WO2008028860A1 - Benzotriazole kinase modulators - Google Patents
Benzotriazole kinase modulators Download PDFInfo
- Publication number
- WO2008028860A1 WO2008028860A1 PCT/EP2007/059040 EP2007059040W WO2008028860A1 WO 2008028860 A1 WO2008028860 A1 WO 2008028860A1 EP 2007059040 W EP2007059040 W EP 2007059040W WO 2008028860 A1 WO2008028860 A1 WO 2008028860A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- hydrogen
- methyl
- pyrimidin
- lower alkyl
- Prior art date
Links
- SCLWTHCFOIFAIL-ZKCHVHJHSA-N COCC(N[C@H](CC1)CC[C@@H]1N)=O Chemical compound COCC(N[C@H](CC1)CC[C@@H]1N)=O SCLWTHCFOIFAIL-ZKCHVHJHSA-N 0.000 description 1
- HCBFJJAPVTVBKW-HDJSIYSDSA-N COCC(N[C@H](CC1)CC[C@@H]1Nc1nccc(-[n]2nnc3ccccc23)n1)=O Chemical compound COCC(N[C@H](CC1)CC[C@@H]1Nc1nccc(-[n]2nnc3ccccc23)n1)=O HCBFJJAPVTVBKW-HDJSIYSDSA-N 0.000 description 1
- NHVOSNPGPQLGJR-UHFFFAOYSA-N CS(c1nccc(-[n]2nnc3c2cccc3)n1)(=O)=O Chemical compound CS(c1nccc(-[n]2nnc3c2cccc3)n1)(=O)=O NHVOSNPGPQLGJR-UHFFFAOYSA-N 0.000 description 1
- DBGBJUHGJKXFRD-UHFFFAOYSA-N CS(c1nccc(N2NNc3c2cccc3)n1)=O Chemical compound CS(c1nccc(N2NNc3c2cccc3)n1)=O DBGBJUHGJKXFRD-UHFFFAOYSA-N 0.000 description 1
- POCASOBJVCKFLS-UHFFFAOYSA-N CSc1nccc(-[n]2nnc3c2cccc3)n1 Chemical compound CSc1nccc(-[n]2nnc3c2cccc3)n1 POCASOBJVCKFLS-UHFFFAOYSA-N 0.000 description 1
- DFOHHQRGDOQMKG-UHFFFAOYSA-N CSc1nccc(Cl)n1 Chemical compound CSc1nccc(Cl)n1 DFOHHQRGDOQMKG-UHFFFAOYSA-N 0.000 description 1
- ZGQKJRTXVYBRNW-UHFFFAOYSA-N NCC(CC1)CCC1Nc1nccc(-[n]2nnc3ccccc23)n1 Chemical compound NCC(CC1)CCC1Nc1nccc(-[n]2nnc3ccccc23)n1 ZGQKJRTXVYBRNW-UHFFFAOYSA-N 0.000 description 1
- IHLKJLTYSPNTFW-UHFFFAOYSA-N O=C(c1c2cccc1)N(CC(CC1)CCC1Nc1nccc(-[n]3nnc4ccccc34)n1)C2=O Chemical compound O=C(c1c2cccc1)N(CC(CC1)CCC1Nc1nccc(-[n]3nnc4ccccc34)n1)C2=O IHLKJLTYSPNTFW-UHFFFAOYSA-N 0.000 description 1
- BVXZHJSCVDXJFH-UHFFFAOYSA-N OCC(CC1)CCC1Nc1nccc(-[n]2nnc3ccccc23)n1 Chemical compound OCC(CC1)CCC1Nc1nccc(-[n]2nnc3ccccc23)n1 BVXZHJSCVDXJFH-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4192—1,2,3-Triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
Definitions
- the present invention relates to a method for modulating c-Jun N-terminal kinases (JNK) and cyclin-dependent kinases (CDK), and a method for treating a subject afflicted with a disease or condition that can be alleviated by modulating JNKs or CDKs with heterocyclic compounds, more particularly, to benzotriazole derivatives.
- JNK c-Jun N-terminal kinases
- CDK cyclin-dependent kinases
- the invention further relates to novel heterocyclic compounds and pharmaceutical compositions comprising said compound.
- JNKs The c-Jun N-terminal kinases
- ERKs extracellular signal-regulated kinases
- JNKl and JNK2 are expressed in a wide variety of tissues, whereas JNK3 is mainly expressed in neurons, and to a lesser extent in heart and testes (D.D. Yang et al, Nature (1997) 389:865-70).
- JNKs are activated by pro -inflammatory cytokines such as tumor necrosis factor ⁇ (TNF- ⁇ ) and interleukin-l ⁇ (IL- l ⁇ ), as well as environmental stresses.
- TNF- ⁇ tumor necrosis factor ⁇
- IL- l ⁇ interleukin-l ⁇
- the activation of JNKs is mediated by its upstream kinases, MKK4 and MKK7, via dual phosphorylation of Thr-183 and Tyr-185 (B. Derijard et al., Cell (1994) 76:1025-37). It has been shown that MKK4 and MMK7 can be activated by the diverse upstream kinases, including MEKKl and MEKK4, depending upon the external stimuli and cellular context (D. Boyle et al., Arthritis Rheum (2003) 48:2450-24).
- JNK-interacting proteins The specificity of JNK signaling is achieved by forming a JNK-specif ⁇ c signaling complex containing multiple components of the kinase cascade using scaffold proteins called JNK-interacting proteins (J. Yasuda et al., MoI. Cell. Biol. (1999) 19:7245-54). JNKs have been shown to play important roles in inflammation, T cell functions, apoptosis and cellular survival by phosphorylating specific substrates, including transcription factors such as c-Jun, the component of activator protein- 1 (API) family, and ATF2, as well as non-transcription factors such as IRS-I and Bcl-2 (A.M. Manning and R.J. Davis, Nat. Rev. Drug Discov. (2003) 2:554-65). Over-activation of JNK is believed to be an important mechanism in autoimmune, inflammatory, metabolic, neurological diseases as well as cancer.
- RA Rheumatoid arthritis
- JNK activation by selective JNK inhibitors blocked proinflammatory cytokines and MMP production in human synoviocytes, macrophages and lymphocytes (Z. Han et al., (2001) supra).
- administration of the selective JNK inhibitors in rats with adjuvant arthritis (Z. Han et al., (2001) supra) or in mice with collagen- induced arthritis (P. Gaillard et al., J Med Chem. (2005) 14:4596-607) effectively protected joints from destruction and significantly reduced paw swelling by inhibiting cytokine and collagenase expression.
- JNK2 deficient mice were partially protected from joint destruction, but showed little effect on paw swelling and inflammation in the passive collagen- induced arthritis model.
- JNK2 is functionally redundant with JNKl in regard to their roles in matrix degradation, inflammation and paw swelling. Therefore, combined inhibition of both JNKl and JNK2 activities is required for effective therapy for RA (Z. Han et al., Arthritis Rheum. (2002) 46:818-23).
- Asthma is a chronic inflammatory disease of airways, characterized by the presence of a cellular inflammatory process and by bronchial hyper-responsiveness associated with structural changes of the airways (B. Bradley et al., J. Allergy Clin. Immunol. (1991) 88:661-74). This disorder has been shown to be driven by many cell types in the airways, including T lymphocytes, eosinophils, mast cells, neutrophils and epithelial cells (J. Bousquet et al., Am. J. Respir. Crit. Care Med. (2000) 161:1720-45).
- JNKs have emerged as promising therapeutic targets for asthma based upon the recent proof-of- s in the cellular and animal models of asthma using selective JNK inhibitors (K. Blease et al, Expert Opin. Emerg. Drugs (2003) 8:71-81). It was shown that JNK inhibitors significantly blocked RANTES production in activated human airway smooth cells (K Kujime et al., J. Immunol. (2000) 164:3222-28). More importantly, the JNK inhibitors showed good efficacy in chronic rat and mouse models for their abilities to reduce cellular infiltration, inflammation, hyper-responsiveness, smooth muscle proliferation, and IgE production (P. Nath et al., Eur. J. Pharmacol. (2005) 506:273-83; P.
- Type 2 diabetes is the most serious and prevalent metabolic disease characterized by insulin resistance and insulin secretion impairment as a result of chronic low- level inflammation and abnormal lipid metabolism associated with oxidative stress. It has been reported that JNK activity is abnormally elevated in various diabetic target tissues under obese and diabetic conditions (J. Hirosumi et al., Nature (2002) 420:333-36; H. Kaneto, Expert. Opin. Ther. Targets (2005) 9:581-92). Activation of the JNK pathway by pro-inflammatory cytokines and oxidative stresses negatively regulates insulin signaling via phosphorylation of insulin receptor substrate- 1 (IRS-I) at Ser 307 , therefore contributes to insulin resistance and glucose tolerance (J. Hirosumi et al., Nature (2002) supra; Y. Lee et al., J. Biol. Chem. (2003) 278:2896-902; Y. Nakatani et al., J. Biol.
- JIP-I JNK inhibitory peptide I(JIP) derived from the JNK binding domain of the JNK- interacting protein- 1
- T T o ⁇ 102:6931-35 revealed that JNK2 plays an important role in type 1 diabetes caused by autoimmune destruction of insulin-producing ⁇ cells.
- Non-obese diabetic mice deficient in JNK2 expression showed reduced destructive insulitis and less disease progression to diabetes, probably due to biased polarization toward the Th2 phenotype.
- these studies demonstrated the utility of JNK inhibitors in the treatment of obesity/type 2 diabetes.
- Neurodegenerative diseases such as Alzheimer's (AD), Parkinson's (PD) and stroke are characterized by synaptic loss, neuronal atrophy and death.
- the JNK pathway leading to c-Jun activation has been shown to play a causal role in apoptosis of isolated primary embryonic neurons and multiple neuronal cell lines upon induction of a variety of stimuli (D. Bozyczko-Coyne et al, Curr. Drug Targets CNS Neurol. Disord. (2002) 1:31-49).
- Over-activation of JNK was observed in human brains from AD patients (J. Pei et al., J. Alzheimers Dis. (2001) 3:41-48) or rodent brain sections derived from animal models of neurodegenerative diseases (M.
- JIP-I peptide JNK inhibitory peptide
- JNK3 was mainly responsible for glutamate excitotoxicity, an important component in ischemic conditions. Taken together, the data suggests that JNKs are an attractive target for multiple CNS diseases associated with neuronal cell death.
- JNK signal transduction pathway may not act exclusively in apoptosis, sustained JNK activation leading to API recently been implicated to contribute to the cellular survival of specific cancer types such as glial tumors and BCL-ABL transformed B lymphoblasts (M. Antonyak et al, Oncogene (2002) 21:5038-46; P. Hess et al, Nat. Genet. (2002) 32:201- 05).
- glial tumors enhanced JNK/ API activity was seen in most of the primary brain tumor samples.
- BCL-ABL was shown to activate the JNK pathway which in turn up-regulated expression of anti- apoptotic bcl-2 gene.
- the multi-drug resistance and hyper-proliferation seen in treatment-refractory AML patients has been causally linked to the sustained JNK activity present in these AML samples (L. Cripe et al., Leukemia (2002) 16:799-812).
- Activation of JNK in leukemic cells resulted in induced expression of efflux pumps such as mdrl and MRPl responsible for multidrug resistance.
- genes with a survival benefit in response to oxidative stress including glutathione-S-transferase ⁇ and ⁇ - glutamyl cysteine synthase were also upregulated by the activated JNK pathway.
- JNK modulators are useful in treating a variety of diseases and/or conditions.
- cyclin-dependent kinases cyclin-dependent kinases
- Inhibitors of cellular proliferation act as reversible cytostatic agents that are useful in the treatment of disease processes which feature abnormal cellular growth, such as cancers and other cell proliferative disorders including, for example inflammation (e.g.
- Parkinson's disease amyotrophic lateral sclerosis, retinitis pigmentosa, spinal muscular erebral degeneration.
- One aspect of the invention provides a compound of formula I:
- R is lower alkyl, hydroxy lower alkyl, or a radical selected from:
- R 1 is hydrogen, halo, alkyl, Or NH 2 ; each of R 3 is independently halo, -NO 2 , lower alkyl, -CN, -OR 7 , -NR 8 R 9 , - C(O)-R 7 ,
- R 4 is hydrogen, lower alkyl, cyano, -(CH 2 ) n OR 7 , -(CH 2 ) n NR 8 R 9 , -(CH 2 ) n -C(O)- NR 8 R 9 ,
- R 5 is hydrogen or alkyl; or R 4 and R 5 together form alkylene dioxy;
- R 6 is hydrogen, lower alkyl, heteroalkyl, cycloalkyl, heterocyclylalkyl, or - NR 8 R 9 ;
- R 10 is alkyl, cycloalkyl, heterocyclylalkyl, or -NR 8 R 9 ;
- R 11 is alkyl, cycloalkyl, heteroalkyl, or (heterocyclyl)alkyl;
- R 2 and R 7 are each independently hydrogen or lower alkyl;
- R 8 is hydrogen, lower alkyl, or acyl;
- R 9 is hydrogen, lower alkyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl; or R 8 and R 9 together with the nitrogen atom to which they are connected to form a heterocyclyl comprising at least one nitrogen ring atom, optionally substituted with OH, oxo, lower alkyl, lower alkoxy, or acyl; each of m and x is independently an integer from
- Y is hydrogen, -(CH 2 ) n -OR 7 , -(CH 2 ) n -C(O)-R 7 or -(CH 2 ) n -C(O)-OR 7 ; each of y and z is independently 0 or 1; and n is an integer from 0 to 4.
- the invention also provides pharmaceutical compositions, methods of using, and methods of preparing the aforementioned compounds.
- Compounds and compositions of the invention are useful in the treatment and/or prevention of a c-Jun N-terminal kinase mediated disorder, such as autoimmune disorders, inflammatory disorders, metabolic disorders, neurological diseases, and cancer.
- a c-Jun N-terminal kinase mediated disorder such as autoimmune disorders, inflammatory disorders, metabolic disorders, neurological diseases, and cancer.
- compounds and compositions of the invention are useful in treating and/or preventing rheumatoid arthritis, asthma, type II diabetes, Alzheimer's disease, Parkinson's disease and/or stroke.
- CDK mediated disorder which are generally disease processes which feature abnormal cellular growth, such as cancers and other cell proliferative disorders including, for example inflammation (e.g. benign prostate hyperplasia, familial adenomauosis, polyposis, neuro-f ⁇ bromatosis, atherosclerosis, pulmonary fibrosis, arthritis, psoriasis, inflammatory bowel disease, transplantation rejections infections), viral infections (including, without limitation, herpesvirus, poxvirus, Epstein-Barr virus), autoimmune disease (e.g.
- inflammation e.g. benign prostate hyperplasia, familial adenomauosis, polyposis, neuro-f ⁇ bromatosis, atherosclerosis, pulmonary fibrosis, arthritis, psoriasis, inflammatory bowel disease, transplantation rejections infections
- viral infections including, without limitation, herpesvirus, poxvirus, Epstein-Barr virus
- autoimmune disease e.g.
- Alkyl means the monovalent linear or branched saturated hydrocarbon moiety, consisting solely of carbon and hydrogen atoms, having from one to twelve carbon atoms.
- “Lower alkyl” refers to an alkyl group of one to six carbon atoms, i.e. Ci-C 6 alkyl. Examples of alkyl groups include, but are not limited to, methyl, ethyl, propyl, isopropyl, isobutyl, sec-butyl, tert-butyl, pentyl, n-hexyl, octyl, dodecyl.
- Branched alkyl refers to an alkyl moiety having at least one branch, for example, isopropyl, isobutyl, tert-butyl, and the like.
- lower alkoxy refers to a moiety of the form -OR
- acyl refers to a moiety of the form -C(O)R, where R is lower alkyl.
- Alkylene means a linear saturated divalent hydrocarbon moiety of one to six carbon atoms or a branched saturated divalent hydrocarbon radical of three to six carbon atoms, e.g., methylene, ethylene, 2,2-dimethylethylene, propylene, 2-methylpropylene, butylene, pentylene.
- Alkylene dioxy means a divalent moiety of the formula -O-R-O-, where R is alkylene as defined herein.
- Aryl means a monovalent cyclic aromatic hydrocarbon moiety consisting of a mono-, bi- or tricyclic aromatic ring. Phenyl or naphthyl is preferred. The aryl group can be optionally substituted as defined herein.
- aryl moieties include, but are not limited to, optionally substituted phenyl, naphthyl, phenanthryl, fluorenyl, indenyl, pentalenyl, azulenyl, oxydiphenyl, biphenyl, methylenediphenyl, aminodiphenyl, diphenylsulf ⁇ dyl, diphenylsulfonyl, diphenylisopropylidenyl, benzodioxanyl, benzofuranyl, benzodioxylyl, benzopyranyl, benzoxazinyl, benzoxazinonyl, benzopiperadinyl, benzopiperazinyl, benzopyrrolidinyl, benzomorpholinyl, methylenedioxyphenyl, ethylenedioxyphenyl, including partially hydrogenated derivatives thereof.
- Cycloalkyl means a monovalent saturated carbocyclic moiety consisting of mono- or bicyclic rings. Cycloalkyl can optionally be substituted with one or more substituents, mbstituent is independently hydroxy, alkyl, alkoxy, halo, haloalkyl, amino, monoalkylamino, or dialkylamino, unless otherwise specifically indicated. Preferred cycloalkyl is C3_7 mono-cyclic cycloalkyl. Examples of cycloalkyl moieties include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, including partially unsaturated derivatives thereof.
- Cycloalkylalkyl mean a moiety of the formula -R a -R b , where R a is alkylene and R b is cycloalkyl as defined herein.
- Heteroalkyl means an alkyl moiety as defined herein, including a branched C 4 -C 7 alkyl, wherein one, two or three hydrogen atoms have been replaced with a substituent independently selected from the group consisting of-OR a , -NR b R c , and -S(O) n R d (where n is an integer from 0 to 2), with the understanding that the point of attachment of the heteroalkyl radical is through a carbon atom, wherein R a is hydrogen, acyl, alkyl, cycloalkyl, or cycloalkylalkyl; R b and R c are independently of each other hydrogen, acyl, alkyl, cycloalkyl, or cycloalkylalkyl; and when n is 0, R d is hydrogen, alkyl, cycloalkyl, or cycloalkylalkyl; when n is 1, R d is alkyl, cycloalkyl,
- Representative examples include, but are not limited to, 2-hydroxyethyl, 3-hydroxypropyl, 2-hydroxy-l -hydro xymethylethyl, 2,3-dihydroxypropyl, 1- hydroxymethylethyl, 3-hydroxybutyl, 2,3-dihydroxybutyl, 2-hydroxy-l -methylpropyl, 2- aminoethyl, 3-aminopropyl, 2-methylsulfonylethyl, aminosulfonylmethyl, aminosulfonylethyl, aminosulfonylpropyl, methylaminosulfonylmethyl, methylamino- sulfonylethyl, methylaminosulfonylpropyl.
- Heteroaryl means a monocyclic or bicyclic moiety of 5 to 12 ring atoms having at least one aromatic ring containing one, two, or three ring heteroatoms selected from N, O, or S, the remaining ring atoms being C, with the understanding that the attachment point of the heteroaryl radical will be on an aromatic ring.
- the heteroaryl ring may be optionally substituted as defined herein.
- heteroaryl moieties include, but are not limited to, optionally substituted imidazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, oxadiazolyl, thiadiazolyl, pyrazinyl, thienyl, thiophenyl, furanyl, pyranyl, pyridinyl, pyrrolyl, pyrazolyl, pyrimidyl, pyridazinyl, quinolinyl, isoquinolinyl, benzofuryl, benzo furanyl, benzo thiophenyl, benzothiopyranyl, benzimidazolyl, benzoxazolyl, benzooxadiazolyl, benzo thiazolyl, benzo thiadiazolyl, benzopyranyl, indolyl, isoindolyl, ⁇ o IyI, triazinyl, quinoxalinyl
- halo halogen
- halide a substituent fluoro, chloro, bromo, or iodo.
- Haloalkyl means alkyl as defined herein in which one or more hydrogen has been replaced with same or different halogen.
- exemplary haloalkyls include -CH 2 Cl, - CH 2 CF 3 ,
- perfiuoroalkyl e.g., -CF 3
- Heterocyclyl means a monovalent saturated moiety, consisting of one to three rings, incorporating one, two, or three or four heteroatoms (chosen from nitrogen, oxygen or sulfur). A monocyclic heterocyclyl, having 3 to 8 ring atoms, is preferred. The heterocyclyl ring may be optionally substituted as defined herein.
- heterocyclyl moieties include, but are not limited to, optionally substituted piperidinyl, piperazinyl, homopiperazinyl, azepinyl, pyrrolidinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, pyridinyl, pyridazinyl, pyrimidinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl, isothiazolidinyl, quinuclidinyl, quinolinyl, isoquinolinyl, benzimidazolyl, thiadiazolylidinyl, benzo thiazolidinyl, benzoazolylidinyl, dihydrofuryl, tetrahydrofuryl, dihydropyranyl, tetrahydropyranyl, thiamorpholinyl, thiamorpholinylsulfoxide,
- Leaving group means a group with the meaning conventionally associated with it in synthetic organic chemistry, i.e., an atom or group displaceable under substitution reaction conditions.
- Examples of leaving groups include, but are not limited to, halogen, alkane- or arylenesulfonyloxy, such as methanesulfonyloxy, ethanesulfonyloxy, thiomethyl, benzenesulfonyloxy, tosyloxy, and thienyloxy, dihalophosphinoyloxy, optionally substituted benzyloxy, isopropyloxy, acyloxy.
- Disease and Disease state means any disease, condition, symptom, disorder or indication.
- Inert organic solvent or “inert solvent” means the solvent is inert under the conditions of the reaction being described in conjunction therewith, including for example, benzene, toluene, acetonitrile, tetrahydrofuran, N,N-dimethylformamide, chloroform, methylene chloride or dichloromethane, dichloroethane, diethyl ether, ethyl acetate, acetone, methyl ethyl ketone, methanol, ethanol, propanol, isopropanol, tert-butanol, dioxane, pyridine.
- the solvents used in the reactions of the present invention are inert solvents.
- “Pharmaceutically acceptable” means that which is useful in preparing a pharmaceutical composition that is generally safe, non-toxic, and neither biologically nor otherwise undesirable and includes that which is acceptable for veterinary as well as human pharmaceutical use.
- “Pharmaceutically acceptable salts” of a compound means salts that are pharmaceutically acceptable, as defined herein, and that possess the desired pharmacological activity of the parent compound. Such salts include:
- acid addition salts formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid; or formed with organic acids such as acetic acid, benzenesulfonic acid, benzoic, camphorsulfonic acid, citric acid, ethanesulfonic acid, fumaric acid, glucoheptonic acid, gluconic acid, glutamic acid, rrK TM i; ⁇ C ⁇ A hydroxynaphtoic acid, 2-hydroxyethanesulfonic acid, lactic acid, maleic acid, malic acid, malonic acid, mandelic acid, methanesulfonic acid, muconic acid, 2- naphthalenesulfonic acid, propionic acid, salicylic acid, succinic acid, tartaric acid, p- toluenesulfonic acid, trimethylacetic acid; or salts formed when an acidic proton present in the parent compound either is replaced by
- Acceptable organic bases include diethanolamine, ethanolamine, N-methylglucamine, triethanolamine, tromethamine.
- Acceptable inorganic bases include aluminum hydroxide, calcium hydroxide, potassium hydroxide, sodium carbonate, sodium hydroxide.
- the preferred pharmaceutically acceptable salts are the salts formed from acetic acid, hydrochloric acid, sulfuric acid, methanesulfonic acid, maleic acid, phosphoric acid, tartaric acid, citric acid, sodium, potassium, calcium, zinc, and magnesium.
- Protecting group means the group which selectively blocks one reactive site in a multifunctional compound such that a chemical reaction can be carried out selectively at another unprotected reactive site in the meaning conventionally associated with it in synthetic chemistry. Certain processes of this invention rely upon the protective groups to block reactive nitrogen and/or oxygen atoms present in the reactants.
- the terms "amino -protecting group” and “nitrogen protecting group” are used interchangeably herein and refer to those organic groups intended to protect the nitrogen atom against undesirable reactions during synthetic procedures.
- Exemplary nitrogen protecting groups include, but are not limited to, trifluoro acetyl, acetamido, benzyl (Bn), benzyloxycarbonyl (carbobenzyloxy, CBZ), p- methoxybenzyloxycarbonyl, p-nitrobenzyloxycarbonyl, te/t-butoxycarbonyl (BOC). Skilled persons will know how to choose a group for the ease of removal and for the ability to withstand the following reactions.
- Subject means mammals and non-mammals. Mammals means any member of the mammalia class including, but not limited to, humans; non-human primates such as chimpanzees and other apes and monkey species; farm animals such as cattle, horses, sheep, goats, and swine; domestic animals such as rabbits, dogs, and cats; laboratory animals including rodents, such as rats, mice, and guinea pigs. Examples of non- mammals include, but are not limited to, birds.
- the term “subject” does not denote a particular age or sex.
- “Therapeutically effective amount” means an amount of a compound that, when administered to a subject for treating a disease state, is sufficient to effect such treatment for the disease state. The “therapeutically effective amount” will vary depending on the compound, disease state being treated, the severity or the disease treated, the age and relative health of the subject, the route and form of administration, the judgment of the attending medical or veterinary practitioner, and other factors.
- Treating" or “treatment” of a disease state includes:
- treating when referring to a chemical reaction means adding or mixing two or more reagents under appropriate conditions to produce the indicated and/or the desired product. It should be appreciated that the reaction which produces the indicated and/or the desired product may not necessarily result directly from the combination of two reagents which were initially added, i.e., there may be one or more intermediates which are produced in the mixture which ultimately leads to the formation of the indicated and/or the desired product.
- One aspect of the invention provides compounds of formula I:
- R is lower alkyl, hydroxy lower alkyl, or a radical selected from:
- each R a is independently H, lower alkyl, OH, or hydroxy-lower alkyl
- each R b is independently H, lower alkyl, halo, nitro, or halo-lower alkyl
- p is 2, 3, or 4;
- R 1 is hydrogen, halo, alkyl, Or NH 2 ; each of R 3 is independently halo, -NO 2 , lower alkyl, -CN, -OR 7 , -NR 8 R 9 , - C(O)-R 7 ,
- R 4 is hydrogen, lower alkyl, cyano, -(CH 2 ) n OR 7 , -(CH 2 ) n NR 8 R 9 , -(CH 2 ) n -C(O)- NR 8 R 9 ,
- R 6 is hydrogen, lower alkyl, heteroalkyl, cycloalkyl, heterocyclylalkyl, or -
- R 10 is alkyl, cycloalkyl, heterocyclylalkyl, or -NR 8 R 9 ;
- R 11 is alkyl, cycloalkyl, heteroalkyl, or (heterocyclyl)alkyl;
- R 2 and R 7 are each independently hydrogen or lower alkyl;
- R 8 is hydrogen, lower alkyl, or acyl;
- R 9 is hydrogen, lower alkyl, heteroalkyl, aryl, heteroaryl, heterocyclyl, cycloalkyl; or R 8 and R 9 together with the nitrogen atom to which they are connected to form a heterocyclyl comprising at least one nitrogen ring atom, optionally substituted with OH, oxo, lower alkyl, lower alkoxy, or acyl;
- each of m and x is independently an integer from 0 to 2;
- Y is hydrogen, -(CH 2 ) n -OR 7 , -(CH 2 ) n -C(O)-R 7 or -(CH 2 ) n -C(O)-OR 7 ;
- each of y and z is independently 0 or 1; and
- n is an integer from 0 to 4.
- R 2 is hydrogen or methyl.
- R is , where z is 1 and X is O or CR R .
- R 4 is OH, -C(O)NR 8 R 9 , -NR 8 R 9 , -NR 7 SO 2 R 10 , or -OR 7 .
- n 0.
- R 1 is hydrogen, methyl, chloro, or fluoro.
- X is CR 4 R 5 , where R 4 and R 5 are those defined herein.
- R 5 is hydrogen or methyl.
- z is 1.
- R 4 is -NR 7 -SO 2 -R 10 , where R 7 , and R 10 are those defined herein.
- x is 2, R 7 is hydrogen or methyl, and R 10 is methyl, ethyl, or -N(CHs) 2 .
- z is 1, and R 4 is hydrogen, lower alkyl, cyano, -(CH 2 ) n OR 7 , or -(CH 2 ) n NR 8 R 9 , or R 4 and R 5 together form alkylene dioxy, where n, R 7 , R 8 , and R 9 are those defined herein.
- R 4 is -(CH 2 ) n OR 7 , n is 0 or 1, and R 7 is hydrogen or methyl.
- R 4 is -(CH 2 ) n NR 8 R 9 , where n, R 8 , and R 9 are those defined herein.
- n is 0 and R 8 is hydrogen
- R 9 is hydrogen, pyrimidin-2-yl, or pyridin-2-yl.
- n is 0 and R 8 and R 9 together with the nitrogen-atom to which they are connected to form 2,5-dioxo-pyrrolidin- 1 -yl.
- R 4 is hydrogen, methyl, ethyl, or cyano.
- R 4 and R 5 together form ethylene dioxy.
- compounds of formula I include those where z is 1, and R 4 is -(CH 2 )n-NR 8 -C(O)-R 1 * , where n, R 8 , and R 1 * are those defined herein.
- n is 0, R 8 is hydrogen or methyl, and R 11 is methyl, ethyl, methoxymethyl, hydro xymethyl, (morpholin-4-yl)methyl, or (4-methyl-piperazin-l- yl)methyl.
- z is 1, and R 4 is -(CH 2 ) n -C(O)-NR 8 R 9 , wherein n, R 8 , and R 9 are those defined herein.
- n is 0, and R 8 and R 9 together with the nitrogen-atom to which they are connected to form morpholin- 4-yl, pyrrolidin-1-yl, or 4-methyl-piperazin-l-yl.
- R 8 is hydrogen or methyl
- R 9 is (2-amino-2-methyl)propyl, (2-hydroxy)ethyl, tetrahydropyran-4-yl, cyclopropyl, or ethyl.
- z is 1, and R 4 is -(CH 2 ) n -C(O)-OR 7 , wherein n and R 7 are those defined herein.
- n is 0 and R 7 is hydrogen or methyl.
- R 4 and R 5 are hydrogen, z is 0, y is 1, and Y is hydroxy on of the cyclopentyl ring moiety. Yet in other embodiments, R 4 and R 5 are hydrogen, z is 1, y is 1, and Y is hydroxy, hydroxymethyl, or -CO 2 CH 2 CH 3 group on the 2-position of the cyclohexyl ring moiety.
- X is NR 6 , wherein R 6 is that defined in Claim 1.
- R 6 is hydrogen, -S(O) 2 CHs, or -CH 2 C(O)NH 2 .
- X is CR 4 R 5 , where R 4 and R 5 are those defined herein.
- R 5 is hydrogen or methyl.
- z is 1, and R 4 is - NR 7 -SO 2 -R 10 , where R 7 and R 10 are those defined herein.
- R 7 is hydrogen or methyl
- R 10 is methyl, ethyl, or -N(CHs) 2 .
- z is 1, and R 4 is hydrogen, lower alkyl, cyano, - (CH 2 ) n OR 7 , or -(CH 2 ) n NR 8 R 9 , or R 4 and R 5 together form alkylene dioxy, where n, R 7 , R 8 , and R 9 are those defined herein.
- R 4 is - (CH 2 ) n OR 7 , n is 0 or 1, and R 7 is hydrogen or methyl.
- R 4 is -(CH 2 ) n NR 8 R 9 , where n, R 8 , and R 9 are those defined herein.
- some of the particular compounds include those where n is 0 and R 8 is hydrogen, and R 9 is hydrogen, pyrimidin-2-yl, or pyridin-2-yl.
- Other particular compounds within these cases include those where n is 0 and R 8 and R 9 together with the nitrogen-atom to which they are connected to form 2,5-dioxo-pyrrolidin-l-yl.
- R 4 is hydrogen, methyl, ethyl, or cyano. In some instances within this embodiment, R 4 and R 5 together form ethylene dioxy.
- compounds of formula I include those where z is 1, and R 4 is -(CH 2 ) n -NR 8 -C(O)-R ⁇ , where n, R 8 , and R 11 are those defined herein.
- n is 0, R 8 is hydrogen or methyl, and R 11 is methyl, ethyl, methoxymethyl, hydroxymethyl, (morpholin-4-yl)methyl, or (4-methyl-piperazin-l- yl)methyl.
- z is 1
- R 4 is -(CH 2 ) n -C(O)-NR 8 R 9 , wherein n, R 8 , and R 9 Led herein.
- n is 0, and R 8 and R 9 together with the nitrogen-atom to which they are connected to form morpholin- 4-yl, pyrrolidin-1-yl, or 4-methyl-piperazin-l-yl.
- R 8 is hydrogen or methyl
- R 9 is (2-amino-2-methyl)propyl, (2-hydroxy)ethyl, tetrahydropyran-4-yl, cyclopropyl, or ethyl.
- z is 1, and R 4 is -(CH 2 )D-C(O)-OR 7 , wherein n and R 7 are those defined herein. Within these embodiments, in some instances n is 0 and R 7 is hydrogen or methyl.
- R 4 and R 5 are hydrogen, z is 0, y is 1, and Y is hydroxy on the 3-position of the cyclopentyl ring moiety.
- R 4 and R 5 are hydrogen, z is 1, y is 1, and Y is hydroxy, hydroxymethyl, or -CO 2 CH 2 CH 3 group on the 2-position of the cyclohexyl ring moiety.
- X is NR 6 , wherein R 6 is that defined in Claim 1.
- R 6 is hydrogen, -S(O) 2 CHs, or - CH 2 C(O)NH 2 .
- compounds of formula I are of the formula IA:
- the starting materials and the intermediates of the synthetic reaction schemes can be isolated and purified if desired using conventional techniques, including but not limited to, filtration, distillation, crystallization, chromatography, and the like. Such materials can be characterized using conventional means, including physical constants and spectral data.
- the reaction described herein preferably are conducted under inert atmosphere, at atmospheric pressure, at a reaction temperature range of from about -78 0 C to about 23O 0 C, and most preferably and conveniently at room (or ambient) temperature, e.g., about 2O 0 C.
- Step A a substituted 4-chloropyrimidine undergoes a S N AT reaction with a variably substituted lH-benzotriazole in the presence of a base such as sodium hydride and in a polar aprotic solvent such as N, ⁇ /-dimethylformamide at a temperature ranging between O 0 C and about RT.
- a base such as sodium hydride
- a polar aprotic solvent such as N, ⁇ /-dimethylformamide
- Step B the thio methyl group Y is converted to a leaving group by oxidation with 3- chloroperoxybenzoic acid in aprotic solvents such as chloroform, or by chlorination with JV-chloro succinimide .
- Step C the leaving group Y or Z (Cl or MeSO 2 ) is displaced by a primary amine a thermally, by heating the mixture in a polar aprotic solvent such as l-methyl-2- pyrrolidinone at a temperature ranging between about 100 0 C and about 13O 0 C or by treatment with a base such as triethylamine at a temperature ranging between RT and r aprotic solvent such as tetrahydrofuran.
- a polar aprotic solvent such as l-methyl-2- pyrrolidinone
- Amines a may comprise, for example: cycloalkylamines such as variably substituted cyclohexylamines and cyclopentylamines; alkyl amines such as isobutylamine; hydroxyalkylamines such as 4- amino-1-butanol; heterocyclic amines such as 4-aminotetrahydropyran, 4-amino-l-BOC- piperidine.
- cycloalkylamines such as variably substituted cyclohexylamines and cyclopentylamines
- alkyl amines such as isobutylamine
- hydroxyalkylamines such as 4- amino-1-butanol
- heterocyclic amines such as 4-aminotetrahydropyran, 4-amino-l-BOC- piperidine.
- Numerous variably substituted alkyl, cycloalkyl and heterocyclic amines a are commercially available or are readily prepared by techniques well known to those skilled in the art
- the products can then be purified, for example, by extraction, crystallization, preparative HPLC, flash chromatography, thin layer chromatography, and the like.
- a compound of generic formula (iv) can undergo the transformations shown in Scheme II to give compounds that are the object of this invention.
- Ra is CORn, CH 2 CONR 8 R 9 , SO 2 Rn, or SO 2 NR 8 R 9 .
- Rb is alkyl, heteroalkyl, (heterocyclic)alkyl.
- R c and Rj are independently H, alkyl, cycloalkyl, alkoxyalkyl, hydroxyalkyl, or heterocyclic.
- Z is heterocyclic.
- R g is COR n , NR 5 R 8 .
- R h is alkyl or aryl. Step F: b or c, NMP, heating or d, NMP, MW or NaBH 4 , e, MeOH.
- Step G NaH, f, NMP.
- Step H NaOH, THF.
- Step I g, BOP, DIPEA, THF.
- Step N 1. IBX, DMSO; 2. h, NaBH(OAc) 3 , AcOH, DCE.
- Step K PPh 3 , DIAD, i, PhMe.
- Step L i, N 2 H 4 , EtOH, heating.
- Step M 1. HCl, THF; 2. k, THF.
- a compound of generic formula (iv) can under undergo an acylation or a sulfonylation reaction, as described in Step F, using for example an acylating agent b such as acetic anhydride in a polar aprotic solvent such as l-methyl-2-pyrrolidinone at a temperature ranging between about RT and 7O 0 C; under the same conditions (iv) can be sulfonylated using, for example, methanesulfonic anhydride as sulfonylating agent c.
- (iv) can undergo an arylation reaction using an heteroarylhalide d, for example, 2-fluoropyridine under microwave conditions in a polar aprotic solvent such as l-methyl-2-pyrrolidinone at high temperature.
- the acylating and sulfonylating agents b and c may comprise for example alkyl anhydrides, cyclic anhydrides, acyl and benzoyl chlorides, alkylsulfonyl anhydrides and alkylsulfonyl and benzoyl sulfonyl chlorides.
- an amine, amide or sulfonamide of generic formula (v) is alkylated using a Ddium hydride and an alkylating agent f such as methyl iodide in a polar aprotic solvent such as l-methyl-2-pyrrolidinone.
- Alkylating agents f may comprise alkyl halides, heteroalkyl and (heterocyclic)alkyl halides.
- an ester of formula (iv) can be hydro lyzed to the corresponding carboxylic acid using an aqueous solution of an inorganic base such as sodium hydroxide in a polar aprotic solvent such as tetrahydrofuran as described in Step H.
- a carboxylic acid (vi) can be coupled with a primary or secondary amine g in the presence of a coupling agent such as BOP and a base such as diisopropylethylamine in a polar aprotic solvent such as tetrahydrofuran as described in Step I to give an amide of generic formula (vii).
- Amines g may comprise for example alkylamines, alkoxyalkylamines, hydro xyalkylamines, cycloalkylamines and heterocyclic amines.
- R x is COOEt or COOMe an ester of formula (iv) can be reduced to the corresponding alcohol by treatment with lithium aluminum hydride in a polar aprotic solvent such as THF at temperatures ranging between about -78 0 C and about RT as described in Step J.
- the alcohol of generic formula (ix) can be oxidized to the corresponding aldehyde by treatment with an oxidizing agent such as o-iodoxybenzoic acid in a polar aprotic solvent such as DMSO.
- the aldehyde obtained in this way can subsequently undergo a reductive amination reaction with a primary or secondary amine h such as morpholine in the presence of sodium triacetoxyborohydride and glacial acetic acid in an apolar solvent such as 1,2-dichloroethane (Step N).
- Amines h may comprise for example alkylamines, cycloalkylamines and heterocyclic amines.
- the alcohol (ix) can undergo a Mitsunobu reaction with an imide i such as phthalimide in the presence of triphenylphosphine and DIAD in an apolar aprotic solvent such as toluene as described in Step K.
- the imines i may comprise cyclic and heterocyclic imines.
- a compound of generic formula (viii) when Z is phthalimide can be treated with hydrazine in a polar protic solvent such as ethanol at high temperature to give the corresponding primary amine which can then be acylated or sulfonylated by treatment with an acylating or sulfonylating agent such as acetyl chloride in presence of a base such as triethylamine as described in Step L.
- Acylating and sulfonylating agents may include acyl and aryl chlorides, sulfonyl and benzenesulfonyl chloride which are either commercially available or readily prepared through techniques known to those of ordinary skill in the art.
- a ketal (iv) can be converted in the corresponding ketone by treatment with an aqueous solution of HCl in a polar aprotic solvent such as tetrahydrofuran at high temperature; the ketone obtained in this manner can undergo an addition reaction with a Grignard reactant k in a polar aprotic solvent such as tetrahydrofuran at low temperature to give the corresponding tertiary alcohol as described in Step M.
- a Grignard reactant k are commercially available or are readily prepared by techniques known to those of ordinary skill in the art.
- the products can then be purified, e.g., by extraction, crystallization, preparative HPLC, flash chromatography, thin layer chromatography and the like.
- the compounds of this invention are CDK and JNK modulators and as such are expected to be effective in the treatment of a wide range of CDK and JNK mediated disorders.
- exemplary JNK mediated disorders include, but are not limited to, autoimmune disorder, inflammatory disorder, metabolic disorder, neurological disease, and cancer.
- compounds of the invention can be used to treat one or more of such disorders.
- compounds of the invention can be used to treat a JNK mediated disorder such as rheumatoid arthritis, asthma, type II diabetes, Alzheimer's disease, Parkinson's disease or stroke.
- a JNK mediated disorder such as rheumatoid arthritis, asthma, type II diabetes, Alzheimer's disease, Parkinson's disease or stroke.
- CDK mediated disorders include, without limitation, inflammation (e.g. benign prostate hyperplasia, familial adenomauosis, polyposis, neuro-f ⁇ bromatosis, atherosclerosis, pulmonary fibrosis, arthritis, psoriasis, inflammatory bowel disease, transplantation rejections infections), viral infections (including, without limitation, herpesvirus, poxvirus, Epstein-Barr virus), autoimmune disease (e.g.
- neurodegenerative disorders including, without limitation, Alzheimer's disease
- neurodegenerative diseases e.g. Parkinson's disease, amyotrophic lateral sclerosis, retinitis pigmentosa, spinal muscular atrophy, and cerebral degeneration.
- compositions comprising at least one compound of the present invention, or an individual isomer, racemic or non-racemic mixture of isomers or a pharmaceutically acceptable salt or solvate thereof, together with at least one pharmaceutically acceptable carrier, and optionally other therapeutic and/or prophylactic ingredients.
- Compounds of the invention may be administered as pharmaceutical formulations including those suitable for oral (including buccal and sub-lingual), rectal, nasal, topical, pulmonary, vaginal, or parenteral (including intramuscular, intraarterial, intrathecal, sub- cutaneous and intravenous) administration or in a form suitable for administration by inhalation or insufflation.
- the preferred manner of administration is generally oral using a convenient daily dosage regimen which can be adjusted according to the degree of affliction.
- a compound or compounds of the invention, together with one or more conventional adjuvants, carriers, or diluents, may be placed into the form of pharmaceutical compositions and unit dosages.
- the pharmaceutical compositions and unit dosage forms may be comprised of conventional ingredients in conventional proportions, with or without additional active compounds or principles, and the unit dosage forms may contain any suitable effective amount of the active ingredient commensurate with the intended daily dosage range to be employed.
- compositions may be em ⁇ loved as solids, such as tablets or filled capsules, semisolids, powders, sustained ations, or liquids such as solutions, suspensions, emulsions, elixirs, or f ⁇ lled capsules for oral use; or in the form of suppositories for rectal or vaginal administration; or in the form of sterile injectable solutions for parenteral use.
- Formulations containing about one (1) mg of active ingredient or, more broadly, about 0.01 to about one hundred (100) mg, per tablet, are accordingly suitable representative unit dosage forms.
- the compounds of the invention may be formulated in a wide variety of oral administration dosage forms.
- the pharmaceutical compositions and dosage forms may comprise a compound or compounds of the present invention or pharmaceutically acceptable salts thereof as the active component.
- the pharmaceutically acceptable carriers may be either solid or liquid. Solid form preparations include powders, tablets, pills, capsules, cachets, suppositories, and dispersible granules.
- a solid carrier may be one or more substances which may also act as diluents, flavoring agents, solubilizers, lubricants, suspending agents, binders, preservatives, tablet disintegrating agents, or an encapsulating material.
- the carrier In powders, the carrier generally is a finely divided solid which is a mixture with the finely divided active component.
- the active component In tablets, the active component generally is mixed with the carrier having the necessary binding capacity in suitable proportions and compacted in the shape and size desired.
- the powders and tablets preferably contain from about one (1) to about seventy (70) percent of the active compound.
- Suitable carriers include but are not limited to magnesium carbonate, magnesium stearate, talc, sugar, lactose, pectin, dextrin, starch, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose, a low melting wax, cocoa butter, and the like.
- the term "preparation" is intended to include the formulation of the active compound with encapsulating material as carrier, providing a capsule in which the active component, with or without carriers, is surrounded by a carrier, which is in association with it.
- cachets and lozenges are included. Tablets, powders, capsules, pills, cachets, and lozenges may be as solid forms suitable for oral administration.
- liquid form preparations including emulsions, syrups, elixirs, aqueous solutions, aqueous suspensions, or solid form preparations which are intended to be converted shortly before use to liquid form preparations.
- Emulsions may be prepared in solutions, for example, in aqueous propylene glycol solutions or may contain emulsifying agents, for example, such as an monooleate, or acacia.
- Aqueous solutions can be prepared by dissolving the active component in water and adding suitable colorants, flavors, stabilizers, and thickening agents.
- Aqueous suspensions can be prepared by dispersing the finely divided active component in water with viscous material, such as natural or synthetic gums, resins, methylcellulose, sodium carboxymethylcellulose, and other well known suspending agents.
- Solid form preparations include solutions, suspensions, and emulsions, and may contain, in addition to the active component, colorants, flavors, stabilizers, buffers, artificial and natural sweeteners, dispersants, thickeners, solubilizing agents, and the like.
- the compounds of the invention may be formulated for parenteral administration (e.g., by injection, for example bolus injection or continuous infusion) and may be presented in unit dose form in ampoules, pre-filled syringes, small volume infusion or in multi-dose containers with an added preservative.
- the compositions may take such forms as suspensions, solutions, or emulsions in oily or aqueous vehicles, for example solutions in aqueous polyethylene glycol.
- oily or nonaqueous carriers, diluents, solvents or vehicles examples include propylene glycol, polyethylene glycol, vegetable oils (e.g., olive oil), and injectable organic esters (e.g., ethyl oleate), and may contain formulatory agents such as preserving, wetting, emulsifying or suspending, stabilizing and/or dispersing agents.
- the active ingredient may be in powder form, obtained by aseptic isolation of sterile solid or by lyophilization from solution for constitution before use with a suitable vehicle, e.g., sterile, pyrogen-free water.
- the compounds of the invention may be formulated for topical administration to the epidermis as ointments, creams or lotions, or as a transdermal patch.
- Ointments and creams may, for example, be formulated with an aqueous or oily base with the addition of suitable thickening and/or gelling agents.
- Lotions may be formulated with an aqueous or oily base and will in general also containing one or more emulsifying agents, stabilizing agents, dispersing agents, suspending agents, thickening agents, or coloring agents.
- Formulations suitable for topical administration in the mouth include lozenges comprising active agents in a flavored base, usually sucrose and acacia or tragacanth; pastilles comprising the active ingredient in an inert base such as gelatin and glycerin or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier.
- the compounds of the invention may be formulated for administration as suppositories.
- a low melting wax such as a mixture of fatty acid glycerides or cocoa butter is first melted and the active component is dispersed homogeneously, for example, by stirring. The molten homogeneous mixture is then poured into convenient sized molds, allowed to cool, and to solidify.
- the compounds of the invention may be formulated for vaginal administration. Pessaries, tampons, creams, gels, pastes, foams or sprays containing in addition to the active ingredient such carriers as are known in the art to be appropriate.
- the subject compounds may be formulated for nasal administration.
- the solutions or suspensions are applied directly to the nasal cavity by conventional means, for example, with a dropper, pipette or spray.
- the formulations may be provided in a single or multidose form. In the latter case of a dropper or pipette, this may be achieved by the patient administering an appropriate, predetermined volume of the solution or suspension. In the case of a spray, this may be achieved for example by means of a metering atomizing spray pump .
- the compounds of the invention may be formulated for aerosol administration, particularly to the respiratory tract and including intranasal administration.
- the compound will generally have a small particle size for example of the order of five (5) microns or less. Such a particle size may be obtained by means known in the art, for example by micronization.
- the active ingredient is provided in a pressurized pack with a suitable propellant such as a chlorofluoro carbon (CFC), for example, dichlorodifluoromethane, trichlorofluoromethane, or dichlorotetrafluoroethane, or carbon dioxide or other suitable gas.
- CFC chlorofluoro carbon
- the aerosol may conveniently also contain a surfactant such as lecithin.
- the dose of drug may be controlled by a metered valve.
- the active ingredients may be provided in a form of a dry powder, for example a powder mix of the compound in a suitable powder base such as lactose, starch, starch derivatives such as hydroxypropylmethyl cellulose and polyvinylpyrrolidine (PVP).
- the powder carrier will form a gel in the nasal cavity.
- the powder composition may be presented in unit dose form for example in capsules or cartridges of e.g., gelatin or blister packs from which the powder may be administered by means of an inhaler.
- formulations can be prepared with enteric coatings adapted for sustained or controlled release administration of the active ingredient.
- the compounds of the present invention can be formulated in transdermal or subcutaneous drug delivery devices.
- transdermal delivery systems are advantageous when sustained release of the compound is necessary and when patient compliance with a treatment regimen is crucial.
- Compounds in transdermal delivery systems are frequently attached to an skin- adhesive solid support.
- the compound of interest can also be combined with a penetration enhancer, e.g., Azone (l-dodecylazacycloheptan-2-one).
- Azone l-dodecylazacycloheptan-2-one
- Sustained release delivery systems are inserted subcutaneously into the subdermal layer by surgery or injection.
- the subdermal implants encapsulate the compound in a lipid soluble membrane, e.g., silicone rubber, or a biodegradable polymer, e.g., polylactic acid.
- the pharmaceutical preparations are preferably in unit dosage forms.
- the preparation is subdivided into unit doses containing appropriate quantities of the active component.
- the unit dosage form can be a packaged preparation, the package containing discrete quantities of preparation, such as packeted tablets, capsules, and powders in vials or ampoules.
- the unit dosage form can be a capsule, tablet, cachet, or lozenge itself, or it can be the appropriate number of any of these in packaged form.
- the compounds of this invention may be used in combination (administered in combination or sequentially) with known anti-cancer treatments such as radiation therapy or with cytostatic or cytotoxic agents, such as for example, but not limited to, DNA interactive agents, such as cisplatin or doxorubicin; topoisomerase II inhibitors such as etoposide: topoisomerase I inhibitors such as CPT-11 or topotecan; tublin interacting agents, such as paclitaxel, docetaxel or epothilones; hormonal agents such as tamoxifen: thymidilaate synthaes inhibitors, such as 5-fluorouracil; and anti-metabolites such as methotrexate.
- DNA interactive agents such as cisplatin or doxorubicin
- topoisomerase II inhibitors such as etoposide: topoisomerase I inhibitors such as CPT-11 or topotecan
- tublin interacting agents such as paclitaxel, do
- Compounds of formula I may also be useful in combination with modulators of p53 transactivation. If formulated as a fixed dose, the above-described combination products include the compounds of this invention within the dosage range described above and the other pharmaceutically active agent or treatment within its approved dose range. For example, an early cdkl inhibitor olomucine has been found to act synergistically with well known cytotoxic agents in inducing apoptosis. (J. Cell ScL (1995) 108:2897-904). Compounds of formula I may also be administered sequentially with known anticancer or cytoxic agents when concommitant administration or a combination is inappropriate.
- compounds of formula I may be administered either prior to or after administration of the known anticancer or cytotoxic agent.
- the cytotoxic activity of the cdk inhibitor flavopiridol is affected by the sequence of administration with anticancer agents. ⁇ Cancer Res (1997) 57:3375).
- the pharmacological properties of the compounds of this invention may be confirmed by a number of pharmacological assays.
- the exemplified pharmacological assays which follow have been carried out with the compounds according to the invention and their salts.
- the compounds of the invention exhibited cdk4/cyclin D activity with IC50 values and Ki values of less than 1.0 ⁇ M.
- the antiproliferative potency of some compounds of the invention was tested in the human colon tumor cell line HCTl 16 with IC90 values reported from an MTT assay of less than 30 ⁇ M, preferably less than 5 ⁇ M.
- Step 1 synthesis of l-(2-methylsulfanyl-pyrimidin-4-yl)-lH-benzotriazole
- a l L round bottom flask was loaded with sodium hydride dispersion (10.0 g, 250 mmol, 60% in mineral oil) and 200 mL of DMF, and the resulting slurry cooled with an ice bath.
- Benzotriazole (18.02 g, 151 mmol) was added portionwise over a 10-12 min period.
- the reaction mixture was stirred for 10 min to allow gas evolution to subside; the ice bath was then removed and 4-chloro-2-methylthiopyrimidine (24.07 g, 150 mmol) was added.
- the resulting mixture was stirred at RT for 15 minutes, then placed in a 90 0 C oil bath for 1.5 hour. The heating bath was turned off and the reaction mixture was allowed to slowly cool with stirring overnight.
- reaction mixture was then poured into 500 mL of water and stirred for 20 min, and then filtered.
- Step 2 synthesis of l-(2-methanesulfonyl-pyrimidin-4-yl)-lH-benzotriazole
- Example 4 Synthesis of ⁇ /-Pyrimidin-2-yl-cyclohexane-tr ⁇ /?5-l,4-diamine
- the synthesis of ⁇ /-Pyrimidin-2-yl-cyclohexane-tr ⁇ /?5-l,4-diamine was carried out according to the process shown in Scheme D.
- Step 1 Synthesis of methanesulfonic acid (S)-2-tert-butoxycarbonylamino-propyl ester 2-(5)-Boc-amino-propanol (10 g, 57.1 mmol, 1.00 equivalents) was dissolved in DCM (200 mL) under nitrogen atmosphere; and triethylamine (7.49g, 10.32 mL, 74.2 mmol, 1.30 eq) was added at RT. The reaction mixture was cooled to 0 0 C and mesyl chloride (7.39g, 4.99 mL, 64.5 mmol, 1.13 eq) was added under nitrogen atmosphere.
- Methanesulfonic acid (S)-2-tert-butoxycarbonylamino-propyl ester (1.0Og, 3.94 mmol, 1 eq) was dissolved in DMF (16 mL), under nitrogen atmosphere, K 2 CO 3 (1.09g, 7.89 mmol, 2 eq) and the appropriate amine of the formula RR'NH (3.95 mmol, 1 equiv,) were then added at RT. The reaction mixture was stirred at 90 0 C for 1 hour. The resulting mixture was concentrated to provide a crude solid which was dissolved with a 1 :1 iPrOH/CHCl 3 mixture, washed twice with H 2 O (10 mL). The aqueous phases were combined, extracted 3 times with a 1 :1 iPrOH/CHCl 3 mixture (20 mL). The organic extracts were combined, dried over Na 2 SO 4 , filtered and evaporated to provide the crude desired amine.
- the (iS)-boc-amino derivative A (3.5 mmol, 1 equivalent) was dissolved in a solution of 4N HCl in 1,4-dioxane (5 mL). The reaction mixture was stirred for 18 hours at RT. The reaction mixture was concentrated under reduced pressure providing a crude solid which was used without further purification in the next step.
- Example 7 Synthesis of trans-4-(4-benzotriazol-l-yl-pyrimidin-2-ylamino)- cvclohexanol
- the synthesis of tr ⁇ /?5-4-(4-benzotriazol-l-yl-pyrimidin-2-ylamino)-cyclohexanol was carried out according to the process shown in Scheme G.
- JV-(4-Benzotriazol- 1 -yl-pyrimidin-2-yl)-N'-pyrimidin-2-yl-cyclohexane-tr ⁇ /?5- 1 ,A- diamine bis-methane sulfonate salt which was prepared adding an excess of methanesulfonic acid to a solution of JV-(4-Benzotriazol-l-yl- pyrimidin-2-yl)-N'-pyrimidin-2-yl-cyclohexane-tr ⁇ /?5- 1 ,4-diamine bis- methane (150 mg) in 100 mL OfCH 2 Cl 2 and approximately 20 mL of
- Step 2 Synthesis of (IS, 2i? y )-[2-(4-benzotriazol-l-yl-pyrimidin-2-ylamino)-cyclohexyl]- methanol
- Lithium aluminum hydride (1.0 M in THF, 0.52 mL, 0.52 mmol) was added dropwise to a -78 0 C solution OfCZS-(I 1 S, 2i?)-2-(4benzotriazole-l-yl-pyrimidin-2-ylamino)- cyclohexanecarboxylic acid ethyl ester (0.19 g, 0.52 mmol) in 5 mL of THF.
- the reaction mixture was stirred for 2 hours, then warmed to RT over a period of 10 minutes. After a standard Fieser work-up (1 :1 :3 H 2 O, 15% NaOH, H 2 O), the reaction mixture was allowed to stir at RT for about 18 hours.
- Example 10 Synthesis of ⁇ /-(4-Benzotriazol-l-yl-pyrimidin-2-yl)-cyclohexane-tr ⁇ /?5- 1,4-diamine Synthesis of JV-(4-Benzotriazol- 1 -yl-pyrimidin ⁇ -yrj-cyclohexane-trans- 1 ,4-diamine was carried out according to the process shown in Scheme J.
- Step 1 Synthesis of (4-benzotriazol-yl-pyrimidin-2-yl)-N'-methyl-cyclohexane-tr ⁇ /?5- 1 ,4-diamine
- Step 2 Synthesis of ⁇ /-[tr ⁇ /?5-4-(4-benzotriazol-l-yl-pyrimidin-2-ylamino)-cyclohexyl]- JV-methyl-acetamide
- Example 13 Synthesis of JV-(4-benzotriazo 1-1 -yl-pyrimidin-2-yl)- ⁇ f'-pyridin-2-yl- cyclohexane-trans- 1 ,4-diamine hydrochloride Synthesis of JV-(4-benzotriazol- 1 -yl-pyrimidin ⁇ -y ⁇ -N'-pyridin ⁇ -yl-cyclohexane-trans- 1,4-diamine hydrochloride was carried out according to the process shown in Scheme M.
- Example 16 Synthesis of 4-(4-benzotriazol-l-yl-pyrimidin-2-ylamino)-cyclohexanone Synthesis of 4-(4-benzotriazol-l-yl-pyrimidin-2-ylamino)-cyclohexanone was carried out according to the process shown in Scheme P.
- Step 1 Synthesis of 4-(4-benzotriazol-l-yl-5-chloro-pyrimidin-2-ylamino)-cyclohexane- carboxylic acid methyl ester
- trans-4-Amino-cyclohexanecarboxylic acid methyl ester (727 mg, 3.76 mmol) was added in portions, at RT under nitrogen atmosphere, to a solution of l-(2,5-dichloro-pyrimidin- 4-yl)-l/f-benzotriazole (500 mg, 1.88 mmol) in THF (50 mL) followed by TEA (0.79 mL, 5.64 mmol). The resultant white suspension was stirred at RT overnight, and then at 60 0 C for 2.5 days. The mixture was partitioned between water (100 mL) and EtOAc (100 mL).
- Step 2 Synthesis of tr ⁇ /?5-4-(4-benzotriazol-l-yl-5-chloro-pyrimidin-2-ylamino)- cyclohexanecarboxylic acid
- Example 19 Synthesis of trans- 4-(4-benzotriazol-l-yl-5-chloro-pyrimidin-2-ylamino)- cyclohexanecarboxylic acid ethylamide Synthesis of trans- 4-(4-benzotriazol-l-yl-5-chloro-pyrimidin-2-ylamino)-cyclohexane- carboxylic acid ethylamide was carried out according to the process shown in Scheme S.
- Step 1 Synthesis of tr ⁇ /?5-2-[4-(4-Benzotriazol-l-yl-pyrimidin-2-ylamino)- cyclohexylmethyl]-iso indole- 1 ,3-dione
- Triphenylphosphine (972 mg, 1.2 eq) was added to a solution of trans-[4-(4- benzotriazol-l-yl-pyrimidin-2-ylamino)-cyclohexyl]-methanol (Ig, 1 equivalent; prepared in a similar manner as described in Example 4) in toluene (100 mL), it was then followed by the dropwise addition of DIAD (0.73 mL, 1.2 eq). The reaction mixture was stirred at RT for 10 minutes and then phthalimide (545 mg, 1.2 eq) was added. The mixture was stirred overnight at RT, water was then added and the resulting mixture was extracted 3 times with EtOAc.
- Step 2 Synthesis of trans-(4-aminomethyl-cyclohexyl)-(4-benzotriazol-l-yl-pyrimidin- 2-yl)-amine
- the solvent was then evaporated under reduced pressure, the residue was dissolved in a mixture of isopropanol and chloroform and it was acidified until pH 3 by the addition of HCl (1 M).
- the organic phase was washed 3 times with water.
- the aqueous phase was basified to pH 8-9 by the addition of NaOH (1 M) and it was extracted 5 times with a mixture of isopropanol and chloroform.
- the combined organic extracts were dried over Na 2 SO 4 , filtered and the solvent was evaporated under reduced pressure. This acid/base extraction procedure was repeated 3 times in order to remove phthalazinone.
- trans-N-N ' - [4-(4-benzotriazo 1- 1 -yl-pyrimidin-2-ylamino)-cyclohexylmethyl] -dimethyl- sulfanoyl urea was prepared in a similar manner using dimethylsulfamoyl chloride as the acylating agent.
- Step 1 Synthesis of 4-(4-benzotriazol-l-yl-pyrimidin-2-ylamino)-cyclohexane- carbaldehyde
- Step 2 Synthesis of (4-benzotriazol-l-yl-pyrimidin-2-yl)-(4-morpholin-4-ylmethyl- cyclohexyl)-amine
- Step 1 Synthesis of tr ⁇ /?5-[4-(2-Chloro-acetylamino)-cyclohexyl]-carbamic acid tert- butyl ester
- Step 2 Synthesis of tr ⁇ /?5-[4-(2-chloro-acetylamino)-cyclohexyl]-carbamic acid tert-bvXy ⁇ ester wi+v.mi+ purification.
- Step 2 Synthesis of tr ⁇ /?5- ⁇ 4-[2-(4-Methyl-piperazin-l-yl)-acetylamino]-cyclohexyl ⁇ - carbamic acid tert-butyi ester
- Step 3 Synthesis of trans- ⁇ /-(4-Amino-cyclohexyl)-2-(4-methyl-piperazin-l-yl)- acetamide
- Example 24 Synthesis of trans- ⁇ /-[4-(4-Benzotriazol-l-yl-pyrimidin-2-ylamino)- cyclohexyll-2-methoxy-acetamide Synthesis of trans-N-[4-(4-BGnzotriazol- 1 -yl-pyrimidin-2-ylamino)-cyclohexyl]-2- methoxy-acetamide was carried out according to the process shown in Scheme X.
- the ingredients are mixed and dispensed into capsules containing about 100 mg each; one capsule would approximate a total daily dosage.
- the ingredients are combined and granulated using a solvent such as methanol.
- the formulation is then dried and formed into tablets (containing about 20 mg of active compound) with an appropriate tablet machine.
- the ingredients are mixed to form a suspension for oral administration.
- the active ingredient is dissolved in a portion of the water for injection. A sufficient quantity of sodium chloride is then added with stirring to make the solution isotonic. The solution is made up to weight with the remainder of the water for injection, filtered through a 0.2 micron membrane filter and packaged under sterile conditions.
- the ingredients are melted together and mixed on a steam bath, and poured into molds containing 2.5 g total weight.
- nasal spray formulations Several aqueous suspensions containing from about 0.025-0.5 percent active compound are prepared as nasal spray formulations.
- the formulations optionally contain inactive ingredients such as, for example, micro crystalline cellulose, sodium carboxymethyl- cellulose, dextrose, and the like. Hydrochloric acid may be added to adjust pH.
- the nasal spray formulations may be delivered via a nasal spray metered pump typically delivering about 50-100 microliters of formulation per actuation. A typical dosing schedule is 2-4 sprays every 4-12 hours.
- kinase assays were conducted using FlashPlateTM assays (NENTM-Life Science Products). FlashPlate assays were performed using recombinant human cyclin B-CDKl , human cyclin E-CDK2 or human cyclin D1-CDK4 complexes.
- GST-cyclinE (GST-cycE), CDK2, GST-cyclinB (GST- cycB), CDKl, GST-CDK4 and cyclin Dl (cycDl) cDNA clones in baculo virus vectors were provided by Dr. W. Harper at the Baylor College of Medicine, Houston, TX.
- Rb protein was used as the substrate for the cycDl-CDK4, cycB-CDKl and the cycE-CDK2 assays (the expression plasmid was provided by Dr. Veronica Sullivan, Department of Molecular Virology, Roche Research /n Garden City, United Kingdom).
- the Rb protein is a natural substrate for phosphorylation by CDK4, CDK2 and CDKl (see Herwig and Strauss Eur. J. Biochem. (1997) 246:581-601 and the references cited therein).
- the expression of the 62 Kd protein was under the control of an IPTG inducible promoter in an M 15 E. coli strain.
- Cells were lysed by sonication and purification was carried out by binding lysates at pH 8.0 to a Ni-chelated agarose column pretreated with 1 mM imidazole. The resin was then washed several times with incrementally decreasing pH buffers to pH 6.0, and eluted with 500 mM imidazole.
- Eluted protein was dialysed against 20 mM HEPES pH 7.5, 30% glycerol, 200 mM NaCl, and 1 mM DTT. Purified Rb fusion protein stocks were quantitated for protein concentration, aliquoted, and stored at -7O 0 C.
- test compounds were added to the wells at 5x final concentration.
- Reactions were initiated by immediate addition of 40 ⁇ l reaction mix (25 mM HEPES, 20 mM MgCl 2 , 0.002% Tween 20, 2 mM DTT, 1 ⁇ M ATP, 4 nM 33 P-ATP) and a sufficient amount of enzyme to give counts that were at least 10-fold above background. Plates were incubated at RT on a shaker for 30 minutes. Plates were washed four times with the wash buffer, sealed, and counted on the TopCount scintillation counter (Packard Instrument Co., Downers Grove, IL]. The percent inhibition of Rb phosphorylation, which is a measure of the inhibition of CDK activity, was determined according to the following formula:
- test compound refers to the average counts per minute of the test duplicates
- nonspecific refers to the average counts per minute when no CyclinD/Cdk4, etc., was ' ' ' ' ' tal” refers to the average counts per minute when no compound was added.
- the IC50 value is the concentration of test compound that reduces by 50% the protein- kinase induced incorporation of the radio label under the test conditions described.
- inhibition activity may be measured using Ki.
- CDKl, CDK2, and CDK4 HTRF assays were set up. These were done in 96-well format and read in 384-well plate format. The assays were run at 3x their respective Rms for ATP.
- test compounds were diluted to 3x their final concentrations in 25 mM Hepes, pH 7.0, 6.25 mM MgCl 2 , 1.5 mM DTT, 135 ⁇ M ATP.
- the DMSO concentration was no greater than 4.76%.
- Twenty microliters were added to the wells of a 96-well plate.
- the kinase reaction was initiated by the addition of 40 ⁇ l /well of a solution containing 0.185 ⁇ M Rb and 2.25 ⁇ g/ml CDK4 in 25 mM HEPES, pH 7.0, 6.25 mM MgCl 2 , 0.003% Tween-20, 0.3 mg/ml BSA, 1.5 mM DTT.
- IC50 values (the concentration of test compounds reducing the assay control fluorescence read-out by 50%) were first calculated from net readings at 665 nm, normalized for europium readings at 615 nm.
- Ki values were calculated according to the following equation:
- Ki IC50/(l + S/Rm) where S refers to the substrate concentration and Km refers to the Michaelis-Menten constant.
- the CDKl and CDK2 assays were similarly run except for small differences in reagent and protein concentrations:
- the compound and enzyme buffers for both assays contained 10 mM MgCl 2 .
- the respective reagent ATP concentrations were 162 ⁇ M and 90 ⁇ M.
- CDKl at a reagent concentration of 0.15 ng/ ⁇ l and CDK2 at a reagent concentration of 0.06 ng/ ⁇ l were used.
- Reagent concentrations of detection reagents were adjusted between 3- 12 nM Eu-Ab and 60-90 nM APC-antiHis 6 to give signal to background ratios of at least 10 to 1.
- Tetrazolium dye proliferation assay (Tetrazolium dye proliferation assay)("MTT Assay") Proliferation was evaluated by the tetrazolium dye assay according to the procedure of Denizot and Lang (F. Denizot and R. Lang, J Immunol Meth (1986) 89:271-77).
- the cell line used was HCTl 16, a colorectal carcinoma cell line obtained from the American Type Cell Culture Collection (ATCC; Rockville, MD). The cells were grown in McCoy's 5 A medium supplemented with 10% FCS and L-glutamine.
- test compounds were serially diluted to four times the final concentration in the appropriate medium containing 1.2% DMSO. One-fourth final volume of each dilution was added in duplicate to the plates containing cells. The same volume of 1.2% DMSO in medium was added to a row of
- control wells such that the final concentration of DMSO in each well was 0.3%.
- Wells to which no cells were added served as the “blank.”
- Wells to which no inhibitor was added served as “no inhibitor control.”
- the plates were returned to the incubator, and at set time points (determined by their growth curves) plates were analyzed as described below.
- SWl 353 cells purchased from American Tissue Culture Collection (ATCC) are grown in a 6-well plate at a density of 3 x 10 5 cells per well containing 2 ml of Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum
- Cell lysate samples (15 ⁇ g of total proteins per sample) are loaded on 10% NuPAGE Bis- Tris gel (Invitrogen) and transferred to nitrocellulose membrane (Invitrogen). The membrane is blocked in 5% dry milk in IxTBS for 1 hour at RT. To determine the levels of both phosphorylated and total cJun in the samples, the membrane is simultaneously probed with rabbit anti-p-cjun and mouse anti- total cJun antibodies (Cell Signaling) in Odyssey blocking buffer (Li-cor) with 0.1% Tween 20(Roche Biochem) for overnight at 4 0 C.
- the membrane is washed 3 times in IxPBS with 0.1% Tween® 20.
- IRDye 700 goat anti-mouse IgG (Rockland) and IRDye 800 goat anti-rabbit 1) are used in a dilution of 1 :6500 in Odyssey blocking buffer.
- the membrane Blot is scanned and quantified using the Odyssey Infrared Imager (Li-cor Cat. No.9201).
- the normalized intensities of p-c-Jun vs total c-Jun are used for IC50 calculation with the Xlfit3 program of Microsoft Excel.
- the IC50 value is interpolated from a graph of inhibitor concentration vs. percent inhibition.
- JNK activity is measured by phosphorylation of GST-ATF2 (19-96) with [ ⁇ - 33 P] ATP.
- the enzyme reaction is conducted at Km concentrations of ATP and the substrate at final volume of 40 ⁇ l in buffer containing 25 mM Hepes, pH 7.5, 2 mM dithiothreitol, 150 mM NaCl, 20 mM MgCl 2 , 0.001% Tween® 20, 0.1% BSA and 10% DMSO.
- Human JNK2 ⁇ 2 assay contains InM enzyme, 1 ⁇ M ATF2, 8 ⁇ M ATP with IuCi [ ⁇ - 33 P] ATP.
- Human JNKl ⁇ l assay contains 2 nM enzyme, l ⁇ M ATF2, 6 ⁇ M ATP with 1 ⁇ Ci [ ⁇ - 33 P] ATP.
- Human JNK3 (Upstate Biotech #14-501 M) assay contains 2 nM enzyme, 1 ⁇ M ATF2, 4 ⁇ M ATP with 1 ⁇ Ci [ ⁇ - 33 P] ATP.
- the enzyme assay is carried out in the presence or absence often compound concentrations. JNK and compound are pre- incubated for 10 minutes. Then, the enzymatic reaction is initiated by addition of ATP and the substrate. The reaction mixture is incubated at 3O 0 C for 30 minutes.
- the reaction is terminated by transferring 25 ⁇ l of the reaction mixture to 150 ⁇ l of 10% glutathione sepharose slurry (Amersham # 27-4574-01) containing 135 mM EDTA.
- the reaction product is captured on the affinity resin and washed on a filtration plate (Millipore, MABVNOB50) with phosphate buffered saline for six times to remove free radio nucleotide. Then the incorporation of 33 P into ATF2 is quantified on a microplate scintillation counter (Packard Topcount).
- Rats Female Wistar-Han rats procured from Charles River Laboratories are allowed to acclimate for one week prior to use and achieve an approximate body weight of 95-13Og Rats are administered test compound via oral gavage, subcutaneous injection or intravenous injection (tail vein) 30 min prior to an intra-peritoneal challenge of 0.5 ⁇ g recombinant rat TNF- ⁇ (Biosource). Blood is collected via cardiocentesis 90 min after TNF- ⁇ challenge. Plasma is prepared using lithium heparin separation tubes (BD microtainer") and frozen at -8O 0 C until analyzed. IL-6 levels are determined using a rat iLISA kit (Biosource). The percent inhibition and ED50 values (calculated as the dose of compound at which TNF- ⁇ production is 50% of the control value) are determined.
- mice Female Lewis rats procured from Harlan Laboratories at 7-8 weeks of age are allowed to acclimate for one week prior to use and achieve an approximate body weight of 120-140 g .
- rats are primed intradermally (i.d.) on several sites on the back with an emulsion of 100 ⁇ g Bovine Type II Collagen (Chondrex) in Incomplete Freund's adjuvant (IFA; total of 0.1 ml in 2-3 sites).
- IFA Incomplete Freund's adjuvant
- Arthritis induction is generally observed 12- 14 days from priming; however a booster injection of 100 ⁇ g collagen/IFA is given around days 7-10 (i.d. up to 0.1 ml total) at base of tail or an alternate site on back to synchronize disease induction.
- Compound dosing can be prophylactic (starting at time of boost or 1-2 days prior) or therapeutic (beginning after boost and coinciding with initial disease scores of 1-2 -see clinical scoring below). Animals are evaluated for the development and progression of disease over the next 21 days.
- Rats are evaluated using a scoring system (described below), paw volume measurements using a plethysmometer for each paw, or measuring paw or joint thickness with a caliper. Baseline measurements are performed on day 0 and starting again at the first signs or swelling for up to three times per week until the end of the experiment. Scoring was evaluated as follows for each paw:
- the hind paws are harvested for weight histology, cellular and/or molecular analysis. Additionally, blood is collected via cardiocentesis, plasma is prepared using lithium heparin separation tubes (BD microtainer) and frozen at -8O 0 C until analyzed. Inflammatory cytokine levels (e.g., TNF- ⁇ , IL-I and IL-6) from the plasma or from homogenized joint tissue are determined using rat-specific ELISA kits (R&D). The level of disease protection or inhibition is determined as a composite of changes in clinical scores, paw volumes and histopathology compared to control animals.
- BD microtainer lithium heparin separation tubes
- R&D rat-specific ELISA kits
- SWl 353 cells are purchased from the American Tissue Culture Collection and maintained in growth media consisting of DMEM medium (Invitrogen) with 10% fetal bovine serum (Invitrogen), ascorbic acids (Sigma) and penicillin (Invitrogen) under the culture condition of 37 0 C in 5% CO 2 .
- Cells are plated at a density of 1.0 x 10 4 cells per well in 100 ⁇ l of media 48 hours before the compound treatment. Immediately before the compound treatment, media is replaced with 160 ⁇ l of fresh media.
- Compound stock (10 mM) is diluted in growth media and added to each well as a 10x concentrated solution in a volume of 20 ⁇ l, mixed and allowed to pre-incubate with cells for 30 minutes.
- the compound vehicle (DMSO) is maintained at a final concentration of 1% in all samples. After 30 minutes the cells are activated with 10 ng/ml of TNF- ⁇ (Roche Biochem). TNF- ⁇ is added as a 10x concentrated solution made up in growth media and added in a volume of 20 ⁇ l per well. Cell plates are cultured for 5 hours. Cell media are harvested and stored at -2O 0 C. Media aliquots are analyzed by sandwich ELISA for the presence of IL-8 as per the manufacturer's instructions (BD Bioscience). The IC50 values are calculated as the concentration of the compound at which the IL-8 production was reduced to 50% of the control value using Xlfit3 in Microsoft Excel program. Certain compounds have an IC50 value ranging from 0.1-20 ⁇ M in this assay.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Immunology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Pulmonology (AREA)
- Rheumatology (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Epidemiology (AREA)
- Pain & Pain Management (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Psychology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description







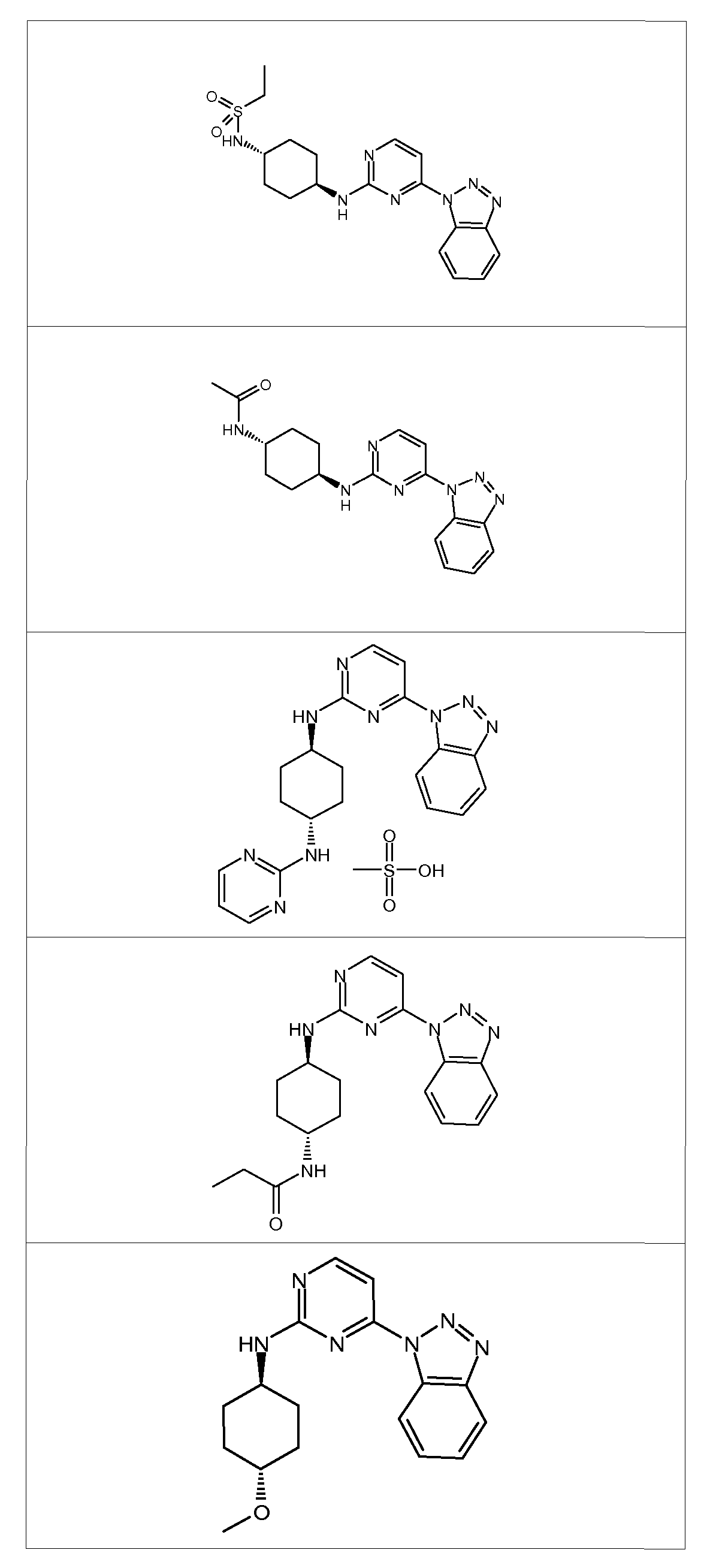







































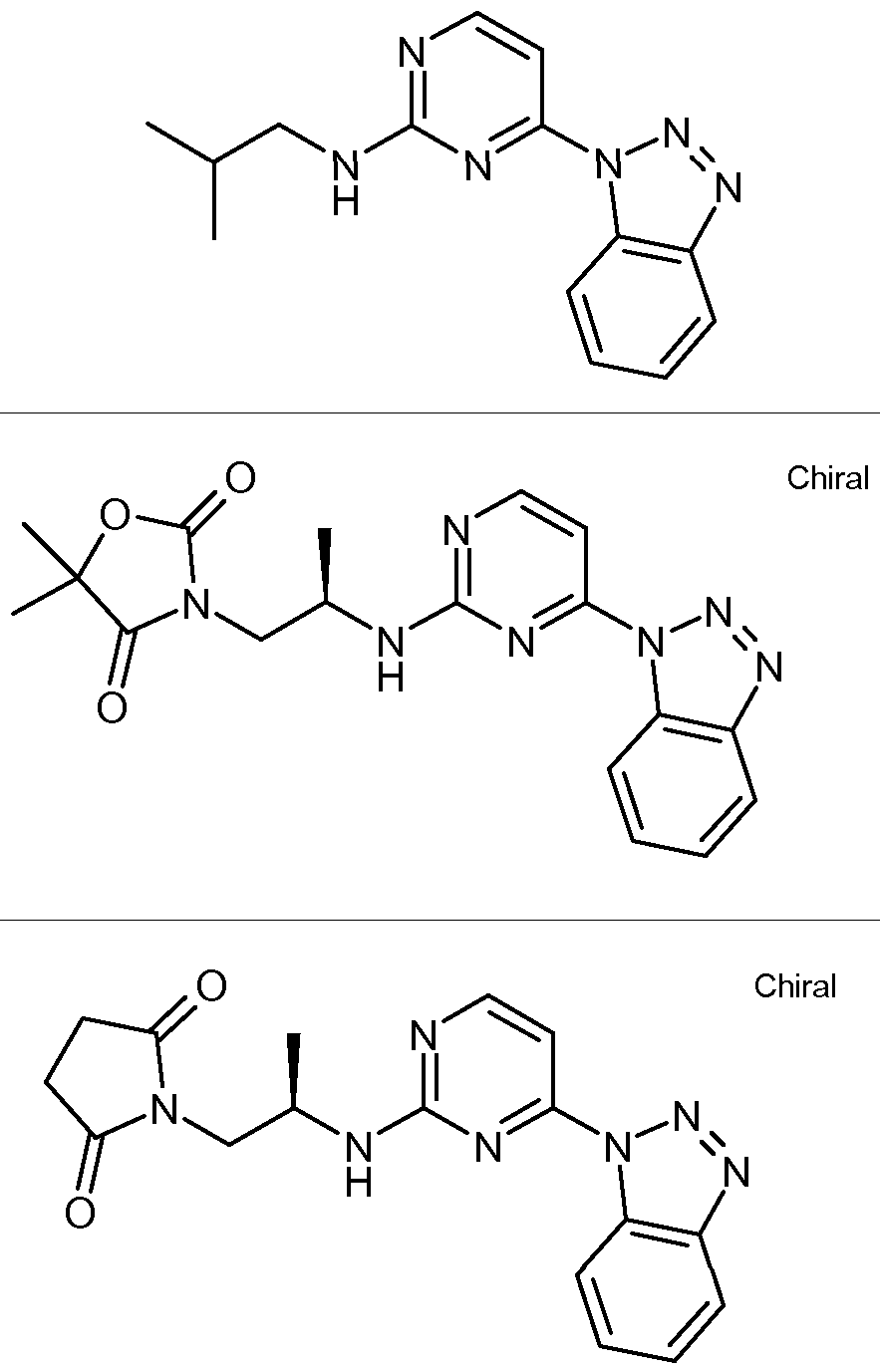













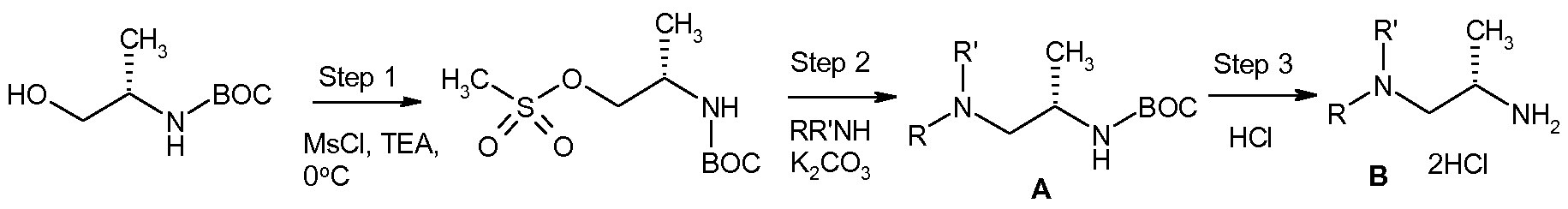



























Claims






Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT07803049T ATE543498T1 (en) | 2006-09-08 | 2007-08-30 | BENZOTRIAZOLE KINASE MODULATORS |
| CN2007800334496A CN101511359B (en) | 2006-09-08 | 2007-08-30 | Benzotriazole kinase modulators |
| KR1020097004741A KR101177729B1 (en) | 2006-09-08 | 2007-08-30 | Benzotriazole kinase modulators |
| AU2007293917A AU2007293917B2 (en) | 2006-09-08 | 2007-08-30 | Benzotriazole kinase modulators |
| EP07803049A EP2066319B1 (en) | 2006-09-08 | 2007-08-30 | Benzotriazole kinase modulators |
| ES07803049T ES2378577T3 (en) | 2006-09-08 | 2007-08-30 | Benzotriazole as kinase modulators |
| BRPI0717035A BRPI0717035B8 (en) | 2006-09-08 | 2007-08-30 | benzotriazole kinase modulators |
| CA2662998A CA2662998C (en) | 2006-09-08 | 2007-08-30 | Benzotriazole kinase modulators |
| JP2009527118A JP5325783B2 (en) | 2006-09-08 | 2007-08-30 | Benzotriazole kinase modulator |
| MX2009002229A MX2009002229A (en) | 2006-09-08 | 2007-08-30 | Benzotriazole kinase modulators. |
| IL197015A IL197015A (en) | 2006-09-08 | 2009-02-12 | Benzotriazole kinase modulators, processes for their preparation, pharmaceutical compositions comprising them and their use in the preparation of medicaments for the treatment of diseases |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US84309006P | 2006-09-08 | 2006-09-08 | |
| US60/843,090 | 2006-09-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008028860A1 true WO2008028860A1 (en) | 2008-03-13 |
Family
ID=38893293
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/059040 WO2008028860A1 (en) | 2006-09-08 | 2007-08-30 | Benzotriazole kinase modulators |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US8962622B2 (en) |
| EP (1) | EP2066319B1 (en) |
| JP (1) | JP5325783B2 (en) |
| KR (1) | KR101177729B1 (en) |
| CN (1) | CN101511359B (en) |
| AR (1) | AR062666A1 (en) |
| AT (1) | ATE543498T1 (en) |
| AU (1) | AU2007293917B2 (en) |
| BR (1) | BRPI0717035B8 (en) |
| CA (1) | CA2662998C (en) |
| CL (1) | CL2007002572A1 (en) |
| ES (1) | ES2378577T3 (en) |
| IL (1) | IL197015A (en) |
| MX (1) | MX2009002229A (en) |
| PE (1) | PE20080659A1 (en) |
| TW (1) | TW200819440A (en) |
| WO (1) | WO2008028860A1 (en) |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009138340A1 (en) * | 2008-05-16 | 2009-11-19 | F. Hoffmann-La Roche Ag | Inhibitors of jnk |
| WO2009143865A1 (en) * | 2008-05-30 | 2009-12-03 | Xigen S.A. | Use of cell-permeable peptide inhibitors of the jnk signal transduction pathway for the treatment of various diseases |
| WO2011018417A1 (en) * | 2009-08-10 | 2011-02-17 | F. Hoffmann-La Roche Ag | Inhibitors of jnk |
| US8080517B2 (en) | 2005-09-12 | 2011-12-20 | Xigen Sa | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| US8183339B1 (en) | 1999-10-12 | 2012-05-22 | Xigen S.A. | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| US8236924B2 (en) | 1999-10-12 | 2012-08-07 | Xigen Sa | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| WO2012109311A3 (en) * | 2011-02-09 | 2012-10-11 | The Lubrizol Corporation | Asphaltene dispersant containing lubricating compositions |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8399462B2 (en) | 2006-12-08 | 2013-03-19 | Roche Palo Alto Llc | JNK modulators |
| US8748395B2 (en) | 2005-09-12 | 2014-06-10 | Xigen Inflammation Ltd. | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| US8981052B2 (en) | 2010-06-21 | 2015-03-17 | Xigen Inflammation Ltd. | JNK inhibitor molecules |
| US9150618B2 (en) | 2010-10-14 | 2015-10-06 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of chronic or non-chronic inflammatory eye diseases |
| US9180159B2 (en) | 2008-05-30 | 2015-11-10 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of chronic or non-chronic inflammatory digestive diseases |
| WO2015193281A1 (en) * | 2014-06-16 | 2015-12-23 | Allinky Biopharma | P38 and jnk mapk inhibitors for the treatment and prophylaxis of degenerative diseases of the nervous system |
| RU2572247C2 (en) * | 2010-06-04 | 2016-01-10 | Ф. Хоффманн-Ля Рош Аг | 2-amino-pyrimidine derivatives as jnk inhibitors |
| WO2014128669A3 (en) * | 2013-02-25 | 2017-01-12 | Aurigene Discovery Technologies Limited | Trisubstituted benzotriazole derivatives as dihydroorotate oxygenase inhibitors |
| WO2017204626A1 (en) | 2016-05-24 | 2017-11-30 | Stichting Het Nederlands Kanker Instituut-Antoni van Leeuwenhoek Ziekenhuis | Combination therapy - combined map2k4/map3k1 and mek/erk inhibition |
| WO2018029336A1 (en) | 2016-08-12 | 2018-02-15 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for determining whether a subject was administered with an activator of the ppar beta/delta pathway. |
| US10023615B2 (en) | 2008-12-22 | 2018-07-17 | Xigen Inflammation Ltd. | Efficient transport into white blood cells |
| US10059690B2 (en) | 2014-04-04 | 2018-08-28 | Syros Pharmaceuticals, Inc. | Inhibitors of cyclin-dependent kinase 7 (CDK7) |
| US10596223B2 (en) | 2011-12-21 | 2020-03-24 | Xigen Inflammation Ltd. | JNK inhibitor molecules for treatment of various diseases |
| US10624948B2 (en) | 2013-06-26 | 2020-04-21 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of various diseases |
| US11147801B2 (en) | 2017-04-24 | 2021-10-19 | Aurigene Discovery Technologies Limited | Methods of use for trisubstituted benzotriazole derivatives as dihydroorotate oxygenase inhibitors |
| US11331364B2 (en) | 2014-06-26 | 2022-05-17 | Xigen Inflammation Ltd. | Use for JNK inhibitor molecules for treatment of various diseases |
| CN115677595A (en) * | 2022-10-26 | 2023-02-03 | 江苏睿实生物科技有限公司 | Preparation method of 2,4, 5-trichloropyrimidine |
| US11717512B2 (en) | 2018-02-20 | 2023-08-08 | Servier Pharmaceuticals Llc | Methods of use for trisubstituted benzotriazole derivatives |
| US11779628B2 (en) | 2013-06-26 | 2023-10-10 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of various diseases |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2499017T3 (en) * | 2007-10-23 | 2014-09-26 | F. Hoffmann-La Roche Ag | New kinase inhibitors |
| CN102325771B (en) | 2009-02-24 | 2014-10-29 | 霍夫曼-拉罗奇有限公司 | Imidazo [1, 2 -a] pyridines as jnk modulators |
| CA2799817A1 (en) * | 2010-06-04 | 2011-12-08 | Leyi Gong | Inhibitors of jnk |
| CN108117523B (en) * | 2016-11-29 | 2021-05-07 | 上海医药工业研究院 | Preparation method of halogenated uracil compound |
| JPWO2022059700A1 (en) * | 2020-09-15 | 2022-03-24 | ||
| CN114456126A (en) * | 2022-02-26 | 2022-05-10 | 郑州萃智医药科技有限公司 | Synthesis method of compound |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6498165B1 (en) * | 1999-06-30 | 2002-12-24 | Merck & Co., Inc. | Src kinase inhibitor compounds |
| WO2006075152A1 (en) * | 2005-01-11 | 2006-07-20 | Cyclacel Limited | 4- (1h-indol-3-yl) -pyrimidin-2-ylamine derivates and their use in therapy |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003503351A (en) | 1999-06-30 | 2003-01-28 | メルク エンド カムパニー インコーポレーテッド | SRC kinase inhibitory compounds |
| HUP0301117A3 (en) | 2000-02-17 | 2004-01-28 | Amgen Inc Thousand Oaks | Imidazole derivatives kinase inhibitors, their use, process for their preparation and pharmaceutical compositions containing them |
| EP1554269A1 (en) | 2002-07-09 | 2005-07-20 | Vertex Pharmaceuticals Incorporated | Imidazoles, oxazoles and thiazoles with protein kinase inhibiting activities |
| GB0308466D0 (en) | 2003-04-11 | 2003-05-21 | Novartis Ag | Organic compounds |
| RU2401265C2 (en) | 2004-06-10 | 2010-10-10 | Айрм Ллк | Compounds and compositions as protein kinase inhibitors |
| WO2006038001A1 (en) | 2004-10-06 | 2006-04-13 | Celltech R & D Limited | Aminopyrimidine derivatives as jnk inhibitors |
| US20060281764A1 (en) | 2005-06-10 | 2006-12-14 | Gaul Michael D | Aminopyrimidines as kinase modulators |
| WO2007111904A2 (en) | 2006-03-22 | 2007-10-04 | Vertex Pharmaceuticals Incorporated | C-met protein kinase inhibitors for the treatment of proliferative disorders |
-
2007
- 2007-08-30 KR KR1020097004741A patent/KR101177729B1/en active IP Right Grant
- 2007-08-30 MX MX2009002229A patent/MX2009002229A/en active IP Right Grant
- 2007-08-30 BR BRPI0717035A patent/BRPI0717035B8/en active IP Right Grant
- 2007-08-30 ES ES07803049T patent/ES2378577T3/en active Active
- 2007-08-30 JP JP2009527118A patent/JP5325783B2/en active Active
- 2007-08-30 EP EP07803049A patent/EP2066319B1/en active Active
- 2007-08-30 CA CA2662998A patent/CA2662998C/en active Active
- 2007-08-30 CN CN2007800334496A patent/CN101511359B/en active Active
- 2007-08-30 AU AU2007293917A patent/AU2007293917B2/en active Active
- 2007-08-30 AT AT07803049T patent/ATE543498T1/en active
- 2007-08-30 WO PCT/EP2007/059040 patent/WO2008028860A1/en active Application Filing
- 2007-09-05 TW TW096133095A patent/TW200819440A/en unknown
- 2007-09-05 CL CL200702572A patent/CL2007002572A1/en unknown
- 2007-09-06 PE PE2007001197A patent/PE20080659A1/en active IP Right Grant
- 2007-09-06 AR ARP070103923A patent/AR062666A1/en active IP Right Grant
- 2007-09-07 US US11/899,758 patent/US8962622B2/en active Active
-
2009
- 2009-02-12 IL IL197015A patent/IL197015A/en active IP Right Grant
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6498165B1 (en) * | 1999-06-30 | 2002-12-24 | Merck & Co., Inc. | Src kinase inhibitor compounds |
| WO2006075152A1 (en) * | 2005-01-11 | 2006-07-20 | Cyclacel Limited | 4- (1h-indol-3-yl) -pyrimidin-2-ylamine derivates and their use in therapy |
Cited By (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8183339B1 (en) | 1999-10-12 | 2012-05-22 | Xigen S.A. | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| US8569447B2 (en) | 1999-10-12 | 2013-10-29 | Xigen Sa | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| US8278413B2 (en) | 1999-10-12 | 2012-10-02 | Xigen Sa | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| US8236924B2 (en) | 1999-10-12 | 2012-08-07 | Xigen Sa | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| US8080517B2 (en) | 2005-09-12 | 2011-12-20 | Xigen Sa | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| US9290538B2 (en) | 2005-09-12 | 2016-03-22 | Xigen Inflammation Ltd. | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| US8748395B2 (en) | 2005-09-12 | 2014-06-10 | Xigen Inflammation Ltd. | Cell-permeable peptide inhibitors of the JNK signal transduction pathway |
| US8399462B2 (en) | 2006-12-08 | 2013-03-19 | Roche Palo Alto Llc | JNK modulators |
| WO2009138340A1 (en) * | 2008-05-16 | 2009-11-19 | F. Hoffmann-La Roche Ag | Inhibitors of jnk |
| CN102015689A (en) * | 2008-05-16 | 2011-04-13 | 霍夫曼-拉罗奇有限公司 | Inhibitors of JNK |
| KR101368079B1 (en) | 2008-05-16 | 2014-03-14 | 에프. 호프만-라 로슈 아게 | Inhibitors of jnk |
| RU2504545C2 (en) * | 2008-05-16 | 2014-01-20 | Ф. Хоффманн-Ля Рош Аг | Jnk inhibitors |
| JP2011520824A (en) * | 2008-05-16 | 2011-07-21 | エフ.ホフマン−ラ ロシュ アーゲー | Inhibitor of JNK |
| US9006185B2 (en) | 2008-05-30 | 2015-04-14 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of various diseases |
| EP2985031A1 (en) * | 2008-05-30 | 2016-02-17 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the jnk signal transduction pathway for the treatment of various diseases |
| US9180159B2 (en) | 2008-05-30 | 2015-11-10 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of chronic or non-chronic inflammatory digestive diseases |
| WO2009144037A1 (en) * | 2008-05-30 | 2009-12-03 | Xigen S.A. | Use of cell-permeable peptide inhibitors of the jnk signal transduction pathway for the treatment of various diseases |
| EP2491942A1 (en) * | 2008-05-30 | 2012-08-29 | Xigen S.A. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of various diseases |
| EP2650007A1 (en) * | 2008-05-30 | 2013-10-16 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of various cardiovascular diseases |
| EP2650008A1 (en) * | 2008-05-30 | 2013-10-16 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of various cardiovascular diseases |
| WO2009143865A1 (en) * | 2008-05-30 | 2009-12-03 | Xigen S.A. | Use of cell-permeable peptide inhibitors of the jnk signal transduction pathway for the treatment of various diseases |
| EP2489361A1 (en) * | 2008-05-30 | 2012-08-22 | Xigen S.A. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of various diseases |
| US9610330B2 (en) | 2008-05-30 | 2017-04-04 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of various diseases |
| US10023615B2 (en) | 2008-12-22 | 2018-07-17 | Xigen Inflammation Ltd. | Efficient transport into white blood cells |
| CN102471319B (en) * | 2009-08-10 | 2014-06-18 | 霍夫曼-拉罗奇有限公司 | Inhibitors of JNK |
| US8536172B2 (en) | 2009-08-10 | 2013-09-17 | Roche Palo Alto Llc | Inhibitors of JNK |
| WO2011018417A1 (en) * | 2009-08-10 | 2011-02-17 | F. Hoffmann-La Roche Ag | Inhibitors of jnk |
| JP2013501746A (en) * | 2009-08-10 | 2013-01-17 | エフ.ホフマン−ラ ロシュ アーゲー | JNK inhibitor |
| CN102471319A (en) * | 2009-08-10 | 2012-05-23 | 霍夫曼-拉罗奇有限公司 | Inhibitors of JNK |
| RU2572247C2 (en) * | 2010-06-04 | 2016-01-10 | Ф. Хоффманн-Ля Рош Аг | 2-amino-pyrimidine derivatives as jnk inhibitors |
| US8981052B2 (en) | 2010-06-21 | 2015-03-17 | Xigen Inflammation Ltd. | JNK inhibitor molecules |
| US9624267B2 (en) | 2010-06-21 | 2017-04-18 | Xigen Inflammation Ltd. | JNK inhibitor molecules |
| US9150618B2 (en) | 2010-10-14 | 2015-10-06 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of chronic or non-chronic inflammatory eye diseases |
| WO2012109311A3 (en) * | 2011-02-09 | 2012-10-11 | The Lubrizol Corporation | Asphaltene dispersant containing lubricating compositions |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US10596223B2 (en) | 2011-12-21 | 2020-03-24 | Xigen Inflammation Ltd. | JNK inhibitor molecules for treatment of various diseases |
| WO2014128669A3 (en) * | 2013-02-25 | 2017-01-12 | Aurigene Discovery Technologies Limited | Trisubstituted benzotriazole derivatives as dihydroorotate oxygenase inhibitors |
| US10080740B2 (en) | 2013-02-25 | 2018-09-25 | Aurigene Discovery Technologies Limited | Trisubstituted benzotriazole derivatives as dihydroorotate oxygenase inhibitors |
| US9937155B2 (en) | 2013-02-25 | 2018-04-10 | Aurigene Discovery Technologies Limited | Methods of use for trisubstituted benzotriazole derivatives as dihydroorotate oxygenase inhibitors |
| US10624948B2 (en) | 2013-06-26 | 2020-04-21 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of various diseases |
| US11779628B2 (en) | 2013-06-26 | 2023-10-10 | Xigen Inflammation Ltd. | Use of cell-permeable peptide inhibitors of the JNK signal transduction pathway for the treatment of various diseases |
| US10106526B2 (en) | 2014-04-04 | 2018-10-23 | Syros Pharmaceuticals, Inc. | Inhibitors of cyclin-dependent kinase 7 (CDK7) |
| US10059690B2 (en) | 2014-04-04 | 2018-08-28 | Syros Pharmaceuticals, Inc. | Inhibitors of cyclin-dependent kinase 7 (CDK7) |
| US9908896B2 (en) | 2014-06-16 | 2018-03-06 | Allinky Biopharma | P38 and JNK MAPK inhibitors for the treatment and prophylaxis of degenerative diseases of the nervous system |
| WO2015193281A1 (en) * | 2014-06-16 | 2015-12-23 | Allinky Biopharma | P38 and jnk mapk inhibitors for the treatment and prophylaxis of degenerative diseases of the nervous system |
| US11331364B2 (en) | 2014-06-26 | 2022-05-17 | Xigen Inflammation Ltd. | Use for JNK inhibitor molecules for treatment of various diseases |
| WO2017204626A1 (en) | 2016-05-24 | 2017-11-30 | Stichting Het Nederlands Kanker Instituut-Antoni van Leeuwenhoek Ziekenhuis | Combination therapy - combined map2k4/map3k1 and mek/erk inhibition |
| WO2018029336A1 (en) | 2016-08-12 | 2018-02-15 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for determining whether a subject was administered with an activator of the ppar beta/delta pathway. |
| US11147801B2 (en) | 2017-04-24 | 2021-10-19 | Aurigene Discovery Technologies Limited | Methods of use for trisubstituted benzotriazole derivatives as dihydroorotate oxygenase inhibitors |
| US11717512B2 (en) | 2018-02-20 | 2023-08-08 | Servier Pharmaceuticals Llc | Methods of use for trisubstituted benzotriazole derivatives |
| CN115677595A (en) * | 2022-10-26 | 2023-02-03 | 江苏睿实生物科技有限公司 | Preparation method of 2,4, 5-trichloropyrimidine |
Also Published As
| Publication number | Publication date |
|---|---|
| BRPI0717035B1 (en) | 2021-05-04 |
| AR062666A1 (en) | 2008-11-26 |
| TW200819440A (en) | 2008-05-01 |
| ES2378577T3 (en) | 2012-04-16 |
| EP2066319B1 (en) | 2012-02-01 |
| PE20080659A1 (en) | 2008-05-17 |
| US8962622B2 (en) | 2015-02-24 |
| MX2009002229A (en) | 2009-03-16 |
| KR101177729B1 (en) | 2012-09-07 |
| AU2007293917B2 (en) | 2013-01-31 |
| CA2662998C (en) | 2015-10-06 |
| CN101511359A (en) | 2009-08-19 |
| CA2662998A1 (en) | 2008-03-13 |
| JP5325783B2 (en) | 2013-10-23 |
| BRPI0717035B8 (en) | 2021-05-25 |
| ATE543498T1 (en) | 2012-02-15 |
| AU2007293917A1 (en) | 2008-03-13 |
| BRPI0717035A2 (en) | 2014-11-25 |
| CL2007002572A1 (en) | 2008-04-18 |
| KR20090040908A (en) | 2009-04-27 |
| CN101511359B (en) | 2012-09-05 |
| JP2010502668A (en) | 2010-01-28 |
| EP2066319A1 (en) | 2009-06-10 |
| US20080103142A1 (en) | 2008-05-01 |
| IL197015A (en) | 2013-12-31 |
| IL197015A0 (en) | 2009-11-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2066319B1 (en) | Benzotriazole kinase modulators | |
| AU2007328981B2 (en) | Substituted pyrimidines and their use as JNK modulators | |
| CN111171000B (en) | EGFR inhibitor and preparation and application thereof | |
| RU2637947C2 (en) | Pyrazolaminopirimidine derivatives as modulators of leucine-repeating kinase 2 | |
| JP5518905B2 (en) | Imidazo [1,2-a] pyridine as a JNK modulator | |
| PT1899329E (en) | Pyrimidine-substituted benzimidazole derivatives as protein kinase inhibitors | |
| KR20070084066A (en) | Compounds and compositions as protein kinase inhibitors | |
| WO2020107987A1 (en) | Erk inhibitor containing isoindoline, preparation method therefor and application thereof | |
| JP2010512405A (en) | Compounds and compositions as kinase inhibitors | |
| US8227478B2 (en) | JNK modulators | |
| EP2283009B1 (en) | Inhibitors of jnk | |
| CA2799817A1 (en) | Inhibitors of jnk | |
| EP2576537B1 (en) | 2-amino-pyrimidine derivatives useful as inhibitors of jnk |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780033449.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07803049 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2007803049 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 197015 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007293917 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2009/002229 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2007293917 Country of ref document: AU Date of ref document: 20070830 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2009527118 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020097004741 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2662998 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1329/CHENP/2009 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: PI0717035 Country of ref document: BR Kind code of ref document: A2 Effective date: 20090306 |