WO2008081191A1 - Improved method for the wittig reaction in the preparation of carboprost - Google Patents
Improved method for the wittig reaction in the preparation of carboprost Download PDFInfo
- Publication number
- WO2008081191A1 WO2008081191A1 PCT/GB2008/000020 GB2008000020W WO2008081191A1 WO 2008081191 A1 WO2008081191 A1 WO 2008081191A1 GB 2008000020 W GB2008000020 W GB 2008000020W WO 2008081191 A1 WO2008081191 A1 WO 2008081191A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- reaction
- carboprost
- triethylsilyloxy
- methyl ester
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 55
- 238000002360 preparation method Methods 0.000 title claims abstract description 8
- 238000007239 Wittig reaction Methods 0.000 title claims description 11
- DLJKPYFALUEJCK-MRVZPHNRSA-N carboprost Chemical compound CCCCC[C@](C)(O)\C=C\[C@H]1[C@H](O)C[C@H](O)[C@@H]1C\C=C\CCCC(O)=O DLJKPYFALUEJCK-MRVZPHNRSA-N 0.000 title claims description 10
- 229960003395 carboprost Drugs 0.000 title claims description 6
- QQCOAAFKJZXJFP-XAYIDPIISA-N 15-methyl-15R-PGF2alpha methyl ester Chemical compound CCCCC[C@](C)(O)\C=C\[C@H]1[C@H](O)C[C@H](O)[C@@H]1C\C=C/CCCC(=O)OC QQCOAAFKJZXJFP-XAYIDPIISA-N 0.000 claims abstract description 20
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 42
- 238000006243 chemical reaction Methods 0.000 claims description 34
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 32
- 239000002904 solvent Substances 0.000 claims description 19
- SIPUZPBQZHNSDW-UHFFFAOYSA-N diisobutylaluminium hydride Substances CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 claims description 18
- DFGFNBAGHJOLER-CTDMZEHRSA-N (3ar,4r,5r,6as)-4-[(e,3s)-3-hydroxy-3-methyloct-1-enyl]-5-triethylsilyloxy-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-2-one Chemical compound O1C(=O)C[C@@H]2[C@@H](/C=C/[C@@](C)(O)CCCCC)[C@H](O[Si](CC)(CC)CC)C[C@@H]21 DFGFNBAGHJOLER-CTDMZEHRSA-N 0.000 claims description 17
- 230000032050 esterification Effects 0.000 claims description 17
- 238000005886 esterification reaction Methods 0.000 claims description 17
- 239000007818 Grignard reagent Substances 0.000 claims description 16
- 150000004795 grignard reagents Chemical class 0.000 claims description 16
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 15
- 239000011734 sodium Substances 0.000 claims description 15
- 229910052708 sodium Inorganic materials 0.000 claims description 15
- MLOSJPZSZWUDSK-UHFFFAOYSA-N 4-carboxybutyl(triphenyl)phosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(CCCCC(=O)O)C1=CC=CC=C1 MLOSJPZSZWUDSK-UHFFFAOYSA-N 0.000 claims description 14
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 13
- 229960001760 dimethyl sulfoxide Drugs 0.000 claims description 13
- DCFKHNIGBAHNSS-UHFFFAOYSA-N chloro(triethyl)silane Chemical compound CC[Si](Cl)(CC)CC DCFKHNIGBAHNSS-UHFFFAOYSA-N 0.000 claims description 12
- ODZPKZBBUMBTMG-UHFFFAOYSA-N sodium amide Chemical compound [NH2-].[Na+] ODZPKZBBUMBTMG-UHFFFAOYSA-N 0.000 claims description 12
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 12
- -1 triethylsilyloxy group Chemical group 0.000 claims description 12
- 238000004128 high performance liquid chromatography Methods 0.000 claims description 11
- DICZUTMNXOMHQD-UHFFFAOYSA-N 3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-2-one Chemical compound C1CCC2OC(=O)CC21 DICZUTMNXOMHQD-UHFFFAOYSA-N 0.000 claims description 10
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 claims description 10
- 239000008096 xylene Substances 0.000 claims description 10
- 238000003747 Grignard reaction Methods 0.000 claims description 9
- AZWXAPCAJCYGIA-UHFFFAOYSA-N bis(2-methylpropyl)alumane Chemical compound CC(C)C[AlH]CC(C)C AZWXAPCAJCYGIA-UHFFFAOYSA-N 0.000 claims description 9
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 claims description 7
- 229940125758 compound 15 Drugs 0.000 claims description 7
- 238000000926 separation method Methods 0.000 claims description 5
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 claims description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 4
- 229940125797 compound 12 Drugs 0.000 claims description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 claims description 4
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 claims description 2
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 claims description 2
- 229940126543 compound 14 Drugs 0.000 claims description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 2
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 30
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 24
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 24
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 22
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 21
- 239000012071 phase Substances 0.000 description 21
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 19
- 239000000243 solution Substances 0.000 description 19
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 16
- 239000000203 mixture Substances 0.000 description 16
- 239000012312 sodium hydride Substances 0.000 description 16
- 229910000104 sodium hydride Inorganic materials 0.000 description 16
- 230000015572 biosynthetic process Effects 0.000 description 15
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 15
- 239000000047 product Substances 0.000 description 15
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 14
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 14
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- 239000012044 organic layer Substances 0.000 description 12
- 229910000027 potassium carbonate Inorganic materials 0.000 description 11
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 11
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 10
- 238000003786 synthesis reaction Methods 0.000 description 10
- DLJKPYFALUEJCK-WJDSMEDOSA-N carboprost Chemical compound CCCCC[C@](C)(O)\C=C\[C@H]1[C@@H](O)C[C@H](O)[C@@H]1C\C=C/CCCC(O)=O DLJKPYFALUEJCK-WJDSMEDOSA-N 0.000 description 9
- 239000012535 impurity Substances 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- CCERQOYLJJULMD-UHFFFAOYSA-M magnesium;carbanide;chloride Chemical compound [CH3-].[Mg+2].[Cl-] CCERQOYLJJULMD-UHFFFAOYSA-M 0.000 description 8
- 239000011541 reaction mixture Substances 0.000 description 7
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000012267 brine Substances 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- 150000003180 prostaglandins Chemical class 0.000 description 6
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- 238000002835 absorbance Methods 0.000 description 5
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 239000000377 silicon dioxide Substances 0.000 description 5
- 125000004469 siloxy group Chemical group [SiH3]O* 0.000 description 5
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- 239000012043 crude product Substances 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 4
- 238000002953 preparative HPLC Methods 0.000 description 4
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 3
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 150000001875 compounds Chemical class 0.000 description 3
- XWCQLLDGXBLGMD-UHFFFAOYSA-M magnesium;pentane;bromide Chemical compound [Mg+2].[Br-].CCCC[CH2-] XWCQLLDGXBLGMD-UHFFFAOYSA-M 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- QGCVCXUCRBGHGQ-UPPVYHDHSA-N (3ar,4r,5r,6as)-5-hydroxy-4-[(e)-3-oxobut-1-enyl]-3,3a,4,5,6,6a-hexahydrocyclopenta[b]furan-2-one Chemical compound O1C(=O)C[C@@H]2[C@@H](/C=C/C(=O)C)[C@H](O)C[C@@H]21 QGCVCXUCRBGHGQ-UPPVYHDHSA-N 0.000 description 2
- IFUYEJWYVORTEU-UHFFFAOYSA-N 3,3a,4,5,6,6a-hexahydro-2h-cyclopenta[b]furan-2-ol Chemical compound C1CCC2OC(O)CC21 IFUYEJWYVORTEU-UHFFFAOYSA-N 0.000 description 2
- 230000005526 G1 to G0 transition Effects 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 239000011449 brick Substances 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- 239000013058 crude material Substances 0.000 description 2
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 2
- 238000011067 equilibration Methods 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- QQCOAAFKJZXJFP-GANRVJIKSA-N methyl (Z)-7-[(1S,2S,3S,5R)-3,5-dihydroxy-2-[(E,3R)-3-hydroxy-3-methyloct-1-enyl]cyclopentyl]hept-5-enoate Chemical group CCCCC[C@@](C)(O)\C=C\[C@@H]1[C@@H](O)C[C@@H](O)[C@H]1C\C=C/CCCC(=O)OC QQCOAAFKJZXJFP-GANRVJIKSA-N 0.000 description 2
- 239000012053 oil suspension Substances 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- 239000012047 saturated solution Substances 0.000 description 2
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 2
- 238000004611 spectroscopical analysis Methods 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 229940086542 triethylamine Drugs 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- IFYARVOVAASOIE-HIUAYJIISA-N (3ar,4r,5r,6as)-4-[(e,3s)-3-hydroxy-3-methyloct-1-enyl]-5-triethylsilyloxy-3,3a,4,5,6,6a-hexahydro-2h-cyclopenta[b]furan-2-ol Chemical compound O1C(O)C[C@@H]2[C@@H](/C=C/[C@@](C)(O)CCCCC)[C@H](O[Si](CC)(CC)CC)C[C@@H]21 IFYARVOVAASOIE-HIUAYJIISA-N 0.000 description 1
- 108010051913 15-hydroxyprostaglandin dehydrogenase Proteins 0.000 description 1
- 102100030489 15-hydroxyprostaglandin dehydrogenase [NAD(+)] Human genes 0.000 description 1
- HERPQHFQUUPWPS-IIWOPCFASA-N CC(/C=C/[C@H]([C@@H](C1)[C@H](C2)OC1=O)[C@@H]2OC)=O Chemical compound CC(/C=C/[C@H]([C@@H](C1)[C@H](C2)OC1=O)[C@@H]2OC)=O HERPQHFQUUPWPS-IIWOPCFASA-N 0.000 description 1
- URQUOJQQUFEMPL-CPBAPSRBSA-N CC[N](CC)(CC)O[C@H](C[C@@H]([C@@H]1C2)OC2=O)[C@@H]1/C=C/C(C)=O Chemical compound CC[N](CC)(CC)O[C@H](C[C@@H]([C@@H]1C2)OC2=O)[C@@H]1/C=C/C(C)=O URQUOJQQUFEMPL-CPBAPSRBSA-N 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- JOOMLFKONHCLCJ-UHFFFAOYSA-N N-(trimethylsilyl)diethylamine Chemical compound CCN(CC)[Si](C)(C)C JOOMLFKONHCLCJ-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 208000018525 Postpartum Hemorrhage Diseases 0.000 description 1
- 125000000746 allylic group Chemical group 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- DLJKPYFALUEJCK-IIELGFQLSA-N carboprost Chemical compound CCCCC[C@](C)(O)\C=C\[C@H]1[C@H](O)C[C@H](O)[C@@H]1C\C=C/CCCC(O)=O DLJKPYFALUEJCK-IIELGFQLSA-N 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 238000006345 epimerization reaction Methods 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- UQEAIHBTYFGYIE-UHFFFAOYSA-N hexamethyldisiloxane Chemical compound C[Si](C)(C)O[Si](C)(C)C UQEAIHBTYFGYIE-UHFFFAOYSA-N 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 239000012454 non-polar solvent Substances 0.000 description 1
- 239000004533 oil dispersion Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000003198 secondary alcohol group Chemical group 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- JLTRXTDYQLMHGR-UHFFFAOYSA-N trimethylaluminium Chemical compound C[Al](C)C JLTRXTDYQLMHGR-UHFFFAOYSA-N 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 238000000825 ultraviolet detection Methods 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C405/00—Compounds containing a five-membered ring having two side-chains in ortho position to each other, and having oxygen atoms directly attached to the ring in ortho position to one of the side-chains, one side-chain containing, not directly attached to the ring, a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, and the other side-chain having oxygen atoms attached in gamma-position to the ring, e.g. prostaglandins ; Analogues or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/347—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups
- C07C51/367—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups by introduction of functional groups containing oxygen only in singly bound form
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
Definitions
- Prostaglandins are a family of 20- carbon fatty acids found in virtually all mammalian cells 1 . They are highly biologically active. Natural prostaglandins possess 5 an allylic, secondary alcohol group at C- 15. This group can be oxidized into a ketone
- the first compound of this type was 15(S)-15-methyl-prostaglandin F 2 ⁇ synthesized by Upjohn chemists (Ernest W. Yankee, Udo Axen and Gordon L. Bundy: Journal of the American Chemical Society / 96:18 / September 4, 1974). 15(S)-15-methyl-prostaglandin F 2 ⁇ as its tromethamine
- Benzoate enone (2) was treated with methyl magnesium bromide at -78°c in ether or tetrahydrofuran as solvent or with trimethyl aluminium in benzene at ambient temperature to give 3(RS) with an epimeric ratio of 1 : 1.
- carboprost methyl ester (10), lactol (13) and carboprost (9) relates in each case to the racemate (RS) and/or each individual R or S isomer. That is to say the process may be used to prepare the racemate of (10) and/or the individual R or S isomer, or any combination thereof.
- the separation of the racemate may occur at any stage of the process, for example by separation of compound 13, of compound 9 or compound 10 (each selected independently) into individual R and S isomers.
- the method of the invention may be performed using either the R or S isomer of lactol 13 (each selected independently).
- sodium methylsulf ⁇ nylmethide is conveniently obtained by the reaction of sodium hydride with dimethylsulphoxide.
- sodium methylsulfinylmethide can be prepared using sodium amide in place of sodium hydride. There are a number of safety hazards associated with the use of sodium hydride; in contrast sodium amide is relatively easy to handle on a large scale.
- the esterification is conveniently effected using dimethyl sulphate and potassium carbonate or methyl iodide and potassium carbonate, more conveniently using methyl iodide and potassium carbonate to yield carboprost methyl esterl 0.
- Use of ethereal diazomethane on large scale is cumbersome and also poses a major safety hazard.
- Convenient solvents used in the esterification include acetone.
- the lactol 13 is conveniently prepared by reduction of (3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -4-[(1E, 3 S)-3-hydroxy-3 -methyl oct-1-en-l-yl] hexahydro — 2H-cyclopenta [b] furan-2-one (12) (12)
- diisobutylaluminium hydride 1.5M solution in toluene
- diisobutylaluminium hydride 1.5M solution in toluene
- about 3.5 moles of diisobutylaluminium hydride and about one molar equivalent of product 12 may be used for the reduction of 12 to 13 as compared to 4.6moles to 5.4 moles of diisobutylaluminium hydride as described by Yankee et al. (op cit).
- furan-2-one (12) is conveniently prepared by Grignard reaction of, for example methyl magnesium chloride with (3aR, 4R, 5R, 6aS)-5-triethylsilyloxy-4- [(lE)-3-oxooct-l-en-l-yl] hexahydro-2H-cyclopenta [b] furan-2-one (11) (triethylsilyloxy PG enone) that had been dissolved in a convenient solvent such as tetrahydrofuran, and cooled at between -6O 0 C and -78 0 C.
- a convenient solvent such as tetrahydrofuran
- the ratio of compound 11 to Grignard reagent used can be about 1 :5 moles. We have found that this ratio is required for the reaction to go to completion. A slight reduction in the molar quantity of Grignard reagent eg. a molar ratio of 1 :4 does not take the reaction to completion. We have carried out this reaction with higher ratio of Grignard reagent and observed that higher quantity of Grignard reagent added does not play any significant role, on the contrary it may increases the impurity formation. This all contrasts with the method of Yankee et al. (op cit) who used a ratio of 1 :16 moles.
- compound 12 is obtained as an isomeric mixture and then separated into individual isomers 12a and 12b.
- Methyl magnesium chloride (Grignard Reagent) as 3.0M solution in tetrahydrofuran was added to (3aR, 4R, 5R, 6aS)-5-triethylsilyloxy-4- [(lE)-3-oxooct- 1-en-l-yl] hexahydro-2H-cyclopenta [b] furan-2-one (11) (triethylsilyloxy PG enone) that had been dissolved in tetrahydrofuran and cooled to -78°c.
- the ratio of 11 to Grignard reagent used was 1:5 moles, as compared to Yankee et al. where the authors used a ratio of 1 : 16 moles.
- the crude product was obtained as RS mixture (12a, 12b).
- the R: S ratio was assigned to be 40:60 that was unambiguously confirmed, the details of this are discussed in relation to Scheme III hereinafter. Based on the HPLC data we decided to look into the possibility of reducing the impurities and consequently increase the yield.
- the ratio of 11 to Grignard reagent used is 1 :5 moles.
- solvents like hexane, pentane and heptane the reaction did not go to completion using, 5.0 moles of Grignard reagent.
- solvents like xylene (o, m, p or mixed) and toluene the reaction proceeded to completion.
- Tetrahydrofuran, Toluene or Xylene (o, m, p or mixed) was used to dissolve 5 the lactol product.
- the 5-trans isomer was formed to an extent of less than 3.0%.
- Yankee et al describe the addition of lactol 13(RS) to ylide at ambient temperature; our observation is that when the lactol is added to the ylide at ambient temperature the amount of 5- trans isomer obtained was around 6.0 to 8.0%.
- the Pharmacopoeia states the limits of 5- trans isomer to be not more than 4.0%.
- the Grignard product (12) thus obtained is reduced with diisobutyl aluminium hydride to yield (13), which is then subjected to Wittig reaction by reacting the ylide obtained from 4-carboxybutyltriphenyl-phosphonium bromide and sodium methylsulfinylmethide (obtained by reaction of sodium hydride and dimethyl sulphoxide) and the lactol (13) at -25°c to 10°c, preferably at -5°c to 5°c to yield 9(RS).
- the second method describes the Grignard reaction of triethyl silyloxy PG enone (11) with the Grignard reagent in xylene (o, m, p or mixed) or toluene as solvent at -78°c to yield 12(RS).
- the Grignard product (12) thus obtained is reduced with diisobutyl aluminium hydride to yield (13), which is then subjected to Wittig reaction by reacting the ylide obtained from 4-carboxybutyltriphenyl-phosphonium bromide and sodium methylsulfinylmethide (obtained by reaction of Sodium amide and dimethyl sulphoxide) with the lactol (13) at -25°c to 10°c preferably at-5°c to 5°c to yield 9(RS).
- the invention describes the use of preparative scale HPLC for the separation of R and S isomers. Once the initial purification is done on silica column, the fractions obtained as RS mixture is taken up for separation on a preparative column using a preparative HPLC.
- the invention describes normal phase and a reverse phase method to separate the isomers. In the normal phase the column was packed with Chiralpak AD material supplied from Diacel, Japan. Mobile phase used was either Heptane: Ethanol, Heptane: Isopropyl alcohol, Hexane: Ethanol and Hexane: isopropyl alcohol. Heptane: Ethanol or Heptane: Isopropyl alcohol gave the best results.
- UV detection is 200 -220 nm, preferably 216nm.
- Inertsil Prep ODS 50mm (LD) column mobile phase water/ methanol/ acetonitrile
- YMC C8 column mobile phase water/ methanol
- Kromasil C 18 column mobile phase water/methanol/acetonitrile
- Merck Lichroprep mobile phase water/ methanol
- the methods of this invention for the synthesis of 15-Methyl-PGF 2 ⁇ Methyl ester 10(RS) as disclosed above may include one or more of the following features:
- the method provides a feasible, production scale synthesis of 15-Methyl- PGF 2 ⁇ Methyl ester 10(RS).
- the invention further describes a novel route (Scheme II) for the synthesis of the intermediate 12.
- the invention further describes a new approach (Scheme III) to the synthesis of either (15R)-15-Methyl-PGF 2 ⁇ Methyl ester (10a) or (15S)-15-Methyl-PGF 2 ⁇ Methyl ester i.e. Carboprost methyl ester (10b).
- the Grignard product (12) is obtained as an isomeric (RS) mixture i.e. 12a and
- the lactol 13a or 13b was further reacted at -15°c to 10°c preferably at -5°c to 2°c with the ylide obtained either from 4-carboxybutyltriphenyl-phosphonium bromide and sodium methylsulfinylmethide (obtained by reaction of sodium hydride and dimethyl sulphoxide) or from 4-carboxybutyltriphenyl-phosphonium bromide and sodium methylsulfinylmethide (obtained by reaction of sodium amide and dimethyl sulphoxide) to yield 9a or 9b.
- the ylide obtained either from 4-carboxybutyltriphenyl-phosphonium bromide and sodium methylsulfinylmethide (obtained by reaction of sodium hydride and dimethyl sulphoxide) or from 4-carboxybutyltriphenyl-phosphonium bromide and sodium methylsulfinylmethide (obtained by reaction
- HPLC instrument used for analysis is Shimadzu SPD - 1OA.
- the instruments used were Agilent 1100 series supplied by Agilent Technologies and Hipersep LAB LC 50 supplied by Novasep.
- Optical rotations were recorded using Jasco DIP- 370 digital polarimeter.
- 1 H or 13 C NMR were recorded using a Bruker Avance DPX 200 NMR instrument.
- Anhydrous solvents were generally prepared by known procedure .
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
Abstract
A process for the preparation of carboprost methyl ester (figure (10)).
Description
IMPROVED METHOD FOR THE WITTIG REACTION IN THE PREPARATION OF CARBOPROST
Prostaglandins are a family of 20- carbon fatty acids found in virtually all mammalian cells1. They are highly biologically active. Natural prostaglandins possess 5 an allylic, secondary alcohol group at C- 15. This group can be oxidized into a ketone

10 Thus a number of prostaglandins with different substitutions at C- 15 were synthesized in order to maintain biological activity. The first compound of this type was 15(S)-15-methyl-prostaglandin F2α synthesized by Upjohn chemists (Ernest W. Yankee, Udo Axen and Gordon L. Bundy: Journal of the American Chemical Society / 96:18 / September 4, 1974). 15(S)-15-methyl-prostaglandin F2αas its tromethamine
15 salt is used for post partum haemorrhage indication.
The method described by Yankee et al starts with the Grignard reaction of benzoate-protected enone (2). This benzoate enone (2) was synthesized from optically pure iodo lactone (1) analogous to a known literature process2.

Benzoate enone (2) was treated with methyl magnesium bromide at -78°c in ether or tetrahydrofuran as solvent or with trimethyl aluminium in benzene at ambient temperature to give 3(RS) with an epimeric ratio of 1 : 1.

The reduction of 3(RS) with diisobutylaluminium hydride gave a lactol product 4(RS).

Alternatively 3 was subjected to hydrolysis using sodium methoxide in methanol to give 5 (RS), this 5 is then protected as trimethyl silyl ether using tri methylsilyldiethylamine to yield a trimethyl silyl protected ether 6 (RS).

(5)

Diisobutylaluminium hydride reduction of 5 and 6 gave lactol product 7(RS) and (RS) respectively.

The reaction of each of these lactols i.e. 4(RS), 7(RS) and 8(RS) with the ylide prepared from 4-carboxybutyltriphenylphosphonium bromide and sodium methylsulfinylmethide gave prostaglandin 9(RS). Esterification of 9(RS) with diazomethane gave Methyl (Z)-7- (IR, 2R, 3R, 5S)- 3,5-dihydroxy-2- [(E)- (3 RS)-3- hydroxy-3 -methyl- 1-octenyl) cyclopentyl]-5- heptenoate i.e. carboprost methyl ester 10(RS) in 28-37% overall yield starting from benzoate enone (2). The two epimers 15R and 15S are separated by column chromatography using silica gel as stationary phase and methylene chloride: acetone as the mobile phase.

(9)

(10)
We have now devised improved processes for the production of carboprost methyl ester. In a first aspect of the invention we provide a process for the preparation of carboprost methyl ester 10
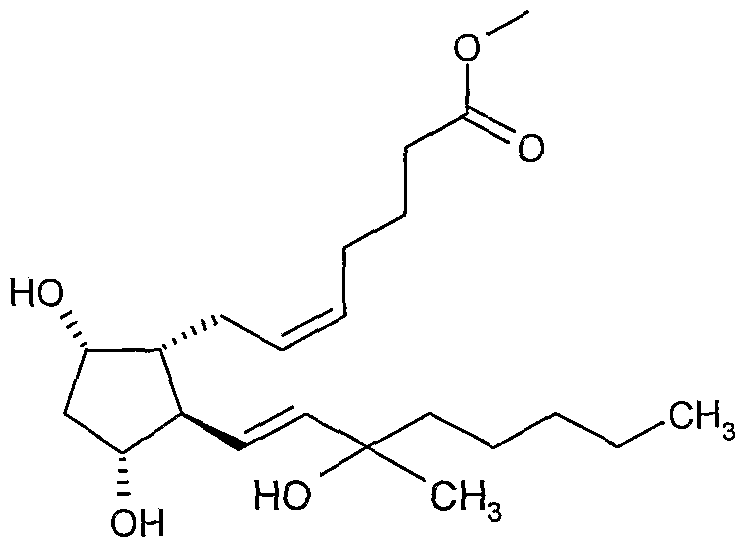
(10)
which process comprises the Wittig reaction of lactol 13

wherein OTES is a triethylsilyloxy group with an ylide of formula Ph3P=CH-(CH2)3-COOH at between -250C and +10°C to give carboprost 9

followed by esterification to give carboprost methyl ester 10.
It is to be understood that reference above to carboprost methyl ester (10), lactol (13) and carboprost (9) relates in each case to the racemate (RS) and/or each individual R or S isomer. That is to say the process may be used to prepare the racemate of (10) and/or the individual R or S isomer, or any combination thereof. The separation of the racemate may occur at any stage of the process, for example by separation of compound 13, of compound 9 or compound 10 (each selected independently) into individual R and S isomers.
Any convenient method may be used to separate a racemic mixture, a particular method is preparative scale HPLC. Convenient HPLC methods are as set out in reference 3 incorporated herein.
For the avoidance of doubt the method of the invention may be performed using either the R or S isomer of lactol 13 (each selected independently).
Yankee et al (op cit) conduct the Wittig reaction at ambient temperature (200C or more). We have found that this results in formation of about 6-8% of the unwanted trans-isomer. In contrast we have found that use of a lower temperature i.e. between -250C and +1O0C results in less than 3% of the unwanted trans-isomer. The Wittig reaction is more conveniently carried out at between -50C and +5°C.
The ylide of formula Ph3P=CH-(CH2)3-COOH is conveniently formed by the reaction of 4-carboxybutyltriphenyl phosphonium bromide and sodium methylsulfinylmethide. In turn sodium methylsulfϊnylmethide is conveniently obtained by the reaction of sodium hydride with dimethylsulphoxide. In a further aspect of the invention we have found that sodium methylsulfinylmethide can be prepared using sodium amide in place of sodium hydride. There are a number of safety hazards associated with the use of sodium hydride; in contrast sodium amide is relatively easy to handle on a large scale.
The esterification is conveniently effected using dimethyl sulphate and potassium carbonate or methyl iodide and potassium carbonate, more conveniently using methyl iodide and potassium carbonate to yield carboprost methyl esterl 0. This contrasts with the method of Yankee et.al who used ethereal diazomethane for esterification. Use of ethereal diazomethane on large scale is cumbersome and also poses a major safety hazard.
Convenient solvents used in the esterification include acetone.
In a further aspect of the invention where the process is used to prepare carboprost methyl ester IORS this is conveniently further separated into individual carboprost methyl esters 10a and 10b


dissolved in a convenient solvent such as tetrahydrofuran, toluene or xylene, preferably tetrahydrofuran, using diisobutylaluminium hydride (1.5M solution in toluene) at between -60°c and -78°c. We have found that about 3.5 moles of diisobutylaluminium hydride and about one molar equivalent of product 12 may be used for the reduction of 12 to 13 as compared to 4.6moles to 5.4 moles of diisobutylaluminium hydride as described by Yankee et al. (op cit). (3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -4-[(1E, 3 S)-3-hydroxy-3 -methyl oct-1- en-l-yl] hexahydro -2H-cyclopenta [b] furan-2-one (12) is conveniently prepared by Grignard reaction of, for example methyl magnesium chloride with (3aR, 4R, 5R, 6aS)-5-triethylsilyloxy-4- [(lE)-3-oxooct-l-en-l-yl] hexahydro-2H-cyclopenta [b] furan-2-one (11) (triethylsilyloxy PG enone) that had been dissolved in a convenient solvent such as tetrahydrofuran, and cooled at between -6O0C and -780C. We have found that the ratio of compound 11 to Grignard reagent used can be about 1 :5 moles. We have found that this ratio is required for the reaction to go to completion. A slight reduction in the molar quantity of Grignard reagent eg. a molar ratio of 1 :4 does not take the reaction to completion. We have carried out this reaction with higher ratio of Grignard reagent and observed that higher quantity of Grignard reagent added does not play any significant role, on the contrary it may increases the impurity formation. This all contrasts with the method of Yankee et al. (op cit) who used a ratio of 1 :16 moles.
In a further aspect of the invention (3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -A- [(1E, 3S)-3-hydroxy-3-methyl oct-1-en-l-yl] hexahydro -2H-cyclopenta [b] furan-2- one (12) is prepared as shown in Scheme II hereinafter by Grignard reaction with compound 15. In turn compound 15 is prepared by protection of the hydroxyl group of compound 14 with triethyl silyl chloride (triethylchlorosilane) in for example pyridine or triethyl amine to yield compound 15 (in 95 - 98 % yield.). Reaction of
Grignard reagent (pentyl magnesium bromide, 2.0M solution in diethyl ether) with compound 15 gave compound 12RS, the structure of which was confirmed by spectroscopic data. , In this case the epimeric ratio of R: S isomer obtained was 50:50.
In a further aspect of the invention compound 12 is obtained as an isomeric mixture and then separated into individual isomers 12a and 12b.
The invention will now be illustrated but not limited by reference to the following specific description and Examples.
Example 1

(11) (12a) (12b)

(10b)
SCHEME I (above)
Methyl magnesium chloride (Grignard Reagent) as 3.0M solution in tetrahydrofuran was added to (3aR, 4R, 5R, 6aS)-5-triethylsilyloxy-4- [(lE)-3-oxooct- 1-en-l-yl] hexahydro-2H-cyclopenta [b] furan-2-one (11) (triethylsilyloxy PG enone) that had been dissolved in tetrahydrofuran and cooled to -78°c. The ratio of 11 to
Grignard reagent used was 1:5 moles, as compared to Yankee et al. where the authors used a ratio of 1 : 16 moles.
The crude product was obtained as RS mixture (12a, 12b). The HPLC analysis of (12) using a Supelcosil silica column and mobile phase as Heptane :Isopropyl alcohol (94:6) at 200 nm absorbance revealed two peaks due to two epimers 12a and 12b in a ratio of 40:60 and a bunch of impurities formed to an extent of 30% (by area %). These impurities were not characterized. The R: S ratio was assigned to be 40:60 that was unambiguously confirmed, the details of this are discussed in relation to Scheme III hereinafter. Based on the HPLC data we decided to look into the possibility of reducing the impurities and consequently increase the yield. HPLC data revealed an impurity formation at an retention time of 7.5 to 8.5 to an extent of 30% (by area) so it was clear that if we were able to control or bring down this impurity, the yield of 12(RS) would subsequently increase. We also wanted to investigate increasing the percentage of S isomer.
Analyzing the various parameters it appeared that solvent was playing a major role. Various non-polar solvents like heptane, pentane, hexane, xylene (o, m, p or mixed) and toluene were used to dissolve the substrate i.e. triethylsilyloxy PG enone (11). Methyl magnesium chloride (Grignard Reagent) as 3.0M solution in tetrahydrofuran was added at -78°c to the solution of 11 which was dissolved in heptane, pentane, hexane, xylene (o, m, p or mixed) or toluene. The ratio of 11 to Grignard reagent used is 1 :5 moles. In solvents like hexane, pentane and heptane the reaction did not go to completion using, 5.0 moles of Grignard reagent. However, in solvents like xylene (o, m, p or mixed) and toluene the reaction proceeded to completion. The HPLC analysis [Supelcosil silica column and mobile phase as Heptane :Isopropyl alcohol (94:6) at 200 nm] of (12) obtained as a RS mixture by changing the solvent revealed that the two epimers 12a and 12b were formed in a ratio of 30:70 and the impurities were formed only to an extent of 4-6% (by area %). The crude product 12 as a RS mixture (12a & 12b) dissolved in tetrahydrofuran was reduced to lactol 13 (RS) using diisobutylaluminium hydride (1.5M solution in toluene) at -78°c. We used 3.5 moles of diisobutylaluminium hydride for the reduction of 12 to 13(RS) [as compared to 4.6moles to 5.4 moles of diisobutylaluminium hydride used for the reduction of 12 as described by Yankee et al.]
Treatment of the lactol 13(RS) with the ylide prepared from 4- carboxybutyltriphenyl-phosphonium bromide and sodium methylsulfmylmethide (obtained by reaction of sodium hydride and dimethyl sulphoxide) at -5°c to 2°c gave 9(RS). Tetrahydrofuran, Toluene or Xylene (o, m, p or mixed) was used to dissolve 5 the lactol product. The 5-trans isomer was formed to an extent of less than 3.0%. Yankee et al describe the addition of lactol 13(RS) to ylide at ambient temperature; our observation is that when the lactol is added to the ylide at ambient temperature the amount of 5- trans isomer obtained was around 6.0 to 8.0%. The Pharmacopoeia states the limits of 5- trans isomer to be not more than 4.0%.
10 Yankee et al have reported the synthesis of sodium methylsulfinylmethide, which is then reacted with 4-carboxybutyltriphenyl-phosphonium bromide to give an ylide. The sodium methylsulfinylmethide is obtained by the reaction of sodium hydride and dimethyl sulphoxide. However handling of sodium hydride on large scale poses a major safety hazard. Sodium hydride is usually available as a 55-60%
15 suspension in oil. Sodium hydride (as a suspension in oil) is slurried with petroleum ether (60-80) to remove the oil suspension. On a large scale the petroleum ether used to free the sodium hydride from the oil suspension has to be siphoned prior to the reaction, which is very cumbersome. We used sodium amide in place of sodium hydride. Sodium amide is easy to handle on large scale. Treatment of the lactol
20 13(RS) at -25°c to 10°c preferably at -5°c to 5°c with the ylide prepared from 4- carboxybutyltriphenyl-phosphonium bromide, sodium amide and dimethyl sulphoxide gave 9(RS) with the formation of 5- trans isomer below 3.0%.
The cleavage of the triethyl silyl group occurred during the product isolation (work up).
25 The carboprost acid 9(RS) obtained after Wittig reaction is subjected to esterification using dimethyl sulphate and potassium carbonate or methyl iodide and potassium carbonate preferably methyl iodide and potassium carbonate to yield 10(RS). These contrasts with that reported by Yankee etal who used ethereal diazomethane for esterification. Use of ethereal diazomethane on large scale is
30 cumbersome and also poses a major safety hazard.
Thus, 15-Methy l-PGF2α Methyl ester [(Carboprost methyl ester) 10(RS)] was synthesized as shown in Scheme I in two ways. The first method started with the Grignard reaction of triethyl silyloxy PG enone (11) and methyl magnesium chloride in THF as solvent at -78°c. The Grignard product (12) thus obtained is reduced with
diisobutyl aluminium hydride to yield (13), which is then subjected to Wittig reaction by reacting the ylide obtained from 4-carboxybutyltriphenyl-phosphonium bromide and sodium methylsulfinylmethide (obtained by reaction of sodium hydride and dimethyl sulphoxide) and the lactol (13) at -25°c to 10°c, preferably at -5°c to 5°c to yield 9(RS). Esterification of 9(RS) using methyl iodide and potassium carbonate in acetone as solvent gave 10 (RS) in 55% overall yield starting from Triethyl silyloxy PG enone (11) with an R: S ratio of 40:60.
The second method describes the Grignard reaction of triethyl silyloxy PG enone (11) with the Grignard reagent in xylene (o, m, p or mixed) or toluene as solvent at -78°c to yield 12(RS). The Grignard product (12) thus obtained is reduced with diisobutyl aluminium hydride to yield (13), which is then subjected to Wittig reaction by reacting the ylide obtained from 4-carboxybutyltriphenyl-phosphonium bromide and sodium methylsulfinylmethide (obtained by reaction of Sodium amide and dimethyl sulphoxide) with the lactol (13) at -25°c to 10°c preferably at-5°c to 5°c to yield 9(RS). Esterification of 9(RS) using methyl iodide and potassium carbonate in acetone as solvent gave 10 (RS) in 75% overall yield starting from triethyl silyl PG enone (11) with an R: S ratio of 30:70.
In contrast Yankee et al separated the 15-Methyl-PGF2α Methyl ester obtained as a RS mixture into individual R and S isomer on silica column using methylene chloride: acetone as mobile phase. Column chromatography of the RS mixture gave various fractions containing either pure R isomer or mixture of RS isomers and pure S isomer. To further separate the RS mixture again into R isomer and S isomer completely one has to carry out column chromatography repeatedly. As evident on large-scale synthesis this is not feasible. Also routine column chromatography using silica stationary phase it was not possible to isolate the S trans isomer.
The invention describes the use of preparative scale HPLC for the separation of R and S isomers. Once the initial purification is done on silica column, the fractions obtained as RS mixture is taken up for separation on a preparative column using a preparative HPLC. The invention describes normal phase and a reverse phase method to separate the isomers. In the normal phase the column was packed with Chiralpak AD material supplied from Diacel, Japan. Mobile phase used was either Heptane: Ethanol, Heptane: Isopropyl alcohol, Hexane: Ethanol and Hexane: isopropyl alcohol. Heptane: Ethanol or Heptane: Isopropyl alcohol gave the best results. The ratios ranging from anywhere between 80:20 to 95:5 were used but preferably 90:10. UV
detection is 200 -220 nm, preferably 216nm. In the reverse phase method Inertsil Prep ODS 50mm (LD) column (mobile phase water/ methanol/ acetonitrile) or YMC C8 column (mobile phase water/ methanol) or Kromasil C 18 column (mobile phase water/methanol/acetonitrile) or Merck Lichroprep (mobile phase water/ methanol) was used. The best results were however obtained with Inertsil Prep ODS 50mm (LD) column the mobile phase was water: methanol: acetonitrile with ratios ranging from 50:15:35 to 70:15:15. However best results were obtained with a ratio of water: methanol: acetonitrile as 60:05:35 at an absorbance 200 -220 nm preferably 200nm. Using Prep HPLC the R and S isomer were efficiently separated. Also the 15(S) -5- trans isomer which otherwise was never isolated before in pure form was also isolated and characterised.
The methods of this invention for the synthesis of 15-Methyl-PGF2α Methyl ester 10(RS) as disclosed above may include one or more of the following features:
1. The method provides a feasible, production scale synthesis of 15-Methyl- PGF2α Methyl ester 10(RS).
2. Improved ratio of R: S isomer of 15-Methyl-PGF2α Methyl ester (10) can be obtained 30:70.
3. The isomeric purification is efficient which in turn leads to higher yields of (15S)-15-Methy -PGF2α Methyl ester i.e. Carboprost methyl ester. 4. The impurity (other side products) formation is only to the extent of 4-6%.
5. The use of sodium amide during Wittig reaction and/or use of methyl iodide during esterification step, thus making it a safer production scale synthesis of 15-Methyl-PGF2α Methyl ester 10(RS).
6. The quantity of reagents i.e. Grignard reagent and/or Diisobutyl aluminium hydride used is less than used in the prior art leading to a more complete consumption of starting material(s).
The disadvantages of the method for the synthesis of 15-Methyl-PGF2α Methyl ester (10) described in the prior art by Ernest W. Yankee, Udo Axen and Gordon L. Bundy: Journal of the American Chemical Society / 96:18 / September 4, 1974 include:
1. The inherent drawbacks do not readily make it a feasible method for industrial scale synthesis.
2. Use of reagents like of Sodium hydride or diazomethane does not make it a safe process on industrial scale.
3. Large excess of reagents like Grignard reagent or Diisobutyl aluminium hydride is used in the reaction.
4. The efficiency of isomers purification is very low.
5. The ratio of R: S isomer of 15-Methyl-PGF2α Methyl ester (10) obtained is 50:50, thus leading to lower yields of (15 S)- 15 -Methyl -PGF2α Methyl ester i.e. Carboprost methyl ester.
Example 2
The invention further describes a novel route (Scheme II) for the synthesis of the intermediate 12.


SCHEME II
The scheme II starts with the protection of the hydroxy 1 group of compoundl4 with triethyl silyl chloride (triethylchlorosilane) in pyridine or triethyl amine to yield compoundl5 in 95 - 98 % yield. Reaction of Grignard reagent (pentyl magnesium bromide, 2.0M solution in diethyl ether) with compound 15 gave compound 12(RS)5 the structure of which was confirmed by spectroscopic data. , In this case the epimeric ratio of R: S isomer obtained was 50:50.
Example 3
The invention further describes a new approach (Scheme III) to the synthesis of either (15R)-15-Methyl-PGF2α Methyl ester (10a) or (15S)-15-Methyl-PGF2α Methyl ester i.e. Carboprost methyl ester (10b).

(11) (12a) (12b)

(9a)
(10a)

(10b) (9b)
SCHEME III
5 The Grignard product (12) is obtained as an isomeric (RS) mixture i.e. 12a and
12b respectively by the reaction of triethyl silyl protected PG enone (11) with the Grignard reagent (Methyl magnesium chloride). In this approach the invention describes the isomers purification at this stage rather than separating the isomers (10a and 10b) at the end. Product 12 obtained as a RS mixture is separated into individual
10 isomers i.e. 12a and 12b respectively using preparative HPLC. Herein we describe two methods a normal phase and a reverse phase to separate the isomers 12a and 12b respectively. In the normal phase, the column was packed with Chiralpak AD material supplied from Diacel, Japan. Mobile phase used was Heptane: Isopropyl alcohol or Heptane: ethanol. The ratio ranging anywhere between 80:20 was used at absorbance
15 200 -220 nm. However the best result was obtained with the mobile phase as Heptane: isopropyl alcohol in a ratio of 94:6 at absorbance 210nm. In the reverse phase method Zorbax ODS 5μm column was used. The mobile phase was water: methanol in a ratio 20:80 at an absorbance of 200nm.
The isomers 12a and 12b thus separated were then reduced with diisobutyl aluminium hydride to yield either 13a or 13b respectively. The lactol 13a or 13b was further reacted at -15°c to 10°c preferably at -5°c to 2°c with the ylide obtained either from 4-carboxybutyltriphenyl-phosphonium bromide and sodium methylsulfinylmethide (obtained by reaction of sodium hydride and dimethyl sulphoxide) or from 4-carboxybutyltriphenyl-phosphonium bromide and sodium methylsulfinylmethide (obtained by reaction of sodium amide and dimethyl sulphoxide) to yield 9a or 9b. The esterification of 9a or 9b with methyl iodide and potassium carbonate gave (15R)-15-Methyl -PGF2α Methyl ester (10a) or (15S)-15- Methyl -PGF2α Methyl ester i.e. Carboprost methyl ester (10b). It is interesting to note that under the reaction conditions employed no epimerization of 10a to epimeric mixture of 10(RS) or 10b to an isomeric mixture of 10(RS) occurred. It was observed that in this case the chemical purification of 10a or 10b was much easy as the other side products or impurities were formed to a lesser extent.
Experimental:
All melting points reported are corrected. The HPLC instrument used for analysis is Shimadzu SPD - 1OA. For preparative work the instruments used were Agilent 1100 series supplied by Agilent Technologies and Hipersep LAB LC 50 supplied by Novasep. Optical rotations were recorded using Jasco DIP- 370 digital polarimeter. 1H or 13C NMR were recorded using a Bruker Avance DPX 200 NMR instrument. Anhydrous solvents were generally prepared by known procedure .
(3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -4- [(1E, 3S)-3-hydroxy-3-methyI oct-1-en-l-yl] hexahydro -2H-cyclopenta [b] furan-2-one, [12(RS)]. 1.011 Kg (2.646 mol) of Triethyl silyloxy PG enone (11) was dissolved in dry toluene or xylene (o, m, p or mixed). To this was slowly added 4.41 litres (13.23mol) of methyl magnesium chloride in tetrahydrofuran (3.0 molar solution) under nitrogen atmosphere at -60°c to -80°c. TLC monitored the progress of the reaction. On complete consumption of starting material the reaction mass is dumped in a saturated solution of ammonium chloride. The reaction mass was then filtered on hyflo bed. The organic layer was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were washed with brine and concentrated to give oil.
o
Yield 1.032 Kg (98.47%). The 1H and 13C NMR values matched with the reported values, [α] D -18°(c 2.62, methanol).
(3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -4-[(1E, 3S)-3-hydroxy-3-methyI oct-l-en-l-yl] hexahydro -2H-cyclopenta [b] furan-2-one, [12(RS)] was also synthesized by following the same procedure mentioned above but instead using Dry THF as a solvent.
(3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -4-[(1E, 3S)-3-hydroxy-3-methyI oct-l-en-l-yl] hexahydro -2H-cyclopenta [b] furan-2-ol, [13(RS)]. To a stirred solution of 775.0gms (1.95 Moles) of (3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -4-[(1E, 3S)-3-hydroxy-3-methyl oct-l-en-l-yl] hexahydro -2H-cyclopenta [b] furan-2-one, [12(RS)] in 9.0 litres of dry THF was added 4.566 litres (6.84 moles) of diisobutyl aluminium hydride (1.5M solution in toluene) at -78°c. The completion of the reaction was monitored by TLC. The reaction mixture was then dumped in ice-cold water. The reaction mass was then filtered on hyflo bed. The organic layer was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were washed with brine and concentrated to give an oil. Yield 774.0 Gms (99.48%).
Methyl (Z)-7- (IR, 2R, 3R, 5S)- 3,5-dihydroxy-2- [(E)- (3 RS)-3-hydroxy-
3-methyl-l-octenyl) cyclopentyl]-5- heptenoate, [10(RS)]. Sodium hydride (60% dispersion in oil) 473.0gms (11.82 moles) was treated with pet ether to make it free from oil dispersion. To this was added 4.73 litres of dimethyl sulphoxide and heated to 75°c under nitrogen atmosphere. The resulting dark clear solution was cooled to ambient temperature and to this was added 2.588 Kg (5.83 moles) of 4-carboxybutyl triphenyl-phosphonium bromide, the resulting brick red coloured solution was cooled to -5°c to 5°c. To this ylide, 774.0 gms (1.94 moles) of (3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -4-[(lE, 3S)-3-hydroxy-3-methyl oct-l-en-l-yl] hexahydro -2H- cyclopenta [b] furan-2-ol, [13(RS)] was added and the solution stirred overnight at - 2°c. The reaction was dumped in ice water and acidified with 10% solution of sodium bisulphate. After equilibration, the aqueous phase was extracted with ethyl acetate. The organic layers were combined and washed with brine. Concentration of this organic layer under vacuum gave crude product 9(RS). The product was not purified
at this stage and taken up for esterification directly. The product was dissolved in acetone and to this was added 703.0 (5.0 moles) Gms of potassium carbonate followed by methyl iodide 636.0 ml (10.216moles). The reaction mixture was stirred overnight. The reaction mixture is filtered through celite bed and is then dumped in water and extracted with ethyl acetate. The organic layer is then concentrated to give crude 10(RS). This crude material is then purified by column chromatography to yield chemically pure 10(RS).
Methyl (Z)-7- (IR, 2R, 3R, 5S)- 3,5-dihydroxy-2- [(E)- (3 RS)-3-hydroxy- 3-methyl-l-octenyl) cyclopentyl]-5- heptenoate, [10(RS)]. Sodium amide
245.0gms (6.28 moles) was added to 2.5 litres of dimethyl sulphoxide followed by 1.4 Kg (3.16 moles) of 4-carboxybutyl triphenyl-phosphonium bromide, the resulting brick red solution was heated to 50°c under nitrogen atmosphere for 1.0 to 2.0 hrs. The resulting dark clear solution was cooled to -5°c to 5°c. To this ylide, 252.0 gms (0.633 moles) of (3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -4-[(1E, 3S)-3-hydroxy-3- methyl oct-1-en-l-yl] hexahydro -2H-cyclopenta [b] furan-2-ol, [13(RS)] was added and the solution stirred overnight at -5°c to 5°c.The reaction was dumped in ice water and acidified with 10% solution of sodium bisulphate. After equilibration, the aqueous phase was extracted with ethyl acetate. The organic layers were combined and washed with brine. Concentration of this organic layer under vacuum gave crude product. The product 9(RS) was not purified at this stage and taken up for esterification directly. The product was dissolved in acetone and to this was added 703.0 (5.0 moles) Gms of potassium carbonate followed by methyl iodide 636.0 ml (10.216moles). The reaction mixture was stirred overnight. The reaction mixture is filtered through celite bed and is then dumped in water and extracted with ethyl acetate. The organic layer is then concentrated to give crude 10(RS). This crude material is then purified by column chromatography to give chemically pure 10(RS).
The synthesis of (15RS)-15-Methyl-PGF2α methyl ester [10(RS)] was carried out in two ways. In the first method the Grignard reaction of Triethyl silyloxy PG enone (11) with methyl magnesium chloride was carried out in THF as a solvent. The product thus obtained was reduced with diisobutyl aluminium hydride and subsequently reacted with ylide formed using 4-carboxybutyl triphenyl-phosphonium bromide and sodium methylsulfϊnylmethide (obtained by reaction of sodium hydride
or sodium amide and dimethyl sulphoxide). The free acid obtained, on esterification gave 10(RS) in 55% overall yield starting from Triethyl silyl PG enone (11) with a ratio of R: S being 40:60. In The second method Grignard reaction of Triethyl silyl PG enone (11) with methyl magnesium chloride was carried out in Xylene (o, m, p or mixed) or toluene as a solvent. The product thus obtained was reduced with diisobutyl aluminium hydride and subsequently reacted with ylide formed using 4-carboxybutyl triphenyl-phosphonium bromide and sodium methylsulfinylmethide (obtained by reaction of sodium hydride or sodium amide and dimethyl sulphoxide). The free acid obtained, on esterification gave 10(RS) in 75% overall yield starting from Triethyl silyl PG enone (11) with a ratio of R: S being 30:70. However in both the cases the 5- trans isomer was formed below 3%.
(3aR, 4R, 5R, 6aS)- 5-triethylsiIyloxy -4- [l(E)-3- oxobut-1-en-l-yl] hexahydro-2H cyclopenta [b] furan -2-one (15). 20.0 Gms (0.0956 moles) of (3aR, 4R, 5R, 6aS) -5- Hydroxy -4- (E)-3-oxo-but-l-enyl-hexahydro-cyclopenta [b] furan- 2-one (14) was added to 200 ml of dry pyridine. To this was added 26.0gms (0.172 moles) of triethylchlorosilane and the reaction mixture heated at 60°c. The progress of the reaction was monitored by TLC. The reaction mixture was dumped in water and extracted with ethyl acetate. The organic layer was then washed with brine and concentrated to yield 30.3 gms (97.74 %) of (15).
(3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -4-[(1E, 3S)-3-hydroxy-3-methyl oct-1-en-l-yl] hexahydro -2H-cyclopenta [b] furan-2-one, [12(RS)]. lOgms (0.030 moles) of (3aR, 4R, 5R, 6aS)- 5 -triethylsilyloxy -4- [l(E)-3- oxobut-1-en-l-yl] hexahydro-2H cyclopenta [b] furan -2-one (15) was dissolved in 100ml of toluene and cooled to -78°c.To this was added 77.0 ml (0.15 moles) of pentyl magnesium bromide (2.0 molar solution in diethyl ether) under nitrogen atmosphere. Completion of the reaction was monitored by TLC. The reaction was then quenched in a saturated solution of ammonium chloride and extracted with ethyl acetate. The organic layer was washed with brine and concentrated to yield 12(RS). The 1H and 13C NMR values matched with the reported values, [α] D-17° (C 2.57, methanol).
(3aR, 4R, 5R, 6as)-5- triethylsilyloxy -4-[(1E, 3S)-3-hydroxy-3-methyl oct- 1-en-l-yl] hexahydro -2H-cyclopenta [b] furan-2-one, [12(RS)] obtained as a mixture of R: S isomer is separated into individual isomers using a preparative HPLC. The individual isomers 12a and 12b thus obtained are then reduced to 13a and 13b using diisobutyl aluminium hydride. Wittig reaction and esterification gave 10a and 10b respectively. The experimental conditions including molar proportions and solvents used for synthesizing 10a and 10b from 12a and 12b respectively (Scheme III) remain the same as that used to synthesize 10 as an RS mixture from 12(RS). The yield of Methyl (Z)-I- (IR, 2R, 3R, 5S)- 3,5-dihydroxy-2- [(E)- (3 R)-3-hydroxy-3- methyl- 1-octenyl) cyclopentyl]-5- heptenoate, (10a) and Methyl (Z)-7- (IR, 2R, 3R, 5S)- 3,5-dihydroxy-2- [(E)- (3 S)-3-hydroxy-3-methyl-l-octenyl) cyclopentyl]-5- heptenoate (10b) obtained was 70-75% Starting from 12a and 12b respectively.
References: 1. (a) S. Bergstrδm, Science, 157,382 (1967); (b) "Prostaglandins" in
Proceedings of Second Nobel Symposium, Stockholm, June, 1966, S. Bergstrδm, and B Samuelsson, Ed., Almquist and Wiksell, Gebers Forlag AB, Stockholm, 1967.
2. (a) EJ Corey, N.M. Weinshenker, T.K. Schaaf, and W. Huber, J.Amer.Chem. Soc, 91, 5677 (1969). (b) EJ. Corey, T.K. Schaaf, W.Huber, U. Koelleker and N.M. Weinshenker, ibid., 92, 397 (1970). (c) EJ. Corey, S.M. Albonico, T.K. Schaaf, U. Koelleker and R.K. Varma, ibid., 93, 1491 (1971).
3. Practical HPLC method development - L.R. Synder, J.Kirkland and J.L. Glajch, John Wiley & sons Inc.
4. Vogel's Textbook of Practical Organic Chemistry, Fifth Edition. Longman Group UK Ltd. 1989.
Claims
1. A process for the preparation of carboprost methyl ester (10)

(10)
which process comprises the Wittig reaction of lactol (13)

wherein OTES is a triethylsilyloxy group, with an ylide of formula Ph3P=CH-(CEE)3-COOH at between -25°C and +10°C to give carboprost (9)

followed by esterification to give carboprost methyl ester 10.
2. A method as claimed in claim 1 wherein the ylide of formula Ph3P=CH-(CHE)3-COOH is formed by the reaction of 4-carboxybutyltriphenyl- phosphonium bromide and sodium methylsulfmylmethide.
3. A method as claimed in claim 2 wherein sodium methylsulfmylmethide is prepared by the reaction of sodium amide and dimethyl sulphoxide.
4. A process as claimed in claim 1 wherein carboprost methyl ester 10(RS) is produced and then separated into carboprost methyl esters 1OR and 1OS

5. A process as claimed in claim 1 wherein the lactol 13 is prepared by reduction of (3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -4-[(lE, 3 S)-3-hydroxy-3 -methyl oct-l-en- 1-yl] hexahydro -2H-cyclopenta [b] furan-2-one (12) 
6. A process as claimed in claim 5 wherein (3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -4-[(1E, 3 S)-3-hydroxy-3 -methyl oct-1-en-l-yl] hexahydro -2H-cyclopenta [b] furan- 2-one (12) is dissolved in a solvent selected from tetrahydrofuran, toluene or xylene, and about 3.5 molar equivalents of diisobutylaluminium hydride are used at between -60°c and -78°c.
7. A process as claimed in claim 6 (3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -4- [(1E, 3S)-3-hydroxy-3-methyl oct-1-en-l-yl] hexahydro -2H-cyclopenta [b] furan-2- one (12) is prepared by Grignard reaction of triethylsilyloxy PG enone (11)

8. A process as claimed in claim 7 wherein the ratio of triethylsilyloxy PG enone (11) to Grignard reagent is about 1:5 moles.
9. A process as claimed in any one of claims 5-8 wherein (3aR, 4R, 5R, 6aS)-5- triethylsilyloxy -4-[(1E, 3 S)-3-hydroxy-3 -methyl oct-1-en-l-yl] hexahydro -2H- cyclopenta [b] furan-2-one (12) is prepared by Grignard reaction with compound 15 
10. A process as claimed in claim 9 wherein compound 15 is prepared by protection of the hydroxyl group of compound 14 with triethyl silyl chloride 5 (triethylchlorosilane).

11. A process as claimed in any one of claims 1-3 wherein either the R or S isomer of compound 13 is used in the process.
10 12. A process as claimed in claim 5 or claim 6 wherein either the R or S isomer of compound 12 is used in the process.
13. A process as claimed in claim 12 wherein compound 12 is firstly separated into individual isomers and either the R or S isomer is used in the process.
14. A process according to any of the previous claims wherein any separation into 15 individual isomers is carried out using preparative scale HPLC.
15. A process according to any one of the previous claims for the preparation of carboprost methyl ester 1OR.
16. A process according to any one of the previous claims for the preparation of carboprost methyl ester 1OS.
20 17. A process according to any one of claims 1 -3 for the preparation of carboprost methyl ester lORS.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/521,821 US20100041912A1 (en) | 2007-01-05 | 2007-01-05 | Method for the wittig reaction in the preparation of carboprost |
| EP08701739A EP2118046A1 (en) | 2007-01-05 | 2008-01-04 | Improved method for the wittig reaction in the preparation of carboprost |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN29CH2007 | 2007-01-05 | ||
| IN29/CHE/2007 | 2007-01-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008081191A1 true WO2008081191A1 (en) | 2008-07-10 |
Family
ID=39205016
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2008/000020 WO2008081191A1 (en) | 2007-01-05 | 2008-01-04 | Improved method for the wittig reaction in the preparation of carboprost |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20100041912A1 (en) |
| EP (1) | EP2118046A1 (en) |
| WO (1) | WO2008081191A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011095990A3 (en) * | 2010-02-03 | 2012-01-12 | Fdc Limited | Process for the purification of prostaglandins and analogues thereof |
| WO2012010089A1 (en) * | 2010-07-21 | 2012-01-26 | 上海天伟生物制药有限公司 | Preparation method for and uses of carboprost tromethamine crystal |
| WO2017093770A1 (en) | 2015-12-01 | 2017-06-08 | CHINOIN Gyógyszer és Vegyészeti Termékek Gyára Zrt. | Process for the preparation of carboprost and its tromethamine salt |
| CN110117242A (en) * | 2018-02-06 | 2019-08-13 | 广州楷模生物科技有限公司 | The synthetic method of Carboprost, tromethamine Carboprost |
| CN115160202A (en) * | 2022-07-30 | 2022-10-11 | 广州楷石医药有限公司 | Preparation method of carboprost tromethamine and intermediate thereof |
| CN115710209A (en) * | 2021-08-23 | 2023-02-24 | 佳和桂科技股份有限公司 | Method and intermediates for preparing carboprost and carboprost tromethamine, and carboprost tromethamine prepared therefrom |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3673263A (en) * | 1968-03-29 | 1972-06-27 | Procter & Gamble | Dihydro-{62 -santalol and process for preparing dihydro-{62 -santalol from 3-endo-methyl-3-exo(4{40 -methyl-5{40 -hydroxypentyl) norcamphor |
| US3872149A (en) * | 1973-06-25 | 1975-03-18 | Syntex Inc | 9-Hydroxy prosta-5-cis, 11,13-trans-trienoic acids and derivatives thereof |
| US3917644A (en) * | 1973-08-03 | 1975-11-04 | Lilly Co Eli | Azetidinesulfenic acids |
| DE2435331C2 (en) * | 1974-07-23 | 1983-08-18 | Hoechst Ag, 6230 Frankfurt | Analogs of prostanoic acids and processes for their preparation, as well as pharmaceuticals containing these analogs of prostanoic acids |
| US4415501A (en) * | 1981-12-16 | 1983-11-15 | American Cyanamid Company | Alkenylzirconium reagents useful for prostaglandin analog synthesis |
| US5274130A (en) * | 1991-09-03 | 1993-12-28 | R-Tech Ueno Ltd. | Process for production of prostaglandin intermediates |
| US5478945A (en) * | 1992-07-15 | 1995-12-26 | Taisho Pharmaceutical Co., Ltd. | Thiazoline derivatives |
| EP1598336A1 (en) * | 2004-05-20 | 2005-11-23 | Laboratorios Del Dr. Esteve, S.A. | Regioselective hydroxylation, functionalisation and protection of spirolactams |
-
2007
- 2007-01-05 US US12/521,821 patent/US20100041912A1/en not_active Abandoned
-
2008
- 2008-01-04 EP EP08701739A patent/EP2118046A1/en not_active Withdrawn
- 2008-01-04 WO PCT/GB2008/000020 patent/WO2008081191A1/en active Application Filing
Non-Patent Citations (13)
| Title |
|---|
| "Almquist and Wiksell", 1967, GEBERS FORLAG AB |
| "Prostaglandins", PROCEEDINGS OF SECOND NOBEL SYMPOSIUM, June 1966 (1966-06-01) |
| "Vogel's Textbook of Practical Organic Chemistry", 1989, LONGMAN GROUP UK LTD. |
| CAREY, F.A. AND SUNDBERG, R.J.: "Advanced Organic Chemistry, second edition, Part B: Reactions and Synthesis", 1985, PLENUM PRESS, NEW Y0RK, XP002474590 * |
| CAREY, F.A., SUNDBERG, R .J: "Advanced Organic Chemistry, second edition, Part B: Reactions and Synthesis", 1985, PLENUM PRESS, pages: 71 - 73 |
| E.J COREY, N.M. WEINSHENKER, T.K. SCHAAF, W. HUBER, J.AMER.CHEM. SOC., vol. 91, 1969, pages 5677 |
| ERNEST W. YANKEE, UDO AXEN, GORDON L. BUNDY, JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 96, 4 September 1974 (1974-09-04), pages 18 |
| ERNEST W. YANKEE, UDO AXEN, GORDON L. BUNDY, JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 96, no. 18, 4 September 1974 (1974-09-04), pages 5865 - 76 |
| L.R. SYNDER, J.KIRKLAND, J.L. GLAJCH: "Practical HPLC method development", JOHN WILEY & SONS INC. |
| MARCH, JERRY: "Advanced Organic Chemistry; Reactions, Mechanisms, and Structure", 1985, WILEY-INTERSCIENCE, pages: 850 - 851 |
| MARCH, JERRY: "Advanced Organic Chemistry; Reactions, Mechanisms, and Structure; Third edition", 1985, WILEY-INTERSCIENCE, NEW YORK, XP002474591 * |
| S. BERGSTR6M, SCIENCE, vol. 157, 1967, pages 382 |
| YANKEE, ERNEST W. ET AL: "Total synthesis of 15-methylprostaglandins", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY , 96(18), 5865-76 CODEN: JACSAT; ISSN: 0002-7863, 1974, XP002474589 * |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011095990A3 (en) * | 2010-02-03 | 2012-01-12 | Fdc Limited | Process for the purification of prostaglandins and analogues thereof |
| WO2012010089A1 (en) * | 2010-07-21 | 2012-01-26 | 上海天伟生物制药有限公司 | Preparation method for and uses of carboprost tromethamine crystal |
| CN102336693A (en) * | 2010-07-21 | 2012-02-01 | 上海天伟生物制药有限公司 | Carboprost tromethamine crystals, preparation method and application thereof |
| GB2495458A (en) * | 2010-07-21 | 2013-04-10 | Shanghai Techwell Biopharm Co | Preparation method for and uses of carboprost tromethamine crystal |
| CN102336693B (en) * | 2010-07-21 | 2014-01-22 | 上海天伟生物制药有限公司 | Carboprost tromethamine crystals, preparation method and application thereof |
| GB2495458B (en) * | 2010-07-21 | 2016-03-09 | Shanghai Techwell Biopharm Co | Crystals of carboprost tromethamine and the preparation method as well as the uses thereof |
| JP2019502666A (en) * | 2015-12-01 | 2019-01-31 | キノイン・ジヨージセル・エーシユ・ベジエーセテイ・テルメーケク・ジヤーラ・ゼー・エル・テー | Carboprost and method for producing tromethamine salt thereof |
| CN108602769A (en) * | 2015-12-01 | 2018-09-28 | 奇诺因药物和化学工厂私人有限公司 | The method for preparing Carboprost and its amino butanetriol salt |
| WO2017093770A1 (en) | 2015-12-01 | 2017-06-08 | CHINOIN Gyógyszer és Vegyészeti Termékek Gyára Zrt. | Process for the preparation of carboprost and its tromethamine salt |
| US10442762B2 (en) | 2015-12-01 | 2019-10-15 | Chinoin Gyogyszer Es Vegyeszeti Termekek Gyara Zrt. | Process for the preparation of carboprost and its tromethamine salt |
| CN108602769B (en) * | 2015-12-01 | 2020-11-13 | 奇诺因药物和化学工厂私人有限公司 | Method for preparing carboprost and tromethamine salt thereof |
| TWI738687B (en) * | 2015-12-01 | 2021-09-11 | 匈牙利商齊諾應醫藥及化學品股份有限公司 | Novel process for the preparation of carboprost tromethamine |
| CN110117242A (en) * | 2018-02-06 | 2019-08-13 | 广州楷模生物科技有限公司 | The synthetic method of Carboprost, tromethamine Carboprost |
| CN115710209A (en) * | 2021-08-23 | 2023-02-24 | 佳和桂科技股份有限公司 | Method and intermediates for preparing carboprost and carboprost tromethamine, and carboprost tromethamine prepared therefrom |
| EP4140982A2 (en) | 2021-08-23 | 2023-03-01 | Chirogate International Inc. | Processes and intermediates for the preparations of carboprost and carboprost tromethamine, and carboprost tromethamine prepared therefrom |
| JP2023031276A (en) * | 2021-08-23 | 2023-03-08 | チャイロゲート インターナショナル インク. | Method and intermediate for preparation of carboprost and carboprost tromethamine, and carboprost tromethamine prepared therefrom |
| EP4140982A3 (en) * | 2021-08-23 | 2023-05-03 | Chirogate International Inc. | Processes and intermediates for the preparations of carboprost and carboprost tromethamine, and carboprost tromethamine prepared therefrom |
| CN115160202A (en) * | 2022-07-30 | 2022-10-11 | 广州楷石医药有限公司 | Preparation method of carboprost tromethamine and intermediate thereof |
| CN115160202B (en) * | 2022-07-30 | 2023-10-31 | 广州楷石医药有限公司 | Preparation method of carboprost tromethamine and intermediate thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2118046A1 (en) | 2009-11-18 |
| US20100041912A1 (en) | 2010-02-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1385819B1 (en) | Process for preparing prostaglandin derivatives and stereospecific starting material thereof | |
| CN108602769B (en) | Method for preparing carboprost and tromethamine salt thereof | |
| JP4475943B2 (en) | Prostaglandins and their analogs | |
| KR101634818B1 (en) | Process for the preparation of prostaglandin analogues and intermediates thereof | |
| WO2008081191A1 (en) | Improved method for the wittig reaction in the preparation of carboprost | |
| CA2859923C (en) | Process for the preparation of travoprost | |
| CN112645861A (en) | Method for separating carboprost 15-position isomer | |
| ES2331205T3 (en) | ENZYMATIC TRANSFORMATION OF AN INTERMEDIATE PRODUCT OF PROSTAGLANDINA (BIMATOPROST). | |
| US6437152B1 (en) | Intermediate for the synthesis of prostaglandins | |
| AU595533B2 (en) | 6,9-epoxy-7-fluoro-11-hydroxy-prost-5-en-1-oic acid derivatives | |
| CZ278597A3 (en) | Process for preparing prostaglandin e1, e2 and analog by making use of furyl-copper compounds | |
| KR20010041632A (en) | Novel Process | |
| KR920004190B1 (en) | Process for the preparation of 7-thiaprostaglandin e1 | |
| US5159102A (en) | 7-thiaprostaglandins E, and process for producing same | |
| JPH02167284A (en) | Prostaglandin precursor and production thereof | |
| JPH025746B2 (en) | ||
| EP1085012B1 (en) | Process for producing a purified prostaglandin derivative | |
| Fox et al. | 30 Commercial Synthesis of the Antiglaucoma Prostanoid Travoprost | |
| JPH025745B2 (en) | ||
| EP1169325B1 (en) | Novel intermediate for the synthesis of prostaglandins | |
| JPH0430952B2 (en) | ||
| JPH0651676B2 (en) | (5E) -Prostaglandin E (2) Manufacturing method | |
| GB2198131A (en) | 7-fluoro-pgi derivatives | |
| JPH06739B2 (en) | 5-Thia-Δ7-prostaglandin Es and method for producing the same | |
| EP0190620A1 (en) | Method for 5-fluoroprostacyclins |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08701739 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008701739 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12521821 Country of ref document: US |