WO2008134300A1 - Compounds and compositions as inhibitors of cannabinoid receptor 1 activity - Google Patents
Compounds and compositions as inhibitors of cannabinoid receptor 1 activity Download PDFInfo
- Publication number
- WO2008134300A1 WO2008134300A1 PCT/US2008/061116 US2008061116W WO2008134300A1 WO 2008134300 A1 WO2008134300 A1 WO 2008134300A1 US 2008061116 W US2008061116 W US 2008061116W WO 2008134300 A1 WO2008134300 A1 WO 2008134300A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- purine
- dione
- chlorophenyl
- ethyl
- Prior art date
Links
- 0 *=C1N(c(cc2)ccc2Cl)C(c(ccc(Cl)c2)c2Cl)=NC(N2c3ccccc3)=C1NC2=O Chemical compound *=C1N(c(cc2)ccc2Cl)C(c(ccc(Cl)c2)c2Cl)=NC(N2c3ccccc3)=C1NC2=O 0.000 description 2
- WYFFMGNGCDQLCV-UHFFFAOYSA-N N#CCN(C1=C(N2c3ccccc3)N=C(c(ccc(Cl)c3)c3Cl)N(c(cc3)ccc3Cl)C1=O)C2=O Chemical compound N#CCN(C1=C(N2c3ccccc3)N=C(c(ccc(Cl)c3)c3Cl)N(c(cc3)ccc3Cl)C1=O)C2=O WYFFMGNGCDQLCV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/28—Oxygen atom
- C07D473/30—Oxygen atom attached in position 6, e.g. hypoxanthine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
Definitions
- the invention provides compounds, pharmaceutical compositions comprising such compounds and methods of using such compounds to treat or prevent diseases or disorders associated with the activity of cannabinoid receptor 1 (CBl).
- CBDl cannabinoid receptor 1
- Cannabinoids are psychoactive ingredients of marijuana, principally delta-9- tetrahydromaynabinol.
- Two cannabinoid receptors have been cloned, CBl and CB2.
- CBl is predominantly expressed in the central nervous system whereas CB2 is expressed in peripheral tissues, principally in the immune system. Both receptors are members of the G-protein coupled class and their inhibition is linked to adenylate cyclase activity.
- the invention provides compounds and pharmaceutical compositions thereof, which may be useful as inhibitors of cannabinoid receptor 1 activity.
- the invention provides compounds of Formula (1):
- Y is O, S or NR 5 ;
- X is O or S
- R 1 and R 2 are independently L-R 9 ;
- L is a bond, S or O
- R 3 and R 4 are independently H, an optionally halogenated Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, cyano, halo, nitro, NR 6 R 7 , CO(CR 2 ) k NR 6 R 7 , OR 8 , SOi_ 2 R 8 , NR 7 SOi_ 2 R 8 , COi_ 2 R 8 or R 9 ;
- R 5 , R 6 , R 7 and R 8 are independently H, Ci_ 6 alkyl, C 2 - 6 alkenyl, C 2 _ 6 alkynyl, or (CR 2 ) k R 9 ; each R 9 is an optionally substituted C 3 _ 7 cycloalkyl, or a 5-12 membered aryl, heteroaryl or heterocyclic ring containing N, O or S;
- R is H or Ci-6 alkyl; and j and k are independently 0-6.
- X and Y may independently be O or S.
- L is a bond.
- R 1 , R 2 and R 4 may independently be an optionally substituted cycloalkyl, or a 5-7 membered aryl, heteroaryl or heterocyclic ring containing N, O or S.
- R 1 , R 2 and R 4 are independently phenyl, piperazinyl, morpholino, benzthiazolyl, pyridinyl or pyrazolyl.
- R 1 , R 2 and R 4 are independently an optionally substituted phenyl.
- the invention provides compounds of Formula (2):
- X and Y are independently O or S;
- R 10 , R 11 and R 12 are independently halo, an optionally substituted alkyl, C 2 -6 alkenyl, C 2 -6 alkynyl, cycloalkyl, or an optionally substituted aryl, heteroaryl or heterocyclic ring; and m, n and p are independently 0-5.
- R 10 may be halo.
- n 1-2
- R 11 is halo, an optionally halogenated Ci_ 6 alkyl, or an optionally substituted cycloalkyl, phenyl, pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, imidazolyl, pyrazolyl, oxazolyl, oxadiazolyl, isoxazolyl, furanyl, thiophenyl, or triazolyl.
- p is 0.
- each optionally substituted moiety may optionally be substituted with an optionally halogenated C]_ 6 alkyl, C 2 - 6 alkenyl, C 2 - 6 alkynyl, cyano, halo, nitro, NR 6 R 7 , CO(CR 2 ) k NR 6 R 7 , OR 8 , SOi_ 2 R 8 , NR 7 SOi_ 2 R 8 , COi_ 2 R 8 or R 9 , wherein R 6 , R 7 , R 8 and R 9 are substituents as defined in Formula (1).
- the invention provides a compound selected from the group consisting of l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8- dione; l-(4-bromo-phenyl)-9-phenyl-8-thioxo-2-p-tolyl-l,7,8,9-tetrahydro-purin-6-one; l-(4-chlorophenyl)-9-phenyl-2-(4-(trifluoromethyl)phenyl)-lH-purine-6,8(7H,9H)- dione; l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-7-methyl-9-phenyl-7,9-dihydro-lH-purine- 6,8-dione; l-(4-chloro-phenyl)
- the present invention provides pharmaceutical compositions comprising a compound of Formula (1) or (2), and a pharmaceutically acceptable carrier.
- the invention provides methods for inhibiting a cannabinoid- 1 receptor, comprising administering to a system or a subject in need thereof, a therapeutically effective amount of a compound comprising Formula (1) or (2), or pharmaceutically acceptable salts or pharmaceutical compositions thereof, thereby inhibiting a cannabinoid- 1 receptor.
- Compounds of the invention may be administered to a cell or tissue system, or to a human or animal subject.
- the invention provides methods for treating a condition mediated by a cannabinoid- 1 receptor, comprising administering to a system or a subject in need thereof, a therapeutically effective amount of a compound comprising Formula (1) or (2), or pharmaceutically acceptable salts or pharmaceutical compositions thereof, thereby treating said condition.
- compounds of Formula (1) or (2) may be used for treating an eating disorder associated with excessive food intake, obesity, diabetes, insulin resistance, hypercholesterolemia, hypertriglyceridemia, substance abuse, osteoporosis, or smoking abuse.
- compounds of Formula (1) or (2) are useful for treating obesity, and may be administered to a subject at risk for obesity at a dosage of about 0.001 mg to about 100 mg per kg of body weight.
- the invention also provides the use of compounds of Formula (1) or (2), or pharmaceutically acceptable salts or pharmaceutical compositions thereof, for inhibiting a cannabinoid-1- receptor, or in the manufacture of a medicament for treating a condition mediated by a cannabinoid-1 receptor.
- the condition includes, but is not limited to, an eating disorder associated with excessive food intake, obesity, diabetes, insulin resistance, hypercholesterolemia, hypertriglyceridemia, substance abuse, osteoporosis, or smoking abuse.
- the condition is obesity.
- Alkyl refers to a moiety and as a structural element of other groups, for example halo-substituted-alkyl and alkoxy, and may be straight-chained or branched.
- An optionally substituted alkyl, alkenyl or alkynyl as used herein may be optionally halogenated (e.g., CF 3 ), or may have one or more carbons that is substituted or replaced with a heteroatom, such as NR, O or S (e.g., -OCH 2 CH 2 O-, alkylthiols, thioalkoxy, alkylamines, etc).
- Aryl refers to a monocyclic or fused bicyclic aromatic ring containing carbon atoms.
- aryl may be phenyl or naphthyl.
- Arylene means a divalent radical derived from an aryl group.
- Heteroaryl as used herein is as defined for aryl above, where one or more of the ring members is a heteroatom.
- heteroaryls include but are not limited to pyridyl, indolyl, indazolyl, quinoxalinyl, quinolinyl, benzofuranyl, benzopyranyl, benzothiopyranyl, benzo[l,3]dioxole, imidazolyl, benzo-imidazolyl, pyrimidinyl, furanyl, oxazolyl, isoxazolyl, triazolyl, tetrazolyl, pyrazolyl, thienyl, etc.
- Examples of carbocyclic rings include but are not limited to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclopropylene, cyclohexanone, etc.
- a "heterocyclic ring” as used herein is as defined for a carbocyclic ring above, wherein one or more ring carbons is a heteroatom.
- heterocyclic rings include but are not limited to morpholino, pyrrolidinyl, pyrrolidinyl-2-one, piperazinyl, piperidinyl, piperidinylone, l,4-dioxa-8-aza-spiro[4.5]dec-8-yl, etc.
- co-administration or “combined administration” or the like as used herein are meant to encompass administration of the selected therapeutic agents to a single patient, and are intended to include treatment regimens in which the agents are not necessarily administered by the same route of administration or at the same time.
- the term "pharmaceutical combination” as used herein refers to a product obtained from mixing or combining active ingredients, and includes both fixed and non-fixed combinations of the active ingredients.
- the term "fixed combination” means that the active ingredients, e.g. a compound of Formula (1) and a co-agent, are both administered to a patient simultaneously in the form of a single entity or dosage.
- the term “non-fixed combination” means that the active ingredients, e.g. a compound of Formula (1) and a co-agent, are both administered to a patient as separate entities either simultaneously, concurrently or sequentially with no specific time limits, wherein such administration provides therapeutically effective levels of the active ingredients in the body of the patient.
- cocktail therapy e.g. the administration of three or more active ingredients.
- terapéuticaally effective amount means the amount of the subject compound that will elicit a biological or medical response in a cell, tissue, organ, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician.
- administration means providing a compound of the invention and prodrugs thereof to a subject in need of treatment.
- the invention provides compounds and pharmaceutical compositions thereof, which may be useful as inhibitors of cannabinoid receptor 1 activity. [0027] In one aspect, the invention provides compounds of Formula (1):
- Y is O, S or NR 5 ;
- X is O or S
- R 1 and R 2 are independently L-R 9 ;
- L is a bond, S or O
- R 3 and R 4 are independently H, an optionally halogenated Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, cyano, halo, nitro, NR 6 R 7 , CO(CR 2 ) k NR 6 R 7 , OR 8 , SOi_ 2 R 8 , NR 7 SOi_ 2 R 8 , COi_ 2 R 8 or R 9 ;
- R 5 , R 6 , R 7 and R 8 are independently H, Ci_ 6 alkyl, C 2 _ 6 alkenyl, C 2 _ 6 alkynyl, or (CR 2 ) k R 9 ; each R 9 is an optionally substituted C 3 - 7 cycloalkyl, or a 5-12 membered aryl, heteroaryl or heterocyclic ring containing N, O or S;
- R is H or Ci_6 alkyl; and j and k are independently 0-6.
- R 1 , R 2 and R 4 are independently an optionally substituted C 3 _ 7 cycloalkyl, or a 5-7 membered aryl, heteroaryl or heterocyclic ring containing N, O or S.
- R 1 , R 2 and R 4 are independently phenyl, piperazinyl, morpholino, benzthiazolyl, pyridinyl or pyrazolyl.
- R 4 may be oxadiazolyl, furanyl, pyridinyl, indolyl, morpholino, piperazinyl, C 3 - 7 cycloalkyl, tetrahydrothiopyranyl or quinolinyl.
- R 3 and R 4 are independently an optionally substituted Ci_ 6 alkyl.
- the invention provides compounds of Formula (2):
- X and Y are independently O or S;
- R 10 , R 11 and R 12 are independently halo, an optionally substituted alkyl, C 2 -6 alkenyl, C 2 -6 alkynyl, cycloalkyl, or an optionally substituted aryl, heteroaryl or heterocyclic ring; and m, n and p are independently 0-5.
- R 10 , R 11 and R 12 may independently be halo, an optionally halogenated Ci_ 6 alkyl, or an optionally substituted cycloalkyl, phenyl, pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, imidazolyl, pyrazolyl, oxazolyl, oxadiazolyl, isoxazolyl, furanyl, thiophenyl, or triazolyl.
- R 10 , R 11 and R 12 may be an optionally substituted Ci_ 6 alkyl, C 2 - 6 alkenyl, C 2 - 6 alkynyl, piperazinyl, piperidinyl, morpholinyl, piperidin- 2-one, pyrrolidin-2-one, phenyl, OR 13 , SR 13 , SO ⁇ 2 R 13 , CO ⁇ 2 R 13 , CONR 13 2 wherein R 13 is an optionally substituted Ci_ 6 alkyl, cycloalkyl, aryl, heteroaryl or heterocyclic ring.
- R 11 may be phenyl or pyridyl, each optionally substituted with C 1-6 alkyl, C 1-6 alkoxy, amino, NHSO 2 (Ci_6 alkyl) and the like.
- p is 0.
- the present invention also includes all suitable isotopic variations of the compounds of the invention, or pharmaceutically acceptable salts thereof.
- An isotopic variation of a compound of the invention or a pharmaceutically acceptable salt thereof is defined as one in which at least one atom is replaced by an atom having the same atomic number but an atomic mass different from the atomic mass usually found in nature.
- isotopes that may be incorporated into the compounds of the invention and pharmaceutically acceptable salts thereof include but are not limited to isotopes of hydrogen, carbon, nitrogen and oxygen such as as 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 17 0, 18 0, 35 S, 18 F, 36 Cl and 123 I.
- isotopic variations of the compounds of the invention and pharmaceutically acceptable salts thereof are useful in drug and/or substrate tissue distribution studies.
- 3 H and 14 C isotopes may be used for their ease of preparation and detectability.
- substitution with isotopes such as 2 H may afford certain therapeutic advantages resulting from greater metabolic stability, such as increased in vivo half-life or reduced dosage requirements.
- Isotopic variations of the compounds of the invention or pharmaceutically acceptable salts thereof can generally be prepared by conventional procedures using appropriate isotopic variations of suitable reagents.
- Compounds of the invention may inhibit the activity of CB 1 and, as such, may be useful for treating diseases or disorders in which the activity of CB 1 contributes to the pathology and/or symptomology of the disease.
- This invention further provides compounds of this invention for use in the preparation of medicaments for the treatment of diseases or disorders in which CB 1 activity contributes to the pathology and/or symptomology of the disease.
- CBl mediated diseases or conditions include, but are not limited to, metabolic disorders as well as conditions associated with metabolic disorders including eating disorders associated with excessive food intake, obesity, bulimia nervosa, and compulsive eating disorders; diabetes, arteriosclerosis, hypertension, polycystic ovary disease, osteoporosis, cardiovascular disease, osteoarthritis, dermatological disorders, hypertension, insulin resistance, hypercholesterolemia, hypertriglyceridemia, cholelithiasis, sleep disorders, and hyperlipidemic conditions; psychiatric disorders such as substance abuse, psychosis, depression, anxiety, stress, epilepsy, mania and schizophrenia; cognitive disorders such as dementia including Alzheimer's disease, memory deficits, short term memory loss and attention deficit disorders; neurodegenerative disorders such as Parkinson's Disease, cerebral apoplexy, craniocerebral trauma, hypotension, catabolism in connection with pulmonary dysfunction and ventilator dependency; cardiac dysfunction including valvular disease, myocardial infarction, cardiac hypertrophy and
- the compounds of the invention may be useful for the treatment of eating disorders by inhibiting excessive food intake and the resulting obesity and complications associated therewith, including left ventricular hypertrophy.
- the compounds of the invention may also be useful for the treatment of substance abuse disorders, such as substance abuse of opiates, alcohol, marijuana, and nicotine; and may also be useful as a smoking cessation aid for treating or ameliorating smoking addictions.
- Marijuana and its derivatives have been used for centuries for medicinal and recreational purposes.
- a major active ingredient in marijuana and hashish has been determined to be ⁇ 9- Tetrahydromaynabinol ( ⁇ 9-THC).
- ⁇ 9-THC ⁇ 9- Tetrahydromaynabinol
- the biological action of ⁇ 9-THC and other members of the cannabinoid family occurs through two G-protein coupled receptors termed CBl and CB2.
- the CB 1 receptor is primarily found in the central and peripheral nervous systems and to a lesser extent in several peripheral organs.
- the CB2 receptor is found primarily in lymphoid tissues and cells.
- Three endogenous ligands for the cannabinoid receptors derived from arachidonic acid have been identified (anandamide, 2- arachidonoyl glycerol, and 2-arachidonyl glycerol ether). Each is an agonist with activities similar to ⁇ 9-THC, including sedation, hypothermia, intestinal immobility, antinociception, analgesia, catalepsy, anti-emesis, and appetite stimulation.
- mice The genes for the respective cannabinoid receptors have each been disrupted in mice.
- the CBl receptor knockout mice appeared normal and fertile. They were resistant to the effects of ⁇ 9-THC and demonstrated a strong reduction in the reinforcing properties of morphine and the severity of withdrawal syndrome. They also demonstrated reduced motor activity and hypoalgesia.
- the CB2 receptor knockout mice were also healthy and fertile. They were not resistant to the central nervous system mediated effects of administered ⁇ 9-THC. There were some effects on immune cell activation, reinforcing the role for the CB 2 receptor in immune system functions. Excessive exposure to ⁇ 9-THC may lead to overeating, psychosis, hypothermia, memory loss, and sedation.
- the compounds may also be useful for the treatment of constipation and chronic intestinal pseudo-obstruction, as well as for the treatment of asthma, osteoporosis, and cirrhosis of the liver.
- Treatment of asthma with CB 1 receptor modulators (such as CB 1 inverse agonists) is supported by the finding that presynaptic cannabinoid CBl receptors mediate the inhibition of noradrenalin release.
- Treatment of cirrhosis of the liver with CB 1 receptor modulators is supported by the finding that a CBl receptor modulator will reverse the low blood pressure observed in rats with carbon tetrachloride-induced liver cirrhosis and will lower the elevated mesenteric blood flow and portal vein pressure.
- the present invention further provides a method for preventing or treating any of the diseases or disorders described above in a subject in need of such treatment, which method comprises administering to said subject a therapeutically effective amount (See, "Administration and Pharmaceutical Compositions," infra) of a compound of Formula (1) or (2), or a pharmaceutically acceptable salt thereof.
- a therapeutically effective amount See, "Administration and Pharmaceutical Compositions," infra
- the required dosage will vary depending on the mode of administration, the particular condition to be treated and the effect desired.
- compounds of the invention will be administered in therapeutically effective amounts via any of the usual and acceptable modes known in the art, either singly or in combination with one or more therapeutic agents.
- a therapeutically effective amount may vary widely depending on the severity of the disease, the age and relative health of the subject, the potency of the compound used and other factors. In general, satisfactory results may be obtained systemically at daily dosages of from about 0.03 to 2.5 mg/kg per body weight.
- An indicated daily dosage in the larger mammal, e.g. humans, is in the range from about 0.5 mg to about 100 mg, conveniently administered, e.g. in divided doses up to four times a day or in retard form.
- Suitable unit dosage forms for oral administration comprise from ca. 1 to 50 mg active ingredient.
- Compounds of the invention may be administered as pharmaceutical compositions by any conventional route, in particular enterally, e.g., orally, e.g., in the form of tablets or capsules, or parenterally, e.g., in the form of injectable solutions or suspensions, topically, e.g., in the form of lotions, gels, ointments or creams, or in a nasal or suppository form.
- Pharmaceutical compositions comprising a compound of the present invention in free form or in a pharmaceutically acceptable salt form in association with at least one pharmaceutically acceptable carrier or diluent may be manufactured in a conventional manner by mixing, granulating or coating methods.
- oral compositions may be tablets or gelatin capsules comprising the active ingredient together with a) diluents, e.g., lactose, dextrose, sucrose, mannitol, sorbitol, cellulose and/or glycine; b) lubricants, e.g., silica, talcum, stearic acid, its magnesium or calcium salt and/or polyethyleneglycol; for tablets also c) binders, e.g., magnesium aluminum silicate, starch paste, gelatin, tragamayth, methylcellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone; if desired d) disintegrants, e.g., starches, agar, alginic acid or its sodium salt, or effervescent mixtures; and/or e) absorbents, colorants, flavors and sweeteners.
- diluents e.g., lactose, dextrose, sucrose
- compositions may be aqueous isotonic solutions or suspensions, and suppositories may be prepared from fatty emulsions or suspensions.
- the compositions may be sterilized and/or contain adjuvants, such as preserving, stabilizing, wetting or emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or buffers. In addition, they may also contain other therapeutically valuable substances.
- Suitable formulations for transdermal applications include an effective amount of a compound of the present invention with a carrier.
- a carrier may include absorbable pharmacologically acceptable solvents to assist passage through the skin of the host.
- transdermal devices are in the form of a bandage comprising a backing member, a reservoir containing the compound optionally with carriers, optionally a rate controlling barrier to deliver the compound to the skin of the host at a controlled and predetermined rate over a prolonged period of time, and means to secure the device to the skin.
- Matrix transdermal formulations may also be used.
- Suitable formulations for topical application, e.g., to the skin and eyes, are preferably aqueous solutions, ointments, creams or gels well-known in the art. Such may contain solubilizers, stabilizers, tonicity enhancing agents, buffers and preservatives.
- Compounds of the invention may be administered in therapeutically effective amounts in combination with one or more therapeutic agents (pharmaceutical combinations).
- therapeutic agents such as, psychosis, memory deficit, cognitive disorders, migraine, neuropathy, neuroinflammatory disorders, cerebral vascular accidents, head trauma, anxiety disorders, stress, epilepsy, Parkinson's disease, schizophrenia, substance abuse disorders such as smoking cessation, osteoporosis, constipation, chronic intestinal pseudo-obstruction, cirrhosis of the liver, asthma, obesity, and other eating disorders associated with excessive food intake, obesity, etc. (see “Pharmacology and Utility", supra).
- a combined preparation or pharmaceutical composition may comprise a compound of the invention as defined above or a pharmaceutical acceptable salt thereof and at least one active ingredient selected from: a) anti-diabetic agents such as insulin, insulin derivatives and mimetics; insulin secretagogues such as the sulfonylureas, e.g., glipizide, glyburide and AMAR YL®; insulinotropic sulfonylurea receptor ligands such as meglitinides, e.g., nateglinide and repaglinide; insulin sensitizer such as protein tyrosine phosphatase- IB (PTP-IB) inhibitors such as PTP-112; GSK3 (glycogen synthase kinase-3)
- GI-262570 diacylglycerol acetyltransferase (DGAT) inhibitors such as those disclosed in WO 2005044250, WO 2005013907, WO 2004094618 and WO 2004047755; b) hypolipidemic agents such as 3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase inhibitors, e.g., lovastatin and related compounds such as those disclosed in U.S. Pat. No. 4,231,938, pitavastatin, simvastatin and related compounds such as those disclosed in U.S. Pat. Nos. 4,448,784 and 4,450,171, pravastatin and related compounds such as those disclosed in U.S. Pat.
- phosphinic acid compounds useful in inhibiting HMG CoA reductase suitable for use herein are disclosed in GB 2205837; squalene synthase inhibitors; FXR (farnesoid X receptor) and LXR (liver X receptor) ligands; cholestyramine; fibrates; nicotinic acid and aspirin; c) an anti-obesity agent or appetite regulating agent such as melanocortin receptor (MC4R) agonists, melanin-concentrating hormone receptor (MCHR) antagonists, growth hormone secretagogue receptor (GHSR) antagonists, galanin receptor modulators, orexin antagonists, CCK agonists, GLP-I agonists, and other Pre-proglucagon-derived peptides; NPYl or NPY5 antagonsist, NPY2 and NPY4 modulators, corticotropin releasing factor agonists, histamine receptor-3 (H3) modulators,
- a thyroid receptor beta modulator such as a thyroid receptor ligand as disclosed in WO 97/21993 (U. CaI SF), WO 99/00353 (KaroBio) and GB98/284425 (KaroBio), a SCD-I inhibitor as disclosed in WO2005011655, a lipase inhibitor, such as orlistat or ATL-962 (Alizyme), serotonin receptor agonists, (e.g., BVT- 933 (Biovitrum)), monoamine reuptake inhibitors or releasing agents, such as fenfluramine, dexfenfluramine, fluvoxamine, fluoxetine, paroxetine, sertraline, chlorphentermine, cloforex, clortermine, picilorex, sibutramine, dexamphetamine, phentermine, phenylpropanolamine or
- ECE inhibitors e.g. SLV306
- ACE/NEP inhibitors such as omapatrilat, sampatrilat and fasidotril
- angiotensin II antagonists such as maydesartan, eprosartan, irbesartan, losartan, telmisartan and valsartan, in particular valsartan
- renin inhibitors such as aliskiren, terlakiren, ditekiren, RO 66-1132, RO-66-1168
- beta- adrenergic receptor blockers such as acebutolol, atenolol, betaxolol, bisoprolol, metoprolol, nadolol, propranolol, sotalol and timolol
- inotropic agents such as digoxin, dobutamine and milrinone
- calcium channel blockers such as digoxin, dobutamine and milrin
- a chemotherapeutic agent such as aromatase inhibitors e.g. femara, anti-estrogens, topoisomerase I inhibitors, topoisomerase II inhibitors, microtubule active agents, alkylating agents, antineoplastic antimetabolites, platin compounds, compounds decreasing the protein kinase activity such as a PDGF receptor tyrosine kinase inhibitor preferably imatinib ( ⁇ N- ⁇ 5- [4-(4-methyl-piperazino-methyl)-benzoylamido]-2-methylphenyl ⁇ -4-(3-pyridyl)-2-pyrimidine- amine ⁇ ) described in the European patent application EP-A-O 564 409 as example 21 or 4- Methyl-N-[3-(4-methyl-imidazol-l-yl)-5-trifluoromethyl-phenyl]-3-(4-pyridin-3-yl-pyrimidin-2- ylamin
- tegaserod hydrogen maleate cisapride, cilansetron
- an agent for treating tobacco abuse e.g., nicotine receptor partial agonists, bupropion hypochloride (also known under the tradename ZYB AN®) and nicotine replacement therapies
- an agent for treating erectile dysfunction e.g., dopaminergic agents, such as apomorphine
- ADD/ADHD agents e.g., RITALIN®, STRATTERA®, CONCERTA® and ADDERALL®
- an agent for treating alcoholism such as opioid antagonists (e.g., naltrexone (REVIA®) and nalmefene), disulfiram (ANTABUSE®), and acamprosate (CAMPRAL®)).
- opioid antagonists e.g., naltrexone (REVIA®) and nalmefene
- disulfiram ANTABUSE®
- acamprosate CAMPRAL®
- agents for reducing alcohol withdrawal symptoms may also be co-administered, such as benzodiazepines, beta- blockers, clonidine, carbamazepine, pregabalin, and gabapentin (NEURONTIN®); q) other agents that are useful including anti-inflammatory agents (e.g., COX-2 inhibitors) ; antidepressants (e.g., fluoxetine hydrochloride (PROZAC®)); cognitive improvement agents (e.g., donepezil hydrochloride (AIRCEPT®) and other acetylcholinesterase inhibitors); neuroprotective agents (e.g., memantine) ; antipsychotic medications (e.g., ziprasidone (GEODON®), risperidone (RISPERDAL®), and olanzapine (ZYPREXA®)); or, in each case a pharmaceutically acceptable salt thereof; and optionally a pharmaceutically acceptable carrier.
- anti-inflammatory agents e.g., COX-2 inhibitors
- the invention also provides for a pharmaceutical combinations, e.g. a kit, comprising a) a first agent which is a compound of the invention as disclosed herein, in free form or in pharmaceutically acceptable salt form, and b) at least one co-agent.
- a pharmaceutical combination e.g. a kit, comprising a) a first agent which is a compound of the invention as disclosed herein, in free form or in pharmaceutically acceptable salt form, and b) at least one co-agent.
- the kit may comprise instructions for its administration.
- the present invention also includes processes for the preparation of compounds of the invention.
- reactive functional groups for example hydroxy, amino, imino, thio or carboxy groups, where these are desired in the final product, to avoid their unwanted participation in the reactions.
- Conventional protecting groups may be used in accordance with standard practice, for example, see T.W. Greene and P. G. M. Wuts in "Protective Groups in Organic Chemistry", John Wiley and Sons, 1991.
- a compound of the invention may be prepared as a pharmaceutically acceptable acid addition salt by reacting the free base form of the compound with a pharmaceutically acceptable inorganic or organic acid.
- a pharmaceutically acceptable base addition salt of a compound of the invention may be prepared by reacting the free acid form of the compound with a pharmaceutically acceptable inorganic or organic base.
- the salt forms of the compounds of the invention may be prepared using salts of the starting materials or intermediates.
- the free acid or free base forms of the compounds of the invention may be prepared from the corresponding base addition salt or acid addition salt from, respectively.
- a compound of the invention in an acid addition salt form may be converted to the corresponding free base by treating with a suitable base (e.g., ammonium hydroxide solution, sodium hydroxide, and the like).
- a suitable base e.g., ammonium hydroxide solution, sodium hydroxide, and the like.
- a compound of the invention in a base addition salt form may be converted to the corresponding free acid by treating with a suitable acid (e.g., hydrochloric acid, etc.).
- Compounds of the invention in unoxidized form may be prepared from N-oxides of compounds of the invention by treating with a reducing agent (e.g., sulfur, sulfur dioxide, triphenyl phosphine, lithium borohydride, sodium borohydride, phosphorus trichloride, tribromide, or the like) in a suitable inert organic solvent (e.g. acetonitrile, ethanol, aqueous dioxane, or the like) at 0 to 8O 0 C.
- a reducing agent e.g., sulfur, sulfur dioxide, triphenyl phosphine, lithium borohydride, sodium borohydride, phosphorus trichloride, tribromide, or the like
- a suitable inert organic solvent e.g. acetonitrile, ethanol, aqueous dioxane, or the like
- Prodrug derivatives of the compounds of the invention may be prepared by methods known to those of ordinary skill in the art (e.g., for further details see Saulnier et al., (1994), Bioorganic and Medicinal Chemistry Letters, Vol. 4, p. 1985).
- appropriate prodrugs may be prepared by reacting a non-derivatized compound of the invention with a suitable carbamylating agent (e.g., 1,1-acyloxyalkylcarbanochloridate, para-nitrophenyl carbonate, or the like).
- Protected derivatives of the compounds of the invention may be made by means known to those of ordinary skill in the art. A detailed description of techniques applicable to the creation of protecting groups and their removal may be found in T. W. Greene, "Protecting Groups in Organic Chemistry", 3 rd edition, John Wiley and Sons, Inc., 1999.
- Hydrates of compounds of the present invention may be conveniently prepared or formed during the process of the invention, as solvates (e.g., hydrates). Hydrates of compounds of the present invention may be conveniently prepared by recrystallization from an aqueous/organic solvent mixture, using organic solvents such as dioxin, tetrahydrofuran or methanol.
- Compounds of the invention may be prepared as their individual stereoisomers by reacting a racemic mixture of the compound with an optically active resolving agent to form a pair of diastereoisomeric compounds, separating the diastereomers and recovering the optically pure enantiomers. While resolution of enantiomers may be carried out using covalent diastereomeric derivatives of the compounds of the invention, dissociable complexes are preferred (e.g., crystalline diastereomeric salts). Diastereomers have distinct physical properties (e.g., melting points, boiling points, solubilities, reactivity, etc.) and may be readily separated by taking advantage of these dissimilarities.
- the diastereomers may be separated by chromatography, or by separation/resolution techniques based upon differences in solubility.
- the optically pure enantiomer is then recovered, along with the resolving agent, by any practical means that would not result in racemization.
- a more detailed description of the techniques applicable to the resolution of stereoisomers of compounds from their racemic mixture may be found in Jean Jacques, Andre Collet, Samuel H. Wilen, "Enantiomers, Racemates and Resolutions", John Wiley And Sons, Inc., 1981.
- the invention encompasses methods of making compounds of Formula (1) and Formula (2), and:
- Step A l-(4-Chloro-phenyl)-2-(2,4-dichloro-phenyl)-9-phenyl-l,9-dihydro-purin-6- one is prepared by the methods reported in WO2006/047516.
- the purinone (96.0 mg, 0.205 mmol) is dissolved in a mixture of 2 mL of acetic acid, 250 ⁇ L of chloroform and 250 ⁇ L of water.
- Sodium acetate 200 mg, 2.44 mmol
- Bromine 250 ⁇ L
- the reaction is checked for completion by reverse phase LC/MS.
- Step B The 8-bromo-purinone (45.0 mg, 82.3 ⁇ mol) is dissolved in 1 mL of tetrahydrofuran and 200 ⁇ L of a 1 M lithium hydroperoxide solution, freshly prepared by mixing lithium hydroxide (18.9 mg, 0.823 mmol) and 36% hydrogen peroxide ( 93.0 ⁇ L, 1.23 mmol) and 103 ⁇ L of water. The reaction is allowed to stir overnight. The reaction is then quenched with 1 M HCl and extracted with ethyl acetate. The organic layer is dried over magnesium sulfate, filtered, and rotary evaporated to dryness. The crude material is purified on silica gel, giving an off white solid.
- Step A Amino-cyano-acetic acid ethyl ester (1.00 g, 7.79 mmol) is dissolved in 100 mL of dichloromethane. Triethylamine (1 mL) and phenyl isothiocyanate (1.05 g, 7.79 mmol) are added sequentially to the reaction mixture, and the mixture is stirred 2 h. The crude is purified by chromatography on silica gel to give 5-amino-l-phenyl-2-thioxo-2,3-dihydro-lH- imidazole-4-carboxylic acid ethyl ester as a white solid.
- Step B N-(4-Bromo-phenyl)-4-methyl-benzamide (29.0 mg, 0.100 mmol) is dissolved in 0.5 mL of thionyl chloride and heated to reflux for 30 min. The reaction is then allowed to cool, and the thionyl chloride is removed under a dry stream of nitrogen to yield imadoyl chloride which is used without further purification. Amino ester prepared from step A (26.4 mg, 0.100 mmol) and vacuum oven dried potassium carbonate (250 mg, 1.81 mmol) are added to the freshly prepared imadoyl chloride, and 2 mL of dry acetonitrile is added to the reaction mixture.
- Step A 2-(4-Bromo-phenyl)-l-(4-chloro-phenyl)-9-phenyl-7,9-dihydro-lH-purine- 6,8-dione (15.0 mg, 30.4 ⁇ mol), anhydrous potassium carbonate (75.0 mg, 0.540 mmol), cyclopropyl boronic acid (20.0 mg, 0.230 mmol), and dichloro[l,l'-bis(diphenylphosphino)- ferrocene]palladium(II) dichloromethane adduct (1.2 mg, 1.47 ⁇ mol) are added to a microwave reaction tube with 0.5 mL of water.
- the tube is sonicated for 2 min, and the tube is capped and purged with an inert atmosphere for 5 min.
- Dimethylformamide (0.5 mL) is added, and the tube is purged for an additional 5 min.
- the tube is then placed in a microwave reactor and heated to 200 0 C for 120 seconds.
- the resulting solution is neutralized with TFA, and an additional 1 mL of dimethylformamide is added.
- the crude product is purified by mass directed reverse phase HPLC.
- Step A 2-(4-Bromo-phenyl)-l-(4-chloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH- purine-6,8-dione (40.0 mg, 76.7 ⁇ mol), potassium acetate (90.0 mg, 0.917 mmol), bispinacolatodiboron (22.3 mg, 88.1 ⁇ mol), and dichloro[l,l'-bis(diphenylphosphino) ferrocene]palladium(II) dichloromethane adduct (3 mg, 3.67 ⁇ mol) are added to a microwave tube and 2 mL of dry dimethylformamide is added.
- Step B A microwave tube is charged with l-(4-chloro-phenyl)-7-ethyl-9-phenyl-2- [4-(4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-phenyl]-7,9-dihydro-lH-purine-6,8-dione (43.0 mg, 75.6 ⁇ mol), potassium carbonate (250 mg, 1.81 mmol), 5-bromo-pyridin-2-ylamine (32.6 mg, 0.189 mmol), and dichloro[l,l'-bis(diphenylphosphino)ferrocene]palladium(II) dichloromethane adduct (4 mg, 4.87 ⁇ mol).
- Step A (5-Bromo-3-methyl-pyridin-2-yl)-carbamic acid tert-butyl ester is prepared by the method of Nantermet et al Bioorg. Med. Chem. Lett. 2004, 2141.
- Step B 2-(4-Bromo-phenyl)-l-(4-chloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH- purine-6,8-dione (52.0 mg, 30.4 ⁇ mol), sodium hydroxide (IM, 200 ⁇ L, 0.200 mmol), [3- methyl-5-(4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-pyridin-2-yl]-carbamic acid tert-butyl ester (47.0 mg, 0.201 mmol), and dichloro[l,l'-bis(diphenylphosphino)ferrocene]palladium(II) dichloromethane adduct (4.00 mg, 4.90 ⁇ mol) are added to a microwave tube with 0.5 mL of water and 2.0 mL of 1 ,2-dimethoxyethane.
- the tube is capped and purged with an inert atmosphere for 5 min.
- the reaction mixture is then heated to 100 0 C for 3 h.
- the reaction is then cooled and the volatiles are removed.
- the crude is then dissolved in a mixture of 1 mL of dichloromethane and 1 mL of trifluoroacetic acid, and the reaction is then stirred for 6 h.
- the resulting crude material is purified by mass directed HPLC.
- Homogenized membranes are prepared from CHO cell clones stably expressing a human cannabinoid receptor 1 (CBl) or human cannabinoid receptor 2 (CB2). Cells are grown and scrapped from 15 cm tissue culture plates, and then subsequently centrifuged down. Cells are washed once with cold PBS, and resuspended in ⁇ 20 ml of Buffer A (20 mM HEPES, pH 7.4, 10 mM EDTA, EDTA-free complete protease inhibitor cocktail [1 tablet/25 ml]). The cell suspension is homogenized on ice, using a Polytron homogenizer at 25,000 rpm at three intervals of 15 seconds each.
- CBDl cannabinoid receptor 1
- CB2 human cannabinoid receptor 2
- the homogenate is first centrifuged at 2,000 rpm on a tabletop low speed centrifuge for 10 minutes.
- the supernatant after passing through a cell strainer, is then centrifuged at 50,000 x g for 25 minutes at 4 0 C.
- the pellet is resuspended into buffer B (15% glycerol, 20 mM HEPES, pH 7.4, 0.1 mM EDTA, EDTA-free complete protease inhibitor cocktail [1 tablet/10 ml]). Protein concentration is determined using the BCA Protein Assay kit using BSA as standard.
- the membranes are aliquoted and kept frozen at -8O 0 C.
- test compounds ranging from 100 ⁇ M to 0.01 nM are prepared in DMSO.
- the desired amount of membrane prep is diluted with ice-cold assay buffer (5OmM Tris-HCl, 2.5mM EDTA, 5 mM MgCl 2 , 0.05% BSA, pH 7.4) and vortexed well.
- Two ⁇ l or less of compound is distributed into each well of a round-bottom 96-well polystyrene assay plate, followed by addition of 100 ⁇ l of diluted membranes (3-10 ⁇ g/well). The mixture is kept on ice until the addition of hot CP55940 (final concentration of 0.5nM).
- [ 3 H]-CP55940 is diluted 1:6300 (v/v) with cold assay buffer and 100 ⁇ l is added into each well. The reaction is carried out at room temperature for 120 minutes before the membranes are harvested onto a PerkinElmer Unifilter GF/B-96 filter plate using a Packard Filtermate Harvester. After nine washes with wash buffer (5OmM Tris-HCl, 2.5mM EDTA, 5 mM MgCl 2 , 0.05% BSA, pH 7), the filter is dried in a 37°C oven for 30 minutes. MicroScint-20 is added and the plate sealed for scintillation counting on TopCount. EC 50 values are obtained by fitting the data with the sigmoidal dose response curve-fitting tool of GraphPad Prism. Eight or twelve different concentrations are used to generate a concentration response curve (using three data points per concentration).
- test compounds ranging from 100 ⁇ M to 0.01 nM are prepared in DMSO.
- the desired amount of membrane prep is diluted with ice-cold assay buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl 2 , 0.1% Fatty acid-free BSA, 5 ⁇ M GDP) and vortexed well.
- Two ⁇ l or less of compound is distributed into each well of a round-bottom 96- well polystyrene assay plate, followed by addition of 100 ⁇ l of diluted membranes (3-10 ⁇ g/well), and the mixture is kept on ice until the addition of hot GTP ⁇ S.
- [ 35 S]-GTP ⁇ S (Perkin Elmer NEG030H; 1 ⁇ Ci/ ⁇ l, 1250 Ci/mmol) is diluted 1: 1000 (v/v) with cold assay buffer and 100 ⁇ l is added into each well. The reaction is carried out at room temperature for 90 minutes before the membranes are harvested onto PerkinElmer Unifilter GF/B-96 filter plate using a Packard Filtermate Harvester. After several washes with wash buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl 2 ), and a rinse with 95% ethanol, the filter is dried in a 37°C oven for 30 minutes. MicroScint-20 is added and the plate sealed for scintillation counting on TopCount. EC 50 values are obtained by fitting the GTP [ ⁇ - 3 S] binding data with the sigmoidal dose response curve-fitting tool of GraphPad Prism. Six or twelve different concentrations are used to generate a concentration response curve (using three data points per concentration).
- mice Male ob/ob mice (age 7-8 weeks old, Jackson Labs, Bar Harbor, Maine) are housed in groups of four and fed commercial standard pellet diet (Lab Diet 5001, PMI Nutrition International, LLC). Diet-induced obese mice are generated using 6-7 weeks old C57BL6 mice (Jackson Labs, Bar Harbor, Maine) placed on high fat diet (D12331, Research Diets) for 12-17 weeks. All mice are maintained on a 12-hour light/dark cycle (lights on at 06:00) in a humidity and temperature-controlled environment with free access to food and water.
- Commercial standard pellet diet Lab Diet 5001, PMI Nutrition International, LLC
- Diet-induced obese mice are generated using 6-7 weeks old C57BL6 mice (Jackson Labs, Bar Harbor, Maine) placed on high fat diet (D12331, Research Diets) for 12-17 weeks. All mice are maintained on a 12-hour light/dark cycle (lights on at 06:00) in a humidity and temperature-controlled environment with free access to food and water.
- mice are singly housed and a habituation to treatment is performed to establish baseline food consumption and body weight. Animals are randomized into treatment groups based on their initial body weight and food consumption.
- test compound a compound of the invention
- DIO mice are treated with either vehicle, a known antagonist as a positive control, or with test compound(s) for up to 7-35 days.
- Test compounds are dosed at ranges between 0.1 up to 100 mg/kg. Animals are treated one hour prior to the start of the dark cycle. Food intake and body weight are recorded manually using an electronic balance prior to treatment, 16 hours post- treatment, followed by daily measurements for up to 7-35 days after the start of study. Compound efficacy is determined by comparing food intake and body weight data between vehicle treated, standard positive control treated, and test compound treated mice.
- compounds of Formula (1) and (2) in free form or in pharmaceutically acceptable salt form, exhibit valuable pharmacological properties, for example, as indicated by the in vitro tests described in this application.
- compounds of the invention have a K 1 of between IxIO "5 and Ix 10 "10 M.
- the compounds of the invention have a K 1 of less than 500 nM; and in other embodiments, a K 1 of less than 10OnM.
- the compounds of the invention exhibit at least 10 fold selectivity for CBl over CB 2. In some instances, the compounds of the invention exhibit at least 20-fold, 50-fold or 100-fold selectivity for CBl over CB2.
Abstract
The invention provides compounds and pharmaceutical compositions, and methods for using such compounds to treat, ameliorate or prevent a condition associated with activity of cannabinoid receptor 1 (CB1).
Description
COMPOUNDS AND COMPOSITIONS AS INHIBITORS OF CANNABINOID
RECEPTOR 1 ACTIVITY
Cross-Reference to Related Applications
[0001] This application claims the benefit of U.S. provisional application serial number 60/914,304, filed April 26, 2007, which is incorporated herein by reference in its entirety.
Technical Field
[0002] The invention provides compounds, pharmaceutical compositions comprising such compounds and methods of using such compounds to treat or prevent diseases or disorders associated with the activity of cannabinoid receptor 1 (CBl).
Background Art
[0003] Cannabinoids are psychoactive ingredients of marijuana, principally delta-9- tetrahydromaynabinol. Two cannabinoid receptors have been cloned, CBl and CB2. CBl is predominantly expressed in the central nervous system whereas CB2 is expressed in peripheral tissues, principally in the immune system. Both receptors are members of the G-protein coupled class and their inhibition is linked to adenylate cyclase activity.
Disclosure of the Invention
[0004] The invention provides compounds and pharmaceutical compositions thereof, which may be useful as inhibitors of cannabinoid receptor 1 activity.
[0005] In one aspect, the invention provides compounds of Formula (1):

and pharmaceutically acceptable salts thereof; wherein Y is O, S or NR5;
X is O or S;
R1 and R2 are independently L-R9;
L is a bond, S or O;
R3 and R4 are independently H, an optionally halogenated Ci_6 alkyl, C2_6 alkenyl, C2_6 alkynyl, cyano, halo, nitro, NR6R7, CO(CR2)kNR6R7, OR8, SOi_2R8, NR7SOi_2R8, COi_2R8 or R9;
R5, R6, R7 and R8 are independently H, Ci_6 alkyl, C2-6 alkenyl, C2_6 alkynyl, or (CR2)kR9; each R9 is an optionally substituted C3_7 cycloalkyl, or a 5-12 membered aryl, heteroaryl or heterocyclic ring containing N, O or S;
R is H or Ci-6 alkyl; and j and k are independently 0-6.
[0006] In the above Formula (1), X and Y may independently be O or S. In some examples, L is a bond.
[0007] In the above Formula (1), R1, R2 and R4 may independently be an optionally substituted cycloalkyl, or a 5-7 membered aryl, heteroaryl or heterocyclic ring containing N, O or S. For example, R1, R2 and R4 are independently phenyl, piperazinyl, morpholino, benzthiazolyl, pyridinyl or pyrazolyl. In particular examples, R1, R2 and R4 are independently an optionally substituted phenyl.
[0008] In one embodiment, the invention provides compounds of Formula (2):

wherein X and Y are independently O or S;
R10, R11 and R12 are independently halo, an optionally substituted alkyl, C2-6 alkenyl, C2-6 alkynyl, cycloalkyl, or an optionally substituted aryl, heteroaryl or heterocyclic ring; and m, n and p are independently 0-5.
[0009] In the above Formula (2), R10 may be halo. In other examples, n is 1-2, and R11 is halo, an optionally halogenated Ci_6 alkyl, or an optionally substituted
 cycloalkyl, phenyl, pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, imidazolyl, pyrazolyl, oxazolyl, oxadiazolyl, isoxazolyl, furanyl, thiophenyl, or triazolyl. In other examples, p is 0.
cycloalkyl, phenyl, pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, imidazolyl, pyrazolyl, oxazolyl, oxadiazolyl, isoxazolyl, furanyl, thiophenyl, or triazolyl. In other examples, p is 0.
[0010] In the above compounds of Formula (1) and (2), each optionally substituted moiety may optionally be substituted with an optionally halogenated C]_6 alkyl, C2-6 alkenyl, C2-6 alkynyl, cyano, halo, nitro, NR6R7, CO(CR2)kNR6R7, OR8, SOi_2R8, NR7SOi_2R8, COi_2R8 or R9, wherein R6, R7, R8 and R9 are substituents as defined in Formula (1).
[0011] In another embodiment, the invention provides a compound selected from the group consisting of l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8- dione; l-(4-bromo-phenyl)-9-phenyl-8-thioxo-2-p-tolyl-l,7,8,9-tetrahydro-purin-6-one; l-(4-chlorophenyl)-9-phenyl-2-(4-(trifluoromethyl)phenyl)-lH-purine-6,8(7H,9H)- dione; l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-7-methyl-9-phenyl-7,9-dihydro-lH-purine- 6,8-dione; l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-6,8-dioxo-9-phenyl-l,6,8,9-tetrahydro- purine-7-carboxylic acid dimethylamide;
[l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-6,8-dioxo-9-phenyl- 1,6,8, 9-tetrahydro- purin-7-yl]-acetonitrile; methyl 2-(l-(4-chlorophenyl)-2-(2,4-dichlorophenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)acetate;
2-(l-(4-chlorophenyl)-2-(2,4-dichlorophenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)acetamide; l-(4-chlorophenyl)-2-(2,4-dichlorophenyl)-9-phenyl-7-(prop-2-ynyl)-lH-purine- 6,8(7H,9H)-dione; l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH-purine- 6,8-dione; l-(4-chlorophenyl)-2-(2,4-dichlorophenyl)-7-isopropyl-9-phenyl-lH-purine-6,8(7H,9H)- dione;
2-(4-bromophenyl)-l-(4-chlorophenyl)-9-phenyl-lH-purine-6,8(7H,9H)-dione; l-(4-chloro-phenyl)-2-(4-cyclopropyl-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8- dione;
2-(4-bromophenyl)-l-(4-chlorophenyl)-7-ethyl-9-phenyl-lH-purine-6,8(7H,9H)-dione;
2-[4-(6-amino-pyridin-3-yl)-phenyl]-l-(4-chloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro- 1 H-purine- 6, 8 -dione ; l-(4-chloro-phenyl)-2-(4-cyclopropyl-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH-purine- 6,8-dione; l-(4-bromophenyl)-9-phenyl-2-p-tolyl-lH-purine-6,8(7H,9H)-dione;
2-(4-(2-aminopyridin-4-yl)phenyl)-l-(4-chlorophenyl)-7-ethyl-9-phenyl-lH-purine- 6,8(7H,9H)-dione;
2-(4-(5-aminopyridin-2-yl)phenyl)-l-(4-chlorophenyl)-7-ethyl-9-phenyl-lH-purine- 6,8(7H,9H)-dione;
2-(4-(6-amino-2-methylpyridin-3-yl)phenyl)-l-(4-chlorophenyl)-7-ethyl-9-phenyl-lH- purine-6,8(7H,9H)-dione; l-(4-chlorophenyl)-2-(4-isopropylphenyl)-9-phenyl-7-(pyridin-4-ylmethyl)-lH-purine- 6,8(7H,9H)-dione; methyl 3-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)propanoate;
methyl 4-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)butanoate;
3-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)propanoic acid;
2-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)propanamide; l-(4-chlorophenyl)-2-(4-isopropylphenyl)-9-phenyl-lH-purine-6,8(7H,9H)-dione;
4-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)butanoic acid; l-(4-chlorophenyl)-7-ethyl-2-(4-isopropylphenyl)-9-phenyl-lH-purine-6,8(7H,9H)- dione;
7-(2-Amino-ethyl)-l-(4-chloro-phenyl)-2-(4-isopropyl-phenyl)-9-phenyl-7,9-dihydro- 1 H-purine- 6, 8 -dione ; l-(4-chlorophenyl)-7-ethyl-2-(4-(6-methoxypyridin-3-yl)phenyl)-9-phenyl-lH-purine- 6,8(7H,9H)-dione;
N-(4'-(l-(4-chlorophenyl)-7-ethyl-6,8-dioxo-9-phenyl-6,7,8,9-tetrahydro-lH-purin-2- yl)biphenyl-4-yl)methanesulfonamide; and
2-[4-(6-amino-5-methyl-pyridin-3-yl)-phenyl]-l-(4-chloro-phenyl)-7-ethyl-9-phenyl-7,9- dihydro- 1 H-purine- 6, 8 -dione ; and pharmaceutical compositions thereof, comprising said compound and a pharmaceutically acceptable carrier.
[0012] In another aspect, the present invention provides pharmaceutical compositions comprising a compound of Formula (1) or (2), and a pharmaceutically acceptable carrier.
[0013] In yet another aspect, the invention provides methods for inhibiting a cannabinoid- 1 receptor, comprising administering to a system or a subject in need thereof, a therapeutically effective amount of a compound comprising Formula (1) or (2), or pharmaceutically acceptable salts or pharmaceutical compositions thereof, thereby inhibiting a cannabinoid- 1 receptor. Compounds of the invention may be administered to a cell or tissue system, or to a human or animal subject.
[0014] Furthermore, the invention provides methods for treating a condition mediated by a cannabinoid- 1 receptor, comprising administering to a system or a subject in need thereof, a therapeutically effective amount of a compound comprising Formula (1) or (2), or
pharmaceutically acceptable salts or pharmaceutical compositions thereof, thereby treating said condition. For example, compounds of Formula (1) or (2) may be used for treating an eating disorder associated with excessive food intake, obesity, diabetes, insulin resistance, hypercholesterolemia, hypertriglyceridemia, substance abuse, osteoporosis, or smoking abuse. In some examples, compounds of Formula (1) or (2) are useful for treating obesity, and may be administered to a subject at risk for obesity at a dosage of about 0.001 mg to about 100 mg per kg of body weight.
[0015] The invention also provides the use of compounds of Formula (1) or (2), or pharmaceutically acceptable salts or pharmaceutical compositions thereof, for inhibiting a cannabinoid-1- receptor, or in the manufacture of a medicament for treating a condition mediated by a cannabinoid-1 receptor.
[0016] In the above uses the condition includes, but is not limited to, an eating disorder associated with excessive food intake, obesity, diabetes, insulin resistance, hypercholesterolemia, hypertriglyceridemia, substance abuse, osteoporosis, or smoking abuse. In particular, the condition is obesity.
Definitions
[0017] "Alkyl" refers to a moiety and as a structural element of other groups, for example halo-substituted-alkyl and alkoxy, and may be straight-chained or branched. An optionally substituted alkyl, alkenyl or alkynyl as used herein may be optionally halogenated (e.g., CF3), or may have one or more carbons that is substituted or replaced with a heteroatom, such as NR, O or S (e.g., -OCH2CH2O-, alkylthiols, thioalkoxy, alkylamines, etc).
[0018] "Aryl" refers to a monocyclic or fused bicyclic aromatic ring containing carbon atoms. For example, aryl may be phenyl or naphthyl. "Arylene" means a divalent radical derived from an aryl group.
[0019] "Heteroaryl" as used herein is as defined for aryl above, where one or more of the ring members is a heteroatom. Examples of heteroaryls include but are not limited to pyridyl, indolyl, indazolyl, quinoxalinyl, quinolinyl, benzofuranyl, benzopyranyl, benzothiopyranyl, benzo[l,3]dioxole, imidazolyl, benzo-imidazolyl, pyrimidinyl, furanyl, oxazolyl, isoxazolyl, triazolyl, tetrazolyl, pyrazolyl, thienyl, etc.
[0020] A "carbocyclic ring" as used herein refers to a saturated or partially unsaturated, monocyclic, fused bicyclic or bridged polycyclic ring containing carbon atoms, which may
optionally be substituted, for example, with =0. Examples of carbocyclic rings include but are not limited to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclopropylene, cyclohexanone, etc.
[0021] A "heterocyclic ring" as used herein is as defined for a carbocyclic ring above, wherein one or more ring carbons is a heteroatom. For example, a heterocyclic ring may contain N, O, S, -N=, -S-, -S(O), -S(O)2-, or -NR- wherein R may be hydrogen, Ci^alkyl or a protecting group. Examples of heterocyclic rings include but are not limited to morpholino, pyrrolidinyl, pyrrolidinyl-2-one, piperazinyl, piperidinyl, piperidinylone, l,4-dioxa-8-aza-spiro[4.5]dec-8-yl, etc.
[0022] The terms "co-administration" or "combined administration" or the like as used herein are meant to encompass administration of the selected therapeutic agents to a single patient, and are intended to include treatment regimens in which the agents are not necessarily administered by the same route of administration or at the same time.
[0023] The term "pharmaceutical combination" as used herein refers to a product obtained from mixing or combining active ingredients, and includes both fixed and non-fixed combinations of the active ingredients. The term "fixed combination" means that the active ingredients, e.g. a compound of Formula (1) and a co-agent, are both administered to a patient simultaneously in the form of a single entity or dosage. The term "non-fixed combination" means that the active ingredients, e.g. a compound of Formula (1) and a co-agent, are both administered to a patient as separate entities either simultaneously, concurrently or sequentially with no specific time limits, wherein such administration provides therapeutically effective levels of the active ingredients in the body of the patient. The latter also applies to cocktail therapy, e.g. the administration of three or more active ingredients.
[0024] The term "therapeutically effective amount" means the amount of the subject compound that will elicit a biological or medical response in a cell, tissue, organ, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician.
[0025] The term "administration" or "administering" of the subject compound means providing a compound of the invention and prodrugs thereof to a subject in need of treatment.
Modes of Carrying Out the Invention
[0026] The invention provides compounds and pharmaceutical compositions thereof, which may be useful as inhibitors of cannabinoid receptor 1 activity.
[0027] In one aspect, the invention provides compounds of Formula (1):

and pharmaceutically acceptable salts thereof; wherein Y is O, S or NR5;
X is O or S;
R1 and R2 are independently L-R9;
L is a bond, S or O;
R3 and R4 are independently H, an optionally halogenated Ci_6 alkyl, C2_6 alkenyl, C2_6 alkynyl, cyano, halo, nitro, NR6R7, CO(CR2)kNR6R7, OR8, SOi_2R8, NR7SOi_2R8, COi_2R8 or R9;
R5, R6, R7 and R8 are independently H, Ci_6 alkyl, C2_6 alkenyl, C2_6 alkynyl, or (CR2)kR9; each R9 is an optionally substituted C3-7 cycloalkyl, or a 5-12 membered aryl, heteroaryl or heterocyclic ring containing N, O or S;
R is H or Ci_6 alkyl; and j and k are independently 0-6.
[0028] In the above Formula (1), R1, R2 and R4 are independently an optionally substituted C3_7 cycloalkyl, or a 5-7 membered aryl, heteroaryl or heterocyclic ring containing N, O or S. For example, R1, R2 and R4 are independently phenyl, piperazinyl, morpholino, benzthiazolyl, pyridinyl or pyrazolyl. In some examples, R4 may be oxadiazolyl, furanyl, pyridinyl, indolyl, morpholino, piperazinyl, C3-7 cycloalkyl, tetrahydrothiopyranyl or quinolinyl. In other examples, R3 and R4 are independently an optionally substituted Ci_6 alkyl.
[0029] In one embodiment, the invention provides compounds of Formula (2):

wherein X and Y are independently O or S;
R10, R11 and R12 are independently halo, an optionally substituted alkyl, C2-6 alkenyl, C2-6 alkynyl, cycloalkyl, or an optionally substituted aryl, heteroaryl or heterocyclic ring; and m, n and p are independently 0-5.
[0030] In the above Formula (2), R10, R11 and R12 may independently be halo, an optionally halogenated Ci_6 alkyl, or an optionally substituted
 cycloalkyl, phenyl, pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, imidazolyl, pyrazolyl, oxazolyl, oxadiazolyl, isoxazolyl, furanyl, thiophenyl, or triazolyl. In some examples, R10, R11 and R12 may be an optionally substituted Ci_6 alkyl, C2-6 alkenyl, C2-6 alkynyl, piperazinyl, piperidinyl, morpholinyl, piperidin- 2-one, pyrrolidin-2-one, phenyl, OR13, SR13, SO^2R13, CO^2R13, CONR13 2 wherein R13 is an optionally substituted Ci_6 alkyl,
cycloalkyl, phenyl, pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, imidazolyl, pyrazolyl, oxazolyl, oxadiazolyl, isoxazolyl, furanyl, thiophenyl, or triazolyl. In some examples, R10, R11 and R12 may be an optionally substituted Ci_6 alkyl, C2-6 alkenyl, C2-6 alkynyl, piperazinyl, piperidinyl, morpholinyl, piperidin- 2-one, pyrrolidin-2-one, phenyl, OR13, SR13, SO^2R13, CO^2R13, CONR13 2 wherein R13 is an optionally substituted Ci_6 alkyl,
 cycloalkyl, aryl, heteroaryl or heterocyclic ring. For example, R11 may be phenyl or pyridyl, each optionally substituted with C1-6 alkyl, C1-6 alkoxy, amino, NHSO2(Ci_6 alkyl) and the like. In other examples, p is 0.
cycloalkyl, aryl, heteroaryl or heterocyclic ring. For example, R11 may be phenyl or pyridyl, each optionally substituted with C1-6 alkyl, C1-6 alkoxy, amino, NHSO2(Ci_6 alkyl) and the like. In other examples, p is 0.
[0031] The present invention also includes all suitable isotopic variations of the compounds of the invention, or pharmaceutically acceptable salts thereof. An isotopic variation of a compound of the invention or a pharmaceutically acceptable salt thereof is defined as one in which at least one atom is replaced by an atom having the same atomic number but an atomic mass different from the atomic mass usually found in nature. Examples of isotopes that may be incorporated into the compounds of the invention and pharmaceutically acceptable salts thereof include but are not limited to isotopes of hydrogen, carbon, nitrogen and oxygen such as as 2H, 3H, 11C, 13C, 14C, 15N, 170, 180, 35S, 18F, 36Cl and 123I. Certain isotopic variations of the compounds of the invention and pharmaceutically acceptable salts thereof, for example, those in which a radioactive isotope such as 3H or 14C is incorporated, are useful in drug and/or substrate
tissue distribution studies. In particular examples, 3H and 14C isotopes may be used for their ease of preparation and detectability. In other examples, substitution with isotopes such as 2H may afford certain therapeutic advantages resulting from greater metabolic stability, such as increased in vivo half-life or reduced dosage requirements. Isotopic variations of the compounds of the invention or pharmaceutically acceptable salts thereof can generally be prepared by conventional procedures using appropriate isotopic variations of suitable reagents.
Pharmacology and Utility
[0032] Compounds of the invention may inhibit the activity of CB 1 and, as such, may be useful for treating diseases or disorders in which the activity of CB 1 contributes to the pathology and/or symptomology of the disease. This invention further provides compounds of this invention for use in the preparation of medicaments for the treatment of diseases or disorders in which CB 1 activity contributes to the pathology and/or symptomology of the disease.
[0033] CBl mediated diseases or conditions include, but are not limited to, metabolic disorders as well as conditions associated with metabolic disorders including eating disorders associated with excessive food intake, obesity, bulimia nervosa, and compulsive eating disorders; diabetes, arteriosclerosis, hypertension, polycystic ovary disease, osteoporosis, cardiovascular disease, osteoarthritis, dermatological disorders, hypertension, insulin resistance, hypercholesterolemia, hypertriglyceridemia, cholelithiasis, sleep disorders, and hyperlipidemic conditions; psychiatric disorders such as substance abuse, psychosis, depression, anxiety, stress, epilepsy, mania and schizophrenia; cognitive disorders such as dementia including Alzheimer's disease, memory deficits, short term memory loss and attention deficit disorders; neurodegenerative disorders such as Parkinson's Disease, cerebral apoplexy, craniocerebral trauma, hypotension, catabolism in connection with pulmonary dysfunction and ventilator dependency; cardiac dysfunction including valvular disease, myocardial infarction, cardiac hypertrophy and congestive heart failure; pulmonary dysfunction, transplant rejection, rheumatoid arthritis, migraine, neuropathy, multiple sclerosis, Guillain-Barre syndrome, the inflammatory sequelae of viral encephalitis, cerebral vascular accidents, inflammatory bowel disease, lupus, graft vs. host disease, T-cell mediated hypersensitivity disease, psoriasis, asthma, Hashimoto's thyroiditis, maycer, contact dermatitis, allergic rhinitis, ischemic or reperfusion injury, head trauma and movement disorders.
[0034] In one embodiment, the compounds of the invention may be useful for the treatment of eating disorders by inhibiting excessive food intake and the resulting obesity and complications associated therewith, including left ventricular hypertrophy.
[0035] The compounds of the invention may also be useful for the treatment of substance abuse disorders, such as substance abuse of opiates, alcohol, marijuana, and nicotine; and may also be useful as a smoking cessation aid for treating or ameliorating smoking addictions. Marijuana and its derivatives have been used for centuries for medicinal and recreational purposes. A major active ingredient in marijuana and hashish has been determined to be Δ9- Tetrahydromaynabinol (Δ9-THC). The biological action of Δ9-THC and other members of the cannabinoid family occurs through two G-protein coupled receptors termed CBl and CB2. The CB 1 receptor is primarily found in the central and peripheral nervous systems and to a lesser extent in several peripheral organs.
[0036] The CB2 receptor is found primarily in lymphoid tissues and cells. Three endogenous ligands for the cannabinoid receptors derived from arachidonic acid have been identified (anandamide, 2- arachidonoyl glycerol, and 2-arachidonyl glycerol ether). Each is an agonist with activities similar to Δ9-THC, including sedation, hypothermia, intestinal immobility, antinociception, analgesia, catalepsy, anti-emesis, and appetite stimulation.
[0037] The genes for the respective cannabinoid receptors have each been disrupted in mice. The CBl receptor knockout mice appeared normal and fertile. They were resistant to the effects of Δ9-THC and demonstrated a strong reduction in the reinforcing properties of morphine and the severity of withdrawal syndrome. They also demonstrated reduced motor activity and hypoalgesia. The CB2 receptor knockout mice were also healthy and fertile. They were not resistant to the central nervous system mediated effects of administered Δ9-THC. There were some effects on immune cell activation, reinforcing the role for the CB 2 receptor in immune system functions. Excessive exposure to Δ9-THC may lead to overeating, psychosis, hypothermia, memory loss, and sedation.
[0038] The compounds may also be useful for the treatment of constipation and chronic intestinal pseudo-obstruction, as well as for the treatment of asthma, osteoporosis, and cirrhosis of the liver. Treatment of asthma with CB 1 receptor modulators (such as CB 1 inverse agonists) is supported by the finding that presynaptic cannabinoid CBl receptors mediate the inhibition of noradrenalin release. Treatment of cirrhosis of the liver with CB 1 receptor modulators is supported by the finding that a CBl receptor modulator will reverse the low blood pressure
observed in rats with carbon tetrachloride-induced liver cirrhosis and will lower the elevated mesenteric blood flow and portal vein pressure.
[0039] In accordance with the foregoing, the present invention further provides a method for preventing or treating any of the diseases or disorders described above in a subject in need of such treatment, which method comprises administering to said subject a therapeutically effective amount (See, "Administration and Pharmaceutical Compositions," infra) of a compound of Formula (1) or (2), or a pharmaceutically acceptable salt thereof. For any of the above uses, the required dosage will vary depending on the mode of administration, the particular condition to be treated and the effect desired.
Administration and Pharmaceutical Compositions
[0040] In general, compounds of the invention will be administered in therapeutically effective amounts via any of the usual and acceptable modes known in the art, either singly or in combination with one or more therapeutic agents. A therapeutically effective amount may vary widely depending on the severity of the disease, the age and relative health of the subject, the potency of the compound used and other factors. In general, satisfactory results may be obtained systemically at daily dosages of from about 0.03 to 2.5 mg/kg per body weight. An indicated daily dosage in the larger mammal, e.g. humans, is in the range from about 0.5 mg to about 100 mg, conveniently administered, e.g. in divided doses up to four times a day or in retard form. Suitable unit dosage forms for oral administration comprise from ca. 1 to 50 mg active ingredient.
[0041] Compounds of the invention may be administered as pharmaceutical compositions by any conventional route, in particular enterally, e.g., orally, e.g., in the form of tablets or capsules, or parenterally, e.g., in the form of injectable solutions or suspensions, topically, e.g., in the form of lotions, gels, ointments or creams, or in a nasal or suppository form. Pharmaceutical compositions comprising a compound of the present invention in free form or in a pharmaceutically acceptable salt form in association with at least one pharmaceutically acceptable carrier or diluent may be manufactured in a conventional manner by mixing, granulating or coating methods. For example, oral compositions may be tablets or gelatin capsules comprising the active ingredient together with a) diluents, e.g., lactose, dextrose, sucrose, mannitol, sorbitol, cellulose and/or glycine; b) lubricants, e.g., silica, talcum, stearic acid, its magnesium or calcium salt and/or polyethyleneglycol; for tablets also c) binders, e.g.,
magnesium aluminum silicate, starch paste, gelatin, tragamayth, methylcellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone; if desired d) disintegrants, e.g., starches, agar, alginic acid or its sodium salt, or effervescent mixtures; and/or e) absorbents, colorants, flavors and sweeteners.
[0042] Injectable compositions may be aqueous isotonic solutions or suspensions, and suppositories may be prepared from fatty emulsions or suspensions. The compositions may be sterilized and/or contain adjuvants, such as preserving, stabilizing, wetting or emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or buffers. In addition, they may also contain other therapeutically valuable substances. Suitable formulations for transdermal applications include an effective amount of a compound of the present invention with a carrier. A carrier may include absorbable pharmacologically acceptable solvents to assist passage through the skin of the host. For example, transdermal devices are in the form of a bandage comprising a backing member, a reservoir containing the compound optionally with carriers, optionally a rate controlling barrier to deliver the compound to the skin of the host at a controlled and predetermined rate over a prolonged period of time, and means to secure the device to the skin. Matrix transdermal formulations may also be used. Suitable formulations for topical application, e.g., to the skin and eyes, are preferably aqueous solutions, ointments, creams or gels well-known in the art. Such may contain solubilizers, stabilizers, tonicity enhancing agents, buffers and preservatives.
[0043] Compounds of the invention may be administered in therapeutically effective amounts in combination with one or more therapeutic agents (pharmaceutical combinations). For example, synergistic effects may occur with other substances used in the treatment of diseases or disorders, such as, psychosis, memory deficit, cognitive disorders, migraine, neuropathy, neuroinflammatory disorders, cerebral vascular accidents, head trauma, anxiety disorders, stress, epilepsy, Parkinson's disease, schizophrenia, substance abuse disorders such as smoking cessation, osteoporosis, constipation, chronic intestinal pseudo-obstruction, cirrhosis of the liver, asthma, obesity, and other eating disorders associated with excessive food intake, obesity, etc. (see "Pharmacology and Utility", supra). Where the compounds of the invention are administered in conjunction with other therapies, dosages of the co- administered compounds will of course vary depending on the type of co-drug employed, on the specific drug employed, on the condition being treated and so forth.
[0044] A combined preparation or pharmaceutical composition may comprise a compound of the invention as defined above or a pharmaceutical acceptable salt thereof and at least one active ingredient selected from: a) anti-diabetic agents such as insulin, insulin derivatives and mimetics; insulin secretagogues such as the sulfonylureas, e.g., glipizide, glyburide and AMAR YL®; insulinotropic sulfonylurea receptor ligands such as meglitinides, e.g., nateglinide and repaglinide; insulin sensitizer such as protein tyrosine phosphatase- IB (PTP-IB) inhibitors such as PTP-112; GSK3 (glycogen synthase kinase-3) inhibitors such as SB-517955, SB-4195052, SB-216763, NN-57-05441 and NN-57-05445; RXR ligands such as GW-0791 and AGN- 194204; sodium-dependent glucose co-transporter inhibitors such as T-1095; glycogen phosphorylase A inhibitors such as BAY R3401; biguanides such as metformin; alpha- glucosidase inhibitors such as acarbose; GLP-I (glucagon like peptide-1), GLP-I analogs such as Exendin-4 and GLP-I mimetics; DPPIV (dipeptidyl peptidase IV) inhibitors such as DPP728, LAF237 (vildagliptin - Example 1 of WO 00/34241), MK-0431, saxagliptin, GSK23A ; an AGE breaker; a thiazolidone derivative (glitazone) such as pioglitazone, rosiglitazone, or (R)-l-{4-[5- methyl-2-(4-trifluoromethyl-phenyl)-oxazol-4-ylmethoxy]-benzenesulfonyl}-2,3-dihydro-lH- indole-2-carboxylic acid described in the patent application WO 03/043985, as compound 19 of Example 4, a non-glitazone type PPAR gamma agonist e.g. GI-262570; diacylglycerol acetyltransferase (DGAT) inhibitors such as those disclosed in WO 2005044250, WO 2005013907, WO 2004094618 and WO 2004047755; b) hypolipidemic agents such as 3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase inhibitors, e.g., lovastatin and related compounds such as those disclosed in U.S. Pat. No. 4,231,938, pitavastatin, simvastatin and related compounds such as those disclosed in U.S. Pat. Nos. 4,448,784 and 4,450,171, pravastatin and related compounds such as those disclosed in U.S. Pat. No.4,346,227, cerivastatin, mevastatin and related compounds such as those disclosed in U.S. Pat. No. 3,983,140, velostatin, fluvastatin, dalvastatin, atorvastatin, rosuvastatin and related statin compounds disclosed in U.S. Pat. No. 5,753,675, rivastatin, pyrazole analogs of mevalonolactone derivatives as disclosed in U.S. Pat. No. 4,613,610, indene analogs of mevalonolactone derivatives as disclosed in PCT application WO 86/03488, 6- [2- (substituted- pyrrol-l-yl)-alkyl)pyran-2-ones and derivatives thereof as disclosed in U.S. Pat. No. 4,647,576, Searle's SC-45355 (a 3- substituted pentanedioic acid derivative) dichloroacetate, imidazole analogs of mevalonolactone as disclosed in PCT application WO 86/07054, 3- carboxy-2-
hydroxy-propane-phosphonic acid derivatives as disclosed in French Patent No. 2,596,393, 2,3- disubstituted pyrrole, furan and thiophene derivatives as disclosed in European Patent Application No. 0221025, naphthyl analogs of mevalonolactone as disclosed in U.S. Pat. No. 4,686,237, octahydronaphthalenes such as disclosed in U.S. Pat. No. 4, 499,289, keto analogs of mevinolin (lovastatin) as disclosed in European Patent Application No.0,142,146 A2, and quinoline and pyridine derivatives disclosed in U.S. Pat. Nos. 5,506,219 and 5,691,322. In addition, phosphinic acid compounds useful in inhibiting HMG CoA reductase suitable for use herein are disclosed in GB 2205837; squalene synthase inhibitors; FXR (farnesoid X receptor) and LXR (liver X receptor) ligands; cholestyramine; fibrates; nicotinic acid and aspirin; c) an anti-obesity agent or appetite regulating agent such as melanocortin receptor (MC4R) agonists, melanin-concentrating hormone receptor (MCHR) antagonists, growth hormone secretagogue receptor (GHSR) antagonists, galanin receptor modulators, orexin antagonists, CCK agonists, GLP-I agonists, and other Pre-proglucagon-derived peptides; NPYl or NPY5 antagonsist, NPY2 and NPY4 modulators, corticotropin releasing factor agonists, histamine receptor-3 (H3) modulators, aP2 inhibitors, PPAR gamma modulators, PPAR delta modulators, acetyl-CoA carboxylase (ACC) inihibitors, 11-β-HSD-l inhibitors, adinopectin receptor modulators; beta 3 adrenergic agonists, such as AJ9677 (Takeda/Dainippon), L750355 (Merck), or CP331648 (Pfizer) or other known beta 3 agonists as disclosed in U.S. Pat. Nos. 5,541,204, 5,770,615, 5, 491,134, 5,776,983 and 5,488,064, a thyroid receptor beta modulator, such as a thyroid receptor ligand as disclosed in WO 97/21993 (U. CaI SF), WO 99/00353 (KaroBio) and GB98/284425 (KaroBio), a SCD-I inhibitor as disclosed in WO2005011655, a lipase inhibitor, such as orlistat or ATL-962 (Alizyme), serotonin receptor agonists, (e.g., BVT- 933 (Biovitrum)), monoamine reuptake inhibitors or releasing agents, such as fenfluramine, dexfenfluramine, fluvoxamine, fluoxetine, paroxetine, sertraline, chlorphentermine, cloforex, clortermine, picilorex, sibutramine, dexamphetamine, phentermine, phenylpropanolamine or mazindol, anorectic agents such as topiramate (Johnson & Johnson), CNTF (ciliary neurotrophic factor)/ AXOKINE® (Regeneron), BDNF (brain-derived neurotrophic factor), leptin and leptin receptor modulators, phentermine, leptin, bromocriptine, dexamphetamine, amphetamine, fenfluramine, dexfenfluramine, sibutramine, orlistat, dexfenfluramine, mazindol, phentermine, phendimetrazine, diethylpropion, fluoxetine, bupropion, topiramate, diethylpropion, benzphetamine, phenylpropanolamine or ecopipam, ephedrine, pseudoephedrine;
d) anti-hypertensive agents such as loop diuretics such as ethacrynic acid, furosemide and torsemide; diuretics such as thiazide derivatives, chlorithiazide, hydrochlorothiazide, amiloride; angiotensin converting enzyme (ACE) inhibitors such as benazepril, captopril, enalapril, fosinopril, lisinopril, moexipril, perinodopril, quinapril, ramipril and trandolapril; inhibitors of the Na-K-ATPase membrane pump such as digoxin; neutralendopeptidase (NEP) inhibitors e.g. thiorphan, terteo-thiorphan, SQ29072; ECE inhibitors e.g. SLV306; ACE/NEP inhibitors such as omapatrilat, sampatrilat and fasidotril; angiotensin II antagonists such as maydesartan, eprosartan, irbesartan, losartan, telmisartan and valsartan, in particular valsartan; renin inhibitors such as aliskiren, terlakiren, ditekiren, RO 66-1132, RO-66-1168; beta- adrenergic receptor blockers such as acebutolol, atenolol, betaxolol, bisoprolol, metoprolol, nadolol, propranolol, sotalol and timolol; inotropic agents such as digoxin, dobutamine and milrinone; calcium channel blockers such as amlodipine, bepridil, diltiazem, felodipine, nicardipine, nimodipine, nifedipine, nisoldipine and verapamil; aldosterone receptor antagonists; aldosterone synthase inhibitors; and dual ET/ All antagonist such as those disclosed in WO 00/01389; e) a HDL increasing compound; f) Cholesterol absorption modulator such as ZETIA® and KT6-971; g) Apo-Al analogues and mimetics; h) thrombin inhibitors such as ximelagatran; i) aldosterone inhibitors such as anastrazole, fadrazole, eplerenone; j) Inhibitors of platelet aggregation such as aspirin, clopidogrel bisulfate; k) estrogen, testosterone, a selective estrogen receptor modulator, a selective androgen receptor modulator;
1) a chemotherapeutic agent such as aromatase inhibitors e.g. femara, anti-estrogens, topoisomerase I inhibitors, topoisomerase II inhibitors, microtubule active agents, alkylating agents, antineoplastic antimetabolites, platin compounds, compounds decreasing the protein kinase activity such as a PDGF receptor tyrosine kinase inhibitor preferably imatinib ( { N- {5- [4-(4-methyl-piperazino-methyl)-benzoylamido]-2-methylphenyl}-4-(3-pyridyl)-2-pyrimidine- amine }) described in the European patent application EP-A-O 564 409 as example 21 or 4- Methyl-N-[3-(4-methyl-imidazol-l-yl)-5-trifluoromethyl-phenyl]-3-(4-pyridin-3-yl-pyrimidin-2- ylamino)-benzamide described in the patent application WO 04/005281 as example 92; and
m) an agent interacting with a 5-HT3 receptor and/or an agent interacting with 5-HT4 receptor such as tegaserod described in the US patent No. 5510353 as example 13, tegaserod hydrogen maleate, cisapride, cilansetron; n) an agent for treating tobacco abuse, e.g., nicotine receptor partial agonists, bupropion hypochloride (also known under the tradename ZYB AN®) and nicotine replacement therapies; o) an agent for treating erectile dysfunction, e.g., dopaminergic agents, such as apomorphine), ADD/ADHD agents (e.g., RITALIN®, STRATTERA®, CONCERTA® and ADDERALL®); p) an agent for treating alcoholism, such as opioid antagonists (e.g., naltrexone (REVIA®) and nalmefene), disulfiram (ANTABUSE®), and acamprosate (CAMPRAL®)). In addition, agents for reducing alcohol withdrawal symptoms may also be co-administered, such as benzodiazepines, beta- blockers, clonidine, carbamazepine, pregabalin, and gabapentin (NEURONTIN®); q) other agents that are useful including anti-inflammatory agents (e.g., COX-2 inhibitors) ; antidepressants (e.g., fluoxetine hydrochloride (PROZAC®)); cognitive improvement agents (e.g., donepezil hydrochloride (AIRCEPT®) and other acetylcholinesterase inhibitors); neuroprotective agents (e.g., memantine) ; antipsychotic medications (e.g., ziprasidone (GEODON®), risperidone (RISPERDAL®), and olanzapine (ZYPREXA®)); or, in each case a pharmaceutically acceptable salt thereof; and optionally a pharmaceutically acceptable carrier.
[0045] The invention also provides for a pharmaceutical combinations, e.g. a kit, comprising a) a first agent which is a compound of the invention as disclosed herein, in free form or in pharmaceutically acceptable salt form, and b) at least one co-agent. The kit may comprise instructions for its administration.
Processes for Making Compounds of the Invention
[0046] The present invention also includes processes for the preparation of compounds of the invention. In the reactions described, it may be necessary to protect reactive functional groups, for example hydroxy, amino, imino, thio or carboxy groups, where these are desired in the final product, to avoid their unwanted participation in the reactions. Conventional protecting groups may be used in accordance with standard practice, for example, see T.W. Greene and P. G. M. Wuts in "Protective Groups in Organic Chemistry", John Wiley and Sons, 1991.
[0047] A compound of the invention may be prepared as a pharmaceutically acceptable acid addition salt by reacting the free base form of the compound with a pharmaceutically acceptable inorganic or organic acid. Alternatively, a pharmaceutically acceptable base addition salt of a compound of the invention may be prepared by reacting the free acid form of the compound with a pharmaceutically acceptable inorganic or organic base. Alternatively, the salt forms of the compounds of the invention may be prepared using salts of the starting materials or intermediates.
[0048] The free acid or free base forms of the compounds of the invention may be prepared from the corresponding base addition salt or acid addition salt from, respectively. For example, a compound of the invention in an acid addition salt form may be converted to the corresponding free base by treating with a suitable base (e.g., ammonium hydroxide solution, sodium hydroxide, and the like). A compound of the invention in a base addition salt form may be converted to the corresponding free acid by treating with a suitable acid (e.g., hydrochloric acid, etc.).
[0049] Compounds of the invention in unoxidized form may be prepared from N-oxides of compounds of the invention by treating with a reducing agent (e.g., sulfur, sulfur dioxide, triphenyl phosphine, lithium borohydride, sodium borohydride, phosphorus trichloride, tribromide, or the like) in a suitable inert organic solvent (e.g. acetonitrile, ethanol, aqueous dioxane, or the like) at 0 to 8O0C.
[0050] Prodrug derivatives of the compounds of the invention may be prepared by methods known to those of ordinary skill in the art (e.g., for further details see Saulnier et al., (1994), Bioorganic and Medicinal Chemistry Letters, Vol. 4, p. 1985). For example, appropriate prodrugs may be prepared by reacting a non-derivatized compound of the invention with a suitable carbamylating agent (e.g., 1,1-acyloxyalkylcarbanochloridate, para-nitrophenyl carbonate, or the like).
[0051] Protected derivatives of the compounds of the invention may be made by means known to those of ordinary skill in the art. A detailed description of techniques applicable to the creation of protecting groups and their removal may be found in T. W. Greene, "Protecting Groups in Organic Chemistry", 3rd edition, John Wiley and Sons, Inc., 1999.
[0052] Compounds of the present invention may be conveniently prepared or formed during the process of the invention, as solvates (e.g., hydrates). Hydrates of compounds of the present
invention may be conveniently prepared by recrystallization from an aqueous/organic solvent mixture, using organic solvents such as dioxin, tetrahydrofuran or methanol.
[0053] Compounds of the invention may be prepared as their individual stereoisomers by reacting a racemic mixture of the compound with an optically active resolving agent to form a pair of diastereoisomeric compounds, separating the diastereomers and recovering the optically pure enantiomers. While resolution of enantiomers may be carried out using covalent diastereomeric derivatives of the compounds of the invention, dissociable complexes are preferred (e.g., crystalline diastereomeric salts). Diastereomers have distinct physical properties (e.g., melting points, boiling points, solubilities, reactivity, etc.) and may be readily separated by taking advantage of these dissimilarities. The diastereomers may be separated by chromatography, or by separation/resolution techniques based upon differences in solubility. The optically pure enantiomer is then recovered, along with the resolving agent, by any practical means that would not result in racemization. A more detailed description of the techniques applicable to the resolution of stereoisomers of compounds from their racemic mixture may be found in Jean Jacques, Andre Collet, Samuel H. Wilen, "Enantiomers, Racemates and Resolutions", John Wiley And Sons, Inc., 1981.
[0054] In summary, the invention encompasses methods of making compounds of Formula (1) and Formula (2), and:
(a) optionally converting a compound of the invention into a pharmaceutically acceptable salt;
(b) optionally converting a salt form of a compound of the invention to a non-salt form;
(c) optionally converting an unoxidized form of a compound of the invention into a pharmaceutically acceptable N-oxide;
(d) optionally converting an N-oxide form of a compound of the invention to its unoxidized form;
(e) optionally resolving an individual isomer of a compound of the invention from a mixture of isomers;
(f) optionally converting a non-derivatized compound of the invention into a pharmaceutically acceptable prodrug derivative; and
(g) optionally converting a prodrug derivative of a compound of the invention to its non- derivatized form.
[0055] Detailed examples of the synthesis of a compound of Formula (1) or (2) may be found in the Examples, infra. In the following examples, several methods of preparing the compounds of the present invention are illustrative. One of skill in the art will appreciate that these methods are representative, and in no way inclusive of all methods for preparing the compounds of the present invention. Insofar as the production of the starting materials is not particularly described, the compounds are known or may be prepared analogously to methods known in the art or as disclosed in the Examples hereinafter.
[0056] The following examples are offered to illustrate but not to limit the invention.
Example 1 l-(4-Chloro-phenyl)-2-(2,4-dichloro-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8-dione (1)

[0057] Step A: l-(4-Chloro-phenyl)-2-(2,4-dichloro-phenyl)-9-phenyl-l,9-dihydro-purin-6- one is prepared by the methods reported in WO2006/047516. The purinone (96.0 mg, 0.205 mmol) is dissolved in a mixture of 2 mL of acetic acid, 250 μL of chloroform and 250 μL of water. Sodium acetate (200 mg, 2.44 mmol) is added to the reaction mixture and stirred until dissolved. Bromine (250 μL) is added to the reaction mixture and the reaction is stirred for 30 min. The reaction is checked for completion by reverse phase LC/MS. The volatiles are then removed by rotary evaporation, and the resulting solid is extracted with water and dichloromethane. The organic layer is concentrated, and the resulting red material is purified by silica gel chromatography yielding 8-bromo-l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-9- phenyl-l,9-dihydro-purin-6-one as a yellow solid.
[0058] Step B: The 8-bromo-purinone (45.0 mg, 82.3 μmol) is dissolved in 1 mL of tetrahydrofuran and 200 μL of a 1 M lithium hydroperoxide solution, freshly prepared by mixing lithium hydroxide (18.9 mg, 0.823 mmol) and 36% hydrogen peroxide ( 93.0μL, 1.23 mmol) and 103 μL of water. The reaction is allowed to stir overnight. The reaction is then quenched with 1 M HCl and extracted with ethyl acetate. The organic layer is dried over magnesium sulfate, filtered, and rotary evaporated to dryness. The crude material is purified on silica gel, giving an off white solid. 1H NMR (CDCl3, 400 MHz) δ 8.74 (br s, IH), 7.57 (d, 2H), 7.50 (t, 2H), 7.39 (m, IH), 7.30 (m, 2H), 7.29 (d, 2H), 7.14 (dd, IH), 7.09 (d, IH), 7.04 (br m, IH). Calculated HPLC /MS for C23H13Cl3N4O2 (M+H+) 483.0, found 483.0.
Example 2 l-(4-Bromo-phenyl)-9-phenyl-8-thioxo-2-p-tolyl-l,7,8,9-tetrahvdro-purin-6-one (2)

[0059] Step A: Amino-cyano-acetic acid ethyl ester (1.00 g, 7.79 mmol) is dissolved in 100 mL of dichloromethane. Triethylamine (1 mL) and phenyl isothiocyanate (1.05 g, 7.79 mmol) are added sequentially to the reaction mixture, and the mixture is stirred 2 h. The crude is purified by chromatography on silica gel to give 5-amino-l-phenyl-2-thioxo-2,3-dihydro-lH- imidazole-4-carboxylic acid ethyl ester as a white solid. 1H NMR (CDCl3, 400 MHz) δ 7.60 (br s, 5H), 4.95 (br s, 2H), 4.37 (m, 3H), 1.38 (t, 3H). Calculated HPLC /MS for Ci2H13N3O2S (M+ H+) 264.1, found 264.1.
[0060] Step B: N-(4-Bromo-phenyl)-4-methyl-benzamide (29.0 mg, 0.100 mmol) is dissolved in 0.5 mL of thionyl chloride and heated to reflux for 30 min. The reaction is then allowed to cool, and the thionyl chloride is removed under a dry stream of nitrogen to yield imadoyl chloride which is used without further purification. Amino ester prepared from step A
(26.4 mg, 0.100 mmol) and vacuum oven dried potassium carbonate (250 mg, 1.81 mmol) are added to the freshly prepared imadoyl chloride, and 2 mL of dry acetonitrile is added to the reaction mixture. The resulting mixture is heated to 180 0C for 30 min. The resulting mixture is purified by mass directed reverse phase HPLC to give l-(4-bromo-phenyl)-9-phenyl-8-thioxo-2- p-tolyl-l,7,8,9-tetrahydro-purin-6-one as a colorless amorphous solid. 1H NMR (DMSO-Dό, 400 MHz) δ 7.69 (m, 7H), 7.44 (d, 2H), 7.28 (d, 2H), 7.17 (d, 2H), 2.35 (s, 3H). Calculated HPLC /MS for C24H17BrN4OS (M+ H+) 489.0, found 489.0.
Example 3 l-(4-Chloro-phenyl)-2-(2,4-dichloro-phenyl)-7-methyl-9-phenyl-7,9-dihydro-lH-purine-6,8-

[0061] l-(4-Chloro-phenyl)-2-(2,4-dichloro-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8- dione (10.0 mg, 20.7 μmol) is dissolved in 3 mL of dry acetonitrile, and the resulting solution is transferred to a microwave tube containing anhydrous potassium carbonate (300 mg, 2.17 mmol). Dimethylsulfate (150 mg, 1.19 mmol) is added to the tube, and the reaction tube is capped and heated to 200 0C for 15 min in a microwave reactor. The resulting solution is filtered, stripped of the volatiles and partitioned with dichloromethane and water. The organic layer is separated, dried over magnesium sulfate and concentrated. The crude material is purified on silica gel to give l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-7-methyl-9-phenyl- 7,9-dihydro-lH-purine-6,8-dione. 1H NMR (CDCl3, 400 MHz) δ 7.66 (d, 2H), 7.49 (t, 2H), 7.38 (m, IH), 7.30 (m, 3H), 7.14 (dd, IH), 7.08 (d, IH), 7.03 (br s, IH), 3.77 (s, 3H). Calculated HPLC /MS for C24H15Cl3N4O2 (M+ H+) 497.0, found 497.0.
Example 4 l-(4-Chloro-phenyl)-2-(2,4-dichloro-phenyl)-6,8-dioxo-9-phenyl-l,6,8,9-tetrahydro-purine-7- carboxylic acid dimethylamide (5)

[0062] l-(4-Chloro-phenyl)-2-(2,4-dichloro-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8- dione (8.0 mg, 16.5 μmol) is dissolved in 1.5 mL of dry acetonitrile, and the resulting solution is transferred to a microwave tube containing anhydrous potassium carbonate (250 mg, 1.81 mmol). Dimethylcarbamyl chloride (117 mg, 1.09 mmol) is added to the tube, and the reaction tube is capped and heated to 200 0C for 30 min in a microwave. The resulting solution is filtered, stripped of the volatiles and partitioned with dichloromethane and water. The organic layer is separated, dried over magnesium sulfate and concentrated. The crude material is purified on silica gel to give l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-6,8-dioxo-9-phenyl- l,6,8,9-tetrahydro-purine-7-carboxylic acid dimethylamide. 1H NMR (CDCl3, 400 MHz) δ 7.62 (d, 2H), 7.50 (t, 2H), 7.40 (m, IH), 7.30 (m, 3H), 7.13 (m, 2H), 7.03 (br s, IH), 1.26 (s, 6H). Calculated HPLC /MS for C25Hi4Cl3N5O2 (M+ H+) 554.0, found 554.0.
Example 5 ri-(4-Chloro-phenyl)-2-(2,4-dichloro-phenyl)-6,8-dioxo-9-phenyl-l,6,8,9-tetrahydro-purin-7- yll-acetonitrile (6)

[0063] l-(4-Chloro-phenyl)-2-(2,4-dichloro-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8- dione (10.0 mg, 20.7 μmol) is dissolved in 1.5 mL of dry tetrahydrofuran, and the resulting solution is transferred to a microwave tube containing anhydrous potassium carbonate (400 mg, 2.90 mmol). Bromoacetonitrile (344 mg, 2.87 mmol) is diluted in 1.5 mL of dry acetonitrile and added to the tube, and the reaction flask is capped and heated to 180 0C for 30 min in a microwave. The resulting solution is filtered, stripped of the volatiles and partitioned with dichloromethane and water. The organic layer is separated, dried over magnesium sulfate and concentrated. The crude material is purified on silica gel to give [l-(4-chloro-phenyl)-2-(2,4- dichloro-phenyl)-6,8-dioxo-9-phenyl-l,6,8,9-tetrahydro-purin-7-yl]-acetonitrile. 1H NMR (CDCl3, 400 MHz) δ 7.63 (d, 2H), 7.51 (t, 2H), 7.42 (m, IH), 7.32 (m, 3H), 7.16 (dd, IH), 7.09 (d, IH), 7.03 (br s, IH), 5.16 (s, 2H). Calculated HPLC /MS for C24H12Cl3N5O2 (M+ H+) 522.0, found 522.0.
Example 6 l-(4-Chloro-phenyl)-2-(2,4-dichloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH-purine-6,8-dione
(10)

[0064] l-(4-Chloro-phenyl)-2-(2,4-dichloro-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8- dione (25.0 mg, 51.7 μmol) is dissolved in 1.5 mL of dry dimethylformamide, and the resulting solution is transferred to a microwave tube containing anhydrous potassium carbonate (500 mg, 3.62 mmol). Bromoethane (375 mg, 3.47 mmol) is added to the tube, and the reaction flask is capped and heated to 70 0C for overnight in an oil bath. The resulting solution is filtered and partitioned with dichloromethane and water. The organic layer is separated, dried over magnesium sulfate and concentrated. The crude material is purified on silica gel to yield l-(4- chloro-phenyl)-2-(2,4-dichloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH-purine-6,8-dione. 1H NMR (CDCl3, 400 MHz) δ 7.60 (d, 2H), 7.49 (t, 2H), 7.37 (m, IH), 7.30 (m, 3H), 7.14 (dd, IH), 7.09 (d, IH), 7.05 (br s, IH), 4.26 (q, 2H), 1.47 (t, 3H). Calculated HPLC /MS for C25H17Cl3N4O2 (M+ H+) 511.0, found 511.0.
Example 7 l-(4-Chloro-phenyl)-2-(4-cvclopropyl-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8-dione (13)

[0065] Step A: 2-(4-Bromo-phenyl)-l-(4-chloro-phenyl)-9-phenyl-7,9-dihydro-lH-purine- 6,8-dione (15.0 mg, 30.4 μmol), anhydrous potassium carbonate (75.0 mg, 0.540 mmol), cyclopropyl boronic acid (20.0 mg, 0.230 mmol), and dichloro[l,l'-bis(diphenylphosphino)- ferrocene]palladium(II) dichloromethane adduct (1.2 mg, 1.47 μmol) are added to a microwave reaction tube with 0.5 mL of water. The tube is sonicated for 2 min, and the tube is capped and purged with an inert atmosphere for 5 min. Dimethylformamide (0.5 mL) is added, and the tube is purged for an additional 5 min. The tube is then placed in a microwave reactor and heated to 200 0C for 120 seconds. The resulting solution is neutralized with TFA, and an additional 1 mL of dimethylformamide is added. The crude product is purified by mass directed reverse phase HPLC. 1H NMR (DMSO-D6, 400 MHz) δ 10.9 (s, IH), 8.18 (d, 2H), 7.97 (m, 2H), 7.84 (m, 5H), 7.69 (d, 2H), 7.38 (d, 2H), 2.29 (m, IH), 1.38 (m, 2H), 1.08 (m, 2H). Calculated HPLC /MS for C26H19ClN4O2 (M+ H+) 455.1, found 455.0.
Example 8
2-r4-(6-Amino-pyridin-3-yl)-phenyll-l-(4-chloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH- purine-6,8-dione (15)
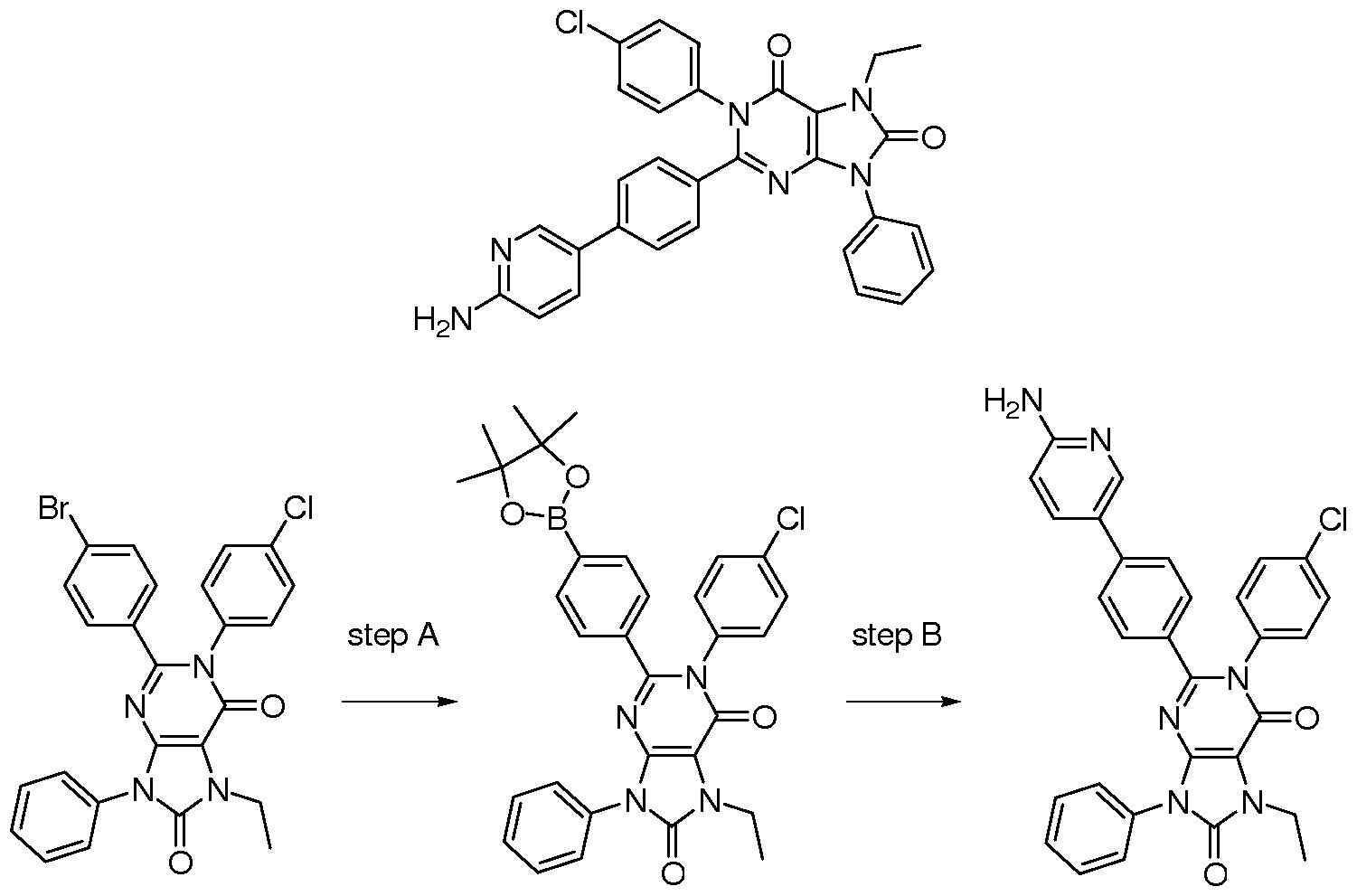
[0066] Step A: 2-(4-Bromo-phenyl)-l-(4-chloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH- purine-6,8-dione (40.0 mg, 76.7 μmol), potassium acetate (90.0 mg, 0.917 mmol), bispinacolatodiboron (22.3 mg, 88.1 μmol), and dichloro[l,l'-bis(diphenylphosphino) ferrocene]palladium(II) dichloromethane adduct (3 mg, 3.67 μmol) are added to a microwave tube and 2 mL of dry dimethylformamide is added. The tube is capped and purged with an inert atmosphere for 5 min. The tube is then heated to 100 0C for 2.5 h. The resulting solution is filtered and partitioned with dichloromethane and water. The organic layer is separated, dried over magnesium sulfate and concentrated. The crude material is purified on silica gel to yield 1- (4-chloro-phenyl)-7-ethyl-9-phenyl-2-[4-(4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-phenyl]- 7,9-dihydro-lH-purine-6,8-dione. Calculated HPLC /MS for C3IH30BClN4O4 (M+ H+) 569.2, found 569.2.
[0067] Step B: A microwave tube is charged with l-(4-chloro-phenyl)-7-ethyl-9-phenyl-2- [4-(4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-phenyl]-7,9-dihydro-lH-purine-6,8-dione (43.0 mg, 75.6 μmol), potassium carbonate (250 mg, 1.81 mmol), 5-bromo-pyridin-2-ylamine (32.6 mg, 0.189 mmol), and dichloro[l,l'-bis(diphenylphosphino)ferrocene]palladium(II)
dichloromethane adduct (4 mg, 4.87 μmol). Water (0.75 mL) is added to the tube and the sample is sonicated for 2 min. Dimethylformamide (2 mL) is added, the tube is capped and purged with an inert atmosphere for 5 min. The tube is then heated to 200 0C for 3 min. The resulting crude product is then purified by mass directed HPLC. 1H NMR (CDCl3, 400 MHz) δ 8.26 (br s, IH), 7.71 (d, 2H), 7.61 (dd, IH), 7.52 (t, 2H), 7.36 (m, 5H), 7.29 (d, 2H), 7.16 (dd, IH), 6.57 (d, IH), 4.26 (q, 2H), 1.47 (t, 3H). Calculated HPLC /MS for C30H23ClN6O2 (M+ H+) 535.2, found 535.2.
Example 9 l-(4-Chloro-phenyl)-2-(4-cvclopropyl-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH-purine-6,8- dione (16)

[0068] 2-(4-Bromo-phenyl)- l-(4-chloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro- lH-purine- 6,8-dione (30.0 mg, 57.5 μmol), anhydrous potassium phosphate (40.0 mg, 0.189 mmol), cyclopropyl boronic acid (30.0 mg, 0.353 mmol), tricyclohexylphosphine (3.55 mg, 12.65 μmol) and palladium acetate (1.43 mg, 6.73 μmol) are added to a microwave tube, capped, and purged with inert atmosphere for 5 min. Water (200 μL) and toluene (1.00 mL) are added to the purging tube, and the resulting slurry is purged and additional 3 min. The tube is then heated to 100 0C for 1.5 h. The crude product is purified by mass directed reverse phase HPLC. 1H NMR (CDCl3, 400 MHz) δ 7.69 (d, 2H), 7.50 (t, 2H), 7.38 (t, IH), 7.35 (d, 2H), 7.12 (d, 2H), 6.87 (d, 2H), 4.25 (q, 2H), 1.81 (m, IH), 1.45 (t, 3H), 0.97 (m, 2H), 0.65 (m, 2H). Calculated HPLC /MS for C28H23ClN4O2 (M+ H+) 483.1, found 483.1.
Example 10
Methyl 3-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin-
7(6H,8H,9H)-yl)propanoate (22)

[0069] l-(4-Chloro-phenyl)-2-(4-isopropyl-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8- dione (25.0 mg, 54.7 μmol), anhydrous potassium carbonate (250 mg, 1.81 mmol), and 3- bromo-propionic acid methyl ester (1.43 mg, 1.37 mmol) are added to a microwave tube with 1.5 mL of dry dimethylformamide, capped and heated to 60 0C for overnight. The crude product is purified by mass directed reverse phase HPLC. 1H NMR (CDCl3, 400 MHz) δ 7.68 (d, 2H), 7.51 (t, 2H), 7.39 (t, IH), 7.34 (d, 2H), 7.15 (d, 2H), 7.12 (d, 2H), 7.05 (d, 2H), 4.50 (t, 2H), 3.69 (s, 3H), 2.95 (t, 2H), 2.83 (sep, IH), 1.17 (d, 6H). Calculated HPLC /MS for C30H27ClN4O4 (M+ H+) 543.2, found 543.2.
Example 11
3-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin-7(6H,8H,9H)- vDpropanoic acid (24)

[0070] 3-[l-(4-Chloro-phenyl)-2-(4-isopropyl-phenyl)-6,8-dioxo-9-phenyl-l,6,8,9- tetrahydro-purin-7-yl] -propionic acid methyl ester (12.0 mg, 22.1 μmol) and lithium hydroxide (20.0 mg, 1.81 mmol) are added to a vial with 1 mL of tetrahydrofuran and 250 μL of water. The reaction is stirred overnight. The crude product is purified by mass directed reverse phase HPLC. 1H NMR (CDCl3, 400 MHz) δ 7.66 (d, 2H), 7.51 (t, 2H), 7.40 (t, IH), 7.34 (d, 2H), 7.15 (d, 2H), 7.12 (d, 2H), 7.06 (d, 2H), 4.49 (br m, 2H), 2.97 (br m, 2H), 2.83 (sep, IH), 1.18 (d, 6H). Calculated HPLC /MS for C29H25ClN4O4 (M+ H+) 529.1, found 529.1.
Example 12
7-(2-Amino-ethyl)-l-(4-chloro-phenyl)-2-(4-isopropyl-phenyl)-9-phenyl-7,9-dihydro-lH- purine-6,8-dione (29)


[0071] l-(4-Chloro-phenyl)-2-(4-isopropyl-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8- dione (30.0 mg, 65.7 μmol) and N-(2-bromoethyl)phthalimide (75.0 mg, 0.295 mmol) are added to a microwave tube with 2 mL of dimethylformamide and potassium carbonate (400 mg, 2.90 mmol). The tube is capped and heated at 70 0C overnight. The reaction is allowed to cool to room temperature and 0.5 mL of hydrazine is added, and the reaction is stirred for 4 h. The crude product is purified by mass directed reverse phase HPLC. 1H NMR (CDCI3, 400 MHz) δ 7.68 (m, 2H), 7.50 (m, 2H), 7.34 (m, 3H), 7.13 (m, 4H), 7.04 (d, 2H), 4.15 (br m, 2H), 2.83 (sep, IH), 1.27 (m, 2H), 1.17 (d, 6H). Calculated HPLC /MS for C28H26ClN5O2 (M+ H+) 500.2, found 500.2.
Example 13
2-r4-(6-Amino-5-methyl-pyridin-3-yl)-phenyll-l-(4-chloro-phenyl)-7-ethyl-9-phenyl-7,9- dihydro-lH-purine-6,8-dione (32)

[0073] Step B: 2-(4-Bromo-phenyl)-l-(4-chloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH- purine-6,8-dione (52.0 mg, 30.4 μmol), sodium hydroxide (IM, 200 μL, 0.200 mmol), [3- methyl-5-(4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-pyridin-2-yl]-carbamic acid tert-butyl ester (47.0 mg, 0.201 mmol), and dichloro[l,l'-bis(diphenylphosphino)ferrocene]palladium(II) dichloromethane adduct (4.00 mg, 4.90 μmol) are added to a microwave tube with 0.5 mL of water and 2.0 mL of 1 ,2-dimethoxyethane. The tube is capped and purged with an inert atmosphere for 5 min. The reaction mixture is then heated to 100 0C for 3 h. The reaction is then cooled and the volatiles are removed. The crude is then dissolved in a mixture of 1 mL of dichloromethane and 1 mL of trifluoroacetic acid, and the reaction is then stirred for 6 h. The resulting crude material is purified by mass directed HPLC. 1H NMR (CDCl3, 400 MHz) δ 8.26 (br s, IH), 8.12 (br s, IH), 7.71 (d, 2H), 7.52 (m, 3H), 7.38 (m, 5H), 7.29 (d, 2H), 7.16 (d, IH), 4.7 (br s, 2H), 4.26 (q, 2H), 2.19 (s, 3H), 1.46 (t, 3H). Calculated HPLC /MS for C3]H25ClN6O2 (M+ H+) 549.2, found 549.2.
[0074] Illustrative compounds of the invention are shown in Table 1.
Table 1

found
(d,
found
(d, IH),
(d,
found
(d,
3.81 found
 MS (m/z) (d,
MS (m/z) (d,
5.62 (M+
(d,
found
(d,
(t, found
found
2H),

IH),
H+)
(d, (d,
found
found
(d,
2H),
found
 MS (m/z)
MS (m/z)
found
(d, (d,
(d, found
(d, (d, m,
found
found
 MS (m/z) (d,
MS (m/z) (d,
found
found
(d, (d,
H+)
m,
found


CBl Biological Assays
[0075] Homogenized membranes are prepared from CHO cell clones stably expressing a human cannabinoid receptor 1 (CBl) or human cannabinoid receptor 2 (CB2). Cells are grown and scrapped from 15 cm tissue culture plates, and then subsequently centrifuged down. Cells are washed once with cold PBS, and resuspended in <20 ml of Buffer A (20 mM HEPES, pH 7.4, 10 mM EDTA, EDTA-free complete protease inhibitor cocktail [1 tablet/25 ml]). The cell suspension is homogenized on ice, using a Polytron homogenizer at 25,000 rpm at three intervals of 15 seconds each. The homogenate is first centrifuged at 2,000 rpm on a tabletop low speed centrifuge for 10 minutes. The supernatant, after passing through a cell strainer, is then centrifuged at 50,000 x g for 25 minutes at 40C. The pellet is resuspended into buffer B (15% glycerol, 20 mM HEPES, pH 7.4, 0.1 mM EDTA, EDTA-free complete protease inhibitor cocktail [1 tablet/10 ml]). Protein concentration is determined using the BCA Protein Assay kit using BSA as standard. The membranes are aliquoted and kept frozen at -8O0C.
r3Hl-CP55940 ligand binding assay
[0076] Solutions of test compounds ranging from 100 μM to 0.01 nM are prepared in DMSO. The desired amount of membrane prep is diluted with ice-cold assay buffer (5OmM Tris-HCl, 2.5mM EDTA, 5 mM MgCl2, 0.05% BSA, pH 7.4) and vortexed well. Two μl or less of compound is distributed into each well of a round-bottom 96-well polystyrene assay plate, followed by addition of 100 μl of diluted membranes (3-10 μg/well). The mixture is kept on ice until the addition of hot CP55940 (final concentration of 0.5nM). [3H]-CP55940 is diluted 1:6300 (v/v) with cold assay buffer and 100 μl is added into each well. The reaction is carried out at room temperature for 120 minutes before the membranes are harvested onto a PerkinElmer Unifilter GF/B-96 filter plate using a Packard Filtermate Harvester. After nine
washes with wash buffer (5OmM Tris-HCl, 2.5mM EDTA, 5 mM MgCl2, 0.05% BSA, pH 7), the filter is dried in a 37°C oven for 30 minutes. MicroScint-20 is added and the plate sealed for scintillation counting on TopCount. EC50 values are obtained by fitting the data with the sigmoidal dose response curve-fitting tool of GraphPad Prism. Eight or twelve different concentrations are used to generate a concentration response curve (using three data points per concentration).
GTPγS binding assay
[0077] Solutions of test compounds ranging from 100 μM to 0.01 nM are prepared in DMSO. The desired amount of membrane prep is diluted with ice-cold assay buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl2, 0.1% Fatty acid-free BSA, 5 μM GDP) and vortexed well. Two μl or less of compound is distributed into each well of a round-bottom 96- well polystyrene assay plate, followed by addition of 100 μl of diluted membranes (3-10 μg/well), and the mixture is kept on ice until the addition of hot GTPγS. [35S]-GTPγS (Perkin Elmer NEG030H; 1 μCi/μl, 1250 Ci/mmol) is diluted 1: 1000 (v/v) with cold assay buffer and 100 μl is added into each well. The reaction is carried out at room temperature for 90 minutes before the membranes are harvested onto PerkinElmer Unifilter GF/B-96 filter plate using a Packard Filtermate Harvester. After several washes with wash buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl2), and a rinse with 95% ethanol, the filter is dried in a 37°C oven for 30 minutes. MicroScint-20 is added and the plate sealed for scintillation counting on TopCount. EC50 values are obtained by fitting the GTP [γ-3 S] binding data with the sigmoidal dose response curve-fitting tool of GraphPad Prism. Six or twelve different concentrations are used to generate a concentration response curve (using three data points per concentration).
[0078] For each assay, a Cheng-Prusoff correction (Cheng and Prusoff, 1973, Biochem. Pharmacol., 22: 3099-3103) is used to convert the EC50 to inhibition constant K1. Thus,
EC50
Ki =
1 + [L] / Kd where [L] is the concentration of the radio-ligand used in the assay, and Kd is the equilibrium binding dissociation constant for the radio-ligand.
Food Intake and Body Weight Gain
[0079] To evaluate the efficacy of compounds of the invention on inhibition of food intake and body weight gain, genetically obese (Lepob/Lepob) mice and diet-induced obese (DIO) mice are used in acute and sub-chronic models, respectively.
[0080] Male ob/ob mice (age 7-8 weeks old, Jackson Labs, Bar Harbor, Maine) are housed in groups of four and fed commercial standard pellet diet (Lab Diet 5001, PMI Nutrition International, LLC). Diet-induced obese mice are generated using 6-7 weeks old C57BL6 mice (Jackson Labs, Bar Harbor, Maine) placed on high fat diet (D12331, Research Diets) for 12-17 weeks. All mice are maintained on a 12-hour light/dark cycle (lights on at 06:00) in a humidity and temperature-controlled environment with free access to food and water.
[0081] The week prior to the start of each study, mice are singly housed and a habituation to treatment is performed to establish baseline food consumption and body weight. Animals are randomized into treatment groups based on their initial body weight and food consumption.
[0082] To determine the acute effects of a single administration of a compound of the invention (test compound) on food consumption, ob/ob mice are treated with either vehicle, a known antagonist as a positive control, or with test compound(s). Similarly, to determine more chronic effects of test compound on food consumption and body weight gain, DIO mice are treated with either vehicle, a known antagonist as a positive control, or with test compound(s) for up to 7-35 days. Test compounds are dosed at ranges between 0.1 up to 100 mg/kg. Animals are treated one hour prior to the start of the dark cycle. Food intake and body weight are recorded manually using an electronic balance prior to treatment, 16 hours post- treatment, followed by daily measurements for up to 7-35 days after the start of study. Compound efficacy is determined by comparing food intake and body weight data between vehicle treated, standard positive control treated, and test compound treated mice.
[0083] Compounds of Formula (1) and (2), in free form or in pharmaceutically acceptable salt form, exhibit valuable pharmacological properties, for example, as indicated by the in vitro tests described in this application. In general, compounds of the invention have a K1 of between IxIO"5 and Ix 10"10M. In some embodiments, the compounds of the invention have a K1 of less than 500 nM; and in other embodiments, a K1 of less than 10OnM. In addition, the compounds of the invention exhibit at least 10 fold selectivity for CBl over CB 2. In some instances, the compounds of the invention exhibit at least 20-fold, 50-fold or 100-fold selectivity for CBl over CB2.
[0084] It is understood that the examples and embodiments described herein are for illustrative purposes only and that various modifications or changes in light thereof will be suggested to persons skilled in the art and are to be included within the spirit and purview of this application and scope of the appended claims. All publications, patents, and patent applications cited herein are hereby incorporated by reference for all purposes.
Claims
Claims
1. A compound of Formula ( 1) :

and pharmaceutically acceptable salts thereof; wherein Y is O, S or NR5;
X is O or S;
R1 and R2 are independently L-R9;
L is a bond, S or O;
R3 and R4 are independently H, an optionally halogenated C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, cyano, halo, nitro, NR6R7, CO(CR2)kNR6R7, OR8, SOi_2R8, NR7SOi_2R8, COi_2R8 or R9;
R5, R6, R7 and R8 are independently H, C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, or (CR2)kR9; each R9 is an optionally substituted Cj1-I cycloalkyl, or a 5-12 membered aryl, heteroaryl or heterocyclic ring containing N, O or S;
R is H or Ci-6 alkyl; and j and k are independently 0-6.
2. The compound of claim 1, wherein X and Y are independently O or S.
3. The compound of claim 1, wherein L is a bond.
4. The compound of claim 1, wherein R1, R2 and R4 are independently an optionally substituted C3-7 cycloalkyl, or a 5-7 membered aryl, heteroaryl or heterocyclic ring containing N, O or S.
5. The compound of claim 1, wherein R1, R2 and R4 are independently phenyl, piperazinyl, morpholino, benzthiazolyl, pyridinyl or pyrazolyl.
6. The compound of claim 4, wherein R1, R2 and R4 are independently an optionally substituted phenyl.
7. The compound of claim 1, wherein said compound is of Formula (2):

wherein X and Y are independently O or S;
R10, R11 and R12 are independently halo, an optionally substituted alkyl, C2-6 alkenyl, C2-6 alkynyl, cycloalkyl, or an optionally substituted aryl, heteroaryl or heterocyclic ring; and m, n and p are independently 0-5.
The compound of claim 7, wherein m is 1, and R io i •s halo.
9. The compound of claim 7, wherein n is 1-2, and R11 is halo, an optionally halogenated C1-6 alkyl, or an optionally substituted C3-7 cycloalkyl, phenyl, pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, imidazolyl, pyrazolyl, oxazolyl, oxadiazolyl, isoxazolyl, furanyl, thiophenyl, or triazolyl.
10. The compound of claim 7, wherein p is 0.
11. The compound of claim 1, wherein each optionally substituted moiety is optionally substituted with an optionally halogenated Ci_6 alkyl, C2-6 alkenyl, C2_6 alkynyl, cyano, halo, nitro, NR6R7, CO(CR2)kNR6R7, OR8, SOi_2R8, NR7SOi_2R8, COi_2R8 or R9.
12. The compound of claim 1, wherein said compound is selected from the group consisting of l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8- dione; l-(4-bromo-phenyl)-9-phenyl-8-thioxo-2-p-tolyl-l,7,8,9-tetrahydro-purin-6-one; l-(4-chlorophenyl)-9-phenyl-2-(4-(trifluoromethyl)phenyl)-lH-purine-6,8(7H,9H)- dione; l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-7-methyl-9-phenyl-7,9-dihydro-lH-purine- 6,8-dione; l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-6,8-dioxo-9-phenyl-l,6,8,9-tetrahydro- purine-7-carboxylic acid dimethylamide;
[l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-6,8-dioxo-9-phenyl- 1,6,8, 9-tetrahydro- purin-7-yl]-acetonitrile; methyl 2-(l-(4-chlorophenyl)-2-(2,4-dichlorophenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)acetate;
2-(l-(4-chlorophenyl)-2-(2,4-dichlorophenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)acetamide; l-(4-chlorophenyl)-2-(2,4-dichlorophenyl)-9-phenyl-7-(prop-2-ynyl)-lH-purine- 6,8(7H,9H)-dione; l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH-purine- 6,8-dione; l-(4-chlorophenyl)-2-(2,4-dichlorophenyl)-7-isopropyl-9-phenyl-lH-purine-6,8(7H,9H)- dione;
2-(4-bromophenyl)-l-(4-chlorophenyl)-9-phenyl-lH-purine-6,8(7H,9H)-dione; l-(4-chloro-phenyl)-2-(4-cyclopropyl-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8- dione;
2-(4-bromophenyl)-l-(4-chlorophenyl)-7-ethyl-9-phenyl-lH-purine-6,8(7H,9H)-dione;
2-[4-(6-amino-pyridin-3-yl)-phenyl]-l-(4-chloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro- 1 H-purine- 6, 8 -dione ;
l-(4-chloro-phenyl)-2-(4-cyclopropyl-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH-purine- 6,8-dione; l-(4-bromophenyl)-9-phenyl-2-p-tolyl-lH-purine-6,8(7H,9H)-dione;
2-(4-(2-aminopyridin-4-yl)phenyl)-l-(4-chlorophenyl)-7-ethyl-9-phenyl-lH-purine- 6,8(7H,9H)-dione;
2-(4-(5-aminopyridin-2-yl)phenyl)-l-(4-chlorophenyl)-7-ethyl-9-phenyl-lH-purine- 6,8(7H,9H)-dione;
2-(4-(6-amino-2-methylpyridin-3-yl)phenyl)-l-(4-chlorophenyl)-7-ethyl-9-phenyl-lH- purine-6,8(7H,9H)-dione; l-(4-chlorophenyl)-2-(4-isopropylphenyl)-9-phenyl-7-(pyridin-4-ylmethyl)-lH-purine- 6,8(7H,9H)-dione; methyl 3-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)propanoate; methyl 4-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)butanoate;
3-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)propanoic acid;
2-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)propanamide; l-(4-chlorophenyl)-2-(4-isopropylphenyl)-9-phenyl-lH-purine-6,8(7H,9H)-dione;
4-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)butanoic acid; l-(4-chlorophenyl)-7-ethyl-2-(4-isopropylphenyl)-9-phenyl-lH-purine-6,8(7H,9H)- dione;
7-(2-Amino-ethyl)-l-(4-chloro-phenyl)-2-(4-isopropyl-phenyl)-9-phenyl-7,9-dihydro- lH-purine-6,8-dione; l-(4-chlorophenyl)-7-ethyl-2-(4-(6-methoxypyridin-3-yl)phenyl)-9-phenyl-lH-purine- 6,8(7H,9H)-dione;
N-(4'-(l-(4-chlorophenyl)-7-ethyl-6,8-dioxo-9-phenyl-6,7,8,9-tetrahydro-lH-purin-2- yl)biphenyl-4-yl)methanesulfonamide; and
2-[4-(6-amino-5-methyl-pyridin-3-yl)-phenyl]-l-(4-chloro-phenyl)-7-ethyl-9-phenyl-7,9- dihydro- lH-purine-6,8-dione.
13. A pharmaceutical composition comprising a therapeutically effective amount of a compound of claim 1 and a pharmaceutically acceptable carrier.
14. A pharmaceutical composition comprising a therapeutically effective amount of a compound of claim 12, and a pharmaceutically acceptable carrier.
15. A method for treating a condition mediated by a cannabinoid- 1 receptor, comprising administering to a system or a subject in need thereof, a therapeutically effective amount of a compound of claim 1, or pharmaceutically acceptable salts or pharmaceutical compositions thereof, thereby treating said disease.
16. The method of claim 15, wherein said condition is an eating disorder associated with excessive food intake, obesity, diabetes, insulin resistance, hypercholesterolemia, hypertriglyceridemia, substance abuse, osteoporosis, or smoking abuse.
17. The method according to claim 15, wherein said condition is obesity.
18. The method of claim 17, comprising administering to a subject at risk for obesity a dosage of about 0.001 mg to about 100 mg per kg of body weight.
19. The method of claim 15, wherein said compound is selected from the group consisting of l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8- dione; l-(4-bromo-phenyl)-9-phenyl-8-thioxo-2-p-tolyl-l,7,8,9-tetrahydro-purin-6-one; l-(4-chlorophenyl)-9-phenyl-2-(4-(trifluoromethyl)phenyl)-lH-purine-6,8(7H,9H)- dione; l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-7-methyl-9-phenyl-7,9-dihydro-lH-purine- 6,8-dione; l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-6,8-dioxo-9-phenyl-l,6,8,9-tetrahydro- purine-7-carboxylic acid dimethylamide;
[l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-6,8-dioxo-9-phenyl- 1,6,8, 9-tetrahydro- purin-7-yl]-acetonitrile;
methyl 2-(l-(4-chlorophenyl)-2-(2,4-dichlorophenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)acetate;
2-(l-(4-chlorophenyl)-2-(2,4-dichlorophenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)acetamide; l-(4-chlorophenyl)-2-(2,4-dichlorophenyl)-9-phenyl-7-(prop-2-ynyl)-lH-purine- 6,8(7H,9H)-dione; l-(4-chloro-phenyl)-2-(2,4-dichloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH-purine- 6,8-dione; l-(4-chlorophenyl)-2-(2,4-dichlorophenyl)-7-isopropyl-9-phenyl-lH-purine-6,8(7H,9H)- dione;
2-(4-bromophenyl)-l-(4-chlorophenyl)-9-phenyl-lH-purine-6,8(7H,9H)-dione; l-(4-chloro-phenyl)-2-(4-cyclopropyl-phenyl)-9-phenyl-7,9-dihydro-lH-purine-6,8- dione;
2-(4-bromophenyl)-l-(4-chlorophenyl)-7-ethyl-9-phenyl-lH-purine-6,8(7H,9H)-dione;
2-[4-(6-amino-pyridin-3-yl)-phenyl]-l-(4-chloro-phenyl)-7-ethyl-9-phenyl-7,9-dihydro- 1 H-purine- 6, 8 -dione ; l-(4-chloro-phenyl)-2-(4-cyclopropyl-phenyl)-7-ethyl-9-phenyl-7,9-dihydro-lH-purine- 6,8-dione; l-(4-bromophenyl)-9-phenyl-2-p-tolyl-lH-purine-6,8(7H,9H)-dione;
2-(4-(2-aminopyridin-4-yl)phenyl)-l-(4-chlorophenyl)-7-ethyl-9-phenyl-lH-purine- 6,8(7H,9H)-dione;
2-(4-(5-aminopyridin-2-yl)phenyl)-l-(4-chlorophenyl)-7-ethyl-9-phenyl-lH-purine- 6,8(7H,9H)-dione;
2-(4-(6-amino-2-methylpyridin-3-yl)phenyl)-l-(4-chlorophenyl)-7-ethyl-9-phenyl-lH- purine-6,8(7H,9H)-dione; l-(4-chlorophenyl)-2-(4-isopropylphenyl)-9-phenyl-7-(pyridin-4-ylmethyl)-lH-purine- 6,8(7H,9H)-dione; methyl 3-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)propanoate; methyl 4-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)butanoate;
3-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)propanoic acid;
2-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)propanamide; l-(4-chlorophenyl)-2-(4-isopropylphenyl)-9-phenyl-lH-purine-6,8(7H,9H)-dione;
4-(l-(4-chlorophenyl)-2-(4-isopropylphenyl)-6,8-dioxo-9-phenyl-lH-purin- 7(6H,8H,9H)-yl)butanoic acid; l-(4-chlorophenyl)-7-ethyl-2-(4-isopropylphenyl)-9-phenyl-lH-purine-6,8(7H,9H)- dione;
7-(2-Amino-ethyl)-l-(4-chloro-phenyl)-2-(4-isopropyl-phenyl)-9-phenyl-7,9-dihydro- 1 H-purine- 6, 8 -dione ; l-(4-chlorophenyl)-7-ethyl-2-(4-(6-methoxypyridin-3-yl)phenyl)-9-phenyl-lH-purine- 6,8(7H,9H)-dione;
N-(4'-(l-(4-chlorophenyl)-7-ethyl-6,8-dioxo-9-phenyl-6,7,8,9-tetrahydro-lH-purin-2- yl)biphenyl-4-yl)methanesulfonamide; and
2-[4-(6-amino-5-methyl-pyridin-3-yl)-phenyl]-l-(4-chloro-phenyl)-7-ethyl-9-phenyl-7,9- dihydro-lH-purine-6,8-dione.
20. The use of a compound of any one of claims 1-12, or pharmaceutically acceptable salts or pharmaceutical compositions thereof, for treating a condition mediated by a cannabinoid-1 receptor.
21. The use of a compound of any one of claims 1-12, or pharmaceutically acceptable salts or pharmaceutical compositions thereof, for the manufacture of a medicament for treating a condition mediated by a cannabinoid-1 receptor.
22. The use of claim 20 or 21, wherein said condition is an eating disorder associated with excessive food intake, obesity, diabetes, insulin resistance, hypercholesterolemia, hypertriglyceridemia, substance abuse, osteoporosis, or smoking abuse.
23. The use of claim 20 or 21, wherein said condition is obesity.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US91430407P | 2007-04-26 | 2007-04-26 | |
| US60/914,304 | 2007-04-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008134300A1 true WO2008134300A1 (en) | 2008-11-06 |
Family
ID=39629097
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/061116 WO2008134300A1 (en) | 2007-04-26 | 2008-04-22 | Compounds and compositions as inhibitors of cannabinoid receptor 1 activity |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008134300A1 (en) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006047516A2 (en) * | 2004-10-26 | 2006-05-04 | Irm Llc | Compounds and compositions as inhibitors of cannabinoid receptor 1 activity |
-
2008
- 2008-04-22 WO PCT/US2008/061116 patent/WO2008134300A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006047516A2 (en) * | 2004-10-26 | 2006-05-04 | Irm Llc | Compounds and compositions as inhibitors of cannabinoid receptor 1 activity |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8722691B2 (en) | Azolopyrimidines as inhibitors of cannabinoid 1 activity | |
| US8334288B2 (en) | 4-phenoxymethylpiperidines as modulators of GPR119 activity | |
| EP2134704B1 (en) | Compounds and compositions as modulators of gpr119 activity | |
| US20110190263A1 (en) | Compounds and compositions as modulators of gpr119 activity | |
| US8158634B2 (en) | Compounds and compositions as inhibitors of cannabinoid receptor 1 activity | |
| AU2009217359B2 (en) | Compounds and compositions as modulators of GPR119 activity | |
| US20100022515A1 (en) | Compounds and compositions as modulators of gpr119 activity | |
| EP2252586A1 (en) | Compounds and compositions as modulators of gpr119 activity | |
| WO2008112674A1 (en) | Compounds and compositions as inhibitors of cannabinoid receptor 1 activity | |
| WO2014052619A1 (en) | Piperidine derivatives and compositions as modulators of gpr119 activity | |
| WO2008134300A1 (en) | Compounds and compositions as inhibitors of cannabinoid receptor 1 activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08769153 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08769153 Country of ref document: EP Kind code of ref document: A1 |