WO2008002244A2 - Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) - Google Patents
Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) Download PDFInfo
- Publication number
- WO2008002244A2 WO2008002244A2 PCT/SE2007/000620 SE2007000620W WO2008002244A2 WO 2008002244 A2 WO2008002244 A2 WO 2008002244A2 SE 2007000620 W SE2007000620 W SE 2007000620W WO 2008002244 A2 WO2008002244 A2 WO 2008002244A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- imidazol
- fluoro
- pyrimidin
- amino
- Prior art date
Links
- XGOXJRQESOESLO-UHFFFAOYSA-N CC(C)N(Cc1ccccc1)C(c(nc1)ccc1Nc(nc1)nc(-c2cnc(C)[n]2C2CCOCC2)c1F)=O Chemical compound CC(C)N(Cc1ccccc1)C(c(nc1)ccc1Nc(nc1)nc(-c2cnc(C)[n]2C2CCOCC2)c1F)=O XGOXJRQESOESLO-UHFFFAOYSA-N 0.000 description 1
- CAPUIXOUSBYPPX-UHFFFAOYSA-N CC(C)Sc(nc1)ccc1Br Chemical compound CC(C)Sc(nc1)ccc1Br CAPUIXOUSBYPPX-UHFFFAOYSA-N 0.000 description 1
- ULLAFWIGAOOHJV-UHFFFAOYSA-N CC(c1cnc(C)[n]1C1CCOCC1)=O Chemical compound CC(c1cnc(C)[n]1C1CCOCC1)=O ULLAFWIGAOOHJV-UHFFFAOYSA-N 0.000 description 1
- WEQSMXWTBRYPDN-UHFFFAOYSA-N Cc1cnc(C)[n]1C Chemical compound Cc1cnc(C)[n]1C WEQSMXWTBRYPDN-UHFFFAOYSA-N 0.000 description 1
- NJHQUYCNRKNNIL-UHFFFAOYSA-N Cc1ncc(-c(nc(Nc2cc(Cl)c(C(N3CCCCC3)=O)nc2)nc2)c2F)[n]1C Chemical compound Cc1ncc(-c(nc(Nc2cc(Cl)c(C(N3CCCCC3)=O)nc2)nc2)c2F)[n]1C NJHQUYCNRKNNIL-UHFFFAOYSA-N 0.000 description 1
- WDRSDPKRIOMBFL-UHFFFAOYSA-N Cc1ncc(-c(nc(Nc2ccc(C(N(CC3)CCC3(F)F)=O)nc2)nc2)c2F)[n]1C1CCOCC1 Chemical compound Cc1ncc(-c(nc(Nc2ccc(C(N(CC3)CCC3(F)F)=O)nc2)nc2)c2F)[n]1C1CCOCC1 WDRSDPKRIOMBFL-UHFFFAOYSA-N 0.000 description 1
- IVFUCSKFJNKPSF-UHFFFAOYSA-N Cc1ncc(-c(nc(Nc2cnc(C(N3CCCCC3)=O)c(Cl)c2)nc2)c2F)[n]1C1CCOCC1 Chemical compound Cc1ncc(-c(nc(Nc2cnc(C(N3CCCCC3)=O)c(Cl)c2)nc2)c2F)[n]1C1CCOCC1 IVFUCSKFJNKPSF-UHFFFAOYSA-N 0.000 description 1
- UXAHSQDTQLJDMM-UHFFFAOYSA-N Nc(nc1)nc(-c2cnc(C(F)(F)F)[n]2C2CCOCC2)c1F Chemical compound Nc(nc1)nc(-c2cnc(C(F)(F)F)[n]2C2CCOCC2)c1F UXAHSQDTQLJDMM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/14—Drugs for dermatological disorders for baldness or alopecia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
Definitions
- the present invention relates to new compounds of formula (I), as a free base or a pharmaceutically acceptable salt thereof, to pharmaceutical formulations containing said compounds and to the use of said compounds in therapy.
- the present invention further relates to a process for the preparation of compounds of formula (I) and to new intermediates used therein.
- Glycogen synthase kinase 3 is a serine / threonine protein kinase composed of two isoforms ( ⁇ and ⁇ ), which are encoded by distinct genes but are highly homologous within the catalytic domain. GSK3 is highly expressed in the central and peripheral nervous system. GSK3 phosphorylates several substrates including tau, ⁇ -catenin, glycogen synthase, pyruvate dehydrogenase and elongation initiation factor 2b (eIF2b). Insulin and growth factors activate protein kinase B, which phosphorylates GSK.3 on serine 9 residue and inactivates it.
- eIF2b elongation initiation factor 2b
- AD dementias Alzheimer's Disease (AD) dementias, and taupathies AD is characterized by cognitive decline, cholinergic dysfunction and neuronal death, neurofibrillary tangles and senile plaques consisting of amyloid- ⁇ deposits.
- Glycogen synthase kinase 3 ⁇ (GSK3 ⁇ ) or Tau phosphorylating kinase selectively phosphorylates the microtubule associated protein Tau in neurons at sites that are hyperphosphorylated in AD brains.
- Hyperphosphorylated tau has lower affinity for microtubules and accumulates as paired helical filaments, which are the main components that constitute neurofibrillary tangles and neuropil threads in AD brains.
- Neurofibrillary tangles are consistently found in diseases such as AD, amyotrophic lateral sclerosis, parkinsonism- dementia of Gaum, corticobasal degeneration, dementia pugilistica and head trauma, Down's syndrome, postencephalatic parkinsonism, progressive supranuclear palsy, Niemann-Pick's Disease and Pick's Disease.
- GSK3 ⁇ preferentially labels neurofibrillary tangles and has been shown to be active in pre-tangle neurons in AD brains. GSK3 protein levels are also increased by 50% in brain tissue from AD patients.
- GSK3 ⁇ phosphorylates pyruvate dehydrogenase, a key enzyme in the glycolytic pathway and prevents the conversion of pyruvate to acetyl-Co-A (Hoshi et al., PNAS 1996, 93: 2719-2723).
- Acetyl-Co-A is critical for the synthesis of acetylcholine, a neurotransmitter with cognitive functions.
- Accumulation of amyloid- ⁇ is an early event in AD.
- GSK Tg mice show increased levels of amyloid- ⁇ in brain.
- PDAPP mice fed with Lithium show decreased amyloid- ⁇ levels in hippocampus and decreased amyloid plaque area (Su et al., Biochemistry 2004, 43: 6899-6908).
- GSK3 ⁇ inhibition may have beneficial effects in progression as well as the cognitive deficits associated with Alzheimer's disease and other above-referred to diseases.
- GSK3 ⁇ activity is increased in cellular and animal models of neurodegeneration such as cerebral ischemia or after growth factor deprivation.
- the active site phosphorylation was increased in neurons vulnerable to apoptosis, a type of cell death commonly thought to occur in chronic and acute degenerative diseases such as cognitive disorders, Alzheimer's Disease, Parkinson's Disease, amyotrophic lateral sclerosis, Huntington's Disease and HIV dementia and traumatic brain injury; and as in ischemic stroke.
- Lithium was neuroprotective in inhibiting apoptosis in cells and in the brain at doses that resulted in the inhibition of GSK3 ⁇ .
- GSK3 ⁇ inhibitors could be useful in attenuating the course of neurodegenerative diseases.
- Bipolar Disorders (BD) BD
- Bipolar Disorders are characterised by manic episodes and depressive episodes. Lithium has been used to treat BD based on its mood stabilising effects. The disadvantage of lithium is the narrow therapeutic window and the danger of overdosing that can lead to lithium intoxication. The discovery that lithium inhibits GSK3 at therapeutic concentrations has raised the possibility that this enzyme represents a key target of lithium's action in the brain (Stambolic et al., Curr. Biol. 1996, 68(12):1664-1668, 1996; Klein and Melton; PNAS 1996, 93:8455-8459; Gould et al., Neuropsychopharmacology, 2005, 30:1223-1237).
- GSK3 inhibitor has been shown to reduce immobilisation time in forced swim test, a model to assess on depressive behavior (O'Brien et al., J Neurosci 2004, 24(30): 6791-6798).
- GSK3 has been associated with a polymorphism found in bipolar II disorder (Szczepankiewicz et al., Neuropsychobiology. 2006, 53: 51-56). Inhibition of GSK3 ⁇ may therefore be of therapeutic relevance in the treatment of BD as well as in AD patients that have affective disorders.
- GSK3 is involved in signal transduction cascades of multiple cellular processes, particularly during neural development.
- GSK3 ⁇ levels were 41% lower in the schizophrenic patients than in comparison subjects.
- This study indicates that schizophrenia involves neurodevelopmental pathology and that abnormal GSK3 regulation could play a role in schizophrenia.
- reduced ⁇ -catenin levels have been reported in patients exhibiting schizophrenia (Cotter et al., Neuroreport 1998, 9(7): 1379-1383).
- Atypical antipsychotic such as olanzapine, clozapine, quetiapine, and ziprasidone, inhibits GSK3 by increasing ser9 phosphorylation suggesting that antipsychotics may exert their beneficial effects via GSK3 inhibition (Li X. et al., Int. J.of Neuropsychopharmacol, 2007, 10: 7-19, Epubl. 2006, May 4).
- Insulin stimulates glycogen synthesis in skeletal muscles via the dephosphorylation and thus activation of glycogen synthase.
- GSK3 phosphorylates and inactivates glycogen synthase via dephosphorylation.
- GSK3 is also over-expressed in muscles from Type II diabetic patients (Nikoulina et al., Diabetes 2000 Feb; 49(2): 263- 71). Inhibition of GSK3 increases the activity of glycogen synthase thereby decreasing glucose levels by its conversion to glycogen.
- GSK3 inhibitors lowered plasma glucose levels up to 50 % (Cline et al., Diabetes, 2002, 51:
- GSK3 inhibition may therefore be of therapeutic relevance in the treatment of Type I and Type II diabetes and diabetic neuropathy.
- GSK3 phosphorylates and degrades ⁇ -catenin.
- ⁇ -catenin is an effector of the pathway for keratonin synthesis, ⁇ -catenin stabilisation may be lead to increase hair development.
- Mice expressing a stabilised ⁇ -catenin by mutation of sites phosphorylated by GSK3 undergo a process resembling de novo hair morphogenesis (Gat et al., Cell, 1998, 95(5): 605-14)).
- the new follicles formed sebaceous glands and dermal papilla, normally established only in embryogenesis.
- GSK3 inhibition may offer treatment for baldness.
- GSK3 inhibitors provide anti-inflammatory effects.
- Inflammation is a common feature of a broad range of conditions including Alzheimer's Disease and mood disorders.
- GSK3 is overexpressed in ovarian, breast and prostate cancer cells and recent data suggests that GSK3b may have a role in contributing to cell proliferation and survival pathways in several solid tumor types.
- GSK3 plays an important role in several signal transduction systems which influence cell proliferation and survival such as WNT, PI3 Kinase and NFkB.
- GSK3b deficient MEFs indicate a crucial role in cell survival mediated NFkB pathway (Ougolkov AV and Billadeau DD., Future Oncol. 2006 Feb; 2(1): 91-100.).
- GSK3 inhibitors may inhibit growth and survival of solid tumors, including pancreatic, colon and prostate cancer.
- GSK3 inhibitors could be used for treatment of bone-related disorders. This has been discussed in e.g. Tobias et al., Expert Opinion on Therapeutic Targets, Feb 2002, pp 41-56.GSK3 inhibitors could be used for treatment of bone-related disorders or other conditions, which involves a need for new and increased bone formation. Remodeling of the skeleton is a continuous process, controlled by systemic hormones such as parathyroid hormone (PTH), local factors (e.g. prostaglandin E2), cytokines and other biologically active substances. Two cell types are of key importance: osteoblasts (responsible for bone formation) and osteoclasts (responsible for bone resorption).
- PTH parathyroid hormone
- prostaglandin E2 local factors
- cytokines cytokines and other biologically active substances.
- Two cell types are of key importance: osteoblasts (responsible for bone formation) and osteoclasts (responsible for bone resorption).
- Osteoporosis is a skeletal disorder in which low bone mass and deterioration of bone microarchitecture lead to increased bone fragility and fracture risk.
- the two main strategies are to either inhibit bone resorption or to stimulate bone formation.
- the majority of drugs currently on the market for the treatment of osteoporosis act to increase bone mass by inhibiting osteoclastic bone resorption. It is recognized that a drug with the capacity to increase bone formation would be of great value in the treatment of osteoporosis as well as having the potential to enhance fracture healing in patients.
- the present invention relates to a compound of formula (I):
- R 1 is selected from sulphamoyl, carbamoyl, a group -R 5 -R 6 and a nitrogen linked 4-7 membered saturated ring which optionally contains an additional nitrogen, oxygen or sulphur atom; wherein said ring is optionally substituted on carbon by one or more R 7 ; and wherein if said ring contains an additional nitrogen atom that nitrogen is optionally substituted by R 8 ;
- At least one of X 1 , X 2 , X 3 and X 4 is selected from N, the other three X 1 , X 2 , X 3 or X 4 are independently selected from N or C(R 9 ), provided that not more than two of X 1 , X 2 , X 3 or X 4 are selected from N;
- R 2 is halo or cyano
- R 3 is methyl, 3-tetrahydropyranyl or 4-tetrahydropyranyl, wherein the tetrahydropyranyl group is optionally substituted on carbon by one or more R 10 ;
- R 4 is selected from hydrogen, halo, cyano and C 1-3 alkyl, wherein C 1-3 alkyl is optionally substituted with one or more halo;
- R 5 is selected from -0-, -C(O)-, -C(O)O-, -C(O)N(R 11 )-, -S(OV and -SO 2 N(R 12 )-; wherein R 11 and R 12 are independently selected from hydrogen or C 1-6 alkyl and said alkyl is optionally substituted by one or more R 13 ; and r is O, 1 or 2;
- R 6 is selected from C 1-6 alkyl, carbocyclyl and heterocyclyl; wherein R 6 is optionally substituted on carbon by one or more R 14 ; and wherein if said heterocyclyl contains an - NH- moiety that nitrogen is optionally substituted by a group selected from R 15 ;
- R 7 is selected from halo, cyano, hydroxy, trifluoromethoxy, C 1-3 alkoxy and C 1-3 alkyl, wherein said C 1-3 alkyl is optionally substituted by one or more halo;
- R 9 is selected from hydrogen, halo, cyano, hydroxy, amino, C 1-3 alkyl and C 1-3 alkoxy;
- R 1 , R 13 and R 14 are independently selected from halo, cyano, hydroxy, amino, sulphamoyl, Ci.6alkanoylamino, N-(C 1-(5 alkyr)carbanioyl, N,N-(Ci -6 alkyl) 2 carbamoyl, C 1-6 alkylS(O) a wherein a is O to 2, iV-(C 1-6 alkyl)sulphamoyl, N,iy-(C 1-6 alkyl) 2 Sulphamoyl, C 1- ⁇ alkylsulphonylamino, carbocyclyl, heterocyclyl, carbocyclylCi-salkyl-R 16 -, heterocyclylCi-salkyl-R 17 -, carbocyclyl-R 18 - and heterocyclyl-R 19 -; wherein R 10 , R 13 and R 1 are independently of each other substituted on carbon by one or more R 20 ; and
- R 16 , R 17 , R 18 and R 19 are independently selected from -O-, -N(R 22 )-, -C(O)-, -N(R 23 )C(O)- 5 -C(O)N(R 24 )-, -S(O)s-, -SO 2 N(R 25 )- and -N(R 26 )SO 2 -; wherein R 22 , R 23 , R 24 , R 25 and R 26 are independently selected from hydrogen and Ci -6 alkyl; and s is O, 1 or 2;
- R 8 , R 15 and R 21 are independently selected from C 1-4 alkyl, carbocyclyl, heterocyclyl, -C 1- 4 alkylcarbocyclyl, and C 1- 4 alkoxycarbonyl; wherein R 8 , R 15 and R 21 independently of each other may be optionally substituted on carbon by one or more R 27 ; and R 20 and R 27 are independently selected from halo, cyano, hydroxy, trifluoromethoxy, trifluoromethyl, amino, methyl, ethyl, phenyl, cyclopropyl, cyclobutyl, methoxy, ethoxy, methylamino, ethylamino, dimethylamino, diethylamino, mesyl, ethylsulphonyl and phenyl;
- One aspect of the present invention relates to a compound of formula (I), wherein
- R 1 is a group -R 5 -R 6 or a nitrogen linked 4-7 membered saturated ring which optionally contains an additional nitrogen, oxygen or sulphur atom; wherein said ring may be optionally substituted on carbon by one or more R 7 ; and wherein if said ring contains an additional nitrogen atom that nitrogen is optionally substituted by R 8 ; at least one of X 1 , X 2 , X 3 and X 4 is selected from N, the other three X 1 , X 2 , X 3 or X 4 are independently selected from N or C(R 9 ) provided that not more than two of X 1 , X 2 , X 3 or X 4 are selected from N;
- R 2 is halo or cyano
- R 3 is methyl or 4-tetrahydropyranyl, wherein said tetrahydropyranyl group is optionally substituted on carbon by one or more R 10 ;
- R 4 is selected from hydrogen, halo, cyano and C 1-3 alkyl, wherein said C 1-3 alkyl is optionally substituted with one or more halo;
- R 5 is selected from -O-, -C(O)-, -C(O)O-,-C(O)N(R n )-, -S(O) 1 - and -SO 2 N(R 12 )-;
- R 11 and R 12 are independently selected from hydrogen or C 1-6 alkyl and said alkyl is optionally substituted by one or more R 13 ; and r is O or 2;
- R 6 is selected from carbocyclyl and heterocyclyl; wherein R 6 is optionally substituted on carbon by one or more R 14 ; and wherein if said heterocyclyl contains an -
- R 7 is selected from halo, cyano, hydroxy, trifluoromethoxy, C 1-3 alkoxy and C 1-3 alkyl, wherein said C 1-3 alkyl is optionally substituted by one or more halo;
- R 9 is selected from hydrogen, halo, cyano, hydroxy, C h alky! and C 1-3 alkoxy;
- R 10 , R 13 and R 14 are independently selected from halo, cyano, hydroxy, amino, sulphamoyl,
- R 16 , R 17 , R 18 and R 19 are independently selected from -O-, -N(R 22 )-, -C(O)-,-N(R 23 )C(O)-, -C(O)N(R 24 )-, -S(O)s-, -SO 2 N(R 25 )- and -N(R 26 )SO 2 -; wherein R 22 , R 23 , R 24 , R 25 and R 26 are independently selected from hydrogen or C ⁇ alkyl; and s is 0, 1 or 2; R 8 , R 15 and R 21 are independently selected from C 1-4 alkyl, carbocyclyl, heterocyclyl, -C 1- 4 alkylcarbocyclyl, -Ci -4 alkylheterocyclyl, C 1-4 alkanoyl, C 1-4 alkylsulphonyl and C 1- 4 alkoxycarbonyl; wherein R 8 , R 15 and R 21 independently of each other may be optionally substitute
- any or all of the compounds of the present invention have a potent inhibiting effect at GSK3 in addition to a selective inhibiting effect at GSK3.
- Another aspect of the present invention relates to a compound of formula (I), wherein R 2 is halo.
- Yet another aspect of the present invention relates to a compound of formula (I), wherein R 2 is fluoro.
- Another aspect of the present invention relates to a compound of formula (I), wherein R 3 is 4-tetrahydropyranyl or methyl.
- Yet another aspect of the present invention relates to a compound of formula (I), wherein R 4 is hydrogen or C 1-3 alkyl, wherein said C 1-3 alkyl is optionally substituted with one or more halo.
- R 4 is C 1-3 alkyl.
- R 4 is methyl.
- R 4 is trifluoromethyl.
- One aspect of the present invention relates to a compound of formula (I), wherein R 5 is -C(O)-or -S(O) 1 -; and r is 0 or 2. According to one embodiment of the present invention is -C(O)-. According to one embodiment of the present invention, -S(O) 1 -; and r is 2.
- One aspect of the present invention relates to a compound of formula (I), wherein R 5 is -O- or C(O)O-.
- Another aspect of the present invention relates to a compound of formula (I), wherein R 5 is -C(O)N(R 1 J )- or -SO 2 N(R 12 )-; wherein R 1 ⁇ and R 12 are independently selected from hydrogen or C 1-6 alkyl.
- Yet another aspect of the present invention relates to a compound of formula (I), wherein R 6 is C 1-6 alkyl or heterocyclyl; wherein R 6 is optionally substituted on carbon by one or more R 14 ; and wherein if said heterocyclyl contains an -NH- moiety that nitrogen is optionally substituted by a group selected from R 15 .
- said C 1-6 alkyl is methyl, ethyl, butan-2-yl, butan-3-yl, propan-2-yl or tert-butyl.
- said heterocyclyl is selected from morpholinyl, homomorpholinyl, piperidinyl, pyrrolidinyl, azetidinyl, piperazinyl, , homopiperidinyl and homopiperazinyl.
- said heterocyclyl is selected from piperidinyl, pyrrolidinyl, azetidinyl and piperazinyl
- R 14 is Ci -6 alkoxy, halo, C 1-6 alkyl, carbocyclyl, heterocyclyl and N,N-(C 1-6 alkyl) 2 amino; wherein R 14 is optionally substituted on carbon by one or more R 20 .
- R is C 1-4 alkyl or carbocycle; wherein R 15 is optionally substituted on carbon by one or more R 27 .
- R 8 is C 1- 4 alkyl, and wherein R 8 may be optionally substituted on carbon by one or more R 27 .
- R 7 is hydroxy, halo, ethoxy, methoxy or phenyl.
- Another aspect of the present invention relates to a compound of formula (I), wherein at least one of X 2 , X 3 and X 4 is selected from N, the other two X 2 , X 3 or X 4 are independently selected from N or C(R 9 ). According to one embodiment of the present invention, X 3 or X 4 is N.
- R 9 is hydrogen, methyl, trifluoromethyl, trifluoromethoxy or halo.
- R 9 is hydrogen.
- one of R 9 is halo.
- said halo is chloro.
- R 10 are for example fluoro, cyano, methyl and ethyl and other sutiable values of R 11 and R 12 are for example hydrogen and C ⁇ alkyl.
- One aspect of the present invention relates to a compound of formula (I), wherein
- R 1 is a group -R 5 -R 6 ; at least one of X 1 , X 2 , X 3 and X 4 is selected from N, the other three X 1 , X 2 , X 3 or X 4 are independently selected from N or C(R 9 ), provided that not more than two of X 1 , X 2 , X 3 or
- X 4 are selected from N;
- R 2 is halo
- R 3 is methyl or 4-tetrahydropyranyl
- R 4 is C 1-3 alkyl, wherein said C 1-3 alkyl is optionally substituted with one or more halo
- R 5 is selected from -O-, -C(O)-, -C(O)O-, -C(O)N(R 11 )-, -S(O) 1 - and -SO 2 N(R 12 )-; wherein
- R 11 and R 12 are independently selected from hydrogen or C 1-6 alkyl and said alkyl is optionally substituted by one or more R 13 and r is 2;
- R 6 is C 1-6 alkyl or heterocyclyl; wherein R 6 is optionally substituted on carbon by one or more R 14 ; and wherein if said heterocyclyl contains an -NH- moiety that nitrogen is optionally substituted by a group selected from R 15 ;
- R 9 is hydrogen or halo
- R 14 is selected from halo, C 1-6 alkyl, carbocycle, N,N-(C 1-6 alkyl) 2 amino, heterocyclyl and
- R 14 is optionally on carbon by one or more R 20 ;
- R 15 is C 1-4 alkyl or carbocycle; wherein R 15 is optionally substituted on carbon by one or more R ; and
- R and R are independently selected from halo, methoxy, ethoxy, and phenyl.
- R 1 is a group -R 5 -R 6 ; at least one of X 1 , X 2 , X 3 and X 4 is selected from N, the other three X 1 , X 2 , X 3 or X 4 are independently selected from N or C(R 9 ), provided that not more than two of X 1 , X 2 , X 3 or X 4 are selected from N;
- R 2 is halo;
- R 3 is 4-tetrahydropyranyl;
- R 4 is C 1-3 alkyl;
- R 5 is -C(O) or -S(0) r - and -SO 2 N(R 12 )-; and r is 2;
- R 6 is C 1-6 alkyl or heterocyclyl; wherein if said heterocyclyl contains an -NH- moiety that nitrogen is optionally substituted by a group selected from R 15 ;
- R 9 is hydrogen; and
- R 15 is
- the present invention also provides a compound selected from:
- the present invention also provides a compound selected from:
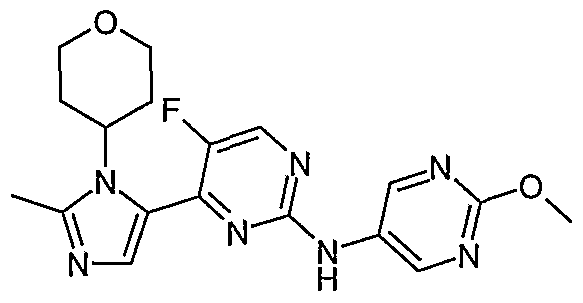
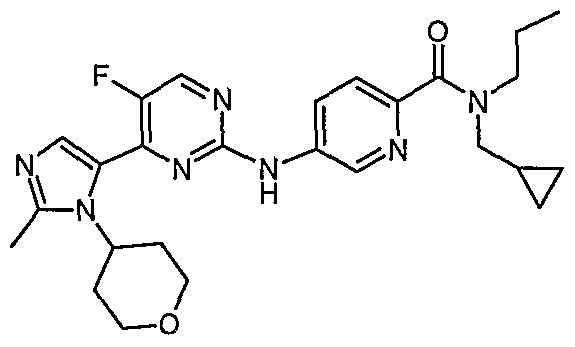
- the present invention also provides a compound selected from: Lithium 5-[[5-fluoro-4-[2-methyl-3-(oxan-4-yl)imidazol-4-yl]pyrimidin-2- yl]amino]pyridine-2-carboxylate;
- Said compound(s) can be used as intermediates in processes for obtaining a compound of formula (I).
- alkyl includes both straight and branched chain alkyl groups but references to individual alkyl groups such as “propyl” are specific for the straight chain version only.
- C 1-6 alkyl and “C 1-4 alkyl” include methyl, ethyl, propyl, isopropyl and t-butyl.
- references to individual alkyl groups such as 'propyl' are specific for the straight-chained version only and references to individual branched chain alkyl groups such as 'isopropyl' are specific for the branched chain version only.
- halo refers to fluoro, chloro, bromo and iodo.
- a “4-7 membered saturated heterocyclic group” is a saturated monocyclic ring containing 4-7 atoms of which at least one atom is chosen from nitrogen, sulphur or oxygen, which may, unless otherwise specified, be carbon or nitrogen linked, wherein a -CH 2 - group can optionally be replaced by a -C(O)- and a sulphur atom may be optionally oxidised to form the S-oxides.
- Examples and suitable values of the term "4-7 membered saturated heterocyclic group" are morpholino, piperidyl, 1,4-dioxanyl, 1,3-dioxolanyl, 1,2- oxathiolanyl, imidazolidinyl, pyrazolidinyl, piperazinyl, thiazolidinyl, pyrrolidinyl, thiomorpholino, homopiperazinyl and tetrahydropyranyl.
- a "nitrogen linked 4-7 membered saturated ring which optionally contains an additional nitrogen, oxygen or sulphur atom” is a saturated monocyclic ring containing 4-7 atoms linked to the X*-X 4 containing ring of formula (I) via a nitrogen atom contained in the ring.
- the ring optionally contains an additional heteroatom selected from nitrogen, sulphur or oxygen, wherein a -CH 2 - group can optionally be replaced by a -C(O)-, and the optional sulphur atom may be optionally oxidised to form the S-oxides.
- an additional heteroatom selected from nitrogen, sulphur or oxygen
- a -CH 2 - group can optionally be replaced by a -C(O)-
- the optional sulphur atom may be optionally oxidised to form the S-oxides.
- Particular examples of a "nitrogen linked 4-7 membered saturated ring which optionally contains an additional nitrogen, oxygen or sulphur atom” are piperazin-1-yl and morpholino, particularly morpholino.
- a “heterocyclyl” is a saturated, partially saturated or unsaturated, mono or bicyclic ring containing 4-12 atoms of which at least one atom is chosen from nitrogen, sulphur or oxygen, which may, unless otherwise specified, be carbon or nitrogen linked, wherein a -CH 2 - group can optionally be replaced by a -C(O)-, a ring nitrogen atom may optionally bear a C 1-6 alkyl group and form a quaternary compound or a ring nitrogen and/or sulphur atom may be optionally oxidised to form the N-oxide and or the S-oxides.
- heterocyclyl examples and suitable values of the term "heterocyclyl” are morpholino, piperidyl, pyridyl, pyranyl, pyrrolyl, isothiazolyl, indolyl, quinolyl, thienyl, 1,3-benzodioxolyl, thiadiazolyl, piperazinyl, thiazolidinyl, pyrrolidinyl, thiomorpholino, pyrrolinyl, homopiperazinyl, 3,5- dioxapiperidinyl, tetrahydropyranyl, imidazolyl, pyrimidyl, pyrazinyl, pyridazinyl, isoxazolyl, iV-methylpyrrolyl, 4-pyridone, 1-isoquinolone, 2-pyrrolidone, 4-thiazolidone, pyridine-JV-oxide and quinoline-JV-oxide.
- a “heterocyclyl” is a saturated, partially saturated or unsaturated, mono or bicyclic ring containing 5 or 6 atoms of which at least one atom is chosen from nitrogen, sulphur or oxygen, it may, unless otherwise specified, be carbon or nitrogen linked, a -CH 2 - group can optionally be replaced by a -C(O)-and a ring sulphur atom may be optionally oxidised to form the S-oxides.
- a “carbocyclyl” is a saturated, partially saturated or unsaturated, mono or bicyclic carbon ring that contains 3-12 atoms; wherein a -CH 2 - group can optionally be replaced by a -C(O)-. Particularly “carbocyclyl” is a monocyclic ring containing 5 or 6 atoms or a bicyclic ring containing 9 or 10 atoms.
- Suitable values for "carbocyclyl” include cyclopropyl, cyclobutyl, 1-oxocyclopentyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, phenyl, naphthyl, tetralinyl, indanyl or 1-oxoindanyl.
- Examples of “C 1-6 alkoxy” include methoxy, ethoxy and propoxy.
- Examples of “Ci -6 alkanoylamino” include formamido, acetamido and propionylamino.
- Examples of "C 1-6 alkylS(O)a wherein a is 0,1 or 2” include methylthio, ethylthio, methylsulphinyl, ethylsulphinyl, mesyl and ethylsulphonyl.
- Examples of “C 1-6 alkanoyl” include propionyl and acetyl.
- Examples of "iV-(C 1-6 alkyl)amino” include methylamino and ethylamino.
- JV,JV-(C 1-6 alkyl) 2 amino examples include di-iV-methylamino, di-(JV-ethyl)amino and N-ethyl-JV-methylamino.
- JV-(C 1-6 alkyl)sulphamoyl examples include JV- (methyl)sulphamoyl and JV-(ethyl)sulphamoyl. are N,JV-(dimethyl)sulphamoyl and N-(methyl)-JV-(ethyl)sulphamoyl.
- JV-(C 1 examples include di-iV-methylamino, di-(JV-ethyl)amino and N-ethyl-JV-methylamino.
- JV-(C 1-6 alkyl)sulphamoyl examples include JV- (methyl)sulphamoyl and JV-(ethyl)sulphamoyl. are N,JV-
- 6 alkyl)carbamoyl are methylaminocarbonyl and ethylaminocarbonyl.
- Examples of “N,JV- (C 1-6 alkyl) 2 carbamoyl” are dimethylaminocarbonyl and methylethylaminocarbonyl.
- Examples of "C 1-6 alkylsulphonylamino” include methylsulphonylamino, isopropylsulphonylamino and t-butylsulphonylamino.
- Examples of “C ⁇ ealkylsulphonyl” include methylsulphonyl, isopropylsulphonyl and t-butylsulphonyl.
- -C M alkylcarbocyclyl and “-C 1-4 alkylheterocyclyl” includes both straight and branched chain alkyl groups of between one and four carbon atoms that then link to a carbocycle or heterocycle respectively.
- carbocycle and heterocycle are as defined above.
- Non-limiting examples of Ci ⁇ ahkylcarbocyclyl therefore include benzyl, 2- phenylethyl, 1-phenylethyl, cyclopropylmethyl and cyclohexylethyl.
- Non-limiting examples of C 1-4 alkylheterocyclyl include pyridin-3-ylmethyl, oxolan-2yl-methyl, 2-(4- piperidyl)ethyl and l-thiophen-2-ylethyl.
- a suitable pharmaceutically acceptable salt of a compound of the present invention is, for example, an acid-addition salt of a compound of the present invention which is sufficiently basic, for example, an acid-addition salt with, for example, an inorganic or organic acid, for example hydrochloric, hydrobromic, sulphuric, phosphoric, trifluoroacetic, citric or maleic acid.
- a suitable pharmaceutically acceptable salt of a compound of the present invention which is sufficiently acidic is an alkali metal salt, for example a sodium or potassium salt, an alkaline earth metal salt, for example a calcium or magnesium salt, an ammonium salt or a salt with an organic base which affords a physiologically-acceptable cation, for example a salt with methylamine, dimethylamine, trimethylamine, piperidine, morphorine or tris-(2-hydroxyethyl)amine.
- an alkali metal salt for example a sodium or potassium salt
- an alkaline earth metal salt for example a calcium or magnesium salt
- an ammonium salt or a salt with an organic base which affords a physiologically-acceptable cation
- a salt with methylamine, dimethylamine, trimethylamine, piperidine, morphorine or tris-(2-hydroxyethyl)amine for example a salt with methylamine, dimethylamine, trimethylamine, piperidine, morphorine or tris-(2-hydroxy
- Some compounds of the formula (I) may have stereogenic centres and/or geometric isomeric centres (E- and Z-isomers), and it is to be understood that the present invention encompasses all such optical, diastereoisomers and geometric isomers that possess GSK3 inhibitory activity.
- the present invention relates to any and all tautomeric forms of the compounds of the formula (I) that possess GSK3 inhibitory activity.
- the definition of compounds of formula (I) also includes in vivo hydrolysable esters, solvates or solvates of salts thereof. It is also to be understood that certain compounds of the formula (I) can exist in solvated as well as unsolvated forms such as, for example, hydrated forms. It is to be understood that the present invention encompasses all such solvated forms that possess GSK3 inhibitory activity.
- the present invention also provides a process for preparing a compound of formula (I), or a pharmaceutically acceptable salt thereof or in vivo hydrolysable ester thereof, which process comprises: a) reacting a pyrimidine of formula (II):
- Y is a displaceable group
- R 1 , R 2 , R 3 , R 4 , X 1 , X 2 , X 3 and X 4 are, unless otherwise specified, as defined in formula (I); and thereafter optionally: b) converting a compound of the formula (I) into another compound of formula (I); c) removing any protecting groups; and d) forming a pharmaceutically acceptable salt or in vivo hydrolysable ester.
- Y is, as mentioned above, a displaceable group. Suitable values for Y are, for example, a halo (such as a chloro, bromo or iodo) or sulphonyloxy group (such as trifluoromethanesulphonyloxy group). According to one embodiment of the present invention, Y is chloro, bromo or iodo.
- Specific reaction conditions for the above reactions are as follows: Step a): Amines of formula (II) and compounds of formula (III) may be reacted together under standard Buchwald-Hartwig conditions, (for example see J. Am. Chem. Soc, 118, 7215; J. Am. Chem. Soc, 119, 8451; J. Am. Chem.
- a suitable solvent for example an aromatic solvent such as toluene, benzene or xylene
- a suitable base for example an inorganic base such as caesium carbonate or an organic base such
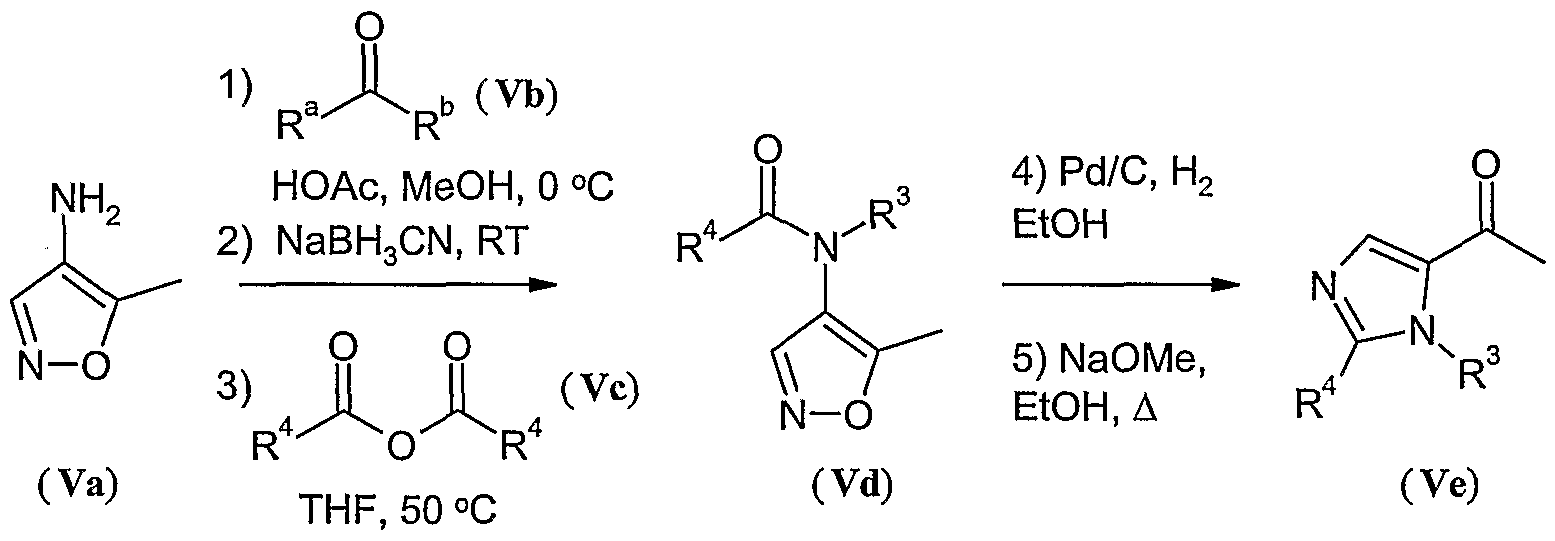
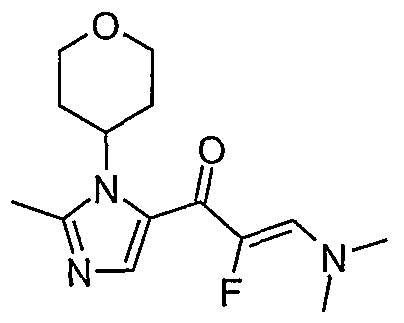
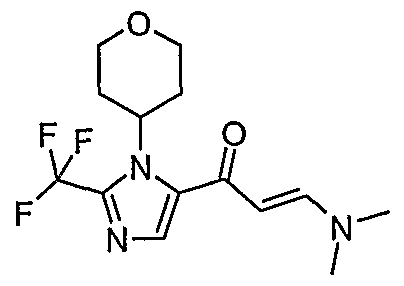
- R 3 has the general structure R a -CH-R b , wherein R a and R b are hydrogen or form together a tetrahydropyran ring, wherein R 4 is hydrogen or C ⁇ aHcyl, wherein said C ⁇ alkyl may optionally be substituted with one or more halo and wherein R 2 is fluoro and R x is as defined above, may be prepared according to Scheme 3, wherein,
- a compound of formula (Ia) can be prepared by reacting an acid intermediate (VI) with primary or secondary amines as shown in Scheme 4. This reaction can be achieved by mixing the acid or carboxylate salt with a coupling agent in a polar, aprotic solvent followed by addition of the primary or secondary amine.
- the amidation conditions involve, for example, taking a mixture of the carboxylate or acid, a coupling agent (such as HBTU or CDI), a base, such as DIPEA, together in a solvent such as DCM, N-methyl pyrrolidinone or dimethylformamide and then adding the amine at room temperature.
- a coupling agent such as HBTU or CDI
- DIPEA a base
- C(O)NR 28 R 29 is defined as -R 5 -R 6 above.
- aromatic substitution reactions include the introduction of a nitro group using concentrated nitric acid, the introduction of an acyl group using, for example, an acyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; the introduction of an alkyl group using an alkyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; and the introduction of a halo group.
- modifications include the reduction of a nitro group to an amino group by for example, catalytic hydrogenation with a nickel catalyst or treatment with iron in the presence of hydrochloric acid with heating; oxidation of alkylthio to alkylsulphinyl or alkylsulphonyl.
- a suitable protecting group for an amino or alkylamino group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an alkoxycarbonyl group, for example a methoxycarbonyl, ethoxycarbonyl or t-butoxycarbonyl group, an arylmethoxycarbonyl group, for example benzyloxycarbonyl, or an aroyl group, for example benzoyl.
- the deprotection conditions for the above protecting groups necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or alkoxycarbonyl group or an aroyl group may be removed for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an acyl group such as a t-butoxycarbonyl group may be removed, for example, by treatment with a suitable acid as hydrochloric, sulphuric or phosphoric acid or trifluoroacetic acid and an arylmethoxycarbonyl group such as a benzyloxycarbonyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon, or by treatment with a Lewis acid for example boron tris(trifluoroacetate).
- a suitable alternative protecting group for a primary amino group is, for example, a phthaloyl group that may be removed by treatment with an alkylamine, for example dimethylaminopropylamine, or with hydrazine.
- a suitable protecting group for a hydroxy group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an aroyl group, for example benzoyl, or an arylmethyl group, for example benzyl.
- the deprotection conditions for the above protecting groups will necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or an aroyl group may be removed, for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an arylmethyl group such as a benzyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on- carbon.
- a suitable protecting group for a carboxy group is, for example, an esterifying group, for example a methyl or an ethyl group which may be removed, for example, by hydrolysis with a base such as sodium hydroxide, or for example a /-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as trifluoroacetic acid, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- a base such as sodium hydroxide
- a /-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as trifluoroacetic acid, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- the protecting groups may be removed at any convenient stage in the synthesis using conventional techniques well-known in the chemical art.
- spectra were recorded at 400 MHz for proton, 376 MHz for fluorine-19 and 100 MHz for carbon-13.
- the following reference signals were used: the middle line of DMSO-Je ⁇ 2.50 (IH), ⁇ 39.51 (13C); the middle line of CD 3 OD ⁇ 3.31 (IH) or ⁇ 49.15 (13C); CDCl 3 ⁇ 7.26 (IH) and the middle line OfCDCl 3 ⁇ 77.16 (13C) (unless otherwise indicated).
- NMR spectra are reported either from high to low field or from low to high field.
- Mass spectra were recorded on a Waters LCMS consisting of an Alliance 2795 (LC), Waters PDA 2996 and a ZQ single quadrupole mass spectrometer.
- the mass spectrometer was equipped with an electrospray ion source (ESI) operated in a positive or negative ion mode.
- the capillary voltage was 3 kV and cone voltage was 30 V.
- the mass spectrometer was scanned between m/z 100-700 with a scan time of 0.3s.
- mass spectra were recorded on a Waters LCMS consisting of an Alliance 2690 Separations Module, Waters 2487 Dual 1 Absorbance Detector (220 and 254 nm) and a Waters ZQ single quadrupole mass spectrometer.
- the mass spectrometer was equipped with an electrospray ion source (ESI) operated in a positive or negative ion mode.
- the capillary voltage was 3 kV and cone voltage was 30 V.
- the mass spectrometer was scanned between m/z 97-800 with a scan time of 0.3 or 0.8 s. Separations were performed on a Chromolith Performance RP-18e (100 x 4.6 mm). A linear gradient was applied starting at 95% A (A: 0.1% HCOOH (aq.)) ending at 100% B (MeCN) in 5 minutes. Flow rate: 2.0 mL/min.
- Microwave heating was performed in a single-mode microwave cavity producing continuous irradiation at 2450 MHz.
- HPLC analyses were performed on a Gynkotek P580 HPG consisting of gradient pump with a Gynkotek UVD 170S UV-vis.-detector equipped with a Chromolith Performance RP column (Cl 8, 100 mm x 4.6 mm). The column temperature was set to +25 0 C. A linear gradient was applied using MeCN/0.1 trifluoroacetic acid in MiUiQ water, run from 10% to 100% MeCN in 5 minutes. Flow rate: 3 ml/min.
- a typical workup procedure after a reaction consisted of extraction of the product with a solvent such as ethyl acetate, washing with water followed by drying of the organic phase over MgSO 4 or Na 2 SO 4 , filtration and concentration of the solution in vacuo.
- TLC Thin layer chromatography
- Merck TLC-plates Silica gel 60 F 254
- Flash chromatography was performed on a Combi Flash ® CompanionTM using RediSepTM normal-phase flash columns.
- Typical solvents used for flash chromatography were mixtures of chloroform/methanol, DCM/methanol, heptane/ethyl acetate, chloroform/methanol/ NH 3 (aq.) and DCM/methanol/NH 3 (aq.).
- SCX ion exchange columns were performed on Isolute ® columns. Chromatography through ion exchange columns were typically performed in solvents such a methanol.
- Preparative chromatography was run on a Waters autopurif ⁇ cation HPLC with a diode array detector. Column: XTerra MS C8, 19 x 300 mm, 10 ⁇ m. Narrow gradients with MeCN/(95:5 0.1M NH 4 OAcMeCN) were used at a flow rate of 20 ml/min. Alternatively, purification was achieved on a semi preparative Shimadzu LC-8A HPLC with a Shimadzu SPD-IOA UV-vis.-detector equipped with a Waters Symmetry ® column (C18, 5 ⁇ m, 100 mm x 19 mm).
- Narrow gradients with MeCN/0.1% trifluoroacetic acid in MiIIiQ Water were used at a flow rate of 10 ml/min.
- the formation of hydrochloride salts of the final products were typically performed in solvents or solvents mixtures such as diethyl ether, tetrahydrofuran, DCM/toluene, DCM/methanol, followed by addition of IM hydrogen chloride in diethyl ether.
- PrOH propan-1-ol r.t. or RT room temperature
- the groups R 1 , R 2 , R 3 , R 4 , X 1 , X 2 , X 3 , X 4 and Y are used independently to indicate the diversity of substitution within each structure.
- the identity of R 1 , R 2 , R 3 , R 4 , X 1 , X 2 , X 3 , X 4 and Y will be clear to a person skilled in the art based on the starting materials and intermediates for each specific example.
- Example 1 which refers to General method A, Al is 5-fluoro-4-[2-methyl-l-(tetrahydro- 2H-pyran-4-yl)-lH-imidazol-5-yl]pyrimidin-2-amine such that R 3 is 4-tetrahydropyranyl and R 4 is methyl and A2 is 2-bromo-5-(methylsulfonyl)pyridine such that X 1 is N, X 2 , X 3 and X 4 are CH and R 1 is sulphonylmethane.
- the title compound was prepared in accordance with general method A using 5-fluoro-4- [2-methyl- 1 -(tetrahydro-2H-pyran-4-yl)- lH-imidazol-5-yl]pyrimidin-2-amine (as described in Example 6) (50 mg, 0.18 mmol) and 5-bromo-2-(methylsulfonyl)pyridine (42 mg, 0.18 mmol) to give the title compound (36 mg, 46%).
- the title compound was prepared in accordance with general method A using 5-fluoro-4- [2-methyl- 1 -(tetrahydro-2H-pyran-4-yl)- lH-imidazol-5-yl]pyrirnidin-2-amine (as described in Example 6) (26 mg, 0.095 mmol) and l-[(5-bromopyridin-2-yl)carbonyl]-4- methylpiperazine (obtained from Example 4b) (27 mg, 0.095 mmol) to give the title compound in 61% (28 mg) yield.
- the title compound was prepared in accordance with general method A, with the exception that a second purification on a silica gel column was necessary to obtain a pure material, using 5-fluoro-4-[2-methyl- 1 -(tetrahydro-2H-pyran-4-yl)- lH-imidazol-5-yl]pyrimidin-2- amine (as described in Example 6) (36 mg, 0.13 mmol) and 2-(azetidin-l-ylcarbonyl)-5- bromopyridine (reported in WO 2005014571) (32 mg, 0.13 mmol) to give the title compound in 18% (10 mg) yield.
- 1,2-Dimethylimidazole (0.960 g, 10.0 mmol) was diluted in dry THF (50 mL) under an argon atmosphere and the solution was cooled to -78°C.
- tert-Butyllithium (1.7M in pentane, 6.47 mL, 11.0 mmol) was added dropwise over 5 minutes.
- the reaction mixture was stirred for 1 h at -78 °C and then treated with a solution of trimethyltin chloride (2.2 g, 11.0 mmol) in anhydrous THF (10 mL). The mixture was stirred for 60 h from -78°C to r.t.. The solvent was then evaporated in vacuo to give the title compound (1.29 g, 50%).
- the crude product was used in the next step without further purification.
- the methanol was removed from the reaction mixture, and the intermediate amine was extracted with ethyl acetate (3x80 mL). The combined organic layers were dried (Na 2 SO 4 ), concentrated to dryness, dissolved in toluene and re-concentrated.
- the crude intermediate amine was dissolved in CH 2 Cl 2 (20 mL) and pyridine (2 mL, 26 mmol) was added. The mixture was cooled to 0°C and trifluoroacetic anhydride (4.35 g, 20.7 mmol) was added dropwise. The mixture was continued stirring for 2 h at r.t. and was then washed with water and saturated NaHCO 3 .
- Trifluoroacetic anhydride (10 mL, 71 mmol) in CH 2 Cl 2 (100 mL) was added to N,5- dimethylisoxazol-4-amine (Reiter, L.A., J. Org. Chem. 1987, 52, 2714-2726) (6.68g, 59.6 mmol) in DCM (200 mL) and pyridine (6 mL, 74 mmol) at 0 °C. The mixture was stirred at 0 °C for 30 min and at r.t. for 2 h. The reaction mixture was diluted with CH 2 Cl 2 (100 mL) and washed with H 2 O and saturated NaHCO 3 (aq).
- the title compound was prepared in accordance with the general method A using 5-fluoro- 4-[2-methyl-l-(tetrahydro-2H-pyran-4-yl)-lH-imidazol-5-yl]pyrimidin-2-amine (as described in Example 6) (50 mg, 0.18 mmol) and 5-bromo-2-ethoxy-pyridine (36 mg, 0.18 5 mmol) to give the title compound (27 mg, 38%).
- Examples 12-40 were prepared by the general procedure B using the appropriate starting materials which include: lithium 5-[[5-fluoro-4-[2-methyl-3-(oxan-4- yl)imidazol-4-yl]pyrimidin-2-yl]amino]pyridine-2-carboxylate (as described below) and the amine necessary to deliver the amide shown in the table below.
- Methyl 5-[[5-fluoro-4-[2-methyl-3-(oxan-4-yl)imidazol-4-yl]pyrimidin-2- yl]amino]pyridine-2-carboxylate (prepared as described in Example 41) (1.49 g, 3.61 mmol) in MeOH (70 mL) was heated at 60 0 C for 30 min. The flask was removed from the oil bath and a solution of LiOH monohydrate (167 mg, 3.97 mmol) in water (13 mL) was added dropwise during one minute. The mixture was heated at 60°C during 4h, allowed to cool and concentrated to a yellow powder which was dried under vacuum to yield 1.44 g (99%) of the title compound. The isolated material was used in amidation reactions without further purification.
- the title compound was prepared in accordance with the general method A using 4-(l,2- dimethyl-lH-imidazol-5-yl)-5-fluoropyrimidin-2-amine (as described in Example 7) (30 mg, 0.145 mmol) and 2-(azetidin-l-ylcarbonyl)-5-bromopyridine (41 mg, 0.17 mmol) (reported in WO 2005014571) to give the title compound (22 mg, 41%).
- the title compound was prepared in accordance with the general method A using 4-(l,2- dimethyl-lH-imidazol-5-yl)-5-fluoropyrimidin-2-amine (as described in Example 7) (40 mg, 0.193 mmol) and l-[(5-bromopyridin-2-yl)carbonyl]-4-methylpiperazine (as described in Example 4(b)) (55 mg, 0.23 mmol) to give the title compound (45 mg, 57%).
- the title compound was prepared in accordance with the general method C using 3,5- dichloropyridine-2-carboxylic acid (500 mg, 2.6 mmol) and azetidine (150 mg, 2.6 mmol) to give the title compound (430 mg, 72%).
- the title compound was prepared in accordance with the general method C using 3,5- dichloropyridine-2-carboxylic acid (555 mg, 2.89 mmol) and 1-methylpiperazine (320 ⁇ L, 2.89 mmol) to give the title compound (417 mg, 53 %).
- the title compound was prepared in accordance with the general method A using 4-(l,2- dimethyl-lH " -imidazol-5-yl)-5-fluoropyrimidin-2-amine (as described in Example 7) (50 mg, 0.24 mmol) and (3,5-dichloropyridin-2-yl)-(4-methylpiperazin-l-yl)methanone (as described above) (66 mg, 0.24 mmol) to give the title compound (29 mg, 27%).
- 3,5-Dichloro-2-pyridine carboxylic acid (1.25 g, 6.5 mmol) was suspended in thionyl chloride (10 ml). DMF (2 drops) was added and the mixture was refluxed for 15 minutes under an atmosphere of nitrogen. The solvent was evaporated. Toluene was added and the solvent was evaporated to give a solid. The solid was dissolved in DCM (8 ml) and the mixture was cooled to 0° C. Piperidine (0.64 ml, 6.5 mmol) was added dropwise followed by triethylamine (0.91 ml, 6.5 mmol). The cooling bath was removed. The mixture was stirred under nitrogen atmosphere until RT was reached and then for an additional 15 minutes.
- a pharmaceutical0 formulation comprising the compound of formula (I) as a free base or a pharmaceutically acceptable salt thereof, in an essentially pure and isolated form, for use in the prevention and/or treatment of conditions associated with glycogen synthase kinase-3.
- the formulation used in accordance with the present invention may be in a form suitables for oral administration, for example as a tablet, pill, syrup, powder, granule or capsule, for parenteral injection (including intravenous, subcutaneous, intramuscular, intravascular or infusion) as a sterile solution, suspension or emulsion, for topical administration as an ointment, patch or cream, for rectal administration as a suppository and for local administration in a body cavity or in a bone cavity.
- parenteral injection including intravenous, subcutaneous, intramuscular, intravascular or infusion
- a sterile solution suspension or emulsion
- topical administration as an ointment, patch or cream
- rectal administration as a suppository and for local administration in a body cavity or in a bone cavity.
- the formulation may be in a form suitable for oral administration, for example as a tablet, for parenteral injection as a sterile solution or suspension.
- the above formulation may be prepared in a conventional manner using pharmaceutically carriers or diluents.
- Suitable daily doses of the compound of formula (I) as a free base and pharmaceutically acceptable salts thereof in the treatment of a mammal, including human are approximately 0.01 to 250 mg/kg bodyweight at per oral administration and about 0.001 to 250 mg/kg bodyweight at parenteral administration.
- the typical daily dose of the active ingredients varies within a wide range and will depend on various factors such as the relevant indication, the route of administration, the age, weight and sex of the patient and may be determined by a physician.
- the compound of formula (I) as a free base or a pharmaceutically acceptable salt thereof, in an essentially pure and isolated form, may be used on its own but will usually be administered in the form of a pharmaceutical formulation in which the active ingredient is in association with pharmaceutically acceptable diluents, excipients or inert carrier.
- the pharmaceutical formulation may comprise from 0.05 to 99 %w (per cent by weight), for example from 0.10 to 50 %w, of active ingredient, all percentages by weight being based on total composition.
- a diluent or carrier includes water, aqueous poly(ethylene glycol), magnesium carbonate, magnesium stearate, talc, a sugar (such as lactose), pectin, dextrin, starch, tragacanth, microcrystalline cellulose, methyl cellulose, sodium carboxymethyl cellulose or cocoa butter.
- a formulation of the present invention can be in a unit dosage form such as a tablet or an injectable solution.
- the tablet may additionally comprise a disintegrant and/or may be coated (for example with an enteric coating or coated with a coating agent such as hydroxypropyl methylcellulose).
- the present invention further provides a process for the preparation of a pharmaceutical formulation of the present invention which comprises mixing of the compound of formula (I) or a pharmaceutically acceptable salt thereof, a hereinbefore defined, with pharmaceutically acceptable diluents, excipients or inert carriers.
- An example of a pharmaceutical formulations of the present invention is an injectable solution comprising the compound of formula (I) as a free base or a pharmaceutically acceptable salt thereof, as hereinbefore defined, and sterile water, and, if necessary, either a base sodium hydroxide or an acid hydrochloric acid to bring the pH of the final formulation to about pH in the range of about 4 to 6 , particularly about 5, and optionally a surfactant to aid dissolution.
- a suitable base is sodium hydroxide.
- a suitable acid is hydrochloric acid.
- a suitable pharmaceutically acceptable salt of the compound of formula (I) useful in accordance to the present invention is, for example, an acid-addition salt, which is sufficiently basic, for example an inorganic or organic acid.
- a suitable pharmaceutically acceptable salt of the compounds of the present invention, which is sufficiently acidic is an alkali metal salt, an alkaline earth metal salt or a salt with an organic base, which affords a physiologically-acceptable cation.
- the compounds of formula (I) defined in the present invention are well suited for inhibiting glycogen synthase kinase-3 (GSK3). Accordingly, said compound of the present invention is expected to be useful in the prevention and/or treatment of conditions associated with glycogen synthase kinase-3 activity, i.e. the compounds may be used to produce an inhibitory effect of GSK3 in mammals, including human, in need of such prevention and/or treatment.
- GSK3 is highly expressed in the central and peripheral nervous system and in other tissues. Thus, it is expected that compound of the present invention is well suited for the prevention and/or treatment of conditions associated with glycogen synthase kinase-3 in the central and peripheral nervous system.
- the compound of the present invention is expected to be suitable for prevention and/or treatment of conditions associated with cognitive disorders and predemented states, especially dementia, Alzheimer's Disease (AD), Cognitive Deficit in Schizophrenia (CDS), Mild Cognitive Impairment (MCI), Age- Associated Memory Impairment (AAMI), Age-Related Cognitive Decline (ARCD) and Cognitive Impairement No Dementia (CIND), diseases associated with neurofibrillar tangle pathologies, Frontotemporal dementia (FTD), Frontotemporal dementia Parkinson's Type (FTDP), progressive supranuclear palsy (PSP), Pick's Disease, Niemann-Pick's Disease, corticobasal degeneration (CBD), traumatic brain injury (TBI) and dementia pugilistica.
- AD Alzheimer's Disease
- CDS Cognitive Deficit in Schizophrenia
- MCI Mild Cognitive Impairment
- AAMI Age- Associated Memory Impairment
- ARCD Age-Related Cognitive Decline
- CIND
- One embodiment of the present invention relates to the prevention and/or treatment of Alzheimer's Disease, especially the use in the delay of the disease progression of Alzheimer's Disease.
- PD Parkinson's Disease
- ALS amyotrophic lateral sclerosis
- MND motor neuron diseases
- ADD attention deficit hyperactivity disorder
- ADHD attention deficit hyperactivity disorder
- affective disorders wherein the affective disorders are Bipolar Disorder including acute mania, bipolar depression, bipolar maintenance, major depressive disorders (MDD) including depression, major depression, mood stabilization, schizoaffective disorders including schizophrenia, and dysthymia.
- Bipolar Disorder including acute mania, bipolar depression, bipolar maintenance, major depressive disorders (MDD) including depression, major depression, mood stabilization, schizoaffective disorders including schizophrenia, and dysthymia.
- MDD major depressive disorders
- schizoaffective disorders including schizophrenia, and dysthymia.
- Type I diabetes Type II diabetes
- diabetic neuropathy diabetic neuropathy
- alopecia inflammatory diseases and cancer
- One embodiment of the present invention relates to the use of a compound of the formula (I), as defined in the present invention, in the prevention and/or treatment of bone-related disorders or conditions in mammals.
- One aspect of the present invention is directed to the use of a compound of the formula (I) , as defined in the present invention to treat osteoporosis.
- One aspect of the present invention is directed to the use of a compound of the formula (I), as defined in the present invention to increase and promote bone formation in mammals.
- One aspect of the present invention is directed to the use of a compound of the formula (I), as defined in the present invention to increase bone mineral density in mammals.
- Another aspect of the present invention is directed to the use of a compound of the formula (I), as defined in the present invention to reduce the rate of fracture and/or increase the rate of fracture healing in mammals.
- Another aspect of the present invention is directed to the use of a compound of the formula (I), as defined in the present invention to increase cancellous bone formation and/or new bone formation in mammals.
- Another aspect of the present invention is directed to a method of prevention and/or treatment of bone-related disorders comprising administering to a mammal in need of such prevention and/or treatment, a therapeutically effective amount of a compound of the formula (I) as defined in the present invention.
- Another aspect of the present invention is directed to a method of prevention and/or treatment of osteoporosis comprising administering to a mammal in need of such prevention and/or treatment, a therapeutically effective amount of a compound of the formula (I) as defined in the present invention.
- Another aspect of the present invention is directed to a method of increasing bone formation comprising administering to a mammal in need of such treatment, a therapeutically effective amount of a compound of the formula (I) as defined in the present invention.
- Another aspect of the present invention is directed to a method of increasing bone mineral density comprising administering to a mammal in need of such treatment, a therapeutically effective amount of a compound of the formula (I) as defined in the present invention.
- Another aspect of the present invention is directed to a method of reducing the incidence of fracture comprising administering to a mammal in need of such treatment, a therapeutically effective amount of a compound of the formula (I) as defined in the present invention.
- Another aspect of the present invention is directed to a method of enhancing fracture healing comprising administering to a mammal in need of such treatment, a therapeutically effective amount of a compound of the formula (I) as defined in the present invention.
- Another aspect of the present invention is directed to said methods and wherein said mammal is a human.
- Another aspect of the present invention is directed to said methods and wherein said mammal is a vertibrate animal, preferably but not limited to bigger animals such as horses, camels, dromedars but not limited thereto.
- GSK3 inhibitors the compounds of formula (I) hereinbefore defined, in primary and secondary ostopeorosis, where primary osteoporosis includes postmenopausal osteoporosis and senile osteoporosis in both men and women, and secondary osteoporosis includes cortison induced osteoporosis, as well as any other type of induced secondary osteoporosis, are included in the term osteoporosis.
- these GSK3 inhibitors may also be used in treatments of myeloma. These GSK3 inhibitors may be administered locally or systemically, in different formulation regimes, to treat these conditions.
- the promotion and increasing of bone formation makes the compounds of the formula (I) hereinbefore defined, suitable to reducing the incidence of fracture, to reduce the rate of fracture and/or increase the rate of fracture healing, to increase cancellous bone formation and/or new bone formation in mammals.
- the use to promote and increase new bone formation may be in connection with surgery.
- This present invention can be used during surgery, where the treating surgeon will place the present invention locally in an appropriate formulation, near the deficient bone and/or in the body cavity.
- the bone may for instance have been broken, and utilizing the present invention as described and claimed herein will then be placed in or near the fracture during open fracture repair. In some instances bone pieces may be missing (e.g. after tumour removal or severe casualties), and utilizing the present invention as described and claimed herein will then be placed near the site of constructive bone surgery.
- the present invention relates also to the use of the compound of formula (I) as as defined in the present invention in the manufacture of a medicament for the prevention and/or treatment of conditions associated with glycogen synthase kinase-3.
- the present invention also provides for a method of treatment and/or prevention of conditions associated with glycogen synthase kinase-3 comprising administering to a mammal, including human in need of such treatment and/or prevention a therapeutically effective amount of the compound of formula (I) as as defined in the present invention.
- the dose required for the therapeutic or preventive treatment of a particular disease will necessarily be varied depending on the host treated, the route of administration and the severity of the illness being treated.
- the dosage form and the dose of the medicament may vary and will depend on various factors such as, for example the individual requirement of the animal treated.
- the term “therapy” also includes “prevention” unless there are specific indications to the contrary.
- the terms “therapeutic” and “therapeutically” should be construed accordingly.
- disorder also includes “condition” unless there are specific indications to the contrary.
- condition also includes “condition” unless there are specific indications to the contrary.
- the compounds of formula (I) as a free base or a pharmaceutically acceptable salt thereof are also useful as pharmacological tools in the development and standardisation of in vitro and in vivo test systems for the evaluation of the effects of inhibitors of GSK3 related activity in laboratory animals such as cats, dogs, rabbits, monkeys, rats and mice, as part of the search for new therapeutics agents.
- the reaction was initiated by the addition of 0.04 ⁇ Ci [ ⁇ - 33 P]ATP (Amersham, UK) and unlabelled ATP at a final concentration of 1 ⁇ M and assay volume of 25 ⁇ l. After incubation for 20 minutes at room temperature, each reaction was terminated by the addition of 25 ⁇ l stop solution containing 5 mM EDTA, 50 ⁇ M ATP, 0.1 % Triton X-100 and 0.25 mg streptavidin coated Scintillation Proximity Assay (SPA) beads (Amersham, UK). After 6 hours the radioactivity was determined in a liquid scintillation counter (1450 MicroBeta Trilux, Wallac). The inhibition curves were analysed by non-linear regression using GraphPad Prism, USA. The K m value of ATP for GSK3 ⁇ , used to calculate the inhibition constants (K;) of the various compounds, was 20 ⁇ M.
- Typical Ki values for the compounds of the present invention are in the range of about 0.001 to about 10,000 nM.
- Other values for Kj are in the range of about 0.001 to about 1000 nM.
- Further values for Ki are in the range of about 0.001 nM to about 700 nM.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Diabetes (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Hematology (AREA)
- Epidemiology (AREA)
- Psychology (AREA)
- Hospice & Palliative Care (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Dermatology (AREA)
- Obesity (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pyridine Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description





































































































Claims



Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BRPI0713576-9A BRPI0713576A2 (en) | 2006-06-27 | 2007-06-26 | compound, pharmaceutical formulation, use of a compound, and methods for preventing and / or treating condition, disorder and disease, for enhancing bone formation, for enhancing spongy bone formation and / or new bone formation, for increasing density bone mineral, to reduce the incidence of fracture and to improve fracture healing, and process for preparing a compound |
| MX2008015720A MX2008015720A (en) | 2006-06-27 | 2007-06-26 | Imidazol-pyrimidine derivatives for treatment of diseases relates to glycogen synthase kinase 3 (gsk3). |
| AU2007265731A AU2007265731A1 (en) | 2006-06-27 | 2007-06-26 | Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (GSK3) |
| JP2009518044A JP2009542638A (en) | 2006-06-27 | 2007-06-26 | Imidazole-pyrimidine derivatives for the treatment of diseases associated with glycogen synthase kinase (GSK3) |
| EP07748281A EP2046782A4 (en) | 2006-06-27 | 2007-06-26 | Imidazolylpyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) |
| CA002655441A CA2655441A1 (en) | 2006-06-27 | 2007-06-26 | New compounds 384 |
| IL195666A IL195666A0 (en) | 2006-06-27 | 2008-12-02 | Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) |
| NO20090327A NO20090327L (en) | 2006-06-27 | 2009-01-21 | New connections 384 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US81675606P | 2006-06-27 | 2006-06-27 | |
| US60/816,756 | 2006-06-27 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| WO2008002244A2 true WO2008002244A2 (en) | 2008-01-03 |
| WO2008002244A3 WO2008002244A3 (en) | 2008-02-14 |
| WO2008002244A8 WO2008002244A8 (en) | 2008-10-09 |
Family
ID=38846127
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/SE2007/000620 WO2008002244A2 (en) | 2006-06-27 | 2007-06-26 | Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) |
Country Status (20)
| Country | Link |
|---|---|
| US (1) | US20080188502A1 (en) |
| EP (1) | EP2046782A4 (en) |
| JP (1) | JP2009542638A (en) |
| KR (1) | KR20090024817A (en) |
| CN (1) | CN101511825A (en) |
| AR (1) | AR061652A1 (en) |
| AU (1) | AU2007265731A1 (en) |
| BR (1) | BRPI0713576A2 (en) |
| CA (1) | CA2655441A1 (en) |
| CL (1) | CL2007001881A1 (en) |
| CO (1) | CO6140059A2 (en) |
| EC (1) | ECSP088973A (en) |
| IL (1) | IL195666A0 (en) |
| MX (1) | MX2008015720A (en) |
| NO (1) | NO20090327L (en) |
| RU (1) | RU2008148902A (en) |
| TW (1) | TW200815418A (en) |
| UY (1) | UY30440A1 (en) |
| WO (1) | WO2008002244A2 (en) |
| ZA (1) | ZA200810484B (en) |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| EP2046783A2 (en) * | 2006-06-27 | 2009-04-15 | AstraZeneca AB | Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) |
| WO2010103438A1 (en) | 2009-03-11 | 2010-09-16 | Pfizer Inc. | Substituted indazole amides and their use as glucokinase activators |
| WO2010120237A1 (en) * | 2009-04-15 | 2010-10-21 | Astrazeneca Ab | Imidazole substituted pyrimidines useful in the treatment of glycogen synthase kinase 3 related disorders such as alzheimer's disease |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2016008966A1 (en) | 2014-07-17 | 2016-01-21 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for treating neuromuscular junction-related diseases |
| WO2016124557A1 (en) | 2015-02-05 | 2016-08-11 | Bayer Cropscience Aktiengesellschaft | 2-(het)aryl-substituted condensed bicyclic heterocycle derivatives as pest control agents |
| WO2016207366A1 (en) | 2015-06-26 | 2016-12-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for the treatment of viral infections |
| US10799501B2 (en) | 2015-11-05 | 2020-10-13 | King's College Hospital Nhs Foundation Trust | Combination of an inhibitor of PARP with an inhibitor of GSK-3 or DOT1L |
| EP3896066A2 (en) | 2015-08-07 | 2021-10-20 | Bayer CropScience Aktiengesellschaft | 2-(het)aryl-substituted condensed heterocycle derivatives as pesticides |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2623374A1 (en) | 2005-09-30 | 2007-04-05 | Astrazeneca Ab | Imidazo [1,2-a] pyridine having anti-cell-proliferation activity |
| AR058073A1 (en) * | 2005-10-03 | 2008-01-23 | Astrazeneca Ab | IMIDAZOL 5-IL-PYRIMIDINE DERIVATIVES, OBTAINING PROCESSES, PHARMACEUTICAL COMPOSITIONS AND USES |
| TW200811169A (en) * | 2006-05-26 | 2008-03-01 | Astrazeneca Ab | Chemical compounds |
| CN102731451B (en) * | 2006-06-27 | 2015-07-29 | 武田药品工业株式会社 | Fused ring compound |
| CN103724207B (en) * | 2013-12-20 | 2016-07-06 | 北京智博高科生物技术有限公司 | Phenylbenzyl ether derivative and its preparation method and application |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0021726D0 (en) * | 2000-09-05 | 2000-10-18 | Astrazeneca Ab | Chemical compounds |
| JP4570955B2 (en) * | 2002-07-09 | 2010-10-27 | バーテクス ファーマスーティカルズ インコーポレイテッド | Imidazoles with protein kinase inhibitory activity |
| EP1648887A1 (en) * | 2003-07-30 | 2006-04-26 | Cyclacel Limited | Pyridinylamino-pyrimidine derivatives as protein kinase inhibitors |
| BRPI0415759A (en) * | 2003-10-21 | 2006-12-19 | Cyclacel Ltd | pyrimidin-4-yl-3,4-thione compounds and their use in therapy |
| GB0402653D0 (en) * | 2004-02-06 | 2004-03-10 | Cyclacel Ltd | Compounds |
| NZ555474A (en) * | 2004-12-17 | 2010-10-29 | Astrazeneca Ab | 4-(4-(imidazol-4-yl) pyrimidin-2-ylamino) benzamides as CDK inhibitors |
| GB0504753D0 (en) * | 2005-03-08 | 2005-04-13 | Astrazeneca Ab | Chemical compounds |
| AR058073A1 (en) * | 2005-10-03 | 2008-01-23 | Astrazeneca Ab | IMIDAZOL 5-IL-PYRIMIDINE DERIVATIVES, OBTAINING PROCESSES, PHARMACEUTICAL COMPOSITIONS AND USES |
| TW200815417A (en) * | 2006-06-27 | 2008-04-01 | Astrazeneca Ab | New compounds II |
-
2007
- 2007-06-21 TW TW096122338A patent/TW200815418A/en unknown
- 2007-06-26 MX MX2008015720A patent/MX2008015720A/en not_active Application Discontinuation
- 2007-06-26 RU RU2008148902/04A patent/RU2008148902A/en not_active Application Discontinuation
- 2007-06-26 CN CNA2007800322107A patent/CN101511825A/en active Pending
- 2007-06-26 EP EP07748281A patent/EP2046782A4/en not_active Withdrawn
- 2007-06-26 KR KR1020097001642A patent/KR20090024817A/en not_active Application Discontinuation
- 2007-06-26 CA CA002655441A patent/CA2655441A1/en not_active Abandoned
- 2007-06-26 AU AU2007265731A patent/AU2007265731A1/en not_active Abandoned
- 2007-06-26 JP JP2009518044A patent/JP2009542638A/en active Pending
- 2007-06-26 BR BRPI0713576-9A patent/BRPI0713576A2/en not_active IP Right Cessation
- 2007-06-26 CL CL200701881A patent/CL2007001881A1/en unknown
- 2007-06-26 UY UY30440A patent/UY30440A1/en unknown
- 2007-06-26 WO PCT/SE2007/000620 patent/WO2008002244A2/en active Application Filing
- 2007-06-26 AR ARP070102831A patent/AR061652A1/en not_active Application Discontinuation
- 2007-06-27 US US11/769,102 patent/US20080188502A1/en not_active Abandoned
-
2008
- 2008-12-02 IL IL195666A patent/IL195666A0/en unknown
- 2008-12-10 ZA ZA200810484A patent/ZA200810484B/en unknown
- 2008-12-12 EC EC2008008973A patent/ECSP088973A/en unknown
- 2008-12-17 CO CO08134161A patent/CO6140059A2/en unknown
-
2009
- 2009-01-21 NO NO20090327A patent/NO20090327L/en not_active Application Discontinuation
Non-Patent Citations (1)
| Title |
|---|
| See references of EP2046782A4 * |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2046783A2 (en) * | 2006-06-27 | 2009-04-15 | AstraZeneca AB | Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) |
| EP2046783A4 (en) * | 2006-06-27 | 2010-08-04 | Astrazeneca Ab | Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010103438A1 (en) | 2009-03-11 | 2010-09-16 | Pfizer Inc. | Substituted indazole amides and their use as glucokinase activators |
| WO2010120237A1 (en) * | 2009-04-15 | 2010-10-21 | Astrazeneca Ab | Imidazole substituted pyrimidines useful in the treatment of glycogen synthase kinase 3 related disorders such as alzheimer's disease |
| US8178529B2 (en) | 2009-04-15 | 2012-05-15 | Astrazeneca Ab | Imidazole substituted pyrimidines |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2016008966A1 (en) | 2014-07-17 | 2016-01-21 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for treating neuromuscular junction-related diseases |
| WO2016124557A1 (en) | 2015-02-05 | 2016-08-11 | Bayer Cropscience Aktiengesellschaft | 2-(het)aryl-substituted condensed bicyclic heterocycle derivatives as pest control agents |
| WO2016207366A1 (en) | 2015-06-26 | 2016-12-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for the treatment of viral infections |
| EP3896066A2 (en) | 2015-08-07 | 2021-10-20 | Bayer CropScience Aktiengesellschaft | 2-(het)aryl-substituted condensed heterocycle derivatives as pesticides |
| EP3896065A1 (en) | 2015-08-07 | 2021-10-20 | Bayer CropScience Aktiengesellschaft | 2-(het)aryl-substituted condensed heterocycle derivatives as pesticides |
| US10799501B2 (en) | 2015-11-05 | 2020-10-13 | King's College Hospital Nhs Foundation Trust | Combination of an inhibitor of PARP with an inhibitor of GSK-3 or DOT1L |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2007265731A1 (en) | 2008-01-03 |
| JP2009542638A (en) | 2009-12-03 |
| TW200815418A (en) | 2008-04-01 |
| WO2008002244A8 (en) | 2008-10-09 |
| WO2008002244A3 (en) | 2008-02-14 |
| CA2655441A1 (en) | 2008-01-03 |
| RU2008148902A (en) | 2010-08-10 |
| NO20090327L (en) | 2009-02-09 |
| ZA200810484B (en) | 2009-08-26 |
| UY30440A1 (en) | 2008-01-31 |
| KR20090024817A (en) | 2009-03-09 |
| CL2007001881A1 (en) | 2008-02-08 |
| MX2008015720A (en) | 2008-12-19 |
| CN101511825A (en) | 2009-08-19 |
| EP2046782A4 (en) | 2010-10-13 |
| EP2046782A2 (en) | 2009-04-15 |
| IL195666A0 (en) | 2009-09-01 |
| AR061652A1 (en) | 2008-09-10 |
| ECSP088973A (en) | 2009-01-30 |
| US20080188502A1 (en) | 2008-08-07 |
| BRPI0713576A2 (en) | 2012-10-23 |
| CO6140059A2 (en) | 2010-03-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008002244A2 (en) | Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) | |
| US20080188503A1 (en) | New Compounds II | |
| AU2006297890B2 (en) | New pyrimidine derivatives and their use in therapy as well as the use of pyrimidine derivatives in the manufacture of a medicament for prevention and/or treatment of Alzheimer's disease | |
| US20090018130A1 (en) | Derivatives of 5-Aryl-1H-Pyrrolo [2, 3B] Pyridine-3-Carboxamide or 5-Aryl-1H-Pyrrolo [2, 3B] Pyridine-3-Carboxylic Acid | |
| US20100234593A1 (en) | Imidazo[4,5-B]Pyridine-7-Carboxamides 704 | |
| EP3684776A1 (en) | Heterocyclyl substituted pyrrolopyridines that are inhibitors of the cdk12 kinase | |
| US20080255106A1 (en) | Novel 2-Phenyl-Imidazo[4,5-B]Pyridine Derivatives as Inhibitors of Glycogen Synthase Kinase for the Treatment of Dementia and Neurodegenerative Disorders | |
| AU2004273771B2 (en) | 3-heterocyclyl-indole derivatives as inhibitors of glycogen synthase kinase-3 (GSK-3) | |
| US20110028489A1 (en) | Pyrimidine Derivatives and Their Use for Treating Bone-Related Disorders | |
| CN115583946A (en) | Heterocyclic compounds and their use as CDK inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780032210.7 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07748281 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 195666 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10118/DELNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/015720 Country of ref document: MX Ref document number: 12008502722 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2655441 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08134161 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007265731 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008122072 Country of ref document: EG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009518044 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2007265731 Country of ref document: AU Date of ref document: 20070626 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 574372 Country of ref document: NZ Ref document number: 1020097001642 Country of ref document: KR Ref document number: 2007748281 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2008148902 Country of ref document: RU Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: a200813929 Country of ref document: UA |
|
| ENP | Entry into the national phase |
Ref document number: PI0713576 Country of ref document: BR Kind code of ref document: A2 Effective date: 20081226 |