WO2008059207A1 - Cannabinoid receptor modulators - Google Patents
Cannabinoid receptor modulators Download PDFInfo
- Publication number
- WO2008059207A1 WO2008059207A1 PCT/GB2007/004225 GB2007004225W WO2008059207A1 WO 2008059207 A1 WO2008059207 A1 WO 2008059207A1 GB 2007004225 W GB2007004225 W GB 2007004225W WO 2008059207 A1 WO2008059207 A1 WO 2008059207A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydrogen
- phenyl
- formula
- chloro
- compound
- Prior art date
Links
- 0 Cc(cc1)ccc1-c1c(C*(N)=O)c(C(O)=O)n[n]1-c1ccccc1C Chemical compound Cc(cc1)ccc1-c1c(C*(N)=O)c(C(O)=O)n[n]1-c1ccccc1C 0.000 description 3
- RIWRFSMVIUAEBX-UHFFFAOYSA-N CNCc1ccccc1 Chemical compound CNCc1ccccc1 RIWRFSMVIUAEBX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to compounds which are modulators of cannabinoid receptor CB1 and which suppress the normal signalling activity of such receptors.
- the invention further relates to compositions and methods using said compounds for the treatment of obesity and overweight, and diseases for which obesity is a risk factor, such ⁇ as metabolic syndrome, type 2 diabetes, cardiovascular disease, osteoarthritis, and some cancers; mental disorders; addictive disorders; neurological disorders; sexual dysfunctions; reproductive dysfunctions; osteoporosis; liver cirrhosis.and liver fibrosis; and, epilepsy.
- the invention also relates to pharmaceutical compositions containing the compounds of the invention, and to the use of the compounds in combination with other treatments for obesity and overweight and for obesity-related diseases.
- the "identifiable signs and symptoms" of obesity include an excess accumulation of fat or adipose tissue, an increase in the size or number of fat cells (adipocyte differentiation), insulin resistance, increased glucose levels (hyperglycemia), increased blood pressure, elevated cholesterol and triglyceride levels and decreased levels of high-density lipoprotein.
- Obesity is associated with a significantly elevated risk for type 2 diabetes, coronary heart disease, strokes, hypertension, various types of cancer and numerous other major illnesses, and overall mortality from all causes (Must et al, 1999, JAMA 282:1523-1529, CaIIe et al, 1999, N. Engl. J. Med. 341 :1097-1105).
- anti-obesity agents such as i) central nervous system agents that affect neurotransmitters or neural ion channels (e.g. antidepressants (bupropion), noradrenaline reuptake inhibitors (GW320659), selective 5HT 2c receptor agonists, antiseizure agents (topiramate, zonisamide), some dopamine antagonists, cannabinoid CB-1 receptor antagonists (rimonabant); ii) leptin/insulin/central nervous system pathway agents (e.g.
- leptin analogues leptin transport and/or receptor promoters
- CNTF Axokine
- NPY antagonists AgRP antagonists
- POMC promoters CART promoters
- MSH analogues MSH analogues
- MC4 receptor agonists agents that affect insulin metabolism/activity [PTP-1 B inhibitors, PPAR receptor antagonists, short-acting bromocriptine (ergoset), somatostatin agonists (octreotide), and adiponectin/Acrp30 (Famoxin or Fatty Acid Metabolic oxidation INducer)]) ; iii) gastrointestinal-neural pathway agents (e.g.
- agents that increase CCK and PYY activity agents that increase GLP-1 activity (extendin 4, liraglutide, dipeptidyl peptidase IV inhibitor), agents that decrease ghrelin activity, amylin (pramlinitide), neuropeptide Y agonists) ; iv) agents that may increase resting metabolic rate (beta-3 agonists, UCP homologues, thyroid receptor agonists) ; and v) other more diverse agents, such as for example including (MCH) melanin concentrating hormone antagonists, phytostanol analogues, functional oils, P57, amylase inhibitors, growth hormone fragments, synthetic analogues of DHEAS (fluasterone), antagonists of adipocyte 11 beta- hydroxysteroid dehydrogenase type 1 activity, CRH agonists, carboxypeptidase inhibitors, inhibitors of fatty acid synthesis (cerulenin and C75), indanones/indanols, aminosterols (trodusquemine
- Drugs effective in obesity treatment may act by different mechanisms such as reduction in food intake (e.g. by inducing satiety), drugs altering metabolism (such as agents modifying the absorption of nutrients e.g. inhibition of fat absorption), drugs that increase energy expenditure (e.g. increase of thermogenesis), drugs that inhibit lipogenesis or that stimulate adipocyte apoptosis.
- drugs altering metabolism such as agents modifying the absorption of nutrients e.g. inhibition of fat absorption
- drugs that increase energy expenditure e.g. increase of thermogenesis
- drugs that inhibit lipogenesis or that stimulate adipocyte apoptosis are available for obesity treatment (for reviews, see Gadde and Allison, 2006, Circulation, 114, 974-984; Weigle, 2003, J Clin Endocrinol Metab., 88, 2462-2469; Schi ⁇ th, 2006, CNS Neurol. Disorders Drug Targets, 5, 241-249).
- Sibutramine is a centrally acting mixed inhibitor of serotonin and norepinephrine presynaptic re-uptake.
- Orlistat is an inhibitor of gastrointestinal lipases that reduces fat absorption in the gut.
- Rimonabant SR141716, Acomplia ®
- Rimonabant is a centrally and peripherally acting cannabinoid CB1 modulator (antagonist and inverse agonist) that recently has been approved for treatment of obesity (for a review see Pagotto et al, 2006, Endocrine Reviews, 27, 73-100; for reports on phase III clinical trials see despres et al, 2005, N. Engl. J. Med. 353, 212; van Gaal et al, 2005, Lancet, 16, 1389; Pi-Sunyer et al, 2006, JAMA, 295, 761 ).
- CB1 cannabinoid receptor 1
- CB2 a peripheral receptor found principally in cells related to the immune system.
- CB 1 antagonists are able to modulate energy homeostasis and that CB 1 antagonists are able to modulate food intake as well as peripherally block lipogenic processes ( Pagotto et al, 2006, Endocrine Reviews, 27, 73-100; Tucci et al, 2006, Curr. Med. Chem. 13, 2669-2680; Lange and Kruse, 2004, Current Opinion in Drug Discovery & Dev., 7, 498-506).
- the peripheral effects of CB1 antagonists can be mediated by several target organs and mechanisms, e.g.
- liver block of de novo lipogenesis
- muscles increase in glucose uptake
- adipose tissue adiponectin stimulation, inhibition of lipogenic enzymes, stimulation of GLUT4, generation of futile cycles
- pancreas insulin regulation
- gastrointestinal tract stimulation of satiety signals.
- Rimonabant (Acomplia ®) is approved as an adjunct to diet and exercise for treatment of obesity. While the effects on body weight and other risk factors (plasma triglycerides, HDL cholesterol, plasma insulin, insulin resistance, adiponectin) are very encouraging, there are also undesirable side effects, possibly centrally mediated (psychiatric and nervous system disorders), such as anxiety, depressive disorders, sleep disorders, nausea, and vomiting (cf. http://emc.medicines.org.uk; http://www.emea.europa.eu/humandocs/PDFs/EPAR/acomplia/AcompliaEparScientifi cD-en.pdf). Accordingly, there still exists a need for alternative CB 1 receptor antagonists associated with differing pharmacokinetic, pharmacological and side- effect profiles.
- the CB1 receptor has been invoked in many disease states (cf. review by Pacher et al, 2006, Pharmacol. Rev, 58, 389-462).
- Modulators of CB1 receptor activity can be useful in the treatment of diseases associated with a CB1 receptor regulation such as obesity and overweight, prevention of weight gain (e.g. induced by medications or smoking cessation), diseases associated with obesity as risk factor (e.g. metabolic syndrome, type 2 diabetes, cardiovascular disease, osteoarthritis, and some cancers), metabolic disorders (e.g.
- dyslipidemia hyperlipidemia, low HDL and/or high LDL cholesterol levels, hypertriglycerideemia, low adiponectin levels, impaired glucose tolerance, insulin resistance, HbAIc [glycosylated haemoglobin], diabetes mellitus, type 2 diabetes, reduced metabolic activity, fatty liver), eating disorders, addictive disorders (e.g. to marijuana, psychostimulants, nicotine, alcohol, cocaine, opiates), mental disorders (e.g. schizophrenia, schizo-affective disorder, bipolar disorders, anxiety, panic disorder), neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis, liver cirrhosis, liver fibrosis,, and epilepsy.
- addictive disorders e.g. to marijuana, psychostimulants, nicotine, alcohol, cocaine, opiates
- mental disorders e.g. schizophrenia, schizo-affective disorder, bipolar disorders, anxiety, panic disorder
- neurological disorders sexual dysfunctions, reproductive dysfunctions, osteoporosis, liver cirrhosis, liver fibrosis,
- cardiovascular hypertension, congestive cardiomyopathy, varicosities, pulmonary embolism, coronary heart disease [CHD], liver cirrhosis
- neurological stroke, idiopathic intracranial hypertension, meralgia parethetica
- respiratory dyspnea, obstructive sleep apnea, hypoventilation syndrome, Pickwickian syndrome, asthma
- musculoskeletal immobility, degenerative osteoarthritis, low back pain
- skin striae distensae or "stretch marks," venous stasis of the lower extremities, lymphedema, cellulitis, intertrigo, carbuncles, acanthosis nigricans, skin tags
- gastrointestinal gastroesoph
- the present invention makes available a class of pyrazole compounds which modulate the activity of the cannabinoid receptor CB1.
- the following publications relate to other pyrazole compounds having CB1 modulatory activity: WO1997021682, WO1997019063, WO2000046209, WO2001058869, WO200129007, WO2003088968, WO2003020217, WO2004052864, , WO2005080343, WO2006067443, WO2006087480, EP00576357, EP00658546, US20030199536, US20040119972, US20040192667, US20050261281 , US20050624941 , US2006028084, US20060509367, J. Med.
- the compounds of the invention are useful for the treatment of obesity and overweight, prevention of weight gain, obesity-related diseases (e.g. metabolic syndrome, type 2 diabetes, cardiovascular disease, osteoarthritis, and some cancers), metabolic disorders, dyslipidemia, eating disorders, addictive disorders, mental disorders, neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis, liver cirrhosis, liver fibrosis, and epilepsy.
- obesity-related diseases e.g. metabolic syndrome, type 2 diabetes, cardiovascular disease, osteoarthritis, and some cancers
- metabolic disorders e.g. metabolic syndrome, type 2 diabetes, cardiovascular disease, osteoarthritis, and some cancers
- dyslipidemia eating disorders
- addictive disorders mental disorders
- neurological disorders e.g., neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis
- liver cirrhosis cirrhosis
- liver fibrosis fibrosis
- They are useful for modulating body weight and energy consumption in mammals and for modulating plasma parameters involved in the metabolic syndrome such as low HDL and/or high LDL cholesterol levels, high triglyceride levels, low adiponectin levels and high HbAIc [glycosylated haemoglobin] and for modulating other characteristics of the metabolic syndrome such as impaired glucose tolerance, insulin resistance, excessive fat tissue in and around the abdomen and high blood pressure.
- the compounds of the invention display varying physicochemical properties and are useful for modulating peripheral CB1 receptors and to varying degree central CB1 receptors. Those compounds of the invention associated with a lowered central action on CB1 receptors may have a reduced propensity to induce psychiatric and nervous system side-effects.
- X is a bond, or a divalent radical selected from -C(R 1O )(Rn)- * , -C(RioXRii)-0-*, -C(R 10 )(R 11 )CH 2 -*, -C(R 10 )(R 11 )CH 2 -O-*, -CH 2 C(R 10 )(R 11 )-*, -CH 2 C(R 10 )(R 1 ⁇ -O- * , and -CH 2 -O-C(R 10 )(R 11 )-*, wherein the bond indicated by an asterisk is attached to the pyrazole ring;
- Z is a bond, -O- or -NH-;
- Ri and R 2 are independently selected from hydrogen, (CrC 3 )alkyl, (C 3 -C 6 )cycloalkyl and monocyclic heterocyclic radicals having 4 to 6 ring atoms, each of said radicals being optionally substituted by fluoro, hydroxy, methoxy, -NH 2 or mono- or di-(Cr C 3 )alkylamino; or Ri and R 2 taken together with the nitrogen to which they are attached form a cyclic amino ring of 4 to 6 ring atoms which is optionally substituted by fluoro, hydroxy, methoxy, -NH 2 , or mono- or di-(Ci-C 3 )alkylamino;
- R 3 is hydrogen or a radical of formula -(O) 3 -AIk 3 wherein a is O or 1 and AIk 3 is (C 1 - C 3 )alkyl or (C r C 3 )fluoroalkyl;
- R 4 is a radical of formula -(O) b -(Alk-i) p -(Qi) r -(L) s -Q 2 wherein
- b, p, r and s are independently O or 1 , provided that (i) at least one of p, r and s is 1 , and (ii) a in R 3 and b in R 4 are not simultaneously 1 ;
- AIk 1 is a divalent (CrC 4 )alkylene radical which (a) is optionally substituted on one carbon by Ri 0 and/or R 11 or by one or two optional substituents, and/or (b) optionally contains a -0-, -S-, -CO-, -SO-, -SO 2 -, or -NR 9 - link;
- L is a divalent radical of formula -(Alk 2 ) n -(W) m -, in either orientation, wherein
- n and m are independently O or 1 ;
- AIk 2 is -C(R 10 )(R 11 )-; and W is -CO-, -SO 2 -, -O-, -NR 9 - or -SO-; provided that when W and/or Alk2 are linked to a heteroatom W is not -O-, -NR 9 - or -SO-;
- Qi is a monocyclic carbocyclic ring of 3 to 7 ring atoms, a bicyclic carbocyclic ring system of 7 to 10 ring atoms, a monocyclic heterocyclic ring of 4 to 7 ring atoms or a bicyclic carbocyclic ring system of 8 to 10 ring atoms, any of which rings or ring systems being optionally substituted;
- Q 2 is (a) in the case where s in -(L) 5 -Q 2 is O or 1 , a monocyclic carbocyclic ring of 3 to 7 ring atoms, a bicyclic carbocyclic ring system of 7 to 10 ring atoms, a monocyclic heterocyclic ring of 4 to 7 ring atoms or a bicyclic carbocyclic ring system of 8 to 10 ring atoms, any of which rings or ring systems being optionally substituted; or (b) only in the case where s in -(L) s -Q 2 is 0, hydrogen;
- R 3 and R 4 taken together with the nitrogen to which they are attached form a cyclic amino ring of 4 to 7 ring atoms which is optionally substituted by a radical of formula -(L) 3 -Q 2 wherein s, L and Q 2 are as defined above, or by an optional substituent selected from hydroxy, methoxy, -NH 2 -, or mono- or di-(C r C 3 )alkylamino;
- R 5 is selected from hydrogen -F, -Cl 1 -Br, -CN, (C r C 3 )alkyl, (Ci-C 3 )fluoroalkyl, cyclopropyl, -COOH, tetrazole and -OR 9 , the R 9 part being optionally substituted with tetrazole or -COOH;
- R 6 , R 7 and R 8 are each independently selected from hydrogen -F, -Cl, -Br, -CN, (CrC 3 )alkyl, (CrC 3 )fluoroalkyl, cyclopropyl, and -OR 9 ;
- R 9 is hydrogen, (Ci-C 3 )alkyl, (C r C 3 )fluoroalkyl, or (C 3 -C 6 )cycloalkyl;
- R 10 and R 11 are independently selected from hydrogen and (C r C 3 )alkyl; or R 10 and R 11 taken together with the carbon atom to which they are attached form a (C 3 - C 5 )cycloalkyl ring.
- the compounds of the invention should have a molecular weight of less than 650.
- Another aspect of the invention is a pharmaceutical composition comprising a compound of formula (I) or a salt, hydrate, solvate or N-oxide thereof, together with one or more pharmaceutically acceptable carriers or excipients.
- compositions for treatment of obesity are: (i) The use of a compound of formula (I) or a salt, hydrate, solvate or N-oxide thereof in the preparation of a composition for treatment of obesity, overweight, diseases for which obesity is a risk factor, mental disorders, addictive disorders, neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis, liver cirrhosis, liver fibrosis, epilepsy, or pathologic conditions mediated by or related to CB 1 mediated mechanisms; and
- a method for the treatment of treatment of obesity, overweight, diseases for which obesity is a risk factor, mental disorders, addictive disorders, neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis, liver cirrhosis, liver fibrosis,,, or pathologic conditions mediated by or related to CB1 mediated mechanisms comprises administering to a subject suffering such disease or condition an effective amount of a compound of formula (I) or a salt, hydrate, solvate or N-oxide thereof.
- Metabolic syndrome is associated with pathological symptoms such as low HDL and/or high LDL cholesterol levels, high triglyceride levels, low adiponectin levels and high HbAIc [glycosylated haemoglobin], impaired glucose tolerance, insulin resistance, excessive fat tissue in and around the abdomen and high blood pressure.
- (C a -C b )alkyl wherein a and b are integers refers to a straight or branched chain alkyl radical having from a to b carbon atoms.
- a 1 and b is 6, for example, the term includes methyl, ethyl, n-propyl, isopropyl, n- butyl, isobutyl, sec-butyl, t-butyl, n-pentyl and n-hexyl.
- divalent (C a -C b )alkylene radical wherein a and b are integers refers to a saturated straight or branched hydrocarbon chain having from a to b carbon atoms and two unsatisfied valences.
- Carbocyclic refers to a mono-, bi- or tricyclic radical having up to 16 ring atoms, all of which are carbon, and includes aryl and cycloalkyl.
- cycloalkyl refers to a monocyclic saturated carbocyclic radical having from 3-8 carbon atoms and includes, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
- aryl refers to a mono-, bi- or tri-cyclic carbocyclic aromatic radical, and includes radicals having two monocyclic carbocyclic aromatic rings which are directly linked by a covalent bond.
- Illustrative of such radicals are phenyl, biphenyl and napthyl.
- heteroaryl refers to a mono-, bi- or tri-cyclic aromatic radical containing one or more heteroatoms selected from S, N and O, and includes radicals having two such monocyclic rings, or one such monocyclic ring and one monocyclic aryl ring, which are directly linked by a covalent bond.
- Illustrative of such radicals are thienyl, benzthienyl, furyl, benzfuryl, pyrrolyl, imidazolyl, benzimidazolyl, thiazolyl, benzthiazolyl, isothiazolyl, benzisothiazolyl, pyrazolyl, oxazolyl, benzoxazolyl, isoxazolyl, benzisoxazolyl, isothiazolyl, triazolyl, benztriazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyridazinyl, pyrimidinyl, triazinyl, indolyl and indazolyl.
- heterocyclyl or “heterocyclic” includes “heteroaryl” as defined above, and in addition means a mono-, bi- or tri-cyclic non- aromatic radical containing one or more heteroatoms selected from S, N and O, and to groups consisting of a monocyclic non-aromatic radical containing one or more such heteroatoms which is covalently linked to another such radical or to a monocyclic carbocyclic radical.
- radicals are pyrrolyl, furanyl, thienyl, piperidinyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, pyrazolyl, pyridinyl, pyrrolidinyl, pyrimidinyl, morpholinyl, piperazinyl, indolyl, morpholinyl, benzfuranyl, pyranyl, isoxazolyl, benzimidazolyl, methylenedioxyphenyl, ethylenedioxyphenyl, maleimido and succinimido groups.
- substituted as applied to any moiety herein means substituted with up to four compatible substituents, each of which independently may be, for example, (CrC 6 )alkyl, (C 1 - C 6 )alkoxy, hydroxy, hydroxy(Ci-C 6 )alkyl, mercapto, mercapto(Ci-C 6 )alkyl, (C 1 - C 6 )alkylthio, halo (including fluoro, bromo and chloro), fully or partially fluorinated (C 1 - C 3 )alkyl, (CrC 3 )alkoxy or (CrCsJalkylthio such as trifluoromethyl, trifluoromethoxy, and trifluoromethylthio, nitro, nitrile (-CN), oxo, phenyl, phenoxy, monocyclic heteroaryl or heteroaryloxy with 5 or 6 ring atoms
- substituent is phenyl, phenoxy or monocyclic heteroaryl or heteroaryloxy with 5 or 6 ring atoms
- the phenyl or heteroaryl ring thereof may itself be substituted by any of the above substituents except phenyl, phenoxy, heteroaryl or heteroaryloxy.
- An “optional substituent” may be one of the foregoing substituent groups.
- salt includes base addition, acid addition and quaternary salts.
- Compounds of the invention which are acidic can form salts, including pharmaceutically acceptable salts, with bases such as alkali metal hydroxides, e.g. sodium and potassium hydroxides; alkaline earth metal hydroxides e.g. calcium, barium and magnesium hydroxides; with organic bases e.g. N-methyl-D-glucamine, choline tris(hydroxymethyl)amino-methane, L-arginine, L-lysine, N-ethyl piperidine, dibenzylamine and the like.
- bases such as alkali metal hydroxides, e.g. sodium and potassium hydroxides; alkaline earth metal hydroxides e.g. calcium, barium and magnesium hydroxides; with organic bases e.g. N-methyl-D-glucamine, choline tris(hydroxymethyl)amino-methane, L-arginine, L-lysine, N-ethyl pipe
- hydrohalic acids such as hydrochloric or hydrobromic acids, sulphuric acid, nitric acid or phosphoric acid and the like
- organic acids e.g. with acetic, tartaric, succinic, fumaric, maleic, malic, salicylic, citric, methanesulphonic, p-toluenesulphonic, benzoic, benzenesunfonic, glutamic, lactic, and mandelic acids and the like.
- 'solvate' is used herein to describe a molecular complex comprising the compound of the invention and a stoichiometric amount of one or more pharmaceutically acceptable solvent molecules, for example, ethanol.
- solvent molecules for example, ethanol.
- 'hydrate' is employed when said solvent is water.
- Compounds with which the invention is concerned which may exist in one or more stereoisomeric form, because of the presence of asymmetric atoms or rotational restrictions, can exist as a number of stereoisomers with R or S stereochemistry at each chiral centre or as atropisomeres with R or S stereochemistry at each chiral axis.
- the invention includes all such enantiomers and diastereoisomers and mixtures thereof.
- the compounds of the invention include compounds of formula (I) as hereinbefore defined, including all polymorphs and crystal habits thereof, prodrugs and isomers thereof (including optical, geometric and tautomeric isomers) as hereinafter defined and isotopically-labeled compounds of formula (I).
- So-called 'pro-drugs' of the compounds of formula (I) are also within the scope of the invention.
- certain derivatives of compounds of formula (I) which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into compounds of formula (I) having the desired activity, for example, by hydrolytic cleavage.
- Such derivatives are referred to as 'prodrugs'.
- Further information on the use of prodrugs may be found in Pro-drugs as Novel Delivery Systems. Vol. 14, ACS Symposium Series (T. Higuchi and V.J. Stella) and Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (ed. E. B. Roche, American Pharmaceutical Association; CS. Larsen and J. ⁇ stergaard, Design and application of prodrugs, In Textbook of Drug Design and Discovery, 3 rd Edition, 2002, Taylor and Francis ).
- Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the compounds of formula (I) with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in Design of Prodrugs by H. Bundgaard (Elsevier, 1985).
- metabolites of compounds of formula (I) that is, compounds formed in vivo upon administration of the drug.
- the divalent linker radical -X- is a bond, or a divalent radical selected from -C(R 10 )(R 11 )-*, -C(R 1O )(Rn)-O-*, -C(R 10 )(R 1I )CH 2 -*, -C(R 10 )(R 11 )CH 2 -O-*, -CH 2 C(R 10 XR 11 )-*, -CH 2 C(R 10 )(R 1 0-O-*, and -CH 2 -O-C(R 10 )(R 11 )-*, wherein the bond indicated by an asterisk is attached to the pyrazole ring.
- Ri 0 and R 11 are independently selected from hydrogen and (C r C 3 )alkyl (ie. methyl, ethyl and n- or isopropyl); or R 10 and R 11 taken together with the carbon atom to which they are attached form a (C 3 -C 5 )cycloalkyl ring such as a cyclopropyl or cyclopentyl ring.
- -X- may be, for example, a bond, -CH 2 - , -CH 2 O-*, or -CH 2 OCH 2 -*, wherein the bond indicated by an asterisk is attached to the pyrazole ring.
- Z is a bond, -O- or -NH-
- specific examples of the combination linker radical -Z- X- are -CH 2 - , -CH 2 O-*, -CH 2 OCH 2 -*, -NH- Or -NH-CH 2 -* wherein the bond indicated by an asterisk is attached to the pyrazole ring.
- R 9 is hydrogen, (C r C 3 )alkyl such as methyl or ethyl, (C r C 3 )fluoroalkyl such as di- or trifluoromethyl, or (C 3 -C 6 )cycloalkyl such as cyclopropyl;
- Ri and R 2 are independently selected from hydrogen, (C-i-C 3 )alkyl such as methyl (C 3 -C 6 )cycloalkyl such as cyclopropyl, cyclopentyl or cyclohexyl, and monocyclic heterocyclic radicals having 4 to 6 ring atoms such as morpholinyl, 4-(Cr C 6 )alkylpiperazinyl, 4-acetylpiperazinyl, 4- (C r C 6 )alkylsulfonylpiperazinyl, azepanyl, pyrrolidinyl or piperidinyl, each of said radicals being optionally substituted by fluoro, hydroxy, methoxy, -NH 2 or mono- or di-(Ci-C 3 )alkylamino such as methylamino, dimethylamino, ethyl amino and diethylamino; or R 1 and R 2 taken together with the nitrogen to which they are attached form
- R-i and R 2 are each hydrogen.
- R 1 is hydrogen and R 2 is methyl, cyclopropyl or optionally substituted cyclohexyl.
- R 1 and R 2 taken together with the nitrogen to which they are attached form a morpholine or an optionally substituted piperidine or piperazine ring.
- optional substituents include one or two substituents selected from hydroxy, methoxy and -NH 2 .
- R 3 is hydrogen or a radical of formula -(O) a -Alk 3 wherein a is O or 1 and AIk 3 is (Cr C 3 )alkyl such as methyl or ethyl or (C r C 3 )fluoroalkyl such as di- or trifluoromethyl. R 3 will often be hydrogen.
- the substituent R* is (Cr C 3 )alkyl such as methyl or ethyl or (C r C 3 )fluoroalkyl such as di- or trifluoromethyl.
- R 3 will often be hydrogen.
- R 4 has formula -(OV(AIk 1 ) p -(Qi X-(L) 5 -Q 2 wherein b, p, r and s are 0 or 1 in any compatible combination, provided that (i) at least one of p, r and s is 1 , and (ii) a in R3 and b in R 4 are not simultaneously 1 , the latter proviso being necessary to avoid incompatible substitutions on the relevant nitrogen.
- b, p and s are 0, and r is 1 , and in another p and r are 1 and b and s are 0.
- AIk 1 when present is a divalent (C-
- R 10 and R- ⁇ may be, for example, methyl; or R 10 and R-n taken together with the carbon atom to which they are attached may form, for example, a cyclopropyl or cyclopentyl ring.
- Optional substituents in this context include fluoro and hydroxyl.
- radicals include -CH 2 -, -CH 2 CH 2 - -CH 2 CH 2 CH 2 -, - CH 2 CH 2 CH 2 CH 2 -, -CH 2 T-, -CH 2 TCH 2 - -CH 2 CH 2 TCH 2 -, -CH 2 CH 2 TCH(CHs)-, — CH 2 TCH 2 CH 2 -, and -CH 2 TCH 2 CH 2 WCH 2 -, where T is -O-, -S-, -NH-, or -N(CH 3 )-, and any of the foregoing wherein one or more hydrogens are exchanged for fluorines, and/or wherein one or two carbons are substituted by methyl or, trifluoromethyl; or wherein one carbon is substituted by a spiro-linked cyclopropyl substituent.
- L when present is a divalent radical of formula -(Alk 2 ) n -(W) m -, in either orientation, wherein n and m are independently 0 or 1 ; AIk 2 is -C(Ri O )(Rn)-; and W is -CO-, -SO 2 -, -0-, -NRg- or -SO-; provided that when W and/or AIk 2 are linked to a heteroatom W is not -0-, -NRg- or -SO-.
- Rg may be, for example, hydrogen or methyl.
- AIk 2 when present are -CH 2 -, -CH(CH 3 )-, a cyclopropyl ring which is linked to each adjacent atom via the same ring carbon, and, in either orientation -CH 2 O-, -CH 2 NH-, -CH(CH 3 )O- and -CH(CH 3 )NH-.
- Q 1 (when present) and Q 2 are each independently a monocyclic carbocyclic radical of 3 to 7 ring atoms, a bicyclic carbocyclic ring system of 7 to 10 ring atoms, a monocyclic heterocyclic ring of 4 to 7 ring atoms or a bicyclic carbocyclic ring system of 8 to 10 ring atoms, any of which rings or ring systems being optionally substituted;
- Q 2 may also be hydrogen when s in -(L) 5 -Q 2 IS 0;
- Examples of Qi, when present, include optionally substituted divalent phenyl, pyridine, piperidine, piperazine or (C 3 - C 7 )cycloalkyl, eg cyclohexyl, cyclopentyl or cyclopropyl, radicals.
- Q 2 examples include hydrogen, or optionally substituted phenyl or pyridyl.
- Optional substituents in rings Q 1 and Q 2 include -F, -Cl, -Br, -CN, -CF 3 , -CH 3 , cyclopropyl, and -OCH 3 , and may be present on, for example 1 or 2 ring atoms.
- R 3 and R 4 taken together with the nitrogen to which they are attached form a cyclic amino ring of 4 to 7 ring atoms.
- examples of such rings include morpholinyl, pyrrolidinyl, piperidinyl, azepanyl and piperazinyl.
- the ring formed by R 3 and R 4 taken together with the nitrogen to which they are attached may be substituted by a radical of formula -(L) 3 -Q 2 wherein s, L and Q 2 are as defined and discussed above, or by an optional substituent selected from hydroxy, methoxy, -NH 2 -, or mono- or di-(C 1 -C 3 )alkylamino such as methylamino, ethylamino, dimethylamino and diethylamino.
- R 10 and R 11 are as defined in claim 1
- Ri2 is selected from hydrogen, -CH 3 , -OH, -CN and -COOH;
- R 13 is selected from hydrogen, -F, -CF 3 , -OCF 3 , -Br. -Cl, -OCH 3 , -CH 3 , -CN and
- R 14 is selected from hydrogen, -F, -CF 3 , -OCF 3 , -Br. -Cl, -OCH 3 , -CH 3 , -CN, -OH, and -COOH;
- Ri 5 and Ri 6 are independently selected from hydrogen and (Ci-C 6 )alkyl or Ri 5 and Ri 6 taken together with the nitrogen to which they are attached form a cyclic amino ring of 4 to 7 ring atoms;
- Ri 8 is selected from hydrogen, -F and -CN;
- R 19 is selected from F, -CF 3 , -OCF 3 , -Br. -Cl, -OCH 3 , -CH 3 , -CN and -COOH; and R 20 is selected from F, -CF 3 , -OCF 3 , -Br. -Cl, -OCH 3 , -CH 3 , -CN, -OH and -COOH.
- R 1S is hydrogen, 3-, 4- or 5-fluoro, 3-, 4- or 5-cyano
- R 20 is methyl, trifluoromethyl, trifluoromethoxy, 3-, 4- or 5-fluoro, 3-, 4- or 5-chloro, or 3-, 4- and 5-cyano, or 6-hydroxy;
- R7 and R 8 are each independently selected from hydrogen -F, -Cl, -Br, -CN, (Ci-C 3 )alkyl such as methyl, (Ci-C 3 )fluoroalkyl such as trifluoromethyl, cyclopropyl, and -OR 9 wherein R 9 is methyl or ethyl.
- R 5 , R 6 , R 7 and R 8 are independently selected from hydrogen, fluoro and chloro.
- R 5 is selected from -COOH, tetrazole or -OR 9 having the Rg part substituted with tetrazole or -COOH;
- the compounds of the present invention act on central and peripheral cannabinoid receptor CBl Some compounds distribute to a lesser extent to the central nervous system, i.e. the compound less readily crosses the blood-brain barrier and will be associated with fewer central nervous system mediated side-effects.
- the compounds of the invention modulate the cannabinoid receptor CB1 by suppressing its natural signalling function.
- the compounds are therefore CB1 receptor antagonists, inverse agonists, or partial agonists.
- CB1 antagonist or "cannabinoid receptor CB1 antagonist” refers to a compound which binds to the receptor, or in its vicinity, and lacks any substantial ability to activate the receptor itself.
- a CB1 antagonist can thereby prevent or reduce the functional activation or occupation of the receptor by a CB1 agonist such as for example the endogenous agonist N-Arachidonylethanolamine (anandamide). This term is well known in the art.
- CB1 inverse agonist or "cannabinoid receptor CB1 inverse agonist” refers to a compound which binds to the receptor and exerts the opposite pharmacological effect as a CB1 receptor agonist does.
- Inverse agonists are effective against certain types of receptors which have intrinsic activity without the acting of a ligand upon them (also referred to as 'constitutive activity'). This term is well known in the art. It is also well known in the art that such a CB1 inverse agonist can also be named a CB1 antagonist as the general properties of both types are equivalent. Accordingly, in the context of the present invention the term “CB1 antagonist” in general is understood as including both the "CB1 antagonist” as defined above and the "CB1 inverse agonist”.
- CB 1 partial agonist or “cannabinoid receptor CB 1 partial agonist” refers to a compound which acts upon the same receptor as the full agonist but that produces a weak maximum pharmacological response and has a low level of intrinsic activity. This term is well known in the art.
- the "CB1 modulator” or “cannabinoid receptor CB1 modulator” is a CB1 antagonist or inverse agonist compound.
- the compounds of the invention are useful for the treatment of obesity and overweight, prevention of weight gain, obesity-related diseases (e.g. metabolic syndrome, type2 diabetes, cardiovascular disease, osteoarthritis, and some cancers), metabolic disorders, dyslipidemia, eating disorders, addictive disorders, mental disorders, neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis, liver cirrhosis, liver fibrosis, and epilepsy.
- obesity-related diseases e.g. metabolic syndrome, type2 diabetes, cardiovascular disease, osteoarthritis, and some cancers
- metabolic disorders e.g. metabolic syndrome, type2 diabetes, cardiovascular disease, osteoarthritis, and some cancers
- dyslipidemia eating disorders
- addictive disorders mental disorders
- neurological disorders e.g., neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis
- liver cirrhosis cirrhosis
- liver fibrosis fibrosis
- the compounds of the invention are useful for modulating body weight and energy consumption in mammals and for modulating plasma parameters involved in the metabolic syndrome such as low HDL and/or high LDL cholesterol levels, high triglyceride levels, low adiponectin levels and high HbAIc [glycosylated haemoglobin] and for modulating other characteristics of the metabolic syndrome such as impaired glucose tolerance, insulin resistance, excessive fat tissue in and around the abdomen and high blood pressure.
- the compounds of the invention display varying physicochemical properties and are useful for modulating peripheral CB1 receptors and to varying degree central CB1 receptors. Those compounds of the invention associated with a lowered central action on CB 1 receptors may have a reduced propensity to induce psychiatric and nervous system side-effects.
- the compounds of the invention may be combined with another therapeutic agent used in treatment of obesity acting by a different mode of action such as central action on satiety or hunger signals, craving mechanisms, appetite regulation, leptin/insulin/central nervous system pathways, gastrointestinal-neural pathways, metabolic rate, energy expenditure, food intake, fat storage, fat excretion, gastrointestinal motility, lipogenesis, glucose transport, glucogenolysis, glycolysis, lipolysis, etc including modulators (inhibitors, agonists, antagonists, analogues) of monoaminergic (NA (noradrenaline), 5-HT (serotonin), DA (dopamine)) receptors or transporters, neural ion channels, leptin or leptin receptor, neuropeptide Y receptors, PP (pancreatic polypeptide), PYY, Protein YY3-36, ghrelin or ghrelin receptor, motilin or motilin receptor, orexins or orexin receptors, bombesin or bombe
- the compounds of the invention may be combined with another therapeutic agent used in treatment of metabolic syndrome or obesity-related diseases such as cardiovascular (hypertension, congestive cardiomyopathy, varicosities, pulmonary embolism, coronary heart disease [CHD], liver cirrhosis), neurological (stroke, idiopathic intracranial hypertension, meralgia parethetica), respiratory (dyspnea, obstructive sleep apnea, hypoventilation syndrome, Pickwickian syndrome, asthma), musculoskeletal (immobility, degenerative osteoarthritis, low back pain, osteoporosis), skin (striae distensae or "stretch marks," venous stasis of the lower extremities, lymphedema, cellulitis, intertrigo, carbuncles, acanthosis nigricans, skin tags), gastrointestinal (gastro-esophageal reflux disorder, nonalcoholic fatty liver/steatohepatitis, cholelithiasis, herni
- the compounds of the invention may be combined with proper reduction in dietary calorie intake and physical exercise.
- the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, route of administration, rate of excretion, drug combination and the severity of the particular disease undergoing treatment. Optimum dose levels and frequency of dosing will be determined by clinical trial, as is required in the pharmaceutical art.
- the total daily dose of the compounds of the invention may typically be in the range 1 mg to 1000 mg depending, of course, on the mode of administration.
- oral administration may require a total daily dose of from 10 mg to 1000 mg, while an intravenous dose may only require from 1 mg to 500 mg.
- the total daily dose may be administered in single or divided doses and may, at the physician's discretion, fall outside of the typical range given herein.
- These dosages are based on an average human subject having a weight of about 60kg to 100kg.
- the physician will readily be able to determine doses for subjects whose weight falls outside this range, such as infants and the elderly, and especially obese patients.
- the compounds with which the invention is concerned may be prepared for administration by any route consistent with their pharmacokinetic properties.
- the orally administrable compositions may be in the form of tablets, capsules, powders, granules, lozenges, liquid or gel preparations, such as oral, topical, or sterile parenteral solutions or suspensions.
- Tablets and capsules for oral administration may be in unit dose presentation form, and may contain conventional excipients such as binding agents, for example syrup, acacia, gelatin, sorbitol, tragacanth, or polyvinylpyrrolidone; fillers for example lactose, sugar, maize-starch, calcium phosphate, sorbitol or glycine; tabletting lubricant, for example magnesium stearate, talc, polyethylene glycol or silica; disintegrants for example potato starch, or acceptable wetting agents such as sodium lauryl sulphate.
- the tablets may be coated according to methods well known in normal pharmaceutical practice.
- Oral liquid preparations may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, or may be presented as a dry product for reconstitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives such as suspending agents, for example sorbitol, syrup, methyl cellulose, glucose syrup, gelatin hydrogenated edible fats; emulsifying agents, for example lecithin, sorbitan monooieate, or acacia; nonaqueous vehicles (which may include edible oils), for example almond oil, fractionated coconut oil, oily esters such as glycerine, propylene glycol, or ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid, and if desired conventional flavouring or colouring agents.
- suspending agents for example sorbitol, syrup, methyl cellulose, glucose syrup, gelatin hydrogenated edible fats
- emulsifying agents for example lecithin, sorbitan monooieate, or acacia
- nonaqueous vehicles which may include edible oils
- almond oil fractionated coconut oil
- oily esters such as glycerine, propylene
- the active ingredient may also be administered parenterally in a sterile medium.
- the drug can either be suspended or dissolved in the vehicle.
- adjuvants such as a local anaesthetic, preservative and buffering agents can be dissolved in the vehicle.
- Step A-V Experimental conditions given in Step A-V are generic and can be found in standard literature sources such as those cited above. Specific references are cited for information and conditions may apply to a given substrate with or without modification/optimization.
- Step A Nitriles can be converted to amides in acidic conditions e.g. sulphuric acid in acetic acid -
- acidic conditions e.g. sulphuric acid in acetic acid -
- Step B Activation of carboxylic acids, followed by nucleophilic displacement with R 3 NHR 4 to give the amides, according to well know methods e.g. 1-ethyl-3-(3- dimethylaminopropyl)carbodiimide, N-hydroxybentriazole in dichloromethane.
- Step C Nitriles can be converted to amides in basic conditions e.g. sodium hydroxide, hydrogen peroxide in ethanol -
- basic conditions e.g. sodium hydroxide, hydrogen peroxide in ethanol -
- Step D Nitriles can be converted to alkyls esters using e.g. trimethylsilyl chloride and an appropriate alcohol e.g. ethanol -
- an appropriate alcohol e.g. ethanol -
- Step E Nitriles can be converted to amidines according to Pinner synthesis e.g. gas hydrogen chloride, ethanol (amidates formation) followed by displacement with R 3 NHR 4 , or alternative methods e.g.
- Step F Esters can be hydrolysed in e.g. basic/acidic conditions according to well known methods described in Protective Groups in Organic Synthesis (T.W. Greene).
- Step Ga Protection of functional groups with a suitable PG, according to well known methods described in Protective Groups in Organic Synthesis (T.W. Greene).
- Step Gb Deprotection of functional groups, according to well known methods described in Protective Groups in Organic Synthesis (T.W. Greene).
- Step H Conversion of amines to ureas can be achieved via reaction of amines with a suitable isocyanate of formula R 3 NCO, leading to monosubstituted ureas. Unsubstituted ureas can be synthesised by reaction of amines with potassium isocyanate. Alternatively, mono or disubstituted ureas can be synthesised via the reaction of amines with an activated agent of formula LG1-(CO)-LG1 , followed by the addition of an amine of formula R 3 NHR 4 .
- Substituted guanidines can be synthesised via reaction of amines with various activated agents e.g. thioisocyanates of formula R 3 NCS (followed by addition of R 4 NH 2 ), thiophosgene (followed by addition of R 3 NH 2 and reaction with R 4 I), ureas of formula R 3 NH(CO)NHR 4 ( activated first with POCI 3 ) or thioureas of formula R 3 NH(CS)NHR 4 -
- activated agents e.g. thioisocyanates of formula R 3 NCS (followed by addition of R 4 NH 2 ), thiophosgene (followed by addition of R 3 NH 2 and reaction with R 4 I), ureas of formula R 3 NH(CO)NHR 4 ( activated first with POCI 3 ) or thioureas of formula R 3 NH(CS)NHR 4 -
- R 3 NCS followeded by addition of
- Step J Bromination of methyl can be achieved with a brominating agent e.g. N- bromosuccinimide under radical conditions, e.g. 2,2'-azobisisobutyronitrile in carbon tetrachloride -
- a brominating agent e.g. N- bromosuccinimide under radical conditions, e.g. 2,2'-azobisisobutyronitrile in carbon tetrachloride -
- radical conditions e.g. 2,2'-azobisisobutyronitrile in carbon tetrachloride
- Step K Nitriles can be synthesised by nucleophilic displacement of bromo methyl derivatives with e.g. sodium/potassium/copper cyanide in polar solvents (e.g. dimethylformamide, ethanol, acetonitrile, dimethylsulfoxid) -
- polar solvents e.g. dimethylformamide, ethanol, acetonitrile, dimethylsulfoxid
- Step L The alpha position of nitriles can be substituted after deprotonation with a strong base e.g. lithium diisopropylamide in tetrahydrofuran at low temperature, followed by reaction of the carbanions with R 10 HaI and/or R ⁇ HaI or R- I0 CHO -
- a strong base e.g. lithium diisopropylamide in tetrahydrofuran at low temperature
- Step M Primary amines can be synthesised by nucleophilic displacement of bromo methyl derivatives with either a protected amino group e.g. phtalamide, followed by removal of the protecting group (Gabriel synthesis), or a masked amino group e.g. sodium azide followed by catalytic hydrogenation -
- a protected amino group e.g. phtalamide
- a masked amino group e.g. sodium azide followed by catalytic hydrogenation
- Step N 4-Pyrazolyloxy acetic acid derivatives can be synthesised by reacting the hydroxyl moiety with an appropriate protected acetic acid of formula PG3O 2 C- C(Rio)(Rii)-LG4 or PGSO 2 C-C(R 10 )(Rn)-OH.
- Typical literature references are: 1) EtO 2 C-C(RIO)(RI I)-Br in presence of a base e.g.
- Step O 4-Pyrazolyloxy acetonitrile derivatives can be synthesised by reacting the hydroxyl moiety with an appropriate alpha substituted acetonitrile of formula NC- C(Rio)(Rii)-LG4 or NC-C(R 10 )(Rn)-OH.
- Typical literature references are: 1) NC- C(R 10 )(Rn)-CI in presence of KF in dimethylformamide; 2) NC-C(Ri 0 )(Ri i)-OH in presence of triphenylphosphine/ diethyl azidodicarboxylate (Mitsunobu synthesis) -
- NC-C(Ri 0 )(Ri i)-OH in presence of triphenylphosphine/ diethyl azidodicarboxylate (Mitsunobu synthesis)
- 1 Nucleosides and Nucleotides 9(8) 1021-1043, 1990 and 2) Pest Management Science 57(9), 844-851 , 2001.
- Step P Synthesis of the ether linker can be achieved by displacement of a suitable leaving group LG attached to the methyl in position 4 by a suitable nucleophile of formula PGSO 2 C-C(Ri 0 )(Rn)-OH or NC-C(Ri 0 )(Rn)-OH, in the presence of a base e.g. potassium carbonate in dimethylformamide or acetone -
- a base e.g. potassium carbonate in dimethylformamide or acetone -
- Step Q 5-substitution of pyrazoles with a suitable aryl ring can be achieved via Suzuki coupling reactions -
- Suzuki coupling reactions For literature references see: Journal of Organometallic Chemistry 576(1-2), 147-168, 1999.
- Step R Aldehydes can be converted to alcohols by reaction with a suitable reducing agent e.g. sodium borohydride in methanol -
- a suitable reducing agent e.g. sodium borohydride in methanol -
- Step S Secondary alcohols can be synthesised by reaction of aldehydes with a suitable organometallic reagent of formula R 10 MX e.g. Ri 0 MgBr in tetrahydrofuran or diethyl ether -
- organometallic reagent of formula R 10 MX e.g. Ri 0 MgBr in tetrahydrofuran or diethyl ether -
- R 10 MX e.g. Ri 0 MgBr in tetrahydrofuran or diethyl ether
- Step T Conversion of hydroxyl groups to corresponding leaving groups LG can be achieved by reacting the hydroxyl moiety with e.g. methane sulfonyl chloride, tosyl chloride, in a presence of pyridine or triethylamine in tetrahydrofuran or by converting the hydroxy! moiety to a suitable halide e.g. bromine with e.g. phosphorus tribromide in dichloromethane -
- a suitable halide e.g. bromine with e.g. phosphorus tribromide in dichloromethane -
- a) Amides/ureas are converted to thioamides/thioureas with e.g. Lawessons reagent; b) Thioamides/thioureas
- a suitable O-Alkyl hydroxylamine of formula RgO-NH 2 e.g. ethanol or dimethylsulfoxide.
- O-Alkyl hydroxylamines are available as their hydrochloride salt; therefore, a base e.g. potassium tert-butoxide is required -
- a base e.g. potassium tert-butoxide is required -
- literature references see: 1 ) Journal of Labelled Compounds & Radiopharmaceuticals 47(4), 233-242, 2004; 2) Journal of Medicinal Chemistry 47(14), 3658-3664, 2004 and 3) WO06090153.
- R1, R2, R3, R4, R5 and R6 are defined as in claim 1
- R9 is H
- R1, R2, R3, R4, R5, R6 and R9 are defined as in claim 1
- PG1 is a suitable protecting group e.g. Ethyl
- PG2 is a suitable protecting group e.g. Boc
- LG1 is a suitable leaving group e.g. Cl or imidazole
- LG2 is a suitable leaving group e.g. pyrazole or -SMe
- R1, R2, R3, R4, R5 and R6 are defined as in claim 1
- Hal is a halide e.g. Cl 1 Br or I.
- PG1 is a suitable protecting group e.g. Ethyl.
- R1 , R2, R3, R4, R5, R6 and R9 are defined as in claim 1
- LG1 is a suitable leaving group e.g. Cl or imidazole
- LG2 is a suitable leaving group e.g. pyrazole or -SMe PG1 is a suitable protecting group e.g. Ethyl.
- X is CH2-O* , CH(RI O)-O* or C(R10)(R11 )-O*
- R1, R2, R3, R4, R5 and R6 are defined as in claim 1
- LG4 is a suitable leaving group e.g. Cl , Br, OMs, OTs or OTf.
- PG1 and PG3 are orthogonal protecting groups.
- X is -C(R10)(R11 )-O-CH2- * , -CH2-0-C(R10)(R11 ) - *
- R1, R2, R3, R4, R5 and R6 are defined as in claim 1
- PG1 and PG3 are orthogonal protecting groups.
- R1 , R2, R3, R4, R5 and R6 are defined as in claim 1
- Compounds of formula (In) can be synthesised from 30 as described in scheme 6b, according to generic steps Q, R, T, P, D, F and B or Q, R, N, D, F and B.
- Compounds of formula (lo) can be synthesised from 30 as described in scheme 6b, according to generic steps Q, R, T, P and E or Q 1 R, T, O and E.
- Compounds of formula (Ip) can be synthesised from 30 as described in scheme 6b, according to generic steps Q, S, N, F and B or Q, S, T, P, F and B.
- Compounds of formula (Iq) can be synthesised from 30 as described in scheme 6b, according to generic steps Q, S, O and E or Q 1 S, T, P and E.
- LG4 is a suitable leaving group e.g. Cl , Br, OMs, OTs or OTf.
- Fg is PG3O 2 C or CN PG3 is a suitable protecting group (see Step N)
- R1, R2, R3, R4, R5 and R6 are defined as in claim 1
- R9 is CN
- R1, R2, R3, R4, R5, R6 and R9 are defined as in claim 1
- ZX is -O- or -O-CH(RIO)*- (wherein the bond indicated by an asterisk is attached to the pyrazole ring)
- R1, R2, R3, R4, R5 and R6 are defined as in claim 1 ,
- R5 and Rs are hydrogen.
- Esters of formula [A] were obtained by well known methods (Ruoxi et al., J. Med. Chem, 1999, 42, 769-776). Esters of formula [A] were brominated by treatment with N-bromosuccinimide (1.1eq.) and 1 ,1 '-azobis(isobutyronitrile) (O.OIeq.) in carbon tetrachloride as described in US 2004/0192667. The resulting bromo compounds were not purified but treated directly with potassium cyanide (2eq.) and 18-crown-6 ether (0.4eq.) in acetonitrile at reflux for 15 hours.
- bromo compounds could be treated with sodium cyanide in a mixture of ethanol and water at reflux, which gave directly the acids of formula [B].
- Compounds of formula [B] (1mmol) were treated in acetic acid/concentrated sulphuric acid mixtures (8ml/4ml) at 10O 0 C, for 30 minutes. The cooled mixtures were poured onto ice/water to yield precipitates which were filtered off, washed with water and dried in vacuum to give amides of formula [C].
- R 5 and R 8 are hydrogen.
- Nitrile intermediates of formula [B] were prepared as described in scheme 1 above.
- Amides of formula [E] were synthesized by well known methods using the appropriate amine R 3 NHR 4 and coupling reagents.
- Compounds of formula [E] were converted to amidine derivatives of formula [F] according to well known procedures (Journal of the American Chemical Society, 126(39), 12220-12221 ; 2004; Bioorganic & Medicinal Chemistry Letters, 14(21), 5263-5267; 2004)
- R 5 , R 8 and Rg are hydrogen.
- Nitrile intermediates of formula [B] were prepared as described in scheme 1 above.
- Amides of formula [E] were synthesized by well known methods using the appropriate amine R 3 NHR 4 and coupling reagents.
- Compounds of formula [E] were converted to amidoxime derivatives of formula [G] according to well known procedures (Bioorganic & Medicinal Chemistry, 14(5), 1506-1517; 2006. Journal of Medicinal Chemistry, 50(7), 1618-1626; 2007. Journal of Medicinal Chemistry, 48(18), 5705-5720; 2005).
- the compounds are grouped in three classes:
- Table 1 and 3 give the biological test results for the compounds synthesised above.
- R 5 and R 8 are hydrogen.
- R 5 and R 8 are hydrogen.
- R 5 , R 8 and R 9 are hydrogen.
- the cDNA encoding the human CB1 (Cannabinoid Receptor-1) receptor was cloned from a human adipose tissue cDNA library and cloned into the eukaryotic expression vector pcDNA3.1 (Invitrogen).
- Chinese Hamster Ovary cells (CHO-K1) stably expressing recombinant human CB1 were generated by transfecting the plasmid containing the coding sequence of the human CB1 receptor in CHO-K1 cells, using lipofectamin, according to the manufacturer instructions. Resistant clones were selected in the presence of 600 ⁇ g/ml G418 (Life technology). Stably transfected CHO-K1 cells were maintained in Ham's F-12 culture medium (Invitrogen), supplemented with 10 % fetal calf serum (Invitrogen), 100 U/ml penicillin, 100 ⁇ g/ml streptomycin (Life Technology), and 600 ⁇ g/ml G418.
- CP55940-induced [ 35 S]GTP ⁇ S binding to membranes prepared from CHO-K1 cells expressing the human CB1 receptor (described in Transfection and Cell Culture).
- CP55940 is a well known nonselective CB1 and CB2 receptor agonist (e.g Felder et al., 1995, Molecular Pharmacology, (48) 443-50).
- Membranes were prepared by a standard procedure. Briefly, cells were harvested using 10 mM EDTA and collected by centrifugation.
- Pelleted cells were homogenized in ice-cold 20 mM Hepes (pH 7.4), 10 mM EDTA and protease inhibitors (Complete protease inhibitor cocktail tablet, Roche) using an Ultra Turrax Homogenizer. The homogenate was centrifuged at 14 000 rpm for 45 min. at 4 0 C. The resultant pellet was resuspended in the same buffer but with only 0.1 mM EDTA and was again centrifuged at 14 000 rpm for 45 min. at 4 0 C.
- the resulting pellet (membranes) was resuspended in 20 mM Hepes (pH 7.4), 0.1 mM EDTA, 2 mM MgCb and protease inhibitors and protein concentration was determined by Micro BCA Protein Assay Reagent Kit (Pierce Biotechnology) according to the manufacturer instructions.
- the [ 35 S]GTP ⁇ S SPA (Scintillation Proximity Assay) binding assay was performed by incubating 5 ⁇ g/well hCB1- membranes with 1 nM [ 35 S]GTP ⁇ S (Perkin Elmer - NEG 030H) in the presence of 3 nM of CP55940 and various concentrations of the test compounds at room temperature for 1hr in 96-well microtiter plates. 0.4mg/well SPA beads (PVT-WGA; RPNQ0001 Amersham Pharmacia Biotech) were then added and the incubation continued for further 30 min. on an orbital shaker.
- the assay buffer contained 5OmM HEPES (pH 7.5), 50 mM NaCI, 2.5 mM MgCI 2 , 0.1% BSA, 1 ⁇ M GDP and 100 ⁇ g/ml Saponin.
- Microtiter plates were centrifuged at 1500 rpm for 5 min. and radioactivity was read immediately using a Topcounter (PerkinElmer Life Sciences). Data were analyzed and IC50 values determined by non-linear regression using the Prism software (GraphPad Software, San Diego).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Child & Adolescent Psychology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Compounds of formula (I) are modulators of cannabinoid receptor CB1, useful for the treatment of obesity and diseases for which obesity is a risk factor: (I) wherein: X is a bond, Or-C(R10)(R11)-*. -C(R10)(R11)-O-*, -C(R10)(R11)CH2-*, -C(R10)(R11)CH2-O-*, -CH2C(R10)(R11)-*, -CH2C(R10)(R11)-O-*, or -CH2-O-C(R10)(R11)-*; Y is =O, =N(R9), =N(OR9), =CH(CN), =C(CN)2 or =N(CN); Z is a bond, -O- or -NH-; R1 and R2 are hydrogen, (CrC3)alkyl, (C3-C6)cycloalkyl or monocyclic heterocyclic; or R1 and R2 taken together with the nitrogen to which they are attached form a cyclic amino ring; R3 is hydrogen or a radical of formula -(O)a- AIk3 wherein a and AIk3 are as defined in the claims; R4 is a radical of formula -(O)b-(Alk1)p-(Q)r-(L)s-Q2 wherein b, p, r, s AIk1, L, Q1 and Q2 are as defined in the claims; or R3 and R4 taken together with the nitrogen to which they are attached form a cyclic amino ring; R5 is hydrogen -F, -Cl, -Br, -CN, (C1-C3)alkyl, (C1-C3)fluoroalkyl, cyclopropyl, -COOH, tetrazole or -OR9; R6, R7 and R8 are hydrogen -F, -Cl, -Br, -CN, (C1-C3)alkyl, (C1-C3)fluoroalkyl, cyclopropyl, or -OR9; and R9 R10 and R11 are as defined in the claims.
Description
CANNABINOID RECEPTOR MODULATORS
The present invention relates to compounds which are modulators of cannabinoid receptor CB1 and which suppress the normal signalling activity of such receptors. The invention further relates to compositions and methods using said compounds for the treatment of obesity and overweight, and diseases for which obesity is a risk factor, such^as metabolic syndrome, type 2 diabetes, cardiovascular disease, osteoarthritis, and some cancers; mental disorders; addictive disorders; neurological disorders; sexual dysfunctions; reproductive dysfunctions; osteoporosis; liver cirrhosis.and liver fibrosis; and, epilepsy. The invention also relates to pharmaceutical compositions containing the compounds of the invention, and to the use of the compounds in combination with other treatments for obesity and overweight and for obesity-related diseases.
Background to the invention
The prevalence of obesity in North America and in most European countries have more than doubled in the last 20 years and over half the adult population are now either overweight or obese. Obesity is now recognized as a chronic disease and a critical global health issue (Fiegal et al, 1998, Int. J. Obesity 22:39-47, Mokdad et al, 1999, JAMA 282:1519-1522; Halford, 2006, Appetite, 46, 6-10). The "identifiable signs and symptoms" of obesity include an excess accumulation of fat or adipose tissue, an increase in the size or number of fat cells (adipocyte differentiation), insulin resistance, increased glucose levels (hyperglycemia), increased blood pressure, elevated cholesterol and triglyceride levels and decreased levels of high-density lipoprotein. Obesity is associated with a significantly elevated risk for type 2 diabetes, coronary heart disease, strokes, hypertension, various types of cancer and numerous other major illnesses, and overall mortality from all causes (Must et al, 1999, JAMA 282:1523-1529, CaIIe et al, 1999, N. Engl. J. Med. 341 :1097-1105).
Like obesity, the prevalence of obesity-related diseases such as diabetes also continues to rise. Weight reduction is critical for the obese patient as it can improve cardiovascular and metabolic values to reduce obesity-related morbidity and mortality (Blackburn, 1999, Am. J. Clin. Nujtr. 69:347-349, Galuska et al, 1999, JAMA 282:1576). It has been shown that 5-10% loss of body weight can substantially improve metabolic parameters and reduce risk factors for diabetes, cancer and cardiovascular disease such as high fasting and post-prandial blood glucose, HbAIc
(glycosylated haemoglobin), insulin, total plasma cholesterol, low density lipoproteins (LDL), triglycerides, uric acid and blood pressure (Goldstein, 1992, J. Obesity, 6, 397-415).
Thus, the primary aim of treatment for obesity is weight loss. Initially, treatments have been proposed which were based on diet and lifestyle changes augmented by therapy with pharmacological therapies. However, while physical exercise and reductions in dietary intake of calories can improve the obese condition, compliance with this treatment is very poor because of sedentary lifestyles and excess food consumption, especially high fat containing food. Additionally, treatment with the available pharmacological therapies to facilitate weight loss fail to provide adequate benefit to many obese patients because of side effects, contraindications or lack of positive response. Hence, there is impetus for developing new and alternative treatments for management of obesity.
Several potential anti-obesity agents are currently investigated (for a review, see Bays, 2004, Obesity Research, 12, 1197-1211) such as i) central nervous system agents that affect neurotransmitters or neural ion channels (e.g. antidepressants (bupropion), noradrenaline reuptake inhibitors (GW320659), selective 5HT 2c receptor agonists, antiseizure agents (topiramate, zonisamide), some dopamine antagonists, cannabinoid CB-1 receptor antagonists (rimonabant); ii) leptin/insulin/central nervous system pathway agents (e.g. leptin analogues, leptin transport and/or receptor promoters, CNTF (Axokine), NPY antagonists, AgRP antagonists, POMC promoters, CART promoters, MSH analogues, MC4 receptor agonists, agents that affect insulin metabolism/activity [PTP-1 B inhibitors, PPAR receptor antagonists, short-acting bromocriptine (ergoset), somatostatin agonists (octreotide), and adiponectin/Acrp30 (Famoxin or Fatty Acid Metabolic oxidation INducer)]) ; iii) gastrointestinal-neural pathway agents (e.g. agents that increase CCK and PYY activity, agents that increase GLP-1 activity (extendin 4, liraglutide, dipeptidyl peptidase IV inhibitor), agents that decrease ghrelin activity, amylin (pramlinitide), neuropeptide Y agonists) ;
iv) agents that may increase resting metabolic rate (beta-3 agonists, UCP homologues, thyroid receptor agonists) ; and v) other more diverse agents, such as for example including (MCH) melanin concentrating hormone antagonists, phytostanol analogues, functional oils, P57, amylase inhibitors, growth hormone fragments, synthetic analogues of DHEAS (fluasterone), antagonists of adipocyte 11 beta- hydroxysteroid dehydrogenase type 1 activity, CRH agonists, carboxypeptidase inhibitors, inhibitors of fatty acid synthesis (cerulenin and C75), indanones/indanols, aminosterols (trodusquemine), other gastrointestinal lipase inhibitors (ATL962 ).
Drugs effective in obesity treatment may act by different mechanisms such as reduction in food intake (e.g. by inducing satiety), drugs altering metabolism (such as agents modifying the absorption of nutrients e.g. inhibition of fat absorption), drugs that increase energy expenditure (e.g. increase of thermogenesis), drugs that inhibit lipogenesis or that stimulate adipocyte apoptosis. However, only few drugs are available for obesity treatment (for reviews, see Gadde and Allison, 2006, Circulation, 114, 974-984; Weigle, 2003, J Clin Endocrinol Metab., 88, 2462-2469; Schiδth, 2006, CNS Neurol. Disorders Drug Targets, 5, 241-249). Sibutramine is a centrally acting mixed inhibitor of serotonin and norepinephrine presynaptic re-uptake. Orlistat is an inhibitor of gastrointestinal lipases that reduces fat absorption in the gut. Rimonabant (SR141716, Acomplia ®) is a centrally and peripherally acting cannabinoid CB1 modulator (antagonist and inverse agonist) that recently has been approved for treatment of obesity ( for a review see Pagotto et al, 2006, Endocrine Reviews, 27, 73-100; for reports on phase III clinical trials see Despres et al, 2005, N. Engl. J. Med. 353, 212; van Gaal et al, 2005, Lancet, 16, 1389; Pi-Sunyer et al, 2006, JAMA, 295, 761 ).
Presently, two cannabinoid receptors have been characterized: CB1 , a receptor found in the mammalian brain and in a number of other sites in peripheral tissues; and CB2, a peripheral receptor found principally in cells related to the immune system. For a reviews on cannabinoid CB1 and CB2 receptor modulators, see Pertwee, 2000, Exp. Opin. Invest. Drugs, 9, 1553-1571 and Muccioli, 2005, Cur. Med. Chem., 12, 1361-1394. A substantial body of evidence indicates that CB1 antagonists (e.g. rimonabant) are able to modulate energy homeostasis and that
CB 1 antagonists are able to modulate food intake as well as peripherally block lipogenic processes ( Pagotto et al, 2006, Endocrine Reviews, 27, 73-100; Tucci et al, 2006, Curr. Med. Chem. 13, 2669-2680; Lange and Kruse, 2004, Current Opinion in Drug Discovery & Dev., 7, 498-506). The peripheral effects of CB1 antagonists can be mediated by several target organs and mechanisms, e.g. i) liver: block of de novo lipogenesis, ii) muscles: increase in glucose uptake, iii) adipose tissue: adiponectin stimulation, inhibition of lipogenic enzymes, stimulation of GLUT4, generation of futile cycles, iv) pancreas: insulin regulation and v) gastrointestinal tract: stimulation of satiety signals.
Rimonabant (Acomplia ®) is approved as an adjunct to diet and exercise for treatment of obesity. While the effects on body weight and other risk factors (plasma triglycerides, HDL cholesterol, plasma insulin, insulin resistance, adiponectin) are very encouraging, there are also undesirable side effects, possibly centrally mediated (psychiatric and nervous system disorders), such as anxiety, depressive disorders, sleep disorders, nausea, and vomiting (cf. http://emc.medicines.org.uk; http://www.emea.europa.eu/humandocs/PDFs/EPAR/acomplia/AcompliaEparScientifi cD-en.pdf). Accordingly, there still exists a need for alternative CB 1 receptor antagonists associated with differing pharmacokinetic, pharmacological and side- effect profiles.
The CB1 receptor has been invoked in many disease states (cf. review by Pacher et al, 2006, Pharmacol. Rev, 58, 389-462). Modulators of CB1 receptor activity can be useful in the treatment of diseases associated with a CB1 receptor regulation such as obesity and overweight, prevention of weight gain (e.g. induced by medications or smoking cessation), diseases associated with obesity as risk factor (e.g. metabolic syndrome, type 2 diabetes, cardiovascular disease, osteoarthritis, and some cancers), metabolic disorders (e.g. dyslipidemia, hyperlipidemia, low HDL and/or high LDL cholesterol levels, hypertriglycerideemia, low adiponectin levels, impaired glucose tolerance, insulin resistance, HbAIc [glycosylated haemoglobin], diabetes mellitus, type 2 diabetes, reduced metabolic activity, fatty liver), eating disorders, addictive disorders (e.g. to marijuana, psychostimulants, nicotine, alcohol, cocaine, opiates), mental disorders (e.g. schizophrenia, schizo-affective disorder, bipolar disorders, anxiety, panic disorder), neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis, liver cirrhosis, liver fibrosis,, and epilepsy.
Since obesity leads to, or significantly increases the risk of, co-morbidities involving various body systems (see Bays, 2004, Obesity Research, 12, 1197-1211 ) including: i) cardiovascular (hypertension, congestive cardiomyopathy, varicosities, pulmonary embolism, coronary heart disease [CHD], liver cirrhosis), ii) neurological (stroke, idiopathic intracranial hypertension, meralgia parethetica), iii) respiratory (dyspnea, obstructive sleep apnea, hypoventilation syndrome, Pickwickian syndrome, asthma), iv) musculoskeletal (immobility, degenerative osteoarthritis, low back pain), v) skin (striae distensae or "stretch marks," venous stasis of the lower extremities, lymphedema, cellulitis, intertrigo, carbuncles, acanthosis nigricans, skin tags), vi) gastrointestinal (gastroesophageal reflux disorder, nonalcoholic fatty liver/steatohepatitis, cholelithiasis, hernias, colon cancer), vii) genitourinary (stress incontinence, obesity-related glomerulopathy, breast and uterine cancer), viii) psychological (depression and low self-esteem, impaired quality of life), and ix) endocrine (metabolic syndrome, type 2 diabetes, dyslipidemia, hyperandrogenemia in women, polycystic ovarian syndrome, dysmenorrhea, infertility, pregnancy complications, male hypogonadism) it is also useful to combine a CB1 modulator with medications used for treatment of such diseases.
Brief Description of the Invention
The present invention makes available a class of pyrazole compounds which modulate the activity of the cannabinoid receptor CB1. The following publications relate to other pyrazole compounds having CB1 modulatory activity: WO1997021682, WO1997019063, WO2000046209, WO2001058869, WO200129007, WO2003088968, WO2003020217, WO2004052864, , WO2005080343, WO2006067443, WO2006087480, EP00576357, EP00658546, US20030199536, US20040119972, US20040192667, US20050261281 ,
US20050624941 , US2006028084, US20060509367, J. Med. Chem. 1999 42, 769- 776, Biochem. Pharmacol, 2000, 60, 1315-1323, J. Med. Chem. 2003, 46, 642-645, Bioorg & Med. Chem. Lett. 2004, 14, 2393-2395, Current Med. Chem. 2005, 12, 1361-1394.
As described herein, the compounds of the invention are useful for the treatment of obesity and overweight, prevention of weight gain, obesity-related diseases (e.g. metabolic syndrome, type 2 diabetes, cardiovascular disease, osteoarthritis, and some cancers), metabolic disorders, dyslipidemia, eating disorders, addictive disorders, mental disorders, neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis, liver cirrhosis, liver fibrosis, and epilepsy. They are useful for modulating body weight and energy consumption in mammals and for modulating plasma parameters involved in the metabolic syndrome such as low HDL and/or high LDL cholesterol levels, high triglyceride levels, low adiponectin levels and high HbAIc [glycosylated haemoglobin] and for modulating other characteristics of the metabolic syndrome such as impaired glucose tolerance, insulin resistance, excessive fat tissue in and around the abdomen and high blood pressure.
The compounds of the invention display varying physicochemical properties and are useful for modulating peripheral CB1 receptors and to varying degree central CB1 receptors. Those compounds of the invention associated with a lowered central action on CB1 receptors may have a reduced propensity to induce psychiatric and nervous system side-effects.
Detailed Description of the Invention
According to the invention there is provided a compound of formula (I) or a salt, hydrate, solvate or N-oxide thereof:

X is a bond, or a divalent radical selected from -C(R1O)(Rn)-*, -C(RioXRii)-0-*, -C(R10)(R11)CH2-*, -C(R10)(R11)CH2-O-*, -CH2C(R10)(R11)-*, -CH2C(R10)(R1 ^-O-*, and -CH2-O-C(R10)(R11)-*, wherein the bond indicated by an asterisk is attached to the pyrazole ring;
Y is =0, =N(Rβ), =N(OR9), =CH(CN), =C(CN)2 or =N(CN);
Z is a bond, -O- or -NH-;
Ri and R2 are independently selected from hydrogen, (CrC3)alkyl, (C3-C6)cycloalkyl and monocyclic heterocyclic radicals having 4 to 6 ring atoms, each of said radicals being optionally substituted by fluoro, hydroxy, methoxy, -NH2 or mono- or di-(Cr C3)alkylamino; or Ri and R2 taken together with the nitrogen to which they are attached form a cyclic amino ring of 4 to 6 ring atoms which is optionally substituted by fluoro, hydroxy, methoxy, -NH2, or mono- or di-(Ci-C3)alkylamino;
R3 is hydrogen or a radical of formula -(O)3-AIk3 wherein a is O or 1 and AIk3 is (C1- C3)alkyl or (CrC3)fluoroalkyl;
R4 is a radical of formula -(O)b-(Alk-i)p-(Qi)r-(L)s-Q2 wherein
b, p, r and s are independently O or 1 , provided that (i) at least one of p, r and s is 1 , and (ii) a in R3 and b in R4 are not simultaneously 1 ;
AIk1 is a divalent (CrC4)alkylene radical which (a) is optionally substituted on one carbon by Ri0 and/or R11 or by one or two optional substituents, and/or (b) optionally contains a -0-, -S-, -CO-, -SO-, -SO2-, or -NR9- link;
L is a divalent radical of formula -(Alk2)n-(W)m-, in either orientation, wherein
n and m are independently O or 1 ;
AIk2 is -C(R10)(R11)-; and
W is -CO-, -SO2-, -O-, -NR9- or -SO-; provided that when W and/or Alk2 are linked to a heteroatom W is not -O-, -NR9- or -SO-;
Qi is a monocyclic carbocyclic ring of 3 to 7 ring atoms, a bicyclic carbocyclic ring system of 7 to 10 ring atoms, a monocyclic heterocyclic ring of 4 to 7 ring atoms or a bicyclic carbocyclic ring system of 8 to 10 ring atoms, any of which rings or ring systems being optionally substituted;
Q2 is (a) in the case where s in -(L)5-Q2 is O or 1 , a monocyclic carbocyclic ring of 3 to 7 ring atoms, a bicyclic carbocyclic ring system of 7 to 10 ring atoms, a monocyclic heterocyclic ring of 4 to 7 ring atoms or a bicyclic carbocyclic ring system of 8 to 10 ring atoms, any of which rings or ring systems being optionally substituted; or (b) only in the case where s in -(L)s-Q2 is 0, hydrogen;
or R3 and R4 taken together with the nitrogen to which they are attached form a cyclic amino ring of 4 to 7 ring atoms which is optionally substituted by a radical of formula -(L)3-Q2 wherein s, L and Q2 are as defined above, or by an optional substituent selected from hydroxy, methoxy, -NH2-, or mono- or di-(CrC3)alkylamino;
R5 is selected from hydrogen -F, -Cl1 -Br, -CN, (CrC3)alkyl, (Ci-C3)fluoroalkyl, cyclopropyl, -COOH, tetrazole and -OR9, the R9 part being optionally substituted with tetrazole or -COOH;
R6, R7 and R8 are each independently selected from hydrogen -F, -Cl, -Br, -CN, (CrC3)alkyl, (CrC3)fluoroalkyl, cyclopropyl, and -OR9;
R9 is hydrogen, (Ci-C3)alkyl, (CrC3)fluoroalkyl, or (C3-C6)cycloalkyl; and
R10 and R11 are independently selected from hydrogen and (CrC3)alkyl; or R10 and R11 taken together with the carbon atom to which they are attached form a (C3- C5)cycloalkyl ring.
In accordance with general principles of medicinal chemistry, it is preferred that the compounds of the invention should have a molecular weight of less than 650.
Another aspect of the invention is a pharmaceutical composition comprising a compound of formula (I) or a salt, hydrate, solvate or N-oxide thereof, together with one or more pharmaceutically acceptable carriers or excipients.
The compounds with which the invention is concerned suppress the normal signalling activity of cannabinoid receptor CB 1. Therefore, further aspects of the invention are: (i) The use of a compound of formula (I) or a salt, hydrate, solvate or N-oxide thereof in the preparation of a composition for treatment of obesity, overweight, diseases for which obesity is a risk factor, mental disorders, addictive disorders, neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis, liver cirrhosis, liver fibrosis, epilepsy, or pathologic conditions mediated by or related to CB 1 mediated mechanisms; and
(ii) A method for the treatment of treatment of obesity, overweight, diseases for which obesity is a risk factor, mental disorders, addictive disorders, neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis, liver cirrhosis, liver fibrosis,,, or pathologic conditions mediated by or related to CB1 mediated mechanisms, which method comprises administering to a subject suffering such disease or condition an effective amount of a compound of formula (I) or a salt, hydrate, solvate or N-oxide thereof.
Diseases or conditions for which obesity is a risk factor, include metabolic syndrome, type 2 diabetes, cardiovascular disease, osteoarthritis, and some cancers. Metabolic syndrome is associated with pathological symptoms such as low HDL and/or high LDL cholesterol levels, high triglyceride levels, low adiponectin levels and high HbAIc [glycosylated haemoglobin], impaired glucose tolerance, insulin resistance, excessive fat tissue in and around the abdomen and high blood pressure.
Terminology
As used herein, the term "(Ca-Cb)alkyl" wherein a and b are integers refers to a straight or branched chain alkyl radical having from a to b carbon atoms. Thus when a is 1 and b is 6, for example, the term includes methyl, ethyl, n-propyl, isopropyl, n- butyl, isobutyl, sec-butyl, t-butyl, n-pentyl and n-hexyl.
As used herein the term "divalent (Ca-Cb)alkylene radical" wherein a and b are integers refers to a saturated straight or branched hydrocarbon chain having from a to b carbon atoms and two unsatisfied valences.
As used herein the unqualified term "carbocyclic" refers to a mono-, bi- or tricyclic radical having up to 16 ring atoms, all of which are carbon, and includes aryl and cycloalkyl.
As used herein the unqualified term "cycloalkyl" refers to a monocyclic saturated carbocyclic radical having from 3-8 carbon atoms and includes, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
As used herein the unqualified term "aryl" refers to a mono-, bi- or tri-cyclic carbocyclic aromatic radical, and includes radicals having two monocyclic carbocyclic aromatic rings which are directly linked by a covalent bond. Illustrative of such radicals are phenyl, biphenyl and napthyl.
As used herein the unqualified term "heteroaryl" refers to a mono-, bi- or tri-cyclic aromatic radical containing one or more heteroatoms selected from S, N and O, and includes radicals having two such monocyclic rings, or one such monocyclic ring and one monocyclic aryl ring, which are directly linked by a covalent bond. Illustrative of such radicals are thienyl, benzthienyl, furyl, benzfuryl, pyrrolyl, imidazolyl, benzimidazolyl, thiazolyl, benzthiazolyl, isothiazolyl, benzisothiazolyl, pyrazolyl, oxazolyl, benzoxazolyl, isoxazolyl, benzisoxazolyl, isothiazolyl, triazolyl, benztriazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyridazinyl, pyrimidinyl, triazinyl, indolyl and indazolyl.
As used herein the unqualified term "heterocyclyl" or "heterocyclic" includes "heteroaryl" as defined above, and in addition means a mono-, bi- or tri-cyclic non- aromatic radical containing one or more heteroatoms selected from S, N and O, and to groups consisting of a monocyclic non-aromatic radical containing one or more such heteroatoms which is covalently linked to another such radical or to a monocyclic carbocyclic radical. Illustrative of such radicals are pyrrolyl, furanyl, thienyl, piperidinyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, pyrazolyl, pyridinyl, pyrrolidinyl, pyrimidinyl, morpholinyl, piperazinyl, indolyl, morpholinyl,
benzfuranyl, pyranyl, isoxazolyl, benzimidazolyl, methylenedioxyphenyl, ethylenedioxyphenyl, maleimido and succinimido groups.
Unless otherwise specified in the context in which it occurs, the term "substituted" as applied to any moiety herein means substituted with up to four compatible substituents, each of which independently may be, for example, (CrC6)alkyl, (C1- C6)alkoxy, hydroxy, hydroxy(Ci-C6)alkyl, mercapto, mercapto(Ci-C6)alkyl, (C1- C6)alkylthio, halo (including fluoro, bromo and chloro), fully or partially fluorinated (C1- C3)alkyl, (CrC3)alkoxy or (CrCsJalkylthio such as trifluoromethyl, trifluoromethoxy, and trifluoromethylthio, nitro, nitrile (-CN), oxo, phenyl, phenoxy, monocyclic heteroaryl or heteroaryloxy with 5 or 6 ring atoms, tetrazolyl, -COORA, -CORA, -OCORA, -SO2RA, -CONRARB, -SO2NRARB, -NRARB, OCONRARB, -NRBCORA, -NRBCOORA, -NRBSO2ORA or -NRACONRARB wherein RA and RB are independently hydrogen or a (CrC6)alkyl group or, in the case where RA and RB are linked to the same N atom, RA and RB taken together with that nitrogen may form a cyclic amino ring, such as a morpholine, piperidinyl or piperazinyl ring. Where the substituent is phenyl, phenoxy or monocyclic heteroaryl or heteroaryloxy with 5 or 6 ring atoms, the phenyl or heteroaryl ring thereof may itself be substituted by any of the above substituents except phenyl, phenoxy, heteroaryl or heteroaryloxy. An "optional substituent" may be one of the foregoing substituent groups.
As used herein the term "salt" includes base addition, acid addition and quaternary salts. Compounds of the invention which are acidic can form salts, including pharmaceutically acceptable salts, with bases such as alkali metal hydroxides, e.g. sodium and potassium hydroxides; alkaline earth metal hydroxides e.g. calcium, barium and magnesium hydroxides; with organic bases e.g. N-methyl-D-glucamine, choline tris(hydroxymethyl)amino-methane, L-arginine, L-lysine, N-ethyl piperidine, dibenzylamine and the like. Those compounds (I) which are basic can form salts, including pharmaceutically acceptable salts with inorganic acids, e.g. with hydrohalic acids such as hydrochloric or hydrobromic acids, sulphuric acid, nitric acid or phosphoric acid and the like, and with organic acids e.g. with acetic, tartaric, succinic, fumaric, maleic, malic, salicylic, citric, methanesulphonic, p-toluenesulphonic, benzoic, benzenesunfonic, glutamic, lactic, and mandelic acids and the like.
For a review on suitable salts, see Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
The term 'solvate' is used herein to describe a molecular complex comprising the compound of the invention and a stoichiometric amount of one or more pharmaceutically acceptable solvent molecules, for example, ethanol. The term 'hydrate' is employed when said solvent is water.
Compounds with which the invention is concerned which may exist in one or more stereoisomeric form, because of the presence of asymmetric atoms or rotational restrictions, can exist as a number of stereoisomers with R or S stereochemistry at each chiral centre or as atropisomeres with R or S stereochemistry at each chiral axis. The invention includes all such enantiomers and diastereoisomers and mixtures thereof.
The compounds of the invention include compounds of formula (I) as hereinbefore defined, including all polymorphs and crystal habits thereof, prodrugs and isomers thereof (including optical, geometric and tautomeric isomers) as hereinafter defined and isotopically-labeled compounds of formula (I).
So-called 'pro-drugs' of the compounds of formula (I) are also within the scope of the invention. Thus certain derivatives of compounds of formula (I) which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into compounds of formula (I) having the desired activity, for example, by hydrolytic cleavage. Such derivatives are referred to as 'prodrugs'. Further information on the use of prodrugs may be found in Pro-drugs as Novel Delivery Systems. Vol. 14, ACS Symposium Series (T. Higuchi and V.J. Stella) and Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (ed. E. B. Roche, American Pharmaceutical Association; CS. Larsen and J. østergaard, Design and application of prodrugs, In Textbook of Drug Design and Discovery, 3rd Edition, 2002, Taylor and Francis ).
Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the compounds of formula (I) with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in Design of Prodrugs by H. Bundgaard (Elsevier, 1985). Such examples could be a prodrug of a carboxyl group (such as -CO-O-CH2-O-COtBu as used in the pivampicillin prodrug of ampicillin), an amide (-CO-NH-CH2-NAIk2) or an amidine ( -C(=N-O-CH3)-NH2).
Also included within the scope of the invention are metabolites of compounds of formula (I), that is, compounds formed in vivo upon administration of the drug. Some examples of metabolites include
(i) where the compound of formula I contains a methyl group, an hydroxymethyl derivative thereof (-CH3 -> -CH2OH):
(ii) where the compound of formula I contains an alkoxy group, an hydroxy derivative thereof (-OR -> -OH);
(iii) where the compound of formula I contains a tertiary amino group, a secondary amino derivative thereof (-NR1R2 -> -NHR1 or -NHR2);
(iv) where the compound of formula I contains a secondary amino group, a primary derivative thereof (-NHR1 -> -NH2);
(v) where the compound of formula I contains a phenyl moiety, a phenol derivative thereof (-Ph -> -PhOH); and
(vi) where the compound of formula I contains an amide group, a carboxylic acid derivative thereof (-CONH2 -> COOH).
For use in accordance with the invention, the following structural characteristics are currently contemplated, in any compatible combination, in the compounds (I):
The variables X, Y and Z:
The divalent linker radical -X- is a bond, or a divalent radical selected from -C(R10)(R11)-*, -C(R1O)(Rn)-O-*, -C(R10)(R1I)CH2-*, -C(R10)(R11)CH2-O-*, -CH2C(R10XR11)-*, -CH2C(R10)(R10-O-*, and -CH2-O-C(R10)(R11)-*, wherein the bond indicated by an asterisk is attached to the pyrazole ring. Ri0 and R11 are independently selected from hydrogen and (CrC3)alkyl (ie. methyl, ethyl and n- or isopropyl); or R10 and R11 taken together with the carbon atom to which they are attached form a (C3-C5)cycloalkyl ring such as a cyclopropyl or cyclopentyl ring. Specifically, -X- may be, for example, a bond, -CH2- , -CH2O-*, or -CH2OCH2-*, wherein the bond indicated by an asterisk is attached to the pyrazole ring.
Z is a bond, -O- or -NH-, and specific examples of the combination linker radical -Z- X- are -CH2- , -CH2O-*, -CH2OCH2-*, -NH- Or -NH-CH2-* wherein the bond indicated by an asterisk is attached to the pyrazole ring.
Y is =0, S=N(R9), =N(ORβ), =CH(CN), =C(CN)2 or =N(CN), wherein R9 is hydrogen, (CrC3)alkyl such as methyl or ethyl, (CrC3)fluoroalkyl such as di- or trifluoromethyl, or (C3-C6)cycloalkyl such as cyclopropyl; In particular sub-classes of compounds of the invention, Y is =0, =NH, =N(OH) or =N(CN).
In one currently preferred subclass of compounds of the invention -Z-X- is -CH2- and Y is =0, =NH, or =N(OH)
The substituents R1 and Rz
Ri and R2 are independently selected from hydrogen, (C-i-C3)alkyl such as methyl (C3-C6)cycloalkyl such as cyclopropyl, cyclopentyl or cyclohexyl, and monocyclic heterocyclic radicals having 4 to 6 ring atoms such as morpholinyl, 4-(Cr C6)alkylpiperazinyl, 4-acetylpiperazinyl, 4- (CrC6)alkylsulfonylpiperazinyl, azepanyl, pyrrolidinyl or piperidinyl, each of said radicals being optionally substituted by fluoro, hydroxy, methoxy, -NH2 or mono- or di-(Ci-C3)alkylamino such as methylamino, dimethylamino, ethyl amino and diethylamino; or R1 and R2 taken together with the nitrogen to which they are attached form a cyclic amino ring of 4 to 6 ring atoms such as morpholinyl, piperidinyl or piperazinyl, which is optionally substituted by fluoro, hydroxy, methoxy, -NH2, or mono- or di-(CrC3)alkylamino such as methylamino, dimethylamino, ethyl amino and diethylamino.
In particular embodiments of the compounds of the invention, R-i and R2 are each hydrogen. In other particular embodiments, R1 is hydrogen and R2 is methyl, cyclopropyl or optionally substituted cyclohexyl. In other particular embodiments R1 and R2 taken together with the nitrogen to which they are attached form a morpholine or an optionally substituted piperidine or piperazine ring. In both contexts, examples of optional substituents include one or two substituents selected from hydroxy, methoxy and -NH2.
The substituent R?
R3 is hydrogen or a radical of formula -(O)a-Alk3 wherein a is O or 1 and AIk3 is (Cr C3)alkyl such as methyl or ethyl or (CrC3)fluoroalkyl such as di- or trifluoromethyl. R3 will often be hydrogen.
The substituent R*
As will be apparent from its definition above, a great diversity of substituents may be present in compounds of the invention.
R4 has formula -(OV(AIk1 )p-(Qi X-(L)5-Q2 wherein b, p, r and s are 0 or 1 in any compatible combination, provided that (i) at least one of p, r and s is 1 , and (ii) a in R3 and b in R4 are not simultaneously 1 , the latter proviso being necessary to avoid incompatible substitutions on the relevant nitrogen. In one particular type of compound of the invention b, p and s are 0, and r is 1 , and in another p and r are 1 and b and s are 0.
In R4. AIk1 when present is a divalent (C-|-C4)alkylene radical which (a) is optionally substituted on one carbon by R10 and/or R11 or by one or two optional substituents, and/or (b) optionally contains a -O-, -S-, -CO-, -SO-, -SO2-, or -NR9- link. In this context, R10 and R-π may be, for example, methyl; or R10 and R-n taken together with the carbon atom to which they are attached may form, for example, a cyclopropyl or cyclopentyl ring. Optional substituents in this context include fluoro and hydroxyl.
Examples of such radicals include -CH2-, -CH2CH2- -CH2CH2CH2-, - CH2CH2CH2CH2-, -CH2T-, -CH2TCH2- -CH2CH2TCH2-, -CH2CH2TCH(CHs)-, — CH2TCH2CH2-, and -CH2TCH2CH2WCH2-, where T is -O-, -S-, -NH-, or -N(CH3)-, and any of the foregoing wherein one or more hydrogens are exchanged for fluorines, and/or wherein one or two carbons are substituted by methyl or, trifluoromethyl; or wherein one carbon is substituted by a spiro-linked cyclopropyl substituent.
In the compounds of the invention, Alki, when present will often be simply -CH2-. Thus in one type of compound of the invention p and r are 1 and b and s are 0.
In R4, L when present is a divalent radical of formula -(Alk2)n-(W)m-, in either orientation, wherein n and m are independently 0 or 1 ; AIk2 is -C(RiO)(Rn)-; and W is -CO-, -SO2-, -0-, -NRg- or -SO-; provided that when W and/or AIk2 are linked to a heteroatom W is not -0-, -NRg- or -SO-. The latter restriction is in order to avoid unstable or undesirable potential structures. In this context, Rg may be, for example, hydrogen or methyl. Examples Of AIk2, when present are -CH2-, -CH(CH3)-, a
cyclopropyl ring which is linked to each adjacent atom via the same ring carbon, and, in either orientation -CH2O-, -CH2NH-, -CH(CH3)O- and -CH(CH3)NH-.
In R4, Q1 (when present) and Q2 are each independently a monocyclic carbocyclic radical of 3 to 7 ring atoms, a bicyclic carbocyclic ring system of 7 to 10 ring atoms, a monocyclic heterocyclic ring of 4 to 7 ring atoms or a bicyclic carbocyclic ring system of 8 to 10 ring atoms, any of which rings or ring systems being optionally substituted; Q2 may also be hydrogen when s in -(L)5-Q2 IS 0; Examples of Qi, when present, include optionally substituted divalent phenyl, pyridine, piperidine, piperazine or (C3- C7)cycloalkyl, eg cyclohexyl, cyclopentyl or cyclopropyl, radicals. Examples Of Q2 include hydrogen, or optionally substituted phenyl or pyridyl. Optional substituents in rings Q1 and Q2 include -F, -Cl, -Br, -CN, -CF3, -CH3, cyclopropyl, and -OCH3, and may be present on, for example 1 or 2 ring atoms.
The groups R? and R4 together
In other types of compound of the invention, R3 and R4 taken together with the nitrogen to which they are attached form a cyclic amino ring of 4 to 7 ring atoms. Examples of such rings include morpholinyl, pyrrolidinyl, piperidinyl, azepanyl and piperazinyl.
The ring formed by R3 and R4 taken together with the nitrogen to which they are attached may be substituted by a radical of formula -(L)3-Q2 wherein s, L and Q2 are as defined and discussed above, or by an optional substituent selected from hydroxy, methoxy, -NH2-, or mono- or di-(C1-C3)alkylamino such as methylamino, ethylamino, dimethylamino and diethylamino.
Specific examples of the radical -C(=O)NR3R4 include those of formulae (A)-(T):

(G) (H) (J)

(Q) (R)

(S) (T) wherein
R10 and R11 are as defined in claim 1
Ri2 is selected from hydrogen, -CH3, -OH, -CN and -COOH;
R13 is selected from hydrogen, -F, -CF3, -OCF3, -Br. -Cl, -OCH3, -CH3, -CN and
-COOH;
R14 is selected from hydrogen, -F, -CF3, -OCF3, -Br. -Cl, -OCH3, -CH3, -CN, -OH, and -COOH;
Ri5 and Ri6 are independently selected from hydrogen and (Ci-C6)alkyl or Ri5 and Ri6 taken together with the nitrogen to which they are attached form a cyclic amino ring of 4 to 7 ring atoms;
R17 is selected from hydrogen, (CrC6)alkyl, (CrC6)alkylC(=O)-, (CrC6)alkylSO2-, benzyloxycarbonyl-, and -C(=O)OCH3; Ri8 is selected from hydrogen, -F and -CN;
R19 is selected from F, -CF3, -OCF3, -Br. -Cl, -OCH3, -CH3, -CN and -COOH; and R20 is selected from F, -CF3, -OCF3, -Br. -Cl, -OCH3, -CH3, -CN, -OH and -COOH.
In one group of compounds of the invention, the radical -C(=O)NR3R4 is selected from the group consisting of:
(i) Formula D, wherein R12 is hydrogen and R14 is methyl, trifluoromethyl, trifluoromethoxy, 3-, 4- or 5-fluoro, 3-, 4- or 5-chloro, or 3-, 4- or 5-cyano, or 6- hydroxy;
(ii) Formula E, wherein R12 is hydrogen and R14 is methyl, trifluoromethyl, , 4- or 5- fluoro, , 4- or 5-chloro, or, 4- or 5-cyano, or 2-, 6-hydroxy; (iii) Formula H;
(iv) Formula L, wherein R1S is hydrogen, 3-, 4- or 5-fluoro, 3-, 4- or 5-cyano, and R20 is methyl, trifluoromethyl, trifluoromethoxy, 3-, 4- or 5-fluoro, 3-, 4- or 5-chloro, or 3-, 4- and 5-cyano, or 6-hydroxy;
(v) Formula M, wherein R18 is hydrogen, 4- or 5-fluσro, , 4- or 5-cyano, and R2o is methyl, trifluoromethyl, trifluoromethoxy, , 4- or 5-fluoro, , 4- or 5-chloro, or, 4- and 5- cyano, or 2-, 6-hydroxy;
(vi) Formula P wherein Ri8 is hydrogen, 3-, 4- or 5-fluoro, 3-, 4- or 5-nitrile, R20 is methyl, trifluoromethyl, trifluoromethoxy, 3-, 4- or 5-fluoro, 3-, 4- or 5-chloro, or 3-, 4- and 5-cyano, or 6-hydroxy, R10 is hydrogen and R11 is methyl, the configuration of the asymmetric carbon carrying R10 and R1-I being (R); and (vii) Formula Q wherein Ri8 is hydrogen, , 4- or 5-fluoro, , 4- or 5-cyano, R20 is methyl, trifluoromethyl, trifluoromethoxy, , 4- or 5-fluoro, , 4- or 5-chloro, or, 4- and 5- cyano, or 2-, 6-hydroxy, R10 is hydrogen and R11 is methyl, the configuration of the asymmetric carbon carrying R10 and Ri1 being (R).
In another group of compounds of the invention, the radical the radical -C(=O)NR3R4 is selected from the group consisting of:
(a) Formula A;
(b) Formula B;
(c) Formula C, wherein Ri2 is hydroxyl, cyano or -COOH, and R13 is fluoro, methyl, trifluoromethyl, cyano, or methoxy;
(d) Formula K, wherein Ri8 is hydrogen or fluoro and R-ig is cyano , methyl or methoxy;, and
(e) Formula O, wherein Ri8 is hydrogen or fluoro , Ri9 is cyano, methyl or methoxy, Rio is hydrogen and Rn is methyl, the configuration of the asymmetric carbon carrying Ri0 and Rn being (R)., (f) Formula T
The substituents RB, RR, RI and RR
R5> Re, R7 and R8 are each independently selected from hydrogen -F, -Cl, -Br, -CN, (Ci-C3)alkyl such as methyl, (Ci-C3)fluoroalkyl such as trifluoromethyl, cyclopropyl, and -OR9 wherein R9 is methyl or ethyl. In one type of compound of the invention, R5, R6, R7 and R8 are independently selected from hydrogen, fluoro and chloro. In another type of compound R5 is selected from -COOH, tetrazole or -OR9 having the Rg part substituted with tetrazole or -COOH;
Specific compounds of the invention include those of the Examples herein, and in particular the following:
4-Carbamoylmethyl-5-(4-chloro-phenyl)-1-(2-chloro-phenyl)-1 H-pyrazole-3-carboxylic acid (4-methyl-pyridin-3-yl)-amide,
4-Carbamoylmethyl-1-(2-chloro-phenyl)-5-(4-chloro-phenyl)-1 H-pyrazole-3-carboxylic acid (i-phenyl-ethyl)-amide
2-{1-(2-Chloro-phenyl)-5-(4-chloro-phenyl)-3-[4-(4-fluoro-phenyl)-4-hydroxy- piperidine-1 -carbonyl]-1 H-pyrazol-4-yl}-acetamide,
i-^-Carbamoylmethyl-i^-chloro-phenyO-δ^-chloro-phenyO-I H-pyrazole-S- carbonyl]-4-phenyl-piperidine-4-carboxylic acid methyl ester,
i-^-Carbamoylmethyl-δ^-chioro-phenyO-i^-chloro-phenyO-I H-pyrazole- 3carbonyl]-4-phenyl-piperidine-4-carboxylic acid
^Carbamimidoylmethyl-δ^-chloro-phenyl^i^Σ-chloro-phenyO-I H-pyrazole-S- carboxylic acid (4-fluoro-phenyl)-amide,
2-{1-(2-Chloro-phenyl)-5-(4-chloro-phenyl)-3-[4-(4-fluoro-phenyl)-4-hydroxy- piperidine-1 -carbonyl]-1 H-pyrazol-4-yl}-acetamidine,
2-{5-(4-Chloro-phenyl)-1-(2-chloro-phenyl)-3-[4-(2-fluoro-phenyl)-piperidine-1- carbonyl]-1 H-pyrazol-4-yl}-N-hydroxyacetamidine,
5-(4-Chloro-phenyl)-1-(2-chloro-phenyl)-4-(N-hydroxycarbamimidoylmethyl)-1 H- pyrazole-3-carboxylic acid piperidin-1-ylamide,
5-(4-Chloro-phenyl)-1-(2-chloro-phenyl)-4-(N-hydroxycarbamimidoylmethyl)-1 H- pyrazole-3-carboxylic acid [(R)-1-(4-trifluoromethyl-phenyl)-ethyl]-amide,
and salts hydrates, solvates and N-oxides thereof.
The compounds of the present invention act on central and peripheral cannabinoid receptor CBl Some compounds distribute to a lesser extent to the central nervous system, i.e. the compound less readily crosses the blood-brain barrier and will be associated with fewer central nervous system mediated side-effects.
The compounds of the invention modulate the cannabinoid receptor CB1 by suppressing its natural signalling function. The compounds are therefore CB1 receptor antagonists, inverse agonists, or partial agonists.
The term "CB1 antagonist" or "cannabinoid receptor CB1 antagonist" refers to a compound which binds to the receptor, or in its vicinity, and lacks any substantial ability to activate the receptor itself. A CB1 antagonist can thereby prevent or reduce the functional activation or occupation of the receptor by a CB1 agonist such as for
example the endogenous agonist N-Arachidonylethanolamine (anandamide). This term is well known in the art.
The term "CB1 inverse agonist" or "cannabinoid receptor CB1 inverse agonist" refers to a compound which binds to the receptor and exerts the opposite pharmacological effect as a CB1 receptor agonist does. Inverse agonists are effective against certain types of receptors which have intrinsic activity without the acting of a ligand upon them (also referred to as 'constitutive activity'). This term is well known in the art. It is also well known in the art that such a CB1 inverse agonist can also be named a CB1 antagonist as the general properties of both types are equivalent. Accordingly, in the context of the present invention the term "CB1 antagonist" in general is understood as including both the "CB1 antagonist" as defined above and the "CB1 inverse agonist".
The term "CB 1 partial agonist" or "cannabinoid receptor CB 1 partial agonist" refers to a compound which acts upon the same receptor as the full agonist but that produces a weak maximum pharmacological response and has a low level of intrinsic activity. This term is well known in the art.
According to a preferred embodiment of the present invention, the "CB1 modulator" or "cannabinoid receptor CB1 modulator" is a CB1 antagonist or inverse agonist compound.
The compounds of the invention are useful for the treatment of obesity and overweight, prevention of weight gain, obesity-related diseases (e.g. metabolic syndrome, type2 diabetes, cardiovascular disease, osteoarthritis, and some cancers), metabolic disorders, dyslipidemia, eating disorders, addictive disorders, mental disorders, neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis, liver cirrhosis, liver fibrosis, and epilepsy. The compounds of the invention are useful for modulating body weight and energy consumption in mammals and for modulating plasma parameters involved in the metabolic syndrome such as low HDL and/or high LDL cholesterol levels, high triglyceride levels, low adiponectin levels and high HbAIc [glycosylated haemoglobin] and for modulating other characteristics of the metabolic syndrome such as impaired
glucose tolerance, insulin resistance, excessive fat tissue in and around the abdomen and high blood pressure. The compounds of the invention display varying physicochemical properties and are useful for modulating peripheral CB1 receptors and to varying degree central CB1 receptors. Those compounds of the invention associated with a lowered central action on CB 1 receptors may have a reduced propensity to induce psychiatric and nervous system side-effects.
The compounds of the invention may be combined with another therapeutic agent used in treatment of obesity acting by a different mode of action such as central action on satiety or hunger signals, craving mechanisms, appetite regulation, leptin/insulin/central nervous system pathways, gastrointestinal-neural pathways, metabolic rate, energy expenditure, food intake, fat storage, fat excretion, gastrointestinal motility, lipogenesis, glucose transport, glucogenolysis, glycolysis, lipolysis, etc including modulators (inhibitors, agonists, antagonists, analogues) of monoaminergic (NA (noradrenaline), 5-HT (serotonin), DA (dopamine)) receptors or transporters, neural ion channels, leptin or leptin receptor, neuropeptide Y receptors, PP (pancreatic polypeptide), PYY, Protein YY3-36, ghrelin or ghrelin receptor, motilin or motilin receptor, orexins or orexin receptors, bombesin or bombesin-like peptide receptors, somatostatin or somatostatin receptors, MCHR1 (melanin concentrating hormone receptor 1), CNTF (ciliary neurotrophic factor), AgRP (agouti-related peptide), POMC (proopiomelanocortin), CART (cocaine and amphetamine regulated transcript), alpha-MSH (alpha-melanocyte-stimulating hormone), MC4 (melanocortin-4) or MC3 (melanocortin-3) receptor, galanin receptors, relaxin-3 receptor, GPR7 receptor, GPR119 receptor, GPR10 receptor, neuromedin U receptors, free-fatty-acid receptors, growth hormone, nesfatin-1, opioid receptors, neuropeptide FF receptors, PTP-1 B (protein-tyrosine phosphatase), PPAR (peroxisome proliferators activated receptors) receptors, retinoid X receptor heterodimers, adiponectin also known as Acrp30 (adipocyte complement-related protein of 3OkDa), fatty acid metabolism, H (histamine) receptors, CCK-A (Cholecystokinin-A) or CCK-A receptor, GLP-1 (glucagon-like peptide-1 ) or GLP-1 receptor, oxyntomodulin, adrenomedullin, DPP-IV (dipeptidyl peptidase IV), amylin, beta-3-adrenergic receptor, UCP (uncoupling protein), thyroid receptor, thyroid- stimulating hormone receptor, 11 beta-hydroxysteroid dehydrogenase type 1, amylase, DHEAS (dehydroepiandrosterone sulfate), CRH (corticotropin releasing hormone) or CRH receptors, carboxypeptidase, fatty acid synthesis, HMG-CoA reductase, ileal bile acid transport, gastrointestinal lipase, P57, AMP-activated protein kinase (AMPK).
The compounds of the invention may be combined with another therapeutic agent used in treatment of metabolic syndrome or obesity-related diseases such as cardiovascular (hypertension, congestive cardiomyopathy, varicosities, pulmonary embolism, coronary heart disease [CHD], liver cirrhosis), neurological (stroke, idiopathic intracranial hypertension, meralgia parethetica), respiratory (dyspnea, obstructive sleep apnea, hypoventilation syndrome, Pickwickian syndrome, asthma), musculoskeletal (immobility, degenerative osteoarthritis, low back pain, osteoporosis), skin (striae distensae or "stretch marks," venous stasis of the lower extremities, lymphedema, cellulitis, intertrigo, carbuncles, acanthosis nigricans, skin tags), gastrointestinal (gastro-esophageal reflux disorder, nonalcoholic fatty liver/steatohepatitis, cholelithiasis, hernias, colon cancer), genitourinary (stress incontinence, obesity-related glomerulopathy, breast and uterine cancer), psychological (depression and low self-esteem, impaired quality of life), and endocrine (metabolic syndrome, type 2 diabetes, dyslipidemia, hyperandrogenemia in women, polycystic ovarian syndrome, dysmenorrhea, infertility, pregnancy complications, male hypogonadism) diseases.
The compounds of the invention may be combined with proper reduction in dietary calorie intake and physical exercise.
It will be understood that the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, route of administration, rate of excretion, drug combination and the severity of the particular disease undergoing treatment. Optimum dose levels and frequency of dosing will be determined by clinical trial, as is required in the pharmaceutical art. However, for administration to human patients, the total daily dose of the compounds of the invention may typically be in the range 1 mg to 1000 mg depending, of course, on the mode of administration. For example, oral administration may require a total daily dose of from 10 mg to 1000 mg, while an intravenous dose may only require from 1 mg to 500 mg. The total daily dose may be administered in single or divided doses and may, at the physician's discretion, fall outside of the typical range given herein.
These dosages are based on an average human subject having a weight of about 60kg to 100kg. The physician will readily be able to determine doses for subjects
whose weight falls outside this range, such as infants and the elderly, and especially obese patients.
The compounds with which the invention is concerned may be prepared for administration by any route consistent with their pharmacokinetic properties. The orally administrable compositions may be in the form of tablets, capsules, powders, granules, lozenges, liquid or gel preparations, such as oral, topical, or sterile parenteral solutions or suspensions. Tablets and capsules for oral administration may be in unit dose presentation form, and may contain conventional excipients such as binding agents, for example syrup, acacia, gelatin, sorbitol, tragacanth, or polyvinylpyrrolidone; fillers for example lactose, sugar, maize-starch, calcium phosphate, sorbitol or glycine; tabletting lubricant, for example magnesium stearate, talc, polyethylene glycol or silica; disintegrants for example potato starch, or acceptable wetting agents such as sodium lauryl sulphate. The tablets may be coated according to methods well known in normal pharmaceutical practice. Oral liquid preparations may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, or may be presented as a dry product for reconstitution with water or other suitable vehicle before use. Such liquid preparations may contain conventional additives such as suspending agents, for example sorbitol, syrup, methyl cellulose, glucose syrup, gelatin hydrogenated edible fats; emulsifying agents, for example lecithin, sorbitan monooieate, or acacia; nonaqueous vehicles (which may include edible oils), for example almond oil, fractionated coconut oil, oily esters such as glycerine, propylene glycol, or ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid, and if desired conventional flavouring or colouring agents.
The active ingredient may also be administered parenterally in a sterile medium. Depending on the vehicle and concentration used, the drug can either be suspended or dissolved in the vehicle. Advantageously, adjuvants such as a local anaesthetic, preservative and buffering agents can be dissolved in the vehicle.
Synthesis
There are multiple synthetic strategies for the synthesis of the compounds (I) with which the present invention is concerned, but all rely on known chemistry, known to the synthetic organic chemist. Thus, compounds according to formula (I) can be synthesised according to procedures described in the standard literature and are
well-known to the one skilled in the art. Typical literature sources are "Advanced organic chemistry", 4th Edition (Wiley), J March, "Comprehensive Organic Transformation", 2nd Edition (Wiley), R.C. Larock , "Handbook of Heterocyclic Chemistrf, 2nd Edition (Pergamon), A.R. Katritzky), P.G.M. Wuts and T. W. Greene "Greene's Protective Groups in Organic Chemistry" 4th Edition (Wiley) review articles such as found in "Synthesis", "Ace. Chem. Res." , "Chem. Rev", or primary literature sources identified by standard literature searches online or from secondary sources such as "Chemical Abstracts" or "Beilstein".
General synthetic routes
Routes listed below are not exhaustive.
Experimental conditions given in Step A-V are generic and can be found in standard literature sources such as those cited above. Specific references are cited for information and conditions may apply to a given substrate with or without modification/optimization.
Steps definition:
Step A: Nitriles can be converted to amides in acidic conditions e.g. sulphuric acid in acetic acid - For literature reference see: 1 ) Journal of Medicinal Chemistry 17 (24), 5894-5911 , 1990.
Step B: Activation of carboxylic acids, followed by nucleophilic displacement with R3NHR4 to give the amides, according to well know methods e.g. 1-ethyl-3-(3- dimethylaminopropyl)carbodiimide, N-hydroxybentriazole in dichloromethane.
Step C: Nitriles can be converted to amides in basic conditions e.g. sodium hydroxide, hydrogen peroxide in ethanol - For literature references see: Journal of Medicinal Chemistry 33(1 ), 336-344, 1990 and 2) J. Heterocyclic Chemistry 27(3), 795-801, 1990.
Step D: Nitriles can be converted to alkyls esters using e.g. trimethylsilyl chloride and an appropriate alcohol e.g. ethanol - For literature references see: 1) Journal of Organic Chemistry (2005), 70(20), 8093-8095 and 2) Organic & Biomolecular Chemistry 3(19), 3482-3487, 2005.
Step E: Nitriles can be converted to amidines according to Pinner synthesis e.g. gas hydrogen chloride, ethanol (amidates formation) followed by displacement with R3NHR4, or alternative methods e.g. ammonium chloride or R3NHR4, trimethylaluminium in toluene - For literature references see: 1 ) Bioorganic & Medicinal Chemistry Letters 13(4), 693-696, 2003; 2) Bioorganic & Medicinal Chemistry Letters, 15(1 ), 93-98, 2005 and 3) Journal of Medicinal Chemistry, 48(7), 2638-2645; 2005.
Step F: Esters can be hydrolysed in e.g. basic/acidic conditions according to well known methods described in Protective Groups in Organic Synthesis (T.W. Greene).
Step Ga: Protection of functional groups with a suitable PG, according to well known methods described in Protective Groups in Organic Synthesis (T.W. Greene).
Step Gb: Deprotection of functional groups, according to well known methods described in Protective Groups in Organic Synthesis (T.W. Greene).
Step H: Conversion of amines to ureas can be achieved via reaction of amines with a suitable isocyanate of formula R3NCO, leading to monosubstituted ureas. Unsubstituted ureas can be synthesised by reaction of amines with potassium isocyanate. Alternatively, mono or disubstituted ureas can be synthesised via the reaction of amines with an activated agent of formula LG1-(CO)-LG1 , followed by the addition of an amine of formula R3NHR4. For literature references see: 1) Journal of Medicinal Chemistry 49(7), 2186-2192, 2006; 2) Journal of Medicinal Chemistry 48(6), 2184-2193, 2005; 3) Organometallics 25(4), 954-960, 2006 and 4) Journal of Organic Chemistry 71(8), 3093-3102, 2006.
Step I: Conversion of amines to guanidines: a) Unsubstituted guanidines (R-i and R9 are Hydrogen), can be synthesised via reaction of amines with an activated intermediate of formula LG2-C(=NPG)-NHPG, or
LG3=C(NHPG)2, followed by removal of the protecting group PG - For literature references see: 1 ) Tetrahedron Asymmetry 17(2) 167-170, 2006; 2) Journal of
Medicinal Chemistry 48(17) 5480-5488, 2005 and 3) Letters in organic Chemistry
2(1) 83-88, 2005.
b) Substituted guanidines can be synthesised via reaction of amines with various activated agents e.g. thioisocyanates of formula R3NCS (followed by addition of R4NH2), thiophosgene (followed by addition of R3NH2 and reaction with R4I), ureas of formula R3NH(CO)NHR4 ( activated first with POCI3) or thioureas of formula R3NH(CS)NHR4- For literature references see: 1) Bioorganic & Medicinal Chemistry Letters 12(20) 2931-2934, 2002; 2) Bioorganic & Medicinal Chemistry Letters 5(15) 1745-1750, 1995 and 3) Phosphorus, Sulfur, Silicon and the related elements 178(10) 2235-2240, 2003.
Step J: Bromination of methyl can be achieved with a brominating agent e.g. N- bromosuccinimide under radical conditions, e.g. 2,2'-azobisisobutyronitrile in carbon tetrachloride - For literature reference see: 1 ) US 2004/0192667
Step K: Nitriles can be synthesised by nucleophilic displacement of bromo methyl derivatives with e.g. sodium/potassium/copper cyanide in polar solvents (e.g. dimethylformamide, ethanol, acetonitrile, dimethylsulfoxid) - For literature references see: 1) Experimental section; 2) Tetrahedron 62(20), 4907-4916, 2006 and 3) Journal of Heterocyclic Chemistry 41(6), 963-970, 2004.
Step L: The alpha position of nitriles can be substituted after deprotonation with a strong base e.g. lithium diisopropylamide in tetrahydrofuran at low temperature, followed by reaction of the carbanions with R10HaI and/or R^HaI or R-I0CHO - For literature references see: 1 ) Journal of the American Chemical Society 99(5), 1612- 19, 1977; 2) Tetrahedron Letters (52), 4647-50, 1975 and 3) Journal of Organic Chemistry 67(2), 426-430, 2002.
Step M: Primary amines can be synthesised by nucleophilic displacement of bromo methyl derivatives with either a protected amino group e.g. phtalamide, followed by removal of the protecting group (Gabriel synthesis), or a masked amino group e.g. sodium azide followed by catalytic hydrogenation - For literature references see: 1 ) Chemical Communications (Cambridge, United Kingdom) (14), 1557-1559, 2006; 2) Bioorganic & Medicinal Chemistry Letters 16(4), 771-774, 2006 and 3) Journal of Organic Chemistry 70(21), 8400-8408, 2005.
Step N: 4-Pyrazolyloxy acetic acid derivatives can be synthesised by reacting the hydroxyl moiety with an appropriate protected acetic acid of formula PG3O2C- C(Rio)(Rii)-LG4 or PGSO2C-C(R10)(Rn)-OH. Typical literature references are: 1)
EtO2C-C(RIO)(RI I)-Br in presence of a base e.g. potassium carbonate in acetone; 2) EtO2C-C(Ri0)(Rn)-OH in presence of triphenylphosphine/diethyl azidodicarboxylate (Mitsunobu synthesis) - For literature references see: 1 ) Journal of Heterocyclic Chemistry 28(8), 1971-6, 1991 and 2) Journal of Medicinal Chemistry 48(17), 5509-5519, 2005.
Step O: 4-Pyrazolyloxy acetonitrile derivatives can be synthesised by reacting the hydroxyl moiety with an appropriate alpha substituted acetonitrile of formula NC- C(Rio)(Rii)-LG4 or NC-C(R10)(Rn)-OH. Typical literature references are: 1) NC- C(R10)(Rn)-CI in presence of KF in dimethylformamide; 2) NC-C(Ri0)(Ri i)-OH in presence of triphenylphosphine/ diethyl azidodicarboxylate (Mitsunobu synthesis) - For literature references see: 1 ) Nucleosides and Nucleotides 9(8) 1021-1043, 1990 and 2) Pest Management Science 57(9), 844-851 , 2001.
Step P: Synthesis of the ether linker can be achieved by displacement of a suitable leaving group LG attached to the methyl in position 4 by a suitable nucleophile of formula PGSO2C-C(Ri0)(Rn)-OH or NC-C(Ri0)(Rn)-OH, in the presence of a base e.g. potassium carbonate in dimethylformamide or acetone - For literature references see: 1) Bulletin of the chemical Society of Japan 62(9) 3038-40, 1989; 2) Andgewandt Chemie 100(12) 1775-77, 1998 and 3) Heterocycles 60(1 ), 167-175, 2003.
Step Q: 5-substitution of pyrazoles with a suitable aryl ring can be achieved via Suzuki coupling reactions - For literature references see: Journal of Organometallic Chemistry 576(1-2), 147-168, 1999.
Step R: Aldehydes can be converted to alcohols by reaction with a suitable reducing agent e.g. sodium borohydride in methanol - For literature reference see: Bioorganic & Medicinal Chemistry 9(4), 961-982, 2001.
Step S: Secondary alcohols can be synthesised by reaction of aldehydes with a suitable organometallic reagent of formula R10MX e.g. Ri0MgBr in tetrahydrofuran or diethyl ether - For literature reference see: Bioorganic & Medicinal Chemistry 12(20), 5465-5483, 2004.
Step T: Conversion of hydroxyl groups to corresponding leaving groups LG can be achieved by reacting the hydroxyl moiety with e.g. methane sulfonyl chloride, tosyl
chloride, in a presence of pyridine or triethylamine in tetrahydrofuran or by converting the hydroxy! moiety to a suitable halide e.g. bromine with e.g. phosphorus tribromide in dichloromethane - For literature references see: 1 ) Tetrahedron 45(6), 1639-46, 1989 and 2) Journal of the American Chemical Society 128(21 ), 7055-7064, 2006.
Step U: Amides and ureas can be converted to known bioisosteres of corresponding formula -NH-C(=NR9)- and -N=C(NHRg)-NH- in a 3-step sequence: a) Amides/ureas are converted to thioamides/thioureas with e.g. Lawessons reagent; b) Thioamides/thioureas are S-methylated by treatment with e.g. dimethyl sulphate in presence of potassium carbonate in acetone; c) Methyl thioimidates/Methyl isothioureas are reacted with a amine nucleophile of formula R9NH2 in presence of e.g. triethylamine in methanol - For literature references see: 1 ) Journal of Medicinal Chemistry 48(5), 1359-1366, 2005 and 2) Bioorganic & Medicinal Chemistry Letters 14, 2323-2326, 2004.
Step V: Amidines can be converted to known amidoxime ester prodrugs of formula - C(=N-ORg)-NH2 by reacting the nitrile moiety with a suitable O-Alkyl hydroxylamine of formula RgO-NH2 in e.g. ethanol or dimethylsulfoxide. Typically, O-Alkyl hydroxylamines are available as their hydrochloride salt; therefore, a base e.g. potassium tert-butoxide is required - For literature references see: 1 ) Journal of Labelled Compounds & Radiopharmaceuticals 47(4), 233-242, 2004; 2) Journal of Medicinal Chemistry 47(14), 3658-3664, 2004 and 3) WO06090153.
Scheme 1 - Synthetic scheme leading to compounds of general formulas (Ia), (Ib) and (Ic):
Wherein:
X is a bond
Z is a bond
Y is =0 or =NH
R1, R2, R3, R4, R5 and R6 are defined as in claim 1
R9 is H

Scheme 1
• Starting materials of formula 1 can be synthesised according to the method described in WO05000820.
• Compounds of formula (Ia) can be synthesised from 1 as described in scheme 1, either according to generic steps A and B, or B and C.
• Compounds of formula (Ib) can be synthesised from 1 as described in scheme 1, according to generic steps B, D, F and B.
• Compounds of formula (Ic) can be synthesised from 1 as described in scheme 1 , according to generic steps B and E.
Scheme 2 - Synthetic scheme leading to compounds of general formulas (Id) and (Ie):
Wherein:
X is a bond
Y is =O or =NR9
Z is -NH-
R1, R2, R3, R4, R5, R6 and R9 are defined as in claim 1



Scheme 2
• Starting materials of formula 5 can be synthesised according to the method described in US2005/0101592.
• Compounds of formula (Id) can be synthesised from 5 as described in scheme 2, according to generic steps Ga, F, Gb, B and H.
• Compounds of formula (Ie) can be synthesised from 5 as described in scheme 2, according to generic steps Ga, F, Gb, B and I.
Wherein:
PG1 is a suitable protecting group e.g. Ethyl
PG2 is a suitable protecting group e.g. Boc
LG1 is a suitable leaving group e.g. Cl or imidazole
LG2 is a suitable leaving group e.g. pyrazole or -SMe
LG3 is S
Scheme 3 - Synthetic scheme leading to compounds of general formulas (If), (Ig) and (Ih):
Wherein:
X is CH2 or C(R10)(R11 )
Y iS =O or =NH
Z is a bond
R1, R2, R3, R4, R5 and R6 are defined as in claim 1

Scheme 3
• Starting materials of formula 8 can be synthesised according to the method described J. Med. Chem, 1999, 42, 769-776
• Compounds of formula (If) can be synthesised from 8 as described in scheme 3, according to generic steps J, K, (L), F, A and B or J, K, (L), F, B and C.
• Compounds of formula (Ig) can be synthesised from 8 as described in scheme 3, according to generic steps J, K, (L), F1 B and D.
• Compounds of formula (Ih) can be synthesised from 8 as described in scheme 3, according to generic steps J, K, (L), F, B and E.
Wherein:
Hal is a halide e.g. Cl1 Br or I.
PG1 is a suitable protecting group e.g. Ethyl.
Scheme 4 - Synthetic scheme leading to compounds of general formulas (Ii) and (Ij): Wherein:
X is CH2
Y is =0 or =NH
Z is -NH-
R1 , R2, R3, R4, R5, R6 and R9 are defined as in claim 1

Scheme 4
Starting materials of formula 9 can be synthesised according to scheme 3.
Compounds of formula (Ii) can be synthesised from 9 as described in scheme 4, according to generic steps M, H, F and B.
Compounds of formula (Ij) can be synthesised from 9 as described in scheme 4, according to generic steps M1 I1 F and B.
Wherein:
LG1 is a suitable leaving group e.g. Cl or imidazole
LG2 is a suitable leaving group e.g. pyrazole or -SMe
PG1 is a suitable protecting group e.g. Ethyl.
Scheme 5 - Synthetic scheme leading to compounds of general formulas (Ik), (II) and (Im):
Wherein:
X is CH2-O* , CH(RI O)-O* or C(R10)(R11 )-O*
Y is =0 or =NH
Z is a bond
R1, R2, R3, R4, R5 and R6 are defined as in claim 1
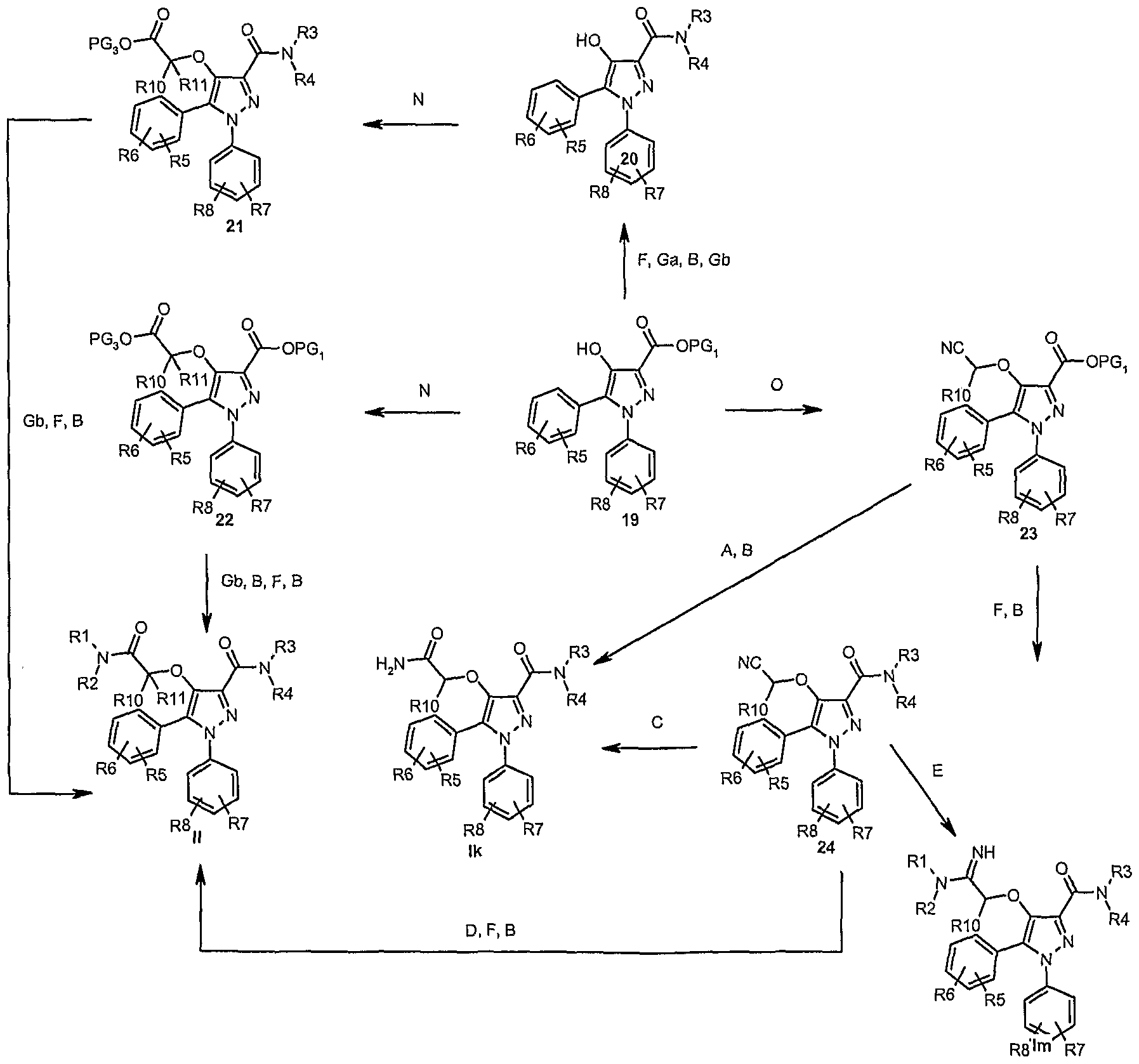
Scheme 5
• Starting materials of formula 19 can be synthesised according to the method described in WO06035310.
• Compounds of formula (Ik) can be synthesised from 19 as described in scheme 5, according to generic steps O, A and B or O, F, B and C.
• Compounds of formula (II) can be synthesised from 19 as described in scheme 5, according to generic steps N, Gb, B and F or F1 Ga, B, Gb and N.
• Compounds of formula (Im) can be synthesised as described in scheme 5, according to generic steps O, F, B and E.
Wherein:
LG4 is a suitable leaving group e.g. Cl , Br, OMs, OTs or OTf.
PG1 and PG3 are orthogonal protecting groups.
Scheme 6a - Synthetic scheme leading to compounds of general formulas (In) and (lo):
Wherein:
X is -C(R10)(R11 )-O-CH2-*, -CH2-0-C(R10)(R11 ) -*
Y is =0 or =NH
Z is a bond
R1, R2, R3, R4, R5 and R6 are defined as in claim 1

Scheme 6a
• Starting materials of formula 9 can be synthesised according to scheme 3.
• Compounds of formula (In) can be synthesised from 9 as described in scheme 6a, according to generic steps P, F and B either via intermediate 26 or 29. Alternatively, (In) can be synthesised from 9 according to generic steps P, F, B and D.
• Compounds of formula (lo) can be synthesised from 9 as described in scheme 6a, according to generic steps P, F, B and E.
Wherein:
PG1 and PG3 are orthogonal protecting groups.
Scheme 6b - Synthetic scheme leading to compounds of general formulas (In), (lo), (Ip) and (Iq):
Wherein:
X is -C(R10)(R11 )-O-CH2-*, -CH2-O-C(R10)(R11 ) -*
Y is =0 or =NH
Z is a bond
R1 , R2, R3, R4, R5 and R6 are defined as in claim 1

Scheme 6b
• Starting materials of formula 30 can be synthesised according to the method described in WO04099157.
Compounds of formula (In) can be synthesised from 30 as described in scheme 6b, according to generic steps Q, R, T, P, D, F and B or Q, R, N, D, F and B. Compounds of formula (lo) can be synthesised from 30 as described in scheme 6b, according to generic steps Q, R, T, P and E or Q1 R, T, O and E. Compounds of formula (Ip) can be synthesised from 30 as described in scheme 6b, according to generic steps Q, S, N, F and B or Q, S, T, P, F and B. Compounds of formula (Iq) can be synthesised from 30 as described in scheme 6b, according to generic steps Q, S, O and E or Q1 S, T, P and E.
Wherein:
LG4 is a suitable leaving group e.g. Cl , Br, OMs, OTs or OTf. Fg is PG3O2C or CN PG3 is a suitable protecting group (see Step N)
Scheme 7 - Synthetic scheme leading to ureas and amides known bioisosteres of general formulas (Ir) and (Is):
Wherein:
X is defined as in claim 1
Y is =N(R9)
Z is define as in claim 1
R1, R2, R3, R4, R5 and R6 are defined as in claim 1
R9 is CN

Ir

Is Scheme 7
Compounds of formula (Ir) can be synthesised from suitable amides as described in scheme 7, according to generic steps Ua, Ub and Uc.
Compounds of formula (Is) can be synthesised from suitable ureas as described in scheme 7, according to generic steps Ua, Ub and Uc.
Scheme 8 - Synthetic scheme leading to known amidine prodrugs of general formula (It):
OR0
N •+ V
H2 >N V
It
Scheme 8
Wherein:
X is define as in claim 1
Y is =N(OR9)
Z is define as in claim 1
R1, R2, R3, R4, R5, R6 and R9 are defined as in claim 1
Compounds of formula (It) can be synthesised from suitable nitriles as described in scheme 8, according to generic step V.
Scheme 9 - Synthetic routes leading to compounds of general formula (Iu) and (Iv):

Scheme 9
Wherein:
ZX is -O- or -O-CH(RIO)*- (wherein the bond indicated by an asterisk is attached to the pyrazole ring)
Y iS =O
R1, R2, R3, R4, R5 and R6 are defined as in claim 1 ,
are analogous to the ones described in scheme 5 and 6b:
Compounds of formula (Iu) can be synthesised from 20 as described in scheme 5, according to generic step H.
Compounds of formula (Iv) can be synthesised from 32 or 33 as described in scheme 6b, according to generic step H.
The following examples of compounds of the invention, their preparation and properties are presented by way of illustration only:
General comments:
Microwave reactions were performed in a Personal Chemistry Emrys Optimizer. NMR spectra were obtained on a Bruker Avance AMX 300 MHz instrument. LC/MS was performed on an Agilent 1100-series instrument. LC/MS methods are as follows:
(A) tfa20p5: Column; Gemini C18, 5μm, 2.0x50mm. Flow: 1.2 ml/min. Gradient: 0-31/2 min: 10-95% acetonitrile in water, 31/2-41/2 min: 95% acetonitrile. Modifier: 0.1% TFA. MS-ionisation mode: API-ES (pos.)
(B) tfa20n5: Column; Gemini C18, 5μm, 2.0x50mm. Flow: 1.2 ml/min. Gradient: 0-31/2 min: 10-95% acetonitrile in water, 31/2-41/2 min: 95% acetonitrile. Modifier: 0.1% TFA. MS-ionisation mode: API-ES (neg.)
(C) HCO.^Qpδ: Column; Gemini C18, 5μm, 2.0x50mm. Flow: 1.2 ml/min. Gradient: 0-4 min:33-90% acetonitrile in water, 4-41/2 min: 95% acetonitrile. Modifier: 5mM NH4HCO3. MS-ionisation mode: API-ES (pos.)
(D) HCO?20p5: Column; Gemini C18, 5μm, 2.0x50mm. Flow: 1.2 ml/min. Gradient: 0-4 min:33-90% acetonitrile in water, 4-41/2 min: 95% acetonitrile. Modifier: 5mM NH4HCO3. MS-ionisation mode: API-ES (neg.)
(E) Preparative Method: Column: YMC 19x100 mm; Flow: 20 mL/min.
Gradient: 0-8 min: 10-70% MeCN in water, 8-9 min: 70-95% MeCN in water, 9-12 min: 95% MeCN. Modifier: 0.1% TFA; MS-ionisation mode: API-ES (pos.)
Data is quoted for all compounds as molecular ion and retention time (RT) using method (A) unless otherwise stated.
Compounds of general formula F11

Wherein R5 and Rs are hydrogen.





Synthesis:

Scheme 1
Compounds of formula [D] were prepared as described in scheme 1 above. Esters of formula [A] were obtained by well known methods (Ruoxi et al., J. Med. Chem, 1999, 42, 769-776). Esters of formula [A] were brominated by treatment with N-bromosuccinimide (1.1eq.) and 1 ,1 '-azobis(isobutyronitrile) (O.OIeq.) in carbon tetrachloride as described in US 2004/0192667. The resulting bromo compounds were not purified but treated directly with potassium cyanide (2eq.) and 18-crown-6 ether (0.4eq.) in acetonitrile at reflux for 15 hours. The reaction mixtures were partitioned between 1 M sodium hydroxide solution and ethyl acetate. The organic phases were dried over sodium sulphate, filtered and evaporated. The residues were purified by chromatography over silica, eluting with ethyl acetate/cyclohexane mixtures to give ethyl ester derivatives of compounds of formula [B]. The esters were converted to their corresponding acids of formula [B]by treatment with sodium hydroxide under conditions familiar to those skilled in the art.
Alternatively, the bromo compounds could be treated with sodium cyanide in a mixture of ethanol and water at reflux, which gave directly the acids of formula [B]. Compounds of formula [B] (1mmol) were treated in acetic acid/concentrated sulphuric acid mixtures (8ml/4ml) at 10O0C, for 30 minutes. The cooled mixtures were
poured onto ice/water to yield precipitates which were filtered off, washed with water and dried in vacuum to give amides of formula [C].
Compounds of formula [C] were converted to compounds of formula [D] by well known methods using the appropriate amine R3NHR4 and coupling reagents.
Compound 1.01
Prepared according to the procedure outlined in scheme 2:

Scheme 2
4-Carbamoylmethyl-5-(4-chloro-phenyl)-1-(2-chloro-phenyl)-1 H-pyrazole-3- carboxylic acid [C1] was obtained according to the procedure outlined in scheme 1 using 1-(4-Chloro-phenyl)-propan-1-one and (2-Chloro-phenyl)-hydrazine.
To a solution of [CI](0.1g, 0.256mmol), 1-hydroxybenzotriazole (0.051 mg, 0.333mmol) and 1-ethyl-3 (3-dimethylaminopropyl) carbodiimid hydrochloride (0.074mg, 0.384mmol) in Dimethylformamide (3ml) was added a solution of 4- cyanobenzylamine hydrochloride (0.052mg, 0.307mmol) and diisopropylethylamine (53.5ul, 0.307mmol) in dimethylformamide (1ml). The reaction mixture was stirred overnight at room temperature. Solvent was removed in vacuo. The residue was dissolved in dichloromethane, washed with aqueous 1 N HCI, sat. aqueous NaHCO3, brine. The organic phase was dried over MgSO4 and concentrated in vacuo. The crude was purified by flash chromatography (4g silica; Eluent:EtOAc/Heptane:1/1) to give the title compound [1.01] as a white solid(0.04g, 0.079mmol, 31%).
1H NMR (CDCI3): δ 3.53 (s,2H), 4.63 (d,2H), 5.36 (bs,1H), 7.19-7.40 (m,10H), 7.55 (d+bs,3H), 7.80 (bs,1 H).
Compound 1.07 was synthesised according to the procedure described for compound 1.01 , with the following modification:
After overnight stirring, a precipitate was formed in the reaction mixture. The precipitate was filtered off, washed with water and dried in vacuo to give the title compound [1.07] as a white powder.
Compound 1.02
Prepared according to the procedure outlined in scheme 3:

Scheme 3
To a suspension of [C1](0.025g, 0.064mmol) in dichloromethane (1ml) were successively added 1-hydroxy-7-azabenzotriazole (0.5M DMF solution, 15OuI, 0.077mmol) and 1-ethyl-3 (3-dimethylaminopropyl) carbodiimid hydrochloride (14.7mg, 0.077mmol). After stirring for 20 minutes at room temperature, benzyl amine (8.4ul, 0.077mmol) and diisopropylethylamine (13.4ul, 0.077mmol) were added to the homogeneous solution. The reaction mixture was stirred for 3 days at room temperature. Sat. aq. NaHCO3 (1ml) was added. The reaction mixture was extracted with dichloromethane (3x1 ml). The phases were separated and the organic phase was dried through a "phase separation" column and concentrated in vacuo. The crude was purified by flash chromatography (1g silica; EluenfcDichloromethane; then EtOAc/Heptane:5/1 containing 5% of MeOH/NH3 (10/1 ) solution) to give the title compound [1.02] (10.7mg, 0.022mmol, 34%).
1H NMR (DMSO): δ 3.52 (s,2H), 4.44 (d,2H), 6.86 (bs,1 H), 7.2-7.6 (m,12H), 7.7 (dd,1H), 8.99 (t,1H).
Compounds 1.03 to 1.27 were synthesized as described for compound 1.02. In these examples, the purification step could either be according to the procedure described for compound 1.02 or via preparative LCMS as described in the analysis section, method E.
Compounds of general formula [21

Wherein R5 and R8 are hydrogen.


Synthesis:
[F]

Scheme 4
Nitrile intermediates of formula [B] were prepared as described in scheme 1 above. Amides of formula [E] were synthesized by well known methods using the appropriate amine R3NHR4 and coupling reagents. Compounds of formula [E] were converted to amidine derivatives of formula [F] according to well known procedures (Journal of the American Chemical Society, 126(39), 12220-12221 ; 2004; Bioorganic & Medicinal Chemistry Letters, 14(21), 5263-5267; 2004)
Compound 2.01
Prepared according to the procedure outlined in scheme 5:

Scheme 5
1-(2-Chloro-phenyl)-5-(4-chloro-phenyl)-4-cyanomethyl-1 H-pyrazole-3- carboxylic acid (4-fluoro-phenyl)-amide [E1] was synthesised from compound of formula [B1] according to the procedure outlined in scheme 4 using para fluoro aniline.
1-(2-Chloro-phenyl)-5-(4-chloro-phenyl)-4-cyanomethyl-1 H-pyrazole-3- carboxylic acid [B1] was obtained according to the procedure outlined in scheme 1 using 1-(4-Chloro-phenyl)-propan-1-one and (2-Chloro-phenyl)-hydrazine.
To a flame-dried flask, charged with ammonium chloride (0.105g, 1.93mmol)was added dry toluene (0.5ml). To the cooled (0-5°C) suspension was dropwise added a 2M solution of trimethylaluminium (0.905ml, 1.82mmol) in toluene. After completion, the mixture was allowed to stir for 2hours at room temperature. A solution of compound [E1](0.5g, 1.074mmol) in toluene (6.5ml)was then added and the reaction mixture was heated (16O0C) in a microwave oven for 17 minutes. After cooling, the mixture was concentrated in vacuo. Methanol was added and the non dissolved ammonium chloride was filtered off. The filtrate was concentrated in vacuo and ethyl acetate was added to yield a precipitate which was filtered and dried to give 600mg of crude product.
The crude product was purified by flash silica chromatography (12g silica; Eluent: EtOAc/Methanol:8/1 to 5/1) to give the title compound [2.01] (120mg, 0.25mmol, 23%).
1H NMR (CDCI3): δ 3.91 (s,2H), 7.05 (t,2H), 7.18 (d,2H), 7.36 (d,2H), 7.39 (d,2H), 7.42 (t,1 H), 7.49 (d, 1 H), 7.58 (q, 2H), 8.39 (bs, 2H), 8.87 (s, 1 H), 9.44 (bs, 2H).
Compounds 2.02 to 2.12 were synthesized as described for compound 2.01.
Compounds of general formula f31
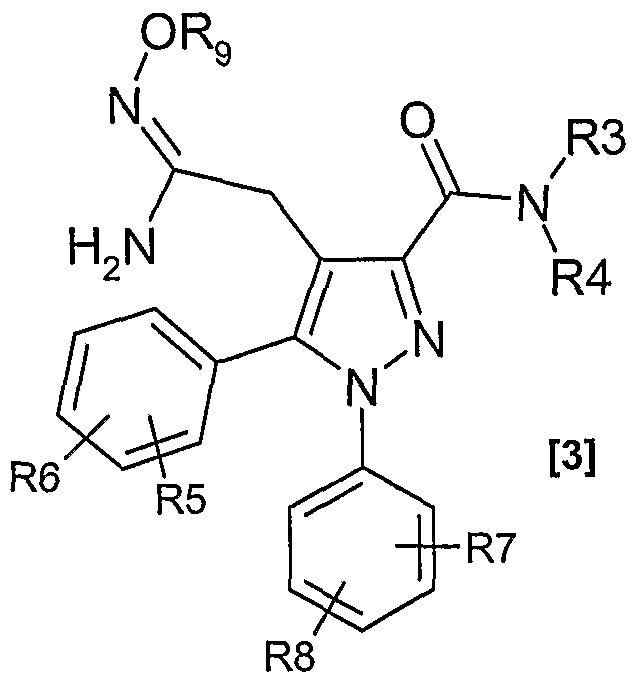
Wherein R5, R8 and Rg are hydrogen.


Synthesis:
[G]
 Scheme 6
Scheme 6
Nitrile intermediates of formula [B] were prepared as described in scheme 1 above. Amides of formula [E] were synthesized by well known methods using the appropriate amine R3NHR4 and coupling reagents. Compounds of formula [E] were converted to amidoxime derivatives of formula [G] according to well known procedures (Bioorganic & Medicinal Chemistry, 14(5), 1506-1517; 2006. Journal of Medicinal Chemistry, 50(7), 1618-1626; 2007. Journal of Medicinal Chemistry, 48(18), 5705-5720; 2005).
Compound 3.01
Prepared according to the procedure outlined in scheme 7:
NH2OKHCI, Et3N Methanol, Δ


Scheme 7
1-(2-Chloro-phenyl)-5-(4-chloro-phenyl)-4-cyanomethyl-1 H-pyrazole-3- carboxylic acid (4-fluoro-phenyl)-amide [E1] was synthesised from compound of formula [B1] according to the procedure outlined in scheme 4 using para fluoro aniline.
1-(2-Chloro-phenyl)-5-(4-chloro-phenyl)-4-cyanomethyl-1H-pyrazole-3- carboxylic acid [B1] was obtained according to the procedure outlined in scheme 1 using 1-(4-Chloro-phenyl)-propan-1-one and (2-Chloro-phenyl)-hydrazine.
To a mixture of compound [E1] (50mg; 0.108mmol) and hydroxylamine hydrochloride (30mg; 0.43mmol) in anhydrous methanol (1ml) was added triethylamine (0.06ml; 0.43mmol). The reaction mixture was heated at 4O0C overnight. LCMS showed product formation but starting material [E1] left. Stirring was continued for a further 24
hours. Solvent was removed in vacuo to give crude product as a solid. The crude material was purified by preparative LCMS (method E) to give pure title compound [3.01] as a white solid (15.2mg; 0.03mmol; 28%)
1H NMR (DMSO): δ 3.87 (m,2H), 7.18 (t,2H), 7.28 (d,2H), 7.49 (d, 2H), 7.51- 7.68 (m,4H), 7.83 (dd,2H), 8.64 (bs,1H), 10.52 (s, 1 H), 10.69 (bs, 1 H), 12.06 (bs, 1 H).
Compounds 3.02 to 3.09 were synthesized as described for compound
3.01.
Biological data:
Compounds were tested in the functional Cannabinoid Receptor-1 assay described below, and their IC50 values for antagonizing a CB1 receptor agonist were assessed.
The compounds are grouped in three classes:
A: IC50 value lower than 0.5 μM
B: IC50 value between 0.5 μM and 5 μM
C: IC5O value higher than 5 μM
Table 1 and 3 give the biological test results for the compounds synthesised above. Table 1 - Compounds of general formula [1]

Wherein R5 and R8 are hydrogen.




Table 2 - Compounds of general formula [2]

Wherein R5 and R8 are hydrogen.



Table 3 - Compounds of general formula F31

Wherein R5, R8 and R9 are hydrogen.


Biological evaluation
Transfection and Cell Culture - The cDNA encoding the human CB1 (Cannabinoid Receptor-1) receptor (GenBank accession number NM_016083) was cloned from a human adipose tissue cDNA library and cloned into the eukaryotic expression vector pcDNA3.1 (Invitrogen).
Chinese Hamster Ovary cells (CHO-K1) stably expressing recombinant human CB1 were generated by transfecting the plasmid containing the coding sequence of the human CB1 receptor in CHO-K1 cells, using lipofectamin, according to the manufacturer instructions. Resistant clones were selected in the presence of 600 μg/ml G418 (Life technology). Stably transfected CHO-K1 cells were maintained in
Ham's F-12 culture medium (Invitrogen), supplemented with 10 % fetal calf serum (Invitrogen), 100 U/ml penicillin, 100 μg/ml streptomycin (Life Technology), and 600 μg/ml G418.
Cannabinoid Receptor- 1 Functional assay.
Functional activities of the above examples of compounds of the invention were assessed in vitro by measuring their ability to inhibit CP55940-induced [35S]GTPγS binding to membranes prepared from CHO-K1 cells expressing the human CB1 receptor (described in Transfection and Cell Culture). CP55940 is a well known nonselective CB1 and CB2 receptor agonist (e.g Felder et al., 1995, Molecular Pharmacology, (48) 443-50). Membranes were prepared by a standard procedure. Briefly, cells were harvested using 10 mM EDTA and collected by centrifugation. Pelleted cells were homogenized in ice-cold 20 mM Hepes (pH 7.4), 10 mM EDTA and protease inhibitors (Complete protease inhibitor cocktail tablet, Roche) using an Ultra Turrax Homogenizer. The homogenate was centrifuged at 14 000 rpm for 45 min. at 40C. The resultant pellet was resuspended in the same buffer but with only 0.1 mM EDTA and was again centrifuged at 14 000 rpm for 45 min. at 40C. The resulting pellet (membranes) was resuspended in 20 mM Hepes (pH 7.4), 0.1 mM EDTA, 2 mM MgCb and protease inhibitors and protein concentration was determined by Micro BCA Protein Assay Reagent Kit (Pierce Biotechnology) according to the manufacturer instructions. The [35S]GTPγS SPA (Scintillation Proximity Assay) binding assay was performed by incubating 5μg/well hCB1- membranes with 1 nM [35S]GTPγS (Perkin Elmer - NEG 030H) in the presence of 3 nM of CP55940 and various concentrations of the test compounds at room temperature for 1hr in 96-well microtiter plates. 0.4mg/well SPA beads (PVT-WGA; RPNQ0001 Amersham Pharmacia Biotech) were then added and the incubation continued for further 30 min. on an orbital shaker. The assay buffer contained 5OmM HEPES (pH 7.5), 50 mM NaCI, 2.5 mM MgCI2, 0.1% BSA, 1 μM GDP and 100 μg/ml Saponin. Microtiter plates were centrifuged at 1500 rpm for 5 min. and radioactivity was read immediately using a Topcounter (PerkinElmer Life Sciences). Data were analyzed and IC50 values determined by non-linear regression using the Prism software (GraphPad Software, San Diego).
GASTROINTESTINAL TRANSIT ASSAY AND DATE TO BE INSERTED (See J- MR's email of 30/10/07)
Claims
Claims:
1. A compound of formula (I) or a salt, hydrate, solvate or N-oxide thereof:

X is a bond, or a divalent radical selected from -C(R10)(Rn)-*, -C(Ri0)(Rn)-O-*, -C(Ri0)(Ri1)CH2-*, -C(R10)(R11)CH2-O-*, -CH2C(R10)(R11)-*, -CH2C(R10)(Rn)-O-*, and -CH2-O-C(R10XR11)-*, wherein the bond indicated by an asterisk is attached to the pyrazole ring;
Y is =0, =N(R9), =N(OR9), =CH(CN), =C(CN)2 or =N(CN);
Z is a bond, -O- or -NH-;
R1 and R2 are independently selected from hydrogen, (CrC3)alkyl, (C3-C6)cycloalkyl and monocyclic heterocyclic radicals having 4 to 6 ring atoms, each of said radicals being optionally substituted by fluoro, hydroxy, methoxy, -NH2 or mono- or CIi-(C1- C3)alkylamino ;or R1 and R2 taken together with the nitrogen to which they are attached form a cyclic amino ring of 4 to 6 ring atoms which is optionally substituted by fluoro, hydroxy, methoxy, -NH2, or mono- or di-(CrC3)alkylamino;
R3 is hydrogen or a radical of formula -(O)3-AIk3 wherein a is O or 1 and AIk3 is (C1- C3)alkyl or (CrC3)fluoroalkyl;
R4 is a radical of formula -(OV(AIk1)P-(QOr(L)S-Q2 wherein
b, p, r and s are independently 0 or 1, provided that (i) at least one of p, r and s is 1, and (ii) a in R3 and b in R4 are not simultaneously 1 ;
Alki is a divalent (CrC4)alkylene radical which (a) is optionally substituted on one carbon by Rio and/or Rn or by one or two optional substituents, and/or (b) optionally contains a -O-, -S-, -CO-, -SO-, -SO2-, or -NR9- link;
L is a divalent radical of formula -(Alk2)n-(W)m-, in either orientation, wherein
n and m are independently O or 1 ;
AIk2 is -C(Ri0)(R11)-; and
W is -CO-, -SO2-, -0-, -NR9- or -SO-; provided that when W and/or Alk2 are linked to a heteroatom W is not -0-, -NRg- or -SO-;
Qi is a monocyclic carbocyclic ring of 3 to 7 ring atoms, a bicyclic carbocyclic ring system of 7 to 10 ring atoms, a monocyclic heterocyclic ring of 4 to 7 ring atoms or a bicyclic carbocyclic ring system of 8 to 10 ring atoms, any of which rings or ring systems being optionally substituted;
Q2 is (a) in the case where s in -(L)5-Q2 is O or 1 , a monocyclic carbocyclic ring of 3 to 7 ring atoms, a bicyclic carbocyclic ring system of 7 to 10 ring atoms, a monocyclic heterocyclic ring of 4 to 7 ring atoms or a bicyclic carbocyclic ring system of 8 to 10 ring atoms, any of which rings or ring systems being optionally substituted; or (b) only in the case where s in -(L)s-Q2 is O, hydrogen;
or R3 and R4 taken together with the nitrogen to which they are attached form a cyclic amino ring of 4 to 7 ring atoms which is optionally substituted by a radical of formula -(L)3-Qa wherein s, L and Q2 are as defined above, or by an optional substituent selected from hydroxy, methoxy, -NH2-, or mono- or di-(CrC3)alkylamino;
R5 is selected from hydrogen -F, -Cl, -Br, -CN, (CrC3)alkyl, (CrC3)fluoroalkyl, cyclopropyl, -COOH, tetrazole and -OR9, the R9 part being optionally substituted with tetrazole or -COOH;
R6, R7 and R8 are each independently selected from hydrogen -F1 -Cl, -Br, -CN, (CrC3)alkyl, (CrC3)fluoroalkyl, cyclopropyl, and -OR9;
R9 is hydrogen, (CrC3)alkyl, (CrC3)fluoroalkyi, or (C3-C6)cycloalkyl; and
Rio and R1I are independently selected from hydrogen and (CrC3)alkyl; or R10 and R11 taken together with the carbon atom to which they are attached form a (C3- C5)cycloalkyl ring.
2. A compound as claimed in claim 1 wherein -Z-X- is -CH2- , -CH2O-*, -CH2OCH2-*, -NH- or -NH-CH2-* wherein the bond indicated by an asterisk is attached to the pyrazole ring.
3. A compound as claimed in claim 1 or claim 2 wherein Y is =0, =NH, =N(OH) or =N(CN).
4. A compound as claimed in claim 1 wherein -Z-X- is -CH2- and Y is =0, =NH, or =N(OH).
5. A compound as claimed in any of the preceding claims wherein R1 and R2 are each hydrogen.
6. A compound as claimed in any of claims 1 to 4 wherein R1 is hydrogen and R2 is methyl, cyclopropyl or optionally substituted cyclohexyl.
7. A compound as claimed in claim 6 wherein R2 is cyclohexyl, substituted with one or two substituents selected from hydroxy, methoxy and -NH2.
8. A compound as claimed in any of claims 1 to 4 wherein R1 and R2 taken together with the nitrogen to which they are attached form a morpholine or an optionally substituted piperidine or piperazine ring
9. A compound as claimed in claim 8 wherein R1 and R2 taken together with the nitrogen to which they are attached form a piperidine or piperazine ring substituted with one or two substituents selected from hydroxy, methoxy and -NH2.
10. A compound as claimed in any of the preceding claims wherein R3 is hydrogen and, in the radical R4, b, p and s are 0, and r is 1.
11. A compound as claimed in any of claims 1 to 9 wherein R3 is hydrogen and, in the radical R4, p and r are 1 and b and s are 0.
12. A compound as claimed in claim 11 wherein, in R4, Alki is -CH2-.
13. A compound as claimed in claim 10 or claim 11 wherein, in R4, Q-i is an optionally substituted divalent phenyl or pyridyl radical.
14. A compound as claimed in claim 10 or claim 11 wherein, in R4, Q-i is an optionally substituted divalent piperidine, piperazine or (C3-C6)cycloalkyl radical.
15. A compound as claimed in any of the preceding claims wherein, in R4, Q2 is hydrogen, or optionally substituted phenyl or pyridyl.
16. A compound as claimed in any of the preceding claims wherein one or two optional ring substituents in are present in ring Q-i and/or ring Q2, said substituents being selected from -F, -Cl, -Br, -CN, -CF3, -CH3, cyclopropyl, and -OCH3
17. A compound as claimed in any of claims 1 to 10 wherein one or both of rings Qi and Q2 are unsubstituted.
18. A compound as claimed in any of the preceding claims wherein R5, RQ, R7 and R8 are independently selected from hydrogen and -Cl,
19. A compound as claimed in any of claims 1 to 11 wherein the radical -C(=O)NR3R4 in formula (I) is selected from those of formulae (A)-(T):

(G) (H) (J)

(K) (L) (M)

(N) (O) (P)

(Q) (R)

(S) (T) wherein
R10 and R11 are as defined in claim 1
R12 is selected from hydrogen, -CH3, -OH, -CN and -COOH;
R13 is selected from hydrogen, -F, -CF3, -OCF3, -Br. -Cl, -OCH3, -CH3, -CN and
-COOH;
R14 is selected from hydrogen, -F1 -CF3, -OCF3, -Br. -Cl, -OCH3, -CH3, -CN, -OH, and -COOH;
R15 and Ri6 are independently selected from hydrogen and (CrC6)alkyl or Ri5 and R-I6 taken together with the nitrogen to which they are attached form a cyclic amino ring of 4 to 7 ring atoms;
R17 is selected from hydrogen, (CrC6)alkyl, (CrC6)alkylC(=O)-, (CrC6)alkylSθ2-, benzyloxycarbonyl-, and -C(=O)OCH3; R18 is selected from hydrogen, -F and -CN;
R19 is selected from F, -CF3, -OCF3, -Br. -Cl, -OCH3, -CH3, -CN and -COOH; and R20 is selected from F, -CF3, -OCF3, -Br. -Cl, -OCH3, -CH3, -CN, -OH and -COOH.
20. A compound as claimed in claim 19 wherein the radical -C(=O)NR3R4 is selected from the group consisting of:
(i) Formula D, wherein R12 is hydrogen and Ri4 is methyl, trifluoromethyl, trifluoromethoxy, 3-, 4- or 5-fluoro, 3-, 4- or 5-chloro, or 3-, 4- or 5-cyano, or 6- hydroxy;
(ii) Formula E, wherein Ri2 is hydrogen and R14 is methyl, trifluoromethyl, , 4- or 5- fluoro, , 4- or 5-chloro, or, 4- or 5-cyano, or 2-, 6-hydroxy; (iii) Formula H;
(iv) Formula L, wherein R13 is hydrogen, 3-, 4- or 5-fluoro, 3-, 4- or 5-cyano, and R2o is methyl, trifluoromethyl, trifluoromethoxy, 3-, 4- or 5-fluoro, 3-, 4- or 5-chloro, or 3-, 4- and 5-cyano, or 6-hydroxy;
(v) Formula M, wherein R1S is hydrogen, 4- or 5-fluoro, , 4- or 5-cyano, and R2o is methyl, trifluoromethyl, trifluoromethoxy, , 4- or 5-fluoro, , 4- or 5-chloro, or, 4- and 5- cyano, or 2-, 6-hydroxy;
(vi) Formula P wherein Ri8 is hydrogen, 3-, 4- or 5-fluoro, 3-, 4- or 5-nitrile, R20 is methyl, trifluoromethyl, trifluoromethoxy, 3-, 4- or 5-fluoro, 3-, 4- or 5-chloro, or 3-, 4- and 5-cyano, or 6-hydroxy, Ri0 is hydrogen and Rn is methyl, the configuration of the asymmetric carbon carrying Ri0 and Rn being (R); and (vii) Formula Q wherein R18 is hydrogen, , 4- or 5-fluoro, , 4- or 5-cyano, R20 is methyl, trifluoromethyl, trifluoromethoxy, , 4- or 5-fluoro, , 4- or 5-chloro, or, 4- and 5- cyano, or 2-, 6-hydroxy, Ri0 is hydrogen and Rn is methyl, the configuration of the asymmetric carbon carrying Rio and Rn being (R).
21. A compound as claimed in claim 19 wherein the radical -C(=O)NR3R4 is selected from the group consisting of:
(a) Formula A;
(b) Formula B;
(c) Formula C, wherein Ri2 is hydroxyl, cyano or -COOH, and Ri3 is fluoro, methyl, trifluoromethyl, cyano, or methoxy;
(d) Formula K, wherein Ri8 is hydrogen or fluoro and R19 is cyano , methyl or methoxy;, and
(e) Formula O, wherein Ri8 is hydrogen or fluoro , R19 is cyano, methyl or methoxy, R10 is hydrogen and Rn is methyl, the configuration of the asymmetric carbon carrying R10 and Rn being (R)., (f) Formula T
23. A compound as claimed in claim 1 , selected from the group consisting of:
4-Carbamoylmethyl-5-(4-chloro-phenyl)-1-(2-chloro-phenyl)-1 H-pyrazole-3-carboxylic acid (4-methyl-pyridin-3-yl)-amide,
4-Carbamoylmethyl-1-(2-chloro-phenyl)-5-(4-chloro-phenyl)-1 H-pyrazole-3-carboxylic acid (i-phenyl-ethyl)-amide
2-{1-(2-Chloro-phenyl)-5-(4-chloro-phenyl)-3-[4-(4-fluoro-phenyl)-4-hydroxy- piperidine-1 -carbonyl]-1 H-pyrazol-4-yl}-acetamide,
1 -[4-Carbamoylmethyl-i -(2-chloro-phenyl)-5-(4-chloro-phenyl)-1 H-pyrazole-3- carbonyl]-4-phenyl-piperidine-4-carboxylic acid methyl ester,
1 -[4-Carbamoylmethyl-5-(4-chloro-phenyl)-1 -(2-chloro-phenyl)-1 H-pyrazole- 3carbonyl]-4-phenyl-piperidine-4-carboxylic acid
4-Carbamimidoylmethyl-5-(4-chloro-phenyl)-1-(2-chloro-phenyl)-1 H-pyrazole-3- carboxylic acid (4-fluoro-phenyl)-amide,
2-{1-(2-Chloro-phenyl)-5-(4-chloro-phenyl)-3-[4-(4-fluoro-phenyl)-4-hydroxy- piperidine-1 -carbonyl]-1 H-pyrazol-4-yl}-acetamidine,
2-{5-(4-Chloro-phenyl)-1-(2-chloro-phenyl)-3-[4-(2-fluoro-phenyl)-piperidine-1- carbonyl]-1H-pyrazol-4-yl}-N-hydroxyacetamidine,
5-(4-Chloro-phenyl)-1-(2-chloro-phenyl)-4-(N-hyciroxycarbannimicloylmethyl)-1 H- pyrazole-3-carboxylic acid piperidin-1-ylamide,
S^-Chloro-phenyO-i^-chloro-phenylH^N-hydroxycarbamimidoylmethyO-IH- pyrazole-3-carboxylic acid [(R)-I -(4-trifluoromethyl-phenyl)-ethyl]-amide,
and salts hydrates, solvates and N-oxides thereof.
24. A pharmaceutical composition comprising a compound as claimed in any of the preceding claims, together with one or more pharmaceutically acceptable carriers or excipients.
25. The use of a compound as claimed in any of claims 1 to 22 in the preparation of a composition for treatment of-obesity, overweight, diseases for which obesity is a risk factor, mental disorders, addictive disorders, neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis, liver cirrhosis, liver fibrosis, epilepsy, or pathologic conditions mediated by or related to CB1 mediated mechanisms.
26. A method for the treatment of treatment of obesity, overweight, diseases for which obesity is a risk factor, mental disorders, addictive disorders, neurological disorders, sexual dysfunctions, reproductive dysfunctions, osteoporosis, liver cirrhosis, liver fibrosis, epilepsy, or pathologic conditions mediated by or related to CB1 mediated mechanisms, which method comprises administering to a subject suffering such disease or condition an effective amount of a compound as claimed in any of claims 1 to 22.
27. The use as claimed in claim 24 or a method as claimed in claim 25 wherein the diseases for which obesity is a risk factor is metabolic syndrome, type 2 diabetes, cardiovascular disease, osteoarthritis, or cancer.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07824461A EP2094666A1 (en) | 2006-11-11 | 2007-11-07 | Cannabinoid receptor modulators |
| CA002669080A CA2669080A1 (en) | 2006-11-11 | 2007-11-07 | Cannabinoid receptor modulators |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0622569.2 | 2006-11-11 | ||
| GBGB0622569.2A GB0622569D0 (en) | 2006-11-11 | 2006-11-11 | Cannabinoid receptor modulators |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008059207A1 true WO2008059207A1 (en) | 2008-05-22 |
Family
ID=37594807
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2007/004225 WO2008059207A1 (en) | 2006-11-11 | 2007-11-07 | Cannabinoid receptor modulators |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP2094666A1 (en) |
| CA (1) | CA2669080A1 (en) |
| GB (1) | GB0622569D0 (en) |
| WO (1) | WO2008059207A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2009074782A1 (en) * | 2007-12-10 | 2009-06-18 | 7Tm Pharma A/S | Cannabinoid receptor modulators |
| US8088798B2 (en) | 2006-03-10 | 2012-01-03 | Jensen Discovery, Inc. | Cannabinoid receptor antagonists/inverse agonists useful for treating obesity |
| CN102358735A (en) * | 2011-08-23 | 2012-02-22 | 范如霖 | Low toxicity multiple independently target able drugs-nitrogen aromatic ring-containing CB1 receptor inhibitor with hydroxyl or substituted hydroxyl, and pharmaceutical use thereof |
| US8133904B2 (en) | 2007-09-07 | 2012-03-13 | Jenrin Discovery, Inc. | Cannabinoid receptor antagonists/inverse agonists useful for treating obesity |
| US11091447B2 (en) | 2020-01-03 | 2021-08-17 | Berg Llc | UBE2K modulators and methods for their use |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040192667A1 (en) * | 2001-08-31 | 2004-09-30 | University Of Connecticut | Novel pyrazole analogs acting on cannabinoid receptors |
| WO2007046550A1 (en) * | 2005-10-21 | 2007-04-26 | Mitsubishi Tanabe Pharma Corporation | Pyrazole compounds having cannabinoid receptor (cb1) antagonizing activity |
| WO2007106721A2 (en) * | 2006-03-10 | 2007-09-20 | Jenrin Discovery | Cannabinoid receptor antagonists/inverse agonists useful for treating obesity |
-
2006
- 2006-11-11 GB GBGB0622569.2A patent/GB0622569D0/en not_active Ceased
-
2007
- 2007-11-07 CA CA002669080A patent/CA2669080A1/en not_active Abandoned
- 2007-11-07 EP EP07824461A patent/EP2094666A1/en not_active Withdrawn
- 2007-11-07 WO PCT/GB2007/004225 patent/WO2008059207A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040192667A1 (en) * | 2001-08-31 | 2004-09-30 | University Of Connecticut | Novel pyrazole analogs acting on cannabinoid receptors |
| WO2007046550A1 (en) * | 2005-10-21 | 2007-04-26 | Mitsubishi Tanabe Pharma Corporation | Pyrazole compounds having cannabinoid receptor (cb1) antagonizing activity |
| WO2007106721A2 (en) * | 2006-03-10 | 2007-09-20 | Jenrin Discovery | Cannabinoid receptor antagonists/inverse agonists useful for treating obesity |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8088798B2 (en) | 2006-03-10 | 2012-01-03 | Jensen Discovery, Inc. | Cannabinoid receptor antagonists/inverse agonists useful for treating obesity |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| US8133904B2 (en) | 2007-09-07 | 2012-03-13 | Jenrin Discovery, Inc. | Cannabinoid receptor antagonists/inverse agonists useful for treating obesity |
| WO2009074782A1 (en) * | 2007-12-10 | 2009-06-18 | 7Tm Pharma A/S | Cannabinoid receptor modulators |
| US8173680B2 (en) | 2007-12-10 | 2012-05-08 | 7Tm Pharma A/S | Cannabinoid receptor modulators |
| EA018900B1 (en) * | 2007-12-10 | 2013-11-29 | 7ТиЭм ФАРМА А/С | Compounds modulating cannabinoid cb1 receptor activity, compositions based thereon and use thereof |
| CN102358735A (en) * | 2011-08-23 | 2012-02-22 | 范如霖 | Low toxicity multiple independently target able drugs-nitrogen aromatic ring-containing CB1 receptor inhibitor with hydroxyl or substituted hydroxyl, and pharmaceutical use thereof |
| US11091447B2 (en) | 2020-01-03 | 2021-08-17 | Berg Llc | UBE2K modulators and methods for their use |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2094666A1 (en) | 2009-09-02 |
| CA2669080A1 (en) | 2008-05-22 |
| GB0622569D0 (en) | 2006-12-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2121617B1 (en) | Cb1 receptor modulators | |
| EP2120931A1 (en) | Pyrazole derivatives as modulators of cannabinoid receptor | |
| US8173680B2 (en) | Cannabinoid receptor modulators | |
| US20060019997A1 (en) | Triazole compounds and their use as metabotropic glutamate receptor antagonists | |
| EP1928859A1 (en) | Pyrazole derivates as cannabinoid receptor modulators | |
| EP2094666A1 (en) | Cannabinoid receptor modulators | |
| WO2007057687A1 (en) | Piperazine derivatives and their use in therapy | |
| JP2009533400A (en) | Aminomethylpyridine derivatives, methods for their preparation, and therapeutic uses thereof | |
| WO2009153569A2 (en) | Cb1 receptor modulators | |
| CN101568525A (en) | Cb1 receptor modulators | |
| CA2816804A1 (en) | Substituted bicyclic carboxamide and urea derivatives as vanilloid receptor ligands |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07824461 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2669080 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007824461 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3318/CHENP/2009 Country of ref document: IN |