WO2008000729A1 - Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases - Google Patents
Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases Download PDFInfo
- Publication number
- WO2008000729A1 WO2008000729A1 PCT/EP2007/056342 EP2007056342W WO2008000729A1 WO 2008000729 A1 WO2008000729 A1 WO 2008000729A1 EP 2007056342 W EP2007056342 W EP 2007056342W WO 2008000729 A1 WO2008000729 A1 WO 2008000729A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- phenyl
- piperazinyl
- pyridinecarboxamide
- fluorophenyl
- Prior art date
Links
- 0 CC1(*)C=C(N)N=CC(Br)=C1 Chemical compound CC1(*)C=C(N)N=CC(Br)=C1 0.000 description 2
- FWOYPABKVAHKKD-LBPRGKRZSA-N C[C@@H](CN(Cc(c(F)c1)ccc1[N+]([O-])=O)CC1)N1C(OC(C)(C)C)=O Chemical compound C[C@@H](CN(Cc(c(F)c1)ccc1[N+]([O-])=O)CC1)N1C(OC(C)(C)C)=O FWOYPABKVAHKKD-LBPRGKRZSA-N 0.000 description 1
- OUNWNHDOYZMWFG-QFIPXVFZSA-N C[C@@H](CN(Cc(cc1)ccc1N(C)C(c(nc1)ccc1Oc1cc(C#N)ccc1)=O)CC1)N1C(OC(C)(C)C)=O Chemical compound C[C@@H](CN(Cc(cc1)ccc1N(C)C(c(nc1)ccc1Oc1cc(C#N)ccc1)=O)CC1)N1C(OC(C)(C)C)=O OUNWNHDOYZMWFG-QFIPXVFZSA-N 0.000 description 1
- IEVCSQGVQLSAAF-ZDUSSCGKSA-N C[C@@H](CN(Cc(cc1F)ccc1NC)CC1)N1C(OC(C)(C)C)=O Chemical compound C[C@@H](CN(Cc(cc1F)ccc1NC)CC1)N1C(OC(C)(C)C)=O IEVCSQGVQLSAAF-ZDUSSCGKSA-N 0.000 description 1
- UXQOGRDHPLLKKX-INIZCTEOSA-N C[C@@H](CN(Cc(ccc(N(C)C(c(cn1)ccc1Cl)=O)c1)c1F)CC1)N1C(OC(C)(C)C)=O Chemical compound C[C@@H](CN(Cc(ccc(N(C)C(c(cn1)ccc1Cl)=O)c1)c1F)CC1)N1C(OC(C)(C)C)=O UXQOGRDHPLLKKX-INIZCTEOSA-N 0.000 description 1
- IDAOMSMUYRIJOP-ZDUSSCGKSA-N C[C@@H](CN(Cc(nc1)ccc1NC)CC1)N1C(OC(C)(C)C)=O Chemical compound C[C@@H](CN(Cc(nc1)ccc1NC)CC1)N1C(OC(C)(C)C)=O IDAOMSMUYRIJOP-ZDUSSCGKSA-N 0.000 description 1
- RHGSMELZFSYVGP-DQUNLGLBSA-N C[C@@H]1NCCN(Cc(c(C)c2)cnc2N(C)CC(C=N2)=CCC2(C)Oc(cc2)ccc2F)C1 Chemical compound C[C@@H]1NCCN(Cc(c(C)c2)cnc2N(C)CC(C=N2)=CCC2(C)Oc(cc2)ccc2F)C1 RHGSMELZFSYVGP-DQUNLGLBSA-N 0.000 description 1
- SBCSWSYSTDCLQQ-IBGZPJMESA-N C[C@@H]1NCCN(Cc(cc2)ccc2N(C)C(C(CC2)CCN2c(cc2)ccc2F)=O)C1 Chemical compound C[C@@H]1NCCN(Cc(cc2)ccc2N(C)C(C(CC2)CCN2c(cc2)ccc2F)=O)C1 SBCSWSYSTDCLQQ-IBGZPJMESA-N 0.000 description 1
- MBLOVZIAFQVXIN-UHFFFAOYSA-N [O-][N+](c1cc(Cl)c(CBr)cc1)=O Chemical compound [O-][N+](c1cc(Cl)c(CBr)cc1)=O MBLOVZIAFQVXIN-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/12—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly or doubly bound nitrogen atoms
- C07D295/135—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly or doubly bound nitrogen atoms with the ring nitrogen atoms and the substituent nitrogen atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/10—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D241/14—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
Definitions
- the present invention relates to novel benzylpiperazine derivatives having pharmacological activity, processes for their preparation, pharmaceutical compositions containing them and their use in the treatment of various disorders.
- GPR38 is a 7-transmembrane, G-protein coupled receptor, with high affinity for the peptide motilin [Feighner et al., Science 1999, 284, 2184], suggesting that endogenous motilin exerts all or most of its activity via this receptor.
- Motilin is a 22 amino acid peptide found in large amounts within endocrine-like cells of the gastrointestinal tract, and especially in the duodenum-jejunum areas. During fasting, the peptide is known to be associated with the onset of Phase III migrating complex activity within the stomach [Boivin et al., Dig. Dis. Sci. 1992, 37, 1562], suggesting a role in the mechanisms of prokinetic activity. Motilin is also released from the gut during feeding, sham feeding, gastric distension or by oral or intravenous nutrient application [Christofides et al., Gut 1979, 20, 102; Bormans et al., Scand. J. Gastroenterol. 1987, 22, 781], suggesting additional roles for this peptide in the modulation of motility patterns during feeding.
- motilin In animals or in man, motilin has long been known to increase gastrointestinal motility, and promote gastric emptying and intestinal propulsion in an anal direction, during both fasting and fed conditions. This activity is thought to be primarily due to a facilitation of at least the cholinergic excitatory function of the gut [Van Assche et al., Eur. J. Pharmacol. 1997, 337, 267], perhaps also involving the activation of the vagus nerve [Mathis & Malbert, Am. J. Physiol. 1998, 274, G80]. In addition, higher concentrations of motilin directly evoke a small contraction of the muscle [Van Assche et al., Eur. J. Pharmacol. 1997, 337, 267].
- the antibiotic erythromycin was shown to mimic the gastrointestinal activity of motilin, in addition to its previously-described antibiotic properties [see Peeters, in Problems of the Gastrointestinal Tract in Anaesthesia Ed., Herbert MK et al. Springer- Verlag, Berlin, Heidelberg 1999, pp 39-51]. More recently, erythromycin has been shown to activate the GPR38 receptor, confirming its ability to mimic the function of motilin [Carreras et al., Analyt. Biochem. 2002, 300, 146]. In addition, the availability of this non-peptide motilin receptor agonist has allowed at least some clinical studies to be undertaken in order to examine the clinical potential of motilin receptor agonists.
- agonists at the GPR38 receptor will mimic the activity of motilin or of other substances acting at this receptor, such as erythromycin, and find clinical utility in the treatment of gastrointestinal disorders associated with hypomotility, especially the functional bowel disorders such as GERD, functional dyspepsia (FD) and irritable bowel syndrome (IBS).
- the compounds will also be useful for the treatment of other Gl conditions where the cause is known and in which Gl motility is reduced.
- Such conditions include constipation, caused by various diseases such as those associated with neuropathy, and/ or by the administration of other drugs, intestinal pseudo-obstruction, paralytic ileus following surgery or some other manipulation, gastric stasis or hypomotility caused by various diseases such as diabetes and/ or by the administration of other drugs, or in enterally fed patients.
- motilin or erythromycin to activate the vagus nerve, the association of this nerve with changes in feeding behaviour [e.g. Furness et al., Auton. Neurosci.
- GPR38 [based on Ensembl: 13q21.1 (58.46 - 59.46 Mb)] within the markers (D13S257- 13q14.11 to D13S258 at 13q21.33) of a locus associated with obesity [Feitosa et al, Am. J. Hum. Genet. 2002, 70, 72] also suggests that agonists active at the GPR38 receptor will, in addition to promoting gastrointestinal motility, facilitate eating behaviours in at least those patients in which some degree of appetite suppression or cachexia is present. Such activity indicates that agonists at this receptor will find clinical utility in the treatment of symptoms associated with - for example - the treatment of cancer or by the presence of the cancer itself.
- motilin receptor agonists In addition to the ability of motilin receptor agonists to promote gastrointestinal motility, the association of motilin gene polymorphism with Crohn's disease [Annese et al., Dig. Dis. ScL 1998, 43, 715-710] and the changes in motilin receptor density during colitis [Depoortere et al., Neurogastroenterol. Motil. 2001 , 13, 55] suggests a utility for agonists at the motilin receptor for the treatment of inflammatory bowel conditions in general.
- GPR38 is also found in regions outside the gastrointestinal tract. These areas include the pituitary, adipose tissue, urinary bladder and certain areas of the brain. The former suggests clinical utility in the promotion of pituitary function, such as the release of growth hormone secretagogues, the presence within adipose tissue again suggests a role in the control of body weight, and the presence within the urinary bladder suggests a role for agonists at this receptor in the treatment of incontinence. The presence of GPR38 within the brain supports the gastrointestinal and feeding utilities already mentioned, but in addition, suggests an involvement of the receptor in a greater spectrum of vagal-hypothalamic functions.
- WO9410185, EP838469, WO9823629, DE19805822, and US6165985 claim erythromycin derivatives targeting GPR38 for use in disorders relating to gastrointestinal motility.
- WO9921846, WO0185694, WO0168620, WO0168621 , and WO0168622 disclose a series of small molecule antagonists of the GPR38 receptor.
- JP07138284 and EP807639 disclose peptide agonists.
- JP09249620, WO02092592, WO05027637, US2005065156 and Li et al., (2004, Journal of Medicinal Chemistry, 47(7) p1704-1708) disclose series of small molecule agonists.
- WO 05012331 and WO 05012332 disclose macrocyclic compounds which are agonists or antagonists of mammalian motilin or ghrelin receptors.
- WO 06127252 discloses erythromycin derivatives.
- WO07/007018 describes compounds of formula (A), which have activity as agonists of the GPR38 receptor
- WO07/012479 describes compounds of formula (B), which have activity as agonists of the GPR38 receptor
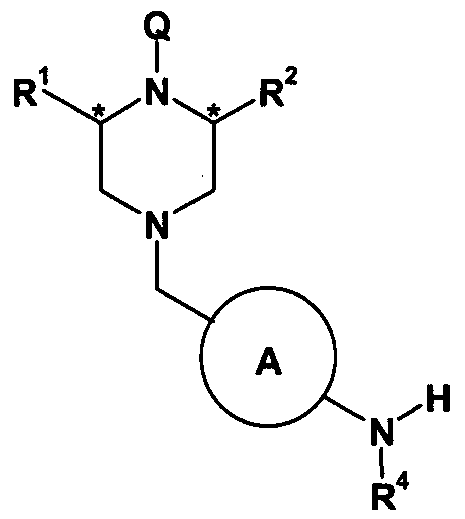
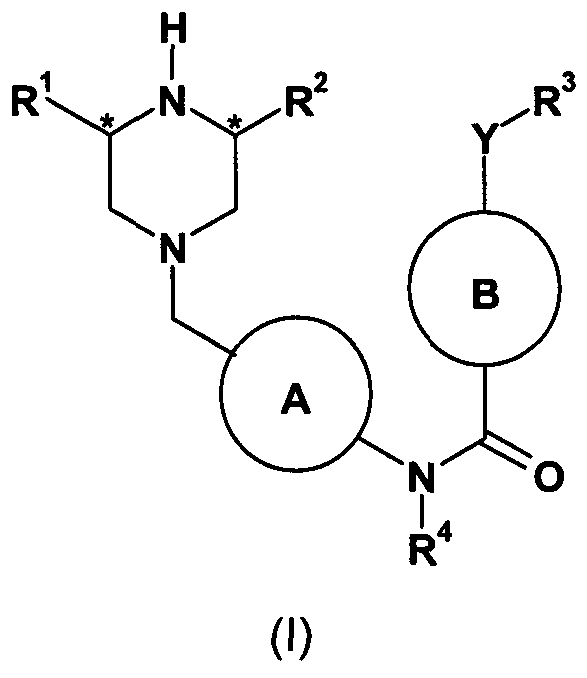
- the present invention therefore provides compounds of formula (I) and salts thereof (hereinafter known as “compounds of the invention”):
- A is phenyl or a 6-membered heteroaryl ring, optionally substituted with one susbsituent selected from halogen, C (1-4) alkyl and C (1 ⁇ )alkoxy;
- R 1 and R 2 are independently H or C (1-4) alkyl
- R 3 is an optionally substituted phenyl, heteroaryl ring, or heterocyclic ring
- B is an optionally substituted phenyl, 6-membered heteroaryl ring or 6-membered heterocyclic ring connected to the amide carbon via a carbon atom;
- R 4 is hydrogen, C (1-4) alkyl or C (1-4) alkoxyalkyl.
- the present invention also provides compounds of formula (IA) or a pharmaceutically acceptable salt or solvate thereof:
- A is phenyl or a 6-membered heteroaryl ring, optionally substituted with halogen, C (1-
- R 1 and R 2 are independently H or C 0-4) alky!;
- R 3 is an optionally substituted phenyl, heteroaryl ring, or heterocyclic ring
- B is an optionally substituted phenyl.a 6-membered heteroaryl ring or a 6-membered heterocyclic ring connected to the amide carbon via a carbon atom;
- R 3 or B When R 3 or B is substituted, it may have 1 , 2 or 3 substituents, each independently selected from halogen, C (1-4) alkyl, C ⁇ jalkoxy, C (3 . 7 )Cycloalkyl, hydroxy, trifluoromethoxy, trifluoromethyl, nitro, cyano, phenyl, NH 2 , NHR 5 , NR 5 R 6 , NHCOR 5 , NHSO 2 R 5 , C(O)CF 3 , C(O)C (1-4) alkyl, C(O)C (3-7 )Cycloalkyl, C(O)OC (1-4) alkyl, C(O)OC ((3- 7) cycloalkyl, OC(O)C (1-4) alkyl, OC(O)C (3-7) cycloalkyl, CONH 2 , CONHR 5 , CONR 5 R 6 , SOR 6 , SO 2 CF 3 , SO 2 R 6 , OSO 2
- R 3 is substituted by fluorine, chlorine, cyano, CONH 2 , methyl, methoxy or trifluoromethoxy.
- alkyl as a group or part of a group e.g. alkoxy or hydroxyalkyl refers to a straight or branched alkyl group in all isomeric forms.
- C (1-4) alkyl refers to an alkyl group, as defined above, containing at least 1 , and at most 4 carbon atoms Examples of such alkyl groups include methyl, ethyl, propyl, /so-propyl, n-butyl, iso- ⁇ butyl, sec-butyl, or te/f-butyl, Examples of such alkoxy groups include methoxy, ethoxy, propoxy, /so-propoxy, butoxy, /so-butoxy, sec-butoxy and tert-butoxy.
- Suitable C( 3-7 )Cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl, cycloheptyl.
- halogen refers to fluorine (F), chlorine (Cl), bromine (Br), or iodine (I) and the term “halo” refers to the halogen: fluoro (-F), chloro (-Cl), bromo (-Br) and iodo (-I).
- heteroaryl ring represents a 5 or 6 membered unsaturated aromatic ring which comprises one or more heteroatoms.
- heteroaryl represents a 5 membered group it contains a heteroatom selected from O, N or S and may optionally contain a further 1 to 3 nitrogen atoms.
- heteroaryl represents a 6- membered group it contains from 1 to 3 nitrogen atoms.
- Examples of such 5 or 6 membered heteroaryl rings include pyrrolyl, triazolyl, thiadiazolyl, tetrazolyl, imidazolyl, pyrazolyl, isothiazolyl, thiazolyl, isoxazolyl, oxazolyl, oxadiazolyl, furazanyl, furanyl, thienyl, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl and triazinyl.
- heterocyclic ring represents a saturated or partially saturated 5 or 6 membered ring which comprises one or more heteroatoms selected from nitrogen, oxygen and sulphur.
- heterocyclyl groups include pyrrolidinyl, piperidinyl, piperazinyl and morpholinyl.
- A is optionally substituted phenyl or pyridyl.
- R 1 is hydrogen or methyl.
- R 2 is hydrogen or methyl.
- R 3 is optionally substituted phenyl, morpholinyl, piperidinyl, oxadiazolyl, pyridyl, pyrimidinyl, imidazolyl, pyrrolyl. In a further embodiment R 3 is optionally substituted phenyl, morpholinyl or piperidinyl.
- B is optionally substituted phenyl, piperidinyl, pyrimidinyl or pyridyl.
- R 4 is hydrogen, methyl, ethyl, methoxyethyl or isopropyl. In a further emodiment R 4 is methyl.
- A is optionally substituted phenyl or pyridyl; and/or R 1 is hydrogen or methyl; and/or R 2 is hydrogen or methyl; and/or
- R 3 is optionally substituted phenyl, morpholinyl or piperidinyl; and/or B is optionally substituted phenyl, piperidinyl, pyrimidinyl or pyridyl; and/or
- A is optionally substituted phenyl or pyridyl; and/or R 1 is hydrogen or methyl; and/or R 2 is hydrogen or methyl; and/or
- R 3 is optionally substituted phenyl, morpholinyl, piperidinyl, oxadiazolyl, pyridyl, pyrimidinyl, imidazolyl, pyrrolyl; and/or
- B is optionally substituted phenyl, piperidinyl, pyrimidinyl or pyridyl; and/or
- compounds of formula (I) may exist as stereoisomers.
- the invention extends to all optical isomers such as stereoisomeric forms of the compounds of formula (I) including enantiomers, diastereoisomers and mixtures thereof, such as racemates.
- the different stereoisomeric forms may be separated or resolved one from the other by conventional methods or any given isomer may be obtained by conventional stereoselective or asymmetric syntheses.
- Preferred compounds of formula (I) wherein R 1 and R 2 are both methyl are those wherein the piperazine C* carbons have the 3R,5S-configuration.
- Preferred compounds of formula (I) wherein one of R 1 and R 2 is methyl and the other is hydrogen are those wherein the piperazine C* carbon has the S-configuration
- Suitable compounds of the invention are:
- One embodiment of the invention is 6-[(4-fluorophenyl)oxy]- ⁇ /-methyl- ⁇ /-(4- ⁇ [(3S)-3- methyl-1-piperazinyl]methyl ⁇ phenyl)-3-pyridinecarboxamide or a salt thereof.
- a further embodiment of the invention is 6-[(4-fluorophenyl)oxy]- ⁇ /-methyl- ⁇ /-(4- ⁇ [(3S)-3-methyl-1-piperazinyl]methyl ⁇ phenyl)-3-pyridinecarboxamide fumarate salt.
- One embodiment of the invention is 6-[(4-fluorophenyl)oxy]- ⁇ /-methyl- ⁇ /-(4-methyl-5- ⁇ [(3S)-3-methyl-1-piperazinyl]methyl ⁇ -2-pyridinyl)-3-pyridinecarboxamide or a salt thereof.
- the compounds of formula (I) can form acid addition salts thereof. It will be appreciated that for use in medicine the salts of the compounds of formula (I) should be pharmaceutically acceptable. Suitable pharmaceutically acceptable salts will be apparent to those skilled in the art and include those described in J. Pharm. ScL, 1977, 66, 1-19, such as acid addition salts formed with inorganic acids e.g. hydrochloric, hydrobromic, sulfuric, nitric or phosphoric acid; and organic acids e.g. succinic, maleic, acetic, fumaric, citric, tartaric, benzoic, p-toluenesulfonic, methanesulfonic or naphthalenesulfonic acid. Certain of the compounds of formula (I) may form acid addition salts with one or more equivalents of the acid.
- the present invention includes within its scope all possible stoichiometric and non-stoichiometric forms.
- the compounds of formula (I) and salts thereof may be prepared in crystalline or non-crystalline form, and, if crystalline, may optionally be hydrated or solvated.
- This invention includes within its scope stoichiometric hydrates or solvates as well as compounds containing variable amounts of water and/or solvent.
- Salts and solvates having non-pharmaceutically acceptable counter-ions or associated solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of formula (I) and their pharmaceutically acceptable salts.
- a prodrug of a compound of formula (I) is a functional derivative of the compound which, upon administration to a patient, eventually liberates the compound of formula (I) in vivo.
- Administration of a compound of formula (I) as a prodrug may enable the skilled artisan to do one or more of the following: (a) modify the onset of action of the compound in vivo; (b) modify the duration of action of the compound in vivo; (c) modify the transportation or distribution of the compound in vivo; (d) modify the solubility of the compound in vivo; and (e) overcome a side effect or other difficulty encountered with the compound.
- Typical .functional derivatives used to prepare prodrugs include modifications of the compound that are chemically or enzymatically cleaved in vivo. Such modifications, which include the preparation of phosphates, amides, esters, thioesters, carbonates, and carbamates, are well known to those skilled in the art.
- the invention also includes isotopically-labeled compounds, which are identical to those described herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine, iodine, and chlorine, such as 3 H, 11 C, 14 C, 18 F, 123 I and 125 I.
- Compounds of the invention that contain the aforementioned isotopes and/or other isotopes of other atoms are within the scope of the present invention.
- Isotopically-labeled compounds of the present invention for example those into which radioactive isotopes such as 3 H, 14 C are incorporated, are useful in drug and/or substrate tissue distribution assays. Tritiated, i.e., 3 H, and carbon-14, i.e., 14 C, isotopes are particularly preferred for their ease of preparation and detectability. 11 C and 18 F isotopes are particularly useful in PET (positron emission tomography), and 125 I isotopes are particularly useful in SPECT (single photon emission computerized tomography), all useful in brain imaging.
- lsotopically labeled compounds of the invention can generally be prepared by carrying out the procedures disclosed in the Schemes and/or in the Examples below, then substituting a readily available isotopically labeled reagent for a non-isotopically labeled reagent.
- this invention provides a process for the preparation of a compound of formula (I)
- the reaction may be carried out using a suitable base such as triethylamine in an inert solvent such as dichloromethane.
- a suitable coupling reagent such as ⁇ /, ⁇ /'- dicyclohexylcarbodiimide (DCC), /V-benzyl-W-cyclohex
- R 7 represents optional substitution in the phenylene moiety as defined for A above and Q is hydrogen or a suitable nitrogen protecting group such as tert-butyloxycarbonyl (Boc) or benzyloxycarbonyl (Cbz) with an appropriate aldehyde, ketone or enol ether to provide R 4 , using conditions suitable for a reductive amination; for example in the presence of a suitable reducing agent such as sodium borohydride and in a suitable solvent such as methanol and optionally in the presence of a suitable base such as sodium methoxide.
- a suitable nitrogen protecting group such as tert-butyloxycarbonyl (Boc) or benzyloxycarbonyl (Cbz) with an appropriate aldehyde, ketone or enol ether to provide R 4 , using conditions suitable for a reductive amination; for example in the presence of a suitable reducing agent such as sodium borohydride and in a suitable solvent such as
- R 1 , R 2 , R 7 and Q are as defined above, using conditions suitable for a reduction; for example when Q is Boc, hydrogenation in the presence of a suitable catalyst such as palladium on charcoal or platinum on charcoal, in a suitable solvent such as methanol and optionally in the presence of a suitable base such as potassium hydroxide or triethylamine.
- a suitable catalyst such as palladium on charcoal or platinum on charcoal
- a suitable solvent such as methanol
- a suitable base such as potassium hydroxide or triethylamine.
- the reduction may be carried out using a suitable metal reducing agent such as iron powder, in the presence of a suitable proton source such as ammonium chloride and in a suitable solvent such as aqueous methanol.
- Compounds of formula (IV) may be prepared by reaction of a compound of formula (V)
- Compounds of formula (VII) are commercially available or may be prepared by methods similar to those described in the literature (see for example WO 03/053972, WO 03/037898.
- An alternative process for preparation of compounds of formula (I) comprises reaction of a compound of formula (VIII)
- R 1 , R 2 , R 4 , A, B and Q are as defined above and L 3 represents a leaving group such as halogen or trifluoromethylsulfonyloxy, and a compound of formula M 1 - Y-R 3 wherein R 3 and Y are as defined above and M 1 represents hydrogen, a metal residue (e.g.
- alkali metal salt, trialkylstannyl) or a boronic acid optionally in the presence of a suitable base such as potassium carbonate, cesium carbonate, sodium carbonate, sodium hydride or triethylamine and optionally using a suitable transition metal catalyst system such as palladium acetate/BINAP, copper (I) chloride/2, 2,6, 6-tetramethyl-3,5-heptanedione or tefra/ «s(triphenylphosphine) palladium (0).
- a suitable base such as potassium carbonate, cesium carbonate, sodium carbonate, sodium hydride or triethylamine
- a suitable transition metal catalyst system such as palladium acetate/BINAP, copper (I) chloride/2, 2,6, 6-tetramethyl-3,5-heptanedione or tefra/ «s(triphenylphosphine) palladium (0).
- An alternative process for preparation of compounds of formula (III) comprises reaction of a compound of formula (Vl) as defined above with a compound of formula
- An alternative process for preparation of compounds of formula (II) wherein A represents an optionally substituted 1 ,4-phenylene group, an optionally substituted 2,5-pyridyl group or an optionally substituted 3,6-pyridyl group comprises reaction of a compound of formula (Vl) as defined above with a compound of formula (X):
- A represents an optionally substituted 1 ,4-phenylene group or an optionally substituted 2,5-pyridyl group and L 3 is as defined above with a compound of formula R 4 NHQ 1 , wherein R 4 is as defined above and Q 1 is a suitable nitrogen protecting group such as tert-butyloxycarbonyl (Boc), in the presence of a suitable transition metal catalyst sytem such as tris(dibenzylideneacetone) dipalladium(0)/xantphos, in the presence of a suitable base such as cesium carbonate and in a suitable solvent such as dioxane; followed by a suitable deprotection step.
- a suitable nitrogen protecting group such as tert-butyloxycarbonyl (Boc)
- a suitable transition metal catalyst sytem such as tris(dibenzylideneacetone) dipalladium(0)/xantphos
- a suitable base such as cesium carbonate
- a suitable solvent such as dioxane
- A represents an optionally substituted 1 ,4-phenylene group or an optionally substituted 2,5-pyridyl group and L 3 is as defined above with a suitable reducing agent such as diisobutylaluminium hydride in a suitable solvent such as toluene.
- a suitable reducing agent such as diisobutylaluminium hydride in a suitable solvent such as toluene.
- R 7 represents optional substitution of the pyridine moiety with C ⁇ alkyl or C d ⁇ alkoxy and L 3 is as defined above with a compound of formula R 4 NH 2 in a suitable solvent such as THF and optionally in the presence of a suitable base.
- An alternative process for preparation of compounds of formula (III) wherein A represents an optionally substituted 3,6-pyridyl group comprises reaction of a compound of formula (Vl) as defined above with a compound of formula (XV):
- R 7 represents optional substitution of the pyridine moiety with C ⁇ - ⁇ alkyl or C ⁇ alkoxy, under conditions suitable for reductive amination as described above.
- R 7 represents optional substitution of the pyridine moiety with C( 1-4) alkyl or C (1-4) alkoxy and Q 1 is a suitable protecting group such as trifluoroacetyl, with a suitable reducing agent such as nickel/aluminium alloy in the presence of formic acid and in a suitable solvent such water.
- Aldehyde or ketone groups can be protected as acetals, ketals, thioacetals or thioketals. Deprotection of such groups is achieved using conventional procedures well known in the art. For example, protecting groups such as tert-butyloxycarbonyl may be removed using an acid such as hydrochloric or trifluoroacetic acid in a suitable solvent such as dichloromethane, diethyl ether, 1 ,4-dioxane, isopropanol or mixtures thereof. Salts may be prepared conventionally by reaction with the appropriate acid or acid derivative.
- the present invention also provides compounds of formula (II) and (VIII) as shown above wherein R 1 , R 2 , R 4 , A and B are as defined for formula (I), Q is hydrogen or a suitable protecting group such as tert-butyloxycarbonyl (BOC) or benzyloxycarbonyl (CBZ) and L 3 is a leaving group such as halogen or trifluoromethylsulfonyloxy.
- Q is hydrogen or a suitable protecting group such as tert-butyloxycarbonyl (BOC) or benzyloxycarbonyl (CBZ)
- L 3 is a leaving group such as halogen or trifluoromethylsulfonyloxy.
- potencies and efficacies of the compounds of this invention for GPR38 can be determined by FLIPR assay performed on the human cloned receptor as described herein.
- Compounds of formula (I) have demonstrated partial or full agonist activity at the GPR38 receptor, using the FLIPR (FLuorometric Imaging Plate Reader) functional assays described herein.
- Compounds of formula (I) and pharmaceutically acceptable salts thereof are therefore of use in the treatment of conditions or disorders which are mediated via the GPR38 receptor.
- the compounds of formula (I) and pharmaceutically acceptable salts thereof are of use in the treatment of certain gastrointestinal disorders such as gastroesophageal reflux disorders, functional dyspepsia, irritable bowel syndrome, constipation, intestinal pseudo-obstruction, paralytic ileus following surgery or other manipulation, emesis, gastric stasis or hypomotility caused by various diseases such as diabetes and/ or by the administration of other drugs, or in enterally fed patients, Crohn's disease, colitis, cachexia associated with advanced diseases such as cancer and/or the treatment thereof, and other disorders such as incontinence (herein after referred to as the "Disorders of the Invention").
- treatment includes prophylaxis as well as alleviation of established symptoms.
- the invention also provides compounds of formula (I) and pharmaceutically acceptable salts thereof for use as a therapeutic substance, in particular in the treatment of conditions or disorders mediated via the GPR38 receptor.
- the invention provides compounds of formula (I) and pharmaceutically acceptable salts thereof for use as a therapeutic substance in the treatment of gastrointestinal disorders such as gastroesophageal reflux disorders, functional dyspepsia, irritable bowel syndrome, constipation, intestinal pseudo-obstruction, paralytic ileus following surgery or other manipulation, emesis, gastric stasis or hypomotility caused by various diseases such as diabetes and/ or by the administration of other drugs, or in enterally fed patients, Crohn's disease, colitis, cachexia associated with advanced diseases such as cancer and/or the treatment thereof, and other disorders such as incontinence.
- gastrointestinal disorders such as gastroesophageal reflux disorders, functional dyspepsia, irritable bowel syndrome, constipation, intestinal pseudo-obstruction, paralytic ileus following surgery or other manipulation, emes
- the invention further provides a method of treatment of conditions or disorders in mammals including humans which can be mediated via the GPR38 receptor, which comprises administering to the sufferer a therapeutically safe and effective amount of a compounds of formula (I) or a pharmaceutically acceptable salt thereof.
- the invention provides for the use of compounds of formula (I) and pharmaceutically acceptable salts thereof in the manufacture of a medicament for use in the treatment of the conditions or disorders mediated via the GPR38 receptor.
- the present invention also provides a pharmaceutical composition, which comprises a compound of formula (I) or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier or excipient.
- the present invention provides a process for preparing a pharmaceutical composition, the process comprising mixing a compound of formula (I) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier or excipient.
- a pharmaceutical composition of the invention which may be prepared by admixture, suitably at ambient temperature and atmospheric pressure, is usually adapted for oral, parenteral or rectal administration and, as such, may be in the form of tablets, capsules, oral liquid preparations, powders, granules, lozenges, reconstitutable powders, injectable or infusible solutions or suspensions or suppositories. Orally administrate compositions are generally preferred. Tablets and capsules for oral administration may be in unit dose form, and may contain conventional excipients, such as binding agents (e.g. pregelatinised maize starch, polyvinylpyrrolidone or hydroxypropyl methylcellulose); fillers (e.g.
- binding agents e.g. pregelatinised maize starch, polyvinylpyrrolidone or hydroxypropyl methylcellulose
- fillers e.g.
- lactose microcrystalline cellulose or calcium hydrogen phosphate
- tabletting lubricants e.g. magnesium stearate, talc or silica
- disintegrants e.g. potato starch or sodium starch glycollate
- acceptable wetting agents e.g. sodium lauryl sulphate.
- the tablets may be coated according to methods well known in normal pharmaceutical practice.
- Oral liquid preparations may be in the form of, for example, aqueous or oily suspension, solutions, emulsions, syrups or elixirs, or may be in the form of a dry product for reconstitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives such as suspending agents (e.g. sorbitol syrup, cellulose derivatives or hydrogenated edible fats), emulsifying agents
- non-aqueous vehicles which may include edible oils e.g. almond oil, oily esters, ethyl alcohol or fractionated vegetable oils, preservatives
- Preparations for oral administration may be suitably formulated to give controlled release of the active compound or pharmaceutically acceptable salt thereof.
- fluid unit dosage forms are prepared utilising a compound of formula (I) or pharmaceutically acceptable salt thereof and a sterile vehicle.
- Formulations for injection may be presented in unit dosage form e.g. in ampoules or in multi-dose, utilising a compound of formula (I) or pharmaceutically acceptable salt thereof and a sterile vehicle, optionally with an added preservative.
- the compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilising and/or dispersing agents.
- the active ingredient may be in powder form for constitution with a suitable vehicle, e.g. sterile pyrogen-free water, before use.
- the compound depending on the vehicle and concentration used, can be either suspended or dissolved in the vehicle.
- the compound can be dissolved for injection and filter sterilised before filling into a suitable vial or ampoule and sealing.
- adjuvants such as a local anaesthetic, preservatives and buffering agents are dissolved in the vehicle.
- the composition can be frozen after filling into the vial and the water removed under vacuum.
- Parenteral suspensions are prepared in substantially the same manner, except that the compound is suspended in the vehicle instead of being dissolved, and sterilisation cannot be accomplished by filtration.
- the compound can be sterilised by exposure to ethylene oxide before suspension in a sterile vehicle.
- a surfactant or wetting agent is included in the composition to facilitate uniform distribution of the compound.
- Lotions may be formulated with an aqueous or oily base and will in general also contain one or more emulsifying agents, stabilising agents, dispersing agents, suspending agents, thickening agents, or colouring agents. Drops may be formulated with an aqueous or non-aqueous base also comprising one or more dispersing agents, stabilising agents, solubilising agents or suspending agents. They may also contain a preservative.
- the compounds of formula (I) or pharmaceutically acceptable salts thereof may also be formulated in rectal compositions such as suppositories or retention enemas, e.g. containing conventional suppository bases such as cocoa butter or other glycerides.
- the compounds of formula (I) or pharmaceutically acceptable salts may also be formulated as depot preparations. Such long acting formulations may be administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection.
- the compounds of formula (I) or pharmaceutically acceptable salts may be formulated with suitable polymeric or hydrophobic materials (for example as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- the compounds formula (I) or pharmaceutically acceptable salts thereof may be formulated as solutions for administration via a suitable metered or unitary dose device or alternatively as a powder mix with a suitable carrier for administration using a suitable delivery device.
- compounds of formula (I) or pharmaceutically acceptable salts thereof may be formulated for oral, buccal, parenteral, topical (including ophthalmic and nasal), depot or rectal administration or in a form suitable for administration by inhalation or insufflation (either through the mouth or nose).
- the compounds of formula (I) and pharmaceutically acceptable salts thereof may be formulated for topical administration in the form of ointments, creams, gels, lotions, pessaries, aerosols or drops (e.g.
- Ointments and creams may, for example, be formulated with an aqueous or oily base with the addition of suitable thickening and/or gelling agents.
- Ointments for administration to the eye may be manufactured in a sterile manner using sterilised components.
- the composition may contain from 0.1% to 99% by weight, preferably from 10 to 60% by weight, of the active material, depending on the method of administration.
- the dose of the compound used in the treatment of the aforementioned disorders will vary in the usual way with the seriousness of the disorders, the weight of the sufferer, and other similar factors.
- suitable unit doses may be 0.05 to 1000 mg, more suitably 1.0 to 500mg or 1.0 to 200 mg, and such unit doses may be administered more than once a day, for example two or three times a day.
- Compounds of formula (I) and pharmaceutically acceptable salts thereof may be used in combination preparations.
- the compounds of formula (I) and pharmaceutically acceptable salts thereof may be used in combination with one or more compounds with activity in reducing gastric acid; one or more compounds with activity in reducing gastro-esophageal reflux; one or more compounds with activity in reducing esophago-gastric irritancy or inflammation, especially when used to alleviate erosive or non-erosive esophagitis; one or more compounds with analgesic activity; and/or one or more compounds with mixed activity on motility and pain.
- Examples of compounds with activity in reducing gastric acid include H2 receptor antagonists, acid pump antagonists and proton pump inhibitors.
- Examples of compounds with activity in reducing gastro-esophageal reflux include agonists at GABA-B.
- Examples of compounds with analgesic activity include compounds active at Neurokinin receptors (NK1 , 2, 3), TRPV1 and sodium-channels.
- Examples of compounds with mixed activity on motility and pain include CRF2 antagonists, 5-HT3 antagonists or octreotide or other molecules active at sst2 receptors.
- the column used is a Waters Atlantis, the dimensions of which are 4.6mm x 50mm.
- the stationary phase particle size is 3 ⁇ m.
- Aqueous solvent Water + 0.05% Formic Acid
- the above method has a flow rate of 3mL/mins
- the column used is a Waters Acquity BEH UPLC C18, the dimensions of which are 2.1mm x 50mm.
- the stationary phase particle size is 1.7 ⁇ m.
- Aqueous solvent Water + 0.05% Formic Acid
- Organic solvent Acetonitrile + 0.05%
- the generic method used has a 2 minute runtime.
- the above method has a flow rate of 1 ml/min.
- the UV detection range is from 220 to 330nm Conditions For Open Access Mass Directed Auto Prep System (MDAP) Hardware
- Open Access Mass Directed Prep instruments consist of the following:
- the column used is typically a Supelco LCABZ++ column whose dimensions are 20mm internal diameter by 100mm in length.
- the stationary phase particle size is
- Aqueous solvent Water + 0.1 % Formic Acid
- Needle rinse solvent MeOH: Water: DMSO 80:10:10
- One of five methods may be used depending on the analytical retention time of the compound of interest. All have a 15-minute runtime, which comprises of a 10-minute gradient followed by a
- All of the above MDAP methods have a flow rate of either 20mls/min (Small Scale) or 40mls/min (Large Scale).
- the title compound was prepared from 1 ,1 -dimethylethyl (2f?)-4-[(4- aminophenyl)methyl]-2-methyl-1 -piperazinecarboxylate (D5) using a method similar to that described for D3 in Description 3A although the reaction was heated at 50 0 C for 48h prior to addition of sodium borohydride.
- the title compound may be prepared using a method similar to that described for D1.
- the title compound was prepared from 1 ,1 -dimethylethyl (2R,6S)-4-[(4- aminophenyl)methyl]-2,6-dimethyl-1 -piperazinecarboxylate (D8) using a method similar to that described for D3 in Description 3A although the reaction was heated at 5O 0 C for 48h prior to addition of sodium borohydride then for 1h after addition. Further paraformaldehyde (1eq) and sodium methoxide (1eq) were added; the reaction was heated at 5O 0 C for 12 h; further sodium borohydride (1eq) was added and the reaction heated at 5O 0 C for 1 h.
- Step 1 6-(4-Fluorophenyl)-2-methyl-3-pyridinecarboxylic acid (0.2 g, 0.866 mol) was suspended in DCM (14 mL) and DMF (1 drop) was added. The mixture was cooled in an ice-bath and oxalyl chloride (0.226 mL, 2.598 mmol) was added portion wise over 5 minutes. The mixture was heated to 40 0 C for 90 minutes. The mixture was allowed to cool and the solvent removed under vacuum to give 6-(4-fluorophenyl)-2-methyl-3- pyridinecarbonyl chloride as a yellow solid (0.27Og) which was used directly in step 2.
- Step 2 The acid chloride from step 1 was taken up in DCM (3 mL) and added to a solution of 1 ,1 -dimethylethyl (2R,6S)-2,6-dimethyl-4- ⁇ [4-(methylamino)phenyl] methyl ⁇ -1 -piperazinecarboxylate (D9) (0.23 g, 0.693 mmol) in DCM (2 mL). Triethylamine (0.193 mL, 1.386 mmol) was added and the mixture was stirred overnight at room temperature. The reaction mixture was diluted with DCM and washed with water. The aqueous layer was re-extracted with DCM (x2) and the combined organics were dried and concentrated.
- D9 1 ,1 -dimethylethyl (2R,6S)-2,6-dimethyl-4- ⁇ [4-(methylamino)phenyl] methyl ⁇ -1 -piperazinecarboxylate
- D9 1 ,1 -di
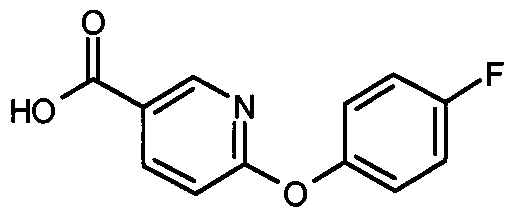
- Step 1 6-[(4-Fluorophenyl)oxy]-3-pyridinecarboxylic acid (D14) (0.232 g, 0.994 mmol) was dissolved in 1 ,4-dioxane (6 mL) and thionyl chloride (0.363 ml_, 4.972 mmol) added dropwise. The mixture was heated at reflux for 3.5 h, then cooled and concentrated in vacuo. DCM was added to the residue which was then re- concentrated to yield 6-[(4-fluorophenyl)oxy]-3-pyridinecarbonyl chloride as a yellow oil (0.252g) which was used directly in step 2.
- Step 2 The acid chloride from step 1 was taken up in DCM (3 mL) and added to 1 ,1- dimethylethyl (2S)-2-methyl-4- ⁇ [4-(methylamino)phenyl]methyl ⁇ -1- piperazinecarboxylate (D3) (0.288 g, 0.904 mmol) in DCM (3 mL). Triethylamine (0.251 mL, 1.808 mmol) was added and the mixture was stirred at room temperature overnight. The reaction mixture was diluted with DCM and washed with water. The aqueous was extracted with DCM (x2) and the combined organic layers were dried and concentrated. The crude product was purified by column chromatography.
- D3 1 ,1- dimethylethyl (2S)-2-methyl-4- ⁇ [4-(methylamino)phenyl]methyl ⁇ -1- piperazinecarboxylate
- D3 1 ,1- dimethylethyl (2S)-2-methyl-4- ⁇ [4-(methylamin
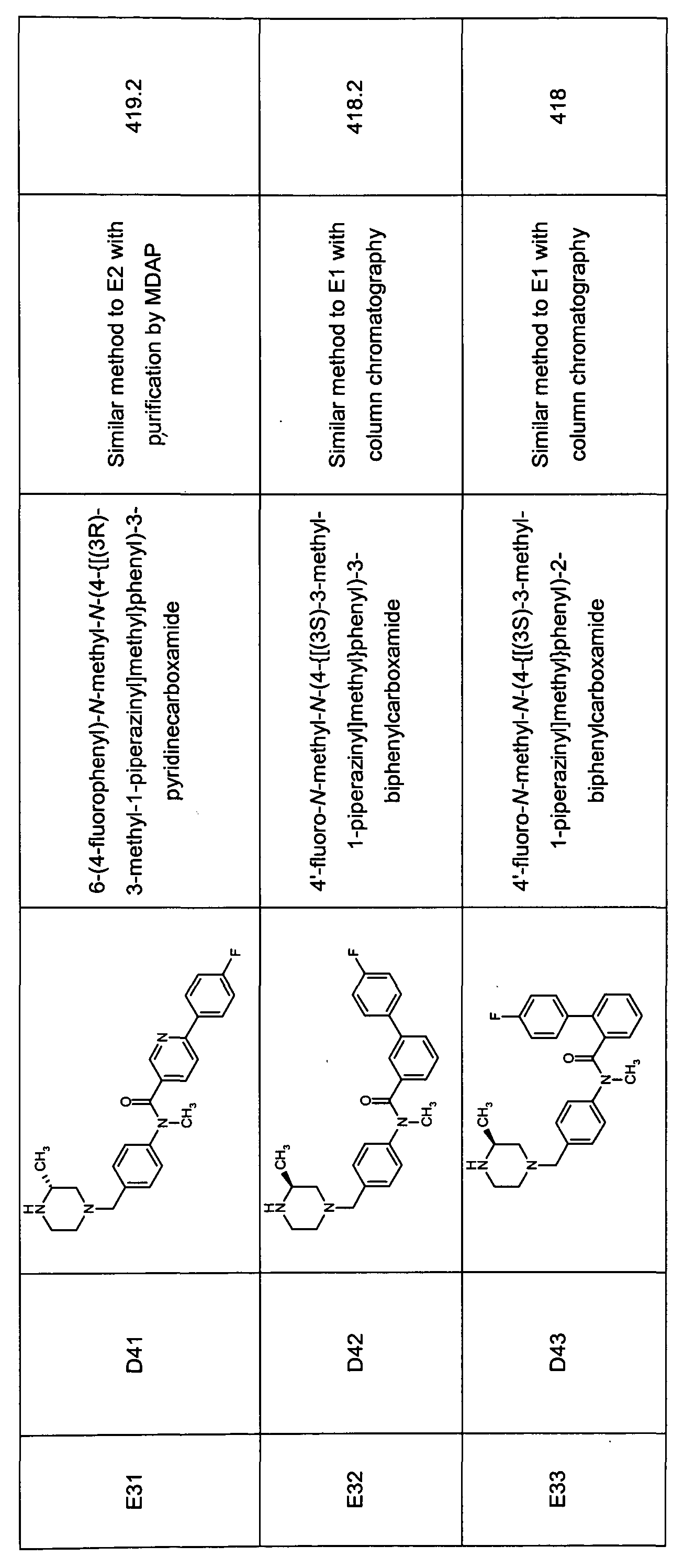
- Tabulated compounds D17, D40 and D41 were prepared using methods similar to those described in Description 15 using the appropriate aniline precursor and appropriate carboxylic acid.
- Tabulated compounds D18 - D53 (excepting D40 and D41 ) were prepared using methods similar to those described in Description 16 using the appropriate aniline precursor and appropriate carboxylic acid.
- the organic layer was dried and concentrated to produce a brown oil.
- the crude product was purified by MDAP to yield the formic acid salt.
- the salt was taken up in DCM and washed with sat. NaHCO 3 .
- the DCM layer was dried and concentrated to yield the title compound as a colourless oil (0.038 g).
- BINAP 0.041 g, 0.0654 mmol
- cesium carbonate 0.213 g, 0.654 mmol
- palladium acetate 0.009 g, 0.0436 mmol
- Step 1 1-(4-Fluorophenyl)-4-piperidinecarboxylic acid (0.1 g, 0.45 mmol) was stirred in dioxane (5 mL) and thionyl chloride (0.165 ml_, 2.25 mmol) was added drop-wise. After stirring for 1 h the solvent was removed by evaporation, DCM was added to the residue which was then re-concentrated to give 1-(4-fluorophenyl)-4- piperidinecarbonyl chloride which was used directly in step 2.
- Step 2 The acid chloride from step 1 was taken up in DCM (2.5 mL) and added drop-wise to 1 ,1 -dimethylethyl (2S)-2-methyl-4- ⁇ [4-(methylamino)phenyl]methyl ⁇ -1- piperazinecarboxylate (D3) (0.121 g, 0.38 mmol) in DCM (2.5 mL), followed by triethylamine (0.080 mL, 0.57 mmol). The reaction mixture was stirred for 3h under argon, then the solvent was removed by evaporation. The residue was partitioned between DCM (30 mL) and water (30 mL).
- Step 1 6-(3-Fluorophenyl)-3-pyridinecarboxylic acid (82 mg, 0.376 mmol) was stirred in dioxane (4 mL) and thionyl chloride (0.137 ml_, 1.88 mmol) added dropwise. The reaction mixture was heated at reflux for 40 min then concentrated in vacuo to give 6-(3-fluorophenyl)-3-pyridinecarbonyl chloride as a white solid (0.088 g) which was used directly in step 2.
- Step 2 The acid chloride from step 1 was taken up in DCM and added drop-wise to a mixture of 1 ,1 -dimethylethyl (2S)-2-methyl-4- ⁇ [4-(methylamino)phenyl]methyl ⁇ -1- piperazinecarboxylate (D3) (0.1 g, 0.313 mmol) and triethylamine (0.065 mL, 0.47 mmol) in DCM (5 mL). The reaction mixture was stirred at room temperature under argon for -15 h, then diluted with water and DCM. The organic layer was dried and concentrated to give the crude product, which was purified by column chromatography.
- D3 1 ,1 -dimethylethyl-2S)-2-methyl-4- ⁇ [4-(methylamino)phenyl]methyl ⁇ -1- piperazinecarboxylate
- D3 1 ,1 -dimethylethyl-2S)-2-methyl-4- ⁇ [4-(methylamino)phenyl]
- H-CubeTM continuous flow hydrogenator and triethylamine was used in place of solid KOH.
- ⁇ H (CDCI 3 , 400MHz) 6.99 (1H, dd), 6.86 (1H, dd), 6.71 (1 H, dd), 4.17 (1 H, br.s), 3.79 (1 H, d), 3.67 (2H, br.s), 3.40 (1 H, d), 3.27 (1 H, d), 3.09 (1 H, td), 2.73 (1H, m), 2.56 (1 H, m), 2.08 (1H, dd), 1.96 (1 H, m), 1.45 (9H, S) 1 1.22 (3H, d).
- the title compound was prepared from 1 ,1 -dimethylethyl (2S)-4-[(4-amino-3- fluorophenyl)methyl]-2-methyl-1 -piperazinecarboxylate (D63) using a method similar to that described for D3 in Description 3A although the reaction was heated at 5O 0 C overnight prior to and after addition of sodium borohydride.
- Step 1 A mixture of 2-fluoro-4-nitrotoluene (1.55 g, 10 mmol), ⁇ /-bromosuccinimide (1.96 g, 11 mmol) and benzoyl peroxide (0.121 g, 0.5 mmol) in CCI 4 (60 ml.) was irradiated with a 500W lamp overnight. The reaction mixture was filtered, concentrated and eluted through a silica column with EtOAc/pentane to give a crude product mixture (2.34g) which was used in step 2.
- Step 2 A mixture of crude 4-(bromomethyl)-3-fluoro-1-nitrobenzene (2.11 g) from step 1 , Hunig's base (1.9 ml_, 10.913 mmol) and 1 ,1-dimethylethyl (2S)-2-methyl-1- piperazinecarboxylate (2g, 10 mmol) in DMF (15 mL) was stirred at room temperature for 40 minutes. The reaction mixture was concentrated, re-dissolved in DCM and washed with water (x2) and brine, then dried and concentrated. The crude product was purified by column chromatography. Elution with EtOAc/pentane yielded the title compound as a yellow gum which crystallised on standing (2.11 g).
- the title compound was prepared from 1 ,1 -dimethylethyl (2S)-4-[(4-amino-2- fluorophenyl)methyl]-2-methyl-1 -piperazinecarboxylate (D66) using a method similar to that described for D3 in Description 3A although the reaction was heated at 5O 0 C overnight prior to addition of sodium borohydride and for 24h after addition.
- the title compound was prepared from 1 ,1 -dimethylethyl (2S)-4-( ⁇ 4-[[(6-chloro-3- pyridinyl)carbonyl](methyl)amino]-3-fluorophenyl ⁇ methyl)-2-methyl-1- piperazinecarboxylate (D68) and 4-fluorophenol using a method similar to that described for D55 in Description 55 although only 1eq of 4-fluorophenol was added initially, the reaction was heated at 13O 0 C over-weekend, further 4-fluorophenol (2eq) and potassium carbonate (4eq) were added and the reaction was heated at 13O 0 C overnight. The product was purified by column chromatography.
- the title compound was prepared from 1,1 -dimethylethyl (2S)-4-( ⁇ 4-[[(6-chloro-3- pyridinyl)carbonyl](methyl)amino]-2-fluorophenyl ⁇ methyl)-2-methyl-1 - piperazinecarboxylate (D70) and 4-fluorophenol using a method similar to that described for D55 in Description 55 although the reaction temp./time was 13O 0 C for 8h and purification was carried out by column chromatography.
- Step 1 5-Bromo-2-pyridinecarbonitrile (1 g, 5.464 mmol) was dissolved in EtOH (20 mL) and water (20 ml.) and treated with potassium hydroxide (1.53 g, 27.32 mmol). The reaction mixture was heated to 80 0 C for 24 h. The solvent was removed under vacuum and the residue was taken up in water and acidified to pH 4 with 2M HCI. The aqueous layer was extracted with ethyl acetate (x3) and the combined organic layers dried and concentrated to give 5-bromo-2-pyridinecarboxylic acid (0.704 g) as an orange solid which was used in step 2.
- Step 2 The acid from step 1 (0.702 g, 3.475 mmol) was suspended in DCM (40 mL) under argon, DMF (1 drop) was added and the mixture was cooled in an ice-bath. Oxalyl chloride was added portion-wise over 5 minutes and the mixture was then heated to 4O 0 C for 90 minutes. On cooling, the solvent was removed to produce 5- bromo-2-pyridinecarbonyl chloride (0.801 g) as a brown solid which was used in step 3.
- Step 3 The acid chloride from step 2 (0.801 g, 3.633 mmol) in DCM (10 mL) was added to a solution of 1,1-dimethylethyl (2S)-2-methyl-4- ⁇ [4- (methylamino)phenyl]methyl ⁇ -1-piperazinecarboxylate (D3) (0.928 g, 2.906 mmol) in DCM (20 mL). Triethylamine (1.009 mL, 7.266 mmol) was added and the mixture was stirred at room temperature overnight. The reaction mixture was diluted with DCM and washed with water.
- D3 1,1-dimethylethyl (2S)-2-methyl-4- ⁇ [4- (methylamino)phenyl]methyl ⁇ -1-piperazinecarboxylate
- the title compound was prepared from 1 , 1 -dimethylethyl (2S)-4-( ⁇ 4-[[(5-bromo-2- pyridinyl)carbonyl](methyl)amino]phenyl ⁇ methyl)-2-methyl-1-piperazinerarboxylate (D72) and 3-fluorophenol using a method similar to that described for D73 in Description 73 although 1eq. CuCI and 0.25eq. THMD were used. MS (ES): MH + 535.3.
- the title compound was prepared from 1 ,1-dimethylethyl (2S)-4-[(4-amino-2- methylphenyl)methyl]-2-methyl-1-piperazinecarboxylate (D80) using a method similar to that described for D3 in Description 3A although the reaction was heated at 5O 0 C for 16h prior to addition of sodium borohydride and 5.5h after addition.
- the title compound was prepared from 6-(methylamino)-3-pyridinecarbaldehyde (D95) and 1 ,1-dimethylethyl (2S)-2-methyl-1-piperazinecarboxylate using a method similar to that described for D81 in Description D81A.
- Step 1 ⁇ -bromo ⁇ -pyridinecarboxylic acid (0.348 g, 1.72 mmol) was dissolved in dry dioxane (15 mL) and thionyl chloride (0.570 mL, 7.84 mmol) was added. The reaction mixture was stirred at reflux for 3 h then concentrated in vacuo, re-dissolved in dioxane (15 mL) and re-concentrated to give 6-chloro-2-pyridinecarbonyl chloride as a pale yellow solid which was used directly in step 2.
- Step 2 The acid chloride from step 1 was dissolved in DCM (15 mL) and added to a mixture of 1 ,1-dimethylethyl (2S)-2-methyl-4- ⁇ [4-(methylamino)phenyl]methyl ⁇ -1- piperazinecarboxylate (D3) (0.500 g, 1.57 mmol) and triethylamine (0.284 mL, 2.04 mmol) in DCM (15 mL). The reaction mixture was stirred at room temperature overnight and then washed with water (x2) and brine. The organic layer was dried and concentrated to give the crude product, which was purified by column chromatography.
- D3 1 ,1-dimethylethyl-2S)-2-methyl-4- ⁇ [4-(methylamino)phenyl]methyl ⁇ -1- piperazinecarboxylate
- D3 1 ,1-dimethylethyl-2S)-2-methyl-4- ⁇ [4-(methylamino)phenyl]methyl ⁇ -1-
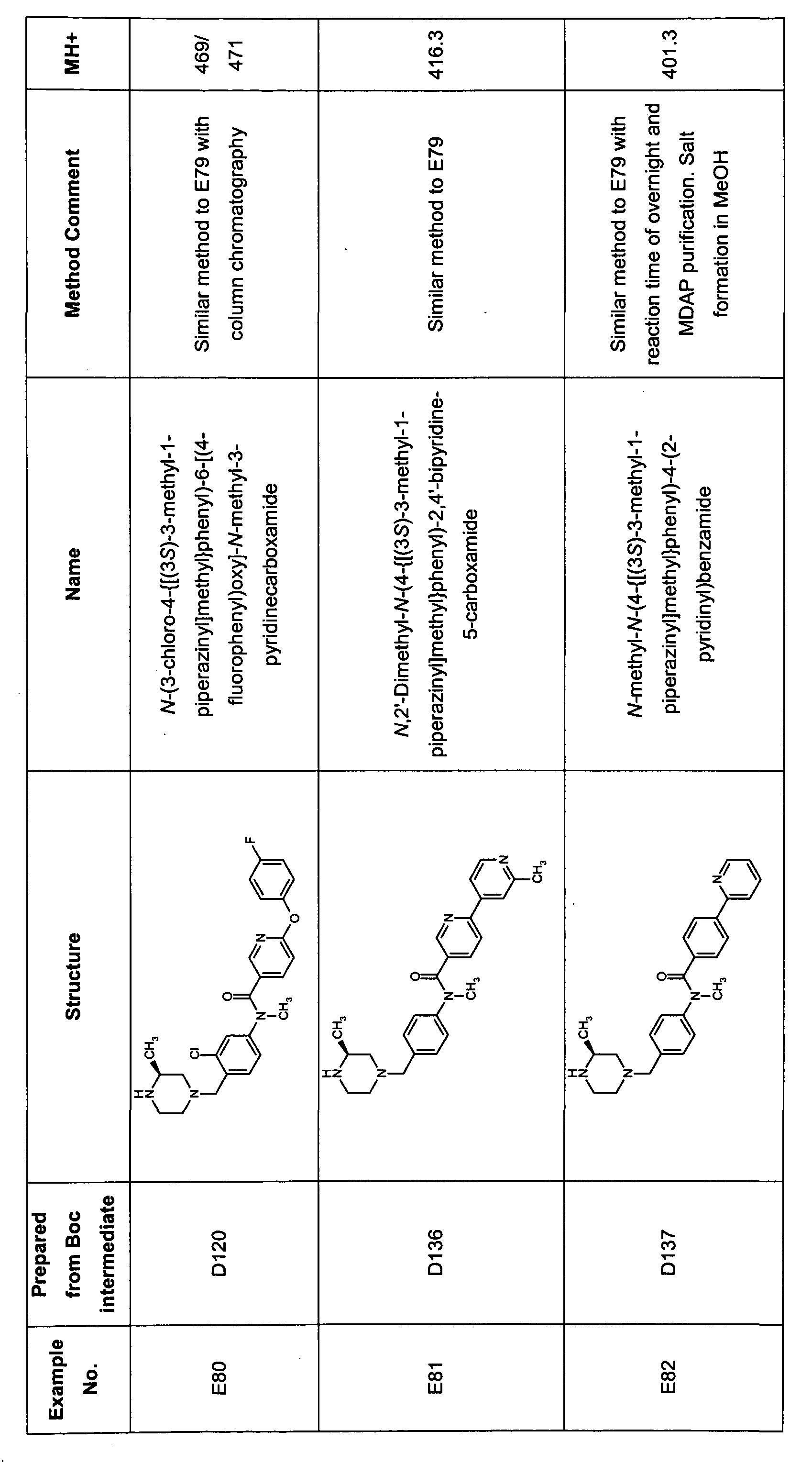
- Tabulated compounds D109 - D110 were prepared using methods similar to those described in Description 15 using the appropriate aniline precursor and appropriate carboxylic acid. PB62033
- Tabulated compounds D111 - D117 were prepared using methods similar to those described in Description 16 using the appropriate aniline precursor and appropriate carboxylic acid.
- Step 1 ⁇ - ⁇ -FluorophenyOoxyl-S-pyridinecarboxylic acid (D14) (0.032 g, 0.14 mmol) was solubilised in dry dioxane (2 ml_). Thionyl chloride (0.081 mg, 0.14 mmol) was added drop-wise and the reaction mixture heated at reflux for 1.25 h.. The reaction mixture was concentrated, and then re-concentrated from DCM to give 6-[(4- fluorophenyl)oxy]-3-pyridinecarbonyl chloride which was used directly in step 2
- Step 2 1 ,1 -Dimethylethyl (2S)-2-methyl-4- ⁇ [4-methyl-6-(methylamino)-3- pyridinyl]methyl ⁇ -1-piperazinecarboxylate (D101 ) (0.038 g, 0.11 mmol) was dissolved in dry DCM (2 ml_). Triethylamine (0.016 g, 0.16 mmol) was added dropwise and after 5 minutes a solution of the acid chloride from step 1 in dry DCM (2mL) was added dropwise. The reaction mixture was then left stirring under argon at room temperature overnight. Saturated aqueous NaHCO 3 (15ml_) was added and the aqueous layer extracted with DCM (3 x 5mL).
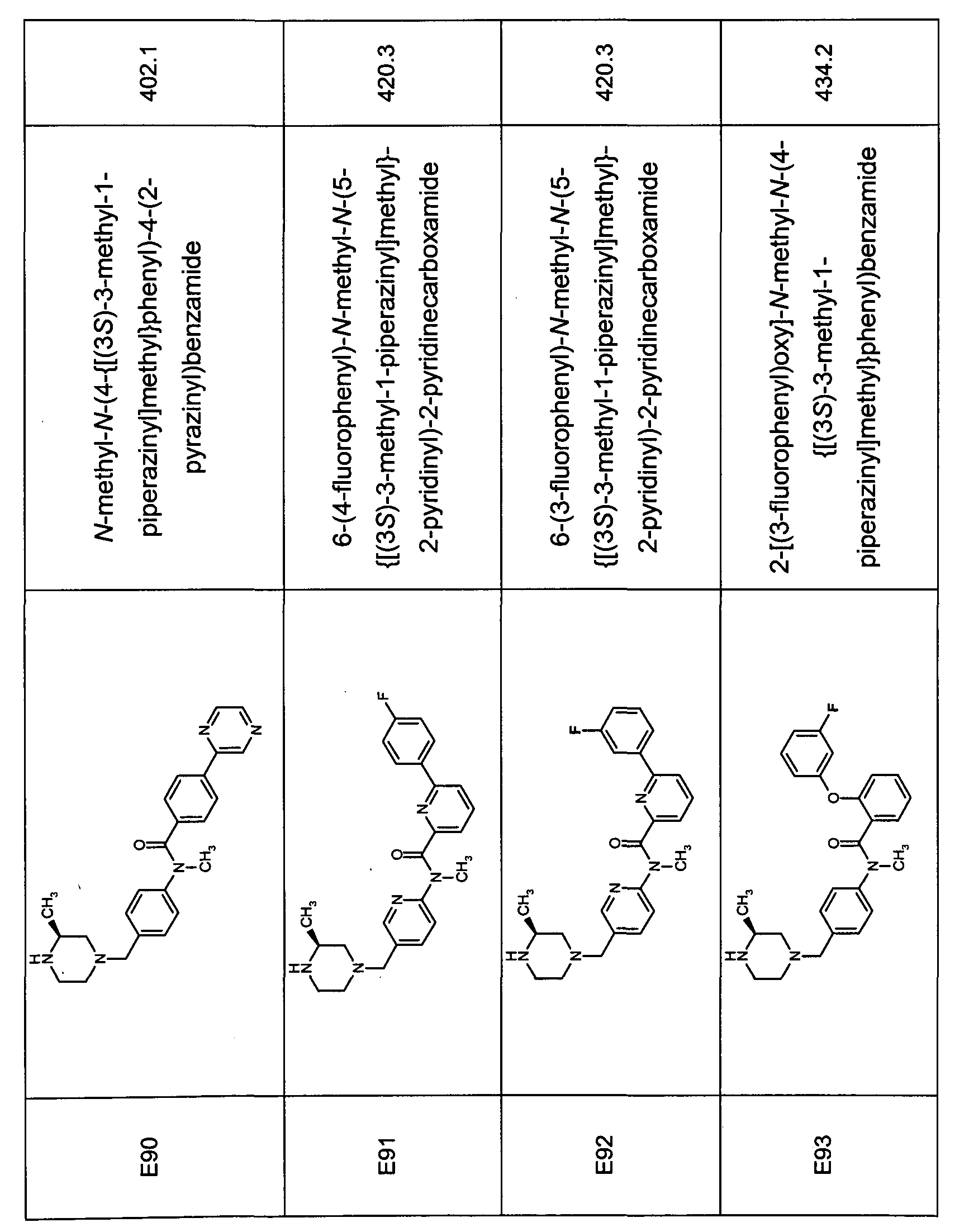
- Tabulated compounds D119 - D122 were prepared using methods similar to those described in Description 16 using the appropriate aniline precursor and appropriate carboxylic acid.
- Step 1 4- ⁇ [4-(Trifluoromethyl)phenyl]carbonyl ⁇ benzoic acid (200 mg, 0.68 mmol) was stirred in dioxane (5 mL) and thionyl chloride (0.496 ml_, 6.8 mmol) added. The reaction mixture was heated at reflux for 1h. Further thionyl chloride (0.496 mL, 6.8 mmol) was added and heating continued for 3h. On cooling, the reaction mixture was concentrated in vacuo to give 4- ⁇ [4-(trifluoromethyl)phenyl]carbonyl ⁇ benzoyl chloride as an oily white liquid (0.286 g) which was used directly in step 2.
- Step 2 The acid chloride from step 1 was taken up in DCM (2 mL) and added drop- wise to a mixture of 1 ,1 -dimethylethyl (2S)-2-methyl-4- ⁇ [4- (methylamino)phenyl]methyl ⁇ -1-piperazinecarboxylate (D3) (0.170 g, 0.53 mmol) and triethylamine (0.11 mL) in DCM (2 mL). The reaction mixture was stirred at room temperature under argon overnight then diluted with water (20 mL) and DCM (20 mL). The aqueous layer was extracted with DCM (20 mL) and the combined organic layers were dried, concentrated and purified by column chromatography. Elution with 0-30% EtOAc/pentane gave the title compound as a colourless oil (0.244 g). MS (ES): MH + 596.2.
- Tabulated compounds D130 - D136 were prepared from the appropriate bromo- pyridine precursor as indicated and the appropriate boronic acid using methods similar to that described for D 129 in Description 129.
- N- benzyl-/V-cyclohexylcarbodiimide resin 0.294 g, 1.6 mmol/g, 0.47 mmol
- 1- hydroxybenzotriazole 0.064 g, 0.47 mmol
- DCM DCM
- Tabulated compounds D138 - D140 were prepared from dimethylethyl (2S)-2- methyl-4- ⁇ [4-(methylamino)phenyl]methyl ⁇ -1-piperazinecarboxylate (D3) and the appropriate carboxylic acid using methods similar to that described in for D137 in Description 137.
- the XRPD data were acquired on a PANalytical X'Pert Pro powder diffractometer, model PW3040/60, serial number DY1850 using an XCelerator detector.
- the acquisition conditions were: radiation: Cu Ka, generator tension: 40 kV, generator current: 45 nriA, start angle: 2.0°2 ⁇ , end angle: 40.0°2 ⁇ , step size: 0.0167 °2 ⁇ , time per step: 31.75 seconds.
- the sample was prepared by mounting a few milligrams of sample on a Si wafer (zero background) plates, resulting in a thin layer of powder. Characteristic XRPD angles and d-spacings are recorded in the table below. Peak positions were measured using Highscore software.
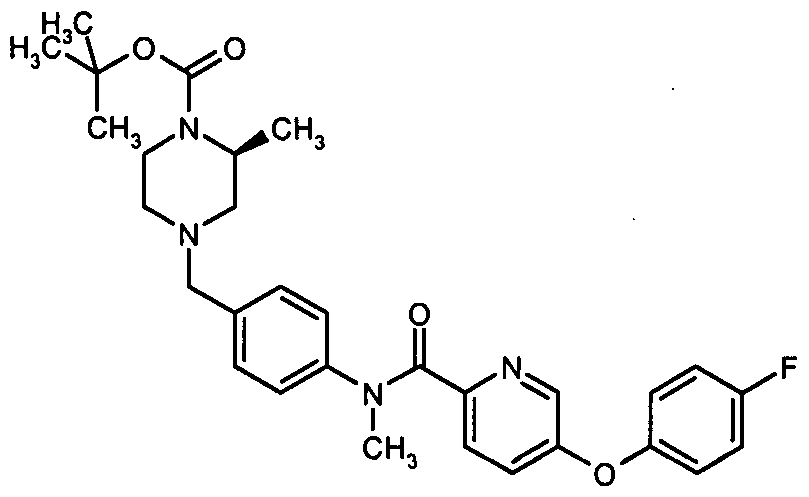
- Example 2B Method 4 6-[(4-Fluorophenyl)oxy]-W-methyl- ⁇ /-(4- ⁇ [(3S)-3-methyl-1 - piperazinyl]methyl ⁇ phenyl)-3-pyridinecarboxamide fumarate (E2B)
- the DSC thermogram of the product was obtained using a TA Q1000 calorimeter, serial number Q1000-0264.
- the sample was weighed into an aluminium pan, a pan lid placed on top and lightly crimped without sealing the pan.
- the experiment was conducted using a heating rate of 1O 0 C min '1 .
- XRPD X-Ray Powder Diffraction
- XRPD data for Example 2B Method 4 were acquired on a PANalytical X'Pert MPD powder diffractometer, model PW3040/60, serial number DY667 using an XCelerator detector.
- the acquisition conditions were: radiation: Cu Ka, generator tension: 40 kV, generator current: 45 mA, start angle: 2.0°2 ⁇ , end angle: 40.0°2 ⁇ , step size: 0.0167 °2 ⁇ , time per step: 48.26 seconds.
- the sample was prepared by mounting a few milligrams of sample on a Si wafer (zero background) plates, resulting in a thin layer of powder. Characteristic XRPD angles and d-spacings are recorded in the table below. Peak positions were measured using Highscore software.
- E39 formate salt was passed through an SCX cartridge eluting with methanol then 2M NH 3 in methanol yielding crude E39 free base (0.026 g) as a colourless oil which was further purified by column chromatography. Elution with 0-10% (2M NH 3 in methanol)/DCM yielded E39 free base as a colourless oil (0.024 g).
- CHO-K1 cells stably expressing the GPR38 receptor were seeded (10,000 cells/well) into poly-D-lysine coated 384-well black-wall, clear-bottom microtitre plates (Greiner). On the day of assay, media was aspirated from cell plates using a cell washer (leaving 10ul of media).
- Master compound plates were prepared in 100% DMSO. A top concentration of 3mM was used (giving 12 ⁇ M final concentration in assay) and this was serially diluted 1 in 4. 1 ul from the master plate was transferred to a daughter plate, to which 50 ⁇ l of compound dilution buffer (Tyrodes + 1mg/mL BSA + 1.5mM CaCI 2 ) was added. In the FLIPR 1 10ul of test compound was added to the cells and changes in fluorescence measured over a 1 minute timeframe. Maximum change in fluorescence over baseline was used to determine agonist response and concentration response curves were constructed, using a 4-parameter logistic equation.
- the loading buffer was HBSS ⁇ Elga water + 137mM NaCI + 5mM KCI + 0.41 mMa KH2PO4(anhyd) + 2OmM HEPES + 5mM glucose + 0.81 mM MgSO4(anhyd) + 1.3mM CaCI2 + 4.16mM NaHCO3 ⁇ + 0.25mM brilliant black + 2uM Fluo 4 dye and the CHO-K1 cells thawed from frozen aliquots and seeded 24 hours prior to the assay.
- Examples 1 to 86 of the invention have a pEC50 ⁇ 6.0 in one or more of the FLIPR assays described above.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Obesity (AREA)
- Hospice & Palliative Care (AREA)
- Otolaryngology (AREA)
- Nutrition Science (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description































































































































































Claims


Priority Applications (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE602007006019T DE602007006019D1 (en) | 2006-06-28 | 2007-06-26 | PIPERAZINE DERIVATIVES SUITABLE FOR THE TREATMENT OF DISEASES MEDIATED BY THE GPR38 RECEPTOR |
| NZ573450A NZ573450A (en) | 2006-06-28 | 2007-06-26 | Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases |
| JP2009517160A JP5028484B2 (en) | 2006-06-28 | 2007-06-26 | Piperazinyl derivatives useful for the treatment of GPR38 receptor mediated diseases |
| AT07765616T ATE465150T1 (en) | 2006-06-28 | 2007-06-26 | PIPERAZINYL DERIVATIVES SUITABLE FOR THE TREATMENT OF DISEASES MEDIATED BY THE GPR38 RECEPTOR |
| DK07765616.3T DK2041093T3 (en) | 2006-06-28 | 2007-06-26 | For the treatment of GPR38 receptor, diseases mediate useful piperazinyl derivatives |
| CA002655540A CA2655540A1 (en) | 2006-06-28 | 2007-06-26 | Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases |
| EA200970065A EA015820B1 (en) | 2006-06-28 | 2007-06-26 | Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases |
| EP07765616A EP2041093B1 (en) | 2006-06-28 | 2007-06-26 | Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases |
| BRPI0713443-6A BRPI0713443A2 (en) | 2006-06-28 | 2007-06-26 | PIPERAZINILY DERIVATIVES USEFUL IN THE TREATMENT OF MEDICAL DISEASES BY GPR38 RECEIVER, USE AND PHARMACEUTICAL COMPOSITION CONTAINING SUCH COMPOUNDS AND PREPARATION PROCESSES |
| CN2007800324070A CN101511791B (en) | 2006-06-28 | 2007-06-26 | Piperazinyl derivatives useful in the treatment of GPR38 receptor mediated diseases |
| AU2007263712A AU2007263712B2 (en) | 2006-06-28 | 2007-06-26 | Piperazinyl derivatives useful in the treatment of GPR38 receptor mediated diseases |
| MX2009000110A MX2009000110A (en) | 2006-06-28 | 2007-06-26 | Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases. |
| SI200730257T SI2041093T1 (en) | 2006-06-28 | 2007-06-26 | Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases |
| PL07765616T PL2041093T3 (en) | 2006-06-28 | 2007-06-26 | Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases |
| IL195900A IL195900A0 (en) | 2006-06-28 | 2008-12-11 | Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases |
| NO20090266A NO20090266L (en) | 2006-06-28 | 2009-01-16 | Piperazinyl derivatives useful in the treatment of GPR38 receptor-mediated diseases |
| HK09106727.8A HK1127608A1 (en) | 2006-06-28 | 2009-07-22 | Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases |
| HR20100284T HRP20100284T1 (en) | 2006-06-28 | 2010-05-21 | Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0612844.1 | 2006-06-28 | ||
| GB0612844A GB0612844D0 (en) | 2006-06-28 | 2006-06-28 | Compounds |
| GB0711525.6 | 2007-06-14 | ||
| GB0711525A GB0711525D0 (en) | 2007-06-14 | 2007-06-14 | Compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008000729A1 true WO2008000729A1 (en) | 2008-01-03 |
Family
ID=38476358
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/056342 WO2008000729A1 (en) | 2006-06-28 | 2007-06-26 | Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases |
Country Status (30)
| Country | Link |
|---|---|
| US (2) | US7700599B2 (en) |
| EP (1) | EP2041093B1 (en) |
| JP (1) | JP5028484B2 (en) |
| KR (1) | KR20090031688A (en) |
| AR (1) | AR061656A1 (en) |
| AT (1) | ATE465150T1 (en) |
| AU (1) | AU2007263712B2 (en) |
| BR (1) | BRPI0713443A2 (en) |
| CA (1) | CA2655540A1 (en) |
| CR (1) | CR10497A (en) |
| CY (1) | CY1110190T1 (en) |
| DE (1) | DE602007006019D1 (en) |
| DK (1) | DK2041093T3 (en) |
| EA (1) | EA015820B1 (en) |
| ES (1) | ES2344484T3 (en) |
| HK (1) | HK1127608A1 (en) |
| HR (1) | HRP20100284T1 (en) |
| IL (1) | IL195900A0 (en) |
| JO (1) | JO2645B1 (en) |
| MA (1) | MA30539B1 (en) |
| MX (1) | MX2009000110A (en) |
| MY (1) | MY147677A (en) |
| NO (1) | NO20090266L (en) |
| NZ (1) | NZ573450A (en) |
| PE (1) | PE20080345A1 (en) |
| PL (1) | PL2041093T3 (en) |
| PT (1) | PT2041093E (en) |
| SI (1) | SI2041093T1 (en) |
| TW (1) | TWI391386B (en) |
| WO (1) | WO2008000729A1 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009068552A1 (en) * | 2007-11-28 | 2009-06-04 | Glaxo Group Limited | Piperazinyl-sulfonamide derivatives useful in the treatment of gpr38 receptor mediated diseases |
| WO2010098145A1 (en) | 2009-02-27 | 2010-09-02 | Raqualia Pharma Inc. | Oxyindole derivatives with motilin receptor agonistic activity |
| US7964602B2 (en) | 2005-12-05 | 2011-06-21 | Glaxo Group Limited | Biaryl compounds useful as agonists of the Gpr38 receptor |
| WO2011092293A2 (en) | 2010-02-01 | 2011-08-04 | Novartis Ag | Cyclohexyl amide derivatives as crf receptor antagonists |
| WO2011092290A1 (en) | 2010-02-01 | 2011-08-04 | Novartis Ag | Pyrazolo[5,1b]oxazole derivatives as crf-1 receptor antagonists |
| WO2011095450A1 (en) | 2010-02-02 | 2011-08-11 | Novartis Ag | Cyclohexyl amide derivatives as crf receptor antagonists |
| US8012981B2 (en) | 2006-06-15 | 2011-09-06 | Glaxo Group Limited | Benzylpiperazine derivatives as motilin receptor agonists |
| WO2012043445A1 (en) | 2010-09-27 | 2012-04-05 | 第一三共株式会社 | Cyclohexane derivative compound |
| US8536182B2 (en) | 2005-07-26 | 2013-09-17 | Glaxo Group Limited | Benzylpiperazine derivatives and their medical use |
| US9321727B2 (en) | 2011-06-10 | 2016-04-26 | Hoffmann-La Roche Inc. | Pyridine derivatives as agonists of the CB2 receptor |
| US9452980B2 (en) | 2009-12-22 | 2016-09-27 | Hoffmann-La Roche Inc. | Substituted benzamides |
| US10508107B2 (en) | 2016-03-17 | 2019-12-17 | Hoffmann-La Roche Inc. | Morpholine derivative |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PE20080345A1 (en) * | 2006-06-28 | 2008-05-29 | Glaxo Group Ltd | PIPERAZINE DERIVATIVES AS AGONISTS OF THE GPR38 RECEPTOR |
| KR20230049673A (en) * | 2020-09-25 | 2023-04-13 | 상하이 메이유에 바이오테크 디벨롭먼트 컴퍼니 리미티드 | Pyrimidine carboxamide compounds and uses thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001085694A2 (en) * | 2000-05-05 | 2001-11-15 | Ortho-Mcneil Pharmaceutical, Inc. | Substituted diamide derivatives useful as motilin antagonists |
| US20030203922A1 (en) * | 2001-01-12 | 2003-10-30 | Amgen Inc. | Substituted amine derivatives and methods of use |
| WO2005063720A1 (en) * | 2003-12-25 | 2005-07-14 | Nippon Shinyaku Co., Ltd. | Amide derivative and medicine |
| WO2007012479A2 (en) * | 2005-07-26 | 2007-02-01 | Glaxo Group Limited | Benzylpiperazine derivates and their medical use |
Family Cites Families (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5385912A (en) | 1991-03-08 | 1995-01-31 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Multicyclic tertiary amine polyaromatic squalene synthase inhibitors |
| JPH06211886A (en) | 1992-11-04 | 1994-08-02 | Chugai Pharmaceut Co Ltd | Erythromycin derivative |
| TW355711B (en) | 1992-11-04 | 1999-04-11 | Chugai Pharmaceutical Co Ltd | Erythromycin derivatives |
| US5593994A (en) | 1994-09-29 | 1997-01-14 | The Dupont Merck Pharmaceutical Company | Prostaglandin synthase inhibitors |
| JP3901239B2 (en) | 1996-03-13 | 2007-04-04 | 大正製薬株式会社 | Arylalkane derivatives |
| US5965578A (en) | 1996-04-03 | 1999-10-12 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| DE19644195A1 (en) | 1996-10-24 | 1998-04-30 | Solvay Pharm Gmbh | 10,13,15-Trioxatricyclo [9.2.1.1. · 9 ·. · 6 ·] -pentadecanone derivatives, processes for their preparation and pharmaceuticals containing these compounds |
| US6100239A (en) | 1996-11-26 | 2000-08-08 | Chugai Seiyaku Kabushiki Kaisha | 13-membered ring macrolide compound, medicine containing the same, and process for producing the same |
| US6060491A (en) | 1997-06-19 | 2000-05-09 | Dupont Pharmaceuticals | 6-membered aromatics as factor Xa inhibitors |
| US5972939A (en) | 1997-10-28 | 1999-10-26 | Ortho-Mcneil Pharmaceutical, Inc. | Cyclopentene derivatives useful as antagonists of the motilin receptor |
| DE19805822B4 (en) | 1998-02-13 | 2009-02-05 | Solvay Pharmaceuticals Gmbh | 11-Acetyl-12,13-dioxabicyclo [8.2.1] tridecenone derivatives, process for their preparation and medicaments containing these compounds |
| US6165985A (en) | 1998-02-13 | 2000-12-26 | Solvay Pharmaceuticals Gmbh | 11-acetyl-12,13-dioxabicyclo[8.2.1]-tridecenone derivatives, processes for their preparation and pharmaceutical compositions comprising them |
| GB9804734D0 (en) | 1998-03-05 | 1998-04-29 | Pfizer Ltd | Compounds |
| RU2260592C9 (en) * | 1999-04-15 | 2017-04-07 | Бристол-Маерс Сквибб Ко. | Cyclic inhibitors of protein-tyrosine kinases |
| US20020010184A1 (en) | 2000-02-18 | 2002-01-24 | Dinsmore Christopher J. | Inhibitors of prenyl-protein transferase |
| US20020052380A1 (en) | 2000-02-18 | 2002-05-02 | Dinsmore Christopher J. | Inhibitors of prenyl-protein transferase |
| WO2001068620A1 (en) | 2000-03-13 | 2001-09-20 | Ortho-Mcneil Pharmaceutical, Inc. | Novel cyclopentene derivatives useful as antagonists of the motilin receptor |
| US6423714B2 (en) | 2000-03-13 | 2002-07-23 | Ortho Mcneil-Pharmaceutical, Inc.. | Cyclohexene derivatives useful as antagonists of the motilin receptor |
| WO2001068622A1 (en) | 2000-03-13 | 2001-09-20 | Ortho-Mcneil Pharmaceutical, Inc. | Novel cyclobutene derivatives useful as antagonists of the motilin receptor |
| GB0013378D0 (en) * | 2000-06-01 | 2000-07-26 | Glaxo Group Ltd | Use of therapeutic benzamide derivatives |
| US7105682B2 (en) * | 2001-01-12 | 2006-09-12 | Amgen Inc. | Substituted amine derivatives and methods of use |
| US6878714B2 (en) * | 2001-01-12 | 2005-04-12 | Amgen Inc. | Substituted alkylamine derivatives and methods of use |
| ATE308537T1 (en) * | 2001-05-10 | 2005-11-15 | Solvay Pharm Gmbh | NEW 1-AMIDOMETHYLCARBONYL-PIPERIDEDINE DERIVATIVES, METHOD FOR THE PRODUCTION THEREOF AND MEDICINAL PRODUCTS CONTAINING THESE COMPOUNDS |
| US6977264B2 (en) | 2001-07-25 | 2005-12-20 | Amgen Inc. | Substituted piperidines and methods of use |
| GB0129015D0 (en) * | 2001-12-04 | 2002-01-23 | Glaxo Group Ltd | Compounds |
| GB0129013D0 (en) * | 2001-12-04 | 2002-01-23 | Glaxo Group Ltd | Compounds |
| SE0203302D0 (en) * | 2002-11-07 | 2002-11-07 | Astrazeneca Ab | Novel Compounds |
| US7223788B2 (en) * | 2003-02-14 | 2007-05-29 | Sanofi-Aventis Deutschland Gmbh | Substituted N-aryl heterocycles, process for their preparation and their use as medicaments |
| US7338954B2 (en) | 2003-09-17 | 2008-03-04 | Bristol-Myers Squibb Company | Compounds useful as motilin agonists and method |
| US7262195B2 (en) | 2003-09-17 | 2007-08-28 | Bristol-Myers Squibb Company | Compounds useful as motilin agonists and method |
| KR20060060047A (en) * | 2003-10-01 | 2006-06-02 | 더 프록터 앤드 갬블 캄파니 | Melanin concentrating hormone antagonists |
| WO2005077368A2 (en) | 2004-02-03 | 2005-08-25 | Astrazeneca Ab | Treatment of gastro-esophageal reflux disease (gerd) |
| WO2005077373A2 (en) | 2004-02-03 | 2005-08-25 | Astrazeneca Ab | Treatment of gastro-esophageal reflux disease (gerd) |
| WO2005077345A1 (en) | 2004-02-03 | 2005-08-25 | Astrazeneca Ab | Compounds for the treatment of gastro-esophageal reflux disease |
| WO2005089502A2 (en) | 2004-03-18 | 2005-09-29 | The Brigham And Women's Hospital, Inc. | Methods for the treatment of synucleinopathies |
| US20070293539A1 (en) | 2004-03-18 | 2007-12-20 | Lansbury Peter T | Methods for the treatment of synucleinopathies |
| SE0401345D0 (en) | 2004-05-25 | 2004-05-25 | Astrazeneca Ab | Therapeutic compounds: Pyridine as scaffold |
| GB0611907D0 (en) | 2006-06-15 | 2006-07-26 | Glaxo Group Ltd | Compounds |
| EP1902022A1 (en) | 2005-07-12 | 2008-03-26 | Glaxo Group Limited | Piperazine heteroaryl derivates as gpr38 agonists |
| PE20080345A1 (en) * | 2006-06-28 | 2008-05-29 | Glaxo Group Ltd | PIPERAZINE DERIVATIVES AS AGONISTS OF THE GPR38 RECEPTOR |
-
2007
- 2007-06-26 PE PE2007000819A patent/PE20080345A1/en not_active Application Discontinuation
- 2007-06-26 EP EP07765616A patent/EP2041093B1/en active Active
- 2007-06-26 CA CA002655540A patent/CA2655540A1/en not_active Abandoned
- 2007-06-26 US US11/768,339 patent/US7700599B2/en not_active Expired - Fee Related
- 2007-06-26 EA EA200970065A patent/EA015820B1/en not_active IP Right Cessation
- 2007-06-26 AT AT07765616T patent/ATE465150T1/en active
- 2007-06-26 WO PCT/EP2007/056342 patent/WO2008000729A1/en active Application Filing
- 2007-06-26 BR BRPI0713443-6A patent/BRPI0713443A2/en not_active IP Right Cessation
- 2007-06-26 MX MX2009000110A patent/MX2009000110A/en active IP Right Grant
- 2007-06-26 PT PT07765616T patent/PT2041093E/en unknown
- 2007-06-26 ES ES07765616T patent/ES2344484T3/en active Active
- 2007-06-26 JP JP2009517160A patent/JP5028484B2/en not_active Expired - Fee Related
- 2007-06-26 AR ARP070102835A patent/AR061656A1/en not_active Application Discontinuation
- 2007-06-26 NZ NZ573450A patent/NZ573450A/en not_active IP Right Cessation
- 2007-06-26 JO JO2007261A patent/JO2645B1/en active
- 2007-06-26 DK DK07765616.3T patent/DK2041093T3/en active
- 2007-06-26 KR KR1020087031612A patent/KR20090031688A/en not_active Application Discontinuation
- 2007-06-26 TW TW096122969A patent/TWI391386B/en not_active IP Right Cessation
- 2007-06-26 AU AU2007263712A patent/AU2007263712B2/en not_active Ceased
- 2007-06-26 DE DE602007006019T patent/DE602007006019D1/en active Active
- 2007-06-26 SI SI200730257T patent/SI2041093T1/en unknown
- 2007-06-26 PL PL07765616T patent/PL2041093T3/en unknown
-
2008
- 2008-12-11 CR CR10497A patent/CR10497A/en not_active Application Discontinuation
- 2008-12-11 IL IL195900A patent/IL195900A0/en unknown
- 2008-12-24 MY MYPI20085319A patent/MY147677A/en unknown
- 2008-12-30 MA MA31526A patent/MA30539B1/en unknown
-
2009
- 2009-01-16 NO NO20090266A patent/NO20090266L/en not_active Application Discontinuation
- 2009-04-02 US US12/417,176 patent/US8853218B2/en not_active Expired - Fee Related
- 2009-07-22 HK HK09106727.8A patent/HK1127608A1/en unknown
-
2010
- 2010-05-21 HR HR20100284T patent/HRP20100284T1/en unknown
- 2010-06-11 CY CY20101100521T patent/CY1110190T1/en unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001085694A2 (en) * | 2000-05-05 | 2001-11-15 | Ortho-Mcneil Pharmaceutical, Inc. | Substituted diamide derivatives useful as motilin antagonists |
| US20030203922A1 (en) * | 2001-01-12 | 2003-10-30 | Amgen Inc. | Substituted amine derivatives and methods of use |
| WO2005063720A1 (en) * | 2003-12-25 | 2005-07-14 | Nippon Shinyaku Co., Ltd. | Amide derivative and medicine |
| WO2007012479A2 (en) * | 2005-07-26 | 2007-02-01 | Glaxo Group Limited | Benzylpiperazine derivates and their medical use |
Non-Patent Citations (1)
| Title |
|---|
| DATABASE CAPLUS [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; XP002452868, retrieved from STN Database accession no. 2005:612264 * |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8236953B2 (en) | 2004-12-29 | 2012-08-07 | Glaxo Group Limited | Process for preparing piper azine derivatives |
| US8536182B2 (en) | 2005-07-26 | 2013-09-17 | Glaxo Group Limited | Benzylpiperazine derivatives and their medical use |
| US7964602B2 (en) | 2005-12-05 | 2011-06-21 | Glaxo Group Limited | Biaryl compounds useful as agonists of the Gpr38 receptor |
| US8012981B2 (en) | 2006-06-15 | 2011-09-06 | Glaxo Group Limited | Benzylpiperazine derivatives as motilin receptor agonists |
| US7964597B2 (en) | 2007-11-28 | 2011-06-21 | Glaxo Group Limited | Piperazinyl-sulfonamide derivatives useful in the treatment of GPR38 receptor mediated diseases |
| WO2009068552A1 (en) * | 2007-11-28 | 2009-06-04 | Glaxo Group Limited | Piperazinyl-sulfonamide derivatives useful in the treatment of gpr38 receptor mediated diseases |
| WO2010098145A1 (en) | 2009-02-27 | 2010-09-02 | Raqualia Pharma Inc. | Oxyindole derivatives with motilin receptor agonistic activity |
| RU2533116C2 (en) * | 2009-02-27 | 2014-11-20 | Раквалиа Фарма Инк. | Oxyindole derivatives possessing agonist activity on motilin receptor |
| CN102317280A (en) * | 2009-02-27 | 2012-01-11 | 拉夸里亚创药株式会社 | Oxyindole derivatives with motilin receptor agonistic activity |
| US8697877B2 (en) | 2009-02-27 | 2014-04-15 | Raqualia Pharma Inc. | Oxyindole derivatives with motilin receptor agonistic activity |
| US9452980B2 (en) | 2009-12-22 | 2016-09-27 | Hoffmann-La Roche Inc. | Substituted benzamides |
| WO2011092290A1 (en) | 2010-02-01 | 2011-08-04 | Novartis Ag | Pyrazolo[5,1b]oxazole derivatives as crf-1 receptor antagonists |
| WO2011092293A2 (en) | 2010-02-01 | 2011-08-04 | Novartis Ag | Cyclohexyl amide derivatives as crf receptor antagonists |
| WO2011095450A1 (en) | 2010-02-02 | 2011-08-11 | Novartis Ag | Cyclohexyl amide derivatives as crf receptor antagonists |
| WO2012043445A1 (en) | 2010-09-27 | 2012-04-05 | 第一三共株式会社 | Cyclohexane derivative compound |
| AU2011309212B2 (en) * | 2010-09-27 | 2014-06-19 | Daiichi Sankyo Company, Limited | Cyclohexane derivative compound |
| JP5753179B2 (en) * | 2010-09-27 | 2015-07-22 | 第一三共株式会社 | Cyclohexane derivative compound |
| TWI501953B (en) * | 2010-09-27 | 2015-10-01 | Daiichi Sankyo Co Ltd | Cyclohexane derivatives |
| US9321727B2 (en) | 2011-06-10 | 2016-04-26 | Hoffmann-La Roche Inc. | Pyridine derivatives as agonists of the CB2 receptor |
| EA027247B1 (en) * | 2011-06-10 | 2017-07-31 | Ф.Хоффманн-Ля Рош Аг | Novel pyridin derivatives |
| US10508107B2 (en) | 2016-03-17 | 2019-12-17 | Hoffmann-La Roche Inc. | Morpholine derivative |
| US11312711B2 (en) | 2016-03-17 | 2022-04-26 | Hoffmann-La Roche Inc. | Morpholine derivative |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2041093B1 (en) | Piperazinyl derivatives useful in the treatment of gpr38 receptor mediated diseases | |
| AU2016202010B2 (en) | Guanidine compound | |
| JP5232143B2 (en) | Benzylpiperazine derivatives as motilin receptor antagonists | |
| US7964602B2 (en) | Biaryl compounds useful as agonists of the Gpr38 receptor | |
| WO2007095024A1 (en) | Metabotropic glutamate receptor-potentiating γsoindolones | |
| US20090054456A1 (en) | Benzylpiperazine derivatives and their medical use | |
| JPH08506354A (en) | Process for producing 5- [2- (4- (benzisothiazol-3-yl) -piperazin-1-yl) ethyl-6-chloro-1,3-dihydro-indol-2-one and intermediate for the production | |
| US7964597B2 (en) | Piperazinyl-sulfonamide derivatives useful in the treatment of GPR38 receptor mediated diseases | |
| CN101511791B (en) | Piperazinyl derivatives useful in the treatment of GPR38 receptor mediated diseases | |
| JPS6153268A (en) | Anthranilamide derivative | |
| JPH0193568A (en) | Amide compound and enterokinesis-activation agent containing said compound as active component | |
| AU2012251943A1 (en) | Isoxazole derivatives and their use as metabotropic glutamate receptor potentiators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780032407.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07765616 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2007263712 Country of ref document: AU Ref document number: 573450 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10202/DELNP/2008 Country of ref document: IN Ref document number: 2007765616 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 195900 Country of ref document: IL Ref document number: CR2008-010497 Country of ref document: CR |
|
| ENP | Entry into the national phase |
Ref document number: 2655540 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12008502819 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008122076 Country of ref document: EG Ref document number: 08136724 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009517160 Country of ref document: JP Ref document number: 1020087031612 Country of ref document: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2009/000110 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2007263712 Country of ref document: AU Date of ref document: 20070626 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2009000051 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200970065 Country of ref document: EA |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0713443 Country of ref document: BR Kind code of ref document: A2 Effective date: 20081223 |