahydro- 1 -methyl-4-pyridinyl)- 1 H-indol-5-yl]ethenesulfonamide (G). The compound (G) is hydrogenated in presence of 10% palladium oxide on charcoal in a mixture of dimethylformamide, water and 2N hydrochloric acid to get crude Naratriptan hydrochloride, which is crystallized with hot IMS(Industrial methylated spirit)/water to get pure Naratriptan hydrochloride (I).The main disadvantage of this process is the use of high quantity of catalyst. The reaction is represented by the scheme below.
US 5659040 describes process for preparation of Naratriptan hydrochloride comprising heating the mixture of compound (C), N-methyl vinyl sulfonamide, Pd(OAc)2, tri-o- tolyphosphine and triethylamine in dimethylformamide to get (E)-N-Methyl-2-[3- ( 1 ,2,3,6-tetrahydro- 1 -methyl-4-pyridinyl)- 1 H-indol-5-yl]ethenesulfonamide (G). The compound (G) is hydrogenated in presence of 10% palladium oxide on charcoal in a mixture of dimethylformamide, water and 2N hydrochloric acid to get crude Naratriptan hydrochloride, which is crystallized with hot IMS(Industrial methylated spirit)/water to get pure Naratriptan hydrochloride (I).The main disadvantage of this process is the use of high quantity of catalyst. The reaction is represented by the scheme below.
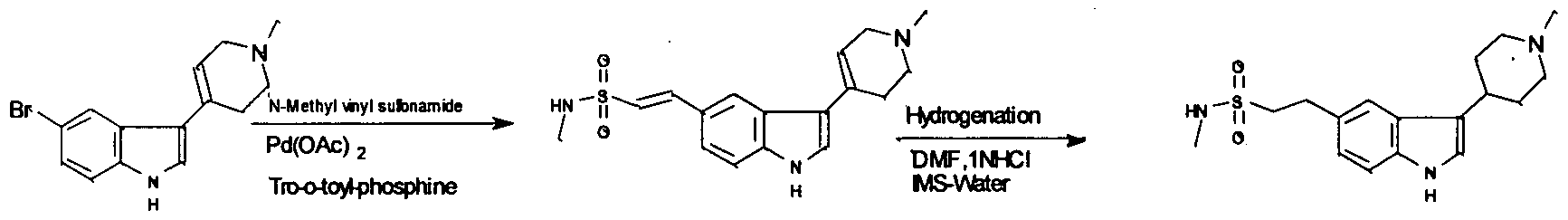
(C) (G) 0
Scheme 5
WO2006010079 describes process for preparation of Naratriptan hydrochloride which comprises reaction of diazonium salt of N-Methyl-2-(4-aminophenyl)ethane sulfonamide with β-ketoester compound , which further upon cyclization, quaternization, reduction (twice), saponification and decarboxylation gives Naratriptan hydrochloride (I). This process is time consuming and economically not viable as it involves many steps.

Scheme 6
Advantages of the present process for preparation of Naratriptan hydrochloride are
1. Provides Naratriptan hydrochloride in high yield and high purity.
2. More suitable for scaled-up operations as it involves less number of steps.
3. Avoids use of column chromatography.
4. Avoid use of expensive reagents like N-methyl-4-piperidine acetaldehyde.
5. It is simple, economical and industrially feasible process for the preparation
Naratriptan hydrochloride.
6. Involves use of easily available raw materials and low operating cost, overcoming the problems of handling the reactants and reagents as used in the prior art.
7. Conversion of compound (VIII) to Naratriptan hydrochloride which reduces number of steps of the process and results in high yield of the desired product.
Object of the invention:
The object of the present invention is to provide a simple and cost effective process for the preparation of Naratriptan hydrochloride i.e. N-methyl-3-(l-methyl-4-piperidinyl)-l
H-indole-5-ethanesulfonamide hydrochloride and intermediates thereof.
Another object of the invention is to provide novel amorphous Naratriptan hydrochloride and process for preparation thereof.
Another object of the invention is to provide desmethyl Naratriptan (IA) as a reference standard to quantify the amount of impurity in a sample of Naratriptan or salts thereof.
Another object of the invention is to provide pharmaceutical compositions comprising
Naratriptan or pharmaceutically acceptable salts thereof along with at least one pharmaceutically acceptable excipient.
Summary of the invention:
According to one aspect of the present invention there is provided a process for preparation of N-methyl-2-[3-(l-methyl-4-piperidinyl)- 1 H-indole-5-ethanesulfonamide salts, particularly hydrochloride of formula (I) which comprises the following steps; a) combining compound (VII) with N-Methyl-4-piperidone to produce compound (VIII); b) combining compound (VIII) in suitable solvent with catalyst under reduced condition and adding acid to the obtained reaction mixture to get Naratriptan salt; a) optionally purifying the obtained salt to get pure compound with purity more than 90%.
According to another aspect of the present invention there is provided process for preparation of key intermediate indole sulfonamide (VII) used for the preparation of Naratriptan hydrochloride which comprises the steps of;
a) combining 5-bromo indole (B) with aryl vinyl sulfonate wherein aryl is substituted or unsubstituted phenyl compound to get aryl lH-indole-5-ethenesulfonate (X). b) hydrogenating the compound (X) to get lH-indole-5-aryl ethanesulfonamide (XI); c) combining compound (XI) with methyamine to obtain desired compound (VII). According to another embodiment the present invention provides process for preparation of key intermediate indole sulfonamide (VII) comprising a) combining compound (X) with methylamine to obtain N-methyl- 1 H- indole-5- ethenesulfonamide (F); b) hydrogenating the compound (X) to get lH-indole-5-aryl ethanesulfonamide (XI) c) treating the obtained compound (XI) with methylamine to get compound (VII).
According to another aspect of the present invention there is provided a process for preparation of key intermediate indole sulfonamide (VII) used for the preparation of Naratriptan hydrochloride which comprises the steps of; a) combining isethionic acid or salts thereof with chlorinating agent to get chloroethane sulfonyl chloride; b) combining obtained compound of step a) with methylamine in suitable solvent to get N-methyl vinyl sulfonamide;
/ O
N-methyl vinyl sulfonamide c) combining N-methyl vinyl sulfonamide with 5-bromoindole (B) in presence of Pd(OAc)2, tri-o-tolylphosphine, and triethylamine in suitable solvent to get compound (F); d) hydrogenating compound (F) using suitable catalyst in suitable solvent to produce compound(VII).

(F) (VH)
Scheme 7
According to another aspect, the present invention provides process for preparation of Naratriptan hydrochloride comprising the steps of; a) combining hydrazino derivative (II) with a pyruvic acid ester compound (III) in ethanol and methane sulphonic acid at 25- 1000C for 2 to 4 hours to obtain hydrazone compound (IV);

(H) (III)

(IV) b) combining the obtained hydrazone compound (IV) with methanesulphonic acid in acetic acid for 2-3 hours at 110-1150C to get an indole carboxylic ester compound (V);

R= an alkyl group having 1 to 5 carbon atoms (V)
c) refluxing a mixture of carboxylic ester (V) and base for 2-3 hours to get carboxylic acid compound (VI);

(VI) d) decarboxylating compound (VI) to obtain indole sulfonamide(VII);

(VII)
According to another aspect, the present invention provides process for preparation of desmethyl Naratriptan (IA).
Another aspect of the invention is to provide desmethyl Naratriptan (IA) as a reference standard to quantify the amount of impurity in a sample of Naratriptan or salts thereof.
According to another aspect, the present invention provides amorphous Naratriptan hydrochloride and process for preparation thereof.
According to another aspect of the present invention there is provided pharmaceutical compositions comprising Naratriptan or pharmaceutically acceptable salts thereof.
Brief description of figures:
Fig.1 is a characteristic X-ray Powder diffraction pattern of Naratriptan base.
Fig.2 is a characteristic X-ray Powder diffraction pattern of Naratriptan hydrochloride
Fig.3 is a characteristic X-ray Powder diffraction pattern of pure Naratriptan hydrochloride.
Fig. 4 is a characteristic X-ray Powder diffraction pattern of Naratriptan hydrochloride,
(USP reference standard).
Fig. 5 is a characteristic X-ray Powder diffraction pattern of Amorphous Naratriptan hydrochloride
Description of the invention:
The present invention describes process for preparation of Naratriptan hydrochloride (I) having less than 0.15% area by HPLC of 3-(l-methyl-4-piperidinyl)-lH-indole-5- ethanesulfonamide (IA) or desmethyl Naratriptan.

Another embodiment of the present invention provides process for preparation of Naratriptan salts comprising the steps of: c) combining compound (VII) with N-Methyl-4-piperidone to produce compound (VIII); d) combining compound (VIII) in suitable solvent under reducing condition with catalyst and adding acid to the obtained reaction mixture to get Naratriptan salt; e) optionally purifying the obtained salt to get pure compound with purity more than 90%.
Particularly, Naratriptan hydrochloride is prepared by condensing compound (VII) with N-methyl-4-piperidone at temperature of about 60 to 1000C for about 8 to 14 hours, preferably 8 to 10 hours in presence of base in suitable solvent to obtain compound (VIII). The obtained compound (VIII) is optionally further dissolved in suitable solvent and refluxed for 1-2 hours and filtered. The solvent is partially distilled out and the reaction mass is cooled at 10-150C to get pure compound (VIII). The compound (VIII) dissolved in suitable solvent is reduced in presence of catalyst and hydrogen gas at temperature of about 25 to 800C, preferably 50 to 600C for about 5 to 12 hours, preferably 6 to 8 hours. The compound (VIII) may be reduced using catalytic transfer hydrogenation. Optionally the reaction is carried out in a solvent or in a mixture of solvent and acid. After completion of reaction, the reaction mixture is filtered and distilled to get an oil, which is stripped with suitable solvent followed by addition of acid and refluxing the obtained reaction mixture for 1-2 hours. The precipitated solid is stirred
at 5-2O0C. The solid thus obtained is washed with solvent and dried to get Naratriptan salt with high yield. The process of producing Naratriptan salt according to the present invention i.e. without isolating Naratriptan base is economically and industrially feasible.
Naratriptan hydrochloride obtained according to the present invention is optionally purified by dissolving in suitable solvent at 70-800C for 30 minutes to 1 hour. The solution is filtered and the filtrate is stirred for 30 minutes at 5-200C to get solid of pure Naratriptan hydrochloride having purity more than 90% by HPLC. The above reaction is represented by the scheme below,

(VII) (VIII) (I)
Scheme 8
According to another embodiment, the present invention provides process for preparation of N-methyl-lH-indole-5-ethanesulfonamide (VII) used for the preparation of Naratriptan hydrochloride which comprises the steps of; a) combining 5-bromo indole (B) with aryl vinyl sulfonate compound wherein aryl is substituted or unsubstituted phenyl to get lH-indole-5-aryl ethenesulfonamide (X). b) hydrogenating the compound (X) to get lH-indole-5-aryl ethanesulfonamide
(XI); c) combining compound (XI) with methylamine to obtain compound (VII). Preferably 5-bromo indole (B) is treated with aryl vinyl sulfonate wherein wherein aryl is substituted or unsubstituted phenyl compound in the presence of Pd(OAc)2, tri-o- tolyphosphine and triethylamine in suitable solvent at temperature of about 80 to 1200C, preferably about 800C for a period of about 2 to 4 hours to get compound (X). The compound (X) is reduced to compound (XI) in the presence of hydrogen gas and catalyst at temperature about 25 to 800C, preferably 40 to 500C for about 8 to 24 hours, preferably 14 to 16 hour. The reduction can be carried out in solvent or mixtures of solvents, preferably mixture of methanol and terahydrofuran. The compound (XI) is further
converted into indole sulfonamide compound (VII) using methylamine in water or organic solvent or mixtures thereof at temperature of about 25 to 800C preferably about 40 to 500C. The reaction is carried out for 2 to 6 hours preferably about 3 to 4 hours. The above reaction is represented in the scheme below,

Scheme 9
According to another embodiment, the present invention provides process for preparation of N-methyl-lH-indole-5-ethanesulfonamide (VII) which comprises the steps of; a) combining compound (B) with N-methyl vinyl sulfonamide in suitable solvent to get compound (F). b) reducing obtained compound (F) in the presence of catalyst at temperature about 25 to 800C for about 8 to 24 hours to get compound (VII).
Preferably a mixture of palladium acetate and tri-o-tolyl phosphine in suitable solvent is stirred under nitrogen atmosphere and a mixture of compound (B), N-methyl vinyl sulfonamide and base is added to the reaction mixture followed by heating for 8 to 10 hrs. After completion of the reaction, the mixture is cooled to 35 to 40° C and filtered The filtrate is distilled off to yield thick mas to which water is slowly added to get compound (F) which may be further crystallized from solvent to get pure 2-(lH-indol-5-yl)-N- methyl ethene sulfonamide (F) with high yield of 98%.The obtained compound (F) is further reduced at temperature about 25 to 800C, preferably 40 to 500C for about 8 to 24 hours, preferably 14 to 16 hours in the presence of hydrogen gas using a catalyst to get compound (VII). The reduction is performed in solvent or mixtures of solvents. The above reaction is represented in the scheme below, coupling >

(B)
 According to another embodiment, the present invention provides process for preparation of N-methyl-lH-indole-5-ethanesulfonamide (VII) comprises the steps of; a) combining lH-indole-5-aryl ethenesulfonamide (X) with methylamine to obtain N-methyl- 1 H-indole-5-ethenesulfonamide (F); b) reducing obtained compound (F) in the presence of catalyst at temperature about 25 to 800C for about 8 to 24 hours to get compound (VII).
According to another embodiment, the present invention provides process for preparation of N-methyl-lH-indole-5-ethanesulfonamide (VII) comprises the steps of; a) combining lH-indole-5-aryl ethenesulfonamide (X) with methylamine to obtain N-methyl- 1 H-indole-5-ethenesulfonamide (F); b) reducing obtained compound (F) in the presence of catalyst at temperature about 25 to 800C for about 8 to 24 hours to get compound (VII).
Prefereably, treating lH-indole-5-aryl ethenesulfonamide (X) with methylamine in water or suitable solvent or mixtures thereof, preferably mixture of methanol and tetrahydrofuran at temperature of about 25 to 800C preferably about 40 to 500C and high pressure to get compound (F). The reaction is carried out for 2 to 6 hours preferably about 3 to 4 hours. The obtained compound (F) is further reduced in the presence of hydrogen gas using a catalyst at temperature about 25 to 800C, preferably 40 to 500C for about 8 to 24 hours, preferably 14 to 16 hours to get compound (VII). The reduction can be carried out in solvent or mixtures of solvents. The above reaction is represented in the scheme below,

Scheme 11
According to another embodiment of the present invention compound (F) is purified by refluxing in suitable solvent to get clear solution. The solvent is partially distilled out and reaction mass is cooled at 1O0C with stirring to get pure compound (F). The obtained compound (F) is optionally further refluxed in a suitable solvent to obtain pure compound (F) in high yield. The suitable solvent used is selected from ethanol, methanol, isopropanol, hexane, ethylacetate and acetone or mixtures thereof, preferably mixture of hexane: ethyl acetate.
N-methyl vinyl sulfonamide is an unstable compound, expensive and not easily available. Also it is difficult to produce N-methyl vinyl sulfonamide at a commercial scale due to its unstability. The present invention provides simple process for preparation of N-methyl
vinyl sulfonamide which comprises the steps of; a) combining isethionic acid salt with chlorinating agent to get chloroethane sulfonyl chloride; b) optionally distilling the compound obtained in step a) under vacuum; c) treating obtained pure with methylamine to get N- methyl vinyl sulfonamide and d) optionally distilling N-methyl vinyl sulfonamide under high vacuum or short path distillation to get pure N- methyl vinyl sulfonamide.
In a preferred embodiment, thionyl chloride is added dropwise to a mixture of isethionic acid salt in DMF and dichloromethae at 20-30°C. The reaction mixture is heated slowly from 30-60°C. After completion of the reaction chilled water is added. The reaction mixture is extracted with dichloromethane. The solvent is distilled out from the reaction mixture to get an oil which is distilled under vacuum to get pure chloroethane sulfonyl chloride with yield of 90%. A mixture of chloroethane sulfonyl chloride in suitable solvent is stirred under nitrogen atmosphere. The reaction mixture is cooled to -50 to -60°C followed by addition of hydroquinone in the catalytic amount and purging N- methyl amine gas (aqueous methylamine was added in potassium hydroxide) in the reaction mixture maintaining temperature - 40 to - 60°C. The reaction reaction mixture is filtered and filtrate is evaporated under vacuum not more than 500C to get thick oil which is stored under nitrogen atmosphere and distilled using short path distillation or high vacuum distillation to get pure N-methyl vinyl sulfonamide. N-methyl vinyl sulfonamide thus obtained has less than about 0.5% of vinyl sulfonamide.
According to the present invention process for preparation of aryl vinyl sulfonate which comprises the steps of; a) combining isethionic acid or salt thereof with chlorinating agent to get chloroethane sulphonyl chloride; b) optionally distilling the obtained compound of step a) under vacuum; c) adding aryl-OH, wherein aryl is substituted or unsubstituted phenyl to the obtained pure chloroethane sulphonyl chloride to get aryl vinyl sulfonate; d) optionally distilling aryl vinyl sulphonate under high vacuum or short path distillation to get pure aryl vinyl sulfonate.
In preferred embodiment thionyl chloride is added to a mixture of isethionic acid salt in DMF and dichloromethane at 20-30°C. The obtained reaction mixture is heated slowly at temperature range from 30-60°C. After completion of the reaction chilled water is added to the reaction mixture and the reaction mixture is extracted with dichloromethane. The solvent is distilled out from the reaction mixture to get oil.
The obtained oil is distilled under vacuum to get pure chloroethane sulfonyl chloride. A mixture of chloroethane sulfonyl chloride and dichloromethane is stirred under nitrogen atmosphere and hydroquinone is added in the catalytic amount. The reaction mixture is chilled at -15 to -200C and a mixture of phenol and triethylamine in dichloromethane is added to it maintaining the same temperature followed by stirring at 25 to 30°C for one hour. After completion of reaction dichloromethane is added. The separated organic layer is treated with hydrochloric acid followed by aqueous sodium hydroxide and water. The organic layer is distilled out to get an oil which is distilled using short path distillation or high vacuum distillation to get pure desired compound.
In the process for preparation of N-methyl vinyl sulfonamide or aryl vinyl sulfonate, the reaction is performed in presence of solvent selected from the group consisting of dimethylformamide, dichloromethane, ethylacetate, tetrahydrofuran. The isethionic acid salt is isethionic acid sodium salt. The high vacuum used is in the range of 0.1mm to 0.5mm preferably 0.1mm to 0.2 mm optionally using glass bids. The methylamine used herein is in the form of solution or gas. The treatment of chloroethane sulfonyl chloride with methylamine is performed in presence of catalytic amount of hydroquinone.
According to another embodiment, the present invention provides process for preparation of key intermediate N-methyl- lH-indole-5-ethanesulfonamide (VII) comprising the steps of; a) reacting hydrazino derivative (II) with a pyruvic acid ester compound (III) to obtain hydrazone compound (FV); ft
(II) ^^NHNH 2 O (m) b) cyclizing the hydrazone compound (IV) in presence of an acid to give the indole
derivative (V); c) hydrolyzing the obtained compound (V) with base and acidifying with acid to produce compound (VI); d) decarboxylating the obtained compound (VI) to produce indole sulfonamide compound (VII);
The reaction of the present invention is represented by the following scheme; wherein R represents an alkyl group having 1 to 5 carbon atoms
condcnsstion


Scheme 12
According to preferred embodiment of the present invention the hydrazino derivative (III) is condensed with pyruvic acid ester (II), maintaining the temperature at about 25 to 1000C, preferably about 800C for a period of about 2 to 4 hours to obtain hydrazone compound (IV). The compound (IV) can be prepared by reacting the compound (II) with compound (III) in presence or absence of catalytic amount of acid in suitable solvent, preferably methanesulphonic acid in ethanol is used. The hydrazone compound (IV) is cyclized using an acid in presence or absence of solvent, preferably methane sulphonic acid in acetic acid, for 2-6 hours at temperature 100-1200C. After the completion of reaction the obtained solid is neutralized with base to get indole derivative (V).
The obtained indole derivative (V) is hydrolyzed by refluxing a mixture of compound (V) and suitable base in water or water-solvent mixture for 2-3 hours. The obtained reaction mixture is acidified with cone, or dil hydrochloric acid to precipitate out the compound
(VI).
The compound (VI) is decarboxylated in the presence of copper catalyst such as cuprous
oxide, copper oxide, copper bronze in the presence of quinoline at temperature in the range of about 140 to 2200C for 2 to 6 hours to obtain compound (VII).
Another embodiment of the present invention provides a process for preparation of Naratriptan or salts thereof comprising the steps of: a) condensing indole sulfonamide (VII) with N-methyl-4-piperidone at 60 to 1000C in presence of base to obtain compound (VIII);


Preferably, compound (VII) is further condensed with N-methyl-4-piperidone at temperature of about 60 to 1000C for about 8 to 14 hours, preferably 8 to 10 hours in presence of base in suitable solvent to obtain compound (VIII). The obtained compound (VIII) is optionally further dissolved in suitable solvent and refluxed for 1-2 hours and filtered. The solvent is partially distilled out and the reaction mass is cooled at 10-150C to get pure compound (VIII). The obtained compound (VIII) is further reduced using catalyst at temperature of about 25 to 800C, preferably 40 to 500C for about 5 to 12 hours, preferably 8 to 12 hours. The reaction mixture is then filtered and filtrate is concentrated. The obtained reaction mass is treated with base followed by extraction with ethyl acetate. The organic layer is concentrated to get Naratriptan base (IX) with high yield of 60-70
%. The reduction can be carried out in presence of solvent or solvent -acid mixture preferably mixture of methanol and acetic acid and hydrogen gas. Naratriptan base (IX) obtained by the above process is characterized by XRPD having the following characteristic peaks at 2Θ angle positions ± 0.2 degrees represented in below table (Fig. 1):

Naratriptan base (IX) thus obtained is further converted into its acid addition salt (I). Preferably, Naratriptan base (IX) is dissolved in suitable solvent at refluxed temperature for 1-2 hours and cooled at room temperature. An acid is added to the cooled solution and the mixture is refluxed. A mixture of suitable solvent is added to it and refluxed further for 1 hour. The hot solution is filtered and the filtrate is stirred for 1 hour at 5-1O0C to get pure desired salt of Naratriptan.
Alternatively, Naratriptan base (IX) is dissolved in suitable solvent at refluxed temperature for 30 minutes and solution is filtered. The filtrate is cooled to 30-350C and acid is added slowly to it followed by cooling at 10-150C to get crude desired salt of Naratriptan. The crude salt of Naratriptan is further dissolved in suitable solvent at refluxed temperature of about 25 to 100°C, preferably 60 to 80°C and the solution is filtered followed by cooling and stirring at 10-150C for 1 hour to get pure Naratriptan salt with high yield of more than 85% and purity more than 90%, more preferably more than 99.8%.
Naratriptan hydrochloride obtained according to the present invention has purity of more than 99.8% by HPLC and characterized by XRPD having the following characteristic peaks at 2Θ angle positions ± 0.2 degrees represented in below table (Fig. 2):

7.4147 3.27
9.2622 22.54
14.3126 100.00
15.1924 2.39
16.6609 1.46
17.3266 37.13
17.6457 16.50
17.8079 5.59
18.0817 8.83
18.5294 53.22
19.6801 10.03
21.3530 33.77
22.0322 2.81
22.9458 47.67
23.2010 14.94
23.6875 67.06
24.0644 26.58
24.7339 2.93
25.0341 19.05
27.9145 8.26
28.7774 83.83
29.2462 12.24
29.7791 4.75
30.7447 8.17
31.1299 11.77
31.6946 1.12
32.9671 3.85
34.2612 4.55
35.0620 3.90
35.4402 4.87
36.4140 14.66
Naratriptan hydrochloride thus obtained may be further purified and characterized by XRPD having the following characteristic peaks at 2Θ angle positions ± 0.2 degrees (Fig. 3):
Pos. r°2Th.l ReI. Int. [%1
7.3842 47.20
9.2255 34.16
14.2628 5.27
14.7809 10.18
15.1251 39.28
16.6427 2.66
17.3066 9.71
17.6113 62.80
18.0623 3.44
18.4864 100.00
19.7568 7.16
21.1604 5.96
21.9868 44.83
22.2297 18.47
22.9190 13.68
23.1499 3.00
23.6384 24.22
24.0295 27.56
24.6353 3.10
24.9934 9.04
25.3342 1.93
26.2211 1.13
27.8667 15.91
28.2065 4.44
28.4916 2.30
28.7449 8.54
29.2325 4.52
29.7337 4.84
30.4851 34.53
31.1088 1.04
31.7752 5.69
Naratriptan hydrochloride is listed in US pharmacopoeia. The USP reference standard shows XRPD having the following characteristic peaks at 2Θ angle positions ± 0.2 degrees (Fig. 4).
Pos. [°2Th.l ReI. Int. \%]
7.3051 6.46
9.1605 31.63
14.2029 30.15
15.0674 10.78
16.5645 7.54
17.2391 28.58
17.5476 43.19
17.6947 14.08
18.0021 18.87
18.4277 100.00
19.5801 20.12
21.0944 11.31
21.2556 12.66
21.9285 10.16
22.8526 63.30
23.1047 17.58
23.5813 58.16
23.9918 20.32
24.9561 33.19
27.8099 13.33
28.2049 5.82
28.6777 25.57
29.1821 8.68
29.8559 3.28
30.4497 7.99
31.0370 3.70
31.6564 1.46
32.9380 2.97
33.6327 3.98
34.1305 5.26
35.3946 2.83
Naratriptan HCl obtained by the process of the present invention has particle size cUo less than about 250 μ preferably less than about lOOμ more preferably less than about 80μ which may be further micronized using conventional micronization techniques to get desired less particle size of Naratriptan or salts thereof.
According to another embodiment of the present invention the process for preparation of desmethyl Naratriptan comprises the steps of; a) treating chloroethane sulfonyl chloride with ammonia in suitable solvent to get vinyl sulfonamide;
9?
H2 *N- S M "^
° . Vinyl sulfonamide b) treating vinyl sulfonamide with 5-bromoindole (B) in presence of coupling catalyst, ligand, and base in suitable solvent to get compound (IXa); f?
° I i[ /
" (IXa) c) hydrogenating compound (IXa) using catalyst in suitable solvent to produce compound (IXb)



The process for preparation of desmethyl Naratriptan is described in steps below; Step a) : A mixture of chloroethane sulfonyl chloride and tetrahydrofuran is stirred under nitrogen atmosphere with cooling to -30 to -60°C followed by purging ammonia gas slowly to the reaction mixture at same temperature. The reaction is monitored on GC. After completion of reaction, the temperature of the reaction is maintained at 25 °C and filtered through hyflow. The solvent is distilled out from the obtained filtrate to get thick oil which is treated with treated with suitable solvent to get vinyl sulfonamide 98% pure on GC in high yield.
Step b): A mixture of palladium acetate and tri-o-tolyl phosphine in dimethylformamide is stirred under nitrogen atmosphere. A mixture of 5-bromo indole , vinyl sulfonamide and triethylamine in dimethylformamide is added to the reaction mixture. The reaction mixture is heated to 100-1100C for 8 to 10 hrs. After completion of the reaction, the mixture is cooled to 35 to 400C and filtered. The filtrate was distilled off completely to yield thick mass to which water is slowly added to get solid which is crystallized from mixture of mixture of solvent to get 2-(lH-indol-5-yl)-ethene sulfonamide (IXa).
Step c) A mixture of lH-indole-5-ethenesulfonamide (IXa) is hydrogenated using 5-60 % of catalyst (50% wet palladium on charcoal) in a mixture of suitable solvent preferably methanol: ethyl acetate for 2 to 8h at 25°-30° C under hydrogen pressure to get 2-(1H- indol-5yl)-ethane sulfonamide (IXb).
Step d): A mixture of 2-(lH-indol-5yl)-ethane sulfonamide (IXb), N-methyl-4- piperidone in presence of base preferably potassium hydroxide in ethanol is refluxed for 2-10 hrs at temperature 50-800C. The reaction mixture is cooled to 25- 40°C and
quenched in water at temperature belowlO°C to precipitate out solid of 2-[3-(l, 2,3,6- tetrahydro-l-methyl-4-pyridinyl)-lH-indol-5-yl]ethanesulfonamide (IXc) or extracting the reaction mixture with 1-butanol.
Step e) : A mixture of compound (IXc) is reduced using platinum oxide in a mixture of 50% acetic acid in methanol for 8h at .70 psi at 500C. After completion of reaction, the reaction mixture is filtered and filtrate is concentrated, basified with aqueous sodium carbonate solution and extracted with solvent. The organic layer is separated, washed with EDTA, water and concentrated to get the 2[3-(l,2,3,6-tetrahydro-l-methyl-4- piperidinyl)- 1 H-indol-5-yl]ethanesulfonamide (IA).
According to another embodiment, the invention provides desmethyl Naratriptan (IA) as a reference standard to quantify the amount of impurity in a sample of Naratriptan or salts thereof.
The acid used is selected from the group consisting of methane sulfonic acid, acetic acid, phosphoric acid, hydrochloric acid, sulfuric acid, para-toluenesulphonic acid and lewis acid such as boron trifluoride and the like optionally in presence or absence of solvent.
The suitable solvent used is alcohol such as methanol, ethanol, propanol, isopropanol, chlorinated solvent such as dichloromethane, chloroform, ethers such as 1,4-dioxane, tetrahydrofuran, esters such as ethyl acetate, isopropyl acetate, butyl acetate, hydrocarbons such as pentane, hexane, heptane, amide such as dimethylformamide, water, acetonitrile or mixtures thereof.
The base used is selected from the group consisting of sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate potassium carbonate, triethylamine, diisopropylethylamine and the like.
The catalyst used is selected from the group consisting of palladium oxide/charcoal, palladium/charcoal, platinum oxide/charcoal, platinum/charcoal, rhodium/charcoal in presence of hydrogen gas and used in an amount less than about 60%, preferably 20% more preferably 10%.
The said methylamine used in above processes is in the form of solution or gas optionally in presence of catalytic amount of hydroquinone.
The coupling catalyst used is palladium acetate and others which are conventionally known.The ligand used is tri-o-tolylphosphine or tri phenyl phosphine.
According to another embodiment, the present invention provides amorphous Naratriptan hydrochloride. The XRPD of amorphous Naratriptan hydrochloride is shown in Fig.5.
Another embodiment of the present invention provides process for preparation of amorphous Naratriptan hydrochloride which comprises the steps of ; a) dissolving Naratriptan HCl in a suitable solvent to get clear solution or suspending Naratriptan in suitable solvent and adding conc.HCl to the suspension and b) lyophilizing the resultant solution of step a) to obtain the amorphous Naratriptan hydrochloride.
The solvent used in step a) is water. The concentration of Naratriptan in solution used for lyophilization is 4-10% preferably 4-8 %. The solution of Naratriptan is lyophilized for 24 hrs to get an amorphous Form.
Another embodiment of the present invention provides process for preparation of amorphous Naratriptan hydrochloride which comprises the steps of ; a) dissolving Naratriptan HCl in a suitable solvent to get clear solution or suspending Naratriptan in suitable solvent and adding conc.HCl to the suspension Naratriptan HCl and b) spray drying the resultant clear solution to get the amorphous form.
The solvent used in step a) is polar protic solvent such as water or Ci -Ci alcohols or mixture thereof, preferably water. The alcohols used is selected from methanol and ethanol preferably methanol. The concentration of Naratriptan in solution used for spray drying is 4-10% preferably 4-8 %. The solution of Naratriptan is spray dried at inlet temperature of 40 to 1700C and outlet temperature of 35-850C.
Another embodiment of the present invention provides process for preparation of amorphous Naratriptan hydrochloride which comprises the steps of ; a) dissolving Naratriptan HCl in a suitable solvent to get clear solution or suspending Naratriptan in suitable solvent and adding conc.HCl to the suspension to get Naratriptan HCl. b) evaporating the solvent from the solution of step a) under vacuum to get amorphous Naratriptan HCl.
The dissolving solvent used is polar protic solvent such as water or Ci-C4 alcohols or mixtures thereof, preferably water. The alcohols used is methanol, ethanol preferably methanol. The solvent from the solution of Naratriptan is evaporated at 650C, under vacuum to get the amorphous Naratriptan HCl.
Another embodiment of the present invention provides pharmaceutical compositions comprising an active ingredient and at least one pharmaceutical excipient. The pharmaceutical compositions are normally employed as tablets, capsules, powders, syrups, solutions, suspensions and the like, optionally containing flavoring agents, sweeteners, etc., in suitable solid or liquid carriers or diluents; and in suitable sterile media to form injectable solutions or suspensions.
The pharmaceutical composition, preferably a tablet, comprises Naratriptan or pharmaceutically acceptable salts thereof in the range of 0.5-2 % by weight of the composition and suitable pharmaceutically acceptable excipients such as fillers, disintegrants, glidants, lubricants and others known in the pharmaceutical field. The filler and disintegrants may include Lactose, Microcrystalline cellulose, dibasic calcium phosphate, monobasic calcium phosphate, Mannitol and the like. The glidants may include croscarmellose sodium, sodium starch glycolate and the like. The lubricants may include stearic acid, calcium stearate and the like.
The amount of fillers may be in the range of about 40-80 % based on the total weight of the formulation. The amount of disintegrants may be in the range of about 20-40 % based on the total weight of the formulation. The amount of glidants may be in the range of about 1-10 % based on the total weight of the formulation. The amount of lubricants may be in the range of about 0.1-2.5 % based on the total weight of the formulation. A
method of formulating the composition into tablets may be selected from any conventional methods including wet granulation method, dry granulation method and direct compression method. The tablet formed may be optionally film coated. The coating can be performed according to a conventional coating method. Naratriptan hydrochloride prepared according to the present invention is used in the manufacture of pharmaceutical composition for the treatment of migraine.
The compound, N-Methyl-3-(l-methyl-4-piperidinyl)-l H-indole-5- ethanesulfonamide hydrochloride (Naratriptan hydrochloride) used in the pharmaceutical compositions can be prepared by any processes known in the art and the present invention is not dependent on a particular synthetic method described herein. Further, any polymorphic form of the Naratriptan is suitable for use to prepare Naratriptan hydrochloride of this invention. The crystallization of the polymorphic form of this invention can be performed as a final step in a synthesis of the compound, and it is not always necessary to isolate a product having another polymorphic form before performing the crystallization.
Another embodiment the present invention provides a method of treating a patient suffering from migraine administering to the patient a pharmaceutical compositions comprising Naratriptan or pharmaceutically acceptable salts thereof or its amorphous form prepared according to the present invention.
The polymorphic forms of Naratriptan or pharmaceutically acceptable salts thereof are characterized by X-ray powder diffraction pattern obtained on Xpert'PRO, Panalytical, diffractometer equipped with accelerator detector using Copper Ka (λ=1.5406A°) radiation with scanning range between 4-50 2Θ at scanning speed of 2°/min.
The following examples are for illustrative purposes only and are not intended, nor should they be interpreted, to limit the scope of the invention.
EXAMPLES:
Example 1: Preparation of hydrazone of 4-hydrazino-N-Methyl benzene ethane sulfonamide (IV):
A mixture of 4-hydrazino-N-methyl benzene ethane sulfonamide hydrochloride (II) (150 g, 56.49 mmol), ethyl pyruvate (III) (65.59 g, 56.49 mmol) in ethanol (1.5 lit) and
methane sulphonic acid (7ml) was stirred for 2 to 3 h at temperature 80°C. The solvent was partially distilled from reaction mixture. The reaction mixture was further cooled to temperature 10°C for one hour to precipitate out the solid. The solid obtain was filtered, washed with chilled ethanol and dried. [Yield: 130 g, 70.58 %]
Example 2
Preparation of Ethyl-5-[2-(methylsulfamoyl)ethyl]-lH-indole-2carboxylate (Indole-
2- ester) (V):
A mixture of (130 g, 39.75 mmol) compound (IV) and methane sulphonic acid (3.06g,
44.78 mmol ) in acetic acid (1 lit) was refluxed for 2 to 3 hrs at temperature 115°C. The acetic acid was distilled out from the reaction mixture and the solid obtained was neutralized with aqueous sodium carbonate, separated solid was filtered, washed with water and dried. [Yield: 105 g, 85 %]
Example 3
Preparation of 5-[2-(methyIsulfamoyl)ethyl]-lH-indoIe-2carboxylic acid (Indole -2- carboxylic acid) (VI) :
A mixture of compound (V) ( 95g, 30.64 mmol) and sodium hydroxide (36.78g,91.92 mmol) in water (570 ml) was refluxed for 2 h. The obtained reaction mixture was cooled and acidified with cone. HCl . The precipitated solid was filtered, washed with water and dried. [ Yield: 85.5g, 90%]
Example 4
Preparation of N-methyl-lH-indole-5-ethanesulfonamide (VII) :
A mixture of compound (VI) (85g, 30.14mmol) and cuprous oxide (4.25 g) in quinoline
(425 ml) was refluxed for 6h at temperature 180-2000C. The obtained reaction mixture was cooled followed by addition of ethyl acetate (400 ml). The reaction mixture was filtered through hyflow. The filtrate was washed with dilute hydrochloric acid followed by water wash and the filtrate was concentrated to get solid of compound (VII). [Yield :
55g,76.66%]
Example 5
Method 1: Preparation of (E)-N-Methyl-2-[3-(l,2,3,6-tetrahydro-l-methyl-4- pyridinyl)-lH-indol-5-yl]ethanesulfonamide (VIII)
A mixture of compound (VII) (51 g, 21.42mmol), N-methyl-4-piperidone ( 60.6 Ig,
53.56mmol) and potassium hydroxide ( 3Og, 53.57mmol) in ethanol (0.357 lit) was refluxed for 12 hr. The obtained reaction mixture was cooled to 10°C and precipitated solid was filtered washed with ethanol followed by water wash and dried. [Yield:
55g,77.07% ]
Method 2: A mixture of the compound (VII) (51 g, 21.42mmol), N-Methyl-4- pipperidone (60.61g, 53.56mmol) and potassium hydroxide (30g, 53.57mmol) in ethanol
(0.357 lit) was refluxed for 6-8 hrs. The obtained reaction mixture was cooled to 20°C and quenched slowly in 1.5 lit chilled water (at 10-15°C). The precipitated solid was stirred for one hour. The obtained residue was washed with water till pH 7 to 8. and then wash with chilled ethanol to get compound (VIII) (55g).
The compound (VIII) (55 g) in methanol (1.65 L) was refluxed for one hour till to get clear solution. The obtained solution was filtered through hyflow. The solvent was partially distilled out. The remaining mixture was cooled to 10°C. The solid obtained was filtered and washed with chilled methanol and dried. [Yield: 49.5g, 90%].
Example 6
Preparation of N-methyl-3-(l-methyl-4-piperidinyl)-lH-indole-5-ethanesulfon- amide (IX):
The compound (VIII) (38g, 11.14mmol) was reduced using platinum oxide (7.6g) in a mixture of 50% acetic acid in methanol (600 ml) for 8h under hydrogen pressure at 40 psi for 8 hours at temperature 400C. After completion of reaction, the reaction mixture was filtered through hyflow. The filtrate was concentrated, basified with aqueous sodium carbonate solution and extracted with ethyl acetate (1 lit). The ethyl acetate layer was separated, washed with EDTA, water and concentrated to get the solid. [ Yield 24.54g,
64.20%]
Example 7
Method 1: Preparation of Naratriptan Hydrochloride (I)
The compound (IX) (24g, 716mmol) was dissolved in ethanol (150 ml) at refluxed temperature. The obtained solution was cooled at room temperature, 24 ml of ethanolic hydrochloride was slowly added to it and refluxed fro 1 hour. A mixture of ethanol- water
(120 ml / 72 ml) was added to the reaction mixture and refluxed for 1 h. followed by addition of (0.24 g) activated charcoal. The hot solution was filtered through hyflow. The
filtrate was stirred for 1 h at temperature 10°C, the separated solid was filtered, washed with cold ethanol and dried. [Yield : 14.4 g, 54.11%]
Method 2: The compound (IX) (24g, 716 mmol) obtained in example 6 was dissolved in ethanol (1.92 lit) at refluxed temperature. Activated charcoal (0.24 g) was added and refluxed for 30 min. The mixture was filtered and washed with ethyl alcohol. The filtrate was cooled to 30-35°C. 24 ml of ethanolic hydrochloride was slowly added to the obtained solution and cooled at 10-15°C. The solid obtained was filtered, washed with ethanol to get titled compound (I) [Yield 24g, 89.78%]
The compound (I) (24g, 646 mmol) was dissolved in ethanol (0.240 lit) and water (0.048 lit) at refluxed temperature. The reaction mixture was filtered through hyflow and cooled at 10-15 °C followed by stirring for one hour. The solid obtained was filtered, washed with mixture of ethanol- water and dried. [Yield 20.4 g, 85%]
Example 8
Preparation of Naratriptan hydrochloride (I) :-
The compound (VIII) (50 g, 1501 mmol) was dissolved in mixture of methanol: acetic acid (0.2 lit:0.2 lit) and 2.5 g platinum oxide was added. The mixture was hydrogenated using 5kg/cm2 at 50 to 60°C for 6 to 8 hrs. After completion of the reaction the mixture was filtered and distilled to get an oil. The obtained oil was stripped with isopropyl alcohol followed by addition of 125 ml ethanolic HCl and the reaction mixture was refluxed for 1 hour. The precipitated solid was stirred at 5 to 20°C for 1 hour, filtered and washed with chilled ethanol to titled compound (I). [Yield 50g,89.76%]
The obtained compound (I) (50g, 1347mmol) was further dissolved in 400 ml of ethanol and 100 ml of water at 78 to 80°C and 2g of activated charcoal was added. The reaction mixture was refluxed for half an hour. The obtained solution was filtered through hyflow and washed with 100 ml of ethanol. The filtrate was stirred for half an hour at 5 to 20°C to precipitates out the solid which was filtered, washed with chilled ethanol and dried.
[Yield 42.5 g, 85%]. Purity : 99.8%
Example 9
Preparation of N-Methyl vinyl sulfonamide
PART A: A mixture of isethionic acid sodium salt (0.250 Kg, 1689 mmol) was taken in
DMF (0.32L) and dichloromethae (0.3L). To this mixture thionyl chloride (0.375L,
5186mmol) was added dropwise at 20-30°C. The reaction mixture was heated slowly from 30-60 "C. After completion of the reaction 3.5 lit of chilled water was added. The reaction mixture was extracted with 3x0.5L dichloromethane. Then dichloromethane was distilled out from the reaction mixture to get an oil. [ Yield 0.250Kg, 90%] which was distilled under vacuum to get pure chloroethane sulfonyl chloride. (0.225kg, 90%). PART B: A mixture of chloroethane sulfonyl chloride (10Og, 613 mmole) and tetrahydrofuran (IL) was stirred under nitrogen atmosphere. The reaction mixture was cooled to -50 to -60°C. To the reaction mixture hydroquinone was added in the catalytic amount and N-methyl amine gas (142ml, 1839mmol) was purged (Aqueous methyl amine was added in potassium hydroxide) in the reaction mixture maintaining temperature -50 to -6O0C. The reaction was monitored on GC. After completion of the reaction, the reaction mixture was filtered. The obtained filtrate was evaporated under vacuum not more than 50°C to get thick oil which was stored under nitrogen atmosphere. Crude N- Methyl vinyl sulfonamide (50gm, 413mmol) was distilled using short path distillation to get pure compound. [Yield 33.61g, 67.35%] Example 10
Preparation of Aryl vinyl sulfonate
PART A: A mixture of isethionic acid sodium salt (0.250 Kg, 1689 mmol) was taken in DMF (0.32L) and dichloromethane (0.3L). To this mixture thionyl chloride (0.375L, 5186mmol) was added dropwise at 20-300C. The obtained reaction mixture was heated slowly from 30-60°C. After completion of the reaction 3.5 lit of chilled water was added to the reaction mixture and the reaction mixture was extracted with 3x0.5L dichloromethane. The solvent was distilled out from the reaction mixture to get oil. The obtained oil ( 0.250 Kg, 1533mmole) was distilled under vacuum to get pure chloroethane sulfonyl chloride [ Yield 0.225Kg, 90%].
PART B: A mixture of chloroethane sulfonyl chloride (10Og, 613 mmole) and dichloromethane (0.4L) was stirred under nitrogen atmosphere. To the obtained reaction mixture hydroquinone was added in the catalytic amount and reaction mixture was chilled to -15 to -20°C. A mixture of phenol (57.6gm, 613mmol) and triethylamine (0.171 L, 813mmol) in dichloromethane (0.4L) was added to the reaction mixture maintaining the same temperature. The mixture was then stirred at 25 to 30°C for one hour and monitored
on GC. After completion of reaction dichloromethane ( 0.2 L) was added. The separated organic layer was treated with hydrochloric acid (2%, 1.5L) followed by aqueous sodium hydroxide ( IL, 5%) and water (2xlL). Then the organic layer was distilled out to get an oil (86gm, 76.78%) which was distilled under high vacuum or shortpath distillation to get pure compound which gets solidified after cooling. [Yield 64gm, 74.41%].
Example 11
Preparation of 2-(lH-indol-5-yl)-N-methyl ethene sulfonamide (F)
A mixture of palladium acetate (1.5 g, 6.375 mmol) and tri-o-tolyl phosphine (9 g, 29.25 mmol) in dimethylformamide (0.2 L) was stirred under nitrogen atmosphere. A mixture of 5-bromo indole (50 g, 255 mmol), N-methyl vinyl sulfonamide (7.5 g, 310 mmol) and triethylamine (0.035 L, 251 mmol) in dimethylformamide (0.05 L) was added to the reaction mixture. The reaction mixture was heated to 100-110°C for 8 to 10 hrs. After completion of the reaction, the mixture was cooled to 35 to 4O0C and filtered through hyflow. The filtrate was distilled off completely to yield thick mass. 500 ml of water was slowly added to the obtained thick mass to get solid which was filtered, dried and crystallized from mixture of ethyl acetate : hexane to get 2-(lH-indol-5-yl)-N-methyl ethene sulfonamide (F) [Yield: 59 g; 98.00%].
Purification of 2-(lH-indol-5-yl)-N-methyl ethene sulfonamide (F):
The compound (F) (29.5 g, 125 mmol) in isopropanol (0.6 lit) was refluxed till solution becomes clear. Partially isopropanol was distilled out from reaction mixture followed by cooling to 10° C and stirring for one hour. The solid obtained was filtered and washed with chilled isopropanol (0.120 lit) and dried in oven. [Yield 19.175 g, 65%]
The mixture of compound (F) (29.5 g, 125 mmol) in hexane : ethyl acetate (0.236 lit :
0.06 lit) was refluxed for one hour. The reaction mixture was filtered, washed with hexane and dried in oven. [Yield 0.25 g, 85%]
Example 12
Preparation of 2-(lH-indol-5-yl)-N-methyl ethane sulfonamide (VΗ):"
Method I: A mixture of N-methyl- lH-indole- 5 -ethenesulfonamide (4Og, 169mmol) was hydrogenated using 50 % wet 10% palladium on charcoal (40 g) in a mixture of
(Methanol: THF 0.2 L each) for 6 h at 70-80"C at 70 psi Kg/cm2 hydrogen pressure. After completion of reaction the reaction mixture was filtered through hyflow and filtrate was
concentrated to obtain solid. This solid was treated with ethylacetate and filtered. The filtrate was concentrated to get residue. The residue was treated with hexane to obtain solid. [Yield 32 gm, 79.34%]
Method II: A mixture of N-methyl-lH-indole-5-ethenesulfonamide (4Og, 169mmol) was hydrogenated using 50 % wet 10% palladium on charcoal (40 g) in a mixture of
(Methanol: ethyl acetate 500: 250 ml) for 6 h at 70-80°C at 70 psi Kg/cm2 hydrogen pressure. After completion of reaction the reaction mixture was filtered through hyflow and filtrate was concentrated to obtain solid. This solid was treated with ethylacetate and filtered. The filtrate was concentrated to get residue. The residue was treated with hexane to obtain solid. [Yield 36 gm, 90%]
Example 13
Preparation of Phenyl-2-(lH-indol-5-yl) ethene sulfonate :-
A mixture of palladium acetate ( 4gm, 17 mmol) and tri-o-tolyl phosphine (12gm,
39mmol) in dimethylformamide (0.2L) was stirred under nitrogen atmosphere. A mixture of 5-bromo indole( 50gm, 255mmol), phenyl vinyl sulfonate ( 42.50gm, 231 mmol) and triethylamine (0.035L, 251mmol) in dimethylformamide (0.05L) was added to the reaction mixture. The reaction mixture was heated to 100-110°C for 8 to lOhrs. After completion of the reaction, the reaction mixture was cooled to 35 to 400C and filtered through hyflow. The filtrate was treated with hexane (0.2L) and then mixture was quenched in water (IL) with stirring. The separated water layer was treated with ethyl acetate (3 x 0.2 L) followed by water. The organic layer was evaporated to get solid which was treated with ethyl acetate and hexane ( 80:20, 0.2L) to get pure solid. [ Yield
59.5gm, 78%]
Example 14
Preparation of Phenyl -2-(lH-indoI-5-yl) ethane sulfonate: -
A mixture of compound (X) (4Og, 133mmol) was hydrogenated using 50 % wet palladium on charcoal (63gm) in a mixture of methanol: tetrahydrofuran (0.2 L each) for
6 to 8h at 70-80°C at 70 psi Kg/cm2 hydrogen pressure. After completion of reaction, the reaction mixture was filtered through hyflow and obtained filtrate was distilled out to get solid.[ Yield 28 gm, 69.54%]
Example 15
Preparation of 2-(lH-indol-5-yl)-N-methyl ethane sulfonamide:-
A mixture of aryl vinyl sulfonate (28gm, 93mmol) and methylamine gas (8.65 gm,
280mmol) in pyridine (7.53ml, 93 mmol) was heated in autoclave at 100°C for 3 hrs.
After reaction mixture was over the mixture was filtered and filtrate concentrated to get oil. [Yield 3gm, 13.55%].
Example 16
Preparation of desmethyl Naratriptan:-
Step I: Preparation of vinyl sulfonamide
A mixture of chloroethane sulfonyl chloride (10Og, 613 mmole) and tetrahydrofuran (IL) was stirred under nitrogen atmosphere. The reaction mixture was cooled to -30 to -60°C.
To the reaction mixture an ammonia gas (35 gm, 2144mmol) was slowly purged at the same temperature. The reaction was monitored on GC. After completion of reaction, the temperature of the reaction was maintained at 25°C. Filtered through hyflow, DMF was distilled to get thick oil which was treated with MDC, and MDC distilled to get oil as vinyl sulfonamide 98% pure on GC. [ Yield 65gm]
Step II: Preparation of 2-(lH-indol-5-yl)-ethenesulfonamide (IXa)
A mixture of palladium acetate (3.0 g, 2.9 mmol) and tri-o-tolyl phosphine (9.09 g, 4.5 mmol) in dimethylformamide (0.2 L) was stirred under nitrogen atmosphere. A mixture of 5-bromo indole (50 g, 255 mmol), vinyl sulfonamide (34.1 g, 318.7 mmol) and triethylamine (0.035 L, 252 mmol) in dimethylformamide (0.05 L) was added to the reaction mixture. The reaction mixture was heated to 100-110°C for 8 to 10 hrs. After completion of the reaction, the mixture was cooled to 35 to 40°C and filtered through hyflo. The filtrate was distilled off completely to yield thick mass. 500 ml of water was slowly added to the obtained thick mass to get solid which was filtered, dried and crystallized from mixture of ethyl acetate : hexane to get 2-(lH-indol-5-yl)-ethene sulfonamide [Yield: 45.00 g; 78.75%].
Step III: Preparation of 2-(lH-indol-Syl)-ethanesulfonamide (IXb)
A mixture of lH-indole-5-ethenesulfonamide (IXa)(40g, 180mmol) was hydrogenated using 50 % wet palladium on charcoal (80gm)) in a mixture of (Methanol: Ethyl acetate
0.4 L: 0.8 L) for 6 to 8h at 25°-30° C at 140 psi Kg/cm2 hydrogen pressure. After
completion of reaction the reaction mixture was filtered through hyflo and filtrate was distilled to get oil. [ Yield 32 gm, 79.30 %].
Step IV: Preparation of 2-[3-(l,2,3,6-tetrahydro-l-methyl-4-pyridinyl)-lH-indol-5- yl]ethanesulfonamide (IXc)
A mixture of 2-(lH-indol-5yl)-ethane sulfonamide (3Og, 132.7mmol), N-methyl-4- piperidone (37.5g, 331.86mmol) and potassium hydroxide (18.6g, 331.85mmol) in ethanol (0.21 L) was refluxed for 8-10 hrs at temperature 78-8O0C. The reaction mixture was cooled to 40°C and quenched in 0.42 L water at temperature 10°C precipitated solid was filtered washed with water followed by chilled ethanol and dried. [Yield:25g,58.67
% ]
Step V: Preparation of 2[3-(l,2,3,6-tetrahydro-l-methyl-4-piperidinyl)-lH-indol-5- yl]ethanesulfonamide (IA)
A mixture of compound (2Og, 62.30mmol) was reduced using platinum oxide (5.0 g) in a mixture of 50% acetic acid in methanol (320 ml) for 8h at 70 psi at 500C. After completion of reaction, the reaction mixture was filtered through hyflo. The filtrate was concentrated, basified with aqueous sodium carbonate solution and extracted with ethyl acetate (1 lit). The ethyl acetate layer was separated, washed with EDTA, water and concentrated to get the solid. [ Yield 12g, 59.64%] Preparation of amorphous Naratriptan hydrchloride Example 17
Ig Naratriptan HCl was dissolved in 40 ml water at 50-600C. The hot solution was filtered to remove insoluble matter. The clear solution was lyophilized for 24 h to get amorphous Naratriptan HCl.
Example 18
1Og Naratriptan was suspended in 250 ml water at 40-500C. 2.6 ml cone. HCl was added to the obtained suspension. The hot solution was filtered to remove insoluble matter. The clear solution was lyophilized for 24hr to get amorphous Naratriptan HCl
Example 19
2Og of Naratriptan HCl was dissolved in 400 ml water at 50-600C. The solution was filtered to get clear solution. The clear solution was spray dried. Spray drying was carried
out at the inlet temperature 1200C and outlet temperature 650C to get amorphous Naratriptan HCl.
Example 20
2Og of Naratriptan was dissolved in 400 ml water at 50-600C. 5.2 ml cone. HCl was added to the obtained suspension. The solution was filtered to get clear solution. The clear solution was spray dried. Spray drying was carried out at the inlet temperature 1200C and outlet temperature 650C to get amorphous Naratriptan HCl.
Example 21
5g Naratriptan HCl was dissolved in 150 ml water at a temperature range of 50-600C. The clear solution was concentrated under vacuum at 650C to get amorphous Naratriptan HCl.
Example 22
1Og Naratriptan was suspended in 250 ml water at 40-500C. 2.6 ml cone. HCl was added to the obtained suspension. The hot solution was filtered to remove insoluble matter. The clear solution was concentrated under vacuum at 650C to get amorphous Naratriptan. HCl.
Claims
1. A process for preparation of N-methyl-2-[3-(l-methyl-4-piperidinyl)-lH-indole-5- yl ethanesulfonamide salts comprising the steps of; a) combining compound (VII) with N-Methyl-4-piperidone to produce compound (VIII); b) combining compound (VIII) in suitable solvent with catalyst under reducing condition and adding acid to the obtained reaction mixture to get Naratriptan salt (I). c) optionally purifying Naratriptan salts to get pure compound with purity more than 90%.
2. Naratriptan or salts thereof having less than 0.15% area by HPLC of 3-(l-methyl- 4-piperidinyl)-lH-indole-5-ethanesulfonamide (IA) or desmethyl Naratriptan.

(IA)
3. Desmethyl Naratriptan (IA) as a reference standard to quantify the amount of impurity in a sample of Naratriptan or salts thereof.
4. A process for preparation of desmethyl Naratriptan which comprises the steps of f) treating chloroethane sufonyl chloride with ammonia in suitable solvent to get vinyl sulfonamide; 
Vinyl sulfonamide g) coupling vinyl sulfonamide with 5-bromoindole (B) in presence of coupling agent, ligand, and base to get compound (IXa);



5. A process for preparation of N-methyl-lH-indole-5-ethanesulfonamide (VII) used for the preparation of Naratriptan hydrochloride which comprises the steps of; a) combining compound (VII) with N-Methyl-4-piperidone to produce compound (VIII); b) combining the obtained compound of step a) with 5-bromo indole, aryl vinyl sulfonate compound wherein aryl is substituted or unsubstituted phenyl to get lH-indole-5-aryl ethenesulfonamide (X). c) hydrogenating the compound (X) to get compound (XI); d) treating compound (XI) with methylamine to obtain compound (VII).
6. A process for preparation of N-methyl-lH-indole-5-ethanesulfonamide (VII) which comprises the steps of a) treating compound (B) with N-methyl vinyl sulfonamide in suitable solvent to get compound (F). b) optionally purifying compound (F); c) reducing obtained compound (F) in the presence of catalyst to get compound
reduction . 

7. A process for preparation of the key intermediate N-methyl- lH-indole-5- ethanesulfonamide (VII) which comprises the steps of; a) combining 5-bromo indole (B) with aryl vinyl sulfonate compound wherein aryl is substituted or unsubstituted phenyl to get lH-indole-5-aryl ethenesulfonamide (X); b) hydrogenating the compound (X) to get lH-indole-5-aryl ethanesulfonamide
(XI); c) treating compound (XI) with methylamine to obtain compound (VII).
8. A process for preparation of the key intermediate N-methyl- 1 H- indole-5- ethanesulfonamide (VII) which comprises the steps of; a) combinig compound (B) with N-methyl vinyl sulfonamide in suitable solvent to get compound (F); b) optionally purifying compound (F); c) reducing obtained compound (F) in the presence of catalyst to get compound (VII). reduction 

9. A process for preparation of N-methyl- lH-indole-5-ethanesulfonamide (VII) comprises the steps of; a) combining lH-indole-5-aryl ethenesulfonamide (X) with methylamine to obtain N-methyl- lH-indole-5-ethenesulfonamide (F); b) optionally purifying compound (F); c) hydrogenating the compound (F) using catalyst to get compound (VII).

10. A process for preparation of N-methyl vinyl sulfonamide or aryl vinyl sulfonate which comprises the steps of; a) combining isethionic acid salt with chlorinating agent to get chloroethane sulfonyl chloride; b) optionally distilling the compound chloroethane sulfonyl chloride under vacuum; c) combining the obtained pure chloroethane sulfonyl chloride with methylamine to get N- methyl vinyl sulfonamide or combining obtained pure chloroethane sulphonyl chloride with aryl OH, wherein aryl is substituted or unsubstituted phenyl compound to get aryl vinyl sulfonate and d) optionally purifying N-methyl vinyl sulfonamide or aryl vinyl sulfonate under high vacuum or short path distillation to get pure N- methyl vinyl sulfonamide.
11. The process as claimed in claim 10 wherein isethionic acid salt is isethionic acid alkali salt.
12. The process as claimed in claim 10 wherein said methylamine is in the form of solution or gas optionally in presence of catalytic amount of hydroquinone.
13. The process as claimed in claim 10 wherein purification of N-methyl vinyl sulfonamide and aryl vinyl sulfonate is performed by vacuum or short path distillation at temperature 90-1700C and at 0.1- 0.5 mm vacuum to get N-methyl vinyl sulfonamide having less than about 0.5% of vinyl sulfonamide.
14. A process for preparation of N-methyl- lH-indole-5-ethanesulfonamide (VII) comprises the steps of; a) reacting hydrazino derivative (II) with a pyruvic acid ester compound (III) to obtain hydrazone compound (IV); b)cyclizing the hydrazone compound (IV) in the presence of an acid to give the indole derivative (V); c)hydrolyzing the obtained compound (V) with base and acidifying with acid to produce compound (VI); d) decarboxylating the obtained compound (VI) to produce indole sulfonamide compound (VII);
15. The process as claimed in in any of the preceding claims wherein said catalyst is selected from the group consisting of palladium oxide on charcoal, palladium on charcoal, platinum oxide on charcoal, platinum on charcoal and said acid used is selected from methane sulfonic acid, acetic acid, phosphoric acid, hydrochloric acid, sulfuric acid, p-toluenesulphonic acid and boron trifluoride optionally using in presence of solvent.
16. The process as claimed in any of the preceding claims wherein said solvent is selected from dimethylformamide, dimethylacetamide, tetrahydrofuran, methylene chloride, methanol, ethanol, isopropanol, propanol, methyl acetate, ethyl acetate, isopropyl acetate, butyl acetate, pentane, hexane, heptane, acetonitrile, dimethylformamide, water or mixture thereof and base used is selected from sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate potassium carbonate, triethylamine and diisopropylethylamine.
17. Naratriptan characterized by the X-ray powder diffraction pattern as shown in Figure 1.
18. Amorphous Naratriptan hydrochloride.
19. A process for preparation of amorphous Naratriptan HCl comprising a) dissolving Naratriptan HCl in a suitable solvent or suspending Naratriptan in suitable solvent and adding conc.HCl to the suspension b) spray drying the resultant clear solution of step a) to get amorphous form.
20. A process for preparation of amorphous Naratriptan HCl comprises a) dissolving Naratriptan HCl in a suitable solvent or suspending Naratriptan in in solvent and adding cone. HCl to get the suspension and b) lyophilizing the resultant clear solution of step a) to get amorphous form.
21. The process as claimed in claim 19 or 20 wherein said solvent is water and polar protic solvent or mixtures thereof.
22. The process as claimed in claim 19 or 20 wherein concentration of Naratriptan HCl used in lyophilization as well as in spray drying is 4-10% preferably 4-8 %.
23. Naratriptan HCl having particle size d^ less than about 250 μ preferably less than about lOOμ more preferably less than about 65μ.
24. A pharmaceutical composition comprising Naratriptan hydrochloride having less than 0.15% area by HPLC of 3-(l-methyl-4-piperidinyl)-lH-indole-5- ethanesulfonamide (IA) or desmethyl Naratriptan along with pharmaceutically acceptable excipients.
25. The process for preparation of Naratriptan and pharmaceutical acceptable salts thereof, substantially as described herein with respect to examples 1 to 22.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN00464/MUM/2008 | 2008-03-07 | ||
| IN464MU2008 | 2008-03-07 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009118753A2 true WO2009118753A2 (en) | 2009-10-01 |
| WO2009118753A3 WO2009118753A3 (en) | 2010-01-28 |
Family
ID=40933289
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2009/000165 WO2009118753A2 (en) | 2008-03-07 | 2009-03-09 | Process for preparation of naratriptan hydrochloride |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009118753A2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011021000A3 (en) * | 2009-08-20 | 2011-09-15 | Cipla Limited | A process for the synthesis of naratriptan |
| WO2013047162A1 (en) * | 2011-09-26 | 2013-04-04 | 東洋紡株式会社 | Method for producing chloroalkylsulfonyl chloride |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0633252B2 (en) * | 1987-08-13 | 1994-05-02 | グラクソ、グループ、リミテッド | Indole derivative |
| GB9320115D0 (en) * | 1993-09-29 | 1993-11-17 | Glaxo Group Ltd | Process |
| WO2008056378A2 (en) * | 2006-11-09 | 2008-05-15 | Natco Pharma Limited | Novel process for the preparation of naratriptan hydrochloride |
| WO2008072257A2 (en) * | 2006-12-12 | 2008-06-19 | Ind-Swift Laboratories Limited | Process for the preparation of indole derivatives |
| WO2009016466A2 (en) * | 2007-07-30 | 2009-02-05 | Orchid Chemicals & Pharmaceuticals Limited | A process for the preparation of naratriptan hydrochloride |
-
2009
- 2009-03-09 WO PCT/IN2009/000165 patent/WO2009118753A2/en active Application Filing
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011021000A3 (en) * | 2009-08-20 | 2011-09-15 | Cipla Limited | A process for the synthesis of naratriptan |
| US8735589B2 (en) | 2009-08-20 | 2014-05-27 | Cipla Limited | Process for the synthesis of naratriptan |
| WO2013047162A1 (en) * | 2011-09-26 | 2013-04-04 | 東洋紡株式会社 | Method for producing chloroalkylsulfonyl chloride |
| JPWO2013047162A1 (en) * | 2011-09-26 | 2015-03-26 | 東洋紡株式会社 | Method for producing chloroalkylsulfonyl chloride |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009118753A3 (en) | 2010-01-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5366163B2 (en) | A novel crystalline form of pitavastatin calcium | |
| EP0421946B1 (en) | 3-indolepyruvic acid derivatives their method of production and therapeutic use | |
| CA2646368A1 (en) | Substantially pure o-desmethylvenlafaxine and processes for preparing it | |
| EP1790635A2 (en) | Processes for preparing amorphous fluvastatin sodium | |
| AU2015254949A1 (en) | Novel polymorphic forms of Vortioxetine and its pharmaceutically acceptable salts | |
| CN113874359B (en) | Process for preparing 1-deoxy-1-methylamino-D-glucitol 2- (3, 5-dichlorophenyl) -6-benzoxazole carboxylate | |
| SK8872002A3 (en) | Novel sertraline hydrochloride polymorphs, processes for preparing them, compositions containing them and methods of using them | |
| KR20130038258A (en) | Saxagliptin intermediates, saxagliptin polymorphs, and processes for preparation thereof | |
| KR20060111675A (en) | Polymorph of Tegaserod Base and Its Salts | |
| EP3649116A1 (en) | A process for preparing alectinib or a pharmaceutically acceptable salt thereof | |
| WO2016146990A1 (en) | Improved process for the preparation of apremilast | |
| CA2719526A1 (en) | Conversion of tryptophan into .beta.-carboline derivatives | |
| CN111285847A (en) | Method for preparing alectinib | |
| US20070254941A1 (en) | Subtantially pure ropinirole hydrochloride, polymorphic form of ropinirole and process for their preparation | |
| WO2009118753A2 (en) | Process for preparation of naratriptan hydrochloride | |
| EP4313987A2 (en) | Process for the preparation of ribociclib and pharmaceutically acceptable salts thereof | |
| CZ20022302A3 (en) | Novel tetrahydropyridines, process of their preparation and pharmaceutical compositions in which these tetrahydropyridines are comprised | |
| KR20070062504A (en) | Polymorph of Tegase Road Maleate | |
| EP3990113A1 (en) | Solid state forms of roluperidone and salts thereof | |
| EP2162434B1 (en) | Salts of perindopril | |
| WO2015075749A1 (en) | Novel processes for the preparation of vemurafenib | |
| Finch et al. | Synthesis of 4-azaoxindole | |
| WO2008072257A2 (en) | Process for the preparation of indole derivatives | |
| US20110245290A1 (en) | Alternative Forms of the Phosphodiesterase-4 Inhibitor N-Cyclopropyl-1--4-Oxo-1,4-Dihydro-1,8-Naphthyridine-3-Carboxyamide | |
| JP4324381B2 (en) | Phenyl- and pyridyl-piperidines having TNF activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09725037 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09725037 Country of ref document: EP Kind code of ref document: A2 |