WO2009109608A1 - Novel compounds - Google Patents
Novel compounds Download PDFInfo
- Publication number
- WO2009109608A1 WO2009109608A1 PCT/EP2009/052567 EP2009052567W WO2009109608A1 WO 2009109608 A1 WO2009109608 A1 WO 2009109608A1 EP 2009052567 W EP2009052567 W EP 2009052567W WO 2009109608 A1 WO2009109608 A1 WO 2009109608A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- dichlorophenyl
- methyl
- pharmaceutically acceptable
- acceptable salt
- Prior art date
Links
- 0 *C(C1)N(*)C[C@]2(*)[C@@]1(*)C2(*)* Chemical compound *C(C1)N(*)C[C@]2(*)[C@@]1(*)C2(*)* 0.000 description 8
- QBNFQHNWDRCADR-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CC(C[n]2c(C)cc(C)c2)(C2)C12c(cc1Cl)ccc1Cl)=O Chemical compound CC(C)(C)OC(N(CC1)CC(C[n]2c(C)cc(C)c2)(C2)C12c(cc1Cl)ccc1Cl)=O QBNFQHNWDRCADR-UHFFFAOYSA-N 0.000 description 1
- MOJRMPLENJBVSM-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CC(C[n]2ncc(C)c2)(C2)C12c(cc1)cc(Cl)c1Cl)=O Chemical compound CC(C)(C)OC(N(CC1)CC(C[n]2ncc(C)c2)(C2)C12c(cc1)cc(Cl)c1Cl)=O MOJRMPLENJBVSM-UHFFFAOYSA-N 0.000 description 1
- FRNOLPFDQXXJGJ-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1(C[n]1c(C)ccc1C)c(cc1Cl)ccc1Cl)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1(C[n]1c(C)ccc1C)c(cc1Cl)ccc1Cl)=O FRNOLPFDQXXJGJ-UHFFFAOYSA-N 0.000 description 1
- KSIBEXHQWYOQEQ-FSJBWODESA-N CC(C)(C)OC(N(CCC1(C2)c(cc3)cc(Cl)c3Cl)CC12/C(/C)=N/N)=O Chemical compound CC(C)(C)OC(N(CCC1(C2)c(cc3)cc(Cl)c3Cl)CC12/C(/C)=N/N)=O KSIBEXHQWYOQEQ-FSJBWODESA-N 0.000 description 1
- CDWPJCDITPWCCS-UHFFFAOYSA-N CC(C)(C)OC(N(CCC1(C2)c(cc3Cl)ccc3Cl)CC12c1n[o]c(C)c1)=O Chemical compound CC(C)(C)OC(N(CCC1(C2)c(cc3Cl)ccc3Cl)CC12c1n[o]c(C)c1)=O CDWPJCDITPWCCS-UHFFFAOYSA-N 0.000 description 1
- DZADYAMWXBEXOJ-UHFFFAOYSA-N CC(C)(C)OC(N(CCC1)CC1(CN=[N+]=[N-])c(cc1Cl)ccc1Cl)=O Chemical compound CC(C)(C)OC(N(CCC1)CC1(CN=[N+]=[N-])c(cc1Cl)ccc1Cl)=O DZADYAMWXBEXOJ-UHFFFAOYSA-N 0.000 description 1
- XXBGLDVRLMPFSC-UHFFFAOYSA-N CC(C)c1n[n](CC(C2)(CN(CC3)C(OC(C)(C)C)=O)C23c(cc2)cc(Cl)c2Cl)nn1 Chemical compound CC(C)c1n[n](CC(C2)(CN(CC3)C(OC(C)(C)C)=O)C23c(cc2)cc(Cl)c2Cl)nn1 XXBGLDVRLMPFSC-UHFFFAOYSA-N 0.000 description 1
- XBOMFAHBQGMFRQ-UHFFFAOYSA-N CC(C)c1nnn[n]1CC(C1)(CN(CC2)C(OC(C)(C)C)=O)C12c(cc1)cc(Cl)c1Cl Chemical compound CC(C)c1nnn[n]1CC(C1)(CN(CC2)C(OC(C)(C)C)=O)C12c(cc1)cc(Cl)c1Cl XBOMFAHBQGMFRQ-UHFFFAOYSA-N 0.000 description 1
- HRCVSENKVJEEJB-UHFFFAOYSA-N Clc1ccc(C2(C3)C3(C[n]3nc(cccc4)c4c3)CNCC2)cc1Cl Chemical compound Clc1ccc(C2(C3)C3(C[n]3nc(cccc4)c4c3)CNCC2)cc1Cl HRCVSENKVJEEJB-UHFFFAOYSA-N 0.000 description 1
- GRCVTMNCDBAKJY-UHFFFAOYSA-N Clc1ccc(C2(C3)C3(C[n]3ncc4c3cccc4)CNCC2)cc1Cl Chemical compound Clc1ccc(C2(C3)C3(C[n]3ncc4c3cccc4)CNCC2)cc1Cl GRCVTMNCDBAKJY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
Definitions
- the present invention relates to novel compounds, processes for their preparation, intermediates used in these processes, pharmaceutical compositions containing them and their use in therapy, as serotonin (5-HT), dopamine (DA) and norepinephrine (NE), re-uptake inhibitors.
- serotonin 5-HT
- DA dopamine
- NE norepinephrine
- Brain tissue is constituted of neuronal cells which are able to communicate with each other via specific cellular structures named synapses.
- the exchange of signals between neurons in the synapses happens through neurochemical messengers named neurotransmitters, acting on specific target protein molecules, both post and pre-synaptic, referred to as receptors.
- Monoamines represent a family of small neurotransmitter molecules sharing common chemical features, and include serotonin (5-HT), dopamine (DA) and norepinephrine (NE).
- Monoamine neurotransmitters are released into the synaptic cleft between neurons and interact with receptors present on the membrane of the target cells.
- the switch of the neurochemical signal occurs mainly by removal of the neurotransmitter molecules through other protein molecules referred to as monoamine transporters (SERT for 5-HT, DAT for DA and NET for NE).
- Transporters are able to bind neurotransmitter molecules and move them into the presynaptic terminals, this cellular mechanism referred to as re-uptake.
- Pharmacological inhibition of the reuptake process can cause an increase of monoamine at synaptic level and as a consequence an enhancement of the physiological activity of neurotransmitters.
- Serotonergic neurotransmission in the brain is mediated by a large family of receptors comprising both the G-protein coupled receptors and ligand-gated ion channels including 14 subtypes, and is involved in a vast variety of physiologic functions.
- Compounds endowed of inhibitory properties at the SERT are predicted to have the ability to treat in mammals, including humans, a variety of disorders associated with this neural system, for example eating disorders, major depression and mood disorders, obsessive compulsive disorders, panic disorders, alcoholism, pain, memory deficits and anxiety.
- disorders related to depression such as pseudodementia or Ganser's syndrome, migraine pain, bulimia, obesity, pre-menstrual syndrome or late luteal phase syndrome, tobacco abuse, panic disorder, post-traumatic syndrome, memory loss, dementia of ageing, acquired immunodeficiency syndrome dementia complex, memory dysfunction in ageing, social phobia, attention deficit hyperactivity disorder, chronic fatigue syndrome, premature ejaculation, erectile difficulty, anorexia nervosa, disorders of sleep, autism, mutism or trichotillomania.
- depression such as pseudodementia or Ganser's syndrome, migraine pain, bulimia, obesity, pre-menstrual syndrome or late luteal phase syndrome, tobacco abuse, panic disorder, post-traumatic syndrome, memory loss, dementia of ageing, acquired immunodeficiency syndrome dementia complex, memory dysfunction in ageing, social phobia, attention deficit hyperactivity disorder, chronic fatigue syndrome, premature ejaculation, erectile difficulty, anorexia nervosa, disorders of sleep, autism,
- Major depression is an affective disorder, or disorder of mood, characterized by several symptoms including feeling of profound sadness, worthlessness, despair and loss of interest in all pleasures (anhedonia), recurrent thoughts of death, mental slowing, loss of energy, an inability to take decision, often associated with anxiety and agitation. These symptoms are persistent and can range from mild to severe.
- the pathophysiology of major depression is poorly understood being a multifactorial syndrome and, due to this, several neurotransmitter systems have been implicated. However, it is generally believed that the disorder stems from a decrease in the synaptic concentration of monoamine neurotransmitters, mainly NE and 5-HT, in critical brain areas, leading to the "monoamine theory" of depression.
- NET potency > SERT many compounds, including old tricyclic antidepressants, have a mixed NET and SERT inhibition profile, like lmipramine and Amitriptyline (with SERT potency > NET) and Desipramine, Nortriptyline, and Protriptyline (NET potency > SERT).
- the pharmacological manipulation of the DAT can in principle have the ability to elevate DA levels in the mesolimbic system, reversing the anhedonia that is a core symptom of major depression.
- a DAT inhibition component in combination with a blockade of SERT and NET, can also have the ability to improve the lack of motivation and attention and enhance cognitive deficits seen in depressed patients.
- blockade of DAT has to be carefully managed in order to avoid potential reinforcing effects and abuse liability.
- compounds with DAT inhibition in their pharmacology such as Dexmethylphenidate, Methylphenidate and Bupropion, have been successfully marketed.
- the compounds of the present invention are considered useful for the treatment of Parkinsonism, depression, obesity, narcolepsy, drug addiction or misuse, including cocaine abuse, attention-deficit hyperactivity disorders, Gilles de Ia Tourettes disease and senile dementia.
- Dopamine re-uptake inhibitors enhance indirectly via the dopamine neurones the release of acetylcholine and are therefore also useful for the treatment of memory deficits, e.g. in Alzheimers disease, presenile dementia, memory dysfunction in ageing, and chronic fatigue syndrome.
- Noradrenaline re-uptake inhibitors are considered useful for enhancing attention, alertness, arousal, vigilance and for treating depression.
- An object of the present invention is to provide novel compounds which are serotonin (5-HT), dopamine (DA) and norepinephrine (NE) re-uptake inhibitors.
- the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof:
- R 1 is H or C-
- R2 is phenyl optionally substituted by one or more groups independently selected from halo, cyano, C-
- R ⁇ is naphthyl, optionally substituted by 1 or 2 groups independently selected from halo, cyano and C-
- R3 and R ⁇ which may be the same or different, are H, fluoro or C-
- R 5 is H or C ⁇
- R8 is phenyl or heteroaryl, either of which is optionally substituted by one or more groups independently selected from halogen, C-
- R ⁇ and R ⁇ which may the same or different, are H, D, C-
- _4alkyl substituent is a univalent radical derived by removal of a hydrogen atom from an acyclic C-
- _4alkyl substituents include methyl and ethyl, may be straight chain (i.e. n-propyl and n-butyl) or branched chain (for example, isopropyl, isobutyl and secbutyl).
- _4alkyl substituent is methyl, ethyl, n-propyl or isopropyl.
- _4alkoxy substituent is a group of formula "R-O-" where R is C-
- alkoxy substituents include methoxy and ethoxy and may be straight chain (i.e. n-propoxy and n-butoxy) or branched chain (for example, isopropoxy, isobutoxy, secbutoxy and tert-butoxy).
- _4alkoxy substituent is methoxy, ethoxy, n-propoxy or isopropoxy.
- _4haloalkyl substituent is a C-
- _4haloalkyl substituents include monofluoromethyl, difluoromethyl, trifluoromethyl and 1-chloro-2-fluoroethyl.
- _4haloalkyl substituent is monofluoromethyl, difluoromethyl or trifluoromethyl.
- _4haloalkoxy substituent is of formula "R x -O-" where R x is C- ⁇ 4haloalkyl as defined above.
- _4haloalkoxy substituents include monofluoromethoxy, difluoromethoxy, trifluoromethoxy and 1-chloro-2-fluoroethoxy and may be straight chain or branched chain.
- .ghaloalkoxy substituent is monofluoromethoxy, difluoromethoxy or trifluoromethoxy.
- a halo substituent refers to fluoro, chloro, bromo and iodo radicals. In an embodiment, unless otherwise indicated, any halo substituent is fluoro or chloro.
- _4alkanoyl substituent refers to C-
- _4alkanoyl substituent is acetyl, ethylcarbonyl, n-propylcarbonyl, i-propylcarbonyl, n-butylcarbonyl or t-butylcarbonyl.
- H and D refers to hydrogen and deuterium radicals respectively.
- R ⁇ and -(CR6R7) ⁇ R8 possess a cis relationship, i.e. both groups R ⁇ and -(CR6R7) ⁇ R8 are on the same face of the bicyclic ring system. It will also be appreciated that two alternatives exist with respect to the geometry at the ring carbon atoms attached to R ⁇ and -(CR6R7) n R8, na mely compounds of formula (IA) and IB).
- the compound of formula (I) is a mixture of compounds of formula (IA) and (IB). In a further embodiment, the compound of formula (I) is a racemic mixture of compounds of formula (IA) and (IB).
- the compound of formula (I) is of formula (IA).
- the enantiomeric excess (e.e.) of (IA) over (IB) is greater than or equal to 90%.
- the e.e. of (IA) over (IB) is greater than or equal to 95%.
- the e.e. of (IA) over (IB) is greater than or equal to 99%.
- the compound of formula (I) is of formula (IB).
- the enantiomeric excess (e.e.) of (IB) over (IA) is greater than or equal to 90%.
- the e.e. of (IB) over (IA) is greater than or equal to 95%.
- the e.e. of (IB) over (IA) is greater than or equal to 99%.
- R-I is hydrogen or methyl. In a further embodiment, R-I is hydrogen.
- R ⁇ is phenyl substituted by one or two groups independently selected from halo and haloC-1.4 alkyl; or R ⁇ is unsubstituted naphthyl. In an embodiment, R ⁇ is hydrogen.
- n is 1 or 2. In a further embodiment n is 1.
- R8 is heteroaryl, it refers to a univalent radical derived by removal of a hydrogen atom from a heteroaromatic ring system.
- the heteroaromatic ring system may be monocyclic or bicyclic.
- the heteroaryl substituent is monocyclic, it comprises one or more carbon atoms and 1 to 4 heteroatoms interconnected to form a ring.
- the heteroatoms are independently selected from nitrogen, oxygen and sulphur.
- the monocyclic heteroaryl substituent is furyl, thienyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, azepinyl, oxazepinyl, thiazepinyl or diazepinyl.
- the monocyclic heteroaryl substituent is furyl, thienyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl or tetrazolyl.
- the heteroaryl substituent is bicyclic, one of the rings may contain from 5 to 7 atoms interconnected to form a ring and the other ring may contain from 5 or 6 carbons interconnected to form a ring.
- the rings may contain 1 to 4 heteroatoms independently selected from nitrogen, oxygen and sulphur.
- the heteroaryl substituent is fused bicyclic, one of the rings is aromatic and the other is saturated, unsaturated or aromatic.
- the bicyclic heteroaryl substituent is indolizinyl; indolyl; isoindolyl; 3H-indolyl; indolinyl; indolizinyl; benzo[b]furyl; benzo[b]thienyl; 1 H-indazolyl; 2H-indazolyl; benzimidazolyl; benzthiazolyl; purinyl; 4H-quninolinyl; quinolinyl; isoquinolinyl; cinnolinyl; phthalazinyl; quinazolinyl; quinoxalinyl; 1 ,8-naththyridinyl; pteridyl;
- the bicyclic heteroaryl substituent is indolizinyl; indolyl; isoindolyl; 3H- indolyl; indolinyl; benzo[b]furyl; benzo[b]thienyl; 1 H-indazolyl; 2H-indazolyl; benzimidazolyl; benzthiazolyl; 2,4,6,7-tetrahydropyrano[4,3-c]pyrazolyl; 1 ,3a,4,6,7,7a-hexahydropyrano[4,3-c]pyrazolyl; 4,5,6,7-tetrahydro-1 H-indazolyl; or 4,5 ! 6,7-tetrahydro-2H-indazolyl.
- R ⁇ is a monocyclic heteroaryl substituent selected from the list furyl, thienyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl or tetrazolyl, any of which substituents are optionally substituted by one or more groups independently selected from halogen, C-
- R ⁇ is pyrazolyl, tetrazolyl, triazolyl, pyrrolyl or oxazolyl any of which substituents are optionally substituted by one or more groups independently selected from halogen, C-
- the compound defined in the first aspect is selected from the list
- the compounds of formula (I) as defined in the first aspect contain a basic centre and may form non-toxic acid addition salts formed with inorganic acids such as hydrochloric, hydrobromic, hydroiodic, sulfuric and phosphoric acid, with carboxylic acids or with organo-sulfonic acids.
- inorganic acids such as hydrochloric, hydrobromic, hydroiodic, sulfuric and phosphoric acid, with carboxylic acids or with organo-sulfonic acids.
- Examples include the HCI, HBr, HI, sulfate or bisulfate, nitrate, phosphate or hydrogen phosphate, acetate, benzoate, succinate, saccharate, fumarate, maleate, lactate, citrate, tartrate, gluconate, camsylate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate and pamoate salts.
- suitable pharmaceutical salts see Berge et al, J.
- the salt is pharmaceutically acceptable.
- pro-drugs examples include Drugs of Today, Volume 19, Number 9, 1983, pp 499 - 538 and in Topics in Chemistry, Chapter 31 , pp 306 - 316 and in "Design of Prodrugs" by H. Bundgaard, Elsevier, 1985, Chapter 1 (the disclosures in which documents are incorporated herein by reference). It will further be appreciated by those skilled in the art, that certain moieties, known to those skilled in the art as “pro- moieties”, for example as described by H. Bundgaard in "Design of Prodrugs” (the disclosure in which document is incorporated herein by reference) may be placed on appropriate functionalities when such functionalities are present within the compounds defined in the first aspect. Therefore, in a further aspect, the invention provides a prodrug of a compound defined in the first aspect.
- the compounds defined in the first aspect, their salts or prodrugs, may exist in solvated or hydrated form. Therefore, in a further aspect, the invention provides a solvate or hydrate of a compound defined in the first aspect or a salt thereof.
- the compounds of formula (I) and their salts, as defined in the first aspect or solvates or hydrates of either, may exist in one or more polymorphic form. Therefore, in a further aspect, the invention provides a polymorph of a compound of formula (I) defined in the first aspect or their salts, or a polymorph of a solvate or hydrate of a compound of formula (I) defined in the first aspect, or a salt thereof.
- compounds of formula (I) as defined in the first aspect their salts and prodrugs; any solvates or hydrates of any salt or prodrug; and any polymorph of any compound, salt, solvate or hydrate are referred to as "compounds of the invention”.
- the term “compounds of the invention” also includes all embodiments of the first aspect.
- the compounds of the invention may possess one or more chiral centres and so exist in a number of stereoisomeric forms. All stereoisomers and mixtures thereof are included in the scope of the present invention. Racemic compounds may either be separated using preparative HPLC and a column with a chiral stationary phase or resolved to yield individual enantiomers utilising methods known to those skilled in the art. In addition, chiral intermediate compounds may be resolved and used to prepare chiral compounds of the invention. In addition, the chiral compounds of the invention may be prepared by chiral synthesis.
- the compounds of the invention may exist in one or more tautomeric forms. All tautomers and mixtures thereof are included in the scope of the present invention. For example, a claim to 2-hydroxyquinolinyl would also cover its tautomeric form, ⁇ - quinolinonyl.
- the invention also includes all suitable isotopic variations of a compound of the invention.
- An isotopic variation of a compound of the invention is defined as one in which at least one atom is replaced by an atom having the same atomic number but an atomic mass different from the atomic mass usually found in nature.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulphur, fluorine and chlorine such as 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 17 O, 18 0, 31 P, 32 P, 35 S, 18 F and 36 CI, respectively.
- isotopic variations of the invention are useful in drug and/or substrate tissue distribution studies. Tritiated, i.e., 3 H, and carbon-14, i.e., 14 C, isotopes are particularly preferred for their ease of preparation and detectability. Further, substitution with isotopes such as deuterium, i.e., 2 H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements and hence may be preferred in some circumstances.
- Isotopic variations of the compounds of the invention can generally be prepared by conventional procedures such as by the illustrative methods or by the preparations described in the Experimental section hereafter using appropriate isotopic variations of suitable reagents.
- compounds of formula (Ib) may be obtained through reductive amination using a suitable aldehyde R ⁇ CHO, a reducing agent such as NaCNBH 3, in aprotic or protic solvent (e.g. toluene, THF or MeOH), at temperature between 80 0 C and room temperature.
- a suitable aldehyde R ⁇ CHO a reducing agent such as NaCNBH 3
- aprotic or protic solvent e.g. toluene, THF or MeOH
- Suitable protecting groups (Pg) include Boc and benzyl. Removal of a Boc protecting group may be achieved by reaction with TFA in DCM at a temperature between 0 0 C and room temperature. Removal of a benzyl protecting group may be achieved by hydrogenation over Pd/C. Alternatively removal of a benzyl protecting group may be achieved using ⁇ -chloroethyl chloroformate at reflux in DCE and then in MeOH. Scheme 2
- Compounds of formula (Na), i.e. compounds of general formula (II) where n is 1 , R ⁇ is a N-linked monocyclic or bicyclic heteroaryl substituent and Pg is a suitable N- protecting group, typically Boc or benzyl, may be obtained from compounds of formula (III), wherein L is a suitable leaving group (e.g. mesylate) according to reaction scheme 3.
- Typical reaction conditions comprise, nucleophilic displacement of L with the pre-formed anion from the N-heteroaryl group using base (such as NaH) in aprotic solvent (e.g. DMF) at between 0 0 C and room temperature followed by heating to a temperature between 40 and 80 0 C.
- Compounds of formula (V) where R ⁇ and R ⁇ are hydrogen or deuterium may be obtained by reduction of compounds of formula (Vl), wherein Pg is a suitable N- protecting group (such as Boc) and R an alkyl group (such as methyl or ethyl) according to reaction scheme 8.
- Suitable regucing agents are LiAID 4 or LiAIH 4 , in aprotic solvent, e. g. diethyl ether or THF, at temperature between -40 and -10 0 C.
- Compounds of formula (Va), i.e. compounds of formula (V) where R ⁇ and R ⁇ are both methyl, may be obtained from compounds of formula (Vl), wherein Pg is a suitable N-protecting group (typically Boc) and R an alkyl group (such as methyl or ethyl) according to reaction scheme 9.
- Suitable reaction conditions comprise reacting (Vl) with methyl magnesium bromide in aprotic solvent, e. g. diethyl ether or THF, at temperature between -40 and -10 0 C.
- Compounds of formula (Vl) may be obtained from compounds of formula (VII), by reaction with an appropriate boronic acid R2B(OH)2 according to reaction scheme 10.
- Suitable reaction conditions comprise reacting (VII) with R2 ⁇ (OH)2 in the presence of Pd(OAc)2 , PPh3 and diisopropylethylamine in a mixture of toluene and water at a temperature ranging from room temperature to 80 0 C.
- Alternative catalysts may be used, for example Pd(PPhi3)4 and PdCl2(dppf).
- R7 are hydrogen, may be obtained directly from compounds of formula (IX) according to reaction scheme 12.
- Suitable reaction conditions comprise treatment with BH 3 or LiAIH 4 in aprotic solvent (such as THF) at reflux, followed by protection of the nitrogen using Boc anhydride under basic conditions at room temperature.
- Compounds of formula (IX) may be obtained according to reaction scheme 13 from compounds of formula (X) wherein R is alkyl and L is a suitable leaving group (such as mesylate) by reaction with ammonia.
- Typical conditions comprise reaction under pressure in a suitable solvent (such as methanol) in a hydrogenation apparatus (for example Parr). Nitrogen protection gives compounds of formula (IX).
- Compounds of formula (X) may be obtained according to reaction scheme 14 from compounds of formula (Xl) by carbene mediated cyclopropanation with dimethyl diazopropandioate and rhodium catalyst (for example Rh2(OAc)2) in chlorinated solvent (e.g, chlorobenzene or DCE) at a temperature between 4O 0 C and 8O 0 C.
- rhodium catalyst for example Rh2(OAc)2
- chlorinated solvent e.g, chlorobenzene or DCE
- Use of asymmetric rhodium catalysts may be used to provide stereospecific compounds of formula (X).
- Compounds of formula (XIVa) may be obtained according to reaction scheme 17 from compounds offormula (XV) by reduction of the olefinic double bond.
- Reaction conditions comprise treatment with triethylsilane and TFA in aprotic solvent (such as toluene).
- aprotic solvent such as toluene.
- Alternative reducing agents may be used, for example sodium triacetoxyborohydride or sodium borohydride.
- Alternative solvents may also be used, for example dichloromethane, trifluorotoluene or chlorobenzene.
- Compounds of formula (XIVa) may then be reduced to give compounds of formula (IVd).
- Compounds of formula (XV) may be obtained from compounds of formula (Vl) by reaction with an appropriate base in the presence of an appropriate alkylating agent according to reaction scheme 18.
- Reaction conditions may comprise treatment with lithium t-butoxide and CH 2 ICI in N-methyl pyrrolidone at low temperature (for example -20 to +10 0 C).
- Alternative alkylating agents may be used, for example CH 2 I 2 -
- Alternative solvents may be used, for example DMF or THF.
- Alternative bases may be used, for example LDA or NaH.
- Compounds of formula (XVIII) may be obtained from compounds of formula (XVI), according to reacytion scheme 21 , by hydroboration of the alkene with borane-THF complex in THF at a temperature between 0 0 C and room temperature followed by oxidation with hydrogen peroxide and sodium hydroxide at 0 0 C.
- Compounds of formula (XIX) may be obtained from compounds of formula (XX), according to reaction scheme 22, by Wittig reaction using methylenetriphenylphosphorane in THF at room temperature.
- Compounds of formula (XX) may be obtained from compounds of formula (IVe), wherein R ⁇ and R ⁇ are hydrogen and Pg is a suitable N-protecting group (typically Boc) according to reaction scheme 23, by oxidation with Dess-Martin periodinane in DCM at a temperature between 0 0 C and room temperature.
- R ⁇ and R ⁇ are hydrogen and Pg is a suitable N-protecting group (typically Boc) according to reaction scheme 23, by oxidation with Dess-Martin periodinane in DCM at a temperature between 0 0 C and room temperature.
- Compounds of formula (XXI) may be obtained according to reaction scheme 24 from compounds of formula (XXII) by reaction of the Weinreb amide with methyl magnesium bromide in aprotic solvent at low temperature (typically between -78°C and 0 0 C).
- Compounds of formula (XXII) may be obtained according to reaction scheme 25 from compounds of formula (XIVb) by reaction with N,O-dimethylhydroxylamine hydrochloride and LiHMDS in aprotic solvent (such as THF) at low temperature (typically between -50 0 C and 0 0 C).
- aprotic solvent such as THF
- reaction scheme 30 An alternative procedure for the preparation of compounds of formula (NIb) is shown in reaction scheme 30.
- a specific enantiomer or diastereoisomer of a compound of the invention may be obtained for example by optical resolution of a mixture of enantiomers or diastereoisomers using conventional methods, such as chiral chromatography.
- the affinity of compounds of the invention for SERT, NET and DAT may be tested in one or other of the following affinity assays.
- LLCPK Porcine tubule Kidney
- Stable cell lines may be generated as follows: i) hSERT - generated by transfecting LLC-PK1 or LLCPK cells with hSERT cloned into the mammalian expression vector pCDNA3.1 Hygro(+); ii) hNET - generated by transfecting LLCPK cells with hNET cloned into the mammalian expression vector pRC/CMV; iii) hDAT- generated by transfecting LLCPK cells with hDAT cloned into the mammalian expression vector pDESTCDNA3.1 (an example of a procedure for transfecting LLCPK cells with hDAT, hSERT and hNET may be found in H. Gu, S. C. Wall and G. Rudnick, J. Biol. Chem. (1994) 269 : 7124-7130.)
- Each cell line is cultured independently in Dulbecco's modified Eagle's medium (DMEM) containing 10% of Foetal Bovine Serum (FBS) supplemented with 400 ⁇ g/ml hygromicin (hSERT) or geneticin at 500 ⁇ g/ml (hNET) or at 1000 ⁇ g/ml (hDAT).
- DMEM Dulbecco's modified Eagle's medium
- FBS Foetal Bovine Serum
- hSERT hygromicin
- hNET ⁇ g/ml
- hDAT ⁇ g/ml
- the culture medium is removed and the cells harvested with phosphate buffered saline (PBS) containing 5 mM EDTA.
- PBS phosphate buffered saline
- the cell suspension is centrifuged at 90Og for 5 minutes at 4 0 C.
- the resultant pellets are re-suspended in 30-50 volumes of Assay Buffer (5OmM Tris pH 7.7 containing 12OmM NaCI, 5mM KCI, 10 ⁇ M pargyline and 0.1% ascorbic acid) and homogenized using a glass-teflon Potter homogeniser and centrifuged at 4800Og for 20 minutes at 4 0 C.
- Assay Buffer 5OmM Tris pH 7.7 containing 12OmM NaCI, 5mM KCI, 10 ⁇ M pargyline and 0.1% ascorbic acid
- the affinity of the compounds of the invention to bind the re-uptake site of SERT may be assessed using [3
- the competition binding assay is conducted in deep-well 96 well plates (1 ml, NUNC, cod.260252) in a total volume of 400 ⁇ l, with each concentration in duplicate.
- test compound 100X solution in neat DMSO as 7 point curve ranging from 10 "6 to 10 "12 M, final concentration
- DMSO to define total binding
- 10 ⁇ M fluoxetine in DMSO to define non-specific binding, NSB
- 200 ⁇ l of [N-Methyl- 3 H]citalopram Amersham Biosciences, 80 Ci/mmol
- the reaction is started by adding 200 ⁇ l/well of membranes diluted 1 :80 in Assay Buffer at concentration of about 2.5 ⁇ g/well of protein.
- reaction is carried out at room temperature for 2 hours and then stopped by rapid filtration through GF/B Unifilter 96-filterplate (Perkin-Elmer) pre-soaked in 0.5% polyethylenimmine (PEI) using a Perkin-Elmer FilterMat-196 harvester.
- GF/B Unifilter 96-filterplate Perkin-Elmer
- PEI polyethylenimmine
- Filterplate is washed 3 times with 1 ml/well ice-cold 0.9% NaCI solution. The plate is dried in an oven for 60 min at 50 0 C then opaque bottom-seal is placed on the underside of the plate and 50 ⁇ l of Microscint 20 (Perkin-Elmer) added to each well. Plate is sealed with a TopSeal and the radioactivity in the samples is counted for 4 min using TopCount liquid scintillation counter (Packard-Perkin-Elmer) and recorded as counts per minute (CPM).
- TopCount liquid scintillation counter Packard-Perkin-Elmer
- Competition binding assay for hNET may be conducted essentially as previously reported for hSERT in 96 well format and in a final assay volume of 400 ⁇ l, except for the use of hNET-LLCPK cell membranes (1 :40 dilution i.e. 4.8 ⁇ g of protein/well) and [ 3 H]nisoxetine as radioligand (1.5nM [N-methyl- 3 H]nisoxetine, Amersham Biosciences, 84 Ci/mmol). 10 ⁇ M desipramine is used for NSB.
- Competition binding assay for hDAT may also be conducted essentially as previously reported for hSERT and hNET in 96 well format and in a final assay volume of 400 ⁇ l, except for the use of hDAT-LLCPK cell membranes (1 :20 i.e. 9.6 ⁇ g of protein/well) and [ 3 H]WI N-35,428 as radioligand (1OnM [N-Methyl- 3 H]WIN-35,428, Perkin Elmer, 85.6 Ci/mmol). Furthermore, 10 ⁇ M GBR-12909 is used for NSB and the incubation time of the binding reaction is 1 hour at room temperature.
- Membranes for the SPA-binding assays are produced by HEK-293F cell infection with BacMam viruses generated for each single human SERT, NET, and DAT transporter.
- hSERT and hDAT are cloned into pFBMRfA vector whereas hNET is cloned into pFASTBacMami vector.
- the generation and use of BacMam viruses is described in Condreay JP et al, Proc. Natl. Acad. Sci. USA, 1999, 96:127-132 and Hassan NJ et al, Protein Expression and Purification, 47(2): 591-598, 2006.
- the HEK-293F suspension cell line (Invitrogen) is routinely grown in 293_Freestyle Expression media (Invitrogen) in shake flask suspension culture.
- the culture is transduced with the appropriate transporter BacMam at a MOI (multiplicity of infection) of 100 virus particles per cell and incubated for 48hrs at 37 0 C, 5% CO 2 in air, shaken at 90rpm in a humidified shaker incubator.
- the culture is then harvested by centrifugation at 100Og, 4 0 C, for 10 minutes and the cell pellet stored at -8O 0 C until required.
- Transduced cell pellets are re-suspended to 10x volume with buffer-A (5OmM HEPES, 1 mM EDTA, 1 mM leupeptin, 25ug/ml_ bacitracin, 1 mM phenylmethylsulfonylfluoride, PMSF, 2 ⁇ M pepstatin A, pH 7.7) and homogenised with 2x 15 second bursts in a glass Waring blender. The homogenate is then centrifuged for 20 minutes at 50Og. Following this, the supernatant is pooled and centrifuged at 13,00Og for 30 minutes.
- buffer-A 5OmM HEPES, 1 mM EDTA, 1 mM leupeptin, 25ug/ml_ bacitracin, 1 mM phenylmethylsulfonylfluoride, PMSF, 2 ⁇ M pepstatin A, pH 7.7
- the homogenate is then centrifuged for 20 minutes at 50O
- the affinity of the compounds of the invention for hSERT, hNET or hDAT may also be assessed by using the [ 3 H]citalopram, [ 3 H]nisoxetine or [ 3 H]WI N-35, 428 binding assays with the SPA technology on BacMam-recombinant human SERT, NET and DAT membranes produced as described before.
- SPA technology GE Healthcare, Amersham
- transporter-bound radioactivity can elicit bead excitation thus no separation of the bound/ unbound radioligand is required.
- hSERT binding SPA is based on Trilux beta-counter (Wallac, Perkin- Elmer). Briefly, 0.5 ⁇ l_ of test compound in neat DMSO (or 1 ⁇ M fluoxetine as positive control) is added by 50 ⁇ l_ of the SPA mixture, containing 2mg/ml_ SPA beads
- hDAT hNET and hSERT SPA-binding assays are performed by using a Viewlux beta-counter (Wallac, Perkin-Elmer) with imaging PS-WGA beads (Amersham RPNQ0260) in a final assay volume of 30 ⁇ l_ and in a 384-well plate format (Greiner 781075).
- Viewlux beta-counter Wallac, Perkin-Elmer
- PS-WGA beads Amersham RPNQ0260
- the affinity of the compounds of the invention for a particular transporter may be calculated from the IC 5 O obtained in competition experiments as the concentration of a compound necessary to displace 50% of the radiolabeled ligand from the transporter, and is reported as a "K,” value calculated by the following equation: IC 50
- a number of compounds supporting the invention have been prepared (see below) and may be tested in assay a) and/or b).
- the following compounds supporting the invention were tested in assay b) 1 , 2, 5-7, 10-12, 15-18, 20, 24-26, 29, 30, 35-39, 42, 43, 47-65, 82-91 , 94-126 and 137-140.
- These compounds gave a pKi against SERT from 7.2 to 10; against NET from 5.0 to 9.4; and against DAT from 6.2 to 9.8.
- the compounds of the invention may be used to treat diseases or conditions mediated by inhibition of monoamine neurotransmitter re-uptake, i.e. inhibition of one or more of SERT, hNET and hDAT. Therefore according to a further aspect, the invention provides a compound of the invention for use in treating a disease or condition.
- the disease or condition is a human disease or condition.
- the disease or condition is mediated by inhibition of monoamine neurotransmitter re-uptake, i.e. inhibition of one or more of SERT, hNET and hDAT.
- the disease or condition mediated by inhibition of monoamine neurotransmitter re-uptake i.e. inhibition of one or more of SERT, hNET and hDAT
- DSM-IV Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, published by the American Psychiatric Association (DSM-IV) and/or the International Classification of Diseases, 10th Edition (ICD-10).
- DSM-IV American Psychiatric Association
- ICD-10 International Classification of Diseases, 10th Edition
- Depression and mood disorders including Major Depressive Episode, Manic Episode, Mixed Episode and Hypomanic Episode; Depressive Disorders including Major Depressive Disorder, Dysthymic Disorder (300.4), Depressive Disorder Not Otherwise Specified (311 ); Other Mood Disorders including Mood Disorder Due to a General Medical Condition (293.83) which includes the subtypes With Depressive Features, With Major Depressive-like Episode, With Manic Features and With Mixed Features), Substance-Induced Mood Disorder (including the subtypes With
- Bipolar Disorders including Bipolar I Disorder, Bipolar Il Disorder (Recurrent Major Depressive Episodes with Hypomanic Episodes) (296.89), Cyclothymic Disorder (301.13) and Bipolar Disorder Not Otherwise Specified (296.80);
- anxiety disorders includes:
- subject related disorder includes:
- Substance-related disorders including Substance Use Disorders such as Substance Dependence, Substance Craving and Substance Abuse; Substance-Induced Disorders such as Substance Intoxication, Substance Withdrawal, Substance- Induced Delirium, Substance-Induced Persisting Dementia, Substance-Induced Persisting Amnestic Disorder, Substance-Induced Psychotic Disorder, Substance- Induced Mood Disorder, Substance-Induced Anxiety Disorder, Substance-Induced sexual Dysfunction, Substance-Induced Sleep Disorder and Hallucinogen Persisting Perception Disorder (Flashbacks); Alcohol-Related Disorders such as Alcohol Dependence (303.90), Alcohol Abuse (305.00), Alcohol Intoxication (303.00), Alcohol Withdrawal (291.81 ), Alcohol Intoxication Delirium, Alcohol Withdrawal Delirium, Alcohol-Induced Persisting Dementia, Alcohol-Induced Persisting Amnestic Disorder, Alcohol-Induced Psychotic Disorder,
- Sleep disorder includes:
- Sleep disorders including primary sleep disorders such as Dyssomnias such as Primary Insomnia (307.42), Primary Hypersomnia (307.44), Narcolepsy (347), Breathing-Related Sleep Disorders (780.59), Circadian Rhythm Sleep Disorder (307.45) and Dyssomnia Not Otherwise Specified (307.47); primary sleep disorders such as Parasomnias such as Nightmare Disorder (307.47), Sleep Terror Disorder (307.46), Sleepwalking Disorder (307.46) and Parasomnia Not Otherwise Specified (307.47); Sleep Disorders Related to Another Mental Disorder such as Insomnia Related to Another Mental Disorder (307.42) and Hypersomnia Related to Another Mental Disorder (307.44); Sleep Disorder Due to a General Medical Condition; and Substance-Induced Sleep Disorder including the subtypes Insomnia Type, Hypersomnia Type, Parasomnia Type and Mixed Type;
- treating disorder includes:
- Eating disorders such as Anorexia Nervosa (307.1 ) including the subtypes Restricting Type and Binge-Eating/Purging Type; Bulimia Nervosa (307.51 ) including the subtypes Purging Type and Nonpurging Type; Obesity; Compulsive Eating Disorder; Binge Eating Disorder; and Eating Disorder Not Otherwise Specified (307.50):
- Attention-Deficit/Hyperactivity Disorder includes:
- Attention-Deficit/Hyperactivity Disorder including the subtypes Attention-Deficit /Hyperactivity Disorder Combined Type (314.01 ), Attention-Deficit /Hyperactivity Disorder Predominantly Inattentive Type (314.00), Attention-Deficit /Hyperactivity Disorder Hyperactive-Impulse Type (314.01 ) and Attention-Deficit /Hyperactivity Disorder Not Otherwise Specified (314.9); Hyperkinetic Disorder; Disruptive Behaviour Disorders such as Conduct Disorder including the subtypes childhood- onset type (321.81 ), Adolescent-Onset Type (312.82) and Unspecified Onset (312.89), Oppositional Defiant Disorder (313.81 ) and Disruptive Behaviour Disorder Not Otherwise Specified; and Tic Disorders such as Tourette's Disorder (307.23);
- Cognitive impairment includes:
- Cognition impairment including cognition impairment in other diseases such as schizophrenia, bipolar disorder, depression, other psychiatric disorders and psychotic conditions associated with cognitive impairment, e.g. Alzheimer's disease;
- Sexual dysfunctions including sexual Desire Disorders such as Hypoactive Sexual Desire Disorder (302.71 ), and sexual Aversion Disorder (302.79); sexual arousal disorders such as Female sexual Arousal Disorder (302.72) and Male Erectile Disorder (302.72); orgasmic disorders such as Female Orgasmic Disorder (302.73), Male Orgasmic Disorder (302.74) and Premature Ejaculation (302.75); sexual pain disorder such as Dyspareunia (302.76) and Vaginismus (306.51 ); Sexual Dysfunction Not Otherwise Specified (302.70); paraphilias such as Exhibitionism (302.4), Fetishism (302.81 ), Frotteurism (302.89), Pedophilia (302.2), Sexual Masochism (302.83), sexual Sadism (302.84), Transvestic Fetishism (302.3), Voyeurism (302.82) and Paraphilia Not Otherwise Specified (302.9); gender identity disorders such as Gender Identity Disorder in Children (302.6) and Gender Identity Disorder in Adolescents or Adults (302.85); and
- Obsessive compulsive spectrum disorder includes:
- Obsessive compulsive spectrum disorder including Obsessive compulsive disorders (300.3), somatoform disorders including body dysmorphic disorder (300.7) and hyperchondriasis (300.7), bulimia nervosa (307.51 ), anorexia nervosa (307.1 ), eating disorders not elsewhere classified (307.50) such as binge eating, impulse control disorders not elsewhere classified (including intermitted explosive disorder (312.34), compulsive buying or shopping, repetitive self-mutilation, onychophagia, psychogenic excoriation, kleptomania (312.32), pathological gambling (312.31 ), trichotillomania (312.39) and internet addiction), paraphilia (302.70) and nonparaphilic sexual addictions, Sydeham's chorea, torticollis, autistic disorders (299.0), compulsive hoarding, and movement disorders, including Tourette's syndrome (307.23).
- somatoform disorders including body dysmorphic disorder (300.7) and hyperchondriasis (300.7
- compounds of the invention may be useful as analgesics.
- they may be useful in the treatment of chronic inflammatory pain (e.g. pain associated with rheumatoid arthritis, osteoarthritis, rheumatoid spondylitis, gouty arthritis and juvenile arthritis); musculoskeletal pain; lower back and neck pain; sprains and strains; neuropathic pain; sympathetically maintained pain; myositis; pain associated with cancer and fibromyalgia; pain associated with migraine; pain associated with influenza or other viral infections, such as the common cold; rheumatic fever; pain associated with functional bowel disorders such as non-ulcer dyspepsia, non-cardiac chest pain and irritable bowel syndrome; pain associated with myocardial ischemia; post operative pain; headache; toothache; and dysmenorrhea.
- chronic inflammatory pain e.g. pain associated with rheumatoid arthritis, osteoarthritis, rheumatoid
- Neuropathic pain syndromes can develop following neuronal injury and the resulting pain may persist for months or years, even after the original injury has healed.
- Neuronal injury may occur in the peripheral nerves, dorsal roots, spinal cord or certain regions in the brain.
- Neuropathic pain syndromes are traditionally classified according to the disease or event that precipitated them.
- Neuropathic pain syndromes include: diabetic neuropathy; sciatica; non-specific lower back pain; multiple sclerosis pain; fibromyalgia; HIV-related neuropathy; post-herpetic neuralgia; trigeminal neuralgia; and pain resulting from physical trauma, amputation, cancer, toxins or chronic inflammatory conditions.
- neuropathic pain are incredibly heterogeneous and are often described as spontaneous shooting and lancinating pain, or ongoing, burning pain.
- pain associated with normally non-painful sensations such as "pins and needles" (paraesthesias and dysesthesias), increased sensitivity to touch (hyperesthesia), painful sensation following innocuous stimulation (dynamic, static or thermal allodynia), increased sensitivity to noxious stimuli (thermal, cold, mechanical hyperalgesia), continuing pain sensation after removal of the stimulation (hyperpathia) or an absence of or deficit in selective sensory pathways (hypoalgesia).
- Compounds of the invention may also be useful in the amelioration of inflammatory disorders, for example in the treatment of skin conditions (e.g. sunburn, burns, eczema, dermatitis, psoriasis); ophthalmic diseases such as glaucoma, retinitis, retinopathies, uveitis and of acute injury to the eye tissue (e.g. conjunctivitis); lung disorders (e.g. asthma, bronchitis, emphysema, allergic rhinitis, respiratory distress syndrome, pigeon fancier's disease, farmer's lung, chronic obstructive pulmonary disease, (COPD); gastrointestinal tract disorders (e.g.
- compounds of the invention are useful in the treatment of depression and anxiety disorders.
- compounds of the invention are useful in the treatment of depression.
- Treatment includes prophylaxis, where this is appropriate for the relevant condition(s).
- the compounds of the invention may also be used in combination with other therapeutic agents.
- the invention thus provides, in a further aspect, a combination comprising a compound of the invention together with a further therapeutic agent.
- the compounds of the invention may be used in combination with the following agents to treat or prevent psychotic disorders: i) antipsychotics; ii) drugs for extrapyramidal side effects, for example anticholinergics (such as benztropine, biperiden, procyclidine and trihexyphenidyl), antihistamines (such as diphenhydramine) and dopaminergics (such as amantadine); iii) antidepressants; iv) anxiolytics; and v) cognitive enhancers for example cholinesterase inhibitors (such as tacrine, donepezil, rivastigmine and galantamine).
- anticholinergics such as benztropine, biperiden, procyclidine and trihexyphenidyl
- antihistamines such as diphenhydramine
- dopaminergics such as amantadine
- antidepressants such as amantadine
- iv) anxiolytics such as anxio
- the compounds of the invention may be used in combination with antidepressants to treat or prevent depression and mood disorders.
- the compounds of the invention may be used in combination with the following agents to treat or prevent bipolar disease: i) mood stabilisers; ii) antipsychotics; and iii) antidepressants.
- the compounds of the invention may be used in combination with the following agents to treat or prevent anxiety disorders: i) anxiolytics; and ii) antidepressants.
- the compounds of the invention may be used in combination with the following agents to improve nicotine withdrawal and reduce nicotine craving: i) nicotine replacement therapy for example a sublingual formulation of nicotine beta- cyclodextrin and nicotine patches; and ii) bupropion.
- the compounds of the invention may be used in combination with the following agents to improve alcohol withdrawal and reduce alcohol craving: i) NMDA receptor antagonists for example acamprosate; ii) GABA receptor agonists for example tetrabamate; and iii) Opioid receptor antagonists for example naltrexone.
- the compounds of the invention may be used in combination with the following agents to improve opiate withdrawal and reduce opiate craving: i) opioid mu receptor agonist/opioid kappa receptor antagonist for example buprenorphine; ii) opioid receptor antagonists for example naltrexone; and iii) vasodilatory antihypertensives for example lofexidine.
- opioid mu receptor agonist/opioid kappa receptor antagonist for example buprenorphine
- opioid receptor antagonists for example naltrexone
- vasodilatory antihypertensives for example lofexidine.
- the compounds of the invention may be used in combination with the following agents to treat or prevent sleeping disorders: i) benzodiazepines for example temazepam, lormetazepam, estazolam and triazolam; ii) non-benzodiazepine hypnotics for example Zolpidem, zopiclone, zaleplon and indiplon; iii) barbiturates for example aprobarbital, butabarbital, pentobarbital, secobarbita and phenobarbital; iv) antidepressants; v) other sedative-hypnotics for example chloral hydrate and chlormethiazole.
- benzodiazepines for example temazepam, lormetazepam, estazolam and triazolam
- non-benzodiazepine hypnotics for example Zolpidem, zopiclone, zaleplon and indiplon
- barbiturates for example
- the compounds of the invention may be used in combination with the following agents to treat anorexia: i) appetite stimulants for example cyproheptidine; ii) antidepressants; iii) antipsychotics; iv) zinc; and v) premenstral agents for example pyridoxine and progesterones.
- the compounds of the invention may be used in combination with the following agents to treat or prevent bulimia: i) antidepressants; ii) opioid receptor antagonists; iii) antiemetics for example ondansetron; iv) testosterone receptor antagonists for example flutamide; v) mood stabilisers; vi) zinc; and vii) premenstral agents.
- the compounds of the invention may be used in combination with the following agents to treat or prevent autism: i) antipsychotics; ii) antidepressants; iii) anxiolytics; and iv) stimulants for example methylphenidate, amphetamine formulations and pemoline.
- the compounds of the invention may be used in combination with the following agents to treat or prevent ADHD: i) stimulants for example methylphenidate, amphetamine formulations and pemoline; and ii) non-stimulants for example norepinephrine reuptake inhibitors (such as atomoxetine), alpha 2 adrenoceptor agonists (such as clonidine), antidepressants, modafinil, and cholinesterase inhibitors (such as galantamine and donezepil).
- stimulants for example methylphenidate, amphetamine formulations and pemoline
- non-stimulants for example norepinephrine reuptake inhibitors (such as atomoxetine), alpha 2 adrenoceptor agonists (such as clonidine), antidepressants, modafinil, and cholinesterase inhibitors (such as galantamine and donezepil).
- the compounds of the invention may be used in combination with the following agents to treat personality disorders: i) antipsychotics; ii) antidepressants; iii) mood stabilisers; and iv) anxiolytics.
- the compounds of the invention may be used in combination with the following agents to treat or prevent male sexual dysfunction: i) phosphodiesterase V inhibitors, for example vardenafil and sildenafil; ii) dopamine agonists/dopamine transport inhibitors for example apomorphine and buproprion; iii) alpha adrenoceptor antagonists for example phentolamine; iv) prostaglandin agonists for example alprostadil; v) testosterone agonists such as testosterone; vi) serotonin transport inhibitors for example serotonin reuptake inhibitors; vii) noradrenaline transport inhibitors for example reboxetine and viii) 5-HT1A agonists, for example flibanserine.
- phosphodiesterase V inhibitors for example vardenafil and sildenafil
- dopamine agonists/dopamine transport inhibitors for example apomorphine and buproprion
- the compounds of the invention may be used in combination with the same agents specified for male sexual dysfunction to treat or prevent female sexual dysfunction, and in addition an estrogen agonist such as estradiol.
- Antipsychotic drugs include Typical Antipsychotics (for example chlorpromazine, thioridazine, mesoridazine, fluphenazine, perphenazine, prochlorperazine, trifluoperazine, thiothixine, haloperidol, molindone and loxapine); and Atypical Antipsychotics (for example clozapine, olanzapine, risperidone, quetiapine, aripirazole, ziprasidone and amisulpride).
- Typical Antipsychotics for example chlorpromazine, thioridazine, mesoridazine, fluphenazine, perphenazine, prochlorperazine, trifluoperazine, thiothixine, haloperidol, molindone and loxapine
- Atypical Antipsychotics for example clozapine, olanzapine, risperidone, quetiapine,
- Antidepressant drugs include serotonin reuptake inhibitors (such as citalopram, escitalopram, fluoxetine, paroxetine and sertraline); dual serotonin/noradrenaline reuptake inhibitors (such as venlafaxine, duloxetine and milnacipran); Noradrenaline reuptake inhibitors (such as reboxetine); tricyclic antidepressants (such as amitriptyline, clomipramine, imipramine, maprotiline, nortriptyline and trimipramine); monoamine oxidase inhibitors (such as isocarboxazide, moclobemide, phenelzine and tranylcypromine); and others (such as bupropion, mianserin, mirtazapine, nefazodone and trazodone).
- serotonin reuptake inhibitors such as citalopram, escitalopram, fluoxetine, parox
- Mood stabiliser drugs include lithium, sodium valproate/valproic acid/divalproex, carbamazepine, lamotrigine, gabapentin, topiramate and tiagabine.
- Anxiolytics include benzodiazepines such as alprazolam and lorazepam.
- the compounds of the invention will normally, but not necessarily, be formulated into pharmaceutical compositions prior to administration to a patient by an appropriate route. Accordingly, in another aspect, the invention provides pharmaceutical compositions comprising a compound of the invention and one or more pharmaceutically-acceptable excipients.
- pharmaceutically acceptable excipient means any pharmaceutically acceptable material present in the pharmaceutical composition or dosage form other than the compound or compounds of the invention. Typically the material gives form, consistency and performance to the pharmaceutical composition.
- compositions of the invention typically contain one compound of the invention. However, in certain embodiments, the pharmaceutical compositions of the invention contain more than one compound of the invention. In addition, the pharmaceutical compositions of the invention may comprise one or more additional pharmaceutically active compounds.
- compositions of the invention may be prepared and packaged in bulk form wherein a safe and effective amount of a compound of the invention can be dispensed and then given to the patient such as with powders or syrups.
- the pharmaceutical compositions of the invention may be prepared and packaged as dosage forms wherein each physically discrete dosage form contains a safe and effective amount of a compound of the invention.
- the invention provides dosage forms comprising pharmaceutical compositions of the invention. Each discrete dosage form typically contains from 1 mg to 500 mg of a compound of the invention.
- the compositions of the invention will typically be formulated into dosage forms which are adapted for administration to the patient by the desired route of administration.
- dosage forms include those adapted for (1 ) oral administration such as tablets, capsules, caplets, pills, lozenges, powders, syrups, elixirs, suspensions, solutions, emulsions, sachets and cachets; (2) parenteral administration such as sterile solutions, suspensions, implants and powders for reconstitution; (3) transdermal administration such as transdermal patches; (4) rectal and vaginal administration such as suppositories, pessaries and foams; (5) inhalation and intranasal such as dry powders, aerosols, suspensions and solutions (sprays and drops); (6) topical administration such as creams, ointments, lotions, solutions, pastes, drops, sprays, foams and gels; (7) ocular administration such as drops, ointment, sprays, suspensions and inserts; (8) buccal and sublingual administration such as lozenges, patches, sprays, drops, chewing gums and tablets.
- parenteral administration such as ster
- Suitable pharmaceutically acceptable excipients will vary depending upon the particular dosage form chosen.
- suitable pharmaceutically acceptable excipients may be chosen for a particular function that they may serve in the composition.
- certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of uniform dosage forms.
- Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of stable dosage forms.
- Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the carrying or transporting of the compound or compounds of the invention once administered to the patient from one organ, or portion of the body, to another organ, or portion of the body.
- Certain pharmaceutically acceptable excipients may be chosen for their ability to enhance patient compliance.
- Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the release of the compound of the invention at the appropriate rate to treat the condition.
- Suitable pharmaceutically acceptable excipients include the following types of excipients: diluents, fillers, binders, disintegrants, lubricants, glidants, granulating agents, coating agents, wetting agents, solvents, co-solvents, suspending agents, emulsifiers, sweeteners, flavouring agents, flavour masking agents, colouring agents, anticaking agents, humectants, chelating agents, plasticizers, viscosity increasing agents, rate modifying agents, antioxidants, preservatives, stabilizers, surfactants and buffering agents.
- excipients may serve more than one function and may serve alternative functions depending on how much of the excipient is present in the formulation and what other ingredients are present in the formulation.
- the compounds of the combination or composition may be administered simultaneously (either in the same or different pharmaceutical formulations), separately or sequentially.
- a disease or condition mediated by inhibition of monoamine neurotransmitter re-uptake, i.e. inhibition of one or more of SERT, hNET and hDAT.
- the disease or condition is depression or an anxiety disorder.
- a method of treating a disease or condition mediated by inhibition of monoamine neurotransmitter re-uptake, i.e. inhibition of one or more of SERT, hNET and hDAT in a mammal comprising administering an effective amount of a compound of the invention.
- the disease or condition is a depression or an anxiety disorder.
- Proton Magnetic Resonance ( ⁇ H NMR) spectra are typically recorded either on a Varian instrument at 300, 400 or 500 MHz or on a Bruker instrument at 300 and 400 MHz. Chemical shifts are reported in ppm ( ⁇ ) using the residual solvent line as internal standard. Splitting patterns are designed as s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; b, broad. The NMR spectra were recorded at a temperature ranging from 25 to 9O 0 C. When more than one conformer is detected the chemical shifts for the most abundant one is reported.
- DAD chromatographic traces, mass chromatograms and mass spectrums may be taken on a on a UPLC/MS AcquityTM system coupled with a Micromass ZQTM mass spectrometer operating in ESI positive or negative.
- the phases used are: A) H 2 O/ACN 95/5 + 0,1 % TFA; B) H 2 O/ACN 5/95 + 0,1% TFA.
- Flash silica gel chromatography was carried out on silica gel 230-400 mesh (supplied by Merck AG Darmstadt, Germany) or over Varian Mega Be-Si pre-packed cartridges or over pre-packed Biotage silica cartridges.
- SPE-SCX cartridges are ion exchange solid phase extraction columns supplied by Varian.
- the eluent used with SPE-SCX cartridges is methanol followed by 2N ammonia solution in methanol.
- SPE-Si cartridges are silica solid phase extraction columns supplied by Varian.
- a number of the supporting compounds have been prepared as racemic mixtures and a number have been prepared as single enantiomers.
- the absolute stereochemistry of those compounds prepared as single enantiomers have not been assigned, but may be assigned using ab initio vibrational circular dichroism (VCD).
- VCD ab initio vibrational circular dichroism
- VCD is the differentia! interaction of a chiral molecule with left and right circularly polarized infrared radiation during vibrational excitation.
- VCD The VCD spectrum of a chiral molecule is dependent on its three-dimensional structure. Most importantly, the VCD spectrum of a chiral molecule is a function of its absolute configuration and, in the case of flexible molecules, of its conformation. In principle, therefore, VCD permits the determination of the structure of a chiral molecule.
- VCD spectra were first measured in the 1970s. Subsequently, VCD instrumentation has developed enormously in spectral range and in sensitivity.
- IR fundamental infrared
- FT Fourier Transform
- VCD VCD
- Freedman TB et al., HeIv Chim Acta 2002; 85:1160-1 165
- Dyatkin AB et al. Chirality 2002;14:215-219
- Solladie ' -Cavallo A Balaz Met al., Tetrahedron Assym 2001 ;12:2605-2611 ; Nafie LA, et al. Circular dichroism, principles and applications, 2nd ed. New York: John Wiley & Sons; 2000.
- the method entails comparison of observed IR and VCD spectra with calculations of the spectra for a specific configuration and provides information both on the absolute configuration and on the solution conformation.
- VCD spectra are always measured simultaneously with vibrational unpolarized absorption spectra ("infrared (IR) spectra") and the two vibrational spectra together provide more information than does the VCD spectrum alone.
- vibrational unpolarized absorption spectra are automatically predicted simultaneously with VCD spectra.
- VCD and unpolarized IR spectra were calculated using the Gaussian 98 software package.
- SCX strong cation exchanger dried refers to a solution dried over anhydrous sodium sulphate r.t./RT room temperature
- the layers were then allowed to separate and the aqueous layer removed.
- the organic layer was then washed with 1 N HCI (25 L) and water (22 L).
- the resulting organic layer was then concentrated to an oil by vacuum distillation, with a jacket temperature of 20 0 C. Once concentrated to an oil, ethanol (13.7 kg)/water (17.5 kg) was then charged to the reaction and warmed to 50 0 C. Once reaction temperature has stabilized, the reaction was then cooled to -10 0 C at a rate of 0.25 °C/min. The reaction was held at -10 0 C for greater than 6 hours.
- the resulting solids were then filtered, and the filtrate used to rinse the reactor and wash the filter cake.
- a 50 L jacketed laboratory reactor was charged with lithium te/f-butoxide (1.85 Kg, 23.1 mol, 3 eq) and 1-methyl-2-pyrrolidinone (19.1 Kg). The mixture was stirred for -30 min, and the resulting solution was charged into a pressure vessel for later use.
- a 50 L jacketed laboratory reactor was charged with ( ⁇ )-3-(1 ,1 -dimethylethyl) 1 -ethyl (1 S,6R)-6-(3,4-dichlorophenyl)-3-azabicyclo[4.1.0]hept-4-ene-1 ,3-dicarboxylate (from step a, 4.40 Kg, 1 1.0 moles, 1 eq) and heptane (15.1 Kg, 22.1 L, 5 vol).
- the resulting slurry is heated to approximately 80 0 C and filtered into a clean 50-L jacketed laboratory reactor.
- the filter and lines were rinsed with heptane (3.0 Kg, 4.4 L, 1 vol) and the rinse was combined with the filtrate.
- the solution was heated to 80 0 C and then cooled to 22 0 C over 107 min. No crystals had formed so a small aliquot was taken out. Crystals formed spontaneously in the aliquot and were returned to the reactor which caused rapid crystallization.
- the slurry was heated back to 80 0 C and cooled to 22 0 C over 105 min. During the cool, a small aliquot was pulled at 52 0 C. The wall of the vial holding the aliquot was scratched to initiate crystallization and the resulting slurry was combined with the bulk solution when its temperature had reached 47 0 C.
- CDI (0.151 g) was added to a stirred solution of (1 R.6S/1 S,6R)-6-(3,4- dichlorophenyl)-3- ⁇ [(1 , 1 -dimethylethyOoxyJcarbonylJ-S-azabicyclo ⁇ .1.0]heptane-1 - carboxylic acid (Intermediate 7, 0.300 g) in ethyl acetate (5 mL) at RT. The mixture was stirred at RT for 1.5 h and then cooled to 0 0 C in an ice-bath. Concentrated ammonium hydroxide (1.464 mL) was then added. The ice bath was removed and the reaction mixture stirred at room temperature for 18h.
- Method A ((I R. ⁇ S/IS.eRJ- ⁇ -CS ⁇ -DichlorophenyO-S-tKI .I-climethylethyOoxylcarbonylJ-S- azabicyclo[4.1.0]hept-1-yl)acetic acid (Intermediate 15, 145 mg) was dissolved in DMF (5 ml.) under nitrogen at rt. DIPEA (0.070 ml) was added followed by TBTU (128 mg). The mixture was stirred at room temperature for 30 min and then HMDS (0.084 ml.) was added. The mixture was stirred at rt for 1.5h.
- Method B ((I R. ⁇ S/IS.eRJ- ⁇ -CS ⁇ -DichlorophenyO-S-tKI .I-climethylethyOoxylcarbonylJ-S- azabicyclo[4.1.0]hept-1-yl)acetic acid (Intermediate 15, 100 mg) was dissolved in DMF (5 ml.) under nitrogen at room temperature and DIPEA (0.048 ml.) was added followed by TBTU (88 mg). The mixture was stirred at rt for 30 min and then HMDS (0.058 ml.) was added. The mixture was stirred at rt for 1.5h and then quenched with water (10 ml_).
- reaction mixture (suspension) was quenched by careful addition of aqueous ammonium chloride sat. solution (400ml) keeping the internal temperature below +10 0 C with an ice bath. At the end of the addition, the pH of the aqueous phase was nearly 1. The two layers were separated. The aqueous layer was back-extracted with DCM (3 x 300 ml_).
- Triethylamine (0.280 ml.) and methanesulfonyl chloride (0.086 ml.) were added at 0 0 C to a solution of 1 ,1-dimethylethyl (1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-1- (hydroxymethyl)-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 27, Enantiomer 1 , 374 mg) in dry dichloromethane (10 ml_). After 5min stirring, the mixture was allowed to warm to room temperature and stirred overnight. A saturated aqueous NH 4 CI solution was then added and the mixture was extracted with dichloromethane.
- Acetamide oxime (32 mg) was added to a suspension of 3A molecular sieves in dry THF (2.5ml_) under N2 at room temperature. After 5min sodium hydride (60% suspension in mineral oil, 17 mg) was added and the mixture was stirred for further 40min. A solution of 3-(1 ,1-dimethylethyl) 1 -ethyl (1 R,6S/1S,6R)-6-(3,4- dichlorophenyl)-3-azabicyclo[4.1.0]heptane-1 ,3-dicarboxylate (Intermediate 6, 120 mg) in THF (1.5ml_) was then added and the mixture was heated to 65 0 C.
- N-Chlorosuccinimmide (94 mg) was added at 0 0 C to a solution of 1 ,1-dimethylethyl (1 R.6S/1 S,6R)-6-(3,4-dichlorophenyl)-1 -[(hydroxyimino)methyl]-3- azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 32, 257 mg) in dry N, N- dimethylformamide (3 ml_). The mixture was then heated to 40 0 C. After 1 h 30min stirring, water was added and the mixture extracted with ethyl acetate.
- Methylmagnesium bromide (3M in diethyl ether, 1.046 mL) was added at -78 0 C to a solution of 1 ,1-dimethylethyl (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-1- ⁇ [methyl(methyloxy)amino]carbonyl ⁇ -3-azabicyclo[4.1.0]heptane-3-carboxylate
- Racemic Compound 7 (19 mg) was submitted to semi-preparative chiral HPLC (Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30 v/v, flow rate 13 ml/min., detection UV 220 nm.
- Analytical conditions chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30 v/v, flow rate 0.8 ml/min., DAD 210-340 nm) obtaining:
- Racemic Compound 12 (55 mg) was submitted to semi-preparative chiral HPLC (Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30 v/v, flow rate 14 ml/min., detection UV 235 nm. Analytical conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30 v/v, flow rate 0.8 ml/min., DAD 210-340 nm) obtaining:
- Compound 20 (1 R6S or 7S.6RV6-(3.4-dichlorophenylV1-r(5-methyl-1 H-pyrazol-1- yl)methyl1-3-azabicvclo[4.1.01heptane (single enantiomer); and Compound 21 : (1S.6R or 1R6S)-6-(3,4-dichlorophenyl)-1-[(5-methyl-1 H-pyrazol-1- yl)methyl1-3-azabicvclo[4.1.01heptane (single enantiomer).
- Second eluting peak Rt 7.31 min, colourless oil (73 mg).
- the oil was taken up in 2 mL of DCM and TFA (0.3 mL) was added.
- the reaction mixture was stirred for 3h at room temperature then the volatiles were evaporated under reduced pressure.
- the residue was purified by SCX cartrige eluting first with MeOH and then with 2.0N NH3 in MeOH.
- Second eluting compound The colourless oil (27 mg) was dissolved in dry DCM (0.6ml_) and TFA (22 ⁇ l_) was added at room temperature. After 30min, toluene (2ml_) was added and the mixture was concentrated in vacuo. The residue was purified by a SCX cartridge (1g) eluting with MeOH and then NH 3 0.5M in MeOH.
- Compound 30 2- ⁇ [(1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-3-azabicvclor4.1.Olhept-1- yl1methyl ⁇ -3-(trifluoromethyl)-2,4,6,7-tetrahvdropyrano[4,3-c1pyrazole (Racemate);
- Compound 31 1- ⁇ [(1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-3-azabicvclor4.1.0lhept- 1-yl1methyl ⁇ -3-(trifluoromethyl)-1 ,3a,4,6,7,7a-hexahvdropyrano[4,3-c1pyrazole (Single enantiomer); and
- Chiral HPLC separation conditions see Table C: A: Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: (ethanol + 0.1 % isopropylamine) 75/25% v/v, flow rate 13 ml/min., detection UV 230 nm. Analytical conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 0.1% isopropylamine 75/25% v/v, flow rate 0.8 ml/min., DAD 210-340 nm; CD at 230nm
- E Preparative conditions: chiral column Chiralcel OJ-H, eluent A: n-hexane; B: ethanol 80/20% v/v, flow rate 13 ml/min., detection UV 225 nm.
- Analytical conditions chiral column Chiralcel OJ-H, eluent A: n-hexane; B: ethanol 80/20% v/v, flow rate 0.8 ml/min., DAD 210-340 nm; CD at 230nm
- G Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30% v/v, flow rate 13 ml/min., detection UV 225 nm.
- H Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: (2- propanol + 0.1% isopropylamine) 80/20% v/v, flow rate 16 ml/min., detection UV 225 nm.
- Analytical conditions enantiomer 1 chiral column Chiralpak AD-H, eluent A: n- hexane; B: (2-propanol + 1% isopropylamine) 98/2% v/v, flow rate 1 ml/min., DAD 210-340 nm; CD at 230nm; enantiomer 2: eluent A: n-hexane; B: (2-propanol + 1% isopropylamine) 80/20% v/v, flow rate 1 ml/min., DAD 210-340 nm; CD at 230nm;
- M Preparative conditions: chiral column Chiralcel AD-H, eluent A: n-hexane; B: (ethanol + 0.1 % isopropylamine) 90/10% v/v, flow rate 14 ml/min., detection UV 225 nm.
- N Preparative conditions: chiral column Chiralcel OJ-H, eluent A: n-hexane; B: (2- propanol + 0.1% isopropylamine) 90/10% v/v, flow rate 14 ml/min., detection UV 225 nm.
Abstract
The invention relates to compounds of formula (I), processes for their preparation, intermediates used in these processes, pharmaceutical compositions containing them and their use in therapy, as serotonin (5-HT), dopamine (DA) and norepinephrine (NE), re-uptake inhibitors.
Description
Novel Compounds
The present invention relates to novel compounds, processes for their preparation, intermediates used in these processes, pharmaceutical compositions containing them and their use in therapy, as serotonin (5-HT), dopamine (DA) and norepinephrine (NE), re-uptake inhibitors.
Brain tissue is constituted of neuronal cells which are able to communicate with each other via specific cellular structures named synapses. The exchange of signals between neurons in the synapses happens through neurochemical messengers named neurotransmitters, acting on specific target protein molecules, both post and pre-synaptic, referred to as receptors. Monoamines represent a family of small neurotransmitter molecules sharing common chemical features, and include serotonin (5-HT), dopamine (DA) and norepinephrine (NE).
Monoamine neurotransmitters are released into the synaptic cleft between neurons and interact with receptors present on the membrane of the target cells. The switch of the neurochemical signal occurs mainly by removal of the neurotransmitter molecules through other protein molecules referred to as monoamine transporters (SERT for 5-HT, DAT for DA and NET for NE). Transporters are able to bind neurotransmitter molecules and move them into the presynaptic terminals, this cellular mechanism referred to as re-uptake. Pharmacological inhibition of the reuptake process can cause an increase of monoamine at synaptic level and as a consequence an enhancement of the physiological activity of neurotransmitters.
Serotonergic neurotransmission in the brain is mediated by a large family of receptors comprising both the G-protein coupled receptors and ligand-gated ion channels including 14 subtypes, and is involved in a vast variety of physiologic functions.
Compounds endowed of inhibitory properties at the SERT are predicted to have the ability to treat in mammals, including humans, a variety of disorders associated with this neural system, for example eating disorders, major depression and mood disorders, obsessive compulsive disorders, panic disorders, alcoholism, pain, memory deficits and anxiety. Included among these disorders are disorders related to depression, such as pseudodementia or Ganser's syndrome, migraine pain,
bulimia, obesity, pre-menstrual syndrome or late luteal phase syndrome, tobacco abuse, panic disorder, post-traumatic syndrome, memory loss, dementia of ageing, acquired immunodeficiency syndrome dementia complex, memory dysfunction in ageing, social phobia, attention deficit hyperactivity disorder, chronic fatigue syndrome, premature ejaculation, erectile difficulty, anorexia nervosa, disorders of sleep, autism, mutism or trichotillomania.
Major depression is an affective disorder, or disorder of mood, characterized by several symptoms including feeling of profound sadness, worthlessness, despair and loss of interest in all pleasures (anhedonia), recurrent thoughts of death, mental slowing, loss of energy, an inability to take decision, often associated with anxiety and agitation. These symptoms are persistent and can range from mild to severe. The pathophysiology of major depression is poorly understood being a multifactorial syndrome and, due to this, several neurotransmitter systems have been implicated. However, it is generally believed that the disorder stems from a decrease in the synaptic concentration of monoamine neurotransmitters, mainly NE and 5-HT, in critical brain areas, leading to the "monoamine theory" of depression.
Several lines of preclinical and clinical evidence indicate that an enhancement of serotonin-mediated neurotransmission might be effective in the treatment of major depression and actually the selective serotonin re-uptake inhibitors (SSRIs ) have come to dominate the therapy of depression over the last two decades. Fluoxetine, the first SSRI to be introduced, is the prototype of this group. Other members include Paroxetine, Sertraline, Fluvoxamine, Citalopram.
However, it is not clear exactly how these agents act to relieve depression. As with other classes of antidepressant, there is a lag of several weeks before the onset of the mood-elevating effect, despite the rapid blockade of the serotonin re-uptake. It is presumed that secondary adaptive changes must occur at serotonergic synapses after chronic administration of SSRIs i.e. down-regulation of release-regulating autoreceptors and increased neurotransmitter release. The delayed onset of antidepressant effect is considered to be a serious drawback to currently used SSRIs. Moreover, although there is generally good tolerability of SSRIs, the elevation of 5- HT levels at central and peripheral synapses leads to stimulation of receptor subtypes like 5-HT2c and 5-HT3, which contributes to agitation and restless, along with gastrointestinal and sexual side-effects.
The success of the SSRIs rekindled interest in the development of selective norepinephrine re-uptake inhibitors (SNRIs) as potential antidepressants. A number of such compounds have been synthesized, e.g. Nisoxetine, Maprotiline, Tomoxetine and Reboxetine. Furthermore, many compounds, including old tricyclic antidepressants, have a mixed NET and SERT inhibition profile, like lmipramine and Amitriptyline (with SERT potency > NET) and Desipramine, Nortriptyline, and Protriptyline (NET potency > SERT).
The pharmacological manipulation of the DAT can in principle have the ability to elevate DA levels in the mesolimbic system, reversing the anhedonia that is a core symptom of major depression. A DAT inhibition component, in combination with a blockade of SERT and NET, can also have the ability to improve the lack of motivation and attention and enhance cognitive deficits seen in depressed patients. On the other hand, blockade of DAT has to be carefully managed in order to avoid potential reinforcing effects and abuse liability. However compounds with DAT inhibition in their pharmacology, such as Dexmethylphenidate, Methylphenidate and Bupropion, have been successfully marketed.
Clinical studies indicate that patients with poor response to SSRIs benefit from combination therapy with agents that enhance dopaminergic tone. As a result, compounds with a strong SERT inhibiting activity combined with a well balanced NET blockade and moderate DAT inhibiting activity may therefore provide a replacement for current combination therapies for treating unresponsive patients, providing greater efficacy and therapeutic flexibility with a more rapid onset of antidepressant effect.
Due to their valuable DAT inhibition, the compounds of the present invention are considered useful for the treatment of Parkinsonism, depression, obesity, narcolepsy, drug addiction or misuse, including cocaine abuse, attention-deficit hyperactivity disorders, Gilles de Ia Tourettes disease and senile dementia. Dopamine re-uptake inhibitors enhance indirectly via the dopamine neurones the release of acetylcholine and are therefore also useful for the treatment of memory deficits, e.g. in Alzheimers disease, presenile dementia, memory dysfunction in ageing, and chronic fatigue syndrome. Noradrenaline re-uptake inhibitors are considered useful for enhancing attention, alertness, arousal, vigilance and for treating depression.
An object of the present invention is to provide novel compounds which are serotonin (5-HT), dopamine (DA) and norepinephrine (NE) re-uptake inhibitors.
According to a first aspect, the invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof:

(I) wherein R1 is H or C-|_4alkyl;
R2 is phenyl optionally substituted by one or more groups independently selected from halo, cyano, C-|_4alkyl, haloC-|_4alkyl, C-|_4alkoxy, haloC-|_4alkoxy, C-|. 4alkanoyl and SF5; or R^ is naphthyl, optionally substituted by 1 or 2 groups independently selected from halo, cyano and C-|_4alkyl; R3 and R^, which may be the same or different, are H, fluoro or C-|_4alkyl; wherein when R3 or R^ is fluoro, the other R^ or R^ is H or fluoro; R5 is H or C<|_4alkyl; n is 0, 1 or 2;
R8 is phenyl or heteroaryl, either of which is optionally substituted by one or more groups independently selected from halogen, C-|_4alkyl, C-|_4haloalkyl,
C-|_4alkoxy and cyano; wherein when n is 0, the heteroaryl substituent is attached to the rest of the compound of formula (I) by a carbon atom of the heteroaryl; and in the instance when n is 1 or 2, R^ and R^, which may the same or different, are H, D, C-|_4alkyl or fluoro, wherein when R6 or R? is fluoro, the other R6 or R? is
H, D or fluoro.
As used herein, a C-|_4alkyl substituent is a univalent radical derived by removal of a hydrogen atom from an acyclic C-|_4alkane. Such C-|_4alkyl substituents include
methyl and ethyl, may be straight chain (i.e. n-propyl and n-butyl) or branched chain (for example, isopropyl, isobutyl and secbutyl). In an embodiment, unless otherwise indicated, any C-|_4alkyl substituent is methyl, ethyl, n-propyl or isopropyl.
As used herein, a C-|_4alkoxy substituent is a group of formula "R-O-" where R is C-|. 4alkyl as defined above. Such alkoxy substituents include methoxy and ethoxy and may be straight chain (i.e. n-propoxy and n-butoxy) or branched chain (for example, isopropoxy, isobutoxy, secbutoxy and tert-butoxy). In an embodiment, unless otherwise indicated, any C-|_4alkoxy substituent is methoxy, ethoxy, n-propoxy or isopropoxy.
As used herein, a C-|_4haloalkyl substituent is a C-|_4alkyl group substituted by one or more halo substituents, which halo substituents may be the same or different. Such C-|_4haloalkyl substituents include monofluoromethyl, difluoromethyl, trifluoromethyl and 1-chloro-2-fluoroethyl. In an embodiment, unless otherwise indicated any C-|_4haloalkyl substituent is monofluoromethyl, difluoromethyl or trifluoromethyl.
As used herein, a C-|_4haloalkoxy substituent is of formula "Rx-O-" where Rx is C-μ 4haloalkyl as defined above. Such C-|_4haloalkoxy substituents include monofluoromethoxy, difluoromethoxy, trifluoromethoxy and 1-chloro-2-fluoroethoxy and may be straight chain or branched chain. In an embodiment, unless otherwise indicated, any C-|.ghaloalkoxy substituent is monofluoromethoxy, difluoromethoxy or trifluoromethoxy.
As used herein, a halo substituent refers to fluoro, chloro, bromo and iodo radicals. In an embodiment, unless otherwise indicated, any halo substituent is fluoro or chloro.
As used herein, a C-|_4alkanoyl substituent refers to C-|_4alkylcarbonyl. In an embodiment, the C-|_4alkanoyl substituent is acetyl, ethylcarbonyl, n-propylcarbonyl, i-propylcarbonyl, n-butylcarbonyl or t-butylcarbonyl.
As used herein, H and D, refers to hydrogen and deuterium radicals respectively.
It will be appreciated from formula (I) that R^ and -(CR6R7)ΠR8 possess a cis relationship, i.e. both groups R^ and -(CR6R7)ΠR8 are on the same face of the
bicyclic ring system. It will also be appreciated that two alternatives exist with respect to the geometry at the ring carbon atoms attached to R^ and -(CR6R7)nR8, namely compounds of formula (IA) and IB).

(IA) (IB)
It will be appreciated that compounds of formula (IA) and (IB) have an enantiomeric relationship. Enantiomeric mixtures of compounds of formula (IA) and (IB) may be separated by appropriate optical resolution techniques (for example chiral HPLC).
Therefore in an embodiment, the compound of formula (I) is a mixture of compounds of formula (IA) and (IB). In a further embodiment, the compound of formula (I) is a racemic mixture of compounds of formula (IA) and (IB).
In a further embodiment, the compound of formula (I) is of formula (IA). In a further embodiment, the enantiomeric excess (e.e.) of (IA) over (IB) is greater than or equal to 90%. In another embodiment, the e.e. of (IA) over (IB) is greater than or equal to 95%. In another embodiment, the e.e. of (IA) over (IB) is greater than or equal to 99%.
In a further embodiment, the compound of formula (I) is of formula (IB). In a further embodiment, the enantiomeric excess (e.e.) of (IB) over (IA) is greater than or equal to 90%. In another embodiment, the e.e. of (IB) over (IA) is greater than or equal to 95%. In another embodiment, the e.e. of (IB) over (IA) is greater than or equal to 99%.
It will be appreciated from formula (I) that the carbon atom attached to R^ and R^! the carbon atom attached to R^; and the carbon atom attached to R^ and R^ may be
chiral. It will be appreciated that diastereomeric mixtures of compounds of formula (I) may be obtained.
In an embodiment, R-I is hydrogen or methyl. In a further embodiment, R-I is hydrogen.
In an embodiment, R^ is phenyl substituted by one or two groups independently selected from halo and haloC-1.4 alkyl; or R^ is unsubstituted naphthyl. In an embodiment, R^ is hydrogen.
In an embodiment, n is 1 or 2. In a further embodiment n is 1.
When R8 is heteroaryl, it refers to a univalent radical derived by removal of a hydrogen atom from a heteroaromatic ring system. The heteroaromatic ring system may be monocyclic or bicyclic. When the heteroaryl substituent is monocyclic, it comprises one or more carbon atoms and 1 to 4 heteroatoms interconnected to form a ring. The heteroatoms are independently selected from nitrogen, oxygen and sulphur. In an embodiment, unless otherwise indicated, the monocyclic heteroaryl substituent is furyl, thienyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, azepinyl, oxazepinyl, thiazepinyl or diazepinyl. In a further embodiment, the monocyclic heteroaryl substituent is furyl, thienyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl or tetrazolyl. When the heteroaryl substituent is bicyclic, one of the rings may contain from 5 to 7 atoms interconnected to form a ring and the other ring may contain from 5 or 6 carbons interconnected to form a ring. Either or both of the rings may contain 1 to 4 heteroatoms independently selected from nitrogen, oxygen and sulphur. When the heteroaryl substituent is fused bicyclic, one of the rings is aromatic and the other is saturated, unsaturated or aromatic. In an embodiment, unless otherwise indicated, the bicyclic heteroaryl substituent is indolizinyl; indolyl; isoindolyl; 3H-indolyl; indolinyl; indolizinyl; benzo[b]furyl; benzo[b]thienyl; 1 H-indazolyl; 2H-indazolyl; benzimidazolyl; benzthiazolyl; purinyl; 4H-quninolinyl; quinolinyl; isoquinolinyl; cinnolinyl; phthalazinyl; quinazolinyl; quinoxalinyl; 1 ,8-naththyridinyl; pteridyl; 2,4,6,7- tetrahydropyrano[4,3-c]pyrazolyl; 1 ,3a,4,6,7,7a-hexahydropyrano[4,3-c]pyrazolyl; 4,5,6,7-tetrahydro-1 H-indazolyl; or 4,5,6,7-tetrahydro-2H-indazolyl. In a further
embodiment, the bicyclic heteroaryl substituent is indolizinyl; indolyl; isoindolyl; 3H- indolyl; indolinyl; benzo[b]furyl; benzo[b]thienyl; 1 H-indazolyl; 2H-indazolyl; benzimidazolyl; benzthiazolyl; 2,4,6,7-tetrahydropyrano[4,3-c]pyrazolyl; 1 ,3a,4,6,7,7a-hexahydropyrano[4,3-c]pyrazolyl; 4,5,6,7-tetrahydro-1 H-indazolyl; or 4,5!6,7-tetrahydro-2H-indazolyl.
In an embodiment, R^ is a monocyclic heteroaryl substituent selected from the list furyl, thienyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl or tetrazolyl, any of which substituents are optionally substituted by one or more groups independently selected from halogen, C-|_4alkyl, C-|_4haloalkyl, C-|_4alkoxy and cyano. In a further embodiment, R^ is pyrazolyl, tetrazolyl, triazolyl, pyrrolyl or oxazolyl any of which substituents are optionally substituted by one or more groups independently selected from halogen, C-|_4alkyl, C-|_4haloalkyl, C-|_4alkoxy and cyano.
In an embodiment, the compound defined in the first aspect is selected from the list
(1 R.6S/1 S,6R)-6-(3,4-dichlorophenyl)-1 -[(1 S)-1 -(3,5-dimethyl-1 H-pyrazol-1 -yl)ethyl]-
3-azabicyclo[4.1.0]heptane (Racemic Compound 25); (1 R.6S/1 S,6R)-6-(3,4-dichlorophenyl)-1 -[(3,5-dimethyl-1 H-pyrazol-1 -yl)methyl]-3- azabicyclo[4.1.0]heptane (Racemic Compound 48);
(1 S,6R/1 R,6R)-6-(3,4-dichlorophenyl)-1-{[5-(1-methylethyl)-2H-tetrazol-2-yl]methyl}-
3-azabicyclo[4.1.0]heptane (Racemic Compound 60);
(1 S,6R/1 R,6S)-6-(3,4-dichlorophenyl)-1 -[(3,5-dimethyl-1 H-1 ,2,4-triazol-1 -yl)methyl]- 3-azabicyclo[4.1.0]heptane (Racemic Compound 61 );
(1 S,6R/1 R,6S)-6-(3,4-dichlorophenyl)-1 -{[5-methyl-3-(trifluoromethyl)-1 H-pyrazol-1 - yl]methyl}-3-azabicyclo[4.1.0]heptane (Racemic Compound 62);
(1 R,6S or 1 S,6R)-6-(3,4-dichlorophenyl)-1-[(3,5-dimethyl-1 H-pyrazol-1 -yl)methyl]-3- azabicyclo[4.1.0]heptane (Compound 68 - single enantiomer); (1 R,6S or 1 S,6R)-6-(3,4-dichlorophenyl)-1-{[3-(trifluoromethyl)-1 H-pyrazol-1 - yl]methyl}-3-azabicyclo[4.1.0]heptane (Compound 74 - single enantiomer);
(1 S,6R or 1 R,6S)-6-(3,4-dichlorophenyl)-1-{[5-(1-methylethyl)-2H-tetrazol-2- yl]methyl}-3-azabicyclo[4.1.0]heptane (Compound 84 - single enantiomer);
(1 S,6R or 1 R,6S)-6-(3,4-dichlorophenyl)-1-{[5-methyl-3-(trifluoromethyl)-1 H-pyrazol- 1-yl]methyl}-3-azabicyclo[4.1.0]heptane (Compound 88 - single enantiomer);
(1S,6R or 1 R!6S)-6-(3!4-dichlorophenyl)-1-{[3-methyl-5-(trifluoromethyl)-1 H-pyrazol- 1-yl]methyl}-3-azabicyclo[4.1.0]heptane (Compound 90 - single enantiomer); (I R^S or i S^RJ-β-CS^-clichlorophenylJ-i-^-methyl-I H-pyrazol-i-yOmethyll-S- azabicyclo[4.1.0]heptane hydrochloride (Compound 96 - single enantiomer); (1 R.6S or ^^^^-(S^-dichlorophenyO-i-p.δ-dimethyl-I H-pyrazol-i-yOmethyO-S- azabicyclo[4.1.0]heptane hydrochloride (Compound 98 - single enantiomer); (1 R,6S or 1 S,6R)-6-(3,4-dichlorophenyl)-1-{[3-(trifluoromethyl)-1 H-pyrazol-1- yl]methyl}-3-azabicyclo[4.1.0]heptane hydrochloride (Compound 108 - single enantiomer); 1-{[(1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-3-azabicyclo[4.1.0]hept-1-yl]methyl}-1 H- pyrazole-5-carbonitrile hydrochloride (Compound 1 10 - single enantiomer); (1S,6R or 1 R!6S)-6-(3!4-dichlorophenyl)-1-[(2,4-dimethyl-1 H-pyrrol-1-yl)methyl]-3- azabicyclo[4.1.0]heptane hydrochloride (Compound 115 - single enantiomer); 1-{[(1 R!6S or 1S,6R)-6-(3,4-dichlorophenyl)-3-azabicyclo[4.1.0]hept-1-yl]methyl}-1 H- pyrrole-3-carbonitrile hydrochloride (Compound 120 - single enantiomer); and (1 R!6S or 1 S,6R)-6-(3!4-dichlorophenyl)-1-[(4-methyl-1 ,3-oxazol-5-yl)methyl]-3- azabicyclo[4.1.0]heptane hydrochloride (Compound 140 - single enantiomer); or a pharmaceutically acceptable salt of any of the compounds.
The compounds of formula (I) as defined in the first aspect contain a basic centre and may form non-toxic acid addition salts formed with inorganic acids such as hydrochloric, hydrobromic, hydroiodic, sulfuric and phosphoric acid, with carboxylic acids or with organo-sulfonic acids. Examples include the HCI, HBr, HI, sulfate or bisulfate, nitrate, phosphate or hydrogen phosphate, acetate, benzoate, succinate, saccharate, fumarate, maleate, lactate, citrate, tartrate, gluconate, camsylate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate and pamoate salts. For reviews on suitable pharmaceutical salts see Berge et al, J. Pharm, ScL, 66, 1-19, 1977; P L Gould, International Journal of Pharmaceutics, 33 (1986), 201-217; and Bighley et al, Encyclopedia of Pharmaceutical Technology, Marcel Dekker Inc, New York 1996, Volume 13, page 453-497. In an embodiment, the salt is pharmaceutically acceptable.
It will be appreciated by those skilled in the art that certain protected derivatives of the compounds of formula (I) as defined in the first aspect, which may be made prior to a final deprotection stage, may not possess pharmacological activity as such, but may, in certain instances, be administered orally or parenterally and thereafter metabolised in
the body to form compounds defined in the first aspect which are pharmacologically active. Such derivatives may therefore be described as "prodrugs". All protected derivatives and prodrugs of compounds defined in the first aspect are included within the scope of the invention. Examples of suitable pro-drugs for the compounds of the present invention are described in Drugs of Today, Volume 19, Number 9, 1983, pp 499 - 538 and in Topics in Chemistry, Chapter 31 , pp 306 - 316 and in "Design of Prodrugs" by H. Bundgaard, Elsevier, 1985, Chapter 1 (the disclosures in which documents are incorporated herein by reference). It will further be appreciated by those skilled in the art, that certain moieties, known to those skilled in the art as "pro- moieties", for example as described by H. Bundgaard in "Design of Prodrugs" (the disclosure in which document is incorporated herein by reference) may be placed on appropriate functionalities when such functionalities are present within the compounds defined in the first aspect. Therefore, in a further aspect, the invention provides a prodrug of a compound defined in the first aspect.
The compounds defined in the first aspect, their salts or prodrugs, may exist in solvated or hydrated form. Therefore, in a further aspect, the invention provides a solvate or hydrate of a compound defined in the first aspect or a salt thereof.
The compounds of formula (I) and their salts, as defined in the first aspect or solvates or hydrates of either, may exist in one or more polymorphic form. Therefore, in a further aspect, the invention provides a polymorph of a compound of formula (I) defined in the first aspect or their salts, or a polymorph of a solvate or hydrate of a compound of formula (I) defined in the first aspect, or a salt thereof.
Hereinafter, compounds of formula (I) as defined in the first aspect, their salts and prodrugs; any solvates or hydrates of any salt or prodrug; and any polymorph of any compound, salt, solvate or hydrate are referred to as "compounds of the invention". The term "compounds of the invention" also includes all embodiments of the first aspect.
The compounds of the invention may possess one or more chiral centres and so exist in a number of stereoisomeric forms. All stereoisomers and mixtures thereof are included in the scope of the present invention. Racemic compounds may either be separated using preparative HPLC and a column with a chiral stationary phase or resolved to yield individual enantiomers utilising methods known to those skilled in
the art. In addition, chiral intermediate compounds may be resolved and used to prepare chiral compounds of the invention. In addition, the chiral compounds of the invention may be prepared by chiral synthesis.
The compounds of the invention may exist in one or more tautomeric forms. All tautomers and mixtures thereof are included in the scope of the present invention. For example, a claim to 2-hydroxyquinolinyl would also cover its tautomeric form, α- quinolinonyl.
The invention also includes all suitable isotopic variations of a compound of the invention. An isotopic variation of a compound of the invention is defined as one in which at least one atom is replaced by an atom having the same atomic number but an atomic mass different from the atomic mass usually found in nature. Examples of isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulphur, fluorine and chlorine such as 2H, 3H, 11C, 13C, 14C, 15N, 17O, 180, 31P, 32P, 35S, 18F and 36CI, respectively. Certain isotopic variations of the invention, for example, those in which a radioactive isotope such as 3H or 14C is incorporated, are useful in drug and/or substrate tissue distribution studies. Tritiated, i.e., 3H, and carbon-14, i.e., 14C, isotopes are particularly preferred for their ease of preparation and detectability. Further, substitution with isotopes such as deuterium, i.e., 2H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements and hence may be preferred in some circumstances. Isotopic variations of the compounds of the invention can generally be prepared by conventional procedures such as by the illustrative methods or by the preparations described in the Experimental section hereafter using appropriate isotopic variations of suitable reagents.
Compounds of the invention may be prepared in a variety of ways. In the following reaction schemes and hereafter, unless otherwise stated R^ to R^ and n are as defined in the first aspect.
Throughout the specification, general formulae are designated by Roman numerals (I), (II), (III), (IV) etc. Subsets of these general formulae are defined as (Ia), (Ib), (Ic) etc .... (IVa), (IVb), (I Vc) etc.
Compounds of formula (Ib), i.e. compounds of formula (I) where R^ is C-|_4alkyl, may be obtained from compounds of formula (Ia) wherein R-I is hydrogen according to reaction scheme 1 following standard alkylation procedures. Typical reaction conditions comprise reaction with methyl iodide, a trialkylamine (such as TEA) in DCM, at temperature between 0 0C and room temperature.
Alternatively, compounds of formula (Ib) may be obtained through reductive amination using a suitable aldehyde R^ CHO, a reducing agent such as NaCNBH3, in aprotic or protic solvent (e.g. toluene, THF or MeOH), at temperature between 80 0C and room temperature.
Scheme 1

(Ia) (Ib)
Compounds of formula (Ia) may be obtained by deprotecting compounds of formula (II) (see reaction scheme 2). Suitable protecting groups (Pg) include Boc and benzyl. Removal of a Boc protecting group may be achieved by reaction with TFA in DCM at a temperature between 0 0C and room temperature. Removal of a benzyl protecting group may be achieved by hydrogenation over Pd/C. Alternatively removal of a benzyl protecting group may be achieved using α-chloroethyl chloroformate at reflux in DCE and then in MeOH.
Scheme 2

(H) (Ia)
Compounds of formula (Na), i.e. compounds of general formula (II) where n is 1 , R^ is a N-linked monocyclic or bicyclic heteroaryl substituent and Pg is a suitable N- protecting group, typically Boc or benzyl, may be obtained from compounds of formula (III), wherein L is a suitable leaving group (e.g. mesylate) according to reaction scheme 3. Typical reaction conditions comprise, nucleophilic displacement of L with the pre-formed anion from the N-heteroaryl group using base (such as NaH) in aprotic solvent (e.g. DMF) at between 0 0C and room temperature followed by heating to a temperature between 40 and 80 0C.
Scheme 3

Compounds of formula (Ilia), i.e. compounds of general formula (III) where R^ is hydrogen, may be obtained according to reaction scheme 4 from compounds of formula (IV). Typical reaction conditions for forming the mesylate comprises reaction with methansulfonyl chloride in DCM under basic condition at O0C or room temperature.
Scheme 4

(IV) (Ilia)
Compounds of formula (IVa), i.e. compounds of formula (IV) where R^ and R^ are both fluoro, may be obtained from compounds of formula (V), according to reaction scheme 5 using dibromodifluoromethane following the method reported in the Journal of Fluorine Chemistry (2003), 119(1 ), 75-80.
Scheme 5

Compounds of formula (IVb), i.e. compounds of formula (IV) where R^ and R^ are methyl, may be obtained from compounds of formula (V) according to reaction scheme 6, using the procedure described in Synlett (2002), (1 ), 176-178.
Scheme 6

(V) (IVb)
Compounds of formula (IVc), i.e. compounds of general formula (IV) where R^ and R4 are hydrogen, may be obtained by Simmons-Smith cyclopropanation from compounds of formula (V) wherein Pg is a suitable N-protecting group, typically Boc, according to Scheme 7. Typical reaction condition comprise reacting (V) with methyl iodide and zinc chloride in DCM.
Scheme 7

(V) (IVc)
Compounds of formula (V) where R^ and R^ are hydrogen or deuterium may be obtained by reduction of compounds of formula (Vl), wherein Pg is a suitable N- protecting group (such as Boc) and R an alkyl group (such as methyl or ethyl) according to reaction scheme 8. Suitable regucing agents are LiAID4 or LiAIH4, in aprotic solvent, e. g. diethyl ether or THF, at temperature between -40 and -10 0C.
Scheme 8

(Vl) (V)
Compounds of formula (Va), i.e. compounds of formula (V) where R^ and R^ are both methyl, may be obtained from compounds of formula (Vl), wherein Pg is a suitable N-protecting group (typically Boc) and R an alkyl group (such as methyl or ethyl) according to reaction scheme 9. Suitable reaction conditions comprise reacting
(Vl) with methyl magnesium bromide in aprotic solvent, e. g. diethyl ether or THF, at temperature between -40 and -10 0C.
Scheme 9

(Vl) (Va)
Compounds of formula (Vl) may be obtained from compounds of formula (VII), by reaction with an appropriate boronic acid R2B(OH)2 according to reaction scheme 10. Suitable reaction conditions comprise reacting (VII) with R2β(OH)2 in the presence of Pd(OAc)2 , PPh3 and diisopropylethylamine in a mixture of toluene and water at a temperature ranging from room temperature to 800C. Alternative catalysts may be used, for example Pd(PPhi3)4 and PdCl2(dppf).
Scheme 10

(VII) (Vl)
Compounds of formula (VII) may be obtained from compounds of formula (VIII) according to reaction scheme 11. Suitable reaction conditions comprise treatment with triflic anhydride, in the presence of diisopropylethylamine, in toluene solvent at a temperature ranging from 0 0C to room temperature.
Scheme 1 1

Compounds of formula (IVd), i.e. compounds of formula (IV) (see reaction scheme 4) where R^, R4, R6 anc| R7 are hydrogen, may be obtained directly from compounds of formula (IX) according to reaction scheme 12. Suitable reaction conditions comprise treatment with BH3 or LiAIH4 in aprotic solvent (such as THF) at reflux, followed by protection of the nitrogen using Boc anhydride under basic conditions at room temperature.
Scheme 12

Compounds of formula (IX) may be obtained according to reaction scheme 13 from compounds of formula (X) wherein R is alkyl and L is a suitable leaving group (such as mesylate) by reaction with ammonia. Typical conditions comprise reaction under pressure in a suitable solvent (such as methanol) in a hydrogenation apparatus (for example Parr). Nitrogen protection gives compounds of formula (IX).
Scheme 13

Compounds of formula (X) may be obtained according to reaction scheme 14 from compounds of formula (Xl) by carbene mediated cyclopropanation with dimethyl diazopropandioate and rhodium catalyst (for example Rh2(OAc)2) in chlorinated solvent (e.g, chlorobenzene or DCE) at a temperature between 4O0C and 8O0C. Use of asymmetric rhodium catalysts may be used to provide stereospecific compounds of formula (X).
Scheme 14

(Xl) (X)
Compounds of formula (Xl) where L is a mesylate, may be obtained according to reaction scheme 15 from compounds of formula (XII) by treatment with methansulfonyl chloride in DCM under basic conditions at O0C or room temperature.
Scheme 15

(XII) (Xi)
Compounds of formula (XII) may be obtained according to reaction scheme 16 by coupling compounds of formula (XIII) with boronic acids of formula R2B(OH)2- Typical reaction conditions comprise reaction of (XIII) with the boronic acid in the presence of Pd(PPhi3)4 and a base (e.g. sodium carbonate) in a mixture of solvents (e.g. toluene, ethanol and water) at 8O0C.
Scheme 16

(XIII) (XII)
Compounds of formula (XIVa) may be obtained according to reaction scheme 17 from compounds offormula (XV) by reduction of the olefinic double bond. Reaction conditions comprise treatment with triethylsilane and TFA in aprotic solvent (such as toluene). Alternative reducing agents may be used, for example sodium triacetoxyborohydride or sodium borohydride. Alternative solvents may also be used, for example dichloromethane, trifluorotoluene or chlorobenzene.
Scheme 17

Compounds of formula (XIVa) may then be reduced to give compounds of formula (IVd).
Compounds of formula (XV) may be obtained from compounds of formula (Vl) by reaction with an appropriate base in the presence of an appropriate alkylating agent according to reaction scheme 18. Reaction conditions may comprise treatment with lithium t-butoxide and CH2ICI in N-methyl pyrrolidone at low temperature (for example -20 to +100C). Alternative alkylating agents may be used, for example CH2I2- Alternative solvents may be used, for example DMF or THF. Alternative bases may be used, for example LDA or NaH.
Scheme 18

Compounds of formula (XVI) wherein R^ and R^ are methyl or fluorine and R is an alkyl group (typically methyl or ethyl) may be obtained according to reaction scheme 19 starting from compounds of formula (XVII) by esterification followed by functionalisation at the alpha position of the ester. Mono- or bis-deprotonation at the alpha position may be achieved using a strong base (such as LDA) in an aprotic solvent at low temperature (-90 and -400C) followed by addition of an electrophilic species, such as methyl iodide or N-Fluorobenzenesulfonimide.
Scheme 19

(XVII) (XVI)
Compounds of formula (XVII) may be obtained according to reaction scheme 20 from compounds of formula (XVIII) by a two step oxidation process. Oxidation to the aldehyde may be achieved with Dess-Martin periodinane, followed by treatment with an aqueous solution of sodium chlorite and sodium dihydrogen phosphate.
Scheme 20
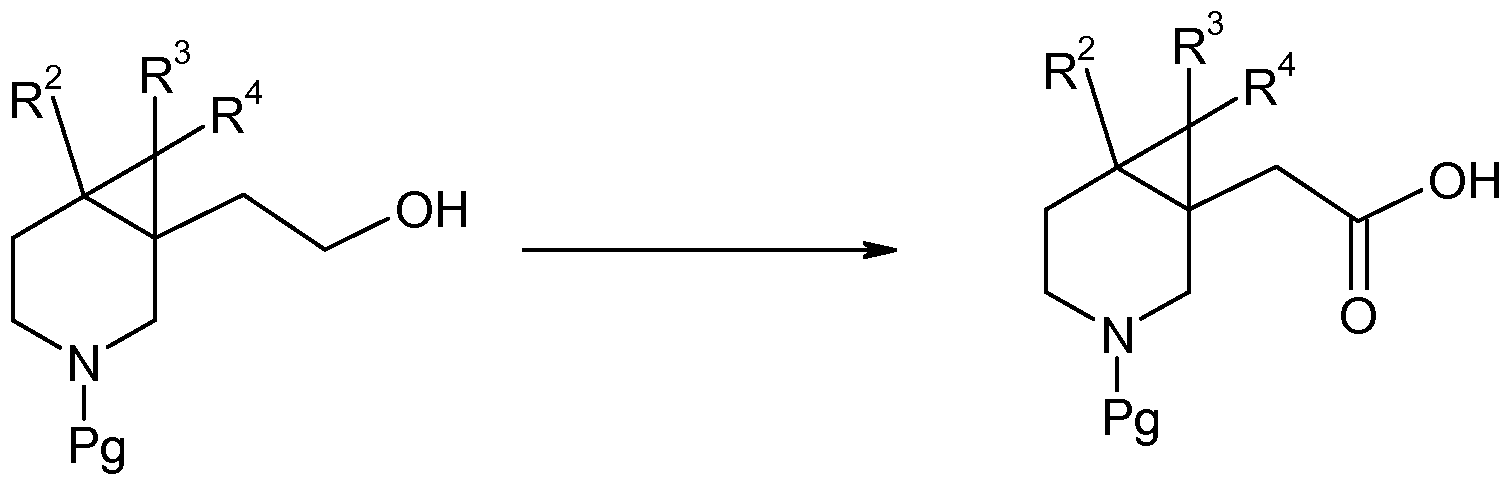
(XVII)
(XVIII)
Compounds of formula (XVIII) may be obtained from compounds of formula (XVI), according to reacytion scheme 21 , by hydroboration of the alkene with borane-THF complex in THF at a temperature between 0 0C and room temperature followed by oxidation with hydrogen peroxide and sodium hydroxide at 0 0C.
Scheme 21

(XIX) (XVIII)
Compounds of formula (XIX) may be obtained from compounds of formula (XX), according to reaction scheme 22, by Wittig reaction using methylenetriphenylphosphorane in THF at room temperature.
Scheme 22

(XX) (XIX)
Compounds of formula (XX) may be obtained from compounds of formula (IVe), wherein R^ and R^ are hydrogen and Pg is a suitable N-protecting group (typically Boc) according to reaction scheme 23, by oxidation with Dess-Martin periodinane in DCM at a temperature between 0 0C and room temperature.
Scheme 23

Compounds of formula (XXI) may be obtained according to reaction scheme 24 from compounds of formula (XXII) by reaction of the Weinreb amide with methyl magnesium bromide in aprotic solvent at low temperature (typically between -78°C and 00C).
Scheme 24

Compounds of formula (XXII) may be obtained according to reaction scheme 25 from compounds of formula (XIVb) by reaction with N,O-dimethylhydroxylamine hydrochloride and LiHMDS in aprotic solvent (such as THF) at low temperature (typically between -500C and 00C).
Scheme 25

Compounds of formula (lib), i.e. compounds of general formula (II) where n is 0 and R8 a C-linked mono or bicyclic heterocycle, may be obtained according to reraction scheme 26 via standard cyclization processes reported in the literature starting from the suitable intermediate (i.e. ester, amide, thioamide) prepared from compounds of formula (XIVc).
Scheme 26

Compounds of formula (lie), i.e. compounds of general formula (II) where n is 1 , R^ and R^ are hydrogen and R^ is a C or N-linked heterocycle, may be obtained through standard cyclization processes reported in the literature starting from the suitable intermediate (i.e. ester, amide, thioamide) prepared from compounds of formula (XVII) (see reaction scheme 27).
Scheme 27

(XVII) (lie)
Compounds of formula (Nd), i.e. compounds of general formula (II) where n is 1 and R8 is a C- or N-linked heterocycle, may be obtained according to reaction scheme 28 via standard cyclization processes reported in the literature starting from the suitable intermediate (i.e. ester, amide, thioamide) prepared from compounds of formula (XVII).
Scheme 28

(XVII) (Md)
Compounds of formula (lib), i.e. compounds of general formula (II) wherein R^ is hydrogen, may be obtained via standard cyclization processes reported in the literature starting from compounds of formula (XXI) (see reaction scheme 29).
Scheme 29

(XXI) (Hb)
An alternative procedure for the preparation of compounds of formula (NIb) is shown in reaction scheme 30.
Scheme 30

(XXII)

(XXlIl) (XXIV)

(XXV) (XXVI)
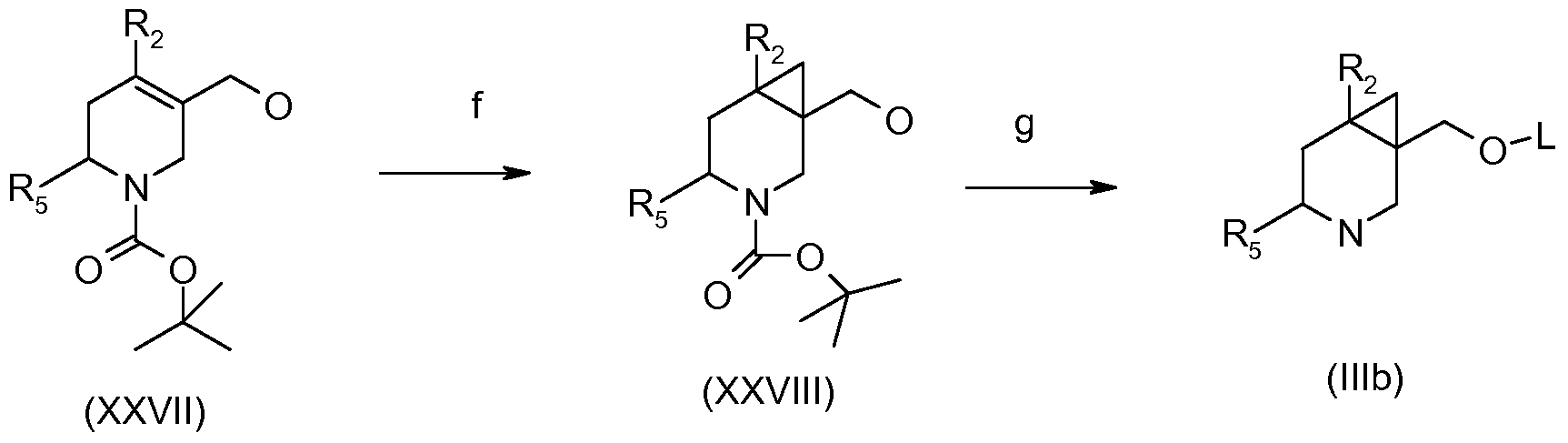
Example of Reaction conditions: a = EtOH, 8 hrs, RT; b = BOC2O; K2CO3, from O 0C to RT for 48 hrs; c = EtONa, Toluene, from O 0C to RT, overnight; d = 1 ) NaH and 1 ,1 , 1-trifluoro-N- phenyl-N-[(trifluoromethyl)sulfonyl]methanesulfonamide, DMF, 1 h; 2) toluene/ EtOH; R2B(OH)2, , K2CO3, Pd(Ph3P)4 , 80 0C, 1 h. ; e= LiAIH4 ,THF from -20 0C to RT, 2 h.; f = Et2Zn7CH2I2 DCM , RT, overnight.; g =.Mes-CI, TEA, DCM
A specific enantiomer or diastereoisomer of a compound of the invention may be obtained for example by optical resolution of a mixture of enantiomers or diastereoisomers using conventional methods, such as chiral chromatography.
The affinity of compounds of the invention for SERT, NET and DAT may be tested in one or other of the following affinity assays.
a) Filtration Binding Assay using membranes from hSERT, hNET, and hDAT Lewis Lung Carcinoma Porcine tubule Kidney (LLCPK) cell lines - Membrane preparation hSERT-LLCPK, hDAT-LLCPK or hNET-LLCPK cell lines are used for the membrane preparations for radioligand binding assays. Stable cell lines may be generated as follows: i) hSERT - generated by transfecting LLC-PK1 or LLCPK cells with hSERT cloned into the mammalian expression vector pCDNA3.1 Hygro(+); ii) hNET - generated by transfecting LLCPK cells with hNET cloned into the mammalian expression vector pRC/CMV; iii) hDAT- generated by transfecting LLCPK cells with hDAT cloned into the mammalian expression vector pDESTCDNA3.1 (an example of a procedure for transfecting LLCPK cells with hDAT, hSERT and hNET may be found in H. Gu, S. C. Wall and G. Rudnick, J. Biol. Chem. (1994) 269 : 7124-7130.)
Each cell line is cultured independently in Dulbecco's modified Eagle's medium (DMEM) containing 10% of Foetal Bovine Serum (FBS) supplemented with 400 μg/ml hygromicin (hSERT) or geneticin at 500 μg/ml (hNET) or at 1000 μg/ml (hDAT). Cells are maintained at 37°C in a humidified environment containing 5% CO2 in air.
When cells are at 70-80% of confluence, the culture medium is removed and the cells harvested with phosphate buffered saline (PBS) containing 5 mM EDTA. The cell suspension is centrifuged at 90Og for 5 minutes at 40C. The resultant pellets are re-suspended in 30-50 volumes of Assay Buffer (5OmM Tris pH 7.7 containing 12OmM NaCI, 5mM KCI, 10μM pargyline and 0.1% ascorbic acid) and homogenized using a glass-teflon Potter homogeniser and centrifuged at 4800Og for 20 minutes at 40C. The resultant membrane pellets are re-suspended in the same volume of Assay Buffer, incubated for 20 minutes at 370C and centrifuged as before at 4800Og. The final protein concentration for each preparation is adjusted to give approximately 480μg protein/ml for hSERT-LLCPK, hDAT-LLCPK and hNET-LLCPK, as determined by the Bio-Rad Protein Assay kit. Membranes are stored at -800C as 1 ml aliquots until required.
- Assay Protocol (For general references to monoamine transporters filtration binding assays see: Michael J. Owens, et al, Neurotrasmitter receptor and transporter binding profile of antidepressants and their metabolites, JPET, 283:1305-1322, 1997; PerAllard, Jan O. Marcusson, Svate B. Ross, [3H]WIN-35,428 binding in the human brain, Brain Res., 706 :347-350, 1996.)
The affinity of the compounds of the invention to bind the re-uptake site of SERT may be assessed using [3|H]citalopram filtration binding assay performed on hSERT- LLCPK cell membranes. The competition binding assay is conducted in deep-well 96 well plates (1 ml, NUNC, cod.260252) in a total volume of 400μl, with each concentration in duplicate. 4μl of test compound (100X solution in neat DMSO as 7 point curve ranging from 10"6 to 10"12M, final concentration) or DMSO (to define total binding) or a final concentration of 10μM fluoxetine in DMSO (to define non-specific binding, NSB) are added to wells; after this, 200μl of [N-Methyl-3H]citalopram (Amersham Biosciences, 80 Ci/mmol) at the final concentration of 0.25nM in Assay Buffer, is added to all wells and finally the reaction is started by adding 200μl/well of membranes diluted 1 :80 in Assay Buffer at concentration of about 2.5μg/well of protein. The reaction is carried out at room temperature for 2 hours and then stopped by rapid filtration through GF/B Unifilter 96-filterplate (Perkin-Elmer) pre-soaked in 0.5% polyethylenimmine (PEI) using a Perkin-Elmer FilterMat-196 harvester.
Filterplate is washed 3 times with 1 ml/well ice-cold 0.9% NaCI solution. The plate is dried in an oven for 60 min at 500C then opaque bottom-seal is placed on the underside of the plate and 50μl of Microscint 20 (Perkin-Elmer) added to each well. Plate is sealed with a TopSeal and the radioactivity in the samples is counted for 4 min using TopCount liquid scintillation counter (Packard-Perkin-Elmer) and recorded as counts per minute (CPM).
Competition binding assay for hNET may be conducted essentially as previously reported for hSERT in 96 well format and in a final assay volume of 400μl, except for the use of hNET-LLCPK cell membranes (1 :40 dilution i.e. 4.8μg of protein/well) and [3H]nisoxetine as radioligand (1.5nM [N-methyl-3H]nisoxetine, Amersham Biosciences, 84 Ci/mmol). 10μM desipramine is used for NSB.
Competition binding assay for hDAT may also be conducted essentially as previously reported for hSERT and hNET in 96 well format and in a final assay volume of 400μl,
except for the use of hDAT-LLCPK cell membranes (1 :20 i.e. 9.6μg of protein/well) and [3H]WI N-35,428 as radioligand (1OnM [N-Methyl-3H]WIN-35,428, Perkin Elmer, 85.6 Ci/mmol). Furthermore, 10μM GBR-12909 is used for NSB and the incubation time of the binding reaction is 1 hour at room temperature.
b) Scintillation Proximity Assay (SPA) for human DAT, NET and SERT binding
- Generation of BacMam viruses for the expression of hSERT, hNET, and hDAT in mammalian cells
Membranes for the SPA-binding assays are produced by HEK-293F cell infection with BacMam viruses generated for each single human SERT, NET, and DAT transporter. hSERT and hDAT are cloned into pFBMRfA vector whereas hNET is cloned into pFASTBacMami vector. The generation and use of BacMam viruses is described in Condreay JP et al, Proc. Natl. Acad. Sci. USA, 1999, 96:127-132 and Hassan NJ et al, Protein Expression and Purification, 47(2): 591-598, 2006.
- Transduction of HEK-293F cells with hSERT/hDAT/hNET BacMam viruses
The HEK-293F suspension cell line (Invitrogen) is routinely grown in 293_Freestyle Expression media (Invitrogen) in shake flask suspension culture. The culture is transduced with the appropriate transporter BacMam at a MOI (multiplicity of infection) of 100 virus particles per cell and incubated for 48hrs at 370C, 5% CO2 in air, shaken at 90rpm in a humidified shaker incubator. The culture is then harvested by centrifugation at 100Og, 40C, for 10 minutes and the cell pellet stored at -8O0C until required.
- Preparation of BacMam hSERT/hDAT/hNET-HEL293F cell membranes
Transduced cell pellets are re-suspended to 10x volume with buffer-A (5OmM HEPES, 1 mM EDTA, 1 mM leupeptin, 25ug/ml_ bacitracin, 1 mM phenylmethylsulfonylfluoride, PMSF, 2μM pepstatin A, pH 7.7) and homogenised with 2x 15 second bursts in a glass Waring blender. The homogenate is then centrifuged for 20 minutes at 50Og. Following this, the supernatant is pooled and centrifuged at 13,00Og for 30 minutes. Pellets are then re-suspended to 4x original pellet volume with buffer-B (5OmM TRIS pH 7.4, 13OmM NaCI) and forced through a 0.8mm needle to give a homogeneous suspension. Membrane aliquots are stored at -8O0C until required. The protein concentration is quantified by Bradford assay.
- Assay Protocol
The affinity of the compounds of the invention for hSERT, hNET or hDAT may also be assessed by using the [3H]citalopram, [3H]nisoxetine or [3H]WI N-35, 428 binding assays with the SPA technology on BacMam-recombinant human SERT, NET and DAT membranes produced as described before. With the SPA technology (GE Healthcare, Amersham) only transporter-bound radioactivity can elicit bead excitation thus no separation of the bound/ unbound radioligand is required.
The protocol for hSERT binding SPA is based on Trilux beta-counter (Wallac, Perkin- Elmer). Briefly, 0.5μl_ of test compound in neat DMSO (or 1 μM fluoxetine as positive control) is added by 50μl_ of the SPA mixture, containing 2mg/ml_ SPA beads
(Amersham RPNQ0001 ), 4μg/ml_ hSERT Bacmam membranes, 0.01% pluronic F- 127, 2.5nM [3H]citalopram in the assay buffer (2OmM HEPES, 145mM NaCI, 5mM KCI, pH 7.3). Incubation are performed at room temperature for at least 2 hours. Counts are stable and could be read up to 3 days.
Alternatively, hDAT hNET and hSERT SPA-binding assays are performed by using a Viewlux beta-counter (Wallac, Perkin-Elmer) with imaging PS-WGA beads (Amersham RPNQ0260) in a final assay volume of 30μl_ and in a 384-well plate format (Greiner 781075). Briefly, 0.3μl_ of test compound in neat DMSO and 0% and 100% effect controls (DMSO for total binding and 10 or 1 μM indatraline as positive control) are added to the wells by using a Hummingbird (Genomic Solutions), followed by the addition of 30μl_ of the SPA mixture, containing 1 mg/mL SPA beads (hSERT) or 2mg/ml SPA beads (hDAT and hNET), 40μg/ml or 20μg/ml or 6 μg/ml of hDAT or hNET or hSERT BacMam membranes, 0.02% pluronic F-127, 1OnM [3H]WI N-35,428 or 1OnM [3H]nisoxetine or 3nM [3H]citalopram for hDAT or hNET or hSERT binding SPA in the assay buffer (2OmM HEPES, 145mM NaCI, 5mM KCI, pH 7.3-7.4). Incubation is performed at room temperature for at least 2 hours, best overnight in the dark. Bound radioactivity is recorded by using a 600s 6x binning and 613nm emission filter with the Viewlux instrument.
Calculation
The affinity of the compounds of the invention for a particular transporter may be calculated from the IC5O obtained in competition experiments as the concentration of a compound necessary to displace 50% of the radiolabeled ligand from the transporter, and is reported as a "K," value calculated by the following equation:
IC50
K1 =
1 + L / KD where L = radioligand and KD = affinity of radioligand for transporter (Cheng and Prusoff, Biochem. Pharmacol. 22:3099, 1973). Results are presented below as the pKi (i.e. the antilogarithm of Ki).
A number of compounds supporting the invention have been prepared (see below) and may be tested in assay a) and/or b). The following compounds supporting the invention were tested in assay b) 1 , 2, 5-7, 10-12, 15-18, 20, 24-26, 29, 30, 35-39, 42, 43, 47-65, 82-91 , 94-126 and 137-140. These compounds gave a pKi against SERT from 7.2 to 10; against NET from 5.0 to 9.4; and against DAT from 6.2 to 9.8.
The compounds of the invention may be used to treat diseases or conditions mediated by inhibition of monoamine neurotransmitter re-uptake, i.e. inhibition of one or more of SERT, hNET and hDAT. Therefore according to a further aspect, the invention provides a compound of the invention for use in treating a disease or condition. In an embodiment the disease or condition is a human disease or condition. In an embodiment the disease or condition is mediated by inhibition of monoamine neurotransmitter re-uptake, i.e. inhibition of one or more of SERT, hNET and hDAT.
In an embodiment the disease or condition mediated by inhibition of monoamine neurotransmitter re-uptake, i.e. inhibition of one or more of SERT, hNET and hDAT, is selected from the list consisting of: Parkinsonism, depression, eating disorders, sleep disorders, substance related disorders, attention-deficit hyperactivity disorders, anxiety disorders, cognition impairment, sexual dysfunctions, obsessive compulsive spectrum disorders, Gilles de Ia Tourettes disease and senile dementia, as well as other disorders sensitive to the monoamine neurotransmitter re-uptake-inhibiting activity of the compounds.
The terms describing some indications used hereinabove are classified in the
Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, published by the American Psychiatric Association (DSM-IV) and/or the International Classification of Diseases, 10th Edition (ICD-10). The various subtypes of the disorders mentioned hereinabove are contemplated as part of the present invention. Numbers in brackets after the listed diseases below refer to the classification code in DSM-IV.
The term "depression" includes:
Depression and mood disorders including Major Depressive Episode, Manic Episode, Mixed Episode and Hypomanic Episode; Depressive Disorders including Major Depressive Disorder, Dysthymic Disorder (300.4), Depressive Disorder Not Otherwise Specified (311 ); Other Mood Disorders including Mood Disorder Due to a General Medical Condition (293.83) which includes the subtypes With Depressive Features, With Major Depressive-like Episode, With Manic Features and With Mixed Features), Substance-Induced Mood Disorder (including the subtypes With
Depressive Features, With Manic Features and With Mixed Features) and Mood Disorder Not Otherwise Specified (296.90). Bipolar Disorders including Bipolar I Disorder, Bipolar Il Disorder (Recurrent Major Depressive Episodes with Hypomanic Episodes) (296.89), Cyclothymic Disorder (301.13) and Bipolar Disorder Not Otherwise Specified (296.80);
The term "anxiety disorders" includes:
Anxiety disorders including Panic Attack; Panic Disorder including Panic Disorder without Agoraphobia (300.01 ) and Panic Disorder with Agoraphobia (300.21 ); Agoraphobia; Agoraphobia Without History of Panic Disorder (300.22), Specific Phobia (300.29, formerly Simple Phobia) including the subtypes Animal Type, Natural Environment Type, Blood-lnjection-lnjury Type, Situational Type and Other Type), Social Phobia (Social Anxiety Disorder, 300.23), Obsessive-Compulsive Disorder (300.3), Posttraumatic Stress Disorder (309.81 ), Acute Stress Disorder (308.3), Generalized Anxiety Disorder (300.02), Anxiety Disorder Due to a General Medical Condition (293.84), Substance-Induced Anxiety Disorder, Separation Anxiety Disorder (309.21 ), Adjustment Disorders with Anxiety (309.24) and Anxiety Disorder Not Otherwise Specified (300.00):
The term "substance related disorder" includes:
Substance-related disorders including Substance Use Disorders such as Substance Dependence, Substance Craving and Substance Abuse; Substance-Induced Disorders such as Substance Intoxication, Substance Withdrawal, Substance- Induced Delirium, Substance-Induced Persisting Dementia, Substance-Induced
Persisting Amnestic Disorder, Substance-Induced Psychotic Disorder, Substance- Induced Mood Disorder, Substance-Induced Anxiety Disorder, Substance-Induced Sexual Dysfunction, Substance-Induced Sleep Disorder and Hallucinogen Persisting Perception Disorder (Flashbacks); Alcohol-Related Disorders such as Alcohol Dependence (303.90), Alcohol Abuse (305.00), Alcohol Intoxication (303.00), Alcohol Withdrawal (291.81 ), Alcohol Intoxication Delirium, Alcohol Withdrawal Delirium, Alcohol-Induced Persisting Dementia, Alcohol-Induced Persisting Amnestic Disorder, Alcohol-Induced Psychotic Disorder, Alcohol-Induced Mood Disorder, Alcohol- Induced Anxiety Disorder, Alcohol-Induced Sexual Dysfunction, Alcohol-Induced Sleep Disorder and Alcohol-Related Disorder Not Otherwise Specified (291.9); Amphetamine (or Amphetamine-I_ike)-Related Disorders such as Amphetamine Dependence (304.40), Amphetamine Abuse (305.70), Amphetamine Intoxication (292.89), Amphetamine Withdrawal (292.0), Amphetamine Intoxication Delirium, Amphetamine Induced Psychotic Disorder, Amphetamine-Induced Mood Disorder, Amphetamine-Induced Anxiety Disorder, Amphetamine-Induced Sexual Dysfunction, Amphetamine-Induced Sleep Disorder and Amphetamine-Related Disorder Not Otherwise Specified (292.9); Caffeine Related Disorders such as Caffeine Intoxication (305.90), Caffeine-Induced Anxiety Disorder, Caffeine-Induced Sleep Disorder and Caffeine-Related Disorder Not Otherwise Specified (292.9); Cannabis- Related Disorders such as Cannabis Dependence (304.30), Cannabis Abuse
(305.20), Cannabis Intoxication (292.89), Cannabis Intoxication Delirium, Cannabis- lnduced Psychotic Disorder, Cannabis-lnduced Anxiety Disorder and Cannabis- Related Disorder Not Otherwise Specified (292.9); Cocaine-Related Disorders such as Cocaine Dependence (304.20), Cocaine Abuse (305.60), Cocaine Intoxication (292.89), Cocaine Withdrawal (292.0), Cocaine Intoxication Delirium, Cocaine- Induced Psychotic Disorder, Cocaine-Induced Mood Disorder, Cocaine-Induced Anxiety Disorder, Cocaine-Induced Sexual Dysfunction, Cocaine-Induced Sleep Disorder and Cocaine-Related Disorder Not Otherwise Specified (292.9); Hallucinogen-Related Disorders such as Hallucinogen Dependence (304.50), Hallucinogen Abuse (305.30), Hallucinogen Intoxication (292.89), Hallucinogen Persisting Perception Disorder (Flashbacks) (292.89), Hallucinogen Intoxication Delirium, Hallucinogen-Induced Psychotic Disorder, Hallucinogen-Induced Mood Disorder, Hallucinogen-Induced Anxiety Disorder and Hallucinogen-Related Disorder Not Otherwise Specified (292.9); Inhalant-Related Disorders such as Inhalant Dependence (304.60), Inhalant Abuse (305.90), Inhalant Intoxication (292.89), Inhalant Intoxication Delirium, Inhalant-Induced Persisting Dementia, Inhalant-
Induced Psychotic Disorder, Inhalant-Induced Mood Disorder, Inhalant-Induced Anxiety Disorder and Inhalant-Related Disorder Not Otherwise Specified (292.9); Nicotine-Related Disorders such as Nicotine Dependence (305.1 ), Nicotine Withdrawal (292.0) and Nicotine-Related Disorder Not Otherwise Specified (292.9); Opioid-Related Disorders such as Opioid Dependence (304.00), Opioid Abuse
(305.50), Opioid Intoxication (292.89), Opioid Withdrawal (292.0), Opioid Intoxication Delirium, Opioid-lnduced Psychotic Disorder, Opioid-lnduced Mood Disorder, Opioid- lnduced Sexual Dysfunction, Opioid-lnduced Sleep Disorder and Opioid-Related Disorder Not Otherwise Specified (292.9); Phencyclidine (or Phencyclidine-Like)- Related Disorders such as Phencyclidine Dependence (304.60), Phencyclidine Abuse (305.90), Phencyclidine Intoxication (292.89), Phencyclidine Intoxication Delirium, Phencyclidine-lnduced Psychotic Disorder, Phencyclidine-lnduced Mood Disorder, Phencyclidine-lnduced Anxiety Disorder and Phencyclidine-Related Disorder Not Otherwise Specified (292.9); Sedative-, Hypnotic-, or Anxiolytic-Related Disorders such as Sedative, Hypnotic, or Anxiolytic Dependence (304.10), Sedative, Hypnotic, or Anxiolytic Abuse (305.40), Sedative, Hypnotic, or Anxiolytic Intoxication (292.89), Sedative, Hypnotic, or Anxiolytic Withdrawal (292.0), Sedative, Hypnotic, or Anxiolytic Intoxication Delirium, Sedative, Hypnotic, or Anxiolytic Withdrawal Delirium, Sedative-, Hypnotic-, or Anxiolytic-Persisting Dementia, Sedative-, Hypnotic-, or Anxiolytic- Persisting Amnestic Disorder, Sedative-, Hypnotic-, or Anxiolytic-lnduced Psychotic Disorder, Sedative-, Hypnotic-, or Anxiolytic-lnduced Mood Disorder, Sedative-, Hypnotic-, or Anxiolytic-lnduced Anxiety Disorder Sedative-, Hypnotic-, or Anxiolytic-lnduced Sexual Dysfunction, Sedative-, Hypnotic-, or Anxiolytic-lnduced Sleep Disorder and Sedative-, Hypnotic-, or Anxiolytic-Related Disorder Not Otherwise Specified (292.9); Polysubstance-Related Disorder such as Polysubstance Dependence (304.80); and Other (or Unknown) Substance-Related Disorders such as Anabolic Steroids, Nitrate Inhalants and Nitrous Oxide;
The term "Sleep disorder" includes:
Sleep disorders including primary sleep disorders such as Dyssomnias such as Primary Insomnia (307.42), Primary Hypersomnia (307.44), Narcolepsy (347), Breathing-Related Sleep Disorders (780.59), Circadian Rhythm Sleep Disorder (307.45) and Dyssomnia Not Otherwise Specified (307.47); primary sleep disorders such as Parasomnias such as Nightmare Disorder (307.47), Sleep Terror Disorder (307.46), Sleepwalking Disorder (307.46) and Parasomnia Not Otherwise Specified
(307.47); Sleep Disorders Related to Another Mental Disorder such as Insomnia Related to Another Mental Disorder (307.42) and Hypersomnia Related to Another Mental Disorder (307.44); Sleep Disorder Due to a General Medical Condition; and Substance-Induced Sleep Disorder including the subtypes Insomnia Type, Hypersomnia Type, Parasomnia Type and Mixed Type;
The term "eating disorder" includes:
Eating disorders such as Anorexia Nervosa (307.1 ) including the subtypes Restricting Type and Binge-Eating/Purging Type; Bulimia Nervosa (307.51 ) including the subtypes Purging Type and Nonpurging Type; Obesity; Compulsive Eating Disorder; Binge Eating Disorder; and Eating Disorder Not Otherwise Specified (307.50):
The term "Attention-Deficit/Hyperactivity Disorder" includes:
Attention-Deficit/Hyperactivity Disorder including the subtypes Attention-Deficit /Hyperactivity Disorder Combined Type (314.01 ), Attention-Deficit /Hyperactivity Disorder Predominantly Inattentive Type (314.00), Attention-Deficit /Hyperactivity Disorder Hyperactive-Impulse Type (314.01 ) and Attention-Deficit /Hyperactivity Disorder Not Otherwise Specified (314.9); Hyperkinetic Disorder; Disruptive Behaviour Disorders such as Conduct Disorder including the subtypes childhood- onset type (321.81 ), Adolescent-Onset Type (312.82) and Unspecified Onset (312.89), Oppositional Defiant Disorder (313.81 ) and Disruptive Behaviour Disorder Not Otherwise Specified; and Tic Disorders such as Tourette's Disorder (307.23);
The term "Cognition impairment" includes:
Cognition impairment including cognition impairment in other diseases such as schizophrenia, bipolar disorder, depression, other psychiatric disorders and psychotic conditions associated with cognitive impairment, e.g. Alzheimer's disease;
The term "Sexual dysfunctions" includes:
Sexual dysfunctions including Sexual Desire Disorders such as Hypoactive Sexual Desire Disorder (302.71 ), and Sexual Aversion Disorder (302.79); sexual arousal
disorders such as Female Sexual Arousal Disorder (302.72) and Male Erectile Disorder (302.72); orgasmic disorders such as Female Orgasmic Disorder (302.73), Male Orgasmic Disorder (302.74) and Premature Ejaculation (302.75); sexual pain disorder such as Dyspareunia (302.76) and Vaginismus (306.51 ); Sexual Dysfunction Not Otherwise Specified (302.70); paraphilias such as Exhibitionism (302.4), Fetishism (302.81 ), Frotteurism (302.89), Pedophilia (302.2), Sexual Masochism (302.83), Sexual Sadism (302.84), Transvestic Fetishism (302.3), Voyeurism (302.82) and Paraphilia Not Otherwise Specified (302.9); gender identity disorders such as Gender Identity Disorder in Children (302.6) and Gender Identity Disorder in Adolescents or Adults (302.85); and Sexual Disorder Not Otherwise Specified (302.9);
The term Obsessive compulsive spectrum disorder" includes:
Obsessive compulsive spectrum disorder including Obsessive compulsive disorders (300.3), somatoform disorders including body dysmorphic disorder (300.7) and hyperchondriasis (300.7), bulimia nervosa (307.51 ), anorexia nervosa (307.1 ), eating disorders not elsewhere classified (307.50) such as binge eating, impulse control disorders not elsewhere classified (including intermitted explosive disorder (312.34), compulsive buying or shopping, repetitive self-mutilation, onychophagia, psychogenic excoriation, kleptomania (312.32), pathological gambling (312.31 ), trichotillomania (312.39) and internet addiction), paraphilia (302.70) and nonparaphilic sexual addictions, Sydeham's chorea, torticollis, autistic disorders (299.0), compulsive hoarding, and movement disorders, including Tourette's syndrome (307.23).
All of the various forms and sub-forms of the disorders mentioned herein are contemplated as part of the present invention.
In an embodiment, compounds of the invention may be useful as analgesics. For example they may be useful in the treatment of chronic inflammatory pain (e.g. pain associated with rheumatoid arthritis, osteoarthritis, rheumatoid spondylitis, gouty arthritis and juvenile arthritis); musculoskeletal pain; lower back and neck pain; sprains and strains; neuropathic pain; sympathetically maintained pain; myositis; pain associated with cancer and fibromyalgia; pain associated with migraine; pain associated with influenza or other viral infections, such as the common cold; rheumatic fever; pain associated with functional bowel disorders such as non-ulcer
dyspepsia, non-cardiac chest pain and irritable bowel syndrome; pain associated with myocardial ischemia; post operative pain; headache; toothache; and dysmenorrhea.
Compounds of the invention may be useful in the treatment of neuropathic pain. Neuropathic pain syndromes can develop following neuronal injury and the resulting pain may persist for months or years, even after the original injury has healed. Neuronal injury may occur in the peripheral nerves, dorsal roots, spinal cord or certain regions in the brain. Neuropathic pain syndromes are traditionally classified according to the disease or event that precipitated them. Neuropathic pain syndromes include: diabetic neuropathy; sciatica; non-specific lower back pain; multiple sclerosis pain; fibromyalgia; HIV-related neuropathy; post-herpetic neuralgia; trigeminal neuralgia; and pain resulting from physical trauma, amputation, cancer, toxins or chronic inflammatory conditions. These conditions are difficult to treat and although several drugs are known to have limited efficacy, complete pain control is rarely achieved. The symptoms of neuropathic pain are incredibly heterogeneous and are often described as spontaneous shooting and lancinating pain, or ongoing, burning pain. In addition, there is pain associated with normally non-painful sensations such as "pins and needles" (paraesthesias and dysesthesias), increased sensitivity to touch (hyperesthesia), painful sensation following innocuous stimulation (dynamic, static or thermal allodynia), increased sensitivity to noxious stimuli (thermal, cold, mechanical hyperalgesia), continuing pain sensation after removal of the stimulation (hyperpathia) or an absence of or deficit in selective sensory pathways (hypoalgesia).
Compounds of the invention may also be useful in the amelioration of inflammatory disorders, for example in the treatment of skin conditions (e.g. sunburn, burns, eczema, dermatitis, psoriasis); ophthalmic diseases such as glaucoma, retinitis, retinopathies, uveitis and of acute injury to the eye tissue (e.g. conjunctivitis); lung disorders (e.g. asthma, bronchitis, emphysema, allergic rhinitis, respiratory distress syndrome, pigeon fancier's disease, farmer's lung, chronic obstructive pulmonary disease, (COPD); gastrointestinal tract disorders (e.g. aphthous ulcer, Crohn's disease, atopic gastritis, gastritis varialoforme, ulcerative colitis, coeliac disease, regional ileitis, irritable bowel syndrome, inflammatory bowel disease, gastroesophageal reflux disease); other conditions with an inflammatory component such as migraine, multiple sclerosis, myocardial ischemia.
In one embodiment, compounds of the invention are useful in the treatment of depression and anxiety disorders.
In another embodiment, compounds of the invention are useful in the treatment of depression.
"Treatment" includes prophylaxis, where this is appropriate for the relevant condition(s).
The compounds of the invention may also be used in combination with other therapeutic agents. The invention thus provides, in a further aspect, a combination comprising a compound of the invention together with a further therapeutic agent.
The compounds of the invention may be used in combination with the following agents to treat or prevent psychotic disorders: i) antipsychotics; ii) drugs for extrapyramidal side effects, for example anticholinergics (such as benztropine, biperiden, procyclidine and trihexyphenidyl), antihistamines (such as diphenhydramine) and dopaminergics (such as amantadine); iii) antidepressants; iv) anxiolytics; and v) cognitive enhancers for example cholinesterase inhibitors (such as tacrine, donepezil, rivastigmine and galantamine).
The compounds of the invention may be used in combination with antidepressants to treat or prevent depression and mood disorders.
The compounds of the invention may be used in combination with the following agents to treat or prevent bipolar disease: i) mood stabilisers; ii) antipsychotics; and iii) antidepressants.
The compounds of the invention may be used in combination with the following agents to treat or prevent anxiety disorders: i) anxiolytics; and ii) antidepressants.
The compounds of the invention may be used in combination with the following agents to improve nicotine withdrawal and reduce nicotine craving: i) nicotine replacement therapy for example a sublingual formulation of nicotine beta- cyclodextrin and nicotine patches; and ii) bupropion.
The compounds of the invention may be used in combination with the following agents to improve alcohol withdrawal and reduce alcohol craving: i) NMDA receptor antagonists for example acamprosate; ii) GABA receptor agonists for example tetrabamate; and iii) Opioid receptor antagonists for example naltrexone.
The compounds of the invention may be used in combination with the following agents to improve opiate withdrawal and reduce opiate craving: i) opioid mu receptor agonist/opioid kappa receptor antagonist for example buprenorphine; ii) opioid receptor antagonists for example naltrexone; and iii) vasodilatory antihypertensives for example lofexidine.
The compounds of the invention may be used in combination with the following agents to treat or prevent sleeping disorders: i) benzodiazepines for example temazepam, lormetazepam, estazolam and triazolam; ii) non-benzodiazepine hypnotics for example Zolpidem, zopiclone, zaleplon and indiplon; iii) barbiturates for example aprobarbital, butabarbital, pentobarbital, secobarbita and phenobarbital; iv) antidepressants; v) other sedative-hypnotics for example chloral hydrate and chlormethiazole.
The compounds of the invention may be used in combination with the following agents to treat anorexia: i) appetite stimulants for example cyproheptidine; ii) antidepressants; iii) antipsychotics; iv) zinc; and v) premenstral agents for example pyridoxine and progesterones.
The compounds of the invention may be used in combination with the following agents to treat or prevent bulimia: i) antidepressants; ii) opioid receptor antagonists; iii) antiemetics for example ondansetron; iv) testosterone receptor antagonists for example flutamide; v) mood stabilisers; vi) zinc; and vii) premenstral agents.
The compounds of the invention may be used in combination with the following agents to treat or prevent autism: i) antipsychotics; ii) antidepressants; iii) anxiolytics; and iv) stimulants for example methylphenidate, amphetamine formulations and pemoline.
The compounds of the invention may be used in combination with the following agents to treat or prevent ADHD: i) stimulants for example methylphenidate, amphetamine formulations and pemoline; and ii) non-stimulants for example norepinephrine reuptake inhibitors (such as atomoxetine), alpha 2 adrenoceptor agonists (such as clonidine), antidepressants, modafinil, and cholinesterase inhibitors (such as galantamine and donezepil).
The compounds of the invention may be used in combination with the following agents to treat personality disorders: i) antipsychotics; ii) antidepressants; iii) mood stabilisers; and iv) anxiolytics.
The compounds of the invention may be used in combination with the following agents to treat or prevent male sexual dysfunction: i) phosphodiesterase V inhibitors, for example vardenafil and sildenafil; ii) dopamine agonists/dopamine transport inhibitors for example apomorphine and buproprion; iii) alpha adrenoceptor antagonists for example phentolamine; iv) prostaglandin agonists for example alprostadil; v) testosterone agonists such as testosterone; vi) serotonin transport inhibitors for example serotonin reuptake inhibitors; vii) noradrenaline transport inhibitors for example reboxetine and viii) 5-HT1A agonists, for example flibanserine.
The compounds of the invention may be used in combination with the same agents specified for male sexual dysfunction to treat or prevent female sexual dysfunction, and in addition an estrogen agonist such as estradiol.
Antipsychotic drugs include Typical Antipsychotics (for example chlorpromazine, thioridazine, mesoridazine, fluphenazine, perphenazine, prochlorperazine, trifluoperazine, thiothixine, haloperidol, molindone and loxapine); and Atypical Antipsychotics (for example clozapine, olanzapine, risperidone, quetiapine, aripirazole, ziprasidone and amisulpride).
Antidepressant drugs include serotonin reuptake inhibitors (such as citalopram, escitalopram, fluoxetine, paroxetine and sertraline); dual serotonin/noradrenaline reuptake inhibitors (such as venlafaxine, duloxetine and milnacipran); Noradrenaline reuptake inhibitors (such as reboxetine); tricyclic antidepressants (such as amitriptyline, clomipramine, imipramine, maprotiline, nortriptyline and trimipramine); monoamine oxidase inhibitors (such as isocarboxazide, moclobemide, phenelzine
and tranylcypromine); and others (such as bupropion, mianserin, mirtazapine, nefazodone and trazodone).
Mood stabiliser drugs include lithium, sodium valproate/valproic acid/divalproex, carbamazepine, lamotrigine, gabapentin, topiramate and tiagabine.
Anxiolytics include benzodiazepines such as alprazolam and lorazepam.
The compounds of the invention will normally, but not necessarily, be formulated into pharmaceutical compositions prior to administration to a patient by an appropriate route. Accordingly, in another aspect, the invention provides pharmaceutical compositions comprising a compound of the invention and one or more pharmaceutically-acceptable excipients.
As used herein, "pharmaceutically acceptable excipient" means any pharmaceutically acceptable material present in the pharmaceutical composition or dosage form other than the compound or compounds of the invention. Typically the material gives form, consistency and performance to the pharmaceutical composition.
The pharmaceutical compositions of the invention typically contain one compound of the invention. However, in certain embodiments, the pharmaceutical compositions of the invention contain more than one compound of the invention. In addition, the pharmaceutical compositions of the invention may comprise one or more additional pharmaceutically active compounds.
Such pharmaceutical compositions of the invention may be prepared and packaged in bulk form wherein a safe and effective amount of a compound of the invention can be dispensed and then given to the patient such as with powders or syrups. Alternatively, the pharmaceutical compositions of the invention may be prepared and packaged as dosage forms wherein each physically discrete dosage form contains a safe and effective amount of a compound of the invention. Accordingly, in another aspect, the invention provides dosage forms comprising pharmaceutical compositions of the invention. Each discrete dosage form typically contains from 1 mg to 500 mg of a compound of the invention.
The compositions of the invention will typically be formulated into dosage forms which are adapted for administration to the patient by the desired route of administration. For example, dosage forms include those adapted for (1 ) oral administration such as tablets, capsules, caplets, pills, lozenges, powders, syrups, elixirs, suspensions, solutions, emulsions, sachets and cachets; (2) parenteral administration such as sterile solutions, suspensions, implants and powders for reconstitution; (3) transdermal administration such as transdermal patches; (4) rectal and vaginal administration such as suppositories, pessaries and foams; (5) inhalation and intranasal such as dry powders, aerosols, suspensions and solutions (sprays and drops); (6) topical administration such as creams, ointments, lotions, solutions, pastes, drops, sprays, foams and gels; (7) ocular administration such as drops, ointment, sprays, suspensions and inserts; (8) buccal and sublingual administration such as lozenges, patches, sprays, drops, chewing gums and tablets.
Suitable pharmaceutically acceptable excipients will vary depending upon the particular dosage form chosen. In addition, suitable pharmaceutically acceptable excipients may be chosen for a particular function that they may serve in the composition. For example, certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of uniform dosage forms. Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of stable dosage forms. Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the carrying or transporting of the compound or compounds of the invention once administered to the patient from one organ, or portion of the body, to another organ, or portion of the body. Certain pharmaceutically acceptable excipients may be chosen for their ability to enhance patient compliance. Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the release of the compound of the invention at the appropriate rate to treat the condition.
Suitable pharmaceutically acceptable excipients include the following types of excipients: diluents, fillers, binders, disintegrants, lubricants, glidants, granulating agents, coating agents, wetting agents, solvents, co-solvents, suspending agents, emulsifiers, sweeteners, flavouring agents, flavour masking agents, colouring agents, anticaking agents, humectants, chelating agents, plasticizers, viscosity increasing agents, rate modifying agents, antioxidants, preservatives, stabilizers, surfactants and buffering agents. The skilled artisan will appreciate that certain pharmaceutically
acceptable excipients may serve more than one function and may serve alternative functions depending on how much of the excipient is present in the formulation and what other ingredients are present in the formulation.
Skilled artisans possess the knowledge and skill in the art to enable them to determine suitable pharmaceutically acceptable excipients in appropriate amounts for use with the compounds of the invention. In addition, there are a number of resources that are available to the skilled artisan which describe pharmaceutically acceptable excipients and may be useful in selecting suitable pharmaceutically acceptable excipients. Examples include Remington's Pharmaceutical Sciences (Mack Publishing Company), The Handbook of Pharmaceutical Additives (Gower Publishing Limited), and The Handbook of Pharmaceutical Excipients (the American Pharmaceutical Association and the Pharmaceutical Press). The pharmaceutical compositions of the invention may be prepared using techniques and methods known to those skilled in the art. Some of the methods commonly used in the art are described in Remington's Pharmaceutical Sciences (Mack Publishing Company).
It will be appreciated that the compounds of the combination or composition may be administered simultaneously (either in the same or different pharmaceutical formulations), separately or sequentially.
All publications, including, but not limited to, patents and patent applications cited in this specification, are herein incorporated by reference as if each individual publication were specifically and individually indicated to be incorporated by reference herein as though fully set forth.
It will be appreciated that the invention includes the following further aspects. The diseases and conditions described above extend, where appropriate, to these further aspects.
i) The use of a compound of the invention in the manufacture of a medicament in treating a disease or condition mediated by inhibition of monoamine neurotransmitter re-uptake, i.e. inhibition of one or more of SERT, hNET and hDAT. In an embodiment the disease or condition is depression or an anxiety disorder.
ii) A method of treating a disease or condition mediated by inhibition of monoamine neurotransmitter re-uptake, i.e. inhibition of one or more of SERT, hNET and hDAT in a mammal comprising administering an effective amount of a compound of the invention. In an embodiment the disease or condition is a depression or an anxiety disorder.
Supporting Compounds
The preparation of a number of the compounds of the invention are described below.
In the procedures that follow, after each starting material, reference to an intermediate is typically provided. This is provided merely for assistance to the skilled chemist. The starting material may not necessarily have been prepared from the batch referred to.
Compounds of the invention and intermediates are named using ACD/Name PRO 6.02 chemical naming software (Advanced Chemistry Development Inc., Toronto, Ontario, M5H2L3, Canada).
Where reference is made to the use of a "similar" or "analogous" procedure, as will be appreciated by those skilled in the art, such a procedure may involve minor variation, for example reaction temperature, reagent/solvent amount, reaction time, work-up conditions or chromatographic purification conditions.
All temperatures refer to 0C.
Proton Magnetic Resonance (^ H NMR) spectra are typically recorded either on a Varian instrument at 300, 400 or 500 MHz or on a Bruker instrument at 300 and 400 MHz. Chemical shifts are reported in ppm (δ) using the residual solvent line as internal standard. Splitting patterns are designed as s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; b, broad. The NMR spectra were recorded at a temperature ranging from 25 to 9O0C. When more than one conformer is detected the chemical shifts for the most abundant one is reported.
Mass spectra (MS) are typically taken on a 4 Il triple quadrupole Mass Spectrometer (Micromass UK) or on a Agilent MSD 1100 Mass Spectrometer, operating in ES (+) and ES (-) ionization mode or on an Agilent LC/MSD 1100 Mass Spectrometer,
operating in ES (+) and ES (-) ionization mode coupled with HPLC instrument Agilent 1100 Series [LC/MS - ES (+):analysis performed on a Supelcosil ABZ +Plus (33x4.6 mm, 3μm) (mobile phase: 100% [water +0.1% HCO2H] for 1 min, then from 100% [water +0.1% HCO2H] to 5% [water +0.1% HCO2H] and 95% [CH3CN ] in 5 min, finally under these conditions for 2 min; T=40°C; flux= 1 mL/min; LC/MS - ES
(-):analysis performed on a Supelcosil ABZ +Plus (33x4.6 mm, 3μm) (mobile phase: 100% [water +0.05% NH3] for 1 min, then from 100% [water +0.05% NH3 to 5% [water +0.05% NH3] and 95% [CH3CN ] in 5 min, finally under these conditions for 2 min; T=40°C; flux= 1 mL/min] ; in the mass spectra only one peak in the molecular ion cluster is reported.
DAD chromatographic traces, mass chromatograms and mass spectrums may be taken on a on a UPLC/MS AcquityTM system coupled with a Micromass ZQTM mass spectrometer operating in ESI positive or negative. The phases used are: A) H2O/ACN 95/5 + 0,1 % TFA; B) H2O/ACN 5/95 + 0,1% TFA. The gradient is: t=0min) 95%A 5%B, t=0,25) 95%A 5%B, t=3,30) 100%B, t=4,0) 100%B, followed by 1 min of reconditioning.
Flash silica gel chromatography was carried out on silica gel 230-400 mesh (supplied by Merck AG Darmstadt, Germany) or over Varian Mega Be-Si pre-packed cartridges or over pre-packed Biotage silica cartridges.
SPE-SCX cartridges are ion exchange solid phase extraction columns supplied by Varian. The eluent used with SPE-SCX cartridges is methanol followed by 2N ammonia solution in methanol.
In a number of preparations, purification was performed using either Biotage manual flash chromatography (Flash+) or automatic flash chromatography (Horizon, SP1 ) systems. All these instruments work with Biotage Silica cartridges.
SPE-Si cartridges are silica solid phase extraction columns supplied by Varian.
A number of the supporting compounds have been prepared as racemic mixtures and a number have been prepared as single enantiomers. The absolute
stereochemistry of those compounds prepared as single enantiomers have not been assigned, but may be assigned using ab initio vibrational circular dichroism (VCD).
Chiral molecules exhibit VCD. VCD is the differentia! interaction of a chiral molecule with left and right circularly polarized infrared radiation during vibrational excitation.
The VCD spectrum of a chiral molecule is dependent on its three-dimensional structure. Most importantly, the VCD spectrum of a chiral molecule is a function of its absolute configuration and, in the case of flexible molecules, of its conformation. In principle, therefore, VCD permits the determination of the structure of a chiral molecule. VCD spectra were first measured in the 1970s. Subsequently, VCD instrumentation has developed enormously in spectral range and in sensitivity. Currently, VCD spectra of liquids and solutions can be measured over the majority of the fundamental infrared (IR) spectral range (v≥ 650 cm-1 ) with high sensitivity at acceptable resolution (1-5 cm-1 ) using both dispersive and Fourier Transform (FT) VCD instrumentation. Very recently, commercial FT VCD instrumentation has become available, greatly enhancing the accessibility of VCD spectra.
The use of VCD as a reliable method for the determination of absolute configuration of chiral molecules is now well established (see for example Shah RD, et al.. Curr Opin Drug Disc Dev 2001 ;4:764-774; Freedman TB, et al., HeIv Chim Acta 2002; 85:1160-1 165; Dyatkin AB, et al. Chirality 2002;14:215-219; Solladie'-Cavallo A, Balaz Met al., Tetrahedron Assym 2001 ;12:2605-2611 ; Nafie LA, et al. Circular dichroism, principles and applications, 2nd ed. New York: John Wiley & Sons; 2000. p 97-131 ; Nafie LA, et al. in: Yan B, Gremlish H-U, editors. Infrared and Raman spectroscopy of biological materials. New York: Marcel Dekker; 2001. p 15-54; Polavarapu PL, et al., J Anal Chem 2000;366:727-734; Stephens PJ, et al., Chirality 2000:12:172-179; Solladie' -Cavallo A, et al., Eur J Org Chem 2002: 1788-1796).
The method entails comparison of observed IR and VCD spectra with calculations of the spectra for a specific configuration and provides information both on the absolute configuration and on the solution conformation.
Given an experimental spectrum of a chiral molecule whose absolute configuration and/or conformation are unknown and to be determined, the general procedure is as follows: 1 ) all possible structures are defined; 2) the spectra of these structures are
predicted; and 3) predicted spectra are compared to the experimental spectrum. The correct structure will give a spectrum in agreement with experiment; incorrect structures will give spectra in disagreement with experiment.
VCD spectra are always measured simultaneously with vibrational unpolarized absorption spectra ("infrared (IR) spectra") and the two vibrational spectra together provide more information than does the VCD spectrum alone. In addition, vibrational unpolarized absorption spectra are automatically predicted simultaneously with VCD spectra.
For ab initio assignments, VCD and unpolarized IR spectra were calculated using the Gaussian 98 software package.
Abreviations
EtOAc/EA ethyl acetate
DCM dichloromethane
Et2O diethyl ether
THF tetrahydrofuran
TFA trifluoroacetic acid
MeOH Methanol
DMSO dimethylsulfoxide
DMF N,N-dimethylformamide
TEA triethylamine
BOC2O di-t-butyldicarbonate
SCX strong cation exchanger dried refers to a solution dried over anhydrous sodium sulphate r.t./RT room temperature
Rt retention time h hour
FC flash chromatography
NH secondary amine functionalised silica cartridge column
CDI carbonyldiimidazole min minutes
cone concentrated vo I volume eq equivalent
LiHMDS lithium hexamethyldisilazide
TosMIC Toluenesulfonylmethyl isocyanide
HOBT 1-hydroxybenzotriazole
EDC N-(3-dimethylaminopropyl)-N'ethylcarbodiimide
TBTU O-(Benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate
DIPEA N,N'-Diisopropylethylamine
HMDS Hexamethyldisilazane
Intermediate 1 : 1-(1 ,1-Dimethvlethvl) 3-ethvl 4-oxo-1 ,3-Dioeridinedicarboxvlate

To a suspension of 3-ethylcarboxylate-4-piperidone hydrochloride (5 kg, 24.08 moles, Alfa Aesar) in heptane (12.7, kg) was charged triethylamine (7.25 kg, Alfa Aesar) at room temperature and the suspension was then stirred for 15 minutes. Di- tert-butyldicarbonate (6.3 kg, 28.89 moles, 1.2 eq., Alfa Aesar) was then added to the reaction over 20 minutes as a solution in heptane (4.1 Kg) at room temperature. The reaction was stirred at room temperature for approximately 40 min. Then water (25 L) was charged to the reaction at room temperature and stirred for 15 minutes. The layers were then allowed to separate and the aqueous layer removed. The organic layer was then washed with 1 N HCI (25 L) and water (22 L). The resulting organic layer was then concentrated to an oil by vacuum distillation, with a jacket temperature of 20 0C. Once concentrated to an oil, ethanol (13.7 kg)/water (17.5 kg) was then charged to the reaction and warmed to 50 0C. Once reaction temperature has stabilized, the reaction was then cooled to -10 0C at a rate of 0.25 °C/min. The reaction was held at -10 0C for greater than 6 hours. The resulting solids were then filtered, and the filtrate used to rinse the reactor and wash the filter cake. The recovered solids were then dried at room temperature under full vacuum with a N2 bleed to give the title compound; 1 H NMR (300 MHz, DMSO-c/6) δ ppm 1.13 - 1.30
(m, 3 H) 1.40 (s, 9 H) 2.32 (t, J=5.98 Hz, 2 H) 3.48 (t, J=5.98 Hz, 2 H) 3.95 (s, 2 H) 4.21 (q, J=7.08 Hz, 2 H).
Intermediate 2: 1-(1 ,1-dimethylethyl) 3-ethyl 4-{[(trifluoromethyl)sulfonyl1oxy)-5,6- dihydro-1 ,3(2/-/)-pyridinedicarboxylate

1 -(1 , 1 -Dimethylethyl) 3-ethyl 4-oxo-1 ,3-piperidinedicarboxylate (Intermediate 1 , 380.48 g, 1.40 moles) was dissolved in toluene (2.97 Kg). The solution was stirred for 10 mins and then cooled to -7 0C and then treated with N,N-diisopropylethylamine (271.56 g, 2.10 mol) while maintaining the reaction below -7 0C. After stirring the reaction mixture for approximately 10 minutes, trifluoromethanesulfonic anhydride (436.29 g, 1.55 mol) was added while maintaining the temperature below 5 0C. The reaction mixture was stirred at 1 0C for 31 minutes. The product was used in the next step without purification (see preparation of Intermediate 3); HPLC: Rt= 2.69 min (HPLC instrument Agilent 1 100 Series analysis performed on a Agilent Zorbax SB C18 (50x3.0 mm, 1.8um), mobile phase: water:acetonitrile:TFA (0.05%), gradient from 0 to 95% in 2.5 min, hold for 0.2 min, then re-equilibrate; T=60; flow= 1.5 mL/min)
Intermediate 3: 1-(1.1-dimethylethvn 3-ethyl 4-(3.4-dichlorophenvn-5.6-dihvdro- 1 ,3(2H)-pyridinedicarboxylate

Intermediate 4: 3-(1 ,1-dimethylethyl) 1-ethyl-6-(3,4-dichlorophenyl)-3- azabicyclo[4.1.01hept-4-ene-1 ,3-dicarboxylate (Racemate)

Step b) (Recrvstallization)
A 50 L jacketed laboratory reactor was charged with (±)-3-(1 ,1 -dimethylethyl) 1 -ethyl (1 S,6R)-6-(3,4-dichlorophenyl)-3-azabicyclo[4.1.0]hept-4-ene-1 ,3-dicarboxylate (from step a, 4.40 Kg, 1 1.0 moles, 1 eq) and heptane (15.1 Kg, 22.1 L, 5 vol). The resulting slurry is heated to approximately 80 0C and filtered into a clean 50-L jacketed laboratory reactor. The filter and lines were rinsed with heptane (3.0 Kg, 4.4 L, 1 vol) and the rinse was combined with the filtrate. The solution was heated to 80 0C and then cooled to 22 0C over 107 min. No crystals had formed so a small aliquot was taken out. Crystals formed spontaneously in the aliquot and were returned to the reactor which caused rapid crystallization. The slurry was heated back to 80 0C and cooled to 22 0C over 105 min. During the cool, a small aliquot was pulled at 520C.
The wall of the vial holding the aliquot was scratched to initiate crystallization and the resulting slurry was combined with the bulk solution when its temperature had reached 47 0C. The solids were collected by filtration, the reactor was rinsed with heptane (3.0 Kg, 4.4 L, 1 vol), and the rinse was used to wash the filter cake. The solids were dried to a constant weight in a 50-60 0C vacuum oven to provide 2.954 Kg of the title compound as an off-white solid; 1 H NMR (CDCI3, 400 MHz) δ 7.35 (2H, m), 7.27 (CDCI3), 7.11 (1 H, m), 6.65 (1 H, br m), 5.16 (1 H, br m), 4.27 (1 H, br m), 3.79 (3H, br m), 2.32 (1 H, m), 1.59 (1 H, m), 1.52 (9H, s), 0.94 (3H, br m).
Intermediate 5: ethyl (1 R6S/7S.6ffl-6-(3.4-dichlorophenvn-3- azabicvclor4.1.0lheptane-1-carboxylate (Racemate)

3-(1 ,1-Dimethylethyl) 1 -ethyl e-^-dichlorophenyO-S-azabicyclo^.i .OJhepM-ene- 1 ,3-dicarboxylate (Intermediate 4, 1.600 g) was dissolved in dry toluene (25 ml.) and triethylsilane (0.744 ml.) was added followed by TFA (2.242 ml_). The reaction mixture was stirred at rt for 18h until completion. It was then quenched with 1.0N NaOH until pH 13. After stirring for 10 min, the organic phase was separated, washed with brine, dried and concentrated. Purification by SCX cartridge eluting first with MeOH and then with 2.0N NH3 in MeOH afforded the title compound as a yellow oil, 1.27g; MS(m/z): 314 [MH]+.
Intermediate 6: 3-(1.1-dimethylethvn 1-ethyl (1 R6S/7S.6/?V6-(3.4-dichlorophenvn-3- azabicvclor4.1.Olheptane-1 ,3-dicarboxylate (Racemate)

Ethyl (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-3-azabicyclo[4.1.0]heptane-1-carboxylate (Intermediate 5, 1.270 g) was dissolved in DCM (15 ml.) and cooled to 0 0C. BOC- anhydride (1.032 mL) in DCM (5 mL) was added dropwise and the mixture was stirred at 0 0C for 3h. The reaction mixture was quenched with NH4CI sat solution, washed with NaHCC>3 sat solution, brine, dried and concentrated. Purification by chromatography eluting with a gradient from 0 to 20% ethyl acetate in cyclohexane yielded the title compound (1.5 g); MS(m/z): 358 [MH -56]+.
Intermediate 7: (1 R6S/7S.6/?V6-(3.4-dichloroDhenvn-3-fr(1.1- dimethylethyl)oxylcarbonyl)-3-azabicvclor4.1.Olheptane-1-carboxylic acid (Racemate)

3-(1 ,1-Dimethylethyl) 1 -ethyl (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-3- azabicyclo[4.1.0]heptane-1 ,3-dicarboxylate (Intermediate 6, 1.330 g) was dissolved in ethanol (3 mL); potassium tert-butoxide (2.88 g) was added followed by water (1.157 mL). The reaction mixture was stirred at 50 0C for 18h before cooling to RT. Volatiles were evaporated under reduced pressure and the residue was partitioned between NH4CI (10 ml) and DCM (10 ml). The organic phase was dried and concentrated to yield the title compound as a white-yellow foam (1.18 g); 1H NMR
(CDCI3, 400 MHz) δ ppm 7.32 (2 H, dd), 7.01 - 7.06 (1 H, m), 4.03 (2 H, br. s.), 3.32 (2 H, br. s.), 1.79 - 2.20 (3 H, m), 1.48 (9 H, s), 1.21 - 1.29 (1 H, m).
Intermediate 8: 1 ,1-dimethylethyl (1 R6S/7S.6/?)-6-(3,4-dichlorophenyl)-1-[(2-propyn- 1-ylamino)carbonyl1-3-azabicvclo[4.1.01heptane-3-carboxylate (Racemate)

To a stirred solution of (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-3-{[(1 ,1- dimethylethyl)oxy]carbonyl}-3-azabicyclo[4.1.0]heptane-1 -carboxylic acid (Intermediate 7, 0.400 g) in DCM (6 mL) at RT were added HOBT (0.167 g) and EDC (0.208 g). After 0.5 h, propargylamine (0.080 mL) in DCM (1 mL) was added dropwise and the reaction mixture was stirred at RT till complete consumption of the starting material was observed. The reaction mixture was then diluted with ethyl acetate, washed first with a saturated solution of NaHCC>3 and then with brine. The organic phase was dried and concentrated. The crude product was purified by chromatography on silica eluting with a gradient from 0 to 40% ethyl acetate in cyclohexane to afford the title compound as a white foam (0.348 g); MS(m/z): 423 [MH]+ and 367 [MH -56]+.
Intermediate 9: 1.1-dimethylethyl (1 R6S/7S.6ffl-1-(aminocarbonyl>6-(3.4- dichlorophenyl)-3-azabicvclo[4.1.01heptane-3-carboxylate (Racemate)

Intermediate 10: 1.1-dimethylethyl (1 R6S/7S.6ft>1-(aminocarbonothioyl>6-(3.4- dichlorophenyl)-3-azabicvclo[4.1.01heptane-3-carboxylate (Racemate)

To a stirred solution of 1 ,1-dimethylethyl (1 R,6S/1 S,6R)-1-(aminocarbonyl)-6-(3,4- dichlorophenyl)-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 9, 0.190 g) in THF (5 mL) at room temperature was added Lawesson's reagent (0.199 g). The mixture was heated to reflux temperature for 4h and then slowly allowed to reach room temperature. The mixture was concentrated under reduced pressure. The crude material was dissolved in ethyl acetate, washed with concentrated NaHCC>3, brine, dried over Na2SC>4 and concentrated under reduced pressure. The residue was purified by chromatography on silica gel eluting with a gradient from 20 to 60% ethyl acetate in cyclohexane to yield the title compound as a colourless foam 93 mg; UPLC: Rt = 0.88 min, MS(m/z): 401 [MH]+ and 345 [MH -56]+.
Intermediate 11 : 1.1-dimethylethyl (1 R6S/7S.6/?)-6-(3,4-dichlorophenyl)-1-(4- methyl-1 ,3-thiazol-2-yl)-3-azabicyclo[4.1.OIheptane-3-carboxylate (Racemate)
1 ,1-Dimethylethyl (1 R!6S/1 S,6R)-1-(aminocarbonothioyl)-6-(3,4-dichlorophenyl)-3- azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 10, 93 mg) was dissolved in toluene (3.00 ml.) and chloroacetone (0.024 ml.) was added. The mixture was heated to 80 0C for 5h. Further 12 μl of chloroacetone were added and the mixture was refluxed for 3 additional hours. The mixture was cooled to room temperature, the solvent was evaporated under reduced pressure and the residue dissolved in DCM, washed with cone NaHCC>3 and brine, then dried and concentrated. Purification by chromatography on silica gel eluting with a gradient from 0 to 60% ethyl acetate in cyclohexane afforded the title compound as a colourless oil (80 mg); UPLC: Rt = 1.03 min; MS(m/z): 439 [MH]+; 1H NMR (CDCI3, 400 MHz) δ ppm 7.25 - 7.28 (1 H, m), 7.17 - 7.21 (1 H, m), 6.97 (1 H, dd), 6.55 (1 H, br. s.), 4.01 - 4.51 (2 H, m), 3.61 (1 H, br. s.), 3.36 (1 H, ddd), 2.29 (3 H, s), 2.04 - 2.24 (2 H, m), 1.99 (1 H, d), 1.45 - 1.54 (10 H, m).
Intermediate 12: 1.1-dimethylethyl (1 R6S/7S.6/?V6-(3.4-dichlorophenvn-1-formyl-3- azabicvclor4.1.Olheptane-3-carboxylate (Racemate)

Intermediate 13: 1.1-dimethylethyl (1 R6S/7S.6/?y6-(3.4-dichlorophenvn-1-ethenyl-3- azabicvclo[4.1.OIheptane-3-carboxylate (Racemate)

To a stirred suspension of methyltriphenylphosphonium bromide (1.179 g) in THF (15 mL) at 0 0C, BuLi 1.6M in hexane (2.063 mL) was added dropwise. The dark yellow reaction mixture was allowed to reach room temperature and stirred for 20 min. It was cooled to 0 0C and 1 ,1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-1- formyl-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 12, 0.940 g) in THF (2 mL) was added dropwise. After removal of the ice bath, the mixture was stirred at room temperature for 18h. It was then quenched with water (10 mL) and the residue was diluted with diethyl ether (3 x 20 ml). The organic phase was separated, dried and concentrated affording a yellow oil which then solidified. The residue was purified by chromatography on silica gel eluting with a gradient from 0 to 25% ethyl acetate in cyclohexene affording the title compound as a colourless oil (610 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.36 - 7.39 (1 H, d), 7.31 (1 H, d), 7.06 (1 H, dd), 4.90 - 5.17 (3 H, m), 3.89 - 4.20 (1 H, m), 3.41 - 3.71 (2 H, m), 3.27 (1 H, ddd), 1.97 -
2.15 (2 H, m), 1.51 (9 H, s), 1.22 - 1.27 (2 H, m); UPLC: Rt= 1.09 min; MS(m/z): 312 [MH -56]+.
Intermediate 14: 1 ,1-dimethylethyl (1 R6S/7S.6/?)-6-(3,4-dichlorophenyl)-1-(2- hvdroxyethvD-S-azabicvcW.I .OIheptane-S-carboxylate (Racemate)

To a stirred solution of 1 ,1-dimethylethyl (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-1- ethenyl-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 13, 0.610 g) in THF (4 ml.) at 0 0C, under nitrogen, borane tetrahydrofuran complex 1.0M in THF (1.988 ml.) was added dropwise. The ice bath was removed and the mixture was stirred at room temperature for 4h. The mixture was cooled again to 0 0C and quenched with water (1 ml_), then sodium hydroxide 2.0M (1.6 ml.) and hydrogen peroxide (30%(w/w), 1.692 ml.) were added. The resulting mixture was stirred for 1/2h and then diluted with water (10 ml.) and ethyl acetate (20 ml_). The organic layer was separated, dried and concentrated to give the title compound as crude material (640 mg); UPLC: Rt= 0.90 min, MS(m/z): 386 [MH]+ and 330 [MH -56]+.
Intermediate 15: ((1 R6S/7S.6/?)-6-(3,4-dichlorophenyl)-3-{[(1 ,1- dimethylethyl)oxy1carbonyl)-3-azabicvclo[4.1.01hept-1-yl)acetic acid (Racemate)

To a stirred solution of 1 ,1-dimethylethyl (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-1-(2- hydroxyethyl)-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 14, 640 mg) in acetone (13 ml.) at 0 0C, was added Jones reagent (prepared dissolving chromium trioxide, 398 mg, in water, 0.7 ml_, and sulphuric acid, 0.212 ml.) dropwise and the mixture was stirred at 0 0C for 1 h. The reaction mixture was quenched at 0 0C with 5 ml. of isopropanol and stirred at this temperature for 10 min. The mixture was diluted with water (20 ml.) and DCM (30 ml_); the organic layer dried and concentrated in vacuo to give a brown foam. 370 mg of this material was purified by chromatography on silica gel eluting with a gradient from 20 to 100% ethyl acetate in cyclohexane to yield the title compound as a colourless solid (160 mg); UPLC: Rt= 0.87 min; MS(m/z): 344 [MH -56]-.
Method B: To a solution of 1 ,1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-1-(2- oxoethyl)-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 57, 350 mg) in tert- butanol (2 ml.) at 0 0C was added an aqueous solution of sodium chlorite (247 mg) and sodium dihydrogen phosphate (328 mg) over a period of 5 min. A yellow suspension formed. The mixture was warmed to rt and stirred at rt for 4h. Volatiles were then evaporated under reduced pressure and the residue was partitioned between DCM (20 ml.) and brine (20 ml_). The organic layer was dried and concentrated. The title compound was obtained as a white foam (300 mg); MS(m/z): 344 [MH -56]+.
Intermediate 16: 1.1-dimethylethyl (1 R6S/7S.6/?)-1-(2-amino-2-oxoethyl)-6-(3,4- dichlorophenyl)-3-azabicvclo[4.1.0lheptane-3-carboxylate (Racemate)

Method A:
((I R.βS/IS.eRJ-β-CS^-DichlorophenyO-S-tKI .I-climethylethyOoxylcarbonylJ-S- azabicyclo[4.1.0]hept-1-yl)acetic acid (Intermediate 15, 145 mg) was dissolved in DMF (5 ml.) under nitrogen at rt. DIPEA (0.070 ml) was added followed by TBTU (128 mg). The mixture was stirred at room temperature for 30 min and then HMDS (0.084 ml.) was added. The mixture was stirred at rt for 1.5h. It was then quenched with water (10 ml.) and diluted with diethyl ether (30 ml_). The organic layer was washed with water (20ml x 3), dried and concentrated. A light brown foam was obtained (140 mg) which was purified by chromatography on silica gel eluting with a gradient from 20 to 70% ethyl acetate in cyclohexane. The title compound was obtained as a white foam (105 mg); UPLC: Rt= 0.81 min, MS(m/z): 399 [MH]+ and 343 [MH -56]+.
Method B: ((I R.βS/IS.eRJ-β-CS^-DichlorophenyO-S-tKI .I-climethylethyOoxylcarbonylJ-S- azabicyclo[4.1.0]hept-1-yl)acetic acid (Intermediate 15, 100 mg) was dissolved in DMF (5 ml.) under nitrogen at room temperature and DIPEA (0.048 ml.) was added followed by TBTU (88 mg). The mixture was stirred at rt for 30 min and then HMDS (0.058 ml.) was added. The mixture was stirred at rt for 1.5h and then quenched with water (10 ml_). It was diluted with diethyl ether (30 ml_), washed with water (20ml x 3), dried and concentrated in vacuo. The residue was purified by chromatography on silica gel eluting with a gradient from 20 to 70% ethyl acetate in cyclohexane affording the title compound as a colourless oil (12 mg); MS(m/z): 399 [MH]+ and 343 [MH -56]+.
Intermediate 17: 1.1-dimethylethyl (1 R6S/7S.6/?)-1-(2-amino-2-thioxoethyl)-6-(3,4- dichlorophenyl)-3-azabicvclo[4.1.01heptane-3-carboxylate (Racemate)

To a stirred solution of 1 ,1-dimethylethyl (1 R,6S/1 S,6R)-1-(2-amino-2-oxoethyl)-6- (3,4-dichlorophenyl)-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 16, 105
mg) in THF (5 ml.) at room temperature was added Lawesson's Reagent (106 mg). The mixture was heated to reflux temperature for 6h and then allowed to reach room temperature before concentrating under reduced pressure. The crude material was dissolved in ethyl acetate, washed with cone NaHCC>3, brine, dried over Na2SC>4 and concentrated under reduced pressure. The residue was purified by chromatography on silica gel eluting with a gradient from 20 to 70% ethyl acetate in cyclohexane to yield the title compound as a colouless oil (50 mg); UPLC: Rt= 0.89 min, MS(m/z): 415 [MH]+ and 359 [MH -56]+.
Intermediate 18: 1.1-dimethylethyl (1 R6SV6-(3.4-dichloroDhenyl)-1-r(4-methyl-1.3- thiazol-2-yl)methyl1-3-azabicvclo[4.1.OIheptane-3-carboxylate

1 ,1-Dimethylethyl (1 R,6S/1 S,6R)-1-(2-amino-2-thioxoethyl)-6-(3,4-dichlorophenyl)-3- azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 17, 50 mg) was dissolved in toluene (3.00 ml.) and chloroacetone (0.014 ml.) was added. The mixture was heated to 80 0C for 5h. It was cooled to room temperature, the solvent evaporated under reduced pressure and the residue dissolved in DCM, washed with cone NaHCC>3, brine, dried and concentrated. Purification by chromatography on silica gel eluting with a gradient from 0 to 60% ethyl acetate in cyclohexane afforded the title compound as a colourless oil (26 mg); UPLC: Rt= 1.05 min, MS(m/z): 453 [MH]+.
Intermediate 19: 1.1-dimethylethyl (1 R6S/7S.6/?V6-(3.4-dichlorophenvn-1-(2-fr(1 ftV
2-hvdroxy-1-methylethyl1amino)-2-oxoethyl)-3-azabicvclo[4.1.01heptane-3- carboxvlate

To a stirred solution of ((1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-3-{[(1 ,1- dimethylethyl)oxy]carbonyl}-3-azabicyclo[4.1.0]hept-1-yl)acetic acid (Intermediate 15, 0.200 g) in DCM (7 ml_), at room temperature were added HOBT (0.084 g) and EDC (0.105 g). After 0.5 h, (R)-2-amino-1-propanol (0.047 ml.) in DCM (1 ml) was added dropwise and the reaction mixture was stirred at RT overnight until complete conversion to the intermediate amide was observed. The reaction mixture was diluted with DCM, it was washed first with NaHCC>3, then with brine and finally concentrated under reduced pressure. The title compound was obtained as a mixture of diasteroisomers in unknown ratio as a pale yellow foam (0.25 g); UPLC: Rt= 0.78 min; MS(m/z): 457 [MH]+.
Intermediate 20: 1.1-dimethylethyl (1 R6S/7S.6/?)-6-(3,4-dichlorophenyl)-1-r(4- methyl-1 ,3-oxazol-2-yl)methyl1-3-azabicvclo[4.1.OIheptane-3-carboxylate (Racemate)

Intermediate 19 (0.25 g) was dissolved in DCM (7 ml.) and cooled to 0 0C. Dess- Martin Periodinane (0.348 g) was added and the reaction mixture was slowly warmed to room temperature. After 1 h stirring, it was diluted with DCM (10 ml_), NaHCC>3 sat solution was added followed by sodium thiosulfate (3 g in 5 ml. of water) and the two phases were stirred for 30 min. The organic phase was separated, washed with brine, dried and concentrated. The crude mixture of diasteroisomers, present in
unknown ratio, was dissolved in DCM (7 mL) and cooled to 0 0C. Triphenylphosphine (0.432 g), 2!6-bis(1 ,1-dimethylethyl)-4-methylpyridine (0.902 g) and 1 ,2-dibromo- 1 ,1 ,2,2-tetrachloroethane (0.536 g) were then added. The reaction mixture was slowly warmed to RT and stirred at RT overnight. It was then diluted with DCM, washed with 0.1 N HCI, with brine and finally concentrated under reduced pressure. The crude product was purified by chromatography on silica gel eluting with a gradient from 0 to 40% ethyl acetate in cyclohexane affording the title compound (70 mg) as a pale yellow oil; UPLC: Rt= 0.99 min; MS(m/z): 437 [MH]+.
Intermediate 21 : 3-(3,4-dichlorophenyl)-3-buten-1-ol

In a round-bottomed flask equipped with mechanical stirrer, under an argon atmosphere, 3,4-dichlorophenylboronic acid (128 g, 672 mmol) and 3-bromo-3- buten-1-ol (78g, 517 mmol) were dissolved in toluene (1230 ml) and ethanol (492 ml). To this solution, tetrakis(triphenylphosphine)palladium(0) (29.8 g, 25.8 mmol) was added followed by sodium carbonate 2M aqueous solution (517 ml, 1033 mmol). The resulting mixture was heated at an internal temperature of 75°C. After 30 minutes, a thick precipitate formed. After 1 h, water (50 ml) was added to make the solid re-dissolved and the reaction mixture turned slightly yellow opalescent. At 3h the reaction was worked-up. The flask was cooled to RT (a precipitate was formed) and the mixture was taken up with aqueous NaHCO3 sat. solution (468ml_), water (468ml_) and ethyl acetate (468ml_). The phases were separated and the aqueous phase was back-extracted with ethyl acetate (2x936ml_). The combined organics were dried (Is^SCv) and concentrated under vacuum to give crude material (20Og) as black thick oil. This oil was purified by silica gel flash chromatography, eluting with cyclohexane/ethyl acetate from 8/2 to 7/3. Evaporation of solvent afforded the title compound (73g) as dark thick oil; 1H NMR (400 MHz, CDCI3) δ ppm 7.51 (d, 1 H), 7.41 (d, 1 H), 7.26 (dd, 1 H), 5.34 - 5.53 (m, 1 H), 5.02 - 5.29 (m, 1 H), 3.60 - 3.95 (m, 2 H), 2.57 - 2.94 (m, 2 H), 1.50 (t, 1 H).
Intermediate 22: 3-(3,4-dichlorophenyl)-3-buten-1-yl methanesulfonate

In a round-bottomed flask, 3-(3,4-dichlorophenyl)-3-buten-1-ol (Intermediate 21 , 73 g, 336mmol) was dissolved in DCM (900 ml) to give a yellow solution. Then triethylamine (69.9 ml, 504 mmol) was added keeping the internal temperature below +5°C with an ice bath. Methanesulfonyl chloride (36.7 ml, 471 mmol) was then added dropwise over 30 min keeping the internal temperature below +5°C with an ice bath. The mixture was allowed to reach room temperature while stirring. After 3 h, the reaction mixture (suspension) was quenched by careful addition of aqueous ammonium chloride sat. solution (400ml) keeping the internal temperature below +100C with an ice bath. At the end of the addition, the pH of the aqueous phase was nearly 1. The two layers were separated. The aqueous layer was back-extracted with DCM (3 x 300 ml_). The combined organic layers were washed with water (2 x 200 ml_), dried (Na2SO4), and evaporated to give a crude product (101g) that was purified over a silica gel pad (1000g) eluting with cyclohexane/EtOAc from 9/1 to 1/1 to afford the title compound (90.8 g) as a dark yellow oil; 1H NMR (400 MHz, CDCI3) δ ppm 7.49 (d, 1 H), 7.44 (d, 1 H), 7.24 (dd, 1 H), 5.46 (d, 1 H), 5.25 (d, 1 H), 4.32 (t, 2 H), 2.98 (s, 3 H), 2.93 (t, 2 H); HPLC (walk-up): Rt=5.37 min.
Intermediate 23: dimethyl 2-(3,4-dichlorophenyl)-2-{2-r(methylsulfonyl)oxylethyl)-1 ,1- cyclopropanedicarboxylate

In a round-bottomed flask, 3-(3,4-dichlorophenyl)-3-buten-1-yl methanesulfonate (Intermediate 22, 90.8 g, 308 mmol) was dissolved in chlorobenzene (200ml) to give a green solution. Rhodium acetate dimer (6.80 g, 15.38 mmol) was added. The suspension was warmed to an internal temperature of +65°C and dimethyl diazopropanedioate (78g, 492mmol, for a reference procedure of preparation see Synthetic Communication 1987, 17 (14), 1709-1716) dissolved in chlorobenzene (150ml) was added dropwise (during 2.5hrs), keeping the internal temperature below 65-67°C. At the end of the addition, the mixture was cooled to room temperature. It was diluted with DCM (300 ml) and filtered over a celite pad to separate the catalyst. The solution was evaporated in vacuo to 1/3 of the volume and the crude (277g) purified over a silica pad (silica gel 1.3 Kg) eluting with cyclohexane /ethyl acetate from 7/3 to 1/1 to afford the title compound (128.25 g); 1H NMR (400 MHz, CDCI3) δ ppm 7.41 (d, 1 H), 7.39 (d, 1 H), 7.15 (dd, 1 H), 4.08 - 4.22 (m, 1 H), 3.94 - 4.06 (m, 1 H), 3.85 (s, 3 H), 3.48 (s, 3 H), 2.95 (s, 3 H), 2.42 (dt, 1 H), 2.21 (d, 1 H), 1.89 - 2.03 (m, 1 H), 1.82 (d, 1 H); HPLC (walk-up): Rt = 5.15 min.
Intermediate 24: methyl 6-(3,4-dichlorophenyl)-2-oxo-3-azabicvclo[4.1.01heptane-1- carboxylate (Racemate)

In a 5 L Parr reactor, dimethyl 2-(3,4-dichlorophenyl)-2-{2-[(methylsulfonyl)oxy]ethyl}- 1 ,1-cyclopropanedicarboxylate (Intermediate 23, 158 g, 372 mmol) was dissolved in
ammonia 2M in methanol (3000 ml) to give a yellow solution. The solution was warmed to +75°C and the resulting mixture was stirred at this temperature overnight (internal pressure = 2 atm). After 24 hrs, reaction was complete. The solution was concentrated to obtain a residue that was mixed with a residue (89.3g) coming from an analogous preparation performed on another batch of 2-(3,4-dichlorophenyl)-2-{2- [(methylsulfonyl)oxy]ethyl}-1 ,1-cyclopropanedicarboxylate. The solvent was evaporated to obtain a crude oil (28Og). To this oil, ethyl acetate (5L) and 1 M aqueous HCI (2.5L) were added and the mixture was vigorously stirred for 30 min in a 10L reactor. A diluted suspension (a mixture of organic phase, aqueous phase and solid) was obtained. The solid was filtered, washed with ethyl acetate and dried to afford a first batch of title compound (54.9g). The organic and aqueous layers were separated. The organic phase was then washed with aqueous HCH M (2L), dried (Na2SC>4) and concentrated to 1/10 of the volume. A solid precipitated out. It was filtered, washed with diethyl ether (150ml) and dried under vacuum to afford a second batch of title compound (34.4 g). The mother liquours of filtration were concentrated in vacuo to obtain a brown oil. This residue was triturated with ethyl ether (1 x 50 mL). The resulting solid was filtered, washed with cold ethyl ether and dried to give a third batch of title compound (7.27g) as a off white solid. The mother liquours were concentrated and chromatographed on Biotage 75M (silica gel) eluting with ethyl acetate to give a solid that was triturated in ethyl ether (35ml) to give a fourth batch of title compound (6.2g). An overall amount of 102.7g of product was thus obtained; 1H NMR (400 MHz, CDCI3) δ ppm 7.47 (d, 1 H), 7.41 (d, 1 H), 7.20 (dd, 1 H), 5.73 (br. s., 1 H), 3.54 (s, 3 H), 3.07 - 3.41 (m, 2 H), 2.32 - 2.45 (m, 1 H), 2.28 (d, 1 H), 2.17 (d, 1 H), 1.93 (d, 1 H).
Intermediate 25: 1 ,1-dimethylethyl 6-(3,4-dichlorophenyl)-1-(hvdroxymethyl)-3- zabicvclor4.1.Olheptane-3-carboxylate (Racemate)

In a round-bottomed flask, methyl 6-(3,4-dichlorophenyl)-2-oxo-3- azabicyclo[4.1.0]heptane-1-carboxylate (Intermediate 24, 90 g, 286 mmol) was dissolved in THF (1450ml) to give a grey suspension. Borane tetrahydrofuran compex 1 M (1633 ml, 1633 mmol) was added dropwise keeping the internal temperature below + 5°. The resulting mixture was gently refluxed for 7hrs. The mixture was cooled to +3°C and quenched by careful addition of methanol (200ml). Aqueous HCI 6M (450ml) was added keeping the internal temperature below +6°C . The mixture was stirred at room temperature for 5h. The acidic solution was concentrated under vacuum to remove THF, then water (900ml) was added . The pH of the final solution was nearly 1. This solution was washed with ethyl ether (2x200 ml). The aqueous solution was basified by portionwise addition of potassium carbonate until pH 8-9, then THF (1200ml) was added and the resulting mixture used directly in the step b).
Step b)
A round-bottomed flask was charged with the mixture coming from step a) (approximately 2700ml, pH=8-9). Di-te/f-butyl dicarbonate (80 ml, 343 mmol) was added portionwise at room temperature and the mixture was stirred overnight. The organic and aqueous layers were separated. The aqueous layer was back-extracted with ethyl acetate (3 x 600 ml_). The combined organic layers were dried (Na2SC>4) and concentrated to obtain a crude oil (165g) that was purified over a silica pad (silica gel 1500 g), eluting with cyclohexane/ethyl acetate 8/2 to give the title compound (1 10 g) as a foamy colourless oil; 1H NMR (400 MHz, CDCI3) δ ppm 7.39 - 7.42 (m, 1 H), 7.37 (d, 1 H), 7.17 (d, 1 H), 3.73 - 3.90 (m, 2 H), 2.99 - 3.49 (m, 4 H), 1.91 - 2.13 (m, 2 H), 1.48 (s, 9 H), 0.92 - 1.07 (m, 2 H); HPLC (walk-up): Rt = 6.12 min.
Intermediate 26: 1.1-dimethylethyl (1 R6SV6-(3.4-dichlorophenylV1- {r(methylsulfonyl)oxylmethyl)-3-azabicvclor4.1.Olheptane-3-carboxylate (Racemate)

1 ,1-Dimethylethyl 6-(3,4-dichlorophenyl)-1-(hydroxymethyl)-3- azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 25, 0.070 g) was dissolved in DCM (3 ml.) and cooled to 0 0C. Triethylamine (0.039 mL) was added followed by mesyl-CI (0.021 mL) and the mixture was stirred at this temperature for 1 h. It was quenched with a sat solution NH4CI and the organic layer was separated, washed with brine, dried and concentrated in vacuo; 1H NMR (400 MHz, CDCI3) δ ppm 7.37 - 7.42 (2 H, m), 7.13 (1 H, dd), 3.66 - 4.01 (4 H, m), 3.30 - 3.48 (2 H, m), 2.86 - 2.93 (3 H, br.s), 1.88 - 2.14 (2 H, m), 1.49 (9 H, s), 1.20 (1 H, d), 1.12 (1 H, d).
Intermediate 27: 1 ,1-dimethylethyl (1 R.6S or 1S,6R)-6-(3,4-dichlorophenyl)-1- (hvdroxymethylKS-azabicvclol^.i .O'lheptane-S-carboxylate (single enantiomer); and Intermediate 28: 1 ,1-dimethylethyl (1 S,6R or 1 R,6S)-6-(3,4-dichlorophenyl)-1- (hydroxymethyl)-3-azabicvclo[4.1.OIheptane-3-carboxylate (single enantiomer)

1 ,1-Dimethylethyl (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-1-(hydroxymethyl)-3- azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 25, 3.2 g) was submitted to preparative HPLC using a chiral column Chiralpack AD-H, eluent n- hexane/isopropanol 90/10, flow rate 14 mL/min, detection UV at 215 nm, obtaining: Intermediate 27 (Enantiomer 1 ): Rt. = 10.3 min, e.e. >99.5%, 1.51 g, colourless oil; MS(m/z): 316 [MH-56]+, 1H NMR (400 MHz, CDCI3) δ ppm 7.39 - 7.42 (m, 1 H), 7.37
(d, 1 H), 7.17 (d, 1 H), 3.73 - 3.90 (m, 2 H), 2.99 - 3.49 (m, 4 H), 1.91 - 2.13 (m, 2 H), 1.48 (s, 9 H), 0.92 - 1.07 (m, 2 H); and
Intermediate 28 (Enantiomer 2): Rt. = 16.1 min, e.e. > 99.5%, 1.65 mg, colourless oil; MS(m/z): 316 [MH-56]+, 1 H NMR (400 MHz, CDCI3) δ ppm 7.39 - 7.42 (m, 1 H), 7.37 (d, 1 H), 7.17 (d, 1 H), 3.73 - 3.90 (m, 2 H), 2.99 - 3.49 (m, 4 H), 1.91 - 2.13 (m, 2 H), 1.48 (s, 9 H), 0.92 - 1.07 (m, 2 H)). Retention times (analytical) and e.e. were obtained using a chiral column Chiralpack AD-H, eluent n-hexane/isopropanol 90/10, flow rate 1.0 mL/min, detection UV at 210-340 nm, CD at 230 nm.
Intermediate 29: 1.1-dimethylethyl (1 R.6S or 1S.6RV6-(3.4-dichlorophenvn-1- {r(methylsulfonyl)oxylmethyl)-3-azabicvclor4.1.Olheptane-3-carboxylate (Single enantiomer)

Triethylamine (0.280 ml.) and methanesulfonyl chloride (0.086 ml.) were added at 0 0C to a solution of 1 ,1-dimethylethyl (1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-1- (hydroxymethyl)-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 27, Enantiomer 1 , 374 mg) in dry dichloromethane (10 ml_). After 5min stirring, the mixture was allowed to warm to room temperature and stirred overnight. A saturated aqueous NH4CI solution was then added and the mixture was extracted with dichloromethane. The organic phase was washed with brine, dried over Na2SO4 and concentrated in vacuo to give 469 mg of crude title compound; 1 H NMR (400 MHz, CDCI3) δ ppm 1.13 (d, 1 H), 1.22 (d, 1 H), 1.50 (s, 9 H), 1.90 - 2.18 (m, 2 H), 2.90 (bs, 3 H), 3.30 - 3.48 (m, 2 H), 3.56 - 4.13 (m, 4 H), 7.13 (d, 1 H), 7.36 - 7.44 (m, 2 H); MS(m/z): 450 [MH]+.
Intermediate 30: 1 ,1-dimethylethyl (1 R.6S or 1S,6R)-6-(3,4-dichlorophenyl)-1- {[(methylsulfonyl)oxy1methyl)-3-azabicvclo[4.1.OIheptane-3-carboxylate (single enantiomer)

The title compound was obtained in a similar fashion to the preparation of Intermediate 29 starting from (1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-1- (hydroxymethyl)-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 28, Enantiomer 2); 1 H NMR (400 MHz, CDCI3) δ ppm 1.13 (d, 1 H), 1.22 (d, 1 H), 1.50 (s, 9 H), 1.90 - 2.18 (m, 2 H), 2.90 (bs, 3 H), 3.30 - 3.48 (m, 2 H), 3.56 - 4.13 (m, 4 H), 7.13 (d, 1 H), 7.36 - 7.44 (m, 2 H).
Intermediate 31 : 1 ,1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-1-(3-methyl- 1 ,2,4-oxadiazol-5-yl)-3-azabicvclo[4.1.OIheptane-3-carboxylate (Racemate)

Acetamide oxime (32 mg) was added to a suspension of 3A molecular sieves in dry THF (2.5ml_) under N2 at room temperature. After 5min sodium hydride (60% suspension in mineral oil, 17 mg) was added and the mixture was stirred for further 40min. A solution of 3-(1 ,1-dimethylethyl) 1 -ethyl (1 R,6S/1S,6R)-6-(3,4- dichlorophenyl)-3-azabicyclo[4.1.0]heptane-1 ,3-dicarboxylate (Intermediate 6, 120 mg) in THF (1.5ml_) was then added and the mixture was heated to 65 0C. After 6h30min, MeOH (50μl) was added and the mixture was filtered through a cotton layer. A saturated NH4CI solution was added and the mixture was extracted with
ethyl acetate (2x). The organic phase was washed with brine, dried over Na2SC>4 and concentrated in vacuo to give 115mg of crude compound, which was purified by silica gel flash chromatography (gradient ethyl acetate in cyclohexane from 5 to 40%) to give 87mg of the title compound as a colourless oil; MS(m/z): 368 [MH -56]+. UPLC: Rt= 0.94 min
Intermediate 32: 1 ,1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-1- r(hvdroxyimino)methyll-3-azabicvclor4.1.0lheptane-3-carboxylate (Racemate)

A solution of 1 ,1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-1-formyl-3- azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 12, 277 mg) in absolute ethanol (8.5 ml.) was added to a solution of hydroxylamine hydrochloride (260 mg) and sodium acetate (491 mg) in water (3.4 ml_). The mixture was stirred at room temperature for 18h and then partitioned between water and ethyl acetate. The organic phase was washed with brine, dried over Na2SO4, filtered and concentrated in vacuo to give 257mg of the title compound as a white solid; MS(m/z): 328 [MH - 56]+; UPLC: Rt= 0.87 min.
Intermediate 33: 1.1-dimethylethyl (1 R.6S/1S.6RV1-r(chloro(hvdroxyimino)methyll-6- (3,4-dichlorophenyl)-3-azabicvclo[4.1.01heptane-3-carboxylate (Racemate)

Intermediate 34: 1.1-dimethylethyl (1 R.6S/1S.6RV6-(3.4-dichloroDhenvn-1-(5-methyl- 3-isoxazolyl)-3-azabicvclor4.1.0lheptane-3-carboxylate (Racemate)

1 ,1 -Dimethylethyl (1 R,6S/1 S,6R)-1 -[chloro(hydroxyimino)methyl]-6-(3,4- dichlorophenyl)-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 33, 342 mg) was dissolved in dichloromethane (8 ml.) and cooled to 0 0C. Pre-cooled 2-chloro-1- propene was added through a gas-tight syringe. Triethylamine (0.227 ml.) was then added dropwise and the mixture was allowed to warm up slowly to room temperature. Additional quantities of 2-chloro-1-propene and TEA were added until complete consumption of the starting material was achieved. After 24h, water was added and the mixture was extracted with dichloromethane. The organic phase was washed with brine, dried over Na2SO4, filtered and concentrated in vacuo to give 350mg of crude compound, which was purified by silica gel flash chromatography (gradient ethyl acetate in cyclohexane from 5 to 40%) to give 1 17mg of the title compound as a colourless oil; MS(m/z): 367 [MH -56]+. UPLC: Rt= 0.97 min.
Intermediate 35: 1 ,1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-1-{r5-
(trifluoromethvD-I H-pyrazol-i-ylimethyll-S-azabicvcloK.I .OIheptane-S-carboxylate
(Racemate); and
Intermediate 36: 1 ,1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-1-{r3-
(trifluoromethvD-I H-pyrazol-i-ylimethyll-S-azabicvcloK.I .OIheptane-S-carboxylate
(Racemate)

Sodium hydride 60% dispersed in mineral oil (38.2 mg) was added at 0 0C to a solution of 3-(trifluoromethyl)-1 H-pyrazole (130 mg) in dry DMF (3ml_). The mixture was stirred at this temperature for 15min, then for an additional 5min at room temperature. A solution of 1 ,1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-1- {[(methylsulfonyOoxylmethylJ-S-azabicyclo^.i .Olheptane-S-carboxylate (Intermediate 26, 215 mg) in dry DMF (2ml_) was then added and the mixture was stirred at 50 0C for 3h 30min. After cooling to rt, a saturated NH4CI solution was added and the mixture was extracted with diethyl ether (2x). The organic phase was washed with brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by silica gel flash chromatography (gradient ethyl acetate in cyclohexane from 5 to 40%) to give 31 mg of Intermediate 35 (colourless oil, Rf = 0.44, cyclohexane/ethyl acetate 8/2), MS(m/z): 434 [MH-56]+; and 152mg of Intermediate 36 (colorless oil , Rf = 0.33, cyclohexane/ethyl acetate 8/2); 1 H NMR (400 MHz, DMSOd6) δ ppm 0.90 (d, 1 H), 1.34 (s, 9 H), 1.47 - 1.57 (m, 1 H), 1.84 - 1.97 (m, 1 H), 2.04 - 2.15 (m, 1 H), 3.12 - 3.28 (m, 2 H), 3.34 - 3.62 (m, 3 H), 4.30 (d, 1 H), 6.70 (d, 1 H), 7.38 (dd, 1 H), 7.60 (d, 1 H), 7.67 (d, 1 H), 7.87 (d, 1 H); MS(m/z): 434 [MH-56]+.
Intermediate 37: 1.1-dimethylethyl (1 R.6S/1S.6R)-1-r(5-cvano-1 H-pyrazol-1- yl)methyll-6-(3,4-dichlorophenyl)-3-azabicvclor4.1.0lheptane-3-carboxylate (Racemate); and
Intermediate 38: 1 ,1-dimethylethyl (1 R,6S/1S,6R)-1-r(3-cvano-1 H-pyrazol-1- yl)methyl1-6-(3,4-dichlorophenyl)-3-azabicvclo[4.1.01heptane-3-carboxylate
(Racemate)

Sodium hydride 60% in mineral oil (38.5 mg) was added at 0 0C to a solution of 1 H- pyrazole-3-carbonitrile (90 mg) in dry DMF (3ml_). The mixture was stirred at this temperature for 15min, then for additional 5min at room temperature. A solution of 1 , 1 -dimethylethyl (1 R.6S/1 S,6R)-6-(3,4-dichlorophenyl)-1 - {[(methylsulfonyl)oxy]methyl}-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 26, 217 mg) in dry DMF (2ml_) was then added and the mixture was stirred at 50 0C for 3h 30min. After cooling to room temperature, a saturated NH4CI solution was added and the mixture was extracted with diethyl ether (2x). The organic phase was washed with brine, dried over Na2SO4 and concentrated in vacuo to give 271 mg of crude compound, which was purified by silica gel flash chromatography (gradient ethyl acetate in cyclohexane from 5 to 40%) to give;
Intermediate 37 (33 mg); Rf = 0.31 (cyclohexane/ethyl acetate 8/2); MS(m/z): 391 [MH-56]+; and Intermediate 38 (113 mg); Rf = 0.19 (cyclohexane/ethyl acetate 8/2); 1 H NMR (400 MHz, DMSOd6) δ ppm 0.89 (d, 1 H), 1.34 (s, 9 H), 1.47 - 1.56 (m, 1 H), 1.83 - 1.96 (m, 1 H), 2.04 - 2.17 (m, 1 H), 3.17 - 3.29 (m, 2 H), 3.33 - 3.39 (m, 1 H), 3.42 - 3.59 (m, 2 H), 4.36 (d, 1 H), 6.96 (d, 1 H), 7.38 (dd, 1 H), 7.61 (d, 1 H), 7.66 (d, 1 H), 7.94 (d, 1 H); MS(m/z): 391 [MH-56]+.
Intermediate 39: 1.1-dimethylethyl (1 S.6S/1 R.6RV6-(3.4-dichlorophenvn-1-r(2.4- dimethyl-1 H-pyrrol-1-yl)methyll-3-azabicvclor4.1.0lheptane-3-carboxylate (Racemate)

Sodium hydride 60% in mineral oil (21.3 mg) was added at 0 0C to a solution of 2,4- dimethyl-1 H-pyrrole (50.7 mg) in dry DMF (1.4ml_). The mixture was stirred at this temperature for 15min, then for an additional 5min at room temperature. A solution of 1 , 1 -dimethylethyl (1 R.6S/1 S,6R)-6-(3,4-dichlorophenyl)-1 -
{[(methylsulfonyOoxylmethylJ-S-azabicyclo^.i .Olheptane-S-carboxylate (Intermediate 26, 120 mg) in dry DMF (1 ml.) was then added and the mixture was stirred overnight at 50 0C. After cooling to RT, a saturated NH4CI solution was added and the mixture was extracted with diethyl ether (2x). The organic phase was washed with brine, dried over Na2SO4 and concentrated in vacuo to give 133mg of crude compound, which was purified by silica gel flash chromatography (gradient ethyl acetate in cyclohexane from 5 to 40%) to give 69mg of the title compound as a brown oil; MS(m/z): 449 [MH]+. Rf = 0.55 (cyclohexane/ethyl acetate 8/2).
Intermediate 40: 1.1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-1- {rmethyl(methyloxy)aminolcarbonyl}-3-azabicvclor4.1.0lheptane-3-carboxylate (Racemate)

To a suspension of 3-(1 ,1 -dimethylethyl) 1 -ethyl (1 R,6S/1S,6R)-6-(3,4- dichlorophenyl)-3-azabicyclo[4.1.0]heptane-1 ,3-dicarboxylate (Intermediate 6, 466 mg) and N,O-dimethylhydroxylamine hydrochloride (165 mg) in dry THF (2ml_) at -50
0C was added dropwise LiHMDS (1 M in THF, 3.82 ml_). The mixture was allowed to warm slowly to -2O0C. After 1 h 10min a saturated aqueous NH4CI solution was added and the mixture was stirred for 30min at room temperature. The mixture was extracted with ethyl acetate (2x), the organic phase was washed with brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by silica gel flash chromatography (gradient ethyl acetate in cyclohexane from 7 to 40%) to give 450mg of the title compound as a colorless oil; MS(m/z): 373 [MH-56]+. UPLC: Rt= 0.88 min.
Intermediate 41 : 1.1-dimethylethyl (1 R.6S/1S.6RV1-acetyl-6-(3.4-dichlorophenvn-3- azabicvclor4.1.Olheptane-3-carboxylate (Racemate)

Methylmagnesium bromide (3M in diethyl ether, 1.046 mL) was added at -78 0C to a solution of 1 ,1-dimethylethyl (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-1- {[methyl(methyloxy)amino]carbonyl}-3-azabicyclo[4.1.0]heptane-3-carboxylate
(Intermediate 40, 449 mg) in dry THF (5.5 mL). After 1 h stirring at -78 0C, the mixture was allowed to warm to 0 0C and stirred at 0 0C for 2h. Finally it was warmed to rt and stirred for 2h. A saturated aqueous NH4CI solution was then added at 0 0C and the mixture was stirred for 30min at room temperature. The mixture was extracted with ethyl acetate (2x), the organic phase was washed with brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by silica gel flash chromatography (gradient ethyl acetate in cyclohexane from 7 to 60%) to give the title compound (376mg) as a colorless oil; MS(m/z): 328 [MH-56]+. UPLC: Rt= 0.89 min.
Intermediate 42: 1 ,1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-1- [ethanehydrazonoyli-S-azabicvcloK.I .OIheptane-S-carboxylate (Racemate)

Hydrazine (0.084 mL) was added to a solution of 1 ,1-dimethylethyl (1 R,6S/1 S,6R)-1- acetyl-6-(3,4-dichlorophenyl)-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 41 , 189 mg) in dry methanol (1 mL). The mixture was stirred at 65 0C. After 4h additional hydrazine (0.084 mL) was added and the mixture heated for 2 further hour. The mixture was then concentrated in vacuo and the residue partitioned between water and ethyl acetate. The organic phase was dried over Na2SC>4 and concentrated in vacuo to give 182mg of the crude title compound; MS(m/z): 398 [MH]+.
Intermediate 43: 4-methyl-1 /-/-pyrrole-3-carbonitrile

To a stirred suspension of sodium hydride 60% in mineral oil (0.358 g) in dry THF (25 mL) at 0 0C was added a solution of TosMIC (1.45 g) and crotonitrile (0.5 g) in dry THF (35 mL). The reaction mixture was heated under reflux for 1 h and then the solution was quenched with water, concentrated in vacuo and extracted with DCM. The organic phase was dried and concentrated under vacuum and the residue was purified by flash-chromatography on silica geleluting with ethyl acetate in cyclohexane with a gradient from 10 to 40% to give title compound (0.322 g); 1 H NMR (CDCI3): δ ppm 8.35 (bs, 1 H) 7.23 (s, 1 H) 6.59 (s, 1 H) 2.22 (s, 3 H); MS(m/z): 107 [MH]+.
Intermediates 44 and 45: 1 ,1-dimethylethyl (1 S,6R/7R6S)-6-(3,4-dichlorophenyl)-1- (4,5,6,7-tetrahvdro-1 /-/-indazol-1-ylmethyl)-3-azabicvclo[4.1.01heptane-3-carboxylate (Racemate); and
1.1-dimethylethyl (1 S.6/ytR6S)-6-(3.4-dichlorophenyl)-1-(4.5.6.7-tetrahvdro-2H- indazol-2-ylmethyl)-3-azabicvclo[4.1.OIheptane-3-carboxylate (Racemate)

1 ,1-Dimethylethyl (1 S,6R/7R,6S)-6-(3,4-dichlorophenyl)-1- {[(methylsulfonyl)oxy]methyl}-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate
26, 250 mg) was reacted with 4,5,6,7-tetrahydro-1H-indazole (136 mg) in a similar fashion to the preparation of Intermediate 39. Purification by chromatography on silica gel (ethyl acetate in cyclohexane from 0 to 30 %) gave:
Intermediate 44; 1H NMR (CDCI3, 400 MHz) δ ppm 7.36 - 7.41 (m, 2 H) 7.26 (br. s., 1 H) 7.13 - 7.20 (m, 1 H) 3.86 (d, 1 H) 3.67 (m, 1 H) 3.37 - 3.46 (m, 1 H) 3.28 (d, 1 H)
2.45 (m, 2 H) 2.40 (m, 1 H) 2.25 (m, 1 H) 2.05 (m, 1 H) 1.64 - 1.82 (m, 4 H) 1.62 (br. s., 2 H) 1.45 (d, 1 1 H) 1.13 (br. s., 1 H); MS(m/z): 476 [MH]+; and then
Intermediate 45; 1H NMR (CDCI3, 400 MHz) δ ppm 7.36 - 7.45 (m, 2 H) 7.14 (dd, 1 H)
6.94 (m, 1 H) 4.03 (d, 1 H) 3.77 - 3.90 (m, 1 H) 3.52 (d, 1 H) 3.43 (m, 1 H) 3.31 (m, 2 H) 2.65 (m, 2 H) 2.5 (m, 2 H) 1.95 - 2.17 (m, 2 H) 1.8 (m, 2 H) 1.75 (m, 2 H) 1.62 (s,
1 H) 1.39 - 1.53 (m, 9 H) 1.09 (dd, 1 H); MS(m/z): 476 [MH]+
Intermediate 46: 1.1-dimethylethyl (1 S,6/?/7R6S)-6-(3,4-dichlorophenyl)-1-{[5-(1- methylethyl)-1 /-/-tetrazol-1-yl1methyl}-3-azabicvclo[4.1.01heptane-3-carboxylate (Racemate); and
Intermediate 47:1.1-dimethylethyl (1 S.6/?/yR6S)-6-(3.4-dichlorophenyl)-1-fr5-(1- methylethyl)-2/-/-tetrazol-2-yl1methyl}-3-azabicvclo[4.1.01heptane-3-carboxylate
(Racemate)

1 ,1-Dimethylethyl (1 S,6R/7R,6S)-6-(3,4-dichlorophenyl)-1-
{[(methylsulfonyOoxyJmethylJ-S-azabicyclo^.i .OJheptane-S-carboxylate (Intermediate 26, 250 mg) was reacted with 5-(1-methylethyl)-1 H-tetrazole (124 mg) in a similar fashion to the preparation of intermediate 39. Purification by chromatography on silica gel (eluting with ethyl acetate in cyclohexane from 0 to 50%) gave: Intermediate 46 (33mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.37 - 7.44 (m, 2 H) 7.15 (dd, 1 H) 3.57 - 4.18 (m, 4 H) 3.29 - 3.50 (m, 2 H) 2.69 - 2.89 (m, 1 H) 1.97 - 2.30 (m, 2 H) 1.45 (s, 9 H) 1.29 - 1.36 (m, 7 H) 1.20 (d, 1 H); MS(m/z): 466 [MH]+; and then Intermediate 47 (75 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.39 - 7.47 (m, 2 H) 7.17 (dd, 1 H) 4.43 (d, 1 H) 3.85 - 4.01 (m, 1 H) 3.72 - 3.84 (m, 0 H) 3.60 (d, 1 H) 3.39 - 3.52 (m, 1 H) 3.19 - 3.36 (m, 2 H) 2.11 - 2.21 (m, 1 H) 1.99 - 2.1 1 (m, 1 H) 1.56 (s, 4 H) 1.42 - 1.48 (m, 9 H) 1.39 (dd, 4 H) 1.10 (d, 1 H); MS(m/z): 466 [MH]+.
Intermediate 48: 1.1-dimethylethyl (1 S.6R/1 R.6SV6-(3.4-dichlorophenvn-1-r(3.5- dimethyl-1 H-1 ,2,4-triazol-1-yl)methyl1-3-azabicvclo[4.1.OIheptane-3-carboxylate (Racemate)

The title compound (40 mg) was obtained in a similar fashion to the preparation of intermediate 39, starting from 1 ,1-dimethylethyl (1 S,6R/7R,6S)-6-(3,4-
dichlorophenyl)-1-{[(methylsulfonyl)oxy]methyl}-3-azabicyclo[4.1.0]heptane-3- carboxylate (Intermediate 26, 250 mg) and 3,5-dimethyl-1 H-1 ,2,4-triazole (108 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.36 - 7.49 (m, 2 H) 7.15 (d, 1 H) 3.92 (d, 1 H) 3.77 (d, 1 H) 3.51 - 3.62 (m, 1 H) 3.21 - 3.46 (m, 3 H) 2.32 (s, 3 H) 2.26 (s, 3 H) 2.08 - 2.20 (m, 1 H) 1.97 - 2.08 (m, 1 H) 1.39 - 1.50 (m, 10 H) 1.09 - 1.15 (m, 1 H); MS(m/z): 451 [MH]+.
Intermediate 49: 1.1-dimethylethyl (1 S.6R/1 R.6SV6-(3.4-dichloroDhenvn-1-fr3- methyl-5-(trifluoromethyl)-1 H-pyrazol-1-yllmethyl}-3-azabicvclor4.1.0lheptane-3- carboxylate (Racemate); and
Intermediate 50: 1.1-dimethylethyl (1 S.6/?/7R6SV6-(3.4-dichlorophenvn-1-fr5- methyl-3-(trifluoromethyl)-1 /-/-pyrazol-1-yllmethyl}-3-azabicvclor4.1.0lheptane-3- carboxylate (Racemate)

1 ,1-Dimethylethyl (1 S,6R/7R6S)-6-(3,4-dichlorophenyl)-1-
{[(methylsulfony^oxyJmethylJ-S-azabicyclo^.i .OJheptane-S-carboxylate (Intermediate 26, 250 mg) was reacted with 5-methyl-3-(trifluoromethyl)-1 H-pyrazole (167 mg) in a similar fashion to the preparation of intermediate 39. Purification by chromatography using silica gel (eluting with ethyl acetate in cyclohexane from 0 to 20%) gave: Intermediate 49 (456 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.36 - 7.47 (m, 2 H) 7.15 - 7.23 (m, 1 H) 6.24 (br. s., 1 H) 3.94 - 4.04 (m, 1 H) 3.83 - 3.91 (m, 1 H) 3.74 - 3.81 (m, 1 H) 3.40 - 3.54 (m, 1 H) 3.26 - 3.35 (m, 1 H) 2.1 1 - 2.20 (m, 4 H) 2.07 (s, 1 H) 1.61 (s, 1 H) 1.45 (dd, 10 H) 1.12 (dt, 1 H); MS(m/z): 504 [MH]+; and then
Intermediate 50 (146 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.48 (d, 1 H) 7.41 (d, 1 H) 7.19 (dd, 1 H) 6.29 (s, 1 H) 4.00 (d, 1 H) 3.40 - 3.55 (m, 2 H) 2.55 (dd, 2 H) 2.27 - 2.31 (m, 2 H) 2.11 - 2.21 (m, 2 H) 1.58 - 1.60 (m, 2 H) 1.51 (d, 1 H) 1.46 (s, 9 H) 1.08 - 1.14 (m, 1 H); MS(m/z): 504 [MH]+.
Intermediate 51 : 1.1-dimethylethyl (1 R6S/7S.6/?)-1-(azidomethyl)-6-(3,4- dichlorophenyl)-3-azabicvclo[4.1.01heptane-3-carboxylate (Racemate)

To a solution of 1 ,1-dimethylethyl (1 S,6R/yR,6S)-6-(3,4-dichlorophenyl)-1- {[(methylsulfonyOoxylmethylJ-S-azabicyclo^.i .Olheptane-S-carboxylate (Intermediate 26, 212 mg) in DMF (5 ml.) was added sodium azide (92 mg). The mixture was heated to 50 0C for 4 hr and then cooled to room temperature. It was partitioned between DCM and water, then the volatiles were removed in vacuo affording the title compound as colourless oil (150 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.42 (d, 1 H) 7.38 (d, 1 H) 7.14 (dd, 1 H) 3.8 - 4.0 (m, 2 H) 3.57 - 3.65 (m, 1 H) 3.28 - 3.45 (m, 2 H) 2.98 (d, 1 H) 1.94 - 2.12 (m, 2 H) 1.51 (s, 9 H) 1.02 - 1.12 (m, 2 H); MS(m/z): 340 [MH]+.
Intermediate 52: 1.1-dimethylethyl (1 R6S/7S.6RV6-(3,4-dichlorophenyl)-1-r(2,5- dimethyl-1 /-/-pyrrol-1-yl)methyl1-3-azabicvclo[4.1.01heptane-3-carboxylate
(Racemate)

To a stirred solution of 1 ,1-dimethylethyl (1 R6S/7S,6R)-1-(azidomethyl)-6-(3,4- dichlorophenyl)-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 51 , 0.1 g) in DCM (10 ml.) at room temperature was added triphenylphosphine (123 mg). After 20hr stirring at room temperature, the reaction mixture was heated to 40 0C for 3 hr
and then left at room temperature for 15 days. The solvent was removed in vacuo and the residue was submitted to chromatographic purification on silica gel (eluting with DCM/MeOH/NH3 (2M MeOH) 9/0.9/0.1 ). The purified residue was suspended in ethanol (10 ml.) and heated to 80 0C. 2,5-Hexanedione (0.035 ml) was added at this temperature followed by HCI (37% in water, catalytic amount). The mixture was heated to 80 0C for 2 hr and, after cooling to room temperature, water and DCM were added. The organic layer was separated and purified by chromatography on silica gel with a gradient from 0 to 25% ethyl acetate in cyclohexane to give 36 mg of title compound as colourless oil; 1H NMR (CDCI3, 400 MHz) δ ppm 7.46 (d, 1 H) 7.42 (d, 1 H) 7.18 (dd, 1 H) 5.75 (br. s., 2 H) 3.85 (d, 1 H) 3.27 - 3.46 (m, 2 H) 3.25 (d, 1 H) 2.88 (d, 1 H) 2.08 - 2.20 (m, 8 H) 1.96 - 2.08 (m, 1 H) 1.39 - 1.48 (m, 9 H) 1.25 - 1.33 (m, 1 H) 1.16 - 1.21 (m, 1 H); MS(m/z): 449 [MH]+.
Intermediate 53: 1.1-dimethylethyl (1 R6S/7S.6/?>6-(3.4-dichlorophenvn-1-(1 H-1.2.4- triazol-1-ylmethyl)-3-azabicvclor4.1.0lheptane-3-carboxylate (Racemate)

1 ,2,4-Triazole (0.026 g, 0.377 mmol) was dissolved in DMF (4 ml) and cooled to 0 0C. Sodium hydride 60% in mineral oil (0.015 g) was added portionwise and the mixture stirred for 10 min until hydrogen evolution ceased. 1 ,1-Dimethylethyl (1 R,6S)-6-(3,4-dichlorophenyl)-1-{[(methylsulfonyl)oxy]methyl}-3- azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 26, 0.1 g) in DMF (1 ml_) was then added and mixture stirred at 50 0C for 12h. After cooling to room temperature, the mixture was quenched with a saturated solution of NH4CI (2 ml.) and partitioned between diethyl ether (20 ml.) and water (20 ml_), the organic phase was washed with brine (20 ml_), dried and concentrated under reduced pressure. Purification by chromatography on silica gel eluting with a gradient from 0 to 50% ethyl acetate in cyclohexane afforded the title compound as a colourless oil (50 mg); UPLC: Rt= 0.86 min, MS(m/z): 423 [MH]+.
Intermediate 54: 1.1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-1-[(4- methyl-I H-pyrazol-i-vDmethyli-S-azabicvcloK.I .OIheptane-S-carboxylate
(Racemate)

The title compound (100 mg) was obtained in a similar fashion to the preparation of Intermediate 39 starting from 1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)- ^{[(methylsulfony^oxylmethylJ-S-azabicyclo^.i .Olheptane-S-carboxylate (Intermediate 26, 0.120 g) and 4-methyl-1 H-pyrazole (0.031 ml_); MS(m/z): 436 [MH]+.
Intermediate 55: 1.1-dimethylethyl (1 R6S/7S.6/?)-6-(3.4-dichlorophenyl)-14(3.5- dimethyl-1 /-/-pyrazol-1-yl)methyll-3-azabicvclor4.1.0lheptane-3-carboxylate
(Racemate)

The title compound (45 mg) was obtained in a similar fashion to the preparation of Intermediate 39 starting from 1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)- ^{[(methylsulfony^oxyJmethylJ-S-azabicyclo^.i .OJheptane-S-carboxylate (Intermediate 26, 0.1 g) and 3,5-dimethyl-1 H-pyrazole (0.036 g); 1H NMR (CDCI3, 400 MHz) δ ppm 7.35 - 7.48 (2 H, m), 7.17 (1 H, d), 5.69 - 5.80 (1 H, m), 3.94 (1 H, d), 3.48 (5 H, d), 2.17 - 2.25 (3 H, m), 1.96 - 2.16 (5 H, m), 1.31 - 1.52 (10 H, m), 1.06 - 1.16 (1 H, m); UPLC: Rt= 1.01 min, MS(m/z): 450 [MH]+.
Intermediate 56: 1.1-dimethylethyl (1 S,6S/7R6/?)-6-(3,4-dichlorophenyl)-1-(1 H- pyrrol-i-ylmethylKS-azabicvcloK.I .O'lhept.ane-S-carboxylat.e

The title compound (45 mg) was obtained in a similar fashion to the preparation of Intermediate 39 starting from 1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)- ^{[(methylsulfony^oxylmethylJ-S-azabicyclo^.i .Olheptane-S-carboxylate (Intermediate 26, 84 mg) and pyrrole (16.3 mg); UPLC: Rt= 1.07 min, MS(m/z): 365 [MH-56]+.
Intermediate 57: 1.1-dimethylethyl (1 R.6S/1S.6RV6-(3.4-dichlorophenvn-1-(2- oxoethyl)-3-azabicvclor4.1.Olheptane-3-carboxylate (Racemate)

Intermediate 58: 1 ,1-dimethylethyl (1 R6S/7S.6/?)-6-(3,4-dichlorophenyl)-1-r(4- methyl-1 ,3-oxazol-5-yl)methyl1-3-azabicvclo[4.1.OIheptane-3-carboxylate (Racemate)

1 ,1-Dimethylethyl (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-1-(2-oxoethyl)-3- azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 57, 0.170 g) was dissolved in methanol. K2CO3 (0.061 g) was then added followed by 1-[(1- isocyanoethyl)sulfonyl]-4-methylbenzene (0.093 g; for a reference procedure of preparation see Tetrahedron, 44 (23), 7243-7254, 1988). The mixture was heated to reflux temperature and stirred for 6h. It was left stirring at 50 ° C overnight before removing the volatiles under reduced pressure. The residue was partitioned between DCM and water. 0.1 N HCI was added to obtain clear phases. The organic phase was washed with water, dried and concentrated. Purification by chromatography on silica eluting with a gradient from 10 to 50% ethyl acetate in cyclohexane afforded the title compound as a pale yellow oil (58 mg); UPLC: Rt = 0.86 min; MS(m/z): 328 [MH- 56]+.
Intermediate 59: 1 ,1-dimethylethyl (1 S,6S or 7R6/?)-6-(3,4-dichlorophenyl)-1-(1 H- pyrrol-1-ylmethyl)-3-azabicvclo[4.1.OIheptane-3-carboxylate (single enantiomer)

The title compound (30 mg) was obtained in a similar fashion to the preparation of Intermediate 56 starting from 1-dimethylethyl (1 R.6S or 1S,6R)-6-(3,4- dichlorophenyl)-1-{[(methylsulfonyl)oxy]methyl}-3-azabicyclo[4.1.0]heptane-3- carboxylate (Intermediate 29, 53 mg) and pyrrole (16 mg); MS(m/z): 365 [MH-56]+.
Supporting Compounds
Compound 1 : π R6S/7S.6/?V6-(3.4-dichlorophenylV1-(5-methyl-1.3-oxazol-2-ylV3- azabicvclor4.1.Olheptane (Racemate)

To a stirred solution of 1 , 1-dimethylethyl (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-1-[(2- propyn-1-ylamino)carbonyl]-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 8, 0.270 g) in acetic acid (8 ml_), mercuric acetate (0.025 g) was added and the solution was heated to 120 0C and stirred for 2.5h. The solvent was removed under reduced pressure and the residue dissolved in ethyl acetate, washed with a saturated solution of NaHCC>3 then with brine. The organic phase was dried and concentrated under reduced pressure. The residue was purified by chromatography on silica gel eluting with a gradient from 30 to 70% ethyl acetate in cyclohexane to afford a colourless oil (60 mg). To this oil was added DCM (8 ml) and then TFA (0.37
ml_). The reaction mixture was stirred at RT for 2h, then the solvent was evaporated under reduced pressure and the residue purified by SCX eluting first with MeOH and then with 2.0N NH3 in MeOH to afford the title compound as a colourless oil. HPLC purification (Preparative conditions: column Luna C18 AXIA 100 x 21 mm, 5 μm, eluent A: H2O + 0.1% TFA; B: CH3CN + 0.1% TFA, gradient: 15% to 35% (B) in 15 min, 35% to 100% (B) in 3 min, 100% (B) for 2 min, flow rate 17.0 ml/min., detection UV 210-350 nm; Analytical conditions: column Acquity UPLC™ BEH C18 50 x 2.1 mm, 1.7 μm, eluent A: H2O + 0.1% TFA; B: CH3CN + 0.1% TFA, gradient: 15% to 30% (B) in 4 min, 30% to 100% (B) in 0.5 min, 100% to 15% (B) in 0.25 min, flow rate 1.0 ml/min., detection UV 210-350 nm) and the product containing fractions were evaporated to dryness. SCX purification afforded the title compound as a free base (8.0 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.27 (1 H, d), 7.20 - 7.24 (1 H, d), 6.98 (1 H, dd), 6.40 (1 H, q), 3.95 (1 H, d), 3.28 (1 H, d), 2.76 - 2.93 (2 H, m), 2.12 (1 H, d), 2.05 - 2.07 (4 H, m), 1.89 - 1.98 (1 H, m), 1.42 - 1.45 (1 H, d); MS(m/z): 323 [MH]+.
Compound 2: (1 R6S/yS.6ffl-6-(3.4-dichloroDhenvn-1-(4-methyl-1.3-thiazol-2-vn-3- azabicyclo[4.1.Oiheptane (Racemate)

1 ,1-Dimethylethyl (1 R,6S/7S,6R)-6-(3,4-dichlorophenyl)-1-(4- methyl-1 ,3-thiazol-2-yl)-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 11 ) (80 mg) was dissolved in DCM (2 mL) and TFA (0.3 mL) added. The mixture was stirred at room temperature for 2h and then the solvent was evaporated under reduced pressure. Purification of the residue with SCX, eluting first with MeOH and then with 2.0N NH3 in MeOH afforded the title compound as a pale yellow oil (68 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.26 - 7.31 (1 H, m), 7.18 (1 H, d), 6.99 (1 H, dd), 6.51 (1 H, q), 3.89 (1 H, d), 3.39 (1 H, d), 2.82 - 2.99 (2 H, m), 2.28 (3 H, d), 1.91 - 2.13 (3 H, m), 1.56 (1 H, d); UPLC: Rt = 0.63 min, MS(m/z): 339 [MH]+.
Compound 2 (50 mg) was submitted to semi-preparative chiral HPLC (Preparative conditions: chiral column Chiralcel OD-H, 25 x 0.46 cm, eluent A: n-hexane; B: ethanol 85/15 v/v, flow rate 13 ml/min., detection UV at 225 nm. Analytical conditions: chiral column Chiralcel OD-H, eluent A: n-hexane; B: ethanol 85/15 v/v, flow rate 0.8 ml/min., DAD 210-340 nm) obtaining:
Compound 3: (1 R6S or 7S.6RV6-(3,4-dichlorophenyl)-1-(4-methyl-1 ,3-thiazol-2-yl)- 3-azabicyclo[4.1.01heptane, Enantiomer 1 , Rt. = 7.73 min, 18 mg, colourless oil; 1H NMR (CDCI3, 400 MHz) δ ppm 7.25 - 7.34 (1 H, m), 7.19 (1 H, d), 6.99 (1 H, dd), 6.52 - 6.53 (1 H, m), 3.90 (1 H, d), 3.40 (1 H, d), 2.85 - 2.98 (2 H, m), 2.28 - 2.30 (3 H, m), 1.93 - 2.13 (3 H, m), 1.57 (1 H. d): MS(m/z): 339 [MH1+: and Compound 4: (1 S.6/? or tR6SV6-(3.4-dichloroDhenyl)-1-(4-methyl-1.3-thiazol-2-vn-3- azabicvclor4.1.0lheptane, Enantiomer 2, Rt. = 8.62 min, 18 mg, colourless oil; 1H NMR (CDCI3, 400 MHz) δ ppm 7.23 - 7.32 (1 H, m), 7.19 (1 H, d), 7.00 (1 H, dd), 6.51 - 6.56 (1 H, m), 3.90 (1 H, d), 3.41 (1 H, d), 2.83 - 2.99 (2 H, m), 2.28 - 2.30 (3 H, m), 1.94 - 2.14 (3 H, m), 1.58 (1 H, d); MS(m/z): 339 [MH]+.
Compound 5: (1 R6S or 7S.6RV6-(3,4-dichlorophenyl)-1-(4-methyl-1 ,3-thiazol-2-yl)-
3-azabicyclo[4.1.01heptane dihydrochloride (Single enatiomer)
Compound 3 (18 mg) was dissolved in 0.1 ml of diethyl ether and was treated with 2.2 eq of 1.25 M HCI in MeOH (93 μl) to give the title compound as a colourless solid which was triturated with diethyl ether and dried in vacuo (18 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 11.06 (1 H, br. s.), 9.67 (1 H, br. s.), 7.71 (1 H, d), 7.60 (1 H, dd), 7.33 (1 H, d), 6.93 - 6.99 (1 H, m), 4.43 - 4.58 (1 H, m), 3.97 - 4.12 (1 H, m), 3.57 - 3.69 (1 H, m), 3.18 - 3.39 (1 H, m), 2.68 - 2.81 (1 H, m), 2.60 (3 H, s), 2.41 - 2.52 (2 H, m), 2.31 - 2.40 (1 H, m); UPLC: Rt = 0.63 min, MS(m/z): 339 [MH]+.
Compound 6: (1 S.6Z? or 7R6SV6-(3.4-dichlorophenylV1-(4-methyl-1.3-thiazol-2-ylV
3-azabicvclo[4.1.01heptane dihvdrochloride (Single enantiomer)
Compound 4 (18 mg) was dissolved in 0.1 mL of diethyl ether and was treated with 2.2 eq of 1.25 M HCI in MeOH (93 μl) to give the title compound as a colourless solid which was triturated with diethyl ether and dried in vacuo (18 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 11.06 (1 H, br. s.), 9.53 (1 H, br. s.), 7.58 (1 H, d), 7.46 (1 H, dd), 7.22 (1 H, d), 6.77 - 6.86 (1 H, m), 4.29 - 4.41 (1 H, m), 3.83 - 3.94 (1 H, m), 3.46 - 3.57 (1 H, m), 3.08 - 3.24 (1 H, m), 2.58 - 2.73 (1 H, m), 2.49 (3 H, s), 2.31 - 2.42 (2 H, m), 2.16 - 2.29 (1 H, m); UPLC: Rt = 0.63 min, MS(m/z): 339 [MH]+.
Compound 7: (1 R6S/yS,6/?)-6-(3,4-dichlorophenyl)-1-r(4-methyl-1 ,3-thiazol-2- yl)methyl1-3-azabicvclo[4.1.Oiheptane (Racemate)

i .i-DimethylethyKI R.eS/I S.βRJ-β-CS^-dichlorophenylJ-i-^-methyl-I .S-thiazol^- y^methylJ-S-azabicyclo^.i .OJheptane-S-carboxylate (Intermediate 18, 26 mg) was dissolved in DCM (2 ml.) and TFA (0.203 mL) was added. The mixture was stirred at room temperature for 2h and then the solvent was evaporated under reduced pressure. Purification of the residue with SCX eluting first with MeOH and then with 2.0N NH3 in MeOH afforded the title compound as a pale yellow oil (22 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.38 - 7.41 (2 H, m), 7.15 (1 H, dd), 6.71 - 6.75 (1 H, m), 3.07 - 3.16 (2 H, m), 3.02 (1 H, d), 2.71 - 2.83 (2 H, m), 2.41 (3 H, s), 2.22 (1 H, d), 1.99 - 2.08 (1 H, m), 1.90 - 1.98 (1 H, m), 1.29 (1 H, d), 1.15 - 1.19 (1 H, m); ); UPLC: Rt= 0.64 min; MS(m/z): 353 [MH]+.
Racemic Compound 7 (19 mg) was submitted to semi-preparative chiral HPLC (Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30 v/v, flow rate 13 ml/min., detection UV 220 nm. Analytical conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30 v/v, flow rate 0.8 ml/min., DAD 210-340 nm) obtaining:
Compound 8: (1 R6S or 7S.6/?)-6-(3,4-dichlorophenyl)-1-[(4-methyl-1 ,3-thiazol-2- yl)methyl1-3-azabicvclo[4.1.01heptane, Enantiomer 1 , Rt. = 8.95 min, 7.5 mg, as a colourless oil; 1H NMR (CDCI3, 400 MHz) δ ppm 7.36 - 7.42 (2 H, m), 7.15 (1 H, dd), 6.73 (1 H, d), 3.06 - 3.16 (2 H, m), 3.02 (1 H, dd), 2.71 - 2.83 (2 H, m), 2.41 (3 H, s), 2.21 (1 H, d), 1.89 - 2.09 (2 H, m), 1.28 (1 H, d), 1.16 (1 H, d); MS(m/z): 353 [MH]+; and Compound 9: (1S.6R or 1R6S)-6-(3,4-dichlorophenyl)-1-[(4-methyl-1 ,3-thiazol- 2-yl)methyl1-3-azabicvclo[4.1.Oiheptane, Enantiomer 2, Rt. = 10.92 min, 7.6 mg, colourless oil; 1H NMR (CDCI3, 400 MHz) δ ppm 7.37 - 7.41 (2 H, m), 7.15 (1 H, dd), 6.73 (1 H, d), 3.06 - 3.15 (2 H, m), 2.99 - 3.05 (1 H, m), 2.70 - 2.84 (2 H, m), 2.39 -
2.42 (3 H, m), 2.21 (1 H, d), 1.89 - 2.09 (2 H, m), 1.29 (1 H, d), 1.16 (1 H, dd); MS(m/z): 353 [MH]+.
Compound 10: (1 R6S or 7S.6/?)-6-(3,4-dichlorophenyl)-1-[(4-methyl-1 ,3-thiazol-2- yl)methyll-3-azabicvclo[4.1.0lheptane hydrochloride (Single enantiomer)
Compound 8 (7.5 mg) was dissolved in 0.2 ml of diethyl ether and was treated with 1.1 eq of 1.25 M HCI in MeOH (19 μl) to give the title compound as a colourless solid which was triturated with diethyl ether and dried in vacuo (7.5 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 9.66 - 10.09 (2 H, m), 7.47 - 7.51 (1 H, m), 7.34 - 7.43 (2 H, m), 6.75 (1 H, s), 3.81 (1 H, d), 3.35 - 3.45 (1 H, m), 3.21 - 3.31 (1 H, m), 3.08 (1 H, d), 2.87 - 3.00 (1 H, m), 2.49 - 2.60 (1 H, m), 2.44 - 2.34 (4 H, m), 2.24 - 2.34 (1 H, m), 1.46 (1 H, d), 1.32 (1 H, d).
Compound 1 1 : (1 R6S or 7S.6RV6-(3.4-dichlorophenylV1-r(4-methyl-1.3-thiazol-2- yl)methyl1-3-azabicvclo[4.1.01heptane hydrochloride (Single enantiomer) Compound 9 (7.6 mg) was dissolved in 0.2 ml. of diethyl ether and was treated with 1.1 eq of 1.25 M HCI in MeOH (19 μl) to give the title compound as a colourless solid which was triturated with diethyl ether and dried in vacuo (7.6 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.46 - 7.49 (1 H, m), 7.39 (1 H, d), 7.31 - 7.36 (1 H, m), 6.71 - 6.78 (1 H, m), 3.73 (1 H, d), 3.28 - 3.37 (1 H, m), 3.24 (1 H, d), 3.07 (1 H, d), 2.86 - 2.97 (1 H, m), 2.35 - 2.52 (5 H, m), 2.20 - 2.31 (1 H, m), 1.43 (1 H, d), 1.29 (1 H, d); MS(m/z): 353 [MH]+.
Compound 12: (1 R6S/7S.6/?V6-(3.4-dichlorophenylV1-r(4-methyl-1.3-oxazol-2- yl)methyl1-3-azabicvclo[4.1.Oiheptane (Racemate)

To a stirred solution of 1 ,1-dimethylethyl (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-1-[(4- methyl-i ^-oxazol^-y^methylJ-S-azabicyclo^.i .OJheptane-S-carboxylate
(Intermediate 20, 70.0 mg) in DCM (7 ml_), at 0 0C was added TFA (0.123 ml). The reaction mixture was slowly warmed to RT and stirred at RT for 1 h. The volatiles were evaporated in vacuo and the residue was purified by SCX eluting first with MeOH and then with 2.0N NH3 in MeOH. The title compound was obtained as a yellow oil (55 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.34 - 7.38 (2 H, m), 7.24 (1 H, d), 7.12 (1 H, dd), 3.07 - 3.19 (2 H, m), 2.71 - 2.82 (2 H, m), 2.58 (1 H, d), 2.10 - 2.17 (4 H, m), 1.88 - 2.05 (2 H, m), 1.24 (1 H, d), 1.10 (1 H, d); MS(m/z): 337 [MH]+.
Racemic Compound 12 (55 mg) was submitted to semi-preparative chiral HPLC (Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30 v/v, flow rate 14 ml/min., detection UV 235 nm. Analytical conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30 v/v, flow rate 0.8 ml/min., DAD 210-340 nm) obtaining:
Compound 13: (1 R6S or 7S.6ftV6-(3.4-dichlorophenylV1-r(4-methyl-1.3-oxazol-2- vnmethyll-3-azabicvclor4.1.0lheptane. Enantiomer 1. Rt = 7.72 min (23 mq): 1H NMR (CDCI3, 400 MHz) δ ppm 7.35 - 7.39 (2 H, m), 7.23 - 7.25 (1 H, m), 7.12 (1 H, dd), 3.07 - 3.19 (2 H, m), 2.71 - 2.82 (2 H, m), 2.58 (1 H, d), 2.11 - 2.17 (4 H, m), 1.89 - 2.06 (2 H, m), 1.25 (1 H, d), 1.10 (1 H, d); MS(m/z): 337 [MH]+; and Compound 14: (1S.6R or 1R6S)-6-(3,4-dichlorophenyl)-1-[(4-methyl-1 ,3-oxazol-2- yl)methyl1-3-azabicvclo[4.1.01heptane Enantiomer 2, Rt = 10.48 min (21 mq); 1H NMR (CDCI3, 400 MHz) δ ppm 7.36 (2 H, dd), 7.23 - 7.26 (1 H, m), 7.12 (1 H, dd), 3.07 - 3.18 (2 H, m), 2.71 - 2.83 (2 H, m), 2.58 (1 H, d), 2.10 - 2.17 (4 H, m), 1.88 - 2.06 (2 H, m), 1.24 (1 H, d), 1.10 (1 H, d); ); MS(m/z): 337 [MH]+.
Compound 15: (1 R6S or 7S.6RV6-(3,4-dichlorophenyl)-1-[(4-methyl-1 ,3-oxazol-2- yl)methyl1-3-azabicvclo[4.1.01heptane hydrochloride (Single enantiomer) Compound 13 (23 mg) was dissolved in 0.3 ml. of diethyl ether and was treated with 1.1 eq of HCI 1.25M in MeOH (60 μl) to give the title compound as a colourless solid which was triturated with diethyl ether and dried in vacuo (21 mg); 1H NMR (DMSO, 400 MHz) δ ppm 8.86 - 9.34 (2 H, m), 7.79 (1 H, s), 7.66 (1 H, d), 7.60 (1 H, d), 7.44 - 7.50 (1 H, m), 3.44 - 3.53 (1 H, m), 3.14 - 3.25 (2 H, m), 2.76 - 2.90 (2 H, m), 2.17 - 2.30 (1 H, m), 2.07 - 2.17 (1 H, m), 2.04 (3 H, d), 1.80 - 1.88 (1 H, m), 1.34 (2 H, m); MS(m/z): 337 [MH]+.
Compound 16: (1S.6R or 1 R6S)-6-(3,4-dichlorophenyl)-1-[(4-methyl-1 ,3-oxazol-2- yl)methyl1-3-azabicvclo[4.1.01heptane hydrochloride (Single enantiomer)
Compound 14 (21 mg) was dissolved in 0.3 ml. of diethyl ether and was treated with 1.1 eq of HCI 1.25M in MeOH (60 μl) to give the title compound as a colourless solid which was triturated with diethyl ether and dried in vacuo (21 mg); 1H NMR (DMSO, 400 MHz) δ ppm 8.74 - 9.08 (2 H, m), 7.73 - 7.81 (1 H, m), 7.57 - 7.69 (2 H, m), 7.40 - 7.50 (1 H, m), 3.42 - 3.56 (1 H, m), 3.10 - 3.26 (2 H, m), 2.73 - 2.92 (2 H, m), 2.17 - 2.31 (1 H, m), 2.07 - 2.16 (1 H, m), 2.04 (3 H, d), 1.79 - 1.90 (1 H, m), 1.34 (2 H, m), MS(m/z): 337 [MH]+.
Compound 17: π R6S/7S.6RV6-(3.4-dichlorophenylV1-π H-1.2.4-triazol-1-ylmethvn- 3-azabicvclor4.1.Olheptane (Racemate)

1 ,1-Dimethylethyl (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-1-(1 H-1 ,2,4-triazol-1- ylmethyl)-3-azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 53, 50 mg) was dissolved in DCM (1.0 ml.) and TFA (0.513 ml.) was added. The reaction mixture was stirred at room temperature for 2h. Volatiles were removed under reduced pressure and the residue purified by SCX, eluting first with MeOH and then with 2.0N NH3 in MeOH. The title compound was obtained as a colourless oil (40 mg); 1H NMR (CDCI3, 500 MHz) δ ppm 7.92 (1 H, s), 7.92 (1 H, s), 7.42 (1 H, d), 7.41 (1 H, d), 7.16 (1 H, dd), 4.12 (1 H, d), 3.41 (1 H, d), 3.14 (1 H, d), 2.95 (1 H, d), 2.68 - 2.83 (2 H, m), 1.99 - 2.09 (1 H, m), 1.90 - 1.99 (1 H, m), 1.37 (1 H, d), 1.22 (1 H, d); MS(m/z): 323 [MH]+.
Compound 18: (1 R6S or 7S.6RV6-(3,4-dichlorophenyl)-1-[(3-methyl-1 H-pyrazol-1- yl)methyl1-3-azabicvclo[4.1.01heptane (single enantiomer);
Compound 19: (1S.6R or 7R6SV6-(3.4-dichlorophenylV1-r(3-methyl-1 H-pyrazol-1- yl)methyl1-3-azabicvclo[4.1.01heptane (single enantiomer);
Compound 20: (1 R6S or 7S.6RV6-(3.4-dichlorophenylV1-r(5-methyl-1 H-pyrazol-1- yl)methyl1-3-azabicvclo[4.1.01heptane (single enantiomer); and
Compound 21 : (1S.6R or 1R6S)-6-(3,4-dichlorophenyl)-1-[(5-methyl-1 H-pyrazol-1- yl)methyl1-3-azabicvclo[4.1.01heptane (single enantiomer).

3-Methyl-1 H-pyrazole (0.125 ml.) was dissolved in DMF (4 ml.) and cooled to 0 0C. Sodium hydride 60% in mineral oil (0.062 g) was added portionwise and the mixture was stirred for 10 min until hydrogen evolution ceased. 1 ,1-Dimethylethyl (1 R.6S/1 S,6R)-6-(3,4-dichlorophenyl)-1 -{[(methylsulfonyl)oxy]methyl}-3- azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 26, 0.41 g) in DMF (1 ml.) was then added and the mixture was stirred for at 50 0C for 12h. The mixture was quenched with a saturated solution of NH4CI (2 ml.) and partitioned between Et20 (20 ml.) and water (20 ml_). The organic layer was washed with brine (20 ml_), dried and concentrated. Purification by chromatography on silica gel eluting with a gradient from 0 to 40% AcOEt in cyclohexane afforded a colourless oil (270 mg) which was submitted to chiral HPLC (Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: 2-propanol 90/10 v/v, flow rate 14 ml/min., detection UV 230 nm. Analytical conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: 2- propanol 90/10 v/v, flow rate 1 ml/min., DAD 210-340 nm) obtaining:
First eluting peak: Rt 6.18 min, colourless oil (71 mg) UPLC: Rt= 0.96 min, MS(m/z): 436 [MH]+. The oil was taken up in 2 mL of DCM and TFA (0.3 mL) was added. The reaction mixture was stirred for 3h at room temperature then the volatiles were evaporated under reduced pressure. The residue was purified by SCX cartrige eluting first with MeOH and then with 2.0N NH3 in MeOH. The ammonia fractions were concentrated under vacuum to give Compound 18 (Single enantiomer) as a colourless oil (58 mg) Rt = 15.24 min (Analytical conditions: chiral column Chiralcel OD-H, eluent A: n-hexane; B: 2-propanol 95/5 v/v, flow rate 1 ml/min., DAD 210-340 nm) >99% ee; UPLC: Rt= 0.63 min; MS(m/z): 336 [MH]+; 1H NMR (CDCI3, 400 MHz) δ ppm 7.33 - 7.43 (2 H, m), 7.10 - 7.21 (2 H, m), 5.97 (1 H, s), 4.01 (1 H, d), 3.25 (1 H, d), 3.12 (1 H, d), 2.91 (1 H, d), 2.65 - 2.82 (2 H, m), 2.25 (3 H, s), 1.82 - 2.07 (2 H, m), 1.36 (1 H, d), 1.16 (1 H, d).
Second eluting peak: Rt 7.31 min, colourless oil (73 mg). The oil was taken up in 2 mL of DCM and TFA (0.3 mL) was added. The reaction mixture was stirred for 3h at room temperature then the volatiles were evaporated under reduced pressure. The residue was purified by SCX cartrige eluting first with MeOH and then with 2.0N NH3 in MeOH. The ammonia fractions were concentrated under vacuum to give Compound 19 (Single enantiomer) as a colourless oil (55 mg) Rt = 18.02 min, >99% ee (Analytical conditions: chiral column Chiralcel OD-H, eluent A: n-hexane; B: 2- propanol 95/5 v/v, flow rate 1 ml/min., DAD 210-340 nm); UPLC: Rt= 0.63 min; MS(m/z): 336 [MH]+; 1H NMR (CDCI3, 400 MHz) δ ppm 7.33 - 7.43 (2 H, m), 7.10 - 7.21 (2 H, m), 5.97 (1 H, s), 4.01 (1 H, d), 3.25 (1 H, d), 3.12 (1 H, d), 2.91 (1 H, d), 2.65 - 2.82 (2 H, m), 2.25 (3 H, s), 1.82 - 2.07 (2 H, m), 1.36 (1 H, d), 1.16 (1 H, d).
Third eluting peak: Rt 8.44 min, colourless oil (44 mg). The oil was taken up in 2 mL of DCM and TFA (0.3 mL) was added. The reaction mixture was stirred for 3h at room temperature then the volatiles were evaporated under reduced pressure. The residue was purified by SCX cartrige eluting first with MeOH and then with 2.0N NH3 in MeOH. The ammonia fractions were concentrated under vacuum affording Compound 20 (Single enantiomer) as a colourless oil (36 mg), Rt = 26.23 min, >99% ee, (Analytical conditions: chiral column Chiralcel OD-H, eluent A: n-hexane; B: 2- propanol 95/5 v/v, flow rate 1 ml/min., DAD 210-340 nm); UPLC: Rt= 0.64 min; MS(m/z): 336 [MH]+; 1H NMR (CDCI3, 400 MHz) δ ppm 7.34 - 7.41 (3 H, m), 7.16 (1 H, dd), 5.96 (1 H, d), 3.90 (1 H, dd), 3.15 - 3.33 (2 H, m), 2.90 (1 H, d), 2.71 - 2.82 (2 H, m), 2.10 (3 H, s), 1.86 - 2.06 (2 H, m), 1.43 (1 H, d), 1.18 (1 H, d).
Fourth eluting peak: Rt 9.20 min, colourless oil (51 mg). The oil was taken up in 2 mL of DCM and TFA (0.3 mL) was added. The reaction mixture was stirred for 3h at room temperature then the volatiles were evaporated under reduced pressure. The residue was purified by SCX cartrige eluting first with MeOH and then with 2.0N NH3 in MeOH. The ammonia fractions were concentrated under vacuum to give
Compound 21 (Single enantiomer) as a colourless oil (38 mg), Rt = 19.27 min, 98% ee, (Analytical conditions: chiral column Chiralcel OD-H, eluent A: n-hexane; B: 2- propanol 95/5 v/v, flow rate 1 ml/min., DAD 210-340 nm); UPLC: Rt= 0.64 min; MS(m/z): 336 [MH]+; 1H NMR (CDCI3, 400 MHz) δ ppm 7.34 - 7.41 (3 H, m), 7.16 (1 H, dd), 5.96 (1 H, d), 3.90 (1 H, dd), 3.15 - 3.33 (2 H, m), 2.90 (1 H, d), 2.71 - 2.82 (2 H, m), 2.10 (3 H, s), 1.86 - 2.06 (2 H, m), 1.43 (1 H, d), 1.18 (1 H, d).
Compound 22: 1-{[(1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-3-azabicvclor4.1.0lhept- 1-yl1methyl}-1 H-indazole (Single enantiomer); and
Compound 23: 2-{[(1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-3-azabicvclor4.1.0lhept- 1-yl1methyl}-2H-indazole (Single enantiomer)

Sodium hydride 60% in mineral oil (17.8 mg) was added at 0 0C to a solution of indazole (52.5 mg) in dry DMF (0.5ml_). The mixture was stirred at this temperature for 15min and then for additional 15min at room temperature. A solution of 1 ,1- dimethylethyl (1 R.6S or 1S,6R)-6-(3,4-dichlorophenyl)-1-
{[(methylsulfonyOoxylmethylJ-S-azabicyclo^.i .Olheptane-S-carboxylate (Intermediate 29, 100 mg) in dry DMF (0.5 ml.) and sodium iodide (16.6 mg) were then added and the mixture was stirred at 50 0C. After 3h, water was added and the mixture was extracted with ethyl acetate (2x). The organic phase was dried over Na2SO4 and concentrated in vacuo to give a crude residue, which was purified by silica gel flash chromatography (gradient ethyl acetate in cyclohexane from 5 to 40%).
First eluting compound: The colourless oil (62mg) was dissolved in dry DCM (1.3ml_) and TFA (50μl_) was added at room temperature. After 30min, toluene (2ml_) was added and the mixture was concentrated in vacuo. The residue was purified by a SCX cartridge (1g) eluting with MeOH and then NH3 0.5M in MeOH. The basic fractions were combined and concentrated in vacuo to give Compound 22 (Single Enantiomer) (36mg) as a colourless oil; 1 H NMR (400 MHz, CDCI3) δ ppm 1.17 (d, 1 H), 1.51 (d, 1 H), 1.92 - 2.09 (m, 2 H), 2.71 - 2.84 (m, 2 H), 2.88 (d, 1 H), 3.25 (d, 1 H), 3.62 (d, 1 H), 4.30 (d, 1 H), 7.13 (t, 1 H), 7.17 (d, 1 H), 7.22 (dd, 1 H), 7.31 (t, 1 H), 7.39 (d, 1 H), 7.46 (d, 1 H), 7.71 (d, 1 H), 7.99 (s, 1 H); MS(m/z): 372 [MH]+.
Second eluting compound: The colourless oil (27 mg) was dissolved in dry DCM (0.6ml_) and TFA (22μl_) was added at room temperature. After 30min, toluene (2ml_) was added and the mixture was concentrated in vacuo. The residue was purified by
a SCX cartridge (1g) eluting with MeOH and then NH3 0.5M in MeOH. The basic fractions were combined and concentrated in vacuo to give Compound 23 (Single Enantiomer) (18mg) as a colourless oil; 1 H NMR (400 MHz, CDCI3) δ ppm 1.25 (d, 1 H), 1.51 (d, 1 H), 1.91 - 2.12 (m, 2 H), 2.69 - 2.85 (m, 2 H), 2.93 (d, 1 H), 3.20 (d, 1 H), 3.65 (d, 1 H), 4.40 (d, 1 H), 7.07 (t, 1 H), 7.19 (dd, 1 H), 7.24 - 7.31 (m, 1 H), 7.39 - 7.47 (m, 2 H), 7.62 (d, 1 H), 7.69 (d, 1 H), 7.79 (s, 1 H); MS(m/z): 372 [MH]+.
Compound 24: 1-f[(1 R.6S or 1S.6RV6-(3.4-dichlorophenvn-3-azabicvclor4.1.0lhept- 1-yllmethyl}-1 H-indole (Single enantiomer)

Sodium hydride 60% in mineral oil (6.22 mg) was added at 0 0C to a solution of indole (18.21 mg) in dry DMF (0.3ml_). The mixture was stirred at this temperature for 15min, then for an additional 15min at room temperature. A solution of 1 ,1- dimethylethyl (1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-1-
{[(methylsulfony^oxylmethylJ-S-azabicyclo^.i .Olheptane-S-carboxylate (Intermediate 29, 35 mg) in dry DMF (0.6ml_) and sodium iodide (5.82 mg) were added and the mixture was stirred at 50 0C. After 6h, water was added and the mixture was extracted with ethyl acetate (2x). The organic phase was dried over Na2SO4 and concentrated in vacuo to give 62mg of crude compound, which was purified by silica gel flash chromatography (gradient ethyl acetate in cyclohexane from 5 to 40%) to give a pale yellow oil (23 mg). This was dissolved in dichloromethane (0.5 ml.) and TFA (0.01 1 ml.) was added at room temperature. Four additions of TFA (0.01 1 ml_ each) were made during 26h. After 4Oh, toluene (2ml_) was added and the mixture was concentrated in vacuo. The residue was purified by silica gel flash chromatography (gradient methanol in dichloromethane from 0 to 10%) to give 8mg of a pink solid. This was dissolved in ethyl acetate and washed with saturated aqueous NaHCθ3 solution and brine. The organic phase was dried over Na2SOφ filtered and concentrated in vacuo to give 7.5mg of the title compound as a pink solid; 1 H NMR (400 MHz, CDCI3) δ ppm 1.22 (d, 1 H), 1.45 (d, 1 H), 1.95 - 2.12 (m,
2 H), 2.69 - 2.90 (m, 3 H), 3.15 (d, 1 H), 3.25 (d, 1 H), 4.21 (d, 1 H), 6.49 (d, 1 H), 7.02 - 7.30 (m, 5 H), 7.43 - 7.50 (m, 2 H), 7.60 (d, 1 H), NH not detected; MS(m/z): 371 [MH]+.
Compounds 25: (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-1-r(1S)-1-(3,5-dimethyl-1 H- pyrazol-1-yl)ethyl1-3-azabicvclo[4.1.01heptane (Racemate); and Compound 26: (1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-1-r(1 R)-1-(3,5-dimethyl-1 H- pyrazol-1-yl)ethyll-3-azabicvclor4.1.Olheptane (Racemate)

Borane (1 M in THF, 0.485 ml.) was added at 0 0C to a solution of 1 ,1-dimethylethyl (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-1 -[ethanehydrazonoyl]-3- azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 42, 161 mg) in dry THF (3.5 ml_). The mixture was stirred at room temperature for 2h, then heated to 60 0C. After 6h, further borane (1 mL) was added and the mixture was stirred overnight at room temperature. The following day, the mixture was heated again to reflux temperature. After 28h, MeOH (0.5ml_) was added at 0 0C followed by HCI 1 M in diethyl ether (2ml_) and the mixture was stirred overnight at room temperature. The following day, the mixture was concentrated in vacuo. HCI 4M in dioxane (4ml_) was added and the mixture was heated to 60 0C for 1 h. The mixture was then concentrated in vacuo and the residue was purified on a SCX cartridge (2g) eluting with MeOH and then NH3 1 M in MeOH. The basic fractions were combined and concentrated in vacuo to give 120mg of a pale yellow oil. This oil was dissolved in ethanol (1 ml.) and acetylacetone (0.041 ml.) and hydrochloric acid (2M in water, 0.300 ml.) were added. The mixture was heated to 60 0C for 1 h 30min, then at room temperature for additional 3h. The mixture was then concentrated in vacuo, basified to pH10 with an aqueous 1 N NaOH solution and extracted with dichloromethane (3x). The organic phase was dried over Na2SO4 and concentrated in vacuo. The residue was purified by silica gel flash chromatography (eluant A: dichloromethane, B: dichloromethane/methanol 9/1 + 0.1 % of 33% aq. NH3 solution, gradient A/B from
100/0 to 0/100) to give 26mg of a colourless oil which was further purified by preparative HPLC (column: XBridge Phenyl, 100 x 19 mm, 5 μm, mobile phase A: NH4HCO3 SoI. 1O mM pH 10; B: CH3CN, gradient: 10%(B) for 1 min to 100%(B) in 1 1 min, 100%(B) for 2 min, flow rate 17ml/min, UV range 210-350 nm, ionization ES+) to give Compound 25 (Diastereoisomer 1 ) (7 mg) (Rt = 8.7min, contaiminated with about 30% of the Compound 26 (Diastereoisomer 2); 1 H NMR (400 MHz, CDCI3) δ ppm 0.92 - 1.05 (m, 2 H), 1.42 (d, 3 H), 1.91 (s, 3 H), 1.95 - 2.06 (m, 2 H), 2.19 (s, 3 H), 2.61 - 2.81 (m, 2 H), 3.15 (q, 1 H), 3.30 (d, 1 H), 3.71 (d, 1 H) , 5.68 (s, 1 H), 7.23 (dd, 1 H), 7.43 (d, 1 H), 7.47 (d, 1 H), NH not observed; MS(m/z): 364 [MH]+.
A further batch of crude material obtained from the silica gel purification (16mg) was further purified by a NH chromatographic column (gradient ethyl acetate in cyclohexane from 0 to 100%) to give Compound 26 (Diastereoisomer 2) (3.5 mg); 1 H NMR (500 MHz, CDCI3) δ ppm 1.09 (d, 1 H), 1.22 (d, 1 H), 1.35 (d, 6 H), 1.66 - 2.05 (m, 2 H), 2.29 (s, 3 H), 2.52 - 2.69 (m, 1 H), 2.85 (d, 1 H), 2.94 (q, 1 H), 3.06 (ddd, 1 H), 4.42 (d, 1 H), 5.70 (s, 1 H), 6.30 - 6.86 (m, 2 H), 7.15 (d, 1 H), NH not observed; MS(m/z): 364 [MH]+.
Compound 27: (1 R.6S or 1 S,6R)-6-(3,4-dichlorophenyl)-1-{[4-methyl-3-
(trifluoromethyl)-i H-pyrazol-1-yl1methyl}-3-azabicvclo[4.1.Oiheptane (Single enantiomer); and
Compound 28: (1 R.6S or 1 S.6RV6-(3.4-dichlorophenylV1-{r4-methyl-5-
(trifluoromethyl)-i H-pyrazol-1-yl1methyl}-3-azabicvclo[4.1.Oiheptane (Single enantiomer)

To a solution of sodium hydride 60% in mineral oil (26.6 mg) in dry DMF (4 ml_), at 0 0C was added 3-(trifluoromethyl)-1 H-pyrazole (100 mg) and the reaction mixture was stirred at rt for 30 min. 1 ,1-Dimethylethyl (1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-1-
{[(methylsulfony^oxyJmethylJ-S-azabicyclo^.i .OJheptane-S-carboxylate (Intermediate 29, 150 mg) was added and the solution was heated to 50 0C for 4h and then RT overnight. After cooling to room temperature, Et2O was added to the reaction mixture and the organic phase was washed with water (2X). The organic layer was dried and concentrated in vacuo. The residue (MS(m/z): 504 [MH]+ ) was dissolved in DCM (1 ml.) and TFA (0.128 ml.) was added. The reaction mixture was stirred at room temperature overnight. The volatiles were evaporated under reduced pressure and the residue purified by SCX, eluting first with MeOH and then with 2.0N NH3 in MeOH. The crude was further purified by Liquid Chromatography under the following conditions: (Preparative conditions: column Gemini C18 AXIA 50 x 21 mm, 5 μm, eluent A: 10 mM aq.sol. NH4HCO3, pH 10; B: CH3CN, gradient: 40% to 45% (B) in 1 min, 45% to 80% (B) in 12 min, 80% to 100% (B) in 0.1 min, 100% (B) for 1.9 min, flow rate 17.0 ml/min. , detection UV 210-350 nm; analytical conditions: column Gemini C18 AXIA 50 x 4.6 mm, 5 μm, eluent A: 10 mM aq.sol. NH4HCO3, pH 10; B: CH3CN, gradient: 35% (B) for 0.5 min, 35% to 95% (B) in 4.5 min, 95% (B) for 1.5 min, flow rate 2.0 ml/min. , detection UV 210-350 nm) obtaining: Compound 27 (Regioisomer 1 ), Rt. = 4.06 min (21.5 mg by LC); NMR (1H, CDCI3):δ ppm 7.41-7.39 (m, 2 H) 7.16 (d, 1 H) 7.08 (s, 1 H) 4.04 (d, 1 H) 3.35 (d, 1 H) 3.13 (d, 1 H) 2.91 (d, 1 H) 2.81 - 2.72 (m, 2 H) 2.13 (s, 3 H) 2.12-1.90 (m, 2 H) 1.35 (d, 1 H) 1.21 (d, 1 H); MS(m/z): 404 [MH]+.
Compound 28 was obtained impure from LC chromatography purification (Rt = 4.33 min). It was submitted for chiral HPLC chromatography (Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30% v/v, flow rate 14 ml/min., detection UV 225 nm. Analytical conditions: chiral column Chiralpak AD- H, eluent A: n-hexane; B: ethanol 70/30% v/v, flow rate 0.8 ml/min., DAD 210-340 nm; CD at 230nm) to give Compound 28. Rt = 5.80 min (4.4 mg by chiral HPLC); 1 H NMR (CDCI3): δ ppm 7.37-7.35 (m, 3 H) 7.16 (d, 1 H) 3.90 (d, 1 H) 3.58 (d, 1 H) 3.20 (d, 1 H) 2.92 (d, 1 H) 2.81 - 2.72 (m, 2 H) 2.15 (s, 3 H) 2.10-1.90 (m, 2 H) 1.43 (d, 1 H) 1.18 (d, 1 H); MS(m/z): 404 [MH]+
Compound 29: (1 R6S/7S.6/?)-6-(3,4-dichlorophenyl)-1-[(2,5-dimethyl-1 H-pyrrol-1- yl)methyl1-3-azabicvclo[4.1.Oiheptane (Racemate)

2,5-Dimethyl-i H-pyrrole (0.045 ml.) was dissolved in DMF (4 ml.) and cooled to 0 0C. Sodium hydride 60% in mineral oil (0.018 g) was added portionwise and the mixture stirred for 10 min until hydrogen evolution ceased. 1 ,1-diMethylethyl (1 R,6S)-6-(3,4- dichlorophenyl)-1-{[(methylsulfonyl)oxy]methyl}-3-azabicyclo[4.1.0]heptane-3- carboxylate (Intermediate 26, 0.1 g) in DMF (1 ml.) was then added and mixture stirred at 50 0C for 12h. After cooling to room temperature, the mixture was quenched with a saturated solution of NH4CI (2 ml.) and partitioned between diethyl ether (20 ml.) and water (20 ml_). The organic phase was washed with brine (20 ml_), dried and concentrated under reduced pressure. Purification by chromatography on silica gel eluting with a gradient from 0 to 50% ethyl acetate in cyclohexane afforded a brown oil (45 mg). The oil was taken up dissolved in DCM (1.00 ml) and TFA (0.017 ml.) was added. The reaction mixture was stirred at room temperature for 2h. The volatiles were evaporated under reduced pressure and the residue purified by SCX, eluting first with MeOH and then with 2.0N NH3 in MeOH. It was further purified by chromatography on silica gel eluting with a gradient from 100% DCM to 5% MeOH in DCM. The title compound was obtained as a yellow oil (8.3 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.41 - 7.50 (2 H, m), 7.22 (1 H, dd), 5.75 (2 H, s), 3.87 (1 H, d), 2.61 - 2.90 (5 H, m), 2.16 - 2.24 (6 H, m), 2.02 - 2.12 (1 H, m), 1 .92 - 2.01 (1 H, m), 1.25 - 1.39 (2 H, m); MS(m/z): 349 [MH]+.
Compound 30: 2-{[(1 R,6S/1S,6R)-6-(3,4-dichlorophenyl)-3-azabicvclor4.1.Olhept-1- yl1methyl}-3-(trifluoromethyl)-2,4,6,7-tetrahvdropyrano[4,3-c1pyrazole (Racemate); Compound 31 : 1-{[(1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-3-azabicvclor4.1.0lhept- 1-yl1methyl}-3-(trifluoromethyl)-1 ,3a,4,6,7,7a-hexahvdropyrano[4,3-c1pyrazole (Single enantiomer); and
Compound 32: 1-{[(1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-3-azabicvclor4.1.0lhept- 1-yllmethyl}-3-(trifluoromethyl)-1 ,3a,4,6,7,7a-hexahvdropyranor4,3-clpyrazole (Single enantiomer)

Race mate
1 ,1-Dimethylethyl (1 R,6S)-6-(3,4-dichlorophenyl)-1-{[(methylsulfonyl)oxy]methyl}-3- azabicyclo[4.1.0]heptane-3-carboxylate (Intermediate 26, 250 mg) was reacted with 3-(trifluoromethyl)-1 ,4,6,7-tetrahydropyrano[4,3-c]pyrazole (213 mg) using a similar procedure to the preparation of Compound 29. The crude material thus obtained was purified by chiral HPLC chromatography (Preparative conditions: column chiralcel OJ-H 25 x 2.0 cm, eluent A: n-hexane; B: ethanol 85/15 % v/v, flow rate 14.0 ml/min. , detection UV at 225 nm; analytical conditions: column chiralcel OJ-H 25 x 0.46 cm, eluent A: n-hexane; B: ethanol 85/15 % v/v, flow rate 0.8 ml/min. , detection UV at 230 nm) obtaining:
Compound 30 (Regioisomer 1 ), as racemic mixture, Rt = 7.364-7.987 min. (8mg); "Η NMR (DMSO-d6): δ ppm 7.52 - 7.60 (m, 2 H) 7.32 (dd, 1 H) 4.55 - 4.66 (m, 2 H) 4.02 (d, 1 H) 3.79 - 3.90 (m, 2 H) 3.28 (d, 1 H) 3.03 (d, 1 H) 2.83 (d, 1 H) 2.65 - 2.74 (m, 2 H) 2.57 - 2.65 (m, 2 H) 1.72 - 1.92 (m, 2 H) 1.35 (d, 1 H) 1.16 (d, 1 H); MS(m/z): 446 [MH]+; and
Compound 31 (Regioisomer 2 Enantiomer 1 ), Rt. = 9.931 min (18 mg); 1 H NMR (CDCI3): δ ppm 7.40-7.36 (m, 2 H) 7.18 (d, 1 H) 4.63 - 4.74 (m, 2 H) 3.97 - 3.86 (m, 1 H) 3.83 - 3.71 (m, 2 H) 3.42 (d, 1 H) 3.36 (d, 1 H) 2.97 (d, 1 H) 2.89 - 2.74 (m, 2 H) 2.62 - 2.55 (m, 1 H) 2.32 - 2.24 (m, 1 H) 2.06 - 1.88 (m, 2 H) 1.40 (d, 1 H) 1.24 (d, 1 H); MS(m/z): 446 [MH]+; and
Compound 32 (Reqioisomer 2, Enantiomer 2), Rt. = 13.583 min (11 mg); 1 H NMR (CDCI3): δ ppm 7.40-7.36 (m, 2 H) 7.18 (d, 1 H) 4.63 - 4.74 (m, 2 H) 3.97 - 3.86 (m, 1 H) 3.83 - 3.71 (m, 2 H) 3.42 (d, 1 H) 3.36 (d, 1 H) 2.97 (d, 1 H) 2.89 - 2.74 (m, 2 H) 2.62 - 2.55 (m, 1 H) 2.32 - 2.24 (m, 1 H) 2.06 - 1.88 (m, 2 H) 1.40 (d, 1 H) 1.24 (d, 1 H); MS(m/z): 446 [MH]+.
Compound 33: 2-{[(1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-3-azabicvclor4.1.0lhept-
1-yl1methyl}-3-(trifluoromethyl)-2,4,6,7-tetrahvdropyrano[4,3-c1pyrazole (Single enantiomer); and
Compound 34: 2-{[(1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-3-azabicvclor4.1.0lhept-
1-yl1methyl}-3-(trifluoromethyl)-2,4,6,7-tetrahvdropyrano[4,3-c1pyrazole (Single enantiomer)

Compound 30 (8 mg) was submitted for chiral chromatography (Preparative conditions: column chiralpak AD-H, 25 x 2.0 cm, eluent A: n-hexane; B: ethanol
70/30 % v/v, flow rate 14.0 ml/min. , detection UV at 225 nm; analytical conditions: column chiralpak AD-H 25 x 0.46 cm, eluent A: n-hexane; B: ethanol 70/30 % v/v, flow rate 0.8 ml/min., detection UV at 230 nm) obtaining:
Compound 33 (Enantiomer 1 ), Rt. = 6.554 min (3.8 mg); and Compound 34 (Enantiomer 2), Rt. = 8.792 min (3.4 mg); NMR (1H, MeOD): δ 7.46 (d,
1 H) 7.24 (s, 2 H) 4.70 (s, 2 H) 4.05-3.98 (m, 4 H) 3.66 (d, 1 H) 3.43 (d, 1 H) 3.03-
2.85 (m, 2 H) 2.40-2.23 (m, 2 H) 1.72-1.69 (m, 2 H) 1.47- 1.23 (m, 2 H); MS(m/z):
446 [MH]+
Compound 35: (1 R6S/yS.6/?)-6-(3,4-dichlorophenyl)-1-{r3-(1 ,1-dimethylethyl)-1 H- pyrazol-1-yllmethyl}-3-azabicvclor4.1.Olheptane (Racemate)

Compound 35 was submitted for chiral HPLC (Preparative conditions: Column = Chiracel OD-H (25 x 0.46 cm), mobile phase = n-Hexane/(2-propanol + 0.1% isopropylamine) 97/3 % v/v, flow rate = 14 ml/min; Analytical conditions: Column = Chiracel OD-H (25 x 0.46 cm), mobile phase = n-Hexane/(2-propanol + 0.1% isopropylamine) 95/5 % v/v, flow rate = 1.0 ml/min, DAD = 210 - 340 nm, CD = 230 nm) obtaining:
Compound 36: (1 R6S or 7S.6RV6-(3.4-dichlorophenylV1-{r3-π .1-dimethylethvn-1 H- PVrazol-1-yl1methyl}-3-azabicvclo[4.1.01heptane (Single enantiomer) (Enantiomer 1 ), Rt = 13.25 min (14 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.43 (d, 1 H) 7.37 (d, 1 H) 7.18 (dd, 1 H) 7.14 (d, 1 H) 6.05 (d, 1 H) 3.97 (d, 1 H) 3.40 (d, 1 H) 3.15 (d, 1 H) 2.88 (d, , 1 H) 2.72 - 2.79 (m, 2 H) 1.97 - 2.07 (m, 1 H) 1.87 - 1.96 (m, 1 H) 1.31 (m, 10 H) 1.14 - 1.18 (m, 1 H); MS(m/z): 378 [MH]+; and
Compound 37: (1 S.6ff or W6S)-6-(3,4-dichlorophenyl)-1-{[3-(1 ,1-dimethylethyl)-1 H- PVrazol-1-yl1methyl}-3-azabicvclo[4.1.01heptane (Sigle enantiomer) (Enantiomer 2), Rt = 15.08 min (17.8 mg); 1H NMR (CDCI3, 400 MHz) δ ppm 7.43 (d, 1 H) 7.37 (d, 1 H) 7.18 (dd, 1 H) 7.14 (d, 1 H) 6.05 (d, 1 H) 3.96 (d, 1 H) 3.39 (d, 1 H) 3.14 (d, 1 H) 2.87 (d, 1 H) 2.72 - 2.78 (m, 2 H) 1.97 - 2.06 (m, 1 H) 1.87 - 1.95 (m, 1 H) 1.27 - 1.33 (m, 10 H) 1.16 (d, 1 H); MS(m/z): 378 [MH]+
Compound 38: (1 R6S or 7S.6RV6-(3.4-dichlorophenylV1-r(2.5-dimethyl-1 H-pyrrol-1- yl)methyl1-3-azabicvclo[4.1.01heptane (Single enantiomer); and
Compound 39: (1S.6R or 1R6SV6-(3.4-dichlorophenylV1-r(2.5-dimethyl-1 H-pyrrol-1- yl)methyl1-3-azabicvclo[4.1.01heptane (Single enantiomer)

Compound 29 (21 mg) was prepared from Intermediate 52 according to procedure described for Compound 17; MS(m/z): 349 [MH]+; The racemic compound was submitted for chiral HPLC (Preparative conditions: Column = Chiracel OD-H (25 x
0.46 cm), mobile phase = n-Hexane/(2-propanol + 0.1 % isopropylamine) 95/5 % v/v, flow rate = 14 ml/min; Analytical conditions: Column = Chiracel OD-H (25 x 0.46 cm), mobile phase = n-Hexane/(2-propanol + 0.1 % isopropylamine) 95/5 % v/v, flow rate
= 1.0 ml/min, DAD = 210 - 340 nm, CD = 230 nm) obtaining:
First elutinq compound: Compound 38 (Enantiomer 1 ), Rt = 9.73 min (3 mg);
MS(m/z): 348 [MH]+ ; and
Second elutinq compound: Compound 39 Enantiomer 2, Rt = 1 1.227 min, 2.4 mg,
MS(m/z): 348 [MH]+.
Compound 40: (1 R6S or 1S.6RV6-(3.4-dichlorophenylV1-r(3-methyl-1 H-pyrrol-1- yl)methyl1-3-azabicvclo[4.1.01heptane (Single enantiomer); and Compound 41 : (1 R6S or 1S.6RV6-(3.4-dichlorophenylV1-r(3-methyl-1 H-pyrrol-1- yl)methyl1-3-azabicvclo[4.1.01heptane (Single enantiomer)

Intermediate 26 (150 mg) was reacted with 3-methyl-1 H-pyrrole (54 mg) according to the procedure described for Compound 29. The racemic product thus obtained was submitted to chiral HPLC separation (Preparative conditions: chiral column Chiralcel
OD-H, eluent A: n-hexane; B: ethanol 90/10 v/v, flow rate 15 ml/min., detection UV 225 nm. Analytical conditions: chiral column Chiralcel OD-H, eluent A: n-hexane; B: ethanol 90/10% v/v, flow rate 1.0 ml/min., DAD 210-340 nm; CD at 230nm) obtaining:
Compound 40 (Enantiomer 1 ) Rt. = 9.538 min (11 mg), 1 H NMR (CDCI3): δ ppm 7.43-7.40 (m, 2 H) 7.15 (d, 1 H) 6.45 (s, 1 H) 6.33 (s, 1 H) 5.93 (s, 1 H) 3.77 (d, 1 H) 3.02-2.91 (m, 3 H) 2.80-2.70 (m, 2 H) 2.08 (s, 3 H) 2.03-1.92 (m, 2 H) 1.31 (d, 1 H) 1.18 (d, 1 H); MS(m/z): 335 [MH]+;
Compound 41 (Enantiomer 2) Rt. = 13.495 min (12 mg), 1 H NMR (CDCI3): δ ppm 7.43-7.40 (m, 2 H) 7.15 (d, 1 H) 6.45 (s, 1 H) 6.33 (s, 1 H) 5.93 (s, 1 H) 3.77 (d, 1 H) 3.02-2.91 (m, 3 H) 2.80-2.70 (m, 2 H) 2.08 (s, 3 H) 2.03-1.92 (m, 2 H) 1.31 (d, 1 H) 1.18 (d, 1 H); MS(m/z): 335 [MH]+.
The following compounds of formula (I) (Cmp) were prepared following a similar procedure to that described for Compound 29 starting from the Intermediate (Int) indicated in the table and the appropriate heterocycle.
Table A
H,
H,
(d, 1 H),

1 1.29
(s, 3 H);

The following compounds of formula (I) were prepared following a similar procedure to that described for the preparation of Compound 17 using the starting material indicated.
Table B

 - (d, 1
- (d, 1
1 (s, 1
1 -
1

- 1
not
1 H), 1 H);
1 - 2 (m, 1 H)

(dd,
1 H) 3 (m,

1 3.50 (d, 1 H) 1.18
H, -


The following compounds of formula (I) were prepared by chiral HPLC separation of the racemic compounds indicated in Table C. Specific chiral HPLC conditions are also given.
Table C










Chiral HPLC separation conditions (see Table C):
A: Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: (ethanol + 0.1 % isopropylamine) 75/25% v/v, flow rate 13 ml/min., detection UV 230 nm. Analytical conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 0.1% isopropylamine 75/25% v/v, flow rate 0.8 ml/min., DAD 210-340 nm; CD at 230nm
B: Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30% v/v, flow rate 13 ml/min., detection UV 225 nm. Analytical conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30% v/v, flow rate 0.8 ml/min., DAD 210-340 nm; CD at 230nm
C: Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 80/20% v/v, flow rate 13 ml/min., detection UV 230 nm. Analytical conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 80/20% v/v, flow rate 0.8 ml/min., DAD 210-340 nm; CD at 230nm
D: Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30% v/v, flow rate 14 ml/min., detection UV 225 nm. Analytical conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30% v/v, flow rate 0.8 ml/min., DAD 210-340 nm; CD at 230nm
E: Preparative conditions: chiral column Chiralcel OJ-H, eluent A: n-hexane; B: ethanol 80/20% v/v, flow rate 13 ml/min., detection UV 225 nm. Analytical conditions: chiral column Chiralcel OJ-H, eluent A: n-hexane; B: ethanol 80/20% v/v, flow rate 0.8 ml/min., DAD 210-340 nm; CD at 230nm
F: Preparative conditions: chiral column Chiralpack AS-H, eluent A: n-hexane; B: ethanol 85/15% v/v, flow rate 14 ml/min., detection UV 230 nm. Analytical conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 85/15% v/v, flow rate 0.8 ml/min., DAD 210-340 nm; CD at 230nm
G: Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 70/30% v/v, flow rate 13 ml/min., detection UV 225 nm. Analytical conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: ethanol 60/40 v/v, flow rate 0.8 ml/min., DAD 210-340 nm; CD at 225nm
H: Preparative conditions: chiral column Chiralpak AD-H, eluent A: n-hexane; B: (2- propanol + 0.1% isopropylamine) 80/20% v/v, flow rate 16 ml/min., detection UV 225 nm. Analytical conditions:enantiomer 1 chiral column Chiralpak AD-H, eluent A: n- hexane; B: (2-propanol + 1% isopropylamine) 98/2% v/v, flow rate 1 ml/min., DAD 210-340 nm; CD at 230nm; enantiomer 2: eluent A: n-hexane; B: (2-propanol + 1% isopropylamine) 80/20% v/v, flow rate 1 ml/min., DAD 210-340 nm; CD at 230nm;
I: Preparative conditions: chiral column Chiralcel OD-H, eluent A: n-hexane; B: 2- propanol 90/10% v/v, flow rate 14 ml/min., detection UV 225 nm. Analytical conditions: chiral column Chiralcel OD-H, eluent A: n-hexane; B: 2-propanol 90/10% v/v, flow rate 1.0 ml/min., DAD 210-340 nm; CD at 230nm
L: Preparative conditions: chiral column Chiralcel OJ-H, eluent A: n-hexane; B: (2- propanol + 0.1% isopropylamine) 95/5% v/v, flow rate 14 ml/min., detection UV 225 nm. Analytical conditions: chiral column Chiralcel OJ-H, eluent A: n-hexane; B: (2- propanol + 0.1% isopropylamine) 95/5% v/v, flow rate 1.0 ml/min., DAD 210-340 nm; CD at 230nm
M: Preparative conditions: chiral column Chiralcel AD-H, eluent A: n-hexane; B: (ethanol + 0.1 % isopropylamine) 90/10% v/v, flow rate 14 ml/min., detection UV 225 nm. Analytical conditions: chiral column Chiralcel AD-H, eluent A: n-hexane; B: (ethanol + 0.1 % isopropylamine) 90/10% v/v, flow rate 1.0 ml/min., DAD 210-340 nm; CD at 230nm
N: Preparative conditions: chiral column Chiralcel OJ-H, eluent A: n-hexane; B: (2- propanol + 0.1% isopropylamine) 90/10% v/v, flow rate 14 ml/min., detection UV 225 nm. Analytical conditions: chiral column Chiralcel OJ-H, eluent A: n-hexane; B: (2- propanol + 0.1% isopropylamine) 90/10% v/v, flow rate 1.0 ml/min., DAD 210-340 nm; CD at 230nm
The following compounds of formula (I) (Cmp) were prepared following a similar procedure to that described for Compound 16 starting form the corresponding Starting Compound.








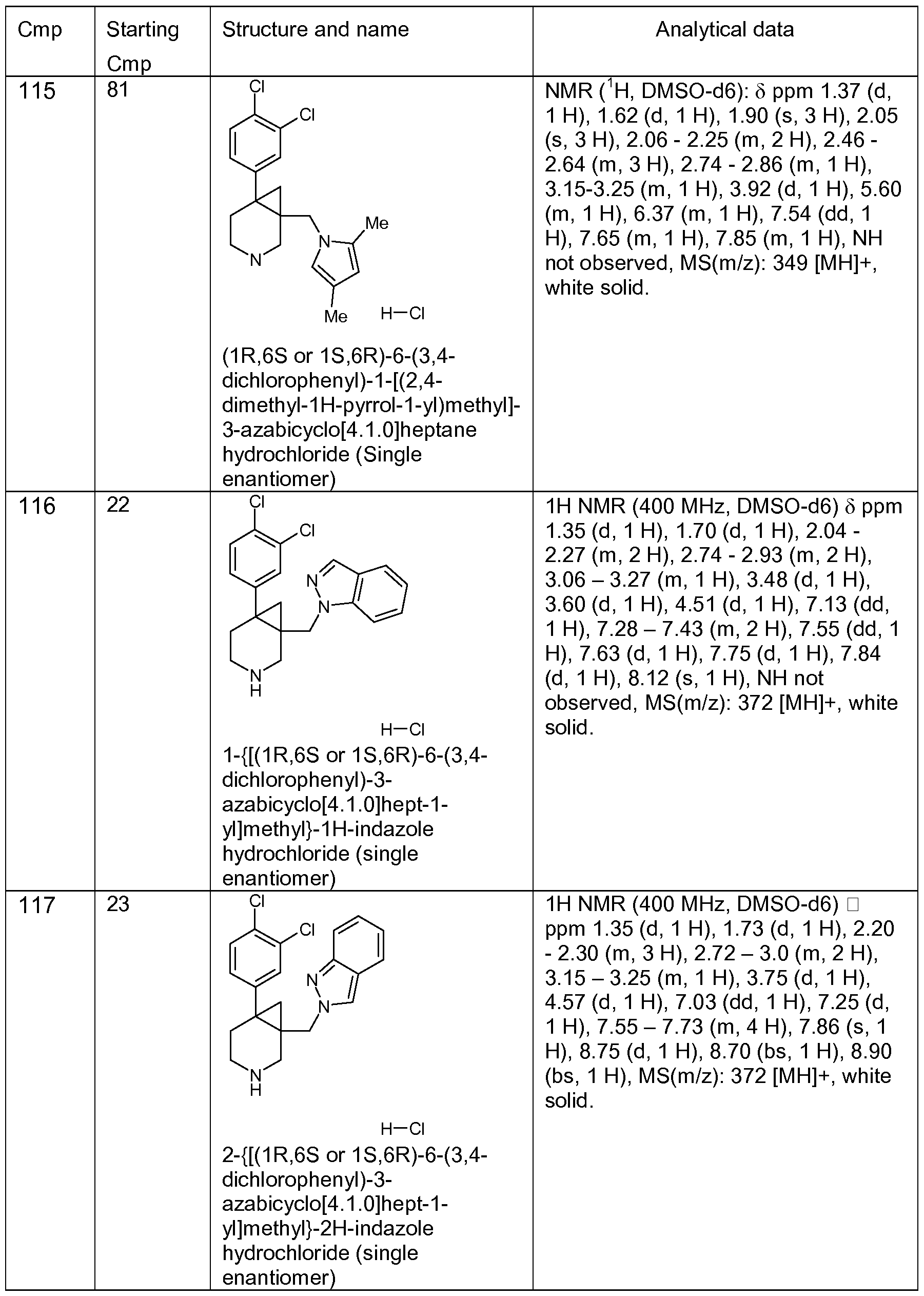








Claims
Claims
1. A compound of formula (I) or a pharmaceutically acceptable salt thereof:

R1 is H or C-|_4alkyl;
R2 is phenyl optionally substituted by one or more groups independently selected from halo, cyano, C-|_4alkyl, haloC-|_4alkyl, C-|.4alkoxy, haloC-|_4alkoxy, C-^alkanoyl and SF5; or R2 is naphthyl, optionally substituted by 1 or 2 groups independently selected from halo, cyano and C-|_4alkyl;
R3 and R^, which may be the same or different, are H, fluoro or C-|_4alkyl; wherein when R3 or R^ is fluoro, the other R3 or R^ is H or fluoro; R5 is H or C-|_4alkyl; n is O, 1 or 2;
R^ is phenyl or heteroaryl, either of which is optionally substituted by one or more groups independently selected from halogen, C-^alkyl, C-|_4haloalkyl, C-|.4alkoxy and cyano; wherein when n is 0, the heteroaryl substituent is attached to the rest of the compound of formula (I) by a carbon atom of the heteroaryl; and in the instance when n is 1 or 2, R^ and R7, which may the same or different, are H, D, C-|.4alkyl or fluoro, wherein when R^ or R7 is fluoro, the other R6 or R7 is H, D or fluoro.
2. The compound according to claim 1 or a pharmaceutically acceptable salt thereof, wherein the compound of formula (I) is a mixture of compounds of formula (IA) and (IB)

(IA) (IB)
3. The compound according to claim 2, or a pharmaceutically acceptable salt thereof, wherein the compound of formula (I) is a racemic mixture of compounds of formula (IA) and (IB).
4. The compound according to claim 3 or a pharmaceutically acceptable salt thereof, wherein the enantiomeric excess (e.e.) of (IA) over (IB) is greater than or equal to 90%
5. The compound according to claim 4 or a pharmaceutically acceptable salt thereof, wherein the e.e. of (IA) over (IB) is greater than or equal to 95%.
6. The compound according to claim 5 or a pharmaceutically acceptable salt thereof wherein the e.e. of (IA) over (IB) is greater than or equal to 99%.
The compound according to claim 1 or a pharmaceutically acceptable salt thereof, wherein the compound of formula (I) is of formula (IA)

(IA)
The compound according to claim 3 or a pharmaceutically acceptable salt thereof, wherein the enantiomeric excess (e.e.) of (IB) over (IA) is greater than or equal to 90%
9. The compound according to claim 9 or a pharmaceutically acceptable salt thereof, wherein the e.e. of (IB) over (IA) is greater than or equal to 95%.
10. The compound according to claim 9 or a pharmaceutically acceptable salt thereof wherein the e.e. of (IB) over (IA) is greater than or equal to 99%.
11. The compound according to claim 1 or a pharmaceutically acceptable salt thereof, wherein the compound of formula (I) is of formula (IB)

(I B)
12. The compound according to any preceding claim, or a pharmaceutically acceptable salt thereof, wherein R^ is hydrogen or methyl.
13. The compound according to any preceding claim, or a pharmaceutically acceptable salt thereof, wherein R-I is hydrogen.
14. The compound according to any preceding claim, or a pharmaceutically acceptable salt thereof, wherein R^ is phenyl substituted by one or two groups independently selected from halo and haloC-1.4 alkyl; or R2 is unsubstituted naphthyl.
15. The compound according to any preceding claim, or a pharmaceutically acceptable salt thereof, wherein R^ is hydrogen.
16. The compound according to any preceding claim, or a pharmaceutically acceptable salt thereof, wherein n is 1 or 2.
17. The compound according to anypreceding claim, or a pharmaceutically acceptable salt thereof, wherein n is 1.
18. The compound according to any preceding claim, or a pharmaceutically acceptable salt thereof, wherein when R^ is heteroaryl, it is monocyclic or bicyclic.
19. The compound according to any preceding claim, or a pharmaceutically acceptable salt thereof, wherein when R^ is monocyclic heteroaryl it is selected from the list furyl, thienyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, azepinyl, oxazepinyl, thiazepinyl and diazepinyl.
20. The compound according to any preceding claim, or a pharmaceutically acceptable salt thereof, wherein when R^ is monocyclic heteroaryl it is selected from the list furyl, thienyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl and tetrazolyl.
21. The compound according to any one of claims 1 to 18, or a pharmaceutically acceptable salt thereof, wherein when R^ is bicyclic heteroaryl, it is selected from the list indolizinyl; indolyl; isoindolyl; 3H-indolyl; indolinyl; indolizinyl; benzo[b]furyl; benzo[b]thienyl; 1 H-indazolyl; 2H-indazolyl; benzimidazolyl; benzthiazolyl; purinyl; 4H-quninolinyl; quinolinyl; isoquinolinyl; cinnolinyl; phthalazinyl; quinazolinyl; quinoxalinyl; 1 ,8-naththyridinyl; pteridyl; 2,4,6,7- tetrahydropyrano[4,3-c]pyrazolyl; 1 ,3a, 4,6,7, 7a-hexahydropyrano[4, 3- c]pyrazolyl; 4,5,6,7-tetrahydro-1 H-indazolyl; and 4,5,6,7-tetrahydro-2H- indazolyl.
22. The compound according to any one of claims 1 to 18, or a pharmaceutically acceptable salt thereof, wherein when R^ is bicyclic heteroaryl, it is selected from the list indolizinyl; indolyl; isoindolyl; 3H-indolyl; indolinyl; benzo[b]furyl; benzo[b]thienyl; 1 H-indazolyl; 2H-indazolyl; benzimidazolyl; benzthiazolyl; 2,4,6,7-tetrahydropyrano[4,3-c]pyrazolyl; 1 ,3a,4,6,7,7a-hexahydropyrano[4,3-
c]pyrazolyl; 4,5,6,7-tetrahydro-1 H-indazolyl; or 4,5,6,7-tetrahydro-2H- indazolyl.
23. The compound according to any one of claims 1 to 18, or a pharmaceutically acceptable salt thereof, wherein R^ is a monocyclic heteroaryl substituent selected from the list furyl, thienyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl and tetrazolyl; any of which substituents are optionally substituted by one or more groups independently selected from halogen, C-|_4alkyl, C-|_4haloalkyl, C-|.4alkoxy and cyano.
24. The compound according to any one of claims 1 to 18, or a pharmaceutically acceptable salt thereof, wherein R^ is a monocyclic heteroaryl substituent selected from the list pyrazolyl, tetrazolyl, triazolyl, pyrrolyl and oxazolyl; any of which substituents are optionally substituted by one or more groups independently selected from halogen, C-|_4alkyl, C-|.4haloalkyl, C-|.4alkoxy and cyano.
25. A compound according to claim 1 selected from the list: (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-1-[(1 S)-1-(3,5-dimethyl-1 H-pyrazol-1- yl)ethyl]-3-azabicyclo[4.1.0]heptane (Racemic Compound 25); (1 R,6S/1 S,6R)-6-(3,4-dichlorophenyl)-1-[(3,5-dimethyl-1 H-pyrazol-1- yl)methyl]-3-azabicyclo[4.1.0]heptane (Racemic Compound 48); (1S,6R/1 R,6R)-6-(3,4-dichlorophenyl)-1-{[5-(1-methylethyl)-2H-tetrazol-2- yl]methyl}-3-azabicyclo[4.1.0]heptane (Racemic Compound 60);
(1S,6R/1 R,6S)-6-(3,4-dichlorophenyl)-1-[(3,5-dimethyl-1 H-1 ,2,4-triazol-1- yl)methyl]-3-azabicyclo[4.1.0]heptane (Racemic Compound 61 ); (1S,6R/1 R,6S)-6-(3,4-dichlorophenyl)-1-{[5-methyl-3-(trifluoromethyl)-1 H- pyrazol-1-yl]methyl}-3-azabicyclo[4.1.0]heptane (Racemic Compound 62);
(1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-1-[(3,5-dimethyl-1 H-pyrazol-1- yl)methyl]-3-azabicyclo[4.1.0]heptane (Compound 68 - single enantiomer);
(1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-1-{[3-(trifluoromethyl)-1 H-pyrazol-1- yl]methyl}-3-azabicyclo[4.1.0]heptane (Compound 74 - single enantiomer);
(1S,6R or 1 R,6S)-6-(3,4-dichlorophenyl)-1-{[5-(1-methylethyl)-2H-tetrazol-2- yl]methyl}-3-azabicyclo[4.1.0]heptane (Compound 84 - single enantiomer);
(1S,6R or 1 R,6S)-6-(3,4-dichlorophenyl)-1-{[5-methyl-3-(trifluoromethyl)-1 H- pyrazol-1-yl]methyl}-3-azabicyclo[4.1.0]heptane (Compound 88 - single enantiomer); (1S,6R or 1 R,6S)-6-(3,4-dichlorophenyl)-1-{[3-methyl-5-(trifluoromethyl)-1 H- pyrazol-1-yl]methyl}-3-azabicyclo[4.1.0]heptane (Compound 90 - single enantiomer); (1 R.6S or 1S,6R)-6-(3,4-dichlorophenyl)-1-[(4-methyl-1 H-pyrazol-1-yl)methyl]-
3-azabicyclo[4.1.0]heptane (Comound 66 - single enantiomer); 1-{[(1 R,6S or 1 S,6R)-6-(3,4-dichlorophenyl)-3-azabicyclo[4.1.0]hept-1- yl]methyl}-1 H-pyrazole-5-carbonitrile (Compound 76 - single enantiomer); (1S.6R or 1 R,6S)-6-(3,4-dichlorophenyl)-1-[(2,4-dimethyl-1 H-pyrrol-1- yl)methyl]-3-azabicyclo[4.1.0]heptane (Compound 81 - single enantiomer); 1-{[(1 R,6S or 1 S,6R)-6-(3,4-dichlorophenyl)-3-azabicyclo[4.1.0]hept-1- yl]methyl}-1 H-pyrrole-3-carbonitrile (Compound 40 - single enantiomer); and
(1 R,6S or 1S,6R)-6-(3,4-dichlorophenyl)-1-[(4-methyl-1 ,3-oxazol-5-yl)methyl]-
3-azabicyclo[4.1.0]heptane (Compound 93 - single enantiomer); or a pharmaceutically acceptable salt thereof.
26. A compound defined in any preceding claim or a pharmaceutically acceptable salt thereof, for use in therapy.
27. A compound defined in any one of claims 1 to 25 or a pharmaceutically acceptable salt thereof, for use in treating depression or an anxiety disorder.
28. The use of a compound defined in any one of claims 1 to 25 or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating depression or an anxiety disorder.
29. A method of treating depression or an anxiety disorder in a mammal comprising administering an effective amount of a compound defined in any one of claims 1 to 25 or a pharmaceutically acceptable salt thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0804326.7 | 2008-03-07 | ||
| GBGB0804326.7A GB0804326D0 (en) | 2008-03-07 | 2008-03-07 | Novel compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009109608A1 true WO2009109608A1 (en) | 2009-09-11 |
Family
ID=39327761
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/052567 WO2009109608A1 (en) | 2008-03-07 | 2009-03-05 | Novel compounds |
Country Status (2)
| Country | Link |
|---|---|
| GB (1) | GB0804326D0 (en) |
| WO (1) | WO2009109608A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020048828A1 (en) | 2018-09-03 | 2020-03-12 | Bayer Pharma Aktiengesellschaft | 5-heteroaryl-3,9-diazaspiro[5.5]undecane compounds |
| WO2020048829A1 (en) | 2018-09-03 | 2020-03-12 | Bayer Aktiengesellschaft | 3,9-diazaspiro[5.5]undecane compounds |
| CN113219094A (en) * | 2021-05-07 | 2021-08-06 | 湖北欣泽霏药业有限公司 | Liquid chromatography detection method for optical isomer of tomoxetine hydrochloride oral solution |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992006079A1 (en) * | 1990-09-28 | 1992-04-16 | Pfizer Inc. | Fused ring analogs of nitrogen containing nonaromatic heterocycles |
| WO2007016155A2 (en) * | 2005-07-27 | 2007-02-08 | Dov Pharmaceutical, Inc. | Novel 1-aryl-3-azabicyclo[3.1.0]hexanes: preparation and use to treat neuropsychiatric disorders |
| WO2008031772A1 (en) * | 2006-09-11 | 2008-03-20 | Glaxo Group Limited | Azabicyclic compounds as inhibitors of monoamines reuptake |
| WO2008031771A2 (en) * | 2006-09-11 | 2008-03-20 | Glaxo Group Limited | 3-azabicyclo [4. 1. 0] heptane derivatives for the treatment of depression |
-
2008
- 2008-03-07 GB GBGB0804326.7A patent/GB0804326D0/en active Pending
-
2009
- 2009-03-05 WO PCT/EP2009/052567 patent/WO2009109608A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992006079A1 (en) * | 1990-09-28 | 1992-04-16 | Pfizer Inc. | Fused ring analogs of nitrogen containing nonaromatic heterocycles |
| WO2007016155A2 (en) * | 2005-07-27 | 2007-02-08 | Dov Pharmaceutical, Inc. | Novel 1-aryl-3-azabicyclo[3.1.0]hexanes: preparation and use to treat neuropsychiatric disorders |
| WO2008031772A1 (en) * | 2006-09-11 | 2008-03-20 | Glaxo Group Limited | Azabicyclic compounds as inhibitors of monoamines reuptake |
| WO2008031771A2 (en) * | 2006-09-11 | 2008-03-20 | Glaxo Group Limited | 3-azabicyclo [4. 1. 0] heptane derivatives for the treatment of depression |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020048828A1 (en) | 2018-09-03 | 2020-03-12 | Bayer Pharma Aktiengesellschaft | 5-heteroaryl-3,9-diazaspiro[5.5]undecane compounds |
| WO2020048829A1 (en) | 2018-09-03 | 2020-03-12 | Bayer Aktiengesellschaft | 3,9-diazaspiro[5.5]undecane compounds |
| CN113219094A (en) * | 2021-05-07 | 2021-08-06 | 湖北欣泽霏药业有限公司 | Liquid chromatography detection method for optical isomer of tomoxetine hydrochloride oral solution |
Also Published As
| Publication number | Publication date |
|---|---|
| GB0804326D0 (en) | 2008-04-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7691893B2 (en) | Chemical compounds | |
| US7855218B2 (en) | Compounds | |
| JP2019520398A (en) | Spirolactams as ROCK inhibitors | |
| BRPI0617189A2 (en) | quarternary alpha-aminocarboxamide derivatives, pharmaceutical composition containing them, preparation process, as well as voltage-dependent sodium channel modulating uses | |
| WO2008122546A1 (en) | Prolinamide derivatives as modulators of voltage-gated sodium channels | |
| MX2011003533A (en) | Novel compounds as calcium channel blockers. | |
| CA2724219C (en) | 5-[5-[2-(3,5-bis(trifluoromethyl)phenyl)-2-methylpropanoylmethylamino]-4-(4-fluoro-2-methylphenyl)]-2-pyridinyl-2-alkyl-prolinamide as nk1 receptor antagonists | |
| WO2013093496A1 (en) | 2 - (pyridin- 2yl) - 1, 7 -diaza-spiro [4.4] nonane- 6 -one compound as voltage-gated sodium channels modulator | |
| US20160184306A1 (en) | Novel Pyrimidinyl-DiazoSpiro Compounds | |
| EP2061461B1 (en) | 3-azabicyclo[4.1.0]heptane derivatives for the treatment of depression | |
| WO2009109608A1 (en) | Novel compounds | |
| US20100029740A1 (en) | Azabicyclic compounds as serotonin, dopamine and norepinephrine re-uptake inhibitors | |
| WO2009027293A1 (en) | Compounds | |
| WO2009056520A1 (en) | Azabicyclo[3.2.1]octane derivatives | |
| WO2009141412A1 (en) | 1,6-disubstituted 3-azabicyclo [3.1.0] hexane derivatives for use as triple reuptake inhibitors | |
| WO2009027295A1 (en) | 3-azabicyclo (4.1.0) heptane derivatives useful as norepinephrine, serotonin or dopamine reuptake inhibitors | |
| WO2010130672A1 (en) | Azabicyclo [4.1.0] heptane derivatives and their use as monoamine reuptake inhibitors | |
| WO2010133569A1 (en) | Azabicyclo[4.1.0]heptane derivatives | |
| WO2010146025A1 (en) | Tricyclic azabicyclo [4.1.0] heptane derivatives as inhibitors of serotonin, dopamine and norepinephrine re-uptake | |
| WO2010125033A1 (en) | Azabicyclo[4.1.0]heptane derivatives | |
| EP2182946A1 (en) | Substituted azabicyclo [4.1.0]heptane compounds for use as monoamine reuptake inhibitors | |
| KR20060118477A (en) | Beta-lactams for treatment of cns disorders | |
| CN101541754A (en) | Azabicyclic compounds as inhibitors of monoamines reuptake | |
| EP2178823A1 (en) | Spiro cyclopentane compounds useful as antagonists of the h1-receptor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09717025 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09717025 Country of ref document: EP Kind code of ref document: A1 |