WO2009058300A1 - Biphenyl derivatives as modulators of the histamine-h3 receptor useful for the treatment of disorders related thereto - Google Patents
Biphenyl derivatives as modulators of the histamine-h3 receptor useful for the treatment of disorders related thereto Download PDFInfo
- Publication number
- WO2009058300A1 WO2009058300A1 PCT/US2008/012272 US2008012272W WO2009058300A1 WO 2009058300 A1 WO2009058300 A1 WO 2009058300A1 US 2008012272 W US2008012272 W US 2008012272W WO 2009058300 A1 WO2009058300 A1 WO 2009058300A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ethyl
- biphenyl
- compound according
- mmol
- methylpyrrolidin
- Prior art date
Links
- 0 C*C(Cc(cc1)ccc1O)=O Chemical compound C*C(Cc(cc1)ccc1O)=O 0.000 description 5
- XQXPVVBIMDBYFF-UHFFFAOYSA-N OC(Cc(cc1)ccc1O)=O Chemical compound OC(Cc(cc1)ccc1O)=O XQXPVVBIMDBYFF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/10—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing aromatic rings
Definitions
- the present invention relates to certain compounds of Formula (Ia) and pharmaceutical compositions thereof that modulate the activity of the histamine H3 -receptor.
- Compounds of the present invention and pharmaceutical compositions thereof are directed to methods useful in the treatment of histamine H3 -associated disorders, such as, cognitive disorders, epilepsy, brain trauma, depression, obesity, disorders of sleep and wakefulness such as narcolepsy, shift-work syndrome, drowsiness as a side effect from a medication, maintenance of vigilance to aid in completion of tasks and the like, cataplexy, hypersomnia, somnolence syndrome, jet lag, sleep apnea and the like, attention deficit hyperactivity disorder (ADHD), schizophrenia, allergies, allergic responses in the upper airway, allergic rhinitis, nasal congestion, dementia, Alzheimer's disease, pain and the like.
- ADHD attention deficit hyperactivity disorder
- One aspect of the present invention encompasses certain biphenyl derivatives selected from compounds of Formula (Ia) and pharmaceutically acceptable salts, solvates and hydrates thereof:
- ring A is heterocyclyl optionally substituted with one, two or three substituents selected from Ci-C 6 alkyl and oxo; wherein each Ci-C 6 alkyl is optionally substituted with a C r C 6 alkoxy substituent;
- R 1 is H, Ci-C 6 alkoxy, C]-C 6 alkyl or halogen
- R 2 is H, Ci-C 6 alkoxy, C]-C 6 alkyl or halogen
- R 3 is H, Ci-C 6 alkoxy, Cj-C 6 alkyl or halogen
- R 4 is H or C 1 -C 4 alkyl; and n is 0, 1 or 2.
- One aspect of the present invention pertains to methods for treating a histamine H3- receptor associated disorder in an individual comprising administering to said individual in need thereof a therapeutically effective amount of a compound of the present invention or a pharmaceutical composition thereof.
- One aspect of the present invention pertains to methods for treating a histamine H3- receptor associated disorder selected from a cognitive disorder, epilepsy, brain trauma, depression, obesity, disorders of sleep and wakefulness, narcolepsy, cataplexy, hypersomnia, somnolence syndrome, jet lag, sleep apnea and the like, attention deficit hyperactivity disorder (ADHD), schizophrenia, allergies, allergic responses in the upper airway, allergic rhinitis, nasal congestion, dementia, Alzheimer's disease and pain.
- a histamine H3- receptor associated disorder selected from a cognitive disorder, epilepsy, brain trauma, depression, obesity, disorders of sleep and wakefulness, narcolepsy, cataplexy, hypersomnia, somnolence syndrome, jet lag, sleep apnea and the like, attention deficit hyperactivity disorder (ADHD), schizophrenia, allergies, allergic responses in the upper airway, allergic rhinitis, nasal congestion, dementia, Alzheimer's disease and pain.
- ADHD attention deficit hyperactivity disorder
- One aspect of the present invention pertains to methods for treating a disorder of sleep and wakefulness.
- One aspect of the present invention pertains to methods for treating a cognitive disorder.
- One aspect of the present invention pertains to methods for treating cataplexy.
- One aspect of the present invention pertains to methods for inducing wakefulness.
- One aspect of the present invention pertains to methods for treating pain.
- One aspect of the present invention pertains to the use of a compound of the present invention in the manufacture of a medicament for the treatment of a histamine H3-receptor associated disorder.
- One aspect of the present invention pertains to the use of a compound of the present invention in the manufacture of a medicament for the treatment of a disorder selected from a cognitive disorder, epilepsy, brain trauma, depression, obesity, disorders of sleep and wakefulness, narcolepsy, cataplexy, hypersomnia, somnolence syndrome, jet lag, sleep apnea and the like, attention deficit hyperactivity disorder (ADHD), schizophrenia, allergies, allergic responses in the upper airway, allergic rhinitis, nasal congestion, dementia, Alzheimer's disease and pain.
- a cognitive disorder selected from a cognitive disorder, epilepsy, brain trauma, depression, obesity, disorders of sleep and wakefulness, narcolepsy, cataplexy, hypersomnia, somnolence syndrome, jet lag, sleep apnea and the like, attention deficit hyperactivity disorder (ADHD), schizophrenia, allergies, allergic responses in the upper airway, allergic rhinitis, nasal congestion, dementia, Alzheimer's disease and pain.
- ADHD attention deficit hyperactivity disorder
- One aspect of the present invention pertains to the use of a compound of the present invention in the manufacture of a medicament for the treatment of a cognitive disorder.
- One aspect of the present invention pertains to the use of a compound of the present invention in the manufacture of a medicament for the treatment of cataplexy.
- One aspect of the present invention pertains to the use of a compound of the present invention in the manufacture of a medicament for inducing wakefulness.
- One aspect of the present invention pertains to the use of a compound of the present invention in the manufacture of a medicament for the treatment of pain.
- One aspect of the present invention pertains to compounds of the present invention for use in a method of treatment of the human or animal body by therapy.
- One aspect of the present invention pertains to compounds of the present invention for use in a method for the treatment of a histamine H3 -receptor associated disorder.
- One aspect of the present invention pertains to compounds of the present invention for use in a method for the treatment of a histamine H3-receptor associated disorder selected from a cognitive disorder, epilepsy, brain trauma, depression, obesity, disorders of sleep and wakefulness, narcolepsy, cataplexy, hypersomnia, somnolence syndrome, jet lag, sleep apnea, attention deficit hyperactivity disorder (ADHD), schizophrenia, allergies, allergic responses in the upper airway, allergic rhinitis, nasal congestion, dementia, Alzheimer's disease and pain.
- a histamine H3-receptor associated disorder selected from a cognitive disorder, epilepsy, brain trauma, depression, obesity, disorders of sleep and wakefulness, narcolepsy, cataplexy, hypersomnia, somnolence syndrome, jet lag, sleep apnea, attention deficit hyperactivity disorder (ADHD), schizophrenia, allergies, allergic responses in the upper airway, allergic rhinitis, nasal congestion, dementia, Alzheimer's disease and pain.
- One aspect of the present invention pertains to compounds of the present invention for use in a method for the treatment of a disorder of sleep and wakefulness.
- One aspect of the present invention pertains to compounds of the present invention for use in a method for the treatment of a cognitive disorder.
- One aspect of the present invention pertains to compounds of the present invention for use in a method for the treatment of cataplexy.
- One aspect of the present invention pertains to compounds of the present invention for use in a method of inducing wakefulness.
- One aspect of the present invention pertains to compounds of the present invention for use in a method of treating pain.
- One aspect of the present invention pertains to compounds for preparing a composition comprising admixing a compound of the present invention and a pharmaceutically acceptable carrier.
- Figure 1 shows two representative methods for preparing intermediates that are useful in the synthesis of the compounds of the present invention.
- the first method involves the reduction of an aryl amino acid derivative followed by cyclization of the resulting amino alcohol with triphosgene to give a 4-substituted cyclic carbamate.
- the second method involves the reduction of an aryl amino ketone derivative followed by cyclization of the resulting amino alcohol with triphosgene to give a 5 -substituted oxazolidinone.
- Figure 2 shows two representative methods of introducing a substituent R 5 onto the nitrogen of a heterocycle, ring A. Both processes involve the reaction of a cyclic carbamate with an alkyl or alkoxyalkyl derivative in the presence of a base. The step can be carried out either before or after the formation of the biphenyl moiety.
- Figure 3 shows a general synthesis of compounds of Formula (Ia).
- the first step is the conversion of an aryl halide derivative into a boronic acid with a trialkyl borate in the presence of a base.
- a palladium-catalyzed coupling is used to form the biphenyl moiety.
- the silyl protecting group is removed and replaced with a leaving group.
- reaction between the biphenyl derivative and a cyclic amine in the presence of base gives the molecule of Formula (Ia).
- Figure 4 shows a general method for preparing an aryl triflate for use in the palladium- catalyzed coupling reaction.
- FIG. 5 shows a general method for preparing a molecule of Formula (Ia) in which the right hand side of the molecule is (i?)-2-methylpyrolidine. First, (i?)-2-methylpyrolidine is reacted in the presence of a base with an arylalkyl derivative activated with a leaving group.
- FIG. 6 shows a general method for preparing an aryl triflate for use in the palladium- catalyzed coupling reaction. Reaction of a phenol derivative with 2-benzamidoacetic acid first provides an aryl amino acid. This is esterified and Boc protected followed by reduction to the alcohol. Cyclization is achieved by reaction with thionyl chloride and the resulting oxazolidinyl- phenol is converted to the aryl triflate with triflic anhydride.
- Figure 7 shows a general synthesis of compounds of Formula (Ia).
- a 2-(4- hydroxyphenyl)acetic acid derivative is esterified and then reduced to the diol.
- the pyrrolidine moiety is introduced by reaction of the primary alcohol with a secondary amine in the presence of triflic anhydride, with concomitant triflation of the phenol.
- This triflate is coupled to a boronic acid derivative, which itself is prepared from another aryl triflate or an aryl halide.
- Figure 8 shows three methods for preparing intermediates useful in preparing the compounds of the present invention.
- 5-oxopyrrolidine-2-carboxylic acid is reacted with anisole in the presence of phosphorus pentoxide and methanesulfonic acid to give 5-(4-methoxyphenyl)pyrrolidin-2-one, which in turn is converted to the triflate.
- the second of these methods involves the conversion of a 4-amino-3-(4-chlorophenyl)butanoic acid derivative to a 4-(4-chlorophenyl)pyrrolidin-2-one by aluminum oxide-mediated cyclization.
- chlorinated aryl triflate intermediates useful in the preparation of chlorinated compounds of the present invention may be prepared by treating phenol derivatives with N- chlorosuccinimide followed by triflation.
- Figure 9 shows a representative synthesis of aryl triflate intermediates useful in the preparation of compounds of the present invention.
- a 2-(4-hydroxyphenyl)acetic acid derivative is esterified and then the phenol is protected with a/j-methoxybenzyl group. The ester is then reduced and the primary alcohol is converted to a leaving group which is displace by a pyrrolidine derivative to give a tertiary amine.
- the PMB group is then removed with TFA and the phenol converted to the triflate.
- Figure 10 shows a representative synthesis of aryl triflate intermediates useful in the preparation of compounds of the present invention.
- a 2-(4- methoxyphenyl)acetic acid derivative is reduced to the corresponding primary alcohol.
- the free hydroxyl is activated with a leaving group which is displaced with a secondary amine.
- the methoxy group is removed with boron tribromide and the free phenol is converted to the triflate.
- Figure 11 shows a general synthesis of compounds of Formula (Ia), wherein Ring A is
- the first method provides 2-oxo-l,3- oxazinan-6-yl intermediates by the reduction aryl-3-oxopropanenitrile derivatives followed by treatment of the resulting hydroxy amine with CDI.
- the second method provides 2-oxo-l,3- oxazinan-4-yl intermediates via reaction of l-amino-3-hydroxypropyl aryl derivatives with CDI.
- the third method provides 5-oxomorpholin-3-yl intermediates from 1 ,2-dihroxyethyl aryl derivatives via the secondary amine by treatment first with sulfuric acid and acetonitrile and then ethyl chloroacetate in the presence of sodium hydride.
- Figure 13 shows a general method for preparing compounds of Formula (Ia), wherein Ring A is 2-oxo-l,3-dioxolan-4-yl of the present invention.
- Palladium catalyzed coupling of a 4- hydroxymethyl aryl halide with a boronic acid derivative gives a biphenyl intermediate which is oxidized and reacted with ethyl acetate in the presence of LDA.
- the resulting ester is reduced to the 1,3-diol and converted to the 2-oxo-l,3-dioxan-4-yl derivative by treatment with CDI.
- agonists is intended to mean moieties that interact and activate the receptor, such as the H3 histamine receptor and initiate a physiological or pharmacological response characteristic of that receptor. For example, when moieties activate the intracellular response upon binding to the receptor, or enhance GTP binding to membranes.
- antagonists is intended to mean moieties that competitively bind to the receptor at the same site as agonists (for example, the endogenous ligand), but which do not activate the intracellular response initiated by the active form of the receptor and can thereby inhibit the intracellular responses by agonists or partial agonists. Antagonists do not diminish the baseline intracellular response in the absence of an agonist or partial agonist.
- contacting is intended to mean bringing the indicated moieties together, whether in an in vitro system or an in vivo system.
- "contacting" a H3 histamine receptor with a compound of the invention includes the administration of a compound of the present invention to an individual, preferably a human, having a H3 histamine receptor, as well as, for example, introducing a compound of the invention into a sample containing a cellular or more purified preparation containing a H3 histamine receptor.
- in need of treatment and the term “in need thereof when referring to treatment are used interchangeably to mean a judgment made by a caregiver (e.g. physician, nurse, nurse practitioner, etc. in the case of humans; veterinarian in the case of animals, including non-human mammals) that an individual or animal requires or will benefit from treatment. This judgment is made based on a variety of factors that are in the realm of a caregiver's expertise, but that includes the knowledge that the individual or animal is ill, or will become ill, as the result of a disease, condition or disorder that is treatable by the compounds of the invention. Accordingly, the compounds of the invention can be used in a protective or preventive manner; or compounds of the invention can be used to alleviate, inhibit or ameliorate the disease, condition or disorder.
- a caregiver e.g. physician, nurse, nurse practitioner, etc. in the case of humans; veterinarian in the case of animals, including non-human mammals
- the term "individual” is intended to mean any animal, including mammals, preferably mice, rats, other rodents, rabbits, dogs, cats, swine, cattle, sheep, horses, or primates and most preferably humans.
- inverse agonists is intended to mean moieties that bind to the endogenous form of the receptor or to the constitutively activated form of the receptor and which inhibit the baseline intracellular response initiated by the active form of the receptor below the normal base level of activity which is observed in the absence of agonists or partial agonists, or decrease GTP binding to membranes.
- the baseline intracellular response is inhibited in the presence of the inverse agonist by at least 30%, more preferably by at least 50% and most preferably by at least 75%, as compared with the baseline response in the absence of the inverse agonist.
- modulate or modulating is intended to mean an increase or decrease in the amount, quality, response or effect of a particular activity, function or molecule.
- composition is intended to mean a composition comprising at least one active ingredient; including but not limited to, salts, solvates and hydrates of compounds of the present invention; whereby the composition is amenable to investigation for a specified, efficacious outcome in a mammal (for example, without limitation, a human).
- a mammal for example, without limitation, a human.
- terapéuticaally effective amount is intended to mean the amount of active compound or pharmaceutical agent that elicits the biological or medicinal response in a tissue, system, animal, individual or human that is being sought by a researcher, veterinarian, medical doctor or other clinician or caregiver; or by an individual, which includes one or more of the following:
- Preventing the disease for example, preventing a disease, condition or disorder in an individual that may be predisposed to the disease, condition or disorder but does not yet experience or display the pathology or symptomatology of the disease,
- Inhibiting the disease for example, inhibiting a disease, condition or disorder in an individual that is experiencing or displaying the pathology or symptomatology of the disease, condition or disorder (i.e., arresting further development of the pathology and/or symptomatology) and (3) Ameliorating the disease; for example, ameliorating a disease, condition or disorder in an individual that is experiencing or displaying the pathology or symptomatology of the disease, condition or disorder (i.e., reversing the pathology and/or symptomatology).
- Ci-Ce alkoxy is intended to mean a Ci-C 6 alkyl radical, as defined herein, attached directly to an oxygen atom, some embodiments are 1 to 5 carbons, some embodiments are 1 to 4 carbons, some embodiments are 1 to 3 carbons and some embodiments are 1 or 2 carbons. Examples include methoxy, ethoxy, n-propoxy, iso-propoxy, n-butoxy, f-butoxy, iso- butoxy, sec-butoxy and the like.
- the term "Ci-C 6 alkyl” is intended to mean a straight or branched carbon radical containing 1 to 6 carbons. Some embodiments are 1 to 5 carbons.
- Some embodiments are 1 to 4 carbons. Some embodiments are 1 to 3 carbons. Some embodiments are 1 or 2 carbons. Some embodiments are 1 carbon.
- Examples of an alkyl include, but are not limited to, methyl, ethyl, n- propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, /-butyl, pentyl, is ⁇ -pentyl, f-pentyl, Tieo-pentyl, 1-methylbutyl [i.e., -CH(CH 3 )CH 2 CH 2 CH 3 ], 2-methylbutyl [i.e., -CH 2 CH(CH 3 )CH 2 CH 3 ], n- hexyl and the like.
- C 1 -C 4 alkyl is intended to mean a straight or branched carbon radical containing 1 to 4 carbons. Some embodiments are 1 to 3 carbons. Some embodiments are 1 or 2 carbons. Some embodiments are 1 carbon. Examples of a C 1 -C 4 alkyl include methyl, ethyl, n- propyl, iso-propyl, n-butyl, sec-butyl, /s ⁇ -butyl and /-butyl.
- aryl is intended to mean an aromatic ring radical containing 6 to 10 ring carbons. Examples include phenyl and naphthyl.
- halogen or “halo” is intended to mean to a fluoro, chloro, bromo or iodo group.
- the ring carbon atoms are optionally substituted with oxo thus forming a carbonyl group.
- ring carbon atoms are optionally substituted with thioxo thus forming a thiocarbonyl group.
- the ring carbon atoms are optionally substituted with C]-C 6 alkyl.
- the ring nitrogen atoms are optionally substituted with C]-C 6 alkyl.
- the C 1 -C 6 alkyl substituent is optionally substituted with a C]-C 6 alkoxy.
- a ring carbon is substituted with Cj-C 6 alkyl.
- a ring nitrogen is substituted with C]-C 6 alkyl.
- the C]-C 6 alkyl substituent is optionally substituted with a Cj-C 6 alkoxy.
- the heterocyclic group is a 3-, A-, 5-, 6- or 7-membered ring.
- heterocyclic group examples include, but are not limited to, aziridin-2-yl, azetidin-2-yl, azetidin-3-yl, piperidin-2-yl, piperidin-3-yl, piperidin-4-yl, morpholin-2-yl, morpholin-3-yl, piperzin-2-yl, piperzin-3-yl, pyrrolidin-2-yl, pyrrolidin-3-yl, [l,3]-dioxolan-2-yl, azepan-2-yl, azepan-3-yl, azepan-4-yl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, 2-oxooxazolidin-4-yl, 2- oxooxazolidin-5-yl, 4-oxooxazolidin-2-yl, 4-oxooxazolidin-5-yl, 5-oxooxox
- the heterocyclic group is a 5- or 6-membered heterocyclic group.
- a 5- or 6-membered heterocyclic group include, but are not limited to, oxooxazolidinyl, oxopyrrolidinyl, oxoimidazolidinyl, oxopiperidinyl, oxomorpholinyl and oxo- 1,3-oxazinanyl.
- examples of a 5- or 6-membered heterocyclic groups include, but are not limited to, 2-oxooxazolidinyl, 2-oxopyrrolidinyl, 2-oxoimidazolidinyl, 2-oxo-l,3- oxazinanyl, 2-oxopiperidinyl, 3 -oxomorpholinyl and the like as shown in Table 1.
- Table 1 2-oxooxazolidinyl, 2-oxopyrrolidinyl, 2-oxoimidazolidinyl, 2-oxo-l,3- oxazinanyl, 2-oxopiperidinyl, 3 -oxomorpholinyl and the like as shown in Table 1.
- examples of a 5- or 6-membered heterocyclic group include, but are not limited to, 2-oxooxazolidinyl, 2-oxo-l,3-oxazinanyl and the like.
- examples of a 5- or 6-membered heterocyclic group include, but are not limited to, 2-oxooxazolidin-4-yl, 2-oxooxazolidin-5-yl, 2-oxo-l,3-oxazinan-4-yl, 2-oxo- l,3-oxazinan-5-yl, 2-oxo-l,3-oxazinan-6-yl and the like.
- examples of a 5- or 6-membered heterocyclic group include, but are not limited to, oxo-l,3-dioxolanyl, oxo-l,3-dioxanyl and the like.
- examples of a 5- or 6-membered heterocyclic group include, but are not limited to, 2-oxo-l,3-dioxolanyl, 2-oxo-l,3-dioxanyl and the like as shown in Table 2.
- Table 2
- examples of a 5- or 6-membered heterocyclic group include, but are not limited to, 2-oxo-l ,3 -dioxolan-4-yl, 2-oxo-l ,3-dioxan-4-yl and the like.
- hydrate as used herein means a compound of the invention or a salt thereof, that further includes a stoichiometric or non-stoichiometric amount of water bound by non- covalent intermolecular forces.
- solvate means a compound of the invention or a salt, thereof, that further includes a stoichiometric or non-stoichiometric amount of a solvent bound by non- covalent intermolecular forces.
- Preferred solvents are volatile, non-toxic, and/or acceptable for administration to humans in trace amounts.
- One aspect of the present invention pertains to certain compounds as shown in Formula (Ia): (Ia) and pharmaceutically acceptable salts, solvates and hydrates thereof; wherein:
- R 1 , R 2 , R 3 , R 4 , ring A and n have the same definitions as described herein, supra and infra.
- substituted indicates that at least one hydrogen atom of the chemical group is replaced by a non-hydrogen substituent or group, the non-hydrogen substituent or group can be monovalent or divalent. When the substituent or group is divalent, then it is understood that this group is further substituted with another substituent or group.
- a chemical group herein when a chemical group herein is "substituted" it may have up to the full valance of substitution; for example, a methyl group can be substituted by 1, 2, or 3 substituents, a methylene group can be substituted by 1 or 2 substituents, a phenyl group can be substituted by 1, 2, 3, 4, or 5 substituents, a naphthyl group can be substituted by 1, 2, 3, 4, 5, 6, or 7 substituents and the like.
- substituted with one or more substituents refers to the substitution of a group with one substituent up to the total number of substituents physically allowed by the group. Further, when a group is substituted with more than one group they can be identical or they can be different.
- Compounds of the invention can also include tautomeric forms, such as keto-enol tautomers and the like. Tautomeric forms can be in equilibrium or sterically locked into one form by appropriate substitution. It is understood that the various tautomeric forms are within the scope of the compounds of the present invention.
- Compounds of the invention can also include all isotopes of atoms occurring in the intermediates and/or final compounds. Isotopes include those atoms having the same atomic number but different mass numbers. For example, isotopes of hydrogen include deuterium and tritium.
- R 1 is selected from the group consisting of H, Ci-C 6 alkoxy, C 1 - C 6 alkyl and halogen.
- R 1 is selected from the group consisting Of C 1 -C 6 alkoxy, C 1 -C 6 alkyl and halogen.
- R 1 is selected from the group consisting of methoxy, methyl, chloro and fluoro.
- R 1 is H. In some embodiments, R 1 is C 1 -C 6 alkoxy. In some embodiments, R 1 is Cj-C 6 alkyl.
- R 1 is halogen
- R 2 is selected from the group consisting of H, C 1 -C 6 alkoxy, C 1 - C 6 alkyl and halogen.
- R 2 is selected from the group consisting Of C 1 -C 6 alkoxy, C 1 -C 6 alkyl and halogen.
- R 2 is selected from the group consisting of methoxy, methyl, chloro and fluoro. In some embodiments, R 2 is H.
- R 2 is C 1 -C 6 alkoxy.
- R 2 is C 1 -C 6 alkyl. In some embodiments, R 2 is halogen.
- R 3 is selected from the group consisting of H, Ci-C 6 alkoxy, Q- C 6 alkyl and halogen.
- R 3 is selected from the group consisting Of Ci-C 6 alkoxy, C]-C 6 alkyl and halogen.
- R 3 is selected from the group consisting of methoxy, methyl, chloro and fluoro. In some embodiments, R 3 is H.
- R 3 is Ci-C 6 alkoxy.
- R 3 is Cj -C 6 alkyl.
- R 3 is halogen
- R 4 is selected from the group consisting of H or Ci-C 4 alkyl. In some embodiments, R 4 is H. In some embodiments, R 4 is C 1 -C 4 alkyl. In some embodiments, R 4 is methyl
- ring A is heterocyclyl optionally substituted with one substituent selected from Ci-C 6 alkyl and oxo; wherein each Q-C 6 alkyl is optionally substituted with a Q- C 6 alkoxy substituent. In some embodiments, ring A is heterocyclyl optionally substituted with two substituents selected from Ci-C 6 alkyl and oxo; wherein each C]-C 6 alkyl is optionally substituted with a C]-C 6 alkoxy substituent.
- ring A is heterocyclyl optionally substituted with three substituents selected from Cj-C 6 alkyl and oxo; wherein each Cj-C 6 alkyl is optionally substituted with a Cj-C 6 alkoxy substituent.
- ring A is heterocyclyl optionally substituted with one or two substituents selected from Cj-C 6 alkyl and oxo; wherein each Cj-C 6 alkyl is optionally substituted with a Cj-C 6 alkoxy substituent.
- ring A is heterocyclyl optionally substituted with one, two or three substituents selected from Cj-C 6 alkyl and oxo; wherein each Q-C 6 alkyl is optionally substituted with a Cj-C 6 alkoxy substituent.
- ring A is selected from oxooxazolidinyl, oxopyrrolidinyl, oxoimidazolidinyl, oxopiperidinyl, oxomorpholinyl and oxo-l,3-oxazinanyl; wherein each ring A is optionally substituted with a Ci-C 6 alkyl substituent; and wherein the Ci-C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent.
- ring A is selected from oxooxazolidinyl, oxopyrrolidinyl, oxoimidazolidinyl, oxopiperidinyl, oxomorpholinyl and oxo-l,3-oxazinanyl; wherein each ring A is optionally substituted with a substituent selected from methyl, isopropyl and 2- methoxyethyl.
- ring A is selected from oxooxazolidinyl and oxo-l,3-oxazinanyl; wherein each ring A is optionally substituted with a Q-C 6 alkyl substituent; and wherein the Q- C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent.
- ring A is selected from oxooxazolidinyl and oxo-l,3-oxazinanyl; wherein each ring A is optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is selected from 2-oxooxazolidinyl, 2-oxopyrrolidinyl, 2- oxoimidazolidinyl, 2-oxopiperidinyl, 3 -oxomorpholinyl and 2-oxo-l,3-oxazinanyl; wherein each ring A is optionally substituted with a Ci -C 6 alkyl substituent; and wherein the Ci -C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent.
- ring A is selected from 2-oxooxazolidinyl, 2-oxopyrrolidinyl, 2- oxoimidazolidinyl, 2-oxopiperidinyl, 3 -oxomorpholinyl and 2-oxo-l,3-oxazinanyl; wherein each ring A is optionally substituted with a substituent selected from methyl, isopropyl and 2- methoxyethyl.
- ring A is selected from 2-oxooxazolidinyl and 2-oxo-l,3- oxazinanyl; wherein each ring A is optionally substituted with a Ci-C 6 alkyl substituent; and wherein the Q-C 6 alkyl substituent is optionally substituted with a Q-C 6 alkoxy substituent.
- ring A is selected from 2-uoxooxazolidinyl and 2-oxo-l,3- oxazinanyl; wherein each ring A is optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is selected from 2-oxooxazolidin-4-yl, 2-oxooxazolidin-5- yl and 2-oxo-l,3-oxazinan-4-yl; wherein each ring A is optionally substituted with a Cj-C 6 alkyl substituent; and wherein the Ci -C 6 alkyl substituent is optionally substituted with a Ci -C 6 alkoxy substituent.
- ring A is selected from 2-oxooxazolidin-4-yl, 2-oxooxazolidin-5- yl and 2-oxo-l,3-oxazinan-4-yl; wherein each ring A is optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is selected from 2-oxooxazolidin-4-yl, 3-methyl-2- oxooxazolidin-4-yl, 3-isopropyl-2-oxooxazolidin-4-yl, 3-(2-methoxyethyl)-2-oxooxazolidin-4- yl, 2-oxooxazolidin-5-yl and 2-oxo-l,3-oxazinan-4-yl.
- ring A is oxooxazolidinyl optionally substituted with a C 1 -C 6 alkyl substituent; wherein the Ci-C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent.
- ring A is oxopyrrolidinyl optionally substituted with a Q-C 6 alkyl substituent; wherein the Ci-C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent.
- ring A is oxoimidazolidinyl optionally substituted with a Ci-C 6 alkyl substituent; wherein the Q-C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent.
- ring A is oxopiperidinyl optionally substituted with a Q-C 6 alkyl substituent; wherein the Ci-C 6 alkyl substituent is optionally substituted with a Q-C 6 alkoxy substituent.
- ring A is oxomorpholinyl optionally substituted with a Ci-C 6 alkyl substituent; wherein the C]-C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent.
- ring A is oxo-l,3-oxazinanyl optionally substituted with a Cj-C 6 alkyl substituent; wherein the Q-C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent.
- ring A is oxooxazolidinyl optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is oxopyrrolidinyl optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is oxoimidazolidinyl optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is oxopiperidinyl optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl. In some embodiments, ring A is oxomorpholinyl optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is oxo-l,3-oxazinanyl optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is 2-oxooxazolidinyl optionally substituted with a Ci-C 6 alkyl substituent; wherein the Q-C 6 alkyl substituent is optionally substituted with a Q-C 6 alkoxy substituent.
- ring A is 2-oxopyrrolidinyl optionally substituted with a C]-C 6 alkyl substituent; wherein the Ci-C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent.
- ring A is 2-oxoimidazolidinyl optionally substituted with a Ci-C 6 alkyl substituent; wherein the Ci-C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent.
- ring A is 2-oxopiperidinyl optionally substituted with a Ci-C 6 alkyl substituent; wherein the Ci-C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent.
- ring A is 3-oxomorpholinyl optionally substituted with a Ci-C 6 alkyl substituent; wherein the Ci-C 6 alkyl substituent is optionally substituted with a Q-C 6 alkoxy substituent.
- ring A is 2-oxo-l,3-oxazinanyl optionally substituted with a Q- C 6 alkyl substituent; wherein the Ci-C 6 alkyl substituent is optionally substituted with a C]-C 6 alkoxy substituent.
- ring A is 2-oxooxazolidinyl optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is 2-oxopyrrolidinyl optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is 2-oxoimidazolidinyl optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is 2-oxopiperidinyl optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is 3-oxomorpholinyl optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is 2-oxo-l,3-oxazinanyl optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is 2-oxooxazolidin-4-yl, optionally substituted with a Q- C 6 alkyl substituent; wherein the C]-C 6 alkyl substituent is optionally substituted with a C]-C 6 alkoxy substituent.
- ring A is 2-oxooxazolidin-5-yl, optionally substituted with a Q- C 6 alkyl substituent; wherein the C]-C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent.
- ring A is 2-oxo-l,3-oxazinan-4-yl, optionally substituted with a C]-C 6 alkyl substituent; wherein the Ci-C 6 alkyl substituent is optionally substituted with a Q- C 6 alkoxy substituent.
- ring A is 2-oxooxazolidin-4-yl, optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl.
- ring A is 2-oxooxazolidin-5-yl, optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl. In some embodiments, ring A is 2-oxo-l ,3-oxazinan-4-yl, optionally substituted with a substituent selected from methyl, isopropyl and 2-methoxyethyl. In some embodiments, ring A is 2-oxooxazolidin-4-yl. In some embodiments, ring A is 3-methyl-2-oxooxazolidin-4-yl. In some embodiments, ring A is 3-isopropyl-2-oxooxazolidin-4-yl. In some embodiments, ring A is 3-(2-methoxyethyl)-2-oxooxazolidin-4-yl.
- ring A is 2-oxooxazolidin-5-yl. In some embodiments, ring A is 2-oxo-l, 3 -oxazinan-4-yl.
- ring A is selected from oxopyrrolidinyl, oxo-l,3-dioxolanyl, oxo-l,3-dioxanyl, oxomorpholinyl and oxo-l,3-oxazinanyl; wherein each ring A is optionally substituted with a Ci -C 6 alkyl substituent; and wherein the Ci-C 6 alkyl substituent is optionally substituted with a Cj-C 6 alkoxy substituent.
- ring A is selected from oxopyrrolidinyl, oxo-l,3-dioxolanyl, oxo-1, 3-dioxanyl, oxomorpholinyl and oxo-l,3-oxazinanyl.
- ring A is selected from 2-oxopyrrolidin-4-yl, 2-oxopyrrolidm-5- yl, 2-oxo-l, 3-dioxolan-4-yl, 2-oxo-l, 3 -dioxan-4-yl, 3-oxomorpholin-5-yl and 2-oxo-l, 3- oxazinan-6-yl.
- ring A is 2-oxopyrrolidin-4-yl. In some embodiments, ring A is 2-oxopyrrolidin-5-yl. In some embodiments, ring A is 2-oxo-l, 3-dioxolan-4-yl. In some embodiments, ring A is 2-oxo-l ,3-dioxan-4-yl.
- ring A is 3-oxomorpholin-5-yl. In some embodiments, ring A is 2-oxo-l, 3-oxazinan-6-yl.
- n 0, 1 or 2.
- n is 0 or 1.
- n is 0 or 2.
- n 1 or 2.
- n is 0. In some embodiments, n is 1.
- n is 2. Certain Combinations of the Present Invention: In some embodiments, R 1 and R 2 are both H. In some embodiments, R 1 and R 3 are both H. In some embodiments, R 2 and R 3 are both H. In some embodiments, R 1 , R 2 and R 3 are all H.
- Some embodiments of the present invention pertain to compounds of Formula (Ic) and pharmaceutically acceptable salts, solvates and hydrates thereof:
- R 4 is H or C 1 -C 4 alkyl
- ring A is selected from oxooxazolidinyl and oxo-l,3-oxazinanyl; wherein each ring A is optionally substituted with a Ci-C 6 alkyl substituent; and wherein the C 1 -C 6 alkyl substituent is optionally substituted with a Ci -C 6 alkoxy substituent; and n is 0, 1 or 2.
- R 4 is H or methyl;
- ring A is selected from 2-oxooxazolidin-4-yl, 3-methyl-2-oxooxazolidin-4-yl, 3- isopropyl-2-oxooxazolidin-4-yl, 3 -(2-methoxyethyl)-2-oxooxazolidin-4-yl, 2-oxooxazolidin-5 - yl and 2-oxo-l,3-oxazinan-4-yl; and n is 0, 1 or 2.
- Some embodiments of the present invention pertain to compounds of Formula (Ie) and pharmaceutically acceptable salts, solvates and hydrates thereof:
- ring A is selected from oxooxazolidinyl and oxo-l,3-oxazinanyl; wherein each ring A is optionally substituted with a Ci-C 6 alkyl substituent; and wherein the Ci-C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent; and n is 0, 1 or 2.
- ring A is selected from 2-oxooxazolidin-4-yl, 3-methyl-2-oxooxazolidin-4-yl, 3- isopropyl-2-oxooxazolidin-4-yl, 3-(2-methoxyethyl)-2-oxooxazolidin-4-yl, 2-oxooxazolidin-5- yl and 2-oxo-l,3-oxazinan-4-yl; and n is 0, 1 or 2.
- Some embodiments of the present invention pertain to compounds of Formula (Ig) and pharmaceutically acceptable salts, solvates and hydrates thereof:
- ring A is selected from oxooxazolidinyl and oxo-l,3-oxazinanyl; wherein each ring A is optionally substituted with a Ci-C 6 alkyl substituent; and wherein the C]-C 6 alkyl substituent is optionally substituted with a Ci-C 6 alkoxy substituent; and n is 0, 1 or 2.
- Some embodiments of the present invention pertain to compounds of Formula (Ig) and pharmaceutically acceptable salts, solvates and hydrates thereof:
- ring A is selected from 2-oxooxazolidin-4-yl, 3-methyl-2-oxooxazolidin-4-yl, 3- isopropyl-2-oxooxazolidin-4-yl, 3-(2-methoxyethyl)-2-oxooxazolidin-4-yl, 2-oxooxazolidin-5- yl and 2-oxo-l,3-oxazinan-4-yl; and n is 0, 1 or 2.
- R 1 , R 2 and R 3 are each independently selected from H, C 1 -C 6 alkoxy, Ci-C 6 alkyl and halogen; ring A is selected from oxooxazolidinyl, oxo-l,3-oxazinanyl, oxopyrrolidinyl, oxo-1,3- dioxolanyl, oxo-l,3-dioxanyl, oxomorpholinyl and oxo-l,3-oxazinanyl; wherein each ring A is optionally substituted with a C 1 -C 6 alkyl substituent; and wherein the C 1 -C 6 alkyl substituent is optionally substituted with a C 1 -C 6 alkoxy substituent; and n is 0, 1 or 2.
- Some embodiments of the present invention pertain to compounds of Formula (Ii) and pharmaceutically acceptable salts, solvates and hydrates thereof:
- R 1 , R 2 and R 3 are each independently selected from H, methoxy, methyl, chloro and fluoro; ring A is selected from 2-oxooxazolidin-4-yl, 3-methyl-2-oxooxazolidin-4-yl, 3- isopropyl-2-oxooxazolidin-4-yl, 3 -(2-methoxyethyl)-2-oxooxazolidin-4-yl, 2-oxooxazolidin-5 - yl, 2-oxo-l,3-oxazinan-4-yl, 2-oxopyrrolidin-4-yl, 2-oxopyrrolidin-5-yl, 2-oxo-l,3-dioxolan-4- yl, 2-oxo-l,3-dioxan-4-yl, 3-oxomorpholin-5-yl and 2-oxo-l,3-oxazinan-6-yl; and n is 0, 1 or
- Some embodiments of the present invention include every combination of one or more compounds selected from the following group shown in TABLE A and TABLE B.
- individual compounds and chemical genera of the present invention encompass all pharmaceutically acceptable salts, solvates and particularly hydrates, thereof.
- the compounds of the Formula (Ia) of the present invention may be prepared according to relevant published literature procedures that are used by one skilled in the art. Exemplary reagents and procedures for these reactions appear hereinafter in the working Examples. Protection and deprotection may be carried out by procedures generally known in the art (see, for example, Greene, T. W. and Wuts, P. G. M., Protecting Groups in Organic Synthesis, 3 rd Edition, 1999 [Wiley]; incorporated herein by reference in its entirety).
- the present invention embraces each diastereomer, each enantiomer and mixtures thereof of each compound and generic formulae disclosed herein just as if they were each individually disclosed with the specific stereochemical designation for each chiral carbon. Separation of the individual isomers (such as, by chiral HPLC, recrystallization of diastereomeric mixtures and the like) or selective synthesis (such as, by enantiomeric selective syntheses and the like) of the individual isomers is accomplished by application of various methods which are well known to practitioners in the art. INDICATIONS AND METHODS OF PROPHYLAXIS AND/OR TREATMENT
- GPCRs G-protein coupled receptors
- Rat and human histamine H3-receptors also show constitutive activity which means that they can transduce a signal even in the absence of a ligand. Histamine H3 -receptors also function as heteroceptors, modulating the release of a number of other transmitter substances including serotonin, acetylcholine, dopamine and noradrenaline (see: Brown et al. Prog. Neurobiol. 2001, 63, 637-672).
- the ligand functions as either an antagonist or inverse agonist (for reviews see: Lews et al. Nat. Rev. Drug. Discov. 2005, 4, 107-120; Passani et al. Trends Pharmacol. ScL 2004, 25, 618-625).
- H3-receptor antagonists have been shown to increase wakefulness (e.g. Lin J. S. et al. Brain Research 1990, 523, 325-330). This effect demonstrates that H3-receptor antagonists can be useful for disorders of sleep and wakefulness (Parmentier et al. J. Neurosci. 2002, 22, 7695-7711; Ligneau et al. J. Pharmacol. Exp. Ther. 1998, 287, 658-666).
- histamine H3-receptor antagonists and inverse agonists can be used to treat the somnolence syndrome associated with different pathological conditions, for example, sleep apnea and Parkinson's disease or circumstances associated with lifestyle, for example, daytime somnolence from sleep deprivation as a result of nocturnal jobs, overwork, or jet-lag (see Passani et al., Trends Pharmacol. ScL 2004, 25, 618-625). Somnolence is one of the major problems of public health because of its high prevalence (19-37% of the general population) and risk for causing work and traffic accidents.
- Sleep apnea is a common sleep disorder characterized by brief interruptions of breathing during sleep. These episodes, called apneas, last 10 seconds or more and occur repeatedly throughout the night. People with sleep apnea partially awaken as they struggle to breathe, but in the morning they may not be aware of the disturbances in their sleep.
- the most common type of sleep apnea is obstructive sleep apnea (OSA), caused by relaxation of soft tissue in the back of the throat that blocks the passage of air.
- OSA obstructive sleep apnea
- CSA Central sleep apnea
- the hallmark symptom of the disorder is excessive daytime sleepiness.
- sleep apnea Additional symptoms of sleep apnea include restless sleep, loud snoring (with periods of silence followed by gasps), falling asleep during the day, morning headaches, trouble concentrating, irritability, forgetfulness, mood or behaviour changes, weight gain, increased heart rate, anxiety, and depression.
- methylxanthine theophylline (chemically similar to caffeine) can reduce the number of episodes of apnea, but can also produce side effects such as palpitations and insomnia.
- Theophylline is generally ineffective in adults with OSA, but is sometimes used to treat CSA, and infants and children with apnea.
- some neuroactive drugs particularly modern-generation antidepressants including mirtazapine, have been reported to reduce incidences of obstructive sleep apnea.
- histamine H3-receptor antagonists and inverse agonists can be used to treat narcolepsy (Tedford et al. Soc. Neurosci. Abstr. 1999, 25, 460.3).
- Narcolepsy is a neurological condition most often characterized by Excessive Daytime Sleepiness (EDS), episodes of sleep and disorder of REM or rapid eye movement sleep.
- EDS Excessive Daytime Sleepiness
- the main characteristic of narcolepsy is overwhelming Excessive Daytime Sleepiness (EDS), even after adequate nighttime sleep.
- a person with narcolepsy is likely to become drowsy or to fall asleep, often at inappropriate times and places.
- nighttime sleep may be fragmented with frequent wakenings.
- Classic symptoms of narcolepsy include, for example, cataplexy which is sudden episodes of loss of muscle function, ranging from slight weakness (such as limpness at the neck or knees, sagging facial muscles, or inability to speak clearly) to complete body collapse. Episodes may be triggered by sudden emotional reactions such as laughter, anger, surprise, or fear, and may last from a few seconds to several minutes.
- Another symptom of narcolepsy is sleep paralysis, which is the temporary inability to talk or move when waking up.
- hypnagogic hallucinations which are vivid, often frightening, dream-like experiences that occur while dozing, falling asleep and/or while awakening, and automatic behaviour which occurs when a person continues to function (talking, putting things away, etc.) during sleep episodes, but awakens with no memory of performing such activities.
- Daytime sleepiness, sleep paralysis, and hypnagogic hallucinations also occur in people who do not have narcolepsy, such as in people who are suffering from extreme lack of sleep. Cataplexy is generally considered unique to narcolepsy.
- narcolepsy treat the symptoms, but not the underlying cause.
- antidepressant medications and other drugs that suppress REM sleep are prescribed.
- the drowsiness is normally treated using stimulants such as methylphenidate (Ritalin), amphetamines (Adderall), dextroamphetamine (Dexedrine), methamphetamine (Desoxyn), modafinil (Provigil), etc.
- Other medications used are codeine and selegiline.
- the cataplexy is treated using clomipramine, imipramine, or protriptyline but this need only be done in severe cases.
- the drug gamma-hydroxybutyrate (GHB) (Xyrem) is approved in the USA by the Food and Drug Administration to treat both the cataplexy and excessive daytime sleepiness associated with narcolepsy.
- histamine H3-receptor antagonists and inverse agonists can be used for the treatment and/or prevention of conditions associated with excessive daytime sleepiness such as hypersomnia, narcolepsy, sleep apnea, time zone change disorder, and other disorders which are associated with excessive daytime sleepiness such as fibromyalgia, and multiple sclerosis (Parmentier et al., J. Neurosci. 2002, 22, 7695-7711; Ligneau et al. J. Pharmacol. Exp. Ther. 1998, 287, 658-666).
- Other conditions include excessive sleepiness due to shift work, medical disorders, psychiatric disorders, narcolepsy, primary hypersomnia, and the like.
- Histamine H3- receptor antagonists and inverse agonists can also be used occasionally to promote wakefulness or vigilance in shift workers, sleep deprivation, post anesthesia grogginess, drowsiness as a side effect from a medication, military use and the like.
- histamine H3-receptor antagonists and inverse agonists have been shown to improve cognitive performance in various animal models (Hancock and Fox in Milestones in Drug Therapy, ed. Buccafusco, 2003). These compounds can be used as pro-cognitive agents and can increase vigilance. Therefore, histamine H3-receptor antagonists and inverse agonists can be used in aging or degenerative disorders in which vigilance, attention and memory are impaired, for example, as in Alzheimer's disease or other dementias.
- AD Alzheimer's disease
- cognitive impairment extends to the domains of language, skilled movements, recognition and functions closely related to the frontal and temporal lobes of the brain such as decision-making and planning.
- drugs which offer symptomatic benefit, specifically with respect to short-term memory impairment.
- acetylcholinesterase inhibitors such as donepezil (Aricept), galantamine (Razadyne) and rivastigmine (Exelon) and NMDA antagonists such as memantine.
- Histamine H3 -receptor antagonists and inverse agonists can be used to treat or prevent cognitive disorders (Passani et al. Trends Pharmacol. Sci. 2004, 25, 618-625), epilepsy (Vohora et al. Pharmacol. Biochem. Behav. 2001, 68, 735-741), depression (Perez-Garcia et al. Psychopharmacol. 1999, 142, 215-220), attention deficit hyperactivity disorder (ADHD), (Fox et al. Behav. Brain Res.
- ADHD attention deficit hyperactivity disorder
- Histamine H3 -receptor antagonists or inverse agonists can also be used as a novel therapeutic approach to restore cortical activation in comatose or brain-traumatized patients (Passani et al., Trends in Pharmacol. Sci. 2004, 25, 618-625).
- histamine H3 -receptor antagonists and inverse agonists can be used to treat or prevent epilepsy.
- Epilepsy (often referred to as a seizure disorder) is a chronic neurological condition characterized by recurrent unprovoked seizures. In terms of their pattern of activity, seizures may be described as either partial (focal) or generalized. Partial seizures only involve a localized part of the brain, whereas generalized seizures involve the entire cortex.
- epilepsy syndromes each presenting with its own unique combination of seizure type, typical age of onset, EEG findings, treatment, and prognosis.
- Some common seizure syndromes include, for example, infantile spasms (West syndrome), childhood absence epilepsy, and benign focal epilepsy of childhood (Benign Rolandic epilepsy), juvenile myoclonic epilepsy, temporal lobe epilepsy, frontal lobe epilepsy and Lennox-Gastaut syndrome.
- compounds of the present invention can be used in combination with various known drugs.
- compounds of the present invention can be used with one or more drugs that prevent seizures or reduce seizure frequency: these include carbamazepine (common brand name Tegretol), clobazam (Frisium), clonazepam (Klonopin), ethosuximide (Zarontin), felbamate (Felbatol), fosphenytoin (Cerebyx), flurazepam (Dalmane), gabapentin (Neurontin), lamotrigine (Lamictal), levetiracetam (Keppra), oxcarbazepine (Trileptal), mephenytoin (Mesantoin), phenobarbital (Luminal), phenytoin (Dilantin), pregabalin (Lyrica), primidone (Mysoline), sodium valproate (Epilim), tiagabine (Gabitril), topiramate (
- Drugs used only in the treatment of refractory status epilepticus include paraldehyde (Paral) and pentobarbital (Nembutal).
- a histamine H3-receptor antagonist or inverse agonist can be used as the sole agent of treatment or can be used in combination with other agents.
- Vohora et al. show that a histamine H3-receptor antagonist can work as an anti-epilepsy, antiseizure drug and also showed effect with sub-effective doses of the H3-receptor antagonist in combination with sub-effective doses of known anti-epileptic drugs (Vohora et al. Pharmacol. Biochem. Behav. 2001, 68, 735-741).
- Perez-Garcia et al. tested the ability of a histamine H3 -receptor agonist and antagonist on experimental mouse models of anxiety (elevated plus-maze) and depression (forced swimming test). They found that while the compounds did not have a significant effect on the model of anxiety, a H3-receptor antagonist did have a significant dose-dependent effect in the model of depression. Thus, histamine H3- receptor antagonists or inverse agonists can have antidepressant effects.
- Clinical depression is a state of sadness or melancholia that has advanced to the point of being disruptive to an individual's social functioning and/or activities of daily living. Clinical depression affects about 16% of the population on at least one occasion in their lives. Clinical depression is currently the leading cause of disability in the U.S. as well as other countries, and is expected to become the second leading cause of disability worldwide (after heart disease) by the year 2020, according to the World Health Organization.
- compounds of the present invention can be used in combination with various known drugs.
- compounds of the present invention can be used with one or more of the drugs currently available that can relieve the symptoms of depression.
- They include, for example, monoamine oxidase inhibitors (MAOIs) such as Nardil or Moclobemide (Manerix), tricyclic antidepressants, selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine (Prozac), paroxetine (Paxil), escitalopram (Lexapro), and sertraline (Zoloft), norepinephrine reuptake inhibitors such as reboxetine (Edronax), and serotonin-norepinephrine reuptake inhibitors (SNRIs) such as venlafaxine (Effexor) and duloxetine (Cymbalta).
- MAOIs monoamine oxidase inhibitors
- SSRIs selective serotonin reuptake inhibitor
- histamine H3-receptor antagonists and inverse agonists can be used to treat or prevent attention deficit hyperactivity disorder (ADHD).
- ADHD attention deficit hyperactivity disorder
- Diagnostic and Statistical Manual of Mental Disorders-FV-TR ADHD is a developmental disorder that arises in childhood, in most cases before the age of 7 years, is characterized by developmentally inappropriate levels of inattention and/or hyperactive-impulsive behavior, and results in impairment in one or more major life activities, such as family, peer, educational, occupational, social, or adaptive functioning. ADHD can also be diagnosed in adulthood.
- the first-line medications used to treat ADHD are mostly stimulants, which work by stimulating the areas of the brain responsible for focus, attention, and impulse control.
- the use of stimulants to treat a syndrome often characterized by hyperactivity is sometimes referred to as a paradoxical effect, but there is no real paradox in that stimulants activate brain inhibitory and self-organizing mechanisms permitting the individual to have greater self-regulation.
- the stimulants used include, for example, methylphenidate (sold as Ritalin, Ritalin SR and Ritalin LA), Metadate, Metadate ER, Metadate CD, Concerta, Focalin, Focalin XR or Methylin.
- the stimulants also include, for example, amphetamines such dextroamphetamine, sold as
- Dexedrine, Dexedrine Spansules, Adderall, and Adderall XR a trade name for a mixture of dextroamphetamine and laevoamphetamine salts, methamphetamine sold as Desoxyn, bupropion, a dopamine and norepinephrine reuptake inhibitor, marketed under the brand name Wellbutrin.
- a non-stimulant medication to treat ADHD is Atomoxetine (sold as Strattera) a norepinephrine reuptake inhibitor.
- Other drugs sometimes used for ADHD include, for example, benzphetamine, Provigil/Alertec/modafinil and clonidine.
- a histamine H3-receptor antagonist was at least as effective as methylphenidate (Ritalin) (Hancock and Fox in Milestones in Drug Therapy, ed. Buccafusco, 2003).
- Compounds of the present invention can be used in combination with various known drugs.
- compounds of the present invention can be used with one or more of the drugs used to treat ADHD and related disorders.
- histamine H3 -receptor antagonists and inverse agonists can be used to treat or prevent schizophrenia.
- Schizophrenia is a psychiatric diagnosis that describes a mental disorder characterized by impairments in the perception or expression of reality and by significant social or occupational dysfunction.
- a person experiencing untreated schizophrenia is typically characterized as demonstrating disorganized thinking, and as experiencing delusions or auditory hallucinations.
- the disorder is primarily thought to affect cognition, it can also contribute to chronic problems with behavior and emotion.
- Schizophrenia is often described in terms of "positive” and "negative” symptoms. Positive symptoms include delusions, auditory hallucinations and thought disorder, and are typically regarded as manifestations of psychosis.
- Negative symptoms are so named because they are considered to be the loss or absence of normal traits or abilities, and include features such as flat, blunted or constricted affect and emotion, poverty of speech and lack of motivation.
- Some models of schizophrenia include formal thought disorder and planning difficulties in a third group, a "disorganization syndrome.”
- the first line pharmacological therapy for schizophrenia is usually the use of antipsychotic medication.
- Antipsychotic drugs are only thought to provide symptomatic relief from the positive symptoms of psychosis.
- the newer atypical antipsychotic medications are usually preferred over older typical antipsychotic medications (such as chlorpromazine and haloperidol) due to their favorable side-effect profile. While the atypical antipsychotics are associated with less extra pyramidal side-effects and tardive dyskinesia than the conventional antipsychotics, some of the agents in this class (especially olanzapine and clozapine) appear to be associated with metabolic side effects such as weight gain, hyperglycemia and hypertriglyceridemia that must be considered when choosing appropriate pharmacotherapy.
- Histamine H3 -receptor antagonists or inverse agonists can be used to treat obesity (Hancock, Curr. Opin. Investig. Drugs 2003, 4, 1190-1197).
- the role of neuronal histamine in food intake has been established for many years and neuronal histamine release and/or signalling has been implicated in the anorectic actions of known mediators in the feeding cycle such as leptin, amylin and bombesin, m the brain, the H3 -receptor is implicated in the regulation of histamine release in the hypothalamus.
- thermogenesis a role in the regulation of thermogenesis (Karlstedt et al., MoI. Cell. Neurosci. 2003, 24, 614-622).
- histamine H3-receptor antagonists have been investigated in various preclinical models of obesity and have shown to be effective in reducing food intake, reducing weight, and decreasing total body fat in mice (Hancock, et al. Eur. J. Pharmacol. 2004, 487, 183-197).
- the most common drugs used for the treatment of obesity are sibutramine (Meridia) and orlistat (Xenical), both of which have limited effectiveness and significant side effects. Therefore, novel anti-obesity agents, such as histamine H3-receptor antagonists or inverse agonists, are needed. Histamine H3-receptor antagonists or inverse agonists can also be used to treat upper airway allergic responses (U.S. Pat. Nos.
- histamine H3 -receptor antagonists in a cat model of nasal decongestion, a combination of histamine H3 -receptor antagonists with the Hl receptor antagonist chlorpheniramine resulted in significant nasal decongestion without the hypertensive effect seen with adrenergic agonists.
- histamine H3-receptor antagonists or inverse agonists can be used alone or in combination with Hl receptor blockage for the treatment of allergic rhinitis and nasal congestion.
- Histamine H3-receptor antagonists or inverse agonists have therapeutic potential for the treatment of pain (Medhurst et al. Biochemical Pharmacology (2007), 73(8), 1182-1194).
- a further aspect of the present invention pertains to pharmaceutical compositions comprising one or more compounds as described herein and one or more pharmaceutically acceptable carriers. Some embodiments pertain to pharmaceutical compositions comprising a compound of the present invention and a pharmaceutically acceptable carrier. Some embodiments of the present invention include a method of producing a pharmaceutical composition comprising admixing at least one compound according to any of the compound embodiments disclosed herein and a pharmaceutically acceptable carrier.
- Formulations may be prepared by any suitable method, typically by uniformly mixing the active compound(s) with liquids or finely divided solid carriers, or both, in the required proportions and then, if necessary, forming the resulting mixture into a desired shape.
- Liquid preparations for oral administration may be in the form of solutions, emulsions, aqueous or oily suspensions and syrups.
- the oral preparations may be in the form of dry powder that can be reconstituted with water or another suitable liquid vehicle before use. Additional additives such as suspending or emulsifying agents, non-aqueous vehicles (including edible oils), preservatives and flavorings and colorants may be added to the liquid preparations.
- Parenteral dosage forms may be prepared by dissolving the compound of the invention in a suitable liquid vehicle and filter sterilizing the solution before filling and sealing an appropriate vial or ampule. These are just a few examples of the many appropriate methods well known in the art for preparing dosage forms.
- a compound of the present invention can be formulated into pharmaceutical compositions using techniques well known to those in the art. Suitable pharmaceutically- acceptable carriers, outside those mentioned herein, are known in the art; for example, see Remington, The Science and Practice of Pharmacy, 20 th Edition, 2000, Lippincott Williams & Wilkins, (Editors: Gennaro et al.)
- a compound of the invention may, in an alternative use, be administered as a raw or pure chemical, it is preferable however to present the compound or active ingredient as a pharmaceutical formulation or composition further comprising a pharmaceutically acceptable carrier.
- the invention thus further provides pharmaceutical formulations comprising a compound of the invention or a pharmaceutically acceptable salt, solvate, hydrate or derivative thereof together with one or more pharmaceutically acceptable carriers thereof and/or prophylactic ingredients.
- the carrier(s) must be "acceptable” in the sense of being compatible with the other ingredients of the formulation and not overly deleterious to the recipient thereof.
- Transdermal patches dispense a drug at a controlled rate by presenting the drug for absorption in an efficient manner with a minimum of degradation of the drug.
- transdermal patches comprise an impermeable backing layer, a single pressure sensitive adhesive and a removable protective layer with a release liner.
- the compounds of the invention may thus be placed into the form of pharmaceutical formulations and unit dosages thereof and in such form may be employed as solids, such as tablets or filled capsules, or liquids such as solutions, suspensions, emulsions, elixirs, gels or capsules filled with the same, all for oral use, in the form of suppositories for rectal administration; or in the form of sterile injectable solutions for parenteral (including subcutaneous) use.
- Such pharmaceutical compositions and unit dosage forms thereof may comprise conventional ingredients in conventional proportions, with or without additional active compounds or principles and such unit dosage forms may contain any suitable effective amount of the active ingredient commensurate with the intended daily dosage range to be employed.
- the pharmaceutical composition may be in the form of, for example, a tablet, capsule, suspension or liquid.
- the pharmaceutical composition is preferably made in the form of a dosage unit containing a particular amount of the active ingredient.
- Examples of such dosage units are capsules, tablets, powders, granules or a suspension, with conventional additives such as lactose, mannitol, corn starch or potato starch; with binders such as crystalline cellulose, cellulose derivatives, acacia, corn starch or gelatins; with disintegrators such as corn starch, potato starch or sodium carboxymethyl-cellulose; and with lubricants such as talc or magnesium stearate.
- the active ingredient may also be administered by injection as a composition wherein, for example, saline, dextrose or water may be used as a suitable pharmaceutically acceptable carrier.
- active ingredient is defined in the context of a "pharmaceutical composition” and is intended to mean a component of a pharmaceutical composition that provides the primary pharmacological effect, as opposed to an "inactive ingredient” which would generally be recognized as providing no pharmaceutical benefit.
- the dose when using the compounds of the present invention can vary within wide limits and as is customary and is known to the physician, it is to be tailored to the individual conditions in each individual case.
- doses of the present invention include, but not limited to, about 0.001 mg to about 5000 mg, about 0.001 mg to about 2500 mg, about 0.001 mg to about 1000 mg, 0.001 mg to about 500 mg, 0.001 mg to about 250 mg, about 0.001 mg to 100 mg, about 0.001 mg to about 50 mg and about 0.001 mg to about 25 mg. Multiple doses may be administered during the day, especially when relatively large amounts are deemed to be needed, for example 2, 3 or 4 doses.
- the amount of active ingredient, or an active salt or derivative thereof, required for use in treatment will vary not only with the particular salt selected but also with the route of administration, the nature of the condition being treated and the age and condition of the patient and will ultimately be at the discretion of the attendant physician or clinician. In general, one skilled in the art understands how to extrapolate in vivo data obtained in a model system, typically an animal model, to another, such as a human.
- these extrapolations may merely be based on the weight of the animal model in comparison to another, such as a mammal, preferably a human, however, more often, these extrapolations are not simply based on weights, but rather incorporate a variety of factors. Representative factors include the type, age, weight, sex, diet and medical condition of the patient, the severity of the disease, the route of administration, pharmacological considerations such as the activity, efficacy, pharmacokinetic and toxicology profiles of the particular compound employed, whether a drug delivery system is utilized, on whether an acute or chronic disease state is being treated or prophylaxis is conducted or on whether further active compounds are administered in addition to the compounds of the present invention and as part of a drug combination.
- the dosage regimen for treating a disease condition with the compounds and/or compositions of this invention is selected in accordance with a variety factors as cited above.
- the actual dosage regimen employed may vary widely and therefore may deviate from a preferred dosage regimen and one skilled in the art will recognize that dosage and dosage regimen outside these typical ranges can be tested and, where appropriate, may be used in the methods of this invention.
- the desired dose may conveniently be presented in a single dose or as divided doses administered at appropriate intervals, for example, as two, three, four or more sub-doses per day.
- the sub-dose itself may be further divided, e.g., into a number of discrete loosely spaced administrations.
- the daily dose can be divided, especially when relatively large amounts are administered as deemed appropriate, into several, for example 2, 3 or 4 part administrations. If appropriate, depending on individual behavior, it may be necessary to deviate upward or downward from the daily dose indicated.
- a suitable pharmaceutically acceptable carrier can be either solid, liquid or a mixture of both. Solid form preparations include powders, tablets, pills, capsules, cachets, suppositories and dispersible granules.
- a solid carrier can be one or more substances which may also act as diluents, flavoring agents, solubilizers, lubricants, suspending agents, binders, preservatives, tablet disintegrating agents, or an encapsulating material.
- the carrier is a finely divided solid which is in a mixture with the finely divided active component.
- the active component is mixed with the carrier having the necessary binding capacity in suitable proportions and compacted to the desire shape and size.
- the powders and tablets may contain varying percentage amounts of the active compound.
- a representative amount in a powder or tablet may contain from 0.5 to about 90 percent of the active compound; however, an artisan would know when amounts outside of this range are necessary.
- Suitable carriers for powders and tablets are magnesium carbonate, magnesium stearate, talc, sugar, lactose, pectin, dextrin, starch, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose, a low melting wax, cocoa butter and the like.
- the term "preparation" is intended to include the formulation of the active compound with encapsulating material as carrier providing a capsule in which the active component, with or without carriers, is surrounded by a carrier, which is thus in association with it.
- cachets and lozenges are included. Tablets, powders, capsules, pills, cachets and lozenges can be used as solid forms suitable for oral administration.
- a low melting wax such as an admixture of fatty acid glycerides or cocoa butter
- the active component is dispersed homogeneously therein, as by stirring.
- the molten homogenous mixture is then poured into convenient sized molds, allowed to cool and thereby to solidify.
- Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or sprays containing in addition to the active ingredient such carriers as are known in the art to be appropriate.
- Liquid form preparations include solutions, suspensions and emulsions, for example, water or water-propylene glycol solutions.
- parenteral injection liquid preparations can be formulated as solutions in aqueous polyethylene glycol solution.
- injectable preparations for example, sterile injectable aqueous or oleaginous suspensions may be formulated according to the known art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol.
- Suitable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectables.
- the compounds according to the present invention may thus be formulated for parenteral administration (e.g. by injection, for example bolus injection or continuous infusion) and may be presented in unit dose form in ampoules, pre-filled syringes, small volume infusion or in multi-dose containers with an added preservative.
- the pharmaceutical compositions may take such forms as suspensions, solutions, or emulsions in oily or aqueous vehicles and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
- the active ingredient may be in powder form, obtained by aseptic isolation of sterile solid or by lyophilization from solution, for constitution with a suitable vehicle, e.g. sterile, pyrogen-free water, before use.
- Aqueous formulations suitable for oral use can be prepared by dissolving or suspending the active component in water and adding suitable colorants, flavors, stabilizing and thickening agents, as desired.
- Aqueous suspensions suitable for oral use can be made by dispersing the finely divided active component in water with viscous material, such as natural or synthetic gums, resins, methylcellulose, sodium carboxymethylcellulose, or other well-known suspending agents.
- viscous material such as natural or synthetic gums, resins, methylcellulose, sodium carboxymethylcellulose, or other well-known suspending agents.
- solid form preparations which are intended to be converted, shortly before use, to liquid form preparations for oral administration.
- liquid forms include solutions, suspensions and emulsions.
- These preparations may contain, in addition to the active component, colorants, flavors, stabilizers, buffers, artificial and natural sweeteners, dispersants, thickeners, solubilizing agents and the like.
- the compounds according to the invention may be formulated as ointments, creams or lotions, or as a transdermal patch.
- Ointments and creams may, for example, be formulated with an aqueous or oily base with the addition of suitable thickening and/or gelling agents.
- Lotions may be formulated with an aqueous or oily base and will in general also contain one or more emulsifying agents, stabilizing agents, dispersing agents, suspending agents, thickening agents, or coloring agents.
- Formulations suitable for topical administration in the mouth include lozenges comprising active agent in a flavored base, usually sucrose and acacia or tragacanth; pastilles comprising the active ingredient in an inert base such as gelatin and glycerin or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier.
- Solutions or suspensions are applied directly to the nasal cavity by conventional means, for example with a dropper, pipette or spray.
- the formulations may be provided in single or multi-dose form. In the latter case of a dropper or pipette, this may be achieved by the patient administering an appropriate, predetermined volume of the solution or suspension. In the case of a spray, this may be achieved for example by means of a metering atomizing spray pump.
- Administration to the respiratory tract may also be achieved by means of an aerosol formulation in which the active ingredient is provided in a pressurized pack with a suitable propellant.
- aerosol formulation in which the active ingredient is provided in a pressurized pack with a suitable propellant.
- the compounds of the present invention or pharmaceutical compositions comprising them are administered as aerosols, for example as nasal aerosols or by inhalation, this can be carried out, for example, using a spray, a nebulizer, a pump nebulizer, an inhalation apparatus, a metered inhaler or a dry powder inhaler.
- Pharmaceutical forms for administration of the compounds of the present invention as an aerosol can be prepared by processes well known to the person skilled in the art.
- solutions or dispersions of the compounds of the present invention in water, water/alcohol mixtures or suitable saline solutions can be employed using customary additives, for example benzyl alcohol or other suitable preservatives, absorption enhancers for increasing the bioavailability, solubilizers, dispersants and others and, if appropriate, customary propellants, for example include carbon dioxide, CFCs, such as, dichlorodifluoromethane, trichlorofluoromethane, or dichlorotetrafluoroethane; and the like.
- the aerosol may conveniently also contain a surfactant such as lecithin.
- the dose of drug may be controlled by provision of a metered valve.
- the compound In formulations intended for administration to the respiratory tract, including intranasal formulations, the compound will generally have a small particle size for example of the order of 10 microns or less. Such a particle size may be obtained by means known in the art, for example by micronization. When desired, formulations adapted to give sustained release of the active ingredient may be employed.
- the active ingredients may be provided in the form of a dry powder, for example, a powder mix of the compound in a suitable powder base such as lactose, starch, starch derivatives such as hydroxypropylmethyl cellulose and polyvinylpyrrolidone (PVP).
- a powder mix of the compound in a suitable powder base such as lactose, starch, starch derivatives such as hydroxypropylmethyl cellulose and polyvinylpyrrolidone (PVP).
- the powder carrier will form a gel in the nasal cavity.
- the powder composition may be presented in unit dose form for example in capsules or cartridges of, e.g., gelatin, or blister packs from which the powder may be administered by means of an inhaler.
- the pharmaceutical preparations are preferably in unit dosage forms.
- the preparation is subdivided into unit doses containing appropriate quantities of the active component.
- the unit dosage form can be a packaged preparation, the package containing discrete quantities of preparation, such as packeted tablets, capsules and powders in vials or ampoules.
- the unit dosage form can be a capsule, tablet, cachet, or lozenge itself, or it can be the appropriate number of any of these in packaged form. Tablets or capsules for oral administration and liquids for intravenous administration are preferred compositions.
- the compounds according to the invention may optionally exist as pharmaceutically acceptable salts including pharmaceutically acceptable acid addition salts prepared from pharmaceutically acceptable non-toxic acids including inorganic and organic acids.
- Representative acids include, but are not limited to, acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethenesulfonic, dichloroacetic, formic, fumaric, gluconic, glutamic, hippuric, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, oxalic, pamoic, pantothenic, phosphoric, succinic, sulf ⁇ ric, tartaric, oxalic, p-toluenesulfonic and the like, such as those pharmaceutically acceptable salts listed in Journal of Pharmaceutical Sciences, 66:1-19 (1977), incorporated herein by reference in its entirety.
- the acid addition salts may be obtained as the direct products of compound synthesis.
- the free base may be dissolved in a suitable solvent containing the appropriate acid and the salt isolated by evaporating the solvent or otherwise separating the salt and solvent.
- the compounds of this invention may form solvates with standard low molecular weight solvents using methods known to the skilled artisan.
- Pro-drugs refers to compounds that have been modified with specific chemical groups known in the art and when administered into an individual these groups undergo biotransformation to give the parent compound. Pro-drugs can thus be viewed as compounds of the invention containing one or more specialized non-toxic protective groups used in a transient manner to alter or to eliminate a property of the compound. Tn one general aspect, the "pro-drug” approach is utilized to facilitate oral absorption. A thorough discussion is provided in T. Higuchi and V. Stella, Prodrugs as Novel Delivery Systems Vol. 14 of the A.C.S. Symposium Series; and in Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, both of which are hereby incorporated by reference in their entirety.
- Some embodiments of the present invention include a method of producing a pharmaceutical composition for "combination-therapy" comprising admixing at least one compound according to any of the compound embodiments disclosed herein, together with at least one known pharmaceutical agent as described herein and a pharmaceutically acceptable carrier.
- H3 histamine receptor modulators are utilized as active ingredients in a pharmaceutical composition, these are not intended for use only in humans, but in other non-human mammals as well. Indeed, recent advances in the area of animal health-care mandate that consideration be given for the use of active agents, such as H3 histamine receptor modulators, for the treatment of a H3-associated disease or disorder in companionship animals (e.g., cats, dogs, etc.) and in livestock animals (e.g., cows, chickens, fish, etc.) Those of ordinary skill in the art are readily credited with understanding the utility of such compounds in such settings.
- companionship animals e.g., cats, dogs, etc.
- livestock animals e.g., cows, chickens, fish, etc.
- HYDRATES AND SOLVATES The compounds of the present invention can be administered in a wide variety of oral and parenteral dosage forms. It will be obvious to those skilled in the art that the following dosage forms may comprise, as the active component, either a compound of the invention or a pharmaceutically acceptable salt, solvate or hydrate of a compound of the invention. Typical procedures for making and identifying suitable hydrates and solvates, outside those mentioned herein, are well known to those in the art; see for example, pages 202-209 of KJ. Guillory,
- one aspect of the present invention pertains to hydrates and solvates of compounds of Formula (Ia), as described herein, that can be isolated and characterized by methods known in the art, such as, thermogravimetric analysis (TGA), TGA-mass spectroscopy, TGA-Infrared spectroscopy, powder X-ray diffraction (XRPD), Karl Fisher titration, high resolution X-ray diffraction, and the like.
- TGA thermogravimetric analysis
- TGA-mass spectroscopy TGA-mass spectroscopy
- TGA-Infrared spectroscopy powder X-ray diffraction (XRPD)
- Karl Fisher titration high resolution X-ray diffraction, and the like.
- Another object of the present invention relates to radio-labeled compounds of the present invention that would be useful not only in radio-imaging but also in assays, both in vitro and in vivo, for localizing and quantitating the H3 histamine receptor in tissue samples, including human and for identifying H3 histamine receptor ligands by inhibition binding of a radio-labeled compound. It is a further object of this invention to develop novel H3-receptor assays of which comprise such radio-labeled compounds.
- the present invention embraces isotopically-labeled compounds of the present invention.
- Isotopically or radio-labeled compounds are those which are identical to compounds disclosed herein, but for the fact that one or more atoms are replaced or substituted by an atom having an atomic mass or mass number different from the atomic mass or mass number most commonly found in nature.
- Suitable radionuclides that may be incorporated in compounds of the present invention include but are not limited to 2 H (also written as D for deuterium), 3 H (also written as T for tritium), 11 C, 13 C, 14 C, 13 N, 15 N, 15 0, 17 O, 18 0, 18 F, 35 S, 36 Cl, 75 Br, 76 Br, 77 Br, 82 Br, 123 1, 124 I, 125 I and 131 I.
- the radionuclide that is incorporated in the instant radio-labeled compounds will depend on the specific application of that radio-labeled compound.
- a "radio-labeled " or "labeled compound” is a compound of Formula (Ia), (Ic), (Ie), (Ig) or (Ii) that has incorporated at least one radionuclide; in some embodiments the radionuclide is selected from the group consisting of 3 H, 14 C, 125 1 , 35 S and 82 Br.
- isotopically-labeled compounds of the present invention are useful in compound and/or substrate tissue distribution assays.
- the radionuclide 3 H and/or 14 C isotopes are useful in these studies.
- substitution with heavier isotopes such as deuterium [i.e., 2 H) may afford certain therapeutic advantages resulting from greater metabolic stability [e.g., increased in vivo half-life or reduced dosage requirements) and hence may be preferred in some circumstances.
- Isotopically labeled compounds of the present invention can generally be prepared by following procedures analogous to those disclosed in the Drawings and Examples infra, by substituting an isotopically labeled reagent for a non-isotopically labeled reagent.
- Synthetic methods for incorporating radio-isotopes into organic compounds are applicable to compounds of the invention and are well known in the art. These synthetic methods, for example, incorporating activity levels of tritium into target molecules, are as follows:
- Tritium Gas Exposure Labeling This procedure involves exposing precursors containing exchangeable protons to tritium gas in the presence of a suitable catalyst.
- Aryl and heteroaryl bromide exchange with 125 I This method is generally a two step process.
- the first step is the conversion of the aryl or heteroaryl bromide to the corresponding tri-alkyltin intermediate using for example, a Pd catalyzed reaction [i.e. Pd(Ph 3 P) 4 ] or through an aryl or heteroaryl lithium, in the presence of a tri-alkyltinhalide or hexaalkylditin [e.g., (CHs) 3 SnSn(CHs) 3 ].
- Pd catalyzed reaction i.e. Pd(Ph 3 P) 4
- a tri-alkyltinhalide or hexaalkylditin e.g., (CHs) 3 SnSn(CHs) 3 ].
- a radiolabeled H3 histamine receptor compound of Formula (Ia) can be used in a screening assay to identify/evaluate compounds.
- a newly synthesized or identified compound i.e., test compound
- test compound can be evaluated for its ability to reduce binding of the "radio-labeled compound of Formula (Ia)" to the H3-receptor. Accordingly, the ability of a test compound to compete with the "radio-labeled compound of Formula (Ia)" for the binding to the H3 histamine receptor directly correlates to its binding affinity.
- the labeled compounds of the present invention bind to the H3 histamine receptor.
- the labeled compound has an ICs 0 less than about 500 ⁇ M, in another embodiment the labeled compound has an ICs 0 less than about 100 ⁇ M, in yet another embodiment the labeled compound has an IC 50 less than about 10 ⁇ M, in yet another embodiment the labeled compound has an IC 50 less than about 1 ⁇ M and in still yet another embodiment the labeled inhibitor has an IC 50 less than about 0.1 ⁇ M.
- Microwave irradiations were carried out using a Smith SynthesizerTM or an Emrys OptimizerTM (Biotage).
- TLC Thin-layer chromatography
- PK6F silica gel 60 A 1 mm plates (Whatman) and column chromatography was carried out on a silica gel column using Kieselgel 60, 0.063-0.200 mm (Merck). Evaporation was done under reduced pressure on a B ⁇ chi rotary evaporator.
- LCMS spec HPLC-pumps: LC-IOAD VP, Shimadzu Inc.; HPLC system controller: SCL-IOA VP, Shimadzu Inc; UV-Detector: SPD-IOA VP, Shimadzu Inc; Autosampler: CTC HTS, PAL, Leap Scientific; Mass spectrometer: API 150EX with Turbo Ion Spray source, AB/MDS Sciex; Software: Analyst 1.2.
- Example 1.2 Preparation of ( ⁇ )-4-(4'-(2-((l.)-2-MethyIpyrrolidin-l-yl)ethyl)biphenyl-4- yl)oxazolidin-2-one (Compound 3).
- the reaction mixture was filtered and rinsed with acetonitrile.
- the filtrate was concentrated under reduced pressure and purified by preparative HPLC using a 25 x 250 mm C 18 column, eluting with 10-75% acetonitrile in water containing 0.1% trifluoroacetic acid over 55 min.
- the resulting HPLC fractions were combined and acetonitrile was evaporated.
- the remaining aqueous solution was made basic with 2 M Na 2 CO 3 , saturated with sodium chloride and extracted with ethyl acetate three times.
- the combined ethyl acetate extracts were dried with magnesium sulfate and filtered.
- HCl (1 M in ether, 1 mL) was added to the filtrate and the solvent was removed under reduced pressure.
- Example 1.3 Preparation of (l?)-4-(4-Bromobenzyl)oxazolidin-2-one. From (R)-2-amino-3-(4-bromophenyl)propanoic acid (2.026 g, 8.30 mmol), using a similar method to the one described in Example 1.1, the title compound was obtained as a white solid (929 mg, 44%).
- Example 1.4 Preparation of ( ⁇ )-4-((4'-(2-((i?)-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4- yl)methyl)oxazolidin-2-one (Compound 4).
- Example 1.5 Preparation of (/?)-3-Methyl-4-(4'-(2-(( ⁇ )-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yl)oxazolidin-2-one (Compound 5).
- Example 1.6 Preparation of ( ⁇ )-3-(2-Methoxyethyl)-4-(4'-(2-((/?)-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yl)oxazolidin-2-one (Compound 6).
- Example 1.8 Preparation of (l?)-3-Methyl-4-((4'-(2-((/?)-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yl)methyl)oxazolidin-2-one (Compound 8). From (i?)-4-(4-bromobenzyl)oxazolidin-2-one (0.204 g, 0.797 mmol), iodomethane
- Example 1.11 Preparation of (5)-3-(2-Methoxyethyl)-4-((4'-(2-(( J R)-2-methylpyrrolidin-l- yl)ethyI)biphenyl-4-yl)methyl)oxazolidin-2-one (Compound 12).
- Example 1.12 Preparation of ( ⁇ )-4-(4-Bromobenzyl)-l,3-oxazinan-2-one. From (R)-3-amino-4-(4-bromophenyl)butanoic acid (1.000 g, 3.87 mmol), using a similar method to the one described in Example 1.1, the title compound was obtained as a white solid (749 mg, 72%).
- Example 1.17 Preparation of 5-((4'-(2-((/?)-2-MethylpyrroIidin-l-yl)ethyl)biphenyl-4- yl)oxazolidin-2-one (Compound 14). From 5-(4-bromophenyl)oxazolidin-2-one (0.142 g, 0.554 mmol) and (R)-4-(2-(2- methylpyrrolidin-l-yl)ethyl)phenylboronic acid (0.129 g, 0.554 mmol), using a similar method to the one described in Example 1.13, the hydrochloride salt of the title compound was obtained as a white solid (149 mg, 67%).
- reaction mixture was then diluted with ethyl acetate (400 mL) and the aqueous layer was separated. The ethyl acetate layer was washed with brine, dried with sodium sulfate and concentrated. The residue was purified by chromatography on a silica column eluting with 20-50% ethyl acetate in dichloromethane to afford the title compound as a colorless oil (2.31 g, 53%).
- reaction mixture was filtered and the solid was rinsed with acetonitrile.
- the filtrate was concentrated and the residue was purified by chromatography on a silica column eluting with 20-60% ethyl acetate in hexanes to afford the title compound as a colorless oil (134 mg, 30%).
- Example 1.20 Preparation of ( J R)-4-(4'-(2-HydroxyethyI)biphenyI-4-yl)oxazolidin-2-one. To a solution of (i?)-4-(4'-(2-(tert-butyldimethylsilyloxy)ethyl)biphenyl-4-yl)oxazolidin-
- Example 1.21 Preparation of ( ⁇ )-2-(4'-(2-Oxooxazolidin-4-yl)biphenyl-4-yl)ethyl methanesulfonate.
- Step A Preparation of (5)-4-(4-Chlorophenyl)-3-methyloxazolidin-2-one.
- (5)-4-(4-chlorophenyl)oxazolidin-2-one 250 mg, 1.27 mmol
- DMF 5.0 mL
- sodium hydride 60% dispersion in mineral oil
- iodomethane 0.159 mL, 2.53 mmol
- the reaction mixture was stirred for 18 h at room temperature.
- the reaction mixture was diluted with EtOAc (50 mL), washed with 1 M HCl (2 x 10 mL), extracted twice with EtOAc and washed with brine.
- Step B Preparation of ( 1 S)-3-Methyl-4-(4'-(2-(( ⁇ )-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yl)oxazolidin-2-one (Compound 15).
- the resulting reaction mixture was heated under microwave irradiation at 120 0 C for 1 hr. After 1 hr, LCMS indicated the reaction was complete.
- the reaction mixture was diluted with water and the organics separated. The aqueous layer was extracted with EtOAc. The combined organics were concentrated, dissolved in ACN/H 2 O (with AcOH) and purified by HPLC (0.1% TFA in acetonitrile/0.1% TFA in water). The combined fractions were basified with 2 M Na 2 CO 3 and extracted 3 times with EtOAc. The combined organics were dried over MgSO 4 , filtered, and concentrated. The residue was dissolved in MeOH (5 mL).
- Example 1.24 Preparation of (5)-3-(2-Methoxyethyl)-4-(4'-(2-(( ⁇ )-2-methylpyrrolidin-l- y.)ethyI)biphenyl-4-yl)oxazolidin-2-one (Compound 16).
- Step A Preparation of (.S)-4-(4-Chlorophenyl)-3-(2-inethoxyethyl)oxazolidin-2-one.
- DMF 5.0 mL
- sodium hydride 60% dispersion in mineral oil
- l-bromo-2-methoxyethane 0.235 mL, 2.53 mmol
- reaction mixture was diluted with EtOAc (50 mL) washed with 1 M HCl (2 x 10 mL), extracted twice with EtOAc and washed with Brine. The combined organics were dried over MgSO 4 , filtered, and concentrated to give a yellow oil in 100% crude yield.
- Step B Preparation of (5)-3-(2-Methoxyethyl)-4-(4'-(2-(( ⁇ )-2-methylpyrrolidin-l- yl)ethyI)biphenyl-4-yl)oxazolidin-2-one (Compound 16).
- Example 1.25 Preparation of (£)-4-(4'-(2-((l?)-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4- yl)oxazolidin-2-one (Compound 2).
- Step A Preparation of (S)-2-Amino-2-(4-chIorophenyl)ethanoI.
- Step B Preparation of (5)-4-(4-Chlorophenyl)oxazolidin-2-one.
- (S)-2-amino-2-(4-chlorophenyl)ethanol hydrochloride (2.00 g, 9.61 mmol) and diisopropylethylamine (6.71 mL, 38.4 mmol) in DCM (40 mL) was added triphosgene (3.14 g, 10.6 mmol) at 25 0 C resulting in an evolution of a large volume of gas.
- the mixture was stirred for 18 h and concentrated under reduced pressure.
- the solid residue was dissolved in 5% HCl (50 mL) solution and extracted with EtOAc (2 x 75 mL).
- Step C Preparation of (5)-4-(4'-(2-((jR)-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4- yl)oxazoIidin-2-one (Compound 2).
- the reaction was heated at 120 0 C under microwave irradiation for 45 min.
- the reaction mixture was filtered through CeliteTM and the filter cake was washed with EtOAc (30 mL).
- the filtrate was concentrated.
- the residue was purified by preparative HPLC.
- the combined fractions were lyophilized to give the trifluoroacetic acid salt of the title compound (0.100 g, 29% yield).
- the trifluoroacetic acid salt was converted to the hydrochloride salt, which was a white solid.
- Example 1.26 Preparation of (5)-4-((4'-(2-((/?)-2-Methylpyrrolidin-l-yl)ethyl)biphenyI-4- yl)methyl)oxazolidin-2-one (Compound 1).
- Step A Preparation of (5)-2-Amino-3-(4-bromophenyl)propan-l-ol.
- Step B Preparation of (5)-4-(4-Bromobenzyl)oxazolidin-2-one.
- Step C Preparation of (5)-4-((4'-(2-(( ⁇ )-2-MethylpyrroUdin-l-yl)ethyl)biphenyl-4- yl)methyl)oxazolidin-2-one (Compound 1).
- Example 1.27 Preparation of (i?)-4-(4'-(2-(/?)-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4- yl)oxazolidin-2-one Hydrochloride Salt (Compound 3).
- Step A Preparation of (jR)-Methyl 2-(tert-Butoxycarbonylamino)-2-(4- hydroxyphenyl)acetate.
- Step B Preparation of (R)-tert-Butyl 2-Hydroxy-l-(4- hydroxyphenyl)ethylcarbamate. A solution of ( ⁇ )-methyl 2-(ter/-butoxycarbonylamino)-2-(4-hydroxyphenyl)acetate
- Step C Preparation of ( ⁇ )-4-(4-Hydroxyphenyl)oxazolidin-2-one.
- Triisopropyl borate (104 mL, 447 mmol) was added at -72 0 C to -66 0 C. Additional THF (50 mL) was again used to rinse the funnel. After addition of triisopropyl borate was completed, the reaction mixture was gradually brought to room temperature (over 20 min) and stirred at the room temperature for 1 h. HCl (2 M, 130 mL) was added dropwise until an off-white suspension formed and the mixture was acidic. The mixture was stirred overnight.
- Step G Preparation of ( ⁇ )-4-(4'-(2-(( ⁇ )-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4- yl)oxazolidin-2-one.
- Step H Preparation of (/?)-4-(4'-(2-( ⁇ )-2-MethyIpyrrolidin-l-yl)ethyl)biphenyl-4- yl)oxazolidin-2-one Hydrochloride Salt.
- Example 1.28 Preparation of ( ⁇ )-3-(2-Methoxyethyl)-4-(4'-(2-(( ⁇ )-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yl)oxazolidin-2-one Hydrochloride (Compound 6).
- Step A Preparation of ( ⁇ )-3-(2-Methoxyethyl)-4-(4'-(2-(( ⁇ )-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yl)oxazolidin-2-one.
- the aqueous phase was cooled (ice water bath) and basified to pH 12-14 by slow addition of 50% aqueous NaOH solution and extracted with ethyl acetate (2 x 25 mL).
- the combined organic phases were washed with water (2 x 25 mL), dried (Na 2 SO 4 ) and the solvent was removed under reduced pressure to obtain a brown oil.
- Step B Preparation of (i?)-3-(2-methoxyethyl)-4-(4'-(2-(( ⁇ )-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yl)oxazolidin-2-one Hydrochloride Salt.
- the product of Example 1.28, Step A was dissolved in ethanol (1.5 mL) and the solution was stirred well.
- Hydrochloric acid (6.20 mL, 7.75 mmol) (1.25 M in ethanol) was added and the mixture was stirred overnight.
- MTBE was added and the mixture was left at —4 0 C for two days.
- Step A Preparation of ( ⁇ )-3-Isopropyl-4-(4'-(2-((i?)-2-methyIpyrrolidin-l- yl)ethyl)biphenyl-4-yl)oxazolidin-2-one.
- the aqueous phase was cooled (ice bath) and slowly basif ⁇ ed with 50% aqueous NaOH and extracted with ethyl acetate (2 x 25 mL).
- the ethyl acetate layer was washed with water (2 x 25 mL), dried (Na 2 SO 4 ) and the solvent was removed under reduced pressure to obtain the product (free base) as a waxy solid, 1.02 g.
- Step B Preparation of ( J R)-3-Isopropyl-4-(4'-(2-((i?)-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yl)oxazolidin-2-one Hydrochloride.
- Example 1.29, Step A The free base of Example 1.29, Step A was dissolved in ethanol (1.5 mL) and HCl (1.25 M in ethanol; 4.15 mL; 2 eq.) was added slowly with stirring. MTBE was added until the solution was cloudy. The mixture was stirred overnight at room temperature. The solvents were removed under reduced pressure and the residue was dried under reduced pressure, to obtain the HCl salt as a foamy solid (1.09 g 65.5%, 95.5% purity by LCMS).
- Step A Preparation of 2-Benzamido-2-(4-hydroxy-3-methylphenyl)acetic Acid.
- o-cresol 5.5 g, 51 mmol.
- the cold bath was allowed to expire while stirring continued overnight.
- the reaction was quenched by pouring into a beaker containing ice and NaHCO 3 (40 g).
- the reaction was extracted with EtOAc (thrice), and the aqueous phase was acidified with concentrated HCl to pH 2.
- Step B Preparation of 2-Amino-2-(4-hydroxy-3-methylphenyl)acetic Acid.
- Step C Preparation of Methyl 2-(tert-ButoxycarbonyIamino)-2-(4-hydroxy-3- methylphenyl)acetate.
- a stirring suspension of 2-amino-2-(4-hydroxy-3-methylphenyl)acetic acid hydrochloride (0.70 g, 3.2 mmol) in MeOH (4 mL) was cooled to 0 0 C.
- Thionyl chloride (0.25 mL, 3.4 mmol) was added dropwise, and then the ice bath was allowed to expire while stirring the red solution overnight.
- N-ethyl-N-isopropylpropan-2 -amine (2.8 mL, 16 mmol) was added dropwise, then the mixture was placed into a 0 0 C ice bath.
- Di-/ert-butyldicarbonate (0.77 g, 3.5 mmol) was added in portions. The mixture was stirred for 2 h at 0 0 C, before removing the ice bath and stirring for another 2 h at room temperature. The mixture was concentrated to give a solid residue.
- the solid material was loaded onto a fritted funnel and washed with water while under reduced pressure. The solid which remained was partitioned between EtOAc and water at pH 8. The aqueous phase was extracted twice more with EtOAc.
- Step D Preparation of te/Y-Butyl 2-Hydroxy-l-(4-hydroxy-3- methylphenyl)ethylcarbamate.
- methyl 2-(ter*-butoxycarbonylamino)-2-(4-hydroxy-3- methylphenyl)acetate (0.87 g, 3.0 mmol) in THF (20 mL) was added lithium aluminum hydride (3.0 mL of 1 M THF solution, 3.0 mmol). The cold bath was removed and the mixture was stirred overnight. The reaction was quenched by pouring onto ice. The slurry was diluted with EtOAc and the solution was adjusted to pH 2 with 10% aqueous HCl.
- Step E Preparation of 4-(4-Hydroxy-3-methylphenyl)oxazolidin-2-one.
- a stirring 0 0 C solution of tert-buXy ⁇ 2-hydroxy-l-(4-hydroxy-3- methylphenyl)ethylcarbamate (0.60 g, 2.2 mmol) in THF (7 mL) was added thionyl chloride
- Trifluoromethanesulfonate To a stirred slurry of 4-(4-hydroxy-3-methylphenyl)oxazolidin-2-one (0.40 g, 2.1 mmol) in acetonitrile (6 mL) was added pyridine (0.67 mL, 8.3 mmol). The mixture was cooled to 0 0 C and trifluoromethanesulfonic anhydride (0.53 mL, 3.1 mmol) was added dropwise. The ice bath was removed and the clear red solution was stirred at room temperature. LCMS after 2 h indicated the reaction was incomplete, so another equivalent OfTf 2 O (0.35 mL, 2.1 mmol) was added. The reaction was stirred for another 30 min.
- Step G Preparation of 4-(2-Methyl-4'-(2-((/?)-2-methylpyrrolidin-l-yl)ethyl)biphenyl- 4-yl)oxazolidin-2-one.
- Example 1.31 Preparation of 4-(3-MethyI-4'-(2-((l?)-2-methylpyrrolidin-l- yl)ethyI)biphenyl-4-yl)oxazolidin-2-one (Compound 24).
- Step A Preparation of 2-Benzamido-2-(4-hydroxy-2-methylphenyl)acetic Acid.
- Step C Preparation of Methyl 2-(terf-Butoxycarbonylamino)-2-(4-hydroxy-2- methylphenyl)acetate.
- Step D Preparation of tert-Butyl 2-Hydroxy-l-(4-hydroxy-2- methylphenyl)ethylcarbamate.
- methyl 2-(tert-butoxycarbonylamino)-2-(4-hydroxy-2- methylphenyl)acetate (0.55 g, 1.9 mmol) in THF (12 mL) was added lithium aluminum hydride (1.9 mL of 1 M THF solution, 1.9 mmol).
- the cold bath was removed and the mixture was stirred overnight.
- the reaction was quenched by pouring onto ice.
- the slurry was diluted with EtOAc and the solution was adjusted to pH 2 with 10% aqueous HCl.
- Step E Preparation of 4-(4-Hydroxy-2-methylphenyl)oxazolidin-2-one.
- a stirring 0 0 C solution of terf-butyl 2-hydroxy-l-(4-hydroxy-2- methylphenyl)ethylcarbamate (0.46 g, 1.7 mmol) in THF (6 mL) was added thionyl chloride (0.14 mL, 1.9 mmol). The ice bath was removed and the mixture was stirred overnight at room temperature. The solvents were evaporated and the residue was washed with MTBE. The MTBE solution was collected and concentrated to give the title compound (with minor impurities, 0.37 g).
- Step G Preparation of 4-(3-Methyl-4'-(2-(( ⁇ )-2-methylpyrrolidin-l-yl)ethyI)biphenyl- 4-yl)oxazolidin-2-one.
- Example 1.32 Preparation of (SJ- ⁇ C'-Methyl ⁇ '- ⁇ -t ⁇ yrrolidin-l-ylJethyObiphenyl ⁇ - yl)oxazolidin-2-one (Compound 32).
- Step A Preparation of Ethyl 2-(4-Hydroxy-3-methylphenyl)acetate.
- 2-(4-hydroxy-3-methylphenyl)acetic acid 5.0 g, 30 mmol
- absolute ethanol 150 mL
- concentrated sulfuric acid 1.6 mL, 30 mmol
- the mixture was heated to 75 0 C for 3 h, and then the heat was removed and the mixture was stirred at room temperature overnight. After 18 h, the volatiles were evaporated and the residue was diluted with water and extracted into EtOAc (thrice). The combined organic extract was washed with brine, dried over sodium sulfate, and the solvents were evaporated to give the title compound as an oil (5.8 g, 100%).
- Step B Preparation of 4-(2-Hydroxyethyl)-2-methyIphenol. To a stirring solution of ethyl 2-(4-hydroxy-3-methylphenyl)acetate (5.8 g, 30 mmol) in
- Step C Preparation of 2-Methyl-4-(2-(pyrrolidin-l-yl)ethyl)phenyl trifluoromethanesulfonate.
- Example 1.33 Preparation of ( ⁇ )-4-(2,6-Dichloro-4'-(2-(( ⁇ )-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yl)oxazolidin-2-one (Compound 18).
- Step A Preparation of ( ⁇ )-4-(3,5-Dichloro-4-hydroxyphenyl)oxazolidin-2-one.
- Step B Preparation of (jR)-2,6-Dichloro-4-(2-oxooxazolidin-4-yl)phenyl Trifluoromethanesulfonate.
- (/?)-4-(3,5-dichloro-4-hydroxyphenyl)oxazolidin-2-one (0.40 g, 1.6 mmol) in acetonitrile (5 mL) was added pyridine (0.52 mL, 6.5 mmol).
- pyridine 0.52 mL, 6.5 mmol
- the mixture was cooled to 0 0 C and trifiuoromethanesulfonic anhydride (0.41 mL, 2.4 mmol) was added dropwise.
- Step C Preparation of (/?)-4-(2,6-Dichloro-4'-(2-((l?)-2-methylpyrroIidin-l- yl)ethyl)biphenyl-4-yl)oxazoIidin-2-one.
- Example 1.34 Preparation of (jR)-4-(2-Chloro-4'-(2-((l?)-2-methylpyrroIidin-l- yl)ethyl)biphenyl-4-yl)oxazoIidin-2-one (Compound 20) Step A: Preparation of (i?)-4-(3-Chloro-4-hydroxyphenyl)oxazolidin-2-one.
- Step B Preparation of (/?)-2-Chloro-4-(2-oxooxazolidin-4-yl)phenyl trifiuoromethanesulfonate.
- To a stirred slurry of impure (i?)-4-(3-chloro-4-hydroxyphenyl)oxazolidin-2-one (0.26 g, 1.2 mmol) in acetonitrile (4 mL) was added pyridine (0.39 mL, 4.9 mmol). The mixture was cooled to 0 °C and trifluoromethanesulfonic anhydride (0.31 mL, 1.8 mmol) was added dropwise. The clear red solution was stirred at 0 0 C.
- Step C Preparation of ( ⁇ )-4-(2-Chloro-4'-(2-(( ⁇ )-2-methylpyrroIidin-l- yl)ethyl)biphenyl-4-yl)oxazolidin-2-one.
- Example 1.35 Preparation of (S)-4-(2'-Chloro-4'-(2-((/?)-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yI)oxazolidin-2-one (Compound 22).
- Step A Preparation of Methyl 2-(3-Chloro-4-hydroxyphenyl)acetate.
- Step C Preparation of 2-(3-Chloro-4-(4-methoxybenzyloxy)phenyl)ethanol.
- methyl 2-(3-chloro-4-(4- methoxybenzyloxy)phenyl)acetate (0.50 g, 1.6 mmol) in THF (15 mL) was added lithium aluminum hydride (1.6 mL of 1 M THF solution, 1.6 mmol).
- the cold bath was allowed to expire while the reaction stirred overnight.
- the reaction was quenched by pouring onto ice, then the slurry was extracted with EtOAc (thrice). The combined organics were washed with brine, dried over sodium sulfate, and concentrated to give the title compound (with minor impurities, 0.49 g).
- Step D Preparation of 3-Chloro-4-(4-methoxybenzyloxy)phenethyl Methanesulfonate.
- Step E Preparation of (l?)-l-(3-Chloro-4-(4-methoxybenzyIoxy)phenethyl)-2- methylpyrrolidine.
- 3-chloro-4-(4-methoxybenzyloxy)phenethyl methanesulfonate (0.60 g, 1.6 mmol) was added (i?)-2-methylpyrrolidine, benzene sulfonate (0.47, 1.9 mmol) and potassium carbonate (0.49 g, 3.6 mmol).
- the mixture was heated at 60 0 C for 18 h.
- the white heterogeneous mixture was cooled to room temperature, filtered, and the filtrate was concentrated.
- Step F Preparation of (i?)-2-Chloro-4-(2-(2-methylpyrrolidin-l-yl)ethyl)phenol.
- Step G Preparation of ( ⁇ )-2-Chloro-4-(2-(2-methylpyrrolidin-l-yl)ethyl)phenyl Trifluoromethanesulfonate. To a stirred slurry of (i?)-2-chloro-4-(2-(2-methylpyrrolidin-l -yl)ethyl)phenol (0.070 g,
- Step H Preparation of (S)-4-(2 ? -Chloro-4'-(2-(( ⁇ )-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yl)oxazoIidin-2-one.
- Example 1.36 Preparation of (5)-4-(3'-Fluoro-4'-(2-((i?)-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yl)oxazolidin-2-one (Compound 23).
- Step A Preparation of 2-(2-Fluoro-4-methoxyphenyl)ethanol.
- Step C Preparation of ( ⁇ )-l-(2-Fluoro-4-methoxyphenethyl)-2-methylpyrrolidine.
- 2-fluoro-4-methoxyphenethyl methanesulfonate 1.5 g, 6.0 mmol
- acetonitrile 15 mL
- (R)-2-methylpyrrolidine benzene sulfonate 1.8, 7.3 mmol
- potassium carbonate 1.8 g, 13 mmol
- Step E Preparation of (/?)-3-Fluoro-4-(2-(2-methylpyrrolidin-l-yl)ethyl)phenyI.
- To a stirred slurry of (R)-3-fluoro-4-(2-(2-methylpyrrolidin-l-yl)ethyl)phenol (0.10 g, 0.45 mmol) in acetonitrile (1.5 mL) was added pyridine (0.15 mL, 1.8 mmol). The mixture was cooled to 0 0 C and trifluoromethanesulfonic anhydride (0.11 mL, 0.67 mmol) was added drop wise.
- Step F Preparation of (5)-4-(3'-Fluoro-4'-(2-(( J R)-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yl)oxazolidin-2-one.
- Example 1.37 Preparation of (i?)-4-(2-Methoxy-4'-(2-((i?)-2-methyIpyrrolidin-l- yl)ethyl)biphenyl-4-yl)oxazolidin-2-one (Compound 21).
- Step A Preparation of Methyl 2-(terf-Butoxycarbonylamino)-2-(4-hydroxy-3- methoxyphenyl)acetate.
- Step B Preparation of tert-Butyl 2-Hydroxy-l-(4-hydroxy-3- methoxyphenyl)ethylcarbamate.
- Step D Preparation of 2-Methoxy-4-(2-oxooxazolidin-4-yl)phenyl trifluoromethanesulfonate.
- Step E Preparation of 4-(2-Methoxy-4'-(2-((i?)-2-methylpyrro-idin-l- yI)ethyl)biphenyl-4-yl)oxazolidin-2-one.
- Example 1.38 Preparation of (S)-4-(2'-Methoxy-4'-(2-(pyrrolidin-l-yI)ethyl)biphenyI-4- yl)oxazolidin-2-one (Compound 33).
- Step A Preparation of 2-Methoxy-4-(2-(pyrrolidin-l-yl)ethyl)phenyl
- Step B Preparation of (S)-4-(2'-Methoxy-4'-(2-(pyrrolidin-l-yl)ethyl)biphenyl-4- yl)oxazolidin-2-one.
- Example 1.39 Preparation of 4-(4'-(2-(( ⁇ )-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4-yl)- l,3-dioxolan-2-one (Compound 26).
- Step A Preparation of 4-(4-Bromophenyl)-2,2-dimethyl-l,3-dioxolane.
- a mixture of l-(4-bromophenyl)ethane-l,2-diol (1.0 g, 4.6 mmol) in acetone (10 mL) and 2,2-dimethoxypropane (11 mL, 92 mmol) was added pyridiniumj ⁇ -toluenesulfonate (PPTs) (0.12 g, 0.46 mmol). After 16 h, the reaction was quenched with water. The volatiles were evaporated and the aqueous slurry was extracted with DCM (thrice).
- PPTs pyridiniumj ⁇ -toluenesulfonate
- Step B Preparation of (2 ⁇ )-l-(2-(4'-(2,2-Dimethyl-l,3-dioxolan-4-yl)biphenyl-4- yl)ethyl)-2-methylpyrrolidine.
- Step C Preparation of l-(4'-(2-((i?)-2-Methylpyrrolidin-l-yl)ethyl)biphenyI-4- yl)ethane-l ,2-dioI.
- Step D Preparation of 4-(4'-(2-(( ⁇ )-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4-yl)- l,3-dioxolan-2-one.
- Example 1.40 Preparation of 4-(4'-(2-(( ⁇ )-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4- yI)pyrrolidin-2-one (Compound 25).
- Step A Preparation of 4-(4-Chlorophenyl)pyrrolidin-2-one.
- Step B Preparation of 4-(4'-(2-(( ⁇ )-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4- yl)pyrrolidin-2-one.
- Example 1.41 Preparation of 5-(4'-(2-(( ⁇ )-2-Methylpyrrolidin-l-yl)ethyI)biphenyl-4- yl)pyrrolidin-2-one (Compound 30).
- Step A Preparation of 5-(4-Methoxyphenyl)pyrrolidin-2-one.
- Step C Preparation of 4-(5-Oxopyrrolidin-2-yl)phenyl trifluoromethanesulfonate.
- 5-(4-hydroxyphenyl)pyrrolidin-2-one (0.20 g, 1.1 mmol) in acetonitrile (4 mL) was added pyridine (0.37 mL, 4.5 mmol).
- pyridine (0.37 mL, 4.5 mmol
- the mixture was cooled to 0 0 C and trifluoromethanesulfonic anhydride (0.29 mL, 1.7 mmol) was added dropwise. After 30 min the solvent was evaporated and the dark residue was washed with (10: 1) MTBE/EtOAc. A solid precipitated which contained no desired product.
- Step D Preparation of 5-(4'-(2-((i?)-2-MethylpyrroIidin-l-yl)ethyl)biphenyI-4- yl)pyrrolidin-2-one.
- Example 1.42 Preparation of 6-(4'-(2-((/?)-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4-yl)- l,3-oxazinan-2-one (Compound 27).
- Step A Preparation of 3-Amino-l-(4-bromophenyl)propan-l-ol.
- a solution of 3-(4-bromophenyl)-3-oxopropanenitrile (1.0 g, 4.5 mmol) in THF (89 mL) was heated to reflux.
- Borane-THF complex 13 mL of 1 M solution, 13 mmol was added dropwise. After 3.5 h the mixture was cooled to room temperature, then MeOH (20 mL) was added. The volatiles were evaporated to give a white residue, which was then dissolved in MTBE. The clear solution was treated with a 1 M ethereal HCl solution.
- Step B Preparation of 6-(4-BromophenyI)-l,3-oxazinan-2-one.
- a solution of impure 3-amino-l-(4-bromophenyl)propan-l-ol (0.85 g, 3.7 mmol) and l,r-carbonyldiimidazole (3.6 g, 22 mmol) in THF (74 mL) was heated to 40 0 C. After 18 h the solvent was evaporated and the residue was treated with water and extracted with DCM (thrice). The combined organic extract was washed with brine, dried over sodium sulfate, and the solvent was evaporated.
- Example 1.43 Preparation of 4-(4'-(2-(( ⁇ )-2-Methylpyrrolidin-l-yl)ethyl)biphenyI-4-yl)- l,3-oxazinan-2-one (Compound 28).
- Step A Preparation of 4-(4-Bromophenyl)-l,3-oxazinan-2-one.
- Step B Preparation of 4-(4 t -(2-((/?)-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4-yl)- 1 ,3-oxazinan-2-one.
- Example 1.44 Preparation of 4-(4'-(2-((J?)-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4-yl)- l,3-dioxan-2-one (Compound 29).
- Step A Preparation of (l?)-(4'-(2-(2-Methylpyrrolidin-l-yl)ethyl)biphenyI-4- yl)methanol.
- Step B Preparation of ( ⁇ )-4'-(2-(2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4- carbaldehyde.
- Step C Preparation of Ethyl 3-Hydroxy-3-(4'-(2-((/?)-2-methylpyrrolidin-l- yl)ethyl)biphenyl-4-yl)propanoate.
- Step D Preparation of l-(4'-(2-(( ⁇ )-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4- yl)propane-l,3-diol.
- Step E Preparation of 4-(4'-(2-((i?)-2-MethyIpyrrolidin-l-yl)ethyl)biphenyl-4-yl)- l,3-dioxan-2-one.
- Example 1.45 Preparation of 5-(4'-(2-((l?)-2-Methylpyrrolidin-l-yl)ethyl)biphenyI-4- yl)morpholin-3-one (Compound 31).
- Step A Preparation of 2-Amino-2-(4-bromophenyl)ethanol.
- Step C Preparation of 5-(4'-(2-((i?)-2-Methylpyrrolidin-l-yl)ethyl)biphenyl-4- yl)morpholin-3-one.
- Example 1.46 Preparation of (5)-4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl)oxazolidin-2-one.
- the mixture was stirred under N 2 and potassium acetate (13.87 g, 141 mmol) was added followed by 4,4,4 1 ,4',5,5,5',5'-octamethyl-2,2 1 -bi(l,3,2-dioxaborolane) (24.48 g, 96 mmol), triphenylphosphine (1.011 g, 3.86 mmol) and PdCl 2 (PPh 3 ) 2 (1.353 g, 1.928 mmol).
- the reaction mixture was gradually heated (by an oil bath) to 100 0 C (internal). There was a mild exotherm initially and the internal temperature went up to 110 0 C.
- the reaction was cooled to ⁇ 100 0 C and maintained at that temperature for 2 h.
- LCMS analysis showed there was less than 1% of starting material.
- the starting material and the product have the same retention time by LCMS; the product formation was analyzed by extracted ion chromatogram data.
- the histamine receptor binding assay was conducted using standard laboratory procedures as described below.
- Imetit was used as an assay positive control at varying concentrations. The plate was incubated for 30 min at room temperature. The assay was terminated by rapid filtration through a 96-well glass fiber filtration plate (GF/C) using a cell harvester (Perkin-Elmer). Captured membranes were washed three times with cold assay buffer and plates were dried at 50 0 C. 35 microliters ( ⁇ L) of scintillation cocktail was added to each well and membrane-bound radioactivity was recorded using a TopCount 96-well plate scintillation counter (Perkin-Elmer). The following table shows the observed activities for certain compounds of the present invention.
- Certain other compounds of the invention had activity values ranging from 0.6 nM to 24.0 nM in this assay.
- Rats Male Sprague-Dawley rats (225-350 g) (Harlan, San Diego, CA) were singly housed and maintained in a humidity- (30-70%) and temperature- (20-22 0 C) controlled facility on a 12 h: 12 h light/dark cycle (lights on at 6:30 A.M.) with free access to food (Harlan-Teklad Western Res., Orange, CA, Rodent Diet 8604) and water. Rats were allowed at least three days of habituation to the animal facility before surgery.
- Rats were anaesthetized with a ketamine/xylazine mixture, and surgically prepared for EEG and EMG recording. After 2-3 weeks of post-surgical recovery, rats were habituated to polypropylene test cages for at least three days. On test days, the rats were placed in the test chambers and habituated overnight. At 10 am the next day, the rats were administered the test compound, connected to the recording apparatus, and placed back into the test chambers for 3 h.
- EEG and EMG data were digitized and stored in 10 s epochs over the three hour test period. These data were then visually scored, and each 10 s epoch characterized as either a non- REM sleep, REM sleep, or waking episode. Total wake time over the three hour period was calculated for each rat after either vehicle administration or test compound. Percent increase in wakefulness was then derived for each rat.
- the following table shows the observed percent increase in wakefulness over 1 h after oral administration of a representative compound at 1.0 mg/kg.
- Example 4 Human Histamine H3-Receptor Binding Assay - MDS Pharma Services (Taiwan).
- Certain other compounds of the invention had activity values ranging from about 4.4 nM to about 23 nM in this assay.
- Example 5 Blockade of RAMH-Induced Drinking Assay.
- H3 agonists such as R- ⁇ -methyl-histamine (RAMH) induce a drinking response that is sensitive to reversal with an H3 antagonist.
- Blockade of RAMH-induced drinking can therefore be utilized as an in vivo assay for functional H3 antagonist activity.
- male Sprague Dawley rats 250-35Og were housed three per cage and maintained under a reverse 12 h light cycle (lights off at 1130 h). At 1030 h on the day of test, rats were individually housed in new cages and food was removed. 120 min later, rats were administered test article (vehicle or H3 antagonist, 0.3 mg/kg PO).
- RAMH vehicle or RAMH 3 mg/kg salt SC
- 10 min after administration of RAMH weighed water bottles were placed in the cages, and drinking was allowed for 20 min. Water consumption was determined for each animal by weighing each bottle to the nearest 0.1 g. Data is expressed as percentage reduction in water intake according to the following formula:
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Pulmonology (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Immunology (AREA)
- Hospice & Palliative Care (AREA)
- Child & Adolescent Psychology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Otolaryngology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Pyrrole Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description

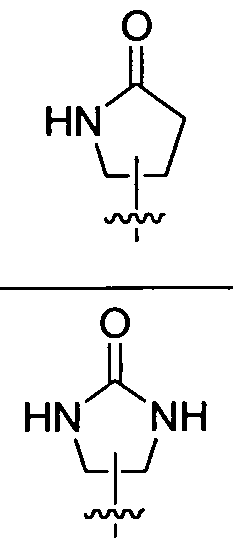

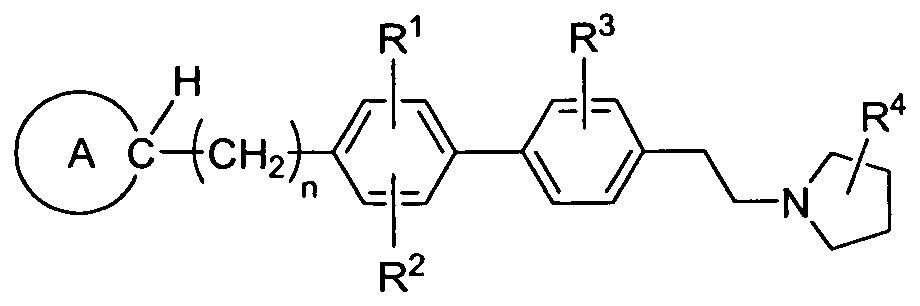


















Claims







Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2702320A CA2702320A1 (en) | 2007-10-30 | 2008-10-29 | Biphenyl derivatives as modulators of the histamine-h3 receptor useful for the treatment of disorders related thereto |
| AU2008319310A AU2008319310A1 (en) | 2007-10-30 | 2008-10-29 | Biphenyl derivatives as modulators of the histamine-H3 receptor useful for the treatment of disorders related thereto |
| CN2008801234112A CN101910155A (en) | 2007-10-30 | 2008-10-29 | Be used for the treatment of biphenyl derivatives with histamine-obstacle that the H3 receptor modulators is relevant as histamine-H3 receptor modulators |
| JP2010531070A JP2011502122A (en) | 2007-10-30 | 2008-10-29 | Biphenyl derivatives as modulators of histamine H3-receptors useful for the treatment of histamine H3-related disorders |
| EP08844595A EP2217592A1 (en) | 2007-10-30 | 2008-10-29 | Biphenyl derivatives as modulators of the histamine-h3 receptor useful for the treatment of disorders related thereto |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US101707P | 2007-10-30 | 2007-10-30 | |
| US61/001,017 | 2007-10-30 | ||
| US18842008P | 2008-08-08 | 2008-08-08 | |
| US61/188,420 | 2008-08-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009058300A1 true WO2009058300A1 (en) | 2009-05-07 |
Family
ID=40242626
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/012272 WO2009058300A1 (en) | 2007-10-30 | 2008-10-29 | Biphenyl derivatives as modulators of the histamine-h3 receptor useful for the treatment of disorders related thereto |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP2217592A1 (en) |
| JP (1) | JP2011502122A (en) |
| CN (1) | CN101910155A (en) |
| AU (1) | AU2008319310A1 (en) |
| CA (1) | CA2702320A1 (en) |
| WO (1) | WO2009058300A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012102562A2 (en) * | 2011-01-26 | 2012-08-02 | 주식회사 바이오랜드 | Composition containing novel 4-o-methylhonokiol derivative as active ingredient for treatment of dementia |
| WO2013151982A1 (en) | 2012-04-03 | 2013-10-10 | Arena Pharmaceuticals, Inc. | Methods and compounds useful in treating pruritus, and methods for identifying such compounds |
| WO2014110103A1 (en) * | 2013-01-09 | 2014-07-17 | Arena Pharmaceuticals, Inc. | Biphenyl-ethyl-pyrrolidine derivatives as histamine h3 receptor modulators for the treatment of cognitive disorders |
| US9346811B2 (en) | 2011-03-01 | 2016-05-24 | Janssen Pharmaceutica Nv | 6,7-dihydro-pyrazolo[1,5-a]pyrazin-4-ylamine derivatives useful as inhibitors of beta-secretase (BACE) |
| US9580433B2 (en) | 2013-06-12 | 2017-02-28 | Janssen Pharmaceutica Nv | 4-amino-6-phenyl-5,6-dihydroimidazo[1,5-A]pyrazine derivatives as inhibitors of beta-secretase (BACE) |
| US9751886B2 (en) | 2013-06-12 | 2017-09-05 | Janssen Pharmaceutica Nv | 4-amino-6-phenyl-6,7-dihydro[1,2,3]triazolo[1,5-A]pyrazine derivatives as inhibitors of beta-secretase (BACE) |
| US9828350B2 (en) | 2010-06-09 | 2017-11-28 | Janssen Pharmaceutica Nv | 5,6-dihydro-2H-[1,4]oxazin-3-yl-amine derivatives useful as inhibitors of beta-secretase (BACE) |
| US9834559B2 (en) | 2013-06-12 | 2017-12-05 | Janssen Pharmaceutica Nv | 4-Amino-6-phenyl-5,6-dihydroimidazo[1,5-a]pyrazin-3(2H)-one derivatives as inhibitors of beta-secretase (BACE) |
| US9840507B2 (en) | 2010-12-22 | 2017-12-12 | Janssen Pharmaceutica, Nv | 5,6-dihydro-imidazo[1,2-a]pyrazin-8-ylamine derivatives useful as inhibitors of beta-secretase (BACE) |
| US9845326B2 (en) | 2011-03-09 | 2017-12-19 | Janssen Pharmaceutica Nv | Substituted 3,4-dihydropyrrolo[1,2-A]pyrazines as beta-secretase (BACE) inhibitors |
| US10106524B2 (en) | 2014-12-18 | 2018-10-23 | Janssen Pharmaceutica Nv | 2,3,4,5-tetrahydropyridin-6-amine and 3,4-dihydro-2H-pyrrol-5-amine compound inhibitors of beta-secretase |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050245543A1 (en) * | 2004-04-30 | 2005-11-03 | Pfizer Inc | Histamine-3 receptor antagonists |
| WO2006011042A1 (en) * | 2004-07-19 | 2006-02-02 | Pfizer Products Inc. | 1, 3 substituted cycloamino derivatives and their use as histamine-3 receptor antagonists |
-
2008
- 2008-10-29 CN CN2008801234112A patent/CN101910155A/en not_active Withdrawn
- 2008-10-29 JP JP2010531070A patent/JP2011502122A/en not_active Withdrawn
- 2008-10-29 CA CA2702320A patent/CA2702320A1/en not_active Withdrawn
- 2008-10-29 AU AU2008319310A patent/AU2008319310A1/en not_active Withdrawn
- 2008-10-29 WO PCT/US2008/012272 patent/WO2009058300A1/en active Application Filing
- 2008-10-29 EP EP08844595A patent/EP2217592A1/en not_active Withdrawn
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050245543A1 (en) * | 2004-04-30 | 2005-11-03 | Pfizer Inc | Histamine-3 receptor antagonists |
| WO2006011042A1 (en) * | 2004-07-19 | 2006-02-02 | Pfizer Products Inc. | 1, 3 substituted cycloamino derivatives and their use as histamine-3 receptor antagonists |
Non-Patent Citations (1)
| Title |
|---|
| MORINI G ET AL: "Dibasic non-imidazole histamine H3 receptor antagonists with a rigid biphenyl scaffold", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, PERGAMON, ELSEVIER SCIENCE, GB, vol. 16, no. 15, 1 August 2006 (2006-08-01), pages 4063 - 4067, XP025107130, ISSN: 0960-894X, [retrieved on 20060801] * |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9828350B2 (en) | 2010-06-09 | 2017-11-28 | Janssen Pharmaceutica Nv | 5,6-dihydro-2H-[1,4]oxazin-3-yl-amine derivatives useful as inhibitors of beta-secretase (BACE) |
| US9840507B2 (en) | 2010-12-22 | 2017-12-12 | Janssen Pharmaceutica, Nv | 5,6-dihydro-imidazo[1,2-a]pyrazin-8-ylamine derivatives useful as inhibitors of beta-secretase (BACE) |
| WO2012102562A3 (en) * | 2011-01-26 | 2013-03-07 | 주식회사 바이오랜드 | Composition containing novel 4-o-methylhonokiol derivative as active ingredient for treatment of dementia |
| WO2012102562A2 (en) * | 2011-01-26 | 2012-08-02 | 주식회사 바이오랜드 | Composition containing novel 4-o-methylhonokiol derivative as active ingredient for treatment of dementia |
| US9346811B2 (en) | 2011-03-01 | 2016-05-24 | Janssen Pharmaceutica Nv | 6,7-dihydro-pyrazolo[1,5-a]pyrazin-4-ylamine derivatives useful as inhibitors of beta-secretase (BACE) |
| US9845326B2 (en) | 2011-03-09 | 2017-12-19 | Janssen Pharmaceutica Nv | Substituted 3,4-dihydropyrrolo[1,2-A]pyrazines as beta-secretase (BACE) inhibitors |
| WO2013151982A1 (en) | 2012-04-03 | 2013-10-10 | Arena Pharmaceuticals, Inc. | Methods and compounds useful in treating pruritus, and methods for identifying such compounds |
| US20150353488A1 (en) * | 2013-01-09 | 2015-12-10 | Arena Pharmaceuticals, Inc. | Biphenyl-ethyl-pyrrolidine derivatives as histamine h3 receptor modulators for the treatment of cognitive disorders |
| US9365511B2 (en) * | 2013-01-09 | 2016-06-14 | Arena Pharmaceuticals, Inc. | Biphenyl-ethyl-pyrrolidine derivatives as histamine H3 receptor modulators for the treatment of cognitive disorders |
| WO2014110103A1 (en) * | 2013-01-09 | 2014-07-17 | Arena Pharmaceuticals, Inc. | Biphenyl-ethyl-pyrrolidine derivatives as histamine h3 receptor modulators for the treatment of cognitive disorders |
| EP3309150A1 (en) * | 2013-01-09 | 2018-04-18 | Arena Pharmaceuticals, Inc. | Biphenyl-ethyl-pyrrolidine derivatives as histamine h3 receptor modulators for the treatment of cognitive disorders |
| EP3889141A1 (en) * | 2013-01-09 | 2021-10-06 | Arena Pharmaceuticals, Inc. | (r)-3-(4'-(2-(2-methylpyrrolidin-1-yl)ethyl)biphenyl-4-yl)propanoic acid as histamine h3 receptor modulators for the treatment of cognitive disorders |
| US9751886B2 (en) | 2013-06-12 | 2017-09-05 | Janssen Pharmaceutica Nv | 4-amino-6-phenyl-6,7-dihydro[1,2,3]triazolo[1,5-A]pyrazine derivatives as inhibitors of beta-secretase (BACE) |
| US9580433B2 (en) | 2013-06-12 | 2017-02-28 | Janssen Pharmaceutica Nv | 4-amino-6-phenyl-5,6-dihydroimidazo[1,5-A]pyrazine derivatives as inhibitors of beta-secretase (BACE) |
| US9834559B2 (en) | 2013-06-12 | 2017-12-05 | Janssen Pharmaceutica Nv | 4-Amino-6-phenyl-5,6-dihydroimidazo[1,5-a]pyrazin-3(2H)-one derivatives as inhibitors of beta-secretase (BACE) |
| US10106524B2 (en) | 2014-12-18 | 2018-10-23 | Janssen Pharmaceutica Nv | 2,3,4,5-tetrahydropyridin-6-amine and 3,4-dihydro-2H-pyrrol-5-amine compound inhibitors of beta-secretase |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2008319310A1 (en) | 2009-05-07 |
| CN101910155A (en) | 2010-12-08 |
| JP2011502122A (en) | 2011-01-20 |
| EP2217592A1 (en) | 2010-08-18 |
| CA2702320A1 (en) | 2009-05-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009058300A1 (en) | Biphenyl derivatives as modulators of the histamine-h3 receptor useful for the treatment of disorders related thereto | |
| EP2074086B1 (en) | Biphenyl sulfonyl and phenyl-heteroaryl sulfonyl modulators of the histamine h3-receptor useful for the treatment of disorders related thereto | |
| JP2022523774A (en) | N-substituted indole and other heterocyclic compounds for the treatment of brain disorders | |
| US20100331312A1 (en) | Modulators of the histamine h3 receptor useful for the treatment of disorders related thereto | |
| EP2035372A1 (en) | Modulators of the histamine h3-receptor useful for the treatment of disorders related thereto | |
| JP2005255685A (en) | New pyrazole analog acting on cannabinoid receptor | |
| JP2014509660A (en) | Substituted 3- (1H-benzo {d} imidazol-2-yl) -1H-indazole analogs as inhibitors of PDK1 kinase | |
| MX2011005119A (en) | Alkylcyclohexylethers of dihydrotetraazabenzoazulenes. | |
| JP2017519772A (en) | Neurotensin receptor 1 small molecule agonist | |
| US20100292288A1 (en) | Crystalline forms of (r)-1-{2-[4`- (3-methoxy-propane-1- sulfonyl)-biphenyl-4-yl]-ethyl}-2-methyl-pyrrolidine, and compositions, and methods related thereto | |
| EP1948605A2 (en) | Modulators of the h3 receptor useful for the treatment of disorders related thereto | |
| US9365511B2 (en) | Biphenyl-ethyl-pyrrolidine derivatives as histamine H3 receptor modulators for the treatment of cognitive disorders | |
| KR20090019847A (en) | Pyrrolidine derivatives having activity at the glyt1 transporter | |
| WO2014028322A1 (en) | Modulators of the histamine h3 receptor and the treatment of disorders related thereto | |
| JP2024512992A (en) | Aryl Heterocycle Compounds as Kv1.3 Potassium Shaker Channel Blockers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880123411.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08844595 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2008319310 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2702320 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010531070 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3093/DELNP/2010 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2008319310 Country of ref document: AU Date of ref document: 20081029 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008844595 Country of ref document: EP |