WO2008112651A2 - Aminopyrimidines useful as inhibitors of protein kinases - Google Patents
Aminopyrimidines useful as inhibitors of protein kinases Download PDFInfo
- Publication number
- WO2008112651A2 WO2008112651A2 PCT/US2008/056433 US2008056433W WO2008112651A2 WO 2008112651 A2 WO2008112651 A2 WO 2008112651A2 US 2008056433 W US2008056433 W US 2008056433W WO 2008112651 A2 WO2008112651 A2 WO 2008112651A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- agent
- disease
- treating
- compound
- compounds
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Definitions
- the present invention relates to compounds useful as inhibitors of protein kinases.
- the invention also provides pharmaceutically acceptable compositions comprising the compounds of the invention and methods of using the compositions in the treatment of various disorders.
- the invention also provides processes for preparing the compounds of the invention.
- Glycogen synthase kinase-3 (GSK-3) is a serine/threonine protein kinase comprised of ⁇ and ⁇ isoforms that are each encoded by distinct genes [Coghlan et al., Chemistry & Biology 2000, 7, 793-803; and Kim and Kimmel, Curr. Opinion Genetics Dev., 2000 10, 508-514]. Protein kinases, particularly GSK-3, have been implicated in various diseases, disorders, and conditions including Diabetes, Alzheimer's, Huntington' s, Amyotrophic Lateral Sclerosis, Parkinson's, Bipolar disorder, Schizophrenia, Cerebral stroke, and Cardiac Hypertrophy.
- Inhibiting GSK-3 is the desired approach for treating these diseases, disorders, and conditions.
- active GSK-3 may be important for inhibiting hypertrophy.
- blocking GSK-3 appears to be important for protecting against apoptosis in hypertrophied cardiac myoctyes.
- GSK-3 regulates multiple downstream effectors associated with a variety of signaling pathways. These proteins include glycogen synthase, which is the rate limiting enzyme necessary for glycogen synthesis, the microtubule associated protein Tau, the gene transcription factor ⁇ -catenin, the translation initiation factor elF2B, as well as ATP citrate lyase, axin, heat shock factor-1, c- Jun, c-myc, c-myb, CREB, and CEPB ⁇ . These diverse protein targets implicate GSK-3 in many aspects of cellular metabolism, proliferation, differentiation, and development.
- GSK-3 is a negative regulator of the insulin-induced signal.
- the presence of insulin causes inhibition of GSK-3 mediated phosphorylation and deactivation of glycogen synthase.
- the inhibition of GSK-3 leads to increased glycogen synthesis and glucose uptake [Klein et al . , PNAS 1996, 93, 8455-8459; Cross et al . , Blochem. J. 1994, 303, 21-26); Cohen, Blochem. Soc. Trans. 1993, 21, 555-567; and Massillon et al., Blochem J.
- GSK-3 activity is associated with Alzheimer's disease.
- the hallmarks of this disease are the extracellular plaques formed by aggregated ⁇ -amyloid peptides and the formation of intracellular neurofibrillary tangles via the tau protein.
- the neurofibrillary tangles contain hyperphosphorylated Tau protein, in which Tau is phosphorylated on abnormal sites.
- GSK-3 is known to phosphorylate these abnormal sites in cell and animal models.
- Conditional transgenic mice that over-express GSK-3 develop aspects of AD including tau hyperphosphorylation, neuronal apoptosis and spatial learning deficit. Turning off GSK-3 in these mice restores normal behavior, reduces Tau hyperphosphorylation and neuronal apoptosis.
- GSK-3 as a target for psychosis and mood disorders, such as schizophrenia and bipolar disease, respectively, have been reported in the literature.
- AKT haplotype deficiency was identified in a subset of schizophrenic patients which correlated with increased GSK-3 activity.
- a single allele knockout of GSK-3 ⁇ resulted in attenuated hyperactivity in response to amphetamine in a behavior model of mania.
- GSK-3 activity is associated with stroke.
- IGF-I insulin growth factor-1
- MCAO transient middle cerebral artery occlusion
- This invention provides compounds and pharmaceutically acceptable compositions thereof that are useful as inhibitors of GSK-3 protein kinases. [0014] These compounds are represented by formula I:
- These compounds have surprising selectivity in blocking the tyrosine autophosphorylation form of the GSK-3 enzyme over the serine/threonine kinase form. These compounds are also surprisingly effective in increasing axonal and dendritic branching in neuronal cells, which is useful in the treatment of degenerative conditions such as stroke, Alzheimer's Disease, Parkinson's Disease, Huntington' s Disease, Amyotrophic Lateral Sclerosis (ALS) Multiple Sclerosis (MS) , Spinal Cord Injury, Traumatic Brain Injury, Charcot-Marie-Tooth, Leukocytopenia, Diabetes, Diabetic Neuropathy, and Osteoporosis.
- ALS Amyotrophic Lateral Sclerosis
- MS Multiple Sclerosis
- These compounds are also effective as chemomodulators of repair, regeneration, and cellular differentiation.
- the present invention also provides processes for preparing these compounds, compositions, pharmaceutical compositions, and methods of using such compounds and compositions for inhibiting protein kinases. These compounds are particularly useful as GSK-3 inhibitors.
- These compounds and pharmaceutically acceptable compositions thereof are also useful for treating or preventing a variety of diseases, disorders or conditions, including, but not limited to, an autoimmune, inflammatory, proliferative, or hyperproliferative disease, a neurodegenerative disease, or an immunologically-mediated disease .
- the compounds provided by this invention are useful for inhibiting kinases in vitro, in vivo, and ex vivo. These compounds also useful for the study of kinases in biological and pathological phenomena; the study of intracellular signal transduction pathways mediated by such kinases; and the comparative evaluation of new kinase inhibitors .
- R x is Ci-3 alkyl
- R ⁇ is Ci-3 alkyl.
- R x is methyl or ethyl. In some embodiments, R x is methyl. In some embodiment R ⁇ is methyl.
- R x and R ⁇ are both methyl. In some embodiments, R x is ethyl and R ⁇ is methyl.
- a specified number range of atoms includes any integer therein.
- a group having from 1-4 atoms could have 1, 2, 3, or 4 atoms.
- compounds of the invention may optionally be substituted with one or more substituents, such as are illustrated generally above, or as exemplified by particular classes, subclasses, and species of the invention. It will be appreciated that the phrase “optionally substituted” is used interchangeably with the phrase “substituted or unsubstituted. " In general, the term "substituted”, whether preceded by the term “optionally” or not, refers to the replacement of hydrogen radicals in a given structure with the radical of a specified substituent.
- an optionally substituted group may have a substituent at each substitutable position of the group, and when more than one position in any given structure may be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at every position.
- Combinations of substituents envisioned by this invention are preferably those that result in the formation of stable or chemically feasible compounds.
- stable refers to compounds that are not substantially altered when subjected to conditions to allow for their production, detection, recovery, purification, and use for one or more of the purposes disclosed herein.
- a stable compound or chemically feasible compound is one that is not substantially altered when kept at a temperature of 4O 0 C or less, in the absence of moisture or other chemically reactive conditions, for at least a week.
- aliphatic or "aliphatic group”, as used herein, means a straight-chain (i.e., unbranched) branched or unbranched, substituted or unsubstituted hydrocarbon chain that is completely saturated or that contains one or more units of unsaturation that has a single point of attachment to the rest of the molecule. Unless otherwise specified, aliphatic groups contain 1-20 aliphatic carbon atoms. In some embodiments, aliphatic groups contain 1-10 aliphatic carbon atoms. In other embodiments, aliphatic groups contain 1-8 aliphatic carbon atoms.
- aliphatic groups contain 1-6 aliphatic carbon atoms, and in yet other embodiments aliphatic groups contain 1-4 aliphatic carbon atoms.
- Suitable aliphatic groups include, but are not limited to, linear or branched, substituted or unsubstituted alkyl, alkenyl, or alkynyl groups. Specific examples include, but are not limited to, methyl, ethyl, isopropyl, n-propyl, sec-butyl, vinyl, n- butenyl, ethynyl, and tert-butyl.
- alkyl as used herein, means a straight- chain (i.e., unbranched) , branched or unbranched, substituted or unsubstituted, hydrocarbon chain that is completely saturated and has a single point of attachment to the rest of the molecule.
- alkyl groups contain 1-6 alkyl carbon atoms. In some embodiments, alkyl groups contain 1-4 alkyl carbon atoms. In other embodiments, alkyl groups contain 1-3 alkyl carbon atoms. Examples include, but are not limited to, methyl, ethyl, isopropyl, n-propyl, sec-butyl, n-butyl, and n-pentyl .
- cycloaliphatic refers to a monocyclic C3-C8 hydrocarbon or bicyclic C8-C12 hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic, that has a single point of attachment to the rest of the molecule wherein any individual ring in said bicyclic ring system has 3-7 members.
- Suitable cycloaliphatic groups include, but are not limited to, cycloalkyl and cycloalkenyl groups. Specific examples include, but are not limited to, cyclohexyl, cyclopropenyl, and cyclobutyl.
- heterocycle means non-aromatic, monocyclic, bicyclic, or tricyclic ring systems in which one or more ring members are an independently selected heteroatom.
- the "heterocycle”, “heterocyclyl”, or “heterocyclic” group has three to fourteen ring members in which one or more ring members is a heteroatom independently selected from oxygen, sulfur, nitrogen, or phosphorus, and each ring in the system contains 3 to 7 ring members .
- Suitable heterocycles include, but are not limited to, 3-lH-benzimidazol-2-one, 3- (1-alkyl) -benzimidazol-2-one, 2-tetrahydrofuranyl, 3-tetrahydrofuranyl, 2- tetrahydrothiophenyl, 3-tetrahydrothiophenyl, 2-morpholino, 3-morpholino, 4-morpholino, 2-thiomorpholino, 3- thiomorpholino, 4-thiomorpholino, 1-pyrrolidinyl, 2- pyrrolidinyl, 3-pyrrolidinyl, 1-tetrahydropiperazinyl, 2- tetrahydropiperazinyl, 3-tetrahydropiperazinyl, 1- piperidinyl, 2-piperidinyl, 3-piperidinyl, 1-pyrazolinyl, 3-pyrazolinyl, 4-pyrazolinyl, 5-pyrazolinyl, 1-piperidinyl, 2- piperidinyl
- Cyclic groups (e.g. cycloaliphatic and heterocycles) , can be linearly fused, bridged, or spirocyclic .
- heteroatom means one or more of oxygen, sulfur, nitrogen, or phosphorus, (including, any oxidized form of nitrogen, sulfur, or phosphorus; the quaternized form of any basic nitrogen or; a substitutable nitrogen of a heterocyclic ring, for example N (as in 3, 4-dihydro-2H- pyrrolyl) , NH (as in pyrrolidinyl) or NR + (as in N- substituted pyrrolidinyl) ) .
- alkoxy or thioalkyl
- alkoxy refers to an alkyl group, as previously defined, attached to the principal carbon chain through an oxygen (“alkoxy”) or sulfur (“thioalkyl”) atom.
- haloalkyl mean alkyl, alkenyl or alkoxy, as the case may be, substituted with one or more halogen atoms.
- halogen means F, Cl, Br, or I.
- aryl used alone or as part of a larger moiety as in “aralkyl”, “aralkoxy”, or “aryloxyalkyl”, refers to monocyclic, bicyclic, and tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic and wherein each ring in the system contains 3 to 7 ring members.
- aryl may be used interchangeably with the term “aryl ring”.
- aryl also refers to heteroaryl ring systems as defined hereinbelow.
- heteroaryl used alone or as part of a larger moiety as in “heteroaralkyl” or “heteroarylalkoxy”, refers to monocyclic, bicyclic, or tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic, at least one ring in the system contains one or more heteroatoms, and wherein each ring in the system contains 3 to 7 ring members.
- heteroaryl may be used interchangeably with the term “heteroaryl ring” or the term “heteroaromatic” .
- Suitable heteroaryl rings include, but are not limited to, 2-furanyl, 3-furanyl, N-imidazolyl, 2-imidazolyl, 4-imidazolyl, 5- imidazolyl, benzimidazolyl, 3-isoxazolyl, 4-isoxazolyl, 5- isoxazolyl, 2-oxazolyl, 4-oxazolyl, 5-oxazolyl, N-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2- pyrimidinyl, 4-pyrimidinyl, 5-pyrimidinyl, pyridazinyl (e.g., 3-pyridazinyl) , 2-thiazolyl, 4-thiazolyl, 5- thiazolyl, tetrazolyl (e.g., 5-tetrazolyl) , triazolyl (e.g., 2-triazolyl and 5-triazo
- a protecting group and “protective group” as used herein, are interchangeable and refer to an agent used to temporarily block one or more desired reactive sites in a multifunctional compound.
- a protecting group has one or more, or preferably all, of the following characteristics: a) is added selectively to a functional group in good yield to give a protected substrate that is b) stable to reactions occurring at one or more of the other reactive sites; and c) is selectively removable in good yield by reagents that do not attack the regenerated, deprotected functional group.
- Exemplary protecting groups are detailed in Greene, T. W., Wuts, P.
- nitrogen protecting group refers to an agents used to temporarily block one or more desired nitrogen reactive sites in a multifunctional compound. Preferred nitrogen protecting groups also possess the characteristics exemplified above, and certain exemplary nitrogen protecting groups are also detailed in Chapter 7 in Greene, T. W., Wuts, P. G in "Protective Groups in Organic Synthesis", Third Edition, John Wiley & Sons, New York: 1999, the entire contents of which are hereby incorporated by reference.
- an alkyl or aliphatic chain can be optionally replaced with another atom or group.
- the optional replacements form a chemically stable compound.
- Optional replacements can occur both within the chain and at either end of the chain; i.e. both at the point of attachment and/or also at the terminal end.
- Two optional replacements can also be adjacent to each other within a chain so long as it results in a chemically stable compound.
- the optional replacements can also completely replace all of the carbon atoms in a chain.
- a C3 aliphatic can be optionally interrupted or replaced by -NR-, -C(O)-, and -NR- to form -NRC(O)NR- (a urea).
- the replacement atom is bound to an H on the terminal end.
- the replacement atom could be -OCH 2 CH 3 , -CH 2 OCH 3 , or -CH 2 CH 2 OH.
- structures depicted herein are also meant to include all isomeric (e.g., enantiomeric, diastereomeric, and geometric (or conformational) ) forms of the structure; for example, the R and S configurations for each asymmetric center, (Z) and (E) double bond isomers, and (Z) and (E) conformational isomers. Therefore, single stereochemical isomers as well as enantiomeric, diastereomeric, and geometric (or conformational) mixtures of the present compounds are within the scope of the invention.
- a substituent can freely rotate around any rotatable bonds.
- a substituent can freely rotate around any rotatable bonds.
- structures depicted herein are also meant to include compounds that differ only in the presence of one or more isotopically enriched atoms.
- compounds having the present structures except for the replacement of hydrogen by deuterium or tritium, or the replacement of a carbon by a 13 C- or 14 C-enriched carbon are within the scope of this invention.
- Such compounds are useful, for example, as analytical tools or probes in biological assays.
- the compounds of the present invention can exist in free form for treatment, or where appropriate, as a pharmaceutically acceptable salt, salts, or mixtures thereof.
- the term "pharmaceutically acceptable salt” refers to salts of a compound which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio.
- Pharmaceutically acceptable salts are well known in the art. For example, S. M. Berge et al . , describe pharmaceutically acceptable salts in detail in J. Pharmaceutical Sciences, 1977, 66, 1-19, incorporated herein by reference.
- Pharmaceutically acceptable salts of the compounds of this invention include those derived from suitable inorganic and organic acids and bases. These salts can be prepared in situ during the final isolation and purification of the compounds. Acid addition salts can be prepared by 1) reacting the purified compound in its free- based form with a suitable organic or inorganic acid and 2) isolating the salt thus formed.
- Examples of pharmaceutically acceptable, nontoxic acid addition salts are salts of an amino group formed with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid or by using other methods used in the art such as ion exchange.
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid
- organic acids such as acetic acid, oxalic acid, maleic acid, tartaric acid, citric acid, succinic acid or malonic acid or by using other methods used in the art such as ion exchange.
- salts include adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptonate, glycerophosphate, glycolate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide, 2-hydroxy- ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate, maleate, malonate, methanesulfonate, 2- naphthalenesulfonate, nicotinate, nitrate, oleate, oxalate, palmitate, palmoate, pe
- Salts derived from appropriate bases include alkali metal, alkaline earth metal, ammonium and N + (Ci_4alkyl) 4 salts.
- This invention also envisions the quaternization of any basic nitrogen-containing groups of the compounds disclosed herein. Water or oil-soluble or dispersible products may be obtained by such quaternization ,
- Base addition salts can be prepared by 1) reacting the purified compound in its acid form with a suitable organic or inorganic base and 2) isolating the salt thus formed.
- Base addition salts include alkali or alkaline earth metal salts.
- Representative alkali or alkaline earth metal salts include sodium, lithium, potassium, calcium, magnesium, and the like.
- Further pharmaceutically acceptable salts include, when appropriate, nontoxic ammonium, quaternary ammonium, and amine cations formed using counterions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, loweralkyl sulfonate and aryl sulfonate.
- Other acids and bases while not in themselves pharmaceutically acceptable, may be employed in the preparation of salts useful as intermediates in obtaining the compounds of the invention and their pharmaceutically acceptable acid or base addition salts.
- TCF/LEF T cell factor/lymphoid enhancer factor DIPEA diisopropylethylamine
- the present invention provides compounds and compositions that are useful as inhibitors of protein kinases.
- the protein kinases are GSK-3 kinases .
- the compounds and compositions of this invention are particularly useful for treating or lessening the severity of a disease, condition, or disorder where a protein kinase is implicated in the disease, condition, or disorder.
- the present invention provides a method for treating or lessening the severity of a disease, condition, or disorder where a protein kinase is implicated in the disease state.
- the present invention provides a method for treating or lessening the severity of a disease, condition, or disorder where inhibition of enzymatic activity is implicated in the treatment of the disease.
- this invention provides a method for treating or lessening the severity of a disease, condition, or disorder with compounds that inhibit enzymatic activity by binding to the protein kinase.
- Another aspect provides a method for treating or lessening the severity of a kinase disease, condition, or disorder by inhibiting enzymatic activity of the kinase with a protein kinase inhibitor.
- said protein kinase inhibitor is a GSK-3 inhibitor.
- the compounds and compositions of this invention are also useful in biological samples.
- One aspect of the invention relates to inhibiting protein kinase activity in a biological sample, which method comprises contacting said biological sample with a compound of formula I or a composition comprising said compound.
- biological sample means an in vitro or an ex vivo sample, including, without limitation, cell cultures or extracts thereof; biopsied material obtained from a mammal or extracts thereof; and blood, saliva, urine, feces, semen, tears, or other body fluids or extracts thereof.
- biological sample does not refer to in vivo samples.
- Inhibition of protein kinase activity in a biological sample is useful for a variety of purposes that are known to one of skill in the art. Examples of such purposes include, but are not limited to, blood transfusion, organ- transplantation, and biological specimen storage.
- Another aspect of this invention relates to the study of protein kinases in biological and pathological phenomena; the study of intracellular signal transduction pathways mediated by such protein kinases; and the comparative evaluation of new protein kinase inhibitors. Examples of such uses include, but are not limited to, biological assays such as enzyme assays and cell-based assays.
- the activity of the compounds as protein kinase inhibitors may be assayed in vitro, in vivo or in a cell line.
- In vitro assays include assays that determine inhibition of either the kinase activity or ATPase activity of the activated kinase. Alternate in vitro assays quantitate the ability of the inhibitor to bind to the protein kinase and may be measured either by radiolabelling the inhibitor prior to binding, isolating the inhibitor/kinase complex and determining the amount of radiolabel bound, or by running a competition experiment where new inhibitors are incubated with the kinase bound to known radioligands.
- Inhibition of GSK-3 activity has been linked to stem cell proliferation, differentiation and neuronal plasticity, and angiogenesis . These various functions are implicated in repair and regeneration. Inhibitors of GSK-3 have been shown to sustain self-renewal of embryonic stem cells, promote neuron, beta-cell, myeloid and osteoblast differentiation.
- one aspect of this invention provides compounds that are useful in cell repair and regeneration.
- said compounds are used to promote cell proliferation, cell differentiation, neuronal plasticity, or angiogenesis.
- said compounds are chemomodulators of cell differentiation.
- said compounds are chemomodulators of repair and regeneration.
- the compounds are used in increasing axonal and dendritic branching in neuronal cells. In some embodiments, the compounds are used to promote neuroplasticity . In other embodiments, the compounds are used to promote angiogenesis. In yet other embodiments, the compounds are used to promote neurogenesis. In yet other embodiments, the compounds are used to treat neuropsychiatric disorders, such as mania and depression. [0061] Another embodiment provides compounds that are used to treat diabetes by promoting beta cell regeneration. [0062] Yet another embodiment provides compounds that are used to treat osteoporosis by osteoblastogenesis .
- GSK-3 functions as both a tyrosine and a serine/threonine kinase, similar to the DYRK kinase family. Like the DYRK kinase family, GSK-3 auto-phosphorylates a key tyrosine residue in its kinase domain (GSK-3a, Tyr 279 and GSK-3b, Tyr 216) . This tyrosine phosphorylation has been shown to be important for positively modulating kinase activity. Locheed et al, demonstrated that this autophosphorylation occurs intramolecularly at a post- translationally intermediate step prior to maturation and is chaperones-dependent (Lochhead et al, Molecular Cell 24, (2006), pp.
- GSK-3 After maturation, GSK-3 loses its tyrosine kinase activity and acts exclusively as a serine and threonine kinase towards exogenous substrates.
- ⁇ -catenin is one of the exogenous serine/threonine substrates that GSK-3 phosphorylates . Inhibition of ⁇ - catenin phosphorylation leads to an increase in b-catenin levels that in turn translocate to the nucleus and transcriptionally control many genes involved in cellular response and function.
- One potential safety concern for GSK- 3 inhibitors is that use of the inhibitors could lead to hyperproliferation via ⁇ -catenin induction.
- GSK-3 is central to many signaling pathways that control multiple cellular activities such as proliferation, differentiation and metabolism.
- one aspect of this invention provides compounds that can partially attenuate GSK-3 activity without completely blocking the enzyme and affecting multiple substrates such as ⁇ -catenin.
- One embodiment provides compounds that selectively inhibit the tyrosine autophosphorylation form of the enzyme over the serine/threonine kinase form.
- said enzyme is GSK-3 ⁇ ; in other embodiments, GSK-3 ⁇ .
- the compounds have a ⁇ -catenin : GSK-3 ⁇ window of at least 30 fold.
- said compounds have a ⁇ -catenin : GSK-3 ⁇ window of at least 400 fold and up to 500 fold.
- compounds that selectively inhibit the auto-phosphorylation of the tyrosine form of the GSK-3 enzyme relative to the serine/threonine kinase form promote neuron growth and dendrite formation, such as by increasing axonal and dendritic branching in neuronal cells.
- Increasing neuron growth and dendrite formation is advantageous and provides and unexpected and improved therapeutic efficacy when treating many types of degenerative conditions such as Stroke, Post stroke, Spinal Cord Injury, Traumatic Brain Injury, Alzheimer's, Parkinson's, Huntington' s, Multiple Sclerosis, Amyotrophic Lateral Sclerosis, Diabetic Neuropathy, Charcot-Marie-Tooth, Leukocytopenia, Diabetes and Osteoporosis.
- Another aspect of this invention provides compounds that are useful for the treatment of diseases, disorders, and conditions including, but not limited to, autoimmune diseases, inflammatory diseases, proliferative and hyperproliferative diseases, immunologically-mediated diseases, immunodeficiency disorders, immunomodulatory or immunosuppressive disorder, bone diseases, metabolic diseases, neurological and neurodegenerative diseases, neurotrophic factor, cardiovascular diseases, hormone related diseases, diabetes, allergies, asthma, and Alzheimer's disease.
- Another aspect of this invention provides compounds that are inhibitors of protein kinases, and thus are useful for the treatment of the diseases, disorders, and conditions, along with other uses described herein .
- compositions comprising any of the compounds described herein and optionally comprising a pharmaceutically acceptable carrier, adjuvant or vehicle.
- these compositions optionally further comprise one or more additional therapeutic agents.
- One aspect of this invention provides a method for the treatment or lessening the severity of a disease, disorder, or condition selected from an autoimmune disease, an inflammatory disease, a proliferative or hyperproliferative disease, such as cancer, an immunologically-mediated disease, an immunodeficiency disorders, a bone disease, a metabolic disease, a neurological or neurodegenerative disease, a cardiovascular disease, allergies, diabetes, asthma, Alzheimer's disease, or a hormone related disease, comprising administering an effective amount of a compound, or a pharmaceutically acceptable composition comprising a compound, to a subject in need thereof.
- a disease, disorder, or condition selected from an autoimmune disease, an inflammatory disease, a proliferative or hyperproliferative disease, such as cancer, an immunologically-mediated disease, an immunodeficiency disorders, a bone disease, a metabolic disease, a neurological or neurodegenerative disease, a cardiovascular disease, allergies, diabetes, asthma, Alzheimer's disease, or a hormone related disease
- cancer includes, but is not limited to, the following cancers: epidermoid Oral : buccal cavity, lip, tongue, mouth, pharynx; Cardiac : sarcoma (angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma) , myxoma, rhabdomyoma, fibroma, lipoma and teratoma; Lung: bronchogenic carcinoma (squamous cell or epidermoid, undifferentiated small cell, undifferentiated large cell, adenocarcinoma) , alveolar (bronchiolar) carcinoma, bronchial adenoma, sarcoma, lymphoma, chondromatous hamartoma, mesothelioma; Gastrointestinal : esophagus (squamous cell carcinoma, larynx, adenocarcinoma, leio
- kidney adenocarcinoma, WiIm' s tumor [nephroblastoma] , lymphoma, leukemia) , bladder and urethra (squamous cell carcinoma, transitional cell carcinoma, adenocarcinoma) , prostate (adenocarcinoma, sarcoma) , testis (seminoma, teratoma, embryonal carcinoma, teratocarcinoma, choriocarcinoma, sarcoma, interstitial cell carcinoma, fibroma, fibroadenoma, adenomatoid tumors, lipoma) ; Liver : hepatoma (hepatocellular carcinoma) , cholangiocarcinoma
- cancer includes a cell afflicted by any one of the above- identified conditions.
- the cancer is selected from colorectal, thyroid, or breast cancer.
- an "effective amount" of the compound or pharmaceutically acceptable composition is that amount effective in order to treat said disease.
- the compounds and compositions, according to the method of the present invention may be administered using any amount and any route of administration effective for treating or lessening the severity of said disease.
- said disease is chosen from allergic or type I hypersensitivity reactions, asthma, diabetes, Alzheimer's disease, Huntington' s disease, Parkinson's disease, AIDS- associated dementia, bipolar disorder, amyotrophic lateral sclerosis (ALS, Lou Gehrig's disease), multiple sclerosis (MS) , schizophrenia, leukocytopenia, cardiomyocyte hypertrophy, reperfusion/ischemia, stroke, baldness, transplant rejection, graft versus host disease, rheumatoid arthritis, and solid and hematologic malignancies.
- said disease is chosen from diabetes, bipolar disorder, schizophrenia, stroke, Huntington' s disease, leukocytopenia and cardiomyocyte hypertrophy.
- said disease is a protein- kinase mediated condition.
- protein kinase-mediated condition means any disease or other deleterious condition in which a protein kinase plays a role.
- Such conditions include, without limitation, autoimmune diseases, inflammatory diseases, proliferative and hyperproliferative diseases, immunologically-mediated diseases, immunodeficiency disorders, immunomodulatory or immunosuppressive disorder, bone diseases, metabolic diseases, neurological and neurodegenerative diseases, cardiovascular diseases, hormone related diseases, diabetes, allergies, asthma, and Alzheimer's disease.
- GSK-3-mediated condition means any disease or other deleterious condition in which GSK-3 plays a role.
- Such conditions include, without limitation, diabetes, diabetic neuropathy, osteoporosis, Alzheimer's disease, Huntington' s disease, Parkinson's disease, AIDS-associated dementia, bipolar disorder, amyotrophic lateral sclerosis (ALS, Lou Gehrig's disease), multiple sclerosis (MS) , schizophrenia, leukocytopenia, cardiomyocyte hypertrophy, stroke, spinal cord injury, traumatic brain injury, Charcot-Marie-Tooth, and rheumatoid arthritis .
- ALS amyotrophic lateral sclerosis
- MS multiple sclerosis
- said disease is a degenerative condition.
- said degenerative condition is chosen from stroke, post-stroke recovery, Alzheimer's Disease, Parkinson's Disease, Huntington' s Disease, Amyotrophic Lateral Sclerosis (ALS) , multiple sclerosis (MS) , spinal cord injury, traumatic brain injury, Charcot- Marie-Tooth, leukocytopenia, diabetes, diabetic neuropathy, and osteoporosis.
- said disease is a neurodegenerative condition.
- said neurodegenerative conditions is selected from stroke, post- stroke recovery, Alzheimer's disease, Parkinson's disease, Huntington' s disease, Amyotrophic Lateral Sclerosis (ALS), multiple sclerosis (MS) , spinal cord injury, traumatic brain injury, and Charcot-Marie-Tooth.
- One embodiment provides a method of increasing axonal and dendritic branching in neuronal cells comprising the step of contacting said cells with a compound described herein. Another embodiment provides a method of promoting neuroplasticity comprising the step of contacting said cells with a compound described herein. Another embodiment provides a method of promoting angiogenesis comprising the step of contacting said cells with a compound described herein. Yet another embodiment provides a method of treating neuropsychiatric disorders, such as mania and depression, comprising administering to a patient a compound described herein.
- said neurodegenerative disease is stroke.
- the compounds are used to treat stroke patients during stroke recovery.
- the compounds are used in post-stroke administration.
- the length of treatment can range from 1 month to one year.
- the compound is administered after the stroke has occurred.
- said administration occurs immediately after ischemia.
- said administration occurs 48 hours after ischemia to 6 months after ischemia.
- the compounds are used in combination with other forms of stroke recovery treatment, such as physical therapy.
- Another embodiment provides a method of treating diabetes comprising the step of contacting a beta cell with a compound described herein. In some embodiments, the compound promotes beta cell regeneration. [0081] Another embodiment provides a method of treating osteoporosis comprising the step of contacting a bone cell with a compound described herein. In some embodiments, said compound promotes osteoblastogenesis in the cell. [0082] It will also be appreciated that certain of the compounds of present invention can exist in free form for treatment, or where appropriate, as a pharmaceutically acceptable salt or pharmaceutically acceptable derivative thereof .
- compositions of the present invention additionally comprise a pharmaceutically acceptable carrier, adjuvant, or vehicle, which, as used herein, includes any and all solvents, diluents, or other liquid vehicle, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, solid binders, lubricants and the like, as suited to the particular dosage form desired.
- a pharmaceutically acceptable carrier, adjuvant, or vehicle which, as used herein, includes any and all solvents, diluents, or other liquid vehicle, dispersion or suspension aids, surface active agents, isotonic agents, thickening or emulsifying agents, preservatives, solid binders, lubricants and the like, as suited to the particular dosage form desired.
- Martin (Mack Publishing Co., Easton, Pa., 1980) discloses various carriers used in formulating pharmaceutically acceptable compositions and known techniques for the preparation thereof. Except insofar as any conventional carrier medium is incompatible with the compounds of the invention, such as by producing any undesirable biological effect or otherwise interacting in a deleterious manner with any other component (s) of the pharmaceutically acceptable composition, its use is contemplated to be within the scope of this invention.
- Some examples of materials which can serve as pharmaceutically acceptable carriers include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, or potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, polyacrylates, waxes, polyethylene- polyoxypropylene-block polymers, wool fat, sugars such as lactose, glucose and sucrose; starches such as corn starch and potato starch; cellulose and its derivatives such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; powdered tragacanth; malt; gelatin;
- the protein kinase inhibitors or pharmaceutical salts thereof may be formulated into pharmaceutical compositions for administration to animals or humans.
- These pharmaceutical compositions which comprise an amount of the protein inhibitor effective to treat or prevent a protein kinase-mediated condition and a pharmaceutically acceptable carrier, are another embodiment of the present invention.
- said protein kinase-mediated condition is a GSK-3-mediated condition.
- a GSK-3-mediated condition is another embodiment of the present invention.
- the exact amount of compound required for treatment will vary from subject to subject, depending on the species, age, and general condition of the subject, the severity of the infection, the particular agent, its mode of administration, and the like.
- the compounds of the invention are preferably formulated in dosage unit form for ease of administration and uniformity of dosage.
- dosage unit form refers to a physically discrete unit of agent appropriate for the patient to be treated. It will be understood, however, that the total daily usage of the compounds and compositions of the present invention will be decided by the attending physician within the scope of sound medical judgment.
- the specific effective dose level for any particular patient or organism will depend upon a variety of factors including the disorder being treated and the severity of the disorder; the activity of the specific compound employed; the specific composition employed; the age, body weight, general health, sex and diet of the patient; the time of administration, route of administration, and rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination or coincidental with the specific compound employed, and like factors well known in the medical arts.
- patient means an animal, preferably a mammal, and most preferably a human.
- compositions of this invention can be administered to humans and other animals orally, rectally, parenterally, intracisternally, intravaginally, intraperitoneally, topically (as by powders, ointments, or drops) , bucally, as an oral or nasal spray, or the like, depending on the severity of the infection being treated.
- the compounds of the invention may be administered orally or parenterally at dosage levels of about 0.01 mg/kg to about 50 mg/kg and preferably from about 1 mg/kg to about 25 mg/kg, of subject body weight per day, one or more times a day, to obtain the desired therapeutic effect.
- Liquid dosage forms for oral administration include, but are not limited to, pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs.
- the liquid dosage forms may contain inert diluents commonly used in the art such as, for example, water or other solvents, solubilizing agents and emulsifiers such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, dimethylformamide, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor, and sesame oils) , glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, and mixtures thereof.
- the oral compositions can also
- Injectable preparations for example, sterile injectable aqueous or oleaginous suspensions may be formulated according to the known art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution, suspension or emulsion in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1, 3-butanediol .
- acceptable vehicles and solvents that may be employed are water, Ringer's solution, U. S. P. and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium prior to use.
- a compound of the present invention In order to prolong the effect of a compound of the present invention, it is often desirable to slow the absorption of the compound from subcutaneous or intramuscular injection. This may be accomplished by the use of a liquid suspension of crystalline or amorphous material with poor water solubility. The rate of absorption of the compound then depends upon its rate of dissolution that, in turn, may depend upon crystal size and crystalline form. Alternatively, delayed absorption of a parenterally administered compound form is accomplished by dissolving or suspending the compound in an oil vehicle. Injectable depot forms are made by forming microencapsule matrices of the compound in biodegradable polymers such as polylactide- polyglycolide .
- compositions for rectal or vaginal administration are preferably suppositories which can be prepared by mixing the compounds of this invention with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- Solid dosage forms for oral administration include capsules, tablets, pills, powders, and granules.
- the active compound is mixed with at least one inert, pharmaceutically acceptable excipient or carrier such as sodium citrate or dicalcium phosphate and/or a) fillers or extenders such as starches, lactose, sucrose, glucose, mannitol, and silicic acid, b) binders such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidinone, sucrose, and acacia, c) humectants such as glycerol, d) disintegrating agents such as agar-- agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate, e) solution retarding agents such as paraffin, f) absorption accelerators such as quaternary ammonium compounds, g) wetting agents such as, for example, cety
- Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethylene glycols and the like.
- the solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings and other coatings well known in the pharmaceutical formulating art. They may optionally contain opacifying agents and can also be of a composition that they release the active ingredient (s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner.
- Examples of embedding compositions that can be used include polymeric substances and waxes.
- Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polethylene glycols and the like.
- the active compounds can also be in microencapsulated form with one or more excipients as noted above.
- the solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings, release controlling coatings and other coatings well known in the pharmaceutical formulating art. In such solid dosage forms the active compound may be admixed with at least one inert diluent such as sucrose, lactose or starch.
- Such dosage forms may also comprise, as is normal practice, additional substances other than inert diluents, e.g., tableting lubricants and other tableting aids such a magnesium stearate and microcrystalline cellulose.
- additional substances other than inert diluents e.g., tableting lubricants and other tableting aids such a magnesium stearate and microcrystalline cellulose.
- the dosage forms may also comprise buffering agents. They may optionally contain opacifying agents and can also be of a composition that they release the active ingredient (s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner.
- embedding compositions that can be used include polymeric substances and waxes.
- Dosage forms for topical or transdermal administration of a compound of this invention include ointments, pastes, creams, lotions, gels, powders, solutions, sprays, inhalants or patches.
- the active component is admixed under sterile conditions with a pharmaceutically acceptable carrier and any needed preservatives or buffers as may be required.
- Ophthalmic formulation, eardrops, and eye drops are also contemplated as being within the scope of this invention.
- the present invention contemplates the use of transdermal patches, which have the added advantage of providing controlled delivery of a compound to the body.
- Such dosage forms can be made by dissolving or dispensing the compound in the proper medium.
- Absorption enhancers can also be used to increase the flux of the compound across the skin. The rate can be controlled by either providing a rate controlling membrane or by dispersing the compound in a polymer matrix or gel.
- compositions to treat or prevent the above-identified disorders .
- a "pharmaceutically acceptable derivative or prodrug” means any pharmaceutically acceptable ester, salt of an ester or other derivative of a compound of this invention which, upon administration to a recipient, is capable of providing, either directly or indirectly, a compound of this invention or an inhibitorily active metabolite or residue thereof.
- Particularly favoured derivatives or prodrugs are those that increase the bioavailability of the compounds of this invention when such compounds are administered to a patient (e.g., by allowing an orally administered compound to be more readily absorbed into the blood) or which enhance delivery of the parent compound to a biological compartment (e.g., the brain or lymphatic system) relative to the parent species .
- Pharmaceutically acceptable prodrugs of the compounds of this invention include, without limitation, esters, amino acid esters, phosphate esters, metal salts and sulfonate esters.
- Pharmaceutically acceptable carriers that may be used in these pharmaceutical compositions include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene- polyoxypropylene-block polymers, polyethylene glycol and wool fat.
- ion exchangers alumina, aluminum stearate, lecithin
- serum proteins such as human serum albumin
- buffer substances such as phosphates, glycine, sorbic
- compositions of the present invention may be administered orally, parenterally, by inhalation spray, topically, rectally, nasally, buccally, vaginally or via an implanted reservoir.
- parenteral as used herein includes, but is not limited to, subcutaneous, intravenous, intramuscular, intra-articular, intra-synovial, intrasternal, intrathecal, intrahepatic, intralesional and intracranial injection or infusion techniques.
- the compositions are administered orally, intraperitoneally or intravenously.
- Sterile injectable forms of the compositions of this invention may be aqueous or oleaginous suspension. These suspensions may be formulated according to techniques known in the art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example as a solution in 1, 3-butanediol .
- the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose, any bland fixed oil may be employed including synthetic mono- or di-glycerides .
- Fatty acids such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions.
- These oil solutions or suspensions may also contain a long- chain alcohol diluent or dispersant, such as carboxymethyl cellulose or similar dispersing agents which are commonly used in the formulation of pharmaceutically acceptable dosage forms including emulsions and suspensions.
- Other commonly used surfactants such as Tweens, Spans and other emulsifying agents or bioavailability enhancers which are commonly used in the manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms may also be used for the purposes of formulation.
- compositions of this invention may be orally administered in any orally acceptable dosage form including, but not limited to, capsules, tablets, aqueous suspensions or solutions.
- carriers commonly used include, but are not limited to, lactose and corn starch.
- Lubricating agents such as magnesium stearate, are also typically added.
- useful diluents include lactose and dried cornstarch.
- aqueous suspensions are required for oral use, the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening, flavoring or coloring agents may also be added.
- the pharmaceutical compositions of this invention may be administered in the form of suppositories for rectal administration. These can be prepared by mixing the agent with a suitable non-irritating excipient which is solid at room temperature but liquid at rectal temperature and therefore will melt in the rectum to release the drug. Such materials include, but are not limited to, cocoa butter, beeswax and polyethylene glycols.
- a suitable non-irritating excipient which is solid at room temperature but liquid at rectal temperature and therefore will melt in the rectum to release the drug.
- suitable non-irritating excipient which is solid at room temperature but liquid at rectal temperature and therefore will melt in the rectum to release the drug.
- Such materials include, but are not limited to, cocoa butter, beeswax and polyethylene glycols.
- the pharmaceutical compositions of this invention may also be administered topically, especially when the target of treatment includes areas or organs readily accessible by topical application, including diseases of the eye, the skin, or the lower intestinal tract. Suitable
- Topical application for the lower intestinal tract can be effected in a rectal suppository formulation (see above) or in a suitable enema formulation. Topically- transdermal patches may also be used.
- the pharmaceutical compositions may be formulated in a suitable ointment containing the active component suspended or dissolved in one or more carriers.
- Carriers for topical administration of the compounds of this invention include, but are not limited to, mineral oil, liquid petrolatum, white petrolatum, propylene glycol, polyoxyethylene, polyoxypropylene compound, emulsifying wax and water.
- the pharmaceutical compositions can be formulated in a suitable lotion or cream containing the active components suspended or dissolved in one or more pharmaceutically acceptable carriers.
- Suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water.
- the pharmaceutical compositions may be formulated as micronized suspensions in isotonic, pH adjusted sterile saline, or, preferably, as solutions in isotonic, pH adjusted sterile saline, either with or without a preservative such as benzylalkonium chloride.
- the pharmaceutical compositions may be formulated in an ointment such as petrolatum.
- compositions of this invention may also be administered by nasal aerosol or inhalation.
- Such compositions are prepared according to techniques well- known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other conventional solubilizing or dispersing agents.
- the amount of protein kinase inhibitor that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated, the particular mode of administration.
- the compositions should be formulated so that a dosage of between 0.01 - 100 mg/kg body weight/day of the inhibitor can be administered to a patient receiving these compositions .
- the invention provides methods for treating or preventing a protein kinase-mediated condition (in some embodiments, a GSK-3- mediated condition) comprising the step of administering to a patient one of the above-described pharmaceutical compositions.
- a protein kinase-mediated condition in some embodiments, a GSK-3- mediated condition
- patient means an animal, preferably a human.
- that method is used to treat or prevent a condition selected from cancers such as cancers of the breast, colon, prostate, skin, pancreas, brain, genitourinary tract, lymphatic system, stomach, larynx and lung, including lung adenocarcinoma and small cell lung cancer; stroke, diabetes, myeloma, hepatomegaly, cardiomegaly, Alzheimer's disease, Parkinson's Disease, Huntington' s Disease, Amyotrophic Lateral Sclerosis (ALS), multiple sclerosis (MS) , spinal cord injury, traumatic brain injury, Charcot-Marie-Tooth, leukocytopenia, diabetic neuropathy, osteoporosis, cystic fibrosis, and viral disease, or any specific disease described above.
- cancers such as cancers of the breast, colon, prostate, skin, pancreas, brain, genitourinary tract, lymphatic system, stomach, larynx and lung, including lung adenocarcinoma and small cell lung cancer
- stroke diabetes, mye
- Another aspect of the invention relates to inhibiting protein kinase activity in a patient, which method comprises administering to the patient a compound of formula I or a composition comprising said compound.
- additional drugs which are normally administered to treat or prevent that condition, may be administered together with the inhibitors of this invention.
- chemotherapeutic agents or other anti-proliferative agents may be combined with the protein kinase inhibitors of this invention to treat proliferative diseases.
- those additional agents may be administered separately, as part of a multiple dosage regimen, from the protein kinase inhibitor-containing compound or composition.
- those agents may be part of a single dosage form, mixed together with the protein kinase inhibitor in a single composition.
- said protein kinase inhibitor is a GSK-3 kinase inhibitor.
- This invention may also be used in methods other than those involving administration to a patient.
- the compounds of this invention may be prepared in general by methods known to those skilled in the art. Those compounds may be analyzed by known methods, including but not limited to LCMS (liquid chromatography mass spectrometry) and NMR (nuclear magnetic resonance) . Compounds of this invention may be also tested according to these examples. It should be understood that the specific conditions shown below are only examples, and are not meant to limit the scope of the conditions that can be used for making, analyzing, or testing the compounds of this invention. Instead, this invention also includes conditions known to those skilled in that art for making, analyzing, and testing the compounds of this invention.
- LCMS Liquid Chromatography Mass Spectrometry
- samples were analyzed on a MicroMass Quattro Micro mass spectrometer operated in single MS mode with electrospray ionization. Samples were introduced into the mass spectrometer using chromatography. Mobile phase for all mass spec, analysis consisted of acetonitrile-water mixtures with either 0.2% formic acid or 0.1% TFA as a modifier. Column gradient conditions are 10%-90% acetonitrile over 3 mins gradient time and 5 mins run time on a Waters YMC Pro- C18 4.6x50mm column. Flow rate is 1.5 ml/min.
- 1 H-NMR spectra were recorded at 400 MHz using a Bruker DPX 400 instrument. The following compounds of formula I were prepared and analyzed as follows.
- Reagents and conditions i . Pd (OAc) 2 , PPh 3 , Et 3 N, H 2 CO 2 ; ii . 1 ) (COCl ) 2 , CH 2 Cl 2 , cat . DMF; 2 ) NH 3 (g) , dioxane , iii . TFAA, Et 3 N, CH 2 Cl 2 , 0 ° C ; iv . H 2 NNH 2 . H 2 O, n-butanol , reflux
- An assay stock buffer solution was prepared containing all of the reagents listed above with the exception of ATP and the test compound of the present invention.
- the assay stock buffer solution (175 ⁇ l) was incubated in a 96 well plate with 5 ⁇ l of the test compound of the present invention at final concentrations spanning 0.002 ⁇ M to 30 ⁇ M at 3O 0 C for 10 min.
- a 12-point titration was conducted by preparing serial dilutions (from 10 mM compound stocks) with DMSO of the test compounds of the present invention in daughter plates. The reaction was initiated by the addition of 20 ⁇ l of ATP (final concentration 20 ⁇ M) .
- Rates of reaction were obtained using a Molecular Devices Spectramax plate reader (Sunnyvale, CA) over 10 min at 3O 0 C.
- the K 1 values were determined from the rate data as a function of inhibitor concentration.
- Compounds of the invention were found to inhibit GSK-3.
- Compounds 1-1 and 1-2 were found to inhibit GSK-3 at a Ki value of ⁇ 5 nM.
- Example 3 GSK-3 ⁇ and GSK3 ⁇ p-TYR Inhibition Assay [00143] Compounds are screened for their ability to inhibit the phosphorylation of tyrosine (TYR) residues through the use of western blotting of Jurkat cells dosed with the compounds. The phosphorylation of the specific TYR residues tested are GSK3 ⁇ TYR 279 and GSK3 ⁇ TYR 216.
- Jurkat cells are seeded at a density of 2xl ⁇ 5 cells/well in a 12 well dish in starvation media
- PBS containing 0.1% Tween 20, such as ImI Tween per IL of PBS is then made up and used for all washes and antibody incubations. The blot is blocked in 5% nonfat milk PBST for one hour.
- the primary antibody (GSK-3 ⁇ / ⁇ pTYR 279/216 at 1:1000 dilution Upstate cat#05-413) is then added in 5%- nonfat milk PBST overnight at 4 0 C with gentle rocking. The blot is then washed in PBST for 5 min. This is then repeated 4 times. A secondary anti-mouse-HRP conjugated antibody (1:5000 dilution) is added for 60min in 5%milk PBST. The blot is then washed in PBST for 5min. This is also repeated 4 times. 3.OmL of the developing solution (ECL plus Western Blotting Detection System from Amersham/GE cat# RPN2132) is made and then added.
- ECL plus Western Blotting Detection System from Amersham/GE cat# RPN2132
- GAPDH expression level is used as a loading control, (GAPDH antibody: santa cruz 25-778) at 1:10000 dilution.
- IC50 For determination of GSK-3 ⁇ and GSK-3 ⁇ pTYR IC50, the density of the respective bands for each protein at specific compound concentration is compared to a no compound DMSO treated control cell sample present on each exposure. IC50 numbers are defined as the concentration of compound in which the density of the GSK-3 ⁇ or GSK-3 ⁇ band is 50% of the no compound control.
- Example 4 ⁇ -Catenin Stabilization Protocol
- GSK-3 phosphorylation of ⁇ -catenin targets it to the proteosome for degradation. Inhibition of GSK-3 results in accumulation of ⁇ -catenin in the cytosol of cells which through interaction with the transcription factor TCF/LEF translocates to the nucleus and drives the transcription of Wnt-dependent genes.
- the assay is designed to determine the level of ⁇ -catenin dependent TCF/LEF transcriptional activity in a quantitative manner through the use of a ⁇ - lactamase reporter assay in Jurkat cells dosed with a compound.
- Jurkat ⁇ -catenin cells are starved overnight in assay media (1% FBS, Ix Penstrep, RPMI) in the flask. The next day Jurkat ⁇ -catenin cells are seeded in 96 well flat bottom plates at a density of 50,000 cells/well in assay media in a volume of lOOul. The compound is added to the well at a final DMSO concentration of 0.75% and incubated at 37 0 C o/n. The next day, 2OuL of 6x CCF4 dye is added to the wells and incubated at room temperature for 1-2 hours. Plates are read on the Cytofluor 4000 series multiwell plate reader and the 460/530 ratio is determined.
- assay media 1% FBS, Ix Penstrep, RPMI
- the GSK-3 IC50 for induction of ⁇ -catenin is determined by plotting the 460/530 ratio against the concentration of compound (Log scale) and using the equation of the slope to calculate the point at which the ratio is 50% of the maximum effect.
- ⁇ -catenin GSK-3 windows were calculated by dividing the ⁇ -catenin IC50 value obtained in Example 4 by the GSK-3 ⁇ or GSK3 ⁇ p-TYR IC50 value obtained in Example 3.
- Both compounds 1-1 and 1-2 were found to have a ⁇ -catenin : GSK-3 ⁇ window between 400 and 500 fold.
- Compound 1-1 was found to have a ⁇ -catenin : GSK-3 ⁇ window between 75 and 100 fold and compound 2 was found to have a ⁇ -catenin : GSK-3 ⁇ window between 25 and 50 fold.
- Table 3 shows GSK-3 ⁇ pTYR, GSK-3 ⁇ pTYR, and ⁇ -catenin IC50 data for select compounds of Table 1.
- Hypotonic Lysis Buffer consists of 1OmM HEPES, 1OmM KCL, 1.5mM MgCl 2 , 1.OmM EDTA, 1.OmM DTT, Ix Roche protease inhibitor cocktail, and 1.OmM AEBSF Calbiochem protease inhibitor cocktail (cat#539134) . All concentrations are final and are diluted in water.
- hypotonic lysis buffer is added to the tissue at 5x' s weight.
- the tissue is then broken up on ice using the end of a syringe plunger.
- the samples are freeze-thawed five times.
- the lysates are then transferred to ultracentrifuge tubes and spun at 100,00Og for 35 min at 4 0 C.
- the supernatants are then collected and an aliquot is taken to measure protein with the Pierce BCA protein assay kit (cat#23225) , using BSA standard curve.
- the remaining protein lysates are diluted 1:1 with laemmli sample buffer containing ⁇ -mercaptoethanol .
- the protein concentration is then normalized.
- the samples are boiled for 5 min at 95 0 C followed by a spin at 14,000 rpm in a mini-centrifuge for 1 min.
- the samples are then snap frozen with dry ice and stored at -20 0 C.
- the samples are first loaded on 10% Tris-glycine gel (lOuL/well) .
- the gel is then run at 120V until the dye marker runs off of gel.
- the PVDF membrane is cut and soaked in methanol for 5 min before use.
- the protein is then transferred to a PVDF membrane at 100V for 75 min (the transfer rig is kept on ice) .
- the membrane is then blocked with 5% non-fat dry milk dissolved in PBS 0.1% Tween 20 for 1 hr at RT (room temperature) .
- a primary antibody (SC-7199, Santa Cruz rabbit polyclonal anti-human ⁇ -catenin) is added at a 1:2000 dilution in block buffer.
- Beta tubulin loading control SC-9104
- SC-9104 Beta tubulin loading control
- the membrane is washed 4 times for 5 min each with PBS 0.1% Tween.
- Anti-rabbit secondary HRP-conjugated antibody (1:5000 dilution in block buffer) is then added.
- the membrane is washed 4 times for 5 min each with PBS 0.1% Tween.
- ECL reagent (Pierce) is added.
- the film is exposed. Induction of ⁇ -catenin is determined by comparison of the density of protein band in samples obtained from compound treated animals with that of vehicle treated animals.
- RIPA consists of 0.5 ml 10% SDS, 2.5 ml 10% Sodium Deoxycholate, 0.5 ml NP40, and 46.5 ml PBS.
- the Lysis buffer consists of 8.3 ml of RIPA, 1.0 ml of ⁇ - glycerolphosphate (50OmM), 0.1 ml NaF (500 mM) , 0.1 ml of Sodium Vanadate (200 mM) , 0.4 ml of protease inhibitor (Roche protease inhibitor cocktail tablets: dissolve 1 tablet in 2 ml of water to obtain 25x stock. cat# 11873580001), and 0.1 ml of PBS.
- lysis buffer modified RIPA
- tissue lml/1/2 brain
- the lysates are then centrifuged at 10000 rpm for lOmin at 4 0 C.
- the supernatant is then transferred to a new tube and kept on ice.
- the supernatant samples are respun at 10000 rpm for 10 min at 4 0 C.
- the supernatant is then transferred to a new tube and kept on ice.
- An aliquot of the supernatant is taken and the protein is measured with the Pierce BCA protein assay kit (cat#23225) , using BSA standard curve (do a 1:25 dilution in PBS).
- Cortical or hippocampal lobes are combined with 9mL of Base media (Neurobasal + Pen/Strep) and put on ice. ImL of 1OX trypsin solution is added and the mixture is swirled gently. The tissue is then digested via incubation in a 37 0 C waterbath for 20 minutes. After 20 minutes, lO ⁇ l/ml DNase (lOO ⁇ L DNase) is added and the mixture is incubated for another 5 minutes.
- the cells are spun at lOOOrpm for 1 minute. The enzyme solution is then removed without removing any of the brain fragments sitting on the bottom. The solid is washed 3 times with Wash media (Neurobasal + 10% and Pen/Strep) . After the 3 rd wash, the cells are re-suspended in 5ml of Culture Media (Neurobasal + B27, L-Glutamine and Pen/Strep). Mechanical dissociation is performed by gently pipetting several times through a flame-narrowed glass pipet, taking care not to make bubbles. The cells are then filtered through a 70 ⁇ m cell strainer. The cells are counted in a hemacytometer and seeded at 5000-10000 cells/well in a 24 well plate with glass coverslip inserts coated with PDL. The cells are incubated at 37 0 C o/n.
- PBS Phosphate Buffered Saline
- Blocking buffer PBS-T + 5% normal donkey serum or HBSS-T + 5% normal donkey serum
- Neurofilament antibody 1:250 Abeam
- MAP2 antibody 1:250 Abeam 6
- the media or PBS is first removed. Then, 500 uL of HistoChoice is added to cover the cells. The cells are incubated at room temperature for 10 minutes. They are then washed 2 times with PBS, with a 5 minute incubation after each wash. Amounts are shown below:
- the cells are incubated with blocking buffer for 30 minutes at room temperature.
- the tissue is then incubated with blocking buffer for 1 hour at room temperature.
- 1° antibodies are diluted in PBS + 0.1% Tween + 5% Donkey serum.
- the blocking solution is removed and sufficient volume of 1° antibody in blocking buffer is added to cover the cells.
- 1° antibody is incubated at 4 0 C overnight.
- the next day 1° antibody is removed and coverslips are washed twice with PBS-T with a 5 minute incubation between each wash.
- the PBS-T is removed and blocking buffer is added.
- the cells are incubated for 30 minutes.
- the 2° antibody is diluted in PBS + 0.1% Tween + 5% Donkey serum. The mixture is incubated for 30 mins at room temperature. The slides are washed three times with PBS-T and once with PBS. Mounting media is added to reduce quenching of fluorochromes . The glass coverslips are removed and placed on a slide for visualization. Analysis
- axonal branching is determined by quantification of area under threshold fluorescence of neurofilament Alexa 488 per cell.
- Dendritic branching is determined by quantification of area under threshold fluorescence of MAP2 Alexa 568 per cell.
- branching can be determined by manual counting of branch points per cell.
- Compound effects are assessed by comparing the area under threshold fluorescence in compound treated cultures to that of a DMSO control at the same time point. Treatment of E16 hippocampal neurons with 10 nM of Compound 1-1 for 7 days resulted in increased axonal and dendritic branching.
- Example 8 CRMP2 phosphorylation assay.
- Cortical or hippocampal lobes are combined with 9mL of Base media (Neurobasal + Pen/Strep) and put on ice. ImL of 1OX trypsin solution is added and the mixture is swirled gently. The tissue is then digested via incubation in a 37 0 C waterbath for 20 minutes. After 20 minutes, lO ⁇ l/ml DNase (lOO ⁇ L DNase) is added and the mixture is incubated for another 5 minutes.
- the cells are spun at lOOOrpm for 1 minute. The enzyme solution is then removed without removing any of the brain fragments sitting on the bottom. The solid is washed 3 times with Wash media (Neurobasal + 10% and Pen/Strep) . After the 3 rd wash, the cells are re-suspended in 5ml of Culture Media (Neurobasal + B27, L-Glutamine and Pen/Strep) . Mechanical dissociation is performed by gently pipetting several times through a flame-narrowed glass pipet, taking care not to make bubbles. The cells are then filtered through a 70 ⁇ m cell strainer. The cells are counted in a hemacytometer and seeded at 50,000 cells/well in a 12 well plate. The cells are incubated at 37 0 C o/n.
- the primary antibody (1:10,000 CRMP2 rabbit polyclonal Abeam #ab36201) is then added in 5%-nonfat milk PBST overnight at 4 0 C with gentle rocking. The blot is then washed in PBST for 5 min. This is then repeated 4 times.
- a secondary anti-mouse-HRP conjugated antibody (1:5000 dilution) is added for 60min in 5%milk PBST. The blot is then washed in PBST for 5min. This is also repeated 4 times .
- HUVEC are used between P3 and P4. HUVEC are mixed with dextran coated cytodex 3 micro-carriers (Amersham
- beads with cells are transferred to a T-25 tissue culture flask and left for 12-16 hr in 5 ml of EGM-2 at 37 0 C 5% CO 2 .
- Angiogenesis is scored by quantification of images captured on an inverted microscope at 1Ox and 2Ox magnification for vessel length, number of vessels and branches per bead using NIH Image J software.
- HUVEC can be spin transduced with a retroviral vector expressing yellow fluorescent protein (YFP) under the control of a constitutively active minimal TK promoter, and sorted for YFP expression to enhance visualization.
- YFP positive HUVEC are then cultured as described above and quantification of vessel formation is determined by calculating the area under the threshold fluorescence using NIH Image J software.
- enhanced angiogenesis is determined by comparing compound treated cultures with a DMSO control culture at the same time point.
- the rats receive surgery during which a stroke is induced.
- the first group receives sham surgery with vehicle as treatment.
- Administration of the test compound and vehicle is determined by the sponsor.
- the core body temperature is maintained at 37° C
- each animal is weighed (average weights were 340 g) and then anesthetized with isoflurane (4% isoflurane carried on 2 1/min medical grade oxygen to induce surgical plane and then 2% with 2 1/min oxygen to maintain a surgical plane) .
- isoflurane 4% isoflurane carried on 2 1/min medical grade oxygen to induce surgical plane and then 2% with 2 1/min oxygen to maintain a surgical plane.
- each rat is individually marked with an ear-punch and administered a subcutaneous dose of buprenorphine (0.025 mg/kg) .
- Rectal temperature is monitored and maintained at 37 C +/- I 0 C for the duration of the surgery and until the rat is awake and mobile (approximately 3 hr) .
- the rat is then placed into a stereotaxic apparatus positioned such that the lateral aspect of the head was facing up.
- the skin between the eye left eye and ear is shaved and washed with surgical antiseptic scrub.
- a vertical incision is made midway between the right orbit and external auditory canal.
- the underlying temporalis muscle is incised, detached from the skull and retracted with care to preserve the facial nerve.
- Two sutures hold the temporal muscle away from the lateral aspect of the skull.
- a craniotomy is performed from the posterior zygoma and along the temporal ridge of the cranium extending ventrally to expose the middle cerebral artery (MCA) and olfactory tract.
- MCA middle cerebral artery
- the dura is opened, and the base of the MCA and the anterior portion of the first branch is electrocoagulated ventral to the olfactory tract, resulting in infarction of the right dorsolateral cerebral cortex.
- Forepaw use is measured with a procedure that is adapted from the method devised by Whishaw, O'Connor, and Dunnett (1986). Each animal is food restricted such that feeding time occurs after testing each day. The animals are placed in test cages (10 X 18 X 10 cm high) with floors and fronts constructed of 2-mm bars, 9 mm apart edge to edge. A 4-cm wide and 5-cm deep tray, containing chicken feed pellets, is mounted outside of each box. The rats are required to extend a forelimb through the gap in the bars, grasp, and retract the food.
- a ⁇ hit' is defined as the successful grasping and retrieval of a food pellet that results in consumption of the pellet.
- a ⁇ miss' is defined as the unsuccessful retrieval of a food pellet (either failed to properly grasp pellet, or lost the pellet during the retrieval such that the pellet was not consumed) .
- Percent of hits is calculated as the total number of hits during a session divided by the total number of reaches. This is calculated separately for left and right paw (or affected and unaffected following stoke) . Once the criterion of 50% success (involving reaches from both paws) is reached, each rat is video-taped during a 5-min reaching session.
- the results of this session serve as pre-surgical baseline.
- the pre-surgical test session are also be used to determine hand dominance of each rat.
- the stroke damage is administered within the brain hemisphere that is contralateral to the dominant hand used for reaching.
- Post surgical testing consists of a 5-min reaching test each week that the animals are tested.
- Each session is observed on a monitor using a frame by frame analysis of each reach. Each session is scored by 2 different scorers. The final calculation of percent hits for each scorer is within a 5% range of one another. If a greater disparity between final scores occurs then the session is rescored by both observers. [00198] The percentage of hits for affected and non affected paws for each group is compared among groups using a one-way analysis of variance.
- Compound 1-1 was tested in the above model. The stroke reliably produced a deficit in reaching performance at 7 and 14 days after surgery. Addition of compound 1-1 significantly ameliorated this deficit at 7 and 14 days after stroke compared with vehicle treatment.
- Forelimb and hindlimb coordination are measured using an apparatus that consists of two Plexiglas panels 1 m long and 25 cm wide (5 mm thick) with holes drilled 1 cm apart along one long edge. The panels are placed 2.5 cm apart and connected via several metal bars (3 mm diameter) through the holes. The bars are randomly placed 1, 2, or 3 cm apart. The apparatus is suspended and oriented such that a narrow alley (2.5 cm wide) is formed 1 m long with walls 25 cm high. The bars form the floor.
- Each animal is introduced to the apparatus using 3 trials in which the rat is placed at greater distances from the goal box on each subsequent trial. That is, on the first trial the rat is placed on the end of the grid near and facing the goal box. Once the rat has entered the goal box it is placed at the half way point on the grid, again facing the goal box. On the third and final trial the rat is placed at the entrance and allowed to traverse the entire grid to reach the goal box.
- One test session of 3 trials are conducted before stroke. Each test session after stroke includes 3 trials. Each trial is videotaped at close range from a horizontal plane. The tapes are scored by 2 observers using frame-by- frame analysis. The number of right and left (affected and unaffected) forelimb and hindlimb placement errors through the mid 80% of the grid are counted. The mid 80% of the grid is marked on the outside of the apparatus with masking tape. An error is whenever a limb extends (either partially or fully, i.e., just the paw or the entire leg) through the horizontal plane of the bars. The forelimb and hindlimb errors are summed separately for ipsilateral and contralateral limbs over the three trials and analyzed independently.
- Forepaw asymmetry of the animals is measured by placing a rat into a clear acrylic cylinder 25 cm in diameter.
- the cylinder is placed on a clear table with a mirror positioned such that the animal can be filmed from below. This vantage point provides a clear picture of the animal's forepaws as it explores the cylinder.
- rats tend to rear a great deal. With each rear the rat places its forepaws against the side of the cylinder to provide balance and support while investigating the cylinder. Investigation involves leans (while rearing) both to the left and right of the body as well as straight up. A normal rat uses equally both left and right forepaws to brace against the wall.
- the rat When investigating straight up the wall the rat uses both paws to brace. During the first 20 rears the bracing paw is noted. The first paw that touches the cylinder wall during the rear is counted. Testing continues until 20 rears have been recorded. The video recording is scored by 2 observers. Left and right paw wall touches are counted. Thus, for each brace the score could be L or R or L&R.
- One test session occurs prior to surgery and then on the designated post surgical test dates thereafter.
- the pre-surgery test is used to determine that the rats do not have a preexisting paw bias (that is, more than 15/20 wall touches to one side) . If they do show a side bias they are removed from the study.
- the post surgical scores are expressed as percentage of the touches using the affected (contralateral to stroke insult) paw. Groups are compared on this score using one-way analysis of variance.
- Compound 1-1 was tested in the above model. The stroke reliably produced a deficit in reaching performance at 7 and 14 days after surgery. Compound 1-1 at low dose significantly ameliorated this deficit at 14 days after stroke .
- Example 6 To track the activity of compound 1-1 in the CNS of rats subjected to MCAO, brains were removed and protein lysate was obtained (as described in Example 6 herein) from both vehicle treated and compound treated animals at the termination of the study. Lysates were analyzed by Western blot (see Example 3) and probed for GSK-3 ⁇ / ⁇ pTYR levels. Compound 1-1 showed a significant reduction in pTYR signal at all doses in the brain compared to vehicle treated rats with no induction of ⁇ -catenin.
- Rats are tested for forelimb somatosensory deficits with the adhesive removal test (Schallert T, et . al . , 1984 # 3) . Each animal receives 3 trials by placing round strips of packing tape (approx. 1.2 cm in diameter) at each testing day and the mean time (seconds) required to remove stimuli from the left forelimb is recorded.
- Protein lysate is obtained from the brains of all vehicle and compound treated animals and is processed for biomarker analysis of GSK3 ⁇ / ⁇ pTYR and ⁇ -catenin by Western blot assay to ensure compound activity on the target.
- Cerebral spinal fluid is obtained from all vehicle and compound treated animals and is analyzed for BDNF levels by ELISA as a surrogate marker for neuronal plasticity. Histological Analysis
- Paraffin embedded brain samples are obtained from Neuroinvestigations and cut into 6 urn sections onto glass slides and analyzed by immunohistochemistry or immunofluorescence for markers/phenotypes that correlate with beneficial outcomes in post-stroke recovery: Stem cell mobilization/proliferation: staining for BrdU and analysis using the Aperio system for quantitation of BrdU positive cells in the subventricular zone (SVZ) .
- Neurogenesis immunofluorescent staining for doublecortin
- DCX DCX in conjunction with BrdU in the SVZ using manual counting for quantitation.
- Angiogenesis staining for von Willebrand factor VIII (vWF) and analysis using the Aperio system for quantitation of vWF positive cells in the peri-infarct .
- vWF von Willebrand factor VIII
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Diabetes (AREA)
- Rheumatology (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Hematology (AREA)
- Psychology (AREA)
- Emergency Medicine (AREA)
- Epidemiology (AREA)
- Endocrinology (AREA)
- Obesity (AREA)
- Hospice & Palliative Care (AREA)
- Heart & Thoracic Surgery (AREA)
- Immunology (AREA)
- Cardiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Enzymes And Modification Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description


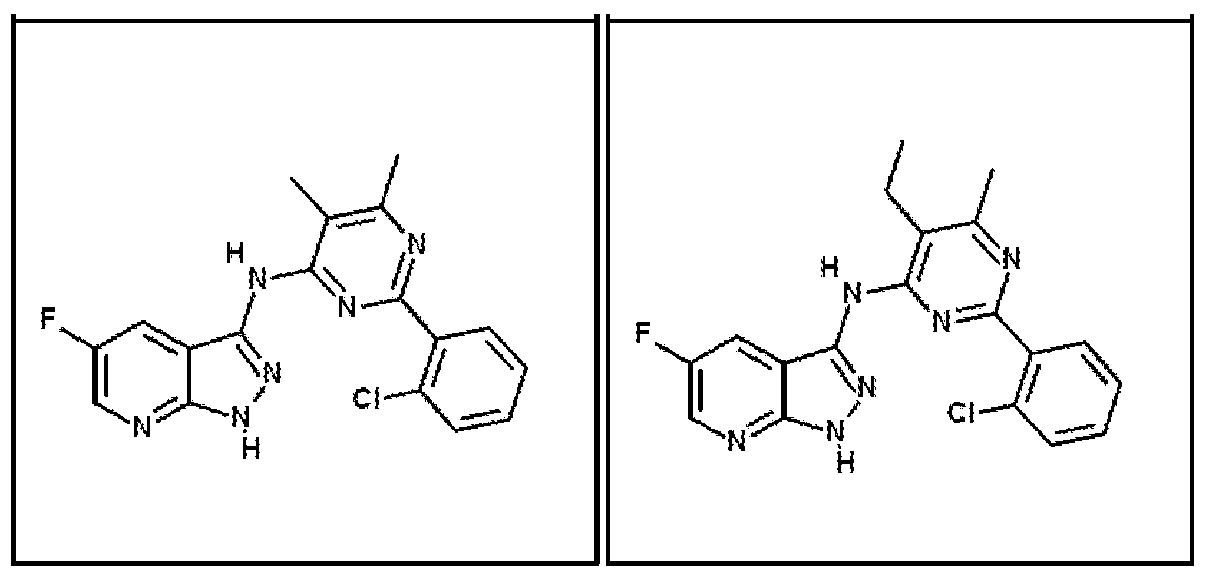










Claims
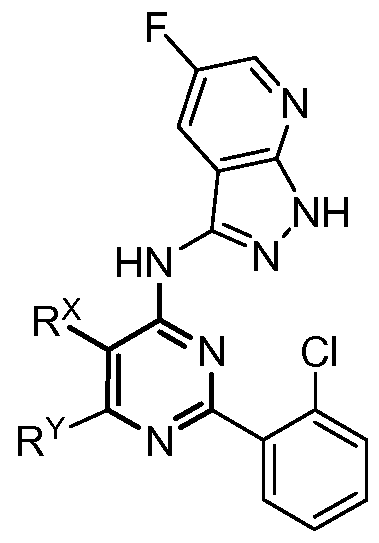


Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08731839A EP2137183B1 (en) | 2007-03-09 | 2008-03-10 | Aminopyrimidines useful as inhibitors of protein kinases |
| AU2008226466A AU2008226466B2 (en) | 2007-03-09 | 2008-03-10 | Aminopyrimidines useful as inhibitors of protein kinases |
| CN2008800138739A CN101668760B (en) | 2007-03-09 | 2008-03-10 | Aminopyrimidines useful as inhibitors of protein kinases |
| CA002680029A CA2680029A1 (en) | 2007-03-09 | 2008-03-10 | Aminopyrimidines useful as inhibitors of protein kinases |
| AT08731839T ATE526328T1 (en) | 2007-03-09 | 2008-03-10 | AMINOPYRIMIDINES SUITABLE AS INHIBITORS OF PROTEIN KINASES |
| MX2009009591A MX2009009591A (en) | 2007-03-09 | 2008-03-10 | Aminopyrimidines useful as inhibitors of protein kinases. |
| ES08731839T ES2374335T3 (en) | 2007-03-09 | 2008-03-10 | USEFUL AMINOPIRIMIDINS AS INHIBITORS OF PROTEIN KINASES. |
| NZ579485A NZ579485A (en) | 2007-03-09 | 2008-03-10 | Aminopyrimidines useful as inhibitors of protein kinases |
| JP2009552930A JP5393489B2 (en) | 2007-03-09 | 2008-03-10 | Aminopyrimidines useful as inhibitors of protein kinases |
| IL200793A IL200793A0 (en) | 2007-03-09 | 2009-09-07 | Aminopyrimidines, compositions comprising the same and use thereof for inhibiting protein kinases |
| ZA2009/06264A ZA200906264B (en) | 2007-03-09 | 2009-09-09 | Aminopyrimidines useful as inhibitors of protein kinases |
| HK10103874.3A HK1138572A1 (en) | 2007-03-09 | 2010-04-21 | Aminopyrimidines useful as inhibitors of protein kinases |
| US12/792,050 US8518953B2 (en) | 2007-03-09 | 2010-06-02 | Aminopyrimidines useful as inhibitors of protein kinases |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US90592907P | 2007-03-09 | 2007-03-09 | |
| US60/905,929 | 2007-03-09 | ||
| US95302407P | 2007-07-31 | 2007-07-31 | |
| US60/953,024 | 2007-07-31 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/792,050 Continuation US8518953B2 (en) | 2007-03-09 | 2010-06-02 | Aminopyrimidines useful as inhibitors of protein kinases |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008112651A2 true WO2008112651A2 (en) | 2008-09-18 |
| WO2008112651A3 WO2008112651A3 (en) | 2008-11-27 |
Family
ID=39745397
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/056433 WO2008112651A2 (en) | 2007-03-09 | 2008-03-10 | Aminopyrimidines useful as inhibitors of protein kinases |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US8518953B2 (en) |
| EP (1) | EP2137183B1 (en) |
| JP (2) | JP5393489B2 (en) |
| KR (1) | KR20090120510A (en) |
| CN (1) | CN101668760B (en) |
| AT (1) | ATE526328T1 (en) |
| AU (1) | AU2008226466B2 (en) |
| CA (1) | CA2680029A1 (en) |
| ES (1) | ES2374335T3 (en) |
| HK (1) | HK1138572A1 (en) |
| IL (1) | IL200793A0 (en) |
| MX (1) | MX2009009591A (en) |
| NZ (1) | NZ579485A (en) |
| RU (1) | RU2009137390A (en) |
| WO (1) | WO2008112651A2 (en) |
| ZA (1) | ZA200906264B (en) |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009018415A1 (en) * | 2007-07-31 | 2009-02-05 | Vertex Pharmaceuticals Incorporated | Process for preparing 5-fluoro-1h-pyrazolo [3, 4-b] pyridin-3-amine and derivatives thereof |
| WO2009145814A3 (en) * | 2008-03-10 | 2010-03-25 | Vertex Pharmaceuticals Incorporated | Pyrimidines and pyridines useful as inhibitors of protein kinases |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| US8247421B2 (en) | 2006-12-21 | 2012-08-21 | Vertex Pharmaceuticals Incorporated | 5-cyano-4-(pyrrolo [2,3B] pyridine-3-yl)-pyrimidine derivatives useful as protein kinase inhibitors |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8871774B2 (en) | 2010-12-16 | 2014-10-28 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US8937064B2 (en) | 2007-12-19 | 2015-01-20 | Vertex Pharmaceuticals Incorporated | Pyrazolo[1,5-a]pyrimidines useful as JAK2 inhibitors |
| US9051319B2 (en) | 2011-08-01 | 2015-06-09 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US9345708B2 (en) | 2009-06-17 | 2016-05-24 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US9771361B2 (en) | 2013-11-13 | 2017-09-26 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US10023569B2 (en) | 2013-11-13 | 2018-07-17 | Vertex Pharmaceuticals Incorporated | Methods of preparing inhibitors of influenza viruses replication |
| US10273233B2 (en) | 2015-05-13 | 2019-04-30 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US10533004B2 (en) | 2015-05-13 | 2020-01-14 | Vertex Pharmaceuticals Incorporated | Methods of preparing inhibitors of influenza viruses replication |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100355750C (en) | 2000-09-15 | 2007-12-19 | 沃泰克斯药物股份有限公司 | Pyrazole compounds useful as protein kinase inhibitors |
| WO2004072029A2 (en) | 2003-02-06 | 2004-08-26 | Vertex Pharmaceuticals Incorporated | Pyrazolopyridazines useful as inhibitors of protein kinases |
| SG166827A1 (en) | 2005-11-03 | 2010-12-29 | Vertex Pharma | Aminopyrimidines useful as kinase inhibitors |
| JP2010509231A (en) | 2006-11-02 | 2010-03-25 | バーテックス ファーマシューティカルズ インコーポレイテッド | Aminopyridines and aminopyrimidines useful as inhibitors of protein kinases |
| WO2008077086A1 (en) | 2006-12-19 | 2008-06-26 | Vertex Pharmaceuticals Incorporated | Aminopyrimidines useful as inhibitors of protein kinases |
| JP5520057B2 (en) | 2007-03-09 | 2014-06-11 | バーテックス ファーマシューティカルズ インコーポレイテッド | Aminopyrimidines useful as inhibitors of protein kinases |
| JP5393489B2 (en) | 2007-03-09 | 2014-01-22 | バーテックス ファーマシューティカルズ インコーポレイテッド | Aminopyrimidines useful as inhibitors of protein kinases |
| CA2679701A1 (en) | 2007-03-09 | 2008-09-18 | Vertex Pharmaceuticals Incorporated | Aminopyridines useful as inhibitors of protein kinases |
| WO2008128009A2 (en) | 2007-04-13 | 2008-10-23 | Vertex Pharmaceuticals Incorporated | Aminopyrimidines useful as kinase inhibitors |
| WO2008137621A1 (en) | 2007-05-02 | 2008-11-13 | Vertex Pharmaceuticals Incorporated | Aminopyrimidines useful as kinase inhibitors |
| JP5389785B2 (en) | 2007-05-02 | 2014-01-15 | バーテックス ファーマシューティカルズ インコーポレイテッド | Thiazoles and pyrazoles useful as kinase inhibitors |
| NZ731797A (en) | 2012-04-24 | 2018-08-31 | Vertex Pharma | Dna-pk inhibitors |
| ME03336B (en) | 2013-03-12 | 2019-10-20 | Vertex Pharma | Dna-pk inhibitors |
| RU2675270C2 (en) | 2013-10-17 | 2018-12-18 | Вертекс Фармасьютикалз Инкорпорейтед | Co-crystals and pharmaceutical compositions containing same |
| CA3038657A1 (en) | 2016-09-27 | 2018-04-05 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of dna-damaging agents and dna-pk inhibitors |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999065897A1 (en) | 1998-06-19 | 1999-12-23 | Chiron Corporation | Inhibitors of glycogen synthase kinase 3 |
| WO2000038675A1 (en) | 1998-12-23 | 2000-07-06 | Smithkline Beecham Plc | Treatment of conditions with a need for the inhibition of gsk-3 |
| US20040039007A1 (en) | 2002-08-02 | 2004-02-26 | Forster Cornelia J. | Compositions useful as inhibitors of GSK-3 |
Family Cites Families (75)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3133081A (en) | 1964-05-12 | J-aminoindazole derivatives | ||
| US3935183A (en) | 1970-01-26 | 1976-01-27 | Imperial Chemical Industries Limited | Indazole-azo phenyl compounds |
| BE754242A (en) | 1970-07-15 | 1971-02-01 | Geigy Ag J R | DIAMINO-S-TRIAZINES AND DINITRO-S-TRIAZINES |
| US3998951A (en) | 1974-03-13 | 1976-12-21 | Fmc Corporation | Substituted 2-arylquinazolines as fungicides |
| DE2458965C3 (en) | 1974-12-13 | 1979-10-11 | Bayer Ag, 5090 Leverkusen | 3-Amino-indazole-N-carboxylic acid derivatives, process for their preparation and pharmaceuticals containing them |
| MA18829A1 (en) | 1979-05-18 | 1980-12-31 | Ciba Geigy Ag | PYRIMIDINE DERIVATIVES, METHODS FOR THEIR PREPARATION, PHARMACEUTICAL COMPOSITIONS CONTAINING THESE COMPOUNDS AND THEIR THERAPEUTIC USE |
| DOP1981004033A (en) | 1980-12-23 | 1990-12-29 | Ciba Geigy Ag | PROCEDURE TO PROTECT CROP PLANTS FROM PHYTOTOXIC ACTION OF HERBICIDES. |
| SE8102194L (en) | 1981-04-06 | 1982-10-07 | Pharmacia Ab | THERAPEUTIC ACTIVE ORGANIC ASSOCIATION AND PHARMACEUTICAL PREPARATION INCLUDING THIS |
| SE8102193L (en) | 1981-04-06 | 1982-10-07 | Pharmacia Ab | THERAPEUTIC ACTIVE ORGANIC ASSOCIATION AND ITS USE |
| JPS58124773A (en) | 1982-01-20 | 1983-07-25 | Mitsui Toatsu Chem Inc | 5-methylthiopyrimidine derivative, its preparation and fungicide for agricultural and horticultural purposes |
| DE3725638A1 (en) | 1987-08-03 | 1989-02-16 | Bayer Ag | NEW ARYLOXY (OR THIO) AMINOPYRIMIDINE |
| US5710158A (en) | 1991-05-10 | 1998-01-20 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Aryl and heteroaryl quinazoline compounds which inhibit EGF and/or PDGF receptor tyrosine kinase |
| DE69532817T2 (en) | 1994-11-10 | 2005-01-13 | Millenium Pharmaceuticals, Inc., Cambridge | USE OF PYRAZOLE COMPOUNDS FOR THE TREATMENT OF GLOMERULONEPHRITIS, CANCER, ATHEROSCLEROSIS OR RESTENOSIS |
| IL117659A (en) | 1995-04-13 | 2000-12-06 | Dainippon Pharmaceutical Co | Substituted 2-phenyl pyrimidino amino acetamide derivative process for preparing the same and a pharmaceutical composition containing same |
| US6716575B2 (en) | 1995-12-18 | 2004-04-06 | Sugen, Inc. | Diagnosis and treatment of AUR1 and/or AUR2 related disorders |
| GB9619284D0 (en) | 1996-09-16 | 1996-10-30 | Celltech Therapeutics Ltd | Chemical compounds |
| US6267952B1 (en) | 1998-01-09 | 2001-07-31 | Geltex Pharmaceuticals, Inc. | Lipase inhibiting polymers |
| JP2000026421A (en) | 1998-01-29 | 2000-01-25 | Kumiai Chem Ind Co Ltd | Diaryl sulfide derivative and pest controlling agent |
| IL137922A0 (en) | 1998-02-17 | 2001-10-31 | Tularik Inc | Anti-viral pyrimidine derivatives |
| AU5777299A (en) | 1998-08-21 | 2000-03-14 | Du Pont Pharmaceuticals Company | Isoxazolo(4,5-d)pyrimidines as CRF antagonists |
| US6184226B1 (en) | 1998-08-28 | 2001-02-06 | Scios Inc. | Quinazoline derivatives as inhibitors of P-38 α |
| GB9828511D0 (en) | 1998-12-24 | 1999-02-17 | Zeneca Ltd | Chemical compounds |
| CA2367856C (en) | 1999-03-29 | 2013-10-15 | Uutech Limited | Analogs of gastric inhibitory peptide and their use for treatment of diabetes |
| GB9914258D0 (en) | 1999-06-18 | 1999-08-18 | Celltech Therapeutics Ltd | Chemical compounds |
| US20020065270A1 (en) | 1999-12-28 | 2002-05-30 | Moriarty Kevin Joseph | N-heterocyclic inhibitors of TNF-alpha expression |
| CN1362953A (en) | 2000-02-05 | 2002-08-07 | 沃泰克斯药物股份有限公司 | Pyrazole compositions useful as inhibitors of ERK |
| AU3704101A (en) | 2000-02-17 | 2001-08-27 | Amgen Inc | Kinase inhibitors |
| GB0004887D0 (en) | 2000-03-01 | 2000-04-19 | Astrazeneca Uk Ltd | Chemical compounds |
| NZ522696A (en) | 2000-06-28 | 2004-08-27 | Astrazeneca Ab | Substituted quinazoline derivatives and their use as inhibitors |
| CN100355750C (en) | 2000-09-15 | 2007-12-19 | 沃泰克斯药物股份有限公司 | Pyrazole compounds useful as protein kinase inhibitors |
| US6660731B2 (en) | 2000-09-15 | 2003-12-09 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
| US6613776B2 (en) | 2000-09-15 | 2003-09-02 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
| WO2002022610A1 (en) | 2000-09-15 | 2002-03-21 | Vertex Pharmaceuticals Incorporated | Isoxazoles and their use as inhibitors of erk |
| US7473691B2 (en) | 2000-09-15 | 2009-01-06 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
| US6610677B2 (en) | 2000-09-15 | 2003-08-26 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
| US6716851B2 (en) | 2000-12-12 | 2004-04-06 | Cytovia, Inc. | Substituted 2-aryl-4-arylaminopyrimidines and analogs as activators or caspases and inducers of apoptosis and the use thereof |
| DE10061863A1 (en) | 2000-12-12 | 2002-06-13 | Basf Ag | Preparation of triethylenediamine, useful for making pharmaceuticals and polymers, by reacting ethylenediamine over specific zeolite catalyst |
| US20040048849A1 (en) * | 2000-12-20 | 2004-03-11 | Gregoire Prevost | Cyclin-dependent kinase (cdk) and glycolene synthase kinase-3 (gsk-3) inhibitors |
| ATE326462T1 (en) | 2000-12-21 | 2006-06-15 | Vertex Pharma | PYRAZOLE COMPOUNDS AS PROTEIN KINASE INHIBITORS |
| MY130778A (en) | 2001-02-09 | 2007-07-31 | Vertex Pharma | Heterocyclic inhibitiors of erk2 and uses thereof |
| ES2292753T4 (en) | 2001-03-29 | 2009-02-16 | Vertex Pharmaceuticals Incorporated | INHIBITORS OF N-TERMINAL KINASES C-JUN (JNK) AND OTHER PROTEIN KINASES. |
| WO2002083667A2 (en) | 2001-04-13 | 2002-10-24 | Vertex Pharmaceuticals Incorporated | Inhibitors of c-jun n-terminal kinases (jnk) and other protein kinases |
| US20030096813A1 (en) | 2001-04-20 | 2003-05-22 | Jingrong Cao | Compositions useful as inhibitors of GSK-3 |
| US6884804B2 (en) | 2001-05-16 | 2005-04-26 | Vertex Pharmaceuticals Incorporated | Inhibitors of Src and other protein kinases |
| DE60232510D1 (en) | 2001-06-15 | 2009-07-16 | Vertex Pharma | 5- (2-AMINOPYRIMIDIN-4-YL) BENZOXOXOLEA AS PROTEIN KINASE INHIBITOR |
| WO2003004492A1 (en) | 2001-07-03 | 2003-01-16 | Vertex Pharmaceuticals Incorporated | Isoxazolyl-pyrimidines as inhibitors of src and lck protein kinases |
| US6698980B2 (en) | 2001-07-30 | 2004-03-02 | Stewart Mining Products Inc. | Rock stabilizing apparatus and method |
| EP2198867A1 (en) | 2001-12-07 | 2010-06-23 | Vertex Pharmaceuticals, Inc. | Pyrimidine-based compounds useful as GSK-3 inhibitors |
| US6846928B2 (en) | 2002-03-15 | 2005-01-25 | Vertex Pharmaceuticals Incorporated | Compositions useful as inhibitors of protein kinases |
| EP1485100B1 (en) | 2002-03-15 | 2010-05-05 | Vertex Pharmaceuticals Incorporated | Azinylaminoazoles as inhibitors of protein kinases |
| US7863282B2 (en) | 2003-03-14 | 2011-01-04 | Vertex Pharmaceuticals Incorporated | Compositions useful as inhibitors of protein kinases |
| ATE433973T1 (en) | 2002-03-15 | 2009-07-15 | Vertex Pharma | AZOLYLAMINOAZINES AS INHIBITORS OF PROTEIN KINASES |
| AU2003218215A1 (en) | 2002-03-15 | 2003-09-29 | Vertex Pharmaceuticals, Inc. | Azolylaminoazines as inhibitors of protein kinases |
| US20030207873A1 (en) | 2002-04-10 | 2003-11-06 | Edmund Harrington | Inhibitors of Src and other protein kinases |
| AU2003237121A1 (en) | 2002-04-26 | 2003-11-10 | Vertex Pharmaceuticals Incorporated | Pyrrole derivatives as inhibitors of erk2 and uses thereof |
| MY141867A (en) | 2002-06-20 | 2010-07-16 | Vertex Pharma | Substituted pyrimidines useful as protein kinase inhibitors |
| US7361665B2 (en) | 2002-07-09 | 2008-04-22 | Vertex Pharmaceuticals Incorporated | Inhibitors of c-Jun N-terminal kinases (JNK) and other protein kinases |
| AU2003286711A1 (en) | 2002-10-25 | 2004-05-13 | Vertex Pharmaceuticals Incorporated | Indazolinone compositions useful as kinase inhibitors |
| ES2328820T3 (en) | 2003-10-17 | 2009-11-18 | Astrazeneca Ab | DERIVATIVES OF 4- (PIRAZOL-3-ILAMINO) PYRIMIDINE FOR USE IN CANCER TREATMENT. |
| EP1917259B1 (en) | 2005-08-18 | 2012-01-25 | Vertex Pharmaceuticals Incorporated | Pyrazine kinase inhibitors |
| WO2007023382A2 (en) | 2005-08-25 | 2007-03-01 | Pfizer Inc. | Pyrimidine amino pyrazole compounds, potent kinase inhibitors |
| ES2535854T3 (en) | 2005-09-30 | 2015-05-18 | Miikana Therapeutics, Inc. | Substituted Pyrazole Compounds |
| SG166827A1 (en) | 2005-11-03 | 2010-12-29 | Vertex Pharma | Aminopyrimidines useful as kinase inhibitors |
| ATE508126T1 (en) | 2005-11-16 | 2011-05-15 | Vertex Pharma | AMINOPYRIMIDINES SUITABLE AS KINASE INHIBITORS |
| JP2010509231A (en) | 2006-11-02 | 2010-03-25 | バーテックス ファーマシューティカルズ インコーポレイテッド | Aminopyridines and aminopyrimidines useful as inhibitors of protein kinases |
| WO2008077086A1 (en) | 2006-12-19 | 2008-06-26 | Vertex Pharmaceuticals Incorporated | Aminopyrimidines useful as inhibitors of protein kinases |
| CA2679701A1 (en) | 2007-03-09 | 2008-09-18 | Vertex Pharmaceuticals Incorporated | Aminopyridines useful as inhibitors of protein kinases |
| JP5393489B2 (en) | 2007-03-09 | 2014-01-22 | バーテックス ファーマシューティカルズ インコーポレイテッド | Aminopyrimidines useful as inhibitors of protein kinases |
| JP5520057B2 (en) | 2007-03-09 | 2014-06-11 | バーテックス ファーマシューティカルズ インコーポレイテッド | Aminopyrimidines useful as inhibitors of protein kinases |
| CN101675041A (en) | 2007-03-20 | 2010-03-17 | 沃泰克斯药物股份有限公司 | Amino-metadiazine compound as kinase inhibitor |
| JP2010524962A (en) | 2007-04-17 | 2010-07-22 | バーテックス ファーマシューティカルズ インコーポレイテッド | Drug discovery for aurora kinase inhibitors |
| WO2008137621A1 (en) | 2007-05-02 | 2008-11-13 | Vertex Pharmaceuticals Incorporated | Aminopyrimidines useful as kinase inhibitors |
| MX2009011811A (en) | 2007-05-02 | 2010-01-14 | Vertex Pharma | Aminopyrimidines useful as kinase inhibitors. |
| JP5389785B2 (en) | 2007-05-02 | 2014-01-15 | バーテックス ファーマシューティカルズ インコーポレイテッド | Thiazoles and pyrazoles useful as kinase inhibitors |
| AU2008257044A1 (en) | 2007-05-24 | 2008-12-04 | Vertex Pharmaceuticals Incorporated | Thiazoles and pyrazoles useful as kinase inhibitors |
-
2008
- 2008-03-10 JP JP2009552930A patent/JP5393489B2/en not_active Expired - Fee Related
- 2008-03-10 NZ NZ579485A patent/NZ579485A/en not_active IP Right Cessation
- 2008-03-10 WO PCT/US2008/056433 patent/WO2008112651A2/en active Application Filing
- 2008-03-10 EP EP08731839A patent/EP2137183B1/en not_active Not-in-force
- 2008-03-10 ES ES08731839T patent/ES2374335T3/en active Active
- 2008-03-10 CA CA002680029A patent/CA2680029A1/en not_active Abandoned
- 2008-03-10 MX MX2009009591A patent/MX2009009591A/en active IP Right Grant
- 2008-03-10 RU RU2009137390/04A patent/RU2009137390A/en not_active Application Discontinuation
- 2008-03-10 KR KR1020097021206A patent/KR20090120510A/en not_active Application Discontinuation
- 2008-03-10 AU AU2008226466A patent/AU2008226466B2/en not_active Ceased
- 2008-03-10 AT AT08731839T patent/ATE526328T1/en active
- 2008-03-10 CN CN2008800138739A patent/CN101668760B/en not_active Expired - Fee Related
-
2009
- 2009-09-07 IL IL200793A patent/IL200793A0/en unknown
- 2009-09-09 ZA ZA2009/06264A patent/ZA200906264B/en unknown
-
2010
- 2010-04-21 HK HK10103874.3A patent/HK1138572A1/en not_active IP Right Cessation
- 2010-06-02 US US12/792,050 patent/US8518953B2/en active Active
-
2013
- 2013-08-09 JP JP2013165986A patent/JP2013227348A/en not_active Withdrawn
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999065897A1 (en) | 1998-06-19 | 1999-12-23 | Chiron Corporation | Inhibitors of glycogen synthase kinase 3 |
| WO2000038675A1 (en) | 1998-12-23 | 2000-07-06 | Smithkline Beecham Plc | Treatment of conditions with a need for the inhibition of gsk-3 |
| US20040039007A1 (en) | 2002-08-02 | 2004-02-26 | Forster Cornelia J. | Compositions useful as inhibitors of GSK-3 |
Non-Patent Citations (15)
| Title |
|---|
| "Synthesis,Third Edition,", 1999, JOHN WILEY & SONS |
| BEAULIEU ET AL., PNAS, vol. 101, 2004, pages 5099 - 5104 |
| COGHLANET, CHEMISTRY & BIOLOGY, vol. 7, 2000, pages 793 - 803 |
| EMAMIAN ET AL., NAT GENET, vol. 36, 2004, pages 131 - 137 |
| FOX ET AL., ROTEIN SCI., vol. 7, 1998, pages 2249 |
| GOULD TD, EXPERT OPIN THER TARGETS, vol. 10, 2006, pages 377 - 392 |
| HAQ ET AL., J. CELL BIOL., vol. 151, 2000, pages 117 |
| HAQ ET AL., J. CELL BIOL., vol. 151, 2000, pages 117 - 130 |
| HASHIMOTO ET AL., J. BIOL. CHEM, vol. 277, 2002, pages 32985 - 32991 |
| HIROTANI ET AL., CIRCULATION RESEARCH, vol. 101, 2007, pages 1164 - 1174 |
| KIM; KIMMEL, CURR. OPINION GENETICS DEV., vol. 10, 2000, pages 508 - 514 |
| LI ET AL., INT J NEUROPSYCHOPHARMACOL, 2006, pages 1 - 13 |
| OBRIEN ET AL., J NEUROSCI, vol. 24, 2004, pages 6791 - 6798 |
| SASAKI ET AL., NEUROL RES, vol. 23, 2001, pages 588 - 92 |
| WANG ET AL., BRAIN RES, vol. 859, 2000, pages 381 - 5 |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8962642B2 (en) | 2006-12-21 | 2015-02-24 | Vertex Pharmaceuticals Incorporated | 5-cyano-4- (pyrrolo [2,3B] pyridine-3-yl) -pyrimidine derivatives useful as protein kinase inhibitors |
| US8530489B2 (en) | 2006-12-21 | 2013-09-10 | Vertex Pharmaceuticals Incorporated | 5-cyano-4-(pyrrolo [2,3B] pyridine-3-yl)-pyrimidine derivatives useful as protein kinase inhibitors |
| US8247421B2 (en) | 2006-12-21 | 2012-08-21 | Vertex Pharmaceuticals Incorporated | 5-cyano-4-(pyrrolo [2,3B] pyridine-3-yl)-pyrimidine derivatives useful as protein kinase inhibitors |
| WO2009018415A1 (en) * | 2007-07-31 | 2009-02-05 | Vertex Pharmaceuticals Incorporated | Process for preparing 5-fluoro-1h-pyrazolo [3, 4-b] pyridin-3-amine and derivatives thereof |
| JP2010535241A (en) * | 2007-07-31 | 2010-11-18 | バーテックス ファーマシューティカルズ インコーポレイテッド | Process for preparing 5-fluoro-1H-pyrazolo [3,4-b] pyridin-3-amine and its derivatives |
| US8937064B2 (en) | 2007-12-19 | 2015-01-20 | Vertex Pharmaceuticals Incorporated | Pyrazolo[1,5-a]pyrimidines useful as JAK2 inhibitors |
| WO2009145814A3 (en) * | 2008-03-10 | 2010-03-25 | Vertex Pharmaceuticals Incorporated | Pyrimidines and pyridines useful as inhibitors of protein kinases |
| US10874673B2 (en) | 2009-06-17 | 2020-12-29 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US10039762B2 (en) | 2009-06-17 | 2018-08-07 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US9808459B2 (en) | 2009-06-17 | 2017-11-07 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US9345708B2 (en) | 2009-06-17 | 2016-05-24 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| US8871774B2 (en) | 2010-12-16 | 2014-10-28 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US9908878B2 (en) | 2011-08-01 | 2018-03-06 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US9394302B2 (en) | 2011-08-01 | 2016-07-19 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US10875855B2 (en) | 2011-08-01 | 2020-12-29 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US9051319B2 (en) | 2011-08-01 | 2015-06-09 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US9771361B2 (en) | 2013-11-13 | 2017-09-26 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US10023569B2 (en) | 2013-11-13 | 2018-07-17 | Vertex Pharmaceuticals Incorporated | Methods of preparing inhibitors of influenza viruses replication |
| US10640501B2 (en) | 2013-11-13 | 2020-05-05 | Vertex Pharmaceuticals Incorporated | Methods of preparing inhibitors of influenza viruses replication |
| US11345700B2 (en) | 2013-11-13 | 2022-05-31 | Vertex Pharmaceuticals Incorporated | Methods of preparing inhibitors of influenza viruses replication |
| US10273233B2 (en) | 2015-05-13 | 2019-04-30 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US10533004B2 (en) | 2015-05-13 | 2020-01-14 | Vertex Pharmaceuticals Incorporated | Methods of preparing inhibitors of influenza viruses replication |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2008226466A1 (en) | 2008-09-18 |
| CA2680029A1 (en) | 2008-09-18 |
| JP2013227348A (en) | 2013-11-07 |
| KR20090120510A (en) | 2009-11-24 |
| CN101668760B (en) | 2013-07-24 |
| ATE526328T1 (en) | 2011-10-15 |
| WO2008112651A3 (en) | 2008-11-27 |
| EP2137183A2 (en) | 2009-12-30 |
| HK1138572A1 (en) | 2010-08-27 |
| JP2010520889A (en) | 2010-06-17 |
| AU2008226466B2 (en) | 2013-06-13 |
| CN101668760A (en) | 2010-03-10 |
| EP2137183B1 (en) | 2011-09-28 |
| US20110020377A1 (en) | 2011-01-27 |
| NZ579485A (en) | 2012-02-24 |
| IL200793A0 (en) | 2010-05-17 |
| RU2009137390A (en) | 2011-04-20 |
| ZA200906264B (en) | 2010-11-24 |
| ES2374335T3 (en) | 2012-02-15 |
| JP5393489B2 (en) | 2014-01-22 |
| US8518953B2 (en) | 2013-08-27 |
| MX2009009591A (en) | 2009-11-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2137183B1 (en) | Aminopyrimidines useful as inhibitors of protein kinases | |
| US8735593B2 (en) | Aminopyridines useful as inhibitors of protein kinases | |
| US8664219B2 (en) | Aminopyrimidines useful as inhibitors of protein kinases | |
| US20110224197A1 (en) | Pyrimidines and pyridines useful as inhibitors of protein kinases | |
| EP2102210B1 (en) | Compounds useful as protein kinase inhibitors | |
| JP2010513567A (en) | Aminopyrimidines useful as inhibitors of protein kinases | |
| JP2010509231A (en) | Aminopyridines and aminopyrimidines useful as inhibitors of protein kinases | |
| JP2009515984A (en) | Azaindazole useful as a kinase inhibitor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880013873.9 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2680029 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 579485 Country of ref document: NZ Ref document number: 2008226466 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200793 Country of ref document: IL Ref document number: 3161/KOLNP/2009 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009552930 Country of ref document: JP Ref document number: MX/A/2009/009591 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2008226466 Country of ref document: AU Date of ref document: 20080310 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008731839 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20097021206 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009137390 Country of ref document: RU |