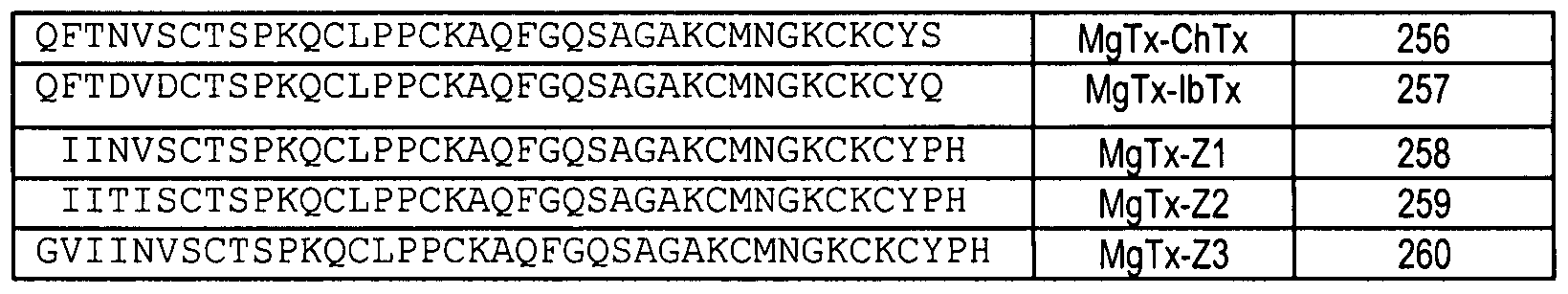Toxin Peptide Therapeutic Agents
This application claims priority from U.S. Provisional Application no. 60/854,674, filed October 25, 2006, and U.S. Application no. 60/995,370, filed September 25, 2007, both of which are hereby incorporated by reference. The instant application contains a "lengthy" Sequence Listing which has been submitted via CD-R in lieu of a printed paper copy, and is hereby incorporated by reference in its entirety. Four copies of said CD-R1 recorded on October 19, 2007 are labeled CRF, "Copy 1" , "Copy 2" and "Copy 3", respectively, and each contains only one identical 2.60 Mb file (A-1186-WO-PCT SeqList.txt). Throughout this application various publications are referenced within parentheses or brackets. The disclosures of these publications in their entireties are hereby incorporated by reference in this application in order to more fully describe the state of the art to which this invention pertains.
Background of the Invention
1. Field of the Invention
The present invention is related to the biochemical arts, in particular to therapeutic peptides and conjugates.
2. Discussion of the Related Art
Ion channels are a diverse group of molecules that permit the exchange of small inorganic ions across membranes. All cells require ion channels for function, but this is especially so for excitable cells such as those present in the nervous system and the heart. The electrical signals orchestrated by ion channels control the thinking brain, the beating heart and the contracting muscle. Ion channels play a role in regulating cell volume, and they control a wide variety of signaling processes.
The ion channel family includes Na+, K+, and Ca2+ cation and Ch anion channels. Collectively, ion channels are distinguished as either ligand-gated or voltage-gated. Ligand-gated channels include both extracellular and intracellular ligand-gated channels. The extracellular ligand-gated channels include the nicotinic acetylcholine receptor (nAChR), the serotonin (5- hdroxytryptamine, 5-HT) receptors, the glycine and γ-butyric acid receptors (GABA) and the glutamate-activated channels including kanate, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and N-methyl-D-aspartate receptors (NMDA) receptors. (Harte and Ouzounis (2002),
FEBS Lett. 514: 129-34). Intracellular ligand gated channels include those activated by cyclic nucleotides (e.g. cAMP, cGMP), Ca2+ and G-proteins. (Harte and Ouzounis (2002), FEBS Lett. 514: 129-34). The voltage-gated ion channels are categorized by their selectivity for inorganic ion species, including sodium, potassium, calcium and chloride ion channels. (Harte and Ouzounis (2002), FEBS Lett. 514: 129-34).
A unified nomenclature for classification of voltage-gated channels was recently presented. (Catterall et al. (2000), Pharmacol. Rev. 55: 573-4; Gutman et al. (2000), Pharmacol. Rev. 55, 583-6; Catterall et al. (2000) Pharmacol. Rev. 55: 579-81; Catterall et al. (2000), Pharmacol. Rev. 55: 575-8; Hofmann et al. (2000), Pharmacol. Rev. 55: 587-9; Clapham et al. (2000), Pharmacol Rev. 55: 591-6; Chandy (1991), Nature 352: 26; Goldin et al. (2000), Neuron 28: 365-8; Ertel et al. (2000), Neuron 25: 533-5).
The K+ channels constitute the largest and best characterized family of ion channels described to date. Potassium channels are subdivided into three general groups: the 6 transmembrane (6TM) K+ channels, the 2TM-2TM/leak K+ channels and the 2TM/Kir inward rectifying channels. (Tang et al. (2004), Ann. Rev. Physiol. 66, 131-159). These three groups are further subdivided into families based on sequence similarity. The voltage-gated K+ channels, including (Kv1-6, Kv8-9), EAG1 KQT, and SIo (BKCa), are family members of the 6TM group. The 2TM-2TM group comprises TWIK1 TREK, TASK, TRAAK, and THIK, whereas the 2TM/Kir group consists of Kir1-7. Two additional classes of ion channels include the inward rectifier potassium (IRK) and ATP-gated purinergic (P2X) channels. (Harte and Ouzounis (2002), FEBS Lett. 514: 129-34).
Toxin peptides produced by a variety of organisms have evolved to target ion channels. Snakes, scorpions, spiders, bees, snails and sea anemone are a few examples of organisms that produce venom that can serve as a rich source of small bioactive toxin peptides or "toxins" that potently and selectively target ion channels and receptors. In most cases, these toxin peptides have evolved as potent antagonists or inhibitors of ion channels, by binding to the channel pore and physically blocking the ion conduction pathway. In some other cases, as with some of the tarantula toxin peptides, the peptide is found to antagonize channel function by binding to a region outside the pore (e.g., the voltage sensor domain). The toxin peptides are usually between about 20 and about 80 amino acids in length, contain 2-5 disulfide linkages and form a very compact structure (see, e.g., Figure 10). Toxin peptides (e.g., from the venom of scorpions, sea anemones and cone snails) have been isolated and characterized for their impact on ion channels. Such peptides appear to have evolved from a
relatively small number of structural frameworks that are particularly well suited to addressing the critical issues of potency and stability. The majority of scorpion and Conus toxin peptides, for example, contain 10-40 amino acids and up to five disulfide bonds, forming extremely compact and constrained structure (microproteins) often resistant to proteolysis. The conotoxin and scorpion toxin peptides can be divided into a number of superfamilies based on their disulfide connections and peptide folds. The solution structure of many of these has been determined by NMR spectroscopy, illustrating their compact structure and verifying conservation of their family fold. (E.g., Tudor et al., lonisation behaviour and solution properties of the potassium-channel blocker ShK toxin, Eur. J. Biochem. 251 (1-2):133-41 (1998); Pennington et al., Role of disulfide bonds in the structure and potassium channel blocking activity of ShK toxin, Biochem. 38(44): 14549-58 (1999); Jaravine et al., Three-dimensional structure of toxin OSK1 from Orthochirus scrobiculosus scorpion venom, Biochem. 36(6): 1223-32 (1997); del Rio-Portillo et al.; NMR solution structure of Cn12, a novel peptide from the Mexican scorpion Centruroides noxius with a typical beta-toxin sequence but with alpha-like physiological activity, Eur. J. Biochem. 271(12): 2504-16 (2004); Prochnicka-Chalufour et al., Solution structure of discrepin, a new K+-channel blocking peptide from the alpha-KTx15 subfamily, Biochem. 45(6):1795-1804 (2006)).
Conserved disulfide structures can also reflect the individual pharmacological activity of the toxin family. (Nicke et al. (2004), Eur. J. Biochem. 271 : 2305-19, Table 1 ; Adams (1999), Drug Develop. Res.46: 219-34). For example, α-conotoxins have well-defined four cysteine/two disulfide loop structures (Loughnan, 2004) and inhibit nicotinic acetylcholine receptors. In contrast, ω-conotoxins have six cysteine/three disulfide loop consensus structures (Nielsen, 2000) and block calcium channels. Structural subsets of toxins have evolved to inhibit either voltage-gated or calcium-activated potassium channels. Figure 9 shows that a limited number of conserved disulfide architectures shared by a variety of venomous animals from bee to snail and scorpion to snake target ion channels. Figure 7A-7B shows alignment of alpha-scorpion toxin family and illustrates that a conserved structural framework is used to derive toxins targeting a vast array of potassium channels.
Due to their potent and selective blockade of specific ion channels, toxin peptides have been used for many years as tools to investigate the pharmacology of ion channels. Other than excitable cells and tissues such as those present in heart, muscle and brain, ion channels are also important to non-excitable cells such as immune cells. Accordingly, the potential therapeutic utility of toxin peptides has been considered for treating various immune disorders, in particular by inhibition of potassium channels such as Kv1.3 and IKCaI since these channels indirectly control
calcium signaling pathway in lymphocytes, [e.g., Kern et al., ShK toxin compositions and methods of use, US Patent No. 6,077,680; Lebrun et al., Neuropeptides originating in scorpion, US Patent No. 6,689,749; Beeton et al., Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channnels for therapy of autoimmune diseases, Molec. Pharmacol. 67(4): 1369-81 (2005); Mouhat et al., K+ channel types targeted by synthetic OSK1, a toxin from Orthochirus scrobiculosus scorpion venom, Biochem. J. 385:95-104 (2005); Mouhat et al., Pharmacological profiling of Orthochirus scrobiculosus toxin 1 analogs with a trimmed N-terminal domain, Molec. Pharmacol. 69:354- 62 (2006); Mouhat et al., OsK1 derivatives, WO 2006/002850 A2; B.S. Jensen et al. The Ca2+-activated K+ Channel of Intermediate Conductance: A Molecular Target for Novel Treatments?, Current Drug Targets 2:401-422 (2001); Rauer et al., Structure-guided
Transformation of Charybdotoxin Yields an Analog That Selectively Targets Ca2*-activated over Voltage-gated K+ Channels, J. Biol. Chem. 275: 1201-1208 (2000); Castle et al., Maurotoxin: A Potent Inhibitor of Intermediate Conductance Ca2+-Activated Potassium Channels, Molecular Pharmacol. 63: 409-418 (2003); Chandy et al., K+ channels as targets for specific Immunomodulation, Trends in Pharmacol. Sciences 25: 280-289 (2004); Lewis & Garcia, Therapeutic Potential of Venom Peptides, Nat. Rev. Drug Discov. 2: 790-802 (2003)].
Small molecules inhibitors of Kv1.3 and IKCaI potassium channels and the major calcium entry channel in T cells, CRAC, have also been developed to treat immune disorders [A. Schmitz et al. (2005) Molecul. Pharmacol. 68, 1254; K.G. Chandy et al. (2004) TIPS 25, 280; H. Wulff et al. (2001) J. Biol. Chem. 276, 32040; C. Zitt et al. (2004) J. Biol. Chem. 279, 12427], but obtaining small molecules with selectivity toward some of these targets has been difficult.
Calcium mobilization in lymphocytes is known to be a critical pathway in activation of inflammatory responses [M.W. Winslow et al. (2003) Current Opinion Immunol. 15, 299]. Compared to other cells, T cells show a unique sensitivity to increased levels of intracellular calcium and ion channels both directly and indirectly control this process. Inositol triphosphate
(IP3) is the natural second messenger which activates the calcium signaling pathway. IP3 is produced following ligand-induced activation of the T cell receptor (TCR) and upon binding to its intracellular receptor (a channel) causes unloading of intracellular calcium stores. The endoplasmic reticulum provides one key calcium store. Thapsigargin, an inhibitor of the sarcoplasmic-endoplasmic reticulum calcium ATPase (SERCA), also causes unloading of intracellular stores and activation of the calcium signaling pathway in lymphocytes. Therefore, thapsigargin can be used as a specific stimulus of the calcium signaling pathway in T cells. The unloading of intracellular calcium stores in T cells is known to cause activation of a calcium
channel on the cell surface which allows for influx of calcium from outside the cell. This store operated calcium channel (SOCC) on T cells is referred to as "CRAC" (calcium release activated channel) and sustained influx of calcium through this channel is known to be critical for full T cell activation [S. Feske et al. (2005) J. Exp. Med. 202, 651 and N. Venkatesh et al. (2004) PNAS 101, 8969]. For many years it has been appreciated that in order to maintain continued calcium influx into T cells, the cell membrane must remain in a hyperpolarized condition through efflux of potassium ions. In T cells, potassium efflux is accomplished by the voltage-gated potassium channel Kv1.3 and the calcium-activated potassium channel IKCaI [K.G. Chandy et al. (2004) TIPS 25, 280], These potassium channels therefore indirectly control the calcium signaling pathway, by allowing for the necessary potassium efflux that allows for a sustained influx of calcium through CRAC.
Sustained increases in intracellular calcium activate a variety of pathways in T cells, including those leading to activation of NFAT, NF-kB and AP-1 [Quintana-A (2005) Pflugers Arch. - Eur. J. Physiol. 450, 1]. These events lead to various T cell responses including alteration of cell size and membrane organization, activation of cell surface effector molecules, cytokine production and proliferation. Several calcium sensing molecules transmit the calcium signal and orchestrate the cellular response. Calmodulin is one molecule that binds calcium, but many others have been identified (MJ. Berridge et al. (2003) Nat. Rev. MoI. Cell. Biol. 4,517). The calcium-calmodulin dependent phosphatase calcineurin is activated upon sustained increases in intracellular calcium and dephosphorylates cytosolic NFAT. Dephosphorylated NFAT quickly translocates to the nucleus and is widely accepted as a critical transcription factor for T cell activation (F. Macian (2005) Nat. Rev. Immunol. 5, 472 and N. Venkatesh et al. (2004) PNAS 101, 8969). Inhibitors of calcineurin, such as cyclosporin A (Neoral, Sandimmune) and FK506 (Tacrolimus) are a main stay for treatment of severe immune disorders such as those resulting in rejection following solid organ transplant (LM. Gonzalez-Pinto et al. (2005) Transplant. Proc. 37, 1713 and D.R.J. Kuypers (2005)
Transplant International 18, 140). Neoral has been approved for the treatment of transplant rejection, severe rheumatoid arthritis (D.E. Yocum et al. (2000) Rheumatol. 39, 156) and severe psoriasis (J. Koo (1998) British J. Dermatol. 139, 88). Preclinical and clinical data has also been provided suggesting calcineurin inhibitors may have utility in treatment of inflammatory bowel disease (IBD; Baumgart DC (2006) Am. J. Gastroenterol. Mar 30; Epub ahead of print), multiple sclerosis (Ann. Neurol. (1990) 27, 591) and asthma (S. Rohatagi et al. (2000) J. Clin. Pharmacol. 40, 1211). Lupus represents another disorder that may benefit from agents blocking activation of helper T cells. Despite the importance of calcineurin in regulating NFAT in T cells, calcineurin is
also expressed in other tissues (e.g. kidney) and cyclosporine A & FK506 have a narrow safety margin due to mechanism based toxicity. Renal toxicity and hypertension are common side effects that have limited the promise of cyclosporine & FK506. Due to concerns regarding toxicity, calcineurin inhibitors are used mostly to treat only severe immune disease (Bissonnette-R et al. (2006) J. Am. Acad. Dermatol. 54, 472). Kv1.3 inhibitors offer a safer alternative to calcineurin inhibitors for the treatment of immune disorders. This is because Kv1.3 also operates to control the calcium signaling pathway in T cells, but does so through a distinct mechanism to that of calcineurin inhibitors, and evidence on Kv1.3 expression and function show that Kv1.3 has a more restricted role in T cell biology relative to calcineurin, which functions also in a variety of non- lymphoid cells and tissues.
Calcium mobilization in immune cells also activates production of the cytokines interleukin 2 (IL-2) and interferon gamma (IFNg) which are critical mediators of inflammation. IL-2 induces a variety of biological responses ranging from expansion and differentiation of CD4+ and CD8+ T cells, to enhancement of proliferation and antibody secretion by B cells, to activation of NK cells [S.L Gaffen & K.D. Liu (2004) Cytokine 28, 109]. Secretion of IL-2 occurs quickly following T cell activation and T cells represent the predominant source of this cytokine. Shortly following activation, the high affinity IL-2 receptor (IL2-R) is upregulated on T cells endowing them with an ability to proliferate in response to IL-2. T cells, NK cells, B cells and professional antigen presenting cells (APCs) can all secrete IFNg upon activation. T cells represent the principle source of IFNg production in mediating adaptive immune responses, whereas natural killer (NK) cells & APCs are likely an important source during host defense against infection [K. Schroder et al. (2004) J. Leukoc. Biol. 75, 163]. IFNg, originally called macrophage-activating factor, upregulates antigen processing and presentation by monocytes, macrophages and dendritic cells. IFNg mediates a diverse array of biological activities in many cell types [U. Boehm et al. (1997) Annu. Rev. Immunol. 15, 749] including growth & differentiation, enhancement of NK cell activity and regulation of B cell immunoglobulin production and class switching.
CD40L is another cytokine expressed on activated T cells following calcium mobilization and upon binding to its receptor on B cells provides critical help allowing for B cell germinal center formation, B cell differentiation and antibody isotype switching. CD40L-mediated activation of CD40 on B cells can induce profound differentiation and clonal expansion of immunoglobulin (Ig) producing B cells [S. Quezada et al. (2004) Annu. Rev. Immunol. 22, 307]. The CD40 receptor can also be found on dendritic cells and CD40L signaling can mediate dendritic cell activation and differentiation as well. The antigen presenting capacity of B cells and dendritic cells is promoted
by CD40L binding, further illustrating the broad role of this cytokine in adaptive immunity. Given the essential role of CD40 signaling to B cell biology, neutralizing antibodies to CD40L have been examined in preclinical and clinical studies for utility in treatment of systemic lupus erythematosis (SLE), - a disorder characterized by deposition of antibody complexes in tissues, inflammation and organ damage [J. Yazdany and J Davis (2004) Lupus 13, 377].
Production of toxin peptides is a complex process in venomous organisms, and is an even more complex process synthetically. Due to their conserved disulfide structures and need for efficient oxidative refolding, toxin peptides present challenges to synthesis. Although toxin peptides have been used for years as highly selective pharmacological inhibitors of ion channels, the high cost of synthesis and refolding of the toxin peptides and their short half-life in vivo have impeded the pursuit of these peptides as a therapeutic modality. Far more effort has been expended to identify small molecule inhibitors as therapeutic antagonists of ion channels, than has been given the toxin peptides themselves. One exception is the recent approval of the small ω- conotoxin MVIIA peptide (Ziconotide™) for treatment of intractable pain. The synthetic and refolding production process for Ziconotide™, however, is costly and only results in a small peptide product with a very short half-life in vivo (about 4 hours).
A cost-effective process for producing therapeutics, such as but not limited to, inhibitors of ion channels, is a desideratum provided by the present invention, which involves toxin peptides fused, or otherwise covalently conjugated to a vehicle.
Summary of the Invention
The present invention relates to a composition of matter of the formula:
(X1)a-(FV(X2)b-(F2)e-(X3)c (D and multimers thereof, wherein: F1 and F2 are half-life extending moieties, and d and e are each independently O or 1 , provided that at least one of d and e is 1 ;
X1, X2, and X3 are each independently -(LJf-P-(L)9-, and f and g are each independently O or 1;
P is a toxin peptide of no more than about 80 amino acid residues in length, comprising at least two intrapeptide disulfide bonds, and at least one P is an 0SK1 peptide analog; L is an optional linker (present when f=1 and/or g =1); and a, b, and c are each independently O or 1 , provided that at least one of a, b and c is 1. The present invention thus concerns molecules having variations on Formula I, such as the formulae: (II) P-(L)9-F1 (i.e., b, c, and e equal to O);
(III) F1-(L)rP (i.e., a, c, and e equal to O);
(IV) P-(L)g-F1-(L)f-P or (X1)a-F1-(X2)b (i.e., c and e equal to 0);
(V) F1-(L)rP-(L)g-F2 (i.e., a and c equal to 0);
(VI) F1-(L)rP-(L)g-F2-(L)rP (i.e., a equal to 0); (VII) F1-F2-(L)rP (i.e., a and b equal to 0);
(VIII) P-(L)g-F1-F2 (i.e., b and c equal to 0);
(IX) P-(L)g-F1-F2-(L)rP (i.e., b equal to 0); and any multimers of any of these, when stated conventionally with the N-terminus of the peptide(s) on the left. All of such molecules of Formulae H-IX are within the meaning of Structural Formula I. Within the meaning of Formula I, the toxin peptide (P), if more than one is present, can be independently the same or different from the OSK1 peptide analog, or any other toxin peptide(s) also present in the inventive composition, and the linker moiety ((L)f and/or (L)9), if present, can be independently the same or different from any other linker, or linkers, that may be present in the inventive composition. Conjugation of the toxin peptide(s) to the half-life extending moiety, or moieties, can be via the N-terminal and/or C-terminal of the toxin peptide, or can be intercalary as to its primary amino acid sequence, F1 being linked closer to the toxin peptide's N-terminus than is linked F2. Examples of useful half-life extending moieties (F1 or F2) include immunoglobulin Fc domain (e.g., a human immunoglobulin Fc domain, including Fc of IgGI , lgG2, lgG3 or lgG4) or a
portion thereof, human serum albumin (HSA)1 or polyethylene glycol) (PEG). These and other half-life extending moieties described herein are useful, either individually or in combination. A monovalent dimeric Fc-toxin peptide fusion (as represented schematically in Figure 2B), for example, an Fc-OSKI peptide analog fusion or Fc-ShK peptide analog fusion, is an example of the inventive composition of matter encompassed by Formula VII above.
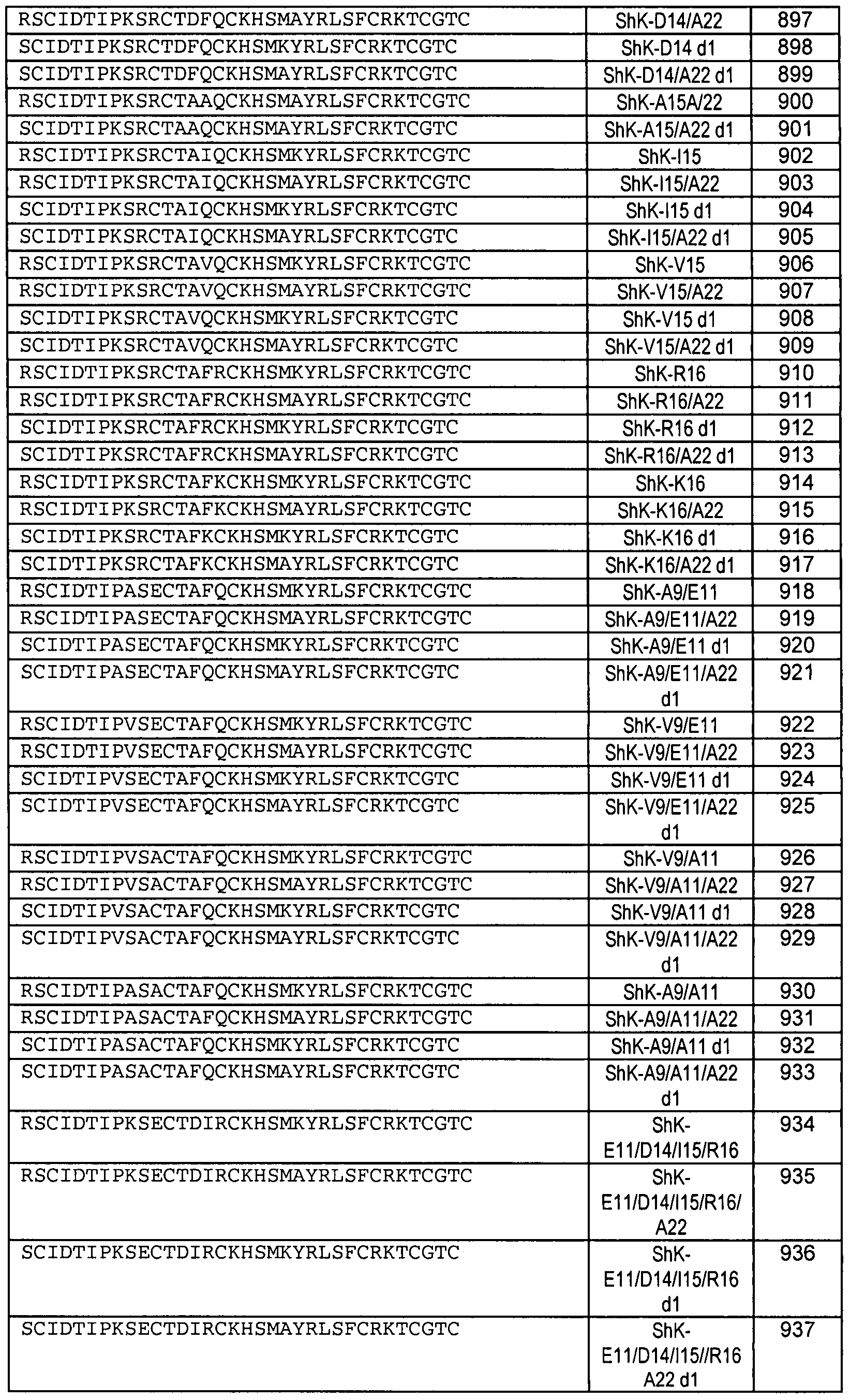
The present invention also relates to a composition of matter, which includes, conjugated or unconjugated, a toxin peptide analog of ShK, 0SK1 , ChTx, or Maurotoxin modified from the native sequences at one or more amino acid residues, having greater Kv1.3 or IKCaI antagonist activity, and/or target selectivity, compared to a ShK, 0SK1, or Maurotoxin (MTX) peptides having a native sequence. The toxin peptide analogs comprise an amino acid sequence selected from any of the following:
SEQ ID NOS: 88, 89, 92, 148 through 200, 548 through 561, 884 through 949, or 1295 through 1300 as set forth in Table 2; or
SEQ ID NOS: 980 through 1274, 1303, or 1308 as set forth in Table 7; or any of SEQ ID NOS: 1391 through 4912, 4916, 4920 through 5006, 5009, 5010, and 5012 through 5015 as set forth in Table 7A, Table 7B, Table 7C1 Table 7D, Table 7E, Table 7F, Table 7G, Table 7H, Table 71 or Table 7J.
SEQ ID NOS: 330 through 337, 341 , 1301 , 1302, 1304 through 1307, 1309, 1311 , 1312, and 1315 through 1336 as set forth in Table 13; or SEQ ID NOS: 36, 59, 344-346, or 1369 through 1390 as set forth in Table 14.
The present invention also relates to other toxin peptide analogs that comprise an amino acid sequence selected from, or comprise the amino acid primary sequence of, any of the following:
SEQ ID NOS: 201 through 225 as set forth in Table 3; or SEQ ID NOS: 242 through 248 or 250 through 260 as set forth in Table 4; or
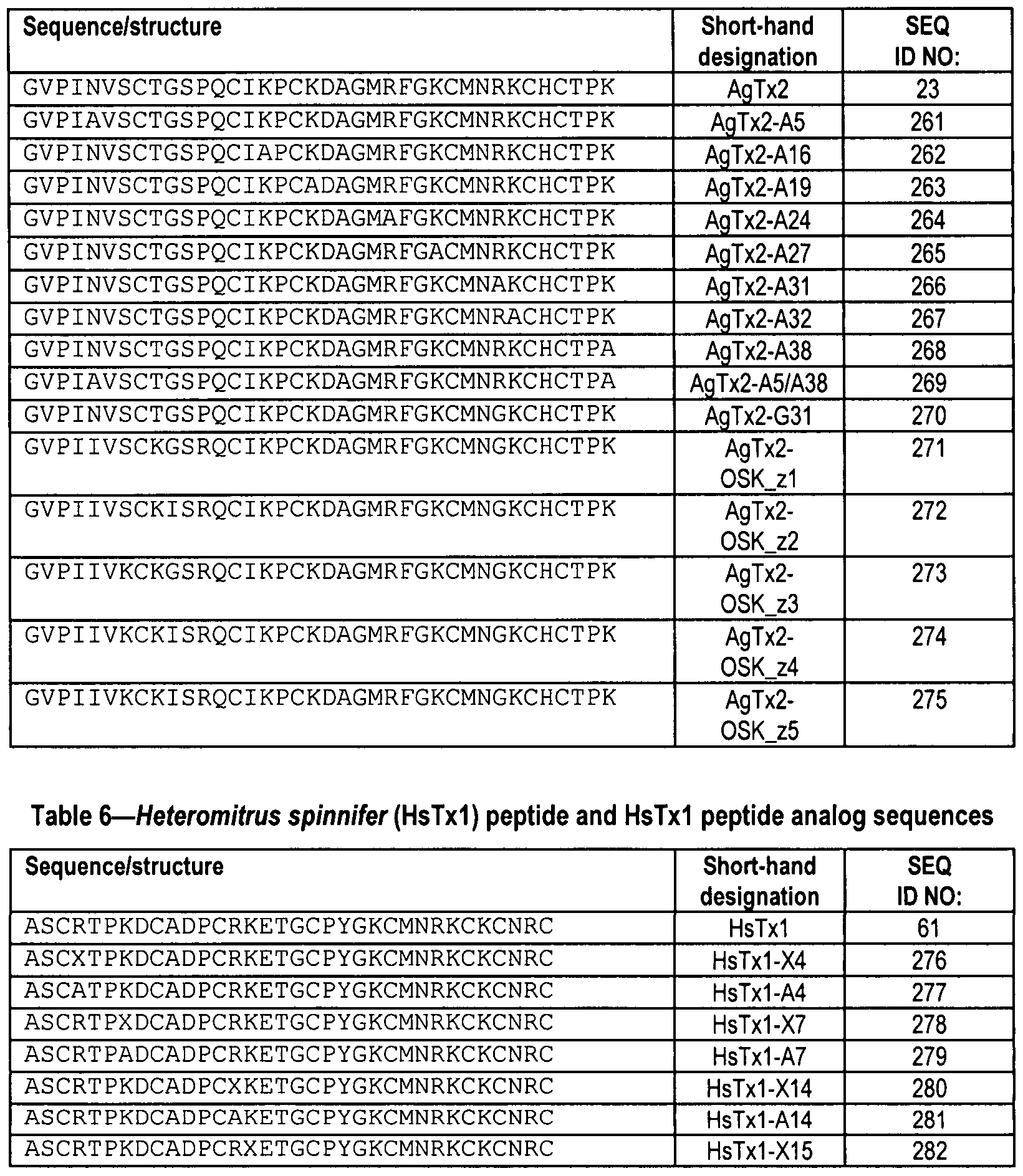
SEQ ID NOS: 261 through 275 as set forth in Table 5; or SEQ ID NOS: 276 through 293 as set forth in Table 6; or SEQ ID NOS: 299 through 315 as set forth in Table 8; or SEQ ID NOS: 316 through 318 as set forth in Table 9; or SEQ ID NO: 319 as set forth in Table 10; or
SEQ ID NO: 327 or 328 as set forth in Table 11 ; or
SEQ ID NOS: 330 through 337, 341, 1301, 1302, 1304 through 1307, 1309, 1311, 1312, or 1315 through 1336 as set forth in Table 13;
SEQ ID NOS: 1369 through 1390 as set forth in Table 14; or
SEQ ID NOS: 348 through 353 as set forth in Table 16; or
SEQ ID NOS: 357 through 362, 364 through 368, 370, 372 through 385, or 387 through 398 as set forth in Table 19; or SEQ ID NOS: 399 through 408 as set forth in Table 20; or
SEQ ID NOS: 410 through 421 as set forth in Table 22; or
SEQ ID NOS: 422, 424, 426, or 428 as set forth in Table 23; or
SEQ ID NOS: 430 through 437 as set forth in Table 24; or
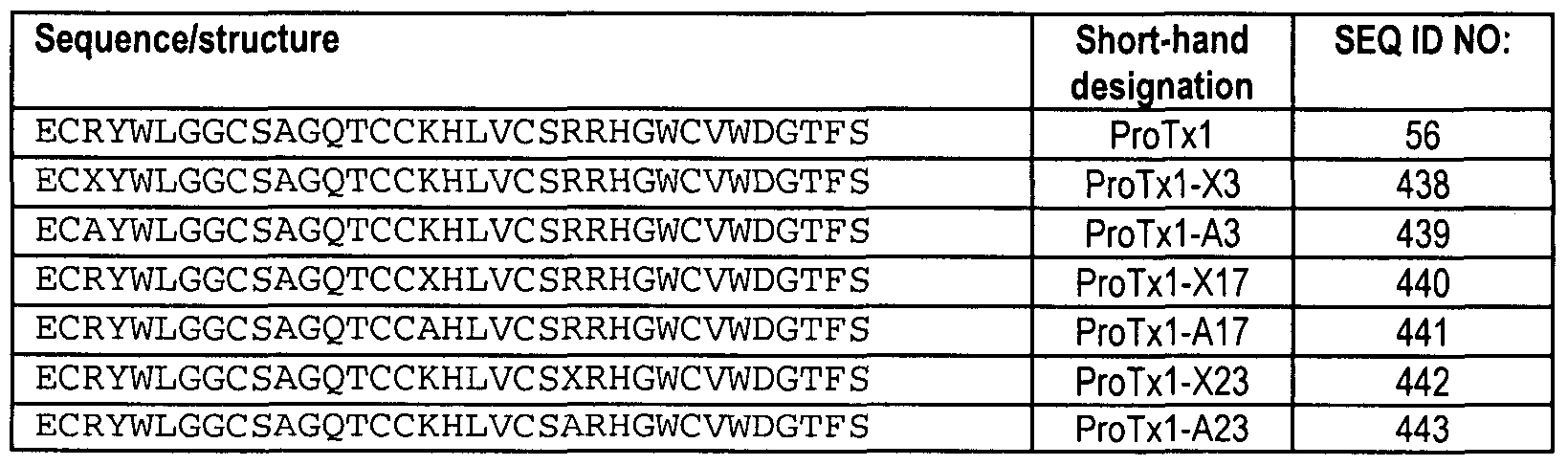
SEQ ID NOS: 438 through 445 as set forth in Table 25; or SEQ ID NOS: 447, 449, 451 , 453, 455, or 457 as set forth in Table 26; or
SEQ ID NOS: 470 through 482 or 484 through 493 as set forth in Table 28; or
SEQ ID NOS: 495 through 506 as set forth in Table 29; or
SEQ ID NOS: 507 through 518 as set forth in Table 30.
The present invention is also directed to a pharmaceutical composition that includes the inventive composition of matter and a pharmaceutically acceptable carrier.
The compositions of this invention can be prepared by conventional synthetic methods, recombinant DNA techniques, or any other methods of preparing peptides and fusion proteins well known in the art. Compositions of this invention that have non-peptide portions can be synthesized by conventional organic chemistry reactions, in addition to conventional peptide chemistry reactions when applicable. Thus the present invention also relates to DNAs encoding the inventive compositions and expression vectors and host cells for recombinant expression.
The primary use contemplated is as therapeutic and/or prophylactic agents. The inventive compositions incorporating the toxin peptide can have activity and/or ion channel target selectivity comparable to— or even greater than— the unconjugated peptide. Accordingly, the present invention includes a method of treating an autoimmune disorder, which involves administering to a patient who has been diagnosed with an autoimmune disorder, such as multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease (IBD, including Crohn's Disease and ulcerative colitis), contact-mediated dermatitis, rheumatoid arthritis, psoriatic arthritis, asthma, allergy, restinosis, systemic sclerosis, fibrosis, scleroderma, glomerulonephritis, Sjogren syndrome, inflammatory bone resorption, transplant rejection, graft-versus-host disease, or lupus, a therapeutically effective amount of the inventive composition of matter (preferably comprising a Kv1.3 antagonist peptide or IKCaI antagonist peptide), whereby at least one symptom of the disorder is alleviated in the patient. In addition, the present invention also relates
to the use of one or more of the inventive compositions of matter in the manufacture of a medicament for the treatment or prevention of an autoimmune disorder, such as, but not limited to, any of the above-listed autoimmune disorders, e.g. multiple sclerosis, type 1 diabetes or IBD.
The present invention is further directed to a method of preventing or mitigating a relapse of a symptom of multiple sclerosis, which method involves administering to a patient, who has previously experienced at least one symptom of multiple sclerosis, a prophylactically effective amount of the inventive composition of matter (preferably comprising a Kv1.3 antagonist peptide or IKCaI antagonist peptide), such that the at least one symptom of multiple sclerosis is prevented from recurring or is mitigated. Although mostly contemplated as therapeutic agents, compositions of this invention can also be useful in screening for therapeutic or diagnostic agents. For example, one can use an Fc- peptide in an assay employing anti-Fc coated plates. The half-life extending moiety, such as Fc, can make insoluble peptides soluble and thus useful in a number of assays.
Numerous additional aspects and advantages of the present invention will become apparent upon consideration of the figures and detailed description of the invention.
United States Nonprovisional Patent Application No. 11/406,454, filed April 17, 2006, is hereby incorporated by reference in its entirety.
Brief Description of the Figures
Figure 1 shows schematic structures of some exemplary Fc dimers that can be derived from an IgGI antibody. "Fc" in the figure represents any of the Fc variants within the meaning of "Fc domain" herein. "X1" and "X2" represent peptides or linker-peptide combinations as defined hereinafter. The specific dimers are as follows:
Figure 1A and Figure 1D: Single disulfide-bonded dimers;
Figure 1B and Figure 1E: Doubly disulfide-bonded dimers;
Figure 1C and Figure 1F: Noncovalent dimers.
Figure 2A-C show schematic structures of some embodiments of the composition of the 0 invention that shows a single unit of the pharmacologically active toxin peptide. Figure 2A shows a single chain molecule and can also represent the DNA construct for the molecule. Figure 2B shows a dimer in which the linker-peptide portion is present on only one chain of the dimer (i.e., a "monovalent" dimer). Figure 2C shows a dimer having the peptide portion on both chains. The dimer of Figure 2C will form spontaneously in certain host cells upon expression of a DNA 5 construct encoding the single chain shown in Figure 2A. In other host cells, the cells could be placed in conditions favoring formation of dimers or the dimers can be formed in vitro.
Figure 3A-3B shows exemplary nucleic acid and amino acid sequences (SEQ ID NOS: 1 and 2, respectively) of human IgGI Fc that is optimized for mammalian expression and can be used in this invention. O Figure 4A-4B shows exemplary nucleic acid and amino acid sequences (SEQ ID NOS: 3 and 4, respectively) of human IgGI Fc that is optimized for bacterial expression and can be used in this invention.
Figure 5A shows the amino acid sequence of the mature ShK peptide (SEQ ID NO: 5), which can be encoded for by a nucleic acid sequence containing codons optimized for expression5 in mammalian cell, bacteria or yeast.
Figure 5B shows the three disulfide bonds (-S— S-) formed by the six cysteines within the ShK peptide (SEQ ID NO: 10).
Figure 6 shows an alignment of the voltage-gated potassium channel inhibitor Stichodactyla helianthus (ShK) with other closely related members of the sea anemone toxin O family. The sequence of the 35 amino acid mature ShK toxin (Accession #P29187) isolated from the venom of Stichodactyla helianthus is shown aligned to other closely related members of the sea anemone family. The consensus sequence and predicted disulfide linkages are shown, with highly conserved residues being shaded. The HmK peptide toxin sequence shown (Swiss-Protein
Accession #097436) is of the immature precursor from the Magnificent sea anemone (Radianthus maqnifica; Heteractis magnifica) and the putative signal peptide is underlined. The mature HmK peptide toxin would be predicted to be 35 amino acids in length and span residues 40 through 74. AeK is the mature peptide toxin, isolated from the venom of the sea anemone Actinia equine (Accession #P81897). The sequence of the mature peptide toxin AsKS (Accession #Q9TWG1) and BgK (Accession #P29186) isolated from the venom of the sea anemone Anemonia sulcata and Bunodosoma qranulifera, respectively, are also shown. Figure 6A shows the amino acid alignment (SEQ ID NO: 10) of ShK to other members of the sea anemone family of toxins, HmK (SEQ ID NO: 6 (Mature Peptide), (SEQ ID NO: 542, Signal and Mature Peptide portions)), AeK (SEQ ID NO: 7), AsKs (SEQ ID NO: 8), and BgK (SEQ ID NO: 9). The predicted disulfide linkages are shown and conserved residues are highlighted. (HmK, SEQ ID NO: 543; ShK, SEQ ID NO: 10; AeK, SEQ ID NO: 544; AsKS, SEQ ID NO: 545). Figure 6B shows a disulfide linkage map for this family having 3 disulfide linkages (C1-C6, C2-C4, C3-C5).
Figure 7A-7B shows an amino acid alignment of the alpha-scorpion toxin family of potassium channel inhibitors. (BmKKI , SEQ ID NO: 11 ; BmKK4, SEQ ID NO: 12; PBTxI , SEQ ID NO: 14; Tc32, SEQ ID NO: 13; BmKK6, SEQ ID NO: 15; P01, SEQ ID NO: 16; Pi2, SEQ ID NO: 17; Pi3, SEQ ID NO: 18; Pi4, SEQ ID NO: 19; MTX, SEQ ID NO: 20; Pi1, SEQ ID NO: 21 ; HsTxI, SEQ ID NO: 61; AgTx2, SEQ ID NO: 23; KTX1, SEQ ID NO: 24; OSK1 , SEQ ID NO: 25; BmKTX, SEQ ID NO: 22; HgTXI , SEQ ID NO: 27; MgTx, SEQ ID NO: 28; C11Tx1, SEQ ID NO: 29; NTX, SEQ ID NO: 30; Tc30, SEQ ID NO: 31 ; TsTX-Ka, SEQ ID NO: 32; PBTx3, SEQ ID NO: 33; Lqh 15- 1 , SEQ ID NO: 34; MartenTx, SEQ ID NO: 37; ChTx, SEQ ID NO:36; ChTx-Lq2, SEQ ID NO: 42; IbTx, SEQ ID NO: 38; SIoTx, SEQ ID NO: 39; BmTxI; SEQ ID NO: 43; BuTx, SEQ ID NO: 41 ; AmmTx3, SEQ ID NO: 44; AaTXI, SEQ ID NO: 45; BmTX3, SEQ ID NO: 46; Td1 SEQ ID NO: 48; OSK2, SEQ ID NO: 49; TsK, SEQ ID NO: 54; CoTxI, SEQ ID NO:55; CoTx2, SEQ ID NO: 871; BmPo5, SEQ ID NO: 60; ScyTx, SEQ ID NO: 51 ; P05, SEQ ID NO: 52; Tamapin, SEQ ID NO: 53; and TmTx, SEQ ID NO: 691. Highly conserved residues are shaded and a consensus sequence is listed. Subfamilies of the α-KTx are listed and are from Rodriguez de Ia Vega, R.C. et al. (2003) TIPS 24: 222-227. A list of some ion channels reported to antagonized is listed (IK = IKCa, BK=BKCa1 SK=SKCa, Kv=voltage-gated K+ channels). Although most family members in this alignment represent the mature peptide product, several represent immature or modified forms of the peptide and these include: BmKKI, BmKK4, BmKK6, BmKTX1 MartenTx, ChTx, ChTx-Lq2, BmTxI, AaTxI, BmTX3, TsK, CoTxI, BmP05.
Figure 8 shows an alignment of toxin peptides converted to peptibodies in this invention (Apamin, SEQ ID NO: 68; HaTxI, SEQ ID NO: 494; ProTxi, SEQ ID NO: 56; PaTx2, SEQ ID NO: 57; ShK[2-35], SEQ ID NO: 92; ShK[1-35], SEQ ID NO: 5; HmK1 SEQ ID NO: 6; ChTx (K32E), SEQ ID NO: 59; ChTx, SEQ ID NO: 36; IbTx, SEQ ID NO: 38; 0SK1 (E16K, K20D), SEQ ID NO: 296; 0SK1 , SEQ ID NO: 25; AgTx2, SEQ ID NO: 23; KTX1 , SEQ ID NO: 24; MgTx, SEQ ID NO: 28; NTX, SEQ ID NO: 30; MTX, SEQ ID NO: 20; Pi2, SEQ ID NO: 17; HsTxI, SEQ ID NO: 61 ; Anuroctoxin [AnTx], SEQ ID NO: 62; BeKmI1 SEQ ID NO: 63; ScyTx, SEQ ID NO: 51; ωGVIA, SEQ ID NO: 64; ωMVIIa, SEQ ID NO: 65; Ptu1, SEQ ID NO: 66; and CTX, SEQ ID NO: 67). The original sources of the toxins is indicated, as well as, the number of cysteines in each. Key ion channels targeted are listed. The alignment shows clustering of toxin peptides based on their source and ion channel target impact.
Figure 9 shows disulfide arrangements within the toxin family. The number of disulfides and the disulfide bonding order for each subfamily is indicated. A partial list of toxins that fall within each disulfide linkage category is presented. Figure 10 illustrates that solution structures of toxins reveal a compact structure.
Solution structures of native toxins from sea anemone (ShK), scorpion (MgTx1 MTX1 HsTxI), marine cone snail (wGVIA) and tarantula (HaTxI) indicate the 28 to 39 amino acid peptides all form a compact structure. The toxins shown have 3 or 4 disulfide linkages and fall within 4 of the 6 subfamilies shown in Figure 9. The solution structures of native toxins from sea anemone (ShK), scorpion (MgTx, MTX1 HsTxI), marine cone snail (wGVIA) and tarantula (HaTxI) were derived from Protein Data Bank (PDB) accession numbers 1ROO (mmdbld:5247), 1MTX (mmdbld:4064), 1TXM (mmdbld:6201), 1QUZ (mmdbld:36904), 1OMZ (mmdbld:1816) and 1D1H (mmdbld:14344) using the MMDB Entrez 3D-structure database [J. Chen et al. (2003) Nucleic Acids Res. 31, 474] and viewer. Figure 11A-C shows the nucleic acid (SEQ ID NO: 69 and SEQ ID NO: 1358) and encoded amino acid (SEQ ID NO:70, SEQ ID NO:1359 and SEQ ID NO: 1360) sequences of residues 5131-6660 of pAMG21ampR-Fc-pep. The sequences of the Fc domain (SEQ ID NOS: 71 and 72) exclude the five C-terminal glycine residues. This vector enables production of peptibodies in which the peptide-linker portion is at the C-terminus of the Fc domain. Figure 11 D shows a circle diagram of a peptibody bacterial expression vector pAMG21ampR-Fc-pep having a chloramphenicol acetyltransferase gene (cat; "CmR" site) that is replaced with the peptide-linker sequence.
Figure 12A-C shows the nucleic acid (SEQ ID NO: 73 and SEQ ID NO: 1361) and encoded amino acid (SEQ ID NO:74, SEQ ID NO: 1362 and SEQ ID NO: 1363) sequences of residues 5131-6319 of pAMG21ampR-Pep-Fc. The sequences of the Fc domain (SEQ ID NOS: 75 and 76) exclude the five N-terminal glycine residues. This vector enables production of peptibodies in which the peptide-linker portion is at the N-terminus of the Fc domain.
Figure 12D shows a circle diagram of a peptibody bacterial expression vector having a zeocin resistance (bje; "ZeoR") site that is replaced with the peptide-linker sequence.
Figure 12E-G shows the nucleic acid (SEQ ID NO: 1339) and encoded amino acid sequences of pAMG21ampR-Pep-Fc (SEQ ID NO:1340, SEQ ID NO:1341 , and SEQ ID NO:1342). The sequences of the Fc domain (SEQ ID NOS: 75 and 76) exclude the five N-terminal glycine residues. This vector enables production of peptibodies in which the peptide-linker portion is at the N-terminus of the Fc domain.
Figure 13A is a circle diagram of mammalian expression vector pCDNA3.1(+) CMVi.
Figure 13B is a circle diagram of mammalian expression vector pCDNA3.1(+)CMVi-Fc- 2xG4S-Activin RIIb that contains a Fc region from human IgGI, a 10 amino acid linker and the activin RIIb gene.
Figure 13C is a circle diagram of the CHO expression vector pDSRa22 containing the Fc- L10-ShK[2-35] coding sequence.
Figure 14A-14B shows the nucleotide and encoded amino acid sequences (SEQ. ID. NOS: 77 and 78, respectively) of the molecule identified as "Fc-LI 0-ShK[1 -35]" in Example 1 hereinafter. The L10 linker amino acid sequence (SEQ ID NO: 79) is underlined.
Figure 15A-15B shows the nucleotide and encoded amino acid sequences (SEQ. ID. NOS: 80 and 81, respectively) of the molecule identified as "Fc-LI 0-ShK[2-35]" in Example 2 hereinafter. The same L10 linker amino acid sequence (SEQ ID NO: 79) as used in Fc-LIO- ShK[1-35] (Figure 14A-14B) is underlined.
Figure 16A-16B shows the nucleotide and encoded amino acid sequences (SEQ. ID. NOS: 82 and 83, respectively) of the molecule identified as "Fc-L25-ShK[2-35]" in Example 2 hereinafter. The L25 linker amino acid sequence (SEQ ID NO: 84) is underlined.
Figure 17 shows a scheme for N-terminal PEGylation of ShK peptide (SEQ ID NO: 5 and SEQ ID NO:10) by reductive amination, which is also described in Example 32 hereinafter.
Figure 18 shows a scheme for N-terminal PEGylation of ShK peptide (SEQ ID NO: 5 and SEQ ID NO:10) via amide formation, which is also described in Example 34 hereinafter.
Figure 19 shows a scheme for N-terminal PEGylation of ShK peptide (SEQ ID NO: 5 and SEQ ID NO:10) by chemoselective oxime formation, which is also described in Example 33 hereinafter.
Figure 2OA shows a reversed-phase HPLC analysis at 214 nm and Figure 2OB shows electrospray mass analysis of folded ShK[2-35], also described as folded-"Des-Arg1-ShK" (Peptide 2).
Figure 21 shows reversed phase HPLC analysis at 214 nm of N-terminally PEGylated ShK[2-35], also referred to as N-Terminally PEGylated-"Des-Arg1-ShK".
Figure 22A shows a reversed-phase HPLC analysis at 214 nm of folded ShK[1-35], also referred to as "ShK".
Figure 22B shows electrospray mass analysis of folded ShK[1-35], also referred to as "ShK".
Figure 23 illustrates a scheme for N-terminal PEGylation of ShK[2-35] (SEQ ID NO: 92 and SEQ ID NO: 58, also referred to as "Des-Arg1-ShK" or "ShK d1") by reductive amination, which is also described in Example 31 hereinafter.
Figure 24A shows a western blot of conditioned medium from HEK 293 cells transiently transfected with Fc-LI 0-ShK[1 -35]. Lane 1 : molecular weight markers; Lane 2: 15 μl Fc-LIO-ShK; Lane 3: 10 μl Fc-LIO-ShK; Lane 4: 5 μl Fc-LIO-ShK; Lane 5; molecular weight markers; Lane 6: blank; Lane 7: 15 μl No DNA control; Lane 8: 10 μl No DNA control; Lane 9: 5 μl No DNA control; Lane 10; molecular weight markers.
Figure 24B shows a western blot of with 15 μl ofconditioned medium from clones of Chinese Hamster Ovary (CHO) cells stably transfected with Fc-L-ShK[I -35]. Lanes 1 - 15 were loaded as follows: blank, BB6, molecular weight markers, BB5, BB4, BB3, BB2, BB1, blank, BD6, BD5, molecular weight markers, BD4, BD3, BD2. Figure 25A shows a western blot of a non-reducing SDS-PAGE gel containing conditioned medium from 293T cells transiently transfected with Fc-L-SmIIIA.
Figure 25B shows a western blot of a reducing SDS-PAGE gel containing conditioned medium from 293T cells transiently transfected with Fc-L-SmIIIA.
Figure 26A shows a Spectral scan of 10 μl purified bivalent dimeric Fc-LI 0-ShK[1 -35] product from stably transfected CHO cells diluted in 700 μl PBS (blanking buffer) using a Hewlett
Packard 8453 spectrophotometer and a 1-cm path length quartz cuvette.
Figure 26B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final bivalent dimeric Fc-LI 0-ShK[1 -35] product. Lanes 1 - 12 were loaded as follows: Novex
Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non- reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 26C shows size exclusion chromatography on 20 μg of the final bivalent dimeric Fc-LI 0-ShK[1 -35] product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NahhPCM, 250 mM NaCI, and pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 26D shows a MALDI mass spectral analysis of the final sample of bivalent dimeric Fc-LI 0-ShK[1 -35] analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
Figure 26E shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final monovalent dimeric Fc-LI 0-ShK[1 -35] product. Lanes 1 - 12 were loaded as follows: Novex Mark12 wide range protein standards (10 μL), 0.5 μg product non-reduced (1.3 μL), blank, 2.0 μg product non-reduced (5 μL), blank, 10 μg product non-reduced (25 μL), Novex Mark12 wide range protein standards (10 μL), 0.5 μg product reduced (1.3 μL), blank, 2.0 μg product reduced (5 μL), blank, and 10 μg product reduced (25 μL).
Figure 26F shows size exclusion chromatography on 20 μg of the final monovalent dimeric Fc-LI 0-ShK[1 -35] product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x
300 mm) in 50 mM NahtøPCM, 250 mM NaCI, and pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 27A shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final purified bivalent dimeric Fc-LI 0-ShK[2-35] product from stably transfected CHO cells. Lane 1 - 12 were loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non- reduced, blank, 2.0 μg product non-reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 27B shows size exclusion chromatography on 50 μg of the purified bivalent dimeric Fc-LI 0-ShK[2-35] injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NahtøPCM, 250 mM NaCI, and pH 6.9 at 1 ml/min observing the absorbance at 280 nm. Figure 27C shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of bivalent dimeric Fc-L5-ShK[2-35] purified from stably transfected CHO cells. Lane 1 - 12 are
loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non-reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced. Figure 27D shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of bivalent dimeric Fc-L25-ShK[2-35] purified from stably transfected CHO cells. Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non-reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 27E shows a spectral scan of 10 μl of the bivalent dimeric Fc-LI 0-ShK[2-35] product diluted in 700 μl PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 27F shows a MALDI mass spectral analysis of the final sample of bivalent dimeric Fc-LI 0-ShK[2-35] analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from about 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses. Figure 27G shows a spectral scan of 10 μl of the bivalent dimeric Fc-L5-ShK[2-35] product diluted in 700 μl PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 27H shows the size exclusion chromatography on 50 mg of the final bivalent dimeric Fc-L5-ShK[2-35] product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 27I shows a MALDI mass spectral analysis of the final sample of bivalent dimeric Fc-L5-ShK[2-35] analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots.
External mass calibration was accomplished using purified proteins of known molecular masses. Figure 27J shows a Spectral scan of 20 μl of the product diluted in 700 μl PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 27K shows the size exclusion chromatography on 50 μg of the final bivalent dimeric Fc-L25-ShK[2-35] product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm. Figure 27L shows a MALDI mass spectral analysis of the final sample of bivalent dimeric
Fc-L25-ShK[2-35] analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from about 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
Figure 28A shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of Fc-LI 0-KTX1 purified and refolded from bacterial cells. Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non- reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 28B shows size exclusion chromatography on 45 μg of purified Fc-LI 0-KTX1 injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 28C shows a Spectral scan of 20 μl of the Fc-L10-KTX1 product diluted in 700 μl PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 28D shows a MALDI mass spectral analysis of the final sample of Fc-L10-KTX1 analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
Figure 29A shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of Fc-L-AgTx2 purified and refolded from bacterial cells. Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non- reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 29B shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of Fc-LI 0-HaTxI purified and refolded from bacterial cells. Lane 1 - 12 are loaded as follows: Novex
Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non- reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced, spectral scan of the purified material. Figure 29C shows a Spectral scan of 20 μl of the Fc-LI 0-AgTx2 product diluted in 700 μl
PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 29D shows the Size exclusion chromatography on 20 μg of the final Fc-LI 0-AgTx2 product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 29E shows a MALDI mass spectral analysis of the final sample of Fc-LI 0-AgTx2 analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from about 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
Figure 29F shows a Spectral scan of 20 μl of the Fc-LI 0-HaTxI product diluted in 700 μl PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 29G shows the size exclusion chromatography on 20 μg of the final Fc-LI 0-HaTxI product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 29H shows a MALDI mass spectral analysis of the final sample of Fc-LI 0-HaTxI analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
Figure 3OA shows Fc-LI 0-ShK[1 -35] purified from CHO cells produces a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing the human Kv1.3 channel. Figure 3OB shows the time course of potassium current block by Fc-LI 0-ShK[1 -35] at various concentrations. The ICso was estimated to be 15 ± 2 pM (n = 4 cells).
Figure 3OC shows synthetic ShK[I -35] (also referred to as "ShK" alone) produces a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel.
Figure 3OD shows the time course of ShK[I -35] block at various concentrations. The IC50 for ShK was estimated to be 12 ± 1 pM (n =4 cells).
Figure 31A shows synthetic peptide analog ShK[2-35] producing a concentration dependent block of the outward potassium current as recorded from HEK293 cells stably expressing human Kv1.3 channel with an IC50 of 49 ± 5 pM (n = 3 cells).
Figure 31 B shows the CHO-derived Fc-LI 0-ShK[2-35] peptibody producing a concentration dependent block of the outward potassium current as recorded from HEK293 cell stably expressing human Kv1.3 channel with an IC50 of 115 ± 18 pM (n = 3 cells).
Figure 31 C shows the Fc-L5-ShK[2-35] peptibody produces a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel with an IC50 of 100 pM (n = 3 cells). Figure 32A shows Fc-L-KTXI peptibody purified from bacterial cells producing a concentration dependent block of the outward potassium current as recorded from HEK293 cell stably expressing human Kv1.3 channel.
Figure 32B shows the time course of potassium current block by Fc-LI 0-KTX1 at various concentrations. Figure 33 shows by immunohistochemistry that CHO-derived Fc-LI 0-ShK[1 -35] peptibody stains HEK 293 cells stably transfected with human Kv1.3 (Figure 33A)1 whereas untransfected HEK 293 cells are not stained with the peptibody (Figure 33B).
Figure 34 shows results of an enzyme-immunoassay using fixed HEK 293 cells stably transfected with human Kv1.3. Figure 34A shows the CHO-derived Fc-LI 0-ShK[1 -35] (referred to here simply as "Fc-LI 0-ShK") peptibody shows a dose-dependent increase in response, whereas the CHO-Fc control ("Fc control") does not. Figure 34B shows the Fc-LI 0-ShK[1 -35] peptibody (referred to here as "Fc-ShK") does not elicit a response from untransfected HEK 293 cells using similar conditions and also shows other negative controls.
Figure 35 shows the CHO-derived Fc-LI 0-ShK[1 -35] peptibody shows a dose-dependent inhibition of IL-2 (Figure 35A) and IFNγ (Figure 35B) production from PMA and αCD3 antibody stimulated human PBMCs. The peptibody shows a novel pharmacology exhibiting a complete inhibition of the response, whereas the synthetic ShK[1-35] peptide alone shows only a partial inhibition.
Figure 36 shows the mammalian-derived Fc-LI 0-ShK[1 -35] peptibody inhibits T cell proliferation (3H-thymidine incorporation) in human PBMCs from two normal donors stimulated with antibodies to CD3 and CD28. Figure 36A shows the response of donor 1 and Figure 36B the response of donor 2. Pre-incubation with the anti-CD32 (FcgRII) blocking antibody did not alter the sensitivity toward the peptibody.
Figure 37 shows the purified CHO-derived Fc-LI 0-ShK[1 -35] peptibody causes a dose- dependent inhibition of IL-2 (Figure 37A) and IFNγ (Figure 37B) production from αCD3 and αCD28 antibody stimulated human PBMCs.
Figure 38A shows the PEGylated ShK[2-35] synthetic peptide produces a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel and the time course of potassium current block at various concentrations is shown in Figure 38B.
Figure 39A shows a spectral scan of 50 μl of the Fc-L10-ShK(1-35) product diluted in 700 μl PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 39B shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-LI 0-ShK(1 -35) product. Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non-reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 39C shows the Size exclusion chromatography on 50 μg of the final Fc-LIO- ShK(1-35) product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NahkPCM, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 4OA shows a Spectral scan of 20 μl of the Fc-LI 0-ShK(2-35) product diluted in 700 μl PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 4OB shows a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-LI 0-ShK(2-35) product. Lanes 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non-reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank,
2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 4OC shows the size exclusion chromatography on 50 μg of the final Fc-UO- ShK(2-35) product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 rriM NahbPCM, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 4OD shows a MALDI mass spectral analysis of the final sample of Fc-UO- ShK(2-35) analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
Figure 41 A shows spectral scan of 50 μl of the Fc-UO-OSKI product diluted in 700 μl Formulation Buffer using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 41 B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-L10-OSK1 product. Lanes 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non-reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 41 C shows size exclusion chromatography on 123 μg of the final Fc-LI 0-OSK1 product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaHkPCM, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm. Figure 41 D shows liquid chromatography - mass spectral analysis of approximately 4 μg of the final Fc-LI 0-OSK1 sample using a Vydac C4 column with part of the effluent directed into a LCQ ion trap mass spectrometer. The mass spectrum was deconvoluted using the Bioworks software provided by the mass spectrometer manufacturer.
Figure 42A-B shows nucleotide and amino acid sequences (SEQ ID NO: 1040 and SEQ ID NO: 1041, respectively) of Fc-LI 0-OSK1.
Figure 43A-B shows nucleotide and amino acid sequences (SEQ ID NO: 1042 and SEQ ID NO: 1043, respectively) of Fc-LI 0-OSK1[K7S].
Figure 44A-B shows nucleotide and amino acid sequences (SEQ ID NO: 1044 and SEQ ID NO: 1045, respectively) of Fc-L10-OSK1[E16K,K20D], Figure 45A-B shows nucleotide and amino acid sequences (SEQ ID NO: 1046 and SEQ
ID NO: 1047, respectively) of Fc-L10-OSK1[K7S,E16K,K20D].
Figure 46 shows a Western blot (from tris-glycine 4-20% SDS-PAGE) with anti-human Fc antibodies. Lanes 1 - 6 were loaded as follows: 15μl of Fc-L10-OSK1 [K7S,E16K,K20D];15μl of
Fc-U 0-OSK1 [E16K.K20D]; 15μl of Fc-LI 0-OSK1 [K7S];15μl of Fc-U 0-OSK1 ; 15μl of "No Decontrol; molecular weight markers.
Figure 47 shows a Western blot (from tris-glycine 4-20% SDS-PAGE) with anti-human Fc antibodies. Lanes 1-5 were loaded as follows: 2μl of Fc-LI 0-OSK1 ; 5μl of Fc-LI 0-OSK1;10μl of Fc-LI 0-OSK1 ; 20ng Human IgG standard; molecular weight markers.
Figure 48 shows a Western blot (from tris-glycine 4-20% SDS-PAGE) with anti-human Fc antibodies. Lanes 1-13 were loaded as follows: 20 ng Human IgG standard; D1; C3; C2; B6; B5; B2; B1; A6; A5; A4; A3; A2 (5 μl of clone-conditioned medium loaded in lanes 2-13).
Figure 49A shows a spectral scan of 50 μl of the Fc-LI 0-OSK1 product diluted in 700 μl PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 49B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-LI 0-OSK1 product. Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non-reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 49C shows Size exclusion chromatography on 149 μg of the final Fc-L10-OSK1 product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PCM, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm. Figure 49D shows MALDI mass spectral analysis of the final sample of Fc-LI 0-OsK1 analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses. Figure 5OA shows a spectral scan of 50 μl of the Fc-LI 0-OsK1 (K7S) product diluted in
700 μl PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 5OB shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-LI 0-OsK1(K7S) product. Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non-reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 5OC shows size exclusion chromatography on 50 μg of the final Fc-L10-OsK1(K7S) product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PCM, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 5OD shows MALDI mass spectral analysis of a sample of the final product Fc-U 0-OsK1 (K7S) analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from - 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses. Figure 51 A shows a spectral scan of 50 μl of the Fc-LI 0-OsK1(E16K, K20D) product diluted in 700 μl PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 51 B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-L10-OsK1(E16K, K20D) product. Lane 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non-reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 51 C shows size exclusion chromatography on 50 μg of the final Fc-L10-OsK1(E16K, K20D) product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 51 D shows MALDI mass spectral analysis of a sample of the final product Fc-LIO- OsK1(E16K, K20D) analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
Figure 52A shows a spectral scan of 50 μl of the Fc-LI 0-OsK1(K7S, E16K, K20D) product diluted in 700 μl PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 52B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final Fc-LI 0-OsK1(K7S, E16K, K20D) product. Lanes 1 - 12 are loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non-reduced,
blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 52C shows size exclusion chromatography on 50 μg of the final Fc-UO- OsK1(K7S, E16K, K20D) product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 52D shows MALDI mass spectral analysis of a sample of the final product Fc-LI 0-OsK1(K7S, E16K, K20D) analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
Figure 53 shows inhibition of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel by synthetic Osk1 , a 38-residue toxin peptide of the Asian scorpion Orthochirus scrobiculosus venom. Figure 53A shows a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel by the synthetic Osk1 toxin peptide. Figure 53B shows the time course of the synthetic Osk1 toxin peptide block at various concentrations. The IC50 for the synthetic Osk1 toxin peptide was estimated to be 39 ± 12 pM (n =4 cells). Figure 54 shows that modification of the synthetic OSK1 toxin peptide by fusion to the Fc- fragment of an antibody (OSK1-peptibody) retained the inhibitory activity against the human Kv1.3 channel. Figure 54A shows a concentration dependent block of the outward potassium current recorded from HEK293 cells stably expressing human Kv1.3 channel by OSK1 linked to a human IgGI Fc-fragment with a linker chain length of 10 amino acid residues (Fc-LI 0-OSK1). The fusion construct was stably expressed in Chinese Hamster Ovarian (CHO) cells. Figure 54B shows the time course of the Fc-LI 0-OSK1 block at various concentrations. The IC50 for Fc-LI 0-OSK1 was estimated to be 198 ± 35 pM (n = 6 cells), approximately 5-fold less potent than the synthetic OSK1 toxin peptide.
Figure 55 shows that a single amino-acid residue substitution of the OSK1-peptibody retained the inhibitory activity against the human Kv1.3 channel. Figure 55A shows a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel by OSK1-peptibody with a single amino acid substitution (lysine to serine at the 7th position from N-terminal, [K7S]) and linked to a human IgGI Fc-fragment with a
linker chain length of 10 amino acid residues (Fc-LI 0-OSK1 [K7S]). The fusion construct was stably expressed in Chinese Hamster Ovarian (CHO) cells. Figure 55B shows the time course of potassium current block by Fc-LI 0-OSK1[K7S] at various concentrations. The IC50 was estimated to be 372 ± 71 pM (n = 4 cells), approximately 10-fold less potent than the synthetic 0SK1 toxin peptide.
Figure 56 shows that a two amino-acid residue substitution of the OSK1-peptibody retained the inhibitory activity against the human Kv1.3 channel. Figure 56A shows a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel by OSK1-peptibody with two amino acid substitutions (glutamic acid to lysine and lysine to aspartic acid at the 16th and 20th position from N-terminal respectively, [E16KK20D]) and linked to a human IgGI Fc-fragment with a linker chain length of 10 amino acid residues (Fc-LI 0-OSK1[E16KK20D]). The fusion construct was stably expressed in Chinese Hamster Ovarian (CHO) cells. Figure 56B shows the time course of potassium current block by Fc-LI 0-OSK1[E16KK20D] at various concentrations. The IC50 was estimated to be 248 ± 63 pM (n = 3 cells), approximately 6-fold less potent than the synthetic OSK1 toxin peptide.
Figure 57 shows that a triple amino-acid residue substitution of the OSK1-peptibody retained the inhibitory activity against the human Kv1.3 channel, but the potency of inhibition was significantly reduced when compared to the synthetic OSK1 toxin peptide. Figure 57A shows a concentration dependent block of the outward potassium current recorded from HEK293 cell stably expressing human Kv1.3 channel by OSK1-peptibody with triple amino acid substitutions (lysine to serine, glutamic acid to lysine and lysine to aspartic acid at the 7th, 16th and 20th position from N- terminal respectively, [K7SE16KK20D]) and linked to a human IgGI Fc-fragment with a linker chain length of 10 amino acid residues (Fc-LI 0-OSK1[K7SE16KK20D]). The fusion construct was stably expressed in Chinese Hamster Ovarian (CHO) cells. Figure 57B shows the time course of potassium current block by Fc-LI 0-OSK1[K7SE16KK20D] at various concentrations. The IC50 was estimated to be 812 + 84 pM (n = 3 cells), approximately 21 -fold less potent than the synthetic OSK1 toxin peptide.
Figure 58 shows Standard curves for ShK (Figure 58A) and 2OK PEG-ShK[I -35] (Figure 58B) containing linear regression equations for each Standard at a given percentage of serum. Figure 59 shows the pharmacokinetic profile in rats of 2OK PEG ShK[I -35] molecule after
IV injection.
Figure 60 shows Kv1.3 inhibitory activity in serum samples (5%) of rats receiving a single equal molar IV injection of Kv1.3 inhibitors ShK versus 2OK PEG-ShK[I -35].
Figure 61 illustrates an Adoptive Transfer EAE model experimental design (n = 5 rats per treatment group). Dosing values in microgram per kilogram (mg/kg) are based on peptide content.
Figure 62 shows that treatment with PEG-ShK ameliorated disease in rats in the adoptive transfer EAE model. Clinical scoring: O = No signs, 0.5 = distal limp tail, 1.0 = limp tail, 2.0 = mild paraparesis, ataxia, 3.0 = moderate paraparesis, 3.5 = one hind leg paralysis, 4.0 = complete hind leg paralysis, 5.0 = complete hind leg paralysis and incontinence, 5.5 = tetraplegia, 6.0 = moribund state or death. Rats reaching a score of 5.5 to 6 died or were euthanized. Mean ± sem values are shown, (n = 5 rats per treatment group.)
Figure 63 shows that treatment with PEG-ShK prevented loss of body weight in the adoptive transfer EAE model. Rats were weighed on days -1 , 4, 6, and 8 (for surviving rats). Mean ± sem values are shown.
Figure 64 shows that thapsigargin-induced IL-2 production in human whole blood was suppressed by the Kv1.3 channel inhibitors ShK[I -35] and Fc-LI 0-ShK[2-35]. The calcineurin inhibitor cyclosporine A also blocked the response. The BKCa channel inhibitor iberiotoxin (IbTx) showed no significant activity. The response of whole blood from two separate donors is shown in Figure 64A and Figure 64B.
Figure 65 shows that thapsigargin-induced IFN-g production in human whole blood was suppressed by the Kv1.3 channel inhibitors ShK[I -35] and Fc-LI 0-ShK[2-35]. The calcineurin inhibitor cyclosporine A also blocked the response. The BKCa channel inhibitor iberiotoxin (IbTx) showed no significant activity. The response of whole blood from two separate donors is shown in Figure 65A and Figure 65B.
Figure 66 shows that thapsigargin-induced upregulation of CD40L on T cells in human whole blood was suppressed by the Kv1.3 channel inhibitors ShK[I -35] and Fc-LI 0-ShK[1 -35] (Fc-ShK). The calcineurin inhibitor cyclosporine A (CsA) also blocked the response. Figure 66A shows results of an experiment looking at the response of total CD4+ T cells. Figure 66B shows results of an experiment that looked at total CD4+ T cells, as well as CD4+CD45+ and CD4+CD45- T cells. In Figure 66B, the BKCa channel inhibitor iberiotoxin (IbTx) and the Kv1.1 channel inhibitor dendrotoxin-K (DTX-K) showed no significant activity.
Figure 67 shows that thapsigargin-induced upregulation of the IL-2R on T cells in human whole blood was suppressed by the Kv1.3 channel inhibitors ShK[I -35] and Fc-LI 0-ShK[1 -35]
(Fc-ShK). The calcineurin inhibitor cyclosporine A (CsA) also blocked the response. Figure 67A shows results of an experiment looking at the response of total CD4+ T cells. Figure 67B shows results of an experiment that looked at total CD4+ T cells, as well as CD4+CD45+ and CD4+CD45-
T cells. In Figure 67B, the BKCa channel inhibitor iberiotoxin (IbTx) and the Kv1.1 channel inhibitor dendrotoxin-K (DTX-K) showed no significant activity.
Figure 68 shows cation exchange chromatograms of PEG-peptide purification on SP Sepharose HP columns for PEG-Shk purification (Figure 68A) and PEG-OSK-1 purification (Figure 68B).
Figure 69 shows RP-HPLC chromatograms on final PEG-peptide pools to demonstrate purity of PEG-Shk purity >99% (Figure 69A) and PEG-0sk1 purity >97% (Figure 69B).
Figure 70 shows the amino acid sequence (SEQ ID NO: 976) of an exemplary FcLoop- L2-OsK1-L2 having three linked domains: Fc N-terminal domain (amino acid residues 1-139); OsK1 (underlined amino acid residues 142-179); and Fc C-terminal domain (amino acid residues 182-270).
Figure 71 shows the amino acid sequence (SEQ ID NO: 977) of an exemplary FcLoop- L2-ShK-L2 having three linked domains: Fc N-terminal domain (amino acid residues 1-139); ShK (underlined amino acid residues 142-176); and Fc C-terminal domain (amino acid residues 179-267).
Figure 72 shows the amino acid sequence (SEQ ID NO: 978) of an exemplary FcLoop- L2-ShK-L4 having three linked domains: Fc N-terminal domain (amino acid residues 1-139); ShK (underlined amino acid residues 142-176); and Fc C-terminal domain (amino acid residues 181-269). Figure 73 shows the amino acid sequence (SEQ ID NO: 979) of an exemplary FcLoop-
L4-OsK1-L2 having three linked domains: Fc N-terminal domain (amino acid residues 1-139); OsK1 (underlined amino acid residues 144-181); and Fc C-terminal domain (amino acid residues 184-272).
Figure 74 shows that the 2OK PEGylated ShK[I -35] provided potent blockade of human Kv1.3 as determined by whole cell patch clamp electrophysiology on HEK293/Kv1.3 cells. The data represents blockade of peak current.
Figure 75 shows schematic structures of some other exemplary embodiments of the composition of matter of the invention. "X2" and "X3" represent toxin peptides or linker-toxin peptide combinations (i.e., -(LJrP-(L)9-) as defined herein. As described herein but not shown in Figure 75, an additional X1 domain and one or more additional PEG moieties are also encompassed in other embodiments. The specific embodiments shown here are as follows:
Figure 75C, Figure 75D, Figure 75G and Figure 75H: show a single chain molecule and can also represent the DNA construct for the molecule.
Figure 75A, Figure 75B, Figure 75E and Figure 75F: show doubly disulfide-bonded Fc dimers (in position F2); Figure 75A and Figure 75B show a dimer having the toxin peptide portion on both chains in position X3; Figure 75E and Figure 75F show a dimer having the toxin peptide portion on both chains In position X2. Figure 76A shows a spectral scan of 50 μl of the ShK[2-35]-Fc product diluted in 700 μl
PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 76B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final ShK[2-35]-Fc product. Lanes 1 - 12 were loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non-reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 76C shows size exclusion chromatography on 70 μg of the final ShK[2-35]-Fc product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 76D shows LC-MS analysis of the final ShK[2-35]-Fc sample using an Agilent 1100 HPCL running reverse phase chromatography, with the column effluent directly coupled to an electrospray source of a Thermo Finnigan LCQ ion trap mass spectrometer. Relevant spectra were summed and deconvoluted to mass data with the Bioworks software package. Figure 77A shows a spectral scan of 20 μl of the met-ShK[1 -35]-Fc product diluted in 700 μl PBS (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 77B shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final met-ShK[1-35]-Fc product. Lanes 1 - 12 were loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non-reduced, blank, 10 μg product non-reduced, Novex Mark12 wide range protein standards, 0.5 μg product reduced, blank, 2.0 μg product reduced, blank, and 10 μg product reduced.
Figure 77C shows size exclusion chromatography on 93 μg of the final met-ShK[1-35]-Fc product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 77D shows MALDI mass spectral analysis of the final met-ShK[1-35]-Fc sample analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25
kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses.
Figure 78 shows a spectral scan of 10 μl of the CH2-OSK1 fusion protein product diluted in 150 μl water (blanking buffer) using a Hewlett Packard 8453 spectrophotometer and a 1 cm path length quartz cuvette.
Figure 79 shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final CH2-OSK1 fusion protein product. Lane 1 - 7 were loaded as follows: Novex Mark12 wide range protein standards, 0.5 μg product non-reduced, blank, 2.0 μg product non-reduced, blank, 10 μg product non-reduced, and Novex Mark12 wide range protein standards. Figure 80 shows size exclusion chromatography on 50 μg of the final CH2-OSK1 fusion protein product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm.
Figure 81 shows liquid chromatography - mass spectral analysis of the CH2-OSK1 fusion protein sample using a Vydac C4 column with part of the effluent directed into a LCQ ion trap mass spectrometer. The mass spectrum was deconvoluted using the Bioworks software provided by the mass spectrometer manufacturer.
Figure 82 shows cation exchange chromatogram of PEG-CH2-OSK1 reaction mixture. Vertical lines delineate fractions pooled to obtain mono-PEGylated CH2-OSK1.
Figure 83 shows Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE of the final PEGylated CH2-OSK1 pool. Lane 1 - 2 were loaded as follows: Novex Mark12 wide range protein standards, 2.0 μg product non-reduced.
Figure 84 shows whole cell patch clamp (WCPC) and PatchXpress (PX) electrophysiology comparing the activity of OSK1[Ala-12] (SEQ ID No:1410) on human Kv1.3 and human Kv1.1 heterologously overexpressed on CHO and HEK293 cells, respectively. The table summarizes the calculated IC50 values and the plots show the individual traces of the impact of various concentrations of analog on the relative Kv1.3 or Kv1.1 current (percent of control, POC).
Figure 85 shows whole cell patch clamp electrophysiology comparing the activity of OSK1[Ala-29] (SEQ ID No:1424) on human Kv1.3 and human Kv1.1 heterologously overexpressed on CHO and HEK293 cells, respectively. Concentration response curves of OSK1[Ala-29] on CHO/Kv1.3 (circle, square and diamond,
0.033nM, n = 3) and on HEK/Kv1.1 (filled triangle,
IC5O= 2.7nM, n=1).
Figure 86 shows a dose-response curve for OSK1[Ala-29] (SEQ ID No:1424) against human Kv1.3 (CHO) (panel A) and human Kv1.1 (HEK293) (panel B) as determined by high- throughput 384-well planar patch clamp electrophysiology using the IonWorks Quattro system. Figure 87 A-B show Western blots of Tris-glycine 4-20% SDS-PAGE (Figure 87A with longer exposure time and Figure 87B with shorter exposure time) of a monovalent dimeric Fc-L- ShK(2-35) molecule product expressed by and released into the conditioned media from mammalian cells transiently transfected with pTT5-Fc-Fc-L10-Shk(2-35), which was sampled after the indicated number of days. Lanes 3-8 were loaded with 20 μL of conditioned medium per lane. The immunoblot was probed with anti-human IgG-Fc-HRP (Pierce). The lanes were loaded as follows: 1) MW Markers; 2) purified Fc-LI 0-ShK(2-35), 100ng; 3) 293-6E-HD (5-day); 4) 293-6E- HD (6-day); 5) 293-6E-PEI (5-day); 6) 293-6E-PEI (6-day); 7) CHO-S (5-day); 8) CHO-S (6-day). Four bands were expected in the reduced gel: Linker-Fc-Shk(2-35) (one cut at 3' furin site; predicted MW: 33.4 kDa); Fc-ShK(2-35) (both furin sites cut; predicted MW: 30.4 kDa); Fc-linker (one cut at 5' furin site; predicted MW: 29.1 kDa); Fc (both furin sites cut; predicted MW: 25.8 kDa). Further mass spec or amino acid sequence analysis of the individual bands is needed to identify these bands and their relative ratios.
Figure 88 shows a western blot of serum samples from a pharmacokinetic study on monovalent dimeric Fc-ShK(I -35) in SD rats. Various times (0.083 - 168 hours) after a single 1 mg/kg intravenous injection of monovalent dimeric Fc-LI 0-ShK(1 -35) (see, Example 2), blood was drawn, and serum was collected. A Costar EIA/RIA 96 well plate was coated with 2μg/ml polyclonal goat anti-human Fc antibody overnight at 4°C. Capture antibody was removed and the plate was washed with PBST and then blocked with Blotto. After the plate was washed, serum samples diluted in PBST/0.1% BSA were added. Binding was allowed to occur at room temperature for several hours, and then the plate was again washed. Samples were eluted from the plate with reducing Laemmle buffer, heated, then run on SDS-Page gels. Run in an adjacent lane ("5 ng Control") of the gel as a standard was 5 ng of the purified monovalent dimeric Fc-LI 0-ShK(1 -35) fusion protein used in the pharmacokinetic study. Proteins were transferred to PVDF membranes by western blot. Membranes were blocked with Blotto followed by incubation with goat anti-Human Fc-HRP conjugated antibody. After the membranes were washed, signal was detected via chemiluminescence using a CCD camera.
Figure 89 shows the NMR solution structure of OSK1 and sites identified by analoging to be important for Kv1.3 activity and selectivity. Space filling structures are shown in Fig. 89A, 89B and 89D. The color rendering in Fig. 89A depicts amino acid charge. In Fig. 89B, several key
0SK1 amino acid residues found to be important for Kv1.3 activity (Tables 37 - 40) are lightly shaded and labeled Phe25, Gly26, Lys27, Met29 and Asn30. In Fig. 89D residues SeM 1, Met29 and His34 are labeled. Some analogues of these residues were found to result in improved Kv1.3 selectivity over Kv1.1 (Tables 41). Fig. 89C shows the three beta strands and single alpha helix of OSK1. The amino acid sequence of native 0SK1 (SEQ ID No: 25) is shown in Fig. 89E, with residues forming the molecules beta strands (β1, β2, β3) and alpha helix (α1) underlined. The 0SK1 structures shown were derived from PDB:1SC0, and were rendered using Cn3D vers4.1. Figure 90A-D illustrates that toxin peptide inhibitors of Kv1.3 provide potent blockade of the whole blood inflammatory response. The activity of the calcineurin inhibitor cyclosporin A (Fig. 90A) and Kv1.3 peptide inhibitors ShK-Ala22 (Fig. 9OB; SEQ ID No: 123), OSK1 -Ala29 (Fig. 9OC; SEQ ID No: 1424) and OSK1-Ala12 (Fig. 9OD; SEQ ID No: 1410) were compared in the whole blood assay of inflammation (Example 46) using the same donor blood sample. The potency (IC50) of each molecule is shown, where for each panel the left curve is the impact on IL-2 production and the right curve is the impact on IFNγ production. Figure 91 A-B shows an immunoblot analysis of expression of monovalent dimeric IgGI-
Fc-L-ShK(2-35) from non-reduced SDS-PAGE. Figure 91A shows detection of human Fc expression with goat anti-human lgG(H+L)-HRP. Figure 91 B shows detection of ShK(2-35) expression with a goat anti-mouse IgG (H+L)-HRP that cross reacts with human IgG. Lane 1: purified Fc-LI 0-Shk(2-35); Lane 2: conditioned medium from 293EBNA cells transiently transfected with pTT5-hulgG1 +pTT5-hKappa+pCMVi-Fc-L10-ShK(2-35); Lane 3: conditioned media from
293EBNA cells transiently transfected with pTT5-hulgG2+pTT5-hKappa+pCMVi-Fc-L10-Shk(2-35); Lane 4: conditioned media from 293EBNA cells transiently transfected with pTT14 vector alone. The two arrows point to the full length hulgG (mol. wt. ~ 150 kDa) and monovalent dimeric hulgG- FcShK(2-35) (mol. wt. ~ 100 kDa); the abundant 60-kDa band is the bivalent dimeric Fc-ShK(2-35). Figure 92A-C shows schematic representations of an embodiment of a monovalent
"hemibody"-toxin peptide fusion protein construct; the single toxin peptide is represented by an oval. Figure 92A, which can also represent the DNA construct for the fusion protein, represents an immunoglobulin light chain (LC, open rectangle), an immunoglobulin heavy chain (HC, longer cross-hatched rectangle), and an immunoglobulin Fc domain (Fc, shorter cross-hatched rectangle), each separated by an intervening peptidyl linker sequence (thick lines) comprising at least one protease cleavage site (arrows), e.g., a furin cleavage site. Figure 92 illustrates the association of the recombinantly expressed LC, HC, and Fc-toxin peptide components connected by the peptidyl linker sequences (thick lines) and, in Figure 92C, the final monovalent chimeric
immunoglobulin(LC+HC)-Fc (i.e., "hemibody")-toxin peptide fusion protein after cleavage (intracellular^ or extracellularly) at the protease cleavage sites, to release the linkers, and formation of disulfide bridges between the light and heavty chains and between the heavy chain and the Fc components (shown as thin horizontal lines between the LC, HC, and Fc components in Figure 92C).
Detailed Description of Embodiments of the Invention
Definition of Terms
The terms used throughout this specification are defined as follows, unless otherwise limited in specific instances. As used in the specification and the appended claims, the singular 5 forms "a", "an", and "the" include plural referents unless the context clearly dictates otherwise.
"Polypeptide" and "protein" are used interchangeably herein and include a molecular chain of two or more amino acids linked through peptide bonds. The terms do not refer to a specific length of the product. Thus, "peptides," and "oligopeptides," are included within the definition of polypeptide. The terms include post-translational modifications of the polypeptide, for 0 example, glycosylates, acetylations, phosphorylations and the like. In addition, protein fragments, analogs, mutated or variant proteins, fusion proteins and the like are included within the meaning of polypeptide. The terms also include molecules in which one or more amino acid analogs or non-canonical or unnatural amino acids are included as can be synthesized, or expressed recombinantly using known protein engineering techniques. In addition, inventive 5 fusion proteins can be derivatized as described herein by well-known organic chemistry techniques.
The term "fusion protein" indicates that the protein includes polypeptide components derived from more than one parental protein or polypeptide. Typically, a fusion protein is expressed from a fusion gene in which a nucleotide sequence encoding a polypeptide sequence O from one protein is appended in frame with, and optionally separated by a linker from, a nucleotide sequence encoding a polypeptide sequence from a different protein. The fusion gene can then be expressed by a recombinant host cell as a single protein.
A "domain" of a protein is any portion of the entire protein, up to and including the complete protein, but typically comprising less than the complete protein. A domain can, but need 5 not, fold independently of the rest of the protein chain and/or be correlated with a particular biological, biochemical, or structural function or location (e.g., a ligand binding domain, or a cytosolic, transmembrane or extracellular domain).
A "secreted" protein refers to those proteins capable of being directed to the ER, secretory vesicles, or the extracellular space as a result of a secretory signal peptide sequence, as well as O those proteins released into the extracellular space without necessarily containing a signal sequence. If the secreted protein is released into the extracellular space, the secreted protein can undergo extracellular processing to produce a "mature" protein. Release into the extracellular space can occur by many mechanisms, including exocytosis and proteolytic cleavage.
The term "signal peptide" refers to a relatively short (3-60 amino acid residues long) peptide chain that directs the post-translational transport of a protein, e.g., its export to the extracellular space. Thus, secretory signal peptides are encompassed by "signal peptide". Signal peptides may also be called targeting signals, signal sequences, transit peptides, or localization 5 signals.
The term "recombinant" indicates that the material (e.g., a nucleic acid or a polypeptide) has been artificially or synthetically (i.e., non-naturally) altered by human intervention. The alteration can be performed on the material within, or removed from, its natural environment or state. For example, a "recombinant nucleic acid" is one that is made by recombining nucleic 0 acids, e.g., during cloning, DNA shuffling or other well known molecular biological procedures. A "recombinant DNA molecule," is comprised of segments of DNA joined together by means of such molecular biological techniques. The term "recombinant protein" or "recombinant polypeptide" as used herein refers to a protein molecule which is expressed using a recombinant DNA molecule. A "recombinant host cell" is a cell that contains and/or expresses a recombinant nucleic acid. 5 A "polynucleotide sequence" or "nucleotide sequence" or "nucleic acid sequence," as used interchangeably herein, is a polymer of nucleotides, including an oligonucleotide, a DNA, and RNA, a nucleic acid, or a character string representing a nucleotide polymer, depending on context. From any specified polynucleotide sequence, either the given nucleic acid or the . complementary polynucleotide sequence can be determined. Included are DNA or RNA of O genomic or synthetic origin which may be single- or double-stranded, and represent the sense or antisense strand.
As used herein, the terms "nucleic acid molecule encoding," "DNA sequence encoding," and "DNA encoding" refer to the order or sequence of deoxyribonucleotides along a strand of deoxyribonucleic acid. The order of these deoxyribonucleotides determines the order of 5 ribonucleotides along the mRNA chain, and also determines the order of amino acids along the polypeptide (protein) chain. The DNA sequence thus codes for the RNA sequence and for the amino acid sequence.
"Expression of a gene" or "expression of a nucleic acid" means transcription of DNA into RNA (optionally including modification of the RNA, e.g., splicing), translation of RNA into a O polypeptide (possibly including subsequent post-translational modification of the polypeptide), or both transcription and translation, as indicated by the context.
The term "gene" is used broadly to refer to any nucleic acid associated with a biological function. Genes typically include coding sequences and/or the regulatory sequences required for
expression of such coding sequences. The term "gene" applies to a specific genomic or recombinant sequence, as well as to a cDNA or mRNA encoded by that sequence. A "fusion gene" contains a coding region that encodes a fusion protein. Genes also include non-expressed nucleic acid segments that, for example, form recognition sequences for other proteins. Non- 5 expressed regulatory sequences including transcriptional control elements to which regulatory proteins, such as transcription factors, bind, resulting in transcription of adjacent or nearby sequences.
As used herein the term "coding region" when used in reference to a structural gene refers to the nucleotide sequences which encode the amino acids found in the nascent polypeptide0 as a result of translation of an mRNA molecule. The coding region is bounded, in eukaryotes, on the 5' side by the nucleotide triplet "ATG" which encodes the initiator methionine and on the 3' side by one of the three triplets which specify stop codons (i.e., TAA, TAG, TGA).
Transcriptional control signals in eukaryotes comprise "promoter" and "enhancer" elements. Promoters and enhancers consist of short arrays of DNA sequences that interact 5 specifically with cellular proteins involved in transcription (Maniatis, et al., Science 236:1237
(1987)). Promoter and enhancer elements have been isolated from a variety of eukaryotic sources including genes in yeast, insect and mammalian cells and viruses (analogous control elements, i.e., promoters, are also found in prokaryotes). The selection of a particular promoter and enhancer depends on what cell type is to be used to express the protein of interest. Some eukaryotic O promoters and enhancers have a broad host range while others are functional in a limited subset of cell types (for review see Voss, et al., Trends Biochem. Sci., 11 :287 (1986) and Maniatis, et al., Science 236:1237 (1987)).
The term "expression vector" as used herein refers to a recombinant DNA molecule containing a desired coding sequence and appropriate nucleic acid sequences necessary for the 5 expression of the operably linked coding sequence in a particular host cell. Nucleic acid sequences necessary for expression in prokaryotes include a promoter, optionally an operator sequence, a ribosome binding site and possibly other sequences. Eukaryotic cells are known to utilize promoters, enhancers, and termination and polyadenylation signals. A secretory signal peptide sequence can also, optionally, be encoded by the expression vector, operably linked to the O coding sequence for the inventive recombinant fusion protein, so that the expressed fusion protein can be secreted by the recombinant host cell, for more facile isolation of the fusion protein from the cell, if desired. Such techniques are well known in the art. (E.g., Goodey, Andrew R.; et al., Peptide and DNA sequences, U.S. Patent No. 5,302,697; Weiner et al., Compositions and
methods for protein secretion, U.S. Patent No. 6,022,952 and U.S. Patent No. 6,335,178; Uemura et al., Protein expression vector and utilization thereof, U.S. Patent No. 7,029,909; Ruben et al., 27 human secreted proteins, US 2003/0104400 A1).
The terms "in operable combination", "in operable order" and "operably linked" as used herein refer to the linkage of nucleic acid sequences in such a manner or orientation that a nucleic acid molecule capable of directing the transcription of a given gene and/or the synthesis of a desired protein molecule is produced. The term also refers to the linkage of amino acid sequences in such a manner so that a functional protein is produced and/or transported.
Recombinant DNA- and/or RNA-mediated protein expression techniques, or any other methods of preparing peptides or, are applicable to the making of the inventive recombinant fusion proteins. For example, the peptides can be made in transformed host cells. Briefly, a recombinant DNA molecule, or construct, coding for the peptide is prepared. Methods of preparing such DNA molecules are well known in the art. For instance, sequences encoding the peptides can be excised from DNA using suitable restriction enzymes. Any of a large number of available and well- known host cells may be used in the practice of this invention. The selection of a particular host is dependent upon a number of factors recognized by the art. These include, for example, compatibility with the chosen expression vector, toxicity of the peptides encoded by the DNA molecule, rate of transformation, ease of recovery of the peptides, expression characteristics, bio- safety and costs. A balance of these factors must be struck with the understanding that not all hosts may be equally effective for the expression of a particular DNA sequence. Within these general guidelines, useful microbial host cells in culture include bacteria (such as Escherichia coli sp.), yeast (such as Saccharomyces sp.) and other fungal cells, insect cells, plant cells, mammalian (including human) cells, e.g., CHO cells and HEK293 cells. Modifications can be made at the DNA level, as well. The peptide-encoding DNA sequence may be changed to codons more compatible with the chosen host cell. For E. coli, optimized codons are known in the art.
Codons can be substituted to eliminate restriction sites or to include silent restriction sites, which may aid in processing of the DNA in the selected host cell. Next, the transformed host is cultured and purified. Host cells may be cultured under conventional fermentation conditions so that the desired compounds are expressed. Such fermentation conditions are well known in the art. The term "half-life extending moiety" (i.e., F1 or F2 in Formula I) refers to a pharmaceutically acceptable moiety, domain, or "vehicle" covalently linked ("conjugated") to the toxin peptide directly or via a linker, that prevents or mitigates in vivo proteolytic degradation or other activity-diminishing chemical modification of the toxin peptide, increases half-life or other
pharmacokinetic properties such as but not limited to increasing the rate of absorption, reduces toxicity, improves solubility, increases biological activity and/or target selectivity of the toxin peptide with respect to a target ion channel of interest, increases manufacturability, and/or reduces immunogenicity of the toxin peptide, compared to an unconjugated form of the toxin peptide. By "PEGylated peptide" is meant a peptide or protein having a polyethylene glycol (PEG) moiety covalently bound to an amino acid residue of the peptide itself or to a peptidyl or non- peptidyl linker (including but not limited to aromatic or aryl linkers) that is covalently bound to a residue of the peptide.
By "polyethylene glycol" or "PEG" is meant a polyalkylene glycol compound or a derivative thereof, with or without coupling agents or derivatization with coupling or activating moieties (e.g., with aldehyde, hydroxysuccinimidyl, hydrazide, thiol, triflate, tresylate, azirdine, oxirane, orthopyridyl disulphide, vinylsulfone, iodoacetamide or a maleimide moiety). In accordance with the present invention, useful PEG includes substantially linear, straight chain PEG, branched PEG, or dendritic PEG. (See, e.g., Merrill, US Patent No. 5,171 ,264; Harris et al., Multiarmed, monofunctional, polymer for coupling to molecules and surfaces, US Patent No. 5,932,462; Shen, N-maleimidyl polymer derivatives, US Patent No. 6,602,498).
The term "peptibody" refers to molecules of Formula I in which F1 and/or F2 is an immunoglobulin Fc domain or a portion thereof, such as a CH2 domain of an Fc, or in which the toxin peptide is inserted into a human IgGI Fc domain loop, such that F1 and F2 are each a portion of an Fc domain with a toxin peptide inserted between them (See, e.g., Figures 70-73 and
Example 49 herein). Peptibodies of the present invention can also be PEGylated as described further herein, at either an Fc domain or portion thereof, or at the toxin peptide(s) portion of the inventive composition, or both.
The term "native Fc" refers to molecule or sequence comprising the sequence of a non- antigen-binding fragment resulting from digestion of whole antibody, whether in monomeric or multimeric form. The original immunoglobulin source of the native Fc is preferably of human origin and can be any of the immunoglobulins, although IgGI or lgG2 are preferred. Native Fc's are made up of monomeric polypeptides that can be linked into dimeric or multimeric forms by covalent (i.e., disulfide bonds) and non-covalent association. The number of intermolecular disulfide bonds between monomeric subunits of native Fc molecules ranges from 1 to 4 depending on class (e.g.,
IgG, IgA, IgE) or subclass (e.g., IgGI, lgG2, lgG3, lgG4, IgAI, lgGA2). One example of a native Fc is a disulfide-bonded dimer resulting from papain digestion of an IgG (see Ellison et al. (1982),
Nucleic Acids Res, 10: 4071-9). The term "native Fc" as used herein is generic to the monomeric, dimeric, and multimeric forms.
The term "Fc variant" refers to a molecule or sequence that is modified from a native Fc but still comprises a binding site for the salvage receptor, FcRn. Several published patent documents describe exemplary Fc variants, as well as interaction with the salvage receptor. See International Applications WO 97/34 631 (published 25 September 1997; WO 96/32 478, corresponding to US Pat. No. 6,096,891, issued August 1, 2000, hereby incorporated by reference in its entirety; and WO 04/110 472. Thus, the term "Fc variant" includes a molecule or sequence that is humanized from a non-human native Fc. Furthermore, a native Fc comprises sites that can be removed because they provide structural features or biological activity that are not required for the fusion molecules of the present invention. Thus, the term "Fc variant" includes a molecule or sequence that lacks one or more native Fc sites or residues that affect or are involved in (1) disulfide bond formation, (2) incompatibility with a selected host cell (3) N-terminal heterogeneity upon expression in a selected host cell, (4) glycosylate, (5) interaction with complement, (6) binding to an Fc receptor other than a salvage receptor, or (7) antibody-dependent cellular cytotoxicity (ADCC). Fc variants are described in further detail hereinafter.
The term "Fc domain" encompasses native Fc and Fc variant molecules and sequences as defined above. As with Fc variants and native Fc's, the term "Fc domain" includes molecules in monomeric or multimeric form, whether digested from whole antibody or produced by other means. The term "multimer" as applied to Fc domains or molecules comprising Fc domains refers to molecules having two or more polypeptide chains associated covalently, noncovalently, or by both covalent and non-covalent interactions. IgG molecules typically form dimers; IgM, pentamers; IgD, dimers; and IgA, monomers, dimers, trimers, or tetramers. One skilled in the art can form multimers by exploiting the sequence and resulting activity of the native Ig source of the Fc or by derivatizing (as defined below) such a native Fc.
The term "dimer" as applied to Fc domains or molecules comprising Fc domains refers to molecules having two polypeptide chains associated covalently or non-covalently. Thus, exemplary dimers within the scope of this invention are as shown in Figure 2. A "monovalent dimeric" Fc- toxin peptide fusion, or "monovalent dimer", is a Fc-toxin peptide fusion that includes a toxin peptide conjugated with only one of the dimerized Fc domains (e.g., as represented schematically in Figure 2B) . A "bivalent dimeric" Fc-toxin peptide fusion, or "bivalent dimer", is a Fc-toxin peptide fusion having both of the dimerized Fc domains each conjugated separately with a toxin peptide (e.g., as represented schematically in Figure 2C).
The terms "derivatizing" and "derivative" or "derivatized" comprise processes and resulting compounds respectively in which (1) the compound has a cyclic portion; for example, cross-linking between cysteinyl residues within the compound; (2) the compound is cross-linked or has a cross- linking site; for example, the compound has a cysteinyl residue and thus forms cross-linked dimers in culture or in vivo; (3) one or more peptidyl linkage is replaced by a non-peptidyl linkage; (4) the N-terminus is replaced by -NRR1, NRC(O)R1, -NRC(O)OR1, -NRS(O)2R1, -NHC(O)NHR, a succinimide group, or substituted or unsubstituted benzyloxycarbonyl-NH-, wherein R and R1 and the ring substituents are as defined hereinafter; (5) the C-terminus is replaced by -C(O)R2 or - NR3R4 wherein R2, R3 and R4 are as defined hereinafter; and (6) compounds in which individual 0 amino acid moieties are modified through treatment with agents capable of reacting with selected side chains or terminal residues. Derivatives are further described hereinafter.
The term "peptide" refers to molecules of 2 to about 80 amino acid residues, with molecules of about 10 to about 60 amino acid residues preferred and those of about 30 to about 50 amino acid residuess most preferred. Exemplary peptides can be randomly generated by any 5 known method, carried in a peptide library (e.g., a phage display library), or derived by digestion of proteins. In any peptide portion of the inventive compositions, for example a toxin peptide or a peptide linker moiety described herein, additional amino acids can be included on either or both of the N- or C- termini of the given sequence. Of course, these additional amino acid residues should not significantly interfere with the functional activity of the composition. O "Toxin peptides" include peptides having the same amino acid sequence of a naturally occurring pharmacologically active peptide that can be isolated from a venom, and also include modified peptide analogs (spelling used interchangeably with "analogues") of such naturally occurring molecules.
The term "peptide analog" refers to a peptide having a sequence that differs from a 5 peptide sequence existing in nature by at least one amino acid residue substitution, internal addition, or internal deletion of at least one amino acid, and/or amino- or carboxy- terminal end truncations, or additions). An "internal deletion" refers to absence of an amino acid from a sequence existing in nature at a position other than the N- or C-terminus. Likewise, an "internal addition" refers to presence of an amino acid in a sequence existing in nature at a position other O than the N- or C-terminus. "Toxin peptide analogs", such as, but not limited to, an OSK1 peptide analog, ShK peptide analog, or ChTx peptide analog, contain modifications of a native toxin peptide sequence of interest (e.g., amino acid residue substitutions, internal additions or insertions, internal deletions, and/or amino- or carboxy- terminal end truncations, or additions as previously
described above) relative to a native toxin peptide sequence of interest, which is in the case of 0SK1: GVIINVKCKISRQCLEPCKKAGMRFGKCMNGKCHCTPK (SEQ ID NO:25).
Examples of toxin peptides useful in practicing the present invention are listed in Tables 1-32. The toxin peptide ("P", or equivalently shown as "P1" in Figure 2) comprises at least two 5 intrapeptide disulfide bonds, as shown, for example, in Figure 9. Accordingly, this invention concerns molecules comprising: a) C1-C3 and C2-C4 disulfide bonding in which C1, C2, C3, and C4 represent the order in which cysteine residues appear in the primary sequence of the toxin peptide stated conventionally with the N-terminus of the peptide on the left, with the first and third 0 cysteines in the amino acid sequence forming a disulfide bond, and the second and fourth cysteines forming a disulfide bond. Examples of toxin peptides with such a C1- C3, C2-C4 disulfide bonding pattern include, but are not limited to, apamin peptides, α- conopeptides, PnIA peptides, PnIB peptides, and Mil peptides, and analogs of any of the foregoing. 5 b) C1-C6, C2-C4 and C3-C5 disulfide bonding in which, as described above, C1, C2, C3, C4,
C5 and C6 represent the order of cysteine residues appearing in the primary sequence of the toxin peptide stated conventionally with the N-terminus of the peptide(s) on the left, with the first and sixth cysteines in the amino acid sequence forming a disulfide bond, the second and fourth cysteines forming a disulfide bond, O and the third and fifth cysteines forming a disulfide bond. Examples of toxin peptides with such a C1-C6, C2-C4, C3-C5 disulfide bonding pattern include, but are not limited to, ShK, BgK, HmK, AeKS, AsK1 and DTX1, and analogs of any of the foregoing, c) C1-C4, C2-C5 and C3-C6 disulfide bonding in which, as described above, C1, C2, C3, C4, C5 and C6 represent the order of cysteine residues appearing in the primary sequence 5 of the toxin peptide stated conventionally with the N-terminus of the peptide(s) on the left, with the first and fourth cysteines in the amino acid sequence forming a disulfide bond, the second and fifth cysteines forming a disulfide bond, and the third and sixth cysteines forming a disulfide bond. Examples of toxin peptides with such a C1-C4, C2- C5, C3-C6 disulfide bonding pattern include, but are not limited to, ChTx, MgTx, 0SK1, O KTX1 , AgTx2, Pi2, Pi3, NTX, HgTxI , BeKMI , BmKTX, P01 , BmKK6, Tc32, Td ,
BmTxI, BmTX3, IbTx, P05, ScyTx, TsK, HaTxI, ProTXI, PaTX2, Ptu1, coGVIA, ωMVIIA, and SmIIIa, and analogs of any of the foregoing.
d) C1-C5, C2-C6, C3-C7, and C4-C8 disulfide bonding in which C1, C2, C3, C4, C5, C5, C7 and C8 represent the order of cysteine residues appearing in the primary sequence of the toxin peptide stated conventionally with the N-terminus of the peptide(s) on the left, with the first and fifth cysteines in the amino acid sequence forming a disulfide bond, the second and sixth cysteines forming a disulfide bond, the third and seventh cysteines forming a disulfide bond, and the fourth and eighth cysteines forming a disulfide bond. Examples of toxin peptides with such a C1-C5, C2-C6, C3-C7, C4-C8 disulfide bonding pattern include, but are not limited to, Anuoroctoxin (AnTx), Pi1, HsTxI1 MTX (P12A, P20A), and Pi4 peptides, and analogs of any of the foregoing.0 e) C1-C4, C2-C6, C3-C7, and C5-C8 disulfide bonding in which C1, C2, C3, C4, C5, C6, C7 and C8 represent the order of cysteine residues appearing in the primary sequence of the toxin peptide stated conventionally with the N-terminus of the peptide(s) on the left, with the first and fourth cysteines in the amino acid sequence forming a disulfide bond, the second and sixth cysteines forming a disulfide bond, the third and seventh 5 cysteines forming a disulfide bond, and the fifth and eighth cysteines forming a disulfide bond. Examples of toxin peptides with such a C1-C4, C2-C6, C3-C7, C5-C8 disulfide bonding pattern include, but are not limited to, Chlorotoxin, Bm-12b, and, and analogs of either, f) C1-C5, C2-C6, C3-C4, and C7-C8 disulfide bonding in which C1, C2, C3, C4, C5, C6, C7 O and C8 represent the order of cysteine residues appearing in the primary sequence of the toxin peptide stated conventionally with the N-terminus of the peptide(s) on the left, with the first and fifth cysteines in the amino acid sequence forming a disulfide bond, the second and sixth cysteines forming a disulfide bond, the third and fourth cysteines forming a disulfide bond, and the seventh and eighth cysteines forming a 5 disulfide bond. Examples of toxin peptides with such a C1-C5, C2-C6, C3-C4, C7-C8 disulfide bonding pattern include, but are not limited to, Maurotoxin peptides and analogs thereof.
The term "randomized" as used to refer to peptide sequences refers to fully random sequences (e.g., selected by phage display methods) and sequences in which one or more O residues of a naturally occurring molecule is replaced by an amino acid residue not appearing in that position in the naturally occurring molecule. Exemplary methods for identifying peptide sequences include phage display, E. coli display, ribosome display, yeast-based screening, RNA- peptide screening, chemical screening, rational design, protein structural analysis, and the like.
The term "pharmacologically active" means that a substance so described is determined to have activity that affects a medical parameter (e.g., blood pressure, blood cell count, cholesterol level) or disease state (e.g., cancer, autoimmune disorders). Thus, pharmacologically active peptides comprise agonistic or mimetic and antagonistic peptides as defined below. 5 The terms "-mimetic peptide" and "-agonist peptide" refer to a peptide having biological activity comparable to a naturally occurring toxin peptide molecule, e.g., naturally occurring ShK toxin peptide. These terms further include peptides that indirectly mimic the activity of a naturally occurring toxin peptide molecule, such as by potentiating the effects of the naturally occurring molecule. 0 The term "antagonist peptide" or "inhibitor peptide" refers to a peptide that blocks or in some way interferes with the biological activity of a receptor of interest, or has biological activity comparable to a known antagonist or inhibitor of a receptor of interest (such as, but not limited to, an ion channel).
The term "acidic residue" refers to amino acid residues in D- or L-form having sidechains 5 comprising acidic groups. Exemplary acidic residues include D and E.
The term "amide residue" refers to amino acids in D- or L-form having sidechains comprising amide derivatives of acidic groups. Exemplary residues include N and Q.
The term "aromatic residue" refers to amino acid residues in D- or L-form having sidechains comprising aromatic groups. Exemplary aromatic residues include F, Y, and W. O The term "basic residue" refers to amino acid residues in D- or L-form having sidechains comprising basic groups. Exemplary basic residues include H, K, R, N-methyl-arginine, ω- aminoarginine, ω-methyl-arginine, 1-methyl-histidine, 3-methyl-histidine, and homoarginine (hR) residues.
The term "hydrophilic residue" refers to amino acid residues in D- or L-form having 5 sidechains comprising polar groups. Exemplary hydrophilic residues include C1 S, T, N, Q, D, E, K, and citrulline (Cit) residues.
The term "nonfunctional residue" refers to amino acid residues in D- or L-form having sidechains that lack acidic, basic, or aromatic groups. Exemplary nonfunctional amino acid residues include M, G, A, V, I, L and norleucine (NIe). O The term "neutral polar residue" refers to amino acid residues in D- or L-form having sidechains that lack basic, acidic, or polar groups. Exemplary neutral polar amino acid residues include A, V, L, I1 P, W, M, and F.
The term "polar hydrophobic residue" refers to amino acid residues in D- or L-form having sidechains comprising polar groups. Exemplary polar hydrophobic amino acid residues include T, G, S1 Y1 C1 Q1 and N.
The term "hydrophobic residue" refers to amino acid residues in D- or L-form having sidechains that lack basic or acidic groups. Exemplary hydrophobic amino acid residues include A, V1 L1 11 P, W, M1 F1 T1 G1 S, Y, C, Q1 and N.
In some useful embodiments of the compositions of the invention, the amino acid sequence of the toxin peptide is modified in one or more ways relative to a native toxin peptide sequence of interest, such as, but not limited to, a native ShK or 0SK1 sequence, their peptide analogs, or any other toxin peptides having amino acid sequences as set for in any of Tables 1-32. The one or more useful modifications can include amino acid additions or insertions, amino acid deletions, peptide truncations, amino acid substitutions, and/or chemical derivatization of amino acid residues, accomplished by known chemical techniques. Such modifications can be, for example, for the purpose of enhanced potency, selectivity, and/or proteolytic stability, or the like. Those skilled in the art are aware of techniques for designing peptide analogs with such enhanced properties, such as alanine scanning, rational design based on alignment mediated mutagenesis using known toxin peptide sequences and/or molecular modeling. For example, ShK analogs can be designed to remove protease cleavage sites (e.g., trypsin cleavage sites at K or R residues and/or chymotrypsin cleavage sites at F1 Y, or W residues) in a ShK peptide- or ShK analog- containing composition of the invention, based partially on alignment mediated mutagenesis using HmK (see, e.g., Figure 6) and molecular modeling. (See, e.g., Kalman et al., ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide, J. Biol. Chem. 273(49):32697-707 (1998); Kern et al., US Patent No. 6,077,680; Mouhat et al., 0sK1 derivatives, WO 2006/002850 A2)).
The term "protease" is synonymous with "peptidase". Proteases comprise two groups of enzymes: the endopeptidases which cleave peptide bonds at points within the protein, and the exopeptidases, which remove one or more amino acids from either N- or C-terminus respectively. The term "proteinase" is also used as a synonym for endopeptidase. The four mechanistic classes of proteinases are: serine proteinases, cysteine proteinases, aspartic proteinases, and metallo- proteinases. In addition to these four mechanistic classes, there is a section of the enzyme nomenclature which is allocated for proteases of unidentified catalytic mechanism. This indicates that the catalytic mechanism has not been identified.
Cleavage subsite nomenclature is commonly adopted from a scheme created by Schechter and Berger (Schechter I. & Berger A., On the size of the active site in proteases.
I. Papain, Biochemical and Biophysical Research Communication, 27:157 (1967); Schechter I. & Berger A., On the active site of proteases. 3. Mapping the active site of papain; specific inhibitor peptides of papain, Biochemical and Biophysical Research Communication, 32:898 (1968)). According to this model, amino acid residues in a substrate undergoing cleavage are designated 5 P1 ,P2, P3, P4 etc. in the N-terminal direction from the cleaved bond. Likewise, the residues in the C-terminal direction are designated PV1 P2', P3', P41. etc.
The skilled artisan is aware of a variety of tools for identifying protease binding or protease cleavage sites of interest. For example, the PeptideCutter software tool is available through the ExPASy (Expert Protein Analysis System) proteomics server of the Swiss Institute of 0 Bioinformatics (SIB; www.expasy.org/tools/peptidecutter). PeptideCutter searches a protein sequence from the SWISS-PROT and/or TrEMBL databases or a user-entered protein sequence for protease cleavage sites. Single proteases and chemicals, a selection or the whole list of proteases and chemicals can be used. Different forms of output of the results are available: tables of cleavage sites either grouped alphabetically according to enzyme names or sequentially 5 according to the amino acid number. A third option for output is a map of cleavage sites. The sequence and the cleavage sites mapped onto it are grouped in blocks, the size of which can be chosen by the user. Other tools are also known for determining protease cleavage sites. (E.g., Turk, B. et al., Determination of protease cleavage site motifs using mixture-based oriented peptide libraries, Nature Biotechnology, 19:661-667 (2001); Barrett A. et al., Handbook of proteolytic O enzymes, Academic Press (1998)).
The serine proteinases include the chymotrypsin family, which includes mammalian protease enzymes such as chymotrypsin, trypsin or elastase or kallikrein. The serine proteinases exhibit different substrate specificities, which are related to amino acid substitutions in the various enzyme subsites interacting with the substrate residues. Some enzymes have an extended 5 interaction site with the substrate whereas others have a specificity restricted to the P1 substrate residue.
Trypsin preferentially cleaves at R or K in position P1. A statistical study carried out by Keil (1992) described the negative influences of residues surrounding the Arg- and Lys- bonds (i.e. the positions P2 and P1', respectively) during trypsin cleavage. (Keil, B., Specificity of proteolysis, O Springer-Verlag Berlin-Heidelberg-NewYork, 335 (1992)). A proline residue in position PV normally exerts a strong negative influence on trypsin cleavage. Similarly, the positioning of R and K in PV results in an inhibition, as well as negatively charged residues in positions P2 and PV.
Chymotrypsin preferentially cleaves at a W1 Y or F in position P1 (high specificity) and to a lesser extent at L, M or H residue in position P1. (Keil, 1992). Exceptions to these rules are the following: When W is found in position P1, the cleavage is blocked when M or P are found in position PV at the same time. Furthermore, a proline residue in position PV nearly fully blocks the cleavage independent of the amino acids found in position P1. When an M residue is found in position P1, the cleavage is blocked by the presence of a Y residue in position PV. Finally, when H is located in position P1 , the presence of a D, M or W residue also blocks the cleavage.
Membrane metallo-endopeptidase (NEP; neutral endopeptidase, kidney-brush-border neutral proteinase, enkephalinase, EC 3.4.24.11) cleaves peptides at the amino side of hydrophobic amino acid residues. (Connelly, JC et al., Neutral Endopeptidase 24.11 in Human Neutrophils: Cleavage of Chemotactic Peptide, PNAS, 82(24):8737-8741 (1985)).
Thrombin preferentially cleaves at an R residue in position P1. (Keil, 1992). The natural substrate of thrombin is fibrinogen. Optimum cleavage sites are when an R residue is in position P1 and GIy is in position P2 and position PV. Likewise, when hydrophobic amino acid residues are found in position P4 and position P3, a proline residue in position P2, an R residue in position P1, and non-acidic amino acid residues in position PV and position P2'. A very important residue for its natural substrate fibrinogen is a D residue in P10.
Caspases are a family of cysteine proteases bearing an active site with a conserved amino acid sequence and which cleave peptides specifically following D residues. (Earnshaw WC et al., Mammalian caspases: Structure, activation, substrates, and functions during apoptosis, Annual Review of Biochemistry, 68:383-424 (1999)).
The Arg-C proteinase preferentially cleaves at an R residue in position P1. The cleavage behavior seems to be only moderately affected by residues in position PV. (Keil, 1992). The Asp- N endopeptidase cleaves specifically bonds with a D residue in position PV. (Keil, 1992). Furin is a ubiquitous subtilisin-like proprotein convertase. It is the major processing enzyme of the secretory pathway and intracellular^ is localized in the trans-golgi network (van den Ouweland, A.M.W. et al. (1990) Nucl. Acids Res., 18, 664; Steiner, D.F. (1998) Curr. Opin. Chem. Biol., 2, 31-39). The minimal furin cleavage site is Arg-X-X-Arg1. However, the enzyme prefers the site Arg-X-(Lys/Arg)-Arg'. An additional arginine at the P6 position appears to enhance cleavage (Krysan, D.J. et al. (1999) J. Biol. Chem., 274, 23229-23234).
The foregoing is merely exemplary and by no means an exhaustive treatment of knowledge available to the skilled artisan concerning protease binding and/or cleavage sites that the skilled artisan may be interested in eliminating in practicing the invention.
Additional useful embodiments of the toxin peptide, e.g., the 0SK1 peptide analog, can result from conservative modifications of the amino acid sequences of the peptides disclosed herein. Conservative modifications will produce peptides having functional, physical, and chemical characteristics similar to those of the parent peptide from which such modifications are made. 5 Such conservatively modified forms of the peptides disclosed herein are also contemplated as being an embodiment of the present invention.
In contrast, substantial modifications in the functional and/or chemical characteristics of the toxin peptides may be accomplished by selecting substitutions in the amino acid sequence that differ significantly in their effect on maintaining (a) the structure of the molecular backbone in the 0 region of the substitution, for example, as an α-helical conformation, (b) the charge or hydrophobicity of the molecule at the target site, or (c) the size of the molecule.
For example, a "conservative amino acid substitution" may involve a substitution of a native amino acid residue with a nonnative residue such that there is little or no effect on the polarity or charge of the amino acid residue at that position. Furthermore, any native residue in the 5 polypeptide may also be substituted with alanine, as has been previously described for "alanine scanning mutagenesis" (see, for example, MacLennan et al., Acta Physiol. Scand. Suppl., 643:55- 67 (1998); Sasaki et al., 1998, Adv. Biophys. 35:1-24 (1998), which discuss alanine scanning mutagenesis).
Desired amino acid substitutions (whether conservative or non-conservative) can be O determined by those skilled in the art at the time such substitutions are desired. For example, amino acid substitutions can be used to identify important residues of the peptide sequence, or to increase or decrease the affinity of the peptide or vehicle-conjugated peptide molecules described herein.
Naturally occurring residues may be divided into classes based on common side chain 5 properties:
1) hydrophobic: norleucine (Nor), Met, AIa1 VaI, Leu, He;
2) neutral hydrophilic: Cys, Ser, Thr, Asn, GIn;
3) acidic: Asp, GIu;
4) basic: His, Lys, Arg; 0 5) residues that influence chain orientation: GIy, Pro; and
6) aromatic: Trp, Tyr, Phe.
Conservative amino acid substitutions may involve exchange of a member of one of these classes with another member of the same class. Conservative amino acid substitutions may encompass
non-naturally occurring amino acid residues, which are typically incorporated by chemical peptide synthesis rather than by synthesis in biological systems. These include peptidomimetics and other reversed or inverted forms of amino acid moieties.
Non-conservative substitutions may involve the exchange of a member of one of these classes for a member from another class. Such substituted residues may be introduced into regions of the human antibody that are homologous with non-human antibodies, or into the nonhomologous regions of the molecule.
In making such changes, according to certain embodiments, the hydropathic index of amino acids may be considered. Each amino acid has been assigned a hydropathic index on the basis of its hydrophobicity and charge characteristics. They are: isoleucine (+4.5); valine (+4.2); leucine (+3.8); phenylalanine (+2.8); cysteine/cystine (+2.5); methionine (+1.9); alanine (+1.8); glycine (-0.4); threonine (-0.7); serine (-0.8); tryptophan (-0.9); tyrosine (-1.3); proline (-1.6); histidine (-3.2); glutamate (-3.5); glutamine (-3.5); aspartate (-3.5); asparagine (-3.5); lysine (-3.9); and arginine (-4.5). The importance of the hydropathic amino acid index in conferring interactive biological function on a protein is understood in the art (see, for example, Kyte ef a/., 1982, J. MoI. Biol. 157:105-131). It is known that certain amino acids may be substituted for other amino acids having a similar hydropathic index or score and still retain a similar biological activity. In making changes based upon the hydropathic index, in certain embodiments, the substitution of amino acids whose hydropathic indices are within ±2 is included. In certain embodiments, those that are within ±1 are included, and in certain embodiments, those within ±0.5 are included.
It is also understood in the art that the substitution of like amino acids can be made effectively on the basis of hydrophilicity, particularly where the biologically functional protein or peptide thereby created is intended for use in immunological embodiments, as disclosed herein. In certain embodiments, the greatest local average hydrophilicity of a protein, as governed by the hydrophilicity of its adjacent amino acids, correlates with its immunogenicity and antigenicity, i.e., with a biological property of the protein.
The following hydrophilicity values have been assigned to these amino acid residues: arginine (+3.0); lysine (+3.0); aspartate (+3.0 ± 1); glutamate (+3.0 ± 1); serine (+0.3); asparagine (+0.2); glutamine (+0.2); glycine (0); threonine (-0.4); proline (-0.5 ± 1); alanine (-0.5); histidine
(-0.5); cysteine (-1.0); methionine (-1.3); valine (-1.5); leucine (-1.8); isoleucine (-1.8); tyrosine (-2.3); phenylalanine (-2.5) and tryptophan (-3.4). In making changes based upon similar hydrophilicity values, in certain embodiments, the substitution of amino acids whose hydrophilicity
values are within ±2 is included, in certain embodiments, those that are within ±1 are included, and in certain embodiments, those within ±0.5 are included. One may also identify epitopes from primary amino acid sequences on the basis of hydrophilicity. These regions are also referred to as "epitopic core regions."
Examples of conservative substitutions include the substitution of one non-polar (hydrophobic) amino acid residue such as isoleucine, valine, leucine norleucine, alanine, or methionine for another, the substitution of one polar (hydrophilic) amino acid residue for another such as between arginine and lysine, between glutamine and asparagine, between glycine and serine, the substitution of one basic amino acid residue such as lysine, arginine or histidine for another, or the substitution of one acidic residue, such as aspartic acid or glutamic acid for another. The phrase "conservative amino acid substitution" also includes the use of a chemically derivatized residue in place of a non-derivatized residue, provided that such polypeptide displays the requisite biological activity. Other exemplary amino acid substitutions that can be useful in accordance with the present invention are set forth in Table 1 A.
Table 1A. Some Useful Amino Acid Substitutions
In other examples, a toxin peptide amino acid sequence, e.g., an 0SK1 peptide analog sequence, modified from a naturally occurring toxin peptide amino acid sequence includes at least one amino acid residue inserted or substituted therein, relative to the amino acid sequence of the native toxin peptide sequence of interest, in which the inserted or substituted amino acid residue has a side chain comprising a nucleophilic or electrophilic reactive functional group by which the peptide is conjugated to a linker or half-life extending moiety. In accordance with the invention, useful examples of such a nucleophilic or electrophilic reactive functional group include, but are not limited to, a thiol, a primary amine, a seleno, a hydrazide, an aldehyde, a carboxylic acid, a ketone, an aminooxy, a masked (protected) aldehyde, or a masked (protected) keto functional group.
Examples of amino acid residues having a side chain comprising a nucleophilic reactive functional group include, but are not limited to, a lysine residue, an α,β-diaminopropionic acid residue, an α,γ-diaminobutyric acid residue, an ornithine residue, a cysteine, a homocysteine, a glutamic acid residue, an aspartic acid residue, or a selenocysteine residue. In some embodiments, the toxin peptide amino acid sequence (or "primary sequence") is modified at one, two, three, four, five or more amino acid residue positions, by having a residue substituted therein different from the native primary sequence (e.g., 0SK1 SEQ ID NO:25) or omitted (e.g., an 0SK1 peptide analog optionally lacking a residue at positions 36, 37, 36-38, 37-38, or 38).
In further describing toxin peptides herein, a one-letter abbreviation system is frequently applied to designate the identities of the twenty "canonical" amino acid residues generally incorporated into naturally occurring peptides and proteins (Table 1B). Such one-letter
abbreviations are entirely interchangeable in meaning with three-letter abbreviations, or non- abbreviated amino acid names. Within the one-letter abbreviation system used herein, an uppercase letter indicates a L-amino acid, and a lower case letter indicates a D-amino acid, unless otherwise noted herein. For example, the abbreviation "R" designates L-arginine and the abbreviation "r" designates D-arginine.
Table 1 B. One-letter abbreviations for the canonical amino acids Three-letter abbreviations are in parentheses Alanine (Ala) A
Glutamine (GIn) Q
Leucine (Leu) L
Serine (Ser) S
Arginine (Arg) R Glutamic Acid (GIu) E
Lysine (Lys) K
Threonine (Thr) T
Asparagine (Asn) N
Glycine (GIy) G Methionine (Met) M
Tryptophan (Trp) W
Aspartic Acid (Asp) D
Histidine (His) H
Phenylalanine (Phe) F Tyrosine (Tyr) Y
Cysteine (Cys) C lsoleucine (He) I
Proline (Pro) P
Valine (VaI) V
An amino acid substitution in an amino acid sequence is typically designated herein with a one-letter abbreviation for the amino acid residue in a particular position, followed by the numerical amino acid position relative to the native toxin peptide sequence of interest, which is then followed by the one-letter symbol for the amino acid residue substituted in. For example, "T30D" symbolizes a substitution of a threonine residue by an aspartate residue at amino acid position 30, relative to a hypothetical native toxin peptide sequence. By way of further example, "R18hR" or "R18Cit" indicates a substitution of an arginine residue by a homoarginine or a citrulline residue, respectively, at amino acid position 18, relative to the hypothetical native toxin peptide. An amino acid position within the amino acid sequence of any particular toxin peptide (or peptide analog) described herein may differ from its position relative to the native sequence, i.e., as determined in an alignment of the N-terminal or C-terminal end of the peptide's amino acid sequence with the N-terminal or C-terminal end, as appropriate, of the native toxin peptide sequence. For example,
amino acid position 1 of the sequence SCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC ShK(2-35); SEQ ID NO:92), a N-terminal truncation of the native ShK sequence, thus aligned with the C-terminal of native ShK(I -35) (SEQ ID N0:5), corresponds to amino acid position 2 relative to the native sequence, and amino acid position 34 of SEQ ID NO:92 corresponds to amino acid 5 position 35 relative to the native sequence (SEQ ID N0:5).
In certain embodiments of the present invention, amino acid substitutions encompass, non-canonical amino acid residues, which include naturally rare (in peptides or proteins) amino acid residues or unnatural amino acid residues. Non-canonical amino acid residues can be incorporated into the peptide by chemical peptide synthesis rather than by synthesis in biological 0 systems, such as recombinantly expressing cells, or alternatively the skilled artisan can employ known techniques of protein engineering that use recombinantly expressing cells. (See, e.g., Link et al., Non-canonical amino acids in protein engineering, Current Opinion in Biotechnology, 14(6):603-609 (2003)). The term "non-canonical amino acid residue" refers to amino acid residues in D- or L-form that are not among the 20 canonical amino acids generally incorporated into 5 naturally occurring proteins, for example, β-amino acids, homoamino acids, cyclic amino acids and amino acids with derivatized side chains. Examples include (in the L-form or D-form; abbreviated as in parentheses): citrulline (Cit), homocitrulline (hCit), Nα-methylcitrulline (NMeCit), Nα-methylhomocitrulline (Nα-MeHoCit), ornithine (Orn), Nα-Methylornithine (Nα-Me0rn or NMeOrn), sarcosine (Sar), homolysine (hLys or hK), homoarginine (hArg or hR), homoglutamine (hQ), O Nα-methylarginine (NMeR), Nα-methylleucine (Nα-MeL or NMeL), N-methylhomolysine (NMeHoK), Nα-methylglutamine (NMeQ), norleucine (NIe), norvaline (Nva), 1,2,3,4-tetrahydroisoquinoline (Tic), Octahydroindole-2-carboxylic acid (Oic), 3-{1-naphthyl)alanine (1-NaI), 3-(2-naphthyl)alanine (2-NaI), 1,2,3,4-tetrahydroisoquinoline (Tic), 2-indanylglycine (IgI), para-iodophenylalanine (pl-Phe), para-aminophenylalanine (4AmP or 4-Amino-Phe), 4-guanidino phenylalanine (Guf), 5 glycyllysine (abbreviated herein "K(Nε-glycyl)" or "K(glycyl)" or "K(gly)"), nitrophenylalanine
(nitrophe), aminophenylalanine (aminophe or Amino-Phe), benzylphenylalanine (benzylphe), γ-carboxyglutamic acid (γ-carboxyglu), hydroxyproline (hydroxypro), p-carboxyl-phenylalanine (Cpa), α-aminoadipic acid (Aad), Nα-methyl valine (NMeVaI), N-α-methyl leucine (NMeLeu), Nα-methylnorleucine (NMeNIe), cyclopentylglycine (Cpg), cyclohexylglycine (Chg), acetylarginine O (acetylarg), α, β-diaminopropionoic acid (Dpr), α, γ-diaminobutyric acid (Dab), diaminopropionic acid (Dap), cyclohexylalanine (Cha), 4-methyl-phenylalanine (MePhe), β, β-diphenyl-alanine (BiPhA), aminobutyric acid (Abu), 4-phenyl-phenylalanine (or biphenylalanine; 4Bip), α-amino- isobutyric acid (Aib), beta-alanine, beta-aminopropionic acid, piperidinic acid, aminocaprioic acid,
aminoheptanoic acid, aminopimelic acid, desmosine, diaminopimelic acid, N-ethylglycine, N-ethylaspargine, hydroxyzine, allo-hydroxylysine, isodesmosine, allo-isoleucine, N-methylglycine, N-methylisoleucine, N-methylvaline, 4-hydroxyproline (Hyp), γ-carboxyglutamate, ε-N,N,N-trimethyllysine, ε-N-acetyllysine, O-phosphoserine, N-acetylserine, N-formylmethionine, 3-methylhistidine, 5-hydroxylysine, ω-methylarginine, 4-Amino-O-Phthalic Acid (4APA), and other similar amino acids, and derivatized forms of any of these as described herein. Table 1 B contains some exemplary non-canonical amino acid residues that are useful in accordance with the present invention and associated abbreviations as typically used herein, although the skilled practitioner will understand that different abbreviations and nomenclatures may be applicable to the same substance and my appear interchangeably herein.
Table 1B. Useful non-canonical amino acids for amino acid addition, insertion, or substitution into toxin peptide sequences, including 0SK1 peptide analog sequences, in accordance with the present invention. In the event an abbreviation listed in Table 1 B differs from another abbreviation for the same substance disclosed elsewhere herein, both abbreviations are understood to be applicable.
Abbreviation Amino Acid
Sar Sarcosine
NIe norleucine
He isoleucine
1-Nal 3- (1-naphthyl) alanine
2-Nal 3- (2-naphthyl) alanine
Bip 4,4' -biphenyl alanine
Dip 3, 3-diphenylalanine
NvI norvaline
NMe-VaI Nα-methyl valine
NMe-Leu Nα-methyl leucine
NMe-NIe Nα-methyl norleucine
Cpg cyclopentyl glycine
Chg cyclohexyl glycine
Hyp hydroxy proline
Oic Octahydroindole-2-Carboxylic Acid
IgI Indanyl glycine
Aib aminoisobutyric acid
Aic 2-aminoindane-2-carboxylic acid
Pip pipecolic acid
BhTic β-homo Tic
BhPro β-homo proline
Sar Sarcosine
Cpg cyclopentyl glycine
1,2,3, 4-L-Tetrahydroisoquinoline-l-Carboxylic
Tiα acid
Nip Nipecotic Acid
Thz Thiazolidine-4-carboxylic acid
Thi 3-thienyl alanine
4GuaPr 4-guanidino proline
4Pip 4-Amino-l-piperidine-4-carboxylic acid
Idc indoline-2-carboxylic acid
1,-2, 3, 4-Tetrahydroisoquinoline-7-hydroxy-3-
Hydroxyl-Tic carboxylic acid Bip 4,4' -biphenyl alanine
Ome-Tyr 0-methyl tyrosine I-Tyr Iodotyrosine
1,2,3, 4-L-Tetrahydroisoquinoline-3-Carboxylic
Tic acid
IgI Indanyl glycine
BhTic β-homo Tic
BhPhe β-homo phenylalanine
AMeF α-methyl Phenyalanine
BPhe β-phenylalanine
Phg phenylglycine
Anc 3-amino-2-naphthoic acid
Ate 2-aminotetraline-2-carboxylic acid
NMe-Phe Nα-methyl phenylalanine
NMe-Lys Nα-methyl lysine
Tpi 1,2,3, 4-Tetrahydronorharman-3-Carboxylic acid
Cpg cyclopentyl glycine
Dip 3, 3-diphenylalanine
4PaI 4-pyridinylalanine
3PaI 3-pyridinylalanine
2PaI 2-pyridinylalanine
4Pip 4-Amino-l-piperidine-4-carboxylic acid
4AmP 4-amino-phenylalanine
Idc indoline-2-carboxylic acid
Chg cyclohexyl glycine hPhe homophenylalanine
BhTrp β-homotryptophan pl-Phe 4-iodophenylalanine
Aic 2-aminoindane-2-carboxylic acid
NMe-Lys Nα-methyl lysine
Orn ornithine
Dpr 2, 3-Diaminopropionic acid
Dbu 2, 4-Diaminobutyric acid homoLys homolysine
N-eMe-K Nε-methyl-lysine
N-eEt-K Nε-ethyl-lysine
N-eIPr-K Nε-isopropyl-lysine bhomoK β-homolysine rLys Lys ψ (CH2NH) -reduced amide bond rOrn Orn ψ (CH2NH) -reduced amide bond
Acm acetamidomethyl
Ahx 6-aminohexanoic acid
ε Ahx 6-aminohexanoic acid
Nε- (O- (aminoethyl) -O' - (2-propanoyl) -
K (NPegll) undecaethyleneglycol) -Lysine
Nε- (0- (aminoethyl) -0' - (2-propanoyl) -
K(NPeg27) (ethyleneglycol) 27-Lysine
Cit Citrulline hArg homoarginine hCit homocitrulline
NMe-Arg Nα-methyl arginine (NMeR)
Guf 4-guanidinyl phenylalanine bhArg β-homoarginine
3G-Dpr 2-amino-3-guanidinopropanoic acid
4AmP 4-amino-phenylalanine
4AmPhe 4-amidino-phenylalanine
2-amino-2- (l-carbamimidoylpiperidin-4-
4AmPig yl) acetic acid
4GuaPr 4-guanidino proline
N-Arg Na- [ (CH2J3NHCH(NH)NH2] substituted glycine rArg Arg ψ (CH2NH) -reduced amide bond
4PipA 4-Piperidinyl alanine
NMe-Arg Nα-methyl arginine (or NMeR)
NMe-Thr Nα-methyl threonine (or NMeThr)
Nomenclature and Symbolism for Amino Acids and Peptides by the UPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN) have been published in the following documents: Biochem. J., 1984, 219, 345-373; Eur. J. Biochem., 1984, 138, 9-37; 1985, 152, 1 ; 1993, 213, 2; Internat. J. Pept. Prot. Res., 1984, 24, following p 84; J. Biol. Chem., 1985,
260,14-42; Pure Appl. Chem., 1984, 56, 595-624; Amino Acids and Peptides, 1985, 16, 387-410; Biochemical Nomenclature and Related Documents, 2nd edition, Portland Press, 1992, pages 39-69].
As stated herein, in accordance with the present invention, peptide portions of the inventive compositions, such as the toxin peptide or a peptide linker, can also be chemically derivatized at one or more amino acid residues. Peptides that contain derivatized amino acid residues can be synthesized by known organic chemistry techniques. "Chemical derivative" or "chemically derivatized" in the context of a peptide refers to a subject peptide having one or more residues chemically derivatized by reaction of a functional side group. Such derivatized molecules include, for example, those molecules in which free amino groups have been derivatized to form amine hydrochlorides, p-toluene sulfonyl groups, carbobenzoxy groups, t-butyloxycarbonyl groups, chloroacetyl groups or formyl groups. Free carboxyl groups may be derivatized to form salts, methyl and ethyl esters or other types of esters or hydrazides. Free hydroxyl groups may be derivatized to form O-acyl or O-alkyl derivatives. The imidazole nitrogen of histidine may be
derivatized to form N-im-benzylhistidine. Also included as chemical derivatives are those peptides which contain one or more naturally occurring amino acid derivatives of the twenty canonical amino acids, whether in L- or D- form. For example, 4-hydroxyproline may be substituted for proline; 5-hydroxylysine maybe substituted for lysine; 3-methylhistidine may be substituted for histidine; homoserine may be substituted for serine; and ornithine may be substituted for lysine.
Useful derivatizations include, in some embodiments, those in which the amino terminal of the toxin peptide, such as but not limited to the 0SK1 peptide analog, is chemically blocked so that conjugation with the vehicle will be prevented from taking place at an N-terminal free amino group. There may also be other beneficial effects of such a modification, for example a reduction in the 0 toxin peptide's susceptibility to enzymatic proteolysis. The N-terminus of the toxin peptide, e.g., the 0SK1 peptide analog, can be acylated or modified to a substituted amine, or derivatized with another functional group, such as an aromatic or aryl moiety (e.g., an indole acid, benzyl (BzI or Bn), dibenzyl (DiBzI or Bn2), benzoyl, or benzyloxycarbonyl (Cbz or Z)), Λ/,Λ/-dimethylglycine or creatine. For example, in some embodiments, an acyl moiety, such as, but not limited to, a formyl, 5 acetyl (Ac), propanoyl, butanyl, heptanyl, hexanoyl, octanoyl, or nonanoyl, can be covalently linked to the N-terminal end of the peptide, e.g., the 0SK1 peptide analog, which can prevent undesired side reactions during conjugation of the vehicle to the peptide. Alternatively, a fatty acid (e.g. butyric, caproic, caprylic, capric, lauric, myristic, palmitic, stearic or the like) or polyethylene glycol moiety can be covalently linked to the N-terminal end of the peptide, e.g., the 0SK1 peptide O analog. Other exemplary N-terminal derivative groups include -NRR1 (other than -NH2), -
NRC(O)R1, -NRC(O)OR1, -NRS(O)2R1, -NHC(O)NHR1, succinimide, or benzyloxycarbonyl-NH- (Cbz-NH-), wherein R and R1 are each independently hydrogen or lower alkyl and wherein the phenyl ring may be substituted with 1 to 3 substituents selected from C1-C4 alkyl, C1-C4 alkoxy, chloro, and bromo. 5 In some embodiments of the present invention, basic residues (e.g., lysine) of the toxin peptide of interest can be replaced with other residues (nonfunctional residues preferred). Such molecules will be less basic than the molecules from which they are derived and otherwise retain the activity of the molecules from which they are derived, which can result in advantages in stability and immunogenicity; the present invention should not, however, be limited by this theory. O Additionally, physiologically acceptable salts of the inventive compositions are also encompassed, including when the inventive compositons are referred to herein as "molecules" or "compounds.". By "physiologically acceptable salts" is meant any salts that are known or later discovered to be pharmaceutically acceptable. Some non-limiting examples of pharmaceutically
acceptable salts are: acetate; trifluoroacetate; hydrohalides, such as hydrochloride and hydrobromide; sulfate; citrate; maleate; tartrate; glycolate; gluconate; succinate; mesylate; besylate; salts of gallic acid esters (gallic acid is also known as 3,4, 5 trihydroxybenzoic acid) such as PentaGalloylGlucose (PGG) and epigallocatechin gallate (EGCG), salts of cholesteryl sulfate, pamoate, tannate and oxalate salts.
Structure of compounds:
In general. Recombinant proteins have been developed as therapeutic agents through, among other means, covalent attachment to half-life extending moieties. Such moieties include the "Fc" domain of an antibody, as is used in Enbrel® (etanercept) , as well as biologically suitable polymers (e.g., polyethylene glycol, or "PEG"), as is used in Neulasta® (pegfilgrastim). Feige ef a/, described the use of such half-life extenders with peptides in U.S. Patent No. 6,660,843, issued December 9, 2003 (hereby incorporated by reference in its entirety).
The present inventors have determined that molecules of this invention— peptides of about 80 amino acids or less with at least two intrapeptide disulfide bonds— possess therapeutic advantages when covalently attached to half-life extending moieties. Molecules of the present invention can further comprise an additional pharmacologically active, covalently bound peptide, which can be bound to the half-life extending moiety (F1and/or F2) or to the peptide portion (P). Embodiments of the inventive compositions containing more than one half-life extending moiety (F1 and F2) include those in which F1 and F2 are the same or different half-life extending moieties. Examples (with or without a linker between each domain) include structures as illustrated in Figure 75 as well as the following embodiments (and others described herein and in the working Examples):
20KPEG - toxin peptide - Fc domain, consistent with the formula [(F1)i-(X2)i-(F2)i];
20KPEG - toxin peptide - Fc CH2 domain, consistent with the formula [(F1)i-(X2)i-(F2)i]; 20KPEG - toxin peptide - HSA, consistent with the formula [(F1)i-(X2)i-(F2)i];
20KPEG - Fc domain- toxin peptide, consistent with the formula [(F1)i-(F2)i-(X3)i]; 20KPEG - Fc CH2 domain- toxin peptide, consistent with the formula [(F1)i-(F2)i-(X3)i]; and
20KPEG - HSA - toxin peptide, consistent with the formula [(F1)i-(F2)i-(X3)i]. Toxin peptides. Any number of toxin peptides (i.e., "P", or equivalently shown as "P1" in
Figure 2) can be used in conjunction with the present invention. Of particular interest are the toxin peptides ShK, HmK, MgTx, AgTx2, Agatoxins, and HsTxI, as well as modified analogs of these, in particular OsK1 (also referred to as "OSK1") peptide analogs of the present invention, and other
peptides that mimic the activity of such toxin peptides. As stated herein above, if more than one toxin peptide "P" is present in the inventive composition, "P" can be independently the same or different from any other toxin peptide(s) also present in the inventive composition. For example, in a composition having the formula P-(L)g-F1-(L)rP, both of the toxin peptides, "P", can be the same peptide analog of ShK, different peptide analogs of ShK, or one can be a peptide analog of ShK and the other a peptide analog of 0SK1. In a preferred embodiment, at least one P is a an 0SK1 peptide analog as further described herein.
In some embodiments of the invention, other peptides of interest are especially useful in molecules having additional features over the molecules of structural Formula I. In such molecules, the molecule of Formula I further comprises an additional pharmacologically active, covalently bound peptide, which is an agonistic peptide, an antagonistic peptide, or a targeting peptide; this peptide can be conjugated to F1 or F2 or P. Such agonistic peptides have activity agonistic to the toxin peptide but are not required to exert such activity by the same mechanism as the toxin peptide. Peptide antagonists are also useful in embodiments of the invention, with a preference for those with activity that can be complementary to the activity of the toxin peptide. Targeting peptides are also of interest, such as peptides that direct the molecule to particular cell types, organs, and the like. These classes of peptides can be discovered by methods described in the references cited in this specification and other references. Phage display, in particular, is useful in generating toxin peptides for use in the present invention. Affinity selection from libraries of random peptides can be used to identify peptide ligands for any site of any gene product. Dedman et al. (1993), J. Biol. Chem. 268: 23025-30. Phage display is particularly well suited for identifying peptides that bind to such proteins of interest as cell surface receptors or any proteins having linear epitopes. Wilson et_aL (1998), Can. J. Microbiol. 44: 313-29; Kay et_al. (1998), Drug Disc. Today 3: 370-8. Such proteins are extensively reviewed in Herz et_a[. (1997), J. Receptor and Signal Transduction Res. 17(5): 671-776, which is hereby incorporated by reference in its entirety.
Such proteins of interest are preferred for use in this invention.
Particularly preferred peptides appear in the following tables. These peptides can be prepared by methods disclosed in the art or as described hereinafter. Single letter amino acid abbreviations are used. Unless otherwise specified, each X is independently a nonfunctional residue.
Table 1— Kv1.3 inhibitor peptide sequences
Many peptides as described in Table 2 can be prepared as described in U.S. Pat. No. 6,077,680 issued June 20, 2000 to Kem et al., which is hereby incorporated by reference in its entirety. Other peptides of Table 2 can be prepared by techniques known in the art. For example, ShK(L5) (SEQ ID NO: 950) can be prepared as described in Beeton et al., Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases, Molec. Pharmacol.67(4): 1369- 81 (2005), which is hereby incorporated by reference in its entirety. In Table 2 and throughout the specification, X
s15, X
s21, X
s22, X
s23 and X
s27 each independently refer to nonfunctional amino acid residues.
Table 3— HmK, BgK, AeK and AsKS peptide and peptide analog sequences
In Table 3 and throughout the specification, Xh6, Xh22, Xh26 are each independently nonfunctional residues.
Table 4— MgTx peptide and MgTx peptide analog sequences
Many peptides as described in Table 4 can be prepared as described in WO 95/03065, published February 2, 1995, for which the applicant is Merck & Co., Inc. That application corresponds to U.S. Ser. No. 07/096,942, filed 22 July 1993, which is hereby incorporated by reference in its entirety.
Table 5— AgTx2 peptide and AgTx2 peptide analog sequences
Peptides as described in Table 5 can be prepared as described in U.S. Pat. No. 6,689,749, issued February 10, 2004 to Lebrun et al., which is hereby incorporated by reference in its entirety.
Table 7— Orthochirus scrobiculosus (OSK1) peptide and OSK1 peptide analog sequences
Table 7A. Additional useful 0SK1 peptide analog sequences
1613 Ac-GVIINVKCKISRQCLEPCKKAGMRFGKCMNGKCECTPK
1614 Ac-GVIINVKCKISRQCLEPCKKAGMRFGKCMNGKCHCEPK
1615 Ac-GVIINVKCKISRQCLEPCKKAGMRFGKCMNGKCHCTEK
1616 Ac-GVIINVKCKISRQCLEPCKKAGMRFGKCMNGKCHCTPE
1617 Ac- [ 1-Nal ] VIINVKCKISRQCLEPCKKAGMRFGKCMNGKCHCTPK
1618 AC-G[I-NaI]IINVKCKISRQCLEPCKKAGMRFGKCMNGKCHCTPK
1619 Ac-GV[l-Nal]INVKCKISRQCLEPCKKAGMRFGKCMNGKCHCTPK
1620 Ac-GVI [1-NaI]NVKCKISRQCLEPCKKAGMRFGKCMNGKCHCTPK
1621 AC-GVII[I-NaI]VKCKISRQCLEPCKKAGMRFGKCMNGKCHCTPK
1622 AC-GVIIN[I-NaI]KCKISRQCLEPCKKAGMRFGKCMNGKCHCTPK
1623 Ac-GVIINV [1-NaI]CKISRQCLEPCKKAGMRFGKCMNGKCHCTPK
1624 AC-GVIINVKC[I-NaI]ISRQCLEPCKKAGMRFGKCMNGKCHCTPK
1625 AC-GVIINVKCK[I-NaI]SRQCLEPCKKAGMRFGKCMNGKCHCTPK
1626 Ac-GVIINVKCKI [1-Nal] RQCLEPCKKAGMRFGKCMNGKCHCTPK
1627 AC-GVIINVKCKIS[I-NaI]QCLEPCKKAGMRFGKCMNGKCHCTPK
1628 AC-GVIINVKCKISR[I-NaI]CLEPCKKAGMRFGKCMNGKCHCTPK
1629 AC-GVIINVKCKISRQC[I-NaI]EPCKKAGMRFGKCMNGKCHCTPK
1630 AC-GVIINVKCKISRQCL[I-NaI]PCKKAGMRFGKCMNGKCHCTPK
1631 AC-GVIINVKCKISRQCLE[I-NaI]CKKAGMRFGKCMNGKCHCTPK
1632 AC-GVIINVKCKISRQCLEPC[I-NaI]KAGMRFGKCMNGKCHCTPK
1633 Ac-GVI INVKCKISRQCLEPCK [1-Nal ] AGMRFGKCMNGKCHCTPK
1634 AC-GVIINVKCKISRQCLEPCKK[I-NaI]GMRFGKCMNGKCHCTPK
1635 AC-GVIINVKCKISRQCLEPCKKA[I-NaI]MRFGKCMNGKCHCTPK
1636 AC-GVIINVKCKISRQCLEPCKKAG[I-NaI]RFGKCMNGKCHCTPK
1637 Ac-GVIINVKCKISRQCLEPCKKAGM[l-Nal]FGKCMNGKCHCTPK
1638 AC-GVIINVKCKISRQCLEPCKKAGMR[I-NaI]GKCMNGKCHCTPK
1639 AC-GVIINVKCKISRQCLEPCKKAGMRF[I-NaI]KCMNGKCHCTPK
1640 Ac-GVIINVKCKISRQCLEPCKKAGMRFG[1-NaI] CMNGKCHCTPK
Table 7B —Additional useful 0SK1 peptide analog sequences: Ala-12 Substituted Series
Table 7C —Additional useful OSK1 peptide analog sequences: Ala-12 & Ala27
Substituted Series
Table 7D —Additional useful OSK1 peptide analog sequences: Ala-12 & Ala29
Substituted Series
Table 7E - Additional useful 0SK1 peptide analogs: Ala-12 & Ala30 Substituted Series
Table 7G - Additional useful OSK1 peptide analogs: Ala 29 Substituted Series
Table 7I - Additional useful OSK1 peptide analogs: Combined Ala-11,12, 27,29,30
Substituted Series
Table 7 J - Additional useful OSK1 peptide analogs
Any OSK1 peptide analog that comprises an amino acid sequence selected from SEQ ID NOS: 1391 through 4912, 4916, 4920 through 5006, 5009, 5010, and 5012 through 5015 as set forth in Tables 7, 7A, 7B, 7C1 7D, 7E, 7F, 7G, 7H or 71, is useful in accordance with the present invention. Any of these can also be derivatized at either its N-terminal or C-terminal, e.g., with a fatty acid having from 4 to 10 carbon atoms and from 0 to 2 carbon-carbon double bonds, or a derivative thereof such as an ω-amino-fatty acid. (E.g., Mouhat et a/., WO 2006/002850 A2, which is incorporated by reference in its entirety). Examples of such fatty acids include valeric acid or (for the C-terminal) ω-amino-valeric acid.
Among useful OSK1 peptide analog sequences of the present invention are analog sequences that introduce amino acid residues that can form an intramolecular covalent bridge (e.g., a disulfide bridge) or non-covalent interactions (e.g. hydrophobic, ionic, stacking) between the first and last beta strand, which may enhance the stability of the structure of the unconjugated or conjugated (e.g., PEGylated) OSK1 peptide analog molecule. Examples of such sequences include SEQ ID NOS: 4985-4987 and 5012-5015.
In some embodiments of the composition of matter, the C-terminal carboxylic acid moiety of the 0SK1 peptide analog is replaced with a moiety selected from:
(A) -COOR, where R is independently (Ci-C8)alkyl, haloalkyl, aryl or heteroaryl;
(B) -C(=O)NRR, where R is independently hydrogen, (Ci-Cβ)alkyl, haloalkyl, aryl or heteroaryl; and
(C) -CH2OR where R is hydrogen, (d-Cβ) alkyl, aryl or heteroaryl.
"Aryl" is phenyl or phenyl vicinally-fused with a saturated, partially-saturated, or unsaturated 3-, 4-, or 5 membered carbon bridge, the phenyl or bridge being substituted by 0, 1, 2 or 3 substituents selected from Ci 8 alkyl, Ci 4 haloalkyl or halo. "Heteroaryl" is an unsaturated 5 , 6 or 7 membered monocyclic or partially-saturated or unsaturated 6-, 7-, 8-, 9-, 10- or 11 membered bicyclic ring, wherein at least one ring is unsaturated, the monocyclic and the bicyclic rings containing 1, 2, 3 or 4 atoms selected from N1 0 and S, wherein the ring is substituted by 0, 1 , 2 or 3 substituents selected from Ci β alkyl, Ci 4 haloalkyl and halo. In other embodiments of the composition of matter comprising a half-life extending moiety, the 0SK1 peptide analog comprises an amino acid sequence of the formula:
G1V2l3l4N5V6K7C8K9l10Xaa11Xaa12Q13C14Xaa15Xaal6P17Cl8Xaa19Xaa20A21G22M23R24F25G26 Xaa27C28Xaa29Xaa30G31 Xaa32C33Xaa34C35Xaa 36Xaa37Xaa38 SEQ ID NO:5011
wherein: amino acid residues 1 through 7 are optional (Thus, the 0SK1 peptide analog optionally includes residues 1-7 as indicated above in SEQ ID NO:5011, or a N-terminal truncation leaving present residues 2-7, 3-7, 4-7, 5-7, 6-7, or 7, or alternatively, a N-terminal truncation wherein all of residues 1-7 are entirely absent.);
Xaa11 is a neutral, basic, or acidic amino acid residue (e.g., Ser, Thr, Ala, GIy, Leu, He, VaI, Met, Cit, Homocitrulline, Oic, Pro, Hyp, Tic, D-Tic, D-Pro, Guf, and 4-Amino-Phe,Thz, Aib, Sar, Pip, Bip, Phe, Tyr, Lys, His, Trp, Arg, Nα Methyl-Arg; homoarginine, 1-NaI, 2-NaI, Om, D-Orn, Asn, GIn, GIu, Asp, α-aminoadipic acid, and para-carboxyl-phenylalanine); Xaa12 is a neutral or acidic amino acid residue (e.g., Ala, GIy, Leu, He, VaI, Met, Oic, Pro,
Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Tyr, Ser, Thr, Asn, GIn, GIu, Asp, α- aminoadipic acid, and para-carboxyl-phenylalanine);
Xaa15 is a neutral or acidic amino acid residue (e.g., Ala, GIy, Leu, He, VaI, Met, Oic, Pro,
Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Tyr, Ser, Thr, Asn, GIn, GIu, Asp, α- aminoadipic acid, and para-carboxyl-phenylalanine);
Xaa16 is a neutral or basic amino acid residue (e.g., Lys, His, Arg, Trp, Arg, Nα Methyl-Arg; 5 homoarginine, 1-NaI, 2-NaI, Om, D-Om, Cit, Nα-Methyl-Cit, Homocitrulline, His, Ala, GIy,
Leu, He, VaI, Met, Oic, Pro, Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Ser, Thr,
Guf, and 4-Amino-Phe);
Xaa19 is a neutral or basic amino acid residue (e.g., Lys, His, Arg, Trp, Arg, Nα Methyl-Arg; homoarginine, 1-NaI, 2-NaI, Orn, D-Om, Cit, Nα-Methyl-Cit, Homocitrulline, His, Ala, GIy, 0 Leu, He, VaI, Met, Oic, Pro, Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Ser, Thr,
Guf, and 4-Amino-Phe);
Xaa20 is a neutral or basic amino acid residue (e.g., Lys, His, Arg, Trp, Arg, Nα Methyl-Arg; homoarginine, 1-NaI, 2-NaI, Om, D-Orn, Cit, Nα-Methyl-Cit, Homocitrulline, His, Ala, GIy,
Leu, He, VaI, Met, Oic, Pro, Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Ser, Thr,5 Guf, and 4-Amino-Phe);
Xaa27 is a neutral, basic, or acidic amino acid residue (e.g., Ser, Thr, Ala, GIy, Leu, He, VaI,
Met, Cit, Homocitrulline, Oic, Pro, Hyp, Tic, D-Tic, D-Pro, Guf, and 4-Amino-Phe,Thz, Aib,
Sar, Pip, Bip, Phe, Tyr, Lys, His, Trp, Arg, Nα Methyl-Arg; homoarginine, 1-NaI, 2-NaI,
Om, D-Orn, Asn, GIn, GIu, Asp, α-aminoadipic acid, and para-carboxyl-phenylalanine); O Xaa29 is a neutral or acidic amino acid residue (e.g., Ala, GIy, Leu, He, VaI, Met, Oic, Pro,
Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Tyr, Ser, Thr, Asn, GIn, GIu, Asp, α- aminoadipic acid, and para-carboxyl-phenylalanine);
Xaa30 is a neutral or acidic amino acid residue (e.g., Ala, GIy, Leu, He, VaI, Met, Oic, Pro,
Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Tyr, Ser, Thr, Asn, GIn, GIu, Asp, α- 5 aminoadipic acid, and para-carboxyl-phenylalanine);
Xaa32 is a neutral, basic, or acidic amino acid residue (e.g., Ser, Thr, Ala, GIy, Leu, He, VaI, Met, Cit, Homocitrulline, Oic, Pro, Hyp, Tic, D-Tic, D-Pro, Guf, and 4-Amino-Phe,Thz, Aib, Sar, Pip, Bip, Phe, Tyr, Lys, His, Trp, Arg, Nα Methyl-Arg; homoarginine, 1-NaI, 2-NaI, Orn, D-Orn, Asn, GIn, GIu, Asp, α-aminoadipic acid, and para-carboxyl-phenylalanine); O Xaa34 is a neutral or acidic amino acid residue (e.g., Ala, GIy, Leu, He, VaI, Met, Oic, Pro,
Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Tyr, Ser, Thr, Asn, GIn, GIu, Asp, α- aminoadipic acid, and para-carboxyl-phenylalanine);
Xaa36 is optional, and if present, is a neutral amino acid residue (e.g., Pro, Ala, GIy, Leu, He, VaI, Met, Oic, Hyp, Tic, D-Tic, D-Pro, Thz, Nα-Methyl-Cit, Homocitrulline, Aib, Sar, Pip, Tyr, Thr, Ser, Phe, Trp, 1-NaI, 2-NaI1 and Bip;
Xaa37 is optional, and if present, is a neutral amino acid residue (e.g., Pro, Ala, GIy, Leu, 5 He, VaI, Met, Oic, Hyp, Tic, D-Tic, D-Pro, Thz, Nα-Methyl-Cit, Homocitrulline, Aib, Sar, Pip,
Tyr, Thr, Ser, Phe, Trp, 1-NaI, 2-NaI, and Bip); and
Xaa38 is optional, and if present, is a basic amino acid residue (e.g., Lys, His, Orn, Trp, D- Om, Arg, Nα Methyl-Arg; homoarginine, Cit, Nα-Methyl-Cit, Homocitrulline, His, Guf, and 4- Amino-Phe). 0 In some other embodiments of the composition of matter comprising a half-life extending moiety, the 0SK1 peptide analog comprises an amino acid sequence of the formula:
GiV2I3I4N5V6K7C8K9I10Xa3 11Xa3 12Q13C14L15Xa3 16P17C18K19Xa3 20A21G22M23R24F25G26
Xaa27C28χaa29χaa30G31K32C33χaa34C35χaa36Xaa37χaa38 SEQ ID NO:4913 5 wherein: amino acid residues 1 to 7 are optional (Thus, the 0SK1 peptide analog optionally includes residues 1-7 as indicated above in SEQ ID NO:4913, or a N-terminal truncation leaving present residues 2-7, 3-7, 4-7, 5-7, 6-7, or 7, or alternatively, a N-terminal O truncation wherein all of residues 1-7 are entirely absent.);
Xaa11 is a neutral, basic or acidic amino acid residue (e.g., Ser, Thr, Ala, GIy, Leu, He, VaI, Met, Cit, Homocitrulline, Oic, Pro, Hyp, Tic, D-Tic, D-Pro, Guf, and 4-Amino-Phe,Thz, Aib, Sar, Pip, Bip, Phe, Tyr, Lys, His, Trp, Arg, Nα Methyl-Arg; homoarginine, 1-NaI, 2-NaI, Orn, D-Om, Asn, GIn, GIu, Asp, α-aminoadipic acid, and para-carboxyl-phenylalanine); 5 Xaa12 is a neutral or acidic amino acid residue (e.g., Ala, GIy, Leu, He, VaI, Met, Oic, Pro,
Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Tyr, Ser, Thr, Asn, GIn, GIu, Asp, α- aminoadipic acid, and para-carboxyl-phenylalanine);
X33 16 is a neutral or basic amino acid residue (e.g., Lys, His, Arg, Trp, Arg, Nα Methyl-Arg; homoarginine, 1-NaI, 2-NaI, Om, D-Om, Cit, Nα-Methyl-Cit, Homocitrulline, His, Ala, GIy, O Leu, He, VaI, Met, Oic, Pro, Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Ser, Thr,
Guf, and 4-Amino-Phe);
Xaa20 is a neutral or basic amino acid residue (e.g., Lys, His, Arg, Trp, Arg, Nα Methyl-Arg; homoarginine, 1-NaI1 2-NaI, Om, D-Om, Cit, Nα-Methyl-Cit, Homocitrulline, His, Ala, GIy,
Leu, He, VaI1 Met, Oic, Pro, Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Ser, Thr,
Guf, and 4-Amino-Phe);
Xaa27 is a neutral, basic, or acidic amino acid residue (e.g., Ser, Thr, Ala, GIy, Leu, He, VaI,
Met, Cit, Homocitrulline, Oic, Pro, Hyp, Tic, D-Tic, D-Pro, Guf, and 4-Amino-Phe,Thz, Aib,
Sar, Pip, Bip, Phe, Tyr, Lys, His, Trp, Arg, Nα Methyl-Arg; homoarginine, 1-NaI, 2-NaI,
Om, D-Om, Asn, GIn, GIu, Asp, α-aminoadipic acid, and para-carboxyl-phenylalanine);
Xaa29 is a neutral or acidic amino acid residue (e.g., Ala, GIy, Leu, He, VaI, Met, Oic, Pro,
Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Tyr, Ser, Thr, Asn, GIn1 GIu, Asp, α- aminoadipic acid, and para-carboxyl-phenylalanine);
Xaa30 is a neutral or acidic amino acid residue (e.g., Ala, GIy, Leu, He, VaI, Met, Oic, Pro,
Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Tyr, Ser, Thr, Asn, GIn, GIu1 Asp, α- aminoadipic acid, and para-carboxyl-phenylalanine);
Xaa34 is a neutral or acidic amino acid residue (e.g., Ala, GIy, Leu, He, VaI, Met, Oic, Pro,
Hyp, Tic, D-Tic, D-Pro, Thz, Aib, Sar, Pip, Bip, Phe, Tyr, Ser, Thr, Asn, GIn, GIu, Asp, α- aminoadipic acid, and para-carboxyl-phenylalanine);
Xaa36 is optional, and if present, is a neutral amino acid residue (e.g., Pro, AIa1 GIy1 Leu,
He, VaI, Met, Oic, Hyp, Tic, D-Tic, D-Pro, Thz, Nα-Methyl-Cit, Homocitrulline, Aib, Sar, Pip,
Tyr, Thr, Ser, Phe, Trp, 1-NaI, 2-NaI, and Bip;
Xaa37 is optional, and if present, is a neutral amino acid residue (e.g., Pro, Ala, GIy, Leu,
He, VaI, Met, Oic, Hyp, Tic, D-Tic, D-Pro, Thz, Nα-Methyl-Cit, Homocitrulline, Aib, Sar, Pip,
Tyr, Thr, Ser, Phe, Trp, 1-NaI, 2-NaI, and Bip); and
Xaa38 is optional, and if present, is a basic amino acid residue (e.g., Lys, His, Orn, Trp, D-
Orn, Arg, Nα Methyl-Arg; homoarginine, Cit, Nα-Methyl-Cit, Homocitrulline, His, Guf, and 4-Amino-
Phe).
Table 8— Pi2 peptide and Pi P2 s peptide analog equences
Table 9— Anuroctoxin (AnTx) peptide and peptide analog sequences
Table 10— Noxiustoxin (NTX) peptide and NTX peptide analog sequences
Table 11— Kaliotoxini (KTX1) peptide and KTX1 peptide analog sequences
Table 12— IKCaI inhibitor peptide sequences
Table 13— Maurotoxin (MTx) peptide amd MTx peptide analog sequences
In Table 13 and throughout this specification, X
m19 and X
m34 are each independently nonfunctional residues.
Table 14— Charybdotoxin(ChTx) peptide and ChTx peptide analog sequences
Table 15— SKCa inhibitor peptide sequences
Table 16— Apamin peptide and peptide analog inhibitor sequences
Table 17— Scyllatoxin (ScyTx), BmP05, P05, Tamapin, P01 peptide and peptide analog inhibitor sequences
Table 18— BKCa inhibitor peptide sequences
Table 19— IbTx, Slotoxin, BmTxI, & BuTX (Slotoxin family) peptide and peptide analog inhibitor sequences
Table 20— Martentoxin peptide and peptide analog inhibitor sequences
Table 21— N type Ca
2+ channel inhibitor peptide sequences
Table 22— ωMVIIA peptide and peptide analog inhibitor sequences
Table 23— OGVIA peptide and peptide analog inhibitor sequences
Table 24— Ptu1 peptide and peptide analog inhibitor sequences
Table 25— Thrixopelma pruriens (ProTxi) and ProTxi peptide analogs and other T type Ca2+ channel inhibitor peptide sequences
Table 26— BeKMI M current inhibitor peptide and BeKMI peptide analog sequences
Table 27— Na
+ channel inhibitor peptide sequences
Table 28— Cl- channel inhibitor peptide sequences
Table 29— Kv2.1 inhibitor peptide sequences
Table 30— Kv4.3 & Kv4.2 inhibitor peptide sequences
Table 32- Agelenopsis aperta (Agatoxin) toxin peptides and peptide analogs and other Ca2+ channel inhibiter peptides
In accordance with this invention are molecules in which at least one of the toxin peptide (P) portions of the molecule comprises a Kv1.3 antagonist peptide. Amino acid sequences selected from ShK, HmK, MgTx, AgTxI, AgTx2, Heterometrus spinnifer (HsTxI), OSK1, Anuroctoxin(AnTx),
Noxiustoxin (NTX), KTX1, Hongotoxin, ChTx, Titystoxin, BgK, BmKTX, BmTx, AeK, AsKS Tc30, Tc32, PM1 Pi2, and/or Pi3 toxin peptides and peptide analogs of any of these are preferred. Examples of useful Kv1.3 antagonist peptide sequences include those having any amino acid sequence set forth in Table 1, Table 2, Table 3, Table 4, Table 5, Table 6, Table 7, Table 8, Table 5 9, Table 10, and/or Table 11 herein above;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is an IKCaI antagonist peptide. Useful IKCaI antagonist peptides include Maurotoxin (MTx), ChTx.peptides and peptide analogs of either of these, examples of which include those having any amino acid sequence set forth in Table 12, Table 13, and/or Table 14; 0 Other embodiments of the inventive composition include at least one toxin peptide (P) that is a
SKCa inhibitor peptide. Useful SKCa inhibitor peptides include, Apamin, ScyTx, BmP05, P01, P05, Tamapin, TsK, and peptide analogs of any of these, examples of which include those having any amino acid sequence set forth in Table 15;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is5 an apamin peptide, and peptide analogs of apamin, examples of which include those having any amino acid sequence set forth in Table 16;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is a Scyllotoxin family peptide, and peptide analogs of any of these, examples of which include those having any amino acid sequence set forth in Table 17; O Other embodiments of the inventive composition include at least one toxin peptide (P) that is a
BKCa inhibitor peptide, examples of which include those having any amino acid sequence set forth in Table 18;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is a Slotoxin family peptide, and peptide analogs of any of these, examples of which include those 5 having any amino acid sequence set forth in Table 19;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is a Martentoxin peptide, and peptide analogs thereof, examples of which include those having any amino acid sequence set forth in Table 20;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is a O N-type Ca2+ channel inhibitor peptide, examples of which include those having any amino acid sequence set forth in Table 21;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is a ωMVIIA peptide, and peptide analogs thereof, examples of which include those having any amino acid sequence set forth in Table 22;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is a coGVIA peptide, and peptide analogs thereof, examples of which include those having any amino acid sequence set forth in Table 23;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is a Ptu1 peptide, and peptide analogs thereof, examples of which include those having any amino acid sequence set forth in Table 24; Other embodiments of the inventive composition include at least one toxin peptide (P) that is a
ProTxi peptide, and peptide analogs thereof, examples of which include those having any amino acid sequence set forth in Table 25;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is a BeKMI peptide, and peptide analogs thereof, examples of which include those having any amino acid sequence set forth in Table 26;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is a Na+ channel inhibitor peptide, examples of which include those having any amino acid sequence set forth in Table 27;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is a Cl- channel inhibitor peptide, examples of which include those having any amino acid sequence set forth in Table 28;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is a Kv2.1 inhibitor peptide, examples of which include those having any amino acid sequence set forth in Table 29; Other embodiments of the inventive composition include at least one toxin peptide (P) that is a
Kv4.2/Kv4.3 inhibitor peptide, examples of which include those having any amino acid sequence set forth in Table 30;
Other embodiments of the inventive composition include at least one toxin peptide (P) that is a nACHR inhibitor peptide, examples of which include those having any amino acid sequence set forth in Table 31; and
Other embodiments of the inventive composition include at least one toxin peptide (P) that is an Agatoxin peptide, a peptide analog thereof or other calcium channel inhibitor peptide, examples of which include those having any amino acid sequence set forth in Table 32.
Half-life extending moieties. This invention involves the presence of at least one half-life extending moiety (F1 and/or F2 in Formula I) attached to a peptide through the N-terminus, C-terminus or a sidechain of one of the intracalary amino acid residues. Multiple half-life extending moieties can also be used; e.g., Fc's at each terminus or an Fc at a terminus and a PEG group at the other terminus or at a sidechain. In other embodiments the Fc domain can be PEGylated (e.g., in accordance with the formulae F1-F2-(L>P; P-(L)g-F1-F2; or P-(L)g-F1-F2-(L)f-P).
The half-life extending moiety can be selected such that the inventive composition achieves a sufficient hydrodynamic size to prevent clearance by renal filtration in vivo. For example, a half-life extending moiety can be selected that is a polymeric macromolecule, which is substantially straight chain, branched-chain, or dendritic in form. Alternatively, a half-life extending moiety can be selected such that, in vivo, the inventive composition of matter will bind to a serum protein to form a complex, such that the complex thus formed avoids substantial renal clearance. The half-life extending moiety can be, for example, a lipid; a cholesterol group (such as a steroid); a carbohydrate or oligosaccharide; or any natural or synthetic protein, polypeptide or peptide that binds to a salvage receptor.
Exemplary half-life extending moieties that can be used, in accordance with the present invention, include an immunoglobulin Fc domain, or a portion thereof, or a biologically suitable polymer or copolymer, for example, a polyalkylene glycol compound, such as a polyethylene glycol or a polypropylene glycol. Other appropriate polyalkylene glycol compounds include, but are not limited to, charged or neutral polymers of the following types: dextran, polylysine, colominic acids or other carbohydrate based polymers, polymers of amino acids, and biotin derivatives. In some monomeric fusion protein embodiments an immunoglobulin (including light and heavy chains) or a portion thereof, can be used as a half-life-extending moiety, preferably an immunoglobulin of human origin, and including any of the immunoglobulins, such as, but not limited to, IgGI , lgG2, lgG3 or lgG4.
Other examples of the half-life extending moiety, in accordance with the invention, include a copolymer of ethylene glycol, a copolymer of propylene glycol, a carboxymethylcellulose, a polyvinyl pyrrolidone, a poly-1 ,3-dioxolane, a poly-1 ,3,6-trioxane, an ethylene/maleic anhydride copolymer, a polyaminoacid (e.g., polylysine), a dextran n-vinyl pyrrolidone, a poly n-vinyl pyrrolidone, a propylene glycol homopolymer, a propylene oxide polymer, an ethylene oxide polymer, a polyoxyethylated polyol, a polyvinyl alcohol, a linear or branched glycosylated chain, a polyacetal, a long chain fatty acid, a long chain hydrophobic aliphatic group, an immunoglobulin
light chain and heavy chain, an immunoglobulin F0 domain or a portion thereof (see, e.g., Feige et al., Modified peptides as therapeutic agents, US Patent No. 6,660,843), a CH2 domain of Fc, an albumin (e.g., human serum albumin (HSA)); see, e.g., Rosen et al., Albumin fusion proteins, US Patent No. 6,926,898 and US 2005/0054051 ; Bridon et al., Protection of endogenous therapeutic peptides from peptidase activity through conjugation to blood components, US 6,887,470), a transthyretin (TTR; see, e.g., Walker et al. Use of transthyretin peptide/protein fusions to increase the serum half-life of pharmacologically active peptides/proteins, US 2003/0195154 A1; 2003/0191056 A1), or a thyroxine-binding globulin (TBG). Thus, exemplary embodiments of the inventive compositions also include HSA fusion constructs such as but not limited to: HSA fusions 0 with ShK, OSK1 , or modified analogs of those toxin peptides. Examples include HSA-L10-ShK(2- 35); HSA-L10-OsK1(1-38); HSA-L10-ShK(2-35); and HSA-L10-OsK1(1-38).
Other embodiments of the half-life extending moiety, in accordance with the invention, include peptide ligands or small (organic) molecule ligands that have binding affinity for a long half- life serum protein under physiological conditions of temperature, pH, and ionic strength. Examples 5 include an albumin-binding peptide or small molecule ligand, a transthyretin-binding peptide or small molecule ligand, a thyroxine-binding globulin-binding peptide or small molecule ligand, an antibody-binding peptide or small molecule ligand, or another peptide or small molecule that has an affinity for a long half-life serum protein. (See, e.g., Blaney et al. Method and compositions for increasing the serum half-life of pharmacologically active agents by binding to transthyretin- 0 selective ligands, US Patent. No. 5,714,142; Sato et al. Serum albumin binding moieties,
US 2003/0069395 A1; Jones et al, Pharmaceutical active conjugates, US Patent No. 6,342,225). A "long half-life serum protein" is one of the hundreds of different proteins dissolved in mammalian blood plasma, including so-called "carrier proteins" (such as albumin, transferrin and haptoglobin), fibrinogen and other blood coagulation factors, complement components, immunoglobulins, 5 enzyme inhibitors, precursors of substances such as angiotensin and bradykinin and many other types of proteins. The invention encompasses the use of any single species of pharmaceutically acceptable half-life extending moiety, such as, but not limited to, those described herein, or the use of a combination of two or more different half-life extending moieties, such as PEG and immunoglobulin Fc domain or a CH2 domain of Fc, albumin (e.g., HSA), an albumin-binding O protein, transthyretin or TBG, or a combination such as immunoglobulin(light chain+heavy chain) and Fc domain (the combination so-called "hemibody").
In some embodiments of the invention an Fc domain or portion thereof, such as a CH2 domain of Fc, is used as a half-life extending moiety. The Fc domain can be fused to the N-
terminal (e.g., in accordance with the formula F1-(L)rP) or C-terminal (e.g., in accordance with the formula P-(L)9-F1) of the toxin peptides or at both the N and C termini (e.g., in accordance with the formulae F1-(L)t-P-(L)g-F2 or P-(L)g-F1-(L)rP)- A peptide linker sequence can be optionally included between the Fc domain and the toxin peptide, as described herein. Examples of the formula F1-(L)rP include: Fc-LI 0-ShK(K22A)[2-35]; Fc-LI 0-ShK(R1K/K22A)[1 -35]; Fc-LIO-
ShK(RI H/K22A)[1 -35]; Fc-L10-ShK(R1Q /K22A)[1-35]; Fc-L10-ShK(R1Y /K22A)[1-35]; Fc-LIO-PP- ShK(K22A) [1-35]; and any other working examples described herein. Examples of the formula P- (L)9-F1 include: ShK(1-35)-L10-Fc; OsK1(1-38)-L10-Fc; Met-ShK(1-35)-L10-Fc; ShK(2-35)-L10-Fc; GIy-ShK(I -35)-L10-Fc; Osk1(1-38)-L10-Fc; and any other working examples described herein. Fc variants are suitable half-life extending moieties within the scope of this invention. A native Fc can be extensively modified to form an Fc variant in accordance with this invention, provided binding to the salvage receptor is maintained; see, for example WO 97/34631 , WO 96/32478, and WO 04/110472. In such Fc variants, one can remove one or more sites of a native Fc that provide structural features or functional activity not required by the fusion molecules of this invention. One can remove these sites by, for example, substituting or deleting residues, inserting residues into the site, or truncating portions containing the site. The inserted or substituted residues can also be altered amino acids, such as peptidomimetics or D-amino acids. Fc variants can be desirable for a number of reasons, several of which are described below. Exemplary Fc variants include molecules and sequences in which: 1. Sites involved in disulfide bond formation are removed. Such removal can avoid reaction with other cysteine-containing proteins present in the host cell used to produce the molecules of the invention. For this purpose, the cysteine-containing segment at the N-terminus can be truncated or cysteine residues can be deleted or substituted with other amino acids (e.g., alanyl, seryl). In particular, one can truncate the N-terminal 20-amino acid segment of SEQ ID NO: 2 or delete or substitute the cysteine residues at positions 7 and 10 of SEQ ID NO: 2. Even when cysteine residues are removed, the single chain Fc domains can still form a dimeric Fc domain that is held together non-covalently. 2. A native Fc is modified to make it more compatible with a selected host cell. For example, one can remove the PA sequence near the N-terminus of a typical native Fc, which can be recognized by a digestive enzyme in E. coli such as proline iminopeptidase. One can also add an N-terminal methionine residue, especially when the molecule is expressed recombinantly in a bacterial cell such as E. coli. The Fc domain of SEQ ID NO: 2 (Figure 4A-4B) is one such Fc variant.
3. A portion of the N-terminus of a native Fc is removed to prevent N-terminal heterogeneity when expressed in a selected host cell. For this purpose, one can delete any of the first 20 amino acid residues at the N-terminus, particularly those at positions 1, 2, 3, 4 and 5.
4. One or more glycosylate sites are removed. Residues that are typically glycosylated 5 (e.g., asparagine) can confer cytolytic response. Such residues can be deleted or substituted with unglycosylated residues (e.g., alanine).
5. Sites involved in interaction with complement, such as the C1q binding site, are removed. For example, one can delete or substitute the EKK sequence of human IgGL Complement recruitment may not be advantageous for the molecules of this invention and0 so can be avoided with such an Fc variant.
6. Sites are removed that affect binding to Fc receptors other than a salvage receptor. A native Fc can have sites for interaction with certain white blood cells that are not required for the fusion molecules of the present invention and so can be removed.
7. The ADCC site is removed. ADCC sites are known in the art; see, for example, Molec. 5 Immunol. 29 (5): 633-9 (1992) with regard to ADCC sites in IgGI . These sites, as well, are not required for the fusion molecules of the present invention and so can be removed.
8. When the native Fc is derived from a non-human antibody, the native Fc can be humanized. Typically, to humanize a native Fc, one will substitute selected residues in the non-human native Fc with residues that are normally found in human native Fc. O Techniques for antibody humanization are well known in the art.
Preferred Fc variants include the following. In SEQ ID NO: 2, the leucine at position 15 can be substituted with glutamate; the glutamate at position 99, with alanine; and the lysines at positions 101 and 103, with alanines. In addition, phenyalanine residues can replace one or more tyrosine residues. 5 An alternative half-life extending moiety would be a protein, polypeptide, peptide, antibody, antibody fragment, or small molecule (e.g., a peptidomimetic compound) capable of binding to a salvage receptor. For example, one could use as a half-life extending moiety a polypeptide as described in U.S. Pat. No. 5,739,277, issued April 14, 1998 to Presta eiaj. Peptides could also be selected by phage display for binding to the FcRn salvage receptor. Such 0 salvage receptor-binding compounds are also included within the meaning of "half-life extending moiety" and are within the scope of this invention. Such half-life extending moieties should be selected for increased half-life (e.g., by avoiding sequences recognized by proteases) and
decreased immunogenicity (e.g., by favoring non-immunogenic sequences, as discovered in antibody humanization).
As noted above, polymer half-life extending moieties can also be used for F1 and F2. Various means for attaching chemical moieties useful as half-life extending moieties are currently 5 available, see, e.g., Patent Cooperation Treaty ("PCT") International Publication No. WO 96/11953, entitled "N-Terminally Chemically Modified Protein Compositions and Methods," herein incorporated by reference in its entirety. This PCT publication discloses, among other things, the selective attachment of water-soluble polymers to the N-terminus of proteins.
In some embodiments of the inventive compositions, the polymer half-life extending 0 moiety is polyethylene glycol (PEG), as F1 and/or F2, but it should be understood that the inventive composition of matter, beyond positions F1 and/or F2, can also include one or more PEGs conjugated at other sites in the molecule, such as at one or more sites on the toxin peptide. Accordingly, some embodiments of the inventive composition of matter further include one or more PEG moieties conjugated to a non-PEG half-life extending moiety, which is F1 and/or F2, or to the 5 toxin peptide(s) ( P), or to any combination of any of these. For example, an Fc domain or portion thereof (as F1 and/or F2) in the inventive composition can be made mono-PEGylated, di- PEGylated, or otherwise multi-PEGylated, by the process of reductive alkylation.
Covalent conjugation of proteins and peptides with polyethylene glycol) (PEG) has been widely recognized as an approach to significantly extend the in vivo circulating half-lives of O therapeutic proteins. PEGylation achieves this effect predominately by retarding renal clearance, since the PEG moiety adds considerable hydrodynamic radius to the protein. (Zalipsky, S., et al., Use of functionalized polyethylene glycol)s for modification of polypeptides., in polyethylene glycol) chemistry: Biotechnical and biomedical applications., J.M. Harris, Ed., Plenum Press: New York., 347-370 (1992)). Additional benefits often conferred by PEGylation of proteins and peptides 5 include increased solubility, resistance to proteolytic degradation, and reduced immunogenicity of the therapeutic polypeptide. The merits of protein PEGylation are evidenced by the commercialization of several PEGylated proteins including PEG-Adenosine deaminase (Adagen™/Enzon Corp.), PEG-L-asparaginase (Oncaspar™/Enzon Corp.), PEG-lnterferon α-2b (PEG-lntron™/Schering/Enzon), PEG-lnterferon α-2a (PEGASYS™/Roche) and PEG-G-CSF O (Neulasta™/Amgen) as well as many others in clinical trials.
Briefly, the PEG groups are generally attached to the peptide portion of the composition of the invention via acylation or reductive alkylation (or reductive amination) through a reactive group
on the PEG moiety (e.g., an aldehyde, amino, thiol, or ester group) to a reactive group on the inventive compound (e.g., an aldehyde, amino, or ester group).
A useful strategy for the PEGylation of synthetic peptides consists of combining, through forming a conjugate linkage in solution, a peptide and a PEG moiety, each bearing a special functionality that is mutually reactive toward the other. The peptides can be easily prepared with conventional solid phase synthesis (see, for example, Figures 5 and 6 and the accompanying text herein). The peptides are "preactivated" with an appropriate functional group at a specific site. The precursors are purified and fully characterized prior to reacting with the PEG moiety. Ligation of the peptide with PEG usually takes place in aqueous phase and can be easily monitored by reverse phase analytical HPLC. The PEGylated peptides can be easily purified by preparative HPLC and characterized by analytical HPLC, amino acid analysis and laser desorption mass spectrometry.
PEG is a well-known, water soluble polymer that is commercially available or can be prepared by ring-opening polymerization of ethylene glycol according to methods well known in the art (Sandler and Kara, Polymer Synthesis, Academic Press, New York, Vol. 3, pages 138-161). In the present application, the term "PEG" is used broadly to encompass any polyethylene glycol molecule, in mono-, bi-, or poly- functional form, without regard to size or to modification at an end of the PEG, and can be represented by the formula:
X-O(CH2CH2O)n-ICH2CH2OH, (X)
where n is 20 to 2300 and X is H or a terminal modification, e.g., a Cu alkyl. In some useful embodiments, a PEG used in the invention terminates on one end with hydroxy or methoxy, i.e., X is H or CH3 ("methoxy PEG"). It is noted that the other end of the PEG, which is shown in formula (II) terminating in OH, covalently attaches to an activating moiety via an ether oxygen bond, an amine linkage, or amide linkage. When used in a chemical structure, the term "PEG" includes the formula (II) above without the hydrogen of the hydroxyl group shown, leaving the oxygen available to react with a free carbon atom of a linker to form an ether bond. More specifically, in order to conjugate PEG to a peptide, the peptide must be reacted with PEG in an "activated" form. Activated PEG can be represented by the formula:
(PEG)-(A) (Xl)
where PEG (defined supra) covalently attaches to a carbon atom of the activation moiety (A) to form an ether bond, an amine linkage, or amide linkage, and (A) contains a reactive group which can react with an amino, azido, alkyne, imino, maleimido, N-succinimidyl, carboxyl, aminooxy, seleno, or thiol group on an amino acid residue of a peptide or a linker moiety covalently attached to the peptide, e.g., the 0SK1 peptide analog. Residues baring chemoselective reactive groups can be introduced into the toxin peptide, e.g., an 0SK1 peptide analog during assembly of the peptide sequence solid-phase synthesis as protected derivatives. Alternatively, chemoselective reactive groups can be introduced in the toxin peptide after assembly of the peptide sequence by solid-phase synthesis via the use of orthogonal protecting groups at specific sites. Examples of 0 amino acid residues useful for chemoselective reactions include, but are not limited to, (amino- oxyacetyl)-L-diaminopropionic acid, p-azido-phenylalanine, azidohomolalanine, para-propargyloxy- phenylalanine, selenocysteine, para-acetylphenylalanine, (NNevulinyl)-Lysine, (Nε-pyruvyl)-Lysine, selenocysteine, and orthogonally protected cysteine and homocysteine.
Accordingly, in some embodiments of the composition of matter, the toxin peptide, e.g., 5 the 0SK1 peptide analog, is conjugated to a polyethylene glycol (PEG) at:
(a) 1, 2, 3 or 4 amino functionalized sites in the toxin peptide;
(b) 1 , 2, 3 or 4 thiol functionalized sites in the toxin peptide; (c) 1 or 2 ketone functionalized sites in the toxin peptide; (d) 1or 2 azido functionalized sites of the toxin peptide; (e) 1 or 2 carboxyl functionalized sites in the toxin peptide; (f) 1 or 2 aminooxy functionalized sites in O the toxin peptide; or (g) 1 or 2 seleno functionalized sites in the toxin peptide.
In other embodiments of the composition of matter, the toxin peptide, e.g., the 0SK1 peptide analog, is conjugated to a polyethylene glycol (PEG) at:
(a) 1, 2, 3 or 4 amino functionalized sites of the PEG;
(b) 1 , 2, 3 or 4 thiol functionalized sites of the PEG; 5 (c) 1 , 2, 3 or 4 maleimido functionalized sites of the PEG;
(d) 1, 2, 3 or 4 N-succinimidyl functionalized sites of the PEG;
(e) 1 , 2, 3 or 4 carboxyl functionalized sites of the PEG; or
(f) 1 , 2, 3 or 4 p-nitrophenyloxycarbonyl functionalized sites of the PEG.
Techniques for the preparation of activated PEG and its conjugation to biologically active O peptides are well known in the art. (E.g., see U.S. Pat. Nos. 5,643,575, 5,919,455, 5,932,462, and
5,990,237; Thompson et al., PEGylation of polypeptides, EP 0575545 B1; Petit, Site specific protein modification, US Patent Nos. 6,451,986, and 6,548,644; S. Herman et al., Polyethylene glycol) with reactive endgroups: I. Modification of proteins, J. Bioactive Compatible Polymers,
10:145-187 (1995); Y. Lu et al., Pegylated peptides III: Solid-phase synthesis with PEGylating reagents of varying molecular weight: synthesis of multiply PEGylated peptides, Reactive Polymers, 22:221-229 (1994); A.M. Felix et al., PEGylated Peptides IV: Enhanced biological activity of site-directed PEGylated GRF analogs, Int. J. Peptide Protein Res., 46:253-264 (1995); A.M. Felix, Site-specific polyethylene glycol)ylation of peptides, ACS Symposium Series
680(poly(ethylene glycol)): 218-238 (1997); Y. lkeda et al., Polyethylene glycol derivatives, their modified peptides, methods for producing them and use of the modified peptides, EP 0473084 B1; G. E. Means et al., Selected techniques for the modification of protein side chains, in: Chemical modification of proteins, Holden Day, Inc., 219 (1971)). Activated PEG, such as PEG-aldehydes or PEG-aldehyde hydrates, can be chemically synthesized by known means or obtained from commercial sources, e.g., Shearwater Polymers, (Huntsville, Al) or Enzon, Inc. (Piscataway, N.J.).
An example of a useful activated PEG for purposes of the present invention is a PEG- aldehyde compound (e.g., a methoxy PEG-aldehyde), such as PEG-propionaldehyde, which is commercially available from Shearwater Polymers (Huntsville, Al). PEG-propionaldehyde is represented by the formula PEG-CH2CH2CHO. (See, e.g., U.S. Pat. No. 5,252,714). Other examples of useful activated PEG are PEG acetaldehyde hydrate and PEG bis aldehyde hydrate, which latter yields a bifunctionally activated structure. (See., e.g., Bentley et al., Polyethylene glycol) aldehyde hydrates and related polymers and applications in modifying amines, US Patent No. 5,990,237).
Another useful activated PEG for generating the PEG-conjugated peptides of the present invention is a PEG-maleimide compound, such as, but not limited to, a methoxy PEG-maleimide, such as maleimido monomethoxy PEG, are particularly useful for generating the PEG-conjugated peptides of the invention. (E.g., Shen, Λ/-maleimidyl polymer derivatives, US Patent No. 6,602,498; C. Delgado et al. The uses and properties of PEG-linked proteins, Crit. Rev. Therap. Drug Carrier
Systems, 9:249-304 (1992); S. Zalipsky et al. Use of functionalized polyethylene glycol)s for modification of polypeptides, in: Polyethylene glycol) chemistry: Biotechnical and biomedical applications (J. M. Harris, Editor, Plenum Press: New York, 347-370 (1992); S. Herman et al, Poly(ethylene glycol) with reactive endgroups: I. Modification of proteins, J. Bioactive Compatible Polymers, 10:145-187 (1995); P.J. Shadle et al, Conjugation of polymer to colony stimulating factor-1, U.S. Patent No. 4,847,325; G. Shaw et al. Cysteine added variants IL-3 and chemical modifications thereof, U.S. Patent No. 5,166,322 and EP 0469074 B1 ; G. Shaw et al. Cysteine added variants of EPO and chemical modifications thereof, EP 0668353 A1; G. Shaw et al.
Cysteine added variants G-CSF and chemical modifications thereof, EP 0668354 A1; N.V. Katre et al., lnterleukin-2 muteins and polymer conjugation thereof, U.S. Patent No. 5,206,344; R.J. Goodson and N.V. Katre, Site-directed pegylation of recombinant interleukin-2 at its glycosylate site, Biotechnology, 8:343-346 (1990)). A polyethylene glycol) vinyl sulfone is another useful activated PEG for generating the
PEG-conjugated peptides of the present invention by conjugation at thiolated amino acid residues, e.g., at C residues. (E.g., M. Morpurgo et al., Preparation and characterization of polyethylene glycol) vinyl sulfone, Bioconj. Chem., 7:363-368 (1996); see also Harris, Functionalization of polyethylene glycol for formation of active sulfone-terminated PEG derivatives for binding to proteins and biologically compatible materials, U.S. Patent Nos. 5,446,090; 5,739,208; 5,900,461 ; 6,610,281 and 6,894,025; and Harris, Water soluble active sulfones of polyethylene glycol), WO 95/13312 A1).
Another activated form of PEG that is useful in accordance with the present invention, is a PEG-N-hydroxysuccinimide ester compound, for example, methoxy PEG-N-hydroxysuccinimidyl (NHS) ester.
Heterobifunctionally activated forms of PEG are also useful. (See, e.g., Thompson et al., PEGylation reagents and biologically active compounds formed therewith, U.S. Patent No. 6,552,170).
Typically, a toxin peptide or, a fusion protein comprising the toxin peptide, is reacted by known chemical techniques with an activated PEG compound, such as but not limited to, a thiol- activated PEG compound, a diol-activated PEG compound, a PEG-hydrazide compound, a PEG- oxyamine compound, or a PEG-bromoacetyl compound. (See, e.g., S. Herman, Polyethylene glycol) with Reactive Endgroups: I. Modification of Proteins, J. Bioactive and Compatible Polymers, 10:145-187 (1995); S. Zalipsky, Chemistry of Polyethylene Glycol Conjugates with Biologically Active Molecules, Advanced Drug Delivery Reviews, 16:157-182 (1995); R. Greenwald et al.,
Poly(ethylene glycol) conjugated drugs and prodrugs: a comprehensive review, Critical Reviews in Therapeutic Drug Carrier Systems, 17:101-161 (2000)).
Methods for N-terminal PEGylation are exemplified herein in Examples 31-34, 45 and 47-48, but these are in no way limiting of the PEGylation methods that can be employed by one skilled in the art.
Any molecular mass for a PEG can be used as practically desired, e.g., from about 1,000 or 2,000 Daltons (Da) to about 100,000 Da (n is 20 to 2300). Preferably, the combined or total molecular mass of PEG used in a PEG-conjugated peptide of the present invention is from about
3,000 Da or 5,000 Da, to about 50,000 Da or 60,000 Da (total n is from 70 to 1,400), more preferably from about 10,000 Da to about 40,000 Da (total n is about 230 to about 910). The most preferred combined mass for PEG is from about 20,000 Da to about 30,000 Da (total n is about 450 to about 680). The number of repeating units "n" in the PEG is approximated for the molecular mass described in Daltons. It is preferred that the combined molecular mass of PEG on an activated linker is suitable for pharmaceutical use. Thus, the combined molecular mass of the PEG molecule should not exceed about 100,000 Da.
Polysaccharide polymers are another type of water-soluble polymer that can be used for protein modification. Dextrans are polysaccharide polymers comprised of individual subunits of glucose predominantly linked by α1-6 linkages. The dextran itself is available in many molecular weight ranges, and is readily available in molecular weights from about 1 kDa to about 70 kDa. Dextran is a suitable water-soluble polymer for use in the present invention as a half-life extending moiety by itself or in combination with another half-life extending moiety (e.g., Fc). See, for example, WO 96/11953 and WO 96/05309. The use of dextran conjugated to therapeutic or diagnostic immunoglobulins has been reported; see, for example, European Patent Publication No. 0 315456, which is hereby incorporated by reference in its entirety. Dextran of about 1 kDa to about 20 kDa is preferred when dextran is used as a half-life extending moiety in accordance with the present invention.
Linkers. Any "linker" group or moiety (i.e., "-(L)f-° or "-(L)9-" in Formulae I-IX) is optional. When present, its chemical structure is not critical, since it serves primarily as a spacer. As stated herein above, the linker moiety (-(L)f - and/or -(L)9-), if present, can be independently the same or different from any other linker, or linkers, that may be present in the inventive composition. For example, an "(L)/' can represent the same moiety as, or a different moiety from, any other "(L)1" or any "(L)9" in accordance with the invention. The linker is preferably made up of amino acids linked together by peptide bonds. Some of these amino acids can be glycosylated, as is well understood by those in the art. For example, a useful linker sequence constituting a sialylation site is X1X2NX4X5G (SEQ ID NO: 637), wherein Xi1 X2, X4 and Xsare each independently any amino acid residue.
As stated above, in some embodiments, a peptidyl linker is present (i.e., made up of amino acids linked together by peptide bonds) that is made in length, preferably, of from 1 up to about 40 amino acid residues, more preferably, of from 1 up to about 20 amino acid residues, and most preferably of from 1 to about 10 amino acid residues. Preferably, but not necessarily, the amino acid residues in the linker are from among the twenty canonical amino acids, more
preferably, cysteine, glycine, alanine, proline, asparagine, glutamine, and /or serine. Even more preferably, a peptidyl linker is made up of a majority of amino acids that are sterically unhindered, such as glycine, serine, and alanine linked by a peptide bond. It is also desirable that, if present, a peptidyl linker be selected that avoids rapid proteolytic turnover in circulation in vivo. Thus, preferred linkers include polyglycines (particularly (GIy)4(SEQ ID NO: 4918), (GIy)5) (SEQ ID NO: 4919), poly(Gly-Ala), and polyalanines. Other preferred linkers are those identified herein as "L5" (GGGGS; SEQ ID NO: 638), 110" (GGGGSGGGGS; SEQ ID NO:79), "L25" GGGGSGGGGSGGGGSGGGGSGGGGS; SEQ ID NO:84) and any linkers used in the working examples hereinafter. The linkers described herein, however, are exemplary; linkers within the scope of this invention can be much longer and can include other residues.
In some embodiments of the compositions of this invention, which comprise a peptide linker moiety (L), acidic residues, for example, glutamate or aspartate residues, are placed in the amino acid sequence of the linker moiety (L). Examples include the following peptide linker sequences: GGEGGG (SEQ ID NO: 639);
GGEEEGGG (SEQ ID NO: 640);
GEEEG (SEQ ID NO: 641);
GEEE (SEQ ID NO: 642);
GGDGGG (SEQ ID NO: 643); GGDDDGG (SEQ ID NO: 644);
GDDDG (SEQ ID NO: 645); GDDD (SEQ ID NO: 646);
GGGGSDDSDEGSDGEDGGGGS (SEQ ID NO: 647); WEWEW (SEQ ID NO: 648); FEFEF (SEQ ID NO: 649);
EEEWWW (SEQ ID NO: 650); EEEFFF (SEQ ID NO: 651); WWEEEWW (SEQ ID NO: 652); or FFEEEFF (SEQ ID NO: 653). In other embodiments, the linker constitutes a phosphorylation site, e.g., X1X2YX3X4G
(SEQ ID NO: 654), wherein Xi, X2,X3 and X4 are each independently any amino acid residue; XiX2SXaX4G (SEQ ID NO: 655), wherein Xi, X2,X3 and X4 are each independently any amino acid
residue; or X1X2TX3X4G (SEQ ID NO: 656), wherein Xi, X2lX3 and X4 are each independently any amino acid residue.
Non-peptide linkers are also possible. For example, alkyl linkers such as -NH-(CH2)S- C(O)-, wherein s = 2-20 could be used. These alkyl linkers can further be substituted by any non- sterically hindering group such as lower alkyl (e.g., C1-C6) lower acyl, halogen (e.g., Cl, Br), CN, NH2, phenyl, etc. An exemplary non-peptide linker is a PEG linker,
(XII)
wherein n is such that the linker has a molecular weight of 100 to 5000 kDa, preferably 100 to 500 kDa. The peptide linkers can be altered to form derivatives in the same manner as described above.
Useful linker embodiments also include aminoethyloxyethyloxy-acetyl linkers as disclosed by Chandy et al. (Chandy et al., WO 2006/042151 A2, incorporated herein by reference in its entirety).
Derivatives. The inventors also contemplate derivatizing the peptide and/or half-life extending moiety portion of the compounds. Such derivatives can improve the solubility, absorption, biological half-life, and the like of the compounds. The moieties can alternatively eliminate or attenuate any undesirable side-effect of the compounds and the like. Exemplary derivatives include compounds in which: 1. The compound or some portion thereof is cyclic. For example, the peptide portion can be modified to contain two or more Cys residues (e.g., in the linker), which could cyclize by disulfide bond formation. 2. The compound is cross-linked or is rendered capable of cross-linking between molecules.
For example, the peptide portion can be modified to contain one Cys residue and thereby be able to form an intermolecular disulfide bond with a like molecule. The compound can also be cross-linked through its C-terminus, as in the molecule shown below.
3. Non-peptidyl linkages (bonds) replace one or more peptidyl [-C(O)NR-] linkages. Exemplary non-peptidyl linkages are -CH2-carbamate [-CH2-0C(0)NR-], phosphonate , -
5 CH2-sulfonamide [-CH2-S(O)2NR-], urea [-NHC(O)NH-], -CH2-secondary amine, and alkylated peptide [-C(O)NR6- wherein R6 is lower alkyl].
4. The N-terminus is chemically derivatized. Typically, the N-terminus can be acylated or modified to a substituted amine. Exemplary N-terminal derivative groups include -NRR1 (other than -NH2), -NRC(O)R1, 0 -NRC(O)OR1, -NRS(O)2R1, -NHC(O)NHR1, succinimide, or benzyloxycarbonyl-NH- (CBZ-
NH-), wherein R and R1 are each independently hydrogen or lower alkyl and wherein the phenyl ring can be substituted with 1 to 3 substituents selected from the group consisting of C1-C4 alkyl, CrC4 alkoxy, chloro, and bromo.
5. The free C-terminus is derivatized. Typically, the C-terminus is esterified or amidated. For 5 example, one can use methods described in the art to add (NH-CH2-CH2-NH2)2 to compounds of this invention having any of SEQ ID NOS: 504 to 508 at the C-terminus. Likewise, one can use methods described in the art to add -NH2 to compounds of this invention having any of SEQ ID NOS: 924 to 955, 963 to 972, 1005 to 1013, or 1018 to 1023 at the C-terminus. Exemplary C-terminal derivative groups include, for example, - 0 C(O)R2 wherein R2 is lower alkoxy or -NR3R4 wherein R3 and R4 are independently hydrogen or CI-CB alkyl (preferably Ci-C4 alkyl).
6. A disulfide bond is replaced with another, preferably more stable, cross-linking moiety (e.g., an alkylene). See, e.g., Bhatnagar et aL (1996), J. Med. Chem. 39: 3814-9; Alberts et al. (1993) Thirteenth Am. Pep. Svmp., 357-9. 5 7. One or more individual amino acid residues are modified. Various derivatizing agents are known to react specifically with selected sidechains or terminal residues, as described in detail below.
Lysinyl residues and amino terminal residues can be reacted with succinic or other carboxylic acid anhydrides, which reverse the charge of the lysinyl residues. Other suitable reagents for derivatizing O alpha-amino-containing residues include imidoesters such as methyl picolinimidate; pyridoxal phosphate;
pyridoxal; chloroborohydride; trinitrobenzenesulfonic acid; O-methylisourea; 2,4 pentanedione; and transaminase-catalyzed reaction with glyoxylate.
Arginyl residues can be modified by reaction with any one or combination of several conventional reagents, including phenylglyoxal, 2,3-butanedione, 1,2-cyclohexanedione, and ninhydrin. 5 Derivatization of arginyl residues requires that the reaction be performed in alkaline conditions because of the high pKa of the guanidine functional group. Furthermore, these reagents can react with the groups of lysine as well as the arginine epsilon-amino group.
Specific modification of tyrosyl residues has been studied extensively, with particular interest in introducing spectral labels into tyrosyl residues by reaction with aromatic diazonium compounds or 0 tetranitromethane. Most commonly, N-acetylimidizole and tetranitromethane are used to form O-acetyl tyrosyl species and 3-nitro derivatives, respectively.
Carboxyl sidechain groups (aspartyl or glutamyl) can be selectively modified by reaction with carbodiimides (R'-N=C=N-R') such as 1-cyclohexyl-3-(2-morpholinyl-(4-ethyl) carbodiimide or 1-ethyl-3-(4- azonia-4,4-dimethylpentyl) carbodiimide. Furthermore, aspartyl and glutamyl residues can be converted5 to asparaginyl and glutaminyl residues by reaction with ammonium ions.
Glutaminyl and asparaginyl residues can be deamidated to the corresponding glutamyl and aspartyl residues. Alternatively, these residues are deamidated under mildly acidic conditions. Either form of these residues falls within the scope of this invention.
Cysteinyl residues can be replaced by amino acid residues or other moieties either to eliminate O disulfide bonding or, conversely, to stabilize cross-linking. See, e.g., Bhatnagar et_aj. (1996), J. Med. Chem. 39: 3814-9.
Derivatization with bifunctional agents is useful for cross-linking the peptides or their functional derivatives to a water-insoluble support matrix or to other macromolecular half-life extending moieties. Commonly used cross-linking agents include, e.g., 1,1-bis(diazoacetyl)-2-phenylethane, glutaraldehyde, 5 N-hydroxysuccinimide esters, for example, esters with 4-azidosalicylic acid, homobifunctional imidoesters, including disuccinimidyl esters such as 3,3'-dithiobis(succinimidylpropionate), and bifunctional maleimides such as bis-N-maleimido-1,8-octane. Derivatizing agents such as methyl-3- [(p-azidophenyl)dithio]propioimidate yield photoactivatable intermediates that are capable of forming crosslinks in the presence of light. Alternatively, reactive water-insoluble matrices such as cyanogen O bromide-activated carbohydrates and the reactive substrates described in U.S. Pat. Nos. 3,969,287;
3,691,016; 4,195,128; 4,247,642; 4,229,537; and 4,330,440 are employed for protein immobilization.
Carbohydrate (oligosaccharide) groups can conveniently be attached to sites that are known to be glycosylation sites in proteins. Generally, O-linked oligosaccharides are attached to
serine (Ser) or threonine (Thr) residues while N-linked oligosaccharides are attached to asparagine (Asn) residues when they are part of the sequence Asn-X-Ser/Thr, where X can be any amino acid except proline. X is preferably one of the 19 naturally occurring amino acids other than proline. The structures of N-linked and O-linked oligosaccharides and the sugar residues found in each type are different. One type of sugar that is commonly found on both is N-acetylneuraminic acid (referred to as sialic acid). Sialic acid is usually the terminal residue of both N-linked and O-linked oligosaccharides and, by virtue of its negative charge, can confer acidic properties to the glycosylated compound. Such site(s) can be incorporated in the linker of the compounds of this invention and are preferably glycosylated by a cell during recombinant production of the 0 polypeptide compounds (e.g., in mammalian cells such as CHO1 BHK, COS). However, such sites can further be glycosylated by synthetic or semi-synthetic procedures known in the art.
Other possible modifications include hydroxylation of proline and lysine, phosphorylation of hydroxyl groups of seryl or threonyl residues, oxidation of the sulfur atom in Cys, methylation of the alpha-amino groups of lysine, arginine, and histidine side chains. Creighton, Proteins: Structure5 and Molecule Properties (W. H. Freeman and Co., San Francisco), pp. 79-86 (1983).
Compounds of the present invention can be changed at the DNA level, as well. The DNA sequence of any portion of the compound can be changed to codons more compatible with the chosen host cell. For E. coli, which is the preferred host cell, optimized codons are known in the art. Codons can be substituted to eliminate restriction sites or to include silent restriction sites, O which can aid in processing of the DNA in the selected host cell. The half-life extending moiety, linker and peptide DNA sequences can be modified to include any of the foregoing sequence changes.
A process for preparing conjugation derivatives is also contemplated. Tumor cells, for example, exhibit epitopes not found on their normal counterparts. Such epitopes include, for 5 example, different post-translational modifications resulting from their rapid proliferation. Thus, one aspect of this invention is a process comprising: a) selecting at least one randomized peptide that specifically binds to a target epitope; and b) preparing a pharmacologic agent comprising (i) at least one half-life extending O moiety (Fc domain preferred), (ii) at least one amino acid sequence of the selected peptide or peptides, and (iii) an effector molecule.
The target epitope is preferably a tumor-specific epitope or an epitope specific to a pathogenic organism. The effector molecule can be any of the above-noted conjugation partners and is preferably a radioisotope.
Methods of Making
5 The present invention also relates to nucleic acids, expression vectors and host cells useful in producing the polypeptides of the present invention. Host cells can be eukaryotic cells, with mammalian cells preferred and CHO cells most preferred. Host cells can also be prokaryotic cells, with E. coli cells most preferred.
The compounds of this invention largely can be made in transformed host cells using 0 recombinant DNA techniques. To do so, a recombinant DNA molecule coding for the peptide is prepared. Methods of preparing such DNA molecules are well known in the art. For instance, sequences coding for the peptides could be excised from DNA using suitable restriction enzymes. Alternatively, the DNA molecule could be synthesized using chemical synthesis techniques, such as the phosphoramidate method. Also, a combination of these techniques could be used. 5 The invention also includes a vector capable of expressing the peptides in an appropriate host. The vector comprises the DNA molecule that codes for the peptides operatively linked to appropriate expression control sequences. Methods of effecting this operative linking, either before or after the DNA molecule is inserted into the vector, are well known. Expression control sequences include promoters, activators, enhancers, operators, ribosomal binding sites, start O signals, stop signals, cap signals, polyadenylation signals, and other signals involved with the control of transcription or translation.
The resulting vector having the DNA molecule thereon is used to transform an appropriate host. This transformation can be performed using methods well known in the art.
Any of a large number of available and well-known host cells can be used in the practice5 of this invention. The selection of a particular host is dependent upon a number of factors recognized by the art. These include, for example, compatibility with the chosen expression vector, toxicity of the peptides encoded by the DNA molecule, rate of transformation, ease of recovery of the peptides, expression characteristics, bio-safety and costs. A balance of these factors must be struck with the understanding that not all hosts can be equally effective for the O expression of a particular DNA sequence. Within these general guidelines, useful microbial hosts include bacteria (such as E. coli sp.), yeast (such as Saccharomvces sp.) and other fungi, insects, plants, mammalian (including human) cells in culture, or other hosts known in the art.
Next, the transformed host is cultured and purified. Host cells can be cultured under conventional fermentation conditions so that the desired compounds are expressed. Such fermentation conditions are well known in the art. Finally, the peptides are purified from culture by methods well known in the art. In some embodiments of the inventive DNA, the DNA encodes a recombinant fusion protein composition of the invention, preferably, but not necessarily, monovalent with respect to the toxin peptide, for expression in a mammalian cell, such as, but not limited to, CHO or HEK293. The encoded fusion protein includes (a)-(c) immediately below, in the N-terminal to C-terminal direction: (a) an immunoglobulin, which includes the constant and variable regions of the immunoglobulin light and heavy chains, or a portion of an immunoglobulin (e.g., an Fc domain, or the variable regions of the light and heavy chains); if both immunoglobulin light chain and heavy chain components are to be included in the construct, then a peptidyl linker, as further described in (b) immediately below, is also included to separate the immunoglobulin components (See, e.g., Figure 92A-C); useful coding sequences for immunoglobulin light and heavy chains are well known in the art;
(b) a peptidyl linker, which is at least 4 (or 5) amino acid residues long and comprises at least one protease cleavage site (e.g., a furin cleavage site, which is particularly useful for intracellular cleavage of the expressed fusion protein); typically, the peptidyl linker sequence can be up to about 35 to 45 amino acid residues long (e.g., a 7x L5 linker modified to include the desired protease cleavage site(s)), but linkers up to about 100 to about 300 amino acid residues long are also useful; and
(c) an immunoglobulin Fc domain or a portion thereof. The Fc domain of (c) can be from the same type of immunoglobulin in (a), or different. In such embodiments, the DNA encodes a toxin peptide covalently linked to the N-terminal or C-terminal end of (a) or (c) above, either directly or indirectly via a peptidyl linker (a linker minus a protease cleavage site). Any toxin peptide or peptide analog thereof as described herein can be encoded by the DNA (e.g., but not limited to, ShK, HmK1 MgTx, AgTxI , AgTx2, HsTxI, OSK1 , Anuroctoxin, Noxiustoxin, Hongotoxin, HsTxI1 ChTx, MTx, Titystoxin, BgK, BmKTX, BmTx, Tc30, Tc32, PM, Pi2, Pi3 toxin peptide, or a peptide analog of any of these). For example, an OSK1 peptide analog comprising an amino acid sequence selected from SEQ ID NOS: 25, 294 through 298, 562 through 636, 980 through 1274, 1303, 1308, 1391 through 4912, 4916, 4920 through 5006, 5009, 5010, and 5012 through 5015, as set forth in Tables 7 and Tables 7A-J, can be employed. Alternatively, an ShK peptide analog
comprising an amino acid sequence selected from SEQ ID NOS: 5, 88 through 200, 548 through 561 , 884 through 950, and 1295 through 1300 as set forth in Table 2, can be employed. Any other toxin peptide sequence described herein that can alternatively be expressed recombinantly using recombinant and protein engineering techniques known in the art can also be used. The immunoglobulin of (a) and (c) above can be in each instance independently selected from any desired type, such as but not limited to, IgGI , lgG2, lgG3, and lgG4. The variable regions can be non-functional in vivo (e.g., CDRs specifically binding KLH)1 or alternatively, if targeting enhancement function is also desired, the variable regions can be chosen to specifically bind (non- competitvely) the ion channel target of the toxin peptide (e.g., Kv1.3) or specifically bind another antigen typically found associated with, or in the vicinity of, the target ion channel. In addition, the inventive DNA optionally further encodes, 5' to the coding region of (a) above, a signal peptide sequence (e.g., a secretory signal peptide) operably linked to the expressed fusion protein. An example of the inventive DNA encoding a recombinant fusion protein for expression in a mammalian cell, described immediately above, is a DNA that encodes a fusion protein comprising, in the N-terminal to C-terminal direction:
(a) an immunoglobulin light chain;
(b) a first peptidyl linker at least 4 amino acid residues long comprising at least one protease cleavage site, as described above;
(c) an immunoglobulin heavy chain; (d) a second peptidyl linker at least 4 amino acid residues long comprising at least one protease cleavage site, as described above; and
(e) an immunoglobulin Fc domain or a portion thereof. Here, the Fc domain of (e) can be from the same type of immunoglobulin as the heavy chain in (c), or different. The DNA encodes a toxin peptide covalently linked to the N-terminal or C-terminal end of (a), (c), or (e) of the expressed fusion protein, either directly or indirectly via a peptidyl linker (a linker minus a protease cleavage site). Figure 92A-C illustrates schematically an embodiment, in which the toxin peptide (e.g., an OSK1 , ShK, or a peptide analog of either of these) is covalently linked to the C-terminal end of the Fc domain of (e). In Figure 92A-C, a linker is shown covalently linking the toxin peptide to the rest of the molecule, but as previously described, this linker is optional. In some embodiments particularly suited for the recombinant expression of monovalent dimeric Fc-toxin peptide fusions or "peptibodies" (see, Figure 2B and Example 56) by mammalian cells, such as, but not limited to, CHO or HEK293, the inventive DNA encodes a recombinant expressed fusion protein that comprises, in the N-terminal to C-terminal direction:
(a) a first immunoglobulin Fc domain or portion thereof;
(b) a peptidyl linker at least 4 (or 5) amino acid residues long comprising at least one protease cleavage site (e.g., a furin cleavage site, which is particularly useful for intracellular cleavage of the expressed fusion protein); typically, the peptidyl linker sequence can be up to about 35 to 45 amino acid residues long (e.g., a 7x L5 linker modified to include the desired protease cleavage site(s)), but linkers up to about 100 to about 300 amino acid residues long are also useful; and
(c) a second immunoglobulin Fc domain or portion thereof (which may be the same or different from the first Fc domain, but should be expressed in the same orientation as the first Fc domain).
For such embodiments, the DNA encodes a toxin peptide covalently linked to the N-terminal or C- terminal end of (a) or (c) of the expressed fusion protein, either directly or indirectly via a peptidyl linker (a linker minus a protease cleavage site); Example 56 describes an embodiment in which the toxin peptide is conjugated to the C-terminal end of the second immunoglobulin Fc domain (c). Any toxin peptide or peptide analog thereof as described herein can be encoded by the DNA (e.g., but not limited to, ShK, HmK1 MgTx, AgTxI, AgTx2, HsTxI, OSK1, Anuroctoxin, Noxiustoxin, Hongotoxin, HsTxI1 ChTx, MTx1 Titystoxin, BgK, BmKTX1 BmTx1 Tc30, Tc32, PM 1 Pi2, Pi3 toxin peptide, or a peptide analog of any of these). For example, an OSK1 peptide analog comprising an amino acid sequence selected from SEQ ID NOS: 25, 294 through 298, 562 through 636, 980 through 1274, 1303, 1308, 1391 through 4912, 4916, 4920 through 5006, 5009, 5010, and 5012 through 5015, as set forth in Tables 7 and Tables 7A-J, can be employed. Alternatively, an ShK peptide analog comprising an amino acid sequence selected from SEQ ID NOS: 5, 88 through 200, 548 through 561, 884 through 950, and 1295 through 1300 as set forth in Table 2, can be employed. Any other toxin peptide sequence described herein that can alternatively be expressed using recombinant and protein engineering techniques known in the art can also be used. In addition, the inventive DNA optionally further encodes, 5' to the coding region of (a) above, a signal peptide sequence (e.g., a secretory signal peptide) operably linked to the expressed fusion protein.
DNA constructs similar to those described above are also useful for recombinant expression by mammalian cells of other dimeric Fc fusion proteins ("peptibodies") or chimeric immunoglobulin(light chain + heavy chain)-Fc heterotrimers ("hemibodies"), conjugated to pharmacologically active peptides (e.g., agonist or antagonist peptides) other than toxin peptides.
Peptide compositions of the present invention can also be made by synthetic methods. Solid phase synthesis is the preferred technique of making individual peptides since it is the most
cost-effective method of making small peptides. For example, well known solid phase synthesis techniques include the use of protecting groups, linkers, and solid phase supports, as well as specific protection and deprotection reaction conditions, linker cleavage conditions, use of scavengers, and other aspects of solid phase peptide synthesis. Suitable techniques are well known in the art. (E.g., Merrifield (1973), Chem. Polypeptides, pp. 335-61 (Katsoyannis and Panayotis eds.); Merrifield (1963), J. Am. Chem. Soc. 85: 2149; Davis et a!. (1985), Biochem. Intl. 10: 394-414; Stewart and Young (1969), Solid Phase Peptide Synthesis: U.S. Pat. No. 3,941,763; Finn et_al. (1976), The Proteins (3rd ed.) 2: 105-253; and Erickson et aL (1976), The Proteins (3rd ed.) 2: 257-527; "Protecting Groups in Organic Synthesis," 3rd Edition, T. W. Greene and P. G. M. Wuts, Eds., John Wiley & Sons, Inc., 1999; NovaBiochem Catalog, 2000; "Synthetic Peptides, A User's Guide," G. A. Grant, Ed., W.H. Freeman & Company, New York, N.Y., 1992; "Advanced Chemtech Handbook of Combinatorial & Solid Phase Organic Chemistry," W. D. Bennet, J. W. Christensen, L. K. Hamaker, M. L. Peterson, M. R. Rhodes, and H. H. Saneii, Eds., Advanced Chemtech, 1998; "Principles of Peptide Synthesis, 2nd ed.," M. Bodanszky, Ed., Springer-Verlag, 1993; "The Practice of Peptide Synthesis, 2nd ed.," M. Bodanszky and A. Bodanszky, Eds.,
Springer-Verlag, 1994; "Protecting Groups," P. J. Kocienski, Ed., Georg Thieme Verlag, Stuttgart, Germany, 1994; "Fmoc Solid Phase Peptide Synthesis, A Practical Approach," W. C. Chan and P. D. White, Eds., Oxford Press, 2000, G. B. Fields et al., Synthetic Peptides: A User's Guide, 1990, 77-183). Whether the compositions of the present invention are prepared by synthetic or recombinant techniques, suitable protein purification techniques can also be involved, when applicable. In some embodiments of the compositions of the invention, the toxin peptide portion and/or the half-life extending portion, or any other portion, can be prepared to include a suitable isotopic label (e.g., 1251, 14C, 13C1 35S, 3H, 2H, 13N, 15N, 180, 17O, efc.), for ease of quantification or detection.
Compounds that contain derivatized peptides or which contain non-peptide groups can be synthesized by well-known organic chemistry techniques. Uses of the Compounds In general. The compounds of this invention have pharmacologic activity resulting from their ability to bind to proteins of interest as agonists, mimetics or antagonists of the native ligands of such proteins of interest. Heritable diseases that have a known linkage to ion channels ("channelopathies") cover various fields of medicine, some of which include neurology, nephrology, myology and cardiology. A list of inherited disorders attributed to ion channels includes:
• cystic fibrosis (Cl- channel; CFTR),
• Dent's disease (proteinuria and hypercalciuria; Cl- channel; CLCN5),
• osteopetrosis (Ch channel; CLCN7),
• familial hyperinsulinemia (SUR1 ; KCNJ11 ; K channel), • diabetes (KATP / SUR channel),
• Andersen syndrome (KCNJ2, Kir2.1 K channel),
• Bartter syndrome (KCNJ1 ; KiM .1/ROMK; K channel),
• hereditary hearing loss (KCNQ4; K channel),
• hereditary hypertension (Liddle's syndrome; SCNN1 ; epithelial Na channel), • dilated cardiomyopathy (SUR2, K channel),
• long-QT syndrome or cardiac arrhythmias (cardiac potassium and sodium channels),
• Thymothy syndrome (CACNA1 C, Cav1.2),
• myasthenic syndromes (CHRNA.CHRNB.CNRNE; nAChR), and a variety of other myopathies, • hyperkalemic periodic paralysis (Na and K channels),
• epilepsy (Na+ and K+ channels),
• hemiplegic migraine (CACNA1A, Cav2.1 Ca2+ channel and ATP1A2),
• central core disease (RYR1 , RyR1 ; Ca2+ channel), and
• paramyotonia and myotonia (Na+, Ch channels) See LJ. Ptacek and Y-H Fu (2004). Arch. Neurol. 61 : 166-8; BA Niemeyer et al. (2001), EMBO reports 21 : 568-73; F. Lehmann-Hom and K. Jurkat-Rott (1999), Physiol. Rev. 79: 1317-72. Although the foregoing list concerned disorders of inherited origin, molecules targeting the channels cited in these disorders can also be useful in treating related disorders of other, or indeterminate, origin. In addition to the aforementioned disorders, evidence has also been provided supporting ion channels as targets for treatment of:
• sickle cell anemia (IKCaI) - in sickle cell anemia, water loss from erythrocytes leads to hemoglobin polymerization and subsequent hemolysis and vascular obstruction. The water loss is consequent to potassium efflux through the so-called Gardos channel i.e., IKCaI . Therefore, block of IKCaI is a potential therapeutic treatment for sickle cell anemia.
• glaucoma (BKCa), - in glaucoma the intraocular pressure is too high leading to optic nerve damage, abnormal eye function and possibly blindness. Block of BKCa
potassium channels can reduce intraocular fluid secretion and increase smooth muscle contraction, possibly leading to lower intraocular pressure and neuroprotection in the eye.
• multiple sclerosis (Kv1 KCa), 5 • psoriasis (Kv1 KCa),
• arthritis (Kv, KCa)1
• asthma (KCa, Kv)1
• allergy(KCa, Kv)1
• COPD (KCa1 Kv1 Ca), 0 • allergic rhinitis (KCa1 Kv)1
• pulmonary fibrosis,
• lupus (IKCaI, Kv)1
• transplantation, GvHD (KCa1 Kv)1
• inflammatory bone resorption (KCa1 Kv), 5 • periodontal disease (KCa, Kv),
• diabetes, type I (Kv), - type I diabetes is an autoimmune disease that is characterized by abnormal glucose, protein and lipid metabolism and is associated with insulin deficiency or resistance. In this disease, Kv1.3-expressing T-lymphocytes attack and destroy pancreatic islets leading to loss of beta-cells. Block of Kv1.3 decreases O inflammatory cytokines. In addition block of Kv1.3 facilitates the translocation of GLUT4 to the plasma membrane, thereby increasing insulin sensitivity.
• obesity (Kv), - Kv1.3 appears to play a critical role in controlling energy homeostasis and in protecting against diet-induced obesity. Consequently, Kv1.3 blockers could increase metabolic rate, leading to greater energy utilization and decreased body 5 weight.
• restenosis (KCa1 Ca2+), - proliferation and migration of vascular smooth muscle cells can lead to neointimal thickening and vascular restenosis. Excessive neointimal vascular smooth muscle cell proliferation is associated with elevated expression of IKCaL Therefore, block of IKCaI could represent a therapeutic strategy to prevent O restenosis after angioplasty.
• ischaemia (KCa1 Ca2+), - in neuronal or cardiac ischemia, depolarization of cell membranes leads to opening of voltage-gated sodium and calcium channels. In turn this can lead to calcium overload, which is cytotoxic. Block of voltage-gated sodium
and/or calcium channels can reduce calcium overload and provide cytoprotective effects. In addition, due to their critical role in controlling and stabilizing cell membrane potential, modulators of voltage- and calcium-activated potassium channels can also act to reduce calcium overload and protect cells. • renal incontinence (KCa), renal incontinence is associated with overactive bladder smooth muscle cells. Calcium-activated potassium channels are expressed in bladder smooth muscle cells, where they control the membrane potential and indirectly control the force and frequency of cell contraction. Openers of calcium-activated potassium channels therefore provide a mechanism to dampen electrical and contractile activity in bladder, leading to reduced urge to urinate.
• osteoporosis (Kv),
• pain, including migraine (NaV) TRP [transient receptor potential channels], P2X, Ca2+), N-type voltage-gated calcium channels are key regulators of nociceptive neurotransmission in the spinal cord. Ziconotide, a peptide blocker of N-type calcium channels reduces nociceptive neurotransmission and is approved worldwide for the symptomatic alleviation of severe chronic pain in humans. Novel blockers of nociceptor- specific N-type calcium channels would be improved analgesics with reduced side- effect profiles.
• hypertension (Ca2+), - L-type and T-type voltage-gated calcium channels are expressed in vascular smooth muscle cells where they control excitation-contraction coupling and cellular proliferation. In particular, T-type calcium channel activity has been linked to neointima formation during hypertension. Blockers of L-type and T-type calcium channels are useful for the clinical treatment of hypertension because they reduce calcium influx and inhibit smooth muscle cell contraction. • wound healing, cell migration serves a key role in wound healing. Intracellular calcium gradients have been implicated as important regulators of cellular migration machinery in keratinocytes and fibroblasts. In addition, ion flux across cell membranes is associated with cell volume changes. By controlling cell volume, ion channels contribute to the intracellular environment that is required for operation of the cellular migration machinery. In particular, IKCaI appears to be required universally for cell migration. In addition, Kv1.3, Kv3.1, NMDA receptors and N-type calcium channels are associated with the migration of lymphocytes and neurons.
• stroke,
• Alzheimer's,
• Parkenson's Disease (nACHR, Nav)
• Bipolar Disorder (Nav, Cav)
• cancer, many potassium channel genes are amplified and protein subunits are upregulated in many cancerous condition. Consistent with a pathophysiological role for potassium channel upregulation, potassium channel blockers have been shown to suppress proliferation of uterine cancer cells and hepatocarcinoma cells, presumably through inhibition of calcium influx and effects on calcium-dependent gene expression.
• a variety of neurological, cardiovascular, metabolic and autoimmune diseases.
Both agonists and antagonists of ion channels can achieve therapeutic benefit. Therapeutic benefits can result, for example, from antagonizing Kv1.3, IKCaI, SKCa, BKCa, N- type or T-type Ca2+ channels and the like. Small molecule and peptide antagonists of these channels have been shown to possess utility in vitro and in vivo. Limitations in production efficiency and pharmacokinetics, however, have largely prevented clinical investigation of inhibitor peptides of ion channels.
Compositions of this invention incorporating peptide antagonists of the voltage-gated potassium channel Kv1.3, in particular 0SK1 peptide analogs, whether or not conjugated to a half- life extending moiety, are useful as immunosuppressive agents with therapeutic value for autoimmune diseases. For example, such molecules are useful in treating multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, and rheumatoid arthritis. (See, e.g., H. Wulff et al. (2003) J. Clin. Invest. 111 , 1703-1713 and H. Rus et al. (2005) PNAS 102, 11094-11099; Beeton et al., Targeting effector memory T cells with a selective inhibitor peptide of Kv1.3 channnels for therapy of autoimmune diseases, Molec. Pharmacol. 67(4):1369-81 (2005);1 Beeton et al. (2006), Kv1.3: therapeutic target for cell-mediated autoimmune disease, electronic preprint at
//webfiles. uci.edu/xythoswfs/webui/ 2670029.1). Inhibitors of the voltage-gated potassium channel Kv1.3 have been examined in a variety of preclinical animal models of inflammation. Small molecule and peptide inhibitors of Kv1.3 have been shown to block delayed type hypersensitivity responses to ovalbumin [C. Beeton et al. (2005) MoI. Pharmacol. 67, 1369] and tetanus toxoid [G. C. Koo et al. (1999) Clin. Immunol. 197, 99]. In addition to suppressing inflammation in the skin, inhibitors also reduced antibody production [G.C. Koo et al. (1997) J. Immunol. 158, 5120]. Kv1.3 antagonists have shown efficacy in a rat adoptive-transfer experimental autoimmune encephalomyelitis (AT-EAE) model of multiple sclerosis (MS). The Kv1.3 channel is
overexpressed on myelin-specific T cells from MS patients, lending further support to the utility Kv1.3 inhibitors may provide in treating MS. Inflammatory bone resorption was also suppressed by Kv1.3 inhibitors in a preclinical adoptive-transfer model of periodontal disease [P. Valverde et al. (2004) J. Bone Mineral Res. 19, 155]. In this study, inhibitors additionally blocked antibody production to a bacterial outer membrane protein, - one component of the bacteria used to induce gingival inflammation. Recently in preclinical rat models, efficacy of Kv1.3 inhibitors was shown in treating pristane-induced arthritis and diabetes [C. Beeton et al. (2006) preprint available at //webfιles.uci.edu/xythoswfs/webui/_xy-2670029_1.]. The Kv1.3 channel is expressed on all subsets of T cells and B cells, but effector memory T cells and class-switched memory B cells are particularly dependent on Kv1.3 [H. Wulff et al. (2004) J. Immunol. 173, 776]. Gad5 / insulin- specific T cells from patients with new onset type 1 diabetes, myelin-specific T cells from MS patients and T cells from the synovium of rheumatoid arthritis patients all overexpress Kv1.3 [C. Beeton et al. (2006) preprint at //webfiles.uci.edu/xythoswfs/webui/_xy-2670029_1.]. Because mice deficient in Kv1.3 gained less weight when placed on a high fat diet [J. Xu et al. (2003) Human MoI. Genet. 12, 551] and showed altered glucose utilization [J. Xu et al. (2004) Proc. Natl. Acad. Sci. 101, 3112], Kv1.3 is also being investigated for the treatment of obesity and diabetes. Breast cancer specimens [M. Abdul et al. (2003)Anticancer Res. 23, 3347] and prostate cancer cell lines [S.P. Fraser et al. (2003) Pflugers Arch. 446, 559] have also been shown to express Kv1.3, and Kv1.3 blockade may be of utility for treatment of cancer. Disorders that can be treated in accordance with the inventive method of treating an autoimmune disorder, involving Kv1.3 inhibitor toxin peptide(s), include multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, contact-mediated dermatitis, rheumatoid arthritis, psoriatic arthritis, asthma, allergy, restinosis, systemic sclerosis, fibrosis, scleroderma, glomerulonephritis, Sjogren syndrome, inflammatory bone resorption, transplant rejection, graft-versus-host disease, and systemic lupus erythematosus (SLE) and other forms of lupus.
Some of the cells that express the calcium-activated potassium of intermediate conductance IKCaI include T cells, B cells, mast cells and red blood cells (RBCs). T cells and RBCs from mice deficient in IKCaI show defects in volume regulation [T. Begenisich et al. (2004) J. Biol. Chem. 279, 47681], Preclinical and clinical studies have demonstrated IKCaI inhibitors utility in treating sickle cell anemia [J. W. Stocker et al. (2003) Blood 101, 2412; www.icagen.com].
Blockers of the IKCaI channel have also been shown to block EAE, indicating they may possess utility in treatment of MS [E. P. Reich et al. (2005) Eur. J. Immunol. 35, 1027]. IgE-mediated histamine production from mast cells is also blocked by IKCaI inhibitors [S. Mark Duffy et al.
(2004) J. Allergy Clin. Immunol. 114, 66], therefore they may also be of benefit in treating asthma. The IKCaI channel is overexpressed on activated T and B lymphocytes [H. Wulff et al. (2004) J. Immunol. 173, 776] and thus may show utility in treatment of a wide variety of immune disorders. Outside of the immune system, IKCaI inhibitors have also shown efficacy in a rat model of vascular restinosis and thus might represent a new therapeutic strategy to prevent restenosis after angioplasty [R. Kohler et al. (2003) Circulation 108, 1119]. It is also thought that IKCaI antagonists are of utility in treatment of tumor angiogenesis since inhibitors suppressed endothelial cell proliferation and angionenesis in vivo [I. Grgic et al. (2005) Arterioscler. Thromb. Vase. Biol. 25, 704]. The IKCaI channel is upregulated in pancreatic tumors and inhibitors blocked proliferation of pancreatic tumor cell lines [H. Jager et al. (2004) MoI Pharmacol. 65, 630]. IKCaI antagonists may also represent an approach to attenuate acute brain damage caused by traumatic brain injury [F. Mauler (2004) Eur. J. Neurosci. 20, 1761]. Disorders that can be treated with IKCaI inhibitors include multiple sclerosis, asthma, psoriasis, contact-mediated dermatitis, rheumatoid & psoriatic arthritis, inflammatory bowel disease, transplant rejection, graft-versus-host disease, Lupus, restinosis, pancreatic cancer, tumor angiogenesis and traumatic brain injury.
Accordingly, molecules of this invention incorporating peptide antagonists of the calcium- activated potassium channel of intermediate conductance, IKCa can be used to treat immune dysfunction, multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, contact- mediated dermatitis, rheumatoid arthritis, psoriatic arthritis, asthma, allergy, restinosis, systemic sclerosis, fibrosis, scleroderma, glomerulonephritis, Sjogren syndrome, inflammatory bone resorption, transplant rejection, graft-versus-host disease, and lupus.
Accordingly, the present invention includes a method of treating an autoimmune disorder, which involves administering to a patient who has been diagnosed with an autoimmune disorder, such as multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, contact- mediated dermatitis, rheumatoid arthritis, psoriatic arthritis, asthma, allergy, restinosis, systemic sclerosis, fibrosis, scleroderma, glomerulonephritis, Sjogren syndrome, inflammatory bone resorption, transplant rejection, graft-versus-host disease, or lupus, a therapeutically effective amount of the inventive composition of matter, whereby at least one symptom of the disorder is alleviated in the patient. "Alleviated" means to be lessened, lightened, diminished, softened, mitigated (i.e., made more mild or gentle), quieted, assuaged, abated, relieved, nullified, or allayed, regardless of whether the symptom of interest is entirely erased, eradicated, eliminated, or prevented in a particular patient.
The present invention is further directed to a method of preventing or mitigating a relapse of a symptom of multiple sclerosis, which method involves administering to a patient, who has previously experienced at least one symptom of multiple sclerosis, a prophylactically effective amount of the inventive composition of matter, such that the at least one symptom of multiple sclerosis is prevented from recurring or is mitigated.
The inventive compositions of matter preferred for use in practicing the inventive method of treating an autoimmune disorder, e.g., inflammatory bowel disease (IBD, including Crohn's Disease and ulcerative colitis), and the method of preventing or mitigating a relapse of a symptom of multiple sclerosis include as P (conjugated as in Formula I), a Kv1.3 or IKCaI antagonist peptide, such as a ShK peptide, an 0SK1 peptide or an 0SK1 peptide analog, a ChTx peptide and/or a Maurotoxin (MTx) peptide, or peptide analogs of any of these.
For example, the conjugated ShK peptide peptide or ShK peptide analog can comprise an amino acid sequence selected from the following:
SEQ ID NOS: 5, 88 through 200, 548 through 561, 884 through 950, or 1295 through 1300 as set forth in Table 2.
The conjugated OSK1 peptide, or conjugated or unconjugated OSK1 peptide analog, can comprise an amino acid sequence selected from the following:
SEQ ID NOS: 25, 294 through 298, 562 through 636, 980 through 1274,
GVIINVSCKISRQCLEPCKKAGMRFGKCMNGKCHCTPK (OSK1-S7)(SEQ ID NO: 1303), or GVIINVSCKISRQCLKPCKDAGMRFGKCMNGKCHCTPK (OSK1-S7,K16,D20)(SEQ ID
NO: 1308) as set forth in Table 7, or any of SEQ ID NOS: 1391 through 4912, 4916, 4920 through 5006, 5009, 5010, and 5012 through 5015 as set forth in Table 7A, Table 7B,
Table 7C, Table 7D1 Table 7E, Table 7F, Table 7G, Table 7H, Table 71, or Table 7J.
Also by way of example, a the conjugated MTX peptide, MTX peptide analog, ChTx peptide or ChTx peptide analog can comprise an amino acid sequence selected from:
SEQ ID NOS: 20, 330 through 343, 1301, 1302, 1304 through 1307, 1309, 1311, 1312, or 1315 through 1336 as set forth in Table 13; or SEQ ID NOS: 36, 59, 344 through 346, or 1369 through 1390 as set forth in Table 14. Also useful in these methods conjugated, or unconjugated, are a Kv1.3 or IKCaI inhibitor toxin peptide analog that comprises an amino acid sequence selected from:
SEQ ID NOS: 88, 89, 92, 148 through 200, 548 through 561, 884 through 949, or 1295 through 1300 as set forth in Table 2; or
SEQ ID NOS: 980 through 1274,
GVIINVSCKISRQCLEPCKKAGMRFGKCMNGKCHCTPK (0SK1-S7)(SEQ ID NO: 1303), or GVIINVSCKISRQCLKPCKDAGMRFGKCMNGKCHCTPK (OSK1-S7,K16,D20)(SEQ ID NO: 1308) as set forth in Table 7; or SEQ ID NOS: 330 through 337, 341, 1301, 1302, 1304 through 1307, 1309, 1311, 1312, and 1315 through 1336 as set forth in Table 13.
In accordance with these inventive methods, a patient who has been diagnosed with an autoimmune disorder, such as, but not limited to multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, contact-mediated dermatitis, rheumatoid arthritis, psoriatic arthritis, asthma, allergy, restinosis, systemic sclerosis, fibrosis, scleroderma, glomerulonephritis, Sjogren syndrome, inflammatory bone resorption, transplant rejection, graft-versus-host disease, or lupus, or a patient who has previously experienced at least one symptom of multiple sclerosis, are well- recognizable and/or diagnosed by the skilled practitioner, such as a physician, familiar with autoimmune disorders and their symptoms. For example, symptoms of multiple sclerosis can include the following: visual symptoms, such as, optic neuritis (blurred vision, eye pain, loss of color vision, blindness); diplopia (double vision); nystagmus (jerky eye movements); ocular dysmetria (constant under- or overshooting eye movements); internuclear ophthalmoplegia (lack of coordination between the two eyes, nystagmus, diplopia); movement and sound phosphenes (flashing lights when moving eyes or in response to a sudden noise); afferent pupillary defect (abnormal pupil responses); motor symptoms, such as, paresis, monoparesis, paraparesis, hemiparesis, quadraparesis (muscle weakness - partial or mild paralysis); plegia, paraplegia, hemiplegia, tetraplegia, quadraplegia (paralysis - total or near total loss of muscle strength); spasticity (loss of muscle tone causing stiffness, pain and restricting free movement of affected limbs); dysarthria (slurred speech and related speech problems); muscle atrophy (wasting of muscles due to lack of use); spasms, cramps (involuntary contraction of muscles); hypotonia, clonus (problems with posture); myoclonus, myokymia (jerking and twitching muscles, tics); restless leg syndrome (involuntary leg movements, especially bothersome at night); footdrop (foot drags along floor during walking); dysfunctional reflexes (MSRs, Babinski's, Hoffman's, Chaddock's); sensory symptoms, such as, paraesthesia (partial numbness, tingling, buzzing and vibration sensations); anaesthesia (complete numbness/loss of sensation); neuralgia,
neuropathic and neurogenic pain (pain without apparent cause, burning, itching and electrical shock sensations); L'Hermitte's (electric shocks and buzzing sensations when moving head); proprioceptive dysfunction (loss of awareness of location of body parts); trigeminal neuralgia (facial pain); coordination and balance symptoms, such as, ataxia (loss of coordination); intention tremor (shaking when performing fine movements); dysmetria (constant under- or overshooting limb movements); vestibular ataxia (abnormal balance function in the inner ear); vertigo (nausea/vomitting/sensitivity to travel sickness from vestibular ataxia); speech ataxia (problems coordinating speech, stuttering); dystonia (slow limb position 0 feedback); dysdiadochokinesia (loss of ability to produce rapidly alternating movements, for example to move to a rhythm); bowel, bladder and sexual symptoms, such as, frequent micturation, bladder spasticity (urinary urgency and incontinence); flaccid bladder, detrusor-sphincter dyssynergia (urinary hesitancy and retention); erectile dysfunction (male and female impotence); 5 anorgasmy (inability to achieve orgasm); retrograde ejaculation (ejaculating into the bladder); frigidity (inability to become sexually aroused); constipation (infrequent or irregular bowel movements); fecal urgency (bowel urgency); fecal incontinence (bowel incontinence); cognitive symptoms, such as, depression; cognitive dysfunction (short-term and long-term O memory problems, forgetfulness, slow word recall); dementia; mood swings, emotional lability, euphoria; bipolar syndrome; anxiety; aphasia, dysphasia (impairments to speech comprehension and production); and other symptoms, such as, fatigue; Uhthoff s Symptom (increase in severity of symptoms with heat); gastroesophageal reflux (acid reflux); impaired sense of taste and smell; 5 epileptic seizures; swallowing problems, respiratory problems; and sleeping disorders.
By way of further example, symptoms of inflammatory bowel disease can include the following symptoms of Crohn's Disease or ulcerative colitis:
A. Symptoms of Crohn's disease can include: O Abdominal pain. The pain often is described as cramping and intermittent, and the abdomen may be sore when touched. Abdominal pain may turn to a dull, constant ache as the condition progresses.
Diarrhea. Some patients may have diarrhea 10 to 20 times per day. They may wake up at night and need to go to the bathroom. Crohn's disease may cause blood in stools, but not always. Loss of appetite. Fever. In severe cases, fever or other symptoms that affect the entire body may develop. A high fever may indicate a complication involving infection, such as an abscess.
Weight loss. Ongoing symptoms, such as diarrhea, can lead to weight loss. Too few red blood cells (anemia). Some patients with Crohn's disease develop anemia because of low iron levels caused by bloody stools or the intestinal inflammation itself.
B. The symptoms of ulcerative colitis may include:
Diarrhea or rectal urgency. Some patients may have diarrhea 10 to 20 times per day. The urge to defecate may wake patients at night.
Rectal bleeding. Ulcerative colitis usually causes bloody diarrhea and mucus.
Patients also may have rectal pain and an urgent need to empty the bowels.
Abdominal pain, often described as cramping. The patient's abdomen may be sore when touched. Constipation. This symptom may develop depending on what part of the colon is affected.
Loss of appetite.
Fever. In severe cases, fever or other symptoms that affect the entire body may develop. Weight loss. Ongoing (chronic) symptoms, such as diarrhea, can lead to weight loss.
Too few red blood cells (anemia). Some patients develop anemia because of low iron levels caused by bloody stools or intestinal inflammation. The symptoms of multiple sclerosis and inflammatory bowel disease (including Crohn's Disease and ulcerative colitis) enumerated above, are merely illustrative and are not intended to be an exhaustive description of all possible symptoms experienced by a single patient or by several sufferers in composite, and to which the present invention is directed. Those skilled in the art are aware of various clinical symptoms and constellations of symptoms of autoimmune disorders
suffered by individual patients, and to those symptoms are also directed the present inventive methods of treating an autoimmune disorder or of preventing or mitigating a relapse of a symptom of multiple sclerosis.
The therapeutically effective amount, prophylactically effective amount, and dosage regimen involved in the inventive methods of treating an autoimmune disorder or of preventing or mitigating a relapse of a symptom of multiple sclerosis, will be determined by the attending physician, considering various factors which modify the action of therapeutic agents, such as the age, condition, body weight, sex and diet of the patient, the severity of the condition being treated, time of administration, and other clinical factors. Generally, the daily amount or regimen should be in the range of about 1 to about 10,000 micrograms (μg) of the vehicle-conjugated peptide per kilogram (kg) of body mass, preferably about 1 to about 5000 μg per kilogram of body mass, and most preferably about 1 to about 1000 μg per kilogram of body mass.
Molecules of this invention incorporating peptide antagonists of the voltage-gated potassium channel Kv2.1 can be used to treat type Il diabetes. Molecules of this invention incorporating peptide antagonists of the M current (e.g.,
BeKm-1) can be used to treat Alzheimer's disease and enhance cognition.
Molecules of this invention incorporating peptide antagonists of the voltage-gated potassium channel Kv4.3 can be used to treat Alzheimer's disease.
Molecules of this invention incorporating peptide antagonists of the calcium-activated potassium channel of small conductance, SKCa can be used to treat epilepsy, memory, learning, neuropsychiatric, neurological, neuromuscular, and immunological disorders, schizophrenia, bipolar disorder, sleep apnea, neurodegeneration, and smooth muscle disorders.
Molecules of this invention incorporating N-type calcium channel antagonist peptides are useful in alleviating pain. Peptides with such activity (e.g., Ziconotide™, ω-conotoxin-MVIIA) have been clinically validated.
Molecules of this invention incorporating T-type calcium channel antagonist peptides are useful in alleviating pain. Several lines of evidence have converged to indicate that inhibition of Cav3.2 in dorsal root ganglia may bring relief from chronic pain. T-type calcium channels are found at extremely high levels in the cell bodies of a subset of neurons in the DRG; these are likely mechanoreceptors adapted to detect slowly-moving stimuli (Shin et al., Nature Neuroscience
6:724-730, 2003), and T-type channel activity is likely responsible for burst spiking (Nelson et al., J Neurosci 25:8766-8775, 2005). Inhibition of T-type channels by either mibefradil or ethosuximide reverses mechanical allodynia in animals induced by nerve injury (Dogrul et al., Pain 105:159-168,
2003) or by chemotherapy (Flatters and Bennett, Pain 109:150-161 , 2004). Antisense to Cav3.2, but not Cav3.1 or Cav3.3, increases pain thresholds in animals and also reduces expression of Cav3.2 protein in the DRG (Bourinet et al., EMBO J 24:315-324, 2005). Similarly, locally injected reducing agents produce pain and increase Cav3.2 currents, oxidizing agents reduce pain and inhibit Cav3.2 currents, and peripherally administered neurosteroids are analgesic and inhibit T- type currents from DRG (Todorovic et al., Pain 109:328-339, 2004; Pathirathna et al., Pain 114:429-443, 2005). Accordingly, it is thought that inhibition of Cav3.2 in the cell bodies of DRG neurons can inhibit the repetitive spiking of these neurons associated with chronic pain states.
Molecules of this invention incorporating L-type calcium channel antagonist peptides are useful in treating hypertension. Small molecules with such activity (e.g., DHP) have been clinically validated.
Molecules of this invention incorporating peptide antagonists of the Nav1 (TTXs-type) channel can be used to alleviate pain. Local anesthetics and tricyclic antidepressants with such activity have been clinically validated. Such molecules of this invention can in particular be useful as muscle relaxants.
Molecules of this invention incorporating peptide antagonists of the Navi(TTXR-type) channel can be used to alleviate pain arising from nerve and or tissue injury.
Molecules of this invention incorporating peptide antagonists of glial & epithelial cell Ca2+- activated chloride channel can be used to treat cancer and diabetes. Molecules of this invention incorporating peptide antagonists of NMDA receptors can be used to treat pain, epilepsy, brain and spinal cord injury.
Molecules of this invention incorporating peptide antagonists of nicotinic receptors can be used as muscle relaxants. Such molecules can be used to treat pain, gastric motility disorders, urinary incontinence, nicotine addiction, and mood disorders. Molecules of this invention incorporating peptide antagonists of 5HT3 receptor can be used to treat Nausea, pain, and anxiety.
Molecules of this invention incorporating peptide antagonists of the norepinephrine transporter can be used to treat pain, anti-depressant, learning, memory, and urinary incontinence.
Molecules of this invention incorporating peptide antagonists of the Neurotensin receptor can be used to treat pain.
In addition to therapeutic uses, the compounds of the present invention can be useful in diagnosing diseases characterized by dysfunction of their associated protein of interest. In one embodiment, a method of detecting in a biological sample a protein of interest (e.g., a receptor)
that is capable of being activated comprising the steps of: (a) contacting the sample with a compound of this invention; and (b) detecting activation of the protein of interest by the compound. The biological samples include tissue specimens, intact cells, or extracts thereof. The compounds of this invention can be used as part of a diagnostic kit to detect the presence of their associated 5 proteins of interest in a biological sample. Such kits employ the compounds of the invention having an attached label to allow for detection. The compounds are useful for identifying normal or abnormal proteins of interest.
The therapeutic methods, compositions and compounds of the present invention can also be employed, alone or in combination with other molecules in the treatment of disease. 0 Pharmaceutical Compositions
In General. The present invention also provides pharmaceutical compositions comprising the inventive composition of matter and a pharmaceutically acceptable carrier. Such pharmaceutical compositions can be configured for administration to a patient by a wide variety of delivery routes, e.g., an intravascular delivery route such as by injection or infusion, subcutaneous, intramuscular, intraperitoneal, 5 epidural, or intrathecal delivery routes, or for oral, enteral, pulmonary (e.g., inhalant), intranasal, transmucosal (e.g., sublingual administration), transdermal or other delivery routes and/or forms of administration known in the art. The inventive pharmaceutical compositions may be prepared in liquid form, or may be in dried powder form, such as lyophilized form. For oral or enteral use, the pharmaceutical compositions can be configured, for example, as tablets, troches, lozenges, aqueous or O oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, syrups, elixirs or enteral formulas.
In the practice of this invention the "pharmaceutically acceptable carrier" is any physiologically tolerated substance known to those of ordinary skill in the art useful in formulating pharmaceutical compositions, including, any pharmaceutically acceptable diluents, excipients, dispersants, binders, 5 fillers, glidants, anti-frictional agents, compression aids, tablet-disintegrating agents (disintegrants), suspending agents, lubricants, flavorants, odorants, sweeteners, permeation or penetration enhancers, preservatives, surfactants, solubilizers, emulsifiers, thickeners, adjuvants, dyes, coatings, encapsulating material(s), and/or other additives singly or in combination. Such pharmaceutical compositions can include diluents of various buffer content (e.g., Tris-HCI, acetate, phosphate), pH and ionic strength; O additives such as detergents and solubilizing agents (e.g., Tween® 80, Polysorbate 80), anti-oxidants
(e.g., ascorbic acid, sodium metabisulfite), preservatives (e.g., Thimersol®, benzyl alcohol) and bulking substances (e.g., lactose, mannitol); incorporation of the material into particulate preparations of polymeric compounds such as polylactic acid, polyglycolic acid, etc. or into liposomes. Hyaluronic acid
can also be used, and this can have the effect of promoting sustained duration in the circulation. Such compositions can influence the physical state, stability, rate of in vivo release, and rate of in vivo clearance of the present proteins and derivatives. See, e.g., Remington's Pharmaceutical Sciences, 18th Ed. (1990, Mack Publishing Co., Easton, PA 18042) pages 1435-1712, which are herein incorporated by reference in their entirety. The compositions can be prepared in liquid form, or can be in dried powder, such as lyophilized form. Implantable sustained release formulations are also useful, as are transdermal or transmucosal formulations. Additionally (or alternatively), the present invention provides compositions for use in any of the various slow or sustained release formulations or microparticle formulations known to the skilled artisan, for example, sustained release microparticle formulations, which can be administered via pulmonary, intranasal, or subcutaneous delivery routes.
One can dilute the inventive compositions or increase the volume of the pharmaceutical compositions of the invention with an inert material. Such diluents can include carbohydrates, especially, mannitol, α-lactose, anhydrous lactose, cellulose, sucrose, modified dextrans and starch. Certain inorganic salts may also be used as fillers, including calcium triphosphate, magnesium carbonate and sodium chloride. Some commercially available diluents are Fast-Flo, Emdex, STA-Rx 1500, Emcompress and Avicell.
A variety of conventional thickeners are useful in creams, ointments, suppository and gel configurations of the pharmaceutical composition, such as, but not limited to, alginate, xanthan gum, or petrolatum, may also be employed in such configurations of the pharmaceutical composition of the present invention. A permeation or penetration enhancer, such as polyethylene glycol monolaurate, dimethyl sulfoxide, N-vinyl-2-pyrrolidone, N-(2-hydroxyethyl)-pyrrolidone, or 3-hydroxy-N-methyl-2- pyrrolidone can also be employed. Useful techniques for producing hydrogel matrices are known. (E.g., Feijen, Biodegradable hydrogel matrices for the controlled release of pharmacologically active agents, U.S. Patent No. 4,925,677; Shah et al., Biodegradable pH/thermosensitive hydrogels for sustained delivery of biologically active agents, WO 00/38651 A1). Such biodegradable gel matrices can be formed, for example, by crosslinking a proteinaceous component and a polysaccharide or mucopolysaccharide component, then loading with the inventive composition of matter to be delivered.
Liquid pharmaceutical compositions of the present invention that are sterile solutions or suspensions can be administered to a patient by injection, for example, intramuscularly, intrathecal^, epidurally, intravascularly (e.g., intravenously or intraarterially), intraperitoneally or subcutaneously. (See, e.g., Goldenberg et al., Suspensions for the sustained release of proteins, U.S. Patent No. 6,245,740 and WO 00/38652 A1). Sterile solutions can also be administered by intravenous infusion. The inventive composition can be included in a sterile solid pharmaceutical composition, such as a lyophilized powder,
which can be dissolved or suspended at a convenient time before administration to a patient using sterile water, saline, buffered saline or other appropriate sterile injectable medium.
Implantable sustained release formulations are also useful embodiments of the inventive pharmaceutical compositions. For example, the pharmaceutically acceptable carrier, being a biodegradable matrix implanted within the body or under the skin of a human or non-human vertebrate, can be a hydrogel similar to those described above. Alternatively, it may be formed from a poly-alpha- amino acid component. (Sidman, Biodegradable, implantable drug delivery device, and process for preparing and using same, U.S. Patent No. 4,351,337). Other techniques for making implants for delivery of drugs are also known and useful in accordance with the present invention. In powder forms, the pharmaceutically acceptable carrier is a finely divided solid, which is in admixture with finely divided active ingredient(s), including the inventive composition. For example, in some embodiments, a powder form is useful when the pharmaceutical composition is configured as an inhalant. (See, e.g., Zeng et al. Method of preparing dry powder inhalation compositions, WO 2004/017918; Trunk et al., Salts of the CGRP antagonist BIBN4096 and inhalable powdered medicaments containing them, U.S. Patent No. 6,900,317).
One can dilute or increase the volume of the compound of the invention with an inert material. These diluents could include carbohydrates, especially mannitol, α-lactose, anhydrous lactose, cellulose, sucrose, modified dextrans and starch. Certain inorganic salts can also be used as fillers including calcium triphosphate, magnesium carbonate and sodium chloride. Some commercially available diluents are Fast-Flo™, Emdex™, STA-Rx™ 1500, Emcompress™ and Avicell™.
Disintegrants can be included in the formulation of the pharmaceutical composition into a solid dosage form. Materials used as disintegrants include but are not limited to starch including the commercial disintegrant based on starch, Explotab™. Sodium starch glycolate, Amberlite™, sodium carboxymethylcellulose, ultramylopectin, sodium alginate, gelatin, orange peel, acid carboxymethyl cellulose, natural sponge and bentonite can all be used. Insoluble cationic exchange resin is another form of disintegrant. Powdered gums can be used as disintegrants and as binders and these can include powdered gums such as agar, Karaya or tragacanth. Alginic acid and its sodium salt are also useful as disintegrants. Binders can be used to hold the therapeutic agent together to form a hard tablet and include materials from natural products such as acacia, tragacanth, starch and gelatin. Others include methyl cellulose (MC), ethyl cellulose (EC) and carboxymethyl cellulose (CMC). Polyvinyl
pyrrolidone (PVP) and hydroxypropylmethyl cellulose (HPMC) could both be used in alcoholic solutions to granulate the therapeutic.
An antifrictional agent can be included in the formulation of the therapeutic to prevent sticking during the formulation process. Lubricants can be used as a layer between the therapeutic and the die wall, and these can include but are not limited to; stearic acid including its magnesium and calcium salts, polytetrafluoroethylene (PTFE)1 liquid paraffin, vegetable oils and waxes. Soluble lubricants can also be used such as sodium lauryl sulfate, magnesium lauryl sulfate, polyethylene glycol of various molecular weights, Carbowax 4000 and 6000.
Glidants that might improve the flow properties of the drug during formulation and to aid rearrangement during compression might be added. The glidants can include starch, talc, pyrogenic silica and hydrated silicoaluminate.
To aid dissolution of the compound of this invention into the aqueous environment a surfactant might be added as a wetting agent. Surfactants can include anionic detergents such as sodium lauryl sulfate, dioctyl sodium sulfosuccinate and dioctyl sodium sulfonate. Cationic detergents might be used and could include benzalkonium chloride or benzethonium chloride. The list of potential nonionic detergents that could be included in the formulation as surfactants are lauromacrogol 400, polyoxyl 40 stearate, polyoxyethylene hydrogenated castor oil 10, 50 and 60, glycerol monostearate, polysorbate 40, 60, 65 and 80, sucrose fatty acid ester, methyl cellulose and carboxymethyl cellulose. These surfactants could be present in the formulation of the protein or derivative either alone or as a mixture in different ratios.
Oral dosage forms. Also useful are oral dosage forms of the inventive compositionss. If necessary, the composition can be chemically modified so that oral delivery is efficacious. Generally, the chemical modification contemplated is the attachment of at least one moiety to the molecule itself, where said moiety permits (a) inhibition of proteolysis; and (b) uptake into the blood stream from the stomach or intestine. Also desired is the increase in overall stability of the compound and increase in circulation time in the body. Moieties useful as covalently attached half- life extending moieties in this invention can also be used for this purpose. Examples of such moieties include: PEG, copolymers of ethylene glycol and propylene glycol, carboxymethyl cellulose, dextran, polyvinyl alcohol, polyvinyl pyrrolidone and polyproline. See, for example, Abuchowski and Davis (1981), Soluble Polymer-Enzyme Adducts, Enzymes as Drugs (Hocenberg and Roberts, eds.), Wiley-lnterscience, New York, NY, pp 367-83; Newmark, et al. (1982), J. Appl. Biochem. 4:185-9. Other polymers that could be used are poly-1,3-dioxolane and poly-1,3,6- tioxocane. Preferred for pharmaceutical usage, as indicated above, are PEG moieties.
For oral delivery dosage forms, it is also possible to use a salt of a modified aliphatic amino acid, such as sodium N-(8-[2-hydroxybenzoyl] amino) caprylate (SNAC), as a carrier to enhance absorption of the therapeutic compounds of this invention. The clinical efficacy of a heparin formulation using SNAC has been demonstrated in a Phase Il trial conducted by Emisphere Technologies. See US Patent No. 5,792,451 , "Oral drug delivery composition and methods."
In one embodiment, the pharmaceutically acceptable carrier can be a liquid and the pharmaceutical composition is prepared in the form of a solution, suspension, emulsion, syrup, elixir or pressurized composition. The active ingredient(s) (e.g., the inventive composition of matter) can be dissolved, diluted or suspended in a pharmaceutically acceptable liquid carrier such as water, an organic solvent, a mixture of both, or pharmaceutically acceptable oils or fats. The liquid carrier can contain other suitable pharmaceutical additives such as detergents and/or solubilizers (e.g., Tween 80, Polysorbate 80), emulsifiers, buffers at appropriate pH (e.g., Tris-HCI, acetate, phosphate), adjuvants, anti-oxidants (e.g., ascorbic acid, sodium metabisulfite), preservatives (e.g., Thimersol, benzyl alcohol), sweeteners, flavoring agents, suspending agents, thickening agents, bulking substances (e.g., lactose, mannitol), colors, viscosity regulators, stabilizers, electrolytes, osmolutes or osmo-regulators. Additives can also be included in the formulation to enhance uptake of the inventive composition. Additives potentially having this property are for instance the fatty acids oleic acid, linoleic acid and linolenic acid. Useful are oral solid dosage forms, which are described generally in Remington's
Pharmaceutical Sciences (1990), supra, in Chapter 89, which is hereby incorporated by reference in its entirety. Solid dosage forms include tablets, capsules, pills, troches or lozenges, cachets or pellets. Also, liposomal or proteinoid encapsulation can be used to formulate the present compositions (as, for example, proteinoid microspheres reported in U.S. Patent No. 4,925,673). Liposomal encapsulation can be used and the liposomes can be derivatized with various polymers
(e.g., U.S. Patent No. 5,013,556). A description of possible solid dosage forms for the therapeutic is given in Marshall, K., Modern Pharmaceutics (1979), edited by G. S. Banker and C. T. Rhodes, in Chapter 10, which is hereby incorporated by reference in its entirety. In general, the formulation will include the inventive compound, and inert ingredients that allow for protection against the stomach environment, and release of the biologically active material in the intestine.
The composition of this invention can be included in the formulation as fine multiparticulates in the form of granules or pellets of particle size about 1 mm. The formulation of
the material for capsule administration could also be as a powder, lightly compressed plugs or even as tablets. The therapeutic could be prepared by compression.
Colorants and flavoring agents can all be included. For example, the protein (or derivative) can be formulated (such as by liposome or microsphere encapsulation) and then further contained within an edible product, such as a refrigerated beverage containing colorants and flavoring agents.
In tablet form, the active ingredient(s) are mixed with a pharmaceutically acceptable carrier having the necessary compression properties in suitable proportions and compacted in the shape and size desired. 0 The powders and tablets preferably contain up to 99% of the active ingredient(s). Suitable solid carriers include, for example, calcium phosphate, magnesium stearate, talc, sugars, lactose, dextrin, starch, gelatin, cellulose, polyvinylpyrrolidine, low melting waxes and ion exchange resins.
Controlled release formulation can be desirable. The composition of this invention could be incorporated into an inert matrix that permits release by either diffusion or leaching mechanisms 5 e.g., gums. Slowly degenerating matrices can also be incorporated into the formulation, e.g., alginates, polysaccharides. Another form of a controlled release of the compositions of this invention is by a method based on the Oros™ therapeutic system (Alza Corp.), i.e., the drug is enclosed in a semipermeable membrane which allows water to enter and push drug out through a single small opening due to osmotic effects. Some enteric coatings also have a delayed release O effect.
Other coatings can be used for the formulation. These include a variety of sugars that could be applied in a coating pan. The therapeutic agent could also be given in a film-coated tablet and the materials used in this instance are divided into 2 groups. The first are the nonenteric materials and include methylcellulose, ethyl cellulose, hydroxyethyl cellulose, methylhydroxy-ethyl 5 cellulose, hydroxypropyl cellulose, hydroxypropyl-methyl cellulose, sodium carboxymethyl cellulose, providone and the polyethylene glycols. The second group consists of the enteric materials that are commonly esters of phthalic acid.
A mix of materials might be used to provide the optimum film coating. Film coating can be carried out in a pan coater or in a fluidized bed or by compression coating. O Pulmonary delivery forms. Pulmonary delivery of the inventive compositions is also useful. The protein (or derivative) is delivered to the lungs of a mammal while inhaling and traverses across the lung epithelial lining to the blood stream. (Other reports of this include Adjei et al., Pharma. Res. (1990) 7: 565-9; Adjei et_al. (1990), Internatl. J. Pharmaceutics 63: 135-44
(leuprolide acetate); Braquet et_aj. (1989), J. Cardiovasc. Pharmacol. 13 (suppl.5): s.143-146 (endothelin-1); Hubbard et_al. (1989), Annals Int. Med, 3: 206-12 (α1 -antitrypsin); Smith et_aj. (1989), J. Clin. Invest. 84: 1145-6 (α1 -proteinase); Oswein et al. (March 1990), "Aerosolization of Proteins," Proc. Svmp. Resp. Drug Delivery II, Keystone, Colorado (recombinant human growth hormone); Debs et_a|. (1988), J. Immunol. 140: 3482-8 (interferon-γ and tumor necrosis factor α) and Platz et al., U.S. Patent No. 5,284,656 (granulocyte colony stimulating factor).
Useful in the practice of this invention are a wide range of mechanical devices designed for pulmonary delivery of therapeutic products, including but not limited to nebulizers, metered dose inhalers, and powder inhalers, all of which are familiar to those skilled in the art. Some specific examples of commercially available devices suitable for the practice of this invention are the Ultravent nebulizer, manufactured by Mallinckrodt, Inc., St. Louis, Missouri; the Acorn Il nebulizer, manufactured by Marquest Medical Products, Englewood, Colorado; the Ventolin metered dose inhaler, manufactured by Glaxo Inc., Research Triangle Park, North Carolina; and the Spinhaler powder inhaler, manufactured by Fisons Corp., Bedford, Massachusetts. (See, e.g., Helgesson et al., Inhalation device, U.S. Patent No. 6,892,728; McDerment et al., Dry powder inhaler, WO 02/11801 A1; Ohki et al., Inhalant medicator, U.S. Patent No. 6,273,086).
All such devices require the use of formulations suitable for the dispensing of the inventive compound. Typically, each formulation is specific to the type of device employed and can involve the use of an appropriate propellant material, in addition to diluents, adjuvants and/or carriers useful in therapy.
The inventive compound should most advantageously be prepared in particulate form with an average particle size of less than 10 μm (or microns), most preferably 0.5 to 5 μm, for most effective delivery to the distal lung.
Pharmaceutically acceptable carriers include carbohydrates such as trehalose, mannitol, xylitol, sucrose, lactose, and sorbitol. Other ingredients for use in formulations can include DPPC, DOPE, DSPC and DOPC. Natural or synthetic surfactants can be used. PEG can be used (even apart from its use in derivatizing the protein or analog). Dextrans, such as cyclodextran, can be used. Bile salts and other related enhancers can be used. Cellulose and cellulose derivatives can be used. Amino acids can be used, such as use in a buffer formulation. Also, the use of liposomes, microcapsules or microspheres, inclusion complexes, or other types of carriers is contemplated.
Formulations suitable for use with a nebulizer, either jet or ultrasonic, will typically comprise the inventive compound dissolved in water at a concentration of about 0.1 to 25 mg of
biologically active protein per mL of solution. The formulation can also include a buffer and a simple sugar (e.g., for protein stabilization and regulation of osmotic pressure). The nebulizer formulation can also contain a surfactant, to reduce or prevent surface induced aggregation of the protein caused by atomization of the solution in forming the aerosol. Formulations for use with a metered-dose inhaler device will generally comprise a finely divided powder containing the inventive compound suspended in a propellant with the aid of a surfactant. The propellant can be any conventional material employed for this purpose, such as a chlorofluorocarbon, a hydrochlorofluorocarbon, a hydrofluorocarbon, or a hydrocarbon, including trichlorofluoromethane, dichlorodifluoromethane, dichlorotetrafluoroethanol, and 1 ,1,1,2- tetrafluoroethane, or combinations thereof. Suitable surfactants include sorbitan trioleate and soya lecithin. Oleic acid can also be useful as a surfactant. (See, e.g., Backstrόm et al., Aerosol drug formulations containing hydrofluoroalkanes and alkyl saccharides, U.S. Patent No. 6,932,962). Formulations for dispensing from a powder inhaler device will comprise a finely divided dry powder containing the inventive compound and can also include a bulking agent, such as lactose, sorbitol, sucrose, mannitol, trehalose, or xylitol in amounts which facilitate dispersal of the powder from the device, e.g., 50 to 90% by weight of the formulation.
Nasal delivery forms. In accordance with the present invention, intranasal delivery of the inventive composition of matter and/or pharmaceutical compositions is also useful, which allows passage thereof to the blood stream directly after administration to the inside of the nose, without the necessity for deposition of the product in the lung. Formulations suitable for intransal administration include those with dextran or cyclodextran, and intranasal delivery devices are known. (See, e.g, Freezer, Inhaler, U.S. Patent No. 4,083,368).
Transdermal and transmucosal (e.g., buccal) delivery forms). In some embodiments, the inventive composition is configured as a part of a pharmaceutically acceptable transdermal or transmucosal patch or a troche. Transdermal patch drug delivery systems, for example, matrix type transdermal patches, are known and useful for practicing some embodiments of the present pharmaceutical compositions. (E.g., Chien et al., Transdermal estrogen/progestin dosage unit, system and process, U.S. Patent Nos. 4,906,169 and 5,023,084; Cleary et al., Diffusion matrix for transdermal drug administration and transdermal drug delivery devices including same, U.S. Patent No. 4,911,916; Teillaud et al., EVA-based transdermal matrix system for the administration of an estrogen and/or a progestogen, U.S. Patent No. 5.605,702; Venkateshwaran et al., Transdermal drug delivery matrix for coadministering estradiol and another steroid, U.S. Patent No. 5,783,208; Ebert et al., Methods for providing testosterone and optionally estrogen replacement
therapy to women, U.S. Patent No. 5,460,820). A variety of pharmaceutically acceptable systems fortransmucosal delivery of therapeutic agents are also known in the art and are compatible with the practice of the present invention. (E.g., Heiber et al., Transmucosal delivery of macromolecular drugs, U.S. Patent Nos. 5,346,701 and 5,516,523; Longenecker et al., Transmembrane formulations for drug administration, U.S. Patent No. 4,994,439).
Buccal delivery of the inventive compositions is also useful. Buccal delivery formulations are known in the art for use with peptides. For example, known tablet or patch systems configured for drug delivery through the oral mucosa (e.g., sublingual mucosa), include some embodiments that comprise an inner layer containing the drug, a permeation enhancer, such as a bile salt or fusidate, and a hydrophiϋc polymer, such as hydroxypropyl cellulose, hydroxypropyl methylcellulose, hydroxyethyl cellulose, dextran, pectin, polyvinyl pyrrolidone, starch, gelatin, or any number of other polymers known to be useful for this purpose. This inner layer can have one surface adapted to contact and adhere to the moist mucosal tissue of the oral cavity and can have an opposing surface adhering to an overlying non-adhesive inert layer. Optionally, such a transmucosal delivery system can be in the form of a bilayer tablet, in which the inner layer also contains additional binding agents, flavoring agents, or fillers. Some useful systems employ a non-ionic detergent along with a permeation enhancer. Transmucosal delivery devices may be in free form, such as a cream, gel, or ointment, or may comprise a determinate form such as a tablet, patch or troche. For example, delivery of the inventive composition can be via a transmucosal delivery system comprising a laminated composite of, for example, an adhesive layer, a backing layer, a permeable membrane defining a reservoir containing the inventive composition, a peel seal disc underlying the membrane, one or more heat seals, and a removable release liner. (E.g., Ebert et al., Transdermal delivery system with adhesive overlay and peel seal disc, U.S. Patent No. 5,662,925; Chang et al., Device for administering an active agent to the skin or mucosa, U.S. Patent Nos. 4,849,224 and 4,983,395). These examples are merely illustrative of available transmucosal drug delivery technology and are not limiting of the present invention.
Dosages. The dosage regimen involved in a method for treating the above-described conditions will be determined by the attending physician, considering various factors which modify the action of drugs, e.g. the age, condition, body weight, sex and diet of the patient, the severity of any infection, time of administration and other clinical factors. Generally, the daily regimen should be in the range of 0.1-
1000 micrograms of the inventive compound per kilogram of body weight, preferably 0.1-150 micrograms per kilogram.
Working examples
The compositions described above can be prepared as described below. These examples are not to be construed in any way as limiting the scope of the present invention.
Example 1 Fc-LI Q-ShK[I -351 mammalian expression
Fc-L10-ShK[1-35], also referred to as "Fc-2xL-ShK[1-35]", an inhibitor of Kv1.3. A DNA sequence coding for the Fc region of human IgGI fused in-frame to a linker sequence and a monomer of the Kv1.3 inhibitor peptide ShK[I -35] was constructed as described below. Methods for expressing and purifying the peptibody from mammalian cells (HEK 293 and Chinese Hamster Ovary cells) are disclosed herein.
The expression vector pcDNA3.1(+)CMVi (Figure 13A) was constructed by replacing the CMV promoter between MIuI and Hindlll in pcDNA3.1(+) with the CMV promoter plus intron (Invitrogen). The expression vector pcDNA3.1(+)CMVi-hFc-ActivinRIIB (Figure 13B) was generated by cloning a Hindlll-Notl digested PCR product containing a 5' Kozak sequence, a signal peptide and the human Fc-linker— ActivinRIIB fusion protein with the large fragment of
Hindlll-Notl digested pcDNA3.1(+)CMVi. The nucleotide and amino acid sequence of the human IgGI Fc region in pcDNA3.1 (+)CMVi-hFc-ActivinRI IB is shown in Figure 3A-3B. This vector also has a GGGGSGGGGS ("L10"; SEQ ID NO:79) linker split by a BamHI site thus enabling with the oligo below formation of the 10 amino acid linker between Fc and the ShK[I -35] peptide (see Figure 14A-14B) for the final Fc-LI 0-ShK[1 -35] nucleotide and amino acid sequence (Figure 14A- 14B and SEQ ID NO: 77 and SEQ ID NO:78).
The Fc-LI 0-ShK[1 -35] expression vector was constructing using PCR stategies to generate the full length ShK gene linked to a four glycine and one serine amino acid linker (lower case letters here indicate linker sequence of L-form amino acid residues) with two stop codons and flanked by BamHI and Notl restriction sites as shown below.
BamHI
GGATCCGGAGGAGGAGGAAGCCGCAGCTGCATCGACACCATCCCCAAGAGCCGCTGCACCGCCTTCCAG g g g g s R S C I D T I P K S R C T A F Q
TGCAAGCACAGCATGAAGTACCGCCTGAGCTTCTGCCGCAAGACCTGCGGCACCTGCTAATGAGCGGCCGC//SEQ ID NO: 657 C K H S M K Y R L S F C R K T C G T C Notl
//SEQ ID NO: 658
Two oligos with the sequence as depicted below were used in a PCR reaction with
Herculase™ polymerase (Stratagene) at 94°C-30sec, 50°C-30sec, and 72°C-1min for 30 cycles.
cat gga tec gga gga gga gga age cgc age tgc ate gac ace ate ccc aag age cgc tgc ace gee ttc cag tgc aag cac //SEQ ID NO: 659
cat gcg gcc get cat tag cag gtg ccg cag gtc ttg egg cag aag etc agg egg tac ttc atg ctg tgc ttg cac tgg aag g //SEQ ID NO: 660
The resulting PCR products were resolved as the 150bp bands on a one percent agarose gel. The 150bp PCR product was digested with BamHI and Notl (Roche) restriction enzymes and agarose gel purified by Gel Purification Kit (Qiagen). At the same time, the pcDNA3.1(+)CMVi- hFc-ActivinRIIB vector ( Figure 13B ) was digested with BamHI and Notl restriction enzymes and the large fragment was purified by Gel Purification Kit. The gel purified PCR fragment was ligated to the purified large fragment and transformed into XL-1blue bacteria (Stratagene). DNAs from transformed bacterial colonies were isolated and digested with BamHI and Notl restriction enzyme digestion and resolved on a one percent agarose gel. DNAs resulting in an expected pattern were submitted for sequencing. Although, analysis of several sequences of clones yielded a 100% percent match with the above sequence, only one clone was selected for large scaled plasmid purification. The DNA from Fc-2xL-ShK in pcDNA3.1(+)CMVi clone was resequenced to confirm the Fc and linker regions and the sequence was 100% identical to the predicted coding sequence, which is shown in Figure 14A-14B.
HEK-293 cells used in transient transfection expression of Fc-2xL-ShK[1-35] in pcDNA3.1(+)CMVi protein were cultured in growth medium containing DMEM High Glucose (Gibco), 10% fetal bovine serum (FBS from Gibco) and 1X non-essential amino acid (NEAA from Gibco). 5.6ug of Fc-2xL-ShK[1-35] in pcDNA3.1(+)CMVi plasmid that had been phenol/chloroform extracted was transfected into HEK-293 cells using Fugene 6 (Roche). The cells recovered for 24 hours, and then placed in DMEM High Glucose and 1x NEAA medium for 48 hours. The conditioned medium was concentrated 5OX by running 30ml through Centriprep YM-10 filter (Amicon) and further concentrated by a Centricon YM-10 (Amicon) filter. Various amounts of concentrated medium were mixed with an in-house 4x Loading Buffer (without B-mercaptoethanol) and electrophoresed on a Novex 4-20% tris-glycine gel using a Novex Xcell Il apparatus at
101V/46mA for 2 hours in a 5x Tank buffer solution (0.123 Tris Base, 0.96M Glycine) along with 1OuI of BenchMark Pre-Stained Protein ladder (Invitrogen). The gel was then soaked in Electroblot buffer (35mM Tris base, 20%methanol, 192mM glycine) for 30 minutes. A PVDF membrane from Novex (Cat. No. LC2002, 0.2um pores size) was soaked in methanol for 30 seconds to activate the PVDF, rinsed with deionized water, and soaked in Electroblot buffer. The pre-soaked gel was
blotted to the PVDF membrane using the XCeII Il Blot module according to the manufacturer instructions (Novex) at 4OmA for 2 hours. Then, the blot was first soaked in a 5% milk (Carnation) in Tris buffered saline solution pH7.5 (TBS) for 1 hour at room temperature and incubated with 1 :500 dilution in TBS with 0.1%Tween-20 (TBST Sigma) and 1% milk buffer of the HRP- conjugated murine anti-human Fc antibody (Zymed Laboratores Cat. no. 05-3320) for two hours shaking at room temperature. The blot was then washed three times in TBST for 15 minutes per wash at room temperature. The primary antibody was detected using Amersham Pharmacia Biotech's ECL western blotting detection reagents according to manufacturer's instructions. Upon ECL detection, the western blot analysis displayed the expected size of 66kDa under non-reducing gel conditions (Figure 24A).
AM1 CHOd- (Amgen Proprietary) cells used in the stable expression of Fc-LI 0-ShK[1 -35] protein were cultured in AM1 CHOd- growth medium containing DMEM High Glucose, 10% fetal bovine serum, 1x hypoxantine/thymidine (HT from Gibco) and 1X NEAA. 6.5ug of pcDNA3.1(+)CMVi-Fc-ShK plasmid was also transfected into AM1 CHOd- cells using Fugene 6. The following day, the transfected cells were plated into twenty 15cm dishes and selected using DMEM high glucose, 10%FBS, 1xHT, IxNEAA and Geneticin (800 μg/ml G418 from Gibco) for thirteen days. Forty-eight surviving colonies were picked into two 24-well plates. The plates were allowed to grow up for a week and then replicated for freezing. One set of each plate was transferred to AM1 CHOd- growth medium without 10% FBS for 48 hours and the conditioned media were harvested. Western Blot analysis similar to the transient Western blot analysis with detection by the same anti-human Fc antibody was used to screen 15ul of conditioned medium for expressing stable CHO clones. Of the 48 stable clones, more than 50% gave ShK expression at the expected size of 66kDa. The BB6, BD5 and BD6 clones were selected with BD5 and BD6 as a backup to the primary clone BB6 (Figure 24B). The BB6 clone was scaled up into ten roller bottles (Corning) using AM1 CHOd- growth medium and grown to confluency as judged under the microscope. Then, the medium was exchanged with a serum-free medium containing to 50% DMEM high glucose and 50% Ham's F12 (Gibco) with 1xHT and IxNEAA and let incubate for one week. The conditioned medium was harvested at the one-week incubation time, filtered through 0.45 μm filter (Corning) and frozen. Fresh serum-free medium was added and incubated for an additional week. The conditioned serum-free medium was harvested like the first time and frozen.
Purification of monovalent and bivalent dimeric Fc-L10-ShK(1-35). Approximately 4 L of conditioned medium was thawed in a water bath at room temperature. The medium was
concentrated to about 450 ml using a Satorius Sartocon Polysulfon 10 tangential flow ultra-filtration cassette (0.1 m2) at room temperature. The retentate was then filtered through a 0.22 μm cellulose acetate filter with a pre-filter. The retentate was then loaded on to a 5 ml Amersham HiTrap Protein A column at 5 ml/min 7 0C, and the column was washed with several column volumes of Dulbecco's phosphate buffered saline without divalent cations (PBS) and sample was eluted with a step to 100 mM glycine pH 3.0. The protein A elution pool (approximately 9 ml) was diluted to 50 ml with water and loaded on to a 5 ml Amersham HiTrap SP-HP column in S-Buffer A (20 mM NaH2PO4, pH 7.0) at 5 ml/min and 7 0C. The column was then washed with several column volumes S-Buffer A, and then developed using a linear gradient from 25% to 75% S-Buffer B (20 mM NaH2PO4, 1 M NaCI, pH 7.0) at 5 ml/min followed by a step to 100% S-Buffer B at 7 0C. Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS- PAGE, and the fractions containing the desired product were pooled based on these data. The pooled material was then concentrated to about 3.4 ml using a Pall Life Sciences Macrosep 10K Omega centrifugal ultra-filtration device and then filtered though a Costar 0.22 μm cellulose acetate syringe filter.
A spectral scan was then conducted on 10 μl of the filtered material diluted in 700 μl PBS using a Hewlett Packard 8453 spectrophotometer (Figure 26A). The concentration of the filtered material was determined to be 5.4 mg/ml using a calculated molecular mass of 32,420 g/mol and extinction coefficient of 47,900 M 1 cm 1. The purity of the filtered bivalent dimeric Fc-UO-ShK(I -35) product was assessed using a
Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 26B). The monvalent dimeric Fc-L10-ShK(1-35) product was analyzed using reducing and non-reducing sample buffers by SDS-PAGE on a 1.0 mm TRIS-glycine 4-20% gel developed at 220 V and stained with Boston Biologicals QuickBlue (Figure 26E). The endotoxin levels were then determined using a Charles River Laboratories Endosafe-PTS system (0.05 - 5 EU/ml sensitivity) using a 108-fold dilution of the sample in PBS yielding a result of <1 EU/mg protein. The macromolecular state of the products was then determined using size exclusion chromatography on 20 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 26C, bivalent dimeric Fc-LI 0-ShK(1 -35); Figure 26F1 monovalent dimeric Fc-LI 0-SHK(1 -35)). The product was then subject to mass spectral analysis by diluting 1 μl of the sample into 10 μl of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile). The resultant solution (1 μl) was spotted onto a MALDI sample plate. The sample was allowed to dry before being analyzed using a Voyager
DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses (Figure 26D) and confirmed (within experimental error) the integrity of the purified peptibody. The product was then stored at -80 0C.
Purified bivalent dimeric Fc-LI 0-ShK[1 -35] potently blocked human Kv1.3 (Figure 3OA and Figure 30B) as determined by electrophysiology (see Example 36). The purified bivalent dimeric Fc-LI 0-ShK[1 -35] molecule also blocked T cell proliferation (Figure 36A and Figure 36B) and production of the cytokines IL-2 (Figure 35A and Figure 37A) and IFN-g (Figure 35B and Figure 37B).
Example 2 Fc-L-ShK[2-351 mammalian expression A DNA sequence coding for the Fc region of human IgGI fused in-frame to a monomer of the Kv1.3 inhibitor peptide ShK[2-35] was constructed using standard PCR technology. The ShK[2-35] and the 5, 10, or 25 amino acid linker portion of the molecule were generated in a PCR reaction using the original Fc-2xL-ShK[1-35] in pcDNA3.1(+)CMVi as a template (Example 1, Figure 14A-14B). All ShK constructs should have the following amino acid sequence of SCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC
(SEQ ID NO: 92) with the first amino acid of the wild-type sequence deleted.
The sequences of the primers used to generate Fc-L5-ShK[2-35], also referred to as "Fc- 1xL-ShK[2-35]", are shown below: cat gga tec age tgc ate gac ace atc//sEQ ID NO: 661,- cat gcg gcc get cat tag c// SEQ ID NO: 662;
The sequences of the primers used to generate Fc-LI 0-ShK[2-35], also referred to as "Fc- 2xL-ShK[2-35]" are shown below: cat gga tec gga gga gga gga age age tgc a //SEQ ID NO:663,- cat gcg gcc get cat tag cag gtg c //SEQ ID NO:664;
The sequences of the primers used to generate Fc-L25-ShK[2-35], also referred to as "Fc- 5xL-ShK[2-35]", are shown below: cat gga tec ggg ggt ggg ggt tct ggg ggt ggg ggt tct gga gga gga gga age gga gga gga gga age age tgc a//SEQ ID NO:665; cat gcg gcc get cat tag cag gtg C//SEQ ID NO:666;
The PCR products were digested with BamHI and Notl (Roche) restriction enzymes and agarose gel purified by Gel Purification Kit. At the same time, the pcDNA3.1(+)CMVi-hFc- ActivinRIIB vector was digested with BamHI and Notl restriction enzymes and the large fragment was purified by Gel Purification Kit. Each purified PCR product was ligated to the large fragment and transformed into XL-1 blue bacteria. DNAs from transformed bacterial colonies were isolated and subjected to BamHI and Notl restriction enzyme digestions and resolved on a one percent agarose gel. DNAs resulting in an expected pattern were submitted for sequencing. Although, analysis of several sequences of clones yielded a 100% percent match with the above sequence, only one clone was selected for large scaled plasmid purification. The DNA from this clone was resequenced to confirm the Fc and linker regions and the sequence was 100% identical to the expected sequence.
Plasmids containing the Fc-1xL-Shk[2-35], Fc-2xL-Shk[2-35] and Fc-5xL-Shk[2-35] inserts in pcDNA3.1(+)CMVi vector were digested with Xba1 and Xho1 (Roche) restriction enzymes and gel purified. The inserts were individually ligated into Notl and Sail (Roche) digested pDSRα-22 (Amgen Proprietary) expression vector. Integrity of the resulting constructs were confirmed by DNA sequencing. The final plasmid DNA expression vector constructs were pDSRα-22-Fc-1xL-Shk[2- 35], pDSRα-22- Fc-2xL-Shk[2-35] (Figure 13C and Figure 15A-15B) and pDSRα-22- Fc-5xL-Shk[2- 35] (Figure 16A-16B) and contained 5, 10 and 25 amino acid linkers, respectively.
Twenty-four hours prior to transfection, 1.2e7 AM-1/D CHOd- (Amgen Proprietary) cells were plated into a T-175 cm sterile tissue culture flask, to allow 70-80% confluency on the day of transfection. The cells had been maintained in the AM-1/D CHOd- culture medium containing DMEM High Glucose, 5% FBS, 1X Glutamine Pen/Strep (Gibco), 1X HT1 1X NEAA's and 1X sodium pyruvate (Gibco). The following day, eighteen micrograms of each of the linearized pDSRα22:Fc-1xL-ShK[2-35], pDSRα22:Fc-2xL-ShK[2-35] and pDSRα22:Fc-5xL-ShK[2-35] (RDS's # 20050037685, 20050053709, 20050073295) plasmids were mixed with 72 μg of linearized Selexis MAR plasmid and pPAGOI (RDS 20042009896) and diluted into 6ml of OptiMEM in a 50ml conical tube and incubate for five minutes. LF2000 (210 μl) was added to 6ml of OptiMEM and incubated for five minutes. The diluted DNA and LF2000 were mixed together and incubated for 20 minutes at room temperature. In the meantime, the cells were washed one time with PBS and then 30ml OptiMEM without antibiotics were added to the cells. tThe OptiMEM was aspirated off, and the cells were incubated with 12ml of DNA/LF2000 mixture for 6 hours or overnight in the 370C incubator with shaking. Twenty-four hours post transfection, the cells were split 1 :5 into AM-1/D CHOd- culture medium and at differing dilutions for colony selection. Seventy-
two hours post transfection, the cell medium was replaced with DHFR selection medium containing 10% Dialyzed FBS (Gibco) in DMEM High Glucose, plus 1X Glutamine Pen/Strep, 1X NEAA's and 1X Na Pyrto allow expression and secretion of protein into the cell medium. The selection medium was changed two times a week until the colonies are big enough to pick. The pDSRa22 expression vector contains a DHFR expression cassette, which allows transfected cells to grow in the absence of hypoxanthine and thymidine. The five T- 175 pools of the resulting colonies were scaled up into roller bottles and cultured under serum free conditions. The conditioned media were harvested and replaced at one-week intervals. The resulting 3 liters of conditioned medium was filtered through a 0.45 μm cellulose acetate filter (Corning, Acton, MA) and transferred to Protein Chemistry for purification. As a backup, twelve colonies were selected from the 10 cm plates after 10-14 days on DHFR selection medium and expression levels evaluated by western blot using HRP conjugated anti human IgGFc as a probe. The three best clones expressing the highest level of each of the different linker length Fc-L-ShK[2-35] fusion proteins were expanded and frozen for future use. Purification of Fc-LI 0-ShK(2-35). Approximately 1 L of conditioned medium was thawed in a water bath at room temperature. The medium was loaded on to a 5 ml Amersham HiTrap Protein A column at 5 ml/min 7 0C, and the column was washed with several column volumes of Dulbecco's phosphate buffered saline without divalent cations (PBS) and sample was eluted with a step to 100 mM glycine pH 3.0. The protein A elution pool (approximately 8.5 ml) combined with 71 μl 3 M sodium acetate and then diluted to 50 ml with water. The diluted material was then loaded on to a 5 ml Amersham HiTrap SP-HP column in S-Buffer A (20 mM NaH2PCM, pH 7.0) at 5 ml/min 7 0C. The column was then washed with several column volumes S-Buffer A, and then developed using a linear gradient from 0% to 75% S-Buffer B (20 mM NaH2PO4, 1 M NaCI, pH 7.0) at 5 ml/min followed by a step to 100% S-Buffer B at 7 0C. Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE, and the fractions containing the desired product were pooled based on these data. The pooled material was then filtere through a 0.22 μm cellulose acetate filter and concentrated to about 3.9 ml using a Pall Life Sciences Macrosep 10K Omega centrifugal ultra-filtration device. The concentrated material was then filtered though a Pall Life Sciences Acrodisc with a 0.22 μm, 25 mm Mustang E membrane at 2 ml/min room temperature. A spectral scan was then conducted on 10 μl of the filtered material diluted in 700 μl PBS using a Hewlett Packard 8453 spectrophotometer (Figure 27E). The concentration of the filtered material was determined to be 2.76 mg/ml using a calculated molecular mass of 30,008 g/mol and extinction coefficient of 36,900 M 1 cm 1. Since material was
found in the permeate, repeated concentration step on the permeate using a new Macrosep cartridge. The new batch of concentrated material was then filtered though a Pall Life Sciences Acrodisc with a 0.22 μm, 25 mm Mustang E membrane at 2 ml/min room temperature. Both lots of concentrated material were combined into one pool. A spectral scan was then conducted on 10 μl of the combined pool diluted in 700 μl PBS using a Hewlett Packard 8453 spectrophotometer. The concentration of the filtered material was determined to be 3.33 mg/ml using a calculated molecular mass of 30,008 g/mol and extinction coefficient of 36,900 M"1 cm 1. The purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 27A). The endotoxin level was then determined using a Charles River Laboratories Endosafe-PTS system (0.05 - 5 EU/ml sensitivity) using a 67-fold dilution of the sample in PBS yielding a result of <1 EU/mg protein. The macromolecular state of the product was then determined using size exclusion chromatography on 50 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PCM, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 27B). The product was then subject to mass spectral analysis by diluting 1 μl of the sample into 10 μl of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile). The resultant solution (1 μl) was spotted onto a MALDI sample plate. The sample was allowed to dry before being analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from - 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses (Figure 27F) and the experiment confirmed the itegrity of the peptibody, within experimental error. The product was then stored at -80 0C.
Figure 31 B shows that purified Fc-LI 0-ShK[2-35] potently blocks human Kv1.3 current (electrophysiology was done as described in Example 36). The purified Fc-LI 0-ShK[2-35] molecule also blocked IL-2 (Figure 64A and Figure 64B) and IFN-g (Figure 65A and Figure 65B) production in human whole blood, as well as, upregulation of CD40L (Figure 66A and Figure 66B) and IL-2R (Figure 67A and Figure 67B) on T cells.
Purification of Fc-L5-ShK(2-35). Approximately 1 L of conditioned medium was loaded on to a 5 ml Amersham HiTrap Protein A column at 5 ml/min 7 0C, and the column was washed with several column volumes of Dulbecco's phosphate buffered saline without divalent cations (PBS) and sample was eluted with a step to 100 mM glycine pH 3.0. The protein A elution pool (approximately 9 ml) combined with 450 μl 1 M tris HCI pH 8.5 followed by 230 μl 2 M acetic acid
and then diluted to 50 ml with water. The pH adjusted material was then filtered through a 0.22 μm cellulose acetate filter and loaded on to a 5 ml Amersham HiTrap SP-HP column in S-Buffer A (20 mM NaH2PO4, pH 7.0) at 5 ml/min 7 0C. The column was then washed with several column volumes S-Buffer A, and then developed using a linear gradient from 0% to 75% S-Buffer B (20 mM NaH2PO4, 1 M NaCI, pH 7.0) at 5 ml/min followed by a step to 100% S-Buffer B at 7 0C. Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS- PAGE, and the fractions containing the desired product were pooled based on these data. The pooled material was then concentrated to about 5.5 ml using a Pall Life Sciences Macrosep 1OK Omega centrifugal ultra-filtration device. The concentrated material was then filtered though a Pall Life Sciences Acrodisc with a 0.22 μm, 25 mm Mustang E membrane at 2 ml/min room temperature.
A spectral scan was then conducted on 10 μl of the combined pool diluted in 700 μl PBS using a Hewlett Packard 8453 spectrophotometer (Figure 27G). The concentration of the filtered material was determined to be 4.59 mg/ml using a calculated molecular mass of 29,750 g/mol and extinction coefficient of 36,900 M -1 cm-1. The purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 27C). The endotoxin level was then determined using a Charles River Laboratories Endosafe-PTS system (0.05 - 5 EU/ml sensitivity) using a 92-fold dilution of the sample in Charles Rivers Endotoxin Specific Buffer BG120 yielding a result of <1 EU/mg protein. The macromolecular state of the product was then determined using size exclusion chromatography on 50 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 27H). The product was then subject to mass spectral analysis by diluting 1 μl of the sample into 10 μl of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile). The resultant solution (1 μl) was spotted onto a MALDI sample plate. The sample was allowed to dry before being analyzed using a
Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses (Figure 27I) and confirmed the integrity of the peptibody, within experimental error. The product was then stored at -800C.
Figure 31 C shows that purified Fc-L5-ShK[2-35] is highly active and blocks human Kv1.3 as determined by whole cell patch clamp electrophysiology (see Example 36).
Purification of Fc-L25-ShK(2-35). Approximately 1 L of conditioned medium was loaded on to a 5 ml Amersham HiTrap Protein A column at 5 ml/min 7 0C, and the column was washed with several column volumes of Dulbecco's phosphate buffered saline without divalent cations (PBS) and sample was eluted with a step to 100 mM glycine pH 3.0. The protein A elution pool (approximately 9.5 ml) combined with 119 μl 3 M sodium acetate and then diluted to 50 ml with water. The pH adjusted material was then loaded on to a 5 ml Amersham HiTrap SP-HP column in S-Buffer A (20 mM NaH2PO1J, pH 7.0) at 5 ml/min 7 °C. The column was then washed with several column volumes S-Buffer A1 and then developed using a linear gradient from 0% to 75% S-Buffer B (20 mM NaH2PO4, 1 M NaCI, pH 7.0) at 5 ml/min followed by a step to 100% S-Buffer B at 7 0C. Fractions containing the main peak from the chromatogram were pooled and filtered through a 0.22 μm cellulose acetate filter.
A spectral scan was then conducted on 20 μl of the combined pool diluted in 700 μl PBS using a Hewlett Packard 8453 spectrophotometer Figure 27J. The concentration of the filtered material was determined to be 1.40 mg/ml using a calculated molecular mass of 31,011 g/mol and extinction coefficient of 36,900 M 1 cm-1. The purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 27D). The endotoxin level was then determined using a Charles River Laboratories Endosafe-PTS system (0.05 - 5 EU/ml sensitivity) using a 28-fold dilution of the sample in Charles Rivers Endotoxin Specific Buffer BG120 yielding a result of <1 EU/mg protein. The macromolecular state of the product was then determined using size exclusion chromatography on 50 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 27K). The product was then subject to mass spectral analysis by diluting 1 μl of the sample into 10 μl of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile). The resultant solution (1 μl) was spotted onto a MALDI sample plate. The sample was allowed to dry before being analyzed using a
Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses (Figure 27L) and this confirmed the itegrity of the peptibody, within experimental error. The product was then stored at
-800C.
Purified Fc-L25-ShK[2-35] inhibited human Kv1.3 with an IC5O of -150 pM by whole cell patch clamp electrophysiology on HEK293/Kv1.3 cells (Example 36).
Example 3 Fc-L-ShK[I -351 bacterial expression
Description of bacterial peptibodv expression vectors and procedures for cloning and expression of peptibodies. The cloning vector used for bacterial expression (Examples 3-30) is based on pAMG21 (originally described in U.S. Patent 2004/0044188). It has been modified in that the kanamycin resistance component has been replaced with ampicillin resistance by excising the DNA between the unique BstBI and Nsil sites of the vector and replacing with an appropriately digested PCR fragment bearing the beta-lactamase gene using PCR primers CCA ACA CAC TTC GAA AGA CGT TGA TCG GCA C ( S EQ ID NO : 667 ) and CAC CCA ACA ATG CAT CCT TAA AAA AAT TAC GCC C (SEQ I D NO : 668 ) with pUC19 DNA as the template source of the beta-lactamase gene conferring resistance to ampicillin. The new version is called pAMG21ampR.
Description of cloning vector pAMG21ampR-Fc-Pep used in examples 3 to 30, excluding 15 and 16. Figure 11A-C and Figure 11D (schematic diagram) show the ds-DNA that has been added to the basic vector pAMG21ampR to permit the cloning of peptide fusions to the C-terminus of the Fc gene. The DNA has been introduced between the unique Ndel and BamHI sites in the pAMG21ampR vector. This entire region of DNA is shown in Figurei 1A-C. The coding region for Fc extends from nt 5134 to 5817 and the protein sequence appears below the DNA sequence. This is followed in frame by a glyX5 linker (nt's 5818-5832). A BsmBI site (GAGACG) spansnucleotides 5834-5839. DNA cleavage occurs between nucleotides 5828 and 5829 on the upper DNA strand and between nucleotides 5832 and 5833 on the lower DNA strand. Digestion creates 4 bp cohesive termini as shown here. The BsmBI site is underlined.
AGGTGG TGGTTGAGACG SEQ I D NO : 683
TCCACCACCA ACTCTGC
SEQ ID NO : 684
A second BsmBI site occurs at nucleotides 6643 through 6648; viz., CGTCTC. DNA cleavage occurs between nucleotides 6650 and 6651 on the upper strand and between 6654 and
6655 on the lower strand.
CGTCTCT TAAGGATCCG SEQ ID NO: 685
GCAGAGAATTC CTAGGC SEQ ID NO: 686
Between the two BsmBI sites is a dispensable chloramphenicol resistance cassette constitutively expressing chloramphenicol acetyltransferase (cat gene). The cat protein sequence:
1 MEKKITGYTT VDISQWHRKE HFEAFQSVAQ CTYNQTVQLD ITAFLKTVKK
51 NKHKFYPAFI HILARLMNAH PEFRMAMKDG ELVIWDSVHP CYTVFHEQTE
101 TFSSLWSEYH DDFRQFLHIY SQDVACYGEN LAYFPKGFIE NMFFVSANPW
151 VSFTSFDLNV ANMDNFFAPV FTMGKYYTQG DKVLMPLAIQ VHHAVCDGFH 201 VGRMLNELQQ YCDEWQGGA //SEQ ID NO: 1337
is shown in Fig. 11A-C and extends from nucleotides 5954 to 6610. The peptide encoding duplexes in each example (except Examples 15 and 16) bear cohesive ends complementary to those presented by the vector. Description of the cloning vector pAMG21ampR-Pep-Fc used in examples 15 and 16.
Figure 12A-C1 and the schematic diagram in Figure 12D, shows the ds-DNA sequence that has been added to the basic vector pAMG21ampR to permit the cloning of peptide fusions to the N- terminus of the Fc gene. The DNA has been introduced between the unique Ndel and BamHI sites in the pAMG21ampR vector. The coding region for Fc extends from nt 5640 to 6309 and the protein sequence appears below the DNA sequence. This is preceded in frame by a glyX5 linker
(nt's 5614-5628). A BsmBI site spans nucleotides 5138 to 5143; viz., GAGACG. The cutting occurs between nucleotides 5132 and 5133 on the upper DNA strand and between 5136 and 5137 on the lower DNA strand.
Digestion creates 4 bp cohesive termini as shown. The BsmBI site is underlined.
AATAACA TATGCGAGACG SEQ ID NO: 687
TTATTGTATAC GCTCTGC
SEQ ID NO : 688 A second BsmBI site occurs at nucleotides 5607 through 5612; viz., CGTCTC. Cutting occurs between nucleotides 5613 and 5614 on the upper strand and between 5617 and 5618 on the lower strand.
CGTCTCA GGTGGTGGT GCAGAGTCCAC CACCA
SEQ ID NO : 689
Between the BsmBI sites is a dispensable zeocin resistance cassette constitutively expressing the Shigella ble protein. The b|e protein sequence:
1 MAKLTSAVPV LTARDVAGAV EFWTDRLGFS RDFVEDDFAG WRDDVTLFI 51 SAVQDQWPD NTLAWVWVRG LDELYAEWSE WSTNFRDAS GPAMTEIGEQ
101 PWGREFALRD PAGNCVHFVA EEQD //SEQ ID NO: 1338
is shown extending from nucleotides 5217 to 5588 in Figure 12A-C. The peptide encoding duplexes in Examples 15 and 16 bear cohesive ends complementary to those presented by the vector.
Description of the cloning vector pAMG21ampR-Pep-Fc used in Examples 52 and 53. Figure 12E-G shows the ds-DNA sequence that has been added to the basic vector pAMG21ampR to permit the cloning of peptide fusions to the N-terminus of the Fc gene in which the first two codons of the peptide are to be met-gly. The DNA has been introduced between the unique Ndel and BamHI sites in the pAMG21ampR vector. The coding region for Fc extends from nt 5632 to 6312 and the protein sequence appears below the DNA sequence. This is preceded in frame by a glyX5 linker (nt's 5617-5631). A BsmBI site spans nucleotides 5141 to 5146; viz., GAGACG. The cutting occurs between nucleotides 5135 and 5136 on the upper DNA strand and between 5139 and 5140 on the lower DNA strand. Digestion creates 4 bp cohesive termini as shown. The BsmBI site is underlined.
MTAACATAT GGGTCGAGACG SEQ ID NO : 1344
SEQ I D NO : 1343
TTATTGTATACCCA GCTCTGC SEQ ID NO : 1345
A second BsmB! site occurs at nucleotides 5607 through 5612; viz, CGTCTC. Cutting occurs between nucleotides 5613 and 5614 on the upper strand and between 5617 and 5618 on the lower strand.
CGTCTCA GGTGGTGGT GCAGAGTCCAC CACCA SEQ ID NO: 1346
Between the BsmBI sites is a dispensable zeocin resistance cassette constitutively expressing the Shigella b|e protein. The ble protein sequence, as described above, is shown extending from nucleotide positions 5220 to 5591. The peptide encoding duplexes in Examples 52 and 53 herein below bear cohesive ends complementary to those presented by the vector.
For Examples 3 to 30 for which all are for bacterial expression, cloned peptide sequences are all derived from the annealing of oligonucleotides to create a DNA duplex that is directly ligated into the appropriate vector. Two oligos suffice for Example 20, four are required for all other
examples. When the duplex is to be inserted at the N-terminus of Fc (see, Examples 15, 16, 52, and 53 herein) the design is as follows with the ordinal numbers matching the listing of oligos in each example:
First Oligo Second Oligo
TATG CCAC
Fourth Oligo Third Oligo
When the duplex is to be inserted at the C-terminus of Fc (Examples 3, 4, 5, 10, 11 , 12, 13, and 30) the design is as follows:
First Oligo Second Oligo
TGGT
-ATTC
Fourth Oligo Third Oligo
All remaining examples have the duplex inserted at the C-terminus of Fc and utilize the following design.
First Oligo Second Oligo
TGGT
-ATTC
Fourth Oligo Third Oligo
No kinasing step is required for the phosphorylation of any of the oligos. A successful insertion of a duplex results in the replacement of the dispensable antibiotic resistance cassette (Zeocin resistance for pAMG21ampR-Pep-Fc and chloramphenicol resistance for pAMG21ampR- Fc-Pep). The resulting change in phenotype is useful for discriminating recombinant from nonrecombinant clones.
The following description gives the uniform method for carrying out the cloning of all 30 bacterially expressed recombinant proteins exemplified herein. Only the set of oligonucleotides and the vector are varied. These spefications are given below in each example. An oligonucleotide duplex containing the coding region for a given peptide was formed by annealing the oligonucleotides listed in each example. Ten picomoles of each oligo was mixed in a final volume of 10 μl containing 1X ligation buffer along with 0.3 μg of appropriate vector that had been previously digested with restriction endonuclease BsmBI. The mix was heated to 800C and allowed to cool at 0.1 degree/sec to room temperature. To this was added 10 μl of 1X ligase
buffer plus 400 units of T4 DNA ligase. The sample was incubated at 14C for 20 min. Ligase was inactivated by heating at 65°C for 10 minutes. Next, 10 units of restriction endonucleases BsmBI were added follwed by incubation at 55C for one hour to cleave any reformed parental vector molecules. Fifty ul of chemically competent E. coli cells were added and held at 2C for 20 minutes followed by heat shock at 42C for 5 second. The entire volume was spread onto Luria Agar plates supplemented with carbenicillin at 200 μg/ml and incubated overnight at 37C. Colonies were tested for the loss of resistance to the replaceable antibiotic resistance marker. A standard PCR test can be used to confirm the expected size of the duplex insert. Plasmid preparations were obtained and the recombinant insert was verified by DNA sequencing. Half liter cultures of a sequence confirmed construct were grown in Terrific Broth, expression of the peptibody was induced by addition of N-(3-oxo-hexanoyl)-homoserine lactone at 50 ng/ml and after 4-6 hours of shaking at 37C the cells were centrifuged and the cell paste stored at -20C.
The following gives for each example the cloning vector and the set of oligonucleotides used for constructing each fusion protein. Also shown is a DNA/protein map. Bacterial expression of Fc-L-ShK[I -351 inhibitor of Kv1.3. The methods to clone and express the peptibody in bacteria are described above. The vector used was pAMG21ampR-Fc- Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-ShK[I -35]. Oligos used to form the duplex:
TGGTTCCGGTGGTGGTGGTTCCCGTTCCTGCATCGACACCAT //SEQ ID NO: 669;
CCCGAAATCCCGTTGCACCGCTTTCCAGTGCAAACACTCCATGAAATACCGTCTGTCCTTCTGCCGTAAAACC TGCGGTACCTGC //SEQ ID NO: 670;
CTTAGCAGGTACCGCAGGTTTTACGGCAGAAGGACAGACGGT //SEQ ID NO: 671;
ATTTCATGGAGTGTTTGCACTGGAAAGCGGTGCAACGGGATTTCGGGATGGTGTCGATGCAGGAACGGGAACC ACCACCACCGGA //SEQ ID NO: 672;
The oligo duplex is shown below:
TGGTTCCGGTGGTGGTGGTTCCCGTTCCTGCATCGACACCATCCCGAAATCCCGTTGCAC 1 + + + + + + 60 AGGCCACCACCACCAAGGGCAAGGACGTAGCTGTGGTAGGGCTTTAGGGCAACGTG
G S G G G G S R S C I D T I P K S R C T - CGCTTTCCAGTGCAAACACTCCATGAAATACCGTCTGTCCTTCTGCCGTAAAACCTGCGG 61 + + + + + + 120
GCGAAAGGTCACGTTTGTGAGGTACTTTATGGCAGACAGGAAGACGGCATTTTGGACGCC
A F Q C K H S M K Y R L S F C R K T C G -
TACCTGC //SEQ ID NO: 673 121
5 ATGGACGATTC //SEQ ID NO: 675
T C - //SEQ ID NO: 674
Bacterial expression of the peptibody was as described in Example 3 and paste was 0 stored frozen. Purification of bacterially expressed Fc-U O-ShK( 1-35) is further described in Example 38 herein below.
Example 4 Fc-L-ShKf2-351 bacterial expression 5
Bacterial expression of Fc-L-ShK[2-351. The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-ShKβ-35]. O Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGG1: T //SEQ ID NO: 676; 5 ACCGTCTGTCCTTCTGCCGTAAAACCTGCGGTACCTGC //SEQ ID NO: 677;
CTTAGCAGGTACCGCA( //SEQ ID NO: 678; 0 TTTCGGGATGGTGTCGATGCAGGAACCACCACCACCGGA //SEQ ID NO: 679;
The oligo duplex formed is shown below:
TGGTTCCGGTGGTGGTGGTTCCTGCATCGACACCATCCCGAAATCCCGTTGCACCGCTTT 5 1 + + + + + + 60
AGGCCACCACCACCAAGGACGTAGCTGTGGTAGGGCTTTAGGGCAACGTGGCGAAA G S G G G G S C I D T I P K S R C T A F - O CCAGTGCAAACACTCCATGAAATACCGTCTGTCCTTCTGCCGTAAAACCTGCGGTACCTG
61 + + + + + + 120
GGTCACGTTTGTGAGGTACTTTATGGCAGACAGGAAGACGGCATTTTGGACGCCATGGAC
Q C K H S M K Y R L S F C R K T C G T C - //SEQ ID NO:6815
C //SEQ ID NO:680 121 -
GATTC //SEQ ID NO: 682 O Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen. Purification of bacterially expressed Fc-LI 0-ShK(2-35) is further described in Example 39 herein below.
Example 5 Fc-L-HmK bacterial expression
Bacterial expression of Fc-L-HmK. The methods to clone and express the peptibody in 5 bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-HmK.
Oligos used to form duplex are shown below: 0 TGGTTCCGGTGGTGGTGGTTCCCGTACCTGCAAAGACCTGAT //SEQ ID NO: 690;
TGC SEQ ID NO: 692; 5 CTTAGCAGGAACCGCAGGTTTTACGGCACAGGTTCAGACGGT //SEQ ID NO: 693;
ATTTCATGGAGGTACGGCAACGGATGTCGGTGCATTCGGAAACCGGGATCAGGTCTTTGCAGGTACGGGAACCACCACCACC GGA //SEQ ID NO: 694 O The oligo duplex formed is shown below:
TGGTTCCGGTGGTGGTGGTTCCCGTACCTGCAAAGACCTGATCCCGGTTTCCGAATGCAC
1 + Y + + + + 60
AGGCCACCACCACCAAGGGCATGGACGTTTCTGGACTAGGGCCAAAGGCTTACGTG
G S G G G G S R T C K D L I P V S E C T - CGACATCCGTTGCCGTACCTCCATGAAATACCGTCTGAACCTGTGCCGTAAAACCTGCGG
61 + + + + + + 120 O GCTGTAGGCAACGGCATGGAGGTACTTTATGGCAGACTTGGACACGGCATTTTGGACGCC
D I R C R T S M K Y R L N L C R K T C G - //SEQ ID NO:696 TTCCTGC //SEQ ID NO: 695 5 121
AAGGACGATTC //SEQ ID NO: 697 S C - O Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 6 Fc-L-KTXI bacterial expression 5 Bacterial expression of Fc-L-KTXI . The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-KTXL
Oligos used to form duplex are shown below: 0
TGGTTCCGGTGGTGGTGGTTCCGGTGTTGAAATCAACGTTAAATGCT //SEQ ID NO: 698;
CCCGAAA //SEQ ID NO: 699;
CTTATTTCGGGGTGCAGTGGCATTTACGGTTCATGCATTTACCGAAA //SEQ ID NO: 700;
CACCGGA //SEQ ID NO: 701;
The oligo duplex formed is shown below:
TGGTTCCGGTGGTGGTGGTTCCGGTGTTGAAATCAACGTTAAATGCTCCGGTTCCCCGCA 1 + + + + + + 60
AGGCCACCACCACCAAGGCCACAACTTTAGTTGCAATTTACGAGGCCAAGGGGCGT
G S G G G G S G V E I N V K C S G S P Q -
GTGCCTGAAACCGTGCAAAGACGCTGGTATGCGTTTCGGTAAATGCATGAACCGTAAATG
61 + + + + + + 120
CACGGACTTTGGCACGTTTCTGCGACCATACGCAAAGCCATTTACGTACTTGGCATTTAC
C L K P C K D A G M R F G K C M N R K C -
CCACTGCACCCCGAAA //SEQ ID NO:702 121 + GGTGACGTGGGGCTTTATTC //SEQ ID NO: 704
H C T P K - //SEQ ID NO:703
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Purification and refolding of Fc-L-KTXI expressed in bacteria. Frozen, E. coli paste (28 g) was combined with 210 ml of room temperature 50 mM tris HCI, 5 mM EDTA, pH 8.0 and was brought to about 0.1 mg/ml hen egg white lysozyme. The suspended paste was passed through a chilled microfluidizer twice at 12,000 PSI. The cell lysate was then centrifuged at 22,000 g for 20 min at 40C. The pellet was then resuspended in 200 ml 1% deoxycholic acid using a tissue grinder and then centrifuged at 22,000 g for 20 min at 40C. The pellet was then resuspended in 200 ml water using a tissue grinder and then centrifuged at 22,000 g for 20 min at 40C. The pellet (4.8 g) was then dissolved in 48 ml 8 M guanidine HCI, 50 mM tris HCI, pH 8.0. The dissolved pellet was then reduced by adding 30 μl 1 M dithiothreitol to 3 ml of the solution and incubating at
370C for 30 minutes. The reduced pellet solution was then centrifuged at 14,000 g for 5 min at room temperature, and then 2.5 ml of the supernatant was transferred to 250 ml of the refolding buffer (2 M urea, 50 mM tris, 160 mM arginine HCI, 5 mM EDTA, 1 mM cystamine HCI, 4 mM cysteine, pH 8.5) at 40C with vigorous stirring. The stirring rate was then slowed and the
incubation was continued for 2 days at 4 0C. The refolding solution was then filtered through a 0.22 μm cellulose acetate filter and stored at 4 0C for 3 days.
The stored refold was then diluted with 1 L of water and the pH was adjusted to 7.5 using 1 M H3PO4. The pH adjusted material was then loaded on to a 10 ml Amersham SP-HP HiTrap column at 10 ml/min in S-Buffer A (20 mM NaH2PO4, pH 7.3) at 7 0C. The column was then washed with several column volumes of S-Buffer A, followed by elution with a linear gradient from 0% to 60% S-Buffer B (20 mM NaH2PO4, 1 M NaCI, pH 7.3) followed by a step to 100% S-Buffer B at 5 ml/min 7 0C. Fractions were then analyzed using a Coomassie brilliant blue stained tris- glycine 4-20% SDS-PAGE, and the fractions containing the desired product were pooled based on these data (45 ml). The pool was then loaded on to a 1 ml Amersham rProtein A HiTrap column in PBS at 2 ml/min 7 0C. Then column was then washed with several column volumes of PBS and eluted with 100 mM glycine pH 3.0. To the elution peak (2.5 ml), 62.5 μl 2 M tris base was added, and then the pH adjusted material was filtered though a Pall Life Sciences Acrodisc with a 0.22 μm, 25 mm Mustang E membrane at 2 ml/min room temperature. A spectral scan was then conducted on 20 μl of the combined pool diluted in 700 μl PBS using a Hewlett Packard 8453 spectrophotometer (Figure 28C). The concentration of the filtered material was determined to be 2.49 mg/ml using a calculated molecular mass of 30,504 g/mol and extinction coefficient of 35,410 M 1 cm 1. The purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 28A). The endotoxin level was then determined using a Charles River Laboratories Endosafe-PTS system
(0.05 - 5 EU/ml sensitivity) using a 50-fold dilution of the sample in Charles Rivers Endotoxin Specific Buffer BG120 yielding a result of <1 EU/mg protein. The macromolecular state of the product was then determined using size exclusion chromatography on 45 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 28B). The product was then subject to mass spectral analysis by diluting 1 μl of the sample into 10 μl of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile). The resultant solution (1 μl) was spotted onto a MALDI sample plate. The sample was allowed to dry before being analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses (Figure 28D) and these
studies confirmed the integrity of the purified peptibody, within experimental error. The product was then stored at -80°C.
Purified Fc-L-KTXI blocked the human Kv1.3 current in a dose-dependent fashion (Figure 32A and Figure 32B) by electrophysiology (method was as described in Example 36).
Example 7
Fc-L-HsTxI bacterial expression
Bacterial expression of Fc-L-HsTI . The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-HsTxI.
Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCGCTTCCTGCCGTACCCCGAAAGAC //SEQ ID NO: 705;
TGCGCTGACCCGTGCCGTAAAGAAACCGGTTGCCCGTACGGTAAATGCATGAACCGTAAATGCAAATGCAACC GTTGC //SEQ ID NO: 706;
CTTAGCAACGGTTGCATTTGCATTTACGGTTCATGCATTTACCGTACG //SEQ ID NO: 707;
GGCAACCGGTTTCTTTACGGCACGGGTCAGCGCAGTCTTTCGGGGTACGGCAGGAAGCGGAACCACCACCACC GGA //SEQ ID NO:708;
The duplex formed by the oligos above is shown below:
TGGTTCCGGTGGTGGTGGTTCCGCTTCCTGCCGTACCCCGAAAGACTGCGCTGACCCGTG 1 + + + + H + go
AGGCCACCACCACCAAGGCGAAGGACGGCATGGGGCTTTCTGACGCGACTGGGCAC G S G G G G S A S C R T P K D C A D P C -
CCGTAAAGAAACCGGTTGCCCGTACGGTAAATGCATGAACCGTAAATGCAAATGCAACCG 61 + + + + + + 120
GGCATTTCTTTGGCCAACGGGCATGCCATTTACGTACTTGGCATTTACGTTTACGTTGGC R K E T G C P Y G K C M N R K C K C N R -
TTGC SEQ ID NO: 709
121 124
AACGATTC SEQ ID NO: 711
C - SEQ ID NO:710
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 8
Fc-L-MgTx bacterial expression
Bacterial expression of Fc-L-MqTx . The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-MgTx.
Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCACCATCATCAACGTTAAATGCACCTC //SEQ ID NO: 712; 0 CCCGAAACAGTGCCTGCCGCCGTGCAAAGCTCAGTTCGGTCAGTCCGCTGGTGCTAAATGCATGAACGGTAAA TGCAAATGCTACCCGCAC //SEQ ID NO: 713;
CTTAGTGCGGGTAGCATTTGCATTTACCGTTCATGCATTTAGCACCAG //SEQ ID NO: 714; 5 CGGACTGACCGAACTGAGCTTTGCACGGCGGCAGGCACTGTTTCGGGGAGGTGCATTTAACGTTGATGATGGT GGAACCACCACCACCGGA //SEQ ID NO: 715;
The oligos above were used to form the duplex shown below: 0
TGGTTCCGGTGGTGGTGGTTCCACCATCATCAACGTTAAATGCACCTCCCCGAAACAGTG 1 + + + + + + 60
AGGCCACCACCACCAAGGTGGTAGTAGTTGCAATTTACGTGGAGGGGCTTTGTCAC 5 G S G G G G S T I I N V K C T S P K Q C - CCTGCCGCCGTGCAAAGCTCAGTTCGGTCAGTCCGCTGGTGCTAAATGCATGAACGGTAA
61 + + + + + + 120
GGACGGCGGCACGTTTCGAGTCAAGCCAGTCAGGCGACCACGATTTACGTACTTGCCATT L P P C K A Q F G Q S A G A K C M N G K - ATGCAAATGCTACCCGCAC SEQ ID NO: 716
121 + D TACGTTTACGATGGGCGTGATTC SEQ ID NO: 718
C K C Y P H - SEQ ID NO:717
Bacterial expression of the peptibody was as described in Example 3 and paste was O stored frozen.
Example 9 Fc-L-AgTx2 bacterial expression
Bacterial expression of Fc-L-AqTx2. The methods to clone and express the peptibody in 5 bacteria are described in Example 3. The vector used was pAMG21 ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-AgTx2.
Oligos used to form duplex are shown below: 0 TGGTTCCGGTGGTGGTGGTTCCGGTGTTCCGATCAACGTTTCCTGCACCGGT //SEQ ID NO: 719;
TCCCCGCAGTGCATCAAACCGTGCAAAGACGCTGGTATGCGTTTCGGTAAATGCATGAACCGTAAATGCCACT GCACCCCGAAA //SEQ ID NO: 720;
CTTATTTCGGGGTGCAGTGGCATTTACGGTTCATGCATTTACCGAAACGCATA //SEQ ID NO: 721;
CCAGCGTCTTTGCACGGTTTGATGCACTGCGGGGAACCGGTGCAGGAAACGTTGATCGGAACACCGGAACCAC CACCACCGGA //SEQ ID NO: 722;
The oligos listed above were used to form the duplex shown below: 0
TGGTTCCGGTGGTGGTGGTTCCGGTGTTCCGATCAACGTTTCCTGCACCGGTTCCCCGCA 1 + + + + + + 6Q
AGGCCACCACCACCAAGGCCACAAGGCTAGTTGCAAAGGACGTGGCCAAGGGGCGT 5 G S G G G G S G V P I N V S C T G S P Q -
GTGCATCAAACCGTGCAAAGACGCTGGTATGCGTTTCGGTAAATGCATGAACCGTAAATG
61 + + + + + + 120
CACGTAGTTTGGCACGTTTCTGCGACCATACGCAAAGCCATTTACGTACTTGGCATTTAC 0
C I K P C K D A G M R F G K C M N R K C -
CCACTGCACCCCGAAA_SEQ ID NO: 723 121 + 5 GGTGACGTGGGGCTTTATTC SEQ ID NO: 725
H C T P K - SEQ ID NO:724
Bacterial expression of the peptibody was as described in Example 3 and paste was O stored frozen.
Refolding and purification of Fc-L-AgTx2 expressed in bacteria. Frozen. E. coli paste (15 g) was combined with 120 ml of room temperature 50 mM tris HCI, 5 mM EDTA, pH 8.0 and was brought to about 0.1 mg/ml hen egg white lysozyme. The suspended paste was passed through a 5 chilled microfluidizer twice at 12,000 PSI. The cell lysate was then centrifuged at 22,000 g for 20 min at 40C. The pellet was then resuspended in 200 ml 1% deoxycholic acid using a tissue grinder and then centrifuged at 22,000 g for 20 min at 40C. The pellet was then resuspended in 200 ml water using a tissue grinder and then centrifuged at 22,000 g for 20 min at 40C. The pellet (4.6 g) was then dissolved in 46 ml 8 M guanidine HCI, 50 mM tris HCI, pH 8.0. The dissolved 0 pellet was then reduced by adding 30 μl 1 M dithiothreitol to 3 ml of the solution and incubating at
370C for 30 minutes. The reduced pellet solution was then centrifuged at 14,000 g for 5 min at room temperature, and then 2.5 ml of the supernatant was transferred to 250 ml of the refolding buffer (2 M urea, 50 mM tris, 160 mM arginine HCI, 5 mM EDTA, 1 mM cystamine HCI, 4 mM cysteine, pH 9.5) at 40C with vigorous stirring. The stirring rate was then slowed and the 5 incubation was continued for 2 days at 40C. The refolding solution was then filtered through a 0.22 μm cellulose acetate filter and stored at -700C.
The stored refold was defrosted and then diluted with 1 L of water and the pH was adjusted to 7.5 using 1 M H3PO4. The pH adjusted material was then filtered through a 0.22 μm cellulose acetate filter and loaded on to a 10 ml Amersham SP-HP HiTrap column at 10 ml/min in S-Buffer A (20 mM Na^PO4, pH 7.3) at 7 0C. The column was then washed with several column volumes of S-Buffer A, followed by elution with a linear gradient from 0% to 60% S-Buffer B (20 mM NaH2PO4, 1 M NaCI, pH 7.3) followed by a step to 100% S-Buffer B at 5 ml/min 7 0C. Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS- PAGE, and the fractions containing the desired product were pooled based on these data (15 ml). The pool was then loaded on to a 1 ml Amersham rProtein A HiTrap column in PBS at 2 ml/min 7 0C. Then column was then washed with several column volumes of 20 mM NaHsPO4 pH 6.5, 1 M NaCI and eluted with 100 mM glycine pH 3.0. To the elution peak (1.5 ml), 70 μl 1 M tris HCI pH 8.5 was added, and then the pH -adjusted material was filtered though a 0.22 μm cellulose acetate filter.
A spectral scan was then conducted on 20 μl of the combined pool diluted in 700 μl PBS using a Hewlett Packard 8453 spectrophotometer (Figure 29C). The concentration of the filtered material was determined to be 1.65 mg/ml using a calculated molecular mass of 30,446 g/mol and extinction coefficient of 35,410 M 1 cm-1. The purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 29A). The endotoxin level was then determined using a Charles River Laboratories Endosafe-PTS system (0.05 - 5 EU/ml sensitivity) using a 33-fold dilution of the sample in Charles Rivers Endotoxin Specific Buffer BG120 yielding a result of <4 EU/mg protein. The macromolecular state of the product was then determined using size exclusion chromatography on 20 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 29D). The product was then subject to mass spectral analysis by diluting 1 μl of the sample into 10 μl of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile) . The resultant solution (1 μl) was spotted onto a MALDI sample plate. The sample was allowed to dry before being analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses (Figure 29E) and these studies confirmed the integrity of the purified peptibody, within experimental error. The product was then stored at -80 °C.
Example 10 Fc-L-OSKI bacterial expression
Bacterial expression of Fc-L-OSKI , The methods used to clone and express the 5 peptibody in bacteria were as described in Example 3. The vector used was pAMG21ampR-Fc- Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-OSKI .
Oligos used to form duplex are shown below: 0 TGGTTCCGGTGGTGGTGGTTCCGGTGTTATCATCAACGTTAAATGCAAAATCTCCCGTCAGTGCCTGGAACCG TGCAAAAAAG //SEQ ID NO: 726;
CTGGTATGCGTTTCGGTAAATGCATGAACGGTAAATGCCACTGCACCCCGAAA //SEQ ID NO: 727; 5 CTTATTTCGGGGTGCAGTGGCATTTACCGTTCATGCATTTACCGAAACGCATACCAGCTTTTTTGCACGGTTC CAGGCACTGA //SEQ ID NO: 728;
CGGGAGATTTTGCATTTAACGTTGATGATAACACCGGAACCACCACCACCGGA //SEQ ID NO: 729;
O The oligos shown above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCGGTGTTATCATCAACGTTAAATGCAAAATCTCCCGTCA I + + + + + + so
AGGCCACCACCACCAAGGCCACAATAGTAGTTGCAATTTACGTTTTAGAGGGCAGT 5
G S G G G G S G V I I N V K C K I S R Q -
GTGCCTGGAACCGTGCAAAAAAGCTGGTATGCGTTTCGGTAAATGCATGAACGGTAAATG
61 + + + + + + 120 O CACGGACCTTGGCACGTTTTTTCGACCATACGCAAAGCCATTTACGTACTTGCCATTTAC
C L E P C K K A G M R F G K C M N G K C -
CCACTGCACCCCGAAA SEQ ID NO: 730 5 121 +
GGTGACGTGGGGCTTTATTC SEQ ID NO: 732 H C T P K - SEQ ID NO:731
O Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen for later use. Purification of Fc-LI 0-OSK1 from E. coli paste is described in Example 40 herein below.
Example 11 5 Fc-L-OSKI (E16K.K20D) bacterial expression
Bacterial expression of Fc-L-0SK1(E16K, K20D). The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep
and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-OSK1(E16K,K20D).
Oligos used to form duplex are shown below: TGGTTCCGGTGGTGGTGGTTCCGGTGTTATCATCAACGTTAAATGCAAAATCTCCCGTCAGTGCCTGAAACCG TGCAAAGACG //SEQ ID NO: 733;
CTGGTATGCGTTTCGGTAAATGCATGAACGGTAAATGCCACTGCACCCCGAAA //SEQ ID NO: 734;
CTTATTTCGGGGTGCAGTGGCATTTACCGTTCATGCATTTACCGAAACGCATACCAGCGTCTTTGCACGGTTT CAGGCACTGA //SEQ ID NO: 735;
CGGGAGATTTTGCATTTAACGTTGATGATAACACCGGAACCACCACCACCGGA //SEQ ID NO: 736; The oligos shown above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCGGTGTTATCATCAACGTTAAATGCAAAATCTCCCGTCA 1 + + + + + + go AGGCCACCACCACCAAGGCCACAATAGTAGTTGCAATTTACGTTTTAGAGGGCAGT
G S G G G G S G V I I N V K C K I S R Q - GTGCCTGAAACCGTGCAAAGACGCTGGTATGCGTTTCGGTAAATGCATGAACGGTAAATG 61 + + + + + + 120
CACGGACTTTGGCACGTTTCTGCGACCATACGCAAAGCCATTTACGTACTTGCCATTTAC C L K P C K D A G M R F G K C M N G K C - CCACTGCACCCCGAAA SEQ ID NO: 737
121 +
GGTGACGTGGGGCTTTATTC SEQ ID NO: 739
H C T P K - SEQ ID NO:738
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen for later use.
Example 12 Fc-L-Anuroctoxin bacterial expression
Bacterial expression of Fc-L-Anuroctoxin, The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-Anuroctoxin.
Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCAAAGAATGCACCGGTCCGCAGCACTGCACCAACTTCTGCCGTAAAAACAAA TGCACCCACG //SEQ ID NO: 740;
GTAAATGCATGAACCGTAAATGCAAATGCTTCAACTGCAAA //SEQ ID NO : 741 ;
CTTATTTGCAGTTGAAGCATTTGCATTTACGGTTCATGCATTTACCGTGGGTGCATTTGTTTTTACGGCAGAA GTTGGTGCAG //SEQ ID NO: 742;
TGCTGCGGACCGGTGCATTCTTTGGAACCACCACCACCGGA //SEQ ID NO: 743;
The oligos shown above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCAAAGAATGCACCGGTCCGCAGCACTGCACCAACTTCTG 1 + + 1- + + + 60 AGGCCACCACCACCAAGGTTTCTTACGTGGCCAGGCGTCGTGACGTGGTTGAAGAC
G S G G G G S K E C T G P Q H C T N F C - CCGTAAAAACAAATGCACCCACGGTAAATGCATGAACCGTAAATGCAAATGCTTCAACTG 61 + + + + + + 120
GGCATTTTTGTTTACGTGGGTGCCATTTACGTACTTGGCATTTACGTTTACGAAGTTGAC R K N K C T H G K C M N R K C K C F N C - CAAA SEQ ID NO:744
121
GTTTATTC SEQ ID NO: 746
K - SEQ ID NO:745
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 13 Fc-L-Noxiustoxin bacterial expression
Bacterial expression of Fc-L-Noxiustoxin or Fc-L-NTX. The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-NTX. Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCACCATCATCAACGTTAAATGCACCTCCCCGAAACAGTGCTCCAAACCGTGC AAAGAACTGT //SEQ ID NO: 747;
ACGGTTCCTCCGCTGGTGCTAAATGCATGAACGGTAAATGCAAATGCTACAACAAC //SEQ ID NO: 748;
CTTAGTTGTTGTAGCATTTGCATTTACCGTTCATGCATTTAGCACCAGCGGAGGAACCGTACAGTTCTTTGCA CGGTTTGGAG //SEQ ID NO: 749;
CACTGTTTCGGGGAGGTGCATTTAACGTTGATGATGGTGGAACCACCACCACCGGA //SEQ ID NO: 750;
The oligos shown above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCACCATCATCAACGTTAAATGCACCTCCCCGAAACAGTG 1 + + + + + + 60
AGGCCACCACCACCAAGGTGGTAGTAGTTGCAATTTACGTGGAGGGGCTTTGTCAC
G S G G G G S T I I N V K C T S P K Q C -
CTCCAAACCGTGCAAAGAACTGTACGGTTCCTCCGCTGGTGCTAAATGCATGAACGGTAA 61 + + + + + + 120
GAGGTTTGGCACGTTTCTTGACATGCCAAGGAGGCGACCACGATTTACGTACTTGCCATT S K P C K E L Y G S S A G A K C M N G K -
ATGCAAATGCTACAACAAC SEQ ID NO: 751 121 +
TACGTTTACGATGTTGTTGATTC SEQ ID NO: 753
C K C Y N N - SEQ ID NO:752
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 14
Fc-L-PJ2 bacterial expression
Bacterial expression of Fc-L-Pi2. The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-Pi2.
Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCACCATCTCCTGCACCAACCCG //SEQ ID NO: 754;
AAACAGTGCTACCCGCACTGCAAAAAAGAAACCGGTTACCCGAACGCTAAATGCATGAACCGTAAATGCAAAT GCTTCGGTCGT //SEQ ID NO: 755;
CTTAACGACCGAAGCATTTGCATTTACGGTTCATGCATTTAGCG //SEQ ID NO: 756;
TTCGGGTAACCGGTTTCTTTTTTGCAGTGCGGGTAGCACTGTTTCGGGTTGGTGCAGGAGATGGTGGAACCAC CACCACCGGA //SEQ ID NO: 757;
The oligos above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCACCATCTCCTGCACCAACCCGAAACAGTGCTACCCGCA 1 + + + + + + go
AGGCCACCACCACCAAGGTGGTAGAGGACGTGGTTGGGCTTTGTCACGATGGGCGT G S G G G G S T I S C T N P K Q C Y P H -
CTGCAAAAAAGAAACCGGTTACCCGAACGCTAAATGCATGAACCGTAAATGCAAATGCTT
61 + + + + + + 120
GACGTTTTTTCTTTGGCCAATGGGCTTGCGATTTACGTACTTGGCATTTACGTTTACGAA
C K K E T G Y P N A K C M N R K C K C F -
CGGTCGT SEQ ID NO: 758
121 GCCAGCAATTC SEQ ID NO: 760
G R - SEQ ID NO:759
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 15 ShKM -351-L-Fc bacterial expression
Bacterial expression of ShK[I -351-L-Fc, The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Pep-Fc and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of ShK[I -35]-L-Fc.
Oligos used to form duplex are shown below: TATGCGTTCTTGTATTGATACTATTCCAAAATCTCGTTGTACTGCTTTTCAATGTAAACATTCTATGAAATAT CGTCTTTCTT //SEQ ID NO: 761;
TTTGTCGTAAAACTTGTGGTACTTGTTCTGGTGGTGGTGGTTCT //SEQ ID NO: 762;
CACCAGAACCACCACCACCAGAACAAGTACCACAAGTTTTACGACAAAAAGAAAGACGATATTTCATAGAATG TTTACATTGA //SEQ ID NO: 763;
AAAGCAGTACAACGAGATTTTGGAATAGTATCAATACAAGAACG //SEQ ID NO: 764;
The oligos shown above were used to form the duplex shown below:
TATGCGTTCTTGTATTGATACTATTCCAAAATCTCGTTGTACTGCTTTTCAATGTAAACA 1 + + + + + + 60 GCAAGAACATAACTATGATAAGGTTTTAGAGCAACATGACGAAAAGTTACATTTGT
M R S C I D T I P K S R C T A F Q C K H - TTCTATGAAATATCGTCTTTCTTTTTGTCGTAAAACTTGTGGTACTTGTTCTGGTGGTGG 61 + + + + + + 120
AAGATACTTTATAGCAGAAAGAAAAACAGCATTTTGAACACCATGAACAAGACCACCACC
S M K Y R L S F C R K T C G T C S G G G - TGGTTCT SEQ ID NO: 765
121 127
ACCAAGACCAC SEQ ID NO: 767
G S - SEQ ID NO:766
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen. Purification of met-ShK[1-35]-Fc was as described in Example 51 herein below.
Example 16 ShK[2-351-L-Fc bacterial expression
Bacterial expression of ShK[2-351-L-Fc. The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Pep-Fc and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of ShK[2-35]-L-Fc.
Oligos used to form duplex are shown below:
TATGTCTTGTATTGATACTATTCCAAAATCTCGTTGTACTGCTTTTCAATGTAAACATTCTATGAAATATCGT CTTTCTT //SEQ ID NO: 768;
TTTGTCGTAAAACTTGTGGTACTTGTTCTGGTGGTGGTGGTTCT //SEQ ID NO: 769;
CACCAGAACCACCACCACCAGAACAAGTACCACAAGTTTTACGACAAAAAGAAAGACGATATTTCATAGAATG TTTACATTGA //SEQ ID NO: 770;
AAAGCAGTACAACGAGATTTTGGAATAGTATCAATACAAGA SEQ ID NO: 771;
The oligos above were used to form the duplex shown below:
TATGTCTTGTATTGATACTATTCCAAAATCTCGTTGTACTGCTTTTCAATGTAAACATTC
1 + + + + + + 60
AGAACATAACTATGATAAGGTTTTAGAGCAACATGACGAAAAGTTACATTTGTAAG
M S C I D T I P K S R C T A F Q C K H S -
TATGAAATATCGTCTTTCTTTTTGTCGTAAAACTTGTGGTACTTGTTCTGGTGGTGGTGG 61 + + + + + + 120
ATACTTTATAGCAGAAAGAAAAACAGCATTTTGAACACCATGAACAAGACCACCACCACC M K Y R L S F C R K T C G T C S G G G G -
TTCT SEQ ID NO: 772
121
AAGACCAC SEQ ID NO: 774
S - SEQ ID NO:773
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen. Purification of the ShK[2-35]-Fc was as described in Example 50 herein below.
Example 17
Fc-L-ChTx bacterial expression
Bacterial expression of Fc-L-ChTx. The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-ChTx.
Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCCAGTTCACCAACGTT //SEQ ID NO: 775;
ACAAAAAATGCCGTTGCTACTCC //SEQ ID NO: 776;
CTTAGGAGTAGCAACGGCATTTTTTGTTCATGCATTTA //SEQ ID NO: 777;
ACTGGGAACCACCACCACCGGA //SEQ ID NO: 778;
The oligos shown above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCCAGTTCACCAACGTTTCCTGCACCACCTCCAAAGAATG 1 + + + + + + 60
AGGCCACCACCACCAAGGGTCAAGTGGTTGCAAAGGACGTGGTGGAGGTTTCTTAC G S G G G G S Q F T N V S C T T S K E C - CTGGTCCGTTTGCCAGCGTCTGCACAACACCTCCCGTGGTAAATGCATGAACAAAAAATG
6i + + + + + + I20
GACCAGGCAAACGGTCGCAGACGTGTTGTGGAGGGCACCATTTACGTACTTGTTTTTTAC
W S V C Q R L H N T S R G K C M N K K C - CCGTTGCTACTCC SEQ ID NO: 779
121 +---
GGCAACGATGAGGATTC SEQ ID NO: 781 R C Y S - SEQ ID NO:780
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 18
Fc-L-IVITX bacterial expression
Bacterial expression of Fc-L-MTX. The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-MTX.
Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCGTTTCCTGCACCGGT //SEQ ID NO: 782;
TCCAAAGACTGCTACGCTCCGTGCCGTAAACAGACCGGTTGCCCGAACGCTAAATGCATCAACAAATCCTGCA AATGCTACGGTTGC //SEQ ID NO: 783;
CTTAGCAACCGTAGCATTTGCAGGATTTGTTGATGCAT //SEQ ID NO: 784;
TTAGCGTTCGGGCAACCGGTCTGTTTACGGCACGGAGCGTAGCAGTCTTTGGAACCGGTGCAGGAAACGGAAC CACCACCACCGGA //SEQ ID NO: 785;
The oligos above were used to form the duplex shown below: TGGTTCCGGTGGTGGTGGTTCCGTTTCCTGCACCGGTTCCAAAGACTGCTACGCTCCGTG
1 + + + + + + 50
AGGCCACCACCACCAAGGCAAAGGACGTGGCCAAGGTTTCTGACGATGCGAGGCAC G S G G G G S V S C T G S K D C Y A P C - CCGTAAACAGACCGGTTGCCCGAACGCTAAATGCATCAACAAATCCTGCAAATGCTACGG
51 + + + + + + 120
GGCATTTGTCTGGCCAACGGGCTTGCGATTTACGTAGTTGTTTAGGACGTTTACGATGCC R K Q T G C P N A K C I N K S C K C Y G -
TTGC SEQ ID NO: 786
121
AACGATTC SEQ ID NO: 788
C - SEQ ID NO:787
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 19
Fc-L-ChTx(K32E) bacterial expression
Bacterial expression of Fc-L-ChTx(K32E), The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-ChTx(K32E).
Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCCAGTTCACCAACGTTTCCTG //SEQ ID NO: 789;
CACCACCTCCAAAGAATGCTGGTCCGTTTGCCAGCGTCTGCACAACACCTCCCGTGGTAAATGCATGAACAAA GAATGCCGTTGCTACTCC //SEQ ID NO: 790;
CTTAGGAGTAGCAACGGCATTCTTTGTTCATGCATTTACCACG //SEQ ID NO: 791;
GGAGGTGTTGTGCAGACGCTGGCAAACGGACCAGCATTCTTTGGAGGTGGTGCAGGAAACGTTGGTGAACTGG GAACCACCACCACCGGA //SEQ ID NO: 792;
The oligos shown above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCCAGTTCACCAACGTTTCCTGCACCACCTCCAAAGAATG
1 + + + + + + 60
AGGCCACCACCACCAAGGGTCAAGTGGTTGCAAAGGACGTGGTGGAGGTTTCTTAC G S G G G G S Q F T N V S C T T S K E C -
CTGGTCCGTTTGCCAGCGTCTGCACAACACCTCCCGTGGTAAATGCATGAACAAAGAATG
61 + + + + + + 120
GACCAGGCAAACGGTCGCAGACGTGTTGTGGAGGGCACCATTTACGTACTTGTTTCTTAC
W S V C Q R L H N T S R G K C M N K E C -
CCGTTGCTACTCC SEQ ID NO: 793 121 + — GGCAACGATGAGGATTC SEQ ID NO : 795
R C Y S - SEQ ID NO:794
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 20 Fc-L-Apamin bacterial expression
Bacterial expression of Fc-L-Apamin, The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-Apamin.
Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCTGCAACTGCAAAGCTCCGGAAACCGCTCTGTGCGCTCGTCGTTGCCAGCAG CACGGT //SEQ ID NO: 796;
CTTAACCGTGCTGCTGGCAACGACGAGCGCACAGAGCGGTTTCCGGAGCTTTGCAGTTGCAGGAACCACCACC ACCGGA //SEQ ID NO: 797; The oligos above were used to form the duplex shown below:
TGGTTCCGGTGGTGGTGGTTCCTGCAACTGCAAAGCTCCGGAAACCGCTCTGTGCGCTCG I + + + + + + go
AGGCCACCACCACCAAGGACGTTGACGTTTCGAGGCCTTTGGCGAGACACGCGAGC
G S G G G G S C N C K A P E T A L C A R -
TCGTTGCCAGCAGCACGGT SEQ ID NO : 798 61 + AGCAACGGTCGTCGTGCCAATTC SEQ ID NO : 800
R C Q Q H G - SEQ I D NO : 799
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 21 Fc-L-Scyllatoxin bacterial expression
Bacterial expression of Fc-L-Scyllatoxin or Fc-L-ScvTx. The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-ScyTx.
Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCGCTTTCTGCAACCTGCG //SEQ ID NO: 801;
TATGTGCCAGCTGTCCTGCCGTTCCCTGGGTCTGCTGGGTAAATGCATCGGTGACAAATGCGAATGCGTTAAA CAC //SEQ ID NO: 802;
CTTAGTGTTTAACGCATTCGCATTTGTCACCGATGCATTT //SEQ ID NO: 803;
ACCCAGCAGACCCAGGGAACGGCAGGACAGCTGGCACATACGCAGGTTGCAGAAAGCGGAACCACCACCACCG GA //SEQ ID NO: 804;
The oligos above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCGCTTTCTGCAACCTGCGTATGTGCCAGCTGTCCTGCCG
1 1 1 j. 1 1 (- gO
AGGCCACCACCACCAAGGCGAAAGACGTTGGACGCATACACGGTCGACAGGACGGC G S G G G G S A F C N L R M C Q L S C R - TTCCCTGGGTCTGCTGGGTAAATGCATCGGTGACAAATGCGAATGCGTTAAACAC SEQ ID NO: 805
61 + + + + + AAGGGACCCAGACGACCCATTTACGTAGCCACTGTTTACGCTTACGCAATTTGTGATTC SEQ ID NO: 807
S L G L L G K C I G D K C E C V K H - SEQ ID NO:806
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 22 Fc-L-IbTx bacterial expression
Bacterial expression of Fc-L-IbTx. The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21 ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-IbTx.
Oligos used to form duplex are shown below: TGGTTCCGGTGGTGGTGGTTCCCAGTTCACCGACGTTGACTGCTCCGT //SEQ ID NO: 808;
TTCCAAAGAATGCTGGTCCGTTTGCAAAGACCTGTTCGGTGTTGACCGTGGTAAATGCATGGGTAAAAAATGC CGTTGCTACCAG //SEQ ID NO: 809; CTTACTGGTAGCAACGGCATTTTTTACCCATGCATTTACCACGGTCAA //SEQ ID NO: 810;
CACCGAACAGGTCTTTGCAAACGGACCAGCATTCTTTGGAAACGGAGCAGTCAACGTCGGTGAACTGGGAACC ACCACCACCGGA //SEQ ID NO: 811; The oligos above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCCAGTTCACCGACGTTGACTGCTCCGTTTCCAAAGAATG
1 + + + + + + 60
AGGCCACCACCACCAAGGGTCAAGTGGCTGCAACTGACGAGGCAAAGGTTTCTTAC
G S G G G G S Q F T D V D C S V S K E C -
CTGGTCCGTTTGCAAAGACCTGTTCGGTGTTGACCGTGGTAAATGCATGGGTAAAAAATG 61 + + + + + + 120 GACCAGGCAAACGTTTCTGGACAAGCCACAACTGGCACCATTTACGTACCCATTTTTTAC
W S V C K D L F G V D R G K C M G K K C -
CCGTTGCTACCAG SEQ ID NO: 812 121 +—
GGCAACGATGGTCATTC SEQ ID NO: 814 R C Y Q - SEQ ID NO:813 Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 23 Fc-L-HaTxI bacterial expression
Bacterial expression of Fc-L-HaTxL The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-HaTxL
Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCGAATGCCGTTACCTGTTCGGTGGTTG //SEQ ID NO: 815;
CAAAACCACCTCCGACTGCTGCAAACACCTGGGTTGCAAATTCCGTGACAAATACTGCGCTTGGGACTTCACC TTCTCC //SEQ ID NO: 816;
CTTAGGAGAAGGTGAAGTCCCAAGCGCAGTATTTGTCACGGAATTTGC //SEQ ID NO: 817;
AACCCAGGTGTTTGCAGCAGTCGGAGGTGGTTTTGCAACCACCGAACAGGTAACGGCATTCGGAACCACCACC ACCGGA //SEQ ID NO: 818;
The oligos above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCGAATGCCGTTACCTGTTCGGTGGTTGCAAAACCACCTC 1 + + + + + + go
AGGCCACCACCACCAAGGCTTACGGCAATGGACAAGCCACCAACGTTTTGGTGGAG G S G G G G S E C R Y L F G G C K T T S - CGACTGCTGCAAACACCTGGGTTGCAAATTCCGTGACAAATACTGCGCTTGGGACTTCAC
61 + + + + + + 120
GCTGACGACGTTTGTGGACCCAACGTTTAAGGCACTGTTTATGACGCGAACCCTGAAGTG
D C C K H L G C K F R D K Y C A W D F T -
CTTCTCC SEQ ID NO: 819 121 GAAGAGGATTC SEQ ID NO:821
F S - SEQ ID NO:820
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Refolding and purification of Fc-L-HaTxI expressed in bacteria. Frozen, £ coli paste (13 g) was combined with 100 ml of room temperature 50 mM tris HCI15 mM EDTA, pH 8.0 and was brought to about 0.1 mg/ml hen egg white lysozyme. The suspended paste was passed through a chilled microfluidizer twice at 12,000 PSI. The cell lysate was then centrifuged at 22,000 g for 20 min at 40C. The pellet was then resuspended in 200 ml 1% deoxycholic acid using a tissue grinder and then centrifuged at 22,000 g for 20 min at 40C. The pellet was then resuspended in 200 ml water using a tissue grinder and then centrifuged at 22,000 g for 20 min at 40C. The pellet (2.6 g) was then dissolved in 26 ml 8 M guanidine HCI, 50 mM tris HCI, pH 8.0. The dissolved
pellet was then reduced by adding 30 μl 1 M dithiothreitol to 3 ml of the solution and incubating at 37 0C for 30 minutes. The reduced pellet solution was then centrifuged at 14,000 g for 5 min at room temperature, and then 2.5 ml of the supernatant was transferred to 250 ml of the refolding buffer (2 M urea, 50 mM tris, 160 mM arginine HCI1 5 mM EDTA1 1 mM cystamine HCI, 4 mM cysteine, pH 8.5) at 4 0C with vigorous stirring. The stirring rate was then slowed and the incubation was continued for 2 days at 4 0C. The refolding solution was then filtered through a 0.22 μm cellulose acetate filter and stored at -70 0C.
The stored refold was defrosted and then diluted with 1 L of water and the pH was adjusted to 7.5 using 1 M H3PO4. The pH adjusted material was then filtered through a 0.22 μm cellulose acetate filter and loaded on to a 10 ml Amersham SP-HP HiTrap column at 10 ml/min in S-Buffer A (20 mM NaHaPCM, pH 7.3) at 7 0C. The column was then washed with several column volumes of S-Buffer A1 followed by elution with a linear gradient from 0% to 60% S-Buffer B (20 mM NaH2PO4, 1 M NaCI, pH 7.3) followed by a step to 100% S-Buffer B at 5 ml/min 7 0C. Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS- PAGE, and the fractions containing the desired product were pooled based on these data (15 ml). The pool was then loaded on to a 1 ml Amersham rProtein A HiTrap column in PBS at 2 ml/min 7 0C. Then column was then washed with several column volumes of 20 mM NaH2PO4 pH 6.5, 1 M NaCI and eluted with 100 mM glycine pH 3.0. To the elution peak (1.4 ml), 70 μl 1 M tris HCI pH 8.5 was added, and then the pH adjusted material was filtered though a 0.22 μm cellulose acetate filter.
A spectral scan was then conducted on 20 μl of the combined pool diluted in 700 μl PBS using a Hewlett Packard 8453 spectrophotometer (Figure 29F). The concentration of the filtered material was determined to be 1.44 mg/ml using a calculated molecular mass of 30,469 g/mol and extinction coefficient of 43,890 M 1 cm 1. The purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 29B). The endotoxin level was then determined using a Charles River Laboratories Endosafe-PTS system (0.05 - 5 EU/ml sensitivity) using a 33-fold dilution of the sample in Charles Rivers Endotoxin Specific Buffer BG120 yielding a result of <4 EU/mg protein. The macromolecular state of the product was then determined using size exclusion chromatography on 20 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 29G). The product was then subject to mass spectral analysis by diluting 1 μl of the sample into 10 μl of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile) . The resultant solution (1 μl) was spotted
onto a MALDI sample plate. The sample was allowed to dry before being analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from - 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses (Figure 29H) and these studies confirmed the integrity of the purified peptibody, within experimental error. The product was then stored at -80 0C.
Example 24 Fc-L-PaTx2 bacterial expression
Bacterial expression of Fc-l_-PaTx2. The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-PaTx2. Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCTACTGCCAGAAATGGA //SEQ ID NO: 822;
TGTGGACCTGCGACGAAGAACGTAAATGCTGCGAAGGTCTGGTTTGCCGTCTGTGGTGCAAACGTATCATCAA CATG //SEQ ID NO: 823;
CTTACATGTTGATGATACGTTTGCACCACAGACGGCAAA //SEQ ID NO: 824;
CCAGACCTTCGCAGCATTTACGTTCTTCGTCGCAGGTCCACATCCATTTCTGGCAGTAGGAACCACCACCACC GGA //SEQ ID NO: 825;
The oligos above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCTACTGCCAGAAATGGATGTGGACCTGCGACGAAGAACG
I \. 1- y (- + y 50 AGGCCACCACCACCAAGGATGACGGTCTTTACCTACACCTGGACGCTGCTTCTTGC
G S G G G G S Y C Q K W M W T C D E E R -
TAAATGCTGCGAAGGTCTGGTTTGCCGTCTGTGGTGCAAACGTATCATCAACATG SEQ ID NO : 826 6i + + + + +
ATTTACGACGCTTCCAGACCAAACGGCAGACACCACGTTTGCATAGTAGTTGTACATTC SEQ ID NO : 828 K C C E G L V C R L W C K R I I N M - SEQ ID NO : 827
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 25 Fc-L-wGVIA bacterial expression Bacterial expression of Fc-L-wGVIA. The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos
listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-wGVIA.
Oligos used to form duplex are shown below: TGGTTCCGGTGGTGGTGGTTCCTGCAAATCCCCGGGTT //SEQ ID NO: 829;
CCTCCTGCTCCCCGACCTCCTACAACTGCTGCCGTTCCTGCAACCCGTACACCAAACGTTGCTACGGT SEQ ID NO:830; 0 CTTAACCGTAGCAACGTTTGGTGTACGGGTTGCAGGAA //SEQ ID NO: 831;
CGGCAGCAGTTGTAGGAGGTCGGGGAGCAGGAGGAACCCGGGGATTTGCAGGAACCACCACCACCGGA //SEQ ID NO: 832; 5 The oligos above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCTGCAAATCCCCGGGTTCCTCCTGCTCCCCGACCTCCTA I + + + + + + 60
AGGCCACCACCACCAAGGACGTTTAGGGGCCCAAGGAGGACGAGGGGCTGGAGGAT 0
G S G G G G S C K S P G S S C S P T S Y -
CAACTGCTGCCGTTCCTGCAACCCGTACACCAAACGTTGCTACGGT SEQ ID NO: 833
61 + + + + 5 GTTGACGACGGCAAGGACGTTGGGCATGTGGTTTGCAACGATGCCAATTC SEQ I D NO : 835
N C C R S C N P Y T K R C Y G SEQ I D NO : 834
Bacterial expression of the peptibody was as described in Example 3 and paste was O stored frozen.
Example 26 Fc-L-ωMVIIA bacterial expression
Bacterial expression of Fc-L-ωMVIIA. The methods to clone and express the peptibody in 5 bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-ωMVIIA.
Oligos used to form duplex are shown below: 0 TGGTTCCGGTGGTGGTGGTTCCTGCAAAGGTAAA //SEQ ID NO: 836;
GGTGCTAAATGCTCCCGTCTGATGTACGACTGCTGCACCGGTTCCTGCCGTTCCGGTAAATGCGGT //SEQ ID NO: 837; 5 CTTAACCGCATTTACCGGAACGGCAGGAACCGGT //SEQ ID NO: 838;
GCAGCAGTCGTACATCAGACGGGAGCATTTAGCACCTTTACCTTTGCAGGAACCACCACCACCGGA //SEQ ID NO: 839; O The oligos above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCTGCAAAGGTAAAGGTGCTAAATGCTCCCGTCTGATGTA
1 H + H H h h 60
AGGCCACCACCACCAAGGACGTTTCCATTTCCACGATTTACGAGGGCAGACTACAT
G S G G G G S C K G K G A K C S R L M Y -
CGACTGCTGCACCGGTTCCTGCCGTTCCGGTAAATGCGGT SEQ ID NO: 840 61 + + + +
GCTGACGACGTGGCCAAGGACGGCAAGGCCATTTACGCCAATTC SEQ ID NO: 842
D C C T G S C R S G K C G - SEQ ID NO:841 Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 27 Fc-L-RuI bacterial expression Bacterial expression of Fc-L-Ptu1 , The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-RuL
Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCGCTGAAAAAGACTGCATC //SEQ ID NO: 843;
GCTCCGGGTGCTCCGTGCTTCGGTACCGACAAACCGTGCTGCAACCCGCGTGCTTGGTGCTCCTCCTACGCTA ACAAATGCCTG //SEQ ID NO: 844;
CTTACAGGCATTTGTTAGCGTAGGAGGAGCACCAAGCACG //SEQ ID NO: 845;
CGGGTTGCAGCACGGTTTGTCGGTACCGAAGCACGGAGCACCCGGAGCGATGCAGTCTTTTTCAGCGGAACCA CCACCACCGGA //SEQ ID NO: 846;
The oligos above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCGCTGAAAAAGACTGCATCGCTCCGGGTGCTCCGTGCTT 1 + + + + + + 60
AGGCCACCACCACCAAGGCGACTTTTTCTGACGTAGCGAGGCCCACGAGGCACGAA G S G G G G S A E K D C I A P G A P C F - CGGTACCGACAAACCGTGCTGCAACCCGCGTGCTTGGTGCTCCTCCTACGCTAACAAATG
61 + + + + + + 120
GCCATGGCTGTTTGGCACGACGTTGGGCGCACGAACCACGAGGAGGATGCGATTGTTTAC
G T D K P C C N P R A W C S S Y A N K C -
CCTG SEQ ID NO: 847
121
GGACATTC SEQ ID NO: 849 L - SEQ ID NO:848
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 28 Fc-L-ProTx1 bacterial expression
Bacterial expression of Fc-L-ProTx1. The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-ProTxL
Oligos used to form duplex are shown below:
TGGTTCCGGTGGTGGTGGTTCCGAATGCCGTTACTGGCTGG //SEQ ID NO: 850;
GTGGTTGCTCCGCTGGTCAGACCTGCTGCAAACACCTGGTTTGCTCCCGTCGTCACGGTTGGTGCGTTTGGGA CGGTACCTTCTCC //SEQ ID NO: 851;
CTTAGGAGAAGGTACCGTCCCAAACGCACCAACCGTGACGA //SEQ ID NO: 852;
CGGGAGCAAACCAGGTGTTTGCAGCAGGTCTGACCAGCGGAGCAACCACCCAGCCAGTAACGGCATTCGGAAC CACCACCACCGGA //SEQ ID NO: 853;
The oligos above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCGAATGCCGTTACTGGCTGGGTGGTTGCTCCGCTGGTCA I + + + + + + go
AGGCCACCACCACCAAGGCTTACGGCAATGACCGACCCACCAACGAGGCGACCAGT G S G G G G Ξ E C R Y W L G G C Ξ A G Q -
GACCTGCTGCAAACACCTGGTTTGCTCCCGTCGTCACGGTTGGTGCGTTTGGGACGGTAC gl + + + + + + 120
<- CTGGACGACGTTTGTGGACCAAACGAGGGCAGCAGTGCCAACCACGCAAACCCTGCCATG
T C C K H L V C S R R H G W C V W D G T -
CTTCTCC SEQ ID NO: 854
121 GAAGAGGATTC SEQ ID NO: 856
F S - SEQ ID NO: 855
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 29 Fc-L-BeKMI bacterial expression
Bacterial expression of Fc-L-BeKML The methods to clone and express the peptibody in bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-BeKMI.
Oligos used to form duplex are shown below: TGGTTCCGGTGGTGGTGGTTCCCGTCCGACCGACATCAAATG //SEQ ID NO: 857;
CTCCGAATCCTACCAGTGCTTCCCGGTTTGCAAATCCCGTTTCGGTAAAACCAACGGTCGTTGCGTTAACGGT TTCTGCGACTGCTTC //SEQ ID NO: 858;
CTTAGAAGCAGTCGCAGAAACCGTTAACGCAACGACCGTTGG //SEQ ID NO: 859;
TTTTACCGAAACGGGATTTGCAAACCGGGAAGCACTGGTAGGATTCGGAGCATTTGATGTCGGTCGGACGGGA ACCACCACCACCGGA //SEQ ID NO: 860;
The oligos above were used to form the duplex below: 0
TGGTTCCGGTGGTGGTGGTTCCCGTCCGACCGACATCAAATGCTCCGAATCCTACCAGTG I + + + + + + go
AGGCCACCACCACCAAGGGCAGGCTGGCTGTAGTTTACGAGGCTTAGGATGGTCAC 5 G S G G G G S R P T D I K C S E S Y Q C - CTTCCCGGTTTGCAAATCCCGTTTCGGTAAAACCAACGGTCGTTGCGTTAACGGTTTCTG
61 + + + + + + 120
GAAGGGCCAAACGTTTAGGGCAAAGCCATTTTGGTTGCCAGCAACGCAATTGCCAAAGAC 0
F P V C K S R F G K T N G R C V N G F C -
CGACTGCTTC SEQ ID NO: 861
121 + 5 GCTGACGAAGATTC SEQ ID NO:863
D C F - SEQ ID NO:862
Bacterial expression of the peptibody was as described in Example 3 and paste was O stored frozen.
Example 30 Fc-L-CTX bacterial expression
Bacterial expression of Fc-L-CTX. The methods to clone and express the peptibody in 5 bacteria are described in Example 3. The vector used was pAMG21ampR-Fc-Pep and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Fc-L-CTX.
Oligos used to form duplex are shown below: 0 TGGTTCCGGTGGTGGTGGTTCCATGTGCATGCCGTGCTTCAC //SEQ ID NO:864;
CACCGACCACCAGATGGCTCGTAAATGCGACGACTGCTGCGGTGGTAAAGGTCGTGGTAAATGCTACGGTCCG CAGTGCCTGTGCCGT //SEQ ID NO: 865; 5 CTTAACGGCACAGGCACTGCGGACCGTAGCATTTACCACGAC //SEQ ID NO: 866;
CTTTACCACCGCAGCAGTCGTCGCATTTACGAGCCATCTGGTGGTCGGTGGTGAAGCACGGCATGCACATGGA ACCACCACCACCGGA //SEQ ID NO: 867; O The oligos above were used to form the duplex below:
TGGTTCCGGTGGTGGTGGTTCCATGTGCATGCCGTGCTTCACCACCGACCACCAGATGGC
1 + + + + + + 60
AGGCCACCACCACCAAGGTACACGTACGGCACGAAGTGGTGGCTGGTGGTCTACCG 5
G S G G G G S M C M P C F T T D H Q M A -
TCGTAAATGCGACGACTGCTGCGGTGGTAAAGGTCGTGGTAAATGCTACGGTCCGCAGTG
61 + + + + + + 120
AGCATTTACGCTGCTGACGACGCCACCATTTCCAGCACCATTTACGATGCCAGGCGTCAC R K C D D C C G G K G R G K C Y G P Q C - CCTGTGCCGT SEQ ID NO: 868
121 +
GGACACGGCACCAC SEQ ID NO : 870
L C R - SEQ ID NO : 869
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 31 N-Terminallv PEGylated-Des-Arg1-ShK Peptide Synthesis of reduced Des-Arq1-ShK. Des-Arg1-ShK, having the sequence
SCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC
(Peptide 1, SEQ ID NO: 92) was synthesized in a stepwise manner on a Symphony™ multi-peptide synthesizer by solid-phase peptide synthesis (SPPS) using 2-(1H-benzotriazole-1-yl)-1)1,3,3-tetramethyluronium hexafluorophosphate (HBTU)/ N-methyl morpholine (NMM)/N,N-dimethyl-formamide (DMF) coupling chemistry at 0.1 mmol equivalent resin scale on Tentagel™-S PHB Fmoc-Cys(Trt)-resin. N-alpha-(9-fluorenylmethyloxycarbonyl)- and side-chain protected amino acids were purchased from Midwest Biotech Incorporated. Fmoc-Cys(Trt)-Tentagel™ resin was purchased from Fluka. The following side-chain protection strategy was employed: ASp(O1Bu), Arg(Pbf), Cys(Trt), Gln(Trt), His(Trt), Lys(NE-Boc), Ser(O'Bu), Thr(O'Bu) and Tyr(O'Bu). Two Oxazolidine dipeptides, Fmoc-Gly-
Thr(ΨMe'MβPro)-OH and Fmoc-Leu-Ser(fMe MePro)-OH, were used in the chain assembly and were obtained from NovaBiochem and used in the synthesis of the sequence. The protected amino acid derivatives (20mmol) were dissolved in 100 ml 20% dimethyl sulfoxide (DMSO) in DMF (v/v). Protected amino acids were activated with 20 mM HBTU, 400 mM NMM in 20%DMSO in DMF, and coupling were carried out using two treatments with O.δmmol protected amino acid, O.δmmol
HBTU, 1 mmol NMM in 20% DMF/DMSO for 25 minutes and then 40 minutes. Fmoc deprotection reactions were carried out with two treatments using a 20% piperidine in DMF (v/v) solution for 10 minutes and then 15 minutes. Following synthesis, the resin was then drained, and washed with DCM, DMF, DCM, and then dried in vacuo. The peptide-resin was deprotected and released from the resin by treatment with a TFA/EDT/TIS/H2O (92.5:2.5:2.5:2.5 (v/v)) solution at room temperature for 1 hour. The volatiles were then removed with a stream of nitrogen gas, the crude peptide precipitated twice with cold diethyl ether and collected by centrifugation. The crude peptide was then analyzed on a Waters 2795 analytical RP-HPLC system using a linear gradient (0-60% buffer B in 12 minutes, A:0.1% TFA in water, B: 0.1% TFA in acetonitrile) on a Jupiter 4μm
Proteo™ 9OA column. A PE-Sciex™ API Electro-spray mass spectrometer was used to confirm correct peptide product mass. Crude peptide was obtained in 143 mg yield at approximately 70% pure based as estimated by analytical RP-HPLC analysis. Reduced Des-Arg1-ShK (Peptide 1) Retention time (Rt) = 5.31 minutes, calculated molecular weight = 3904.6917 Da (average); Experimental observed molecular weight 3907.0 Da.
Folding of Des-Arq1-ShK (Disulphide bond formation). Following TFA cleavage and peptide precipitation, reduced Des-Arg1-ShK was then air-oxidized to give the folded peptide. The crude cleaved peptide was extracted using 20% AcOH in water (v/v) and then diluted with water to a concentration of approximately 0.15 mg reduced Des-Arg1-ShK per mL, the pH adjusted to about 8.0 using NH4OH (28-30%), and gently stirred at room temperature for 36 hours. Folding process was monitored by LC-MS analysis. Following this, folded Des-Arg1-ShK peptide was purified using reversed phase HPLC using a 1" Luna 5 μm C18 100 A Proteo™ column with a linear gradient 0-40% buffer B in 120 min (A=O.1% TFA in water, B=O.1% TFA in acetonitrile). Folded Des-Arg1-ShK crude peptide eluted earlier (when compared to the elution time in its reduced form) at approximately 25% buffer B. Folded Des-Arg1-ShK (Peptide 2) was obtained in 23.2 mg yield in >97% purity as estimated by analytical RP-HPLC analysis (Figure 20A). Calculated molecular weight = 3895.7693 Da (monoisotopic), experimental observed molecular weight = 3896.5 Da(analyzed on a Waters LCT Premier Micromass MS Technologies). (Figure 20B). Des-Arg1- ShK disulfide connectivity was C1-C6, C2-C4, C3-C5.
SCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC
(Peptide 2, SEQ ID NO: 58)
N-terminal PEGylation of Folded Des-Arq1-ShK. Folded Des-Arg1-ShK, (Peptide 2) was dissolved in water at 1 mg/ml concentration. A 2 M MeO-PEG-Aldehyde, CH3O-[CH2CH2OJn- CH2CH2CHO (average molecular weight 20 kDa), solution in 50 mM NaOAc, pH 4.5, and a separate 1 M solution of NaCNBH3 were freshly prepared. The peptide solution was then added to the MeO-PEG-Aldehyde containing solution and was followed by the addition of the NaCNBH3 solution. The reaction stoichiometry was peptide:PEG:NaCNBH3 (1 :2:0.02), respectively. The reaction was left for 48 hours, and was analyzed on an Agilent 1100 RP-HPLC system using Zorbax™ 300SB-C8 5 μm column at 40 °C with a linear gradient (6-60% B in 16 minutes, A: 0.1% TFA in water, B: 0.1% TFA/90% ACN in water). Mono-pegylated folded Des-Arg1-ShK constituted
approximately 58% of the crude product by analytical RP-HPLC. Mono Pegylated Des-Arg1-ShK was then isolated using a HiTrap™ 5 ml SP HP cation exchange column on AKTA FPLC system at 4 'C at 1 mL/min using a gradient of 0-50% B in 25 column volumes (Buffers: A = 20 mM sodium acetate pH 4.0, B = 1 M NaCI1 20 mM sodium acetate, pH 4.0). The fractions were analyzed using a 4-20 tris-Gly SDS-PAGE gel and RP-HPLC (as described for the crude). SDS-PAGE gels were run for 1.5 hours at 125 V, 35 mA, 5 W. Pooled product was then dialyzed at 4 °C in 3 changes of 1 L of A4S buffer(10 mM NaOAc, 5% sorbitol, pH 4.0). The dialyzed product was then concentrated in 10 K microcentrifuge filter to 2 mL volume and sterile-filtered using 0.2 μM syringe filter to give the final product. N-Terminally PEGylated-Des-Arg1-ShK (Peptide 3) was isolated in 1.7 mg yield with 85% purity as estimated by analytical RP-HPLC analysis (Figure 23).
The N-Terminally PEGylated-Des-Arg1-ShK, also referred to as "PEG-ShK[2-35]", was active in blocking human Kv1.3 (Figure 38A and Figure 38B) as determined by patch clamp electrophysiology (Example 36).
Example 32 N-Terminallv PEGylated ShK
The experimental procedures of this working example correspond to the results shown in Figure 17.
Peptide Synthesis of reduced ShK. ShK, having the amino acid sequence
RSCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC (Peptide 4, SEQ ID NO:5) was synthesized in a stepwise manner on a Symphony™ multi-peptide synthesizer by solid-phase peptide synthesis (SPPS) using 2-(1 H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU)/ N-methyl morpholine (NMM)/N,N-dimethyl-formamide (DMF) coupling chemistry at 0.1 mmol equivalent resin scale on Tentagel™-S PHB Fmoc-Cys(Trt)-resin. N-alpha-9-fluorenylmethyloxycarbonyl) and side-chain protected amino acids were purchased from
Midwest Biotech Incorporated. Fmoc-Cys(Trt)-Tentagel™ resin was purchased from Fluka. The following side-chain protection strategy was employed: Asp(O'Bu), Arg(Pbf), Cys(Trt), Gln(Trt), His(Trt), Lys(Nε-Boc), Ser(O'Bu), Thr(O'Bu) and Tyr(O'Bu). Two Oxazolidine dipeptides, Fmoc-Gly- Thr(fMe MePro)-OH and Fmoc-Leu-Ser(fMe MePro)-OH, were used in the chain assembly and were obtained from NovaBiochem and used in the synthesis of the sequence. The protected amino acid derivatives (20 mmol) were dissolved in 100 ml 20% dimethyl sulfoxide (DMSO) in DMF {v/v). Protected amino acids were activated with 200 mM HBTU, 400 mM NMM in 20%DMSO in DMF, and coupling were carried out using two treatments with 0.5 mmol protected amino acid, 0.5 mmol
HBTU1 1 mmol NMM in 20%DMF/DMSO for 25 minutes and then 40 minutes. Fmoc deprotections were carried out with two treatments using a 20% piperidine in DMF (v/v) solution for 10 minutes and then 15 minutes. Following synthesis, the resin was then drained, and washed with DCM, DMF, DCM, and then dried in vacuo. The peptide-resin was deprotected and released from the resin by treatment with a TFA/EDT/TIS/H2O (92.5:2.5:2.5:2.5 (v/v)) solution at room temperature for 1 hour. The volatiles were then removed with a stream of nitrogen gas, the crude peptide precipitated twice with cold diethyl ether and collected by centrifugation. The crude peptide was then analyzed on a Waters 2795 analytical RP-HPLC system using a linear gradient (0-60% buffer B in 12 minutes, A:0.1% TFA in water, B: 0.1% TFA in acetonitrile) on a Jupiter 4μm Proteo™ 90 A column. A PE-Sciex API Electro-spray mass spectrometer was used to confirm correct peptide product mass. Crude peptide was approximately was obtained 170 mg yield at about 45% purity as estimated by analytical RP-HPLC analysis. Reduced ShK (Peptide 4) Retention time (Rt) = 5.054 minutes, calculated molecular weight = 4060.8793 Da (average); experimental observed molecular weight = 4063.0 Da. Folding of ShK (Disulphide bond formation). Following TFA cleavage and peptide precipitation, reduced ShK was then air oxidized to give the folded peptide. The crude cleaved peptide was extracted using 20% AcOH in water (v/v) and then diluted with water to a concentration of approximately 0.15 mg reduced ShK per mL, the pH adjusted to about 8.0 using NH4OH (28-30%), and gently stirred at room temperature for 36 hours. Folding process was monitored by LC-MS analysis. Following this, folded ShK peptide was purified by reversed phase HPLC using a 1" Luna 5 μm C18 100 A Proteo™ column with a linear gradient 0-40% buffer B in 120 min (A=0.1% TFA in water, B=O.1% TFA in acetonitrile). Folded ShK crude peptide eluted earlier (when compared to the elution time in its reduced form) at approximately 25% buffer B. Folded ShK (Peptide 5) was obtained in 25.5 mg yield in >97% purity as estimated by analytical RP-HPLC analysis. See Figure 60. Calculated molecular weight = 4051.8764 Da (monoisotopic); experimental observed molecular weight = 4052.5 Da (analyzed on Waters LCT Premier
Micromass MS Technologies). ShK disulfide connectivity was C1-C6, C2-C4, and C3-C5.
RSCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC (Peptide 5, SEQ ID NO:10)
N-terminal PEGylation of Folded ShK. Folded ShK, having the amino acid sequence
RSCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC
(SEQ ID N0:5) can be dissolved in water at 1 mg/ml concentration. A 2 M MeO-PEG-Aldehyde, CH3O- CH2CH2OIn-CH2CH2CHO (average molecular weight 20 kDa), solution in 50 mM NaOAc, pH 4.5 and a separate 1 M solution of NaCNBH3 can be freshly prepared. The peptide solution can be then added to the MeO-PEG-Aldehyde containing solution and can be followed by the addition of the NaCNBH3 solution. The reaction stoichiometry can be peptide:PEG:NaCNBH3 (1:2:0.02), respectively. The reaction can be left for 48 hours, and can be analyzed on an Agilent™ 1100 RP- HPLC system using Zorbax™ 300SB-C85 μm column at 40 °C with a linear gradient (6-60% B in 16 minutes, A: 0.1% TFA in water, B: 0.1% TFA/90% ACN in water). Mono-pegylated Shk (Peptide 6) can be then isolated using a HiTrap™ 5 mL SP HP cation exchange column on AKTA FPLC system at 4 0C at 1 mL/min using a gradient of 0-50 % B in 25 column volumes (Buffers: A = 20 mM sodium acetate pH 4.0, B = 1 M NaCI, 20 mM sodium acetate, pH 4.0). The fractions can be analyzed using a 4-20 tris-Gly SDS-PAGE gel and RP-HPLC. SDS-PAGE gels can be run for 1.5 hours at 125 V, 35 mA, 5 W. Pooled product can be then dialyzed at 4 'C in 3 changes of 1 L of A4S buffer (10 mM sodium acetate, 5% sorbitol, pH 4.0). The dialyzed product can be then concentrated in 10 K microcentrifuge filter to 2 mL volume and sterile-filtered using 0.2 μM syringe filter to give the final product.
Example 33
N-Terminallv PEGylated ShK by oxime formation
Peptide Synthesis of reduced ShK. ShK, having the sequence
RSCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC
(SEQ ID NO: 5) can be synthesized in a stepwise manner on a Symphony™ multi-peptide synthesizer by solid- phase peptide synthesis (SPPS) using 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU)/ N-methyl morpholine (NMM)/N,N-dimethyl-formamide (DMF) coupling chemistry at 0.1 mmol equivalent resin scale on Tentagel™-S PHB Fmoc-Cys(Trt)-resin. N-alpha-(9-fluorenylmethyloxycarbonyl)- and side-chain protected amino acids can be purchased from Midwest Biotech Incorporated. Fmoc-Cys(Trt)-Tentagel™ resin can be purchased from Fluka.
The following side-chain protection strategy can be employed: Asp(O'Bu), Arg(Pbf), Cys(Trt), GIn(TiI), His(Trt), Lys(Nε-Boc), Ser(O'Bu), Thr(O'Bu) and Tyr(O'Bu). Two Oxazolidine dipeptides, Fmoc-Gly-Thr(ΨMe.MePro)-OH and Fmoc-Leu-Ser(ψMe MePro)-OH, can be used in the chain
assembly and can be obtained from NovaBiochem and used in the synthesis of the sequence. The protected amino acid derivatives (20 mmol) can be dissolved in 100 ml 20% dimethyl sulfoxide (DMSO) in DMF {v/v). Protected amino acids can be activated with 200 mM HBTU, 400 mM NMM in 20% DMSO in DMF1 and coupling can be carried out using two treatments with 0.5 mmol protected amino acid, 0.5 mmol HBTU, 1 mmol NMM in 20% DMF/DMSO for 25 minutes and then 40 minutes. Fmoc deprotection reactions can be carried out with two treatments using a 20% piperidine in DMF (v/v) solution for 10 minutes and then 15 minutes. Following the chain-assembly of the Shk peptide, Boc-amionooxyacetic acid (1.2 equiv) can be coupled at the N-terminus using 0.5 M HBTU in DMF with 4 equiv collidine for 5 minutes. Following synthesis, the resin can be then drained, and washed with DCM, DMF, DCM, and then dried in vacuo. The peptide-resin can be deprotected and released from the resin by treatment with a TFA/amionooxyacetic acid/TIS/EDT/H2O (90:2.5:2.5:2.5:2.5) solution at room temperature for 1 hour. The volatiles can be then removed with a stream of nitrogen gas, the crude peptide precipitated twice with cold diethyl ether and collected by centrifugation. The aminooxy-Shk peptide (Peptide 7) can be then analyzed on a Waters 2795 analytical RP-HPLC system using a linear gradient (0-60% buffer B in 12 minutes, A: 0.1% TFA in water also containing 0.1% aminooxyacetic acid, B: 0.1% TFA in acetonitrile) on a Jupiter 4 μm Proteo™ 90 A column.
Reversed-Phase HPLC Purification. Preparative Reversed-phase high-performance liquid chromatography can be performed on C18, 5 μm, 2.2 cm * 25 cm) column. Chromatographic separations can be achieved using linear gradients of buffer B in A (A = 0.1 % aqueous TFA; B = 90% aq. ACN containing 0.09% TFA and 0.1% aminooxyacetic acid), typically 5-95% over 90 minutes at 15 mL/min. Preparative HPLC fractions can be characterized by ESMS and photodiode array (PDA) HPLC, combined and lyophilized.
N-Terminal PEGylation of Shk by Oxime Formation. Lyophilized aminooxy-Shk (Peptide 7) can be dissolved in 50% HPLC buffer A/B (5 mg/mL) and added to a two-fold molar excess of MeO-PEG-Aldehyde, CH3O-[CH2CH2O]n-CH2CH2CHO (average molecular weight 20 kDa). The reaction can be left for 24 hours, and can be analyzed on an Agilent™ 1100 RP-HPLC system using Zorbax™ 300SB-C8 5 μm column at 40 0C with a linear gradient (6-60 % B in 16 minutes, A: 0.1% TFA in water, B: 0.1% TFA/90% ACN in water). Mono-pegylated reduced Shk constituted approximately 58% of the crude product by analytical RP-HPLC. Mono PEGylated (oximated) Shk
(Peptide 8) can be then isolated using a HiTrap™ 5 mL SP HP cation exchange column on AKTA FPLC system at 4 °C at 1 mL/min using a gradient of 0-50 % B in 25 column volumes (Buffers: A = 20 mM sodium acetate pH 4.0, B = 1 M NaCI, 20 mM sodium acetate, pH 4.0). The fractions can
be analyzed using a 4-20 tris-Gly SDS-PAGE gel and RP-HPLC. SDS-PAGE gels can be run for 1.5 hours at 125 V, 35 mA, 5 W. Pooled product can be then dialyzed at 4 °C in 3 changes of 1 L of A4S buffer (10 mM NaOAc, 5% sorbitol, pH 4.0). The dialyzed product can be then concentrated in 10 K microcentrifuge filter to 2 mL volume and sterile-filtered using 0.2 μM syringe filter to give the final product.
Folding of ShK (Disulphide bond formation). The mono-PEGylated (oximated) Shk can be dissolved in 20% AcOH in water (v/v) and can be then diluted with water to a concentration of approximately 0.15 mg peptide mL, the pH adjusted to about 8.0 using NH4OH (28-30%), and gently stirred at room temperature for 36 hours. Folding process can be monitored by LC-MS analysis. Following this, folded mono-PEGylated (oximated) Shk (Peptide 9) can be purified using by reversed phase HPLC using a 1" Luna 5 μm C18 100 A Proteo™ column with a linear gradient 0-40% buffer B in 120 min (A=0.1% TFA in water, B=0.1% TFA in acetonitrile). Mono-PEGylated (oximated) ShK disulfide connectivity can be C1-C6, C2-C4, and C3-C5.
MeO- PEG-CH2CH2CH2NHOCH2CO-RSCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC
(Peptide 9, SEQ ID NO:10)
Example 34 N-Terminallv PEGylated ShK (amidation)
The experimental procedures of this working example correspond to the results shown in Figure 18.
N-Terminal PEGylation of Shk by Amide Formation. A 10 mg/mL solution of folded Shk (Peptide 5), in 100 mM Bicine pH 8.0, can be added to solid succinimidyl ester of 20 kDa PEG propionic acid (mPEG-SPA; CH3O-[CH2CH2OJn-CH2CH2CO-NHS) at room temperature using a 1.5 molar excess of the mPEG-SPA to Shk. After one hour with gentle stirring, the mixture can be diluted to 2 mg/mL with water, and the pH can be adjusted to 4.0 with dilute HCI. The extent of mono-pegylated Shk (Peptide 10), some di-PEGylated Shk or tri-PEGylated Shk, unmodified Shk and succinimidyl ester hydrolysis can be determined by SEC HPLC using a Superdex™ 75 HR 10/30 column (Amersham) eluted with 0.05 M NaH2PO4, 0.05 M Na2HPO4, 0.15 M NaCI, 0.01 M NaN3, pH 6.8, at 1 mL/min. The fractions can be analyzed using a 4-20 tris-Gly SDS-PAGE gel and RP-HPLC. SDS-PAGE gels can be run for 1.5 hours at 125 V, 35 mA, 5 W. Pooled product
can be then dialyzed at 4 °C in 3 changes of 1 L of A4S buffer (10 mM NaOAc1 5% sorbitol, pH 4.0). The dialyzed N-terminally PEGylated (amidated) ShK (Peptide 10) can be then concentrated in 10 K microcentrifuge filter to 2 mL volume and sterile-filtered using 0.2 μM syringe filter to give the final product.
I
PEG 2OkDa
MeO-PBG-CH2CH2CO-Mf-RSCIDTIPKSRCTAFQCKHSMKYRLSFCRKTCGTC
(Peptide 10, SEQ ID NO:10)
Example 35 Fc-L-SmIIIA
Fc-SmIIIA expression vector. A 104 bp BamHI-Notl fragment containing partial linker sequence and SmIIIA peptide encoded with human high frequency codons was assembled by PCR with overlapping primers 3654-50 and 3654-51 and cloned into to the 7.1 kb Notl-BamHI back bone to generate pcDNA3.1(+)CMVi-hFc-SmlllA as described in Example 1.
BamHI 5' GGATCCGGAGGAGGAGGAAGCTGCTGCAACGGCCGCCGCGGCTGCAGCAGCCGCTGG
C C N G R R G C S S R W
TGCCGCGACCACAGCCGCTGCTGCTGAGCGGCCGCS' //SEQ ID NO: 872 C R D H S R C C Notl SEQ ID NO: 873
Forward 5' -3' :
GGAGGAGGATCCGGAGGAGGAGGAAGCTGCTGCAACGGCCGCCGCGGCTGCAGCAGC CGC //SEQ ID NO:874
Reverse 5' -3' :
ATTATTGCGGCCGCTCAGCAGCAGCGGCTGTGGTCGCGGCACCAGCGGCTGCTGCAG CCGC SEQ ID NO:875 The sequences of the BamHI to Notl fragments in the final constructs were verified by sequencing.
Transient expression of Fc-L-SmIIIa. 7.5 ug of the toxin peptide Fc fusion construct pcDNA3.1(+)CMVi-hFc-SmlllA were transfected into 293-T cells in 10 cm tissue culture plate with FuGENE 6 as transfection reagent. Culture medium was replaced with serum-free medium at 24 hours post-transfection and the conditioned medium was harvested at day 5 post-transfection. Transient expression of Fc-SmIIIA from 293-T cells was analyzed by Western blot probed with anti- hFc antibody (Figure 25A and Figure 25B). Single band of expressed protein with estimated MW was shown in both reduced and non-reduced samples. Transient expression level of Fc-SmIIIA was further determined to be 73.4 μg/ml according to ELISA.
Example 36
Electrophvsiology experiments
Cell Culture. Stable cell line expressing human KV1.3 channel was licensed from Biofocus. Cells were kept at 37 0C in 5% CO2 environment. Culture medium contains DMEM with GlutaMax™ (Invitrogen), 1X non-essential amino acid, 10% fetal bovine serum and 500 μg/mL geneticin. Cells were plated and grown at low confluence on 35 mm culture dishes for at least 24 hours prior to electrophysiology experiments.
Electrophysioloqy Recording by Patch Clamping. Whole-cell currents were recorded from single cells by using tight seal configuration of the patch-clamp technique. A 35 mm culture dish was transferred to the recording stage after rinsing and replacing the culture medium with recording buffer containing 135 mM NaCI, 5 mM KCI, 1.8 mM CaCb, 1O mM HEPES, and 5 mM
Glucose. pH was adjusted to 7.4 with NaOH and the osmolarity was set at 300 mOsm. Cells were perfused continuously with the recording buffer via one of the glass capillaries arranged in parallel and attached to a motorized rod, which places the glass capillary directly on top of the cell being recorded. Recording pipette solution contained 90 mM K-gluconate, 20 mM KF, 10 mM NaCI1 1 mM MgCI2-6H20, 10 mM EGTA, 5 mM K2-ATP, and 10 mM HEPES. The pH for the internal solution was adjusted to 7.4 with KOH and the osmolarity was set at 280 mOsm. Experiments were performed at room temperature (20-22 0C) and recorded using Multiclamp™ 700A amplifier (Molecular Devices Inc.). Pipette resistances were typically 2-3 MΩ.
Protein toxin potency determination on Kv1.3 current: HEK293 cells stably expressing human Kv1.3 channel were voltage clamped at -80 mV holding potential. Outward KV1.3 currents were activated by giving 200 msec long depolarizing steps to +30 mV from the holding potential of -80 mV and filtered at 3 kHz. Each depolarizing step was separated from the subsequent one with a 10 s interval. Analogue signals were digitized by Digidata™ 1322A digitizer (Molecular Devices)
and subsequently stored on computer disk for offline analyses using Clampfit™ 9 (Molecular Devices Inc.). In all studies, stable baseline Kv1.3 current amplitudes were established for 4 minutes before starting the perfusion of the protein toxin at incremental concentrations. A steady state block was always achieved before starting the perfusion of the subsequent concentration of the protein toxin.
Data analysis. Percent of control (POC) is calculated based on the following equation: (Kv1.3 current after protein toxin addition/Kv1.3 current in control)* 100. At least 5 concentrations of the protein toxin (e.g. 0.003, 0.01, 0.03, 0.1 , 0.3, 100 nM) were used to calculate the IC5O value. ICso values and curve fits were estimated using the four parameter logistic fit of XLfit software (Microsoft Corp.). IC50 values are presented as mean value ± s.e.m. (standard error of the mean). Drug preparations. Protein toxins (typically 10 -100 μM) were dissolved in distilled water and kept frozen at -80 0C. Serial dilutions of the stock protein toxins were mixed into the recording buffer containing 0.1% bovine serum albumin (BSA) and subsequently transferred to glass perfusion reservoirs. Electronic pinch valves controlled the flow of the protein toxin from the reservoirs onto the cell being recorded.
Example 37 lmmunobiology and Channel Binding
Inhibition of T cell cytokine production following PMA and anti-CD3 antibody stimulation of PBMCs. PBMCs were previously isolated from normal human donor Leukophoresis packs, purified by density gradient centrifugation (Ficoll Hypaque), cryopreserved in CPZ Cryopreservation Medium Complete (INCELL, MCPZF-100 plus 10% DMSO final). PBMCs were thawed (95% viability), washed, and seeded at 2x105 cells per well in culture medium (RPMI medium 1640; GIBCO) supplemented with 10% fetal calf serum, 100U/ml penicillin, 100mg/ml streptomycin 2mM L-glutamine, 10OuM non-essential amino acids, and 2OuM 2-ME) in 96-well flat- bottom tissue culture plates. Cells were pre-incubated with serially diluted (100nM-0.001nM final) ShK[I -35], Fc-LI 0-ShK[1 -35] or fc control for 90 min before stimulating for 48hr with PMA/anti-CD3 (1ng/ml and 50ng/ml, respectively) in a final assay volume of 200 ul. Analysis of the assay samples was performed using the Meso Scale Discovery (MSD) SECTOR™ Imager 6000 (Meso Scale Discovery, Gaithersbury, MD) to measure the IL-2 and IFNg protein levels by utilizing electrochemiluminescence (ECL). The conditioned medium (5OuI) was added to the MSD Multi- spot 96-well plates (each well containing three capture antibodies; IL-2, TNF, IFNγ). The plates were sealed, wrapped in tin foil, and incubated at room temperature on a plate shaker for 2 hr. The
wells were washed 1 X with 20OuI PBST (BIOTEK, Elx405 Auto Plate Washer). For each well, 20 ul of Ruthenium-labeled detection antibodies (1μg/ml final in Antibody Dilution Buffer; IL-1, TNF1 IFNγ) and 130 ul of 2X MSD Read Buffer added, final volume 15OuI. The plates were sealed, wrapped in tin foil, and incubated at room temperature on a plate shaker for 1 hr. The plates were then read on the SECTOR™ Imager 6000. Figure 35A & 35B shows the CHO-derived Fc-UO- ShK[1-35] peptibody potently inhibits IL-2 and IFNg production from T cells in a dose-dependent manner. Compared to native ShK[I -35] peptide, the peptibody produces a greater extent of inhibition (POC = Percent Of Control of response in the absence of inhibitor).
Inhibition of T cell cytokine production following anti-CD3 and anti-CD28 antibody stimulation of PBMCs. PBMCs were previously isolated from normal human donor Leukopheresis packs, purified by density gradient centrifugation (Ficoll Hypaque), and cryopreserved using INCELL Freezing Medium. PBMCs were thawed (95% viability), washed, and seeded (in RPMI complete medium containing serum replacement, PSG) at 2x105 cells per well into 96-well flat bottom plates. Cells were pre-incubated with serially diluted (100nM-0.003nM final) ShK[I -35], Fc- L10-ShK[1-35], or Fc control for 1 hour before the addition of aCD3 and aCD28 (2.5 ng/mL and 100 ng/mL respectively) in a final assay volume of 200 mL. Supernatants were collected after 48 hours, and analyzed using the Meso Scale Discovery (MSD) SECTOR™ Imager 6000 (Meso Scale Discovery, Gaithersbury, MD) to measure the IL-2 and IFNg protein levels by utilizing electrochemiluminescence (ECL). 20 mL of supernatant was added to the MSD multi-spot 96-well plates (each well containing IL-2, TNFa, and IFNg capture antibodies). The plates were sealed and incubated at room temperature on a plate shaker for 1 hour. Then 20 mL of Ruthenium- labeled detection antibodies (1μg/ml final of IL-2, TNFα, and IFNγ in Antibody Dilution Buffer) and 110 mL of 2X MSD Read Buffer were added. The plates were sealed, covered with tin foil, and incubated at room temperature on a plate shaker for 1 hour. The plates were then read on the SECTOR™ Imager 6000. Figure 37A & 37B shows the CHO-derived Fc-LI 0-ShK[1 -35] peptibody potently inhibits IL-2 and IFNg production from T cells in a dose-dependent manner. Compared to native ShK[I -35] peptide which shows only partial inhibtion, the peptibody produces nearly complete inhibitiono of the inflammatory cytokine response. (POC = Percent Of Control of response in the absence of inhibitor).
Inhibition of T cell proliferation following anti-CD3 and anti-CD28 antibody stimulation of PBMCs. PBMCs were previously isolated from normal human donor Leukophoresis packs, purified by density gradient centrifugation (Ficoll Hypaque), cryopreserved in CPZ Cryopreservation Medium Complete (INCELL1 MCPZF-100 plus 10% DMSO final). PBMCs were
thawed (95% viability), washed, and seeded at 2x105 cells per well in culture medium (RPMI medium 1640; GIBCO) supplemented with 10% fetal calf serum, 100U/ml penicillin, 100mg/ml streptomycin, 2mM L-glutamine, 100 μM non-essential amino acids, and 20 μM 2-ME) in 96-well flat-bottom tissue culture plates. Cells were pre-incubated with either anti-human CD32 (FcyRII) blocking antibody (per manufacturers instructions EASY SEP Human Biotin Selection Kit #18553, StemCell Technologies Vancouver, BC) or Fc-LIO-ShK (100nM-0.001nM final) for 45 min. Fc- LIO-ShK (100nM-0.001nM final) was then added to the cells containing anti-human CD32 blocking antibody while medium was added to the cells containing Fc-LIO-ShK. Both sets were incubated for an additional 45 min before stimulating for 48hr with aCD3/aCD28 (0.2 ng/ml and 100 ng/ml, respectively). Final assay volume was 200 ul. [3H]TdR (1 uCi per well) was added and the plates were incubated for an additional 16 hrs. Cells were then harvested onto glass fiber filters and radioactivity was measured in a B-scintillation counter. Figure 36A & 36B shows the CHO-derived Fc-LI 0-ShK[1 -35] peptibody potently inhibits proliferation of T cells in a dose-dependent manner. Pre-blocking with the anti-CD32 (FcR) blocking antibody has little effect on the peptibodies ability to inhibit T cell proliferation suggesting Kv1.3 inhibition and not FcR binding is the mechanism for the inhibition observed (POC = Percent Of Control of response in the absence of inhibitor).
Immunohistochemistrv analysis of Fc-LI Q-ShK[I -351 binding to HEK 293 cells overexpressinq human Kv1.3. HEK 293 cells overexpressing human Kv1.3 (HEK Kv1.3) were obtained from BioFocus pic (Cambridge, UK) and maintained per manufacturer's recommendation. The parental HEK 293 cell line was used as a control. Cells were plated on Poly-D-Lysine 24 well plates (#35-4414; Becton-Dickinson, Bedford, MA) and allowed to grow to approximately 70% confluence. HEK KV1.3 were plated at 0.5 x 10e5 cells/well in 1 ml/well of medium. HEK 293 cells were plated at a density of 1.5 x 10e5 cells/well in 1ml/well of medium. Before staining, cells were fixed with formalin (Sigma HT50-1-1 Formalin solution, diluted 1 :1 with PBS/0.5% BSA before use) by removing cell growth medium, adding 0.2ml/well formalin solution and incubating at room temperature for ten minutes. Cells were stained by incubating with 0.2ml/well of 5 μg/ml Fc-LIO- ShK[I -35] in PBS/BSA for 30' at room temperature. Fc-LI 0-ShK[1 -35] was aspirated and then the cells were washed one time with PBS/0.5% BSA. Detection antibody (Goat F(ab)2 anti-human IgG-phycoerythrin; Southern Biotech Associates, Birmingham, AL) was added to the wells at 5 μg/ml in PBS/0.5% BSA and incubated for 30' at room temperature. Wash cells once with
PBS/0.5% BSA and examine using confocal microscopy (LSM 510 Meta Confocal Microscope; Carl Zeiss AG, Germany). Figure 33B shows the Fc-LI 0-ShK[1 -35] peptibody retains binds to Kv1.3 overexpressing HEK 293 cells but shows little binding to untransfected cells(Figure 33A)
indicating the Fc-LI 0-ShK[1 -35] peptibody can be used as a reagent to detect cells overexpressing the Kv1.3 channel. In disease settings where activated T effector memory cells have been reported to overproduce Kv1.3, this reagent can find utility in both targeting these cells and in their detection. An ELISA assay demonstrating Fc-LI Q-ShKH -351 binding to fixed HEK 293 cells overexpressing Kv1.3. Figure 34A shows a dose-dependent increase in the peptibody binding to fixed cells that overexpress Kv1.3, demonstrating that the peptibody shows high affinity binding to its target and the utility of the Fc-LI 0-ShK[1 -35] molecule in detection of cells expressing the channel. Antigen specific T cells that cause disease in patients with multiple sclerosis have been shown to overexpress Kv1.3 by whole cell patch clamp electrophysiology, - a laborius approach. Our peptibody reagent can be a useful and convenient tool for monitoring Kv1.3 channel expression in patients and have utility in diagnostic applications. The procedure shown in Figure 34A and Figure 34B follows.
Figure 34A. A whole cell immunoassay was performed to show binding of intact Fc-LIO- ShK[I -35] to Kv1.3 transfected HEK 293 cells (BioFocus pic, Cambridge, UK). Parent HEK 293 cells or HEK Kv1.3 cells were plated at 3 x 10e4 cells/well in poly-D-Lysine coated ninety-six well plates (#35-4461; Becton-Dickinson, Bedford, MA). Cells were fixed with formalin (Sigma HT50-1- 1 Formalin solution, diluted 1 :1 with PBS/0.5% BSA before use) by removing cell growth medium, adding 0.2ml/well formalin solution and incubating at room temperature for 25 minutes and then washing one time with 10O μl/well of PBS/0.5% BSA. Wells were blocked by addition of 0.3ml/well of BSA blocker (50-61-00; KPL 10% BSA Diluent/Blocking Solution, diluted 1:1 with PBS; KPL, Gaithersburg, MD) followed by incubation at room temperature, with shaking, for 3hr. Plates were washed 2 times with 1x KP Wash Buffer (50-63-00; KPL). Samples were diluted in Dilution Buffer (PBS/0.5% Tween-20) or Dilution Buffer with 1% Male Lewis Rat Serum (RATSRM-M; Bioreclamation Inc., Hicksville, NY) and 0.1 ml/well was added to blocked plates, incubating for 1hr at room temperature with shaking. Plates were washed 3 times with 1xKP Wash Buffer and then incubated with HRP-Goat anti-human IgG Fc (#31416; Pierce, Rockford, IL) diluted 1 :5000 in PBS/0.1% Tween-20 for 1hr at room temperature, with shaking. Plates were washed plates 3 times with 1xKP Wash Buffer, and then 0.1 ml/well TMB substrate (52-00-01; KPL) was added. The reactions were stopped by addition of 0.1 ml/well 2 N Sulfuric Acid. Absorbance was read at
450nm on a Molecular Devices SpectroMax 340 (Sunnyvale, CA).
Figure 34B. Whole cell immunoassay was performed as above with the following modifications. HEK 293 cells were plated at 1 x 10e5 cells/well and HEK Kv1.3 cells were plated
at 6 x 10e4 cells/well in poly-D-Lysine coated 96 well plates. Fc Control was added at 500ng/ml in a volume of 0.05ml/well. HRP-Goat anti-human IgG Fc (#31416; Pierce, Rockford, IL) was diluted 1:10,000 in PBS/0.1% Tween-20. ABTS (50-66-00, KPL) was used as the substrate. Absorbances were read at 405nm after stopping reactions by addition of 0.1 ml/well of 1% SDS.
Example 38
Purification of Fc-U 0-ShK(1 -35)
Expression of Fc-LI 0-ShK[1 -35] was as described in Example 3 herein above. Frozen, E. coli paste (18 g) was combined with 200 ml of room temperature 50 mM tris HCI, 5 mM EDTA1 pH 8.0 and was brought to about 0.1 mg/ml hen egg white lysozyme. The suspended paste was passed through a chilled microfluidizer twice at 12,000 PSI. The cell lysate was then centrifuged at 22,000 g for 15 min at 4 0C. The pellet was then resuspended in 200 ml 1% deoxycholic acid using a tissue grinder and then centrifuged at 22,000 g for 15 min at 4 0C. The pellet was then resuspended in 200 ml water using a tissue grinder and then centrifuged at 22,000 g for 15 min at 4 0C. The pellet (3.2 g) was then dissolved in 32 ml 8 M guanidine HCI, 50 mM tris HCI, pH 8.0. The pellet solution was then centrifuged at 27,000 g for 15 min at room temperature, and then 5 ml of the supernatant was transferred to 500 ml of the refolding buffer (3 M urea, 20% glycerol, 50 mM tris, 160 mM arginine HCI1 5 mM EDTA1 1 mM cystamine HCI, 4 mM cysteine, pH 9.5) at 4 0C with vigorous stirring. The stirring rate was then slowed and the incubation was continued for 2 days at 4 0C. The refolding solution was then stored at -70 0C.
The stored refold was defrosted and then diluted with 2 L of water and the pH was adjusted to 7.3 using 1 M H3PO4. The pH adjusted material was then filtered through a 0.22 μm cellulose acetate filter and loaded on to a 60 ml Amersham SP-FF (2.6 cm I. D.) column at 20 ml/min in S-Buffer A (20 mM NaH2PO4, pH 7.3) at 7 0C. The column was then washed with several column volumes of S-Buffer A, followed by elution with a linear gradient from 0% to 60% S-
Buffer B (20 mM NaH2PO4, 1 M NaCI1 pH 7.3) followed by a step to 100% S-Buffer B at 10 ml/min 7 0C. Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE, and the fractions containing the desired product were pooled based on these data. The pool was then loaded on to a 1 ml Amersham rProtein A HiTrap column in PBS at 1 ml/min 7 0C. Then column was then washed with several column volumes of 20 mM Na^PCM pH 6.5, 1 M
NaCI and eluted with 100 mM glycine pH 3.0. To the elution peak, 0.0125 volumes (25 ml) of 3 M sodium acetate was added.
A spectral scan was then conducted on 50 μl of the combined pool diluted in 700 μl water using a Hewlett Packard 8453 spectrophotometer (Figure 46A). The concentration of the filtered material was determined to be 2.56 mg/ml using a calculated molecular mass of 30,410 g/mol and extinction coefficient of 36,900 M-1 cm-1. The purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 46B). The macromolecular state of the product was then determined using size exclusion chromatography on 20 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH∑PO-i, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 46C). The product was then subject to mass spectral analysis by diluting 1 μl of the sample into 10 μl of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile). One milliliter of the resultant solution was spotted onto a MALDI sample plate. The sample was allowed to dry before being analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from - 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses. The product was then stored at -80 0C.
The IC50 for blockade of human Kv1.3 by purified £co//-derived Fc-LI 0-ShK[1 -35], also referred to as "Fc-L-ShK[I -35]", is shown in Table 35 (in Example 50 herein below).
Example 39
Purification of bacterially expressed Fc-LI Q-Sh K(2-35)
Expression of Fc-LI 0-ShK[2-35] was as described in Example 4 herein above. Frozen, E. coli paste (16.5 g) was combined with 200 ml of room temperature 50 mM tris HCI, 5 mM EDTA, pH 8.0 and was brought to about 0.1 mg/ml hen egg white lysozyme. The suspended paste was passed through a chilled microfluidizer twice at 12,000 PSI. The cell lysate was then centrifuged at
22,000 g for 15 min at 4 0C. The pellet was then resuspended in 200 ml 1% deoxycholic acid using a tissue grinder and then centrifuged at 22,000 g for 15 min at 4 0C. The pellet was then resuspended in 200 ml water using a tissue grinder and then centrifuged at 22,000 g for 15 min at 4 0C. The pellet (3.9 g) was then dissolved in 39 ml 8 M guanidine HCI, 50 mM tris HCI, pH 8.0. The pellet solution was then centrifuged at 27,000 g for 15 min at room temperature, and then 5 ml of the supernatant was transferred to 500 ml of the refolding buffer (3 M urea, 20% glycerol, 50 mM tris, 160 mM arginine HCI, 5 mM EDTA, 1 mM cystamine HCI, 4 mM cysteine, pH 9.5) at 4 0C with
vigorous stirring. The stirring rate was then slowed and the incubation was continued for 2 days at 4 0C. The refolding solution was then stored at -70 0C.
The stored refold was defrosted and then diluted with 2 L of water and the pH was adjusted to 7.3 using 1 M H3PO4. The pH adjusted material was then filtered through a 0.22 μm cellulose acetate filter and loaded on to a 60 ml Amersham SP-FF (2.6 cm I. D.) column at 20 ml/min in S-Buffer A (20 mM NahbPO-i, pH 7.3) at 7 0C. The column was then washed with several column volumes of S-Buffer A, followed by elution with a linear gradient from 0% to 60% S-Buffer B (20 mM NaH2PO4, 1 M NaCI, pH 7.3) followed by a step to 100% S-Buffer B at 10 ml/min 7 0C. The fractions containing the desired product were pooled and filtered through a 0.22 μm cellulose acetate filter. The pool was then loaded on to a 1 ml Amersham rProtein A HiTrap column in PBS at 2 ml/min 7 0C. Then column was then washed with several column volumes of 20 mM NaH2PO4 pH 6.5, 1 M NaCI and eluted with 100 mM glycine pH 3.0. To the elution peak, 0.0125 volumes (18 ml) of 3 M sodium acetate was added, and the sample was filtered through a 0.22 μm cellulose acetate filter. A spectral scan was then conducted on 20 μl of the combined pool diluted in 700 μl water using a Hewlett Packard 8453 spectrophotometer (Figure 40A). The concentration of the filtered material was determined to be 3.20 mg/ml using a calculated molecular mass of 29,282 g/mol and extinction coefficient of 36,900 M"1 cm-1. The purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 40B). The macromolecular state of the product was then determined using size exclusion chromatography on 50 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO-1, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 40C). The product was then subject to mass spectral analysis by diluting 1 μl of the sample into 10 μl of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile). One milliliter of the resultant solution was spotted onto a MALDI sample plate. The sample was allowed to dry before being analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses (Figure 40D). The product was then stored at -80 0C.
The IC50 for blockade of human Kv1.3 by purified £co//-derived Fc-LI 0-ShK[2-35], also referred to as "Fc-L-ShK[2-35]", is shown in Table 35 (in Example 50 herein below).
Example 40 Purification of bacterially expressed Fc-LI 0-OsK1
Frozen, E. coli paste (129 g; see Example 10) was combined with 1290 ml of room temperature 50 mM tris HCI, 5 mM EDTA, pH 7.8 and was brought to about 0.1 mg/ml hen egg white lysozyme. The suspended paste was passed through a chilled microfluidizer twice at 12,000 PSI. The cell lysate was then centrifuged at 17,700 g for 15 min at 40C. The pellet was then resuspended in 1290 ml 1% deoxycholic acid using a tissue grinder and then centrifuged at 17,700 g for 15 min at 40C. The pellet was then resuspended in 1290 ml water using a tissue grinder and then centrifuged at 17,700 g for 15 min at 4 0C. 8 g of the pellet (16.3 g total) was then dissolved in 160 ml 8 M guanidine HCI, 5O mM tris HCI, pH 8.0. 100 ml of the pellet solution was then incubated with 1 ml of 1 M DTT for 60 min at 37 0C. The reduced material was transferred to 5000 ml of the refolding buffer (1 M urea, 50 mM tris, 160 mM arginine HCI, 2.5 mM EDTA, 1.2 mM cystamine HCI, 4 mM cysteine, pH 10.5) at 2 ml/min , 4°C with vigorous stirring. The stirring rate was then slowed and the incubation was continued for 3 days at 40C. The pH of the refold was adjusted to 8.0 using acetic acid. The pH adjusted material was then filtered through a 0.22 μm cellulose acetate filter and loaded on to a 50 ml Amersham Q Sepharose-FF (2.6 cm I. D.) column at 10 ml/min in Q-Buffer A (20 mM Tris, pH 8.5) at 8 0C with an inline 50 Amersham Protein A column (2.6 cm I. D.). After loading, the Q Sepharose column was removed from the circuit, and the remaining chromatography was carried out on the protein A column. The column was washed with several column volumes of Q-Buffer A, followed by elution using a step to 100 mM glycine pH 3.0. The fractions containing the desired product were pooled and immediately loaded on to a 50 ml Amersham SP-Sepharose HP column (2.6 cm I. D.) at 20 ml/min in S-Buffer A (20 mM NaH2PCM, pH 7.0) at 8 0C. The column was then washed with several column volumes of S-Buffer A followed by a linear gradient from 5% to 60% S-Buffer B (20 mM NaH2PO4, 1 M NaCI, pH 7.0) followed by a step to 100% S-Buffer B. Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE. The fractions containing the bulk of the desired product were pooled and then applied to a 75 ml MEP Hypercel column (2.6 cm I. D.) at 5 ml/min in MEP Buffer A (20 mM tris, 200 mM NaCI, pH 8.0) at 8 0C. Column was eluted with a linear gradient from 5% to 50% MEP Buffer B(50 mM sodium citrate pH 4.0) followed by a step to 100% MEP Buffer B. Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE, and the fractions containing the bulk of the desired product were pooled.
The MEP pool was then concentrated to about 20 ml using a Pall Jumbo-Sep with a 10 kDa membrane followed by buffer exchange with Formulation Buffer (20 mM NaH2PO-1, 200 mM NaCI, pH 7.0) using the same membrane. A spectral scan was then conducted on 50 μl of the combined pool diluted in 700 μl Formulation Buffer using a Hewlett Packard 8453 spectrophotometer (Figure 41A). The concentration of the material was determined to be 4.12 mg/ml using a calculated molecular mass of 30,558 g/mol and extinction coefficient of 35,720 M 1 cm-1. The purity of the material was then assessed using a Coomassie brilliant blue stained tris- glycine 4-20% SDS-PAGE (Figure 41 B). The macromolecular state of the product was then determined using size exclusion chromatography on 123 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 41C). The product was then subject to mass spectral analysis by chromatographing approximately 4 μg of the sample through a RP- HPLC column (Vydac C4, 1 x 150 mm). Solvent A was 0.1 % trifluoroacetic acid in water and solvent B was 0.1 % trifluoroacetic acid in 90 % acetonitrile, 10 % water. The column was pre- equilibrated in 10 % solvent B at a flow rate of 80 μl per min. The protein was eluted using a linear gradient of 10 % to 90% solvent B over 30 min. Part of the effluent was directed into a LCQ ion trap mass spectrometer. The mass spectrum was deconvoluted using the Bioworks software provided by the mass spectrometer manufacturer. (Figure 41 D). The product was filtered through a 0.22 μm cellulose acetate filter and then stored at -80 0C. The yield for the E. coli -expressed Fc-LI 0-OSK1 prep was 81 mg from 40 g of cell paste
(129 g x (8g / 16.3g) x (100 ml / 160 ml) = 39.6 g which was rounded to 40 g), the purity was greater than 80% judging by SDS-PAGE, it is running as the expected dimer judging by SEC- HPLC, and the mass was within the expected molecular weight range judging by MS.
The IC50 for blockade of human Kv1.3 by purified £.co//-derived Fc-LI 0-OSK1, also referred to as "Fc-L-OSKI", is shown in Table 35 (in Example 50 herein below).
Example 41
FC-L10-OSK1, Fc-LI Q-OSKIf K7S1. Fc-LI 0-OSK1 TE16K.K20D1. and Fc-L10-OSK1 [K7S,E16K,K2OD1 expressed by mammalian cells FC-L10-OSK1, Fc-LI 0-OSK1[K7S], Fc-L10-OSK1[E16K,K20D], and Fc-L10-OSK1
[K7S,E16K,K20D], inhibitors of Kv1.3, were expressed in mammalian cells. A DNA sequence coding for the Fc region of human IgGI fused in-frame to a linker sequence and a monomer of the Kv1.3 inhibitor peptide OSK1, OSK1[K7S], OSK1[E16K,K20D], or OSK1[K7S,E16K,K20D] was
constructed as described below. Methods for expressing and purifying the peptibody from mammalian cells (HEK 293 and Chinese Hamster Ovary cells) are disclosed herein.
For construction of Fc-LI 0-OSK1, Fc-LI 0-OSK1[K7S], Fc-LI 0-OSK1 [E16K.K20D], and Fc-LI 0-OSK1[K7S,E16K,K20D] expression vectors, a PCR strategy was employed to generate the full length genes, 0SK1, 0SK1[K7S], OSK1[E16K,K20D], and OSK1[K7S,E16K,K20D], each linked to a four glycine and one serine amino acid linker with two stop codons and flanked by BamHI and Notl restriction sites as shown below.
Two oligos for each of 0SK1, 0SK1[K7S], OSK1[E16K,K20D], and [K7S,E16K,K20D]OSK1 with the sequence as depicted below were used in a PCR reaction with PfuTurbo HotStart DNA polymerase (Stratagene) at 95°C-30sec, 55°C-30sec, 75°C-45sec for 35 cycles; BamHI (ggatcc) and Notl (gcggccgc) restriction sites are underlined.
0SK1:
Forward primer: cat qqa tec gga gga gga gga age ggc gtg ate ate aac gtg aag tgc aag ate age cgc cag tgc ctg gag ccc tgc aag aag gcc g (SEQ ID NO: 876);
Reverse primer: cat gcg gcc get tac tac ttg ggg gtg cag tgg cac ttg ccg ttc atg cac ttg ccg aag cgc atg ccg gcc ttc ttg cag ggc tec a (SEQ ID NO:877);
OSK1[K7S]:
Forward primer: cat gga tec gga gga gga gga age ggc gtg ate ate aac gtg age tgc aag ate age cgc cag tgc ctg gag ccc tgc aag aag gcc g (SEQ ID NO:878);
Reverse primer: cat qcq gcc get tac tac ttg ggg gtg cag tgg cac ttg ccg ttc atg cac ttg ccg aag cgc atg ccg gcc ttc ttg cag ggc tec a (SEQ ID NO:879);
OSK1 [E16K,K20D]:
Forward primer: cat qqa tec gga gga gga gga age ggc gtg ate ate aac gtg aag tgc aag ate age cgc cag tgc ctg aag ccc tgc aag gac gcc g (SEQ ID NO:880);
Reverse primer: cat qcq gee get tac tac ttg ggg gtg cag tgg cac ttg ccg ttc atg cac ttg ccg aag cgc atg ccg gcg tec ttg cag ggc ttc a (SEQ ID NO:881);
OSK1[K7S,E16K,K20D]:
Forward primer: cat qqa tec gga gga gga gga age ggc gtg ate ate aac gtg age tgc aag ate age cgc cag tgc ctg aag ccc tgc aag gac gcc g (SEQ ID NO:882);
Reverse primer: cat qcq gcc get tac tac ttg ggg gtg cag tgg cac ttg ccg ttc atg cac ttg ccg aag cgc atg ccg gcg tec ttg cag ggc ttc a (SEQ ID NO:883).
The resulting PCR products were resolved as the 155bp bands on a four percent agarose gel. The 155bp PCR product was purified using PCR Purification Kit (Qiagen), then digested with BamHI and Notl (Roche) restriction enzymes, and agarose gel was purified by Gel Extraction Kit (Qiagen). At the same time, the pcDNA3.1(+)CMVi-hFc-Shk[2-35] vector was digested with BamHI and Notl restriction enzymes and the large fragment was purified by Gel Extraction Kit. The gel purified PCR fragment was ligated to the purified large fragment and transformed into One Shot® ToplOF' (Invitrogen). DNAs from transformed bacterial colonies were isolated and digested with BamHI and Notl restriction enzymes and resolved on a two percent agarose gel. DNAs resulting in an expected pattern were submitted for sequencing. Although, analysis of several sequences of clones yielded a 100% percent match with the above sequences, only one clone from each gene was selected for large scaled plasmid purification. The DNA of Fc-LI 0-OSK1, Fc-LI 0-OSK1[K7S], Fc-L10-OSK1[E16K,K20D], and Fc-L10-OSK1[K7S,E16K,K20D] in pCMVi vector was resequenced to confirm the Fc and linker regions and the sequence was 100% identical to the above sequences. The sequences and pictorial representations of Fc-LI 0-OSK1, Fc-LI 0-OSK1[K7S], Fc- L10-OSK1 [E16K,K20D], and Fc-LI 0-OSK1 [K7S,E16K,K20D] are depicted in Figure 42A-B, Figure 43A-B, Figure 44A-B and Figure 45A-B, respectively.
HEK-293 cells used in transient transfection expression of Fc-LI 0-OSK1, Fc-LIO- OSK1[K7S], Fc-L10-OSK1[E16K,K20D], and Fc-L10-OSK1[K7S,E16K,K20D] in pCMVi protein were cultured in growth medium containing DMEM High Glucose (Gibco), 10% fetal bovine serum
(FBS from Gibco), 1X non-essential amino acid (NEAA from Gibco)and 1X Penicillin/Streptomycine/Glutamine (Pen/Strep/Glu from Gibco). 5.6 μg each of Fc-LI 0-OSK1, Fc- L10-OSK1[K7S], Fc-L10-OSK1[E16K,K20D], and Fc-L10-OSK1[K7S,E16K,K20D] in pCMVi plasmid that had been phenol/chloroform extracted was transfected into HEK-293 cells using FuGENE 6 (Roche). The cells were recovered for 24 hours, and then placed in DMEM High
Glucose, 1x NEAA and 1X Pen/Strep/Glu medium for 48 hours. Fc-LI 0-OSK1[K7S], Fc-LIO- OSK1[E16K,K20D], and Fc-L10-OSK1[K7S,E16K,K20D] were purified from medium
conditioned by these transfected HEK-293 cells using a protocol described in Example 50 herein below.
Fifteen μl of conditioned medium was mixed with an in-house 4x Loading Buffer (without β-mercaptoethanol) and electrophoresed on a Novex 4-20% tris-glycine gel using a Novex Xcell Il apparatus at 101V/46mA for 2 hours in a 1x Gel Running solution (25mM Tris Base, 192mM Glycine, 3.5mM SDS) along with 20μl of BenchMark Pre-Stained Protein ladder (Invitrogen). The gel was then soaked in Electroblot buffer (25mM Tris base, 192mM glycine, 20%methanol,) for 5 minutes. A nitrocellulose membrane from Invitrogen (Cat. No. LC200, 0.2 μm pores size) was soaked in Electroblot buffer. The pre-soaked gel was blotted to the nitrocellulose membrane using the Mini Trans-Blot Cell module according to the manufacturer instructions (Bio-Rad Laboratories) at 30OmA for 2 hours. The blot was rinsed in Tris buffered saline solution pH7.5 with 0.1% Tween20 (TBST). Then, the blot was first soaked in a 5% milk (Carnation) in TBST for 1 hour at room temperature, followed by washing three times in TBST for 10 minutes per wash. Then, incubated with 1 :1000 dilution of the HRP-conjugated Goat anti-human IgG, (Fcγ) antibody (Pierece Biotechnology Cat. no. 31413) in TBST with 5% milk buffer for 1 hour with shaking at room temperature. The blot was then washed three times in TBST for 15 minutes per wash at room temperature. The primary antibody was detected using Amersham Pharmacia Biotech's ECL western blotting detection reagents according to manufacturer's instructions. Upon ECL detection, the western blot analysis displayed the expected size of 66kDa under non-reducing gel conditions (Figure 46).
Plasmids containing the Fc-LI 0-OSK1 , Fc-LI 0-OSK1[K7S], Fc-L10-OSK1 [E16K.K20D], and Fc-LI 0-OSK1[K7S,E16K,K20D] inserts in pCMVi vector were digested with Xbal and Notl (Roche) restriction enzymes and gel purified. The inserts were individually ligated into Spel and Notl (Roche) digested pDSRα24 (Amgen Proprietary) expression vector. Integrity of the resulting constructs were confirmed by DNA sequencing. Although, analysis of several sequences of clones yielded a 100% percent match with the above sequence, only one clone was selected for large scaled plasmid purification.
AM1 CHOd- (Amgen Proprietary) cells used in the stable expression of Fc-LI 0-OSK1 protein were cultured in AM1 CHOd- growth medium containing DMEM High Glucose, 10% fetal bovine serum, 1x hypoxantine/thymidine (HT from Gibco), 1X NEAA and 1X Pen/Strep/Glu. 5.6 μg of pDSRα-24-Fc-L10-OSK1 plasmid was transfected into AM1 CHOd- cells using FuGene 6. Twenty-four hours post transfection, the cells were split 1:11 into DHFR selection medium (DMEM High Glucose plus 10% Dialyzed Fetal Bovine Serum (dFBS), 1x NEAA and 1X Pen/Strep/Glu)
and at 1 :50 dilution for colony selection. The cells were selected in DHFR selection medium for thirteen days. The ten 10-cm2 pools of the resulting colonies were expanded to ten T-175 flasks, then were scaled up ten roller bottles and cultured under AM1 CHOd- production medium (DMEM/F12 (1:1), 1X NEAA1 1X Sodium Pyruvate (Na Pyruvate), 1X Pen/Strep/Glu and 1.5% DMSO). The conditioned medium was harvested and replaced at one-week intervals. The resulting six liters of conditioned medium were filtered through a 0.45 μm cellulose acetate filter (Corning, Acton, MA), and characterized by SDS-PAGE analysis as shown in Figure 47. Then, transferred to Protein Chemistry for purification.
Twelve colonies were selected after 13 days on DHFR selection medium and picked into one 24-well plate. The plate was allowed to grow up for one week, and then was transferred to AM1 CHOd- production medium for 48-72 hours and the conditioned medium was harvested. The expression levels were evaluated by Western blotting similar to the transient Western blot analysis with detection by the same HRP-conjugated Goat anti-human IgG, (Fcγ) antibody to screen 5μl of conditioned medium. All 12 stable clones exhibited expression at the expected size of 66 kDa. Two clones, A3 and C2 were selected and expanded to T175 flask for freezing with A3 as a backup to the primary clone C2 (Figure 48).
The C2 clone was scaled up into fifty roller bottles (Corning) using selection medium and grown to confluency. Then, the medium was exchanged with a production medium, and let incubate for one week. The conditioned medium was harvested and replaced at the one-week interval. The resulting fifty liters of conditioned medium were filtered through a 0.45 μm cellulose acetate filter (Corning, Acton, MA), and characterized by SDS-PAGE analysis (data not shown). Further purification was accomplished as described in Example 42 herein below.
Example 42 Purification of Fc-L10-OSK1, Fc-LI 0-OSK1(K7S), Fc-L10-OSK1(E16K,K20D), and
Fc-LI 0-OSK1(K7S,E16K,K20D) expressed by mammalian cells
Purification of Fc-L10-OSK1. Approximately 6 L of CHO (AM1 CHOd-) cell-conditioned medium (see, Example 41 above) was loaded on to a 35 ml MAb Select column (GE Healthcare) at 10 ml/min 70C, and the column was washed with several column volumes of Dulbecco's phosphate buffered saline without divalent cations (PBS) and sample was eluted with a step to 100 mM glycine pH 3.0. The MAb Select elution was directly loaded on to an inline 65 ml SP-HP column (GE Healthcare) in S-Buffer A (20 mM NaH2PO4, pH 7.0) at 10 ml/min 7°C. After disconnecting the MAb select column, the SP-HP column was then washed with several column
volumes S-Buffer A1 and then developed using a linear gradient from 5% to 60% S-Buffer B (20 mM NaH2PO4, 1 M NaCI1 pH 7.0) at 10 ml/min followed by a step to 100% S-Buffer B at 7 0C. Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS- PAGE, and the fractions containing the desired product were pooled based on these data. The pooled material was then concentrated to about 20 ml using a Pall Life Sciences Jumbosep 1OK Omega centrifugal ultra-filtration device. The concentrated material was then buffer exchanged by diluting with 20 ml of 20 mM NaH2PO4, pH 7.0 and reconcentrated to 20 ml using the Jumbosep 1OK Omega filter. The material was then diluted with 20 ml 20 mM NaH2PO4, 200 mM NaCI, pH 7.0 and then reconcentrated to 22 ml. The buffer exchanged material was then filtered though a Pall Life Sciences Acrodisc with a 0.22 μm, 25 mm Mustang E membrane at 1 ml/min room temperature. A spectral scan was then conducted on 50 μl of the filtered material diluted in 700 μl PBS using a Hewlett Packard 8453 spectrophotometer (Figure 49A1 black trace). The concentration of the filtered material was determined to be 4.96 mg/ml using a calculated molecular mass of 30,371 g/mol and extinction coefficient of 35,410 M 1 cm 1. The purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 49B). The endotoxin level was then determined using a Charles River Laboratories Endosafe-PTS system (0.05 - 5 EU/ml sensitivity) using a 30-fold dilution of the sample in Charles Rivers Laboratories Endotoxin Specific Buffer yielding a result of 1.8 EU/mg protein. The macromolecular state of the product was then determined using size exclusion chromatography on 149 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI1 pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 49C). The product was then subject to mass spectral analysis by diluting 1 μl of the sample into 10 μl of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile) . One milliliter of the resultant solution was spotted onto a MALDI sample plate. The sample was allowed to dry before being analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from about 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses. (Figure 49D). The product was then stored at -800C. The yield for the mammalian Fc-LI 0-OSK1 prep was 115 mg from 6 L, the purity was
>90% judging by SDS-PAGE; Fc-LI 0-OSK1 ran as the expected dimer judging by SEC-HPLC, and the mass is with the expected range judging by MS.
The activity of purified Fc-LI 0-OSK1 in blocking human Kv1.3 and human Kv1.1 is described in Example 43 herein below.
Purification of Fc-LI 0-OSKKK7S), Fc-LI 0-OSKKE16K.K20D), and Fc-UO- OSK1(K7S,E16K,K20D). Approximately 500 mL of medium conditioned by transfected HEK-293 (see, Example 41 above) was combined with a 65 % slurry of MAb Select resin (1.5 ml) (GE Healthcare) and 500 μl 20% Natø. The slurry was then gently agitated for 3 days at 4 0C followed by centrifugation at 1000 g for 5 minutes at 4 0C using no brake. The majority of the supernatant was then aspirated and the remaining slurry in the pellet was transferred to a 14 ml conical tube and combined with 12 ml of Dulbecco's phosphate buffered saline without divalent cations (PBS). The slurry was centrifuged at 2000 g for 1 minute at 4 0C using a low brake and the supernatant was aspirated. The PBS wash cycle was repeated an additional 3 times. The bound protein was then eluted by adding 1 ml of 100 mM glycine pH 3.0 and gently agitating for 5 min at room temperature. The slurry was then centrifuged at 2000 g for 1 minute at 4 0C using a low brake and the supernatant was aspirated as the first elution. The elution cycle was repeated 2 more times, and all 3 supernatants were combined into a single pool. Sodium acetate (37.5 μl of a 3 M solution) was added to the elution pool to raise the pH, which was then dialyzed against 10 mM acetic acid, 5% sorbitol, pH 5.0 for 2 hours at room temperature using a 10 kDa SlideAlyzer (Pierce). The dialysis buffer was changed, and the dialysis continued over night at 40C. The dialyzed material was then filtered through a 0.22 μm cellulose acetate filter syringe filter. Then concentration of the filtered material was determined to be 1.27 mg/ml using a calculated molecular mass of 30,330 and extinction coefficient of 35,410 M 1 cm 1 (Figure 50A). The purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 50B). The endotoxin level was then determined using a Charles River Laboratories Endosafe-PTS system (0.05 - 5 EU/ml sensitivity) using a 25-fold dilution of the sample in Charles Rivers Laboratories Endotoxin Specific Buffer yielding a result of <1 EU/mg protein. The macromolecular state of the product was then determined using size exclusion chromatography on 50 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 50C). The product was then subject to mass spectral analysis by diluting 1 μl of the sample into 10 μl of sinapinic acid (10 mg per ml in 0.05% trifluroacetic acid, 50% acetonitrile).
One milliter of the resultant solution was spotted onto a MALDI sample plate. The sample was allowed to dry before being analyzed using a Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). The positive ion/linear mode was used, with
an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots. External mass calibration was accomplished using purified proteins of known molecular masses. (Figure 50D). The product was then stored at -80 0C.
Figures 51A-D show results from the purification and analysis for Fc-LI 0-OsK1(E16K, K20D), which was conducted using the same protocol as that for the Fc-LI 0-OsK1 (K7S) molecule (described above) with the following exceptions: the concentration was found to be 1.59 mg/ml using a calculated molecular mass of 30,357 g/mol and a calculated extinction coefficient of 35,410; the pyrogen level was found to be < 1 EU/mg using a 32-fold dilution.
Figures 52A-D show results from the purification and analysis for Fc-LIO- OsK1(K7S,E16K, K20D), which was conducted using the same protocol as that for the Fc-LIO- OsK1(K7S) molecule (described above) with the following exceptions: the concentration was found to be 0.81 mg/ml using a calculated molecular mass of 30,316 g/mol and a calculated extinction coefficient of 35,410; the pyrogen level was found to be < 1 EU/mg using a 16-fold dilution. The activity of purified Fc-LI 0-OSK1 [K7S], Fc-L10-OSK1[E16K, K20D] and Fc-LIO-
OSK1[K7S, E16K, K20D] in blocking human Kv1.3 and human Kv1.1 is described in Example 43 herein below.
Example 43 Electrophysiology of OSK1 and OSK1 peptibodv analogs
A 38-residue peptide toxin of the Asian scorpion Orthochirus scrobiculosus venom (OSK1) was synthesized (see, Examples 41) to evaluate its impact on the human Kv1.1 and Kv1.3 channels, subtypes of the potassium channel family. The potency and selectivity of synthetic OSK1 in inhibiting the human Kv1.1 and Kv1.3 channels was evaluated by the use of HEK293 cell expression system and electrophysiology (Figure 53). Whole cell patch clamp recording of stably expressed Kv1.3 channels revealed that the synthetic OSK1 peptide is more potent in inhibiting human Kv1.3 when compared to Kv1.1 (Table 33).
Fusion of OSK1 peptide toxin to antibody to generate OSK1 peptibodv. To improve plasma half-life and prevent OSK1 peptide toxin from penetrating the CNS, the OSK1 peptide toxin was fused to the Fc-fragment of a human antibody IgGI via a linker chain length of 10 amino acid residues (Fc-LI 0-OSK1), as described in Example 41 herein. This fusion resulted in a decrease in the potency of Kv1.3 by 5-fold when compared to the synthetic OSK1 peptide. However, it
significantly improved the selectivity of 0SK1 against Kv1.1 by 210-fold when compared to that of the synthetic peptide alone (4-fold; Table 33 and Figure 54).
Modification of OSK1-peptibodv (Fc-U Q-OSKH. 0SK1 shares 60 to 80% sequence homology to other members of scorpion toxins, which are collectively termed α-KTx3. Sequence alignment of OSK1 and other members of α-KTx3 family revealed 4 distinct structural differences at positions 12, 16, 20, and 36. These structural differences of OSK1 have been postulated to play an important role in its wide range of activities against other potassium channels, which is not observed with other members of α-KTx3 family. Hence, two amino acid residues at position 16 and 20 were restored to the more conserved amino acid residues within the OSK1 sequence in order to evaluate their impact on selectivity against other potassium channels such as Kv1.1 , which is predominantly found in the CNS as a heterotetromer with Kv1.2. By substituting for glutamic acid at position 16, and for lysine at position 20, the conserved lysine and aspartic acid residues, respectively (i.e., Fc-L10-OSK1[E16K, K20D]), we did not observe a significant change in potency when compared to that of Fc-L10-OSK1 (1.3-fold difference; Figure 56 and Table 33). However, this double mutation removed the blocking activity against Kv1.1. The selectivity ratio of Kv1.1/Kv1.3 was 403-fold, which was a significant improvement over the selectivity ratio for Fc- L10-OSK1 (210-fold). A single amino acid mutation at position 7 from lysine to serine (Fc-UO- OSK1[K7S]) produced a slight change in potency and selectivity by 2- and 1.3-fold, respectively, when compared to those of Fc-LI 0-OSK1 (Figure 55 and Table 33). There was a significant decrease in potency as well as selectivity when all three residues were mutated to generate Fc- L10-OSK1[K7S, E16K, K20D] (Figure 57 and Table 33).
As demonstrated by the results in Table 33, we dramatically improved selectivity against Kv1.1 by fusing the OSK1 peptide toxin to the Fc-fragment of the human antibody IgGI , but reduced target potency against Kv1.3. The selectivity against Kv1.1 was further improved when 2 residues at two key positions were restored to the conserved residues found in other members of the α-KTx3 family.
Table 33 shows a summary of IC50 values for OSK1 and OSK1 analogs against hKv1.3 and hKv1.1 channels. All analogues are ranked based on their potency against hKv1.3. Also shown in the table is the selectivity ratio of hKv1.1/hKv1.3 for all OSK1 analogs.
Example 44 Pharmacokinetic Study of PEG-ShK[I -351 molecule in Rats
The intravenous (IV) pharmacokinetic profile was determined of a about 24-kDa 2OK PEG-ShK[I -35] molecule and the about 4-kDa small native ShK peptide was determined in
Spraque Dawley rats. The IV dose for the native ShK peptide and our novel 20K PEG-ShK[I -35] molecule was 1 mg/kg. This dose represented equal molar amounts of these two molecules. The average weight of the rats was about 0.3 kg and two rats were used for each dose & molecule. At various times following IV injection, blood was drawn and about 0.1 ml of serum was collected. Serum samples were stored frozen at -8O0C until analysis.
Assay Plate preparation for electrophysiology. Rat serum samples containing the 2OK PEG-ShK[I -35] molecule or the native ShK peptide from pharmacokinetic studies were received frozen. Before experiments, each sample was thawed at room temperature and an aliquot (70 to 80 μl) was transferred to a well in a 96-well polypropylene plate. In order to prepare the Assay Plate, several dilutions were made from the pharmacokinetic serum samples to give rise to Test Solutions. Dilutions of serum samples from the pharmacokinetic study were into 10% Phosphate Buffered Saline (PBS, with Ca2+ and Mg2+). For determination of the amount of our novel 2OK PEG- ShK[I -35] molecule in serum samples from the pharmacokinetic study, the final serum concentrations in the Test Solutions were 90%, 30%, 10%, 3.3% and 1.1%. Purified 20K PEG-
Shk[1-35] Standard inhibition curves were also prepared in the Assay Plate. To do this, 8-point serial dilutions of the purified 2OK PEG-ShK[1-35] molecule (Standard) were prepared in either 90%, 30%, 10%, 3.3% or 1.1% rat serum and the final concentration of standard was 50, 16.7, 5.5, 1.85, 0.62, 0.21, 0.068 and 0.023 nM.
Cell preparation for electrophvsioloqy, CHO cells stably expressing the voltage-activated
K+ channel, KV1.3 were plated in T-175 tissue culture flasks (at a density of 5x106) 2 days before experimentation and allowed to grow to around 95% confluence. Immediately prior to the experiment, the cells were washed with PBS and then detached with a 2 ml mixture (1 :1 volume ratio) of trypsin (0.25%) and versene (1:5000) at 37°C (for 3 minutes). Subsequently, the cells were re-suspended in the flask in 10 ml of tissue culture medium (HAM's F-12 with Glutamax,
InVitrogen, Cat#31765) with 10% FBS, 1x NEAA and 750 μg/ml of G418) and centrifuged at about 1000 rpm for VA minutes. The resultant cell pellet was re-suspended in PBS at 3-5x106CeIIs/ ml.
IonWorks electrophysiology and data analysis. The ability of Test solutions or Standards in serum to inhibit K+ currents in the CHO-KvI .3 cells was investigated using the automated electrophysiology system, IonWorks Quattro. Re-suspended cells, the Assay Plate, a Population Patch Clamp (PPC) PatchPlate as well as appropriate intracellular (90mMK-Gluconate, 2OmMKF, 2 mM NaCI, 1mM MgCI2, 1OmM EGTA1 1OmM HEPES, pH 7.35) and extracellular (PBS1 with Ca2+ and Mg2+) buffers were positioned on IonWorks Quattro. Electrophysiology recordings were made from the CHO-KvI .3 cells using an amphotericin-based perforated patch-clamp method. Using the voltage-clamp circuitry of the IonWorks Quattro, cells were held at a membrane potential of -80 mV and voltage-activated K+ currents were evoked by stepping the membrane potential to +30 mV for 400 ms. K+ currents were evoked under control conditions i.e., in the absence of inhibitor at the beginning of the experiment and after 10-minute incubation in the presence of the Test Solution or Standard. The mean K+ current amplitude was measured between 430 and 440ms and the data were exported to a Microsoft Excel spreadsheet. The amplitude of the K+ current in the presence of each concentration of the Test Solution or Standard was expressed as a percentage of the K+ current in control conditions in the same well.
Standard inhibition curves were generated for each standard in various levels of rat serum and expressed as current percent of control (POC) versus log of nM concentration. Percent of control (POC) is inversely related to inhibition, where 100 POC is no inhibition and 0 POC is 100% inhibition. Linear regression over a selected region of the curve was used to derive an equation to enable calculation of drug concentrations within Test solutions. Only current values within the linear portion of the Standard curve were used to calculate the concentration of drug in Test solutions. The corresponding Standard curve in a given level of serum, was always compared to the same level of serum of Test solution when calculating drug level. The Standard curves for ShK and 2OK PEG-ShK[I -35] are shown in Figure 58A and Figure 58B1 respectively, and each figure contains linear regression equations for each Standard at a given percentage of serum. For the 2OK PEG-ShK[1-35] standard curve the linear portion of the Standard curve was from 20 POC to 70 POC and only current values derived from the Test solution which fell within this range were used to calculate drug concentration within the Test solution.
The pharmacokinetic profile of our novel 2OK PEG ShK[1-35] molecule after IV injection is shown in Figure 59. The terminal half-life (ti«b) of this molecule is estimated from this curve to be between 6 to 12 hours long. Beyond 48 hours, the level of drug falls outside the linear range of the
Standard curve and is not calculated. The calculated 6 to 12 hour half-life of our novel 2OK PEG- ShK[I -35] molecule was substantially longer than the approximately 0.33 hour (or 20 min) half-life of the native ShK molecule reported earlier by C. Beeton et al. [C. Beeton et al. (2001) Proc. Natl Acad. Sci. 98, 13942-13947], and is a desirable feature of a therapeutic molecule. A comparison of the relative levels of Kv1.3 inhibitor after an equal molar IV injection of ShK versus 2OK PEG- ShK[I -35] is shown in Figure 60. As can be seen from this figure examining 5% serum Test solutions, the 2OK PEG-ShK[I -35] molecule showed significant suppression of Kv1.3 current (<70 POC) for more than 24 hours, whereas the native ShK peptide only showed a significant level of inhibition of Kv1.3 current for the first hour and beyond 1 hour showed no significant blockade. These data again demonstrate a desirable feature of the 2OK PEG ShK[I -35] molecule as a therapeutic for treatment of autoimmune disease.
Example 45
PEGylated Toxin Peptide suppressed severe autoimmune encephalomyelitis in animal model
The 20KPEG-ShK inhibitor of Kv1.3 shows improved efficacy in suppressing severe autoimmune encephalomyelitis in rats. Using an adoptive transfer experimental autoimmune encephalomyelitis (AT-EAE) model of multiple sclerosis described earlier [C. Beeton et al. (2001) J. Immunol. 166, 936], we examined the activity in vivo of our novel 20KPEG-ShK molecule and compared its efficacy to that of the ShK toxin peptide alone. The study design is illustrated in Figure 61. The results from this in vivo study are provided in Figure 62 and Figure 63. The 20KPEG-ShK molecule delivered subcutaneously (SC) at 10 μg/kg daily from day -1 to day 3 significantly reduced disease severity and increased survival, whereas animals treated with an equal molar dose (10 μg/kg) of the small ShK peptide developed severe disease and died. The 35-amino acid toxin peptide ShK (Stichodactyla helianthus neurotoxin) was purchased from Bachem Bioscience lnc and confirmed by electrophysiology to potently block Kv1.3 (see Example 36 herein). The synthesis, PEGylation and purification of the 20KPEG ShK molecule was as described herein above. The encephalomyelogenic CD4+ rat T cell line, PAS, specific for myelin-basic protein (MBP) originated from Dr. Evelyne Beraud. The maintenance of these cells in vitro and their use in the AT-EAE model has been described earlier [C. Beeton et al.
(2001) PNAS 98, 13942]. PAS T cells were maintained in vitro by alternating rounds of antigen stimulation or activation with MBP and irradiated thymocytes (2 days), and propagation with T cell growth factors (5 days). Activation of PAS T cells (3 * 105/ml) involved incubating the cells for
2 days with 10 μg/ml MBP and 15 * 106/ml syngeneic irradiated (3500 rad) thymocytes. On day 2 after in vitro activation, 10-15 * 106 viable PAS T cells were injected into 6-12 week old female Lewis rats (Charles River Laboratories) by tail IV. Daily subcutaneous injections of vehicle (2% Lewis rat serum in PBS)1 20KPEG-ShK or ShK were given from days -1 to 3 (Figure 61), where day -1 represent 1 day prior to injection of PAS T cells (day 0). In vehicle treated rats, acute EAE developed 4 to 5 days after injection of PAS T cells (Figure 62). Serum was collected by retro- orbital bleeding at day 4 and by cardiac puncture at day 8 (end of the study) for analysis of levels of inhibitor. Rats were weighed on days -1, 4, 6, and 8. Animals were scored blinded once a day from the day of cell transfer (day 0) to day 3, and twice a day from day 4 to day 8. Clinical signs were evaluated as the total score of the degree of paresis of each limb and tail. Clinical scoring: 0 = No signs, 0.5 = distal limp tail, 1.0 = limp tail, 2.0 = mild paraparesis, ataxia, 3.0 = moderate paraparesis, 3.5 = one hind leg paralysis, 4.0 = complete hind leg paralysis, 5.0 = complete hind leg paralysis and incontinence, 5.5 = tetraplegia, 6.0 = moribund state or death. Rats reaching a score of 5.5 were euthanized. Treatment of rats with the Kv1.3 blocker PEG-ShK prior to the onset of EAE caused a lag in the onset of disease, inhibited the progression of disease, and prevented death in a dose- dependent manner (Figure 62). Onset of disease in rats that were treated with the vehicle alone, 10 μg/kg ShK or 1 μg/kg of PEG-ShK was observed on day 4, compared to day 4.5 in rats treated with 10 μg/kg PEG-ShK or 100 μg/kg PEG-ShK. In addition, rats treated with vehicle alone, 10 μg/kg ShK or 1 μg/kg of PEG-ShK all developed severe disease by the end of the study with an
EAE score of 5.5 or above. In contrast, rats treated with 10 μg/kg PEG-ShK or 100 μg/kg PEG- ShK, reached a peak clinical severity score average of < 2, and all but one rat survived to the end of the study. Furthermore, we found that rat body weight correlated with disease severity (Figure 63). Rats treated with vehicle alone, 10 μg/kg ShK or 1 μg/kg of PEG-ShK all lost an average of 31 g, 3Og, and 3Og, respectively, while rats treated with 10 μg/kg PEG-ShK or 100 μg/kg PEG-ShK lost 18g and 11g, respectively. Rats in the latter two groups also appeared to be gaining weight by the end of the study, a sign of recovery. It should be noted that rats treated with 10 μg/kg ShK and 10 μg/kg PEG-ShK received molar equivalents of the ShK peptide. The significantly greater efficacy of the PEG-ShK molecule relative to unconjugated ShK, is likely due to the PEG-ShK molecule's greater stability and prolonged half-life in vivo (see, Example 44).
Example 46
Compositions including Kv1.3 Antagonist Peptides Block Inflammation in
Human Whole Blood
Ex vivo assay to examine impact of Kv1.3 inhibitors on secretion of IL-2 and IFN-q. Human whole blood was obtained from healthy, non-medicated donors in a heparin vacutainer. DMEM complete media was Iscoves DMEM (with L-glutamine and 25 mM Hepes buffer) containg 0.1% human albumin (Bayer #68471), 55 μM 2-mercaptoethanol (Gibco), and 1X Pen-Strep-Gin (PSG, Gibco, Cat#10378-016). Thapsigargin was obtained from Alomone Labs (Israel). A 10 mM stock solution of thapsigargin in 100% DMSO was diluted with DMEM complete media to a 40 μM, 4X solution to provide the 4X thapsigargin stimulus for calcium mobilization. The Kv1.3 inhibitor peptide ShK (Stichodacytla helianthus toxin, Cat# H2358) and the BKCaI inhibitor peptide IbTx (Iberiotoxin, Cat# H9940) were purchased from Bachem Biosciences, whereas the Kv1.1 inhibitor peptide DTX-k (Dendrotoxin-K) was from Alomone Labs (Israel). The CHO-derived Fc-LI 0-ShK[2- 35] peptibody inhibitor of Kv1.3 was obtained as described herein at Example 4 and Example 39. The calcineurin inhibitor cyclosporin A was obtained from the Amgen sample bank, but is also available commercially from a variety of vendors. Ten 3-fold serial dilutions of inhibitors were prepared in DMEM complete media at 4X final concentration and 50 μl of each were added to wells of a 96-well Falcon 3075 flat-bottom microtiter plate. Whereas columns 1-5 and 7-11 of the microtiter plate contained inhibitors (each row with a separate inhibitor dilution series), 50 μl of DMEM complete media alone was added to the 8 wells in column 6 and 100 μl of DMEM complete media alone was added to the 8 wells in column 12. To initiate the experiment, 100 μl of whole blood was added to each well of the microtiter plate. The plate was then incubated at 370C, 5% CO2 for one hour. After one hour, the plate was removed and 50 μl of the 4X thapsigargin stimulus (40 μM) was added to all wells of the plate, except the 8 wells in column 12. The plates were placed back at 370C, 5% CO2 for 48 hours. To determine the amount of IL-2 and IFN-g secreted in whole blood, 100 μl of the supernatant (conditioned media) from each well of the 96- well plate was transferred to a storage plate. For MSD electrochemilluminesence analysis of cytokine production, 20 μl of the supematants (conditioned media) were added to MSD Multi-Spot Custom Coated plates (www.meso-scale.com). The working electrodes on these plates were coated with four Capture Antibodies (hlL-5, hlL-2, hlFNg and hlL-4) in advance. After addition of
20 μl of conditioned media to the MSD plate, 150 μl of a cocktail of Detection Antibodies and P4 Buffer were added to each well. The 150 μl cocktail contained 20 ul of four Detection Antibodies (hlL-5, hlL-2, hlFNg and hlL-4) at 1 μg/ml each and 130 ul of 2X P4 Buffer. The plates were
covered and placed on a shaking platform overnight (in the dark). The next morning the plates were read on the MSD Sector Imager. Since the 8 wells in column 6 of each plate received only the thapsigargin stimulus and no inhibitor, the average MSD response here was used to calculate the "High" value for a plate. The calculate "Low" value for the plate was derived from the average MSD response from the 8 wells in column 12 which contained no thapsigargin stimulus and no inhibitor. Percent of control (POC) is a measure of the response relative to the unstimulated versus stimulated controls, where 100 POC is equivalent to the average response of thapsigargin stimulus alone or the "High" value. Therefore, 100 POC represents 0% inhibition of the response. In contrast, 0 POC represents 100% inhibition of the response and would be equivalent to the response where no stimulus is given or the "Low" value. To calculate percent of control (POC), the following formula is used: [(MSD response of well) - ("Low")] / [("High") - ("Low")] x 100. The potency of the molecules in whole blood was calculated after curve fitting from the inhibition curve (IC) and IC50 was derived using standard curve fitting software. Although we describe here measurement of cytokine production using a high throughput MSD electrochemillumenescence assay, one of skill in the art can readily envision lower throughput ELISA assays are equally applicable for measuring cytokine production.
Ex vivo assay demonstrating Kv1.3 inhibitors block cell surface activation of CD40L & IL- 2R. Human whole blood was obtained from healthy, non-medicated donors in a heparin vacutainer. DMEM complete media was Iscoves DMEM (with L-glutamine and 25 mM Hepes buffer) containing 0.1% human albumin (Bayer #68471), 55 μM 2-mercaptoethanol (Gibco), and 1X Pen-Strep-Gin (PSG1 Gibco, Cat#10378-016). Thapsigargin was obtained from Alomone Labs (Israel). A 10 mM stock solution of thapsigargin in 100% DMSO was diluted with DMEM complete media to a 40 μM, 4X solution to provide the 4X thapsigargin stimulus for calcium mobilization. The Kv1.3 inhibitor peptide peptide ShK (Stichodacytla helianthus toxin, Cat# H2358) and the BKCaI inhibitor peptide IbTx (Iberiotoxin, Cat# H9940) were purchased from Bachem Biosciences, whereas the Kv1.1 inhibitor peptide DTX-k (Dendrotoxin-K) was from Alomone Labs (Israel). The CHO-derived Fc-LI 0-ShK[2-35] peptibody inhibitor of Kv1.3 was obtained as described in Example 4 and Example 39. The calcineurin inhibitor cyclosporin A was obtained from the Amgen sample bank, but is also available commercially from a variety of vendors. The ion channel inhibitors ShK, IbTx or DTK-k were diluted into DMEM complete media to 4X of the final concentration desired (final = 50 or 100 nM). The calcineurin inhibitor cyclosporin A was also diluted into DMEM complete media to 4X final concentration (final = 10 μM). To appropriate wells of a 96-well Falcon 3075 flat-bottom microtiter plate, 50 μl of either DMEM complete media or the 4X inhibitor solutions
were added. Then, 100 μl of human whole blood was added and the plate was incubated for 1 hour at 370C, 5% CO2. After one hour, the plate was removed and 50 μl of the 4X thapsigargin stimulus (40 μM) was added to all wells of the plate containing inhibitor. To some wells containing no inhibitor but just DMEM complete media, thapsigargin was also added whereas others wells with just DMEM complete media had an additional 50 μl of DMEM complete media added. The wells with no inhibitor and no thapsigargin stimulus represented the untreated "Low" control. The wells with no inhibitor but which received thapsigargin stimulus represented the control for maximum stimulation or "High" control. Plates were placed back at 370C, 5% CO2 for 24 hours. After 24 hours, plates were removed and wells were process for FACS analysis. Cells were removed from the wells and washed in staining buffer (phosphate buffered saline containing 2% heat-inactivated fetal calf serum). Red blood cells were lysed using BD FACS Lysing Solution containing 1.5% formaldehyde (BD Biosciences) as directed by the manufacturer. Cells were distributed at a concentration of 1 million cells per 100 microliters of staining buffer per tube. Cells were first stained with 1 microliter of biotin-labeled anti-human CD4, washed, then stained simultaneously 1 microliter each of streptavidin-APC, FITC-labeled anti-human CD45RA, and phycoerythrin (PE)-labeled anti-human CD25 (IL-2Ra) or PE-labeled anti-human CD40L Cells were washed with staining buffer between antibody addition steps. All antibodies were obtained from BD Biosciences (San Diego, CA). Twenty to fifty thousand live events were collected for each sample on a Becton Dickinson FACSCaliber (Mountain View, CA) flow cytometer and analyzed using FlowJo software (Tree Star Inc., San Carlos, CA). Dead cells, monocytes, and granulocytes were excluded from the analysis on the basis of forward and side scatter properties.
Figure 64 and Figure 67 demonstrate that Kv1.3 inhibitors ShK and Fc-LI 0-ShK[2-35] potently blocked IL-2 secretion in human whole blood, in addition to suppressing activation of the IL-2R on CD4+ T cells. The Kv1.3 inhibitor Fc-LI 0-ShK[2-35] was more than 200 times more potent in blocking IL-2 production in human whole blood than cyclosporine A (Figure 64) as reflected by the IC50. Figure 65 shows that Kv1.3 inhibitors also potently blocked secretion of IFNg in human whole blood, and Figure 66 demonstrates that upregulation of CD40L on T cells was additionally blocked. The data in Figures 64-67 show that the Fc-L10-ShK[2-35] molecule was stable in whole blood at 370C for up to 48 hours, providing potent blockade of inflammatory responses. Toxin peptide therapeutic agents that target Kv1.3 and have prolonged half-life, are sought to provide sustained blockade of these responses in vivo over time. In contrast, despite the fact the Kv1.3 inhibitor peptide ShK also showed potent blockade in whole blood, the ShK peptide has a short (-20 min) half-life in vivo (C. Beeton et al. (2001) Proc. Natl. Acad. Sci. 98, 13942), and
cannot, therefore, provide prolonged blockade. Whole blood represents a physiologically relevant assay to predict the response in animals. The whole blood assays described here can also be used as a pharmacodynamic (PD) assay to measure target coverage and drug exposure following dosing of patients. These human whole blood data support the therapeutic usefulness of the compositions of the present invention for treatment of a variety immune disorders, such as multiple sclerosis, type 1 diabetes, psoriasis, inflammatory bowel disease, contact-mediated dermatitis, rheumatoid arthritis, psoriatic arthritis, asthma, allergy, restinosis, systemic sclerosis, fibrosis, scleroderma, glomerulonephritis, Sjogren syndrome, inflammatory bone resorption, transplant rejection, graft-versus-host disease, and lupus.
Example 47 PEGylated Peptibodies
By way of example, PEGylated peptibodies of the present invention were made by the following method. CHO-expressed FcL10-OsK1(19.2 mg; MW 30,371 Da, 0.63 micromole) in 19.2 ml A5S, 2OmM NaBH3CN, pH 5, was treated with 38 mg PEG aldehyde (MW 20 kDa; 3x, Lot 104086). The sealed reaction mixture was stirred in a cold room overnight. The extent of the protein modification during the course of the reaction was monitored by SEC HPLC using a Superose 6 HR 10/30 column (Amersham Pharmacia Biotech) eluted with a 0.05 M phosphate buffer, 0.5 M NaCI1 pH 7.0 at 0.4 ml/min. The reaction mixture was dialyzed with A5S, pH 5 overnight. The dialyzed material was then loaded onto an SP HP FPLC column (16/10) in A5S pH 5 and eluted with a 1 M NaCI gradient. The collected fractions were analyzed by SEC HPLC, pooled into 3 pools, exchanged into DPBS, concentrated and submitted for functional testing (Table 34).
In another example, FcL10-ShK1 (16.5 mg; MW 30,065 Da, 0.55 micro mole) in 16.5 ml A5S, 2OmM NaBH3CN, pH 5 was treated with 44 mg PEG aldehyde (MW 20 kDa; 4x, Lot 104086).
The sealed reaction mixture was stirred in a cold room overnight. The extent of the protein modification during the course of the reaction was monitored by SEC HPLC using a Superose 6 HR 10/30 column (Amersham Pharmacia Biotech) eluted with a 0.05 M phosphate buffer, 0.5 M NaCI, pH 7.0 at 0.4 ml/min. The reaction mixture was dialyzed with A5S, pH 5 overnight. The dialyzed material was loaded onto an SP HP FPLC column (16/10) in A5S pH 5 and was eluted with a 1 M NaCI gradient. The collected fractions were analyzed by SEC HPLC, pooled into 3 pools, exchanged into DPBS, concentrated and submitted for functional testing (Table 34).
The data in Table 34 demonstrate potency of the PEGylated peptibody molecules as Kv1.3 inhibitors.
Table 34 shows determinations of IC50 made by whole cell patch clamp electrophysiology with HEK 293 as described in Example 36 herein above. The sustained IC50 was derived from the current 400 msecs after voltage ramp from -8OmV to +30 mV. Pool #2 samples comprised di- PEGylated peptibodies and Pool #3 samples comprised mono-PEGylated peptibodies.
Example 48
PEGylated Toxin Peptides
Shk and Osk-1 PEGylation, purification and analysis. Synthetic Shk or OSK1-1 toxin peptides were selectively PEGylated by reductive alkylation at their N-termini. Conjugation was achieved, with either Shk or OSK-1 toxin peptides, at 2 mg/ml in 5OmM NaH2PO-I1 pH 4.5 reaction buffer containing 2OmM sodium cyanoborohydride and a 2 molar excess of 20 kDa monomethoxy- PEG-aldehyde (Nektar Therapeutics, Huntsville, AL). Conjugation reactions were stirred overnight at room temperature, and their progress was monitored by RP-HPLC. Completed reactions were quenched by 4-fold dilution with 2OmM NaOAc, pH 4, adjusted to pH 3.5 and chilled to 4°C. The PEG-peptides were then purified chromatographically at 40C; using SP Sepharose HP columns (GE Healthcare, Piscataway, NJ) eluted with linear 0-1 M NaCI gradients in 2OmM NaOAc, pH 4.0. (Figure 68A and Figure 68B) Eluted peak fractions were analyzed by SDS-PAGE and RP-HPLC and pooling determined by purity >97%. Principle contaminants observed were di-PEGylated toxin peptide and unmodified toxin peptide. Selected pools were concentrated to 2-5 mg/ml by centrifugal filtration against 3kDa MWCO membranes and dialyzed into 1OmM NaOAc1 pH 4 with 5% sorbitol. Dialyzed pools were then sterile filtered through 0.2 micron filters and purity determined to be >97% by SDS-PAGE and RP-HPLC (Figure 69A and Figure 69B). Reverse- phase HPLC was performed on an Agilent 1100 model HPLC running a Zorbax 5μm 300SB-C84.6
x 50 mm column (Phenomenex) in 0.1% TFA/H2O at 1 ml/min and column temperature maintained at 40°C. Samples of PEG-peptide (20 μg) were injected and eluted in a linear 6-60% gradient while monitoring wavelengths 215 nm and 280 nm.
Electrophysiology performed by patch clamp on whole cells (see, Example 36) yielded a peak IC50 of 1.285 nM for PEG-OSK1 and 0.169 nM for PEG-ShK[I -35] (Figure 74), in a concentration dependent block of the outward potassium current recorded from HEK293 cells stably expressing human Kv1.3 channel. The purified PEG-ShK[I -35] molecule, also referred to as "2OK PEG-ShK[I -35]" and "PEG-ShK", had a much longer half-life in vivo than the small ShK peptide (Figure 59 and Figure 60). PEG-ShK[I -35] suppressed severe autoimmune encephalomyelitis in rats (Example 45, Figures 61 - 63) and showed greater efficacy than the small native ShK peptide.
PEG conjugates of OSK1 peptide analogs were also generated and tested for activity in blocking T cell inflammation in the human whole blood assay (Example 46). As shown in Table 43, OSK1[Ala12], OSK1[Ala29], OSK1[1Nal11] and OSK1[Ala29, 1Nal34] analogs containing an N- terminal 2OK PEG conjugate, all provided potent blockade of the whole blood cytokine response in this assay. The 2OK PEG-ShK was also highly active (Table 43).
Example 49 Fc loop insertions of ShK and OSK1 toxin peptides As exemplified in Figure 70, Figure 71 , Figure 72, and Figure 73, disulphide-constrained toxin peptides were inserted into the human IgGI Fc-loop domain, defined as the sequence D137E138L139T140K141 , according to the method published in Example 1 in Gegg et al., Modified Fc molecules, WO 2006/036834 A2 [PCT/US2005/034273]). Exemplary FcLoop-L2-OsK1-L2, FcLoop-L2-ShK-L2, FcLoop-L2-ShK-L4, and FcLoop-L4-OsK1-L2 were made having three linked domains. These were collected, purified and submitted for functional testing.
The peptide insertion for these examples was between Fc residues Leui39 and Thr^o and included 2-4 GIy residues as linkers flanking either side of the inserted peptide. However, alternate insertion sites for the human IgGI Fc sequence, or different linkers, are also useful in the practice of the present invention, as is known in the art, e.g., as described in Example 13 of Gegg et al., Modified Fc molecules, WO 2006/036834 A2 [PCT/US2005/034273]).
Purified FcLoop OSK1 and FcLoop ShK1 molecules were tested in the whole blood assay of inflammation (see, Example 46). FcLoop-L2-OsK1-L2, FcLoop-L4-OsK1-L4 and FcLoop-
L2-ShK-L2 toxin conjugates all provided potent blockade of the whole blood cytokine response in this assay, with IC50 values in the pM range (Table 43).
Example 50 Purification of ShK(2-35)-L-Fc from E. coli
Frozen, E. coli paste (117 g), obtained as described in Example 16 herein above, was combined with 1200 ml of room temperature 50 mM tris HCI, 5 mM EDTA, pH 7.5 and was brought to about 0.1 mg/ml hen egg white lysozyme. The suspended paste was passed through a chilled microfluidizer twice at 12,000 PSI. The cell lysate was then centrifuged at 17,700 g for 30 min at 4 0C. The pellet was then resuspended in 1200 ml 1% deoxycholic acid using a tissue grinder and then centrifuged at 17,700 g for 30 min at 4 0C. The pellet was then resuspended in 1200 ml water using a tissue grinder and then centrifuged at 17,700 g for 30 min at 4 0C. 6.4 g of the pellet (total 14.2g) was then dissolved in 128 ml 8 M guanidine HCI, 50 mM tris HCI1 pH 8.0. 120 ml of the pellet solution was then incubated with 0.67 ml of 1 M DTT for 60 min at 37°c The reduced material was transferred to 5500 ml of the refolding buffer (3 M urea, 50 mM tris, 160 mM arginine HCI, 2.5 mM EDTA, 2.5 mM cystamine HCI, 4 mM cysteine, pH 9.5) at 2 ml/min , 4 0C with vigorous stirring. The stirring rate was then slowed and the incubation was continued for 3 days at 40C.
The refold was diluted with 5.5L of water, and the pH was adjusted to 8.0 using acetic acid, then the solution was filtered through a 0.22 μm cellulose acetate filter and loaded on to a 35 ml Amersham Q Sepharose-FF (2.6 cm I. D.) column at 10 ml/min in Q-Buffer A (20 mM Tris, pH 8.5) at 8 0C with an inline 35 ml Amersham Mab Select column (2.6 cm I. D.). After loading, the Q Sepharose column was removed from the circuit, and the remaining chromatography was carried out on the Mab Select column. The column was washed with several column volumes of Q-Buffer A, followed by elution using a step to 100 mM glycine pH 3.0. The fractions containing the desired product immediately loaded on to a 5.0 ml Amersham SP-Sepharose HP column at 5.0 ml/min in S-Buffer A (10 mM NaH2PO4, pH 7.0) at 8 0C. The column was then washed with several column volumes of S-Buffer A followed by a linear gradient from 5% to 60% S-Buffer B (IO mM NaH2PO4, 1 M NaCI, pH 7.0) followed by a step to 100% S-Buffer B. Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE. The fractions containing the bulk of the desired product were pooled and then applied to a 50 ml MEP Hypercel column (2.6 cm I. D.) at 10 ml/min in MEP Buffer A (20 mM tris, 200 mM NaCI1 pH 8.0) at 80C. Column was eluted with a linear gradient from 5% to 50% MEP Buffer B(50 mM sodium citrate pH 4.0) followed by a step to
100% MEP Buffer B. Fractions were then analyzed using a Coomassie brilliant blue stained tris- glycine 4-20% SDS-PAGE1 and the fractions containing the bulk of the desired product were pooled.
The MEP-pool was then concentrated to about 10 ml using a Pall Jumbo-Sep with a 10 kDa membrane. A spectral scan was then conducted on 50 μl of the combined pool diluted in 700 μl PBS using a Hewlett Packard 8453 spectrophotometer (Figure 76A). Then concentration of the material was determined to be 3.7 mg/ml using a calculated molecular mass of 30,253 and extinction coefficient of 36,900 W cm 1. The purity of the material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 76B). The macromolecular state of the product was then determined using size exclusion chromatography on 70 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 76C). The product was then subject to mass spectral analysis by chromatographing approximately 4 μg of the sample through a RP-HPLC column (Vydac C4, 1 x 150 mm). Solvent A was 0.1 % trifluoroacetic acid in water and solvent B was 0.1 % trifluoroacetic acid in 90 % acetonitrile, 10 % water. The column was pre-equilibrated in 10 % solvent B at a flow rate of 80 μl per min. The protein was eluted using a linear gradient of 10 % to 90% solvent B over 30 min. Part of the effluent was directed into a LCQ ion trap mass spectrometer. The mass spectrum was deconvoluted using the Bioworks software provided by the mass spectrometer manufacturer. (Figure 76D). The product was filtered through a 0.22 μm cellulose acetate filter and then stored at -80 0C.
In Table 35, IC50 data for the purified E. coli-derived ShK[2-35]-L-Fc are compared to some other embodiments of the inventive composition of matter.
Table 35. E.coli-derived recombinant Fc-L-ShK[I -35], Fc-L-ShK[2-35], Fc-L-OSKI , Shk[1-35]-L-
Fc and ShK[2-35]-L-Fc peptibodies containing Fc at either the N-terminus or C-terminus show potent blockade of human Kv1.3. The activity of the CHO-derived Fc-LI 0-ShK[1 -35] R1 Q mutant is also shown. Whole cell patch clamp electrophysiology (WCVC), by methods described in Example 36, was performed using HEK293 / Kv1.3 cells and the IC50 shown is the average from dose-response curves from 3 or more cells. IonWorks™ (IWQ) planar patch clamp electrophysiology by methods described in Example 44 was on CHO / Kv1.3 cells and the average IC50 is shown. The inventive molecules were obtained by methods as described in the indicated Example: E.coli-derived Fc-L-ShK[I -35] (Example 3 and Example 38), E.coli-derived Fc-L-ShK[2- 35] (Example 4 and Example 39), E.coli Fc-L-OSKI (Example 10 and Example 40), ShK[I -35]-L- Fc (Example 15 and Example 51 ), and ShK[2-35]-L-Fc (Example 16 and this Example 50). CHO- derived Fc-LI 0-ShK[1 -35] R1Q molecule was generated using methods similar to those described for CHO-derived Fc-LI 0-ShK[1 -35].

Example 51 Purification of Met-ShK(1-35)-Fc from E. coli
Frozen, E. coli paste (65 g), obtained as described in Example 15 herein above was combined with 660 ml of room temperature 50 mM tris HCI1 5 mM EDTA, pH 7.5 and was brought to about 0.1 mg/ml hen egg white lysozyme. The suspended paste was passed through a chilled microfluidizer twice at 12,000 PSI. The cell lysate was then centrifuged at 17,700 g for 30 min at 4 0C. The pellet was then resuspended in 660 ml 1% deoxycholic acid using a tissue grinder and then centrifuged at 17,700 g for 30 min at 4 0C. The pellet was then resuspended in 660 ml water using a tissue grinder and then centrifuged at 17,700 g for 30 min at 4 0C. 13 g of the pellet was then dissolved in 130 ml 8 M guanidine HCI, 50 mM tris HCI1 pH 8.0. 10 ml of the pellet solution was then incubated with 0.1 ml of 1 M DTT for 60 min at 37 °C The reduced material was transferred to 1000 ml of the refolding buffer (2 M urea, 50 mM tris, 160 mM arginine HCI1 2.5 mM EDTA, 1.2 mM cystamine HCI, 4 mM cysteine, pH 8.5) at 2 ml/min , 40C with vigorous stirring. The stirring rate was then slowed and the incubation was continued for 3 days at 40C.
The refold was diluted with 1 L of water, and filtered through a 0.22 μm cellulose acetate filter then loaded on to a 35 ml Amersham Q Sepharose-FF (2.6 cm I. D.) column at 10 ml/min in Q- Buffer A (20 mM Tris, pH 8.5) at 8 0C with an inline 35 ml Amersham Mab Select column (2.6 cm I. D.). After loading, the Q Sepharose column was removed from the circuit, and the remaining chromatography was carried out on the Mab Select column. The column was washed with several column volumes of Q-Buffer A1 followed by elution using a step to 100 mM glycine pH 3.0. The fractions containing the desired product immediately loaded on to a 5.0 ml Amersham SP- Sepharose HP column at 5.0 ml/min in S-Buffer A (20 mM NaH2PO4, pH 7.0) at 8 0C. The column was then washed with several column volumes of S-Buffer A followed by a linear gradient from 5% to 60% S-Buffer B (20 mM NaH2PO4, 1 M NaCI, pH 7.0) followed by a step to 100% S-Buffer B. Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS- PAGE. The fractions containing the bulk of the desired product were pooled.
The S-pool was then concentrated to about 10 ml using a Pall Jumbo-Sep with a 10 kDa membrane. A spectral scan was then conducted on 20 μl of the combined pool diluted in 700 μl PBS using a Hewlett Packard 8453 spectrophotometer (Figure 77A). Then concentration of the material was determined to be 3.1 mg/ml using a calculated molecular mass of 30,409 and extinction coefficient of 36,900 M 1 cm-1. The purity of the material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 77B). The macromolecular state of the product was then determined using size exclusion chromatography on 93 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 77C). The product was then subject to mass spectral analysis by MALDI mass spectrometry.
An aliquot of the sample was spotted with the MALDI matrix sinapinic acid on sample plate. A Voyager DE-RP time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse) was used to collect spectra. The positive ion/linear mode was used, with an accelerating voltage of 25 kV. Each spectrum was produced by accumulating data from ~ 200 laser shots (Figure 77D). External mass calibration was accomplished using purified proteins of known molecular masses.
The IC50 for blockade of human Kv1.3 by purified £.co//-derived Met-ShK(1-35)-Fc, also referred to as "ShK[I -35]-L-Fc", is shown in Table 35 herein above.
Example 52
Bacterial expression of OsKI-L-Fc inhibitor of Kv1,3
The methods to clone and express the peptibody in bacteria were as described in Example 3. The vector used was pAMG21amgR-pep-Fc and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of OsKI-L-Fc. Oligos used to form duplex are shown below:
GGGTGTTATCATCAACGTTAAATGCAAAATCTCCCGTCAGTGCCTGGAACCGTGCAAAAAAGCTGGTATGCGT //SEQ ID NO: 1347;
TTCGGTAAATGCATGAACGGTAAATGCCACTGCACCCCGAAATCTGGTGGTGGTGGTTCT //SEQ ID NO: 1348;
CACCAGAACCACCACCACCACCAGATTTCGGGGTGCAGTGGCATTTACCGTTCATGCATTTACCGAAACGCAT //SEQ ID NO: 1349;
ACCAGCTTTTTTGCACGGTTCCAGGCACTGACGGGAGATTTTGCATTTAACGTTGATGATAAC //SEQ ID NO: 1310;
The oligos shown above were used to form the duplex shown below:
GGGTGTTATCATCAACGTTAAATGCAAAATCTCCCGTCAGTGCCTGGAACCGTGCAAAAA 5 i + + + + + + 50
CAATAGTAGTTGCAATTTACGTTTTAGAGGGCAGTCACGGACCTTGGCACGTTTTT G V I I N V K C K I S R Q C L E P C K K - O AGCTGGTATGCGTTTCGGTAAATGCATGAACGGTAAATGCCACTGCACCCCGAAATCTGG
61 + + + + + + 120
TCGACCATACGCAAAGCCATTTACGTACTTGCCATTTACGGTGACGTGGGGCTTTAGACC
A G M R F G K C M N G K C H C T P K S G -5
TGGTGGTGGTTCT //SEQ ID NO: 1350 121 + 137
ACCACCACCAAGACCAC //SEQ ID NO: 1352 0 G G G S G - //SEQ ID NO:1351
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen. 5 Example 53
Bacterial expression of GIy-Sh Kd -35)-L-Fc inhibitor of Kv1.3
The methods to clone and express the peptibody in bacteria were as described in Example 3. The vector used was pAMG21amgR-pep-Fc and the oligos listed below were used to generate a duplex (see below) for cloning and expression in bacteria of Gly-ShK(1-35) -L-Fc. O Oligos used to form duplex are shown below:
GGGTCGTTCTTGTATTGATACTATTCCAAAATCTCGTTGTACTGCTTTTCAATGTAAACATTCTA TGAAATATCGTCTTTCTT //SEQIDNO:1313; 5 TTTGTCGTAAAACTTGTGGTACTTGTTCTGGTGGTGGTGGTTCT //SEQ ID NO:1314;
CACCAGAACCACCACCACCAGAACAAGTACCACAAGTTTTACGACAAAAAGAAAGACGATATTT CATAGAATGTTTACATTGA //SEQ ID NO:1353; 0 AAAGCAGTACAACGAGATTTTGGAATAGTATCAATACAAGAACG //SEQ ID NO: 1354 The oligos shown above were used to form the duplex shown below:
GGGTCGTTCTTGTATTGATACTATTCCAAAATCTCGTTGTACTGCTTTTCAATGTAAACA 5 1 + + + + + + 60
GCAAGAACATAACTATGATAAGGTTTTAGAGCAACATGACGAAAAGTTACATTTGT
G R S C I D T I P K S R C T A F Q C K H -
TTCTATGAAATATCGTCTTTCTTTTTGTCGTAAAACTTGTGGTACTTGTTCTGGTGGTGG 61 + + + + + + 120
AAGATACTTTATAGCAGAAAGAAAAACAGCATTTTGAACACCATGAACAAGACCACCACC S M K Y R L S F C R K T C G T C S G G G -
TGGTTCT //SEQ ID NO: 1355 121 +- 131
ACCAAGACCAC //SEQ ID NO: 1357
G S G - //SEQ ID NO: 1356
Bacterial expression of the peptibody was as described in Example 3 and paste was stored frozen.
Example 54 Bacterial expression of CH2-0SK1 inhibitor of Kv1.3
The methods to clone and express the fusion of a CH2 domain of an Fc with 0SK1 in bacteria were generally as described in Example 3. The vector used was pAMG21.G2.H6.G3.CH2.(G4S)2.OSK. Briefly, the pAMG21 vector was modified to remove the multi-cloning site's BamHI. This allowed the BamHI in front of the OSK as a site to swap out different sequences for fusion with the OSK. The sequence upstream of the 0SK1 coding sequence was ligated between the Ndel and BamHI sites. The sequence of the entire vector, including the insert was the following: gtcgtcaacgaccccccattcaagaacagcaagcagcattgagaactttggaatccagtccctcttccacctgctgaccggatcagcagt ccccggaacatcgtagctgacgccttcgcgttgctcagttgtccaaccccggaaacgggaaaaagcaagttttccccgctcccggcgtttc aataactgaaaaccatactatttcacagtttaaatcacattaaacgacagtaatccccgttgatttgtgcgccaacacagatcttcgtcacaat tctcaagtcgctgatttcaaaaaactgtagtatcctctgcgaaacgatccctgtttgagtattgaggaggcgagatgtcgcagacagaaaat gcagtgacttcctcattgagtcaaaagcggtttgtgcgcagaggtaagcctatgactgactctgagaaacaaatggccgttgttgcaagaa aacgtcttacacacaaagagataaaagtttttgtcaaaaatcctctgaaggatctcatggttgagtactgcgagagagaggggataacac aggctcagttcgttgagaaaatcatcaaagatgaactgcaaagactggatatactaaagtaaagactttactttgtggcgtagcatgctaga ttactgatcgtttaaggaattttgtggctggccacgccgtaaggtggcaaggaactggttctgatgtggatttacaggagccagaaaagcaa aaaccccgataatcttcttcaacttttgcgagtacgaaaagattaccggggcccacttaaaccgtatagccaacaattcagctatgcgggg agtatagttatatgcccggaaaagttcaagacttctttctgtgctcgctccttctgcgcattgtaagtgcaggatggtgtgactgatcttcaccaa acgtattaccgccaggtaaagaacccgaatccggtgtttacaccccgtgaaggtgcaggaacgctgaagttctgcgaaaaactgatgga aaaggcggtgggcttcacttcccgttttgatttcgccattcatgtggcgcacgcccgttcgcgtgatctgcgtcgccgtatgccaccagtgctg cgtcgtcgggctattgatgcgctcttgcaggggctgtgtttccactatgacccgctggccaaccgcgtccagtgctccatcaccacgctggcc attgagtgcggactggcgacggagtctgctgccggaaaactctccatcacccgtgccacccgtgccctgacgttcctgtcagagctggga ctgattacctaccagacggaatatgacccgcttatcgggtgctacattccgaccgatatcacgttcacatctgcactgtttgctgccctcgatgt atcagaggaggcagtggccgccgcgcgccgcagccgtgtggtatgggaaaacaaacaacgcaaaaagcaggggctggataccctg ggcatggatgaactgatagcgaaagcctggcgttttgttcgtgagcgttttcgcagttatcagacagagcttaagtcccgtggaataaagcg tgcccgtgcgcgtcgtgatgcggacagggaacgtcaggatattgtcaccctggtgaaacggcagctgacgcgcgaaatcgcggaagg gcgcttcactgccaatcgtgaggcggtaaaacgcgaagttgagcgtcgtgtgaaggagcgcatgattctgtcacgtaaccgtaattacag ccggctggccacagcttccccctgaaagtgacctcctctgaataatccggcctgcgccggaggcttccgcacgtctgaagcccgacagc gcacaaaaaatcagcaccacatacaaaaaacaacctcatcatccagcttctggtgcatccggccccccctgttttcgatacaaaacacg cctcacagacggggaattttgcttatccacattaaactgcaagggacttccccataaggttacaaccgttcatgtcataaagcgccatccgc
cagcgttacagggtgcaatgtatcttttaaacacctgtttatatctcctttaaactacttaattacattcatttaaaaagaaaacctattcactgcct gtccttggacagacagatatgcacctcccaccgcaagcggcgggcccctaccggagccgctttagttacaacactcagacacaaccac cagaaaaaccccggtccagcgcagaactgaaaccacaaagcccctccctcataactgaaaagcggccccgccccggtccgaaggg ccggaacagagtcgcttttaattatgaatgttgtaactacttcatcatcgctgtcagtcttctcgctggaagttctcagtacacgctcgtaagcgg ccctgacggcccgctaacgcggagatacgccccgacttcgggtaaaccctcgtcgggaccactccgaccgcgcacagaagctctctca tggctgaaagcgggtatggtctggcagggctggggatgggtaaggtgaaatctatcaatcagtaccggcttacgccgggcttcggcggttt tactcctgtttcatatatgaaacaacaggtcaccgccttccatgccgctgatgcggcatatcctggtaacgatatctgaattgttatacatgtgta tatacgtggtaatgacaaaaataggacaagttaaaaatttacaggcgatgcaatgattcaaacacgtaatcaatatcgggggtgggcgaa gaactccagcatgagatccccgcgctggaggatcatccagccggcgtcccggaaaacgattccgaagcccaacctttcatagaaggcg gcggtggaatcgaaatctcgtgatggcaggttgggcgtcgcttggtcggtcatttcgaaccccagagtcccgctcagaagaactcgtcaag aaggcgatagaaggcgatgcgctgcgaatcgggagcggcgataccgtaaagcacgaggaagcggtcagcccattcgccgccaagct cttcagcaatatcacgggtagccaacgctatgtcctgatagcggtccgccacacccagccggccacagtcgatgaatccagaaaagcg gccattttccaccatgatattcggcaagcaggcatcgccatgagtcacgacgagatcctcgccgtcgggcatgcgcgccttgagcctggc gaacagttcggctggcgcgagcccctgatgctcttcgtccagatcatcctgatcgacaagaccggcttccatccgagtacgtgctcgctcg atgcgatgtttcgcttggtggtcgaatgggcaggtagccggatcaagcgtatgcagccgccgcattgcatcagccatgatggatactttctc ggcaggagcaaggtgagatgacaggagatcctgccccggcacttcgcccaatagcagccagtcccttcccgcttcagtgacaacgtcg agcacagctgcgcaaggaacgcccgtcgtggccagccacgatagccgcgctgcctcgtcctgcaattcattcaggacaccggacaggt cggtcttgacaaaaagaaccgggcgcccctgcgctgacagccggaacacggcggcatcagagcagccgattgtctgttgtgcccagtc atagccgaatagcctctccacccaagcggccggagaacctgcgtgcaatccatcttgttcaatcatgcgaaacgatcctcatcctgtctctt g atctg atcttg atcccctg eg ccatcagatccttggcggcaagaaag ccatccagtttactttgcagggcttcccaaccttaccagagg gc gccccagctggcaattccggttcgcttgctgtccataaaaccgcccagtctagctatcgccatgtaagcccactgcaagctacctgctttctct ttgcgcttgcgttttcccttgtccagatagcccagtagctgacattcatccggggtcagcaccgtttctgcggactggctttctacgtgttccgctt cctttagcagcccttgcgccctgagtgcttgcggcagcgtgaagctacatatatgtgatccgggcaaatcgctgaatattccttttgtctccga ccatcaggcacctgagtcgctgtctttttcgtgacattcagttcgctgcgctcacggctctggcagtgaatgggggtaaatggcactacaggc gccttttatggattcatgcaaggaaactacccataatacaagaaaagcccgtcacgggcttctcagggcgttttatggcgggtctgctatgtg gtgctatctgactttttgctgttcagcagttcctgccctctgattttccagtctgaccacttcggattatcccgtgacaggtcattcagactggctaa tgcacccagtaaggcagcggtatcatcaacaggcttacccgtcttactgtcgaagacgtgcgtaacgtatgcatggtctccccatgcgaga gtagggaactgccaggcatcaaataaaacgaaaggctcagtcgaaagactgggcctttcgttttatctgttgtttgtcggtgaacgctctcct gagtaggacaaatccgccgggagcggatttgaacgttgcgaagcaacggcccggagggtggcgggcaggacgcccgccataaact gccaggcatcaaattaagcagaaggccatcctgacggatggcctttttgcgtttctacaaactcttttgtttatttttctaaatacattcaaatatg gacgtcgtacttaacttttaaagtatgggcaatcaattgctcctgttaaaattgctttagaaatactttggcagcggtttgttgtattgagtttcatttg cgcattggttaaatggaaagtgaccgtgcgcttactacagcctaatatttttgaaatatcccaagagctttttccttcgcatgcccacgctaaac attctttttctcttttggttaaatcgttgtttgatttattatttgctatatttatttttcgataattatcaactagagaaggaacaattaatggtatgttcatac acgcatgtaaaaataaactatctatatagttgtctttctctgaatgtgcaaaactaagcattccgaagccattattagcagtatgaatagggaa actaaacccagtgataagacctgatgatttcgcttctttaattacatttggagattttttatttacagcattgttttcaaatatattccaattaatcggt gaatgattggagttagaataatctactataggatcatattttattaaattagcgtcatcataatattgcctccattttttagggtaattatccagaatt gaaatatcagatttaaccatagaatgaggataaatgatcgcgagtaaataatattcacaatgtaccattttagtcatatcagataagcattgat taatatcattattgcttctacaggctttaattttattaattattctgtaagtgtcgtcggcatttatgtctttcatacccatctctttatccttacctattgtttg tcgcaagttttgcgtgttatatatcattaaaacggtaatagattgacatttgattctaataaattggatttttgtcacactattatatcgcttgaaatac aattgtttaacataagtacctgtaggatcgtacaggtttacgcaagaaaatggtttgttatagtcgattaatcgatttgattctagatttgttttaact aattaaaggaggaataacatatgggcggccatcatcatcatcatcatggcgggggaccgtcagttttcctcttccccccaaaacccaagg acaccctcatgatctcccggacccctgaggtcacatgcgtggtggtggacgtgagccacgaagaccctgaggtcaagttcaactggtac gtggacggcgtggaggtgcataatgccaagacaaagccgcgggaggagcagtacaacagcacgtaccgtgtggtcagcgtcctcacc gtcctgcaccaggactggctgaatggcaaggagtacaagtgcaaggtctccaacaaagccctcccagcccccatcgagaaaaccatct ccggcggcggcggcagcggcggcggcggatccggtgttatcatcaacgttaaatgcaaaatctcccgtcagtgcctggaaccgtgcaa aaaagctggtatgcgtttcggtaaatgcatgaacggtaaatgccactgcaccccgaaataatgaattcgagctcactagtgtcgacctgca gggtaccatggaagcttactcgaagatccgcggaaagaagaagaagaagaagaaagcccgaaaggaagctgagttggctgctgcc accgctgagcaataactagcataaccccttggggcctctaaacgggtcttgaggggttttttgctgaaaggaggaaccgctcttcacgctctt cacgcggataaataagtaacgatccggtccagtaatgacctcagaactccatctggatttgttcagaacgctcggttgccgccgggcgttttt tattggtgagaatcgcagcaacttgtcgcgccaatcgagccatgtc//SEQ ID NO:4914.
The insert DNA sequence was the following: atgggcggccatcatcatcatcatcatggcgggggaccgtcagttttcctcttccccccaaaacccaaggacaccctcatgatctcccgga cccctgaggtcacatgcgtggtggtggacgtgagccacgaagaccctgaggtcaagttcaactggtacgtggacggcgtggaggtgcat aatgccaagacaaagccgcgggaggagcagtacaacagcacgtaccgtgtggtcagcgtcctcaccgtcctgcaccaggactggctg aatggcaaggagtacaagtgcaaggtctccaacaaagccctcccagcccccatcgagaaaaccatctccggcggcggcggcagcgg cggcggcggatccggtgttatcatcaacgttaaatgcaaaatctcccgtcagtgcctggaaccgtgcaaaaaagctggtatgcgtttcggt aaatgcatgaacggtaaatgccactgcaccccgaaa//SEQ ID NO:4915.
The amino acid sequence of the CH2-0SK1 fusion protein product was the following:
MGGHHHHHHGGGPSVFLFPPKPKDTLMISRTPEVTCWVDVSHEDPEVKFNWYVDGVEVHNAKT KPREEQYNSTYRWSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISGGGGSGGGGS GVIINVKCKISRQCLEPCKKAGMRFGKCMNGKCHCTPK//SEQ ID NO:4917.
SEQ ID NO:4917 includes the 0SK1 sequence
GVIINVKCKISRQCLEPCKKAGMRFGKCMNGKCHCTPK (SEQ ID NO:25).
Purification and refolding of CH2-0SK1 expressed in bacteria. Frozen, E. coli paste (13.8 g) was combined with 180 ml of room temperature 50 mM tris HCI1 5 mM EDTA1 pH 8.0 and was brought to about 0.1 mg/ml hen egg white lysozyme. The suspended paste was passed through a chilled microfluidizer twice at 12,000 PSI. The cell lysate was then centrifuged at 17,700 g for 50 min at 4 0C. The pellet was then resuspended in 90 ml 1 % deoxycholate using a tissue grinder and then centrifuged at 15,300 g for 40 min at 4 °C. The pellet was then resuspended in 90 ml water using a tissue grinder and then centrifuged at 15,300 g for 40 min at 4 0C. The pellet (3.2 g) was then dissolved in 64 ml 8 M guanidine HCI, 50 mM tris HCI, pH 8.0. The suspension was then incubated at room temperature (about 23 0C) for 30 min with gentle agitation followed by centrifugation at 15,300 g for 30 min at 4 0C. The supernatant (22 ml) was then reduced by adding
220 μl 1 M dithiothreitol and incubating at 37 0C for 30 minutes. The reduced suspension (20 ml) was transferred to 2000 ml of the refolding buffer (1 M urea, 50 mM ethanolamine, 160 mM arginine HCI, 0.02% NaN3, 1.2 mM cystamine HCI14 mM cysteine, pH 9.8) at 4 0C with vigorous stirring. The stirring rate was then slowed and the incubation was continued for approximately 2.5 days at 4 0C.
Ten milliliters of 500 mM imidazole was added to the refolding solution and the pH was adjusted to pH to 8.0 with 5 M acetic acid. The refold was then filtered through a 0.45 μm cellulose acetate filter with two pre-filters. This material was then loaded on to a 50 ml Qiagen Ni-NTA Superflow column (2.6 cm ID) in Ni-Buffer A (50 mM NaH2PO4, 300 mM NaCI1 pH 7.5) at 15 ml/min
13 0C. The column was then washed with 10 column volumes of Ni-Buffer A followed by 8% Ni- Buffer B (250 mM imidazole, 50 mM NaH2PO4, 300 mM NaCI, pH 7.5) at 25 ml/min. The column was then eluted with 60% Ni-Buffer B followed by 100% Ni-Buffer B at 10 ml/min. The peak fractions were collected and dialyzed against S-Buffer A (10 mM NaH2PO4, pH 7.1) The dialyzed sample was then loaded on to a 5 ml Amersham SP-HP HiTrap column at 5 ml/min in S-Buffer A at 13 0C. The column was then washed with several column volumes of S- Buffer A, followed by elution with a linear gradient from 0% to 60% S-Buffer B (10 mM NaH2PO4, 1 M NaCI, pH 7.1) followed by a step to 100% S-Buffer B at 1.5 ml/min 13 0C. Fractions were then analyzed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE, and the fractions containing the desired product were pooled based on these data. The pool was then concentrated to about 1.6 ml using a Pall Macrosep with a 10 kDa membrane at 4 0C. The concentrated sample was then filtered through a 0.22 μm cellulose acetate centrifugal filter.
A spectral scan was then conducted on 10 μl of the combined pool diluted in 150 μl water using a Hewlett Packard 8453 spectrophotometer (Figure 78). The concentration of the filtered material was determined to be 3.35 mg/ml using a calculated molecular mass of 17,373 g/mol and extinction coefficient of 17,460 M-1 cm 1. The purity of the filtered material was then assessed using a Coomassie brilliant blue stained tris-glycine 4-20% SDS-PAGE (Figure 79). The endotoxin level was then determined using a Charles River Laboratories Endosafe-PTS system (0.05 - 5 EU/ml sensitivity) using a 67-fold dilution of the sample in Charles Rivers Endotoxin Specific Buffer BG120 yielding a result of <1 EU/mg protein. The macromolecular state of the product was then determined using size exclusion chromatography on 50 μg of the product injected on to a Phenomenex BioSep SEC 3000 column (7.8 x 300 mm) in 50 mM NaH2PO4, 250 mM NaCI, pH 6.9 at 1 ml/min observing the absorbance at 280 nm (Figure 80). The product was then subject to mass spectral analysis by chromatographing approximately 4 μg of the sample through a RP- HPLC column (Vydac C4, 1 x 150 mm). Solvent A was 0.1 % trifluoroacetic acid in water and solvent B was 0.1 % trifluoroacetic acid in 90 % acetonitrile, 10 % water. The column was pre- equilibrated in 10 % solvent B at a flow rate of 80 μl per min. The protein was eluted using a linear gradient of 10 % to 90% solvent B over 30 min. Part of the effluent was directed into a LCQ ion trap mass spectrometer. The mass spectrum was deconvoluted using the Bioworks software provided by the mass spectrometer manufacturer. (Figure 81). The product was then stored at
-8O0C.
PEGylation of CH2-0SK1. The CH2-0SK1 fusion protein was diluted to 2 mg/ml in 50 mM sodium acetate, 1OmM sodium cyanoborohdride, pH 4.8 with a 4-fold molar excess of 2OkD methoxy-PEG- aldehyde (Nektar Therapeutics, Huntsville, AL). The reaction was allowed to proceed overnight (-18 hrs) at 4 0C. Upon completion, reaction was quenched with 4 volumes of 10 mM sodium acetate, 50 mM NaCI, pH 5, then loaded at 0.7 mg protein/ml resin to an SP Sepharose HP column (GE Healthcare, Piscataway, NJ) equilibrated in 10 mM sodium acetate, 50 mM NaCI, pH 5. The mono-PEGylated CH2-Osk fusion was eluted with a linear 50 mM - 1 M NaCI gradient (Figure 82). Peak fractions were evaluated by SDS-PAGE and the mono-PEG-CH2-OSK1 fractions pooled, concentrated and dialyzed into Dulbecco's Phosphate Buffered Saline. The final product was analyzed by SDS-PAGE (Figure 83).
As shown in Table 43, the purified CH2-OSK1 ("L2-6H-L3-CH2-L10-OsK1(1-38)") and PEGylated CH2-OSK1 ("20 k PEG-L2-6H-L3-CH2-L10-OSK1(1-38)") molecules were active in blocking inflammation in the human whole blood assay (See, Example 46).
Example 55
Bioactivitv of OSK1 peptide analogs
The activity of OSK1 peptide analogs in blocking human Kv1.3 versus human Kv1.1 current is shown in Figures 84 through 86 and Tables 37-41. Three electrophysiology techniques were used (See, e.g., Example 36 and Example 44). Whole cell patch clamp (Figure 84 & 85 and Table 41) represents a low throughput technique which is well established in the field and has been available for many years. We also used two new planar patch clamp techniques, PatchXpress and IonWorks Quattro, with improved throughput which facilitate assessment of potency and selectivity of novel OSK1 analogs described in this application. The PatchXpress technique is of moderate throughput and the novel OSK1[Ala-12] analog (SEQ ID No:1410) had similar Kv1.3 potency and selectivity over Kv1.1 to that observed by whole cell patch clamp (Figure
84 and Table 41). IonWorks Quattro represents a 384-well planar patch clamp electrophysiology system of high throughput. Using this IonWorks system, the novel OSK1 [Ala-29] analog (SEQ ID No: 1424) showed potent inhibition of the Kv1.3 current and improved selectivity over Kv1.1 (Figure 86). The OSK1 [Ala-29] analog (SEQ ID No:1424) showed similar Kv1.3 activity and selectivity over Kv1.1 by whole cell patch clamp electrophysiology (Figure 85) to that observed by IonWorks
(Figure 86). The Kv1.3 and Kv1.1 activities of Alanine, Arginine, Glutamic acid and
1 -Naphthylalanine analogs of OSK1 were determined by IonWorks electrophysiology and is reported in Tables 37 - 40. OSK1 peptide analogs identified by IonWorks to have good potency or
Kv1.3 selectivity, were tested further in whole cell patch clamp studies (see, Table 41). The Kv1.3 IC50 of the His34Ala analog of 0SK1 (SEQ ID No:1428) was 797 fold lower than its IC50 against Kv1.1 (Table 41), demonstrating that this analog is a highly selective Kv1.3 inhibitor. In this same assay, native 0SK1 (SEQ ID NO: 25) showed only slight Kv1.3 selectivity, with the Kv1.1 IC50 being only 5 fold higher than Kv1.3.
The novel 0SK1 peptide analogs described in this application which inhibit Kv1.3 are useful in the treatment of autoimmune disease and inflammation. Kv1.3 is expressed on T cells and Kv1.3 inhibitors suppress inflammation by these cells. As one measure of inflammation mediated by T cells, we examined the impact of 0SK1 analogs on IL-2 and IFN-g production in human whole blood following addition of a pro-inflammatory stimulus (Tables 36-40 and Table 42). "WB / IL-2" in these tables refers to the assay measuring IL-2 response of whole blood (see, Example 46), whereas "WB / IFNg" refers to the assay measuring IFNg response of whole blood (see, Example 46). The IC50 values listed in Tables 36 - 40 and 42, represent the average IC50 value determined from experiments done with two or more blood donors. The whole blood assay (see Example 46) allows for a combined measurement of the potency of the analogs in blocking inflammation and Kv1.3, as well as an assessment of the stability of the molecules in a complex biological fluid. Using this assay, several OSK1 analogs were examined and found to potently suppress inflammation (Figure 9OC & 90D1 Tables 36 - 40). Some of these analogs showed reduced activity in this whole blood assay, which may indicate that these residues play an important role in binding Kv1.3. Relative to the immunosuppressive agent cyclosporin A, Kv1.3 peptide inhibitors ShK-Ala22, OSK1-Ala29, and OSK1-Ala12 were several orders of magnitude more potent in blocking the cytokine response in human whole blood (Figure 90).
The solution NMR structure of OSK1 has been solved and is provided as pdb accession number "1SCO" in Entrez's Molecular Modeling Database (MMDB) [J. Chen et al. (2003) Nucleic Acids Res. 31 , 474-7]. Figure 89 shows space filling (Fig. 89A, 89B, 89D) and worm (Fig. 89C)
Cn3D rendering of the OSK1 structure. Light colored OSK1 amino acid residues Phe25, Gly26, Lys27, Met29 and Asn30 are shown in Figure 89B. Some analogs of these residues were found to significantly reduce Kv1.3 activity (Tables 37 - 40), implying that these residues may make important contacts with the Kv1.3 channel. The molecular structure shown in Figure 89A indicates these amino acids reside on a common surface of the OSK1 three-dimensional (3D) structure.
Figure 89D shows OSK1 residues (light shading) Ser11, Met29 and His34. These residues when converted to some amino acid analogs, provide improved Kv1.3 selectivity over Kv1.1 (Table 41). Although about 23 amino acids are between residues Ser11 and His34 in the contiguous
polypeptide chain, the structure shown in Figure 89D illustrates that in the 3D structure of the folded molecule these residues are relatively close to one another. Upon comparing Figures 89D and 89B, one can see that residues His34 and SeM 1 (Fig. 89D) are on the left and right side, respectively, and adjacent to the major Kv1.3 contact surface displayed in Figure 89B. It is 5 envisioned that molecular modeling can be used to identify 0SK1 analogs with improved Kv1.3 activity and selectivity, upon considering the Kv1.3 and Kv1.1 bioactivity information provided in Tables 37 through 42 and the solution NMR structure of 0SK1 described above. Figure 89C shows a worm rendering of the 0SK1 structure with secondary structure elements (beta strands and alpha helices) depicted. The primary amino acid sequence of 0SK1 is provided in Figure 89E 0 and amino acid residues comprising the beta strands & alpha helix are underlined. Wiggly lines in Figure 89C indicate amino acid residues between or beyond these secondary structure elements, whereas straight lines depict the three disulfide bridges in 0SK1. The first beta strand (β1) shown in Figure 89C and 89E contains no disulfide bridges to link it covalently to other secondary structure elements of the 0SK1 molecule, unlike beta strand 3 (β3) that has two disulfide bridges5 with the alpha helix (α1). As shown in Table 42, 0SK1 analogs without beta strand 1 (labeled "des 1-7") still retain activity in blocking inflammation (see SEQ ID No: 4989 of Table 42) suggesting that this region of 0SK1 is not essential for the molecules Kv1.3 bioactivity.
0SK1 analogs containing multiple amino acid changes were generated and their activity in the human whole blood assay of inflammation is provided in Table 42. Several analogs retain O high potency in this assay despite as many as 12 amino acid changes. Based on the improved Kv1.3 selectivity of analogs with single amino acid changes, anologs with multiple amino acid changes may result in additional improvements in selectivity. It is also envisioned that analogs with multiple amino acid changes may have improved activity or stability in vivo, alone or in the context of a peptide conjugate to a half-life prolonging moiety. 5 Kv1.3 peptide toxins conjugated to half-life prolonging moieties are provided within this application. The bioactivity of several toxin conjugates is described in Table 43. 0SK1 analogs with a N-terminal half-life prolonging 2OK PEG moiety (see Example 48) were found to provide potent suppression of the whole blood IL-2 ("WB / IL-2") and IFNg ("WB / IFNg") response (Table 43). The 2OK PEG-ShK conjugate, shown earlier to have prolonged half-life in vivo (see Examples0 44 and 48), was also highly active in this whole blood assay. The FcLoop-OSK1 conjugates (see Example 49) were highly active in blocking inflammation (Table 43), and the CH2-OSK1 or PEG- CH2-OSK1 conjugates (see Example 54) provided modest blockade of the whole blood cytokine response (Table 43). The IL-2 and IFNg cytokine response measured in this whole blood assay
results from T cell activation. Since this cytokine response is Kv1.3 dependent and potently blocked by the Kv1.3 peptide and peptide-conjugate inhibitors described herein, these whole blood studies illustrate the therapeutic utility of these molecules in treatment of immune disorders.
Table 36. Activity of 0SK1 analogs in blocking thapsigargin-induced IL-2 and IFNg production in 50% human whole blood as described in Example 46.
Table 37.0SK1 Alanine Analogs.
Table 38. 0SK1 Arginine Analogs.
Table 39. 0SK1 Glutamic Acid Analogs.
Table 40. OSK1 Naphthylalanine Analogs.
Table 41. 0SK1 Analogues with Improved Selectivity at Kv1.3 over Kv1.1 (whole cell patch clamp ePhys).
FfetchXpπessdarta
Table 42. 0SK1 Analogues with Multiple Amino Acid Substitutions.
Table 43. Bioactivity of 0SK1 and 0SK1 peptide analog conjugates with half-life-extending moieties as indicated. Fcloop structures G2-OSK1-G2 (SEQ ID NO:976), G4-OSK1-G2 (SEQ ID NO:979), and G2-ShK-G2 (SEQ ID NO:977) are described in Example 49, and CH2-L10- OSK1(1-38) SEQ ID NO:4917 is described in Example 54.
Example 56 Design and expression of monovalent Fc-fusion molecules There may be pharmacokinetic or other reasons, in some cases, to prefer a monovalent dimeric Fc-toxin peptide fusion (as represented schematically in Figure 2B) to a ("bivalent") dimer (as represented schematically in Figure 2C). However, conventional Fc fusion constructs typically result in a mixture containing predominantly dimeric molecules, both monovalent and bivalent. Monovalent dimeric Fc-toxin peptide fusions (or "peptibodies"), including monovalent dimeric Fc- OSK1 peptide analog fusions and Fc-ShK peptide analog fusions, can be isolated from conditioned media which also contains bivalent dimeric Fc-toxin peptide, and dimeric Fc lacking the toxin peptide fusion. Separation of all three species can be accomplished using ion exchange chromatography, for example, as described in Examples 1, 2, and 41 herein.
A number of other exemplary ways that a monovalent dimeric Fc-toxin peptide fusion can be produced with greater efficiency are provided here, including for the production of monovalent dimeric Fc-OSKI peptide analog fusions:
(1) Co-expressing equal amounts of Fc and Fc-toxin peptide in the same cells (e.g. mammalian cells). With the appropriate design, a mixture of bivalent dimeric Fc-toxin peptide fusion, monovalent dimeric Fc-toxin peptide fusion and dimeric Fc will be produced and released into the conditioned media. The monovalent dimeric Fc-toxin peptide can be purified from the mixture using conventional purification methods, for example, methods described in Examples 1, 2, and 41 herein.
(2) Engineering and recombinantly expressing in mammalian cells a single polypeptide construct represented by the following schematic:
Signal peptide - Fc - furin cleavage site - linker -furin cleavage site --Fc-toxin peptide
Furin cleavage occurs as the molecule travels through the endoplasmic reticulum and the intra- molecular Fc pairing (resulting in monovalent dimeric Fc-toxin peptide fusion) can occur preferentially to intermolecular Fc pairing (resulting in dimeric Fc-toxin peptide being expressed into conditioned medium; Figure 87A-B).
By way of example of method (2) above, a DNA construct was produced for recombinant expression in mammalian cells of the following schematic polypeptide construct:
Signal peptide - Fc - furin cleavage site - linker - furin cleavage site - Fc-ShK(2-35)
The DNA construct had the following nucleotide coding sequence:
atggaatggagctgggtctttctcttcttcctgtcagtaacgactggtgtccactccgacaaaactcacacatgcccaccgtgcc cagcacctgaactcctggggggaccgtcagtcttcctcttccccccaaaacccaaggacaccctcatgatctcccggacccc tgaggtcacatgcgtggtggtggacgtgagccacgaagaccctgaggtcaagttcaactggtacgtggacggcgtggaggt gcataatgccaagacaaagccgcgggaggagcagtacaacagcacgtaccgtgtggtcagcgtcctcaccgtcctgcac caggactggctgaatggcaaggagtacaagtgcaaggtctccaacaaagccctcccagcccccatcgagaaaaccatct ccaaagccaaagggcagccccgagaaccacaggtgtacaccctgcccccatcccgggatgagctgaccaagaaccag gtcagcctgacctgcctggtcaaaggcttctatcccagcgacatcgccgtggagtgggagagcaatgggcagccggagaa caactacaagaccacgcctcccgtgctggactccgacggctccttcttcctctacagcaagctcaccgtggacaagagcag gtggcagcaggggaacgtcttctcatgctccgtgatgcatgaggctctgcacaaccactacacgcagaagagcctctccctg tctccgggtaaacgaggcaagagggctgtggggggcggtgggagcggcggcgggggctcaggtggcgggggaagtgg
cgggggagggagtggagggggagggagtggaggcgggggatccggcggggggggtagcaagcgtcgcgagaagcg ggataagacccatacctgccccccctgtcccgcgcccgagttgctcgggggccccagcgtgtttttgtttcctcccaagcctaa agatacattgatgattagtagaacacccgaagtgacctgtgtcgtcgtcgatgtctctcatgaggatcccgaagtgaaattcaa ttggtatgtcgatggggtcgaagtccacaacgctaaaaccaaacccagagaagaacagtataattctacctatagggtcgtg 5 tctgtgttgacagtgctccatcaagattggctcaacgggaaagaatacaaatgtaaagtgagtaataaggctttgcccgctcct attgaaaagacaattagtaaggctaagggccaacctagggagccccaagtctatacactccctcccagtagagacgagct gaccaagaaccaggtcagcctgacctgcctggtcaaaggcttctatcccagcgacatcgccgtggagtgggagagcaatg ggcagccggagaacaactacaagaccacgcctcccgtgctggactccgacggctccttcttcctctacagcaagctcaccg tggacaagagcaggtggcagcaggggaacgtcttctcatgctccgtgatgcatgaggctctgcacaaccactacacgcag0 aagagcctctccctgtctccgggtaaaggaggaggaggatccggaggaggaggaagcagctgcatcgacaccatcccc aagagccgctgcaccgccttccagtgcaagcacagcatgaagtaccgcctgagcttctgccgcaagacctgcggcacctg ctaa //SEQ ID NO:5007.
The resulting expressed polypeptide (from vector pTT5-Fc-Fc-L10-Shk(2-35)) had the 5 following amino acid sequence before furin cleavage (the first 19 residues are a signal peptide sequence; furin cleavage sites are underlined):
mewswvflfflsvttgvhsdkthtcppcpapellggpsvflfppkpkdtlmisrtpevtcvvvdvshedpevkfnwyvdgvev hnaktkpreeqynstyrvvsvltvlhqdwlngkeykckvsnkalpapiektiskakgqprepqvytlppsrdeltknqvsltclv0 kgfypsdiavewesngqpennykttppvldsdgsfflyskltvdksrwqqgnvfscsvmhealhnhytqkslslspgkrgkr avggggsggggsggggsggggsggggsggggsggggskrrekrdkthtcppcpapellggpsvflfppkpkdtlmisrtp evtcvvvdvshedpevkfnwyvdgvevhnaktkpreeqynstyrvvsvltvlhqdwlngkeykckvsnkalpapiektiska kgqprepqvytlppsrdeltknqvsltclvkgfypsdiavewesngqpennykttppvldsdgsfflyskltvdksrwqqgnvfs csvmhealhnhytqkslslspgkggggsggggsscidtipksrctafqckhsmkyrlsfcrktcgtc //SEQ ID5 NO:5008.
Figure 87A-B demonstrates recombinant expression of a monovalent dimeric Fc-L-ShK(2- 35) molecule product expressed by and released into the conditioned media from transiently transfected mammalian cells. Figure 88 shows results from a pharmacokinetic study on the O monovalent dimeric Fc-ShK(I -35) in SD rats. Serum samples were added to microtiter plates coated with an anti-human Fc antibody to enable affinity capture. Plates were then washed, captured samples were released by SDS and run on a polyacrylamide gel. Samples were then visualized by western blot using an anti-human Fc-specific antibody and secondary-HRP conjugate. The MW of bands from serum samples is roughly identical to the original purified 5 material, suggesting little, if any, degradation of the protein occurred in vivo over a pro-longed half- life, in spite of the presence of Arg at position 1 of the ShK(1-35) sequence.
(3) Similar to (2) above, a Fc-toxin peptide fusion monomer can be conjugated with an immunoglobulin light chain and heavy chain resulting in a monovalent chimeric immunoglobulin-Fc-toxin peptide molecule. We have termed an immunoglobulin(light chain + heavy chain)-Fc construct a "hemibody"; such "hemibodies" containing a dimeric Fc portion can provide the long half-life typical of a dimeric antibody. The schematic representation in Figure 92A-C illustrates an embodiment of a hemibody- toxin peptide fusion protein and its recombinant expression by mammalian cells.
If the antibody chosen is a target specific antibody (e.g., an anti-Kv1.3 or anti- IKCaI antibody), the chimeric molecule may also enhance the targeting efficiency of the toxin peptide. Figure 91 A-B demonstrates that such chimeric molecules, in this example Fc-LI 0-ShK(2-35) dimerized with human IgGI or human lgG2 light and heavy chains, can be expressed and released into the conditioned media from transfected mammalian cells.
Example 57 Osk1 PEGylated at residue 4 by oxime formation:
fDpr<A0A)-pEG>4l0sk1 Peptide Synthesis of reduced [Dpr<AOA'4lOsk1. [DpHAOA>4]Osk1, having the sequence:
GVI [Dpr (AOA) ] NVKCKI SRQCLEPCKKAGMRFGKCMNGKCHCTPK (SEQ ID NO:5009)
can be synthesized in a stepwise manner on a Symphony™ multi-peptide synthesizer by solid- phase peptide synthesis (SPPS) using 2-(1 H-benzotriazole-1 -yl)-1 , 1 ,3,3-tetramethyluronium hexafluorophosphate (HBTU)/ N-methyl morpholine (NMM)/N,N-dimethyl-formamide (DMF) coupling chemistry at 0.1 mmol equivalent resin scale on Fmoc-Lys(Boc)-Wang resin
(Novabiochem). N-alpha-(9-fluorenylmethyloxycarbonyl)- and side-chain protected amino acids can be purchased from Novabiochem. The following side-chain protection strategy can be employed: Asp(O'Bu), Arg(Pbf), Cys(Trt), Gln(Trt), His(Trt), Lys(Nε-Boc), Ser(O'Bu), Thr(O'Bu) and Tyr(O'Bu). Dpr(AOA), i.e., N-α-Fmoc-N-b-(N-t.-Boc-amino-oxyacetyl)-L-diaminopropionic acid, can be purchased from Novabiochem (Cat. No. 04-12-1185). The protected amino acid derivatives (20 mmol) can be dissolved in 100 ml 20% dimethyl sulfoxide (DMSO) in DMF (v/v). Protected amino acids can be activated with 200 mM HBTU, 400 mM NMM in 20% DMSO in DMF, and coupling can be carried out using two treatments with 0.5 mmol protected amino acid, 0.5 mmol HBTU, 1 mmol NMM in 20% DMF/DMSO for 25 minutes and then 40 minutes. Fmoc deprotection reactions can be carried out with two treatments using a 20% piperidine in DMF (v/v) solution for 10 minutes
and then 15 minutes. Following synthesis and removal of the N-terminal Fmoc group, the resin can be then drained, and washed with DCM, DMF, DCM, and then dried in vacuo. The peptide- resin can be deprotected and released from the resin by treatment with a TFA/amionooxyacetic acid/TIS/EDT/H20 (90:2.5:2.5:2.5:2.5) solution at room temperature for 1 hour. The volatiles can be then removed with a stream of nitrogen gas, the crude peptide precipitated twice with cold diethyl ether and collected by centrifugation. The [Dpr<AOA'4]Osk1 peptide can be then analyzed on a Waters 2795 analytical RP-HPLC system using a linear gradient (0-60% buffer B in 12 minutes, A: 0.1% TFA in water also containing 0.1% aminooxyacetic acid, B: 0.1% TFA in acetonitrile) on a Jupiter 4 μm Proteo™ 90 A column. Reversed-Phase HPLC Purification. Preparative Reversed-phase high-performance liquid chromatography can be performed on C18, 5 μm, 2.2 cm * 25 cm) column. The [Dpr<AOA>4]Osk1 peptide is dissolved in 50% aqueous acetronitrile containing acetic acid and amionooxyacetic acid and loaded onto a preparative HPLC column. Chromatographic separations can be achieved using linear gradients of buffer B in A (A = 0.1% aqueous TFA; B = 90% aq. ACN containing 0.09% TFA), typically 5-95% over 90 minutes at 15 mL/min. Preparative HPLC fractions can be characterized by ESMS and photodiode array (PDA) HPLC, combined and lyophilized.
Osk1 peptide analog PEGylated at residue 4 by oxime formation: [DprJA0A) pEG>4l0sk1 (i.e., GVI [Dpr(A0A~PEG)]NVKCKISRQCLEPCKKAGMRFGKCMNGKCHCTPK//SEQ ID NO:5010) can be made as follows. Lyophilized [Dpr<AOA>4]Osk1 peptide can be dissolved in 50% HPLC buffer A/B (5 mg/mL) and added to a two-fold molar excess of MeO-PEG-aldehyde, CH3O- [CH2CH2O]n-CHaCH2CHO (average molecular weight 20 kDa). The aminoxyacetyl group within the peptide at residue 4 reacts with the aldehyde group of the PEG to form a covalent oxime linkage. The reaction can be left for 24 hours, and can be analyzed on an Agilent™ 1100 RP-HPLC system using Zorbax™ 300SB-C8 5 μm column at 40 °C with a linear gradient (6-60 % B in 16 minutes, A:
0.1% TFA in water, B: 0.1% TFA/90% ACN in water). Mono PEGylated [Dpr<A0A> PEG>4]0sk1 peptide can be then isolated using a HiTrap™ 5 mL SP HP cation exchange column on AKTA FPLC system at 4 'C at 1 mL/min using a gradient of 0-50 % B in 25 column volumes (Buffers: A = 20 mM sodium acetate pH 4.0, B = 1 M NaCI, 20 mM sodium acetate, pH 4.0). The fractions can be analyzed using a 4-20 tris-Gly SDS-PAGE gel and RP-HPLC. SDS-PAGE gels can be run for 1.5 hours at 125 V, 35 mA, 5 W. Pooled product, mono-PEGylated [Dpr<AOA>-pEG>4]Osk1 peptide, can be then dialyzed at 4 0C in 3 changes of 1 L of A4S buffer (10 mM NaOAc, 5% sorbitol, pH 4.0).
The dialyzed product can be then concentrated in 10 K microcentrifuge filter to 2 ml. volume and sterile-filtered using 0.2 μM syringe filter to give the final product.
Folding of [Dpr<A0A>-pEG>4l0sk1 (Disυlphide bond formation). The mono-PEGylated [Dpc(A0A)-PEG)4]0sk1 peptide can be dissolved in 20% AcOH in water (v/v) and can be then diluted with water to a concentration of approximately 0.15 mg peptide mL, the pH adjusted to about 8.0 using NH4OH (28-30%), and gently stirred at room temperature for 36 hours. Folding process can be monitored by LC-MS analysis. Following this, folded mono-PEGylated [Dpr<A0A) pEG'4]0sk1 can be purified using by reversed phase HPLC using a 1" Luna 5 μm C18 100 A Proteo™ column with a linear gradient 0-40% buffer B in 120 min (A=O.1% TFA in water, B=O.1% TFA in acetonitrile). Mono-PEGylated (oximated) [Dpr<A0A>-pEG>4]0sk1 peptide disulfide connectivity can be C1-C4, C2-C5, and C3-C6.
Abbreviations
Abbreviations used throughout this specification are as defined below, unless otherwise defined in specific circumstances.
Ac acetyl (used to refer to acetylated residues)
AcBpa acetylated p-benzoyl-L-phenylalanine
ADCC antibody-dependent cellular cytotoxicity
Aib aminoisobutyric acid bA beta-alanine
Bpa p-benzoyl-L-phenylalanine
BrAc bromoacetyl (BrCH2C(O)
BSA Bovine serum albumin
BzI Benzyl Cap Caproic acid
COPD Chronic obstructive pulmonary disease
CTL Cytotoxic T lymphocytes
DCC Dicylcohexylcarbodiimide
Dde 1 -(4,4-dimethyl-2,6-dioxo-cyclohexylidene)ethyl ESI-MS Electron spray ionization mass spectrometry
Fmoc fluorenylmethoxycarbonyl
HOBt 1-Hydroxybenzotriazole
HPLC high performance liquid chromatography
HSL homoseπne lactone
IB inclusion bodies
KCa calcium-activated potassium channel (including IKCa1 BKCa1 SKCa)
Kv voltage-gated potassium channel
Lau Laurie acid
LPS lipopolysaccharide
LYMPH lymphocytes
MALDI-MS Matrix-assisted laser desorption ionization mass spectrometry
Me methyl
MeO methoxy
MHC major histocompatibility complex
MMP matrix metalloproteinase
1-Nap 1-napthylalanine
NEUT neutrophils
NIe norleucine
NMP N-methyl-2-pyrrolidinone
PAGE polyacrylamide gel electrophoresis
PBMC peripheral blood mononuclear cell
PBS Phosphate-buffered saline
Pbf 2,2,4,6,7-pendamethyldihydrobenzofuran-5-sulfonyl
PCR polymerase chain reaction
Pec pipecolic acid
PEG Polyethylene glycol) pGlu pyroglutamic acid
Pic picolinic acid pY phosphotyrosine
RBS ribosome binding site
RT room temperature (25 0C)
Sar sarcosine
SDS sodium dodecyl sulfate
STK serine-threonine kinases t-Boc tert-Butoxycarbonyl tBu tert-Butyl
THF thymic humoral factor
Trt trityl