WO2008024391A1 - Pharmaceutical formulations of an indole-type derivative and related methods of use - Google Patents
Pharmaceutical formulations of an indole-type derivative and related methods of use Download PDFInfo
- Publication number
- WO2008024391A1 WO2008024391A1 PCT/US2007/018555 US2007018555W WO2008024391A1 WO 2008024391 A1 WO2008024391 A1 WO 2008024391A1 US 2007018555 W US2007018555 W US 2007018555W WO 2008024391 A1 WO2008024391 A1 WO 2008024391A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- day
- dose
- pharmaceutically acceptable
- drug
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/286—Polysaccharides, e.g. gums; Cyclodextrin
- A61K9/2866—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
Definitions
- the invention relates to dosage forms and methods of using (2R-trans)-6-chloro-5-[[4- [(4-fluorophenyl)methyl]-2,5-dimethyl-l-piperazinyl]carbonyl]-N,N, 1-trimethyl-alpha-oxo-lH- indole-3-acetamide.
- the invention relates to dosage forms, methods, and novel pharmaceutical compositions.
- P38 MAP kinase has been identified as a drug target given its role in mediating the intracellular response to pro-inflammatory stress. Inadequate regulation of p38 kinase has been associated with inflammation related disorders including but not limited to rheumatoid arthritis, osteoarthritis, inflammatory bowel disease, asthma, psoriasis, and congestive heart failure.
- x > represents a single or double bond
- one Z 2 is CA or CR 8 A and the other is CR 1 , CR l 2 , NR 6 or N wherein each R 1 , R 6 and R 8 is independently hydrogen or noninterfering substituent
- A is -CO(X)jY wherein Y is COR 2 or an isostere thereof and R 2 is hydrogen or a noninterfering substituent, X is a spacer of 2-6 A, and j is 0 or 1
- Z 1 is CR 5 or N wherein R 5 is hydrogen or a noninterfering substituent; each of 1 and k is an integer from 0-2 wherein the sum of 1 and k is 0-3; Ar is an aryl group substituted with 0-5 noninterfering substituents, wherein two noninterfering substituents can form a fused ring; and the distance between the atom of Ar linked to L 2 and the center of the ⁇ ring is 4.5-24 A
- indole based compounds have been identified as useful antagonists of p38 kinase, an indole typed compound, and proper dosage form thereof, with properties suitable for use as a pharmaceutical in humans has yet to be particularly identified and developed.
- the invention is directed to methods and compounds useful in treating conditions that are characterized by enhanced p38- ⁇ activity. These conditions include inflammation (e.g., rheumatoid arthritis), proliferative diseases, and certain cardiovascular disorders, as further described below.
- inflammation e.g., rheumatoid arthritis
- proliferative diseases e.g., rheumatoid arthritis
- cardiovascular disorders e.g., rheumatoid arthritis
- the invention provides methods to treat a condition mediated by p38- ⁇ kinase comprising administering to a subject in need of such treatment a compound of the invention.
- a condition ameliorated, treated or prevented by a composition of the invention, or practicing the methods of the invention is a proinflammation response.
- the invention provides formulations, e.g., liquid, solid, powder, aerosol, spray, and the like (including solid oral formulations), and methods of using them, comprising a therapeutically effective amount of a compound of the invention, or pharmaceutically acceptable salts thereof (e.g., a hydrochloride salt), and a pharmaceutically acceptable excipient.
- formulations e.g., liquid, solid, powder, aerosol, spray, and the like (including solid oral formulations), and methods of using them, comprising a therapeutically effective amount of a compound of the invention, or pharmaceutically acceptable salts thereof (e.g., a hydrochloride salt), and a pharmaceutically acceptable excipient.
- a pharmaceutically acceptable excipient such as lactose, Avicel, Prosolv, pregelatinized starch, HPMC, NaCMC, croscarmellose sodium (in low amounts), sodium starch glycolate, magnesium stearate, stearic acid or a combination thereof.
- a formulation of the invention including liquid, solid, powder, aerosol, spray formulations (including solid oral formulations) comprises the compound having the formula:
- a formulation of the invention e.g., a solid oral formulation
- a hydrochloride salt e.g., a compound of the invention
- a compound of the invention is formulated for extended release with an excipient comprising calcium phosphate dibasic, povidone, sodium lauryl sulfate or a combination thereof.
- a compound of the invention e.g., a solid oral formulation, is formulated as a powder, a tablet, a pill or a capsule, or included in an aerosol or spray.
- the invention provides pharmaceutical compositions (and methods of using them) comprising a therapeutically effective amount of the compound or pharmaceutically acceptable salts thereof (e.g., a hydrochloride salt), and a pharmaceutically acceptable excipient.
- the compound is formulated with a pharmaceutically acceptable excipient as set forth in Table 2.1 , Table 2.2, Table 2.3 or Table 3.1.
- the invention provides pharmaceutical composition comprising a therapeutically effective amount of the compound as set forth in any one of claims 1 to 35, or pharmaceutically acceptable salts or pharmaceutically acceptable forms thereof, wherein the compound is formulated as an immediate release tablet with a pharmaceutically acceptable excipient as set forth in Table 3.1 or Table 3.2.
- a compound of the invention is formulated with a pharmaceutically acceptable excipient comprising lactose anhydrous microcrystalline cellulose, crosslinked cellulose, a starch, colloidal silicon dioxide, magnesium stearate or a combination thereof, wherein in one aspect the starch comprises partially pre-gelatinized starch, and in one aspect the crosslinked cellulose comprises carboxymethyl cellulose.
- the invention also provides methods for treating, ameliorating, preventing or delaying disease progression in an inflammatory disease or condition, or a disease or condition having an inflammatory component, or ameliorating the inflammatory disease or condition or disease or condition having an inflammatory component, comprising administering to a subject in need of such treatment a compound of the invention, a pharmaceutical formulation comprising the compound or a pharmaceutically acceptable salt thereof (e.g., a hydrochloride salt), a therapeutically effective amount of a compound of the invention.
- the inflammatory disease or condition, or the disease or condition having an inflammatory component is arthritis, and in one aspect the arthritis is rheumatoid arthritis (RA).
- the compound, pharmaceutical formulation or pharmaceutically acceptable salt is administered at doses of 1 mg/kg once daily (qd), twice daily (bid), and three times daily (tid). In one aspect, the compound, pharmaceutical formulation or pharmaceutically acceptable salt is administered at single-doses of between about 0.25, 0.5, 0.75, 1.0 and 5, 6, 7, 8, 9, or 10 or more mg/kg, wherein in one aspect the dose levels are 1 , 2, 3, 4 or 5 or more mg/kg dose levels. In one aspect, the compound, pharmaceutical formulation or pharmaceutically acceptable salt is administered in dosages of between about 1 and 3, 4 or 5 or more mg/kg once daily (qd), about 1 and 3 mg/kg twice daily (bid), or about 1 and 3 mg/kg three times daily (tid).
- the compound, pharmaceutical formulation or pharmaceutically acceptable salt is administered in dosages of 1, 2, 3, 4 or 5 or more mg/kg once daily (qd), 1, 2, 3, 4 or 5 or more mg/kg twice daily (bid), or 1 , 2, 3, 4 or 5 or more mg/kg three times daily (tid).
- the compound, pharmaceutical formulation or pharmaceutically acceptable salt is administered in dosages of between about 10 mg and 120 mg single dosages once daily (qd). twice daily (bid), or three times daily (tid).
- the compound, pharmaceutical formulation or pharmaceutically acceptable salt is administered in dosages of about 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110 or 120 or more mg single dosages once daily (qd), twice daily (bid), or three times daily (tid).
- Figure 1 illustrates the manufacture of the Immediate Release (IR) tablet in Flow diagram, as described in detail Example 2, below.
- Figure 2 illustrates Table 3.4, which describes exemplary formulations of the invention, as described in detail Example 2, below.
- Figure 3 illustrates Table 3.5, summarizing data for exemplary formulations (tablets) of the invention, as described in detail Example 2, below.
- Figure 4 illustrates Table 3.6, a characterization of exemplary formulations (tablets) of the invention, as described in detail Example 2, below.
- Figure 5 illustrates a summary of data for pre-milled granulation, milled granulation and final blend, as described in detail Example 2, below.
- Figure 6 illustrates a summary of data for stability studies for the compound IR tablets at
- Figure 7 is an illustration of an exemplary manufacturing process of the invention: a flow chart of a manufacturing process for exemplary Extended Release (ER) formulations of the invention, as described in detail Example 2, below.
- Figure 8 summarizes data regarding exemplary formulations of the invention, as described in detail Example 2, below.
- Figure 10 illustrates a summary of the disposition of subjects of the clinical study described in detail Example 2, below.
- Figure 1 1 and Figure 12 illustrate a summary of treatment group responses in the clinical study described in detail Example 2, below.
- Figure 13 is a chart showing the pK effect of the compound in fasted males.
- Figure 14 illustrates the correlation of an oral dosage formulation of the compound relative to ex vivo TNF ⁇ as measured in blood plasma post dosing.
- Figure 15 illustrates the correlation of an oral dosage formulation of the compound relative to ex vivo TNF ⁇ as measured in blood plasma post dosing.
- Figure 17 provides the plasma profile of Compound following administration of a single
- Figure 18 provides the plasma profile of Compound following administration of a single
- Figure 23 provides the plasma profile of Compound following administration of a single
- Figure 24 illustrates the effect of Compound dosage form on tmax in the dog.
- Figure 25 illustrates the effect of Compound dosage form on exposure in the dog (doses are normalized)
- Figure 26 illustrates the effect of Compound dosage form on Cmax in the dog.
- Figures 27 and 28 are charts providing correlative data in human subjects between relative Cmax levels of the Compound and reported episodes of dizziness.
- this molecule exhibits a specific conformational shape and rigidity that enables it to uniquely interact with p38 kinase, the alpha isoform in particular, making it unusually selective and effective at modulating enzymatic activity.
- administering means providing a drug to a patient in a manner that is pharmacologically useful.
- AUC Absolute under the curve
- AUCo-48 refers to the AUC obtained from integrating the plasma concentration curve over a period of zero to 48 hours, where zero is conventionally the time of administration of the drug or dosage form comprising the drug to a patient.
- AUQ refers to area under the plasma concentration curve from hour 0 to the last detectable concentration at time t, calculated by the trapezoidal rule.
- AUCj nf refers to the AUC value extrapolated to infinity, calculated as the sum of AUC 1 and the area extrapolated to infinity, calculated by the concentration at time t (Ct) divided by k. (If the ti/, value was not estimable for a subject, the mean t> / , value of that treatment was used to calculate AUQ nf .).
- "Mean, single dose, area under a plasma concentration-time curve AUC, n f" means the mean AU C mf obtained over several patients or multiple administrations to the same patient on different occasions with sufficient washout in between dosings to allow drug levels to subside to pre-dose levels, etc., following a single administration of a dosage form to each patient.
- “Ascending plasma concentration” means a drug plasma concentration profile over about the first 12 to 24 hours following initial dosing, wherein the profile shows an increase to a maximum concentration, wherein said maximum occurs more than about 6 hours following the initial dose, preferably, more than about 10 hours following initial dose, more preferably, more than about 12 hours after dose.
- a drug plasma concentration is listed, the value listed is the calculated mean value based on values obtained from a groups of subjects tested.
- Ascending rate of release or “ascending release rate” means a rate of release wherein the amount of drug released from a dosage form as a function of time increases over a period of time, preferably continuously and gradually.
- the rate of drug released as a function of time increases in a steady (rather than step- wise) manner. More preferably, an ascending rate of release may be characterized as follows. The rate of release as a function of time for a dosage form is measured and plotted as % drug release versus time or as milligrams of drug released / hour versus time.
- An ascending rate of release is preferably characterized by an average rate (expressed in mg of drug per hour) wherein the rate within a given two hour span is higher as compared with the previous two hour time span, over the period of time of about 2 hours to about 12 hours, preferably, about 2 hours to about 18 hours, more preferably about 4 hours to about 12 hours, more preferably still, about 4 hours to about 18 hours.
- the increase in average rate is gradual such that less than about 30% of the dose is delivered during any 2 hour interval, more preferably, less than about 25% of the dose is delivered during any 2 hour interval.
- the ascending release rate is maintained until at least about 50%, more preferably until at least about 75% of the drug in the dosage form has been released.
- ascending rates of release may be defined with reference to specific release rates measured at specified times following administration of the dosage form in question. Preferably such release rates are determined in vitro.
- C means the concentration of drug in blood plasma, or serum, of a subject, generally expressed as mass per unit volume, typically nanograms per milliliter. For convenience, this concentration may be referred to herein as “drug plasma concentration", “plasma drug concentration” or “plasma concentration”.
- the plasma drug concentration at any time following drug administration is referenced as Ctime, as in C9h or C24h, etc.
- a maximum plasma concentration obtained following administration of a dosage form obtained directly from the experimental data without interpolation is referred to as Cmax.
- the average or mean plasma concentration obtained during a period of interest is referred to as Cavg or Cmean.
- Mean, single dose, maximum plasma concentration Cmax means the mean Cmax obtained over several patients or multiple administrations to the same patient with sufficient washout in between dosings to allow drug levels to subside to pre-dose levels, etc., etc., following a single administration of a dosage form to each patient.
- Composition means a product containing a compound of the present invention (such as a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from such combinations of the specified ingredients in the specified amounts).
- Compound or “drug” means the compound (2R-trans)-6-chloro-5-[[4-[(4- fluorophenyl)methyl]-2,5-dimethyl-l-piperazinyl]carbonyl]-N,N, 1-trimethyl-alpha-oxo-lH- indole-3-acetamide as represented by Formula (I) below
- the compounds of the invention may be present as racemates, enantiomers and enantiomeric mixtures thereof.
- Examples of a compound selected from Formula (I) for use in the present invention include an enantiomer of Formula (I) in an enantiomeric mixture wherein the enantiomer of Formula (I) predominates.
- the enantiomer represented by Formula (I) preferably predominates to the extent of about 90% or greater.
- examples of the present invention also include enantiomeric mixtures wherein said enantiomer preferably predominates to the extent of about 98% or greater.
- Dosage form means one or more compounds in a medium, carrier, vehicle, or device suitable for administration to a patient.
- Oral dosage form means a dosage form suitable for oral administration.
- Dose means a unit of drug. Conventionally, a dose is provided as a dosage form.
- Doses may be administered to patients according to a variety of dosing regimens. Common dosing regimens include once daily orally (qd), twice daily orally (bid), and thrice daily orally (tid).
- Effective amount means that amount of compound that elicits the biological or medicinal response in a tissue system, animal or human, that is being sought by a researcher, veterinarian, medical doctor, or other clinician, which includes therapeutic alleviation of the symptoms of the disease or disorder being treated and prophylactic.
- Enantiomer means one of a pair of molecular species that are mirror images of each other and are not superposable.
- the term “diastereomer” refers to stereoisomers that are not related as mirror images.
- the symbols “R” and “S” represent the configuration of substituents around a chiral carbon atom(s).
- the symbols “R*” and “S*” denote the relative configurations of of substituents around a chiral carbon atom(s).
- isomers refers to compounds that have the same composition and molecular weight but differ in physical and/or chemical properties. Such substances have the same number and kind of atoms but differ in structure. The structural difference may be in constitution (geometric isomers) or in an ability to rotate the plane of polarized light (stereoisomers).
- stereoisomer refers to isomers of identical constitution that differ in the arrangement of their atoms in space. Enantiomers and diastereomers are stereoisomers wherein an asymmetrically substituted carbon atom acts as a chiral center.
- Frat plasma curve means a plasma concentration curve that reaches and maintains a substantially constant value after a defined period of time following administration of a dosage form according to the invention.
- “Immediate-release dosage form” means a dosage form that releases greater than or equal to about 80% of the drug in less than or equal to about 1 hour following administration of the dosage form to a patient. "Initiation of release” means the beginning of a release rate test, when the dosage form is placed in a liquid and the sequence of events begins that leads to release of the compounds of Formula (I).
- “Medicament” means a product for use in preventing, treating or ameliorating substance related disorders such as substance dependence, substance abuse or substance induced disorders in a subject in need thereof.
- Patient means an animal, preferably a mammal, more preferably a human, in need of therapeutic intervention.
- “Pharmaceutically acceptable” means molecular entities and compositions that are of sufficient purity and quality for use in the formulation of a composition or medicament of the present invention. Since both human use (clinical and over-the-counter) and veterinary use are equally included within the scope of the present invention, a formulation would include a composition or medicament for either human or veterinary use.
- “Pharmaceutically acceptable salt” means an acid or base salt of the compounds of the invention that are of sufficient purity and quality for use in the formulation of a composition or medicament of the present invention and are tolerated and sufficiently non toxic to be used in a pharmaceutical preparation.
- Suitable pharmaceutically acceptable salts include acid addition salts which may, for example, be formed by reacting the drug compound with a suitable pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, fumaric acid, maleic acid, succinic acid, acetic acid, benzoic acid, citric acid, tartaric acid, carbonic acid or phosphoric acid.
- representative pharmaceutically acceptable salts include, but are not limited to, the following: acetate, alpha-ketoglutarate, alpha-glycerophosphate, ascorbate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide, calcium edetate, camsylate, carbonate, chloride, clavulanate, citrate, dihydrochloride, edetate, edisylate, estolate, esylate, ethanesulfonate, fumarate, gluceptate, gluconate, glutamate, glycollylarsanilate, hexylresorcinate, hydrabamine, hydrobromide, hydrochloride, hydroxynaphthoate, iodide, isothionate, lactate, lactobionate, laurate, malate, maleate, malonate, mandelate, mesylate, methane
- salts may be obtained using standard procedures well known in the art, for example by reacting a sufficiently basic compound such as an amine with a suitable acid affording a physiologically acceptable anion.
- a sufficiently basic compound such as an amine
- a suitable acid affording a physiologically acceptable anion.
- Alkali metal for example; sodium, potassium or lithium, or alkaline earth metals, for example calcium salts of carboxylic acids can also be made.
- “Plasma drug concentration curve” or “drug plasma concentration curve”, or “plasma concentration curve” or “plasma profile” or “plasma concentration profile” refer to the curve obtained by plotting plasma drug concentration or drug plasma concentration, or plasma concentration versus time.
- the convention is that the zero point on the time scale (conventionally on the x axis) is the time of administration of the drug or dosage form comprising the drug to a patient.
- Rate of release or “release rate” means to the quantity of compound released from a dosage form per unit time, e.g., milligrams of drug released per hour (mg/hr).
- Drug release rates for dosage forms may be measured as an in vitro rate of drug release, i.e., a quantity of drug released from the dosage form per unit time measured under appropriate conditions and in a suitable fluid.
- Relative bioavailability means AUQ nf for inventive dosage form/AUCj nf for immediate release dosage form; wherein both dosage forms comprise the same or substantially the same amount of drug, expressed in units of mass.
- Tmax is the mean time elapsed from administration to a patient of a dosage form comprising a drug to the time at which the Cmax for that drug is obtained over several patients or multiple administrations to the same patient with sufficient washout in between dosings to allow drug levels to subside to pre-dose levels, etc., following a single administration of the dosage form to each patient, and obtained directly from the experimental data without interpolation.
- “Therapeutically effective amount” means that amount of drug that elicits the biological or medicinal response in a tissue system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician, which includes alleviation of the symptoms of the disease or disorder being treated.
- "Zero order rate of release” or “zero order release rate” means a rate of release wherein the amount of drug released as a function of time is substantially constant. In other words, the dosage form exhibits zero order or substantially zero order release kinetics.
- the rate of release of drug as a function of time shall vary by less than about 30%, preferably, less than about 20%, more preferably, less than about 10%, most preferably, less than about 5%, wherein the measurement is taken over the period of time wherein the cumulative release is between about 25% and about 75%, preferably, between about 25% and about 90%.
- the Compound can be provided in different formulations adapted to achieve pharmacologically effective levels in the blood.
- the Compound is provided in an oral formulation adapted to provide an immediate release profile.
- particular benefit may be provided through such a formulation when used to treat conditions requiring rapid onset and/or rapid clearance of the Compound into and from the patient's plasma.
- it may be desired to have a sustained release formulation which can be tailored to provide a more constant or . prolonged exposure of the Compound.
- the Compound is formulated into dosage forms suitable for administration to patients in need thereof.
- Oral dosage may be provided in any pharmaceutically acceptable form.
- a solid form may involve any of a tablet, capsule, and the like.
- a liquid form may involve delivery of the compound through any of an oral suspension, solution, gel, liquid capsule and the like.
- the Compound is provided in a solid dosage form.
- Standard solid dosage forms may comprise the Compound in combination with various pharmaceutically acceptable excipients, said dosage form adapted to provide a release profile of the Compound in a manner to obtain the desired clinical effect through oral administration to the patient.
- Pharmaceutically acceptable excipients are known in the art and can be provided according to considerations of desired functionality and processability. Roles for the excipients in the oral dosage form include but are not limited to fillers, binders, disintegrants, release- controlling agents, glidants, lubricants, coatings and the like. For example, in one embodiment of the invention, it is desired to have an immediate release profile for the dosage form.
- the dosage form should preferably comprise disintegrant in an amount between 3 and 20% of the total form wherein the Compound also comprises a pharmaceutically effective amount of the form.
- a controlled or sustained release formulation of the Compound is desired. Such a formulation can be achieved by varying the amounts, concentrations and ratios of certain release controlling polymers.
- an oral extended release formulation is provided that in tablet form comprising approximately 250 mg of the Compound, hydro xypropyl methylcellulose, carboxymethylcellulose sodium (NaGMC), lactose monohydrate, magnesium stearate, and OPADRY II film-coating.
- the ratio of HPMC to NaCMC is varied from 1 :1 to 3 : 1 to drive the release profile of the formulation towards a zero order dissolution rate, as measured through appropriate plasma samples.
- the ratio of HPMC to NaCMC is 2: 1.
- fillers used in the art include but are not limited to sugars such as lactose, dextrose, glucose, sucrose, cellulose, starches and carbohydrate derivatives, calcium carbonates, magnesium carbonates and the like.
- binders include hydroxypropyl methylcellulose, methylcellulose, starches, and the like.
- Useful disintegrants may be selected from starches, clays, celluloses, algins and gums and crosslinked starches, celluloses and polymers.
- Representative disintegrants include microcrystalline cellulose, crosscarmellose sodium, alginic acid, sodium alginate, crosprovidone, cellulose, agar and related gums, sodium starch glycolate, corn starch, potato starch, sodium starch glycolate, Veegum HV, methylcellulose, agar, bentonite, carboxymethylcellulose, alginic acid, guar gum and the like.
- Glidants commonly used in the art include magnesium carbonate, magnesium lauryl sulphate, calcium silicate, talc, fumed silicon dioxide and the like.
- Useful lubricants include but are not limited to magnesium stearate, calcium stearate, stearic acid, sodium stearyl fumarate, polyethylene glycol, sodium lauryl sulphate, magnesium lauryl sulphate, sodium benzoate, and the like.
- Polymers commonly used as excipients include but are not limited to methylcellulose (MC), ethylcellulose (EC), hydroxyethylcellulose (HEC), methyl hydroxyethylcellulose (MHEC), hydroxypropyl cellulose (HPC), hydroxypropyl methylcellulose (HPMC), sodium carboxymethylcellulose (NaCMC), and the like. These polymers, either alone or in various combinations, can serve multiple purposes including but not limited to controlling release of the the Compound.
- the appropriate excipients should be selected such that they are compatible with other excipients and do not bind with the Compound or cause drug degradation.
- the dosage form may be manufactured by the wet granulation technique.
- the drug and carrier are blended using an aqueous or organic solvent, such as denatured anhydrous ethanol, as the granulation fluid.
- the remaining ingredients can be dissolved in a portion of the granulation fluid, such as the solvent described above, and this latter prepared wet blend is slowly added to the drug blend with continual mixing in the blender.
- the granulating fluid is added until a wet blend is produced, which wet mass blend is then forced through a predetermined screen and dried in a fluid bed dryer. The dried granules are then sized.
- magnesium stearate, or another suitable lubricant and other excipient materials are added to the drug granulation, and the granulation is put into milling jars and mixed on a jar mill for 10 minutes.
- the composition is pressed into a layer, for example, in a Manesty® press or a Korsch LCT press.
- granules or powders of the drug layer compositions and push layer composition are sequentially placed in an appropriately-sized die with intermediate compression steps being applied to each of the first two layers, followed by a final compression step after the last layer is added to the die to form the trilayered core.
- the intermediate compression typically takes place under a force of about 50-100 newtons.
- Final stage compression typically takes place at a force of 3500 newtons or greater, often 3500- 5000 newtons.
- the compressed cores are fed to a dry coater press, e.g., Kilian® Dry Coater press, and subsequently coated with the wall materials as described herein.
- Pan coating may be conveniently used to provide the completed dosage form.
- the wall-forming composition for the inner wall or the outer wall is deposited by successive spraying of the appropriate wall composition onto the compressed core accompanied by tumbling in a rotating pan.
- a pan coater is used because of its availability at commercial scale.
- Other techniques can be used for coating the compressed core.
- the wall is dried in a forced-air oven or in a temperature and humidity controlled oven to free the dosage form of solvent(s) used in the manufacturing. Drying conditions will be conventionally chosen on the basis of available equipment, ambient conditions, solvents, coatings, coating thickness, and the like.
- one alternative technique uses an air-suspension procedure. This procedure consists of suspending and tumbling the compressed core in a current of air, until a coating is applied to the core.
- the air-suspension procedure is described in U.S. Patent No. 2,799,241; in J. Am. Pharm. Assoc, Vol. 48, pp. 451- 459 (1959); and, ibid., Vol. 49, pp. 82-84 (1960).
- the dosage form also can be coated with a Wurster ⁇ air-suspension coater using, for example, methylene dichloride methanol as a cosolvent for the wall forming material.
- An Aeromatic® air-suspension coater can be used employing a cosolvent.
- the drug and other ingredients comprising the drug layer are blended and pressed into a solid layer.
- the layer possesses dimensions that correspond to the internal dimensions of the area the layer is to occupy in the dosage form, and it also possesses dimensions corresponding to the push layer, if included, for forming a contacting arrangement therewith.
- the drug and other ingredients can also be blended with a solvent and mixed into a solid or semisolid form by conventional methods, such as ballmilling, calendering, stirring or rollmilling, and then pressed into a preselected shape.
- the compressed cores then may be coated with the inner wall material and the semipermeable wall material as described herein.
- Another manufacturing process that can be used comprises blending the powdered ingredients in a fluid bed granulator. After the powdered ingredients are dry blended in the granulator, a granulating fluid, for example, poly(vinylpyrrolidone) in water, is sprayed onto the powders. The coated powders are then dried in the granulator. This process granulates all the ingredients present therein while adding the granulating fluid. After the granules are dried, a lubricant, such as stearic acid or magnesium stearate, is mixed into the granulation using a blender e.g., V-blender or tote blender. The granules are then pressed and coated in the manner described above.
- a granulating fluid for example, poly(vinylpyrrolidone) in water
- Exemplary solvents suitable for manufacturing the dosage form components comprise aqueous or inert organic solvents that do not adversely harm the materials used in the system.
- the solvents broadly include members selected from the group consisting of aqueous solvents, alcohols, ketones, esters, ethers, aliphatic hydrocarbons, halogenated solvents, cycloaliphatics, aromatics, heterocyclic solvents and mixtures thereof.
- Typical solvents include acetone, diacetone alcohol, methanol, ethanol, isopropyl alcohol, butyl alcohol, methyl acetate, ethyl acetate, isopropyl acetate, n-butyl acetate, methyl isobutyl ketone, methyl propyl ketone, n- hexane, n-heptane, ethylene glycol monoethyl ether, ethylene glycol monoethyl acetate, methylene dichloride, ethylene dichloride, propylene dichloride, carbon tetrachloride nitroethane, nitropropane tetrachloroethane, ethyl ether, isopropyl ether, cyclohexane, cyclooctane, benzene, toluene, naphtha, 1,4-dioxane, tetrahydrofuran, diglyme, water,
- Exemplary liquid carriers for the present invention include lipophilic solvents (e.g., oils and lipids), surfactants, and hydrophilic solvents.
- Exemplary lipophilic solvents include, but are not limited to, Capmul PG-8, Caprol MPGO, Capryol 90, Plurol Oleique CC 497, Capmul MCM, Labrafac PG, N-Decyl Alcohol, Caprol 1 OGlOO, Oleic Acid, Vitamin E, Maisine 35-1, Gelucire 33/01, Gelucire 44/14, Lauryl Alcohol, Captex 355EP, Captex 500, Capylic/Caplic Triglyceride, Peceol, Caprol ET, Labrafil M2125 CS, Labrafac CC, Labrafil M 1944 CS, Captex 8277, Myvacet 9-45, Isopropyl Nyristate, Caprol PGE 860, Olive Oil, Plurol Oleique, Peanut Oil, Captex 300 Low C6, and Capric Acid.
- Exemplary surfactants include, but are not limited to, Vitamin E TPGS, Cremophor (grades EL, EL-P, and RH40), Labrasol, Tween (grades 20, 60, 80), Pluronic (grades L-31, L-35, L-42, L-64, and L-121), Acconon S-35, Solutol HS-15, and Span (grades 20, and 80).
- Exemplary hydrophilic solvents for example, include, but are not limited to, Isosorbide Dimethyl Ether, Polyethylene Glycol (PEG grades 300, 400, 600, 3000, 4000, 6000, and 8000) and Propylene Glycol (PG).
- any formulation comprising a sufficient dosage of the Compound solubilized in a liquid carrier suitable for administration to a subject.
- Drug may be provided in particles by comminution that produces the size of the drug and the size of one or more accompanying polymers used in the fabrication of the dosage form, typically with a core containing the compound, according to the mode and the manner of the invention.
- the means for producing particles include granulation, spray drying, sieving, lyophilization, crushing, grinding, jet milling, micronizing and chopping to produce the intended micron particle size.
- the process can be performed by size reduction equipment, such as a micropulverizer mill, a fluid energy grinding mill, a grinding mill, a roller mill, a hammer mill, an attrition mill, a chaser mill, a ball mill, a vibrating ball mill, an impact pulverizer mill, a centrifugal pulverizer, a coarse crusher and a fine crusher.
- size reduction equipment such as a micropulverizer mill, a fluid energy grinding mill, a grinding mill, a roller mill, a hammer mill, an attrition mill, a chaser mill, a ball mill, a vibrating ball mill, an impact pulverizer mill, a centrifugal pulverizer, a coarse crusher and a fine crusher.
- the size of the particle can be ascertained by screening, including a grizzly screen, a flat screen, a vibrating screen, a revolving screen, a shaking screen, an oscillating screen and a reciprocating screen.
- Drug emulsified formulations of the present invention can initially comprise an oil and a non-ionic surfactant.
- the oil phase of the emulsion comprises any pharmaceutically acceptable oil which is not immiscible with water.
- the oil can be an edible liquid such as a non-polar ester of an unsaturated fatty acid, derivatives of such esters, or mixtures of such esters.
- the oil can be vegetable, mineral, animal or marine in origin.
- non-toxic oils can also include, for example, in addition to the surfactants listed above, a member selected from the group consisting of peanut oil, cottonseed oil, sesame oil, corn oil, almond oil, mineral oil, castor oil, coconut oil, palm oil, cocoa butter, saf ⁇ lower, a mixture of mono- and diglycerides of 16 to 18 carbon atoms, unsaturated fatty acids, fractionated triglycerides derived from coconut oil, fractionated liquid triglycerides derived from short chain 10 to 15 carbon atoms fatty acids, acetylated monoglycerides, acetylated diglycerides, acetylated triglycerides, olein known also as glyceral trioleate, palmitin known as glyceryl tripalmitate, stearin known also as glyceryl tristearate, lauric acid hexylester, oleic acid oleylester, glycolyzed eth,
- the concentration of oil, or oil derivative in the emulsion formulation can be from about 1 wt % to about 40 wt %, with the wt % of all constituents in the emulsion preparation equal to 100 wt %.
- the oils are disclosed in Pharmaceutical Sciences by Remington, 17th Ed., pp. 403-405, (1985) published by Mark Publishing Co., in Encyclopedia of Chemistry, by Van Nostrand Reinhold, 4th Ed., pp. 644-645, (1984) published by Van Nostrand Reinhold Co.; and in U.S. Pat. No. 4,259,323.
- the amount of Compound incorporated in the dosage forms of the present invention is generally from about 10% to about 90% by weight of the composition depending upon the therapeutic indication and the desired administration period, e.g., every 12 hours, every 24 hours, and the like. Depending on the dose of Compound desired to be administered, one or more of the dosage forms can be administered. Depending upon the formulation, the Compound will preferably be in the form of an HCl salt or free base form.
- An oral liquid formulation of the Compound may be in the form of a capsule.
- the capsule can be made conveniently in two parts, with one part (the “cap") slipping over and capping the other part (the “body”) as long as the capsule is deformable under the forces exerted by the expandable layer and seals to prevent leakage of the liquid drug formulation from between the telescoping portions of the body and cap.
- the two parts completely surround and capsulate the internal lumen that contains the liquid drug formulation, which can contain useful additives.
- the two parts can be fitted together after the body is filled with a preselected formulation.
- the assembly can be done by slipping or telescoping the cap section over the body section, and sealing the cap and body, thereby completely surrounding and encapsulating the formulation of drug.
- Soft capsules typically have a wall thickness that is greater than the wall thickness of hard capsules.
- soft capsules can, for example, have a wall thickness on the order of 10-40 mils, about 20 mils being typical, whereas hard capsules can, for example, have a wall thickness on the order of 2-6 mils, about 4 mils being typical.
- a soft capsule in one embodiment, can be of single unit construction and can be surrounded by an unsymmetrical hydro-activated layer as the expandable layer.
- the expandable layer will generally be unsymmetrical and have a thicker portion remote from the exit orifice.
- the presence of an unsymmetrical layer functions to assure that the maximum dose of drug is delivered from the dosage form.
- a barrier layer can be first coated onto the capsule and then the tableted, expandable layer is attached to the barrier-coated capsule with a biologically compatible adhesive.
- Suitable adhesives include, for example, starch paste, aqueous gelatin solution, aqueous gelatin/glycerin solution, aery 1 ate- vinylacetate based adhesives such as Duro- Tak adhesives (National Starch and Chemical Company), aqueous solutions of water soluble hydrophilic polymers such as hydroxypropyl methyl cellulose, hydroxymethyl cellulose, hydroxyethyl cellulose, and the like. That intermediate dosage form can be then coated with a semipermeable layer. Hard capsules are also contemplated by the invention.
- Hard capsules are typically composed of two parts, a cap and a body, which are fitted together after the larger body is filled with a preselected appropriate formulation. This can be done by slipping or telescoping the cap section over the body section, thus completely surrounding and encapsulating the drug formulation.
- Hard capsules can be made, for example, by dipping stainless steel molds into a bath containing a solution of a capsule lamina-forming material to coat the mold with the material. Then, the molds are withdrawn, cooled, and dried in a current of air. The capsule is stripped from the mold and trimmed to yield a lamina member with an internal lumen.
- the engaging cap that telescopically caps the formulation receiving body is made in a similar manner.
- the closed and filled capsule can be encapsulated with a semipermeable lamina.
- the semipermeable lamina can be applied to capsule parts before or after parts and are joined into the final capsule.
- the hard capsules can be made with each part having matched locking rings near their opened end that permit joining and locking together the overlapping cap and body after filling with formulation.
- a pair of matched locking rings are formed into the cap portion and the body portion, and these rings provide the locking means for securely holding together the capsule.
- the capsule can be manually filled with the drug formulation, or they can be machine filled with the drug formulation.
- the hard capsule is encapsulated with a semipermeable lamina permeable to the passage of fluid and substantially impermeable to the passage of drug.
- Methods of forming hard cap dosage forms are described in U.S. Patent No. 6,174,547, U.S. Patent Nos. 6,596,314, 6,419,952, and 6, 174,547.
- the hard and soft capsules can comprise, for example, gelatin; gelatin having a viscosity of 15 to 30 millipoises and a bloom strength up to 150 grams; gelatin having a bloom value of 160 to 250; a composition comprising gelatin, glycerine, water and titanium dioxide; a composition comprising gelatin, erythrosin, iron oxide and titanium dioxide; a composition comprising gelatin, glycerine, sorbitol, potassium sorbate and titanium dioxide; a composition comprising gelatin, acacia glycerine, and water; and the like.
- Materials useful for forming capsule wall are known in U.S. Pat. Nos. 4,627,850; and in 4,663,148.
- the capsules can be made out of materials other than gelatin (see for example, products made by BioProgres pic).
- the capsules typically can be provided, for example, in sizes from about 3 to about 22 minims (1 minim being equal to 0.0616 ml) and in shapes of oval, oblong or others. They can be provided in standard shape and various standard sizes, conventionally designated as (000), (00), (0), (1), (2), (3), (4), and (5). The largest number corresponds to the smallest size. Non-standard shapes can be used as well. In either case of soft capsule or hard capsule, non-conventional shapes and sizes can be provided if required for a particular application.
- dosage forms described herein are merely exemplary of a variety of dosage forms designed for and capable of achieving administration of the inventive substance(s). Those of skill in the pharmaceutical arts can identify other dosage forms that would be suitable.
- inventive methods, compositions, and dosage forms are useful in treating a variety of indications that are treatable using the Compound.
- the invention provides a method for treating an indication, such as a disease or disorder, in a patient by administering an inventive composition or dosage form that comprises the Compound.
- a composition or dosage form comprising the Compound is administered to the patient via oral administration.
- the dose administered is generally adjusted in accord with the age, weight, and condition of the patient, taking into consideration the dosage form and the desired result.
- Inventive dosage forms may comprise the Compound or pharmacologically active metabolites in combination.
- any dose or frequency of administration that provides the therapeutic or prophylactic effect described herein is suitable for use in the present invention.
- Dosage regimens may be varied depending upon the requirement of the subjects (including factors associated with the particular subject being treated, including subject age, weight and diet, strength of the preparation, the advancement of the disease condition and the mode and time of administration) and the use of a particular compound of Formula (I) or pharmaceutical composition thereof or a pharmaceutically acceptable salt thereof.
- Optimal dosages to be administered may be readily determined by those skilled in the art and will result in the need to adjust the dose to an appropriate therapeutic or prophylactic level.
- the use of either daily administration or post-periodic dosing may be employed.
- dosage forms according to the invention comprise an amount of the Compound ranging from about 5 mg to about 1000 mg, preferably from about 10 mg to about 600 mg, and more preferably from about 50 rng to about 300 mg.
- the S-carboxylic- ⁇ -chloro indole acid (1.56 g, 7.44 mmol) was dissolved in dry methylene chloride 60 mL to this was added the EDACHCl (1.57 g, 8.18 mmol) and DMAP(IO % mol). After stirring under nitrogen for 10 min. the amine (2.19 g, 7. 5 mmol) was added, followed by triethylamine (3 mL, 21.52 mmol). After overnight at room temperature, the reaction mixture was concentrated and the residue was taken up in ethyl acetate and washed with 10% aq. sodium carbonate, saturated sodium chloride, dried over anhydrous sodium sulfate and filtered.
- Example 2 A Phase 2, Multicenter, Randomized. Double-Blind, Placebo-Controlled,
- the following example describes a study the demonstrates that the compound of the invention is effective in treating a condition involving selective inhibition of the activity of a p38- ⁇ isoform; in particular in ameliorating active rheumatoid arthritis.
- the following example describes a Phase 2, multicenter, randomized, double-blind, placebo-controlled, dose-escalating study of Compound in patients with active rheumatoid arthritis receiving methotrexate.
- pH/Solubilitv Profile of Compound (in Table 2) at ca. 25°C: The solubility of the Compound was determined over the pH range of 1.0 to 12.0 at ca. 25°C using various concentrations of NaOH and HCl. The maximum solubility for Compound was determined to be 40.6 mg/mL at pH 3.5. In the experiment solubility decreased below pH 3.5 due to a common ion effect.
- pKa The pKa of Compound was determined to be 5.85 ⁇ 0.02 at an average ionic strength of 0.174 M, using a co-solvent potentiometric titration procedure. This corresponds to a thermodynamic standard state value at zero total ionic strength of 6.10 ⁇ 0.02.
- Solubility of Compound in Selected Solvents at 25°C The solubility of Compound was determined in hexane, methanol (MeOH), ethanol (EtOH), acetonitrile, methyl ethyl ketone (2-butanone, MEK), acetone, isopropyl alcohol (IPA), 1 :1 Propylene Glycol (PG): Polyethylene Glycol (PEG 400), 1 : 1 EtOH:PG, and 1 :9 to 9:1 EtOH:water. Solubilities are shown in Table
- Hveroscopicitv by dynamic vapor sorption Compound is a crystalline solid. Upon heating, Compound exhibits a complex DSC scan with very weak endotherms. Optical hot stage analysis shows that Compound exhibits a very broad melting range from about 154°C to 170 0 C at a heating rate of l°C/minute. Capillary melting point data shows the melting point of the Compound to be dependent upon heating rate.
- Dynamic vapor sorption measurements show Compound to be slightly hygroscopic above 70%RH. When dry, Compound quickly gains ⁇ 3% water. At very low relative humidity ( ⁇ 4%RH), the water content levels briefly ( ⁇ 3%), then quickly increases to ⁇ 6% water content when humidity is raised to 18 to 21%. From ⁇ 21 % RH to ⁇ 70% RH the estimated water content remains in the range of 6 - 7%. Above ⁇ 71 %RH approximately 1.5% water weight is gained, and above ⁇ 90%RH an additional estimated 2% water is gained.
- Compound API particle size distribution and bulk density Particle size (by laser light scattering) and bulk density of several lots of Compound API are summarized in Table 1.2, below. Particle size distribution of all the API lots produced at one facility was homogeneous. The two lots differ in particle size and drying time varied greatly. This difference is not expected to have an effect during drug substance manufacturing.
- Adsorption of Compound on Filter Media Adsorption of Compound was studied on the following filters: LC PVDF, Nylon, Polysulfone, and Polypropylene. When polypropylene syringes were used in conjunction with the four types of filters, the results indicated that Compound is adsorbed. At pH 1.5, the range of adsorption was 0.60 ⁇ g/cm 2 to 2.04 ⁇ /cm 2 . The glass and polypropylene syringes without filters were 0.06 and 0.07 ⁇ g/cm 2 , respectively. At pH 7.4, the range of adsorption is 10.4 ⁇ g/cm 2 to 11.6 ⁇ g/cm 2 .
- the glass and polypropylene syringes without filters were 0.37 and 0.29 ⁇ g/cm 2 , respectively. At pH 10.8, the range of adsorption is 2.34 ⁇ g/cm 2 to 2.47 ⁇ g/cm 2 . Slight adsorption, 0.07 ⁇ g/cm 2 , was found for the glass and polypropylene syringes.
- Excipient Compatibility Compound compatibility with excipients commonly used in solid oral formulations was studied. Drug was mixed with each of fifteen excipients (see Table 2.1), and compared to a drug-only control, in both dry and wet blends. Study samples were stored at 40°C/75% RH conditions for 12 weeks. Samples were tested for appearance, recovery of active drug and impurity profile at 0, 2, 4, 8 and 12 weeks. Based on the results of this study, appropriate excipients were selected for further capsule and tablet formulation development.
- Calcium phosphate dibasic, povidone and sodium lauryl sulfate should not be used in Compound products, unless calcium phosphate dibasic is employed for extended release.
- the invention also provides an immediate release tablet having a formulation as set forth in Table 3.1, below:
- COMPOUND IR Tablet Manufacturing An exemplary Compound IR tablet manufacturing process is illustrated in Figure 1, as a Flow chart. Exemplary equipment that can be used to make compounds of the invention include examples described in Table 3.3, below:
- Figure 2 illustrates Table 3.4, which describes exemplary formulations of the invention.
- Table 3.4 describes tablet feasibility lots at a nominal 2-Kg scale.
- A intragranular
- B extragranular
- Constant parameters for Batches 64-194 impeller speed 500 ⁇ m, chopper speed 1500 (max) rpm.
- Figure 3 illustrates Table 3.5, summarizing data for exemplary formulations (tablets) of the invention, in this aspect: Compound IR feasibility tablets — Particle Size Analysis.
- Table 3.5 summarizing data for exemplary formulations (tablets) of the invention, in this aspect: Compound IR feasibility tablets — Particle Size Analysis.
- Figure 4 illustrates Table 3.6, a characterization of exemplary formulations (tablets) of the invention, in this example - using 2-Kg Feasibility Lots.
- Table 3.6 Tooling: #11-2, 1 l/32"rd std concave, target weight 340 mg.
- IR Tablets at 12-Kg scale Exemplary methods of making exemplary pharmaceutical formulations of the invention are summarized in Table 5.4, below.
- Table 5.4 granulation size was 12 kg, total batch size was ⁇ 14 kg. The amount of water needed for granulation at that scale was studied, and measured as % of granulation weight. For all the 4 batches, tablets were compressed at 3 compressions forces to achieve target hardness's of 10 kp, 13 kp and 16 kp. Based on the Process DOE study, it was estimated that the amount of water needed for granulation would be about 45% of granulation weight. Two lots were produced with 45% water; one lot was produced with 40% water (LW for "low weight”) and one with 50% water (HW for "high weight”). The two 45% lots were coated. The results are summarized in Table 5.4: Compound IR Tablets, 12-Kg scale: In-process data:
- IR Tablets at 60-Kg scale Exemplary tablets were made at 60-Kg (nominal) scale at three strengths, 30-, 60- and 90-mg, in August and September 2004.
- the granulation size was 60 Kg
- uncoated tablet batch size was ⁇ 68 Kg.
- 50 Kg of tablets were coated. All lots were successful.
- 45% water (as % of granulation weight) was the target amount for granulation; addition of water was stopped when satisfactory granulation was achieved. Actual amount of water used in granulation was:
- Tablets were compressed at 3 compressions forces to achieve target hardnesses of 10 kp, 13 kp and 16 kp.
- Blend uniformity results for the 90-mg lot were broad (one tablet at 112); note that this lot was produced with a lot with the largest particle size.
- Exemplary tablets of the invention can have various particle size distribution for (2) pre- milled granulation (3) milled granulation and/or (4) final blend;
- Figure 5 illustrates a summary of data for pre-milled granulation, milled granulation and final blend at 60 mg tabs, 60 kg scale).
- the invention also provides methods for packaging, and various packaged forms of the compositions of the invention.
- bulk tablets can be stored at a double- lined with polyethylene bags drum with no desiccant.
- Compound IR tablets can be packaged in 75 cc (12.5g), Blake HDPE OB, rectangular, white bottles with 33/400 closure (CR, CLIC-LOC III, Selig M-I; PHILLIPS-SUMIKA) polypropylene caps.
- the bottles are sealed with heat- induction seals.
- Ninety (90) tablets are packaged in each bottle.
- Stability studies for Compound IR tablets at 30 and 90 mg. 18-month stability data is available for 30 and 90 mg tablets at 25C/60%RH, and 9-month stability study for 30 and 90 mg tablets at 40C/75%RH conditions, where the data is summarized in Figure 6 (Table 7.1); in Figure 6, stability of Compound IR 30mg and 90 mg exemplary tablets, at 25C/60%RH and 40C/60%RH conditions.
- Product/Package Appearance Description Code
- the invention provides extended release (ER) tablets comprising at least one composition of the invention.
- ER extended release
- the invention provides ER tablets with approximately zero- order release for up to 12 hours, suitable for once- or twice-daily dosing.
- the ER tablets are formulated as a polymer matrix, wet granulation with high shear mixing:
- Tablet Trade Dress Oval, convex, coated tablet with debossing. 50 to 120 mg Compound in 600-mg core. Clinical tablets to date have been white-coated and non-debossed. Packaging; 30 tablets/ 75 cc (12.5g), Blake HDPE OB, rectangular, white bottles with 33/400 closure (CR, CLIC-LOC III, Selig M-I ; PHILLIPS-SUMIKA) polypropylene caps. Heat- induction seals, no cotton or dessicant.
- Two exemplary formulations of the invention comprise Hydroxypropyl Methylcellulose (“HPMC”) and Carboxymethylcellulose Sodium (“NaCMC”) selected and manufactured as 100- mg/ tablets.
- HPMC Hydroxypropyl Methylcellulose
- NaCMC Carboxymethylcellulose Sodium
- Exemplary Extended Release (ER) formulations of the invention are summarized in Table 4-1 and Table 4-2, below: Table 4-1 : Extended Release (ER) Tablet Formulations
- Figure 7 is an illustration of an exemplary manufacturing process of the invention: a flow chart of a manufacturing process for exemplary Extended Release (ER) formulations of the invention.
- Table 4-3 summarizes exemplary equipment that can be used in these exemplary manufacturing process of the invention.
- Figure 8 summarizes data regarding exemplary formulations of the invention.
- COMPOUND Clinical Study design This was a placebo-controlled, dose-escalating study designed to assess the safety, tolerabiiity, efficacy, pharmacokinetics (PK), and pharmacodynamics of Compound in subjects with active RA who were also receiving methotrexate.
- Subjects 132 total
- dose regimens A-F or placebo dose regimens A-F or placebo
- Dose levels for each successive treatment period were increased over the preceding period, with the exception of treatment period 4 that was amended to escalate to 60 mg tid (see the table below).
- Safety and PK data from the previous treatment period were reviewed before initiating the next treatment period. Each subject was followed for up to 58 days (30-day treatment period plus 28-day follow-up).
- Placebo Placebo (8; 8) a
- Placebo Placebo (5; 5) a
- Numbers in parentheses (Number randomized; number treated). Target number treated was 16 subjects per Compound treatment group, and 24 total subjects in the placebo group. aTotal enrollment in all placebo groups was 28 subjects, with a total of 26 subjects treated.
- bDosing regimen for 60 mg qbt group was 60 mg qd for Days 1—7, bid for Days 8—14, and tid for Days 15-30.
- DIAGNOSIS AND Men and women > 18 years of age with evidence of active RA MAIN CRITERIA FOR and receiving stable doses of methotrexate therapy patients INCLUSION receiving stable doses of non-steroidal anti-inflammatory drugs [NSAIDs] and/or low-dose prednisone were allowed in the study).
- NSAIDs non-steroidal anti-inflammatory drugs
- test product was COMPOUND, supplied as opaque white, NO(S) hard gelatin capsules containing 30 mg of COMPOUND free base equivalent plus the following excipients: lactose SYNOPSIS OF STUDY monohydrate, croscarmellose sodium, colloidal silicon dioxide, and magnesium stearate.
- Placebo REFERENCE DRUG/ Placebo was supplied as capsules that were identical in BATCH NO(S). appearance to those containing active drug. Placebo capsules contained no COMPOUND but were otherwise identical in composition to those containing active drug.
- DOSE/ROUTE/REGIM Oral COMPOUND as 30 mg capsules in total doses ranging EN/ DURATION from 0 to 180 mg per day, for 30 days (see Table in Study Design section).
- EFFICACY RA and diagnosis of active RA [according to the American College of Rheumatology (ACR) response criteria], ACR 20 response consisting of: tender and swollen joint count, Health Assessment Questionnaire (HAQ), visual analog scale for pain, patient global assessment, physician global assessment; C-reactive protein.
- ACR 20 response consisting of: tender and swollen joint count, Health Assessment Questionnaire (HAQ), visual analog scale for pain, patient global assessment, physician global assessment; C-reactive protein.
- HAQ Health Assessment Questionnaire
- PHARMACOKINETICS Plasma concentrations for COMPOUND and its metabolites were assessed at selected time points. In treatment period 4, plasma concentrations of methotrexate and its 7-hydroxy metabolite were assessed with and without COMPOUND coadministration in subjects who volunteered for this procedure.
- TNF- ⁇ tumor necrosis factor alpha
- PHARMACODYNAMICS interleukin-l ⁇ (IL- l ⁇ ) were measured.
- SAFETY Physical examination, medical history, vital signs, orthostatic vital signs, chest radiograph, 12-lead electrocardiogram (ECG), clinical laboratory evaluations (including serum chemistry, hematology, qualitative urinalysis, and liver function tests), purified protein derivative test for tuberculosis, neurological tests, adverse events, and concomitant medications.
- Subjects in the 30 mg tid and 90 mg qd groups showed trends for the greatest rate of response to treatment according to the ACR20 criteria.
- the response in the 30 mg tid group peaked early (Day 8) with 50% of subjects in that group responding; the level of improvement was fairly well maintained throughout the 30-day treatment period.
- the response in the 90 mg qd group gradually increased over time to a maximum response (53% responders) at Day 30.
- the maximum response in the placebo group was 23% responders at Day 30.
- Plasma concentrations of COMPOUND assessed in all subjects, increased with increasing doses and declined in parallel across all groups.
- MTX and 7-OH-MTX concentrations assessed in a small subset of subjects, were approximately 20% larger when the patients received MTX concomitantly with COMPOUND versus MTX alone.
- composition of the invention used in this study was a novel, orally active chemical entity with anti-inflammatory and arthritis disease modifying properties. It is an inhibitor of p38 ⁇ mitogen activated protein kinase (MAPK), an intracellular enzyme that mediates cellular responses to inflammatory stimuli. Activation of p38 ⁇ MAPK in acute and chronic inflammatory states leads to the production of proinflammatory mediators, such as interleukin (IL)- l ⁇ , tumor necrosis factor (TNF) ⁇ , and prostaglandin E2 (PGE2).
- IL interleukin
- TNF tumor necrosis factor
- PGE2 prostaglandin E2
- Compound blocks the synthesis and activity of TNF- ⁇ , and the synthesis of IL- 1 ⁇ and cyclooxygenase (COX)-2, a key inducible enzyme involved in the synthesis of inflammatory PGE2. Compound reduced signs and symptoms in preclinical models of acute inflammation.
- RA rheumatoid arthritis
- TNF- ⁇ rheumatoid arthritis
- IL-I ⁇ IL-I ⁇
- COX-2 rheumatoid arthritis
- the second Phase 1 trial was a placebo-controlled, ascending multiple-dose study in healthy subjects receiving treatment with Compound for 10 consecutive days.
- Compound was well tolerated at doses of 1 mg/kg once daily (qd), twice daily (bid), and three times daily (tid), but was less well tolerated at the highest dose examined, 2 mg/kg bid.
- the most frequently reported drug-related AE was mild, transient dizziness that occurred with increased incidence at the 2 mg/kg bid dose level; frequency of recurrence decreased markedly after the first five days of dosing.
- One subject in the highest dose group discontinued dosing because of alanine aminotransferase (ALT) levels elevated to three times the upper limit of normal (ULN).
- ALT alanine aminotransferase
- the primary objective of this study was to assess the safety and tolerability of multiple oral doses of Compound in patients with active RA who were also receiving stable doses of MTX. Secondary objectives included: To assess the efficacy of multiple oral doses of Compound using the American College of Rheumatology (ACR) response criteria; To determine the PK of multiple oral doses of Compound in patients with active RA who are also receiving MTX; To assess the effects of multiple oral doses of Compound on TNFct and IL- l ⁇ levels in patients with active RA who are also receiving MTX. Materials and Methods:
- Study Design This multi-center, randomized, double-blind, placebo-controlled, dose- escalating study assessed the safety, tolerability, efficacy, PK, and pharmacodynamics of Compound in patients with active RA who were also receiving MTX.
- a total of 132 subjects were randomized during one of four treatment periods.
- Subjects were assigned to one of seven treatment groups, depending on the treatment period during which they were randomized, with the total daily dose of Compound ranging from 0 to 180 mg over the course of the study. Dosing of the groups was staggered over four treatment periods, with placebo group assignments for each period.
- Safety and available PK data from the previous treatment period were reviewed before initiating higher dose regimens in the next treatment period.
- Subjects received study drug for 30 consecutive days and were followed for an additional 28 days after treatment. Subjects were not allowed to re-enroll for participation in a second treatment period.
- Study drug was supplied in the form of capsules (30 mg of
- Each blister card was configured with 7 or 21 wells with each well containing two or three capsules. Each well contained a combination of active study drug and placebo that was determined by the treatment to which the subject was randomized. Five cards were packaged per study drug kit. One study drug kit was supplied to each subject. Individual study drug kits and blister cards were labeled with a unique identifier number and subject number.
- Study Drug Subjects received study drug as oral capsules containing either 30 mg Compound or placebo. Doses were self-administered except during all scheduled visits, when the dose was administered at the site by the site staff after the appropriate blood samples were obtained.
- the dose regimen was two capsules once a day in the morning.
- the regimen was three capsules three times a day, approximately 4 to 6 hours apart.
- the regimen was two capsules once a day in the morning on Days 1-7, two capsules twice a day approximately 10 hours apart on Days 8-14, and two capsules three times daily, approximately 4 to 6 hours apart on Days 15- 30.
- Study drug was taken at the assigned regimen for Days 2-29 inclusive.
- Treatment Period 1 subjects were randomized to 30 mg qd, 60 mg qd, or placebo.
- Treatment Period 2 subjects were randomized to 90 mg qd, 30 mg tid, or placebo.
- subjects were randomized to 60 mg tid or placebo. During Treatment Period 3, subjects were randomized to 60 mg tid or placebo. During Treatment Period 3, subjects were randomized to 60 mg tid or placebo. During Treatment Period 3, subjects were randomized to 60 mg tid or placebo. During Treatment Period 3, subjects were randomized to 60 mg tid or placebo. During Treatment Period 3, subjects were randomized to 60 mg tid or placebo. During Treatment Period 3, subjects were randomized to 60 mg tid or placebo. During Treatment Period 3, subjects were randomized to 60 mg tid or placebo. During Treatment Period 3, subjects were randomized to 60 mg tid or placebo. During Treatment Period 3, subjects were randomized to 60 mg tid or placebo. During Treatment Period 3, subjects were randomized to 60 mg tid or placebo. During Treatment Period 3, subjects were randomized to 60 mg tid or placebo. During Treatment Period 3, subjects were randomized to 60 mg tid or placebo. During Treatment Period 3, subjects were randomized to 60 mg
- Treatment Period 4 subjects were randomized to 60 mg qbt or placebo. Each of the four consecutive treatment periods was comprised of a separate cohort of subjects; subjects were not allowed to re-enroll for a second treatment period. Study Assessments and Procedures
- Schedule of Assessments Clinical assessments were performed at baseline, at regularly scheduled intervals during the study, and post-treatment for the safety, efficacy, and pharmacokinetic parameters described in the following sections (see Figure 9, which provides a summary of the Schedule of Assessments, as discussed herein). Concomitant medications and AEs were monitored and recorded throughout the study and the follow-up period. All study-specific procedures and the results of all study evaluations were recorded in the subject's medical record and on the appropriate CRF.
- Subjects participated in the following procedures at the screening visit: signed informed consent form (ICF), medical history (including medications within the previous 14 days), physical examination, vital signs, 12-lead ECG, confirmation of RA diagnosis and of active RA, and clinical laboratory evaluations (including urinalysis, hematology, and biochemistry), chest radiograph, hepatitis panel, pregnancy test, and purified protein derivative test for tuberculosis.
- ICF informed consent form
- medical history including medications within the previous 14 days
- physical examination including vital signs, 12-lead ECG, confirmation of RA diagnosis and of active RA, and clinical laboratory evaluations (including urinalysis, hematology, and biochemistry), chest radiograph, hepatitis panel, pregnancy test, and purified protein derivative test for tuberculosis.
- ICF informed consent form
- medical history including medications within the previous 14 days
- vital signs including 12-lead ECG, confirmation of RA diagnosis and of active RA
- clinical laboratory evaluations including urinalysis, he
- Visits on Days 2, 31, and 32 were determined by the requirement for a PK blood draw as indicated by Schedule I or Il in Protocol Appendix Ib (provided in Appendix 6). 10 On Study Days 1 and 30 in Treatment Periods 2, 3, and 4, subjects were to receive only the First morning dose to allow for PK blood collections, "in a subset of subjects for MTX profiling; samples were taken on the subject's usual day of MTX dosing. 12 Collected only serious AEs since last visit. Study Procedures
- ACR Response The ACR response consisted of the following individual components: 68-count tender/painful joint count, 66-count swollen joint count, physician global assessment, patient global assessment, visual analog scale for pain, HAQ, and a measure of acute phase reactants (specifically, CRP). The Sponsor performed the scoring of the ACR response.
- Tender/swollen joint count The investigator determined the number of painful or tender joints (68 joints) and the number of swollen joints (66 joints), and assessed each joint for tenderness, pain, and swelling (Appendix 7 of the Protocol, provided in Appendix 6).
- HAQ This questionnaire was self-administered by the subject; the coordinator ensured that each question on the questionnaire had a response (Appendix 6 of the Protocol, provided in Appendix 6).
- Visual analog scale for pain This assessment was done using a 10-cm horizontal visual analog scale. The subject placed a mark on the horizontal line indicating the severity of pain, with the left end of the line representing no pain and the right end representing unbearable pain. Site staff measured the length of the line using a standard ruler.
- Patient global assessment This assessment was done used a 10-cm horizontal visual analog scale. The subject was asked to answer the question "Considering all ways arthritis affects you, how well are you doing today?" by placing a mark on the horizontal line, with the left end of the line representing very well and the right end representing very poor. Site staff measured the length of the line using a standard ruler.
- Physician global assessment This assessment was done using a 10-cm horizontal visual analog scale. The investigator placed a mark on the horizontal line indicating his/her assessment of the subject's current disease activity, with the left end of the line representing no disease activity and the right end representing maximum disease activity. Site staff measured the length of the line using a standard ruler.
- Clinical Laboratory Assessments Clinical laboratory evaluations, performed at screening and on Days 1, 8, 15, 22, 30, and
- Plasma COMPOUND and metabolites concentrations were determined using a validated LC/MS/MS method (Appendix 3.1). Blood samples for PK analyses were drawn at regularly scheduled intervals. Pharmacokinetic sampling on Days 1 and 30 consisted of two different schedules: subjects on Schedule I provided samples at 0 (pre-dose), 0.5, 2, and 8 hours post-dose on Day 1 and at 0, 1 , 4, and 22-24 hours on Day 30. In a reverse schedule, subjects in Schedule II gave samples at 0, 1, 4, and 22—24 hours on Day 1, and at 0, 0.5, 2, and 8 hours post-dose on Day 30 (Appendix Ib of Protocol, provided in Appendix 6). Each site was assigned to either Schedule I or II. Assignment of a PK sampling schedule could be adjusted with agreement of the investigator and the Sponsor.
- Plasma samples for PK analysis of COMPOUND levels were collected via an indwelling catheter and/or via direct venipuncture, using 7-mL-draw green-top Vacutainer ® collection tubes containing sodium heparin solution. Samples were centrifuged at 3,000 rpm in a refrigerated centrifuge (4°C) for 15 minutes. (If the site did not have access to a refrigerated centrifuge, the sample was to be stored on crushed ice for at least 30 minutes, but no longer than 1 hour before centrifugation.) The separated plasma was then aliquotted into three suitably labeled, 5-mL polypropylene tubes provided by the Sponsor. Plasma samples were frozen within 1 hour of collection and stored at -20 0 C or, if available, at -70 0 C.
- Plasma concentrations of COMPOUND and its metabolites were measured by the Sponsor using validated analytical procedures.
- IL-I ⁇ was performed by the Mayo Central Laboratory for Clinical Trials (Rochester, MN). Blood samples were collected via an indwelling catheter and/or via direct venipuncture, using 7- mL red-top collection tubes. Blood samples were allowed to clot for 15 to 30 minutes at room temperature and subsequently centrifuged at 3,000 rpm for 10 minutes. The separated serum was aliquotted into separate tubes for analysis of TNF- ⁇ and IL- l ⁇ and stored frozen until shipping. A 2-mL archival serum sample was also collected for exploratory analyses of other biomarkers of inflammation. Evaluation of these additional biomarkers is currently underway; results of these analyses will be summarized in a separate report.
- Methotrexate Assessments (in a Subset of Subjects): Blood samples from a subset of subjects were drawn on the usual day of the subject's MTX dosing during the last week of study drug administration (between Days 22 and 30, inclusive; i.e., first MTX profiling). These subjects had a second MTX profiling performed on the usual day of MTX dosing between 7 and 21 days after the last day of study drug administration. Subjects were to bring their weekly oral MTX dose (in its original bottle, if possible) with them to the site on each of these visits. On the first visit for MTX profiling during the last week of study drug administration, subjects were to also bring their full daily doses of study drug with them to the clinic. Subjects had the option to have an indwelling catheter to facilitate blood draws. Measurement of MTX and 7-OH-MTX concentrations were performed using a validated HPLC method by Advion Biosciences (Appendix 3.2).
- the primary efficacy endpoint of the study was the rate of
- ACR20 response defined as a 20% reduction in tender and swollen joint counts and 20% improvement in three of the five remaining ACR core set measures: patient and physician global assessments, visual analog scale for pain, HAQ, and CRP. Secondary Efficacy Endpoints: Secondary efficacy endpoints of the study were as follows: Rate of ACR50 response (a 50% reduction/improvement, using the same assessments used for ACR20).
- IL-I ⁇ levels was assessed as a secondary objective. Percent change and change from baseline for the two cytokines at the Day 8, 30, and 58 visits were the endpoints measured. Efficacy Analysis
- Secondary efficacy variables consisted of the proportion of subjects exhibiting improvement according to the ACR50 response criteria and all individual items of ACR response assessments (number of tender joints, number of swollen joints, physician's assessment, subject's assessment, pain VAS, HAQ, and CRP). For each ACR component, actual value, change from baseline, and percentage change from baseline were summarized for each visit.
- Interim efficacy analyses were performed for each of Treatment Periods 1—3 after the last subject in the treatment period completed study procedures through Day 31. Efficacy variables were descriptively analyzed. Data were listed and/or summarized by treatment group but not by subject number. Sponsor personnel responsible for data management and analysis (Associate Director, Biostatistics; Associate Director, Clinical Information Systems; and Senior Clinical Applications Programmer) were unblinded to individual subject numbers and treatment assignments. Sponsor personnel responsible for monitoring the study remained blinded to individual subject numbers and treatment assignments.
- ACR20 Response The 30 mg tid and 90 mg qd groups showed a greater rate of response to treatment and a greater level of improvement, according to the ACR20 criteria, at all time points compared to subjects in the placebo group (
- the 30 mg qd and 60 mg qd dose groups showed modest improvement relative to placebo, except at Day 30, when responses were similar to that of placebo.
- the greatest improvement in the ACR20 response was at Day 30; until that time, responses generally were similar to placebo.
- Subjects in the 30 mg tid group showed the most consistent improvement at the level of ACR20.
- ACR20 response was greater than placebo at all time points.
- Figure 11 illustrates ACR20 Responders by Treatment Group and Study Day — Efficacy Population.
- Figure 12 illustrates individual ACR Response Components on Day 30 by Treatment Group — Efficacy Population.
- ACR50 Response Subjects in the 30 mg tid group showed the most consistent ACR50 improvement, with peak improvement in this group at Days 15 and 30 (25% of subjects improved at each time point) ( Table IV For all COMPOUND treatment groups combined, ACR50 response was greater than placebo at all time points through the Day 58 assessment (28 days after the end of study drug treatment, 4% ACR50 response in the placebo group versus 10% in the combined COMPOUND treatment groups).
- Individual Components of the ACR Response Criteria The ACR response criteria consist of the following individual components: number of tender joints, number of swollen joints, physician's assessment, subject's assessment, pain VAS, HAQ, and CRP. Each variable was assessed as a secondary efficacy endpoint.
- Tender Joint Count Median baseline values for tender joint count (TJC) were similar among treatment groups (range, 21.5 to 31.0; maximum possible count, 68). All treatment groups, including placebo, experienced an improvement in TJC (Table 2). At Days 8 and 15, all COMPOUND groups had a greater reduction in TJC compared to placebo as assessed by median percentage change from baseline (refer to Appendix 1 , Table 10.1.1). The median percentage improvement from baseline was consistently better than placebo for the 60 mg qd, 90 mg qd, 30 mg tid, and 60 mg qbt groups (Table 2).
- Swollen Joint Count Median baseline values for swollen joint count (SJC) were similar among treatment groups (range, 13.0 to 22.0; maximum possible count, 66). All groups, including the placebo group, had a decrease in median swollen joint count from baseline at all time points. The median percentage improvement from baseline was consistently better than placebo on all test days for the 90 mg qd and 60 mg qbt groups, and on all test days except Day 58 for the 30 mg tid and 60 mg tid groups. Table 3: Swollen Joint Count — Median Percentage Change from Baseline (Efficacy Population)
- Physician's Assessment Median baseline values for the Physician's Assessment were similar between treatment groups (range, 57.0 to 67.5; range of possible values, 0 to 100).
- Table 4 which lists the median percentage change from baseline for each time point, which depicts median percentage change from baseline on Day 30, show that all groups, including placebo, showed some improvement with this assessment. For the majority of treatment groups, the peak response occurred at Day 30.
- Table 4 Physician and Subject Global Assessment — Median Percentage Change from Baseline (Efficacy Population) Placebo 30 mg qd 60 mg qd 90 mg qd 30 mg tid 60 mg tid 60 mg qbt
- Subject's Assessment Median baseline values for the Subject's Assessment were similar among treatment groups (range, 42.5 to 65.0; range of possible values, 0 to 100).
- Table 4 which lists the median percentage change from baseline for each time point, which depicts median percentage change from baseline on Day 30, show that most groups, including placebo, showed some improvement in this assessment. However, these changes were generally small.
- Pain Visual Analog Scale Median baseline values for the pain VAS were similar among treatment groups (range, 42.0 to 60.0; range of possible values, 0 to 100).
- Table 5 which lists the median percentage change from baseline for each time point, shows that all groups, including placebo, experienced at least modest relief from pain with treatment. Maximal improvement in pain VAS scores for most of the active groups were generally similar to that of placebo, with the 60 mg qd and 30 mg tid groups showing a moderately greater improvement over other treatment groups.
- Table 6 which lists the median percentage change from baseline for each time point, shows that active groups generally responded better than placebo with respect to this assessment. Only the 60 mg tid group failed to perform better than placebo. The 30 mg tid group showed some degree of improvement over placebo in median percentage change from baseline HAQ at all time points through Day 58. Similarly, the 60 mg qbt group performed better than placebo on all test days after Day 8.
- Table 6 Health Assessment Questionnaire — Median Percentage Change from Baseline (Efficacy Population)
- C-Reactive Protein Median baseline CRP values were similar among treatment groups (range, 0.4 to 1.3; normal range, 0.02 to 0.8 mg/dL). The median change from baseline in CRP was generally greatest at Day 8 or Day 15; on both of these test days, all groups displayed greater median percentage reduction from baseline compared to placebo. However, the changes in all groups declined by Day 30, with the exception of the 60 mg qbt group, which displayed its greatest decrease at this time point.
- the 30 mg tid and 60 mg qbt groups also consistently performed better than placebo throughout the treatment period, with a maximum decrease of 1.1 on Day 15 for the 60 mg qbt group and a maximum decrease of 0.9 on Day 8 for the 30 mg tid group.
- a greater proportion of subjects in all active groups fell into the highest category of improvement in the DAS28 score (> 1.2) compared to the placebo group, with the 90 mg qd, 30 mg tid, and 60 mg qbt groups exhibiting particularly improved responses over placebo at this time point (43%, 38%, and 38%, respectively, compared to 20% in the placebo group)
- Mean plasma concentration of COMPOUND over time during the 48 hours after administration of the last dose of study drug on Day 30 is plotted on a log-linear scale for each treatment group. Error bars are not included in the graph for clarity; CV% generally ranged between 30% and 100%, with greater error occurring at the latter two time points.
- COMPOUND plasma concentrations of COMPOUND generally increased with increasing daily dose of COMPOUND . After maximum concentrations were achieved, COMPOUND concentrations declined in parallel across all dose groups. COMPOUND plasma concentrations for all subjects receiving placebo were below the level of quantitation
- TNF- ⁇ Due to high intra-group variability, most notably within the 60 mg qbt group, median TNF- ⁇ values and median changes in TNF- ⁇ levels from baseline best summarize the trends in TNF- ⁇ levels during treatment. Overall, the median change in TNF- ⁇ levels at each time point was small for each treatment group. A majority of subjects in all treatment groups exhibited a small decrease in TNF- ⁇ levels from baseline to Day 8, as illustrated by a negative median percent change in TNF- ⁇ levels for all treatment groups at this time point (
- TNF- ⁇ Median Percentage Change from Baseline (Efficacy Population)
- the 30 mg qd and 30 mg tid groups each experienced a 100% decrease in IL-Ib levels on Day 8, with levels increasing to near-baseline levels by Day 30.
- the 90 mg qd group experienced a decrease of the same magnitude at the post-treatment assessment on Day 58.
- Safety results are summarized descriptively for all subjects who received at least one dose of study drug. Overall, safety results indicate that COMPOUND was well tolerated at all doses compared with placebo.
- the upper range of the cumulative dose received in the 30 mg qd and 90 mg qd groups exceeded that expected for a 30-day treatment period. This occurred because eight subjects were permitted to receive study drug beyond the 30-day treatment period. These exceptions were made to accommodate scheduling problems with respect to the Day 30 visit. For example, one subject (30 mg qd) missed the visit on the last day of dosing due to a viral syndrome, and was therefore permitted to continue dosing until she could make the last visit (34 total days).
- the cumulative dose for the latter subject did not exceed the expected cumulative dose for a 30-day treatment with 90 mg qd since this subject had to temporarily discontinue study drug from Day 12 to Day 21 due to an AE.
- COMPOUND assessed in all subjects, increased with increasing doses and declined in parallel across all groups.
- MTX and 7-OH-MTX concentrations assessed in a small subset of subjects, were approximately 20% larger when the patients received MTX concomitantly with COMPOUND versus MTX alone.
- COMPOUND is generally well tolerated. With the exception of elevations in liver transaminases, there were no clinically significant, drug-related changes in laboratory values and no clear trend indicating a dose-relationship in the incidence of AEs. Of the ten subjects with AEs related to abnormal liver function tests, seven were in one of the two highest dose groups. These data suggest that the 60 mg tid dose regimen may be the highest tolerated dose level for rheumatoid arthritis patients receiving concomitant MTX, a patient population particularly susceptible to elevations in liver transaminases.
- IR capsules were also developed for clinical studies.
- Formulation development included an excipient compatibility study to identify suitable excipients in several functional categories and a feasibility evaluation of formulations and processes including wet granulation with high shear mixing, direct blending, and dry granulation with slugging. Trial lots were evaluated for content uniformity and powder flow. Optimal uniformity and flow for capsule filling were obtained with the dry granulation process. Below is a description of the capsules and manufacturing process and that was developed.
- the components are dry granulated together by "slugging" in a press and then milled to an appropriately sized granulation by an oscillating mill.
- the milled material is blended to ensure uniformity before the encapsulation process.
- the dog was used as a model species to evaluate the effect of dosage form on the pharmacokinetics of Compound.
- a study group of 6 animals (3 male, 3 female) was tested. Single doses, either as IR capsules (multiples of 30-mg capsules), IR tablets (30 or 90-mg) and ER tablets (60 or 120-mg, two formulation types) were administered in the fed state; fasted state effects were evaluated for a smaller group of formulations.
- the study demonstrated that (1) AUC (area-under-the-curve, exposure, bioavailability) in the fed state is dose-proportional and independent of dosage form, (2) extended-release formulations blunt Cmax (maximum concentration), and (3) extended-release formulations prolong tmax (time of maximum concentration).
- one embodiment of the invention is to provide a dosing strategy for the Compound which will result in Cmax levels less than 2000 ng/mL yet still achieve pharmacologically effective levels in the blood.
- This objective can be accomplished through the use of an extended or sustained release formulation of the Compound such as those that are provided herein.
Abstract
The invention is directed to pharmaceutical compositions comprising the compound and methods of use of said compositions for the treatment of conditions mediated by p38 MAP kinase. The invention provides formulations and dosage schedules, and it also provides methods to use these compounds in conjunction with other therapies commonly used for inflammatory diseases.
Description
PHARMACEUTICAL FORMULATIONS OF AN INDOLE-TYPE DERIVATIVE AND
RELATED METHODS OF USE
FIELD OF THE INVENTION
The invention relates to dosage forms and methods of using (2R-trans)-6-chloro-5-[[4- [(4-fluorophenyl)methyl]-2,5-dimethyl-l-piperazinyl]carbonyl]-N,N, 1-trimethyl-alpha-oxo-lH- indole-3-acetamide. In particular, the invention relates to dosage forms, methods, and novel pharmaceutical compositions.
BACKGROUND
Substituted indole based compounds have been described in US Pat. 6,864,260 to Mavunkel, et al (incorporated herein by reference) as useful for inhibiting the activity of p38 MAP kinase. P38 MAP kinase has been identified as a drug target given its role in mediating the intracellular response to pro-inflammatory stress. Inadequate regulation of p38 kinase has been associated with inflammation related disorders including but not limited to rheumatoid arthritis, osteoarthritis, inflammatory bowel disease, asthma, psoriasis, and congestive heart failure.
A method for treating or preventing pain has been described in US Patent application serial no. 10/655,745 to Protter, et al (incorporated herein by reference) by administering a compound of the formula:

and the pharmaceutically acceptable salts thereof, or a pharmaceutical composition thereof, wherein
x> represents a single or double bond; one Z2 is CA or CR8A and the other is CR1, CRl 2, NR6 or N wherein each R1, R6 and R8 is independently hydrogen or noninterfering substituent;
A is -CO(X)jY wherein Y is COR2 or an isostere thereof and R2 is hydrogen or a noninterfering substituent, X is a spacer of 2-6 A, and j is 0 or 1; Z3 is NR7 or O; each R3 is independently a noninterfering substituent; n is 0-3; each of L1 and L2 is a linker; each R4 is independently a noninterfering substituent; m is 0-4;
Z1 is CR5 or N wherein R5 is hydrogen or a noninterfering substituent; each of 1 and k is an integer from 0-2 wherein the sum of 1 and k is 0-3; Ar is an aryl group substituted with 0-5 noninterfering substituents, wherein two noninterfering substituents can form a fused ring; and the distance between the atom of Ar linked to L2 and the center of the α ring is 4.5-24 A
While indole based compounds have been identified as useful antagonists of p38 kinase, an indole typed compound, and proper dosage form thereof, with properties suitable for use as a pharmaceutical in humans has yet to be particularly identified and developed.
SUMMARY OF THE INVENTION
In one aspect, the invention is directed to methods and compounds useful in treating conditions that are characterized by enhanced p38-α activity. These conditions include inflammation (e.g., rheumatoid arthritis), proliferative diseases, and certain cardiovascular disorders, as further described below.
In one aspect, the invention provides methods to treat a condition mediated by p38-α kinase comprising administering to a subject in need of such treatment a compound of the invention. In one aspect, a condition ameliorated, treated or prevented by a composition of the invention, or practicing the methods of the invention, is a proinflammation response.
The invention provides formulations, e.g., liquid, solid, powder, aerosol, spray, and the like (including solid oral formulations), and methods of using them, comprising a therapeutically effective amount of a compound of the invention, or pharmaceutically acceptable salts thereof (e.g., a hydrochloride salt), and a pharmaceutically acceptable excipient.
In one aspect, a formulation, e.g., a solid oral formulation, of the invention comprises a pharmaceutically acceptable excipient such as lactose, Avicel, Prosolv, pregelatinized starch,
HPMC, NaCMC, croscarmellose sodium (in low amounts), sodium starch glycolate, magnesium stearate, stearic acid or a combination thereof.
In one aspect, a formulation of the invention, including liquid, solid, powder, aerosol, spray formulations (including solid oral formulations) comprises the compound having the formula:

In one aspect, a formulation of the invention, e.g., a solid oral formulation, is formulated as a hydrochloride salt. In one aspect, a compound of the invention is formulated for extended release with an excipient comprising calcium phosphate dibasic, povidone, sodium lauryl sulfate or a combination thereof. In one aspect, a compound of the invention, e.g., a solid oral formulation, is formulated as a powder, a tablet, a pill or a capsule, or included in an aerosol or spray.
The invention provides pharmaceutical compositions (and methods of using them) comprising a therapeutically effective amount of the compound or pharmaceutically acceptable salts thereof (e.g., a hydrochloride salt), and a pharmaceutically acceptable excipient. In one aspect, the compound is formulated with a pharmaceutically acceptable excipient as set forth in Table 2.1 , Table 2.2, Table 2.3 or Table 3.1. The invention provides pharmaceutical composition comprising a therapeutically effective amount of the compound as set forth in any one of claims 1 to 35, or pharmaceutically acceptable salts or pharmaceutically acceptable forms thereof, wherein the compound is formulated as an immediate release tablet with a pharmaceutically acceptable excipient as set forth in Table 3.1 or Table 3.2.
In one aspect, a compound of the invention is formulated with a pharmaceutically acceptable excipient comprising lactose anhydrous microcrystalline cellulose, crosslinked cellulose, a starch, colloidal silicon dioxide, magnesium stearate or a combination thereof, wherein in one aspect the starch comprises partially pre-gelatinized starch, and in one aspect the crosslinked cellulose comprises carboxymethyl cellulose.
The invention also provides methods for treating, ameliorating, preventing or delaying disease progression in an inflammatory disease or condition, or a disease or condition having an inflammatory component, or ameliorating the inflammatory disease or condition or disease or
condition having an inflammatory component, comprising administering to a subject in need of such treatment a compound of the invention, a pharmaceutical formulation comprising the compound or a pharmaceutically acceptable salt thereof (e.g., a hydrochloride salt), a therapeutically effective amount of a compound of the invention. In one aspect, the inflammatory disease or condition, or the disease or condition having an inflammatory component is arthritis, and in one aspect the arthritis is rheumatoid arthritis (RA).
In one aspect, the compound, pharmaceutical formulation or pharmaceutically acceptable salt is administered at doses of 1 mg/kg once daily (qd), twice daily (bid), and three times daily (tid). In one aspect, the compound, pharmaceutical formulation or pharmaceutically acceptable salt is administered at single-doses of between about 0.25, 0.5, 0.75, 1.0 and 5, 6, 7, 8, 9, or 10 or more mg/kg, wherein in one aspect the dose levels are 1 , 2, 3, 4 or 5 or more mg/kg dose levels. In one aspect, the compound, pharmaceutical formulation or pharmaceutically acceptable salt is administered in dosages of between about 1 and 3, 4 or 5 or more mg/kg once daily (qd), about 1 and 3 mg/kg twice daily (bid), or about 1 and 3 mg/kg three times daily (tid). In one aspect, the compound, pharmaceutical formulation or pharmaceutically acceptable salt is administered in dosages of 1, 2, 3, 4 or 5 or more mg/kg once daily (qd), 1, 2, 3, 4 or 5 or more mg/kg twice daily (bid), or 1 , 2, 3, 4 or 5 or more mg/kg three times daily (tid). In one aspect, the compound, pharmaceutical formulation or pharmaceutically acceptable salt is administered in dosages of between about 10 mg and 120 mg single dosages once daily (qd). twice daily (bid), or three times daily (tid). In one aspect, the compound, pharmaceutical formulation or pharmaceutically acceptable salt is administered in dosages of about 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110 or 120 or more mg single dosages once daily (qd), twice daily (bid), or three times daily (tid).
The details of one or more embodiments of the invention are set forth in the accompanying drawings and the description below. Other features, objects, and advantages of the invention will be apparent from the description and drawings, and from the claims. All publications, patents and patent applications cited herein are hereby expressly incorporated by reference for all purposes.
BRIEF DESCRIPTION OF THE DRAWINGS
The following drawings are illustrative of aspects of the invention and are not meant to limit the scope of the invention as encompassed by the claims.
Figure 1 illustrates the manufacture of the Immediate Release (IR) tablet in Flow diagram, as described in detail Example 2, below.
Figure 2 illustrates Table 3.4, which describes exemplary formulations of the invention, as described in detail Example 2, below.
Figure 3 illustrates Table 3.5, summarizing data for exemplary formulations (tablets) of the invention, as described in detail Example 2, below.
Figure 4 illustrates Table 3.6, a characterization of exemplary formulations (tablets) of the invention, as described in detail Example 2, below.
Figure 5 illustrates a summary of data for pre-milled granulation, milled granulation and final blend, as described in detail Example 2, below. Figure 6 illustrates a summary of data for stability studies for the compound IR tablets at
30 and 90 mg, as described in detail Example 2, below.
Figure 7 is an illustration of an exemplary manufacturing process of the invention: a flow chart of a manufacturing process for exemplary Extended Release (ER) formulations of the invention, as described in detail Example 2, below. Figure 8 summarizes data regarding exemplary formulations of the invention, as described in detail Example 2, below.
Figure 9, which provides a summary of the Schedule of Assessments, as described in detail Example 2, below.
Figure 10 illustrates a summary of the disposition of subjects of the clinical study described in detail Example 2, below.
Figure 1 1 and Figure 12 illustrate a summary of treatment group responses in the clinical study described in detail Example 2, below.
Figure 13 is a chart showing the pK effect of the compound in fasted males.
Figure 14 illustrates the correlation of an oral dosage formulation of the compound relative to ex vivo TNFα as measured in blood plasma post dosing.
Figure 15 illustrates the correlation of an oral dosage formulation of the compound relative to ex vivo TNFα as measured in blood plasma post dosing.
Figure 16 provides the plasma profile of Compound following administration of a single 60-mg IR capsule dose in the dog (n=3 each, male and female).
Figure 17 provides the plasma profile of Compound following administration of a single
60-mg ER tablet dose in the dog (n=3 each, male and female)
Figure 18 provides the plasma profile of Compound following administration of a single
120-mg IR capsule dose in the dog (n=3 each, male and female)
Figure 19 provides the plasma profile of Compound following administration of a single 30-mg IR tablet dose in the dog (n=3 each, male and female)
Figure 20 provides the plasma profile of Compound following administration of a single 90-mg IR tablet dose in the dog (n=3 each, male and female)
Figure 21 provides the plasma profile of Compound following administration of a single 60-mg ER tablet dose in the dog (n=3 each, male and female)
Figure 22 provides the plasma profile of Compound following administration of a single 120-mg ER tablet dose in the dog (n=3 each, male and female)
Figure 23 provides the plasma profile of Compound following administration of a single
120-mg ER tablet dose in the dog (n=3 each, male and female)
Figure 24 illustrates the effect of Compound dosage form on tmax in the dog.
Figure 25 illustrates the effect of Compound dosage form on exposure in the dog (doses are normalized)
Figure 26 illustrates the effect of Compound dosage form on Cmax in the dog.
Figures 27 and 28 are charts providing correlative data in human subjects between relative Cmax levels of the Compound and reported episodes of dizziness.
DETAILED DESCRIPTION OF THE INVENTION
(2R-trans)-6-chloro-5-[[4-[(4-fluorophenyl)methyl]-2,5-dimethyl-l- piperazinyl]carbonyl]-N,N, l-trimethyl-alpha-oxo-lH-indole-3-acetaniide has been identified as being useful as a medicament suitable for pharmaceutical administration. Represented by the formula

this molecule exhibits a specific conformational shape and rigidity that enables it to uniquely interact with p38 kinase, the alpha isoform in particular, making it unusually selective and effective at modulating enzymatic activity.
In recent studies a crystallization of (2R-trans)-6-chloro-5-[[4-[(4-fluorophenyl)methyl]- 2,5-dimethyl-l-piperazinyl]carbonyl]-N,N, l-trimethyl-alpha-oxo-lH-indole-3-acetamide bound to the alpha isoform of p38 kinase was resolved. Given the unique binding properties between this small molecule and the drug target, the Compound has demonstrated unique and remarkable efficacy in pro-inflammatory disease models such as arthritis.
In certain studies, humans have safely received doses of the Compound. From these studies, it has been demonstrated that the Compound is orally bioavailable and well tolerated at dose levels that suggest efficacy for certain inflammatory disorders and/or achieve pharmacologically active concentrations in the blood.
The invention will now be described in more detail below. All references cited herein are incorporated herein by reference in their entirety and for all purposes to the same extent as if each individual publication or patent or patent application was specifically and individually indicated to be incorporated by reference in its entirety for all purposes.
DEFINITIONS
The present invention is best understood by reference to the following definitions, the drawings and exemplary disclosure provided herein.
"Administering" or "administration" means providing a drug to a patient in a manner that is pharmacologically useful.
"Apparent terminal half-life" (t/, ) is calculated as 0.693/k, wherein "k" means the apparent elimination rate constant, estimated by linear regression of the log-transformed plasma concentration during the terminal log-linear elimination phase.
"Area under the curve" or "AUC" is the area as measured under a plasma drug concentration curve. Often, the AUC is specified in terms of the time interval across which the plasma drug concentration curve is being integrated, for instance AUCstan-nnish- Thus, AUCo-48 refers to the AUC obtained from integrating the plasma concentration curve over a period of zero to 48 hours, where zero is conventionally the time of administration of the drug or dosage form comprising the drug to a patient. AUQ refers to area under the plasma concentration curve from hour 0 to the last detectable concentration at time t, calculated by the trapezoidal rule.
AUCjnf refers to the AUC value extrapolated to infinity, calculated as the sum of AUC1 and the area extrapolated to infinity, calculated by the concentration at time t (Ct) divided by k. (If the ti/, value was not estimable for a subject, the mean t>/, value of that treatment was used to calculate AUQnf.). "Mean, single dose, area under a plasma concentration-time curve AUC,nf" means the mean AU Cmf obtained over several patients or multiple administrations to the same patient on different occasions with sufficient washout in between dosings to allow drug levels to subside to pre-dose levels, etc., following a single administration of a dosage form to each patient.
"Ascending plasma concentration" means a drug plasma concentration profile over about the first 12 to 24 hours following initial dosing, wherein the profile shows an increase to a maximum concentration, wherein said maximum occurs more than about 6 hours following the initial dose, preferably, more than about 10 hours following initial dose, more preferably, more than about 12 hours after dose.
Persons of skill in the art will appreciate that blood plasma drug concentrations obtained in individual subjects will vary due to interpatient variability in the many parameters affecting drug absorption, distribution, metabolism and excretion. For this reason, unless otherwise indicated, when a drug plasma concentration is listed, the value listed is the calculated mean value based on values obtained from a groups of subjects tested.
"Ascending rate of release" or "ascending release rate" means a rate of release wherein the amount of drug released from a dosage form as a function of time increases over a period of time, preferably continuously and gradually. Preferably, the rate of drug released as a function of time increases in a steady (rather than step- wise) manner. More preferably, an ascending rate of release may be characterized as follows. The rate of release as a function of time for a dosage form is measured and plotted as % drug release versus time or as milligrams of drug released / hour versus time. An ascending rate of release is preferably characterized by an average rate (expressed in mg of drug per hour) wherein the rate within a given two hour span is higher as compared with the previous two hour time span, over the period of time of about 2 hours to about 12 hours, preferably, about 2 hours to about 18 hours, more preferably about 4 hours to about 12 hours, more preferably still, about 4 hours to about 18 hours. Preferably, the increase in average rate is gradual such that less than about 30% of the dose is delivered during any 2 hour interval, more preferably, less than about 25% of the dose is delivered during any 2 hour interval. Preferably, the ascending release rate is maintained until at least about 50%, more preferably until at least about 75% of the drug in the dosage form has been released.
In other preferred embodiments, ascending rates of release may be defined with reference to specific release rates measured at specified times following administration of the dosage form in question. Preferably such release rates are determined in vitro.
"C" means the concentration of drug in blood plasma, or serum, of a subject, generally expressed as mass per unit volume, typically nanograms per milliliter. For convenience, this concentration may be referred to herein as "drug plasma concentration", "plasma drug concentration" or "plasma concentration". The plasma drug concentration at any time following drug administration is referenced as Ctime, as in C9h or C24h, etc. A maximum plasma concentration obtained following administration of a dosage form obtained directly from the experimental data without interpolation is referred to as Cmax. The average or mean plasma concentration obtained during a period of interest is referred to as Cavg or Cmean. "Mean, single dose, maximum plasma concentration Cmax" means the mean Cmax obtained over
several patients or multiple administrations to the same patient with sufficient washout in between dosings to allow drug levels to subside to pre-dose levels, etc., etc., following a single administration of a dosage form to each patient.
"Composition" means a product containing a compound of the present invention (such as a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from such combinations of the specified ingredients in the specified amounts).
"Compound" or "drug" means the compound (2R-trans)-6-chloro-5-[[4-[(4- fluorophenyl)methyl]-2,5-dimethyl-l-piperazinyl]carbonyl]-N,N, 1-trimethyl-alpha-oxo-lH- indole-3-acetamide as represented by Formula (I) below

Formula (I)
or pharmaceutically acceptable forms thereof.
Compounds of the present invention may be prepared as described generally in United States Patents 6,864,260 and 6,867,209 to Mavunkel et al.
It is apparent to those skilled in the art that the compounds of the invention may be present as racemates, enantiomers and enantiomeric mixtures thereof. Examples of a compound selected from Formula (I) for use in the present invention include an enantiomer of Formula (I) in an enantiomeric mixture wherein the enantiomer of Formula (I) predominates.
For an enantiomeric mixture wherein one enantiomer predominates, the enantiomer represented by Formula (I) preferably predominates to the extent of about 90% or greater. Examples of the present invention also include enantiomeric mixtures wherein said enantiomer preferably predominates to the extent of about 98% or greater.
"Dosage form" means one or more compounds in a medium, carrier, vehicle, or device suitable for administration to a patient. "Oral dosage form" means a dosage form suitable for oral administration. "Dose" means a unit of drug. Conventionally, a dose is provided as a dosage form.
Doses may be administered to patients according to a variety of dosing regimens. Common
dosing regimens include once daily orally (qd), twice daily orally (bid), and thrice daily orally (tid).
"Effective amount" means that amount of compound that elicits the biological or medicinal response in a tissue system, animal or human, that is being sought by a researcher, veterinarian, medical doctor, or other clinician, which includes therapeutic alleviation of the symptoms of the disease or disorder being treated and prophylactic.
"Enantiomer" means one of a pair of molecular species that are mirror images of each other and are not superposable. The term "diastereomer" refers to stereoisomers that are not related as mirror images. The symbols "R" and "S" represent the configuration of substituents around a chiral carbon atom(s). The symbols "R*" and "S*" denote the relative configurations of of substituents around a chiral carbon atom(s). The isomeric descriptors "R," "S," "S*" or "R*" are used as described herein for indicating atom configuration(s) relative to a core molecule and are intended to be used as defined in the literature (IUPAC Recommendations for Fundamental Stereochemistry (Section E), Pure Appl. Chem., 1976, 45:13-30)(incorporated by reference herein). "Forms" means various isomers and mixtures thereof for a compound of Formula (I).
The term "isomer" refers to compounds that have the same composition and molecular weight but differ in physical and/or chemical properties. Such substances have the same number and kind of atoms but differ in structure. The structural difference may be in constitution (geometric isomers) or in an ability to rotate the plane of polarized light (stereoisomers). The term "stereoisomer" refers to isomers of identical constitution that differ in the arrangement of their atoms in space. Enantiomers and diastereomers are stereoisomers wherein an asymmetrically substituted carbon atom acts as a chiral center. The term "chiral" refers to a molecule that is not superposable on its mirror image, implying the absence of an axis and a plane or center of symmetry. "Flat plasma curve" means a plasma concentration curve that reaches and maintains a substantially constant value after a defined period of time following administration of a dosage form according to the invention.
"Immediate-release dosage form" means a dosage form that releases greater than or equal to about 80% of the drug in less than or equal to about 1 hour following administration of the dosage form to a patient.
"Initiation of release" means the beginning of a release rate test, when the dosage form is placed in a liquid and the sequence of events begins that leads to release of the compounds of Formula (I).
"Medicament" means a product for use in preventing, treating or ameliorating substance related disorders such as substance dependence, substance abuse or substance induced disorders in a subject in need thereof.
"Patient" means an animal, preferably a mammal, more preferably a human, in need of therapeutic intervention.
"Pharmaceutically acceptable" means molecular entities and compositions that are of sufficient purity and quality for use in the formulation of a composition or medicament of the present invention. Since both human use (clinical and over-the-counter) and veterinary use are equally included within the scope of the present invention, a formulation would include a composition or medicament for either human or veterinary use.
"Pharmaceutically acceptable salt" means an acid or base salt of the compounds of the invention that are of sufficient purity and quality for use in the formulation of a composition or medicament of the present invention and are tolerated and sufficiently non toxic to be used in a pharmaceutical preparation. Suitable pharmaceutically acceptable salts include acid addition salts which may, for example, be formed by reacting the drug compound with a suitable pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, fumaric acid, maleic acid, succinic acid, acetic acid, benzoic acid, citric acid, tartaric acid, carbonic acid or phosphoric acid.
Thus, representative pharmaceutically acceptable salts include, but are not limited to, the following: acetate, alpha-ketoglutarate, alpha-glycerophosphate, ascorbate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide, calcium edetate, camsylate, carbonate, chloride, clavulanate, citrate, dihydrochloride, edetate, edisylate, estolate, esylate, ethanesulfonate, fumarate, gluceptate, gluconate, glutamate, glycollylarsanilate, hexylresorcinate, hydrabamine, hydrobromide, hydrochloride, hydroxynaphthoate, iodide, isothionate, lactate, lactobionate, laurate, malate, maleate, malonate, mandelate, mesylate, methanesulfonate, methylbromide, methylnitrate, methyl sulfate, mucate, napsylate, nitrate, N- methylglucamine ammonium salt, oleate, pamoate (embonate), palmitate, pantothenate, phosphate/diphosphate, polygalacturonate, salicylate, stearate, sulfate, subacetate, succinate, tannate, tartrate, teoclate, tosylate, triethiodide and valerate.
Pharmaceutically acceptable salts may be obtained using standard procedures well known in the art, for example by reacting a sufficiently basic compound such as an amine with a suitable acid affording a physiologically acceptable anion. Alkali metal, for example; sodium, potassium or lithium, or alkaline earth metals, for example calcium salts of carboxylic acids can also be made.
"Plasma drug concentration curve" or "drug plasma concentration curve", or "plasma concentration curve" or "plasma profile" or "plasma concentration profile" refer to the curve obtained by plotting plasma drug concentration or drug plasma concentration, or plasma concentration versus time. Usually, the convention is that the zero point on the time scale (conventionally on the x axis) is the time of administration of the drug or dosage form comprising the drug to a patient.
"Rate of release" or "release rate" means to the quantity of compound released from a dosage form per unit time, e.g., milligrams of drug released per hour (mg/hr). Drug release rates for dosage forms may be measured as an in vitro rate of drug release, i.e., a quantity of drug released from the dosage form per unit time measured under appropriate conditions and in a suitable fluid.
"Relative bioavailability" means AUQnf for inventive dosage form/AUCjnf for immediate release dosage form; wherein both dosage forms comprise the same or substantially the same amount of drug, expressed in units of mass.
"Mean, single dose, time to maximum plasma concentration Tmax" is the mean time elapsed from administration to a patient of a dosage form comprising a drug to the time at which the Cmax for that drug is obtained over several patients or multiple administrations to the same patient with sufficient washout in between dosings to allow drug levels to subside to pre-dose levels, etc., following a single administration of the dosage form to each patient, and obtained directly from the experimental data without interpolation.
"Therapeutically effective amount" means that amount of drug that elicits the biological or medicinal response in a tissue system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician, which includes alleviation of the symptoms of the disease or disorder being treated.
"Zero order rate of release" or "zero order release rate" means a rate of release wherein the amount of drug released as a function of time is substantially constant. In other words, the dosage form exhibits zero order or substantially zero order release kinetics. More particularly, the rate of release of drug as a function of time shall vary by less than about 30%, preferably, less than about 20%, more preferably, less than about 10%, most preferably, less than about 5%, wherein the measurement is taken over the period of time wherein the cumulative release is between about 25% and about 75%, preferably, between about 25% and about 90%.
PLASMA PROFILES
With a plasma half-life of approximately 5 to 8 hours, the Compound can be provided in different formulations adapted to achieve pharmacologically effective levels in the blood. Depending upon the intended treatment, it may be desired to provide the Compound in different dosage forms. For instance, in one embodiment of the invention, the Compound is provided in an oral formulation adapted to provide an immediate release profile. Depending upon considerations such as required dosing and safety, particular benefit may be provided through such a formulation when used to treat conditions requiring rapid onset and/or rapid clearance of the Compound into and from the patient's plasma. In another embodiment, it may be desired to have a sustained release formulation which can be tailored to provide a more constant or . prolonged exposure of the Compound.
DOSAGE FORMS
In embodiments, the Compound is formulated into dosage forms suitable for administration to patients in need thereof.
Oral dosage may be provided in any pharmaceutically acceptable form. For example, a solid form may involve any of a tablet, capsule, and the like. A liquid form may involve delivery of the compound through any of an oral suspension, solution, gel, liquid capsule and the like. Preferably the Compound is provided in a solid dosage form.
Standard solid dosage forms may comprise the Compound in combination with various pharmaceutically acceptable excipients, said dosage form adapted to provide a release profile of the Compound in a manner to obtain the desired clinical effect through oral administration to the patient. Pharmaceutically acceptable excipients are known in the art and can be provided
according to considerations of desired functionality and processability. Roles for the excipients in the oral dosage form include but are not limited to fillers, binders, disintegrants, release- controlling agents, glidants, lubricants, coatings and the like. For example, in one embodiment of the invention, it is desired to have an immediate release profile for the dosage form. To help achieve this profile in a solid dosage form, the dosage form should preferably comprise disintegrant in an amount between 3 and 20% of the total form wherein the Compound also comprises a pharmaceutically effective amount of the form. In another preferred embodiment of the invention, a controlled or sustained release formulation of the Compound is desired. Such a formulation can be achieved by varying the amounts, concentrations and ratios of certain release controlling polymers. For example, in one preferred embodiment of the invention, an oral extended release formulation is provided that in tablet form comprising approximately 250 mg of the Compound, hydro xypropyl methylcellulose, carboxymethylcellulose sodium (NaGMC), lactose monohydrate, magnesium stearate, and OPADRY II film-coating. In this embodiment, the ratio of HPMC to NaCMC is varied from 1 :1 to 3 : 1 to drive the release profile of the formulation towards a zero order dissolution rate, as measured through appropriate plasma samples. Preferably, the ratio of HPMC to NaCMC is 2: 1.
Examples of fillers used in the art include but are not limited to sugars such as lactose, dextrose, glucose, sucrose, cellulose, starches and carbohydrate derivatives, calcium carbonates, magnesium carbonates and the like. Examples of binders include hydroxypropyl methylcellulose, methylcellulose, starches, and the like.
Useful disintegrants may be selected from starches, clays, celluloses, algins and gums and crosslinked starches, celluloses and polymers. Representative disintegrants include microcrystalline cellulose, crosscarmellose sodium, alginic acid, sodium alginate, crosprovidone, cellulose, agar and related gums, sodium starch glycolate, corn starch, potato starch, sodium starch glycolate, Veegum HV, methylcellulose, agar, bentonite, carboxymethylcellulose, alginic acid, guar gum and the like.
Glidants commonly used in the art include magnesium carbonate, magnesium lauryl sulphate, calcium silicate, talc, fumed silicon dioxide and the like. Useful lubricants include but are not limited to magnesium stearate, calcium stearate, stearic acid, sodium stearyl fumarate, polyethylene glycol, sodium lauryl sulphate, magnesium lauryl sulphate, sodium benzoate, and the like. Polymers commonly used as excipients include but are not limited to methylcellulose (MC), ethylcellulose (EC), hydroxyethylcellulose (HEC), methyl hydroxyethylcellulose
(MHEC), hydroxypropyl cellulose (HPC), hydroxypropyl methylcellulose (HPMC), sodium carboxymethylcellulose (NaCMC), and the like. These polymers, either alone or in various combinations, can serve multiple purposes including but not limited to controlling release of the the Compound.
In any case, the appropriate excipients should be selected such that they are compatible with other excipients and do not bind with the Compound or cause drug degradation.
Dosage forms in accordance with the embodiments depicted herein are manufactured by standard techniques. For example, the dosage form may be manufactured by the wet granulation technique. In the wet granulation technique, the drug and carrier are blended using an aqueous or organic solvent, such as denatured anhydrous ethanol, as the granulation fluid. The remaining ingredients can be dissolved in a portion of the granulation fluid, such as the solvent described above, and this latter prepared wet blend is slowly added to the drug blend with continual mixing in the blender. The granulating fluid is added until a wet blend is produced, which wet mass blend is then forced through a predetermined screen and dried in a fluid bed dryer. The dried granules are then sized. Next, magnesium stearate, or another suitable lubricant and other excipient materials are added to the drug granulation, and the granulation is put into milling jars and mixed on a jar mill for 10 minutes. The composition is pressed into a layer, for example, in a Manesty® press or a Korsch LCT press. For a trilayered core, granules or powders of the drug layer compositions and push layer composition are sequentially placed in an appropriately-sized die with intermediate compression steps being applied to each of the first two layers, followed by a final compression step after the last layer is added to the die to form the trilayered core. The intermediate compression typically takes place under a force of about 50-100 newtons. Final stage compression typically takes place at a force of 3500 newtons or greater, often 3500- 5000 newtons. The compressed cores are fed to a dry coater press, e.g., Kilian® Dry Coater press, and subsequently coated with the wall materials as described herein.
Pan coating may be conveniently used to provide the completed dosage form. In the pan coating system, the wall-forming composition for the inner wall or the outer wall, as the case may be, is deposited by successive spraying of the appropriate wall composition onto the compressed core accompanied by tumbling in a rotating pan. A pan coater is used because of its availability at commercial scale. Other techniques can be used for coating the compressed core. Once coated, the wall is dried in a forced-air oven or in a temperature and humidity controlled oven to free the dosage form of solvent(s) used in the manufacturing. Drying conditions will be
conventionally chosen on the basis of available equipment, ambient conditions, solvents, coatings, coating thickness, and the like.
Other coating techniques can also be employed. For example, one alternative technique uses an air-suspension procedure. This procedure consists of suspending and tumbling the compressed core in a current of air, until a coating is applied to the core. The air-suspension procedure is described in U.S. Patent No. 2,799,241; in J. Am. Pharm. Assoc, Vol. 48, pp. 451- 459 (1959); and, ibid., Vol. 49, pp. 82-84 (1960). The dosage form also can be coated with a Wursterφ air-suspension coater using, for example, methylene dichloride methanol as a cosolvent for the wall forming material. An Aeromatic® air-suspension coater can be used employing a cosolvent.
In another embodiment, the drug and other ingredients comprising the drug layer are blended and pressed into a solid layer. The layer possesses dimensions that correspond to the internal dimensions of the area the layer is to occupy in the dosage form, and it also possesses dimensions corresponding to the push layer, if included, for forming a contacting arrangement therewith. The drug and other ingredients can also be blended with a solvent and mixed into a solid or semisolid form by conventional methods, such as ballmilling, calendering, stirring or rollmilling, and then pressed into a preselected shape. The compressed cores then may be coated with the inner wall material and the semipermeable wall material as described herein.
Another manufacturing process that can be used comprises blending the powdered ingredients in a fluid bed granulator. After the powdered ingredients are dry blended in the granulator, a granulating fluid, for example, poly(vinylpyrrolidone) in water, is sprayed onto the powders. The coated powders are then dried in the granulator. This process granulates all the ingredients present therein while adding the granulating fluid. After the granules are dried, a lubricant, such as stearic acid or magnesium stearate, is mixed into the granulation using a blender e.g., V-blender or tote blender. The granules are then pressed and coated in the manner described above.
Exemplary solvents suitable for manufacturing the dosage form components comprise aqueous or inert organic solvents that do not adversely harm the materials used in the system. The solvents broadly include members selected from the group consisting of aqueous solvents, alcohols, ketones, esters, ethers, aliphatic hydrocarbons, halogenated solvents, cycloaliphatics, aromatics, heterocyclic solvents and mixtures thereof. Typical solvents include acetone,
diacetone alcohol, methanol, ethanol, isopropyl alcohol, butyl alcohol, methyl acetate, ethyl acetate, isopropyl acetate, n-butyl acetate, methyl isobutyl ketone, methyl propyl ketone, n- hexane, n-heptane, ethylene glycol monoethyl ether, ethylene glycol monoethyl acetate, methylene dichloride, ethylene dichloride, propylene dichloride, carbon tetrachloride nitroethane, nitropropane tetrachloroethane, ethyl ether, isopropyl ether, cyclohexane, cyclooctane, benzene, toluene, naphtha, 1,4-dioxane, tetrahydrofuran, diglyme, water, aqueous solvents containing inorganic salts such as sodium chloride, calcium chloride, and the like, and mixtures thereof such as acetone and water, acetone and methanol, acetone and ethyl alcohol, methylene dichloride and methanol, and ethylene dichloride and methanol.
Exemplary liquid carriers for the present invention include lipophilic solvents (e.g., oils and lipids), surfactants, and hydrophilic solvents. Exemplary lipophilic solvents, for example, include, but are not limited to, Capmul PG-8, Caprol MPGO, Capryol 90, Plurol Oleique CC 497, Capmul MCM, Labrafac PG, N-Decyl Alcohol, Caprol 1 OGlOO, Oleic Acid, Vitamin E, Maisine 35-1, Gelucire 33/01, Gelucire 44/14, Lauryl Alcohol, Captex 355EP, Captex 500, Capylic/Caplic Triglyceride, Peceol, Caprol ET, Labrafil M2125 CS, Labrafac CC, Labrafil M 1944 CS, Captex 8277, Myvacet 9-45, Isopropyl Nyristate, Caprol PGE 860, Olive Oil, Plurol Oleique, Peanut Oil, Captex 300 Low C6, and Capric Acid.
Exemplary surfactants for example, include, but are not limited to, Vitamin E TPGS, Cremophor (grades EL, EL-P, and RH40), Labrasol, Tween (grades 20, 60, 80), Pluronic (grades L-31, L-35, L-42, L-64, and L-121), Acconon S-35, Solutol HS-15, and Span (grades 20, and 80). Exemplary hydrophilic solvents for example, include, but are not limited to, Isosorbide Dimethyl Ether, Polyethylene Glycol (PEG grades 300, 400, 600, 3000, 4000, 6000, and 8000) and Propylene Glycol (PG).
The skilled practitioner will understand that any formulation comprising a sufficient dosage of the Compound solubilized in a liquid carrier suitable for administration to a subject.
Drug may be provided in particles by comminution that produces the size of the drug and the size of one or more accompanying polymers used in the fabrication of the dosage form, typically with a core containing the compound, according to the mode and the manner of the invention. The means for producing particles include granulation, spray drying, sieving, lyophilization, crushing, grinding, jet milling, micronizing and chopping to produce the intended micron particle size. The process can be performed by size reduction equipment, such as a
micropulverizer mill, a fluid energy grinding mill, a grinding mill, a roller mill, a hammer mill, an attrition mill, a chaser mill, a ball mill, a vibrating ball mill, an impact pulverizer mill, a centrifugal pulverizer, a coarse crusher and a fine crusher. The size of the particle can be ascertained by screening, including a grizzly screen, a flat screen, a vibrating screen, a revolving screen, a shaking screen, an oscillating screen and a reciprocating screen. The processes and equipment for preparing drug and carrier particles are disclosed in Pharmaceutical Sciences,
Remington, 17th Ed., pp. 1585-1594 (1985); Chemical Engineers Handbook, Perry, 6th Ed., pp. 21-13 to 21-19 (1984); Journal of Pharmaceutical Sciences, Parrot, Vol. 61, No. 6, pp. 813-829 (1974); and Chemical Engineer, Hixon, pp. 94-103 (1990).
Drug emulsified formulations of the present invention can initially comprise an oil and a non-ionic surfactant. The oil phase of the emulsion comprises any pharmaceutically acceptable oil which is not immiscible with water. The oil can be an edible liquid such as a non-polar ester of an unsaturated fatty acid, derivatives of such esters, or mixtures of such esters. The oil can be vegetable, mineral, animal or marine in origin. Examples of non-toxic oils can also include, for example, in addition to the surfactants listed above, a member selected from the group consisting of peanut oil, cottonseed oil, sesame oil, corn oil, almond oil, mineral oil, castor oil, coconut oil, palm oil, cocoa butter, safϊlower, a mixture of mono- and diglycerides of 16 to 18 carbon atoms, unsaturated fatty acids, fractionated triglycerides derived from coconut oil, fractionated liquid triglycerides derived from short chain 10 to 15 carbon atoms fatty acids, acetylated monoglycerides, acetylated diglycerides, acetylated triglycerides, olein known also as glyceral trioleate, palmitin known as glyceryl tripalmitate, stearin known also as glyceryl tristearate, lauric acid hexylester, oleic acid oleylester, glycolyzed ethoxylated glycerides of natural oils, branched fatty acids with 13 molecules of ethyleneoxide, and oleic acid decylester. The concentration of oil, or oil derivative in the emulsion formulation can be from about 1 wt % to about 40 wt %, with the wt % of all constituents in the emulsion preparation equal to 100 wt %. The oils are disclosed in Pharmaceutical Sciences by Remington, 17th Ed., pp. 403-405, (1985) published by Mark Publishing Co., in Encyclopedia of Chemistry, by Van Nostrand Reinhold, 4th Ed., pp. 644-645, (1984) published by Van Nostrand Reinhold Co.; and in U.S. Pat. No. 4,259,323.
The amount of Compound incorporated in the dosage forms of the present invention is generally from about 10% to about 90% by weight of the composition depending upon the therapeutic indication and the desired administration period, e.g., every 12 hours, every 24 hours, and the like. Depending on the dose of Compound desired to be administered, one or
more of the dosage forms can be administered. Depending upon the formulation, the Compound will preferably be in the form of an HCl salt or free base form.
An oral liquid formulation of the Compound may be in the form of a capsule. The capsule can be made conveniently in two parts, with one part (the "cap") slipping over and capping the other part (the "body") as long as the capsule is deformable under the forces exerted by the expandable layer and seals to prevent leakage of the liquid drug formulation from between the telescoping portions of the body and cap. The two parts completely surround and capsulate the internal lumen that contains the liquid drug formulation, which can contain useful additives. The two parts can be fitted together after the body is filled with a preselected formulation. The assembly can be done by slipping or telescoping the cap section over the body section, and sealing the cap and body, thereby completely surrounding and encapsulating the formulation of drug.
Soft capsules typically have a wall thickness that is greater than the wall thickness of hard capsules. For example, soft capsules can, for example, have a wall thickness on the order of 10-40 mils, about 20 mils being typical, whereas hard capsules can, for example, have a wall thickness on the order of 2-6 mils, about 4 mils being typical.
In one embodiment of the dosage system, a soft capsule can be of single unit construction and can be surrounded by an unsymmetrical hydro-activated layer as the expandable layer. The expandable layer will generally be unsymmetrical and have a thicker portion remote from the exit orifice. As the hydro-activated layer imbibes and/or absorbs external fluid, it expands and applies a push pressure against the wall of capsule and optional barrier layer and forces drug formulation through the exit orifice. The presence of an unsymmetrical layer functions to assure that the maximum dose of drug is delivered from the dosage form.
In some embodiments, a barrier layer can be first coated onto the capsule and then the tableted, expandable layer is attached to the barrier-coated capsule with a biologically compatible adhesive. Suitable adhesives include, for example, starch paste, aqueous gelatin solution, aqueous gelatin/glycerin solution, aery 1 ate- vinylacetate based adhesives such as Duro- Tak adhesives (National Starch and Chemical Company), aqueous solutions of water soluble hydrophilic polymers such as hydroxypropyl methyl cellulose, hydroxymethyl cellulose, hydroxyethyl cellulose, and the like. That intermediate dosage form can be then coated with a semipermeable layer.
Hard capsules are also contemplated by the invention. Hard capsules are typically composed of two parts, a cap and a body, which are fitted together after the larger body is filled with a preselected appropriate formulation. This can be done by slipping or telescoping the cap section over the body section, thus completely surrounding and encapsulating the drug formulation. Hard capsules can be made, for example, by dipping stainless steel molds into a bath containing a solution of a capsule lamina-forming material to coat the mold with the material. Then, the molds are withdrawn, cooled, and dried in a current of air. The capsule is stripped from the mold and trimmed to yield a lamina member with an internal lumen. The engaging cap that telescopically caps the formulation receiving body is made in a similar manner. Then, the closed and filled capsule can be encapsulated with a semipermeable lamina. The semipermeable lamina can be applied to capsule parts before or after parts and are joined into the final capsule. In another embodiment, the hard capsules can be made with each part having matched locking rings near their opened end that permit joining and locking together the overlapping cap and body after filling with formulation. In this embodiment, a pair of matched locking rings are formed into the cap portion and the body portion, and these rings provide the locking means for securely holding together the capsule. The capsule can be manually filled with the drug formulation, or they can be machine filled with the drug formulation. In the final manufacture, the hard capsule is encapsulated with a semipermeable lamina permeable to the passage of fluid and substantially impermeable to the passage of drug. Methods of forming hard cap dosage forms are described in U.S. Patent No. 6,174,547, U.S. Patent Nos. 6,596,314, 6,419,952, and 6, 174,547.
The hard and soft capsules can comprise, for example, gelatin; gelatin having a viscosity of 15 to 30 millipoises and a bloom strength up to 150 grams; gelatin having a bloom value of 160 to 250; a composition comprising gelatin, glycerine, water and titanium dioxide; a composition comprising gelatin, erythrosin, iron oxide and titanium dioxide; a composition comprising gelatin, glycerine, sorbitol, potassium sorbate and titanium dioxide; a composition comprising gelatin, acacia glycerine, and water; and the like. Materials useful for forming capsule wall are known in U.S. Pat. Nos. 4,627,850; and in 4,663,148. Alternatively, the capsules can be made out of materials other than gelatin (see for example, products made by BioProgres pic).
The capsules typically can be provided, for example, in sizes from about 3 to about 22 minims (1 minim being equal to 0.0616 ml) and in shapes of oval, oblong or others. They can be provided in standard shape and various standard sizes, conventionally designated as (000), (00),
(0), (1), (2), (3), (4), and (5). The largest number corresponds to the smallest size. Non-standard shapes can be used as well. In either case of soft capsule or hard capsule, non-conventional shapes and sizes can be provided if required for a particular application.
Other approaches to achieving oral dosage forms are known in the art. Dosage forms that operate in accord with these other approaches are encompassed by the scope of the disclosure herein to the extent that the drug release characteristics and/or the blood plasma concentration characteristics as recited herein and in the claims describe those dosage forms either literally or equivalently.
It will be appreciated the dosage forms described herein are merely exemplary of a variety of dosage forms designed for and capable of achieving administration of the inventive substance(s). Those of skill in the pharmaceutical arts can identify other dosage forms that would be suitable.
METHODS OF USE
The inventive methods, compositions, and dosage forms are useful in treating a variety of indications that are treatable using the Compound. In an aspect, the invention provides a method for treating an indication, such as a disease or disorder, in a patient by administering an inventive composition or dosage form that comprises the Compound. In an embodiment, a composition or dosage form comprising the Compound is administered to the patient via oral administration. The dose administered is generally adjusted in accord with the age, weight, and condition of the patient, taking into consideration the dosage form and the desired result. Inventive dosage forms may comprise the Compound or pharmacologically active metabolites in combination.
It will be readily apparent to those skilled in the art that any dose or frequency of administration that provides the therapeutic or prophylactic effect described herein is suitable for use in the present invention. Dosage regimens may be varied depending upon the requirement of the subjects (including factors associated with the particular subject being treated, including subject age, weight and diet, strength of the preparation, the advancement of the disease condition and the mode and time of administration) and the use of a particular compound of Formula (I) or pharmaceutical composition thereof or a pharmaceutically acceptable salt thereof. Optimal dosages to be administered may be readily determined by those skilled in the art and will result in the need to adjust the dose to an appropriate therapeutic or prophylactic level. The use of either daily administration or post-periodic dosing may be employed.
In a preferred embodiment, dosage forms according to the invention comprise an amount of the Compound ranging from about 5 mg to about 1000 mg, preferably from about 10 mg to about 600 mg, and more preferably from about 50 rng to about 300 mg.
While there has been described and pointed out features and advantages of the invention, as applied to present embodiments, those skilled in the medical art will appreciate that various modifications, changes, additions, and omissions in the method described in the specification can be made without departing from the spirit of the invention.
The present invention is not limited to the particular embodiments described in this application, which are intended as single illustrations of individual aspects of the invention. Many modifications and variations of this invention can be made without departing from its spirit and scope, as will be apparent to those skilled in the art. Functionally equivalent methods within the scope of the invention, in addition to those enumerated herein, will be apparent to those skilled in the art from the foregoing description. Such modifications and variations are intended to fall within the scope of the appended claims. The present invention is to be limited only by the terms of the appended claims, along with the full scope of equivalents to which such claims are entitled.
The following Examples are meant to be illustrative of the claimed invention, and not limiting in any way.
EXAMPLES
Example 1: Synthesis of the Compound


The S-carboxylic-β-chloro indole acid (1.56 g, 7.44 mmol) was dissolved in dry methylene chloride 60 mL to this was added the EDACHCl (1.57 g, 8.18 mmol) and DMAP(IO % mol). After stirring under nitrogen for 10 min. the amine (2.19 g, 7. 5 mmol) was added, followed by triethylamine (3 mL, 21.52 mmol). After overnight at room temperature, the reaction mixture was concentrated and the residue was taken up in ethyl acetate and washed with 10% aq. sodium carbonate, saturated sodium chloride, dried over anhydrous sodium sulfate and filtered. Concentration gives the crude product that was chromatographed on silica gel using a gradient of EtOAc/Hexane 2/8 - 6/4. TLC Rf 0.435 (EtOAc:Hexane, 1 :1), EIMS M+ 413.
1.02 g of the product from the above step was dissolved in 30 mL dry DCM. The reaction mixture was purged with nitrogen and placed in an ice bath. To this was added 4 mL of 2M oxalyl chloride in DCM. The reaction mixture was stirred at room temperature for 1 hour and then at room temperature for 2 hours. Reaction mixture was concentrated on a rotary evaporator . After drying on a vacuum pump for 15 min. the residue (a yellow solid) was dissolved in dry DCM, 30 mL, to which was added 4 mL of a 2M solution of
dimethylamine in THF. 30 minutes later the reaction mixture was concentrated and the residue was taken up in ethyl acetate and washed with 10% aq. sodium carbonate, saturated sodium chloride, dried over anhydrous sodium sulfate and filtered. Concentration gives the crude product that was chromatographed on silica gel using a gradient of EtOAc 100% - EtOAc/MeOH 9:1. TLC Rf 0.5 (EtOAc:MeOH, 9:1), EIMS M+ 513. The white solid from the above step was dissolved in 10 mL dry DCM. To this was added sufficient 2 M HCl in ether, till a precipitate persists. The mixture was then concentrated on a rotary evaporator to dryness and then dried overnight under high vacuum to give the final product (1.08 g).
Example 2: A Phase 2, Multicenter, Randomized. Double-Blind, Placebo-Controlled,
Dose-Escalating Study of Compound in Patients with Active Rheumatoid Arthritis Receiving Methotrexate
The following example describes a study the demonstrates that the compound of the invention is effective in treating a condition involving selective inhibition of the activity of a p38-α isoform; in particular in ameliorating active rheumatoid arthritis.
In particular, the following example describes a Phase 2, multicenter, randomized, double-blind, placebo-controlled, dose-escalating study of Compound in patients with active rheumatoid arthritis receiving methotrexate.
The synthesis of the Compound is described above. In the following studies, the HCl salt-free base of the Compound was used.
pH/Solubilitv Profile of Compound (in Table 2) at ca. 25°C: The solubility of the Compound was determined over the pH range of 1.0 to 12.0 at ca. 25°C using various concentrations of NaOH and HCl. The maximum solubility for Compound was determined to be 40.6 mg/mL at pH 3.5. In the experiment solubility decreased below pH 3.5 due to a common ion effect. pKa: The pKa of Compound was determined to be 5.85 ± 0.02 at an average ionic strength of 0.174 M, using a co-solvent potentiometric titration procedure. This corresponds to a thermodynamic standard state value at zero total ionic strength of 6.10 ± 0.02.
Solubility of Compound in Selected Solvents at 25°C: The solubility of Compound was determined in hexane, methanol (MeOH), ethanol (EtOH), acetonitrile, methyl ethyl ketone (2-butanone, MEK), acetone, isopropyl alcohol (IPA), 1 :1 Propylene Glycol (PG): Polyethylene Glycol (PEG 400), 1 : 1 EtOH:PG, and 1 :9 to 9:1 EtOH:water. Solubilities are shown in Table
1.1 :

Octanol-water partition coefficient: The n-octanol/water partition coefficients for Compound , were determined over the pH range of 2 to 8. From the observed partitioning coefficients, the intrinsic partitioning coefficients were calculated to be: ko = 0.09, ki = 1594. The increase in the observed partition coefficient above pH 4.5 is due to the increase in the fraction of uncharged species present.
Hveroscopicitv by dynamic vapor sorption: Compound is a crystalline solid. Upon heating, Compound exhibits a complex DSC scan with very weak endotherms. Optical hot stage analysis shows that Compound exhibits a very broad melting range from about 154°C to 1700C at a heating rate of l°C/minute. Capillary melting point data shows the melting point of the Compound to be dependent upon heating rate.
Dynamic vapor sorption measurements show Compound to be slightly hygroscopic above 70%RH. When dry, Compound quickly gains ~ 3% water. At very low relative humidity (~ 4%RH), the water content levels briefly (~ 3%), then quickly increases to ~ 6% water content when humidity is raised to 18 to 21%. From ~ 21 % RH to ~ 70% RH the
estimated water content remains in the range of 6 - 7%. Above ~ 71 %RH approximately 1.5% water weight is gained, and above ~ 90%RH an additional estimated 2% water is gained. The fact that Compound rapidly gains water weight to about 6 - 7%, and the water content remains between 6 -7% over a humidity range from ~ 21-70%RH, suggest hydration of Compound (2 moles of water per molecule of Compound would be 6.15% water). This is in reasonably good agreement with the observed TGA weight loss, i.e., 6.67% weight loss through 165°C.
Variable humidity XRPD studies showed that subtle but reversible changes in the XRPD pattern will occur at either very low (<20%) or very high (>95%) relative humidity. Since water absorption is reversible, with only subtle changes observed in the XRPD pattern as a function of water content, changes can be considered to be due to an "isomorphic desolvate" of the same crystal form, rather than different polymorphs.
Only two forms of Compound, one crystalline and one amorphous, were found during the recrystallization studies. Recrystallization of Compound from ethanol, acetonitrile, isopropanol, acetone, and water yielded either crystalline or amorphous drug substance or mixtures of the two forms. Only ethyl acetate was found to give consistently crystalline drug substance from the recrystallization method used.
Based upon the hygroscopicity data, bulk substance packaging protection from high humidity conditions may not be necessary unless bulk drug handling or flow characteristics of the bulk drug substance suggest otherwise.
Compound API particle size distribution and bulk density: Particle size (by laser light scattering) and bulk density of several lots of Compound API are summarized in Table 1.2, below. Particle size distribution of all the API lots produced at one facility was homogeneous. The two lots differ in particle size and drying time varied greatly. This difference is not expected to have an effect during drug substance manufacturing.
Table 1.2 Results for Different Lot - one l t er

NT = Not tested
Adsorption of Compound on Filter Media: Adsorption of Compound was studied on the following filters: LC PVDF, Nylon, Polysulfone, and Polypropylene. When polypropylene syringes were used in conjunction with the four types of filters, the results indicated that Compound is adsorbed. At pH 1.5, the range of adsorption was 0.60 μg/cm2 to 2.04 μ/cm2. The glass and polypropylene syringes without filters were 0.06 and 0.07 μg/cm2, respectively. At pH 7.4, the range of adsorption is 10.4 μg/cm2 to 11.6 μg/cm2. The glass and polypropylene syringes without filters were 0.37 and 0.29 μg/cm2, respectively. At pH 10.8, the range of adsorption is 2.34 μg/cm2 to 2.47 μg/cm2. Slight adsorption, 0.07 μg/cm2, was found for the glass and polypropylene syringes.
Excipient Compatibility: Compound compatibility with excipients commonly used in solid oral formulations was studied. Drug was mixed with each of fifteen excipients (see Table 2.1), and compared to a drug-only control, in both dry and wet blends. Study samples were stored at 40°C/75% RH conditions for 12 weeks. Samples were tested for appearance, recovery of active drug and impurity profile at 0, 2, 4, 8 and 12 weeks. Based on the results of this study, appropriate excipients were selected for further capsule and tablet formulation development.
Apparent binding was observed beginning at Time Zero in the binary mixtures of Compound with either calcium phosphate dibasic or sodium lauryl sulfate. A lesser decrease in recovery was observed with croscarmellose sodium or magnesium stearate at >4 weeks of stability.
Degradation (as increase in % total peak area of impurity peaks) was none to very slight (<0.3%) in all samples except povidone (ca. 4% degradation at 12 weeks in both wet and dry samples) and sodium lauryl sulfate (0.5% in dry, 0.9% in wet samples at 12 weeks).
Based on this study, the following excipients are recommended for use in Compound solid oral formulations: lactose, Avicel, Prosolv, pregelatinized starch, HPMC, croscarmellose sodium (in low amounts), sodium starch glycolate, magnesium stearate and stearic acid.
Calcium phosphate dibasic, povidone and sodium lauryl sulfate should not be used in Compound products, unless calcium phosphate dibasic is employed for extended release.
Table 2.1 : Excipient Compatibility Mixtures


Table 2.2: Component origins

NA = Not applicable
Table 2.3: % Recovery of Compound in Excipient Compatibility Study

Table 2.3 (continued)
 Table 2.3 (continued)
Table 2.3 (continued)
♦Sample 7 (HPMC) was re-analyzed after 1 week stored at RT (same vial), as well as the original (diluted)/ϊ/rera/ sample with recoveries of 65.4 % and 66.4 % (avg. of 2 injections), respectively. The original unfiltered sample (stored at RT) was diluted, filtered, and analyzed after 1 week. The recovery was determined to be 105.5 % (avg. of 2 injections). The low recovery values from the experiments performed first, seem to be related to an extraction problem. Extraction of drug/HPMC samples is being improved.
**Sample Preparation: 30 min shaking, then 30 min sonication.
Table 2.4: Total amount of impurities formed (as % degradation of Compound )


Immediate Release Tablet
The invention also provides an immediate release tablet having a formulation as set forth in Table 3.1, below:
Table 3.1: IR Tablet Formulations

Table 3.1 : (continued)
* Quantities of Compound adjusted to achieve free base content of Compound in tablet. Concentrations of intragranular lactose and Avicel PH 101 are adjusted to compensate for higher API content. ** Removed during processing.
Table 3.2: Excipient Functions

COMPOUND IR Tablet Manufacturing: An exemplary Compound IR tablet manufacturing process is illustrated in Figure 1, as a Flow chart. Exemplary equipment that can be used to make compounds of the invention include examples described in Table 3.3, below:
Table 3.3: Equipment

Figure 2 illustrates Table 3.4, which describes exemplary formulations of the invention.
Table 3.4, describes tablet feasibility lots at a nominal 2-Kg scale. In Figure 2, A: intragranular; B: extragranular; Constant parameters for Batches 64-194: impeller speed 500 φm, chopper speed 1500 (max) rpm.
Figure 3 illustrates Table 3.5, summarizing data for exemplary formulations (tablets) of the invention, in this aspect: Compound IR feasibility tablets — Particle Size Analysis. In Table
3.5: The granulation is passed through a 30 mesh screen. Then coarse material is milled through a Fitzmill equipped with 20 Mesh screen at medium speed and knives forward. Sieve analysis is performed on 100 g granulate. For Batches 115-194: The granulation is passed through a
Fitzmill equipped with 30 Mesh screen.
Figure 4 illustrates Table 3.6, a characterization of exemplary formulations (tablets) of the invention, in this example - using 2-Kg Feasibility Lots. In Table 3.6, Tooling: #11-2, 1 l/32"rd std concave, target weight 340 mg.
IR Tablets at 12-Kg scale: Exemplary methods of making exemplary pharmaceutical formulations of the invention are summarized in Table 5.4, below. In Table 5.4, granulation size was 12 kg, total batch size was ~14 kg. The amount of water needed for granulation at that scale was studied, and measured as % of granulation weight. For all the 4 batches, tablets were compressed at 3 compressions forces to achieve target hardness's of 10 kp, 13 kp and 16 kp. Based on the Process DOE study, it was estimated that the amount of water needed for granulation would be about 45% of granulation weight. Two lots were produced with 45% water; one lot was produced with 40% water (LW for "low weight") and one with 50% water (HW for "high weight"). The two 45% lots were coated. The results are summarized in Table 5.4: Compound IR Tablets, 12-Kg scale: In-process data:
Table 5.4

In Table 5.4:
* Unable to achieve hardness above 10 kp using typical compression force. Increasing amount of water changed granulation endpoint and properties of granulation (this batch had much harder granules). Run was performed using target compression force and recording obtained tablet hardness. Hardness reached plateau at 12 kp. **NC - data not collected.
IR Tablets at 60-Kg scale: Exemplary tablets were made at 60-Kg (nominal) scale at three strengths, 30-, 60- and 90-mg, in August and September 2004. The granulation size was 60 Kg, uncoated tablet batch size was ~68 Kg. 50 Kg of tablets were coated. All lots were successful. 45% water (as % of granulation weight) was the target amount for granulation; addition of water was stopped when satisfactory granulation was achieved. Actual amount of water used in granulation was:
30 mg: 25.84 kg - 43.1% 60 mg: 27.30 -45.5% 90 mg: 27.30 -45.5%
Tablets were compressed at 3 compressions forces to achieve target hardnesses of 10 kp, 13 kp and 16 kp.
In-process testing:

Blend uniformity results for the 90-mg lot were broad (one tablet at 112); note that this lot was produced with a lot with the largest particle size.
Exemplary methods of making exemplary pharmaceutical formulations of the invention are summarized in Table 5.5: Compound IR Tablets, at a 60-Kg scale: In-process data:
Table 5.5

* NC-Not collected
Exemplary tablets of the invention can have various particle size distribution for (2) pre- milled granulation (3) milled granulation and/or (4) final blend; Figure 5 illustrates a summary of data for pre-milled granulation, milled granulation and final blend at 60 mg tabs, 60 kg scale).
Packaging: The invention also provides methods for packaging, and various packaged forms of the compositions of the invention. For example, bulk tablets can be stored at a double- lined with polyethylene bags drum with no desiccant. Compound IR tablets can be packaged in
75 cc (12.5g), Blake HDPE OB, rectangular, white bottles with 33/400 closure (CR, CLIC-LOC III, Selig M-I; PHILLIPS-SUMIKA) polypropylene caps. The bottles are sealed with heat- induction seals. Ninety (90) tablets are packaged in each bottle.
Stability for clinical studies: Stability studies for Compound IR tablets at 30 and 90 mg.: 18-month stability data is available for 30 and 90 mg tablets at 25C/60%RH, and 9-month stability study for 30 and 90 mg tablets at 40C/75%RH conditions, where the data is summarized in Figure 6 (Table 7.1); in Figure 6, stability of Compound IR 30mg and 90 mg exemplary tablets, at 25C/60%RH and 40C/60%RH conditions. Product/Package Appearance Description Code:
0 = No change from initial, I = Change but acceptable, 2 = Change not acceptable NT = Not Tested, NS = Not Scheduled, NO = None Detected, ATST = At Storage = Time Zero.
Extended Release Tablets
In one aspect, the invention provides extended release (ER) tablets comprising at least one composition of the invention. The invention provides ER tablets with approximately zero- order release for up to 12 hours, suitable for once- or twice-daily dosing. In one aspect, the ER tablets are formulated as a polymer matrix, wet granulation with high shear mixing:
Tablet Trade Dress: Oval, convex, coated tablet with debossing. 50 to 120 mg Compound in 600-mg core. Clinical tablets to date have been white-coated and non-debossed. Packaging; 30 tablets/ 75 cc (12.5g), Blake HDPE OB, rectangular, white bottles with 33/400 closure (CR, CLIC-LOC III, Selig M-I ; PHILLIPS-SUMIKA) polypropylene caps. Heat- induction seals, no cotton or dessicant.
Also studied on stability: bulk tablets in double-polyethylene-lined drums; alternate HDPE bottle size; PVC/Aclar blister with foil lining.
Two exemplary formulations of the invention comprise Hydroxypropyl Methylcellulose ("HPMC") and Carboxymethylcellulose Sodium ("NaCMC") selected and manufactured as 100- mg/ tablets. Exemplary Extended Release (ER) formulations of the invention are summarized in Table 4-1 and Table 4-2, below:
Table 4-1 : Extended Release (ER) Tablet Formulations

For Table 4-1: * Compound quantity expressed as free base content. lntragranular lactose monohydrate is adjusted to compensate for actual Compound API content.
** Water is used as granulating and coating suspension agent. It is removed during processing.
Table 4-2: Excipients description and properties

Figure 7 is an illustration of an exemplary manufacturing process of the invention: a flow chart of a manufacturing process for exemplary Extended Release (ER) formulations of the
invention. Table 4-3 summarizes exemplary equipment that can be used in these exemplary manufacturing process of the invention.
Table 4-3: Manufacturing Equipment

Figure 8 summarizes data regarding exemplary formulations of the invention.
Other exemplary formulations of the invention include: Clinical Tablet Lots (10-Kg scale): Two 10-Kg lots of Compound 100-mg ER tablets (two coating runs from a common core; One 10-Kg lot of Compound ; 50-mg ER tablets:
50 % water (as % of granulation weight) was used for granulation.
In-process testing:

50-Kg lot of Compound 100-mg ER Tablets was manufactured: Granulation size was 60 kg, total batch size was -~73 kg. 50 Kg of tablets were coated. 46.3 % water (as % of granulation weight) was used for granulation. Tablets were compressed at 3 compressions forces to achieve target hardnesses of 10 kp, 13 kp and 16 kp. The following Table 4-5, summarizes exemplary manufacturing parameters for the 10 kg, and 60 kg Compound ER tablets, 50 and 100 mg.:
Table 4-5: Clinical Tablet Lots (10- to 50-Ke scale)
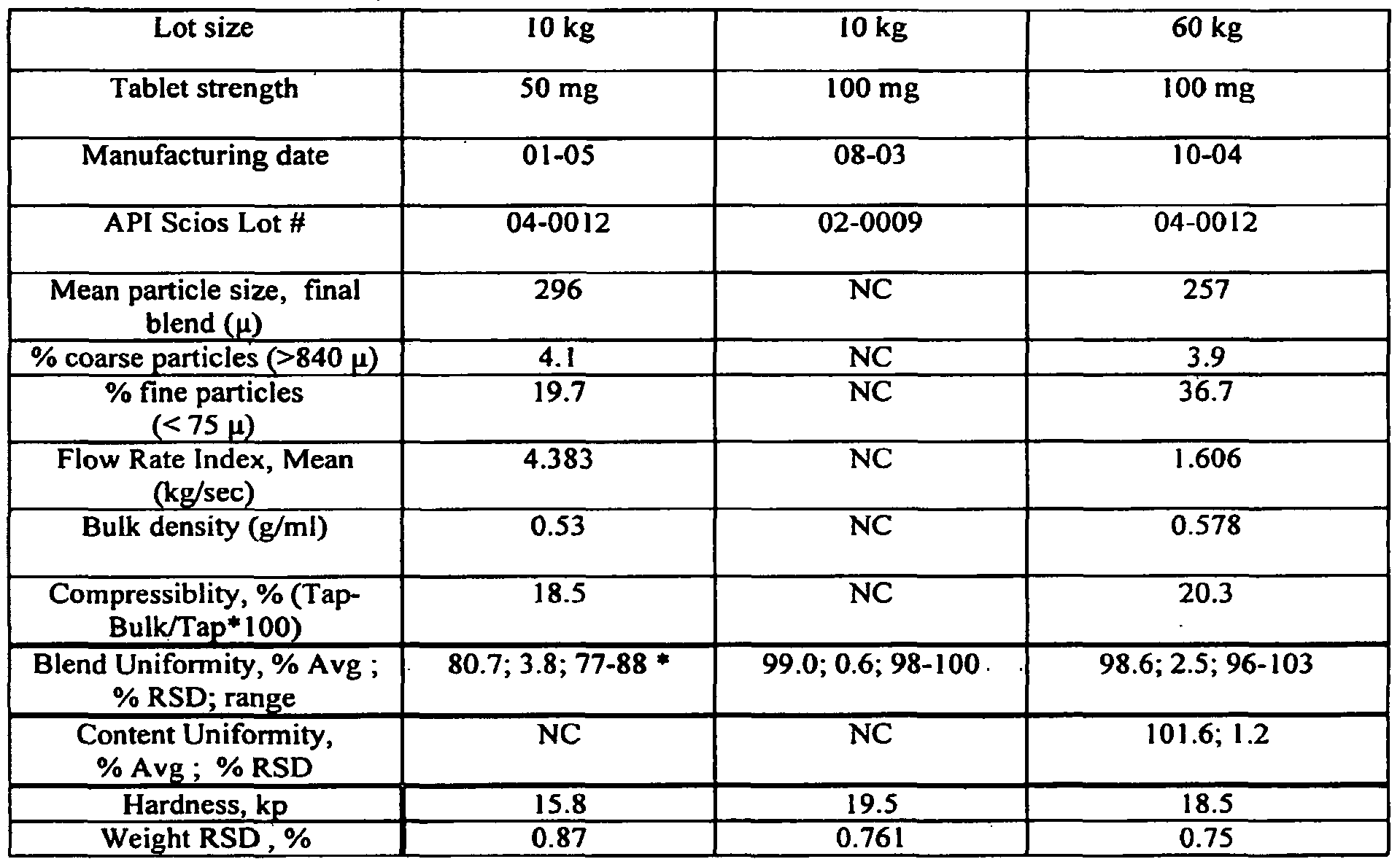
* Tablet potency and uniformity was satisfactory.
Stability: Stability studies on coated tablets; 1 -month 40/ 75% RH data show no change. 9 month stability data is available on 100 mg Compound ER uncoated prototype tablets at 25/60%RH and 40/75%RH conditions. Tablets were stable.
COMPOUND Clinical Study design: This was a placebo-controlled, dose-escalating study designed to assess the safety, tolerabiiity, efficacy, pharmacokinetics (PK), and pharmacodynamics of Compound in subjects with active RA who were also receiving methotrexate. Subjects (132 total) were randomly assigned to one of seven treatment groups (dose regimens A-F or placebo) over four separate treatment periods, each 30 days in length.
Dose levels for each successive treatment period were increased over the preceding period, with the exception of treatment period 4 that was amended to escalate to 60 mg tid (see the table below). Safety and PK data from the previous treatment period were reviewed before initiating the next treatment period. Each subject was followed for up to 58 days (30-day treatment period plus 28-day follow-up). In summary:
Treatment Treatment Treatment Treatment Treatment Group Period 1 Period 2 Period 3 Period 4
A 30 mg qd B (19; 16)
Placebo 60 mg qd (18; 16)
Placebo (10; 10)a
C 90 mg qd (16; 15)
D 30 mg tid (16; 16)
Placebo Placebo (8; 8)a
E 60 mg tid (18; Placebo 16)
Placebo (5; 3)a
F 60 mg qbt (17; 16)b
Placebo Placebo (5; 5)a
Numbers in parentheses = (Number randomized; number treated). Target number treated was 16 subjects per Compound treatment group, and 24 total subjects in the placebo group. aTotal enrollment in all placebo groups was 28 subjects, with a total of 26 subjects treated. bDosing regimen for 60 mg qbt group was 60 mg qd for Days 1—7, bid for Days 8—14, and tid for Days 15-30.
SYNOPSIS OF STUDY
NUMBER OF 132 randomized; 121 treated SUBJECTS
DIAGNOSIS AND Men and women > 18 years of age with evidence of active RA MAIN CRITERIA FOR and receiving stable doses of methotrexate therapy (patients INCLUSION receiving stable doses of non-steroidal anti-inflammatory drugs [NSAIDs] and/or low-dose prednisone were allowed in the study).
TRIAL DRUG/BATCH The test product was COMPOUND, supplied as opaque white, NO(S) hard gelatin capsules containing 30 mg of COMPOUND free base equivalent plus the following excipients: lactose
SYNOPSIS OF STUDY monohydrate, croscarmellose sodium, colloidal silicon dioxide, and magnesium stearate.
REFERENCE DRUG/ Placebo was supplied as capsules that were identical in BATCH NO(S). appearance to those containing active drug. Placebo capsules contained no COMPOUND but were otherwise identical in composition to those containing active drug.
DOSE/ROUTE/REGIM Oral COMPOUND as 30 mg capsules in total doses ranging EN/ DURATION from 0 to 180 mg per day, for 30 days (see Table in Study Design section).
CRITERIA FOR
EVALUATION
EFFICACY: RA and diagnosis of active RA [according to the American College of Rheumatology (ACR) response criteria], ACR 20 response consisting of: tender and swollen joint count, Health Assessment Questionnaire (HAQ), visual analog scale for pain, patient global assessment, physician global assessment; C-reactive protein.
PHARMACOKINETICS: Plasma concentrations for COMPOUND and its metabolites were assessed at selected time points. In treatment period 4, plasma concentrations of methotrexate and its 7-hydroxy metabolite were assessed with and without COMPOUND coadministration in subjects who volunteered for this procedure.
Serum levels of tumor necrosis factor alpha (TNF-α) and
PHARMACODYNAMICS: interleukin-lβ (IL- lβ) were measured. SAFETY: Physical examination, medical history, vital signs, orthostatic vital signs, chest radiograph, 12-lead electrocardiogram (ECG), clinical laboratory evaluations (including serum chemistry, hematology, qualitative urinalysis, and liver function tests), purified protein derivative test for tuberculosis, neurological tests, adverse events, and concomitant medications.
STATISTICAL The safety data, including AEs, laboratory data, vital signs, METHODS ECGs, concomitant medications, and reasons for withdrawal from study, are listed and/or summarized by treatment.
All plasma concentrations and nominal sampling times are summarized for COMPOUND and its metabolites by treatment, with mean, standard deviation, geometric mean (if appropriate), coefficient of variation, minimum, maximum, and sample size. A two-way ANOVA was used to analyze CL/F of COMPOUND and AUC of COMPOUND metabolites.
A posthoc analysis of ECGs was performed by a contract research organization.
SYNOPSIS OF STUDY
EFFICACY RESULTS:
Subjects in the 30 mg tid and 90 mg qd groups showed trends for the greatest rate of response to treatment according to the ACR20 criteria. The response in the 30 mg tid group peaked early (Day 8) with 50% of subjects in that group responding; the level of improvement was fairly well maintained throughout the 30-day treatment period. In the 90 mg qd group, the response gradually increased over time to a maximum response (53% responders) at Day 30. In comparison, the maximum response in the placebo group was 23% responders at Day 30. Maximum ACR20 responses in the other treatment groups were intermediate, with a maximum of 27% responders in the 30 mg qd group at Day 22, 31% in the 60 mg qd group at Day 22, and 38% in the 60 mg tid and 60 mg qbt groups at Day 30. Subjects in the 30 mg tid and 90 mg qd groups generally did better on the individual components of the ACR response, particularly with respect to tender and swollen joint counts, the subject assessment, and performance on the HAQ. The median change from baseline in CRP was generally greatest at Day 8 or Day 15; on both of these test days, all groups displayed greater median percentage reduction from baseline compared to placebo. However, the changes in all groups declined by Day 30, with the exception of the 60 mg qbt group, which displayed its greatest decrease at this time point.
PHARMACOKINETIC RESULTS:
Plasma concentrations of COMPOUND , assessed in all subjects, increased with increasing doses and declined in parallel across all groups. MTX and 7-OH-MTX concentrations, assessed in a small subset of subjects, were approximately 20% larger when the patients received MTX concomitantly with COMPOUND versus MTX alone.
CONCLUSIONS:
Overall, the safety results from this study indicate that COMPOUND is generally well tolerated.
The efficacy data demonstrated that a total daily dose of 90 mg COMPOUND , administered as 90 mg qd or 30 mg tid, can be effective in the treatment of signs and symptoms of rheumatoid arthritis. For this reason, and because this dose level is associated with a favorable safety profile, this daily dose appears to be an optimal dose to evaluate in studies of the safety and efficacy of long-term administration of COMPOUND to patients with rheumatoid arthritis who are also receiving methotrexate.
The exemplary composition of the invention used in this study, Compound (see description, above), was a novel, orally active chemical entity with anti-inflammatory and arthritis disease modifying properties. It is an inhibitor of p38α mitogen activated protein kinase (MAPK), an intracellular enzyme that mediates cellular responses to inflammatory stimuli. Activation of p38α MAPK in acute and chronic inflammatory states leads to the production of proinflammatory mediators, such as interleukin (IL)- lβ, tumor necrosis factor (TNF) α, and prostaglandin E2 (PGE2). Compound blocks the synthesis and activity of TNF-α, and the synthesis of IL- 1 β and cyclooxygenase (COX)-2, a key inducible enzyme involved in the synthesis of inflammatory PGE2.
Compound reduced signs and symptoms in preclinical models of acute inflammation.
Furthermore, in rats with established experimental arthritis, treatment with Compound resulted in a dose-related reduction in signs and symptoms and in disease progression. The preclinical pharmacological profile of Compound , blocking three independent and clinically validated therapeutic targets for rheumatoid arthritis (RA), TNF-α, IL-I β, and COX-2, supports its evaluation as a treatment for reducing signs and symptoms and for delaying disease progression in RA.
In a Phase 1 placebo-controlled, ascending single-dose study in healthy subjects, Compound was well tolerated at doses up to 3 mg/kg by young male and female, and elderly male subjects. The only adverse event (AE) considered to be drug-related was mild dizziness, which was reported only at the 3 and 5 mg/kg dose levels.
The second Phase 1 trial was a placebo-controlled, ascending multiple-dose study in healthy subjects receiving treatment with Compound for 10 consecutive days. Compound was well tolerated at doses of 1 mg/kg once daily (qd), twice daily (bid), and three times daily (tid), but was less well tolerated at the highest dose examined, 2 mg/kg bid. The most frequently reported drug-related AE was mild, transient dizziness that occurred with increased incidence at the 2 mg/kg bid dose level; frequency of recurrence decreased markedly after the first five days of dosing. One subject in the highest dose group discontinued dosing because of alanine aminotransferase (ALT) levels elevated to three times the upper limit of normal (ULN). One study was designed to provide information regarding the safety, tolerability, efficacy, pharmacokinetic (PK), and pharmacodynamic, of Compound in patients with active RA who were taking a stable dose of methotrexate (MTX). Different dosing regimens were used to examine the effects of modifying the pharmacokinetic profile on the efficacy, safety, and tolerability of Compound . The maximum dose level examined in this study was similar to the maximum total daily dose studied in the Phase 1 ascending multiple-dose trial. The 30-day duration of dosing was chosen to extend the safety and tolerability experience with Compound without exposing subjects to undue risk.
The primary objective of this study was to assess the safety and tolerability of multiple oral doses of Compound in patients with active RA who were also receiving stable doses of MTX. Secondary objectives included: To assess the efficacy of multiple oral doses of Compound using the American College of Rheumatology (ACR) response criteria; To determine the PK of multiple oral doses of Compound in patients with active RA who are also receiving MTX; To assess the effects of multiple oral doses of Compound on TNFct and IL- lβ levels in patients with active RA who are also receiving MTX.
Materials and Methods:
Study Design: This multi-center, randomized, double-blind, placebo-controlled, dose- escalating study assessed the safety, tolerability, efficacy, PK, and pharmacodynamics of Compound in patients with active RA who were also receiving MTX. In order to achieve approximately 120 subjects randomized and treated, a total of 132 subjects were randomized during one of four treatment periods. Subjects were assigned to one of seven treatment groups, depending on the treatment period during which they were randomized, with the total daily dose of Compound ranging from 0 to 180 mg over the course of the study. Dosing of the groups was staggered over four treatment periods, with placebo group assignments for each period. Safety and available PK data from the previous treatment period were reviewed before initiating higher dose regimens in the next treatment period. Subjects received study drug for 30 consecutive days and were followed for an additional 28 days after treatment. Subjects were not allowed to re-enroll for participation in a second treatment period.
Overview of Study Design
Formulation and Packaging: Study drug was supplied in the form of capsules (30 mg of
Compound or placebo) in child-resistant blister cards. Each blister card was configured with 7 or 21 wells with each well containing two or three capsules. Each well contained a combination of active study drug and placebo that was determined by the treatment to which the subject was randomized. Five cards were packaged per study drug kit. One study drug kit was supplied to each subject. Individual study drug kits and blister cards were labeled with a unique identifier number and subject number.
Administration of Study Drug: Subjects received study drug as oral capsules containing either 30 mg Compound or placebo. Doses were self-administered except during all scheduled visits, when the dose was administered at the site by the site staff after the appropriate blood samples were obtained. For Treatment Period 1, the dose regimen was two capsules once a day in the morning. For Treatment Periods 2 and 3, the regimen was three capsules three times a day, approximately 4 to 6 hours apart. For Treatment Period 4, the regimen was two capsules once a day in the morning on Days 1-7, two capsules twice a day approximately 10 hours apart on Days 8-14, and two capsules three times daily, approximately 4 to 6 hours apart on Days 15- 30.
Subjects were instructed to take study drug with a meal or snack including solid food (e.g., fruit or crackers). On Days 1 and 30, only the first morning dose was taken, followed by
blood draws for PK assessment. Study drug was taken at the assigned regimen for Days 2-29 inclusive.
Assignment to Treatment Group/Sequence: Subjects were randomly assigned to one of seven treatment groups, depending on the treatment period during which they were randomized.
During Treatment Period 1, subjects were randomized to 30 mg qd, 60 mg qd, or placebo. During Treatment Period 2, subjects were randomized to 90 mg qd, 30 mg tid, or placebo.
During Treatment Period 3, subjects were randomized to 60 mg tid or placebo. During
Treatment Period 4, subjects were randomized to 60 mg qbt or placebo. Each of the four consecutive treatment periods was comprised of a separate cohort of subjects; subjects were not allowed to re-enroll for a second treatment period. Study Assessments and Procedures
Schedule of Assessments: Clinical assessments were performed at baseline, at regularly scheduled intervals during the study, and post-treatment for the safety, efficacy, and pharmacokinetic parameters described in the following sections (see Figure 9, which provides a summary of the Schedule of Assessments, as discussed herein). Concomitant medications and AEs were monitored and recorded throughout the study and the follow-up period. All study-specific procedures and the results of all study evaluations were recorded in the subject's medical record and on the appropriate CRF.
Screening Procedures: Before enrollment, subjects were to have at least two evaluations confirming active RA, as defined in the inclusion criteria. The two evaluations were to be at least 7 and no more than 28 days apart, with the second evaluation performed before the administration of study drug on Day 1. The second evaluation assessed tender and swollen joint counts only.
Subjects participated in the following procedures at the screening visit: signed informed consent form (ICF), medical history (including medications within the previous 14 days), physical examination, vital signs, 12-lead ECG, confirmation of RA diagnosis and of active RA, and clinical laboratory evaluations (including urinalysis, hematology, and biochemistry), chest radiograph, hepatitis panel, pregnancy test, and purified protein derivative test for tuberculosis.
Abnormal laboratory evaluations deemed clinically significant by the investigator could be repeated once within the 28-day period before first dose of study drug administration to meet eligibility requirements.
In Figure 9: * Screening procedures were to be conducted prior to the start of study drug on Day
1. Oral temperature, respiratory rate, seated pulse, and BP. 2Joint count only. 3B lood samples were drawn predose. Blood samples were drawn in the fasted state at screening, Day 1, Day 15, Day 30, and
Day 58. "Hematology and liver function tests only. 5HbsAg, anti-HBs, anti-HBc, anti-HCV. 6For women of childbearing potential only; serum test at screening and urine test on Day 1. 7Blood samples for PK trough analysis were collected predose during Days 2, 8, 15, and 22; and at approximately the same time as the morning dose during Days 31 and 32. 8PK blood samples drawn per schedules in Protocol Appendix Ib (provided in Appendix 6). Visits on Days 2, 31, and 32 were determined by the requirement for a PK blood draw as indicated by Schedule I or Il in Protocol Appendix Ib (provided in Appendix 6). 10On Study Days 1 and 30 in Treatment Periods 2, 3, and 4, subjects were to receive only the First morning dose to allow for PK blood collections, "in a subset of subjects for MTX profiling; samples were taken on the subject's usual day of MTX dosing. 12Collected only serious AEs since last visit. Study Procedures
ACR Response: The ACR response consisted of the following individual components: 68-count tender/painful joint count, 66-count swollen joint count, physician global assessment, patient global assessment, visual analog scale for pain, HAQ, and a measure of acute phase reactants (specifically, CRP). The Sponsor performed the scoring of the ACR response. Tender/swollen joint count: The investigator determined the number of painful or tender joints (68 joints) and the number of swollen joints (66 joints), and assessed each joint for tenderness, pain, and swelling (Appendix 7 of the Protocol, provided in Appendix 6).
HAQ: This questionnaire was self-administered by the subject; the coordinator ensured that each question on the questionnaire had a response (Appendix 6 of the Protocol, provided in Appendix 6).
Visual analog scale for pain: This assessment was done using a 10-cm horizontal visual analog scale. The subject placed a mark on the horizontal line indicating the severity of pain, with the left end of the line representing no pain and the right end representing unbearable pain. Site staff measured the length of the line using a standard ruler. Patient global assessment: This assessment was done used a 10-cm horizontal visual analog scale. The subject was asked to answer the question "Considering all ways arthritis affects you, how well are you doing today?" by placing a mark on the horizontal line, with the left end of the line representing very well and the right end representing very poor. Site staff measured the length of the line using a standard ruler. Physician global assessment: This assessment was done using a 10-cm horizontal visual analog scale. The investigator placed a mark on the horizontal line indicating his/her assessment of the subject's current disease activity, with the left end of the line representing no disease activity and the right end representing maximum disease activity. Site staff measured the length of the line using a standard ruler.
Clinical Laboratory Assessments
Clinical laboratory evaluations, performed at screening and on Days 1, 8, 15, 22, 30, and
58, included serum chemistry, hematology, urinalysis, liver function, and other tests (C-reactive protein, hepatitis panel, purified protein derivative test for tuberculosis, and urine drug screening tests). In addition, female subjects of childbearing potential had serum and urine pregnancy tests performed at specific visits. Routine clinical laboratory evaluations, sample collection, processing, handling, and storage, were provided by the Mayo Central Laboratory for Clinical Trials (Rochester, MN). A local laboratory performed additional laboratory assessments ordered by the investigator that were not part of the protocol.
Pharmacokinetic Assessments: Plasma COMPOUND and metabolites concentrations were determined using a validated LC/MS/MS method (Appendix 3.1). Blood samples for PK analyses were drawn at regularly scheduled intervals. Pharmacokinetic sampling on Days 1 and 30 consisted of two different schedules: subjects on Schedule I provided samples at 0 (pre-dose), 0.5, 2, and 8 hours post-dose on Day 1 and at 0, 1 , 4, and 22-24 hours on Day 30. In a reverse schedule, subjects in Schedule II gave samples at 0, 1, 4, and 22—24 hours on Day 1, and at 0, 0.5, 2, and 8 hours post-dose on Day 30 (Appendix Ib of Protocol, provided in Appendix 6). Each site was assigned to either Schedule I or II. Assignment of a PK sampling schedule could be adjusted with agreement of the investigator and the Sponsor.
Blood samples for PK analysis of COMPOUND levels were collected via an indwelling catheter and/or via direct venipuncture, using 7-mL-draw green-top Vacutainer® collection tubes containing sodium heparin solution. Samples were centrifuged at 3,000 rpm in a refrigerated centrifuge (4°C) for 15 minutes. (If the site did not have access to a refrigerated centrifuge, the sample was to be stored on crushed ice for at least 30 minutes, but no longer than 1 hour before centrifugation.) The separated plasma was then aliquotted into three suitably labeled, 5-mL polypropylene tubes provided by the Sponsor. Plasma samples were frozen within 1 hour of collection and stored at -200C or, if available, at -700C.
Samples for Days 1 and 2 for each individual subject were batched and shipped together overnight to the Sponsor within 48 hours of processing. Individual subject samples for Days 8, 15, 22, and 30—32 similarly were batched and shipped to the Sponsor within 48 hours of processing. Plasma concentrations of COMPOUND and its metabolites were measured by the Sponsor using validated analytical procedures.
Assessments of Cytokines: The analysis of blood samples for the cytokines TNF-α and
IL-I β was performed by the Mayo Central Laboratory for Clinical Trials (Rochester, MN).
Blood samples were collected via an indwelling catheter and/or via direct venipuncture, using 7- mL red-top collection tubes. Blood samples were allowed to clot for 15 to 30 minutes at room temperature and subsequently centrifuged at 3,000 rpm for 10 minutes. The separated serum was aliquotted into separate tubes for analysis of TNF-α and IL- lβ and stored frozen until shipping. A 2-mL archival serum sample was also collected for exploratory analyses of other biomarkers of inflammation. Evaluation of these additional biomarkers is currently underway; results of these analyses will be summarized in a separate report.
Methotrexate Assessments (in a Subset of Subjects): Blood samples from a subset of subjects were drawn on the usual day of the subject's MTX dosing during the last week of study drug administration (between Days 22 and 30, inclusive; i.e., first MTX profiling). These subjects had a second MTX profiling performed on the usual day of MTX dosing between 7 and 21 days after the last day of study drug administration. Subjects were to bring their weekly oral MTX dose (in its original bottle, if possible) with them to the site on each of these visits. On the first visit for MTX profiling during the last week of study drug administration, subjects were to also bring their full daily doses of study drug with them to the clinic. Subjects had the option to have an indwelling catheter to facilitate blood draws. Measurement of MTX and 7-OH-MTX concentrations were performed using a validated HPLC method by Advion Biosciences (Appendix 3.2).
Efficacy Endpoints
Primary Efficacy Endpoint: The primary efficacy endpoint of the study was the rate of
ACR20 response, defined as a 20% reduction in tender and swollen joint counts and 20% improvement in three of the five remaining ACR core set measures: patient and physician global assessments, visual analog scale for pain, HAQ, and CRP. Secondary Efficacy Endpoints: Secondary efficacy endpoints of the study were as follows: Rate of ACR50 response (a 50% reduction/improvement, using the same assessments used for ACR20).
All individual variables of the ACR response assessment: Number of tender joints;
Number of swollen joints; Physician's assessment; Subject's assessment; Pain VAS; HAQ; CRP. Pharmacokinetic Analysis: The PK profile of multiple oral doses of COMPOUND was determined as a secondary objective.
Cytokine Analysis: The effect of multiple oral doses of COMPOUND on TNF-α and
IL-I β levels was assessed as a secondary objective. Percent change and change from baseline for the two cytokines at the Day 8, 30, and 58 visits were the endpoints measured. Efficacy Analysis
Primary Efficacy Variable: The proportion of subjects exhibiting improvement according to the ACR20 response criteria was calculated and summarized for each treatment group for the Day 8, 15, 22, 30, 44, and 58 visits. A pooled proportion was also calculated for all COMPOUND subjects combined.
Secondary Efficacy Variables: Secondary efficacy variables consisted of the proportion of subjects exhibiting improvement according to the ACR50 response criteria and all individual items of ACR response assessments (number of tender joints, number of swollen joints, physician's assessment, subject's assessment, pain VAS, HAQ, and CRP). For each ACR component, actual value, change from baseline, and percentage change from baseline were summarized for each visit.
Interim Efficacy Analysis: Interim efficacy analyses were performed for each of Treatment Periods 1—3 after the last subject in the treatment period completed study procedures through Day 31. Efficacy variables were descriptively analyzed. Data were listed and/or summarized by treatment group but not by subject number. Sponsor personnel responsible for data management and analysis (Associate Director, Biostatistics; Associate Director, Clinical Information Systems; and Senior Clinical Applications Programmer) were unblinded to individual subject numbers and treatment assignments. Sponsor personnel responsible for monitoring the study remained blinded to individual subject numbers and treatment assignments.
The interim efficacy analyses were conducted for administrative purposes only, i.e., to assist in planning future studies, and were not used to alter the conduct of the trial. Results of the analyses were not shared with investigators or other site personnel.
Primary Efficacy Variable
ACR20 Response: The 30 mg tid and 90 mg qd groups showed a greater rate of response to treatment and a greater level of improvement, according to the ACR20 criteria, at all time points compared to subjects in the placebo group (
Table 1). For the 30 mg tid group, improvement could be seen as early as Day 8 (ACR20, 50%); the level of improvement was fairly well maintained throughout the 30-day treatment period. The 90 mg qd group displayed a more gradual response to treatment, but by
Day 30 the proportion of its subjects meeting the ACR20 response criteria was the greatest for all groups at any time point (53%). ACR20 responses (Days 44 and 58) for both groups declined after Day 30 (last day of study drug administration).
At most times measured, the 30 mg qd and 60 mg qd dose groups showed modest improvement relative to placebo, except at Day 30, when responses were similar to that of placebo. For the 60 mg tid and 60 mg qbt groups, the greatest improvement in the ACR20 response was at Day 30; until that time, responses generally were similar to placebo. Subjects in the 30 mg tid group showed the most consistent improvement at the level of ACR20. For all COMPOUND treatment groups combined, ACR20 response was greater than placebo at all time points.
Table 1: Subjects with ACR20 and ACRSO Responses — Efficacy Population
All
Placebo 30 mg qd 60 mg qd 90 mg qd 30 mg tid 60 mg tid 60 mg qbt COMPOUND
(n = 26) (n = 15) (n = 16) (π = 15) (n = 16) (n = 16) (n = 16) (n = 94)
ACR20 Response
Day 8 i (4%) 2 (13%) 2 (13%) 2 (13%) 8 (50%) 3 (19%) 2 (13%) 19 (20%)
Day 15 4 (15%) 3 (20%) 4 (25%) 5 (33%) 7 (44%) 3 (19%) 2 (13%) 24 (26%)
Day 22 5 (19%) 4 (27%) 5 (31%) 4 (27%) 5 (31%) 2 ( 13%) 2 (13%) 22 (24%)
Day 30 6 (23%) 3 (20%) 4 (25%) 8 (53%) 7 (44%) 6 (38%) 6 (38%) 34 (36%)
Day 44 1 (4%) 3 (20%) 3 (19%) 4 (27%) 5 (31%) 2 (13%) 5 (31%)" 22 (23%)
Day 58 3 (12%) 3 (20%) 4 (25%) 5 (33%) 4 (25%) 3 (19%) 4 (25%)" 23 (24%)
ACR50 Response
Day 8 0 (0%) 1 (7%) 1 (6%) 0 (0%) 3 (19%) 0 (0%) 0 (0%) 5 (5%)
Day 15 0 (0%) 1 (7%) 0 (0%) 1 (7%) 4 (25%) 0 (0%) 1 (6%) 7 (7%)
Day 22 0 (0%) 0 (0%) 1 (6%) 1 (7%) 1 (6%) 0 (0%) 0 (0%) 3 (3%)
Day 30 2 (8%) 2 (13%) 1 (6%) 2 (13%) 4 (25%) 0 (0%) 1 (6%) 10 (11%)
Day 44 1 (4%) 1 (7%) 2 (13%) 3 (20%) 3 (19%) 0 (0%) 4 (25%)" 13 (14%)
Day 58 1 (4%)° 1 (7%) 1 (6%) 2 (13%) 2 (13%) 1 (6%) 2 (13%)" 9 (10%)
Numbers in bold indicate values for which COMPOUND group performed better than placebo.
" Includes one subject who withdrew from study drug on Day 11 and received Enbrel on Day 39.
Note: Subjects who terminated from the study prematurely were treated as nonresponders in the analyses of ACR20 and ACR50.
Source: Appendix 1 , Table 9 A.I
Figure 11 illustrates ACR20 Responders by Treatment Group and Study Day — Efficacy Population. Figure 12 illustrates individual ACR Response Components on Day 30 by Treatment Group — Efficacy Population.
Secondary Efficacy Variables
ACR50 Response: Subjects in the 30 mg tid group showed the most consistent ACR50 improvement, with peak improvement in this group at Days 15 and 30 (25% of subjects improved at each time point) (
Table IV For all COMPOUND treatment groups combined, ACR50 response was greater than placebo at all time points through the Day 58 assessment (28 days after the end of study drug treatment, 4% ACR50 response in the placebo group versus 10% in the combined COMPOUND treatment groups). Individual Components of the ACR Response Criteria: The ACR response criteria consist of the following individual components: number of tender joints, number of swollen joints, physician's assessment, subject's assessment, pain VAS, HAQ, and CRP. Each variable was assessed as a secondary efficacy endpoint. Because of a few subjects with extreme values that skewed the mean at several time points for several of the variables, median baseline values and median percentage change from baseline, which better reflect the overall trend for each treatment group, have been presented for all ACR components in tables. Mean values are presented in Appendix 1, Tables 10.1 through 10.7.
Tender Joint Count: Median baseline values for tender joint count (TJC) were similar among treatment groups (range, 21.5 to 31.0; maximum possible count, 68). All treatment groups, including placebo, experienced an improvement in TJC (Table 2). At Days 8 and 15, all COMPOUND groups had a greater reduction in TJC compared to placebo as assessed by median percentage change from baseline (refer to Appendix 1 , Table 10.1.1). The median percentage improvement from baseline was consistently better than placebo for the 60 mg qd, 90 mg qd, 30 mg tid, and 60 mg qbt groups (Table 2).
Table 2- Tender Joint Count — Median Percentage Change from Baseline (Efficacy Population')
Placebo 30 mg qd 60 mg qd 90 mg qd 30 mg tid 60 mg tid 60 mg qbt
(n = 26) (n = 15) (n = 16) (n = 15) (n = 16) (n = 16) (n = 16)
Day 1 Baseline
Median value 21 5 31 0 300 30.8 21 5 290 30 5
Interquartile range 14 O to 31 0 190to 420 18 O to 360 180 to 430 19 O to 28 5 13 O to 43 5 220 to 370
Day 8
Median % change -106 -12.2 -35.0 -36.4 -32.1 -33.9 -35.4
Interquartile range -27 3 to 4 0 -548 to 60 -474 10 -187 -6401O-10 5 -74 3 to -1 8 -45 1 to86 -52 I to 00
Day 15
Median % change -8 3 ^35.7 -45.3 -27.3 -26.1" -20.7" -45.8"
Interquartile range -406 IO 23 I -524 to -3 7 -598 to -294 -667 to -167 -778 to 00 -48 1 to 136 -67410 -13 2
Day 22
Median % change -23 2 -25.0 -42.4 -44.2* -37.4 -19 2 -33-3"
Interquartile range -52 4 Io 50 -54 8 to 4 0 -57 Ho -122 -68 O lo -27 9 -509 to -4 4 -45 5 to -1 3 -54 8 IO -90
Day 30
Median % change -31 8C -31 3 -37.4 -46.4* -54.5 -36.1 -43.5
Interquartile range -57 1 to -100 -737 to 00 -704 to -182 -767 IO-21 1 -73 4 to -5 3 -52 3 to -64 -646 to -4 2
Day 44
Median % change -15 4C -26.2 -O9.1 -33.3 -22.7 -12 9 -44.9°
Interquartile range -33 3 to 97 -47 6 to 200 -564 to -45 -769 to 10 5 -46 9 to I l S -404 to 104 -65 l to 12 9
Day 58
Median % change -18 8C -14 0 -25.2 -35.1 -30.5 -28.1" -32.7°
Interquartile range -360 to 16 1 -429 to 7 1 -696 to 04 -880 to 56 -67 6 to -26 -474 to 103 -58 3 to 27
Numbers in bold indicate values for which COMPOUND group performed better than placebo αn = 15, *n = 14. cn = 25 Source Appendix I, Table 10 I I
Swollen Joint Count: Median baseline values for swollen joint count (SJC) were similar among treatment groups (range, 13.0 to 22.0; maximum possible count, 66). All groups, including the placebo group, had a decrease in median swollen joint count from baseline at all time points. The median percentage improvement from baseline was consistently better than placebo on all test days for the 90 mg qd and 60 mg qbt groups, and on all test days except Day 58 for the 30 mg tid and 60 mg tid groups. Table 3: Swollen Joint Count — Median Percentage Change from Baseline (Efficacy Population)
Placebo 30 mg qd 60 mg qd 90 mg qd 30 mg tid 60 mg tid 60 mg qbt
(n = 26) (H = IS) <π = 16) (n = 15) (n = 16) (n = 16) (n - 16)
Day 1 Baseline
Median value 14.0 17.0 14.0 13.0 14.5 13.5 22.0
Interquartile range 10.0 to 25.0 12.0 to 23.0 11.5 to 23.0 9.0 to 22.0 9.5 to 16.5 10.5 to 21.0 14.5 to 31.5
Day 8
Median % change -0.4 -23.1 -17.4 -29.4 -40.4 -15.8 -14.5
Interquartile range -31.6 to 16.7 - 44.4 to 33.3 -47.5 to -1.4 -4S.5 to -4.8 -62.4 10 -11.9 -56.2 to 7.3 - 48.1 to -4.1
Day IS
Median % change -7.1 -22.2 -28.8 -57.1 -15.4 -33.3 -43.2
Interquartile range -44.4 to 16.7 -53.8 to 0.0 -46.8 to 0.0 - 76.5 to 0.0 -53.3 to 0.0 -43.8 to 33.3 -63.6 to -11.8
Day 22
Median % change -16.3 -41.2 -13.9 -36.4 -19.7 -20.7 -27.3
Interquartile range -31.4 to 0.0 -55.6 to 26.1 -43.2 to 1.9 -71.4 10 -23.1 -43.7 to 3.6 -46.9 to 12.5 -57.9 to -7.9
Day 30
Median % change -23.3 -16.7 -24.9 -45.6 -51.7 -33.0 -36.6
Interquartile range -47.4 to 0.0 -50.0 to 15.4 -48.7 to 0.0 -100.0 to -36.4 -65.1 Io 19.3 -51.3 to 20.2 -57.3 to -4.5
Day 44
Median % change -7.7 -8.7 -7.3 -41.2 -23.0 -17.2 -54.5
Interquartile range -33.3 to 7.7 -42.3 to 14.3 -41.7 to 4.5 -69.210-7.1 -49.0 to 30.0 -41.9 to 26.2 -60.0 to 0.0
Day 58
Median % change -16.7 -6.7 -23.2 ^»5.5 -10.5 -7.5 -22.7
Interquartile range -36.8 to 16.0 -42.3 to 17.4 -48.0 to 14.3 -79.2 to -Λ.5 -44.4 to 27.9 -63.6 to 23 I -63.2 to 0.0
Numbers in bold indicate values for which COMPOUND group performed better than placebo. ° n = 15; 6 n = l4; c n = 25. Source: Appendix 1, Table 10.2.1
Physician's Assessment: Median baseline values for the Physician's Assessment were similar between treatment groups (range, 57.0 to 67.5; range of possible values, 0 to 100).
Table 4, which lists the median percentage change from baseline for each time point, which depicts median percentage change from baseline on Day 30, show that all groups, including placebo, showed some improvement with this assessment. For the majority of treatment groups, the peak response occurred at Day 30.
Table 4: Physician and Subject Global Assessment — Median Percentage Change from Baseline (Efficacy Population)
Placebo 30 mg qd 60 mg qd 90 mg qd 30 mg tid 60 mg tid 60 mg qbt
(n = 26) (n = IS) (n = 16) (n = 15) (n = 16) (n = 16) (n = 16)
Physician Global Assessment
Day 1 Baseline (median) 65.0 62.0 58.5 57.0 63.5 67.5 58.0
Median percentage change from baseline
Day 8 -14.0 -12.8 -23.8 -35.0 -36.6 -12.4 -19.1
Day IS -21.1 -22.6 -23.1 -43J" -25.4* -21.9* -18.3*
Day 22 -31.3 -43.3 -27.2 -25.1° -^17.8 -21.9 -6.7*
Day 30 -39.0c -43.1 -37.3 ^»5.9α -39.2 -33.9 -23.9
Day 44 -28.4C ^34.8 -18.9 -9.9° -22.2 -19.7 0.0*
Day 58 -23.9C -30.6 -33.7 -10.3" -19.1 -34.1* -7.5*
Subject Global Assessment
Day 1 Baseline (median) 50.5 58.0 56.5 48.0 42.5 60.0 65.0
Median percentage change from baseline
Day 8 4.7 -2.7 -1.5 -6.7 -14.1 -10.4 -4.3
Day IS -9.5 -4.5 -9.2 -16.9 -13.6 -8.5* -14.7*
Day 22 -5.7 1.4 -12.5 -3.2° -15.9 -12.5 -13.2*
Day 30 -10.6c -2.0 -18.0 -26.8" -28.0 -24.8 -14.0
Day 44 -4.8C 0.0 0.5 -7.1" 29.7 -11.7 -7.5*
Day 58 -15.6C -5.4 -2.5 -22.7° 28.3 -17.1* -11.9*
Numbers in bold indicate values for which COMPOUND group performed better than placebo.
■ 14; V ■■ IS; cn = 25.
Source: Appendix 1, Table 10.3.1, 10.4.1.
Subject's Assessment: Median baseline values for the Subject's Assessment were similar among treatment groups (range, 42.5 to 65.0; range of possible values, 0 to 100).
Table 4, which lists the median percentage change from baseline for each time point, which depicts median percentage change from baseline on Day 30, show that most groups, including placebo, showed some improvement in this assessment. However, these changes were generally small.
Pain Visual Analog Scale: Median baseline values for the pain VAS were similar among treatment groups (range, 42.0 to 60.0; range of possible values, 0 to 100).
Table 5, which lists the median percentage change from baseline for each time point, shows that all groups, including placebo, experienced at least modest relief from pain with treatment. Maximal improvement in pain VAS scores for most of the active groups were generally similar to that of placebo, with the 60 mg qd and 30 mg tid groups showing a moderately greater improvement over other treatment groups.
Table 5: Pain Visual Analog Scale — Median Percentage Change from Baseline (Efficacy Population)
Placebo 30 mg qd 60 mg qd 90 rag qd 30 mg tid 60 mg tid 60 mg qbt (n = 26) (n = 15) (n ° 16) (n ° 15) (π = 16) (n ■= 16) (" ~ 16)
Day 1 Baseline (median) 51.5 60.0 60.0 50.0 42 O 52.0 53.5
Median percentage change from baseline Day 8 -5.3 -13.8 -9.4 -10.8 -21.3 -25.3 -3.4
Day 15 -6.3 -9.2 -15.9 -6.9 -4.8 -8.2» -1.7»
Day 22 -19.1 -13.3 -28.9* -17.4° -22.7 -1 1.6 -42"
Day 30 -20.8c -11.5 -26.8 -18.9° -28.4 -7.8 -15.3
Day 44 -5.9C -13.8 -13.0 -0.9" 18.2 10.3 2.6*
Day 58 -17.4C -8.1 -24.5 -14.3" 16.5 0.0* 5.1*
Numbers in bold indicate values for which COMPOUND group performed better than placebo. °n = 14; *n = 15 cn = 25. Source: Appendix I, Table 10.5.1.
Health Assessment Questionnaire: Median baseline HAQ values were similar among treatment groups (range, 1.3 to 1.9; range of possible values, 0 to 3.
Table 6, which lists the median percentage change from baseline for each time point, shows that active groups generally responded better than placebo with respect to this assessment. Only the 60 mg tid group failed to perform better than placebo. The 30 mg tid group showed some degree of improvement over placebo in median percentage change from baseline HAQ at all time points through Day 58. Similarly, the 60 mg qbt group performed better than placebo on all test days after Day 8. Table 6: Health Assessment Questionnaire — Median Percentage Change from Baseline (Efficacy Population)
Placebo 30 mg qd 60 mg qd 90 mg qd 30 mg tid 60 mg tid 60 mg qbt
(n = 26) (n = 15) (n = 16) (n = 15) (n = 16) (n = 16) (n = 16)
Day 1 Baseline (median) 1.3α 1.9 1.4 1.6 1.4 1.8 1.3
Median percentage change from baseline
Day 8 0.0° -3.8* -7.1C -6.5* -24.7 0.0 0.0c
Day 15 -6.7° -15.0* 0.0 -5.9 -24.7 0.0r -15.6*
Day 22 0.0rf -11.1 -14.3 -13.4* -33.8 0.0 -18.4*
Day 30 0.0" 0.0 -18.2C -33.0* -30.4 -63 -21.4C
Day 44 -3.3" -5.8* -8.7 -17.9* -28.2 0.0 -12.1*
Day 58 0.0d 0.0 -16.2 -22.5* -14.4 -7.1C -10.6*
Numbers in bold indicate values for which COMPOUND group performed better than placebo. " n = 25; * n = 14 c n = 15^ = 24. Source: Appendix 1, Table 10.6.1.
C-Reactive Protein: Median baseline CRP values were similar among treatment groups (range, 0.4 to 1.3; normal range, 0.02 to 0.8 mg/dL). The median change from baseline in CRP was generally greatest at Day 8 or Day 15; on both of these test days, all groups displayed greater median percentage reduction from baseline compared to placebo. However, the changes in all
groups declined by Day 30, with the exception of the 60 mg qbt group, which displayed its greatest decrease at this time point.
Table 7: C-Reactive Protein — Median Percentage Change from Baseline (Efficacy Population)
Placebo 30 mg qd 60 mg qd 90 mg qd 30 mg rid 60 mg ιid 60 mg qbt (n = 26) (n = 15) (n = 16) (n = 15) (n = 16) (n = 16) («• = ««)
Day 1 Baseline (median),
0.7 1.3 0.7* 0.5 0.7 0.7 0.4 mg/dL
Median percentage change from baseline
Day 8 4.0 -21.3 -46.6* -39.8" -25.4 -47.8 -28.4
Day 15 -1.0c -2S.2 -17.1* -25.7" -11.1 -23.6* -34.8*
Day 22 -10.0 . -5.5 -24.6* -14.6" -13.8 -14.9 -32.6*
Day 30 -0.8'' -18.1 5.5* -6.6° -3.2 -21.6 -39.5
Day 44 -13.5* -23.0 6.3* -5.4' 19.8* 20.9 -27.7*
Day 58 -10.9c -14.4" 1.3° 8.1" 36.4 7.3* -1.25*
Numbers in bold indicate values for which COMPOUND group performed better than placebo. °n = 14; *n = 15; cn = 24; dn = 25; 'n = 13. Source: Appendix 1, Table 10.7.1.
Secondary Analysis:
DAS Scores DAS28 scores were calculated according to the following formula: DAS28 = 0.56*sqrt(TJC28) + 0.28*sqrt(SJC28) + 0.36*ln(CRP+l) + 0.014*(SA) + 0.96, where TJC28 is tender joint count based on 28 joints, S JC28 is swollen joint count based on 28 joints, CRP is C-reactive protein, and SA is subject assessment. The following 28 joints were used: bilateral shoulders, elbows, wrists, carpometacarpal joints, metacarpophalangeal joints no. 1-5, proximal interphalangeal joints no. 2—5, and knees.
Mean DAS28 scores at baseline and mean changes in DAS28 over time are summarized by treatment group in Table 8; shifts in DAS28 scores at Day 30 are summarized by treatment group in
Table 9. Mean baseline DAS28 scores indicated moderate (> 3.2 to < 5.1) to severe
(> 5.1) activity in all treatment groups (range: 4.6 to 5.2). All COMPOUND groups experienced a greater DAS28 response on Day 8 compared to placebo (Table 8). Responses in
the 30 mg qd and 60 mg tid groups were generally similar to placebo from Day 15 through the end of the study period. The response in the 60 mg qd group was greater than placebo through Day 22 and then was similar to placebo thereafter. The 90 mg qd treatment group experienced the greatest overall response, with a 1.2-point decrease from baseline at Day 30; responses in this group were better than placebo at all time points. The 30 mg tid and 60 mg qbt groups also consistently performed better than placebo throughout the treatment period, with a maximum decrease of 1.1 on Day 15 for the 60 mg qbt group and a maximum decrease of 0.9 on Day 8 for the 30 mg tid group. On Day 30, a greater proportion of subjects in all active groups fell into the highest category of improvement in the DAS28 score (> 1.2) compared to the placebo group, with the 90 mg qd, 30 mg tid, and 60 mg qbt groups exhibiting particularly improved responses over placebo at this time point (43%, 38%, and 38%, respectively, compared to 20% in the placebo group) (
Table 9). After cessation of treatment on Day 30, mean DAS28 scores increased in all treatment groups.
Table 8: Disease Activity Scores — Mean Change from Baseline (Efficacy Population)
Placebo 30 mg qd 60mgqd 90mgqd 30 mg tid 60 mg tid 60 mg qbt
(n - 26) <n = 15) (n = 16) (n = 15) (n = 16) (n = 16) (n=16)
DΛS Score (Mean ± SE)
Baseline 48±015 52±013 48 ± 022° 50 ± 032 46±019 5 I±O 18 5O±O25
Change in DAS from Baseline (Mean ± SE)
Day 8 -01±OI2 -0.4 ± 0.20 -0.4 ±0.21 -0.8 ±0.17* -0.9 ± 0.24 -0.6 ±0.15 -0.7 ±0.17
Day IS -O5±019c -0.6 ±0.17 -0.7 ± 0.14 -0.8 ±0.23* -0.9 ± OJl" -04±015° -1.1 ± OJl"
Day 22 -06±015 -05 ± 022 -0.7 ± OJl -0.9 ±0J2* -0.8 ± 0.26 -06±020 -0.8 ±0.29"
Day 30 -08±015° -O8±030 -08±023 -1.2 ±0.34* -0.9 ± 0 J4 -07±017 -0.9 ± 0.30
Day 44 -03±016* -0.6 ±0.27 -0.5 ±0.27 -0.5 ± 033d -0.5 ±0.33" -0.4 ± OJl -0.7±0J3*
Day 58 -05 ±020' -0.6 ± OJS* -03 ±022° -0.9 ±0.36* -03 ± 033 -0.6 ±0.24* -0.7 ± OJO'
Numbers in bold indicate values for which COMPOUND group performed better than placebo * n = 15, * n = 14, °n = 24, 'n = 13, *n = 25 Source Appendix 1, Table 7011
Table 9: Shift in DAS28 Score at Day 30 by Treatment Group — Efficacy Population
Baseline Level Improvement in DAS28 at Day 30 (Decrease from Baseline) n (%) <0.6 > 0.6 to <1.2 >1.2 Total
Placebo
>3.2to<5.1 7 (28%) 7 (28%) 1 (4%) 15(60%)
>5.l 4 (16%) 2 (8%) 4(16%) 10(40%)
Total 11 (44%) 9 (36%) 5 (20%) 25 (100%)"
30 mg qd
>3.2to<5.1 4 (27%) 2 (13%) 2 (13%) 8 (53%)
>5.1 5 (33%) 0 2(13%) 7 (47%)
Total 9 (60%) 2 (13%) 4 (27%) 15(100%)
60 mg qd
>3.2to<5.1 5 (33%) I (7%) 1 (7%) 7 (47%)
>5.1 2 (13%) 2(13%) 4 (27%) 8 (53%)
Total 7 (47%) 3 (20%) 5 (33%) 15(100%)°
90 mg qd
<3.2 0 I (7%) 0 1 (7%)
>3.2to<5.1 1 (7%) I (7%) 4 (29%) 6 (43%)
>5.1 2(14%) 3(21%) 2(14%) 7 (50%)
Total 3(21%) 5 (36%) 6 (43%) 14(100%)"
30 mg tid
>3.2to<5.1 5(31%) 3(19%) 3 (19%) 11 (69%)
>5.1 2(12%) 0 3(19%) 5(31%)
Total 7 (44%) 3(19%) 6 (38%) 16(100%)
60 mg tid
>3.2to<5.1 5(31%) 3(19%) 1 (6%) 9 (56%)
>5.1 1 (6%) 3(19%) 3(19%) 7 (44%)
Total 6 (38%) 6 (38%) 4 (25%) 16(100%)
60 mg qbt
<3.2 1 (6%) 0 0 1 (6%)
>3.2to<5.l 2(12%) 4 (25%) 4 (25%) 10 (62%)
>5.1 3(19%) 0 2(12%) 5(31%)
Total 6 (38%) 4 (25%) 6 (38%) 16(100%)
Source: Appendix I , Table 72.
'One subject missing due to lack of baseline (60 mg qd group) or Day 30 (placebo, 90 mg qd groups) assessment
Pharmacokinetic Results: Mean plasma concentration of COMPOUND over time during the first 24 hours after administration of the first dose of study drug on Day 1 is plotted on a log-linear scale by dose administered. Because only a single dose was administered on Day 1 , subjects randomized to the 30 mg qd and 30 mg tid groups received the same single dose, and their data have been combined. For the same reason, data for subjects randomized to the 60 mg qd, 60 mg tid, and 60 mg qbt groups have also been combined. Data plotted for the 90 mg dose
level contains only data from the 90 mg qd group since this group is the only group administered 90 mg on Day 1. Error bars are not included in the graph; the coefficient of variation (CV%) generally ranged between 50% and 200%.
Following administration of a single dose of 30, 60, or 90 mg on Day 1, plasma concentrations of COMPOUND increased with increasing dose administered, and concentrations declined in parallel for all three dose levels. COMPOUND plasma concentrations for all subjects receiving placebo were below the level of quantitation.
Mean plasma concentration of COMPOUND over time during the 48 hours after administration of the last dose of study drug on Day 30 is plotted on a log-linear scale for each treatment group. Error bars are not included in the graph for clarity; CV% generally ranged between 30% and 100%, with greater error occurring at the latter two time points.
Following administration of the last dose of study drug, plasma concentrations of COMPOUND generally increased with increasing daily dose of COMPOUND . After maximum concentrations were achieved, COMPOUND concentrations declined in parallel across all dose groups. COMPOUND plasma concentrations for all subjects receiving placebo were below the level of quantitation
Mean trough plasma COMPOUND concentrations on Days 1, 8, 15, 22, and 30. Error bars are not included in the graph; CV% generally ranged between 50% and 100%.
When looking at mean trough concentrations of COMPOUND from Day 8 through Day 30, no observable pattern in COMPOUND plasma concentrations was observed, suggesting that COMPOUND concentrations are stable during this time period and have achieved steady state.
Results of Methotrexate Profiling: Mean MTX and 7-OH-MTX concentration-time profiles during the last week of COMPOUND administration (with COMPOUND ) and 7 to 21 days after last dose of COMPOUND (without COMPOUND ) are presented on a log-linear scale. When comparing MTX AUC with COMPOUND coadministration to that without
COMPOUND coadministration, the mean ratio was 1.20 (with COMPOUND : 2247.37 ng»hr/mL; without COMPOUND : 1802.77 ng»hr/mL). For 7-OH-MTX, the mean ratio was 1.19 (with COMPOUND : 1035.57 ng»hr/mL; without COMPOUND : 961.97 ng hr/mL). Thus, higher MTX concentrations were generally achieved when patients received concomitant COMPOUND . However, since this substudy included a small number of subjects (n = 9) and a limited sampling scheme that does not allow for a full characterization of MTX disposition, definitive conclusions on the effect of COMPOUND on the PK of MTX cannot be drawn.
Pharmacodynamic Results
TNF-α: Due to high intra-group variability, most notably within the 60 mg qbt group, median TNF-α values and median changes in TNF-α levels from baseline best summarize the trends in TNF-α levels during treatment. Overall, the median change in TNF-α levels at each time point was small for each treatment group. A majority of subjects in all treatment groups exhibited a small decrease in TNF-α levels from baseline to Day 8, as illustrated by a negative median percent change in TNF-α levels for all treatment groups at this time point (
Table 10). Median TNF-α levels continued to decline for the 60 mg qd and 60 mg qbt groups to Day 30, with the 60 mg qbt group exhibiting the greatest overall median percent decrease (-28.6%) at this time point. Table 10: TNF-α — Median Percentage Change from Baseline (Efficacy Population)
Placebo 30 mg qd 60 mg qd 90 mg qd 30 mg tid 60 mg tid 60 mg qbt (n = 26) (n = 15) (π = 16) (π = 15) (n = 16) (n = 16) (n = 16)
Day 1 Baseline (median), pg/mL 2.9 2.7 3.1" 2.8 1.9* 4.0 4.3* Median percentage change from baseli nnee
Day 8 -3.9 -3.7° -10.0" -12.5 -7.7* -3.6 -15.8*
Day 30 0.0rf -5.9 -13.4" 4.7' 7.4* 16.4 -28.6*
Day 58 5.6C -7.5" 15.3" 38.9* 30.8* -14.9"
Numbers in bold indicate values for which COMPOUND group experienced a greater median reduction than placebo. °n = 14; *n = 15; cn = 23; rfn = 25; 'n = 13^n = U. Source: Appendix 1 , Table 11.1.
IL-I β: There was no consistent effect of treatment on serum IL-I β concentration in any of the treatment groups (
Table 11). The 30 mg qd and 30 mg tid groups each experienced a 100% decrease in IL-Ib levels on Day 8, with levels increasing to near-baseline levels by Day 30. The 90 mg qd group experienced a decrease of the same magnitude at the post-treatment assessment on Day 58.
Table 11 : IL-I β — Median Percentage Change from Baseline (Efficacy Population)
Placebo 30 mg qd 60 mg qd 90 mg qd 30 mg tid 60 mg tid 60 mg qbt (n = 26) (n = 15) (n = 16) (n = 15) (n = 16) (n = 16) (n = 16)
Day 1 Baseline (median), pg/mL 0.0 0.0 0.0° 0.0 0.0 0.2 0.0*
Median percentage change from baseline Day 8 -24.1C -56.5^ -100.0'' -67,-f -100.0' 16.-/ -«3.5ff
Day 30 28.3C -33.2C -25.7* -72.2C -9.9' U S/ -35.3ff
Day 58 -20.5c -71.8" -46.1* -100.0c 106.5' -14.y -5.5s
Numbers in bold indicate values for which COMPOUND group experienced a greater median reduction than placebo. °n = 14; *n = !5; cn = 7: ^ = 6; en = 4/n = l l ;en = 3; *n = 5. Source: Appendix I . Table 12.1.
Safety Results: Safety results are summarized descriptively for all subjects who received at least one dose of study drug. Overall, safety results indicate that COMPOUND was
well tolerated at all doses compared with placebo.
Extent of Exposure to Study Drug and Duration of Treatment: Of the 132 subjects randomized into this study, 121 (92%) received at least one dose of study drug. The mean and median cumulative doses calculated for each treatment group reflect the fact that the large majority of subjects (90/121, 74%) remained in the study and received treatment for 30 (or more) days, with 93% (112/121) receiving treatment for at least 25 days.
The upper range of the cumulative dose received in the 30 mg qd and 90 mg qd groups exceeded that expected for a 30-day treatment period. This occurred because eight subjects were permitted to receive study drug beyond the 30-day treatment period. These exceptions were made to accommodate scheduling problems with respect to the Day 30 visit. For example, one subject (30 mg qd) missed the visit on the last day of dosing due to a viral syndrome, and was therefore permitted to continue dosing until she could make the last visit (34 total days). Six other subjects received study drug for 31 days (60 mg qd;, 90 mg qd; 90 mg qd; 90 mg qd; 90 mg qd; 60 mg qbt), and one subject for 32 days (90 mg qd). The cumulative dose for the latter subject did not exceed the expected cumulative dose for a 30-day treatment with 90 mg qd since this subject had to temporarily discontinue study drug from Day 12 to Day 21 due to an AE.
Summary of the Efficacy Analysis: In this study, efficacy was assessed as a secondary objective, and ACR20 responses were analyzed descriptively rather than statistically. Subjects in the 30 mg tid and 90 mg qd groups showed trends for the greatest rate of response to treatment according to the ACR20 criteria. The response in the 30 mg tid group peaked early (Day 8) with 50% of subjects in that group responding; the level of improvement was fairly well maintained throughout the 30-day treatment period. In the 90 mg qd group, the response gradually increased over time to a maximum response (53% responders) at Day 30. In comparison, the maximum response in the placebo group was 23% responders at Day 30. Maximum ACR20 responses in the other treatment groups were intermediate, with a maximum of 27% responders in the 30 mg qd group at Day 22, 31% in the 60 mg qd group at Day 22, and 38% in the 60 mg tid and 60 mg qbt groups at Day 30. Subjects in the 30 mg tid and 90 mg qd groups generally did better on the individual components of the ACR response, particularly with respect to tender and swollen joint counts, the subject assessment, and performance on the HAQ. The median change from baseline in CRP was generally greatest at Day 8 or Day 15; on both of these test days, all groups displayed greater median percentage reduction from baseline compared to placebo. However, the changes in all groups declined by Day 30, with the exception of the 60 mg qbt group, which displayed its greatest decrease at this time point.
Summary of the Pharmacokinetic Analysis: Plasma concentrations of
COMPOUND , assessed in all subjects, increased with increasing doses and declined in parallel across all groups. MTX and 7-OH-MTX concentrations, assessed in a small subset of subjects, were approximately 20% larger when the patients received MTX concomitantly with COMPOUND versus MTX alone.
Summary of the Pharmacodynamic Analysis: No formal statistical analysis of the effect of COMPOUND on pharmacodynamic markers of inflammation was planned or conducted. Conclusions: Overall, the safety results from this study indicate that
COMPOUND is generally well tolerated. With the exception of elevations in liver transaminases, there were no clinically significant, drug-related changes in laboratory values and no clear trend indicating a dose-relationship in the incidence of AEs. Of the ten subjects with AEs related to abnormal liver function tests, seven were in one of the two highest dose groups. These data suggest that the 60 mg tid dose regimen may be the highest tolerated dose level for rheumatoid arthritis patients receiving concomitant MTX, a patient population particularly susceptible to elevations in liver transaminases.
The efficacy data suggest that a total daily dose of 90 mg COMPOUND , administered as 90 mg qd or 30 mg tid, may be effective in alleviating symptoms of rheumatoid arthritis. For this reason, and because this dose level is associated with a favorable safety profile, this daily dose appears to be an optimal dose to evaluate in future studies of the safety and efficacy of long-term administration of COMPOUND to patients with rheumatoid arthritis.
EXAMPLE 3 - IR CAPSULES
IR capsules were also developed for clinical studies. Formulation development included an excipient compatibility study to identify suitable excipients in several functional categories and a feasibility evaluation of formulations and processes including wet granulation with high shear mixing, direct blending, and dry granulation with slugging. Trial lots were evaluated for content uniformity and powder flow. Optimal uniformity and flow for capsule filling were obtained with the dry granulation process. Below is a description of the capsules and manufacturing process and that was developed.
TABLE 4: Compound 30-mg Capsules Formulation Component Weight (mg/capsule)
Compound Drug Substance 30.00
Lactose Moπohydrate Fast-Flo 255.60
Croscarmellose Sodium Type A 51.00
(Ac-Di-SoI)
Colloidal Silicon Dioxide M5P 0.85
Magnesium Stearate Non-Bovine 2.55
The components are dry granulated together by "slugging" in a press and then milled to an appropriately sized granulation by an oscillating mill. The milled material is blended to ensure uniformity before the encapsulation process. The specific steps are described as follows:
1. Add croscarmellose sodium, Compound drug substance, and lactose monohydrate Fast-Flo to a V-blender and blend.
2. Remove material from V-blender and pass material through an oscillating mill. Return milled material to V-blender. 3. Pre-mix a small amount of the blend with colloidal silicon dioxide in a polyethylene bag. Screen the pre-mix through a screen into V-blender containing the blended drug substance. Blend.
4. Pre-mix a small amount of the blend with magnesium stearate in a polyethylene bag. Screen the pre-mix through a screen into a V-blender containing the blended drug substance. Blend.
5. Compress the blend on a press into approximately 2.5 gram tablets/slugs to granulate the powders together.
6. Mill the compressed tablets/slugs down to an acceptable granulation powder in an oscillating mill.
7. Blend the milled material in a V-blender.
8. Fill capsules with 340 mg of blend via a tamping encapsulator.
EXAMPLE 4 - Clinical Study Directed to Evaluation of Compound pK
A human study was conducted investigating the pharmacokinetics of both IR and ER formulations of the Compound. Subjects were treated as outlined below and blood plasma was sampled accordingly. The results are provided in the following table 8.
Table 8: Human Pharmacokinetic Studies
Mean Values (CV%) for Plasma pK Parameters Following Compound Single Dose
Treatments

EXAMPLE 5
In a placebo-controlled, ascending single dose study, the Compound was administered to 35 male subjects. Six different dosages were provided at 0.03, 0.1, 0.3, 1, 3, and 5 mg/kg levels. Results from subjects who were in a fasted state are provided in Figure 13.
EXAMPLE 6 - Double Blind Placebo Controlled Study of Ascending Multiple Oral Doses of Compound
The following study involved ascending multiple oral doses of Compound. Four dose regimens of 1 mg/kg od, bid, and tid and 2mg/kg bid were studied will all dosed being administered in the fed state. Over a period of 12 days, the effect of Compound on ex vivo plasma levels of LPS induced TNFα was measured at the indicated points from dosing. The results of the study are provided in Figures 14 and 15. On day 12, there was a marked inhibition of the ex vivo production of TNFα at all dose levels of the Compound. As shown in Figure 15, the relationship of blood plasma levels of Compound and the inhibition of TNFα indicated an increased inhibition of TNFα production up to plasma concentrations of approximately 200 ng/mL of Compound. Above this level, TNFα values had generally reached a plateau. A summary of related data is included in the table below:

AUClO-24 h) 3628 5170 2894 7849 3585 12951 6705 15631 '
(iiε li/nilj (47.3) (30.2) (23.8) C7.8) (I S S) ( 13.8) (24.0) (24.1)
AUC(O-) 3847 3007 3730 7075 fng.h/inl.) (50.5) (25.6) (16.6) (25.9)
488 585 419 491 797-*-«= 887 1 128-=-»*
(ng/ιiιL) (29.6.1 (20.5) (25.3) (20.6) (17 0) ( 12.3) (28.3) (23 6)
2.50 2.50 200 200-** 2.00 1.50—* 2.50 200««-»
(IO ( 1.00-4.00) (2.00-4.00) (2.00-j.OOι (2.00-200) Q.00-3.UO) ( 1.00-3.00) U .00-U)O) (2.00-2.00)
C, 24.4 48.6 60.3 |46*<"' 253 283*"' 174 288-"~. ma/ml.) ( I 10) (70.3 ) (39.9) (40.0) ( 12.2) (21.9) (33.8) (3.VU
5.85 4.92 5.35 5.75
Ch) (23.2) (38.2) (l O.'J) (19.0)
MRT 8 28 6.94 7.23 7.98
Ih) U 6.6) (20.71 (119) ( 16.1 )
CUF 4-?3 3.22 5.54 4.25 4.47 J 86 4.71 4.26'
IniL/miπ/kgl (50.5) (30.2) (23.6) ( 17.8) (166) ( I38ι (25.9) (24.1 )
V,/F 2 19 2.36 207 234
(UkSl (33.0) (22.1 ) f 13-4) ( 15.6)
RA; I.06- 1.43 1.20" 1.63 1.44- 1.66 1.22' " 1.42'
O. QH) ( 17.9) (7.76) (8.02) (6.77) <3.75> (4.04) (975)
KA. I-OCi' 1.20 I.201 1-51 1.44' 1.50 1.22' - l.2h-
(3-08) (8.821 (7.76) (5.46) (6.77) ( 13.3) (4.94) (31 4 )
RL 1.34 1.36 1.16 1.16s
(20.41 (7.33) (8.58) (7.34)
N= N'umbcr of subjects studied
Geometric mean (CVx) data ore presented
+ Median (inin-mnx). * predicted value:
UAi & RA; = Accumulation ratios based an AUC(O-T) and Cn.. RL = Linearity ratio
S N=3
* VVF = CLVF Day 1 1/?., Day 12. "■"* AUC(H-Tl = AϋC(0-24 h)/3. "•" Overall dose
EXAMPLE 7 - Canine Studies
The dog was used as a model species to evaluate the effect of dosage form on the pharmacokinetics of Compound. A study group of 6 animals (3 male, 3 female) was tested. Single doses, either as IR capsules (multiples of 30-mg capsules), IR tablets (30 or 90-mg) and ER tablets (60 or 120-mg, two formulation types) were administered in the fed state; fasted state effects were evaluated for a smaller group of formulations. The study demonstrated that (1) AUC (area-under-the-curve, exposure, bioavailability) in the fed state is dose-proportional and independent of dosage form, (2) extended-release formulations blunt Cmax (maximum concentration), and (3) extended-release formulations prolong tmax (time of maximum concentration).
Data from the study is provided in Figures 16 to 26.
EXAMPLE 8 - ALLEVIATION OF PATIENT REPORTED SIDE EFFECTS From a review of data generated from different studies where human patients have been administered the Compound, certain trends can be identified with respect to reports of adverse events. In particular, episodes of dizziness have been reported by patients in certain instances. When reviewed in the context of relevant plasma level concentrations of the Compound, there appears to be a correlation between a higher Cmax levels and these incidents of dizziness. The data from this review is provided in Figures 27 and 38.
Based upon this data, one embodiment of the invention is to provide a dosing strategy for the Compound which will result in Cmax levels less than 2000 ng/mL yet still achieve pharmacologically effective levels in the blood. One skilled in the art will recognize that this objective can be accomplished through the use of an extended or sustained release formulation of the Compound such as those that are provided herein.
While the invention has been described in detail with reference to certain preferred aspects thereof, it will be understood that modifications and variations are within the spirit and scope of that which is described and claimed.
Claims
1. A pharmaceutical composition comprising a therapeutically effective amount of a compound of the formula

the pharmaceutically acceptable salts or pharmaceutically acceptable forms thereof wherein said composition is in a solid formulation suitable for oral administration.
2. The pharmaceutical composition of claim 1 further comprising a pharmaceutically acceptable excipient.
3. The pharmaceutical composition of claim 1 further comprising two or more pharmaceutically acceptable excipients.
4. The pharmaceutical composition of claim 1, wherein the compound is admixed with at least one pharmaceutically acceptable excipient selected from lactose, Avicel, Prosolv, pregelatinized starch, HPMC, NaCMC, croscarmellose sodium (in low amounts), sodium starch glycolate, magnesium stearate, stearic acid.
5. The pharmaceutical composition of claim 1 wherein the compound is formulated as a hydrochloride salt.
6. The pharmaceutical composition of claim 1 further comprising at least one excipient selected from calcium phosphate dibasic, povidone, sodium lauryl sulfate or a combination thereof such that said formulation exhibits an extended release profile of compound.
7. The solid oral formulation of claim 1, wherein the compound is formulated as a tablet, a pill or a capsule.
8. An oral dosage form comprising: the compound (2R-trans)-6-chloro-5-[[4-[(4- fluorophenyl)methyl]-2,5-dimethyI- 1 -piperazinyl]carbonyl]-N,N, 1 -trimethyl-alpha-oxo- 1 H- indole-3-acetamide as represented by the formula

an oral dosing structure adapted to release said compound at rates that provide maximum plasma concentration Cmax of the compound which satisfy the relationship :
about 5 ng/mL < Cmax <_ 4500 ng/mL.
9. The oral dosage form of claim 8, wherein the maximum plasma concentration Cmax of the compound satisfies the relationship:
about 5 ng/mL < Cmax < 2000 ng/mL.
10. The oral dosage form of claim 8, wherein the maximum plasma concentration Cmax of the compound satisfies the relationship:
about 20 ng/mL < Cmax < 450 ng/mL.
11. The oral dosage form of claim 8, wherein the maximum plasma concentration Cmax of the compound satisfies the relationship:
about 200 ng/mL < Cmax < 4100 ng/mL.
12. The oral dosage form of claim 8, wherein the mean, single dose, area under a plasma concentration-time curve AUCjnf of the compound which satisfies the relationship:
about 500 ng hr/mL < AUCinf < 25000 ng hr/mL.
13. The oral dosage form of claim 8, wherein the mean, single dose, area under a plasma concentration-time curve AUCιnf of the compound which satisfies the relationship: about 1100 ng hr/mL < AUCinf < 25000 ng hr/mL.
14. A method for treating a p38 kinase mediated disorder, said method comprising administering to a patient in need thereof, the pharmaceutical composition of claim 1.
15. The method of claim 14 wherein said disorder is a proinflammatory disorder.
16. The method of claim 15 wherein said disorder is rheumatoid arthritis.
17. The method of claim 16 wherein the patient is also treated with an additional rheumatoid arthritis medication.
18. The method of claim 17 wherein the additional rheumatoid arthritis medication is methotrexate.
19. The method of claim 14 wherein said disorder involves a bone related disorder.
20. The method of claim 14, wherein the compound or pharmaceutically acceptable salt or form thereof is administered at a dose of about 30 mg, 60 mg, or 90 mg, which dose is administered once daily (qd), twice daily (bid), or three times daily (tid).
21. The method of claim 14, wherein the compound or pharmaceutically acceptable salt is administered at a total daily dose of about 30 to 350 mg.
22. The method of claim 21, wherein the compound or pharmaceutically acceptable salt is administered at a total daily dose of about 90 mg.
23. The method of claim 14 wherein the compound or pharmaceutically acceptable salt or form thereof is administered at dosages of between about 1 and 3 mg/kg once daily (qd), between about 1 and 3 mg/kg twice daily (bid), or between about 1 and 3 mg/kg three times daily (tid).
24. The method of claim 14 wherein the compound or pharmaceutically acceptable salt is administered at a dosage of about 1 mg/kg once daily (qd), about 1 mg/kg twice daily (bid), or about 1 mg/kg three times daily (tid).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US82324806P | 2006-08-22 | 2006-08-22 | |
| US60/823,248 | 2006-08-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008024391A1 true WO2008024391A1 (en) | 2008-02-28 |
Family
ID=38791998
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/018555 WO2008024391A1 (en) | 2006-08-22 | 2007-08-22 | Pharmaceutical formulations of an indole-type derivative and related methods of use |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008024391A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10342786B2 (en) | 2017-10-05 | 2019-07-09 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US11291659B2 (en) | 2017-10-05 | 2022-04-05 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000071535A1 (en) * | 1999-05-21 | 2000-11-30 | Scios Inc. | INDOLE-TYPE DERIVATIVES AS INHIBITORS OF p38 KINASE |
-
2007
- 2007-08-22 WO PCT/US2007/018555 patent/WO2008024391A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000071535A1 (en) * | 1999-05-21 | 2000-11-30 | Scios Inc. | INDOLE-TYPE DERIVATIVES AS INHIBITORS OF p38 KINASE |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10342786B2 (en) | 2017-10-05 | 2019-07-09 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US10537560B2 (en) | 2017-10-05 | 2020-01-21 | Fulcrum Therapeutics. Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US11291659B2 (en) | 2017-10-05 | 2022-04-05 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US11479770B2 (en) | 2017-10-05 | 2022-10-25 | Fulcrum Therapeutics, Inc. | Use of p38 inhibitors to reduce expression of DUX4 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4108980B2 (en) | Sustained release ranolazine formulation | |
| US5631296A (en) | Drugs containing S(+)-ibuprofen | |
| JP2020055816A (en) | Modified release 1-[(3-hydroxy-adamant-1-ylamino)-acetyl]-pyrrolidine-2(s)-carbonitrile formulation | |
| EP3103448B1 (en) | Pharmaceutical compositions comprising an s1p modulator | |
| KR101615108B1 (en) | Compositions and methods for treating centrally mediated nausea and vomiting | |
| CN1420766A (en) | Partial fatty acid oxidation inhibitors in treatment of congestive heart failure | |
| FR2460667A1 (en) | PHARMACEUTICAL COMPOSITION WITH PROLONGED RELEASE BASED ON A SOLID DRUG SUBSTANCE | |
| EP3143990B1 (en) | Formulations comprising 2-amino-2-[2-(4-octylphenyl)ethyl]propane-1,3-diol | |
| CA2600407A1 (en) | Fibrate compositions containing a surfactant mixture of peg-600 and poloxamer 407 | |
| US20120045505A1 (en) | Fixed dose drug combination formulations | |
| JP2017510607A (en) | S1P modulator immediate release dosing regimen | |
| WO2013100630A1 (en) | Fixed dose combination formulation comprising losartan, amlodipine and hydrochlorothiazide | |
| WO2008024391A1 (en) | Pharmaceutical formulations of an indole-type derivative and related methods of use | |
| JP2010528112A (en) | How to treat diabetes | |
| AU2019404552A1 (en) | Amorphous sparsentan compositions | |
| US8183287B2 (en) | Pharmaceutical formulations and compositions of a selective antagonist of either CXCR2 or both CXCR1 and CXCR2 and methods of using the same for treating inflammatory disorders | |
| CN107693516A (en) | A kind of DEFERASIROX pharmaceutical composition and its pharmaceutical preparation, preparation method and purposes | |
| EP1465607B1 (en) | Pharmaceutical formulations with modified release | |
| EP1707197A1 (en) | Formulations containing fenofibrate and a surfactant mixture | |
| JPS6058726B2 (en) | Antiallergic, analgesic, and sedative agent containing purine derivatives as active ingredients | |
| AU2022223224A1 (en) | Combination therapy of obicetrapib and ezetimibe for use in statin intolerant patients suffering from hyperlipidemia or mixed dyslipidaemia | |
| AU2013263769A1 (en) | Paediatric compositions for treating multiple sclerosis | |
| JPH04352723A (en) | Akathisia therapeutic agent |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07811475 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| 32PN | Ep: public notification in the ep bulletin as address of the adressee cannot be established |
Free format text: NOTING OF LOSS OF RIGTS PURSUANT TO RULE112(1) EPC, 1205A DATED 04.05.09 |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07811475 Country of ref document: EP Kind code of ref document: A1 |