WO2009026345A1 - Thiazolidinone compounds, and methods of making and using same - Google Patents
Thiazolidinone compounds, and methods of making and using same Download PDFInfo
- Publication number
- WO2009026345A1 WO2009026345A1 PCT/US2008/073683 US2008073683W WO2009026345A1 WO 2009026345 A1 WO2009026345 A1 WO 2009026345A1 US 2008073683 W US2008073683 W US 2008073683W WO 2009026345 A1 WO2009026345 A1 WO 2009026345A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- quinolin
- compound
- thiazolidine
- dione
- mmol
- Prior art date
Links
- 0 **C=C(C(N1I)=O)SC1=* Chemical compound **C=C(C(N1I)=O)SC1=* 0.000 description 1
- CIPXQJGUSQWSOW-OYKKKHCWSA-N CC(C)(C)NS(c1cc(-c2ccc(-c3ccnc4ccc(/C=C(/C(N5)=O)\SC5=O)cc34)[s]2)ccc1)(=O)=O Chemical compound CC(C)(C)NS(c1cc(-c2ccc(-c3ccnc4ccc(/C=C(/C(N5)=O)\SC5=O)cc34)[s]2)ccc1)(=O)=O CIPXQJGUSQWSOW-OYKKKHCWSA-N 0.000 description 1
- JSVJPFBZMVQCKX-UHFFFAOYSA-N CC(C)(C)S(C)Oc(cc1)ccc1NC(c1ccc(-c2ccnc3c2cc(C=O)cc3)[s]1)=O Chemical compound CC(C)(C)S(C)Oc(cc1)ccc1NC(c1ccc(-c2ccnc3c2cc(C=O)cc3)[s]1)=O JSVJPFBZMVQCKX-UHFFFAOYSA-N 0.000 description 1
- IMWHPXRAFGDRII-UHFFFAOYSA-N CC(C)(C)[Si](C)(C)Oc1cc(NC(c2ccc(-c3ccnc4ccc(C=O)cc34)[s]2)=O)ccc1 Chemical compound CC(C)(C)[Si](C)(C)Oc1cc(NC(c2ccc(-c3ccnc4ccc(C=O)cc34)[s]2)=O)ccc1 IMWHPXRAFGDRII-UHFFFAOYSA-N 0.000 description 1
- MCEWPPORWQCMOB-CFRMEGHHSA-N CC(C)Oc1cc(-c2ccc(-c3ccnc4c3cc(/C=C(/C(N3)=O)\SC3=O)cc4)[s]2)ccc1 Chemical compound CC(C)Oc1cc(-c2ccc(-c3ccnc4c3cc(/C=C(/C(N3)=O)\SC3=O)cc4)[s]2)ccc1 MCEWPPORWQCMOB-CFRMEGHHSA-N 0.000 description 1
- MJOZVYYMDNYWQJ-ZBKNUEDVSA-N N#Cc1ccc(cc(-c2c(cc(/C=C(/C(N3)=O)\SC3=O)cc3)c3ncc2)[s]2)c2c1 Chemical compound N#Cc1ccc(cc(-c2c(cc(/C=C(/C(N3)=O)\SC3=O)cc3)c3ncc2)[s]2)c2c1 MJOZVYYMDNYWQJ-ZBKNUEDVSA-N 0.000 description 1
- NBPGCQLSWSXGJI-UHFFFAOYSA-N OC(c1ccc(-c2ccnc3ccc(C=O)cc23)[s]1)=O Chemical compound OC(c1ccc(-c2ccnc3ccc(C=O)cc23)[s]1)=O NBPGCQLSWSXGJI-UHFFFAOYSA-N 0.000 description 1
- WWSNTKHBLFMHAJ-MTJSOVHGSA-N Oc(cc1)ccc1NC(c1ccc(-c2ccnc3c2cc(/C=C(/C(N2)=O)\SC2=O)cc3)[s]1)=O Chemical compound Oc(cc1)ccc1NC(c1ccc(-c2ccnc3c2cc(/C=C(/C(N2)=O)\SC2=O)cc3)[s]1)=O WWSNTKHBLFMHAJ-MTJSOVHGSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- PI3K phosphatidylinositol-3-kinase pathway plays an important role in cellular signaling. In addition to the key role that PI3Ks play in cell proliferation and survival, they have been implicated in disease states involving vasculostasis, vascularization, membrane trafficking, glucose transport, neurite outgrowth, membrane ruffling, superoxide production, actin reorganization, and chemotaxis. Further, the PI3K pathway is stimulated as a physiological consequence of many growth factors and regulators.
- PI3Ks are lipid kinases, consist of eight identified members, and are divided into three sub-families based on their sub- structures and substrate specificities. Class I PI3Ks are further divided into two sub-classes, Class IA and Class IB. Class IA consists of pi 10a, pi lO ⁇ , and pi 105 as catalytic sub-units and these are activated in tyrosine kinase receptor signaling. Class IB contains only the pi 10 ⁇ sub-unit, which is mostly activated by seven trans-membrane G-protein coupled receptors (GPCRs) via its regulatory sub-unit plOl and G-protein ⁇ sub- units.
- GPCRs trans-membrane G-protein coupled receptors
- Class II PI3Ks have the ⁇ , ⁇ , and ⁇ isoforms.
- Class III PI3Ks include the phophatidylinositol specific 3-kinases. Overall the PI3K pathways play important roles in various disease states on account of their pivotal role in cell signaling.
- PI3K signaling is important to many aspects of cell growth and survival. Because the PI3K pathway is stimulated as a physiological consequence of many growth factors and regulators, it is quite frequently targeted by genomic aberrations including mutation, amplification and rearrangement, The activation of the PI3K pathway results in a disturbance of control of cell growth and survival, which contributes to a competitive growth advantage, metastatic competence and, often resistance to therapy. This makes the PI3Ks an attractive target for the development of novel anticancer agents.
- PI3Ks play a role in vasculostasis.
- Compromised vasculostasis has serious pathologic consequences. Examples where excessive vascular permeability leads to particularly deleterious effects include pulmonary edema, cerebral edema, and cardiac edema. In general, however, edema in any tissue or organ leads to some loss of normal function, and therefore to the risk of morbidity or even mortality.
- excessive endothelial proliferation may damage tissues, such as the retina in proliferative retinopathies, or fuel unwanted tissue growth, such as with tumor growth. Many pathologic and disease situations are marked by multiple dysregulations in vasculostasis.
- Angio genesis encompasses both enhanced vascular proliferation and permeability, as newly-formed blood vessels do not generally exhibit the same level of vascular barrier function as well-established or mature vessels. Examples of such hyper-permeable vasculature can be found in cancers, vasculo proliferative diseases, retinal diseases, and rheumatoid arthritis.
- the connection between angiogenesis and hyperpermeability may partly result from the dual action of factors such as vascular endothelial growth factor (VEGF), which induces both endothelial proliferation and vascular permeability.
- VEGF vascular endothelial growth factor
- the PI3K family may also play an important role in inflammatory responses, and in respiratory diseases. Therefore, in addition to direct roles in regulating vasculostasis, the PI3K family can also influence situations in which vasculostasis is compromised, including ischemia and ischemia-reperfusion injury, through their control of leukocyte functioning. Maintaining or restoring vasculostasis should be beneficial to overall patient outcome where such indications as, for example, inflammation, allergic diseases, cancer, cerebral stroke, myocardial infarction, pulmonary and cardiac insufficiency, renal failure, and retinopathies, are present.
- This disclosure is generally directed to compounds that include a thiazolidinone moiety.
- this disclosure also directed to compounds that are phosphoinositide 3-kinase (PI3K) enzyme or pathway inhibitors or modulators.
- PI3K phosphoinositide 3-kinase
- compounds are provided that inhibit a PI3K, with an IC50 of about 200 nM or less, about 50 nM or less, from about 1 nM to about 50 nM, or about 10 nM or less.
- the compound may inhibit one or more of PI3K ⁇ , PI3K ⁇ , PI3K ⁇ , or PI3K ⁇ .
- a method of treating an respiratory or ocular disorder comprising administering to a patient in need thereof an effective amount of a compound disclosed herein.
- a method of treating cancer comprising administrating to a patient in need thereof an effective amount of a compound of any of the disclosed compounds.
- a method for inhibiting tumor cell growth, tumor cell proliferation, or tumorigenesis comprising administering to a patient in need thereof an effective amount of a compound of any of the disclosed compounds..
- methods for treating pain, diabetes, inflammation, platelet aggregation, ischemic heart disease, sclerosis, restenosis, disorders, HIV, bone resorption, cancer, non-small cell lung cancer, or brain cancer, comprising administering to a patient in need thereof an effective amount of any of the disclosed compounds.
- the present disclosure is directed in part towards novel compounds and compositions that inhibit PI3K and methods of making and using the same.
- Such compounds may inhibit one or more of the phosphoinositide-3-kinase family, including PI3K ⁇ , PI3K ⁇ , PI3K ⁇ , and/or PI3K ⁇ .
- therapeutic agent refers to any chemical moiety that is a biologically, physiologically, or pharmacologically active substance that acts locally or systemically in a subject.
- therapeutic agents also referred to as “drugs”
- drug are described in well-known literature references such as the Merck Index, the Physicians Desk Reference, and The Pharmacological Basis of Therapeutics, and they include, without limitation, medicaments; vitamins; mineral supplements; substances used for the treatment, prevention, diagnosis, cure or mitigation of a disease or illness; substances which affect the structure or function of the body; or pro-drugs, which become biologically active or more active after they have been placed in a physiological environment.
- therapeutic effect is art-recognized and refers to a local or systemic effect in animals, particularly mammals, and more particularly humans caused by a pharmacologically active substance.
- the term thus means any substance intended for use in the diagnosis, cure, mitigation, treatment or prevention of disease or in the enhancement of desirable physical or mental development and/or conditions in an animal or human.
- therapeutically-effective amount means that amount of such a substance that produces some desired local or systemic effect at a reasonable benefit/risk ratio applicable to any treatment.
- the therapeutically effective amount of such substance will vary depending upon the subject and disease condition being treated, the weight and age of the subject, the severity of the disease condition, the manner of administration and the like, which can readily be determined by one of ordinary skill in the art.
- compositions of the present invention may be administered in a sufficient amount to produce a at a reasonable benefit/risk ratio applicable to such treatment.
- modulation is art-recognized and refers to up regulation (i.e., activation or stimulation), down regulation (i.e., inhibition or suppression) of a response, or the two in combination or apart.
- a "patient,” “subject” or “host” to be treated by the subject method may mean either a human or non-human animal.
- the term “treating” is art-recognized and refers to curing as well as ameliorating at least one symptom of any condition or disease.
- the term “prodrug” is art-recognized and is intended to encompass compounds which, under physiological conditions, are converted into the agents of the present invention.
- a common method for making a prodrug is to select moieties which are hydrolyzed under physiological conditions to provide the desired compound. In other embodiments, the prodrug is converted by an enzymatic activity of the host animal or the target organ or cell.
- alkyl is art-recognized, and includes saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl substituted cycloalkyl groups, and cycloalkyl substituted alkyl groups.
- a straight chain or branched chain alkyl has about 30 or fewer carbon atoms in its backbone (e.g., C 1 -C 3 O for straight chain, C3-C30 for branched chain), and alternatively, about 20 or fewer, e.g. from 1 to 6 carbons.
- cycloalkyls have from about 3 to about 10 carbon atoms in their ring structure, and alternatively about 5, 6 or 7 carbons in the ring structure.
- alkyl is also defined to include halosubstituted alkyls.
- alkyl (or “lower alkyl”) includes “substituted alkyls”, which refers to alkyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone.
- Such substituents may include, for example, a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxyl, a phosphoryl, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic moiety.
- a carbonyl such as a carboxyl, an alkoxy
- the moieties substituted on the hydrocarbon chain may themselves be substituted, if appropriate.
- the substituents of a substituted alkyl may include substituted and unsubstituted forms of amino, azido, imino, amido, phosphoryl (including phosphonate and phosphinate), sulfonyl (including sulfate, sulfonamido, sulfamoyl and sulfonate), and silyl groups, as well as ethers, alkylthios, carbonyls (including ketones, aldehydes, carboxylates, and esters), -CN and the like. Exemplary substituted alkyls are described below.
- Cycloalkyls may be further substituted with alkyls, alkenyls, alkoxys, alkylthios, aminoalkyls, carbonyl- substituted alkyls, -CN, and the like.
- aralkyl is art-recognized and refers to an alkyl group substituted with an aryl group (e.g., an aromatic or heteroaromatic group).
- alkenyl and alkynyl are art-recognized and refer to unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but that contain at least one double or triple bond respectively.
- lower alkyl refers to an alkyl group, as defined above, but having from one to about ten carbons, alternatively from one to about six carbon atoms in its backbone structure.
- lower alkenyl and “lower alkynyl” have similar chain lengths.
- heteroatom is art-recognized and refers to an atom of any element other than carbon or hydrogen.
- Illustrative heteroatoms include boron, nitrogen, oxygen, phosphorus, sulfur and selenium.
- aryl is art-recognized and refers to 5-, 6- and 7-membered single-ring aromatic groups that may include from zero to four heteroatoms, for example, benzene, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, triazole, pyrazole, pyridine, pyrazine, pyridazine and pyrimidine, and the like.
- aryl groups having heteroatoms in the ring structure may also be referred to as "heteroaryl” or “heteroaromatics.”
- the aromatic ring may be substituted at one or more ring positions with such substituents as described above, for example, halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, sulfonamido, ketone, aldehyde, ester, heterocyclyl, aromatic or heteroaromatic moieties, -CF3, -CN, or the like.
- aryl or aromatic, also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings (the rings are "fused rings") wherein at least one of the rings is aromatic, e.g., the other cyclic rings may be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyl s.
- ortho, meta and para are art-recognized and refer to 1,2-, 1,3- and 1,4- disubstituted benzenes, respectively.
- 1,2-dimethylbenzene and ortho- dimethylbenzene are synonymous.
- heterocyclyl or “heterocyclic group” are art-recognized and refer to 3- to about 10-membered ring structures, alternatively 3- to about 7-membered rings, whose ring structures include one to four heteroatoms. Heterocycles may also be polycycles.
- Heterocyclyl groups include, for example, thiophene, thianthrene, furan, pyran, isobenzofuran, chromene, xanthene, phenoxanthene, pyrrole, imidazole, pyrazole, isothiazole, isoxazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, pyrimidine, phenanthroline, phenazine, phenarsazine, phenothiazine, furazan, phenoxazine, pyrrolidine, o
- the heterocyclic ring may be substituted at one or more positions with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -CF3, -CN, or the like.
- substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl,
- polycyclyl or “polycyclic group” are art-recognized and refer to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls) in which two or more carbons are common to two adjoining rings, e.g., the rings are "fused rings". Rings that are joined through non-adjacent atoms are termed "bridged" rings.
- Each of the rings of the polycycle may be substituted with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -CF3, -CN, or the like.
- substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl
- carrier is art-recognized and refers to an aromatic or non-aromatic ring in which each atom of the ring is carbon.
- nitro is art-recognized and refers to -NO2; the term “halogen” is art- recognized and refers to -F, -Cl, -Br or -I; the term “sulfhydryl” is art-recognized and refers to - SH; the term “hydroxyl” means -OH; and the term “sulfonyl” is art-recognized and refers to - SO2 " .
- amine and “amino” are art-recognized and refer to both unsubstituted and substituted amines, e.g., a moiety that may be represented by the general formulas:
- R51 R52 wherein R50, R51 and R52 each independently represent a hydrogen, an alkyl, an alkenyl, - (CH2)m-R61, or R50 and R51, taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure; R61 represents an aryl, a cycloalkyl, a cycloalkenyl, a heterocycle or a polycycle; and m is zero or an integer in the range of 1 to 8.
- only one of R50 or R51 may be a carbonyl, e.g., R50, R51 and the nitrogen together do not form an imide.
- R50 and R51 each independently represent a hydrogen, an alkyl, an alkenyl, or -(CH2)m- R61.
- alkylamine includes an amine group, as defined above, having a substituted or unsubstituted alkyl attached thereto, i.e., at least one of R50 and R51 is an alkyl group.
- amino is art recognized as an amino-substituted carbonyl and includes a moiety that may be represented by the general formula:
- acylamino is art-recognized and refers to a moiety that may be represented by the general formula:
- R50 is as defined above
- R54 represents a hydrogen, an alkyl, an alkenyl or - (CH 2 ) m -R61, where m and R61 are as defined above.
- alkylthio refers to an alkyl group, as defined above, having a sulfur radical attached thereto.
- the "alkylthio" moiety is represented by one of -S-alkyl, -S-alkenyl, -S-alkynyl, and -S-(CH2)m-R61, wherein m and R61 are defined above.
- Representative alkylthio groups include methylthio, ethyl thio, and the like.
- carbonyl is art recognized and includes such moieties as may be represented by the general formulas:
- X50 is a bond or represents an oxygen or a sulfur
- R55 and R56 represents a hydrogen, an alkyl, an alkenyl, -(CH 2 ) m -R61or a pharmaceutically acceptable salt
- R56 represents a hydrogen, an alkyl, an alkenyl or -(CH 2 ) m -R61, where m and R61 are defined above.
- X50 is an oxygen and R55 or R56 is not hydrogen
- the formula represents an "ester”.
- X50 is an oxygen
- R55 is as defined above, the moiety is referred to herein as a carboxyl group, and particularly when R55 is a hydrogen, the formula represents a "carboxylic acid".
- X50 is an oxygen, and R56 is hydrogen
- the formula represents a "formate".
- the oxygen atom of the above formula is replaced by sulfur
- the formula represents a "thiolcarbonyl” group.
- X50 is a sulfur and R55 or R56 is not hydrogen
- the formula represents a "thiolester.”
- X50 is a sulfur and R55 is hydrogen
- the formula represents a "thiolcarboxylic acid.”
- X50 is a sulfur and R56 is hydrogen
- the formula represents a "thiolformate.”
- X50 is a bond, and R55 is not hydrogen
- the above formula represents a "ketone” group.
- X50 is a bond, and R55 is hydrogen
- the above formula represents an "aldehyde” group.
- each expression e.g. alkyl, m, n, and the like, when it occurs more than once in any structure, is intended to be independent of its definition elsewhere in the same structure.
- compositions of the present invention may exist in particular geometric or stereoisomeric forms.
- polymers of the present invention may also be optically active.
- the present invention contemplates all such compounds, including cis- and trans-isomers, R- and S-enantiomers, diastereomers, (D)-isomers, (L)- isomers, the racemic mixtures thereof, and other mixtures thereof, as falling within the scope of the invention.
- Additional asymmetric carbon atoms may be present in a substituent such as an alkyl group. All such isomers, as well as mixtures thereof, are intended to be included in this invention.
- a particular enantiomer of compound of the present invention may be prepared by asymmetric synthesis, or by derivation with a chiral auxiliary, where the resulting diastereomeric mixture is separated and the auxiliary group cleaved to provide the pure desired enantiomers.
- the molecule contains a basic functional group, such as amino, or an acidic functional group, such as carboxyl, diastereomeric salts are formed with an appropriate optically- active acid or base, followed by resolution of the diastereomers thus formed by fractional crystallization or chromatographic means well known in the art, and subsequent recovery of the pure enantiomers.
- substitution or “substituted with” includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, or other reaction.
- the term "substituted" is also contemplated to include all permissible substituents of organic compounds.
- the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and nonaromatic substituents of organic compounds.
- Illustrative substituents include, for example, those described herein above.
- the permissible substituents may be one or more and the same or different for appropriate organic compounds.
- the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms. This invention is not intended to be limited in any manner by the permissible substituents of organic compounds.
- the chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 67 th Ed., 1986-87, inside cover.
- the term "hydrocarbon” is contemplated to include all permissible compounds having at least one hydrogen and one carbon atom.
- the permissible hydrocarbons include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and nonaromatic organic compounds that may be substituted or unsubstituted.
- pharmaceutically-acceptable salts is art-recognized and refers to the relatively non-toxic, inorganic and organic acid addition salts of compounds, including, for example, those contained in compositions of the present invention.
- pharmaceutically acceptable carrier refers to a pharmaceutically-acceptable material, composition or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material, involved in carrying or transporting any subject composition or component thereof from one organ, or portion of the body, to another organ, or portion of the body.
- a pharmaceutically-acceptable material such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material, involved in carrying or transporting any subject composition or component thereof from one organ, or portion of the body, to another organ, or portion of the body.
- Each carrier must be “acceptable” in the sense of being compatible with the subject composition and its components and not injurious to the patient.
- materials which may serve as pharmaceutically acceptable carriers include: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as corn starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients, such as cocoa butter and suppository waxes; (9) oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol and polyethylene glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14) buffering agents, such as magnesium hydroxide and aluminum hydroxide;
- systemic administration refers to the administration of a subject composition, therapeutic or other material other than directly into the central nervous system, such that it enters the patient's system and, thus, is subject to metabolism and other like processes, for example, subcutaneous administration.
- parenteral administration and “administered parenterally” are art- recognized and refer to modes of administration other than enteral and topical administration, usually by injection, and includes, without limitation, intravenous, intramuscular, intraarterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intra-articulare, subcapsular, subarachnoid, intraspinal, and intrasternal injection and infusion.
- a 1 is a nine or ten membered bicyclic heterocycle, for example a nine or ten membered bicyclic aromatic heterocycle, containing one to three heteroatoms chosen from nitrogen, oxygen or sulfur optionally substituted by one to four substituents each independently selected from halo, hydroxyl, or alkyl;
- a 2 is an heterocycle group, for example an aromatic heterocyclic group, which may contain an heteroatom chosen from sulfur or oxygen;
- X is O, S or NR 12 ;
- R 1 is selected from the group consisting of H, amide, ester, carbamate or alkyl
- a 2 is optionally substituted by one to four substituents each independently selected from the group consisting of halo, hydroxyl, mercapto, nitro, formyl, formamido, carboxy, cyano, amino, amide, carbamoyl, sulphamoyl, ureido, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, thiocarbonyl, N-alkylureido, N-alkylamino, N-alkylsulphamoyl, N- alkylcarbamoyl, N-arylamide, N-alkylamide, aryl, heterocycle, or cycloalkyl, wherein said alkyl, alkenyl, alkynyl or alkoxy may be optionally substituted by one or more groups selected from R a , and wherein said aryl, heterocycle or cycloalkyl may each be optionally substituted by one or
- R a is independently selected from halo, hydroxyl, nitro, formyl, formamido, alkyl, alkoxy, carboxyl, cyano, amino, amido, carbamoyl, sulphamoyl, ureido, aryl, heterocycle, or cycloalkyl.
- R 12 is selected from the group consisting of alkyl, alkenyl, alkynl, aryl, heterocycle, or cycloalkyl.
- the moieties of R 12 may be optionally substituted by one, two, or three substituents selected from the group consisting of halo, hydroxyl, nitro, formyl, formamido, alkyoxy, alkyl, carboxyl, cyano, amino, amido, carbamoyl, sulphamoyl, ureido, aryl, heterocycle, or cycloalkyl.
- R 12 may be CF 3 .
- a 1 may be, in some embodiments, a nine or ten membered aromatic bicyclic heterocycle containing one to three heteroatoms chosen, for each occurrence, from nitrogen or oxygen.
- a 1 is selected from the group consisting of: indolyl, naphthyl, quinolinyl, isoquinolinyl, quinoxalinyl, quinazolinyl, benzofuryl, benzthienyl, cinnolinyl, and pteridinyl.
- a 1 can be selected from quinolinyl or quinazolinyl.
- a 2 may be selected from thiopene, furan, pyran, chromene, isothiazole, isoxazole, benzothiophene, 2,3-dihydrothieno[3,4-b][l,4]dioxine, 4,5,6,7-tetrahydrothieno[3,4-c]pyridine, or benzofuran.
- a 2 may be substituted on a ring carbon with 1 or 2 substituents each independently chosen from: halo, carboxyl, cyano, amino, amide, formyl, formamido, alkoxy, carbamoyl, sulphamoyl, ureido, an optionally substituted alkyl (for example CF 3 ), an optionally substituted aryl, or an optionally substituted heterocycle group.
- a 2 can be substituted on a ring carbon with 1 or 2 substituents each independently chosen from halo, methyl, phenyl, or isoxazole, wherein said phenyl and isoxazole are optionally substituted on a ring carbon by 1 or 2 substituents chosen from halo, alkyl, aryl or heterocycle.
- a disclosed compound is represented by formula Ia or Ib:
- Y is CR b or N
- Z is S or O
- R 2 , R 3 , R 4 are each independently selected from the group consisting of H, halo, alkyl, alkenyl, alkynyl, hydroxyalkyl, alkoxy, cyano, carboxyl, N-arylamido, aryl, and heterocycle, or R 2 and R 3 , or R 3 and R 4 , form, together with the carbon atoms to which they are attached, a 5 or 6 carbocycle or heterocycle ring, wherein any of said alkyl, alkenyl, alkynyl, carbocycle, aryl, or heterocycle can be optionally substituted with halo, hydroxyl, cyano, alkoxy, ureido, sulphamoyl, N-alkylsulphamoyl, alkyl, aryl, heterocycle or alkylheterocycle.
- R 3 may be phenyl or N-phenylacetamide wherein the phenyl is optionally substituted with hydroxyl, alkyl, N-alkylsulphamoyl, alkoxy, or an alkylmorpholino.
- R b is selected from H, halo, hydroxyl, or alkyl.
- Z is S or O.
- R 2 is H.
- R 3 and R 4 are each independently selected from H, -CH 2 OH, -CH 2 CH 2 OH, CH 3 , phenyl, F, Cl, CO 2 H,-C(O)NH-phenyl, -C(O)NCH 2 CH 2 -pyrrolidine, or dimethylisoxazole, wherein the phenyl is optionally substituted by, for example, one or two substituents selected from the group consisting of halo, cyano, hydroxyl, alkyl, alkoxy, N-alkylsulphamoyl, and sulphamoyl.
- R 3 and R 4 taken together with the ring carbons on which they are attached, form a bicyclic ring such as benzofuran, benzothiophene, 2,3-dihydro- thieno[3,4b][l,4]dioxin or 4,5,6,7-tetrahydrothieno[3,2-c]pyridine.
- Such bicyclic rings may be substituted by, for example, one or two substituents selected from the group consisting of halo, hydroxyl, cyano, and alkyl.
- X may be, for example NR 12 , wherein R 12 is phenyl optionally substituted by one or two halogen moities; or X may be, for example, O.
- Re is selected from the group consisting of:
- X is O, S or NR 12 ;
- W is, independently for each occurrence, CR b , N-alkyl, NH, N-C(O)-alkyl, N-C(O)- heterocycle, N-C(O)-N-alkyl, N-C(O)-N-heterocycle, O or S;
- Z is, independently for each occurrence, S or O;
- R b is selected from H, halo, hydroxyl, or alkyl
- R 1 is H or alkyl
- R 7 is H or alkyl
- Rg and Rg are each independently selected from H, Cl, F, alkyl, alkynl, alkenyl, carboxyl, cyano, hydroxyl, hydroxyalkyl, formyl, formamido, amido, amine, sulphamoyl, N-
- n is an integer from 0 to 8.
- R 1O is independently selected for each occurrence from H, Cl, F, hydroxyl, cynao, or alkyl;
- R 11 is selected from group consisting of heterocycle, aryl, or cycloalkyl; wherein R 11 is optionally substituted at one to four substituents each independently selected from halo, hydroxyl, carboxyl, sulphamoyl, N-alkylsulphamoyl, -alkyl-heterocycle, -alkyl-carbocycle or alkyl;
- R 12 is selected from the group consisting of alkyl, aryl, heterocycle, or cycloalkyl, wherein R 12 may be optionally substituted by one, two, or three substituents selected from the group consisting of halo, hydroxyl, nitro, formyl, formamido, alkyoxy, alkyl, carboxyl, cyano, amino, amido, carbamoyl, sulphamoyl, ureido, aryl, heterocycle, or cycloalkyl;
- R 13 is alkylene, alkenylene, or alkynyl
- formula II may be represented by:
- n is 1 to 3, or 1 to 5, for example, m may be 1, 2, 3, 4, or 5.
- R 1O at each occurrence is H.
- R 11 may be substituted at one or two positions by methyl.
- R 11 is chosen from: pyrrolindyl, phenyl, isoxazole, or thiazolidinedione.
- R 7 is H or methyl.
- Rg may be H, methyl, or ethyl. .
- Rg may be chosen, for example, from: H, Cl, phenyl, co 2H , or 3,5-dimethyl-isoxazol- 4-yl.
- compositions of the present invention will vary depending on the symptoms, age and body weight of the patient, the nature and severity of the disorder to be treated or prevented, the route of administration, and the form of the subject composition. Any of the subject formulations may be administered in a single dose or in divided doses. Dosages for the compositions of the present invention may be readily determined by techniques known to those of skill in the art or as taught herein.
- the dosage of the subject compounds will generally be in the range of about 0.01 ng to about 10 g per kg body weight, specifically in the range of about 1 ng to about 0.1 g per kg, and more specifically in the range of about 100 ng to about 10 mg per kg.
- An effective dose or amount, and any possible affects on the timing of administration of the formulation may need to be identified for any particular composition of the present invention. This may be accomplished by routine experiment as described herein, using one or more groups of animals (preferably at least 5 animals per group), or in human trials if appropriate.
- the effectiveness of any subject composition and method of treatment or prevention may be assessed by administering the composition and assessing the effect of the administration by measuring one or more applicable indices, and comparing the post-treatment values of these indices to the values of the same indices prior to treatment.
- the health of the patient may be monitored by measuring one or more of the relevant indices at predetermined times during the treatment period.
- Treatment including composition, amounts, times of administration and formulation, may be optimized according to the results of such monitoring.
- the patient may be periodically reevaluated to determine the extent of improvement by measuring the same parameters.
- Adjustments to the amount(s) of subject composition administered and possibly to the time of administration may be made based on these reevaluations.
- Treatment may be initiated with smaller dosages which are less than the optimum dose of the compound. Thereafter, the dosage may be increased by small increments until the optimum therapeutic effect is attained.
- compositions may reduce the required dosage for any individual agent contained in the compositions because the onset and duration of effect of the different agents may be complimentary.
- Toxicity and therapeutic efficacy of subject compositions may be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e.g., for determining the LD50 and the ED50.
- the data obtained from the cell culture assays and animal studies may be used in formulating a range of dosage for use in humans.
- the dosage of any subject composition lies preferably within a range of circulating concentrations that include the ED50 with little or no toxicity.
- the dosage may vary within this range depending upon the dosage form employed and the route of administration utilized.
- the therapeutically effective dose may be estimated initially from cell culture assays.
- compositions of the present invention may be administered by various means, depending on their intended use, as is well known in the art.
- compositions of the present invention may be formulated as tablets, capsules, granules, powders or syrups.
- formulations of the present invention may be administered parenterally as injections (intravenous, intramuscular or subcutaneous), drop infusion preparations, suppositories or administration intranasally (for example, to deliver a dosage to the brain via the nose or to deliver a dosage to the nose directly) or by inhalation (e.g. to treat a condition of the respiratory tract or to pretreat or vaccinate via the respiratory tract).
- compositions of the present invention may be formulated as eyedrops or eye ointments. These formulations may be prepared by conventional means, and, if desired, the compositions may be mixed with any conventional additive, such as an excipient, a binder, a disintegrating agent, a lubricant, a corrigent, a solubilizing agent, a suspension aid, an emulsifying agent or a coating agent.
- any conventional additive such as an excipient, a binder, a disintegrating agent, a lubricant, a corrigent, a solubilizing agent, a suspension aid, an emulsifying agent or a coating agent.
- wetting agents such as sodium lauryl sulfate and magnesium stearate, as well as coloring agents, release agents, coating agents, sweetening, flavoring and perfuming agents, preservatives and antioxidants may be present in the formulated agents.
- Subject compositions may be suitable for oral, nasal, topical (including buccal and sublingual), rectal, vaginal, aerosol and/or parenteral administration.
- the formulations may conveniently be presented in unit dosage form and may be prepared by any methods well known in the art of pharmacy.
- the amount of composition that may be combined with a carrier material to produce a single dose vary depending upon the subject being treated, and the particular mode of administration.
- Methods of preparing these formulations include the step of bringing into association compositions of the present invention with the carrier and, optionally, one or more accessory ingredients.
- the formulations are prepared by uniformly and intimately bringing into association agents with liquid carriers, or finely divided solid carriers, or both, and then, if necessary, shaping the product.
- Formulations suitable for oral administration may be in the form of capsules, cachets, pills, tablets, lozenges (using a flavored basis, usually sucrose and acacia or tragacanth), powders, granules, or as a solution or a suspension in an aqueous or non-aqueous liquid, or as an oil-in-water or water-in-oil liquid emulsion, or as an elixir or syrup, or as pastilles (using an inert base, such as gelatin and glycerin, or sucrose and acacia), each containing a predetermined amount of a subject composition thereof as an active ingredient.
- Compositions of the present invention may also be administered as a bolus, electuary, or paste.
- the subject composition is mixed with one or more pharmaceutically acceptable carriers, such as sodium citrate or dicalcium phosphate, and/or any of the following: (1) fillers or extenders, such as starches, lactose, sucrose, glucose, mannitol, and/or silicic acid; (2) binders, such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinyl pyrrolidone, sucrose and/or acacia; (3) humectants, such as glycerol; (4) disintegrating agents, such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate; (5) solution retarding agents, such as paraffin; (6) absorption accelerators, such as quaternary ammonium compounds; (7) wetting agents, such as, for example,
- compositions may also comprise buffering agents.
- Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugars, as well as high molecular weight polyethylene glycols and the like.
- a tablet may be made by compression or molding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared using binder (for example, gelatin or hydroxypropylmethyl cellulose), lubricant, inert diluent, preservative, disintegrant (for example, sodium starch glycolate or cross-linked sodium carboxymethyl cellulose), surface- active or dispersing agent.
- Molded tablets may be made by molding in a suitable machine a mixture of the subject composition moistened with an inert liquid diluent. Tablets, and other solid dosage forms, such as dragees, capsules, pills and granules, may optionally be scored or prepared with coatings and shells, such as enteric coatings and other coatings well known in the pharmaceutical-formulating art.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs.
- the liquid dosage forms may contain inert diluents commonly used in the art, such as, for example, water or other solvents, solubilizing agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, cyclodextrins and mixtures thereof.
- inert diluents commonly used in the art, such as, for example, water or other solvents, solubilizing
- Suspensions in addition to the subject composition, may contain suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
- suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
- Formulations for rectal or vaginal administration may be presented as a suppository, which may be prepared by mixing a subject composition with one or more suitable non- irritating excipients or carriers comprising, for example, cocoa butter, polyethylene glycol, a suppository wax or a salicylate, and which is solid at room temperature, but liquid at body temperature and, therefore, will melt in the body cavity and release the active agent.
- suitable non- irritating excipients or carriers comprising, for example, cocoa butter, polyethylene glycol, a suppository wax or a salicylate, and which is solid at room temperature, but liquid at body temperature and, therefore, will melt in the body cavity and release the active agent.
- Formulations which are suitable for vaginal administration also include pessaries, tampons, creams, gels, pastes, foams or spray formulations containing such carriers as are known in the art to be appropriate.
- Dosage forms for transdermal administration of a subject composition includes powders, sprays, ointments, pastes, creams, lotions, gels, solutions, patches and inhalants.
- the active component may be mixed under sterile conditions with a pharmaceutically acceptable carrier, and with any preservatives, buffers, or propellants which may be required.
- the ointments, pastes, creams and gels may contain, in addition to a subject composition, excipients, such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
- excipients such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
- Powders and sprays may contain, in addition to a subject composition, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and polyamide powder, or mixtures of these substances.
- Sprays may additionally contain customary propellants, such as chlorofluorohydrocarbons and volatile unsubstituted hydrocarbons, such as butane and propane.
- Compositions and compounds of the present invention may alternatively be administered by aerosol. This is accomplished by preparing an aqueous aerosol, liposomal preparation or solid particles containing the compound. A non-aqueous (e.g., fluorocarbon propellant) suspension could be used. Sonic nebulizers may be used because they minimize exposing the agent to shear, which may result in degradation of the compounds contained in the subject compositions.
- an aqueous aerosol is made by formulating an aqueous solution or suspension of a subject composition together with conventional pharmaceutically acceptable carriers and stabilizers.
- the carriers and stabilizers vary with the requirements of the particular subject composition, but typically include non-ionic surfactants (T weens, Pluronics, or polyethylene glycol), innocuous proteins like serum albumin, sorbitan esters, oleic acid, lecithin, amino acids such as glycine, buffers, salts, sugars or sugar alcohols.
- Aerosols generally are prepared from isotonic solutions.
- Dosages for administration by nasal delivery e.g. delivered to or via the nasal cavity, can be applied as drops, ointments, gels, mists/sprays (aqueous or nonaqueous), aerosols
- compositions for inhalation and/or delivery to the nose may contain from 1% to 20% by weight of a penetrator enhancer (for example, surfactants, e.g. sugar esters, sugar ethers, carbohydrate esters) which may allow enhanced nose permeability of the active agent.
- a penetrator enhancer for example, surfactants, e.g. sugar esters, sugar ethers, carbohydrate esters
- Dosages for administration by inhalation or by delivered to or via the lung can be applied as mists/sprays (aqueous or nonaqueous), aerosols (liquids, suspensions or dry powders),liquids or suspensions (aqueous or nonaqueous), powders, or combinations thereof.
- Such delivery can be achieved by commercially available devices such as 1) nebulizers, 2) metered dose inhalers, 3) dry powder inhalers, 4) soft mist inhalers, or by instillation or insufflation, or other mechanisms and/or devices known in the art.
- compositions of this invention suitable for parenteral administration comprise a subject composition in combination with one or more pharmaceutically- acceptable sterile isotonic aqueous or non-aqueous solutions, dispersions, suspensions or emulsions, or sterile powders which may be reconstituted into sterile injectable solutions or dispersions just prior to use, which may contain antioxidants, buffers, bacteriostats, solutes which render the formulation isotonic with the blood of the intended recipient or suspending or thickening agents.
- aqueous and non-aqueous carriers examples include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol, and the like), and suitable mixtures thereof, vegetable oils, such as olive oil, and injectable organic esters, such as ethyl oleate and cyclodextrins.
- polyols such as glycerol, propylene glycol, polyethylene glycol, and the like
- vegetable oils such as olive oil
- injectable organic esters such as ethyl oleate and cyclodextrins.
- Proper fluidity may be maintained, for example, by the use of coating materials, such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants.
- Treatment or amelioration of disease states and pathological conditions that implicate PI3K pathways are contemplated herein, and such treatment comprises administering one or more of the disclosed compounds, such as those recited in Formulas I or II, or a composition as described herein comprising a disclosed compound.
- Methods of treating a patient suffering from diseases such as myocardial infarction, stroke, congestive heart failure, ischemia or reperfusion injury, vascular leakage syndrome (VLS), cancer, arthritis, (for example rheumatoid arthritis), or other arthropathy, eye diseases including uveitis, retinopathy or vitreoretinal disease, macular degeneration, autoimmune diseases, vascular leakage syndrome, inflammatory diseases, edema, transplant rejection, burn, respiratory diseases such as acute respiratory distress syndrome (ARDS), asthma, and chronic obstructive pulmonary disorder (COPD), and transplant rejection are contemplated, and may comprise administering a disclosed compound, such as those recited in Formulas I or II, or a composition comprising a disclosed compound.
- diseases such as myocardial infarction, stroke, congestive heart failure, ischemia or reperfusion injury, vascular leakage syndrome (VLS), cancer, arthritis, (for example rheumatoid arthritis), or other arthropathy,
- Compounds and compositions disclosed herein, e.g. that inhibit vascular permeability may be used in a co-therapy to reduce the deleterious side-effects of such therapies.
- edema formation may cause uneven delivery of therapeutic agents to diseased tissues, therefore vasculostatic agents that inhibit vascular permeability could be used in a co-therapy approach to enhance delivery and efficacy of such therapies.
- exemplary methods of treating cancers include treatment of myeloma.
- Acute and/or prophylactic treatment of the interruption of blood flow by pathologic conditions such as thrombus formation, or medical intervention such as cardioplegia, organ transplantation, and angioplasty, or physical trauma, using disclosed compounds is also contemplated.
- a microwave vial was charged with 4 (0.10 g, 0.40 mmol), thiazolidine-2,4-dione (0.70 g, 0.59 mmol), and Cs 2 CO 3 (0.26 g, 0.79 mmol) in ethanol (2 mL).
- the reaction mixture was heated for 30 min at 150 0 C in a Biotage microwave reactor.
- the resulting mixture was separated by preparative HPLC. Fractions that contained the desired product were combined, neutralized with saturated Na 2 CO 3 , and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO 4 , and concentrated to afford the title compound as a yellow solid (0.082 g, 59%).
- a microwave vial was charged with 5 (0.12 g, 0.42 mmol), thiazolidine-2,4-dione (0.73 g, 0.62 mmol), and Cs 2 CO 3 (0.27 g, 0.83 mmol) in ethanol (3 mL).
- the reaction mixture was heated for 30 min at 150 0 C in a Biotage microwave reactor. Precipitate from the resulting mixture was filtered and washed with ethanol (10 mL) followed by H 2 O (10 mL) to afford the title compound as an orange solid (0.066 g, 41%).
- a microwave vial was charged with 7 (0.2 g, 0.74 mmol), thiazolidine-2,4-dione (0.15 g, 1.11 mmol), Cs 2 CO 3 (0.48 g, 1.49 mmol) in ethanol (4.5 mL).
- the reaction mixture was heated for 30 min at 150 0 C in a Biotage microwave reactor. Upon cooling to room temperature, the reaction mixture added MeOH (50 mL). Precipitate from the resulting mixture was filtered to afford the title compound as a brown solid (0.079 g, 29%).
- reaction mixture was purified on a flash chromatography (SiO 2 , 10% ethyl acetate in hexanes to 100% ethyl acetate over 15 minutes) to afford the title compound (0.37 g, 61%) as a pale yellow solid.
- Example 29 Preparation of: 5- ⁇ 6-[(Z)-(2,4-Dioxo-l,3-thiazolidin-5-ylidine)methyl]quinolin-4- yl ⁇ -,/V-(2-pyrrolidin- 1 -ylethyl)thiophene-2-carboxamide:
- Chlorobenzene (1000 mL) and PPA (280 g) were combined and heated to reflux (130 0 C).
- the solution of compound 20 (130 g, 0.48 mol) in chlorobenzene (300 mL) was added drop wise to the mixture over 1.5 h.
- the reaction mixture was refluxed for 3-4 h and then cooled.
- the solvent was decanted from the residue and toluene (400 mLx2 ) was added to the residue, stirred and decanted.
- the chlorobenzene/toluene extracts were combined and concentrated under vacuum, and the residue was then taken up with PE (800 mL) and water (400 mL).
- Example 34 Preparation of: Benzo[b]thiophene-6-carbonitrile (23) and Benzo[b]thiophene-4- carbonitrile (24)
- the reaction mixture was poured into 2 mL water and 6 mL ethyl acetate. The phases were separated and the aqueous phase was extracted with ethyl acetate (3x18 mL). The combined organic layers were washed with water (2 mL), brine (4 mL), dried (Na 2 SO 4 ), and concentrated in vacuo. The crude residue was triturated in ethyl acetate (3 mL) and then fully precipitated with petroleum ether (6 mL) to afford the title compound as an off-white solid (206 mg, 46%). The material was used as is for the next reaction.
- the reaction mixture was poured into 2 mL water and 6 mL ethyl acetate. The phases were separated and the aqueous phase was extracted with ethyl acetate (3x18 mL). The combined organic layers were washed with water (2 mL), brine (4 mL), dried (Na 2 SO 4 ), and concentrated in vacuo. The crude residue was triturated in ethyl acetate (3 mL) and then fully precipitated with petroleum ether (6 mL) to afford the title compound as a mustard colored solid (168 mg, 48%). The material was used as is for the next reaction.
- reaction mixture Upon completion, the reaction mixture was poured into ice water. The resulting mixture was acidified to pH ⁇ 2 with 1 N HCl. The aqueous layer was extracted with MTBE (1000 mL x X). The combined organic layers were washed with brine, dried over sodium sulfate, and then concentrated. The crude product was crystallized from dichloromethane to give the compound (28 g, 34.5%) as a light yellow solid.
- a microwave vial was charged with 57 (0.130 g, 0.37 mmol), thiazolidine-2,4-dione (0.071 g, 0.55 mmol), and Cs 2 CO 3 (0.24 g, 0.73 mmol) in ethanol (2 mL).
- the reaction mixture was heated 30 min at 150 0 C in a Biotage microwave reactor.
- the resulting mixture concentrated and purified by silica gel chromatography (CH 2 Cl 2 :Me0H 100:0 to 90: 10 gradient) to afford the title compound in a 1:1 ratio of E and Z isomers as an orange solid (0.033 g, 20 %).
- a microwave vial was charged with 58 (0.273 g, 0.73 mmol), thiazolidine-2,4-dione (0.143 g, 1.1 mmol), and Cs 2 CO 3 (0.476 g, 1.46 mmol) in ethanol (4 mL).
- the reaction mixture was heated 30 min at 150 0 C in a Biotage microwave reactor.
- the resulting mixture concentrated and purified by silica gel chromatography (CH 2 Cl 2 :Me0H 100:0 to 90: 10 gradient) to afford the title compound as an orange solid (0.033 g, 20 %).
- a microwave vial was charged with 59 (0.150 g, 0.40 mmol), thiazolidine-2,4-dione (0.070 g, 0.60 mmol), and Cs 2 CO 3 (0.260 g, 0.80 mmol) in ethanol (4 mL).
- the reaction mixture was heated 30 min at 150 0 C in a Biotage microwave reactor.
- the resulting mixture concentrated and purified by HPLC to afford the title compound as a brown solid (0.024 g, 13 %).
- a microwave vial was charged with 65 (0.200 g, 0.44 mmol), thiazolidine-2,4-dione (0.085 g, 0.66 mmol), and Cs 2 CO 3 (0.46 g, 1.3 mmol) in ethanol (4 mL).
- the reaction mixture was heated 30 min at 150 0 C in a Biotage microwave reactor.
- the resulting mixture concentrated and purified by silica gel chromatography (CH 2 Cl 2 :Me0H 100:0 to 50:50 gradient) to afford the title compound a yellow solid (0.103 g, 42%).
- a microwave vial was charged with 65 (0.275 g, 0.58 mmol), thiazolidine-2,4-dione (0.113 g, 0.87 mmol), and Cs 2 CO 3 (0.64 g, 1.8 mmol) in ethanol (3 mL).
- the reaction mixture was heated 30 min at 150 0 C in a Biotage microwave reactor.
- the resulting mixture concentrated and purified by silica gel chromatography (CH 2 Cl 2 IMeOH 100:0 to 50:50 gradient) to afford the title compound a yellow solid (0.180 g, 56%).
- a microwave vial was charged with 66 (0.140 g, 0.52 mmol), thiazolidine-2,4-dione (0.204 g, 1.57 mmol), and Cs 2 CO 3 (1.11 g, 3.14 mmol) in ethanol (4 mL).
- the reaction mixture was heated 30 min at 160 0 C in a Biotage microwave reactor.
- the resulting mixture concentrated and purified by silica gel chromatography (CH 2 Cl 2 :Me0H 100:0 to 0:100 gradient) to afford the title compound a brown solid (0.042 g, 17%).
- a microwave vial was charged with 67 (0.32 g, 0.82 mmol), thiazolidine-2,4-dione (0.161 g, 1.24 mmol), and Cs 2 CO 3 (0.805 g, 2.47 mmol) in ethanol (6 mL).
- the reaction mixture was heated 30 min at 160 0 C in a Biotage microwave reactor.
- the resulting mixture concentrated and purified by preparative HPLC to afford the title compound a brown solid (0.202 g, 50%).
- Example 84 Complicatedple 84103 Preparation of tert-Butyl-(3-ethynyl-phenoxy)-dimethyl- silane (68)
- a microwave vial was charged with 72 (0.50 g, 3.3 mmol), chloro-acetic acid (0.32 g, 3.4 mmol), and sodium acetate (0.30 g, 3.7 mmol) in acetic acid (3 mL).
- the reaction mixture was heated for 20 min at 150 0 C in a Biotage microwave reactor. Water was added (20 mL) and the resulting solid filtered to afford the title compound (a 1/1 mixture of E/Z isomers) as a yellow solid (0.46 g, 73%).
- a microwave vial was charged with 74 (0.85 g, 4.1 mmol), chloro-acetic acid (0.45 g, 4.7 mmol), and sodium acetate (0.40 g, 4.9 mmol) in acetic acid (6 mL).
- the reaction mixture was heated for 20 min at 150 0 C in a Biotage microwave reactor. Water was added (20 mL) to the reaction mixture and the resulting solid filtered to afford the title compound as a white solid (0.71 g, 70%).
- a microwave vial was charged with 3 (50 mg, 0.21 mmol), 75 (70 mg, 0.29 mmol), and Cs 2 CO 3 (0.13 g, 0.40 mmol) in ethanol (4 mL). The reaction mixture was heated for 30 min at 140 0 C in a Biotage microwave reactor. The resulting solid was filtered, washed with ethanol followed by water to afford the title compound as an orange solid (55 mg, 57%).
- IC 50 values for compounds against the isoforms of PI3-Kinase were generated using either luminescence or fluorescence polarization based assays.
- a four order of magnitude serial dilution of the compounds was introduced into a buffered solution containing appropriate amounts of either PI3-kinase isoform, ATP (3 ⁇ M for luminescence or 25 ⁇ M for fluorescence polarization) and PIP3 (50 ⁇ M for luminescence and 10 ⁇ M for fluorescence polarization); the reaction was then allowed to proceed for an appropriate time.
- Example 96 ELISA for C5a-induced phosphorylation of AKT in Raw 264.7 macrophages.
- Raw 264.7 mouse macrophage cells (ATCC#TIB-71) were cultured in DMEM containing 10% heat inactivated serum and IX penicillin/streptomycin. 7.5xl0 4 cells were seeded into a 96 well plate were allowed to adhere overnight. Cells were then washed one time with serum-free medium, and then incubated under serum-free conditions for 3-4 hours at 37°C/5%CO 2 . Cells were pretreated with the indicated compound (10 ⁇ M - 0.0045 ⁇ M) for 1 h, followed by stimulation with 0.5 ⁇ g/ml C5a (Sigma) for 5 min. Cells were immediately placed on ice, and washed one time with cold PBS.
- Cells were lysed with 65 ⁇ L of 1% Triton- X-100 lysis buffer containing protease inhibitors (Cell Signaling Technology) and further incubated in this solubilization buffer for 20-30 min on ice with mild agitation. Lysates were then centrifuged at 5000 rpm at 4 0 C for 10-15 min. Thereafter, an ELISA specific for phosphorylated AKT (pAKT-Ser473) (Cell Signaling Technology) was performed. Absorbance was measured at 450-540 nm.
- Example 97 VEGF-induced hRMVEC proliferation assay
- hRMVECs early passage human retinal microvascular endothelial cells (hRMVECs) cells (Cell Systems, Kirkland, WA) were plated at a density of -1.5 x 10 3 cells/well in a 96-well plate (Corning, Corning, NY), and allowed to adhere for ⁇ 6 hours or overnight. Medium was then changed into CSC-Maintenance Medium (Cell Systems, Kirkland, WA) and further incubated for at least 24 h - 48 h at 37°C/5% CO 2 .
- CSC-Maintenance Medium Cell Systems, Kirkland, WA
- this specialized medium contains 10% FBS but no growth factor, and renders proliferating cells into a quiescent state.
- the cells were then pre-treated with varying concentrations of the indicated compound (20 ⁇ M - 0.00914 ⁇ M or 10 ⁇ M - 0.00457 ⁇ M or DMSO (as a vehicle control) prepared in basal CSC-medium containing 10% FBS and 50 ⁇ g/ml heparin for -60 minutes at 37°C/5% CO 2 .
- Human recombinant VEGF (Peprotech, Rocky Hill, NJ) was then added to a final concentration of 50 ng/ml.
- cellular proliferation was quantified using the Cell Proliferation Kit (Roche, Alameda, CA) as described by the manufacturer. Briefly, for one 96-well plate, 100 ⁇ L of electron-coupling reagent was added to 5 mL of XTT labeling solution. 50 ⁇ L of this solution was then added to each well, and the reaction was allowed to develop at 37°C/5% CO 2 . The colored formazan product that is generated by metabolically active cells was measured spectrophotometrically using the SpectraMax spectrophotometer (Molecular Devices, Sunnyvale, CA) at 492 nm with correction at 690 nm.
- SpectraMax spectrophotometer Molecular Devices, Sunnyvale, CA
- IV and PO intravenous and oral dose formulations were prepared fresh.
- PO intravenous and oral dose formulations were prepared fresh.
- PO intravenous solutions containing appropriate excipients or suspended in 0.5% MC and 0.05% Tween80.
- IV formulation the compounds were solubilized in an aqueous vehicle containing suitable excipients. The IV formulation was aseptically filtered through 0.22um filters.
- Rats Six jugular vein cannulated male Sprague-Dawley rats ( ⁇ 300g) with were divided into three groups with three rats in IV dose group and three rats in the PO dose group. Animals were allowed food and water ad libitum. The study was conducted at TargeGen. (San Diego, CA).
- Blood samples (approximately 500 ⁇ L per time point) were serially collected via jugular vein cannula and transferred into tubes containing sodium heparin anticoagulant at 0.083, 0.25, 0.5, 1, 3, 5, 7, and 24 hours post-dose for IV dose; and at 0.5, 1, 3, 5, 7, and 24 hours post dose for PO doses. Blood was maintained in an ice and water mixture prior to centrifugation to obtain plasma. Plasma samples are transferred to a -2O 0 C freezer and stored until analysis.
- Matrix calibration standards and QC samples were prepared by spiking the compound into blank rat plasma (Valley Biomedical Inc., Lot # L51663). The final concentrations of a selected compound were 0, 1, 5, 10, 50, 100, 500, 1000, 2500 and 5000 ng/niL for calibration standards, and 2.50, 25.0, 250 and 2500 for QC samples.
- Plasma samples were processed using a standard protocol. The samples were analyzed using a LC/MS/MS Waters Quattro LC by standard determined conditions. Chromatogram signals were integrated and calibrated using MassLynx 3.0. Pharmacokinetic parameters were estimated using WinNonlin (version 4.1) from mean plasma concentration- time profiles. The values for the maximum plasma concentration (C max ) and the time to maximum concentration (T max ) were determined from measure plasma concentrations. The area under the curves, AUC ( i ast) and AUC (inf ) were calculated from plasma concentration-time profiles using the linear trapezoidal rule.
- the PK evaluation in mice utilized the same procedures described above except the number of mice was three per time point per dosed group and the blood samples were collected by cardiac puncture.
- Table 2 provides data from a mouse PK model with IV and PO arms.
- Example 99 Protocol for ocular exposure following topical instillation
- Table 3 shows ocular PK data for selected compounds with low oral availability
- Example 101 Murine model of ocular edema and neovascularization
- a murine model of ocular edema and neovasularization was used to assess compound activity. Laser energy is used to rupture the Bruch's membrane, after which edema and neovascularization develop within the choroid. (Tobe T. et al. (1998) Am J Pathol 153:1641-1646). Optical coherence tomography (OCT) was then used 7-9 days post-lasering to quantify the area of laser lesion sites, as a measure of edema and neovascularization.
- OCT optical coherence tomography
- Example 102 Cell data against for selected compounds against various cancer cell lines
Abstract
Provided herein are thiazolidinone compounds, and methods of making and using the same. Such compounds may be used in inflammatory or immune-mediated disorders. The disclosure provides for treating respiratory or ocular disorders, treating arthritis, or may be used to treat cancer, such as prostate or breast cancer, or multiple myeloma.
Description
THIAZOLIDINONE COMPOUNDS, AND METHODS OF MAKING AND USING SAME
RELATED APPLICATIONS
[0001] This application claims priority to U.S.S.N. 60/956,831, filed August 20, 2007; U.S.S.N. 60/977,147, filed October 3, 2007; U.S.S.N. 61/022,217, filed January 18, 2008; U.S.S.N. 61/051,424, filed May 8, 2008; and U.S.S.N. 61/020,786, filed January 14, 2008, each of which is incorporated by reference in its entirety.
BACKGROUND
[0002] The phosphatidylinositol-3-kinase (PI3K) pathway plays an important role in cellular signaling. In addition to the key role that PI3Ks play in cell proliferation and survival, they have been implicated in disease states involving vasculostasis, vascularization, membrane trafficking, glucose transport, neurite outgrowth, membrane ruffling, superoxide production, actin reorganization, and chemotaxis. Further, the PI3K pathway is stimulated as a physiological consequence of many growth factors and regulators.
[0003] PI3Ks are lipid kinases, consist of eight identified members, and are divided into three sub-families based on their sub- structures and substrate specificities. Class I PI3Ks are further divided into two sub-classes, Class IA and Class IB. Class IA consists of pi 10a, pi lOβ, and pi 105 as catalytic sub-units and these are activated in tyrosine kinase receptor signaling. Class IB contains only the pi 10γ sub-unit, which is mostly activated by seven trans-membrane G-protein coupled receptors (GPCRs) via its regulatory sub-unit plOl and G-protein βγ sub- units. Class II PI3Ks have the α, β, and γisoforms. Class III PI3Ks include the phophatidylinositol specific 3-kinases. Overall the PI3K pathways play important roles in various disease states on account of their pivotal role in cell signaling.
[0004] The role of PI3K in cancers has received much attention. PI3K signaling is important to many aspects of cell growth and survival. Because the PI3K pathway is stimulated as a physiological consequence of many growth factors and regulators, it is quite frequently targeted by genomic aberrations including mutation, amplification and rearrangement, The activation of the PI3K pathway results in a disturbance of control of cell growth and survival, which contributes to a competitive growth advantage, metastatic
competence and, often resistance to therapy. This makes the PI3Ks an attractive target for the development of novel anticancer agents.
[0005] Further, PI3Ks play a role in vasculostasis. Compromised vasculostasis has serious pathologic consequences. Examples where excessive vascular permeability leads to particularly deleterious effects include pulmonary edema, cerebral edema, and cardiac edema. In general, however, edema in any tissue or organ leads to some loss of normal function, and therefore to the risk of morbidity or even mortality. Similarly, excessive endothelial proliferation may damage tissues, such as the retina in proliferative retinopathies, or fuel unwanted tissue growth, such as with tumor growth. Many pathologic and disease situations are marked by multiple dysregulations in vasculostasis.
[0006] Angio genesis, for example, encompasses both enhanced vascular proliferation and permeability, as newly-formed blood vessels do not generally exhibit the same level of vascular barrier function as well-established or mature vessels. Examples of such hyper-permeable vasculature can be found in cancers, vasculo proliferative diseases, retinal diseases, and rheumatoid arthritis. The connection between angiogenesis and hyperpermeability may partly result from the dual action of factors such as vascular endothelial growth factor (VEGF), which induces both endothelial proliferation and vascular permeability.
[0007] The PI3K family may also play an important role in inflammatory responses, and in respiratory diseases. Therefore, in addition to direct roles in regulating vasculostasis, the PI3K family can also influence situations in which vasculostasis is compromised, including ischemia and ischemia-reperfusion injury, through their control of leukocyte functioning. Maintaining or restoring vasculostasis should be beneficial to overall patient outcome where such indications as, for example, inflammation, allergic diseases, cancer, cerebral stroke, myocardial infarction, pulmonary and cardiac insufficiency, renal failure, and retinopathies, are present.
SUMMARY
[0008] This disclosure is generally directed to compounds that include a thiazolidinone moiety. In part, this disclosure also directed to compounds that are phosphoinositide 3-kinase (PI3K) enzyme or pathway inhibitors or modulators. [0009] For example, compounds are provided that inhibit a PI3K, with an IC50 of about 200 nM or less, about 50 nM or less, from about 1 nM to about 50 nM, or about 10 nM or less. For example, the compound may inhibit one or more of PI3Kα, PI3Kβ, PI3Kγ, or PI3Kδ.
[0010] In an embodiment, a method of treating an respiratory or ocular disorder is provided, wherein the method comprise administering to a patient in need thereof an effective amount of a compound disclosed herein.
[0011] In another embodiment, a method of treating cancer is provided comprising administrating to a patient in need thereof an effective amount of a compound of any of the disclosed compounds.
[0012] A method is provided herein for inhibiting tumor cell growth, tumor cell proliferation, or tumorigenesis comprising administering to a patient in need thereof an effective amount of a compound of any of the disclosed compounds..
[0013] Further, methods are provided for treating pain, diabetes, inflammation, platelet aggregation, ischemic heart disease, sclerosis, restenosis, disorders, HIV, bone resorption, cancer, non-small cell lung cancer, or brain cancer, comprising administering to a patient in need thereof an effective amount of any of the disclosed compounds.
DETAILED DESCRIPTION
[0014] The present disclosure is directed in part towards novel compounds and compositions that inhibit PI3K and methods of making and using the same. Such compounds may inhibit one or more of the phosphoinositide-3-kinase family, including PI3Kα, PI3Kβ, PI3Kδ, and/or PI3Kγ.
[0015] Before further description of the present invention, certain terms employed in the specification, examples and appended claims are collected here. These definitions should be read in light of the remainder of the disclosure and understood as by a person of skill in the art.
Unless defined otherwise, all technical and scientific terms used herein have the same meaning as commonly understood by a person of ordinary skill in the art.
[0016] The term "therapeutic agent" is art-recognized and refers to any chemical moiety that is a biologically, physiologically, or pharmacologically active substance that acts locally or systemically in a subject. Examples of therapeutic agents, also referred to as "drugs", are described in well-known literature references such as the Merck Index, the Physicians Desk Reference, and The Pharmacological Basis of Therapeutics, and they include, without limitation, medicaments; vitamins; mineral supplements; substances used for the treatment, prevention, diagnosis, cure or mitigation of a disease or illness; substances which affect the structure or function of the body; or pro-drugs, which become biologically active or more active after they have been placed in a physiological environment.
[0017] The term "therapeutic effect" is art-recognized and refers to a local or systemic effect in animals, particularly mammals, and more particularly humans caused by a pharmacologically active substance. The term thus means any substance intended for use in the diagnosis, cure, mitigation, treatment or prevention of disease or in the enhancement of desirable physical or mental development and/or conditions in an animal or human. The phrase "therapeutically-effective amount" means that amount of such a substance that produces some desired local or systemic effect at a reasonable benefit/risk ratio applicable to any treatment. The therapeutically effective amount of such substance will vary depending upon the subject and disease condition being treated, the weight and age of the subject, the severity of the disease condition, the manner of administration and the like, which can readily be determined by one of ordinary skill in the art. For example, certain compositions of the present invention may be administered in a sufficient amount to produce a at a reasonable benefit/risk ratio applicable to such treatment. [0018] The term "modulation" is art-recognized and refers to up regulation (i.e., activation or stimulation), down regulation (i.e., inhibition or suppression) of a response, or the two in combination or apart.
[0019] A "patient," "subject" or "host" to be treated by the subject method may mean either a human or non-human animal. [0020] The term "treating" is art-recognized and refers to curing as well as ameliorating at least one symptom of any condition or disease.
[0021] The term "prodrug" is art-recognized and is intended to encompass compounds which, under physiological conditions, are converted into the agents of the present invention. A common method for making a prodrug is to select moieties which are hydrolyzed under physiological conditions to provide the desired compound. In other embodiments, the prodrug is converted by an enzymatic activity of the host animal or the target organ or cell.
[0022] The term "alkyl" is art-recognized, and includes saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl substituted cycloalkyl groups, and cycloalkyl substituted alkyl groups. In certain embodiments, a straight chain or branched chain alkyl has about 30 or fewer carbon atoms in its backbone (e.g., C1-C3O for straight chain, C3-C30 for branched chain), and alternatively, about 20 or fewer, e.g. from 1 to 6 carbons. Likewise, cycloalkyls have from about 3 to about 10 carbon atoms in their ring structure, and alternatively about 5, 6 or 7 carbons in the ring structure. The term "alkyl" is also defined to include halosubstituted alkyls.
[0023] Moreover, the term "alkyl" (or "lower alkyl") includes "substituted alkyls", which refers to alkyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone. Such substituents may include, for example, a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxyl, a phosphoryl, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic moiety. It will be understood by those skilled in the art that the moieties substituted on the hydrocarbon chain may themselves be substituted, if appropriate. For instance, the substituents of a substituted alkyl may include substituted and unsubstituted forms of amino, azido, imino, amido, phosphoryl (including phosphonate and phosphinate), sulfonyl (including sulfate, sulfonamido, sulfamoyl and sulfonate), and silyl groups, as well as ethers, alkylthios, carbonyls (including ketones, aldehydes, carboxylates, and esters), -CN and the like. Exemplary substituted alkyls are described below. Cycloalkyls may be further substituted with alkyls, alkenyls, alkoxys, alkylthios, aminoalkyls, carbonyl- substituted alkyls, -CN, and the like. [0024] The term "aralkyl" is art-recognized and refers to an alkyl group substituted with an aryl group (e.g., an aromatic or heteroaromatic group).
[0025] The terms "alkenyl" and "alkynyl" are art-recognized and refer to unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but that contain at least one double or triple bond respectively.
[0026] Unless the number of carbons is otherwise specified, "lower alkyl" refers to an alkyl group, as defined above, but having from one to about ten carbons, alternatively from one to about six carbon atoms in its backbone structure. Likewise, "lower alkenyl" and "lower alkynyl" have similar chain lengths.
[0027] The term "heteroatom" is art-recognized and refers to an atom of any element other than carbon or hydrogen. Illustrative heteroatoms include boron, nitrogen, oxygen, phosphorus, sulfur and selenium.
[0028] The term "aryl" is art-recognized and refers to 5-, 6- and 7-membered single-ring aromatic groups that may include from zero to four heteroatoms, for example, benzene, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, triazole, pyrazole, pyridine, pyrazine, pyridazine and pyrimidine, and the like. Those aryl groups having heteroatoms in the ring structure may also be referred to as "heteroaryl" or "heteroaromatics." The aromatic ring may be substituted at one or more ring positions with such substituents as described above, for example, halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, sulfonamido, ketone, aldehyde, ester, heterocyclyl, aromatic or heteroaromatic moieties, -CF3, -CN, or the like. The term "aryl" or aromatic, also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings (the rings are "fused rings") wherein at least one of the rings is aromatic, e.g., the other cyclic rings may be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyl s.
[0029] The terms ortho, meta and para are art-recognized and refer to 1,2-, 1,3- and 1,4- disubstituted benzenes, respectively. For example, the names 1,2-dimethylbenzene and ortho- dimethylbenzene are synonymous.
[0030] The terms "heterocyclyl" or "heterocyclic group" are art-recognized and refer to 3- to about 10-membered ring structures, alternatively 3- to about 7-membered rings, whose ring structures include one to four heteroatoms. Heterocycles may also be polycycles. Heterocyclyl groups include, for example, thiophene, thianthrene, furan, pyran, isobenzofuran, chromene, xanthene, phenoxanthene, pyrrole, imidazole, pyrazole, isothiazole, isoxazole, pyridine,
pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, pyrimidine, phenanthroline, phenazine, phenarsazine, phenothiazine, furazan, phenoxazine, pyrrolidine, oxolane, thiolane, oxazole, piperidine, piperazine, morpholine, lactones, lactams such as azetidinones and pyrrolidinones, sultams, sultones, and the like. The heterocyclic ring may be substituted at one or more positions with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -CF3, -CN, or the like.
[0031] The terms "polycyclyl" or "polycyclic group" are art-recognized and refer to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls) in which two or more carbons are common to two adjoining rings, e.g., the rings are "fused rings". Rings that are joined through non-adjacent atoms are termed "bridged" rings. Each of the rings of the polycycle may be substituted with such substituents as described above, as for example, halogen, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, ketone, aldehyde, ester, a heterocyclyl, an aromatic or heteroaromatic moiety, -CF3, -CN, or the like.
[0032] The term "carbocycle" is art-recognized and refers to an aromatic or non-aromatic ring in which each atom of the ring is carbon.
[0033] The term "nitro" is art-recognized and refers to -NO2; the term "halogen" is art- recognized and refers to -F, -Cl, -Br or -I; the term "sulfhydryl" is art-recognized and refers to - SH; the term "hydroxyl" means -OH; and the term "sulfonyl" is art-recognized and refers to - SO2". [0034] The terms "amine" and "amino" are art-recognized and refer to both unsubstituted and substituted amines, e.g., a moiety that may be represented by the general formulas:
R50 R50 I
/ I + N N R53
R51 R52
wherein R50, R51 and R52 each independently represent a hydrogen, an alkyl, an alkenyl, - (CH2)m-R61, or R50 and R51, taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure; R61 represents an aryl, a cycloalkyl, a cycloalkenyl, a heterocycle or a polycycle; and m is zero or an integer in the range of 1 to 8. In certain embodiments, only one of R50 or R51 may be a carbonyl, e.g., R50, R51 and the nitrogen together do not form an imide. In other embodiments, R50 and R51 (and optionally R52) each independently represent a hydrogen, an alkyl, an alkenyl, or -(CH2)m- R61. Thus, the term "alkylamine" includes an amine group, as defined above, having a substituted or unsubstituted alkyl attached thereto, i.e., at least one of R50 and R51 is an alkyl group.
[0035] The term "amido" is art recognized as an amino-substituted carbonyl and includes a moiety that may be represented by the general formula:

[0036] The term "acylamino" is art-recognized and refers to a moiety that may be represented by the general formula:
O
N u R54
R50
wherein R50 is as defined above, and R54 represents a hydrogen, an alkyl, an alkenyl or - (CH2)m-R61, where m and R61 are as defined above.
[0037] The term "alkylthio" refers to an alkyl group, as defined above, having a sulfur radical attached thereto. In certain embodiments, the "alkylthio" moiety is represented by one of -S-alkyl, -S-alkenyl, -S-alkynyl, and -S-(CH2)m-R61, wherein m and R61 are defined above. Representative alkylthio groups include methylthio, ethyl thio, and the like.
[0038] The term "carbonyl" is art recognized and includes such moieties as may be represented by the general formulas:

[0039] The definition of each expression, e.g. alkyl, m, n, and the like, when it occurs more than once in any structure, is intended to be independent of its definition elsewhere in the same structure.
[0040] Certain compounds contained in compositions of the present invention may exist in particular geometric or stereoisomeric forms. In addition, polymers of the present invention may also be optically active. The present invention contemplates all such compounds, including cis- and trans-isomers, R- and S-enantiomers, diastereomers, (D)-isomers, (L)- isomers, the racemic mixtures thereof, and other mixtures thereof, as falling within the scope of the invention. Additional asymmetric carbon atoms may be present in a substituent such as an alkyl group. All such isomers, as well as mixtures thereof, are intended to be included in this invention.
[0041] If, for instance, a particular enantiomer of compound of the present invention is desired, it may be prepared by asymmetric synthesis, or by derivation with a chiral auxiliary, where the resulting diastereomeric mixture is separated and the auxiliary group cleaved to provide the pure desired enantiomers. Alternatively, where the molecule contains a basic functional group, such as amino, or an acidic functional group, such as carboxyl, diastereomeric salts are formed with an appropriate optically- active acid or base, followed by resolution of the diastereomers thus formed by fractional crystallization or chromatographic means well known in the art, and subsequent recovery of the pure enantiomers.
[0042] It will be understood that "substitution" or "substituted with" includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, or other reaction.
[0043] The term "substituted" is also contemplated to include all permissible substituents of organic compounds. In a broad aspect, the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and nonaromatic substituents of organic compounds. Illustrative substituents include, for example, those described herein above. The permissible substituents may be one or more and the same or different for appropriate organic compounds. For purposes of this invention, the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms. This invention is not intended to be limited in any manner by the permissible substituents of organic compounds.
[0044] For purposes of this invention, the chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 67th Ed., 1986-87, inside cover. Also for purposes of this invention, the term "hydrocarbon" is contemplated to include all permissible compounds having at least one hydrogen and one carbon atom. In a broad aspect, the permissible hydrocarbons include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and nonaromatic organic compounds that may be substituted or unsubstituted.
[0045] The term "pharmaceutically-acceptable salts" is art-recognized and refers to the relatively non-toxic, inorganic and organic acid addition salts of compounds, including, for example, those contained in compositions of the present invention.
[0046] The term "pharmaceutically acceptable carrier" is art-recognized and refers to a pharmaceutically-acceptable material, composition or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material, involved in carrying or transporting any subject composition or component thereof from one organ, or portion of the body, to another organ, or portion of the body. Each carrier must be "acceptable" in the sense of being compatible with the subject composition and its components and not injurious to the patient. Some examples of materials which may serve as pharmaceutically acceptable carriers include: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as corn starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients, such as cocoa butter and suppository waxes; (9) oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol and polyethylene glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14) buffering agents, such as magnesium hydroxide and aluminum hydroxide; (15) alginic acid; (16) pyrogen-free water; (17) isotonic saline; (18) Ringer's solution; (19) ethyl alcohol; (20) phosphate buffer solutions; and (21) other non-toxic compatible substances employed in pharmaceutical formulations.
[0047] The terms "systemic administration," "administered systemically," "peripheral administration" and "administered peripherally" are art-recognized and refer to the administration of a subject composition, therapeutic or other material other than directly into the central nervous system, such that it enters the patient's system and, thus, is subject to metabolism and other like processes, for example, subcutaneous administration.
[0048] The terms "parenteral administration" and "administered parenterally" are art- recognized and refer to modes of administration other than enteral and topical administration, usually by injection, and includes, without limitation, intravenous, intramuscular, intraarterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intra-articulare, subcapsular, subarachnoid, intraspinal, and intrasternal injection and infusion.
Compounds
[0049] Provided herein, in part, is a compound according to formula I:

wherein
A1 is a nine or ten membered bicyclic heterocycle, for example a nine or ten membered bicyclic aromatic heterocycle, containing one to three heteroatoms chosen from nitrogen, oxygen or sulfur optionally substituted by one to four substituents each independently selected from halo, hydroxyl, or alkyl;
A2 is an heterocycle group, for example an aromatic heterocyclic group, which may contain an heteroatom chosen from sulfur or oxygen;
X is O, S or NR12;
R1 is selected from the group consisting of H, amide, ester, carbamate or alkyl;
wherein A2 is optionally substituted by one to four substituents each independently selected from the group consisting of halo, hydroxyl, mercapto, nitro, formyl, formamido, carboxy, cyano, amino, amide, carbamoyl, sulphamoyl, ureido, alkyl, alkenyl, alkynyl, alkoxy, alkanoyl, alkoxycarbonyl, thiocarbonyl, N-alkylureido, N-alkylamino, N-alkylsulphamoyl, N- alkylcarbamoyl, N-arylamide, N-alkylamide, aryl, heterocycle, or cycloalkyl, wherein said alkyl, alkenyl, alkynyl or alkoxy may be optionally substituted by one or more groups selected from Ra, and wherein said aryl, heterocycle or cycloalkyl may each be optionally substituted by one, two or three substituents selected from hydroxyl, halo, amino, cyano, carboxy, nitro, amido, amino, carbamoyl, sulphamoyl, alkoxy, or alkyl, or another heterocycle, aryl, or alkylheterocycle .
[0050] Ra is independently selected from halo, hydroxyl, nitro, formyl, formamido, alkyl, alkoxy, carboxyl, cyano, amino, amido, carbamoyl, sulphamoyl, ureido, aryl, heterocycle, or cycloalkyl.
[0051] R12 is selected from the group consisting of alkyl, alkenyl, alkynl, aryl, heterocycle, or cycloalkyl. In some embodiments, the moieties of R12 may be optionally substituted by one, two, or three substituents selected from the group consisting of halo, hydroxyl, nitro, formyl, formamido, alkyoxy, alkyl, carboxyl, cyano, amino, amido, carbamoyl, sulphamoyl, ureido, aryl, heterocycle, or cycloalkyl. For example, R12 may be CF3.
[0052] The pharmaceutically acceptable salts, prodrugs, N-oxides, and hydrates of the disclosed compounds, including formula I, are contemplated herein.
[0053] A1 may be, in some embodiments, a nine or ten membered aromatic bicyclic heterocycle containing one to three heteroatoms chosen, for each occurrence, from nitrogen or oxygen. In other embodiments, A1 is selected from the group consisting of: indolyl, naphthyl, quinolinyl, isoquinolinyl, quinoxalinyl, quinazolinyl, benzofuryl, benzthienyl, cinnolinyl, and pteridinyl. For example, A1 can be selected from quinolinyl or quinazolinyl.
[0054] A2 may be selected from thiopene, furan, pyran, chromene, isothiazole, isoxazole, benzothiophene, 2,3-dihydrothieno[3,4-b][l,4]dioxine, 4,5,6,7-tetrahydrothieno[3,4-c]pyridine, or benzofuran. In an embodiments, A2 may be substituted on a ring carbon with 1 or 2 substituents each independently chosen from: halo, carboxyl, cyano, amino, amide, formyl, formamido, alkoxy, carbamoyl, sulphamoyl, ureido, an optionally substituted alkyl (for example CF3), an optionally substituted aryl, or an optionally substituted heterocycle group.
[0055] For example, A2 can be substituted on a ring carbon with 1 or 2 substituents each independently chosen from halo, methyl, phenyl, or isoxazole, wherein said phenyl and isoxazole are optionally substituted on a ring carbon by 1 or 2 substituents chosen from halo, alkyl, aryl or heterocycle.
[0056] In some embodiments, a disclosed compound is represented by formula Ia or Ib:

wherein X and R1 are as defined above;
Y is CRb or N;
Z is S or O;
R2, R3, R4 are each independently selected from the group consisting of H, halo, alkyl, alkenyl, alkynyl, hydroxyalkyl, alkoxy, cyano, carboxyl, N-arylamido, aryl, and heterocycle, or R2 and R3, or R3 and R4, form, together with the carbon atoms to which they are attached, a 5 or 6 carbocycle or heterocycle ring, wherein any of said alkyl, alkenyl, alkynyl, carbocycle, aryl, or heterocycle can be optionally substituted with halo, hydroxyl, cyano, alkoxy, ureido, sulphamoyl, N-alkylsulphamoyl, alkyl, aryl, heterocycle or alkylheterocycle. For example, R3 may be phenyl or N-phenylacetamide wherein the phenyl is optionally substituted with hydroxyl, alkyl, N-alkylsulphamoyl, alkoxy, or an alkylmorpholino.
[0057] Rb is selected from H, halo, hydroxyl, or alkyl.
[0058] In some embodiments, Z is S or O. In other embodiments R2 is H. In an embodiment, R3 and R4 are each independently selected from H, -CH2OH, -CH2CH2OH, CH3, phenyl, F, Cl, CO2H,-C(O)NH-phenyl, -C(O)NCH2CH2-pyrrolidine, or dimethylisoxazole, wherein the phenyl is optionally substituted by, for example, one or two substituents selected from the group consisting of halo, cyano, hydroxyl, alkyl, alkoxy, N-alkylsulphamoyl, and sulphamoyl.
[0059] In another embodiment, R3 and R4, taken together with the ring carbons on which they are attached, form a bicyclic ring such as benzofuran, benzothiophene, 2,3-dihydro- thieno[3,4b][l,4]dioxin or 4,5,6,7-tetrahydrothieno[3,2-c]pyridine. Such bicyclic rings may be substituted by, for example, one or two substituents selected from the group consisting of halo, hydroxyl, cyano, and alkyl.
[0060] X may be, for example NR12, wherein R12 is phenyl optionally substituted by one or two halogen moities; or X may be, for example, O.
[0061] Also provided herein is a compound represented by formula II:

Re is selected from the group consisting of:


X is O, S or NR12;
W is, independently for each occurrence, CRb, N-alkyl, NH, N-C(O)-alkyl, N-C(O)- heterocycle, N-C(O)-N-alkyl, N-C(O)-N-heterocycle, O or S;
Z is, independently for each occurrence, S or O;
Rb is selected from H, halo, hydroxyl, or alkyl;
R1 is H or alkyl;
R7 is H or alkyl;
Rg and Rg are each independently selected from H, Cl, F, alkyl, alkynl, alkenyl, carboxyl, cyano, hydroxyl, hydroxyalkyl, formyl, formamido, amido, amine, sulphamoyl, N-

alkylsulphamoyl, ureido, R11, -R13R11, or ;
m is an integer from 0 to 8;
R1O is independently selected for each occurrence from H, Cl, F, hydroxyl, cynao, or alkyl;
R11 is selected from group consisting of heterocycle, aryl, or cycloalkyl; wherein R11 is optionally substituted at one to four substituents each independently selected from halo, hydroxyl, carboxyl, sulphamoyl, N-alkylsulphamoyl, -alkyl-heterocycle, -alkyl-carbocycle or alkyl;
R12 is selected from the group consisting of alkyl, aryl, heterocycle, or cycloalkyl, wherein R12 may be optionally substituted by one, two, or three substituents selected from the group consisting of halo, hydroxyl, nitro, formyl, formamido, alkyoxy, alkyl, carboxyl, cyano, amino, amido, carbamoyl, sulphamoyl, ureido, aryl, heterocycle, or cycloalkyl;
R13 is alkylene, alkenylene, or alkynyl; or
pharmaceutically acceptable salts, prodrugs, N-oxides, and hydrates thereof. [0062] In some embodiments, formula II may be represented by:

[0065] R11 may be substituted at one or two positions by methyl. In some embodiments, R11 is chosen from: pyrrolindyl, phenyl, isoxazole, or thiazolidinedione. [0066] In some embodiments, R7 is H or methyl.
[0067] In some embodiments, Rg may be H, methyl, or ethyl. .
[0068] Rg may be chosen, for example, from: H, Cl, phenyl, co2H, or 3,5-dimethyl-isoxazol- 4-yl.
Dosages [0069] The dosage of any compositions of the present invention will vary depending on the symptoms, age and body weight of the patient, the nature and severity of the disorder to be treated or prevented, the route of administration, and the form of the subject composition. Any of the subject formulations may be administered in a single dose or in divided doses. Dosages for the compositions of the present invention may be readily determined by techniques known to those of skill in the art or as taught herein.
[0070] In certain embodiments, the dosage of the subject compounds will generally be in the range of about 0.01 ng to about 10 g per kg body weight, specifically in the range of about 1 ng to about 0.1 g per kg, and more specifically in the range of about 100 ng to about 10 mg per kg.
[0071] An effective dose or amount, and any possible affects on the timing of administration of the formulation, may need to be identified for any particular composition of the present invention. This may be accomplished by routine experiment as described herein, using one or more groups of animals (preferably at least 5 animals per group), or in human trials if appropriate. The effectiveness of any subject composition and method of treatment or prevention may be assessed by administering the composition and assessing the effect of the administration by measuring one or more applicable indices, and comparing the post-treatment values of these indices to the values of the same indices prior to treatment.
[0072] The precise time of administration and amount of any particular subject composition that will yield the most effective treatment in a given patient will depend upon the activity, pharmacokinetics, and bioavailability of a subject composition, physiological condition of the
patient (including age, sex, disease type and stage, general physical condition, responsiveness to a given dosage and type of medication), route of administration, and the like. The guidelines presented herein may be used to optimize the treatment, e.g., determining the optimum time and/or amount of administration, which will require no more than routine experimentation consisting of monitoring the subject and adjusting the dosage and/or timing.
[0073] While the subject is being treated, the health of the patient may be monitored by measuring one or more of the relevant indices at predetermined times during the treatment period. Treatment, including composition, amounts, times of administration and formulation, may be optimized according to the results of such monitoring. The patient may be periodically reevaluated to determine the extent of improvement by measuring the same parameters.
Adjustments to the amount(s) of subject composition administered and possibly to the time of administration may be made based on these reevaluations.
[0074] Treatment may be initiated with smaller dosages which are less than the optimum dose of the compound. Thereafter, the dosage may be increased by small increments until the optimum therapeutic effect is attained.
[0075] The use of the subject compositions may reduce the required dosage for any individual agent contained in the compositions because the onset and duration of effect of the different agents may be complimentary.
[0076] Toxicity and therapeutic efficacy of subject compositions may be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e.g., for determining the LD50 and the ED50.
[0077] The data obtained from the cell culture assays and animal studies may be used in formulating a range of dosage for use in humans. The dosage of any subject composition lies preferably within a range of circulating concentrations that include the ED50 with little or no toxicity. The dosage may vary within this range depending upon the dosage form employed and the route of administration utilized. For compositions of the present invention, the therapeutically effective dose may be estimated initially from cell culture assays.
Formulations
[0078] The compositions of the present invention may be administered by various means, depending on their intended use, as is well known in the art. For example, if compositions of
the present invention are to be administered orally, they may be formulated as tablets, capsules, granules, powders or syrups. Alternatively, formulations of the present invention may be administered parenterally as injections (intravenous, intramuscular or subcutaneous), drop infusion preparations, suppositories or administration intranasally (for example, to deliver a dosage to the brain via the nose or to deliver a dosage to the nose directly) or by inhalation (e.g. to treat a condition of the respiratory tract or to pretreat or vaccinate via the respiratory tract). AFor application by the ophthalmic mucous membrane route, compositions of the present invention may be formulated as eyedrops or eye ointments. These formulations may be prepared by conventional means, and, if desired, the compositions may be mixed with any conventional additive, such as an excipient, a binder, a disintegrating agent, a lubricant, a corrigent, a solubilizing agent, a suspension aid, an emulsifying agent or a coating agent.
[0079] In formulations of the subject invention, wetting agents, emulsifiers and lubricants, such as sodium lauryl sulfate and magnesium stearate, as well as coloring agents, release agents, coating agents, sweetening, flavoring and perfuming agents, preservatives and antioxidants may be present in the formulated agents.
[0080] Subject compositions may be suitable for oral, nasal, topical (including buccal and sublingual), rectal, vaginal, aerosol and/or parenteral administration. The formulations may conveniently be presented in unit dosage form and may be prepared by any methods well known in the art of pharmacy. The amount of composition that may be combined with a carrier material to produce a single dose vary depending upon the subject being treated, and the particular mode of administration.
[0081] Methods of preparing these formulations include the step of bringing into association compositions of the present invention with the carrier and, optionally, one or more accessory ingredients. In general, the formulations are prepared by uniformly and intimately bringing into association agents with liquid carriers, or finely divided solid carriers, or both, and then, if necessary, shaping the product.
[0082] Formulations suitable for oral administration may be in the form of capsules, cachets, pills, tablets, lozenges (using a flavored basis, usually sucrose and acacia or tragacanth), powders, granules, or as a solution or a suspension in an aqueous or non-aqueous liquid, or as an oil-in-water or water-in-oil liquid emulsion, or as an elixir or syrup, or as pastilles (using an inert base, such as gelatin and glycerin, or sucrose and acacia), each
containing a predetermined amount of a subject composition thereof as an active ingredient. Compositions of the present invention may also be administered as a bolus, electuary, or paste.
[0083] In solid dosage forms for oral administration (capsules, tablets, pills, dragees, powders, granules and the like), the subject composition is mixed with one or more pharmaceutically acceptable carriers, such as sodium citrate or dicalcium phosphate, and/or any of the following: (1) fillers or extenders, such as starches, lactose, sucrose, glucose, mannitol, and/or silicic acid; (2) binders, such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinyl pyrrolidone, sucrose and/or acacia; (3) humectants, such as glycerol; (4) disintegrating agents, such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate; (5) solution retarding agents, such as paraffin; (6) absorption accelerators, such as quaternary ammonium compounds; (7) wetting agents, such as, for example, acetyl alcohol and glycerol monostearate; (8) absorbents, such as kaolin and bentonite clay; (9) lubricants, such a talc, calcium stearate, magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate, and mixtures thereof; and (10) coloring agents. In the case of capsules, tablets and pills, the compositions may also comprise buffering agents. Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugars, as well as high molecular weight polyethylene glycols and the like.
[0084] A tablet may be made by compression or molding, optionally with one or more accessory ingredients. Compressed tablets may be prepared using binder (for example, gelatin or hydroxypropylmethyl cellulose), lubricant, inert diluent, preservative, disintegrant (for example, sodium starch glycolate or cross-linked sodium carboxymethyl cellulose), surface- active or dispersing agent. Molded tablets may be made by molding in a suitable machine a mixture of the subject composition moistened with an inert liquid diluent. Tablets, and other solid dosage forms, such as dragees, capsules, pills and granules, may optionally be scored or prepared with coatings and shells, such as enteric coatings and other coatings well known in the pharmaceutical-formulating art.
[0085] Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs. In addition to the subject composition, the liquid dosage forms may contain inert diluents commonly used in the art, such as, for example, water or other solvents, solubilizing agents and emulsifiers, such as
ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, cyclodextrins and mixtures thereof. [0086] Suspensions, in addition to the subject composition, may contain suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
[0087] Formulations for rectal or vaginal administration may be presented as a suppository, which may be prepared by mixing a subject composition with one or more suitable non- irritating excipients or carriers comprising, for example, cocoa butter, polyethylene glycol, a suppository wax or a salicylate, and which is solid at room temperature, but liquid at body temperature and, therefore, will melt in the body cavity and release the active agent. Formulations which are suitable for vaginal administration also include pessaries, tampons, creams, gels, pastes, foams or spray formulations containing such carriers as are known in the art to be appropriate.
[0088] Dosage forms for transdermal administration of a subject composition includes powders, sprays, ointments, pastes, creams, lotions, gels, solutions, patches and inhalants. The active component may be mixed under sterile conditions with a pharmaceutically acceptable carrier, and with any preservatives, buffers, or propellants which may be required.
[0089] The ointments, pastes, creams and gels may contain, in addition to a subject composition, excipients, such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof. [0090] Powders and sprays may contain, in addition to a subject composition, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and polyamide powder, or mixtures of these substances. Sprays may additionally contain customary propellants, such as chlorofluorohydrocarbons and volatile unsubstituted hydrocarbons, such as butane and propane.
[0091] Compositions and compounds of the present invention may alternatively be administered by aerosol. This is accomplished by preparing an aqueous aerosol, liposomal preparation or solid particles containing the compound. A non-aqueous (e.g., fluorocarbon propellant) suspension could be used. Sonic nebulizers may be used because they minimize exposing the agent to shear, which may result in degradation of the compounds contained in the subject compositions.
[0092] Ordinarily, an aqueous aerosol is made by formulating an aqueous solution or suspension of a subject composition together with conventional pharmaceutically acceptable carriers and stabilizers. The carriers and stabilizers vary with the requirements of the particular subject composition, but typically include non-ionic surfactants (T weens, Pluronics, or polyethylene glycol), innocuous proteins like serum albumin, sorbitan esters, oleic acid, lecithin, amino acids such as glycine, buffers, salts, sugars or sugar alcohols. Aerosols generally are prepared from isotonic solutions.
[0093] Dosages for administration by nasal delivery, e.g. delivered to or via the nasal cavity, can be applied as drops, ointments, gels, mists/sprays (aqueous or nonaqueous), aerosols
(liquids, suspensions or dry powders), powders, or combinations thereof. Such delivery can be achieved by commercially available devices such as droppers, nasal sprayers, metered dose aerosols, or other mechanisms known in the art. Pharmaceutical formulations for inhalation and/or delivery to the nose, may contain from 1% to 20% by weight of a penetrator enhancer (for example, surfactants, e.g. sugar esters, sugar ethers, carbohydrate esters) which may allow enhanced nose permeability of the active agent.
[0094] Dosages for administration by inhalation or by delivered to or via the lung, can be applied as mists/sprays (aqueous or nonaqueous), aerosols (liquids, suspensions or dry powders),liquids or suspensions (aqueous or nonaqueous), powders, or combinations thereof. Such delivery can be achieved by commercially available devices such as 1) nebulizers, 2) metered dose inhalers, 3) dry powder inhalers, 4) soft mist inhalers, or by instillation or insufflation, or other mechanisms and/or devices known in the art.
[0095] Pharmaceutical compositions of this invention suitable for parenteral administration comprise a subject composition in combination with one or more pharmaceutically- acceptable sterile isotonic aqueous or non-aqueous solutions, dispersions, suspensions or emulsions, or sterile powders which may be reconstituted into sterile injectable solutions or dispersions just
prior to use, which may contain antioxidants, buffers, bacteriostats, solutes which render the formulation isotonic with the blood of the intended recipient or suspending or thickening agents.
[0096] Examples of suitable aqueous and non-aqueous carriers which may be employed in the pharmaceutical compositions of the invention include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol, and the like), and suitable mixtures thereof, vegetable oils, such as olive oil, and injectable organic esters, such as ethyl oleate and cyclodextrins. Proper fluidity may be maintained, for example, by the use of coating materials, such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants.
Methods
[0097] Treatment or amelioration of disease states and pathological conditions that implicate PI3K pathways are contemplated herein, and such treatment comprises administering one or more of the disclosed compounds, such as those recited in Formulas I or II, or a composition as described herein comprising a disclosed compound. Methods of treating a patient suffering from diseases such as myocardial infarction, stroke, congestive heart failure, ischemia or reperfusion injury, vascular leakage syndrome (VLS), cancer, arthritis, (for example rheumatoid arthritis), or other arthropathy, eye diseases including uveitis, retinopathy or vitreoretinal disease, macular degeneration, autoimmune diseases, vascular leakage syndrome, inflammatory diseases, edema, transplant rejection, burn, respiratory diseases such as acute respiratory distress syndrome (ARDS), asthma, and chronic obstructive pulmonary disorder (COPD), and transplant rejection are contemplated, and may comprise administering a disclosed compound, such as those recited in Formulas I or II, or a composition comprising a disclosed compound. [0098] Also contemplated is the treatment of e.g. the edema formation that may be the unwanted consequence of other therapeutic interventions, such as immunotherapy, cancer chemotherapy and radiation therapy. Compounds and compositions disclosed herein, e.g. that inhibit vascular permeability may be used in a co-therapy to reduce the deleterious side-effects of such therapies. Furthermore, edema formation may cause uneven delivery of therapeutic agents to diseased tissues, therefore vasculostatic agents that inhibit vascular permeability could be used in a co-therapy approach to enhance delivery and efficacy of such therapies.
[0099] Disclosed herein are methods of treating tumor cell growth, tumor cell proliferation, or tumorigenesis by administering to a patient a disclosed compound. For example, disclosed herein are methods of treating breast or prostate cancer. Other exemplary methods of treating cancers include treatment of myeloma. [0100] Acute and/or prophylactic treatment of the interruption of blood flow by pathologic conditions such as thrombus formation, or medical intervention such as cardioplegia, organ transplantation, and angioplasty, or physical trauma, using disclosed compounds is also contemplated.
[0101] The examples which follow are intended in no way to limit the scope of this invention but are provided to illustrate how to prepare and use compounds of the present invention. Many other embodiments of this invention will be apparent to one skilled in the art.
EXEMPLIFICATION
General Methods
[0102] All synthetic experiments were performed under anhydrous conditions (i.e. dry solvents) in an atmosphere of argon, except where stated, using oven-dried apparatus and employing standard techniques in handling air-sensitive materials. Aqueous solutions of sodium bicarbonate (NaHCO3) and sodium chloride (brine) were saturated. Analytical thin layer chromatography (TLC) was carried out on Merck Kieselgel 60 F254 plates with visualization by ultraviolet and/or anisaldehyde, potassium permanganate or phosphomolybdic acid dips. Reverse-phase HPLC chromatography was carried out on Gilson 215 liquid handler equipped with Waters SymmetryShield™ RP18 7μm (40 x 100mm) Prep-Pak cartridge. Mobile phase consisted of standard acetonitrile (ACN) and DI Water, each with 0.1% TFA added. Purification was carried out at a flow rate of 4OmL/ min. NMR spectra: 1H Nuclear magnetic resonance spectra were recorded at 500 MHz. Data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, qn = quintet, dd = doublet of doublets, m = multiplet, br s = broad singlet), coupling constant (J/Hz) and integration. Coupling constants were taken directly from the spectra and are uncorrected. Low resolution mass spectra: Electrospray (ES+) ionization was used. The protonated parent ion (M+H) or fragment of highest mass is quoted. Analytical gradient consisted of 10% ACN in water ramping up to 100% ACN over 5 min unless otherwise stated.
Example 1: Preparation of 4-Chloro-6-vinylquinoline (1)

[0103] To a solution of 6-bromo-4-chloroquinoline (2.0 g, 8.3 mmol) and Pd(PPh3)4 (0.095 g, 0.83 mmol) in 1,4-dioxane (7.5 mL) was added tributyl(vinyl)tin (2.4 mL, 8.3 mmol). The reaction mixture was heated for 20 min at 150 C in a Biotage microwave reactor. The resulting mixture was concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 70:30 gradient) to afford the title compound as a white solid (1.3 g, 84 %).
[0104] 1H NMR (500 MHz, DMSOd6): δ 5.49 (d, J = 11.0 Hz, IH), 6.10 (d, J = 17.7 Hz, IH), 7.05 (dd, J = 17.7, 11.0 Hz, IH), 7.76 (d, J= 4.8 Hz, IH), 8.06 (d, J= 6.5 Hz, IH), 8.12 (d, J = 6.5 Hz, IH), 8.13 (s, IH), 8.80 (d, J = 4.6 Hz, IH)
MS (ES+): m/z 190 (M+H)+
Example 2: Preparation of 4-Chloroquinoline-6-carbaldehyde (2)

[0105] To a solution of 1 (1.0 g, 5.3 mmol) in 1,4-dioxane (2.8 mL) was sequentially added H2O (1 mL), OsO4 (2.5% solution in tert-butanol, 1.3 mL, 0.11 mmol), 2,6-lutidine (1.3 mL, 10.6 mmol), and and NaIO4 (4.5 g, 21.1 mmol). The reaction mixture was stirred for 2 h at room temperature, quenched by adding H2O (20 mL), and extracted two times with CH2Cl2 (2 x 20 mL). The combined organic layers were dried over MgSO4, concentrated, and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 50:50 gradient) to afford the title compound as a white solid (0.55 g, 55 %).
[0106] 1H NMR (500 MHz, DMSO-d6): δ 7.93 (d, J= 1.0 Hz, IH), 8.24 (s, IH), 8.84 (dd, J = 1.3, 1.1 Hz, IH), 9.00 (d, J= 4.6 Hz, IH), 10.28 (s, IH) MS (ES+): m/z 192 (M+H)+
Example 3: Preparation of 4-(4-Methylthiophen-2-yl)quinoline-6-carbaldehyde (3)

[0107] To a solution of 2 (0.25 g, 1.27 mmol), 4-methylthiophene-2-boronic acid (0.18 g, 1.27 mmol), and Pd(PPh3)4 (0.15 g, 0.13 mmol) in DMF (4 mL) was added Na2CO3 (2.0 M, 1.1 mL). The reaction mixture was heated for 15 min at 150 0C in a Biotage microwave reactor. The resulting mixture was filtered through a pad of silica gel, concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 50:50 gradient) to afford the title compound as a yellow solid (0.29 g, 90 %). [0108] 1H NMR (500 MHz, DMSOd6): δ 2.35 (s, 3H), 7.50 (s, IH), 7.50 (s, IH), 7.67 (d, J = 4.8 Hz, IH), 8.19 (dd, J = 8.9, 1.3 Hz, IH), 8.22 (d, J = 8.8 Hz, IH), 8.91 (d, J= 1.6 Hz, IH), 9.03 (d, J = 5.0 Hz, IH), 10.20 (s, IH)
MS (ES+): m/z 254 (M+H)+
Example 4: Preparation of (Z)-5-((4-(4-Methylthiophen-2-yl)quinolin-6- yl)methylene)thiazolidine-2,4-dione:

[0109] A microwave vial was charged with 3 (0.13 g, 0.51 mmol), thiazolidine-2,4-dione (0.10 g, 0.77 mmol), and Cs2CO3 (0.33 g, 1.03 mmol) in ethanol (3 mL). The reaction mixture was heated for 30 min at 150 0C in a Biotage microwave reactor. The resulting mixture was separated by preparative HPLC. Fractions that contained the desired product were combined, neutralized with saturated Na2CO3, and extracted with EtOAc. The organic layer was washed
with brine, dried over MgSO4, and concentrated to afford the title compound as a yellow solid (0.006 g, 3%).
[0110] 1H NMR (500 MHz, DMSOd6): δ 2.36 (s, 3H), 7.49 (s, IH), 7.51 (s, IH), 7.62 (d, J = 4.5 Hz, IH), 7.95 (s, IH), 8.03 (dd, J = 8.8, 1.9 Hz, IH), 8.18 (d, J = 8.8 Hz, IH), 8.48 (d, J = 1.8 Hz, IH), 8.95 (d, J= 4.5 Hz, IH), 12.60 (br s, IH)
MS (ES+): m/z 353 (M+H)+
Example 5: Preparation of 4-(3-Methylthiophen-2-yl)quinoline-6-carbaldehyde (4)

[0111] To a solution of 2 (0.13 g, 0.63 mmol), 3-methylthiophene-2-boronic acid (0.089 g, 0.63 mmol), and Pd(PPh3)4 (0.072 g, 0.063 mmol) in DMF (2 mL) was added Na2CO3 (2.0 M, 0.55 mL). The reaction mixture was heated for 15 min at 150 0C in a Biotage microwave reactor. The resulting mixture was filtered through a pad of silica gel, concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 70:30 gradient) to afford the title compound as a yellow solid (0.098 g, 62 %).
[0112] 1H NMR (500 MHz, DMSO-d6): δ 7.21 (d, J= 5.1 Hz, IH), 7.64 (d, J= 4.4 Hz, IH), 7.80 (d, J = 5.1 Hz, IH), 8.20 (dd, J = 8.7, 1.7 Hz, IH), 8.26 (d, / = 8.8 Hz, IH), 8.39 (d, / = 1.6 Hz, IH), 9.10 (d, / = 4.5 Hz, IH), 10.15 (s, IH)
MS (ES+): m/z 254 (M+H)+ Example 6: Preparation of (Z)-5-((4-(3-Methylthiophen-2-yl)quinolin-6- yl)methylene)thiazolidine-2,4-dione:

[0114] 1H NMR (500 MHz, DMSO-d6): δ 2.07 (s, 3H), 7.21 (d, J = 5.1 Hz, IH), 7.58 (d, J = 4.3 Hz, IH), 7.80 (d, J = 5.1 Hz, IH), 7.92 (s, IH), 7.94 (d, J = 1.2 Hz, IH), 8.02 (dd, J = 8.8, 1.7 Hz, IH), 8.20 (d, J = 8.7 Hz, IH), 9.01 (d, J = 4.3 Hz, IH), 12.70 (br s, IH)
MS (ES+): m/z 353 (M+H)+
Example 7: Preparation of 4-(Benzo[b]thiophen-2-yl)quinoline-6-carbaldehyde (5)

[0115] To a solution of 2 (0.20 g, 1.04 mmol), benzo(£)thiophen-2-ylboronic acid (0.19 g, 1.04 mmol), and Pd(PPh3)4 (0.12 g, 0.10 mmol) in DMF (3 mL) was added Na2CO3 (2.0 M, 0.9 mL). The reaction mixture was heated for 15 min at 150 0C in a Biotage microwave reactor. The resulting mixture was filtered through a pad of silica gel, concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 65:35 gradient) to afford the title compound as a yellow solid (0.24 g, 79 %).
[0116] 1H NMR (500 MHz, DMSO-d6): δ 7.50-7.52 (m, 2H), 7.82 (d, J = 4.5 Hz, IH), 7.99 (s, IH), 7.99-8.04 (m, IH), 8.13-8.15 (m, IH), 8.21 (dd, J= 8.6, 1.6 Hz, IH), 8.27 (d, J = 8.9 Hz, IH), 8.97 (d, J = 1.7 Hz, IH), 9.13 (d, J = 4.5 Hz, IH), 10.21 (s, IH)
MS (ES+): m/z 290 (M+H)
Example 8: Preparation of (Z)-5-((4-(Benzo[b]thiophen-2-yl)quinolin-6- yl)methylene)thiazolidine-2,4-dione

[0117] A microwave vial was charged with 5 (0.12 g, 0.42 mmol), thiazolidine-2,4-dione (0.73 g, 0.62 mmol), and Cs2CO3 (0.27 g, 0.83 mmol) in ethanol (3 mL). The reaction mixture was heated for 30 min at 150 0C in a Biotage microwave reactor. Precipitate from the resulting mixture was filtered and washed with ethanol (10 mL) followed by H2O (10 mL) to afford the title compound as an orange solid (0.066 g, 41%).
[0118] IH NMR (500 MHz, DMSO-d6): δ 7.47-7.53 (m, 3H), 7.69 (d, J = 4.4 Hz, IH), 7.96 (s, IH), 7.99 (dd, J = 8.8, 1.7 Hz, IH), 8.01 (d, J = 8.4 Hz, IH), 8.12 (d, J= 6.3 Hz, IH), 8.15 (d, J = 8.6 Hz, IH), 8.53 (s, IH), 8.94 (d, J = 4.4 Hz, IH)
MS (ES+): m/z 389 (M+H)+
Example 9: Preparation of 4-(Thiophen-2-yl)quinoline-6-carbaldehyde (6)

[0119] To a solution of 2 (0.30 g, 1.57 mmol), thiophene-2-boronic acid (0.21 g, 1.64 mmol), and Pd(PPh3 )4 (0.09 g, 0.078 mmol) in DMF (3 mL) was added Na2CO3 (2.0 M, 1.4 mL). The reaction mixture was heated for 15 min at 150 0C in a Biotage microwave reactor. The resulting mixture was filtered through a pad of silica gel, concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 70:30 gradient) to afford the title compound as a yellow solid (0.31 g, 84 %).
[0120] MS (ES+): m/z 240 (M +H)+
Example 10: Preparation of (Z)-5-((4-(Thiophen-2-yl)quinolin-6-yl)methylene)thiazolidine- 2,4-dione:

[0121] To a solution of 6 (0.05 g, 0.21 mmol), thiazolidine-2,4-dione (0.27 g, 0.21 mmol), and in ethanol (1 rnL) was added 1-methylpiperazine (0.042 g, 0.42 mmol). The reaction mixture was heated for 30 min at 150 0C in a Biotage microwave reactor. Precipitate from the resulting mixture was filtered and washed with ethanol (10 mL) to afford the title compound as a yellow solid (0.023 g, 33%).
[0122] 1H NMR (500 MHz, DMSOd6): δ 7.39 (dd, J = 5.0, 3.5 Hz, IH), 7.64-7.70 (m, 2H), 7.94 (d, J = 5.0 Hz, IH), 7.99 (s, IH), 8.03 (dd, J = 8.9, 1.9 Hz, IH), 8.20 (d, J= 8.8 Hz, IH), 8.46 (d, J = 1.9 Hz, IH), 8.97 (d, J = 4.5 Hz, IH), 12.70 (br s, IH)
MS (ES+): m/z 339 (M+H)+
Example 11: Preparation of 4-(5-(Hydroxymethyl)thiophen-2-yl)quinoline-6-carbaldehyde (7)
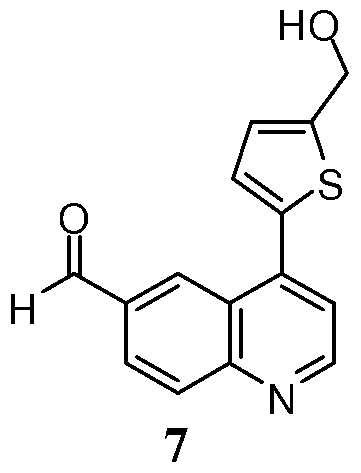
[0123] To a solution of 2 (1.0 g, 5.2 mmol), 5-hydroxymethylthiophene-2-boronic acid (0.82 g, 5.2 mmol), and Pd(PPh3)4 (0.60 g, 0.078 mmol) in DMF (16 mL) was added Na2CO3 (2.0 M, 4.6 mL). The reaction mixture was heated for 15 min at 150 0C in a Biotage microwave reactor. The resulting mixture was filtered through a pad of silica gel, concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 0:100 gradient) to afford the title compound as a yellow solid (0.93 g, 66 %).
[0124] MS (ES+): m/z 270 (M +H)+
Example 12: Preparation of (Z)-5-((4-(5-(Hydroxymethyl)thiophen-2-yl)quinolin-6- yl)methylene)thiazolidine-2,4-dione

[0125] A microwave vial was charged with 7 (0.2 g, 0.74 mmol), thiazolidine-2,4-dione (0.15 g, 1.11 mmol), Cs2CO3 (0.48 g, 1.49 mmol) in ethanol (4.5 mL). The reaction mixture was heated for 30 min at 150 0C in a Biotage microwave reactor. Upon cooling to room temperature, the reaction mixture added MeOH (50 mL). Precipitate from the resulting mixture was filtered to afford the title compound as a brown solid (0.079 g, 29%).
[0126] 1H NMR (500 MHz, DMSO-d6): δ 4.76 (d, J = 5.5 Hz, 2H), 5.70 (t, J = 5.7 Hz, IH), 7.21 (d, J = 3.8 Hz, IH), 7.51 (d, J = 3.6 Hz, IH), 7.61 (d, J = 4.5 Hz, IH), 7.97 (s, IH), 8.02 (dd, J = 8.8, 2.0 Hz, IH), 8.19 (d, J= 8.7 Hz, IH), 8.49 (d, J= 1.9 Hz, IH), 8.96 (d, J= 4.6 Hz, IH), 12.70 (br s, IH)
[0127] MS (ES+): m/z 369 (M+H)+
Example 13: Preparation of 4-(5-Phenyl-thiophen-2-yl)-quinoline-6-carbaldehyde (8)

[0128] To a solution of 2 (0.20 g, 1.0 mmol), 5-phenylthiophene-2-boronic acid (0.30 g, 1.5 mmol), and Pd(PPh3 )4 (0.12 g, 0.10 mmol) in DMF (6 mL) was added Na2CO3 (2.0 M, 1.5 mL) and the reaction mixture heated for 2 h at 130 0C. The resulting mixture was filtered, washed with DCM and the filtrate concentrated. The crude product was purified by silica gel
chromatography (hexanes to 40% EtOAc/hexanes) to afford the title compound as a yellow solid (0.1 I g, 33%).
[0129] MS (ES+) : m/z 316 (M +H)+
Example 14: Preparation of 5-[4-(5-Phenyl-thiophen-2-yl)-quinolin-6-ylmethylene]- thiazolidine-2,4-dione

A suspension of 8 (70 mg, 0.22 mmol), thiazolidine-2,4-dione (40 mg, 0.34 mmol), Cs2CO3 (0.10 g, 0.31 mmol) in ethanol (8 mL) was heated at 120 0C for 4 h. Upon cooling to room temperature, the resulting solid was filtered, washed firstly with water (15 mL) then with EtOH (15 mL). The title compound was obtained as a yellow solid (81 mg, 88%).
[0130] 1H NMR (500 MHz, DMSO-d6): δ 7.38 (t, J = 7.0 Hz, IH), 7.45-7.51 (m, 3H), 7.65 (d, J = 4.6 Hz, IH), 7.68 (d, J = 3.8 Hz, IH), 7.79 (d, J = 3.7 Hz, IH), 7.80 (s, IH), 7.82 (s, IH), 7.97 (dd, J = 8.8, 1.9 Hz, IH), 8.11 (d, J= 8.8 Hz, IH), 8.54 (d, J= 1.8 Hz, IH), 8.90 (d, J = 4.5 Hz, IH) [0131] MS (ES+): m/z 415 (M+H)+
Example 15: Preparation of 4-(5-Chlorothiophen-2-yl)quinoline-6-carbaldehyde (9)

[0132] A solution of 2 (0.2 g, 1.04 mmol), 5-chlorothienyl-2-boronic acid (0.17 g, 1.04 mmol), tetrakis(triphenylphosphine)palladium (0) (0.060 g, 0.052 mmol), and potassium carbonate (0.22 g, 1.56 mmol) in dimethyl formamide (10 mL) under argon was heated at 150
0C for 60 min in a microwave oven. The reaction mixture was filtered, and concentrated under reduced pressure. The concentrated reaction mixture was purified on a flash chromatography (SiO2, 10% ethyl acetate in hexanes to 100% ethyl acetate over 15 minutes) to afford the title compound (0.10 g, 35%) as a pale yellow solid. [0133] 1H NMR (500 MHz, DMSOd6): δ 10.22 (s IH), 9.08 (d, J = 4.5 Hz, IH), 8.88 (d, J = 1.3 Hz, IH), 8.20-8.26 (m, 2H), 7.71 (d, J = 4.5 Hz, IH), 7.58 (d, J = 4.0 Hz, IH), 7.43 (d, J = 4.0 Hz, IH)
MS (ES+): m/z 21 A (M+H)+
Example 16: Preparation of (5Z)-5-{ [4-(5-Chlorothiophen-2-yl)quinolin-6-yl]methylidene}- l,3-thiazolidine-2,4-dione

[0134] A solution of 9 (0.07 g, 0.24 mmol), 2,4-thiazolidinedione (0.11 g, 0.96 mmol), and cesium carbonate (0.31 g, 0.96 mmol) in ethanol (4.8 mL) was heated at 150 0C for 30 min in a microwave oven. The solid was filtered, washed with water, and ethanol to afford the title compound (0.05 g, 57%) as pale yellow solid.
[0135] 1H NMR (500 MHz, DMSOd6): δ 8.89 (d, J = AA Hz, IH), 8.39 (s, IH), 8.10 (d, J = 8.7 Hz, IH), 7.96 (d, J = 8.7 Hz, IH)
[0136] MS (ES+): m/z 373 (M+H)+
Example 17: Preparation of 5-{4-[5-(3,5-Dimethyl-isoxazol-4-yl)-thiophen-2-yl]-quinolin-6- ylmethylene } -thiazolidine-2,4-dione

[0138] 1H NMR (500 MHz, DMSO-d6): δ 2.42 (s, 3H), 2.61 (s, 3H), 7.49 (d, J = 3.8 Hz, IH), 7.70-7.73 (m, 2H), 7.98 (s, IH), 8.04 (dd, J = 8.7, 1.0 Hz, IH), 8.21 (d, J = 8.8 Hz, IH), 8.52 (d, J = 1.0 Hz, IH), 8.99 (d, J = 4.6 Hz, IH), 12.80 (br s, IH) MS (ES+): m/z 434 (M+H)+
Example 18 Preparation of 4-(2,3-Dihydro-thieno[3,4-b][l,4]dioxin-5-yl)-quinoline-6- carbaldehyde (10)

Example 19: 5-[4-(2,3-Dihydro-thieno[3,4-b][l,4]dioxin-5-yl)-quinolin-6-ylmethylene]- thiazolidine-2,4-dione:

[0142] 1H NMR (500 MHz, DMSO-d6): δ 4.28 (d, J = 3.5 Hz, 2H), 4.33 (d, J = 3.4 Hz, 2H), 6.98 (s, IH), 7.46 (s, IH), 7.50 (d, J = 4.4 Hz, IH), 7.92 (dd, J = 8.8, 1.4 Hz, IH), 8.06 (d, J = 8.7 Hz, IH), 8.24 (s, IH), 8.87 (d, J = 4.5 Hz, IH)
[0143] MS (ES+): m/z 397 (M+H)+
Example 20: Preparation of 4-(l-Benzothiophen-3-yl)quinoline-6-carbaldehyde (14)

[0144] A solution of 2 (0.4 g, 2.08 mmol), benzothienyl-3-boronic acid (0.38 g, 2.08 mmol), tetrakis(triphenylphosphine)palladium (0) (0.12 g, 0.1 mmol), potassium carbonate (0.58 g, 4.2 mmol) in dimethyl formamide (20 mL) under argon was heated at 150 0C for 60 min in a microwave oven. The reaction mixture was filtrered, and concentrated under reduced pressure. The reaction mixture was purified on a flash chromatography (SiO2, 10% ethyl acetate in hexanes to 100% ethyl acetate over 15 minutes) to afford the title compound (0.37 g, 61%) as a pale yellow solid.
[0145] 1H NMR (500 MHz, DMSO-d6): δ 10.05 (s, IH), 9.16 (d, J = 4.4 Hz, IH), 8.40 (d, / = 1.7 Hz, IH), 8.29 (d, J = 8.7 Hz, IH), 8.18-8.21 (m, 2H), 8.17 (s, IH), 7.74 (d, J = 4.4 Hz, IH), 7.48-7.51 (m, 2H), 7.38-7.41 (m, IH)
MS (ES+): m/z 290 (M+H)+
Example 21: Preparation of (5Z)-5-{ [4-(l-Benzothiophen-3-yl)quinolin-6-yl]methylidene}- 1 ,3-thiazolidine-2,4-dione:

[0146] A solution of 14 (0.29 g, 1 mmol), 2,4-thiazolidinedione (0.23 g, 2 mmol), cesium carbonate (0.65 g, 2 mmol) in ethanol (10 mL) was heated at 150 0C for 30 min in a microwave oven. The solid was filtered, washed with water, and ethanol to afford the title compound (0.28 g, 72%) as pale yellow solid.
[0147] 1H NMR (500 MHz, DMSOd6): δ 8.99 (d, J= AA Hz, IH), 8.18 (d, J= 8.0 Hz, IH), 8.13 (dd, J = 8.2, 1.2 Hz, IH), 8.10 (s, IH), 7.95 (d, J = 2.0 Hz, IH), 7.93 (s, IH), 7.60 (d, J = AA Hz, IH), 7.36-7.49 (m, 3H), 7.32 (s, IH) MS (ES+): m/z 389 (M+H)+
Example 22: Preparation of 4-Furan-2-ylquinoline-6-carbaldehyde (15)

15
[0148] To a solution of 2 (115 mg, 0.6 mmol) in dimethoxyethane (DME, 10 mL) was added a solution of furan-2-yl-2-boronic acid (74 mg, 0.66 mmol) in EtOH (50 mL), 1.0 M
Na2CO3 (2 mL), and tetrakis(triphenylphosphine)palladium (0) (Pd(PPh3 )4, 69 mg, 0.06 mmol). The reaction mixture was heated at 110 0C for 20 min under μ-wave. The hot solution was
filtered and the solid was washed with EtOAc. The filtrate was washed with brine (100 rnL). The aqueous was extracted with EtOAc (3 x 20 rnL). Combined organic layer was dried (Na2SO4). The solvent was removed in vacuo and the crude material was used for next reaction without further purification.
Example 23: Preparation of (5Z)-5-[(4-Furan-2-ylquinolin-6-yl)methylidene]-l,3-thiazolidine- 2,4-dione:

[0149] To a solution of 15 (0.5 mmol) in EtOH (10 mL) was added thiazolidine-2,4-dione (TZD, 70 mg, 0.6 mmol) and Cs2CO3 (782 mg, 2.4 mmol). The reaction mixture was heated at 120 0C for 20 min under μ-wave. The hot solution was filtered and the solid was washed with EtOAc. The filtrate was concentrated and residue was purified by HPLC. The HPLC fractions containing product were combined and neutralized with saturated NaHCO3 (50 mL). The free base was extracted with EtOAc (2 x 50 mL). The combined organic layer was dried (Na2SO4). The solvent was removed in vacuo. The title compound (10 mg, 5%) was afforded as a yellow solid.
[0150] 1H NMR (500 MHz, DMSO-d6): δ 6.86-6.87 (m, IH), 7.46 (d, J = 3.4 Hz, IH), 7.84 (d, J = 4.7 Hz, IH), 8.00 (dd, J = 8.8, 2.0 Hz, IH), 8.03 (s, IH), 8.11 (d, J = 1.2 Hz, IH), 8.16 (d, J = 8.8 Hz, IH), 8.76 (d, J = 1.8 Hz, IH), 8.97 (d, J = 4.6 Hz, IH), 12.62 (br s, IH) MS
(ES+): m/z 323 (M+H)+ Example 24: Preparation of 4-Benzofuran-2-ylquinoline-6-carbaldehyde (16)

Example 25: Preparation of: (5Z)-5-{ [4-(l-benzofuran-2-yl)quinolin-6-yl]methylidene}-l,3- thiazolidine-2,4-dione

[0152] To a solution of 16 (369 mg, 1.35 mmol) in EtOH (15 mL) was added thiazolidine- 2,4-dione (TZD, 316 mg, 2.7 mmol) and Cs2CO3 (1.3 g, 4.0 mmol). The reaction mixture was heated at 120 0C for 30 min under μ-wave. The solvent was removed in vacuo and the residue was suspended in H2O-EtOAc. The solid was collected and washed with water and then
MeOH. The solid was dried in air and the title compound (327 mg, 65%) was afforded as a yellow solid.
[0153] 1H NMR (500 MHz, DMSO-d6): δ 7.39 (t, J = 7.4 Hz, IH), 7.49 (t, J = 7.1 Hz, IH), 7.55 (s, IH), 7.78 (d, J = 8.3 Hz, IH), 7.83 (s, IH), 7.84 (d, J = 8.3 Hz, IH), 7.97 (d, J = 4.5 Hz, IH), 8.00 (dd, J = 8.8, 2.0 Hz, IH), 8.13 (d, J = 8.8 Hz, IH), 8.84 (d, J = 1.7 Hz, IH), 8.98 (d, J = 4.5 Hz, IH), 12.62 (br s, IH)
MS (ES+): m/z 373 (M+H)+
Example 26: Preparation of: 5-(6-Formylquinolin-4-yl)thiophene-2-carboxylic acid (17)

[0154] To a solution of 2 (192 mg, 1.0 mmol) in dimethoxyethane (DME, 10 rnL) was added a solution of 2-carboxythiophene-5-boronic acid (172 mg, 1.0 mmol) in EtOH (5 mL), 1.0 M Na2CO3 (4 mL), and tetrakis(triphenylphosphine)palladium (0) (Pd(PPh3 )4, 116 mg, 0.1 mmol). The reaction mixture was heated at 110 0C for 20 min under μ-wave. The hot solution was filtered and the solid was washed with water. The filtrate was mixed with H2O (50 mL) and EtOAc (50 mL). The organic layer was separated and the aqueous layer was neutralized with 1 M HCl. The solid was collected by filtration and washed with water. The title compound (70 mg, 25%) was obtained as a white solid.
Example 27: Preparation of: 5-{6-[(Z)-(2,4-Dioxo-l,3-thiazolidin-5-ylidine)methyl]quinolin- 4-yl}thiophene-2-carboxylic acid
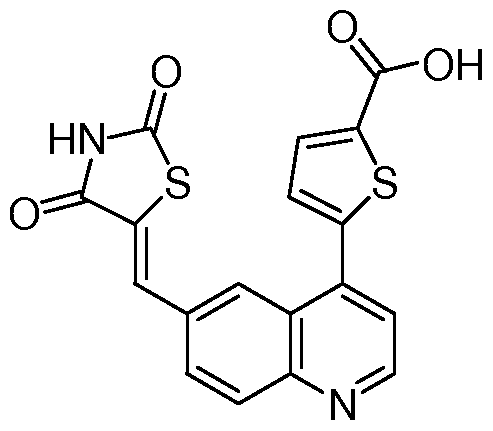
[0155] To a solution of 17 (30 mg, 0.1 mmol) in EtOH (15 mL) was added thiazolidine-2,4- dione (TZD, 24 mg, 0.2 mmol) and Cs2CO3 (130 mg, 0.4 mmol). The reaction mixture was heated at 120 0C for 30 min under μ-wave. The reaction mixture was concentrated and the residue was purified by HPLC. The HPLC fractions containing the product were combined and concentrated. The title compound (4 mg, 8%) was obtained as the TFA salt as a yellow solid.
[0156] 1H NMR (500 MHz, DMSO-d6): δ 7.67 (d, J = 3.8 Hz, IH), 7.72 (d, J = 4.5 Hz, IH), 7.92 (d, J = 3.8 Hz, IH), 8.01 (s, IH), 8.05 (dd, J = 8.8, 1.8 Hz, IH), 8.23 (d, J= 8.8 Hz, IH),
8.40 (d, J = 1.8 Hz, IH), 9.02 (d, J = 4.5 Hz, IH), 12.62 (br s, IH), 13.40 (br s, IH); MS (ES+): m/z 383 (M+H)+
Example 28: reparation of: 5-(6-Formylquinolin-4-yl)-N-(2-pyrrolidin-l-ylethyl)thiophene-2- carboxamide (18)

[0157] The compound 17 (120 mg, 0.42 mmol) was dissolved in SOCl2 (15 mL) and heated under reflux for 2 h. The excess SOCl2 was removed in vacuo. The residue was suspended in PhMe (20 mL) and added l-(2-aminoethyl)-pyrrolidine (57 mg, 0.5 mmol) and Et3N (0.5 mL). The mixture was heated under reflux overnight. After cooling, the saturated NaHCO3 (50 mL) was added and organic layer was separated. The aqueous was extracted with EtOAc (2 x 20 mL). Combined organic layer was dried (Na2SO4). Solvent was removed in vacuo. The crude material was used for next step without further purification.
Example 29: Preparation of: 5-{6-[(Z)-(2,4-Dioxo-l,3-thiazolidin-5-ylidine)methyl]quinolin-4- yl } -,/V-(2-pyrrolidin- 1 -ylethyl)thiophene-2-carboxamide:

[0158] To a solution of 18 (0.42 mmol) in EtOH (15 mL) was added thiazolidine-2,4-dione (TZD, 98 mg, 0.84 mmol) and Cs2CO3 (0.52 g, 1.6 mmol). The reaction mixture was heated at 120 0C for 30 min under μ-wave. The reaction mixture was concentrated and residue was purified by HPLC. The HPLC fractions containing the product were combined and concentrated. The title compound (7 mg, 3%) TFA salt was obtained as a yellow solid.
[0159] 1H NMR (500 MHz, DMSO-d6): δ 1.88-1.89 (m, 2H), 2.03 (br, 2H), 3.08-3.11 (m, 2H), 3.34-3.38 (m 2H), 3.62-3.66 (m, 4H), 7.70-7.71 (m, 2H), 7.98 (d, J = 3.9 Hz, IH), 8.01 (s, IH), 8.06 (dd, J = 8.8, 1.9 Hz, IH), 8.23 (d, J = 8.8 Hz, IH), 8.40 (d, J = 1.9 Hz, IH), 9.00 (s, IH), 9.02 (d, J = 4.5 Hz, IH), 9.65 (br s, IH), 12.80 (br s, IH)
MS (ES+): m/z 479 (M+H)+
Example 30: Preparation of 5-(6-Formyl-quinolin-4-yl)-thiophene-2-carboxylic acid amide (19)

19 [0160] Compound 17 (420 mg, 1.5 mmol) was dissolved in thionyl chloride (10 mL) and heated under reflux for 4 h. The excess thionyl chloride was removed in vacuo. The residue was suspended in NH3-H2O (20 mL) and heated under reflux overnight. The solid was collected by filtration and washed with water. The title compound (100 mg, 24%) was obtained as a yellow solid. Example 31: Preparation of: 5-[6-(2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4-yl]- thiophene-2-carboxylic acid amide

[0161] To a solution of 19 (100 mg, 0.35 mmol) in EtOH (15 mL) was added thiazolidine- 2,4-dione (TZD, 62 mg, 0.53 mmol) and Cs2CO3 (0.46 g, 1.4 mmol). The reaction mixture was heated at 120 0C for 30 min under μ-wave. The solvent was removed in vacuo and the residue was suspended in H2O-EtOAc. The solid was collected and washed with water and then
MeOH. The solid was dried in air and the title compound (80 mg, 60%) was afforded as a brown solid.
[0162] 1H NMR (500 MHz, DMSOd6): δ 7.55 (br s, IH), 7.61-7.63 (m, 2H), 7.96-7.98 (m, 2H), 8.10 (d, J = 8.7 Hz, IH), 8.20 (br s, IH), 8.39 (d, J = 1.8 Hz, IH), 8.90 (d, J = 4.5 Hz, IH)
[0163] MS (ES+): m/z 382 (M+H)+
Example 32: Preparation of: l-Bromo-3-(2,2-dimethoxy-ethylsulfanyl)-benzene (20)

Example 33: Preparation of: 6-Bromo-benzo[b]thiophene (21) and 4-Bromo-benzo[b]thiophene
(22)

22
[0165] Chlorobenzene (1000 mL) and PPA (280 g) were combined and heated to reflux (130 0C). The solution of compound 20 (130 g, 0.48 mol) in chlorobenzene (300 mL) was added drop wise to the mixture over 1.5 h. The reaction mixture was refluxed for 3-4 h and then cooled. The solvent was decanted from the residue and toluene (400 mLx2 ) was added to
the residue, stirred and decanted. The chlorobenzene/toluene extracts were combined and concentrated under vacuum, and the residue was then taken up with PE (800 mL) and water (400 mL). After layers separation the organic phase was washed with saturated NaHCO3, brine, dried over Na2SO4 and concentrated to give a crude product, which was purified by column chromatography (silica gel, elute: PE) to afford a mixture of 21 and 22 (80 g, 70 %) as a colorless oil.
Example 34: Preparation of: Benzo[b]thiophene-6-carbonitrile (23) and Benzo[b]thiophene-4- carbonitrile (24)

[0166] Under Ar atmosphere, to the mixture of compound 21 and compound 22 (40 g, 0.19 mol) and Zn(CN)2 (16 g, 0.132 mol) were added Pd(PPh3)4 (10 g) and DMF (800 mL) and the mixture was stirred at 90 0C -100 0C for 3 h. The reaction solution was cooled to room temperature (30 0C) and filtered. The filtrate was concentrated under reduced pressure and the residue was then taken up with EtOAc (1000 mL) and saturated aq. NaHCO3 (500 mL), the insoluble solid was discarded via filtration. The filtrate layers were separated. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated to give a mixture product, which was purified by careful column chromatography (silica, elute; PE to EtO Ac:PE= 1:40) to afford compound 23 (14 g, 46.2 %) as an oil and compound 24 (7 g, 23%) as a solid.
Example 35: Preparation of 6-Cyano-benzothiophene-2-boronic acid (25)

25
[0167] Compound 23 (14 g, 0.088 mol) in dry THF (350 mL) was purged with nitrogen. The mixture was then cooled (-90 0C) with liquid nitrogen bath and ra-BuLi (2.5 M in hexanes,
42 niL, 0.105 mol) was added dropwise with stirred. The mixture was stirred for a further 30 min, and B(OMe)3 (18 g, 0.173 mol) was added at -90 0C. After 30 min, aqueous HCl (2M, 200 mL) was added. The mixture was stirred for a further 30 min, allowed to warm to room temperature (30 0C), poured into water (100 mL). The mixture was extracted with EtOAc (400 mLx3). The combined organic layers were washed with water, brine, dried over Na2SO4 and concentrated to give a crude product, which was crystallized with EtOAc/PE (1:2, 100 mL) to give the title compound (12 g, 72%) as a white solid.
Example 36: Preparation of: 4-Cyano-benzothiophene-2-boronic acid (26)

[0168] Compound 24 (12 g, 0.076 mol) in dry THF (300 mL) was purged with nitrogen. The mixture was then cooled (-90 0C) with liquid nitrogen bath and ra-BuLi (2.5 M in hexanes, 35 mL, 0.083 mol) was added dropwise with stirred. The mixture was stirred for a further 30 min, and B(OMe)3 (16 g, 0.154 mol) was added at -90 0C. After 30 min, aqueous HCl (2M, 200 mL) was added and solid was precipitated out. The mixture was stirred for a further 30 min, allowed to warm to room temperature (30 0C), poured into water (100 mL). The mixture was extracted with EtOAc (300 mLx3). The combined organic layers were washed with water, brine, dried over Na2SO4 and concentrated to give a crude product, which was crystallized with EtOAc/PE (1:2, 100 mL) to give the title compound (H g, 73%) as a white solid. Example 37: Preparation of Benzo[b]thiophene-7-carbonitrile (27)

27
[0169] Under Ar atmosphere, to a mixture of 7-bromo-benzo[b]thiophene (40 g, 0.19 mol) and Zn(CN)2 (16 g, 0.132 mol) were added Pd(PPh3)4 (10 g) and DMF (800 mL) and the mixture was stirred at 90 0C -100 0C for 3 h. The reaction solution was cooled to room temperature (30 0C) and filtered. The filtrate was concentrated under reduced pressure. The residue was taken up with EtOAc (1000 mL) and saturated aq. NaHCO3 (500 mL) and then
filtered. The filtrate layers were separated. The organic layer was washed with brine, dried over Na2SO4 and concentrated to give a crude product, which was purified by column chromatography (silica, elute; PE to EtOAc:PE=l:30) to afford the title compound (24 g, 80 %) as a white solid.
Example 38: Preparation of 7-Cyano-benzothiophene-2-boronic acid (28)

28
[0170] Compound 27 (20 g, 0.126 mol) in dry THF (500 mL) was purged with nitrogen. The mixture was then cooled (-90 0C) with liquid nitrogen bath and ra-BuLi (2.5 M in hexanes, 60 mL, 0.15 mol) was added dropwise with stirred. The mixture was stirred for a further 30 min, and B(OMe)3 (26 g, 0.25 mol) was added at -90 0C. After 30 min, aqueous HCl (2M, 300 mL) was added and solid was precipitated out. The mixture was stirred for a further 30 min, allowed to warm to room temperature (30 0C), poured into water (200 mL). The solid was filtered, washed with ice- water and dried to give the title compound (18 g, 72%) as a white solid.
Example 39: Preparation of 2-(6-Formyl-quinolin-4-yl)-benzo[b]thiophene-6-carbonitrile (29)

29 [0171] To a solution of 2 (0.30 g, 1.6 mmol), 25 (0.40 g, 2.0 mmol), and Pd(PPh3)4 (0.15 g, 0.13 mmol) in DMF (5 mL) was added Na2CO3 (2.0 M, 2.0 mL). The reaction mixture was heated for 20 min at 140 0C in a Biotage microwave reactor. The resulting mixture was filtered, washed with DCM and the filtrate concentrated. The crude product was purified by
silica gel chromatography (hexanes to 60% EtOAc/hexanes) to afford the title compound as a white solid (0.17 g, 35%).
[0172] MS (ES+) : m/z 315 (M+H)+
Example 40: Preparation of 2-[6-(2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4-yl]- benzo[b]thiophene-6-carbonitrile

[0173] A suspension of 29 (0.17 g, 0.54 mmol), thiazolidine-2,4-dione (0.12 g, 1.0 mmol), Cs2CO3 (0.30 g, 0.92 mmol) in ethanol (10 mL) was heated at 100 0C for 2 h. Upon cooling to room temperature, the resulting solid was filtered and washed with water (15 mL). The title compound was obtained as a brownish orange solid (80 mg, 36%).
[0174] 1H NMR (500 MHz, DMSO-J6): δ 7.48 (s, IH), 7.72 (d, J = 4.4 Hz, IH), 7.88 (dd, J = 8.2, 1.3 Hz, IH), 8.02 (dd, J = 8.8, 1.8 Hz, IH), 8.09 (s, IH), 8.16 (d, J = 8.8 Hz, IH), 8.19 (d, J = 8.3 Hz, IH), 8.46 (d, J = 1.6 Hz, IH), 8.78 (d, J = 0.7 Hz, IH), 8.98 (d, J = 4.4 Hz, IH)
[0175] MS (ES+): m/z 414 (M+H)+ Example 41: Preparation of 2-(6-Formyl-quinolin-4-yl)-benzo[b]thiophene-4-carbonitrile (30)

30
[0176] To a solution of 2 (0.30 g, 1.6 mmol), 26 (0.40 g, 2.0 mmol), and Pd(PPh3)4 (0.15 g, 0.13 mmol) in DMF (4 mL) was added Na2CO3 (2.0 M, 2.0 mL). The reaction mixture was heated for 20 min at 150 0C in a Biotage microwave reactor. The resulting mixture was filtered, washed with DCM and the filtrate concentrated. The crude product was purified by
silica gel chromatography (hexanes to 50% EtOAc/hexanes) to afford the title compound as a white solid (0.14 g, 28%).
[0177] MS (ES+) : m/z 315 (M+H)+
Example 42: Preparation of 2-[6-(2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4-yl]- benzo [b] thiophene-4-carbonitrile

Cs2CO3 (0.20 g, 0.60 mmol) in ethanol (10 mL) was heated at 100 0C for 2 h. Upon cooling to room temperature, the resulting solid was filtered and washed with water (15 mL). The title compound was obtained as a reddish brown solid (75 mg, 41%).
[0179] 1H NMR (500 MHz, DMSO-J6): δ 7.48 (s, IH), 7.67 (t, J = 1.9 Hz, IH), 7.77 (d, J = 4.4 Hz, IH), 7.99 (s, IH), 8.02 (dd, J = 8.8, 1.8 Hz, IH), 8.07 (d, J= 8.0 Hz, IH), 8.16 (d, J = 8.8 Hz, IH), 8.47 (d, J = 1.8 Hz, IH), 8.55 (d, J = 8.2 Hz, IH), 8.98 (d, J = 4.4 Hz, IH)
[0180] MS (ES+): m/z 414 (M+H)+ Example 43: Preparation of 2-(6-Formyl-quinolin-4-yl)-benzo[b]thiophene-7-carbonitrile (31)

31
[0181] To a solution of 2 (0.30 g, 1.6 mmol), 28 (0.45 g, 2.2 mmol), and Pd(PPh3)4 (0.15 g, 0.13 mmol) in DMF (5 mL) was added Na2CO3 (2.0 M, 2.0 mL). The reaction mixture was heated for 30 min at 110 0C in a Biotage microwave reactor. The resulting mixture was filtered, washed with DCM and the filtrate concentrated. The residue was triturated in MeOH and the resulting solid filtered to afford the title compound as a white solid (0.10 g, 20%).
[0182] MS (ES+) : m/z 315 (M+H)+
Example 44: Preparation of 2-[6-(2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4-yl]- benzo[b]thiophene-7-carbonitrile
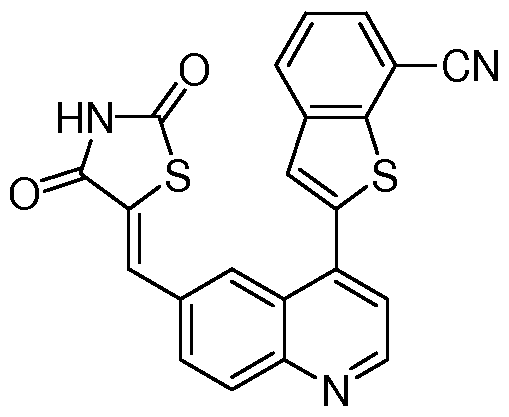
[0185] MS (ES+): m/z 414 (M+H)+
Example 45: Preparation of (3-Bromophenyl)(2,2-dimethoxypropyl)sulfane (40)

40
[0186] A mixture of 3-bromobenzenethiol (630 μL, 5.33 mmol), l-bromo-2,2- dimethoxypropane (715 μL, 5.29 mmol), and K2CO3 (1.10 g, 7.96 mmol) in DMF (11 mL) was heated at 150 0C for 2.5 hours. The crude reaction mixture was cooled, filtered, and concentrated. The residue was purified using flash chromatography (0-30% ethyl acetate in hexanes) to afford the title compound as a clear oil (1.66 g, 108% with some trapped solvent). The material was used as is for the next reaction.
[0187] 1H NMR (500 MHz, DMSO-J6) δ 1.33 (s, 3H), 3.12 (s, 6H), 3.25 (s, 2H), 7.25 (dd, J = 8.4, 7.5 Hz, IH), 7.36 (dd, J = 7.9, 1.9 Hz, 2H), 7.54 (t, J= 1.8 Hz, IH)
Example 46: Preparation of 6-Bromo-3-methylbenzo[b]thiophene (41) and 4-bromo-3- methylbenzo[b]thiophene (42)

41 42
[0188] Polyphosphoric acid (3.92 g) was added to chlorobenzene (12 rnL). The biphasic mixture was heated to 130 0C and a solution of 40 (1.66 g, 5.33 mmol) in chlorobenzene (2.5 mL) was added dropwise over 5 minutes. The reaction was heated at reflux for two hours, then cooled to room temperature. The organic solvent was decanted and the residue washed with toluene (2x5 mL) and in turn decanted. The combined organic layers were concentrated in vacuo. The residue was taken up in petroleum ether (10 mL) and washed with water (5 mL), sat. aq. NaHCO3 (5 mL), dried (Na2SO4), and concentrated in vacuo. The crude material was purified using flash chromatography (petroleum ether) to afford a mixture of the title compound as a clear oil (1.006 g, 77%). The material was used as is for the next reaction.
[0189] 41 and 42: 1H NMR (500 MHz, DMSO-J6) δ 2.39 (d, J = 0.8 Hz, 4.2H), 2.66 (d, J = 0.7 Hz, 3.7H), 7.23 (t, J = 7.8 Hz, 1.1H), 7.42 (d, J= 0.9 Hz, 1.4H), 7.53-7.56 (m, 2.2H) 7.61 (d, J = 7.8 Hz, 1.0H), 7.70 (d, J= 8.5 Hz, 1.3H), 8.00 (d, J = 7.9 Hz, 1. OH), 8.25 (d, J= 1.8 Hz, 1.0H)
Example 47: Preparation of 3-Methylbenzo[b]thiophene-6-carbonitrile (43) and 3- methylbenzo [b] thiophene-4-carbonitrile (44)
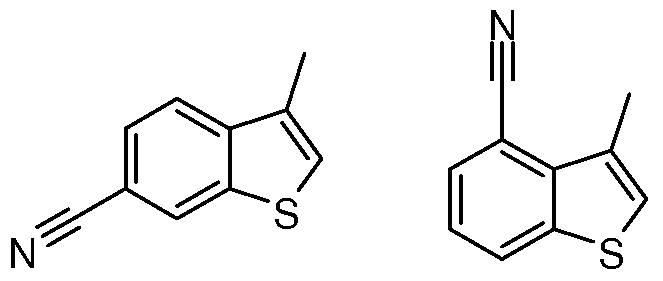
43 44
[0190] To a solution of 41 and 42 ( 1.0Og, 4.40 mmol) in DMF (10 mL) was added zinc cyanide (364 mg, 3.10 mmol) and Pd(PPh3 )4 (253 mg, 0.2 mmol) with a further 12 mL of DMF. The reaction was heated at 95 0C for 5 hours then cooled to room temperature. The
DMF was removed in vacuo and the residue was partitioned between ethyl acetate (25 rnL) and sat. aq. NaHCO3 (12 rnL). The insoluable solids were removed by filtration and the organic layer was separated, washed with brine (5 mL), dried (Na2SO4), and concentrated in vacuo. The crude material was purified using flash chromatography (1:40 ethyl acetate/petroleum ether) to afford each title compound separately: 43 as a clear oil that crystallized on standing (361 mg, 47%) and 44 as a white solid (264 mg, 35%).
[0191] 43: 1H NMR (500 MHz, DMSO-J6) δ 2.44 (d, J = 1.1 Hz, 3H), 7.76 (d, J = 1.2 Hz, IH), 7.78 (dd, J = 8.3, 1.5 Hz, IH), 7.95 (d, J= 8.2 Hz, IH), 8.61 (d, J= 1.2 Hz, IH)
[0192] 44: 1H NMR (500 MHz, DMSO-J6) δ 2.66 (d, J = 1.3 Hz, 3H), 7.51 (t, J = 7.8 Hz, IH), 7.75 (d, J = 1.0 Hz, IH), 7.92 (dd, J = 7.4, 0.9 Hz, IH), 8.36 (dd, J= 8.0, 0.9 Hz, IH)
Example 48: Preparation of 6-Cyano-3-methylbenzo[b]thiophen-2-ylboronic acid (45)

45
[0193] To an argon flushed, -78 0C solution of 43 (358 mg, 2.07 mmol) in anhydrous THF (8.3 mL) was added dropwise 2.5 M nBuLi in hexanes (1.0 mL, 2.5 mmol). The resulting solution was stirred for 30 min. Trimethylborate (0.46 mL, 4.13 mmol) was added dropwise and the cold bath removed. The reaction was allowed to warm naturally to 0 0C over 15 min at which point it was held there with an ice bath for another 15 min. 2M HCl (4.6 mL) was added and the ice bath was removed, and allowed to warm to room temperature over 30 minutes. The reaction mixture was poured into 2 mL water and 6 mL ethyl acetate. The phases were separated and the aqueous phase was extracted with ethyl acetate (3x18 mL). The combined organic layers were washed with water (2 mL), brine (4 mL), dried (Na2SO4), and concentrated in vacuo. The crude residue was triturated in ethyl acetate (3 mL) and then fully precipitated with petroleum ether (6 mL) to afford the title compound as an off-white solid (206 mg, 46%). The material was used as is for the next reaction.
Example 49: Preparation of 2-(6-Formylquinolin-4-yl)-3-methylbenzo[b]thiophene-6- carbonitrile (46)

46
[0194] To a solution of 2 (150 mg, 0.78 mmol) and 45 (202 mg, 0.93 mmol) in anhydrous THF (5.2 niL) was added IM NaHCO3 (4.3 niL, 4.3 mmol) and Pd(PPh3)2Cl2 (26.4 mg, 0.04 mmol). The resulting solution was heated to 50 0C for 1 hour. The reaction mixture was concentrated by half and water used to rinse down sides of flask. The mixture was cooled in an ice bath for 1 hour before filtering to obtain white and black solids. The crude material was purified using preparative HPLC to afford the title compound as a beige solid (160 mg, 62%).
[0195] MS (ES+): m/z 329 (M+H)+ Example 50: Preparation of (Z)-2-(6-((2,4-Dioxothiazolidin-5-ylidene)methyl)quinolin-4-yl)- 3-methylbenzo[b]thiophene-6-carbonitrile

[0197] 1H NMR (500 MHz, DMSO-J6) δ 2.25 (s, 3H), 7.73 (d, J = 4.4 Hz, IH), 7.93 (dd, J = 8.4, 1.4 Hz, IH), 7.96 (s, IH), 7.99 (d, J = 1.8 Hz, IH), 8.03 (dd, J = 8.7, 2.1 Hz, IH), 8.13
(d, J = 8.4 Hz, IH), 8.26 (d, J = 8.8 Hz, IH), 8.74 (d, J= 1.1 Hz, IH), 9.10 (d, J= 4.4 Hz, IH), 12.68 (br s, IH)
[0198] MS (ES+): m/z 428 (M+H)+
Example 51: Preparation of 4-Cyano-3-methylbenzo[b]thiophen-2-ylboronic acid (47)

[0199] To an argon flushed, -78 0C solution of 44 (279 mg, 1.61 mmol) in anhydrous THF (6.4 rnL) was added dropwise 2.5 M nBuLi in hexanes (0.77 rnL, 1.93 mmol). The resulting solution was stirred for 30 min. Trimethylborate (0.36 mL, 3.23 mmol) was added dropwise and the cold bath removed. The reaction was allowed to warm naturally to 0 0C over 15 min at which point it was held there with an ice bath for another 15 min. 2M HCl (3.6 mL) was added and the ice bath was removed, and allowed to warm to room temperature over 30 minutes. The reaction mixture was poured into 2 mL water and 6 mL ethyl acetate. The phases were separated and the aqueous phase was extracted with ethyl acetate (3x18 mL). The combined organic layers were washed with water (2 mL), brine (4 mL), dried (Na2SO4), and concentrated in vacuo. The crude residue was triturated in ethyl acetate (3 mL) and then fully precipitated with petroleum ether (6 mL) to afford the title compound as a mustard colored solid (168 mg, 48%). The material was used as is for the next reaction.
Example 52: Preparation of 2-(6-Formylquinolin-4-yl)-3-methylbenzo[b]thiophene-4- carbonitrile (48)

48
[0200] To a solution of 2 (125 mg, 0.65 mmol) and 47 (156 mg, 0.72 mmol) in anhydrous THF (4.4 mL) was added IM NaHCO3 (3.6 mL, 3.6 mmol) and Pd(PPh3)2Cl2 (22.6 mg, 0.03
mmol). The resulting solution was heated to 50 0C for 1 hour. The reaction mixture was concentrated by half and water used to rinse down sides of flask. The mixture was cooled in an ice bath for 1 hour before filtering to obtain white and black solids. The crude material was purified using preparative HPLC to afford the title compound as a beige solid (104 mg, 49%). [0201] MS (ES+): m/z 329 (M+H)+
Example 53: Preparation of (Z)-2-(6-((2,4-Dioxothiazolidin-5-ylidene)methyl)quinolin-4-yl)- 3-methylbenzo[b]thiophene-4-carbonitrile

[0203] 1H NMR (500 MHz, DMSO-J6) δ 2.45 (d, J = 3.2 Hz, 3H), 7.66 (t, J= I. S Hz, IH), 7.73 (d, J = 4.4 Hz, IH), 8.01 (d, J = 2.8 Hz, 2H), 8.03 (dd, J = 8.8, 1.2 Hz, IH), 8.08 (dd, J = 7.5, 0.8 Hz, IH), 8.27 (d, J= 8.8 Hz, IH), 8.51 (dd, J= 8.0, 0.8 Hz, IH), 9.11 (d, J = 4.3 Hz, IH), 12.65 (br s, IH)
[0204] MS (ES+): m/z 428 (M+H)+
Example 54: Preparation of N-fer?-Butyl-3-[5-(6-formyl-quinolin-4-yl)-thiophen-2-yl]- benzenesulfonamide (49)

[0205] To a solution of 9 (73 mg, 0.27 mmol) in dimethoxyethane (DME, 10 niL) was added a solution of 3-(jV-te/t-butylsulfamoyl)-phenylboronic acid (78 mg, 0.3 mmol) in EtOH (5 mL), 1.0 M Na2CO3 (2 mL), and tetrakis(triphenylphosphine)palladium (0) (Pd(PPh3)4, 35 mg, 0.03 mmol). The reaction mixture was heated at 150 0C for 30 min under μ-wave. The hot solution was filtered and the solid was washed with EtOAc. The filtrate was washed with brine (100 mL). The aqueous was extracted with EtOAc (3 x 20 mL). Combined organic layer was dried (Na2SO4). The solvent was removed in vacuo. The residue was purified by flash column (SiO2/CH2Cl2). The title compound (90 mg, 74%) was obtained as a yellow solid. Example 55: Preparation of N-fer?-Butyl-3-{5-[6-(2,4-dioxo-thiazolidin-5-ylidenemethyl)- quinolin-4-yl] -thiophen-2-yl } -benzenesulfonamide

[0206] To a solution of 49 (90 mg, 0.2 mmol) in EtOH (15 mL) was added thiazolidine-2,4- dione (TZD, 62 mg, 0.53 mmol) and Cs2CO3 (0.35 g, 1.1 mmol). The reaction mixture was heated at 140 0C for 30 min under μ-wave. The solvent was removed in vacuo. The crude product was purified by HPLC. The HPLC fractions containing product were combined and concentrated. The title compound (9 mg, 8%) was obtained as an orange solid.
[0207] 1H NMR (500 MHz, DMSO-J6): δ 1.13 (s, 9H), 7.67-7.69 (m, 2H), 7.74-7.76 (m, 2H), 7.82 (d, J = 7.9 Hz, IH), 7.90 (d, J = 3.8 Hz, IH), 8.04-8.06 (m, 3H), 8.20 (br s, IH), 8.23 (d, J = 8.8 Hz, IH), 8.56 (s, IH), 9.02 (d, J = 4.5 Hz, IH), 12.42 (s, IH)
[0208] MS (ES+): m/z 550 (M+H)+
Example 56: Preparation of 4-(4-Methylthiophen-3-yl)quinoline-6-carbaldehyde (50)
 50
50
[0209] To a solution of 2 (191 mg, 1.0 mmol) in dimethoxyethane (DME, 10 niL) was added a solution of 4-methylthiophen-3-yl-3-boronic acid (156 mg, 1.1 mmol) in EtOH (5 mL), 1.0 M Na2CO3 (2 mL), and tetrakis(triphenylphosphine)palladium (0) (Pd(PPh3 )4, 116 mg, 0.1 mmol). The reaction mixture was heated at 110 0C for 30 min under μ-wave. The hot solution was filtered and the solid was washed with EtOAc. The filtrate was washed with brine (100 mL). The aqueous was extracted with EtOAc (3 x 20 mL). Combined organic layer was dried (Na2SO4). The solvent was removed in vacuo. The residue was added MeOH (10 mL) and sonicated. The solid was collected by centrifuge. The title compound (210 mg, 82%) was obtained as a yellow solid.
Example 57: Preparation of (Z)-5-((4-(4-Methylthiophen-3-yl)quinolin-6- yl)methylene)thiazolidine-2,4-dione

[0212] MS (ES+): m/z 353 (M+H)+
Example 58: Preparation of 5-(fert-Butoxycarbonyl)-4,5,6,7-tetrahydrothieno[J,2-c]pyridin-2- yl-2-boronic acid (52)

Example 59: Preparation of tert-Butyl 2-(6-formylquinolin-4-yl)-6,7-dihydrothieno[3,2- c]pyridine-5(4H)-carboxylate (53)

[0214] To a solution of 2 (193 mg, 1.0 mmol) in dimethoxyethane (DME, 10 mL) was added a solution of 53 (340 mg, 1.1 mmol) in EtOH (5 mL), 1.0 M Na2CO3 (4 mL), and tetrakis(triphenylphosphine)palladium (0) (Pd(PPh3 )4, 116 mg, 0.1 mmol). The reaction mixture was heated at 120 0C for 30 min under μ-wave. The hot solution was filtered and the
solid was washed with EtOAc. The filtrate was washed with brine (100 niL). The aqueous was extracted with EtOAc (3 x 20 rnL). Combined organic layer was dried (Na2SO4). The solvent was removed in vacuo. The crude product was directly used for next step without further purification.
Example 60: Preparation of (Z)-5-((4-(4,5,6,7-Tetrahydrothieno[3,2-c]pyridin-2-yl)quinolin-6- yl)methylene) thiazolidine-2,4-dione

[0215] To a solution of 53 (1 mmol) in EtOH (15 mL) was added thiazolidine-2,4-dione (TZD, 234 mg, 2 mmol) and Cs2CO3 (0.65 g, 2 mmol). The reaction mixture was heated at 150 0C for 30 min under μ-wave. The solvent was removed in vacuo. The 50% TFA in CH2Cl2 was added and the mixture was stirred for 2 h at room temperature. The solvent was removed in vacuo. The crude product was purified by flash column (SiO2/CH2Cl2:MeOH:NH3.H2O = 100:10:1). The title compound (48 mg, 12%) was obtained as a yellow solid.
[0216] 1H NMR (500 MHz, DMSO-J6): δ 3.18 (t, J = 5.8 Hz, 2H), 3.53 (t, J = 5.8 Hz, 2H), 4.33 (s, 2H), 7.52 (s, IH), 7.62 (d, J = 4.5 Hz, IH), 8.00 (s, IH), 8.03 (dd, J = 8.8, 1.9 Hz, IH), 8.22 (d, J = 8.8 Hz, IH), 8.47 (d, J = 1.9 Hz, IH), 8.88 (d, J = 4.5 Hz, IH), 9.29 (br s, IH), 12.36 (br s, IH)
[0217] MS (ES+): m/z 394 (M+H)+
Example 61: Preparation of 5-Cyano-4-methylthiophen-2-yl-2-boronic acid (55)

[0218] To a stirred solution of diisopropylamine (4.8 g, 0.64 mol) in dry THF (1500 rnL) being cooled below -65 0C under N2 was added dropwise ra-BuLi (234 rnL, 0.58 mol). The mixture was warmed to -30 0C slowly, and stirred at this temperature for -30 min, and then re- cooled below -65 0C. The 3-methy\thiophene-2-carbonitrile (60 g, 0.49 mol) was added dropwise, and the mixture was stirred at below -65 0C for an additional 3 h. Then B(OMe)3 was added. The mixture was stirred at 10 0C overnight. Upon completion, the reaction mixture was poured into ice water. The resulting mixture was acidified to pH<2 with 1 N HCl. The aqueous layer was extracted with MTBE (1000 mL x X). The combined organic layers were washed with brine, dried over sodium sulfate, and then concentrated. The crude product was crystallized from dichloromethane to give the compound (28 g, 34.5%) as a light yellow solid.
Example 62: Preparation of 5-(6-Formylquinolin-4-yl)-3-methylthiophene-2-carbonitrile (56)

Example 63: Preparation of 3-Methyl-5-(6-((Z)-(2,4-dioxothiazolidin-5- ylidene)methyl)quinolin-4-yl)thiophene-2-carbonitrile

[0220] To a solution of 56 (1 mmol) in EtOH (15 niL) was added thiazolidine-2,4-dione (TZD, 234 mg, 2 mmol) and Cs2CO3 (0.65 g, 2 mmol). The reaction mixture was heated at 150 0C for 30 min under μ-wave. The solvent was removed in vacuo. The crude product was purified by HPLC. The HPLC fractions containing product were combined and concentrated. The title compound (5 mg, 1%) was obtained as a yellow solid.
[0221] 1H NMR (500 MHz, DMSO-J6): δ 2.54 (s, 3H), 7.68 (s, IH), 7.72 (d, J = 4.5 Hz, IH), 8.04 (d, J = 1.9 Hz, IH), 8.05 (s, IH), 8.24 (d, J = 8.8 Hz, IH), 8.38 (d, J = 1.9 Hz, IH), 9.04 (d, J = 4.5 Hz, IH), 12.73 (s, IH) [0222] MS (ES+): m/z 378 (M+H)+
Example 64: Preparation of 4-(5-(3-Isopropylphenyl)thiophen-2-yl)quinoline-6-carbaldehyde (57)

57 [0223] To a solution of 9 (0.200 g, 0.73 mmol), 3-isopropylphenylboronic acid (0.120 g, 0.73 mmol), and Pd(PPh3)4 (0.084 g, 0.073 mmol) in DMF (3 mL) was added Na2CO3 (2.0 M, 0.6 mL). The reaction mixture was heated 1 h at 180 0C in a Biotage microwave reactor. The resulting mixture was concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 70:30 gradient) to afford the title compound as a yellow solid (0.130 g, 50 %). [0224] 1H NMR (500 MHz, DMSO-J6): δ 1.27 (d, J= 6.9 Hz, 6H), 2.99 (quin, J= 6.9 Hz, IH), 7.32 (d, J = 1.9 Hz, IH), 7.43 (t, J = 5.8 Hz, IH), 7.67 (d, J = 6.2 Hz, IH), 7.68 (s, IH),
7.84 (d, J = 4.1 Hz, IH), 8.17 (d, J = 8.7 Hz, IH), 8.32 (d, J= 4.0 Hz, IH), 8.39 (dd, J= 8.7, 1.7 Hz, IH), 9.23 (d, J= 1.6 Hz, IH), 9.34 (s, IH), 10.32 (s, IH)
[0225] MS (ES+): m/z 359 (M +H)+
Example 65: Preparation of (Z)-5-((4-(5-(3-Isopropylphenyl)thiophen-2-yl)quinolin-6- yl)methylene)thiazolidine-2,4-dione

[0226] A microwave vial was charged with 57 (0.130 g, 0.37 mmol), thiazolidine-2,4-dione (0.071 g, 0.55 mmol), and Cs2CO3 (0.24 g, 0.73 mmol) in ethanol (2 mL). The reaction mixture was heated 30 min at 150 0C in a Biotage microwave reactor. The resulting mixture concentrated and purified by silica gel chromatography (CH2Cl2:Me0H 100:0 to 90: 10 gradient) to afford the title compound in a 1:1 ratio of E and Z isomers as an orange solid (0.033 g, 20 %).
[0227] 1H NMR (500 MHz, DMSO-J6): δ 1.26 (d, J= 7.0 Hz, 6H), 1.27 (d, J= 7.0 Hz, 6H), 2.94 (quin, J = 6.8 Hz, IH), 2.99 (quin, J = 6.7 Hz, IH), 7.31 (d, J= 7.6 Hz, 2H), 7.31 (d, J = 7.6 Hz, 2H), 7.42 (t, J = 7.7 Hz, 2H), 7.66 (d, J = 7.0 Hz, 2H), 6.80 (d, J = 5.8 Hz, 2H), 7.71 (s, 2H), 7.75 (d, J = 4.0 Hz, IH), 7.85 (d, J = 4.0 Hz, IH), 8.03 (d, J = 8.8 Hz, 2H), 8.13 (d, J = 8.8 Hz, IH), 8.22 (dd, J = 8.9, 1.8 Hz, IH), 8.27 (d, J = 4.1 Hz, IH), 8.32 (d, J = 4.1 Hz, IH), 8.40 (dd, J = 8.9, 1.7 Hz, IH), 8.77 (s, IH), 12.65 (br s, 2H)
[0228] MS (ES+): m/z 458 (M +H)+
Example 66: Preparation of 4-(5-(3-Isopropoxyphenyl)thiophen-2-yl)quinoline-6-carbaldehyde (58)

58
[0229] To a solution of 9 (0.200 g, 0.73 mmol), 3-isopropoxyphenylboronic acid (0.132 g, 0.73 mmol), and Pd(PPh3)4 (0.084 g, 0.073 mmol) in DMF (3 mL) was added Na2CO3 (2.0 M, 0.6 mL). The reaction mixture was heated 1 h at 180 0C in a Biotage microwave reactor. The resulting mixture was concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 70:30 gradient) to afford the title compound as a yellow solid (0.261 g, 96 %).
[0230] MS (ES+): m/z 359 (M +H) +
Example 67: Preparation of (Z)-5-((4-(5-(3-Isopropoxyphenyl)thiophen-2-yl)quinolin-6- yl)methylene)thiazolidine-2,4-dione

[0231] A microwave vial was charged with 58 (0.273 g, 0.73 mmol), thiazolidine-2,4-dione (0.143 g, 1.1 mmol), and Cs2CO3 (0.476 g, 1.46 mmol) in ethanol (4 mL). The reaction mixture was heated 30 min at 150 0C in a Biotage microwave reactor. The resulting mixture concentrated and purified by silica gel chromatography (CH2Cl2:Me0H 100:0 to 90: 10 gradient) to afford the title compound as an orange solid (0.033 g, 20 %).
[0232] 1H NMR (500 MHz, DMSO-J6): δ 1.31 (d, J= 6.1 Hz, 6H), 4.76 (quin, J= 3.9 Hz, IH), 6.96-6.99 (m, IH), 7.32-7.43 (m, 3H), 7.84 (d, J= 4.0 Hz, IH), 8.15 (s, IH), 8.19 (dd, / = 8.8, 1.8 Hz, IH), 8.25 (d, /= 4.1 Hz), 8.77 (s, IH), 9.25 (s, 1H)\ [0233] MS (ES+): m/z 474 (M +H)+
Example 68: Preparation of 4-(5-(2,6-Dimethoxyphenyl)thiophen-2-yl)quinoline-6- carbaldehyde (59 )

59 [0234] To a solution of 9 (0.200 g, 0.73 mmol), 3-isopropoxyphenylboronic acid (0.132 g, 0.73 mmol), and Pd(PPh3)4 (0.084 g, 0.073 mmol) in DMF (3 mL) was added Na2CO3 (2.0 M, 0.6 mL). The reaction mixture was heated 1 h at 180 0C in a Biotage microwave reactor. The resulting mixture was concentrated and purified by silica gel chromatography (hexanes/EtOAc 100:0 to 50:50 gradient) to afford the title compound as an orange solid (0.197 g, 72 %). [0235] 1H NMR (500 MHz, DMSO-J6): δ 3.29 (s, 6H), 5.75 (s, IH), 6.85 (d, J = 8.5 Hz,
2H), 7.39 (t, J = 8.5 Hz, IH), 7.72 (d, J = 4.0 Hz, IH), 8.16 (d, J = 8.7 Hz, IH), 8.22 (d, J = 5.6 Hz, IH), 8.38 (dd, J = 8.7, 1.6 Hz, IH), 9.23 (s, IH), 9.32 (s, IH), 10.31 (s, IH)
[0236] MS (ES+): m/z 311 (M +H)+
Example 69: Preparation of (Z)-5-((4-(5-(2,6-Dimethoxyphenyl)thiophen-2-yl)quinolin-6- yl)methylene)thiazolidine-2,4-dione

[0237] A microwave vial was charged with 59 (0.150 g, 0.40 mmol), thiazolidine-2,4-dione (0.070 g, 0.60 mmol), and Cs2CO3 (0.260 g, 0.80 mmol) in ethanol (4 mL). The reaction mixture was heated 30 min at 150 0C in a Biotage microwave reactor. The resulting mixture concentrated and purified by HPLC to afford the title compound as a brown solid (0.024 g, 13 %).
[0238] 1H NMR (500 MHz, DMSO-J6): δ 3.88 (s, 6H), 6.83-6.85 (m, 2H), 7.35-7.40 (m, IH), 7.64 (d, 2.2 Hz, IH), 7.65-7.67 (m, IH), 8.12-8.19 (m, 3H), 8.23 (d, J = 2.2 Hz, IH), 8.80 (s, IH), 9.24 (s, IH)
[0239] MS (ES+): m/z 47 '6 (M +H)+
Example 70: Preparation o/3-(te/t-Butyldimethylsilyloxy)aniline (60)
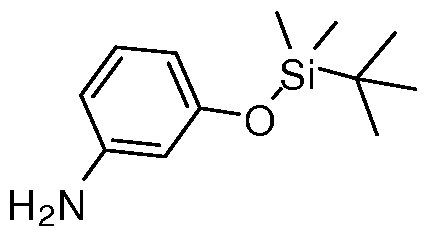
60
[0240] A solution of 3-aminophenol (3.0 g, 27.5 mmol), te/t-butyldimethylsilyl chloride (4.56 g, 30.2 mmol), and imidazole (2.34 g, 34.4 mmol) in DMF (9 mL) was stirred for 4 h at room temperature and EtOAc (15 mL) and H2O (20 mL). The aqueous layer was extracted two times with EtOAc. The organic layers were combined and washed with H2O, washed with brine, and dried over MgSO4. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc 100:0 to 60:40 gradient) to afford the title compound as an orange solid (4.62 g, 75 %). [0241] 1H NMR (500 MHz, DMSO-J6): δ 0.92 (s, 9H), 5.00 (s, 2H), 5.97 (d, 7.9 Hz, IH), 6.07 (t, J = 2.3 Hz, IH), 6.14 (dt, J = 9.5, 0.8 Hz, IH), 6.83 (t, J= 8.1 Hz, IH)
Example 71 : Preparation of 5-(6-Formylquinolin-4-yl)-N-(3-hydroxyphenyl)thiophene-2- carboxamide (61)

[0242] To a solution of 17 (0.50 g, 1.76 mmol) and 2-chloro-4,6-dimethoxy-l,3,5-triazine (0.47 g, 2.68 mmol) in CH2Cl2 (10 mL) was added JV-methylmorpholine (0.78 mL, 7.1 mmol). The reaction mixture was stirred for 2 h at room temperature and 60 (0.60 g, 2.68 mmol) was
added. The reaction mixture was stirred additional 2 h at room temperature and concentrated. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc 100:0 to 0:100 gradient) to afford the title compound as a white solid (0.352 g, 41 %).
[0243] 1H NMR (500 MHz, DMSO-J6): δ 0.23 (s, 6H), 0.97 (s, 9H), 3.96 (s, IH), 6.62 (d, J = 8.0 Hz, IH), 7.24 (t, J = 8.1 Hz, IH), 7.36 (d, J = 1.6 Hz, IH), 7.42 (d, J = 2.1 Hz, IH), 7.76 (d, J = 3.8 Hz, IH), 7.80 (d, J = 4.6 Hz, IH), 8.22-8.28 (m, 3H), 8.88 (d, J = 1.6 Hz, IH), 9.12 (d, J = 4.6 Hz, IH), 10.22 (s, IH), 10.36 (s, IH)
Example 72: Preparation of (Z)-5-(6-((2,4-Dioxothiazolidin-5-ylidene)methyl)quinolin-4-yl)- jV-(3-hydroxyphenyl)thiophene-2-carboxamide

[0244] A microwave vial was charged with 61 (0.150 g, 0.31 mmol), thiazolidine-2,4-dione
(0.054 g, 0.46 mmol), and Cs2CO3 (0.325 g, 0.92 mmol) in ethanol (3 mL). The reaction mixture was heated 30 min at 150 0C in a Biotage microwave reactor. The resulting mixture concentrated and purified by silica gel chromatography (CH2Cl2:Me0H 100:0 to 50:50 gradient) to afford the title compound a brown solid (0.079 g).
[0245] 1H NMR (500 MHz, DMSO-J6): δ 6.54 (d, J = 1.5 Hz, IH), 7.13-7.32 (m, 3H), 7.34 (s, IH), 7.67 (d, J = 4.6 Hz, IH), 7.69 (d, J = 4.5 Hz, IH), 7.76 (s, IH), 8.02 (dd, J = 8.8, 1.8 Hz, IH), 8.18 (d, J = 8.8 Hz, IH), 8.26 (d, J = 3.9 Hz, IH), 8.43 (s, IH), 8.98 (d, J = 4.5 Hz, IH), 9.48 (br s, IH), 10.34 (s, IH) [0246] MS (ES+): m/z AlA (M +H)+
Example 73: Preparation of 4-(te/t-Butyldimethylsilyloxy)aniline (62)

62
[0247] A solution o/4-aminophenol (3.0 g, 27.5 mmol), tert-butyldimethylsilyl chloride (4.56 g, 30.2 mmol), and imidazole (2.34 g, 34.4 mmol) in DMF (9 mL) was stirred for 4 h at room temperature and EtOAc (15 mL) and H2O (20 mL). The aqueous layer was extracted two times with EtOAc. The organic layers were combined and washed with H2O, washed with brine, and dried over MgSO4. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc 100:0 to 60:40 gradient) to afford the title compound yellow oil (4.03 g, 66 %). [0248] 1H NMR (500 MHz, DMSO-J6): δ 0.10 (s, 6H), 0.91 (s, 9H), 4.60 (br s, 2H), 6.45 (d, J = 6.5 Hz, 2H), 6.52 (d, J = 6.5 Hz, 2H)
Example 74: Preparation of N-(4-(fert-Butyldimethylsilyloxy)phenyl)-5-(6-formylquinolin-4- yl)thiophene-2-carboxamide (63)

[0249] To a solution of 17 (0.30 g, 1.06 mmol) and 2-chloro-4,6-dimethoxy-l,3,5-triazine (0.22 g, 1.27 mmol) in CH2Cl2 (6 mL) was added JV-methylmorpholine (0.47 mL, 4.2 mmol). The reaction mixture was stirred for 1 h at room temperature and 62 (0.28 g, 1.27 mmol) was added. The reaction mixture was stirred additional 2 h at room temperature and concentrated. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc 100:0 to 0: 100 gradient) to afford the title compound as a yellow solid (0.333 g, 64 %).
[0250] 1H NMR (500 MHz, DMSO-J6): δ 0.20 (s, 6H), 0.96 (s, 9H), 6.86-6.91 (m, 2H), 7.33 (d, J = 6.7 Hz, 2H), 7.62 (d, J = 8.9 Hz, 2H), 8.17-8.21 (m, IH), 8.43 (dd, J = 8.9, 1.7 Hz, IH), 8.72 (d, J= 1.6 Hz, IH), 8.88 (s, IH), 9.01 (d, J = 4.5 Hz, IH), 10.31 (s, IH)
[0251] MS (ES+): m/z 489 (M +H)+
Example 75: Preparation of (Z)-5-(6-((2,4-Dioxothiazolidin-5-ylidene)methyl)quinolin-4-yl)- jV-(4-hydroxyphenyl)thiophene-2-carboxamide

(0.074 g, 0.57 mmol), and Cs2CO3 (0.408 g, 1.14 mmol) in ethanol (4 mL). The reaction mixture was heated 30 min at 150 0C in a Biotage microwave reactor. The resulting mixture concentrated and purified by silica gel chromatography (CH2Cl2:Me0H 100:0 to 50:50 gradient) to afford the title compound an orange solid (0.068 g).
[0253] 1H NMR (500 MHz, DMSO-J6): δ 6.77 (d, J = 8.5 Hz, 2H), 7.53 (d, J= S3 Hz, 2H), 7.67-7.71 (m, 3H), 8.00 (d, J= 8.6 Hz, IH), 8.15 (d, J= 8.8 Hz, IH), 8.20 (d, J= 3.3 Hz, IH), 8.43 (s, IH), 8.95 (d, J = 4.2 Hz, IH), 9.34 (br s, IH), 10.28 (s, IH)
[0254] MS (ES+): m/z 502 (M +H)+
Example 76: Preparation of 5-(6-Formylquinolin-4-yl)-/V-(4- (morp/zo/mome?/ry/)phenyl)thiophene-2-carboxamide (64)

64
[0255] To a solution of 17 (0.20 g, 0.71 mmol) and 2-chloro-4,6-dimethoxy-l,3,5-triazine (0.15 g, 0.85 mmol) in CH2Cl2 (4 mL) was added N-methylmorpholine (0.31 mL, 2.8 mmol). The reaction mixture was stirred for 1 h at room temperature and 4-(morpholin-4- ylmethyl) aniline (0.163 g, 0.85 mmol) was added. The reaction mixture was stirred additional 2 h at room temperature and concentrated. The crude mixture was purified by silica gel chromatography (CH2Cl2: MeOH 100:0 to 70:30 gradient) to afford the title compound as yellow oil (0.229 g, 11 %).
[0256] MS (ES+): m/z 458 (M +H)+
Example 77: Preparation of (Z)-5-(6-((2,4-Dioxothiazolidin-5-ylidene)methyl)quinolin-4-yl)- N-(4-(morpholinomethyl)phenyl)thiophene-2-carboxamide

[0257] A microwave vial was charged with 65 (0.200 g, 0.44 mmol), thiazolidine-2,4-dione (0.085 g, 0.66 mmol), and Cs2CO3 (0.46 g, 1.3 mmol) in ethanol (4 mL). The reaction mixture was heated 30 min at 150 0C in a Biotage microwave reactor. The resulting mixture concentrated and purified by silica gel chromatography (CH2Cl2:Me0H 100:0 to 50:50 gradient) to afford the title compound a yellow solid (0.103 g, 42%).
[0258] 1H NMR (500 MHz, DMSO-J6): δ 2.39 (br s, 4H), 3.58 (m, 4H), 3.85 (s, 2H), 5.40 (br s, IH), 7.32 (d, J = 8.5 Hz, 2H), 7.70 (d, J = 4.5 Hz, IH), 7.71-7.75 (m, 3H), 8.02 (dd, J = 8.8, 1.9 Hz, IH), 8.17 (d, J = 8.7 Hz, IH), 8.26 (d, J = 3.9 Hz, IH), 8.43 (d, J = 1.8 Hz, IH), 8.98 (d, J= 4.5 Hz, IH), 10.47 (s, IH)
[0259] MS (ES+): m/z 557 (M +H)+
Example 78: Preparation of 5-(6-Formylquinolin-4-yl)-/V-(3- (moφholinomethyl)phenyl)thiophene-2-carboxamide (65)

[0261] MS (ES+): m/z 458 (M +H)+
Example 79: Preparation of (Z)-5-(6-((2,4-Dioxothiazolidin-5-ylidene)methyl)quinolin-4-yl)- N-(3-(morpholinomethyl)phenyl)thiophene-2-carboxamide

[0262] A microwave vial was charged with 65 (0.275 g, 0.58 mmol), thiazolidine-2,4-dione (0.113 g, 0.87 mmol), and Cs2CO3 (0.64 g, 1.8 mmol) in ethanol (3 mL). The reaction mixture was heated 30 min at 150 0C in a Biotage microwave reactor. The resulting mixture concentrated and purified by silica gel chromatography (CH2Cl2IMeOH 100:0 to 50:50 gradient) to afford the title compound a yellow solid (0.180 g, 56%).
[0263] 1H NMR (500 MHz, DMSO-J6): δ 2.42 (br s, 4H), 3.52 (br s, 4H), 3.60 (s, 2H), 7.09 (d, J = 6.8 Hz, IH), 7.35 (t, J = 7.7 Hz, IH), 7.71-7.75 (m, 4H), 8.02 (d, J = 8.5 Hz, IH), 8.17 (d, J = 9.1 Hz, IH), 8.30 (s, IH), 8.43 (s, IH), 8.97 (s, IH), 10.53 (s, IH) [0264] MS (ES+): m/z 557 (M +H)+
Example 80: Preparation of 4-(5-Formylthiophen-2-yl)quinoline-6-carbaldehyde (66)

66
[0265] To a mixture of 5-formyl-2-thiopheneboronic acid (0.50 g, 3.2 mmol), 2 (0.61 g, 3.2 mmol), and PdP(PPh3)4 (0.37 g, 0.32 mmol) in DMF (15 mL) was added 2.0 M Na2CO3 (2.75 mL). The reaction mixture was heated at 160 0C for 30 min in a Biotage microwave reactor. The resulting mixture was added sat Na2CO3 (5 mL) and washed extracted with EtOAc (20 mL). The organic layer was washed with brine, dried over MgSO4, and concentrated. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc 100:0 to 0:100 gradient) to afford the title compound as a peach solid (0.145 g, 17 %).
[0266] 1H NMR (500 MHz, DMSO-J6): δ 7.83 (d, J = 4.6 Hz, IH), 7.86 (d, J = 3.9 Hz, IH), 8.22-8.29 (m, 3H), 8.82 (d, J = 1.5 Hz, IH), 9.13 (d, J = 4.5 Hz, IH), 10.07 (s, IH), 10.21 (s, IH)
[0267] MS (ES+): m/z 268 (M +H)+
Example 81: Preparation of (Z)-5-((5-(6-((Z)-(2,4-Dioxothiazolidin-5-ylidene)methyl)quinolin- 4-yl)thiophen-2-yl)methylene)thiazolidine-2,4-dione

[0268] A microwave vial was charged with 66 (0.140 g, 0.52 mmol), thiazolidine-2,4-dione (0.204 g, 1.57 mmol), and Cs2CO3 (1.11 g, 3.14 mmol) in ethanol (4 mL). The reaction mixture was heated 30 min at 160 0C in a Biotage microwave reactor. The resulting mixture concentrated and purified by silica gel chromatography (CH2Cl2:Me0H 100:0 to 0:100 gradient) to afford the title compound a brown solid (0.042 g, 17%).
[0269] 1H NMR (500 MHz, DMSO-d6): δ 7.69 (d, J = 4.5 Hz, IH), 7.74 (s, 3H), 7.88 (s, IH), 8.00 (dd, J = 8.8, 1.9 Hz, IH), 8.16 (d, J= 8.8 Hz, IH), 8.44 (d, J= 1.8 Hz, IH), 8.95 (d, J = 4.5 Hz, IH), 12.02-12.08 (br s, 2H)
Example 82: Preparation of 4-(5-(3-Methoxyphenyl)thiophen-2-yl)quinoline-6-carbaldehyde (67)

67
[0270] To a solution of 17 (0.25 g, 0.88 mmol) and 2-chloro-4,6-dimethoxy-l,3,5-triazine (0.186 g, 1.06 mmol) in CH2Cl2 (5 mL) was added JV-methylmorpholine (0.39 mL, 3.53 mmol). The reaction mixture was stirred for 1 h at room temperature and m-anisidine (0.13 g, 1.06 mmol) was added. The reaction mixture was stirred additional 3 h at room temperature and concentrated. The crude mixture was purified by silica gel chromatography (hexanes/EtOAc 100:0 to 0:100 gradient) to afford the title compound as a yellow solid (0.326 g, 95 %).
[0271] MS (ES+): m/z 389 (M +H)+
Example 83: Preparation of (Z)-5-((4-(5-(3-Methoxyphenyl)thiophen-2-yl)quinolin-6- yl)methylene)thiazolidine-2,4-dione

[0272] A microwave vial was charged with 67 (0.32 g, 0.82 mmol), thiazolidine-2,4-dione (0.161 g, 1.24 mmol), and Cs2CO3 (0.805 g, 2.47 mmol) in ethanol (6 mL). The reaction mixture was heated 30 min at 160 0C in a Biotage microwave reactor. The resulting mixture concentrated and purified by preparative HPLC to afford the title compound a brown solid (0.202 g, 50%).
[0273] 1H NMR (500 MHz, DMSO-J6): δ 3.77 (s, 3H), 6.72 (dd, J = 8.5, 2.4 Hz, IH), 7.28 (t, J = 8.1 Hz, IH), 7.39 (d, J = 7.8 Hz, IH), 7.47 (s, IH), 7.72 (s, IH), 7.73 (s, IH), 8.00 (s, IH), 8.05 (dd, J = 8.8, 2.0 Hz, IH), 8.22 (d, J = 8.8 Hz, IH), 8.30 (d, J = 4.0 Hz, IH), 8.43 (d, J = 1.8 Hz, IH), 9.02 (d, J = 4.5 Hz, IH), 10.47 (s, IH)
[0274] MS (ES+): m/z 488 (M +H)+
[0275] Exam Paul Van Dyk f. Ashley Tomberlin
Example 84: Complicatedple 84103 Preparation of tert-Butyl-(3-ethynyl-phenoxy)-dimethyl- silane (68)

68
[0276] 3-Ethynyl-phenol (4.0 g, 33.9 mmol), tert-butyldimethylsilyl chloride (5.62 g, 37.3 mmol) and imidazole (2.88 g, 42.4 mmol) were combined and diluted with DMF (25 mL). This was stirred at room temperature for 14h. Reaction contents were then poured onto water and extracted with EtOAc (3 x 100 mL). Organic phase was evaporated to provide desired product as an amber oil (7.9g, 98%).
Example 85: Preparation of te/t-Butyl-dimethyl-(3-thiophen-2-ylethynyl-phenoxy)-silane (69)

[0277] A solution of 2-bromothiophene ( 1.5 g, 9.2 mmol) in TEA (6 mL) and DMF ( 1.2 mL) was treated with copper iodide (0.175 g, 0.92 mmol) and purged with argon for 3 min. 68 (3.2 g, 13.8 mmol) and PdCl2(PPhS)2 (0.323 g, 0.46 mmol) were then added and microwaved at 12O0C for 20 min. The reaction mixture was diluted with DCM (20 mL), filtered and evaporated. This was then purified by silica gel flash chromatography to afford the title compound as a clear oil (2.2 g, 79%).
Example 86: Preparation of te/t-Butyl-dimethyl-(3-thiophen-2-ylethynyl-phenoxy)-silane-2- boronic acid (70)

Example 87: Preparation of 4-[5-(3-Hydroxy-phenylethynyl)-thiophen-2-yl]-quinoline-6- carbaldehyde (71)

71
[0279] A mixture of 2 (0.078 g, 0.406 mmol), 70 (0.160 g, 0.447 mmol), Pd(PPh3)4 (0.047 g, 0.041 mmol) and solid sodium carbonate (0.086 g, 0.813 mmol) were suspended in DMF (5 mL) in a sealed microwave tube and heated in oil bath at 160° C for 5h. The reaction mixture was cooled to room temperature and purified on silica gel column to afford the title compound (0.076 g, 53%).
Example 88: Preparation of 5-{4-[5-(3-Hydroxy-phenylethynyl)-thiophen-2-yl]-quinolin-6- ylmethylene } -thiazolidine-2,4-dione

[0280] A mixture of 71 (0.076 g, 0.214 mmol), thiazolidine dione (0.05 g, 0.428 mmol) and Boc-β-Ala-OH (0.081 g, 0.428 mmol) were suspended in acetic acid (2.2 mL) and heated to
120° C for 3h. Reaction was then cooled to room temperature and poured onto water and filtered (0.015 g, 15%).
[0281] 1H NMR (500 MHz, DMSO-J6): δ 6.86-6.88 (m, IH), 6.94-6.95 (m, IH), 7.01-7.03 (m, IH), 7.26 (t, J = 7.9 Hz, IH), 7.64-7.70 (m, 3H), 8.01-8.05 (m, 2H), 8.19-8.22 (m, IH), 8.45-8.48 (m, IH), 8.99-9.00 (m, IH), 9.78 (s, IH), 12.7 (br s, IH)
[0282] MS (ES+): m/z 455 (M+H)+ Example 89: Preparation of phenyl thiourea 72:

Example 90: Preparation of 2-Phenylimino-thiazolidin-4-one (73)

73
[0285] A microwave vial was charged with 72 (0.50 g, 3.3 mmol), chloro-acetic acid (0.32 g, 3.4 mmol), and sodium acetate (0.30 g, 3.7 mmol) in acetic acid (3 mL). The reaction mixture was heated for 20 min at 150 0C in a Biotage microwave reactor. Water was added (20 mL) and the resulting solid filtered to afford the title compound (a 1/1 mixture of E/Z isomers) as a yellow solid (0.46 g, 73%).
[0286] 1H NMR (500 MHz, DMSO-J6, isomeric mixture): δ 3.96 (s, 2H), 4.01 (s, 2H), 6.99 (br d, J = 5.8 Hz, 2H), 7.15 (t, J = 7.2 Hz, 2H), 7.30-7.43 (m, 4H), 7.69 (d, J = 1.6 Hz, 2H), 11.16 (s, IH), 11.74 (s, IH)
[0287] MS (ES+): m/z 193 (M+H)+
Example 91: Preparation of 2-Phenylimino-5-(4-thiophen-2-yl-quinolin-6-ylmethylene)- thiazolidin-4-one

2-Phenylimino-5-(4-thiophen-2-yl-quinolin-6-ylmethylene)-thiazolidin-4-one [0288] A microwave vial was charged with 3 (55 mg, 0.23 mmol), 73 (50 mg, 0.26 mmol), and Cs2CO3 (0.15 g, 0.46 mmol) in ethanol (4 mL). The reaction mixture was heated for 30 min at 140 0C in a Biotage microwave reactor. The resulting solid was filtered, washed with ethanol followed by water to afford the title compound (a 2/3 mixture of isomers) as a yellow solid (45 mg, 47%). [0289] 1H NMR (500 MHz, DMSO-J6, isomeric mixture): δ 6.87 (d, J = 7.3 Hz, 3H), 7.01 (dd, J = 5.0, 3.7 Hz, IH), 7.09 (t, J= 7.7 Hz, IH), 7.18 (t, J= 7.7 Hz, 2H), 7.19-7.35 (m, 5H), 7.38 (s, IH), 7.39-7.41 (m, IH), 7.47 (d, J= 3.2 Hz, IH), 7.54 (d, J= 4.5 Hz, IH), 7.59 (d, J = 4.5 Hz, IH), 7.67 (d, J = 3.2 Hz, IH), 7.71 (d, J = 4.6 Hz, IH), 7.88 (dd, J = 8.8, 1.8 Hz, IH), 7.93-8.06 (m, 2H), 8.11 (d, J = 8.7 Hz, IH), 8.33 (d, J = 1.5 Hz, IH), 8.44 (d, J = 1.2 Hz, IH), 8.83 (d, J = 4.5 Hz, IH), 8.88 (d, J = 4.5 Hz, IH)
[0290] MS (ES+): m/z 414 (M+H)+
Example 92: Preparation of (2-Chloro-5-fluoro-phenyl)-thiourea (74)

74 [0291] To a solution of l-chloro-4-fluoro-2-isothiocyanato-benzene (1.0 g, 5.3 mmol) in ethanol (10 mL) was added ammonia (7 N in MeOH; 3.0 mL, 21 mmol). The reaction mixture was heated at 70 0C for 1 h. The reaction mixture was concentrated and triturated in water. The
resulting solid was filtered to afford the title compound as a white solid (0.85 g, 78%). MS (ES+): m/z 205 (M+H)+
Example 93: Preparation of 2-(2-Chloro-5-fluoro-phenylimino)-thiazolidin-4-one (75)

75
[0292] A microwave vial was charged with 74 (0.85 g, 4.1 mmol), chloro-acetic acid (0.45 g, 4.7 mmol), and sodium acetate (0.40 g, 4.9 mmol) in acetic acid (6 mL). The reaction mixture was heated for 20 min at 150 0C in a Biotage microwave reactor. Water was added (20 mL) to the reaction mixture and the resulting solid filtered to afford the title compound as a white solid (0.71 g, 70%).
[0293] 1H NMR (500 MHz, DMSO-J6): δ 4.05 (s, 2H), 6.94 (br d, J = 9.0 Hz, IH), 7.00 (t, J = 8.4 Hz, IH), 7.53 (dd, J = 8.8, 5.7 Hz, IH), 12.09 (s, IH)
Example 94: Preparation of 2-(2-Chloro-5-fluoro-phenylimino)-5-(4-thiophen-2-yl-quinolin-6- ylmethylene)-thiazolidin-4-one

2-(2-Chloro-5-fluoro-phenylimino)-5-(4-thiophen-2-yl-quinolin-6-ylmethylene)-thiazolidin-4- one
[0294] A microwave vial was charged with 3 (50 mg, 0.21 mmol), 75 (70 mg, 0.29 mmol), and Cs2CO3 (0.13 g, 0.40 mmol) in ethanol (4 mL). The reaction mixture was heated for 30 min at 140 0C in a Biotage microwave reactor. The resulting solid was filtered, washed with ethanol followed by water to afford the title compound as an orange solid (55 mg, 57%).
[0295] 1H NMR (500 MHz, DMSO-J6): δ 6.82 (t, J = 6.9 Hz, IH), 7.11-7.24 (m, 2H), 7.39 (dd, J = 8.7, 6.0 Hz, IH), 7.49 (s, IH), 7.54 (d, J = 3.8 Hz, IH), 7.56 (d, J = 4.5 Hz, IH), 7.76 (d, J= 4.9 Hz, IH), 7.92 (dd, J= 8.8, 1.9 Hz, IH), 8.10 (d, J = 8.7 Hz, IH), 8.38 (d, J = 1.7 Hz, IH), 8.88 (d, J = 4.5 Hz, IH) MS (ES+): m/z 466 (M+H)+ Example 95: Enzyme Assays
[0296] IC50 values for compounds against the isoforms of PI3-Kinase (PI3K-γ, PI3K-β, PI3K-α) were generated using either luminescence or fluorescence polarization based assays. A four order of magnitude serial dilution of the compounds was introduced into a buffered solution containing appropriate amounts of either PI3-kinase isoform, ATP (3 μM for luminescence or 25 μM for fluorescence polarization) and PIP3 (50 μM for luminescence and 10 μM for fluorescence polarization); the reaction was then allowed to proceed for an appropriate time. These reactions were then terminated by the addition of either the KinaseGlo reagent (Promega) for luminescence or the probe/detector solution for fluorescence polarization (Echelon Biosciences) and then allowed to proceed for an additional 10 min to maximize the luminescence or fluorescence polarization. Values were then measured and IC50 values were derived from experimental data using the non-linear curve fitting capabilities of Prism (Version 4; GraphPad Software), the results, expressed as IC50, are presented in Table 1.
Example 96: ELISA for C5a-induced phosphorylation of AKT in Raw 264.7 macrophages.
[0297] Raw 264.7 mouse macrophage cells (ATCC#TIB-71) were cultured in DMEM containing 10% heat inactivated serum and IX penicillin/streptomycin. 7.5xl04 cells were seeded into a 96 well plate were allowed to adhere overnight. Cells were then washed one time with serum-free medium, and then incubated under serum-free conditions for 3-4 hours at 37°C/5%CO2. Cells were pretreated with the indicated compound (10 μM - 0.0045 μM) for 1 h, followed by stimulation with 0.5 μg/ml C5a (Sigma) for 5 min. Cells were immediately placed on ice, and washed one time with cold PBS. Cells were lysed with 65 μL of 1% Triton- X-100 lysis buffer containing protease inhibitors (Cell Signaling Technology) and further incubated in this solubilization buffer for 20-30 min on ice with mild agitation. Lysates were then centrifuged at 5000 rpm at 4 0C for 10-15 min. Thereafter, an ELISA specific for phosphorylated AKT (pAKT-Ser473) (Cell Signaling Technology) was performed. Absorbance was measured at 450-540 nm. Background was subtracted from each pAKT absorbance and EC50 values were determined using the GraphPad Prism 4.0 software with
ODpAkt plotted in the y-axis and logarithmic values of concentration (μM) on the x-axis. The C5a data give a direct read of blocking signal downstream of PI3K-γ and PI3K-5. Results, expressed as IC50, are presented in Table 1.
Example 97: VEGF- induced hRMVEC proliferation assay [0298] For cellular proliferation assays, early passage human retinal microvascular endothelial cells (hRMVECs) cells (Cell Systems, Kirkland, WA) were plated at a density of -1.5 x 103 cells/well in a 96-well plate (Corning, Corning, NY), and allowed to adhere for ~6 hours or overnight. Medium was then changed into CSC-Maintenance Medium (Cell Systems, Kirkland, WA) and further incubated for at least 24 h - 48 h at 37°C/5% CO2. As indicated by the manufacturer, this specialized medium contains 10% FBS but no growth factor, and renders proliferating cells into a quiescent state. The cells were then pre-treated with varying concentrations of the indicated compound (20 μM - 0.00914 μM or 10 μM - 0.00457 μM or DMSO (as a vehicle control) prepared in basal CSC-medium containing 10% FBS and 50 μg/ml heparin for -60 minutes at 37°C/5% CO2. Human recombinant VEGF (Peprotech, Rocky Hill, NJ) was then added to a final concentration of 50 ng/ml. After 48-72 h of incubation, cellular proliferation was quantified using the Cell Proliferation Kit (Roche, Alameda, CA) as described by the manufacturer. Briefly, for one 96-well plate, 100 μL of electron-coupling reagent was added to 5 mL of XTT labeling solution. 50 μL of this solution was then added to each well, and the reaction was allowed to develop at 37°C/5% CO2. The colored formazan product that is generated by metabolically active cells was measured spectrophotometrically using the SpectraMax spectrophotometer (Molecular Devices, Sunnyvale, CA) at 492 nm with correction at 690 nm. The effective concentration at which VEGF-induced cellular proliferation was inhibited by 50% (EC50) was determined by using the GraphPad Prism 4.0 software (San Diego, CA). Data shown in Table 1 in nM. [0299] Table 1 PI3K enzymatic and cellular data
Table 1 PI3K enzyme and cell data

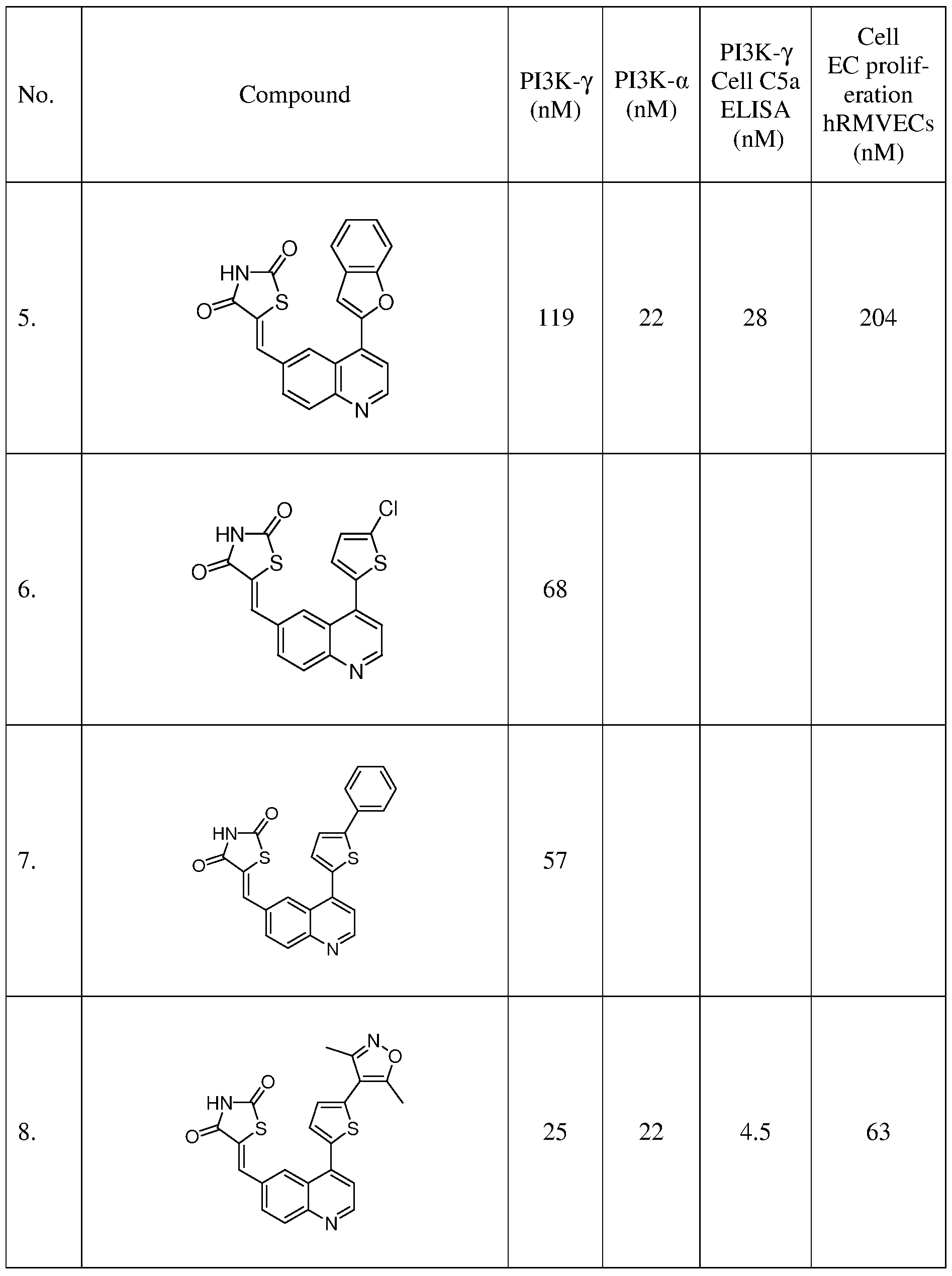


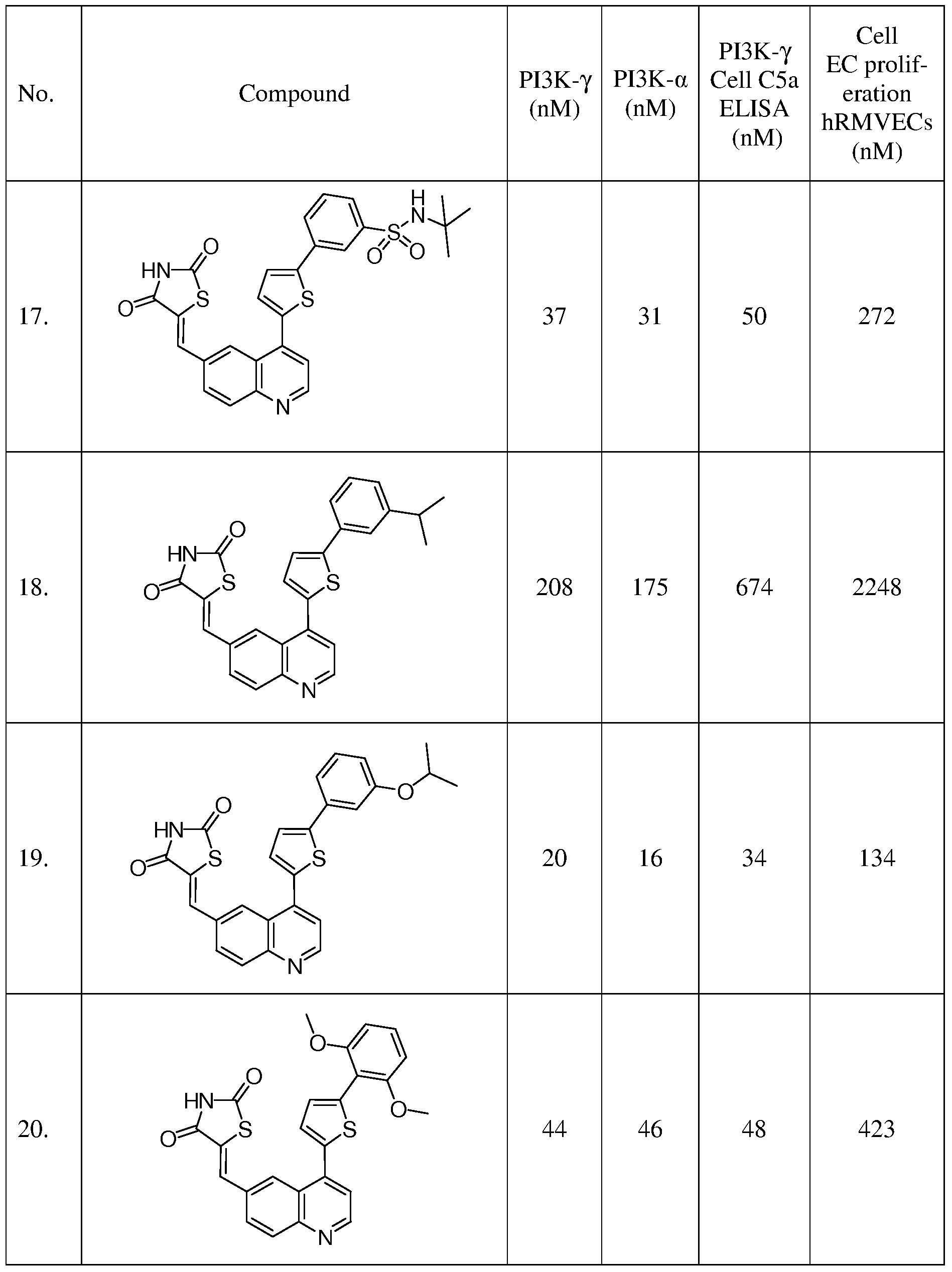




[0300] Pharmacokinetics parameters were determined in rats and mice following a single intravenous (IV) and oral (PO) doses. Intravenous and oral dose formulations were prepared fresh. For the PO formulation, the compounds were dissolved in an aqueous solutions containing appropriate excipients or suspended in 0.5% MC and 0.05% Tween80. For the IV formulation, the compounds were solubilized in an aqueous vehicle containing suitable excipients. The IV formulation was aseptically filtered through 0.22um filters.
[0301] Rats: Six jugular vein cannulated male Sprague-Dawley rats (~300g) with were divided into three groups with three rats in IV dose group and three rats in the PO dose group. Animals were allowed food and water ad libitum. The study was conducted at TargeGen. (San Diego, CA).
[0302] Blood samples (approximately 500 μL per time point) were serially collected via jugular vein cannula and transferred into tubes containing sodium heparin anticoagulant at 0.083, 0.25, 0.5, 1, 3, 5, 7, and 24 hours post-dose for IV dose; and at 0.5, 1, 3, 5, 7, and 24 hours post dose for PO doses. Blood was maintained in an ice and water mixture prior to centrifugation to obtain plasma. Plasma samples are transferred to a -2O0C freezer and stored until analysis.
[0303] Matrix calibration standards and QC samples were prepared by spiking the compound into blank rat plasma (Valley Biomedical Inc., Lot # L51663). The final concentrations of a selected compound were 0, 1, 5, 10, 50, 100, 500, 1000, 2500 and 5000 ng/niL for calibration standards, and 2.50, 25.0, 250 and 2500 for QC samples.
[0304] Plasma samples were processed using a standard protocol. The samples were analyzed using a LC/MS/MS Waters Quattro LC by standard determined conditions. Chromatogram signals were integrated and calibrated using MassLynx 3.0. Pharmacokinetic parameters were estimated using WinNonlin (version 4.1) from mean plasma concentration- time profiles. The values for the maximum plasma concentration (Cmax) and the time to maximum concentration (Tmax) were determined from measure plasma concentrations. The area under the curves, AUC (iast) and AUC (inf) were calculated from plasma concentration-time profiles using the linear trapezoidal rule. The oral bioavailability (F) was calculated using the following equation; F=(AUC(o-inf),po x Div)/(AUC(o-inf),iv x DPO)* 100%
[0305] The PK evaluation in mice utilized the same procedures described above except the number of mice was three per time point per dosed group and the blood samples were collected by cardiac puncture.
[0306] Table 2 provides data from a mouse PK model with IV and PO arms.
Table 2

Example 99: Protocol for ocular exposure following topical instillation
[0307] Twenty male mice were given one 10 μL drop of the 1% formulation of test compound. Composite sampling was employed to generate tissue concentration-time profiles for compound over the following a certain defined time course for sampling post-instillation.
Both eyes were removed from each mouse and dissected to obtain the cornea, retina and eye cup (sclera and choroid). Plasma samples were also obtained from each mouse at the time of euthanasia. Determinations of amounts of compounds in various eye tissues were made by LC/MS/MS. (Z)-5-((4-(Thiophen-2-yl)quinolin-6-yl)methylene)thiazolidine-2,4-dione was evaluated in an ocular PK and showed levels well above cell EC50 in the back of the eye tissues.
Example 100: Ocular PK
[0308] Table 3 shows ocular PK data for selected compounds with low oral availability
Table 3

Example 101: Murine model of ocular edema and neovascularization [0309] A murine model of ocular edema and neovasularization was used to assess compound activity. Laser energy is used to rupture the Bruch's membrane, after which edema and neovascularization develop within the choroid. (Tobe T. et al. (1998) Am J Pathol 153:1641-1646). Optical coherence tomography (OCT) was then used 7-9 days post-lasering to quantify the area of laser lesion sites, as a measure of edema and neovascularization. Topical application of a 0.3% formulation of 5-[4-(5-hydroxymethyl-thiophen-2-yl)-quinolin-6- ylmethylene]-thiazolidine-2,4-dione (twice daily as a 10 μL eyedrop) reduced laser lesion area by 24% vs. vehicle-treated animals (P= 0.02):

Example 102: Cell data against for selected compounds against various cancer cell lines
Table 4a


Compound # PTEN deleted as identified in tumor lines
Table 1 Breast Glioblastoma Multiple Myeloma
BT549 U87 MG I Multiple lines
EC50, nM
502 1370
References
[0310] All publications and patents mentioned herein, including those items listed below, are hereby incorporated by reference in their entirety as if each individual publication or patent was specifically and individually incorporated by reference. In case of conflict, the present application, including any definitions herein, will control.
Equivalents
[0311] While specific embodiments of the subject invention have been discussed, the above specification is illustrative and not restrictive. Many variations of the invention will become apparent to those skilled in the art upon review of this specification. The full scope of the invention should be determined by reference to the claims, along with their full scope of equivalents, and the specification, along with such variations.
[0312] Unless otherwise indicated, all numbers expressing quantities of ingredients, reaction conditions, and so forth used in the specification and claims are to be understood as being modified in all instances by the term "about." Accordingly, unless indicated to the contrary, the numerical parameters set forth in this specification and attached claims are approximations that may vary depending upon the desired properties sought to be obtained by the present invention.
Claims
Claims 1. The compound represented by formula Ia or Ib:

Z is S or O;
X is S, O or NR12; R2 is H or alkyl;
R3 and R4 are each independently selected from the group consisting of H, halo, alkyl, alkynyl, alkenyl, hydroxyalkyl, alkoxy, carboxyl, cyano, N-arylamido, aryl, and heterocycle, or R2 and R3, or R3 and R4, form, together with the carbon atoms to which they are attached, a 5 or 6 membered carbocycle or heterocycle ring, wherein any of said alkyl, alkynyl, alkenyl, carbocycle, aryl, or heterocycle can be optionally substituted with one, two or three groups selected from the group consisting of: halo, cyano, hydroxyl, sulphamoyl, N-alkylsulphamoyl, alkoxy, ureido, alkyl, aryl, heterocycle each optionally substituted with one, two or three substituents selected from halo, cyano, hydroxyl, alkyl, or -alkyl-heterocycle optionally substituted by one, two, or three substituents selected from halo, cyano, hydroxyl, or alkyl; and R12 is selected from the group consisting of alkyl, aryl, heterocycle, or cycloalkyl, wherein R12 may be optionally substituted by one, two, or three substituents selected from the group consisting of halo, hydroxyl, nitro, formyl, formamido, alkyoxy, alkyl, carboxyl, cyano, amino, amido, carbamoyl, sulphamoyl, or ureido; and the pharmaceutically acceptable salts and N-oxides thereof. 2. The compound of claim 1, wherein R3 and R4 are each independently selected from H, - CH2OH, -CH2CH2OH, CH3, -CONH2, -CONH2, -C(O)NH-phenyl, phenyl, -alkynyl-phenyl, F, Cl, CO2H, — C(O)NCH2CH2-pyrrolidine, or dimethylisoxazole, wherein the phenyl is optionally substituted by one or two substitutents selected from the group consisting of halo, cyano, hydroxyl, alkyl, alkoxy, N-alkylsulphamoyl, sulphamoyl, 3. The compound of claim 1, wherein R3 and R4, taken together with the ring carbons on which they are attached, form a bicyclic ring chosen from: benzofuran, benzothiophene,
2,
3-dihydro- thieno[3,4b][l,4]dioxane, or 4,5,6,7-tetrahydrothieno[3,4-c]pyridine; wherein the bicyclic ring is optionally substituted by one or two substituents selected from the group consisting of: halo, hydroxyl, cyano, and alkyl.
4. The compound of claim 1, wherein X is NR12, and R12 is phenyl optionally substituted by one or two halogen moieties.
5. The compound of claim 1, wherein X is O.
6. The compound of claim 1, wherein Z is O.
7. The compound of claim 1, wherein Z is S.
8. The compound of claim 5, wherein R3 is phenyl optionally substituted by a substituent chosen from hydroxyl, alkyl, N-alkylsulphamoyl, alkoxy, or alkylmorpholino.
9. A compound represented by formula II:


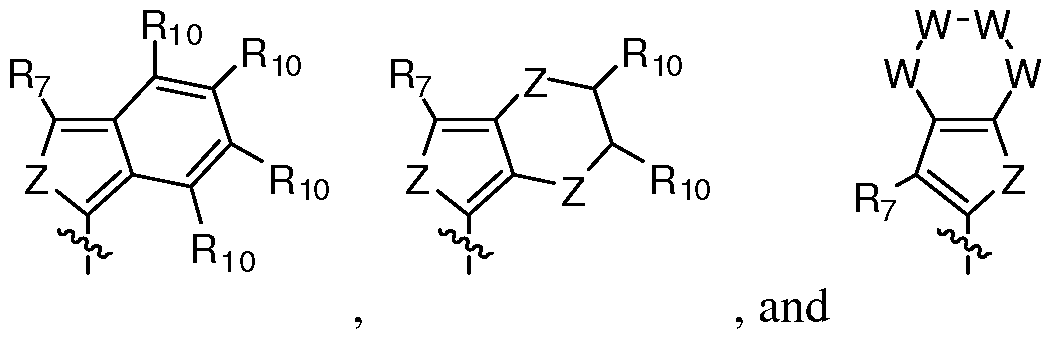
Z is, independently for each occurrence, S or O;
W is, independently for each occurrence, CRb, NH, N-alkyl, N-aryl, N-heterocycle, N-
C(O)-alkyl, N-C(O)-heterocycle, N-C(O)-N-alkyl, N-C(O)-N-heterocycle, O, or S;
R1 is H or alkyl;
R7 is H or alkyl;
Rg and R9 are each independently selected from H, Cl, F, alkyl, alkyne, alkenyl, carboxyl, hydroxyalkyl, hydroxyl, cyano, formyl, formamido, amido, amine,  sulphamoyl, ureido, R11, -R13R11, or m is an integer from 0 to 8; R1O is independently selected for each occurrence from the group consisting of H, Cl, F, hydroxyl, cyano, alkoxy,or alkyl; Rn is selected from group consisting of heterocycle or aryl, wherein R11 is optionally substituted at one to four substituents each independently selected from halo, hydroxyl, alkyl, alkoxy, cyano, sulphamoyl, N-alkylsulphamoyl, and alkyl-heterocycle optionally substituted by one or two substituents chosed from halo, alkyl, hydroxyl and cyano;
sulphamoyl, ureido, R11, -R13R11, or m is an integer from 0 to 8; R1O is independently selected for each occurrence from the group consisting of H, Cl, F, hydroxyl, cyano, alkoxy,or alkyl; Rn is selected from group consisting of heterocycle or aryl, wherein R11 is optionally substituted at one to four substituents each independently selected from halo, hydroxyl, alkyl, alkoxy, cyano, sulphamoyl, N-alkylsulphamoyl, and alkyl-heterocycle optionally substituted by one or two substituents chosed from halo, alkyl, hydroxyl and cyano;
R12 is selected from aryl or alkyl; R13 is chosen from alkylene, alkenylene, and alkynylene; and pharmaceutically acceptable salts, prodrugs, N-oxides, and hydrates thereof.
10. The compound of claim 9, wherein X is O.
11. The compound of claim 9, wherein m is 0.
12. The compound of claim 9, wherein m is an integer from 1 to 3, inclusive.
13. The compound claim of 12, wherein m is 2.
14. The compound of claim 9, wherein R1O at each occurrence is H.
15. The compound of claim 9, wherein one of R7 or Rg is H and one of R7 or Rg is 
; wherein R11 is chosen from: pyrrolindyl, phenyl, or isoxazole.
16. The compound of claim 15, wherein m is 0.
17. The compound of claim 9, wherein R11 is phenyl.
18. The compound claim 17, wherein R11 is substituted at one or two positions by methyl.
19. The compound of claim 9, wherein R7 is H.
20. The compound of claim 9, wherein R7 is methyl.
21. The compound of claim 9, wherein R8 is methyl.
22. The compound of claim 20, wherein R8 is H.
23. The compound of claim 19, wherein R8 is methyl.
24. The compound of claim 9, wherein R9 is chosen from: H, Cl, phenyl, CO2H, or 3,5- dimethyl-isoxazol-4-yl.
25. The compound of claim 9, wherein R13 is alkynylene.
26. A compound selected from: 5-(4-Thiophen-2-yl-quinolin-6-ylmethylene)-thiazolidine-2,4- dione; 5-(4-Benzo[b]thiophen-3-yl-quinolin-6-ylmethylene)-thiazolidine-2,4-dione; 5-[4-(5- Hydroxymethyl-thiophen-2-yl)-quinolin-6-ylmethylene]-thiazolidine-2,4-dione; 5-(4-Furan-2- yl-quinolin-6-ylmethylene)-thiazolidine-2,4-dione; 5-(4-Benzofuran-2-yl-quinolin-6- ylmethylene)-thiazolidine-2,4-dione; 5-[4-(5-Chloro-thiophen-2-yl)-quinolin-6-ylmethylene]- thiazolidine-2,4-dione; 5-[4-(5-Phenyl-thiophen-2-yl)-quinolin-6-ylmethylene]-thiazolidine- 2,4-dione; 5-{4-[5-(3,5-Dimethyl-isoxazol-4-yl)-thiophen-2-yl]-quinolin-6-ylmethylene}- thiazolidine-2,4-dione; 5-[4-(4-Methyl-thiophen-2-yl)-quinolin-6-ylmethylene]-thiazolidine- 2,4-dione; 5-[6-(2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4-yl]-thiophene-2- carboxylic acid; 5-[6-(2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4-yl]-thiophene-2- carboxylic acid (2-pyrrolidin-l-yl-ethyl)-amide; 5-[4-(3-Methyl-thiophen-2-yl)-quinolin-6- ylmethylene]-thiazolidine-2,4-dione; 5-(4-Benzo[b]thiophen-2-yl-quinolin-6-ylmethylene)- thiazolidine-2,4-dione; 5-[4-(2,3-Dihydro-thieno[3,4-b][l,4]dioxin-5-yl)-quinolin-6- ylmethylene]-thiazolidine-2,4-dione; 5-[6-(2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4- yl]-thiophene-2-carboxylic acid amide; 5-(4-Furan-2-yl-quinazolin-6-ylmethylene)- thiazolidine-2,4-dione; 2-[6-(2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4-yl]- benzo[b]thiophene-6-carbonitrile; N-tert-Butyl-3-{5-[6-(2,4-dioxo-thiazolidin-5- ylidenemethyl)-quinolin-4-yl]-thiophen-2-yl}-benzenesulfonamide; 5-{4-[5-(3-Isopropyl- phenyl)-thiophen-2-yl]-quinolin-6-ylmethylene}-thiazolidine-2,4-dione; 5-{4-[5-(3- Isopropoxy-phenyl)-thiophen-2-yl]-quinolin-6-ylmethylene}-thiazolidine-2,4-dione; 5- {4- [5- (2,6-Dimethoxy-phenyl)-thiophen-2-yl]-quinolin-6-ylmethylene}-thiazolidine-2,4-dione; 2-[6- (2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4-yl]-benzo[b]thiophene-4-carbonitrile; 5- [4-(4-Methyl-thiophen-3-yl)-quinolin-6-ylmethylene]-thiazolidine-2,4-dione; 5-[6-(2,4-Dioxo- thiazolidin-5-ylidenemethyl)-quinolin-4-yl]-thiophene-2-carboxylic acid (3-methoxy-phenyl)- amide; 5-[6-(2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4-yl]-thiophene-2-carboxylic acid (3-hydroxy-phenyl)-amide; 5-[6-(2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4-yl]- thiophene-2-carboxylic acid (4-hydroxy-phenyl)-amide; 5-[6-(2,4-Dioxo-thiazolidin-5- ylidenemethyl)-quinolin-4-yl] -thiophene-2-carboxylic acid (4-morpholin-4-ylmethyl-phenyl)- amide; 5-[4-(4,5,6,7-Tetrahydro-thieno[3,2-c]pyridin-2-yl)-quinolin-6-ylmethylene]- thiazolidine-2,4-dione; 5-[4-(4,5,6,7-Tetrahydro-thieno[3,2-c]pyridin-2-yl)-quinazolin-6- ylmethylene] thiazolidine-2,4-dione; 2-[6-(2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4- yl]-benzo[b]thiophene-6-carbonitrile; 2-[6-(2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4- yl]-benzo[b]thiophene-4-carbonitrile; 2-[6-(2,4-Dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4- yl]-benzo[b]thiophene-7-carbonitrile; (Z)-2-(6-((2,4-Dioxothiazolidin-5- ylidene)methyl)quinolin-4-yl)-3-methylbenzo[b]thiophene-6-carbonitrile; (Z)-2-(6-((2,4- Dioxothiazolidin-5-ylidene)methyl)quinolin-4-yl)-3-methylbenzo[b]thiophene-4-carbonitrile; N-fer?-Butyl-3-{5-[6-(2,4-dioxo-thiazolidin-5-ylidenemethyl)-quinolin-4-yl]-thiophen-2-yl}- benzenesulfonamide; (Z)-5-((4-(4-Methylthiophen-3-yl)quinolin-6-yl)methylene)thiazolidine- 2,4-dione; (Z)-5-((4-(4,5,6,7-Tetrahydrothieno[J,2-c]pyridin-2-yl)quinolin-6-yl)methylene) thiazolidine-2,4-dione; 3-Methyl-5-(6-((Z)-(2,4-dioxothiazolidin-5-ylidene)methyl)quinolin-4- yl)thiophene-2-carbonitrile; (Z)-5-((4-(5-(3-Isopropylphenyl)thiophen-2-yl)quinolin-6- yl)methylene)thiazolidine-2,4-dione; (Z)-5-((4-(5-(3-Isopropoxyphenyl)thiophen-2-yl)quinolin- 6-yl)methylene)thiazolidine-2,4-dione; (Z)-5-((4-(5-(2,6-Dimethoxyphenyl)thiophen-2- yl)quinolin-6-yl)methylene)thiazolidine-2,4-dione; (Z)-5-(6-((2,4-Dioxothiazolidin-5- ylidene)methyl)quinolin-4-yl)-N-(3-hydroxyphenyl)thiophene-2-carboxamide; (Z)-5-(6-((2,4- Dioxothiazolidin-5-ylidene)methyl)quinolin-4-yl)-N-(4-hydroxyphenyl)thiophene-2- carboxamide; (Z)-5-(6-((2,4-Dioxothiazolidin-5-ylidene)methyl)quinolin-4-yl)-N-(4- (morpholinomethyl)phenyl)thiophene-2-carboxamide; (Z)-5-(6-((2,4-Dioxothiazolidin-5- ylidene)methyl)quinolin-4-yl)-N-(3-(moφholinomethyl)phenyl)thiophene-2-carboxamide; (Z)- 5-((5-(6-((Z)-(2,4-Dioxothiazolidin-5-ylidene)methyl)quinolin-4-yl)thiophen-2- yl)methylene)thiazolidine-2,4-dione; (Z)-5-((4-(5-(3-Methoxyphenyl)thiophen-2-yl)quinolin-6- yl)methylene)thiazolidine-2,4-dione; 5-{4-[5-(3-Hydroxy-phenylethynyl)-thiophen-2-yl]- quinolin-6-ylmethylene}-thiazolidine-2,4-dione; 2-(2-Chloro-5-fluoro-phenylimino)-5-(4- thiophen-2-yl-quinolin-6-ylmethylene)-thiazolidin-4-one and 2-Phenylimino-5-(4-thiophen-2- yl-quinolin-6-ylmethylene)-thiazolidin-4-one and pharmaceutically acceptable salts, prodrugs, N-oxides, and hydrates thereof.
27 '. A composition comprising a compound of claim 1, and a pharmaceutically acceptable carrier.
28. A composition comprising a compound of claim 9, and a pharmaceutically acceptable carrier.
29. The composition of claim 28, wherein the composition is formulated for one of: oral administration, intraveneous administration, injectable administration, topical application, as a suppository, inhalation administration, or systemic administration.
30. The composition of claim 29 wherein the composition is formulated for use in the eye.
31. The composition of claim 29 wherein the composition is formulated as an eye drop.
32. A method of treating an respiratory or ocular disorder, comprising administering to a patient in need thereof an effective amount of a compound of claim 9.
33. A method of inhibiting tumor cell growth, tumor cell proliferation, or tumorigenesis comprising administering to a patient in need thereof an effective amount of a compound of claim 9.
34. A method of treating pain, diabetes, inflammation, platelet aggregation, ischemic heart disease, sclerosis, restenosis, immune-mediated disease, rheumatoid arthritis, HIV, bone resorption, cancer, non-small cell lung cancer, or brain cancer, comprising administering to a patient in need thereof an effective amount of a compound of claim 9.
35. The method of claim 34, wherein the cancer is breast or prostate cancer, or multiple myeloma.
Applications Claiming Priority (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US95683107P | 2007-08-20 | 2007-08-20 | |
| US60/956,831 | 2007-08-20 | ||
| US97714707P | 2007-10-03 | 2007-10-03 | |
| US60/977,147 | 2007-10-03 | ||
| US2078608P | 2008-01-14 | 2008-01-14 | |
| US61/020,786 | 2008-01-14 | ||
| US2221708P | 2008-01-18 | 2008-01-18 | |
| US61/022,217 | 2008-01-18 | ||
| US5142408P | 2008-05-08 | 2008-05-08 | |
| US61/051,424 | 2008-05-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009026345A1 true WO2009026345A1 (en) | 2009-02-26 |
Family
ID=40378610
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/073684 WO2009026346A1 (en) | 2007-08-20 | 2008-08-20 | Thiazolidine compounds, and methods of making and using same |
| PCT/US2008/073683 WO2009026345A1 (en) | 2007-08-20 | 2008-08-20 | Thiazolidinone compounds, and methods of making and using same |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/073684 WO2009026346A1 (en) | 2007-08-20 | 2008-08-20 | Thiazolidine compounds, and methods of making and using same |
Country Status (1)
| Country | Link |
|---|---|
| WO (2) | WO2009026346A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| US8372971B2 (en) | 2004-08-25 | 2013-02-12 | Targegen, Inc. | Heterocyclic compounds and methods of use |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8481536B2 (en) | 2004-04-08 | 2013-07-09 | Targegen, Inc. | Benzotriazine inhibitors of kinases |
| WO2014068070A1 (en) | 2012-10-31 | 2014-05-08 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for preventing antiphospholipid syndrome (aps) |
| WO2016059220A1 (en) | 2014-10-16 | 2016-04-21 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Tcr-activating agents for use in the treatment of t-all |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2766751T3 (en) | 2012-04-20 | 2020-06-15 | Gb006 Inc | Compositions for the regulation of integrins |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040092561A1 (en) * | 2002-11-07 | 2004-05-13 | Thomas Ruckle | Azolidinone-vinyl fused -benzene derivatives |
| US20040138199A1 (en) * | 2002-12-20 | 2004-07-15 | Gogliotti Rocco Dean | Benzoxazines and derivatives thereof as therapeutic agents |
| WO2007030360A2 (en) * | 2005-09-07 | 2007-03-15 | Laboratoires Serono S.A. | P13k inhibitors for the treatment of endometriosis |
-
2008
- 2008-08-20 WO PCT/US2008/073684 patent/WO2009026346A1/en active Application Filing
- 2008-08-20 WO PCT/US2008/073683 patent/WO2009026345A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040092561A1 (en) * | 2002-11-07 | 2004-05-13 | Thomas Ruckle | Azolidinone-vinyl fused -benzene derivatives |
| US20040138199A1 (en) * | 2002-12-20 | 2004-07-15 | Gogliotti Rocco Dean | Benzoxazines and derivatives thereof as therapeutic agents |
| WO2007030360A2 (en) * | 2005-09-07 | 2007-03-15 | Laboratoires Serono S.A. | P13k inhibitors for the treatment of endometriosis |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8481536B2 (en) | 2004-04-08 | 2013-07-09 | Targegen, Inc. | Benzotriazine inhibitors of kinases |
| US8372971B2 (en) | 2004-08-25 | 2013-02-12 | Targegen, Inc. | Heterocyclic compounds and methods of use |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014068070A1 (en) | 2012-10-31 | 2014-05-08 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for preventing antiphospholipid syndrome (aps) |
| WO2016059220A1 (en) | 2014-10-16 | 2016-04-21 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Tcr-activating agents for use in the treatment of t-all |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009026346A1 (en) | 2009-02-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009026345A1 (en) | Thiazolidinone compounds, and methods of making and using same | |
| US11773110B2 (en) | Heterocycle amines and uses thereof | |
| WO2009046416A1 (en) | Anilinopyrimidines as jak kinase inhibitors | |
| EP1578745B1 (en) | BENZOFURAN DERIVATivES, PROCESS FOR THEIR PREPARATION AND INTERMEDIATES THEREOF | |
| ES2338234T3 (en) | USEFUL COMPOSITIONS AS INHIBITORS OF KINASE PROTEINS. | |
| US8796268B2 (en) | Heteroaryls and uses thereof | |
| AU2006212761B2 (en) | Combination therapy | |
| US20040038992A1 (en) | Inhibitors of Jak protein kinase | |
| WO2009049028A1 (en) | Pyrrolopyrimidine compounds and their use as janus kinase modulators | |
| US20040214868A1 (en) | Amino nicotinate derivatives as glucokinase (GLK) modulators | |
| CN111116492B (en) | Deuterated benzylaminopyrimidinedione derivative and application thereof | |
| CA2832865C (en) | Aminopyrimidine kinase inhibitors | |
| KR101990605B1 (en) | Aminopyrimidine kinase inhibitors | |
| CN102574857A (en) | Heterocyclic compounds as jak receptor and protein tyrosine kinase inhibitors | |
| KR20170095814A (en) | Triazolopyridine compounds and methods for the treatment of cystic fibrosis | |
| JP2010533736A5 (en) | ||
| MX2012007191A (en) | Aminopyrimidine kinase inhibitors. | |
| CN103619841A (en) | Heteroaryl compounds and methods of use thereof | |
| AU2006258461A1 (en) | Thienopyrimidine derivative | |
| JP2010522238A (en) | Indole carboxamides as IKK2 inhibitors | |
| WO2016009297A1 (en) | Pyridine derivatives as muscarinic m1 receptor positive allosteric modulators | |
| KR20130083389A (en) | Heterocyclic compounds as janus kinase inhibitors | |
| CN108558833B (en) | Pyrazole alcohol compound, pharmaceutical composition thereof and application thereof in medicines | |
| AU2021296087B2 (en) | Heterocyclic compound as inhibitor for casein kinase 1δ and/or activin receptor-like kinase 5 | |
| WO2022000031A1 (en) | Novel compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08798247 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08798247 Country of ref document: EP Kind code of ref document: A1 |