WO2008085601A2 - Genemap of the human genes associated with asthma disease - Google Patents
Genemap of the human genes associated with asthma disease Download PDFInfo
- Publication number
- WO2008085601A2 WO2008085601A2 PCT/US2007/083522 US2007083522W WO2008085601A2 WO 2008085601 A2 WO2008085601 A2 WO 2008085601A2 US 2007083522 W US2007083522 W US 2007083522W WO 2008085601 A2 WO2008085601 A2 WO 2008085601A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gene
- cell
- tables
- asthma disease
- sample
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/20—Allele or variant detection, e.g. single nucleotide polymorphism [SNP] detection
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/40—Population genetics; Linkage disequilibrium
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B25/00—ICT specially adapted for hybridisation; ICT specially adapted for gene or protein expression
- G16B25/10—Gene or protein expression profiling; Expression-ratio estimation or normalisation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/106—Pharmacogenomics, i.e. genetic variability in individual responses to drugs and drug metabolism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/136—Screening for pharmacological compounds
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/156—Polymorphic or mutational markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/16—Primer sets for multiplex assays
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/172—Haplotypes
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B25/00—ICT specially adapted for hybridisation; ICT specially adapted for gene or protein expression
Definitions
- the invention relates to the field of genomics and genetics, including genome analysis and the study of DNA variations.
- the invention relates to the fields of pharmacogenomics, diagnostics, patient therapy and the use of genetic haplotype information to predict an individual's susceptibility to asthma disease and/or their response to a particular drug or drugs, so that drugs tailored to genetic differences of population groups may be developed and/or administered to the appropriate population.
- the invention also relates to a GeneMap for asthma disease, which links variations in DNA (including both genie and non-genic regions) to an individual's susceptibility to asthma disease and/or response to a particular drug or drugs.
- the invention further relates to the genes disclosed in the GeneMap (see Table 3 and 4), which is related to methods and reagents for detection of an individual's increased or decreased risk for asthma disease by identifying at least one polymorphism in one or a combination of the genes from the GeneMap. Also related are the candidate regions identified in Table 2, which are associated with asthma disease.
- the invention further relates to nucleotide sequences of those genes including genomic DNA sequences, cDNA sequences, single nucleotide polymorphisms (SNPs), other types of polymorphisms (insertions, deletions, microsatellites), alleles and haplotypes (see Sequence Listing and Tables 1 , 2, 3 and 4).
- the invention further relates to isolated nucleic acids comprising these nucleotide sequences and isolated polypeptides or peptides encoded thereby. Also related are expression vectors and host cells comprising the disclosed nucleic acids or fragments thereof, as well as antibodies that bind to the encoded polypeptides or peptides.
- the present invention further relates to ligands that modulate the activity of the disclosed genes or gene products.
- the invention relates to diagnostics and therapeutics for asthma disease, utilizing the disclosed nucleic acids, polymorphisms, chromosomal regions, gene maps, polypeptides or peptides, antibodies and/or ligands and small molecules that activate or repress relevant signaling events.
- Asthma is generally defined as an inflammatory disorder of the airways, and clinical symptoms arise from intermittent airflow obstruction.
- Two common subdivisions of asthma are atopic (allergic or extrinsic) asthma and non-atopic (intrinsic) asthma.
- atopic asthma activation of the immune system by ubiquitous antigens is generally a response to environmental stimuli, and the disorder is generally characterized by an increased ability of lymphocytes to produce IgE antibodies in response to these antigens.
- Non-atopic asthma may be defined as reversible airflow limitation in the absence of allergies.
- Asthma is a disease that is broadly characterized by this immune activation when pulmonary inflammation ensues.
- Asthma is a disease of reversible bronchial obstruction, characterized by airway inflammation, epithelial damage, airway smooth muscle hypertrophy and bronchial hyperreactivity.
- Certain cells are important in this inflammatory reaction in the airways and they include T cells and antigen presenting cells, B cells that produce IgE, mast cells/basophils that store inflammatory mediators and bind IgE, and eosinophils that release additional mediators. These inflammatory cells accumulate at the site of allergic inflammation, and the toxic products they release contribute to the tissue destruction related to the disorder.
- the agents that can diminish the underlying inflammation have their own known list of side effects that range from immunosuppression to bone loss.
- Other nonsteroid treatments have been proposed to address inflammation, such as Glycophorin A, cyclosporin, and a peptide fragment of IL-2. While these agents may represent alternatives to steroids in the treatment of asthmatics, they all inhibit interleukin-2 dependent T lymphocyte proliferation and potentially critical immune functions associated with homeostasis. What is needed in the art is technology to expedite the development of therapeutics that is specifically designed to treat the cause, and not the symptoms, of atopic asthma.
- the DNA sequences between two human genomes are 99.9% identical.
- the variations in DNA sequence between individuals can be, as an example, deletions of small or large stretches of DNA, insertions of stretches of DNA 1 variations in the number of repetitive DNA elements, and changes in single base positions in the genome called "single nucleotide polymorphisms" (SNPs).
- SNPs single nucleotide polymorphisms
- GWS Genome-wide scans
- ADAM33 gene on 20p13 region (Van Eerdewegh 2002), and PHF11 on 13q14 region (Zhang 2003).
- a GWS searches throughout the genome without any a priori hypothesis and consequently can identify genes that are not obvious candidates for the disease as well as genes that are relevant candidates for the disease, it can also identify chromosomal regions that are structurally important where mutations can influence gene function of specific genes.
- LD linkage disequilibrium
- Family-based linkage mapping methods were initially used for disorder locus identification. This technique locates genes based on the relatively limited number of genetic recombination events within the families used in the study, and results in large chromosomal regions containing hundreds of genes, any one of which could be the disorder-causing gene.
- Population-based, or linkage disequilibrium (LD) mapping is based on the premise that regions adjacent to a gene of interest are co-transmitted through the generations along with the gene. As a result, LD extends over shorter genetic regions than does linkage (Hewett et a/., 2002), and can facilitate detection of genes with lower relative risk than family linkage mapping approaches. LD-based mapping also defines much smaller candidate regions which may contain only a few genes, making the identification of the actual disorder gene much easier.
- identifying susceptibility genes associated with asthma disease and their respective biochemical pathways will facilitate the identification of diagnostic markers as well as novel targets for improved therapeutics. It will also improve the quality of life for those afflicted by this disease and will reduce the economic costs of these afflictions at the individual and societal level.
- the identification of those genetic markers would provide the basis for novel genetic tests and eliminate or reduce the therapeutic methods currently used.
- the identification of those genetic markers will also provide the development of effective therapeutic intervention for the battery of laboratory, radiological, and other medical evaluations typically required to diagnose asthma disease.
- the present invention satisfies this need and provides related advantages as well.
- Figure 1 Method employed by the inventors to permit the identification of genes involved in a particular disorder or trait, such as asthma disease.
- the method can be applied for any given disorder and the end result is the construction of a GeneMap for a particular disorder. Briefly, a disorder or genetic trait is selected followed by in depth literature review on the known genes and candidate regions known in the art, and on the prevalence, incidence and phenotypes of the disorder. A clinical specialist in the field of the disorder is consulted for the definition of phenotype. Inclusion and exclusion criteria are then set and a study protocol is constructed. IRB and ethical approval are sought prior to patient recruitment. A network of physicians is required to recruit the necessary cases and controls for the study from the Quebec Founder Population.
- Ultrafine mapping is performed on all the samples to identify the polymorphisms that are most associated with the disorder phenotype as part of the search for the actual DNA polymorphisms that confer susceptibility to the disorder.
- the genes found associated with the disorder are then corroborated.
- the corroborated genes are used for the construction of a GeneMap.
- the CD-R labeled "GeneMap of the human gene associated with asthma disease” contains the following one file of sequence listing. Each electronic copy of the sequence listing was created on November 2, 2005 with a file size of 7,759 kb. The file name is as follows: 059908-5010 sequence listing.txt. An electronic copy of the Sequence Listing is also being filed herewith. This electronic copy of the Sequence Listing is hereby incorporated by reference in its entirety.
- Allele One of a pair, or series, of forms of a gene or non-genic region that occur at a given locus in a chromosome. Alleles are symbolized with the same basic symbol (e.g., B for dominant and b for recessive; B1 , B2, Bn for n additive alleles at a locus). In a normal diploid cell there are two alleles of any one gene (one from each parent), which occupy the same relative position (locus) on homologous chromosomes. Within a population there may be more than two alleles of a gene. See multiple alleles. SNPs also have alleles, i.e., the two (or more) nucleotides that characterize the SNP.
- Amplification of nucleic acids refers to methods such as polymerase chain reaction (PCR), ligation amplification (or ligase chain reaction, LCR) and amplification methods based on the use of Q-beta replicase. These methods are well known in the art and are described, for example, in U.S. Patent Nos. 4,683,195 and 4,683,202. Reagents and hardware for conducting PCR are commercially available. Primers useful for amplifying sequences from the disorder region are preferably complementary to, and preferably hybridize specifically to, sequences in the disorder region or in regions that flank a target region therein. Genes from Tables 3 and 4 generated by amplification may be sequenced directly. Alternatively, the amplified sequence(s) may be cloned prior to sequence analysis.
- PCR polymerase chain reaction
- LCR ligase chain reaction
- Antigenic component is a moiety that binds to its specific antibody with sufficiently high affinity to form a detectable antigen-antibody complex.
- Antibodies refer to polyclonal and/or monoclonal antibodies and fragments thereof, and immunologic binding equivalents thereof, that can bind to proteins and fragments thereof or to nucleic acid sequences from the disorder region, particularly from the disorder gene products or a portion thereof.
- the term antibody is used both to refer to a homogeneous molecular entity, or a mixture such as a serum product made up of a plurality of different molecular entities.
- Proteins may be prepared synthetically in a protein synthesizer and coupled to a carrier molecule and injected over several months into rabbits. Rabbit sera are tested for immunoreactivity to the protein or fragment.
- Monoclonal antibodies may be made by injecting mice with the proteins, or fragments thereof.
- Monoclonal antibodies can be screened by ELISA and tested for specific immunoreactivity with protein or fragments thereof (Harlow et al. 1988, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). These antibodies will be useful in developing assays as well as therapeutics.
- Associated allele refers to an allele at a polymorphic locus that is associated with a particular phenotype of interest, e.g., a predisposition to a disorder or a particular drug response.
- cDNA refers to complementary or copy DNA produced from an RNA template by the action of RNA-dependent DNA polymerase (reverse transcriptase).
- a cDNA clone means a duplex DNA sequence complementary to an RNA molecule of interest, included in a cloning vector or PCR amplified. This term includes genes from which the intervening sequences have been removed.
- cDNA library refers to a collection of recombinant DNA molecules containing cDNA inserts that together comprise essentially all of the expressed genes of an organism or tissue.
- a cDNA library can be prepared by methods known to one skilled in the art (see, e.g., Cowell and Austin, 1997, "DNA Library Protocols," Methods in Molecular Biology). Generally, RNA is first isolated from the cells of the desired organism, and the RNA is used to prepare cDNA molecules.
- Cloning refers to the use of recombinant DNA techniques to insert a particular gene or other DNA sequence into a vector molecule. In order to successfully clone a desired gene, it is necessary to use methods for generating DNA fragments, for joining the fragments to vector molecules, for introducing the composite DNA molecule into a host cell in which it can replicate, and for selecting the clone having the target gene from amongst the recipient host cells.
- Cloning vector refers to a plasmid or phage DNA or other DNA molecule that is able to replicate in a host cell.
- the cloning vector is typically characterized by one or more endonuclease recognition sites at which such DNA sequences may be cleaved in a determinable fashion without loss of an essential biological function of the DNA, and which may contain a selectable marker suitable for use in the identification of cells containing the vector.
- Coding sequence or a protein-coding sequence is a polynucleotide sequence capable of being transcribed into mRNA and/or capable of being translated into a polypeptide or peptide.
- the boundaries of the coding sequence are typically determined by a translation start codon at the 5'-terminus and a translation stop codon at the 3'-terminus.
- Complement of a nucleic acid sequence refers to the antisense sequence that participates in Watson-Crick base-pairing with the original sequence.
- Disorder region refers to the portions of the human chromosomes displayed in Table 2 bounded by the markers from Tables 1 , 2 and 5.
- Disorder-associated nucleic acid or polypeptide sequence refers to a nucleic acid sequence that maps to region of Table 2 or the polypeptides encoded therein (Tables 3 and 4, nucleic acids, and polypeptides).
- nucleic acids this encompasses sequences that are identical or complementary to the gene sequences from Table 3 and 4, as well as sequence-conservative, function- conservative, and non-conservative variants thereof.
- polypeptides this encompasses sequences that are identical to the polypeptide, as well as function-conservative and non-conservative variants thereof.
- alleles of naturally-occurring polymorphisms causative of asthma disease such as, but not limited to, alleles that cause altered expression of genes of Tables 3 and 4 and alleles that cause altered protein levels or stability (e.g., decreased levels, increased levels, expression in an inappropriate tissue type, increased stability, and decreased stability).
- Expression vector refers to a vehicle or plasmid that is capable of expressing a gene that has been cloned into it, after transformation or integration in a host cell.
- the cloned gene is usually placed under the control of (i.e., operably linked to) a regulatory sequence.
- Function-conservative variants are those in which a change in one or more nucleotides in a given codon position results in a polypeptide sequence in which a given amino acid residue in the polypeptide has been replaced by a conservative amino acid substitution. Function-conservative variants also include analogs of a given polypeptide and any polypeptides that have the ability to elicit antibodies specific to a designated polypeptide.
- Founder population Also called a population isolate, this is a large number of people who have mostly descended, in genetic isolation from other populations, from a much smaller number of people who lived many generations ago.
- Gene refers to a DNA sequence that encodes through its template or messenger RNA a sequence of amino acids characteristic of a specific peptide, polypeptide, or protein.
- the term "gene” also refers to a DNA sequence that encodes an RNA product.
- the term gene as used herein with reference to genomic DNA includes intervening, non-coding regions, as well as regulatory regions, and can include 5' and 3' ends.
- a gene sequence is wild-type if such sequence is usually found in individuals unaffected by the disorder or condition of interest. However, environmental factors and other genes can also play an important role in the ultimate determination of the disorder. In the context of complex disorders involving multiple genes (oligogenic disorder), the wild type, or normal sequence can also be associated with a measurable risk or susceptibility, receiving its reference status based on its frequency in the general population.
- GeneMaps are defined as groups of gene(s) that are directly or indirectly involved in at least one phenotype of a disorder. As such, GeneMaps enable the development of synergistic diagnostic products, creating "theranostics”.
- Genotype Set of alleles at a specified locus or loci.
- Haplotype The allelic pattern of a group of (usually contiguous) DNA markers or other polymorphic loci along an individual chromosome or double helical DNA segment. Haplotypes identify individual chromosomes or chromosome segments. The presence of shared haplotype patterns among a group of individuals implies that the locus defined by the haplotype has been inherited, identical by descent (IBD), from a common ancestor. Detection of identical by descent haplotypes is the basis of linkage disequilibrium (LD) mapping. Haplotypes are broken down through the generations by recombination and mutation. In some instances, a specific allele or haplotype may be associated with susceptibility to a disorder or condition of interest, e.g., asthma disease. In other instances, an allele or haplotype may be associated with a decrease in susceptibility to a disorder or condition of interest, i.e., a protective sequence.
- IBD identical by descent
- Detection of identical by descent haplotypes is
- Host includes prokaryotes and eukaryotes.
- the term includes an organism or cell that is the recipient of an expression vector (e.g., autonomously replicating or integrating vector).
- Hybridizable nucleic acids are hybridizable to each other when at least one strand of the nucleic acid can anneal to another nucleic acid strand under defined stringency conditions.
- hybridization requires that the two nucleic acids contain at least 10 substantially complementary nucleotides; depending on the stringency of hybridization, however, mismatches may be tolerated.
- the appropriate stringency for hybridizing nucleic acids depends on the length of the nucleic acids and the degree of complementarity, and can be determined in accordance with the methods described herein.
- IBD Identity by descent
- Identity is a relationship between two or more polypeptide sequences or two or more polynucleotide sequences, as determined by comparing the sequences. In the art, identity also means the degree of sequence relatedness between polypeptide or polynucleotide sequences, as the case may be, as determined by the match between strings of such sequences. Identity and similarity can be readily calculated by known methods, including but not limited to those described in A.M. Lesk (ed), 1988, Computational Molecular Biology, Oxford University Press, NY; D.W. Smith (ed), 1993, Biocomputing. Informatics and Genome Projects, Academic Press, NY; A.M. Griffin and H. G. Griffin, H.
- Immunogenic component is a moiety that is capable of eliciting a humoral and/or cellular immune response in a host animal.
- Isolated nucleic acids are nucleic acids separated away from other components (e.g., DNA, RNA, and protein) with which they are associated (e.g., as obtained from cells, chemical synthesis systems, or phage or nucleic acid libraries). Isolated nucleic acids are at least 60% free, preferably 75% free, and most preferably 90% free from other associated components. In accordance with the present invention, isolated nucleic acids can be obtained by methods described herein, or other established methods, including isolation from natural sources (e.g., cells, tissues, or organs), chemical synthesis, recombinant methods, combinations of recombinant and chemical methods, and library screening methods.
- natural sources e.g., cells, tissues, or organs
- chemical synthesis e.g., recombinant methods, combinations of recombinant and chemical methods, and library screening methods.
- Isolated polypeptides or peptides are those that are separated from other components (e.g., DNA, RNA, and other polypeptides or peptides) with which they are associated (e.g., as obtained from cells, translation systems, or chemical synthesis systems).
- isolated polypeptides or peptides are at least 10% pure; more preferably, 80% or 90% pure.
- Isolated polypeptides and peptides include those obtained by methods described herein, or other established methods, including isolation from natural sources (e.g., cells, tissues, or organs), chemical synthesis, recombinant methods, or combinations of recombinant and chemical methods.
- Proteins or polypeptides referred to herein as recombinant are proteins or polypeptides produced by the expression of recombinant nucleic acids.
- a portion as used herein with regard to a protein or polypeptide refers to fragments of that protein or polypeptide. The fragments can range in size from 5 amino acid residues to all but one residue of the entire protein sequence. Thus, a portion or fragment can be at least 5, 5-50, 50-100, I00-200, 200-400, 400-800, or more consecutive amino acid residues of a protein or polypeptide.
- LD Linkage disequilibrium
- Markers that are in high LD can be assumed to be located near each other and a marker or haplotype that is in high LD with a genetic trait can be assumed to be located near the gene that affects that trait.
- the physical proximity of markers can be measured in family studies where it is called linkage or in population studies where it is called linkage disequilibrium.
- LD mapping population based gene mapping, which locates disorder genes by identifying regions of the genome where haplotypes or marker variation patterns are shared statistically more frequently among disorder patients compared to healthy controls. This method is based upon the assumption that many of the patients will have inherited an allele associated with the disorder from a common ancestor (IBD), and that this allele will be in LD with the disorder gene.
- IBD common ancestor
- Locus a specific position along a chromosome or DNA sequence.
- a locus could be a gene, a marker, a chromosomal band or a specific sequence of one or more nucleotides.
- MAF Minor allele frequency
- Markers an identifiable DNA sequence that is variable (polymorphic) for different individuals within a population. These sequences facilitate the study of inheritance of a trait or a gene. Such markers are used in mapping the order of genes along chromosomes and in following the inheritance of particular genes; genes closely linked to the marker or in LD with the marker will generally be inherited with it. Two types of markers are commonly used in genetic analysis, microsatellites and SNPs.
- Microsatellite DNA of eukaryotic cells comprising a repetitive, short sequence of DNA that is present as tandem repeats and in highly variable copy number, flanked by sequences unique to that locus.
- Mutant sequence if it differs from one or more wild-type sequences.
- a nucleic acid from a gene listed in Tables 3 and 4 containing a particular allele of a single nucleotide polymorphism may be a mutant sequence.
- the individual carrying this allele has increased susceptibility toward the disorder or condition of interest.
- the mutant sequence might also refer to an allele that decreases the susceptibility toward a disorder or condition of interest and thus acts in a protective manner.
- the term mutation may also be used to describe a specific allele of a polymorphic locus.
- Non-conservative variants are those in which a change in one or more nucleotides in a given codon position results in a polypeptide sequence in which a given amino acid residue in a polypeptide has been replaced by a non- conservative amino acid substitution.
- Non-conservative variants also include polypeptides comprising non-conservative amino acid substitutions.
- Nucleic acid or polynucleotide purine- and pyrimidine-containing polymers of any length, either polyribonucleotides or polydeoxyribonucleotide or mixed polyribo polydeoxyribonucleotides. This includes single-and double-stranded molecules, i.e., DNA-DNA, DNA-RNA and RNA-RNA hybrids, as well as protein nucleic acids (PNA) formed by conjugating bases to an amino acid backbone. This also includes nucleic acids containing modified bases.
- PNA protein nucleic acids
- Nucleotide a nucleotide, the unit of a DNA molecule, is composed of a base, a 2'-deoxyribose and phosphate ester(s) attached at the 5' carbon of the deoxyribose. For its incorporation in DNA, the nucleotide needs to possess three phosphate esters but it is converted into a monoester in the process.
- Operably linked means that the promoter controls the initiation of expression of the gene.
- a promoter is operably linked to a sequence of proximal DNA if upon introduction into a host cell the promoter determines the transcription of the proximal DNA sequence(s) into one or more species of RNA.
- a promoter is operably linked to a DNA sequence if the promoter is capable of initiating transcription of that DNA sequence.
- Ortholog denotes a gene or polypeptide obtained from one species that has homology to an analogous gene or polypeptide from a different species.
- Paralog denotes a gene or polypeptide obtained from a given species that has homology to a distinct gene or polypeptide from that same species.
- Phenotype any visible, detectable or otherwise measurable property of an organism such as symptoms of, or susceptibility to, a disorder.
- Polymorphism occurrence of two or more alternative genomic sequences or alleles between or among different genomes or individuals at a single locus.
- a polymorphic site thus refers specifically to the locus at which the variation occurs.
- an individual carrying a particular allele of a polymorphism has an increased or decreased susceptibility toward a disorder or condition of interest.
- Portion and fragment are synonymous.
- a portion as used with regard to a nucleic acid or polynucleotide refers to fragments of that nucleic acid or polynucleotide. The fragments can range in size from 8 nucleotides to all but one nucleotide of the entire gene sequence.
- the fragments are at least about 8 to about 10 nucleotides in length; at least about 12 nucleotides in length; at least about 15 to about 20 nucleotides in length; at least about 25 nucleotides in length; or at least about 35 to about 55 nucleotides in length.
- Probe or primer refers to a nucleic acid or oligonucleotide that forms a hybrid structure with a sequence in a target region of a nucleic acid due to complementarity of the probe or primer sequence to at least one portion of the target region sequence.
- Protein and polypeptide are synonymous. Peptides are defined as fragments or portions of polypeptides, preferably fragments or portions having at least one functional activity (e.g., proteolysis, adhesion, fusion, antigenic, or intracellular activity) as the complete polypeptide sequence.
- functional activity e.g., proteolysis, adhesion, fusion, antigenic, or intracellular activity
- Recombinant nucleic acids nuclei acids which have been produced by recombinant DNA methodology, including those nucleic acids that are generated by procedures which rely upon a method of artificial replication, such as the polymerase chain reaction (PCR) and/or cloning into a vector using restriction enzymes. Portions of recombinant nucleic acids which code for polypeptides can be identified and isolated by, for example, the method of M. Jasin et al., U.S. Patent No. 4,952,501.
- Regulatory sequence refers to a nucleic acid sequence that controls or regulates expression of structural genes when operably linked to those genes. These include, for example, the lac systems, the trp system, major operator and promoter regions of the phage lambda, the control region of fd coat protein and other sequences known to control the expression of genes in prokaryotic or eukaryotic cells. Regulatory sequences will vary depending on whether the vector is designed to express the operably linked gene in a prokaryotic or eukaryotic host, and may contain transcriptional elements such as enhancer elements, termination sequences, tissue-specificity elements and/or translational initiation and termination sites.
- Sample refers to a biological sample, such as, for example, tissue or fluid isolated from an individual or animal (including, without limitation, plasma, serum, cerebrospinal fluid, lymph, tears, nails, hair, saliva, milk, pus, and tissue exudates and secretions) or from in vitro cell culture-constituents, as well as samples obtained from, for example, a laboratory procedure.
- tissue or fluid isolated from an individual or animal (including, without limitation, plasma, serum, cerebrospinal fluid, lymph, tears, nails, hair, saliva, milk, pus, and tissue exudates and secretions) or from in vitro cell culture-constituents, as well as samples obtained from, for example, a laboratory procedure.
- Single nucleotide polymorphism variation of a single nucleotide. This includes the replacement of one nucleotide by another and deletion or insertion of a single nucleotide.
- SNPs are biallelic markers although tri- and tetra- allelic markers also exist.
- SNP A ⁇ C may comprise allele C or allele A (Table 1).
- a nucleic acid molecule comprising SNP A ⁇ C may include a C or A at the polymorphic position.
- an ambiguity code is used in Tables 1 , 2, 5 and the sequence listing, to represent the variations.
- haplotype is used, e.g.
- haplotype is used to describe a combination of SNP alleles, e.g., the alleles of the SNPs found together on a single DNA molecule.
- the SNPs in a haplotype are in linkage disequilibrium with one another.
- variants are those in which a change of one or more nucleotides in a given codon position results in no alteration in the amino acid encoded at that position (i.e., silent mutation).
- nucleic acid or fragment thereof is substantially homologous to another if, when optimally aligned (with appropriate nucleotide insertions and/or deletions) with the other nucleic acid (or its complementary strand), there is nucleotide sequence identity in at least 60% of the nucleotide bases, usually at least 70%, more usually at least 80%, preferably at least 90%, and more preferably at least 95-98% of the nucleotide bases.
- substantial homology exists when a nucleic acid or fragment thereof will hybridize, under selective hybridization conditions, to another nucleic acid (or a complementary strand thereof). Selectivity of hybridization exists when hybridization which is substantially more selective than total lack of specificity occurs.
- selective hybridization will occur when there is at least about 55% sequence identity over a stretch of at least about nine or more nucleotides, preferably at least about 65%, more preferably at least about 75%, and most preferably at least about 90% (M. Kanehisa, 1984, NucL Acids Res. 11:203-213).
- the length of homology comparison, as described, may be over longer stretches, and in certain embodiments will often be over a stretch of at least 14 nucleotides, usually at least 20 nucleotides, more usually at least 24 nucleotides, typically at least 28 nucleotides, more typically at least 32 nucleotides, and preferably at least 36 or more nucleotides.
- Wild-type gene from Tables 3 and 4 refers to the reference sequence.
- the wild-type gene sequences from Tables 3 and 4 used to identify the variants (polymorphisms, alleles, and haplotypes) described in detail herein.
- Van Eerdewegh et al characterized the ADAM33 gene on 20p13 region (Van Eerdewegh et al., 2002). This gene encodes a metallo protease and has been associated to asthma and bronchial hyperreactivity (BHR). Since, ADAM33 has been evaluated in 6 case-control association studies on samples from different populations, and three studies replicated the association with asthma (Howard et al., 2003; Raby et al., 2004 and Werner et al., 2004). Also, Jongepier et al., (2004) showed an association between a polymorphism in ADAM33 and accelerated lung function decline in asthma patients.
- ADAM33 the largest studies performed in different populations have failed to show an association between ADAM33 and asthma or related phenotypes. Thus, the involvement of ADAM33 in this disease remains controversial.
- G protein-coupled receptor 154 (or GPRA) was identified in the 7p15 region as associated with high serum IgE and asthma in Finnish and French
- the present invention is based on the discovery of genes associated with asthma disease.
- Disease-associated loci (candidate regions; Table 2) are identified by the statistically significant differences in allele or haplotype frequencies between the cases and the controls.
- 231 candidate regions exhibiting a -Iog10 P value of 3.5 or higher are identified, comprises a few which have been previously reported to be associated with asthma disease.
- the invention provides a method for the discovery of genes associated with asthma disease and the construction of a GeneMap for asthma disease in a human population, comprising the following steps (see Figure 1 and Example section herein):
- Step 1 Recruit patients (cases) and controls
- 500 patients diagnosed for asthma disease along with two family members are recruited from the Quebec Founder Population (QFP).
- the preferred trios recruited are parent-parent-child (PPC) trios.
- Trios can also be recruited as parent-child-child (PCC) trios.
- more or less than 500 trios are recruited.
- the present invention is performed as a whole or partially with DNA samples from individuals of another founder population than the Quebec population or from the general population.
- Step 2 DNA extraction and quantitation
- sample comprising cells or nucleic acids from patients or controls may be used.
- Preferred samples are those easily obtained from the patient or control.
- Such samples include, but are not limited to blood, peripheral lymphocytes, buccal swabs, epithelial cell swabs, nails, hair, bronchoalveolar lavage fluid, sputum, or other body fluid or tissue obtained from an individual.
- DNA is extracted from such samples in the quantity and quality necessary to perform the invention using conventional DNA extraction and quantitation techniques.
- the present invention is not linked to any DNA extraction or quantitation platform in particular.
- Step 3 Genotype the recruited individuals
- assay specific and/or locus-specific and/or allele- specific oligonucleotides for every SNP marker of the present invention are organized onto one or more arrays.
- the genotype at each SNP locus is revealed by hybridizing short PCR fragments comprising each SNP locus onto these arrays.
- the arrays permit a high-throughput genome wide association study using DNA samples from individuals of the Quebec founder population.
- Such assay-specific and/or locus-specific and/or allele-specific oligonucleotides necessary for scoring each SNP of the present invention are preferably organized onto a solid support.
- Such supports can be arrayed on wafers, glass slides, beads or any other type of solid support.
- the assay-specific and/or locus-specific and/or allele- specific oligonucleotides are not organized onto a solid support but are still used as a whole, in panels or one by one.
- the present invention is therefore not linked to any genotyping platform in particular.
- one or more portions of the SNP maps are used to screen the whole genome, a subset of chromosomes, a chromosome, a subset of genomic regions or a single genomic region.
- the 1,500 individuals composing the 500 trios are preferably individually genotyped with at least 80,000 markers, generating at least a few million genotypes; more preferable, at least a hundred million.
- Step 4 Exclude the markers that did not pass the quality control of the assay.
- the quality controls consist of, but are not limited to, the following criteria: eliminate SNPs that had a high rate of Mendelian errors (cut-off at 1% Mendelian error rate), that deviate from the Hardy-Weinberg equilibrium, that are non-polymorphic in the Quebec founder population or have too many missing data (cut-off at 1% missing values or higher), or simply because they are non- polymorphic in the Quebec founder population (cut-off at 1% ⁇ 10% minor allele frequency (MAF)).
- Step 5 Perform the genetic analysis on the results obtained using haplotype information as well as single-marker association.
- genetic analysis is performed on all the genotypes from step 3.
- genetic analysis is performed on a total of 80,654 SNPs.
- the genetic analysis consists of, but is not limited to features corresponding to Phase information and haplotype structures.
- Phase information and haplotype structures are preferably deduced from trio genotypes using Phasefinder. Since chromosomal assignment (phase) can not be estimated when all trio members are heterozygous, an Expectation-Maximization (EM) algorithm may be used to resolve chromosomal assignment ambiguities after Phasefinder.
- EM Expectation-Maximization
- the PL-EM algorithm Partition-Ligation EM; Niu et al.., Am. J. Hum. Genet. 70:157 (2002)
- the PL-EM algorithm can be used to estimate haplotypes from the "genotype" data as a measured estimate of the reference allele frequency of a SNP in 15-marker windows that advance in increments of one marker across the data set.
- the results from such algorithms are converted into 15-marker haplotype files.
- the individual 15-marker block files are assembled into one continuous block of haplotypes for the entire chromosome. These extended haplotypes can then be used for further analysis.
- haplotype assembly algorithms take the consensus estimate of the allele call at each marker over all separate estimations (most markers are estimated 15 different times as the 15 marker blocks pass over their position).
- haplotypes for both the controls and the patients are derived in this manner.
- the preferred control of a trio structure is the spouse if the patient is one of the parents or the non-transmitted chromosomes (chromosomes found in parents but not in affected child) if the patient is the child.
- the haplotype frequencies among patients are compared to those among the controls using LDSTATS, a program that assesses the association of haplotypes with the disease.
- Such program defines haplotypes using multi-marker windows that advance across the marker map in one-marker increments. Such windows can be 1 , 3, 5, 7 or 9 markers wide, and all these window sizes are tested concurrently.
- the frequency of haplotypes in cases is compared to the frequency of haplotypes in controls.
- Such allele frequency differences for single marker windows can be tested using Pearson's Chi-square with one degree of freedom.
- Multi-allelic haplotype association can be tested using Smith's normalization of th e square root of Pearson's Chi-square. Such significance of association can be reported in two ways:
- P-values of association for each specific marker can be calculated as a pooled P- value across all haplotype windows in which they occur.
- the pooled P-value is calculated using an expected value and variance calculated using a permutation test that considers covariance between individual windows.
- Such pooled P- values can yield narrower regions of gene location than the window data (see example 3 for details on analysis methods, such as LDSTATs V2.0 and V4.0).
- conditional haplotype analyses can be performed on subsets of the original set of cases and controls using the program LDSTAT. The selection of a subset of cases and their matched controls can be based on the carrier status of cases at a gene or locus of interest (see conditional analysis section in example 3 herein). Various conditional haplotypes can be derived, such as protective haplotypes and risk haplotypes. Step 6: Fine Mapping
- step 4 the candidate regions that were identified by step 4 are further mapped for the purpose of refinement and validation.
- this fine mapping is performed with a density of genetic markers higher than in the genome wide scan (step 3) using any genotyping platform available in the art.
- Such fine mapping can be, but is not limited to, typing the allele via an allele-specific elongation assay that is then ligated to a locus-specific oligonucleotide.
- Such assays can be performed directly on the genomic DNA at a highly multiplex level and the products can be amplified using universal oligonucleotides.
- the density of genetic markers can be, but is not limited to, a set of SNP markers with an average inter-marker distance of 1-4 Kb distributed over about 400 Kb to 1 Mb, roughly centered at the highest point of the GWS association.
- the preferred samples are those obtained from asthma disease PPC trios including the ones used for the GWS.
- the genetic analysis of the results obtained using haplotype information as well as single-marker association is performed as described herein (step 5 and example section).
- the candidate regions that are validated and confirmed after this analysis proceed to a gene mining step described in example 5, herein, to characterize their marker and genetic content.
- Step 7 SNP and DNA polymorphism discovery
- all the candidate genes and regions identified in step 6 are sequenced for polymorphism identification.
- the entire region, including all introns, is sequenced to identify all polymorphisms.
- the candidate genes are prioritized for sequencing, and only functional gene elements (promoters, conserved noncoding sequences, exons and splice sites) are sequenced.
- previously identified polymorphisms in the candidate regions can also be used. For example, SNPs from dbSNP, Perlegen Sciences, Inc., or others can also be used rather than resequencing the candidate regions to identify polymorphisms.
- the discovery of SNPs and DNA polymorphisms generally comprises a step consisting of determining the major haplotypes in the region to be sequenced.
- the preferred samples are selected according to which haplotypes contribute to the association signal observed in the region to be sequenced.
- the purpose is to select a set of samples that covers all the major haplotypes in the given region.
- Each major haplotype is preferably analyzed in at least a few individuals.
- Any analytical procedure may be used to detect the presence or absence of variant nucleotides at one or more polymorphic positions of the invention.
- allelic variation requires a mutation discrimination technique, optionally an amplification reaction and optionally a signal generation system. Any means of mutation detection or discrimination may be used. For instance, DNA sequencing, scanning methods, hybridization, extension based methods, incorporation based methods, restriction enzyme-based methods and ligation-based methods may be used in the methods of the invention.
- Sequencing methods include, but are not limited to, direct sequencing, and sequencing by hybridization.
- Scanning methods include, but are not limited to, protein truncation test (PTT), single-strand conformation polymorphism analysis (SSCP), denaturing gradient gel electrophoresis (DGGE), temperature gradient gel electrophoresis (TGGE), cleavage, heteroduplex analysis, chemical mismatch cleavage (CMC), and enzymatic mismatch cleavage.
- Hybridization-based methods of detection include, but are not limited to, solid phase hybridization such as dot blots, multiple allele specific diagnostic assay (MASDA), reverse dot blots, and oligonucleotide arrays (DNA Chips).
- Solution phase hybridization amplification methods may also be used, such as Taqman.
- Extension based methods include, but are not limited to, amplification refraction mutation systems (ARMS), amplification refractory mutation systems (ALEX), and competitive oligonucleotide priming systems (COPS).
- Incorporation based methods include, but are not limited to, mini-sequencing and arrayed primer extension (APEX).
- Restriction enzyme-based detection systems include, but are not limited to, restriction site generating PCR.
- ligation based detection methods include, but are not limited to, oligonucleotide ligation assays (OLA).
- Signal generation or detection systems that may be used in the methods of the invention include, but are not limited to, fluorescence methods such as fluorescence resonance energy transfer (FRET), fluorescence quenching, fluorescence polarization as well as other chemiluminescence, electrochemiluminescence, Raman, radioactivity, colometric methods, hybridization protection assays and mass spectrometry methods.
- Further amplification methods include, but are not limited to self sustained replication (SSR), nucleic acid sequence based amplification (NASBA), ligase chain reaction (LCR), strand displacement amplification (SDA) and branched DNA (B-DNA).
- SSR self sustained replication
- NASBA nucleic acid sequence based amplification
- LCR ligase chain reaction
- SDA strand displacement amplification
- B-DNA branched DNA
- This step further maps the candidate regions and genes confirmed in the previous step to identify and validate the responsible polymorphisms associated with asthma disease in the human population.
- the discovered SNPs and polymorphisms of step 7 are ultrafine mapped at a higher density of markers than the fine mapping described herein using the same technology described in step 6.
- the confirmed variations in DNA are used to build a GeneMap for asthma disease.
- the gene content of this GeneMap is described in more detail below.
- Such GeneMap can be used for other methods of the invention comprising the diagnostic methods described herein, the susceptibility to asthma disease, the response to a particular drug, the efficacy of a particular drug, the screening methods described herein and the treatment methods described herein.
- all of the above steps or the steps of Figure 1 do not need to be performed, or performed in a given order to practice or use the SNPs 1 genomic regions, genes, proteins, etc. in the methods of the invention.
- the GeneMap consists of genes and targets, in a variety of combinations, identified from the candidate regions listed in Table 2. In the preferred embodiment, all genes from Tables 3 and 4 are present in the GeneMap. In another preferred embodiment, the GeneMap consists of a selection of genes from Table 3 and 4.
- genes of the invention are arranged by candidate regions and by their chromosomal location. Such order is for the purpose of clarity and does not reflect any other criteria of selection in the association of the genes with asthma disease.
- the nucleic acid sequences of the present invention may be derived from a variety of sources including DNA, cDNA, synthetic DNA, synthetic RNA, derivatives, mimetics or combinations thereof. Such sequences may comprise genomic DNA, which may or may not include naturally occurring introns, genie regions, nongenic regions, and regulatory regions. Moreover, such genomic DNA may be obtained in association with promoter regions or poly (A) sequences.
- the sequences, genomic DNA, or cDNA may be obtained in any of several ways. Genomic DNA can be extracted and purified from suitable cells by means well known in the art. Alternatively, mRNA can be isolated from a cell and used to produce cDNA by reverse transcription or other means.
- nucleic acids described herein are used in certain embodiments of the methods of the present invention for production of RNA, proteins or polypeptides, through incorporation into cells, tissues, or organisms.
- DNA containing all or part of the coding sequence for the genes described in Tables 3 and 4, or the SNP markers described in Table 1 is incorporated into a vector for expression of the encoded polypeptide in suitable host cells.
- the invention also comprises the use of the nucleotide sequence of the nucleic acids of this invention to identify DNA probes for the genes described in Tables 3 and 4 or the SNP markers described in Table 1 , PCR primers to amplify the genes described in Tables 3 and 4 or the SNP markers described in Table 1, nucleotide polymorphisms in the genes described in Tables 3 and 4, and regulatory elements of the genes described in Tables 3 and 4.
- nucleic acids of the present invention find use as primers and templates for the recombinant production of asthma disease-associated peptides or polypeptides, for chromosome and gene mapping, to provide antisense sequences, for tissue distribution studies, to locate and obtain full length genes, to identify and obtain homologous sequences (wild-type and mutants), and in diagnostic applications.
- an antisense nucleic acid or oligonucleotide is wholly or partially complementary to, and can hybridize with, a target nucleic acid (either DNA or RNA) having the sequence of SEQ ID NO:1 , NO:3 or any SEQ ID from Tables 1 , 3 or 4.
- a target nucleic acid either DNA or RNA
- an antisense nucleic acid or oligonucleotide comprising 16 nucleotides can be sufficient to inhibit expression of at least one gene from Tables 3 and 4.
- an antisense nucleic acid or oligonucleotide can be complementary to 5' or 3' untranslated regions, or can overlap the translation initiation codon (5' untranslated and translated regions) of at least one gene from Tables 3 and 4, or its functional equivalent.
- the antisense nucleic acid is wholly or partially complementary to, and can hybridize with, a target nucleic acid that encodes a polypeptide from a gene described in Tables 3 and 4.
- oligonucleotides can be constructed which will bind to duplex nucleic acid (i.e., DNA:DNA or DNA:RNA), to form a stable triple helix containing or triplex nucleic acid.
- duplex nucleic acid i.e., DNA:DNA or DNA:RNA
- triplex oligonucleotides can inhibit transcription and/or expression of a gene from Tables 3 and 4, or its functional equivalent (M. D. Frank-Kamenetskii et al., 1995).
- Triplex oligonucleotides are constructed using the basepairing rules of triple helix formation and the nucleotide sequence of the genes described in Tables 3 and 4.
- oligonucleotide refers to naturally-occurring species or synthetic species formed from naturally-occurring subunits or their close homologs.
- the term may also refer to moieties that function similarly to oligonucleotides, but have non-naturally-occurring portions.
- oligonucleotides may have altered sugar moieties or inter-sugar linkages. Exemplary among these are phosphorothioate and other sulfur containing species which are known in the art.
- At least one of the phosphodiester bonds of the oligonucleotide has been substituted with a structure that functions to enhance the ability of the compositions to penetrate into the region of cells where the RNA whose activity is to be modulated is located. It is preferred that such substitutions comprise phosphorothioate bonds, methyl phosphonate bonds, or short chain alkyl or cycloalkyl structures.
- the phosphodiester bonds are substituted with structures which are, at once, substantially non-ionic and non- chiral, or with structures which are chiral and enantiomerically specific. Persons of ordinary skill in the art will be able to select other linkages for use in the practice of the invention.
- Oligonucleotides may also include species that include at least some modified base forms. Thus, purines and pyrimidines other than those normally found in nature may be so employed. Similarly, modifications on the furanosyl portions of the nucleotide subunits may also be effected, as long as the essential tenets of this invention are adhered to. Examples of such modifications are 2'-O-alkyl- and 2'-halogen-substituted nucleotides. Some non- limiting examples of modifications at the 2' position of sugar moieties which are useful in the present invention include OH, SH, SCH3, F, OCH3, OCN, O(CH2), NH2 and O(CH2)n CH3, where n is from 1 to about 10.
- oligonucleotides are functionally interchangeable with natural oligonucleotides or synthesized oligonucleotides, which have one or more differences from the natural structure. All such analogs are comprehended by this invention so long as they function effectively to hybridize with at least one gene from Tables 3 and 4 DNA or RNA to inhibit the function thereof.
- the oligonucleotides in accordance with this invention preferably comprise from about 3 to about 50 subunits. It is more preferred that such oligonucleotides and analogs comprise from about 8 to about 25 subunits and still more preferred to have from about 12 to about 20 subunits.
- a "subunit" is a base and sugar combination suitably bound to adjacent subunits through phosphodiester or other bonds.
- Antisense nucleic acids or oligonucleotides can be produced by standard techniques (see, e.g., Shewmaker et al., U.S. Patent No. 6,107,065).
- oligonucleotides used in accordance with this invention may be conveniently and routinely made through the well-known technique of solid phase synthesis. Any other means for such synthesis may also be employed; however, the actual synthesis of the oligonucleotides is well within the abilities of the practitioner. It is also well known to prepare other oligonucleotides such as phosphorothioates and alkylated derivatives.
- RNA e.g., mRNA
- DNA oligonucleotide
- an oligonucleotide that hybridizes to mRNA from a gene described in Tables 3 and 4 can be used to target the mRNA for RnaseH digestion.
- an oligonucleotide that can hybridize to the translation initiation site of the mRNA of a gene described in Tables 3 and 4 can be used to prevent translation of the mRNA.
- oligonucleotides that bind to the double-stranded DNA of a gene from Tables 3 and 4 can be administered. Such oligonucleotides can form a triplex construct and inhibit the transcription of the DNA encoding polypeptides of the genes described in Tables 3 and 4. Triple helix pairing prevents the double helix from opening sufficiently to allow the binding of polymerases, transcription factors, or regulatory molecules. Recent therapeutic advances using triplex DNA have been described (see, e.g., J. E. Gee et al., 1994, Molecular and Immunologic Approaches, Futura Publishing Co., Mt. Kisco, NY).
- antisense oligonucleotides may be targeted to hybridize to the following regions: mRNA cap region; translation initiation site; translational termination site; transcription initiation site; transcription termination site; polyadenylation signal; 3 1 untranslated region; 5' untranslated region; 5' coding region; mid coding region; and 3' coding region.
- the complementary oligonucleotide is designed to hybridize to the most unique 5' sequence of a gene described in Tables 3 and 4, including any of about 15-35 nucleotides spanning the 5' coding sequence.
- the antisense oligonucleotide can be synthesized, formulated as a pharmaceutical composition, and administered to a subject.
- expression vectors derived from retroviruses, adenovirus, herpes or vaccinia viruses or from various bacterial plasmids may be used for delivery of nucleotide sequences to the targeted organ, tissue or cell population.
- Methods which are well known to those skilled in the art can be used to construct recombinant vectors which will express nucleic acid sequence that is complementary to the nucleic acid sequence encoding a polypeptide from the genes described in Tables 3 and 4. These techniques are described both in Sambrook et al., 1989 and in Ausubel et al., 1992.
- expression of at least one gene from Tables 3 and 4 can be inhibited by transforming a cell or tissue with an expression vector that expresses high levels of untranslatable sense or antisense sequences. Even in the absence of integration into the DNA, such vectors may continue to transcribe RNA molecules until they are disabled by endogenous nucleases. Transient expression may last for a month or more with a nonreplicating vector, and even longer if appropriate replication elements are included in the vector system.
- Various assays may be used to test the ability of gene-specific antisense oligonucleotides to inhibit the expression of at least one gene from Tables 3 and 4.
- mRNA levels of the genes described in Tables 3 and 4 can be assessed by Northern blot analysis (Sambrook et al., 1989; Ausubel et al., 1992; J. C. Alwine et al. 1977; I. M. Bird, 1998), quantitative or semi-quantitative RT-PCR analysis (see, e.g., W.M. Freeman et al., 1999; Ren et al., 1998; J. M. CaIe et al., 1998), or in situ hybridization (reviewed by A.K. Raap, 1998).
- antisense oligonucleotides may be assessed by measuring levels of the polypeptide from the genes described in Tables 3 and 4, e.g., by western blot analysis, indirect immunofluorescence and immunoprecipitation techniques (see, e.g., J. M. Walker, 1998, Protein Protocols on CD-ROM, Humana Press, Totowa, NJ). Any other means for such detection may also be employed, and is well within the abilities of the practitioner.
- mapping technologies may be based on amplification methods, restriction enzyme cleavage methods, hybridization methods, sequencing methods, and cleavage methods using agents.
- Amplification methods include: self sustained sequence replication (Guatelli et al., 1990), transcriptional amplification system (Kwoh et al., 1989), Q-Beta Replicase (Lizardi et al., 1988), isothermal amplification (e.g. Dean et al., 2002; and Hafner ef al., 2001), or any other nucleic acid amplification method, followed by the detection of the amplified molecules using techniques well known to those of ordinary skill in the art. These detection schemes are especially useful for the detection of nucleic acid molecules if such molecules are present in very low number.
- Restriction enzyme cleavage methods include: isolating sample and control DNA, amplification (optional), digestion with one or more restriction endonucleases, determination of fragment length sizes by gel electrophoresis and comparing samples and controls. Differences in fragment length sizes between sample and control DNA indicates mutations in the sample DNA.
- sequence specific ribozymes see, e.g., U.S. Pat. No. 5,498,531 or DNAzyme (e.g. U.S. Pat. No. 5,807,718) can be used to score for the presence of specific mutations by development or loss of a ribozyme or DNAzyme cleavage site.
- SNPs and SNP maps of the invention can be identified or generated by hybridizing sample nucleic acids, e.g., DNA or RNA, to high density arrays or bead arrays containing oligonucleotide probes corresponding to the polymorphisms of Table 1 (see the Affymetrix arrays and lllumina bead sets at www.affymetrix.com and www.illumina.com and see Cronin et a/., 1996; or Kozal et al., 1996).
- sample nucleic acids e.g., DNA or RNA
- sequencing reactions can be used to directly sequence nucleic acids for the presence or the absence of one or more polymorphisms of Table 1. Examples of sequencing reactions include those based on techniques developed by Maxam and Gilbert (1977) or Sanger (1977). It is also contemplated that any of a variety of automated sequencing procedures can be utilized, including sequencing by mass spectrometry (see, e.g. PCT International Publication No. WO 94/16101; Cohen et a/., 1996; and Griffin et a/., 1993), real-time pyrophosphate sequencing method (Ronaghi et a/., 1998; and Permutt et a/., 2001) and sequencing by hybridization (see e.g. Drmanac et a/., 2002).
- mass spectrometry see, e.g. PCT International Publication No. WO 94/16101; Cohen et a/., 1996; and Griffin et a/., 1993
- RNA/RNA, DNA/DNA or RNA/DNA heteroduplexes Other methods of detecting polymorphisms include methods in which protection from cleavage agents is used to detect mismatched bases in RNA/RNA, DNA/DNA or RNA/DNA heteroduplexes (Myers et al., 1985).
- mismatch cleavage starts by providing heteroduplexes formed by hybridizing (labeled) RNA or DNA containing a wild-type sequence with potentially mutant RNA or DNA obtained from a sample.
- the double-stranded duplexes are treated with an agent who cleaves single-stranded regions of the duplex such as which will exist due to basepair mismatches between the control and sample strands.
- RNA/DNA duplexes can be treated with RNase and DNA/DNA hybrids treated with S1 nuclease to enzymatically digest the mismatched regions.
- either DNA/DNA or RNA/DNA duplexes can be treated with hydroxylamine or osmium tetroxide and with piperidine in order to digest mismatched regions. After digestion of the mismatched regions, the resulting material is then separated by size on denaturing polyacrylamide gels to determine the site of a mutation or SNP (see, for example, Cotton et al., 1988; and Saleeba et al., 1992).
- the control DNA or RNA can be labeled for detection.
- the mismatch cleavage reaction employs one or more proteins that recognize mismatched base pairs in double-stranded DNA (so called "DNA mismatch repair" enzymes) in defined systems for detecting and mapping polymorphisms.
- DNA mismatch repair enzymes
- the mutY enzyme of E. coli cleaves A at G/A mismatches (Hsu et al., 1994).
- Other examples include, but are not limited to, the MutHLS enzyme complex of E. coli (Smith and Modrich Proc. 1996) and CeI 1 from the celery (Kulinski et al., 2000) both cleave the DNA at various mismatches.
- a probe based on a polymorphic site corresponding to a polymorphism of Tables 1 , 3 or 4 is hybridized to a cDNA or other DNA product from a test cell or cells.
- the duplex is treated with a DNA mismatch repair enzyme, and the cleavage products, if any, can be detected from electrophoresis protocols or the like. See, for example, U.S. Pat. No. 5,459,039.
- the screen can be performed in vivo following the insertion of the heteroduplexes in an appropriate vector. The whole procedure is known to those ordinary skilled in the art and is referred to as mismatch repair detection (see e.g. Fakhrai-Rad et al., 2004).
- alterations in electrophoretic mobility can be used to identify polymorphisms in a sample.
- SSCP single strand conformation polymorphism
- Single-stranded DNA fragments of case and control nucleic acids will be denatured and allowed to renature.
- the secondary structure of single-stranded nucleic acids varies according to sequence. The resulting alteration in electrophoretic mobility enables the detection of even a single base change.
- the DNA fragments may be labeled or detected with labeled probes.
- RNA rather than DNA
- the method utilizes heteroduplex analysis to separate double stranded heteroduplex molecules on the basis of changes in electrophoretic mobility (Kee et al., 1991).
- the movement of mutant or wild-type fragments in a polyacrylamide gel containing a gradient of denaturant is assayed using denaturing gradient gel electrophoresis (DGGE) (Myers et al., 1985).
- DGGE denaturing gradient gel electrophoresis
- DNA will be modified to insure that it does not completely denature, for example by adding a GC clamp of approximately 40 bp of high-melting GC-rich DNA by PCR.
- a temperature gradient is used in place of a denaturing gradient to identify differences in the mobility of control and sample DNA (Rosenbaum et al., 1987).
- the mutant fragment is detected using denaturing HPLC (see e.g. Hoogendoorn et al., 2000).
- oligonucleotide primers may be prepared in which the polymorphism is placed centrally and then hybridized to target DNA under conditions which permit hybridization only if a perfect match is found (Saiki et al., 1986; Saiki et al., 1989). Such oligonucleotides are hybridized to PCR amplified target DNA or a number of different mutations when the oligonucleotides are attached to the hybridizing membrane and hybridized with labeled target DNA.
- the amplification, the allele-specific hybridization and the detection can be done in a single assay following the principle of the 5' nuclease assay (e.g. see Livak et al., 1995).
- the associated allele, a particular allele of a polymorphic locus, or the like is amplified by PCR in the presence of both allele-specific oligonucleotides, each specific for one or the other allele.
- Each probe has a different fluorescent dye at the 5' end and a quencher at the 3' end.
- the Taq polymerase via its 5' exonuclease activity will release the corresponding dyes. The latter will thus reveal the genotype of the amplified product.
- Hybridization assays may also be carried out with a temperature gradient following the principle of dynamic allele-specific hybridization or like e.g. Jobs et al., (2003); and Bourgeois and Labuda, (2004).
- the hybridization is done using one of the two allele-specific oligonucleotides labeled with a fluorescent dye, and an intercalating quencher under a gradually increasing temperature.
- the probe is hybridized to both the mismatched and full-matched template.
- the probe melts at a lower temperature when hybridized to the template with a mismatch.
- the release of the probe is captured by an emission of the fluorescent dye, away from the quencher.
- the probe melts at a higher temperature when hybridized to the template with no mismatch.
- the temperature-dependent fluorescence signals therefore indicate the absence or presence of an associated allele, a particular allele of a polymorphic locus, or the like ( e.g. Jobs et al., 2003).
- the hybridization is done under a gradually decreasing temperature. In this case, both allele-specific oligonucleotides are hybridized to the template competitively. At high temperature none of the two probes are hybridized. Once the optimal temperature of the full- matched probe is reached, it hybridizes and leaves no target for the mismatched probe (e.g. Bourgeois and Labuda, 2004). In the latter case, if the allele-specific probes are differently labeled, then they are hybridized to a single PCR-amplified target. If the probes are labeled with the same dye, then the probe cocktail is hybridized twice to identical templates with only one labeled probe, different in the two cocktails, in the presence of the unlabeled competitive probe.
- Oligonucleotides used as primers for specific amplification may carry the associated allele, a particular allele of a polymorphic locus, or the like, also referred to as "mutation" of interest in the center of the molecule, so that amplification depends on differential hybridization (Gibbs et al., 1989) or at the extreme 3' end of one primer where, under appropriate conditions, mismatch can prevent, or reduce polymerase extension (Prossner, 1993).
- amplification may also be performed using Taq ligase for amplification (Barany, 1991).
- ligation will occur only if there is a perfect match at the 3 1 end of the 5' sequence making it possible to detect the presence of a known associated allele, a particular allele of a polymorphic locus, or the like at a specific site by looking for the presence or absence of amplification.
- the products of such an oligonucleotide ligation assay can also be detected by means of gel electrophoresis.
- the oligonucleotides may contain universal tags used in PCR amplification and zip code tags that are different for each allele. The zip code tags are used to isolate a specific, labeled oligonucleotide that may contain a mobility modifier (e.g. Grossman et al., 1994).
- allele-specific elongation followed by ligation will form a template for PCR amplification.
- elongation will occur only if there is a perfect match at the 3' end of the allele-specific oligonucleotide using a DNA polymerase.
- This reaction is performed directly on the genomic DNA and the extension/ligation products are amplified by PCR.
- the oligonucleotides contain universal tags allowing amplification at a high multiplex level and a zip code for SNP identification.
- the PCR tags are designed in such a way that the two alleles of a SNP are amplified by different forward primers, each having a different dye.
- the zip code tags are the same for both alleles of a given SNPs and they are used for hybridization of the PCR-amplified products to oligonucleotides bound to a solid support, chip, bead array or like.
- Fan et al. Cold Spring Harbor Symposia on Quantitative Biology, Vol. LXVIII, pp. 69-78 2003.
- Another alternative includes the single-base extension/ligation assay using a molecular inversion probe, consisting of a single, long oligonucleotide (see e.g. Hardenbol et al., 2003).
- the oligonucleotide hybridizes on both side of the SNP locus directly on the genomic DNA, leaving a one-base gap at the SNP locus.
- the gap-filling, one-base extension/ligation is performed in four tubes, each having a different dNTP.
- the oligonucleotide is circularized whereas unreactive, linear oligonucleotides are degraded using an exonuclease such as exonuclease I of E. coli.
- the circular oligonucleotides are then linearized and the products are amplified and labeled using universal tags on the oligonucleotides.
- the original oligonucleotide also contains a SNP-specific zip code allowing hybridization to oligonucleotides bound to a solid support, chip, and bead array or like. This reaction can be performed at a high multiplexed level.
- the associated allele, a particular allele of a polymorphic locus, or the like is scored by single-base extension (see e.g. U.S. Pat. No. 5,888,819).
- the template is first amplified by PCR.
- the extension oligonucleotide is then hybridized next to the SNP locus and the extension reaction is performed using a thermostable polymerase such as ThermoSequenase (GE Healthcare) in the presence of labeled ddNTPs. This reaction can therefore be cycled several times. The identity of the labeled ddNTP incorporated will reveal the genotype at the SNP locus.
- the labeled products can be detected by means of gel electrophoresis, fluorescence polarization (e.g. Chen et a/., 1999) or by hybridization to oligonucleotides bound to a solid support, chip, and bead array or like. In the latter case, the extension oligonucleotide will contain a SNP-specific zip code tag.
- a SNP is scored by selective termination of extension.
- the template is first amplified by PCR and the extension oligonucleotide hybridizes in the vicinity of the SNP locus, close to but not necessarily adjacent to it.
- the extension reaction is carried out using a thermostable polymerase such as ThermoSequenase (GE Healthcare) in the presence of a mix of dNTPs and at least one ddNTP.
- ThermoSequenase GE Healthcare
- ThermoSequenase GE Healthcare
- ThermoSequenase GE Healthcare
- SNPs are detected using an invasive cleavage assay (see U.S. Pat. No. 6,090,543).
- oligonucleotides per SNP to interrogate but these are used in a two step-reaction. During the primary reaction, three of the designed oligonucleotides are first hybridized directly to the genomic DNA. One of them is locus-specific and hybridizes up to the SNP locus (the pairing of the 3' base at the SNP locus is not necessary).
- the present invention provides methods for identifying agents that modulate the expression of a nucleic acid encoding a gene from Tables 3 and 4. Such methods may utilize any available means of monitoring for changes in the expression level of the nucleic acids of the invention.
- an agent is said to modulate the expression of a nucleic acid of the invention if it is capable of up- or down- regulating expression of the nucleic acid in a cell.
- Such cells can be obtained from any parts of the body such as the respiratory system, lung, trachea, esophagus, bronchus and bronchiole, duct, alveoli, alviolar duct, alveolar sacs, stomach, nasal cavity, paranasal sinuses, nasopharynx, larynx, inner and outer lung coatings, inner and outer trachea coatings, mucosa, submucosa, rectum, scalp, blood, dermis, epidermis, skin cells, cutaneous surfaces, intertrigious areas, genitalia, vessels and endothelium.
- the respiratory system such as the respiratory system, lung, trachea, esophagus, bronchus and bronchiole, duct, alveoli, alviolar duct, alveolar sacs, stomach, nasal cavity, paranasal sinuses, nasopharynx, larynx, inner and outer lung coatings, inner and outer
- cells that can be used are: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, dendritic cell, cilated cell, synovial cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina intestinal lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
- the expression of a nucleic acid encoding a gene of the invention in a cell or tissue sample is monitored directly by hybridization to the nucleic acids of the invention.
- Cell lines or tissues are exposed to the agent to be tested under appropriate conditions and time and total RNA or mRNA is isolated by standard procedures such as those disclosed in Sambrook et al., (1989) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press).
- Probes to detect differences in RNA expression levels between cells exposed to the agent and control cells may be prepared as described above. Hybridization conditions are modified using known methods, such as those described by Sambrook et al., and Ausubel et al., as required for each probe. Hybridization of total cellular RNA or RNA enriched for polyA RNA can be accomplished in any available format. For instance, total cellular RNA or RNA enriched for polyA RNA can be affixed to a solid support and the solid support exposed to at least one probe comprising at least one, or part of one of the sequences of the invention under conditions in which the probe will specifically hybridize.
- nucleic acid fragments comprising at least one, or part of one of the sequences of the invention can be affixed to a solid support, such as a silicon chip or a porous glass wafer.
- the chip or wafer can then be exposed to total cellular RNA or polyA RNA from a sample under conditions in which the affixed sequences will specifically hybridize to the RNA.
- agents which up or down regulate expression are identified.
- an agent is said to modulate the expression of a protein of the invention if it is capable of up- or down- regulating expression of the protein in a cell.
- Such cells can be obtained from any parts of the body such as the respiratory system, lung, trachea, esophagus, bronchus and bronchiole, duct, alveoli, alviolar duct, alveolar sacs, stomach, nasal cavity, paranasal sinuses, nasopharynx, larynx, inner and outer lung coatings, inner and outer trachea coatings, mucosa, submucosa, rectum, scalp, blood, dermis, epidermis, skin cells, cutaneous surfaces, intertrigious areas, genitalia, vessels and endothelium.
- the respiratory system such as the respiratory system, lung, trachea, esophagus, bronchus and bronchiole, duct, alveoli, alviolar duct, alveolar sacs, stomach, nasal cavity, paranasal sinuses, nasopharynx, larynx, inner and outer lung coatings, inner and outer
- cells that can be used are: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, synovial cell, cilated cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina intestinal lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
- the specific activity of a protein of the invention may be assayed in a cell population that has been exposed to the agent to be tested and compared to an unexposed control cell population may be assayed.
- Cell lines or populations are exposed to the agent to be tested under appropriate conditions and times.
- Cellular lysates may be prepared from the exposed cell line or population and a control, unexposed cell line or population. The cellular lysates are then analyzed with the probe.
- Antibody probes can be prepared by immunizing suitable mammalian hosts utilizing appropriate immunization protocols using the proteins of the invention or antigen-containing fragments thereof. To enhance immunogenicity, these proteins or fragments can be conjugated to suitable carriers. Methods for preparing immunogenic conjugates with carriers such as BSA, KLH or other carrier proteins are well known in the art. In some circumstances, direct conjugation using, for example, carbodiimide reagents may be effective; in other instances linking reagents such as those supplied by Pierce Chemical Co. (Rockford, IL) may be desirable to provide accessibility to the hapten.
- the hapten peptides can be extended at either the amino or carboxy terminus with a cysteine residue or interspersed with cysteine residues, for example, to facilitate linking to a carrier.
- Administration of the immunogens is conducted generally by injection over a suitable time period and with use of suitable adjuvants, as is generally understood in the art.
- suitable adjuvants as is generally understood in the art.
- titers of antibodies are taken to determine adequacy of antibody formation. While the polyclonal antisera produced in this way may be satisfactory for some applications, for pharmaceutical compositions, use of monoclonal preparations is preferred.
- Immortalized cell lines which secrete the desired monoclonal antibodies may be prepared using standard methods, see e.g., Kohler & Milstein (1992) or modifications which affect immortalization of lymphocytes or spleen cells, as is generally known.
- the immortalized cell lines secreting the desired antibodies can be screened by immunoassay in which the antigen is the peptide hapten, polypeptide or protein.
- the cells can be cultured either in vitro or by production in ascites fluid.
- the desired monoclonal antibodies may be recovered from the culture supernatant or from the ascites supernatant.
- Fragments of the monoclonal antibodies or the polyclonal antisera which contain the immunologically significant portion(s) can be used as antagonists, as well as the intact antibodies.
- Use of immunologically reactive fragments, such as Fab or Fab' fragments, is often preferable, especially in a therapeutic context, as these fragments are generally less immunogenic than the whole immunoglobulin.
- the antibodies or fragments may also be produced, using current technology, by recombinant means.
- Antibody regions that bind specifically to the desired regions of the protein can also be produced in the context of chimeras derived from multiple species.
- Antibody regions that bind specifically to the desired regions of the protein can also be produced in the context of chimeras from multiple species, for instance, humanized antibodies.
- the antibody can therefore be a humanized antibody or a human antibody, as described in U.S. Patent 5,585,089 or Riechmann et al. (1988).
- Agents that are assayed in the above method can be randomly selected or rationally selected or designed.
- an agent is said to be randomly selected when the agent is chosen randomly without considering the specific sequences involved in the association of the protein of the invention alone or with its associated substrates, binding partners, etc.
- An example of randomly selected agents is the use of a chemical library or a peptide combinatorial library, or a growth broth of an organism.
- an agent is said to be rationally selected or designed when the agent is chosen on a non-random basis which takes into account the sequence of the target site or its conformation in connection with the agent's action.
- Agents can be rationally selected or rationally designed by utilizing the peptide sequences that make up these sites.
- a rationally selected peptide agent can be a peptide whose amino acid sequence is identical to or a derivative of any functional consensus site.
- the agents of the present invention can be, as examples, oligonucleotides, antisense polynucleotides, interfering RNA, peptides, peptide mimetics, antibodies, antibody fragments, small molecules, vitamin derivatives, as well as carbohydrates.
- Peptide agents of the invention can be prepared using standard solid phase (or solution phase) peptide synthesis methods, as is known in the art.
- the DNA encoding these peptides may be synthesized using commercially available oligonucleotide synthesis instrumentation and produced recombinant ⁇ using standard recombinant production systems. The production using solid phase peptide synthesis is necessitated if non-gene-encoded amino acids are to be included.
- Another class of agents of the present invention includes antibodies or fragments thereof that bind to a protein encoded by a gene in Tables 3 and 4.
- Antibody agents can be obtained by immunization of suitable mammalian subjects with peptides, containing as antigenic regions, those portions of the protein intended to be targeted by the antibodies (see section above of antibodies as probes for standard antibody preparation methodologies).
- the present invention includes peptide mimetics that mimic the three-dimensional structure of the protein encoded by a gene from Tables 3 and 4.
- peptide mimetics may have significant advantages over naturally occurring peptides, including, for example: more economical production, greater chemical stability, enhanced pharmacological properties (half-life, absorption, potency, efficacy, etc.), altered specificity (e.g., a broad-spectrum of biological activities), reduced antigenicity and others.
- mimetics are peptide-containing molecules that mimic elements of protein secondary structure.
- peptide mimetics The underlying rationale behind the use of peptide mimetics is that the peptide backbone of proteins exists chiefly to orient amino acid side chains in such a way as to facilitate molecular interactions, such as those of antibody and antigen. A peptide mimetic is expected to permit molecular interactions similar to the natural molecule.
- peptide analogs are commonly used in the pharmaceutical industry as non-peptide drugs with properties analogous to those of the template peptide. These types of non-peptide compounds are also referred to as peptide mimetics or peptidomimetics (Fauchere, 1986; Veber & Freidinger, 1985; Evans et a/., 1987) which are usually developed with the aid of computerized molecular modeling.
- Peptide mimetics that are structurally similar to therapeutically useful peptides may be used to produce an equivalent therapeutic or prophylactic effect.
- peptide mimetics are structurally similar to a paradigm polypeptide (i.e., a polypeptide that has a biochemical property or pharmacological activity), but have one or more peptide linkages optionally replaced by a linkage using methods known in the art.
- Labeling of peptide mimetics usually involves covalent attachment of one or more labels, directly or through a spacer (e.g., an amide group), to non-interfering position(s) on the peptide mimetic that are predicted by quantitative structure-activity data and molecular modeling.
- Such non-interfering positions generally are positions that do not form direct contacts with the macromolecule(s) to which the peptide mimetic binds to produce the therapeutic effect.
- Derivitization (e.g., labeling) of peptide mimetics should not substantially interfere with the desired biological or pharmacological activity of the peptide mimetic.
- the use of peptide mimetics can be enhanced through the use of combinatorial chemistry to create drug libraries.
- the design of peptide mimetics can be aided by identifying amino acid mutations that increase or decrease binding of the protein to its binding partners. Approaches that can be used include the yeast two hybrid method (see Chien et al., 1991) and the phage display method.
- the two hybrid method detects protein- protein interactions in yeast (Fields et al., 1989).
- the phage display method detects the interaction between an immobilized protein and a protein that is expressed on the surface of phages such as lambda and M13 (Amberg et al., 1993; Hogrefe et al., 1993). These methods allow positive and negative selection for protein-protein interactions and the identification of the sequences that determine these interactions.
- the present invention also relates to methods for diagnosing inflammatory disease or a related disease, preferably asthma disease, a disposition to such disease, predisposition to such a disease and/or disease progression.
- the steps comprise contacting a target sample with (a) nucleic acid molecule(s) or fragments thereof and comparing the concentration of individual mRNA(s) with the concentration of the corresponding mRNA(s) from at least one healthy donor.
- An aberrant (increased or decreased) mRNA level of at least one gene from Tables 3 and 4, at least 5 or 10 genes from Tables 3 and 4, at least 50 genes from Tables 3 and 4, at least 100 genes from Tables 3 and 4 or at least 200 genes from Tables 3 and 4 determined in the sample in comparison to the control sample is an indication of asthma disease or a related disease or a disposition to such kinds of diseases.
- samples are, preferably, obtained from inflamed lung tissue.
- Samples can also be obtained from any parts of the body such as the respiratory system, lung, trachea, esophagus, bronchus and bronchiole, duct, alveoli, alviolar duct, alveolar sacs, stomach, nasal cavity, paranasal sinuses, nasopharynx, larynx, inner and outer lung coatings, inner and outer trachea coatings, mucosa, submucosa, rectum, scalp, blood, dermis, epidermis, skin cells, cutaneous surfaces, intertrigious areas, genitalia, vessels and endothelium.
- the respiratory system such as the respiratory system, lung, trachea, esophagus, bronchus and bronchiole, duct, alveoli, alviolar duct, alveolar sacs, stomach, nasal cavity, paranasal sinuses, nasopharynx, larynx, inner and outer lung coatings, inner and
- cells that can be used are: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, synovial cell, cilated cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina intestinal lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
- RNA is obtained from cells according to standard procedures and, preferably, reverse-transcribed.
- a DNAse treatment is performed.
- Some non-limiting examples of cells that can be used are: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, synovial cell, cilated cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina basement lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
- the nucleic acid molecule or fragment is typically a nucleic acid probe for hybridization or a primer for PCR.
- the person skilled in the art is in a position to design suitable nucleic acids probes based on the information provided in the Tables of the present invention.
- the target cellular component i.e. mRNA, e.g., in lung tissue
- Detection methods include Northern blot analysis, RNase protection, in situ methods, e.g.
- PCR in situ hybridization
- in vitro amplification methods PCR, LCR, QRNA replicase or RNA- transcription/amplification (TAS, 3SR), reverse dot blot disclosed in EP- B10237362
- RNA- transcription/amplification TAS, 3SR
- reverse dot blot disclosed in EP- B10237362
- products obtained by in vitro amplification can be detected according to established methods, e.g. by separating the products on agarose or polyacrylamide gels and by subsequent staining with ethidium bromide.
- the amplified products can be detected by using labeled primers for amplification or labeled dNTPs.
- detection is based on a microarray.
- the probes (or primers) (or, alternatively, the reverse-transcribed sample mRNAs) can be detectably labeled, for example, with a radioisotope, a bioluminescent compound, a chemiluminescent compound, a fluorescent compound, a metal chelate, or an enzyme.
- the present invention also relates to the use of the nucleic acid molecules or fragments described above for the preparation of a diagnostic composition for the diagnosis of asthma disease or a disposition to such a disease.
- the present invention also relates to the use of the nucleic acid molecules of the present invention for the isolation or development of a compound which is useful for therapy of asthma disease.
- the nucleic acid molecules of the invention and the data obtained using said nucleic acid molecules for diagnosis of asthma disease might allow for the identification of further genes which are specifically dysregulated, and thus may be considered as potential targets for therapeutic interventions.
- the invention further provides prognostic assays that can be used to identify subjects having or at risk of developing asthma disease.
- a test sample is obtained from a subject and the amount and/or concentration of the nucleic acid described in Tables 3 and 4 is determined; wherein the presence of an associated allele, a particular allele of a polymorphic locus, or the likes in the nucleic acids sequences of this invention (see SEQ ID from Tables 1 or 3) can be diagnostic for a subject having or at risk of developing asthma.
- a test sample refers to a biological sample obtained from a subject of interest.
- a test sample can be a biological fluid, a cell sample, or tissue.
- a biological fluid can be, but is not limited to saliva, serum, mucus, urine, stools, spermatozoids, vaginal secretions, lymph, amiotic liquid, pleural liquid and tears.
- Cells can be, but are not limited to: muscle cells, nervous cells, blood and vessels cells, dermis, epidermis and other skin cells, smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, synovial cell, cilated cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina intestinal lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
- the prognostic assays described herein can be used to determine whether a subject can be administered an agent (e.g., an agonist, antagonist, peptidomimetic, polypeptide, nucleic acid such as antisense DNA or interfering RNA (RNAi), small molecule or other drug candidate) to treat asthma disease.
- an agent e.g., an agonist, antagonist, peptidomimetic, polypeptide, nucleic acid such as antisense DNA or interfering RNA (RNAi), small molecule or other drug candidate
- these assays can be used to predict whether an individual will have an efficacious response or will experience adverse events in response to such an agent.
- such methods can be used to determine whether a subject can be effectively treated with an agent that modulates the expression and/or activity of a gene from Tables 3 and 4 or the nucleic acids described herein.
- an association study may be performed to identify polymorphisms from Table 1 that are associated with a given response to the agent, e.g., an efficacious response or the likelihood of one or more adverse events.
- one embodiment of the present invention provides methods for determining whether a subject can be effectively treated with an agent for a disorder associated with aberrant expression or activity of a gene from Tables 3 and 4 in which a test sample is obtained and nucleic acids or polypeptides from Tables 3 and 4 are detected (e.g., wherein the presence of a particular level of expression of a gene from Tables 3 and 4 or a particular allelic variant of such gene, such as polymorphism from Table 1 , is diagnostic for a subject that can be administered an agent to treat a disorder such as asthma disease).
- the method includes obtaining a sample from a subject suspected of having asthma disease or an affected individual and exposing such sample to an agent.
- the expression and/or activity of the nucleic acids and/or genes of the invention are monitored before and after treatment with such agent to assess the effect of such agent. After analysis of the expression values, one skilled in the art can determine whether such agent can effectively treat such subject.
- the method includes obtaining a sample from a subject having or susceptible to developing asthma disease and determining the allelic constitution of polymorphisms from Table 1 that are associated with a particular response to an agent. After analysis of the allelic constitution of the individual at the associated polymorphisms, one skilled in the art can determine whether such agent can effectively treat such subject.
- the methods of the invention can also be used to detect genetic alterations in a gene from Tables 3 and 4, thereby determining if a subject with the lesioned gene is at risk for a disorder associated with asthma disease.
- the methods include detecting, in a sample of cells from the subject, the presence or absence of a genetic alteration characterized by at least one alteration linked to or affecting the integrity of a gene from Tables 3 and 4 encoding a polypeptide or the misexpression of such gene.
- such genetic alterations can be detected by ascertaining the existence of at least one of: (1) a deletion of one or more nucleotides from a gene from Tables 3 and 4; (2) an addition of one or more nucleotides to a gene from Table 3 and 4; (3) a substitution of one or more nucleotides of a gene from Tables 3 and 4; (4) a chromosomal rearrangement of a gene from Tables 3 and 4; (5) an alteration in the level of a messenger RNA transcript of a gene from Tables 3 and 4; (6) aberrant modification of a gene from Tables 3 and 4, such as of the methylation pattern of the genomic DNA, (7) the presence of a non-wild type splicing pattern of a messenger RNA transcript of a gene from Tables 3 and 4; (8) inappropriate post-translational modification of a polypeptide encoded by a gene from Tables 3 and 4; and (9) alternative promoter use.
- a preferred biological sample is a peripheral blood sample obtained by conventional means from a subject.
- Another preferred biological sample is a buccal swab.
- Other biological samples can be, but are not limited to, urine, stools, spermatozoids, respiratory system secretions, vaginal secretions, lymph, amiotic liquid, pleural liquid and tears.
- detection of the alteration involves the use of a probe/primer in a polymerase chain reaction (PCR) (see, e.g., U.S. Pat. Nos. 4,683,195 and 4,683,202), such as anchor PCR or RACE PCR, or alternatively, in a ligation chain reaction (LCR) (see, e.g., Landegran et a/., 1988; and Nakazawa et al., 1994), the latter of which can be particularly useful for detecting point mutations in a gene from Tables 3 and 4 (see Abavaya et al., 1995).
- PCR polymerase chain reaction
- LCR ligation chain reaction
- This method can include the steps of collecting a sample of cells from a patient, isolating nucleic acid (e.g., genomic DNA, mRNA, or both) from the cells of the sample, contacting the nucleic acid sample with one or more primers which specifically hybridize to a gene from Tables 3 and 4 under conditions such that hybridization and amplification of the nucleic acid from Tables 3 and 4 (if present) occurs, and detecting the presence or absence of an amplification product, or detecting the size of the amplification product and comparing the length to a control sample.
- PCR and/or LCR may be desirable to use as a preliminary amplification step in conjunction with some of the techniques used for detecting a mutation, an associated allele, a particular allele of a polymorphic locus, or the like described herein.
- Alternative amplification methods include: self sustained sequence replication (Guatelli et al., 1990), transcriptional amplification system (Kwoh et al., 1989), Q- Beta Replicase (Lizardi et al., 1988), isothermal amplification (e.g. Dean et al., 2002); and Hafner et al., 2001), or any other nucleic acid amplification method, followed by the detection of the amplified molecules using techniques well known to those of ordinary skill in the art. These detection schemes are especially useful for the detection of nucleic acid molecules if such molecules are present in very low number.
- alterations in a gene from Tables 3 and 4, from a sample cell can be identified by identifying changes in a restriction enzyme cleavage pattern.
- sample and control DNA is isolated, amplified (optionally), digested with one or more restriction endonucleases, and fragment length sizes are determined by gel electrophoresis and compared. Differences in fragment length sizes between sample and control DNA indicate a mutation(s), an associated allele, a particular allele of a polymorphic locus, or the like in the sample DNA.
- sequence specific ribozymes see, e.g., U.S. Pat. No. 5,498,531 or DNAzyme e.g. U.S. Pat. No. 5,807,718) can be used to score for the presence of specific associated allele, a particular allele of a polymorphic locus, or the likes by development or loss of a ribozyme or DNAzyme cleavage site.
- the present invention also relates to further methods for diagnosing asthma disease or a related disorder, a disposition to such disorder, predisposition to such a disorder and/or disorder progression.
- the steps comprise contacting a target sample with (a) nucleic molecuie(s) or fragments thereof and determining the presence or absence of a particular allele of a polymorphism that confers a disorder-related phenotype (e.g., predisposition to such a disorder and/or disorder progression).
- the presence of at least one allele from Table 1 that is associated with asthma disease (“associated allele"), at least 5 or 10 associated alleles from Table 1 , at least 50 associated alleles from Table 1 , at least 100 associated alleles from Table 1 , or at least 200 associated alleles from Table 1 , determined in the sample is an indication of asthma disease or a related disorder, a disposition or predisposition to such kinds of disorders, or a prognosis for such disorder progression.
- Such samples can be obtained from any parts of the body such as the respiratory system, lung, trachea, esophagus, bronchus and bronchiole, duct, alveoli, alviolar duct, alveolar sacs, stomach, nasal cavity, paranasal sinuses, nasopharynx, larynx, inner and outer lung coatings, inner and outer trachea coatings, mucosa, submucosa, rectum, scalp, blood, dermis, epidermis, skin cells, cutaneous surfaces, intertrigious areas, genitalia, vessels and endothelium.
- cells that can be used are: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, synovial cell, cilated cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina intestinal lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
- alterations in a gene from Tables 3 and 4 can be identified by hybridizing sample and control nucleic acids, e.g., DNA or RNA, to high density arrays or bead arrays containing tens to thousands of oligonucleotide probes (Cronin et al., 1996; Kozal et a!., 1996).
- alterations in a gene from Tables 3 and 4 can be identified in two dimensional arrays containing light-generated DNA probes as described in Cronin et a/., (1996).
- a first hybridization array of probes can be used to scan through long stretches of DNA in a sample and control to identify base changes between the sequences by making linear arrays of sequential overlapping probes. This step allows the identification of point mutations, associated alleles, particular alleles of a polymorphic locus, or the like. This step is followed by a second hybridization array that allows the characterization of specific mutations by using smaller, specialized probe arrays complementary to all variants, mutations, alleles detected. Each mutation array is composed of parallel probe sets, one complementary to the wild-type gene and the other complementary to the mutant gene.
- any of a variety of sequencing reactions known in the art can be used to directly sequence a gene from Tables 3 and 4 and detect an associated allele, a particular allele of a polymorphic locus, or the like by comparing the sequence of the sample gene from Tables 3 and 4 with the corresponding wild-type (control) sequence.
- Examples of sequencing reactions include those based on techniques developed by Maxam and Gilbert (1977) or Sanger (1977). It is also contemplated that any of a variety of automated sequencing procedures can be utilized when performing the diagnostic assays (Bio/Techniques 19:448, 1995) including sequencing by mass spectrometry (see, e.g. PCT International Publication No.
- RNA/RNA, DNA/DNA or RNA/DNA heteroduplexes Other methods of detecting an associated allele, a particular allele of a polymorphic locus, or the likes in a gene from Tables 3 and 4 include methods in which protection from cleavage agents is used to detect mismatched bases in RNA/RNA, DNA/DNA or RNA/DNA heteroduplexes (Myers et al., 1985).
- the technique of "mismatch cleavage" starts by providing heteroduplexes formed by hybridizing (labeled) RNA or DNA containing the wild-type gene sequence from Tables 3 and 4 with potentially mutant RNA or DNA obtained from a tissue sample.
- RNA/DNA duplexes can be treated with RNase and DNA/DNA hybrids treated with S1 nuclease to enzymatically digest the mismatched regions.
- either DNA/DNA or RNA/DNA duplexes can be treated with hydroxylamine or osmium tetroxide and with piperidine in order to digest mismatched regions.
- control DNA or RNA can be labeled for detection, as described herein.
- the mismatch cleavage reaction employs one or more proteins that recognize mismatched base pairs in double-stranded DNA (so called "DNA mismatch repair" enzymes) in defined systems for detecting and mapping point an associated allele, a particular allele of a polymorphic locus, or the likes in a gene from Tables 3 and 4 cDNAs obtained from samples of cells.
- DNA mismatch repair enzymes
- the mutY enzyme of E. coli cleaves A at G/A mismatches (Hsu et al., 1994).
- Other examples include, but are not limited to, the MutHLS enzyme complex of E.
- a probe based on a gene sequence from Tables 3 and 4 is hybridized to a cDNA or other DNA product from a test cell or cells.
- the duplex is treated with a DNA mismatch repair enzyme, and the cleavage products, if any, can be detected using electrophoresis protocols or the like. See, for example, U.S. Pat. No. 5,459,039.
- the screen can be performed in vivo following the insertion of the heteroduplexes in an appropriate vector. The whole procedure is known to those ordinary skilled in the art and is referred to as mismatch repair detection (see e.g. Fakhrai-Rad et al., 2004).
- alterations in electrophoretic mobility can be used to identify an associated allele, a particular allele of a polymorphic locus, or the likes in genes from Tables 3 and 4.
- SSCP single strand conformation polymorphism
- Single-stranded DNA fragments of sample and control nucleic acids from Tables 3 and 4 will be denatured and allowed to renature.
- the secondary structure of single-stranded nucleic acids varies according to sequence; the resulting alteration in electrophoretic mobility enables the detection of even a single base change.
- the DNA fragments may be labeled or detected with labeled probes.
- the sensitivity of the assay may be enhanced by using RNA (rather than DNA), in which the secondary structure is more sensitive to a change in sequence.
- the method utilizes heteroduplex analysis to separate double stranded heteroduplex molecules on the basis of changes in electrophoretic mobility (Kee ef a/., 1991).
- the movement of mutant or wild-type fragments in a polyacrylamide gel containing a gradient of denaturant is assayed using denaturing gradient gel electrophoresis (DGGE) (Myers et al., 1985).
- DGGE denaturing gradient gel electrophoresis
- DNA will be modified to insure that it does not completely denature, for example by adding a GC clamp of approximately 40 bp of high-melting GC-rich DNA by PCR.
- a temperature gradient is used in place of a denaturing gradient to identify differences in the mobility of control and sample DNA (Rosenbaum et al., 1987).
- the mutant fragment is detected using denaturing HPLC (see e.g. Hoogendoom et al., 2000).
- oligonucleotide primers may be prepared in which the known associated allele, particular allele of a polymorphic locus, or the like is placed centrally and then hybridized to target DNA under conditions which permit hybridization only if a perfect match is found (Saiki et al., 1986; Saiki et al., 1989).
- Such allele specific oligonucleotides are hybridized to PCR amplified target DNA of a number of different associated alleles, a particular allele of a polymorphic locus, or the likes where the oligonucleotides are attached to the hybridizing membrane and hybridized with labeled target DNA.
- the amplification, the allele-specific hybridization and the detection can be done in a single assay following the principle of the 5' nuclease assay (e.g. see Livak et al., 1995).
- the associated allele, a particular allele of a polymorphic locus, or the like locus is amplified by PCR in the presence of both allele-specific oligonucleotides, each specific for one or the other allele.
- Each probe has a different fluorescent dye at the 5' end and a quencher at the 3' end.
- the Taq polymerase via its 5' exonuclease activity will release the corresponding dyes. The latter will thus reveal the genotype of the amplified product.
- the hybridization may also be carried out with a temperature gradient following the principle of dynamic allele-specific hybridization or like (e.g. Jobs et al., 2003; and Bourgeois and Labuda, 2004).
- the hybridization is done using one of the two allele-specific oligonucleotides labeled with a fluorescent dye, an intercalating quencher under a gradually increasing temperature.
- the probe is hybridized to both the mismatched and full-matched template.
- the probe melts at a lower temperature when hybridized to the template with a mismatch.
- the release of the probe is captured by an emission of the fluorescent dye, away from the quencher.
- the probe melts at a higher temperature when hybridized to the template with no mismatch.
- the temperature- dependent fluorescence signals therefore indicate the absence or presence of the associated allele, particular allele of a polymorphic locus, or the like (e.g. Jobs ef al. supra).
- the hybridization is done under a gradually decreasing temperature.
- both allele-specific oligonucleotides are hybridized to the template competitively.
- none of the two probes is hybridized.
- the probe cocktail is hybridized twice to identical templates with only one labeled probe, different in the two cocktails, in the presence of the unlabeled competitive probe.
- allele specific amplification technology that depends on selective PCR amplification may be used in conjunction with the present invention. Oligonucleotides used as primers for specific amplification may carry the associated allele, particular allele of a polymorphic locus, or the like of interest in the center of the molecule, so that amplification depends on differential hybridization (Gibbs et al., 1989) or at the extreme 3' end of one primer where, under appropriate conditions, mismatch can prevent, or reduce polymerase extension (Prossner, 1993).
- amplification may also be performed using Taq ligase for amplification (Barany, 1991). In such cases, ligation will occur only if there is a perfect match at the 3' end of the 5' sequence making it possible to detect the presence of a known associated allele, a particular allele of a polymorphic locus, or the like at a specific site by looking for the presence or absence of amplification.
- oligonucleotide ligation assay can also be detected by means of gel electrophoresis.
- the oligonucleotides may contain universal tags used in PCR amplification and zip code tags that are different for each allele.
- the zip code tags are used to isolate a specific, labeled oligonucleotide that may contain a mobility modifier (e.g. Grossman et a/., 1994).
- allele-specific elongation followed by ligation will form a template for PCR amplification.
- elongation will occur only if there is a perfect match at the 3' end of the allele-specific oligonucleotide using a DNA polymerase.
- This reaction is performed directly on the genomic DNA and the extension/ligation products are amplified by PCR.
- the oligonucleotides contain universal tags allowing amplification at a high multiplex level and a zip code for SNP identification.
- the PCR tags are designed in such a way that the two alleles of a SNP are amplified by different forward primers, each having a different dye.
- the zip code tags are the same for both alleles of a given SNP and they are used for hybridization of the PCR-amplified products to oligonucleotides bound to a solid support, chip, bead array or like.
- Fan et al. Cold Spring Harbor Symposia on Quantitative Biology, Vol. LXVIII, pp. 69-78, 2003.
- Another alternative includes the single-base extension/ligation assay using a molecular inversion probe, consisting of a single, long oligonucleotide (see e.g. Hardenbol et al., 2003).
- the oligonucleotide hybridizes on both sides of the SNP locus directly on the genomic DNA, leaving a one-base gap at the SNP locus.
- the gap-filling, one-base extension/ligation is performed in four tubes, each having a different dNTP.
- the oligonucleotide is circularized whereas unreactive, linear oligonucleotides are degraded using an exonulease such as exonuclease I of E. coli.
- the circular oligonucleotides are then linearized and the products are amplified and labeled using universal tags on the oligonucleotides.
- the original oligonucleotide also contains a SNP-specific zip code allowing hybridization to oligonucleotides bound to a solid support, chip, bead array or the like. This reaction can be performed at a highly multiplexed level.
- the associated allele, particular allele of a polymorphic locus, or the like is scored by single-base extension (see e.g. U.S. Pat. No. 5,888,819).
- the template is first amplified by PCR.
- the extension oligonucleotide is then hybridized next to the SNP locus and the extension reaction is performed using a thermostable polymerase such as ThermoSequenase (GE Healthcare) in the presence of labeled ddNTPs. This reaction can therefore be cycled several times. The identity of the labeled ddNTP incorporated will reveal the genotype at the SNP locus.
- the labeled products can be detected by means of gel electrophoresis, fluorescence polarization (e.g. Chen et al., 1999) or by hybridization to oligonucleotides bound to a solid support, chip, bead array or the like. In the latter case, the extension oligonucleotide will contain a SNP-specific zip code tag.
- the variant is scored by selective termination of extension.
- the template is first amplified by PCR and the extension oligonucleotide hybridizes in vicinity to the SNP locus, close to but not necessarily adjacent to it.
- the extension reaction is carried out using a thermostable polymerase such as ThermoSequenase (GE Healthcare) in the presence of a mix of dNTPs and at least one ddNTP.
- ThermoSequenase GE Healthcare
- ThermoSequenase GE Healthcare
- ThermoSequenase GE Healthcare
- the associated allele, particular allele of a polymorphic locus, or the like is detected using an invasive cleavage assay (see U.S. Pat. No. 6,090,543).
- an invasive cleavage assay see U.S. Pat. No. 6,090,543
- allele-specific oligonucleotides that hybridize in tandem to the locus-specific probe but also contain a 5' flap that is specific for each allele of the SNP.
- this creates a structure that is recognized by a cleavase enzyme (U.S. Pat. No. 6,090,606) and the allele- specific flap is released.
- the flap fragments hybridize to a specific cassette to recreate the same structure as above except that the cleavage will release a small DNA fragment labeled with a fluorescent dye that can be detected using regular fluorescence detector. In the cassette, the emission of the dye is inhibited by a quencher.
- microsatellites can also be useful to detect the genetic predisposition of an individual to a given disorder.
- Microsatellites consist of short sequence motifs of one or a few nucleotides repeated in tandem. The most common motifs are polynucleotide runs, dinucleotide repeats (particularly the CA repeats) and trinucleotide repeats. However, other types of repeats can also be used.
- the microsatellites are very useful for genetic mapping because they are highly polymorphic in their length. Microsatellite markers can be typed by various means, including but not limited to DNA fragment sizing, oligonucleotide ligation assay and mass spectrometry.
- the locus of the microsatellite is amplified by PCR and the size of the PCR fragment will be directly correlated to the length of the microsatellite repeat.
- the size of the PCR fragment can be detected by regular means of gel electrophoresis.
- the fragment can be labeled internally during PCR or by using end-labeled oligonucleotides in the PCR reaction (e.g. Mansfield et al., 1996).
- the size of the PCR fragment is determined by mass spectrometry. In such a case, however, the flanking sequences need to be eliminated. This can be achieved by ribozyme cleavage of an RNA transcript of the microsatellite repeat (Krebs et al., 2001).
- the microsatellite locus is amplified using oligonucleotides that include a T7 promoter on one end and a ribozyme motif on the other end. Transcription of the amplified fragments will yield an RNA substrate for the ribozyme, releasing small RNA fragments that contain the repeated region. The size of the latter is determined by mass spectrometry.
- the flanking sequences are specifically degraded. This is achieved by replacing the dTTP in the PCR reaction by dUTP.
- dUTP nucleosides are then removed by uracyl DNA glycosylases and the resulting abasic sites are cleaved by either abasic endonucleases such as human AP endonuclease or chemical agents such as piperidine.
- Bases can also be modified post-PCR by chemical agents such as dimethyl sulfate and then cleaved by other chemical agents such as piperidine (see e.g. Maxam and Gilbert, 1977; U.S. Pat. No. 5,869,242; and U.S. Patent pending serial No. 60/335,068).
- an oligonucleotide ligation assay can be performed.
- the microsatellite locus is first amplified by PCR.
- different oligonucleotides can be submitted to ligation at the center of the repeat with a set of oligonucleotides covering all the possible lengths of the marker at a given locus (Zirvi et al., 1999).
- Another example of design of an oligonucleotide assay comprises the ligation of three oligonucleotides; a 5' oligonucleotide hybridizing to the 5' flanking sequence, a repeat oligonucleotide of the length of the shortest allele of the marker hybridizing to the repeated region and a set of 3' oligonucleotides covering all the existing alleles hybridizing to the 3' flanking sequence and a portion of the repeated region for all the alleles longer than the shortest one.
- the 3' oligonucleotide exclusively hybridizes to the 3' flanking sequence (U.S. Pat. No. 6,479,244).
- the methods described herein may be performed, for example, by utilizing prepackaged diagnostic kits comprising at least one probe nucleic acid selected from the SEQ ID of Table 1 , or antibody reagent described herein, which may be conveniently used, for example, in a clinical setting to diagnose patient exhibiting symptoms or a family history of a disorder or disorder involving abnormal activity of genes from Tables 3 and 4.
- the present invention provides methods of treating a disorder associated with asthma disease by expressing in vivo the nucleic acids of at least one gene from Tables 3 and 4.
- These nucleic acids can be inserted into any of a number of well- known vectors for the transfection of target cells and organisms as described below.
- the nucleic acids are transfected into cells, ex vivo or in vivo, through the interaction of the vector and the target cell.
- the nucleic acids encoding a gene from Tables 3 and 4 under the control of a promoter, then express the encoded protein, thereby mitigating the effects of absent, partial inactivation, or abnormal expression of a gene from Tables 3 and 4.
- Non-viral vector delivery systems include DNA plasmids, naked nucleic acid, and nucleic acid complexed with a delivery vehicle such as a liposome.
- Viral vector delivery systems include DNA and RNA viruses, which have either episomal or integrated genomes after delivery to the cell.
- RNA or DNA based viral systems for the delivery of nucleic acids take advantage of highly evolved processes for targeting a virus to specific cells in the body and trafficking the viral payload to the nucleus.
- Viral vectors can be administered directly to patients (in vivo) or they can be used to treat cells in vitro and the modified cells are administered to patients (ex vivo).
- Conventional viral based systems for the delivery of nucleic acids could include retroviral, lentivirus, adenoviral, adeno-associated and herpes simplex virus vectors for gene transfer.
- Viral vectors are currently the most efficient and versatile method of gene transfer in target cells and tissues. Integration in the host genome is possible with the retrovirus, lentivirus, and adeno-associated virus gene transfer methods, often resulting in long term expression of the inserted transgene. Additionally, high transduction efficiencies have been observed in many different cell types and target tissues.
- Lentiviral vectors are retroviral vectors that are able to transduce or infect non-dividing cells and typically produce high viral titers. Selection of a retroviral gene transfer system would therefore depend on the target tissue. Retroviral vectors are comprised of c/s-acting long terminal repeats with packaging capacity for up to 6- 10 kb of foreign sequence. The minimum cis-acting LTRs are sufficient for replication and packaging of the vectors, which are then used to integrate the therapeutic gene into the target cell to provide permanent transgene expression.
- Widely used retroviral vectors include those based upon murine leukemia virus (MuLV), gibbon ape leukemia virus (GaLV), Simian lmmuno deficiency virus (SIV), human immuno deficiency virus (HIV) 1 and combinations thereof (see, e.g., Buchscher et al., 1992; Johann et al., 1992; Sommerfelt et a/., 1990; Wilson et a/., 1989; Miller et al., 1999;and PCT/US94/05700).
- MiLV murine leukemia virus
- GaLV gibbon ape leukemia virus
- SIV Simian lmmuno deficiency virus
- HAV human immuno deficiency virus
- Adenoviral based systems are typically used.
- Adenoviral based vectors are capable of very high transduction efficiency in many cell types and do not require cell division. With such vectors, high titer and levels of expression have been obtained. This vector can be produced in large quantities in a relatively simple system.
- Adeno-associated virus (“AAV”) vectors are also used to transduce cells with target nucleic acids, e.g., in the in vitro production of nucleic acids and peptides, and for in vivo and ex vivo gene therapy procedures (see, e.g., West et al., 1987; U.S. Pat. No.
- pLASN and MFG-S are examples are retroviral vectors that have been used in clinical trials (Dunbar et al., 1995; Kohn et al., 1995; Malech et al., 1997).
- PA317/pl_ASN was the first therapeutic vector used in a gene therapy trial (Blaese et al., 1995). Transduction efficiencies of 50% or greater have been observed for MFG-S packaged vectors (Ellem et al., 1997; and Dranoff et al., 1997).
- rAAV Recombinant adeno-associated virus vectors
- All vectors are derived from a plasmid that retains only the AAV 145 bp inverted terminal repeats flanking the transgene expression cassette. Efficient gene transfer and stable transgene delivery due to integration into the genomes of the transduced cell are key features for this vector system (Wagner et al., 1998, Kearns et a/1996).
- Ad vectors Replication-deficient recombinant adenoviral vectors (Ad) are predominantly used in transient expression gene therapy; because they can be produced at high titer and they readily infect a number of different cell types. Most adenovirus vectors are engineered such that a transgene replaces the Ad E1a, E1b, and E3 genes; subsequently the replication defector vector is propagated in human 293 cells that supply the deleted gene function in trans. Ad vectors can transduce multiple types of tissues in vivo, including nondividing, differentiated cells such as those found in the liver, kidney and muscle tissues. Conventional Ad vectors have a large carrying capacity.
- Ad vector An example of the use of an Ad vector in a clinical trial involved polynucleotide therapy for antitumor immunization with intramuscular injection (Sterman et al., 1998). Additional examples of the use of adenovirus vectors for gene transfer in clinical trials include Rosenecker et al., 1996; Sterman et al., 1998; Welsh et al., 1995; Alvarez et al., 1997; Topf et al., 1998.
- Packaging cells are used to form virus particles that are capable of infecting a host cell. Such cells include 293 cells, which package adenovirus, and ⁇ 2 cells or PA317 cells, which package retrovirus.
- Viral vectors used in gene therapy are usually generated by a producer cell line that packages a nucleic acid vector into a viral particle. The vectors typically contain the minimal viral sequences required for packaging and subsequent integration into a host, other viral sequences being replaced by an expression cassette for the protein to be expressed. The missing viral functions are supplied in trans by the packaging cell line. For example, AAV vectors used in gene therapy typically only possess ITR sequences from the AAV genome which are required for packaging and integration into the host genome.
- Viral DNA is packaged in a cell line, which contains a helper plasmid encoding the other AAV genes, namely rep and cap, but lacking ITR sequences.
- the cell line is also infected with adenovirus as a helper.
- the helper virus promotes replication of the AAV vector and expression of AAV genes from the helper plasmid.
- the helper plasmid is not packaged in significant amounts due to a lack of ITR sequences. Contamination with adenovirus can be reduced by, e.g., heat treatment to which adenovirus is more sensitive than AAV.
- a viral vector is typically modified to have specificity for a given cell type by expressing a ligand as a fusion protein with a viral coat protein on the viruses outer surface.
- the ligand is chosen to have affinity for a receptor known to be present on the cell type of interest.
- Moloney murine leukemia virus can be modified to express human heregulin fused to gp70, and the recombinant virus infects certain human breast cancer cells expressing human epidermal growth factor receptor. This principle can be extended to other pairs of viruses expressing a ligand fusion protein and target cells expressing a receptor.
- filamentous phage can be engineered to display antibody fragments (e.g., Fab or Fv) having specific binding affinity for virtually any chosen cellular receptor.
- antibody fragments e.g., Fab or Fv
- Such vectors can be engineered to contain specific uptake sequences thought to favor uptake by specific target cells.
- Gene therapy vectors can be delivered in vivo by administration to an individual patient, typically by systemic administration (e.g., intravenous, intraperitoneal, intramuscular, subdermal, or intracranial infusion) or topical application.
- vectors can be delivered to cells ex vivo, such as cells explanted from an individual patient (e.g., lymphocytes, bone marrow aspirates, and tissue biopsy) or universal donor hematopoietic stem cells, followed by reimplantation of the cells into a patient, usually after selection for cells which have incorporated the vector.
- Ex vivo cell transfection for diagnostics, research, or for gene therapy is well known to those of skill in the art.
- cells are isolated from the subject organism, transfected with a nucleic acid (gene or cDNA), and re-infused back into the subject organism (e.g., patient).
- a nucleic acid gene or cDNA
- Various cell types suitable for ex vivo transfection are well known to those of skill in the art (see, e.g., Freshney et al., 1994; and the references cited therein for a discussion of how to isolate and culture cells from patients).
- stem cells are used in ex vivo procedures for cell transfection and gene therapy.
- stem cells can be differentiated into other cell types in vitro, or can be introduced into a mammal (such as the donor of the cells) where they will engraft in the bone marrow.
- Methods for differentiating CD34+ cells in vitro into clinically important immune cell types using cytokines such a GM-CSF, IFN- ⁇ and TNF- ⁇ are known (see Inaba ef a/., 1992).
- Stem cells are isolated for transduction and differentiation using known methods. For example, stem cells are isolated from bone marrow cells by panning the bone marrow cells with antibodies which bind unwanted cells, such as CD4+ and CD8+ (T cells), CD45+ (panB cells), GR-1 (granulocytes), and lad (differentiated antigen presenting cells).
- T cells CD4+ and CD8+
- CD45+ panB cells
- GR-1 granulocytes
- lad differentiated antigen presenting cells
- Vectors e.g., retroviruses, adenoviruses, liposomes, etc.
- therapeutic nucleic acids can be also administered directly to the organism for transduction of cells in vivo.
- naked DNA can be administered.
- nucleic acids from Tables 3 and 4 are administered in any suitable manner, preferably with the pharmaceutically acceptable carriers described above. Suitable methods of administering such nucleic acids are available and well known to those of skill in the art, and, although more than one route can be used to administer a particular composition, a particular route can often provide a more immediate and more effective reaction than another route (see Samulski et al., 1989).
- the present invention is not limited to any method of administering such nucleic acids, but preferentially uses the methods described herein.
- the present invention further provides other methods of treating asthma disease such as administering to an individual having asthma disease an effective amount of an agent that regulates the expression, activity or physical state of at least one gene from Tables 3 and 4.
- An "effective amount" of an agent is an amount that modulates a level of expression or activity of a gene from Tables 3 and 4, in a cell in the individual at least about 10%, at least about 20%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80% or more, compared to a level of the respective gene from Tables 3 and 4 in a cell in the individual in the absence of the compound.
- the preventive or therapeutic agents of the present invention may be administered, either orally or parenterally, systemically or locally.
- intravenous injection such as drip infusion, intramuscular injection, intraperitoneal injection, subcutaneous injection, suppositories, intestinal lavage, oral enteric coated tablets, and the like can be selected, and the method of administration may be chosen, as appropriate, depending on the age and the conditions of the patient.
- the effective dosage is chosen from the range of 0.01 mg to 100 mg per kg of body weight per administration.
- the dosage in the range of 1 to 1000 mg, preferably 5 to 50 mg per patient may be chosen.
- the therapeutic efficacy of the treatment may be monitored by observing various parts of the respiratory tract, by chest and sinus X-rays, barium, performing a spirometry breathing test, a routine pulmonary function test, or any other monitoring methods known in the art.
- monitoring efficacy can be, but are not limited to monitoring inflammatory conditions involving the respiratory system such as monitoring the amelioration on the lung capacity and function, decrease in pain, improved breathing, decreased breathlessness and wheeze, reduced chest pain, or improvement in vitality and exercise ability.
- the present invention further provides a method of treating an individual clinically diagnosed with asthma disease.
- the methods generally comprises analyzing a biological sample that includes a cell, in some cases, a respiratory system cell, from an individual clinically diagnosed with asthma disease for the presence of modified levels of expression of at least 1 gene, at least 10 genes, at least 50 genes, at least 100 genes, or at least 200 genes from Tables 3 and 4.
- a treatment plan that is most effective for individuals clinically diagnosed as having a condition associated with asthma disease is then selected on the basis of the detected expression of such genes in a cell.
- Treatment may include administering a composition that includes an agent that modulates the expression or activity of a protein from Tables 3 and 4 in the cell. Information obtained as described in the methods above can also be used to predict the response of the individual to a particular agent.
- the invention further provides a method for predicting a patient's likelihood to respond to a drug treatment for a condition associated with asthma disease, comprising determining whether modified levels of a gene from Tables 3 and 4 is present in a cell, wherein the presence of protein is predictive of the patient's likelihood to respond to a drug treatment for the condition.
- Examples of the prevention or improvement of symptoms accompanied by asthma disease that can monitored for effectiveness include improvement of breathing, amelioration on the lung capacity and function, decrease in pain, decreased breath lessness and wheeze, reduced chest pain, or improvement in vitality and exercise ability, and the like, and as a result, a preventing or improving agent for breathing amelioration, a preventing or improving agent for amelioration of lung capacity, an inhibitor breathlessness and wheeze, and the like can be identified.
- the invention also provides a method of predicting a response to therapy in a subject having asthma disease by determining the presence or absence in the subject of one or more markers associated with asthma disease described in Tables 1 , 3 or 4, diagnosing the subject in which the one or more markers are present as having asthma disease, and predicting a response to a therapy based on the diagnosis e.g., response to therapy may include an efficacious response and/or one or more adverse events.
- the invention also provides a method of optimizing therapy in a subject having asthma disease by determining the presence or absence in the subject of one or more markers associated with a clinical subtype of asthma disease, diagnosing the subject in which the one or more markers are present as having a particular clinical subtype of asthma disease, and treating the subject having a particular clinical subtype of asthma disease based on the diagnosis.
- treatment for the exercise- induced asthma subtype of asthma disease currently includes inhaled bronchodilators via a nebulizer.
- Example 1 Identification of cases and controls
- the Quebec founder population has two distinct advantages over general populations for LD mapping. Because it is relatively young (about 12 to 15 generations from the mid 17th century to the present), and because it has a limited but sufficient number of founders (approximately 2600 effective founders, Charbonneau et al. 1987), the Quebec population is characterized both by extended LD and by decreased genetic heterogeneity. The increased extent of LD allows the detection of disorder genes using a reasonable marker density, while still allowing the increased meiotic resolution of population-based mapping.
- the number of founders is small enough to result in increased LD and reduced allelic heterogeneity, yet large enough to insure that all of the major disorder genes involved in general populations are present in Quebec.
- Reduced allelic heterogeneity will act to increase relative risk imparted by the remaining alleles and so increase the power of case/control studies to detect a gene, an associated allele, a particular allele of a polymorphic locus, or the likes involved in complex disorders within the Quebec population.
- the specific combination of age in generations, optimal number of founders and large present population size makes the QFP optimal for LD-based gene mapping.
- Patient inclusion criteria for the study include a diagnosis of asthma by a lung specialist or an allergist. Also, the onset of the disease must have occurred before 35 years old. All human sampling was subject to ethical review procedures.
- the extraction method yielded high molecular weight DNA, and the quality of every DNA sample was verified by agarose gel electrophoresis. Genomic DNA appeared on the gel as a large band of very high molecular weight. The remaining two buffy coats were stored at -80°C as backups.
- trios were Parent, Parent, Child (PPC) trios.
- PPC Parent, Parent, Child
- One member of each trio was affected with asthma disease. These included 252 daughters, 134 sons, 68 mothers and 43 fathers.
- the two non-transmitted parental chromosomes were used as controls, when one of the parents was affected, that person's spouse provided the control chromosomes.
- the recruitment of trios allowed the precise determination of haplotypes.
- Genotyping was performed using Perlegen's ultra-high-throughput platform. Marker loci were amplified by PCR and hybridized to wafers containing arrays of oligonucleotides. Allele discrimination was performed through allele-specific hybridization. In total, 80,654 SNPs, distributed as evenly as possible throughout the genome, were genotyped on the 497 trios for a total of 120,560,328 genotypes. These markers were mostly selected from various databases including the ⁇ 1.6 million SNP database of Perlegen Life Sciences (Patil, 2001); several thousand were obtained from the HapMap consortium database and/or dbSNP at NCBI. The SNPs were chosen to maximize uniformity of genetic coverage and to cover a distribution of allele frequencies.
- the genotyping information was entered into a Unified Genotype Database (a proprietary database under development) from which it was accessed using custom-built programs for export to the genetic analysis pipeline. Analyses of these genotypes were performed with the statistical tools described in Example 3. The GWS permitted the identification of 231 candidate regions. Some of these are further analyzed by the Fine Mapping approach described below.
- the dataset from the GWS was verified for completeness of the trios.
- the program GGFileMod removed any trios with abnormal structure or missing individuals (e.g. trios without a proband, duos, singletons, etc.), and calculated the total number of complete trios in the dataset.
- the trios were also tested to make sure that no subjects within the cohort were related more closely than second cousins (6 meiotic steps).
- Markers or families not meeting these criteria were removed from the dataset in the following step. Analyses of variance were performed using the algorithm GenAnova, to assess whether families or markers have the greater effect on missing values and non-Mendelian segregation. This was used to determine the smallest number of data points to remove from the dataset to meet the requirements for missing values and non-Mendelian segregation. The families and/or markers were removed from the dataset using the program DataPull, which generates an output file that is used for subsequent analysis of the genotype data.
- the Program PhaseFinderSNP2.0 was used to determine phase from trio data on a marker-by-marker, trio-by-trio basis.
- the output file contains haplotype data for all trio members, including ambiguities where all trio members are heterozygous or where data is missing.
- the program FileWriterTemp was then used to determine case and control haplotypes and to prepare the data in the proper input format for the next stage of analysis, using the expectation maximization algorithm, PL-EM, to determine phase on the remaining ambiguities. This stage consists of several modules for resolution of the remaining phase ambiguities.
- PLEMInOuti was first used to recode the haplotypes for input into the PL-EM algorithm in 15-marker blocks.
- the haplotype information was encoded as genotypes, allowing for the entry of known phase into the algorithm, which limits the possible number of estimated haplotypes.
- the PL-EM algorithm was used to estimate haplotypes from the "genotype" data in 15-marker windows, advancing in increments of one marker across the chromosome. The results were then converted into multiple 15-marker haplotype files using the program PLEMInOut2.
- PLEMBIockGroup was used to convert the individual 15-marker block files into one continuous block of haplotypes for the entire chromosome, and to generate files for further analysis by LDSTATS, Hapfreq and HapColor.
- PLEMBIockGroup takes the consensus estimation of the allele call at each marker over all separate estimations (most markers are estimated 15 different times as the 15 marker blocks pass over their position).
- Haplotype sharing analysis was performed using the program LDSTATS.
- LDSTATS tests for association of haplotypes with the disease phenotype.
- the algorithms LDSTATS (v2.0) and LDSTATS (v3.0) define haplotypes using multi- marker windows that advance across the marker map in one-marker increments. Windows can be 1 , 3, 5, 7 or 9 markers wide. At each position the frequency of haplotypes in cases and controls was calculated. Allele frequency differences for single marker and 3-marker windows were then tested using Pearson's Chi- square test with one degree of freedom. Larger windows of multi-allelic haplotype association were tested using Smith's normalization of the square root of Pearson's Chi-square. In addition, LDSTATS calculates Chi-square values for the transmission disequilibrium test (TDT) for single markers in situations where the trios consisted of parents and an affected child. The significance of association for any given marker was calculated in the following manner:
- This permutation test provides the P value for mean significance of each marker over all windows in which it occurs.
- the regions with the most significant P-values were selected for further analyses. For each of these regions, the frequencies in cases and controls for the significant haplotypes were determined by HapFreq, a subroutine of LDSTATS(v3.0). The significantly shared haplotypes that were different between cases and controls were represented in different colors using HapColor.
- Haplotype frequencies were then used to verify that the significance was the result of distinct differences in frequency between cases and controls for specific common haplotypes, and was not the cumulative result of many rare haplotypes that existed only in either cases or controls (see Table 1).
- Conditional haplotype analyses were performed on subsets of the original set of cases and controls using the program LDSTAT. The selection of a subset of cases and their matched controls was based on the carrier status of cases at a gene or locus of interest.
- a reduced haplotype diversity was observed and we selected three sets of haplotypes for conditional analyses.
- the first set contained four protective haplotypes (111121222, 111111112, 111221212 and 212211111).
- the second set contained five risk haplotypes (211111111 , 111122211 , 211111112, 121121111 and 211112122) while the third set contained ten risk haplotypes (111121112, 111112211 , 111112122, 122211111 , 111111111 , and the 5 risk haplotypes above).
- the cases were separated into two groups with the first group consisting of those cases that were carrier of one of the haplotypes in the set and the second group consisting of those cases that were not carrier.
- the resulting sample sizes were 159 and 309 for the protective set, 88 and 380 for the 5-risk set and 288 and 180 for the 10-risk set respectively.
- a separate analysis for association with asthma was then performed with the two groups in each set using LDSTAT.
- Example 4 Fine Mapping 79 of the top regions identified as being associated with asthma disease by the GWS are further analyzed by fine mapping using a denser set of markers, in order to validate and/or refine the signal.
- the fine mapping is carried out using the lllumina BeadStation 500GX SNP genotyping platform.
- Alleles are genotyped using an allele-specific elongation assay that is ligated to a locus-specific oligonucleotide.
- the assay is performed directly on genomic DNA at a highly multiplex level and the products are amplified using universal oligonucleotides.
- a set of SNP markers is selected with an average inter-marker distance of 1-4 Kb distributed over about 400 Kb to 1 Mb and is roughly centered at the highest point of the GWS curves.
- the cohort used for the fine mapping analysis consists of the 497 asthma disease trios used for the GWS.
- the algorithms used for genetic analyses are the same as used in the GWS and are described in Example 3.
- a unique consensus sequence was constructed for each splice variant and a trained reviewer assessed each alignment. This assessment included examination of all putative splice junctions for consensus splice donor/acceptor sequences, putative start codons, consensus Kozak sequences and upstream in- frame stops, and the location of polyadenylation signals. In addition, conserved noncoding sequences (CNSs) that could potentially be involved in regulatory functions were included as important information for each gene. The genomic reference and exon sequences were then archived for future reference. A master assembly that included all splice variants, exons and the genomic structure was used in subsequent analyses (i.e., analysis of polymorphisms).
- the UniGene database contains information regarding the tissue source for ESTs and cDNAs contributing to individual clusters. This information was extracted and summarized to provide an indication in which tissues the gene was expressed. Particular emphasis was placed on annotating the tissue source for bona fide ESTs, since many ESTs mapped to Unigene clusters are artifactual.
- SAGE and microarray data also curated at NCBI (Gene Expression Omnibus), provided information on expression profiles for individual genes. Particular emphasis was placed on identifying genes that were expressed in tissues known to be involved in the pathophysiology of asthma disorder.
- Polymorphisms identified in candidate genes are evaluated for potential function. Initially, polymorphisms are examined for potential impact upon encoded proteins. If the protein is a member of a gene family with reported 3-dimensional structural information, this information is used to predict the location of the polymorphism with respect to protein structure. This information provided insight into the potential role of polymorphisms in altering protein or ligand interactions, as well as suitability as a drug target. In a second phase of analysis we evaluate the potential role of polymorphisms in other biological phenomena, including regulation of transcription, splicing and mRNA stability, etc. There are many examples of the functional involvement of naturally occurring polymorphisms in these processes. As part of this analysis, polymorphisms located in promoter or other regulatory elements, canonical splice sites, exonic and intronic splice enhancers and repressors, conserved noncoding sequences and UTRs are localized.
- Candidate genes and regions are selected for sequencing in order to identify all polymorphisms (Table 5). In cases where the critical interval, identified by fine mapping, was relatively small ( ⁇ 50 kb), the entire region, including all introns, is sequenced to identify polymorphisms. In situations where the region is large (>50 kb), candidate genes are prioritized for sequencing, and/or only functional gene elements (promoters, exons and splice sites) are sequenced.
- the samples sequenced are selected according to which haplotypes contribute to the association signal observed in the region.
- the purpose is to select a set of samples that covered all the major haplotypes in the given region. Each major haplotype must be present in a few copies.
- the first step therefore consisted of determining the major haplotypes in the region to be sequenced.
- the sequencing protocol included the following steps, once a region was delimited:
- the design of the primers was performed using a proprietary primer design tool.
- a primer quality control step was included in the primer design process.
- Primers that successfully passed the control quality process were synthesized by Integrated DNA Technologies (IDT).
- IDT Integrated DNA Technologies
- the sense and anti-sense oligos were separated such that the sense oligos were placed on one plate in the same position as their anti-sense counterparts on another plate.
- Two additional plates were created from each storage plate, one for use in PCR and the other for sequencing.
- the sense and anti-sense oligos of the same pair were combined in the same well to achieve a final concentration of 1.5 ⁇ M for each oligonucleotide.
- PCR conditions were optimized by testing a variety of conditions that included varying salt concentrations and temperatures, as well as including various additives. PCR products were checked for robust amplification and minimal background by agarose gel electrophoresis.
- PCR products used for sequencing were amplified using the conditions chosen during optimization.
- the PCR products were purified free of salts, dNTPs and unincorporated primers by use of a Multiscreen PCR384 filter plate manufactured by Millipore.
- the amplicons were quantified by use of a lambda/Hind III standard curve. This was done to ensure that the quantity of PCR product required for sequencing had been generated. The raw data was measured against the standard curve data in Excel by use of a macro. 4.
- the ABI Prism SeqScape software (Applied Biosystems) was used for SNP identification.
- the chromatogram trace files were imported into a SeqScape sequencing project and the base calling was automatically performed. Sequences are then aligned and compared to each other using the SeqScape program.
- the base calling was checked manually, base by base; editing was performed if needed.
- the SNPs and polymorphisms discovered in this example are listed in Table 5.
- genotyping assays may need to be utilized based on the type of polymorphism identified (i.e., SNP, indel, microsatellite).
- the assay type can be, but is not restricted to, Sentrix Assay Matrix on lllumina BeadStations, microsatellite on MegaBACE, SNP on ABI or Orchid.
- the frequencies of genotypes and haplotypes in cases and controls are analyzed in a similar manner as the GWS and fine mapping data.
- polymorphisms are identified that increase an individual's susceptibility to asthma disease.
- the goal of ultra-fine mapping is to identify the polymorphism that is most associated with disease phenotype as part of the search for the actual DNA polymorphism that confers susceptibility to disease. This statistical identification may need to be corroborated by functional studies.
- Example 8 Confirmation of Candidate regions and genes in a general population The confirmation of any putative associations described in Example 7 is performed in an independent patient sample. These DNA samples consist of one cohort, which consists of 500 PPC trios (500 patients with asthma disease).
- Miller JH, Calos MP editors. Gene Transfer Vectors for Mammalian Cells. New York: Cold Spring Harbor Laboratory Press; 1987. 169 p. Miller JH, Calos MP, editors. Gene Transfer Vectors For Mammalian Cells. New York: Cold Spring Harbor Laboratory; 1987. 169p.
Abstract
The present invention relates to the selection of a set of poymorphism markers for use in genome wide association studies based on linkage disequilibrium mapping. In particular, the invention relates to the fields of pharmacogenomics, diagnostics, patient therapy and the use of genetic haplotype information to predict an individual's susceptibility to asthma disease and/or their response to a particular drug or drugs.
Description
GeneMap of the human genes associated with asthma disease
INVENTORS: Abdelmajid Belouchi, John Verner Raelson, Walter Edward Bradley, Bruno Paquin, Helene Foumier, Quynh Nguyen-Huu, Pascal Croteau, Rene Allard, Sophie Debrus, Paul Van Eerdewegh, Randall David Little, Tim Keith and Jonathan Segal.
PRIORITY
This application is entitled to priority to US Provisional Application No. 60/856,003, filed November 2, 2006, which is herby incorporated by reference in its entirety.
FIELD OF THE INVENTION
The invention relates to the field of genomics and genetics, including genome analysis and the study of DNA variations. In particular, the invention relates to the fields of pharmacogenomics, diagnostics, patient therapy and the use of genetic haplotype information to predict an individual's susceptibility to asthma disease and/or their response to a particular drug or drugs, so that drugs tailored to genetic differences of population groups may be developed and/or administered to the appropriate population.
The invention also relates to a GeneMap for asthma disease, which links variations in DNA (including both genie and non-genic regions) to an individual's susceptibility to asthma disease and/or response to a particular drug or drugs. The invention further relates to the genes disclosed in the GeneMap (see Table 3 and 4), which is related to methods and reagents for detection of an individual's increased or decreased risk for asthma disease by identifying at least one polymorphism in one or a combination of the genes from the GeneMap. Also related are the candidate regions identified in Table 2, which are associated with asthma disease. In addition, the invention further relates to nucleotide sequences of those genes including genomic DNA sequences, cDNA sequences, single nucleotide polymorphisms (SNPs), other types of polymorphisms (insertions,
deletions, microsatellites), alleles and haplotypes (see Sequence Listing and Tables 1 , 2, 3 and 4).
The invention further relates to isolated nucleic acids comprising these nucleotide sequences and isolated polypeptides or peptides encoded thereby. Also related are expression vectors and host cells comprising the disclosed nucleic acids or fragments thereof, as well as antibodies that bind to the encoded polypeptides or peptides.
The present invention further relates to ligands that modulate the activity of the disclosed genes or gene products. In addition, the invention relates to diagnostics and therapeutics for asthma disease, utilizing the disclosed nucleic acids, polymorphisms, chromosomal regions, gene maps, polypeptides or peptides, antibodies and/or ligands and small molecules that activate or repress relevant signaling events.
BACKGROUND OF THE INVENTION
Asthma is generally defined as an inflammatory disorder of the airways, and clinical symptoms arise from intermittent airflow obstruction. Two common subdivisions of asthma are atopic (allergic or extrinsic) asthma and non-atopic (intrinsic) asthma. In atopic asthma, activation of the immune system by ubiquitous antigens is generally a response to environmental stimuli, and the disorder is generally characterized by an increased ability of lymphocytes to produce IgE antibodies in response to these antigens. Non-atopic asthma may be defined as reversible airflow limitation in the absence of allergies. Asthma is a disease that is broadly characterized by this immune activation when pulmonary inflammation ensues. Asthma is a disease of reversible bronchial obstruction, characterized by airway inflammation, epithelial damage, airway smooth muscle hypertrophy and bronchial hyperreactivity. Certain cells are important in this inflammatory reaction in the airways and they include T cells and antigen presenting cells, B cells that produce IgE, mast cells/basophils that store inflammatory mediators and bind IgE, and eosinophils that release additional
mediators. These inflammatory cells accumulate at the site of allergic inflammation, and the toxic products they release contribute to the tissue destruction related to the disorder.
Both the diagnosis and treatment of asthma and related disorders are problematic. In particular, the assessment of inflamed lung tissue is often difficult, and frequently, the cause of the inflammation cannot be determined. Current treatments for asthma disease are primarily aimed at reducing symptoms by suppressing inflammation and by opening up constricted airways, and do not address the root cause of the disease and offer their own set of disadvantage. The main therapeutic agents, β-agonists, reduce the symptoms by bronchodilatation, and these result in a transiently improved pulmonary function state, but do not affect the underlying inflammation. Thus, the lung tissue remains in jeopardy. In addition, constant use of β-agonists results in desensitization which reduces their efficacy and safety. The agents that can diminish the underlying inflammation, the anti-inflammatory steroids, have their own known list of side effects that range from immunosuppression to bone loss. Other nonsteroid treatments have been proposed to address inflammation, such as Glycophorin A, cyclosporin, and a peptide fragment of IL-2. While these agents may represent alternatives to steroids in the treatment of asthmatics, they all inhibit interleukin-2 dependent T lymphocyte proliferation and potentially critical immune functions associated with homeostasis. What is needed in the art is technology to expedite the development of therapeutics that is specifically designed to treat the cause, and not the symptoms, of atopic asthma. Studies have demonstrated a genetic predisposition to asthma by showing, for example, a greater concordance for this trait among monozygotic twins than among dizygotic twins. The genetics of asthma is complex, however, and shows no simple pattern of inheritance. Environment also plays a role in asthma development, for example, children of smokers are more likely to develop asthma than are children of non-smokers.
Despite a preponderance of evidence showing inheritance of a risk for asthma disease through epidemiological studies and genome wide linkage analyses, all the genes affecting asthma disease have yet to be discovered. There is a need in
the art for identifying specific genes related to asthma disease to enable the development of therapeutics that address the causes of the disease rather than relieving its symptoms. The failure in past studies to identify causative genes in complex diseases, such as asthma disease, has been due to the lack of appropriate methods to detect a sufficient number of variations in genomic DNA samples (markers), the insufficient quantity of necessary markers available, and the number of needed individuals to enable such a study. The present invention addresses these issues.
The DNA sequences between two human genomes are 99.9% identical. The variations in DNA sequence between individuals can be, as an example, deletions of small or large stretches of DNA, insertions of stretches of DNA1 variations in the number of repetitive DNA elements, and changes in single base positions in the genome called "single nucleotide polymorphisms" (SNPs). Human DNA sequence variation accounts for a large fraction of observed differences between individuals, including susceptibility to disease.
Many common diseases, like asthma disease, are complex genetic traits and are believed to involve several disease-genes rather than single genes, as is observed for rare diseases. This makes detection of any particular gene substantially more difficult than in a rare disease, where a single gene mutation that segregates according to a Mendelian inheritance pattern is the causative mutation. Any one of the multiple interacting gene mutations involved in the etiology of a complex disease will impart a lower relative risk for the disease than will the single gene mutation involved in a simple genetic disease. Low relative risk alleles are more difficult to detect and, as a result, the success of positional cloning using linkage mapping that was achieved for simple genetic disease genes has not been repeated for complex diseases.
Several approaches have been proposed to discover and characterize multiple genes in complex genetic traits. These gene discovery methods can be subdivided into hypothesis-free disorder association studies and hypothesis- driven candidate gene or region studies. The candidate gene approach relies on the analysis of a gene in patients who have a disorder in which the gene is
thought to play a role. This approach is limited in utility because it only provides for the investigation of genes with known functions. Although variant sequences of candidate genes may be identified using this approach, it is inherently limited by the fact that variant sequences in other genes that contribute to the phenotype will be necessarily missed when the technique is employed. Genome-wide scans (GWS) have been shown to be efficient in identifying asthma disease susceptibility genes, such as ADAM33 gene on 20p13 region (Van Eerdewegh 2002), and PHF11 on 13q14 region (Zhang 2003). In contrast to the candidate gene approach, a GWS searches throughout the genome without any a priori hypothesis and consequently can identify genes that are not obvious candidates for the disease as well as genes that are relevant candidates for the disease, it can also identify chromosomal regions that are structurally important where mutations can influence gene function of specific genes.
Family-based linkage mapping methods were initially used for disorder locus identification. This technique locates genes based on the relatively limited number of genetic recombination events within the families used in the study, and results in large chromosomal regions containing hundreds of genes, any one of which could be the disorder-causing gene. Population-based, or linkage disequilibrium (LD) mapping is based on the premise that regions adjacent to a gene of interest are co-transmitted through the generations along with the gene. As a result, LD extends over shorter genetic regions than does linkage (Hewett et a/., 2002), and can facilitate detection of genes with lower relative risk than family linkage mapping approaches. LD-based mapping also defines much smaller candidate regions which may contain only a few genes, making the identification of the actual disorder gene much easier.
It has been estimated that a GWS that uses a general population and case/control association (LD) analysis would require approximately 700,000 SNP markers (Carlson et al., 2003). The cost of a GWS at this marker density for a sufficient sample size for statistical power is economically prohibitive. The use of a special founder population (genetic isolate), such as the French Canadian population of Quebec, is one solution to the problem with LD analysis. The French Canadian population in Quebec (Quebec Founder Population - QFP)
provides one of the best resources in the world for gene discovery based on its high levels of genetic sharing and genetic homogeneity. By combining DNA collected from the QFP, high throughput genotyping capabilities and proprietary algorithms for genetic analysis, a comprehensive genome-wide association study was facilitated. The present invention relates specifically to a set of asthma disease-causing genes (GeneMap) and targets which present attractive points of therapeutic intervention.
In view of the foregoing, identifying susceptibility genes associated with asthma disease and their respective biochemical pathways will facilitate the identification of diagnostic markers as well as novel targets for improved therapeutics. It will also improve the quality of life for those afflicted by this disease and will reduce the economic costs of these afflictions at the individual and societal level. The identification of those genetic markers would provide the basis for novel genetic tests and eliminate or reduce the therapeutic methods currently used. The identification of those genetic markers will also provide the development of effective therapeutic intervention for the battery of laboratory, radiological, and other medical evaluations typically required to diagnose asthma disease. The present invention satisfies this need and provides related advantages as well.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 Method employed by the inventors to permit the identification of genes involved in a particular disorder or trait, such as asthma disease. The method can be applied for any given disorder and the end result is the construction of a GeneMap for a particular disorder. Briefly, a disorder or genetic trait is selected followed by in depth literature review on the known genes and candidate regions known in the art, and on the prevalence, incidence and phenotypes of the disorder. A clinical specialist in the field of the disorder is consulted for the definition of phenotype. Inclusion and exclusion criteria are then set and a study protocol is constructed. IRB and ethical approval are sought prior to patient recruitment. A network of physicians is required to recruit the necessary cases
and controls for the study from the Quebec Founder Population. Individuals (cases and controls) are then recruited and DNA extraction and quantitation is performed from the blood samples obtained. All case and all controls samples are individually genotyped. A GWS is performed on the case and control samples using at least the required marker density for the Quebec Founder Population (QLD map). The results from the GWS genotyping are analyzed and candidate regions are selected for fine mapping in the same samples, at a higher marker density, in order to validate and/or refine the signal. The gene content of the candidate regions is analyzed and characterized. The representative haplotypes contributing to the association signal are then selected and sequenced. Once polymorphisms are identified by sequencing efforts, the frequencies of genotypes and haplotypes in cases and controls are analyzed in a similar manner as for the fine mapping data. Ultrafine mapping is performed on all the samples to identify the polymorphisms that are most associated with the disorder phenotype as part of the search for the actual DNA polymorphisms that confer susceptibility to the disorder. The genes found associated with the disorder are then corroborated. The corroborated genes are used for the construction of a GeneMap.
SEQUENCE LISTING
The sequence listing submitted with US Provisional Application No. 60/856,003 on November 2, 2006 on compact disc is hereby incorporated by reference in its entirety. Two duplicate copies of the CD-R labeled "copy 1" and "copy 2" were submitted. The material on each of the duplicate CD-R was identical. Thus, descriptions or references herein to the CD-R labeled "GeneMap of the human gene associated with asthma disease" and the files contained thereon apply to both "copy 1" and "copy 2", and both are hereby incorporated by reference in their entirety.
The CD-R labeled "GeneMap of the human gene associated with asthma disease" contains the following one file of sequence listing. Each electronic copy of the sequence listing was created on November 2, 2005 with a file size of 7,759 kb. The file name is as follows: 059908-5010 sequence listing.txt.
An electronic copy of the Sequence Listing is also being filed herewith. This electronic copy of the Sequence Listing is hereby incorporated by reference in its entirety.
TABLES
Tables 1-4, which are being filed electronically herewith, are hereby incorporated by reference in their entirety.
DEFINITIONS
Throughout the description of the present invention, several terms are used that are specific to the science of this field. For the sake of clarity and to avoid any misunderstanding, these definitions are provided to aid in the understanding of the specification and claims:
Allele: One of a pair, or series, of forms of a gene or non-genic region that occur at a given locus in a chromosome. Alleles are symbolized with the same basic symbol (e.g., B for dominant and b for recessive; B1 , B2, Bn for n additive alleles at a locus). In a normal diploid cell there are two alleles of any one gene (one from each parent), which occupy the same relative position (locus) on homologous chromosomes. Within a population there may be more than two alleles of a gene. See multiple alleles. SNPs also have alleles, i.e., the two (or more) nucleotides that characterize the SNP.
Amplification of nucleic acids: refers to methods such as polymerase chain reaction (PCR), ligation amplification (or ligase chain reaction, LCR) and amplification methods based on the use of Q-beta replicase. These methods are well known in the art and are described, for example, in U.S. Patent Nos. 4,683,195 and 4,683,202. Reagents and hardware for conducting PCR are commercially available. Primers useful for amplifying sequences from the disorder region are preferably complementary to, and preferably hybridize
specifically to, sequences in the disorder region or in regions that flank a target region therein. Genes from Tables 3 and 4 generated by amplification may be sequenced directly. Alternatively, the amplified sequence(s) may be cloned prior to sequence analysis.
Antigenic component: is a moiety that binds to its specific antibody with sufficiently high affinity to form a detectable antigen-antibody complex.
Antibodies: refer to polyclonal and/or monoclonal antibodies and fragments thereof, and immunologic binding equivalents thereof, that can bind to proteins and fragments thereof or to nucleic acid sequences from the disorder region, particularly from the disorder gene products or a portion thereof. The term antibody is used both to refer to a homogeneous molecular entity, or a mixture such as a serum product made up of a plurality of different molecular entities. Proteins may be prepared synthetically in a protein synthesizer and coupled to a carrier molecule and injected over several months into rabbits. Rabbit sera are tested for immunoreactivity to the protein or fragment. Monoclonal antibodies may be made by injecting mice with the proteins, or fragments thereof. Monoclonal antibodies can be screened by ELISA and tested for specific immunoreactivity with protein or fragments thereof (Harlow et al. 1988, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). These antibodies will be useful in developing assays as well as therapeutics.
Associated allele: refers to an allele at a polymorphic locus that is associated with a particular phenotype of interest, e.g., a predisposition to a disorder or a particular drug response.
cDNA: refers to complementary or copy DNA produced from an RNA template by the action of RNA-dependent DNA polymerase (reverse transcriptase). Thus, a cDNA clone means a duplex DNA sequence complementary to an RNA molecule of interest, included in a cloning vector or PCR amplified. This term includes genes from which the intervening sequences have been removed.
cDNA library: refers to a collection of recombinant DNA molecules containing cDNA inserts that together comprise essentially all of the expressed genes of an organism or tissue. A cDNA library can be prepared by methods known to one skilled in the art (see, e.g., Cowell and Austin, 1997, "DNA Library Protocols," Methods in Molecular Biology). Generally, RNA is first isolated from the cells of the desired organism, and the RNA is used to prepare cDNA molecules.
Cloning: refers to the use of recombinant DNA techniques to insert a particular gene or other DNA sequence into a vector molecule. In order to successfully clone a desired gene, it is necessary to use methods for generating DNA fragments, for joining the fragments to vector molecules, for introducing the composite DNA molecule into a host cell in which it can replicate, and for selecting the clone having the target gene from amongst the recipient host cells.
Cloning vector: refers to a plasmid or phage DNA or other DNA molecule that is able to replicate in a host cell. The cloning vector is typically characterized by one or more endonuclease recognition sites at which such DNA sequences may be cleaved in a determinable fashion without loss of an essential biological function of the DNA, and which may contain a selectable marker suitable for use in the identification of cells containing the vector.
Coding sequence or a protein-coding sequence: is a polynucleotide sequence capable of being transcribed into mRNA and/or capable of being translated into a polypeptide or peptide. The boundaries of the coding sequence are typically determined by a translation start codon at the 5'-terminus and a translation stop codon at the 3'-terminus.
Complement of a nucleic acid sequence: refers to the antisense sequence that participates in Watson-Crick base-pairing with the original sequence.
Disorder region: refers to the portions of the human chromosomes displayed in Table 2 bounded by the markers from Tables 1 , 2 and 5.
Disorder-associated nucleic acid or polypeptide sequence: refers to a nucleic acid sequence that maps to region of Table 2 or the polypeptides encoded
therein (Tables 3 and 4, nucleic acids, and polypeptides). For nucleic acids, this encompasses sequences that are identical or complementary to the gene sequences from Table 3 and 4, as well as sequence-conservative, function- conservative, and non-conservative variants thereof. For polypeptides, this encompasses sequences that are identical to the polypeptide, as well as function-conservative and non-conservative variants thereof. Included are the alleles of naturally-occurring polymorphisms causative of asthma disease such as, but not limited to, alleles that cause altered expression of genes of Tables 3 and 4 and alleles that cause altered protein levels or stability (e.g., decreased levels, increased levels, expression in an inappropriate tissue type, increased stability, and decreased stability).
Expression vector: refers to a vehicle or plasmid that is capable of expressing a gene that has been cloned into it, after transformation or integration in a host cell. The cloned gene is usually placed under the control of (i.e., operably linked to) a regulatory sequence.
Function-conservative variants: are those in which a change in one or more nucleotides in a given codon position results in a polypeptide sequence in which a given amino acid residue in the polypeptide has been replaced by a conservative amino acid substitution. Function-conservative variants also include analogs of a given polypeptide and any polypeptides that have the ability to elicit antibodies specific to a designated polypeptide.
Founder population: Also called a population isolate, this is a large number of people who have mostly descended, in genetic isolation from other populations, from a much smaller number of people who lived many generations ago.
Gene: Refers to a DNA sequence that encodes through its template or messenger RNA a sequence of amino acids characteristic of a specific peptide, polypeptide, or protein. The term "gene" also refers to a DNA sequence that encodes an RNA product. The term gene as used herein with reference to genomic DNA includes intervening, non-coding regions, as well as regulatory regions, and can include 5' and 3' ends. A gene sequence is wild-type if such sequence is usually found in individuals unaffected by the disorder or condition of
interest. However, environmental factors and other genes can also play an important role in the ultimate determination of the disorder. In the context of complex disorders involving multiple genes (oligogenic disorder), the wild type, or normal sequence can also be associated with a measurable risk or susceptibility, receiving its reference status based on its frequency in the general population.
GeneMaps: are defined as groups of gene(s) that are directly or indirectly involved in at least one phenotype of a disorder. As such, GeneMaps enable the development of synergistic diagnostic products, creating "theranostics".
Genotype: Set of alleles at a specified locus or loci.
Haplotype: The allelic pattern of a group of (usually contiguous) DNA markers or other polymorphic loci along an individual chromosome or double helical DNA segment. Haplotypes identify individual chromosomes or chromosome segments. The presence of shared haplotype patterns among a group of individuals implies that the locus defined by the haplotype has been inherited, identical by descent (IBD), from a common ancestor. Detection of identical by descent haplotypes is the basis of linkage disequilibrium (LD) mapping. Haplotypes are broken down through the generations by recombination and mutation. In some instances, a specific allele or haplotype may be associated with susceptibility to a disorder or condition of interest, e.g., asthma disease. In other instances, an allele or haplotype may be associated with a decrease in susceptibility to a disorder or condition of interest, i.e., a protective sequence.
Host: includes prokaryotes and eukaryotes. The term includes an organism or cell that is the recipient of an expression vector (e.g., autonomously replicating or integrating vector).
Hybridizable: nucleic acids are hybridizable to each other when at least one strand of the nucleic acid can anneal to another nucleic acid strand under defined stringency conditions. In some embodiments, hybridization requires that the two nucleic acids contain at least 10 substantially complementary nucleotides; depending on the stringency of hybridization, however, mismatches may be tolerated. The appropriate stringency for hybridizing nucleic acids depends on the
length of the nucleic acids and the degree of complementarity, and can be determined in accordance with the methods described herein.
Identity by descent (IBD): Identity among DNA sequences for different individuals that is due to the fact that they have all been inherited from a common ancestor. LD mapping identifies IBD haplotypes as the likely location of disorder genes shared by a group of patients.
Identity: as known in the art, is a relationship between two or more polypeptide sequences or two or more polynucleotide sequences, as determined by comparing the sequences. In the art, identity also means the degree of sequence relatedness between polypeptide or polynucleotide sequences, as the case may be, as determined by the match between strings of such sequences. Identity and similarity can be readily calculated by known methods, including but not limited to those described in A.M. Lesk (ed), 1988, Computational Molecular Biology, Oxford University Press, NY; D.W. Smith (ed), 1993, Biocomputing. Informatics and Genome Projects, Academic Press, NY; A.M. Griffin and H. G. Griffin, H. G (eds), 1994, ComputerAnalysis of Sequence Data, Part 1 , Humana Press, NJ; G. von Heinje, 1987, Sequence Analysis in Molecular Biology, Academic Press; and M. Gribskov and J. Devereux (eds), 1991 , Sequence Analysis Primer, M Stockton Press, NY; H. Carillo and D. Lipman, 1988, SIAM J. Applied Math., 48:1073.
Immunogenic component: is a moiety that is capable of eliciting a humoral and/or cellular immune response in a host animal.
Isolated nucleic acids: are nucleic acids separated away from other components (e.g., DNA, RNA, and protein) with which they are associated (e.g., as obtained from cells, chemical synthesis systems, or phage or nucleic acid libraries). Isolated nucleic acids are at least 60% free, preferably 75% free, and most preferably 90% free from other associated components. In accordance with the present invention, isolated nucleic acids can be obtained by methods described herein, or other established methods, including isolation from natural sources (e.g., cells, tissues, or organs), chemical synthesis, recombinant methods, combinations of recombinant and chemical methods, and library screening methods.
Isolated polypeptides or peptides: are those that are separated from other components (e.g., DNA, RNA, and other polypeptides or peptides) with which they are associated (e.g., as obtained from cells, translation systems, or chemical synthesis systems). In a preferred embodiment, isolated polypeptides or peptides are at least 10% pure; more preferably, 80% or 90% pure. Isolated polypeptides and peptides include those obtained by methods described herein, or other established methods, including isolation from natural sources (e.g., cells, tissues, or organs), chemical synthesis, recombinant methods, or combinations of recombinant and chemical methods. Proteins or polypeptides referred to herein as recombinant are proteins or polypeptides produced by the expression of recombinant nucleic acids. A portion as used herein with regard to a protein or polypeptide, refers to fragments of that protein or polypeptide. The fragments can range in size from 5 amino acid residues to all but one residue of the entire protein sequence. Thus, a portion or fragment can be at least 5, 5-50, 50-100, I00-200, 200-400, 400-800, or more consecutive amino acid residues of a protein or polypeptide.
Linkage disequilibrium (LD): the situation in which the alleles for two or more loci do not occur together in individuals sampled from a population at frequencies predicted by the product of their individual allele frequencies. In other words, markers that are in LD do not follow Mendel's second law of independent random segregation. LD can be caused by any of several demographic or population artifacts as well as by the presence of genetic linkage between markers. However, when these artifacts are controlled and eliminated as sources of LD, then LD results directly from the fact that the loci involved are located close to each other on the same chromosome so that specific combinations of alleles for different markers (haplotypes) are inherited together. Markers that are in high LD can be assumed to be located near each other and a marker or haplotype that is in high LD with a genetic trait can be assumed to be located near the gene that affects that trait. The physical proximity of markers can be measured in family studies where it is called linkage or in population studies where it is called linkage disequilibrium.
LD mapping: population based gene mapping, which locates disorder genes by identifying regions of the genome where haplotypes or marker variation patterns are shared statistically more frequently among disorder patients compared to healthy controls. This method is based upon the assumption that many of the patients will have inherited an allele associated with the disorder from a common ancestor (IBD), and that this allele will be in LD with the disorder gene.
Locus: a specific position along a chromosome or DNA sequence. Depending upon context, a locus could be a gene, a marker, a chromosomal band or a specific sequence of one or more nucleotides.
Minor allele frequency (MAF): the population frequency of one of the alleles for a given polymorphism, which is equal or less than 50%. The sum of the MAF and the Major allele frequency equals one.
Markers: an identifiable DNA sequence that is variable (polymorphic) for different individuals within a population. These sequences facilitate the study of inheritance of a trait or a gene. Such markers are used in mapping the order of genes along chromosomes and in following the inheritance of particular genes; genes closely linked to the marker or in LD with the marker will generally be inherited with it. Two types of markers are commonly used in genetic analysis, microsatellites and SNPs.
Microsatellite: DNA of eukaryotic cells comprising a repetitive, short sequence of DNA that is present as tandem repeats and in highly variable copy number, flanked by sequences unique to that locus.
Mutant sequence: if it differs from one or more wild-type sequences. For example, a nucleic acid from a gene listed in Tables 3 and 4 containing a particular allele of a single nucleotide polymorphism may be a mutant sequence. In some cases, the individual carrying this allele has increased susceptibility toward the disorder or condition of interest. In other cases, the mutant sequence might also refer to an allele that decreases the susceptibility toward a disorder or condition of interest and thus acts in a protective manner. The term mutation may also be used to describe a specific allele of a polymorphic locus.
Non-conservative variants: are those in which a change in one or more nucleotides in a given codon position results in a polypeptide sequence in which a given amino acid residue in a polypeptide has been replaced by a non- conservative amino acid substitution. Non-conservative variants also include polypeptides comprising non-conservative amino acid substitutions.
Nucleic acid or polynucleotide: purine- and pyrimidine-containing polymers of any length, either polyribonucleotides or polydeoxyribonucleotide or mixed polyribo polydeoxyribonucleotides. This includes single-and double-stranded molecules, i.e., DNA-DNA, DNA-RNA and RNA-RNA hybrids, as well as protein nucleic acids (PNA) formed by conjugating bases to an amino acid backbone. This also includes nucleic acids containing modified bases.
Nucleotide: a nucleotide, the unit of a DNA molecule, is composed of a base, a 2'-deoxyribose and phosphate ester(s) attached at the 5' carbon of the deoxyribose. For its incorporation in DNA, the nucleotide needs to possess three phosphate esters but it is converted into a monoester in the process.
Operably linked: means that the promoter controls the initiation of expression of the gene. A promoter is operably linked to a sequence of proximal DNA if upon introduction into a host cell the promoter determines the transcription of the proximal DNA sequence(s) into one or more species of RNA. A promoter is operably linked to a DNA sequence if the promoter is capable of initiating transcription of that DNA sequence.
Ortholog: denotes a gene or polypeptide obtained from one species that has homology to an analogous gene or polypeptide from a different species.
Paralog: denotes a gene or polypeptide obtained from a given species that has homology to a distinct gene or polypeptide from that same species.
Phenotype: any visible, detectable or otherwise measurable property of an organism such as symptoms of, or susceptibility to, a disorder.
Polymorphism: occurrence of two or more alternative genomic sequences or alleles between or among different genomes or individuals at a single locus. A
polymorphic site thus refers specifically to the locus at which the variation occurs. In some cases, an individual carrying a particular allele of a polymorphism has an increased or decreased susceptibility toward a disorder or condition of interest. Portion and fragment: are synonymous. A portion as used with regard to a nucleic acid or polynucleotide refers to fragments of that nucleic acid or polynucleotide. The fragments can range in size from 8 nucleotides to all but one nucleotide of the entire gene sequence. Preferably, the fragments are at least about 8 to about 10 nucleotides in length; at least about 12 nucleotides in length; at least about 15 to about 20 nucleotides in length; at least about 25 nucleotides in length; or at least about 35 to about 55 nucleotides in length. Probe or primer: refers to a nucleic acid or oligonucleotide that forms a hybrid structure with a sequence in a target region of a nucleic acid due to complementarity of the probe or primer sequence to at least one portion of the target region sequence.
Protein and polypeptide: are synonymous. Peptides are defined as fragments or portions of polypeptides, preferably fragments or portions having at least one functional activity (e.g., proteolysis, adhesion, fusion, antigenic, or intracellular activity) as the complete polypeptide sequence.
Recombinant nucleic acids: nuclei acids which have been produced by recombinant DNA methodology, including those nucleic acids that are generated by procedures which rely upon a method of artificial replication, such as the polymerase chain reaction (PCR) and/or cloning into a vector using restriction enzymes. Portions of recombinant nucleic acids which code for polypeptides can be identified and isolated by, for example, the method of M. Jasin et al., U.S. Patent No. 4,952,501.
Regulatory sequence: refers to a nucleic acid sequence that controls or regulates expression of structural genes when operably linked to those genes. These include, for example, the lac systems, the trp system, major operator and promoter regions of the phage lambda, the control region of fd coat protein and other sequences known to control the expression of genes in prokaryotic or eukaryotic cells. Regulatory sequences will vary depending on whether the vector
is designed to express the operably linked gene in a prokaryotic or eukaryotic host, and may contain transcriptional elements such as enhancer elements, termination sequences, tissue-specificity elements and/or translational initiation and termination sites.
Sample: as used herein refers to a biological sample, such as, for example, tissue or fluid isolated from an individual or animal (including, without limitation, plasma, serum, cerebrospinal fluid, lymph, tears, nails, hair, saliva, milk, pus, and tissue exudates and secretions) or from in vitro cell culture-constituents, as well as samples obtained from, for example, a laboratory procedure.
Single nucleotide polymorphism (SNP): variation of a single nucleotide. This includes the replacement of one nucleotide by another and deletion or insertion of a single nucleotide. Typically, SNPs are biallelic markers although tri- and tetra- allelic markers also exist. For example, SNP A\C may comprise allele C or allele A (Table 1). Thus, a nucleic acid molecule comprising SNP A\C may include a C or A at the polymorphic position. For clarity purposes, an ambiguity code is used in Tables 1 , 2, 5 and the sequence listing, to represent the variations. For a combination of SNPs, the term "haplotype" is used, e.g. the genotype of the SNPs in a single DNA strand that are linked to one another. In certain embodiments, the term "haplotype" is used to describe a combination of SNP alleles, e.g., the alleles of the SNPs found together on a single DNA molecule. In specific embodiments, the SNPs in a haplotype are in linkage disequilibrium with one another.
Sequence-conservative: variants are those in which a change of one or more nucleotides in a given codon position results in no alteration in the amino acid encoded at that position (i.e., silent mutation).
Substantially homologous: a nucleic acid or fragment thereof is substantially homologous to another if, when optimally aligned (with appropriate nucleotide insertions and/or deletions) with the other nucleic acid (or its complementary strand), there is nucleotide sequence identity in at least 60% of the nucleotide bases, usually at least 70%, more usually at least 80%, preferably at least 90%, and more preferably at least 95-98% of the nucleotide bases. Alternatively,
substantial homology exists when a nucleic acid or fragment thereof will hybridize, under selective hybridization conditions, to another nucleic acid (or a complementary strand thereof). Selectivity of hybridization exists when hybridization which is substantially more selective than total lack of specificity occurs. Typically, selective hybridization will occur when there is at least about 55% sequence identity over a stretch of at least about nine or more nucleotides, preferably at least about 65%, more preferably at least about 75%, and most preferably at least about 90% (M. Kanehisa, 1984, NucL Acids Res. 11:203-213). The length of homology comparison, as described, may be over longer stretches, and in certain embodiments will often be over a stretch of at least 14 nucleotides, usually at least 20 nucleotides, more usually at least 24 nucleotides, typically at least 28 nucleotides, more typically at least 32 nucleotides, and preferably at least 36 or more nucleotides.
Wild-type gene from Tables 3 and 4: refers to the reference sequence. The wild- type gene sequences from Tables 3 and 4 used to identify the variants (polymorphisms, alleles, and haplotypes) described in detail herein.
Technical and scientific terms used herein have the meanings commonly understood by one of ordinary skill in the art to which the present invention pertains, unless otherwise defined. Reference is made herein to various methodologies known to those of skill in the art. Publications and other materials setting forth such known methodologies to which reference is made are incorporated herein by reference in their entireties as though set forth in full. Standard reference works setting forth the general principles of recombinant DNA technology include J. Sambrook et al., 1989, Molecular Cloning: A Laboratory Manual, 2d Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY; P. B. Kaufman et al., (eds), 1995, Handbook of Molecular and Cellular Methods in Biology and Medicine, CRC Press, Boca Raton; MJ. McPherson (ed), 1991 , Directed Mutagenesis: A Practical Approach, IRL Press, Oxford; J. Jones, 1992, Amino Acid and Peptide Synthesis, Oxford Science Publications, Oxford; B. M. Austen and O. M. R. Westwood, 1991, Protein Targeting and Secretion, IRL Press, Oxford; D.N Glover (ed), 1985, DNA Cloning, Volumes I and 11; MJ. Gait (ed), 1984, Oligonucleotide Synthesis; B. D. Hames and SJ. Higgins (eds), 1984,
Nucleic Acid Hybridization; Quirke and Taylor (eds), 1991 , PCR-A Practical Approach; Harries and Higgins (eds), 1984, Transcription and Translation; R.I. Freshney (ed), 1986, Animal Cell Culture; Immobilized Cells and Enzymes, 1986, IRL Press; Perbal, 1984, A Practical Guide to Molecular Cloning, J. H. Miller and M. P. Calos (eds), 1987, Gene Transfer Vectors for Mammalian Cells, Cold Spring Harbor Laboratory Press; M.J. Bishop (ed), 1998, Guide to Human Genome Computing, 2d Ed., Academic Press, San Diego, CA; L.F. Peruski and A. H. Peruski, 1997, The Internet and the New Biology. Tools for Genomic and Molecular Research, American Society for Microbiology, Washington, D. C. Standard reference works setting forth the general principles of immunology include S. Sell, 1996, Immunology, lmmunopathology & Immunity, 5th Ed., Appleton & Lange, Publ., Stamford, CT; D. Male et al., 1996, Advanced Immunology, 3d Ed., Times Mirror Int'l Publishers Ltd., Publ., London; D. P. Stites and A.L Terr, 1991 , Basic and Clinical Immunology, 7th Ed., Appleton & Lange, Publ., Norwalk, CT; and A.K. Abbas et a/., 1991 , Cellular and Molecular Immunology, W. B. Saunders Co., Publ., Philadelphia, PA. Any suitable materials and/or methods known to those of skill can be utilized in carrying out the present invention; however, preferred materials and/or methods are described. Materials, reagents, and the like to which reference is made in the following description and examples are generally obtainable from commercial sources, and specific vendors are cited herein.
DETAILED DESCRIPTION OF THE INVENTION General Description of asthma Disease
Previously identified genes and loci
Genetic studies have previously indicated the presence of several loci predisposing to asthma. In 2002, Van Eerdewegh et al characterized the ADAM33 gene on 20p13 region (Van Eerdewegh et al., 2002). This gene encodes a metallo protease and has been associated to asthma and bronchial hyperreactivity (BHR). Since, ADAM33 has been evaluated in 6 case-control association studies on samples from different populations, and three studies replicated the association with asthma (Howard et al., 2003; Raby et al., 2004
and Werner et al., 2004). Also, Jongepier et al., (2004) showed an association between a polymorphism in ADAM33 and accelerated lung function decline in asthma patients.
However, the largest studies performed in different populations have failed to show an association between ADAM33 and asthma or related phenotypes. Thus, the involvement of ADAM33 in this disease remains controversial.
In 2003, Zhang et al. showed that gene PHF11 was associated with IgE levels and severe clinical asthma in the 13q14 region. This gene encodes a protein containing 2 PHD zinc fingers that may be involved in transcriptional regulation, but its precise function is not known. No study attempting to replicate this finding has been reported to date.
In the 2q14 region, Allen et al., (2003) identified the gene DPP10 as being linked to asthma. This gene encodes a homolog of dipeptidyl peptidases. As for PHF11 , no study attempting to replicate this finding+ for DPP10 has been reported to date.
Finally, the G protein-coupled receptor 154 (or GPRA) was identified in the 7p15 region as associated with high serum IgE and asthma in Finnish and French
Canadian samples (Laitinen et al., 2004). So far, 2 studies replicated these results in other populations (Kormann et al., 2005; Melen et al., 2005), and one study failed to replicate this in a Korean population (Shin et al., 2004).
Most of the genes determining susceptibility in asthma disease remain to be identified. Thus, there is a continuing need in the medical arts for genetic markers of asthma disease and guidance for the use of such markers.
The genetic variants that have been identified so far in asthma explain only a fraction of the genetic predisposition to this disorder. It is clear that multiple components contribute to disease risk, each component having a modest effect on disease susceptibility. Thus the development of GeneMaps for asthma may lead to a better understanding of pathogenesis and to the identification of new pathways involved in the disease, ultimately leading to better treatments for the patients. GeneMaps may also lead to molecular diagnostic tools that will identify subjects at risk for asthma or for serious complications of the disease.
Genome wide association study to construct a GeneMap for asthma disease
The present invention is based on the discovery of genes associated with asthma disease. In the preferred embodiment, Disease-associated loci (candidate regions; Table 2) are identified by the statistically significant differences in allele or haplotype frequencies between the cases and the controls. For the purpose of the present invention, 231 candidate regions exhibiting a -Iog10 P value of 3.5 or higher are identified, comprises a few which have been previously reported to be associated with asthma disease.
The invention provides a method for the discovery of genes associated with asthma disease and the construction of a GeneMap for asthma disease in a human population, comprising the following steps (see Figure 1 and Example section herein):
Step 1: Recruit patients (cases) and controls
In the preferred embodiment, 500 patients diagnosed for asthma disease along with two family members are recruited from the Quebec Founder Population (QFP). The preferred trios recruited are parent-parent-child (PPC) trios. Trios can also be recruited as parent-child-child (PCC) trios. In another preferred embodiment, more or less than 500 trios are recruited.
In another embodiment, the present invention is performed as a whole or partially with DNA samples from individuals of another founder population than the Quebec population or from the general population.
Step 2: DNA extraction and quantitation
Any sample comprising cells or nucleic acids from patients or controls may be used. Preferred samples are those easily obtained from the patient or control. Such samples include, but are not limited to blood, peripheral lymphocytes, buccal swabs, epithelial cell swabs, nails, hair, bronchoalveolar lavage fluid, sputum, or other body fluid or tissue obtained from an individual.
In the preferred embodiment, DNA is extracted from such samples in the quantity and quality necessary to perform the invention using conventional DNA extraction and quantitation techniques. The present invention is not linked to any DNA extraction or quantitation platform in particular.
Step 3: Genotype the recruited individuals
In the preferred embodiment, assay specific and/or locus-specific and/or allele- specific oligonucleotides for every SNP marker of the present invention (Table 1) are organized onto one or more arrays. The genotype at each SNP locus is revealed by hybridizing short PCR fragments comprising each SNP locus onto these arrays. The arrays permit a high-throughput genome wide association study using DNA samples from individuals of the Quebec founder population. Such assay-specific and/or locus-specific and/or allele-specific oligonucleotides necessary for scoring each SNP of the present invention are preferably organized onto a solid support. Such supports can be arrayed on wafers, glass slides, beads or any other type of solid support.
In another embodiment, the assay-specific and/or locus-specific and/or allele- specific oligonucleotides are not organized onto a solid support but are still used as a whole, in panels or one by one. The present invention is therefore not linked to any genotyping platform in particular.
In another embodiment, one or more portions of the SNP maps (publicly available maps, proprietary maps from Perlegen Sciences, Inc. (Mountain View, CA, USA), and our own proprietary QLDM map; see patent application 60/634,555 for details) are used to screen the whole genome, a subset of chromosomes, a chromosome, a subset of genomic regions or a single genomic region.
The 1,500 individuals composing the 500 trios are preferably individually genotyped with at least 80,000 markers, generating at least a few million genotypes; more preferable, at least a hundred million.
Step 4: Exclude the markers that did not pass the quality control of the assay.
Preferably, the quality controls consist of, but are not limited to, the following criteria: eliminate SNPs that had a high rate of Mendelian errors (cut-off at 1% Mendelian error rate), that deviate from the Hardy-Weinberg equilibrium, that are non-polymorphic in the Quebec founder population or have too many missing data (cut-off at 1% missing values or higher), or simply because they are non- polymorphic in the Quebec founder population (cut-off at 1% ≤ 10% minor allele frequency (MAF)).
Step 5: Perform the genetic analysis on the results obtained using haplotype information as well as single-marker association.
In the preferred embodiment, genetic analysis is performed on all the genotypes from step 3.
In another embodiment, genetic analysis is performed on a total of 80,654 SNPs.
In one embodiment, the genetic analysis consists of, but is not limited to features corresponding to Phase information and haplotype structures. Phase information and haplotype structures are preferably deduced from trio genotypes using Phasefinder. Since chromosomal assignment (phase) can not be estimated when all trio members are heterozygous, an Expectation-Maximization (EM) algorithm may be used to resolve chromosomal assignment ambiguities after Phasefinder.
In yet another embodiment, the PL-EM algorithm (Partition-Ligation EM; Niu et al.., Am. J. Hum. Genet. 70:157 (2002)) can be used to estimate haplotypes from the "genotype" data as a measured estimate of the reference allele frequency of a SNP in 15-marker windows that advance in increments of one marker across the data set. The results from such algorithms are converted into 15-marker haplotype files. Subsequently, the individual 15-marker block files are assembled into one continuous block of haplotypes for the entire chromosome. These extended haplotypes can then be used for further analysis. Such haplotype assembly algorithms take the consensus estimate of the allele call at each marker over all separate estimations (most markers are estimated 15 different times as the 15 marker blocks pass over their position).
In the preferred embodiment, the haplotypes for both the controls and the patients are derived in this manner. The preferred control of a trio structure is the spouse if the patient is one of the parents or the non-transmitted chromosomes (chromosomes found in parents but not in affected child) if the patient is the child.
In another embodiment, the haplotype frequencies among patients are compared to those among the controls using LDSTATS, a program that assesses the association of haplotypes with the disease. Such program defines haplotypes using multi-marker windows that advance across the marker map in one-marker increments. Such windows can be 1 , 3, 5, 7 or 9 markers wide, and all these window sizes are tested concurrently. At each position the frequency of haplotypes in cases is compared to the frequency of haplotypes in controls. Such allele frequency differences for single marker windows can be tested using Pearson's Chi-square with one degree of freedom. Multi-allelic haplotype association can be tested using Smith's normalization of th e square root of Pearson's Chi-square. Such significance of association can be reported in two ways:
The significance of association within any one haplotype window is plotted against the marker that is central to that window.
P-values of association for each specific marker can be calculated as a pooled P- value across all haplotype windows in which they occur. The pooled P-value is calculated using an expected value and variance calculated using a permutation test that considers covariance between individual windows. Such pooled P- values can yield narrower regions of gene location than the window data (see example 3 for details on analysis methods, such as LDSTATs V2.0 and V4.0).
In another embodiment, conditional haplotype analyses can be performed on subsets of the original set of cases and controls using the program LDSTAT. The selection of a subset of cases and their matched controls can be based on the carrier status of cases at a gene or locus of interest (see conditional analysis section in example 3 herein). Various conditional haplotypes can be derived, such as protective haplotypes and risk haplotypes.
Step 6: Fine Mapping
In this step, the candidate regions that were identified by step 4 are further mapped for the purpose of refinement and validation.
In the preferred embodiment, this fine mapping is performed with a density of genetic markers higher than in the genome wide scan (step 3) using any genotyping platform available in the art. Such fine mapping can be, but is not limited to, typing the allele via an allele-specific elongation assay that is then ligated to a locus-specific oligonucleotide. Such assays can be performed directly on the genomic DNA at a highly multiplex level and the products can be amplified using universal oligonucleotides. For each candidate region, the density of genetic markers can be, but is not limited to, a set of SNP markers with an average inter-marker distance of 1-4 Kb distributed over about 400 Kb to 1 Mb, roughly centered at the highest point of the GWS association. The preferred samples are those obtained from asthma disease PPC trios including the ones used for the GWS.
In the preferred embodiment, the genetic analysis of the results obtained using haplotype information as well as single-marker association (as performed as in step 5, described herein) is performed as described herein (step 5 and example section). The candidate regions that are validated and confirmed after this analysis proceed to a gene mining step described in example 5, herein, to characterize their marker and genetic content.
Step 7: SNP and DNA polymorphism discovery
In the preferred embodiment, all the candidate genes and regions identified in step 6 are sequenced for polymorphism identification.
In another embodiment, the entire region, including all introns, is sequenced to identify all polymorphisms.
In yet another embodiment, the candidate genes are prioritized for sequencing, and only functional gene elements (promoters, conserved noncoding sequences, exons and splice sites) are sequenced.
In yet another embodiment, previously identified polymorphisms in the candidate regions can also be used. For example, SNPs from dbSNP, Perlegen Sciences, Inc., or others can also be used rather than resequencing the candidate regions to identify polymorphisms.
The discovery of SNPs and DNA polymorphisms generally comprises a step consisting of determining the major haplotypes in the region to be sequenced. The preferred samples are selected according to which haplotypes contribute to the association signal observed in the region to be sequenced. The purpose is to select a set of samples that covers all the major haplotypes in the given region. Each major haplotype is preferably analyzed in at least a few individuals.
Any analytical procedure may be used to detect the presence or absence of variant nucleotides at one or more polymorphic positions of the invention. In general, the detection of allelic variation requires a mutation discrimination technique, optionally an amplification reaction and optionally a signal generation system. Any means of mutation detection or discrimination may be used. For instance, DNA sequencing, scanning methods, hybridization, extension based methods, incorporation based methods, restriction enzyme-based methods and ligation-based methods may be used in the methods of the invention.
Sequencing methods include, but are not limited to, direct sequencing, and sequencing by hybridization. Scanning methods include, but are not limited to, protein truncation test (PTT), single-strand conformation polymorphism analysis (SSCP), denaturing gradient gel electrophoresis (DGGE), temperature gradient gel electrophoresis (TGGE), cleavage, heteroduplex analysis, chemical mismatch cleavage (CMC), and enzymatic mismatch cleavage. Hybridization-based methods of detection include, but are not limited to, solid phase hybridization such as dot blots, multiple allele specific diagnostic assay (MASDA), reverse dot blots, and oligonucleotide arrays (DNA Chips). Solution phase hybridization amplification methods may also be used, such as Taqman. Extension based methods include, but are not limited to, amplification refraction mutation systems (ARMS), amplification refractory mutation systems (ALEX), and competitive oligonucleotide priming systems (COPS). Incorporation based methods include,
but are not limited to, mini-sequencing and arrayed primer extension (APEX). Restriction enzyme-based detection systems include, but are not limited to, restriction site generating PCR. Lastly, ligation based detection methods include, but are not limited to, oligonucleotide ligation assays (OLA). Signal generation or detection systems that may be used in the methods of the invention include, but are not limited to, fluorescence methods such as fluorescence resonance energy transfer (FRET), fluorescence quenching, fluorescence polarization as well as other chemiluminescence, electrochemiluminescence, Raman, radioactivity, colometric methods, hybridization protection assays and mass spectrometry methods. Further amplification methods include, but are not limited to self sustained replication (SSR), nucleic acid sequence based amplification (NASBA), ligase chain reaction (LCR), strand displacement amplification (SDA) and branched DNA (B-DNA).
Step 8: Ultrafine Mapping
This step further maps the candidate regions and genes confirmed in the previous step to identify and validate the responsible polymorphisms associated with asthma disease in the human population.
In a preferred embodiment, the discovered SNPs and polymorphisms of step 7 are ultrafine mapped at a higher density of markers than the fine mapping described herein using the same technology described in step 6.
Step 9: GeneMap construction
The confirmed variations in DNA (including both genie and non-genic regions) are used to build a GeneMap for asthma disease. The gene content of this GeneMap is described in more detail below. Such GeneMap can be used for other methods of the invention comprising the diagnostic methods described herein, the susceptibility to asthma disease, the response to a particular drug, the efficacy of a particular drug, the screening methods described herein and the treatment methods described herein.
As is evident to one of ordinary skill in the art, all of the above steps or the steps of Figure 1 do not need to be performed, or performed in a given order to practice or use the SNPs1 genomic regions, genes, proteins, etc. in the methods of the invention.
Genes from the GeneMap
In the preferred embodiment the GeneMap consists of genes and targets, in a variety of combinations, identified from the candidate regions listed in Table 2. In the preferred embodiment, all genes from Tables 3 and 4 are present in the GeneMap. In another preferred embodiment, the GeneMap consists of a selection of genes from Table 3 and 4.
The genes of the invention (Tables 3 and 4) are arranged by candidate regions and by their chromosomal location. Such order is for the purpose of clarity and does not reflect any other criteria of selection in the association of the genes with asthma disease.
Nucleic acid sequences
The nucleic acid sequences of the present invention may be derived from a variety of sources including DNA, cDNA, synthetic DNA, synthetic RNA, derivatives, mimetics or combinations thereof. Such sequences may comprise genomic DNA, which may or may not include naturally occurring introns, genie regions, nongenic regions, and regulatory regions. Moreover, such genomic DNA may be obtained in association with promoter regions or poly (A) sequences. The sequences, genomic DNA, or cDNA may be obtained in any of several ways. Genomic DNA can be extracted and purified from suitable cells by means well known in the art. Alternatively, mRNA can be isolated from a cell and used to produce cDNA by reverse transcription or other means. The nucleic acids described herein are used in certain embodiments of the methods of the present invention for production of RNA, proteins or polypeptides, through incorporation into cells, tissues, or organisms. In one embodiment, DNA containing all or part of
the coding sequence for the genes described in Tables 3 and 4, or the SNP markers described in Table 1 , is incorporated into a vector for expression of the encoded polypeptide in suitable host cells. The invention also comprises the use of the nucleotide sequence of the nucleic acids of this invention to identify DNA probes for the genes described in Tables 3 and 4 or the SNP markers described in Table 1 , PCR primers to amplify the genes described in Tables 3 and 4 or the SNP markers described in Table 1, nucleotide polymorphisms in the genes described in Tables 3 and 4, and regulatory elements of the genes described in Tables 3 and 4. The nucleic acids of the present invention find use as primers and templates for the recombinant production of asthma disease-associated peptides or polypeptides, for chromosome and gene mapping, to provide antisense sequences, for tissue distribution studies, to locate and obtain full length genes, to identify and obtain homologous sequences (wild-type and mutants), and in diagnostic applications.
Antisense oligonucleotides
In a particular embodiment of the invention, an antisense nucleic acid or oligonucleotide is wholly or partially complementary to, and can hybridize with, a target nucleic acid (either DNA or RNA) having the sequence of SEQ ID NO:1 , NO:3 or any SEQ ID from Tables 1 , 3 or 4. For example, an antisense nucleic acid or oligonucleotide comprising 16 nucleotides can be sufficient to inhibit expression of at least one gene from Tables 3 and 4. Alternatively, an antisense nucleic acid or oligonucleotide can be complementary to 5' or 3' untranslated regions, or can overlap the translation initiation codon (5' untranslated and translated regions) of at least one gene from Tables 3 and 4, or its functional equivalent. In another embodiment, the antisense nucleic acid is wholly or partially complementary to, and can hybridize with, a target nucleic acid that encodes a polypeptide from a gene described in Tables 3 and 4.
In addition, oligonucleotides can be constructed which will bind to duplex nucleic acid (i.e., DNA:DNA or DNA:RNA), to form a stable triple helix containing or triplex nucleic acid. Such triplex oligonucleotides can inhibit transcription and/or
expression of a gene from Tables 3 and 4, or its functional equivalent (M. D. Frank-Kamenetskii et al., 1995). Triplex oligonucleotides are constructed using the basepairing rules of triple helix formation and the nucleotide sequence of the genes described in Tables 3 and 4.
The present invention encompasses methods of using oligonucleotides in antisense inhibition of the function of the genes from Tables 3 and 4. In the context of this invention, the term "oligonucleotide" refers to naturally-occurring species or synthetic species formed from naturally-occurring subunits or their close homologs. The term may also refer to moieties that function similarly to oligonucleotides, but have non-naturally-occurring portions. Thus, oligonucleotides may have altered sugar moieties or inter-sugar linkages. Exemplary among these are phosphorothioate and other sulfur containing species which are known in the art. In preferred embodiments, at least one of the phosphodiester bonds of the oligonucleotide has been substituted with a structure that functions to enhance the ability of the compositions to penetrate into the region of cells where the RNA whose activity is to be modulated is located. It is preferred that such substitutions comprise phosphorothioate bonds, methyl phosphonate bonds, or short chain alkyl or cycloalkyl structures. In accordance with other preferred embodiments, the phosphodiester bonds are substituted with structures which are, at once, substantially non-ionic and non- chiral, or with structures which are chiral and enantiomerically specific. Persons of ordinary skill in the art will be able to select other linkages for use in the practice of the invention. Oligonucleotides may also include species that include at least some modified base forms. Thus, purines and pyrimidines other than those normally found in nature may be so employed. Similarly, modifications on the furanosyl portions of the nucleotide subunits may also be effected, as long as the essential tenets of this invention are adhered to. Examples of such modifications are 2'-O-alkyl- and 2'-halogen-substituted nucleotides. Some non- limiting examples of modifications at the 2' position of sugar moieties which are useful in the present invention include OH, SH, SCH3, F, OCH3, OCN, O(CH2), NH2 and O(CH2)n CH3, where n is from 1 to about 10. Such oligonucleotides are functionally interchangeable with natural oligonucleotides or synthesized
oligonucleotides, which have one or more differences from the natural structure. All such analogs are comprehended by this invention so long as they function effectively to hybridize with at least one gene from Tables 3 and 4 DNA or RNA to inhibit the function thereof.
The oligonucleotides in accordance with this invention preferably comprise from about 3 to about 50 subunits. It is more preferred that such oligonucleotides and analogs comprise from about 8 to about 25 subunits and still more preferred to have from about 12 to about 20 subunits. As defined herein, a "subunit" is a base and sugar combination suitably bound to adjacent subunits through phosphodiester or other bonds. Antisense nucleic acids or oligonucleotides can be produced by standard techniques (see, e.g., Shewmaker et al., U.S. Patent No. 6,107,065). The oligonucleotides used in accordance with this invention may be conveniently and routinely made through the well-known technique of solid phase synthesis. Any other means for such synthesis may also be employed; however, the actual synthesis of the oligonucleotides is well within the abilities of the practitioner. It is also well known to prepare other oligonucleotides such as phosphorothioates and alkylated derivatives.
The oligonucleotides of this invention are designed to be hybridizable with RNA (e.g., mRNA) or DNA from genes described in Tables 3 and 4. For example, an oligonucleotide (e.g., DNA oligonucleotide) that hybridizes to mRNA from a gene described in Tables 3 and 4 can be used to target the mRNA for RnaseH digestion. Alternatively an oligonucleotide that can hybridize to the translation initiation site of the mRNA of a gene described in Tables 3 and 4 can be used to prevent translation of the mRNA. In another approach, oligonucleotides that bind to the double-stranded DNA of a gene from Tables 3 and 4 can be administered. Such oligonucleotides can form a triplex construct and inhibit the transcription of the DNA encoding polypeptides of the genes described in Tables 3 and 4. Triple helix pairing prevents the double helix from opening sufficiently to allow the binding of polymerases, transcription factors, or regulatory molecules. Recent therapeutic advances using triplex DNA have been described (see, e.g., J. E. Gee et al., 1994, Molecular and Immunologic Approaches, Futura Publishing Co., Mt. Kisco, NY).
As non-limiting examples, antisense oligonucleotides may be targeted to hybridize to the following regions: mRNA cap region; translation initiation site; translational termination site; transcription initiation site; transcription termination site; polyadenylation signal; 31 untranslated region; 5' untranslated region; 5' coding region; mid coding region; and 3' coding region. Preferably, the complementary oligonucleotide is designed to hybridize to the most unique 5' sequence of a gene described in Tables 3 and 4, including any of about 15-35 nucleotides spanning the 5' coding sequence. In accordance with the present invention, the antisense oligonucleotide can be synthesized, formulated as a pharmaceutical composition, and administered to a subject. The synthesis and utilization of antisense and triplex oligonucleotides have been previously described (e.g., Simon et al., 1999; Barre et al., 2000; Elez et a/., 2000; Sauter et a/., 2000).
Alternatively, expression vectors derived from retroviruses, adenovirus, herpes or vaccinia viruses or from various bacterial plasmids may be used for delivery of nucleotide sequences to the targeted organ, tissue or cell population. Methods which are well known to those skilled in the art can be used to construct recombinant vectors which will express nucleic acid sequence that is complementary to the nucleic acid sequence encoding a polypeptide from the genes described in Tables 3 and 4. These techniques are described both in Sambrook et al., 1989 and in Ausubel et al., 1992. For example, expression of at least one gene from Tables 3 and 4 can be inhibited by transforming a cell or tissue with an expression vector that expresses high levels of untranslatable sense or antisense sequences. Even in the absence of integration into the DNA, such vectors may continue to transcribe RNA molecules until they are disabled by endogenous nucleases. Transient expression may last for a month or more with a nonreplicating vector, and even longer if appropriate replication elements are included in the vector system. Various assays may be used to test the ability of gene-specific antisense oligonucleotides to inhibit the expression of at least one gene from Tables 3 and 4. For example, mRNA levels of the genes described in Tables 3 and 4 can be assessed by Northern blot analysis (Sambrook et al., 1989; Ausubel et al., 1992; J. C. Alwine et al. 1977; I. M. Bird,
1998), quantitative or semi-quantitative RT-PCR analysis (see, e.g., W.M. Freeman et al., 1999; Ren et al., 1998; J. M. CaIe et al., 1998), or in situ hybridization (reviewed by A.K. Raap, 1998). Alternatively, antisense oligonucleotides may be assessed by measuring levels of the polypeptide from the genes described in Tables 3 and 4, e.g., by western blot analysis, indirect immunofluorescence and immunoprecipitation techniques (see, e.g., J. M. Walker, 1998, Protein Protocols on CD-ROM, Humana Press, Totowa, NJ). Any other means for such detection may also be employed, and is well within the abilities of the practitioner.
Mapping Technologies
The present invention includes various methods which employ mapping technologies to map SNPs and polymorphisms. For purpose of clarity, this section comprises, but is not limited to, the description of mapping technologies that can be utilized to achieve the embodiments described herein. Mapping technologies may be based on amplification methods, restriction enzyme cleavage methods, hybridization methods, sequencing methods, and cleavage methods using agents.
Amplification methods include: self sustained sequence replication (Guatelli et al., 1990), transcriptional amplification system (Kwoh et al., 1989), Q-Beta Replicase (Lizardi et al., 1988), isothermal amplification (e.g. Dean et al., 2002; and Hafner ef al., 2001), or any other nucleic acid amplification method, followed by the detection of the amplified molecules using techniques well known to those of ordinary skill in the art. These detection schemes are especially useful for the detection of nucleic acid molecules if such molecules are present in very low number.
Restriction enzyme cleavage methods include: isolating sample and control DNA, amplification (optional), digestion with one or more restriction endonucleases, determination of fragment length sizes by gel electrophoresis and comparing samples and controls. Differences in fragment length sizes between sample and
control DNA indicates mutations in the sample DNA. Moreover, sequence specific ribozymes (see, e.g., U.S. Pat. No. 5,498,531) or DNAzyme (e.g. U.S. Pat. No. 5,807,718) can be used to score for the presence of specific mutations by development or loss of a ribozyme or DNAzyme cleavage site.
SNPs and SNP maps of the invention can be identified or generated by hybridizing sample nucleic acids, e.g., DNA or RNA, to high density arrays or bead arrays containing oligonucleotide probes corresponding to the polymorphisms of Table 1 (see the Affymetrix arrays and lllumina bead sets at www.affymetrix.com and www.illumina.com and see Cronin et a/., 1996; or Kozal et al., 1996).
A variety of sequencing reactions known in the art can be used to directly sequence nucleic acids for the presence or the absence of one or more polymorphisms of Table 1. Examples of sequencing reactions include those based on techniques developed by Maxam and Gilbert (1977) or Sanger (1977). It is also contemplated that any of a variety of automated sequencing procedures can be utilized, including sequencing by mass spectrometry (see, e.g. PCT International Publication No. WO 94/16101; Cohen et a/., 1996; and Griffin et a/., 1993), real-time pyrophosphate sequencing method (Ronaghi et a/., 1998; and Permutt et a/., 2001) and sequencing by hybridization (see e.g. Drmanac et a/., 2002).
Other methods of detecting polymorphisms include methods in which protection from cleavage agents is used to detect mismatched bases in RNA/RNA, DNA/DNA or RNA/DNA heteroduplexes (Myers et al., 1985). In general, the technique of "mismatch cleavage" starts by providing heteroduplexes formed by hybridizing (labeled) RNA or DNA containing a wild-type sequence with potentially mutant RNA or DNA obtained from a sample. The double-stranded duplexes are treated with an agent who cleaves single-stranded regions of the duplex such as which will exist due to basepair mismatches between the control and sample strands. For instance, RNA/DNA duplexes can be treated with RNase and DNA/DNA hybrids treated with S1 nuclease to enzymatically digest the mismatched regions. In other embodiments, either DNA/DNA or RNA/DNA
duplexes can be treated with hydroxylamine or osmium tetroxide and with piperidine in order to digest mismatched regions. After digestion of the mismatched regions, the resulting material is then separated by size on denaturing polyacrylamide gels to determine the site of a mutation or SNP (see, for example, Cotton et al., 1988; and Saleeba et al., 1992). In a preferred embodiment, the control DNA or RNA can be labeled for detection.
In still another embodiment, the mismatch cleavage reaction employs one or more proteins that recognize mismatched base pairs in double-stranded DNA (so called "DNA mismatch repair" enzymes) in defined systems for detecting and mapping polymorphisms. For example, the mutY enzyme of E. coli cleaves A at G/A mismatches (Hsu et al., 1994). Other examples include, but are not limited to, the MutHLS enzyme complex of E. coli (Smith and Modrich Proc. 1996) and CeI 1 from the celery (Kulinski et al., 2000) both cleave the DNA at various mismatches. According to an exemplary embodiment, a probe based on a polymorphic site corresponding to a polymorphism of Tables 1 , 3 or 4 is hybridized to a cDNA or other DNA product from a test cell or cells. The duplex is treated with a DNA mismatch repair enzyme, and the cleavage products, if any, can be detected from electrophoresis protocols or the like. See, for example, U.S. Pat. No. 5,459,039. Alternatively, the screen can be performed in vivo following the insertion of the heteroduplexes in an appropriate vector. The whole procedure is known to those ordinary skilled in the art and is referred to as mismatch repair detection (see e.g. Fakhrai-Rad et al., 2004).
In other embodiments, alterations in electrophoretic mobility can be used to identify polymorphisms in a sample. For example, single strand conformation polymorphism (SSCP) analysis can be used to detect differences in electrophoretic mobility between mutant and wild type nucleic acids (Orita et al., 1989; Cotton et al., 1993; and Hayashi 1992). Single-stranded DNA fragments of case and control nucleic acids will be denatured and allowed to renature. The secondary structure of single-stranded nucleic acids varies according to sequence. The resulting alteration in electrophoretic mobility enables the detection of even a single base change. The DNA fragments may be labeled or detected with labeled probes. The sensitivity of the assay may be enhanced by
using RNA (rather than DNA), in which the secondary structure is more sensitive to a change in sequence. In a preferred embodiment, the method utilizes heteroduplex analysis to separate double stranded heteroduplex molecules on the basis of changes in electrophoretic mobility (Kee et al., 1991).
In yet another embodiment, the movement of mutant or wild-type fragments in a polyacrylamide gel containing a gradient of denaturant is assayed using denaturing gradient gel electrophoresis (DGGE) (Myers et al., 1985). When DGGE is used as the method of analysis, DNA will be modified to insure that it does not completely denature, for example by adding a GC clamp of approximately 40 bp of high-melting GC-rich DNA by PCR. In a further embodiment, a temperature gradient is used in place of a denaturing gradient to identify differences in the mobility of control and sample DNA (Rosenbaum et al., 1987). In another embodiment, the mutant fragment is detected using denaturing HPLC (see e.g. Hoogendoorn et al., 2000).
Examples of other techniques for detecting polymorphisms include, but are not limited to, selective oligonucleotide hybridization, selective amplification, selective primer extension, selective ligation, single-base extension, selective termination of extension or invasive cleavage assay. For example, oligonucleotide primers may be prepared in which the polymorphism is placed centrally and then hybridized to target DNA under conditions which permit hybridization only if a perfect match is found (Saiki et al., 1986; Saiki et al., 1989). Such oligonucleotides are hybridized to PCR amplified target DNA or a number of different mutations when the oligonucleotides are attached to the hybridizing membrane and hybridized with labeled target DNA. Alternatively, the amplification, the allele-specific hybridization and the detection can be done in a single assay following the principle of the 5' nuclease assay (e.g. see Livak et al., 1995). For example, the associated allele, a particular allele of a polymorphic locus, or the like is amplified by PCR in the presence of both allele-specific oligonucleotides, each specific for one or the other allele. Each probe has a different fluorescent dye at the 5' end and a quencher at the 3' end. During PCR, if one or the other or both allele-specific oligonucleotides are hybridized to the template, the Taq polymerase via its 5' exonuclease activity will release the
corresponding dyes. The latter will thus reveal the genotype of the amplified product.
Hybridization assays may also be carried out with a temperature gradient following the principle of dynamic allele-specific hybridization or like e.g. Jobs et al., (2003); and Bourgeois and Labuda, (2004). For example, the hybridization is done using one of the two allele-specific oligonucleotides labeled with a fluorescent dye, and an intercalating quencher under a gradually increasing temperature. At low temperature, the probe is hybridized to both the mismatched and full-matched template. The probe melts at a lower temperature when hybridized to the template with a mismatch. The release of the probe is captured by an emission of the fluorescent dye, away from the quencher. The probe melts at a higher temperature when hybridized to the template with no mismatch. The temperature-dependent fluorescence signals therefore indicate the absence or presence of an associated allele, a particular allele of a polymorphic locus, or the like ( e.g. Jobs et al., 2003). Alternatively, the hybridization is done under a gradually decreasing temperature. In this case, both allele-specific oligonucleotides are hybridized to the template competitively. At high temperature none of the two probes are hybridized. Once the optimal temperature of the full- matched probe is reached, it hybridizes and leaves no target for the mismatched probe (e.g. Bourgeois and Labuda, 2004). In the latter case, if the allele-specific probes are differently labeled, then they are hybridized to a single PCR-amplified target. If the probes are labeled with the same dye, then the probe cocktail is hybridized twice to identical templates with only one labeled probe, different in the two cocktails, in the presence of the unlabeled competitive probe.
Alternatively, allele specific amplification technology that depends on selective PCR amplification may be used in conjunction with the present invention. Oligonucleotides used as primers for specific amplification may carry the associated allele, a particular allele of a polymorphic locus, or the like, also referred to as "mutation" of interest in the center of the molecule, so that amplification depends on differential hybridization (Gibbs et al., 1989) or at the extreme 3' end of one primer where, under appropriate conditions, mismatch can prevent, or reduce polymerase extension (Prossner, 1993). In addition it may be
desirable to introduce a novel restriction site in the region of the mutation to create cleavage-based detection (Gasparini et al., 1992). It is anticipated that in certain embodiments, amplification may also be performed using Taq ligase for amplification (Barany, 1991). In such cases, ligation will occur only if there is a perfect match at the 31 end of the 5' sequence making it possible to detect the presence of a known associated allele, a particular allele of a polymorphic locus, or the like at a specific site by looking for the presence or absence of amplification. The products of such an oligonucleotide ligation assay can also be detected by means of gel electrophoresis. Furthermore, the oligonucleotides may contain universal tags used in PCR amplification and zip code tags that are different for each allele. The zip code tags are used to isolate a specific, labeled oligonucleotide that may contain a mobility modifier (e.g. Grossman et al., 1994).
In yet another alternative, allele-specific elongation followed by ligation will form a template for PCR amplification. In such cases, elongation will occur only if there is a perfect match at the 3' end of the allele-specific oligonucleotide using a DNA polymerase. This reaction is performed directly on the genomic DNA and the extension/ligation products are amplified by PCR. To this end, the oligonucleotides contain universal tags allowing amplification at a high multiplex level and a zip code for SNP identification. The PCR tags are designed in such a way that the two alleles of a SNP are amplified by different forward primers, each having a different dye. The zip code tags are the same for both alleles of a given SNPs and they are used for hybridization of the PCR-amplified products to oligonucleotides bound to a solid support, chip, bead array or like. For an example of the procedure, see Fan et al. (Cold Spring Harbor Symposia on Quantitative Biology, Vol. LXVIII, pp. 69-78 2003).
Another alternative includes the single-base extension/ligation assay using a molecular inversion probe, consisting of a single, long oligonucleotide (see e.g. Hardenbol et al., 2003). In such an embodiment, the oligonucleotide hybridizes on both side of the SNP locus directly on the genomic DNA, leaving a one-base gap at the SNP locus. The gap-filling, one-base extension/ligation is performed in four tubes, each having a different dNTP. Following this reaction, the oligonucleotide is circularized whereas unreactive, linear oligonucleotides are
degraded using an exonuclease such as exonuclease I of E. coli. The circular oligonucleotides are then linearized and the products are amplified and labeled using universal tags on the oligonucleotides. The original oligonucleotide also contains a SNP-specific zip code allowing hybridization to oligonucleotides bound to a solid support, chip, and bead array or like. This reaction can be performed at a high multiplexed level.
In another alternative, the associated allele, a particular allele of a polymorphic locus, or the like is scored by single-base extension (see e.g. U.S. Pat. No. 5,888,819). The template is first amplified by PCR. The extension oligonucleotide is then hybridized next to the SNP locus and the extension reaction is performed using a thermostable polymerase such as ThermoSequenase (GE Healthcare) in the presence of labeled ddNTPs. This reaction can therefore be cycled several times. The identity of the labeled ddNTP incorporated will reveal the genotype at the SNP locus. The labeled products can be detected by means of gel electrophoresis, fluorescence polarization (e.g. Chen et a/., 1999) or by hybridization to oligonucleotides bound to a solid support, chip, and bead array or like. In the latter case, the extension oligonucleotide will contain a SNP-specific zip code tag.
In yet another alternative, a SNP is scored by selective termination of extension. The template is first amplified by PCR and the extension oligonucleotide hybridizes in the vicinity of the SNP locus, close to but not necessarily adjacent to it. The extension reaction is carried out using a thermostable polymerase such as ThermoSequenase (GE Healthcare) in the presence of a mix of dNTPs and at least one ddNTP. The latter has to terminate the extension at one of the allele of the interrogated SNP, but not both such that the two alleles will generate extension products of different sizes. The extension product can then be detected by means of gel electrophoresis, in which case the extension products need to be labeled, or by mass spectrometry (see e.g. Storm et a/., 2003).
In another alternative, SNPs are detected using an invasive cleavage assay (see U.S. Pat. No. 6,090,543). There are five oligonucleotides per SNP to interrogate but these are used in a two step-reaction. During the primary reaction, three of
the designed oligonucleotides are first hybridized directly to the genomic DNA. One of them is locus-specific and hybridizes up to the SNP locus (the pairing of the 3' base at the SNP locus is not necessary). There are two allele-specific oligonucleotides that hybridize in tandem to the locus-specific probe but also contain a 5' flap that is specific for each allele of the SNP. Depending upon hybridization of the allele-specific oligonucleotides at the base of the SNP locus, this creates a structure that is recognized by a cleavase enzyme (U.S. Pat. No. 6,090,606) and the allele-specific flap is released. During the secondary reaction, the flap fragments hybridize to a specific cassette to recreate the same structure as above except that the cleavage will release a small DNA fragment labeled with a fluorescent dye that can be detected using regular fluorescence detector. In the cassette, the emission of the dye is inhibited by a quencher.
Methods to identify agents that modulate the expression of a nucleic acid encoding a gene involved in asthma disease.
The present invention provides methods for identifying agents that modulate the expression of a nucleic acid encoding a gene from Tables 3 and 4. Such methods may utilize any available means of monitoring for changes in the expression level of the nucleic acids of the invention. As used herein, an agent is said to modulate the expression of a nucleic acid of the invention if it is capable of up- or down- regulating expression of the nucleic acid in a cell. Such cells can be obtained from any parts of the body such as the respiratory system, lung, trachea, esophagus, bronchus and bronchiole, duct, alveoli, alviolar duct, alveolar sacs, stomach, nasal cavity, paranasal sinuses, nasopharynx, larynx, inner and outer lung coatings, inner and outer trachea coatings, mucosa, submucosa, rectum, scalp, blood, dermis, epidermis, skin cells, cutaneous surfaces, intertrigious areas, genitalia, vessels and endothelium. Some non-limiting examples of cells that can be used are: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, dendritic cell, cilated cell, synovial cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina
propria lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
In one assay format, the expression of a nucleic acid encoding a gene of the invention (see Tables 3 and 4) in a cell or tissue sample is monitored directly by hybridization to the nucleic acids of the invention. Cell lines or tissues are exposed to the agent to be tested under appropriate conditions and time and total RNA or mRNA is isolated by standard procedures such as those disclosed in Sambrook et al., (1989) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press).
Probes to detect differences in RNA expression levels between cells exposed to the agent and control cells may be prepared as described above. Hybridization conditions are modified using known methods, such as those described by Sambrook et al., and Ausubel et al., as required for each probe. Hybridization of total cellular RNA or RNA enriched for polyA RNA can be accomplished in any available format. For instance, total cellular RNA or RNA enriched for polyA RNA can be affixed to a solid support and the solid support exposed to at least one probe comprising at least one, or part of one of the sequences of the invention under conditions in which the probe will specifically hybridize. Alternatively, nucleic acid fragments comprising at least one, or part of one of the sequences of the invention can be affixed to a solid support, such as a silicon chip or a porous glass wafer. The chip or wafer can then be exposed to total cellular RNA or polyA RNA from a sample under conditions in which the affixed sequences will specifically hybridize to the RNA. By examining for the ability of a given probe to specifically hybridize to an RNA sample from an untreated cell population and from a cell population exposed to the agent, agents which up or down regulate expression are identified.
Methods to identify agents that modulate the activity of a protein encoded by a gene involved in asthma disease.
The present invention provides methods for identifying agents that modulate at least one activity of the proteins described in Tables 3 and 4. Such methods may utilize any means of monitoring or detecting the desired activity. As used herein, an agent is said to modulate the expression of a protein of the invention if it is capable of up- or down- regulating expression of the protein in a cell. Such cells can be obtained from any parts of the body such as the respiratory system, lung, trachea, esophagus, bronchus and bronchiole, duct, alveoli, alviolar duct, alveolar sacs, stomach, nasal cavity, paranasal sinuses, nasopharynx, larynx, inner and outer lung coatings, inner and outer trachea coatings, mucosa, submucosa, rectum, scalp, blood, dermis, epidermis, skin cells, cutaneous surfaces, intertrigious areas, genitalia, vessels and endothelium. Some non-limiting examples of cells that can be used are: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, synovial cell, cilated cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina propria lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
In one format, the specific activity of a protein of the invention, normalized to a standard unit, may be assayed in a cell population that has been exposed to the agent to be tested and compared to an unexposed control cell population may be assayed. Cell lines or populations are exposed to the agent to be tested under appropriate conditions and times. Cellular lysates may be prepared from the exposed cell line or population and a control, unexposed cell line or population. The cellular lysates are then analyzed with the probe.
Antibody probes can be prepared by immunizing suitable mammalian hosts utilizing appropriate immunization protocols using the proteins of the invention or antigen-containing fragments thereof. To enhance immunogenicity, these proteins or fragments can be conjugated to suitable carriers. Methods for preparing immunogenic conjugates with carriers such as BSA, KLH or other carrier proteins are well known in the art. In some circumstances, direct conjugation using, for example, carbodiimide reagents may be effective; in other instances linking reagents such as those supplied by Pierce Chemical Co.
(Rockford, IL) may be desirable to provide accessibility to the hapten. The hapten peptides can be extended at either the amino or carboxy terminus with a cysteine residue or interspersed with cysteine residues, for example, to facilitate linking to a carrier. Administration of the immunogens is conducted generally by injection over a suitable time period and with use of suitable adjuvants, as is generally understood in the art. During the immunization schedule, titers of antibodies are taken to determine adequacy of antibody formation. While the polyclonal antisera produced in this way may be satisfactory for some applications, for pharmaceutical compositions, use of monoclonal preparations is preferred. Immortalized cell lines which secrete the desired monoclonal antibodies may be prepared using standard methods, see e.g., Kohler & Milstein (1992) or modifications which affect immortalization of lymphocytes or spleen cells, as is generally known. The immortalized cell lines secreting the desired antibodies can be screened by immunoassay in which the antigen is the peptide hapten, polypeptide or protein. When the appropriate immortalized cell culture secreting the desired antibody is identified, the cells can be cultured either in vitro or by production in ascites fluid. The desired monoclonal antibodies may be recovered from the culture supernatant or from the ascites supernatant. Fragments of the monoclonal antibodies or the polyclonal antisera which contain the immunologically significant portion(s) can be used as antagonists, as well as the intact antibodies. Use of immunologically reactive fragments, such as Fab or Fab' fragments, is often preferable, especially in a therapeutic context, as these fragments are generally less immunogenic than the whole immunoglobulin. The antibodies or fragments may also be produced, using current technology, by recombinant means. Antibody regions that bind specifically to the desired regions of the protein can also be produced in the context of chimeras derived from multiple species. Antibody regions that bind specifically to the desired regions of the protein can also be produced in the context of chimeras from multiple species, for instance, humanized antibodies. The antibody can therefore be a humanized antibody or a human antibody, as described in U.S. Patent 5,585,089 or Riechmann et al. (1988).
Agents that are assayed in the above method can be randomly selected or rationally selected or designed. As used herein, an agent is said to be randomly selected when the agent is chosen randomly without considering the specific sequences involved in the association of the protein of the invention alone or with its associated substrates, binding partners, etc. An example of randomly selected agents is the use of a chemical library or a peptide combinatorial library, or a growth broth of an organism. As used herein, an agent is said to be rationally selected or designed when the agent is chosen on a non-random basis which takes into account the sequence of the target site or its conformation in connection with the agent's action. Agents can be rationally selected or rationally designed by utilizing the peptide sequences that make up these sites. For example, a rationally selected peptide agent can be a peptide whose amino acid sequence is identical to or a derivative of any functional consensus site. The agents of the present invention can be, as examples, oligonucleotides, antisense polynucleotides, interfering RNA, peptides, peptide mimetics, antibodies, antibody fragments, small molecules, vitamin derivatives, as well as carbohydrates. Peptide agents of the invention can be prepared using standard solid phase (or solution phase) peptide synthesis methods, as is known in the art. In addition, the DNA encoding these peptides may be synthesized using commercially available oligonucleotide synthesis instrumentation and produced recombinant^ using standard recombinant production systems. The production using solid phase peptide synthesis is necessitated if non-gene-encoded amino acids are to be included.
Another class of agents of the present invention includes antibodies or fragments thereof that bind to a protein encoded by a gene in Tables 3 and 4. Antibody agents can be obtained by immunization of suitable mammalian subjects with peptides, containing as antigenic regions, those portions of the protein intended to be targeted by the antibodies (see section above of antibodies as probes for standard antibody preparation methodologies).
In yet another class of agents, the present invention includes peptide mimetics that mimic the three-dimensional structure of the protein encoded by a gene from Tables 3 and 4. Such peptide mimetics may have significant advantages over
naturally occurring peptides, including, for example: more economical production, greater chemical stability, enhanced pharmacological properties (half-life, absorption, potency, efficacy, etc.), altered specificity (e.g., a broad-spectrum of biological activities), reduced antigenicity and others. In one form, mimetics are peptide-containing molecules that mimic elements of protein secondary structure. The underlying rationale behind the use of peptide mimetics is that the peptide backbone of proteins exists chiefly to orient amino acid side chains in such a way as to facilitate molecular interactions, such as those of antibody and antigen. A peptide mimetic is expected to permit molecular interactions similar to the natural molecule. In another form, peptide analogs are commonly used in the pharmaceutical industry as non-peptide drugs with properties analogous to those of the template peptide. These types of non-peptide compounds are also referred to as peptide mimetics or peptidomimetics (Fauchere, 1986; Veber & Freidinger, 1985; Evans et a/., 1987) which are usually developed with the aid of computerized molecular modeling. Peptide mimetics that are structurally similar to therapeutically useful peptides may be used to produce an equivalent therapeutic or prophylactic effect. Generally, peptide mimetics are structurally similar to a paradigm polypeptide (i.e., a polypeptide that has a biochemical property or pharmacological activity), but have one or more peptide linkages optionally replaced by a linkage using methods known in the art. Labeling of peptide mimetics usually involves covalent attachment of one or more labels, directly or through a spacer (e.g., an amide group), to non-interfering position(s) on the peptide mimetic that are predicted by quantitative structure-activity data and molecular modeling. Such non-interfering positions generally are positions that do not form direct contacts with the macromolecule(s) to which the peptide mimetic binds to produce the therapeutic effect. Derivitization (e.g., labeling) of peptide mimetics should not substantially interfere with the desired biological or pharmacological activity of the peptide mimetic. The use of peptide mimetics can be enhanced through the use of combinatorial chemistry to create drug libraries. The design of peptide mimetics can be aided by identifying amino acid mutations that increase or decrease binding of the protein to its binding partners. Approaches that can be used include the yeast two hybrid method (see Chien et al., 1991) and the phage display method. The two hybrid method detects protein-
protein interactions in yeast (Fields et al., 1989). The phage display method detects the interaction between an immobilized protein and a protein that is expressed on the surface of phages such as lambda and M13 (Amberg et al., 1993; Hogrefe et al., 1993). These methods allow positive and negative selection for protein-protein interactions and the identification of the sequences that determine these interactions.
Method to diagnose asthma disease
The present invention also relates to methods for diagnosing inflammatory disease or a related disease, preferably asthma disease, a disposition to such disease, predisposition to such a disease and/or disease progression. In some methods, the steps comprise contacting a target sample with (a) nucleic acid molecule(s) or fragments thereof and comparing the concentration of individual mRNA(s) with the concentration of the corresponding mRNA(s) from at least one healthy donor. An aberrant (increased or decreased) mRNA level of at least one gene from Tables 3 and 4, at least 5 or 10 genes from Tables 3 and 4, at least 50 genes from Tables 3 and 4, at least 100 genes from Tables 3 and 4 or at least 200 genes from Tables 3 and 4 determined in the sample in comparison to the control sample is an indication of asthma disease or a related disease or a disposition to such kinds of diseases. For diagnosis, samples are, preferably, obtained from inflamed lung tissue. Samples can also be obtained from any parts of the body such as the respiratory system, lung, trachea, esophagus, bronchus and bronchiole, duct, alveoli, alviolar duct, alveolar sacs, stomach, nasal cavity, paranasal sinuses, nasopharynx, larynx, inner and outer lung coatings, inner and outer trachea coatings, mucosa, submucosa, rectum, scalp, blood, dermis, epidermis, skin cells, cutaneous surfaces, intertrigious areas, genitalia, vessels and endothelium. Some non-limiting examples of cells that can be used are: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, synovial cell, cilated cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina propria lymphocyte, intraepithelial lymphocyte,
goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
For analysis of gene expression, total RNA is obtained from cells according to standard procedures and, preferably, reverse-transcribed. Preferably, a DNAse treatment (in order to get rid of contaminating genomic DNA) is performed. Some non-limiting examples of cells that can be used are: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, synovial cell, cilated cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina propria lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
The nucleic acid molecule or fragment is typically a nucleic acid probe for hybridization or a primer for PCR. The person skilled in the art is in a position to design suitable nucleic acids probes based on the information provided in the Tables of the present invention. The target cellular component, i.e. mRNA, e.g., in lung tissue, may be detected directly in situ, e.g. by in situ hybridization or it may be isolated from other cell components by common methods known to those skilled in the art before contacting with a probe. Detection methods include Northern blot analysis, RNase protection, in situ methods, e.g. in situ hybridization, in vitro amplification methods (PCR, LCR, QRNA replicase or RNA- transcription/amplification (TAS, 3SR), reverse dot blot disclosed in EP- B10237362) and other detection assays that are known to those skilled in the art. Products obtained by in vitro amplification can be detected according to established methods, e.g. by separating the products on agarose or polyacrylamide gels and by subsequent staining with ethidium bromide. Alternatively, the amplified products can be detected by using labeled primers for amplification or labeled dNTPs. Preferably, detection is based on a microarray.
The probes (or primers) (or, alternatively, the reverse-transcribed sample mRNAs) can be detectably labeled, for example, with a radioisotope, a
bioluminescent compound, a chemiluminescent compound, a fluorescent compound, a metal chelate, or an enzyme.
The present invention also relates to the use of the nucleic acid molecules or fragments described above for the preparation of a diagnostic composition for the diagnosis of asthma disease or a disposition to such a disease.
The present invention also relates to the use of the nucleic acid molecules of the present invention for the isolation or development of a compound which is useful for therapy of asthma disease. For example, the nucleic acid molecules of the invention and the data obtained using said nucleic acid molecules for diagnosis of asthma disease might allow for the identification of further genes which are specifically dysregulated, and thus may be considered as potential targets for therapeutic interventions.
The invention further provides prognostic assays that can be used to identify subjects having or at risk of developing asthma disease. In such method, a test sample is obtained from a subject and the amount and/or concentration of the nucleic acid described in Tables 3 and 4 is determined; wherein the presence of an associated allele, a particular allele of a polymorphic locus, or the likes in the nucleic acids sequences of this invention (see SEQ ID from Tables 1 or 3) can be diagnostic for a subject having or at risk of developing asthma. As used herein, a "test sample" refers to a biological sample obtained from a subject of interest. For example, a test sample can be a biological fluid, a cell sample, or tissue. A biological fluid can be, but is not limited to saliva, serum, mucus, urine, stools, spermatozoids, vaginal secretions, lymph, amiotic liquid, pleural liquid and tears. Cells can be, but are not limited to: muscle cells, nervous cells, blood and vessels cells, dermis, epidermis and other skin cells, smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, synovial cell, cilated cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina propria lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
Furthermore, the prognostic assays described herein can be used to determine whether a subject can be administered an agent (e.g., an agonist, antagonist, peptidomimetic, polypeptide, nucleic acid such as antisense DNA or interfering RNA (RNAi), small molecule or other drug candidate) to treat asthma disease. Specifically, these assays can be used to predict whether an individual will have an efficacious response or will experience adverse events in response to such an agent. For example, such methods can be used to determine whether a subject can be effectively treated with an agent that modulates the expression and/or activity of a gene from Tables 3 and 4 or the nucleic acids described herein. In another example, an association study may be performed to identify polymorphisms from Table 1 that are associated with a given response to the agent, e.g., an efficacious response or the likelihood of one or more adverse events. Thus, one embodiment of the present invention provides methods for determining whether a subject can be effectively treated with an agent for a disorder associated with aberrant expression or activity of a gene from Tables 3 and 4 in which a test sample is obtained and nucleic acids or polypeptides from Tables 3 and 4 are detected (e.g., wherein the presence of a particular level of expression of a gene from Tables 3 and 4 or a particular allelic variant of such gene, such as polymorphism from Table 1 , is diagnostic for a subject that can be administered an agent to treat a disorder such as asthma disease). In one embodiment, the method includes obtaining a sample from a subject suspected of having asthma disease or an affected individual and exposing such sample to an agent. The expression and/or activity of the nucleic acids and/or genes of the invention are monitored before and after treatment with such agent to assess the effect of such agent. After analysis of the expression values, one skilled in the art can determine whether such agent can effectively treat such subject. In another embodiment, the method includes obtaining a sample from a subject having or susceptible to developing asthma disease and determining the allelic constitution of polymorphisms from Table 1 that are associated with a particular response to an agent. After analysis of the allelic constitution of the individual at the associated polymorphisms, one skilled in the art can determine whether such agent can effectively treat such subject.
The methods of the invention can also be used to detect genetic alterations in a gene from Tables 3 and 4, thereby determining if a subject with the lesioned gene is at risk for a disorder associated with asthma disease. In preferred embodiments, the methods include detecting, in a sample of cells from the subject, the presence or absence of a genetic alteration characterized by at least one alteration linked to or affecting the integrity of a gene from Tables 3 and 4 encoding a polypeptide or the misexpression of such gene. For example, such genetic alterations can be detected by ascertaining the existence of at least one of: (1) a deletion of one or more nucleotides from a gene from Tables 3 and 4; (2) an addition of one or more nucleotides to a gene from Table 3 and 4; (3) a substitution of one or more nucleotides of a gene from Tables 3 and 4; (4) a chromosomal rearrangement of a gene from Tables 3 and 4; (5) an alteration in the level of a messenger RNA transcript of a gene from Tables 3 and 4; (6) aberrant modification of a gene from Tables 3 and 4, such as of the methylation pattern of the genomic DNA, (7) the presence of a non-wild type splicing pattern of a messenger RNA transcript of a gene from Tables 3 and 4; (8) inappropriate post-translational modification of a polypeptide encoded by a gene from Tables 3 and 4; and (9) alternative promoter use. As described herein, there are a large number of assay techniques known in the art which can be used for detecting alterations in a gene from Tables 3 and 4. A preferred biological sample is a peripheral blood sample obtained by conventional means from a subject. Another preferred biological sample is a buccal swab. Other biological samples can be, but are not limited to, urine, stools, spermatozoids, respiratory system secretions, vaginal secretions, lymph, amiotic liquid, pleural liquid and tears.
In certain embodiments, detection of the alteration involves the use of a probe/primer in a polymerase chain reaction (PCR) (see, e.g., U.S. Pat. Nos. 4,683,195 and 4,683,202), such as anchor PCR or RACE PCR, or alternatively, in a ligation chain reaction (LCR) (see, e.g., Landegran et a/., 1988; and Nakazawa et al., 1994), the latter of which can be particularly useful for detecting point mutations in a gene from Tables 3 and 4 (see Abavaya et al., 1995). This method can include the steps of collecting a sample of cells from a patient, isolating nucleic acid (e.g., genomic DNA, mRNA, or both) from the cells of the
sample, contacting the nucleic acid sample with one or more primers which specifically hybridize to a gene from Tables 3 and 4 under conditions such that hybridization and amplification of the nucleic acid from Tables 3 and 4 (if present) occurs, and detecting the presence or absence of an amplification product, or detecting the size of the amplification product and comparing the length to a control sample. PCR and/or LCR may be desirable to use as a preliminary amplification step in conjunction with some of the techniques used for detecting a mutation, an associated allele, a particular allele of a polymorphic locus, or the like described herein.
Alternative amplification methods include: self sustained sequence replication (Guatelli et al., 1990), transcriptional amplification system (Kwoh et al., 1989), Q- Beta Replicase (Lizardi et al., 1988), isothermal amplification (e.g. Dean et al., 2002); and Hafner et al., 2001), or any other nucleic acid amplification method, followed by the detection of the amplified molecules using techniques well known to those of ordinary skill in the art. These detection schemes are especially useful for the detection of nucleic acid molecules if such molecules are present in very low number.
In an alternative embodiment, alterations in a gene from Tables 3 and 4, from a sample cell can be identified by identifying changes in a restriction enzyme cleavage pattern. For example, sample and control DNA is isolated, amplified (optionally), digested with one or more restriction endonucleases, and fragment length sizes are determined by gel electrophoresis and compared. Differences in fragment length sizes between sample and control DNA indicate a mutation(s), an associated allele, a particular allele of a polymorphic locus, or the like in the sample DNA. Moreover, sequence specific ribozymes (see, e.g., U.S. Pat. No. 5,498,531 or DNAzyme e.g. U.S. Pat. No. 5,807,718) can be used to score for the presence of specific associated allele, a particular allele of a polymorphic locus, or the likes by development or loss of a ribozyme or DNAzyme cleavage site.
The present invention also relates to further methods for diagnosing asthma disease or a related disorder, a disposition to such disorder, predisposition to
such a disorder and/or disorder progression. In some methods, the steps comprise contacting a target sample with (a) nucleic molecuie(s) or fragments thereof and determining the presence or absence of a particular allele of a polymorphism that confers a disorder-related phenotype (e.g., predisposition to such a disorder and/or disorder progression). The presence of at least one allele from Table 1 that is associated with asthma disease ("associated allele"), at least 5 or 10 associated alleles from Table 1 , at least 50 associated alleles from Table 1 , at least 100 associated alleles from Table 1 , or at least 200 associated alleles from Table 1 , determined in the sample is an indication of asthma disease or a related disorder, a disposition or predisposition to such kinds of disorders, or a prognosis for such disorder progression. Such samples can be obtained from any parts of the body such as the respiratory system, lung, trachea, esophagus, bronchus and bronchiole, duct, alveoli, alviolar duct, alveolar sacs, stomach, nasal cavity, paranasal sinuses, nasopharynx, larynx, inner and outer lung coatings, inner and outer trachea coatings, mucosa, submucosa, rectum, scalp, blood, dermis, epidermis, skin cells, cutaneous surfaces, intertrigious areas, genitalia, vessels and endothelium. Some non-limiting examples of cells that can be used are: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, synovial cell, cilated cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina propria lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
In other embodiments, alterations in a gene from Tables 3 and 4 can be identified by hybridizing sample and control nucleic acids, e.g., DNA or RNA, to high density arrays or bead arrays containing tens to thousands of oligonucleotide probes (Cronin et al., 1996; Kozal et a!., 1996). For example, alterations in a gene from Tables 3 and 4 can be identified in two dimensional arrays containing light-generated DNA probes as described in Cronin et a/., (1996). Briefly, a first hybridization array of probes can be used to scan through long stretches of DNA in a sample and control to identify base changes between the sequences by making linear arrays of sequential overlapping probes. This step allows the
identification of point mutations, associated alleles, particular alleles of a polymorphic locus, or the like. This step is followed by a second hybridization array that allows the characterization of specific mutations by using smaller, specialized probe arrays complementary to all variants, mutations, alleles detected. Each mutation array is composed of parallel probe sets, one complementary to the wild-type gene and the other complementary to the mutant gene.
In yet another embodiment, any of a variety of sequencing reactions known in the art can be used to directly sequence a gene from Tables 3 and 4 and detect an associated allele, a particular allele of a polymorphic locus, or the like by comparing the sequence of the sample gene from Tables 3 and 4 with the corresponding wild-type (control) sequence. Examples of sequencing reactions include those based on techniques developed by Maxam and Gilbert (1977) or Sanger (1977). It is also contemplated that any of a variety of automated sequencing procedures can be utilized when performing the diagnostic assays (Bio/Techniques 19:448, 1995) including sequencing by mass spectrometry (see, e.g. PCT International Publication No. WO 94/16101 ; Cohen et al., 1996; and Griffin et al. 1993), real-time pyrophosphate sequencing method (Ronaghi et al., 1998; and Permutt et al., 2001) and sequencing by hybridization (see e.g. Drmanac ef a/., 2002).
Other methods of detecting an associated allele, a particular allele of a polymorphic locus, or the likes in a gene from Tables 3 and 4 include methods in which protection from cleavage agents is used to detect mismatched bases in RNA/RNA, DNA/DNA or RNA/DNA heteroduplexes (Myers et al., 1985). In general, the technique of "mismatch cleavage" starts by providing heteroduplexes formed by hybridizing (labeled) RNA or DNA containing the wild-type gene sequence from Tables 3 and 4 with potentially mutant RNA or DNA obtained from a tissue sample. The double-stranded duplexes are treated with an agent that cleaves single-stranded regions of the duplex such as which will exist due to basepair mismatches between the control and sample strands. For instance, RNA/DNA duplexes can be treated with RNase and DNA/DNA hybrids treated with S1 nuclease to enzymatically digest the mismatched regions. In other
embodiments, either DNA/DNA or RNA/DNA duplexes can be treated with hydroxylamine or osmium tetroxide and with piperidine in order to digest mismatched regions. After digestion of the mismatched regions, the resulting material is then separated by size on denaturing polyacrylamide gels to determine the site of an associated allele, a particular allele of a polymorphic locus, or the like (see, for example, Cotton et al., 1988; Saleeba et al., 1992). In a preferred embodiment, the control DNA or RNA can be labeled for detection, as described herein.
In still another embodiment, the mismatch cleavage reaction employs one or more proteins that recognize mismatched base pairs in double-stranded DNA (so called "DNA mismatch repair" enzymes) in defined systems for detecting and mapping point an associated allele, a particular allele of a polymorphic locus, or the likes in a gene from Tables 3 and 4 cDNAs obtained from samples of cells. For example, the mutY enzyme of E. coli cleaves A at G/A mismatches (Hsu et al., 1994). Other examples include, but are not limited to, the MutHLS enzyme complex of E. coli (Smith and Modrich., 1996) and CeI 1 from the celery (Kulinski et a/., 2000) both cleave the DNA at various mismatches. According to an exemplary embodiment, a probe based on a gene sequence from Tables 3 and 4 is hybridized to a cDNA or other DNA product from a test cell or cells. The duplex is treated with a DNA mismatch repair enzyme, and the cleavage products, if any, can be detected using electrophoresis protocols or the like. See, for example, U.S. Pat. No. 5,459,039. Alternatively, the screen can be performed in vivo following the insertion of the heteroduplexes in an appropriate vector. The whole procedure is known to those ordinary skilled in the art and is referred to as mismatch repair detection (see e.g. Fakhrai-Rad et al., 2004).
In other embodiments, alterations in electrophoretic mobility can be used to identify an associated allele, a particular allele of a polymorphic locus, or the likes in genes from Tables 3 and 4. For example, single strand conformation polymorphism (SSCP) analysis can be used to detect differences in electrophoretic mobility between mutant and wild type nucleic acids (Orita et al., 1993; see also Cotton, 1993; and Hayashi et al., 1992). Single-stranded DNA fragments of sample and control nucleic acids from Tables 3 and 4 will be
denatured and allowed to renature. The secondary structure of single-stranded nucleic acids varies according to sequence; the resulting alteration in electrophoretic mobility enables the detection of even a single base change. The DNA fragments may be labeled or detected with labeled probes. The sensitivity of the assay may be enhanced by using RNA (rather than DNA), in which the secondary structure is more sensitive to a change in sequence. In a preferred embodiment, the method utilizes heteroduplex analysis to separate double stranded heteroduplex molecules on the basis of changes in electrophoretic mobility (Kee ef a/., 1991).
In yet another embodiment, the movement of mutant or wild-type fragments in a polyacrylamide gel containing a gradient of denaturant is assayed using denaturing gradient gel electrophoresis (DGGE) (Myers et al., 1985). When DGGE is used as the method of analysis, DNA will be modified to insure that it does not completely denature, for example by adding a GC clamp of approximately 40 bp of high-melting GC-rich DNA by PCR. In a further embodiment, a temperature gradient is used in place of a denaturing gradient to identify differences in the mobility of control and sample DNA (Rosenbaum et al., 1987). In another embodiment, the mutant fragment is detected using denaturing HPLC (see e.g. Hoogendoom et al., 2000).
Examples of other techniques for detecting point mutations, an associated allele, a particular allele of a polymorphic locus, or the like include, but are not limited to, selective oligonucleotide hybridization, selective amplification, selective primer extension, selective ligation, single-base extension, selective termination of extension or invasive cleavage assay. For example, oligonucleotide primers may be prepared in which the known associated allele, particular allele of a polymorphic locus, or the like is placed centrally and then hybridized to target DNA under conditions which permit hybridization only if a perfect match is found (Saiki et al., 1986; Saiki et al., 1989). Such allele specific oligonucleotides are hybridized to PCR amplified target DNA of a number of different associated alleles, a particular allele of a polymorphic locus, or the likes where the oligonucleotides are attached to the hybridizing membrane and hybridized with labeled target DNA. Alternatively, the amplification, the allele-specific
hybridization and the detection can be done in a single assay following the principle of the 5' nuclease assay (e.g. see Livak et al., 1995). For example, the associated allele, a particular allele of a polymorphic locus, or the like locus is amplified by PCR in the presence of both allele-specific oligonucleotides, each specific for one or the other allele. Each probe has a different fluorescent dye at the 5' end and a quencher at the 3' end. During PCR, if one or the other or both allele-specific oligonucleotides are hybridized to the template, the Taq polymerase via its 5' exonuclease activity will release the corresponding dyes. The latter will thus reveal the genotype of the amplified product.
The hybridization may also be carried out with a temperature gradient following the principle of dynamic allele-specific hybridization or like (e.g. Jobs et al., 2003; and Bourgeois and Labuda, 2004). For example, the hybridization is done using one of the two allele-specific oligonucleotides labeled with a fluorescent dye, an intercalating quencher under a gradually increasing temperature. At low temperature, the probe is hybridized to both the mismatched and full-matched template. The probe melts at a lower temperature when hybridized to the template with a mismatch. The release of the probe is captured by an emission of the fluorescent dye, away from the quencher. The probe melts at a higher temperature when hybridized to the template with no mismatch. The temperature- dependent fluorescence signals therefore indicate the absence or presence of the associated allele, particular allele of a polymorphic locus, or the like (e.g. Jobs ef al. supra). Alternatively, the hybridization is done under a gradually decreasing temperature. In this case, both allele-specific oligonucleotides are hybridized to the template competitively. At high temperature none of the two probes is hybridized. Once the optimal temperature of the full-matched probe is reached, it hybridizes and leaves no target for the mismatched probe. In the latter case, if the allele-specific probes are differently labeled, then they are hybridized to a single PCR-amplified target. If the probes are labeled with the same dye, then the probe cocktail is hybridized twice to identical templates with only one labeled probe, different in the two cocktails, in the presence of the unlabeled competitive probe.
Alternatively, allele specific amplification technology that depends on selective PCR amplification may be used in conjunction with the present invention. Oligonucleotides used as primers for specific amplification may carry the associated allele, particular allele of a polymorphic locus, or the like of interest in the center of the molecule, so that amplification depends on differential hybridization (Gibbs et al., 1989) or at the extreme 3' end of one primer where, under appropriate conditions, mismatch can prevent, or reduce polymerase extension (Prossner, 1993). In addition it may be desirable to introduce a novel restriction site in the region of the associated allele, particular allele of a polymorphic locus, or the like to create cleavage-based detection (Gasparini et al., 1992). It is anticipated that in certain embodiments amplification may also be performed using Taq ligase for amplification (Barany, 1991). In such cases, ligation will occur only if there is a perfect match at the 3' end of the 5' sequence making it possible to detect the presence of a known associated allele, a particular allele of a polymorphic locus, or the like at a specific site by looking for the presence or absence of amplification. The products of such an oligonucleotide ligation assay can also be detected by means of gel electrophoresis. Furthermore, the oligonucleotides may contain universal tags used in PCR amplification and zip code tags that are different for each allele. The zip code tags are used to isolate a specific, labeled oligonucleotide that may contain a mobility modifier (e.g. Grossman et a/., 1994).
In yet another alternative, allele-specific elongation followed by ligation will form a template for PCR amplification. In such cases, elongation will occur only if there is a perfect match at the 3' end of the allele-specific oligonucleotide using a DNA polymerase. This reaction is performed directly on the genomic DNA and the extension/ligation products are amplified by PCR. To this end, the oligonucleotides contain universal tags allowing amplification at a high multiplex level and a zip code for SNP identification. The PCR tags are designed in such a way that the two alleles of a SNP are amplified by different forward primers, each having a different dye. The zip code tags are the same for both alleles of a given SNP and they are used for hybridization of the PCR-amplified products to oligonucleotides bound to a solid support, chip, bead array or like. For an
example of the procedure, see Fan et al. (Cold Spring Harbor Symposia on Quantitative Biology, Vol. LXVIII, pp. 69-78, 2003).
Another alternative includes the single-base extension/ligation assay using a molecular inversion probe, consisting of a single, long oligonucleotide (see e.g. Hardenbol et al., 2003). In such an embodiment, the oligonucleotide hybridizes on both sides of the SNP locus directly on the genomic DNA, leaving a one-base gap at the SNP locus. The gap-filling, one-base extension/ligation is performed in four tubes, each having a different dNTP. Following this reaction, the oligonucleotide is circularized whereas unreactive, linear oligonucleotides are degraded using an exonulease such as exonuclease I of E. coli. The circular oligonucleotides are then linearized and the products are amplified and labeled using universal tags on the oligonucleotides. The original oligonucleotide also contains a SNP-specific zip code allowing hybridization to oligonucleotides bound to a solid support, chip, bead array or the like. This reaction can be performed at a highly multiplexed level.
In another alternative, the associated allele, particular allele of a polymorphic locus, or the like is scored by single-base extension (see e.g. U.S. Pat. No. 5,888,819). The template is first amplified by PCR. The extension oligonucleotide is then hybridized next to the SNP locus and the extension reaction is performed using a thermostable polymerase such as ThermoSequenase (GE Healthcare) in the presence of labeled ddNTPs. This reaction can therefore be cycled several times. The identity of the labeled ddNTP incorporated will reveal the genotype at the SNP locus. The labeled products can be detected by means of gel electrophoresis, fluorescence polarization (e.g. Chen et al., 1999) or by hybridization to oligonucleotides bound to a solid support, chip, bead array or the like. In the latter case, the extension oligonucleotide will contain a SNP-specific zip code tag.
In yet another alternative, the variant is scored by selective termination of extension. The template is first amplified by PCR and the extension oligonucleotide hybridizes in vicinity to the SNP locus, close to but not necessarily adjacent to it. The extension reaction is carried out using a
thermostable polymerase such as ThermoSequenase (GE Healthcare) in the presence of a mix of dNTPs and at least one ddNTP. The latter has to terminate the extension at one of the alleles of the interrogated SNP, but not both such that the two alleles will generate extension products of different sizes. The extension product can then be detected by means of gel electrophoresis, in which case the extension products need to be labeled, or by mass spectrometry (see e.g. Storm ef a/., 2003).
In another alternative, the associated allele, particular allele of a polymorphic locus, or the like is detected using an invasive cleavage assay (see U.S. Pat. No. 6,090,543). There are five oligonucleotides per SNP to interrogate but these are used in a two step-reaction. During the primary reaction, three of the designed oligonucleotides are first hybridized directly to the genomic DNA. One of them is locus-specific and hybridizes up to the SNP locus (the pairing of the 3' base at the SNP locus is not necessary). There are two allele-specific oligonucleotides that hybridize in tandem to the locus-specific probe but also contain a 5' flap that is specific for each allele of the SNP. Depending upon hybridization of the allele- specific oligonucleotides at the base of the SNP locus, this creates a structure that is recognized by a cleavase enzyme (U.S. Pat. No. 6,090,606) and the allele- specific flap is released. During the secondary reaction, the flap fragments hybridize to a specific cassette to recreate the same structure as above except that the cleavage will release a small DNA fragment labeled with a fluorescent dye that can be detected using regular fluorescence detector. In the cassette, the emission of the dye is inhibited by a quencher.
Other types of markers can also be used for diagnostic purposes. For example, microsatellites can also be useful to detect the genetic predisposition of an individual to a given disorder. Microsatellites consist of short sequence motifs of one or a few nucleotides repeated in tandem. The most common motifs are polynucleotide runs, dinucleotide repeats (particularly the CA repeats) and trinucleotide repeats. However, other types of repeats can also be used. The microsatellites are very useful for genetic mapping because they are highly polymorphic in their length. Microsatellite markers can be typed by various means, including but not limited to DNA fragment sizing, oligonucleotide ligation
assay and mass spectrometry. For example, the locus of the microsatellite is amplified by PCR and the size of the PCR fragment will be directly correlated to the length of the microsatellite repeat. The size of the PCR fragment can be detected by regular means of gel electrophoresis. The fragment can be labeled internally during PCR or by using end-labeled oligonucleotides in the PCR reaction (e.g. Mansfield et al., 1996). Alternatively, the size of the PCR fragment is determined by mass spectrometry. In such a case, however, the flanking sequences need to be eliminated. This can be achieved by ribozyme cleavage of an RNA transcript of the microsatellite repeat (Krebs et al., 2001). For example, the microsatellite locus is amplified using oligonucleotides that include a T7 promoter on one end and a ribozyme motif on the other end. Transcription of the amplified fragments will yield an RNA substrate for the ribozyme, releasing small RNA fragments that contain the repeated region. The size of the latter is determined by mass spectrometry. Alternatively, the flanking sequences are specifically degraded. This is achieved by replacing the dTTP in the PCR reaction by dUTP. The dUTP nucleosides are then removed by uracyl DNA glycosylases and the resulting abasic sites are cleaved by either abasic endonucleases such as human AP endonuclease or chemical agents such as piperidine. Bases can also be modified post-PCR by chemical agents such as dimethyl sulfate and then cleaved by other chemical agents such as piperidine (see e.g. Maxam and Gilbert, 1977; U.S. Pat. No. 5,869,242; and U.S. Patent pending serial No. 60/335,068).
In another alternative, an oligonucleotide ligation assay can be performed. The microsatellite locus is first amplified by PCR. Then, different oligonucleotides can be submitted to ligation at the center of the repeat with a set of oligonucleotides covering all the possible lengths of the marker at a given locus (Zirvi et al., 1999). Another example of design of an oligonucleotide assay comprises the ligation of three oligonucleotides; a 5' oligonucleotide hybridizing to the 5' flanking sequence, a repeat oligonucleotide of the length of the shortest allele of the marker hybridizing to the repeated region and a set of 3' oligonucleotides covering all the existing alleles hybridizing to the 3' flanking sequence and a portion of the repeated region for all the alleles longer than the shortest one. For
the shortest allele, the 3' oligonucleotide exclusively hybridizes to the 3' flanking sequence (U.S. Pat. No. 6,479,244).
The methods described herein may be performed, for example, by utilizing prepackaged diagnostic kits comprising at least one probe nucleic acid selected from the SEQ ID of Table 1 , or antibody reagent described herein, which may be conveniently used, for example, in a clinical setting to diagnose patient exhibiting symptoms or a family history of a disorder or disorder involving abnormal activity of genes from Tables 3 and 4.
Method to treat an animal suspected of having asthma disease
The present invention provides methods of treating a disorder associated with asthma disease by expressing in vivo the nucleic acids of at least one gene from Tables 3 and 4. These nucleic acids can be inserted into any of a number of well- known vectors for the transfection of target cells and organisms as described below. The nucleic acids are transfected into cells, ex vivo or in vivo, through the interaction of the vector and the target cell. The nucleic acids encoding a gene from Tables 3 and 4, under the control of a promoter, then express the encoded protein, thereby mitigating the effects of absent, partial inactivation, or abnormal expression of a gene from Tables 3 and 4.
Such gene therapy procedures have been used to correct acquired and inherited genetic defects, cancer, and viral infection in a number of contexts. The ability to express artificial genes in humans facilitates the prevention and/or cure of many important human disorders, including many disorders which are not amenable to treatment by other therapies (for a review of gene therapy procedures, see Anderson, 1992; Nabel & Feigner, 1993; Mitani & Caskey, 1993; Mulligan, 1993; Dillon, 1993; Miller, 1992; Van Brunt, 1998; Vigne, 1995; Kremer & Perricaudet 1995; Doerfler & Bohm 1995; and Yu et al., 1994).
Delivery of the gene or genetic material into the cell is the first critical step in gene therapy treatment of a disorder. A large number of delivery methods are
well known to those of skill in the art. Preferably, the nucleic acids are administered for in vivo or ex vivo gene therapy uses. Non-viral vector delivery systems include DNA plasmids, naked nucleic acid, and nucleic acid complexed with a delivery vehicle such as a liposome. Viral vector delivery systems include DNA and RNA viruses, which have either episomal or integrated genomes after delivery to the cell. For a review of gene therapy procedures, see the references included in the above section.
The use of RNA or DNA based viral systems for the delivery of nucleic acids take advantage of highly evolved processes for targeting a virus to specific cells in the body and trafficking the viral payload to the nucleus. Viral vectors can be administered directly to patients (in vivo) or they can be used to treat cells in vitro and the modified cells are administered to patients (ex vivo). Conventional viral based systems for the delivery of nucleic acids could include retroviral, lentivirus, adenoviral, adeno-associated and herpes simplex virus vectors for gene transfer. Viral vectors are currently the most efficient and versatile method of gene transfer in target cells and tissues. Integration in the host genome is possible with the retrovirus, lentivirus, and adeno-associated virus gene transfer methods, often resulting in long term expression of the inserted transgene. Additionally, high transduction efficiencies have been observed in many different cell types and target tissues.
The tropism of a retrovirus can be altered by incorporating foreign envelope proteins, expanding the potential target population of target cells. Lentiviral vectors are retroviral vectors that are able to transduce or infect non-dividing cells and typically produce high viral titers. Selection of a retroviral gene transfer system would therefore depend on the target tissue. Retroviral vectors are comprised of c/s-acting long terminal repeats with packaging capacity for up to 6- 10 kb of foreign sequence. The minimum cis-acting LTRs are sufficient for replication and packaging of the vectors, which are then used to integrate the therapeutic gene into the target cell to provide permanent transgene expression. Widely used retroviral vectors include those based upon murine leukemia virus (MuLV), gibbon ape leukemia virus (GaLV), Simian lmmuno deficiency virus (SIV), human immuno deficiency virus (HIV)1 and combinations thereof (see, e.g.,
Buchscher et al., 1992; Johann et al., 1992; Sommerfelt et a/., 1990; Wilson et a/., 1989; Miller et al., 1999;and PCT/US94/05700).
In applications where transient expression of the nucleic acid is preferred, adenoviral based systems are typically used. Adenoviral based vectors are capable of very high transduction efficiency in many cell types and do not require cell division. With such vectors, high titer and levels of expression have been obtained. This vector can be produced in large quantities in a relatively simple system. Adeno-associated virus ("AAV") vectors are also used to transduce cells with target nucleic acids, e.g., in the in vitro production of nucleic acids and peptides, and for in vivo and ex vivo gene therapy procedures (see, e.g., West et al., 1987; U.S. Pat. No. 4,797,368; WO 93/24641 ; Kotin, 1994; Muzyczka, 1994). Construction of recombinant AAV vectors is described in a number of publications, including U.S. Pat. No. 5,173,414; Tratschin et al., 1985; Tratschin, et al., 1984; Hermonat & Muzyczka, 1984; and Samulski et al., 1989.
In particular, numerous viral vector approaches are currently available for gene transfer in clinical trials, with retroviral vectors by far the most frequently used system. All of these viral vectors utilize approaches that involve complementation of defective vectors by genes inserted into helper cell lines to generate the transducing agent. pLASN and MFG-S are examples are retroviral vectors that have been used in clinical trials (Dunbar et al., 1995; Kohn et al., 1995; Malech et al., 1997). PA317/pl_ASN was the first therapeutic vector used in a gene therapy trial (Blaese et al., 1995). Transduction efficiencies of 50% or greater have been observed for MFG-S packaged vectors (Ellem et al., 1997; and Dranoff et al., 1997).
Recombinant adeno-associated virus vectors (rAAV) are a promising alternative gene delivery systems based on the defective and nonpathogenic parvovirus adeno-associated type 2 virus. All vectors are derived from a plasmid that retains only the AAV 145 bp inverted terminal repeats flanking the transgene expression cassette. Efficient gene transfer and stable transgene delivery due to integration into the genomes of the transduced cell are key features for this vector system (Wagner et al., 1998, Kearns et a/1996).
Replication-deficient recombinant adenoviral vectors (Ad) are predominantly used in transient expression gene therapy; because they can be produced at high titer and they readily infect a number of different cell types. Most adenovirus vectors are engineered such that a transgene replaces the Ad E1a, E1b, and E3 genes; subsequently the replication defector vector is propagated in human 293 cells that supply the deleted gene function in trans. Ad vectors can transduce multiple types of tissues in vivo, including nondividing, differentiated cells such as those found in the liver, kidney and muscle tissues. Conventional Ad vectors have a large carrying capacity. An example of the use of an Ad vector in a clinical trial involved polynucleotide therapy for antitumor immunization with intramuscular injection (Sterman et al., 1998). Additional examples of the use of adenovirus vectors for gene transfer in clinical trials include Rosenecker et al., 1996; Sterman et al., 1998; Welsh et al., 1995; Alvarez et al., 1997; Topf et al., 1998.
Packaging cells are used to form virus particles that are capable of infecting a host cell. Such cells include 293 cells, which package adenovirus, and ψ2 cells or PA317 cells, which package retrovirus. Viral vectors used in gene therapy are usually generated by a producer cell line that packages a nucleic acid vector into a viral particle. The vectors typically contain the minimal viral sequences required for packaging and subsequent integration into a host, other viral sequences being replaced by an expression cassette for the protein to be expressed. The missing viral functions are supplied in trans by the packaging cell line. For example, AAV vectors used in gene therapy typically only possess ITR sequences from the AAV genome which are required for packaging and integration into the host genome. Viral DNA is packaged in a cell line, which contains a helper plasmid encoding the other AAV genes, namely rep and cap, but lacking ITR sequences. The cell line is also infected with adenovirus as a helper. The helper virus promotes replication of the AAV vector and expression of AAV genes from the helper plasmid. The helper plasmid is not packaged in significant amounts due to a lack of ITR sequences. Contamination with adenovirus can be reduced by, e.g., heat treatment to which adenovirus is more sensitive than AAV.
In many gene therapy applications, it is desirable that the gene therapy vector be delivered with a high degree of specificity to a particular tissue type. A viral vector
is typically modified to have specificity for a given cell type by expressing a ligand as a fusion protein with a viral coat protein on the viruses outer surface. The ligand is chosen to have affinity for a receptor known to be present on the cell type of interest. For example, Han et al., 1995, reported that Moloney murine leukemia virus can be modified to express human heregulin fused to gp70, and the recombinant virus infects certain human breast cancer cells expressing human epidermal growth factor receptor. This principle can be extended to other pairs of viruses expressing a ligand fusion protein and target cells expressing a receptor. For example, filamentous phage can be engineered to display antibody fragments (e.g., Fab or Fv) having specific binding affinity for virtually any chosen cellular receptor. Although the above description applies primarily to viral vectors, the same principles can be applied to nonviral vectors. Such vectors can be engineered to contain specific uptake sequences thought to favor uptake by specific target cells.
Gene therapy vectors can be delivered in vivo by administration to an individual patient, typically by systemic administration (e.g., intravenous, intraperitoneal, intramuscular, subdermal, or intracranial infusion) or topical application. Alternatively, vectors can be delivered to cells ex vivo, such as cells explanted from an individual patient (e.g., lymphocytes, bone marrow aspirates, and tissue biopsy) or universal donor hematopoietic stem cells, followed by reimplantation of the cells into a patient, usually after selection for cells which have incorporated the vector.
Ex vivo cell transfection for diagnostics, research, or for gene therapy (e.g., via re-infusion of the transfected cells into the host organism) is well known to those of skill in the art. In a preferred embodiment, cells are isolated from the subject organism, transfected with a nucleic acid (gene or cDNA), and re-infused back into the subject organism (e.g., patient). Various cell types suitable for ex vivo transfection are well known to those of skill in the art (see, e.g., Freshney et al., 1994; and the references cited therein for a discussion of how to isolate and culture cells from patients).
In one embodiment, stem cells are used in ex vivo procedures for cell transfection and gene therapy. The advantage to using stem cells is that they can be differentiated into other cell types in vitro, or can be introduced into a mammal (such as the donor of the cells) where they will engraft in the bone marrow. Methods for differentiating CD34+ cells in vitro into clinically important immune cell types using cytokines such a GM-CSF, IFN-γ and TNF-α are known (see Inaba ef a/., 1992).
Stem cells are isolated for transduction and differentiation using known methods. For example, stem cells are isolated from bone marrow cells by panning the bone marrow cells with antibodies which bind unwanted cells, such as CD4+ and CD8+ (T cells), CD45+ (panB cells), GR-1 (granulocytes), and lad (differentiated antigen presenting cells).
Vectors (e.g., retroviruses, adenoviruses, liposomes, etc.) containing therapeutic nucleic acids can be also administered directly to the organism for transduction of cells in vivo. Alternatively, naked DNA can be administered.
Administration is by any of the routes normally used for introducing a molecule into ultimate contact with blood or tissue cells, as described above. The nucleic acids from Tables 3 and 4 are administered in any suitable manner, preferably with the pharmaceutically acceptable carriers described above. Suitable methods of administering such nucleic acids are available and well known to those of skill in the art, and, although more than one route can be used to administer a particular composition, a particular route can often provide a more immediate and more effective reaction than another route (see Samulski et al., 1989). The present invention is not limited to any method of administering such nucleic acids, but preferentially uses the methods described herein.
The present invention further provides other methods of treating asthma disease such as administering to an individual having asthma disease an effective amount of an agent that regulates the expression, activity or physical state of at least one gene from Tables 3 and 4. An "effective amount" of an agent is an amount that modulates a level of expression or activity of a gene from Tables 3 and 4, in a cell in the individual at least about 10%, at least about 20%, at least
about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80% or more, compared to a level of the respective gene from Tables 3 and 4 in a cell in the individual in the absence of the compound. The preventive or therapeutic agents of the present invention may be administered, either orally or parenterally, systemically or locally. For example, intravenous injection such as drip infusion, intramuscular injection, intraperitoneal injection, subcutaneous injection, suppositories, intestinal lavage, oral enteric coated tablets, and the like can be selected, and the method of administration may be chosen, as appropriate, depending on the age and the conditions of the patient. The effective dosage is chosen from the range of 0.01 mg to 100 mg per kg of body weight per administration. Alternatively, the dosage in the range of 1 to 1000 mg, preferably 5 to 50 mg per patient may be chosen. The therapeutic efficacy of the treatment may be monitored by observing various parts of the respiratory tract, by chest and sinus X-rays, barium, performing a spirometry breathing test, a routine pulmonary function test, or any other monitoring methods known in the art. Other ways of monitoring efficacy can be, but are not limited to monitoring inflammatory conditions involving the respiratory system such as monitoring the amelioration on the lung capacity and function, decrease in pain, improved breathing, decreased breathlessness and wheeze, reduced chest pain, or improvement in vitality and exercise ability.
The present invention further provides a method of treating an individual clinically diagnosed with asthma disease. The methods generally comprises analyzing a biological sample that includes a cell, in some cases, a respiratory system cell, from an individual clinically diagnosed with asthma disease for the presence of modified levels of expression of at least 1 gene, at least 10 genes, at least 50 genes, at least 100 genes, or at least 200 genes from Tables 3 and 4. A treatment plan that is most effective for individuals clinically diagnosed as having a condition associated with asthma disease is then selected on the basis of the detected expression of such genes in a cell. Treatment may include administering a composition that includes an agent that modulates the expression or activity of a protein from Tables 3 and 4 in the cell. Information obtained as described in the methods above can also be used to predict the response of the individual to a
particular agent. Thus, the invention further provides a method for predicting a patient's likelihood to respond to a drug treatment for a condition associated with asthma disease, comprising determining whether modified levels of a gene from Tables 3 and 4 is present in a cell, wherein the presence of protein is predictive of the patient's likelihood to respond to a drug treatment for the condition. Examples of the prevention or improvement of symptoms accompanied by asthma disease that can monitored for effectiveness include improvement of breathing, amelioration on the lung capacity and function, decrease in pain, decreased breath lessness and wheeze, reduced chest pain, or improvement in vitality and exercise ability, and the like, and as a result, a preventing or improving agent for breathing amelioration, a preventing or improving agent for amelioration of lung capacity, an inhibitor breathlessness and wheeze, and the like can be identified.
The invention also provides a method of predicting a response to therapy in a subject having asthma disease by determining the presence or absence in the subject of one or more markers associated with asthma disease described in Tables 1 , 3 or 4, diagnosing the subject in which the one or more markers are present as having asthma disease, and predicting a response to a therapy based on the diagnosis e.g., response to therapy may include an efficacious response and/or one or more adverse events. The invention also provides a method of optimizing therapy in a subject having asthma disease by determining the presence or absence in the subject of one or more markers associated with a clinical subtype of asthma disease, diagnosing the subject in which the one or more markers are present as having a particular clinical subtype of asthma disease, and treating the subject having a particular clinical subtype of asthma disease based on the diagnosis. As an example, treatment for the exercise- induced asthma subtype of asthma disease currently includes inhaled bronchodilators via a nebulizer.
Thus, while there are a number of treatments for asthma disease currently available, they all are accompanied by various side effects, high costs, and long complicated treatment protocols, which are often not available and effective in a large number of individuals. Accordingly, there remains a need in the art for more
effective and otherwise improved methods for treating and preventing asthma disease. Thus, there is a continuing need in the medical arts for genetic markers of asthma disease and guidance for the use of such markers. The present invention fulfills this need and provides further related advantages.
EXAMPLES
Example 1 : Identification of cases and controls
All individuals were sampled from the Quebec founder population (QFP). Membership in the founder population was defined as having four grandparents with French Canadian family names who were born in the Province of Quebec, Canada or in adjacent areas of the Provinces of New Brunswick and Ontario or in New England or New York State. The Quebec founder population has two distinct advantages over general populations for LD mapping. Because it is relatively young (about 12 to 15 generations from the mid 17th century to the present), and because it has a limited but sufficient number of founders (approximately 2600 effective founders, Charbonneau et al. 1987), the Quebec population is characterized both by extended LD and by decreased genetic heterogeneity. The increased extent of LD allows the detection of disorder genes using a reasonable marker density, while still allowing the increased meiotic resolution of population-based mapping. The number of founders is small enough to result in increased LD and reduced allelic heterogeneity, yet large enough to insure that all of the major disorder genes involved in general populations are present in Quebec. Reduced allelic heterogeneity will act to increase relative risk imparted by the remaining alleles and so increase the power of case/control studies to detect a gene, an associated allele, a particular allele of a polymorphic locus, or the likes involved in complex disorders within the Quebec population. The specific combination of age in generations, optimal number of founders and large present population size makes the QFP optimal for LD-based gene mapping.
Patient inclusion criteria for the study include a diagnosis of asthma by a lung specialist or an allergist. Also, the onset of the disease must have occurred before 35 years old. All human sampling was subject to ethical review procedures.
All enrolled QFP subjects (patients and controls) provided a 30 ml blood sample (3 barcoded tubes of 10 ml each). Samples were processed immediately upon arrival at Genizon's laboratory. All samples were scanned and logged into a
LabVantage Laboratory Information Management System (LIMS), which served as a hub between the clinical data management system and the genetic analysis system. Following centrifugation, the buffy coat containing the white blood cells was isolated from each tube. Genomic DNA was extracted from the buffy coat from one of the tubes, and stored at 4°C until required for genotyping. DNA extraction was performed with a commercial kit using a guanidine hydrochloride based method (FlexiGene, Qiagen) according to the manufacturer's instructions. The extraction method yielded high molecular weight DNA, and the quality of every DNA sample was verified by agarose gel electrophoresis. Genomic DNA appeared on the gel as a large band of very high molecular weight. The remaining two buffy coats were stored at -80°C as backups.
The samples were collected as family trios from asthma disease subjects and two close relatives. All of the trios were Parent, Parent, Child (PPC) trios. One member of each trio was affected with asthma disease. These included 252 daughters, 134 sons, 68 mothers and 43 fathers. When a child was the affected member of the trio, the two non-transmitted parental chromosomes (one from each parent) were used as controls, when one of the parents was affected, that person's spouse provided the control chromosomes. The recruitment of trios allowed the precise determination of haplotypes.
Example 2: Genome Wide Association
Genotyping was performed using Perlegen's ultra-high-throughput platform. Marker loci were amplified by PCR and hybridized to wafers containing arrays of oligonucleotides. Allele discrimination was performed through allele-specific hybridization. In total, 80,654 SNPs, distributed as evenly as possible throughout the genome, were genotyped on the 497 trios for a total of 120,560,328 genotypes. These markers were mostly selected from various databases including the ~1.6 million SNP database of Perlegen Life Sciences (Patil, 2001); several thousand were obtained from the HapMap consortium database and/or dbSNP at NCBI. The SNPs were chosen to maximize uniformity of genetic coverage and to cover a distribution of allele frequencies. All SNPs that did not
pass the quality controls for the assay, that is, had a minor allele frequency of less than 10%, a Mendelian error rate within trios greater than 1%, that deviated significantly from the Hardy-Weinberg equilibrium, or that had too many missing data (cut-off at 5% missing values or higher) were removed from the analysis. Genetic analysis was performed on a total of 80,654 SNPs (66,146 SNPs after cleaning). The average gap size was approximately 45 kb.
The genotyping information was entered into a Unified Genotype Database (a proprietary database under development) from which it was accessed using custom-built programs for export to the genetic analysis pipeline. Analyses of these genotypes were performed with the statistical tools described in Example 3. The GWS permitted the identification of 231 candidate regions. Some of these are further analyzed by the Fine Mapping approach described below.
Example 3: Genetic Analysis
1. Dataset quality assessment
Prior to performing any analysis, the dataset from the GWS was verified for completeness of the trios. The program GGFileMod removed any trios with abnormal structure or missing individuals (e.g. trios without a proband, duos, singletons, etc.), and calculated the total number of complete trios in the dataset. The trios were also tested to make sure that no subjects within the cohort were related more closely than second cousins (6 meiotic steps).
Subsequently, the program DataCheck2.1 was used to calculate the following statistics per marker and per family:
Minor allele frequency (MAF) for each marker; Missing values for each marker and family; Hardy Weinberg Equilibrium for each marker; and Mendelian segregation error rate.
The following acceptance criteria were applied for internal analysis purposes:
MAF > 10%; Missing values < 1%; Observed non-Mendelian segregation < 0.33%; and Allele frequencies for controls in Hardy Weinberg Equilibrium.
Markers or families not meeting these criteria were removed from the dataset in the following step. Analyses of variance were performed using the algorithm GenAnova, to assess whether families or markers have the greater effect on missing values and non-Mendelian segregation. This was used to determine the smallest number of data points to remove from the dataset to meet the requirements for missing values and non-Mendelian segregation. The families and/or markers were removed from the dataset using the program DataPull, which generates an output file that is used for subsequent analysis of the genotype data.
2. Phase Determination
The Program PhaseFinderSNP2.0 was used to determine phase from trio data on a marker-by-marker, trio-by-trio basis. The output file contains haplotype data for all trio members, including ambiguities where all trio members are heterozygous or where data is missing. The program FileWriterTemp was then used to determine case and control haplotypes and to prepare the data in the proper input format for the next stage of analysis, using the expectation maximization algorithm, PL-EM, to determine phase on the remaining ambiguities. This stage consists of several modules for resolution of the remaining phase ambiguities. PLEMInOuti was first used to recode the haplotypes for input into the PL-EM algorithm in 15-marker blocks. The haplotype information was encoded as genotypes, allowing for the entry of known phase into the algorithm, which limits the possible number of estimated haplotypes. The PL-EM algorithm was used to estimate haplotypes from the "genotype" data in 15-marker windows, advancing in increments of one marker across the chromosome. The results were then converted into multiple 15-marker haplotype files using the program PLEMInOut2. Subsequently PLEMBIockGroup was used to convert the individual 15-marker block files into one continuous block of haplotypes for the entire chromosome, and to generate files for further analysis
by LDSTATS, Hapfreq and HapColor. PLEMBIockGroup takes the consensus estimation of the allele call at each marker over all separate estimations (most markers are estimated 15 different times as the 15 marker blocks pass over their position).
3. Haplotype sharing analysis
Haplotype sharing analysis was performed using the program LDSTATS. LDSTATS tests for association of haplotypes with the disease phenotype. The algorithms LDSTATS (v2.0) and LDSTATS (v3.0) define haplotypes using multi- marker windows that advance across the marker map in one-marker increments. Windows can be 1 , 3, 5, 7 or 9 markers wide. At each position the frequency of haplotypes in cases and controls was calculated. Allele frequency differences for single marker and 3-marker windows were then tested using Pearson's Chi- square test with one degree of freedom. Larger windows of multi-allelic haplotype association were tested using Smith's normalization of the square root of Pearson's Chi-square. In addition, LDSTATS calculates Chi-square values for the transmission disequilibrium test (TDT) for single markers in situations where the trios consisted of parents and an affected child. The significance of association for any given marker was calculated in the following manner:
1. The X2 value for significance of association within any specific haplotype sliding window was calculated from case-control haplotype frequency tables. The significance of this X2 value was plotted against the position of the central marker
in the sliding window in LDSTATS(ver2.0). In LDSTATS(ver3.0), the for
 each specific sliding window was normalized according to the method of Smith (Ann Hum Genet 50:163-7, 1986). For each marker, the mean normalized
each specific sliding window was normalized according to the method of Smith (Ann Hum Genet 50:163-7, 1986). For each marker, the mean normalized
 value over all windows which contain the marker was calculated. The
value over all windows which contain the marker was calculated. The
statistical significance of this mean normalized
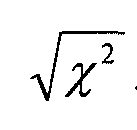 for each marker was determined from a permutation test in which case-control status was permuted across all chromosome wide haplotypes. The P value for significance was derived from the Z test:
for each marker was determined from a permutation test in which case-control status was permuted across all chromosome wide haplotypes. The P value for significance was derived from the Z test:

This permutation test provides the P value for mean significance of each marker over all windows in which it occurs.
The P values for different window sizes generated by LDSTATS (v 3.0) were merged into a single output file containing all haplotype window sizes using the program FileMerger and chromosome wide graphs of - log 10 P value for all marker windows determined from LDSTATS(ver3.0) were plotted against the chromosomal position of the reference marker. Tables of significant - Iog10 P values were also constructed from these analyses
The regions with the most significant P-values were selected for further analyses. For each of these regions, the frequencies in cases and controls for the significant haplotypes were determined by HapFreq, a subroutine of LDSTATS(v3.0). The significantly shared haplotypes that were different between cases and controls were represented in different colors using HapColor.
Haplotype frequencies were then used to verify that the significance was the result of distinct differences in frequency between cases and controls for specific common haplotypes, and was not the cumulative result of many rare haplotypes that existed only in either cases or controls (see Table 1).
5. Conditional Haplotype Analyses
Conditional haplotype analyses were performed on subsets of the original set of cases and controls using the program LDSTAT. The selection of a subset of cases and their matched controls was based on the carrier status of cases at a gene or locus of interest. We selected the gene BACH2 on chromosome 16 based on our association finding using LDSTAT (v2.0). The most significant association in BACH2 was obtained with a haplotype window of size 9 containing SNPs corresponding to SEQ IDs 2358, 2359, 2360, 2361 , 2362, 2363, 2364,
2365, and 2366. (see Table below for conversion to the specific DNA alleles used). A reduced haplotype diversity was observed and we selected three sets of haplotypes for conditional analyses. The first set contained four protective haplotypes (111121222, 111111112, 111221212 and 212211111). The second set contained five risk haplotypes (211111111 , 111122211 , 211111112, 121121111 and 211112122) while the third set contained ten risk haplotypes (111121112, 111112211 , 111112122, 122211111 , 111111111 , and the 5 risk haplotypes above). For a given set, the cases were separated into two groups with the first group consisting of those cases that were carrier of one of the haplotypes in the set and the second group consisting of those cases that were not carrier. The resulting sample sizes were 159 and 309 for the protective set, 88 and 380 for the 5-risk set and 288 and 180 for the 10-risk set respectively. A separate analysis for association with asthma was then performed with the two groups in each set using LDSTAT.

Example 4: Fine Mapping
79 of the top regions identified as being associated with asthma disease by the GWS are further analyzed by fine mapping using a denser set of markers, in order to validate and/or refine the signal. The fine mapping is carried out using the lllumina BeadStation 500GX SNP genotyping platform. Alleles are genotyped using an allele-specific elongation assay that is ligated to a locus-specific oligonucleotide. The assay is performed directly on genomic DNA at a highly multiplex level and the products are amplified using universal oligonucleotides. For each candidate region, a set of SNP markers is selected with an average inter-marker distance of 1-4 Kb distributed over about 400 Kb to 1 Mb and is roughly centered at the highest point of the GWS curves. The cohort used for the fine mapping analysis consists of the 497 asthma disease trios used for the GWS. The algorithms used for genetic analyses are the same as used in the GWS and are described in Example 3.
Example 5: Gene identification and characterization
Any gene or EST mapping to the interval based on public map data or proprietary map data was considered as a candidate asthma gene. The approach used to identify all genes located in the critical regions is described below.
Public gene mining
Once regions were identified using the analyses described above, a series of public data mining efforts were undertaken, with the aim of identifying all genes located within the critical intervals as well as their respective structural elements (i.e., promoters and other regulatory elements, UTRs, exons and splice sites). The initial analysis relied on annotation information stored in public databases (e.g. NCBI, UCSC Genome Bioinformatics, Entrez Human Genome Browser, OMIM - see below for database URL information).


For some genes the available public annotation was extensive, whereas for others very little was known about a gene's function. A more detailed analysis was therefore performed to characterize genes that corresponded to this latter class. Importantly, the presence of rare splice variants and artifactual ESTs was carefully evaluated. The public data derived from the EST clustering software ECgene (Kim et al., 2005) were also examined for additional gene models. This tool combines genome-based EST clustering and transcript assembly to generate a group of alternatively spliced consensi. This analysis of novel ESTs provided
an indication of additional gene content in some regions. The resulting clusters were graphically displayed against the genomic sequence, providing indications of separate clusters that may contribute to the same gene, thereby facilitating development of confirmatory experiments in the laboratory.
A unique consensus sequence was constructed for each splice variant and a trained reviewer assessed each alignment. This assessment included examination of all putative splice junctions for consensus splice donor/acceptor sequences, putative start codons, consensus Kozak sequences and upstream in- frame stops, and the location of polyadenylation signals. In addition, conserved noncoding sequences (CNSs) that could potentially be involved in regulatory functions were included as important information for each gene. The genomic reference and exon sequences were then archived for future reference. A master assembly that included all splice variants, exons and the genomic structure was used in subsequent analyses (i.e., analysis of polymorphisms).
An important component of these efforts was the ability to visualize and store the results of the data mining efforts. A customized version of the highly versatile genome browser GBrowse (http://www.gmod.org/) was implemented in order to permit the visualization of several types of information against the corresponding genomic sequence. In addition, the results of the statistical analyses were plotted against the genomic interval, thereby greatly facilitating focused analysis of gene content.
2. Computational Analysis of Genes and GeneMaps
In order to assist in the prioritization of candidate genes for which minimal annotation existed, a series of computational analyses were performed that included basic BLAST searches and alignments to identify related genes. In some cases this provided an indication of potential function. In addition, protein domains and motifs were identified that further assisted in the understanding of potential function, as well as predicted cellular localization.
A comprehensive review of the public literature was also performed in order to facilitate identification of information regarding the potential role of candidate
genes in the pathophysiology of asthma. In addition to the standard review of the literature, public resources (Medline and other online databases) were also mined for information regarding the involvement of candidate genes in specific signaling pathways. A variety of pathway and yeast two hybrid databases were mined for information regarding protein-protein interactions. These included BIND, MINT, DIP, Interdom, and Reactome, among others. By identifying homologues of genes in the asthma candidate regions and exploring whether interacting proteins had been identified already, knowledge regarding the GeneMaps for asthma was advanced. The pathway information gained from the use of these resources was also integrated with the literature review efforts, as described above.
3. Expression Studies
In order to determine the expression patterns for genes, relevant information was first extracted from public databases. The UniGene database, for example, contains information regarding the tissue source for ESTs and cDNAs contributing to individual clusters. This information was extracted and summarized to provide an indication in which tissues the gene was expressed. Particular emphasis was placed on annotating the tissue source for bona fide ESTs, since many ESTs mapped to Unigene clusters are artifactual. In addition, SAGE and microarray data, also curated at NCBI (Gene Expression Omnibus), provided information on expression profiles for individual genes. Particular emphasis was placed on identifying genes that were expressed in tissues known to be involved in the pathophysiology of asthma disorder.
4. Polymorphism analysis
Polymorphisms identified in candidate genes, including those from the public domain as well as those identified by sequencing candidate genes and regions, are evaluated for potential function. Initially, polymorphisms are examined for potential impact upon encoded proteins. If the protein is a member of a gene family with reported 3-dimensional structural information, this information is used to predict the location of the polymorphism with respect to protein structure. This
information provided insight into the potential role of polymorphisms in altering protein or ligand interactions, as well as suitability as a drug target. In a second phase of analysis we evaluate the potential role of polymorphisms in other biological phenomena, including regulation of transcription, splicing and mRNA stability, etc. There are many examples of the functional involvement of naturally occurring polymorphisms in these processes. As part of this analysis, polymorphisms located in promoter or other regulatory elements, canonical splice sites, exonic and intronic splice enhancers and repressors, conserved noncoding sequences and UTRs are localized.
Example 6: SNP and Polymorphism Discovery (SNPD)
Candidate genes and regions are selected for sequencing in order to identify all polymorphisms (Table 5). In cases where the critical interval, identified by fine mapping, was relatively small (~50 kb), the entire region, including all introns, is sequenced to identify polymorphisms. In situations where the region is large (>50 kb), candidate genes are prioritized for sequencing, and/or only functional gene elements (promoters, exons and splice sites) are sequenced.
The samples sequenced are selected according to which haplotypes contribute to the association signal observed in the region. The purpose is to select a set of samples that covered all the major haplotypes in the given region. Each major haplotype must be present in a few copies. The first step therefore consisted of determining the major haplotypes in the region to be sequenced.
Once a region was defined with the two boundary markers, all the markers used in fine mapping that are located within the region are used to determine the major haplotypes. Long haplotypes covering the whole region are thus inferred using the middle marker as an anchor. The results included two series of haplotype themes that define the major haplotypes, comparing the cases and the controls. This exercise was repeated using an anchor in the peripheral regions to ensure that major haplotype subsets that are not anchored at the original middle marker are not missed.
Once the major haplotypes were determined as described above, appropriate genomic DNA samples were selected such that each major haplotype and haplotype subset were represented in at least two to four copies.
The sequencing protocol included the following steps, once a region was delimited:
1. Primer design
The design of the primers was performed using a proprietary primer design tool. A primer quality control step was included in the primer design process. Primers that successfully passed the control quality process were synthesized by Integrated DNA Technologies (IDT). The sense and anti-sense oligos were separated such that the sense oligos were placed on one plate in the same position as their anti-sense counterparts on another plate. Two additional plates were created from each storage plate, one for use in PCR and the other for sequencing. For PCR, the sense and anti-sense oligos of the same pair were combined in the same well to achieve a final concentration of 1.5 μM for each oligonucleotide.
2. PCR optimization
PCR conditions were optimized by testing a variety of conditions that included varying salt concentrations and temperatures, as well as including various additives. PCR products were checked for robust amplification and minimal background by agarose gel electrophoresis.
3. PCR on selected samples
PCR products used for sequencing were amplified using the conditions chosen during optimization. The PCR products were purified free of salts, dNTPs and unincorporated primers by use of a Multiscreen PCR384 filter plate manufactured by Millipore. Following PCR, the amplicons were quantified by use of a lambda/Hind III standard curve. This was done to ensure that the quantity of PCR product required for sequencing had been generated. The raw data was measured against the standard curve data in Excel by use of a macro.
4. Sequencing
Sequencing of PCR products was performed by DNA Landmarks using ABI 3730 capillary sequencing instruments.
5. Sequence analysis
The ABI Prism SeqScape software (Applied Biosystems) was used for SNP identification. The chromatogram trace files were imported into a SeqScape sequencing project and the base calling was automatically performed. Sequences are then aligned and compared to each other using the SeqScape program. The base calling was checked manually, base by base; editing was performed if needed. The SNPs and polymorphisms discovered in this example are listed in Table 5.
Example 7: Ultra Fine Mapping (UFM)
Once polymorphisms are identified by sequencing efforts as described in Example 6, additional genotyping of all newly found polymorphisms is performed on the samples used in the fine mapping studies. Various types of genotyping assays may need to be utilized based on the type of polymorphism identified (i.e., SNP, indel, microsatellite). The assay type can be, but is not restricted to, Sentrix Assay Matrix on lllumina BeadStations, microsatellite on MegaBACE, SNP on ABI or Orchid. The frequencies of genotypes and haplotypes in cases and controls are analyzed in a similar manner as the GWS and fine mapping data. By examining all SNPs in a region, polymorphisms are identified that increase an individual's susceptibility to asthma disease. The goal of ultra-fine mapping is to identify the polymorphism that is most associated with disease phenotype as part of the search for the actual DNA polymorphism that confers susceptibility to disease. This statistical identification may need to be corroborated by functional studies.
Example 8: Confirmation of Candidate regions and genes in a general population
The confirmation of any putative associations described in Example 7 is performed in an independent patient sample. These DNA samples consist of one cohort, which consists of 500 PPC trios (500 patients with asthma disease).
All publications, patents and patent applications mentioned in the specification and reference list are herein incorporated by reference in their entirety for all purposes. Various modifications and variations of the described method and system of the invention will be apparent to those skilled in the art without departing from the scope and spirit of the invention. Although the invention has been described in connection with specific preferred embodiments, it should be understood that the invention as claimed should not be unduly limited to such specific embodiments. Indeed, various modifications of the described modes for carrying out the invention that are obvious to those skilled in molecular biology, genetics, or related fields are intended to be within the scope of the following claims.
The practice of the present invention will employ, unless otherwise indicated, conventional techniques of cell biology, cell culture, molecular biology, transgenic biology, microbiology, recombinant DNA, and immunology, which are within the skill of the art. Such techniques are explained fully in the literature. See, for example, Molecular Cloning A Laboratory Manual, 2nd Ed., ed. by Sambrook, Fritsch and Maniatis (Cold Spring Harbor Laboratory Press: 1989); DNA Cloning, Volumes I and H (D. N. Glover ed., 4 ); Oligonucleotide Synthesis (M. J. Gait ed., 1984); Mullis et al. U.S. Patent No. 4.683,195; Nucleic Acid Hybridization (B. D. Hames & S. J. Higgins eds. 1984); Transcription And Translation (B. D. Haines & S. J. Higgins eds. 1984); Culture Of Animal Cells (R. 1. Freshney, Alan R. Liss, Inc., 1987); Immobilized Cells And Enzymes (IRL Press, 1986); B. Perbal, A Practical Guide To Molecular Cloning (1984); the treatise, Methods In Enzymology (Academic Press, Inc., N.Y.); Gene Transfer Vectors For Mammalian Cells (J. H. Miller and M. P. Calos eds., 1987, Cold Spring Harbor Laboratory); Methods In Enzymology, VoIs. 154 and 155 (Wu et al. eds.), Immunochemical Methods In Cell And Molecular Biology (Mayer and Walker, eds., Academic Press, London, 1987); Handbook Of Experimental Immunology, Volumes I-IV (D. M. Weir and C. C. Blackwell, eds., 1986); Manipulating the
Mouse Embryo, (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1986).
REFERENCES
Adcock, I. M., M. J. Peters, et al. (1993). "Transcription factor interactions in human lung." Biochem Soc Trans 21(Pt 3)(3): 277S.
Allen, M., A. Heinzmann, et al. (2003). "Positional cloning of a novel gene influencing asthma from Chromosome 2q14." Nat Genet 19: 19.
Alwine, J. C, D. J. Kemp, et al. (1977). "Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes." Proc Natl Acad Sci U S A 74(12): 5350-4.
Anderson, W. F. (1992). "Human gene therapy." Science 256(5058): 808-13.
Barany, F. (1991). "Genetic disease detection and DNA amplification using cloned thermostable ligase." Proc Natl Acad Sci U S A 88(1): 189-93.
Barre, F. X., S. Ait-Si-Ali, et al. (2000). "Unambiguous demonstration of triple- helix-directed gene modification." Proc Natl Acad Sci U S A 97(7): 3084-8.
Bourgeois, S. and D. Labuda (2004). "Dynamic allele-specific oligonucleotide hybridization on solid support." Anal Biochem 324(2): 309-11.
CaIe, J. M., C. E. Shaw, et al. (1998). "Optimization of a reverse transcription- polymerase chain reaction (RT-PCR) mass assay for low-abundance mRNA." Methods MoI Biol 105: 351-71.
Chen, X., L. Levine, et al. (1999). "Fluorescence polarization in homogeneous nucleic acid analysis." Genome Res 9(5): 492-8.
Chien, C. T., P. L. Bartel, et al. (1991). "The two-hybrid system: a method to identify and clone genes for proteins that interact with a protein of interest." Proc Natl Acad Sci U S A 88(21): 9578-82.
Cohen, A. S., D. L. Smisek, et al. (1996). "Emerging technologies for sequencing antisense oligonucleotides: capillary electrophoresis and mass spectrometry." Adv Chromatogr 36: 127-62.
Cotton, R. G., N. R. Rodrigues, et al. (1988). "Reactivity of cytosine and thymine in single-base-pair mismatches with hydroxylamine and osmium tetroxide and its application to the study of mutations." Proc Natl Acad Sci U S A 85(12): 4397- 401.
Cronin, M. T., R. V. Fucini, et al. (1996). "Cystic fibrosis mutation detection by hybridization to light-generated DNA probe arrays." Hum Mutat 7(3): 244-55.
Dean, F. B., S. Hosono, et al. (2002). "Comprehensive human genome amplification using multiple displacement amplification." Proc Natl Acad Sci U S A 99(8): 5261-6.
Dillon, N. (1993). "Regulating gene expression in gene therapy." Trends BiotechnoM 1(5): 167-73.
Drmanac, R., S. Drmanac, et al. (2002). "Sequencing by hybridization (SBH): advantages, achievements, and opportunities." Adv Biochem Eng Biotechnol 77: 75-101.
Elez, R., A. Piiper, et al. (2000). "Polo-like kinasel , a new target for antisense tumor therapy." Biochem Biophys Res Commun 269(2): 352-6.
Evans, B. E., K. E. Rittle, et al. (1987). "Design of nonpeptidal ligands for a peptide receptor: cholecystokinin antagonists." J Med Chem 30(7): 1229-39.
Fackler, O. T. and B. M. Peterlin (2000). "Endocytic entry of HIV-1." Curr Biol 10(16): 1005-8.
Fakhrai-Rad, H., J. Zheng, et al. (2004). "SNP discovery in pooled samples with mismatch repair detection." Genome Res 14(7): 1404-12.
Fields, S. and O. Song (1989). "A novel genetic system to detect protein-protein interactions." Nature 340(6230): 245-6.
Frank-Kamenetskii, M. D. and S. M. Mirkin (1995). "Triplex DNA structures." Annu Rev Biochem 64: 65-95.
Freeman, W. M., S. J. Walker, et al. (1999). "Quantitative RT-PCR: pitfalls and potential." Biotechniques 26(1): 112-22, 124-5.
Gasparini, P., A. Bonizzato, et al. (1992). "Restriction site generating-polymerase chain reaction (RG-PCR) for the probeless detection of hidden genetic variation: application to the study of some common cystic fibrosis mutations." MoI Cell Probes 6(1): 1-7.
Gibbs, R. A., P. N. Nguyen, et al. (1989). "Detection of single DNA base differences by competitive oligonucleotide priming." Nucleic Acids Res 17(7): 2437-48.
Griffin, H. G. and A. M. Griffin (1993). "DNA sequencing. Recent innovations and future trends." Appl Biochem Biotechnol 38(1-2): 147-59.
Grossman, P. D., W. Bloch, et al. (1994). "High-density multiplex detection of nucleic acid sequences: oligonucleotide ligation assay and sequence-coded separation." Nucleic Acids Res 22(21): 4527-34.
Guatelli, J. C, K. M. Whitfield, et al. (1990). "Isothermal, in vitro amplification of nucleic acids by a multienzyme reaction modeled after retroviral replication." Proc Natl Acad Sci U S A 87(5): 1874-8.
Guatelii, J. C1 K. M. Whitfield, et al. (1990). "Isothermal, in vitro amplification of nucleic acids by a multienzyme reaction modeled after retroviral replication." Proc Natl Acad Sci U S A 87(19): 7797.
Hafner, G. J., I. C. Yang, et al. (2001). "Isothermal amplification and multimerization of DNA by Bst DNA polymerase." Biotechniques 30(4): 852-6, 858, 860 passim.
Hardenbol, P., J. Baner, et al. (2003). "Multiplexed genotyping with sequence- tagged molecular inversion probes." Nat Biotechnol 21(6): 673-8. Epub 2003 May 5.
Hayashi, K. (1992). "PCR-SSCP: a method for detection of mutations." Genet Anal Tech Appl 9(3): 73-9.
Howard, T. D., D. S. Postma, et al. (2003). "Association of a disintegrin and metalloprotease 33 (ADAM33) gene with asthma in ethnically diverse populations." J Allergy Clin Immunol 112(4): 717-22.
Heintz, K., K. Palme, et al. (1992). "The Ncypti gene from Neurospora crassa is located on chromosome 2: molecular cloning and structural analysis." MoI Gen Genet 235(2-3): 413-21.
Hermonat, P. L. and N. Muzyczka (1984). "Use of adeno-associated virus as a mammalian DNA cloning vector: transduction of neomycin resistance into mammalian tissue culture cells." Proc Natl Acad Sci U S A 81(20): 6466-70.
Hobbs, M. R., V. Udhayakumar, et al. (2002). "A new NOS2 promoter polymorphism associated with increased nitric oxide production and protection from severe malaria in Tanzanian and Kenyan children." Lancet 360(9344): 1468- 75.
Hogrefe, H. H., R. L. Mullinax, et al. (1993). "A bacteriophage lambda vector for the cloning and expression of immunoglobulin Fab fragments on the surface of filamentous phage." Gene 128(1): 119-26.
Hoogendoom, B., N. Norton, et al. (2000). "Cheap, accurate and rapid allele frequency estimation of single nucleotide polymorphisms by primer extension and DHPLC in DNA pools." Hum Genet 107(5): 488-93.
Hsu, I. C, Q. Yang, et al. (1994). "Detection of DNA point mutations with DNA mismatch repair enzymes." Carcinogenesis 15(8): 1657-62.
Johannesen, J., A. Pie, et al. (2001). "Linkage of the human inducible nitric oxide synthase gene to type 1 diabetes." J Clin Endocrinol Metab 86(6): 2792-6.
Jongepier, H., H. M. Boezen, et al. (2004). "Polymorphisms of the ADAM33 gene are associated with accelerated lung function decline in asthma." Clin Exp Allergy 34(5): 757-60.
Kajita, M., Y. Ezura, et al. (2003). "Association of the -381T/C promoter variation of the brain natriuretic peptide gene with low bone-mineral density and rapid postmenopausal bone loss." J Hum Genet 48(2): 77-81.
Kanazawa, N., M. Kurosaki, et al. (2004). "Regulation of hepatitis C virus replication by interferon regulatory factor 1." J Virol 78(18): 9713-20.
Kanehisa, M. (1984). "Use of statistical criteria for screening potential homologies in nucleic acid sequences." Nucleic Acids Res 12(1 Pt 1): 203-13.
Karason, A., J. E. Gudjonsson, et al. (2003). "A susceptibility gene for psoriatic arthritis maps to chromosome 16q: evidence for imprinting." Am J Hum Genet 72(1): 125-31.
Kohler, G. and C. Milstein (1992). "Continuous cultures of fused cells secreting antibody of predefined specificity. 1975." Biotechnology 24: 524-6.
Kohn, D. B., K. I. Weinberg, et al. (1995). "Engraftment of gene-modified umbilical cord blood cells in neonates with adenosine deaminase deficiency." Nat Med 1 (10): 1017-23.
Kolodziejski, P. J., A. Musial, et al. (2002). "Ubiquitination of inducible nitric oxide synthase is required for its degradation." Proc Natl Acad Sci U S A 99(19): 12315-20. Epub 2002 Sep 9.
Kormann, M. S., D. Carr, et al. (2005). "G-Protein-coupled Receptor Polymorphisms Are Associated with Asthma in a Large German Population." Am J Respir Crit Care Med 171(12): 1358-62.
Kotin, R. M. (1994). "Prospects for the use of adeno-associated virus as a vector for human gene therapy." Hum Gene Ther 5(7): 793-801.
Kozal, M. J., N. Shah, et al. (1996). "Extensive polymorphisms observed in HIV-1 clade B protease gene using high-density oligonucleotide arrays." Nat Med 2(7): 753-9.
Krebs, S., D. Seichter, et al. (2001). "Genotyping of dinucleotide tandem repeats by MALDI mass spectrometry of ribozyme-cleaved RNA transcripts." Nat Biotechnol 19(9): 877-80.
Kremer, E. J. and M. Perricaudet (1995). "Adenovirus and adeno-associated virus mediated gene transfer." Br Med Bull 51(1): 31-44.
Kulinski, J., D. Besack, et al. (2000). "CEL I enzymatic mutation detection assay." Biotechniques 29(1): 44-6, 48.
Kwoh, D. Y., G. R. Davis, et al. (1989). "Transcription-based amplification system and detection of amplified human immunodeficiency virus type 1 with a bead- based sandwich hybridization format." Proc Natl Acad Sci U S A 86(4): 1173-7.
Laitinen, T., A. Polvi, et al. (2004). "Characterization of a common susceptibility locus for asthma-related traits." Science 304(5668): 300-4.
Livak, K. J., J. Marmaro, et al. (1995). "Towards fully automated genome-wide polymorphism screening." Nat Genet 9(4): 341-2.
Lizardi, P. M., X. Huang, et al. (1998). "Mutation detection and single-molecule counting using isothermal rolling-circle amplification." Nat Genet 19(3): 225-32.
Lowenstein, C. J., C. S. Glatt, et al. (1992). "Cloned and expressed macrophage nitric oxide synthase contrasts with the brain enzyme." Proc Natl Acad Sci U S A 89(15): 6711-5.
Malech, H. L., P. B. Maples, et al. (1997). "Prolonged production of NADPH oxidase-corrected granulocytes after gene therapy of chronic granulomatous disease." Proc Natl Acad Sci U S A 94(22): 12133-8.
Mansfield, E. S., M. Vainer, et al. (1996). "Sensitivity, reproducibility, and accuracy in short tandem repeat genotyping using capillary array electrophoresis." Genome Res 6(9): 893-903.
Maxam, A. M. and W. Gilbert (1977). "A new method for sequencing DNA." Proc Natl Acad Sci U S A 74(2): 560-4.
Melen, E., S. Bruce, et al. (2005). "Haplotypes of G-protein-coupled Receptor 154 are Associated with Childhood Allergy and Asthma." Am J Respir Crit Care Med 11 : 11.
Metcalf, D. (1985). "The granulocyte-macrophage colony stimulating factors." Cell 43(1): 5-6.
Miller, A. D. (1992). "Human gene therapy comes of age." Nature 357(6378): 455-60.
Miller, A. D., J. V. Garcia, et al. (1991). "Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus." J Virol 65(5): 2220-4.
Mitani, K. and C. T. Caskey (1993). "Delivering therapeutic genes-matching approach and application." Trends Biotechnol 11(5): 162-6.
Muzyczka, N. (1994). "Adeno-associated virus (AAV) vectors: will they work?" J Clin Invest 94(4): 1351.
Myers, R. M., Z. Larin, et al. (1985). "Detection of single base substitutions by ribonuclease cleavage at mismatches in RNA: DNA duplexes." Science 230(4731): 1242-6.
Myers, R. M., N. Lumelsky, et al. (1985). "Detection of single base substitutions in total genomic DNA." Nature 313(6002): 495-8.
Nabel, G. J. and P. L. Feigner (1993). "Direct gene transfer for immunotherapy and immunization." Trends Biotechnol 11(5): 211-5.
Nagley, P. and Y. H. Wei (1998). "Ageing and mammalian mitochondrial genetics." Trends Genet 14(12): 513-7.
Napolitano, M., F. Miceli, et al. (2000). "Expression and relationship between endothelin-1 messenger ribonucleic acid (mRNA) and inducible/endothelial nitric oxide synthase mRNA isoforms from normal and preeclamptic placentas." J Clin Endocrinol Metab 85(6): 2318-23.
Nathan, C. and M. U. Shiloh (2000). "Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens." Proc Natl Acad Sci U S A 97(16): 8841-8.
Orita, M., H. Iwahana, et al. (1989). "Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms." Proc Natl Acad Sci U S A 86(8): 2766-70.
Raap, A. K. (1998). "Advances in fluorescence in situ hybridization." Mutat Res 400(1-2): 287-98.
Raby, B. A., E. K. Silverman, et al. (2004). "ADAM33 polymorphisms and phenotype associations in childhood asthma." J Allergy Clin Immunol 113(6): 1071-8.
Ronaghi, M., M. Uhlen, et al. (1998). "A sequencing method based on real-time pyrophosphate." Science 281(5375): 363, 365.
Rosenecker, J., K. H. Harms, et al. (1996). "Adenovirus infection in cystic fibrosis patients: implications for the use of adenoviral vectors for gene transfer." Infection 24(1): 5-8.
Saiki, R. K., T. L. Bugawan, et al. (1986). "Analysis of enzymatically amplified beta-globin and HLA-DQ alpha DNA with allele-specific oligonucleotide probes." Nature 324(6093): 163-6.
Saiki, R. K., P. S. Walsh, et al. (1989). "Genetic analysis of amplified DNA with immobilized sequence-specific oligonucleotide probes." Proc Natl Acad Sci U S A 86(16): 6230-4.
Saleeba, J. A. and R. G. Cotton (1993). "Chemical cleavage of mismatch to detect mutations." Methods Enzymol 217: 286-95.
Samulski, R. J., L. S. Chang, et al. (1989). "Helper-free stocks of recombinant adeno-associated viruses: normal integration does not require viral gene expression." J Virol 63(9): 3822-8.
Sanders, L. C, F. Matsumura, et al. (1999). "Inhibition of myosin light chain kinase by p21 -activated kinase." Science 283(5410): 2083-5.
Sandford, A. J., T. Shirakawa, et al. (1993). "Localisation of atopy and beta subunit of high-affinity IgE receptor (Fc epsilon Rl) on chromosome 11q." Lancet 341(8841): 332-4.
Sanger, F., S. Nicklen, et al. (1977). "DNA sequencing with chain-terminating inhibitors." Proc Natl Acad Sci U S A 74(12): 5463-7.
Sauter, E. R., M. Herlyn, et al. (2000). "Prolonged response to antisense cyclin D1 in a human squamous cancer xenograft model." Clin Cancer Res 6(2): 654- 60.
Sells, M. A., U. G. Knaus, et al. (1997). "Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells." Curr Biol 7(3): 202-10.
Shin, H. D., K. S. Park, et al. (2004). "Lack of association of GPRA (G protein- coupled receptor for asthma susceptibility) haplotypes with high serum IgE or asthma in a Korean population." J Allergy Clin Immunol 114(5): 1226-7.
Simon, H., I. Fortsch, et al. (1999). "Triple helix formation inhibits DNA gyrase activity." Antisense Nucleic Acid Drug Dev 9(6): 527-31.
Smith, J. and P. Modrich (1996). "Mutation detection with MutH, MutL, and MutS mismatch repair proteins." Proc Natl Acad Sci U S A 93(9): 4374-9.
Smith, M. J., S. D. Gitlin, et al. (2001). "GLI-2 modulates retroviral gene expression." J Virol 75(5): 2301-13.
Sommerfelt, M. A. and R. A. Weiss (1990). "Receptor interference groups of 20 retroviruses plating on human cells." Virology 176(1): 58-69.
Steinhauer, D. A., S. A. Wharton, et al. (1991). "Amantadine selection of a mutant influenza virus containing an acid-stable hemagglutinin glycoprotein: evidence for virus-specific regulation of the pH of glycoprotein transport vesicles." Proc Natl Acad Sci U S A 88(24): 11525-9.
Storm, N., B. Darnhofer-Patel, et al. (2003). "MALDI-TOF mass spectrometry- based SNP genotyping." Methods MoI Biol 212: 241-62.
Tratschin, J. D., M. H. West, et al. (1984). "A human parvovirus, adeno- associated virus, as a eucaryotic vector: transient expression and encapsidation of the procaryotic gene for chloramphenicol acetyltransferase." MoI Cell Biol 4(10): 2072-81.
Van Eerdewegh, P., R. D. Little, et al. (2002). "Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness." Nature 418(6896): 426-30.
Vojtek, A. B. and C. J. Der (1998). "Increasing complexity of the Ras signaling pathway." J Biol Chem 273(32): 19925-8.
Warabi, K., M. D. Richardson, et al. (2002). "Human substance P receptor undergoes agonist-dependent phosphorylation by G protein-coupled receptor kinase 5 in vitro." FEBS Lett 521(1-3): 140-4.
Werner, M., N. Herbon, et al. (2004). "Asthma is associated with single- nucleotide polymorphisms in ADAM33." Clin Exp Allergy 34(1): 26-31.
West, M. H., J. P. Trempe, et al. (1987). "Gene expression in adeno-associated virus vectors: the effects of chimeric mRNA structure, helper virus, and adenovirus VA1 RNA." Virology 160(1): 38-47.
Wilson, C1 M. S. Reitz, et al. (1989). "Formation of infectious hybrid virions with gibbon ape leukemia virus and human T-cell leukemia virus retroviral envelope glycoproteins and the gag and pol proteins of Moloney murine leukemia virus." J Virol 63(5): 2374-8.
Yu, M., E. Poeschla, et al. (1994). "Progress towards gene therapy for HIV infection." Gene Ther 1(1): 13-26.
Zhang, Y., N. !. Leaves, et al. (2003). "Positional cloning of a quantitative trait locus on chromosome 13q14 that influences immunoglobulin E levels and asthma." Nat Genet 34(2): 181-186.
Zhang, S. X., E. G. Gras, et a/. (2005). "Identification of direct serum response factor gene targets during DMSO induced P19 cardiac cell differentiation." J Biol Chem 28: 28.
BOOKS:
Abbas AK, Litchman AH. Cellular and Molecular Immunology. Philadelphia: Saunders; 1991. 417 p.
Austen BM and. Westwood OMR. Protein Targeting and Secretion. Oxford: IRL Press; 1991. 85 p.
Bishop MJ, editor. Guide to Human Genome Computing, 2d ed. San Diego: Academic Press; 1998. 306 p.
Cowell IG, Austin CA, editors. DNA Library Protocols. Methods in Molecular Biology. Vol. 69 Totowa, N.J.:Humana Press; 1997. 321p.
Freshney Rl, editor. Animal Cell Culture: A Practical Approach. Oxford: IRL Press; 1986.
Freshney Rl. Culture Of Animal Cells: A Manual of Basic Technique. New York: AR Liss; 1987. 397 p.
Glover DM, editor. DNA Cloning: A Pratical Approach. VoIs 1 & 2. Oxford; Washington: IRL Press; 1985.
Gribskov M, Devereux J, editors. Sequence Analysis Primer. Oxford University Press; 1994. 296 p.
Griffin AM, Griffin HG, editors. ComputerAnalysis of Sequence Data, Part 1. Totowa, N.J. Humana Press; 1994. 392 p.
Hames BD, Higgins SJ, editors. Nucleic Acid Hybridization: A Practical Approach. Oxford: IRL Press; 1985. 245 p.
Hames BD, Higgins SJ, editors. Transcription and Translation: A Practical Approach. Oxford: IRL Press; 1984. 328 p.
Harlow Ed, Lane D. Antibodies: A Laboratory Manual. New York: Cold Spring Harbor Laboratory; 1988. 726 p.
Heinje G. von. Sequence Analysis in Molecular Biology. San Diego: Academic Press; 1987. 188 p.
Hogan B, Costantini F, Lacy E, editors. Manipulating the Mouse Embryo: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press; 1986. 332
P-
Huber BE, Carr Bl. Molecular and Immunologic Approaches. Mt. Kisco, NY: Futura Publishing Co; 1994.
Jones J. Amino Acid and Peptide Synthesis. Oxford; New York: Oxford Science Publications; 1992. 86 p.
Kaufman PB, William W, Donghem K1 editors. Handbook of Molecular and Cellular Methods in Biology and Medicine. Boca Raton: CRC Press; 1995. 484 p.
Lesk AM, editor. Computational Molecular Biology: sources and methods for sequence analysis. New York: Oxford University Press; 1988. 254p.
Male D, Cooke A, Owen M, Trowsdale J, Champion B, editors. Advanced Immunology. 3rd ed. London; Baltimore: Mosby; 1996. 273 p.
McPherson MJ, editor. Directed Mutagenesis: A Practical Approach. New York: IRL Press; 1991. 257 p.
McPherson MJ, Quirke P, Taylor JR, editors. PCR: A Practical Approach. Oxford; New York: IRL Press; 1991. 253 p.
Miller JH, Calos MP, editors. Gene Transfer Vectors for Mammalian Cells. New York: Cold Spring Harbor Laboratory Press; 1987. 169 p.
Miller JH, Calos MP, editors. Gene Transfer Vectors For Mammalian Cells. New York: Cold Spring Harbor Laboratory; 1987. 169p.
Pawlowitzki IH, Edwards JH, Thompson EA, editors. Genetic Mapping of Disease Genes. Academic Press London; 1997. 288 p.
Perbal BV. A Practical Guide to Molecular Cloning. 1st ed. New York: Wiley lnterscience Publication; 1984. 554 p.
Perbal BV. A Practical Guide To Molecular Cloning. New York: Wiley; 1984. 554 P-
Peruski LF, Peruski AH. The Internet and the New Biology. Tools for Genomic and Molecular Research. Washington, D. C: American Society for Microbiology Press; 1997.
Sambrook J. Molecular Cloning: A Laboratory Manual. 2nd ed. 3 vols. New York: Cold Spring Harbor Laboratory Press; 1989.
Sell S. Immunology, lmmunopathology & Immunity. 5th ed. Stamford, CT: Appleton & Lange; 1996. 1014 p.
Smith DW, editor. Biocomputing. Informatics and Genome Projects, New York: Academic Press; 1993. 336p.
Stites DP, Terr AT, editors. Basic and Clinical Immunology. 7th ed. Norwalk, CT: Appleton & Lange; 1991. 870 p.
Walker JM. Protein Protocols on CD-ROM, Humana Press, Totowa, NJ.
Weir DM, Herzenberg LA, Blackwell C, editors. Handbook Of Experimental Immunology. 4 vols. Oxford: Blackwell; 1986.
Woodward J. Immobilized Cells And Enzymes: A Practical Approach. Oxford: IRL Press; 1986.
Wu R, Grossman L, editors. Methods in Enzymoloqy: Rexcombinant DNA Part E. Vol. 154. Amsterdam: Elsevier Science; 1987. 576 p.
Wu R, Grossman L, editors. Methods in Enzymoloqy: Rexcombinant DNA Part F. Vol. 155. Amsterdam: Elsevier Science; 1987. 628 p.
Patents
U.S. 4,683,202. U.S. 4,952,501. WO03042661A2 US 20040009479A1 U.S. 5,315,000 WO1997US0005216 U.S. 5,498,531 U.S. 5,807,718 U.S. 5,888,819 U.S. 6,090,543 U.S. 6,090,606 U.S. 5,585,089 U.S. 4,683,195 U.S. 4,683,202 U.S. 5,459,039. U.S. 6,090,543). U.S. 6,090,606 U.S. 5,869,242
U.S. 60/335,068 U.S. 6,479,244 PCT/US94/05700 U.S. 4,797,368 WO 93/24641 U.S. 5,173,414






































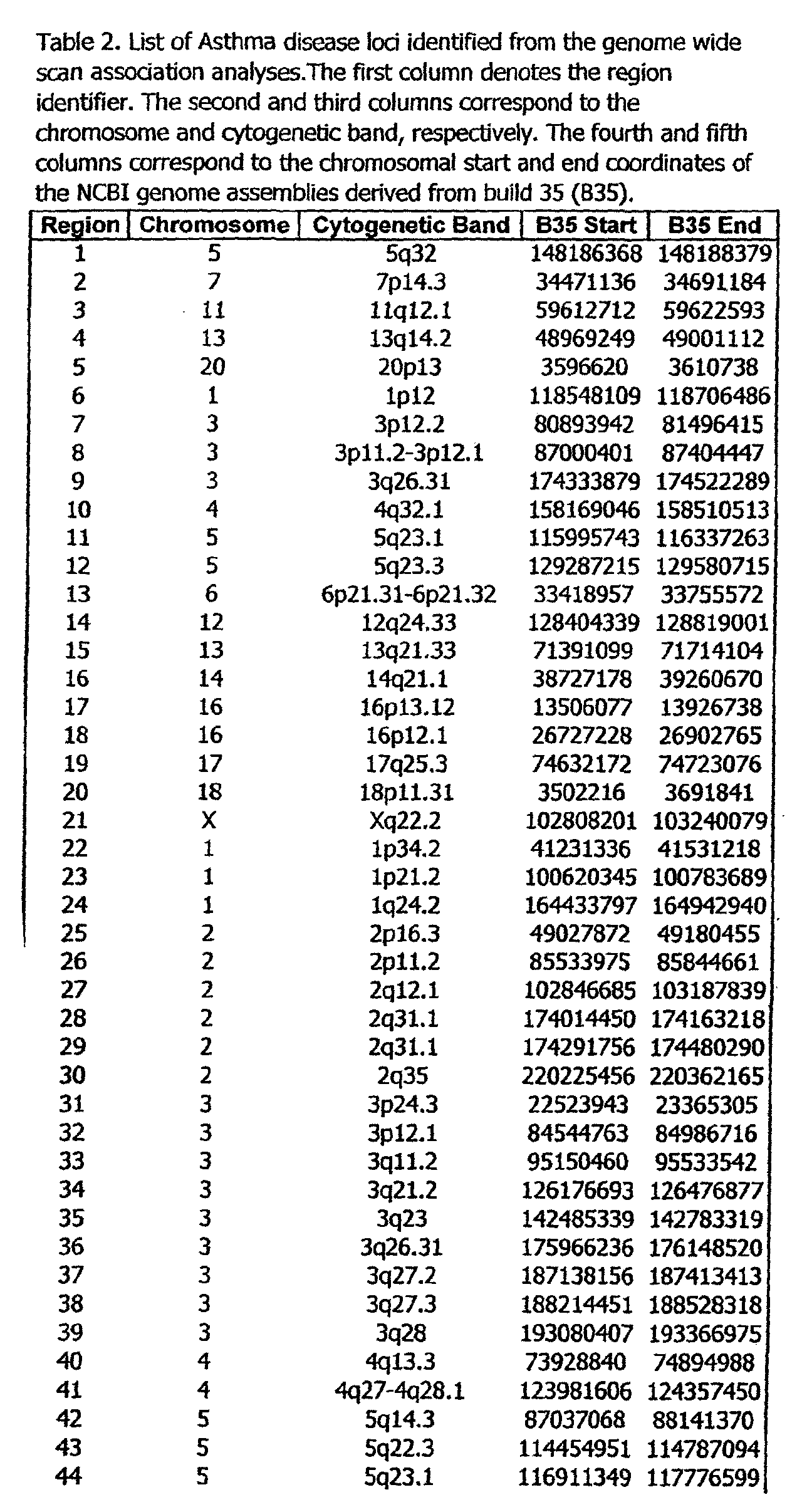








O

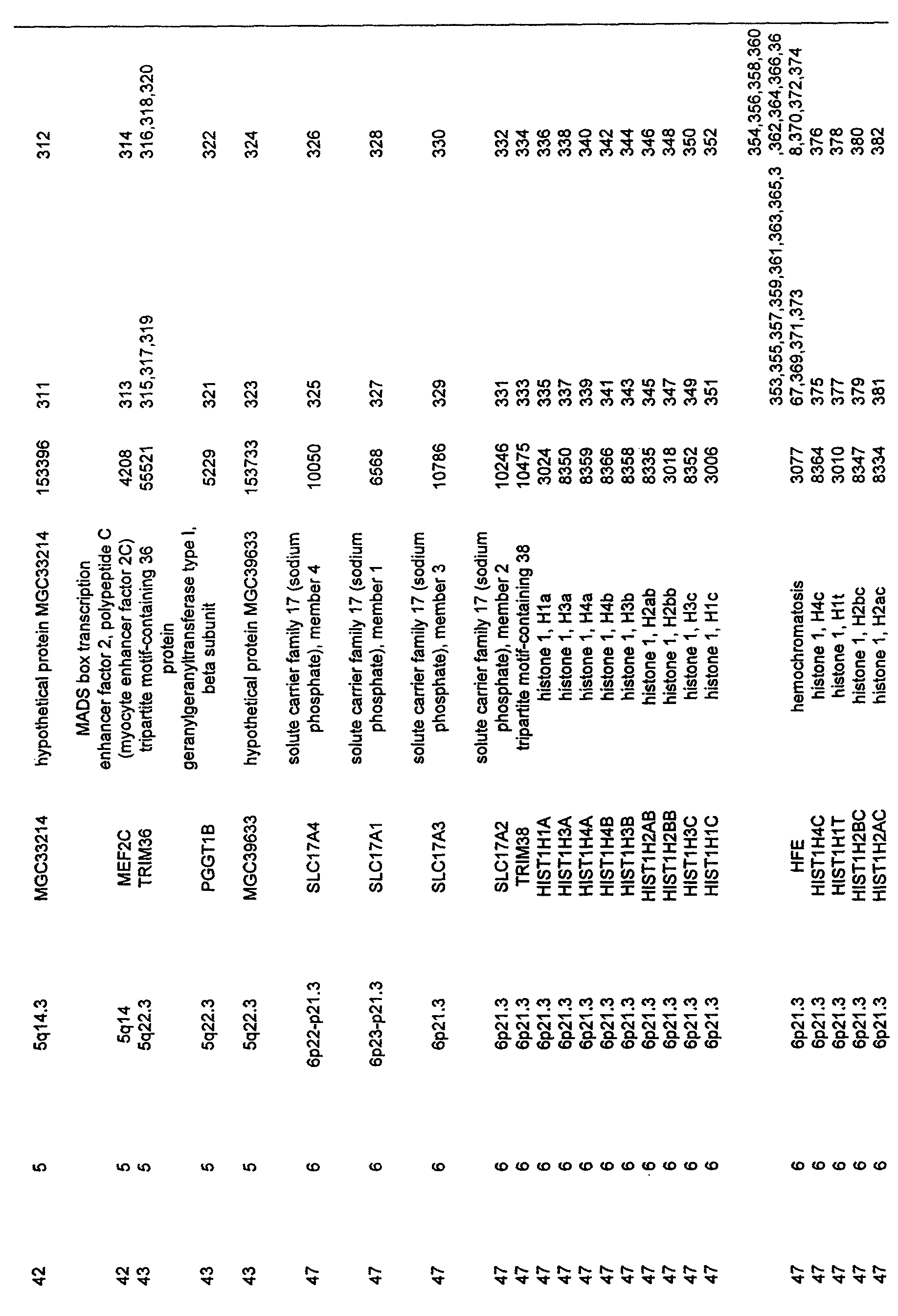

















































































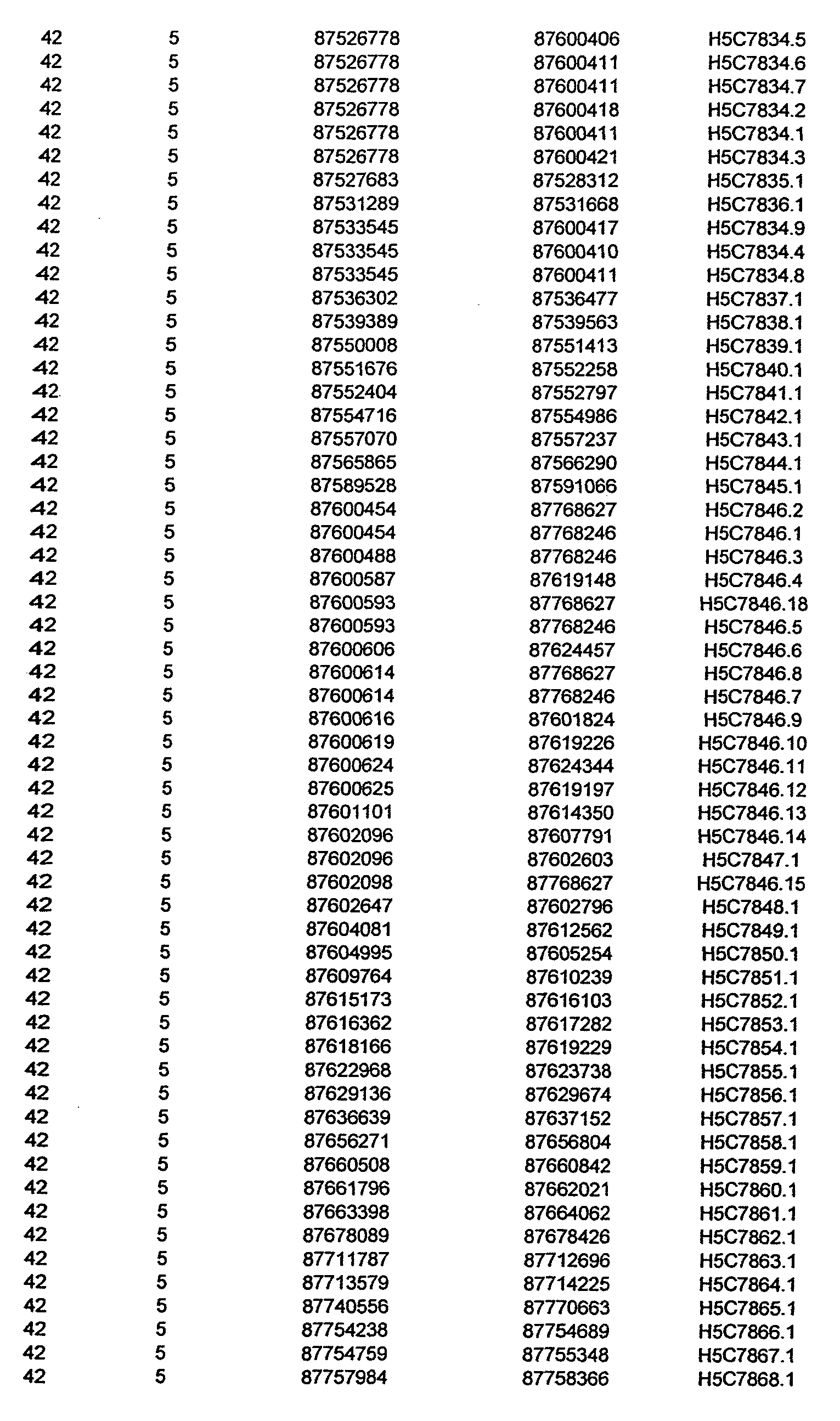
























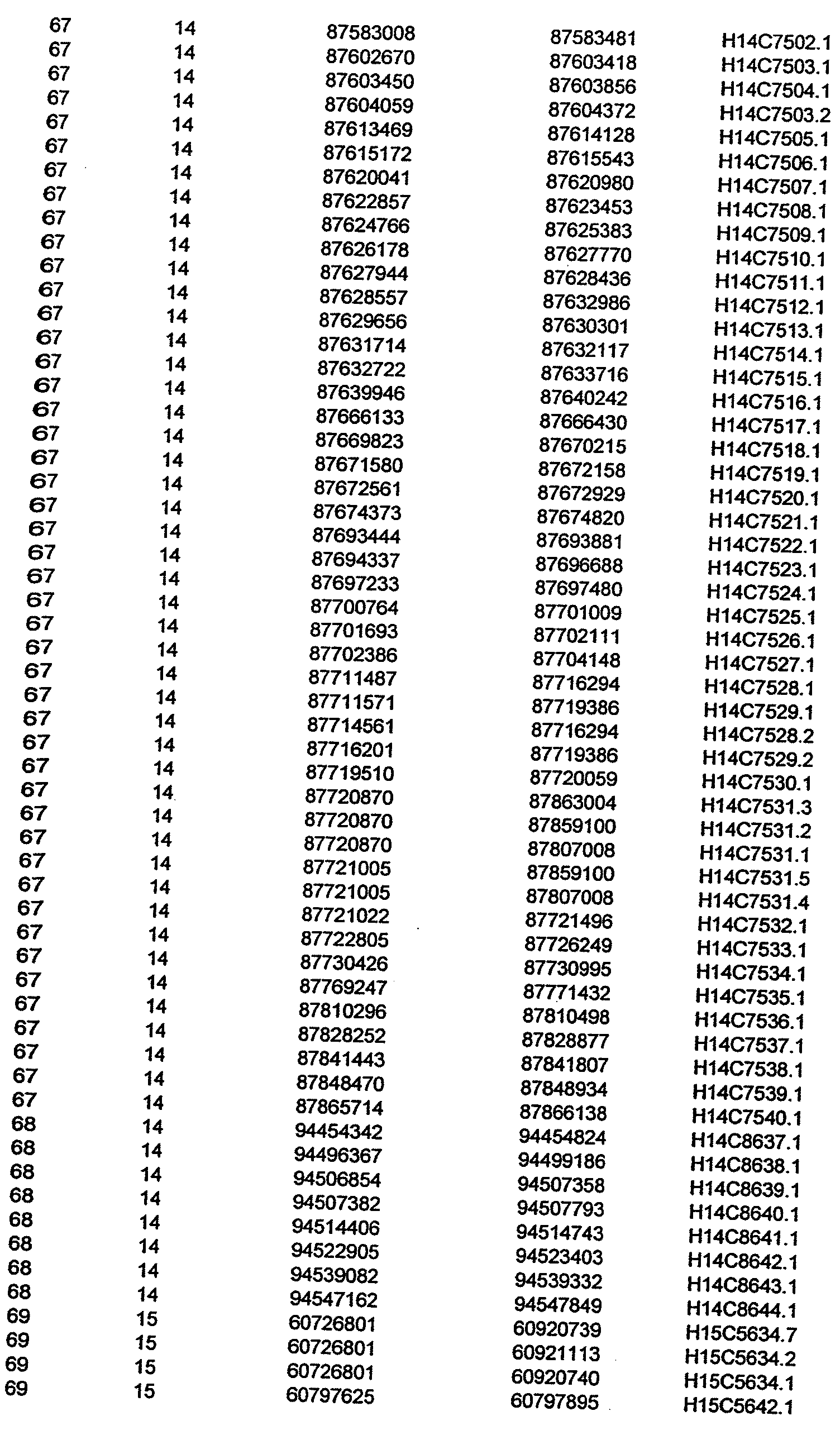










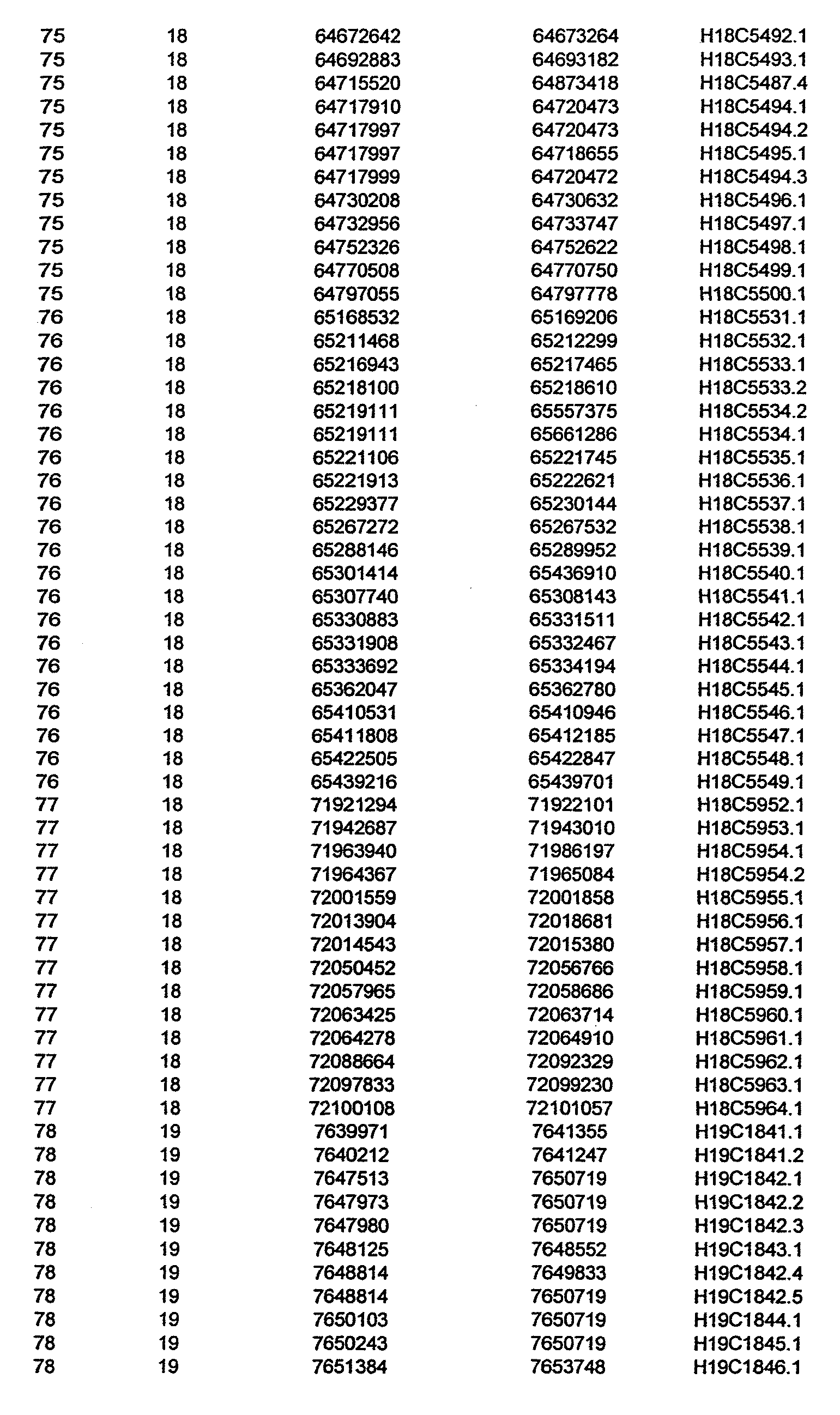




MISSING UPON FILING







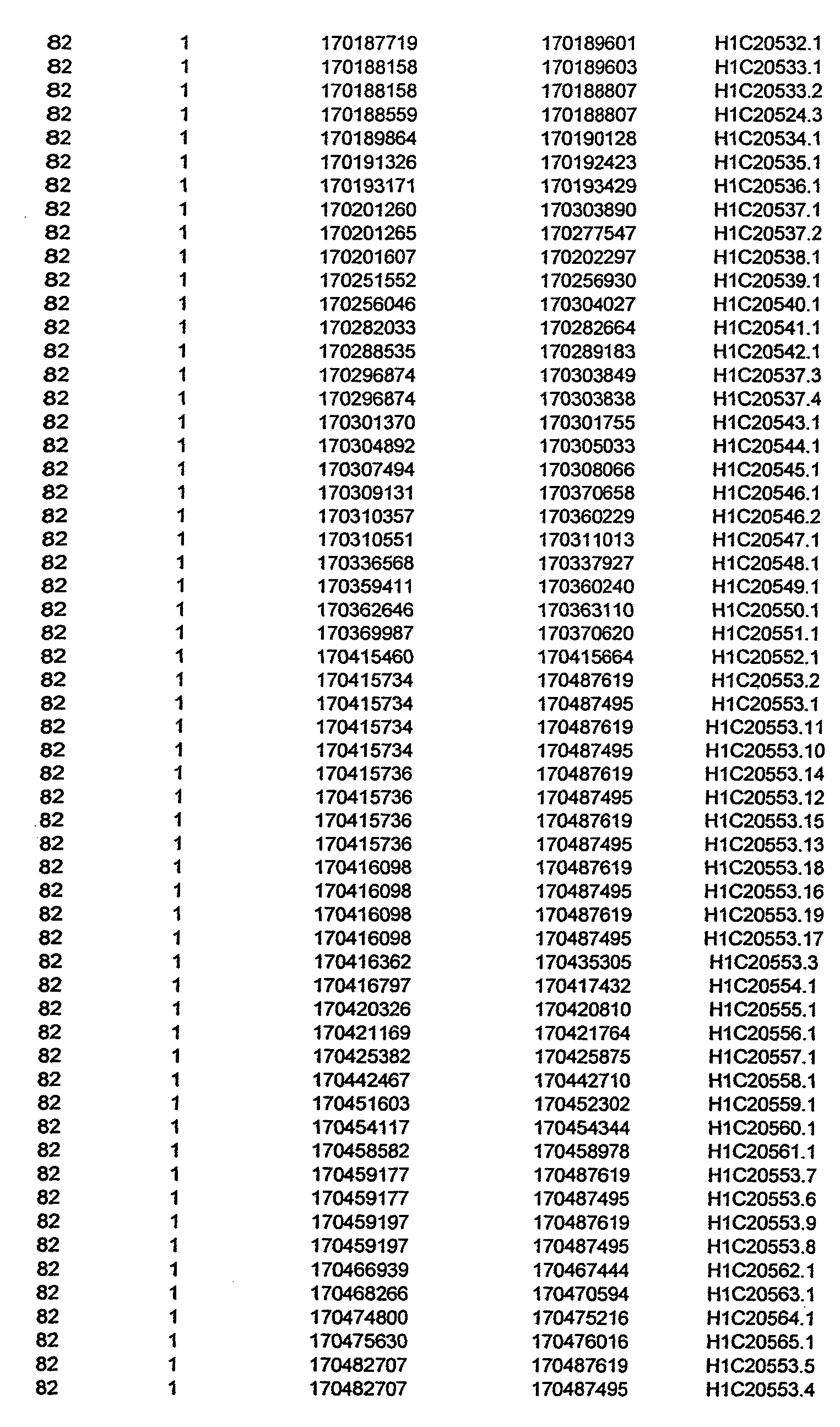







































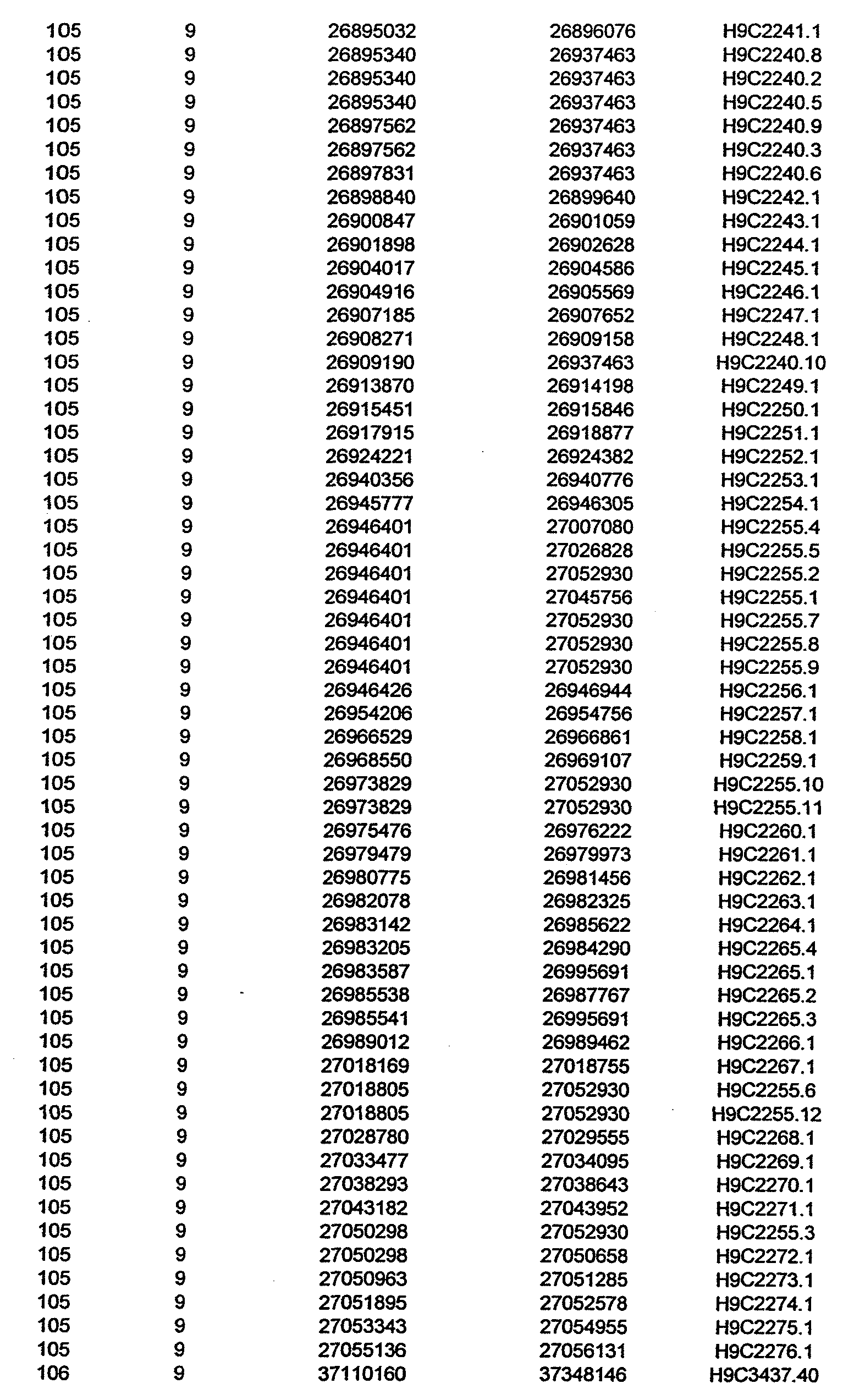












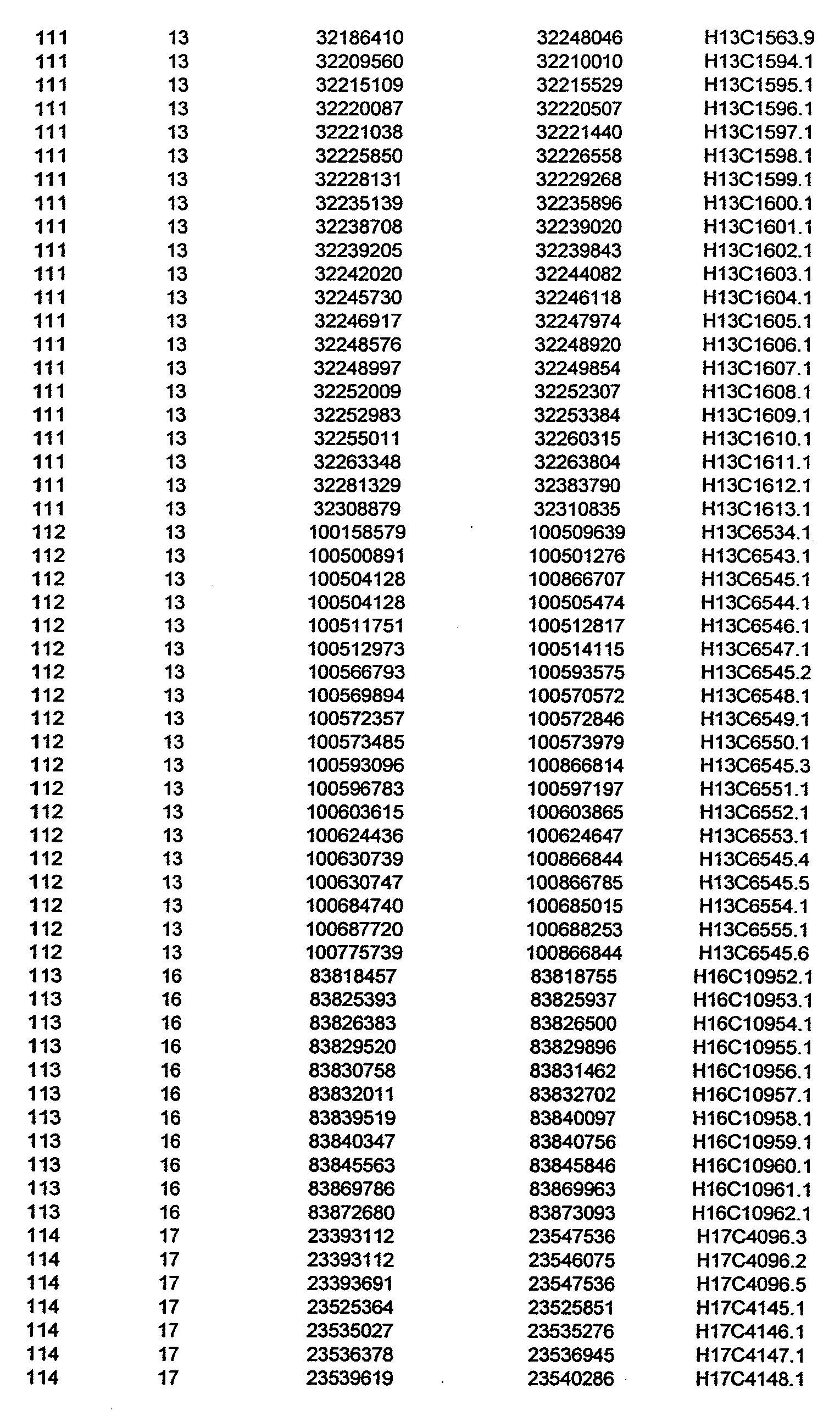





































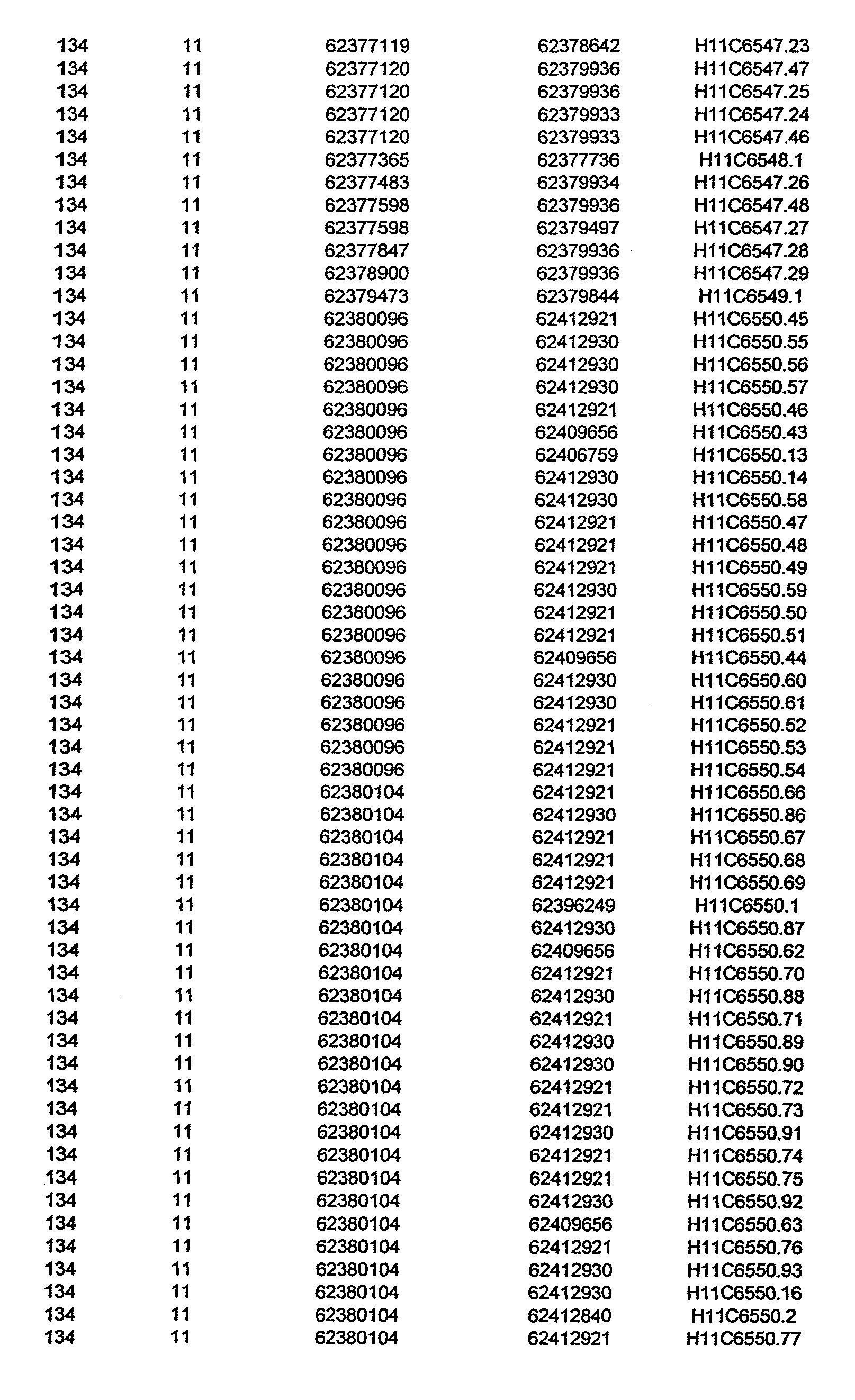


















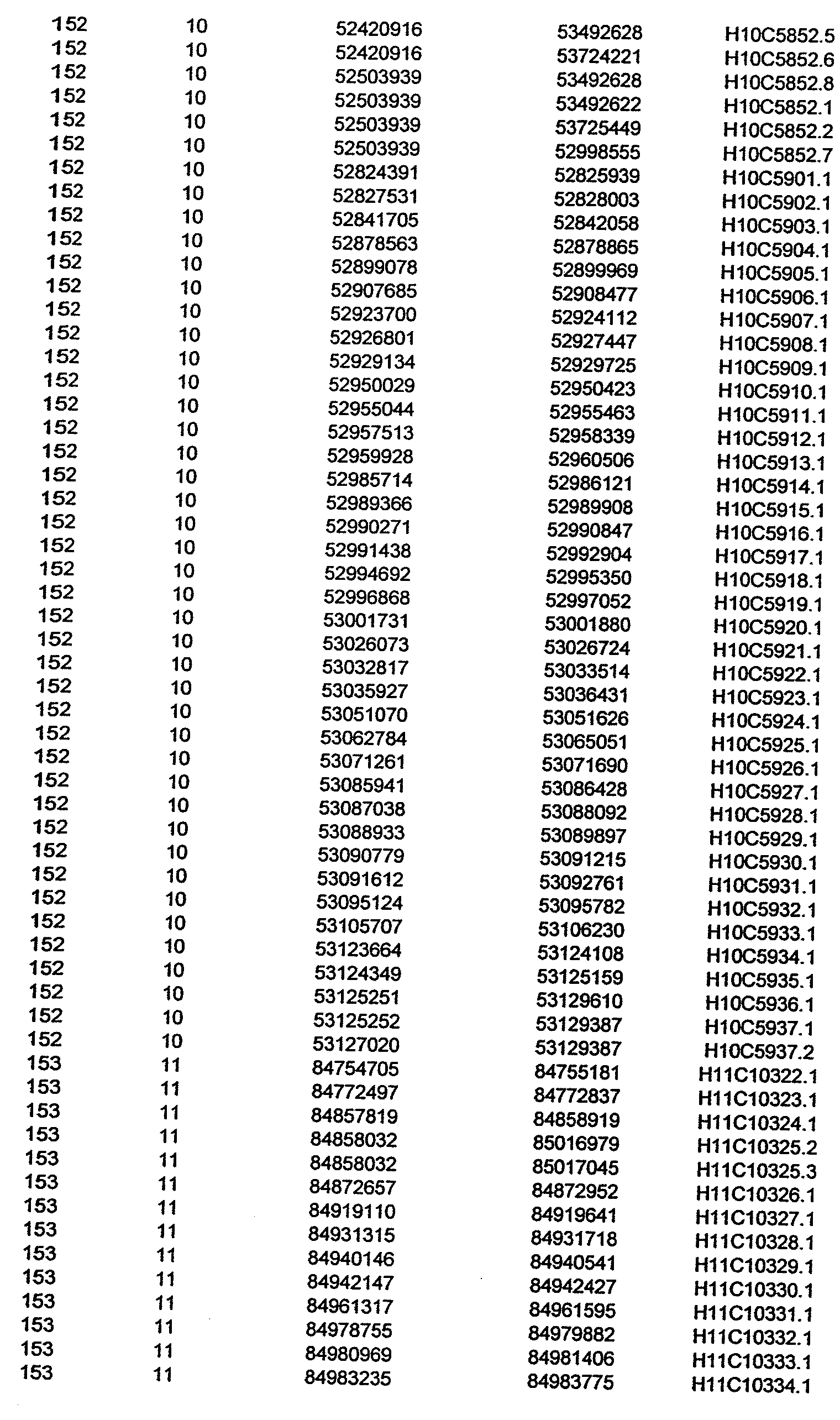













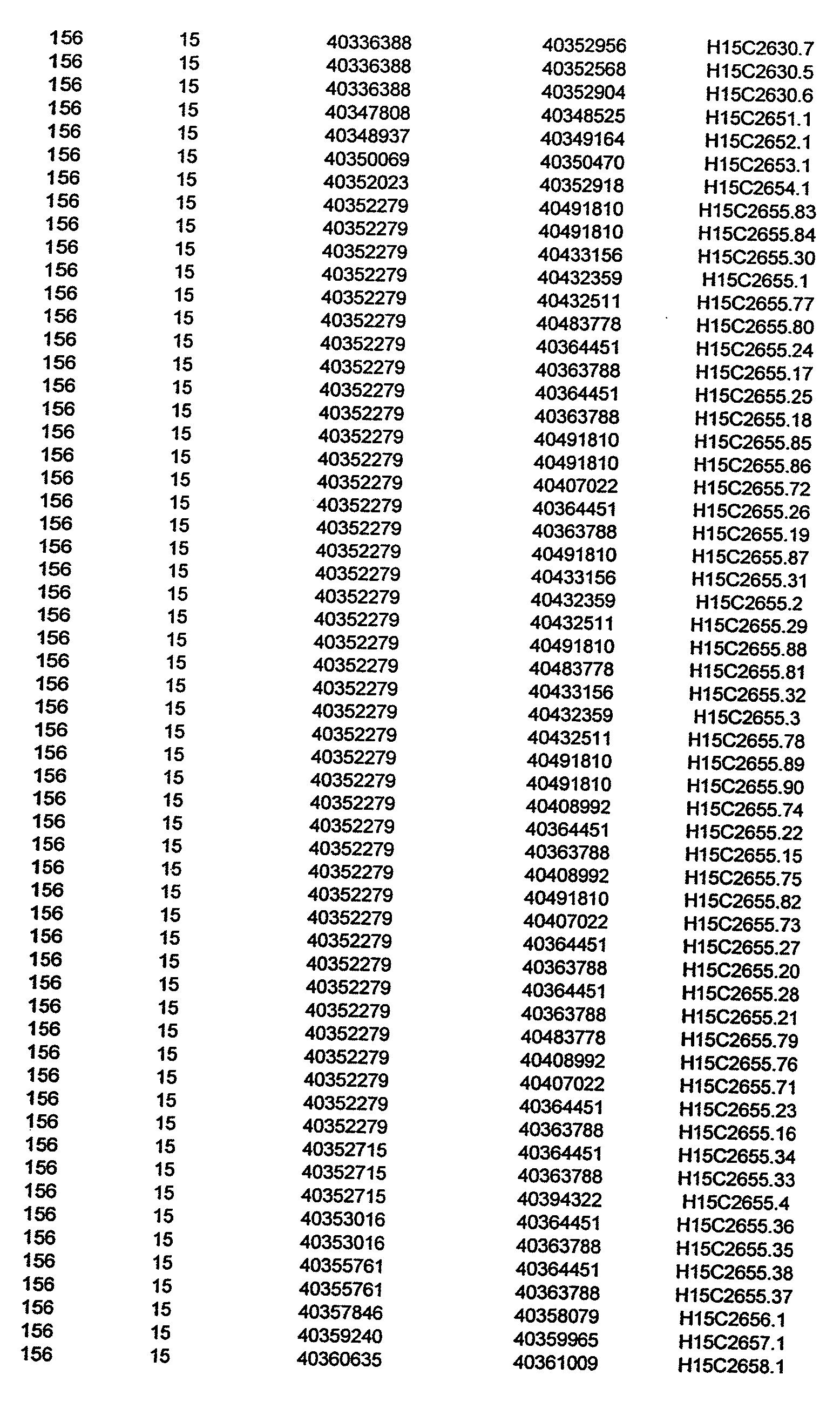





















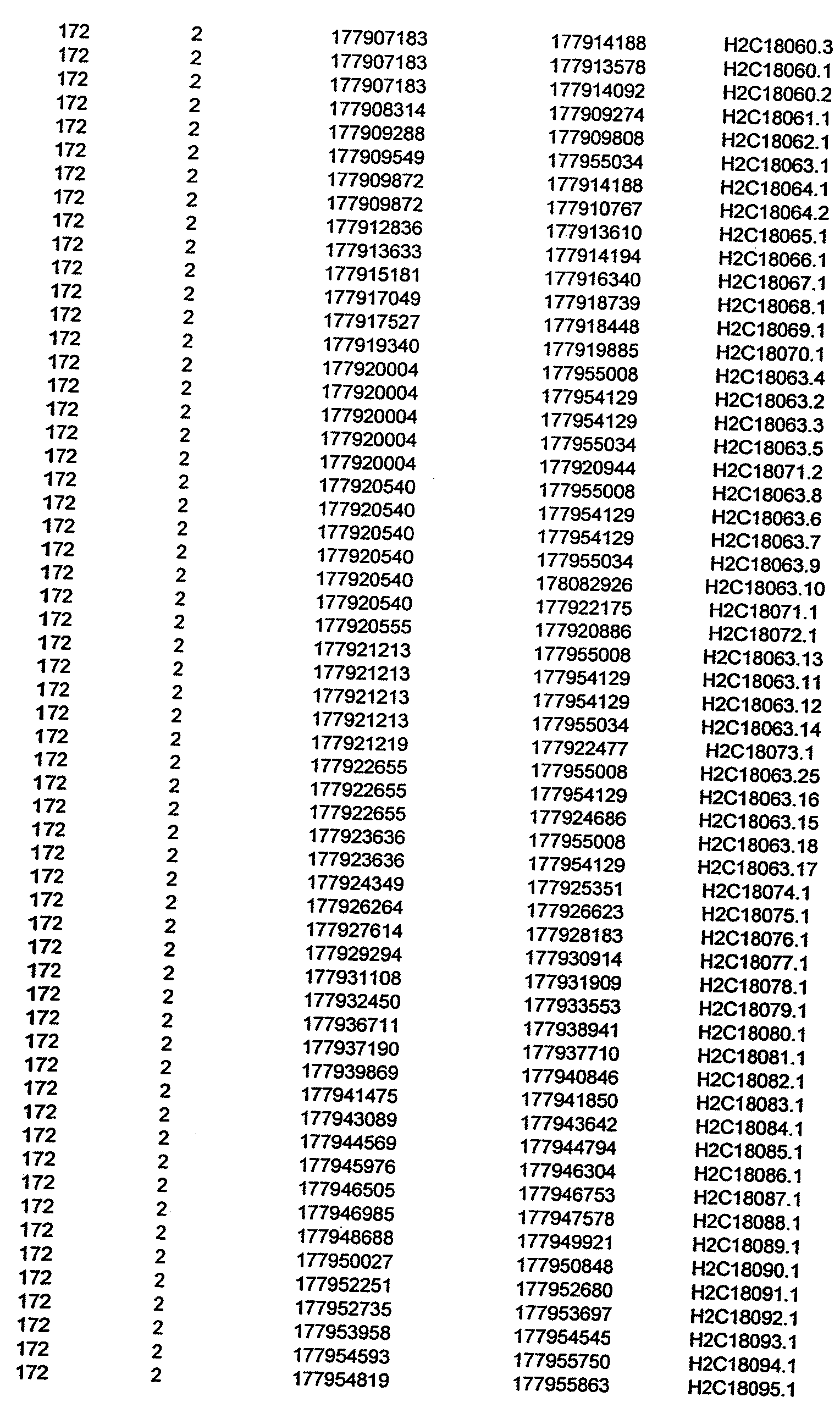





















































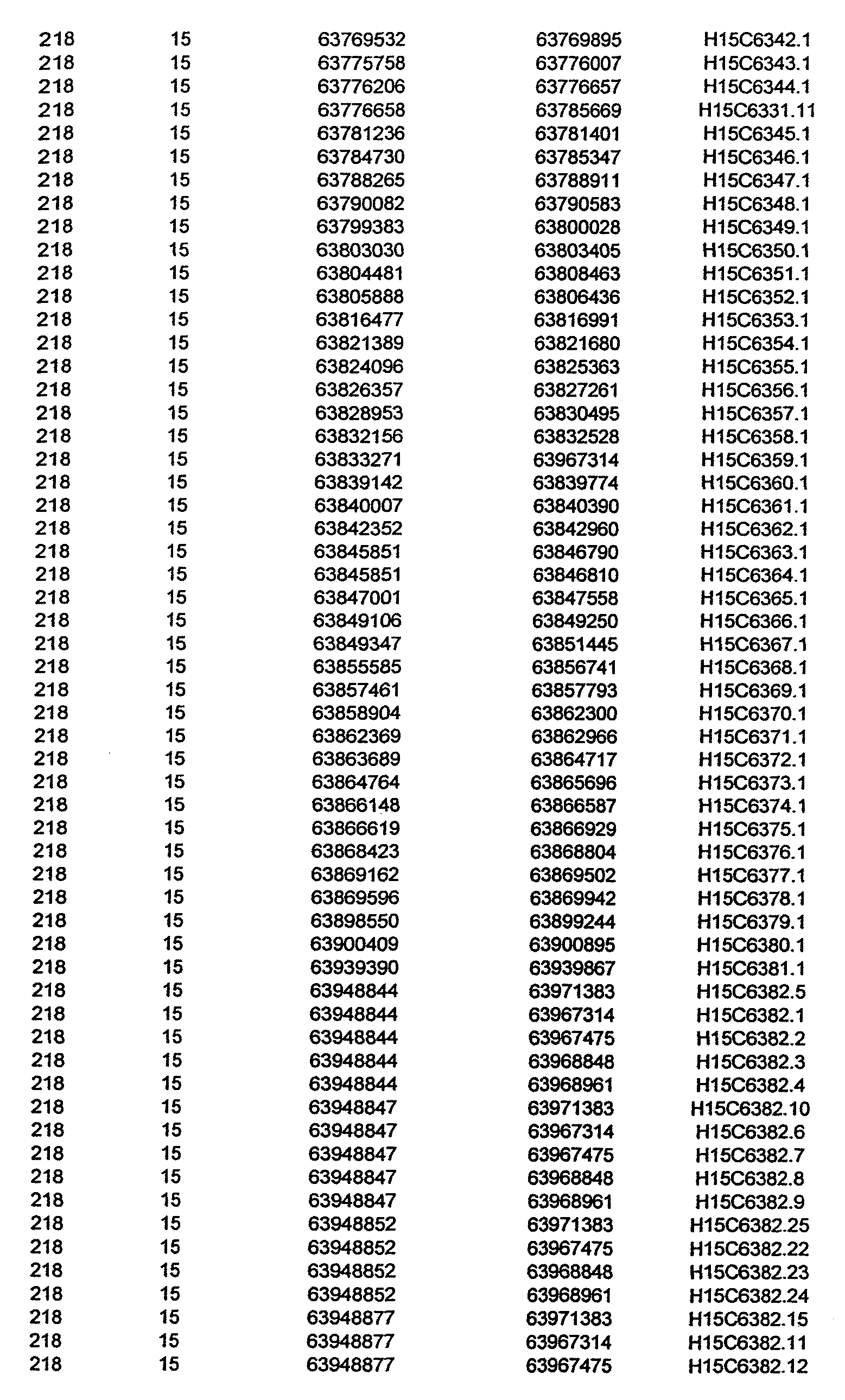









Claims
1. A method of constructing a asthma disease GeneMap in a human population, comprising screening for the expression level of or presence or absence of at least one allele of at least one gene from Tables 3 and 4 in at least one sample.
2. A method of claim 1 , wherein said population is a general population.
3. A method of claim 1 , wherein said population is a founder population.
4. A method of claim 3, wherein said founder population is the population of Quebec.
5. A method of constructing a GeneMap in a human population, comprising screening for the presence or absence of an allele of at least one single nucleotide polymorphisms (SNPs) from Table 1 , in at least one sample.
6. A method of claim 5, wherein said population is a general population.
7. A method of claim 5, wherein said population is a founder population.
8. A method of claim 7, wherein said founder population is the population of Quebec.
9. A method of genetic mapping for detecting the association of at least one marker for asthma disease comprising: a) obtaining biological samples from at least one disease patient; b) screening for the presence or absence of an allele of at least one SNP or a group of SNPs from Table 1 , within each biological sample; and c) evaluating whether said SNP or a group of SNPs shows a statistically significant skewed genotype distribution between a group of patients compared to a group of controls.
10. A method of claim 9, wherein said biological samples are hair, fluid, serum, tissue or buccal swabs, saliva, mucus, urine, stools, spermatozoids, vaginal secretions, lymph, amniotic fluid, pleural liquid or tears.
11. A method of claim 9, wherein said patients and controls are from a human population.
12. A method of claim 11 , wherein said population is the general population.
13. A method of claim 11 , wherein said population is a founder population.
14. A method of claim 13, wherein said founder population is the population of Quebec.
15. A method of claim 9, wherein said patients and controls are recruited independently according to specific phenotypic criteria.
16. A method of claim 9, wherein said patients and controls are recruited in the form of trios, comprising two parents and one child or one parent and two children, or in the form of unrelated individuals.
17. A method of claim 9, wherein said screening is performed by a method selected from the group consisting of an allele-specific hybridization assay, an oligonucleotide ligation assay, an allele-specific elongation/ligation assay, an allele-specific amplification assay, a single-base extension assay, a molecular inversion probe assay, an invasive cleavage assay and a selective termination assay. Other assays include RFLP1 sequencing assay, SSCP, mismatch-cleaving assay and denaturing gradient gel electrophoresis.
18. A method of claim 9, wherein said screening is carried out on each individual of a cohort at each of at least one SNP or a group of SNPs from Table !
19. A method of claim 9, wherein said screening is carried out on pools of patients and pools of controls.
20. A method of claim 9, wherein the genotype distribution is compared one SNP at a time.
21. A method of claim 9, wherein a genotype distribution is compared with a group of markers from Table 1 forming a haplotype.
22. A method of claim 9, wherein a genotype distribution is compared using the allelic frequencies between the patient pools and control pools.
23. A set of genetic markers comprising at least two SNPs of Table 1.
24. A set of nucleic acid probes that specifically detect said SNPs of claim 23.
25. A solid support or collection of solid supports comprising the nucleic acid probes of claim 24.
26. A solid support of claim 25, wherein the support is selected from the group consisting of at least one microarray and a set of beads.
27. A method for predicting the efficacy of a drug for treating asthma disease in a human patient, comprising: a) obtaining a sample of cells from the patient; b) obtaining a gene expression profile from the sample in the absence and presence of in vitro modulation of the cells with specific mediators; the gene expression profile comprising one or more genes from Tables 3 and 4; and c) comparing the gene expression profile of the sample with a reference gene expression profile, wherein similarity between the sample expression profile and the reference expression profile predicts the efficacy of the drug for treating asthma disease in the patient.
28. The method of claim 27, further comprising exposing the sample to the drug for treating asthma disease prior to obtaining the gene expression profile of the sample.
29. The method of claim 27, wherein the sample of cells is derived from a tissue selected from the group consisting of: any component of the respiratory system, inner and outer lung coatings, inner and outer trachea coatings, muscles, epithelial and nervous tissue.
30. The method of claim 29, wherein the cells are selected from the group consisting of: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, dendritic cell, synovial cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina propria lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
31. The method of claim 27, wherein the sample is obtained via lung, trachea, bronchi or any respiratory track component biopsy.
32. The method of claim 27, wherein the gene expression profile comprises expression values for at least two of the genes listed in Tables 3 and 4.
33. The method of claim 32, wherein the gene expression profile of the sample is obtained by detecting the protein products of said genes.
34. The method of claim 27, wherein the gene expression profile of the sample is obtained using a hybridization assay to oligonucleotides contained in a microarray.
35. The method of claim 34, wherein the oligonucleotides comprises nucleic acid molecules at least 95% identical to SEQ ID from Tables 1 and 3.
36. The method of claim 27, wherein the reference expression profile is that of cells derived from patients that do not have asthma disease.
37. The method of claim 27, wherein the drug is selected from the group consisting of symptom relievers and anti-inflammatory drugs for an inflammatory disease condition.
38. A method for inducing a asthma disease-like state in a resident tissue or cell, comprising contacting the tissue or cell with at least one gene from Tables 3 and 4 that induces an asthma disease-like state.
39. The method of claim 38, wherein the resident tissue cell is selected from the group consisting of: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, dendritic cell, synovial cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina propria lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
40. A method for screening drug candidates for treating asthma disease, comprising: a) contacting a resident cell induced by the method of claim 38 with a drug candidate for treating asthma disease; and b) assaying for a pro-inflammatory like state, such that an absence of the pro-inflammatory like state is indicative of the drug candidate being effective in treating asthma disease.
41. A method for inducing a resident tissue cell to mimic asthma disease, comprising modulating the expression of at least one gene from Tables 3 and 4 in the cells.
42. The method of claim 41, wherein the resident tissue cell is selected from the group consisting of: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, dendritic cell, synovial cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina propria lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
43. A method for screening drug candidates for treating asthma disease, comprising: a) contacting the resident tissue cell induced according to the method of claim 41 with a drug candidate for treating asthma disease; and b) assaying for a pro-inflammatory like state, such that an absence of the pro-inflammatory like state is indicative of the drug candidate being effective in treating asthma disease.
44. A method for treating an animal having asthma disease comprising administering a drug identified by the method of claim 43.
45. A drug screening assay comprising: a) administering a test compound to an animal having asthma disease, or a cell composition isolated therefrom; and b) comparing the level of gene expression of at least one gene from Tables 3 and 4 in the presence of the test compound with one or both of the levels of said gene expression in the absence of the test compound or in normal cells; wherein test compounds which cause the level of expression of one or more genes from Tables 3 and 4 to approach normal are candidates for drugs to treat asthma disease.
46. A method for treating an animal having asthma disease comprising administering a compound identified by the assay of claim 45.
47. A pharmaceutical preparation for treating an animal having asthma disease comprising a compound identified by the assay of claim 45 and a pharmaceutically acceptable excipient.
48. A method for identifying a gene that regulates drug response in asthma disease, comprising: a) obtaining a gene expression profile for at least one gene from Tables 3 and 4 in a resident tissue cell induced for a proinflammatory like state in the presence of the candidate drug; and b) comparing the expression profile of said gene to a reference expression profile for said gene in a cell induced for the pro-inflammatory like state in the absence of the candidate drug, wherein genes whose expression relative to the reference expression profile is altered by the drug may identify the gene as a gene that regulates drug response in asthma disease.
49. An expression profile indicative of the presence of asthma disease in a patient, comprising the level of expression of at least one gene of Tables 3 and 4.
50. A microarray comprising probes that hybridize to one or more genes of Tables 3 and 4.
51. A method of diagnosing susceptibility to asthma disease in an individual, comprising screening for an at-risk haplotype of at least one gene or gene region from Tables 3 and 4, or at least one SNP from Table 1 , that is more frequently present in an individual susceptible to asthma disease compared to a control individual, wherein the at-risk haplotype increases risk of asthma disease.
52. The method of claim 51 wherein the risk increase is at least about 20%.
53. A method of diagnosing susceptibility to asthma disease in an individual, comprising screening for an at-risk haplotype of at least one gene from Tables 3 and 4 or comprising at least one SNP from Table 1 , that is more frequently present in an individual susceptible to asthma disease, compared to the frequency of its presence in a control individual, wherein the presence of the at-risk haplotype is indicative of a susceptibility to asthma disease.
54. The method of claim 53 wherein the at-risk haplotype is characterized by the presence of at least one single nucleotide polymorphism from Table 1.
55. The method of claim 53 wherein screening for the presence of an at-risk haplotype in at least one gene from Tables 3 and 4, or comprising at least one SNP from Table 1 , comprises enzymatic amplification of nucleic acid from said individual or amplification using universal oligos on elongation/ligation products.
56. The method of claim 55 wherein the nucleic acid is DNA.
57. The method of claim 55 wherein the DNA is human DNA.
58. The method of claim 53 wherein screening for the presence of an at-risk haplotype in at least one gene from Tables 3 and 4 or comprising at least one SNP from Table 1 comprises: a) obtaining material containing nucleic acid from the individual; (b) amplifying said nucleic acid as for claim 55; and c) determining the presence or absence of an at-risk haplotype in said amplified nucleic acid.
59. The method of claim 58 wherein determining the presence of an at-risk haplotype is performed by electrophoretic analysis.
60. The method of claim 58 wherein determining the presence of an at-risk haplotype is performed by restriction length polymorphism analysis.
61. The method of claim 58 wherein determining the presence of an at-risk haplotype is performed by sequence analysis.
62. The method of claim 58 wherein determining the presence of an at-risk haplotype is performed by hybridization analysis.
63. A kit for diagnosing susceptibility to asthma disease in an individual comprising: primers for nucleic acid amplification of a region of at least one gene from Tables 3 and 4 or a region comprising at least one SNP from Table t
64. The kit of claim 63 wherein the primers comprise a segment of nucleic acids of length suitable for nucleic acid amplification of a target sequence, selected from the group consisting of: single nucleotide polymorphism from Table 1 , primers flanking a SNP from Table 1 , and combinations thereof.
65. A method of diagnosing a susceptibility to asthma disease, comprising detecting an alteration in the expression or composition of a polypeptide encoded by at least one gene from Tables 3 and 4 in a test sample, in comparison with the expression or composition of a polypeptide encoded by said gene in a control sample, wherein the presence of an alteration in expression or composition of the polypeptide in the test sample is indicative of a susceptibility to asthma disease.
66. The method of claim 65, wherein the alteration in the expression or composition of a polypeptide encoded by said gene comprises expression of a splicing variant polypeptide in a test sample that differs from a splicing variant polypeptide expressed in a control sample.
67. A method of treating asthma disease in a patient in need thereof, comprising expressing in vivo at least one gene from Tables 3 and 4 (wild type/non-disease associated allele) in an amount sufficient to treat the disease.
68. A method of claim 67, comprising: a) administering to a patient a vector comprising a gene selected from Tables 3 and 4 that encodes the protein; and b) allowing said protein to be expressed from said gene in said patient in an amount sufficient to treat the disorder.
69. A method of claim 68, wherein said vector is selected from the group consisting of an adenoviral vector, and a lentiviral vector.
70. A method of claim 68, wherein said vector is administered by a route selected from the group consisting of: topical administration, intraocular administration, parenteral administration, intranasal administration, intratracheal administration, intrabronchial administration and subcutaneous administration.
71. A method of claim 68, wherein said vector is a replication-defective viral vector.
72. A method of claim 68, wherein said gene encodes a human protein.
73. A method of treating asthma disease in a patient in need thereof, comprising administering an agent that regulates the expression, activity or physical state of at least one gene or its encoded RNA from Tables 3 and 4 in the patient.
74. A method of claim 73, wherein the encoded protein from said gene comprises an alteration.
75. A method of claim 73, wherein said gene comprises an associated allele, a particular allele of a polymorphic locus, or the like that modulates the expression of the encoded protein.
76. A method of claim 73, wherein said agent is selected from the group consisting of chemical compounds, oligonucleotides, peptides and antibodies.
77. A method of claim 73, wherein said agent is an antisense molecule or interfering RNA.
78. A method of claim 73, wherein said agent is an expression modulator.
79. A method of claim 78, wherein said modulator is an activator.
80. A method of claim 78, wherein said modulator is a repressor.
81. A method of claim 73, wherein said gene comprises an associated allele, a particular allele of a polymorphic locus, or the like that modifies at least one property or function of the encoded protein.
82. A method of claim 81 , wherein the agent modulates at least one property or function of said gene or a polymorphism wherein at least one allele of said polymorphism modifies at least one property or function of the encoded protein.
83. A method for preventing the occurrence of asthma disease in an individual in need thereof, comprising regulating the level of at least one gene from Tables 3 and 4 compared to a control.
84. The method of claim 83 wherein said level is regulated by regulating expression of at least one gene from Tables 3 and 4 using a binding agent, a receptor to said gene, a peptidomimetic, a fusion protein, a prodrug, an antibody or a ribozyme.
85. The method of claim 83 wherein said level is controlled by genetically altering the expression level of at least one gene from Tables 3 and 4, whereby the regulated level of said gene mimics the level in a control individual.
86. A method for monitoring the effectiveness of treatment on the regulation of expression of one or more genes from Tables 3 and 4 at the RNA or protein level, or its enzymatic activity by measuring RNA, protein or enzymatic activity in a sample of peripheral blood or cells derived thereof.
87. A method of diagnosing asthma disease, the predisposition to asthma disease, or the progression of asthma disease, comprising the steps of a) obtaining a biological sample of mammalian body fluid or tissue to be diagnosed; b) determining the amount and/or concentration of at least one polypeptide from Tables 3 and 4 and/or nucleic acids encoding the polypeptide present in said biological sample; and c) comparing the amount and/or concentration of said polypeptide determine in said biological sample with the amount and/or concentration of said polypeptide as determined in a control sample and/or comparing the amount and/or concentration of nucleic acids encoding said polypeptide determined in said biological sample with the amount and/or concentration of nucleic acids encoding said polypeptides measured in a control sample, wherein the difference in the amount of said polypeptides and/or nucleic acids encoding the polypeptides is indicative of asthma disease or the stage of asthma disease.
88. A method according to claim 87, wherein a nucleic acid probe is used for determining the amount and/or concentration of at least one nucleic acid from Tables 3 and 4 encoding the polypeptide.
89. A method according to claim 88, wherein said nucleic acid probe is derived from the nucleic acid sequence depicted in SEQ ID NO: 1 to 4417.
90. A method according to claim 88, wherein said nucleic acid probe comprises nucleic acids hybridizing to the nucleic acid sequence depicted in SEQ ID NO: 1 to 4417, and/or fragments thereof.
91. A method according to claim 88, wherein a PCR technique is employed.
92. A method according to claim 87, wherein a specific antibody is used for determining the amount and/or concentration of at least one polypeptide from Tables 3 and 4.
93. A method according to claim 92, wherein said antibody is selected from the group comprising polyclonal antiserum, polyclonal antibody, monoclonal antibody, antibody fragments, single chain antibodies and diabodies.
94. A method of claim 87, wherein at least five polypeptides or nucleic acids are determined.
95. Use of a method according to claim 87, wherein the diagnosis serves as a basis for prevention and/or monitoring of asthma disease.
96. A method of treatment of asthma disease in a mammal in need thereof, comprising the steps of a) performing steps a) to c) according to claim 87; and b) treating the mammal in need of said treatment; wherein said medical treatment is based on the stage of the disease.
97. A method for determining the phenotype of a cell comprising detecting the differential expression, relative to a normal cell, of at least one gene from Tables 3 and 4.
98. The method of claim 97, wherein said difference in the level of expression of said gene, is of at least a factor of about two.
99. The method of claim 98, including the further step of cloning said genes which are up- or down-regulated.
100. The method of claim 98, including the further step of generating nucleic acid probes for detecting the level of expression of said genes which are up- or down-regulated.
101. A kit for assessing a patient's risk of having or developing asthma disease, comprising: a) detection means for detecting the differential expression, relative to a normal cell, of at least one gene shown in Tables 3 and 4 or the gene product thereof; and b) instructions for correlating the differential expression of said gene or gene product with a patient's risk of having or developing asthma disease.
102. The kit of claim 101 , wherein the detection means includes nucleic acid probes for detecting the level of mRNA of said genes.
103. A kit for assessing a patients risk of having or developing asthma disease, comprising: at least one means for amplifying or detecting a sequence of at least one gene in Tables 3 and 4, or at least one sequence comprising a SNP in Table 1 , wherein the detection means includes nucleic acid probes or primers for detecting the presence or absence of an associated allele, a particular allele of a polymorphic locus, or the like or changes to at least one sequence of Tables 3 and 4 or Table 1 , and (b) instructions for correlating the presence or absence of at least one sequence of Tables 3 and 4 or Table 1 , with a patient's risk of having or developing asthma disease.
104. The kit of claim 101 , wherein the detection means includes an immunoassay for detecting the level of at least one gene product from Tables 3 and 4.
105. A method of assessing a patient's risk of having or developing asthma disease, comprising: a) determining the level of expression of at least one gene from Tables 3 and 4 or gene products thereof, and comparing the level of expression to a normal cell; and b) assessing a patient's risk of having or developing asthma disease, if any, by determining the correlation between the differential expression of said genes or gene products with known changes in expression of said genes measured in at least one patient suffering from asthma disease.
106. A nucleic acid array comprising a solid support comprising nucleic acid probes which selectively hybridize to at least 5 different genes from Tables 3 and 4 or at least 5 different SNPs of Table 1.
107. The array of claim 106, wherein the solid support is selected from the group consisting of paper, membranes, filters, chips, pins, and glass.
108. A method of diagnosing asthma disease in a patient, comprising detecting a nucleic acid molecule encoding at least one protein from Tables 3 and 4 in a fluid or tissue sample from the patient.
109. A method of claim 108, wherein the detection comprises detecting at least one associated allele, particular allele of a polymorphic locus, or the like in the nucleic acid molecule encoding said protein.
110. A method of claim 109, wherein said method comprises hybridizing a probe to said patient's sample of DNA or RNA under stringent conditions which allow hybridization of said probe to nucleic acid comprising said associated allele, a particular allele of a polymorphic locus, or the like, wherein the presence of a hybridization signal indicates the presence of said associated allele, particular allele of a polymorphic locus, or the like, in at least one gene from Tables 3 and 4.
111. A method of claim 110, wherein the patient's DNA or RNA has been amplified and said amplified DNA or RNA is hybridized.
112. A method of claim 111 , wherein said method comprises using a single- stranded conformation polymorphism technique to assay for said associated allele, particular allele of a polymorphic locus, or the like.
113. A method of claim 111 , wherein said method comprises sequencing at least one gene from Tables 3 and 4 in a sample of DNA from a patient.
114. A method of claim 27, wherein said patient's sample of DNA has been amplified or cloned.
115. A method of claim 111 , wherein said method comprises sequencing at least one gene from Tables 3 and 4 in a sample of RNA or DNA from a patient.
116. A method of claim 111 , wherein said method comprises determining the sequence of at least one gene from Tables 3 and 4 by preparing cDNA from RNA taken from said patient and sequencing said cDNA to determine the presence or absence of an associated allele, a particular allele of a polymorphic locus, or the like.
117. A method of claim 110, wherein said method comprises performing an RNAse assay.
118. A method of claim 110, wherein said probe is attached to a microarray or a bead.
119. A method of claim 110, wherein said probes are oligonucleotides.
120. A method of claim 108, wherein said sample is selected from the group consisting of blood, normal tissue and tumor tissue.
121. A method of claim 109, wherein the associated allele, particular allele of a polymorphic locus, or the like is selected from the group consisting of at least one of the SNPs from Table 1 alone or in combination.
122. A method for assaying the presence of a nucleic acid associated with resistance or susceptibility to asthma disease in a sample, comprising: contacting said sample with a nucleic acid recited in claim 1 or claim 5 under stringent hybridization conditions; and detecting a presence of a hybridization complex.
123. A method for diagnosing or determining the predisposition to asthma disease, or the progression of asthma disease, comprising obtaining a sample from a patient; contacting the sample with a nucleic acid of Table 1 ; and detecting the presence or absence of a hybridization complex, wherein the presence or absence of a hybridization complex is a diagnosis of asthma disease.
124. A method for assaying the presence or amount of a polypeptide encoded by a gene of Tables 3 and 4 for use in diagnostics, prognostics, prevention, treatment, or study of asthma disease, comprising: contacting a sample with an antibody of claim that specifically binds to a gene of Tables 3 and 4 under conditions appropriate for binding; and assessing the sample for the presence or amount of binding of the antibody to the polypeptide.
125. A method for diagnosing or prognosticating asthma disease comprising comparing the level of expression or activity of a polypeptide encoded by a gene of Tables 3 and 4 in a test sample from a patient with the level of expression or activity of the same polypeptide in a control sample wherein a difference in the level of expression or activity between the test sample and control sample is indicative of asthma disease.
126. A method for identifying an agent that can alter the level of activity or expression of a polypeptide encoded by a gene of Tables 3 and 4 for use in diagnostics, prognostics, prevention, treatment, or study of asthma disease, comprising: contacting a cell, cell lysate, or the polypeptide, with an agent to be tested; assessing a level of activity or expression of the polypeptide; and comparing the level of activity or expression of the polypeptide with a control sample in an absence of the agent, wherein if the level of activity or expression of the polypeptide in the presence of the agent differs by an amount that is statistically significant from the level in the absence of the agent then the agent alters the activity or expression of the polypeptide.
127. A cosmetic composition for inhibiting asthma disease in a patient, said cosmetic composition comprising a compound that modulates asthma disease.
128. The cosmetic composition of claim 127, wherein said composition is in a form selected from the group consisting of lotions, sprays, ointments, oils, and gels.
129. A method for predicting the efficacy of a drug for treating asthma disease in a human patient, comprising: a) obtaining a sample of cells from the patient; b) obtaining a set of genotypes from the sample, wherein the set of genotypes comprises genotypes of one or more polymorphic loci from Table 1 , 3 or 4; and c) comparing the set of genotypes of the sample with a set of genotypes associated with efficacy of the drug, wherein similarity between the set of genotypes of the sample and the set of genotypes associated with efficacy of the drug predicts the efficacy of the drug for treating asthma disease in the patient.
130. The method of claim 129, wherein the sample of cells is derived from a tissue selected from the group consisting of: the scalp, Gl track, muscle, sebaceous gland, nerve, blood, dermis, epidermis and other skin cells, cutaneous surfaces, respiratory track components, lungs, trachea, bronchi, inner and outer lung coatings, inner and outer trachea coatings, intertrigious areas, genitalia, vessels and endothelium.
131. The method of claim 130, wherein the cells are selected from the group consisting of: smooth muscle cell, neutrophil, dentric cell, T cell, mast cell, CD4+ lymphocyte, B-lymphocyte, eosinophil, monocyte, macrophage, dendritic cell, synovial cell, glial cell, lung cell, basement membrane cell, neutrophilic granulocyte, eosinophilic granulocyte, keratinocyte, lamina propria lymphocyte, intraepithelial lymphocyte, goblet cell, sputum cell, alveolar wall cell, tracheal wall cell, bronchial smooth muscle cell, nerve cell, and epithelial cell.
132. The method of claim 129, wherein the sample is obtained via biopsy.
133. The method of claim 129, wherein the set of genotypes from the sample comprises genotypes of at least two of the polymorphic loci listed in Tables 1 , 3 or 4.
134. The method of claim 129 wherein the set of genotypes from the sample is obtained by hybridization to allele-specific oligonucleotides complementary to the polymorphic loci from Tables 1 , 3 or 4, wherein said allele-specific oligonucleotides are contained on a microarray.
135. The method of claim 134, wherein the oligonucleotides comprise nucleic acid molecules at least 95% identical to SEQ ID from Tables 1 , 3 or 4.
136. The method of claim 129 wherein the set of genotypes from the sample is obtained by sequencing said polymorphic loci in said sample.
137. The method of claim 129, wherein the drug is selected from the group consisting of symptom relievers and drugs for asthma disease.
138. A method of treating asthma disease in a patient in need thereof, comprising administering an agent that regulates the expression, activity or physical state of at least one polypeptide encoded by a gene from Tables 3 and 4 in the patient.
139. A method of claim 138, wherein the encoded protein from said gene comprises an alteration, wherein said alteration is encoded by a polymorphic locus in said gene.
140. A method of claim 138, wherein said gene comprises an associated allele, a particular allele of a polymorphic locus, or the like that modulates the expression of the encoded protein.
141. A method of claim 138, wherein said agent is selected from the group consisting of chemical compounds, oligonucleotides, peptides and antibodies.
142. A method of claim 138, wherein said agent is an antisense molecule or interfering RNA.
143. A method of claim 138, wherein said agent is an expression modulator.
144. A method of claim 143, wherein said modulator is an activator.
145. A method of claim 143, wherein said modulator is a repressor.
146. A method of claim 138, wherein said gene comprises an associated allele, a particular allele of a polymorphic locus, or the like that modifies at least one property or function of the encoded protein.
147. A kit for assessing a patient's risk of having or developing asthma disease, comprising: a) a detection means for detecting the genotype of at least one polymorphic locus shown in Tables 1 , 3 or 4; and b) instructions for correlating the genotype of said at least one polymorphic locus with a patient's risk of having or developing asthma disease.
148. The kit of claim 147, wherein the detection means includes nucleic acid probes for detecting the genotype of said at least one polymorphic locus.
149. A method of assessing a patient's risk of having or developing asthma disease, comprising: a) selecting at least one polymorphic locus from Tables 1 , 3 or 4; b) determining a genotype for at least one polymorphic locus from Tables 1 , 3 or 4 in a patient; c) comparing said genotype of b) to a genotype for at least one polymorphic locus from Tables 1 , 3 or 4 that is associated with asthma disease; and d) assessing the patient's risk of having or developing asthma disease, wherein said patient has a higher risk of having or developing asthma disease if the genotype for at least one polymorphic locus from Table 1 , 3 or 4 in said patient is the same as said genotype for at least one polymorphic locus from Table 1 , 3 or 4 that is associated with asthma disease.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07871349A EP2082343A2 (en) | 2006-11-02 | 2007-11-02 | Genemap of the human genes associated with asthma disease |
| CA002667476A CA2667476A1 (en) | 2006-11-02 | 2007-11-02 | Genemap of the human genes associated with asthma disease |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US85600306P | 2006-11-02 | 2006-11-02 | |
| US60/856,003 | 2006-11-02 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008085601A2 true WO2008085601A2 (en) | 2008-07-17 |
| WO2008085601A3 WO2008085601A3 (en) | 2008-12-11 |
Family
ID=39609231
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/083522 WO2008085601A2 (en) | 2006-11-02 | 2007-11-02 | Genemap of the human genes associated with asthma disease |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP2082343A2 (en) |
| CA (1) | CA2667476A1 (en) |
| WO (1) | WO2008085601A2 (en) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2221066A1 (en) * | 2009-02-18 | 2010-08-25 | Sanofi-Aventis | Use of VgII3 activity modulator for the modulation of adipogenesis |
| WO2011050350A1 (en) * | 2009-10-23 | 2011-04-28 | The Translational Genomics Research Institute | Methods and kits used in identifying glioblastoma |
| WO2011001135A3 (en) * | 2009-07-03 | 2011-06-30 | University College Cardiff Consultants Limited | Diagnosis and treatment of alzheimer's disease |
| WO2011151783A1 (en) * | 2010-05-31 | 2011-12-08 | University Hospital Of Basel | Method for assaying lower respiratory tract infection or inflammation |
| US20130165345A1 (en) * | 2010-08-31 | 2013-06-27 | Universitaet Zuerich | Method for assaying peritonitis in humans |
| US9388215B2 (en) | 2013-03-15 | 2016-07-12 | Shenzhen Hightide Biopharmaceutical, Ltd. | Compositions and methods of using islet neogenesis peptides and analogs thereof |
| DK178879B1 (en) * | 2012-05-26 | 2017-04-24 | Hans Bisgaard | Method of diagnosing asthma and other wheezing disorders. |
| EP3321280A1 (en) * | 2016-11-10 | 2018-05-16 | Deutsches Krebsforschungszentrum Stiftung des Öffentlichen Rechts | Immune modulators for reducing immune-resistance in melanoma and other proliferative diseases |
| WO2018087285A1 (en) * | 2016-11-10 | 2018-05-17 | Deutsches Krebsforschungszentrum Stiftung des öffentlichen Rechts | Or10h1 antigen binding proteins and uses thereof |
| US10426777B2 (en) | 2009-10-23 | 2019-10-01 | The Translational Genomics Research Institute | Methods used to treat cancer |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020119474A1 (en) * | 1997-03-03 | 2002-08-29 | Drazen Jeffrey M. | Diagnosing asthma patients predisposed to adverse beta-agonist reactions |
-
2007
- 2007-11-02 CA CA002667476A patent/CA2667476A1/en not_active Abandoned
- 2007-11-02 EP EP07871349A patent/EP2082343A2/en not_active Withdrawn
- 2007-11-02 WO PCT/US2007/083522 patent/WO2008085601A2/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020119474A1 (en) * | 1997-03-03 | 2002-08-29 | Drazen Jeffrey M. | Diagnosing asthma patients predisposed to adverse beta-agonist reactions |
Non-Patent Citations (4)
| Title |
|---|
| DRYSDALE ET AL.: 'Complex promoter and coding region beta2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness' PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES USA vol. 97, September 2000, pages 10483 - 10488, XP002213210 * |
| HOGERKORP ET AL.: 'CD-44-stimulated human B cells express transcripts specifically involved in immunomodulation and inflammation as analyzed by DNA microarrays' BLOOD vol. 101, March 2003, pages 2307 - 2313, XP008109776 * |
| MILLER ET AL.: 'Life stress and diminished expression of genes encoding glucocorticoid receptor and beta2-adrenergic receptor in children with asthma' PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES USA vol. 103, April 2006, pages 5496 - 5501, XP008110743 * |
| PAHL ET AL.: 'Pharmacogenomics of Asthma' CURRENT PHARMACEUTICAL DESIGN vol. 12, September 2006, pages 3195 - 3206, XP008110720 * |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010095096A1 (en) * | 2009-02-18 | 2010-08-26 | Sanofi-Aventis | Use of vgii3 activity modulator for the modulation of adipogenesis |
| EP2221066A1 (en) * | 2009-02-18 | 2010-08-25 | Sanofi-Aventis | Use of VgII3 activity modulator for the modulation of adipogenesis |
| CN102355911A (en) * | 2009-02-18 | 2012-02-15 | 赛诺菲 | Use of vgii3 activity modulator for the modulation of adipogenesis |
| US8852939B2 (en) | 2009-02-18 | 2014-10-07 | Sanofi | Use of Vgll3 activity modulator for the modulation of adipogenesis |
| EP2891723A1 (en) * | 2009-07-03 | 2015-07-08 | University College Cardiff Consultants Limited | Diagnosis and treatment of Alzheimer's disease |
| WO2011001135A3 (en) * | 2009-07-03 | 2011-06-30 | University College Cardiff Consultants Limited | Diagnosis and treatment of alzheimer's disease |
| WO2011050350A1 (en) * | 2009-10-23 | 2011-04-28 | The Translational Genomics Research Institute | Methods and kits used in identifying glioblastoma |
| US10426777B2 (en) | 2009-10-23 | 2019-10-01 | The Translational Genomics Research Institute | Methods used to treat cancer |
| WO2011151783A1 (en) * | 2010-05-31 | 2011-12-08 | University Hospital Of Basel | Method for assaying lower respiratory tract infection or inflammation |
| US11402390B2 (en) | 2010-05-31 | 2022-08-02 | University Hospital Of Basel | Method for assaying lower respiratory tract infection or inflammation |
| US20130165345A1 (en) * | 2010-08-31 | 2013-06-27 | Universitaet Zuerich | Method for assaying peritonitis in humans |
| DK178879B1 (en) * | 2012-05-26 | 2017-04-24 | Hans Bisgaard | Method of diagnosing asthma and other wheezing disorders. |
| US9388215B2 (en) | 2013-03-15 | 2016-07-12 | Shenzhen Hightide Biopharmaceutical, Ltd. | Compositions and methods of using islet neogenesis peptides and analogs thereof |
| US9738695B2 (en) | 2013-03-15 | 2017-08-22 | Shenzhen Hightide Biopharmaceutical, Ltd. | Compositions and methods of using islet neogenesis peptides and analogs thereof |
| US10899815B2 (en) | 2013-03-15 | 2021-01-26 | Shenzhen Hightide Biopharmaceutical, Ltd. | Compositions and methods of using islet neogenesis peptides and analogs thereof |
| EP3321280A1 (en) * | 2016-11-10 | 2018-05-16 | Deutsches Krebsforschungszentrum Stiftung des Öffentlichen Rechts | Immune modulators for reducing immune-resistance in melanoma and other proliferative diseases |
| WO2018087285A1 (en) * | 2016-11-10 | 2018-05-17 | Deutsches Krebsforschungszentrum Stiftung des öffentlichen Rechts | Or10h1 antigen binding proteins and uses thereof |
| WO2018087276A1 (en) * | 2016-11-10 | 2018-05-17 | Deutsches Krebsforschungszentrum Stiftung des öffentlichen Rechts | Or10h1 modulators and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2008085601A3 (en) | 2008-12-11 |
| EP2082343A2 (en) | 2009-07-29 |
| CA2667476A1 (en) | 2008-07-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20090305900A1 (en) | Genemap of the human genes associated with longevity | |
| US20100291551A1 (en) | Genemap of the human associated with crohn's disease | |
| WO2008024114A1 (en) | Genemap of the human genes associated with schizophrenia | |
| US20100120627A1 (en) | Genemap of the human genes associated with psoriasis | |
| WO2008085601A2 (en) | Genemap of the human genes associated with asthma disease | |
| US20100120628A1 (en) | Genemap of the human genes associated with adhd | |
| US20100144538A1 (en) | Genemap of the human genes associated with schizophrenia | |
| US20100099083A1 (en) | Crohn disease susceptibility gene | |
| US20090081658A1 (en) | Genemap of the human genes associated with crohn's disease | |
| US20090181380A1 (en) | Genemap of the human genes associated with crohn's disease | |
| WO2009026116A2 (en) | Genemap of the human genes associated with longevity | |
| WO2009039244A2 (en) | Genemap of the human genes associated with crohn's disease | |
| CA2676415A1 (en) | Genemap of the human genes associated with endometriosis | |
| US20140011863A1 (en) | Methods and compositions for assessment of pulmonary function and disorders | |
| WO2008055196A2 (en) | Genemap of the human genes associated with male pattern baldness | |
| Baron et al. | DNA sequence variants in epithelium-specific ETS-2 and ETS-3 are not associated with asthma | |
| KR101617612B1 (en) | SNP Markers for hypertension in Korean | |
| KR20150092937A (en) | SNP Markers for hypertension in Korean | |
| BELOUCHI et al. | Patent 2620082 Summary |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07871349 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2667476 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007871349 Country of ref document: EP |