WO2009027679A1 - Process for the preparation of guanidino substituted bi-and polyphenyls that are suitable as small molecule carriers - Google Patents
Process for the preparation of guanidino substituted bi-and polyphenyls that are suitable as small molecule carriers Download PDFInfo
- Publication number
- WO2009027679A1 WO2009027679A1 PCT/GB2008/002911 GB2008002911W WO2009027679A1 WO 2009027679 A1 WO2009027679 A1 WO 2009027679A1 GB 2008002911 W GB2008002911 W GB 2008002911W WO 2009027679 A1 WO2009027679 A1 WO 2009027679A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- formula
- compound
- moiety
- moieties
- Prior art date
Links
- 0 CN**C(*)=* Chemical compound CN**C(*)=* 0.000 description 8
- YJVGSVZSEVAZKT-UHFFFAOYSA-N NC(NCCOc1cc(-c(ccc(CNC(CCSSc2ccccn2)=O)c2)c2OCCNC(N)=N)ccc1)=N Chemical compound NC(NCCOc1cc(-c(ccc(CNC(CCSSc2ccccn2)=O)c2)c2OCCNC(N)=N)ccc1)=N YJVGSVZSEVAZKT-UHFFFAOYSA-N 0.000 description 1
- DRIKGPRGZHUWEG-UHFFFAOYSA-N NCCOc1cc(-c2cc(C#N)ccc2OCCNC(OCc2ccccc2)=O)ccc1 Chemical compound NCCOc1cc(-c2cc(C#N)ccc2OCCNC(OCc2ccccc2)=O)ccc1 DRIKGPRGZHUWEG-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/70—Sulfur atoms
- C07D213/71—Sulfur atoms to which a second hetero atom is attached
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/543—Lipids, e.g. triglycerides; Polyamines, e.g. spermine or spermidine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C279/00—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups
- C07C279/04—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of guanidine groups bound to acyclic carbon atoms of a carbon skeleton
- C07C279/08—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of guanidine groups bound to acyclic carbon atoms of a carbon skeleton being further substituted by singly-bound oxygen atoms
Definitions
- the present invention relates to a new synthetic route to small molecule carriers (SMCs) 5 and also to some new small molecule carriers. More specifically, the invention relates to new routes to SMCs that are useful for the in vitro and in vivo delivery of various cargo moieties into cells. It also relates to some new SMC products, which are particularly effective in achieving the delivery of cargo moieties into cells. 0 Over recent years, studies have shown that a variety of peptides, many of which are present in viral proteins, have the ability to cross biological membranes in various different cell types.
- PTDs protein transduction domains
- SMCs small molecule carriers
- molecular tugs are more amenable than peptide-PTDs due to their in vivo stability by virtue of their resistance to cellular enzymes that degrade peptides.
- WO 2005/123676 which is incorporated herein by reference, describes SMC compounds and a process for their production. However, the synthesis described is laborious and is not suitable for large-scale preparations.
- the inventors have now found a new convergent synthesis for the compounds disclosed in WO 2005/123676. This new route enables an improvement in the overall yield and also allows easy synthesis of a range of different SMC compounds.
- the invention also relates to a number of new SMC compounds.
- the present invention therefore provides a process for the of a compound of formula I, or a pharmaceutically acceptable salt thereof,
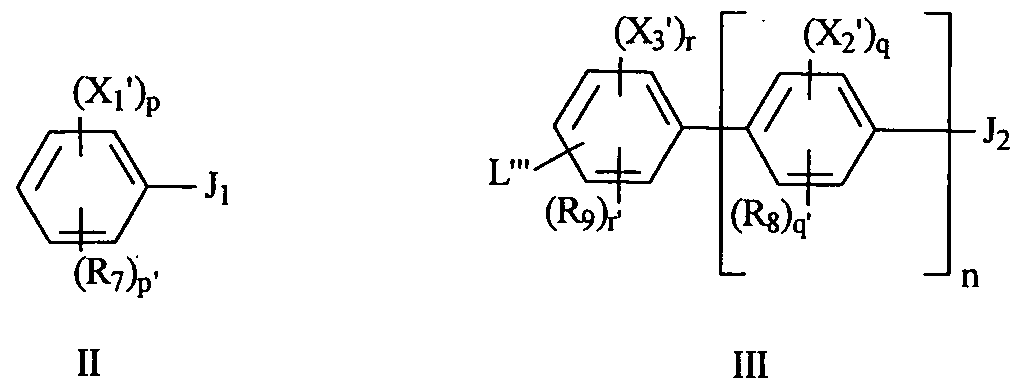
- Xl, X 2 and X 3 are each independently
- Y is an alkylene, alkenylene or alkynylene group, each of which may be optionally substituted with one or more substituents selected from alkyl, halo, CF 3 , OH, alkoxy, NH 2 , CN, NO 2 and COOH; W is absent or is O, S or NH;
- R 1 , R 2 , R 3 and R 4 are each independently selected from H, alkyl, aryl and a protecting group
- R 7 , Rg and R 9 are each independently selected from H, alkyl, halo, CF 3 , OH, alkoxy, NH 2 , CN, NO 2 and COOH;
- q and r are each independently 1, 2, 3 or 4;
- q 1 and r' are each independently 0, 1, 2 or 3, where q + q 1 and r + r' each equal 4;
- p is 1, 2, 3, 4 or 5, and p' is 0, 1, 2, 3 or 4, where p + p 1 is 5;
- n is 0, 1, 2, 3, 4, 5 or 6; and
- L is (Z) m NR 5 R 6 wherein Z is a hydrocarbyl group and m is 0 or 1 wherein R 5 and R 6 are each independently H, CO(CH 2 )JQ 1 or where j and k are each independently 0, 1, 2, 3, 4 or 5, and Q 1 and Q 2 are each independently selected from COOH, a chromophore,
- L"' is L, as defined above, a leaving group LG 2 , -COR 18 , -(Z) 111 NHR 12 , or a group which can be reduced to a moiety -(Z) m NH 2 , where Z and m are defined as above, R 12 is a protecting group P 3 and R 18 is hydrogen or a C 1 -C 4 alkyl group; and
- X 1 , X 3 , R 7 , R 9 and L are as defined above; r is 1, 2, 3 or 4 and r' is 0, 1, 2 or 3, wherein r + r' is 4; p is 1, 2, 3, 4 or 5 and p' is 0, 1, 2, 3 or 4, where p + p' is 5; and p + r is 3.
- hydrocarbyl refers to a saturated or unsaturated, straight-chain, branched, or cyclic group comprising at least C and H that may optionally comprise one or more other suitable substituents.
- substituents may include halo, alkoxy, hydroxy, CF 3 , CN, amino, COOH, nitro or a cyclic group.
- a combination of substituents may form a cyclic group. If the hydrocarbyl group comprises more than one C then those carbons need not necessarily be linked to each other. For example, at least two of the carbons may be linked via a suitable element or group.
- the hydrocarbyl group may contain heteroatoms. Suitable heteroatoms will be apparent to those skilled in the art and include, for instance, sulphur, nitrogen, oxygen, phosphorus and silicon.
- the hydrocarbyl group is an aryl or alkyl group.
- the hydrocarbyl group is unsubstituted. More preferably, the hydrocarbyl group is an unsubstituted Ci -6 alkyl group.
- alkoxy includes both straight chain and branched alkoxy groups which may be substituted (mono- or poly-) or unsubstituted.
- the alkoxy group is a C 1-20 alkoxy group, more preferably a C 1-15 alkoxy group, more preferably still a C 1-I2 alkoxy group, more preferably still, a C 1-6 alkoxy group, more preferably a Cj -3 alkoxy group.
- Particularly preferred alkoxy groups include, for example, methyoxy, ethyoxy, propoxy, isopropoxy, butoxy, isobutoxy, tert-butoxy, pentoxy and hexoxy.
- Suitable substituents include alkyl, halo, CF 3 , OH, alkoxy, NH 2 , CN, NO 2 and COOH.
- the alkoxy group is unsubstituted. More preferably, the alkoxy group is an unsubstituted C 1-4 alkoxy group .
- alkyl includes both saturated straight chain and branched alkyl groups which may be substituted (mono- or poly-) or unsubstituted.
- the alkyl group is a C 1-2O alkyl group, more preferably a C 1-I5 , more preferably still a C 1-I2 alkyl group, more preferably still, a Ci -6 alkyl group, more preferably a C 1-3 alkyl group.
- alkyl groups include, for example, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, pentyl and hexyl.
- Suitable substituents include halo, CF 3 , OH, alkoxy, NH 2 , CN, NO 2 and COOH.
- alkylene should be construed accordingly.
- the alkyl group is unsubstituted. More preferably, the alkyl group is an unsubstituted C 1-4 alkyl group.
- aryl refers to a substituted (mono- or poly-) or unsubstituted monoaromatic or polyaromatic system, wherein said polyaromatic system may be fused or unfused.
- aryl includes groups having from 6 to 10 carbon atoms, e.g. phenyl, naphthyl etc.
- aryl is synonymous with the term “aromatic”. Suitable substituents include alkyl, halo, CF 3 , OH, alkoxy, NH 2 , CN, NO 2 and COOH.
- an aryl group is unsubstituted or is substituted with 1, 2 or 3 substituents selected from C 1-4 alkyl, halo, CF 3 , OH, NH 2 groups. Typically, these substituents are themselves unsubstituted.
- the aryl group is an optionally substituted phenyl group. More typically, the aryl group is an unsubstituted phenyl group.
- alkenyl refers to a group containing one or more carbon-carbon double bonds, which may be branched or unbranched, substituted (mono- or poly-) or unsubstituted.
- the alkenyl group is a C 2-20 alkenyl group, more preferably a C 2-15 alkenyl group, more preferably still a C 2-12 alkenyl group, or preferably a C 2-6 alkenyl group, more preferably a C 2-3 alkenyl group.
- Suitable substituents include alkyl, halo, CF 3 , OH, alkoxy, NH 2 , CN, NO 2 and COOH.
- alkenylene should be construed accordingly.
- the alkenyl group is unsubstituted. More preferably, the alkenyl group is an unsubstituted C 2-4 alkenyl group.
- alkynyl refers to a carbon chain containing one or more triple bonds, which may be branched or unbranched, and substituted (mono- or poly-) or unsubstituted.
- the alkynyl group is a C 2-20 alkynyl group, more preferably a C 2- 15 alkynyl group, more preferably still a C 2-12 alkynyl group, or preferably a C 2-6 alkynyl group or a C 2-3 alkynyl group.
- Suitable substituents include alkyl, halo, CF 3 , OH, alkoxy, NH 2 , CN, NO 2 and COOH.
- alkynylene should be construed accordingly.
- the alkynyl group is unsubstituted. More preferably, the alkynyl group is an unsubstituted C 2-4 alkynyl group.
- chromophore refers to any functional group that absorbs light, giving rise to colour.
- the term refers to a group of associated atoms which can exist in at least two states of energy, a ground state of relatively low energy and an excited state to which it may be raised by the absorption of light energy from a specified region of the radiation spectrum.
- the group of associated atoms contains delocalised electrons.
- the chromophore present in the compounds prepared by the process of the invention can be a conjugated Il system or a metal complex.
- a chromophore is a porphyrin, a polyene, a polyyne or a polyaryl.
- the chromophore is one of.
- Protecting groups P 1 , P 2 and P 3 are protecting groups suitable for protecting a nitrogen atom. Many examples of such protecting groups are known to the person skilled in the art, for example those protecting groups mentioned in "Protecting Group Chemistry” Jeremy Robertson, OUP, 2000, which is incorporated herein by reference.
- P 1 , P 2 and P 3 are selected from benzyl, trityl, 9-phenylfluorenyl, benzydryl, fluorenyl, carbamate, benzylcarbamate (Cbz), t-butyl carbamate (Boc), 9-fluorenylmethyl carbamate (Fmoc), acetamide, p-toluenesulfonate (p-Ts), silyl and triisopropylsilyl (TIPS) groups.
- P 1 is a Boc group.
- P 2 is a Cbz group.
- P 2 and P 3 are different. More typically, P 1 , P 2 and P 3 are all different. Preferably, P 2 and P 3 are orthogonal. More preferably, P 1 and P 2 are orthogonal to P 3 .
- LG 1 is typically any group that can undergo oxidative addition with Pd(O). Those of skill in the art will easily be able to select appropriate leaving groups.
- LGj is preferably a halogen, triflate (OTf), tosylate (OTs), N-hydroxysuccinimide (OSu) or a mesylate (OMs) group.
- LGi is more preferably halogen, most preferably bromide or iodide.
- Leaving group LG 2 is typically a leaving group suitable for an aryl cyanation reaction. Those of skill in the art will easily be able to select appropriate such leaving groups.
- LG 2 is preferably a halogen, triflate (OTf), tosylate (OTs), N-hydroxysuccinimide (OSu) or a mesylate (OMs) group.
- LG 2 is more preferably halogen, most preferably bromide.
- LG 3 is typically a leaving group suitable for a nucleophilic substitution reaction at a saturated carbon centre. Those of skill in the art will easily be able to select appropriate leaving groups. LG 3 is preferably a halogen, triflate (OTf), tosylate (OTs), N- hydroxysuccinimide (OSu) or a mesylate (OMs) group. LG 3 is more preferably a OMs group.
- LG 4 can be any leaving group suitable for a guanidinylation reaction.
- LG 4 represents a moiety such that -NLG 4 is a leaving group in guanidinylation reaction.
- a skilled chemist can easily select appropriate leaving groups in this regard.
- preferred LG 4 groups include triflyl (Tf), tosyl (Ts) and mesyl (Ms) groups.
- LG 4 is most preferably a triflyl group, such that -NLG 4 represents -NTf.
- LG 4 ' can be any leaving group suitable for a guanidinylation reaction. A skilled chemist can easily select appropriate leaving groups in this regard. LG 4 ' is typically a halogen atom, triflate (OTf), tosylate (OTs), mesylate (OMs) or 1-pyrazole group, preferably a 1-pyrazole group.
- OTf triflate
- OTs tosylate
- OMs mesylate
- 1-pyrazole group preferably a 1-pyrazole group.
- LG 5 can be any leaving group suitable for a nucleophilic substitution reaction at a carbonyl or thiocarbonyl group. Those of skill in the art will easily be able to select appropriate leaving groups. LG 5 is typically a halogen, triflate (OTf), tosylate (OTs), N-hydroxysuccinimide (OSu) or a mesylate (OMs) group. LG 5 is preferably a OSu group.
- LG 7 is typically a leaving group suitable for a nucleophilic substitution reaction at a saturated carbon centre. Those of skill in the art will easily be able to select appropriate leaving groups. LG 7 is preferably a halogen, triflate (OTf), tosylate (OTs), N- hydroxysuccinimide (OSu) or a mesylate (OMs) group. LG 7 is more preferably a OMs group.
- Typical reagents and conditions for this process are described in US 6,380,358, and Katritsky, et al ARKIVOC 2005 (iv) 49-87 both of which are incorporated herein by reference.
- Katritsky, et al describes recent advances in the development of guanylating reagents, which are defined as compounds forming a guanidine structure by a chemical transformation.
- This document describes a wide rantge of possible reagents for the preparation of guanidines, including thioureas, isothioureas, carbodiimides and cyanamides, pyrazole-1-carboximidamides, triflyl guanidines, ammoiminomethane-sulfonic and -sulfinic acids and benzotriazole and imidazole-containing reagents.
- the coupling reaction between the compounds of formulae (II) and (III) is typically an organometallic coupling, preferably a coupling mediated by Pd, more preferably Pd(O).
- the coupling is typically a Stille, Suzuki, Negishi, Hiyama or Kumada coupling.
- the coupling is a Suzuki coupling.
- the reaction between the compounds of formulae (II) and (III) is a Suzuki coupling, performed using a Pd(O) catalyst in the presence of a base, preferably a nucleophilic base, more preferably triethylamine.
- the Pd(O) catalyst is typically used in a catalytic amount.
- the reaction is typically carried out at elevated temperature (i.e. above room temperature), preferably between 75 and 125 0 C, more preferably at 82°C or 100 0 C, most preferably at 82°C.
- a reaction time of 1 to 18 hours is typically employed, hi some cases a reaction time of 1, 3 or 18 hours may be used.
- the reaction is typically heated using microwave radiation or conventional heat sources.
- the reaction is typically carried out in an aqueous organic solvent, preferably aqueous toluene or aqueous isopropyl alcohol, more preferably aqueous isopropyl alcohol.
- the solvents used are preferably degassed prior to use.
- the reaction may be carried out by heating at 100 0 C a compound of formula II with a compound of formula III with PdCl 2 dppf. CH 2 Cl 2 and potassium phosphate in aqueous toluene.
- the reaction is carried out by heating at 82°C a compound of formula II with a compound of formula III with Pd Cl 2 dppf.CH 2 Cl 2 and triethylamine in aqueous isopropyl alcohol for 18 hours.
- X 3 1 is -W-Y-NR 1 R 10 . More preferably, all OfX 1 ', X 2 ' and X 3 ' are -W-Y-NR 1 R 10 .
- a boronic acid group is a group of formula -B(OH) 2 .
- the boronic ester is -B(OR 13 )(OR 14 ) or
- R 13 and Ri 4 are each independently selected from C 1 -C 6 alkyl groups and R 15 is a C 1 -C 6 alkyl or phenyl group. More preferably, the boronic ester is
- R 15 is a -CH(CH 3 ) 2 CH(CH 3 ) 2 - group.
- the borane is BR 16 R 17 where R 16 and R 17 are each independently selected from Ci-C 6 alkyl groups.
- a trihalogenoborate salt group is a group of formula -(B(HaI) 3 )TVI + , where each Hal group is the same or different and is independently chosen from halogen atoms.
- Halogen atoms are typically F, Cl, Br or I atoms, preferably F or Cl atoms, more preferably F atoms.
- each Hal group is the same, hi a more preferred embodiment, each Hal group is the same and is a fluorine atom.
- the group M + is any ionic group capable of acting as the counterion to the group - (B(HaI) 3 ) " , but is typically a monovalent metal ion, preferably a monovalent Group I metal ion.
- Monovalent Group I metal ions are typically Li + , Na + , K + or Rb + , preferably Li + , Na + or K + , more preferably K + .
- the group -(B(HaI) 3 )TvI + is -(BF 3 )TC + .
- J 1 is a boronic acid, a boronic ester or a borane group and J 2 is a leaving group LG 1 . More preferably, J 1 is a boronic acid or a boronic ester, more preferably a boronic acid or a compound of formula
- R 15 is as defined above, and J 2 is a halogen, for example bromine or iodine.
- J is a trihalogenoborate salt as defined above.
- compounds of formula II can be prepared by treating a compound of formula IX with a boronating agent in the presence of a catalyst.
- the boronating agent can be a diborane, for example, bispinacolactodiborane.
- the catalyst is preferably Pd.
- the reaction is carried out between 50 and 100 0 C, more preferably at 8O 0 C. A long reaction time of several days is typically employed, for example 72 hours.
- the reaction is typically carried out in an organic solvent, preferably dimethyl sulphoxide (DMSO).
- DMSO dimethyl sulphoxide
- J 1 is a trihalogenoborate salt group
- compounds of formula II where Ji is a trihalogenoborate salt group can be prepared by treating compounds of formula II where J 1 is a boronic acid, boronic ester or borane group with a saturated solution OfMH(HaI) 2 , where M and Hal are as defined above,
- reaction is carried out at room temperature, which is typically between 18°C and 30°C, preferably 21°C.
- a reaction time of between 1 and 5 hours is typically employed, preferably 3 hours.
- the reaction is typically carried out in an organic solvent, preferably methanol.
- Compounds of formula II' may be prepared using similar methodology to that described in Skaff, et al, J.O.C., Vol. 70, No. 18, 2005, 7353-7363 or Eur. J. Org.
- each X 1 ' represents -W-Y-NR 1 Ri 0 and W is O, S or NH
- each Xj represents a hydroxy, thiol or amino group
- W is O
- such compounds can be prepared from corresponding compounds of formula IX in which each X 1 1 represents -W-Y-NR 1 R 1O by deprotecting any protected amine groups on -W-Y-NR 1 R 10 moieties at the Xi' position, and guanidinylating the deprotected moieties.
- the compound of formula X in which each X 1 " is hydroxy, thiol or amino can be generated by deprotecting a corresponding compound in which the or each X]" is a protected hydroxy, thiol or amino group.
- a compound which carries an alkyl ether group at the or each X 1 " position can be converted to a compound of formula X in which the or each Xj" is hydroxy by reaction with BBr 3 .
- the reaction between the compound of formula X and LG 3 -Y-N-RiRi 0 is carried out between 75 and 125 0 C, more preferably at 100°C.
- a reaction time of 1 to 5 hours can be employed, for example 2 hours.
- the reaction is typically carried out in an organic solvent, preferably dimethyl formamide (DMF).
- each Xj' represents -W-Y-NRiR 1O and W is absent can be obtained by a Sonogashira coupling reaction between a suitably chosen alkyne, for example H(CsC)-Y-NR 1 R 1O and a leaving group on the phenyl ring, followed by reduction of the C ⁇ C group.
- a suitably chosen alkyne for example H(CsC)-Y-NR 1 R 1O and a leaving group on the phenyl ring
- W is absent
- a suitably chosen alkyne for example H(CsC)-Y-NR 1 R 10 and a leaving group on the phenyl ring
- such compounds can be prepared from corresponding compounds of formula (III) in which each X 3 ' represents -W-Y-NR 1 R 1O by deprotecting any protected amine groups on -W-Y-NR 1 R 10 moieties at the X 1 ' position, and guanidinylating the deprotected moieties.
- the compound of formula XI in which each X 3 " is hydroxy, thiol or amino can be generated by deprotecting a corresponding compound in which the or each X 3 " is a protected hydroxy, thiol or amino group.
- a compound which carries an alkyl ether group at the or each X 3 " position can be converted to a compound of formula X in which the or each X 3 " is hydroxy by reaction with BBr 3 .
- the reaction between the compound of formula XI and LG 3 -Y-N-R 1 R 1O is carried out between 50 and 100 0 C, more preferably at 8O 0 C.
- a reaction time of 24 hours is typically employed.
- the reaction is typically carried out in an organic solvent, preferably dimethyl formamide (DMF).
- each X 3 ' represents -W-Y-NR 1 R 10 and W is absent can be obtained by a Sonogashira coupling reaction between a suitably chosen alkyne, for example H(C ⁇ C)- Y-NRj R 10 and a leaving group on the phenyl ring, followed by reduction of the C ⁇ C group.
- a suitably chosen alkyne for example H(C ⁇ C)- Y-NRj R 10 and a leaving group on the phenyl ring
- W is absent
- a suitably chosen alkyne for example H(CsC)-Y-NR 1 R 10 and a leaving group on the phenyl ring
- a compound of formula (III) in which J 2 is a trihalogenoborate salt group can be prepared by reacting a corresponding compound in which J 2 is a boronic acid, boronic ester or borane group with a compound of formula MH(HaI) 2 , under the reaction conditions described above for the preparation of the compounds of formula (II).
- a compound of formula (III) in which J 2 is a boronic acid, boronic ester or borane group can be prepared by reacting a corresponding compound in which J 2 is a leaving group with a boronating agent, under the reaction conditions described above for the preparation of the compounds of formula (II).
- a compound of formula (111) or (XI) in which L'" is CN can be prepared, for example, from a corresponding compound in which L'" is a leaving group, for example iodine, by reaction with CN " , preferably with zinc cyanide and Pd(H).
- CN preferably with zinc cyanide and Pd(H).
- J 2 is a leaving group
- no more than 0.5 equivalents of CN " are used in the reaction, to minimise formation of the dicyano compound.
- J 2 is a boronic acid, a boronic ester or a borane group or a trihalogenoborate salt group and J 1 is a leaving group LG 1 .
- any of the X 1 ', X 2 ' and X 3 ' moieties in the formula (IV) are OH, SH or NH 2
- the alkylation of hydroxy, thiol and amino groups at the X 1 ', X 2 ' and X 3 ' positions in the formula IV so that they represent -W-Y-NRiRi 0 is effected with a compound of formula LG 3 -Y-NRiRi 0 where Ri, Y and LG 3 are defined as above and Rio is H or a protecting group P 2 .
- R 1O is a protecting group, for example Cbz. More preferably, R 1 is H and R 10 is a protecting group, for example Cbz.
- the alkylation of hydroxy, thiol and amino groups at the X 1 ', X 2 ' and X 3 ' positions in the formula IV so that they represent -W-Y-NR 1 R 10 is carried out at elevated temperature (i.e. above room temperature).
- the reaction is carried out between 50 and 100 0 C, more preferably at 8O 0 C.
- a reaction time of 1 to 4 hours is typically employed. In some cases, reaction times of 1, 3, 3.5 and 4 hours may be used.
- the reaction is typically carried out in an organic solvent, preferably dimethylformamide (DMF).
- DMF dimethylformamide
- CsCO 3 is typically added to the reaction mixture.
- Deprotection of any amine groups in the -W-Y-NR 1 R 1O moieties present at the X 1 ', X 2 ' and X 3 ' positions can be carried out by standard techniques. The exact conditions employed will depend on the exact nature of the protecting group, hi the case where the amine groups in the -W-Y-NR 1 R 1O moiety are protected with Cbz groups, the protecting groups can be removed with an acid, typically an anhydrous acid, preferably HBr and CH 3 COOH in DCM.
- said guanidinylation of deprotected -W-Y-NRiR 10 moieties at the X]', X 2 ' and X 3 ' positions is effected either by a compound of formula V, or a tautomer thereof, where R 2 , R 3 and R 4 are as defined in claim 1 and LG 4 is a leaving group;
- R 2 , R 3 and R 4 are as defined above and LG 4 " is a leaving group.
- guanidinylation is effected with N,N-di-boc-N'-trifluoromethanesulfonyl guanidine or N j N'-Di-Boc-lH-pyrazole-l-carboxamidine, most preferably N,N'-Di-Boc- 1 H-pyrazole- 1 -carboxamidine.
- tautomer is well known to the person skilled in the art and refers to the result of a migration of a hydrogen atom accompanied by the switch of a single bond and adjacent double bond.
- tautomers of certain molecules such as those represented by formulae V and V above exist in, for example, aqueous media.
- the guanidinylation is carried out at room temperature.
- the reaction mixture may be heated.
- the reaction is typically left to run overnight (i.e. a time in excess of 12 hours, for example, 18 hours).
- the reaction is typically carried out in an organic solvent, preferably DCM and triethylamine or diisopropylethylamine (DIEA), more preferably DCM and DIEA.
- At least one X 1 ', X 2 ' and X 3 1 moiety is -W-Y-NR 1 R 10 and the other X 1 ', X 2 ' and X 3 ' moieties are each independently selected from OH, SH, NH 2 and -W-Y- NR 1 R 10 , wherein and R 1 , W and Y are defined as above and R 10 is H or a protecting group, and in step (b) when any X 1 ', X 2 ' and X 3 ' moieties are OH, SH or NH 2 , alkylation is effected so that all X 1 ', X 2 ' and X 3 ' moieties represent -W-Y-NR 1 R 10 , any protected amine groups on the X 1 ', X 2 ' and X 3 1 moieties are deprotected and the X 1 ', X 2 ' and X 3 ' moieties are guanidinylated so
- step (b) of the process of the invention the alkylation and guanidinylation steps, and the conversion of L'" to a moiety L may be carried out in any order.
- any OfX 1 ', X 2 ' and X 3 ' are OH, SH or NH 2
- alkylation is first effected so that all OfX 1 ', X 2 ' and X 3 ' represent -W-Y-NR 1 R 1O
- any protected amine groups on X 1 ', X 2 ' and X 3 ' moieties represented by -W-Y-NR 1 R 10 are then deprotected and (iii) the Xj', X 2 ' and X 3 ' moieties are then guanidinylated so that they each represent a group of formula
- a catalyst is employed.
- this catalyst is an alkali metal carbonate, preferably sodium or cesium carbonate, more preferably cesium carbonate.
- the amine reacted with the compound of formula (V) or (V) is dried using known techniques before guanidinylation.
- L'" is typically -CN or -CH 2 NO 2 , preferably -CN.
- the reduction of L'" takes place under a hydrogen atmosphere, in the presence of a catalyst.
- reduction is effected with Raney Nickel under a hydrogen atmosphere.
- the reaction typically takes place at room temperature.
- the reaction time is typically 16 hours.
- the reaction typically takes place in an organic solvent, preferably THF. NH 4 OH may optionally be added.
- Cyanation is typically effected with zinc cyanide and Pd(II).
- the reaction typically takes place at elevated temperature (i.e. greater than room temperature).
- the reaction preferably takes place at from 150 to 200 0 C, more preferably 16O 0 C.
- the reaction is preferably heated with microwave irradiation.
- a short reaction time of less than one hour is typically employed, preferably 10 minutes.
- the reaction typically takes place in an organic solvent, preferably DMA.
- This is advantageous because the amount of protection and deprotection necessary on functional groups is minimised. Further, it enables a wide range of different L moieties to be introduced easily.
- R 18 is hydrogen
- reductive amination of the -COR 18 moiety is effected by (a) reaction with NH 2 ORi 9 or NHR 5 R 6 and (b) reduction of carbon nitrogen double bond, wherein R ⁇ is H or an alkyl group.
- reductive amination is effected by NH 2 ORi 9 , followed by reduction of the carbon nitrogen double bond.
- R 19 is hydrogen
- the carbon nitrogen bond is reduced with H 2 /Pd, SmZI 2 , In/NEUCl, LiAH 4 , Bu 3 SnH with BF 3 -OE 2 , Bu 2 SnClH in HMPA, Cl 3 SiH and pyrrolidine carboxaldehyde, SmBr in HMPA, Z-propanol with a ruthenium catalyst, NaCN(BH 3 ) or trithylammonium formate with microwave irradiation.
- a ruthenium catalyst NaCN(BH 3 ) or trithylammonium formate with microwave irradiation.
- Ri 9 is hydrogen
- reduction with H 2 /Pd is preferred.
- L'" is different from J 2 .
- p, q and r are each independently 1, 2, 3 or 4.
- Y is a C 1-1O alkylene group, a C 2-10 alkenylene group or a C 2-10 alkynylene group. More typically, Y is a C 1-12 alkylene group, preferably a C 1-1O alkylene group, even more preferably a C 1-6 alkylene group, and more preferably still, -CH 2 CH 2 -.
- W is O, S or NH. More preferably, W is O.
- L'" is a leaving group LG 2 , -COR 18 (in particular -CHO), or a group which can be reduced to a moiety -(Z) 1n NH 2 .
- the group which can be reduced to a moiety -(Z) m NH 2 is preferably -CN.
- m is 1 and Z is an alkylene group, more preferably, a C 1-12 alkylene group, more preferably still a C 1-1O alkylene group, even more preferably a C 1-6 or Ci -4 alkylene group. More preferably, Z is a CH 2 group.
- L is selected from the following: -CH 2 NH 2 , -CH 2 NHCOCH 2 CH 2 COOH,
- R] R 2 , R 3 and R 4 are each independently selected from H and a protecting group P 1 . More preferably, R 1 and R 3 are hydrogen and R 2 and R 4 represent H or Pi. Most preferably, Ri and R 3 are hydrogen and R 2 and R 4 are each independently selected from H and a butyloxycarbonyl (Boc) protecting group. Preferably, p, q and r are each independently 1 or 2.
- p, q and r are all equal to 1.
- p, q and r are all equal to 2.
- r is equal to 1 and p is equal to 2.
- R 7 , R 8 and R 9 are all H.
- X 1 , X 2 and X 3 are the same and are all
- R 2 and R 3 are each independently H or a Boc protecting group.
- n is 0 or 1. More preferably, n is 0.
- j and k are each independently 0, 1, 2 or 3, preferably 0, 1 or 2.
- the process of the invention is a process for the production of a compound of formula Ia or Ib
- X 1 and X 3 are the same and are both where R 2 and R 3 are each independently H or a Boc protecting group.
- said compound of formula II is of formula Ha, lib, lie or Hd and/or said compound III is of formula Ilia, IEb, IIIc or IHd.
- a further finding of the present invention is that compounds of formula (I) in which Z is para to an X 3 moiety have superior efficiency in delivering a cargo moiety inside a cell.
- the present invention also provides a compound of formula (I), as defined above, or a pharmaceutically acceptable salt thereof, wherein r is 1 or 2 and the phenyl ring which carries L has no substituents ortho to the L moiety.
- the phenyl ring which carries L has a hydrogen atom ortho to the moiety
- r is 1 and L is para to the X 3 moiety.
- the present invention also provides a compound of formula Via, VIb, VIc, VId or Vie
- the present invention also provides a compound of formula VII
- X 1 , X 3 , R 7 , Rg and L are as defined in any one of claims 1, 7, 8, 9 or 11; - r is 1, 2, 3 or 4 and r' is 0, 1, 2 or 3, wherein r + r 1 is 4; p is 1, 2, 3, 4 or 5 and p' is 0, 1, 2, 3 or 4, where p + p 1 is 5; and p + r equals 3.
- the compound of formula (VII) is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl
- the present invention further provides a process for preparing a conjugate, which process comprises: (i) preparing a compound of formula I, or a pharmaceutically acceptable salt thereof, in which L is other than a moiety -(Z) m - ⁇ hthalimide, wherein Z and m are as defined in claim 1, by a process according to the invention; and (ii) reacting said compound of formula I with a cargo moiety selected from a protein, a peptide, an oligonucleotide, a nucleotide, a diagnostic agent, a biologically active compound, an antibody and a drug.
- the cargo moiety can be an oligonucleotide, nucleotide, protein, peptide, biologically active compound, diagnostic agent, or a combination thereof.
- the cargo moiety may be directly or indirectly linked to the carrier moiety, hi the embodiment wherein the cargo moiety is indirectly linked to the carrier, the linkage may be by an intermediary bonding group such as a sulphydryl or carboxyl group or any larger group, all such linking groups are herein referred to as linker moieties as discussed below.
- linker moieties are linked directly.
- the diagnostic agent can be nonbiological, for example a microbead.
- Appropriate processes for preparing a conjugate of a compound of formula I and a nonbiological diagnostic agent such as a microbead are familiar to those of skill in the art.
- oligonucleotide cargo moieties include genes, gene fragments, sequences of DNA, cDNA, RNA, nucleotides, nucleosides, heterocyclic bases, synthetic and non-synthetic, sense or anti-sense oligonucleotides including those with nuclease resistant backbones etc. or any of the above incorporating a radioactive label, that are desired to be delivered into a cell or alternatively to be delivered from a cell to its exterior.
- the oligonucleotide cargo moiety is a gene or gene fragment.
- suitable protein or peptide cargo moieties include; proteins, peptides, and their derivatives such as: antibodies and fragments thereof; cytokines and derivatives or fragments thereof, for example, the interleukins (IL) and especially the IL-I, IL-2, IL-3, IL-4, IL-5, IL- 6, IL-7, IL-8, IL-9, IL-10, IL-Il and IL- 12 subtypes thereof; colony stimulating factors, for example granulocyte-macrophage colony stimulating factor, granulocyte-colony stimulating factor (alpha and beta forms), macrophage colony stimulating factor (also known as CSF-I); haemopoietins, for example erythropoietin, haemopoietin-alpha and kit-ligand (also known as stem cell factor or Steel factor); interferons (IFNS), for example IFN- ⁇ , IFN- ⁇ and EFN- ⁇ ; growth factors and bifunctional
- non-nucleotide/proteinaceous biologically active cargo moieties are drug moieties selected from cytotoxic agents, anti-neoplastic agents, anti-hypertensives, cardioprotective agents, anti-arrhythmics, ACE inhibitors, anti-inflammatory's, diuretics, muscle relaxants, local anaesthetics, hormones, cholesterol lowering drugs, anti-coagulants, anti-depressants, tranquilizers, neuroleptics, analgesics such as a narcotic or anti-pyretic analgesics, anti-virals, anti-bacterials, anti-fungals, bacteriostats, CNS active agents, anticonvulsants, anxiolytics, antacids, narcotics, antibiotics, respiratory agents, antihistamines, immunosuppressants, immunoactivating agents, nutritional additives, anti-tussives, diagnostic agents, emetics and anti-emetics, carbohydrates, glycosoaminoglycans,
- the cargo moiety is selected from a protein, a peptide, an antibody and a drug.
- the cargo moiety is protein A, a bacterially derived protein that binds strongly to conventional antibodies.
- the compound of formula I is linked to commercially available (natural) protein A via a lysine NH 2 group of protein A.
- a cysteine residue may be engineered into the protein to allow conjugation to said compound of formula I. Further details on the preparation of cysteine modified proteins may be found in Neisler et al [Bioconjugate Chem. 2002, 13, 729-736].
- the cargo moiety is covalently attached to the L group of the compound of formula I.
- a reactive group in the cargo moiety reacts with a reactive group in the L moiety of the compound of formula (I).
- a nucleophilic group on the cargo moiety for example an amine, thiol or hydroxy group
- a nucleophilic group on the cargo moiety for example an amine, thiol or hydroxy group
- a thiol-containing protein eg geminin
- Q 1 or Q 2 is -S-S-(2-pyridyl) via thiol exchange.
- a nucleophilic moiety such as the 2'-hydroxy group on the taxol or docetaxel molecule can displace a succinimyl, -S-S-(2-pyridyl), iodine, -S-S(O) 2 -OMe or -CO-O-N-succininyl group in the moiety L in formula (I).
- L when L is nucleophilic (eg when L is -NH 2 ), it can react in step (iii) with an electrophilic site in the cargo moiety.
- the cargo moiety is directly linked to the carrier moiety.
- the cargo moiety is indirectly linked to the carrier moiety by means of a linker moiety.
- the cargo moiety comprises a protein, a peptide, an oligonucleotide, a nucleotide, a diagnostic agent, a biologically active compound or a drug which is attached to a linker moiety.
- a reactive site on the linker group reacts with the moiety L in the formula (I) as explained above.
- Direct linkage may occur through any convenient functional group on the cargo moiety, such as a hydroxy, carboxy or amino group. Indirect linkage will occur through a linking moiety.
- Suitable linking moieties include bi- and multi-functional alkyl, aryl, aralkyl or peptidic moieties, alkyl, aryl or aralkyl aldehydes acids esters and anhydrides, sulphydryl or carboxyl groups, such as maleimido benzoic acid derivatives, maleimido proprionic acid derivatives and succinimido derivatives or may be derived from cyanuric bromide or chloride, carbonyldiimidazole, succinimidyl esters or sulphonic halides and the like.
- the functional group on the linker moiety used to form covalent bonds between the compound of formula I and the cargo moiety may be, for example, amino, hydrazino, hydroxyl, thiol, maleimido, carbonyl, and carboxyl groups, etc.
- the linker moiety may include a short sequence of from 1 to 4 amino acid residues that optionally includes a cysteine residue through which the linker moiety bonds to the compound of formula I.
- the compound of formula I and the cargo moiety may be linked by leucine zippers, dimerisation domains, or an avidin/biotin linker.
- the cargo moiety is selected from a recombinant antibody, a Fab fragment, a F(ab') 2 fragment, a single chain Fv, a diabody, a disulfide linked Fv, a single antibody domain and a CDR.
- CDR complementary determining region
- the antibody may be selected from Herceptin, Rituxan, Theragyn (Pemtumomab), Infliximab, Zenapex, Panorex, Vitaxin, Protovir, EGFRl or MFE-23.
- the cargo moiety is a genetically engineered fragment selected from a Fab fragment, a F(ab') 2 fragment, a single chain Fv, or any other antibody- derived format.
- Fab fragment refers to a protein fragment obtained (together with Fc and Fc 1 fragments) by papain hydrolysis of an immunoglobulin molecule. It consists of one intact light chain linked by a disulfide bond to the N-terminal part of the contiguous heavy chain (the Fd fragment). Two Fab fragments are obtained from each immunoglobulin molecule, each fragment containing one binding site. In the context of the present invention, the Fab fragment may be prepared by gene expression of the relevant DNA sequences.
- F(ab') 2 fragment refers to a protein fragment obtained (together with the pFc' fragment) by pepsin hydrolysis of an immunoglobulin molecule. It consists of that part of the immunoglobulin molecule N-terminal to the site of pepsin attack and contains both Fab fragments held together by disulfide bonds in a short section of the Fc fragment (the hinge region).
- One F(ab') 2 fragment is obtained from_each immunoglobulin molecule; it contains two antigen binding sites, but not the site for complement fixation.
- the F(ab') 2 fragment may be prepared by gene expression of the relevant DNA sequences.
- Fv fragment refers to the N-terminal part of the Fab fragment of an immunoglobulin molecule, consisting of the variable portions of one light chain and one heavy chain.
- Single-chain Fvs (about 30 KDa) are artificial binding molecules derived from whole antibodies, but which contain the minimal part required to recognise antigen.
- the cargo moiety is a synthetic or natural peptide, a growth factor, a hormone, a peptide ligand, a carbohydrate or a lipid.
- the cargo moiety can be designed or selected from a combinatorial library to bind with high affinity and specificity to a target antigen. Typical affinities are in the 10 "6 to 10 ⁇ 15 M K d range.
- Functional amino acid residues present in the cargo moiety may be altered by site-directed mutagenesis where possible, without altering the properties of the cargo moiety. Examples of such changes include mutating any free surface thiol-containing residues (cysteine) to serines or alanines, altering lysines and arginines to asparagines and histidines, and altering serines to alanines.
- the cargo moiety is a drug.
- the conjugate can be described as a delivery system.
- the delivery system is therapeutically active in its intact state.
- the drug moiety is derived from a cytotoxic drug.
- the drug moiety is selected from DNA damaging agents, anti-metabolites, anti-tumour antibiotics, natural products and their analogues, dihydrofolate reductase inhibitors, pyrimidine analogues, purine analogues, cyclin-dependent kinase inhibitors, thymidylate synthase inhibitors, DNA intercalators, DNA cleavers, topoisomerase inhibitors, anthracyclines, vinca drugs, mitomycins, bleomycins, cytotoxic nucleosides, pteridine drugs, diynenes, podophyllotoxins, platinum containing drugs, differentiation inducers and taxanes.
- the drug moiety is selected from methotrexate, methopterin, dichloromethotrexate, 5-fluorouracil, 6-mercaptopurine, tri-substituted purines such as olomoucine, roscovitine and bohemine, flavopiridol, staurosporin, cytosine arabinoside, melphalan, leurosine, actinomycin, daunorubicin, doxorubicin, mitomycin D, mitomycin A, carninomycin, aminopterin, tallysomycin, podophyllotoxin (and derivatives thereof), etoposide, cisplatinum, carboplatinum, vinblastine, vincristine, vindesin, paclitaxel, docetaxel, taxotere retinoic acid, butyric acid, acetyl spermidine, tamoxifen, irinotecan and camptothecin.
- purines
- the drug moiety is directly linked to the carrier moiety.
- the drug moiety is indirectly linked to the carrier moiety by means of a linker moiety.
- each carrier moiety bears more than one drug moiety.
- each carrier moiety bears more than one drug moiety, the drug moieties are different.
- each drug moiety is linked to the carrier moiety by way of a linker moiety
- each drug moiety is linked to the carrier moiety by an identical linker moiety.
- each drug moiety is linked to the carrier moiety by a different linker moiety.
- the delivery system may further comprise a targeting moiety.
- the targeting moiety is capable of directing the delivery system to the specific cell type to which it is preferable for the drug moiety to function.
- the targeting moiety acts as an address system biasing the body's natural distribution of drugs or the delivery system to a particular cell type.
- the targeting moiety may be attached to the drug moiety or alternatively to the carrier moiety.
- the targeting moiety is directly linked to the carrier moiety.
- the targeting moiety is indirectly linked to the carrier moiety by means of a linker moiety.
- Direct linkage may occur through any convenient functional group on the targeting moiety, such as a hydroxy, carboxy or amino group. Indirect linkage will occur through a linking moiety.
- Suitable linking moieties include bi- and multi-functional alkyl, aryl, aralkyl or peptidic moieties, alkyl, aryl or aralkyl aldehydes acids esters and anhydrides, sulphydryl or carboxyl groups, such as maleimido benzoic acid derivatives, maleimido proprionic acid derivatives and succinimido derivatives or may be derived from cyanuric bromide or chloride, carbonyldiimidazole, succinimidyl esters or sulphonic halides and the like.
- the functional groups on the linker moiety used to form covalent bonds to the targeting moiety may be two or more of, e.g., amino, hydrazino, hydroxyl, thiol, maleimido, carbonyl, and carboxyl groups, etc.
- the linker moiety may include a short sequence of from 1 to 4 amino acid residues that optionally includes a cysteine residue through which the linker moiety bonds to the targeting moiety.
- the targeting moiety may be linked by leucine zippers, dimerisation domains, or an avidin/biotin linker.
- the present invention also provides a conjugate obtainable by reacting a compound of formula (I), as defined above, in which r is 1 or 2 and the phenyl ring which carries L has no substituents ortho to the L moiety, or a compound of formula Via, VIb, VIc, VId, VIe or VII, as defined above, with a cargo moiety as defined above, or a pharmaceutically acceptable salt of said conjugate.
- Another aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising a compound or conjugate of the invention admixed with one or more pharmaceutically acceptable diluents, excipients or carriers.
- the compounds and conjugates of the present invention can be administered alone, they will generally be administered in admixture with a pharmaceutical carrier, excipient or diluent, particularly for human therapy.
- the pharmaceutical compositions may be for human or animal usage in human and veterinary medicine.
- Acceptable carriers or diluents for therapeutic use are well known in the pharmaceutical art, and are described, for example, in Remington's Pharmaceutical Sciences, Mack Publishing Co. (A. R. Gennaro edit. 1985).
- suitable carriers include lactose, starch, glucose, methyl cellulose, magnesium stearate, mannitol, sorbitol and the like.
- suitable diluents include ethanol, glycerol and water.
- compositions may comprise as, or in addition to, the carrier, excipient or diluent any suitable binder(s), lubricant(s), suspending agent(s), coating agent(s), solubilising agent(s).
- Suitable binders include starch, gelatin, natural sugars such as glucose, anhydrous lactose, free-flow lactose, beta-lactose, corn sweeteners, natural and synthetic gums, such as acacia, tragacanth or sodium alginate, carboxymethyl cellulose and polyethylene glycol.
- Suitable lubricants include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the like.
- Preservatives, stabilizers, dyes and even flavoring agents may be provided in the pharmaceutical composition.
- preservatives include sodium benzoate, sorbic acid and esters of p-hydroxybenzoic acid.
- Antioxidants and suspending agents may be also used.
- the compounds of the invention can be present as salts or esters, in particular pharmaceutically acceptable salts or esters.
- salts of the compounds of the invention include suitable acid addition or base salts thereof.
- suitable pharmaceutical salts may be found in Berge et al, J Pharm Sci, 66, 1-19 (1977). Salts are formed, for example with strong inorganic acids such as mineral acids, e.g.
- sulphuric acid, phosphoric acid or hydrohalic acids with strong organic carboxylic acids, such as alkanecarboxylic acids of 1 to 4 carbon atoms which are unsubstituted or substituted (e.g., by halogen), such as acetic acid; with saturated or unsaturated dicarboxylic acids, for example oxalic, malonic, succinic, maleic, fumaric, phthalic or tetraphthalic; with hydroxycarboxylic acids, for example ascorbic, glycolic, lactic, malic, tartaric or citric acid; with aminoacids, for example aspartic or glutamic acid; with benzoic acid; or with organic sulfonic acids, such as (C 1 -C 4 )-alkyl- or aryl-sulfonic acids which are unsubstituted or substituted (for example, by a halogen) such as methane- or p-toluene sulfonic acid.
- Esters are formed either using organic acids or alcohols/hydroxides, depending on the functional group being esterif ⁇ ed.
- Organic acids include carboxylic acids, such as alkanecarboxylic acids of 1 to 12 carbon atoms which are unsubstituted or substituted (e.g., by halogen), such as acetic acid; with saturated or unsaturated dicarboxylic acid, for example oxalic, malonic, succinic, maleic, fumaric, phthalic or tetraphthalic; with hydroxycarboxylic acids, for example ascorbic, glycolic, lactic, malic, tartaric or citric acid; with aminoacids, for example aspartic or glutamic acid; with benzoic acid; or with organic sulfonic acids, such as (d-C 4 )-alkyl- or aryl-sulfonic acids which are unsubstituted or substituted (for example, by a halogen) such as methane- or p-
- Suitable hydroxides include inorganic hydroxides, such as sodium hydroxide, potassium hydroxide, calcium hydroxide, aluminium hydroxide.
- Alcohols include alkanealcohols of 1- 12 carbon atoms which may be unsubstituted or substituted, e.g. by a halogen).
- the invention includes, where appropriate all enantiomers and tautomers of compounds of the invention.
- the man skilled in the art will recognise compounds that possess optical properties (one or more chiral carbon atoms) or tautomeric characteristics.
- the corresponding enantiomers and/or tautomers may be isolated/prepared by methods known in the art.
- Some of the compounds of the invention may exist as stereoisomers and/or geometric isomers — e.g. they may possess one or more asymmetric and/or geometric centres and so may exist in two or more stereoisomeric and/or geometric forms.
- the present invention contemplates the use of all the individual stereoisomers and geometric isomers of those compounds, and mixtures thereof.
- the terms used in the claims encompass these forms, provided said forms retain the appropriate functional activity (though not necessarily to the same degree).
- the present invention also includes all suitable isotopic variations of the compound or pharmaceutically acceptable salt thereof.
- An isotopic variation of a compound of the present invention or a pharmaceutically acceptable salt thereof is defined as one in which at least one atom is replaced by an atom having the same atomic number but an atomic mass different from the atomic mass usually found in nature.
- isotopes that can be incorporated into the agent and pharmaceutically acceptable salts thereof include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulphur, fluorine and chlorine such as 2 H, 3 H, 13 C, 14 C, 15 N, 17 0, 18 0, 31 P, 32 P, 35 S, 18 F and 36 Cl, respectively.
- isotopic variations of the agent and pharmaceutically acceptable salts thereof are useful in drug and/or substrate tissue distribution studies. Tritiated, i.e., 3 H, and carbon-14, i.e., 14 C, isotopes are particularly preferred for their ease of preparation and detectability. Further, substitution with isotopes such as deuterium, i.e., 2 H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements and hence may be preferred in some circumstances. Isotopic variations of the agent of the present invention and pharmaceutically acceptable salts thereof of this invention can generally be prepared by conventional procedures using appropriate isotopic variations of suitable reagents.
- the present invention also includes the use of solvate forms of the compounds of the present invention.
- the terms used in the claims encompass these forms.
- the invention furthermore relates to the compounds and/or conjugates of the present invention in their various crystalline forms, polymorphic forms and (an)hydrous forms. It is well established within the pharmaceutical industry that chemical compounds may be isolated in any of such forms by slightly varying the method of purification and or isolation form the solvents used in the synthetic preparation of such compounds.
- the invention further includes the compounds of the present invention in prodrug form.
- prodrugs are generally compounds of the invention wherein one or more appropriate groups have been modified such that the modification may be reversed upon administration to a human or mammalian subject.
- Such reversion is usually performed by an enzyme naturally present in such subject, though it is possible for a second agent to be administered together with such a prodrug in order to perform the reversion in vivo.
- Examples of such modifications include ester (for example, any of those described above), wherein the reversion may be carried out be an esterase etc.
- Other such systems will be well known to those skilled in the art.
- compositions of the present invention may be adapted for oral, rectal, vaginal, parenteral, intramuscular, intraperitoneal, intraarterial, intrathecal, intrabronchial, subcutaneous, intradermal, intravenous, nasal, buccal or sublingual routes of administration.
- parenteral intramuscular, intraperitoneal, intraarterial, intrathecal, intrabronchial, subcutaneous, intradermal, intravenous, nasal, buccal or sublingual routes of administration.
- intramuscular intraperitoneal
- intraarterial intrathecal
- intrabronchial subcutaneous, intradermal, intravenous, nasal, buccal or sublingual routes of administration.
- these compositions contain from 1 to 250 mg and more preferably from 10-100 mg, of active ingredient per dose.
- compositions of the present invention may also be in form of suppositories, pessaries, suspensions, emulsions, lotions, ointments, creams, gels, sprays, solutions or dusting powders.
- the active ingredient can be incorporated into a cream consisting of an aqueous emulsion of polyethylene glycols or liquid paraffin.
- the active ingredient can also be incorporated, at a concentration of between 1 and 10% by weight, into an ointment consisting of a white wax or white soft paraffin base together with such stabilisers and preservatives as may be required.
- Injectable forms may contain between 10-1000 mg, preferably between 10-250 mg, of active ingredient per dose.
- compositions may be formulated in unit dosage form, i.e., in the form of discrete portions containing a unit dose, or a multiple or sub-unit of a unit dose.
- a person of ordinary skill in the art can easily determine an appropriate dose of one of the compositions of the invention to administer to a subject without undue experimentation.
- a physician will determine the actual dosage which will be most suitable for an individual patient and it will depend on a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the individual undergoing therapy.
- the dosages disclosed herein are exemplary of the average case. There can of course be individual instances where higher or lower dosage ranges are merited, and such are within the scope of this invention.
- the agent may be administered at a dose of from 0.01 to 30 mg/kg body weight, such as from 0.1 to 10 mg/kg, more preferably from 0.1 to 1 mg/kg body weight, hi an exemplary embodiment, one or more doses of 10 to 150 mg/day will be administered to the patient.
- the one or more compounds and/or conjugates of the invention are administered in combination with one or more other therapeutically active agents, for example, existing drugs available on the market.
- the compounds of the invention may be administered consecutively, simultaneously or sequentially with the one or more other therapeutically active agents.
- Drugs in general are more effective when used in combination.
- combination therapy is desirable in order to avoid an overlap of major toxicities, mechanism of action and resistance mechanism(s).
- the major advantages of combining chemotherapeutic drugs are that it may promote additive or possible synergistic effects through biochemical interactions and also may decrease the emergence of resistance in cells which would have been otherwise responsive to initial chemotherapy with a single agent.
- the present invention also provides use of a compound or conjugate of the invention in the manufacture of a medicament for use delivering a drug to a patient transdermally.
- the present invention also provides a skin patch which comprises a compound or conjugate of the invention and a pharmaceutically acceptable carrier or diluent.
- Figure 1 shows the cellular uptake, via live microscopy, of three compounds of the invention conjugated to Geminin labelled with AlexFluor 488 (11-g * , 33-g * and 35-g * ) in two cell lines (Wl -38 human diploid fibroblast and human U2OS osteosarcoma cells).
- Figure 2 shows the results of cellular uptake monitored by FACS analysis of eight compounds of the invention conjugated to Geminin labelled with AlexFluor 488 (23-g , 26- g * , 18-g * , 20-g ⁇ 33-g * , 31-g * , 35-g * and 11-g * ) in two cell lines (Wl-38 HDF and U2OS).
- Figure 3 shows the results of a cell proliferation assay monitored via confocal fluorescence microscopy of 3 compounds of the invention coupled to 15 Geminin in Wl-38 and U2OS cell lines.
- n-BuLi 1.6M in hexane 75 ml, 120 mmol was added dropwise at 0 0 C to a solution of veratrole (3.3 g, 24 mmol) and TMEDA (22.5 ml, 150 mmol) in anhydrous ether (250 ml) under nitrogen and stirred at room temperature for 3 days.
- the reaction mixture was cooled to -78 0 C and Br 2 (7.5mL, 147 mmol) was added, after stirring for further day at room temperature.
- the reaction mixture was diluted with ether (150 mL) and washed with water (150 mL), IN HCl (150 mL ⁇ 2) and brine (150 mL), dried over Na 2 SO 4.
- n-BuLi 1.6M in hexane 70 mL, 112 mmol was added dropwise at 0 0 C to a solution of veratrole (10 g, 72.3 mmol) and TMEDA (10.9 mL, 72.3 mmol) in anhydrous ether (50 mL) under nitrogen and stirred at room temperature for 2 h.
- the reaction mixture was cooled to -78 0 C and (CBrCl 2 ) 2 (31.2 g, 112 mmol) was added, after stirring for a further 10 min, the cooling bath was removed and the reaction vessel allowed to warm to room temperature.
- reaction mixture was diluted with ether (50 mL), washed with water (50 mL), IN HCl (2 x 50 mL), brine (50 mL) and dried over Na 2 SO 4.
- the solvent was removed under vacuum and the crude product was purified by silica gel flash chromatography using hexane/DCM as eluent (5/1) to afford l-bromo-2,3-dimethoxy-benzene as a colourless oil (12.1 g, 77% yield).
- the l-bromo-2,3-dimethoxybenzene (2.17 g, 10 mmol) was dissolved in DCM (60 ml) and treated with 1.0 M DCM solution OfBBr 3 (15 mL, 15 mmol) at 0 °C and then allowed to warm to room temperature. After stirring overnight, the reaction mixture was cooled to 0 0 C, 2 mL of MeOH was added, and the solvent was removed under vacuum. The residue was dissolved in 20 mL of EtOAc and washed with IN HCl (2 x 50 mL), water, brine and dried over Na 2 SO 4 . The solvent was removed under vacuum.
- Pd(dppf)Cl 2 (1.5 g, 1.9 mmol), potassium acetate (5.4 g, 56 mmol) and bispinacolatodiboran (10 g, 41 mmol) were weighted in a three necked round bottom flask and DMSO (100 ml) was added. After stirring for 2 minutes, dibenzyl 2,2'-(3-bromo-l,2- phenylene)bis(oxy)bis(ethane-2,l-diyl)dicarbamate (20 g, 37 mmol) was added. The reaction mixture was heated to 80°C for 72 h. After cooling the mixture at room temperature, 1 L of water and 0.5 L of toluene were added.
- a degassed mixture of dibenzyl 2,2'-(3-bromo-6-cyano-l,2-phenylene) bis(oxy)bis(ethane- 2,l-diyl)dicarbamate (0.3 g, 0.5 mmol), dibenzyl 2,2 I -(3-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-l,2-phenylene)bis(oxy)bis(ethane-2,l-diyl)dicarbamate (0.35 g, 0.6 mmol), PdCl 2 dppf.
- the reaction mixture was stirred at 82 °C for 18 h.
- the reaction mixture was diluted with IM HCl (aq) (50 mL) and the product extracted with DCM (3 x 200 mL). The combined organic fractions were dried over MgSO 4 , filtered and concentrated under reduced pressure.
- the product was purified by flash chromatography using a gradient of 0- 50% EtOAc in cyclohexane over 10 CV to afford the title product (530 mg, 79%) as a white foam.
- N-succinimidyl-3-(2-pyridyldithio)propionate 22 mg, 1 eq
- the solvent was removed under vacuum and the crude oil was purified by preparative TLC (ethyl acetate/hexane 1/1) to afford the title compound (13 mg, 11% yield).
- Dibenzyl 2,2'-(4-cyanobiphenyl-2,2'-diyl)bis(oxy)bis(ethane-2, 1 -diyl)dicarbamate (0.12 g, 0.21 mmol) was dissolved in DCM (10 niL) and HBr (30% in acetic acid, 1.2 mL) was added dropwise. After stirring at room temperature for 1.5 h, the solvent was removed under vacuum and the crude diamine was carefully dried under vacuum for several hours.
- N- ⁇ 2, 2'-Bis ⁇ 2 ⁇ [N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy ⁇ -biphenyl-4-ylmethyl ⁇ - 3-[2-pyridyl)dithio]propionamide (15 mg, 0.015 mmol) was dissolved in formic acid (1 mL) and stirred at room temperature overnight. The mixture was then heated at 50 0 C for
- Dibenzyl 2,2'-(4-cyanobiphenyl-2,3'-diyl)bis(oxy)bis(ethane-2,l-diyl)dicarbamate 0.362 g, 0.64 mmol was dissolved in DCM (10 mL) and HBr (30% in acetic acid, 3.5 mL) was added dropwise. After stirring at room temperature for 1.5 h, the solvent was removed under vacuum and the crude diamine was carefully dried under vacuum for several hours.
- Dibenzyl 2,2'-(4-cyanobiphenyl-3,2'-diyl)bis(oxy)bis(ethane-2,l-diyl)dicarbamate (0.21 g, 0.371 mmol) was dissolved in DCM (10 mL) and HBr (30% in acetic acid, 2 niL) was added dropwise. After stirring at room temperature for 1.5 h, the solvent was removed under vacuum and the crude diamine was carefully dried under vacuum for several hours.
- Dibenzyl 2,2'-(4-cyanobiphenyl-3,3 l -diyl)bis(oxy)bis(ethane-2,l-diyl)dicarbamate (0.15 g, 0.26 mmol) was dissolved in DCM (10 mL) and HBr (30% in acetic acid, 1.5 mL) was added dropwise. After stirring at room temperature for 1.5 h, the solvent was removed under vacuum and the crude diamine was carefully dried under vacuum for several hours.
- Dibenzyl 2,2'-(5-cyanobiphenyl-2,2'-diylbis(oxy))bis(ethane-2,l-diyl)tetracarbamate (0.9 g, 1.6 mmol) was dissolved in DCM (50 mL) and HBr (30% in acetic acid, 8 mL) was added dropwise. After stirring at room temperature for 1.5 h, water (25 mL) was added to the mixture, the layers were separated and the aqueous layer was washed with DCM (2 x 25 mL). The water was then removed under vacuum and the crude diamine was carefully dried under vacuum for several hours.
- N- ⁇ 2, 3'-Bis ⁇ 2-[N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy ⁇ -biphenyl-5- ylmethyl ⁇ -3-[2-pyridyl)dithio]propionamide (8.5 mg, 0.0086 mmol) was dissolved in a mixture of TFA/ H 2 O/triisopropylsilane 95/2.5/2.5 (1 mL) and stirred at room temperature for 3 h. The solvent was removed under vacuum to give the title compound as a colourless oil (8 mg, 100%).
- N- ⁇ 2-[5'-Cyano-2'-(2-phenylacetylamino-ethoxy)-biphenyl-3-yloxy]-ethyl ⁇ -2-phenyl- acetamide (1.7 g, 3 mmol) was dissolved in DCM (25 mL) and HBr (30% in acetic acid, 4.0 mL) was added dropwise. After stirring at room temperature for 1.5 h, water (25 mL) was added to the mixture, the layers were separated and the aqueous layer was washed with DCM (2 x 25 mL). The water was then removed under vacuum and the crude diamine was carefully dried under vacuum for several hours.
- Raney Nikel (0.5 g). The mixture was stirred vigorously under H 2 atmosphere for 16 h. The catalyst was filtered off and washed several times with MeOH. Diethylenetriamine (leq) was added and the mixture stirred for 30 minutes then concentrated under vacuum. The residue was dissolved in EtOAc (10 mL) and washed with sat. aq. NaHCO 3 (10 mL). The organic layer was dried over MgSO 4 , filtered and concentrated under vacuum. The residue was then purified by flash chromatography using DCM/MeOH (97/3) as eluent to afford the title compound (0.213 g, 39% yield).
- N-succinimidyl-3-(2-pyridyldithio)propionate 13 mg, 0.045 mmol
- the solvent was removed under vacuum and the crude oil was purified by preparative TLC (ethyl acetate/hexane 35/75) to afford the title compound (35 mg, 60 % yield).
- N- ⁇ 2, 3 '-Bis ⁇ 2-[N, N'-bis ⁇ ert-butoxycarbony ⁇ guanidinoJ-ethyloxy ⁇ -biphenyl-5- ylmethyl ⁇ -3-[2-pyridyl)dithio]propionamide (6 mg) was dissolved in a mixture of TFA/ H 2 O/triisopropylsilane 95/2.5/2.5 (1 mL) and stirred at room temperature for 3 h. The solvent was removed under vacuum to give the title compound as colourless oil (4 mg, 60%).
- Geminin and 15k were expressed and purified according to the method described in R. V. Stevens, G.S. Bisacchi, Journal of Organic Chemistry 1982, 47, 2396-2399, the entirety of which is incorporated herein by reference.
- Geminin was conjugated with Alexa Fluor 488 using a Molecular Probes Protein Labelling kit, according to the manufacturer's protocol (Invitrogen). Coupling reactions of proteins with different SMOCs have been described in Okuyama M. et al, Nat. Methods 2007, 4, 153-159, the entirety of which is incorporated herein by reference.
- Human U2OS and WI-38 human diploid fibroblasts were cultured in DMEM supplemented with 10% FCS, 100 LVmL penicillin and 0.1 mg/mL streptomycin.
- Coverslips were washed extensively in PBS, placed in a plate containing medium without Red Phenol (Gibco) and observed by live confocal fluorescence microscopy (MP-UV, Leica Microsystems GmbH, Wetzlar, Germany) using 40 ⁇ and 63 x water immersion objectives, hi order to obtain similar fluorescence intensities, WI-38, and U2OS cells required incubation with the protein conjugate for 1 and 5 hours respectively.
- Figure 2 shows the mean value of fluorescence obtained from this experiment in both cell lines.
- Cells treated with labelled Geminin conjugated to compounds of the invention show a clear shift in the FACS profile towards higher FLlH values compared to the controls, revealing that proteins conjugated to compounds of the invention can enter efficiently into the cells (approximately 100% of U2OS cells show uptake, a range between 75 and 90% in the case of WI-38 HDF).
- Both cell lines showed similar trend which means that the relative efficiency of compounds of the invention was identical in normal and cancer cells.
- the fluorescence was more intense in WI-38 HDF meaning that the compounds of the invention were more efficient carriers in this cell line.
- Examples 9, 11, 5, 7, 17, 14, 20 and 3 were coupled to Geminin labelled with AlexaFluor488, giving compounds 23-g*, 26-g*, 18-g*, 20-g*, 33- g*, 31-g*, 35-g* and 11-g*, respectively.
- the first six bars of the chart shown in Figure 2 represent the uptake obtained using bis- guanidines (23-g*, 26-g*, 18-g*, 20-g*, 33-g* and 31-g*) as carrier, the seventh and eighth bar represent the data for the tris-guanidine compound 35-g* and the tetra-guanidine compound 11-g* respectively. It appears that when more guanidine moieties are present on the carrier, its efficiency is higher. Using the data obtained from the bis-guanidine, it was possible to make a correlation between the position of the guanidine moiety and the linker moiety and the ability of the compounds to carry a biomolecule.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biophysics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
A process for the production of a compound of formula (I), or a pharmaceutically acceptable salt thereof, Formula (I) which process comprises:(a) coupling a compound of formula (II) to a compound of formula (III) to form a compound of formula (IV).
Description
PROCESS FOR THE PREPARATION OF GUANIDINO SUBSTITUTED BI- AND POLYPHENYLS THAT
ARE SUITABLE AS SMALL MOLECULE CARRIERS
The present invention relates to a new synthetic route to small molecule carriers (SMCs) 5 and also to some new small molecule carriers. More specifically, the invention relates to new routes to SMCs that are useful for the in vitro and in vivo delivery of various cargo moieties into cells. It also relates to some new SMC products, which are particularly effective in achieving the delivery of cargo moieties into cells. 0 Over recent years, studies have shown that a variety of peptides, many of which are present in viral proteins, have the ability to cross biological membranes in various different cell types. These peptides, known as "protein transduction domains" (PTDs), can be linked to a wide variety of molecules with limited ability to cross membranes, (e.g., peptides, proteins, DNA), thereby enabling them to traverse biological membranes. Studies have shown that5 PTD fusion molecules introduced into mice exhibit delivery to all tissues, including the traversal of the blood-brain barrier [Schwarze, SR., Dowdy, SF., Trends Pharmacol. Sd, 2000, 21, 45]. Similar basic peptides are known to have anti-bacterial activity against MDR forms. 0 Most therapeutic drugs are limited to a relatively narrow range of physical properties. By way of example, they must be sufficiently polar for administration and distribution, but sufficiently non-polar so as to allow passive diffusion through the relatively non-polar bilayer of the cell. As a consequence, many promising drug candidates (including many peptide drugs) fail to advance clinically because they fall outside of this range, proving to5 be either too non-polar for administration and distribution, or too polar for passive cellular entry. A novel approach to circumvent this problem is to covalently tether these potential drugs to PTDs. However, it is very costly and time consuming to prepare such peptide- PTDs and their peptide structure often renders them susceptible to rapid degradation by cellular enzymes. 0
One solution to this problem is to use small molecule carriers (SMCs or "molecular tugs") that are more amenable than peptide-PTDs due to their in vivo stability by virtue of their resistance to cellular enzymes that degrade peptides.
WO 2005/123676, which is incorporated herein by reference, describes SMC compounds and a process for their production. However, the synthesis described is laborious and is not suitable for large-scale preparations.
The inventors have now found a new convergent synthesis for the compounds disclosed in WO 2005/123676. This new route enables an improvement in the overall yield and also allows easy synthesis of a range of different SMC compounds. The invention also relates to a number of new SMC compounds.
The present invention therefore provides a process for the of a compound of formula I, or a pharmaceutically acceptable salt thereof,

I wherein
Xl, X2 and X3 are each independently
NR5 N NR1 NR 3R, where Y is an alkylene, alkenylene or alkynylene group, each of which may be optionally substituted with one or more substituents selected from alkyl, halo, CF3, OH, alkoxy, NH2, CN, NO2 and COOH; W is absent or is O, S or NH;
R1, R2, R3 and R4 are each independently selected from H, alkyl, aryl and a protecting group
Pi ;
R7, Rg and R9 are each independently selected from H, alkyl, halo, CF3, OH, alkoxy, NH2, CN, NO2 and COOH; q and r are each independently 1, 2, 3 or 4; q1 and r' are each independently 0, 1, 2 or 3, where q + q1 and r + r' each equal 4; p is 1, 2, 3, 4 or 5, and p' is 0, 1, 2, 3 or 4, where p + p1 is 5; n is 0, 1, 2, 3, 4, 5 or 6; and
L is (Z)mNR5R6 wherein Z is a hydrocarbyl group and m is 0 or 1 wherein R5 and R6 are each independently H, CO(CH2)JQ1 or
 where j and k are each independently 0, 1, 2, 3, 4 or 5, and Q1 and Q2 are each independently selected from COOH, a chromophore,
where j and k are each independently 0, 1, 2, 3, 4 or 5, and Q1 and Q2 are each independently selected from COOH, a chromophore,

or R5, R6 and the nitrogen to which they are attached together form

(a) coupling a compound of formula II to a compound of formula III to form a compound of formula IV

IV
wherein: n, p, p', q, q', r and r' are defined as above; at least one X1 1, X2 1 and X3 1 moiety is -W-Y-NR1R10 or -W-Y-NR1 -C(=NR2)-NR3R4 and the other X1', X2' and X3' moieties are each independently selected from OH, SH, NH2, -W-Y-NR1R10, and -W-Y-NR1 -C(=NR2)-NR3R4 wherein R1, W and Y are defined as above and R10 is H or a protecting group P2; one of Ji and J2 is a leaving group LG1, and the other is a boronic acid, a boronic ester, a borane group or a trihalogenoborate salt;
L"' is L, as defined above, a leaving group LG2, -COR18, -(Z)111NHR12, or a group which can be reduced to a moiety -(Z)mNH2, where Z and m are defined as above, R12 is a protecting group P3 and R18 is hydrogen or a C1-C4 alkyl group; and
(b) if any OfX1', X2' and X3 1 in the formula (IV) is other than -W-Y-NR1 -C(=NR2)- NR3R4, alkylating the X1 1, X2' and X3' moieties so that they each represent a group of formula
where W, Y, R1, R2, R3 and R4 are defined as above, and, if necessary, converting the moiety L'" to a moiety L as defined above, to obtain a compound of formula I.
Also provided is a compound of formula (I), as defined above, or a pharmaceutically acceptable salt thereof, wherein r is 1 or 2 and the phenyl ring which carries L has no substituents ortho to the L moiety.
Further provided is a compound of formula Via, VIb, VIc, VId or Vie, or a pharmaceutically acceptable salt thereof,

VId VIe
wherein X1, X3 and L are as defined above.
Also provided is a compound of formula VII, or a pharmaceutically acceptable salt thereof,

VII wherein:
X1, X3, R7, R9 and L are as defined above; r is 1, 2, 3 or 4 and r' is 0, 1, 2 or 3, wherein r + r' is 4; p is 1, 2, 3, 4 or 5 and p' is 0, 1, 2, 3 or 4, where p + p' is 5; and p + r is 3.
As used herein, the term "hydrocarbyl" refers to a saturated or unsaturated, straight-chain, branched, or cyclic group comprising at least C and H that may optionally comprise one or more other suitable substituents. Examples of such substituents may include halo, alkoxy, hydroxy, CF3, CN, amino, COOH, nitro or a cyclic group. In addition to the possibility of
the substituents being a cyclic group, a combination of substituents may form a cyclic group. If the hydrocarbyl group comprises more than one C then those carbons need not necessarily be linked to each other. For example, at least two of the carbons may be linked via a suitable element or group. Thus, the hydrocarbyl group may contain heteroatoms. Suitable heteroatoms will be apparent to those skilled in the art and include, for instance, sulphur, nitrogen, oxygen, phosphorus and silicon.
Preferably, the hydrocarbyl group is an aryl or alkyl group. Typically, the hydrocarbyl group is unsubstituted. More preferably, the hydrocarbyl group is an unsubstituted Ci-6 alkyl group.
As used herein, the term "alkoxy" includes both straight chain and branched alkoxy groups which may be substituted (mono- or poly-) or unsubstituted. Preferably, the alkoxy group is a C1-20 alkoxy group, more preferably a C1-15 alkoxy group, more preferably still a C1-I2 alkoxy group, more preferably still, a C1-6 alkoxy group, more preferably a Cj-3 alkoxy group. Particularly preferred alkoxy groups include, for example, methyoxy, ethyoxy, propoxy, isopropoxy, butoxy, isobutoxy, tert-butoxy, pentoxy and hexoxy. Suitable substituents include alkyl, halo, CF3, OH, alkoxy, NH2, CN, NO2 and COOH. Preferably, the alkoxy group is unsubstituted. More preferably, the alkoxy group is an unsubstituted C1-4 alkoxy group .
As used herein, the term "alkyl" includes both saturated straight chain and branched alkyl groups which may be substituted (mono- or poly-) or unsubstituted. Preferably, the alkyl group is a C1-2O alkyl group, more preferably a C1-I5, more preferably still a C1-I2 alkyl group, more preferably still, a Ci-6 alkyl group, more preferably a C1-3 alkyl group.
Particularly preferred alkyl groups include, for example, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, pentyl and hexyl. Suitable substituents include halo, CF3, OH, alkoxy, NH2, CN, NO2 and COOH. The term "alkylene" should be construed accordingly. Preferably, the alkyl group is unsubstituted. More preferably, the alkyl group is an unsubstituted C1-4 alkyl group.
As used herein, the term "aryl" refers to a substituted (mono- or poly-) or unsubstituted monoaromatic or polyaromatic system, wherein said polyaromatic system may be fused or unfused. Preferably, the term "aryl" includes groups having from 6 to 10 carbon atoms, e.g. phenyl, naphthyl etc. The term "aryl" is synonymous with the term "aromatic". Suitable substituents include alkyl, halo, CF3, OH, alkoxy, NH2, CN, NO2 and COOH. Preferably, an aryl group is unsubstituted or is substituted with 1, 2 or 3 substituents selected from C1-4 alkyl, halo, CF3, OH, NH2 groups. Typically, these substituents are themselves unsubstituted. Preferably, the aryl group is an optionally substituted phenyl group. More typically, the aryl group is an unsubstituted phenyl group.
As used herein, the term "alkenyl" refers to a group containing one or more carbon-carbon double bonds, which may be branched or unbranched, substituted (mono- or poly-) or unsubstituted. Preferably the alkenyl group is a C2-20 alkenyl group, more preferably a C2-15 alkenyl group, more preferably still a C2-12 alkenyl group, or preferably a C2-6 alkenyl group, more preferably a C2-3 alkenyl group. Suitable substituents include alkyl, halo, CF3, OH, alkoxy, NH2, CN, NO2 and COOH. The term "alkenylene" should be construed accordingly. Preferably, the alkenyl group is unsubstituted. More preferably, the alkenyl group is an unsubstituted C2-4 alkenyl group.
As used herein, the term "alkynyl" refers to a carbon chain containing one or more triple bonds, which may be branched or unbranched, and substituted (mono- or poly-) or unsubstituted. Preferably the alkynyl group is a C2-20 alkynyl group, more preferably a C2- 15 alkynyl group, more preferably still a C2-12 alkynyl group, or preferably a C2-6 alkynyl group or a C2-3 alkynyl group. Suitable substituents include alkyl, halo, CF3, OH, alkoxy, NH2, CN, NO2 and COOH. The term "alkynylene" should be construed accordingly. Preferably, the alkynyl group is unsubstituted. More preferably, the alkynyl group is an unsubstituted C2-4 alkynyl group.
As used herein, the term "chromophore" refers to any functional group that absorbs light, giving rise to colour. Typically, the term refers to a group of associated atoms which can exist in at least two states of energy, a ground state of relatively low energy and an excited state to which it may be raised by the absorption of light energy from a specified region of
the radiation spectrum. Often, the group of associated atoms contains delocalised electrons. The chromophore present in the compounds prepared by the process of the invention can be a conjugated Il system or a metal complex. Typically, a chromophore is a porphyrin, a polyene, a polyyne or a polyaryl. Preferably the chromophore is one of.

Protecting groups P1, P2 and P3 are protecting groups suitable for protecting a nitrogen atom. Many examples of such protecting groups are known to the person skilled in the art, for example those protecting groups mentioned in "Protecting Group Chemistry" Jeremy Robertson, OUP, 2000, which is incorporated herein by reference. Preferably P1, P2 and P3 are selected from benzyl, trityl, 9-phenylfluorenyl, benzydryl, fluorenyl, carbamate, benzylcarbamate (Cbz), t-butyl carbamate (Boc), 9-fluorenylmethyl carbamate (Fmoc), acetamide, p-toluenesulfonate (p-Ts), silyl and triisopropylsilyl (TIPS) groups.
Preferably, P1 is a Boc group. <
Preferably, P2 is a Cbz group.
Typically, P2 and P3 are different. More typically, P1, P2 and P3 are all different. Preferably, P2 and P3 are orthogonal. More preferably, P1 and P2 are orthogonal to P3.
Leaving group LG1 is typically any group that can undergo oxidative addition with Pd(O). Those of skill in the art will easily be able to select appropriate leaving groups. LGj is preferably a halogen, triflate (OTf), tosylate (OTs), N-hydroxysuccinimide (OSu) or a mesylate (OMs) group. LGi is more preferably halogen, most preferably bromide or iodide.
Leaving group LG2 is typically a leaving group suitable for an aryl cyanation reaction. Those of skill in the art will easily be able to select appropriate such leaving groups. LG2 is preferably a halogen, triflate (OTf), tosylate (OTs), N-hydroxysuccinimide (OSu) or a mesylate (OMs) group. LG2 is more preferably halogen, most preferably bromide.
Leaving group LG3 is typically a leaving group suitable for a nucleophilic substitution reaction at a saturated carbon centre. Those of skill in the art will easily be able to select appropriate leaving groups. LG3 is preferably a halogen, triflate (OTf), tosylate (OTs), N- hydroxysuccinimide (OSu) or a mesylate (OMs) group. LG3 is more preferably a OMs group.
Leaving group LG4 can be any leaving group suitable for a guanidinylation reaction. The skilled reader will appreciate that LG4 represents a moiety such that -NLG4 is a leaving group in guanidinylation reaction. A skilled chemist can easily select appropriate leaving groups in this regard. Thus, preferred LG4 groups include triflyl (Tf), tosyl (Ts) and mesyl (Ms) groups. LG4 is most preferably a triflyl group, such that -NLG4 represents -NTf.
Leaving group LG4' can be any leaving group suitable for a guanidinylation reaction. A skilled chemist can easily select appropriate leaving groups in this regard. LG4' is typically a halogen atom, triflate (OTf), tosylate (OTs), mesylate (OMs) or 1-pyrazole group, preferably a 1-pyrazole group.
Leaving group LG5 can be any leaving group suitable for a nucleophilic substitution reaction at a carbonyl or thiocarbonyl group. Those of skill in the art will easily be able to select appropriate leaving groups. LG5 is typically a halogen, triflate (OTf), tosylate (OTs), N-hydroxysuccinimide (OSu) or a mesylate (OMs) group. LG5 is preferably a OSu group.
Leaving group LG7 is typically a leaving group suitable for a nucleophilic substitution reaction at a saturated carbon centre. Those of skill in the art will easily be able to select appropriate leaving groups. LG7 is preferably a halogen, triflate (OTf), tosylate (OTs), N-
hydroxysuccinimide (OSu) or a mesylate (OMs) group. LG7 is more preferably a OMs group.
As used herein, the term "guanidinylating" refers to the process of forming a guanidine moiety -NR1-C(=NR2)-NR3R4. Typical reagents and conditions for this process are described in US 6,380,358, and Katritsky, et al ARKIVOC 2005 (iv) 49-87 both of which are incorporated herein by reference.
US 6,380,358 describes reagents and methods for the synthesis of organic molecules containing a (protected) guanidine group, hi particular it describes processes for the guanidinylation of amines.
Katritsky, et al describes recent advances in the development of guanylating reagents, which are defined as compounds forming a guanidine structure by a chemical transformation. This document describes a wide rantge of possible reagents for the preparation of guanidines, including thioureas, isothioureas, carbodiimides and cyanamides, pyrazole-1-carboximidamides, triflyl guanidines, ammoiminomethane-sulfonic and -sulfinic acids and benzotriazole and imidazole-containing reagents.
The coupling reaction between the compounds of formulae (II) and (III) is typically an organometallic coupling, preferably a coupling mediated by Pd, more preferably Pd(O). The coupling is typically a Stille, Suzuki, Negishi, Hiyama or Kumada coupling. Preferably, the coupling is a Suzuki coupling.
More preferably, the reaction between the compounds of formulae (II) and (III) is a Suzuki coupling, performed using a Pd(O) catalyst in the presence of a base, preferably a nucleophilic base, more preferably triethylamine. The Pd(O) catalyst is typically used in a catalytic amount. The reaction is typically carried out at elevated temperature (i.e. above room temperature), preferably between 75 and 1250C, more preferably at 82°C or 1000C, most preferably at 82°C. A reaction time of 1 to 18 hours is typically employed, hi some cases a reaction time of 1, 3 or 18 hours may be used. The reaction is typically heated
using microwave radiation or conventional heat sources. The reaction is typically carried out in an aqueous organic solvent, preferably aqueous toluene or aqueous isopropyl alcohol, more preferably aqueous isopropyl alcohol. The solvents used are preferably degassed prior to use. The reaction may be carried out by heating at 1000C a compound of formula II with a compound of formula III with PdCl2dppf. CH2Cl2 and potassium phosphate in aqueous toluene. Preferably, the reaction is carried out by heating at 82°C a compound of formula II with a compound of formula III with Pd Cl2dppf.CH2Cl2 and triethylamine in aqueous isopropyl alcohol for 18 hours.
In a preferred embodiment, X3' is -W-Y-NR1R10 or -W-Y-NR1-CC=NR2)^R3R4. More preferably, all OfX1 1, X2 1 and X3 1 are -W-Y-NR1R10 or -W-Y-NR1-CC=NRs)-NR3R4.
In a more preferred embodiment, X3 1 is -W-Y-NR1R10. More preferably, all OfX1', X2' and X3' are -W-Y-NR1R10.
A boronic acid group is a group of formula -B(OH)2.
Preferably, the boronic ester is -B(OR13)(OR14) or
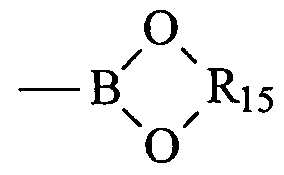
where R13 and Ri4 are each independently selected from C1-C6 alkyl groups and R15 is a C1-C6 alkyl or phenyl group. More preferably, the boronic ester is

where R15 is a -CH(CH3)2CH(CH3)2- group.
Preferably, the borane is BR16R17 where R16 and R17 are each independently selected from Ci-C6 alkyl groups.
A trihalogenoborate salt group is a group of formula -(B(HaI)3)TVI+, where each Hal group is the same or different and is independently chosen from halogen atoms. Halogen atoms are typically F, Cl, Br or I atoms, preferably F or Cl atoms, more preferably F atoms. In a preferred embodiment, each Hal group is the same, hi a more preferred embodiment, each Hal group is the same and is a fluorine atom.
The group M+ is any ionic group capable of acting as the counterion to the group - (B(HaI)3)", but is typically a monovalent metal ion, preferably a monovalent Group I metal ion. Monovalent Group I metal ions are typically Li+, Na+, K+ or Rb+, preferably Li+, Na+ or K+, more preferably K+.
Thus, in a particularly preferred embodiment, the group -(B(HaI)3)TvI+ is -(BF3)TC+.
hi a preferred embodiment, J1 is a boronic acid, a boronic ester or a borane group and J2 is a leaving group LG1. More preferably, J1 is a boronic acid or a boronic ester, more preferably a boronic acid or a compound of formula
O
-<o>5
wherein R15 is as defined above, and J2 is a halogen, for example bromine or iodine.
Most preferably, though, J, is a trihalogenoborate salt as defined above.
The compounds of formulae (II) and (III) are known compounds, or can be prepared by analogy with known methods.
For example, when J1 is a boronic acid, boronic ester, or a borane, compounds of formula II can be prepared by treating a compound of formula IX with a boronating agent in the presence of a catalyst.

(IX) (ii) wherein X1 1, R7, p and p' are defined as above, LG is a leaving group, for example bromine, and B is a boronic acid, boronic ester or borane moiety. The boronating agent can be a diborane, for example, bispinacolactodiborane. The catalyst is preferably Pd. Preferably, the reaction is carried out between 50 and 1000C, more preferably at 8O0C. A long reaction time of several days is typically employed, for example 72 hours. The reaction is typically carried out in an organic solvent, preferably dimethyl sulphoxide (DMSO). KOAc is typically present.
Further, when J1 is a trihalogenoborate salt group, compounds of formula II where Ji is a trihalogenoborate salt group can be prepared by treating compounds of formula II where J1 is a boronic acid, boronic ester or borane group with a saturated solution OfMH(HaI)2, where M and Hal are as defined above,

(H) (U) wherein Xi', R7, p and p' are defined as above, B is a boronic acid, boronic ester or borane moiety and B' is a trihalogenoborate salt group. Preferably, the reaction is carried out at room temperature, which is typically between 18°C and 30°C, preferably 21°C. A reaction time of between 1 and 5 hours is typically employed, preferably 3 hours. The reaction is typically carried out in an organic solvent, preferably methanol. Compounds of formula II' may be prepared using similar methodology to that described in Skaff, et al, J.O.C., Vol. 70, No. 18, 2005, 7353-7363 or Eur. J. Org. Chem., 1999, 1875-1885, the entirety of which are incorporated herein by reference.
The compound of formula IX can also be prepared by known methods. For example compounds of formula IX wherein each X1' represents -W-Y-NR1Ri0 and W is O, S or NH can be obtained by reacting a compound of formula (X) in which each Xj" represents a hydroxy, thiol or amino group, with LG3-Y-N-R1Ri0.
Compounds of formula IX wherein each X1 1 represents -W-Y-NR1 -C(=NR2)-NR3R4 and W is O, S or NH can be obtained by reacting a compound of formula (X) in which each X1" represents a hydroxy, thiol or amino group with LG7-Y-NR1 -C(=NR2)-NR3R4. Alternatively, such compounds can be prepared from corresponding compounds of formula IX in which each X1 1 represents -W-Y-NR1R1O by deprotecting any protected amine groups on -W-Y-NR1 R10 moieties at the Xi' position, and guanidinylating the deprotected moieties.

(X) (IX)
The compound of formula X in which each X1" is hydroxy, thiol or amino can be generated by deprotecting a corresponding compound in which the or each X]" is a protected hydroxy, thiol or amino group. For example, a compound which carries an alkyl ether group at the or each X1" position can be converted to a compound of formula X in which the or each Xj" is hydroxy by reaction with BBr3.
Preferably, the reaction between the compound of formula X and LG3-Y-N-RiRi0 is carried out between 75 and 1250C, more preferably at 100°C. A reaction time of 1 to 5 hours can be employed, for example 2 hours. The reaction is typically carried out in an organic solvent, preferably dimethyl formamide (DMF).
Compounds of formula IX wherein each Xj' represents -W-Y-NRiR1O and W is absent can be obtained by a Sonogashira coupling reaction between a suitably chosen alkyne, for
example H(CsC)-Y-NR1R1O and a leaving group on the phenyl ring, followed by reduction of the C≡C group. Compounds of formula IX wherein each X1' represents -W-Y-NR1- C(=NR2)-NR3R4 and W is absent can be obtained by a Sonogashira coupling reaction between a suitably chosen alkyne, for example H(CsC)-Y-NR1 -C(=NR2)-NR3R4 and a leaving group on the phenyl ring, followed by reduction of the C≡C group. Alternatively, compounds of formula IX wherein each X1' represents -W-Y-NR1 -C(=NR2)-NR3R4 and W is absent can be obtained by a Sonogashira coupling reaction between a suitably chosen alkyne, for example H(CsC)-Y-NR1R10 and a leaving group on the phenyl ring, followed by reduction of the C≡C group, deprotecting any protected amine groups on -W-Y-NR1R10 moieties at the X1' position and guanidinylating the deprotected moieties.
When J2 is a leaving group LG (for example bromine) compounds of formula III in which each X3 represents -Y-N-NR1R10 and W is O, S or NH can be prepared by reacting a compound of formula XI, in which each X3" represents a hydroxy, thiol or amino group, with LG3-Y-N-R1R1O- Compounds of formula El in which X3' represents -Y-NR1-
C(=NR2)-NR3R4 and W is O, S or NH can be obtained by reacting a compound of formula (XI) in which each X3" represents a hydroxy, thiol or amino group with LG7-Y-NR1- C(=NR2)-NR3R4. Alternatively, such compounds can be prepared from corresponding compounds of formula (III) in which each X3' represents -W-Y-NR1R1O by deprotecting any protected amine groups on -W-Y-NR1R10 moieties at the X1' position, and guanidinylating the deprotected moieties.

(XI) m
The compound of formula XI in which each X3" is hydroxy, thiol or amino can be generated by deprotecting a corresponding compound in which the or each X3" is a protected hydroxy, thiol or amino group. For example, a compound which carries an alkyl
ether group at the or each X3" position can be converted to a compound of formula X in which the or each X3" is hydroxy by reaction with BBr3.
Preferably, the reaction between the compound of formula XI and LG3-Y-N-R1R1O is carried out between 50 and 1000C, more preferably at 8O0C. A reaction time of 24 hours is typically employed. The reaction is typically carried out in an organic solvent, preferably dimethyl formamide (DMF).
Compounds of formula III wherein each X3' represents -W-Y-NR1R10 and W is absent can be obtained by a Sonogashira coupling reaction between a suitably chosen alkyne, for example H(C≡C)- Y-NRj R10 and a leaving group on the phenyl ring, followed by reduction of the C≡C group. Compounds of formula III wherein each X3' represents -W-Y-NR1- C(=NR2)-NR3R4 and W is absent can be obtained by a Sonogashira coupling reaction between a suitably chosen alkyne, for example H(C=C)-Y-NR1 -C(=NR2)-NR3R4 and a leaving group on the phenyl ring, followed by reduction of the C≡C group. Alternatively, compounds of formula III wherein each X3' represents -W-Y-NR1 -C(=rNR2)-NR3R4 and W is absent can be obtained by a Sonogashira coupling reaction between a suitably chosen alkyne, for example H(CsC)-Y-NR1R10 and a leaving group on the phenyl ring, followed by reduction of the C≡C group, deprotecting any protected amine groups on -W-Y-NR1Ri0 moieties at the X3' position and guanidinylating the deprotected moieties.
A compound of formular (III) in which J2 is a trihalogenoborate salt group can be prepared by reacting a corresponding compound in which J2 is a boronic acid, boronic ester or borane group with a compound of formula MH(HaI)2, under the reaction conditions described above for the preparation of the compounds of formula (II).
A compound of formula (III) in which J2 is a boronic acid, boronic ester or borane group can be prepared by reacting a corresponding compound in which J2 is a leaving group with a boronating agent, under the reaction conditions described above for the preparation of the compounds of formula (II).
A compound of formula (111) or (XI) in which L'" is CN can be prepared, for example, from a corresponding compound in which L'" is a leaving group, for example iodine, by reaction with CN", preferably with zinc cyanide and Pd(H). Preferably, when J2 is a leaving group, no more than 0.5 equivalents of CN" are used in the reaction, to minimise formation of the dicyano compound.
Compounds of formula (IH) and (XI) in which L'" is -COR18 are commercially available or can be prepared using known methods.
In a further embodiment of the invention, J2 is a boronic acid, a boronic ester or a borane group or a trihalogenoborate salt group and J1 is a leaving group LG1.
In one embodiment, when any of the X1', X2' and X3' moieties in the formula (IV) are OH, SH or NH2, alkylation so that all X1', X2' and X3' moieties represent a group of formula -W- Y-NRI-C(=NR2)-NR3PM is effected by either:
(i) alkylating any X1', X2' and X3' moieties that are OH, SH or NH2 so that they represent -W-Y-NR]R1O, deprotecting any protected amine groups on -W-Y-NRiR10 moieties present at the X1', X2' and X3' positions and guanidinylating the deprotected moieties so that they each represent a group of formula -W-Y-NR1 -C(=NR2)-N NR3R4; or
(ii) alkylating any X1', X2' and X3' moieties that are OH, SH or NH2 so that they each represent a group of formula -W-Y-NRi -C(=NR2)-NR3R4, deprotecting any protected amine groups on -W-Y-NRiRi0 moieties present at the X]', X2' and X3' positions and guanidinylating the deprotected moieties so that they each represent a group of formula -W-Y-NR1 -C(=NR2)-NR3R4.
Preferably, the alkylation of hydroxy, thiol and amino groups at the X1', X2' and X3' positions in the formula IV so that they represent -W-Y-NRiRi0 is effected with a compound of formula LG3-Y-NRiRi0 where Ri, Y and LG3 are defined as above and Rio is H or a protecting group P2.
Preferably, R1O is a protecting group, for example Cbz. More preferably, R1 is H and R10 is a protecting group, for example Cbz.
Preferably, the alkylation of hydroxy, thiol and amino groups at the X1', X2' and X3' positions in the formula IV so that they represent -W-Y-NR1R10 is carried out at elevated temperature (i.e. above room temperature). Preferably, the reaction is carried out between 50 and 1000C, more preferably at 8O0C. A reaction time of 1 to 4 hours is typically employed. In some cases, reaction times of 1, 3, 3.5 and 4 hours may be used. The reaction is typically carried out in an organic solvent, preferably dimethylformamide (DMF). CsCO3 is typically added to the reaction mixture.
Preferably, the alkylation of any X1', X2' and X3 1 moieties that are OH, SH or NH2 so that they each represent a group of formula -W-Y-NR1 -C(=NR2)-NR3R_( is effected by a compound of formula LG7-Y-NR1 -Q=NRz)-NR3R4.
Deprotection of any amine groups in the -W-Y-NR1R1O moieties present at the X1', X2' and X3' positions can be carried out by standard techniques. The exact conditions employed will depend on the exact nature of the protecting group, hi the case where the amine groups in the -W-Y-NR1R1O moiety are protected with Cbz groups, the protecting groups can be removed with an acid, typically an anhydrous acid, preferably HBr and CH3COOH in DCM.
In another embodiment, all of the X1', X2' and X3' moieties are -W-Y-NR1R10 or -W-Y-NR1- C(=NR2)-NR3R4, and any necessary alkylation OfX1', X2' and X3' moieties so that they each represent -W-Y-NR1 -C(=NR2)-NR3R4 is effected by deprotecting any protected amine groups on -W-Y-NR1R10 moieties present at the X1', X2' and X3' positions and guanidinylating the deprotected moieties so that they represent a group of formula -W-Y- NR1-C(^NRz)-NR3R4.
Preferably, said guanidinylation of deprotected -W-Y-NRiR10 moieties at the X]', X2' and X3' positions is effected either by a compound of formula V, or a tautomer thereof, where R2, R3 and R4 are as defined in claim 1 and LG4 is a leaving group;

V
or by a compound of formula V, or a tautomer thereof,
R4R3N
^LG4 1 R2N
V
where R2, R3 and R4 are as defined above and LG4" is a leaving group.
More preferably, guanidinylation is effected with N,N-di-boc-N'-trifluoromethanesulfonyl guanidine or NjN'-Di-Boc-lH-pyrazole-l-carboxamidine, most preferably N,N'-Di-Boc- 1 H-pyrazole- 1 -carboxamidine.
The term tautomer is well known to the person skilled in the art and refers to the result of a migration of a hydrogen atom accompanied by the switch of a single bond and adjacent double bond. The skilled person is aware that tautomers of certain molecules, such as those represented by formulae V and V above exist in, for example, aqueous media.
Preferred tautomers of the compounds of formula (V) are represented by the formula
R4R3N LG4
R2N Typically the guanidinylation is carried out at room temperature. Alternatively, the reaction mixture may be heated. The reaction is typically left to run overnight (i.e. a time in excess of 12 hours, for example, 18 hours). The reaction is typically carried out in an organic solvent, preferably DCM and triethylamine or diisopropylethylamine (DIEA), more preferably DCM and DIEA.
In another embodiment at least one X1', X2' and X3 1 moiety is -W-Y-NR1R10 and the other X1', X2' and X3' moieties are each independently selected from OH, SH, NH2 and -W-Y- NR1R10, wherein and R1, W and Y are defined as above and R10 is H or a protecting group, and in step (b) when any X1', X2' and X3' moieties are OH, SH or NH2, alkylation is effected so that all X1', X2' and X3' moieties represent -W-Y-NR1R10, any protected amine groups on the X1', X2' and X3 1 moieties are deprotected and the X1', X2' and X3' moieties are guanidinylated so that they each represent a group of the formula -W-Y-NR1 -C(=NR2)- NR3R4, and, if necessary, the moiety L'" is converted to a moiety L, to obtain a compound of formula (I).
In this embodiment, in step (b) of the process of the invention, the alkylation and guanidinylation steps, and the conversion of L'" to a moiety L may be carried out in any order. Thus, for example, a first X1', X2' or X3 1 moiety represented by -W-Y-NR1R10 can undergo guanidinylation so that it represents -W-Y-NRi-C(=NR2)-NR3R4, followed by the alkylation of one or more OH, SH or NH2 moieties at other X1', X2' or X3 1 positions so that they represent -W-Y-NRiR1O and further subsequent guanidinylation reactions. Preferably, though, when any OfX1', X2' and X3' are OH, SH or NH2, (i) alkylation is first effected so that all OfX1', X2' and X3' represent -W-Y-NR1R1O, (ϋ) any protected amine groups on X1', X2' and X3' moieties represented by -W-Y-NR1R10 are then deprotected and (iii) the Xj', X2' and X3' moieties are then guanidinylated so that they each represent a group of formula

where W, Y, Ri, R2, R3 and R4 are defined as above.
Typically, in the present invention, when a moiety OH, SH or NH2 is converted to a moiety -W-Y-NRiR10, a catalyst is employed. Typically, this catalyst is an alkali metal carbonate, preferably sodium or cesium carbonate, more preferably cesium carbonate.
Typically, in the present invention, when a moiety -W-Y-NR1R10 is converted to a moiety - W-Y-NR1 -C(=NR2)-NR3R4, the amine reacted with the compound of formula (V) or (V) is dried using known techniques before guanidinylation.
Typically, in step (b) the alkylation of the X1', X2 1 and X3' moieties so that they each represent a group of formula -W-Y-NR1-C(=NR2)-NR3R4is effected before the conversion of the moiety L'" to a moiety L.
Preferably, when L'" is a group which can be reduced to a moiety -(Z)mNH2, the conversion of the moiety L"' to a moiety L to obtain a compound of formula I comprises (i) reducing L'" and (ii) optionally reacting the thus obtained compound with a compound of formula LG5CO(CH2)JQ1 or LG6C=S (NH)(CH2)kQ2 where j, k, Q1 and Q2 are defined as above and LG5 and LG6 are leaving groups. In this embodiment, L'" is typically -CN or -CH2NO2, preferably -CN.
Typically, the reduction of L'" takes place under a hydrogen atmosphere, in the presence of a catalyst. Preferably, reduction is effected with Raney Nickel under a hydrogen atmosphere. The reaction typically takes place at room temperature. The reaction time is typically 16 hours. The reaction typically takes place in an organic solvent, preferably THF. NH4OH may optionally be added.
Preferably, when L'" is a leaving group LG2, the conversion of the moiety L'" to a moiety L in step (b) comprises (i) cyanating with a source of CN" in the presence of a suitable catalyst, (ii) reducing the thus obtained CN moiety as described above and (iii) optionally reacting the thus obtained compound with a compound of formula LG5CO(CH2)JQ1 or LG6C=S(NH)(CH2)IcQ2 where j, k, Q1, Q2, LG5 and LG6 are defined as above.
Cyanation is typically effected with zinc cyanide and Pd(II). The reaction typically takes place at elevated temperature (i.e. greater than room temperature). The reaction preferably takes place at from 150 to 2000C, more preferably 16O0C. The reaction is preferably heated with microwave irradiation. A short reaction time of less than one hour is typically
employed, preferably 10 minutes. The reaction typically takes place in an organic solvent, preferably DMA.
In a preferred embodiment of the invention, L'" is CN or a leaving group LG2 which is cyanated in the process of the invention, and the CN moiety is reduced after the alkylation OfX1', X2' and X3' moieties so that they each represent a group of formula -W-Y-NR1- C(=NR2)-NR3R4. This is advantageous because the amount of protection and deprotection necessary on functional groups is minimised. Further, it enables a wide range of different L moieties to be introduced easily.
Preferably, when L'" is -COR]8, the conversion of the moiety L'" to a moiety L in step (b) comprises (i) reductively aminating the -COR18 moiety and (ii) optionally reacting the thus obtained compound with a compound of formula LG5CO(CH2)JQ1 or LG6C=S(NH)(CH2)kQ2.
Peferably, R18 is hydrogen.
Typically, reductive amination of the -COR18 moiety is effected by (a) reaction with NH2ORi9 or NHR5R6 and (b) reduction of carbon nitrogen double bond, wherein R^ is H or an alkyl group. Preferably, however, reductive amination is effected by NH2ORi 9, followed by reduction of the carbon nitrogen double bond.
Preferably R19 is hydrogen.
Typically, the carbon nitrogen bond is reduced with H2/Pd, SmZI2, In/NEUCl, LiAH4, Bu3SnH with BF3-OE2, Bu2SnClH in HMPA, Cl3SiH and pyrrolidine carboxaldehyde, SmBr in HMPA, Z-propanol with a ruthenium catalyst, NaCN(BH3) or trithylammonium formate with microwave irradiation. Appropriate reagents and conditions for such reductive amination processes are familiar to those of skill in the art. hi the case where Ri 9 is hydrogen, reduction with H2/Pd is preferred.
Preferably, L'" is different from J2. When L'" is different from J2, issues of chemoselectivity in the coupling reaction may be avoided. More preferably, when L'" and J2 are both leaving groups, J2 is more reactive than L1". More preferably, when L'" and J2 are both leaving groups, J2 is iodide and L1" is Br. Oxidative addition between the compounds of formulae (JJ) and (UJ) with Pd(O) can then be effected preferably with iodide.
For compounds of formula I, p, q and r are each independently 1, 2, 3 or 4.
hi a preferred embodiment of the invention, Y is a C1-1O alkylene group, a C2-10 alkenylene group or a C2-10 alkynylene group. More typically, Y is a C1-12 alkylene group, preferably a C1-1O alkylene group, even more preferably a C1-6 alkylene group, and more preferably still, -CH2CH2-.
In a preferred embodiment, W is O, S or NH. More preferably, W is O.
Preferably, L'" is a leaving group LG2, -COR18 (in particular -CHO), or a group which can be reduced to a moiety -(Z)1nNH2. The group which can be reduced to a moiety -(Z)mNH2 is preferably -CN.
Preferably, m is 1 and Z is an alkylene group, more preferably, a C1-12 alkylene group, more preferably still a C1-1O alkylene group, even more preferably a C1-6 or Ci-4 alkylene group. More preferably, Z is a CH2 group.
Preferably, one of R5 and R6 is H and the other is selected from H, CO(CH2)JQ1 or C=S(NH)(CH2)IcQ2, or R5, R6 and the nitrogen to which they are attached together form

In one preferred embodiment, L is selected from the following:
-CH2NH2, -CH2NHCOCH2CH2COOH,


Preferably, R], R2, R3 and R4 are each independently selected from H and a protecting group P1. More preferably, R1 and R3 are hydrogen and R2 and R4 represent H or Pi. Most preferably, Ri and R3 are hydrogen and R2 and R4 are each independently selected from H and a butyloxycarbonyl (Boc) protecting group.
Preferably, p, q and r are each independently 1 or 2.
In one preferred embodiment, p, q and r are all equal to 1.
In another preferred embodiment, p, q and r are all equal to 2.
In a further preferred embodiment, r is equal to 1 and p is equal to 2.
Preferably, R7, R8 and R9 are all H.
hi one particularly preferred embodiment, X1, X2 and X3 are the same and are all

where R2 and R3 are each independently H or a Boc protecting group.
Preferably, n is 0 or 1. More preferably, n is 0.
Typically, j and k are each independently 0, 1, 2 or 3, preferably 0, 1 or 2.
In a more preferred embodiment, the process of the invention is a process for the production of a compound of formula Ia or Ib


More preferably, said compound of formula II is of formula Ha, lib, lie or Hd and/or said compound III is of formula Ilia, IEb, IIIc or IHd.



Die πid
Most preferably, the compound of formula (I) is






A further finding of the present invention is that compounds of formula (I) in which Z is para to an X3 moiety have superior efficiency in delivering a cargo moiety inside a cell.
Thus, the present invention also provides a compound of formula (I), as defined above, or a pharmaceutically acceptable salt thereof, wherein r is 1 or 2 and the phenyl ring which carries L has no substituents ortho to the L moiety. Preferably, the phenyl ring which carries L has a hydrogen atom ortho to the moiety

More preferably, r is 1 and L is para to the X3 moiety.
The present invention also provides a compound of formula Via, VIb, VIc, VId or Vie

VId Vie wherein X1, X3 and L are as defined above.
Compounds of formula VId and VIe are particularly preferred. These compounds, in which the linker position is para to the X3 moiety, show particularly high efficacy.
Also provided is a compound selected from


The present invention also provides a compound of formula VII

VII wherein
X1, X3, R7, Rg and L are as defined in any one of claims 1, 7, 8, 9 or 11; - r is 1, 2, 3 or 4 and r' is 0, 1, 2 or 3, wherein r + r1 is 4; p is 1, 2, 3, 4 or 5 and p' is 0, 1, 2, 3 or 4, where p + p1 is 5; and p + r equals 3.
Preferably, the compound of formula (VII) is

The present invention further provides a process for preparing a conjugate, which process comprises: (i) preparing a compound of formula I, or a pharmaceutically acceptable salt thereof, in which L is other than a moiety -(Z)m-ρhthalimide, wherein Z and m are as defined in claim 1, by a process according to the invention; and
(ii) reacting said compound of formula I with a cargo moiety selected from a protein, a peptide, an oligonucleotide, a nucleotide, a diagnostic agent, a biologically active compound, an antibody and a drug.
The cargo moiety can be an oligonucleotide, nucleotide, protein, peptide, biologically active compound, diagnostic agent, or a combination thereof.
The cargo moiety may be directly or indirectly linked to the carrier moiety, hi the embodiment wherein the cargo moiety is indirectly linked to the carrier, the linkage may be by an intermediary bonding group such as a sulphydryl or carboxyl group or any larger group, all such linking groups are herein referred to as linker moieties as discussed below. Preferably, the carrier and cargo moieties are linked directly.
The diagnostic agent can be nonbiological, for example a microbead. Appropriate processes for preparing a conjugate of a compound of formula I and a nonbiological diagnostic agent such as a microbead are familiar to those of skill in the art.
Examples of suitable oligonucleotide cargo moieties include genes, gene fragments, sequences of DNA, cDNA, RNA, nucleotides, nucleosides, heterocyclic bases, synthetic and non-synthetic, sense or anti-sense oligonucleotides including those with nuclease resistant backbones etc. or any of the above incorporating a radioactive label, that are desired to be delivered into a cell or alternatively to be delivered from a cell to its exterior. Preferably, the oligonucleotide cargo moiety is a gene or gene fragment.
Examples of suitable protein or peptide cargo moieties include; proteins, peptides, and their derivatives such as: antibodies and fragments thereof; cytokines and derivatives or fragments thereof, for example, the interleukins (IL) and especially the IL-I, IL-2, IL-3, IL-4, IL-5, IL- 6, IL-7, IL-8, IL-9, IL-10, IL-Il and IL- 12 subtypes thereof; colony stimulating factors, for example granulocyte-macrophage colony stimulating factor, granulocyte-colony stimulating factor (alpha and beta forms), macrophage colony stimulating factor (also known as CSF-I); haemopoietins, for example erythropoietin, haemopoietin-alpha and kit-ligand (also known as stem cell factor or Steel factor); interferons (IFNS), for example IFN-α, IFN- β and EFN-γ;
growth factors and bifunctional growth modulators, for example epidermal growth factor, platelet derived growth factor, transforming growth factor (alpha and beta forms), amphiregulin, somatomedin-C, bone growth factor, fibroblast growth factors, insulin-like growth factors, heparin binding growth factors and tumour growth factors; differentiation factors and the like, for example macrophage differentiating factor, differentiation inducing factor (DEF) and leukaemia inhibitory factor; activating factors, for example platelet activating factor and macrophage activation factor; coagulation factors such as fibrinolytic/anticoagulant agents including heparin and proteases and their pro-factors, for example clotting factors VII, VEH, DC, X, XI and XII, antithrombin m, protein C, protein S, streptokinase, urokinase, prourokinase, tissue plasminogen activator, fibrinogen and hirudin; peptide hormones, for example insulin, growth hormone, gonadotrophins, follicle stimulating hormone, leutenising hormone, growth hormone releasing hormone and calcitonin; enzymes such as superoxide dismutase, glucocerebrosidase, asparaginase and adenosine deaminase; vaccines or vaccine antigens such as, for example hepatitis-B vaccine, malaria vaccine, melanoma vaccine and HIV-I vaccine; transcription factors and transcriptional modulators.
Examples of a suitable non-nucleotide/proteinaceous biologically active cargo moieties are drug moieties selected from cytotoxic agents, anti-neoplastic agents, anti-hypertensives, cardioprotective agents, anti-arrhythmics, ACE inhibitors, anti-inflammatory's, diuretics, muscle relaxants, local anaesthetics, hormones, cholesterol lowering drugs, anti-coagulants, anti-depressants, tranquilizers, neuroleptics, analgesics such as a narcotic or anti-pyretic analgesics, anti-virals, anti-bacterials, anti-fungals, bacteriostats, CNS active agents, anticonvulsants, anxiolytics, antacids, narcotics, antibiotics, respiratory agents, antihistamines, immunosuppressants, immunoactivating agents, nutritional additives, anti-tussives, diagnostic agents, emetics and anti-emetics, carbohydrates, glycosoaminoglycans, glycoproteins and polysaccharides; lipids, for example phosphatidyl-ethanolamine, phosphtidylserine and derivatives thereof; sphingosine; steroids; vitamins; antibiotics including lantibiotics; bacteristatic and bactericidal agents; antifungal, anthelminthic and other agents effective against infective agents including unicellular pathogens; small effect or molecules such as noradrenalin, alpha adrenergic receptor ligands, dopamine receptor ligands, histamine receptor ligands, GABA/benzodiazepine receptor ligands, serotonin
receptor ligands, leukotrienes and triodothyronine; cytotoxic agents such as doxorubicin, methotrexate and derivatives thereof.
In one preferred embodiment, the cargo moiety is selected from a protein, a peptide, an antibody and a drug.
In another preferred embodiment the cargo moiety is protein A, a bacterially derived protein that binds strongly to conventional antibodies.
Previous studies have demonstrated that a fusion protein containing the protein transduction domain of HIV-I TAT and the B domain of staphylococcal protein A can be used to internalise antibodies into mammalian cells [Mie et al, Biochemical and Biophysical Research Communications 310 (2003); 730-734].
Preferably, the compound of formula I is linked to commercially available (natural) protein A via a lysine NH2 group of protein A.
In one particularly preferred embodiment of the invention, the conjugate is the.reaction product of a protein (such as for example, protein A) and a compound of formula (I) as shown above wherein L1 is (Z)mNR5R6 where Z is a hydrocarbyl group, as defined above, and m is 0 or 1; where R5 and R6 are each independently H, CO(CH2)jQj or C=S(NH)(CH2)kQ2 where j and k are defined above and Q1 and Q2 are each independently

hi an alternative preferred embodiment, a cysteine residue may be engineered into the protein to allow conjugation to said compound of formula I. Further details on the preparation of cysteine modified proteins may be found in Neisler et al [Bioconjugate Chem. 2002, 13, 729-736].
Preferably, the cargo moiety is covalently attached to the L group of the compound of formula I. Thus, in step (ii), typically, a reactive group in the cargo moiety reacts with a reactive group in the L moiety of the compound of formula (I).
Typically, in step (ii) a nucleophilic group on the cargo moiety (for example an amine, thiol or hydroxy group) displaces a leaving group in the moiety L in the formula (I). For example, a thiol-containing protein (eg geminin) can react with a compound of formula (I) in which Q1 or Q2 is -S-S-(2-pyridyl) via thiol exchange. Similarly, a nucleophilic moiety such as the 2'-hydroxy group on the taxol or docetaxel molecule can displace a succinimyl, -S-S-(2-pyridyl), iodine, -S-S(O)2-OMe or -CO-O-N-succininyl group in the moiety L in formula (I).
Alternatively, when L is nucleophilic (eg when L is -NH2), it can react in step (iii) with an electrophilic site in the cargo moiety.
In one preferred embodiment, the cargo moiety is directly linked to the carrier moiety.
In another preferred embodiment, the cargo moiety is indirectly linked to the carrier moiety by means of a linker moiety. In this embodiment, the cargo moiety comprises a protein, a peptide, an oligonucleotide, a nucleotide, a diagnostic agent, a biologically active compound or a drug which is attached to a linker moiety. Typically, a reactive site on the linker group reacts with the moiety L in the formula (I) as explained above.
Direct linkage may occur through any convenient functional group on the cargo moiety, such as a hydroxy, carboxy or amino group. Indirect linkage will occur through a linking moiety. Suitable linking moieties include bi- and multi-functional alkyl, aryl, aralkyl or peptidic moieties, alkyl, aryl or aralkyl aldehydes acids esters and anhydrides, sulphydryl or carboxyl groups, such as maleimido benzoic acid derivatives, maleimido proprionic acid derivatives and succinimido derivatives or may be derived from cyanuric bromide or chloride, carbonyldiimidazole, succinimidyl esters or sulphonic halides and the like. The functional group on the linker moiety used to form covalent bonds between the compound
of formula I and the cargo moiety may be, for example, amino, hydrazino, hydroxyl, thiol, maleimido, carbonyl, and carboxyl groups, etc. The linker moiety may include a short sequence of from 1 to 4 amino acid residues that optionally includes a cysteine residue through which the linker moiety bonds to the compound of formula I. Alternatively, the compound of formula I and the cargo moiety may be linked by leucine zippers, dimerisation domains, or an avidin/biotin linker.
In one preferred embodiment, the cargo moiety is selected from a recombinant antibody, a Fab fragment, a F(ab')2 fragment, a single chain Fv, a diabody, a disulfide linked Fv, a single antibody domain and a CDR.
As used herein, the term "CDR" or "complementary determining region" refers to the hypervariable regions of an antibody molecule, consisting of three loops from the heavy chain and three from the light chain, that together form the antigen-binding site.
By way of example, the antibody may be selected from Herceptin, Rituxan, Theragyn (Pemtumomab), Infliximab, Zenapex, Panorex, Vitaxin, Protovir, EGFRl or MFE-23.
In one preferred embodiment, the cargo moiety is a genetically engineered fragment selected from a Fab fragment, a F(ab')2 fragment, a single chain Fv, or any other antibody- derived format.
Conventionally, the term "Fab fragment" refers to a protein fragment obtained (together with Fc and Fc1 fragments) by papain hydrolysis of an immunoglobulin molecule. It consists of one intact light chain linked by a disulfide bond to the N-terminal part of the contiguous heavy chain (the Fd fragment). Two Fab fragments are obtained from each immunoglobulin molecule, each fragment containing one binding site. In the context of the present invention, the Fab fragment may be prepared by gene expression of the relevant DNA sequences.
Conventionally, the term "F(ab')2" fragment refers to a protein fragment obtained (together with the pFc' fragment) by pepsin hydrolysis of an immunoglobulin molecule. It consists of
that part of the immunoglobulin molecule N-terminal to the site of pepsin attack and contains both Fab fragments held together by disulfide bonds in a short section of the Fc fragment (the hinge region). One F(ab')2 fragment is obtained from_each immunoglobulin molecule; it contains two antigen binding sites, but not the site for complement fixation. In the context of the present invention, the F(ab')2 fragment may be prepared by gene expression of the relevant DNA sequences.
As used herein, the term "Fv fragment" refers to the N-terminal part of the Fab fragment of an immunoglobulin molecule, consisting of the variable portions of one light chain and one heavy chain. Single-chain Fvs (about 30 KDa) are artificial binding molecules derived from whole antibodies, but which contain the minimal part required to recognise antigen.
hi another preferred embodiment, the cargo moiety is a synthetic or natural peptide, a growth factor, a hormone, a peptide ligand, a carbohydrate or a lipid.
The cargo moiety can be designed or selected from a combinatorial library to bind with high affinity and specificity to a target antigen. Typical affinities are in the 10"6 to 10~15 M Kd range. Functional amino acid residues present in the cargo moiety may be altered by site-directed mutagenesis where possible, without altering the properties of the cargo moiety. Examples of such changes include mutating any free surface thiol-containing residues (cysteine) to serines or alanines, altering lysines and arginines to asparagines and histidines, and altering serines to alanines.
In a further embodiment of the invention, the cargo moiety is a drug. In this embodiment, the conjugate can be described as a delivery system. Preferably, the delivery system is therapeutically active in its intact state.
More preferably, the drug moiety is derived from a cytotoxic drug.
More preferably, the drug moiety is selected from DNA damaging agents, anti-metabolites, anti-tumour antibiotics, natural products and their analogues, dihydrofolate reductase inhibitors, pyrimidine analogues, purine analogues, cyclin-dependent kinase inhibitors,
thymidylate synthase inhibitors, DNA intercalators, DNA cleavers, topoisomerase inhibitors, anthracyclines, vinca drugs, mitomycins, bleomycins, cytotoxic nucleosides, pteridine drugs, diynenes, podophyllotoxins, platinum containing drugs, differentiation inducers and taxanes.
Even more preferably, the drug moiety is selected from methotrexate, methopterin, dichloromethotrexate, 5-fluorouracil, 6-mercaptopurine, tri-substituted purines such as olomoucine, roscovitine and bohemine, flavopiridol, staurosporin, cytosine arabinoside, melphalan, leurosine, actinomycin, daunorubicin, doxorubicin, mitomycin D, mitomycin A, carninomycin, aminopterin, tallysomycin, podophyllotoxin (and derivatives thereof), etoposide, cisplatinum, carboplatinum, vinblastine, vincristine, vindesin, paclitaxel, docetaxel, taxotere retinoic acid, butyric acid, acetyl spermidine, tamoxifen, irinotecan and camptothecin.
hi one preferred embodiment, the drug moiety is directly linked to the carrier moiety.
hi another preferred embodiment, the drug moiety is indirectly linked to the carrier moiety by means of a linker moiety.
In another preferred embodiment, each carrier moiety bears more than one drug moiety.
hi one preferred embodiment, where each carrier moiety bears more than one drug moiety, the drug moieties are different.
hi one preferred embodiment, where each carrier moiety bears more than one drug moiety, each drug moiety is linked to the carrier moiety by way of a linker moiety, hi one particularly preferred embodiment, each drug moiety is linked to the carrier moiety by an identical linker moiety. In an alternative embodiment, each drug moiety is linked to the carrier moiety by a different linker moiety.
hi a further preferred embodiment of the invention, the delivery system may further comprise a targeting moiety. The targeting moiety is capable of directing the delivery
system to the specific cell type to which it is preferable for the drug moiety to function. Thus, the targeting moiety acts as an address system biasing the body's natural distribution of drugs or the delivery system to a particular cell type. The targeting moiety may be attached to the drug moiety or alternatively to the carrier moiety.
In one preferred embodiment, the targeting moiety is directly linked to the carrier moiety.
hi another preferred embodiment, the targeting moiety is indirectly linked to the carrier moiety by means of a linker moiety.
Direct linkage may occur through any convenient functional group on the targeting moiety, such as a hydroxy, carboxy or amino group. Indirect linkage will occur through a linking moiety. Suitable linking moieties include bi- and multi-functional alkyl, aryl, aralkyl or peptidic moieties, alkyl, aryl or aralkyl aldehydes acids esters and anhydrides, sulphydryl or carboxyl groups, such as maleimido benzoic acid derivatives, maleimido proprionic acid derivatives and succinimido derivatives or may be derived from cyanuric bromide or chloride, carbonyldiimidazole, succinimidyl esters or sulphonic halides and the like. The functional groups on the linker moiety used to form covalent bonds to the targeting moiety may be two or more of, e.g., amino, hydrazino, hydroxyl, thiol, maleimido, carbonyl, and carboxyl groups, etc. The linker moiety may include a short sequence of from 1 to 4 amino acid residues that optionally includes a cysteine residue through which the linker moiety bonds to the targeting moiety. Alternatively, the targeting moiety may be linked by leucine zippers, dimerisation domains, or an avidin/biotin linker.
The present invention also provides a conjugate obtainable by reacting a compound of formula (I), as defined above, in which r is 1 or 2 and the phenyl ring which carries L has no substituents ortho to the L moiety, or a compound of formula Via, VIb, VIc, VId, VIe or VII, as defined above, with a cargo moiety as defined above, or a pharmaceutically acceptable salt of said conjugate.
Another aspect of the invention relates to a pharmaceutical composition comprising a compound or conjugate of the invention admixed with one or more pharmaceutically
acceptable diluents, excipients or carriers. Even though the compounds and conjugates of the present invention (including their pharmaceutically acceptable salts, esters and pharmaceutically acceptable solvates) can be administered alone, they will generally be administered in admixture with a pharmaceutical carrier, excipient or diluent, particularly for human therapy. The pharmaceutical compositions may be for human or animal usage in human and veterinary medicine.
Examples of such suitable excipients for the various different forms of pharmaceutical compositions described herein may be found in the "Handbook of Pharmaceutical Excipients, 2nd Edition, (1994), Edited by A Wade and PJ Weller.
Acceptable carriers or diluents for therapeutic use are well known in the pharmaceutical art, and are described, for example, in Remington's Pharmaceutical Sciences, Mack Publishing Co. (A. R. Gennaro edit. 1985).
Examples of suitable carriers include lactose, starch, glucose, methyl cellulose, magnesium stearate, mannitol, sorbitol and the like. Examples of suitable diluents include ethanol, glycerol and water.
The choice of pharmaceutical carrier, excipient or diluent can be selected with regard to the intended route of administration and standard pharmaceutical practice. The pharmaceutical compositions may comprise as, or in addition to, the carrier, excipient or diluent any suitable binder(s), lubricant(s), suspending agent(s), coating agent(s), solubilising agent(s).
Examples of suitable binders include starch, gelatin, natural sugars such as glucose, anhydrous lactose, free-flow lactose, beta-lactose, corn sweeteners, natural and synthetic gums, such as acacia, tragacanth or sodium alginate, carboxymethyl cellulose and polyethylene glycol.
Examples of suitable lubricants include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride and the like.
Preservatives, stabilizers, dyes and even flavoring agents may be provided in the pharmaceutical composition. Examples of preservatives include sodium benzoate, sorbic acid and esters of p-hydroxybenzoic acid. Antioxidants and suspending agents may be also used.
The compounds of the invention can be present as salts or esters, in particular pharmaceutically acceptable salts or esters.
Pharmaceutically acceptable salts of the compounds of the invention include suitable acid addition or base salts thereof. A review of suitable pharmaceutical salts may be found in Berge et al, J Pharm Sci, 66, 1-19 (1977). Salts are formed, for example with strong inorganic acids such as mineral acids, e.g. sulphuric acid, phosphoric acid or hydrohalic acids; with strong organic carboxylic acids, such as alkanecarboxylic acids of 1 to 4 carbon atoms which are unsubstituted or substituted (e.g., by halogen), such as acetic acid; with saturated or unsaturated dicarboxylic acids, for example oxalic, malonic, succinic, maleic, fumaric, phthalic or tetraphthalic; with hydroxycarboxylic acids, for example ascorbic, glycolic, lactic, malic, tartaric or citric acid; with aminoacids, for example aspartic or glutamic acid; with benzoic acid; or with organic sulfonic acids, such as (C1-C4)-alkyl- or aryl-sulfonic acids which are unsubstituted or substituted (for example, by a halogen) such as methane- or p-toluene sulfonic acid.
Esters are formed either using organic acids or alcohols/hydroxides, depending on the functional group being esterifϊed. Organic acids include carboxylic acids, such as alkanecarboxylic acids of 1 to 12 carbon atoms which are unsubstituted or substituted (e.g., by halogen), such as acetic acid; with saturated or unsaturated dicarboxylic acid, for example oxalic, malonic, succinic, maleic, fumaric, phthalic or tetraphthalic; with hydroxycarboxylic acids, for example ascorbic, glycolic, lactic, malic, tartaric or citric acid; with aminoacids, for example aspartic or glutamic acid; with benzoic acid; or with organic sulfonic acids, such as (d-C4)-alkyl- or aryl-sulfonic acids which are unsubstituted or substituted (for example, by a halogen) such as methane- or p-toluene sulfonic acid.
Suitable hydroxides include inorganic hydroxides, such as sodium hydroxide, potassium
hydroxide, calcium hydroxide, aluminium hydroxide. Alcohols include alkanealcohols of 1- 12 carbon atoms which may be unsubstituted or substituted, e.g. by a halogen).
In all aspects of the present invention previously discussed, the invention includes, where appropriate all enantiomers and tautomers of compounds of the invention. The man skilled in the art will recognise compounds that possess optical properties (one or more chiral carbon atoms) or tautomeric characteristics. The corresponding enantiomers and/or tautomers may be isolated/prepared by methods known in the art.
Some of the compounds of the invention may exist as stereoisomers and/or geometric isomers — e.g. they may possess one or more asymmetric and/or geometric centres and so may exist in two or more stereoisomeric and/or geometric forms. The present invention contemplates the use of all the individual stereoisomers and geometric isomers of those compounds, and mixtures thereof. The terms used in the claims encompass these forms, provided said forms retain the appropriate functional activity (though not necessarily to the same degree).
The present invention also includes all suitable isotopic variations of the compound or pharmaceutically acceptable salt thereof. An isotopic variation of a compound of the present invention or a pharmaceutically acceptable salt thereof is defined as one in which at least one atom is replaced by an atom having the same atomic number but an atomic mass different from the atomic mass usually found in nature. Examples of isotopes that can be incorporated into the agent and pharmaceutically acceptable salts thereof include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulphur, fluorine and chlorine such as 2H, 3H, 13C, 14C, 15N, 170, 180, 31P, 32P, 35S, 18F and 36Cl, respectively. Certain isotopic variations of the agent and pharmaceutically acceptable salts thereof, for example, those in which a radioactive isotope such as 3H or 14C is incorporated, are useful in drug and/or substrate tissue distribution studies. Tritiated, i.e., 3H, and carbon-14, i.e., 14C, isotopes are particularly preferred for their ease of preparation and detectability. Further, substitution with isotopes such as deuterium, i.e., 2H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or
reduced dosage requirements and hence may be preferred in some circumstances. Isotopic variations of the agent of the present invention and pharmaceutically acceptable salts thereof of this invention can generally be prepared by conventional procedures using appropriate isotopic variations of suitable reagents.
The present invention also includes the use of solvate forms of the compounds of the present invention. The terms used in the claims encompass these forms.
The invention furthermore relates to the compounds and/or conjugates of the present invention in their various crystalline forms, polymorphic forms and (an)hydrous forms. It is well established within the pharmaceutical industry that chemical compounds may be isolated in any of such forms by slightly varying the method of purification and or isolation form the solvents used in the synthetic preparation of such compounds.
The invention further includes the compounds of the present invention in prodrug form. Such prodrugs are generally compounds of the invention wherein one or more appropriate groups have been modified such that the modification may be reversed upon administration to a human or mammalian subject. Such reversion is usually performed by an enzyme naturally present in such subject, though it is possible for a second agent to be administered together with such a prodrug in order to perform the reversion in vivo. Examples of such modifications include ester (for example, any of those described above), wherein the reversion may be carried out be an esterase etc. Other such systems will be well known to those skilled in the art.
The pharmaceutical compositions of the present invention may be adapted for oral, rectal, vaginal, parenteral, intramuscular, intraperitoneal, intraarterial, intrathecal, intrabronchial, subcutaneous, intradermal, intravenous, nasal, buccal or sublingual routes of administration.
For oral administration, particular use is made of compressed tablets, pills, tablets, gellules, drops, and capsules. Preferably, these compositions contain from 1 to 250 mg and more preferably from 10-100 mg, of active ingredient per dose.
Other forms of administration comprise solutions or emulsions which may be injected intravenously, intraarterially, intrathecally, subcutaneously, intradermally, intraperitoneally or intramuscularly, and which are prepared from sterile or sterilisable solutions. The pharmaceutical compositions of the present invention may also be in form of suppositories, pessaries, suspensions, emulsions, lotions, ointments, creams, gels, sprays, solutions or dusting powders.
An alternative means of transdermal administration is by use of a skin patch. For example, the active ingredient can be incorporated into a cream consisting of an aqueous emulsion of polyethylene glycols or liquid paraffin. The active ingredient can also be incorporated, at a concentration of between 1 and 10% by weight, into an ointment consisting of a white wax or white soft paraffin base together with such stabilisers and preservatives as may be required.
Injectable forms may contain between 10-1000 mg, preferably between 10-250 mg, of active ingredient per dose.
Compositions may be formulated in unit dosage form, i.e., in the form of discrete portions containing a unit dose, or a multiple or sub-unit of a unit dose.
A person of ordinary skill in the art can easily determine an appropriate dose of one of the compositions of the invention to administer to a subject without undue experimentation. Typically, a physician will determine the actual dosage which will be most suitable for an individual patient and it will depend on a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the
individual undergoing therapy. The dosages disclosed herein are exemplary of the average case. There can of course be individual instances where higher or lower dosage ranges are merited, and such are within the scope of this invention.
Depending upon the need, the agent may be administered at a dose of from 0.01 to 30 mg/kg body weight, such as from 0.1 to 10 mg/kg, more preferably from 0.1 to 1 mg/kg body weight, hi an exemplary embodiment, one or more doses of 10 to 150 mg/day will be administered to the patient.
In a particularly preferred embodiment, the one or more compounds and/or conjugates of the invention are administered in combination with one or more other therapeutically active agents, for example, existing drugs available on the market. In such cases, the compounds of the invention may be administered consecutively, simultaneously or sequentially with the one or more other therapeutically active agents.
Drugs in general are more effective when used in combination. In particular, combination therapy is desirable in order to avoid an overlap of major toxicities, mechanism of action and resistance mechanism(s). Furthermore, it is also desirable to administer most drugs at their maximum tolerated doses with minimum time intervals between such doses. The major advantages of combining chemotherapeutic drugs are that it may promote additive or possible synergistic effects through biochemical interactions and also may decrease the emergence of resistance in cells which would have been otherwise responsive to initial chemotherapy with a single agent.
By way of example, numerous combinations are used in current treatments of cancer and leukaemia. A more extensive review of medical practices may be found in "Oncologic Therapies" edited by E. E. Vokes and H. M. Golomb, published by Springer.
The present invention also provides use of a compound or conjugate of the invention in the manufacture of a medicament for use delivering a drug to a patient transdermally.
The present invention also provides a skin patch which comprises a compound or conjugate of the invention and a pharmaceutically acceptable carrier or diluent.
Figure 1 shows the cellular uptake, via live microscopy, of three compounds of the invention conjugated to Geminin labelled with AlexFluor 488 (11-g*, 33-g* and 35-g*) in two cell lines (Wl -38 human diploid fibroblast and human U2OS osteosarcoma cells).
Figure 2 shows the results of cellular uptake monitored by FACS analysis of eight compounds of the invention conjugated to Geminin labelled with AlexFluor 488 (23-g , 26- g*, 18-g*, 20-g\ 33-g*, 31-g*, 35-g* and 11-g*) in two cell lines (Wl-38 HDF and U2OS).
Figure 3 shows the results of a cell proliferation assay monitored via confocal fluorescence microscopy of 3 compounds of the invention coupled to 15 Geminin in Wl-38 and U2OS cell lines.
The following Examples illustrate the invention.
Examples
All starting materials were either commercially available or synthesised by methods reported in the literature. 1H and 13C spectra were recorded on a Bruker AMX-300 spectrometer. Chemical shifts are reported as ppm relative to TMS as internal standard. Mass spectra were recorded on either a VG ZAB SE spectrometer (EI, FAB) or a Gilson- Finningan AQA LC-mass spectrometer using C-18 column (Hypersil BDS 100 x 4.6 mm, 5 μm). Microanalysis was carried out by the Analytical Services Section, Department of Chemistry, University College London. Purification was by reverse-phase HPLC (Gilson) using preparative C-18 columns (Hypersil PEP 100 x 21 mm, 5μm). Melting points were determined on a Gallenkamp melting point apparatus and are uncorrected. IR spectra were recorded on a Perkin-Elmer Spectrum One series FT-IR spectrophotometer. The microwave experiments were run on a Biotage Initiator 60 microwave.
Examples 1 to 3: Synthesis of SMOC carrying 4 guanidine moieties
l,4-Dibromo-2,3-dimethoxy-benzene
M. Albrecht, Synthesis - Stuttgart 1996, 230

1, 2-Dimethoxy-3-bromo-6-iodobenzene

13C-NMR (CDCl3): δ-0.63, 60.2, 60.4, 119.7, 127.9, 130.4, 149.4, 158.6.
Theoretical Mass: (M + H) 288.01811. Measured Mass: (M + H) 288.01863
To a solution of l,2-dimethoxy-3-bromo-6-trimethylsilylbenzene (5.8 g, 20 mmol) in DCM
(60 mL) was added dropwise at - 50 °C a solution of iodine chloride (3.6 g, 22 mmol) in DCM (20 mL) keeping the temperature below - 40 0C. After stirring for 2 h at - 50 0C, the reaction mixture was quenched with Na2S2O3 1 M (100 mL). The layers were separated and the aqueous layer was extracted with DCM (100 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated under vacuum. The residue was then
crystallized from MeOH (30 mL) to afford the title compound (1.82 g, 26% yield) as a white solid.
1H-NMR (CDCl3): .53.86 (s, 6H), 7.02 (d, J- 8.7 Hz, IH), 7.35 (d, J= 8.6 Hz, IH). 13C-NMR (CDCl3): 560.8, 60.8, 91.3, 118.4, 129.7, 134.4, 150.7, 154.1. Theoretical Mass: (M + H) 342.88306. Measured Mass: (M + H) 342.88334 M.p. 30-32°C
2-(Benzyloxycarbonylamino)ethyl methanesulfonate
 To a solution of 2-(benzyloxycarbonylamino)ethanol (25 g, 0.128 mol) and Et3N (70 mL,
To a solution of 2-(benzyloxycarbonylamino)ethanol (25 g, 0.128 mol) and Et3N (70 mL,
0.51 mol) in DCM (250 mL) was added methanesulfonyl chloride (25 mL, 0.32 mol) dropwise at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight. The organic layer was washed with HCl IM (2 x 50 mL), sat. aq.
NaHCO3 (2 x 50 mL), brine (2 x 50 mL), and dried over MgSO4 and filtered. The solvent was removed under vacuum to afford the title compound (35 g, 100% yield) as an orange solid which was used in the next step without further purification.
1H-NMR (CDCl3): 52.92 (s, 3H), 3.54 (q, J= 5 Hz, 2H), 4.39 (t, J= 5 Hz, 2H), 5.08 (s,
2H), 5.84(bs, 1 H), 7.3 l(m, 5H).
13C-NMR (CDCl3): 537.2, 40.3, 66.9, 68.6, 128.1, 128.6, 136.3, 156.5. Theoretical Mass: (M + H) 274.07491. Measured Mass: (M + H) 274.07510
Microanalysis: %C 47.86 (48.34), %H 5.66 (5.66), %N 5.08 (5.12)
M.p. 44-46°C
Dibenzyl 2,2'-(3-bromo-6-iodo-l,2-phenyIene)bis(oxy)bis(ethane-2,l-diyl)dicarbamate

To a solution of 1, 2-dimethoxy-3-bromo-6-iodobenzene (5.7 g, 16.5 mmol) in DCM (50 mL) was added a 1.0 M DCM solution OfBBr3 (16.5 mL, 16.5 mmol) dropwise at 0 °C.
The reaction mixture was allowed to warm to room temperature. After stirring overnight, the reaction mixture was cooled to 0 °C and MeOH (2 mL) was added dropwise. After stirring for 10 minutes at room temperature, the organic layer was washed with IN HCl (2 x 20 mL), water (20 mL) and brine (20 mL), dried over MgSO4, filtered and concentrated under vacuum. The resulting 3-bromo-6-iodocatechol was then dissolved in DMF (70 mL) and Na2CO3 (5.25 g, 49.5 mmol) and 2-(benzyloxycarbonylamino)ethyl methanesulfonate (13.5 g, 49.5 mmol) were added. After stirring at 80°C under nitrogen for 24 h, the reaction mixture was poured onto water (200 mL) and extracted with EtOAc (2 x 100 mL). The organic layer was washed with water (100 mL), dried over Na2SO4 and filtered. The solvent was then removed under vacuum and the crude compound was purified by flash chromatography using cyclohexane/EtOAc (8/2) as eluent to afford the title compound (6.5 g, 59% yield) as a white solid.
ESMS; m/z 669, 671 (M+l), 686,688 (M+18)
1H-NMR (CDCl3): .53.53 (q, J= 5 Hz, 4H), 4.05 (m, 4H), 5.12 (s, 4H), 5.63 (bs, IH), 5.65 (bs, IH), 7.03 (d, J= 8.5 Hz, IH), 7.34 (m, HH).
13C-NMR (CDCl3): 541-1, 41.2, 66.8, 72.8, 72.9, 91.4, 118.5, 128.1, 128.5, 130.2, 134.9, 136.6, 149.4, 152.8, 156.5.
Theoretical Mass: (M + Na) 690.99166. Measured Mass: (M + Na) 690.98946 Microanalysis: %C 47.01 (46.66), %H 4.01 (3.92), %N 4.13 (4.19) M.p. 79°C
Dibenzyl 2,2'-(3-bromo-6-cyano-l,2-phenylene)bis(oxy)bis(ethane-2,l- diyl)dicarb amate

A mixture of dibenzyl 2,2'-(3-bromo-6-iodo-l,2-phenylene)bis(oxy)bis(ethane-2,l- diyl)dicarbamate (1.34 g, 2 mmol), zinc cyanide (0.12 g, 1 mmol), Pd2dba3 (37 mg, 0.04 mmol), dppf (44 mg, 0.08 mmol), zinc acetate (75 mg, 0.4 mmol) and zinc (26 mg, 0.4 mmol) in dimethylacetamide (10 mL) was heated at 1400C for 10 minutes under microwave irradiation. The reaction mixture was diluted in EtOAc (25 mL) and water (100 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 x 25 mL). The combined organic layers were washed with water (50 mL) and brine (50 mL), dried over MgSO4, filtered and concentrated under vacuum. The residue was then purified by flash chromatography using cyclohexane/EtOAc (7/3) as eluent to afford the title compound (0.69 g, 61% yield) as a white solid.
ESMS; m/z 568-570 (M+l), 585-587 (M+18). 1H-NMR (CDCl3): 53.54 (bs, 4H), 4.08 (bs, 2H), 4.25 (bs, 2H), 5.10 (s, 2H), 5.11 (s, 2H),
5.52 (bs, 2H), 7.21-7.38 (m, 12H).
13C-NMR (CDCl3): 541-1, 66.9, 73.0, 74.3, 107.4, 115.4, 124.5, 128.1, 128.2, 128.5, 128.6,
128.8, 129.2, 136.4, 150.0, 155.0, 156.4.
Theoretical Mass: (M + Na) 590.09026. Measured Mass: (M + Na) 590.09091 IR v Max KBR/cm" 3322 (br NH, NH2), 2231 (CN), 1694 (C=ON)
M.p. 91-920C
l-Bromo-2,3-dimethoxy-benzene

13C-NMR (CDCl3): 60.3, 60.6, 109.6, 111.7, 117.4, 132.1, 149.4, 151.5.
Dibenzyl 2,2'-(3-bromo-l,2-phenylene)bis(oxy)bis(ethane-2,l-diyl)dicarbamate

1H-NMR (CDCl3): 63.54 (m, 4H), 4.06 (m, 4H), 5.09 (s, 4H), 5.62(bs, IH), 5.75(bs, IH), 6.82 (d, J= 8 Hz, IH), 6.91 (t, J= 8 Hz, IH), 7.13 (dd, J= 8 Hz, J= 1.3 Hz, IH), 7.32 (m, 10H). 13C-NMR (CDCl3): 540.5, 41.3, 66.8, 68.2, 71.8, 112.9, 118.0, 125.4, 128.1, 128.2, 128.5, 136.4, 152.6, 156.5.
Theoretical Mass: (M + Na) 565.09501. Measured Mass: (M + Na) 565.09508 Microanalysis: %C 57.58 (57.47), %H 5.10 (5.01), %N 5.16 (5.04) M.p. 54°C
Dibenzyl 2,2'-(3-(4,4,5,5-tetramethyI-l,3,2-dioxaborolan-2-yl)-l,2- phenylene)bis(oxy)bis(ethane-2,l-diyl)dicarbamate

Pd(dppf)Cl2 (1.5 g, 1.9 mmol), potassium acetate (5.4 g, 56 mmol) and bispinacolatodiboran (10 g, 41 mmol) were weighted in a three necked round bottom flask and DMSO (100 ml) was added. After stirring for 2 minutes, dibenzyl 2,2'-(3-bromo-l,2- phenylene)bis(oxy)bis(ethane-2,l-diyl)dicarbamate (20 g, 37 mmol) was added. The reaction mixture was heated to 80°C for 72 h. After cooling the mixture at room temperature, 1 L of water and 0.5 L of toluene were added. Toluene was separated and washed with brine, dried over MgSO4, filtered and evaporated to dryness. The resulting oil was purified by silica chromatography using DCM to DCM/EtOAc (9/1) as eluent yielding
(6.4g, 30%) as a white solid.
ESMS; m/z 591(M+1)
1H-NMR (CDCl3): .61.30 (s 12H), 3.54 (t, J = 4.6 Hz, 4H), 4.04 (m, 2H), 4.14 (t, J= 4.4 Hz, 2H), 5.11 (s, 4H), 5.12 (bs, IH), 5.25 (bs, IH), 6.49 (bs, IH), 6.98 (m, 2H), 7.33 (bs,
10H).
13C-NMR (CDCl3): 624.6, 40.7, 41.5, 66.7, 66.8, 72.7, 83.9, 117.8, 124.2, 128.0, 128.1,
128.4, 128.5, 129.2, 136.5, 136.7, 151.1, 153.1, 156.5, 156.6
Theoretical Mass: (M + Na) 613.26970. Measured Mass: (M + Na) 613.26890 Microanalysis: %C 65.12 (65.09), %H 6.76 (6.66), %N 4.74 (4.74) M.p. 59°C
Potassium (2,3-bis(2-(benzyIoxycarbonylamino)ethoxy)phenyl) trifluoroborate


A degassed mixture of dibenzyl 2,2'-(3-bromo-6-cyano-l,2-phenylene) bis(oxy)bis(ethane- 2,l-diyl)dicarbamate (0.3 g, 0.5 mmol), dibenzyl 2,2I-(3-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-l,2-phenylene)bis(oxy)bis(ethane-2,l-diyl)dicarbamate (0.35 g, 0.6 mmol), PdCl2dppf. CH2Cl2 (20 mg, 0.025 mmol), potassium phosphate (0.21 g, 1 mmol), toluene (1 mL) and water (0.1 mL) was heated at 100°C for 18 h. The reaction mixture was diluted in EtOAc (25 mL) and water (25 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 x 25 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under vacuum. The residue was then purified by flash chromatography using cyclohexane/EtOAc (1/1) as eluent to afford the title compound (0.48 g, 100% yield) as a yellow oil. ESMS; m/z 952 (M+l), 969 (M+18) 1H-NMR (CDCl3): .53.20 (m, 4H), 3.50 (m, 4H), 3.80 (m, 4H), 3.93 (m, 2H), 4.26 (m, 2H), 4.85 (bs, IH), 4.98 (s, 2H), 5.01 (s, 2H), 5.08 (s, 4H), 5.31(bs, IH), 5.56 (bs, IH), 5.68 (bs, IH), 6.86 (m, 2H), 7.05 (m, 3H), 7.30 (m, 20H).
13C-NMR (CDCl3): 540.6, 41.1, 66.8, 68.0, 70.9, 72.7, 73.9, 75.0, 112.0, 114.1, 116.0, 122.6, 124.5, 127.0, 128.0, 128.2, 128.5, 131.1, 136.4, 136.5, 139.3, 145.8, 149.5, 151.6, 154.2, 156.2, 156.5.
Theoretical Mass: (M + Na) 974.35882. Measured Mass: (M + Na) 974.360673 Microanalysis
 %C 64.92 (64.63), %H 5.51 (5.81), %N 6.81(7.09) IR v Max KBR/cm- 3418 (br NH), 3335 (br NH), 2230 (CN), 1713 (C=O) Mp 72-74°C
Tetrabenzyl 2,2',2",2'"-(4-cyanobiphenyl-2,2',3,3'- tetrayltetrakis(oxy))tetrakis(ethane-2,l-diyl)tetracarbamate (Alternative protocol)
%C 64.92 (64.63), %H 5.51 (5.81), %N 6.81(7.09) IR v Max KBR/cm- 3418 (br NH), 3335 (br NH), 2230 (CN), 1713 (C=O) Mp 72-74°C
Tetrabenzyl 2,2',2",2'"-(4-cyanobiphenyl-2,2',3,3'- tetrayltetrakis(oxy))tetrakis(ethane-2,l-diyl)tetracarbamate (Alternative protocol)

To a degassed solution of potassium (2,3-bis(2-(benzyloxycarbonylamino)ethoxy)phenyl) trifluoroborate (800 mg, 1.40 mmol) in isopropanol/water (2:1) (26 mL) was added PdCl2dppf. CH2Cl2 (40 mg, 0.049 mmol) and triethylamine (600 μL, 4.2 mmol). The mixture was stirred at room temperature for 2 minutes then dibenzyl 2,2'-(3-bromo-6- cyano-l,2-phenylene) bis(oxy)bis(ethane-2,l-diyl)dicarbamate (400 mg, 0.70 mmol) was added. The reaction mixture was stirred at 82 °C for 18 h. The reaction mixture was diluted with IM HCl (aq) (50 mL) and the product extracted with DCM (3 x 200 mL). The combined organic fractions were dried over MgSO4, filtered and concentrated under reduced pressure. The product was purified by flash chromatography using a gradient of 0- 50% EtOAc in cyclohexane over 10 CV to afford the title product (530 mg, 79%) as a white foam.
2, 3, 2', 3'-Tetra{2-[iV, iV'-bis(tert-butoxycarbonyl)guaiiidino]-ethyloxy}-4- cyanobiphenyl

The resulting solid was suspended in DCM (10 mL) and Et3N (2.9 mL, 21 mmol) and N, N- di-boc-ΛT-trifluoromethanesulfonyl-guanidine (3.3 g, 8.4 mmol) were added. The mixture was stirred overnight at room temperature, diluted with DCM (40 mL) then washed with 2M NaHSO4 2M (25 mL) followed by sat. aq. NaHCO3 (25 mL) and brine (25 mL). The organic layer was dried over MgSO4, filtered and concentrated under vacuum. The residue was purified by flash chromatography using cyclohexane/EtOAc (8/2) as eluent to afford the title compound (1.87 g, 65% yield) as a white solid. ESMS; m/z 1385 (M+l), 693 (M+2), 462 (M+3) 1H-NMR (CDCl3): .81.47 (m, 72H), 3.47 (m, 2H), 3.54 (m, 2H), 3.90 (m, 8H), 4.16 (t, J= 5 Hz, 2H), 4.38 (t, J= 5 Hz, 2H), 7.00 (m, 3H), 7.18 (d, J= 8 Hz, IH), 7.29 (d, J= 8 Hz, IH), 8.45 (m, 2H), 8.79 (m, 2H), 11.40 (s, IH), 11.42 (s, IH), 11.48 (bs, 2H). 13C-NMR (CDCl3): 526.8, 28.0, 28.0, 28.2, 39.9, 40.6, 40.8, 41.3, 73.2, 78.8, 78.9, 79.0, 79.2, 82.9, 82.9, 83.1, 107.1, 113.7, 115.7, 123.2, 124.0, 127.6, 127.7, 130.8, 138.4, 144.8, 149.6, 151.6, 152.8, 152.9, 153.0, 153.0, 154.2, 156.0, 156.3, 163.3, 163.4. Theoretical Mass: (M + Na) 1406.71832. Measured Mass: (M + Na) 1406.71565 IR v Max KBR/cm- 3335 (br, NH), 2231 (CN), 1722 (C=O), 1641 (C=N) Mp 101-1030C
2, 3, 2', 3'-Tetra{2-[iV, Λ^-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-4- cyanobiphenyl (Alternative protocol)

To a solution of benzyl 2,2',2",2'"-(4-cyanobiphenyl-2,2',3,3l- tetrayl)tetrakis(oxy)tetrakis(ethane-2,l- diyl)tetracarbamate (550 mg, 0.58 mmol) in DCM (25 mL) was added a 30% solution of HBr in acetic acid (5.5 mL) dropwise. The reaction mixture was stirred at room temperature for 1 h then water (20 mL) was added and the layers separated. The organic fraction was extracted with water (20 mL). The combined aqueous fractions were concentrated under reduced pressure and the crude product was dried under vacuum for 8 hours. The resulting solid was suspended in DCM (25 mL) and DBEA (1 mL, 5.84 mmol) was added. After stirring for 10 min N,N'-Di-Boc-lH-pyrazo Ie-I- carboxamidine (790 mg, 2.54 mmol) was added and the reaction mixture stirred at room temperature for 18 h. The reaction mixture was diluted with DCM (50 mL) then washed with 10% citric acid (aq) (25 mL), followed by sat. aq. NaHCO3 (25 mL) and brine (25 mL). The organic layer was dried over MgSO4, filtered and concentrated under vacuum. The product was purified by flash chromatography using a gradient of 0-50% EtOAc in cyclohexane over 12 CV to afford the title product (570 mg, 71%) as white foam.
2, 3, 2', 3'-Tetra{2-[iV, iV'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-4- (aminomethyl)biphenyl (Example 1)

Tert-butyl (2,2I,2",2'"-(4-cyanobiρhenyl-2,2',3 ,3 '-tetrayl)tetrakis(oxy)tetrakis (ethane-2, 1 - diyl))tetrakis(azanediyl)tetrakis((tert-butoxycarbonylamino)methan- 1 -yl- 1 - ylidene)tetracarbamateylidene)tetracarbamate (0.47 g, 0.34 mmol) was dissolved in THF (7 mL) and NH4OH (1.4 mL) was added followed by Raney Nickel (0.5 g). The mixture was stirred vigorously under H2 atmosphere for 16 h. The catalyst was filtered off and washed several times with MeOH. Diethylenetriamine (leq) was added and the mixture stirred for 30 minutes then concentrated under vacuum. The residue was dissolved in EtOAc (10 mL) and washed with sat. aq. NaHCO3 (10 mL). The organic layer was dried over MgSO4, filtered and concentrated under vacuum. The residue was purified by flash chromatography using DCM/MeOH (97/3) as eluent to afford the title compound (0.21Og, 44% yield) as a white solid. ESMS; m/z 1388 (M+l), 695 (M+2), 464 (M+3)
1H-NMR (CDCl3): δl.41 (m, 72H), 3.44 (m, 4H), 3.78 (m, 10H), 4.13 (m, 4H), 6.93 (m,
5H), 8.43 (bt, IH), 8.49 (bt, IH), 8.78 (m, 2H), 11.38 (m, 2H), 11.44 (bs, IH), 11.48 (bs,
IH).
13C-NMR (CDCl3): 628.1, 28.3, 40.0, 40.9, 41.2, 41.4, 41.7, 67.2, 70.6, 71.0, 71.4, 78.8, 78.9, 79.0, 79.1, 82.6, 82.8, 83.0, 112.8, 123.3, 123.8, 126.8, 131.4, 132.5, 136.9, 145.2, 149.2, 149.4, 151.7, 152.8, 152.9, 153.0, 156.0, 156.2, 156.3, 163.4, 163.5. IR v Max KBR/cm' 3335 (br NH, NH2), 1722 (C=O), 1641 (C=N) Mp 105-1070C
iV-{2, 3, 2', 3'-Tetra{2-[iV, iV-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-biphenyl- 4-ylmethyl}-3-[2-pyridyI)dithio]propionamide (Example 2)

To a solution of 2, 3, 2', 3 '-terra {2- [N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-4-
(aminomethyl)biphenyl (0.10 g, 0.072 mmol) and Z-Pr2EtN (20 μL, 0.10 mmol) in DCM
(1.5 mL) was added N-succinimidyl-3-(2-pyridyldithio)propionate ( 22 mg, 1 eq) and the mixture was stirred in the dark for 24 h. The solvent was removed under vacuum and the crude oil was purified by preparative TLC (ethyl acetate/hexane 1/1) to afford the title compound (13 mg, 11% yield).
ESMS; m/z 1587 (M+l), 794 (M+2), 530 (M+3)
1H-NMR (CDCl3): δ 1.43-1.51 (m, 72H), 2.70 (t, J= 7.2 Hz, 2H), 3.11 (t, J= 7.2 Hz, 2H),
3.45-3.50 (m, 4H), 3.84-3.93 (m, 8H), 4.15-4.21 (m, 4H), 4.46 (d, J= 5.6 Hz, 2H), 6.89-
6.92 (m, 2H), 6.99-7.08 (m, 4H), 7.57-7.68 (m, 2H), 8.36 (m, IH), 8.46 (br t, IH), 8.55 (br t, IH), 8.80 (br t, 2H), 11.35 (bs, IH), 11.41 (bs, IH), 11.47 (bs, IH), 11.53 (bs, IH).
IR v Max KBR/cm" 3336 (NH), 1722 (C=O), 1641 (C=N)
Theoretical Mass: (M + H) 1585.76459. Measured Mass: (M + H) 1585.05043
iV-[2, 3, T, 3'-Tetra(2-guanidino-ethyloxy)-biphenyl-4-yImethyl]-3-[2- pyridyl)dithio]propionamide, trifluoroacetate salt (Example 3)
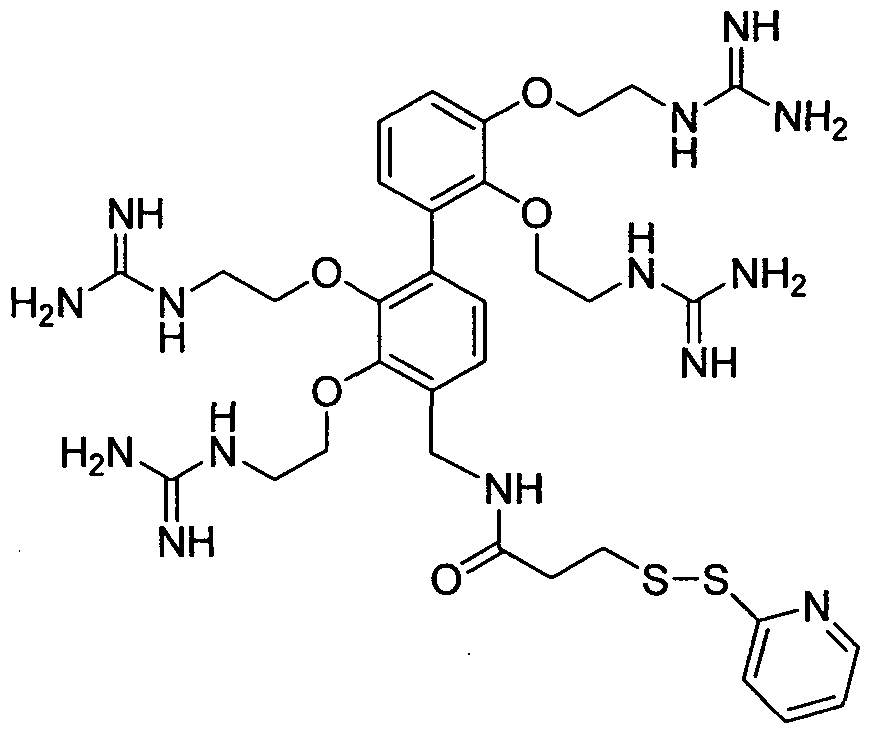
N'-Bis(tert-butoxycarbonyl)guanidino] -ethyloxy } -biphenyl-4-ylmethyl } -3 - [2- pyridyl)dithio]propionamide (15 mg, 0.0094 mmol) was dissolved in a mixture of TFA/
H2O/triisopropylsilane 95/2.5/2.5 (1 mL) and stirred at room temperature for 3 h. The solvent was removed under vacuum to give the title compound as colourless oil (15 mg,
100%yield).
ESMS; m/z 785(M+1), 395 (M+2) 1H-NMR (MeOH-d4): δ 2.73 (t, J= 6.6 Hz, 2H), 3.10 (t, J= 6.6 Hz, 2H), 3.23-3.30 (m,
4H), 3.62-3.67 (m, 4H), 3.88 (m, 2H), 3.96 (m, 2H), 4.22 (m, 4H), 4.47 (s, 2H), 6.87 (dd, J
= 7.4, 1.7 Hz, IH), 7.02 (d, J= 7.9 Hz, IH), 7.08-7.24 (m, 4H), 7.76-7.80 (m, 2H), 8.38 (d,
J= 4.7 Hz, IH), 8.70 (br t, IH).
IR v Max KBR/cm" 3376 (br NH, NH2), 1681 (C=O) Theoretical Mass: 748.33734. Measured Mass: 748.33682
Examples 4 to 17: Synthesis of SMC carrying 2 guanidϊne moieties
Benzyl 2-(2-bromo-5-iodophenoxy)ethylcarb amate

13C-NMR (CDCl3): 540.4, 67.0, 68.7, 92.4, 112.4, 122.7, 128.2, 128.2, 128.6, 131.5, 134.6, 136.4, 155.4, 156.4.
Theoretical Mass: (M + Na) 497.91777. Measured Mass: (M + Na) 497.91638 Microanalysis: %C 40.41 (40.36), %H 3.24 (3.18), %N 2.86 (2.94) M.p. 700C
Benzyl 2-(2-bromo-5-cyanophenoxy)ethylcarbamate

A mixture of benzyl 2-(2-bromo-5-iodophenoxy)ethylcarbarnate (1.9 g, 4 mmol), zinc cyanide (0.23 g, 2 mmol), Pd2dba3 (73 mg, 0.08 mmol), dppf (88 mg, 0.16 mmol), zinc acetate (0.14 g, 0.8 mmol) and zinc (52 mg, 0.8 mmol) in dimethylacetamide (20 mL) was heated at 140 °C for 10 minutes under microwave irradiation. The reaction mixture was diluted in EtOAc (25 mL) and water (100 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 x 25 mL). The combined organic layers were washed with water (50 mL) and brine (50 mL), dried over MgSO4, filtered and concentrated under vacuum. The residue was then purified by flash chromatography using cyclohexane/EtOAc (7/3) as eluent to afford the title compound (1.13 g, 75% yield) as a white solid.
ESMS; m/z 375, 377 (M+l), 392, 394 (M+18)
1H-NMR (CDCl3): 53.69 (q, J= 5.2 Hz, 2H), 4.11 (t, J= 4.7 Hz, 2H), 5.12 (s, 2H), 5.26 (bs, IH), 7.08 (s, IH), 7.15 (dd, J= 8.1 Hz5 J= 1.7 Hz, IH), 7.35 (bs, 5H), 7.64 (d, J= 8.1
Hz5 IH).
13C-NMR (CDCl3): 540.3, 67.0, 68.7, 112.3, 115.8, 118.0, 118.4, 125.9, 128.2, 128.3,
128.6, 134.4, 136.3, 155.3, 156.4.
Microanalysis: %C 54.48 (54.42), %H 4.09 (4.03), %N 7.35 (7.47) Theoretical Mass: (M + Na) 397.01637. Measured Mass: (M + Na) 397.01504
IR v Max KBR/cπT3322 (br NH, NH2), 2231 (CN), 1694 (C=ON)
M.p. 86-87°C
Benzyl 2-(4-cyano-2'-hydroxybiphenyl-2-yloxy)ethylcarbamate
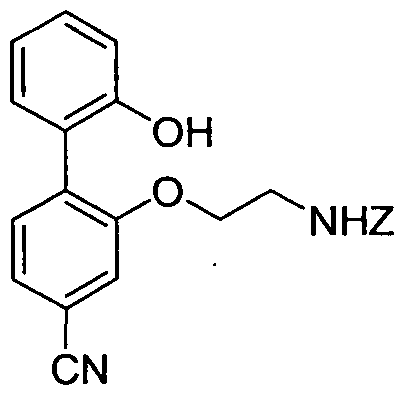
1H-NMR (CDCl3): 63.47 (m, 2H), 4.05 (m, 2H), 5.06 (s, 2H), 5.30 (bs, IH), 6.92-7.04 (m, 2H), 7.15 (m, 2H), 7.28-7.39 (m, 8H).
13C-NMR (CDCl3): 540.3, 66.9, 68.4, 112.5, 115.8, 116.9, 118.5, 121.1, 124.3, 125.8, 128.1, 128.3, 128.6, 130.1, 131.0, 132.8, 133.2, 136.3, 153.2, 155.5, 156.5. Theoretical Mass: (M + Na) 411.13207. Measured Mass: (M + Na) 411.13099
Dibenzyl 2,2'-(4-cyanobiphenyl-2,2'-diyl)bis(oxy)bis(ethane-2,l-diyl)dicarbamate

(benzyloxycarbonylamino)ethyl methanesulfonate (0.21 g, 0.77 mmol) were added. After stirring at 8O0C under nitrogen for 2 h, the reaction mixture was dissolved in EtOAc (25 mL). The organic layer was washed with brine (3 x 20 mL) and dried over MgSO4. The solvent was removed under vacuum and the crude compound was purified by flash chromatography using cyclohexane/EtOAc (65/35) as eluent to afford (0.16g, 54% yield) as colourless oil. ESMS; m/z 566 (M+l), 583 (M+18)
1H-NMR (CDCl3): .53.33 (m, 4H), 3.90 (m, 4H), 4.80 (bs, IH), 4.97 (bs, 5H), 6.88 (d, J= 8.1 Hz, IH), 7.04 (m, 2H), 7.14 (dd, J= 7.5 Hz, J= 1.6 Hz, IH), 7.30 (m, 13H). 13C-NMR (CDCl3): 640.3, 66.9, 68.2, 68.3, 112.4, 113.3, 116.2, 119.9, 121.8, 125.6, 128.3, 128.6, 128.7, 129.9, 130.8, 132.1, 134.5, 136.4, 155.5, 156.0, 156.1. Theoretical Mass: (M + H) 566.22910. Measured Mass: (M + H) 566.22799 IR v Max KBR/cm-3347 (br, NH), 2228 (CN), 1715 (C=O)
2, 2'-Bis{2-[iV, iV-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-4-cyanobiphenyl

1H-NMR (CDCl3): .51.49 (m, 36H), 3.71 (q, J= 5 Hz, 4H), 4.05 (t, J= 4.6 Hz, 4H), 6.96 (d, J= 8.3 Hz, IH), 7.03 (t, J= 7.1 Hz, IH), 7.17 (d, J= 1.2 Hz, IH), 7.30-7.34 (m, 3H), 7.46 (d, J= 7.8 Hz, IH), 8.46 (m, 2H), 11.46 (bs, 2H). 13C-NMR (CDCl3): .628.1, 28.3, 39.9, 40.0, 66.7, 67.3, 79.4, 83.1, 83.3, 111.8, 112.0, 115.5, 118.7, 120.8, 124.9, 125.3, 129.5, 131.6, 132.9, 132.9, 153.0, 155.2, 155.8, 156.2, 163.4. Theoretical Mass: (M + H) 782.40885. Measured Mass: (M + H) 782.40963 Microanalysis (C39H55N7O10^H2O): %C 56.90 (57.27), %H 7.12 (7.09), %N 11.58 (11.99)
IR v Max KBR/cm" 3333 (br NH), 2228 (CN), 1721 (C=O), 1640 (C=N), 1618 (C=N) M.p. 82°C
N-{2, 2'-Bis{2-[iV, iV'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-biphenyl-4- ylmethyl}-3-[2-pyridyl)dithio]propionamide (Example 4)

2, 2'-Bis{2-[N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-4-cyanobiphenyl (0.06 g, 0.077 mmol) was dissolved in THF (2 mL) and NH4OH (0.2 mL) was added followed by Raney Nickel (0.05 g). The mixture was stirred vigorously under H2 atmosphere for 16 h. The catalyst was filtered off and washed several times with MeOH. Diethylenetriamine (leq) was added and the mixture stirred for 30 minutes then concentrated under vacuum. The residue was dissolved in EtOAc (10 mL) and washed with sat. aq. NaHCO3 (10 mL). The organic layer was dried over MgSO4, filtered and concentrated under vacuum. To a solution of the crude amine and J-Pr2EtN (26 μl, 0.15 mmol) in DCM (1.5 mL) was added N-succinimidyl-3-(2-pyridyldithio)propionate (34 mg, 0.11 mmol) and the mixture was stirred in the dark for 24 h. The solvent was removed under vacuum and the crude oil was purified by preparative TLC (ethyl acetate/hexane 1/1) to afford the title compound (20 mg, 26% yield).
1H-NMR (CDCl3): .81.48 (m, 36H), 2.68 (t, J= 5.0 Hz, 2H), 3.03 (t, J= 5 Hz, 2H), 3.67 (m, 4H), 3.99 (m, 4H), 4.49 (d, J= 5.2 Hz, 2H), 6.90-7.05 (m, 4H), 7.21-7.33 (m, 3H), 7.57 (m, 2H), 8.20 (d, J= 4.7 Hz, IH), 8.47 (m, 2), 11.46 (bs, 2H).
13C-NMR (CDCl3): 828.1, 28.3, 29.7, 35.2, 35.8, 40.2, 53.4, 66.9, 79.3, 82.9, 83.1, 112.3, 112.7, 120.4, 121.0, 126.9, 128.5, 132.0, 132.3, 136.9, 138.4, 149.6, 152.9, 155.6, 155.8, 156.2, 159.4, 163.5, 170.6. Theoretical Mass: (M + Na) 1005.41899. Measured Mass: (M + Na) 1005.41622
N-[2, 2'-Bis(2-guanidino-ethyloxy)-biphenyl-4-ylmethyl]-3-[2- pyridyl)dithio]propionamide, formiate salt (Example 5)

N- {2, 2'-Bis {2~[N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy} -biphenyl-4-ylmethyl} - 3-[2-pyridyl)dithio]propionamide (15 mg, 0.015 mmol) was dissolved in formic acid (1 mL) and stirred at room temperature overnight. The mixture was then heated at 500C for
Ih. Water was added and the product was lyophilised to give as colourless oil (13.6 mg,
100%).
1H-NMR (MeOH-d4): 52.76 (t, J= 6.6 Hz, 2H), 3.11 (t, 2H, J= 6.3 Hz, 2H), 3.42 (m, 4H), 4.04 (m, 4H), 4.421 (s, 2H), 6.99-7.24 (m, 6H), 7.35 (t, J= 6.9 Hz, IH), 7.78-7.83 (m, 2H),
8.15 (m, IH), 8.39 (d, J= 4.6 Hz, IH), 8.57 (bs, 2H).
Theoretical Mass: (M + H) 583.22734. Measured Mass: (M + H) 583.22651
Benzyl 2-(4-cy ano-3 '-hydroxybiphenyl-2-yIoxy)ethylcarbamate
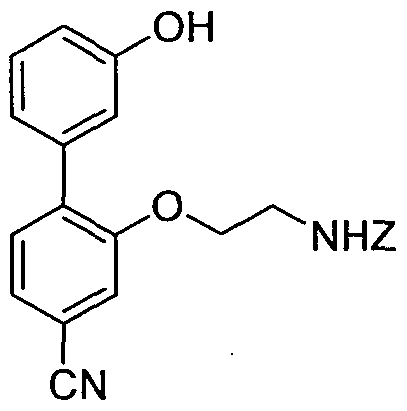
A degassed mixture of benzyl 2-(2-bromo-5-cyanophenoxy)ethylcarbamate (0.7 g, 1.8 mmol), 3-hydroxyphenylboronic acid (0.3 g, 2.2 mmol), PdCl2dppf. CH2Cl2 (73 mg, 0.09 mmol), potassium phosphate (0.83 g, 3.6 mmol), toluene (4 mL) and water (0.4 mL) was heated at 1000C for 18 h. The reaction mixture was diluted in EtOAc (25 mL) and water (25 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 x 25 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under vacuum. The residue was then purified by flash chromatography using
cyclohexane/EtOAc (7/3) as eluent to afford the title compound (0.57 g, 81% yield) as transparent oil.
ESMS; m/z 389 (M+l), 406 (M+18)
1H-NMR (CDCl3): 53.63 (m, 2H), 4.01 (m, 2H), 5.10 (bs, 3H), 5.19 (bs, IH), 6.89 (m, 2H), 6.99 (d, J= 7.6 Hz, IH), 7.09-7.40 (m, 8H), 7.48 (d, J= 7.8 Hz, IH).
13C-NMR (CDCl3): 640.4, 67.3, 67.9, 111.8, 115.6, 116.4, 117.0, 118.7, 121.0, 125.5, 128.2, 128.4, 128.6, 129.6, 131.3, 135.2, 136.0, 137.4, 155.5, 155.9, 156.8. Theoretical Mass: (M + H) 389.15012. Measured Mass: (M + H) 389.15110
Dibenzyl 2,2'-(4-cyanobiphenyl-2,3'-diyl)bis(oxy)bis(ethane-2,l-diyI)dicarbamate

To benzyl 2-(4-cyano-3'-hydroxybiphenyl-2-yloxy)ethylcarbamate (0.49 g, 1.26 mmol) dissolved in DMF (12 mL) was added Cs2CO3 (0.82 g, 2.5 mmol) and 2- (benzyloxycarbonylamino)ethyl methanesulfonate (0.38 g, 1.4 mmol). After stirring at 80 0C under nitrogen for 3 h, further Cs2CO3 (0.41 g, 1.25 mmol) and 2-
(benzyloxycarbonylamino)ethyl methanesulfonate (0.2 g, 0.7 mmol) were added. After stirring for an additional 2 h, the reaction mixture was dissolved in EtOAc (25 mL). The organic layer was washed with brine (3 x 20 mL) and dried over MgSO4. The solvent was removed under vacuum and the crude compound was purified by flash chromatography using cyclohexane/EtOAc (65/35) as eluent to afford the title compound (0.44g, 62% yield) as a colourless oil.
ESMS; m/z 566 (M+l), 583 (M+18)
1H-NMR (CDCl3): 53.51 (bs, 4H), 4.03 (bs, 4H), 5.07 (m, 5H), 5.32 (bs, IH), 6.87 (d, J = 7.6 Hz, IH), 7.04 (m, 2H), 7.21 (s, IH), 7.32-7.38 (m, 13H). 13C-NMR (CDCl3): 540.4, 40.6, 67.0, 68.0, 112.1, 114.0, 115.9, 118.6, 122.0, 125.5, 128.2, 128.3, 128.6, 129.5, 131.5, 135.8, 136.3, 136.4, 138.0, 155.5, 156.3, 156.5, 158.4. Theoretical Mass: (M + H) 566.22910. Measured Mass: (M + H) 566.22854
2, 3'-Bis{2-[N, iV'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-4-cyanobiphenyl

Dibenzyl 2,2'-(4-cyanobiphenyl-2,3'-diyl)bis(oxy)bis(ethane-2,l-diyl)dicarbamate (0.362 g, 0.64 mmol) was dissolved in DCM (10 mL) and HBr (30% in acetic acid, 3.5 mL) was added dropwise. After stirring at room temperature for 1.5 h, the solvent was removed under vacuum and the crude diamine was carefully dried under vacuum for several hours.
The resulting solid was suspended in DCM (5 mL) and Et3N (0.88 mL, 6.4 mmol) and N,
N-di-boc-N'-trifluoromethanesulfonyl-guanidine (0.47 g, 1.2 mmol) were added. The mixture was stirred overnight at room temperature, diluted with DCM (20 mL) then washed with 2M NaHSO4 2M (25 mL), sat. aq. NaHCO3 (25 mL) and brine (25 mL). The organic layer was dried over MgSO4, filtered and concentrated under vacuum. The residue was purified by flash chromatography using cyclohexane/EtOAc (8/2) as eluent to afford the title compound (0.33 g, 66% yield) as a white solid. 1H-NMR (CDCl3): .61.48 (m, 36H), 3.79 (q, J= 5.2 Hz, 2H), 3.84 (q, J= 5 Hz, 2H), 4.09
(m, 4H), 6.94 (dd, J= 8.1 Hz, J= 1.7 Hz, IH), 6.99 (d, J= 1.4 Hz, IH), 7.18-7.22 (m, 2H),
7.34 (m, 2H), 7.40 (d, J= 7.8 Hz, IH), 8.63 (t, J= 5.1Hz, IH), 8.75 (bt, IH), 11.45 (bs,
2H).
13C-NMR (CDCl3): 528.0, 28.3, 39.8, 40.2, 66.5, 67.4, 79.4, 79.5, 83.2, 83.2, 112.0, 114.2, 115.5, 115.8, 118.7, 122.7, 125.3, 129.2, 131.6, 135.8, 137.8, 153.0, 155.5, 156.3, 156.4,
158.6, 163.4, 163.5.
Theoretical Mass: (M + H) 782.40885. Measured Mass: (M + H) 782.40733
M.p. 89-91°C
IR v Max KBR/crn 3333 (br NH), 2229 (CN), 1722 (C=O), 1641 (C=N), 1618 (C=N)
N-{2, 3'-Bis{2-[iV, iV'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-biphenyl-4- yImethyl}-3-[2-pyridyl)dithio]propionamide (Example 6)

2, 3'-Bis{2-[iV, A/'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-4-cyanobiphenyl (0.5Og, 0.7mmol) was dissolved in THF (14 mL) and NH4OH (2.4 mL) was added followed by Raney Nickel (0.50 g). The mixture was stirred vigorously under H2 atmosphere for 16 h. The catalyst was filtered off and washed several times with MeOH. Diethylenetriamine (leq) was added and the mixture stirred for 30 minutes then concentrated under vacuum. The residue was dissolved in EtOAc (10 mL) and washed with sat. aq. NaHCO3 (10 mL). The organic layer was dried over MgSO4, filtered and concentrated under vacuum. To a solution of the crude (0.040 g, 0.050 mmol) and Z-Pr2EtN (10 μL, 0.10 mmol) in DCM (0.7 mL) was added N-succinimidyl-3-(2-pyridyldithio)propionate (15 mg, 0.053 mmol) and the mixture was stirred in the dark for 24 h. The solvent was removed under vacuum and the crude oil was purified by preparative TLC (ethyl acetate/hexane 1/1) to afford the title compound (8.5 mg, 16% yield).
1H-NMR (CDCl3): .51.49 (m, 36H), 2.67 (t, J= 5 Hz, 2H), 3.13 (t, J= 5 Hz, 2H), 3.73 (m, 2H), 3.84 (m, 2H), 4.05 (m, 4H), 4.50 (d, J= 4.1 Hz, 2H), 6.98-6.94 (m, 4H), 7.21-7.730 (m, 3H), 7.55 (m, 2H), 8.19 (d, J= 3.5 Hz, IH), 8.62 (t, J= 4 Hz, IH), 8.76 (m, IH), 11.46 (bs, 2H). 13C-NMR (CDCl3): 527.0, 28.0, 28.2, 28.5, 35.3, 35.8, 39.9, 40.2, 43.6, 66.3, 66.7, 79.3, 79.4, 83.1, 112.0, 113.1, 115.6, 120.6, 120.7, 121.1, 122.9, 128.8, 129.9, 131.1, 137.0, 139.0, 139.3, 149.51, 152.9, 155.47, 156.2, 156.3, 158.4, 159.1, 163.4, 163.5, 170.7, 171.9. Theoretical Mass: (M + H) 983.43705. Measured Mass: (M + H) 983.43839 IR v Max KBR/cm'3332 (NH), 1722 (C=O), 1641 (C=O).
N- [2, 3 '-Bis(2-guanidino-ethyloxy)-biphenyl-4-ylmethyI]-3- [2- pyridyl)dithio]propionamide, formate salt (Example 7)

N- {2, 3'-Bis {2-[N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy} -biphenyl-4-ylmethyl} - 3-[2-pyridyl)dithio]propionamide (11 mg, 0.011 mmol) was dissolved in formic acid (1 mL) and stirred at room temperature overnight. The mixture was then heated at 50°C during Ih. Water was then added and the product was lyophilised to give the title compound as colourless oil (10 mg, 100%).
1H-NMR (MeOH-d4): 52.71 (t, J= 6.6 Hz, 2H), 3.11 (t, 2H, J= 6.9 Hz, 2H), 3.50 (bt, 2H), 3.60 (bt, 2H), 4.12 (m, 4H), 4.41 (s, 2H), 6.92 (d, J= 7.5 Hz, IH), 6.99-7.12 (m, 4H), 7.18-
7.34 (m, 3H), 7.73-7.83 (m, 2H), 8.37 (d, J= 3.9 Hz, IH), 8.44 (bs, 2H).
Theoretical Mass: (M + H) 583.22734. Measured Mass: (M + H) 583.22816
Benzyl 2-(4-bromo-2'-hydroxybiphenyl-3-yloxy)ethylcarbamate

A degassed mixture of benzyl 2-(2-bromo-5-iodophenoxy)ethylcarbamate (1.43 g, 3 mmol), 2-hydroxyphenylboronic acid (0.37 g, 2.7 mmol), PdCl2dppf. CH2Cl2 (0.12 g, 0.15 mmol), potassium phosphate (1.27 g, 6 mmol), toluene (6 mL) and water (0.6 mL) was heated at 100°C for 18 h. The reaction mixture was diluted in EtOAc (25 mL) and water (25 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 x 25 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under vacuum. The residue was then purified by flash chromatography using
cyclohexane/EtOAc (75/25) as eluent to afford the title compound (0.90 g, 75% yield) as a transparent oil.
ESMS; m/z 442, 444 (M+l), 459, 461 (M+18)
1H-NMR (CDCl3): 63.64 (m, 2H), 4.10 (m, 2H), 5.11 (s, 2H), 5.43 (bs, IH), 6.02 (s, IH), 6.97-7 '.03 (m, 4H), 7.25 (m, 2H), 7.35 (bs, 5H), 7.59 (d, J= 8 Hz, IH).
13C-NMR (CDCl3): 540.5, 67.0, 68.4, 111.6, 114.7, 116.2, 120.9, 123.1, 127.2, 128.2, 128.2, 128.6, 129.4, 130.2, 133.6, 136.3, 138.4, 152.6, 155.0, 156.6 Theoretical Mass: (M + Na) 464.04733. Measured Mass: (M + Na) 464.04803
Dibenzyl 2,2'-(4-bromobiphenyl-3,2'-diyl)bis(oxy)bis(ethane-2,l-diyl)dicarbamate

Benzyl 2-(4-bromo-2'-hydroxybiphenyl-3-yloxy)ethylcarbamate (0.8 g, 1.8 mmol) was dissolved in DMF (10 mL) and Cs2CO3 (1.17 g, 3.6 mmol) and 2- (benzyloxycarbonylamino)ethyl methanesulfonate (0.55 g, 2 mmol) were added. After stirring at 80 °C under nitrogen for 3.5 h, the reaction mixture was dissolved in EtOAc (25 mL). The organic layer was washed with brine (3 x 20 mL) and dried over MgSO4. The solvent was removed under vacuum and the crude compound was purified by flash chromatography using cyclohexane/EtOAc (7/3) as eluent to afford the title compound (0.6 g, 54% yield) as a colourless oil. ESMS; m/z 619, 621 (M+l), 636, 638 (M+18)
1H-NMR (CDCl3): .63.50 (q, J= 5.1 Hz, 2H), 3.58 (m, 2H), 4.04 (t, J= 4.8 Hz, 2H), 4.08 (m, 2H), 5.08 (m, 5H), 5.39 (bs, IH), 6.97 (m, 2H), 7.06 (m, 2H), 7.25-7.33 (m, 12H), 7.53 (d, J= 8.1 Hz, IH). 13C-NMR (CDCl3): .640.6, 66.9, 67.7, 68.5, 111.1, 113.2, 115.2, 121.7, 123.6, 128.1, 128.5, 128.6, 129.3, 130.0, 130.7, 132.8, 136.4, 136.5, 139.2, 154.5, 155.2, 156.4, 156.5. Theoretical Mass: (M + Na) 641.12631. Measured Mass: (M + Na) 641.12487
Dibenzyl 2,2'-(4-cyanobiphenyl-3,2'-diyl)bis(oxy)bis(ethane-2,l-diyl)dicarbamate

A mixture of dibenzyl 2,2'-(4-bromobiphenyl-3,2'-diyl)bis(oxy)bis(ethane-2,l- diyl)dicarbamate (0.35 g, 0.56 mmol), zinc cyanide (0.1 g, 0.85 mmol), Pd2dba3 (26 mg, 0.03 mmol), dppf (39 mg, 0.07 mmol), zinc acetate (51 mg, 0.28 mmol) and zinc (18 mg, 0.28 mmol) in dimethylacetamide (4 mL) was heated at 160 0C for 10 minutes under microwave irradiation. The reaction mixture was diluted in EtOAc (25 mL) and water (100 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 x 25 mL). The combined organic layers were washed with water (50 mL) and brine (50 mL), dried over MgSO4, filtered and concentrated under vacuum. The residue was purified by flash chromatography using cyclohexane/EtOAc (65/35) as eluent to afford the title compound (0.24 g, 76% yield) as a colourless oil. ESMS; m/z 566 (M+l), 583 (M+18) 1H-NMR (CDCl3): .53.50 (q, J= 5.2 Hz, 2H), 3.58 (m, 2H), 4.06 (t, J= 4.9 Hz, 2H), 4.14 (m, 2H), 5.07 (m, 5H), 5.30 (bs, IH), 6.97 (d, J= 8.2 Hz, IH), 7.06-7.14 (m, 3H), 7.28-7.38 (m, 12H), 7.53 (d, J= 7.9 Hz, IH).
13C-NMR (CDCl3): 540.3, 40.4, 66.9, 67.5, 68.1, 100.4, 113.0, 113.8, 116.4, 121.7, 122.5, 128.1, 128.2, 128.3, 128.5, 128.6, 129.2, 130.1, 130.7, 133.1, 136.3, 145.2, 155.2, 156.3, 156.4, 159.9. Theoretical Mass: (M + H) 566.22910. Measured Mass: (M + H) 566.22854 IR v wax KBR/cm"3336 (br NH), 2224 (CN), 1712 (C=O)
3, 2'-Bis{2-[./V, 7V'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-4-cyanobiphenyl

1H-NMR (CDCl3): .51.48 (m, 36H), 3.78 (q, J= 4.9Hz, 2H), 3.90 (q, J= 5.1Hz, 2H), 4.08(t, J= 4.8Hz, 2H), 4.23 (t, J= 5Hz, 2H), 6.98 (d, J= 8.2Hz, IH), 7.05 (m, 2H), 7.26-7.37 (m, 4H), 8.60 (t, J= 5.1Hz, IH), 8.82 (t, J= 5Hz, IH), 11.45 (bs, 2H). 13C-NMR (CDCl3): 528.1, 28.3, 39.9, 66.8, 67.5, 79.3, 79.4, 83.3, 83.3, 100.6, 112.3, 113.5, 116.3, 121.5, 123.2, 129.1, 130.0, 130.7, 133.2, 144.9, 153.0, 153.1, 155.1, 156.3, 156.5, 159.9, 163.4.
Theoretical Mass: (M + H) 782.40885. Measured Mass: (M + H) 782.40887 IR v Max KBR/cin 3333 (br NH), 2226 (CN), 1723 (C=O), 1643 (C=N), 1615 (C=N) M.p. 88-90°C
iV-{3, 2'-Bis{2-[iV, N-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-biphenyl-4- ylmethyl}-3-[2-pyridyl)dithio]propionamide (Example 8)

iV-[3, 2'-Bis(2-guanidino-ethyloxy)-biphenyI-4-ylmethyl]-3-[2- pyridyl)dithio]propionamide, formate salt (Example 9)

N- {3, 2'-Bis{2-[N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-biphenyl-4-ylmethyl}- 3-[2-pyridyl)dithio]propionamide (14 mg, 0.014 mmol) was dissolved in formic acid (1 mL) and stirred at room temperature overnight. The mixture was then heated at 50°C during Ih. Water was then added and the product was lyophilised to give the title compound as a colourless oil (11.4 mg, 93%).
1H-NMR (MeOH-(I4): 52.72 (t, J= 6.1 Hz, 2H), 2.97 (t, 2H, J= 6.0 Hz, 2H), 3.49 (bs, 2H), 3.64 (bs, 2H), 4.08 (bs, 2H), 4.17 (bs, 2H), 4.45 (s, 2H), 7.05-7.09 (m, 3H), 7.20 (t, J= 5.7 Hz, IH), 7.28-7.35 (m, 3H), 7.70-7.92 (m, 2H), 8.37 (d, J= 4.5 Hz, IH), 8.40 (bs, 2H). Theoretical Mass: (M + H) 583.22734. Measured Mass: (M + H) 583.22816
Benzyl 2-(4-bromo-3 '-hydroxybiphenyI-3-yIoxy)ethylcarbamate

A degassed mixture of benzyl 2-(2-bromo-5-iodophenoxy)ethylcarbamate (1.43 g, 3 mmol), 2-hydroxyphenylboronic acid (0.37 g, 2.7 mmol), PdCl2dppf. CH2Cl2 (0.12 g, 0.15 mmol), potassium phosphate (1.27 g, 6 mmol), toluene (6 mL) and water (0.6 mL) was heated at 100°C for 18 h. The reaction mixture was diluted in EtOAc (25 mL) and water (25 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 x 25 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under vacuum. The residue was then purified by flash chromatography using cyclohexane/EtOAc (75/25) as eluent to afford the title compound (0.54 g, 45% yield) as a transparent oil.
ESMS; m/z 442, 444 (M+l), 459, 461 (M+18)
1H-NMR (CDCl3): 63.65 (q, J= 5 Hz, 2H), 4.08 (t, J= 4.8 Hz, 2H), 5.14 (s, 2H), 5.43 (bs, IH), 6.77 (s, IH), 6.88 (dd, J= 8.1 Hz, J= 1.7Hz, IH)5 6.98-7.06 (m, 4H), 7.24-7.36 (m, 6H), 7.52 (d, J= 8 Hz, IH).
13C-NMR (CDCl3): .540.6, 67.2, 68.3, 111.5, 112.4, 114.1, 114.9, 119.1, 121.2, 128.2, 128.3, 128.6, 130.1, 133.4, 136.1, 141.6, 141.8, 154.9, 156.6, 156.9. Theoretical Mass: (M + Na) 464.04733. Measured Mass: (M + Na) 464.04628
Dibenzyl 2,2'-(4-bromobiphenyl-3,3'-diyI)bis(oxy)bis(ethane-2,l-diyl)dicarbamate

Benzyl 2-(4-bromo-3'-hydroxybiphenyl-3-yloxy)ethylcarbamate (0.46 g, 1.8 mmol) was dissolved in DMF (10 mL) and Cs2CO3 (1.17 g, 3.6 mmol) and 2-
(benzyloxycarbonylamino)ethyl methanesulfonate (0.55 g, 2 mmol) were added. After stirring at 80 °C under nitrogen for 3.5 h, the reaction mixture was dissolved in EtOAc (25 mL). The organic layer was washed with brine (3 x 20 mL) and dried over MgSO4. The solvent was removed under vacuum and the crude compound was purified by flash chromatography using cyclohexane/EtOAc (7/3) as eluent to afford the title compound (0.33 g, 52 % yield) as a colourless oil. ESMS; m/z 619, 621 (M+l), 636, 638 (M+18)
1H-NMR (CDCl3): 53.65 (m, 4H), 4.10 (t, J= 4.5 Hz, 2H), 4.16 (t, J= 4.4 Hz, 2H), 5.12 (s, 4H), 5.32 (bs, IH), 5.40 (bs, IH), 6.88 (d, J= 6.8 Hz, IH), 7.05 (m, 3H), 7.14 (d, J= 7.7 Hz, IH), 7.35 (bs, 1 IH), 7.57 (d, J= 8.6 Hz, IH).
13C-NMR (CDCl3): 640.6, 66.9, 67.0, 68.5, 111.7, 112.5, 113.5, 119.9, 121.2, 128.2, 128.6, 130.1, 133.5, 136.4, 141.7, 141.8, 155.1, 156.5, 158.9.
Theoretical Mass: (M + Na) 641.12631. Measured Mass: (M + Na) 641.12715 Microanalysis: %C 62.08 (62.04), %H 5.13 (5.04), %N 4.39 (4.52) IR v Max KBR/cm" 3339 (br, NH), 2226 (CN), 1723 (C=O), 1641 (C=N), 1620 (C=N)
Dibenzyl 2,2'-(4-cyanobiphenyl-3,3'-diyl)bis(oxy)bis(ethane-2,l-diyl)dicarbamate

1H-NMR (CDCl3): 53.61-3.71 (m, 4H), 4.10 (t, J= 4.6 Hz, 2H), 4.22 (t, J= 4.7 Hz, 2H), 5.11 (s, 4H), 5.27 (bs, IH), 5.37 (bs, IH), 6.93 (d, J= 8.1 Hz, IH), 7.07-7.22 (m, 4H), 7.29- 7.40 (m, 1 IH), 7.59 (d, J= 8 Hz, IH).
13C-NMR (CDCl3): 540.3, 40.4, 66.9, 67.1, 68.2, 100.9, 111.1, 113.7, 114.5, 120.2, 128.1, 128.2, 128.2, 128.5, 128.6, 130.3, 134.0, 136.3, 140.9, 147.4, 156.5, 159.0, 160.4. Theoretical Mass: (M + H) 566.22910. Measured Mass: (M + H) 566.22964 IR v Max KBR/cm"3339 (br, NH), 2225 (CN), 1713 (C=O)
3, 3'-Bis{2-[iV, iV'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-4-cyanobiphenyl

Dibenzyl 2,2'-(4-cyanobiphenyl-3,3l-diyl)bis(oxy)bis(ethane-2,l-diyl)dicarbamate (0.15 g, 0.26 mmol) was dissolved in DCM (10 mL) and HBr (30% in acetic acid, 1.5 mL) was added dropwise. After stirring at room temperature for 1.5 h, the solvent was removed under vacuum and the crude diamine was carefully dried under vacuum for several hours. The resulting solid was suspended in DCM (5 mL) and Et3N (0.36 mL, 2.6 mmol) and N, N-di-boc-N'-trifluoromethanesulfonyl-guanidine (0.21 g, 0.53 mmol) were added. The
mixture was stirred overnight at room temperature, diluted with DCM (20 mL) then washed with 2M NaHSO42M (25 mL) followed by sat.aq. NaHCO3 (25 mL) and brine (25 mL). The organic layer was dried over MgSO4, filtered and concentrated under vacuum. The residue was then purified by flash chromatography using cyclohexane/EtOAc (8/2) as eluent to afford the title compound (0.18 g, 86% yield) as a white solid.
1H-NMR (CDCl3): 61.50 (m, 36H), 3.86 (q, J= 5.1 Hz, 2H), 3.93 (q, J= 5.2 Hz, 2H), 4.15 (t, J= 5 Hz, 2H), 4.29 (t, J= 5.1 Hz, 2H), 6.99 (dd, J= 8.1 Hz, J=2 Hz, IH), 7.10-7.16 (m, 3H), 7.21 (d, J= 8 Hz, IH), 7.38 (t, J= 8 Hz5 IH), 7.60 (d, J= 8 Hz, IH), 8.62 (bt, IH), 8.82 (bt, IH), 11.45 (bs, 2H). 13C-NMR (CDCl3): 527.8, 28.1, 28.3, 39.9, 40.2, 66.6, 67.6, 79.4, 83.2, 83.3, 101.2, 111.3, 114.1, 114.7, 116.0, 120.2, 120.3, 130.2, 134.1, 141.0, 147.4, 153.0, 153.1, 156.4, 156.5, 159.1, 160.4, 163.5.
Theoretical Mass: (M + H) 782.40885. Measured Mass: (M + H) 782.40656 IR v Max KBR/cnϊ 3332 (br, NH), 2226 (CN), 1723 (C=O), 1641 (C=N), 1620 (C=N) M.p. 82-84°C
N-{3, 3'-Bis{2-[W, iV-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-biphenyl-4- ylmethyl}-3-[2-pyridyl)dithio]propionamide (Example 10)

To a solution of palladium on carbon 10% (25 mg), Raney nickel (25 mg) and NH4OH (20 μL) in dioxane (40 mL) and water (10 mL) was added 3, 3'-Bis{2-[N, N'-bis(tert- butoxycarbonyl)guanidino]-ethyloxy}-5-cyanobiphenyl (0.070 g, 0.09 mmol). The mixture was then hydrogenated (6 bar) overnight. The catalyst was then filtered off and washed several times with MeOH. Diethylenetriamine (leq) was added and the mixture was stirred for 30 minutes then concentrated under vacuum. The residue was dissolved in EtOAc (10
mL) and washed with sat. aq. NaHCO3 10 mL). The organic layer was dried over MgSO4, filtered and concentrated under vacuum. To a solution of the crude and J--Pr2EtN (31 μL, 0.18 mmol) in DCM (2 mL) was added N-succinimidyl-3-(2-pyridyldithio)propionate (39 mg, 0.126 mmol) and the mixture was stirred in the dark for 24 h. The solvent was removed under vacuum and the crude oil was purified by preparative TLC (ethyl acetate/hexane 1/1) to afford the title compound (8 mg, 11% yield).
1H-NMR (CDCl3): .81.50 (m, 36H), 2.64 (t, 2H), 3.08 (t, J= 6.9 Hz, 2H), 3.89 (m , 4H), 4.17 (m, 4H), 4.50 (d, J= 5.7 Hz, 2H), 6.81 (bt, IH), 6.92 (dd, J= 7.8 Hz, J= 2.4 Hz, IH), 7.02-7.15 (m, 5H), 7.32 (t, J= 4.5 Hz, IH), 7.42 (d, J= 8.1 Hz, IH), 7.53-7.63 (m, 2H), 8.34 (d, J= 3.6 Hz, IH), 8.78 (m, 2H), 8.78 (bt, IH), 11.49 (bs, IH), 11.54 (bs, IH).
13C-NMR (CDCl3): 828.0, 28.3, 34.4, 35.3, 38.5, 39.9, 40.1, 66.3, 66.7, 79.3, 79.5, 83.1, 83.4, 111.5, 112.9, 120.1, 120.4, 125.7, 129.2, 130.1, 136.0, 140.9, 141.3, 148.0, 153.7, 156.9, 159.6, 163.5, 170.5. Theoretical Mass: (M + H) 983.43705. Measured Mass: (M + H) 983.43928
iV-[3, 3'-Bis(2-guanidino-ethyloxy)-biphenyl-4-ylmethyl]-3-[2- pyridyl)dithio]propionamide, formate salt (Example 11)

N- {3, 3'-Bis {2-[N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy} -biphenyl-4-ylmethyl} - 3-[2-pyridyl)dithio]propionamide (8 mg, 0.0081 mmol) was dissolved in formic acid (1 mL) and stirred at room temperature overnight. The mixture was then heated at 50°C for
Ih. Water was added and the product was freeze-dried to give the title compound as colourless oil (7.1 mg, 100%).
1H-NMR (MeOH-Cl4): 82.69 (t, J= 6.8 Hz, 2H), 3.09 (t, 2H, J= 6.5 Hz, 2H), 3.60-3.68 (m, 4H), 4.18(t, J= 4.9 Hz, 2H), 4.24 (t, J= 4.8 Hz, 2H), 4.46 (s, 2H), 6.95 (d, J= 7.5 Hz, IH),
7.18-7.24 (m, 4H), 7.31-7.39 (m, 2H), 7.71-7.78 (m, 2H), 8.36 (d, J= 4.6 Hz, IH), 8.43 (bs,
2H).
Theoretical Mass: (M + H) 583.22734. Measured Mass: (M + H) 583.22796
iV-(2-(4-cyanophenoxy)ethyl)-2-phenyIacetamide

3-Bromo-4-hydroxybenzonitrile (7.25 g, 36.6 mmol) was dissolved in DMF (20 mL) and
Na2CO3 (3.7 g, 73.2 mmol) and 2-(benzyloxycarbonylamino)ethyl methanesulfonate (12 g, 42 mmol) were added. After stirring at 80 °C under nitrogen for 15 h, the reaction mixture was dissolved in EtOAc (25 mL). The organic layer was washed with brine (3 x 20 mL), dried over MgSO4 and filtered. The solvent was removed under vacuum and the crude compound was purified by flash chromatography using cyclohexane/EtOAc (8/2) as eluent to afford the title compound (9.4 g, 68% yield) as a white solid. 1H-NMR (CDCl3): 53.70 (q, J= 5.3 Hz, 2H), 4.13 (m, 2H), 5.11 (s, 2H), 5.23 (bs, IH), 6.90
(d, J= 8.5 Hz, IH), 7.35 (m, 5H), 7.56 (d, J= 8.5 Hz, IH), 7.81 (d, J= 1.9 Hz, IH).
13C-NMR (CDCl3): 540.2, 67.0, 68.5, 105.7, 112.7, 113.0, 117.6, 128.2, 128.3, 128.6,
133.2, 136.3, 136.8, 156.4, 158.4.
Theoretical Mass: (M + Na) 397.01637. Measured Mass: (M + Na) 397.01539. Microanalysis: %C 54.10 (54.42), %H 4.23 (4.03), %N 7.19 (7.47)
IR v Max KBR/cm"3351(br NH, NH2), 2227 (CN), 11704 (C=ON)
M.p. 65-68°C
Benzyl 2-(5-cyano-2'-hydroxybiphenyl-2-yIoxy)ethylcarbamate

Dibenzyl 2,2'-(5-cyanobiphenyl-2,2'-diylbis(oxy))bis(ethane-2,l-diyl)tetracarbamate

(benzyloxycarbonylamino)ethyl methanesulfonate (0.75 g, 2.7 mmol) were added. After stirring at 80 0C under nitrogen overnight, further Cs2CO3 (0.9 g, 2.7 mmol) and 2- (benzyloxycarbonylamino)ethyl methanesulfonate (0.4 g, 1.5 mmol) were added. After stirring for a further 4 h, the reaction mixture was dissolved in EtOAc (25 mL). The organic layer was washed with brine (3 x 20 mL) and dried over MgSO4. The solvent was removed under vacuum and the crude compound was purified by flash chromatography using cyclohexane/EtOAc (1/1) as eluent to afford the title compound (1.23 g, 80% yield) as a colourless oil.
1H-NMR (CDCl3): δ3,32(m, 2H), 3.80 (t, J= 8.9 Hz, 2H), 4.00 (m, 2H), 4.39 (t, J= 8.7 Hz, 2H), 5.04 (s, 2H), 5.22 (s, 2H), 5.30 (bs, IH), 5.54 (bs, IH), 6.86 (m, 2H), 7.02 (t, J= 7.3 Hz, IH), 7.13 (dd, J= 8.9 Hz, J= 1.7 Hz, IH), 7.34 (m, HH), 7.46 (d, J= 2 Hz, IH), 7.53 (dd, J= 8.5 Hz, 1.8 Hz, IH). 13C-NMR (CDCl3): 640.1, 40.3, 66.8, 68.0, 68.3, 104.7, 112.8, 113.5, 118.9, 121.9, 126.4,
128.1, 128.3, 128.5, 128.6, 129.7, 129.8, 131.0, 133.5, 134.9, 135.4, 136.4, 155.6, 156.2,
159.2, 163.6.
Theoretical Mass: (M + Na) 588.21104. Measured Mass: (M + Na) 588.21104 IR v Max KBR/cm" 3342 (br, NH), 2224 (CN), 1721 (C=O)
2, 2'-Bis{2-[N, Λ^'-bis(tert-butoxycarbonyI)guanidino]-ethyloxy}-5-cyanobiphenyl

Dibenzyl 2,2'-(5-cyanobiphenyl-2,2'-diylbis(oxy))bis(ethane-2,l-diyl)tetracarbamate (0.9 g, 1.6 mmol) was dissolved in DCM (50 mL) and HBr (30% in acetic acid, 8 mL) was added dropwise. After stirring at room temperature for 1.5 h, water (25 mL) was added to the mixture, the layers were separated and the aqueous layer was washed with DCM (2 x 25 mL). The water was then removed under vacuum and the crude diamine was carefully dried under vacuum for several hours. The residue was suspended in DCM (7 mL) and Et3N (1.2 mL, 8 mmol) and N, N-di-boc- N'-trifluoromethanesulfonyl-guanidine (1.3 g, 3.2 mmol) were added. The mixture was stirred overnight at room temperature, diluted with DCM (20 mL) then washed with 2M NaHSO4 2M (25 mL) follow by sat. aq. NaHCO3 (25 mL) and brine (25 mL). The organic layer was dried over MgSO4, filtered and concentrated under vacuum. The residue was purified by flash chromatography using cyclohexane/EtOAc (8/2) as eluent to afford the title compound (0.28 g, 22% yield) as a white solid. ESMS; m/z 782(M+1), 391(M+2)
1H-NMR (CDCl3): 51.48 (m, 36H), 3.69 (m, 4H), 4.05 (t, J= 5.2 Hz, 2H), 4.13 (t, J= 5.2 Hz, 2H), 6.95-7.06 (m, 3H), 7.22 (dd, 7=7.5, 1.7 Hz, IH), 7.32 (td, J= 7.2, 1.7 Hz, IH), 7.55 (d, J= 2 Hz, IH), 7.59 (dd, J= 8.5, 2.1 Hz, IH), 8.40 (m, 2H), 11.42 (bs, 2H). 13C-NMR (CDCl3): 628.1, 28.3, 39.8, 40.1, 66.7, 66.9, 79.3, 79.4, 83.1, 104.1, 112.2, 112.4, 119.2, 121.0, 125.6, 129.1, 129.4, 131.3, 133.2, 135.2, 152.9, 155.6, 156.3, 159.2, 163.4. Theoretical Mass: (M + Na) 804.39079. Measured Mass: (M + Na) 804.39290 IR v Max KBR/crn 3333 (br, NH), 2226 (CN), 1722 (C=O), 1642 (C=N), 1619 (C=N) M.p. 82°C
2, 2'-Bis{2-[iV, iV-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-5- (aminomethyl)biphenyl (Example 12)

2, 2'-Bis {2-[N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy} -5-cyanobiphenyl (0.1O g, 0.12 mmol) was dissolved in THF (7 mL) and NH4OH (1.4 mL) was added followed by Raney Nickel (0.1 g). The mixture was stirred vigorously under H2 atmosphere for 16 h. The catalyst was filtered off and washed several times with MeOH. Diethylenetriamine (leq) was added and the mixture stirred for 30 minutes then concentrated under vacuum. The residue was dissolved in EtOAc (10 mL) and washed with sat. aq. NaHCO3 (10 mL). The organic layer was dried over MgSO4, filtered and concentrated under vacuum. The residue was then purified by flash chromatography using DCM/MeOH (97/3) as eluent to afford the title compound (45 mg, 48% yield). ESMS; m/z 786(M+1), 394(M+2) 1H-NMR (CDCl3): .51.47 (m, 36H), 3.63 (m, 4H), 3.81 (s, 2H), 4.02 (m, 4H), 6.90-6.99 (m, 3H), 7.22 (m, 4H), , 8.40 (m, 2H), 11.42 (bs, 2H).
13C-NMR (CDCl3): 528.1, 28.3, 40.2, 40.3, 67.1, 79.1, 79.2, 82.8, 82.9, 112.9, 113.0, 121.0, 127.0, 127.8, 128.0, 128.5, 130.6, 131.8, 135.7, 152.8, 154.7, 155.7, 156.2, 163.4. IR v Max KBR/cm~3338 (NH, NH2), 1723 (C=O), 1641(C=N), 1614 (C=N)
N-{2, 2'-Bis{2-[iV, N-bis(tert-butoxycarbonyl)guanidino]-ethyIoxy}-biphenyI-5- yImethyl}-3-[2-pyridyl)dithio]propionamide (Example 13)
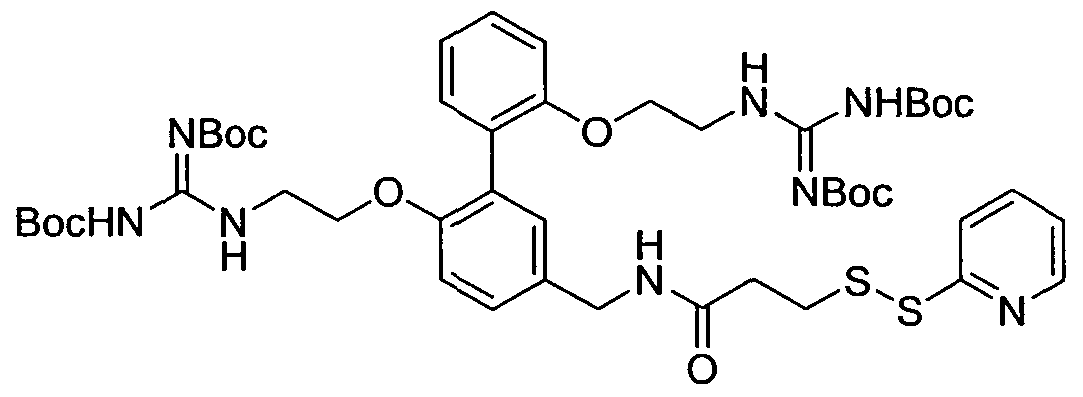
1H-NMR (CDCl3): δl.48 (m, 36H), 2.61 (t, J= 6.8 Hz, 2H), 3.09 (t, J= 6.7 Hz, 2H), 3.65 (m, 4H), 4.02 (m, 4H), 4.42 (d, J= 5.4 Hz, 2H), 6.89-7.03 (m, 6H), 7.23-7.30 (m, 3H), 7.58 (m, 2H), 8.23 (m, 2H), 8.23 (dd, J= 3.8, 1.5Hz, IH), 8.36 (bt, IH), 8.47 (bt, IH), 11.66 (bs, 2H).
Theoretical Mass: (M + H) 983.43705. Measured Mass: (M + H) 983.43763 IR v Max KBR/cm" 3332 (NH), 1722 (C=O), 1641 (C=O).
N-[2, 3'-Bis(2-guanidino-ethyloxy)-biphenyI-5-ylmethyl]-3-[2- pyridyl)dithio]propionamide, trifluoroacetate salt (Example 14)

N- {2, 3'-Bis {2-[N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy} -biphenyl-5- ylmethyl}-3-[2-pyridyl)dithio]propionamide (8.5 mg, 0.0086 mmol) was dissolved in a mixture of TFA/ H2O/triisopropylsilane 95/2.5/2.5 (1 mL) and stirred at room temperature for 3 h. The solvent was removed under vacuum to give the title compound as a colourless oil (8 mg, 100%).
1H-NMR (MeOH-d4): 52.71 (t, J= 6.7Hz, 2H), 3.07 (t, 2H, J= 6.7Hz, 2H), 3.41 (m, 4H), 4.05 (m, 4H), 4.35 (s, 2H), 7.07 (m, 4H), 7.18 (m, 4H), 7.27 (m, 2H), 7.35 (m, 2H), 7.75 (m, 2H), 8.37 (m, IH). Theoretical Mass: (M + H) 583.22734. Measured Mass: (M + H) 583.22566 IR v Max KBR/cm"3336 (NH), 1642 (C=O)
Benzyl 2-(5-cyano-3 '-hydroxybiphenyI-2-yloxy)ethylcarbamate

A degassed mixture of iV-(2-(4-cyanophenoxy)ethyl)-2-phenylacetamide (3.7 g, 9.8 mmol), 3-hydroxyphenyl boronic acid (1.6 g, 11.6 mmol), Pd(OAc)2 (22 mg, 0.1 mmol) 2- dicyclohexylphosphino-2',6'-dimethoxybiphenyl (82 mg, 0.2 mmol) potassium phosphate
(4.2 g, 20 mmol), toluene (20 mL) and water (2 mL) was heated at 1000C for 1 h. The reaction mixture was diluted in EtOAc (50 mL) and water (50 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 x 25 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under vacuum.
The residue was then purified by flash chromatography using cyclohexane/EtOAc (7/3) as eluent to afford the title compound (2.2 g, 59% yield) as a colourless solid.
ESMS; m/z 389 (M+ 1)
1H-NMR (CDCl3): 53.40 (m, 2H), 3.60 (m, 2H), 5.08 (s, 2H), 5.23 (m, 2H), 6.89 (m, 4H), 6.95 (2, IH), 7.18 (m, 56H), 7.53 (m, IH).
Microanalysis: C23H20N2O4- H2O: %C 67.81 (67.97), %H 5.41 (5.46), %N 6.85 (6.89).
IR v Max KBR/cm'3347(br NH, NH2), 2225(CN), 1714 (C=ON)
Mp >200°C
ΛL{2-[5'-Cyano-2'-(2-phenylacetylamino-ethoxy)-biphenyl-3-yloxy]-ethyl}-2-phenyl- acetamide

Benzyl 2-(5-cyano-3'-hydroxybiphenyl-2-yloxy)ethylcarbamate (0.62 g, 1.6 mmol) was dissolved in DMF (8 mL) and Cs2CO3 (1.04 g, 3.2 mmol) and 2- (benzyloxycarbonylamino)ethyl methanesulfonate (0.44 g, 1.6 mmol) were added. After
stirring at 80 0C under nitrogen for 3.5 h, the reaction mixture was dissolved in EtOAc (25 mL). The organic layer was washed with brine (3 x 20 mL) and dried over MgSO4. The solvent was removed under vacuum and the crude compound was purified by flash chromatography using cyclohexane/EtOAc (1/1) as eluent to afford the title compound (0.86 g, 95% yield).
1H-NMR (CDCl3): 63.70 (q, J= 5.3 Hz, 2H), 4.13 (m, 2H), 5.11 (s, 2H), 5.23 (bs, IH), 6.90 (d, J= 8.5 Hz, IH), 7.35 (m, 5H), 7.56 (d, J= 8.5 Hz, IH), 7.81 (d, J= 1.9 Hz, IH). 13C-NMR (CDCl3): D40.2, 67.0, 68.5, 105.7, 112.7, 113.0, 117.6, 128.2, 128.3, 128.6, 133.2, 136.3, 136.8, 156.4, 158.4. IR v wax KBR/crn 3335(br NH, NH2), 2225(CN), 1714 (C=ON)

Microanalysis: C39H55N7O10: %C 60.30 (59.91), %H 7.20 (7.09), %N 12.45 (13.54). IR v Max KBR/crn 3333 (br, NH), 2226 (CN), 1722 (C=O), 1642 (C=N), 1619 (C=N) Mp 120-1210C
2, 3'-BiS(Z-[N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-5- (aminomethyl)biphenyl (Example 15)

2, 3 '-Bis {2- [N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-5-cyanobiphenyl (0.54 g, 0.7 mmol) was dissolved in THF (14 mL) and NH4OH (2.4 mL) was added followed by
Raney Nikel (0.5 g). The mixture was stirred vigorously under H2 atmosphere for 16 h. The catalyst was filtered off and washed several times with MeOH. Diethylenetriamine (leq) was added and the mixture stirred for 30 minutes then concentrated under vacuum. The residue was dissolved in EtOAc (10 mL) and washed with sat. aq. NaHCO3 (10 mL). The organic layer was dried over MgSO4, filtered and concentrated under vacuum. The residue was then purified by flash chromatography using DCM/MeOH (97/3) as eluent to afford the title compound (0.213 g, 39% yield).
ESMS; m/z 787 (M+l), 394 (M+2)
1H-NMR (CDCl3): 51.47 (m, 36H), 3.59 (m, 2H), 3.78 (m, 4H), 4.03 (t, J= 4.9 Hz, 2H), 4.10 (t, J= 4.9 Hz, 2H), 6.86 (dd, J= 8, 2Hz, IH), 6.93 (d, J= 8.3 Hz, IH), 7.20-7.30 (m,
4H), 8.63 (t, J= 5.2 Hz, IH), 8.74 (t, J= 5.1 Hz, IH), 11.54 (bs, 2H).
13C-NMR (CDCl3): 528.1, 28.3, 40.1, 40.3, 45.9, 66.3, 67.1, 79.3, 83.0, 83.1, 112.9, 113.2,
115.6, 123.0, 127.3, 128.9, 129.9, 130.9, 136.2, 139.7, 152.9, 153.0, 154.2, 156.3, 156.3,
158.4, 163.5, 163.5.
Microanalysis: C39H59N7O10- H2O: %C 58.60 (58.27), %H 7.4 (7.65), %N 12.20 (11.91)
IR v Max KBR/crn 3338 (br NH, NH2), 1722 (C=ON), 1641 (C=O)
Mp 124-1250C
iV-{2, 3'-Bis{2-[iV, N'-bis(tert-butoxycarbonyl)guanidino]-ethyIoxy}-biphenyI-5- ylmethyl}-3-[2-pyridyI)dithio]propionamide (Example 16)

To a solution of 2, 3'-Bis{2-[N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-5- (aminomethyl)biphenyl (0.10 g, 0.12 mmol) and Z-Pr2EtN (40 μL, leq) in DCM (1.5 mL) was added N-succinimidyl-3-(2-pyridyldithio)propionate (44 mg, 0.12 mmol) and the mixture was stirred in the dark for 24 h. The solvent was removed under vacuum and the crude oil was purified by preparative TLC (ethyl acetate/hexane 1/1) to afford the title compound (25 mg, 21% yield). ESMS; m/z 983(M+1), 492(M+2)
1H-NMR (CDCl3): 61.49 (m, 36H), 2.62 (t, J= 6.5 Hz, 2H), 2.72 (t, J= 6.5 Hz, 2H), 3.76 (m, 2H), 3.83 (m, 2H), 4.08 (m, 4H), 4.45 (d, J= 5.4 Hz, 2H), 6.95 (m, 4H), 7.21 (m, 4H), 7.55 (m, 2H), 8.10 (d, J= 4.8 Hz, IH), 8.46 (t, J= 4.7 Hz, IH), 8.74 (bs, IH), 11.47 (bs, 2H).
13C-NMR (MeOH-d4): 528.0, 28.3, 34.5, 35.3, 39.5, 39.8, 43.3, 66.3, 67.0, 79.3, 81.3, 83.1, 110.5, 111.2, 113.3, 120.5, 120.9, 122.8, 128.1, 128.9, 130.9, 131.0, 1369, 149.5, 151.2, 153.0, 156.3, 158.8, 159.1, 163.5, 170.5. IR v Max KBR/cm3332 (NH), 1722 (C=O), 1641 (C=O)
iV-[2, 3'-Bis(2-guanidino-ethyloxy)-biphenyl-5-ylmethyl]-3-[2- pyridyl)dithio]propionamide, trifluoroacetate salt (Example 17)

N- {2, 3'-Bis{2-[N, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-biphenyl-5-ylmethyl}- 3-[2-pyridyl)dithio]propionamide (25 mg, 0.025 mmol) was dissolved in a mixture of
TFA/H2O/triisopropylsilane 95/2.5/2.5 (1 mL) and stirred at room temperature for 3 h. The solvent was removed under vacuum to give the title compound as colourless oil (20 mg,
80%).
ESMS; m/z 582(M+1), 292(M+2) 1H-NMR (MeOH-d4): 52.65 (t, J= 6.7 Hz, 2H), 3.07 (t, 2H, J= 6.7 Hz, 2H), 3.50 (m, 2H),
3.60 (m, 2H), 4.10 (m, 4H), 4.35 (s, 2H), 6.91 (d, J= 7.4 Hz, IH), 7.10 (m, 7H), 7.74 (m,
2H), 8.35 (d, J= 4.7 Hz, IH).
13C-NMR (MeOH-d4): 526.9, 29.7, 40.4, 67.2, 67.7, 104.6, 112.8, 115.4, 116.8, 119.0,
121.0, 128.1, 128.3, 128.6, 129.6, 131.4, 133.1, 134.9, 135.9, 137.1, 156.0, 156.8, 158.7. IR v Max KBR/cm"3376 (NH), 1681(C=ON)
Examples 18 to 20: Synthesis of SMOC carrying 3 guanidine moieties
Tribenzyl 2,2',2"-(5-cyanobiphenyl-2,2',3'-triyltris(oxy))tris(ethane-2,l- diyl)tetracarbamate
ZHN
 A degassed mixture of N-(2-(4-cyanophenoxy)ethyl)-2-phenylacetamide (1.0 g, 2.67 mmol) 2,2'-(3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l,2-phenylene)bis(oxy)bis (ethane- 2,l-diyl)dicarbamate (2.07 g, 3.47 mmol), PdCl2dppf.CH2Cl2 (100 mg, 4 mol%), potassium phosphate (1.13 g, 5.34 mmol), toluene (2 mL) and water (0.133 mL) was heated at 100°C for 18 h. The reaction mixture was diluted in EtOAc (25 mL) and water (25 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 x 25 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under vacuum. The residue was then purified by flash chromatography using cyclohexane/EtOAc (75/35) as eluent to afford the title compound (1.9 g, 93% yield). 1H-NMR (CDCl3): 53.15 (m, 2H), 3.38 (m, 2H), 3.46 (m, 2H), 3.60 (m, 2H), 3.68 (m, 2H), 4.12 (m, 4H), 5.00 (bs, 2H), 5.03 (bs, 2H) 5.07 (bs, 2H), 5.25 (bs, IH), 5.32 (bs, IH), 5.97 (bs, IH), 6.61-6.93 (m, 6H), 7.07-7.61 (m, 15H). IR v Max KBR/cm 3335(br, NH), 2225(CN), 1714 (C=O)
A degassed mixture of N-(2-(4-cyanophenoxy)ethyl)-2-phenylacetamide (1.0 g, 2.67 mmol) 2,2'-(3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l,2-phenylene)bis(oxy)bis (ethane- 2,l-diyl)dicarbamate (2.07 g, 3.47 mmol), PdCl2dppf.CH2Cl2 (100 mg, 4 mol%), potassium phosphate (1.13 g, 5.34 mmol), toluene (2 mL) and water (0.133 mL) was heated at 100°C for 18 h. The reaction mixture was diluted in EtOAc (25 mL) and water (25 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 x 25 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under vacuum. The residue was then purified by flash chromatography using cyclohexane/EtOAc (75/35) as eluent to afford the title compound (1.9 g, 93% yield). 1H-NMR (CDCl3): 53.15 (m, 2H), 3.38 (m, 2H), 3.46 (m, 2H), 3.60 (m, 2H), 3.68 (m, 2H), 4.12 (m, 4H), 5.00 (bs, 2H), 5.03 (bs, 2H) 5.07 (bs, 2H), 5.25 (bs, IH), 5.32 (bs, IH), 5.97 (bs, IH), 6.61-6.93 (m, 6H), 7.07-7.61 (m, 15H). IR v Max KBR/cm 3335(br, NH), 2225(CN), 1714 (C=O)
2, 2', 3'-Tris{2-[iV, N'-bis^ert-butoxycarbony^guanidinol-ethyloxyJ-S-cyanobiphenyl

Tribenzyl 2,2',2"-(5-cyanobiphenyl-2,2',3I-triyltris(oxy))tris(ethane-2, 1 -diyl) tetracarbamate
(1.6 g, 2.11 mmol) was dissolved in DCM (10 mL) and HBr (30% in acetic acid, IQmL) was added dropwise. After stirring at room temperature for 2 h, water (25 mL) was added to the mixture, the layers were separated and the aqueous layer was washed with DCM (2 x
25 mL). The water was then removed under vacuum and the crude diamine was carefully dried under vacuum for several hours.
ESMS; m/z 357 (M+l)
1H-NMR (MeOH-Cl4): 53.05 ((t, J= 5.1 Hz, 2H), 3.30 (m, 2H), 3.52 (t, J= 4.7 Hz, 2H), 3.98 (t, J= 5.3 Hz, 2H), 3.68 (m, 2H), 4.12 (m, 4H), 5.00 (bs, 2H), 5.03 (bs, 2H) 5.07 (bs,
2H), 5.25 (bs, IH), 5.32 (bs, IH), 5.97 c, 6.99 (dd, J= 6.9, 2.3Hz, IH), 7.17-7.26 (m, 2H), (d, J= 8.6 Hz, IH), 7.65 (s, IH), 7.80 (dd, J= 8.6, 2.1 Hz, IH).
The resulting solid was suspended in DCM (10 mL) and Z-Pr2EtN (1.6 mL) and N, N-di- boc-N'-trifluoromethanesulfonyl-guanidine (2.47 g, 3eq) were added. The mixture was stirred overnight at room temperature, diluted with DCM (40 mL) then washed with 2M NaHSO4 (25 mL) followed by sat. aq. NaHCO3 (25 mL) and brine (25 mL). The organic layer was dried over MgSO4, filtered and concentrated under vacuum. The residue was then purified by flash chromatography using cyclohexane/EtOAc (80/20) as eluent to afford the title compound (1.3 g, 57% yield) as a white solid. ESMS; m/z 1084(M+l), 542(M+2)
1H-NMR (CDCl3): δl.43-1.53 (m, 54H), 3.43 (m, 2H), 3.73 (m, 2H), 3.84 (m, 2H), 3.91 (m, 2H), 4.07-4.18 (m, 5H), 6.88 (m, IH), 6.97 (m, IH), 7.06 (m, 2H), 7.56 (bs, IH), 8.41 (bt, IH), 8.44 (bt, IH), 8.80 (bt,lH), 11.42 (bs, IH), 11.47 (bs, IH), 11.10 (bs, IH). 13C-NMR (CDCl3): .627.8, 28.0, 28.1, 28.3, 38.9, 39.7, 40.6, 66.5, 71.0, 73.5, 79.3, 82.8, 83.1, 85.9, 104.9, 112.7, 114.7, 124.5, 124.1, 127.7, 130.8, 135.9 , 145.8, 149.0, 151.4, 153.0, 156.2, 156.3, 159.5, 163.4.
IR v Max KBR/cm" 3326 (br NH, NH2), 2226 (CN), 1724 (C=O), 1619 (C=O) Mp 74-750C
2, 2', 3'-Tris{2-[iV, iV'-bis(tert-butoxycarbonyI)guanidino]-ethyIoxy}-5- (aminomethyl)biphenyl (Example 18)
BocHN

2,2',3'-Tris {2-[N,N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy} -5-cyanobiphenyl (0.100 g, 0.092 mmol) was dissolved in THF (1.2 mL) and NH4OH (0.30 mL) was added followed by Raney Nickel (0.010 g). The mixture was stirred vigorously under H2 atmosphere for 16 h. The catalyst was filtered off and washed several times with MeOH.
Diethylenetriamine (leq) was added and the mixture stirred for 30 minutes then concentrated under vacuum. The residue was dissolved in EtOAc (10 mL) and washed with sat. aq. NaHCO3 (10 mL). The organic layer was dried over MgSO4, filtered and concentrated under vacuum. The residue was then purified by flash chromatography using MeOH/DCM (5/95) as eluent to afford the title compound (70 mg, 69% yield).
1H-NMR (CDCl3): .51.47 (m, 54H), 3.38 (m, 2H), 3.70 (m, 2H), 3.79 (m, 4H), 3.89 (m, 2H), 3.70 (m, 2H), 4.01 (m, 2H), 6.70-6.95 (m, 4H), 7.05 (m, 2H), 8.34 (bt, IH), 8.42 (bt, IH), 8.80 (m, IH), 11.43 (m, 2H), 11.46 (bs, IH).
13C-NMR (CDCl3): 528.1, 28.9, 30.34, 38.72, 40.1, 41.5, 41.4, 45.8, 50.6, 66.7, 67.2, 68.1, 71.0, 72.4, 78.8, 79.2, 82.6, 82.8, 83.0, 112.8, 123.8, 124.4, 127.1, 127.2, 128.5, 131.0, 131.4, 132.5, 136.0, 145.2, 151.6, 152.8, 152.9, 154.3, 156.0, 156.3, 156.3, 163.4. IR v Max KBR/cm- 3335 (br NH, NH2), 1722 (C=O), 1641 (C=ON)
N-{2, 2',3'-Tris{2-[N, N'-bis(tert-butoxycarbonyI)guanidino]-ethyloxy}-biphenyl-5- ylmethyl}-3-[2-pyridyl)dithio]propionamide (Example 19)

To a solution of 2,2',3'-Tris{2-[iV, N'-bis(tert-butoxycarbonyl)guanidino]-ethyloxy}-5-
(aminomethyl)biphenyl (50 mg, 0.045 mmol) and Z-Pr2EtN (10 μL, 0.10 mmol) in DCM
(0.7 mL) was added N-succinimidyl-3-(2-pyridyldithio)propionate (13 mg, 0.045 mmol) and the mixture was stirred in the dark for 24 h. The solvent was removed under vacuum and the crude oil was purified by preparative TLC (ethyl acetate/hexane 35/75) to afford the title compound (35 mg, 60 % yield).
ESMS; m/z 1285 (M+l), 643 (M+2), 429 (M+3).
1H-NMR (CDCl3): .51.43-1.53 (m, 54H), 2.64 (t, J= 7.1 Hz, 2H), 3.10 (t, J= 6.8 Hz, 2H), 3.40 (m, 2H), 3.71 (m, 2H), 3.83 (m, 2H), 3.91 (m, 2H), 4.03 (m, 2H), 4.15 (m, 2H), 4.38
(d, J= 5.6 Hz, 2H), 6.84-7.06 (m, 7H), 7.23 (m, IH), 7.61 (m, 2H), 8.31 (dd, J= 4.1, IHz,
IH), 8.39 (bt, IH), 8.52 (bt, IH), 8.80 (bt,lH), 11.40 (bs, IH), 11.45 (bs, IH), 11.05 (bs, IH).
13C-NMR (CDCl3): 521.0, 25.5, 28.0, 28.1, 28.3, 34.7, 35.6, 40.0, 41.3, 43.0, 60.3, 66.7, 67.1, 70.6, 79.1, 79.2, 82.9, 83.0, 112.1, 112.7, 119.9, 120.7, 123.8, 124.3, 127.0, 128.8, 130.5, 131.4, 132.7, 136.9, 145.4, 1495, 151.6, 152.8, 153.0, 154.8, 156.1, 156.2, 156.3, 159.7, 163.3, 163.5, 170.5.
N-[2, 2\ 3'-Tris(2-guanidino-ethyloxy)-biphenyl-5-ylmethyl]-3-[2- pyridyl)dithio]propionamide, trifluroacetate salt (Example 20)
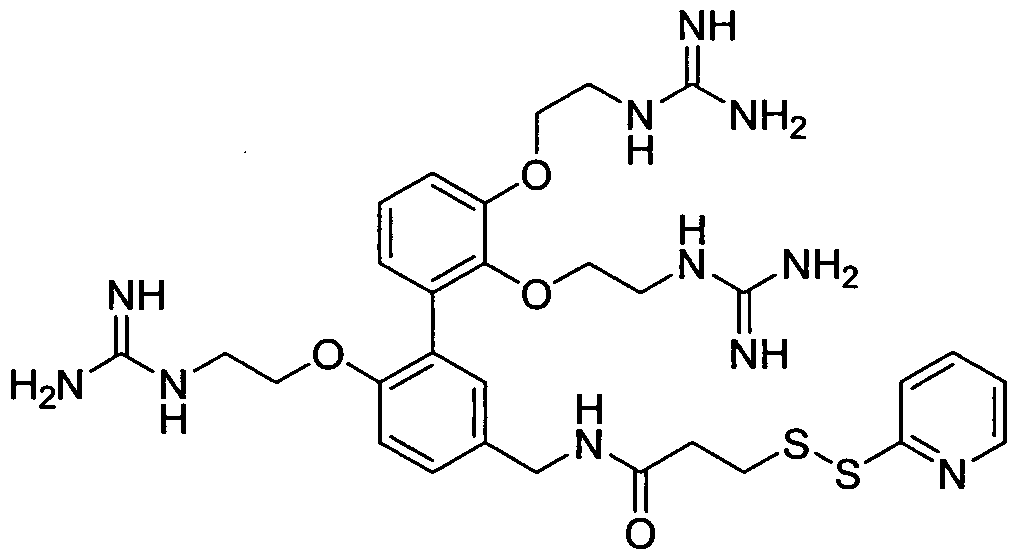
N- {2, 3 '-Bis {2-[N, N'-bis^ert-butoxycarbony^guanidinoJ-ethyloxy} -biphenyl-5- ylmethyl}-3-[2-pyridyl)dithio]propionamide (6 mg) was dissolved in a mixture of TFA/ H2O/triisopropylsilane 95/2.5/2.5 (1 mL) and stirred at room temperature for 3 h. The solvent was removed under vacuum to give the title compound as colourless oil (4 mg, 60%).
ESMS; m/z 343 (M+2)
1H-NMR (MeOH-d4): δ 2.68 (t, J= 6.8Hz, 2H), 3.07 (t, J= 6.7 Hz, 2H), 3.20 (m, 2H), 3.42 (m, 2H), 3.65 (m, 2H), 3.90 (m, 2H), 4.08 (m, 2H), 4.17 (m, 2H), 4.37 (bs, 2H), 6.86 (m, IH), 7.05-7.29 (m, 6H), 7.73 (m, 2H), 8.37 (m, IH). 13C-NMR (CDCl3): δ 35.4, 35.8, 42.3, 43.0, 43.6, 68.3, 68.8 67.1, 72.7, 79.1, 114.1, 114.5, 121.2, 122.4, 125.0, 125.6, 128.9, 129.1, 130.9, 132.7, 134.6, 139.1, 146.5, 150.4, 152.7, 156.1, 159.3, 161.0, 173.6.
Theoretical Mass: (M + H) 684.28572. Measured Mass: (M + H) 684.28417 IR v Max KBR/cm"3412 (br NH, NH2), 1677 (C=ON)
Example 21: Observation of cellular uptake via live microscopy
Geminin and 15k were expressed and purified according to the method described in R. V. Stevens, G.S. Bisacchi, Journal of Organic Chemistry 1982, 47, 2396-2399, the entirety of which is incorporated herein by reference. Geminin was conjugated with Alexa Fluor 488 using a Molecular Probes Protein Labelling kit, according to the manufacturer's protocol (Invitrogen). Coupling reactions of proteins with different SMOCs have been described in Okuyama M. et al, Nat. Methods 2007, 4, 153-159, the entirety of which is incorporated herein by reference. The efficiency of coupling was assessed by spectrophotometric quantification of pyridyl at 343 run and monitoring release of sulfhydryl groups using Ellman's Reagent (Pierce, Rockford, IL, USA) according to Pierce protocol 22582.
Human U2OS and WI-38 human diploid fibroblasts (HDF), were cultured in DMEM supplemented with 10% FCS, 100 LVmL penicillin and 0.1 mg/mL streptomycin.
To determine the ability of compounds of the invention to carry biomolecules into cells, the nuclear protein Geminin, a repressor of origin licensing, was coupled to compounds of the invention via a disulfide bond. Geminin is a 23.5 kDa protein that blocks DNA replication. Three compounds of the invention, Examples 3, 17 and 20 were conjugated to Geminin labelled with AlexaFluor 488 via thiol exchange, giving compounds 11-g*, 33-g* and 35- g* respectively. The cellular uptake, followed via live microscopy in two cell lines (WI-38 human diploid fibroblast and human U2OS osteosarcoma cells) is shown in Figure 1.
When U2OS cell line was treated with the fluorescent tagged protein, a higher uptake was detected when the cells were incubated with 11-g* and a lower when treated with 23-g*. In the case of WI-38 HDF the difference in the uptake was not so obvious by this method, but a preferential perinuclear and nuclear localisation was observed when Geminin was linked to the compounds 35-g* and 11-g* containing 3 and 4 guanidine moieties respectively. These data suggested a correlation between the number of guanidine moieties and efficiency of the delivery and cargo localisation.
For detection of the uptake of compounds of the invention coupled with Geminin- AlexaFluor488 into live cells, exponentially growing cells (WI-38 HDF and U2OS) were cultured on glass coverslips. Cells were washed in PBS, and incubated with fresh medium containing 10 μM solutions of compounds of the invention coupled with Geminin- AlexaFluor488. Coverslips were washed extensively in PBS, placed in a plate containing medium without Red Phenol (Gibco) and observed by live confocal fluorescence microscopy (MP-UV, Leica Microsystems GmbH, Wetzlar, Germany) using 40χ and 63 x water immersion objectives, hi order to obtain similar fluorescence intensities, WI-38, and U2OS cells required incubation with the protein conjugate for 1 and 5 hours respectively.
Example 22: Analysis of cellular uptake by FACS analysis
To evaluate and compare the ability of different compounds of the invention to carry biomolecules into different human cells and provide a measure of the efficiency of cell penetration compounds of the invention coupled with Geminin-AlexaFluor488 molecules were treated with WI-38 and U2OS.
Cells were seeded in 6-well plates and incubated with 10 μM solutions of compounds of the invention coupled with Geminin-AlexaFluor488 for 3 h (WI-38) or 5 h (U2OS). Cells were then washed extensively with PBS, trypsinised, resuspended in medium, washed again in cold PBS and immediately analysed by Flow Cytometry (FACSCalibur). A total of 10,000 cells per sample were counted. The mean fluorescence intensity of treated cell populations was compared with untreated control cells and cells incubated with AF488 or AF488-15k only.
Figure 2 shows the mean value of fluorescence obtained from this experiment in both cell lines. Cells treated with labelled Geminin conjugated to compounds of the invention show a clear shift in the FACS profile towards higher FLlH values compared to the controls, revealing that proteins conjugated to compounds of the invention can enter efficiently into the cells (approximately 100% of U2OS cells show uptake, a range between 75 and 90% in the case of WI-38 HDF).
Both cell lines showed similar trend which means that the relative efficiency of compounds of the invention was identical in normal and cancer cells. However, the fluorescence was more intense in WI-38 HDF meaning that the compounds of the invention were more efficient carriers in this cell line.
Several compounds of the invention, Examples 9, 11, 5, 7, 17, 14, 20 and 3 were coupled to Geminin labelled with AlexaFluor488, giving compounds 23-g*, 26-g*, 18-g*, 20-g*, 33- g*, 31-g*, 35-g* and 11-g*, respectively.
The first six bars of the chart shown in Figure 2 represent the uptake obtained using bis- guanidines (23-g*, 26-g*, 18-g*, 20-g*, 33-g* and 31-g*) as carrier, the seventh and eighth bar represent the data for the tris-guanidine compound 35-g* and the tetra-guanidine compound 11-g* respectively. It appears that when more guanidine moieties are present on the carrier, its efficiency is higher. Using the data obtained from the bis-guanidine, it was possible to make a correlation between the position of the guanidine moiety and the linker moiety and the ability of the compounds to carry a biomolecule. In the case of compounds 18-g*, 20-g*, 33-g* and 31-g* the guanidine moieties are in the same position but the linker moiety is in a different position. The efficiency was higher when the linker was para to the guanidine moiety. Moreover, when the guanidine moiety is ortho (compounds 23-g* and 26-g*) to the linker moiety, a reduction in the ability to carry the cargo into the cells was observed.
Example 22: Cell proliferation assay
To assess whether 15k Geminin retains its ability to inhibit DNA replication in cycling cells when coupled with compounds of the invention, 10 μm solutions of compounds of the invention coupled with 15k were added to WI-38 and U2OS and replenished every 24 h, for a total 72 h treatment. Seventy-one hours after the initial treatment, cells were pulse- labelled for 1 h with 10 μM BrdU, then fixed with 4% paraformaldehyde, and treated as described in Okayama et al, Nat Methods 2007, 4 153-159. Confocal fluorescence microscopy was performed on a Leica TCS DMRE confocal microscope. The results are shown in Figure 3. The functional activity of Geminin was retained.
Claims
1. A process for the production of a compound of formula I, or a pharmaceutically acceptable salt thereof,

X1, X2 and X3 are each independently
NR2 Nv' ^NR, NR3R4 where Y is an alkylene, alkenylene or alkynylene group, each of which may be optionally substituted with one or more substituents selected from alkyl, halo, CF3, OH, alkoxy, NH2, CN, NO2 and COOH; W is absent or is O, S or NH;
R1, R2, R3 and R4 are each independently selected from H, alkyl, aryl and a protecting group
Pi;
R7, R8 and R9 are each independently selected from H, alkyl, halo, CF3, OH, alkoxy, NH2, CN, NO2 and COOH; - q and r are each independently 1, 2, 3 or 4; q1 and r' are each independently O, 1, 2 or 3, where q + q' and r + r' each equal 4; p is 1, 2, 3, 4 or 5, and p' is O, 1, 2, 3 or 4, where p + p' is 5; n is 0, 1, 2, 3, 4, 5 or 6; and
L is (Z)1nNR5R6 wherein Z is a hydrocarbyl group and m is 0 or 1 wherein R5 and R6 are each independently H, CO(CH2)jQi or C=S(NH)(CH2)kQ2, where j and k are each independently 0, 1, 2, 3, 4 or 5, and Q1 and Q2 are each independently selected from COOH, a chromophore, 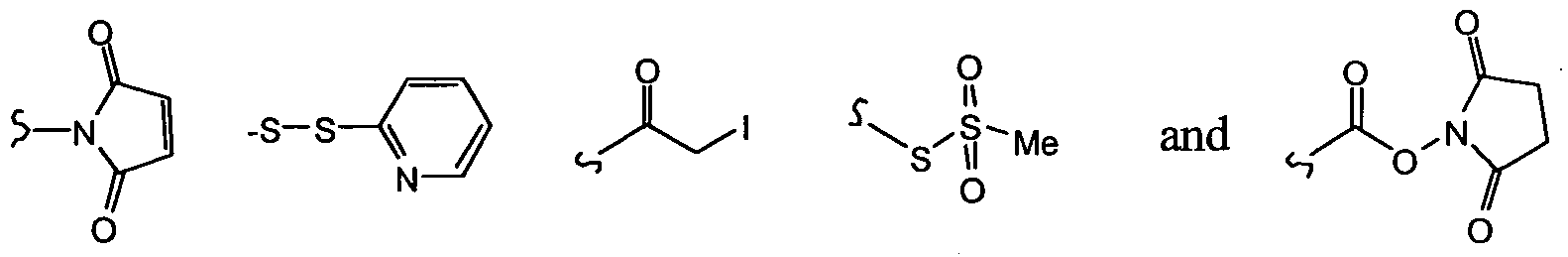
or R5, R6 and the nitrogen to which they are attached together form

(a) coupling a compound of formula II to a compound of formula III to form a compound of formula IV


IV wherein: n, p, p', q, q', r and r' are defined as above; at least one X1 1, X2' and X3' moiety is -W-Y-NR1Ri0 or -W-Y-NR1-CC=NRz)-NR3R4 and the other X1', X2' and X3' moieties are each independently selected from OH, SH, NH2, -W-Y-NR1R10, and -W-Y-NR1 -C(=NR2)-NR3R4 wherein R1, W and Y are defined as above and R10 is H or a protecting group P2; one of J1 and J2 is a leaving group LG1, and the other is a boronic acid, a boronic ester, a borane group or a trihalogenoborate salt group; L'" is L, as defined above, a leaving group LG2, -COR18, -(Z)1nNHR12, or a group which can be reduced to a moiety -(Z)mNH2, where Z and m are defined as above, R12 is a protecting group P3 and R]8 is hydrogen or a C1-C4 alkyl group; and
(b) if any of X1 ', X2' and X3 1 in the formula (IV) is other than -W-Y-NR1 -C(=NR2)- NR3R4, alkylating the X1', X2 1 and X3' moieties so that they each represent a group of formula 
where W, Y, R1, R2, R3 and R4 are defined as above, and, if necessary, converting the moiety L'" to a moiety L as defined above, to obtain a compound of formula I.
2. A process according to claim 1, wherein at least one X3' is -W-Y-NR1Ri0 or -W-Y- NR1-C(=NR2)-NR3R4.
3. A process according to claim 1, wherein when any of the Xi', X2 1 and X3' moieties in the formula (IV) are OH, SH or NH2, alkylation so that all X1', X2' and X3' moieties represent a group of formula -W-Y-NRi -C(=NR2)-NR3R4 is effected by either: (i) alkylating any X1', X2' and X3' moieties that are OH, SH or NH2 so that they represent -W-Y-NR1R1O, deprotecting any protected amine groups on -W-Y-NRiRi0 moieties present at the Xi1, X2' and X3' positions and guanidinylating the deprotected moieties so that they each represent a group of formula -W-Y-NRi -C(=NR2)- NR3R4; or (ii) alkylating any Xi', X2' and X3' moieties that are OH, SH or NH2 so that they each represent a group of formula -W-Y-NR1 -C(=NR2)-NR3R4, deprotecting any protected amine groups on -W-Y-NRiRi0 moieties present at the Xi', X2' and X3' positions and guanidinylating the deprotected moieties so that they each represent a group of formula -W-Y-NR]-C(=NR2)-NR3R4.
4. A process according to claim 3 wherein the alkylation of hydroxy, thiol and amino groups at the Xi', X2' and X3' positions so that they represent -W-Y-NRiRi0 is effected with a compound of formula LG3-Y-NR1R10 where R1 and Y are as defined in claim 1, R10 is H or a protecting group P2 and LG3 is a leaving group.
5. A process according to claim 3 wherein the alkylation of any X1', X2' and X3' moieties that are OH, SH or NH2 so that they each represent a group of formula -W-Y-NR1- C(=NR2)-NR3R4 is effected by a compound of formula LG7-Y-NRj -C(=NR2)-NR3R4.
6. A process according to claim 1 wherein all of the X1', X2' and X3' moieties are -W-Y-NR1R10 or -W-Y-NR1 -C(=NR2)-NR3R4 and any necessary alkylation of Xi1, X2' and X3' moieties so that they each represent -W-Y-NR1 -C(=NR2)-NR3R4 is effected by deprotecting any protected amine groups on -W-Y-NR1Ri0 moieties present at the X1 1, X2' and X3' positions and guanidinylating the deprotected moieties so that they represent a group of formula -W-Y-NR1 -C(=NR2)-NR3R4.
7. A process according to any one of claims 3, 4, 5 and 6, wherein said guanidinylation of deprotected -W-Y-NRiRi0 moieties at the X1', X2' and X3' positions is effected either by a compound of formula V, or a tautomer thereof, where R2, R3 and R4 are
R4R3N LG4
R2HN
V as defined in claim 1 and LG4 is a leaving group; or by a compound of formula V\ or a tautomer thereof,
R4R3N
R2N
V
where R2, R3 and R4 are as defined in claim 1 and LG4' is a leaving group.
8. A process according to claim 7, wherein LG4 is a triflyl group or LG4' is a pyrazole group.
9. A process according to claim 1, wherein at least one X1', X2' and X3 1 moiety is -W- Y-NR1R1O and the other X1', X2' and X3 1 moieties are each independently selected from OH, SH, NH2 and -W-Y-NR1R10 wherein R1, W and Y are as defined in claim 1 and R10 is H or a protecting group P2, and wherein in step (b) when any X1', X2' and X3' moieties are OH, SH or NH2, alkylation is effected so that all X1', X2' and X3' moieties represent -W-Y-NRiR10, any protected amine groups on the X1', X2' and X3 1 moieties are deprotected and the X1', X2' and X3' moieties are guanidinylated so that they each represent a group of the formula -W-Y- NRrC(=NR2)-NR3R4 and, if necessary, the moiety L"' is converted to a moiety L, to obtain a compound of formula (I).
10. A process according to any one of the preceding claims wherein L'" is a group which can be reduced to a moiety -(Z)mNH2, and the conversion of the moiety L'" to a moiety L in step (b) comprises (i) reducing L'" and (ii) optionally reacting the thus obtained compound with a compound of formula LG5CO(CH2)JQJ or LG6C=S (NH)(CH2)kQ2 where j, k, Q1 and Q2 are as defined in any one of the preceding claims and LG5 and LG6 are leaving groups.
11. A process according to claim 10 wherein L'" is CN.
12. A process according to any one of claims 1 to 9 wherein L'" is a leaving group LG2, and the conversion of the moiety L'" to a moiety L in step (b) comprises (i) cyanating with a source of CN' in the presence of a suitable catalyst, (ii) reducing the thus obtained CN moiety and (iii) optionally reacting the thus obtained compound with a compound of formula LG5CO(CH2)JQ1 or LG6C=S (NH)(CH2)kQ2 where j, k, Q1, Q2, LG5 and LG6 are as defined in any one of the preceding claims.
13. A process according to claim 11 or 12, wherein the CN moiety is reduced under a hydrogen atmosphere, in the presence of a catalyst.
14. A process according to any one of claims 1 to 13, wherein, in step (b), the alkylation of the X1', X2' and X3' moieties so that they each represent a group of formula -W-Y-NR1- C(=NR2)-NR3R4 is effected before the conversion of the moiety L'" to a moiety L.
15. A process according to any one of claims 1 to 9 and 14, wherein L'" is -COR18, and the conversion of the moiety L'" to a moiety L in step (b) comprises (i) reductively aminating the -COR18 moiety and (ii) optionally reacting the thus obtained compound with a compound of formula LG5CO(CH2)JQ1 or LG6C=S(NH)(CH2)kQ2.
16. A process according to claim 15 wherein reductive amination of the -COR18 moiety is effected by (a) reaction with NH2OR^ or NHR5R6 and (b) reduction of the carbon nitrogen double bond, wherein R^ is H or an alkyl group.
17. A process according to any one of the preceding claims wherein W is O and/or Y is CH2CH2.
18. A process according to any one of the preceding claims, wherein R1 and R3 are hydrogen, and R2 and R4 are each independently selected from H and a buryloxycarbonyl (Boc) protecting group.
19. A compound according to any one of the preceding claims, wherein p, q and r are each independently 1 or 2.
20. A process according to any one of the preceding claims, wherein in formula IV, R1 is H and R10 is a carbobenzyloxy (Cbz) protecting group.
21. A process according to any one of the preceding claims, wherein n is 0.
22. A process according to any one of the preceding claims, wherein Ji is a boronic acid, a boronic ester, a borane group or a trihalogenoborate salt group and J2 is a leaving group LGi.
23. A process according to any one of the preceding claims wherein the boronic ester is -B(OR13)(OR14) Or 
where R13 and R14 are each independently selected from C1-C6 alkyl groups and R15 is a C1-C6 alkyl or phenyl group.
24. A process according to any one of claims 1 to 22 wherein the borane is -BR16R17 where R16 and R17 are each independently selected from C1-C6 alkyl groups.
25. A process according to any one of claims 1 to 22 wherein said compound of formula II is of formula Ha, lib, Hc or Hd and/or said compound III is of formula Ilia, IHb, IHc, IHd or HIe.

Ha πb Hc πd

IHa nib me  ma me
ma me
26. A compound of formula (I), as defined in any one of the preceding claims, or a pharmaceutically acceptable salt thereof, wherein r is 1 or 2 and the phenyl ring which carries L has no substituents ortho to the L moiety.
27. A compound according to claim 26, wherein the phenyl ring which carries L has a hydrogen atom ortho to the moiety
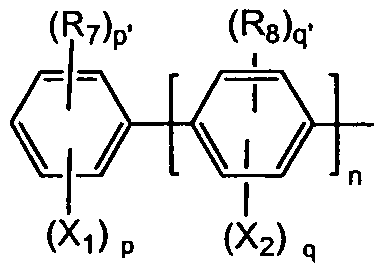
28. A compound according to claim 26, wherein r is 1 and L is para to the X3 moiety.
29. A compound of formula Via, VIb, VIc, VId or VIe or a pharmaceutically acceptable salt thereof,

VId Vie
wherein X1, X3 and L are as defined in any one of the preceding claims.
30. A compound according to claim 29 which is selected from the following: 
31. A compound of formula VII, or a pharmaceutically acceptable salt thereof,

VII wherein:
X], X3, R7, Rg and L are as defined in any one of the preceding claims; r is 1, 2, 3 or 4 and r' is 0, 1, 2 or 3, wherein r + r' is 4; p is 1, 2, 3, 4 or 5 and p1 is 0, 1, 2, 3 or 4, where p + p1 is 5; and p + r is 3.
32. A compound according to claim 31 which is:

33. A process for preparing a conjugate, which process comprises: (i) preparing a compound of formula I, or a pharmaceutically acceptable salt thereof, in which L is other than a moiety -(Z)m-phthalimide, wherein Z and m are as defined in claim 1, by a process according to any one of claims 1 to 25; and (ii) reacting said compound of formula I with a cargo moiety selected from a protein, a peptide, an oligonucleotide, a nucleotide, a diagnostic agent, a biologically active compound, an antibody and a drug.
34. A process according to claim 33, wherein in step (ii) a nucleophilic group on the cargo moiety displaces a leaving group in the moiety L in the compound of formula (I).
35. A process according to claim 33 or 34, wherein the cargo moiety is a drug.
36. A process according to claim 35, wherein the drug is a cytotoxic drug.
37. A process according to claim 35, wherein the drug is selected from DNA damaging agents, anti-metabolites, anti-tumour antibiotics, natural products and their analogues, dihydrofolate reductase inhibitors, pyrimidine analogues, purine analogues, cyclin- dependent kinase inhibitors, thymidylate synthase inhibitors, DNA intercalators, DNA cleavers, topoisomerase inhibitors, anthracyclines, vinca drugs, mitomycins, bleomycins, cytotoxic nucleosides, pteridine drugs, diynenes, podophyllotoxins, platinum containing drugs, differentiation inducers and taxanes.
38. A process according to claim 37, wherein the drug is selected from methotrexate, methopterin, dichloromethotrexate, 5-fluorouracil, 6-mercaptopurine, tri-substituted purines such as olomoucine, roscovitine and bohemine, flavopiridol, staurosporin, cytosine arabinoside, melphalan, leurosine, actinomycin, daunorubicin, doxorubicin, mitomycin D, mitomycin A, carninomycin, aminopterin, tallysomycin, podophyllotoxin (and derivatives thereof), etoposide, cisplatinum, carboplatinum, vinblastine, vincristine, vindesin, paclitaxel, docetaxel, taxotere retinoic acid, butyric acid, acetyl spermidine, tamoxifen, irinotecan and campotothecin.
39. A conjugate obtainable by reacting a compound as defined in any one of claims 26 to 32 with a cargo moiety as defined in any one of claims 33 and 35 to 38, or a pharmaceutically acceptable salt of said conjugate.
40. Use of a compound or conjugate according to any one of claims 26 to 32 or 39 in the manufacture of a medicament for use delivering a drug to a patient transdermally.
41. A skin patch which comprises a compound or conjugate according to any one of claims 26 to 32 or 39 and a pharmaceutically acceptable carrier or diluent.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0716783.6 | 2007-08-29 | ||
| GBGB0716783.6A GB0716783D0 (en) | 2007-08-29 | 2007-08-29 | New Process |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009027679A1 true WO2009027679A1 (en) | 2009-03-05 |
Family
ID=38616921
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2008/002911 WO2009027679A1 (en) | 2007-08-29 | 2008-08-29 | Process for the preparation of guanidino substituted bi-and polyphenyls that are suitable as small molecule carriers |
Country Status (2)
| Country | Link |
|---|---|
| GB (1) | GB0716783D0 (en) |
| WO (1) | WO2009027679A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010097604A3 (en) * | 2009-02-27 | 2010-10-21 | Ucl Business Plc | Guanidino-substituted bi- and polyphenyls as small molecule carriers |
| WO2011018611A1 (en) | 2009-08-10 | 2011-02-17 | Ucl Business Plc | Reversible covalent linkage of functional molecules |
| WO2012022932A1 (en) | 2010-08-20 | 2012-02-23 | Ucl Business Plc | Process for producing radiohalogenated bioconjugates and products thereof |
| WO2013132268A1 (en) | 2012-03-09 | 2013-09-12 | Ucl Business Plc | Chemical modification of antibodies |
| US11827610B2 (en) | 2021-09-15 | 2023-11-28 | Enko Chem, Inc. | Protoporphyrinogen oxidase inhibitors |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005123676A1 (en) * | 2004-06-17 | 2005-12-29 | Ucl Business Plc | Bi- or tetra-guanidino-biphenyl compounds as small molecule carriers |
-
2007
- 2007-08-29 GB GBGB0716783.6A patent/GB0716783D0/en not_active Ceased
-
2008
- 2008-08-29 WO PCT/GB2008/002911 patent/WO2009027679A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005123676A1 (en) * | 2004-06-17 | 2005-12-29 | Ucl Business Plc | Bi- or tetra-guanidino-biphenyl compounds as small molecule carriers |
Non-Patent Citations (1)
| Title |
|---|
| OKUYAMA ET AL.: "Small-molecule mimics of an alpha-helix for efficient transport of proteins into cells", NATURE METHODS, vol. 4, no. 2, February 2007 (2007-02-01), pages 153 - 159, XP002504839 * |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010097604A3 (en) * | 2009-02-27 | 2010-10-21 | Ucl Business Plc | Guanidino-substituted bi- and polyphenyls as small molecule carriers |
| WO2011018611A1 (en) | 2009-08-10 | 2011-02-17 | Ucl Business Plc | Reversible covalent linkage of functional molecules |
| US9295729B2 (en) | 2009-08-10 | 2016-03-29 | Ucl Business Plc | Reversible covalent linkage of functional molecules |
| US10174094B2 (en) | 2009-08-10 | 2019-01-08 | Ucl Business Plc | Reversible covalent linkage of functional molecules |
| US10548982B2 (en) | 2009-08-10 | 2020-02-04 | Ucl Business Ltd | Reversible covalent linkage of functional molecules |
| US10933142B2 (en) | 2009-08-10 | 2021-03-02 | Ucl Business Ltd | Reversible covalent linkage of functional molecules |
| WO2012022932A1 (en) | 2010-08-20 | 2012-02-23 | Ucl Business Plc | Process for producing radiohalogenated bioconjugates and products thereof |
| WO2013132268A1 (en) | 2012-03-09 | 2013-09-12 | Ucl Business Plc | Chemical modification of antibodies |
| US11827610B2 (en) | 2021-09-15 | 2023-11-28 | Enko Chem, Inc. | Protoporphyrinogen oxidase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| GB0716783D0 (en) | 2007-10-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11819532B2 (en) | Peptide macrocycles against Acinetobacter baumannii | |
| JP6883518B2 (en) | Conjugated and therapeutic hemiasterin derivatives | |
| CA2627046C (en) | Methods and compounds for preparing cc-1065 analogs | |
| EP1948242B9 (en) | Cytotoxic compounds | |
| EP0328589B1 (en) | Tetra-aza macrocycles and metal complexes thereof | |
| TWI790996B (en) | Process for the preparation of pegylated drug-linkers and intermediates thereof | |
| EP2750681A1 (en) | Branched discrette peg constructs | |
| KR20160125361A (en) | Var2csa-drug conjugates | |
| WO2009027679A1 (en) | Process for the preparation of guanidino substituted bi-and polyphenyls that are suitable as small molecule carriers | |
| US6458768B1 (en) | Benzoheterocyclic distamycin derivatives, process for preparing them, and their use as antitumor agents | |
| US20080139493A1 (en) | Bi- or Tetra-Guanidino-Biphenyl Compounds as Small Molecule Carriers | |
| US9808447B2 (en) | Inhibitors of Rho associated protein kinases (ROCK) and methods of use | |
| CN113631228A (en) | Macrocyclic compounds as STING agonists | |
| JP7076433B2 (en) | New cytotoxic agents and their conjugates | |
| KR102388412B1 (en) | Phenothiazine derivatives and methods of use thereof | |
| US11453652B2 (en) | Di-macrocycles | |
| TWI820038B (en) | Process for the preparation of tubulysins and intermediates thereof | |
| WO2017096430A1 (en) | Hydroxamic acid-based compounds | |
| US20120077863A1 (en) | Guanidino-substituted bi-and polyphenyls as small molecule carriers | |
| JP6570034B2 (en) | Novel glutamic acid derivatives and uses thereof | |
| TWI839055B (en) | Pegylated drug-linkers and intermediates thereof | |
| TW202320805A (en) | Radiofluorinated agents for pet imaging selectively targeting fibroblast activation protein | |
| TW202024043A (en) | Alternative processes for the preparation of tubulysins and intermediates thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08788465 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08788465 Country of ref document: EP Kind code of ref document: A1 |