WO2008041240A1 - Process for preparing (s)-pramipexole and its intermediates - Google Patents
Process for preparing (s)-pramipexole and its intermediates Download PDFInfo
- Publication number
- WO2008041240A1 WO2008041240A1 PCT/IN2007/000017 IN2007000017W WO2008041240A1 WO 2008041240 A1 WO2008041240 A1 WO 2008041240A1 IN 2007000017 W IN2007000017 W IN 2007000017W WO 2008041240 A1 WO2008041240 A1 WO 2008041240A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- pramipexole
- diamino
- sodium
- benzothiazole
- Prior art date
Links
- DRRYZHHKWSHHFT-UHFFFAOYSA-N NC(CC1)Cc2c1nc(N)[s]2 Chemical compound NC(CC1)Cc2c1nc(N)[s]2 DRRYZHHKWSHHFT-UHFFFAOYSA-N 0.000 description 3
- VVPFOYOFGUBZRY-ZCFIWIBFSA-N CCC(N[C@H](CC1)Cc2c1nc(N)[s]2)=O Chemical compound CCC(N[C@H](CC1)Cc2c1nc(N)[s]2)=O VVPFOYOFGUBZRY-ZCFIWIBFSA-N 0.000 description 1
- 0 C[C@]1CC(S=C(*2)N)=C2CC1 Chemical compound C[C@]1CC(S=C(*2)N)=C2CC1 0.000 description 1
- DRRYZHHKWSHHFT-SCSAIBSYSA-N N[C@H](CC1)Cc2c1nc(N)[s]2 Chemical compound N[C@H](CC1)Cc2c1nc(N)[s]2 DRRYZHHKWSHHFT-SCSAIBSYSA-N 0.000 description 1
- JGSJAYTWMVKMPF-UHFFFAOYSA-N Nc1nc(CCC(C2)N(C(c3c4cccc3)=O)C4=O)c2[s]1 Chemical compound Nc1nc(CCC(C2)N(C(c3c4cccc3)=O)C4=O)c2[s]1 JGSJAYTWMVKMPF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
- C07D277/62—Benzothiazoles
- C07D277/68—Benzothiazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 2
- C07D277/82—Nitrogen atoms
Definitions
- the present invention relates to an improved process for the preparation of (S)- 2,6-diamino-4,5,6,7-tetrahydrobenzothiazole of formula (II) useful in the preparation of pramipexole or (S)-2,6-amino-6-(n-propylamino)-4,5,6,7-tetrahydrobenzothiazole of formula (I) and its pharmaceutically acceptable salts or solvates thereof.
- the present invention further provides a process for the preparation of Pramipexole and its pharmaceutically acceptable salts, hydrates, solvates thereof.
- 2-amino-6-(substituted)amino-4,5,6,7-tetrahydrobenzothiazoles are known pharmaceutically active agents.
- the tetrahydrobenzothiazole derivatives are taught to be useful in treating schizophrenia, Parkinson's disease or Parkinsonism, and/or hypertension.
- Pramipexole is the commercial product marketed, in a form of a dihydrochloride salt in a peroral formulation, under several brand names e.g. Mirapexin[TM].
- the compound of formula (I) has one symmetric carbon and they may exist either as a single enantiomer or in a mixed or racemic form.
- the pharmacological activity of compounds of formula (I) is generally connected only or mainly with one isomer thereof. Accordingly, the dopaminergic activity of the (S) isomer is twice as high as that of the (R) enantiomer.
- the synthesis of pramipexole by the above process yields R,S( ⁇ )-2 ⁇ amino-6- propylamino-4,5,6,7-tetrahydrobenzothiazole.
- the above-mentioned acknowledge that the produced racemic compound may be resolved into single enantiomers by classical methods such as chromatography on a chiral phase or fractional crystallization of a salt with an optically active acid.
- the S(-)-enantiomer of pramipexole was disclosed and characterized therein, no information is provided how it was prepared; i.e. whether it was prepared by a resolution of racemic pramipexole of form some optically active precursor. Further, no information is provided on how to produce the S(-)- enantiomer of pramipexole.
- WO 2006/003677 Al discloses the improved process the preparation of biologically active tetrahydrobenzothiazole derivative.
- the patent application discloses the process that has tried to solve the problems of prior art. However, much improvement over the prior art process has still been achieved.
- the process discloses the formation of 2,6-diamino-4,5,6,7-tetrahydrobenzothiazole via an isolated bromo intermediate, which on reaction with thiourea gets converted to tetrahydrobenzothiazole.
- the isolation of bromo intermediate can also be avoided.
- the halogenation of the protected cyclohexanone derivative is performed in presence of Lewis acid catalysts like AICI 3 , ZnCl 2 or SnCl 2 etc. which will give aluminous waste though increase the yield during the halognation reaction.
- the overall steps of the reaction will increase by performing isolation and work up for bromo intermediate.
- US 6,727,637 B2 discloses the monobasic acid addition salts and the mixed salts of 2-amino-6-propylamino-4,5,6,7-tetrahydrobenzothiazole wherein the monobasic acid includes hydrochloric, hydrobromic, hydroiodic, nitric, benzoic, acetic, methane sulfonic, ethane sulfonic, trifluromethane sulfonic, benzene sulfonic, and p- toluene sulfonic acids.
- patent US '637 B2 discloses the formation of mixed salts like of 2-amino-6-propylamino-4,5,6,7-tetrahydrobenzothiazole monohydrochloride monotartrate, of 2-amino-6-propylamino -4,5,6,7- tetrahydrobenzothiazole monohydrobromide monotartrate or of 2-amino-6- propylamino-4,5,6,7-tetrahydrobenzothiazole. monomethane sulfonate dibenzoyl-D- tartrate.
- US 6,770,761 B2 also discloses the process for preparation of 2-amino-6(alkyl)- amino-4,5,6,7-tetrahydrobenzothiazoles which includes the bromination of 1,4- cyclohexadione by bromine in an alcoholic solvent, followed by treatment of the reaction mixture with thiourea or N-acylthiourea and isolation of compound (A), that is further treated with an amine R 1 -NH 2 or a chiral amine to get an imine intermediate and reducing it by reaction with said reducing agent or by hydrogenation, to yield the compound of formula (B)
- polymorphism is the occurrence of different crystalline forms of a single compound and it is a property of some compounds and complexes. Thus, polymorphs are distinct solids sharing the same molecular formula, yet each polymorph may have distinct physical properties. Therefore, a single compound may give rise to a variety of polymorphic forms where each form has different and distinct physical properties, such as different solubility profiles, different melting point temperatures and/or different x- ray diffraction peaks. Since the solubility of each polymorph may vary, identifying the existence of pharmaceutical polymorphs is essential for providing pharmaceuticals with predicable solubility profiles.
- polymorphic forms of a compound can be distinguished in a laboratory by X-ray diffraction spectroscopy and by other methods such as, infrared spectrometry.
- X-ray diffraction spectroscopy and by other methods such as, infrared spectrometry.
- polymorphs and the pharmaceutical applications of polymorphs see G. M. Wall, Pharm Manuf. 3, 33 (1986); J. K. Haleblian and W. McCrone, J. Pharm. ScL, 58, 911 (1969); and J. K. Haleblian, J. Pharm. ScL, 64, 1269 (1975), all of which are incorporated herein by reference.
- the present invention relates to provide a simple, cost effective, non-hazardous and easily scaleable at large commercial production process for preparation of (S)-2,6-diamino-4,5,6,7- tetrahydrobenzothiazole of formula (JI) and Pramipexole dihydrochloride of formula
- the present invention provides an improved process for preparation of (S)-2,6- diamino-4,5,6,7-tetrahydrobenzothiazole of formula (II), which is an useful intermediate for the preparation of Pramipexole or (S)-2,6-amino-6-(n-propylamino)- 4,5,6,7-tetrahydro benzothiazole of formula (I) and its pharmaceutically acceptable salts, hydrates, solvates thereof.
- a crystalline form of pramipexole dihydrochloride monohydrate in its preferred form characterized by x-ray powder diffraction patter having 20 6.50, 12.00, 12.96, 19.48, 21.34, 24.19, 28.47, 32.74 ⁇ 0.2° as the characteristic peaks and showing the DSC endotherm at 300 0 C (decomposition);
- the present invention further provides an improved process for the preparation of Pramipexole dihydrochloride monohydrate. BRIEF DESCRIPTION OF THE DRAWINGS
- FIG. I is a Differential Scanning Calorimetry (DSC) theromgram of the Pramipexole dihydrochloride monohydrate
- FIG. II is an X-ray powder diffractogram (XRD) of the Pramipexole dihydrochloride monohydrate, measured on Rigaku D/Max-2200/PC Diffractometer with Cu K alpha- 1 radiation source.
- XRD X-ray powder diffractogram
- an improved process for the preparation of Pramipexole its pharmaceutically acceptable salts, solvates, hydrates thereof which comprises the steps of; a) treating a compound 2-amino-6-phthalimido-4,5,6,7-tetrahydrobenzothiazole (III)
- 2-amino-6-phthalimido-4,5,6,7- tetrahydrobenzothiazole of formula (III) is treated with a base at a ratio of 1:1.5-1:5 in an aqueous medium to give racemic 2,6-diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula (IV).
- the reaction is preferably carried out at about 30 to 6O 0 C.
- said base used in step (a) is a deprotecting reagents of phthalimido groups like hydrazine, phenyl hydrazine, sodium sulphide monohydrate, DCC, sodium borohydride, monomethylamine, triethylamine, isopropyl amine, preferably monomethylamine.
- deprotection reaction is selectively carried out in presence of base such as amine selected from monomethylamine, triethylamine with aqueous medium without using hydrazine hydrate.
- the compound of formula (III) is treated with said base at a molar ratio of 1:1.5-1:5, preferably 1:2.5.
- compound of formula (III) is treated with base at temperature 30°C-60°C, preferably 45°C-50°C.
- the compound of formula (III) is treated with base in aqueous medium, wherein said aqueous medium is 40% solution of base in water.
- optically active auxiliary acids used in step (b) for resolution is consisting of L-tartaric acid, ditoloyl-D-tartaric acid, and dibenzoyl-D-tartaric acid, camphor acid, camphor sulfonic acid or ⁇ -methoxy-phenylacetic acid, preferably L-tartaric acid.
- step (c) includes sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, sodium acetate, potassium acetate, sodium hydroxide, potassium hydroxide, sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide.
- Preferably sodium or potassium carbonate is used in the reaction step (c).
- Polar organic solvent used in step (c) is selected from the group including water, methanol, ethanol, isopropanol, n-propanol, n-butanol, methylene dichloride, ethylene dichloride, tetrahydrofuran, dioxan, acetone, acetonitrile, dimethylsulfoxide or mixture thereof.
- the preferred solvent is ethanol. According to the most preferred embodiment of the present invention is converting S-(-)-2-amino-6-(n-propylamino)-4,5,6,7-tetrahydrobenzothiazole (I) to its pharmaceutically acceptable salts or solvates.
- the pharmaceutically acceptable salts includes dibasic acid addition salts like hydrochloric, hydrobromic, hydroiodic, nitric, benzoic, acetic, methane sulfonic, ethane sulfonic, trifluromethane sulfonic, benzene sulfonic, p-toluene sulfonic, sulfuric, phosphoric, lactic, citric, tartaric, succinic, maleic or fumaric acid and hydrate thereof, preferably dihydrochloride monohydrate.
- the present invention provides an improved process for preparation of (S)-2,6- diamino-4,5,6,7-tetrahydrobenzothiazole of formula (II), which is an useful intermediate for the preparation of Pramipexole or (S)-2,6-amino-6-(n-propylamino)-
- said reaction is preferably carried out at about 30 to 6O 0 C.
- said base used in step (a) is a deprotecting reagents of phthalimido groups like hydrazine, phenyl hydrazine, sodium sulphide monohydrate, DCC, sodium borohydride, monomethylamine, triethylamine, isopropyl amine, preferably monomethylamine.
- deprotection reaction is selectively carried out in presence of base such as amine selected from monomethylamine, triethylamine with aqueous medium without using hydrazine hydrate.
- base such as amine selected from monomethylamine, triethylamine with aqueous medium without using hydrazine hydrate.
- the compound of formula (III) is treated with said base at a molar ratio of 1:1.5-1:5, preferably 1 :2.5.
- compound of formula (III) is treated with base at temperature 30°C-60°C, preferably 45°C-50°C.
- the compound of formula (III) is treated with base in aqueous medium, wherein said aqueous medium is 40% solution of base in water.
- optically active auxiliary acids used in step (b) for resolution is consisting of L-tartaric acid, ditoluoyl-D-tartaric acid, and dibenzoyl-D-tartaric acid, camphor acid, camphor sulfonic acid or ⁇ -methoxy-phenylacetic acid, preferably L-tartaric acid.
- (IV) is treated with tartaric acid to provide tartrate salt of 2,6-diamino-4,5,6,7- tetrahydro-l,3-benzothiazole, which upon treatment with base provides (S)-2,6- diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula (II).
- alcoholic solvent used in step (iii) is selected from methanol, ethanol, propanol, isopropanol, butanol or mixtures thereof.
- the present invention further provides an improved process for the preparation of Pramipexole of formula (I), its pharmaceutically acceptable salt, hydrates, solvates thereof, which comprises a) reacting (S)-2,6-diarnino-4,5,6,7-tetrahydro-l,3-berizothiazole of formula (II) with n-propylbromide in presence of base in a polar organic solvent to give Pramipexole of formula (I); b) optionally converting Pramipexole to its pharmaceutically acceptable salts, hydrates, or solvates thereof.
- Base used in step (a) includes sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, sodium acetate, potassium acetate, sodium hydroxide, potassium hydroxide, sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide.
- sodium or potassium carbonate is used in the reaction step (a).
- Polar organic solvent used in step (a) is selected from the group including water, methanol, ethanol, isopropanol, n-propanol, n-butanol, methylene dichloride, ethyelene dichloride, tetrahydrofuran, dioxan, acetone, acetonitrile, dimethylsulfoxide or mixture thereof.
- the preferred solvent is ethanol.
- the pharmaceutically acceptable salts includes dibasic acid addition salts like hydrochloric, hydrobromic, hydroiodic, nitric, benzoic, acetic, methane sulfonic, ethane sulfonic, trifluromethane sulfonic, benzene sulfonic, p-toluene sulfonic, sulfuric, phosphoric, lactic, citric, tartaric, succinic, maleic or fumaric acid and hydrate thereof, preferably dihydrochloride monohydrate .
- a crystalline form of pramipexole dihydrochloride monohydrate in its preferred form characterized by x-ray powder diffraction patter having 2 ⁇ 6.50, 12.00, 12.96, 19.48,
- the present invention further provides an improved process for the preparation of Pramipexole dihydrochloride monohydrate, which comprises (i) reacting pramipexole with solution of hydrochloric acid in C 1 -C 5 alcohols in presence of water (ii) isolating Pramipexole dihydrochloride monohydrate
- the alcohol is selected from methanol, ethanol, isopropanolj propanol, isobutanol, butanol, tert-butanol, and pentanol, preferably isopropanol.
- the present invention further provides an improved process for the preparation of substantially pure Pramipexole dihydrochloride monohydrate, which comprises (i) reacting pramipexole with solution of hydrochloric acid in C1-C5 alcohols (ii) isolating wet cake of Pramipexole dihydrochloride monohydrate (iii) drying wet cake of Pramipexole dihydrochloride monohydrate in tray drier at about 40 to 7O 0 C for about 30 minutes to about 7 hours, (iv) optionally cooling the dried material under airflow.
- Alcohol is selected from methanol, ethanol, isopropanol, propanol, isobutanol, butanol, tert-butanol, pentanol, preferably isopropanol.
- a process for the preparation of substantially pure Pramipexole dihydrochloride monohydrate includes reaction of Pramipexole with alcoholic solution of HCl, preferably isopropanolic hydrochloric acid in molar ratio of Pramipexole to HCl of about 1:1.9 to 2.3 wt/wt at about ambient temperature to reflux temperature and subsequently cooling the solution to provide wet cake, which is dried in tray drier preferably at 50 to 55 0 C for 6 hours and subsequently dried material is cooled under air flow to obtain substantially pure Pramipexole dihydrochloride monohydrate.
- alcoholic solution of HCl preferably isopropanolic hydrochloric acid in molar ratio of Pramipexole to HCl of about 1:1.9 to 2.3 wt/wt at about ambient temperature to reflux temperature and subsequently cooling the solution to provide wet cake, which is dried in tray drier preferably at 50 to 55 0 C for 6 hours and subsequently dried material is cooled under air flow to
- the compound of formula (VII) is treated with bromine in an alcoholic solvent, followed by treatment of the reaction mixture with thiourea to tetrahydrobenzothizole analogue via an bromo intermediate.
- the alcoholic solvent used in step (iii) is selected from the group consisting of Ci-C 6 alcohols preferably, ethanol.
- step (iv) The isolation of compound of formula (III) in step (iv) is achieved by centrifugation, followed by washing and drying.
- Scheme-1 According to another aspect of the present invention there is provided a crystalline form of pramipexole dihydrochloride monohydrate in its preferred form characterized by x-ray powder diffraction patter having 20 values 6.5, 12.0, 12.9, 13.8,
- the most preferred embodiment of the present invention is to prepare pramipexole dihydrochloride having purity 99.5% by HPLC and having impurities like tertiary amine analogues (2-amino-6,6-dipropyl-4,5,6,7-tetrahydrobenzothiazole) not more than 0.1%, diamine analogue (2,6-diamino-4,5,6,7-tetrahydrobenzothiazole) not more than 0.1% and the total impurities not 1.0%.
- Example-1 Preparation of 2-amino-6-phthaIimido-4,5,6,7- tetrahydrobenzothiazole A) Preparation of chromic acid:
- the reaction mass was cooled and treated with 3.36 L ethanol at 45 0 C to 25 0 C while gradual cooling.
- the reaction mixture was further cooled to 15 0 C to 2O 0 C and treated with 0.22 L of bromine and 0.43 Kg of thiourea under stirring for 1 h.
- the reaction mixture was heated to reflux at 75 0 C to 78 0 C for 6 hrs.
- the reaction mixture was cooled and stirred for 1 hr at 5 0 C to 1O 0 C.
- the product was isolated by centrifuge, washing with ethanol 0.66 L and drying under vacuum at 5O 0 C t0 55 0 C. (yield: 0.70 Kg).
- the reaction mixture was cooled to 25 0 C to 35 0 C.
- 40% sodium hydroxide solution (0.108 Kg in 0.27 L water) was added to adjust the constant pH 10.0 to 10.5 followed by treatment with 5.0 L methylene dichloride twice and separating the organic layer.
- the organic layer was treated with 5.0 L of process water and stirred for 30 minutes.
- the separated organic layer was subjected to distillation to remove methylene dichloride under vacuum.
- 5.0 L of isopropanol was added at 4O 0 C to 45 0 C and heated up to 6O 0 C to 65 0 C.
- Acidic isopropanol 0.440L was added to adjust the pH 7.0 to 7.5 and stirred for 1 hour.
- the reaction mass was cooled to 25 0 C to 35°C.
- the product was obtained by centrifuge, washing with 0.5 L of isopropanol and drying at 5O 0 C to 55 0 C followed by cooling. (Yield: 1.0 Kg)
Abstract
The present invention relates to an improved process for the preparation of (S)- 2,6-diamino-4,5,6,7-tetrahydrobenzothiazole of formula (II) useful in the preparation of pramipexole or (S)-2,6-amino-6-(n-propylamino)-4,5,6,7-tetrahydrobenzothiazole of formula (I) and its pharmaceutically acceptable salts or solvates thereof. The present invention further provides a process for the preparation of Pramipexole and its pharmaceutically acceptable salts, hydrates, solvates thereof.
Description
PROCESS FOR PREPARING (S)-PRAMIPEXOLE AND ITS
INTERMEDIATES
FIELD OF THE INVENTION
The present invention relates to an improved process for the preparation of (S)- 2,6-diamino-4,5,6,7-tetrahydrobenzothiazole of formula (II) useful in the preparation of pramipexole or (S)-2,6-amino-6-(n-propylamino)-4,5,6,7-tetrahydrobenzothiazole of formula (I) and its pharmaceutically acceptable salts or solvates thereof. The present invention further provides a process for the preparation of Pramipexole and its pharmaceutically acceptable salts, hydrates, solvates thereof.

(H) (I)
BACKGROUND OF THE INVENTION
Any discussion of the prior art throughout the specification should in no way be considered as an admission that such prior art is widely known or forms part of common general knowledge in the field. 2-amino-6-(substituted)amino-4,5,6,7-tetrahydrobenzothiazoles are known pharmaceutically active agents. The tetrahydrobenzothiazole derivatives are taught to be useful in treating schizophrenia, Parkinson's disease or Parkinsonism, and/or hypertension. Among the known compounds is (S)-2-amino-6-propylaminio-4,5,6,7- tetrahydrobenzothiazole of formula (I), which is more commonly known as Pramipexole. Pramipexole is the commercial product marketed, in a form of a dihydrochloride salt in a peroral formulation, under several brand names e.g. Mirapexin[TM].
The compound of formula (I) has one symmetric carbon and they may exist either as a single enantiomer or in a mixed or racemic form. The pharmacological activity of compounds of formula (I) is generally connected only or mainly with one isomer thereof. Accordingly, the dopaminergic activity of the (S) isomer is twice as high as that of the (R) enantiomer.
A general process for the preparation of Pramipexole dihydrochloride has been described in US 4886812, EP 186087 and EP 207696. The process comprises the
protection of amino function of 4-aminocyclohexanol to give the intermediate compound wherein, Rl is acyl or alkoxycarbonyl and R2 is hydrogen or Rl and R2 together form an amino protective group such as pthalimido group which on oxidation with an oxidising agent, followed by halogenation (preferably bromination) of protected ketone to give alpha halogenatedketone which on reaction with thiourea, followed by deprotection yielded the racemic 2,6-diaminotetrahydrobenzothiazole. Reductive alkylation of diaminotetrahydrobenzothiazole with n-propanal furnished the racemic pramipexole. Although the (S) isomer of pramipexole is mentioned therein, it is not clear at what stage the chiral resolution has been carried out. The general process steps are indicated in Scheme- Ia below.

H2N
Racemate Resolution

n-Propyl Bromide -


Scheme-la
Another process for preparing optically pure pramipexole dihydrochloride was disclosed in J. Med. Chem. 1987, 30, 494-498, wherein, racemic 2,6-diamino-4,5,6,7- tetrahydrobenzo- thiazole was resolved, using L (+) tartaric acid to give optically pure (S)-2,6-diamino-4,5,6,7-tetrahydrobenzothiazole, which was converted to optically pure pramipexole by reacting (S)- 2,6-diamino-4,5,6,7-tetrahydro benzothiazole with propionic anhydride in THF and followed by reduction with borane THF complex . The reaction steps are shown in Scheme-lb as under:

(VIII) (II)
(CH3CH2CO)2O

2HCl Scheme- Ib
However, the variants of the above general process prepare only racemate.
Thus, the synthesis of pramipexole by the above process yields R,S(±)-2~amino-6- propylamino-4,5,6,7-tetrahydrobenzothiazole. The above-mentioned acknowledge that the produced racemic compound may be resolved into single enantiomers by classical methods such as chromatography on a chiral phase or fractional crystallization of a salt with an optically active acid. However, even though the S(-)-enantiomer of pramipexole was disclosed and characterized therein, no information is provided how it was prepared; i.e. whether it was prepared by a resolution of racemic pramipexole of form some optically active precursor. Further, no information is provided on how to produce the S(-)- enantiomer of pramipexole.
The prior art process suffers with some of the disadvantages like using hydrobromic acid in acetic acid, which is corrosive in nature, carries out the bromination and work up of the reaction is tedious. The use of diethyl ether to remove the impurities causes hazards to commercial scale. The isolation and separation of racemic pramipexole and pramipexole dihydrochloride requires techniques like column chromatography, which results in low yields i.e. below 50% and below 26%, respectively.
WO 2006/003677 Al discloses the improved process the preparation of biologically active tetrahydrobenzothiazole derivative. The patent application discloses the process that has tried to solve the problems of prior art. However, much improvement over the prior art process has still been achieved. Moreover, the process discloses the formation of 2,6-diamino-4,5,6,7-tetrahydrobenzothiazole via an isolated bromo intermediate, which on reaction with thiourea gets converted to
tetrahydrobenzothiazole. The isolation of bromo intermediate can also be avoided. The halogenation of the protected cyclohexanone derivative is performed in presence of Lewis acid catalysts like AICI3, ZnCl2 or SnCl2 etc. which will give aluminous waste though increase the yield during the halognation reaction. Moreover, the overall steps of the reaction will increase by performing isolation and work up for bromo intermediate.
US 6,727,637 B2 discloses the monobasic acid addition salts and the mixed salts of 2-amino-6-propylamino-4,5,6,7-tetrahydrobenzothiazole wherein the monobasic acid includes hydrochloric, hydrobromic, hydroiodic, nitric, benzoic, acetic, methane sulfonic, ethane sulfonic, trifluromethane sulfonic, benzene sulfonic, and p- toluene sulfonic acids. Further the patent US '637 B2 discloses the formation of mixed salts like of 2-amino-6-propylamino-4,5,6,7-tetrahydrobenzothiazole monohydrochloride monotartrate, of 2-amino-6-propylamino -4,5,6,7- tetrahydrobenzothiazole monohydrobromide monotartrate or of 2-amino-6- propylamino-4,5,6,7-tetrahydrobenzothiazole. monomethane sulfonate dibenzoyl-D- tartrate. The process as disclosed in US '637 B2 converts the racemic pramipexole into monohydrochloride salt of pramipexole which is then resolved with a optically active auxilliary acid to give mixed salt like of 2-amino-6-propylamino-4,5,6,7-tetrahydro- benzothiazole monohydrochloride monotartrate which is then converted to (S)- pramipexole free base and then to the desired pharmaceutically active ingredient (S)- pramipexole dihydrochloride.
US 6,770,761 B2 also discloses the process for preparation of 2-amino-6(alkyl)- amino-4,5,6,7-tetrahydrobenzothiazoles which includes the bromination of 1,4- cyclohexadione by bromine in an alcoholic solvent, followed by treatment of the reaction mixture with thiourea or N-acylthiourea and isolation of compound (A), that is further treated with an amine R1-NH2 or a chiral amine to get an imine intermediate and reducing it by reaction with said reducing agent or by hydrogenation, to yield the compound of formula (B)


(A) (B)
Polymorphism is the occurrence of different crystalline forms of a single compound and it is a property of some compounds and complexes. Thus, polymorphs are distinct solids sharing the same molecular formula, yet each polymorph may have distinct physical properties. Therefore, a single compound may give rise to a variety of polymorphic forms where each form has different and distinct physical properties, such as different solubility profiles, different melting point temperatures and/or different x- ray diffraction peaks. Since the solubility of each polymorph may vary, identifying the existence of pharmaceutical polymorphs is essential for providing pharmaceuticals with predicable solubility profiles. It is desirable to investigate all solid-state forms of a drug, including all polymorphic forms, and to determine the stability^ dissolution and flow properties of each polymorphic form. Polymorphic forms of a compound can be distinguished in a laboratory by X-ray diffraction spectroscopy and by other methods such as, infrared spectrometry. For a general review of polymorphs and the pharmaceutical applications of polymorphs see G. M. Wall, Pharm Manuf. 3, 33 (1986); J. K. Haleblian and W. McCrone, J. Pharm. ScL, 58, 911 (1969); and J. K. Haleblian, J. Pharm. ScL, 64, 1269 (1975), all of which are incorporated herein by reference.
To over the drawbacks of the prior art process the present invention relates to provide a simple, cost effective, non-hazardous and easily scaleable at large commercial production process for preparation of (S)-2,6-diamino-4,5,6,7- tetrahydrobenzothiazole of formula (JI) and Pramipexole dihydrochloride of formula
(I)-
It is also an object of the present invention to provide a crystalline form of pramipexole dihydrochloride. OBJECTS OF THE INVENTION
It is an object of the present invention to overcome or ameliorate at least one of the disadvantages of the prior art, or to provide a useful alternative.
It is an object of the present invention in its preferred form to provide an improved process for preparation of (S)-2,6-diamino-4,5,6,7-tetrahydrobenzothiazole and to use it for the preparation of (S)-(-)-2-amino-6-(n-propylamino)-4,5,6,7- benzothiazole and its pharamaceutically acceptable salts, solvates etc.
It is also an object of the present invention in its preferred form to provide an improved process for preparation of crystalline form of pramipexole dihydrochloride
monohydrate characterized by its x-ray powder diffraction and differential scanning calorimetry.
Further object of the present invention is to overcome the problems associated with the prior art process and to prepare Pramipexole by simple, cost effective, non- hazardous and easily scaleable way. SUMMARY OF THE INVENTION
The present invention provides an improved process for preparation of (S)-2,6- diamino-4,5,6,7-tetrahydrobenzothiazole of formula (II), which is an useful intermediate for the preparation of Pramipexole or (S)-2,6-amino-6-(n-propylamino)- 4,5,6,7-tetrahydro benzothiazole of formula (I) and its pharmaceutically acceptable salts, hydrates, solvates thereof.
According to the present invention, there is provided an improved process for the preparation of (S)-2,6-diamino-4,5,6-7-tetrahydrobenzothiazole of formula (II);

a) treating a compound 2-amino-6-phthalimido-4,5,6,7-tetrahydrobenzothiazole of formula (III)

with a base at a ratio of 1:1.5-1:5 in an aqueous medium to give racemic 2,6-diamino- 4,5,6,7-tetrahydro-l,3-benzothiazole of formula (IV);


(i) reacting 4-amino cyclohexanol of formula (V) or its acid addition salts with phthalic anhydride to obtain 4-(phthalimido)-cyclohexanol of formula (VI);

(V)
(VI
)
(ii) oxidizing 4-(phthalimido)-cyclohexanol of formula (VI) to give 4-(phthalimido)- cyclohexanone of formula (VII)

(iii) treating compound of formula (VII) by bromine in an alcoholic solvent, followed by treatment of the reaction mixture with thiourea; and (iv) isolating the compound of formula (III).
According to another aspect of the present invention there is provided a crystalline form of pramipexole dihydrochloride monohydrate in its preferred form characterized by x-ray powder diffraction patter having 20 6.50, 12.00, 12.96, 19.48, 21.34, 24.19, 28.47, 32.74 ± 0.2° as the characteristic peaks and showing the DSC endotherm at 3000C (decomposition);
The present invention further provides an improved process for the preparation of Pramipexole dihydrochloride monohydrate.
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. I is a Differential Scanning Calorimetry (DSC) theromgram of the Pramipexole dihydrochloride monohydrate
FIG. II is an X-ray powder diffractogram (XRD) of the Pramipexole dihydrochloride monohydrate, measured on Rigaku D/Max-2200/PC Diffractometer with Cu K alpha- 1 radiation source. DESCRIPTION OF THE INVENTION
According to the present invention, there is provided an improved process for the preparation of Pramipexole its pharmaceutically acceptable salts, solvates, hydrates thereof, which comprises the steps of; a) treating a compound 2-amino-6-phthalimido-4,5,6,7-tetrahydrobenzothiazole (III)

(HI) with a base at a ratio of 1:1.5-1:5 in an aqueous medium to give racemic 2,6-diamino- 4,5,6,7-tetrahydro-l,3-benzothiazole of formula (IV);
¥I JΓ /^NH2
•N (IV)
b) resolving racemic 2,6-diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula (IV) to give (S)-2,6-diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula (II); c) reacting (S)-2,6-diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula (II) with n-propylbromide in presence of base in a polar organic solvent to give Pramipexole of formula (I); d) optionally converting Pramipexole to its pharmaceutically acceptable salts, hydrates, or solvates thereof. According to the preferred embodiment, 2-amino-6-phthalimido-4,5,6,7- tetrahydrobenzothiazole of formula (III) is treated with a base at a ratio of 1:1.5-1:5 in an aqueous medium to give racemic 2,6-diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula (IV). The reaction is preferably carried out at about 30 to 6O0C.
According to the preferred embodiment, said base used in step (a) is a deprotecting reagents of phthalimido groups like hydrazine, phenyl hydrazine, sodium sulphide monohydrate, DCC, sodium borohydride, monomethylamine, triethylamine, isopropyl amine, preferably monomethylamine. In the preferred embodiment, deprotection reaction is selectively carried out in presence of base such as amine selected from monomethylamine, triethylamine with aqueous medium without using hydrazine hydrate.
According to present invention, the compound of formula (III) is treated with said base at a molar ratio of 1:1.5-1:5, preferably 1:2.5. Preferably, compound of formula (III) is treated with base at temperature 30°C-60°C, preferably 45°C-50°C.
According to the present invention, the compound of formula (III) is treated with base in aqueous medium, wherein said aqueous medium is 40% solution of base in water.
Another preferred embodiment of the present invention, resoluation of racemic 2,6-diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula (IV) with an optically active auxiliary acid to give (S)-2,6-diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula (II).
The optically active auxiliary acids used in step (b) for resolution is consisting of L-tartaric acid, ditoloyl-D-tartaric acid, and dibenzoyl-D-tartaric acid, camphor acid, camphor sulfonic acid or α-methoxy-phenylacetic acid, preferably L-tartaric acid.
According to one of the preferred embodiment of the present invention, reacting (S)-2,6-diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula (II) with n- propylbromide in presence of base in a polar organic solvent to give S-(-)-2-amino-6- (n-propylamino)-4,5,6,7-tetrahydrobenzothiazole of formula (I). Base used in step (c) includes sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, sodium acetate, potassium acetate, sodium hydroxide, potassium hydroxide, sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide. Preferably sodium or potassium carbonate is used in the reaction step (c). Polar organic solvent used in step (c) is selected from the group including water, methanol, ethanol, isopropanol, n-propanol, n-butanol, methylene dichloride, ethylene dichloride, tetrahydrofuran, dioxan, acetone, acetonitrile, dimethylsulfoxide or mixture thereof. The preferred solvent is ethanol.
According to the most preferred embodiment of the present invention is converting S-(-)-2-amino-6-(n-propylamino)-4,5,6,7-tetrahydrobenzothiazole (I) to its pharmaceutically acceptable salts or solvates.
The pharmaceutically acceptable salts includes dibasic acid addition salts like hydrochloric, hydrobromic, hydroiodic, nitric, benzoic, acetic, methane sulfonic, ethane sulfonic, trifluromethane sulfonic, benzene sulfonic, p-toluene sulfonic, sulfuric, phosphoric, lactic, citric, tartaric, succinic, maleic or fumaric acid and hydrate thereof, preferably dihydrochloride monohydrate.
The present invention provides an improved process for preparation of (S)-2,6- diamino-4,5,6,7-tetrahydrobenzothiazole of formula (II), which is an useful intermediate for the preparation of Pramipexole or (S)-2,6-amino-6-(n-propylamino)-
4,5,6>7-tetrahydro benzothiazole of formula (I) and its pharmaceutically acceptable salts, hydrates, solvates thereof.
According to the present invention, there is provided an improved process for the preparation of (S)-2,6-diamino-4,5,6-7-tetrahydrobenzothiazole of formula (II);



In the preferred embodiment, deprotection reaction is selectively carried out in presence of base such as amine selected from monomethylamine, triethylamine with aqueous medium without using hydrazine hydrate.
According to present invention, the compound of formula (III) is treated with said base at a molar ratio of 1:1.5-1:5, preferably 1 :2.5. Preferably, compound of formula (III) is treated with base at temperature 30°C-60°C, preferably 45°C-50°C. According to the present invention, the compound of formula (III) is treated with base in aqueous medium, wherein said aqueous medium is 40% solution of base in water.
Another preferred embodiment of the present invention, resoluation of racemic 2,6-diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula (IV) with an optically active auxiliary acid to give (S)-2,6-diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula (II).
The optically active auxiliary acids used in step (b) for resolution is consisting of L-tartaric acid, ditoluoyl-D-tartaric acid, and dibenzoyl-D-tartaric acid, camphor acid, camphor sulfonic acid or α-methoxy-phenylacetic acid, preferably L-tartaric acid. Thus, racemic 2,6-diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula
(IV) is treated with tartaric acid to provide tartrate salt of 2,6-diamino-4,5,6,7- tetrahydro-l,3-benzothiazole, which upon treatment with base provides (S)-2,6- diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula (II).
According to another aspect of the present invention, there is provided an improved process for preparation of compound of formula (III) which comprises the steps of:
(III)
 (i) reacting 4-amino cyclohexanol of formula (V) or its acid addition salts with phthalic
(i) reacting 4-amino cyclohexanol of formula (V) or its acid addition salts with phthalic
anhydride to obtain 4-(phthalimido)-cyclohexanol of formula (VI);

(V)
(ii) oxidizing 4-(phthalimido)-cyclohexanol of formula (VI) to give 4-(phthalimido)- cyclohexanone of formula (VII)

(iii) treating compound of formula (VII) by bromine in an alcoholic solvent, followed by treatment of the reaction mixture with thiourea; and
(iv) isolating the compound of formula (III).
According to the present invention, alcoholic solvent used in step (iii) is selected from methanol, ethanol, propanol, isopropanol, butanol or mixtures thereof. The present invention, further provides an improved process for the preparation of Pramipexole of formula (I), its pharmaceutically acceptable salt, hydrates, solvates thereof, which comprises a) reacting (S)-2,6-diarnino-4,5,6,7-tetrahydro-l,3-berizothiazole of formula (II) with n-propylbromide in presence of base in a polar organic solvent to give Pramipexole of formula (I); b) optionally converting Pramipexole to its pharmaceutically acceptable salts, hydrates, or solvates thereof.
Base used in step (a) includes sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, sodium acetate, potassium acetate, sodium
hydroxide, potassium hydroxide, sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide. Preferably sodium or potassium carbonate is used in the reaction step (a).
Polar organic solvent used in step (a) is selected from the group including water, methanol, ethanol, isopropanol, n-propanol, n-butanol, methylene dichloride, ethyelene dichloride, tetrahydrofuran, dioxan, acetone, acetonitrile, dimethylsulfoxide or mixture thereof. The preferred solvent is ethanol.
According to the most preferred embodiment of the present invention is converting S-(-)-2-amino-6-(n-propylamino)-4,5,6,7-tetrahydrobenzothiazole (I) to its pharmaceutically acceptable salts or solvates.
The pharmaceutically acceptable salts includes dibasic acid addition salts like hydrochloric, hydrobromic, hydroiodic, nitric, benzoic, acetic, methane sulfonic, ethane sulfonic, trifluromethane sulfonic, benzene sulfonic, p-toluene sulfonic, sulfuric, phosphoric, lactic, citric, tartaric, succinic, maleic or fumaric acid and hydrate thereof, preferably dihydrochloride monohydrate .
According to another aspect of the present invention there is provided a crystalline form of pramipexole dihydrochloride monohydrate in its preferred form characterized by x-ray powder diffraction patter having 2Θ 6.50, 12.00, 12.96, 19.48,
21.34, 24.19, 28.47, 32.74 ± 0.2° as the characteristic peaks and showing the DSC endotherm at 3000C (decomposition).
The present invention further provides an improved process for the preparation of Pramipexole dihydrochloride monohydrate, which comprises (i) reacting pramipexole with solution of hydrochloric acid in C1-C5 alcohols in presence of water (ii) isolating Pramipexole dihydrochloride monohydrate
The alcohol is selected from methanol, ethanol, isopropanolj propanol, isobutanol, butanol, tert-butanol, and pentanol, preferably isopropanol.
The present invention further provides an improved process for the preparation of substantially pure Pramipexole dihydrochloride monohydrate, which comprises (i) reacting pramipexole with solution of hydrochloric acid in C1-C5 alcohols (ii) isolating wet cake of Pramipexole dihydrochloride monohydrate (iii) drying wet cake of Pramipexole dihydrochloride monohydrate in tray drier at about 40 to 7O0C for about 30 minutes to about 7 hours, (iv) optionally cooling the dried material under airflow.
Alcohol is selected from methanol, ethanol, isopropanol, propanol, isobutanol, butanol, tert-butanol, pentanol, preferably isopropanol.
According to the present invention, there is provided a process for the preparation of substantially pure Pramipexole dihydrochloride monohydrate includes reaction of Pramipexole with alcoholic solution of HCl, preferably isopropanolic hydrochloric acid in molar ratio of Pramipexole to HCl of about 1:1.9 to 2.3 wt/wt at about ambient temperature to reflux temperature and subsequently cooling the solution to provide wet cake, which is dried in tray drier preferably at 50 to 550C for 6 hours and subsequently dried material is cooled under air flow to obtain substantially pure Pramipexole dihydrochloride monohydrate.
According to another aspect of the present invention, there is provided an improved process for preparation of compound of formula (III)


(V) (VI)
The preparation of compound of 4-(phthalimido)-cyclohexanol of formula (VI) is well know in the art as reported in US 4886812.
Oxidizing 4-(phthalimido)-cyclohexanol of formula (VI) by conventional means to give 4-(phthalimido)-cyclohexanone of formula (VII) is one of the important aspects of the present invention.

The alcoholic solvent used in step (iii) is selected from the group consisting of Ci-C6 alcohols preferably, ethanol.
According to one of the aspect of the present invention is isolating the compound of formula (III), which is important starting material for the preparation of compound of formula (II) .
The isolation of compound of formula (III) in step (iv) is achieved by centrifugation, followed by washing and drying.
According to the present invention, there is provided an improved process for preparation pramipexole and its pharmaceutically acceptable salts via the important intermediate (S)-2,6-diamino-4,5,6,7-tetrahydrobenzothiazole as represented in Scheme- 1 as below.

(Ia)
Scheme-1
According to another aspect of the present invention there is provided a crystalline form of pramipexole dihydrochloride monohydrate in its preferred form characterized by x-ray powder diffraction patter having 20 values 6.5, 12.0, 12.9, 13.8,
15.6, 16.9, 17.2, 19.4, 21.3, 23.3, 24.2, 24.7, 25.6, 26.7, 28.4, 31.6, 32.7 ± 0.2° and showing the DSC endotherm at 3000C (decomposition).
The most preferred embodiment of the present invention is to prepare pramipexole dihydrochloride having purity 99.5% by HPLC and having impurities like tertiary amine analogues (2-amino-6,6-dipropyl-4,5,6,7-tetrahydrobenzothiazole) not more than 0.1%, diamine analogue (2,6-diamino-4,5,6,7-tetrahydrobenzothiazole) not more than 0.1% and the total impurities not 1.0%.
It is one of the preferred embodiments of the present invention to provide process for preparation of pramipexole dihydrochloride monohydrate having mean particle size less than 400 μm, preferably less than 200 μm and more preferably less than 100 μm when measured with Malvern light scattering instruments.
Although the invention has been described with reference to a specific example, it will be appreciated by those skilled in the art that the invention may be embodied in many other forms.
The process of the present invention will be explained in more detail with reference to the following examples, which are provided by way of illustration only and should not be constructed as limit to the scope of the claims in any manner. Examples:
Example-1: Preparation of 2-amino-6-phthaIimido-4,5,6,7- tetrahydrobenzothiazole A) Preparation of chromic acid:
0.278 kg of chromium trioxide was added in 0.428 L of water at 150C to 35°C. The reaction mixture was cooled to 50C to 1O0C. 0.198 L of sulfuric acid Was added slowly within 25 to 30 minutes. 1.0 L of water was added to get the clear solution. B) Preparation of 2-amino-6-phthalimido-4,5,6,7-tetrahydrobenzothiazole via 4- (phthalimido)-cyclohexanone
1.0 Kg of 4-(phthalimido)-cyclohexanol was added in 20.0 L of acetone at 250C to 350C. The reaction mixture was cooled to 50C to 100C and treated with chromic acid solution. 0.2 L of isopropanol was added and stirred for 30 min. The reaction mixture was filtered and washed with acetone (1.0 L). The filtrate was treated with 0.4 kg
sodium bicarbonate at 250C to 350C and stirred for 1 h. The reaction mass was again filtered, washed with acetone (1.0 L). Excess of acetone was distilled under vacuum. The residue was treated with 0.5 L ethanol followed by distillation of ethanol under vacuum. The reaction mass was cooled and treated with 3.36 L ethanol at 450C to 250C while gradual cooling. The reaction mixture was further cooled to 150C to 2O0C and treated with 0.22 L of bromine and 0.43 Kg of thiourea under stirring for 1 h. The reaction mixture was heated to reflux at 750C to 780C for 6 hrs. The reaction mixture was cooled and stirred for 1 hr at 50C to 1O0C. The product was isolated by centrifuge, washing with ethanol 0.66 L and drying under vacuum at 5O0C t0 550C. (yield: 0.70 Kg).
ExampIe-2: Preparation of 2, 6-diamino-4,5,6,7-tetrahydrobenzothiazole
1.595 kg of 2-amino-6-phthalimido-4,5,6,7-tetrahydrobenzothiazole was treated with 40% aqueous solution of monomethylamine at 250C to 350C. The reaction mass was allowed to stir for 5-10 minutes and heated at 45°C to 5O0C for 1 - 1.5 hr. The reaction mixture was cooled gradually to 50C to 1O0C and maintained for 30 minutes. The product thus obtained was filtered, washed with chilled water and dried at 5O0C to 550C to obtained racemic 2,6-diamino-4,5,6,7-tetrahydrobenzothiazole. (Yield: 0.522 kg)
Example-3: Preparation of 2, 6-diamino-4,5,6,7-tetrahydrobenzothiazoIe tartrate salt
1.0 Kg of 2, 6-diamino-4,5,6,7-tetrahydrobenzothiazole was added in 9.5 L of water and heated at 750C to 850C. 0.888 Kg of L-(+)-tartaric acid was added to the reaction mixture and maintained for 30 minutes. The reaction mixture was fine filtered at high temperature and washed with 0.5 L of water. The filtrate was gradually cooled to 250C to 300C and maintained for 16 hours. The product was centrifuge and washed with 1 L water. The wet cake was treated with 6.0 L water and heated at 8O0C to 9O0C with addition of excess water to ensure clear solution. The reaction mass was fine filtered at high temperature and washed with 0.5 L water. The filtrate thus obtained was gradually cooled to 5°C to 1O0C and maintained for 2 hrs. The product was centrifuge and washed with 1 L chilled water. The wet cake was treated with 6.0 L water and heated at 8O0C to 9O0C with addition of excess water to ensure clear solution. The reaction mass was gradually cooled to 950C to 25°C and maintained for 2 hrs. The product was centrifuge, washed with 1 L chilled water, dried at 5O0C to 550C and cooled to 250C to 350C. (Yield: 0.70 Kg).
ExampIe-4: Preparation of (S)-2,6-diamino-4,5,6,7-tetrahydrobenzothiazole
1.0 Kg of 2, 6-diamino-4,5,6,7-tetrahydrobenzothiazole tartrate salt was treated with 1.5 L of water and stirred for 15 minutes at 25°C to 35°C. 0.245 Kg of sodium hydroxide solution in 0.612 L of water was added to adjust the pH 11.0 to 12.0 within 35 to 40 minutes and stirred for 1 hr. The product was centrifuge, washed with 1.0 L water and dried at 500C to 550C. The product was cooled to 20°C-40°C to obtain (S)- 2,6-diamino-4,5,6,7-tetrahydrobenzothiazole. (Yield: 0.37 Kg). Example-5: Preparation of Pramipexole crude
To the solution of 1.0 Kg of (S)-2,6-diamino-4,5,6,7-tetrahydrobenzothiazole and 0.1225 Kg of potassium carbonate in 10.0 L isopropanol was added 0.540 L n- propyl bromide. The reaction mixture was stirred for 15 minutes and heated to reflux on a water bath up to 8O0C and was maintained for 5 hours. 0.3236 L of n-propyl bromide was further added in two portions at 8O0C to 82°C and maintained for 5 hours. The isopropanol was removed completely by distillation under vacuum at 550C to 750C. 7.5 L of process water was added into the reaction mass and stirred for 30 minutes. The reaction mixture was cooled to 250C to 350C. 40% sodium hydroxide solution (0.108 Kg in 0.27 L water) was added to adjust the constant pH 10.0 to 10.5 followed by treatment with 5.0 L methylene dichloride twice and separating the organic layer. The organic layer was treated with 5.0 L of process water and stirred for 30 minutes. The separated organic layer was subjected to distillation to remove methylene dichloride under vacuum. 5.0 L of isopropanol was added at 4O0C to 450C and heated up to 6O0C to 650C. Acidic isopropanol 0.440L was added to adjust the pH 7.0 to 7.5 and stirred for 1 hour. The reaction mass was cooled to 250C to 35°C. The product was obtained by centrifuge, washing with 0.5 L of isopropanol and drying at 5O0C to 550C followed by cooling. (Yield: 1.0 Kg)
ExampIe-6: Preparation of Pramipexole dihydrochloride monohydrate
1.0 Kg of crude Pramipexole was added in 5.0 L of ethanol and heated to reflux using water bath at 800C. The reaction mixture was maintained for 1 hour and cooled to 250C to 35°C and stirred for 1 hour. The product was centrifuge and washed with 0.5 L ethanol. The wet cake thus obtained was further treated with 5.0 L of ethanol and heated to reflux using water bath at 8O0C. The reaction mixture was maintained for 1 hour and cooled to 250C to 350C and stirred for 1 hour. The product was centrifuge and washed with 0.5 L ethanol. The wet cake was treated with 5.0 L isopropanol and heated to 6O0C to 65°C using water bath. Acidic isopropanol 0.35 L was added to adjust the pH
1.7 to 2.3 and maintained for 1 hour. The product was centrifuge and washed with 1 L of isopropanol and dried in hot air oven at 5O0C to 550C to give Pramipexole dihydrochloride pure, which is converted to Pramipexole dihydrochloride monohydrate upon cooling the dried material under airflow. (Purity: 99.5% by HPLC and having known individual impurities less than 0.1% and total impurities less than 1.0%.) Example-7.: Preparation of Pramipexole dihydrochloride monohydrate
1.0 Kg of crude Pramipexole was added in 5.0 L of ethanol and heated to reflux using water bath at 8O0C. The reaction mixture was maintained for 1 hour and cooled to 250C to 350C and stirred for 1 hour. The product was centrifuge and washed with 0.5 L ethanol. The wet cake thus obtained was further treated with 5.0 L of ethanol and heated to reflux using water bath at 800C. The reaction mixture was maintained for 1 hour and cooled to 250C to 350C and stirred for 1 hour. The product was centrifuge and washed with 0.5 L ethanol. The wet cake was treated with 5.0 L isopropanol and heated to 600C to 650C using water bath. Isopropanolic HCl (0.35 L) containing water was added to adjust the pH 1.7 to 2.3 and maintained for 1 hour. The product was centrifuge and washed with 1 L of isopropanol and dried at 4O0C to 5O0C to give Pramipexole dihydrochloride monohydrate
Claims
1. An improved process for the preparation of Pramipexole its pharmaceutically acceptable salts, solvates, hydrates thereof, which comprises the steps of: a) treating a compound 2-amino-6-phthalimido-4,5,6,7-tetrahydrobenzothiazole (III)
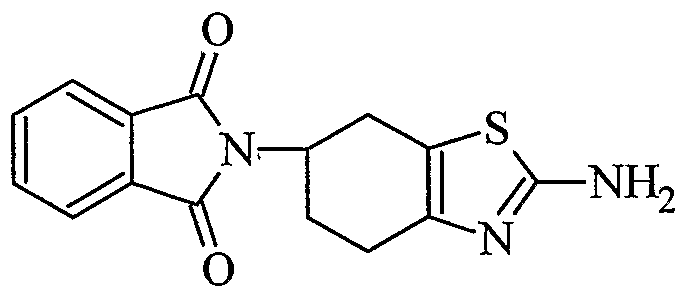
(HI) with a base at a ratio of 1:1.5-1:5 in an aqueous medium to give racemic 2,6-diamino- 4,5,6,7-tetrahydro-l,3-benzothiazole of formula (IV);

2» A process claimed in claim 1, wherein base used in step (a) is selected from hydrazine, phenyl hydrazine, sodium sulphide monohydrate, DCC, sodium borohydride, monomethylamine, triethylamine, isopropyl amine, preferably monomethylamine.
3. A process claimed in claim 1 or 2 wherein base used in step (c) is selected from sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, sodium acetate, potassium acetate, sodium hydroxide, potassium hydroxide, sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide.
4. A process as claimed in any preceding claim wherein said polar solvent is selected from the group consisting of water, methanol, ethanol, isopropanol, n-propanol, n- butanol, methylene dichloride, ethyelene dichloride, tetrahydrofuran, dioxan, acetone, acetonitrile, dimethylsulfoxide or mixture thereof.
5. A process for preparing (S)-2,6-diamino-4,5,6-7-tetrahydrobenzothiazole of formula (H);



6. A process claimed in claim 5, wherein base used in step (a) is selected from hydrazine, phenyl hydrazine, sodium sulphide monohydrate, DCC, sodium borohydride, monomethylamine, triethylamine, isopropyl amine, preferably monomethylamine.
7. A process for preparing compound of formula (III) which comprising the steps of:


(VII)
(VI
) (ii) oxidizing 4-(phthalimido)-cyclohexanol of formula (VI) to give 4-(phthalimido)- cyclohexanone of formula (VII)

(iii) treating compound of formula (VII) by bromine in an alcoholic solvent, followed by treatment of the reaction mixture with thiourea; and (iv) isolating the compound of formula (III).
8. A process claimed in claim 7, wherein the alcoholic solvent is selected from methanol, ethanol, propanol, isopropanol, butanol or mixtures thereof.
9. A process for preparing Pramipexole of formula (I), its pharmaceutically acceptable salt, hydrates, solvates thereof, which comprises a) reacting (S)-2,6-diamino-4,5,6,7-tetrahydro-l,3-benzothiazole of formula (II) with n-propylbromide in presence of base in a polar organic solvent to give Pramipexole of formula (I); b) optionally converting Pramipexole to its pharmaceutically acceptable salts, hydrates, or solvates thereof.
10. A process claimed in claim 9, wherein said base is selected from sodium carbonate, potassium carbonate, sodium bicarbonate, potassium bicarbonate, sodium acetate, potassium acetate, sodium hydroxide, potassium hydroxide, sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide.
11. A process as claimed in claim 9, wherein said polar solvent selected from the group including water, methanol, ethanol, isopropanol, n-propanol, n-butanol, methylene dichloride, ethyelene dichloride, tetrahydrofuran, dioxan, acetone, acetonitrile, dimethylsulfoxide or mixture thereof.
12. A process for preparation of Pramipexole dihydrochloride monohydrate, which comprises (i) reacting pramipexole with solution of hydrochloric acid in C1-C5 alcohols in presence of water (ii) isolating Pramipexole dihydrochloride monohydrate
13. A process as claimed in claim 12, wherein said alcohol is selected from methanol, ethanol, isopropanol, propanol, isobutanol, butanol, tert-butanol, pentanol.
14. A process for preparing substantially pure Pramipexole dihydrochloride monohydrate, which comprises
(i) reacting pramipexole with solution of hydrochloric acid in C1-C5 alcohols (ii) isolating wet cake of Pramipexole dihydrochloride monohydrate (iii) drying wet cake of Pramipexole dihydrochloride monohydrate in tray drier at about 40 to 7O0C for about 30 minutes to about 7 hours.
(iv) optionally cooling the dried material under airflow.
15. A process as claimed in claim 14, wherein said alcohol is selected from methanol, ethanol, isopropanol, propanol, isobutanol, butanol, tert-butanol, pentanol.
16. Substantially pure Pramipexole dihydrochloride monohydrate characterized by x- ray powder diffraction patter having 2Θ values 6.5, 12.0, 12.9, 13.8, 15.6, 16.9, 17.2,
19.4, 21.34, 23.3, 24.2, 24.7, 25.6, 26.7, 28.4, 31.6, 32.7 ± 0.2°
17. Substantially pure Pramipexole dihydrochloride monohydrate characterized by having DSC endotherm at 3000C.
18. Substantially pure Pramipexole dihydrochloride monohydrate having purity greater than 99.5% by HPLC and having impurities, 2-amino-6,6-dipropyl-4,5,6,7- tetrahydrobenzothiazole not more than 0.1%, 2,6-diamino-4,5,6j7- tetrahydrobenzothiazole not more than 0.1%.
19. Pramipexole dihydrochloride monohydrate having mean particle size less than 400 μm.
20. Pramipexole dihydrochloride monohydrate having mean particle size less than 200 μm, preferably less than 100 μm.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1631MU2006 | 2006-10-03 | ||
| IN1631/MUM/2006 | 2006-10-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008041240A1 true WO2008041240A1 (en) | 2008-04-10 |
Family
ID=38283113
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2007/000017 WO2008041240A1 (en) | 2006-10-03 | 2007-01-11 | Process for preparing (s)-pramipexole and its intermediates |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008041240A1 (en) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2137171A2 (en) * | 2007-03-14 | 2009-12-30 | Knopp Neurosciences, Inc. | Synthesis of chirally purified substituted benzothiazole diamines |
| CN104496936A (en) * | 2015-01-07 | 2015-04-08 | 海南康虹医药科技开发有限公司 | Preparation method of pramipexole dihydrochloride |
| US9468630B2 (en) | 2013-07-12 | 2016-10-18 | Knopp Biosciences Llc | Compositions and methods for treating conditions related to increased eosinophils |
| US9512096B2 (en) | 2011-12-22 | 2016-12-06 | Knopp Biosciences, LLP | Synthesis of amine substituted 4,5,6,7-tetrahydrobenzothiazole compounds |
| US9642840B2 (en) | 2013-08-13 | 2017-05-09 | Knopp Biosciences, Llc | Compositions and methods for treating plasma cell disorders and B-cell prolymphocytic disorders |
| US9662313B2 (en) | 2013-02-28 | 2017-05-30 | Knopp Biosciences Llc | Compositions and methods for treating amyotrophic lateral sclerosis in responders |
| US9763918B2 (en) | 2013-08-13 | 2017-09-19 | Knopp Biosciences Llc | Compositions and methods for treating chronic urticaria |
| US9849116B2 (en) | 2008-08-19 | 2017-12-26 | Knopp Biosciences Llc | Compositions and methods of using (R)-pramipexole |
| CN107573301A (en) * | 2017-11-08 | 2018-01-12 | 江苏长青农化股份有限公司 | A kind of preparation method of intermediate of tricyclazole |
| CN109232471A (en) * | 2018-10-31 | 2019-01-18 | 安徽省庆云医药股份有限公司 | A kind of preparation method of body of Pramipexole dihydrochloride |
| US10383857B2 (en) | 2013-07-12 | 2019-08-20 | Knopp Biosciences Llc | Compositions and methods for treating conditions related to elevated levels of eosinophils and/or basophils |
| CN110669024A (en) * | 2019-10-30 | 2020-01-10 | 福建福瑞明德药业有限公司 | Alkali precipitation method of (S) -2, 6-diamino-4, 5,6, 7-tetrahydrobenzothiazole L-tartrate |
| CN113816923A (en) * | 2021-09-30 | 2021-12-21 | 艾希尔(深圳)药物研发有限公司 | Preparation method of pramipexole N-methyl impurity |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0186087A1 (en) * | 1984-12-22 | 1986-07-02 | Dr. Karl Thomae GmbH | Tetrahydro-benzothiazoles, their production and their use as intermediates or drugs |
| EP0207696A1 (en) * | 1985-06-24 | 1987-01-07 | Eli Lilly And Company | Dialkylaminotetrahydrobenzothiazoles and oxazoles |
| WO2006003677A1 (en) * | 2004-07-01 | 2006-01-12 | Alembic Limited | Improved process for the preparation of biologically active tetrahydrobenzthiazole derivative |
| EP1731514A1 (en) * | 2005-06-02 | 2006-12-13 | Sandoz AG | Process for the preparation of Pramipexole |
-
2007
- 2007-01-11 WO PCT/IN2007/000017 patent/WO2008041240A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0186087A1 (en) * | 1984-12-22 | 1986-07-02 | Dr. Karl Thomae GmbH | Tetrahydro-benzothiazoles, their production and their use as intermediates or drugs |
| EP0207696A1 (en) * | 1985-06-24 | 1987-01-07 | Eli Lilly And Company | Dialkylaminotetrahydrobenzothiazoles and oxazoles |
| WO2006003677A1 (en) * | 2004-07-01 | 2006-01-12 | Alembic Limited | Improved process for the preparation of biologically active tetrahydrobenzthiazole derivative |
| EP1731514A1 (en) * | 2005-06-02 | 2006-12-13 | Sandoz AG | Process for the preparation of Pramipexole |
Non-Patent Citations (2)
| Title |
|---|
| BOEHRINGER INGELHEIM: "Mirapex", 2006, pages 4 - 31, XP002444888, Retrieved from the Internet <URL:http://www.fda.gov/medwaTCH/safety/2006/Nov_PIs/Mirapex_PI.pdf> * |
| SCHNEIDER C S ET AL: "Dopamine autoreceptor agonists: resolution and pharmacological activity of 2,6-diaminotetrahydrobenzothiazole and aminothiazole analogue of apomorphine", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 30, no. 3, March 1987 (1987-03-01), pages 494 - 498, XP002186199, ISSN: 0022-2623 * |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2137171A4 (en) * | 2007-03-14 | 2010-05-19 | Knopp Neurosciences Inc | Synthesis of chirally purified substituted benzothiazole diamines |
| AU2008224844B2 (en) * | 2007-03-14 | 2012-08-09 | Knopp Neurosciences, Inc. | Synthesis of chirally purified substituted benzothiazole diamines |
| EP2137171A2 (en) * | 2007-03-14 | 2009-12-30 | Knopp Neurosciences, Inc. | Synthesis of chirally purified substituted benzothiazole diamines |
| US10179774B2 (en) | 2007-03-14 | 2019-01-15 | Knopp Biosciences Llc | Synthesis of chirally purified substituted benzothiazole diamines |
| US9849116B2 (en) | 2008-08-19 | 2017-12-26 | Knopp Biosciences Llc | Compositions and methods of using (R)-pramipexole |
| US10208003B2 (en) | 2011-12-22 | 2019-02-19 | Knopp Biosciences Llc | Synthesis of amine substituted 4,5,6,7-tetrahydrobenzothiazole compounds |
| US9512096B2 (en) | 2011-12-22 | 2016-12-06 | Knopp Biosciences, LLP | Synthesis of amine substituted 4,5,6,7-tetrahydrobenzothiazole compounds |
| US9956206B2 (en) | 2013-02-28 | 2018-05-01 | Knopp Biosciences Llc | Compositions and methods for treating amyotrophic lateral sclerosis in responders |
| US9662313B2 (en) | 2013-02-28 | 2017-05-30 | Knopp Biosciences Llc | Compositions and methods for treating amyotrophic lateral sclerosis in responders |
| US10285981B2 (en) | 2013-02-28 | 2019-05-14 | Knopp Biosciences Llc | Compositions and methods for treating amyotrophic lateral sclerosis in responders |
| US10828284B2 (en) | 2013-07-12 | 2020-11-10 | Knopp Biosciences Llc | Compositions and methods for treating conditions related to elevated levels of eosinophils and/or basophils |
| US11612589B2 (en) | 2013-07-12 | 2023-03-28 | Areteia Therapeutics, Inc. | Compositions and methods for treating conditions related to elevated levels of eosinophils and/or basophils |
| US10383857B2 (en) | 2013-07-12 | 2019-08-20 | Knopp Biosciences Llc | Compositions and methods for treating conditions related to elevated levels of eosinophils and/or basophils |
| US11026928B2 (en) | 2013-07-12 | 2021-06-08 | Knopp Biosciences Llc | Compositions and methods for treating conditions related to elevated levels of eosinophils and/or basophils |
| US9468630B2 (en) | 2013-07-12 | 2016-10-18 | Knopp Biosciences Llc | Compositions and methods for treating conditions related to increased eosinophils |
| US10383856B2 (en) | 2013-07-12 | 2019-08-20 | Knopp Biosciences Llc | Compositions and methods for treating conditions related to increased eosinophils |
| US10980783B2 (en) | 2013-07-12 | 2021-04-20 | Knopp Biosciences Llc | Compositions and methods for treating conditions related to increased eosinophils |
| US9642840B2 (en) | 2013-08-13 | 2017-05-09 | Knopp Biosciences, Llc | Compositions and methods for treating plasma cell disorders and B-cell prolymphocytic disorders |
| US10195183B2 (en) | 2013-08-13 | 2019-02-05 | Knopp Biosciences Llc | Compositions and methods for treating chronic urticaria |
| US10028940B2 (en) | 2013-08-13 | 2018-07-24 | Knopp Biosciences Llc | Compositions and methods for treating plasma cell disorders and B-cell prolymphocytic disorders |
| US10456381B2 (en) | 2013-08-13 | 2019-10-29 | Knopp Biosciences Llc | Compositions and methods for treating plasma cell disorders and B-cell prolymphocytic disorders |
| US9763918B2 (en) | 2013-08-13 | 2017-09-19 | Knopp Biosciences Llc | Compositions and methods for treating chronic urticaria |
| CN104496936A (en) * | 2015-01-07 | 2015-04-08 | 海南康虹医药科技开发有限公司 | Preparation method of pramipexole dihydrochloride |
| CN107573301A (en) * | 2017-11-08 | 2018-01-12 | 江苏长青农化股份有限公司 | A kind of preparation method of intermediate of tricyclazole |
| CN107573301B (en) * | 2017-11-08 | 2020-08-11 | 江苏长青农化股份有限公司 | Preparation method of tricyclazole intermediate |
| CN109232471A (en) * | 2018-10-31 | 2019-01-18 | 安徽省庆云医药股份有限公司 | A kind of preparation method of body of Pramipexole dihydrochloride |
| CN109232471B (en) * | 2018-10-31 | 2022-05-10 | 安徽省庆云医药股份有限公司 | Preparation method of pramipexole dihydrochloride |
| CN110669024A (en) * | 2019-10-30 | 2020-01-10 | 福建福瑞明德药业有限公司 | Alkali precipitation method of (S) -2, 6-diamino-4, 5,6, 7-tetrahydrobenzothiazole L-tartrate |
| CN113816923A (en) * | 2021-09-30 | 2021-12-21 | 艾希尔(深圳)药物研发有限公司 | Preparation method of pramipexole N-methyl impurity |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008041240A1 (en) | Process for preparing (s)-pramipexole and its intermediates | |
| US6770761B2 (en) | Process for preparation of 2-amino-6 (alkyl) amino-4,5,6,7-tetrahydrobenzothiazoles | |
| EP1730129B1 (en) | Intermediates for the preparation of pramipexole | |
| US8952173B2 (en) | Method for the resolution of 2-amino-6-propylamino-4,5,6,7-tetrahydrobenzothiazol and intermediate compounds | |
| US20090105483A1 (en) | Process for the preparation of pramipexole base and/or its salts | |
| CA2669069A1 (en) | Process for the preparation of 2-imino-thiazolidin-4-one derivatives | |
| MX2015002127A (en) | Process for the preparation of|(2z,5z)-5-(3-chloro-4-((r)-2,3-dih ydroxypropoxy)benzylidene)-2-(propylimino)-3-|(o-tolyl)thiazolid in-4-one and intermediate used in said process. | |
| US20070123573A1 (en) | Process for the preparation of biologically active tetrahydrobenzthiazole derivative | |
| WO2011021214A2 (en) | Improved process for the preparation of (s)-2-amino-4,5,6,7-tetrahydro-6 - (propylamino) benzothiazole and its pharmaceutically acceptable salts | |
| WO2013065063A1 (en) | Anhydrous form of dasatinib, process for its preparation and its use | |
| JP5927126B2 (en) | Process for producing 2- (cyclohexylmethyl) -N- {2-[(2S) -1-methylpyrrolidin-2-yl] ethyl} -1,2,3,4-tetrahydroisoquinoline-7-sulfonamide | |
| US6197969B1 (en) | Process for producing substituted alkylamines or salts thereof | |
| WO2006117614A1 (en) | Process for the preparation of pramipexole and new anhydrous forms of its dihydrochloride | |
| WO2007054970A2 (en) | Novel polymorphic forms of (s)-(-)-2-amino-6-(n- propylamino) 4,5,6,7- tetrahydrobenzothiazole | |
| US7875750B2 (en) | Method of obtaining 2-amino-6-alkyl-amino-4,5,6,7-tetrahydrobenzothiazoles | |
| CZ302094B6 (en) | N-(4-benzyloxyphenyl)-alpha-amino-4-benzyloxypropiophenone, process for its preparation and its use | |
| EP1878731B1 (en) | Process for producing pramipexole | |
| WO2015155704A1 (en) | An improved process for the preparation of pramipexole dihydrochloride monohydrate | |
| WO2006128688A1 (en) | Process for the preparation of pramipexole | |
| WO2010073255A1 (en) | Process for preparing ziprasidone |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07736498 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07736498 Country of ref document: EP Kind code of ref document: A1 |