WO2008130669A1 - Benzimidazoles and pharmaceutical compositions thereof - Google Patents
Benzimidazoles and pharmaceutical compositions thereof Download PDFInfo
- Publication number
- WO2008130669A1 WO2008130669A1 PCT/US2008/005084 US2008005084W WO2008130669A1 WO 2008130669 A1 WO2008130669 A1 WO 2008130669A1 US 2008005084 W US2008005084 W US 2008005084W WO 2008130669 A1 WO2008130669 A1 WO 2008130669A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- aryl
- alkyl
- cycloalkyl
- heterocyclic
- halo
- Prior art date
Links
- 150000001556 benzimidazoles Chemical class 0.000 title description 2
- 239000008194 pharmaceutical composition Substances 0.000 title description 2
- 238000000034 method Methods 0.000 claims abstract description 26
- 150000003839 salts Chemical class 0.000 claims abstract description 26
- 241000589601 Francisella Species 0.000 claims abstract description 7
- 241000187479 Mycobacterium tuberculosis Species 0.000 claims abstract description 6
- 125000003118 aryl group Chemical group 0.000 claims description 111
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 80
- 125000000217 alkyl group Chemical group 0.000 claims description 77
- 125000005843 halogen group Chemical group 0.000 claims description 37
- 150000001875 compounds Chemical class 0.000 claims description 36
- 125000001424 substituent group Chemical group 0.000 claims description 33
- -1 fused or unfused Chemical group 0.000 claims description 30
- 125000002837 carbocyclic group Chemical group 0.000 claims description 28
- 125000000623 heterocyclic group Chemical group 0.000 claims description 28
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 24
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical group N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 21
- 125000005343 heterocyclic alkyl group Chemical group 0.000 claims description 19
- 229910052717 sulfur Inorganic materials 0.000 claims description 19
- 229910052760 oxygen Inorganic materials 0.000 claims description 18
- 125000004122 cyclic group Chemical group 0.000 claims description 17
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 13
- 229910052757 nitrogen Inorganic materials 0.000 claims description 10
- XXJGBENTLXFVFI-UHFFFAOYSA-N 1-amino-methylene Chemical compound N[CH2] XXJGBENTLXFVFI-UHFFFAOYSA-N 0.000 claims description 8
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 8
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 8
- 125000001246 bromo group Chemical group Br* 0.000 claims description 8
- 125000004432 carbon atom Chemical group C* 0.000 claims description 8
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 8
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 8
- 125000001153 fluoro group Chemical group F* 0.000 claims description 8
- 125000005842 heteroatom Chemical group 0.000 claims description 8
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 claims description 8
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 8
- 125000006574 non-aromatic ring group Chemical group 0.000 claims description 8
- 239000001301 oxygen Substances 0.000 claims description 8
- 229920006395 saturated elastomer Polymers 0.000 claims description 8
- 239000011593 sulfur Chemical group 0.000 claims description 8
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 4
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims 1
- 101150068906 snr-6 gene Proteins 0.000 claims 1
- 125000003785 benzimidazolyl group Chemical class N1=C(NC2=C1C=CC=C2)* 0.000 abstract description 14
- 229940058303 antinematodal benzimidazole derivative Drugs 0.000 abstract description 9
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 59
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 52
- 238000005160 1H NMR spectroscopy Methods 0.000 description 49
- 235000002639 sodium chloride Nutrition 0.000 description 19
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 18
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 17
- 230000015572 biosynthetic process Effects 0.000 description 17
- 238000003786 synthesis reaction Methods 0.000 description 17
- 239000007787 solid Substances 0.000 description 15
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 14
- 239000011541 reaction mixture Substances 0.000 description 13
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 12
- 239000000203 mixture Substances 0.000 description 12
- 239000000047 product Substances 0.000 description 12
- OKKJLVBELUTLKV-UHFFFAOYSA-N methanol Natural products OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 11
- 201000008827 tuberculosis Diseases 0.000 description 10
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- 239000000243 solution Substances 0.000 description 9
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- 239000000543 intermediate Substances 0.000 description 8
- 235000019439 ethyl acetate Nutrition 0.000 description 7
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- 239000006172 buffering agent Substances 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
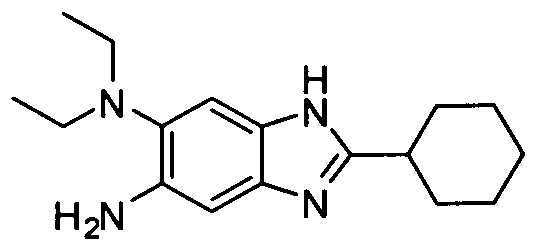
- OARNSPHYTOOVPV-UHFFFAOYSA-N 2-cyclohexyl-5-n,5-n-diethyl-1h-benzimidazole-5,6-diamine Chemical compound N1C=2C=C(N)C(N(CC)CC)=CC=2N=C1C1CCCCC1 OARNSPHYTOOVPV-UHFFFAOYSA-N 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 239000003381 stabilizer Substances 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- NZVYMMMOCWJXAG-UHFFFAOYSA-N methyl n-[7-amino-2-(4-bromophenyl)-3h-benzimidazol-5-yl]carbamate Chemical compound N=1C2=CC(NC(=O)OC)=CC(N)=C2NC=1C1=CC=C(Br)C=C1 NZVYMMMOCWJXAG-UHFFFAOYSA-N 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 3
- RAGRTYREMCPEIV-UHFFFAOYSA-N 5-fluoro-2,4-dinitroaniline Chemical compound NC1=CC(F)=C([N+]([O-])=O)C=C1[N+]([O-])=O RAGRTYREMCPEIV-UHFFFAOYSA-N 0.000 description 3
- WTCKJEJYOVNPPS-UHFFFAOYSA-N 5-n,5-n-diethyl-2-(2-methoxyphenyl)-1h-benzimidazole-5,6-diamine Chemical compound N=1C=2C=C(N)C(N(CC)CC)=CC=2NC=1C1=CC=CC=C1OC WTCKJEJYOVNPPS-UHFFFAOYSA-N 0.000 description 3
- FVPCLKNXCTXPKE-UHFFFAOYSA-N 5-n,5-n-diethyl-2-(4-methylphenyl)-1h-benzimidazole-5,6-diamine Chemical compound N=1C=2C=C(N)C(N(CC)CC)=CC=2NC=1C1=CC=C(C)C=C1 FVPCLKNXCTXPKE-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 208000034784 Tularaemia Diseases 0.000 description 3
- 239000003242 anti bacterial agent Substances 0.000 description 3
- 230000001580 bacterial effect Effects 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- TVCLGQCWXYISPZ-UHFFFAOYSA-N methyl n-(4-amino-3,5-dinitrophenyl)carbamate Chemical compound COC(=O)NC1=CC([N+]([O-])=O)=C(N)C([N+]([O-])=O)=C1 TVCLGQCWXYISPZ-UHFFFAOYSA-N 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- RZNHSEZOLFEFGB-UHFFFAOYSA-N 2-methoxybenzoyl chloride Chemical compound COC1=CC=CC=C1C(Cl)=O RZNHSEZOLFEFGB-UHFFFAOYSA-N 0.000 description 2
- RMVUGEPUWQMQPR-UHFFFAOYSA-N 4-amino-3,5-dinitrobenzoic acid Chemical compound NC1=C([N+]([O-])=O)C=C(C(O)=O)C=C1[N+]([O-])=O RMVUGEPUWQMQPR-UHFFFAOYSA-N 0.000 description 2
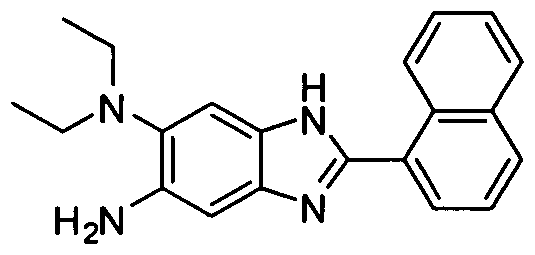
- OGDNMFBQRPLCGG-UHFFFAOYSA-N 5-n,5-n-diethyl-2-naphthalen-1-yl-1h-benzimidazole-5,6-diamine Chemical compound C1=CC=C2C(C=3NC=4C=C(C(=CC=4N=3)N)N(CC)CC)=CC=CC2=C1 OGDNMFBQRPLCGG-UHFFFAOYSA-N 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- SOFUJZVVQNHPTL-UHFFFAOYSA-N CCN(CC)c(cc1[nH]c(C2CCCCC2)nc1c1)c1NC(c(cc1)ccc1OC)=O Chemical compound CCN(CC)c(cc1[nH]c(C2CCCCC2)nc1c1)c1NC(c(cc1)ccc1OC)=O SOFUJZVVQNHPTL-UHFFFAOYSA-N 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 229910006124 SOCl2 Inorganic materials 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 229960004926 chlorobutanol Drugs 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- CLCXAPBVJFIAAT-UHFFFAOYSA-N ethyl n-(7-amino-2-anilino-3h-benzimidazol-5-yl)carbamate Chemical compound N=1C2=CC(NC(=O)OCC)=CC(N)=C2NC=1NC1=CC=CC=C1 CLCXAPBVJFIAAT-UHFFFAOYSA-N 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 150000002460 imidazoles Chemical class 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 201000009671 multidrug-resistant tuberculosis Diseases 0.000 description 2
- ABZWLCLVBIHHEV-UHFFFAOYSA-N n-[5-(diethylamino)-2,4-dinitrophenyl]-2-methoxybenzamide Chemical compound C1=C([N+]([O-])=O)C(N(CC)CC)=CC(NC(=O)C=2C(=CC=CC=2)OC)=C1[N+]([O-])=O ABZWLCLVBIHHEV-UHFFFAOYSA-N 0.000 description 2
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- BKIMMITUMNQMOS-UHFFFAOYSA-N nonane Chemical compound CCCCCCCCC BKIMMITUMNQMOS-UHFFFAOYSA-N 0.000 description 2
- 239000002736 nonionic surfactant Substances 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 238000013268 sustained release Methods 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 238000007910 systemic administration Methods 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- DRTQHJPVMGBUCF-UCVXFZOQSA-N 1-[(2s,3s,4s,5s)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidine-2,4-dione Chemical compound O[C@H]1[C@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UCVXFZOQSA-N 0.000 description 1
- 125000006039 1-hexenyl group Chemical group 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- 125000005273 2-acetoxybenzoic acid group Chemical group 0.000 description 1
- PFSVEICVATWCNL-UHFFFAOYSA-N 2-cyclohexyl-6-piperidin-1-yl-3h-benzimidazol-5-amine Chemical compound NC1=CC=2N=C(C3CCCCC3)NC=2C=C1N1CCCCC1 PFSVEICVATWCNL-UHFFFAOYSA-N 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000003542 3-methylbutan-2-yl group Chemical group [H]C([H])([H])C([H])(*)C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- BNVZFENSTYWDDB-UHFFFAOYSA-N 4-amino-3,5-dinitrobenzamide Chemical compound NC(=O)C1=CC([N+]([O-])=O)=C(N)C([N+]([O-])=O)=C1 BNVZFENSTYWDDB-UHFFFAOYSA-N 0.000 description 1
- ZRYZBQLXDKPBDU-UHFFFAOYSA-N 4-bromobenzaldehyde Chemical compound BrC1=CC=C(C=O)C=C1 ZRYZBQLXDKPBDU-UHFFFAOYSA-N 0.000 description 1
- RKIDDEGICSMIJA-UHFFFAOYSA-N 4-chlorobenzoyl chloride Chemical compound ClC(=O)C1=CC=C(Cl)C=C1 RKIDDEGICSMIJA-UHFFFAOYSA-N 0.000 description 1
- PJEKMNVLUNZFNU-UHFFFAOYSA-N 5-n,5-n-diethyl-2-(4-fluorophenyl)-1h-benzimidazole-5,6-diamine Chemical compound N=1C=2C=C(N)C(N(CC)CC)=CC=2NC=1C1=CC=C(F)C=C1 PJEKMNVLUNZFNU-UHFFFAOYSA-N 0.000 description 1
- QTNCGISIAOMWLA-UHFFFAOYSA-N 5-n,5-n-diethyl-2-(4-methoxyphenyl)-1h-benzimidazole-5,6-diamine Chemical compound N=1C=2C=C(N)C(N(CC)CC)=CC=2NC=1C1=CC=C(OC)C=C1 QTNCGISIAOMWLA-UHFFFAOYSA-N 0.000 description 1
- VKKAEXZEBQIVKU-UHFFFAOYSA-N 5-n,5-n-diethyl-2-phenyl-1h-benzimidazole-5,6-diamine Chemical compound N=1C=2C=C(N)C(N(CC)CC)=CC=2NC=1C1=CC=CC=C1 VKKAEXZEBQIVKU-UHFFFAOYSA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- UMHAOXUYLKMFOI-UHFFFAOYSA-N CCN(CC)c(cc(c([N+]([O-])=O)c1)NC(c2ccc(C(C)(C)C)cc2)=O)c1[N+]([O-])=O Chemical compound CCN(CC)c(cc(c([N+]([O-])=O)c1)NC(c2ccc(C(C)(C)C)cc2)=O)c1[N+]([O-])=O UMHAOXUYLKMFOI-UHFFFAOYSA-N 0.000 description 1
- JQKIMIBQSMWNNW-UHFFFAOYSA-N CCN(CC)c(cc1[nH]c(-c2ccccc2OC)nc1c1)c1NC(c(cc1)ccc1Cl)=O Chemical compound CCN(CC)c(cc1[nH]c(-c2ccccc2OC)nc1c1)c1NC(c(cc1)ccc1Cl)=O JQKIMIBQSMWNNW-UHFFFAOYSA-N 0.000 description 1
- GYPMBQZAVBFUIZ-UHFFFAOYSA-N Cc(cc1)cc(OC)c1OC Chemical compound Cc(cc1)cc(OC)c1OC GYPMBQZAVBFUIZ-UHFFFAOYSA-N 0.000 description 1
- ZBTMRBYMKUEVEU-UHFFFAOYSA-N Cc(cc1)ccc1Br Chemical compound Cc(cc1)ccc1Br ZBTMRBYMKUEVEU-UHFFFAOYSA-N 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010022004 Influenza like illness Diseases 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- BRMSANHPVMRIDK-UHFFFAOYSA-N Nc(c([N+]([O-])=O)c1)cc(N2CCOCC2)c1[N+]([O-])=O Chemical compound Nc(c([N+]([O-])=O)c1)cc(N2CCOCC2)c1[N+]([O-])=O BRMSANHPVMRIDK-UHFFFAOYSA-N 0.000 description 1
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- ZWBTYMGEBZUQTK-PVLSIAFMSA-N [(7S,9E,11S,12R,13S,14R,15R,16R,17S,18S,19E,21Z)-2,15,17,32-tetrahydroxy-11-methoxy-3,7,12,14,16,18,22-heptamethyl-1'-(2-methylpropyl)-6,23-dioxospiro[8,33-dioxa-24,27,29-triazapentacyclo[23.6.1.14,7.05,31.026,30]tritriaconta-1(32),2,4,9,19,21,24,26,30-nonaene-28,4'-piperidine]-13-yl] acetate Chemical compound CO[C@H]1\C=C\O[C@@]2(C)Oc3c(C2=O)c2c4NC5(CCN(CC(C)C)CC5)N=c4c(=NC(=O)\C(C)=C/C=C/[C@H](C)[C@H](O)[C@@H](C)[C@@H](O)[C@@H](C)[C@H](OC(C)=O)[C@@H]1C)c(O)c2c(O)c3C ZWBTYMGEBZUQTK-PVLSIAFMSA-N 0.000 description 1
- 150000001263 acyl chlorides Chemical class 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229940124326 anaesthetic agent Drugs 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- MXMOTZIXVICDSD-UHFFFAOYSA-N anisoyl chloride Chemical compound COC1=CC=C(C(Cl)=O)C=C1 MXMOTZIXVICDSD-UHFFFAOYSA-N 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 229940027983 antiseptic and disinfectant quaternary ammonium compound Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 150000004982 aromatic amines Chemical group 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- TWONWILUDBHKQU-UHFFFAOYSA-N bicyclo[5.2.0]nonane Chemical compound C1CCCCC2CCC21 TWONWILUDBHKQU-UHFFFAOYSA-N 0.000 description 1
- YBZXTUFIYJLTAR-UHFFFAOYSA-N but-3-enyl n-[2-cyclohexyl-6-(diethylamino)-3h-benzimidazol-5-yl]carbamate Chemical compound N=1C=2C=C(NC(=O)OCCC=C)C(N(CC)CC)=CC=2NC=1C1CCCCC1 YBZXTUFIYJLTAR-UHFFFAOYSA-N 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 125000002676 chrysenyl group Chemical group C1(=CC=CC=2C3=CC=C4C=CC=CC4=C3C=CC12)* 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 231100000517 death Toxicity 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000003113 dilution method Methods 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 230000008029 eradication Effects 0.000 description 1
- HAWSPTSRPZKUBS-UHFFFAOYSA-N ethyl n-(7-acetamido-2-phenyl-3h-benzimidazol-5-yl)carbamate Chemical compound N=1C2=CC(NC(=O)OCC)=CC(NC(C)=O)=C2NC=1C1=CC=CC=C1 HAWSPTSRPZKUBS-UHFFFAOYSA-N 0.000 description 1
- RGSMQKYAWZWREZ-UHFFFAOYSA-N ethyl n-(7-amino-2-phenyl-3h-benzimidazol-5-yl)carbamate Chemical compound N=1C2=CC(NC(=O)OCC)=CC(N)=C2NC=1C1=CC=CC=C1 RGSMQKYAWZWREZ-UHFFFAOYSA-N 0.000 description 1
- HFUXINMSXMXTDB-UHFFFAOYSA-N ethyl n-[7-amino-2-(furan-2-yl)-3h-benzimidazol-5-yl]carbamate Chemical compound N=1C2=CC(NC(=O)OCC)=CC(N)=C2NC=1C1=CC=CO1 HFUXINMSXMXTDB-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 208000036984 extensively drug-resistant tuberculosis Diseases 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000003193 general anesthetic agent Substances 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N hydroxymaleic acid group Chemical group O/C(/C(=O)O)=C/C(=O)O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 229960003350 isoniazid Drugs 0.000 description 1
- QRXWMOHMRWLFEY-UHFFFAOYSA-N isoniazide Chemical compound NNC(=O)C1=CC=NC=C1 QRXWMOHMRWLFEY-UHFFFAOYSA-N 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Substances [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- KBENJDJVNOAJCU-UHFFFAOYSA-N methyl 4-[4-amino-6-(methoxycarbonylamino)-1h-benzimidazol-2-yl]benzoate Chemical compound N=1C2=CC(NC(=O)OC)=CC(N)=C2NC=1C1=CC=C(C(=O)OC)C=C1 KBENJDJVNOAJCU-UHFFFAOYSA-N 0.000 description 1
- UBHZIONOLZEBIT-UHFFFAOYSA-N methyl n-[7-acetamido-2-(4-bromophenyl)-3h-benzimidazol-5-yl]carbamate Chemical compound N=1C2=CC(NC(=O)OC)=CC(NC(C)=O)=C2NC=1C1=CC=C(Br)C=C1 UBHZIONOLZEBIT-UHFFFAOYSA-N 0.000 description 1
- FXHDSAJJHYWOBD-UHFFFAOYSA-N methyl n-[7-amino-2-(2,4-dimethoxyphenyl)-3h-benzimidazol-5-yl]carbamate Chemical compound N=1C2=CC(NC(=O)OC)=CC(N)=C2NC=1C1=CC=C(OC)C=C1OC FXHDSAJJHYWOBD-UHFFFAOYSA-N 0.000 description 1
- KROHXUAARQQGNA-UHFFFAOYSA-N methyl n-[7-amino-2-(3-fluorophenyl)-3h-benzimidazol-5-yl]carbamate Chemical compound N=1C2=CC(NC(=O)OC)=CC(N)=C2NC=1C1=CC=CC(F)=C1 KROHXUAARQQGNA-UHFFFAOYSA-N 0.000 description 1
- 244000000010 microbial pathogen Species 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 239000003595 mist Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical class O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 230000036457 multidrug resistance Effects 0.000 description 1
- NUWCWJXGLBLMKV-UHFFFAOYSA-N n-[2-cyclohexyl-6-(diethylamino)-3h-benzimidazol-5-yl]-4-methylbenzamide Chemical compound CCN(CC)C1=CC=2NC(C3CCCCC3)=NC=2C=C1NC(=O)C1=CC=C(C)C=C1 NUWCWJXGLBLMKV-UHFFFAOYSA-N 0.000 description 1
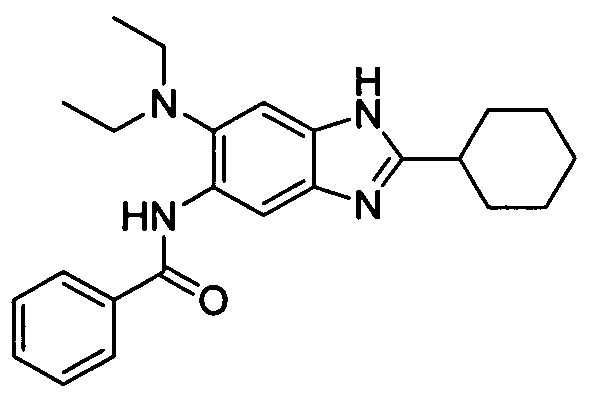
- NCGWFLVWRSAZPK-UHFFFAOYSA-N n-[2-cyclohexyl-6-(diethylamino)-3h-benzimidazol-5-yl]benzamide Chemical compound CCN(CC)C1=CC=2NC(C3CCCCC3)=NC=2C=C1NC(=O)C1=CC=CC=C1 NCGWFLVWRSAZPK-UHFFFAOYSA-N 0.000 description 1
- FJWHEZDYPDKYKR-UHFFFAOYSA-N n-[2-cyclohexyl-6-(diethylamino)-3h-benzimidazol-5-yl]pentanamide Chemical compound N1C=2C=C(N(CC)CC)C(NC(=O)CCCC)=CC=2N=C1C1CCCCC1 FJWHEZDYPDKYKR-UHFFFAOYSA-N 0.000 description 1
- BETHMAHBKZRHKA-UHFFFAOYSA-N n-[6-(diethylamino)-2-(2-methoxyphenyl)-3h-benzimidazol-5-yl]-4-methoxybenzamide Chemical compound CCN(CC)C1=CC=2NC(C=3C(=CC=CC=3)OC)=NC=2C=C1NC(=O)C1=CC=C(OC)C=C1 BETHMAHBKZRHKA-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- UMRZSTCPUPJPOJ-KNVOCYPGSA-N norbornane Chemical compound C1C[C@H]2CC[C@@H]1C2 UMRZSTCPUPJPOJ-KNVOCYPGSA-N 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 229920001542 oligosaccharide Polymers 0.000 description 1
- 150000002482 oligosaccharides Chemical class 0.000 description 1
- 244000039328 opportunistic pathogen Species 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 125000000913 palmityl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920001993 poloxamer 188 Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920002503 polyoxyethylene-polyoxypropylene Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229950008882 polysorbate Drugs 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229920005990 polystyrene resin Polymers 0.000 description 1
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical class [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- YMTCPGQVOXUSCH-UHFFFAOYSA-N prop-2-enyl n-[2-cyclohexyl-6-(diethylamino)-3h-benzimidazol-5-yl]carbamate Chemical compound N=1C=2C=C(NC(=O)OCC=C)C(N(CC)CC)=CC=2NC=1C1CCCCC1 YMTCPGQVOXUSCH-UHFFFAOYSA-N 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- POAUDUUVRRBSRI-UHFFFAOYSA-N propyl n-[2-cyclohexyl-6-(diethylamino)-3h-benzimidazol-5-yl]carbamate Chemical compound N1C=2C=C(N(CC)CC)C(NC(=O)OCCC)=CC=2N=C1C1CCCCC1 POAUDUUVRRBSRI-UHFFFAOYSA-N 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 229960005206 pyrazinamide Drugs 0.000 description 1
- IPEHBUMCGVEMRF-UHFFFAOYSA-N pyrazinecarboxamide Chemical compound NC(=O)C1=CN=CC=N1 IPEHBUMCGVEMRF-UHFFFAOYSA-N 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000001725 pyrenyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 229960000885 rifabutin Drugs 0.000 description 1
- JQXXHWHPUNPDRT-WLSIYKJHSA-N rifampicin Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C([O-])=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N1CC[NH+](C)CC1 JQXXHWHPUNPDRT-WLSIYKJHSA-N 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000003548 sec-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 231100000735 select agent Toxicity 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- QJQRNDGUWQVAEV-AAFSJPGBSA-M sodium bisulfite adduct Chemical compound [Na+].[O-]S(=O)(=O)C([C@H]1N(C(C2=C3)=O)C=C(C1)/C=C/C(=O)N(C)C)NC2=CC1=C3OCO1 QJQRNDGUWQVAEV-AAFSJPGBSA-M 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000005846 sugar alcohols Chemical class 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 125000003960 triphenylenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C3=CC=CC=C3C12)* 0.000 description 1
- 125000004417 unsaturated alkyl group Chemical group 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 235000019871 vegetable fat Nutrition 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/18—Benzimidazoles; Hydrogenated benzimidazoles with aryl radicals directly attached in position 2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/08—Antibacterial agents for leprosy
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
- C07D235/08—Radicals containing only hydrogen and carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- Tuberculosis was one of the first infectious diseases to be identified. More than fifty years of research has been directed to controlling and eliminating this disease. However, the eradication of TB is still one of the most prominent challenges for basic and clinical research scientists.
- MDR-TB Drug resistant tuberculosis in Estonia, IntJTuberc LungDis 2, 130-3.
- MDR-TB is much more difficult to treat than sensitive TB, requiring administration of more expensive, second-line antibiotics for up to two years.
- the frequency of resistance to at least one of the first-line TB drugs ranged from 1.7% in Brazil to 36.9% in Estonia.
- the frequency of resistance is indicative of the global problem involving not only the spread oiMtb, but also treatment.
- Tularemia is primarily enzootic, however, in humans, it causes lesions and flu-like symptoms. Finding new methods of treating F. tulerensis is of great importance because it is one of the most pathogenic microorganisms presently known. As such, it is currently listed as a category A select agent by the Centers for Disease Control and Prevention because of its potential as a bioterrorism agent.
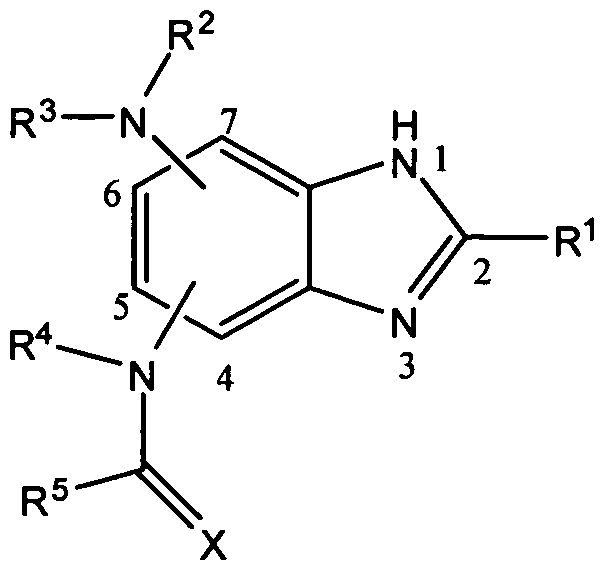
- the invention relates to a molecule having formula I:
- R 1 represents NH 2 , NHR 6 , NR 9 R 10 , NR 6 CONR 9 R 10 , NR 6 CSNR 9 R 10 , OH, OR 6 , SH, SR 6 , CHO, COOR 6 , COR 6 , CH 2 OH, CR 7 R 8 OH, CH 2 OR 6 , CR 7 R 8 OR 6 , CH 2 NH 2 , CR 7 R 8 NH 2 , CR 7 R 8 NR 9 R 10 , alkyl, cycloalkyl, aryl, or halo; R 2 and R 4 independently represent H, alkyl, cycloalkyl, or aryl;
- R 3 represents alkyl, cycloalkyl, or aryl
- R 5 represents H, R 6 , OR 6 , SR 6 , NH 2 , NHR 6 , or NR 9 R 10 ;
- X represents O, S, NH, or NR 6 ;
- R 6 , R 7 , R 8 , R 9 , and R 10 independently represent alkyl, cycloalkyl, aryl, or halo;
- R 2 and R 3 ; R 4 and R 5 ; and R 9 and R 10 independently, may be combined to represent a heterocyclic alkyl or heterocyclic aryl;
- R 7 and R 8 may be combined to represent a cycloalkyl
- alkyl groups are branched or unbranched, saturated or unsaturated, and have 1- 18 carbon atoms in their longest chain;
- cycloalkyl groups are carbocyclic or heterocyclic, fused or unfused, non- aromatic ring systems having a total of 5-16 ring members including substituent rings;
- aryl groups are carbocyclic or heterocyclic
- carbocyclic aryl groups are fused or unfused ring systems having a total of 6-
- heterocyclic aryl groups are fused or unfused ring systems having a total of 5- 16 ring members including substituent rings;
- halo substituents are fluoro, chloro, or bromo
- each alkyl, cycloalkyl, and aryl may be unsubstituted or substituted with one or more substituent at any position;
- alkyl substituents are halo, hydroxyl, OR 6 , SR 6 , NH 2 , NHR 6 , NR 9 R 10 , cycloalkyl, or aryl;
- cycloalkyl substituents are halo, hydroxyl, OR 6 , SR 6 , NH 2 , NHR 6 , NR 9 R 10 , alkyl, cycloalkyl, or aryl; aryl substituents are halo, hydroxyl, OR 6 , SR 6 , NH 2 , NHR 6 , NR 9 R 10 , alkyl, cycloalkyl, aryl, nitro, or carboxyl; and
- heterocyclic alkyl and heterocyclic aryl have at least one heteroatom selected from oxygen, nitrogen and sulfur;
- the invention also relates to a method of treating a patient infected with Mycobaterium tuberculosis or Francisella tulerensis, the method comprising administering to the patient the compound of formula I or a pharmaceutically acceptable salt thereof.
- the invention relates to novel benzimidazole derivatives. These benzimidazole derivatives can be used to treat a patient infected by Mycobacterium tuberculosis or Francisella tulerensis.
- R 1 represents NH 2 , NHR 6 , NR 9 R 10 , NR 6 CONR 9 R 10 ,
- NR 6 CSNR 9 R 10 OH, OR 6 , SH, SR 6 , CHO, COOR 6 , COR 6 , CH 2 OH, CR 7 R 8 OH, CH 2 OR 6 , CR 7 R 8 OR 6 , CH 2 NH 2 , CR 7 R 8 NH 2 , CR 7 R 8 NR 9 R 10 , alkyl, cycloalkyl, aryl, or halo.
- R 2 and R 4 independently represent H, alkyl, cycloalkyl, or aryl.
- R 2 may represent ethyl and R 4 may represent H.
- R 3 represents alkyl, cycloalkyl, or aryl.
- R may represent tetrahydrof ⁇ iranyl or ethyl.
- R 3 represents COR 6 .
- R 3 is not methyl
- R 5 represents H, R 6 , OR 6 , SR 6 , NH 2 , NHR 6 , or NR 9 R 10 .
- R 10 independently represent alkyl, cycloalkyl, aryl, or halo.
- R 6 , R 7 , R 8 , R 9 , and R 10 independently represent alkyl, cycloalkyl, or aryl. More preferably, R 6 , R 7 , R 8 , R 9 , and R 10 independently represent alkyl or aryl.
- R 2 and R 3 ; R 4 and R 5 ; and R 9 and R 10 may be combined to represent a heterocyclic alkyl or heterocyclic aryl ring.
- R 2 and R 3 can be combined to represent a heterocyclic alkyl ring, resulting in the following structure:
- R 4 and R 5 can be combined to represent a heterocyclic alkyl ring, resulting in the following structure:
- R > 7 a __nd j ⁇ R% 8 may be combined to represent a cycloalkyl.
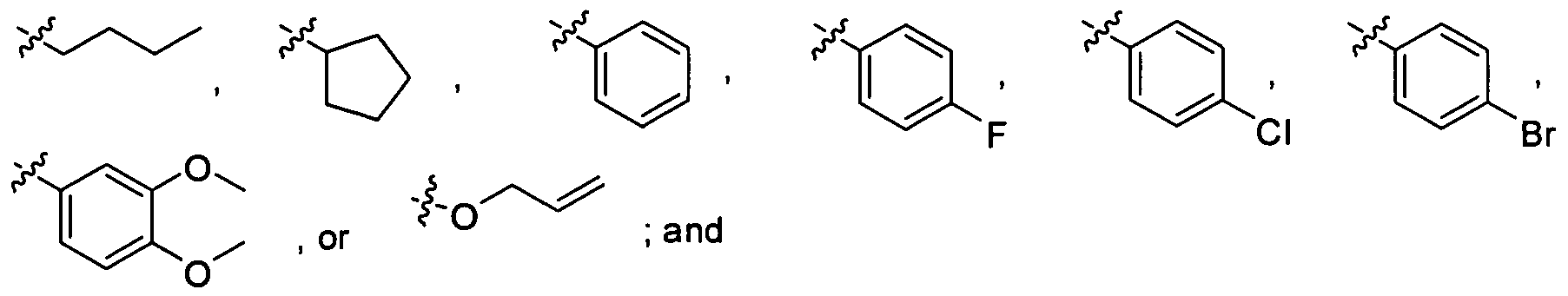
- Alkyl groups are branched or unbranched, saturated or unsaturated, and have 1-18 carbon atoms in their longest chain.
- suitable straight-chained, saturated alkyl groups include methyl, ethyl, n-propyl, «-butyl, w-pentyl, «-hexyl groups and dodecyl and hexadecyl.
- Preferred straight chain, saturated alkyl groups include methyl and ethyl.
- branched, saturated alkyl groups include iso- propyl, iso-butyl, sec-butyl, t-butyl, 1-methylbutyl, 2-methylbutyl, 3-methylbutyl (isopentyl), 1,1-dimethylpropyl, 1,2-dimethylpropyl, 2,2-dimethylpropyl (neopentyl), 1-methylpentyl, 2-methylpentyl, 3-methylpentyl, 4-methylpentyl groups, and 2- methyl,5-ethyldecyl.
- Preferred branched, saturated alkyl groups include isopropyl and t-butyl.
- unsaturated alkyl groups include ethenyl, ethynyl, propenyl, propargyl, isopropenyl, crotyl, 1-hexenyl, and 1-octenyl.
- Cycloalkyl groups are carbocyclic or heterocyclic, fused or unfused, non- aromatic ring systems having a total of 5-16 ring members including substituent rings. Ring systems are monocyclic, bicyclic, tricyclic, or tetracyclic and can be bridged or non-bridged.
- carbocyclic alkyl groups include cyclobutanyl, cyclopentanyl, cyclohexanyl, and cycloheptanyl.
- fused carbocyclic alkyl groups include indenyl, isoindenyl.
- Bridged groups include bicyclo [2.2.1] heptane, bicyclco [5.2.0] nonane, and bicyclo [5.2.0] nonane.
- heterocyclic alkyl groups include pyrrolidinyl, piperidinyl, piperazinyl, tetrahydrofuranyl, morpholino, and oxazolidinyl.
- fused heterocyclic alkyl groups include benzomorpholino, benzopyrrolidinyl, indolinyl, and benzopiperidinyl.
- Aryl groups can be either carbocyclic or heterocyclic.
- Carbocyclic aryl groups are fused or unfused ring systems having a total of 6- 16 ring members including substituent rings.
- a preferred unfused carbocyclic aryl group is phenyl.
- fused carbocyclic aryl groups include naphthyl, phenanthryl, anthracenyl, triphenylenyl, chrysenyl, and pyrenyl.
- Heterocyclic aryl groups are fused or unfused ring systems having a total of 5- 16 ring members including substituent rings.
- unfused heterocyclic aryl groups include thiophenyl, furyl, pyrrolyl, pyrazolyl, imidazolyl, oxazolyl, thiazolyl, pyridinyl, pyridazinyl, pyrimidinyl, and pyrazinyl.
- fused heterocyclic aryl groups include purinyl, 1,4-diazanaphthalenyl, indolyl, benzimidazolyl, 4,5-diazaphenanthrenyl, benzoxazolyl, isoindolyl, quinolinyl, isoquinolinyl, and benzofuranyl.
- Halo substituents are fluoro, chloro, or bromo.
- alkyl, cycloalkyl, and aryl may be unsubstituted or substituted with one or more substituent at any position.
- Alkyl substituents are halo, hydroxyl, OR 6 , SR 6 , NH 2 , NHR 6 , NR 9 R 10 , cycloalkyl, or aryl.
- Cycloalkyl substituents are halo, hydroxyl, OR 6 , SR 6 , NH 2 , NHR 6 , NR 9 R 10 , alkyl, cycloalkyl, or aryl.
- Aryl substituents are halo, hydroxyl, OR 6 , SR 6 , NH 2 , NHR 6 , NR 9 R 10 , alkyl, cycloalkyl, aryl, nitro, or carboxyl.
- Heterocyclic alky and heterocyclic aryl have at least one heteroatom selected from oxygen, nitrogen, and sulfur.
- X represents O, S, NH, or NR 6 .
- R 6 is described above.
- X is identified above as representing O, S, NH, or NR 6 .
- R 5 is identified above as being H, R 6 , OR 6 , SR 6 , NH 2 , NHR 6 , or NR 9 R 10 .
- Each element of X (O, S, NH or NR 6 ) can be combined with each and every element of R 5 (H, R 6 , OR 6 , SR 6 , NH 2 , NHR 6 , or NR 9 R 10 ).
- X may be O and R 5 may be H.
- X may be NH and R 5 may be NR 9 R 10 , etc.
- a third parameter is R 4 , in which the elements are defined as H, alkyl, cycloalkyl, or aryl.
- R 4 may be H (or any other chemical moiety within the element of R 4 ).
- a stable compound or chemically feasible compound is one in which the chemical structure is not substantially altered when kept at a temperature of 40 0 C or less, in the absence of moisture or other chemically reactive conditions, for at least a week.
- the present invention also relates to pharmaceutically acceptable salts of the benzimidazole derivatives.
- the pharmaceutically acceptable salts include the conventional non-toxic salts of the benzimidazole derivatives as formed, e.g., from non-toxic inorganic or organic acids.
- such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like: and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxy-benzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, trifluoroacetic and the like.
- inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like
- organic acids such as acetic, propionic, succinic, glycolic,
- the pharmaceutically acceptable salts of the benzimidazole derivatives of this invention can be synthesized from the compounds of this invention which contain a basic moiety by conventional chemical methods. Generally, the salts are prepared either by ion exchange chromatography or by reacting the free base with stoichiometric amounts or with an excess of the desired salt-forming inorganic or organic acid in a suitable solvent or various combinations of solvents. Synthesis of the benzimidazole derivatives
- the benzimidazoles of the present invention can be synthesized by methods known in the art.
- the following scheme represents one approach to the synthesis of the compounds of the invention.
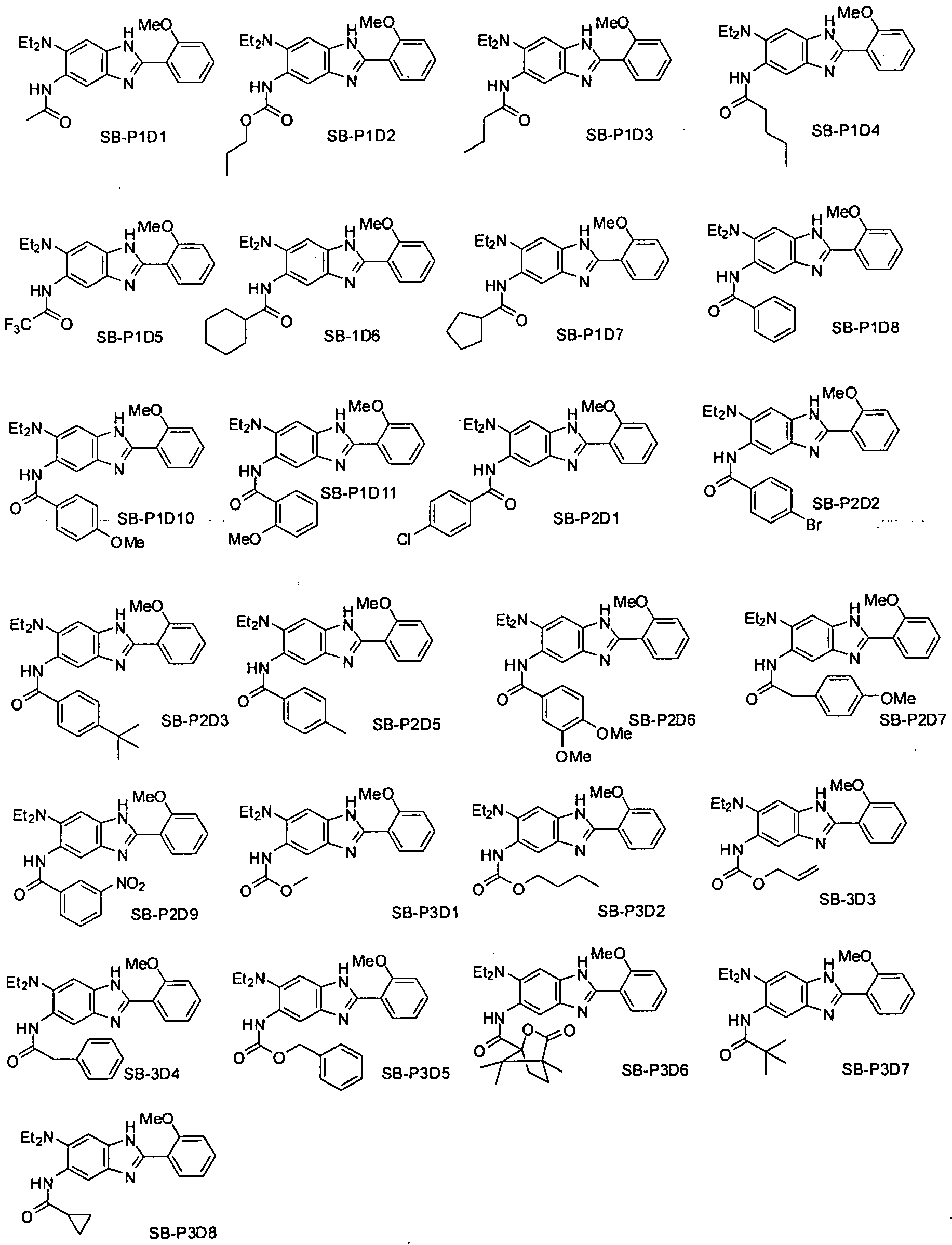
- Scheme I shows an example of a synthesis that yields individual compounds of the invention or a library of compounds of the invention.
- the compounds of the invention may be made using polymer-assisted solution-phase (PASP) synthesis.
- PASP is a parallel synthesis method for creation of a trisubstituted benzimidazoles (BAZ-I) library using 2,4-dinitro-5-fluoroaniline (1) as the starting material.
- the first step involves the nucleophilic substitution of compound 1 with a secondary amine in the presence of N,N-diisopropylethylamine.
- the reaction produces compound 2 in high yields and purity at room temperature.
- the free aromatic amino group of compound 5 is modified in different ways.
- anhydride, acyl chloride, sulfonyl chloride, and isocyanate are used as modifying agents.
- the modification of the aromatic amine moiety takes place smoothly in dry dichloromethane and all excess acylating reagents are scavenged by commercially available aminomethylated polystyrene resin (from nova-biochem) to give the desired product 6 in 80-95% yield.
- the invention also relates to a method of treating a patient infected with Mycobacterium tuberculosis or Francisella tulerensis.
- the method comprises administering to the patient the compound of formula (I) or a pharmaceutically acceptable salt thereof.
- the method and compounds of the invention may be employed alone, or in combination with other anti-bacterial agents.
- Other anti-bacterial agents include isoniazid, rifampin, pyrazinamide, rifabutin, streptomycin and ciprofloxacin.
- the combination of these anti-bacterial agents and the compounds of the invention will provide new agents for the treatment of tuberculosis, including MDR-TB and XDR- TB, and tularemia.
- An effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof as used herein is any amount effective to treat a patient infected by Mtb or F. tulerensis. Modes of administration and doses can be determined by those having skill in the art. An effective amount of the compound will vary with the group of patients (age, sex, weight, etc.), the nature and severity of the condition to be treated, the particular compound administered, and its route of administration. Amounts suitable for administration to humans are routinely determined by physicians and clinicians during clinical trials. The minimum dose of the compound is the lowest dose at which efficacy is observed. For example, the minimum dose of the compound may be about O.lmg/kg/day, about 1 mg/kg/day, or about 3 mg/kg/day.
- the maximum dose of the compound is the highest dose at which efficacy is observed in a patient, and side effects are tolerable.
- the maximum dose of the compound may be about 10 mg/kg/day, about 9 mg/kg/day, or about 8 mg/kg/day.
- a benzimidazole derivative useful in the methods of the present invention may be administered by any method known in the art.
- suitable modes of administration include oral and systemic administration.
- Systemic administration can be enteral or parenteral.
- Liquid or solid (e.g., tablets, gelatin capsules) formulations can be employed.
- Parenteral administration of the benzimidazole derivative include, for example intravenous, intramuscular, and subcutaneous injections.
- a chemical compound may be administered to a patient by sustained release, as is known in the art.
- Sustained release administration is a method of drug delivery to achieve a certain level of the drug over a particular period of time.
- routes of administration include oral, topical, intrabronchial, or intranasal administration.
- oral administration liquid or solid formulations may be used.
- formulations suitable for oral administration include tablets, gelatin capsules, pills, troches, elixirs, suspensions, syrups, and wafers.
- Intrabronchial administration can include an inhaler spray.
- administration of a chemical compound can be accomplished by a nebulizer or liquid mist.
- the chemical compound can be formulated in a suitable pharmaceutical carrier.
- a pharmaceutical carrier is considered to be synonymous with a vehicle or an excipient as is understood by practitioners in the art.
- carriers include starch, milk, sugar, certain types of clay, gelatin, stearic acid or salts thereof, magnesium or calcium stearate, talc, vegetable fats or oils, gums and glycols.
- the chemical compound can be formulated into a composition containing one or more of the following: a stabilizer, a surfactant, preferably a nonionic surfactant, and optionally a salt and/or a buffering agent.
- the stabilizer may, for example, be an amino acid, such as for instance, glycine; or an oligosaccharide, such as for example, sucrose, tetralose, lactose or a dextran.
- the stabilizer may be a sugar alcohol, such as for instance, mannitol; or a combination thereof.
- the stabilizer or combination of stabilizers constitutes from about 0.1% to about 10% weight for weight of the chemical compound.
- the surfactant is preferably a nonionic surfactant, such as a polysorbate.
- Suitable surfactants include Tween 20, Tween 80; a polyethylene glycol or a polyoxyethylene polyoxypropylene glycol, such as Pluronic F-68 at from about 0.001% (w/v) to about 10% (w/v).
- the salt or buffering agent may be any salt or buffering agent, such as for example sodium chloride, or sodium/potassium phosphate, respectively.
- the buffering agent maintains the pH of the chemical compound formulation in the range of about 5.5 to about 7.5.
- the salt and/or buffering agent is also useful to maintain the osmolality at a level suitable for administration to a patient.
- the salt or buffering agent is present at a roughly isotonic concentration of about 150 mM to about 300 mM.
- the chemical compound can be formulated into a composition which may additionally contain one or more conventional additives.
- additives include a solubilizer such as, for example, glycerol; an antioxidant such as for example, benzalkonium chloride (a mixture of quaternary ammonium compounds, known as "quart"), benzyl alcohol, chloretone or chlorobutanol; anaesthetic agent such as, for example a morphine derivative; or an isotonic agent etc.
- a solubilizer such as, for example, glycerol
- an antioxidant such as for example, benzalkonium chloride (a mixture of quaternary ammonium compounds, known as "quart"), benzyl alcohol, chloretone or chlorobutanol
- anaesthetic agent such as, for example a morphine derivative
- the composition may be stored under nitrogen gas in vials sealed with impermeable stoppers.
- MIC values were determined using the microplate dilution method, previously reported [R. A. Slayden and C. E. Barry, III. "The role of KasA and KasB in the biosynthesis of meromycolic acids and isoniazid resistance in Mycobacterium tuberculosis", Tuberculosis (Edinb) 82:149-60 (2002)].
- Bacteria were cultivated in liquid medium to an optical density of ⁇ 0.4 at 600 nm. The bacterial cultures were then prepared for testing by diluting 1:100 in liquid medium. A total of 50 ⁇ L of each culture was added to each well of a 96-well optical plate. Analogs were prepared at 60 ⁇ M in 100% DMSO. Compound stock solutions were diluted 1 :2 in liquid medium and then distributed in the plate as 2-fold serial dilutions to achieve a concentration range of 200-0.2 mg/mL in a total final volume of 100 ⁇ L. The plates were incubated at 37 0 C and evaluated for the presence of bacterial growth or non-growth by optical density using an inverted plate reading method. The MIC99 was determined to be the lowest concentration of compound that inhibited bacterial growth. Reported MIC values represent measurements performed independently in triplicate.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Pharmacology & Pharmacy (AREA)
- Oncology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description


































































Claims

















Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010504113A JP5400032B2 (en) | 2007-04-20 | 2008-04-21 | Benzimidazole and pharmaceutical composition thereof |
| AU2008242488A AU2008242488B2 (en) | 2007-04-20 | 2008-04-21 | Benzimidazoles and pharmaceutical compositions thereof |
| MX2009011281A MX2009011281A (en) | 2007-04-20 | 2008-04-21 | Benzimidazoles and pharmaceutical compositions thereof. |
| EP08799882.9A EP2154966B1 (en) | 2007-04-20 | 2008-04-21 | Benzimidazoles and pharmaceutical compositions thereof |
| PL08799882T PL2154966T3 (en) | 2007-04-20 | 2008-04-21 | Benzimidazoles and pharmaceutical compositions thereof |
| CN200880021064A CN101677553A (en) | 2007-04-20 | 2008-04-21 | benzimidazoles and pharmaceutical compositions thereof |
| ES08799882T ES2434693T3 (en) | 2007-04-20 | 2008-04-21 | Benzimidazoles and pharmaceutical compositions thereof |
| IN3959KON2009 IN2009KN03959A (en) | 2007-04-20 | 2008-04-21 | |
| SI200831097T SI2154966T1 (en) | 2007-04-20 | 2008-04-21 | Benzimidazoles and pharmaceutical compositions thereof |
| EA200901423A EA016726B1 (en) | 2007-04-20 | 2008-04-21 | Benzimidazoles and pharmaceutical compositions thereof |
| DK08799882.9T DK2154966T3 (en) | 2007-04-20 | 2008-04-21 | Benzimidazoles and their pharmaceutical compositions |
| US12/596,347 US8232410B2 (en) | 2007-04-20 | 2008-04-21 | Benzimidazoles and pharmaceutical compositions thereof |
| CA2684594A CA2684594C (en) | 2007-04-20 | 2008-04-21 | Benzimidazoles and pharmaceutical compositions thereof for treating mycobacterium tuberculosis or francisella tulerensis infection |
| BRPI0810244-9A BRPI0810244A2 (en) | 2007-04-20 | 2008-04-21 | METHOD AND METHODS TO TREAT A PATIENT INFECTED WITH MYCOBACTERIUM TUBERCULOSIS AND FRANCISELLA TULERENSIS |
| HRP20131103AT HRP20131103T1 (en) | 2007-04-20 | 2013-11-20 | Benzimidazoles and pharmaceutical compositions thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US91298007P | 2007-04-20 | 2007-04-20 | |
| US60/912,980 | 2007-04-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008130669A1 true WO2008130669A1 (en) | 2008-10-30 |
Family
ID=39875833
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/005084 WO2008130669A1 (en) | 2007-04-20 | 2008-04-21 | Benzimidazoles and pharmaceutical compositions thereof |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US8232410B2 (en) |
| EP (1) | EP2154966B1 (en) |
| JP (1) | JP5400032B2 (en) |
| CN (1) | CN101677553A (en) |
| AU (1) | AU2008242488B2 (en) |
| BR (1) | BRPI0810244A2 (en) |
| CA (1) | CA2684594C (en) |
| DK (1) | DK2154966T3 (en) |
| EA (1) | EA016726B1 (en) |
| ES (1) | ES2434693T3 (en) |
| HR (1) | HRP20131103T1 (en) |
| IN (1) | IN2009KN03959A (en) |
| MX (1) | MX2009011281A (en) |
| PL (1) | PL2154966T3 (en) |
| PT (1) | PT2154966E (en) |
| SI (1) | SI2154966T1 (en) |
| WO (1) | WO2008130669A1 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2246332A1 (en) * | 2009-04-22 | 2010-11-03 | The Ohio State University Research Foundation | Pyrazole derivatives as anti-francisella agents |
| US8039502B2 (en) | 2007-07-24 | 2011-10-18 | The Ohio State University Research Foundation | Anti-infective agents against intracellular pathogens |
| JP2013504544A (en) * | 2009-09-11 | 2013-02-07 | プロビオドルグ エージー | Heterocyclic derivatives as glutaminyl cyclase inhibitors |
| WO2013116823A1 (en) * | 2012-02-02 | 2013-08-08 | The Research Foundation Of State University Of New York | Benzimidazoles and uses thereof |
| WO2013142326A1 (en) * | 2012-03-23 | 2013-09-26 | The Research Foundation Of State University Of New York | 5-carbonylamino-/(sulfonamido-) substituted benz imidazoles and use thereof treatment of tuberculosis |
| US10287617B2 (en) | 2014-03-11 | 2019-05-14 | Colorado State University Research Foundation | Methods for in vitro—in vivo efficacy determination |
| CN111303092A (en) * | 2020-04-07 | 2020-06-19 | 山东昌邑灶户盐化有限公司 | 2, 4-dinitro-6-chloroaniline derivative, synthetic method and application thereof |
| US11878968B2 (en) | 2021-07-09 | 2024-01-23 | Plexium, Inc. | Aryl compounds and pharmaceutical compositions that modulate IKZF2 |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8232310B2 (en) | 2006-12-29 | 2012-07-31 | Georgetown University | Targeting of EWS-FLI1 as anti-tumor therapy |
| NZ723971A (en) * | 2012-04-12 | 2018-01-26 | Univ Georgetown | Methods and compositions for treating ewings sarcoma family of tumors |
| CN103333170B (en) * | 2013-06-14 | 2016-08-10 | 清华大学 | Benzimidazoles compound and application thereof |
| KR20160065986A (en) | 2013-10-24 | 2016-06-09 | 조지타운 유니버시티 | Methods and compositions for treating cancer |
| US9845296B2 (en) | 2013-11-22 | 2017-12-19 | The Research Foundation For The State University Of New York | Benzimidazoles and their use in the treatment of tuberculosis |
| CN107108580B (en) | 2014-10-09 | 2020-06-30 | 英克特诺治疗公司 | Indolone compounds and use thereof |
| JP6864379B2 (en) | 2016-03-31 | 2021-04-28 | オンターナル セラピューティック インコーポレイテッドOncternal Therapeutics, Inc. | Indole analogues and their use |
| US10159660B2 (en) | 2016-07-29 | 2018-12-25 | Oncternal Therapeutics, Inc. | Uses of indolinone compounds |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050124678A1 (en) | 2001-05-04 | 2005-06-09 | Paratek Pharmaceuticals, Inc. | Transcription factor modulating compounds and methods of use thereof |
| US20060116412A1 (en) * | 2004-09-30 | 2006-06-01 | Raymond Ng | Novel benzimidazole derivatives useful as selective androgen receptor modulators (sarms) |
| WO2007105023A1 (en) | 2006-03-15 | 2007-09-20 | Csir | Modulation of phosphoryl transferase activity of glutamine synthetase |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS61170732A (en) * | 1985-01-25 | 1986-08-01 | Fuji Photo Film Co Ltd | Silver halide photographic sensitive material |
-
2008
- 2008-04-21 WO PCT/US2008/005084 patent/WO2008130669A1/en active Application Filing
- 2008-04-21 CA CA2684594A patent/CA2684594C/en active Active
- 2008-04-21 EP EP08799882.9A patent/EP2154966B1/en not_active Not-in-force
- 2008-04-21 ES ES08799882T patent/ES2434693T3/en active Active
- 2008-04-21 US US12/596,347 patent/US8232410B2/en active Active
- 2008-04-21 EA EA200901423A patent/EA016726B1/en not_active IP Right Cessation
- 2008-04-21 MX MX2009011281A patent/MX2009011281A/en active IP Right Grant
- 2008-04-21 PT PT87998829T patent/PT2154966E/en unknown
- 2008-04-21 DK DK08799882.9T patent/DK2154966T3/en active
- 2008-04-21 BR BRPI0810244-9A patent/BRPI0810244A2/en not_active Application Discontinuation
- 2008-04-21 PL PL08799882T patent/PL2154966T3/en unknown
- 2008-04-21 JP JP2010504113A patent/JP5400032B2/en not_active Expired - Fee Related
- 2008-04-21 SI SI200831097T patent/SI2154966T1/en unknown
- 2008-04-21 IN IN3959KON2009 patent/IN2009KN03959A/en unknown
- 2008-04-21 CN CN200880021064A patent/CN101677553A/en active Pending
- 2008-04-21 AU AU2008242488A patent/AU2008242488B2/en not_active Ceased
-
2013
- 2013-11-20 HR HRP20131103AT patent/HRP20131103T1/en unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050124678A1 (en) | 2001-05-04 | 2005-06-09 | Paratek Pharmaceuticals, Inc. | Transcription factor modulating compounds and methods of use thereof |
| US20060116412A1 (en) * | 2004-09-30 | 2006-06-01 | Raymond Ng | Novel benzimidazole derivatives useful as selective androgen receptor modulators (sarms) |
| WO2007105023A1 (en) | 2006-03-15 | 2007-09-20 | Csir | Modulation of phosphoryl transferase activity of glutamine synthetase |
| EP2008210A1 (en) | 2006-03-15 | 2008-12-31 | Csir | Modulation of phosphoryl transferase activity of glutamine synthetase |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2154966A4 |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8039502B2 (en) | 2007-07-24 | 2011-10-18 | The Ohio State University Research Foundation | Anti-infective agents against intracellular pathogens |
| EP2246332A1 (en) * | 2009-04-22 | 2010-11-03 | The Ohio State University Research Foundation | Pyrazole derivatives as anti-francisella agents |
| US8580827B2 (en) | 2009-04-22 | 2013-11-12 | The Ohio State University Research Foundation | Anti-Francisella agents |
| JP2013504544A (en) * | 2009-09-11 | 2013-02-07 | プロビオドルグ エージー | Heterocyclic derivatives as glutaminyl cyclase inhibitors |
| US9173885B2 (en) | 2009-09-11 | 2015-11-03 | Probiodrug Ag | Inhibitors |
| US9650362B2 (en) | 2009-09-11 | 2017-05-16 | Probiodrug Ag | Inhibitors |
| WO2013116823A1 (en) * | 2012-02-02 | 2013-08-08 | The Research Foundation Of State University Of New York | Benzimidazoles and uses thereof |
| WO2013142326A1 (en) * | 2012-03-23 | 2013-09-26 | The Research Foundation Of State University Of New York | 5-carbonylamino-/(sulfonamido-) substituted benz imidazoles and use thereof treatment of tuberculosis |
| US10287617B2 (en) | 2014-03-11 | 2019-05-14 | Colorado State University Research Foundation | Methods for in vitro—in vivo efficacy determination |
| CN111303092A (en) * | 2020-04-07 | 2020-06-19 | 山东昌邑灶户盐化有限公司 | 2, 4-dinitro-6-chloroaniline derivative, synthetic method and application thereof |
| CN111303092B (en) * | 2020-04-07 | 2021-05-25 | 山东昌邑灶户盐化有限公司 | 2, 4-dinitro-6-chloroaniline derivative, synthetic method and application thereof |
| US11878968B2 (en) | 2021-07-09 | 2024-01-23 | Plexium, Inc. | Aryl compounds and pharmaceutical compositions that modulate IKZF2 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2684594C (en) | 2015-10-06 |
| BRPI0810244A2 (en) | 2014-09-30 |
| EP2154966A1 (en) | 2010-02-24 |
| AU2008242488A1 (en) | 2008-10-30 |
| CA2684594A1 (en) | 2008-10-30 |
| HRP20131103T1 (en) | 2013-12-20 |
| PL2154966T3 (en) | 2014-03-31 |
| ES2434693T3 (en) | 2013-12-17 |
| EP2154966A4 (en) | 2010-12-15 |
| MX2009011281A (en) | 2010-03-08 |
| US8232410B2 (en) | 2012-07-31 |
| DK2154966T3 (en) | 2013-11-18 |
| IN2009KN03959A (en) | 2015-08-28 |
| JP2010524947A (en) | 2010-07-22 |
| CN101677553A (en) | 2010-03-24 |
| SI2154966T1 (en) | 2014-02-28 |
| AU2008242488B2 (en) | 2014-10-30 |
| EA200901423A1 (en) | 2010-06-30 |
| US20100256203A1 (en) | 2010-10-07 |
| PT2154966E (en) | 2013-11-13 |
| JP5400032B2 (en) | 2014-01-29 |
| EP2154966B1 (en) | 2013-08-21 |
| EA016726B1 (en) | 2012-07-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2154966B1 (en) | Benzimidazoles and pharmaceutical compositions thereof | |
| CA2821999C (en) | Benzimidazole respiratory syncytial virus inhibitors | |
| TWI393708B (en) | Hydroxamate compounds, use thereof and synthesizing method for the same | |
| EP2947073B1 (en) | Fused ring analogues of anti-fibrotic agents | |
| US20100222340A1 (en) | SUBSTITUTED PYRIDO [1,2-a] ISOQUINOLINE DERIVATIVES | |
| EP2864323B1 (en) | 1,3-dihydro-2h-benzimidazol-2-one derivatives substituted with heterocycles as respiratory syncytial virus antiviral agents | |
| CA2895430A1 (en) | Quinoxalinones and dihydroquinoxalinones as respiratory syncytial virus antiviral agents | |
| EP1558612B1 (en) | Imidazopyridine derivatives, preparation method and pharmaceutical compositions containing same | |
| US5545653A (en) | Anti-viral compounds | |
| WO2003024937A1 (en) | Benzimidazolylalkoxyaryl alkanoic acid derivatives and their use as antihyperglycemics | |
| RU2695383C2 (en) | Bendamustine derivatives, related compounds and their medical application for treating cancer | |
| CN109851614B (en) | Heterocyclic peptide deformylase inhibitor and preparation method and application thereof | |
| WO2013142326A1 (en) | 5-carbonylamino-/(sulfonamido-) substituted benz imidazoles and use thereof treatment of tuberculosis | |
| WO2013116823A1 (en) | Benzimidazoles and uses thereof | |
| HUT70041A (en) | 2-substituted quinolylmethoxy-phenylacetic acid derivatives, pharmaceutical compositions comprising them and the process for preparing thereof | |
| US11834430B2 (en) | Compounds useful as inhibitors of isoprenylcysteine carboxyl methyltransferase | |
| DK168377B1 (en) | N- (1H-indol-4-yl) benzamide derivatives and their salts, process for their preparation, and pharmaceutical compositions containing them | |
| SK62293A3 (en) | Substituted (benzothiazolyl and quinaxalyl-methoxy) phenyl-acetic acid derivatives | |
| KR101252632B1 (en) | Nitroimidazole compounds, process for the preparation thereof, and pharmaceutical composition for treating tuberculosis comprising the same | |
| US3711505A (en) | 2-(2-imidazolin-2-ylthio)-acetanilides | |
| IE853327L (en) | Allylic amines | |
| JP2002161036A (en) | Phosphodiesterase inhibitor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880021064.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08799882 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2684594 Country of ref document: CA Ref document number: MX/A/2009/011281 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010504113 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3959/KOLNP/2009 Country of ref document: IN Ref document number: 2008242488 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008799882 Country of ref document: EP Ref document number: 200901423 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 2008242488 Country of ref document: AU Date of ref document: 20080421 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12596347 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0810244 Country of ref document: BR Kind code of ref document: A2 Effective date: 20091020 |