WO2009071576A1 - 5-aryl-1,3,4-oxadiazole-2-amines as nicotinic acetylcholine receptor agonists - Google Patents
5-aryl-1,3,4-oxadiazole-2-amines as nicotinic acetylcholine receptor agonists Download PDFInfo
- Publication number
- WO2009071576A1 WO2009071576A1 PCT/EP2008/066698 EP2008066698W WO2009071576A1 WO 2009071576 A1 WO2009071576 A1 WO 2009071576A1 EP 2008066698 W EP2008066698 W EP 2008066698W WO 2009071576 A1 WO2009071576 A1 WO 2009071576A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- salt
- disorders
- formula
- phenyl
- Prior art date
Links
- 0 CC*C(/C=C/C=CC=C(C=CC1)C(NNC(OC(C)(C)C)=O)=O)=CC1=C Chemical compound CC*C(/C=C/C=CC=C(C=CC1)C(NNC(OC(C)(C)C)=O)=O)=CC1=C 0.000 description 1
- ACOXURKIHJSMAC-UHFFFAOYSA-N NCCCCN1CCCCC1 Chemical compound NCCCCN1CCCCC1 ACOXURKIHJSMAC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to novel oxadiazole derivatives having pharmacological activity, processes for their preparation, compositions containing them and their use in the treatment of neurological, psychiatric disorders and gastrointestinal disorders.
- Nicotinic acetylcholine receptors are cation-specific, excitatory ligand-gated ion channels that are widely expressed throughout the central and peripheral nervous systems.
- 16 mammalian nAChR subunit genes have been cloned: 5 encoding muscle receptor subunits, and 11 encoding neuronal receptor subunits.
- the nicotinic ⁇ 7 receptor subunit is predominantly expressed in the mammalian central nervous system (CNS), where it is thought to assemble as a functional homopentameric complex, and is also expressed in peripheral tissues including the sympathetic nervous system, immune cells and the Gl tract.
- CNS mammalian central nervous system
- Activation of neuronal nicotinic ⁇ 7 receptors by selective agonists or the endogenous ligand acetylcholine can modulate the release of various neurotransmitters including glutamate, GABA, dopamine, and noradrenaline and, thus, has the potential to modulate a range of neurological functions. Additionally, in vivo studies have shown that ⁇ 7 nAChR agonists can modulate neurotransmitter release in brain areas such as the cortex and hippocampus that are relevant to cognition (Paterson D et al, (2000) Prog Neurobiol 61 :75-111.
- ⁇ 7 nAChRs Activation of ⁇ 7 nAChRs has also been reported to ameliorate sensory gating deficits in both preclinical (Simosky J. K. et al., (2001 ) Biological Psychiatry. 50(7):493-500) and small clinical studies. These data suggest that novel ⁇ 7 nAChR agonists and/or partial agonists such as the current series could be useful for the treatment of cognitive impairments in neurological and psychiatric disorders such as Alzheimer's disease, related neurodegenerative disorders and schizophrenia.
- the present invention provides, in a first aspect, compounds of formula (I) or a salt thereof:
- R 1 and R 2 independently represent hydrogen, C 1-6 alkyl or C 3-6 cycloalkyl; or R 1 and R 2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group which is unsubstituted or substituted with 1 , 2, or 3 substituents selected from methyl or fluoro;
- Q represents -(CH 2 ) n - wherein n represents 3 or 4;
- A represents a phenyl ring and B represents a 5 or 6 membered heteroaryl ring or B represents a phenyl ring and A represents a 5 or 6 membered heteroaryl ring;
- R 3 and R 4 independently represent Ci -6 alkyl, Ci -6 alkoxy and halo;
- t and m independently represent an integer from 0 to 3.
- Ci -6 alkyl' refers to a linear or branched saturated hydrocarbon group containing from 1 to 6 carbon atoms.
- Ci -6 alkyl groups include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl or hexyl.
- the terms propyl, butyl etc include all straight and branched chain forms having the appropriate number of carbon atoms e.g. propyl includes n-propyl and isopropyl.
- Ci -6 alkoxy' refers to an -0-Ci -6 alkyl group wherein Ci -6 alkyl is as defined herein.
- Ci -6 alkoxy groups include methoxy, ethoxy, propoxy, butoxy, pentoxy or hexoxy.
- propoxy, butoxy etc include all straight and branched chain forms having the appropriate number of carbon atoms e.g. propoxy includes n-propoxy and isopropoxy
- 'halo' refers to a fluorine, chlorine, bromine or iodine atom.
- 'C 3-6 cycloalkyl' refers to a saturated monocyclic hydrocarbon ring of 3 to 6 carbon atoms.
- C 3-6 cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl.
- nitrogen containing heterocyclyl group includes a 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system, a 6-9 membered saturated or partially unsaturated bridged ring system or a 4-7 membered saturated or partially unsaturated aliphatic ring fused to a benzene ring containing a nitrogen atom in addition to 1 or 2 optional additional heteroatoms selected from oxygen, nitrogen or sulphur.
- Suitable examples of such monocyclic rings include pyrrolidinyl, azetidinyl, piperidinyl, piperazinyl, morpholinyl, hexahydroazepanyl, hexahydrodiazepanyl and homomorpholinyl.
- bridged ring systems are azabicycloheptanyl and azabicyclononanyl.
- Suitable examples of benzofused heterocyclic rings include indolinyl, isoindolinyl, 2,3,4,5-tetrahydro-1 H-3-benzazepinyl or tetrahydroisoquinolinyl.
- 5 or 6 membered heteroaryl ring represents an aromatic ring which may contain 1 , 2, 3, or 4 hetero atoms. Preferably the hetero atoms are selected from oxygen, nitrogen or sulphur.
- 5-membered heteroaryl rings in this instance include furanyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, oxadiazolyl, thiadiazolyl, triazolyl, triazinyl, isothiazolyl, isoxazolyl, thienyl, pyrazolyl or tetrazolyl.
- 6- membered heteroaryl rings are pyridinyl, pyrizinyl, pyrimidinyl, pyrazinyl, triazinyl, or tetrazinyl.
- Q represents -(CH 2 ) n - wherein n is 4. In another embodiment Q represents -(CH 2 ) n - wherein n is 3.
- R 1 and R 2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group e.g. piperidinyl, morpholinyl, pyrrolidinyl wherein the heterocyclyl group is unsubstituted or substituted with 1 , 2, or 3 substituents selected from methyl or fluoro.
- a nitrogen containing heterocyclyl group e.g. piperidinyl, morpholinyl, pyrrolidinyl wherein the heterocyclyl group is unsubstituted or substituted with 1 , 2, or 3 substituents selected from methyl or fluoro.
- R 1 and R 2 together with the nitrogen atom to which they are attached form a 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system containing a nitrogen atom in addition to 1 or 2 optional additional heteroatoms selected from oxygen, nitrogen or sulphur, wherein the 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system is unsubstituted or substituted with 1 , 2, or 3 substituents selected from methyl or fluoro.
- R 1 and R 2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group e.g. piperidinyl, morpholinyl, pyrrolidinyl wherein the heterocyclyl group is unsubstituted.
- R 1 and R 2 together with the nitrogen atom to which they are attached form a 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system containing a nitrogen atom in addition to 1 or 2 optional additional heteroatoms selected from oxygen, nitrogen or sulphur, wherein the 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system is unsubstituted.
- R 1 and R 2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group selected from morpholinyl and piperidinyl wherein the heterocyclyl group is unsubstituted
- A is phenyl and B represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyridinyl.
- A is phenyl and B represents furanyl, isoxazolyl, thienyl (thiophenyl), pyrazolyl or pyrimidinyl.
- A is phenyl and B represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyridinyl, and wherein B is bonded to the 2- or 4-position of A relative to the attachment to the oxadiazole ring.
- A is phenyl and B represents furan-2-yl, isoxazol-4-yl, thien-2-yl, pyrazol-4-yl, pyrazol-3-yl or pyridin-4-yl, and wherein B is bonded to the 2- or 4-position of A relative to the attachment to the oxadiazole ring.
- B is phenyl and A represents furanyl, isoxazolyl, thienyl (thiophenyl), pyrazolyl or pyridinyl.
- B is phenyl and A represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyrimidinyl.
- B is phenyl and A represents thienyl (thiophenyl), particularly thien- 2-yl. In one aspect of this embodiment, B is bonded to the 2 or 3 position of A relative to the attachment to the oxadiazole ring.
- R 3 is Ci_s alkyl, particularly methyl.
- R 3 is methyl, m is 0, 1 , 2 or 3 and t is 0.
- t 0.
- A is phenyl and B represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyridinyl, R 1 and R 2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group wherein the heterocyclyl group is unsubstituted, Q represents -(CH 2 ) n - wherein n is 3 or 4, R 3 is methyl, m is 0, 1 , 2 or 3 and t is 0.
- R 1 and R 2 together with the nitrogen atom to which they are attached form a 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system containing a nitrogen atom, e.g. piperidinyl, morpholinyl, and wherein the monocyclic ring system is unsubstituted.
- A is phenyl and B represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyridinyl, and wherein B is bonded to the 2- or 4-position of A, R 1 and R 2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group wherein the heterocyclyl group is unsubstituted, Q represents -(CH 2 ) n - wherein n is 3 or 4, R 3 is methyl, m is 0, 1 , 2 or 3 and t is 0.
- R 1 and R 2 together with the nitrogen atom to which they are attached form a 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system containing a nitrogen atom, e.g. piperidinyl, morpholinyl, and wherein the monocyclic ring system is unsubstituted.
- a nitrogen atom e.g. piperidinyl, morpholinyl
- A is phenyl and B represents furan-2-yl, isoxazol-4-yl, thien-2-yl, pyrazol-3-yl, pyrazol-4-yl or pyridin-4-yl, and wherein B is bonded to the 2- or 4-position of A, R 1 and R 2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group selected from piperidinyl and morpholinyl, wherein the heterocyclyl group is unsubstituted, Q represents -(CH 2 ) n - wherein n is 3 or 4, R 3 is methyl, m is 0, 1 , 2 or 3 and t is 0.
- B is phenyl and A represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyridinyl, R 1 and R 2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group e.g. piperidinyl, morpholinyl, pyrrolidinyl wherein the heterocyclyl group is unsubstituted, Q represents -(CH 2 ) n - wherein n is 3 or 4, and m and t are 0.
- A represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyridinyl
- R 1 and R 2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group e.g. piperidinyl, morpholinyl, pyrrolidinyl wherein the heterocyclyl group is unsubstituted
- Q represents -(CH 2 ) n - wherein
- R 1 and R 2 together with the nitrogen atom to which they are attached form a 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system containing a nitrogen atom, e.g. morpholinyl, and wherein the monocyclic ring system is unsubstituted.
- B is phenyl and A represents thienyl, R 1 and R 2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group selected from morpholinyl wherein the heterocyclyl group is unsubstituted, Q represents -(CH 2 ) n - wherein n is 4, and m and t are 0.
- Compounds of formula (I) include a compound of Examples 1 to 9 or a salt thereof.
- a compound of formula (I) or a salt thereof is selected from a compound of Examples 1 to 5, or a salt thereof.
- the present invention encompasses all isomers of formula (I) and their salts including all geometric, tautomeric and optical forms, and mixtures thereof (e.g. racemic mixtures). Where additional chiral centres are present in compounds of formula (I), the present invention includes within its scope all possible diastereoisomers, including mixtures thereof.
- the different isomeric forms may be separated or resolved one from the other by conventional methods, or any given isomer may be obtained by conventional synthetic methods or by stereospecific or asymmetric syntheses.
- the present invention may also include isotopically-labelled compounds, which are identical to the compounds of formula (I), except that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, iodine, and chlorine, such as 2 H, 3 H, 11 C, 14 C, 18 F, 35 S, 123 I and 125 I.
- Compounds of formula (I) and salts of said compounds that contain the aforementioned isotopes and/or other isotopes of other atoms are within the scope of the present invention.
- Isotopically-labelled compounds of the present invention for example those into which radioactive isotopes such as 3 H and/or 14 C are incorporated, are useful in drug and/or substrate tissue distribution assays.
- 3 H and 14 C are considered useful due to their ease of preparation and detectability.
- 11 C and 18 F isotopes are considered useful in PET (positron emission tomography), and 125 I isotopes are considered useful in SPECT (single photon emission computerized tomography), all useful in brain imaging. Substitution with heavier isotopes such as 2 H may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements and, hence, are considered useful in some circumstances.
- the compounds of formula (I) and salts thereof are not isotopically labelled.
- Certain compounds of formula (I) or salts thereof may be prepared in crystalline or non-crystalline form, and may exist in the form of solvates, such as hydrates.
- This invention includes with in its scope stoichiometric solvates as well as compounds containing variable amounts of solvate.
- salts of compounds of formula (I) are preferably pharmaceutically acceptable.
- Pharmaceutically acceptable salts include those described by Berge, Bighley and Monkhouse, J. Pharm. Sci., 1977, 66, 1-19.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable acids, including inorganic and organic acids.
- Such acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, ethanedisulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, pamoic, pantothenic, phosphoric, propionic, succinic, sulfuric, tartaric, p- toluenesulfonic acid, and the like. In some circumstances some salts may be non- stoichiometric.
- the present invention also provides processes for the preparation of a compound of formula (I) or a salt thereof:
- ring A, ring B, Q, R 1 , R 2 , R 3 , R 4 , t and m are as defined for compounds of formula (I).
- Step (a) typically comprises the use of a suitable reagent, such as 1 ,1- thiocarbonyldiimidazole or both carbon disulfide and a coupling agent such as dicyclohexylcarbodiimide in a suitable solvent such as THF or DMF, at a suitable temperature such as room temperature.
- a suitable reagent such as 1 ,1- thiocarbonyldiimidazole or both carbon disulfide and a coupling agent such as dicyclohexylcarbodiimide
- a suitable solvent such as THF or DMF
- Step (b) is a cyclisation reaction that typically comprises the reaction of a compound of formula (IV) with a compound of formula (III) in a suitable solvent such as DMF or THF at a suitable temperature such as from room temperature to 60 0 C.
- Step (c) typically comprises addition of a suitable reagent such as EDACHCI or dicyclohexylcarbodiimide to compound (II) in a suitable solvent such as DMF.
- a suitable reagent such as EDACHCI or dicyclohexylcarbodiimide
- a suitable solvent such as DMF.
- a suitable temperature for example, would be 60 to 80 0 C.
- Step (a) typically comprises the use of a suitable reagent, such as hydrazine monohydrate in the presence of a suitable solvent, such as methanol at a suitable temperature, e.g. from room temperature to reflux.
- a suitable solvent such as methanol
- a suitable temperature e.g. from room temperature to reflux.
- compounds of formula (Vl) are used as the methyl ester.
- Step (b) typically comprises the reaction of a compound of formula (VIII) with BocNHNH 2 in a suitable solvent e.g. CH 2 CI 2 Or DMF using a suitable coupling agent such as EDACHCI and optionally HOBt.
- a suitable solvent e.g. CH 2 CI 2 Or DMF
- a suitable coupling agent such as EDACHCI and optionally HOBt.
- Boc group can be removed by conventional means, e.g. by treatment with 4M HCI in dioxane.
- ring A, ring B, R 3 , R 4 , t, and m are as defined for compounds of formula (I) and LG is a leaving group for example Br.
- Step (a) typically comprises reaction of compound of formula (IX) which is typically used as the methyl ester, with compound of formula (X) over a catalyst e.g. palladium tetrakis(triphenylphoshine) catalyst, heated under microwave conditions.
- a catalyst e.g. palladium tetrakis(triphenylphoshine) catalyst, heated under microwave conditions.
- the reaction takes place in a suitable solvent, e.g. a mixture of toluene and ethanol, in the presence of a suitable base such as sodium carbonate.
- Step (a) is a reaction of NHR 1 R 2 in the presence of a suitable base e.g. triethylamine in a suitable solvent such as ethanol at a suitable temperature, e.g. 8O 0 C.
- a suitable base e.g. triethylamine
- a suitable solvent such as ethanol
- Step (b) typically comprises the use of MeNH 2 in a suitable solvent such as ethanol at a suitable temperature such as room temperature or the use of NH 2 NH 2 -H 2 O in a suitable solvent such as ethanol at a suitable temperature such as reflux.
- a suitable solvent such as ethanol at a suitable temperature such as room temperature
- NH 2 NH 2 -H 2 O in a suitable solvent such as ethanol at a suitable temperature such as reflux.
- R 1 , R 2 , and Q are as defined for compounds of formula (I) and w is 1 or 2.
- Step (a) is a reaction of NHR 1 R 2 in the presence of a suitable reducing agent e.g. NaBH(OAc) 3 in a suitable solvent such as DCM at a suitable temperature, e.g. room temperature.
- a suitable reducing agent e.g. NaBH(OAc) 3
- a suitable solvent such as DCM
- Step (b) typically comprises the use of MeNH 2 in a suitable solvent such as ethanol at a suitable temperature such as room temperature or the use of NH 2 NH 2 -H 2 O in a suitable solvent such as ethanol at a suitable temperature such as reflux.
- ring A, ring B, Q, R 1 , R 2 , R 3 ,R 4 , t and m are as defined for compounds of formula (I) and LG is a leaving group, for example Br.
- Step (a) typically comprises the use of a suitable reagent, such as 1 ,1- thiocarbonyldiimidazole or both carbon disulfide and a coupling agent such as dicyclohexylcarbodiimide in a suitable solvent such as THF or DMF, at a suitable temperature such as from 60 to 80 0 C.
- a suitable reagent such as 1 ,1- thiocarbonyldiimidazole or both carbon disulfide and a coupling agent such as dicyclohexylcarbodiimide
- a suitable solvent such as THF or DMF
- Step (b) typically comprises the reaction of a compound of formula (IV) with a compound of formula (XVIII) in a suitable solvent such as DMF or THF, at a suitable temperature such as from room temperature to 60 0 C.
- a suitable solvent such as DMF or THF
- Step (c) is a cyclisation reaction that typically comprises addition of a suitable reagent such as EDACHCI or dicyclohexylcarbodiimide to a compound of formula (XVII) in a suitable solvent such as a mixture of THF and DMF to give a compound of formula (XV).
- a suitable temperature for example, would be 60 to 80 °C.
- Step (d) typically comprises reaction of a compound of fomula (XV) with a compound of formula (XVI) e.g. phenyl boronic acid over a catalyst e.g a palladium tetrakis(triphenylphoshine) catalyst heated under microwave conditions in a suitable solvent such as DMF. Is sodium carbonate essential?
- ring A, R 4 and t are as defined for compounds of formula (I) and LG is a leaving group for example Br.
- Step (a) typically comprises the use of a suitable reagent, such as hydrazine monohydrate in the presence of a suitable solvent, such as methanol at a suitable temperature, e.g. from room temperature to reflux.
- a suitable reagent such as hydrazine monohydrate
- a suitable solvent such as methanol
- a suitable temperature e.g. from room temperature to reflux.
- compounds of formula (IX) are used as the methyl ester.
- Step (b) typically comprises the reaction of a compound of formula (XX) with BocNHNH 2 in a suitable solvent e.g. CH 2 CI 2 or DMF using a suitable coupling agent such as EDACHCI and optionally HOBt.
- a suitable solvent e.g. CH 2 CI 2 or DMF
- a suitable coupling agent such as EDACHCI and optionally HOBt.
- Boc group can be removed by conventional means, e.g. by treatment with 4M HCI in dioxane.
- Compounds of formula (I) may also be prepared by deprotecting a protected compound of formula (I). Examples of protecting groups and the means for their removal can be found in T. W. Greene 'Protective Groups in Organic Synthesis' (J. Wiley and Sons, 3 rd Ed. 1999). Suitable amine protecting groups include acyl (e.g. acetyl, removed by hydrolysis), carbanoyls (e.g. 2',2',2'-trichloroethoxycarbonyl, removed with zinc in acetic acid, benzyloxycarbonyl, removed by acidolysis or hydrogenolysis or t-butoxycarbonyl removed by acidolysis e.g.
- acyl e.g. acetyl, removed by hydrolysis
- carbanoyls e.g. 2',2',2'-trichloroethoxycarbonyl, removed with zinc in acetic acid
- benzyloxycarbonyl removed by acidolysis or
- arylalkyl e.g. benzyl, removed by hydrogenolysis
- suitable amine protecting groups include trifluoroacetyl (-COCF 3 ) which may be removed by base catalysed hydrolysis or a solid phase resin bound benzyl group, such as a Merrifield resin bound 2,6- dimethoxybenzyl group (Ellman linker), which may be removed by acid catalysed hydrolysis, for example with trifluoroacetic acid.
- protected compound of formula (I) is used herein to refer to a compound which includes a protecting group, for example those referred to above.
- Process (d) may be performed using conventional interconversion procedures such as epimerisation, oxidation, reduction, alkylation, nucleophilic or electrophilic aromatic substitution or amide bond formation.
- a further process of the invention is the preparation of pharmaceutically acceptable salts, solvates and hydrates of compounds of formula (I).
- Compounds of formula (I) and their pharmaceutically acceptable salts may have affinity for and be agonists at the nicotinic ⁇ 7 receptor and are believed to be of potential use in the treatment of neurological diseases including Alzheimer's disease (particularly cognitive deficit of Alzheimer's disease), dementia (including Lewy body dementia and vascular dementia), age-related memory dysfunction, cognitive impairment as listed below, cognitive deficit especially cognitive deficit of schizophrenia, Parkinson's disease and Tourette's syndrome, psychiatric disorders including schizophrenia as listed below, attention deficit/hyperactivity disorder as listed below, depression as listed below, anxiety as listed below and addiction, pain related disorders including pain of neuropathic origin including neuralgias, neuritis and back pain, and inflammatory pain including osteoarthritis, rheumatoid arthritis, acute inflammatory pain and back pain, migraine; and other diseases including obesity, sepsis and gastro-intestinal disorders (including irritable bowel syndrome and inflammatory bowel disease). Further neurological diseases for which these compounds may be of potential use is epilepsy and learning & memory disorders.
- the following disease classification refer to the classification code in Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, published by the American Psychiatric Association (DSM-IV) and/or the International Classification of Diseases, 10th Edition (ICD-10):
- i) Psychotic disorders for example Schizophrenia including the subtypes Paranoid Type (295.30), Disorganised Type (295.10), Catatonic Type (295.20), Undifferentiated Type (295.90) and Residual Type (295.60)); Schizophreniform Disorder (295.40); Schizoaffective Disorder (295.70) (including the subtypes Bipolar Type and Depressive Type); Delusional Disorder (297.1) (including the subtypes Erotomanic Type, Grandiose Type, Jealous Type, Persecutory Type, Somatic Type, Mixed Type and Unspecified Type); Brief Psychotic Disorder (298.8); Shared Psychotic Disorder (297.3); Psychotic Disorder due to a General Medical Condition (including the subtypes with Delusions and with Hallucinations); Substance-Induced Psychotic Disorder (including the subtypes with Delusions (293.81 ) and with Hallucinations (293.82)); and Psychotic Disorder Not Otherwise Specified (298.9).
- cognitive impairment including for example the treatment of impairment of cognitive functions including attention, orientation, learning disorders, memory (i.e. memory disorders, amnesia, amnesic disorders, transient global amnesia syndrome and age- associated memory impairment) and language function; cognitive impairment as a result of stroke, Alzheimer's disease, Huntington's disease, Pick disease, Aids-related dementia or other dementia states such as Multiinfarct dementia, alcoholic dementia, hypothyroidism-related dementia, and dementia associated to other degenerative disorders such as cerebellar atrophy and amyotropic lateral sclerosis; other acute or sub-acute conditions that may cause cognitive decline such as delirium or depression (pseudodementia states) trauma, head trauma, age related cognitive decline, stroke, neurodegeneration, drug-induced states, neurotoxic agents, mild cognitive impairment, age related cognitive impairment, autism related cognitive impairment, Down's syndrome, cognitive deficit related to psychosis, and post-electroconvulsive treatment related cognitive disorders; and dyskinetic disorders such as Parkinson's disease, neurol, memory disorders
- Depression and mood disorders for example Depressive Episodes (including Major Depressive Episode, Manic Episode, Mixed Episode and Hypomanic Episode); Depressive Disorders (including Major Depressive Disorder, Dysthymic Disorder (300.4), Depressive Disorder Not Otherwise Specified (311 )); Bipolar Disorders (including Bipolar I Disorder, Bipolar Il Disorder (i.e.
- Anxiety disorders for example Social Anxiety Disorder; Panic Attack; Agoraphobia, Panic Disorder; Agoraphobia Without History of Panic Disorder (300.22); Specific Phobia (300.29) (including the subtypes Animal Type, Natural Environment Type, Blood-lnjection-lnjury Type, Situational Type and Other Type); Social Phobia (300.23); Obsessive-Compulsive Disorder (300.3); Posttraumatic Stress Disorder (309.81 ); Acute Stress Disorder (308.3); Generalized Anxiety Disorder (300.02); Anxiety Disorder Due to a General Medical Condition (293.84); Substance-Induced Anxiety Disorder; and Anxiety Disorder Not Otherwise Specified (300.00).
- Specific Phobia (300.29) (including the subtypes Animal Type, Natural Environment Type, Blood-lnjection-lnjury Type, Situational Type and Other Type); Social Phobia (300.23); Obsessive-Compulsive Disorder (300.3); Posttraumatic Stress Disorder (309.81 ); Acute Stress Disorder (308.3); Generalized Anxiety
- Attention-Deficit /Hyperactivity Disorder including the subtypes Attention-Deficit /Hyperactivity Disorder Combined Type (314.01 ), Attention-Deficit/Hyperactivity Disorder Predominantly Inattentive Type (314.00), Attention-Deficit/Hyperactivity Disorder Hyperactive-Impulse Type (314.01 ) and Attention-Deficit/Hyperactivity Disorder Not Otherwise Specified (314.9)); Hyperkinetic Disorder; Disruptive Behaviour Disorders such as Conduct Disorder (including the subtypes childhood-onset type (321.81 ), Adolescent-Onset Type (312.82) and Unspecified Onset (312.89), Oppositional Defiant Disorder (313.81 ) and Disruptive Behaviour Disorder Not Otherwise Specified; and Tic Disorders such as Tourette's Disorder (307.23).
- a compound of formula (I) or a pharmaceutically acceptable salt thereof may be useful in the treatment or prophylaxis of pain. More particularly, a compound of formula (I) or a pharmaceutically acceptable salt thereof may be useful in the treatment of pain.
- pain includes pain of neuropathic origin including neuralgias, neuritis and back pain; acute pain, chronic pain, chronic articular pain, musculoskeletal pain, inflammatory pain including osteoarthritis, and rheumatoid arthritis, acute inflammatory pain and back pain, visceral pain, pain associated with cancer, pain associated with migraine, tension headache and cluster headaches, pain associated with functional gastrointestinal disorders, lower back and neck pain, pain associated with sprains and strains, sympathetically maintained pain; myositis, pain associated with influenza or other viral infections such as the common cold, pain associated with rheumatic fever, pain associated with myocardial ischemia, post operative pain, headache, toothache and dysmenorrhea.
- a compound of formula (I) or a pharmaceutically acceptable salt thereof may be useful in the treatment or prophylaxis of chronic pain, post-operative pain, chronic lower back and neck pain, cancer pain, sprains and strains. More particularly, a compound of formula (I) or a pharmaceutically acceptable salt thereof may be useful in a treatment of these pain conditions.
- treatment refers to symptomatic treatment.
- Chronic articular pain conditions include rheumatoid arthritis, osteoarthritis, rheumatoid spondylitis, gouty arthritis and juvenile arthritis.
- Pain associated with functional gastrointestinal disorders includes non-ulcer dyspepsia, non-cardiac chest pain and irritable bowel syndrome.
- the invention also provides a compound of formula (I) or a pharmaceutically acceptable salt thereof, for use in therapy for example in the treatment or prophylaxis of the above disorders, in particular pain, neurological (e.g. cognitive deficit of Alzheimer's disease) and psychiatric disorders (e.g. cognitive deficit of schizophrenia). More particularly, a compound of formula (I) or a pharmaceutically acceptable salt thereof may be useful in the treatment or pain.
- the invention further provides a method of treatment of the above disorders, in mammals including humans, which comprises administering to the sufferer a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof in the manufacture of a medicament for use in the treatment or prophylaxis of the above disorders.
- a compound of formula (I) or a pharmaceutically acceptable salt thereof is usually formulated in a standard pharmaceutical composition.
- Such compositions can be prepared using standard procedures.
- the present invention further provides a pharmaceutical composition for use in the treatment or prophylaxis of the above disorders which comprises a compound of formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier.
- the present invention further provides a pharmaceutical composition which comprises a compound of formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier.
- the compound of formula (I) or a pharmaceutically acceptable salt thereof may be used in combination with other medicaments indicated to be useful as either disease modifying or symptomatic treatments of Alzheimer's disease.
- Suitable examples of such other therapeutic agents may be agents known to modify cholinergic transmission such as 5-HT 1A antagonists, (e.g. lecozotan), 5-HT6 antagonists, M1 muscarinic agonists, M2 muscarinic antagonist, acetylcholinesterase inhibitors (e.g tetrahydroaminoacridine, donepezil or rivastigmine), or allosteric modulators, nicotinic receptor agonists or allosteric modulators, symptomatic agents such as 5-HT6 receptor antagonists, e.g. SB742457, H3 receptor antagonists e.g.
- 5-HT 1A antagonists e.g. lecozotan
- 5-HT6 antagonists e.g. lecozotan
- M1 muscarinic agonists e.g. M2 muscarinic antagonist
- acetylcholinesterase inhibitors e.g tetrahydroaminoacrid
- GSK189254 and GSK239512 5-HT4 receptor agonist, PPAR agonists, also NMDA receptor antagonists or modulators, also disease modifying agents such as ⁇ or v- secretase inhibitors (e.g. R-flurbiprofen), also AMPA positive modulators and Glycine Transporter Reuptake inhibitors.
- ⁇ or v- secretase inhibitors e.g. R-flurbiprofen
- AMPA positive modulators e.g. Glycine Transporter Reuptake inhibitors.
- the compound of formula (I) or a pharmaceutically acceptable salt thereof may be used in combination with other medicaments indicated to be useful in the treatment of pain of neuropathic origin including neuralgias, neuritis and back pain, and inflammatory pain including osteoarthritis, rheumatoid arthritis, acute inflammatory pain, back pain and migraine.
- Such therapeutic agents include for example COX-2 (cyclooxygenase-2 ) inhibitors, such as celecoxib, deracoxib, rofecoxib, valdecoxib, parecoxib, COX-189 or 2-(4- ethoxy-phenyl)-3-(4-methanesulfonyl-phenyl)-pyrazolo[1 ,5-b]pyridazine (WO99/012930); 5-lipoxygenase inhibitors; NSAIDs (non-steroidal anti-inflammatory drugs) such as diclofenac, indomethacin, nabumetone or ibuprofen; bisphosphonates, leukotriene receptor antagonists; DMARDs (disease modifying anti-rheumatic drugs) such as methotrexate; adenosine A1 receptor agonists; sodium channel blockers, such as lamotrigine; NMDA (N-methyl-D-aspartate) receptor
- COX-2 inhibitors are disclosed in US Patent Nos. 5,474,995, US5,633,272; US5,466,823, US6,310,099 and US6.291.523; and in WO 96/25405, WO 97/38986, WO 98/03484, WO 97/14691 , WO99/12930, WO00/26216, WO00/52008, WO00/38311 , WO01/58881 and WO02/18374.
- the compounds may be administered either sequentially or simultaneously by any convenient route.
- the invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a further therapeutic agent or agents.
- compositions comprising a combination as defined above together with a pharmaceutically acceptable carrier or excipient comprise a further aspect of the invention.
- the individual components of such combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical formulations.
- each compound may differ from that when the compound is used alone. Appropriate doses will be readily appreciated by those skilled in the art.
- a pharmaceutical composition of the invention which may be prepared by admixture, suitably at ambient temperature and atmospheric pressure, is usually adapted for oral, parenteral or rectal administration and, as such, may be in the form of tablets, capsules, oral liquid preparations, powders, granules, lozenges, reconstitutable powders, injectable or infusible solutions or suspensions or suppositories. Orally administrable compositions are generally preferred.
- Tablets and capsules for oral administration may be in unit dose form, and may contain conventional excipients, such as binding agents, fillers, tabletting lubricants, disintegrants and acceptable wetting agents.
- the tablets may be coated according to methods well known in normal pharmaceutical practice.
- Oral liquid preparations may be in the form of, for example, aqueous or oily suspension, solutions, emulsions, syrups or elixirs, or may be in the form of a dry product for reconstitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives such as suspending agents, emulsifying agents, non-aqueous vehicles (which may include edible oils), preservatives, and, if desired, conventional flavourings or colorants.
- fluid unit dosage forms may be prepared utilising a compound of the invention or pharmaceutically acceptable salt or solvate thereof and a sterile vehicle.
- the compound depending on the vehicle and concentration used, can be either suspended or dissolved in the vehicle.
- the compound can be dissolved for injection and filter sterilised before filling into a suitable vial or ampoule and sealing.
- adjuvants such as a local anaesthetic, preservatives and buffering agents are dissolved in the vehicle.
- the composition can be frozen after filling into the vial and the water removed under vacuum.
- Parenteral suspensions are prepared in a similar manner, except that the compound is suspended in the vehicle instead of being dissolved, and sterilisation cannot be accomplished by filtration.
- the compound can be sterilised by exposure to ethylene oxide before suspension in a sterile vehicle.
- a surfactant or wetting agent is included in the composition to facilitate uniform distribution of the compound.
- the composition may contain from 0.1% to 99% by weight, preferably from 10% to 60% by weight, of the active material, depending on the method of administration.
- the dose of the compound of formula (I) or a pharmaceutically acceptable salt thereof used in the treatment of the aforementioned disorders will vary in the usual way with the seriousness of the disorders, the weight of the sufferer, and other similar factors.
- suitable unit doses may be 0.05 to 1000 mg, more suitably 1.0 to 200 mg, and such unit doses may be administered more than once a day, for example two or three a day. Such therapy may extend for a number of weeks, months, years or even life.
- a further aspect to the invention is a pharmaceutical composition
- a pharmaceutical composition comprising 0.05 to IOOOmg of a compound of formula (I) or a pharmaceutically acceptable salt thereof, and 0 to 3 g more suitably 0 to 2g of at least one pharmaceutically acceptable carrier.
- LCMS Li ⁇ uid Chromato ⁇ raohv Mass Soectrometrv Biotage SP4: Four column sequential FLASH purification system with expanded fraction bed designed for multiple sample purification, Website: http://www.biotage.com/
- 3-Bromo-2-thiophenecarbohydrazide (0.4g, 1.8mmol) was added to 4-(4- isothiocyanatobutyl)morpholine (0.54g, 2.71 mmol) and dissolved in tetrahydrofuran (10ml). The reaction was heated at 60 0 C for 1.5h. EDACHCI (0.38g, 1.99mmol) was added, followed by DMF (4ml). The mixture was heated at 80 0 C for 2h; however LC-MS indicated presence of starting material (25%). An additional 0.5eq of EDACHCI (0.2g, 0.9mmol) was added to the mixture and heating was continued for 2h. However LC-MS indicated presence of starting material (11%).
- the title compound (405mg) was prepared from the appropriate hydrazide (213mg, 1.O ⁇ mmol) and isothiocyanate (253.5mg).
- the appropriate hydrazide was prepared from the carboxylic acid by reaction with Boc protected hydrazine.
- Assay 1 ⁇ 7 nAChR FLIPR® (Fluorometric Imaging Plate Reader) assay Function of the heterologous expressed ⁇ 7 nAChR was assessed by a FLIPR-Ca 2+ assay. Since nAChRs are non-selective cation channels with high permeability to Ca 2+ , these studies were carried out by measuring changes of intracellular Ca 2+ concentration using the Ca 2+ -chelating fluorescent dye Fluo-4 and FLIPR® (Fluorometric Imaging Plate Reader) technology.
- GH4C1 cells pituitary tumor, immortalized cell line
- human ⁇ 7 nAChR Biocat ID 96986
- growth medium Ham's Nutrient Mixture F10 - Ham's F10, Invitrogen 31550-023, 15% Horse Serum heat inactivated - Invitrogen 26050-047, 2.5% Foetal Bovine Serum - FBS, Gibco 10500-064, 200 ⁇ g/ml Hygromycin B - Invitrogen, 10687-010, 10 mg/L Phenol Red - Sigma, P 0290, 1 mM Glutamine - Invitrogen, 25030-024) and plated in 500 cm 2 Triple Flask.
- cells growing in suspension were harvested, centrifuged, resuspended in growth medium at a density of 1.8 x 10 5 /mL and plated in coated clear bottom black 384 wells plates (Pierce) at 9000 cells/well. Cells were then incubated at 30 0 C, 5% CO 2 for 72 hours.
- the dye was then diluted with AB to a final concentration of 2 ⁇ M and placed on the cells. After 45-60 minutes dye loading incubation at 37°C, the unincorporated dye was removed from the cells by washing (80 ⁇ L, 3 times) with AB, and a final volume of 30 ⁇ L/well of AB was left in each well.
- Examples 1 , 3, 5, 6 to 9 were tested in the ⁇ 7 nAChR FLIPR® assay.
- the results are expressed as pEC50 values.
- a pEC50 is the negative logarithm of the agonist EC50 calculation as determined in the ⁇ 7 nAChR FLIPR® assay. Certain Examples have been tested more than once. Variations in pEC50 may arise between tests.
- ⁇ 7 nAChR FLIPR® Fluorometric Imaging Plate Reader
- Function of the heterologous expressed ⁇ 7 nAChR was assessed by a FLIPR-Ca 2+ assay. Since nAChRs are non-selective cation channels with high permeability to Ca 2+ , these studies were carried out by measuring changes of intracellular Ca 2+ concentration using the Ca 2+ -chelating fluorescent dye Fluo-4 and FLIPR TETRA ® (Fluorometric Imaging Plate Reader) technology.
- GH4C1 cells pituitary tumor, immortalized cell line
- human ⁇ 7 nAChR Biocat ID 96986
- growth medium Ham's Nutrient Mixture F10 - Ham's F10, Invitrogen 11550-043, 15% Horse Serum heat inactivated - Invitrogen 26050-088, 2.5% Foetal Bovine Serum heat inactivated - FBS, Gibco 10500- 064, 200 ⁇ g/ml Hygromycin B - Invitrogen, 10687-010, 10 mg/L Phenol Red - Sigma, P 0290, 1 mM Glutamine - Invitrogen, 25030-024) and seeded in 175 cm 2 Flasks.
- cells growing in semi-suspension were harvested, centrifuged, resuspended in growth medium at a density of 8 x 10 5 /mL and plated in poly-D-lysine coated clear bottom, black wall, 96 well plates (BD Bioscience) at 80,000 cells/well. Cells were then incubated at 30 0 C, 5% CO 2 for 72 hours.
- the dye was then diluted with AB to a final concentration of 2 ⁇ M and placed on the cells. After 45 minutes dye loading incubation at 37°C, the unincorporated dye was removed from the cells by washing (200 ⁇ L, 3 times) with AB, and a final volume of 150 ⁇ L/well of AB was left in each well. Plates containing test compounds, dissolved in AB, at 40 ⁇ M (from 10 mM, 100% DMSO stock) and serially diluted with AB were placed onto the FLIPR TETRA ®. Cell plates were then placed in the FLI PR®, and cell fluorescence was determined before drug addition (30 seconds) and monitored (excitation 488 nm, emission 510-580 nm) immediately following exposure to compounds. Maximum fluorescence values were recorded and fitted for agonist EC50 calculations.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- Neurology (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to novel oxadiazole derivatives having pharmacological activity, processes for their preparation, compositions containing them and their use in the treatment of neurological, psychiatric disorders and gastrointestinal disorders. wherein R1 and R2 independently represent hydrogen, C1-6 alkyl or C3-6 cycloalkyl; or R1 and R2 together with the nitrogen atom to which they are attached form a nitroge containing heterocyclyl group which is unsubstituted or substituted with 1, 2, or 3 substituents selected from methyl or fluoro; Q represents -(CH2)n- wherein n represents 3 or 4; A represents a phenyl ring and B represents a 5 or 6 membered heteroaryl ring or B represents a phenyl ring and A represents a 5 or 6 membered heteroaryl ring; R3 and R4 independently represent C1-6 alkyl, C1-6 alkoxy and halo; t and m independently represent an integer from 0 to 3. Compounds of formula (I) and their pharmaceutically acceptable salts may have affinity for and be agonists at the nicotinic α7 receptor and are believed to be of potential use in the treatment of neurological diseases including Alzheimer's disease.
Description
-ARYL-l,3,4-0XADIAZ0LE-2-AMINES AS NICOTINIC ACETYLCHOLINE RECEPTOR AGONISTS
The present invention relates to novel oxadiazole derivatives having pharmacological activity, processes for their preparation, compositions containing them and their use in the treatment of neurological, psychiatric disorders and gastrointestinal disorders.
Nicotinic acetylcholine receptors (nAChRs) are cation-specific, excitatory ligand-gated ion channels that are widely expressed throughout the central and peripheral nervous systems. To date, 16 mammalian nAChR subunit genes have been cloned: 5 encoding muscle receptor subunits, and 11 encoding neuronal receptor subunits. The nicotinic α7 receptor subunit is predominantly expressed in the mammalian central nervous system (CNS), where it is thought to assemble as a functional homopentameric complex, and is also expressed in peripheral tissues including the sympathetic nervous system, immune cells and the Gl tract. Activation of neuronal nicotinic α7 receptors by selective agonists or the endogenous ligand acetylcholine can modulate the release of various neurotransmitters including glutamate, GABA, dopamine, and noradrenaline and, thus, has the potential to modulate a range of neurological functions. Additionally, in vivo studies have shown that α7 nAChR agonists can modulate neurotransmitter release in brain areas such as the cortex and hippocampus that are relevant to cognition (Paterson D et al, (2000) Prog Neurobiol 61 :75-111.
A number of literature reports have demonstrated the cognitive enhancing properties of α7 nAChR agonists (e.g. GTS-21 (3-(2,4-dimethoxybenzylidene)anabaseine), AR-R 17779 ((-)-spiro[1 -azabicyclo[2.2.2]octane-3,5'-oxazoldin-2'-one] 4-propyl-benzylidene anabaseine) and SSR-180771 (4-bromophenyl 1 ,4-diazabicyclo[3.2.2]nonane-4- carboxylate hydrochloride) in rodent and primate cognition models including the radial arm maze (Levin E. D. et al. (1999), Behavioural Pharmacology. 10(6-7) :675-80), social recognition (Van Kampen M. et al. (2004) Psychopharmacology. 172(4):375-83), elevated plus maze/delayed matching-to-sample test (Briggs CA. et al. (1997) Pharmacology, Biochemistry & Behavior. 57(1-2):231-41 ), active avoidance and radial arm maze (Arendash G.W. et al. (1995) Brain Research. 674(2): 252-9).
Consistent with these animal studies, recent data from small clinical trials demonstrates that the α7 nAChR partial agonist GTS-21 enhanced memory and attention in healthy volunteers (Kitagawa H. et al. (2003) Neuropsychopharmacology. 28(3):542-51 ). Furthermore, beneficial effects of nicotine on attention parameters have also been demonstrated in Alzheimer's disease (Potter A. and Levin E. D. (1997) Drugs & Aging. 11(3):206-28), age-associated memory impairments (White H. K. and Levin E. D. (2004), Psychopharmacology. 171(4):465-71 ) and attention deficit disorder (Levin E. D. et al. (1996) Psychopharmacology. 123(1):55-63). Activation of α7 nAChRs has also been reported to ameliorate sensory gating deficits in both preclinical (Simosky J. K. et al., (2001 ) Biological Psychiatry. 50(7):493-500) and small clinical studies. These data suggest that novel α7 nAChR agonists and/or partial agonists such as the current series
could be useful for the treatment of cognitive impairments in neurological and psychiatric disorders such as Alzheimer's disease, related neurodegenerative disorders and schizophrenia.
The present invention provides, in a first aspect, compounds of formula (I) or a salt thereof:

(I) wherein
R1 and R2 independently represent hydrogen, C1-6 alkyl or C3-6cycloalkyl; or R1 and R2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group which is unsubstituted or substituted with 1 , 2, or 3 substituents selected from methyl or fluoro; Q represents -(CH2)n- wherein n represents 3 or 4;
A represents a phenyl ring and B represents a 5 or 6 membered heteroaryl ring or B represents a phenyl ring and A represents a 5 or 6 membered heteroaryl ring; R3 and R4 independently represent Ci-6 alkyl, Ci-6alkoxy and halo; t and m independently represent an integer from 0 to 3.
The term 'Ci-6 alkyl' as used herein as a group or a part of the group refers to a linear or branched saturated hydrocarbon group containing from 1 to 6 carbon atoms. Ci-6 alkyl groups include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl or hexyl. Unless a particular structure is specified, the terms propyl, butyl etc include all straight and branched chain forms having the appropriate number of carbon atoms e.g. propyl includes n-propyl and isopropyl.
The term 'Ci-6 alkoxy' as used herein refers to an -0-Ci-6 alkyl group wherein Ci-6 alkyl is as defined herein. Ci-6 alkoxy groups include methoxy, ethoxy, propoxy, butoxy, pentoxy or hexoxy. As for alkyl unless a particular stucture is specified the terms propoxy, butoxy etc include all straight and branched chain forms having the appropriate number of carbon atoms e.g. propoxy includes n-propoxy and isopropoxy
The term 'halo' as used herein refers to a fluorine, chlorine, bromine or iodine atom.
The term 'C3-6 cycloalkyl' as used herein refers to a saturated monocyclic hydrocarbon ring of 3 to 6 carbon atoms. C3-6 cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl.
The term "nitrogen containing heterocyclyl group" includes a 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system, a 6-9 membered saturated or partially unsaturated bridged ring system or a 4-7 membered saturated or partially unsaturated aliphatic ring fused to a benzene ring containing a nitrogen atom in addition to 1 or 2 optional additional heteroatoms selected from oxygen, nitrogen or sulphur. Suitable examples of such monocyclic rings include pyrrolidinyl, azetidinyl, piperidinyl, piperazinyl, morpholinyl, hexahydroazepanyl, hexahydrodiazepanyl and homomorpholinyl. Examples of such bridged ring systems are azabicycloheptanyl and azabicyclononanyl. Suitable examples of benzofused heterocyclic rings include indolinyl, isoindolinyl, 2,3,4,5-tetrahydro-1 H-3-benzazepinyl or tetrahydroisoquinolinyl.
The term "5 or 6 membered heteroaryl ring" represents an aromatic ring which may contain 1 , 2, 3, or 4 hetero atoms. Preferably the hetero atoms are selected from oxygen, nitrogen or sulphur. Examples of 5-membered heteroaryl rings in this instance include furanyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, oxadiazolyl, thiadiazolyl, triazolyl, triazinyl, isothiazolyl, isoxazolyl, thienyl, pyrazolyl or tetrazolyl. Examples of 6- membered heteroaryl rings are pyridinyl, pyrizinyl, pyrimidinyl, pyrazinyl, triazinyl, or tetrazinyl.
In one embodiment Q represents -(CH2)n- wherein n is 4. In another embodiment Q represents -(CH2)n- wherein n is 3.
In one embodiment, R1 and R2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group e.g. piperidinyl, morpholinyl, pyrrolidinyl wherein the heterocyclyl group is unsubstituted or substituted with 1 , 2, or 3 substituents selected from methyl or fluoro. In a more particular embodiment, R1 and R2 together with the nitrogen atom to which they are attached form a 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system containing a nitrogen atom in addition to 1 or 2 optional additional heteroatoms selected from oxygen, nitrogen or sulphur, wherein the 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system is unsubstituted or substituted with 1 , 2, or 3 substituents selected from methyl or fluoro.
In one embodiment, R1 and R2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group e.g. piperidinyl, morpholinyl, pyrrolidinyl wherein the heterocyclyl group is unsubstituted. In a more particular embodiment, R1 and R2 together with the nitrogen atom to which they are attached form a 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system containing a nitrogen atom in addition to 1 or 2 optional additional heteroatoms selected
from oxygen, nitrogen or sulphur, wherein the 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system is unsubstituted. In an even more particular embodiment, R1 and R2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group selected from morpholinyl and piperidinyl wherein the heterocyclyl group is unsubstituted
In one embodiment A is phenyl and B represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyridinyl. In an alternative embodiment, A is phenyl and B represents furanyl, isoxazolyl, thienyl (thiophenyl), pyrazolyl or pyrimidinyl.
In one embodiment A is phenyl and B represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyridinyl, and wherein B is bonded to the 2- or 4-position of A relative to the attachment to the oxadiazole ring.
In one embodiment A is phenyl and B represents furan-2-yl, isoxazol-4-yl, thien-2-yl, pyrazol-4-yl, pyrazol-3-yl or pyridin-4-yl, and wherein B is bonded to the 2- or 4-position of A relative to the attachment to the oxadiazole ring.
In one embodiment B is phenyl and A represents furanyl, isoxazolyl, thienyl (thiophenyl), pyrazolyl or pyridinyl. In an alternative embodiment, B is phenyl and A represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyrimidinyl.
In one embodiment B is phenyl and A represents thienyl (thiophenyl), particularly thien- 2-yl. In one aspect of this embodiment, B is bonded to the 2 or 3 position of A relative to the attachment to the oxadiazole ring.
In one embodiment R3 is Ci_s alkyl, particularly methyl.
In one embodiment R3 is methyl, m is 0, 1 , 2 or 3 and t is 0.
In one embodiment t is 0.
In one embodiment A is phenyl and B represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyridinyl, R1 and R2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group wherein the heterocyclyl group is unsubstituted, Q represents -(CH2)n- wherein n is 3 or 4, R3 is methyl, m is 0, 1 , 2 or 3 and t is 0. In a more particular embodiment, R1 and R2 together with the nitrogen atom to which they are attached form a 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system containing a nitrogen atom, e.g. piperidinyl, morpholinyl, and wherein the monocyclic ring system is unsubstituted.
In one embodiment A is phenyl and B represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyridinyl, and wherein B is bonded to the 2- or 4-position of A, R1 and R2 together with
the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group wherein the heterocyclyl group is unsubstituted, Q represents -(CH2)n- wherein n is 3 or 4, R3 is methyl, m is 0, 1 , 2 or 3 and t is 0. In a more particular embodiment, R1 and R2 together with the nitrogen atom to which they are attached form a 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system containing a nitrogen atom, e.g. piperidinyl, morpholinyl, and wherein the monocyclic ring system is unsubstituted.
In one embodiment A is phenyl and B represents furan-2-yl, isoxazol-4-yl, thien-2-yl, pyrazol-3-yl, pyrazol-4-yl or pyridin-4-yl, and wherein B is bonded to the 2- or 4-position of A, R1 and R2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group selected from piperidinyl and morpholinyl, wherein the heterocyclyl group is unsubstituted, Q represents -(CH2)n- wherein n is 3 or 4, R3 is methyl, m is 0, 1 , 2 or 3 and t is 0.
In one embodiment B is phenyl and A represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyridinyl, R1 and R2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group e.g. piperidinyl, morpholinyl, pyrrolidinyl wherein the heterocyclyl group is unsubstituted, Q represents -(CH2)n- wherein n is 3 or 4, and m and t are 0. In a more particular embodiment, R1 and R2 together with the nitrogen atom to which they are attached form a 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system containing a nitrogen atom, e.g. morpholinyl, and wherein the monocyclic ring system is unsubstituted..
In one embodiment B is phenyl and A represents thienyl, R1 and R2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group selected from morpholinyl wherein the heterocyclyl group is unsubstituted, Q represents -(CH2)n- wherein n is 4, and m and t are 0.
Compounds of formula (I) include a compound of Examples 1 to 9 or a salt thereof.
In one embodiment of the invention, a compound of formula (I) or a salt thereof is selected from a compound of Examples 1 to 5, or a salt thereof.
It is to be understood that the present invention encompasses all isomers of formula (I) and their salts including all geometric, tautomeric and optical forms, and mixtures thereof (e.g. racemic mixtures). Where additional chiral centres are present in compounds of formula (I), the present invention includes within its scope all possible diastereoisomers, including mixtures thereof. The different isomeric forms may be separated or resolved one from the other by conventional methods, or any given isomer may be obtained by conventional synthetic methods or by stereospecific or asymmetric syntheses.
The present invention may also include isotopically-labelled compounds, which are identical to the compounds of formula (I), except that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, iodine, and chlorine, such as 2H, 3H, 11C, 14C, 18F, 35S, 123I and 125I. Compounds of formula (I) and salts of said compounds that contain the aforementioned isotopes and/or other isotopes of other atoms are within the scope of the present invention. Isotopically-labelled compounds of the present invention, for example those into which radioactive isotopes such as 3H and/or 14C are incorporated, are useful in drug and/or substrate tissue distribution assays. 3H and 14C are considered useful due to their ease of preparation and detectability. 11C and 18F isotopes are considered useful in PET (positron emission tomography), and 125I isotopes are considered useful in SPECT (single photon emission computerized tomography), all useful in brain imaging. Substitution with heavier isotopes such as 2H may afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements and, hence, are considered useful in some circumstances. In one embodiment of the invention, the compounds of formula (I) and salts thereof are not isotopically labelled.
Certain compounds of formula (I) or salts thereof may be prepared in crystalline or non-crystalline form, and may exist in the form of solvates, such as hydrates. This invention includes with in its scope stoichiometric solvates as well as compounds containing variable amounts of solvate.
Because of the potential use of compounds of formula (I) in medicine, salts of compounds of formula (I) are preferably pharmaceutically acceptable.
Pharmaceutically acceptable salts include those described by Berge, Bighley and Monkhouse, J. Pharm. Sci., 1977, 66, 1-19. The term "pharmaceutically acceptable salts" refers to salts prepared from pharmaceutically acceptable acids, including inorganic and organic acids. Such acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, ethanedisulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, pamoic, pantothenic, phosphoric, propionic, succinic, sulfuric, tartaric, p- toluenesulfonic acid, and the like. In some circumstances some salts may be non- stoichiometric.
Compounds of formula (I) can be prepared as set forth in the following Schemes and in the Examples. The following processes form another aspect of the present invention.
The present invention also provides processes for the preparation of a compound of formula (I) or a salt thereof:
Process (a):
Compounds of formula (I) may be prepared in accordance with the following Scheme 1 :
Scheme 1

wherein ring A, ring B, Q, R1, R2, R3, R4, t and m are as defined for compounds of formula (I).
Step (a) typically comprises the use of a suitable reagent, such as 1 ,1- thiocarbonyldiimidazole or both carbon disulfide and a coupling agent such as dicyclohexylcarbodiimide in a suitable solvent such as THF or DMF, at a suitable temperature such as room temperature.
Step (b) is a cyclisation reaction that typically comprises the reaction of a compound of formula (IV) with a compound of formula (III) in a suitable solvent such as DMF or THF at a suitable temperature such as from room temperature to 600C.
Step (c) typically comprises addition of a suitable reagent such as EDACHCI or dicyclohexylcarbodiimide to compound (II) in a suitable solvent such as DMF. A suitable temperature, for example, would be 60 to 80 0C.
The above steps can be carried out separately with isolation of intermediates (IV) and/or (II) or carried out sequentially in a one pot reaction .
Compounds of formula (III) may be prepared in accordance with the following Scheme 2:
Scheme 2

wherein ring A, ring B, R3, R4, t and m are as defined for compounds of formula (I).
Step (a) typically comprises the use of a suitable reagent, such as hydrazine monohydrate in the presence of a suitable solvent, such as methanol at a suitable temperature, e.g. from room temperature to reflux. Typically compounds of formula (Vl) are used as the methyl ester.
Step (b) typically comprises the reaction of a compound of formula (VIII) with BocNHNH2 in a suitable solvent e.g. CH2CI2Or DMF using a suitable coupling agent such as EDACHCI and optionally HOBt.
In Step (c) the Boc group can be removed by conventional means, e.g. by treatment with 4M HCI in dioxane.
Compounds of formula (Vl) may be prepared as set out in Scheme 3:
Scheme 3

wherein ring A, ring B, R3, R4, t, and m are as defined for compounds of formula (I) and LG is a leaving group for example Br.
Step (a) typically comprises reaction of compound of formula (IX) which is typically used as the methyl ester, with compound of formula (X) over a catalyst e.g. palladium tetrakis(triphenylphoshine) catalyst, heated under microwave conditions. The reaction takes place in a suitable solvent, e.g. a mixture of toluene and ethanol, in the presence of a suitable base such as sodium carbonate.
Compounds of formula (V) may be prepared in accordance with the following Scheme 4:
Scheme 4

wherein Q, R1 and R2 are as defined for compounds of formula (I).
Step (a) is a reaction of NHR1R2 in the presence of a suitable base e.g. triethylamine in a suitable solvent such as ethanol at a suitable temperature, e.g. 8O0C.
Step (b) typically comprises the use of MeNH2 in a suitable solvent such as ethanol at a suitable temperature such as room temperature or the use of NH2NH2-H2O in a suitable solvent such as ethanol at a suitable temperature such as reflux.
Compounds of formula (V) may also be prepared in accordance with the following Scheme 5:
Scheme 5

(XIII)
wherein R1, R2, and Q are as defined for compounds of formula (I) and w is 1 or 2.
Step (a) is a reaction of NHR1R2 in the presence of a suitable reducing agent e.g. NaBH(OAc)3 in a suitable solvent such as DCM at a suitable temperature, e.g. room temperature.
Step (b) typically comprises the use of MeNH2 in a suitable solvent such as ethanol at a suitable temperature such as room temperature or the use of NH2NH2-H2O in a suitable solvent such as ethanol at a suitable temperature such as reflux.
Process (b)
Compounds of formula (I) may also be prepared as set out in Scheme 6:
Scheme 6

wherein ring A, ring B, Q, R1, R2, R3 ,R4, t and m are as defined for compounds of formula (I) and LG is a leaving group, for example Br.
Step (a) typically comprises the use of a suitable reagent, such as 1 ,1- thiocarbonyldiimidazole or both carbon disulfide and a coupling agent such as dicyclohexylcarbodiimide in a suitable solvent such as THF or DMF, at a suitable temperature such as from 60 to 800C.
Step (b) typically comprises the reaction of a compound of formula (IV) with a compound of formula (XVIII) in a suitable solvent such as DMF or THF, at a suitable temperature such as from room temperature to 600C.
Step (c) is a cyclisation reaction that typically comprises addition of a suitable reagent such as EDACHCI or dicyclohexylcarbodiimide to a compound of formula (XVII) in a suitable solvent such as a mixture of THF and DMF to give a compound of formula (XV). A suitable temperature, for example, would be 60 to 80 °C.
The above steps can be carried out separately with isolation of intermediates (IV) and/or (XVII) or carried out sequentially in a one pot reaction.
Step (d) typically comprises reaction of a compound of fomula (XV) with a compound of formula (XVI) e.g. phenyl boronic acid over a catalyst e.g a palladium tetrakis(triphenylphoshine) catalyst heated under microwave conditions in a suitable solvent such as DMF. Is sodium carbonate essential?
Compounds of formula (XVIII) may be prepared in accordance with the following Scheme 7:
Scheme 7

(XIX)
wherein ring A, R4 and t are as defined for compounds of formula (I) and LG is a leaving group for example Br.
Step (a) typically comprises the use of a suitable reagent, such as hydrazine monohydrate in the presence of a suitable solvent, such as methanol at a suitable temperature, e.g. from room temperature to reflux. Typically compounds of formula (IX) are used as the methyl ester.
Step (b) typically comprises the reaction of a compound of formula (XX) with BocNHNH2 in a suitable solvent e.g. CH2CI2 or DMF using a suitable coupling agent such as EDACHCI and optionally HOBt.
In Step (c) the Boc group can be removed by conventional means, e.g. by treatment with 4M HCI in dioxane.
Process (c): Compounds of formula (I) may also be prepared by deprotecting a protected compound of formula (I).
Examples of protecting groups and the means for their removal can be found in T. W. Greene 'Protective Groups in Organic Synthesis' (J. Wiley and Sons, 3rd Ed. 1999). Suitable amine protecting groups include acyl (e.g. acetyl, removed by hydrolysis), carbanoyls (e.g. 2',2',2'-trichloroethoxycarbonyl, removed with zinc in acetic acid, benzyloxycarbonyl, removed by acidolysis or hydrogenolysis or t-butoxycarbonyl removed by acidolysis e.g. using an acid such as HCI or TFA) and arylalkyl (e.g. benzyl, removed by hydrogenolysis) as appropriate. Other suitable amine protecting groups include trifluoroacetyl (-COCF3) which may be removed by base catalysed hydrolysis or a solid phase resin bound benzyl group, such as a Merrifield resin bound 2,6- dimethoxybenzyl group (Ellman linker), which may be removed by acid catalysed hydrolysis, for example with trifluoroacetic acid.
The term "protected compound of formula (I)" is used herein to refer to a compound which includes a protecting group, for example those referred to above.
Process (d): Compounds of formula (I) may also be prepared by interconversion of a compound of formula (I) to another compound of formula (I).
Process (d) may be performed using conventional interconversion procedures such as epimerisation, oxidation, reduction, alkylation, nucleophilic or electrophilic aromatic substitution or amide bond formation.
A further process of the invention is the preparation of pharmaceutically acceptable salts, solvates and hydrates of compounds of formula (I).
Compounds of formula (VIII), (IX), (X), (XII), (XIV) (XVI) and (XX) are either commercially available, or may be prepared by known methods.
Compounds of formula (I) and their pharmaceutically acceptable salts may have affinity for and be agonists at the nicotinic α7 receptor and are believed to be of potential use in the treatment of neurological diseases including Alzheimer's disease (particularly cognitive deficit of Alzheimer's disease), dementia (including Lewy body dementia and vascular dementia), age-related memory dysfunction, cognitive impairment as listed below, cognitive deficit especially cognitive deficit of schizophrenia, Parkinson's disease and Tourette's syndrome, psychiatric disorders including schizophrenia as listed below, attention deficit/hyperactivity disorder as listed below, depression as listed below, anxiety as listed below and addiction, pain related disorders including pain of neuropathic origin including neuralgias, neuritis and back pain, and inflammatory pain including osteoarthritis, rheumatoid arthritis, acute inflammatory pain and back pain, migraine; and other diseases including obesity, sepsis and gastro-intestinal disorders (including irritable bowel syndrome and inflammatory bowel disease). Further
neurological diseases for which these compounds may be of potential use is epilepsy and learning & memory disorders.
The following disease classification refer to the classification code in Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, published by the American Psychiatric Association (DSM-IV) and/or the International Classification of Diseases, 10th Edition (ICD-10):
i) Psychotic disorders for example Schizophrenia (including the subtypes Paranoid Type (295.30), Disorganised Type (295.10), Catatonic Type (295.20), Undifferentiated Type (295.90) and Residual Type (295.60)); Schizophreniform Disorder (295.40); Schizoaffective Disorder (295.70) (including the subtypes Bipolar Type and Depressive Type); Delusional Disorder (297.1) (including the subtypes Erotomanic Type, Grandiose Type, Jealous Type, Persecutory Type, Somatic Type, Mixed Type and Unspecified Type); Brief Psychotic Disorder (298.8); Shared Psychotic Disorder (297.3); Psychotic Disorder due to a General Medical Condition (including the subtypes with Delusions and with Hallucinations); Substance-Induced Psychotic Disorder (including the subtypes with Delusions (293.81 ) and with Hallucinations (293.82)); and Psychotic Disorder Not Otherwise Specified (298.9).
ii) cognitive impairment including for example the treatment of impairment of cognitive functions including attention, orientation, learning disorders, memory (i.e. memory disorders, amnesia, amnesic disorders, transient global amnesia syndrome and age- associated memory impairment) and language function; cognitive impairment as a result of stroke, Alzheimer's disease, Huntington's disease, Pick disease, Aids-related dementia or other dementia states such as Multiinfarct dementia, alcoholic dementia, hypothyroidism-related dementia, and dementia associated to other degenerative disorders such as cerebellar atrophy and amyotropic lateral sclerosis; other acute or sub-acute conditions that may cause cognitive decline such as delirium or depression (pseudodementia states) trauma, head trauma, age related cognitive decline, stroke, neurodegeneration, drug-induced states, neurotoxic agents, mild cognitive impairment, age related cognitive impairment, autism related cognitive impairment, Down's syndrome, cognitive deficit related to psychosis, and post-electroconvulsive treatment related cognitive disorders; and dyskinetic disorders such as Parkinson's disease, neuroleptic-induced parkinsonism, and tardive dyskinesias.
iii) Depression and mood disorders for example Depressive Episodes (including Major Depressive Episode, Manic Episode, Mixed Episode and Hypomanic Episode); Depressive Disorders (including Major Depressive Disorder, Dysthymic Disorder (300.4), Depressive Disorder Not Otherwise Specified (311 )); Bipolar Disorders (including Bipolar I Disorder, Bipolar Il Disorder (i.e. Recurrent Major Depressive Episodes with Hypomanic Episodes) (296.89), Cyclothymic Disorder (301.13) and Bipolar Disorder Not Otherwise Specified (296.80)); Other Mood Disorders (including Mood Disorder due to a General Medical Condition (293.83) which includes the subtypes With Depressive Features, With Major Depressive-like Episode, With Manic Features and With Mixed Features); Substance-Induced Mood Disorder (including the subtypes With Depressive Features, With Manic Features and With Mixed Features); and Mood Disorder Not Otherwise Specified (296.90).
iv) Anxiety disorders for example Social Anxiety Disorder; Panic Attack; Agoraphobia, Panic Disorder; Agoraphobia Without History of Panic Disorder (300.22); Specific Phobia (300.29) (including the subtypes Animal Type, Natural Environment Type, Blood-lnjection-lnjury Type, Situational Type and Other Type); Social Phobia (300.23); Obsessive-Compulsive Disorder (300.3); Posttraumatic Stress Disorder (309.81 ); Acute Stress Disorder (308.3); Generalized Anxiety Disorder (300.02); Anxiety Disorder Due to a General Medical Condition (293.84); Substance-Induced Anxiety Disorder; and Anxiety Disorder Not Otherwise Specified (300.00).
v) Attention-Deficit /Hyperactivity Disorder (including the subtypes Attention-Deficit /Hyperactivity Disorder Combined Type (314.01 ), Attention-Deficit/Hyperactivity Disorder Predominantly Inattentive Type (314.00), Attention-Deficit/Hyperactivity Disorder Hyperactive-Impulse Type (314.01 ) and Attention-Deficit/Hyperactivity Disorder Not Otherwise Specified (314.9)); Hyperkinetic Disorder; Disruptive Behaviour Disorders such as Conduct Disorder (including the subtypes childhood-onset type (321.81 ), Adolescent-Onset Type (312.82) and Unspecified Onset (312.89), Oppositional Defiant Disorder (313.81 ) and Disruptive Behaviour Disorder Not Otherwise Specified; and Tic Disorders such as Tourette's Disorder (307.23).
A compound of formula (I) or a pharmaceutically acceptable salt thereof may be useful in the treatment or prophylaxis of pain. More particularly, a compound of formula (I) or a pharmaceutically acceptable salt thereof may be useful in the treatment of pain.
When used herein the term pain, includes pain of neuropathic origin including neuralgias, neuritis and back pain; acute pain, chronic pain, chronic articular pain, musculoskeletal pain, inflammatory pain including osteoarthritis, and rheumatoid arthritis, acute inflammatory pain and back pain, visceral pain, pain associated with cancer, pain associated with migraine, tension headache and cluster headaches, pain associated with functional gastrointestinal disorders, lower back and neck pain, pain associated with sprains and strains, sympathetically maintained pain; myositis, pain associated with influenza or other viral infections such as the common cold, pain associated with rheumatic fever, pain associated with myocardial ischemia, post operative pain, headache, toothache and dysmenorrhea.
In one embodiment a compound of formula (I) or a pharmaceutically acceptable salt thereof may be useful in the treatment or prophylaxis of chronic pain, post-operative pain, chronic lower back and neck pain, cancer pain, sprains and strains. More particularly, a compound of formula (I) or a pharmaceutically acceptable salt thereof may be useful in a treatment of these pain conditions. In the context of the present invention, treatment refers to symptomatic treatment.
Chronic articular pain conditions include rheumatoid arthritis, osteoarthritis, rheumatoid spondylitis, gouty arthritis and juvenile arthritis.
Pain associated with functional gastrointestinal disorders includes non-ulcer dyspepsia, non-cardiac chest pain and irritable bowel syndrome.
Thus the invention also provides a compound of formula (I) or a pharmaceutically acceptable salt thereof, for use in therapy for example in the treatment or prophylaxis of the above disorders, in particular pain, neurological (e.g. cognitive deficit of Alzheimer's disease) and psychiatric disorders (e.g. cognitive deficit of schizophrenia). More particularly, a compound of formula (I) or a pharmaceutically acceptable salt thereof may be useful in the treatment or pain.
The invention further provides a method of treatment of the above disorders, in mammals including humans, which comprises administering to the sufferer a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
In another aspect, the invention provides the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof in the manufacture of a medicament for use in the treatment or prophylaxis of the above disorders.
When used in therapy, a compound of formula (I) or a pharmaceutically acceptable salt thereof is usually formulated in a standard pharmaceutical composition. Such compositions can be prepared using standard procedures.
Thus, the present invention further provides a pharmaceutical composition for use in the treatment or prophylaxis of the above disorders which comprises a compound of formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier.
The present invention further provides a pharmaceutical composition which comprises a compound of formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier.
When used in the treatment of Alzheimer's disease, the compound of formula (I) or a pharmaceutically acceptable salt thereof may be used in combination with other medicaments indicated to be useful as either disease modifying or symptomatic treatments of Alzheimer's disease.
Suitable examples of such other therapeutic agents may be agents known to modify cholinergic transmission such as 5-HT1A antagonists, (e.g. lecozotan), 5-HT6 antagonists, M1 muscarinic agonists, M2 muscarinic antagonist, acetylcholinesterase inhibitors (e.g tetrahydroaminoacridine, donepezil or rivastigmine), or allosteric modulators, nicotinic receptor agonists or allosteric modulators, symptomatic agents such as 5-HT6 receptor antagonists, e.g. SB742457, H3 receptor antagonists e.g. GSK189254 and GSK239512, 5-HT4 receptor agonist, PPAR agonists, also NMDA receptor antagonists or modulators, also disease modifying agents such as β or v- secretase inhibitors (e.g. R-flurbiprofen), also AMPA positive modulators and Glycine Transporter Reuptake inhibitors.
When used in the treatment of pain, the compound of formula (I) or a pharmaceutically acceptable salt thereof may be used in combination with other medicaments indicated to be useful in the treatment of pain of neuropathic origin including neuralgias, neuritis and back pain, and inflammatory pain including osteoarthritis, rheumatoid arthritis, acute inflammatory pain, back pain and migraine.
Such therapeutic agents include for example COX-2 (cyclooxygenase-2 ) inhibitors, such as celecoxib, deracoxib, rofecoxib, valdecoxib, parecoxib, COX-189 or 2-(4- ethoxy-phenyl)-3-(4-methanesulfonyl-phenyl)-pyrazolo[1 ,5-b]pyridazine (WO99/012930); 5-lipoxygenase inhibitors; NSAIDs (non-steroidal anti-inflammatory drugs) such as diclofenac, indomethacin, nabumetone or ibuprofen; bisphosphonates, leukotriene receptor antagonists; DMARDs (disease modifying anti-rheumatic drugs) such as methotrexate; adenosine A1 receptor agonists; sodium channel blockers, such as lamotrigine; NMDA (N-methyl-D-aspartate) receptor modulators, such as glycine receptor antagonists or memantine; ligands for the α2δ-subunit of voltage gated calcium channels, such as gabapentin and pregabalin; tricyclic antidepressants such as amitriptyline; neurone stabilising antiepileptic drugs; cholinesterase inhibitors such as galantamine; mono-aminergic uptake inhibitors such as venlafaxine; opioid analgesics; local anaesthetics; 5HT1 agonists, such as triptans, for example sumatriptan, naratriptan, zolmitriptan, eletriptan, frovatriptan, almotriptan or rizatriptan; nicotinic acetyl choline (nACh) receptor modulators; glutamate receptor modulators, for example modulators of the NR2B subtype; EP4 receptor ligands; EP2 receptor ligands; EP3 receptor ligands; EP4 agonists and EP2 agonists; EP4 antagonists; EP2 antagonists and EP3 antagonists; cannabinoid receptor ligands; bradykinin receptor ligands; vanilloid receptor ligand; and purinergic receptor ligands, including antagonists at P2X3, P2X2/3, P2X4, P2X7 or P2X4/7,.
Additional COX-2 inhibitors are disclosed in US Patent Nos. 5,474,995, US5,633,272; US5,466,823, US6,310,099 and US6.291.523; and in WO 96/25405, WO 97/38986, WO 98/03484, WO 97/14691 , WO99/12930, WO00/26216, WO00/52008, WO00/38311 , WO01/58881 and WO02/18374.
When a compound of formula (I) or a pharmaceutically acceptable salt thereof is used in combination with another therapeutic agent, the compounds may be administered either sequentially or simultaneously by any convenient route.
The invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with a further therapeutic agent or agents.
The combinations referred to above may conveniently be presented for use in the form of a pharmaceutical formulation and thus pharmaceutical formulations comprising a combination as defined above together with a pharmaceutically acceptable carrier or excipient comprise a further aspect of the invention. The individual components of such
combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical formulations.
When a compound of formula (I) or a pharmaceutically acceptable salt thereof is used in combination with a second therapeutic agent active the dose of each compound may differ from that when the compound is used alone. Appropriate doses will be readily appreciated by those skilled in the art.
A pharmaceutical composition of the invention, which may be prepared by admixture, suitably at ambient temperature and atmospheric pressure, is usually adapted for oral, parenteral or rectal administration and, as such, may be in the form of tablets, capsules, oral liquid preparations, powders, granules, lozenges, reconstitutable powders, injectable or infusible solutions or suspensions or suppositories. Orally administrable compositions are generally preferred.
Tablets and capsules for oral administration may be in unit dose form, and may contain conventional excipients, such as binding agents, fillers, tabletting lubricants, disintegrants and acceptable wetting agents. The tablets may be coated according to methods well known in normal pharmaceutical practice.
Oral liquid preparations may be in the form of, for example, aqueous or oily suspension, solutions, emulsions, syrups or elixirs, or may be in the form of a dry product for reconstitution with water or other suitable vehicle before use. Such liquid preparations may contain conventional additives such as suspending agents, emulsifying agents, non-aqueous vehicles (which may include edible oils), preservatives, and, if desired, conventional flavourings or colorants.
For parenteral administration, fluid unit dosage forms may be prepared utilising a compound of the invention or pharmaceutically acceptable salt or solvate thereof and a sterile vehicle. The compound, depending on the vehicle and concentration used, can be either suspended or dissolved in the vehicle. In preparing solutions, the compound can be dissolved for injection and filter sterilised before filling into a suitable vial or ampoule and sealing. Advantageously, adjuvants such as a local anaesthetic, preservatives and buffering agents are dissolved in the vehicle. To enhance the stability, the composition can be frozen after filling into the vial and the water removed under vacuum. Parenteral suspensions are prepared in a similar manner, except that the compound is suspended in the vehicle instead of being dissolved, and sterilisation
cannot be accomplished by filtration. The compound can be sterilised by exposure to ethylene oxide before suspension in a sterile vehicle. Advantageously, a surfactant or wetting agent is included in the composition to facilitate uniform distribution of the compound.
The composition may contain from 0.1% to 99% by weight, preferably from 10% to 60% by weight, of the active material, depending on the method of administration. The dose of the compound of formula (I) or a pharmaceutically acceptable salt thereof used in the treatment of the aforementioned disorders will vary in the usual way with the seriousness of the disorders, the weight of the sufferer, and other similar factors. However, as a general guide suitable unit doses may be 0.05 to 1000 mg, more suitably 1.0 to 200 mg, and such unit doses may be administered more than once a day, for example two or three a day. Such therapy may extend for a number of weeks, months, years or even life.
A further aspect to the invention is a pharmaceutical composition comprising 0.05 to IOOOmg of a compound of formula (I) or a pharmaceutically acceptable salt thereof, and 0 to 3 g more suitably 0 to 2g of at least one pharmaceutically acceptable carrier.
All publications, including but not limited to patents and patent applications, cited in this specification are herein incorporated by reference as if each individual publication were specifically and individually indicated to be incorporated by reference herein as though fully set forth.
DMF: N,N-dιmethylformamιde
THF: Tetrahydrofuran,
EDACHCI: 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride
HOBt: 1 -Hydroxybenzotriazole
Boc: t-butyl carbonate
DCM: Dichloromethane h: Hour(s)
MDAP: Mass Directed Auto-Purification System
DMSO: Dimethyl sulfoxide
HEPES: 4-(2-hydroxyethyl)-1 -piperazineethanesulfonic acid
NaBH(OAc)3: Sodium triacetoxyborohydride
MeOH: Methanol
LCMS: Liαuid Chromatoαraohv Mass Soectrometrv
Biotage SP4: Four column sequential FLASH purification system with expanded fraction bed designed for multiple sample purification, Website: http://www.biotage.com/
SCX Strong Cationic Exchange Resin used for isolation of amines, Website: http://www.biotage.com/
Conditions on MDAP system: Basic:
Mobile phase: water 0.2% diethylamine - acetonitrile 0.2% diethylamine Column: Xbridge™ C18 30X100 mm - 5 microns Detection : MS and photodiode array detector (PDA)
Acidic:
Mobile phase: water 0.2% formic acid - acetonitrile 0.2% formic acid Column: Xbridge™ C18 30X100 mm - 5 microns Detection : MS and photodiode array detector (PDA)
The following non-limiting Examples illustrate the preparation of pharmacologically active compounds of the invention.
Description 1 : [4-(1-Piperidinyl)butyl]amine

To a solution of N-(4-bromobutyl)phthalimide (7.625g, 27.025mmol) in ethanol (80ml) was added piperidine (3.2ml, 32.4mmol) followed by triethylamine (7.5ml, 54.1 mmol). The reaction mixture was then brought to reflux for 5h. The reaction was cooled to room temperature and hydrazine hydrate (2.6ml, 54.1 mmol) was then added to the mixture. After heating for 2h, an off-white precipitate was produced; the reaction was cooled to room temperature and allowed to stir overnight. The precipitate was then filtered off and the filtrate passed through a SCX column (eluting with 10% ammonia in methanol) to provide [4-(1-piperidinyl)butyl]amine as an oil. (2.4g)
Description 2: 1-(4-lsothiocyanatobutyl)piperidine

[4-(1-Piperidinyl)butyl]amine (300mg, 1.9mmol) was dissolved in DMF (1.5ml) and added dropwise to a stirring solution of 1 ,1-thiocarbonyldiimidazole (342mg, 1.9mmol)
previously dissolved in DMF (1.5ml). After 12h of stirring at room temperature, an LC- MS was taken of the reaction mixture. LC-MS showed the presence of product, (MH+= 199). The solvent of the reaction mixture was evaporated on the rotary evaporator to afford an oil. This oil was then dissolved in 40ml of DCM and extracted with 40ml of water. The aqueous layer was further extracted with DCM (3 x 40ml). The organic layers were combined, dried over magnesium sulfate, filtered and concentrated to afford an oil. LC-MS showed presence of product (MH+=I 99). 1-(4- lsothiocyanatobutyl)piperidine (518mg) was isolated.
Description 3: Methyl 2-(1-methyl-1H-pyrazol-4-yl)benzoate

1 -Methyl-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (750mg, 3.6mmol) was dissolved in toluene (12ml) and ethanol (3.8ml) before palladium- tetrakis(triphenylphosphine) (160mg, 0.14mmol), methyl 2-bromobenzoate (389ul, 2.8mmol) and 2M aqueous sodium carbonate (3.8ml) were added. The reaction mixture was heated at 1500C for 15mins in the microwave after which an LC-MS was taken of the reaction mixture. LC-MS showed presence of product, (MH+=217). The reaction mixture was filtered through SiO2 and washed with ethyl acetate (150ml). The solvent was evaporated off under reduced pressure to afford an oil, which was then dissolved in DCM (75ml) and washed with water (75ml) The aqueous layer was further extracted with DCM (3 x50ml). The organic layers were combined and dried over magnesium sulfate, filtered and concentrated to yield an oil. LC-MS showed the presence of product, (MH+=217). A 1Og SCX column was washed through with methanol (100ml) before loading the oil previously dissolved in 1 :1 methanokDCM (50ml). The column was washed with methanol (200ml),followed by 2M ammonia in methanol solution (200ml). The combined ammonia in methanol fractions were evaporated under reduced pressure to afford an oil. LC-MS showed presence of product, (MH+=217). Methyl 2-(1- methyl-1 H-pyrazol-4-yl)benzoate (842mg) was isolated.
Description 4: 2-(1-Methyl-1H-pyrazol-4-yl)benzohydrazide

Methyl 2-(1-methyl-1 H-pyrazol-4-yl)benzoate (421 mg, 1.9mmol) was dissolved in methanol (3ml) before the addition of hydrazine hydrate (1.4ml, 13.6mmol). The reaction mixture was refluxed for 3h after which a TLC indicated that starting material was still present. The reaction mixture was therefore refluxed for another 12h at which time the LC-MS showed the presence of product, (MH+=217). A 1Og SCX column was washed through with methanol (50ml) before the reaction mixture was pipetted onto the column and loaded under gravity. The loaded SCX was then washed with methanol (100ml) which cleaned the column of any colour. The methanol wash was concentrated to yield a dull pink solid. LC-MS showed presence of product, (MH+=217).The pink solid was triturated with 25ml of diethyl ether to yield a grey solid (311mg). LC-MS showed presence of product, (MH+=217).
Example 1 : 5-[2-(1-Methyl-1H-pyrazol-4-yl)phenyl]-N-[4-(1-piperidinyl)butyl]-1,3,4- oxadiazol-2-amine hydrochloride

2-(1-Methyl-1 H-pyrazol-4-yl)benzohydrazide (153mg, 0.7mmol) was dissolved in DMF (1ml) and added to 1-(4-isothiocyanatobutyl)piperidine (154mg, O.δmmol) previously dissolved in DMF (1 ml). The reaction mixture was stirred at 600C for 12h. After 12h, an LC-MS taken of the reaction mixture showed the presence of the reaction intermediate 2-{[2-(1-methyl-1 H-pyrazol-4-yl)phenyl]carbonyl}-N-[4-(1- piperidinyl)butyl]hydrazinecarbothioamide, (MH+=415). EDACHCI (162mg, 0.9mmol) was dissolved in DMF (2ml) and added to the reaction mixture. The reaction mixture was stirred at 600C for 36h. An LC-MS taken of the reaction mixture showed the presence of the reaction intermediate above, (MH+=415) and the desired product (MH+=381 ). To push the reaction to completion, an additional equivalent of EDACHCI (135mg, 0.7mmol) previously dissolved in DMF (1ml) was added to the reaction mixture. The reaction mixture was stirred for a further 48h at room temperature, after which an LC-MS was taken of the reaction mixture. It showed the presence of product, (MH+=381 ) and the reaction intermediate was no longer present. The solvent of the
reaction mixture was evaporated under reduced pressure to afford an oil; this oil was dissolved in DCM (30ml) and extracted with water (30ml). The aqueous layer was then further extracted with DCM (3x30ml). The organic layers were combined, dried over magnesium sulfate, filtered and concentrated to afford an oil. Purification using a silica column (100% DCM to 100% (10% 2M ammonia in methanol) in DCM) yielded an oil. In an attempt to turn the oil into a solid, it was triturated with diethyl ether (10ml), hexane (10ml) and acetone (10ml) separately. None of the attempts were successful; therefore it was dissolved in methanol (2ml) before the addition of 1 M HCI in diethyl ether (1 ml). The solvent was evaporated under reduced pressure and a solid (82mg) was isolated. LC-MS (MH+=381)
Description 5: 4-(4-Morpholinyl)butyl]amine

A solution of morpholine (3ml, 34.9mmol), N-(4-bromobutyl)phthalimide (8g, 29mmol) and triethylamine (8ml, 58mmol) in ethanol (55ml) was refluxed for 16h, after which the LC-MS showed reaction completion. The reaction was cooled to room temperature. Ethanol (10ml) and hydrazine hydrate (6ml, 58mmol) were added and the mixture refluxed for 20min, and a solid crashed out. The reaction was filtered and the filtrate passed through SCX with methanol followed by 10% 2M ammonium hydroxide in methanol. The latter fractions eluted with 10% ammonium hydroxide in methanol were combined and concentrated in vacuo to afford an oil (3g). ELSD LC-MS, (MH+= 159).
Description 6: 4-(4-lsothiocyanatobutyl)morpholine

4-(4-Morpholinyl)butyl]amine (0.83g, 5.25mmol) was dissolved in tetrahydrofuran (20ml). 1 ,1-Thiocarbonyldiimidazole (0.935g, 5.245mmol) was added and the reaction was stirred for 70min, LC-MS: (MH+=201.1). The solvent was removed in vacuo and the residue was taken up in DCM (30ml) and washed with water (15ml). The aqueous layer was further extracted with DCM (3 x 20ml). The combined organic layers were dried over sodium sulfate and reduced in vacuo to give an oil (1.266g).
Description 7: 3-Bromo-2-thiophenecarbohydrazide

To a solution of methyl 3-bromo-2-thiophenecarboxylate (2.538g, 11.484mmol) in methanol (12ml) was added hydrazine hydrate (1 ml) and the reaction mixture brought to reflux for 1Oh. After 1Oh, the reaction was shown to be incomplete by LC-MS. The reaction was then brought to reflux for a further 5h after which a precipitate was formed. The precipitate was then filtered, washed with ether (30ml) and then dried in vacuo to afford 3-bromo-2-thiophenecarbohydrazide as a solid. (878mg,) LC-MS: (MH+=222).
Description 8: 5-(3-Bromo-2-thienyl)-N-[4-(4-morpholinyl)butyl]-1,3,4-oxadiazol-2- amine

3-Bromo-2-thiophenecarbohydrazide (0.4g, 1.8mmol) was added to 4-(4- isothiocyanatobutyl)morpholine (0.54g, 2.71 mmol) and dissolved in tetrahydrofuran (10ml). The reaction was heated at 600C for 1.5h. EDACHCI (0.38g, 1.99mmol) was added, followed by DMF (4ml). The mixture was heated at 800C for 2h; however LC-MS indicated presence of starting material (25%). An additional 0.5eq of EDACHCI (0.2g, 0.9mmol) was added to the mixture and heating was continued for 2h. However LC-MS indicated presence of starting material (11%). Another 0.5 eq of EDACHCI was added. Heating was continued for 19h, LC-MS: (MH+=389). The solvent was removed in vacuo. The residue was taken up in DCM (30ml) and washed with water (15ml). The aqueous layer was further extracted with DCM (3 x 20ml). The combined organic layers were dried over sodium sulfate and reduced in vacuo to afford 1.07g of an oil. It was purified using Biotage SP4 (4OM silica gel), eluting with DCM and 10% methanol in DCM to afford a solid (420mg). LC-MS: (MH+=389).
Example 2: N-[4-(4-Morpholinyl)butyl]-5-(3-phenyl-2-thienyl)-1,3,4-oxadiazol-2- amine hydrochloride

To a solution of 5-(3-bromo-2-thienyl)-N-[4-(4-morpholinyl)butyl]-1 ,3,4-oxadiazol-2- amine (200mg, 0.5mmol) in DMF (3ml) was added phenylboronic acid (81.8mg, 0.67mmol), palladium-tetrakis(triphenylphosphine) (30mg, 0.03mmol), and 2M aqueous sodium carbonate (0.78ml) and the reaction was brought to 1300C using microwave reactor for 40min. The solvent was removed in vacuo. The crude was filtered through SCX using 100% methanol followed by 2M ammonia in methanol to give an oil (215mg). It was purified using biotage SP4 (25M silica gel), eluting with DCM and 10% methanol in DCM to afford an oil. Further purification was done using MDAP basic conditions. The solvent was removed in vacuo. Toluene (2 x 10ml) was added and removed in vacuo. The sample was dissolved in DCM (1 ml), followed by addition of HCI in ether (0.29ml). The mixture was stirred at room temperature for 5min. The solvent was removed in vacuo and the sample dried in the vacuum oven at 400C overnight to afford a solid (120mg). LC-MS (MH+ 385.2)
Table 1
Examples 3 to 7 were prepared in an analogous manner to Example 2. Where indicated, HCI salts were prepared.


Description 9: 1,1-Dimethylethyl 2-{[4-(4- pyridinyl)phenyl]carbonyl}hydrazinecarboxylate

4-Pyridin-4-yl benzoic acid (250μg, 1.25mmol), EDAC (288μg, 1.51 mmol), HOBt (227μg), and 1 ,1-dimethylethyl hydrazinecarboxylate (199μg), in DMF were stirred at
room temperature for 1 h. The reaction mixture was diluted with water (20μl), extracted with ethyl acetate (30μl x 3), the organic phase washed with bicarbonate, dried over magnesium sulfate and evaporated to give an oil, 520mg. This was used without further purification.
Description 10: 4-(4-Pyridinyl)benzohydrazide

1 ,1-Dimethylethyl 2-{[4-(4-pyridinyl)phenyl]carbonyl}hydrazinecarboxylate (1.25mmol) was dissolved in dioxane (4μl) and HCI (4M in dioxane) (4ml) was added. The resulting mixture (and white solid precipitate) was stirred for 1 h. The solid was filtered off, treated with potassium carbonate solution and extracted with ethyl acetate (20μl x3). The combined extracts were dried over magnesium sulphate and evaporated to give a solid (112mg).
Description 11 : Λ/-[3-(1-Piperidinyl)propyl]-2-{[4-(4- pyridinyl)phenyl]carbonyl}hydrazinecarbothioamide

4-(4-Pyridinyl)benzohydrazide (112mg 0.525mmol) was dissolved in THF (7ul) and 1- (3-isothiocyanatopropyl)piperidine (126mg, 0.68mmol) was added. The solution was stirred at room temperature overnight for about 18h. The solvent was evaporated to give a solid (271 μg).
Example 8. N-[3-(1 -Piperidinyl)propyl]-5-[4-(4-pyridinyl)phenyl]-1 ,3,4-oxadiazol- 2-amine

Λ/-[3-(1-Piperidinyl)propyl]-2-{[4-(4-pyridinyl)phenyl]carbonyl}hydrazinecarbothioamide (0.525mmol) was dissolved in DMF (~5ml), EDAC was added (120mg, 0.63mmol), and the mixture heated at 8O0C for 2 hours. The reaction mixture was cooled, diluted with
water (15ml) and extracted with ethyl acetate (50μl x 3). The organic layer was washed with water (20μl x2), dried over magnesium sulphate and evaporated. The compound was purified on a column using 0-10% 2M NH3 in MeOH/DCM. It was further purified using MDAP. Following concentration of the appropriate fractions, the residue obtained was diluted with ethyl acetate and washed with sodium carbonate solution. The organic layers were dried over magnesium sulphate and evaporated to give a solid (~77mg). LCMS (M+H)+ 364
Description 12: Λ/-[3-(1-Piperidinyl)propyl]-2-{[4-(1H-pyrazol-3- yl)phenyl]carbonyl}hydrazinecarbothioamide
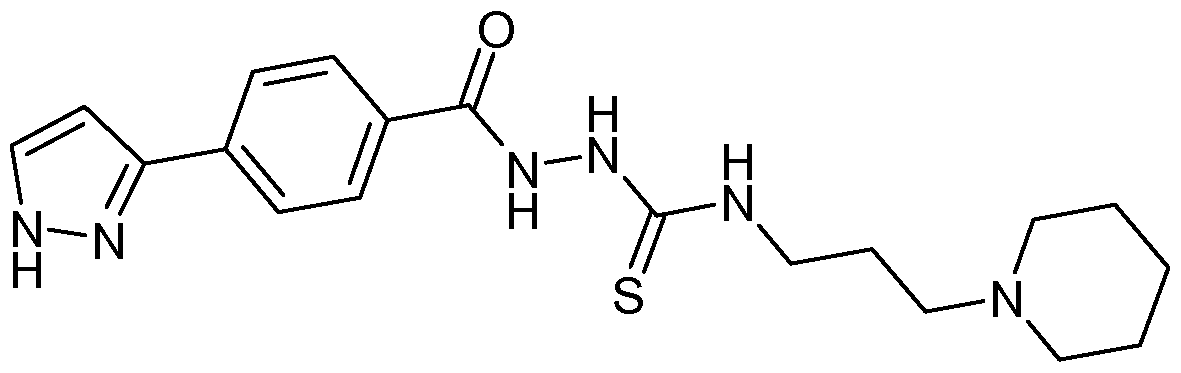
The title compound (405mg) was prepared from the appropriate hydrazide (213mg, 1.Oθmmol) and isothiocyanate (253.5mg). The appropriate hydrazide was prepared from the carboxylic acid by reaction with Boc protected hydrazine.
Example 9: N-[3-(1 -Piperidinyl)propyl]-5-[4-(1 H-pyrazol-3-yl)phenyl]-1 ,3,4- oxadiazol-2 -amine

Λ/-[3-(1 -Piperidinyl)propyl]-2-{[4-(1 H-pyrazol-3- yl)phenyl]carbonyl}hydrazinecarbothioamide (405mg, 1.05mmol) and EDAC
(1.26mmol, 241.1 mg) were stirred at 8O0C under argon in DMF overnight and cooled.
Water (10ml) was added and extracted with ethyl acetate (30ml x 3). The organic phase was washed with water (40mlx3), dried over magnesium sulphate and evaporated.
Purification used a 2Og Si column; solvent 10% 2M NH3 MeOH/DCM. The fractions containing the product were mixed and solvent was evaporated. MDAP was used for further purification, followed by evaporation. The residue was diluted with ethyl acetate, sodium carbonate solution was added and the title product( 89mg) was extracted and dried.
LCMS (M+H)+ 353
ASSAYS FOR DETERMINING BIOLOGICAL ACTIVITY
Assay 1 :
α7 nAChR FLIPR® (Fluorometric Imaging Plate Reader) assay Function of the heterologous expressed α7 nAChR was assessed by a FLIPR-Ca2+ assay. Since nAChRs are non-selective cation channels with high permeability to Ca2+, these studies were carried out by measuring changes of intracellular Ca2+ concentration using the Ca2+-chelating fluorescent dye Fluo-4 and FLIPR® (Fluorometric Imaging Plate Reader) technology.
GH4C1 cells (pituitary tumor, immortalized cell line) stably transfected with human α7 nAChR (Biocat ID 96986), were thawed, suspended in growth medium (Ham's Nutrient Mixture F10 - Ham's F10, Invitrogen 31550-023, 15% Horse Serum heat inactivated - Invitrogen 26050-047, 2.5% Foetal Bovine Serum - FBS, Gibco 10500-064, 200 μg/ml Hygromycin B - Invitrogen, 10687-010, 10 mg/L Phenol Red - Sigma, P 0290, 1 mM Glutamine - Invitrogen, 25030-024) and plated in 500 cm2 Triple Flask. 72 hours before an experiment, cells growing in suspension were harvested, centrifuged, resuspended in growth medium at a density of 1.8 x 105/mL and plated in coated clear bottom black 384 wells plates (Pierce) at 9000 cells/well. Cells were then incubated at 300C, 5% CO2 for 72 hours.
On the day of the experiment, cells were washed twice with Assay Buffer (AB) (145 mM NaCI, 5 mM KCI, 1 mM MgCI2, 2 mM CaCI2, 20 mM HEPES, 5.5 mM Glucose pH=7.3) containing 2.5 mM Probenecid. Changes in the intracellular Ca2+ content of stably transfected cells were measured using the Ca2+ chelating dye Fluo-4 (Tef Labs 0152) in conjunction with a FLIPR® (Molecular Devices). The cell permeant dye Fluo-4 was prepared to a concentration of 1 mM in 100% DMSO and 10% Pluronic acid. The dye was then diluted with AB to a final concentration of 2 μM and placed on the cells. After 45-60 minutes dye loading incubation at 37°C, the unincorporated dye was removed from the cells by washing (80 μL, 3 times) with AB, and a final volume of 30 μL/well of AB was left in each well.
Plates containing test compounds (dissolved in 100% DMSO at 2 mM and serially diluted with DMSO) were copied into "daughter" plates (1 μL/well dispensation). Just prior to starting the assay, the "daughter" plate was diluted with 50 μL/well of AB.
The plates were then placed in the FLIPR®, and cell fluorescence was determined before drug addition (30 seconds) and monitored (excitation 488 nm, emission 510-580 nm) immediately following exposure to compounds. Maximum fluorescence values were recorded and fitted for agonist EC50 calculations.
Results
The compounds of Examples 1 , 3, 5, 6 to 9 were tested in the α7 nAChR FLIPR® assay.
The results are expressed as pEC50 values. A pEC50 is the negative logarithm of the agonist EC50 calculation as determined in the α7 nAChR FLIPR® assay. Certain Examples have been tested more than once. Variations in pEC50 may arise between tests.
The compounds of Examples 1 , 3, 5, 6, 7 were tested as the HCI salt, compounds of Examples 8 and 9 as the free base.
The compounds of Examples 1 , 3, 5, 6 to 9 exhibited a pEC50 of >5
Assay 2:
α7 nAChR FLIPR® (Fluorometric Imaging Plate Reader) assay Function of the heterologous expressed α7 nAChR was assessed by a FLIPR-Ca2+ assay. Since nAChRs are non-selective cation channels with high permeability to Ca2+, these studies were carried out by measuring changes of intracellular Ca2+ concentration using the Ca2+-chelating fluorescent dye Fluo-4 and FLIPRTETRA® (Fluorometric Imaging Plate Reader) technology.
GH4C1 cells (pituitary tumor, immortalized cell line) stably transfected with human α7 nAChR (Biocat ID 96986), were thawed, suspended in growth medium (Ham's Nutrient Mixture F10 - Ham's F10, Invitrogen 11550-043, 15% Horse Serum heat inactivated - Invitrogen 26050-088, 2.5% Foetal Bovine Serum heat inactivated - FBS, Gibco 10500- 064, 200 μg/ml Hygromycin B - Invitrogen, 10687-010, 10 mg/L Phenol Red - Sigma, P 0290, 1 mM Glutamine - Invitrogen, 25030-024) and seeded in 175 cm2 Flasks. 72 hours before an experiment, cells growing in semi-suspension were harvested, centrifuged, resuspended in growth medium at a density of 8 x 105/mL and plated in poly-D-lysine coated clear bottom, black wall, 96 well plates (BD Bioscience) at 80,000 cells/well. Cells were then incubated at 300C, 5% CO2 for 72 hours.
On the day of the experiment, cells were washed twice with Assay Buffer (AB) (145 mM NaCI, 5 mM KCI, 1 mM MgCI2, 2 mM CaCI2, 20 mM HEPES, 5.5 mM Glucose pH 7.4) containing 2.5 mM Probenecid. Changes in the intracellular Ca2+ content of stably transfected cells were measured using the Ca2+ chelating dye Fluo-4 (Tef Labs 0152) in conjunction with a FLIPR® (Molecular Devices). The cell permeant dye Fluo-4 was prepared to a concentration of 1 mM in 100% DMSO and 20% Pluronic acid. The dye was then diluted with AB to a final concentration of 2 μM and placed on the cells. After 45 minutes dye loading incubation at 37°C, the unincorporated dye was removed from the cells by washing (200 μL, 3 times) with AB, and a final volume of 150 μL/well of AB was left in each well.
Plates containing test compounds, dissolved in AB, at 40μM (from 10 mM, 100% DMSO stock) and serially diluted with AB were placed onto the FLIPRTETRA®. Cell plates were then placed in the FLI PR®, and cell fluorescence was determined before drug addition (30 seconds) and monitored (excitation 488 nm, emission 510-580 nm) immediately following exposure to compounds. Maximum fluorescence values were recorded and fitted for agonist EC50 calculations.
The compounds of Examples 1 to 7 were tested in this assay and exhibited a pEC50 value ≥ 5.
The compounds of Examples 1 to 3 and 5 to 7 were tested as the HCI salt, the compound of Example 8 as a free base.
More particularly, the compounds of Examples 1 to 5 exhibited a pEC50 value ≥ 6.
The application of which this description and claims forms part may be used as a basis for priority in respect of any subsequent application. The claims of such subsequent application may be directed to any feature or combination of features described herein. They may take the form of product, composition, process, or use claims and may include, by way of example and without limitation the following claims:
Claims
1. A compound of formula (I) or a salt thereof:

(I) wherein
R1 and R2 independently represent hydrogen, C1-6 alkyl or C3-6cycloalkyl; or R1 and R2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group which is unsubstituted or substituted with 1 , 2, or 3 substituents selected from methyl or fluoro; Q represents -(CH2)n- wherein n represents 3 or 4;
A represents a phenyl ring and B represents a 5 or 6 membered heteroaryl ring or B represents a phenyl ring and A represents a 5 or 6 membered heteroaryl ring; R3 and R4 independently represent Ci-6 alkyl, Ci-6alkoxy and halo; t and m independently represent an integer from 0 to 3.
2. A compound or salt as claimed in claim 1 wherein Q represents -(CH2)n- wherein n is 4.
3. A compound or salt as claimed in claim 1 or 2 wherein R1 and R2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group which is unsubstituted or substituted with 1 , 2, or 3 substituents selected from methyl or fluoro.
4. A compound or salt as claimed in claim 1 or 2 wherein R1 and R2 together with the nitrogen atom to which they are attached form a 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system containing a nitrogen atom in addition to 1 or 2 optional additional heteroatoms selected from oxygen, nitrogen or sulphur, wherein the 4-7 membered monocyclic saturated or partially unsaturated aliphatic ring system is unsubstituted or substituted with 1 , 2, or 3 substituents selected from methyl or fluoro.
5. A compound or salt as claimed in claim 1 or 2 wherein R1 and R2 together with the nitrogen atom to which they are attached form a nitrogen containing heterocyclyl group selected from morpholinyl and piperidinyl wherein the heterocyclyl group is unsubstituted.
6. A compound or salt as claimed in any one of claims 1 to 5 wherein A is phenyl and B represents furanyl, isoxazolyl, thienyl, pyrazolyl or pyridinyl.
7. A compound or salt as claimed in claim 6, wherein B is bonded to the 2- or 4- position of A relative to the attachment to the oxadiazole ring.
8. A compound or salt as claimed in claim 7, wherein A is phenyl and B represents furan-2-yl, isoxazol-4-yl, thien-2-yl, pyrazol-3-yl, pyrazol-4-yl or pyridin-4-yl.
9. A compound or salt as claimed in any one of claims 1 to 5 wherein B is phenyl and A represents thienyl.
10. A compound or salt as claimed in claim 1 selected from the compounds of Example 1 to 9 or a salt thereof.
11. A compound or salt as claimed in any one of claims 1 to 10 wherein the salt is a pharmaceutically acceptable salt.
12. A pharmaceutical composition comprising a compound or salt as claimed in claim 11.
13. A pharmaceutical composition as claimed in claim 12 further comprising a pharmaceutical carrier.
14. A compound or salt as claimed in claim 11 for use as an active therapeutic substance.
15. A compound or salt as claimed in claim 11 for use in the treatment or prophylaxis of neurological diseases, psychiatric disorders, pain related disorders, obesity, sepsis and gastro-intestinal disorders.
16. A method of treating a human or animal subject suffering from neurological diseases, psychiatric disorders, pain related disorders, obesity, sepsis and gastro- intestinal disorders which comprises administering to said subject an effective amount of a compound or salt as claimed in claim 11.
17. Use of a compound or salt as claimed in claim 1 1 , for the manufacture of a medicament for the treatment or prophylaxis of neurological diseases, psychiatric disorders, pain related disorders, obesity, sepsis and gastro-intestinal disorders.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB0723814.0A GB0723814D0 (en) | 2007-12-05 | 2007-12-05 | Compounds |
| GB0723814.0 | 2007-12-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009071576A1 true WO2009071576A1 (en) | 2009-06-11 |
Family
ID=38983045
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/066698 WO2009071576A1 (en) | 2007-12-05 | 2008-12-03 | 5-aryl-1,3,4-oxadiazole-2-amines as nicotinic acetylcholine receptor agonists |
Country Status (2)
| Country | Link |
|---|---|
| GB (1) | GB0723814D0 (en) |
| WO (1) | WO2009071576A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| EP4122923A4 (en) * | 2020-03-17 | 2024-04-24 | Sumitomo Pharma Co., Ltd. | Oxadiazole derivative |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004029053A1 (en) * | 2002-09-30 | 2004-04-08 | Neurosearch A/S | Novel 1,4-diazabicycloalkane derivatives, their preparation and use |
| WO2004076453A1 (en) * | 2003-02-27 | 2004-09-10 | Neurosearch A/S | Novel diazabicyclic aryl derivatives |
-
2007
- 2007-12-05 GB GBGB0723814.0A patent/GB0723814D0/en not_active Ceased
-
2008
- 2008-12-03 WO PCT/EP2008/066698 patent/WO2009071576A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004029053A1 (en) * | 2002-09-30 | 2004-04-08 | Neurosearch A/S | Novel 1,4-diazabicycloalkane derivatives, their preparation and use |
| WO2004076453A1 (en) * | 2003-02-27 | 2004-09-10 | Neurosearch A/S | Novel diazabicyclic aryl derivatives |
Non-Patent Citations (1)
| Title |
|---|
| MICHAEL W. DECKER*, LYNNE E. RUETER, AND R. SCOTT BITNER: "Nicotinic Acetylcholine Receptor Agonists: A Potential New Class of Analgesics", CURRENT TOPICS IN MEDICINAL CHEMISTRY, vol. 4, no. 3, January 2004 (2004-01-01), pages 369 - 384, XP002519705 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| EP4122923A4 (en) * | 2020-03-17 | 2024-04-24 | Sumitomo Pharma Co., Ltd. | Oxadiazole derivative |
Also Published As
| Publication number | Publication date |
|---|---|
| GB0723814D0 (en) | 2008-01-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7820674B2 (en) | Aminomethyl beta-secretase inhibitors for the treatment of alzheimer's disease | |
| TWI410421B (en) | Pyridin-4-yl derivatives | |
| WO2009071577A1 (en) | Oxadiazole derivatives and their use as nicotinic acetylcholine receptor modulators | |
| ES2895124T3 (en) | 6-hydroxy-4-oxo-1,4-dihydropyrimidine-5-carboxamides as APJ agonists | |
| JP4975447B2 (en) | Indole derivatives and their use as kinase inhibitors, in particular as IKK2 inhibitors | |
| EP1709028B1 (en) | Fused heteroaryl derivatives and their use as p38 kinase inhibitors | |
| EA015263B1 (en) | Novel oxadiazole derivatives and their use as positive allosteric modulators of metabotropic glutamate receptors | |
| CA2919783C (en) | Heterobicycloaryl rorc2 inhibitors and methods of use thereof | |
| MXPA06009023A (en) | Polyheterocyclic compounds and their use as metabotropic glutamate receptor antagonists. | |
| JPWO2016153023A1 (en) | Novel oxadiazole derivative and pharmaceutical containing the same | |
| WO2006057945A2 (en) | 2,3,4,6-substituted pyridyl derivative compounds useful as beta-secretase inhibitors for the treatment of alzheimer's disease | |
| AU2008320718B2 (en) | Indol-2-one derivatives disubstituted in the 3-position, preparation thereof and therapeutic use thereof | |
| US20090105217A1 (en) | 2-phenyl-5-amino-1,3,4-oxadiazoles and their use as nicotinic acetylcholine receptor ligands | |
| BR112021015963A2 (en) | BICYCLIC COMPOUNDS SUBSTITUTED AS FARNESOID X RECEPTOR MODULATORS | |
| EP1723126A2 (en) | Novel mch receptor antagonists | |
| WO2009071576A1 (en) | 5-aryl-1,3,4-oxadiazole-2-amines as nicotinic acetylcholine receptor agonists | |
| JP2013540792A (en) | Diaza-spiro [5.5] undecanes useful as orexin receptor antagonists | |
| BR112018008204B1 (en) | COMPOUND, PHARMACEUTICAL COMPOSITION, AND METHOD FOR TREATMENT OF DISORDERS ASSOCIATED WITH IRREGULARITIES OF GLUTAMATERGIC SIGNAL TRANSMISSION AND DISORDERS OF THE CENTRAL NERVOUS SYSTEM | |
| CN101495465A (en) | 2-phenyl-5-amino-1,3,4-oxadiazoles and their use as nicotinic acetylcholine receptor ligands | |
| CA2511360A1 (en) | Novel pharmaceuticals | |
| WO2009071519A1 (en) | Oxadiazole derivatives and their use as nicotinic acetylcholine receptor modulators | |
| MX2008015287A (en) | 2-phenyl-5-amino-1,3,4-oxadiazoles and their use as nicotinic acetylcholine receptor ligands. | |
| US8309584B2 (en) | Sulfur containing pyrazole-heterocycle derivatives as cannabinoid CB1 receptor antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08856997 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08856997 Country of ref document: EP Kind code of ref document: A1 |