WO2008117148A1 - Substituted oxadiazole analogs as calcium channel antagonists - Google Patents
Substituted oxadiazole analogs as calcium channel antagonists Download PDFInfo
- Publication number
- WO2008117148A1 WO2008117148A1 PCT/IB2008/000645 IB2008000645W WO2008117148A1 WO 2008117148 A1 WO2008117148 A1 WO 2008117148A1 IB 2008000645 W IB2008000645 W IB 2008000645W WO 2008117148 A1 WO2008117148 A1 WO 2008117148A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- amine
- ethyl
- oxadiazol
- ylmethyl
- Prior art date
Links
- 0 C*(CC*(CCN)CCNSN)CC(O)=*C(C1(CC*CC1)/C1=C\C(C)(*)/C=C/C(C)(C)/C=C1)=* Chemical compound C*(CC*(CCN)CCNSN)CC(O)=*C(C1(CC*CC1)/C1=C\C(C)(*)/C=C/C(C)(C)/C=C1)=* 0.000 description 1
- PSRHRFNKESVOEL-UHFFFAOYSA-N CC(C)(C)OC(N1CCC(CC=O)CC1)=O Chemical compound CC(C)(C)OC(N1CCC(CC=O)CC1)=O PSRHRFNKESVOEL-UHFFFAOYSA-N 0.000 description 1
- IDXKPWMYURAXTA-UHFFFAOYSA-N CC(CCNC(OC(C)(C)C)=O)O Chemical compound CC(CCNC(OC(C)(C)C)=O)O IDXKPWMYURAXTA-UHFFFAOYSA-N 0.000 description 1
- JKDRUTUCWZXOQO-UHFFFAOYSA-N COc1c(C2CCN(CCNCc3nc(C4(CCOCC4)c(cc4)ccc4Cl)n[o]3)CC2)cccc1 Chemical compound COc1c(C2CCN(CCNCc3nc(C4(CCOCC4)c(cc4)ccc4Cl)n[o]3)CC2)cccc1 JKDRUTUCWZXOQO-UHFFFAOYSA-N 0.000 description 1
- KMSJLAAYQKQUFI-UHFFFAOYSA-N N/C(/C1(CCC1)c(cc1)ccc1Cl)=N\OC(CCl)=O Chemical compound N/C(/C1(CCC1)c(cc1)ccc1Cl)=N\OC(CCl)=O KMSJLAAYQKQUFI-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- This invention relates to oxadiazole derivatives. More particularly, this invention relates to oxadiazole derivatives that are calcium channel antagonists.
- the oxadiazole derivatives of the invention modulate one or more of the L-type, N-type, and T-type calcium channels and are useful as pharmaceutical agents in the treatment of a variety of disorders ranging from pain, hypertension, angina, and/or obesity.
- Voltage-gated calcium channels are membrane-spanning, multi-subunit proteins that open in response to membrane depolarization, allowing calcium ion entry from the extracellular milieu.
- Calcium channels were initially classified based on the time and voltage-dependence of channel opening and on the sensitivity to pharmacological block. The categories were low-voltage activated (primarily T-type) and high-voltage activated (L, N, P, Q or R-type). Recently, an alternative classification scheme was devised based upon the molecular subunit composition, as summarized in Table 1 (Hockerman G H, Peterson B Z, Johnson B D, Catterall W A. 1997. Annu Rev Pharmacol Toxicol 37: 361-96).
- the Cd subunit is the primary determinant of the pharmacological properties and contains the channel pore and voltage sensor (Hockennan G H, Peterson B Z, Johnson B D, Catterall W A. 1997. Annu Rev Pharmacol Toxicol 37: 361-96; Striessnig J. 1999. Cell Physiol Biochem 9: 242-69).
- Ten isoforms of the ⁇ i subunit are known, as indicated in Table 1.
- the ⁇ 2 ⁇ subunit consists of two disulfide linked subunits, ⁇ 2 , which is primarily extracellular and a transmembrane ⁇ subunit.
- the ⁇ subunit is a non-glycosylated cytoplasmic protein that binds to the ⁇ i subunit.
- Four isoforms are known, termed ⁇ i to ⁇ 4 .
- the y subunit is a transmembrane protein that has been biochemically isolated as a component of Ca v 1 and Ca v 2 channels. The nomenclature for voltage-gated calcium channels is based upon the content of the O 1 subunit, as indicated in Table 1.
- Each type of di subunit can associate with a variety of ⁇ , ⁇ 2 ⁇ or y subunits, so that each Ca v type corresponds to many different combinations of subunits.
- Ca v 2 currents are found almost exclusively in the central and peripheral nervous system and in neuroendocrine cells and constitute the predominant forms of presynaptic voltage-gated calcium current. Presynaptic action potentials cause channel opening and neurotransmitter release is steeply dependent upon the subsequent calcium entry.
- Ca v 2 channels play a central role in mediating neurotransmitter release.
- N-type calcium channels (Ca v 2.2) contain high-affinity binding sites for the peptide toxins ⁇ -conotoxin-MVIIC and ⁇ -conotoxin-GVIA, respectively, and these peptides have been used to determine the distribution and function of each channel type.
- Ca v 2.2 is highly expressed at the presynaptic nerve terminals of neurons from the dorsal root ganglion and neurons of lamina I and Il of the dorsal horn (Westenbroek R E, Hoskins L, Catterall W A. 1998. J Neurosci 18: 6319-30; Cizkova D, Marsala J, Lukacova N, Marsala M, Jergova S, et al. 2002. Exp Brain Res 147: 456-63). Ca v 2.2 channels are also found in presynaptic terminals between second and third order interneurons in the spinal cord. Both sites of neurotransmission are very important in relaying pain information to the brain.
- Pain, particularly neuropathic and intractable pain is a large unmet medical need. Millions of individuals suffer from severe pain that is not well controlled by current therapeutics.
- the current drugs used to treat pain include non-steroidal antiinflammatory drugs (NSAIDs), cyclo-oxygenase 2 (COX-2) inhibitors, opioids, tricyclic antidepressants, and anticonvulsants.
- NSAIDs non-steroidal antiinflammatory drugs
- COX-2 cyclo-oxygenase 2
- opioids opioids
- tricyclic antidepressants tricyclic antidepressants
- anticonvulsants anticonvulsants.
- Neuropathic pain has been particularly difficult to treat as it does not respond well to opioids until high doses are reached.
- Gabapentin is currently the most widely used therapeutic for the treatment of neuropathic pain, although additional therapeutic agents are desirable, particularly those with broader ranges of activities.
- the T-type calcium channel (Ca v 3.1 , Ca v 3.2, and Ca v 3.3) may become over- expressed due to genetic or environmental causes, such as epilepsy (Tsakiridou, E. et al., J. Neurosci. 1995, 15, 3110-3117), high blood pressure (Self, D. A. et al., J. Vacs. Res. 1994, 31 , 359-366), ventricular hypertrophy (Nuss, H. B. et al., Circ. Res. 1995, 73, 777-7825), pain (Shin, H. S. et al., Science 2003, 302, 117-119), and angina pectoris (Van der Vring, J. A.
- a representative drug for blocking the T-type calcium channel is mibefradil of Hoffman La Roche Ltd.
- the drug was found to be effective in treating high blood pressure, angina pectoris and cerebral apoplexy. It was approved for treating high blood pressure in May, 1997.
- a side effect caused by a drug-drug interaction due to inhibition of CYP 3A4 hepatic enzyme was discovered. As such, the drug was withdrawn from the market in June, 1999.
- Dihydropyridine (DHP) antagonists of L-type calcium channels are widely used therapeutics in the treatment of hypertension, angina, arrhythmias, congestive heart failure, cardiomyopathy, atheriosclerosis, and cerebral and peripheral vascular disorders (Janis and Triggle, 1990 CRC Press, Cleveland).
- DHPs Dihydropyridine
- some of the DHPs are sensitive to T-type channel activity.
- N. Akaike, H. Kanaide, T, Kuga, M, Nakamura, J. Sadoshima and Tomoike “Low Voltage Activated Calcium Current in rat Aorta Smooth Muscle Cells In Primary Cultur” J Physiol. 416, 141-160, (1989).
- dihydropyridines e.g., amlodipine, felodipine, nifedipine, nicardipine, isradipine, nimodipine
- benzothiazepines e.g., diltiazem
- phenylalkylamines e.g., verapamil
- diarylaminopropylamine ether e.g., bepridil
- the present invention relates to a compound of formula (I)
- R 1 and R 2 are each independently -H, -OH, halo, C 1 -C 6 alkyl, C r C 6 alkoxy, -CF 3 , substituted
- R 3 and R 4 are each independently -H or Ci-C 6 alkyl or R 3 and R 4 taken together with the carbon atom to which they are attached form C 3 -C 6 cycloalkyl, or cycloheteroalkyl, provided that if one of R 3 and R 4 is -H, then the other is C 1 -C 6 alkyl;
- R 5 is -H, C 1 -C 6 alkyl, d-C 6 alkoxy, -(CH 2 ) q -C(0)0-W, wherein W is -H or C r C 6 alkyl and q is 1-6;
- G 1 is methylene or ethylene
- G 2 is C(R 6 ) or N, wherein R 6 is -H, -OH or Ci-C 6 alkyl; Y is -CH 2 -, -CH 2 CH 2 -, -CH 2 CH 2 CH 2 -, -O-, -C(O)-, -C(O)CH 2 -, -S-, -S(O)-, -S(O) 2 -
- Ar 1 is a radical of the formulae
- R P1 , R P2 , R P3 , R P4 , R P5 , R P6 , R P7 , R P8 , R N1 , R N2 , R N3 , and R Z1 are each independently -H, -OH, C 1 -C 6 alkyl, CrC 6 alkoxy, halo, -CN, -CF 3 , or -NR 8 R 9 , wherein R 8 and R 9 are each independently -H or C 1 -C 6 alkyl; R M1 , R M2 , R B1 , R B2 , R B3 , R 84 , R B5 , R B6 R xi R X2 R x3 R x4 R ⁇ i R Y2 R Y3 R Y4 R Y5 an(J R Y6 are each j ndepenc
- ently -H or CrC 6 alkyl; X 1 and X 2 are independently CH or N; X
- the present invention also relates to a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the present invention further relates to a method of blocking calcium channels, the method comprising administering to a patient in need of calcium channel blocking a therapeutically effective amount of a compound of formula (I) to block calcium channels.
- Another embodiment of the invention relates to a method of treating pain in a patient in need thereof comprising administering to said patient a therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- Another embodiment of the invention relates to a method of causing vasodilation in a patient in need thereof comprising administering to said patient a therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- a further embodiment of the invention relates to a method of treating a disease selected from hypertension, congestive heart failure, stroke, ischaemic heart disease, and angina pectoris comprising administering to a patient in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- Another aspect of this invention is directed to methods of reducing myocardial tissue damage (e.g., substantially preventing tissue damage, inducing tissue protection) during surgery (e.g., coronary artery bypass grafting (CABG) surgeries, vascular surgeries, percutaneous transluminal coronary angioplasty (PTCA) or any percutaneous transluminal coronary intervention (PTCI), organ transplantation, or other non-cardiac surgeries) comprising administering to a mammal (e.g., a female or male human) a therapeutically effective amount of a compound of formula (I) or (II), or a pharmaceutically acceptable salt of said compound.
- a mammal e.g., a female or male human
- halogen refers to a fluorine atom, chlorine atom, bromine atom, or iodine atom.
- Ci-C 6 alkyl refers to a branched or straight chained alkyl radical containing from 1 to 6 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec butyl, t-butyl, pentyl, hexyl, and the like.
- d-C 4 alkyl refers to a branched or straight chained alkyl radical containing from 1 to 4 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, t-butyl, and the like.
- substituted CrC 6 alkyl refers to a CrC 6 alkyl substituted with from 1 to 3 substituents selected from halogen and CrC 4 alkoxy. Included within this definition is -CH 2 F, -CHF 2 , -CF 3 ,
- CrC 6 alkoxy refers to a straight or branched alkoxy group containing from 1 to 6 carbon atoms, such as methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy, sec-butoxy, t-butoxy, pentoxy, hexoxy, and the like.
- CrC 4 alkoxy refers to a straight or branched alkoxy group containing from 1 to 4 carbon atoms, such as methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy, sec-butoxy, t-butoxy, and the like.
- substituted Ci-C 6 alkoxy refers to a CrC 6 alkoxy substituted with from 1 to 3 substituents selected from halogen and CrC 4 alkyl.
- C 3 -C 6 cycloalkyl refers to a cyclic alkyl radical containing from 3 to 6 carbon atoms, such as cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl,
- CrC 4 haloalkyl refers to a CrC 4 alkyl is substituted with from 1 to 3 halo atoms per monovalent carbon and 1 to 2 halo atoms per divalent carbons, such as -CF 3 , -CH 2 F, -CHF 2 , -CF 3 , -CHFCH 3 , -CH 2 CF 3 , -CH 2 CH 2 CF 3 , -CH 2 CH 2 CH 2 CF 3 , -CH 2 Br, -CHBr 2 , -CH 2 CHBr 2 , - CCI 3 , -CHCI 2 , -CH 2 CI, -CCI 3 , and the like.
- cycloheteroalkyl refers to a cyclic alkyl moiety containing five carbons and one ring atom of -O- or -N(R H1 ), wherein R H1 is -H or d-C 6 alkyl as is further depicted in the formula (III) compounds disclosed herein. Examples of a
- cycloheteroalkyl include tetrahydro-2H-pyran-4-yl, tetrahydropyridin-4-yl, N- methylpiperidin-4-yl
- R 4 refers to a carboxylic acid (R 4 is H) or an ester (R 4 is CrC 6 alkyl or C 3 -C 6 cycloalkyl) of the formula: wherein R 4 is H, C r C 6 alkyl, or C 3 -C 6 cycloalkyl.
- R 5 and R 6 are each independently -H or C 1 -C 6 alkyl.
- R 7 is -H or CrC 6 alkyl.
- C(R 8 ) refers to a moiety of the formula: R° wherein R 8 is -H, -OH, C r C 6 alkyl.
- R 9 is -H or CrC 4 alkyl.
- the designation "-NHS(O) 2 -" refers to a sulfonamide of the formula: o
- R 10 and R 11 are each independently -H or CrC 6 alkyl.
- the designation refers to a naphthalene (when X 1 and X 2 are both CH), a quinoline (when one of X 1 and X 2 is N and the other is CH), or a quinoxaline (when X 1 and X 2 are both N) and it is understood that the radical is attached at any of the 1 through 8 positions when both X 1 and X 2 are CH, any of positions 2 through 8 when X 1 is N, and any of positions 2, 3 and 5-8 when both X 1 and X 2 are N; it is further understood that when the radical is attached at any given position, the substituents represented by R N1 , R N2 , and R N3 can be attached at any of the other non-nitrogen positions, for example, if the radical is attached at the 1 -position and X 1 and X 2 are both CH, the substituents represented by R N1 , R N2 , or R N3 can be attached at any of the 2-, 3-, 4-, 5-, 6-, 7-, or 8-positions
- R Z1 refers to a pyridazine and it is understood that the radical is attached to any of the 3-, A-, 5-, or 6-positions; it is further understood that when the radical is attached at any given position, the substituent represented by R Z1 can be attached at any of the other non- nitrogen positions, for example, if the radical is attached at the 3-position, the substituent represented by R Z1 can be attached at any of the A-, 5-, or 6-positions.
- the designation refers to a pyrazine and it is understood that the radical is attached to any of the 2-, 3-, 5-, or 6-positions; it is further understood that when the radical is attached at any given position, the substituent represented by R P7 can be attached at any of the other non- nitrogen positions, for example, if the radical is attached at the 2-position, the substituent represented by R P7 can be attached at any of the 3-, 5-, or 6-positions.
- R X2 and R X3 refers to an isoxazole and it is understood that the radical is attached to any of the 3-, A- , or 5-positions; it is further understood that when the radical is attached at any given position, the substituents represented by R X2 and R X3 can be attached at any of the non- nitrogen or non-oxygen positions, for example, if the radical is attached at the 3- position, then the substituents represented by R X2 and R X3 can be attached at either of the 4- or 5- positions.
- the specific stereoisomers can be prepared by stereospecific synthesis using enantiomerically pure or enantiomerically enriched starting materials.
- the specific stereoisomers of either starting materials or products can be resolved and recovered by techniques known in the art, such as chromatography on chiral stationary phases, enzymatic resolution, or fractional recrystallization of addition salts formed by reagents used for that purpose.
- Useful methods of resolving and recovering specific stereoisomers are know in the art and described in Stereochemistry of Organic Compounds, E. L. ENeI and S. H. Wilen, Wiley (1994) and Enantiomers, Racemates, and Resolutions, J. Jacques, A. Collet, and S. H. Wilen, Wiley (1981).
- suitable solvent refers to any solvent, or mixture of solvents, inert to the ongoing reaction that sufficiently solubilizes the reactions to afford a medium within which to effect the desired reaction.
- compositions of formula I include the acid addition and base salts (including disalts) thereof.
- Suitable acid addition salts are formed from acids which form non-toxic salts. Examples include the acetate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, saccharate, stearate, succinate, tartrate, tosylate and trifluor
- Suitable base salts are formed from bases which form non-toxic salts. Examples include the aluminium, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and zinc salts.
- a pharmaceutically acceptable salt of a compound of formula (I) may be readily prepared by mixing together solutions of the compound of formula (I) and the desired acid or base, as appropriate.
- the salt may precipitate from solution and be collected by filtration or may be recovered by evaporation of the solvent.
- the degree of ionisation in the salt may vary from completely ionised to almost non-ionised.
- Compounds of formula (I) containing one or more asymmetric carbon atoms can exist as two or more stereoisomers. Where a compound of formula (I) contains an alkenyl or alkenylene group, geometric cis/trans (or ZJE) isomers are possible. Where the compound contains, for example, a keto or oxime group or an aromatic moiety, tautomeric isomerism ('tautomerism') can occur. It follows that a single compound may exhibit more than one type of isomerism.
- (I) including compounds exhibiting more than one type of isomerism, and mixtures of one or more thereof. Also included are acid addition or base salts wherein the counterion is optically active, for example, D-lactate or L-lysine, or racemic, for example,
- Cisltrans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallisation.
- racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of formula (I) contains an acidic or basic moiety, an acid or base such as tartaric acid or 1-phenylethylamine.
- a suitable optically active compound for example, an alcohol, or, in the case where the compound of formula (I) contains an acidic or basic moiety, an acid or base such as tartaric acid or 1-phenylethylamine.
- the resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to a skilled person.
- Chiral compounds of the invention may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% isopropanol, typically from 2 to 20%, and from 0 to
- the present invention includes all pharmaceutically acceptable isotopically- labelled compounds of formula (I) wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes suitable for inclusion in the compounds of the invention include isotopes of hydrogen, such as 2 H and 3 H, carbon, such as 11 C, 13 C and 14 C, chlorine, such as 36 CI, fluorine, such as 18 F, iodine, such as 123 I and 125 I, nitrogen, such as 13 N and 15 N, oxygen, such as 15 O, 17 O and 18 O, phosphorus, such as 32 P, and sulphur, such as 35 S.
- isotopically-labelled compounds of formula (I), for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies.
- the radioactive isotopes tritium, i.e. 3 H, and carbon-14, i.e. 14 C, are particularly useful for this purpose in view of their ease of incorporation and ready means of detection.
- substitution with heavier isotopes such as deuterium, i.e. 2 H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances.
- Isotopically-labeled compounds of formula (I) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagents in place of the non-labeled reagent previously employed.
- the compounds of the present invention may be administered as prodrugs.
- prodrugs certain derivatives of compounds of formula (I) which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into compounds of formula (I) having the desired activity, for example, by hydrolytic cleavage.
- Such derivatives are referred to as 'prodrugs'.
- Further information on the use of prodrugs may be found in 'Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T Higuchi and W Stella) and 'Bioreversible Carriers in Drug Design 1 , Pergamon Press, 1987 (ed. E B Roche, American Pharmaceutical Association).
- Prodrugs can, for example, be produced by replacing appropriate functionalities present in the compounds of formula (I) with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in "Design of Prodrugs” by H Bundgaard (Elsevier, 1985).
- prodrugs include: (i) where the compound of formula (I) contains a carboxylic acid functionality
- each independently -H, -CF 3 , halo, or CrC 4 alkyl independently -H, fluoro, chloro, -CF 3 , or C r C 4 alkyl; or
- R 3 and R 4 are: (a) taken together with the carbon atom to which they are attached form C3-C6 cycloalkyl;
- R 1 and R 2 are each independently -H, -OH, halo, C r C 6 alkyl, C r C 6 alkoxy, -CF 3 , or substituted CrC 6 alkyl;
- G 2 is C(R 6 ) or N, wherein R 6 is -H, -OH or C 1 -C 6 alkyl;
- Y is -CH 2 -, -CH 2 CH 2 -, -CH 2 CH 2 CH 2 -, -0-, -C(O)-, -C(O)CH 2 -, -S-, -S(O)-, -S(O) 2 -, -NH-, -NHC(O)-,
- R 8 and R 9 are each independently -H or C 1 -C 6 alkyl;
- R M1 , R M2 , R B1 , R B2 , R B3 , R 84 , R B5 , R B6 , R X1 , R X2 , R X3 , R X4 , R Y1 , R Y2 , R Y3 , R Y4 , R Y5 , and R Y6 are each independently -H or C 1 -C 6 alkyl;
- X 1 and X 2 are independently CH or N;
- X 3 , X 4 , and X 5 are each independently NH, O, or S;
- X 6 is CH 2 or O;
- Q is substituted C 1 -C 6 alkyl, pheny
- G 2 is: (a) C(R 6 ), wherein R 6 is -H, -OH, or C 1 -C 6 alkyl;
- further preferred embodiments of the invention can be obtained by combining (1)(a) and (2)(a); (1)(a) and (2)(b); (1)(b) and (2)(a); (1)(b) and (2)(b); (1 )(C) and (2)(a); (1)(c) and (2)(b); (1)(a) and (2)(c); (1)(c) and (2)(c); (2)(a) and (3)(a); (2)(b) and (3)(a); (2)(b) and (3)(a); (1)(b) and (4)(a); (1)(b) and (5)(a); (1)(b), (4)(b) and (5)(b); (1)(b), (3)(a), (4)(b) and (5)(a); (3)(b), (4)(b), and (5)(a); (2)(a), (3)(a), (4)(b), and (5)(a); (2)(a), (3)(a), (4)(b), and (5)(a); (2)(b), (3)(a), (4)(b), and (5)(a); (2)(a), (3)(a), (4)(b), and
- R 1 , R 2 , G 2 , Y, Ar 1 , and n in each of (HA), (MB), (MC), and (MD) are as defined in formula (M).
- Compounds of formula (MA) are those compounds of formula (M) where m is 1.
- Compounds of formula (MB) are those compounds of formula (M) where m is 2.
- Compounds of formula (MC) are those compounds of formula (M) where m is 3.
- Compounds of formula (MD) are those compounds of formula (II) where m is 4.
- Preferred embodiments of compounds of formulae (MA), (MB), (MC), or (ND), or stereoisomers or pharmaceutically acceptable salts thereof are given below: (1) Compounds wherein R 1 and R 2 are: a.
- G 2 is: a. C(R 6 ), wherein R 6 is -H, -OH, or C 1 -C 6 alkyl; or b. N;
- further preferred embodiments of the invention can be obtained by combining (1)(a) and (2)(a); (1)(a) and (2)(b); (1)(b) and (2)(a); (1)(b) and (2)(b); (1)(c) and (2)(a); (1)(c) and (2)(b); (2)(a) and (3); (2)(b) and (3); (1)(b) and (3); (1)(b) and (4)(a); (1)(b), (4)(a) and (5); (1)(b), (4)(b) and (5); (1)(b), (3), (4)(b) and (5); (3), (4)(b), and (5); (2)(a), (3), (4)(b), and (5); (2)(b), (3), (4)(b), and (5); (1)(b), (2)(a), (3), (4)(b), and (5); (1)(b), (2)(a), (3), (4)(b), and (5); (1)(b), (2)(a), (3), (4)(b), and (5); (1)(b), (2)(a), (3), (4)(b), and
- R 1 and R 2 are each independently -H, -OH, halo, C r C 6 alkyl, C r C 6 alkoxy, -CF 3 , or substituted CrC 6 alkyl;
- G 2 is C(R 6 ) or N, wherein R 6 is -H, -OH or C 1 -C 6 alkyl;
- G 3 is -O- or -N(R H1 ), wherein R H1 is -H or C 1 -C 6 alkyl;
- Y is -CH 2 -, -CH 2 CH 2 -, -CH 2 CH 2 CH 2 -, -0-, -C(O)-, -C(O)CH 2 -, -S-, -S(O)-, -S(O) 2 -, -NH-, -NHC(O)-,
- Ar 1 is a radical of the formulae wherein R P1 , R P2 , R P3 , R P4 , R P5 , R P6 , R P7 , R P8 , R N1 , R N2 , R N3 , and R Z1 are each independently -H, -OH, CrC 6 alkyl, Ci-C 6 alkoxy, halo, -CN, -CF 3 , or -NR 8 R 9 , wherein
- R 8 and R 9 are each independently -H or C r C 6 alkyl; R M1 , R M2 , R B1 , R B2 , R B3 , R 84 , R B5 ,
- R B6 R xi R x2 R x3 R x4 R YI R Y2 R Y3 R Y4 R Y5 and R Y6 are egch independently -H or
- X 1 and X 2 are independently CH or N; X 3 , X 4 , and X 5 are each independently NH, O, or S; X 6 is CH 2 or O; Q is substituted C r C 6 alkyl, phenyl, napthyl, 2-pyridyl, or 3-pyridyl; and n is O or 1 ; provided that when Y is O, S, NH, NHC(O), NHS(O) 2 , NHC(O)CH(R 7 ), or NHS(O) 2 , then G 2 is CH.
- R 1 and R 2 are: a. each independently -H -CF 3, halo, or d-C 4 alkyl; b. each independently -H, fluoro, chloro, or CrC 4 alkyl; or c. one of R 1 and R 2 is -H and the other is fluoro;
- G 2 is: a. C(R 6 ), wherein R 6 is -H, -OH, or C r C 6 alkyl; or b. N;
- R P1 , R P2 , R P3 , R P4 , R P5 , R P6 , R Z1 , R P7 and R P8 are each independently -H, halo, CrC 4 alkyl, Ci-C 4 alkoxy, or -CF 3 ;
- step 1 the phenyl carbonitrile of formula (1) is treated with hydroxylamine hydrochloride to provide the oxime of formula (1A).
- the phenyl carbonitrile (1) is dissolved in a suitable solvent, such as ethanol, or an ethanokH ⁇ O mixture, and contacted with hydroxylamine hydrochloride and a suitable base such as potassium carbonate, sodium ethoxide, sodium hydroxide, or mixtures of the base and water.
- a suitable solvent such as ethanol, or an ethanokH ⁇ O mixture
- a suitable base such as potassium carbonate, sodium ethoxide, sodium hydroxide, or mixtures of the base and water.
- the mixture is refluxed and stirred until analysis indicates that the reaction is complete.
- the oxime (1A) can be purified by techniques well known in the art, such as extraction, evaporation, trituration, chromatography, and recrystallization.
- step 2 the oxime (1A) is reacted with chloroacetyl chloride to provide the compound of formula (1B).
- the oxime (1A) is dissolved in a suitable organic solvent such as acetone, a suitable base, such as potassium carbonate, is added and the mixture is cooled.
- Chloroacetyl chloride is then slowly added over a period of time ranging from about 5 to about 60 minutes. The mixture is then warmed to room temperature and stirred until analysis indicates that the reaction is complete.
- the compound of formula (1B) can be isolated and purified by techniques well known in the art, such as extraction, evaporation, trituration, chromatography, and recrystallization.
- step 3 the compound of formula (1B) is refluxed in a suitable organic solvent to provide the 5-chloromethyl-3-benzyl-[1,2,3]oxadiazole (1 C).
- the compound of formula (1B) is refluxed with a Dean-Stark apparatus in a suitable organic solvent such as toluene until analysis indicates that the reaction is complete.
- the solution is then cooled and 5-chloromethyl-3-benzyl-[1 ,2,3]oxadiazole (1C) may be isolated and purified by techniques well known in the art, such as extraction, evaporation, trituration, chromatography, and recrystallization.
- step 4 the benzyl oxadiazole of formula (3) is prepared by coupling the substituted cyclic amine of formula (2) with 5-chloromethyl-3-benzyl- [1 ,2,3]oxadiazole (1C).
- substituted amine (2) and 5-chloromethyl-3- benzyl-[1 ,2,3]oxadiazole (1C) are dissolved in a suitable organic solvent such as ethanol in the presence of a base, such as N-methylmorpholine, sodium carbonate, triethylamine, N,N-diisopropylethylamine, potassium carbonate or sodium carbonate and refluxed until analysis indicates that the reaction is complete.
- a base such as N-methylmorpholine, sodium carbonate, triethylamine, N,N-diisopropylethylamine, potassium carbonate or sodium carbonate and refluxed until analysis indicates that the reaction is complete.
- the benzyl oxadiazole of formula (3) may be isolated and purified
- phenyl carbonitriles of formula (1) are commercially available or are well- known in the art such as 1-phenyl-cyclopropanecarbonitrile, 1-(4-methylphenyl)-1- cyclopropanecarbonitrile, 2-phenylbutyronitrile, 1-phenylcyclobutanecarbonitrile, p- chloro-alpha-methylphenyl acetonitrile, 1-(4-fluorophenyl)cyclopentanecarbonitrile, 1- phenylcyclohexane-1-carbonitrile, and the like.
- phenyl carbonitriles of formula (1) may be synthesized by techniques well known and appreciated by those of ordinary skill in the art.
- 1-(4-fluoro-phenyl)-cyclopropanecarbonitrile may be prepared by reacting (4-fluoro-phenyl)-acetonitrile with bromochloroethane in an aqueous basic solution in the presence of triethylammonium chloride as set forth in the examples herein.
- substituted cyclic amines of formula (2) are commercially available or are well-known in the art such as 4-(phenylmethyl)-1-piperidine ethanamine, 4-(phenylmethyl)-1-piperazine ethanamine, 4-(2-methoxyphenyl)-1- piperazine ethananamine, 4-(4-methoxyphenyl)-1-piperazine ethananamine 4-(4- chlorophenyl)-1-piperazine ethananamine, 4-[(3-methoxyphenylmethyl))-1-piperazine ethananamine, 4-(5-chloro-2-methylphenyl)-1-piperazine ethananamine, 4-[4- (trifluoromethyl)-2-pyrimidyl]-1-piperazine ethananamine,
- substituted amines of formula (2) may be synthesized by the techniques set forth in Scheme A1.
- step 1 the cyclic amine of formula (2A) is treated with chloroacetonitrile in the presence of potassium carbonate to' provide the cyclic amine carbonitrile of formula (2B).
- the cyclic amine of formula (2A) is dissolved in a suitable solvent such as acetonitrile, in the presence of a base such as potassium carbonate, at room temperature. Chloroacetonitrile is then added slowly over a period of time ranging from 25 to 60 minutes. The mixture is then heated to a temperature of about 95 0 C, and stirred until analysis indicates that the reaction is complete.
- the compound of formula (2B) may be isolated and purified by techniques well known in the art such as extraction, evaporation, trituration, chromatography, and recrystallization.
- step 2 the cyclic amine carbonitrile (2B) is treated with borane dimethyl sulfide complex in THF to provide the substituted amine of formula (2).
- the cyclic amine carbonitrile (2B) is dissolved in a suitable solvent such as THF.
- a 2M solution of borane dimethyl sulfide complex is added dropwise over a period of time ranging from 5 to 60 minutes. The mixture is then heated to a temperature of about 65 0 C, and stirred until analysis indicates that the reaction is complete.
- the compound of formula (2) may be isolated and purified by techniques well known in the art such as extraction, evaporation, trituration, chromatography, and recrystallization.
- substituted cyclic amines of formula (2A) are commercially available or are well-known in the art such as 1-phenylpiperazine, 1-(3- (trifluoromethyl)phenyl)piperazine, 1-(2,4-dichlorophenyl)piperazine, 1-(4-chloro-2- fluorophenyl)piperazine, 1-(p-tolyl)piperazine, 1-(3,4-difluorophenyl) piperazine, and the like.
- the substituted cyclic amines of formula (2A) may be synthesized by techniques well known in the art.
- 1-(3-methoxyphenyl)piperazine may be prepared by reacting bis(2-chloroethyl)amine hydrochloride, 3-methoxyaniline and diethylene glycol monomethyl ether at 150 0 C.
- Scheme B provides a synthetic process for making benzyl oxadiazole piperazine aryl compounds of formula (8) which represent compounds of formula (I) wherein n is 1 , G 2 is N, Pg is a suitable amino protecting group, such as t-Boc, and all of the remaining substituents are as defined in formula (I).
- step 1 the N-protected benzyl oxadiazole piperazine of formula (5) is prepared by coupling the N-protected ethyleneaminepiperizine of formula (4) with 5- chloromethyl-3-benzyl-[1,2,3]oxadiazole (1C) according to the procedure set forth in Scheme A 1 step 4.
- step 3 the benzyl oxadiazole piperazine aryl of formula (8) is prepared by coupling the benzyl oxadiazole piperazine of formula (6) with the aryl chloride of formula (7).
- benzyl oxadiazole piperazine (6) is contacted with Ar 1 -Y-Cl (7) in a suitable solvent such as methylene chloride in the presence of a suitable base such as triethylamine, diisopropylethylamine, N-methylmorpholine, Huniq's base, sodium carbonate, sodium bicarbonate, or potassium carbonate.
- a suitable solvent such as methylene chloride
- a suitable base such as triethylamine, diisopropylethylamine, N-methylmorpholine, Huniq's base, sodium carbonate, sodium bicarbonate, or potassium carbonate.
- the reaction is generally carried out at temperatures ranging from about ambient temperature to about 100°C for a period of time until analysis indicates that the reaction is complete.
- the benzyl oxadiazole piperazine aryl or formula (8) may be isolated and purified by techniques well known in the art, such as extraction, evaporation, trituration, chromatography, and recrystallization.
- the N-protected ethyleneaminepiperizines of formula (4) may be synthesized by techniques well known in the art.
- Boc-piperazine is reacted with bromoacetonitrile, in the presence of a base such as potassium carbonate, in a suitable solvent, such as acetonitrile, at temperatures varying from 30 to 65 0 C, for a period of time between 10-24 hours, under a nitrogen atmosphere, and stirred until analysis indicates that the reaction is complete.
- a base such as potassium carbonate
- a suitable solvent such as acetonitrile
- the nitrile product is then reduced with an anhydride, for example lithium aluminum hydride, in a suitable solvent, such as THF, at O 0 C, for 1 hour, and at room temperature for 2 to 3 hours, and stirred until analysis indicates that the reaction is complete.
- anhydride for example lithium aluminum hydride
- THF a suitable solvent
- the compound of formula (4) may be isolated and purified by techniques well known in the art such as extraction, evaporation, trituration, chromatography, and recrystallization
- aryl chlorides of formula (7) are commercially available or are well-known in the art such as benzoyl chloride, 4-methoxybenzoyl chloride, 2-trifluoromethylbenzoyl chloride, 2,4-dichlorobenzoyl chloride, benzenesulfonyl chloride, 3- trifluoromethylbenzenesulfonyl chloride, 3-fluorobenzenesulfonlyl chloride, and the like.
- the aryl chlorides of formula (7) may be synthesized by techniques well known in the art. For example, 2,5-dimethoxybenzenesulfonyl chloride may be prepared from the corresponding sulfonic acid by using thionyl chloride in dimethylformamide.
- Scheme C provides a synthetic process for making benzyl oxadiazole piperidine aryl compounds of formula (13) which represent compounds of formula (I) wherein n is 1, G 2 is CH, Y is -NHC(O)-, -NHS(O) 2 -, or -NHC(O)CH(R 7 )-, and Pg is a suitable amino protecting group, such as t- Boc and all of the remaining substituents are as defined in formula (I).
- step 1 the N-protected benzyl oxadiazole piperidine of formula (10) is prepared by coupling the N-protected piperidine of formula (9) with 5- chloromethyl-3-benzyl-[1,2,3]oxadiazole (1C) according to the procedure set forth in Scheme A, step 4.
- step 2 the benzyl oxadiazole piperidine of formula (11) is prepared by deprotecting the N-protected benzyl oxadiazole piperazine of formula (10) according to procedures set forth in Scheme B, step 2.
- the benzyl oxadiazole piperidine aryl of formula (13) is prepared by coupling the benzyl oxadiazole piperidine of formula (11) with the aryl chloride of formula (12) according to the procedures set forth in Scheme B, step 3.
- the N-protected ethyleneaminepiperidines of formula (9) may be synthesized by techniques well known in the art.
- Boc-4-amino-piperidine is reacted with bromoacetonitrile, in the presence of a base such as potassium carbonate, in a suitable solvent, such as acetonitrile, at temperatures varying from 30 to 65 0 C, for a period of time between 10-24 hours, under a nitrogen atmosphere, and stirred until analysis indicates that the reaction is complete.
- a base such as potassium carbonate
- acetonitrile acetonitrile
- the nitrile product is then reduced with an anhydride, for example lithium aluminum hydride, in a suitable solvent, such as THF, at O 0 C, for 1 hour, and at room temperature for 2 to 3 hours, and stirred until analysis indicates that the reaction is complete.
- anhydride for example lithium aluminum hydride
- a suitable solvent such as THF
- the compound of formula (9) may be isolated and purified by techniques well known in the art such as extraction, evaporation, trituration, chromatography, and recrystallization
- aryl chlorides of formula (12) are commercially available or are well-known in the art such as benzoyl chloride, 4-methoxybenzoyl chloride, 2-trifluoromethylbenzoyl chloride, 2,4-dichlorobenzoyl chloride, benzenesulfonyl chloride, 3- trifluoromethylbenzenesulfonyl chloride, 3-fluorobenzenesulfonlyl chloride, and the like.
- the aryl chlorides of formula (12) may be synthesized by techniques well known in the art. For example, 2,5-dimethoxybenzenesulfonyl chloride may be prepared from the corresponding sulfonic acid by using thionyl chloride in dimethylformamide.
- Scheme D provides a synthetic process for making benzyl oxadiazole piperidine aryl compounds of formula (16) which represent compounds of formula (I) wherein R 5 is CrC 6 alkyl, C r C 6 alkoxy, or CrC 6 -C(O)O-W, wherein W is -H or C r C 6 alkyl; and all of the remaining substituents are as defined in formula (I).
- the compound of formula (14) depicts compounds of formula (I) where R 5 is -H.
- the substituent R 5a is used in compounds (15) and (16) below to depict compounds of formula (I) when R 5 is not -H.
- the aryl oxadiazole of formula (14) is alkylated with an appropriate alkyl bromide (15) to provide alkylated aryl oxadiazole of formula (16).
- An appropriate alkylating agent of formula (15) is one in which R 5a is as desired in the final product of formula (I).
- the aryl oxadiazole of formula (14) is contacted with 2.0 to 3.0 molar equivalents of alkyl bromide (15).
- the reaction is carried out in the presence of a suitable base such as sodium bis(trimethylsilyl)amide or lithium diisopropylamide and in the presence of triethyl borane.
- the reaction is carried out in a suitable solvent such as tetrahydrofuran.
- the reaction is generally carried out at temperatures ranging from about -78°C to about 0 0 C. Generally the reactions require from about 1 to 72 hours.
- the product can be isolated and purified by techniques well known in the art such as extraction, evaporation, trituration, distillation, chromatography, and recrystallization.
- Scheme E provides a synthetic process for making benzyl oxadiazole piperidine aryl compounds of formula (18) which represent compounds of formula (I) wherein G 2 is CH, n is 1 and Y is -CH 2 -,
- the benzyl oxadiazole piperidine of formula (18) is prepared by coupling the aryl piperidine of formula (17) with 5-chloromethyl-3-benzyl- [1 ,2,3]oxadiazole (1C) according to the procedure set forth in Scheme A, step 4.
- the compound of formula (22) is then treated with bromoacetonitrile followed by lithium aluminum hydride, as described for the synthesis of compound (4), as shown above.
- 4-piperidineethanol (23) is N-protected according to standard nitrogen protecting techniques.
- the selection and use of suitable amine protecting groups is described in Protecting Groups in Organic Synthesis by T. Greene and is well known and appreciated in the art.
- the 4-piperidineethanol (23) may be N- protected using (Boc) 2 O in the presence of a tertiary amine, such as triethylamine and a suitable organic solvent such as chloroform to provide the Boc-protected 4- piperidineethanol (24).
- the N-protected aldehyde of formula (25) is obtained by oxidizing the Boc-protected 4- piperidineethanol (24) with a suitable oxidizing agent such as oxalyl chloride in the presence of a tertiary amine such as triethylamine and DMSO in a suitable solvent, such as dichloromethane.
- a suitable oxidizing agent such as oxalyl chloride
- a tertiary amine such as triethylamine and DMSO
- a suitable solvent such as dichloromethane.
- the N-protected aldehyde of formula (25) is then reacted with an aryl Grignard reactant in a suitable organic solvent such as tetrahydrofuran under standard Grignard conditions to provide the aryl- substituted N-protected piperidine of formula (26).
- the hydroxy moiety is then removed from the aryl-substituted N-protected piperidine of formula (26) using a suitable reducing agent such as lithium aluminum hydride to provide the de-hydroxylated N-protected piperidine of formula (27).
- the aryl-substituted piperidine of formula (28) is obtained by deprotecting the de-hydroxylated N-protected piperidine of formula (27).
- the removal of amine protecting groups is well known and appreciated in the art and is described in Protecting Groups in Organic Synthesis by T. Greene.
- the product may be isolated and purified according to art-known techniques such as extraction, evaporation, chromatography, and recrystallization.
- the compound of formula (28) is then treated with bromoacetonitrile followed by lithium aluminum hydride, as described for the synthesis of compound (4), as shown above.
- 4-piperidineethanol (23) is N-protected according to standard nitrogen protecting techniques.
- the selection and use of suitable amine protecting groups is described in Protecting Groups in Organic Synthesis by T. Greene and is well known and appreciated in the art.
- the 4-piperidineethanol (23) may be N- protected using (BoC) 2 O in the presentee of a tertiary amine, such as triethylamine and a suitable organic solvent such as chloroform to provide the Boc-protected 4- piperidineethanol (24).
- the phosphorus ylid (29) is obtained by treating the Boc- protected 4-piperidineethanol (24) with iodine in the presence of triphenylphosphine in a suitable solvent such as diethylether and acetonitrile. The resulting iodo intermediate is then treated in situ with triphenylphosphine in a suitable solvent such as acetonitrile, at reflux, to give the phosphorus ylid (29).
- Scheme F provides a synthetic process for making benzyl oxadiazole piperidine aryl compounds of formula (35) which represent compounds of formula (I) wherein R 3 and R 4 taken together with the carbon atom to which they are attached form cycloheteroaryl, more specifically compounds of formula (III) where G 3 is -O-; and all of the remaining substituents are as defined in formula (I).
- step 1 the phenyl tetrahydro-pyran-4-carbonitrile (32) is prepared by reacting the phenyl acetonitrile of formula (31) with 1-chloro-2-(2-chloro-ethoxy)- ethane in the presence of a suitable base.
- a suitable base such as NaH at a temperature ranging from about 20 to 25 0 C.
- reaction flask is kept in a water bath at a temperature ranging from about 20 to 25 0 C for a period of time ranging from about 0.5 to 2 hours over which time 1-chloro-2-(2-chloro-ethoxy)-ethane is slowly added.
- the resulting mixture is then stirred vigorously at room temperature for a period of time ranging from about 15 to 24 hours.
- the phenyl tetrahydro-pyran-4-carbonitrile (32) is then isolated and purified according to techniques well known in the art such as extraction, evaporation, and chromatograpy.
- step 2 the oxime of formula (33) is prepared by contacting the phenyl tetrahydro-pyran-4-carbonitrile (32) with hydroxylamine hydrochloride in the presence of a suitable base according to the procedures set forth in Scheme A, step 1.
- step 3 the compound of formula (34) is prepared by reacting the oxime of formula (33) with chloroacetyl chloride according to the procedures set forth in Scheme A, step 2.
- step 4 the compound of formula (35) is prepared by refluxing the compound of formula (34) in a suitable organic solvent according to the procedures set forth in Scheme A, step 3.
- AICI 3 (3.4g, 25.5mmol) is added in portions over 2 min to 1 M LiAIH 4 in diethyl ether (55ml, 55mmol) under vigorous stirring at room temperature under nitrogen. The milky solution is stirred for 30 min given an almost solid mixture. THF (40ml) is added and stirring for 10 min to give a clear solution. Compound 1A (2.4Og 1 10.4mmol) in THF (20ml) is added drop-wise over 5 min. The reaction mixture is stirred another 3 hours at room temperature and then cooled on an ice-bath. The reaction is quenched by drop- wise addition of water (15ml). The mixture is poured into 4M NaOH (150ml).
- Chloroacetyl chloride (20.3g, 0.18moles) is added dropwise to a mixture of 1C (27g, 0.12moles), potassium carbonate (18.3g, 0.132moles) and acetone (250ml) at O 0 C under nitrogen. After the addition, the mixture is allowed to warm to room temperature and stirred overnight. TLC analysis [100% DCM] shows the appearance of a higher R F spot and disappearance of starting material. The mixture is evaporated to dryness, extracted with EtOAc (2x250ml) / water (200ml). The organic layer dried with MgSO 4 , filtered and evaporated to dryness yielding the desired product as an off white solid (Total product yield 32g (88%).
- Preparations 2 - 26 represent the Block 1 intermediates or headpieces in the synthesis of compounds of formula (I).
- the Block 1 intermediates may be prepared according to Example 1E (Preparations 1C to 1 D to 1E), using the indicated starting materials in TABLE 2 below in place of 1-(4-chloro-phenyl)-cyclobutanecarbonitrile in Preparation 1C. TABLE 2
- MeO refers to methoxy and “Me” refers to methyl.
- Preparations 27 - 50 represent the Block 2 intermediates or tailpieces in the synthesis of compounds of formula (I).
- the Block 2 intermediates may be prepared according to Example 1B (Preparations 1A to 1B), using the indicated starting materials in TABLE 3 below in place of 4-(2-methoxy-phenyl)-piperidine in Preparation 1A.
- Examples 2 - 100 set forth in Table 4 below represent compounds of formula (I) which may be prepared by combining the Block 1 intermediates or headpieces of Table 2 with the Block 2 intermediates or tailpieces of Table 3 according to procedures substantially similar to that described for Example 1.
- Step A 4-(4-Chloro-phenyl)-tetrahydro-pyran-4-carbonitrile
- Step B 4-(4-Chloro-phenyl)-N-hydroxy-tetrahydro-pyran-4-carboxamidine
- Step D 5-Chloromethyl-3-[4-(4-chloro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazole
- Example 101 3-[4-(4-Chloro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5- ylmethyl ⁇ - ⁇ 2-[4-(2-methoxy-phenyl)-piperidin-1-yl]-ethyl ⁇ -amine, hydrochloride
- PREPARATIONS 51 - 52 represent the Block 1 intermediates or headpieces in the synthesis of compounds of formula (III).
- the Block 1 intermediates may be prepared according to Example 101 (Steps A, B, C, and D), using the indicated starting materials in TABLE 5 below in place of (4-chloro-phenyl)-acetonitrile in Example 101 , Step A.
- Examples 102 - 109 set forth in Table 6 below represent compounds of formula (I) which may be prepared by combining the Block 1 intermediates or headpieces of Table 5 with the Block 2 intermediates or tailpieces of Table 3 according to procedures substantially similar to that described for Example 101.
- compounds of the invention may exist in isomeric form; for example, as tautomers, enantiomers, or diasteromers. Some compounds may exhibit polymorphism. All enantiomers, and diasteromers are incorporated within the definition of the compounds of the invention.
- the present invention encompasses any racemic, optically-active, polymorphic, or stereoisomeric form, or mixtures thereof, of a compound of the invention, which possess the useful properties described herein, it being well known in the art how to prepare optically active forms (for example, by resolution of the racemic form by recrystallization techniques, by synthesis from optically-active starting materials, by chiral synthesis, or by chromatographic separation using a chiral stationary phase) and how to determine activity or cytotoxicity using the standard tests described herein, or using other similar tests which are well known in the art.
- Certain of the compounds of the present invention can exist in unsolvated forms as well as solvated forms, including h yd rated forms. In general, the solvated forms, including hydrated forms, are equivalent to unsolvated forms and are intended to be encompassed within the scope of the present invention.
- the present invention provides a method for causing vasodilation in a patient in need thereof comprising administering a compound of formulae (I), (II), or (III).
- a compound of formulae (I), (II), or (III) for the sake of brevity, all of the sub-formulae of formula (II), i.e. formulae (MA), (NB), (HC), and (MD), are included when mentioning compounds of formula (II).
- the present invention provides a method of blocking calcium channels, the method comprising of administering to a patient in need of calcium channel blocking a therapeutically effective amount of a compound of formulae (I) or (II) to block calcium channels.
- the calcium channels are T-type calcium channels.
- the calcium channels are N-type, T-type, and L-type calcium channels.
- the present invention provides a method of treating a disease selected from hypertension, congestive heart failure, stroke, ischaemic heart disease, and angina pectoris comprising administering to a patient in need thereof a therapeutically effective amount of a compound of formulae (I), (II), or (III).
- Another aspect of this invention is directed to methods of reducing myocardial tissue damage (e.g., substantially preventing tissue damage, inducing tissue protection) during surgery (e.g., coronary artery bypass grafting (CABG) surgeries, vascular surgeries, percutaneous transluminal coronary angioplasty (PTCA) or any percutaneous transluminal coronary intervention (PTCI), organ transplantation, or other non-cardiac surgeries) comprising administering to a mammal (e.g., a female or male human) a therapeutically effective amount of a compound of formulae (I), (II), or (III), or a pharmaceutically acceptable salt of said compound.
- a mammal e.g., a female or male human
- the compounds of formula (I), (II), and (III), which are N-type calcium channel antagonists, are potentially useful in the treatment of a range of disorders.
- the treatment of pain, particularly neuropathic pain is a preferred use.
- Physiological pain is an important protective mechanism designed to warn of danger from potentially injurious stimuli from the external environment.
- the system operates through a specific set of primary sensory neurones and is activated by noxious stimuli via peripheral transducing mechanisms (see Millan, 1999, Prog. Neurobiol., 57, 1-164 for a review). These sensory fibres are known as nociceptors and are characteristically small diameter axons with slow conduction velocities.
- neuropathic pain e.g. painful diabetic neuropathy, postherpetic neuralgia
- carpal tunnel syndrome back pain
- headache cancer pain
- arthritic pain chronic post-surgical pain.
- Clinical pain is present when discomfort and abnormal sensitivity feature among the patient's symptoms. Patients tend to be quite heterogeneous and may present with various pain symptoms. Such symptoms include: 1) spontaneous pain which may be dull, burning, or stabbing; 2) exaggerated pain responses to noxious stimuli (hyperalgesia); and 3) pain produced by normally innocuous stimuli (allodynia - Meyer et al., 1994, Textbook of Pain, 13-44). Although patients suffering from various forms of acute and chronic pain may have similar symptoms, the underlying mechanisms may be different and may, therefore, require different treatment strategies. Pain can also therefore be divided into a number of different subtypes according to differing pathophysiology, including nociceptive, inflammatory and neuropathic pain.
- Nociceptive pain is induced by tissue injury or by intense stimuli with the potential to cause injury. Pain afferents are activated by transduction of stimuli by nociceptors at the site of injury and activate neurons in the spinal cord at the level of their termination. This is then relayed up the spinal tracts to the brain where pain is perceived (Meyer et al., 1994, Textbook of Pain, 13-44). The activation of nociceptors activates two types of afferent nerve fibres. Myelinated A-delta fibres transmit rapidly and are responsible for sharp and stabbing pain sensations, whilst unmyelinated C fibres transmit at a slower rate and convey a dull or aching pain.
- Back pain may be due to herniated or ruptured intervertebral discs or abnormalities of the lumber facet joints, sacroiliac joints, paraspinal muscles or the posterior longitudinal ligament. Back pain may resolve naturally but in some patients, where it lasts over 12 weeks, it becomes a chronic condition which can be particularly debilitating.
- Neuropathic pain is currently defined as pain initiated or caused by a primary lesion or dysfunction in the nervous system. Nerve damage can be caused by trauma and disease and thus the term 'neuropathic pain' encompasses many disorders with diverse aetiologies. These include, but are not limited to, peripheral neuropathy, diabetic neuropathy, post herpetic neuralgia, trigeminal neuralgia, back pain, cancer neuropathy, HIV neuropathy, phantom limb pain, carpal tunnel syndrome, central post-stroke pain and pain associated with chronic alcoholism, hypothyroidism, uremia, multiple sclerosis, spinal cord injury, Parkinson's disease, epilepsy and vitamin deficiency. Neuropathic pain is pathological as it has no protective role.
- neuropathic pain are difficult to treat, as they are often heterogeneous even between patients with the same disease (Woolf & Decosterd, 1999, Pain Supp., 6, S141-S147; Woolf and Mannion, 1999, Lancet, 353, 1959-1964). They include spontaneous pain, which can be continuous, and paroxysmal or abnormal evoked pain, such as hyperalgesia (increased sensitivity to a noxious stimulus) and allodynia (sensitivity to a normally innocuous stimulus).
- the inflammatory process is a complex series of biochemical and cellular events, activated in response to tissue injury or the presence of foreign substances, which results in swelling and pain (Levine and Taiwo, 1994, Textbook of Pain, 45-56).
- Arthritic pain is the most common inflammatory pain.
- Rheumatoid disease is one of the commonest chronic inflammatory conditions in developed countries and rheumatoid arthritis is a common cause of disability. The exact aetiology of rheumatoid arthritis is unknown, but current hypotheses suggest that both genetic and microbiological factors may be important (Grennan & Jayson, 1994, Textbook of Pain, 397-407).
- Visceral pain is pain associated with the viscera, which encompass the organs of the abdominal cavity. These organs include the sex organs, spleen and part of the digestive system. Pain associated with the viscera can be divided into digestive visceral pain and non-digestive visceral pain.
- Gl gastrointestinal
- FBD functional bowel disorder
- IBD inflammatory bowel disease
- Gl disorders include a wide range of disease states that are currently only moderately controlled, including, in respect of FBD, gastro-esophageal reflux, dyspepsia, irritable bowel syndrome (IBS) and functional abdominal pain syndrome (FAPS), and, in respect of IBD, Crohn's disease, ileitis and ulcerative colitis, all of which regularly produce visceral pain.
- Other types of visceral pain include the pain associated with dysmenorrhea, cystitis and pancreatitis and pelvic pain.
- musculoskeletal disorders including myalgia, fibromyalgia, spondylitis, sero-negative (non-rheumatoid) arthropathies, non-articular rheumatism, dystrophinopathy, glycogenosis, polymyositis and pyomyositis; • heart and vascular pain, including pain caused by angina, myocardical infarction, mitral stenosis, pericarditis, Raynaud's phenomenon, scleredoma and skeletal muscle ischemia;
- head pain such as migraine (including migraine with aura and migraine without aura), cluster headache, tension-type headache mixed headache and headache associated with vascular disorders; and
- orofacial pain including dental pain, otic pain, burning mouth syndrome and temporomandibular myofascial pain.
- the term "patient” refers to a warm-blooded animal such as a mammal which is (1) in need of vasodilation, (2) in need of blocking calcium channels, (3) afflicted with or at risk of developing hypertension, congestive heart failure, stroke, ischaemic heart disease, or angina pectoris, or (4) afflicted with pain or a sub-category of pain as described above. It is understood that guinea pigs, dogs, cats, rats, mice, horses, cattle, sheep, and humans are examples of animals within the scope of the meaning of the term.
- a patient is in need of treatment for hypertension, congestive heart failure, stroke, ischaemic heart disease, angina pectoris, or pain when the patient is afflicted within one or more of the diseases or conditions described herein or is at a recognized risk of developing one or more of the diseases or conditions described herein as diagnosed by an attending physician or clinician.
- the term "therapeutically effective amount" of a compound of formulae (I), (II), or (III) refers to an amount which is effective in (1) causing vasodilation in the patient in need thereof, (2) blocking calcium channels, (3) treating hypertension, congestive heart failure, stroke, ischaemic heart disease, or angina pectoris, or (4) treating pain.
- treating is intended to refer to all processes wherein there may be a slowing, interrupting, arresting, or stopping of the progression of the diseases and conditions described herein, but does not necessarily indicate a total elimination of all disease and condition symptoms, and is intended to include prophylactic treatment of the hypertension, congestive heart failure, stroke, ischaemic heart disease, angina pectoris, or pain.
- a therapeutically effective amount can be readily determined by the attending diagnostician, as one skilled in the art, by the use of conventional techniques and by observing results obtained under analogous circumstances.
- the dose a number of factors are considered by the attending diagnostician, including, but not limited to: the species of mammal; its size, age, and general health; the specific disease involved; the degree of involvement or the severity of the disease; the response of the individual patient; the particular compound administered; the mode of administration; the bioavailability characteristic of the preparation administered; the dose regimen selected; the use of concomitant medication; and other relevant circumstances.
- the compounds of the invention intended for pharmaceutical use may be administered alone or in combination with one or more other compounds of the invention or in combination with one or more other drugs (or as any combination thereof). Generally, they will be administered as a formulation in association with one or more pharmaceutically acceptable excipients.
- excipient is used herein to describe any ingredient other than the compound(s) of the invention. The choice of excipient will to a large extent depend on factors such as the particular mode of administration, the effect of the excipient on solubility and stability, and the nature of the dosage form.
- Pharmaceutical compositions suitable for the delivery of compounds of the present invention and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation may be found, for example, in 'Remington's Pharmaceutical Sciences', 19th Edition (Mack Publishing Company, 1995).
- the compounds of the invention may be administered orally.
- Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, or buccal or sublingual administration may be employed by which the compound enters the blood stream directly from the mouth.
- Formulations suitable for oral administration include solid formulations, such as tablets, capsules containing particulates, liquids, or powders; lozenges (including liquid- filled), chews; multi- and nano-particulates; gels, solid solution, liposome, films (including muco-adhesive), ovules, sprays and liquid formulations.
- Liquid formulations include suspensions, solutions, syrups and elixirs. Such formulations may be employed as fillers in soft or hard capsules and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet.
- the compounds of the invention may also be used in fast-dissolving, fast- disintegrating dosage forms such as those described in Expert Opinion in Therapeutic Patents, ⁇ (6), 981-986 by Liang and Chen (2001).
- the drug may make up from 1 weight% to 80 weight% of the dosage form, more typically from 5 weight% to 60 weightt% of the dosage form.
- tablets generally contain a disintegrant.
- disintegrants examples include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl-substituted hydroxypropyl cellulose, starch, pregelatinised starch and sodium alginate.
- the disintegrant will comprise from 1 weight% to 25 weight%, preferably from 5 weight% to 20 weight% of the dosage form.
- Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate.
- lactose monohydrate, spray-dried monohydrate, anhydrous and the like
- mannitol xylitol
- dextrose sucrose
- sorbitol microcrystalline cellulose
- starch dibasic calcium phosphate dihydrate
- Tablets may also optionally comprise surface active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc.
- surface active agents such as sodium lauryl sulfate and polysorbate 80
- glidants such as silicon dioxide and talc.
- surface active agents may comprise from 0.2 weight % to 5 weight% of the tablet, and glidants may comprise from 0.2 weight% to 1 weight% of the tablet.
- Tablets also generally contain lubricants such as magnesium s tea rate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate.
- Lubricants generally comprise from 0.25 weight% to 10 weight%, preferably from 0.5 weight% to 3 weight% of the tablet.
- ingredients include anti-oxidants, colourants, flavoring agents, preservatives and taste-masking agents.
- Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting.
- the final formulation may comprise one or more layers and may be coated or uncoated; it may even be encapsulated.
- the compounds of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ.
- Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intra urethral, intrasternal, intracranial, intramuscular and subcutaneous.
- Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
- Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile non-aqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
- excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9)
- a suitable vehicle such as sterile, pyrogen-free water.
- parenteral formulations under sterile conditions may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
- solubility of compounds of formula (I) used in the preparation of parenteral solutions may be increased by the use of appropriate formulation techniques, such as the incorporation of solubility-enhancing agents.
- Formulations for parenteral administration may be formulated to be immediate and/or modified release.
- compounds of the invention may be formulated as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound.
- examples of such formulations include drug- coated stents and PGLA microspheres.
- the compounds of the invention may also be administered topically to the skin or mucosa, that is, dermally or transdermally.
- Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibres, bandages and microemulsions. Liposomes may also be used.
- Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol. Penetration enhancers may be incorporated; see, for example, J
- compositions of the invention may also be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an aerosol spray from a pressurised container, pump, spray, atomiser (preferably an atomiser using electrohydrodynamics to produce a fine mist), or nebuliser, with or without the use of a suitable propellant, such as 1 ,1 ,1 ,2-tetrafluoroethane or 1,1 ,1,2,3,3,3- heptafluoropropane.
- a suitable propellant such as 1 ,1 ,1 ,2-tetrafluoroethane or 1,1 ,1,2,3,3,3- heptafluoropropane.
- the pressurised container, pump, spray, atomizer, or nebuliser contains a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- the drug product Prior to use in a dry powder or suspension formulation, the drug product is micronised to a size suitable for delivery by inhalation (typically less than 5 microns).
- This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenisation, or spray drying.
- Capsules made, for example, from gelatin or HPMC
- blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the compound of the invention, a suitable powder base such as lactose or starch and a performance modifier such as /-leucine, mannitol, or magnesium stearate.
- the lactose may be anhydrous or in the form of the monohydrate, preferably the latter.
- Other suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
- a suitable solution formulation for use in an atomiser using electrohydrodynamics to produce a fine mist may contain from 1 ⁇ g to 20mg of the compound of the invention per actuation and the actuation volume may vary from 1 ⁇ l to 100 ⁇ l.
- a typical formulation may comprise a compound of formula (I), propylene glycol, sterile water, ethanol and sodium chloride.
- Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
- Suitable flavours such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations of the invention intended for inhaled/intranasal administration.
- Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, poly(DL-lactic-coglycolic acid (PGLA).
- Modified release formulations include delayed-, sustained-, pulsed-, controlled- , targeted and programmed release.
- the dosage unit is determined by means of a valve which delivers a metered amount.
- Units in accordance with the invention are typically arranged to administer a metered dose or "puff' of the compound of formula (I).
- the overall daily may be administered in a single dose or, more usually, as divided doses throughout the day.
- the compounds of the invention may be administered rectally or vaginally, for example, in the form of a suppository, pessary, or enema.
- Cocoa butter is a traditional suppository base, but various well known alternatives may be used as appropriate.
- the compounds of the invention may also be administered directly to the eye or ear, typically in the form of drops of a micronised suspension or solution in isotonic, pH- adjusted, sterile saline.
- Other formulations suitable for ocular and aural administration include ointments, biodegradable (e.g. absorbable gel sponges, collagen) and non- biodegradable (e.g. silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes.
- the compounds of the invention may be combined with soluble macromolecular entities, such as cyclodextrin and suitable derivatives thereof or polyethylene glycol- containing polymers, in order to improve their solubility, dissolution rate, taste-masking, bioavailability and/or stability for use in any of the aforementioned modes of administration.
- soluble macromolecular entities such as cyclodextrin and suitable derivatives thereof or polyethylene glycol- containing polymers
- Drug-cyclodextrin complexes are found to be generally useful for most dosage forms and administration routes. Both inclusion and non-inclusion complexes may be used.
- the cyclodextrin may be used as an auxiliary additive, i.e. as a carrier, diluent, or solubiliser. Most commonly used for these purposes are alpha-, beta- and gamma-cyclodextrins, examples of which may be found in International Patent Applications Nos. WO 91/11172, WO 94/02518 and WO 98/55148.
- a calcium channel blocker of the present invention may be usefully combined with another pharmacologically active compound, or with two or more other pharmacologically active compounds, particularly in the treatment of pain.
- a compound of formulae (I), (II), or (III), or a pharmaceutically acceptable salt thereof, as defined above may be administered simultaneously, sequentially or separately in combination with one or more agents selected from:
- opioid analgesics e.g. morphine, heroin, hydromorphone, oxymorphone, levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine, dihydrocodeine, oxycodone, hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, nalbuphine and pentazocine;
- opioid analgesics e.g. morphine, heroin, hydromorphone, oxymorphone, levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine, dihydrocodeine, oxycodone, hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, n
- nonsteroidal antiinflammatory drugs e.g. aspirin, diclofenac, diflusinal, etodolac, fenbufen, fenoprofen, flufenisal, flurbiprofen, ibuprofen, indomethacin, ketoprofen, ketorolac, meclofenamic acid, mefenamic acid, nabumetone, naproxen, oxaprozin, phenylbutazone, piroxicam, sulindac, tolmetin, zomepirac, and their pharmaceutically acceptable salts;
- NSAIDs nonsteroidal antiinflammatory drugs
- barbiturate sedatives e.g. amobarbital, aprobarbital, butabarbital, butabital, mephobarbital, metharbital, methohexital, pentobarbital, phenobartital, secobarbital, talbutal, theamylal, thiopental and their pharmaceutically acceptable salts;
- benzodiazepines having a sedative action e.g. chlordiazepoxide, clorazepate, diazepam, flurazepam, lorazepam, oxazepam, temazepam, triazolam and their pharmaceutically acceptable salts,
- Hi antagonists having a sedative action e.g. diphenhydramine, pyrilamine, promethazine, chlorpheniramine, chlorcyclizine and their pharmaceutically acceptable salts;
- miscellaneous sedatives such as glutethimide, meprobamate, methaqualone, dichloralphenazone and their pharmaceutically acceptable salts;
- skeletal muscle relaxants e.g. baclofen, carisoprodol, chlorzoxazone, cyclobenzaprine, methocarbamol, orphrenadine and their pharmaceutically acceptable salts
- NMDA receptor antagonists e.g. dextromethorphan ((+)-3-hydroxy-N- methylmorphinan) and its metabolite dextrorphan ((+)-3-hydroxy-N- methylmorphinan), ketamine, memantine, pyrroloquinoline quinone and cis-4-
- alpha-adrenergic active compounds e.g. doxazosin, tamsulosin, clonidine and 4- amino-6,7-dimethoxy-2-(5-methanesulfonamido-1 ,2,3,4-tetrahydroisoquinol-2-yl)- 5-(2-pyridyl) quinazoline;
- tricyclic antidepressants e.g. desipramine, imipramine, amytriptiline and nortriptiline;
- anticonvulsants e.g. carbamazepine and valproate
- Tachykinin (NK) antagonists particularly Nk-3, NK-2 and NK-1 e.g. antagonists, ( ⁇ R,9R)-7-[3,5-bis(trifluoromethyl)benzyl]-8,9, 10,11 -tetrahydro-9- methyl-5-(4-methylphenyl)-7H-[1 ,4]diazocino[2, 1 -g][1 ,7]naphthridine-6-13-dione (TAK-637), 5-[[(2R,3S)-2-[(1 R)-1-[3,5-bis(trifluoromethyl)phenyl]ethoxy-3-(4- fluorophenyl)-4-morpholinyl]methyl]-1 ,2-dihydro-3H-1 ,2,4-triazol-3-one (MK-869), lanepitant, dapitant and 3-[[2-methoxy-5-(trifluoromethoxy)phenyl]methylamino]-
- Muscarinic antagonists e.g oxybutin, tolterodine, propiverine, tropsium chloride and darifenacin;
- COX-2 inhibitors e.g. celecoxib, rofecoxib and valdecoxib;
- Non-selective COX inhibitors e.g. nitroflurbiprofen (HCT-1026);
- coal-tar analgesics in particular, paracetamol
- neuroleptics such as droperidol
- Beta-adrenergic compounds such as propranolol
- Corticosteriods such as dexamethasone
- xxii serotonin receptor agonists and antagonists
- PDEV inhibitors such as sildenafil, vardenafil or taladafil
- serotonin reuptake inhibitors e.g. fluoxetine, paroxetine, citalopram and sertraline
- mixed serotonin-noradrenaline reuptake inhibitors e.g. milnacipran, venlafaxine and duloxetine
- noradrenaline reuptake inhibitors e.g. reboxetine
- alpha-2-delta ligands e.g. gabapentin and pregabalin.
- the compounds of the invention may be usefully combined with one or more agents for reducing the risk of a cardiovascular disorder including anti-inflammatory agents, such as alclofenac, algestone acetonide, alpha amylase, amcinafal, amcinafide, amfenac sodium, amiprilose hydrochloride, anakinra, anirolac, apazone, balsalazide disodium, bendazac, benoxaprofen, benzydamine hydrochloride, bromelains, broperamole, budesonide, carprofen, cicloprofen, cintazone, cliprofen, clobetasol propionate, clobetasone butyrate, clopirac, cloticasone propionate, cortodoxone, deflazacort, desonide, desoximetasone, dexamethasone dipropionate, diclofenac potassium, diclofenac sodium, diflumidone sodium, diflu
- Useful dosages of the compounds of the invention can be determined by comparing their in vitro activity, and in vivo activity in animal models.
- the amount of the compound, or an active salt or derivative thereof, required for use in treatment will vary not only with the particular salt selected but also with the route of administration, the nature of the condition being treated and the age and condition of the patient and will be ultimately at the discretion of the attendant physician or clinician.
- the compounds of the present invention can be administered to a human patient at dosage levels in the range of about 1 to about 2,000 mg per day, preferably from about 1 to about 1 ,000 mg per day, more preferably from about 5 to about 600 mg per day, even more preferably from about 10 to 300 mg per day.
- a dosage in the range of about 0.01 to about 10 mg per kilogram of body weight per day is preferable.
- the specific dosage used can vary. For example, the dosage can depended on a numbers of factors including the requirements of the patient, the severity of the condition being treated, and the pharmacological activity of the compound being used. The determination of optimum dosages for a particular patient is well-known to those skilled in the art.
- the active ingredient should be administered to achieve peak plasma concentrations of the active compound of from about 0.5 to about 75 ⁇ M, preferably, about 1 to 50 ⁇ M, most preferably, about 0.1 to about 5 ⁇ M. This may be achieved, for example, by the intravenous injection of a 0.05 to 5% solution of the active ingredient, optionally in saline, or orally administered as a bolus containing about 10-500 mg of the active ingredient. Desirable blood levels may be maintained by multiple oral dosing, or continuous infusion to provide about 0.01-5.0 mg/kg/hr or by intermittent infusions containing about 0.4-15 mg/kg of the active ingredient(s).
- the desired dose may conveniently be presented in a single dose or as divided doses administered at appropriate intervals, for example, as two, three, four or more sub-doses per day.
- the sub-dose itself may be further divided, e.g., into a number of discrete loosely spaced administrations; such as multiple inhalations from an insufflator or by application of a plurality of drops into the eye.
- a stable tetracycline-inducible TREx-293 cell line is generated expressing recombinant mouse ⁇ 1H.
- mouse ⁇ 1H T-type calcium channel cDNA accession number NM_021415
- cloning techniques e.g. Molecular Cloning Techniques
- This expression vector has a CMV promoter to drive expression of the ⁇ 1H gene.
- This plasmid construct is used to stably transfect TREx-293 cells (Invitrogen, Carlsbad CA), a human embryonic kidney cell line stably expressing the tetracycline repressor protein.
- Cells are maintained at 37° C and 5% CO 2 in Dulbecco's Modified Eagle Media supplemented with 10% fetal bovine serum, 200 ⁇ g/ml Zeocin, and 5 ⁇ g/ml Blasticidin. Cells are induced with 1 ⁇ g/ml tetracycline and plated onto black-sided 384 well PoIy-D Lysine coated plates at 12,000 cells/well for at least twenty-four hours. The cells are incubated with the fluorescent Ca 2+ indicator Fluo-4 AM (50 ⁇ g, Molecular Devices, Sunnyvale, CA) dissolved in pluronic acid and DMEM supplemented with 2.6 mM probenecid for 1 hour at 37°C and 5% CO 2 .
- Fluo-4 AM 50 ⁇ g, Molecular Devices, Sunnyvale, CA
- Cells are then rinsed with assay buffer (consisting of 0.34 mM Na 2 HPO 4 , 4.2 mM NaHCO 3 , 0.44 mM KH 2 PO 4 , 0.41 mM MgSO 4 , 0.49 mM MgCI 2 , 20 mM HEPES, 5.5 mM d-Glucose, 0.1% BSA, 137 mM NaCI, and 2.6 mM probenecid) and incubated at 37°C and 5% CO 2 for 10 minutes. Cells are pretreated with putative antagonists for 5 minutes followed by a rapid increase of 4.8 mM extracellular Ca 2+ .
- assay buffer consisting of 0.34 mM Na 2 HPO 4 , 4.2 mM NaHCO 3 , 0.44 mM KH 2 PO 4 , 0.41 mM MgSO 4 , 0.49 mM MgCI 2 , 20 mM HEPES, 5.5 mM d-Glucose, 0.1% B
- FLIPR fluorometric imaging plate reader
- FLIPR fluorometric imaging plate reader
- the basic principal involves illumination of 384-well microplate with the simultaneous measurement of emitted fluorescence using a CCD camera.
- the cell plate wells contain cells that have been loaded with a fluorescent dye, whose emission characteristics change upon binding with a particular ion (Ca 2+ in this case).
- Ca 2+ a particular ion
- the growth media for A10 cells is Ham's F12/DME high glucose (Irvine Scientific, 9052), supplemented with 20% fetal bovine serum (HyClone Labs, SH30071.02), and 1% each of L-glutamine (Gibco BRL, 25030-032) and antibiotic-antimycotic (Gibco BRL, 15240-096).
- Cells are grown to confluency and replated on black-sided 384-well plates (Falcon, 35 3962) at 12K cells/well for use in FLIPR. Forty-eight hours after replating, growth media is removed and cells are loaded at 37 0 C with 50 ⁇ l media containing 1 ⁇ M Fluo-4 dye (Molecular Probes, F-14201) for 1 hour. The dye-containing media Is then washed away six times with buffer (composition in mM: 1.25 CaCI 2 , 1.2 MgSO 4 , 11 glucose, 10 HEPES, 3.0 KCI, 137.0 NaCI, pH 7.4 with Tris base) in an Embla384 (Molecular Devices, 0200-3906). The residual buffer volume is adjusted to 20 ⁇ l and allowed to incubate at room temp for an additional hour.
- FLIPR protocol A five minute drug-pre-incubation period at is initiated when 20 ⁇ l of drug-containing buffer is pipetted into the cell plate with the 384-well pipettor integrated in the FLIPR apparatus. Fluorescent counts are monitored at two second intervals for 960 seconds, beginning 60 seconds prior to the delivery of drug-containing buffer. Following drug addition, 20 ⁇ l aliquots of a high K+, depolarizing stimulus (composition in mM: 1.25 CaCI 2 , 1.2 MgSO 4 , 11 glucose, 10 HEPES, 140.0 KCI, pH 7.4 with Tris base) are added to each well and fluorescence is monitored at one second intervals for 120 secibds, beginning ten seconds prior to the stimulus addition. CCD camera exposure time is 0.4 seconds, laser excitation is at 488 nm with a power of 0.6W, and a 510 to 560nm bandpass interference filter preceded the camera.
- Data are analyzed as a summation of fluorescent counts above basal during the stimulation period (an approximation of area under the curve), after normalizing the data with a spatial uniformity correction (for variations in laser illumination and cell density), a negative control correction and a bias subtraction (a bias subtraction subtracts the fluorescence value measured at a specific sample point from all the other time points in each well and allows for all data on the y-axis to be zeroed).
- Drug effects are expressed as percent inhibition of fluorescence from an average of 8 K+-stimulated wells that were pre-treated in the drug incubation period with buffer only. Data are analyzed using FLIPR software, Microsoft Excel and Origin. The IC50 calculations are performed and graphed in Origin.
- a stable cell line is generated expressing recombinant rat D I B.
- the coding sequence for rat DIB CDNA (accession number AF055477) is cloned using standard cloning techniques (e.g. Molecular Cloning A Laboratory Manual, 2nd Edition, J.
- This plasmid construct is used to stably transfect HEK- tsA201 cells.
- Cells are maintained at 37° C and 5% CO 2 in Dulbecco's Modified Eagle Media supplemented with 10% fetal bovine serum, 25 ⁇ g/ml Zeocin, and 5 ⁇ g/ml Blasticidin and 25 ⁇ g/ml Hygromycin.
- Cells are plated onto black-sided 384 well PoIy-D Lysine coated plates at 5,000 cells/well and incubate for 24 hours.
- the cells are incubated with the fluorescent Ca 2+ indicator Fluo-4 AM (50 ⁇ g, Molecular Probes, Eugene, OR) dissolved in pluronic acid for 1 hour at 37°C and 5% CO 2 .
- Cells are then rinsed with assay buffer (consisting of 1 mM Na 2 HPO 4 , 26 mM NaHCO 3 , 1.8 mM CaCI 2 , 0.8 mM MgCI 2 , 117 mM NaCI, 20 mM HEPES, 5.6 mM d-Glucose) and incubated at 37°C for 10 minutes.
- Cells are pretreated with putative antagonists for 5 minutes followed by an addition of KCI (30 mM).
- Normotensive and/or hypertensive rats are isoflurane-anesthetized and will be prepared for IV administration of compounds solubilized in 5% NMP/45% PEG400/50%
- Compounds will be administered as a initial IV bolus dose (1 ml/kg over 30 seconds) followed by a constant maintenance infusion (0.1ml/kg/min) for 60 minutes.
- Mean (MBP), systolic (SBP), diastolic (DBP) blood pressures as well as heart rate (HR) will be collected every 30 sec.
- the computer stores each experiment's data and then the data is manually downloaded via intranet to network server for permanent storage and analysis. Blood samples will be collected at 30 and 60 minutes during the compound infusion for rat pharmacokinetic (PK) data.
Abstract
This invention relates to novel compounds of formula (I) (I) wherein R1, R2, R3, R4, R5, G1, G2, Y, n, and Ar1 are as defined in the specification, pharmaceutical compositions containing said compounds useful as calcium channel antagonists, and to methods of causing vasodilation of treating a disease selected from hypertension, congestive heart failure, stroke, ischaemic heart disease, and angina pectoris and of reducing myocardial tissue damage (e.g., substantially preventing tissue damage, inducing tissue protection) during surgery (e.g., coronary artery bypass grafting (CABG) surgeries, vascular surgeries, percutaneous transluminal coronary angioplasty (PTCA) or any percutaneous transluminal coronary intervention (PTCI), organ transplantation, or other non-cardiac surgeries), chronic pain, inflammatory pain, neuropathic pain, visceral pain, nociceptive pain, multiple sclerosis, neurodegenerative disorder, irritable bowel syndrome, osteoarthritis, rheumatoid arthritis, neuropathological disorders, functional bowel disorders, inflammatory bowel diseases, pain associated with dysmenorrhea, pelvic pain, cystitis, pancreatitis, migraine, cluster and tension headaches, diabetic neuropathy, sciatica, fibromyalgia and causalgia.
Description
SUBSTITUTED OXADIAZOLE ANALOGS AS CALCIUM CHANNEL ANTAGONISTS
This invention relates to oxadiazole derivatives. More particularly, this invention relates to oxadiazole derivatives that are calcium channel antagonists. The oxadiazole derivatives of the invention modulate one or more of the L-type, N-type, and T-type calcium channels and are useful as pharmaceutical agents in the treatment of a variety of disorders ranging from pain, hypertension, angina, and/or obesity.
Voltage-gated calcium channels are membrane-spanning, multi-subunit proteins that open in response to membrane depolarization, allowing calcium ion entry from the extracellular milieu. Calcium channels were initially classified based on the time and voltage-dependence of channel opening and on the sensitivity to pharmacological block. The categories were low-voltage activated (primarily T-type) and high-voltage activated (L, N, P, Q or R-type). Recently, an alternative classification scheme was devised based upon the molecular subunit composition, as summarized in Table 1 (Hockerman G H, Peterson B Z, Johnson B D, Catterall W A. 1997. Annu Rev Pharmacol Toxicol 37: 361-96).
There are four primary subunit types that make up calcium channels— Oh, α2δ, β and Y. The Cd subunit is the primary determinant of the pharmacological properties and contains the channel pore and voltage sensor (Hockennan G H, Peterson B Z, Johnson B D, Catterall W A. 1997. Annu Rev Pharmacol Toxicol 37: 361-96; Striessnig J. 1999. Cell Physiol Biochem 9: 242-69). Ten isoforms of the αi subunit are known, as indicated in Table 1. The α2δ subunit consists of two disulfide linked subunits, α2, which is primarily extracellular and a transmembrane δ subunit. Four isoforms of α2δ are known, cfcδ-i, α2δ-2, α 2δ-3 and δ2δ-4. The β subunit is a non-glycosylated cytoplasmic protein that binds to the αi subunit. Four isoforms are known, termed βi to β4. The y subunit is a transmembrane protein that has been biochemically isolated as a component of Cav1 and Cav2 channels. The nomenclature for voltage-gated calcium channels is based upon the content of the O1 subunit, as indicated in Table 1. Each type of di subunit can associate with a variety of β, α2δ or y subunits, so that each Cav type corresponds to many different combinations of subunits.
TABLE 1
Classification of Neuronal Calcium Channels
Cav Nomenclature αi subunit Pharmacological name
Cav1.1 dis L- type
Cav1.2 die L-type

Cav2.1 α1A P- or Q-type
Cav2.2 α iβ N-type
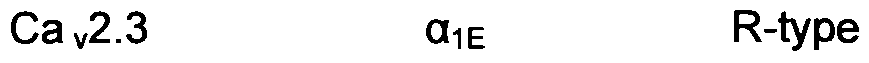
Cav3.1 α iG T-type
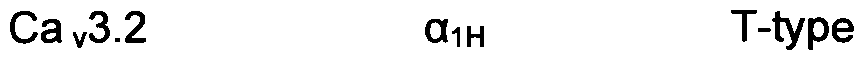
Cav3.3 a n T-type
Cav2 currents are found almost exclusively in the central and peripheral nervous system and in neuroendocrine cells and constitute the predominant forms of presynaptic voltage-gated calcium current. Presynaptic action potentials cause channel opening and neurotransmitter release is steeply dependent upon the subsequent calcium entry.
Thus, Cav2 channels play a central role in mediating neurotransmitter release.
N-type calcium channels (Cav2.2) contain high-affinity binding sites for the peptide toxins ω-conotoxin-MVIIC and ω-conotoxin-GVIA, respectively, and these peptides have been used to determine the distribution and function of each channel
type. Ca v 2.2 is highly expressed at the presynaptic nerve terminals of neurons from the dorsal root ganglion and neurons of lamina I and Il of the dorsal horn (Westenbroek R E, Hoskins L, Catterall W A. 1998. J Neurosci 18: 6319-30; Cizkova D, Marsala J, Lukacova N, Marsala M, Jergova S, et al. 2002. Exp Brain Res 147: 456-63). Cav2.2 channels are also found in presynaptic terminals between second and third order interneurons in the spinal cord. Both sites of neurotransmission are very important in relaying pain information to the brain.
Pain, particularly neuropathic and intractable pain is a large unmet medical need. Millions of individuals suffer from severe pain that is not well controlled by current therapeutics. The current drugs used to treat pain include non-steroidal antiinflammatory drugs (NSAIDs), cyclo-oxygenase 2 (COX-2) inhibitors, opioids, tricyclic antidepressants, and anticonvulsants. Neuropathic pain has been particularly difficult to treat as it does not respond well to opioids until high doses are reached. Gabapentin is currently the most widely used therapeutic for the treatment of neuropathic pain, although additional therapeutic agents are desirable, particularly those with broader ranges of activities.
The T-type calcium channel (Cav3.1 , Cav3.2, and Cav3.3) may become over- expressed due to genetic or environmental causes, such as epilepsy (Tsakiridou, E. et al., J. Neurosci. 1995, 15, 3110-3117), high blood pressure (Self, D. A. et al., J. Vacs. Res. 1994, 31 , 359-366), ventricular hypertrophy (Nuss, H. B. et al., Circ. Res. 1995, 73, 777-7825), pain (Shin, H. S. et al., Science 2003, 302, 117-119), and angina pectoris (Van der Vring, J. A. et al., Am. J. Ther. 1999, 6, 229- 233). A representative drug for blocking the T-type calcium channel is mibefradil of Hoffman La Roche Ltd. The drug was found to be effective in treating high blood pressure, angina pectoris and cerebral apoplexy. It was approved for treating high blood pressure in May, 1997. However, a side effect caused by a drug-drug interaction due to inhibition of CYP 3A4 hepatic enzyme was discovered. As such, the drug was withdrawn from the market in June, 1999.
Dihydropyridine (DHP) antagonists of L-type calcium channels (Cav1.1 , Cav1.2, and Cav1.3) are widely used therapeutics in the treatment of hypertension, angina, arrhythmias, congestive heart failure, cardiomyopathy, atheriosclerosis, and cerebral and peripheral vascular disorders (Janis and Triggle, 1990 CRC Press, Cleveland). In addition to L-type channel activity, some of the DHPs are sensitive to T-type channel
activity. (N. Akaike, H. Kanaide, T, Kuga, M, Nakamura, J. Sadoshima and Tomoike "Low Voltage Activated Calcium Current in rat Aorta Smooth Muscle Cells In Primary Cultur" J Physiol. 416, 141-160, (1989).
Specific calcium channel antagonists approved for cardiovascular use in the United States fall into several chemical classes: the dihydropyridines (e.g., amlodipine, felodipine, nifedipine, nicardipine, isradipine, nimodipine); the benzothiazepines (e.g., diltiazem), phenylalkylamines (e.g., verapamil); and diarylaminopropylamine ether (e.g., bepridil). While there are several alternatives from which a physician may choose, there remains a need for novel calcium channel blockers, particularly in a distinct chemical class. It is an object of this invention to provide a novel class of calcium channel antagonists which inhibit one or more of the N-type, T-type, and L-type calcium channels.
SUMMARY OF THE INVENTION
The present invention relates to a compound of formula (I)

or a pharmaceutically acceptable salt thereof, wherein R1 and R2 are each independently -H, -OH, halo, C1-C6 alkyl, CrC6 alkoxy, -CF3, substituted
C1-C6 alkyl, or substituted CrC6 alkoxy;
R3 and R4 are each independently -H or Ci-C6 alkyl or R3 and R4 taken together with the carbon atom to which they are attached form C3-C6 cycloalkyl, or cycloheteroalkyl, provided that if one of R3 and R4 is -H, then the other is C1-C6 alkyl;
R5 is -H, C1-C6 alkyl, d-C6 alkoxy, -(CH2)q-C(0)0-W, wherein W is -H or CrC6 alkyl and q is 1-6;
G1 is methylene or ethylene;
G2 is C(R6) or N, wherein R6 is -H, -OH or Ci-C6 alkyl; Y is -CH2-, -CH2CH2-, -CH2CH2CH2-, -O-, -C(O)-, -C(O)CH2-, -S-, -S(O)-, -S(O)2-
-NH-, -NHC(O)-, -NHC(O)CH(R7)-, or -NHS(O)2-, wherein R7 is -H or C1-C4 alkyl;
Ar1 is a radical of the formulae

The present invention also relates to a pharmaceutical composition comprising a compound of formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
The present invention further relates to a method of blocking calcium channels, the method comprising administering to a patient in need of calcium channel blocking a therapeutically effective amount of a compound of formula (I) to block calcium channels.
Another embodiment of the invention relates to a method of treating pain in a patient in need thereof comprising administering to said patient a therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
Another embodiment of the invention relates to a method of causing vasodilation in a patient in need thereof comprising administering to said patient a therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
A further embodiment of the invention relates to a method of treating a disease selected from hypertension, congestive heart failure, stroke, ischaemic heart disease, and angina pectoris comprising administering to a patient in need thereof a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
Another aspect of this invention is directed to methods of reducing myocardial tissue damage (e.g., substantially preventing tissue damage, inducing tissue protection) during surgery (e.g., coronary artery bypass grafting (CABG) surgeries, vascular surgeries, percutaneous transluminal coronary angioplasty (PTCA) or any percutaneous transluminal coronary intervention (PTCI), organ transplantation, or other non-cardiac surgeries) comprising administering to a mammal (e.g., a female or male human) a
therapeutically effective amount of a compound of formula (I) or (II), or a pharmaceutically acceptable salt of said compound.
DETAILED DESCRIPTION OF THE INVENTION
Definitions:
The term "halogen" or "halo" refers to a fluorine atom, chlorine atom, bromine atom, or iodine atom.
The term "Ci-C6 alkyl" refers to a branched or straight chained alkyl radical containing from 1 to 6 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec butyl, t-butyl, pentyl, hexyl, and the like.
The term "d-C4 alkyl" refers to a branched or straight chained alkyl radical containing from 1 to 4 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, t-butyl, and the like.
The term "substituted CrC6 alkyl" refers to a CrC6 alkyl substituted with from 1 to 3 substituents selected from halogen and CrC4 alkoxy. Included within this definition is -CH2F, -CHF2, -CF3,
-CH2CH2F, -CH2CHF2, -CH2CF3, -CH2CH2CH2CH2F, — CH2CI, -CHCI2, -CCI3, — CH2CH2CI, -CH2CHCI2,
-CH2CCI3, -CH2CH2CH2CH2CI, -CH2OCH3, -CH2CH2OCH3, -CH2CH2CH2OCH3, and the like.
The term "CrC6 alkoxy" refers to a straight or branched alkoxy group containing from 1 to 6 carbon atoms, such as methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy, sec-butoxy, t-butoxy, pentoxy, hexoxy, and the like.
The term "CrC4 alkoxy" refers to a straight or branched alkoxy group containing from 1 to 4 carbon atoms, such as methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy, sec-butoxy, t-butoxy, and the like.
The term "substituted Ci-C6 alkoxy" refers to a CrC6 alkoxy substituted with from 1 to 3 substituents selected from halogen and CrC4 alkyl.
The term "C3-C6 cycloalkyl" refers to a cyclic alkyl radical containing from 3 to 6 carbon atoms, such as cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl,
The term "CrC4 haloalkyl" refers to a CrC4 alkyl is substituted with from 1 to 3 halo atoms per monovalent carbon and 1 to 2 halo atoms per divalent carbons, such as -CF3, -CH2F, -CHF2, -CF3,
-CHFCH3, -CH2CF3, -CH2CH2CF3, -CH2CH2CH2CF3, -CH2Br, -CHBr2, -CH2CHBr2, - CCI3, -CHCI2, -CH2CI, -CCI3, and the like.
The term "cycloheteroalkyl" refers to a cyclic alkyl moiety containing five carbons and one ring atom of -O- or -N(RH1), wherein RH1 is -H or d-C6 alkyl as is further depicted in the formula (III) compounds disclosed herein. Examples of a
"cycloheteroalkyl" include tetrahydro-2H-pyran-4-yl, tetrahydropyridin-4-yl, N- methylpiperidin-4-yl
The designation " ""^ " or " " refers to a bond for which the stereochemistry is not designated.
The designation " — ~ " refers to a bond that protrudes forward out of the plane of the page.
The designation " refers to a bond that protrudes backward out of the plane of the page. The designation "-C(O)-" or "C(O)" refers to a carbonyl group of the formula: o
The designation "-0C(O)R4" refers to a carboxylic acid (R4 is H) or an ester (R4 is CrC6 alkyl or C3-C6 cycloalkyl) of the formula:
 wherein R4 is H, CrC6 alkyl, or C3-C6 cycloalkyl.
wherein R4 is H, CrC6 alkyl, or C3-C6 cycloalkyl.
The designation "-OC(O)NR5R6" refers to a carbamate of the formula:
R5
O wherein R5 and R6 are each independently -H or C1-C6 alkyl.
The designation "-N(R7)-" refers to a divalent amine of the formula:
?'
wherein R7 is -H or CrC6 alkyl.
The designation "C(R8)" refers to a moiety of the formula:
R° wherein R8 is -H, -OH, CrC6 alkyl.
The designation "-C(O)CH2" refers to a moiety of the formula:
O
H,
The designation "-NHC(O)-" refers to an amide of the formula: o
The designation "-NHC(O)CH(R9)-" refers to an amide of the formula:
O
'N H R9 wherein R9 is -H or CrC4 alkyl. The designation "-NHS(O)2-" refers to a sulfonamide of the formula: o
H O
The designation "-NR10R11" refers to an amine of the formula:
R10
-1S" wherein R10 and R11 are each independently -H or CrC6 alkyl. The designation






The designation


As is appreciated by one of ordinary skill in the art some of the compounds of the formula (I) exist as stereoisomers. Any reference in this application to one of the compounds of the formula (I) is meant to encompass either specific stereoisomers or a mixture of stereoisomers. Where indicated, the compounds follow the (+)- and (-)- designation or the Cahn-lngold-Prelog designation of (R)- and (S)- for the stereochemistry of compounds represented by formula (I) and intermediates thereof.
The specific stereoisomers can be prepared by stereospecific synthesis using enantiomerically pure or enantiomerically enriched starting materials. The specific stereoisomers of either starting materials or products can be resolved and recovered by techniques known in the art, such as chromatography on chiral stationary phases, enzymatic resolution, or fractional recrystallization of addition salts formed by reagents used for that purpose. Useful methods of resolving and recovering specific stereoisomers are know in the art and described in Stereochemistry of Organic
Compounds, E. L. ENeI and S. H. Wilen, Wiley (1994) and Enantiomers, Racemates, and Resolutions, J. Jacques, A. Collet, and S. H. Wilen, Wiley (1981).
In order to prepare one optical isomer over its enantiomer, a number of routes are available. As an example, a mixture of enantiomers may be prepared, and then the two enantiomers may be separated. A commonly employed method for the separation of a racemic mixture is the use of chiral high pressure liquid chromatography. Further details regarding resolution of enantiomeric mixtures may be found in J. Jacques, et al, Enantiomers, Racemates, and Resolutions, (1991).
The term "suitable solvent" refers to any solvent, or mixture of solvents, inert to the ongoing reaction that sufficiently solubilizes the reactions to afford a medium within which to effect the desired reaction.
Pharmaceutically acceptable salts of the compounds of formula I include the acid addition and base salts (including disalts) thereof.
Suitable acid addition salts are formed from acids which form non-toxic salts. Examples include the acetate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, saccharate, stearate, succinate, tartrate, tosylate and trifluoroacetate salts.
Suitable base salts are formed from bases which form non-toxic salts. Examples include the aluminium, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and zinc salts.
For a review on suitable salts, see "Handbook of Pharmaceutical Salts: Properties, Selection, and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002). A pharmaceutically acceptable salt of a compound of formula (I) may be readily prepared by mixing together solutions of the compound of formula (I) and the desired acid or base, as appropriate. The salt may precipitate from solution and be collected by
filtration or may be recovered by evaporation of the solvent. The degree of ionisation in the salt may vary from completely ionised to almost non-ionised.
Compounds of formula (I) containing one or more asymmetric carbon atoms can exist as two or more stereoisomers. Where a compound of formula (I) contains an alkenyl or alkenylene group, geometric cis/trans (or ZJE) isomers are possible. Where the compound contains, for example, a keto or oxime group or an aromatic moiety, tautomeric isomerism ('tautomerism') can occur. It follows that a single compound may exhibit more than one type of isomerism.
Included within the scope of the claimed compounds of the present invention are all stereoisomers, geometric isomers and tautomeric forms of the compounds of formula
(I), including compounds exhibiting more than one type of isomerism, and mixtures of one or more thereof. Also included are acid addition or base salts wherein the counterion is optically active, for example, D-lactate or L-lysine, or racemic, for example,
DL-tartrate or DL-arginine. Cisltrans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallisation.
Conventional techniques for the preparation/isolation of individual enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate (or the racemate of a salt or derivative) using, for example, chiral high pressure liquid chromatography (HPLC).
Alternatively, the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of formula (I) contains an acidic or basic moiety, an acid or base such as tartaric acid or 1-phenylethylamine. The resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to a skilled person.
Chiral compounds of the invention (and chiral precursors thereof) may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% isopropanol, typically from 2 to 20%, and from 0 to
5% of an alkylamine, typically 0.1% diethylamine. Concentration of the eluate affords the enriched mixture.
Mixtures of stereoisomers may be separated by conventional techniques known to those skilled in the art. [see, for example, "Stereochemistry of Organic Compounds" by E L Eliel (Wiley, New York, 1994).]
The present invention includes all pharmaceutically acceptable isotopically- labelled compounds of formula (I) wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
Examples of isotopes suitable for inclusion in the compounds of the invention include isotopes of hydrogen, such as 2H and 3H, carbon, such as 11C, 13C and 14C, chlorine, such as 36CI, fluorine, such as 18F, iodine, such as 123I and 125I, nitrogen, such as 13N and 15N, oxygen, such as 15O, 17O and 18O, phosphorus, such as 32P, and sulphur, such as 35S.
Certain isotopically-labelled compounds of formula (I), for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies. The radioactive isotopes tritium, i.e. 3H, and carbon-14, i.e. 14C, are particularly useful for this purpose in view of their ease of incorporation and ready means of detection.
Substitution with heavier isotopes such as deuterium, i.e. 2H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances.
Substitution with positron emitting isotopes, such as 11C, 18F, 15O and 13N, can be useful in Positron Emission Topography (PET) studies for examining substrate receptor occupancy. Isotopically-labeled compounds of formula (I) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagents in place of the non-labeled reagent previously employed.
The compounds of the present invention may be administered as prodrugs. Thus certain derivatives of compounds of formula (I) which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into compounds of formula (I) having the desired activity, for example, by hydrolytic cleavage. Such derivatives are referred to as 'prodrugs'. Further information
on the use of prodrugs may be found in 'Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T Higuchi and W Stella) and 'Bioreversible Carriers in Drug Design1, Pergamon Press, 1987 (ed. E B Roche, American Pharmaceutical Association). Prodrugs can, for example, be produced by replacing appropriate functionalities present in the compounds of formula (I) with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in "Design of Prodrugs" by H Bundgaard (Elsevier, 1985).
Some examples of such prodrugs include: (i) where the compound of formula (I) contains a carboxylic acid functionality
(-COOH), an ester thereof, for example, replacement of the hydrogen with (Ci-C8)alkyl;
(ii) where the compound of formula (I) contains an alcohol functionality (-OH), an ether thereof, for example, replacement of the hydrogen with (Ci-C6)alkanoyloxymethyl; and (iii) where the compound of formula (I) contains a primary or secondary amino functionality (-NH2 or -NHR where R ≠ H), an amide thereof, for example, replacement of one or both hydrogens with (CrC10)alkanoyl.
Further examples of replacement groups in accordance with the foregoing examples and examples of other prodrug types may be found in the aforementioned references.
Finally, certain compounds of formula (I) may themselves act as prodrugs of other compounds of formula (I).
As with any group of structurally related compounds which possesses a particular utility, certain groups and configurations are preferred for the compounds of formula (I) and their end-use application.
Preferred embodiments of compounds of formula (I) or stereoisomers or pharmaceutically acceptable salts thereof are given below:
(1) Compounds wherein R1 and R2 are:
(a) each independently -H, -CF3, halo, or CrC4 alkyl; (b) each independently -H, fluoro, chloro, -CF3, or CrC4 alkyl; or
(c) one of R1 and R2 is -H and the other is fluoro;
(2) Compounds wherein R3 and R4 are:
(a) taken together with the carbon atom to which they are attached form C3-C6 cycloalkyl;
(b) taken together with the carbon atom to which they are attached form cycloheteroalkyl; (c) taken together with the carbon atom to which they are attached form tetrahyd ro-2 H-py ran-4-y I ;
(3) Compounds wherein R5 is -H;
(4) Compounds wherein G1 is:
(a) methylene; or (b) ethylene;
(5) Compounds wherein G2 is:
(a) C(R6), wherein R6 is -H, -OH, or C1-C6 alkyl;
(b) C(R6), wherein R6 is -H; or
(C) N;
(6) Compounds wherein Y is
(a) -C(O)-, -S(O)2-, -NHC(O)- or -NHS(O)2-;
(b) -C(O)-, or -S(O)2-;
(7) Compounds wherein n is
(a) O; or
(b) 1 ;
(8) Compounds wherein Ar1 is


(9) Compounds wherein RP1 , RP2, RP3, RP4, RP5, RP6, RZ1 , RP7 and RP8 are each independently
-H, halo, CrC4 alkyl, Ci-C4 alkoxy, or -CF3;
(10) Compounds wherein RB4 and RB6 are each independently -H or methyl;
(11 ) Compounds wherein X5 is (a) S; or (b) NH;
(12) Compounds wherein X3 is NH or S.
It is understood that further preferred embodiments of formula (I) can be selected by requiring one or more of the preferred embodiments (1) through (12) above of compounds of formula (I) or stereoisomers or pharmaceutically acceptable salts thereof or by reference to the examples given herein. For example, further preferred embodiments of the invention can be obtained by combining (1)(a) and (2)(a); (1)(b) and
(2)(a); (1)(c) and (2)(a); (1)(a) and (2)(b); (1)(b) and (2)(b); (1)(c) and (2)(b); (1)(b), (2)(a), and (3); (1)(c), (2)(a), and (3); (2)(b) and (3); (2)(a), (3), (4)(a), and (5)(b); (1)(b),
(2)(a), (3), (4)(a), (5)(c); (2)(b), (3), (4)(a), (5)(b), and (6)(a); (2)(a), (3), (4)(a), (5)(b), and (6)(b); (1)(c), (2)(a), (3), (4)(a), (5)(c), and (6)(a); (1)(c), (2)(a), (3), (4)(a), (5)(a), and (6)(b); (1)(b), (2)(a), (3), (6)(a), and (7)(b); (1)(c), (2)(c), (3), (6)(a), and (7)(b); (2)(a), (3), (6)(b), (7)(b), and (8)(a); (2)(a), (3), (6)(b), (7)(b), and (8)(b); (2)(a), (3), (6)(b), (7)(b), and (B)(C); (2)(a), (3), (6)(b), (7)(b), and (8)(d); (2)[(a) or (b)], (6)(a), (7)(b), and (8)(a); (3), (6)(b), (7)(b), and (8)(c); (3), (6)(a), (7)(a), and (8)(b); (2)(a), (3), (7)(a), (8)(a), and (9); (2)(b), (3), (6)(b), (7)(b), (β)(a), and (9); (2)(b), (3), (6)(b), (7)(b), (8)(b), and (9); (2), (7)(a), (8)(a), and (9); (2)(a), (6)(b), (7)(b), (8)(a), and (9); (7)(a), (8)(a), and (9); (6)(a), (7)(b), (8)(d), and (9); (2)(a), (7)(b), (8)(a), and (9); (2)(b), (7)(b), (8)(a), and (9); (7)(b)
and (9); (4)(a), (6)(a), (J)(Jo), (8)(a), and (9); (4)(b), (6)(b), (7)(b), (8)(a), and (9); (2)(a), (4)(a), (6)(a), (7)(b), (8)(a), and (9); (2)(a), (4)(b), (6)(b), (7)(b), (8)(d), and (9); (2)(a), (5)(b), and (7)(b); (2)(a), (5)(c), and (7)(b); (1)(c), (4)(a), and (7)(a); (1)(b), (4)(a), (7)(b), (9), and (10); (1)(b), (2)(a), (4)(a), (7)(a), (9), and (10); (1)(c), (2)(b), (4)(a), (6)(a), (9), (10), and (11)(a); (1)(b), (2)(a), (4)(a), (6)(a), (7)(b), (9), (10), and (11)(b); (1)(b), (2)(a), (4)(a), (6)(b), (7)(b), (9), (10), (11)(a), and (12); (1)(b), (2)(b), (4)(a), (6)(a), (9), (10), (11)(b), and (12); (8)(a), (11)(a), and (12); (8)(a), (11)(b), and (12); (2)(a), (8)(a), (11)(a), and (12); (2)(b), (8)(a), (11)(b), and (12); and the like, or by solely requiring (1)(b); (2)(a); (3); (4)(a); (4)(b); (5)(a); (5)(b); (5)(c); (6)(a); (6)(b); (7)(a); (7)(b); (8)(a); (8)(b); (8)(c); (8)(d); (9); (10); (11)(a); (11)(b); (12) and the like. It is further understood that the stereoisomers and pharmaceutically acceptable salts are included in the term "compound" unless specifically disclaimed.
Additional embodiments of the invention are represented by compounds of formula (II)
-Ar'
 n " (II) or a pharmaceutically acceptable salt thereof, wherein R1 and R2 are each independently -H, -OH, halo, CrC6 alkyl, CrC6 alkoxy, -CF3, or substituted CrC6 alkyl;
n " (II) or a pharmaceutically acceptable salt thereof, wherein R1 and R2 are each independently -H, -OH, halo, CrC6 alkyl, CrC6 alkoxy, -CF3, or substituted CrC6 alkyl;
G2 is C(R6) or N, wherein R6 is -H, -OH or C1-C6 alkyl;
Y is -CH2-, -CH2CH2-, -CH2CH2CH2-, -0-, -C(O)-, -C(O)CH2-, -S-, -S(O)-, -S(O)2-, -NH-, -NHC(O)-,
-NHC(O)CH(R7)-, or -NHS(O)2-, wherein R7 is -H or C1-C4 alkyl; Ar1 is a radical of the formulae


Preferred embodiments of compounds of formula (II) or stereoisomers or pharmaceutically acceptable salts thereof are given below: (1) Compounds wherein R1 and R2 are:
(a) each independently -H, -CF3, halo, or C1-C4 alkyl;
(b) each independently -H, fluoro, chloro, or C1-C4 alkyl; or
(c) one of R1 and R2 is -H and the other is fluoro;
(2) Compounds wherein G2 is: (a) C(R6), wherein R6 is -H, -OH, or C1-C6 alkyl;
(b) C(R6), wherein R6 is -H; or (C) N;
(3) Compounds wherein Y is:
(a) -C(O)-, -S(O)2-, -NHC(O)- or -NHS(O)2-; (b) -C(O)-, or -S(O)2-;
(4) Compounds wherein n is
(a) 0; or
(b) 1 ;
(5) Compounds wherein Ar1 is:

(6) Compounds wherein RP1, RP2, RP3, RP4, RP5, RP6, RZ1, RP7 and RP8 are each independently
-H1 halo, C1-C4 alkyl, CrC4 alkoxy, Or -CF3;
(7) Compounds wherein R64 and RB6 are each independently -H or methyl;
(8) Compounds wherein X5 is
(a) S; or
(b) NH;
(9) Compounds wherein X3 is NH or S.
It is understood that further preferred embodiments of formula (II) can be selected by requiring one or more of the preferred embodiments (1) through (9) above of compounds of formula (II) or stereoisomers or pharmaceutically acceptable salts thereof or by reference to the examples given herein. For example, further preferred embodiments of the invention can be obtained by combining (1)(a) and (2)(a); (1)(a) and (2)(b); (1)(b) and (2)(a); (1)(b) and (2)(b); (1 )(C) and (2)(a); (1)(c) and (2)(b); (1)(a) and (2)(c); (1)(c) and (2)(c); (2)(a) and (3)(a); (2)(b) and (3)(a); (2)(a) and (3)(b); (1)(b) and (3)(a); (1)(b) and (4)(a); (1)(b) and (5)(a); (1)(b), (4)(b) and (5)(b); (1)(b), (3)(a), (4)(b) and (5)(a); (3)(b), (4)(b), and (5)(a); (2)(a), (3)(a), (4)(b), and (5)(a); (2)(b), (3)(a), (4)(b), and (5)(a); (2)(a), (3)(a), (4)(b), and (5)(b); (2)(a), (3)(a), (4)(b), and (5)(c); (2)(a), (3)(a), (4)(b), and (5)(d); (1)(b), (3)(a), (4)(b), and (5)(c); (1)(b), (2)(c), (3)(b), (4)(b), and (5)(b); (1)(b), (3)(b), (5)(a), and (6); (1)(b), (3)(b), (5)(b), and (6); (1)(b), (3)(b), (5)(c), and (6); (1)(b), (3)(b), (5)(d), and (6); (1)(b), (3)(a), (5)(a), and (6); (1)(b), (3)(a), (5)(a), (6), and (7); (1)(b), (3)(a), (5)(a), (6), (7), and (8)(a); (1)(b), (3)(b), (5)(a), (6), (7), and (8)(b); (1)(b), (3)(b), (5)(a), (6), (7), (8)(a) and (9); (1)(b), (3)(a), (5)(a), (6), (7), (8)(b), and (9); (3)(a), (5)(c), (6), (7), (8)(b), and (9); (5)(a), (6), (7), (8)(a) and (9); (5)(a), (6), (7), (8)(b) and (9); (5)(b), (6), (7), (8)(a) and (9); (5)(c), (6), (7), (8)(a) and (9); (5)(d), (6), (7), (8)(a) and (9); (5)(a), (6), (7), and (9); and the like; or by solely requiring (1)(b); (2)(a); (2)(b); (3)(a); (4)(a); (4)(b); (5)(a), (7); and the like. It is further understood that the stereoisomers and pharmaceutically acceptable salts are included in the term "compound" unless specifically disclaimed.
Further embodiments of the invention are represented by compounds or pharmaceutically acceptable salts of formulae (MA), (MB), (MC), and (MD):


wherein R1, R2, G2, Y, Ar1, and n in each of (HA), (MB), (MC), and (MD) are as defined in formula (M). Compounds of formula (MA) are those compounds of formula (M) where m is 1. Compounds of formula (MB) are those compounds of formula (M) where m is 2. Compounds of formula (MC) are those compounds of formula (M) where m is 3. Compounds of formula (MD) are those compounds of formula (II) where m is 4. Preferred embodiments of compounds of formulae (MA), (MB), (MC), or (ND), or stereoisomers or pharmaceutically acceptable salts thereof are given below:
(1) Compounds wherein R1 and R2 are: a. each independently -H, -CF3, halo, or CrC4 alkyl; b. each independently -H, fluoro, chloro, or CrC4 alkyl; or c. one of R1 and R2 is -H and the other is fluoro;
(2) Compounds wherein G2 is: a. C(R6), wherein R6 is -H, -OH, or C1-C6 alkyl; or b. N;
(3) Compounds wherein Y is -C(O)-, -S(O)2-, -NHC(O)- or -NHS(O)2-;
(4) Compounds wherein n is a. O; or b. 1 ;
(5) Compounds wherein Ar1 is

(6) Compounds wherein RP1, RP2, Rp3, RP4, RP5, RP6, RZ1, RP7 and RP8 are each independently
-H, halo, CrC4 alkyl, d-C4 alkoxy, or -CF3;
(7) Compounds wherein R64 and RB6 are each independently -H or methyl;
(8) Compounds wherein X5 is a. S; or b. NH;
(9) Compounds wherein X3 is NH or S.
It is understood that further preferred embodiments of formulae (MA), (MB), (MC), and (MD) can be selected by requiring one or more of the preferred embodiments (1) through (9) above of compounds of any of formulae (MA), (MB), (MC), or (MD), or stereoisomers or pharmaceutically acceptable salts thereof or by reference to the examples given herein. For example, further preferred embodiments of the invention can be obtained by combining (1)(a) and (2)(a); (1)(a) and (2)(b); (1)(b) and (2)(a);
(1)(b) and (2)(b); (1)(c) and (2)(a); (1)(c) and (2)(b); (2)(a) and (3); (2)(b) and (3); (1)(b) and (3); (1)(b) and (4)(a); (1)(b), (4)(a) and (5); (1)(b), (4)(b) and (5); (1)(b), (3), (4)(b) and (5); (3), (4)(b), and (5); (2)(a), (3), (4)(b), and (5); (2)(b), (3), (4)(b), and (5); (1)(b), (2)(a), (3), (4)(b), and (5); (1)(b), (2)(b), (3), (4)(b), and (5); (1)(b), (3), (4)(b), (5), and (6); (1)(b), (3), (5), and (6); (1)(b), (3), (5), (6), and (7); (1)(b), (3), (5), (6), (7), and (8)(a); (1)(b), (3), (5), (6), (7), and (8)(b); (1)(b), (3), (5), (6), (7), (8)(a) and (9); (1)(b), (3), (5), (6), (7), (8)(b), and (9); (5), (6), (7), (8)(a) and (9); (5), (6), (7), (8)(b) and (9); (5), (6), (7), and (9); and the like; or by solely requiring (1)(b); (2)(a); (2)(b); (3); (4)(a); (4)(b); (5), (7); and the like. It is further understood that the stereoisomers and pharmaceutically acceptable salts are included in the term "compound" unless specifically disclaimed.
Further embodiments of the invention are represented by compounds of formula (III)

R1 and R2 are each independently -H, -OH, halo, CrC6 alkyl, CrC6 alkoxy, -CF3, or substituted CrC6 alkyl;
G2 is C(R6) or N, wherein R6 is -H, -OH or C1-C6 alkyl; G3 is -O- or -N(RH1), wherein RH1 is -H or C1-C6 alkyl;
Y is -CH2-, -CH2CH2-, -CH2CH2CH2-, -0-, -C(O)-, -C(O)CH2-, -S-, -S(O)-, -S(O)2-, -NH-, -NHC(O)-,
-NHC(O)CH(R7)-, or -NHS(O)2-, wherein R7 is -H or Ci-C4 alkyl; Ar1 is a radical of the formulae
 wherein RP1, RP2, RP3, RP4, RP5, RP6, RP7, RP8, RN1, RN2, RN3, and RZ1 are each independently -H, -OH, CrC6 alkyl, Ci-C6 alkoxy, halo, -CN, -CF3, or -NR8R9, wherein
wherein RP1, RP2, RP3, RP4, RP5, RP6, RP7, RP8, RN1, RN2, RN3, and RZ1 are each independently -H, -OH, CrC6 alkyl, Ci-C6 alkoxy, halo, -CN, -CF3, or -NR8R9, wherein
R8 and R9 are each independently -H or CrC6 alkyl; RM1, RM2, RB1, RB2, RB3, R84, RB5,
RB6 Rxi Rx2 Rx3 Rx4 RYI RY2 RY3 RY4 RY5 and RY6 are egch independently -H or
CrC6 alkyl; X1 and X2 are independently CH or N; X3, X4, and X5 are each independently NH, O, or S; X6 is CH2 or O; Q is substituted CrC6 alkyl, phenyl, napthyl, 2-pyridyl, or 3-pyridyl; and n is O or 1 ; provided that when Y is O, S, NH, NHC(O), NHS(O)2, NHC(O)CH(R7), or NHS(O)2, then G2 is CH.
Preferred embodiments of compounds of formula (III), or stereoisomers or pharmaceutically acceptable salts thereof are given below: (1) Compounds wherein R1 and R2 are:
a. each independently -H -CF3, halo, or d-C4 alkyl; b. each independently -H, fluoro, chloro, or CrC4 alkyl; or c. one of R1 and R2 is -H and the other is fluoro;
(2) Compounds wherein G2 is: a. C(R6), wherein R6 is -H, -OH, or CrC6 alkyl; or b. N;
(3) Compounds wherein Y is -C(O)-, -S(O)2-, -NHC(O)- or -NHS(O)2-;
(4) Compounds wherein n is a. O; or b. 1 ;
(5) Compounds wherein Ar1 is

(6) Compounds wherein RP1, RP2, RP3, RP4, RP5, RP6, RZ1, RP7 and RP8 are each independently -H, halo, CrC4 alkyl, Ci-C4 alkoxy, or -CF3;
(7) Compounds wherein RB4 and RB6 are each independently -H or methyl;
(8) Compounds wherein X5 is a. S; or b. NH; (9) Compounds wherein X3 is NH or S.
It is understood that further preferred embodiments of formula (III) can be selected by requiring one or more of the preferred embodiments (1) through (9) above of compounds of any of formula (III) or stereoisomers or pharmaceutically acceptable salts thereof or by reference to the examples given herein. For example, further preferred embodiments of the invention can be obtained by combining (1)(a) and (2)(a); (1)(a) and (2)(b); (1)(b) and (2)(a); (1)(b) and (2)(b); (1 )(c) and (2)(a); (1)(c) and (2)(b); (2)(a) and (3); (2)(b) and (3); (1)(b) and (3); (1)(b) and (4)(a); (1)(b), (4)(a) and (5);
(1)(b), (4)(b) and (5); (1)(b), (3), (4)(b) and (5); (3), (4)(b), and (5); (2)(a), (3), (4)(b), and (5); (2)(b), (3), (4)(b), and (5); (1)(b), (2)(a), (3), (4)(b), and (5); (1)(b), (2)(b), (3), (4)(b), and (5); (1)(b), (3), (4)(b), (5), and (6); (1)(b), (3), (5), and (6); (1)(b), (3), (5), (6), and (7); (1)(b), (3), (5), (6), (7), and (8)(a); (1)(b), (3), (5), (6), (7), and (8)(b); (1)(b), (3), (5), (6), (7), (8)(a) and (9); (1)(b), (3), (5), (6), (7), (8)(b), and (9); (5), (6), (7), (8)(a) and (9); (5), (6), (7), (8)(b) and (9); (5), (6), (7), and (9); and the like; or by solely requiring (1)(b); (2)(a); (2)(b); (3); (4)(a); (4)(b); (5), (7); and the like. It is further understood that the stereoisomers and pharmaceutically acceptable salts are included in the term "compound" unless specifically disclaimed. Specific compounds of formula (I) include:
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-methoxy- phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3-trifluoro- methyl-benzyl)-piperazin-1-yl]-ethyl}-amine; {3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2,3- dimethoxy-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5-trifluoro- methyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-chloro- phenyl)-piperidin-1 -yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3-trifluoro- methyl-phenyl)-pipehdin-1-yl]-ethyl}-amine;
[2-(4-Benzenesulfonyl-piperazin-1-yl)-ethyl]-{3-[1-(4-chloro-phenyl)-cyclobutyl]- [1 ,2,4]oxadiazol-5-ylmethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutylH1,2,4]oxadiazol-5-ylmethylH2-[4-(2-methoxy- phenoxy)-piperidin-1-yl]-ethyl}-amine;
[3-(1-p-Tolyl-cyclobutyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(5-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; [3-(1-m-Tolyl-cyclobutyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(5-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
[3-(1-P-ToIyI-CyClOPrOPyI)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(4-fluoro- phenoxy)-piperidin-1-yl]-ethyl}-amine;
[3-(1-p-Tolyl-cyclopropyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(5-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; [3-(1-p-Tolyl-cyclopropyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(5-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1 -yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{2-[4-(2-Methoxy-phenyl)-piperidin-1-yl]-ethyl}-[3-(1-phenyl- cyclopentyl)[1 ,2,4]oxadiazol-5-ylmethyl]-amine; {3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3-methoxy- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-[2-(4-o-tolyl- piperidin-1-yl)-ethyl]-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3-methoxy- phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(4- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine; {3-[1-(3-Chloro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
[3-(1-Phenyl-cyclopentyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(5-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3-Chloro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethylH2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
[3-(1-Phenyl-cyclopentyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- phenyl)-piperidin-1-yl]-ethyl}-amine; {3-[1 -(3-Chloro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclohexyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifiuoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyi)-cyclohexyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclohexyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trfluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; {3-[1-(4-Chloro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
[3-(1-Phenyl-cyclobutyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
[3-(1-p-Tolyl-cyclobutyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- phenyl)-piperidin-1-yl]-ethyl}-amine; [3-(1-m-Tolyl-cyclobutyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- phenyl)-piperidin-1-yl]-ethyl}-amine;
[3-(1-m-Tolyl-cyclopropyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- phenyl)-piperidin-1-yl]-ethyl}-amine;
^-(i-m-Tolyl-cyclopropyO-li ^^Joxadiazol-S-ylmethyll^-^-fS-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(2-Fluoro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{2-[4-(2-Methoxy-phenyl)-piperidin-1-yl]-ethyl}-[3-(1-p-tolyl-cyclobutyl)- [1 ,2,4]oxadiazol-5-ylmethyl]-amine; {2-[4-(2-Methoxy-phenyl)-piperidin-1 -yl]-ethyl}-[3-(1 -m-tolyl-cyclobutyl)-
[1 ,2,4]oxadiazol-5-ylmethyl]-amine;
{2-[4-(2-Methoxy-phenyl)-piperidin-1-yl]-ethyl}-[3-(1-m-tolyl-cyclopropyl)- [1 ,2,4]oxadiazol-5-ylmethyl]-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(4-chloro- phenyl)-piperidin-1-yl]-ethyl}-amine;
{2-[4-(2-Methoxy-phenyl)-piperidin-1-yl]-ethyl}-[3-(1-phenyl-cyclobutyl)- [1 ,2,4]oxadiazol-5-ylmethyl]-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-fluoro- phenoxy)-piperidin-1-yl]-ethyl}-amine; {3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-[2-(4-p-tolyloxy- piperidin-1 -yl)-ethyl]-amine;
[3-(1-Phenyl-cyclobutyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(4,4,4-trifIuoro-3- methyl-1-methylene-but-2-enyl)-piperidin-1-yl]-ethyl}-amine;
[3-(1-P-ToIyI-CyClOPrOPyI)-[1 , 2,4)oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- phenyl)-piperidin-1-yl]-ethyl}-amine;
[S-CI-p-Tolyl-cyclobutyO-ti ^^loxadiazol-S-ylmethyll^-μ^S-trifluoromethyl- benzyl)-piperazin-1-yl]-ethyl}-amine;
[3-(1-m-Tolyl-cyclobutyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- benzyl)-piperazin-1-yl]-ethyl}-amine; [3-(1-m-Tolyl-cyclopropyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3,4-Dichloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(2,4-Dichloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclopropyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(3,4-Dichloro-1-methylene-but-2-enyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5- ylmethylH2-[4-(3-trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine; {3-[1-(4-Methoxy-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethylH2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobυtyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-methoxy- 5-trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-methoxy- 4-trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(3-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
[3-(1-p-Tolyl-cyclopentyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(5-trifIuoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; {3-[1-(4-Methoxy-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3-Chloro-phenyl)-cyclopentyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
[3-(1-Phenyl-cyclopentyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- benzyl)-piperazin-1 -yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-fluoro- phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(2,4-Dichloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine; {3-[1-(3,4-Dichloro-phenyl)-cyclopropyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclopentyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclohexyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethy^{2-[4-(3- trfluoromethyl-phenyl)-pipeidin-1-yl]-ethyl}-amine;
[3-(1-p-Tolyl-cyclopentyl)-[1,2,4]oxadiazol-5-ylmethylH2-[4-(3-trifluoromethyl- phenyl)-piperidin-1-yl]-ethyl}-amine; {3-[1-(3-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperzin-1-yl]-ethyl}-amine;
[3-(1-p-Tolyl-cyclopentyl)-[1 ,2,4]oxadiazol-5-ylmethylH2-[4-(3-trifluoromethyl- benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(2,4-Dichloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(2,4-Dichloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethylH2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3,4-Dimethoxy-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifIuoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; {3-[1-(4-Chloro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3-Fluoro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(4-fluoro- phenyl)-piperidin-1 -yl]-ethyl}-amine;
{3-[1-(2-Fluoro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(2-Fluoro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine; [3-(1-Phenyl-cyclopropyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(5-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{S-II-CS-Fluoro-phenyO-cyclopentyll-li^^loxadiazol-S-ylmethylJ^-K^S- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(4-methoxy- phenyl)-piperidin-1-yl]-ethyl}-aπnine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(4- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3,4-Dichloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5-chloro- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; {3-[1-(3-Methoxy-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3-chloro- phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(3,4-Dimethyl-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3,4-Dichloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{2-[4-(2-Chloro-phenyl)-piperidin-1-yl]-ethyl}-{3-[1-(3,4-dimethyl-phenyl)- cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-amine; {1-[2-({3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1,2,4]oxadiazol-5-ylmethyl}-amino)- ethyl]-piperidin-4-yl}-(5-trifluoromethyl-pyridin-2-yl)-amine;
N-{1-[2-({3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-amino)- ethyl]-piperidin-4-yl}-2,5-dimethoxy-benzenesulfonamide;
{3-[4-(4-Chloro-phenyl)-tetrahydro-pyran-4-yl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4- (2-methoxy-phenyl)-piperidin-1 -yl]-ethyl}-amine;
{3-[4-(4-Chloro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4- (3-trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[4-(4-Chloro-phenyl)-tetrahydro-pyran-4-yl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4- (5-trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; {3-[4-(4-Fluoro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-
(5-trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[4-(4-Chloro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4- (3-trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[4-(4-Fluoro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4- (3-trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[4-(3-Methoxy-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2- [4-(3-trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[4-(4-Fluoro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4- (2-methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
{2-[4-(2-Chloro-phenyl)-piperidin-1-yl]-ethyl}-{3-[4-(4-chloro-phenyl)-tetrahydro- pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-amine; and pharmaceutically acceptable salts thereof.
REACTION SCHEMES
Compounds of formula (I) and intermediates thereof can be prepared as described in Reaction Schemes A through E. All the substituents, unless otherwise indicated, are as previously defined. The reagents and starting materials are readily available to one of ordinary skill in the art. Scheme A provides a synthetic process for making benzyl oxadiazole compounds of formula (3) which represent compounds of formula (I) wherein n is 0 and all of the remaining substituents are as defined in formula (D-
Scheme A

In Scheme A, step 1 , the phenyl carbonitrile of formula (1) is treated with hydroxylamine hydrochloride to provide the oxime of formula (1A). For example, the phenyl carbonitrile (1) is dissolved in a suitable solvent, such as ethanol, or an ethanokH∑O mixture, and contacted with hydroxylamine hydrochloride and a suitable base such as potassium carbonate, sodium ethoxide, sodium hydroxide, or mixtures of the base and water. The mixture is refluxed and stirred until analysis indicates that the
reaction is complete. After cooling, the oxime (1A) can be purified by techniques well known in the art, such as extraction, evaporation, trituration, chromatography, and recrystallization.
In Scheme A, step 2, the oxime (1A) is reacted with chloroacetyl chloride to provide the compound of formula (1B). For example, the oxime (1A) is dissolved in a suitable organic solvent such as acetone, a suitable base, such as potassium carbonate, is added and the mixture is cooled. Chloroacetyl chloride is then slowly added over a period of time ranging from about 5 to about 60 minutes. The mixture is then warmed to room temperature and stirred until analysis indicates that the reaction is complete. The compound of formula (1B) can be isolated and purified by techniques well known in the art, such as extraction, evaporation, trituration, chromatography, and recrystallization.
In Scheme A, step 3, the compound of formula (1B) is refluxed in a suitable organic solvent to provide the 5-chloromethyl-3-benzyl-[1,2,3]oxadiazole (1 C). For example, the compound of formula (1B) is refluxed with a Dean-Stark apparatus in a suitable organic solvent such as toluene until analysis indicates that the reaction is complete. The solution is then cooled and 5-chloromethyl-3-benzyl-[1 ,2,3]oxadiazole (1C) may be be isolated and purified by techniques well known in the art, such as extraction, evaporation, trituration, chromatography, and recrystallization. In Scheme A, step 4, the benzyl oxadiazole of formula (3) is prepared by coupling the substituted cyclic amine of formula (2) with 5-chloromethyl-3-benzyl- [1 ,2,3]oxadiazole (1C). For example, substituted amine (2) and 5-chloromethyl-3- benzyl-[1 ,2,3]oxadiazole (1C) are dissolved in a suitable organic solvent such as ethanol in the presence of a base, such as N-methylmorpholine, sodium carbonate, triethylamine, N,N-diisopropylethylamine, potassium carbonate or sodium carbonate and refluxed until analysis indicates that the reaction is complete. The benzyl oxadiazole of formula (3) may be isolated and purified by techniques well known in the art, such as extraction, evaporation, trituration, chromatography, and recrystallization.
Many phenyl carbonitriles of formula (1) are commercially available or are well- known in the art such as 1-phenyl-cyclopropanecarbonitrile, 1-(4-methylphenyl)-1- cyclopropanecarbonitrile, 2-phenylbutyronitrile, 1-phenylcyclobutanecarbonitrile, p- chloro-alpha-methylphenyl acetonitrile, 1-(4-fluorophenyl)cyclopentanecarbonitrile, 1- phenylcyclohexane-1-carbonitrile, and the like. Alternatively, phenyl carbonitriles of
formula (1) may be synthesized by techniques well known and appreciated by those of ordinary skill in the art. For example, 1-(4-fluoro-phenyl)-cyclopropanecarbonitrile may be prepared by reacting (4-fluoro-phenyl)-acetonitrile with bromochloroethane in an aqueous basic solution in the presence of triethylammonium chloride as set forth in the examples herein.
Additionally, many substituted cyclic amines of formula (2) are commercially available or are well-known in the art such as 4-(phenylmethyl)-1-piperidine ethanamine, 4-(phenylmethyl)-1-piperazine ethanamine, 4-(2-methoxyphenyl)-1- piperazine ethananamine, 4-(4-methoxyphenyl)-1-piperazine ethananamine 4-(4- chlorophenyl)-1-piperazine ethananamine, 4-[(3-methoxyphenylmethyl))-1-piperazine ethananamine, 4-(5-chloro-2-methylphenyl)-1-piperazine ethananamine, 4-[4- (trifluoromethyl)-2-pyrimidyl]-1-piperazine ethananamine,
Alternatively, the substituted amines of formula (2) may be synthesized by the techniques set forth in Scheme A1.
Scheme A1
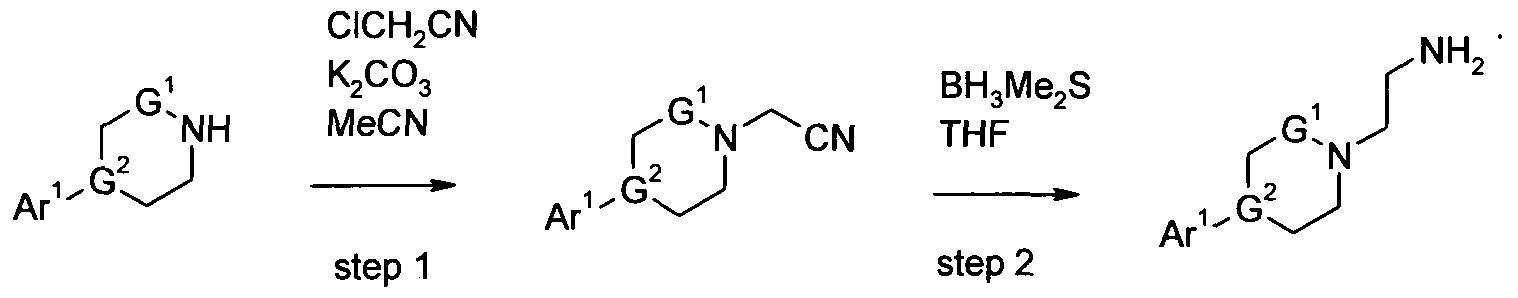
(2A) (2B) (2)
In Scheme A1 , step 1 , the cyclic amine of formula (2A) is treated with chloroacetonitrile in the presence of potassium carbonate to' provide the cyclic amine carbonitrile of formula (2B). For example, the cyclic amine of formula (2A), is dissolved in a suitable solvent such as acetonitrile, in the presence of a base such as potassium carbonate, at room temperature. Chloroacetonitrile is then added slowly over a period of time ranging from 25 to 60 minutes. The mixture is then heated to a temperature of about 950C, and stirred until analysis indicates that the reaction is complete. The compound of formula (2B) may be isolated and purified by techniques well known in the art such as extraction, evaporation, trituration, chromatography, and recrystallization.
In Scheme A1 , step 2, the cyclic amine carbonitrile (2B) is treated with borane dimethyl sulfide complex in THF to provide the substituted amine of formula (2). For example, the cyclic amine carbonitrile (2B) is dissolved in a suitable solvent such as THF. A 2M solution of borane dimethyl sulfide complex is added dropwise over a period of time ranging from 5 to 60 minutes. The mixture is then heated to a temperature of about 650C, and stirred until analysis indicates that the reaction is complete. The compound of formula (2) may be isolated and purified by techniques well known in the art such as extraction, evaporation, trituration, chromatography, and recrystallization.
Many substituted cyclic amines of formula (2A) are commercially available or are well-known in the art such as 1-phenylpiperazine, 1-(3- (trifluoromethyl)phenyl)piperazine, 1-(2,4-dichlorophenyl)piperazine, 1-(4-chloro-2- fluorophenyl)piperazine, 1-(p-tolyl)piperazine, 1-(3,4-difluorophenyl) piperazine, and the like. Alternatively, the substituted cyclic amines of formula (2A) may be synthesized by techniques well known in the art. For example, 1-(3-methoxyphenyl)piperazine may be prepared by reacting bis(2-chloroethyl)amine hydrochloride, 3-methoxyaniline and diethylene glycol monomethyl ether at 1500C.
Scheme B provides a synthetic process for making benzyl oxadiazole piperazine aryl compounds of formula (8) which represent compounds of formula (I) wherein n is 1 , G2 is N, Pg is a suitable amino protecting group, such as t-Boc, and all of the remaining substituents are as defined in formula (I).
Scheme B

(8) (6)
In Scheme B, step 1 , the N-protected benzyl oxadiazole piperazine of formula (5) is prepared by coupling the N-protected ethyleneaminepiperizine of formula (4) with 5- chloromethyl-3-benzyl-[1,2,3]oxadiazole (1C) according to the procedure set forth in Scheme A1 step 4.
In Scheme B, step 2, the benzyl oxadiazole piperazine of formula (6) is prepared by deprotecting the N-protected benzyl oxadiazole piperazine of formula (5). Removal of amino protecting groups is well known and appreciated in the art and is described in Protecting Groups in Organic Synthesis, by T. Green, Wiley-lnterscience (1981). For example, the N-protected benzyl oxadiazole piperazine of formula (5) is mixed with concentrated hydrochloric acid in methanol and heated to reflux until analysis indicates that the reaction is complete. The benzyl oxadiazole piperazine of formula (6) may be isolated and purified by techniques well known in the art, such as extraction, evaporation, trituration, chromatography, and recrystallization.
In Scheme B, step 3, the benzyl oxadiazole piperazine aryl of formula (8) is prepared by coupling the benzyl oxadiazole piperazine of formula (6) with the aryl
chloride of formula (7). For example, benzyl oxadiazole piperazine (6) is contacted with Ar1 -Y-Cl (7) in a suitable solvent such as methylene chloride in the presence of a suitable base such as triethylamine, diisopropylethylamine, N-methylmorpholine, Huniq's base, sodium carbonate, sodium bicarbonate, or potassium carbonate. The reaction is generally carried out at temperatures ranging from about ambient temperature to about 100°C for a period of time until analysis indicates that the reaction is complete. The benzyl oxadiazole piperazine aryl or formula (8) may be isolated and purified by techniques well known in the art, such as extraction, evaporation, trituration, chromatography, and recrystallization. The N-protected ethyleneaminepiperizines of formula (4) may be synthesized by techniques well known in the art. For example, Boc-piperazine is reacted with bromoacetonitrile, in the presence of a base such as potassium carbonate, in a suitable solvent, such as acetonitrile, at temperatures varying from 30 to 650C, for a period of time between 10-24 hours, under a nitrogen atmosphere, and stirred until analysis indicates that the reaction is complete. The nitrile product may be isolated and purified by techniques well known in the art such as extraction, evaporation, trituration, chromatography, and recrystallization. The nitrile product is then reduced with an anhydride, for example lithium aluminum hydride, in a suitable solvent, such as THF, at O0C, for 1 hour, and at room temperature for 2 to 3 hours, and stirred until analysis indicates that the reaction is complete. The compound of formula (4) may be isolated and purified by techniques well known in the art such as extraction, evaporation, trituration, chromatography, and recrystallization
Many aryl chlorides of formula (7) are commercially available or are well-known in the art such as benzoyl chloride, 4-methoxybenzoyl chloride, 2-trifluoromethylbenzoyl chloride, 2,4-dichlorobenzoyl chloride, benzenesulfonyl chloride, 3- trifluoromethylbenzenesulfonyl chloride, 3-fluorobenzenesulfonlyl chloride, and the like. Alternatively, the aryl chlorides of formula (7) may be synthesized by techniques well known in the art. For example, 2,5-dimethoxybenzenesulfonyl chloride may be prepared from the corresponding sulfonic acid by using thionyl chloride in dimethylformamide.
Scheme C provides a synthetic process for making benzyl oxadiazole piperidine aryl compounds of formula (13) which represent compounds of formula (I) wherein n is 1, G2 is CH, Y is -NHC(O)-,
-NHS(O)2-, or -NHC(O)CH(R7)-, and Pg is a suitable amino protecting group, such as t- Boc and all of the remaining substituents are as defined in formula (I).
Scheme C
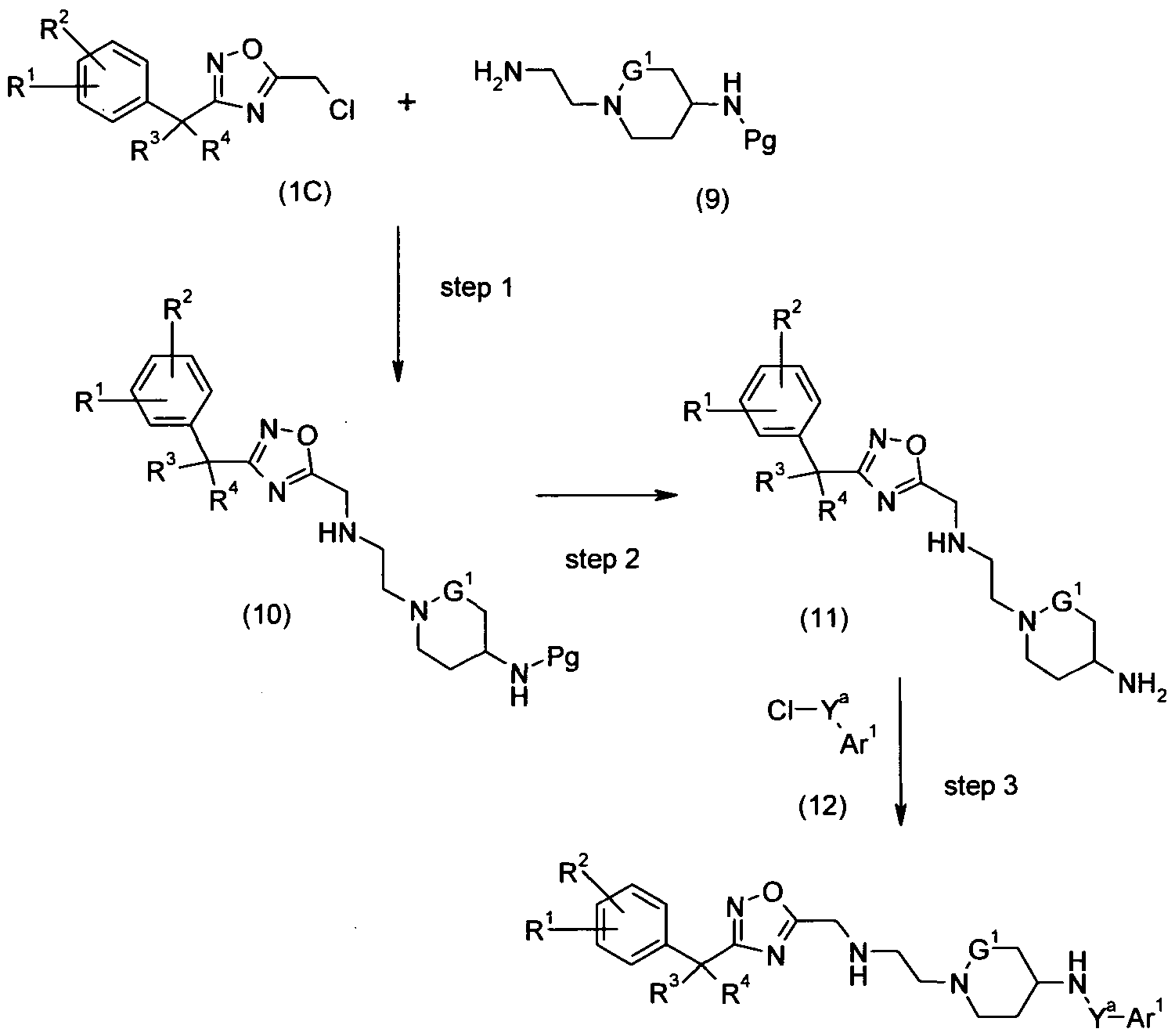
(13)
In Scheme C1 step 1, the N-protected benzyl oxadiazole piperidine of formula (10) is prepared by coupling the N-protected piperidine of formula (9) with 5- chloromethyl-3-benzyl-[1,2,3]oxadiazole (1C) according to the procedure set forth in Scheme A, step 4. In Scheme C, step 2, the benzyl oxadiazole piperidine of formula (11) is prepared by deprotecting the N-protected benzyl oxadiazole piperazine of formula (10) according to procedures set forth in Scheme B, step 2.
In Scheme C1 step 3, the benzyl oxadiazole piperidine aryl of formula (13) is prepared by coupling the benzyl oxadiazole piperidine of formula (11) with the aryl chloride of formula (12) according to the procedures set forth in Scheme B, step 3.
The N-protected ethyleneaminepiperidines of formula (9) may be synthesized by techniques well known in the art. For example, Boc-4-amino-piperidine is reacted with bromoacetonitrile, in the presence of a base such as potassium carbonate, in a suitable solvent, such as acetonitrile, at temperatures varying from 30 to 650C, for a period of time between 10-24 hours, under a nitrogen atmosphere, and stirred until analysis indicates that the reaction is complete. The nitrile product may be isolated and purified by techniques well known in the art such as extraction, evaporation, trituration, chromatography, and recrystallization. The nitrile product is then reduced with an anhydride, for example lithium aluminum hydride, in a suitable solvent, such as THF, at O0C, for 1 hour, and at room temperature for 2 to 3 hours, and stirred until analysis indicates that the reaction is complete. The compound of formula (9) may be isolated and purified by techniques well known in the art such as extraction, evaporation, trituration, chromatography, and recrystallization
Many aryl chlorides of formula (12) are commercially available or are well-known in the art such as benzoyl chloride, 4-methoxybenzoyl chloride, 2-trifluoromethylbenzoyl chloride, 2,4-dichlorobenzoyl chloride, benzenesulfonyl chloride, 3- trifluoromethylbenzenesulfonyl chloride, 3-fluorobenzenesulfonlyl chloride, and the like. Alternatively, the aryl chlorides of formula (12) may be synthesized by techniques well known in the art. For example, 2,5-dimethoxybenzenesulfonyl chloride may be prepared from the corresponding sulfonic acid by using thionyl chloride in dimethylformamide.
Scheme D provides a synthetic process for making benzyl oxadiazole piperidine aryl compounds of formula (16) which represent compounds of formula (I) wherein R5 is CrC6 alkyl, CrC6 alkoxy, or CrC6-C(O)O-W, wherein W is -H or CrC6 alkyl; and all of the remaining substituents are as defined in formula (I). The compound of formula (14) depicts compounds of formula (I) where R5 is -H. The substituent R5a is used in compounds (15) and (16) below to depict compounds of formula (I) when R5 is not -H.
Scheme D

In Scheme D, the aryl oxadiazole of formula (14) is alkylated with an appropriate alkyl bromide (15) to provide alkylated aryl oxadiazole of formula (16). An appropriate alkylating agent of formula (15) is one in which R5a is as desired in the final product of formula (I). For example, the aryl oxadiazole of formula (14) is contacted with 2.0 to 3.0 molar equivalents of alkyl bromide (15). The reaction is carried out in the presence of a suitable base such as sodium bis(trimethylsilyl)amide or lithium diisopropylamide and in the presence of triethyl borane. The reaction is carried out in a suitable solvent such as tetrahydrofuran. The reaction is generally carried out at temperatures ranging from about -78°C to about 00C. Generally the reactions require from about 1 to 72 hours. The product can be isolated and purified by techniques well known in the art such as extraction, evaporation, trituration, distillation, chromatography, and recrystallization.
Scheme E provides a synthetic process for making benzyl oxadiazole piperidine aryl compounds of formula (18) which represent compounds of formula (I) wherein G2 is CH, n is 1 and Y is -CH2-,
-CH2CH2-, or -CH2CH2CH2-; and all of the remaining substituents are as defined in formula (I). In formulae (17) and (18) below, q is 1 , 2, or 3 to provide compounds of formula (I) where Y is -CH2-, -CH2CH2-, Or -CH2CH2CH2-.
Scheme E

In Scheme E, the benzyl oxadiazole piperidine of formula (18) is prepared by coupling the aryl piperidine of formula (17) with 5-chloromethyl-3-benzyl- [1 ,2,3]oxadiazole (1C) according to the procedure set forth in Scheme A, step 4.
Many aryl piperidines of formula (17) are commercially available or are well- known in the art such as 4-(phenylmethyl)-1-piperidineethanamine and the like. Alternatively, the aryl piperidines of formula (17) may be synthesized by techniques well known in the art as set forth in Schemes E1 , E2, and E3.
Scheme E1

1. bromoacetonitrile
2. Lithium aluminum hydride
H2N^x-.
N
Ar1
(17a, q = 1)
In Scheme E1, 4-pyridinecarboxaldehyde (19) is reacted with the aryl Grignard reactant in a suitable organic solvent such as tetrahydrofuran under standard Grignard conditions to provide the aryl-substituted pyridine of formula (21). The aryl-substituted piperidine of formula (22) is obtained by reducing the aryl-substituted pyridine of formula (21) according to standard palladium catalyzed reduction techniques in the presence of a suitable organic solvent such as acetic acid. The product may be isolated and purified according to art-known techniques such as extraction, evaporation, chromatography, and recrystallization. The compound of formula (22) is then treated with bromoacetonitrile followed by lithium aluminum hydride, as described for the synthesis of compound (4), as shown above. The product of formula (17a, q = 1) may be isolated and purified according to art-known techniques such as extraction, evaporation, chromatography, and recrystallization.
Scheme E2

(26) (27) (28)
1. bromoacetonitrile
2. Lithium aluminum hydride
H,N.
Ar1
(17b, q = 2)
In Scheme E2, 4-piperidineethanol (23) is N-protected according to standard nitrogen protecting techniques. The selection and use of suitable amine protecting groups is described in Protecting Groups in Organic Synthesis by T. Greene and is well known and appreciated in the art. For example, the 4-piperidineethanol (23) may be N-
protected using (Boc)2O in the presence of a tertiary amine, such as triethylamine and a suitable organic solvent such as chloroform to provide the Boc-protected 4- piperidineethanol (24). The N-protected aldehyde of formula (25) is obtained by oxidizing the Boc-protected 4- piperidineethanol (24) with a suitable oxidizing agent such as oxalyl chloride in the presence of a tertiary amine such as triethylamine and DMSO in a suitable solvent, such as dichloromethane. The N-protected aldehyde of formula (25) is then reacted with an aryl Grignard reactant in a suitable organic solvent such as tetrahydrofuran under standard Grignard conditions to provide the aryl- substituted N-protected piperidine of formula (26). The hydroxy moiety is then removed from the aryl-substituted N-protected piperidine of formula (26) using a suitable reducing agent such as lithium aluminum hydride to provide the de-hydroxylated N-protected piperidine of formula (27). The aryl-substituted piperidine of formula (28) is obtained by deprotecting the de-hydroxylated N-protected piperidine of formula (27). The removal of amine protecting groups is well known and appreciated in the art and is described in Protecting Groups in Organic Synthesis by T. Greene. The product may be isolated and purified according to art-known techniques such as extraction, evaporation, chromatography, and recrystallization. The compound of formula (28) is then treated with bromoacetonitrile followed by lithium aluminum hydride, as described for the synthesis of compound (4), as shown above. The product of formula (17b, q = 2) may be isolated and purified according to art-known techniques such as extraction, evaporation, chromatography, and recrystallization.
Scheme E3
1. I21 PPh3 imidazole -Boc

1. KHMDS THF

3. HCI MeOH
In Scheme E3, 4-piperidineethanol (23) is N-protected according to standard nitrogen protecting techniques. The selection and use of suitable amine protecting groups is described in Protecting Groups in Organic Synthesis by T. Greene and is well
known and appreciated in the art. For example, the 4-piperidineethanol (23) may be N- protected using (BoC)2O in the presentee of a tertiary amine, such as triethylamine and a suitable organic solvent such as chloroform to provide the Boc-protected 4- piperidineethanol (24). The phosphorus ylid (29) is obtained by treating the Boc- protected 4-piperidineethanol (24) with iodine in the presence of triphenylphosphine in a suitable solvent such as diethylether and acetonitrile. The resulting iodo intermediate is then treated in situ with triphenylphosphine in a suitable solvent such as acetonitrile, at reflux, to give the phosphorus ylid (29). The reaction of an aldehyde with the phosphorus ylid (29) to give an alkene, under standard Wittig reaction conditions, is then carried out, using a suitable base such as potassium hexamethyldisilazamide (KHMDS)1 in a suitable solvent such as tetrahydrofuran. This is followed by reduction of the alkene according to standard palladium catalyzed reduction techniques, in the presence of a suitable organic solvent such as methanol, to give the aryl-substituted N- protected piperidine of formula (30). The compound of formula (30) is then treated with bromoacetonitrile followed by lithium aluminum hydride, as described for the synthesis of compound (4), as shown above. The product of formula (17c, q = 3) may be isolated and purified according to art-known techniques such as extraction, evaporation, chromatography, and recrystallization.
Scheme F provides a synthetic process for making benzyl oxadiazole piperidine aryl compounds of formula (35) which represent compounds of formula (I) wherein R3 and R4 taken together with the carbon atom to which they are attached form cycloheteroaryl, more specifically compounds of formula (III) where G3 is -O-; and all of the remaining substituents are as defined in formula (I).
Scheme F

(31) (32)

In Scheme F, step 1, the phenyl tetrahydro-pyran-4-carbonitrile (32) is prepared by reacting the phenyl acetonitrile of formula (31) with 1-chloro-2-(2-chloro-ethoxy)- ethane in the presence of a suitable base. For example, the phenyl acetonitrile of formula (31) is dissolved in dimethyl sulfoxide. To this solution are added small portions of a suitable base such as NaH at a temperature ranging from about 20 to 25 0C. After the NaH addition, the reaction flask is kept in a water bath at a temperature ranging from about 20 to 25 0C for a period of time ranging from about 0.5 to 2 hours over which time 1-chloro-2-(2-chloro-ethoxy)-ethane is slowly added. The resulting mixture is then stirred vigorously at room temperature for a period of time ranging from about 15 to 24 hours. The phenyl tetrahydro-pyran-4-carbonitrile (32) is then isolated and purified according to techniques well known in the art such as extraction, evaporation, and chromatograpy.
In Scheme F1 step 2, the oxime of formula (33) is prepared by contacting the phenyl tetrahydro-pyran-4-carbonitrile (32) with hydroxylamine hydrochloride in the presence of a suitable base according to the procedures set forth in Scheme A, step 1. In Scheme F, step 3, the compound of formula (34) is prepared by reacting the oxime of formula (33) with chloroacetyl chloride according to the procedures set forth in Scheme A, step 2.
In Scheme F, step 4, the compound of formula (35) is prepared by refluxing the compound of formula (34) in a suitable organic solvent according to the procedures set forth in Scheme A, step 3.
In the preparations and examples the following terms have the indicated meanings; "ng" refers to nanograms; "μg" refers to micrograms; "mg" refers to milligrams; "g" refers to grams; "kg" refers to kilograms; "nmole" or "inmol" refers to nanomoles; "mmol" refers to millimoles; "mol" refers to moles; "μL" refers to microliters; "mL" refers to milliliters; "L" refers to liters; "Rf" refers to retention factor; "0C" refers to degrees Celsius; "bp" refers to boiling point; "mm of Hg" refers to pressure in millimeters of mercury; "mp" refers to melting point; "dec" refers to decomposition; "[α]2 D°" refers to specific rotation of the D line of sodium at 2O0C obtained in a 1 decimeter cell; "c" refers to concentration in g/mL; "nM" refers to nanomolar; "μM" refers to micromolar; "mM" refers to millimolar; "M" refers to molar; "psi" refers to pounds per square inch; "rpm" refers to revolutions per minute; "HPLC" refers to high performance liquid chromatography; "RP-HPLC" refers to reverse phase high performance liquid chromatography; "HRMS" refers to high resolution mass spectrum; "; "DMSO" refers to dimethyl sulfoxide, "brine" refers to a saturated aqueous solution of sodium chloride; "μCi" refers to microcuries; "i.p." refers to intraperitoneally; "i.v." refers to intravenously; "Bn" refers to benzyl; "Boc" refers to t -butyloxycarbonyl; "DCC" refers toΛ/,Λ/'- dicyclohexylcarbodiimide; "DCM" refers to dichloromethane; "DIBAL-H" refers to diisobutylaluminum hydride; "DMF" refers toΛ/,Λ/ -dimethylformamide; "DMSO" refers to dimethyl sulfoxide; "EtOH" refers to ethanol; "IPA" refers to isopropyl alcohol; "LDA" refers to lithium diisopropylamide; "LAH" refers to lithium aluminum hydride; "NaOtBu" refers to sodium t-butoxide; "TEA" refers to triethyl amine; "TFA" referst to trifluoroacetic acid; "THF" refers to tetrahydrofuran; "h" refers to hour or hours; "min" refers to minute or minutes; "s" refers to second or seconds; "Eq" refers to equivalent; or equivalents; N refers to normality (Eq/I); "soln" referst o solutions; "temp" refers to temperature; "cone" refers to concentrate; "vac" refers to vacuum.
EXAMPLE 1
{3-[1-(4-Chloro-phenyl)-cyclobutylH1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-methoxy- phenyl)-piperidin-1-yl]-ethyl}-amine, hydrochloride
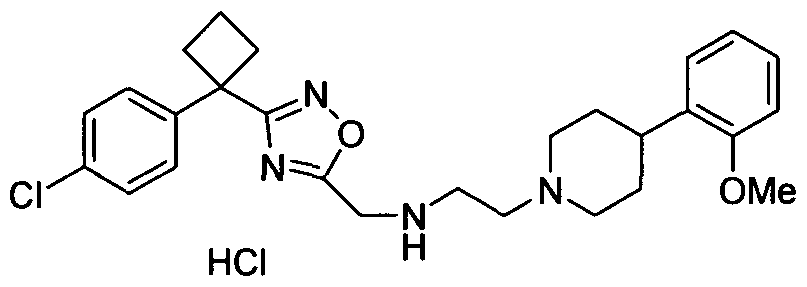
Preparation 1A: [4-(2-Methoxy-phenyl)-piperidin-1-yl]-acetonitrile
MeO
NC ■o-o
4-(2-Methoxy-phenyl)-piperidine (3.Og, 15.7mmol), chloroacetonitrile (1.20ml, 18.8mmol) and K2CO3 (10.8g, 78.5mmol) are stirred in acetonitrile (50ml) overnight at 650C under nitrogen. The reaction mixture is cooled to room temperature and then poured into water (250ml). The resulting mixture is extracted with ethyl acetate (2x200ml). The combined organic phases are washed with brine (100ml) and then dried over MgSO4. Evaporation under reduced pressure gives a solid, which is taken up in dichloromethane (50ml). A dark precipitate is filtered off using filterpaper. Evaporation under reduced pressure gives 3.4Og (94%) of compound 1A as a solid.
Preparation 1 B: 2-[4-(2-Methoxy-phenyl)-piperidin-1-yl]-ethylamine
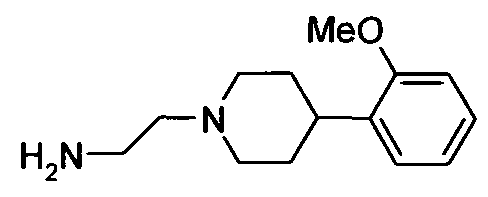
AICI3 (3.4g, 25.5mmol) is added in portions over 2 min to 1 M LiAIH4 in diethyl ether (55ml, 55mmol) under vigorous stirring at room temperature under nitrogen. The milky solution is stirred for 30 min given an almost solid mixture. THF (40ml) is added and stirring for 10 min to give a clear solution. Compound 1A (2.4Og1 10.4mmol) in THF
(20ml) is added drop-wise over 5 min. The reaction mixture is stirred another 3 hours at room temperature and then cooled on an ice-bath. The reaction is quenched by drop- wise addition of water (15ml). The mixture is poured into 4M NaOH (150ml). The resulting mixture is stirred for 10 min and then dichloromethane (200ml) is added. The mixture is stirred vigorously for 5 hours and then transferred to a separation funnel. The water phase is extracted with dichloromethane (300ml). The combined organic phases are washed with brine (150ml) and then dried over MgSO4. Evaporation under reduced pressure gives 2.1g (86%) of compound 1B as a yellow oil.
Preparation 1C: 1-(4-Chloro-phenyl)-N-hydroxy-cyclobutanecarboxamidine

1-(4-Chlorophenyl)-1-cyclobutanecarbonithle (25g, 0.13moles), hydroxylamine hydrochloride (22.7g, 0.33moles), and sodium ethoxide (22.4g, 0.33moles) are dissolved in ethanol (200ml) under nitrogen and refluxed overnight. TLC analysis
[100% DCM] shows the appearance of a lower RF spot and disappearance of starting material. The mixture is cooled to room temperature and evaporated to dryness, extracted with EtOAc (2x250ml) / water (200ml). The organic layer dried with MgSO4, filtered and evaporated to dryness yielding the desired product, as an off white solid
(Total product yield 27.7g (94%).
Preparation 1D

Chloroacetyl chloride (20.3g, 0.18moles) is added dropwise to a mixture of 1C (27g, 0.12moles), potassium carbonate (18.3g, 0.132moles) and acetone (250ml) at O0C under nitrogen. After the addition, the mixture is allowed to warm to room temperature
and stirred overnight. TLC analysis [100% DCM] shows the appearance of a higher RF spot and disappearance of starting material. The mixture is evaporated to dryness, extracted with EtOAc (2x250ml) / water (200ml). The organic layer dried with MgSO4, filtered and evaporated to dryness yielding the desired product as an off white solid (Total product yield 32g (88%).
Preparation 1E 5-Chloromethyl-3-[1-(4-chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazole

1D (32g, 0.038moles) is refluxed in toluene (300ml) using Dean Stark apparatus overnight. TLC analysis [100% DCM] shows the appearance of a higher RF spot and disappearance of starting material. The reaction is cooled to room temperature and evaporated to dryness yielding the desired product as a brown oil, (~70% purity). It is then purified using a silica plug with eluting solvent 100% hexane -> 30%EA, 70% hexane (Total product yield 24.9g (80%).
Preparation 1F {3-[1 -(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-methoxy- phenyl)-piperidin-1-yl]-ethyl}-amine
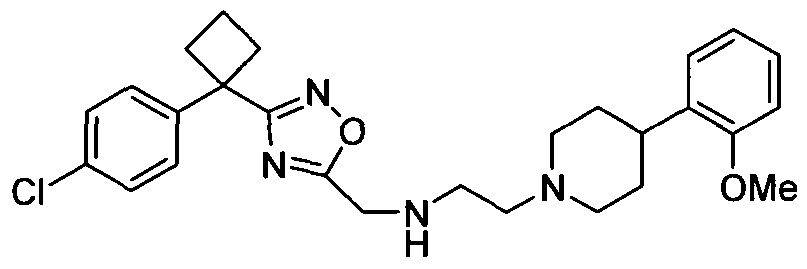
Compound 1B (2.1g, 9.0mmol) and compound 1E (0.64g, 2.25mmol) are stirred in absolute ethanol (100 ml) overnight at reflux under nitrogen. The reaction mixture is evaporated under reduced pressure, which gives an oily material. Purification (silica column, compound pre-adsorbed onto silica, dichloromethane/hexane 0% → 100% and
then methanol/dichloromethane 0% → 5%) gives 0.65g (60%) of compound 1F as a light brown oil.
Example 1 {3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-methoxy- phenyl)-piperidin-1 -yl]-ethyl}-amine hydrochloride

Compound 1 F (0.63g, 1.31mmol) is dissolved in anhydrous diethyl ether (30 ml) by stirring under nitrogen. HCI in diethyl ether (1 M, 3.5ml, 3.5mmol) is added dropwise over 2 min, which gives a precipitate. A solid is collected by filtration. The solid is dried at 4O0C at 1 mbar for 4 hours, which gave 0.6Og of an off-white solid. The solid obtained is given a slurry in boiling 4% methanol/diethyl ether, which gives after filtration and drying 0.48g (66%) of compound 1 as an off-white solid. Mp 208-2100C. 1H-NMR (500 MHz, d-CH3OH) δ 7.36 (s, 4H), 7.24 (m, 2H), 6.97 (m, 2H), 4.73 (s, 2H), 3.87 (s, 3H)1 3.75 (m, 4H), 3.59 (m, 2H), 3.27 (m, 3H), 2.95 (m, 2H), 2.76 (m, 2H), 2.20 (m, 3H), 2.05 (m, 3H). MS (+ ve APCI) m/z (M+1) 481.6. CHNCI analysis: Expected for C27H33CIN4O2, 2HCI; C, 58.34; H, 6.37; N, 10.11 ; Cl, 19.20; Found; C, 58.34; H, 6.41 ; N, 9.97; Cl, 18.90.
PREPARATIONS 2 - 26 Preparations 2 - 26 represent the Block 1 intermediates or headpieces in the synthesis of compounds of formula (I). The Block 1 intermediates may be prepared according to Example 1E (Preparations 1C to 1 D to 1E), using the indicated starting materials in TABLE 2 below in place of 1-(4-chloro-phenyl)-cyclobutanecarbonitrile in Preparation 1C. TABLE 2

Prep. Starting Material Ri R13 R2 R2a m [M+H]*
2 1-p-tolylcyclobutane Me H H H 2 m/z 263 [M+H] carbonitrile
3 1-m-tolylcyclobutane H H Me H 2 m/z 263 [M+H] carbonitrile
4 1-(4-methylphenyl)-1- Me H H H 1 m/z 249 [M+H] cyclopropanecar bonitrile
5 1-phenylcyclobutane- H H H H 2 m/z 249 [M+ 1] carbonitrile
6 1-(4-methoxyphenyl)-1- MeO H H H 3 m/z 293 [M+H] cyclopentanecarb onitrile
7 1-(4-chlorophenyl)-1- Cl H H H 1 m/z 270 [M+H] cyclopropanecar bonitrile
8 1-phenylcyclopentane- H H H H 3 m/z 263 [M+H] carbonitrile
9 1-(4-mthoxyphenyl)-1- MeO H H H 2 m/z 279 [M+H] cyclobutanecarbo nitrile
1-(3-chlorophenyl)-1- H H Cl H 3 m/z 298 [M+H] cyclopentanecarb onitrile
1-(4-chlorophenyl)-1- Cl H H H 4 7n/z 312 [M+H] cyclohexanecarb onitrile
1-(4-chlorophenyl)-1- Cl H H H 3 m/z 298 [M+H] cyclopentanecarb onitrile
1m-tolyl-1- H H Me H 1 m/z 249 [M+H] cyclopropanecar bonitrile
1-(2-fluorophenyl)-1- H H H F 1 m/z 281 [M+H] cyclopropanecar bonitrile
1-(4-methoxyphenyl)-1- MeO H H H 1 m/z 265 [M+H] cyclopropanecar bonitrile
1-(3,4-dichlorophenyl)- Cl H Cl H 1 m/z 304 [M+H]
1- cyclopropanecar bonitrile
1-(2,4-dichlorophenyl)- Cl H H Cl 1 m/z 304 [M+H]
1- cyclopropanecar bonitrile
1-(3-chlorophenyl)-1- H Cl H H 2 m/z 284 [M+H] cyclobutanecarbo nitrile
1-p-tolyl-1- Me H H H 3 m/z 277 [M+H] cyclopentanecarb onitrile
1-(3,4- MeO MeO H H 3 m/z 323 [M+H] dimethoxyphenyl)
-1- cyclopentanecarb onitrile
1-(4-fluorophenyl)-1- H H F H 3 m/z 281 [M+H] cyclopentanecarb onitrile
1-(2-fluorophenyl)-1- H H H F 3 m/z 281 [M+H] cyclopentanecarb onitrile
1-(3,4-dichlorophenyl)- Cl Cl H H 2 m/z 318 [M+H]
1- cyclobutanecarbo nitrile
1-(3-methoxyphenyl)-1- H MeO H H 2 m/z 279 [M+H] cyclobutanecarbo nitrile
1-(3,4-dimethylphenyl)- Me Me H H 2 m/z 277 [M+H]
1-
cyclobutanecarbo nitrile
1-phenyl-1- H H H H 1 m/z 235 [M+H] cyclopropanecar bonitrile
[M+H]* Mass spectrometry data
For use in the table, "MeO" refers to methoxy and "Me" refers to methyl.
PREPARATIONS 27 - 50
Preparations 27 - 50 represent the Block 2 intermediates or tailpieces in the synthesis of compounds of formula (I). The Block 2 intermediates may be prepared according to Example 1B (Preparations 1A to 1B), using the indicated starting materials in TABLE 3 below in place of 4-(2-methoxy-phenyl)-piperidine in Preparation 1A.
TABLE 3
H2N-
-N G— (Y)n— Ar1
Prep. Starting Material Ar1 [M+H]*

28 1-(2,3-dimethoxybenzyl) N CH2 1 m/z 280 piperazine [M+H]

29 1-(5- N m/z 275
CF,
(trifluoromethyl)pyridine- [M+H] 2-yl) piperazine
30 4-(2-chlorophenyl) CH 0 m/z 239 piperidine [M+H]
 (trifluo
(trifluo

i-(phenylsulfonyl) N SO2 1 m/z 270 piperazine [M+H]
4-(2-methoxyphenoxy) CH 0 m/z 251 piperidine [M+H]

4-(4-fluorophenoxy) CH 0 m/z 239 piperidine [M+H]
1-(3-methoxypyridin-2-yl) N m/z 237 piperazine [M+H]

4-o-tolylpiperidine CH

4-(3-methoxyphenvn CH 0 m/z 235 piperidine [M+H]

(trifluoro
 piperidine
piperidine
4-(4-chlorophenyl) CH m/z 239 piperidine
 [M+H]
[M+H]
4-(p-tolyloxy) piperidine CH 0 m/z 235
-CH, [M+H]
4-(2-methoxy-5- CH m/z 303
(trifluormethyl)phenyl) [M+H] piperidine
4-(2-methoxy-4- CH 0 m/z 303
(trifluormethyl)phenyl) [M+H] piperidine
4-(2-fluorophenyl) CH m/z 223 piperidine [M+H]

4-(4-fluorophenyl) CH m/z 223 piperidine
 [M+H]
[M+H]
4-(4-methoxyphenyl) CH m/z 235
-OMe piperidine [M+H]
(trifl

1-(5-chloropyridin-2-yl) N piperazine C/ m/z 241
Cl [M+H]
4-(3-chlorophenyl) CH m/z 239 piperidine [M+H]

N-(piperidin-4-yl)-5- CH NH N=\ m/z 289
(trifluoromethyl) pyridines- CF, amine [M+H]
50 2,5-dimethoxy-N-(piperidin- CH NHSO2 1 4-yl)benzene sulfonamide

EXAMPLES 2 - 100
Examples 2 - 100 set forth in Table 4 below represent compounds of formula (I) which may be prepared by combining the Block 1 intermediates or headpieces of Table 2 with the Block 2 intermediates or tailpieces of Table 3 according to procedures substantially similar to that described for Example 1.
TABLE 4

















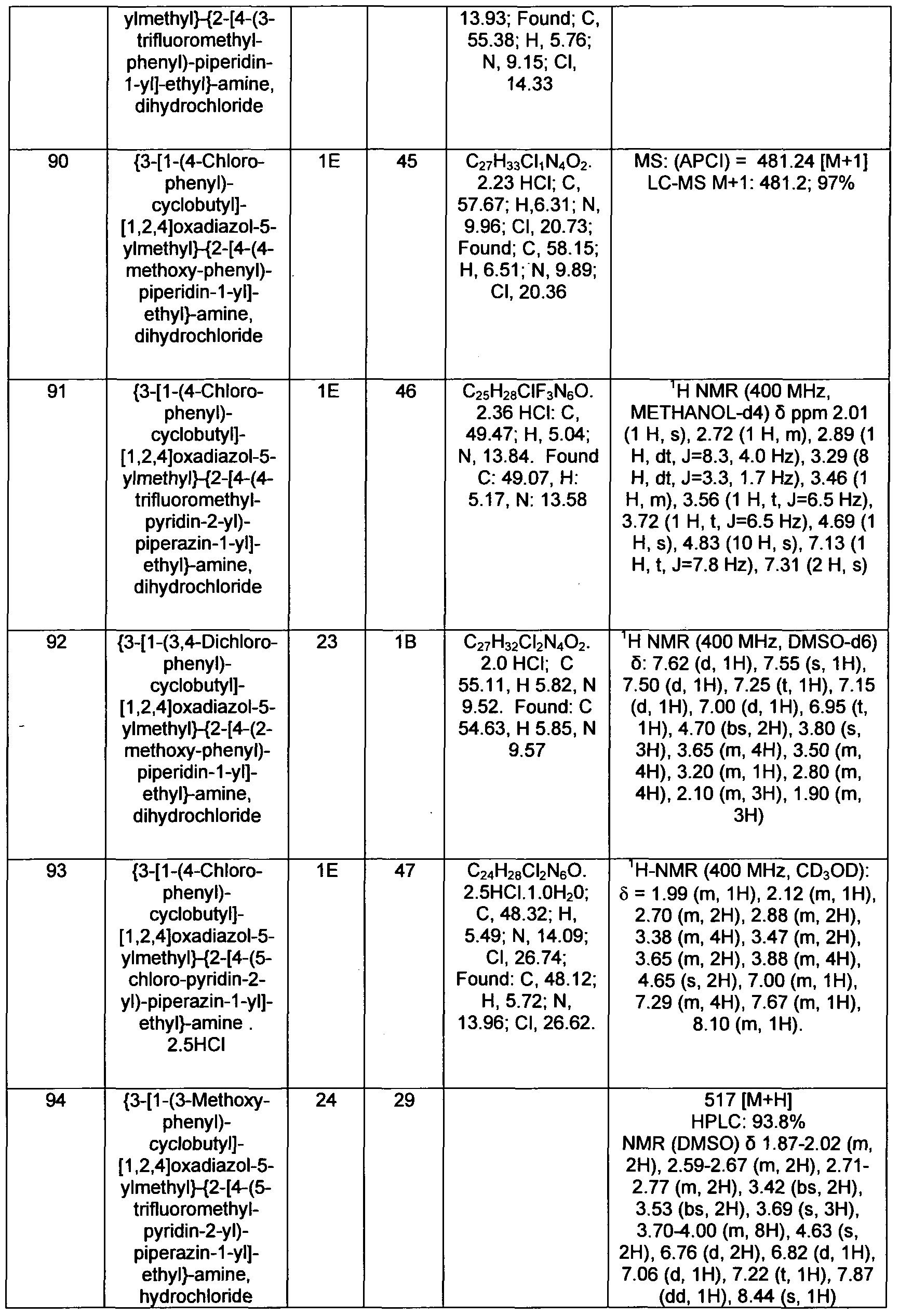


* H-NMR data provided unless otherwise indicated
EXAMPLE 101
{3-[4-(4-Chloro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1 -yl]-ethyl}-amine, hydrochloride

Step A: 4-(4-Chloro-phenyl)-tetrahydro-pyran-4-carbonitrile

To a solution of (4-chloro-phenyl)-acetonitrile (24.0 g, 0.158 mol) in dimethyl sulfoxide (240 ml_), small portions of NaH (60 % paraffin dispersion, 13.3 g, 0.332 mol) is added at 22 0C. After 30 min, the reaction flask is kept on a water bath at 20 0C, and 1-chloro- 2-(2-chloro-ethoxy)-ethane is added very slowly. The dark brown mixture is stirred vigorously for 16 h at it The reaction mixture is poured into ice (700 ml_), and extracted with a mixture of toluene:ethyl acetate (1 :1 ; 3 x 400 mL). The organic layer is washed with brine, dried over Na2SO4, and evaporated. The residue is purified by flash column chromatography on silica (230-400 mesh, 5.5 x 25 cms) using ethyl acetate:hexane (1 :4) as eluant, to give 101A. Yield = 22.5 g (64.1 %). 1H NMR (400 MHz, CDCI3): δ 7.42 (m, 4H), 4.12 (m, 2H), 3.91 (m, 2H), 2.05 (m, 4H).
Step B: 4-(4-Chloro-phenyl)-N-hydroxy-tetrahydro-pyran-4-carboxamidine

Step C:

Chloroacetyl chloride (3.14 ml_, 0.039 mol) is added to a suspension of 101B (8.384 g, 0.033 mol), and K2CO3 (9.09 g, 0.065mol) in acetone (60 mL) at 0 0C. After addition, the suspension is stirred at rt for 2h. The volatiles are evaporated, and the suspension is partitioned in a mixture of water (100 mL), and ethyl acetate (100 mL). The organic layer is separated, and the aqueous layer is extracted with ethyl acetate (100 mL). The organic layer is washed with brine, dried over Na2SO4, and evaporated to give 101 C. Yield = 9.77 g. 1H NMR (400 MHz1 CDCI3): δ 7.38 (m, 4H)1 4.52 (b.s, 2H), 4.29 (s, 2H)1
3.81 (m, 4H), 2.39 (d, 2H), 2.04 (m, 2H).
Step D: 5-Chloromethyl-3-[4-(4-chloro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazole

Step E: 3-[4-(4-Chloro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-
(2-methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine

A mixture of compound 101D (0.3 g, 0.96 mmol), 2-[4-(2-methoxy-phenyl)-piperidin-1- yl]-ethylamine (1B) (0.22 g, 0.96 mmol), and triethyl amine (0.8 mL; 5.7 mmol) in anhydrous ethanol (7 mL) is heated at 100 0C for 16 h. The volatiles are evaporated on a rotavap. The residue is purified by flash column chromatography on silica (230-400 mesh, 3.5 x 11 cms) using a mixture of dichloromethane:methanol:NH4OH (92.5:7.5:0.22) as eluant to give 101E. Yield = 0.235 g (48.0 %). 1H NMR (400 MHz, CDCI3): δ 7.29 (m, 4H), 7.19 (t, 2H), 6.97 (t, 1H), 6.87 (d, 1H), 4.03 (s, 2H), 3.92 (d, 2H), 3.61 (s, 3H)1 3.59 (t, 2H), 2.97 (m, 3H), 2.78 (m, 2H), 2.64 (d, 2H)1 2.51 (t, 2H), 2.21 (m, 2H), 2.09 (t, 2H), 1.77 (m, 4H). ES-MS m/z 511.00 (C28H35CIN4O3 + I)+.
Example 101 : 3-[4-(4-Chloro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5- ylmethyl}-{2-[4-(2-methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine, hydrochloride

PREPARATIONS 51 - 52 Preparations 51 - 52 represent the Block 1 intermediates or headpieces in the synthesis of compounds of formula (III). The Block 1 intermediates may be prepared according to Example 101 (Steps A, B, C, and D), using the indicated starting materials in TABLE 5 below in place of (4-chloro-phenyl)-acetonitrile in Example 101 , Step A.

Prep. Starting Material Ri MS (Cl) Data*
51 (4-fIuoro-phenyl)- F H m/z 297 [M+H] acetonitrile
52 (3-methoxy-phenyl)- H MeO m/z 309 [M+H] acetonitrile
EXAMPLES 102 - 109
Examples 102 - 109 set forth in Table 6 below represent compounds of formula (I) which may be prepared by combining the Block 1 intermediates or headpieces of Table 5 with the Block 2 intermediates or tailpieces of Table 3 according to procedures substantially similar to that described for Example 101.
TABLE 6


[1 ,2,4]oxadiazol-5- 2H), 1.98 (m, 2H) ylmethyl}-amine, dihydrochloride
METHODS AND FORMULATIONS
In some situations, compounds of the invention may exist in isomeric form; for example, as tautomers, enantiomers, or diasteromers. Some compounds may exhibit polymorphism. All enantiomers, and diasteromers are incorporated within the definition of the compounds of the invention. It is further to be understood that the present invention encompasses any racemic, optically-active, polymorphic, or stereoisomeric form, or mixtures thereof, of a compound of the invention, which possess the useful properties described herein, it being well known in the art how to prepare optically active forms (for example, by resolution of the racemic form by recrystallization techniques, by synthesis from optically-active starting materials, by chiral synthesis, or by chromatographic separation using a chiral stationary phase) and how to determine activity or cytotoxicity using the standard tests described herein, or using other similar tests which are well known in the art. Certain of the compounds of the present invention can exist in unsolvated forms as well as solvated forms, including h yd rated forms. In general, the solvated forms, including hydrated forms, are equivalent to unsolvated forms and are intended to be encompassed within the scope of the present invention.
In one embodiment, the present invention provides a method for causing vasodilation in a patient in need thereof comprising administering a compound of formulae (I), (II), or (III). For the sake of brevity, all of the sub-formulae of formula (II), i.e. formulae (MA), (NB), (HC), and (MD), are included when mentioning compounds of formula (II).
In another embodiment, the present invention provides a method of blocking calcium channels, the method comprising of administering to a patient in need of calcium channel blocking a therapeutically effective amount of a compound of formulae (I) or (II) to block calcium channels. In a preferred embodiment of this method, the calcium channels are T-type calcium channels. In another preferred embodiment of this method, the calcium channels are N-type, T-type, and L-type calcium channels.
In a further embodiment, the present invention provides a method of treating a disease selected from hypertension, congestive heart failure, stroke, ischaemic heart disease, and angina pectoris comprising administering to a patient in need thereof a therapeutically effective amount of a compound of formulae (I), (II), or (III). It is also recognized that one skilled in the art may affect the associated diseases and conditions by treating a patient presently afflicted with the diseases or conditions or by prophylactically treating a patient afflicted with the diseases or conditions with a therapeutically effective amount of the compounds of formulae (I), (II), or (III).
Another aspect of this invention is directed to methods of reducing myocardial tissue damage (e.g., substantially preventing tissue damage, inducing tissue protection) during surgery (e.g., coronary artery bypass grafting (CABG) surgeries, vascular surgeries, percutaneous transluminal coronary angioplasty (PTCA) or any percutaneous transluminal coronary intervention (PTCI), organ transplantation, or other non-cardiac surgeries) comprising administering to a mammal (e.g., a female or male human) a therapeutically effective amount of a compound of formulae (I), (II), or (III), or a pharmaceutically acceptable salt of said compound.
The compounds of formula (I), (II), and (III), which are N-type calcium channel antagonists, are potentially useful in the treatment of a range of disorders. The treatment of pain, particularly neuropathic pain, is a preferred use. Physiological pain is an important protective mechanism designed to warn of danger from potentially injurious stimuli from the external environment. The system operates through a specific set of primary sensory neurones and is activated by noxious stimuli via peripheral transducing mechanisms (see Millan, 1999, Prog. Neurobiol., 57, 1-164 for a review). These sensory fibres are known as nociceptors and are characteristically small diameter axons with slow conduction velocities. Nociceptors encode the intensity, duration and quality of noxious stimulus and by virtue of their topographically organised projection to the spinal cord, the location of the stimulus. The nociceptors are found on nociceptive nerve fibres of which there are two main types, A- delta fibres (myelinated) and C fibres (non-myelinated). The activity generated by nociceptor input is transferred, after complex processing in the dorsal horn, either directly, or via brain stem relay nuclei, to the ventrobasal thalamus and then on to the cortex, where the sensation of pain is generated.
Pain may generally be classified as acute or chronic. Acute pain begins suddenly and is short-lived (usually twelve weeks or less). It is usually associated with a specific cause such as a specific injury and is often sharp and severe. It is the kind of pain that can occur after specific injuries resulting from surgery, dental work, a strain or a sprain. Acute pain does not generally result in any persistent psychological response. In contrast, chronic pain is long-term pain, typically persisting for more than three months and leading to significant psychological and emotional problems. Common examples of chronic pain are neuropathic pain (e.g. painful diabetic neuropathy, postherpetic neuralgia), carpal tunnel syndrome, back pain, headache, cancer pain, arthritic pain and chronic post-surgical pain.
When a substantial injury occurs to body tissue, via disease or trauma, the characteristics of nociceptor activation are altered and there is sensitisation in the periphery, locally around the injury and centrally where the nociceptors terminate. These effects lead to a hightened sensation of pain. In acute pain these mechanisms can be useful, in promoting protective behaviours which may better enable repair processes to take place. The normal expectation would be that sensitivity returns to normal once the injury has healed. However, in many chronic pain states, the hypersensitivity far outlasts the healing process and is often due to nervous system injury. This injury often leads to abnormalities in sensory nerve fibres associated with maladaptation and aberrant activity (Woolf & Salter, 2000, Science, 288, 1765-1768).
Clinical pain is present when discomfort and abnormal sensitivity feature among the patient's symptoms. Patients tend to be quite heterogeneous and may present with various pain symptoms. Such symptoms include: 1) spontaneous pain which may be dull, burning, or stabbing; 2) exaggerated pain responses to noxious stimuli (hyperalgesia); and 3) pain produced by normally innocuous stimuli (allodynia - Meyer et al., 1994, Textbook of Pain, 13-44). Although patients suffering from various forms of acute and chronic pain may have similar symptoms, the underlying mechanisms may be different and may, therefore, require different treatment strategies. Pain can also therefore be divided into a number of different subtypes according to differing pathophysiology, including nociceptive, inflammatory and neuropathic pain.
Nociceptive pain is induced by tissue injury or by intense stimuli with the potential to cause injury. Pain afferents are activated by transduction of stimuli by nociceptors at the site of injury and activate neurons in the spinal cord at the level of their termination.
This is then relayed up the spinal tracts to the brain where pain is perceived (Meyer et al., 1994, Textbook of Pain, 13-44). The activation of nociceptors activates two types of afferent nerve fibres. Myelinated A-delta fibres transmit rapidly and are responsible for sharp and stabbing pain sensations, whilst unmyelinated C fibres transmit at a slower rate and convey a dull or aching pain. Moderate to severe acute nociceptive pain is a prominent feature of pain from central nervous system trauma, strains/sprains, burns, myocardial infarction and acute pancreatitis, post-operative pain (pain following any type of surgical procedure), posttraumatic pain, renal colic, cancer pain and back pain. Cancer pain may be chronic pain such as tumour related pain (e.g. bone pain, headache, facial pain or visceral pain) or pain associated with cancer therapy (e.g. postchemotherapy syndrome, chronic postsurgical pain syndrome or post radiation syndrome). Cancer pain may also occur in response to chemotherapy, immunotherapy, hormonal therapy or radiotherapy. Back pain may be due to herniated or ruptured intervertebral discs or abnormalities of the lumber facet joints, sacroiliac joints, paraspinal muscles or the posterior longitudinal ligament. Back pain may resolve naturally but in some patients, where it lasts over 12 weeks, it becomes a chronic condition which can be particularly debilitating.
Neuropathic pain is currently defined as pain initiated or caused by a primary lesion or dysfunction in the nervous system. Nerve damage can be caused by trauma and disease and thus the term 'neuropathic pain' encompasses many disorders with diverse aetiologies. These include, but are not limited to, peripheral neuropathy, diabetic neuropathy, post herpetic neuralgia, trigeminal neuralgia, back pain, cancer neuropathy, HIV neuropathy, phantom limb pain, carpal tunnel syndrome, central post-stroke pain and pain associated with chronic alcoholism, hypothyroidism, uremia, multiple sclerosis, spinal cord injury, Parkinson's disease, epilepsy and vitamin deficiency. Neuropathic pain is pathological as it has no protective role. It is often present well after the original cause has dissipated, commonly lasting for years, significantly decreasing a patient's quality of life (Woolf and Mannion, 1999, Lancet, 353, 1959-1964). The symptoms of neuropathic pain are difficult to treat, as they are often heterogeneous even between patients with the same disease (Woolf & Decosterd, 1999, Pain Supp., 6, S141-S147; Woolf and Mannion, 1999, Lancet, 353, 1959-1964). They include spontaneous pain, which can be continuous, and paroxysmal or abnormal evoked pain, such as
hyperalgesia (increased sensitivity to a noxious stimulus) and allodynia (sensitivity to a normally innocuous stimulus).
The inflammatory process is a complex series of biochemical and cellular events, activated in response to tissue injury or the presence of foreign substances, which results in swelling and pain (Levine and Taiwo, 1994, Textbook of Pain, 45-56). Arthritic pain is the most common inflammatory pain. Rheumatoid disease is one of the commonest chronic inflammatory conditions in developed countries and rheumatoid arthritis is a common cause of disability. The exact aetiology of rheumatoid arthritis is unknown, but current hypotheses suggest that both genetic and microbiological factors may be important (Grennan & Jayson, 1994, Textbook of Pain, 397-407). It has been estimated that almost 16 million Americans have symptomatic osteoarthritis (OA) or degenerative joint disease, most of whom are over 60 years of age, and this is expected to increase to 40 million as the age of the population increases, making this a public health problem of enormous magnitude (Houge & Mersfelder, 2002, Ann Pharmacother., 36, 679-686; McCarthy et al., 1994, Textbook of Pain, 387-395). Most patients with osteoarthritis seek medical attention because of the associated pain. Arthritis has a significant impact on psychosocial and physical function and is known to be the leading cause of disability in later life. Ankylosing spondylitis is also a rheumatic disease that causes arthritis of the spine and sacroiliac joints. It varies from intermittent episodes of back pain that occur throughout life to a severe chronic disease that attacks the spine, peripheral joints and other body organs.
Another type of inflammatory pain is visceral pain which includes pain associated with inflammatory bowel disease (IBD). Visceral pain is pain associated with the viscera, which encompass the organs of the abdominal cavity. These organs include the sex organs, spleen and part of the digestive system. Pain associated with the viscera can be divided into digestive visceral pain and non-digestive visceral pain. Commonly encountered gastrointestinal (Gl) disorders that cause pain include functional bowel disorder (FBD) and inflammatory bowel disease (IBD). These Gl disorders include a wide range of disease states that are currently only moderately controlled, including, in respect of FBD, gastro-esophageal reflux, dyspepsia, irritable bowel syndrome (IBS) and functional abdominal pain syndrome (FAPS), and, in respect of IBD, Crohn's disease, ileitis and ulcerative colitis, all of which regularly produce visceral pain. Other
types of visceral pain include the pain associated with dysmenorrhea, cystitis and pancreatitis and pelvic pain.
It should be noted that some types of pain have multiple aetiologies and thus can be classified in more than one area, e.g. back pain and cancer pain have both nociceptive and neuropathic components.
Other types of pain include:
• pain resulting from musculoskeletal disorders, including myalgia, fibromyalgia, spondylitis, sero-negative (non-rheumatoid) arthropathies, non-articular rheumatism, dystrophinopathy, glycogenosis, polymyositis and pyomyositis; • heart and vascular pain, including pain caused by angina, myocardical infarction, mitral stenosis, pericarditis, Raynaud's phenomenon, scleredoma and skeletal muscle ischemia;
• head pain, such as migraine (including migraine with aura and migraine without aura), cluster headache, tension-type headache mixed headache and headache associated with vascular disorders; and
• orofacial pain, including dental pain, otic pain, burning mouth syndrome and temporomandibular myofascial pain.
As used herein, the term "patient" refers to a warm-blooded animal such as a mammal which is (1) in need of vasodilation, (2) in need of blocking calcium channels, (3) afflicted with or at risk of developing hypertension, congestive heart failure, stroke, ischaemic heart disease, or angina pectoris, or (4) afflicted with pain or a sub-category of pain as described above. It is understood that guinea pigs, dogs, cats, rats, mice, horses, cattle, sheep, and humans are examples of animals within the scope of the meaning of the term. A patient is in need of treatment for hypertension, congestive heart failure, stroke, ischaemic heart disease, angina pectoris, or pain when the patient is afflicted within one or more of the diseases or conditions described herein or is at a recognized risk of developing one or more of the diseases or conditions described herein as diagnosed by an attending physician or clinician.
The identification of those patients who are in need of treatment for hypertension, congestive heart failure, stroke, ischaemic heart disease, angina pectoris, or pain is well within the ability and knowledge of one skilled in the art. A clinician skilled in the art can readily identify, by the use of clinical tests, physical examination and medical/family history, those patients who are in need of such treatment.
As used herein, the term "therapeutically effective amount" of a compound of formulae (I), (II), or (III) refers to an amount which is effective in (1) causing vasodilation in the patient in need thereof, (2) blocking calcium channels, (3) treating hypertension, congestive heart failure, stroke, ischaemic heart disease, or angina pectoris, or (4) treating pain. The term "treating" is intended to refer to all processes wherein there may be a slowing, interrupting, arresting, or stopping of the progression of the diseases and conditions described herein, but does not necessarily indicate a total elimination of all disease and condition symptoms, and is intended to include prophylactic treatment of the hypertension, congestive heart failure, stroke, ischaemic heart disease, angina pectoris, or pain.
A therapeutically effective amount can be readily determined by the attending diagnostician, as one skilled in the art, by the use of conventional techniques and by observing results obtained under analogous circumstances. In determining the therapeutically effective amount, the dose, a number of factors are considered by the attending diagnostician, including, but not limited to: the species of mammal; its size, age, and general health; the specific disease involved; the degree of involvement or the severity of the disease; the response of the individual patient; the particular compound administered; the mode of administration; the bioavailability characteristic of the preparation administered; the dose regimen selected; the use of concomitant medication; and other relevant circumstances.
The compounds of the invention intended for pharmaceutical use may be administered alone or in combination with one or more other compounds of the invention or in combination with one or more other drugs (or as any combination thereof). Generally, they will be administered as a formulation in association with one or more pharmaceutically acceptable excipients. The term "excipient" is used herein to describe any ingredient other than the compound(s) of the invention. The choice of excipient will to a large extent depend on factors such as the particular mode of administration, the effect of the excipient on solubility and stability, and the nature of the dosage form. Pharmaceutical compositions suitable for the delivery of compounds of the present invention and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation may be found,
for example, in 'Remington's Pharmaceutical Sciences', 19th Edition (Mack Publishing Company, 1995).
The compounds of the invention may be administered orally. Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, or buccal or sublingual administration may be employed by which the compound enters the blood stream directly from the mouth.
Formulations suitable for oral administration include solid formulations, such as tablets, capsules containing particulates, liquids, or powders; lozenges (including liquid- filled), chews; multi- and nano-particulates; gels, solid solution, liposome, films (including muco-adhesive), ovules, sprays and liquid formulations.
Liquid formulations include suspensions, solutions, syrups and elixirs. Such formulations may be employed as fillers in soft or hard capsules and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet.
The compounds of the invention may also be used in fast-dissolving, fast- disintegrating dosage forms such as those described in Expert Opinion in Therapeutic Patents, ϋ (6), 981-986 by Liang and Chen (2001). For tablet dosage forms, depending on dose, the drug may make up from 1 weight% to 80 weight% of the dosage form, more typically from 5 weight% to 60 weightt% of the dosage form. In addition to the drug, tablets generally contain a disintegrant. Examples of disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl-substituted hydroxypropyl cellulose, starch, pregelatinised starch and sodium alginate. Generally, the disintegrant will comprise from 1 weight% to 25 weight%, preferably from 5 weight% to 20 weight% of the dosage form.
Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol,
xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate.
Tablets may also optionally comprise surface active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc. When present, surface active agents may comprise from 0.2 weight % to 5 weight% of the tablet, and glidants may comprise from 0.2 weight% to 1 weight% of the tablet.
Tablets also generally contain lubricants such as magnesium s tea rate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate. Lubricants generally comprise from 0.25 weight% to 10 weight%, preferably from 0.5 weight% to 3 weight% of the tablet.
Other possible ingredients include anti-oxidants, colourants, flavoring agents, preservatives and taste-masking agents.
Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting. The final formulation may comprise one or more layers and may be coated or uncoated; it may even be encapsulated.
The formulation of tablets is discussed in "Pharmaceutical Dosage Forms: Tablets, Vol. 1", by H. Lieberman and L. Lachman, Marcel Dekker, N.Y., N.Y., 1980 (ISBN 0-8247-6918-X). The foregoing formulations for the various types of administration discussed above may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
Suitable modified release formulations for the purposes of the invention are described in US Patent No. 6,106,864. Details of other suitable release technologies such as high energy dispersions and osmotic and coated particles are to be found in
Verma et a/, Pharmaceutical Technology On-line, 25(2), 1-14 (2001). The use of chewing gum to achieve controlled release is described in WO 00/35298.
The compounds of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ. Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intra urethral, intrasternal, intracranial, intramuscular and subcutaneous.
Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile non-aqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
The preparation of parenteral formulations under sterile conditions, for example, by lyophilisation, may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
The solubility of compounds of formula (I) used in the preparation of parenteral solutions may be increased by the use of appropriate formulation techniques, such as the incorporation of solubility-enhancing agents.
Formulations for parenteral administration may be formulated to be immediate and/or modified release. Thus, compounds of the invention may be formulated as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound. Examples of such formulations include drug- coated stents and PGLA microspheres.
The compounds of the invention may also be administered topically to the skin or mucosa, that is, dermally or transdermally. Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibres, bandages and microemulsions. Liposomes may also be used. Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol. Penetration enhancers may be incorporated; see, for example, J
Pharm Sci, 88 (10), 955-958 by Finnin and Morgan (October 1999).
Other means of topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and microneedle or needle-free (e.g. Powderject™, Bioject™, etc.) injection. The compounds of the invention may also be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an aerosol
spray from a pressurised container, pump, spray, atomiser (preferably an atomiser using electrohydrodynamics to produce a fine mist), or nebuliser, with or without the use of a suitable propellant, such as 1 ,1 ,1 ,2-tetrafluoroethane or 1,1 ,1,2,3,3,3- heptafluoropropane. For intranasal use, the powder may comprise a bioadhesive agent, for example, chitosan or cyclodextrin.
The pressurised container, pump, spray, atomizer, or nebuliser contains a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
Prior to use in a dry powder or suspension formulation, the drug product is micronised to a size suitable for delivery by inhalation (typically less than 5 microns).
This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenisation, or spray drying.
Capsules (made, for example, from gelatin or HPMC), blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the compound of the invention, a suitable powder base such as lactose or starch and a performance modifier such as /-leucine, mannitol, or magnesium stearate. The lactose may be anhydrous or in the form of the monohydrate, preferably the latter. Other suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
A suitable solution formulation for use in an atomiser using electrohydrodynamics to produce a fine mist may contain from 1 μg to 20mg of the compound of the invention per actuation and the actuation volume may vary from 1μl to 100μl. A typical formulation may comprise a compound of formula (I), propylene glycol, sterile water, ethanol and sodium chloride. Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
Suitable flavours, such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations of the invention intended for inhaled/intranasal administration.
Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, poly(DL-lactic-coglycolic acid
(PGLA). Modified release formulations include delayed-, sustained-, pulsed-, controlled- , targeted and programmed release.
In the case of dry powder inhalers and aerosols, the dosage unit is determined by means of a valve which delivers a metered amount. Units in accordance with the invention are typically arranged to administer a metered dose or "puff' of the compound of formula (I). The overall daily may be administered in a single dose or, more usually, as divided doses throughout the day.
The compounds of the invention may be administered rectally or vaginally, for example, in the form of a suppository, pessary, or enema. Cocoa butter is a traditional suppository base, but various well known alternatives may be used as appropriate.
The compounds of the invention may also be administered directly to the eye or ear, typically in the form of drops of a micronised suspension or solution in isotonic, pH- adjusted, sterile saline. Other formulations suitable for ocular and aural administration include ointments, biodegradable (e.g. absorbable gel sponges, collagen) and non- biodegradable (e.g. silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes. A polymer such as crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid; a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose; or a heteropolysacchahde polymer, for example, gelan gum, may be incorporated together with a preservative, such as benzalkonium chloride. Such formulations may also be delivered by iontophoresis.
The compounds of the invention may be combined with soluble macromolecular entities, such as cyclodextrin and suitable derivatives thereof or polyethylene glycol- containing polymers, in order to improve their solubility, dissolution rate, taste-masking, bioavailability and/or stability for use in any of the aforementioned modes of administration.
Drug-cyclodextrin complexes, for example, are found to be generally useful for most dosage forms and administration routes. Both inclusion and non-inclusion complexes may be used. As an alternative to direct complexation with the drug, the cyclodextrin may be used as an auxiliary additive, i.e. as a carrier, diluent, or solubiliser. Most commonly used for these purposes are alpha-, beta- and gamma-cyclodextrins, examples of which may be found in International Patent Applications Nos. WO 91/11172, WO 94/02518 and WO 98/55148.
A calcium channel blocker of the present invention, particularly those showing N- type calcium channel antagonism, may be usefully combined with another pharmacologically active compound, or with two or more other pharmacologically active compounds, particularly in the treatment of pain. For example, a compound of formulae (I), (II), or (III), or a pharmaceutically acceptable salt thereof, as defined above, may be administered simultaneously, sequentially or separately in combination with one or more agents selected from:
(i) opioid analgesics, e.g. morphine, heroin, hydromorphone, oxymorphone, levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine, dihydrocodeine, oxycodone, hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, nalbuphine and pentazocine;
(ii) nonsteroidal antiinflammatory drugs (NSAIDs), e.g. aspirin, diclofenac, diflusinal, etodolac, fenbufen, fenoprofen, flufenisal, flurbiprofen, ibuprofen, indomethacin, ketoprofen, ketorolac, meclofenamic acid, mefenamic acid, nabumetone, naproxen, oxaprozin, phenylbutazone, piroxicam, sulindac, tolmetin, zomepirac, and their pharmaceutically acceptable salts;
(iii) barbiturate sedatives, e.g. amobarbital, aprobarbital, butabarbital, butabital, mephobarbital, metharbital, methohexital, pentobarbital, phenobartital, secobarbital, talbutal, theamylal, thiopental and their pharmaceutically acceptable salts; (iv) benzodiazepines having a sedative action, e.g. chlordiazepoxide, clorazepate, diazepam, flurazepam, lorazepam, oxazepam, temazepam, triazolam and their pharmaceutically acceptable salts,
(v) Hi antagonists having a sedative action, e.g. diphenhydramine, pyrilamine, promethazine, chlorpheniramine, chlorcyclizine and their pharmaceutically acceptable salts;
(vi) miscellaneous sedatives such as glutethimide, meprobamate, methaqualone, dichloralphenazone and their pharmaceutically acceptable salts;
(vii) skeletal muscle relaxants, e.g. baclofen, carisoprodol, chlorzoxazone, cyclobenzaprine, methocarbamol, orphrenadine and their pharmaceutically acceptable salts,
(viii) NMDA receptor antagonists, e.g. dextromethorphan ((+)-3-hydroxy-N- methylmorphinan) and its metabolite dextrorphan ((+)-3-hydroxy-N- methylmorphinan), ketamine, memantine, pyrroloquinoline quinone and cis-4-
(phosphonomethyl)-2- piperidinecarboxylic acid and their pharmaceutically acceptable salts;
(ix) alpha-adrenergic active compounds, e.g. doxazosin, tamsulosin, clonidine and 4- amino-6,7-dimethoxy-2-(5-methanesulfonamido-1 ,2,3,4-tetrahydroisoquinol-2-yl)- 5-(2-pyridyl) quinazoline;
(x) tricyclic antidepressants, e.g. desipramine, imipramine, amytriptiline and nortriptiline;
(xi) anticonvulsants, e.g. carbamazepine and valproate;
(xii) Tachykinin (NK) antagonists, particularly Nk-3, NK-2 and NK-1 e.g. antagonists, (αR,9R)-7-[3,5-bis(trifluoromethyl)benzyl]-8,9, 10,11 -tetrahydro-9- methyl-5-(4-methylphenyl)-7H-[1 ,4]diazocino[2, 1 -g][1 ,7]naphthridine-6-13-dione (TAK-637), 5-[[(2R,3S)-2-[(1 R)-1-[3,5-bis(trifluoromethyl)phenyl]ethoxy-3-(4- fluorophenyl)-4-morpholinyl]methyl]-1 ,2-dihydro-3H-1 ,2,4-triazol-3-one (MK-869), lanepitant, dapitant and 3-[[2-methoxy-5-(trifluoromethoxy)phenyl]methylamino]- 2-phenyl-pipehdine (2S.3S)
(xiii) Muscarinic antagonists, e.g oxybutin, tolterodine, propiverine, tropsium chloride and darifenacin;
(xiv) COX-2 inhibitors, e.g. celecoxib, rofecoxib and valdecoxib;
(xv) Non-selective COX inhibitors (preferably with Gl protection), e.g. nitroflurbiprofen (HCT-1026);
(xvi) coal-tar analgesics, in particular, paracetamol; (xvii) neuroleptics, such as droperidol;
(xviii) Vanilloid receptor agonists, e.g. resinferatoxin;
(xix) Beta-adrenergic compounds such as propranolol;
(xx) Local anaesthetics, such as mexiletine;
(xxi) Corticosteriods, such as dexamethasone (xxii) serotonin receptor agonists and antagonists;
(xxiii) cholinergic (nicotinic) analgesics;
(xxiv) miscellaneous agents such as Tramadol®;
(xxv) PDEV inhibitors, such as sildenafil, vardenafil or taladafil;
(xxvi) serotonin reuptake inhibitors, e.g. fluoxetine, paroxetine, citalopram and sertraline; (xxvii) mixed serotonin-noradrenaline reuptake inhibitors, e.g. milnacipran, venlafaxine and duloxetine; (xxviii) noradrenaline reuptake inhibitors , e.g. reboxetine; (xxix) alpha-2-delta ligands, e.g. gabapentin and pregabalin.
The compounds of the invention may be usefully combined with one or more agents for reducing the risk of a cardiovascular disorder including anti-inflammatory agents, such as alclofenac, algestone acetonide, alpha amylase, amcinafal, amcinafide, amfenac sodium, amiprilose hydrochloride, anakinra, anirolac, apazone, balsalazide disodium, bendazac, benoxaprofen, benzydamine hydrochloride, bromelains, broperamole, budesonide, carprofen, cicloprofen, cintazone, cliprofen, clobetasol propionate, clobetasone butyrate, clopirac, cloticasone propionate, cortodoxone, deflazacort, desonide, desoximetasone, dexamethasone dipropionate, diclofenac potassium, diclofenac sodium, diflumidone sodium, diflunisal, difluprednate, diftalone, drocinonide, enlimomab, enolicam sodium, epirizole, etodolac, etofenamate, felbinac, fenamole, fenbufen, fenclofenac, fenclorac, fendosal, fenpipalone, fentiazac, flazalone, fluazacort, flufenamic acid, flumizole, flunisolide acetate, flunixin, flunixin meglumine, fluocortin butyl, fluorometholone acetate, fluquazone, flurbiprofen, fluretofen, fluticasone propionate, furaprofen, furobufen, ibufenac, ibuprofen, ibuprofen aluminum, ilonidap, indomethacin, indomethacin sodium, indoprofen, indoxole, intrazole, isoflupredone acetate, isoxepac, isoxicam, ketoprofen, lofemizole hydrochloride, lornoxicam, meclofenamate sodium, meclofenamic acid, mefenamic acid, mesalamine, meseclazone, methylprednisolone suleptanate, morniflumate, nabumetone, naproxen, naproxen sodium, naproxol, nimazone, olsalazine sodium, orgotein, orpanoxin, oxaprozin, oxyphenbutazone, paranyline hydrochloride, pentosan polysulfate sodium, phenbutazone sodium glycerate, pirfenidone, piroxicam, piroxicam cinnamate, piroxicam olamine, pirprofen, prednazate, prifelone, prodolic acid, proquazone, proxazole, proxazole citrate, rimexolone, romazarit, salcolex, salsalate, salycilates, sanguinarium chloride, seclazone, sermetacin, sudoxicam, sulindac, suprofen, talmetacin, talniflumate, talosalate, tebufelone, tenidap, tenidap sodium, tenoxicam, tesicam, tesimide, tetrydamine, tiopinac, tolmetin, tolmetin sodium, triclonide,
triflumidate, zidometacin, zomepirac sodium; anti-thrombotic and/or fibrinolytic agents, such as plasminogen (to plasmin via interactions of prekallikrein, kininogens, Factors XII, XIIIa, plasminogen proactivator, and tissue plasminogen activatorfTPA]) streptokinase, urokinase: anisoylated plasminogen-streptokinase activator complex; pro-urokinase, (Pro-UK); rTPA (altepiase or activase; r denotes recombinant), rPro-UK, abbokinase, eminase, sreptase anagrelide hydrochloride, bivalirudin, dalteparin sodium, danaparoid sodium, dazoxiben hydrochloride, efegatran sulfate, enoxaparin sodium, ifetroban, ifetroban sodium, tinzaparin sodium, retaplase, trifenagrel, warfarin, dextrans; anti-platelet agents, such as clopridogrel, sulfinpyrazone, aspirin; dipyridamole, clofibrate, pyridinol carbamate, PGE, glucagon, antiserotonin drugs, caffeine, theophyllin pentoxifyllin, ticlopidine, anagrelide; lipid reducing agents, such as gemfibrozil, cholystyramine, colestipol, nicotinic acid, probucol, lovastatin, fluvastatin, simvastatin, atorvastatin, pravastatin, cirivastatin; and direct thrombin inhibitors, such as hirudin, hirugen, hirulog, agatroban, PPACK, and thrombin aptamers. Useful dosages of the compounds of the invention can be determined by comparing their in vitro activity, and in vivo activity in animal models. The amount of the compound, or an active salt or derivative thereof, required for use in treatment will vary not only with the particular salt selected but also with the route of administration, the nature of the condition being treated and the age and condition of the patient and will be ultimately at the discretion of the attendant physician or clinician.
The compounds of the present invention can be administered to a human patient at dosage levels in the range of about 1 to about 2,000 mg per day, preferably from about 1 to about 1 ,000 mg per day, more preferably from about 5 to about 600 mg per day, even more preferably from about 10 to 300 mg per day. For a normal human adult having a body weight of about 70 kilograms, a dosage in the range of about 0.01 to about 10 mg per kilogram of body weight per day is preferable. However, the specific dosage used can vary. For example, the dosage can depended on a numbers of factors including the requirements of the patient, the severity of the condition being treated, and the pharmacological activity of the compound being used. The determination of optimum dosages for a particular patient is well-known to those skilled in the art.
Ideally, the active ingredient should be administered to achieve peak plasma concentrations of the active compound of from about 0.5 to about 75 μM, preferably,
about 1 to 50 μM, most preferably, about 0.1 to about 5 μM. This may be achieved, for example, by the intravenous injection of a 0.05 to 5% solution of the active ingredient, optionally in saline, or orally administered as a bolus containing about 10-500 mg of the active ingredient. Desirable blood levels may be maintained by multiple oral dosing, or continuous infusion to provide about 0.01-5.0 mg/kg/hr or by intermittent infusions containing about 0.4-15 mg/kg of the active ingredient(s).
The desired dose may conveniently be presented in a single dose or as divided doses administered at appropriate intervals, for example, as two, three, four or more sub-doses per day. The sub-dose itself may be further divided, e.g., into a number of discrete loosely spaced administrations; such as multiple inhalations from an insufflator or by application of a plurality of drops into the eye.
The ability of a compound of the present invention to modulate the T-, L-, and N- type calcium channels is demonstrated using pharmacological models that are well known to the art, for example, using models such as the tests described below.
T-tvpe Calcium Channel FLIPR Assay
A stable tetracycline-inducible TREx-293 cell line is generated expressing recombinant mouse α1H.
The coding sequence for mouse α1H T-type calcium channel cDNA (accession number NM_021415) is cloned using standard cloning techniques (e.g. Molecular
Cloning A Laboratory Manual, 2nd Edition, J. Sambrook, E. F. Fritsch, T. Maniatis; Cold
Spring Habor Laboratory Press; Cold Spring Habor, N.Y., 1989) and is subcloned into the mammalian expression vector pcDNA4rTO (Invitrogen, Carlsbad CA). This expression vector has a CMV promoter to drive expression of the α1H gene. This plasmid construct is used to stably transfect TREx-293 cells (Invitrogen, Carlsbad CA), a human embryonic kidney cell line stably expressing the tetracycline repressor protein.
Cells are maintained at 37° C and 5% CO2 in Dulbecco's Modified Eagle Media supplemented with 10% fetal bovine serum, 200 μg/ml Zeocin, and 5 μg/ml Blasticidin. Cells are induced with 1 μg/ml tetracycline and plated onto black-sided 384 well PoIy-D Lysine coated plates at 12,000 cells/well for at least twenty-four hours. The cells are incubated with the fluorescent Ca2+ indicator Fluo-4 AM (50 μg, Molecular Devices, Sunnyvale, CA) dissolved in pluronic acid and DMEM supplemented with 2.6 mM probenecid for 1 hour at 37°C and 5% CO2. Cells are then rinsed with assay buffer
(consisting of 0.34 mM Na2HPO4, 4.2 mM NaHCO3, 0.44 mM KH2PO4, 0.41 mM MgSO4, 0.49 mM MgCI2, 20 mM HEPES, 5.5 mM d-Glucose, 0.1% BSA, 137 mM NaCI, and 2.6 mM probenecid) and incubated at 37°C and 5% CO2 for 10 minutes. Cells are pretreated with putative antagonists for 5 minutes followed by a rapid increase of 4.8 mM extracellular Ca2+. The increase in intracellular calcium as determined by an increase in fluorescence is then measured for 5 minutes following the extracellular Ca2+ addition on a Fluorometric Imaging Plate Reader (FLIPR, Molecular Devices, Sunnyvale, Calif.). Fluorescence increase in response to Ca2+ in the presence of test compounds is compared to control responses in the same plate and the IC50 for each compound is determined using Prism 4.0 (GraphPad Software, Inc., San Diego CA). The results of this evaluation are shown in Table 7.
L-Tvpe Calcium Channel FLIPR A10 Assay
FLIPR Instrumentation: FLIPR (fluorometric imaging plate reader) is Molecular Devices's rapid throughput system for cell-based fluorescence assays. In this application, it is used specifically for measuring changes in intracellular calcium flux. The basic principal involves illumination of 384-well microplate with the simultaneous measurement of emitted fluorescence using a CCD camera. The cell plate wells contain cells that have been loaded with a fluorescent dye, whose emission characteristics change upon binding with a particular ion (Ca2+ in this case). As a result, changes in fluorescence can be quantified and thus extrapolated to represent some specific pharmacologic effect (or lack thereof).
Cell Culture and Dye Loading: The A10 smooth muscle cell line derived from embryonic thoracic aorta of the DBIX rat (ATCC, CRL-1476); which endogenously expresses L-type calcium channels; is used for this assay. The growth media for A10 cells is Ham's F12/DME high glucose (Irvine Scientific, 9052), supplemented with 20% fetal bovine serum (HyClone Labs, SH30071.02), and 1% each of L-glutamine (Gibco BRL, 25030-032) and antibiotic-antimycotic (Gibco BRL, 15240-096).
Cells are grown to confluency and replated on black-sided 384-well plates (Falcon, 35 3962) at 12K cells/well for use in FLIPR. Forty-eight hours after replating, growth media is removed and cells are loaded at 370C with 50μl media containing 1μM Fluo-4 dye (Molecular Probes, F-14201) for 1 hour. The dye-containing media Is then washed away six times with buffer (composition in mM: 1.25 CaCI2, 1.2 MgSO4, 11
glucose, 10 HEPES, 3.0 KCI, 137.0 NaCI, pH 7.4 with Tris base) in an Embla384 (Molecular Devices, 0200-3906). The residual buffer volume is adjusted to 20μl and allowed to incubate at room temp for an additional hour.
FLIPR protocol: A five minute drug-pre-incubation period at is initiated when 20μl of drug-containing buffer is pipetted into the cell plate with the 384-well pipettor integrated in the FLIPR apparatus. Fluorescent counts are monitored at two second intervals for 960 seconds, beginning 60 seconds prior to the delivery of drug-containing buffer. Following drug addition, 20μl aliquots of a high K+, depolarizing stimulus (composition in mM: 1.25 CaCI2, 1.2 MgSO4, 11 glucose, 10 HEPES, 140.0 KCI, pH 7.4 with Tris base) are added to each well and fluorescence is monitored at one second intervals for 120 secibds, beginning ten seconds prior to the stimulus addition. CCD camera exposure time is 0.4 seconds, laser excitation is at 488 nm with a power of 0.6W, and a 510 to 560nm bandpass interference filter preceded the camera.
Data Analysis: Data are analyzed as a summation of fluorescent counts above basal during the stimulation period (an approximation of area under the curve), after normalizing the data with a spatial uniformity correction (for variations in laser illumination and cell density), a negative control correction and a bias subtraction (a bias subtraction subtracts the fluorescence value measured at a specific sample point from all the other time points in each well and allows for all data on the y-axis to be zeroed). Drug effects are expressed as percent inhibition of fluorescence from an average of 8 K+-stimulated wells that were pre-treated in the drug incubation period with buffer only. Data are analyzed using FLIPR software, Microsoft Excel and Origin. The IC50 calculations are performed and graphed in Origin.
N-tvpe Calcium Channel FDSS6000 Assay
A stable cell line is generated expressing recombinant rat D I B. The coding sequence for rat DIB CDNA (accession number AF055477) is cloned using standard cloning techniques (e.g. Molecular Cloning A Laboratory Manual, 2nd Edition, J.
Sambrook, E. F. Fritsch, T. Maniatis; Cold Spring Habor Laboratory Press; Cold Spring Habor, N.Y., 1989) and is subcloned into the mammalian expression vector pcDNA
6/V5 (lnvitrogen, Carlsbad CA). This plasmid construct is used to stably transfect HEK- tsA201 cells.
Cells are maintained at 37° C and 5% CO2 in Dulbecco's Modified Eagle Media supplemented with 10% fetal bovine serum, 25 μg/ml Zeocin, and 5 μg/ml Blasticidin and 25 μg/ml Hygromycin. Cells are plated onto black-sided 384 well PoIy-D Lysine coated plates at 5,000 cells/well and incubate for 24 hours. The cells are incubated with the fluorescent Ca2+ indicator Fluo-4 AM (50 μg, Molecular Probes, Eugene, OR) dissolved in pluronic acid for 1 hour at 37°C and 5% CO2. Cells are then rinsed with assay buffer (consisting of 1 mM Na2HPO4, 26 mM NaHCO3, 1.8 mM CaCI2, 0.8 mM MgCI2, 117 mM NaCI, 20 mM HEPES, 5.6 mM d-Glucose) and incubated at 37°C for 10 minutes. Cells are pretreated with putative antagonists for 5 minutes followed by an addition of KCI (30 mM). The influx of intracellular calcium as determined by an increase in fluorescence is then measured for 5 minutes following the extracellular K+ addition on a plate reader (FDSS6000, HAMAMATSU, Japan). Fluorescence increase in response to K+ in the presence of test compounds is compared to control responses in the same plate and the IC50 for each compound is determined using Excel Fit™ (Sigmoidal-600, Microsoft). The results of this evaluation are shown in Table 7.
TABLE 7 Calcium Channel Functional Activity











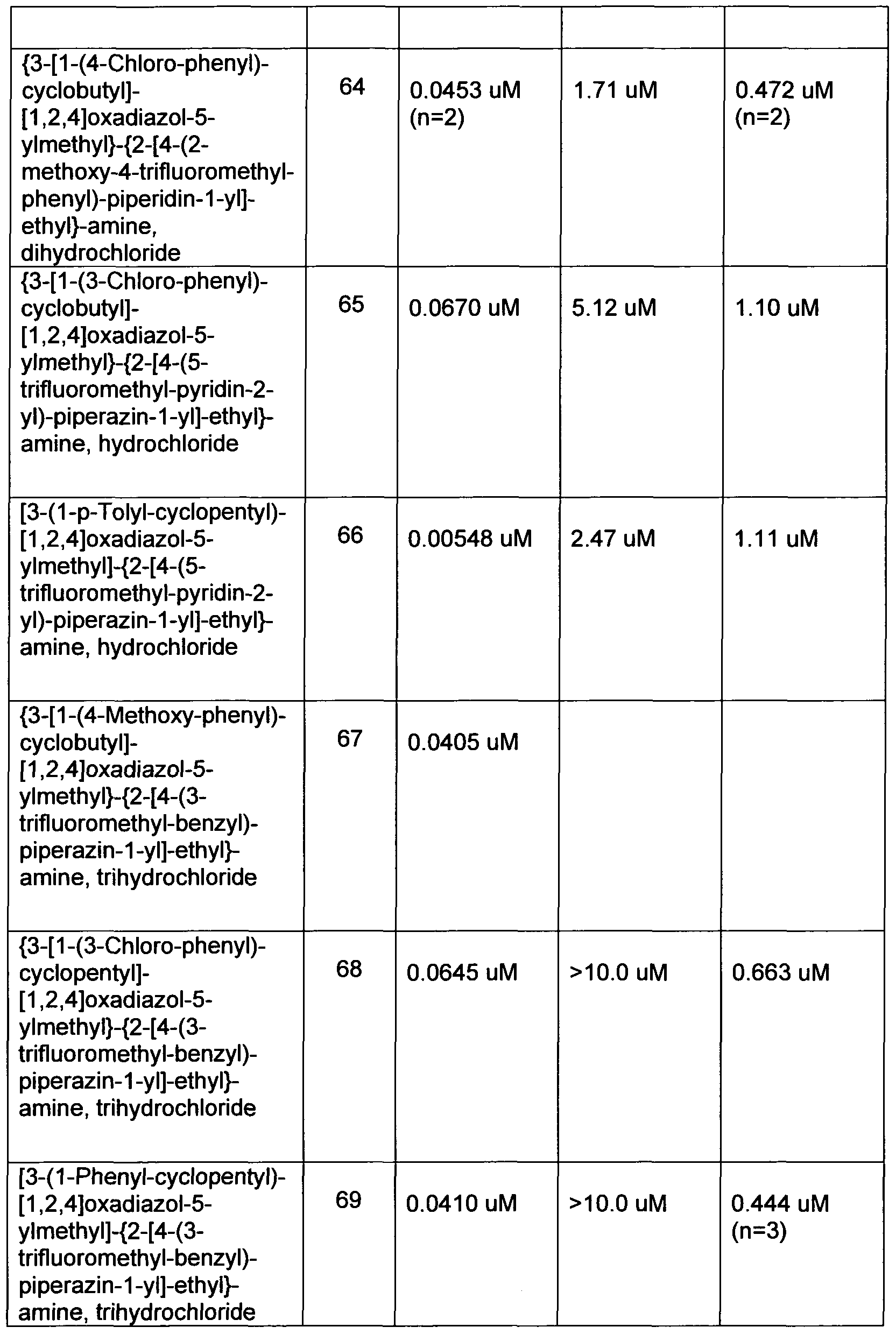







Notes: a: The biological values are from a n=1 , unless otherwise noted.
The ability of a compound of the present invention to lower blood pressure is demonstrated by evaluating compounds for IV antihypertensive efficacy in isoflurane- anesthetized normotensive and/or hypertensive rats.
Rising Dose In Vivo Screen In Anesthetized Hypertensive And/Or Normotensive Male Rats To Identify Compounds Causing A Maximum Reduction Of Mean Arterial Blood Pressure Bv At Least 10%.
Animal Preparation:
Normotensive and/or hypertensive rats are isoflurane-anesthetized and will be prepared for IV administration of compounds solubilized in 5% NMP/45% PEG400/50%
5OmM lactic acid. Two polyethylene PE50 catheters are implanted one into the left carotid artery (BP measurement) and the other into the right jugular vein (compound injection). The raw data will be collected every 5 sec while the summarized data will be digitally recorded every 30 seconds using a PONEMAH acquisition system
(LDS/Gould). After surgery, a 30 min equilibration period (1 % isoflurane setting) is done to allow hemodynamic parameters to stabilize prior to beginning experimental procedure.
Experimental Design:
Compounds will be administered as a initial IV bolus dose (1 ml/kg over 30 seconds) followed by a constant maintenance infusion (0.1ml/kg/min) for 60 minutes. Mean (MBP), systolic (SBP), diastolic (DBP) blood pressures as well as heart rate (HR) will be collected every 30 sec. The computer stores each experiment's data and then the data is manually downloaded via intranet to network server for permanent storage and analysis. Blood samples will be collected at 30 and 60 minutes during the compound infusion for rat pharmacokinetic (PK) data.
Table 8: In Vivo SHR Efficacy Results (IV administration)
Example Number Dose (mg/kg) Max MBP fall (mm Hg)
1 10 -90
2 30
3 10 -51
4 10 -62
6 20 -79
15 10 -64
15 3 -40
44 30 0
The foregoing biological tests establish that the compounds of the present invention are potent calcium channel antagonists.
Claims
1. A compound of the formula (I)

or a pharmaceutically acceptable salt thereof, wherein R1 and R2 are each independently -H, -OH, halo, Ci-C6 alkyl, CrC6 alkoxy, -CF3, substituted CrC6 alkyl, or substituted CrC6 alkoxy;
R3 and R4 are each independently -H or CrC6 alkyl or R3 and R4 taken together with the carbon atom to which they are attached form C3-C6 cycloalkyl, or cycloheteroalkyl, provided that if one of R3 and R4 is -H, then the other is CrC6 alkyl;
R5 is -H, CrC6 alkyl, C1-C6 alkoxy, -(CH2)q-C(0)0-W, wherein W is -H or CrC6 alkyl and q is 1-6;
G1 is methylene or ethylene;
G2 is C(R6) or N, wherein R6 is -H, -OH or Ci-C6 alkyl; Y is -CH2-, -CH2CH2-, -CH2CH2CH2-, -O-, -C(O)-, -C(O)CH2-, -S-, -S(O)-, -S(O)2-,
-NH-, -NHC(O)-, -NHC(O)CH(R7)-, or -NHS(O)2-, wherein R7 is -H or C1-C4 alkyl;
Ar1 is a radical of the formulae  wherein RP1, RP2, RP3, RP4, RP5, RP6, RP7, RP8, RN1, RN2, RN3, and RZ1 are each independently -H, -OH, Ci-C6 alkyl, Ci-C6 alkoxy, halo, -CN, -CF3, or -NR8R9, wherein
wherein RP1, RP2, RP3, RP4, RP5, RP6, RP7, RP8, RN1, RN2, RN3, and RZ1 are each independently -H, -OH, Ci-C6 alkyl, Ci-C6 alkoxy, halo, -CN, -CF3, or -NR8R9, wherein
R8 and R9 are each independently -H or C1-C6 alkyl; RM1, RM2, RB1, RB2, RB3, R64, RB5,
RB6 Rxi Rx2 Rx3 Rx4 RYI RY2 RY3 RY4 RY5 an(J Rγ6 ^ each independently -H or
CrC6 alkyl; X1 and X2 are independently CH or N; X3, X4, and X5 are each independently NH, O, or S; X6 is CH2 or O; Q is substituted CrC6 alkyl, phenyl, napthyl, 2-pyridyl, or 3-pyridyl; and n is O or 1 ; provided that when Y is O, S, NH, NHC(O), NHS(O)2, NHC(O)CH(R7), or NHS(O)2, then G2 is CH.
2. A compound according to claim 1 wherein R1 and R2 are each independently -H, halo, -CF3 or CrC4 alkyl; or a pharmaceutically acceptable salt thereof.
3. A compound according to either claim 1 or claim 2 where R3 and R4 taken together with the carbon atom to which they are attached form C3-C6 cycloalkyl; or a pharmaceutically acceptable salt thereof.
4. A compound according to either claim 1 or 2 where R3 and R4 taken together with the carbon atom to which they are attached form cycloheteroalkyl; or a pharmaceutically acceptable salt thereof.
5. A compound according to any of claims 1 to 4 wherein R5 is -H; or a pharmaceutically acceptable salt thereof.
6. A compound according to any of claims 1 to 5 wherein G1 is methylene; or a pharmaceutically acceptable salt thereof.
7. A compound according to any of claims 1 to 6 wherein n is 0; or a pharmaceutically acceptable salt thereof.
8. A compound according to any of claims 1 to 6 wherein n is 1 and Y is - C(O)-, -S(O)2-,
-NHS(O)2-, -NHC(O)-; or a pharmaceutically acceptable salt thereof.
9. A compound according to any of claims 1 to 8 wherein Ar1 is
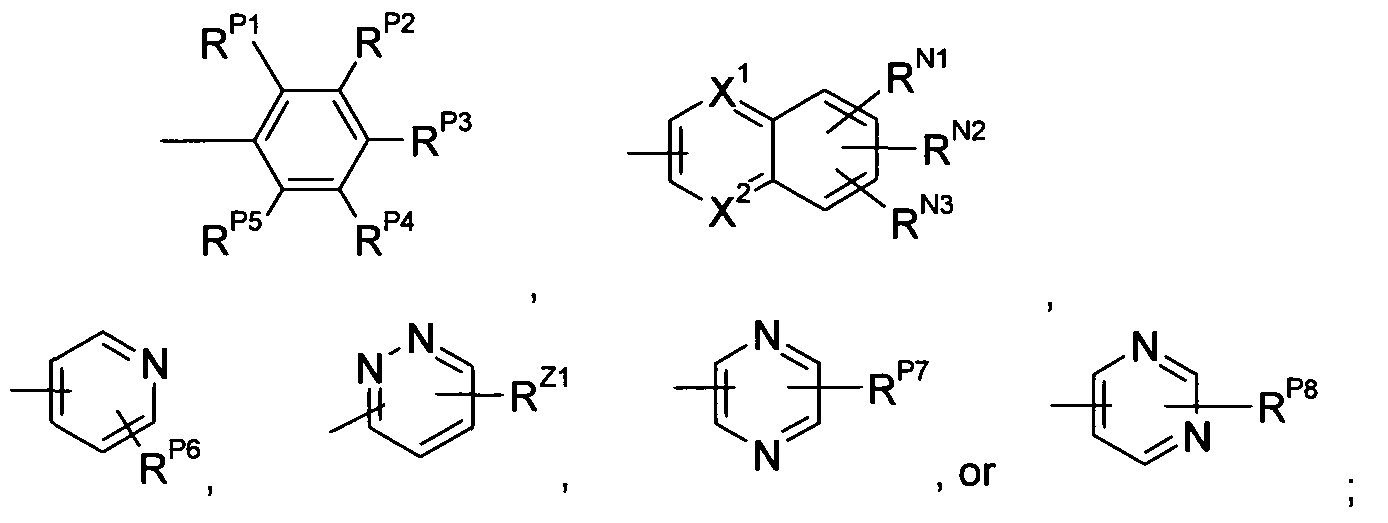
10. A compound according to any of claims 1 to 9 wherein RP1, RP2, RP3, RP4, RP5, RP6, R21, RP7 and RP8 are each independently -H, halo, Ci-C4 alkyl, CrC4 alkoxy, or -CF3; or a pharmaceutically acceptable salt thereof.
11. A compound of the formula

R1 and R2 are each independently -H, -OH, halo, CrC6 alkyl, Ci-C6 alkoxy, -CF3 or substituted C1-C6 alkyl;
G2 is C(R6) or N, wherein R6 is -H, -OH or Ci-C6 alkyl; Y is -CH2-, -CH2CH2-, -CH2CH2CH2-, -O-, -C(O)-, -C(O)CH2-, -S-, -S(O)-, -S(O)2-,
-NH-, -NHC(O)-,
-NHC(O)CH(R7)-, or -NHS(O)2-, wherein R7 is -H or CrC4 alkyl; Ar1 is a radical of the formulae

R8 and R9 are each independently -H or CrC6 alkyl; RM1, RM2, RB1, RB2, RB3, R64, R85,
RB6 Rxi Rx2 Rx3 RX4 RYI RY2 RY3 RY4 RY5 and RY6 we each jndependently -H or
CrC6 alkyl; X1 and X2 are independently CH or N; X3, X4, and X5 are each independently NH, O1 or S; X6 is CH2 or O; Q is substituted CrC6 alkyl, phenyl, napthyl, 2-pyridyl, or 3-pyridyl; m is 1 , 2, 3, or 4; and n is 0 or 1 ; provided that when Y is O, S, NH, NHC(O), NHS(O)2, NHC(O)CH(R7), or NHS(O)2, then G2 is CH.
12. A compound according to claim 11 wherein R1 and R2 are each independently -H, halo, -CF3 or CrC4 alkyl and n is 0; or a pharmaceutically acceptable salt thereof.
13. A compound according to claim 11 wherein R1 and R2 are each independently -H, halo, -CF3 or CrC4 alkyl; n is 1 ; Y is -C(O)-, -S(O)2-, -NHS(O)2-, -NHC(O)-; and Ar1 is

14. A compound according to any of claims 11 to 13 wherein m is 1 ; or a pharmaceutically acceptable salt thereof.
15. A compound according to any of claims 11 to 13 wherein m is 2; or a pharmaceutically acceptable salt thereof.
16. A compound according to any of claims 11 to 13 wherein m is 3; or a pharmaceutically acceptable salt thereof.
17. A compound according to any of claims 11 to 13 wherein m is 4; or a pharmaceutically acceptable salt thereof.
18. A compound of the formula (III)

R1 and R2 are each independently -H, -OH, halo, Ci-C6 alkyl, C1-C6 alkoxy, -CF3, or substituted Ci-C6 alkyl;
G2 is C(R6) or N, wherein R6 is -H, -OH or CrC6 alkyl;
G3 is -O- or -N(RH1), wherein RH1 is -H or CrC6 alkyl;
Y is -CH2-, -CH2CH2-, -CH2CH2CH2-, -0-, -C(O)-, -C(O)CH2-, -S-, -S(O)-, -S(O)2-, -NH-, -NHC(O)-, -NHC(O)CH(R7)-, or -NHS(O)2-, wherein R7 is -H or CrC4 alkyl;
Ar1 is a radical of the formulae


19. A compound according to claim 18 wherein R1 and R2 are each independently -H, halo, -CF3 or C1-C4 alkyl and n is O; or a pharmaceutically acceptable salt thereof.
20. A compound according to claim 19 wherein R1 and R2 are each independently -H, halo,
-CF3 or C1-C4 alkyl; n is 1 ; Y is -C(O)-, -S(O)2-, -NHS(O)2-, -NHC(O)-; and Ar1 is

21. A compound selected from the group consisting of: {3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-methoxy- phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethylH2-[4-(3-trifluoro- methyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2,3- dimethoxy-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5-trifluoro- methyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-chloro- phenyl)-piperidin-1-yl]-ethyl}-amine; {3-[1 -(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3-trifluoro- methyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
[2-(4-Benzenesulfonyl-piperazin-1-yl)-ethyl]-{3-[1-(4-chloro-phenyl)-cyclobutyl]- [1 ,2,4]oxadiazol-5-ylmethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-methoxy- phenoxy)-piperidin-1 -yl]-ethyl}-amine;
^-(i-p-Tolyl-cyclobutyO-Ii ^^loxadiazol-S-ylmethyll^-μ-CS-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
[3-(1-m-Tolyl-cyclobutyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(5-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; [S^I-p-Tolyl-cyclopropyO-Ii ^^loxadiazol-δ-ylmethyll^-μ^S-trifluoromethyl- benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(4-fluoro- phenoxy)-piperidin-1-yl]-ethyl}-amine; [3-(1-p-Tolyl-cyclopropyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(5-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
[3-(1-P-ToIyI-CyClOPrOPyI)-[1 , 2,4]oxadiazol-5-ylmethyl]-{2-[4-(5-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; {3-[1 -(4-Methoxy-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifiuoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{2-[4-(2-Methoxy-phenyl)-piperidin-1-yl]-ethyl}-[3-(1-phenyl- cyclopentyl)[1 ,2,4]oxadiazol-5-ylmethyl]-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3-methoxy- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; {3-[1 -(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-[2-(4-o-tolyl- piperidin-1 -yl)-ethyl]-amine;
{3-[1-(4-ChIoro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3-methoxy- phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethylH2-[4-(4- trifluoromethyl-phenyl)-piperidin-1 -yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(3-Chloro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine; {3-[1-(4-Methoxy-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
[3-(1-Phenyl-cyclopentyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(5-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1 -(3-Chloro-phenyl)-cyclopentyl]-[ 1 ,2 ,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1 -yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine; [3-(1-Phenyl-cyclopentyl)-[1 ,2,4]oxadiazol-5-ylmethylH2-[4-(3-trifluoromethyl- phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(3-Chloro-phenyl)-cyclopentyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine; {3-[1 -(4-Chloro-phenyl)-cyclohexyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclohexyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclohexyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trfluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine; [3-(1-Phenyl-cyclobutyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1 -yl]-ethyl}-amine;
[3-(1-p-Tolyl-cyclobutyl)-[1,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- phenyl)-piperidin-1-yl]-ethyl}-amine;
^-(i-m-Tolyl-cyclobutyO-Ii ^^loxadiazol-S-ylmethyll^-^^S-trifluoromethyl- phenyl)-piperidin-1-yl]-ethyl}-amine; [3-(1-m-Tolyl-cyclopropyl)-[1,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- phenyl)-piperidin-1-yl]-ethyl}-amine;
[3-(1-m-Tolyl-cyclopropy!)-[1,2,4]oxadiazol-5-ylmethyl]-{2-t4-(5-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(2-Fluoro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; {2-[4-(2-Methoxy-phenyl)-piperidin-1-yl]-ethyl}-[3-(1-p-tolyl-cyclobutyl)- [1,2,4]oxadiazol-5-ylmethyl]-amine;
{2-[4-(2-Methoxy-phenyl)-piperidin-1-yl]-ethyl}-[3-(1-m-tolyl-cyclobutyl)- [1,2,4]oxadiazol-5-ylmethyl]-amine; {2-[4-(2-Methoxy-phenyl)-piperidin-1-yl]-ethyl}-[3-(1-m-tolyl-cyclopropyl)-
[1 ,2,4]oxadiazol-5-ylmethyl]-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(4-chloro- phenyl)-piperidin-1-yl]-ethyl}-amine;
{2-[4-(2-Methoxy-phenyl)-piperidin-1-yl]-ethyl}-[3-(1-phenyl-cyclobutyl)- [1 ,2,4]oxadiazol-5-ylmethyl]-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutylH1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-fluoro- phenoxy)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-[2-(4-p-tolyloxy- piperidin-1 -yl)-ethyl]-amine; [3-(1-Phenyl-cyclobutyl)-[1 ,2,4]oxadiazol-5-ylmethylH2-[4-(4,4,4-trifluoro-3- methyl-1-methylene-but-2-enyl)-piperidin-1-yl]-ethyl}-amine;
[3-(1-p-Tolyl-cyclopropyl)-[1,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- phenyl)-piperidin-1-yl]-ethyl}-amine;
[S-CI-p-Tolyl-cyclobutyO-ti ^^loxadiazol-δ-ylmethyll^-μ^S-trifluoromethyl- benzyl)-piperazin-1-yl]-ethyl}-amine;
[3-(1-m-Tolyl-cyclobutyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- benzyl)-piperazin-1-yl]-ethyl}-amine;
^-(i-m-Tolyl-cyclopropyO-Ii ^^loxadiazol-S-ylmethyll^-K-CS-trifluoromethyl- benzyl)-piperazin-1-yl]-ethyl}-amine; {3-[1-(4-Methoxy-phenyl)-cyclopropyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3,4-Dichloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(2,4-Dichloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine; {3-[1-(3,4-Dichloro-1-methylene-but-2-enyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5- ylmethyl}-{2-[4-(3-trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine; {3-[1 -(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-methoxy-
5-trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-methoxy- 4-trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(3-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
[3-(1-p-Tolyl-cyclopentyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(5-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine; {3-[1-(3-Chloro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
[3-(1-Phenyl-cyclopentyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-fluoro- phenyl)-piperidin-1 -yl]-ethyl}-amine;
{3-[1-(2,4-Dichloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3,4-Dichloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine; {3-[1-(4-Methoxy-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethylH2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Methoxy-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclohexyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trfluoromethyl-phenyl)-pipeidin-1-yl]-ethyl}-amine; [3-(1-p-Tolyl-cyclopentyl)-[1,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(3-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperzin-1-yl]-ethyl}-amine; [3-(1-p-Tolyl-cyclopentyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl- benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(2,4-Dichloro-phenyl)-cyclopropyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(2,4-Dichloro-phenyl)-cyclopropyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3,4-Dimethoxy-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine; {3-[1-(3-Fluoro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(4-fluoro- phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(2-Fluoro-phenyl)-cyclopentyl]-[1 ,2I4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1 -yl]-ethyl}-amine;
{3-[1-(2-Fluoro-phenyl)-cyclopentyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifiuoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
^-(i-Phenyl-cyclopropyO-II ^^Joxadiazol-S-ylmethyll^-^-CS-trifluoromethyl- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; {3-[1-(3-Fluoro-phenyl)-cyclopentyl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(4-methoxy- phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(4- trifIuoromethyl-pyridin-2-yl)-piperazin-1 -yl]-ethyl}-amine;
{3-[1-(3,4-Dichloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2- methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine; {3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5-chloro- pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3-Methoxy-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(5- trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; {3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3-chloro- phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[1-(3,4-Dimethyl-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-benzyl)-piperazin-1-yl]-ethyl}-amine;
{3-[1-(3,4-Dichloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(3- trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{2-[4-(2-Chloro-phenyl)-piperidin-1-yl]-ethyl}-{3-[1-(3,4-dimethyl-phenyl)- cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-amine;
{1-[2-({3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-amino)- ethyl]-piperidin-4-yl}-(5-trifluoromethyl-pyridin-2-yl)-amine; N-{1-[2-({3-[1-(4-Chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-amino)- ethyl]-piperidin-4-yl}-2,5-dimethoxy-benzenesulfonamide;
{3-[4-(4-Chloro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4- (2-methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[4-(4-Chloro-phenyl)-tetrahydro-pyran-4-yl]-[1,2,4]oxadiazol-5-ylmethyl}-{2-[4- (3-trifluoromethyl-benzyl)-piperazin-1 -yl]-ethyl}-amine;
{3-[4-(4-Chloro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4- (5-trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine;
{3-[4-(4-Fluoro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4- (5-trifluoromethyl-pyridin-2-yl)-piperazin-1-yl]-ethyl}-amine; {3-[4-(4-Chloro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-
(3-trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[4-(4-Fluoro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4- (3-trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[4-(3-Methoxy-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2- [4-(3-trifluoromethyl-phenyl)-piperidin-1-yl]-ethyl}-amine;
{3-[4-(4-Fluoro-phenyl)-tetrahydro-pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4- (2-methoxy-phenyl)-piperidin-1-yl]-ethyl}-amine; {2-[4-(2-Chloro-phenyl)-piperidin-1-yl]-ethyl}-{3-[4-(4-chloro-phenyl)-tetrahydro- pyran-4-yl]-[1 ,2,4]oxadiazol-5-ylmethyl}-amine; or a pharmaceutically acceptable salts thereof.
22. A compound of claim 1 wherein the compound is {3-[1-(4-chloro-phenyl)- cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-methoxy-phenyl)-piperidin-1-yl]-ethyl}- amine, or a pharmaceutically acceptable salt thereof.
23. A compound of claim 1 wherein the compound is {3-[1-(4-chloro-phenyl)- cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2,3-dimethoxy-benzyl)-piperazin-1-yl]- ethyl}-amine, or a pharmaceutically acceptable salt thereof.
24. A compound of claim 1 wherein the compound is [2-(4-benzenesulfonyl- piperazin-1-yl)-ethyl]-{3-[1-(4-chloro-phenyl)-cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}- amine, or a pharmaceutically acceptable salt thereof.
25. A compound of claim 1 wherein the compound is {3-[1-(4-chloro-phenyl)- cyclobutyl]-[1 ,2,4]oxadiazol-5-ylmethyl}-{2-[4-(2-methoxy-phenoxy)-piperidin-1-yl]-ethyl}- amine, or a pharmaceutically acceptable salt thereof.
26. A compound of claim 1 wherein the compound is [3-(1-p-tolyl- cyclopropyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(3-trifluoromethyl-benzyl)-piperazin-1-yl]- ethyl}-amine, or a pharmaceutically acceptable salt thereof.
27. A compound of claim 1 wherein the compound is [3-(1-phenyl- cyclopropyl)-[1 ,2,4]oxadiazol-5-ylmethyl]-{2-[4-(5-trifluoromethyl-pyridin-2-yl)-piperazin- 1-yl]-ethyl}-amine, or a pharmaceutically acceptable salt thereof.
28. A pharmaceutical composition comprising a compound according to any of claims 1 to 27, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
29. A method of causing vasodilation in a patient in need thereof comprising administering to said patient a therapeutically effective amount of a compound according to any of claims 1 to 27, or a pharmaceutically acceptable salt thereof.
30. A method of treating a disease or condition selected from hypertension, congestive heart failure, stroke, ischaemic heart disease, or angina pectoris in a patient in need thereof comprising administering to said patient a therapeutically effective amount of a compound according to any of claims 1 to 27, or a pharmaceutically acceptable salt thereof.
31. A method of treating a disease or condition selected from the group consisting of chronic pain, inflammatory pain, neuropathic pain, visceral pain, nociceptive pain, multiple sclerosis, neurodegenerative disorder, irritable bowel syndrome, osteoarthritis, rheumatoid arthritis, neuropathological disorders, functional bowel disorders, inflammatory bowel diseases, pain associated with dysmenorrhea, pelvic pain, cystitis, pancreatitis, migraine, cluster and tension headaches, diabetic neuropathy, sciatica, fibromyalgia and causalgia comprising administering to said patient a therapeutically effective amount of a compound according to any of claims 1 to 27, or a pharmaceutically acceptable salt thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US89664407P | 2007-03-23 | 2007-03-23 | |
| US60/896,644 | 2007-03-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008117148A1 true WO2008117148A1 (en) | 2008-10-02 |
Family
ID=39588058
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2008/000645 WO2008117148A1 (en) | 2007-03-23 | 2008-03-10 | Substituted oxadiazole analogs as calcium channel antagonists |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008117148A1 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013119751A1 (en) * | 2012-02-09 | 2013-08-15 | Boehringer Ingelheim International Gmbh | Process for preparing carboxamidine compounds |
| WO2015049616A1 (en) | 2013-10-04 | 2015-04-09 | Pfizer Inc. | Novel bicyclic pyridinones as gamma-secretase modulators |
| WO2017070680A1 (en) | 2015-10-22 | 2017-04-27 | Cavion Llc | Methods for treating angelman syndrome and related disorders |
| WO2020070611A1 (en) * | 2018-10-01 | 2020-04-09 | Pi Industries Ltd | Oxadiazoles as fungicides |
| US11130750B2 (en) | 2017-02-15 | 2021-09-28 | Cavion, Inc. | Calcium channel inhibitors |
| US11311522B1 (en) | 2018-10-03 | 2022-04-26 | Cavion, Inc. | Treating essential tremor using (R)-2-(4-Isopropylphenyl)-N-(1-(5-(2,2,2-trifluoroethoxy)pyridin-2-yl)ethyl)acetamide |
| US11324733B2 (en) | 2017-04-26 | 2022-05-10 | Cavion, Inc. | Methods for improving memory and cognition and for treating memory and cognitive disorders |
| US11427540B2 (en) | 2019-07-11 | 2022-08-30 | Praxis Precision Medicines, Inc. | Formulations of T-type calcium channel modulators and methods of use thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004014370A2 (en) * | 2002-08-09 | 2004-02-19 | Astrazeneca Ab | Oxadiazoles as modulators of metabotropic glutamate receptor-5 |
-
2008
- 2008-03-10 WO PCT/IB2008/000645 patent/WO2008117148A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004014370A2 (en) * | 2002-08-09 | 2004-02-19 | Astrazeneca Ab | Oxadiazoles as modulators of metabotropic glutamate receptor-5 |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013119751A1 (en) * | 2012-02-09 | 2013-08-15 | Boehringer Ingelheim International Gmbh | Process for preparing carboxamidine compounds |
| JP2015511237A (en) * | 2012-02-09 | 2015-04-16 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Method for preparing carboxamidine compounds |
| US9029592B2 (en) | 2012-02-09 | 2015-05-12 | Boehringer Ingelheim International Gmbh | Process for preparing carboxamidine compounds |
| WO2015049616A1 (en) | 2013-10-04 | 2015-04-09 | Pfizer Inc. | Novel bicyclic pyridinones as gamma-secretase modulators |
| US11273218B2 (en) | 2015-10-22 | 2022-03-15 | Cavion, Inc. | Methods for treating Angelman syndrome and related disorders |
| WO2017070680A1 (en) | 2015-10-22 | 2017-04-27 | Cavion Llc | Methods for treating angelman syndrome and related disorders |
| US11130750B2 (en) | 2017-02-15 | 2021-09-28 | Cavion, Inc. | Calcium channel inhibitors |
| US11324733B2 (en) | 2017-04-26 | 2022-05-10 | Cavion, Inc. | Methods for improving memory and cognition and for treating memory and cognitive disorders |
| WO2020070611A1 (en) * | 2018-10-01 | 2020-04-09 | Pi Industries Ltd | Oxadiazoles as fungicides |
| CN113195461A (en) * | 2018-10-01 | 2021-07-30 | Pi工业有限公司 | Novel oxadiazole compound |
| JP2022504040A (en) * | 2018-10-01 | 2022-01-13 | ピーアイ インダストリーズ リミテッド | New oxadiazole |
| US11311522B1 (en) | 2018-10-03 | 2022-04-26 | Cavion, Inc. | Treating essential tremor using (R)-2-(4-Isopropylphenyl)-N-(1-(5-(2,2,2-trifluoroethoxy)pyridin-2-yl)ethyl)acetamide |
| US11427540B2 (en) | 2019-07-11 | 2022-08-30 | Praxis Precision Medicines, Inc. | Formulations of T-type calcium channel modulators and methods of use thereof |
| US11649207B2 (en) | 2019-07-11 | 2023-05-16 | Praxis Precision Medicines, Inc. | Formulations of T-type calcium channel modulators and methods of use thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008050200A1 (en) | Oxadiazole compounds as calcium channel antagonists | |
| JP5685203B2 (en) | Aryl-substituted carboxamide derivatives as calcium channel blockers or sodium channel blockers | |
| WO2008117148A1 (en) | Substituted oxadiazole analogs as calcium channel antagonists | |
| JP4400566B2 (en) | Benzamide derivatives or salts thereof | |
| KR101586966B1 (en) | (4-phenylimidazol-2-yl) ethylamine derivatives useful as sodium channel modulators | |
| US7960377B2 (en) | Substituted pyridoxazines | |
| TWI428334B (en) | Sulfonylpyrazole and sulfonylpyrazoline carboxamidine derivatives as 5-ht6 antagonists | |
| WO2009056934A1 (en) | 1,4-dihydronaphthyridine derivatives | |
| JP2005523310A (en) | Substituted indole | |
| JP2010529117A (en) | Metabotropic glutamate receptor oxadiazole ligands and their use as potentiators | |
| KR20150013500A (en) | BICYCLIC SULFONE COMPOUNDS FOR INHIBITION OF RORgamma ACTIVITY AND THE TREATMENT OF DISEASE | |
| JP7330202B2 (en) | Compounds and their use as PDE4 activators | |
| JP2007509140A (en) | Novel tetrahydrospiro {piperidine-2,7'-pyrrolo [3,2-b] pyridine derivatives and novel indole derivatives useful in the treatment of diseases associated with 5-HT6 receptors | |
| JP6694452B2 (en) | Triazole derivatives and their use as PDE4 activators | |
| US20130045948A1 (en) | Azocyclic inhibitors of fatty acid amide hydrolase | |
| TW201315730A (en) | Methylpyperidine derivatives | |
| EP3519403B1 (en) | Compounds and their use as pde4 activators | |
| AU8249998A (en) | New compounds | |
| CN114728170B (en) | Compounds active on nuclear receptors | |
| MXPA06014025A (en) | Substituted triazole derivatives as oxytocin antagonists. | |
| TW200826927A (en) | Diaryl, dipyridinyl and aryl-pyridinyl derivatives and uses thereof | |
| JP6539749B2 (en) | 3- (4-Ethynylphenyl) hexahydropyrimidine-2,4-dione derivatives as modulators of mGluR4 | |
| JP2015531393A (en) | Tropomyosin-related kinase inhibitor | |
| JP2014514315A (en) | Pyrazolidin-3-one derivatives | |
| EP1656367A1 (en) | 4- (heterocyclyl- fused phenyl)- 3- (phenyl or pyrid -2- yl) pyrazoles as inhibitors of the alk-5- receptor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08719332 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08719332 Country of ref document: EP Kind code of ref document: A1 |