WO2008156614A2 - Imidazopyrazines as protein kinase inhibitors - Google Patents
Imidazopyrazines as protein kinase inhibitors Download PDFInfo
- Publication number
- WO2008156614A2 WO2008156614A2 PCT/US2008/007295 US2008007295W WO2008156614A2 WO 2008156614 A2 WO2008156614 A2 WO 2008156614A2 US 2008007295 W US2008007295 W US 2008007295W WO 2008156614 A2 WO2008156614 A2 WO 2008156614A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- heteroaryl
- aryl
- mmol
- Prior art date
Links
- 0 *c1cnc2[n]1c(*)c(*)nc2N Chemical compound *c1cnc2[n]1c(*)c(*)nc2N 0.000 description 14
- ITPQCPXVHIFHIJ-UHFFFAOYSA-N Cc(nc1Nc2ccc(CN3CCOCC3)cc2)c[n]2c1ncc2-c1c[nH]nc1 Chemical compound Cc(nc1Nc2ccc(CN3CCOCC3)cc2)c[n]2c1ncc2-c1c[nH]nc1 ITPQCPXVHIFHIJ-UHFFFAOYSA-N 0.000 description 2
- YNNHPZRYZQVGRZ-UHFFFAOYSA-N CC(C)(C)CN(C)C(C)(C)C Chemical compound CC(C)(C)CN(C)C(C)(C)C YNNHPZRYZQVGRZ-UHFFFAOYSA-N 0.000 description 1
- GXAWOTJEXZEKFS-UHFFFAOYSA-N CC(C)(C)CN(CC1)CCC1c1ccccc1 Chemical compound CC(C)(C)CN(CC1)CCC1c1ccccc1 GXAWOTJEXZEKFS-UHFFFAOYSA-N 0.000 description 1
- XJBZWNPYLJZWJD-UHFFFAOYSA-N CC(C)CSCCN(C)C Chemical compound CC(C)CSCCN(C)C XJBZWNPYLJZWJD-UHFFFAOYSA-N 0.000 description 1
- NFZFKDVLQOZJPU-UHFFFAOYSA-N CCCOC[n]1ncc(-c2cnc3[n]2cc(C)nc3S(C)(=O)=O)c1 Chemical compound CCCOC[n]1ncc(-c2cnc3[n]2cc(C)nc3S(C)(=O)=O)c1 NFZFKDVLQOZJPU-UHFFFAOYSA-N 0.000 description 1
- UTLDDSNRFHWERZ-UHFFFAOYSA-N CCN(C)C(C)C Chemical compound CCN(C)C(C)C UTLDDSNRFHWERZ-UHFFFAOYSA-N 0.000 description 1
- IPQQFSYETGKMPX-UHFFFAOYSA-N CCN(CC)CCCOCC Chemical compound CCN(CC)CCCOCC IPQQFSYETGKMPX-UHFFFAOYSA-N 0.000 description 1
- UXXLSONRDDIOIL-UHFFFAOYSA-N CCN(CCN(C)C)CCO Chemical compound CCN(CCN(C)C)CCO UXXLSONRDDIOIL-UHFFFAOYSA-N 0.000 description 1
- LPCWDBCEHWHJGX-UHFFFAOYSA-N CCN1C(C)CCCC1 Chemical compound CCN1C(C)CCCC1 LPCWDBCEHWHJGX-UHFFFAOYSA-N 0.000 description 1
- DDPRYTUJYNYJKV-UHFFFAOYSA-N CCN1CCN(CC)CC1 Chemical compound CCN1CCN(CC)CC1 DDPRYTUJYNYJKV-UHFFFAOYSA-N 0.000 description 1
- JWOICHXTXAOTOY-UHFFFAOYSA-N CCS(CCN(C)C)=O Chemical compound CCS(CCN(C)C)=O JWOICHXTXAOTOY-UHFFFAOYSA-N 0.000 description 1
- GFERXXPZQYBEBF-UHFFFAOYSA-N COC(c1c[s]c(N)c1)=O Chemical compound COC(c1c[s]c(N)c1)=O GFERXXPZQYBEBF-UHFFFAOYSA-N 0.000 description 1
- YRGYIZCDBMJMGM-UHFFFAOYSA-N COC(c1n[s]c(N)c1)=O Chemical compound COC(c1n[s]c(N)c1)=O YRGYIZCDBMJMGM-UHFFFAOYSA-N 0.000 description 1
- UMNYDDNJQVGZIS-UHFFFAOYSA-O C[SH+](C)(C)CCOC[n]1ncc(-c2cnc(c(S(C)(=O)=O)n3)[n]2cc3Br)c1 Chemical compound C[SH+](C)(C)CCOC[n]1ncc(-c2cnc(c(S(C)(=O)=O)n3)[n]2cc3Br)c1 UMNYDDNJQVGZIS-UHFFFAOYSA-O 0.000 description 1
- NRTWXBXJSGGTTE-UHFFFAOYSA-N Cc(cc(cc1)N)c1C(OC)=O Chemical compound Cc(cc(cc1)N)c1C(OC)=O NRTWXBXJSGGTTE-UHFFFAOYSA-N 0.000 description 1
- JBOYPHNLYZXVGX-UHFFFAOYSA-N Cc(cc(cc1)Nc2nc(C)c[n]3c2ncc3-c2c[n](COCC[Si](C)(C)C)nc2)c1C(OC)=O Chemical compound Cc(cc(cc1)Nc2nc(C)c[n]3c2ncc3-c2c[n](COCC[Si](C)(C)C)nc2)c1C(OC)=O JBOYPHNLYZXVGX-UHFFFAOYSA-N 0.000 description 1
- IHPUUEXGWJBNDP-UHFFFAOYSA-N Cc1cc(Nc2nc(Br)c[n]3c2ncc3-c2c[n](COCC[Si](C)(C)C)nc2)ccc1C(OC)=O Chemical compound Cc1cc(Nc2nc(Br)c[n]3c2ncc3-c2c[n](COCC[Si](C)(C)C)nc2)ccc1C(OC)=O IHPUUEXGWJBNDP-UHFFFAOYSA-N 0.000 description 1
- WNYFVEFUHMDIRQ-UHFFFAOYSA-N Nc1ccc(CN2CCOCC2)cc1 Chemical compound Nc1ccc(CN2CCOCC2)cc1 WNYFVEFUHMDIRQ-UHFFFAOYSA-N 0.000 description 1
- KBXWFEFORBGBLA-UHFFFAOYSA-N Nc1cnc(CN2CCCCC2)nc1 Chemical compound Nc1cnc(CN2CCCCC2)nc1 KBXWFEFORBGBLA-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to imidazo[1 ,2-a]pyrazine compounds useful as protein kinase inhibitors, regulators or modulators, pharmaceutical compositions containing the compounds, and methods of treatment using the compounds and compositions to treat diseases such as, for example, cancer, inflammation, arthritis, viral diseases, neurodegenerative diseases such as Alzheimer's disease, cardiovascular diseases, and fungal diseases.
- the present compounds are especially useful as Aurora kinase inhibitors.
- Protein kinases are a family of enzymes that catalyze phosphorylation of proteins, in particular the hydroxyl group of specific tyrosine, serine, or threonine residues in proteins. Protein kinases are pivotal in the regulation of a wide variety of cellular processes, including metabolism, cell proliferation, cell differentiation, and cell survival. Uncontrolled proliferation is a hallmark of cancer cells, and can be manifested by a deregulation of the cell division cycle in one of two ways - making stimulatory genes hyperactive or inhibitory genes inactive.
- Protein kinase inhibitors, regulators or modulators alter the function of kinases such as cyclin-dependent kinases (CDKs), mitogen activated protein kinase (MAPK/ERK), glycogen synthase kinase 3 (GSK3beta), Checkpoint (Chk) (e.g., CHK-1 , CHK-2 etc.) kinases, AKT kinases, JNK, and the like.
- CDKs cyclin-dependent kinases
- MAPK/ERK mitogen activated protein kinase
- GSK3beta glycogen synthase kinase 3
- Checkpoint Chk
- Examples of protein kinase inhibitors are described in WO02/22610 A1 and by Y. Mettey et al in J. Med. Chem., (2003) 46 222-236.
- the cyclin-dependent kinases are serine/threonine protein kinases, which are the driving force behind the cell cycle and cell proliferation. Misregulation of CDK function occurs with high frequency in many important solid tumors. Individual CDK's, such as, CDK1 , CDK2, CDK3, CDK4, CDK5, CDK6 and CDK7, CDK8 and the like, perform distinct roles in cell cycle progression and can be classified as either G1 , S, or G2M phase enzymes. CDK2 and CDK4 are of particular interest because their activities are frequently misregulated in a wide variety of human cancers. CDK2 activity is required for progression through G1 to the S phase of the cell cycle, and CDK2 is one of the key components of the G1 checkpoint.
- CDK2 pathway influences tumorgenesis at the level of tumor suppressor function (e.g. p52, RB, and p27) and oncogene activation (cyclin E).
- tumor suppressor function e.g. p52, RB, and p27
- cyclin E oncogene activation
- Many reports have demonstrated that both the coactivator, cyclin E, and the inhibitor, p27, of CDK2 are either over- or underexpressed, respectively, in breast, colon, nonsmall cell lung, gastric, prostate, bladder, non-Hodgkin's lymphoma, ovarian, and other cancers. Their altered expression has been shown to correlate with increased CDK2 activity levels and poor overall survival. This observation makes CDK2 and its regulatory pathways compelling targets for the development of cancer treatments.
- adenosine 5'-triphosphate (ATP) competitive small organic molecules as well as peptides have been reported in the literature as CDK inhibitors for the potential treatment of cancers.
- U.S. 6,413,974, col. 1 , line 23- col. 15, line 10 offers a good description of the various CDKs and their relationship to various types of cancer.
- Flavopiridol (shown below) is a nonselective CDK inhibitor that is currently undergoing human clinical trials, A. M. Senderowicz et al, J. Clin. Oncol. (1998) 16, 2986-2999.
- CDK inhibitors include, for example, olomoucine (J. Vesely et al, Eur. J. Biochem., (1994) 224, 771-786) and roscovitine (I. Meijer et al, Eur. J. Biochem., (1997) 243, 527-536).
- U.S. 6,107,305 describes certain pyrazolo[3,4-b] pyridine compounds as CDK inhibitors.
- An illustrative compound from the '305 patent is:
- Imidazopyrazines are known.
- U.S. 6,919,341 (the disclosure of which is incorporated herein by reference) and US2005/0009832 disclose various imidazopyrazines. Also being mentioned are the following: WO2005/047290;
- imidazopyrazines as protein kinase inhibitors of the following structure:
- R1 is H, halo, aryl or heteroaryl, wherein each of said aryl and heteroaryl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl, heterocyclyl, - CH2OR5, -C(O)NR5R6, -C(O)OH, -C(O)NH2, -NR5R6 (wherein the R5 and R6, together with the N of said -NR5R6, form a heterocyclyl ring), -S(O)R5, -S(O2)R5, -CN, -CHO, -SR5,
- R2 is H, halo, aryl, arylalkyl or heteroaryl, wherein each of said aryl, arylalkyl and heteroaryl can be unsubstituted or optionally independently be substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, amide, alkyl, alkenyl, alkynyl, cycloalkyl, aryl,
- R3 is H, alkyl, cycloalkyl, heterocyclyl, aryl or heteroaryl, wherein:
- - said alkyl shown above for R3 can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of -OR5, alkoxy, heteroaryl, and -NR5R6;
- aryl shown above for R3 is unsubstituted, or optionally substituted, or optionally fused, with halo, heteroaryl, heterocyclyl, cycloalkyl or heteroarylalkyl, wherein each of said heteroaryl, heterocyclyl, cycloalkyl and heteroarylalkyl can be unsubstituted or optionally independently substituted with one or more moieties which can be the same or different each moiety being independently selected from alkyl, -OR5, -N(R5R6) and -S(O2)R5; and
- heteroaryl shown above for R3 can be unsubstituted or optionally substituted, or optionally fused, with one or more moieties which can be the same or different with each moiety being independently selected from the group consisting of halo, amino, alkoxycarbonyl, -OR5, alkyl, - CHO, - NR5R6, -S(O2)N(R5R6),
- R5 is H, alkyl, aminoalkyl, aryl, heteroaryl, heterocyclyl or cycloalkyl
- R6 is H, alkyl, aryl, arylalkyl, heteroaryl, heterocyclyl or cycloalkyl; further wherein in any -NR5R6 in Formula I, said R5 and R6 can optionally be joined together with the N of said -NR5R6 to form a cyclic ring.
- Another series of protein kinases are those that play an important role as a checkpoint in cell cycle progression.
- Checkpoints prevent cell cycle progression at inappropriate times, such as in response to DNA damage, and maintain the metabolic balance of cells while the cell is arrested, and in some instances can induce apoptosis (programmed cell death) when the requirements of the checkpoint have not been met.
- Checkpoint control can occur in the G1 phase (prior to DNA synthesis) and in G2, prior to entry into mitosis.
- Tyrosine kinases can be of the receptor type (having extracellular, transmembrane and intracellular domains) or the non-receptor type (being wholly intracellular).
- Receptor-type tyrosine kinases are comprised of a large number of transmembrane receptors with diverse biological activity. In fact, about 20 different subfamilies of receptor-type tyrosine kinases have been identified.
- One tyrosine kinase subfamily, designated the HER subfamily is comprised of EGFR (HER1 ), HER2, HER3 and HER4.
- Ligands of this subfamily of receptors identified so far include epithelial growth factor, TGF-alpha, amphiregulin, HB-EGF, betacellulin and heregulin.
- Another subfamily of these receptor-type tyrosine kinases is the insulin subfamily, which includes INS-R, IGF-IR, IR, and IR-R.
- the PDGF subfamily includes the PDGF-alpha and beta receptors, CSFIR, c-kit and FLK-II.
- the FLK family is comprised of the kinase insert domain receptor (KDR), fetal liver kinase-1(FLK-1 ), fetal liver kinase-4 (FLK-4) and the fms-like tyrosine kinase-1 (flt-1 ).
- KDR kinase insert domain receptor
- FLK-1 fetal liver kinase-1
- FLK-4 fetal liver kinase-4
- flt-1 fms-like tyrosine kinase-1
- At least one of the non-receptor protein tyrosine kinases is believed to mediate the transduction in T-cells of a signal from the interaction of a cell- surface protein (Cd4) with a cross-linked anti-Cd4 antibody.
- Cd4 cell- surface protein
- the non-receptor type of tyrosine kinases is also comprised of numerous subfamilies, including Src, Frk, Btk, Csk, AbI1 Zap70, Fes/Fps, Fak, Jak, Ack, and LIMK.
- Src subfamily is one of the largest and includes Src, Yes, Fyn, Lyn, Lck, BIk, Hck, Fgr, and Yrk.
- Src subfamily of enzymes has been linked to oncogenesis.
- angiogenesis is the mechanism by which new capillaries are formed from existing vessels.
- the vascular system has the potential to generate new capillary networks in order to maintain the proper functioning of tissues and organs.
- angiogenesis is fairly limited, occurring only in the process of wound healing and neovascularization of the endometrium during menstruation.
- unwanted angiogenesis is a hallmark of several diseases, such as retinopathies, psoriasis, rheumatoid arthritis, age-related macular degeneration, and cancer (solid tumors).
- Protein kinases which have been shown to be involved in the angiogenic process include three members of the growth factor receptor tyrosine kinase family; VEGF-R2 (vascular endothelial growth factor receptor 2, also known as KDR (kinase insert domain receptor) and as FLK 1 ); FGF-R (fibroblast growth factor receptor); and TEK (also known as Tie-2).
- VEGF-R2 vascular endothelial growth factor receptor 2, also known as KDR (kinase insert domain receptor) and as FLK 1
- FGF-R fibroblast growth factor receptor
- TEK also known as Tie-2
- VEGF-R2 which is expressed only on endothelial cells, binds the potent angiogenic growth factor VEGF and mediates the subsequent signal transduction through activation of its intracellular kinase activity.
- VEGF-R2 direct inhibition of the kinase activity of VEGF-R2 will result in the reduction of angiogenesis even in the presence of exogenous VEGF (see Strawn et al, Cancer Research, 56, 3540-3545 (1996)), as has been shown with mutants of VEGF-R2 which fail to mediate signal transduction. Millauer et al, Cancer Research, 56, 1615-1620 (1996).
- VEGF-R2 appears to have no function in the adult beyond that of mediating the angiogenic activity of VEGF. Therefore, a selective inhibitor of the kinase activity of VEGF-R2 would be expected to exhibit little toxicity.
- FGFR binds the angiogenic growth factors aFGF and bFGF and mediates subsequent intracellular signal transduction. Recently, it has been suggested that growth factors such as bFGF may play a critical role in inducing angiogenesis in solid tumors that have reached a certain size. Yoshiji et al., Cancer Research, 57, 3924-3928 (1997).
- FGF-R is expressed in a number of different cell types throughout the body and may or may not play important roles in other normal physiological processes in the adult. Nonetheless, systemic administration of a small molecule inhibitor of the kinase activity of FGF-R has been reported to block bFGF-induced angiogenesis in mice without apparent toxicity. Mohammad et al., EMBO Journal, 17, 5996-5904 (1998).
- TEK also known as Tie-2
- Tie-2 is another receptor tyrosine kinase expressed only on endothelial cells which has been shown to play a role in angiogenesis.
- the binding of the factor angiopoietin-1 results in autophosphorylation of the kinase domain of TEK and results in a signal transduction process which appears to mediate the interaction of endothelial cells with peri-endothelial support cells, thereby facilitating the maturation of newly formed blood vessels.
- the factor angiopoietin-2 appears to antagonize the action of angiopoietin-1 on TEK and disrupts angiogenesis. Maisonpierre et al., Science, 277, 55-60 (1997).
- the kinase, JNK belongs to the mitogen-activated protein kinase (MAPK) superfamily.
- JNK plays a crucial role in inflammatory responses, stress responses, cell proliferation, apoptosis, and tumorigenesis.
- JNK kinase activity can be activated by various stimuli, including the proinflammatory cytokines (TNF-alpha and interleukin- 1 ), lymphocyte costimulatory receptors (CD28 and CD40), DNA-damaging chemicals, radiation, and Fas signaling.
- results from the JNK knockout mice indicate that JNK is involved in apoptosis induction and T helper cell differentiation.
- Pim-1 is a small serine/threonine kinase.
- Pim-1 acts as a cell survival factor and may prevent apoptosis in malignant cells.
- Aurora kinases are serine/threonine protein kinases that have been implicated in human cancer, such as colon, breast and other solid tumors.
- Aurora-A also sometimes referred to as AIK
- Aurora-A is believed to be involved in protein phosphorylation events that regulate the cell cycle.
- Aurora-A may play a role in controlling the accurate segregation of chromosomes during mitosis. Misregulation of the cell cycle can lead to cellular proliferation and other abnormalities.
- Aurora-A, Aurora-B, Aurora-C have been found to be over-expressed (see, Bischoff et al., EMBO J., 17:3052-3065 (1998); Schumacher et al., J. Cell Biol. 143:1635-1646 (1998); Kimura et al., J. Biol. Chem., 272:13766-13771 (1997)).
- kinase inhibitors especially small- molecule compounds that may be readily synthesized.
- the present invention provides a novel class of imidazo[1 ,2-a]pyrazine compounds, methods of preparing such compounds, pharmaceutical compositions comprising one or more such compounds, methods of preparing pharmaceutical formulations comprising one or more such compounds, and methods of treatment, prevention, inhibition or amelioration of one or more diseases associated with protein kinases using such compounds or pharmaceutical compositions.
- the present invention provides compounds represented by Formula I:
- R is H, CN, -NR5R6, cycloalkyl, cycloalkenyl, heterocyclenyl, heteroaryl, -C(O)NR5R6, -N(R5)C(O)R6, heterocyclyl, heteroaryl substituted with (CH2)i.3
- NR5R6 unsubstituted alkyl, or alkyl substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of -OR5, heterocyclyl,
- R1 is H, halo, aryl or heteroaryl, wherein each of said aryl and heteroaryl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl, heterocyclyl, - CH2OR5, -C(O)NR5R6, -C(O)OH, -C(O)NH2, -NR5R6 (wherein the R5 and R6, together with the N of said
- R2 is H, halo, aryl, arylalkyl or heteroaryl, wherein each of said aryl, arylalkyl and heteroaryl can be unsubstituted or optionally independently be substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, amide, alkyl, alkenyl, alkynyl, cycloalkyl, aryl,
- R3 is heterocyclyl-(CR7R8)n-X, heterocyclenyl-(CR7R8)n-X, heteroaryl-(CR7R8)n-X or aryl-(CR7R8)n-X wherein each of the heterocyclyl-, heterocyclenyl-, heteroaryl- or aryl- moieties of said R3 can be unsubstituted or substituted with one or more moieties, independently selected from the group consisting Of -CONR5R6,
- n 1-6
- X is selected from the group consisting of -NR5R6, -OR5, -SO-R5, -SR5, SO2R5, heteroaryl, heterocyclyl and aryl, wherein said heteroaryl or aryl can be unsubstituted or substituted with one or more moieties, independently selected from the group consisting of -O-alkyl, alkyl, halo, or NR5R6;
- R7 and R8 are each independently hydrogen, alkyl, heterocyclyl, aryl, heteroaryl or cycloalkyl;
- R5 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkoxyalkyl,
- -alkyl-S-alkyl aminoalkyl, aryl, heteroaryl, heterocyclenyl, heterocycloalkyl, cycloalkyl, cyclenyl, heterocyclylalkoxyl, -S- alkylheterocyclyl, heterocyclyl, heterocyclenyl, alkylN(alkyl)2, alkylNH(alkyl), alkylN(alkenyl)2, -alkylN(alkoxyl)2, -alkyl-SH, hydroxyalkyl, trihaloalkyl, dihaloalkyl, monohaloalkyl, wherein each of said alkyl, alkenyl, alkoxyalkyl, -alkyl-S-alkyl, aminoalkyl, aryl, heteroaryl, heterocyclenyl, heterocycloalkyl, cycloalkyl, cyclenyl, heterocyclylalkoxyl, -S-alkylheterocyclyl
- R6 is selected from the group consisting of hydrogen, alkyl, alkenyl, aryl, cyclenyl, cycloalkyl, arylalkyl, cyclenylalkyl, cycloalkylalkyl, heteroaryl, heterocyclenyl, heterocyclyl, heteroarylalkyl, heterocyclenylalkyl, heterocycloalkylalkyl, alkoxyalkyl, -alkyl-S-alkyl, -alkylSH, alkoxyl, -S- alkyl, hydroxyalkyl, aminoalkyl, -alkyl-OC(O)alkyl, -alkylOC(O)cycloalkyl, -alkylOC(O)aryl, -alkylOC(O)aralkyl, -alkylOC(O)NR5aryl, - alkylOC(O)NR5alkyl, -alkylOC(O)NR5heterocyclyl
- the compounds of Formula I can be useful as protein kinase inhibitors.
- the compounds of Formula I can also be useful as Aurora kinase inhibitors.
- the compounds of Formula I can be useful in the treatment and prevention of proliferative diseases, for example, cancer, inflammation and arthritis, neurodegenerative diseases such Alzheimer's disease, cardiovascular diseases, viral diseases and fungal diseases.
- the present invention provides imidazopyrazine compounds, especially imidazo[1 ,2-a]pyrazine compounds which are represented by structural Formula I, or pharmaceutically acceptable salts, solvates, esters or prodrug thereof, wherein the various moieties are as described above.
- R is H, CN, -NR5R6, cycloalkenyl, heterocyclenyl, -C(O)NR5R6, -N(R5)C(O)R6, or alkyl substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of -OR5 and - NR5R6;
- R1 is H, halo, aryl or heteroaryl, wherein each of said aryl and heteroaryl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl, heterocyclyl, - C(O)NR5R6 and -OR5;
- R2 is H, halo, or heteroaryl, wherein said heteroaryl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, heteroaryl and heterocyclyl;
- R3 is heterocyclyl-(CR7R8)n-X, heterocyclenyl-(CR7R8)n-X, heteroaryl- ⁇ CR7R8)n-X or aryl-(CR7R8)n-X wherein each of the heterocyclyl-, heterocyclenyl-, heteroaryl- or aryl- moieties of said R3 can be unsubstituted or substituted with one or more moieties, independently selected from the group consisting Of -CONR5R6,
- n 1 ,
- X is selected from the group consisting of, -NR5R6, -OR5, -SO-R5 and -SR5, R7 and R8 are each independently hydrogen or alkyl; R5 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkoxyalkyl,
- -alkyl-S-alkyl aminoalkyl, aryl, heteroaryl, heterocyclenyl, heterocycloalkyl, cycloalkyl, cyclenyl, heterocyclylalkoxyl, -S- alkylheterocyclyl, heterocyclyl, heterocyclenyl, alkylN(alkyl)2, alkylNH(alkyl), alkylN(alkenyl)2, -alkylN(alkoxyl)2, -alkyl-SH and hydroxyalkyl, wherein each of said aryl, heteroaryl, heterocyclenyl, heterocycloalkyl, cycloalkyl, cyclenyl, heterocyclylalkoxyl, -S- alkylheterocyclyl, heterocyclyl, heterocyclenyl can be unsubstituted or substituted with one or more moieties independently selected from the group consisting of alkyl, alkyl, alkenyl, aryl,
- R6 is selected from the group consisting of hydrogen, alkyl, alkenyl, aryl, cyclenyl, cycloalkyl, arylalkyl, cyclenylalkyl, cycloalkylalkyl, heteroaryl, heterocyclenyl, heterocyclyl, heteroarylalkyl, heterocyclenylalkyl, heterocycloalkylalkyl, alkoxyalkyl, -alkyl-S-alkyl, -alkylSH, alkoxyl, -S- alkyl, hydroxyalkyl, and aminoalkyl, wherein each of said aryl, cyclenyl, cycloalkyl, arylalkyl, cyclenylalkyl, cycloalkylalkyl, heteroaryl, heterocyclenyl, heterocyclyl, heteroarylalkyl, heterocyclenylalkyl, heterocycloalkylalkyl can be unsubstituted or substituted with one or
- R, R1 and R2 are not all H simultaneously.
- R2 is unsubstituted heteroaryl or heteroaryl substituted with alkyl.
- R2 is heteroaryl substituted with alkyl. In another embodiment, in Formula I, R2 is pyrazolyl.
- R2 is pyrazolyl substituted with alkyl. In another embodiment, in Formula I, R2 is 1-methyl-pyrazol-4-yl. In another embodiment, in Formula I, R is H. In another embodiment, in Formula I, R is CN. In another embodiment, in Formula I, R is -C(O)NR5R6.
- R is -C(O)NH2. In another embodiment, in Formula I, R is heterocyclenyl. In another embodiment, in Formula I, R is tetrahydropyridinyl.
- R is 1 ,2,3,6-tetrahydropyridinyl.
- R is alkyl substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of -OR1 and -NR5R6.
- R is alkyl substituted with one or more - NR5R6.
- R is alkyl substituted with -NH2.
- R is alkyl substituted with -NH(methyl). In another embodiment, R is unsubstituted alkyl.
- both R and R1 are not H simultaneously.
- R3 is heteroaryl-CH2-X, wherein X is - OR5, -SOR5, -NR5R6, or -SR5; R5 is hydrogen, -alkylN(alkyl)2, heterocyclylalkyl or heterocyclenylalkyl; or R5 and R6 can optionally be joined together with the N of said - NR5R6 to form a cyclic ring or bridged cyclic ring, wherein said cyclic ring or bridged cyclic ring can be unsubstituted or substituted one or more moities, which can be the same or different, independently selected from the group consisting of hydroxy!, alkyl, alkoxyl, alkoxylalkyl, hydroxyalkyl, arylalkyl, aryl, heterospirocyclyl, heterospirocyclenyl, heterospiroaryl and -CO2alkyl.
- R3 is heteroaryl-
- X is -NR5R6, R5 is -alkylN(alkyl)2, alkyl, alkoxyalkyl, hydroxyalkyl, arylalkyl, heterocyclenylalkyl, cycloalkyl, cycloalkylalkyl, heteroarylalkyl, or -alkylSH, R6 is hydrogen, alkyl, hydroxyalkyl, alkoxyalkyl, or -alkylN(alkyl)2; or R5 and R6 can optionally be joined together with the N of said -NR5R6 to form a cyclic ring or bridged cyclic ring, wherein said cyclic ring or bridged cyclic ring can be unsubstituted or substituted one or more moities, which can be the same or different, independently selected from the group consisting of hydroxyl, alkyl, alkoxyl, alkoxylalkyl, hydroxyalkyl, aryl
- R3 is heteroaryl-CH2-X , wherein the heteroaryl of said heteroaryl-CH2-X is substituted with alkyl or -CONR5R6, wherein X is -NR5R6, R5 is alkyl, R6 is alkyl, or R5 and R6 are optionally joined together with the N of said -NR5R6 to form a cyclic ring.
- R3 is aryl-CH2-X , wherein the aryl of said aryl-CH2-X is substituted with alkyl, wherein X is heterocyclyl.
- R5 is s C- ** . or wherein X is selected from the group consisting of, -NR5R6, -OR5 -SO-R5 and -SR5,
- R5 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkoxyalkyl, -alkyl-S-alkyl, aminoalkyl, aryl, heteroaryl, heterocyclenyl, heterocycloalkyl, cycloalkyl, cyclenyl, heterocyclylalkoxyl, -S- alkylheterocyclyl, heterocyclyl, heterocyclenyl, alkylN(alkyl)2, alkylNH(alkyl), alkylN(alkenyl)2, -alkylN(alkoxyl)2, -alkyl-SH and hydroxyalkyl, wherein each of said aryl, heteroaryl, heterocyclenyl, heterocycloalkyl, cycloalkyl, cyclenyl, heterocyclylalkoxyl, -S- alkylheterocyclyl, heterocyclyl, heterocyclenyl can be unsubstituted or substituted with one or
- R3 is isothiazole, thiophene or pyrimidine substituted with:
- R3 is pyrimidinyl substituted with heterocyclylmethyl.
- R3 is pyrimidinyl substituted with morpholinylmethyll or pyrrolidinylmethyl.
- R3 is phenyl substituted with heterocyclylalkyl, wherein said heterocyclylalkyl can be unsubstituted or substituted with one or more moieties, which can be the same or different, each moiety being independently selected from the group consisting of alkyl.
- R3 is phenyl-CHmethyl-X or phenyl-CH2- X , wherein X is piperazinyl, piperadinyl, pyrrolidinyl, morpholinyl or thiomorpholinyl wherein said piperazinyl, piperadinyl, pyrrolidinyl, morpholinyl or thiomorpholinyl can be unsubstituted or substituted with one or more moieties, which can be the same or different, each moiety being independently selected from the group consisting alkyl.
- R3 is phenyl substituted with heterocyclylmethyl, wherein said phenyl group is further substituted with alkyl.
- R3 is phenyl substituted with piperidinylmethyl, morpholinylmethyl or thiomorpholinylmethyl, wherein said phenyl group is further substituted with methyl.
- R3 is x .
- X is heterocyclyl wherein said heterocyclyl can be unsubstituted or substituted with one or more moieties, which can be the same or different, each moiety being independently selected from the group consisting of hydroxyl, alkyl, hydroxyalkyl, alkoxyl, -CO2alkyl, arylalkyl, aryl, alkoxyalkyl, and heterocyclyl.
- R3 is wherein X is heterocyclyl wherein said heterocyclyl can be unsubstituted or substituted with one or more moieties, which can be the same or different, each moiety being independently selected from the group consisting of alkyl.
- this invention discloses a compound of the formula:
- R2 is heteroaryl, wherein said heteroaryl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, amide, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, -C(O)OH, -C(O)NH2, -NR5R6 (where R5 and R6 form a cyclic amine together with the N of said -NR5R6), -CN, arylalkyl, -CH2OR5, -S(O)R5, -S(O2)R5, -CN, -CHO, -SR5, - C(O)OR5, -C(O)R5, heteroaryl and heterocyclyl; R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the same or different each moiety
- R1 is H;
- R3 is heteroaryl-CH2-X or heteroaryl-CHMethyl-X, wherein X is -NR5R6, R5 is - alkylN(alkyl)2, alkyl, alkoxyalkyl, hydroxyalkyl, arylalkyl, heterocyclenylalkyl, cycloalkyl, cycloalkylalkyl, heteroarylalkyl, or-alkylSH, R6 is hydrogen, alkyl, hydroxyalkyl, alkoxyalkyl, or -alkylN(alkyl)2; or R5 and R6 can optionally be joined together with the N of said -NR5R6 to form a cyclic ring or bridged cyclic ring, wherein said cyclic ring or
- this invention discloses a compound of the formula:
- R2 is heteroaryl, wherein said heteroaryl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, amide, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, -C(O)OH, -C(O)NH2, -NR5R6 (where R5 and R6 form a cyclic amine together with the N of said -NR5R6), -CN, arylalkyl, -CH2OR5, -S(O)R5, -S(O2)R5, -CN, -CHO, -SR5, - C(O)OR5, -C(O)R5, heteroaryl and heterocyclyl; R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the same or different each moiety
- this invention discloses a compound of the formula:
- R2 is heteroaryl, wherein said heteroaryl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, amide, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, -C(O)OH, -C(O)NH2, -NR5R6 (where R5 and R6 form a cyclic amine together with the N of said -NR5R6), -CN, arylalkyl, -CH2OR5, -S(O)R5, -S(O2)R5, -CN, -CHO, -SR5, - C(O)OR5, -C(O)R5, heteroaryl and heterocyclyl; R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the same or different each moiety
- R1 is H
- R3 is isothiazole, thiophene or pyrimidine substituted with:
- this invention discloses a compound of the formula:
- R1 is H;
- R3 is phenyl-CHmethyl-X or phenyl-CH2-X , wherein said phenyl of each of said phenyl- CHmethyl-X or phenyl-CH2-X can be unsubstituted or substituted with alkyl, further wherein X is piperazinyl, piperadinyl, pyrrolidinyl, morpholinyl or thiomorpholinyl wherein each of said piperazinyl, piperadinyl, pyrrolidinyl, morpholinyl or thiomorpholinyl can be unsubstituted or substituted with alkyl; wherein R5 and R6 are as defined above.
- this invention discloses a compound of the formula:
- R3 N-H or a pharmaceutically acceptable salt, solvate or ester thereof, wherein R2 is heteroaryl, wherein said heteroaryl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, amide, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, -C(O)OH, -C(O)NH2, -NR5R6 (where R5 and R6 form a cyclic amine together with the N of said -NR5R6), -CN, arylalkyl, -CH2OR5, -S(O)R5, -S(O2)R5, -CN, -CHO, -SR5, - C(O)OR5, -C(O)R5, heteroaryl and heterocyclyl; R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the
- X is selected from the group consisting of, -NR5R6, -OR5 -SO-R5 and -SR5,
- R5 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkoxyalkyl, -alkyl-S-alkyl, aminoalkyl, aryl, heteroaryl, heterocyclenyl, heterocycloalkyl, cycloalkyl, cyclenyl, heterocyclylalkoxyl, -S- alkylheterocyclyl, heterocyclyl, heterocyclenyl, alkylN(alkyl)2, alkylNH(alkyl), alkylN(alkenyl)2, -alkylN(alkoxyl)2, -alkyl-SH and hydroxyalkyl, wherein each of said aryl, heteroaryl, heterocyclenyl, heterocycloalkyl, cycloalkyl, cyclenyl, heterocyclylalkoxyl, -S- alkylheterocyclyl, heterocyclyl and heterocyclenyl can be unsubstituted or substituted with one or
- R2 is heteroaryl, wherein said heteroaryl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, amide, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, -C(O)OH, -C(O)NH2, -NR5R6 (where R5 and R6 form a cyclic amine together with the N of said -NR5R6), -CN, arylalkyl, -CH2OR5, -S(O)R5, -S(O2)R5, -CN, -CHO, -SR5, - C(O)OR5, -C(O)R5, heteroaryl and heterocyclyl; R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the same or different each moiety
- R5 and R6 are as defined above.
- this invention discloses a compound of the formula:
- R1 is H;
- R3 is heteroaryl-CH2-X or heteroaryl-CHMethyl-X, wherein X is -NR5R6, R5 is - alkylN(alkyl)2, alkyl, alkoxyalkyl, hydroxyalkyl, arylalkyl, heterocyclenylalkyl, cycloalkyl, cycloalkylalkyl, heteroarylalkyl, or -alkylSH, R6 is hydrogen, alkyl, hydroxyalkyl, alkoxyalkyl, or -alkylN(alkyl)2; or R5 and R6 can optionally be joined together with the N of said -NR5R6 to form a cyclic ring or bridged cyclic ring, wherein said cyclic ring
- this invention discloses a compound of the formula:
- R2 is pyrazolyl, wherein said pyrazolyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, amide, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, -C(O)OH, - C(O)NH2, -NR5R6 (where R5 and R6 form a cyclic amine together with the N of said - NR5R6), -CN, arylalkyl, -CH2OR5, -S(O)R5, -S(O2)R5, -CN, -CHO, -SR5, -C(O)OR5, - C(O)R5, heteroaryl and heterocyclyl; R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the same
- R2 is pyrazolyl, wherein said pyrazolyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, amide, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, -C(O)OH, - C(O)NH2, -NR5R6 (where R5 and R6 form a cyclic amine together with the N of said - NR5R6), -CN, arylalkyl, -CH2OR5, -S(O)R5, -S(O2)R5, -CN, -CHO, -SR5, -C(O)OR5, - C(O)R5, heteroaryl and heterocyclyl; R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the same
- R1 is H
- R3 is isothiazole, thiophene or pyrimidine substituted with:
- R5 and R6 are as defined above.
- this invention discloses a compound of the formula:
- R2 is pyrazolyl, wherein said pyrazolyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, amide, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, -C(O)OH, - C(O)NH2, -NR5R6 (where R5 and R6 form a cyclic amine together with the N of said - NR5R6), -CN, arylalkyl, -CH2OR5, -S(O)R5, -S(O2)R5, -CN, -CHO, -SR5, -C(O)OR5, - C(O)R5, heteroaryl and heterocyclyl; R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the same
- R2 is pyrazolyl, wherein said pyrazolyl can be unsubstituted or substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of halo, amide, alkyl, alkenyl, alkynyl, cycloalkyl, aryl, -C(O)OH, - C(O)NH2, -NR5R6 (where R5 and R6 form a cyclic amine together with the N of said - NR5R6), -CN, arylalkyl, -CH2OR5, -S(O)R5, -S(O2)R5, -CN, -CHO, -SR5, -C(O)OR5, - C(O)R5, heteroaryl and heterocyclyl; R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the same
- R5 and R6 are as defined above.
- this invention discloses a compound of the formula:
- R2 is 1-H- pyrazol-4-yl
- R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of -OR5, heterocyclyl, -N(R5)C(O)N(R5R6), -N(R5)-C(O)OR6, -(CH2)1-3- N(R5R6) and -NR5R6
- R1 is H
- R3 is heteroaryl-CH2-X or heteroaryl-CHMethyl-X, wherein X is -NR5R6, R5 is -alkylN(alkyl)2, alkyl, alkoxyalkyl, hydroxyalkyl, arylalkyl, heterocyclenylalkyl, cycloalkyl, cycloalkylalkyl, heteroarylalkyl, or -alkylSH,
- this invention discloses a compound of the formula:
- R2 is 1-A7- pyrazol-4-yl
- R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of -OR5, heterocyclyl, -N(R5)C(O)N(R5R6), -N(R5)-C(O)OR6, -(CH2)1-3- N(R5R6) and -NR5R6
- R1 is H
- R3 is heteroaryl-CH2-X , wherein the heteroaryl of said heteroaryl-CH2-X is substituted with alkyl Or -CONR5R6, wherein X is -NR5R6, R5 is alkyl, R6 is alkyl, or R5 and R6 are optionally joined together with the N of said - NR5R6 to form heterocyclyl.
- R5 and R6 are as defined above.
- R2 is 1- H-pyrazol-4-yl
- R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of -OR5, heterocyclyl, -N(R5)C(O)N(R5R6), -N(R5)-C(O)OR6, -(CH2)1-3- N(R5R6) and -NR5R6
- R1 is H
- R3 is isothiazole, thiophene or pyrimidine substituted with:
- this invention discloses a compound of the formula:
- R2 is 1- H-pyrazol-4-yl
- R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of -OR5, heterocyclyl, -N(R5)C(O)N(R5R6), -N(R5)-C(O)OR6, -(CH2)1-3- N(R5R6) and -NR5R6
- R1 is H
- R3 is phenyl-CHmethyl-X or phenyl-CH2-X , wherein said phenyl of each of said phenyl-CHmethyl-X or phenyl-CH2-X can be unsubstituted or substituted with alkyl, further wherein X is piperazinyl, piperadinyl, pyrrolidinyl, morpholinyl or thiomorpholinyl wherein each of said piperazin
- this invention discloses a compound of the formula:
- R2 is 1-H- pyrazol-4-yl
- R is unsubstituted alkyl or alkyl substituted with one or more moieties which can be the same or different each moiety being independently selected from the group consisting of -OR5, heterocyclyl, -N(R5)C(O)N(R5R6), -N(R5)-C(O)OR6, -(CH2)1-3-
- this invention discloses a compound of the formula:
- R2 is 1-H- pyrazol-4-yl; R is unsubstituted alkyl; R1 is H; R3 is heteroaryl-CH2-X or heteroaryl- CHMethyl-X, wherein X is -NR5R6, R5 is -alkylN(alkyl)2) alkyl, alkoxyalkyl, hydroxyalkyl, arylalkyl, heterocyclenylalkyl, cycloalkyl, cycloalkylalkyl, heteroarylalkyl, or -alkylSH, R6 is hydrogen, alkyl, hydroxyalkyl, alkoxyalkyl, or -alkylN(alkyl)2; or R5 and R6 can optionally be joined together with the N of said -NR5R6 to form a cyclic ring or bridged cyclic ring, wherein said cyclic ring or bridged cyclic
- this invention discloses a compound of the formula:
- R2 is 1-H- pyrazol-4-yl; R is unsubstituted alkyl; R1 is H; R3 is heteroaryl-CH2-X , wherein the heteroaryl of said heteroaryl-CH2-X is substituted with alkyl or -CONR5R6, wherein X is -NR5R6, R5 is alkyl, R6 is alkyl, or R5 and R6 are optionally joined together with the N of said -NR5R6 to form heterocyclyl; wherein R5 and R6 are as defined above.
- this invention discloses a compound of the formula:
- R2 is 1- H-pyrazol-4-yl; R is unsubstituted alkyl; R1 is H; R3 is isothiazole, thiophene or pyrimidine substituted with:
- R5 and R6 are as defined above.
- this invention discloses a compound of the formula:
- R2 is 1- H-pyrazol-4-yl; R is unsubstituted alkyl; R1 is H; R3 is phenyl-CHmethyl-X or phenyl- CH2-X , wherein said phenyl of each of said phenyl-CHmethyl-X or phenyl-CH2-X can be unsubstituted or substituted with alkyl, further wherein X is piperazinyl, piperadinyl, pyrrolidinyl, morpholinyl or thiomorpholinyl wherein each of said piperazinyl, piperadinyl, pyrrolidinyl, morpholinyl or thiomorpholinyl can be unsubstituted or substituted with alkyl; wherein R5 and R6 are as defined above.
- this invention discloses a compound of the formula:
- R is unsubstituted alkyl
- R1 is H
- R3 is, , ,
- this invention discloses a compound of the formula:
- R2 is 1-H- pyrazol-4-yl; R is methyl, R1 is H; R3 is heteroaryl-CH2-X or heteroaryl-CHMethyl-X, wherein X is -NR5R6, R5 is -alkylN(alkyl)2, alkyl, alkoxyalkyl, hydroxyalkyl, arylalkyl, heterocyclenylalkyl, cycloalkyl, cycloalkylalkyl, heteroarylalkyl, or -alkylSH, R6 is hydrogen, alkyl, hydroxyalkyl, alkoxyalkyl, or -alkylN(alkyl)2; or R5 and R6 can optionally be joined together with the N of said -NR5R6 to form a cyclic ring or bridged cyclic ring, wherein said cyclic ring or bridged cyclic ring can be unsubsti
- this invention discloses a compound of the formula:
- R2 is 1-H- pyrazol-4-yl
- R is methyl, R1 is H
- R3 is heteroaryl-CH2-X , wherein the heteroaryl of said heteroaryl-CH2-X is substituted with alkyl or -CONR5R6, wherein X is -NR5R6, R5 is alkyl, R6 is alkyl, or R5 and R6 are optionally joined together with the N of said - NR5R6 to form heterocyclyl; wherein R5 and R6 are as defined above.
- this invention discloses a compound of the formula:
- R2 is 1- H-pyrazol-4-yl; R is methyl, R1 is H; R3 is isothiazole, thiophene or pyrimidine substituted with:
- this invention discloses a compound of the formula:
- R is methyl, R1 is H; R3 is phenyl-CHmethyl-X or phenyl-CH2-X , wherein said phenyl of each of said phenyl-CHmethyl-X or phenyl-CH2-X can be unsubstituted or substituted with alkyl, further wherein X is piperazinyl, piperadinyl, pyrrolidinyl, morpholinyl or thiomorpholinyl wherein each of said piperazinyl, piperadinyl, pyrrolidinyl, morpholinyl or thiomorpholinyl can be unsubstituted or substituted with alkyl.
- this invention discloses a compound of the formula:
- this invention discloses a compound of the formula:
- R5 is alkyl
- R6 is selected from the group consisting of alkoxyalkyl, hydroxyalkyl, cycloalkyl, wherein said cycloalkyl is substituted by hydroxyalkyl; or R5 and R6 together with the N of said -NR5R6 to form a cyclic ring, wherein said cyclic ring is substituted by one or more moieties independently selected from the group consisting of alkoxyalkyl, hydroxyalkyl, and alkyl.
- this invention discloses a compound of the formula:
- R5 is methyl, ethyl, or propyl
- R6 is selected from the group consisting of ethoxyethyl, 1 ,1- dimethylhydroxyethyl, cyclopentyl, cyclohexyl, wherein each of said cyclopentyl and cyclohexyl is substituted by hydroxymethyl
- R5 and R6 together with the N of said - NR5R6 to form a cyclic ring, wherein said cyclic ring is substituted by one or more moieties independently selected from the group consisting of ethoxymethyl, methoxymethyl, hydroxymethyl, and methyl.
- Non-limiting examples of compounds of Formula I include:
- alkyl means an aliphatic hydrocarbon group which may be straight or branched and comprising about 1 to about 20 carbon atoms in the chain. Preferred alkyl groups contain about 1 to about 12 carbon atoms in the chain. More preferred alkyl groups contain about 1 to about 6 carbon atoms in the chain. Branched means that one or more, lower alkyl groups such as methyl, ethyl or propyl, are attached to a linear alkyl chain.
- “Lower alkyl” means a group having about 1 to about 6 carbon atoms in the chain which may be straight or branched.
- suitable alkyl groups include methyl, ethyl, n-propyl, isopropyl and t-butyl.
- Alkenyl means an aliphatic hydrocarbon group containing at least one carbon- carbon double bond and which may be straight or branched and comprising about 2 to about 15 carbon atoms in the chain. Preferred alkenyl groups have about 2 to about 12 carbon atoms in the chain; and more preferably about 2 to about 6 carbon atoms in the chain. Branched means that one or more, lower alkyl groups such as methyl, ethyl or propyl, are attached to a linear alkenyl chain. "Lower alkenyl” means about 2 to about 6 carbon atoms in the chain which may be straight or branched.
- alkenyl may be unsubstituted or optionally substituted by one or more substituents which may be the same or different, each substituent being independently selected from the group consisting of halo, alkyl. aryl, cycloalkyl, cyano, alkoxy and -S(alkyl).
- suitable alkenyl groups include ethenyl, propenyl, n-butenyl, 3-methylbut- 2-enyl, n-pentenyl, octenyl and decenyl.
- Alkylene means a difunctional group obtained by removal of a hydrogen atom from an alkyl group that is defined above.
- alkylene include methylene, ethylene and propylene.
- Alkynyl means an aliphatic hydrocarbon group containing at least one carbon- carbon triple bond and which may be straight or branched and comprising about 2 to about 15 carbon atoms in the chain.
- Preferred alkynyl groups have about 2 to about 12 carbon atoms in the chain; and more preferably about 2 to about 4 carbon atoms in the chain.
- Branched means that one or more, lower alkyl groups such as methyl, ethyl or propyl, are attached to a linear alkynyl chain.
- Lower alkynyl means about 2 to about 6 carbon atoms in the chain which may be straight or branched.
- alkynyl groups include ethynyl, propynyl, 2-butynyl and 3- methylbutynyl.
- Alkynyl may be unsubstituted or optionally substituted by one or more substituents which may be the same or different, each substituent being independently selected from the group consisting of alkyl, aryl and cycloalkyl.
- Aryl means an aromatic monocyclic or multicyclic ring system comprising about 6 to about 14 carbon atoms, preferably about 6 to about 10 carbon atoms. The aryl group can be optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined herein.
- Suitable aryl groups include phenyl and naphthyl.
- “Bridged cyclic ring” is a hydrocarbon ring such as cycloalkyl, cyclenyl, or aryl or heteroatom containing ring such as, heterocyclyl, heterocyclenyl, or heteroaryl as described herein, that contains a bridge, which is a valence bond or an atom or an unbranched chain of atoms connecting two different parts of the ring.
- bridgeheads The two tertiary carbon atoms connected through the bridge are termed "bridgeheads".
- Heteroaryl means an aromatic monocyclic or multicyclic ring system comprising about 5 to about 14 ring atoms, preferably about 5 to about 10 ring atoms, in which one or more of the ring atoms is an element other than carbon, for example nitrogen, oxygen or sulfur, alone or in combination. Preferred heteroaryls contain about 5 to about 6 ring atoms.
- the "heteroaryl” can be optionally substituted by one or more "ring system substituents" which may be the same or different, and are as defined herein.
- the prefix aza, oxa or thia before the heteroaryl root name means that at least a nitrogen, oxygen or sulfur atom respectively, is present as a ring atom.
- heteroaryl may also include a heteroaryl as defined above fused to an aryl as defined above.
- suitable heteroaryls include pyridyl, pyrazinyl, furanyl, thienyl, pyrimidinyl, pyridone (including N-substituted pyridones), isoxazolyl, isothiazolyl, oxazolyl, thiazolyl, pyrazolyl, furazanyl, pyrrolyl, pyrazolyl, triazolyl, 1 ,2,4- thiadiazolyl, pyrazinyl, pyridazinyl, quinoxalinyl, phthalazinyl, oxindolyl, imidazo[1 ,2- a]pyridinyl, imidazo[2,1-b]thiazolyl,
- Aralkyl or “arylalkyl” means an aryl-alkyl- group in which the aryl and alkyl are as previously described. Preferred aralkyls comprise a lower alkyl group. Non-limiting examples of suitable aralkyl groups include benzyl, 2-phenethyl and naphthalenylmethyl. The bond to the parent moiety is through the alkyl.
- Alkylaryl means an alkyl-aryl- group in which the alkyl and aryl are as previously described. Preferred alkylaryls comprise a lower alkyl group. Non-limiting example of a suitable alkylaryl group is tolyl. The bond to the parent moiety is through the aryl.
- Cycloalkyl means a non-aromatic mono- or multicyclic ring system comprising about 3 to about 10 carbon atoms, preferably about 5 to about 10 carbon atoms. Preferred cycloalkyl rings contain about 5 to about 7 ring atoms.
- the cycloalkyl can be optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined above.
- suitable monocyclic cycloalkyls include cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl and the like.
- suitable multicyclic cycloalkyls include 1-decalinyl, norbomyl, adamantyl and the like.
- Cycloalkylalkyl means a cycloalkyl moiety as defined above linked via an alkyl moiety (defined above) to a parent core.
- suitable cycloalkylalkyls include cyclohexylmethyl, adamantylmethyl and the like.
- Cycloalkenyl means a non-aromatic mono or multicyclic ring system comprising about 3 to about 10 carbon atoms, preferably about 5 to about 10 carbon atoms which contain at least one carbon-carbon double bond. Preferred cycloalkenyl rings contain about 5 to about 7 ring atoms.
- the cycloalkenyl can be optionally substituted with one or more "ring system substituents" which may be the same or different, and are as defined above.
- suitable monocyclic cycloalkenyls include cyclopentenyl, cyclohexenyl, cyclohepta-1 ,3-dienyl, and the like.
- Non-limiting example of a suitable multicyclic cycloalkenyl is norbornylenyl.
- Cycloalkenylalkyl means a cycloalkenyl moiety as defined above linked via an alkyl moiety (defined above) to a parent core.
- suitable cycloalkenylalkyls include cyclopentenylmethyl, cyclohexenylmethyl and the like.
- Halogen means fluorine, chlorine, bromine, or iodine. Preferred are fluorine, chlorine and bromine.
- Ring system substituent means a substituent attached to an aromatic or non- aromatic ring system which, for example, replaces an available hydrogen on the ring system.
- Ring system substituents may be the same or different, each being independently selected from the group consisting of alkyl, alkenyl, alkynyl, aryl, heteroaryl, aralkyl, alkylaryl, heteroaralkyl, heteroarylalkenyl, heteroarylalkynyl, alkylheteroaryl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, acyl, aroyl, halo, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylthio, arylthio, heteroarylthio, aralkylthio, hetero
- Ring system substituent may also mean a single moiety which simultaneously replaces two available hydrogen on two adjacent carbon atoms (one H on each carbon) on a ring system.
- Examples of such moiety are methylene dioxy, ethylenedioxy, -C(CH3)2- and the like which form moieties such as, for example:
- Heteroarylalkyl means a heteroaryl moiety as defined above linked via an alkyl moiety (defined above) to a parent core.
- suitable heteroaryls include 2-pyridinylmethyl, quinolinylmethyl and the like.
- Heterocyclyl means a non-aromatic saturated monocyclic or multicyclic ring system comprising about 3 to about 10 ring atoms, preferably about 5 to about 10 ring atoms, in which one or more of the atoms in the ring system is an element other than carbon, for example nitrogen, oxygen or sulfur, alone or in combination. There are no adjacent oxygen and/or sulfur atoms present in the ring system.
- Preferred heterocyclyls contain about 5 to about 6 ring atoms.
- the prefix aza, oxa or thia before the heterocyclyl root name means that at least a nitrogen, oxygen or sulfur atom respectively is present as a ring atom.
- Any -NH in a heterocyclyl ring may exist protected such as, for example, as an -N(Boc), -N(CBz), -N(Tos) group and the like; such protections are also considered part of this invention.
- the heterocyclyl can be optionally substituted by one or more "ring system substituents" which may be the same or different, and are as defined herein.
- the nitrogen or sulfur atom of the heterocyclyl can be optionally oxidized to the corresponding N-oxide, S-oxide or S,S- dioxide.
- heterocyclyl rings include piperidyl, pyrrolidinyl, piperazinyl, morpholinyl, thiomorpholinyl, thiazolidinyl, 1 ,4- dioxanyl, tetrahydrofuranyl, tetrahydrothiophenyl, lactam, lactone, and the like.
- Heterocyclyl may also mean a single moiety (e.g., carbonyl) which simultaneously replaces two available hydrogen on the same carbon atom on a ring system. Example of such moiety is pyrrolidone:
- Heterocyclylalkyl means a heterocyclyl moiety as defined above linked via an alkyl moiety (defined above) to a parent core.
- suitable heterocyclylalkyls include piperidinylmethyl, piperazinylmethyl and the like.
- Heterocyclenyl means a non-aromatic monocyclic or multicyclic ring system comprising about 3 to about 10 ring atoms, preferably about 5 to about 10 ring atoms, in which one or more of the atoms in the ring system is an element other than carbon, for example nitrogen, oxygen or sulfur atom, alone or in combination, and which contains at least one carbon-carbon double bond or carbon-nitrogen double bond. There are no adjacent oxygen and/or sulfur atoms present in the ring system.
- Preferred heterocyclenyl rings contain about 5 to about 6 ring atoms.
- the prefix aza, oxa or thia before the heterocyclenyl root name means that at least a nitrogen, oxygen or sulfur atom respectively is present as a ring atom.
- the heterocyclenyl can be optionally substituted by one or more ring system substituents, wherein "ring system substituent" is as defined above.
- the nitrogen or sulfur atom of the heterocyclenyl can be optionally oxidized to the corresponding N-oxide, S-oxide or S,S-dioxide.
- heterocyclenyl groups include 1 ,2,3,4- tetrahydropyridinyl, 1 ,2-dihydropyridinyl, 1 ,4-dihydropyridinyl, 1 ,2,3,6- tetrahydropyridinyl, 1 ,4,5,6-tetrahydropyrimidinyl, 2-pyrrolinyl, 3-pyrrolinyl, 2- imidazolinyl, 2-pyrazolinyl, dihydroimidazolyl, dihydrooxazolyl, dihydrooxadiazolyl, dihydrothiazolyl, 3,4-dihydro-2H-pyranyl, dihydrofuranyl, fluorodihydrofuranyl, 7- oxabicyclo[2.2.1]heptenyl, dihydrothiophenyl, dihydrothiopyranyl, and the like.
- Heterocyclenyl may also mean a single moiety (
- Heterocyclenylalkyl means a heterocyclenyl moiety as defined above linked via an alkyl moiety (defined above) to a parent core.
- hetero-atom containing ring systems of this invention there are no hydroxyl groups on carbon atoms adjacent to a N, O or S, as well as there are no N or S groups on carbon adjacent to another heteroatom.
- N, O or S there are no hydroxyl groups on carbon atoms adjacent to a N, O or S, as well as there are no N or S groups on carbon adjacent to another heteroatom.
- Alkynylalkyl means an alkynyl-alkyl- group in which the alkynyl and alkyl are as previously described. Preferred alkynylalkyls contain a lower alkynyl and a lower alkyl group. The bond to the parent moiety is through the alkyl. Non-limiting examples of suitable alkynylalkyl groups include propargylmethyl.
- Heteroaralkyl means a heteroaryl-alkyl- group in which the heteroaryl and alkyl are as previously described. Preferred heteroaralkyls contain a lower alkyl group. Non-limiting examples of suitable aralkyl groups include pyridylmethyl, and quinolin-3- ylmethyl. The bond to the parent moiety is through the alkyl.
- Spiro ring systems have two or more rings linked by one common atom.
- Preferred spiro ring systems include spiroheteroaryl, spiroheterocyclenyl, spiroheterocyclyl, spirocycloalkyl, spirocyclenyl, and spiroaryl.
- the spiro ring systems can be optionally substituted by one or more ring system substituents, wherein "ring system substituent" is as defined above.
- Hydroxyalkyl means a HO-alkyl- group in which alkyl is as previously defined. Preferred hydroxyalkyls contain lower alkyl. Non-limiting examples of suitable hydroxyalkyl groups include hydroxymethyl and 2-hydroxyethyl.
- acyl means an H-C(O)-, alkyl-C(O)- or cycloalkyl-C(O)-, group in which the various groups are as previously described.
- the bond to the parent moiety is through the carbonyl.
- Preferred acyls contain a lower alkyl.
- suitable acyl groups include formyl, acetyl and propanoyl.
- Aroyl means an aryl-C(O)- group in which the aryl group is as previously described. The bond to the parent moiety is through the carbonyl.
- suitable groups include benzoyl and 1- naphthoyl.
- Alkoxy means an alkyl-O- group in which the alkyl group is as previously described.
- suitable alkoxy groups include methoxy, ethoxy, n-propoxy, isopropoxy and n-butoxy.
- the bond to the parent moiety is through the ether oxygen.
- Aryloxy means an aryl-O- group in which the aryl group is as previously described.
- suitable aryloxy groups include phenoxy and naphthoxy. The bond to the parent moiety is through the ether oxygen.
- Alkyloxy means an aralkyl-O- group in which the aralkyl group is as previously described.
- suitable aralkyloxy groups include benzyloxy and 1- or 2-naphthalenemethoxy.
- the bond to the parent moiety is through the ether oxygen.
- Alkylthio means an alkyl-S- group in which the alkyl group is as previously described.
- suitable alkylthio groups include methylthio and ethylthio.
- the bond to the parent moiety is through the sulfur.
- Arylthio means an aryl-S- group in which the aryl group is as previously described.
- suitable arylthio groups include phenylthio and naphthylthio. The bond to the parent moiety is through the sulfur.
- Alkylthio means an aralkyl-S- group in which the aralkyl group is as previously described.
- Non-limiting example of a suitable aralkylthio group is benzylthio.
- the bond to the parent moiety is through the sulfur.
- Alkoxycarbonyl means an alkyl-O-CO- group. Non-limiting examples of suitable alkoxycarbonyl groups include methoxycarbonyl and ethoxycarbonyl. The bond to the parent moiety is through the carbonyl.
- Aryloxycarbonyl means an aryl-O-C(O)- group. Non-limiting examples of suitable aryloxycarbonyl groups include phenoxycarbonyl and naphthoxycarbonyl. The bond to the parent moiety is through the carbonyl.
- Alkoxycarbonyl means an aralkyl-O-C(O)- group.
- a suitable aralkoxycarbonyl group is benzyloxycarbonyl.
- the bond to the parent moiety is through the carbonyl.
- Alkylsulfonyl means an alkyl-S(O2)- group. Preferred groups are those in which the alkyl group is lower alkyl. The bond to the parent moiety is through the sulfonyl.
- Arylsulfonyl means an aryl-S(O2)- group. The bond to the parent moiety is through the sulfonyl.
- substituted means that one or more hydrogen on the designated atom is replaced with a selection from the indicated group, provided that the designated atom's normal valency under the existing circumstances is not exceeded, and that the substitution results in a stable compound. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- stable compound' or “stable structure” is meant a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
- purified refers to the physical state of said compound after being isolated from a synthetic process (e.g. from a reaction mixture), or natural source or combination thereof.
- purified refers to the physical state of said compound after being obtained from a purification process or processes described herein or well known to the skilled artisan (e.g., chromatography, recrystallization and the like) , in sufficient purity to be characterizable by standard analytical techniques described herein or well known to the skilled artisan.
- protecting groups When a functional group in a compound is termed "protected", this means that the group is in modified form to preclude undesired side reactions at the protected site when the compound is subjected to a reaction. Suitable protecting groups will be recognized by those with ordinary skill in the art as well as by reference to standard textbooks such as, for example, T. W. Greene et al, Protective Groups in organic Synthesis (1991 ), Wiley, New York.
- composition is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- Prodrugs and solvates of the compounds of the invention are also contemplated herein.
- a discussion of prodrugs is provided in T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems (1987) 14 of the A.C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press.
- the term "prodrug” means a compound (e.g., a drug precursor) that is transformed in vivo to yield a compound of Formula (I) or a pharmaceutically acceptable salt, hydrate or solvate of the compound. The transformation may occur by various mechanisms (e.g., by metabolic or chemical processes), such as, for example, through hydrolysis in blood.
- a prodrug can comprise an ester formed by the replacement of the hydrogen atom of the acid group with a group such as, for example, (C1-C ⁇ JalkyI, (C2- C12)alkanoyloxymethyl, 1-(alkanoyloxy)ethyl having from 4 to 9 carbon atoms, 1- methyl-1-(alkanoyloxy)-ethyl having from 5 to 10 carbon atoms, alkoxycarbonyloxymethyl having from 3 to 6 carbon atoms, 1-(alkoxycarbonyloxy)ethyl having from 4 to 7 carbon atoms, 1-methyl-1-(alkoxycarbonyloxy)ethyl having from 5 to 8 carbon atoms, N-(alkoxycarbonyl)aminomethyl having from 3 to 9 carbon atoms, 1-(N-(alkoxycarbonyl)a group such as, for example, (C1-C ⁇ JalkyI, (C2- C12)alkanoyloxymethyl, 1-(alkanoyl
- a prodrug can be formed by the replacement of the hydrogen atom of the alcohol group with a group such as, for example, (C1-C6)alkanoyloxymethyl, 1-((Cr C6)alkanoyloxy)ethyl, 1 -methyl-1 -((C1-C6)alkanoyloxy)ethyl, (Cr C6)alkoxycarbonyloxymethyl, N-(C1-C6)alkoxycarbonylaminomethyl, succinoyl, (C1- C-6)alkanoyl, ⁇ -amino(C1-C4)alkanyl, arylacyl and ⁇ -aminoacyl, or ⁇ -aminoacyl- ⁇ - aminoacyl, where each ⁇ -aminoacyl group is independently selected from the naturally occurring L-amino acids, P(O)(OH)2, -P(O)(O(C1-C6)alky
- a prodrug can be formed by the replacement of a hydrogen atom in the amine group with a group such as, for example, R-carbonyl, RO-carbonyl, NRR'-carbonyl where R and R' are each independently (C1-C- ⁇ O)alkyl, (C3-C7) cycloalkyl, benzyl, or R-carbonyl is a natural ⁇ -aminoacyl or natural ⁇ -aminoacyl, — C(OH)C(O)OY1 wherein Y1 is H, (C1- C6)alkyl or benzyl, — C(OY2)Y3 wherein Y2 is (CrC4) alkyl and Y3 is (C1-C ⁇ )alkyl, carboxy (C1-C6)alkyl, amino(C1-C4)alkyl or mono-N — or di-N,N-(C1-C6)alkyla
- One or more compounds of the invention may exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like, and it is intended that the invention embrace both solvated and unsolvated forms.
- “Solvate” means a physical association of a compound of this invention with one or more solvent molecules. This physical association involves varying degrees of ionic and covalent bonding, including hydrogen bonding. In certain instances the solvate will be capable of isolation, for example when one or more solvent molecules are incorporated in the crystal lattice of the crystalline solid. "Solvate” encompasses both solution-phase and isolatable solvates. Non-limiting examples of suitable solvates include ethanolates, methanolates, and the like.
- “Hydrate” is a solvate wherein the solvent molecule is H2O.
- One or more compounds of the invention may optionally be converted to a solvate.
- Preparation of solvates is generally known.
- M. Caira et al, J. Pharmaceutical Sd., 93(3). 601-611 (2004) describes the preparation of the solvates of the antifungal fluconazole in ethyl acetate as well as from water.
- Similar preparations of solvates, hemisolvate, hydrates and the like are described by E. C. van Tonder et al, AAPS PharmSciTech., 5£ ⁇ , article 12 (2004); and A. L. Bingham et al, Chem. Commun., 603-604 (2001 ).
- a typical, non-limiting, process involves dissolving the inventive compound in desired amounts of the desired solvent (organic or water or mixtures thereof) at a higher than ambient temperature, and cooling the solution at a rate sufficient to form crystals which are then isolated by standard methods.
- Analytical techniques such as, for example I. R. spectroscopy, show the presence of the solvent (or water) in the crystals as a solvate (or hydrate).
- Effective amount or “therapeutically effective amount” is meant to describe an amount of compound or a composition of the present invention effective in inhibiting the above-noted diseases and thus producing the desired therapeutic, ameliorative, inhibitory or preventative effect.
- the compounds of Formula I can form salts which are also within the scope of this invention. Reference to a compound of Formula I herein is understood to include reference to salts thereof, unless otherwise indicated.
- salts when a compound of Formula I contains both a basic moiety, such as, but not limited to a pyridine or imidazole, and an acidic moiety, such as, but not limited to a carboxylic acid, zwitterions ("inner salts") may be formed and are included within the term "salt(s)" as used herein.
- Pharmaceutically acceptable (i.e., non-toxic, physiologically acceptable) salts are preferred, although other salts are also useful. Salts of the compounds of the Formula I may be formed, for example, by reacting a compound of Formula I with an amount of acid or base, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization.
- Exemplary acid addition salts include acetates, ascorbates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, fumarates, hydrochlorides, hydrobromides, hydroiodides, lactates, maleates, methanesulfonates, naphthalenesulfonates, nitrates, oxalates, phosphates, propionates, salicylates, succinates, sulfates, tartarates, thiocyanates, toluenesulfonates (also known as tosylates.) and the like.
- Exemplary basic salts include ammonium salts, alkali metal salts such as sodium, lithium, and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases (for example, organic amines) such as dicyclohexylamines, t-butyl amines, and salts with amino acids such as arginine, lysine and the like.
- Basic nitrogen-containing groups may be quarternized with agents such as lower alkyl halides (e.g. methyl, ethyl, and butyl chlorides, bromides and iodides), dialkyl sulfates (e.g.
- esters of the present compounds include the following groups: (1 ) carboxylic acid esters obtained by esterification of the hydroxy groups, in which the non-carbonyl moiety of the carboxylic acid portion of the ester grouping is selected from straight or branched chain alkyl (for example, acetyl, n- propyl, t-butyl, or n-butyl), alkoxyalkyl (for example, methoxymethyl), aralkyl (for example, benzyl), aryloxyalkyl (for example, phenoxymethyl), aryl (for example, phenyl optionally substituted with, for example, halogen, C-i ⁇ alkyl, or C-i ⁇ alkoxy or amino); (2) sulfonate esters, such as alkyl- or aralkylsulfonyl (for example, methanesulfonyl); (3) amino acid esters (for example, L-valyl or L-isoleucyl);
- the compounds of Formula (I) may contain asymmetric or chiral centers, and, therefore, exist in different stereoisomeric forms. It is intended that all stereoisomeric forms of the compounds of Formula (I) as well as mixtures thereof, including racemic mixtures, form part of the present invention.
- the present invention embraces all geometric and positional isomers. For example, if a compound of Formula (I) incorporates a double bond or a fused ring, both the cis- and trans-forms, as well as mixtures, are embraced within the scope of the invention.
- Diastereomeric mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods well known to those skilled in the art, such as, for example, by chromatography and/or fractional crystallization.
- Enantiomers can be separated by converting the enantiomeric mixture into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g., chiral auxiliary such as a chiral alcohol or Moshei ⁇ s acid chloride), separating the diastereomers and converting (e.g., hydrolyzing) the individual diastereomers to the corresponding pure enantiomers.
- an appropriate optically active compound e.g., chiral auxiliary such as a chiral alcohol or Moshei ⁇ s acid chloride
- the compounds of Formula (I) may be atropisomers (e.g., substituted biaryls) and are considered as part of this invention. Enantiomers can also be separated by use of chiral HPLC column. It is also possible that the compounds of Formula (I) may exist in different tautomeric forms, and all such forms are embraced within the scope of the invention. Also, for example, all keto-enol and imine-enamine forms of the compounds are included in the invention.
- All stereoisomers for example, geometric isomers, optical isomers and the like
- of the present compounds including those of the salts, solvates, esters and prodrugs of the compounds as well as the salts, solvates and esters of the prodrugs
- those which may exist due to asymmetric carbons on various substituents including enantiomeric forms (which may exist even in the absence of asymmetric carbons), rotameric forms, atropisomers, and diastereomeric forms, are contemplated within the scope of this invention, as are positional isomers (such as, for example, 4-pyridyl and 3-pyridyl).
- the use of the terms “salt”, “solvate”, “ester”, “prodrug” and the like, is intended to equally apply to the salt, solvate, ester and prodrug of enantiomers, stereoisomers, rotamers, tautomers, positional isomers, racemates or prodrugs of the inventive compounds.
- the present invention also embraces isotopically-labelled compounds of the present invention which are identical to those recited herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, fluorine and chlorine, such as 2H, 3H, 13C, 14C, 15N, 180, 170, 31P, 32P, 35S, 18F, and 36CI, respectively.
- Certain isotopically-labelled compounds of Formula (I) are useful in compound and/or substrate tissue distribution assays. Tritiated (i.e., 3H) and carbon-14 (i.e., 14C) isotopes are particularly preferred for their ease of preparation and detectability.
- lsotopically labeled compounds of Formula (I) can generally be prepared by following procedures analogous to those disclosed in the Schemes and/or in the Examples herein below, by substituting an appropriate isotopically labeled reagent for a non-isotopically labeled reagent.
- the compounds according to the invention have pharmacological properties; in particular, the compounds of Formula I can be inhibitors, regulators or modulators of protein kinases.
- protein kinases that can be inhibited, regulated or modulated include cyclin-dependent kinases (CDKs), such as, CDK1 , CDK2, CDK3, CDK4, CDK5, CDK6 and CDK7, CDK8, mitogen activated protein kinase (MAPK/ERK), glycogen synthase kinase 3 (GSK3beta), Pim-1 kinases, Chk kinases (such as Chk1 and Chk2), tyrosine kinases, such as the HER subfamily (including, for example, EGFR (HER1 ), HER2, HER3 and HER4), the insulin subfamily (including, for example, INS-R, IGF-IR, IR, and IR-R), the PDGF subfamily (including, for example, PDGF-
- the compounds of Formula I can be inhibitors of protein kinases such as, for example, the inhibitors of the checkpoint kinases such as Chk1 , Chk2 and the like.
- Preferred compounds can exhibit IC5O values of less than about 5 ⁇ m, preferably about 0.001 to about 1.0 ⁇ m, and more preferably about 0.001 to about 0.1 ⁇ m.
- the assay methods are described in the Examples set forth below.
- the compounds of Formula I can be useful in the therapy of proliferative diseases such as cancer, autoimmune diseases, viral diseases, fungal diseases, neurological/neurodegenerative disorders, arthritis, inflammation, anti-proliferative (e.g., ocular retinopathy), neuronal, alopecia and cardiovascular disease.
- proliferative diseases such as cancer, autoimmune diseases, viral diseases, fungal diseases, neurological/neurodegenerative disorders, arthritis, inflammation, anti-proliferative (e.g., ocular retinopathy), neuronal, alopecia and cardiovascular disease.
- proliferative diseases such as cancer, autoimmune diseases, viral diseases, fungal diseases, neurological/neurodegenerative disorders, arthritis, inflammation, anti-proliferative (e.g., ocular retinopathy), neuronal, alopecia and cardiovascular disease.
- the compounds of Formula I can be useful in the treatment of a variety of cancers, including (but not limited to) the following: tumor of the bladder, breast (including BRCA-mutated breast cancer, colorectal, colon, kidney, liver, lung, small cell lung cancer, non-small cell lung cancer, head and neck, esophagus, bladder, gall bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin, including squamous cell carcinoma; leukemia, acute lymphocytic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T- cell lymphoma, Hodgkins lymphoma, non-Hodgkins lymphoma, hairy cell lymphoma, mantle cell lymphoma, myeloma and Burkett's lymphoma; chronic lymphocytic leukemia ("CLL”), acute and chronic myelogenous leukemia, myelodysplastic syndrome and promyelocytic leuk
- inhibitors could act as reversible cytostatic agents which may be useful in the treatment of any disease process which features abnormal cellular proliferation, e.g., benign prostate hyperplasia, familial adenomatosis polyposis, neuro-fibromatosis, atherosclerosis, pulmonary fibrosis, arthritis, psoriasis, glomerulonephritis, restenosis following angioplasty or vascular surgery, hypertrophic scar formation, inflammatory bowel disease, transplantation rejection, endotoxic shock, and fungal infections.
- Compounds of Formula I may also be useful in the treatment of Alzheimer's disease, as suggested by the recent finding that CDK5 is involved in the phosphorylation of tau protein (J. Biochem, (1995) 117, 741-749).
- Compounds of Formula I may induce or inhibit apoptosis.
- the apoptotic response is aberrant in a variety of human diseases.
- Compounds of Formula I, as modulators of apoptosis, will be useful in the treatment of cancer (including but not limited to those types mentioned hereinabove), viral infections (including but not limited to herpevirus, poxvirus, Epstein- Barr virus, Sindbis virus and adenovirus), prevention of AIDS development in HIV-infected individuals, autoimmune diseases (including but not limited to systemic lupus, erythematosus, autoimmune mediated glomerulonephritis, rheumatoid arthritis, psoriasis, inflammatory bowel disease, and autoimmune diabetes mellitus), neurodegenerative disorders (including but not limited to Alzheimer's disease, AIDS-related dementia, Parkinson's disease, amyotrophic lateral sclerosis, retinitis pigmentosa, spinal muscular atrophy and cerebellar degeneration), mye
- Compounds of Formula I can modulate the level of cellular RNA and DNA synthesis. These agents would therefore be useful in the treatment of viral infections (including but not limited to HIV, human papilloma virus, herpesvirus, poxvirus, Epstein-Barr virus, Sindbis virus and adenovirus).
- viral infections including but not limited to HIV, human papilloma virus, herpesvirus, poxvirus, Epstein-Barr virus, Sindbis virus and adenovirus.
- Chemoprevention is defined as inhibiting the development of invasive cancer by either blocking the initiating mutagenic event or by blocking the progression of pre-malignant cells that have already suffered an insult or inhibiting tumor relapse.
- Compounds of Formula I may also be useful in inhibiting tumor angiogenesis and metastasis.
- Compounds of Formula I may also act as inhibitors of cyclin dependent kinases and other protein kinases, e.g., protein kinase C, her2, raf 1 , MEK1 , MAP kinase, EGF receptor, PDGF receptor, IGF receptor, PI3 kinase, weel kinase, Src, AbI and thus be effective in the treatment of diseases associated with other protein kinases.
- protein kinase C her2, raf 1 , MEK1 , MAP kinase, EGF receptor, PDGF receptor, IGF receptor, PI3 kinase, weel kinase, Src, AbI and thus be effective in the treatment of diseases associated with other protein kinases.
- Another aspect of this invention is a method of treating a mammal (e.g., human) having a disease or condition associated with kinases (e.g., CDKs, CHK and Aurora kinases) by administering a therapeutically effective amount of at least one compound of Formula I, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound to the mammal.
- a mammal e.g., human
- kinases e.g., CDKs, CHK and Aurora kinases
- a preferred dosage is about 0.001 to 1000 mg/kg of body weight/day of the compound of Formula I.
- An especially preferred dosage is about 0.01 to 25 mg/kg of body weight/day of a compound of Formula I, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound.
- the compounds of this invention may also be useful in combination (administered together or sequentially) with one or more of anti-cancer treatments such as radiation therapy, and/or one or more anti-cancer agents different from the compound of Formula I.
- the compounds of the present invention can be present in the same dosage unit as the anti-cancer agent or in separate dosage units.
- Another aspect of the present invention is a method of treating one or more diseases associated with a kinase (such as CDK, CHK and Aurora), comprising administering to a mammal in need of such treatment: an amount of a first compound, which is a compound of Formula 1 , or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof; and an amount of at least one second compound, the second compound being an anti-cancer agent different from the compound of Formula 1 , wherein the amounts of the first compound and the second compound result in a therapeutic effect.
- a kinase such as CDK, CHK and Aurora
- Non-limiting examples of suitable anti-cancer agent is selected from the group consisting of a cytostatic agent, cisplatin, doxorubicin, liposomal doxorubicin (e.g., Caelyx®, Myocet®, Doxil®), taxotere, taxol, etoposide, irinotecan, camptostar, topotecan, paclitaxel, docetaxel, epothilones, tamoxifen, 5-fluorouracil, methoxtrexate, temozolomide, cyclophosphamide, SCH 66336, R115777®, L778.123®, BMS 214662®, Iressa®, Tarceva®, antibodies to EGFR, antibodies to IGFR (including, for example, those published in US 2005/0136063 published June 23, 2005), KSP inhibitors (such as, for example, those published in WO 2006/098962 and WO 2006/098961 ;
- such combination products employ the compounds of this invention within the dosage range described herein and the other pharmaceutically active agent or treatment within its dosage range.
- the CDC2 inhibitor olomucine has been found to act synergistically with known cytotoxic agents in inducing apoptosis (J. Cell Sci., (1995) 108, 2897.
- Compounds of Formula I may also be administered sequentially with known anticancer or cytotoxic agents when a combination formulation is inappropriate.
- the invention is not limited in the sequence of administration; compounds of Formula I may be administered either prior to or after administration of the known anticancer or cytotoxic agent.
- cytotoxic activity of the cyclin-dependent kinase inhibitor flavopiridol is affected by the sequence of administration with anticancer agents. Cancer Research, (1997) 57, 3375. Such techniques are within the skills of persons skilled in the art as well as attending physicians.
- this invention includes combinations comprising an amount of at least one compound of Formula I, or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof, and an amount of one or more anti-cancer treatments and anti-cancer agents listed above wherein the amounts of the compounds/ treatments result in desired therapeutic effect.
- Another aspect of the present invention is a method of inhibiting one or more Aurora kinases in a patient in need thereof, comprising administering to the patient a therapeutically effective amount of at least one compound of Formula 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- Another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more Aurora kinases in a patient in need thereof, comprising administering a therapeutically effective amount of at least one compound of Formula 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- Yet another aspect of the present invention is a method of treating one or more diseases associated with Aurora kinase, comprising administering to a mammal in need of such treatment an amount of a first compound, which is a compound of Formula 1 , or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof; and an amount of at least one second compound, the second compound being an anti-cancer agent, wherein the amounts of the first compound and the second compound result in a therapeutic effect.
- a first compound which is a compound of Formula 1 , or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof
- Another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more Aurora kinases in a patient in need thereof, comprising administering a therapeutically effective amount of a pharmaceutical composition comprising in combination at least one pharmaceutically acceptable carrier and at least one compound according to Formula 1 , or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- the Aurora kinase to be inhibited can be Aurora A, Aurora B and/or Aurora C.
- Another aspect of the present invention is a method of inhibiting one or more Checkpoint kinases in a patient in need thereof, comprising administering to the patient a therapeutically effective amount of at least one compound of formula 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- Another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more Checkpoint kinases in a patient in need thereof, comprising administering a therapeutically effective amount of at least one compound of formula 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- Yet another aspect of the present invention is a method of treating one or more diseases associated with Checkpoint kinase, comprising administering to a mammal in need of such treatment an amount of a first compound, which is a compound of formula 1 , or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof; and an amount of at least one second compound, the second compound being an anti-cancer agent, wherein the amounts of the first compound and the second compound result in a therapeutic effect.
- a first compound which is a compound of formula 1 , or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof
- Another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more Checkpoint kinases in a patient in need thereof, comprising administering a therapeutically effective amount of a pharmaceutical composition comprising in combination at least one pharmaceutically acceptable carrier and at least one compound according to formula 1 , or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- the checkpoint kinase to be inhibited can be Chk1 and/or Chk2.
- Another aspect of the present invention is a method of inhibiting one or more cyclin dependent kinases in a patient in need thereof, comprising administering to the patient a therapeutically effective amount of at least one compound of formula 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- Another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more cyclin dependent kinases in a patient in need thereof, comprising administering a therapeutically effective amount of at least one compound of formula 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- Yet another aspect of the present invention is a method of treating one or more diseases associated with cyclin dependent kinase, comprising administering to a mammal in need of such treatment an amount of a first compound, which is a compound of formula 1 , or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof; and an amount of at least one second compound, the second compound being an anti-cancer agent, wherein the amounts of the first compound and the second compound result in a therapeutic effect.
- a first compound which is a compound of formula 1 , or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof
- an amount of at least one second compound the second compound being an anti-cancer agent
- Another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more cyclin dependent kinases in a patient in need thereof, comprising administering a therapeutically effective amount of a pharmaceutical composition comprising in combination at least one pharmaceutically acceptable carrier and at least one compound according to formula 1 , or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- the checkpoint kinase to be inhibited can be CDK1 and/or CDK2.
- Another aspect of the present invention is a method of inhibiting one or more tyrosine kinases in a patient in need thereof, comprising administering to the patient a therapeutically effective amount of at least one compound of Formula 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- Yet another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more tyrosine kinases in a patient in need thereof, comprising administering a therapeutically effective amount of at least one compound of Formula 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- Another aspect of the present invention is a method of treating one or more diseases associated with tyrosine kinase, comprising administering to a mammal in need of such treatment an amount of a first compound, which is a compound of
- Another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more tyrosine kinases in a patient in need thereof, comprising administering a therapeutically effective amount of a pharmaceutical composition comprising in combination at least one pharmaceutically acceptable carrier and at least one compound according to Formula 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- the tyrosine kinase can be VEGFR (VEGF-R2), EGFR,
- HER2 HER2, SRC, JAK and/or TEK.
- Another aspect of the present invention is a method of inhibiting one or more
- Pim-1 kinases in a patient in need thereof comprising administering to the patient a therapeutically effective amount of at least one compound of Formula 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- Yet another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more Pim-1 kinases in a patient in need thereof, comprising administering a therapeutically effective amount of at least one compound of Formula 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- Another aspect of the present invention is a method of treating one or more diseases associated with Pim-1 kinase, comprising administering to a mammal in need of such treatment an amount of a first compound, which is a compound of Formula 1 , or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof; and an amount of at least one second compound, the second compound being an anti-cancer agent, wherein the amounts of the first compound and the second compound result in a therapeutic effect.
- a first compound which is a compound of Formula 1 , or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof
- an amount of at least one second compound the second compound being an anti-cancer agent
- Another aspect of the present invention is a method of treating, or slowing the progression of, a disease associated with one or more Pim-1 kinases in a patient in need thereof, comprising administering a therapeutically effective amount of a pharmaceutical composition comprising in combination at least one pharmaceutically acceptable carrier and at least one compound according to Formula 1 or a pharmaceutically acceptable salt, solvate, ester or prodrug thereof.
- the pharmacological properties of the compounds of this invention may be confirmed by a number of pharmacological assays.
- the exemplified pharmacological assays which are described herein below have been carried out with compounds according to the invention and their salts, solvates, esters or prodrugs.
- compositions which comprise at least one compound of Formula I, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound and at least one pharmaceutically acceptable carrier.
- inert, pharmaceutically acceptable carriers can be either solid or liquid.
- Solid form preparations include powders, tablets, dispersible granules, capsules, cachets and suppositories.
- the powders and tablets may be comprised of from about 5 to about 95 percent active ingredient.
- Suitable solid carriers are known in the art, e.g., magnesium carbonate, magnesium stearate, talc, sugar or lactose. Tablets, powders, cachets and capsules can be used as solid dosage forms suitable for oral administration. Examples of pharmaceutically acceptable carriers and methods of manufacture for various compositions may be found in A.
- Liquid form preparations include solutions, suspensions and emulsions. As an example may be mentioned water or water-propylene glycol solutions for parenteral injection or addition of sweeteners and opacifiers for oral solutions, suspensions and emulsions. Liquid form preparations may also include solutions for intranasal administration.
- Aerosol preparations suitable for inhalation may include solutions and solids in powder form, which may be in combination with a pharmaceutically acceptable carrier, such as an inert compressed gas, e.g. nitrogen.
- a pharmaceutically acceptable carrier such as an inert compressed gas, e.g. nitrogen.
- solid form preparations that are intended to be converted, shortly before use, to liquid form preparations for either oral or parenteral administration.
- liquid forms include solutions, suspensions and emulsions.
- the compounds of the invention may also be deliverable transdermally.
- the transdermal compositions can take the form of creams, lotions, aerosols and/or emulsions and can be included in a transdermal patch of the matrix or reservoir type as are conventional in the art for this purpose.
- the compounds of this invention may also be delivered subcutaneously.
- the compound is administered orally or intravenously.
- the pharmaceutical preparation is in a unit dosage form.
- the preparation is subdivided into suitably sized unit doses containing appropriate quantities of the active component, e.g., an effective amount to achieve the desired purpose.
- the quantity of active compound in a unit dose of preparation may be varied or adjusted from about 1 mg to about 100 mg, preferably from about 1 mg to about 50 mg, more preferably from about 1 mg to about 25 mg, according to the particular application.
- the actual dosage employed may be varied depending upon the requirements of the patient and the severity of the condition being treated. Determination of the proper dosage regimen for a particular situation is within the skill of the art. For convenience, the total daily dosage may be divided and administered in portions during the day as required. The amount and frequency of administration of the compounds of the invention and/or the pharmaceutically acceptable salts thereof will be regulated according to the judgment of the attending clinician considering such factors as age, condition and size of the patient as well as severity of the symptoms being treated.
- a typical recommended daily dosage regimen for oral administration can range from about 1 mg/day to about 500 mg/day, preferably 1 mg/day to 200 mg/day, in two to four divided doses.
- kits comprising a therapeutically effective amount of at least one compound of Formula I, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound and a pharmaceutically acceptable carrier, vehicle or diluent.
- kits comprising an amount of at least one compound of Formula I, or a pharmaceutically acceptable salt, solvate, ester or prodrug of said compound and an amount of at least one anticancer therapy and/or anti-cancer agent listed above, wherein the amounts of the two or more ingredients result in desired therapeutic effect.
- VXR-200 (200 MHz, 1H), Varian Gemini-300 (300 MHz) or XL-400 (400 MHz) and are reported as ppm down field from Me4Si with number of protons, multiplicities, and coupling constants in Hertz indicated parenthetically.
- analyses was performed using an Applied Biosystems API-100 mass spectrometer and Shimadzu SCL-10A LC column: Altech platinum C18, 3 micron, 33mm x 7mm ID; gradient flow: 0 min - 10% CH3CN, 5 min - 95% CH3CN, 7 min - 95% CH3CN, 7.5 min - 10% CH3CN, 9 min - stop. The retention time and observed parent ion are given.
- Part A Prepared according to US20060106023 (A1 ).
- Part B To a solution of compound from Example 1 , Part A (2.00 g, 8.19 mmol) in DMF (50 mL) was added N-iodosuccinimide (1.84 g, 8.19 mmol). The reaction mixture was stirred at 6OC for 16 hours. The mixture was cooled to 25C and concentrated. The residue was dissolved in DCM with a small amount of methanol and then loaded on the column. Purification by column chromatography (Si ⁇ 2, 40% ethyl acetate/hexanes) afforded compound 4 as a white solid 2.30 g (76%).
- Part C A suspension of bromide from Part B (45.6 g), Pd(PPh3)4 (10.8 g), potassium carbonate (77.4 g), trimethylboroxine (46.9 g) and potassium carbonate (77.4 g) in DMF (410 mL) was heated overnight under nitrogen at 105C. After cooling, the mixture was diluted with ethyl acetate (1 L), washed with brine (2 x 500 mL), dried (magnesium sulfate), filtered, concentrated and purified by chromatography on silica gel. The title compound was obtained as a pale yellow solid (21.4 g, 64%).
- Part D To a DMF (400 mL) solution of compound from Example 1 , Part C (21.8 g) was added N-iodosuccinimide (26.9 g) and the resulting mixture was heated overnight at 6OC. The mixture was concentrated and water (400 mL) was added. After stirring 1 hr at rt, saturated sodium carbonate was added (250 mL) and subsequently stirred an additional 30 min at rt. The mixture was filtered, washed with water, methanol (100 mL) and the filter cake was dried overnight under vacuum. A brown solid was obtained (31.4 g, 87%).
- Part E A flask was charged with iodide from Part D (1.00 equiv), Bpin-compound 5a (1.3 equiv), PdCI2(dppf) (0.1 equiv) and potassium phosphate monohydrate (3.0 equiv). After purging the flask with argon, 1 ,4-dioxane (50 mL) and water (5) were added and the resulting mixture was heated at 8OC overnight (23 h). The reaction was cooled to room temperature. EtOAc was added to the reaction mixture and filtered through Celite. After concentration the residue was purified by column chromatography (silica gel, 25% EtOAc/hexane) to give the title compound.
- Part F To a solution of compound from Example 1 , Part E (1.0 equiv) in DCM (10 mL) was added m-CPBA (2.05 equiv) in one portion. The resulting mixture was stirred at room temperature for 30 min. The mixture was concentrated and then partitioned between EtOAc and water. The organic layer was washed with NaHCO3 (sat. aq., twice), brine and dried (Na2SO4). After concentration, the title compound was obtained and used in the next step directly without further purification.
- Step A To a solution of 4 (76 mg, 0.14 mmol) in 6 mL of THF was added Pd(PPh3J4 (16 mg, 0.014 mmol) and 0.35 mL of MeZnCI (2 M solution in THF, 0.69 mmol). The reaction was stirred at 80 °C for 20 min. It was cooled to room temperature and quenched by adding 0.5 mL of MeOH. It was diluted with 30 mL of CH2CI2 and washed with 20 mL of 0.5 N aqueous HCI solution. The solvent was removed under vacuum.
- Step B The above crude material was dissolved in 5 mL of THF. To the solution was added 0.5 mL of LiBHEt3 (1 M solution in THF). The reaction was stirred at room temperature for 30 min.
- Step A Mixture of compound 6 (17 mg, 0.032 mmol) and sodium azide (15 mg, 0.23 mmol) in 1 mL of DMF was heated at 70 °C for 3 h. It was cooled to room temperature and added 10 mL of water. The resulting solid was collected by filtration and purified by flash chromatography eluting with 5% MeOHZCH2CI2 to give 12 mg of (3- azidomethyl-isothiazol-5-yl)- ⁇ 6-methyl-3-[1-(2-trimethylsilanyl-ethoxymethyl)-1 H- pyrazol-4-yl]-imidazo[1 ,2-a]pyrazin-8-yl ⁇ -amine.
- Step B The above material was dissolved in 3 mL of MeOH. To the solution was added 15 mg of 10% wt. Pd/C. The mixture was stirred under H2 (1 atm) for 1 h. It was filtered through celite. The filtrate was concentrated under vacuum to give 12 mg of compound 7.
- NMR (400 MHz, CDCI3) ⁇ 7.88 (s, 1 H), 7.80 (s, 1 H), 7.60 (s, 1 H), 7.47 (s, 1 H), 6.86 (s, 1 H), 5.55 (s, 2H), 4.00 (brs, 2H), 3.65 (t, 2H), 2.50 (s, 3H), 1.00 (t, 2H), 0.00 (s, 9H).
- Step A To a solution of compound 7 (9 mg, 0.02 mmol) in 1 mL of MeOH/CH2Cl2 (1 :1 ), was added formaldehyde (40% wt. in water, 6 mg, 0.2 mmol). It was stirred at room temperature for 15 min when NaBH4 (16 mg, 0.4 mmol) was added in two portions.
- Step B The above material was then dissolved in 2 mL of THF. The resulting solution was heated at 70 °C when 0.5 mL of 4 N HCI in dioxane was added. To the resulting mixture was added 1 mL of MeOH. The reaction was stirred at 70 °C for 1 h and then cooled to room temperature. Most of the solvent was removed under vacuum. To the residue was added 5 mL of ether. The solid was collected by filtration and washed with ether to give 5 mg of compound 9 as its HCI salt form.
- Step A A solution of compound 10 (100 mg, 0.220 mmol) and pyrrolidine (156 mg, 2.20 mmol) in 14 mL of CH2CI2 was stirred at room temperature for 20 min. To the solution was added two drops of acetic acid, followed by NaBH4 (67 mg, 1.8 mmol). The resulting mixture was stirred at room temperature for 5 min when 3 mL of MeOH was added. The stirring was continued for additional 20 min. The reaction was quenched by adding 15 mL of saturated aqueous NaHCO3 solution. After diluted with 20 mL of CH2Ck, the organic was isolated. The solvent was removed under vacuum.
- Step B To a solution of ⁇ 6-methyl-3-[1-(2-trimethylsilanyl-ethoxymethyl)-1 H-pyrazol-4- yl]-imidazo[1 ,2-a]pyrazin-8-yl ⁇ -(3-pyrrolidin-1-ylmethyl-isothiazol-5-yl)-amine (98 mg, 0.19 mmol) in 8 mL of THF heated at 70 °C, was added 2 mL of 4 N HCI in dioxane. To the resulting mixture was added MeOH until it became homogeneous. The reaction was stirred at 70 °C for 1 h and then cooled to room temperature. To the mixture was added 3 mL of ether.
- Part C A solution of alcohol from Part B (0.52 g, 0.88 mmol, 1 equivalent) in DCM (15 mL) was treated with triethylamine (1.5 equivalents) for 15 min at OC (ice-bath), at which time, methanesulfonyl chloride (1.2 equivalents) was added to the reaction at OC. The resulting solution was allowed to slowly warm to rt and continued to stir at rt for a further 3h. LC-MS analysis indicated the reaction was complete. The reaction mixture was diluted with ethyl acetate (10OmL) and washed with water, brine, dried (anh.
- Part D A solution of the respective alcohol (3 equivalents) in THF (1.5 mL) was treated with NaH (60% dispersion in oil, 2 equivalents) for 15 min at rt, at which time, mesylate from Part C (40 mg, 0.06 mmol, 1 equivalent) was added to the reaction mixture. After stirring at rt for 1h, LC-MS analysis indicated the reaction was complete. The reaction was quenched with sat. aq. ammonium chloride and then extracted with ethyl acetate (twice). The combined organic layer was dried (sodium sulfate) and concentrated to afford crude ether, which was used without further purification.
- Part E A solution of compound from Part D in 1 ,4-dioxane (1 mL) was treated with 4N HCI in 1 ,4-dioxane solution (1 mL) at 6OC for 10 min at which time HPLC-MS indicated that the reaction was complete. The solvent was removed and the residue was purified by Prep-LC. Conversion to a hydrochloric salt afforded compounds listed in Table 3.
- Example 19 was prepared in similar manner to Example 4. 1H NMR (300 MHz, DMSO-de) ⁇ 12.4 (bs, 1 H), 7.81 (s, 1 H), 7.75 (s, 1 H), 7.59 (s, 1 H), 3.85 (s, 3H), 2.49 (s, 3H).
- Example 20 was prepared in similar manner to Example 17, Part A.
- Example 23 was prepared in a similar manner to example 22 with the substitution of 3-methylpiperidine for piperidine.
- Example 24 was prepared in a similar manner to example 23 with the substitution of pyrrolidine for piperidine.
- Example 26 was prepared in a similar manner to example 25.
- 1H NMR 300 MHz, CD3OD
- a flask containing the prepared aryl iodide scaffolds (compound from Example 22, 23, or 24, 1 equivalent), commercially available or readily prepared in 1 to 3 steps aryl/heteroaryl/alkyl boronic acid/ester/boroxine or aryl/heteroaryl/alkyl magnesium bromide or aryl/heteroaryl/alkyl zinc chloride (1.5 - 3 equivalents), potassium phosphate or potassium carbonate (2- 3 equivalents) and Pd(PPh3J4 or PdCI2dppf (0.05 - 0.10 equivalents) was evacuated, backfilled with nitrogen and repeated.
- Example 30 was prepared in a similar manner as Example 29.
- 1H NMR 300 MHz, CD3OD
- ⁇ 7.77 (s, 1 H), 7.68 (s, 1 H), 7.20 (s, 1 H), 4.39 (s, 2H), 3.47-3.67 (m, 2H), 2.97 (m, 1 H), 2.71 (m, 1 H), 2.55 (s, 3H)1 1.77-2.01 (m, 4H), 1.20 (m, 1 H), 1.00 (d, J 6.4 Hz, 3H).
- HPLC tR 4.98 min. Mass calculated for formula C17H2iCIN6S 376.12; observed MH+ 377.6 (m/z).
- Example 31 was prepared in a similar manner to compound 29 with the substitution of tetrachlorodibromoethane for hexachloroethane.
- 1H NMR 300 MHz, CD3OD
- HPLC tR 5.19 min. Mass calculated for formula C16Hi9BrN6S 406.06; observed MH+ 407.4 (m/z).
- Example 36 was prepared in a similar manner to Example 31.
- 1H NMR 300 MHz, CD3OD
- HPLC tR 5.00 min (UV 254nm)- Mass calculated for formula C16H19IN6S 454.04; observed MH+ (ESI MS) 455.0 (m/z).
- Part B To a stirred solution of (2-Bromo-thiazol-5-yl)-carbamic acid tert-butyl ester (2.5 g, 8.9928 mmol) in 1 ,4-dioxane (20.0 mL) were added tributy(vinyl)tin (2.9 mL, 9.892 mmol), 2,6-di-tert-butyl-4-methylphenol (cat. amt) and tetrakis(triphenyl phosphine) palladium(O) (506.0 mg, 0.4496 mmol). The reaction mixture was heated to 100 °C and stirred for 12 hrs, LCMS showed the complete disappearance of the starting material.
- Part D To a stirred solution of (2-Formyl-thiazol-5-yl)-carbamic acid tert-butyl ester (0.76 g, 2.857 mmol) in 1 ,2-dichloroethane(10 mL) were added Morpholine (250 mg, 1.1135 mmol) triacetoxysodium borohydride (472 mg, 2.227 mmol) and Cat amount acetic acid (three drops) and stirred for two hrs at room temp. To the reaction mixture was added sodium borohydride (126 mg, 3.3405 mmol) and stirred for one hrs. LCMS showed the disappearance of the starting material.
- Part E To a stirred solution of (2-M ⁇ rpholin-4-ylmethyl-thiazol-5-yl)-carbamic acid tert- butyl ester (80.0 mg, 0.268 mmol) in dichloromethane(5 mL) was added iodotirmethylsilane ( 44 ⁇ l_, 0.321 mmol) and stirred for 10 min. LCMS showed the disappearance of the starting material.
- Part F To a stirred solution of 2-Morpholin-4-ylmethyl-thiazol-5-ylamine (30.0 mg, 0.151 mmol) in DMSO (2.5 mL) was added 8-Methanesulfonyl-6-methyl-3-(1 H- pyrazol-4-yl)-imidazo[1 ,2-a]pyrazine ( 25.0 mg, 0.09045 mmol) followed by NaH 60% in mineral oil (48 mg, 1.206 mmol) and stirred for 30 min. LCMS showed the disappearance of the starting material.
- Et3N (1261.6 uL, 9.05 mmol) was added at O°C to a mixture of 5-tert- Butoxycarbonylamino- thiophene-2-carboxylic acid (550 mg, 2.26mmol), EDCI (1086 mg, 5.65 mmol) , and piperidine (447 uL, 4.52 mmol) in DMF (6ml). The reaction mixture was warmed up to room temperature and stirred at this temperature overnight.
- THF was treated with HCI in dioxane (4M; 0.5 mL) and placed in an oil bath at 70°C After heating for 30 min, a precipitate formed which dissolved upon adding 0.5 mL of methanol.
- the reaction mixture was heated at a bath temperature of 7O°C for an additional 1 hr.
- the contents of the reaction were cooled to RT and all the volatiles were removed on a rotary evaporator.
- the residue was suspended in THF and triturated with ether.
- the precipitate was collected by filtration, washed with ⁇ 10 mL of ether and dried in air (0.5 hr) and in vacuo (16 hr) to furnish 10 mg (93%) of the title compound as a yellow solid.
- Part A Lithium hexamethyldisilazide (1 M in THF; 0.18 mL) was added to an amber solution of 4-morpholin-4-ylmethyl phenylamine (0.013g; 0.068 mmol) and 8- methanesulfonyl-6-methyl-3-[1-(2-trimethylsilanyl-ethoxymethyl)-1/-/-pyrazol-4-yl]- imidazo[1 , 2-a]pyrazine (0.025g; 0.061 mmol) in 2 mL of THF at RT resulting in a burgundy solution. After stirring at RT for 20 minutes, the reaction mixture was quenched with saturated aqueous NH4CI solution.
- the contents were diluted with ethyl acetate and washed with water and brine.
- the crude material from the organic extract was purified by prep TLC (5% methanol-CH2CI2) to obtain the title compound as pale yellow oil (0.025 g; 80%).
- Part B The compound from Part A (0.025g; 0.048 mmol) was suspended in dry THF and treated with HCI in dioxane (4M; 1 mL) and heated in an oil bath set to 7O°C for 15 minutes when a white precipitate was formed. Methanol was added to dissolve some of the solid and the reaction mixture was continued to be heated for 45 minutes more. After cooling to RT, the volatiles were removed on the rotary evaporator. The residue was suspended in THF and the precipitated solid was collected by filtration, washed with ether and dried in vacuo overnight. The title compound was isolated as a beige solid (14 mg; 78%). All the analogues in Table 12 were similarly prepared. TABLE 12
- Part A A solution of 4-Amino-2-methyl-benzoic acid methyl ester (0.33 g; 2 mmol; prepared from commercially available 4-nitro-2-methyl-benzoic acid) and 8- methanesulfonyl-6-bromo-3-[1-(2-trimethylsilanyl-ethoxymethyl)-1/-/-pyrazol-4-yl]- imidazo[1 , 2-a]pyrazine (0.472 g; 1.0 mmol) was treated with LiHMDS (1 M in THF; 2 mL) at RT. The resulting burgundy solution was stirred at RT for 20 minutes and then quenched with saturated aqueous NH4CI solution. Standard work up as described for Example 65 and flash silicagel chromatography (25% EtOAc in CH2CI2) provided the title compound as pale yellow foam (0.48 g; 86%).
- Part B A solution of compound from Part A (0.48 g; 0.86 mmol) in 2 mL of dry THF was treated with a solution of dimethyl zinc (2M; 4 mL) dropwise. After the effervescence ceased, solid Pd(PPh3)4 was added and the reaction was flushed with nitrogen, fitted with a reflux condenser and heated in an oil bath at 65-70°C After 0.5 hr, the reaction mixture had turned from yellow orange to deep red and after 4 more hours, it had become an opaque black. TLC (25% EtOAc- CH2CI2) indicated the formation of a slightly more polar spot. The reaction was cooled to RT, quenched with saturated aqueous NH4CI solution and extracted with EtOAc.
- the substrate (1 g, 5.07 mmol) was dissolved in THF:H2O (12 mL, 1 :1 , v/v) and treated with K2CO3 (1.4 g, 10.15 mmol) at room temperature. Then benzyl chloroformate (0.79 ml, 5.58 mmol) in THF (2 mL) was slowly added. The mixture was stirred for 16 h. It was diluted with ethyl acetate (25 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (2 * 25 mL).
- the crude product was hydrogenated in ethyl acetate using 10% Pd/C at 1 atmosphere hydrogen pressure.
- the catalyst was filtered off, and solvent was evaporated under reduce pressure to give the crude product.
- Part A The substrate (1 eq.), amine (4 eq.), catalytic AcOH, NaB(OAc)3H in 1 ,2- dichloroethane was stirred at room temperature for 2 h. Then sodium borohydride (3 eq.) was added and the mixture was stirred for 30 min at which point LC-MS analysis indicate complete consumption of starting material to product. Then the reaction was quenched with 2N aqueous NaOH, and the mixture was stirred vigorously until two clear layer separated. The organic layer was washed with water, brine, dried (Na2SO4), filtered and concentrated under reduced pressure to give the product.
- Part B The crude product was hydrogenated in ethyl acetate using 10% Pd/C at 1 atmosphere hydrogen pressure. The catalyst was filtered off, and solvent was evaporated under reduce pressure to give the crude product.
- Part A The substrate (1 eq.), amine (4 eq.), catalytic AcOH, NaB(OAc)3H in 1 ,2- dichloroethane was stirred at room temperature for 2 h. Then sodium borohydride (3 eq.) was added and the mixture was stirred for 30 min at which point LC-MS analysis indicate complete consumption of starting material to product. Then the reaction was quenched with 2N aqueous NaOH, and the mixture was stirred vigorously until two clear layer separated. The organic layer was washed with water, brine, dried (Na2SO4), filtered and concentrated under reduced pressure to give the product.
- Part B The crude product was hydrogenated in ethyl acetate using 10% Pd/C at 1 atmosphere hydrogen pressure. The catalyst was filtered off, and solvent was evaporated under reduce pressure to give the crude product.
- Part A The substrate (1 eq.) and amine (1.5-2 eq.) was dissolved in DMSO under argon, and treated with NaH (5 eq., 60% dispersion in oil). After 30 min, LC-MS analysis indicated complete consumption of starting material. The reaction was quenched by addition of saturated aqueous NH4CI-acetonitrile (1 :1 , v/v). The two layers were separated, and the aqueous layer was extracted with ethyl acetate. The combined organic layer was washed with brine, dried (Na2SO4), filtered and concentrated under reduced pressure to give the crude product. Part B: The substrate was dissolved in 4N HCI in dioxane, and stirred at room temperature for 30 min. The solvent was then evaporated, and the residue was purified by Prep-LC. Conversion to hydrochloride salt afforded the product as solid.
- Part C To a solution of compound from Part B (30 mg, 0.105 mmol, 1 equivalent), 3- methylpiperidine (10 equivalents) in dichloromethane:methanol (5:1 ) (3 ml) was added acetic acid (1 drop). The resulting solution was stirred at rt for 30 minutes, and then sodium borohydride (8 equivalents) added to the reaction. The reaction mixture was stirred at rt for 1 hour at which time LC-MS analysis indicated the reaction was complete. The reaction was quenched with sat. aq. sodium bicarbonate and then extracted with dichloromethane (x2). The combined organic layer was dried (sodium sulfate) and concentrated.
- Step A Sodium hydride (60% dispersion in mineral oil, 6.68 g, 3.40 equiv) was slowly added in one portion to a stirring mixture of compound sulfone (20.0 g, 1.00 equiv) and aminoisothiazole (11.5 g, 1.20 equiv, as HCI salt) in DMF (490 mL) at room temperature (with aid of a room temperature water bath). Reaction was allowed to stir for 1 hour at which time HPLC analysis indicated the reaction was complete. The reaction was carefully quenched with saturated aqueous sodium bicarbonate (200 mL) and then diluted with water (1 L).
- Step B A mixture of compound from Step A (4.27g, 3.73 mmol) was dissolved in 180 mL of THF. The resulting solution was cooled to 0 °C and LiAIH4 powder (2.6 g, 68.5 mmol) was carefully added. The cooling bath was removed and the reaction was stirred at RT under a N2 atmosphere for 1.5 hr. The reaction was cooled to 0 °C and carefully quenched by the sequential addition of 2.6 mL of H2O; 2.6 mL of 15 % NaOH (aq); 7.8 mL H2O. After stirring for 10 min, the reaction was filtered through a very thin pad of Celite (rinsing with THF, EtOAc and DCM).
- Step B (Alternative procedure; e.g. Example 76-39): A solution of 4,4- difluoropiperidine hydrochloride (25.1 mg, 0.16 mmol) in THF (2.0 mL) was added NaH (60% dispersion in mineral oil, 12 mg, 0.30 mmol). The mixture was stirred under a N2 atmosphere at room temperature for 10 min, then mesylate (31.4 mg, 0.06 mmol) and NaI (4 mg, 0.03 mmol) were added to the reaction flask.
- the reaction was heated at 80 °C under a N2 atmosphere for 8 hr.
- the reaction was cooled to room temperature and 15 mL of saturated NH4CI (aq) solution was added.
- the reaction was diluted with dichloromethane (20 mL) and the layers were separated.
- the aqueous layer was extracted with dichloromethane (2 x 20 mL).
- the organic phase was washed with 15 mL of saturated NaHCO3 (aq), then brine (15 mL).
- the organic phase was dried over NaSO4 and concentrated in vacuo. Purification via preparative TLC (10% MeOH/CH2CI2) gave 19.7 mg (60% yield) of the title compound.
- Step C A mixture of compound from Step B (2.40 g, 4.49 mmol), amine (1.57 g, 13.46 mmol), and NaI (63.0 mg, 0.449 mmol) in 45 mL of THF was heated at 80 °C for 12 h. It was diluted with 200 mL of CH2CI2, and washed with 100 mL of saturated aqueous NaHCO3 solution, then with brine (100 mL). The solvent was removed under vacuum. The residue was purified by flash chromatography eluting with 5% to 10% MeOH/CH2CI2 to give 1.68 g of the title compound.
- Example 76 Using essentially the same procedures as described for Example 76, the following compounds were prepared.
- Example 76-3 1H NMR (400 MHz, CD3OD) ⁇ 8.16 (s, 2H), 8.13 (s, 1 H), 7.99 (s, 1 H),
- Example 76-7 1H-NMR (400 MHz, CD3OD ) ⁇ 8.33 m (3H), 8.15 s (1H), 7.41 s (1H), 4.80 (d, 2H), 4.15 (d, 2H), 4.06 (d, 2H), 3.62 (d, 2H), 3.58 (m, 1H), 2.68 (d, 3H), 2.21 (m, 1H), 1.81 (m , 6H) and 1.45 (s, 3H).
- HPLC-MS tR 1.80Min (UV 254nm). Mass calculated for formula C21H26N8OS 438.55, observed LC/MS m/z 439.1 (M+H).
- Example 76-8 1H-NMR (400 MHz, DMSO-d6 ) ⁇ 12.73 bs (1H), 9.2 bs (1H), 8.28 s (2H), 8.09 s (1H), 8.08 s (1H), 7.36 s (1H), 4.71 m (1H), 4.05 m (1H), 3.82 m (1H), 3.63 m (1 H), 3.25 m (2H), 1.97 m (1 H), 1.65 m (6H) and 1.30 s (3H).
- Example 76-9 1H-NMR (400 MHz, DMSO-d6 ) ⁇ 8.28 (1H), 8.25 (2H), 8.08 (1 H), 7.32 (1 H), 4.71 (1 H), 4.08 (1 H), 3.84 (1 H), 3.52 (3 H), 3.46 (1 H), 2.63 (3 H), 2.17 (2 H), 1.87-1.73(6H), 1.45(3H).
- Example 76-10 1H-NMR (400 MHz, DMSO-d6 ) ⁇ 8.28 (1H), 8.25 (2H), 8.08 (1 H), 7.32 (1 H), 4.71 (1 H), 4.08 (1 H), 3.84 (1 H), 3.52 (3 H), 3.46 (1 H), 2.63 (3 H), 2.17 (2 H), 1.87-1.73(6H), 1.45(3H).
- Example 76-11 1HNMR (400 MHz, CD3OD) ⁇ 8.20 (s, 2H), 8.14 (s, 1H), 8.03 (s, 1H), 7.25 (s, 1H), 4.48 (d, 1H), 4.37 (d, 1H), 3.46 (s, 3H), 2.91-3.60 (m, 6H), 2.62 (s, 3H),
- Example 76-12 1HNMR (400 MHz, CD3OD) ⁇ 8.20 (s, 2H), 8.14 (s, 1 H)1 8.03 (s, 1 H), 7.25 (s, 1 H)1 4.48 (d, 1 H), 4.37 (d, 1 H)1 3.46 (s, 3H)1 2.91-3.60 (m, 6H)1 2.62 (s, 3H), 1.40 - 1.89 (m, 4H), 0.92 (s, 3H).
- Example 76-40 1H NMR (400 MHz1 CD3OD) ⁇ 8.28 (s, 1 H), 8.25 (s, 2H), 8.10 (s, 1 H), 7.38 (s, 1 H), 4.59 (s, 2H)1 3.3-3.9 (m, 4H), 2.64 (s, 3H), 2.3-2.5 (m, 4H).
- Example 76-42 1H NMR (400 MHz1 CD3OD) ⁇ 8.13 (broad s, 2H), 7.85 (s, 1 H)1 7.78 (s, 1 H)1 7.15 (s, 1 H), 4.10 (d, J ⁇ 14 Hz, 1 H), 3.96 (d, J -14 Hz1 1 H)1 3.54-3.66 (m, 1 H), 3.07-3.17 (m, 1 H), 2.62-2.72 (m, 1 H), 2.53 (s, 3H), 1.82-2.21 (m, 4H).
- Step A The substrate (10 g) was suspended in THF (200 mL). Then lithium aluminum hydride solution (110 mL, 2M in THF) was slowly added. The mixture was stirred at room temperature for 12 h. The solution was cooled to 0 °C, and saturated aqueous Na2SO4 (200 mL) was slowly added. The mixture was filtered through Celite, and filtercake was washed with ethyl acetate (400 mL). The organic layer was washed with water (200 mL) and brine (200 mL). The organic layer was dried (anhydrous Na2SO4), filtered and evaporated to give the amino alcohol (6.9g).
- Step B The alcohol from Step A (1.936 g) was dissolved in dichloromethane (80 mL), and treated with proton sponge (8.32 g) at room temperature. Then trimethyloxonium tetrafluoroborate (5.69 g) was added. The mixture was stirred for 1 h. The reaction was quenched with saturated aqueous ammonium chloride solution (100 mL). The two layers were separated, and the aqueous layer was extracted with dichloromethane (2 x 100 mL).
- Step C The enantiomerically pure methyl ether from Step B in EtOH was treated with Pd(OH)2 on carbon (20% wt) and stirred in hydrogen atmosphere at atmospheric pressure at room temperature for 2 h. The mixture was filtered off, and the filtrate was evaporated under reduced pressure to give the amine.
- Step D The enatiomerically pure isomers from Step C were dissolved in (1 mmol, 277 mg) in EtOH (6 ml) was mixed with 20% Pd(OH)2 (51 mg) and stirred under H2 balloon at room temperature for 2h. Filtration through celite and concentration afforded the title compound, which was used for next step without further purification.
- LCMS tR 0.26 Min. Mass calculated for, M+ 143.1 , observed LC/MS m/z 144.1 (M+H).
- Step A The parent compounds were prepared from Example 76-2 using acid chlorides, acids, ureas and isocyanates using standard reaction conditions.
- Step B Sem-protected material from Step A was dissolved in 1 ,4-dioxane (1 mL) and treated with 4 N HCI in 1 ,4-dioxane (1 mL). then heated at 6OC for 1 hr.. The mixture was concentrated under reduced pressure and the resulting residue was purified by prep-HPLC and conversion to the hydrochloride salt afforded the title compound as a colorless solid.
- Step A The starting sulfone was prepared by essentially the same procedure described in Example 6 except that ethylboronic acid or cyclopropylboronic acid was used. Final products listed in Table 17 were obtained by using the procedures described for Example 76.
- Step A The title compound was prepared using as described for Example 7 except that f-butylamine was used.
- Step B To a solution of the product of Step A (1 equivalent) in THF (3 mL) was added DIEA (3 equivalents), and the respective trifilate (1.2 equivalents) at room temperature. The reaction was heated at reflux until consumption of starting material was observed by LC-MS analysis. The solution was cooled to room temperature and concentrated under reduced pressure. Purification by column chromatography (Si ⁇ 2, 30% ethyl acetate/dichloromethane) afforded the desired coupled intermediate. This material was dissolved in 1 ,4-dioxane, HCI (4N in dioxane) was added and the mixture was sonicated until such time that HPLC indicated no starting material remained. The mixture was concentrated under reduced pressure, purified by prep-HPLC, and conversion to the hydrochloride salt afforded the title compounds as off-white solids in Table 18.
- Step A Sodium thiomethoxide (39 mg, 3.00 equiv) was added to a stirring mixture of mesylate prepared in example 7 (100 mg, 1.00 equiv) and sodium iodide (14 mg, 0.50 equiv) in DMF (6 ml.) at room temperature. The resulting mixture was allowed to stir for 2.5 hours at which time LC-MS analysis indicated the reaction was complete. The reaction was quenched with saturated aqueous sodium bicarbonate (30 mL) and then extracted with dichloromethane (2 * 70 mL). The combined organics were dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to afford the title compound as a yellow solid, 100 mg (>99%).
- Step B m-Chloroperbenzoic acid (66 mg, 2.05 equiv) was added to a stirring solution of compound from Step A (91 mg, 1.00 equiv) in dichloromethane (3 mL) at room temperature. The mixture was allowed to stir for 2 hours at which time thin layer chromatography indicated the reaction was complete. The mixture was diluted with ethyl acetate (40 mL) and then washed with saturated aqueous sodium bicarbonate (15 mL) and brine (10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure.
- Step C To a solution of nitroamide (435.6 mg, 1.69 mmol) in HOAc (20 mL) was added iron powder (471.5 mg, 8.44 mmol). The reaction mixture was heated at 7O°C for 30 min. The mixture was cooled to room temperature and concentrated to dryness. To the residue was added 3OmL of 20% MeOH/CH2CI2 followed by 20 mL of saturated aqueous NaHCO3. The mixture was stirred until it stoped bobbling. The mixture was extracted by EtOAc (x 2), dried over Na2SO4, and then concentrated. The crude amine was used for displacement reaction without further purification.
- Step A To a solution of nitroester (2285 mg, 12.22 mmol) in HOAc (55 mL) was added iron powder (6825 mg, 122.20 mmol). The reaction mixture was heated at 75°C for 10 min. The mixture was cooled to room temperature and then added 200 mL of MeOH. The resulting mixture was filtered through celite (the celite was rinsed with additional amount MeOH). The filtrate was concentrated to remove most of AcOH. To the residue was added 50 mL of 20% MeOH/CH2CI2 followed by saturated aqueous NaHC ⁇ 3 until it stoped bubbling. The mixture was extracted by EtOAc (x 2), dried over Na2SO4, and then concentrated. The crude amine was used without further purification.
- Step C To a solution of the product of Step B (206 mg, 0.4271 mmol) in THF (8 mL) was treated with DIBAL (1.0 M in CH2CI2, 2.56 mL) at -78°C dropwise. After stirring at -78°C for 4.5h, LCMS indicated the existence of small amount of starting material.. Two more equivalents DIBAL (0.85 mL) were added. After stirring at -78°C for another 0.5 h, brine (6 mL) was added portionwise at -78°C to quench the excess reagents. The reaction mixture was extracted with CH2CI2 (3X).
- Step D To a solution of alcohol from Step C (537 mg, 1.17 mmol) in THF (26 mL), was added H2O (0.078 mL) followed by Dess-Martin periodinane (599 mg, 1.41 mmol) at 0°C The reaction was stirred at room temperature until LCMS indicated the reaction was complete.
- Step A To a solution of mesylate (1.1 g, 1.65 mmol) in DMSO (20 mL) at room temperature was added NaI (280 mg, 1.88 mmol) and NaCN (300 mg, 6.12 mmol). The mixture was stirred at 60 °C for 1 h. It was diluted with 200 mL of EtOAc and washed with water (200 mL X 2). The solvent was removed under vacuum. The residue was purified by column chromatography (Si ⁇ 2, 60% EtOAc/hexanes) to afford 980 mg of the title compound.
- Step B A solution of compound from Step A (530 mg, 0.889 mmol) in 30 mL of CH2Cb was cooled to 0 °C.
- Step C A solution of compound from Step B (400 mg, 0.667 mmol) and NaOAc (400 mg, 4.88 mmol) in 20 mL of AcOH was stirred at 60 °C. To this was slowly added t- butyl nitrite (1.40 mL, 11.8 mmol). The reaction was stirred at 60 °C for 20 min. It was cooled to room temperature and added 20 mL of CH2CI2. The solid was filtered off, and the solvent in the filtrate was removed under vacuum. The residue was diluted with 100 mL of CH2CI2 and washed with 50 mL of saturated NaHCO3 aqueous solution. The organic portion was concentrated. The residuw was dissolved in 10 mL of MeOH.
- Step A To a solution of Example 90 (200 mg, 0.333 mol) in 10 mL of THF, was added NEt3 (84 mg, 0.830 mmol) followed by methanesulfonyl chloride (76.4 mg, 0.667 mmol). The reaction was stirred at room temperature for 20 min. It was quenched by adding 10 mL of water and diluted with 50 mL of CH2CI2. The mixture was washed with 20 mL of 0.5 N aqueous HCI solution. The organic was dried over anhydrous Na2SO4. The solvent was removed under vacuum. The residue was purified by column chromatography (SiO2, 70% EtOAc/hexanes) to give 180 mg of the title compound.
- Step B A mixture of mesylate from Step A (42 mg, 0.062 mmol), thiomorpholine (16 mg, 0.16 mmol), K2CO3 (8.5 mg, 0.062 mmol) and a trace amount of NaI in 1.5 mL of THF was stirred at 80 °C for 24 h. It was cooled to room temperature. The solvent was removed under vacuum. The residue was purified by column chromatography (SiO2, 5% 7 N NH3 in MeOH/CH2CI2) to give 37 mg of the title compound.
- Step C To a solution of product from Step B (37 mg, 0.054 mmol) in 2 mL of THF/MeOH (1 :1 ) stirred at 80 °C, was added 0.5 mL of 4 N HCI in dioxane solution. The reaction was stirred at 80 °C for 30 min. It was cooled to room temperature and diluted with 2 mL of THF and 1 mL of ether. The solid was collected by filtration and washed with ether to give 26 mg of the title compound as its HCI salt form.
- HPLC-MS tR 2.21 min (UV 254nm)- Mass calculated for formula C19H2ONsOS 426.1 ; observed MH+ (LCMS) 427.2 (m/z).
- Step A To a solution of carbon tetrabromide (170 mg, 0.512 mmol) in 4 mL of CH2CI2 stirred at 0 °C, was added PPh3 (267 mg, 1.02 mmol). The reaction was stirred at 0 °C for 15 min when the aldehyde (200 mg, 0.341 mmol) was added. The resulting solution was further stirred at 0 °C for 15 min. It was quenched with 10 mL of saturated NaHCO3 aqueous solution. The mixture was extracted by 20 mL of CH2CI2. The aqueous phase was further extracted by CH2CI2 (10 mL X 2). The combined organics were concentrated and further purified by column chromatography (SiO2, 50% EtOAc/hexanes) to give 150 mg of the title compound.
- Step B A stirred solution of compound from Step A (40 mg, 0.054 mmol) and pyrrolidine (30 mg, 0.43 mmol) in 0.6 mL of DMSO and 0.15 mL of water was stirred at 100 °C for 3 h. It was cooled to room temperature and diluted with 15 mL of CH2CI2. The content was washed with water, saturated aqueous NaHCO3 and brine sequentially. The organic was concentrated and purified by column chromatography (SiO2, 3.5% 7 N NH3 in MeOH/ CH2CI2) to give 20 mg of the title compound.
- Step A To a solution of mesylate (560 mg, 0.841 mmol) in 16 mL of acetone was added LiBr (730 mg, 8.41 mmol). The mixture was stirred at room temperature for 1.5 h. It was diluted with 100 mL of CH2Ck and washed with brine (100 mL). The solvent was removed under vacuum.
- Step B To a solution of compound from Step A (40 mg, 0.061 mmol) in 1.5 mL of THF, was added 2-tri-n-butylstannylpyridine (45 mg, 0.12 mmol), and Pd(PPh3J4 (17 mg, 0.015 mmol). The reaction was stirred at 80 °C in a sealed vial for 16 h. The solvent was removed under vacuum. The residue was purified by column chromatography (SiO2, 3% 7 N NH3 in MeOH/ CH2CI2) to give 32 mg of crude title compound contaminated by triphenylphosphine oxide. This material was used in Step C without further purification.
- Step C The product of Step B was dissolved in 2 mL of MeOH/THF (1 :1 ) at 80 °C. To this solution was added 0.5 mL of 4 N HCI in dioxane. The reaction was stirred at 80 °C for 30 min. It was cooled to room temperature and diluted with 1 mL of THF. The solid was collected by filtration and washed with THF and ether to give 15 mg of the title compound as its HCI salt form, ⁇ 8.8(d, 1 H), 8.86 (t, 1 H), 8.10 (s, 3H), 7.92-8.08 (m, 3H), 7.22 (s, 1 H)1 4.60 (s, 2H), 2.58 (s, 3H).
- Step A To a mixture of 10 mL of THF/DMF (1 :1) was added NaH (39.3 mg, 1.64 mmol). It was cooled to -10 °C and a solution of trimethylsulfonium iodide (334 mg, 1.64 mmol) in 5 mL of DMSO was then slowly added. To the resulting mixture was added aldehyde. The reaction was stirred at room temperature for 40 min. It was quenched with ice water, and diluted with 50 mL of CH2Cb. The mixture was washed with water and brine. The solvent was removed under vacuum.
- Step B A solution of compound from Step A (270 mg, 0.450 mmol) in 4 mL of DMF was treated with sodium methanethiolate (100 mg, 1.43 mmol). The reaction was stirred at room temperature for 30 min. It was diluted with 15 mL of water. The mixture was extracted with EtOAc (20 mL x 3). The combined organics were washed with brine (20 mL) and then concentrated.
- Step C To a solution of compound from Step B (30 mg, 0.046 mol) in 1 mL of THF, was added NEt3 (14 mg, 0.14 mmol) followed by methanesulfonyl chloride (16 mg, 0.14 mmol). The reaction was stirred at room temperature for 15 min. It was quenched by adding 2 mL of water and diluted with 15 mL of CH2CI2. The mixture was washed with 10 mL of 0.2 N aqueous HCI solution. The organic was dried over anhydrous Na2SO4 and then concentrated. The residue was treated with NaI (10 mg, 0.071 mmol) and piperidine (13 mg, 0.15 mmol) in 1 mL of THF.
- Step D To a solution of compound from Step C (12 mg, 0.017 mmol) in 1 mL of THF/MeOH (1 :1 ), was added 0.5 mL of 4 N HCI in dioxane. The reaction was stirred at 80 °C for 1 h. It was cooled to room temperature and diluted with 10 mL of ether. The solid was collected by filtration and washed with ether to give 8 mg of the title compound as its HCI salt form.
- HPLC-MS tR 2.88 min (UV 254nm)- Mass calculated for formula C21H26N8S2 454.2; observed MH+ (LCMS) 455.3 (m/z).
- Step A To a stirred solution of compound alcohol (1.00 g, 1.70 mmol) in 20 mL of THF, was added Dess-Martin periodinane (1.84 g, 4.26 mmol) and a trace amount of water. The reaction was stirred at room temperature for 40 min. It was diluted with 200 mL of CH2CI2, and washed with water and brine. The organic was dried over anhydrous Na2SO4. The solvent was removed under vacuum. The residue was purified by column chromatography (SiO2, 40% EtOAc/hexanes) to give 250 mg of the title compound.
- Step B To a solution of compound from Step A (0.05 mmol) in 1 mL of CH2CI2/MeOH (1 :1 ) was added the respective amine (5 equivalent) and a trace amount of trifluoroacetic acid. The mixture was stirred at room temperature for 30 min when NaBH4 (10 equivalent) was added. The stirring was continued for additional 10 min. The reaction was quenched with saturated aqueous NH4CI solution. The mixture was extracted with CH2CI2. The organic was concentrated and the residue was purified by column chromatography (SiO2, 5% 7 N NH3 in MeOH/ CH2CI2) to give title compound.
- Step C To a solution of compound from Step B(0.05 mmol) in 1 mL of THF/MeOH (1 :1), was added 1 mL of 4 N HCI in dioxane. The reaction was stirred at 80 °C for 30 min. It was cooled to room temperature and diluted with 10 mL of ether. The solid was collected by filtration to afford compound 97-1 and 97-2, respectively. TABLE 26
- Part A To a solution of the isothiazole-aldehyde (534 mg; 0.9 mmol) in anhydrous THF (9 mL) was added methyl magnesium bromide (3M; 1.8 mL) at room temperature. After stirring for 20 min, the reaction mixture was quenched with 5 mL of saturated aqueous NH4CI solution and diluted with CH2Ck. The organic layer was washed with water and brine. The aqueous layer was back extracted with CH2CI2. The combined organic layers were dried over sodium sulfate and concentrated to obtain the crude product.
- methyl magnesium bromide 3M
- Part C A solution of the mesylate (40 mg; 0.06 mmol) in 2 mL of anhydrous THF was treated with hexamethyleneimine (15 mg; 0.15 mmol) plus a catalytic amount of NaI and the mixture was heated at reflux in an oil bath (8O°C; 20 h). The reaction mixture was cooled to room temperature and diluted with water and CH2CI2. The organic layer was washed with water, brine and dried over Na2SO4. Concentration in vacuo gave the crude product. Purification was carried out on flash silica gel column, eluting the product with CH2CI2 containing 2-4% of 7N-Ammonia in methanol.
- Part D To a solution of the above di-SEM protected amine from Part C (31 mg; 0.045 mmol) in 0.2 mL of THF and 0.2 mL of CH3OH was added 4N-HCI in dioxane (0.2 mL). The resulting mixture was heated at 8O°C in an oil bath for 30 min and then allowed to cool to room temperature. THF (2 mL) was added to the reaction mixture and the precipitated product was collected by filtration. The filer cake was washed with THF and ether and dried under vacuum to obtain the title product as white solid (23 mg). By using appropriate Grignard reagents in the first step and appropriate amine in the third step in the procedures described above, all the target compounds listed in Table 1 were prepared and characterized. TABLE 27
- the substrate 500 mg was suspended in f-BuOH (30 mL), and Et3N (0.45 mL) and DPPA (0.73 mL) was added sequentially at room temperature. Then the mixture was heated at 85 °C overnight. The reaction mixture was cooled to room temperature, and the solvent was evaporated under reduced pressure. The residue was taken up in ethyl acetate (50 mL) and water (50 mL) was added. The biphasic mixture was stirred for 15 min. Then two layers were separated, and the aqueous layer was extracted with ethyl acetate (2 * 50 mL).
- Diacid (3 g) was suspended in CH2CI2 (50 mL), and N1O- dimethylhydroxylamine hydrochloride (1.69 g), HATU (6.6 g) and diisopropylethylamine (12.12 mL) was added sequentially at room temperature. The reaction mixture was stirred overnight, and quenched by addition of water (100 mL).
- the two layers were separated.
- the aqueous layer was acidified to pH 4.0, and extracted with CHCb (5 * 100 mL).
- the organic layers were combined, and dried
- Part B The substrate from Part A (3.2 g) was suspended in f-BuOH (100 mL), and Et ⁇ N (2.27 mL) and DPPA (3.64 mL) was added sequentially at room temperature. Then the mixture was heated at 85 CC overnight. The reaction mixture was cooled to room temperature, and the solvent was evaporated under reduced pressure. The residue was taken up in ethyl acetate (100 mL) and water (100 mL) was added. The biphasic mixture was stirred for 15 min. Then two layers were separated, and the aqueous layer was extracted with ethyl acetate (2 x 100 mL).
- Part C The substrate from Part B (191 mg) was dissolved in THF/Et2O (3mL/6mL) and cooled to 0 °C. Then methylmagnesium bromide (0.83 mL, 2.0 M solution) was added dropwise. The reaction mixture was warmed to room temperature, and stirred for 12 h and quenched by addition of saturated ammonium chloride (10 mL). The two layers were separated and the aqueous layer was extracted with ethyl acetate (10 mL). The combined organic layer was washed with brine (30 mL), dried (Na2SO4), filtered and evaporated under reduced pressure to give the crude product which was purified by column chromatography (SiO2).
- Part D The substrate from Part C (10 mg) was dissolved in 30% TFA in CH2CI2 (2 mL), and the mixture was stirred for 30 min. Then the solvent was evaporated under reduced pressure and the residue was dried in vacuum. The crude product was used in the next step without further purification.
- Part E The sulfone (574.19 mg) and amine trifluoroacetic acid salt (514 mg) was dissolved in DMF (15 mL) under argon and treated with NaH (432 mg, 60% dispersion in oil). After LCMS indicated complete conversion of starting material to product, the reaction mixture was quenched with saturated ammonium chloride solution (15 mL). Then ethyl acetate (25 mL) was added. The two layers were separated, and the aqueous layer was extracted with ethyl acetate (25 mL).
- Step A To a solution of 2-ethoxyethylamine (2.0 g, 22.4 mmol) in diethyl ether (40 mL) at 0 °C was added trifluoroacetic anhydride (4.7 g, 22.4 mmol) dropwise. The reaction was stirred at room temperature for 1 hr. Potassium carbonate (10 g) was added to the reaction solution. The reaction was stirred at room temperature for 1 hr. The mixture was filtered through Celite and the organic filtrate was concentrated to give 1.5 g (36% yield) of the title compound.
- Step B A solution of the (trifluoromethyl)acetamide from Step A (1.5 g, 8.1 mmol) in Et2 ⁇ (20 mL) was added to a flask charged with lithium aluminum hydride (0.92 g, 24.3 mmol) in Et2 ⁇ (20 mL). The reaction was stirred at room temperature for 30 min, then at reflux for 12 hr. The reaction was cooled to room temperature and quenched with MeOH until the bubbling ceased. The reaction was diluted with Et2 ⁇ (30 mL) and filtered through a pad of Celite. The filtrate was concentrated by distillation to give 0.5 g (36% yield) of title compound as a colorless liquid. The amine was used without further purification.
- Step A To a solution of 2-ethoxyethylamine (1.0 g, 11.2 mmol) in dichloromethane (50 mL) at 0 °C was added pyridine (2.2 g, 28.1 mmol). Difluoroacetic anhydride (2.3 g, 13.5 mmol) was then slowly added to the reaction solution. The reaction was further stirred at 0 °C for 15 min, then at room temperature for 2.5 hr. The reaction was diluted with dichloromethane (50 mL) and H2O (20 mL). The reaction was sequentially washed with 1 N HCI (aq); saturated NaHCO3 (aq); brine. The organic phase was dried over NaSCU and concentrated in vacuo to give 1.03 g (55% yield) of the (difluoromethyl)acetamide as a colorless liquid.
- Step B A solution of the (difluoromethyl)acetamide from Step A (1.04 g, 6.20 mmol) in Et2O (21 mL) was added to a flask charged with lithium aluminum hydride (0.47 g, 12.39 mmol) in Et2O (25 mL) at 0 °C while under a N2 atmosphere. The reaction was stirred under a N2 atmosphere at room temperature for 2 hr. The reaction was quenched by the sequential addition of 0.47 mL of H2O; 0.47 mL of 15 % NaOH (aq) solution; 1.4 mL H2O. The reaction was stirred at room temperature for 15 min then filtered through a pad of Celite. The filtrate was concentrated by distillation to give 0.79 g (83% yield) of the title compound as a colorless liquid. The amine was used without further purification.
- Step A To a solution of 1-amino-1-cyclopentane methanol (1.0 g, 8.68 mmol) in dichloromethane (35 mL) at 0 °C was added pyridine (2.4 g, 30.4 mmol). Trifluoroacetic anhydride (4.6 g, 21.7 mmol) was then slowly added to the reaction solution. The reaction was further stirred at 0 °C for 15 min, then at room temperature for 16 hr. The reaction was sequentially washed with 1 N HCI (aq); saturated NaHCO3 (aq); brine. The organic phase was dried over NaSO4 and concentrated in vacuo to give 1.07 g (60% yield) of the title compound as a light brown liquid.
- Step B A solution of the (trifluoromethyl)acetamide from Step A (0.64 g, 3.06 mmol) in Et2O (10 mL) was added to a flask charged with lithium aluminum hydride (0.35 g, 9.1 mmol) in Et2O (30 mL) at 0 °C while under a N2 atmosphere. The reaction was stirred under a N2 atmosphere at 0 °C for 30 min, then at room temperature for 19 hr. The reaction was cooled to room temperature and stirred for 3 days.
- the reaction was then cooled to 0 °C and quenched by the sequential addition of 0.35 mL of H2O; 0.35 mL of 15 % NaOH (aq) solution; 1.05 mL H2O.
- the reaction was stirred at room temperature for 20 min then filtered through a pad of Celite. The filtercake was washed with Et2O and concentrated in vacuo to give 0.39 g (65% yield) of the title compound as a white solid.
- the amine was used without further purification.
- Step A To a solution of 2-amino-2-methyl-1-propanol (1.0 g, 11.2 mmol) in dichloromethane (100 mL) at 0 °C was added pyridine (3.1 g, 39.6 mmol). Trifluoroacetic anhydride (5.9 g, 28.1 mmol) was then slowly added to the reaction solution. The reaction was further stirred at 0 °C for 15 min, then at room temperature for 16 hr. The reaction was sequentially washed with 1 N HCI (aq); saturated NaHCO3 (aq); brine. The organic phase was dried over NaSO4 and concentrated in vacuo to give 0.79 g (38% yield) of the title compound as a white solid.
- Step B A solution of the (trifluoromethyl)acetamide from Step A (0.79 g, 4.29 mmol) in Et2O (43 mL) was added to a flask charged with lithium aluminum hydride (0.49 g, 12.91 mmol) in Et2O (13 mL) at 0 °C while under a N2 atmosphere. The reaction was stirred under a N2 atmosphere at 0 °C for 30 min, then at reflux for 4 hr. The reaction was cooled to room temperature and stirred for 3 days.
- the reaction was then cooled to 0 °C and quenched by the sequential addition of 0.49 mL of H2O; 0.49 mL of 15 % NaOH (aq) solution; 1.47 mL H2O.
- the reaction was stirred at room temperature for 20 min, then filtered through a pad of Celite. The filtercake was washed with Et2O and concentrated in vacuo to give 0.67 g (92% yield) of the title compound as a white solid.
- the amine was used without further purification.
- Step A To a solution of 2-amino-2-methyl-1-propanol (1.0 g, 11.2 mmol) in dichloromethane (50 mL) at 0 °C was added pyridine (2.7 g, 33.7 mmol). Difluoroacetic anhydride (3.9 g, 22.4 mmol) was then slowly added to the reaction solution. The reaction was further stirred at 0 °C for 15 min, then at room temperature for 2 hr. The reaction was diluted with dichloromethane (50 mL) and H2O (20 mL). The reaction was sequentially washed with 1 N HCI (aq); saturated NaHCO3 (aq); brine. The organic phase was dried over NaSO4 and concentrated in vacuo to give 2.04 g (74% yield) of the title compound as a colorless liquid.
- Step B A solution of the (difluoromethyl)acetamide from Step A (2.04 g, 8.31 mmol) in Et2O (17 mL) was added to a flask charged with lithium aluminum hydride (0.95 g, 24.92 mmol) in Et2O (50 mL) at 0 °C while under a N2 atmosphere. The reaction was stirred under a N2 atmosphere at room temperature for 2 hr. The reaction was quenched by the sequential addition of 0.95 mL of H2O; 0.95 mL of 15 % NaOH (aq) solution; 2.85 mL H2O. The reaction was stirred at room temperature for 15 min then filtered through a pad of Celite. The filtercake was washed with Et2O and concentrated in vacuo to give 1.23 g (97% yield) of the title compound as white needles. The amine was used without further purification.
- Step A Dess-Martin periodinane reagent (1.3 g; 3.1 mmol) was added to a solution of the isothiazole-alcohol (450 mg; 1 mmol) in 30 mL of THF containing 0.06 mL of water and the reaction mixture was stirred at room temperature for 45 min. The reaction was diluted with ether and filtered and washed with more ether. The filtrate was washed with saturated NaHCO3 solution, brine and dried. Concentration in vacuo gave the isothiazole aldehyde (418 mg; 93%).
- Step B To a solution of sodium hydride (60% in mineral oil; 169 mg; 4.2 mmol) in a mixture of 3.6 mL of DMSO and 3.6 mL of THF cooled to -1O°C, was added a solution of trimethyl sulfonium iodide (863 mg; 4.2 mmol) in 3.6 mL of DMSO drop wise. This was followed by the addition of a solution of the aldyhyde (363 mg; 0.84 mmol) in 5.6 mL of anhydrous THF, added in one portion. After stirring at room temperature for one hour, the reaction mixture was quenched with ice water. The organic products were extracted with EtOAc.
- Step C A solution of sodium methoxide in methanol (25% by wt; 4.5 mmol; 1 mL) was added to a solution of the epoxide (201 mg; 0.45 mmol) in a 1 :1 mixture of DMF- methanol (4 mL). The resulting solution was heated at 6O°C for 3.5 hr, then cooled to room temperature and quenched with water. Extracted the organic product with EtOAc, washed the organic extract with water and brine and dried over Na2SO4. Concentration gave the crude product. Purification by flash silica gel chromatography using 1 :1 mixture of CH2CI2 and EtOAc provided the desired methoxymethyl carbinol (180 mg; 84%) as colorless oil.
- Step D-F This sequence of steps was carried out as described for Example 76 in 64% overall yield for the 3 step sequence.
- Aurora A Assay An in vitro assay was developed that utilizes recombinant Aurora A or Aurora B as an enzyme source and a peptide based on PKA as the substrate.
- Aurora A kinase assays were performed in low protein binding 384-well plates (Corning Inc). All reagents were thawed on ice. Compounds were diluted in 100% DMSO to desirable concentrations. Each reaction consisted of 8 nM enzyme (Aurora A, Upstate cat#14-511), 100 nM Tamra-PKAtide (Molecular Devices, 5TAMRA- GRTGRRNSICOOH ), 25 ⁇ M ATP (Roche), 1 mM DTT (Pierce), and kinase buffer (10 mM Tris, 10 mM MgCI2, 0.01 % Tween 20).
- Aurora B kinase assays were performed in low protein binding 384-well plates (Corning Inc). All reagents were thawed on ice. Compounds were diluted in 100% DMSO to desirable concentrations. Each reaction consisted of 26 nM enzyme (Aurora B, Invitrogen cat#pv3970), 100 nM Tamra-PKAtide (Molecular Devices, 5TAMRA-GRTGRRNSICOOH ), 50 ⁇ M ATP (Roche), 1 mM DTT (Pierce), and kinase buffer (10 mM Tris, 10 mM MgCI2, 0.01 % Tween 20).
- Dose-response curves were plotted from inhibition data generated each in duplicate, from 8 point serial dilutions of inhibitory compounds. Concentration of compound was plotted against kinase activity, calculated by degree of fluorescent polarization. To generate IC50 values, the dose-response curves were then fitted to a standard sigmoidal curve and IC50 values were derived by nonlinear regression analysis.
- Compounds of the present invention exhibit Aurora A IC50 values of about 4 nm to about 3000 nM, Aurora B IC50 values of about 13 nM to about 3000 nM, and p-HH3 IC50 values of about 1 nM to about 10,000 nM.
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description







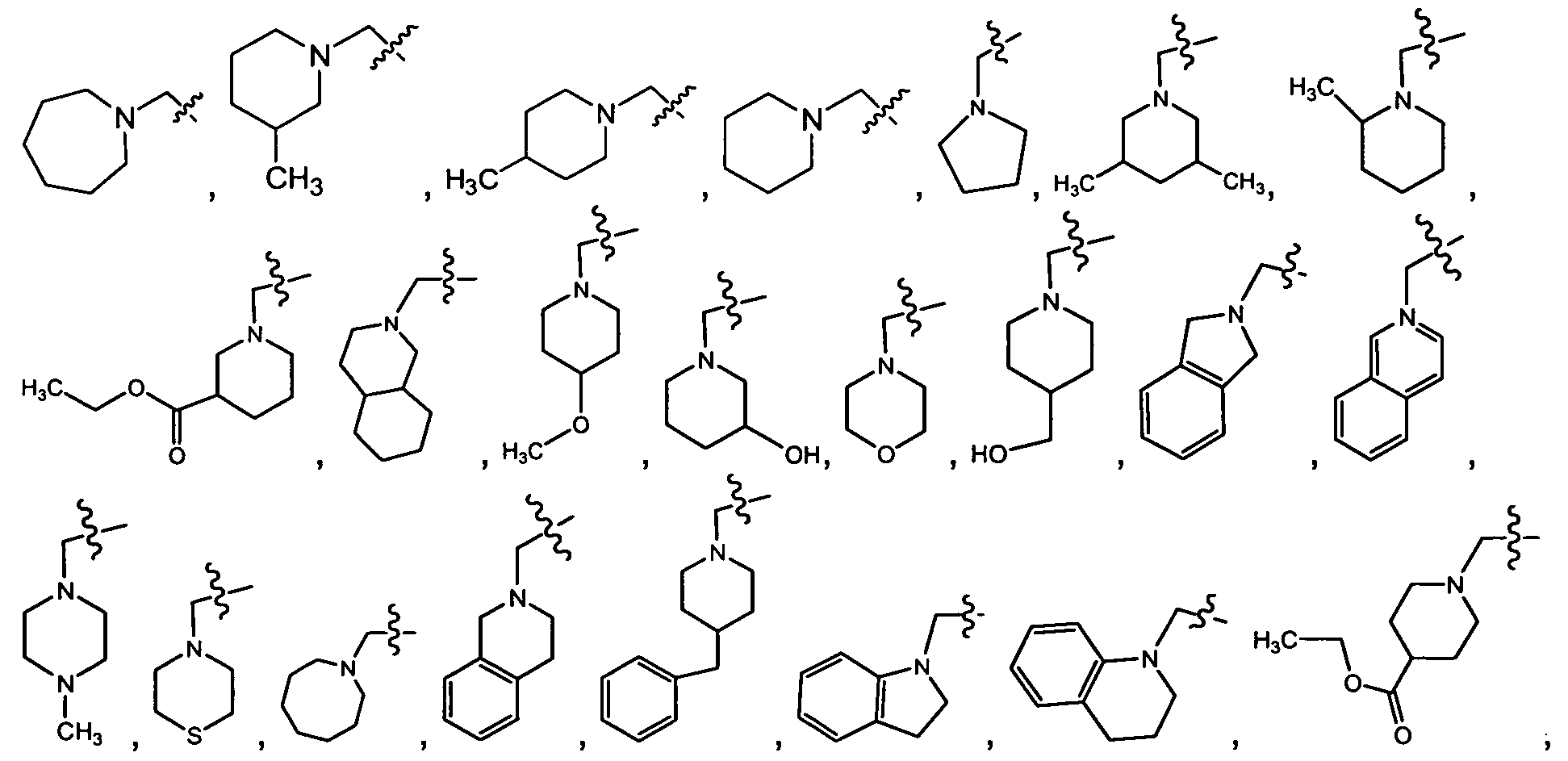












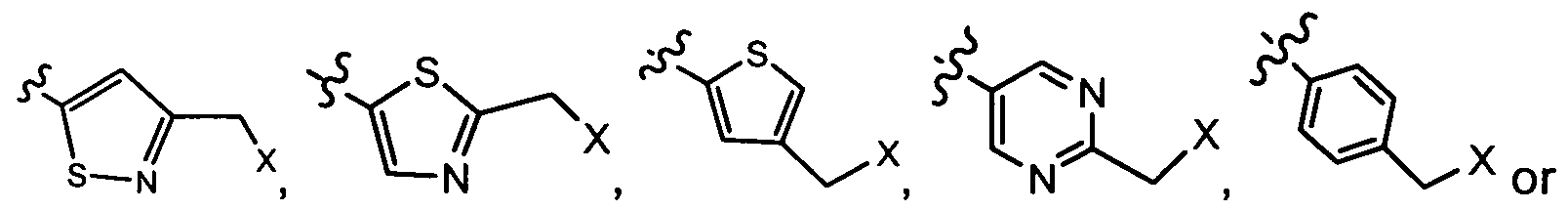









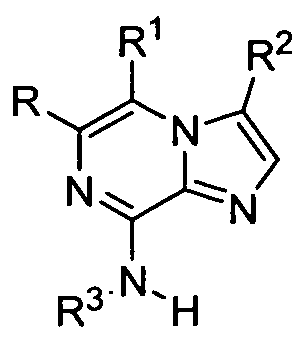




































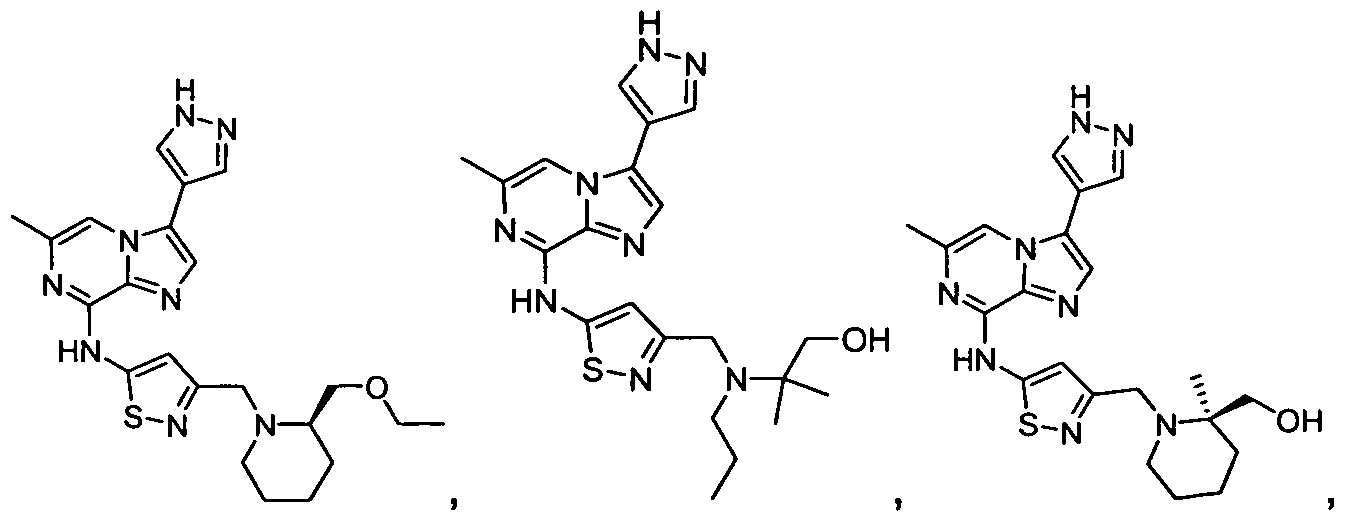























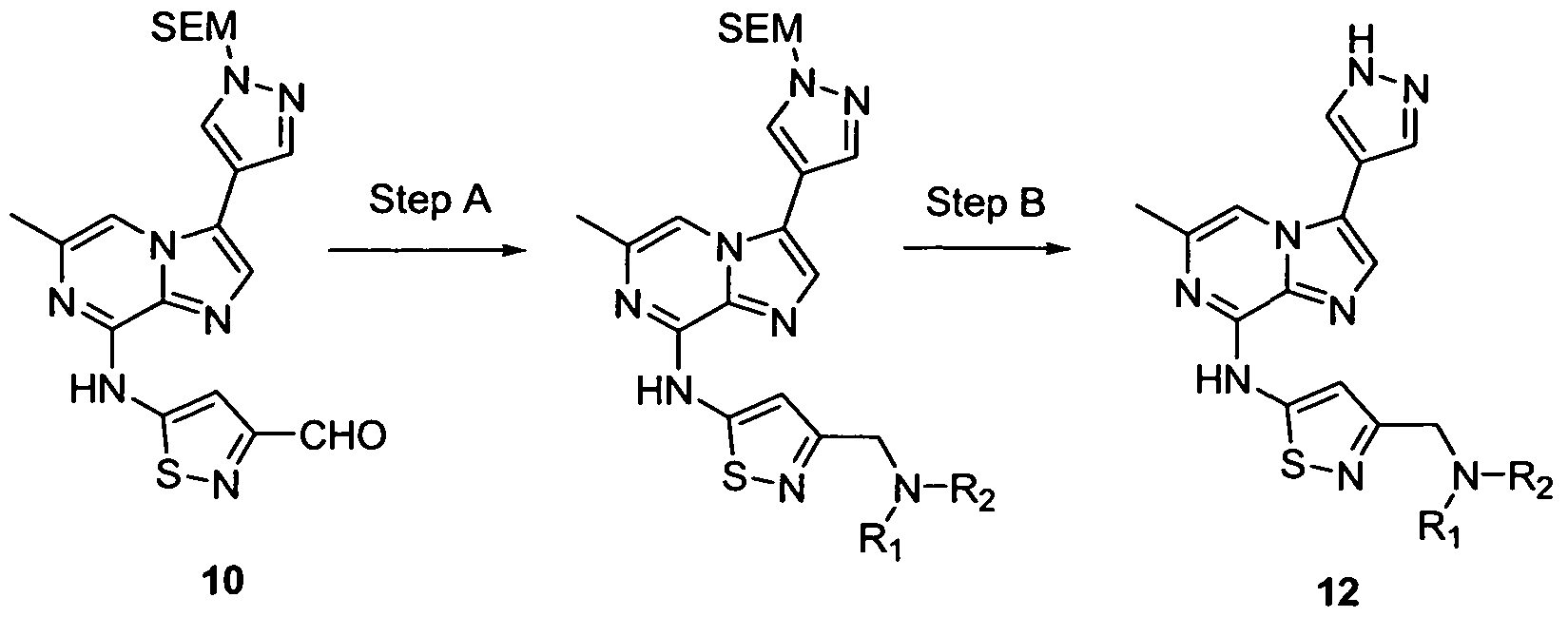
















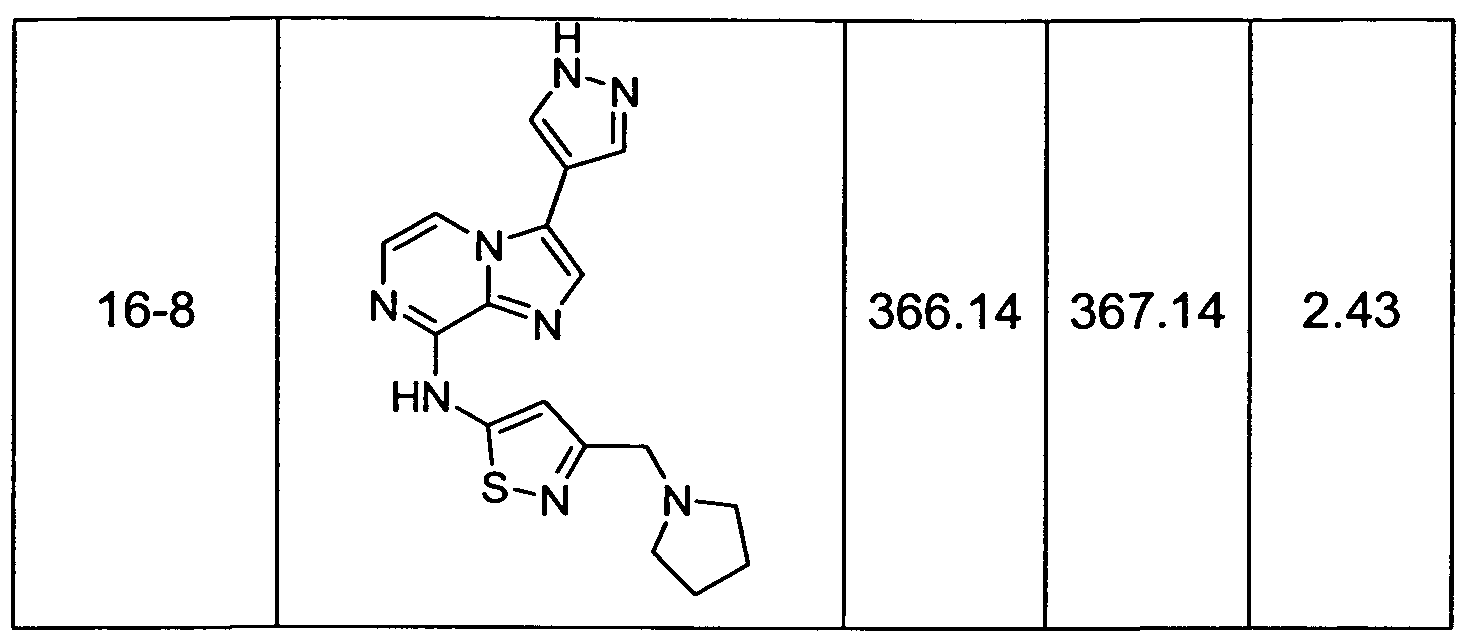


























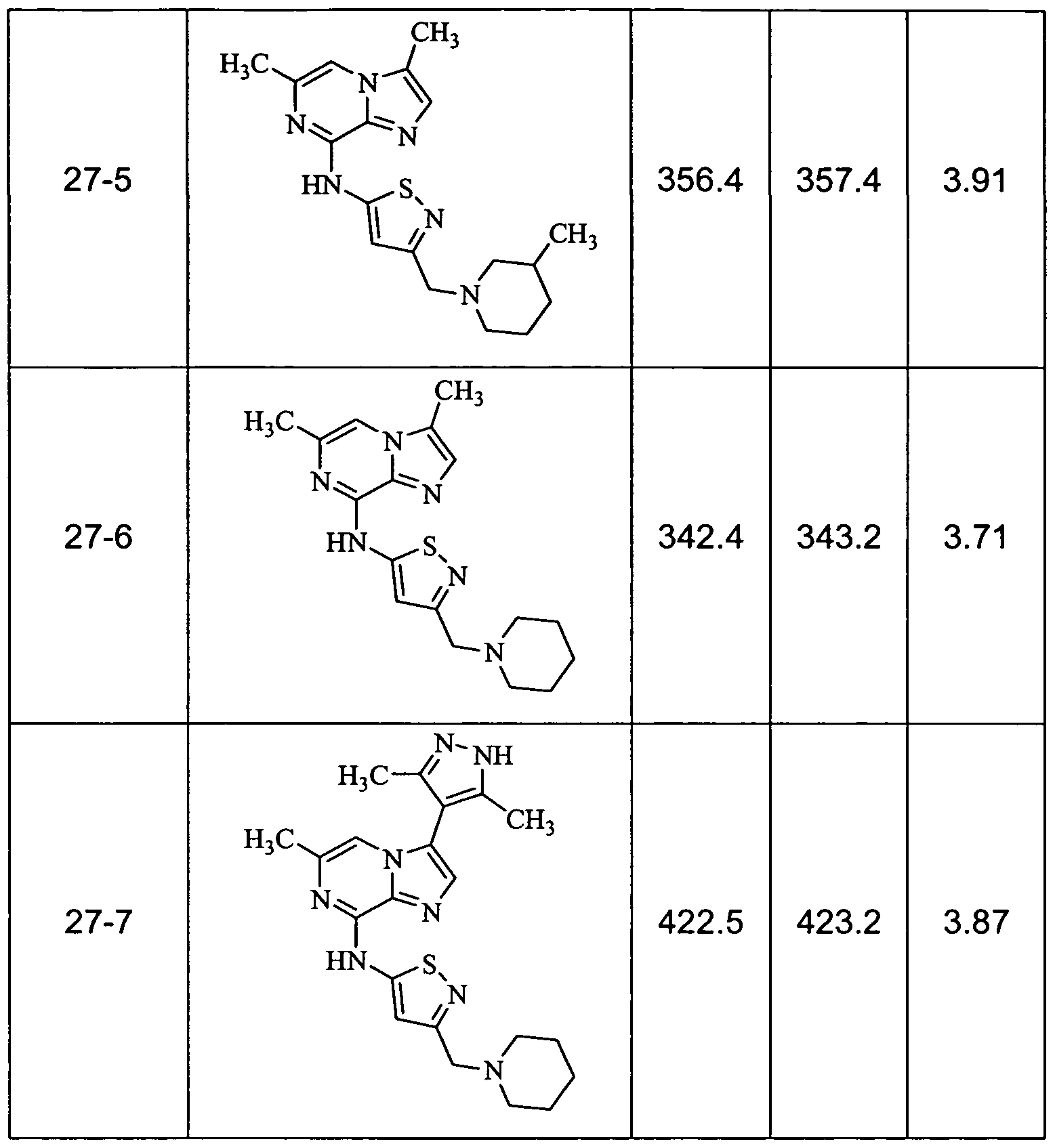







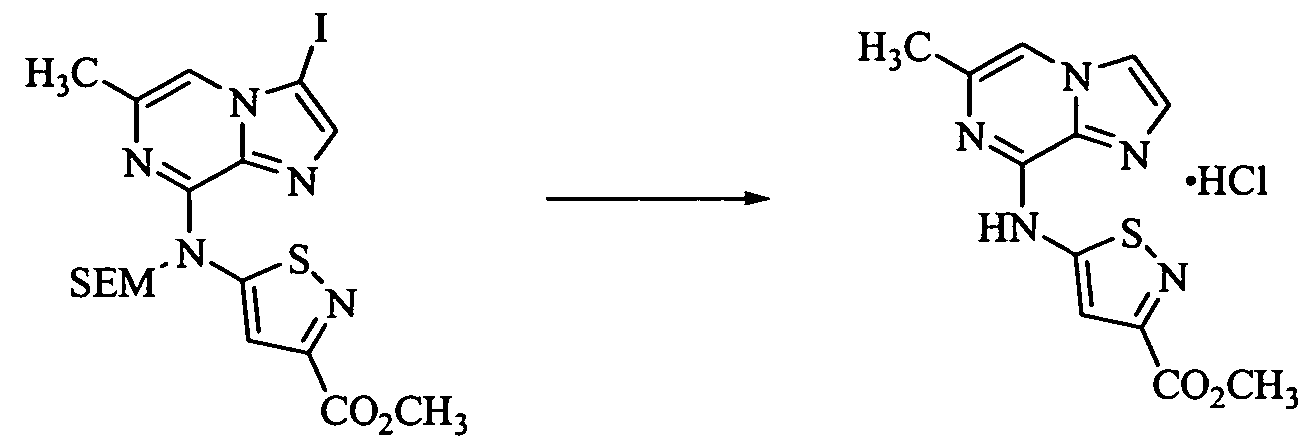





























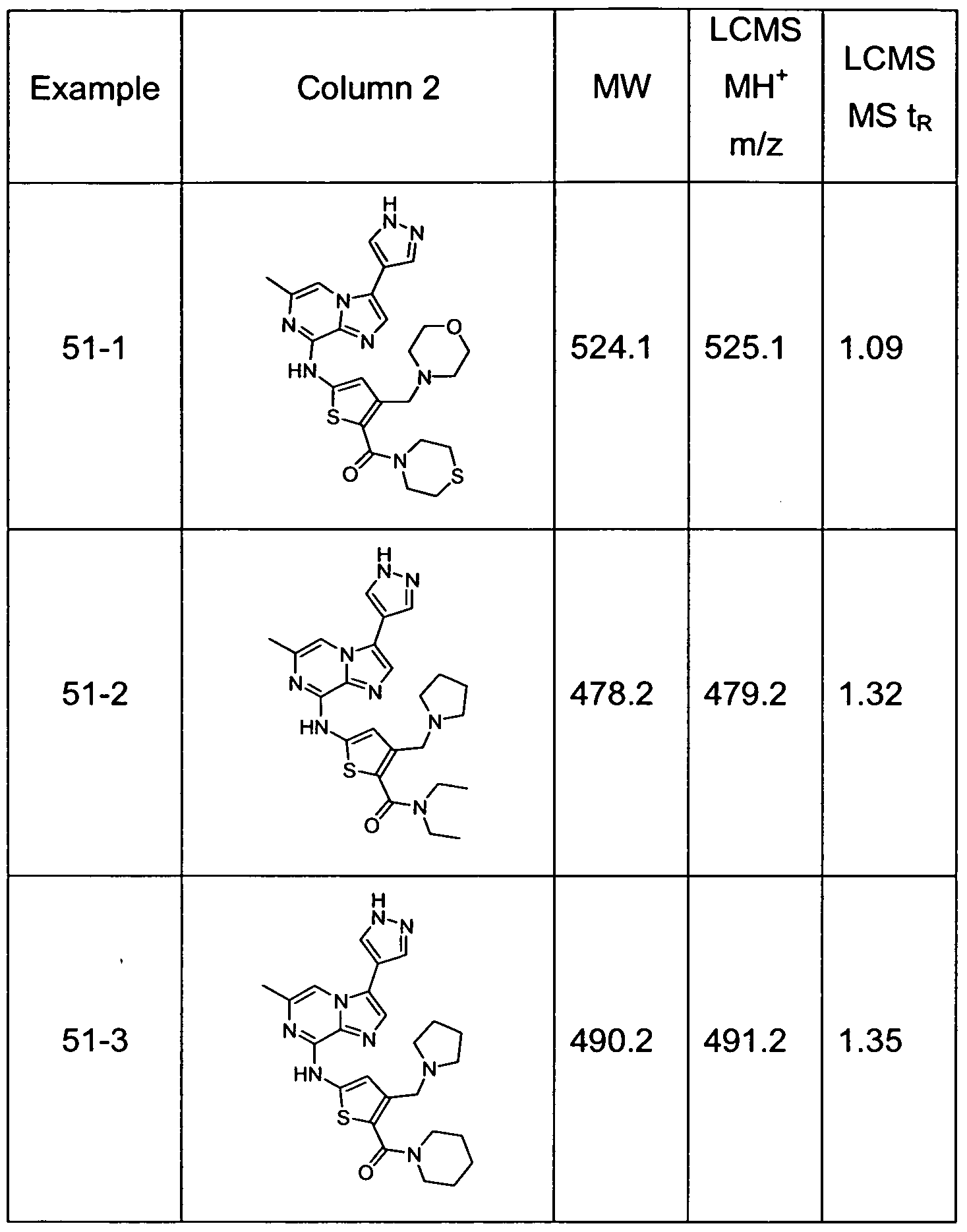





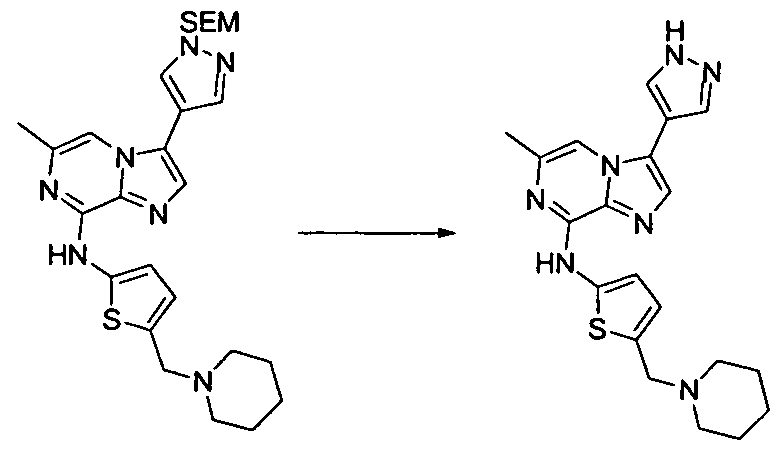















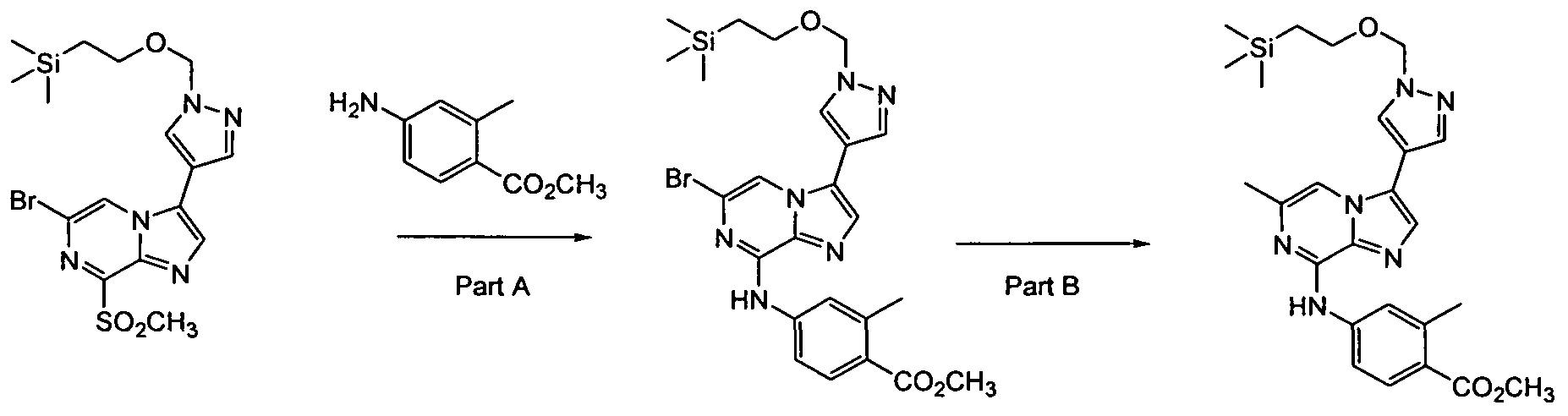
























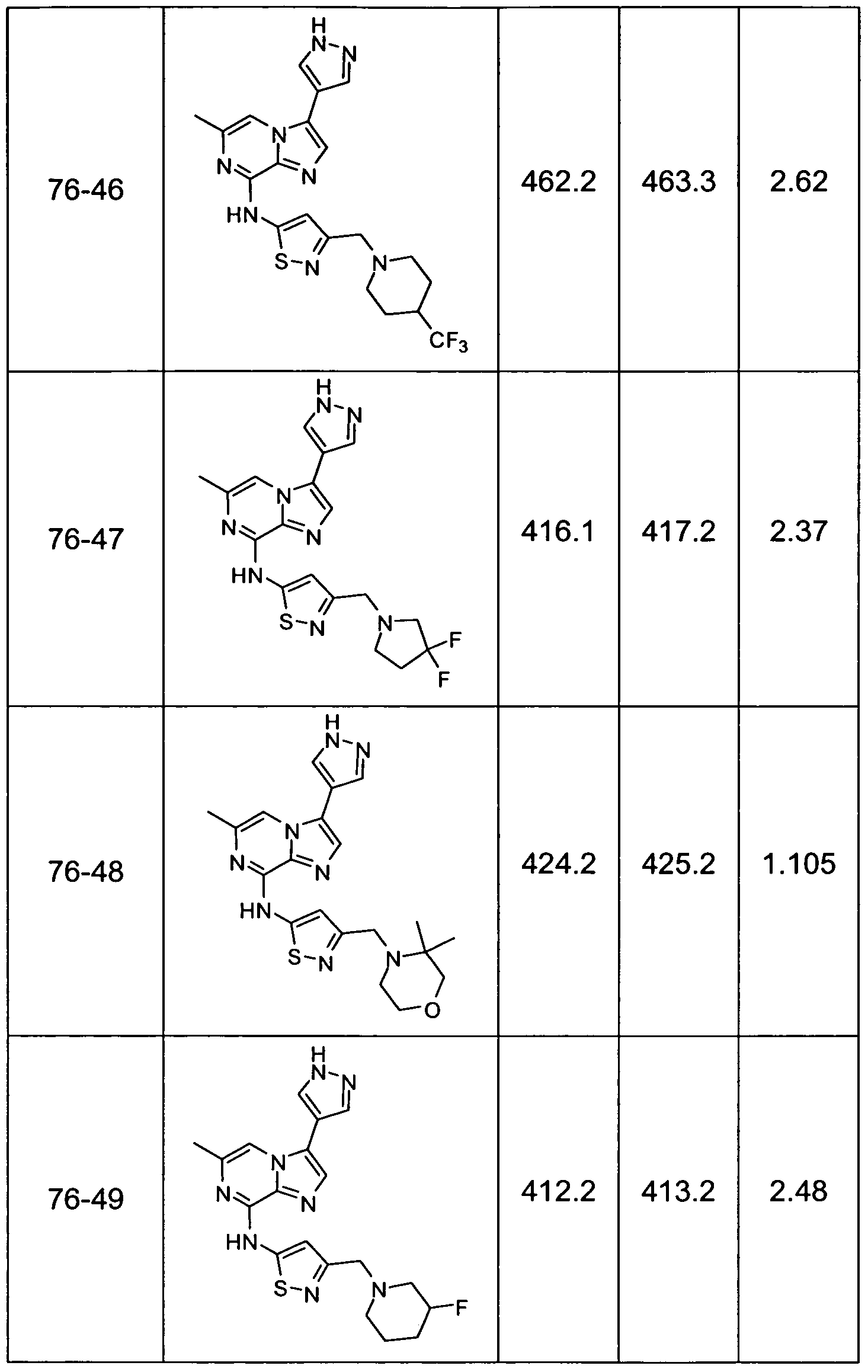
















































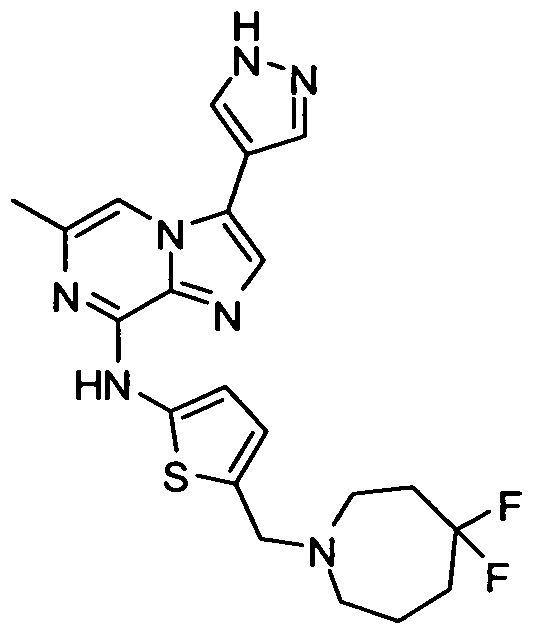







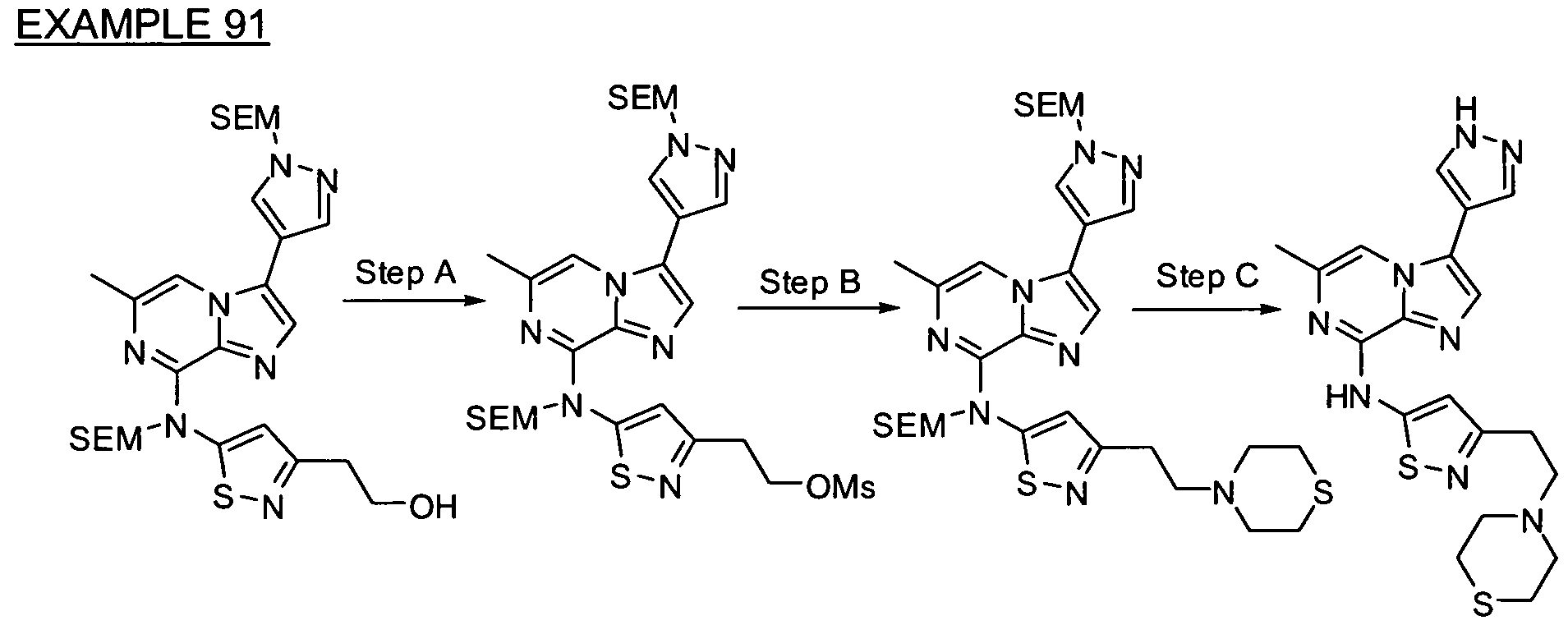








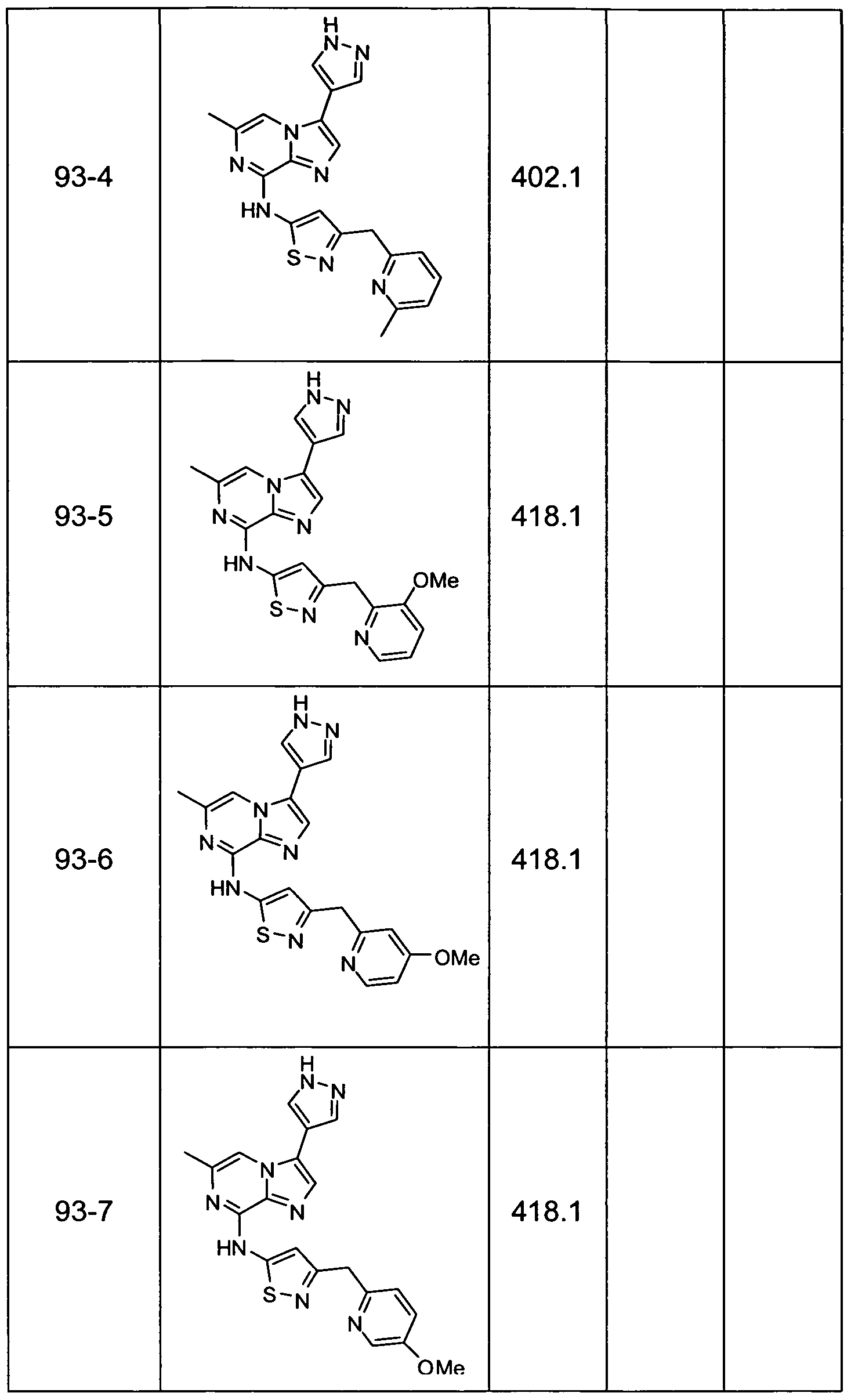

























Claims












Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MX2009013729A MX2009013729A (en) | 2007-06-14 | 2008-06-11 | Imidazopyrazines as protein kinase inhibitors. |
| CN200880101960A CN101772500A (en) | 2007-06-14 | 2008-06-11 | Imidazopyrazines as protein kinase inhibitors |
| CA2690557A CA2690557A1 (en) | 2007-06-14 | 2008-06-11 | Imidazopyrazines as protein kinase inhibitors |
| EP08768351A EP2170892A2 (en) | 2007-06-14 | 2008-06-11 | Imidazopyrazines as protein kinase inhibitors |
| JP2010512177A JP2010529195A (en) | 2007-06-14 | 2008-06-11 | Imidazopyrazine as an inhibitor of protein kinase |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US94399907P | 2007-06-14 | 2007-06-14 | |
| US60/943,999 | 2007-06-14 | ||
| US98793207P | 2007-11-14 | 2007-11-14 | |
| US60/987,932 | 2007-11-14 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008156614A2 true WO2008156614A2 (en) | 2008-12-24 |
| WO2008156614A3 WO2008156614A3 (en) | 2009-02-12 |
Family
ID=40002934
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/007295 WO2008156614A2 (en) | 2007-06-14 | 2008-06-11 | Imidazopyrazines as protein kinase inhibitors |
Country Status (10)
| Country | Link |
|---|---|
| EP (1) | EP2170892A2 (en) |
| JP (1) | JP2010529195A (en) |
| CN (1) | CN101772500A (en) |
| AR (1) | AR066958A1 (en) |
| CA (1) | CA2690557A1 (en) |
| CL (1) | CL2008001745A1 (en) |
| MX (1) | MX2009013729A (en) |
| PE (1) | PE20090365A1 (en) |
| TW (1) | TW200911811A (en) |
| WO (1) | WO2008156614A2 (en) |
Cited By (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011013729A1 (en) * | 2009-07-30 | 2011-02-03 | オンコセラピー・サイエンス株式会社 | Fused imidazole derivative having ttk inhibitory action |
| WO2011028638A1 (en) | 2009-09-04 | 2011-03-10 | Schering Corporation | Modulators of cell cycle checkpoints and their use in combination with checkpoint kinase inhibitors |
| WO2012052954A1 (en) * | 2010-10-20 | 2012-04-26 | Universite Bordeaux Segalen | Signatures of clinical outcome in gastro intestinal stromal tumors and method of treatment of gastroinstestinal stromal tumors |
| WO2012052745A1 (en) | 2010-10-21 | 2012-04-26 | Centro Nacional De Investigaciones Oncológicas (Cnio) | Combinations of pi3k inhibitors with a second anti -tumor agent |
| WO2012162365A1 (en) | 2011-05-25 | 2012-11-29 | Bristol-Myers Squibb Company | Substituted sulfonamides useful as antiapoptotic bcl inhibitors |
| CN103038235A (en) * | 2010-06-01 | 2013-04-10 | 拜耳知识产权有限责任公司 | Substituted imidazopyrazines |
| US8716282B2 (en) | 2009-10-30 | 2014-05-06 | Janssen Pharmaceutica Nv | Imidazo[1,2-b]pyridazine derivatives and their use as PDE10 inhibitors |
| US8778935B2 (en) | 2009-04-16 | 2014-07-15 | Centro Nacional De Investigaciones Oncologicas (Cnio) | Imidazopyrazines for use as kinase inhibitors |
| US8859543B2 (en) | 2010-03-09 | 2014-10-14 | Janssen Pharmaceutica Nv | Imidazo[1,2-a]pyrazine derivatives and their use for the prevention or treatment of neurological, psychiatric and metabolic disorders and diseases |
| US9493450B2 (en) | 2014-02-13 | 2016-11-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9493442B2 (en) | 2014-02-13 | 2016-11-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9527835B2 (en) | 2014-02-13 | 2016-12-27 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9550784B2 (en) | 2012-07-09 | 2017-01-24 | Beerse Pharmaceutica NV | Inhibitors of phosphodiesterase 10 enzyme |
| US9670210B2 (en) | 2014-02-13 | 2017-06-06 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9669035B2 (en) | 2012-06-26 | 2017-06-06 | Janssen Pharmaceutica Nv | Combinations comprising PDE 2 inhibitors such as 1-aryl-4-methyl-[1,2,4]triazolo-[4,3-A]]quinoxaline compounds and PDE 10 inhibitors for use in the treatment of neurological of metabolic disorders |
| US9695168B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted imidazo[1,5-α]pyridines and imidazo[1,5-α]pyrazines as LSD1 inhibitors |
| US9695167B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted triazolo[1,5-a]pyridines and triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| US9695180B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted imidazo[1,2-a]pyrazines as LSD1 inhibitors |
| US9758523B2 (en) | 2014-07-10 | 2017-09-12 | Incyte Corporation | Triazolopyridines and triazolopyrazines as LSD1 inhibitors |
| US9944647B2 (en) | 2015-04-03 | 2018-04-17 | Incyte Corporation | Heterocyclic compounds as LSD1 inhibitors |
| US9993554B2 (en) | 2010-10-21 | 2018-06-12 | Centro Nacional De Investigaciones Oncologicas (Cnio) | Use of P13K Inhibitors for the Treatment of Obesity, Steatosis and ageing |
| US10166221B2 (en) | 2016-04-22 | 2019-01-01 | Incyte Corporation | Formulations of an LSD1 inhibitor |
| US10329255B2 (en) | 2015-08-12 | 2019-06-25 | Incyte Corporation | Salts of an LSD1 inhibitor |
| US10604523B2 (en) | 2011-06-27 | 2020-03-31 | Janssen Pharmaceutica Nv | 1-aryl-4-methyl-[1,2,4]triazolo[4,3-a]quinoxaline derivatives |
| US10968200B2 (en) | 2018-08-31 | 2021-04-06 | Incyte Corporation | Salts of an LSD1 inhibitor and processes for preparing the same |
| CN116113406A (en) * | 2020-07-10 | 2023-05-12 | 密歇根大学董事会 | GAS41 inhibitors and methods of use thereof |
| US11690847B2 (en) | 2016-11-30 | 2023-07-04 | Case Western Reserve University | Combinations of 15-PGDH inhibitors with corticosteroids and/or TNF inhibitors and uses thereof |
| US11718589B2 (en) | 2017-02-06 | 2023-08-08 | Case Western Reserve University | Compositions and methods of modulating short-chain dehydrogenase |
| EP4252853A3 (en) * | 2017-03-31 | 2023-12-13 | Aurigene Oncology Limited | Compounds and compositions for treating hematological disorders |
| WO2024088192A1 (en) * | 2022-10-26 | 2024-05-02 | Js Innopharm (Suzhou) Ltd. | An aurora a inhibitor for use in treatments of cancers |
| US11981685B2 (en) | 2014-01-13 | 2024-05-14 | Aurigene Oncology Limited | Bicyclic heterocyclyl derivatives as IRAK4 inhibitors |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012080230A1 (en) * | 2010-12-17 | 2012-06-21 | Bayer Pharma Aktiengesellschaft | 6 substituted imidazopyrazines for use as mps-1 and tkk inhibitors in the treatment of hyperproliferative disorders |
| US8673905B2 (en) * | 2011-03-17 | 2014-03-18 | Hoffmann-La Roche Inc. | Imidazo pyrazines |
| CN107954977A (en) * | 2017-12-15 | 2018-04-24 | 上海鼎雅药物化学科技有限公司 | The synthetic method of Raltitrexed intermediate |
| WO2022222995A1 (en) * | 2021-04-23 | 2022-10-27 | 南京明德新药研发有限公司 | Picolinamide compound |
| CN116715668A (en) * | 2022-03-07 | 2023-09-08 | 上海凌达生物医药有限公司 | Nitrogen-containing heterocyclic cell cycle inhibitor compound, preparation method and application |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004026877A1 (en) * | 2002-09-23 | 2004-04-01 | Schering Corporation | Imidazopyrazines as cyclin dependent kinase inhibitors |
| WO2006053121A2 (en) * | 2004-11-10 | 2006-05-18 | Cgi Pharmaceuticals, Inc. | Imidazo[1 , 2-a] pyrazin-8-ylamines useful as modulators of kinase activity |
| WO2007058873A2 (en) * | 2005-11-10 | 2007-05-24 | Schering Corporation | Imidazopyrazines as cyclin depentend kinase inhibitors |
| WO2007058942A2 (en) * | 2005-11-10 | 2007-05-24 | Schering Corporation | Imidazopyrazines as protein kinase inhibitors |
-
2008
- 2008-06-11 WO PCT/US2008/007295 patent/WO2008156614A2/en active Application Filing
- 2008-06-11 MX MX2009013729A patent/MX2009013729A/en not_active Application Discontinuation
- 2008-06-11 JP JP2010512177A patent/JP2010529195A/en not_active Withdrawn
- 2008-06-11 EP EP08768351A patent/EP2170892A2/en not_active Withdrawn
- 2008-06-11 CN CN200880101960A patent/CN101772500A/en active Pending
- 2008-06-11 CA CA2690557A patent/CA2690557A1/en not_active Abandoned
- 2008-06-11 AR ARP080102481A patent/AR066958A1/en not_active Application Discontinuation
- 2008-06-11 PE PE2008001001A patent/PE20090365A1/en not_active Application Discontinuation
- 2008-06-12 TW TW097121942A patent/TW200911811A/en unknown
- 2008-06-12 CL CL2008001745A patent/CL2008001745A1/en unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004026877A1 (en) * | 2002-09-23 | 2004-04-01 | Schering Corporation | Imidazopyrazines as cyclin dependent kinase inhibitors |
| WO2006053121A2 (en) * | 2004-11-10 | 2006-05-18 | Cgi Pharmaceuticals, Inc. | Imidazo[1 , 2-a] pyrazin-8-ylamines useful as modulators of kinase activity |
| WO2007058873A2 (en) * | 2005-11-10 | 2007-05-24 | Schering Corporation | Imidazopyrazines as cyclin depentend kinase inhibitors |
| WO2007058942A2 (en) * | 2005-11-10 | 2007-05-24 | Schering Corporation | Imidazopyrazines as protein kinase inhibitors |
Cited By (53)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8778935B2 (en) | 2009-04-16 | 2014-07-15 | Centro Nacional De Investigaciones Oncologicas (Cnio) | Imidazopyrazines for use as kinase inhibitors |
| WO2011013729A1 (en) * | 2009-07-30 | 2011-02-03 | オンコセラピー・サイエンス株式会社 | Fused imidazole derivative having ttk inhibitory action |
| WO2011028638A1 (en) | 2009-09-04 | 2011-03-10 | Schering Corporation | Modulators of cell cycle checkpoints and their use in combination with checkpoint kinase inhibitors |
| US8716282B2 (en) | 2009-10-30 | 2014-05-06 | Janssen Pharmaceutica Nv | Imidazo[1,2-b]pyridazine derivatives and their use as PDE10 inhibitors |
| US8859543B2 (en) | 2010-03-09 | 2014-10-14 | Janssen Pharmaceutica Nv | Imidazo[1,2-a]pyrazine derivatives and their use for the prevention or treatment of neurological, psychiatric and metabolic disorders and diseases |
| CN103038235B (en) * | 2010-06-01 | 2015-07-29 | 拜耳知识产权有限责任公司 | The Imidazopyrazines replaced |
| CN103038235A (en) * | 2010-06-01 | 2013-04-10 | 拜耳知识产权有限责任公司 | Substituted imidazopyrazines |
| WO2012052954A1 (en) * | 2010-10-20 | 2012-04-26 | Universite Bordeaux Segalen | Signatures of clinical outcome in gastro intestinal stromal tumors and method of treatment of gastroinstestinal stromal tumors |
| WO2012052745A1 (en) | 2010-10-21 | 2012-04-26 | Centro Nacional De Investigaciones Oncológicas (Cnio) | Combinations of pi3k inhibitors with a second anti -tumor agent |
| US9993554B2 (en) | 2010-10-21 | 2018-06-12 | Centro Nacional De Investigaciones Oncologicas (Cnio) | Use of P13K Inhibitors for the Treatment of Obesity, Steatosis and ageing |
| WO2012162365A1 (en) | 2011-05-25 | 2012-11-29 | Bristol-Myers Squibb Company | Substituted sulfonamides useful as antiapoptotic bcl inhibitors |
| US10604523B2 (en) | 2011-06-27 | 2020-03-31 | Janssen Pharmaceutica Nv | 1-aryl-4-methyl-[1,2,4]triazolo[4,3-a]quinoxaline derivatives |
| US9669035B2 (en) | 2012-06-26 | 2017-06-06 | Janssen Pharmaceutica Nv | Combinations comprising PDE 2 inhibitors such as 1-aryl-4-methyl-[1,2,4]triazolo-[4,3-A]]quinoxaline compounds and PDE 10 inhibitors for use in the treatment of neurological of metabolic disorders |
| US9550784B2 (en) | 2012-07-09 | 2017-01-24 | Beerse Pharmaceutica NV | Inhibitors of phosphodiesterase 10 enzyme |
| US11981685B2 (en) | 2014-01-13 | 2024-05-14 | Aurigene Oncology Limited | Bicyclic heterocyclyl derivatives as IRAK4 inhibitors |
| US9527835B2 (en) | 2014-02-13 | 2016-12-27 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9670210B2 (en) | 2014-02-13 | 2017-06-06 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US11247992B2 (en) | 2014-02-13 | 2022-02-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US11155532B2 (en) | 2014-02-13 | 2021-10-26 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10717737B2 (en) | 2014-02-13 | 2020-07-21 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10676457B2 (en) | 2014-02-13 | 2020-06-09 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10174030B2 (en) | 2014-02-13 | 2019-01-08 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9994546B2 (en) | 2014-02-13 | 2018-06-12 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9493442B2 (en) | 2014-02-13 | 2016-11-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10513493B2 (en) | 2014-02-13 | 2019-12-24 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10300051B2 (en) | 2014-02-13 | 2019-05-28 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US9493450B2 (en) | 2014-02-13 | 2016-11-15 | Incyte Corporation | Cyclopropylamines as LSD1 inhibitors |
| US10138249B2 (en) | 2014-07-10 | 2018-11-27 | Incyte Corporation | Triazolopyridines and triazolopyrazines as LSD1 inhibitors |
| US10968221B2 (en) | 2014-07-10 | 2021-04-06 | Incyte Corporation | Substituted [1,2,4]triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| US10125133B2 (en) | 2014-07-10 | 2018-11-13 | Incyte Corporation | Substituted [1,2,4]triazolo[1,5-a]pyridines and substituted [1,2,4]triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| US9695168B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted imidazo[1,5-α]pyridines and imidazo[1,5-α]pyrazines as LSD1 inhibitors |
| US10112950B2 (en) | 2014-07-10 | 2018-10-30 | Incyte Corporation | Substituted imidazo[1,2-a]pyrazines as LSD1 inhibitors |
| US10556908B2 (en) | 2014-07-10 | 2020-02-11 | Incyte Corporation | Substituted imidazo[1,2-a]pyrazines as LSD1 inhibitors |
| US10047086B2 (en) | 2014-07-10 | 2018-08-14 | Incyte Corporation | Imidazopyridines and imidazopyrazines as LSD1 inhibitors |
| US10640503B2 (en) | 2014-07-10 | 2020-05-05 | Incyte Corporation | Imidazopyridines and imidazopyrazines as LSD1 inhibitors |
| US9695167B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted triazolo[1,5-a]pyridines and triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| US9758523B2 (en) | 2014-07-10 | 2017-09-12 | Incyte Corporation | Triazolopyridines and triazolopyrazines as LSD1 inhibitors |
| US9695180B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted imidazo[1,2-a]pyrazines as LSD1 inhibitors |
| US9944647B2 (en) | 2015-04-03 | 2018-04-17 | Incyte Corporation | Heterocyclic compounds as LSD1 inhibitors |
| US10800779B2 (en) | 2015-04-03 | 2020-10-13 | Incyte Corporation | Heterocyclic compounds as LSD1 inhibitors |
| US11401272B2 (en) | 2015-04-03 | 2022-08-02 | Incyte Corporation | Heterocyclic compounds as LSD1 inhibitors |
| US11498900B2 (en) | 2015-08-12 | 2022-11-15 | Incyte Corporation | Salts of an LSD1 inhibitor |
| US10723700B2 (en) | 2015-08-12 | 2020-07-28 | Incyte Corporation | Salts of an LSD1 inhibitor |
| US10329255B2 (en) | 2015-08-12 | 2019-06-25 | Incyte Corporation | Salts of an LSD1 inhibitor |
| US10166221B2 (en) | 2016-04-22 | 2019-01-01 | Incyte Corporation | Formulations of an LSD1 inhibitor |
| US11690847B2 (en) | 2016-11-30 | 2023-07-04 | Case Western Reserve University | Combinations of 15-PGDH inhibitors with corticosteroids and/or TNF inhibitors and uses thereof |
| US11718589B2 (en) | 2017-02-06 | 2023-08-08 | Case Western Reserve University | Compositions and methods of modulating short-chain dehydrogenase |
| EP4252853A3 (en) * | 2017-03-31 | 2023-12-13 | Aurigene Oncology Limited | Compounds and compositions for treating hematological disorders |
| US10968200B2 (en) | 2018-08-31 | 2021-04-06 | Incyte Corporation | Salts of an LSD1 inhibitor and processes for preparing the same |
| US11512064B2 (en) | 2018-08-31 | 2022-11-29 | Incyte Corporation | Salts of an LSD1 inhibitor and processes for preparing the same |
| CN116113406A (en) * | 2020-07-10 | 2023-05-12 | 密歇根大学董事会 | GAS41 inhibitors and methods of use thereof |
| EP4178571A4 (en) * | 2020-07-10 | 2024-07-17 | Univ Michigan Regents | Gas41 inhibitors and methods of use thereof |
| WO2024088192A1 (en) * | 2022-10-26 | 2024-05-02 | Js Innopharm (Suzhou) Ltd. | An aurora a inhibitor for use in treatments of cancers |
Also Published As
| Publication number | Publication date |
|---|---|
| TW200911811A (en) | 2009-03-16 |
| CN101772500A (en) | 2010-07-07 |
| EP2170892A2 (en) | 2010-04-07 |
| PE20090365A1 (en) | 2009-04-04 |
| WO2008156614A3 (en) | 2009-02-12 |
| MX2009013729A (en) | 2010-01-25 |
| JP2010529195A (en) | 2010-08-26 |
| CA2690557A1 (en) | 2008-12-24 |
| AR066958A1 (en) | 2009-09-23 |
| CL2008001745A1 (en) | 2008-12-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008156614A2 (en) | Imidazopyrazines as protein kinase inhibitors | |
| US7645762B2 (en) | Substituted pyrazolo[1,5-a] pyrimidines as protein kinase inhibitors | |
| CA2624882C (en) | Pyrazolopyrimidines as protein kinase inhibitors | |
| JP5520325B2 (en) | Use of pyrazolo [1,5-A] pyrimidine derivatives to inhibit protein kinases and methods for inhibiting protein kinases | |
| EP1934225B1 (en) | Pyrazolo [1,5-a] pyrimidines as protein kinase inhibitors | |
| US7642266B2 (en) | Substituted pyrazolo[1,5-a]pyrimidines as protein kinase inhibitors | |
| US7511040B2 (en) | Imidazopyrazines as protein kinase inhibitors | |
| US20090175852A1 (en) | Imidazopyrazines as protein kinase inhibitors | |
| WO2007058942A2 (en) | Imidazopyrazines as protein kinase inhibitors | |
| EP1945216A1 (en) | Methods for inhibiting protein kinases | |
| CA2654202A1 (en) | Imidazopyrazines as protein kinase inhibitors | |
| WO2009097233A9 (en) | Imidazopyrazines as protein kinase inhibitors | |
| EP1945643A2 (en) | Imidazopyrazines as cyclin dependent kinase inhibitors | |
| AU2006302435B2 (en) | Use of pyrazolo [1 , 5 -a] pyrimidine derivatives for inhibiting protein kinases methods for inhibiting protein kinases |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880101960.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08768351 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2690557 Country of ref document: CA Ref document number: 2010512177 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2009/013729 Country of ref document: MX |
|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008768351 Country of ref document: EP |