WO2007127691A2 - Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation - Google Patents
Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation Download PDFInfo
- Publication number
- WO2007127691A2 WO2007127691A2 PCT/US2007/067188 US2007067188W WO2007127691A2 WO 2007127691 A2 WO2007127691 A2 WO 2007127691A2 US 2007067188 W US2007067188 W US 2007067188W WO 2007127691 A2 WO2007127691 A2 WO 2007127691A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkylated
- phosphate ester
- triaryl phosphate
- alkylated triaryl
- phenol
- Prior art date
Links
- XZZNDPSIHUTMOC-UHFFFAOYSA-N triphenyl phosphate Chemical compound C=1C=CC=CC=1OP(OC=1C=CC=CC=1)(=O)OC1=CC=CC=C1 XZZNDPSIHUTMOC-UHFFFAOYSA-N 0.000 title claims abstract description 20
- 230000029936 alkylation Effects 0.000 title abstract description 9
- 238000005804 alkylation reaction Methods 0.000 title abstract description 9
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 title abstract description 6
- WRXFONORSZHETC-UHFFFAOYSA-N phenyl propan-2-yl hydrogen phosphate Chemical class CC(C)OP(O)(=O)OC1=CC=CC=C1 WRXFONORSZHETC-UHFFFAOYSA-N 0.000 title 1
- -1 aryl phosphates Chemical class 0.000 claims abstract description 216
- 229910019142 PO4 Inorganic materials 0.000 claims abstract description 200
- 235000021317 phosphate Nutrition 0.000 claims abstract description 196
- 239000000203 mixture Substances 0.000 claims abstract description 134
- 238000000034 method Methods 0.000 claims abstract description 72
- 239000010452 phosphate Substances 0.000 claims description 171
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 claims description 134
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 claims description 97
- 238000006243 chemical reaction Methods 0.000 claims description 90
- 150000002989 phenols Chemical class 0.000 claims description 78
- 239000007795 chemical reaction product Substances 0.000 claims description 76
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 71
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 65
- 239000003054 catalyst Substances 0.000 claims description 60
- CMPQUABWPXYYSH-UHFFFAOYSA-N phenyl phosphate Chemical class OP(O)(=O)OC1=CC=CC=C1 CMPQUABWPXYYSH-UHFFFAOYSA-N 0.000 claims description 43
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical group [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 claims description 36
- 150000003014 phosphoric acid esters Chemical class 0.000 claims description 35
- 239000000047 product Substances 0.000 claims description 33
- AYEKOFBPNLCAJY-UHFFFAOYSA-O thiamine pyrophosphate Chemical compound CC1=C(CCOP(O)(=O)OP(O)(O)=O)SC=[N+]1CC1=CN=C(C)N=C1N AYEKOFBPNLCAJY-UHFFFAOYSA-O 0.000 claims description 31
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 28
- ASMQGLCHMVWBQR-UHFFFAOYSA-N Diphenyl phosphate Chemical class C=1C=CC=CC=1OP(=O)(O)OC1=CC=CC=C1 ASMQGLCHMVWBQR-UHFFFAOYSA-N 0.000 claims description 24
- 238000004821 distillation Methods 0.000 claims description 24
- 239000003085 diluting agent Substances 0.000 claims description 19
- 150000003013 phosphoric acid derivatives Chemical class 0.000 claims description 18
- 229910001629 magnesium chloride Inorganic materials 0.000 claims description 17
- 229920000642 polymer Polymers 0.000 claims description 17
- 125000000217 alkyl group Chemical group 0.000 claims description 14
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 14
- 229920005989 resin Polymers 0.000 claims description 14
- 239000011347 resin Substances 0.000 claims description 14
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 13
- 125000005037 alkyl phenyl group Chemical group 0.000 claims description 12
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical group Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 claims description 12
- 239000000178 monomer Substances 0.000 claims description 12
- 239000004800 polyvinyl chloride Substances 0.000 claims description 11
- 229920000915 polyvinyl chloride Polymers 0.000 claims description 11
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical group [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 claims description 10
- 125000005233 alkylalcohol group Chemical group 0.000 claims description 10
- 229920001577 copolymer Polymers 0.000 claims description 10
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical group [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 claims description 10
- 125000005461 organic phosphorous group Chemical group 0.000 claims description 10
- 230000000979 retarding effect Effects 0.000 claims description 10
- HPEUEJRPDGMIMY-IFQPEPLCSA-N molybdopterin Chemical compound O([C@H]1N2)[C@H](COP(O)(O)=O)C(S)=C(S)[C@@H]1NC1=C2N=C(N)NC1=O HPEUEJRPDGMIMY-IFQPEPLCSA-N 0.000 claims description 9
- 239000002585 base Substances 0.000 claims description 8
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 8
- 150000004714 phosphonium salts Chemical group 0.000 claims description 8
- 229920005830 Polyurethane Foam Polymers 0.000 claims description 7
- 239000002253 acid Substances 0.000 claims description 7
- 229920001519 homopolymer Polymers 0.000 claims description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 7
- 239000011496 polyurethane foam Substances 0.000 claims description 7
- 150000003242 quaternary ammonium salts Chemical class 0.000 claims description 7
- 239000004604 Blowing Agent Substances 0.000 claims description 6
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 6
- MWKFXSUHUHTGQN-UHFFFAOYSA-N decan-1-ol Chemical compound CCCCCCCCCCO MWKFXSUHUHTGQN-UHFFFAOYSA-N 0.000 claims description 6
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 6
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 6
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 claims description 6
- 235000013824 polyphenols Nutrition 0.000 claims description 6
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical group [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 claims description 5
- 239000004215 Carbon black (E152) Substances 0.000 claims description 5
- 229910021578 Iron(III) chloride Inorganic materials 0.000 claims description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 5
- WDIHJSXYQDMJHN-UHFFFAOYSA-L barium chloride Chemical group [Cl-].[Cl-].[Ba+2] WDIHJSXYQDMJHN-UHFFFAOYSA-L 0.000 claims description 5
- 229910001626 barium chloride Chemical group 0.000 claims description 5
- 239000001110 calcium chloride Chemical group 0.000 claims description 5
- 229910001628 calcium chloride Inorganic materials 0.000 claims description 5
- 229930195733 hydrocarbon Natural products 0.000 claims description 5
- 150000002430 hydrocarbons Chemical class 0.000 claims description 5
- RBTARNINKXHZNM-UHFFFAOYSA-K iron trichloride Chemical group Cl[Fe](Cl)Cl RBTARNINKXHZNM-UHFFFAOYSA-K 0.000 claims description 5
- 239000001301 oxygen Substances 0.000 claims description 5
- 229910052760 oxygen Inorganic materials 0.000 claims description 5
- 229920005862 polyol Polymers 0.000 claims description 5
- 150000003077 polyols Chemical class 0.000 claims description 5
- 239000001103 potassium chloride Chemical group 0.000 claims description 5
- 238000013022 venting Methods 0.000 claims description 5
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 claims description 4
- 239000000654 additive Substances 0.000 claims description 4
- 239000003963 antioxidant agent Substances 0.000 claims description 4
- LQZZUXJYWNFBMV-UHFFFAOYSA-N dodecan-1-ol Chemical compound CCCCCCCCCCCCO LQZZUXJYWNFBMV-UHFFFAOYSA-N 0.000 claims description 4
- 239000007789 gas Substances 0.000 claims description 4
- 239000012948 isocyanate Substances 0.000 claims description 4
- 150000002513 isocyanates Chemical class 0.000 claims description 4
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 4
- 238000000926 separation method Methods 0.000 claims description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical group C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 3
- 229910001508 alkali metal halide Inorganic materials 0.000 claims description 3
- 150000008045 alkali metal halides Chemical class 0.000 claims description 3
- 229910052784 alkaline earth metal Inorganic materials 0.000 claims description 3
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 3
- XPVBPNXEBOVGCP-UHFFFAOYSA-N dichloro hydrogen phosphate Chemical class ClOP(=O)(O)OCl XPVBPNXEBOVGCP-UHFFFAOYSA-N 0.000 claims description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 3
- 239000004094 surface-active agent Substances 0.000 claims description 3
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 claims description 2
- 125000002015 acyclic group Chemical group 0.000 claims description 2
- 230000000996 additive effect Effects 0.000 claims description 2
- 239000000956 alloy Substances 0.000 claims description 2
- 229910045601 alloy Inorganic materials 0.000 claims description 2
- 239000002216 antistatic agent Substances 0.000 claims description 2
- 150000004982 aromatic amines Chemical class 0.000 claims description 2
- 239000000470 constituent Substances 0.000 claims description 2
- 239000000975 dye Substances 0.000 claims description 2
- 239000000945 filler Substances 0.000 claims description 2
- 125000000524 functional group Chemical group 0.000 claims description 2
- 150000004679 hydroxides Chemical class 0.000 claims description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 claims description 2
- 150000005309 metal halides Chemical class 0.000 claims description 2
- 239000011707 mineral Substances 0.000 claims description 2
- 150000007522 mineralic acids Chemical class 0.000 claims description 2
- 239000002667 nucleating agent Substances 0.000 claims description 2
- 238000005191 phase separation Methods 0.000 claims description 2
- 239000000049 pigment Substances 0.000 claims description 2
- 239000004014 plasticizer Substances 0.000 claims description 2
- 150000003230 pyrimidines Chemical class 0.000 claims description 2
- 239000012744 reinforcing agent Substances 0.000 claims description 2
- 239000003381 stabilizer Substances 0.000 claims description 2
- 150000003871 sulfonates Chemical class 0.000 claims description 2
- 150000003467 sulfuric acid derivatives Chemical class 0.000 claims description 2
- 150000003512 tertiary amines Chemical group 0.000 claims description 2
- 229920001169 thermoplastic Polymers 0.000 claims description 2
- 229910019213 POCl3 Inorganic materials 0.000 claims 6
- JREYOWJEWZVAOR-UHFFFAOYSA-N triazanium;[3-methylbut-3-enoxy(oxido)phosphoryl] phosphate Chemical compound [NH4+].[NH4+].[NH4+].CC(=C)CCOP([O-])(=O)OP([O-])([O-])=O JREYOWJEWZVAOR-UHFFFAOYSA-N 0.000 claims 6
- 239000011342 resin composition Substances 0.000 claims 2
- 239000006057 Non-nutritive feed additive Substances 0.000 claims 1
- 239000003063 flame retardant Substances 0.000 abstract description 18
- RNFJDJUURJAICM-UHFFFAOYSA-N 2,2,4,4,6,6-hexaphenoxy-1,3,5-triaza-2$l^{5},4$l^{5},6$l^{5}-triphosphacyclohexa-1,3,5-triene Chemical compound N=1P(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP=1(OC=1C=CC=CC=1)OC1=CC=CC=C1 RNFJDJUURJAICM-UHFFFAOYSA-N 0.000 abstract description 12
- 238000002360 preparation method Methods 0.000 abstract description 4
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 27
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 27
- CRBJBYGJVIBWIY-UHFFFAOYSA-N 2-isopropylphenol Chemical compound CC(C)C1=CC=CC=C1O CRBJBYGJVIBWIY-UHFFFAOYSA-N 0.000 description 26
- 238000013019 agitation Methods 0.000 description 24
- 238000009472 formulation Methods 0.000 description 22
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 18
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- 239000006260 foam Substances 0.000 description 17
- 239000000376 reactant Substances 0.000 description 17
- 238000004458 analytical method Methods 0.000 description 16
- 239000000463 material Substances 0.000 description 16
- 239000000543 intermediate Substances 0.000 description 15
- 229920002635 polyurethane Polymers 0.000 description 15
- 239000004814 polyurethane Substances 0.000 description 15
- 229920000582 polyisocyanurate Polymers 0.000 description 14
- 239000011495 polyisocyanurate Substances 0.000 description 13
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 12
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 12
- 229910052698 phosphorus Inorganic materials 0.000 description 12
- 239000011574 phosphorus Substances 0.000 description 12
- 238000004679 31P NMR spectroscopy Methods 0.000 description 11
- 238000005481 NMR spectroscopy Methods 0.000 description 11
- 239000010410 layer Substances 0.000 description 11
- 238000007792 addition Methods 0.000 description 10
- 239000012043 crude product Substances 0.000 description 10
- NUHSROFQTUXZQQ-UHFFFAOYSA-N isopentenyl diphosphate Chemical compound CC(=C)CCO[P@](O)(=O)OP(O)(O)=O NUHSROFQTUXZQQ-UHFFFAOYSA-N 0.000 description 10
- 238000011084 recovery Methods 0.000 description 10
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 9
- 239000013067 intermediate product Substances 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 8
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 8
- 239000007806 chemical reaction intermediate Substances 0.000 description 8
- WVPKAWVFTPWPDB-UHFFFAOYSA-M dichlorophosphinate Chemical compound [O-]P(Cl)(Cl)=O WVPKAWVFTPWPDB-UHFFFAOYSA-M 0.000 description 7
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 7
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 7
- 239000008399 tap water Substances 0.000 description 7
- 235000020679 tap water Nutrition 0.000 description 7
- 230000000052 comparative effect Effects 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 238000010438 heat treatment Methods 0.000 description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 6
- 239000011541 reaction mixture Substances 0.000 description 6
- 239000000523 sample Substances 0.000 description 6
- JJXNVYMIYBNZQX-UHFFFAOYSA-N diphenyl (2-propan-2-ylphenyl) phosphate Chemical compound CC(C)C1=CC=CC=C1OP(=O)(OC=1C=CC=CC=1)OC1=CC=CC=C1 JJXNVYMIYBNZQX-UHFFFAOYSA-N 0.000 description 5
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 239000003518 caustics Substances 0.000 description 4
- 230000003247 decreasing effect Effects 0.000 description 4
- 238000005755 formation reaction Methods 0.000 description 4
- 150000005826 halohydrocarbons Chemical class 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 238000006116 polymerization reaction Methods 0.000 description 4
- 230000035484 reaction time Effects 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 238000003860 storage Methods 0.000 description 4
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 4
- NHGXDBSUJJNIRV-UHFFFAOYSA-M tetrabutylammonium chloride Chemical group [Cl-].CCCC[N+](CCCC)(CCCC)CCCC NHGXDBSUJJNIRV-UHFFFAOYSA-M 0.000 description 4
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 3
- 239000003377 acid catalyst Substances 0.000 description 3
- 150000001336 alkenes Chemical class 0.000 description 3
- XYLMUPLGERFSHI-UHFFFAOYSA-N alpha-Methylstyrene Chemical compound CC(=C)C1=CC=CC=C1 XYLMUPLGERFSHI-UHFFFAOYSA-N 0.000 description 3
- 239000000538 analytical sample Substances 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 150000001875 compounds Chemical class 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 238000004508 fractional distillation Methods 0.000 description 3
- 150000008282 halocarbons Chemical class 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- UETPRJJVZPVPQI-UHFFFAOYSA-N (2-propan-2-ylphenyl) dihydrogen phosphate Chemical compound CC(C)C1=CC=CC=C1OP(O)(O)=O UETPRJJVZPVPQI-UHFFFAOYSA-N 0.000 description 2
- GJVWMPQFNARTPN-UHFFFAOYSA-N (3-propan-2-ylphenyl) dihydrogen phosphate Chemical compound CC(C)C1=CC=CC(OP(O)(O)=O)=C1 GJVWMPQFNARTPN-UHFFFAOYSA-N 0.000 description 2
- JMEROBIHUFJHHG-UHFFFAOYSA-N (4-propan-2-ylphenyl) dihydrogen phosphate Chemical compound CC(C)C1=CC=C(OP(O)(O)=O)C=C1 JMEROBIHUFJHHG-UHFFFAOYSA-N 0.000 description 2
- UWOKONWYYPRXCH-UHFFFAOYSA-N 1-[chloro-(2-propan-2-ylphenoxy)phosphoryl]oxy-2-propan-2-ylbenzene Chemical compound CC(C)C1=CC=CC=C1OP(Cl)(=O)OC1=CC=CC=C1C(C)C UWOKONWYYPRXCH-UHFFFAOYSA-N 0.000 description 2
- KEUMBYCOWGLRBQ-UHFFFAOYSA-N 2,4-di(propan-2-yl)phenol Chemical compound CC(C)C1=CC=C(O)C(C(C)C)=C1 KEUMBYCOWGLRBQ-UHFFFAOYSA-N 0.000 description 2
- YQUQWHNMBPIWGK-UHFFFAOYSA-N 4-isopropylphenol Chemical compound CC(C)C1=CC=C(O)C=C1 YQUQWHNMBPIWGK-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N Ethylbenzene Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- JPOXNPPZZKNXOV-UHFFFAOYSA-N bromochloromethane Chemical compound ClCBr JPOXNPPZZKNXOV-UHFFFAOYSA-N 0.000 description 2
- 229920001429 chelating resin Polymers 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 150000001993 dienes Chemical class 0.000 description 2
- JUHFQCKQQLMGAB-UHFFFAOYSA-N diphenyl (4-propan-2-ylphenyl) phosphate Chemical compound C1=CC(C(C)C)=CC=C1OP(=O)(OC=1C=CC=CC=1)OC1=CC=CC=C1 JUHFQCKQQLMGAB-UHFFFAOYSA-N 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 230000005484 gravity Effects 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 229920001580 isotactic polymer Polymers 0.000 description 2
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 2
- 239000000347 magnesium hydroxide Substances 0.000 description 2
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 2
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 2
- 229910052753 mercury Inorganic materials 0.000 description 2
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- LPLPPIFARDQDDL-UHFFFAOYSA-N phenyl (2-propan-2-ylphenyl) hydrogen phosphate Chemical compound CC(C)C1=CC=CC=C1OP(O)(=O)OC1=CC=CC=C1 LPLPPIFARDQDDL-UHFFFAOYSA-N 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 229920002959 polymer blend Polymers 0.000 description 2
- OLBCVFGFOZPWHH-UHFFFAOYSA-N propofol Chemical compound CC(C)C1=CC=CC(C(C)C)=C1O OLBCVFGFOZPWHH-UHFFFAOYSA-N 0.000 description 2
- 238000010926 purge Methods 0.000 description 2
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 229920001576 syndiotactic polymer Polymers 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 238000010555 transalkylation reaction Methods 0.000 description 2
- 238000005809 transesterification reaction Methods 0.000 description 2
- LIPMRGQQBZJCTM-UHFFFAOYSA-N tris(2-propan-2-ylphenyl) phosphate Chemical compound CC(C)C1=CC=CC=C1OP(=O)(OC=1C(=CC=CC=1)C(C)C)OC1=CC=CC=C1C(C)C LIPMRGQQBZJCTM-UHFFFAOYSA-N 0.000 description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- UOCLXMDMGBRAIB-UHFFFAOYSA-N 1,1,1-trichloroethane Chemical compound CC(Cl)(Cl)Cl UOCLXMDMGBRAIB-UHFFFAOYSA-N 0.000 description 1
- APQIUTYORBAGEZ-UHFFFAOYSA-N 1,1-dibromoethane Chemical compound CC(Br)Br APQIUTYORBAGEZ-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- QVCUKHQDEZNNOC-UHFFFAOYSA-N 1,2-diazabicyclo[2.2.2]octane Chemical compound C1CC2CCN1NC2 QVCUKHQDEZNNOC-UHFFFAOYSA-N 0.000 description 1
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 1
- MPPPKRYCTPRNTB-UHFFFAOYSA-N 1-bromobutane Chemical compound CCCCBr MPPPKRYCTPRNTB-UHFFFAOYSA-N 0.000 description 1
- KTZVZZJJVJQZHV-UHFFFAOYSA-N 1-chloro-4-ethenylbenzene Chemical compound ClC1=CC=C(C=C)C=C1 KTZVZZJJVJQZHV-UHFFFAOYSA-N 0.000 description 1
- VFWCMGCRMGJXDK-UHFFFAOYSA-N 1-chlorobutane Chemical compound CCCCCl VFWCMGCRMGJXDK-UHFFFAOYSA-N 0.000 description 1
- BKMVKEHNEIKUMZ-UHFFFAOYSA-N 1-dichlorophosphoryloxy-2-propan-2-ylbenzene Chemical compound CC(C)C1=CC=CC=C1OP(Cl)(Cl)=O BKMVKEHNEIKUMZ-UHFFFAOYSA-N 0.000 description 1
- OEVVKKAVYQFQNV-UHFFFAOYSA-N 1-ethenyl-2,4-dimethylbenzene Chemical compound CC1=CC=C(C=C)C(C)=C1 OEVVKKAVYQFQNV-UHFFFAOYSA-N 0.000 description 1
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 1
- IGGDKDTUCAWDAN-UHFFFAOYSA-N 1-vinylnaphthalene Chemical compound C1=CC=C2C(C=C)=CC=CC2=C1 IGGDKDTUCAWDAN-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- ZDXGQHXSMPGQRI-UHFFFAOYSA-N 2,6-ditert-butyl-3-[(2,4-ditert-butyl-3-hydroxyphenyl)methyl]phenol Chemical compound CC(C)(C)C1=C(O)C(C(C)(C)C)=CC=C1CC1=CC=C(C(C)(C)C)C(O)=C1C(C)(C)C ZDXGQHXSMPGQRI-UHFFFAOYSA-N 0.000 description 1
- UPSXAPQYNGXVBF-UHFFFAOYSA-N 2-bromobutane Chemical compound CCC(C)Br UPSXAPQYNGXVBF-UHFFFAOYSA-N 0.000 description 1
- BSPCSKHALVHRSR-UHFFFAOYSA-N 2-chlorobutane Chemical compound CCC(C)Cl BSPCSKHALVHRSR-UHFFFAOYSA-N 0.000 description 1
- JLBJTVDPSNHSKJ-UHFFFAOYSA-N 4-Methylstyrene Chemical compound CC1=CC=C(C=C)C=C1 JLBJTVDPSNHSKJ-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 239000007848 Bronsted acid Substances 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- UUGLJVMIFJNVFH-UHFFFAOYSA-N Hexyl benzoate Chemical compound CCCCCCOC(=O)C1=CC=CC=C1 UUGLJVMIFJNVFH-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- CYTYCFOTNPOANT-UHFFFAOYSA-N Perchloroethylene Chemical group ClC(Cl)=C(Cl)Cl CYTYCFOTNPOANT-UHFFFAOYSA-N 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 229920000265 Polyparaphenylene Polymers 0.000 description 1
- 239000004721 Polyphenylene oxide Substances 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 239000004614 Process Aid Substances 0.000 description 1
- 229910021607 Silver chloride Inorganic materials 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229910003074 TiCl4 Inorganic materials 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical class ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 1
- DZONBDFKZXLORJ-UHFFFAOYSA-N [2,4-di(propan-2-yl)phenyl] diphenyl phosphate Chemical compound CC(C)C1=CC(C(C)C)=CC=C1OP(=O)(OC=1C=CC=CC=1)OC1=CC=CC=C1 DZONBDFKZXLORJ-UHFFFAOYSA-N 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 125000005210 alkyl ammonium group Chemical group 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- 229910001778 ammonium mineral Inorganic materials 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 229910052797 bismuth Inorganic materials 0.000 description 1
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 150000003841 chloride salts Chemical class 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000010908 decantation Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- FJBFPHVGVWTDIP-UHFFFAOYSA-N dibromomethane Chemical compound BrCBr FJBFPHVGVWTDIP-UHFFFAOYSA-N 0.000 description 1
- PLBFTNYDDQQDME-UHFFFAOYSA-N diphenyl (3-propan-2-ylphenyl) phosphate Chemical compound CC(C)C1=CC=CC(OP(=O)(OC=2C=CC=CC=2)OC=2C=CC=CC=2)=C1 PLBFTNYDDQQDME-UHFFFAOYSA-N 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 239000003344 environmental pollutant Substances 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 239000004210 ether based solvent Substances 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 238000001125 extrusion Methods 0.000 description 1
- 238000005187 foaming Methods 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 239000012760 heat stabilizer Substances 0.000 description 1
- 229920005669 high impact polystyrene Polymers 0.000 description 1
- 239000004797 high-impact polystyrene Substances 0.000 description 1
- 210000004124 hock Anatomy 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 238000001746 injection moulding Methods 0.000 description 1
- ULYZAYCEDJDHCC-UHFFFAOYSA-N isopropyl chloride Chemical compound CC(C)Cl ULYZAYCEDJDHCC-UHFFFAOYSA-N 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- KRPXAHXWPZLBKL-UHFFFAOYSA-L magnesium;diphenoxide Chemical compound [Mg+2].[O-]C1=CC=CC=C1.[O-]C1=CC=CC=C1 KRPXAHXWPZLBKL-UHFFFAOYSA-L 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229910001510 metal chloride Inorganic materials 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- YCWSUKQGVSGXJO-NTUHNPAUSA-N nifuroxazide Chemical group C1=CC(O)=CC=C1C(=O)N\N=C\C1=CC=C([N+]([O-])=O)O1 YCWSUKQGVSGXJO-NTUHNPAUSA-N 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 238000010606 normalization Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 125000002524 organometallic group Chemical group 0.000 description 1
- 238000010525 oxidative degradation reaction Methods 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000002530 phenolic antioxidant Substances 0.000 description 1
- XYFCBTPGUUZFHI-UHFFFAOYSA-O phosphonium Chemical compound [PH4+] XYFCBTPGUUZFHI-UHFFFAOYSA-O 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000000865 phosphorylative effect Effects 0.000 description 1
- 231100000719 pollutant Toxicity 0.000 description 1
- 229920005906 polyester polyol Polymers 0.000 description 1
- 229920001225 polyester resin Polymers 0.000 description 1
- 239000004645 polyester resin Substances 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 239000002952 polymeric resin Substances 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 230000006207 propylation Effects 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 150000003222 pyridines Chemical class 0.000 description 1
- 238000010526 radical polymerization reaction Methods 0.000 description 1
- HKZLPVFGJNLROG-UHFFFAOYSA-M silver monochloride Chemical compound [Cl-].[Ag+] HKZLPVFGJNLROG-UHFFFAOYSA-M 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 150000003440 styrenes Chemical class 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- GQSNYNMMDQPIDR-UHFFFAOYSA-M tetrakis(diethylamino)phosphanium;bromide Chemical compound [Br-].CCN(CC)[P+](N(CC)CC)(N(CC)CC)N(CC)CC GQSNYNMMDQPIDR-UHFFFAOYSA-M 0.000 description 1
- 229920005992 thermoplastic resin Polymers 0.000 description 1
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 125000005270 trialkylamine group Chemical group 0.000 description 1
- UBOXGVDOUJQMTN-UHFFFAOYSA-N trichloroethylene Natural products ClCC(Cl)Cl UBOXGVDOUJQMTN-UHFFFAOYSA-N 0.000 description 1
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K21/00—Fireproofing materials
- C09K21/06—Organic materials
- C09K21/12—Organic materials containing phosphorus
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/09—Esters of phosphoric acids
- C07F9/12—Esters of phosphoric acids with hydroxyaryl compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/49—Phosphorus-containing compounds
- C08K5/51—Phosphorus bound to oxygen
- C08K5/52—Phosphorus bound to oxygen only
- C08K5/521—Esters of phosphoric acids, e.g. of H3PO4
- C08K5/523—Esters of phosphoric acids, e.g. of H3PO4 with hydroxyaryl compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L27/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a halogen; Compositions of derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L75/00—Compositions of polyureas or polyurethanes; Compositions of derivatives of such polymers
- C08L75/04—Polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2101/00—Manufacture of cellular products
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2115/00—Oligomerisation
- C08G2115/02—Oligomerisation to isocyanurate groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/014—Additives containing two or more different additives of the same subgroup in C08K
Definitions
- the present invention relates to flame retardant compositions and a process for their preparation. More particularly, the present invention relates to low triphenyl phosphate, high phosphorous content, aryl phosphates, with high ortho alkylation that are suitable for use as flame retardant compositions and a process for their preparation. BACKGROUND OF THE INVENTION
- Alkylated aryl phosphates are known in the art to be useful as flame-retardants. These compounds can be formed by a number of methods commonly used in the art. For example, it is known to prepare mixed synthetic triaryl phosphates by alkylating phenol with alkenes such as propylene or isobutylene to obtain a mixture of phenol and substituted phenols. According to U.S. Pat. No. 4,093,680 this alkylate mixture is then reacted with phosphorus oxychloride (POCl 3 ) to form a mixed triaryl phosphate ester.
- POCl 3 phosphorus oxychloride
- the product mix is a statistical mixture based on the composition of the starting alkylates and always includes some fraction of triphenyl phosphates ("TPP"), usually from 5 to 50 percent.
- TPP triphenyl phosphates
- the product's physical properties are determined by the degree of alkylation of the phenol. A highly alkylated phenol mixture will result in a more viscous phosphate ester product than one less substituted.
- Triphenyl phosphate is a by-product of the alkylated phenyl phosphate formation reaction and is unwanted in the final product because of environmental concerns.
- TPP has been classified as a marine pollutant in some jurisdictions.
- wipe film purification can be used to produce mixed alkylated triphenyl phosphates with TPP concentrations of less than 2 wt%.
- the '404 patent also discloses that an undesirable method of reducing the TPP concentration of alkylated phenyl phosphates is by fractional distillation.
- United States Patent Number 6,232,485 discloses a process for producing a liquid triaryl phosphate ester having low triphenyl phosphate concentrations and low viscosity comprising (a) an alkylation stage wherein a phenol is reacted with an olefin having 2 to 12 carbon atoms in the presence of a strong acid catalyst to give a reaction product comprising a mixture of meta and para alkylated phenols; (b) a transalkylation stage wherein the mixture of alkylated phenols from the alkylation stage is heated in the presence of a strong acid catalyst to increase the meta isomer content of the mixture to at least 20% whilst maintaining a phenol level below 22%; and (c) a phosphorylation stage.
- the mixture of alkylated phenols from the transalkylation stage is reacted with a phosphorylating agent, and the strong acid catalyst used in stages (a) and (b) is a Bronsted acid having an acid strength of less than zero.
- the present invention relates to an alkylated triaryl phosphate ester comprising less than about lwt% triphenyl phosphate, based on the total weight of the alkylated triaryl phosphate ester, and an organic phosphorous content in the range of from about 5 to about 10wt%, based on the total weight of the alkylated triaryl phosphate ester.
- the present invention relates to an alkylated triaryl phosphate ester comprising one or more of the following alkylated phenyl phosphates: a) monoalkylphenyl diphenyl phosphates; b) di-(alkylphenyl) phenyl phosphates; c) dialkylphenyl diphenyl phosphates,; d) trialkylphenyl phosphates; e) alkylphenyl dialkylphenyl phenyl phosphates; and f) less than about lwt.% triphenyl phosphate, based on the total weight of the alkylated triaryl phosphate ester, wherein the alkyl moieties of the alkylated phenyl phosphates are selected from methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tertiary-butyl, amy
- the present invention also relates to a process for making alkylated triaryl phosphate esters comprising: a) reacting an alkylated phenol comprising less than about lmole% phenol and up to about 75mole% dialkyl phenol, both based on the total moles of reactive alkylated phenolics in the alklyated phenol, with POCl 3 in the presence of a first catalyst under first reaction conditions including temperatures ranging from about 8O 0 C to about 21O 0 C thereby producing a first reaction product comprising greater than about 75 mole% monoalkylated phenyl-dichloro phosphates, based on the total moles of the first reaction product; and b) reacting the first reaction product with an alcohol selected from aryl alcohols, alkyl alcohols, alkylated aryl alcohols, and mixtures thereof in the presence of a second catalyst under second reaction conditions including temperatures ranging from about 9O 0 C to about 26O 0 C thereby producing an alcohol selected from
- the present invention also relates to a process for making alkylated triaryl phosphate esters comprising: a) reacting an alkylated phenol comprising less than about 1% phenol and up to about 75% dialkyl phenol, both based on the total weight of the alkylated phenol, with a molar excess Of POCl 3 in the presence of a first catalyst under first reaction conditions including temperatures ranging from about 8O 0 C to about 21O 0 C thereby producing a first reaction product comprising greater than about 50 mole%, based on the total moles of the first reaction product, monoalkylated phenyl-dichloro phosphates and excess POCl 3 ; b) removing at least a portion of the excess POCI 3 from the first reaction product to produce an intermediate product, wherein said intermediate reaction product contains less than 15mole% phosphorus, based on the total moles of the intermediate reaction product, in the form of POCl 3 remains; and c) reacting the first reaction product with an
- IP is meant to refer to isopropylatedphenols
- OIP is meant to refer to or ⁇ o-isopropylphenol
- MIP is meant to refer to met ⁇ -isopropylphenol
- PIP is meant to refer to /? ⁇ r ⁇ -isopropylphenol
- TPP is meant to refer to triphenyl phosphate
- 2,6- DIP is meant to refer to 2,6-diisopropylphenol
- 2,4-DIP is meant to refer to 2,4- diisopropylphenol
- 2,4,6-TIP is meant to refer to 2,4,6-triisoproplylphenol
- 2-IPP is meant to refer to 2-isopropylphenyl diphenyl phosphate
- ⁇ 3-ipp i s meant to refer to 3- isopropylphenyl diphenyl phosphate
- 4-IPP is meant to refer to 4-isopropylphen
- the present invention relates to alkylated triaryl phosphate esters.
- the alkylated triaryl phosphate esters of the present invention are characterized as containing less than about lwt% TPP, based on the total weight of the alkylated triaryl phosphate ester, in some embodiments less than about 0.75wt% TPP, on the same basis, and in other embodiments, less than about 0.5wt% TPP, on the same basis.
- the alkylated triaryl phosphate esters of the present invention still contain a high amount of phosphorus.
- the alkylated triaryl phosphate esters of the present invention contain from about 5 to about 10wt% organic phosphorous, based on the total weight of the alkylated triaryl phosphate ester.
- the organic phosphorus content ranges from about 7 to about 9wt%, on the same basis, and in more preferred embodiments the organic phosphorous content ranges from about 7.5 to about 8.5wt%, most preferably in the range of from about 8.0 to about 8.4%, on the same basis.
- the alkylated triaryl phosphate esters of the present invention are further characterized as containing greater than about 20wt% monalkylphenyl diphenyl phosphates, based on the total weight of the alkylated triaryl phosphate ester.
- the alkylated triaryl phosphate esters contain greater than about 75wt%, on the same basis, monalkylphenyl diphenyl phosphates, more preferably greater than about 90wt%, on the same basis.
- the alkylated triaryl phosphate esters of the present invention can further be characterized as containing less than about 80wt% di-(alkylphenyl) phenyl phosphates, based on the total weight of the alkylated triaryl phosphate ester.
- the alkylated triaryl phosphate esters of the present invention contain less than about 25wt%, more preferably less than about 10wt%, di-(alkylphenyl) phenyl phosphates, on the same basis.
- the alkylated triaryl phosphate esters of the present invention can also be further characterized as containing less than about 50wt%, based on the total weight of the alkylated triaryl phosphate ester, dialkylphenyl diphenyl phosphates.
- the alkylated triaryl phosphate esters of the present invention contain less than about 25wt%, more preferably less than about 10wt%, dialkylphenyl diphenyl phosphates, on the same basis.
- the alkylated triaryl phosphate esters of the present invention contain less than about lwt%, based on the total weight of the alkylated triaryl phosphate ester, dialkylphenyl diphenyl phosphates.
- the inventors hereof have unexpectedly discovered that, in some embodiments, the removal of unreacted alkylated phenols during the production of the alkylated triaryl phosphate esters of the present invention is more efficient for alkylated triaryl phosphate esters having these concentrations of dialkylphenyl diphenyl phosphates.
- the amount of trialkylphenyl phosphates present in the alkylated triaryl phosphate esters of the present invention is generally less than about 20wt%, based on the total weight of the alkylated triaryl phosphate ester.
- the alkylated phenyl phosphates of the present invention can contain less than about 2wt%, on the same basis, of trialkylphenyl phosphates.
- the level of trialkylphenyl phosphates is less than 0.5wt%, on the same basis.
- the alkylated phenyl phosphates according to the present invention also comprise less than about 20wt%, based on the total weight of the alkylated triaryl phosphate ester, alkylphenyl dialkylphenyl phenyl phosphates.
- the alkylated triaryl phosphate esters of the present invention contain less than 0.05 wt%, based on the total weight of the alkylated triaryl phosphate ester, of the alkylphenyl dialkylphenyl phenyl phosphates.
- Exemplary alkylated triaryl phosphate esters of the present invention are those a) those that comprise: in the range of from about 90 to about 92wt.% IPP, in the range of from about 0.5 to about 0.75wt.% TPP, in the range of from about 1 to about 3 wt.% DTPP, in the range of from about 0.05 to about 0.15 wt.% TTPP, and in the range of from about 0.5 to about 0.75wt.% 2,4-DDP; b) in the range of from about 94 to about 96wt.% IPP, in the range of from about 3.5 to about 5.5 wt.% DTPP, and in the range of from about 0.1 to about 0.3 wt.% TTPP; and c) in the range of from about 71 to about 73wt.% IPP, in the range of from about 0.05 to about 0.15wt.% TPP, in the range of from about 26 to about 28 wt.% DT
- Monoalkylphenyl diphenyl phosphates, di- (alkylphenyl) phenyl phosphates, dialkylphenyl diphenyl phosphates, trialkylphenyl phosphates, and alkylphenyl dialkylphenyl phenyl phosphates, present in the alkylated triaryl phosphate esters of the present invention are those wherein the alkyl moieties are selected from methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tertiary-butyl, amyl, isoamyl, tertiary-amyl groups, and cyclohexyl alkyl moieties.
- the alkyl moieties of at least one of, preferably at least two of, more preferably all of, the monoalkylphenyl diphenyl phosphates, di-(alkylphenyl) phenyl phosphates, dialkylphenyl diphenyl phosphates, trialkylphenyl phosphates, and alkylphenyl dialkylphenyl phenyl phosphates, present in the alkylated triaryl phosphate esters are isopropyl moieties.
- the alkylated triaryl phosphate esters according to the present invention are isopropylphenyl diphenyl phosphate esters.
- 0.1 to 99.9 wt% is 2-isopropylphenyl phosphate (2-IPP), 0.1 to 99.9 wt% is 3-isopropylphenyl phosphate (3-IPP), 0.1 to 99.9 wt% is 4-isopropylphenyl phosphate (4-IPP), all based on the total weight of the alkylated triaryl phosphate ester.
- 66 to 100 wt% of the isopropylphenyl phenyl phosphate present in the alkylated triaryl phosphate esters according to the present invention is 2-isopropylphenyl phosphate (2-IPP), 0.1 to 4-wt% is 3-isopropylphenyl phosphate (3-IPP), 0.1 to 40 wt% is 4-isopropylphenyl phosphate (4-IPP). It should be noted that although specific ranges of isopropylphenyl phenyl phosphate have been discussed above, it is within the scope of the present invention to produce an alkylated triaryl phosphate ester having any possible relative ratio of 2-IPP, 3-IPP and 4-IPP.
- the alkylated triaryl phosphate esters according to the present invention are isopropylphenyl diphenyl phosphate esters wherein in the range of about 63 to about 68% of the isopropylphenyl diphenyl phosphate esteris 2-IPP, in the range of from about 0.5 to about 2.5 % is 3-IPP and in the range of from about 30.5 to about 36.5% is 4- IPP.
- the alkylated triaryl phosphate esters according to the present invention are isopropylphenyl diphenyl phosphate esters wherein about 66% of the isopropylphenyl diphenyl phosphate ester is 2-IPP, about 1% is 3- IPP and about 33% is 4-IPP.
- the alkylated triaryl phosphate esters of the present invention can suitably be formed by a process comprising reacting an alkylated phenol with POCl 3 in the presence of a first catalyst, thus forming a first reaction product.
- the first reaction product is then reacted with an alcohol selected from aryl alcohols, alkyl alcohols, alkylated aryl alcohols, and mixtures thereof in the presence of a second catalyst under second reaction conditions including temperatures ranging from about 9O 0 C to about 26O 0 C thereby producing an alkylated triaryl phosphate ester according to the present invention.
- a second catalyst under second reaction conditions including temperatures ranging from about 9O 0 C to about 26O 0 C thereby producing an alkylated triaryl phosphate ester according to the present invention.
- the reaction that produces the first reaction product is sometimes referred to as the first reaction herein, and the reaction of the first reaction product with the alcohol is sometimes referred to herein as the second reaction.
- Alkylated phenols suitable for use in the first reaction include those wherein the alkyl group is selected from methyl, ethyl, n-propyl, isopropyl, isobutyl, tertiary-butyl, amyl, isoamyl, tertiary-amyl alkyl moieties, and cyclohexyl, preferably isopropyl moieties.
- the alkylated phenol that is reacted with the POCI 3 in the presence of the first catalyst contains less than 1 mole% phenol and less than 25 mole% dialkyl phenol, both based on the total moles of reactive alkylated phenols (as described below) in the alklyated phenol.
- the alkylated phenol contains less than 0.5 mole% phenol and less than 15 mole% dialkyl phenol, both based on the total moles of reactive alkylated phenols in the alklyated phenol.
- the alkylated phenol contains less than 0.5 mole% phenol and less than 5 mole% 2,4-diisopropylphenol, both based on the total moles of reactive alkylated phenolics in the alklyated phenol.
- the dialkyl phenol of the alkylated phenol is 2,4-disiopropylphenol.
- the alkylated phenol stream comprises essentially OIP, MIP, and PIP constituents.
- the alkylated phenol stream comprise in the range of from about 64 to about 68 wt.% OIP, in the range of from about 0.5 to about 2.5 wt.% MIP, and in the range of from about 31 to about 35wt.% PIP, all based on the total weight of the alkylated phenol.
- Total moles of reactive alkylated phenols and "Reactive alkylated phenol” as used herein is meant to refer to the total moles of alkylated phenols that are part of the reaction between the alkylated phenol and the POCI 3 .
- This unit of measure is used herein because unreactive alklyated phenols are also present in the alkylated phenol.
- 2,6-DIP and 2,4,6-TIP are common impurities in an IP's stream but are for all intents and purposes unreactive.
- Table 1 describes one example of an alkylated phenol suitable for use herein:
- the amount of POCl 3 used herein can be a molar equivalency, in some embodiments a molar excess, and in other embodiments, less than a molar equivalency.
- a molar equivalency of POCl 3 it is meant that a molar ratio of about 1 moles of POCI 3 to about 1 mole of reactive alkylated phenol.
- a molar excess Of POCl 3 it is meant that a molar ratio of greater than 1 moles Of POCl 3 to 1 mole of reactive alkylated phenol.
- a molar excess is in the range of from about 1.0 to about 5.0 moles Of POCl 3 to about 1 mole of reactive alkylated phenol, and more preferably in the range of from about 1.15 to about 2.5 moles of POCl 3 to about 1 mole of reactive alkylated phenol are used in the practice of this embodiment of the present invention.
- a molar equivalency of POCI 3 it is meant a molar ratio of less than 1 mole of POCI 3 to 1 mole of reactive alkylated phenol.
- a molar excess of alklyated phenol can be used, i.e. less than a molar equivalency Of POCl 3 .
- Reactive alkylated phenol is defined above.
- Catalysts suitable for use as the first catalyst herein can be selected from tertiary amines such as trialkyl amines, dialkylaryl amines, and heterocyclic tertiary amines such as 1,4 Diazabicyclo[2,2,2]octane (DABCO); aromatic amines such as pyridine and substituted pyridines with N,N-dimethylaminopyridine being preferred from this group; pyrimidines and its derivatives; pyrazine and its derivatives; pyroles and its derivatives; imidizoles, its derivatives and their corresponding mineral and organic acid salts with N-methylimidazole, imidiazole and its derivatives being preferred from this group; quaternary ammonium salts; quaternary phosphonium salts; tetrakis dialkylamino phosphonium salts having the general formula P(NRR') 4 + X " especially tetrakis diethylamino
- any alkali metal halide and salts e.g. ammonium, phosphonium, etc., as described above, can be used as long as the salt/halide has appreciable solubility to initiate the reaction with POCl 3 such that co- produced hydrogen chloride ultimately converts the metal catalyst salt to the metal chloride salt.
- alkali metal and alkali earth metal catalysts include AlCl 3, MgCl 2 , CaCl 2 , NaCl, KCl, FeCl 3 , LiCl, TiCl 4 , SbCl 4 , AgCl and BaCl 2 .
- Non-limiting examples of suitable quaternary ammonium salts include tetrabutylammonium halide, tetra alkyl or mixed alkyl ammonium mineral or organic acid salt.
- suitable quaternary phosphonium salts include any tetra alkyl or tetraaryl phosphonium salt.
- the first catalyst is selected from quaternary ammonium salts, quaternary phosphonium salts, tetrabutylammonium chloride, MgCl 2 , and pyridine.
- the first catalyst is tetrabutylammonium chloride.
- the first catalyst is MgCl 2 .
- the first catalyst is pyridine.
- first reaction conditions include temperatures ranging from about 75 0 C to about 21O 0 C.
- first reaction conditions include temperatures ranging from about 8O 0 C to about 15O 0 C, more preferably temperatures ranging from about 9O 0 C to about 14O 0 C.
- the reactants and first catalyst can be combined, contacted, etc. in any order.
- the alkylated phenol reactant be added to the POCl 3 reactant. It has been found that an alkylated phenol phosphate with superior viscosity, i.e. less viscous, can be produced by combining the reactants and catalyst in this order.
- first reaction conditions also include venting of HCl gas. This venting can be conducted by any means known to be effective at venting HCl gas from a reaction vessel. However, in preferred embodiments, the venting is accomplished by conducting the reaction under first reaction conditions that include sub-atmospheric pressures, i.e. under a vacuum.
- the amount of vacuum used is readily selected by one having ordinary skill in the art taking into consideration that too much vacuum would cause the reaction temperature to fall outside of the ranges described above, thus slowing the reaction rate. Further, while vacuum pressures are preferred, the reaction can be conducted at atmospheric pressures up to about 5 psig and still produce a desirable product, albeit at a slightly reduced rate. A pressure significantly above 5 psig would slow the reaction rate somewhat more and potentially lead to the undesirable cleavage and/or transesterification reactions.
- first reaction conditions further include the substantial absence of oxygen.
- a diluent can be added along with the POCI 3 , first catalyst, and alkylated phenol.
- Diluents suitable for use herein are those that i) do not react substantially with the reagents, products and co-products, including HCl, utilized or generated during the first and/or second reactions; and ii) do not substantially reduce the catalytic activity of the first and/or second catalyst.
- diluents suitable for use herein can be further characterized as those that iii) do not lower the reaction temperature such that the reaction rate slows significantly to the point of not being commercially feasible, i.e. below the ranges disclosed herein.
- the diluent can be added as a complex with the first catalyst.
- suitable diluents include a) hydrocarbon solvents, such as heptane, petroleum ethers, methylcyclohexane and boiling point heptane; b) aromatic hydrocarbons such as toluene, xylene(s) and ethyl benzene; c) halo hydrocarbons and halo aromatic hydrocarbons such as chlorobenzene, dibromomethane, dibromoethane, and all isomers of trichloroethylene; d) ether solvents such as, tetrahydrofuran or 1,4-dioxane.
- the diluent is 1,4-dioxane.
- the diluent is toluene.
- the reaction of the POCI 3 and alkylated phenol produces a first reaction product comprising greater than about 50 mole% monoalkylatedphenyl dichloro phosphates, based on the total moles of the first reaction product excluding unreacted POCI 3 and any added diluent.
- the first reaction product can comprise in the range of from about 70 to about 99.9 mole% monoalkylatedphenyl dichloro phosphates, on the same basis, and in the range of from about 0.1 mole % to about 30 mole% bis-(monoalkylated)phenyl- chloro phosphates, on the same basis.
- excess POCl 3 it is meant any POCl 3 that did not react with the alkylated phenol, i.e. unreacted POCI 3 it.
- the first reaction product comprises in the range of from about 5 to about 80 mole % unreacted POCI 3 , based on the total phosphorus in the first reaction product, as determined by some suitable method, preferably quantitative P-31 NMR.
- the amount of unreacted POCI 3 in the first reaction product is obviously dependent on the amount of POCl 3 used in the first reaction stage.
- the first reaction product can contain substantially no excess POCI 3 , depending on the efficiency of the reaction between the alkylated phenol and POCI 3 ; however, if a molar excess Of POCl 3 is used, then the amount of excess POCI 3 will depend on the efficiency of the reaction and on the amount of POCI 3 used.
- the first reaction product can be, and in some embodiments is, directly reacted with an alcohol without removal of unreacted POCl 3 .
- excess POCl 3 is used in producing the first reaction product, then it is preferred that at least a portion of the excess POCI 3 be removed from the first reaction product, thereby producing an intermediate reaction product.
- the amount of excess POCI 3 removed from the first reaction product is that amount necessary to produce an intermediate product containing less than about 15mole%, preferably less than about 10 mole%, more preferably less than about 5mole%, most preferably less than about 1 mole%, POCI 3 , all based on the total phosphorus in the first reaction product.
- substantially all unreacted POCI 3 is removed from the first reaction product, which in some embodiments can produce an intermediate reaction product that contains substantially no unreacted alkylated phenol.
- the amount of unreacted POCI 3 removed from the first reaction product must be that amount necessary to produce an intermediate product containing less than about 1.2 mole%, preferably less than about 1 mole% of total organic phosphorus.
- the method by which the POCI 3 is removed from the first reaction product to produce the intermediate product is not critical to the present invention, and non-limiting examples of suitable removal techniques include vacuum distillation, flashing, stripping, vacuum stripping, and the like.
- POCl 3 is removed by vacuum stripping.
- Vacuum stripping can suitably be carried out by heating the first reaction product to within the range of about 115 0 C to about 17O 0 C, under constant agitation and vacuum in the range of from about 700 mmHg to about 0.001 mmHg. It is within the scope of the present invention that a nitrogen purge accompany the vacuum stripping.
- an inert "chaser” solvent at the end of the vacuum stripping to reduce the POCI 3 in the intermediate reaction product to less than 1 mole%, based on the intermediate reaction product. If a chaser solvent is used, it is preferred to use toluene, methylcyclohexane, boiling-point heptanes or n-heptane.
- the optional POCI 3 removal is accompanied by the removal of a portion of any diluent added during the first reaction.
- conditions can be adjusted to within the above ranges and means selected from those described above to provide for more efficient removal of both the POCl 3 and diluent.
- the first reaction product or intermediate reaction product is reacted with an alcohol selected from aryl alcohols including phenol, alkyl alcohols, alkylated aryl alcohols, and mixtures thereof in the presence of a second catalyst or processed to remove at least a portion of excess POCl 3 .
- an alcohol selected from aryl alcohols including phenol, alkyl alcohols, alkylated aryl alcohols, and mixtures thereof in the presence of a second catalyst or processed to remove at least a portion of excess POCl 3 .
- the first reaction product or intermediate product can be reacted sequentially with more than one alcohol selected from aryl alcohols including phenol, alkyl alcohols, alkylated aryl alcohols, and mixtures thereof.
- the first reaction product or intermediate product be reacted with a first alcohol, and when the first alcohol has been consumed, as determined by a suitable method such as P 31 NMR, a second alcohol, preferably different from the first alcohol, be added.
- a suitable method such as P 31 NMR
- a second alcohol preferably different from the first alcohol
- the first reaction product or intermediate reaction product is reacted with the first portion of the first alcohol until substantially all of the first portion of the first alcohol is consumed, as determined by a suitable method such as P NMR. After substantially all of the first portion of the first alcohol has been consumed, the second portion of the first alcohol is added, and the reaction allowed to continue until substantially all of the second portion of the first alcohol has been consumed, as determined by a suitable method such as P 31 NMR, thus producing a first intermediate reaction product comprising at least aryl dichlorophosphate and chloro diarylphosphate.
- the first intermediate reaction product which is richer, i.e. has a higher concentration, in chloro diarylphosphates then the first reaction product, is then reacted with an effective amount of the second alcohol.
- an effective amount of the second alcohol it is meant that amount of the second alcohol that is effective at converting substantially all of the aryl dichlorophosphate and chloro diarylphosphate to alkylated triaryl phosphate esters according to the present invention.
- the first and second alcohol can be independently selected from aryl alcohols including phenol, alkyl alcohols, alkylated aryl alcohols, and mixtures thereof.
- Non-limiting examples of suitable alkylated aryl alcohols are those wherein the alkyl group contains from about 1 to about 5 carbon atoms such as methyl.
- Non-limiting examples of suitable alkyl alcohols are those wherein the alkyl group contains from about 1 to about 20 carbon atoms such as n-decanol.
- the alcohol is selected from phenol, decanol, dodecanol or mixtures thereof and in a most preferred embodiment, the alcohol is phenol.
- Catalysts suitable for use as the second catalyst herein can be selected from quaternary ammonium salts, quaternary phosphonium salts, MgCl 2 , CaCl 2 , AlCl 3 , KCl, FeCl 3 , LiCl, and BaCl 2 .
- suitable quaternary ammonium salts and quaternary phosphonium salts include are those listed above in relation to the first catalyst.
- the second catalyst is selected from MgCl 2 , CaCl 2 , AlCl 3 , KCl, FeCl 3 , LiCl, and BaCl 2 . More preferably the second catalyst is MgCl 2 .
- the first reaction product or intermediate reaction product and alcohol are reacted in the presence of the second catalyst under second reaction conditions including temperatures ranging from about 75 0 C to about 26O 0 C.
- second reaction conditions include temperatures ranging from about 100 0 C to about 18O 0 C, most preferred from about 14O 0 C to about 150 0 C.
- the first reaction product or intermediate reaction product, alcohol, and second catalyst can be combined, contacted, etc., in any order.
- the first reaction product or intermediate reaction product, alcohol, and second catalyst can be co-fed to a reaction vessel, the first reaction product or intermediate reaction product can be added to a reaction vessel containing the alcohol and second catalyst, etc.
- the alcohol preferably in the molten or liquid state, be added to the first reaction product reactant or intermediate reaction product to which the second catalyst has already been introduced.
- the inventors have unexpectedly discovered that combining, contacting, etc. the first reaction product or intermediate reaction product, second catalyst and alcohol in this manner provides for an alkylated phenol phosphate having TPP concentrations lower than those formed when the reactants are not added in this manner.
- the catalyst is preferably present with the alcohol, but it can be co-fed or fed after the intermediate product.
- second reaction conditions further include the substantial absence of oxygen.
- the reaction of the first reaction product or intermediate reaction product and alcohol produces an alkylated triaryl phosphate ester according to the present invention, as described above.
- alkylated triaryl phosphate ester produced from the present process it may be desirable to further refine the alkylated triaryl phosphate ester produced from the present process, for example to remove any excess alcohol that may be present in the alkylated triaryl phosphate ester. Further processing can also include adding an additional amount of alcohol such as monoisopropylated phenols, diisopropylated phenols, phenol, and mixtures thereof and/or second catalyst to the alkylated triaryl phosphate ester. The alcohol-rich alkylated triaryl phosphate ester product comprising excess alcohol can then be recovered, and at least a portion, preferably substantially all, of the excess alcohol removed by, for example, phase separation and/or stripping and/or distillation. In preferred embodiments, steam stripping is used.
- the alkylated triaryl phosphate ester may also be washed one or more times with an acid, a base, or water.
- the alkylated triaryl phosphate ester can first be washed with an acid and/or base, preferably a base, and then washed with water.
- the alkylated triaryl phosphate ester can also be processed in a wipe film evaporator, a distillation column, or other similar separation device, in conjunction with the above further refinement processes or as a stand-alone refinement.
- a wipe film evaporator e.g., a distillation column
- other similar separation device e.g., a distillation column
- the alkylated triaryl phosphate esters of the present invention are suitable for use as a flame retardant in many applications.
- the alkylated triaryl phosphate esters of the present invention are especially well suited for use in polyurethane foams, polymer resins, and polyester resins.
- the present invention relates to a flame retardant polyvinyl chloride resin formulation comprising at least one, in some embodiments only one, polyvinyl chloride resin and a flame retarding amount of at least one, in some embodiments only one, alkylated triaryl phosphate ester, as described herein.
- a flame retarding amount of alkylated triaryl phosphate ester it is meant in the range of from about 2 to about 150 parts per hundred resin ("phr") of the at least one alkylated triaryl phosphate ester, based on the total weight of the flame retardant polyvinyl chloride resin formulation.
- a flame retarding amount of alkylated triaryl phosphate ester is to be considered in the range of from about 5 to about 70 phr, more preferably in the range of from about 12 to about 45 phr, of the at least one alkylated triaryl phosphate ester, on the same basis.
- Resins suitable for use in this embodiment of the present invention include those comprising a polymer comprised of one or more polymerized monomers having a polymerizable olefinic double bond in the molecule.
- There are three groups of such polymers namely (i) one or more vinylaromatic homopolymers or copolymers, preferably high-impact polystyrene, (ii) one or more acyclic olefinic hydrocarbon homopolymers or copolymers, such as polyethylene, polypropylene, and copolymers of ethylene or propylene with at least one higher olefin and with or without a diene monomer, and (iii) one or more copolymers of at least one vinylaromatic monomer and at least one non-vinylaromatic monomer containing a functional group, such as acrylonitrile, an acrylate monomer, or a methacrylate monomer with or without a diene monomer.
- Vinylaromatic polymers that can be flame retarded in the practice of this invention can be homopolymers, copolymers or block polymers and such polymers can be formed from such vinylaromatic monomers as styrene, ring-substituted styrenes in which the substituents are one or more C 1-6 alkyl groups, alpha-methylstyrene, ring-substituted alpha-methylstyrenes in which the substituents are one or more C 1 ⁇ alkyl groups, vinylnaphthalene, and similar polymerizable styrenic monomers, i.e., styrenic compounds capable of being polymerized, e.g., by means of peroxide or like catalysts, into thermoplastic resins.
- Homopolymers and copolymers of simple styrenic monomers are preferred from the standpoints of cost and availability.
- the vinylaromatic polymers that are flame retarded pursuant to this invention can be homopolymers or copolymers can be produced by free- radical polymerization, cationically-initiated polymerization, or anionically-initiated polymerization.
- the vinylaromatic polymers that are flame retarded in the practice of this invention can be foamable, expanded, or foamed vinylaromatic polymer compositions.
- the vinylaromatic polymers can have various structural configurations. For example they can be isotactic polymers, syndiotactic polymers, or mixtures of isotactic and syndiotactic polymers.
- the vinylaromatic polymers can be in the form of blends or alloys with other thermoplastic polymers, such as polyphenylene ether-styrenic polymer blends and polycarbonate-styrenic polymer blends.
- the vinylaromatic polymers can be impact-modified or rubber-modified polymers.
- the resin is a polyvinyl chloride resin.
- the flame retardant resin formulation can also include conventional additives such as, for example, process aids, acid scavengers, dyes, pigments, fillers, stabilizers, antioxidants, antistatic agents, reinforcing agents, blowing agents, nucleating agents, plasticizers, and the like.
- process aids such as, for example, process aids, acid scavengers, dyes, pigments, fillers, stabilizers, antioxidants, antistatic agents, reinforcing agents, blowing agents, nucleating agents, plasticizers, and the like.
- the amount of these additives used in the flame retardant polyvinyl chloride resin formulation is conventional, and one having ordinary skill in the art can readily select the amount and specific additive depending on the desired characteristics of the flame retardant polyvinyl chloride resin formulation.
- the alkylated triaryl phosphate esters of the present invention are suitable for use as a flame retardant in polyurethane/polyisocyanurate foams and polyurethane/polyisocyanurate foam formulations.
- the present invention relates to polyurethane/polyisocyanurate foams, polyurethane/polyisocyanurate foam formulations, and processes for forming flame retarded polyurethane/polyisocyanurate foam formulations, both rigid and flexible, all containing a flame retarding amount of at least one, in some embodiments only one, alkylated triaryl phosphate ester, as described herein.
- the present invention relates to polyurethane foams, polyurethane foam formulations, and processes for forming flame retarded polyurethane foam formulations, both rigid and flexible, in some embodiments flexible all containing a flame retarding amount of at least one, in some embodiments only one, alkylated triaryl phosphate ester, as described herein.
- a flame retarding amount of alkylated triaryl phosphate ester it is meant in the range of from about 5 to about 75wt.% of the at least one alkylated triaryl phosphate ester, based on the total weight of the polyurethane/polyisocyanurate foams or polyurethane/polyisocyanurate foam formulations.
- a flame retarding amount of alkylated triaryl phosphate ester is to be considered in the range of from about 5 to about 70wt.%.
- flexible polyurethane foams are typically prepared by chemical reaction between two liquids, isocyanates and polyols.
- the polyols are polyether or polyester polyols.
- the reaction readily occurs at room temperature in the presence of a blowing agent such as water, a volatile hydrocarbon, halocarbon, or halohydrocarbon, or mixtures of two or more such materials.
- Catalysts used in effecting the reaction include amine catalysts, tin-based catalysts, bismuth-based catalysts or other organometallic catalysts, and the like.
- Surfactants such as substituted silicone compounds are often used in order to maintain homogeneity of the cells in the polymerization system.
- Hindered phenolic antioxidants e.g., 2,6-di-tert-butyl-para-cresol and methylenebis(2,6-di-tert-butylphenol), can be used to further assist in stabilization against oxidative degradation.
- the present invention also relates to a polyurethane/polyisocyanurate foam formulation comprising a flame retarding amount of at least one, in some embodiments only one, alkylated triaryl phosphate ester, as described herein; at least one, in some embodiments only one, isocyanate or polyol; and at least one, in some embodiments only one, blowing agent, and polyurethane/polyisocyanurate foams, both rigid and flexible, formed therefrom.
- Blowing agents suitable for use herein include water, a volatile hydrocarbon, halocarbon, or halohydrocarbon, or mixtures of two or more such materials.
- the polyurethane/polyisocyanurate foams and foam formulations can contain any other component known in the art and used in the formation for flexible and rigid polyurethane/polyisocyanurate foams, and these other components used in forming polyurethane polymerization formulations or recipes are well known to those of ordinary skill in the art.
- the polyurethane/polyisocyanurate foam formulations can contain surfactants, antioxidants, diluents such as low viscosity liquid Cj -4 halocarbon and/or halohydrocarbon diluents in which the halogen content is 1-4 bromine and/or chlorine atoms can also be included in the compositions of this invention.
- Non-limiting examples of such diluents include bromochloromethane, methylene chloride, ethylene dichloride, ethylene dibromide, isopropyl chloride, n-butyl bromide, sec-butyl bromide, n-butyl chloride, sec- butyl chloride, chloroform, perchloroethylene, methyl chloroform, and carbon tetrachloride.
- the notation “Wt% in Crude” indicates the amount of each component in the ester product recovered from the reactor, and is thus based on the total weight of the product recovered from the reactor.
- "Normalized wt%” indicates the amount of each component calculated by dividing the "Wt% in Crude” values by the "Normalization Factor”, thus indicating the amount of each component in relation to the alkylated triaryl phosphate ester.
- a reaction flask was purged with nitrogen. 15.3g (0.1 mole) of phosphorous oxychloride (“POCl 3 ”) followed by 13.6g(0.1 mole) of ortho-isopropylphenol (“OIP"). The mixture was heated to about HO 0 C for 10 hours under agitation. The content of the flask were analyzed via 1 H-NMR and it was discovered that greater than 50mol.% of the OIP was unreacted.
- POCl 3 phosphorous oxychloride
- OIP ortho-isopropylphenol
- a reaction flask was purged with nitrogen. 15.3g (0.1 mole) of phosphorous oxychloride (“POCI 3 ”) followed by 13.6g(0.1 mole) of orf/20-isopropylphenol (“OIP”) was then added to the flask over a 30 minute period. The mixture was heated to 195 0 C for 5 hours under agitation. The content of the flask were analyzed via Proton NMR, and the presence of unreacted OIP in the flask detected by this analysis indicated that the reaction was incomplete. The content of the flask were then heated to 25O 0 C for 3 hours under agitation until no OIP was detected.
- POCI 3 phosphorous oxychloride
- OIP orf/20-isopropylphenol
- Powdered anhydrous magnesium chloride (0.5 part) was added to catalyze the reaction.
- the reaction mixture was rapidly heated to 13O 0 C and then slowly to 23O 0 C over a period of about 2 hours, after which there was no further appreciable evolution of hydrogen chloride.
- Completion of the reaction was checked by titration tests on the crude product which was then distilled under vacuum to give a fraction of recovered phenols, a small intermediate fraction and a main ester fraction (88% of crude product) boiling at 205 0 C- 225 0 C at 1 mm. of mercury.
- the composition of the recovered phenolic fraction was shown by analysis to be substantially the same as that of the phenolic feedstock mixture, indicating that there had been no appreciable separation of the components due to preferential esterification, which was verified by hydrolyzing a portion of the main ester fraction and analyzing the recovered phenols.
- the distilled phosphate ester had a satisfactory color, content of oxidizable impurities and acidity and was not therefore further purified.
- the viscosity of the distilled phosphate ester was 30cs at 25 0 C and the specific gravity (25°C/25°C) was 1.169.
- the constitution of the distilled phosphate ester is indicated in Table 2, below. The wt.% is based on the total weight of the distilled phosphate ester. TABLE 2
- Example l Example 3 herein
- the product required no further purification and had a viscosity of 58cs at 25 0 C and a specific gravity (25°C/25°C) of
- the wt.% is based on the total weight of the distilled phosphate ester.
- the mixed phosphate esters had a carbon number of 24 and contained 66 mole- percent of the isopropylphenyl group.
- Excess POCl 3 was recovered from the intermediate product (95% of theoretical amount) by first heating it at atmospheric pressure to about 130 0 C and then heating it to about 135°C at 1 mmHg. Heating of the reactor's content was discontinued, the reactor's content allowed to cool, and the reactor charged with 0.3 g OfMgCl 2 and 188 g of phenol (2.0 mole, 99.6%). [0070] After the addition of the MgCl 2 and phenol, the temperature of the reactor's content was increased to about HO 0 C, and the reaction mixture in the reactor was then heated from about 110 to about 130 0 C over a period of about 3 hours under agitation.
- the alkylated triaryl phosphate ester thus produced was analyzed, and the alkylated triaryl phosphate ester was found to have the characteristics outlined in Table 5, below. Normalized, or relative weight percents, are based on the total weight of phenol and the alkylated triaryl phosphate ester as is indicated in the table. TABLE 4
- a 2.0-liter Parr autoclave was charged with 50 grams of dry Amberlyst ® 15 and 1200 g (12.75 mole) of molten Phenol (Mallinckrodt, 99.6% loose crystals).
- the autoclave was sealed, purged with N 2 and heated to 1 10 0 C.
- the headspace of the autoclave was vented to atmospheric pressure and then purged with a 10-gram charge of propylene.
- Propylene, 19O g (4.5 mole) was then fed to the autoclave over a 90-minute period. The feed was such that the autoclave pressure varied from 80-30 psig during the addition.
- reaction temperature was maintained from 1 10 to 118°C for an additional hour, and the autoclave content then cooled to 7O 0 C. The autoclave content was then allowed to settle for 30 minutes prior to transferring (positive N 2 pressure through dip leg) to a nitrogen purged storage bottle.
- a second 1000 g (10.6 mole) charge of molten phenol was made to the autoclave containing the IP's/ Amberlyst ® 15 heel. The propylation reaction was repeated with 156 g (3.7 mole) of propylene. The combined decanted reaction mixture from the first and second molten phenol additions was then fractionally distilled (1 atmosphere).
- the light cut (typically 93% phenol, 7% OIP) was returned to the autoclave with make-up phenol and again propylated. This decantation and distillation process was continued through 8 runs.
- the concentrated crude IP's was distilled at 1 atmosphere to produce 3300 g of material. The analysis of which is reported in Table 5, below. The material was separated via distillation into a light cut (220Og, 93% phenol and 7% OIP), which was combined with (2200 g) fresh phenol and subsequently used as the alcohol of the second step to make IPP crude in Examples 7, 8 and 10-13 below.
- the reactor was then charged with 3.26 g Of MgCl 2 (1.0 mole%) and heated to HO 0 C.
- To the reactor was fed 629.1 g (6.69 moles) of a mixture comprising phenol (96.3wt%) and 2- isopropylphenol (3.7 wt%), recycled from the final light cut of IP's preparation described above (Example 6) with concomitant heating of the reactor's content from 110 0 C to 135°C over a 3 hour period.
- 31 P-NMR analysis indicated complete conversion of the monoaryl dichlorophosphate to triaryl phosphates.
- the pressure was reduced to 10 mmHg and unreacted phenol partially removed overhead at
- the alkylated triaryl phosphate ester thus produced was analyzed, and the alkylated triaryl phosphate ester was found to have the characteristics outlined in Table 7, below.
- the mixture was heated to 118 0 C and refluxed at that temperature until evolution of HCl slowed.
- the temperature was gradually increased to 135°C and left at that temperature until HCl evolution ceased.
- the excess POCI 3 was recovered in vacuo, stripping to an end point of 135°C and ⁇ 1.0mmHg.
- the reactor was allowed to cool, and then charged with 5.5 g of MgCl 2 (1.75 mole%) and heated to HO 0 C.
- the alkylated triaryl phosphate ester was recovered from the reactor and analyzed, and the alkylated triaryl phosphate ester was found to have the characteristics outlined in
- the reactor was allowed to cool, and after cooling to HO 0 C, 612g (6.5 moles) of a phenol/2-isopropylphenol mixture comprising 96.3wt% phenol and 3.7wt% 2- isopropylphenol, was fed to the reactor while gradually increasing the temperature of the reactor's content from 1 10° to 135 0 C over a 3-hour period.
- 31 P-NMR analysis indicated complete conversion of the monoaryl dichlorophosphate to triaryl phosphates.
- the pressure of the reactor was reduced to 10 mmHg and unreacted phenol removed overhead at 140 0 C.
- the alkylated triaryl phosphate ester was recovered from the reactor and analyzed, and the alkylated triaryl phosphate ester was found to have the characteristics outlined in
- the alkylated triaryl phosphate ester phosphate was recovered from the reactor and analyzed, and the alkylated triaryl phosphate ester was found to have the characteristics outlined in Table 11, below. Normalized or relative weight percents are based on the total weight of phenol and the alkylated triaryl phosphate ester as is indicated in the table. TABLE 11
- the alkylated triaryl phosphate ester was recovered from the reactor and analyzed, and the alkylated phenyl phosphate was found to have the characteristics outlined in Table 12, below. Normalized or relative weight percents are based on the total weight of phenol and the alkylated triaryl phosphate ester as is indicated in the table. TABLE 12
- a fully jacketed 2000 ml dry round bottom flask was used as a reactor in this example. It was equipped with an overhead stirrer, thermometer, oil jacketed addition funnel and an efficient condenser/takeoff head. The reactor was vented through a Drierite ® column to a caustic scrubber.
- the jacket temperature was increased to 135°C, within an additional 60 minutes the pot temperature had reached 127 0 C, HCl Evolution was essentially complete, and pyridine ⁇ Cl separated as an oil suspended in the reaction mixture (the resulting turbidity seems to be a good visual indictor of the reaction endpoint).
- a second phase was removed from the bottom of the reactor to yield a 195g less turbid yet still milky aqueous solution having a pH of 10. 530 g of a dilute H 3 PO 4 solution, 0.56 wt%, H 3 PO 4 , based on the solution, was then charged to the reactor, and the bottom turbid product layer was collected (1357 g).
- the alkylated triaryl phosphate ester was recovered from the reactor and analyzed, and the alkylated triaryl phosphate ester was found to have the characteristics outlined in Table 13, below. Normalized or relative weight percents are based on the total weight of phenol and the alkylated phenyl phosphate as is indicated in the table. TABLE 13
- the undistilled bottoms represented the last 2wt%, based on the mixture, of the total mass of the mixture.
- the forerun was analyzed by HPLC, which found 13.2 wt% phenol, 0.7 wt% 4-isopropylphenol, 13.0 wt% 2-isopropylphenol, 7.0 wt% 2,6-diisopropylphenol, 0.0 wt% TPP, 2.3 wt% monoisopropylphenyl diphenyl phosphates, 0.2 wt% diisopropylated triaryl phosphates, and 0.02 wt% triisopropylated triaryl phosphate.
- the product cut was also analyzed for purity and physical properties; the results are presented in the Table 14, below. All weight percents are absolute and are based on the total mass of that which is being analyzed.
- a 5-liter reactor was equipped with an addition funnel, thermal well and distillation apparatus.
- the distillation apparatus was vented to a caustic scrubber through a Drierite column.
- the reactor was purged with N 2 and charged with 3886 g (25.34 mole) of recycled POCl 3 and 6.37 g (0.53 mole%) Of MgCl 2 .
- the content of the reactor was heated to 85 0 C.
- a redistilled 67:1 :32 blend (1725 g, 12.67 mole) of OIP (Aldrich) ) MIP (Aldrich) and PIP (Aldrich) was fed to the reactor over a 3 -hour period. During the feed the reaction temperature was gradually increased to 130 0 C.
- a 5-liter glycol jacketed baffled reactor was charged with 500 g of 11% aqueous Na 2 COs and 2065 g of the alkylated triaryl phosphate ester of Table 13. The mixture was briefly stirred at 85 - 92°C and then left to phase separate. The bottom aqueous layer was removed along with a clear rag layer of intermediate density. The wash procedure was repeated through 4 washes. In order to completely remove the suspended rag layer, comprised primarily of sodium and magnesium phenoxide, 2000 ml of Toluene was added. The IPP/toluene mixture was then washed with tap water (2 X 500ml).
- a 5-liter reactor was equipped with an addition funnel, thermal well and distillation apparatus.
- the distillation apparatus was vented to a caustic scrubber through a Drierite ® column.
- the reactor was purged with N 2 and charged with 3385 g (22.08 mole) of recycled POCl 3 and 8.90 g (0.85 mole%) Of MgCl 2 .
- the content of the reactor was heated to 85 0 C.
- a redistilled 67:32:1 blend 1503.3 g, 11.04 mole) of OIP (Aldrich), PIP (Aldrich) and MIP (Aldrich) was fed to the reactor over a 3-hour period. During the feed the reaction temperature was gradually increased to 130 0 C.
- a 5-liter glycol jacketed baffled reactor was charged with 650 g of tap water, 1750 ml of toluene and 1915 g of the crude product mixture of Table 15. The mixture was briefly stirred at from 85 to 92°C and then left to phase separate. The bottom aqueous layer was removed along with a turbid rag layer of intermediate density. The organic layer was washed (2 X 100Og) with 2% aqueous NaOH at 85°C and then washed (4 X 800g) with tap water until a neutral pH of the wash water was achieved. The above process was repeated with the balance of the crude material (1897 g).
- a 5-liter reactor was equipped with an addition funnel, thermal well and distillation apparatus.
- the distillation apparatus was vented to a caustic scrubber through a Drierite ® column.
- the reactor was purged with N 2 and charged with 900.00 g (5.88 mole) of recycled POCl 3 21.0 g (3.94 mole%) of dry pyridine and 916.1 g (6.73 mole) of the redistilled 67:32:1 blend of OIP (Aldrich), PIP (Aldrich) and MIP (Aldrich).
- the stirred content of the reactor was heated to 114°C, the temperature at which HCl evolution began in earnest. During the course of 7 hours the reaction temperature was gradually increased to 130 0 C. After 8 hours of total reaction time a sample was removed and analyzed by Phosphorus NMR, which indicted a 93.6:18.5 relative ratio of ArOPOCl 2 I(ArO) 2 POCl.
- a 5-liter glycol jacketed baffled reactor was charged with 650 g of tap water, 1750 ml of toluene and 2055 g of the crude product mixture. The mixture was briefly stirred at from 85 to 92°C and then left to phase separate. The bottom aqueous layer was removed along with a turbid rag layer of intermediate density. The organic layer was washed (2 X 100Og) with 2% aqueous NaOH at 85°C and then washed (4 X 80Og) with tap water until a neutral pH of the wash water was achieved.
- test formulations were prepared by combining the materials outlined below in the amounts described below. Each formulation was prepared by mixing the components in a #5 Brabender bowl at 160-165 0 C and compression molded at 165°C into the appropriate thickness test specimens. ASTM type IV tensile bars were die cut from compression-molded plaques at 1/16" thickness. All formulations are based on a 75 Shore A hardness, 6O 0 C flexible PVC formulation. Resin molecular weight is suitable for thick wall injection molding or thin wall profile or wire extrusion or film. Each test formulations contained the following: Component Amounts in Formulation:
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Lubricants (AREA)
- Fireproofing Substances (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description
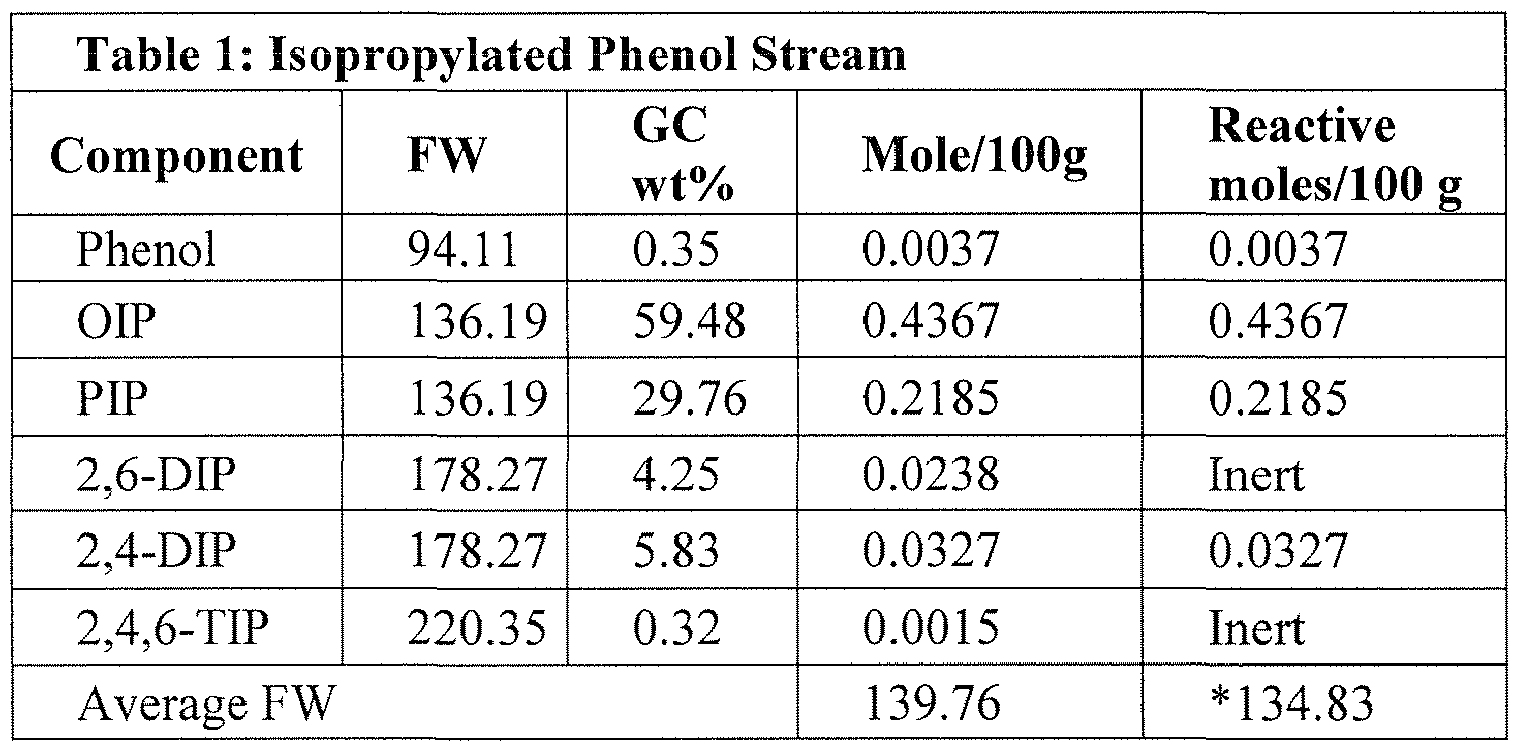


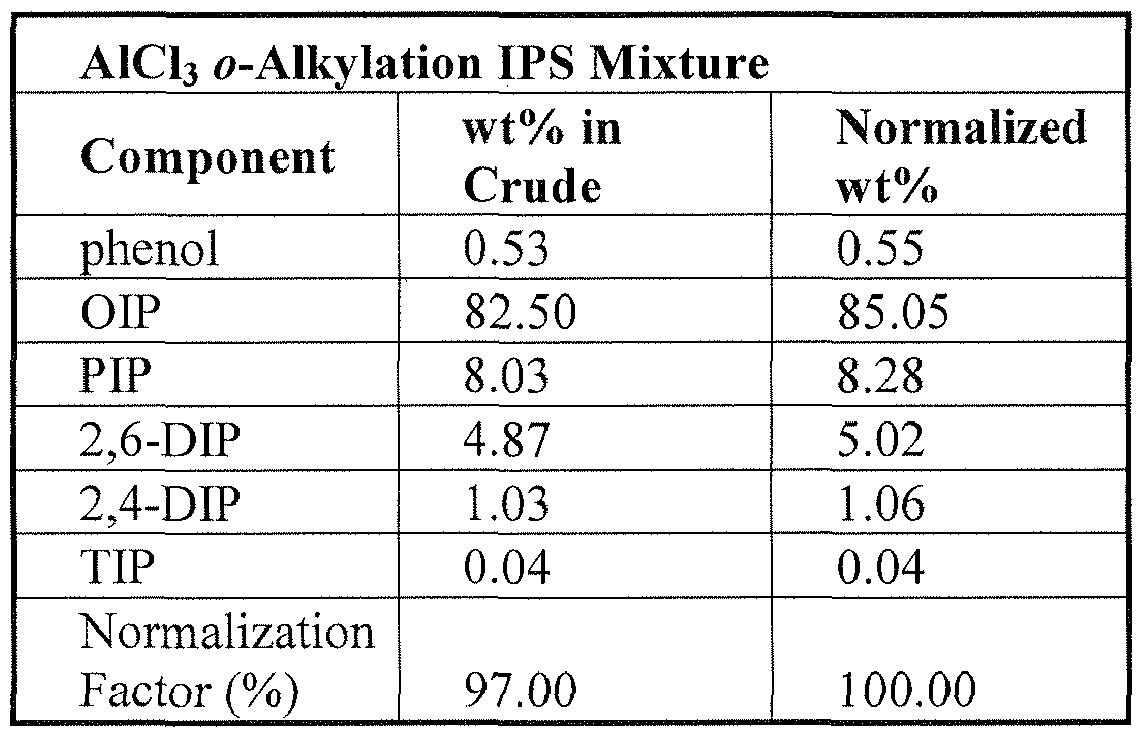



















Claims
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/297,273 US8143433B2 (en) | 2006-04-24 | 2007-04-23 | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation |
| EP07797267A EP2021347A2 (en) | 2006-04-24 | 2007-04-23 | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation |
| MX2008013263A MX2008013263A (en) | 2006-04-24 | 2007-04-23 | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation. |
| CA002649249A CA2649249A1 (en) | 2006-04-24 | 2007-04-23 | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation |
| CN2007800145742A CN101426801B (en) | 2006-04-24 | 2007-04-23 | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation |
| BRPI0711409-5A BRPI0711409A2 (en) | 2006-04-24 | 2007-04-23 | isopropyl phenyl phosphates with low triphenyl phosphate and high phosphorus with high ortho alkylation |
| AU2007244958A AU2007244958A1 (en) | 2006-04-24 | 2007-04-23 | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation |
| JP2009507914A JP2009534469A (en) | 2006-04-24 | 2007-04-23 | Isopropylphenyl phosphate with low triphenyl phosphate content, high phosphorus content and high degree of orthoalkyl substitution |
| IL194529A IL194529A0 (en) | 2006-04-24 | 2008-10-05 | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation |
| US13/232,388 US8350074B2 (en) | 2006-04-24 | 2011-09-14 | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US79478506P | 2006-04-24 | 2006-04-24 | |
| US60/794,785 | 2006-04-24 | ||
| US90829207P | 2007-03-27 | 2007-03-27 | |
| US90828707P | 2007-03-27 | 2007-03-27 | |
| US60/908,287 | 2007-03-27 | ||
| US60/908,292 | 2007-03-27 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/297,273 A-371-Of-International US8143433B2 (en) | 2006-04-24 | 2007-04-23 | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation |
| US13/232,388 Division US8350074B2 (en) | 2006-04-24 | 2011-09-14 | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007127691A2 true WO2007127691A2 (en) | 2007-11-08 |
| WO2007127691A3 WO2007127691A3 (en) | 2008-01-03 |
Family
ID=38610919
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/067188 WO2007127691A2 (en) | 2006-04-24 | 2007-04-23 | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation |
Country Status (11)
| Country | Link |
|---|---|
| US (2) | US8143433B2 (en) |
| EP (1) | EP2021347A2 (en) |
| JP (2) | JP2009534469A (en) |
| KR (1) | KR20080112323A (en) |
| AU (1) | AU2007244958A1 (en) |
| BR (1) | BRPI0711409A2 (en) |
| CA (1) | CA2649249A1 (en) |
| IL (1) | IL194529A0 (en) |
| MX (1) | MX2008013263A (en) |
| TW (1) | TW200745148A (en) |
| WO (1) | WO2007127691A2 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012511089A (en) * | 2008-12-08 | 2012-05-17 | アルベマール・コーポレーシヨン | Phosphorus flame retardant and its use |
| RU2751888C1 (en) * | 2020-10-29 | 2021-07-19 | Общество с ограниченной ответственностью Научно-производственное предприятие КВАЛИТЕТ ООО НПП КВАЛИТЕТ | Method for obtaining fire-resistant hydraulic fluid base |
| EP4431560A1 (en) * | 2023-03-15 | 2024-09-18 | LANXESS Deutschland GmbH | Phosphoric acid ester compositions for flame retardant soft pvc with high colour stability |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8143433B2 (en) * | 2006-04-24 | 2012-03-27 | Albemarle Corporation | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation |
| ES2374858T3 (en) * | 2007-08-07 | 2012-02-22 | Albemarle Corporation | PIRORRETARD RIGID POLYURETHANE FOAMS AND RIGID POLYURETHANE FOAM FORMULATIONS. |
| MX2010001412A (en) * | 2007-08-07 | 2010-03-10 | Albemarle Corp | Flame retarded flexible polyurethane foams and flexible polyurethane foam formulations. |
| US8747173B1 (en) | 2011-06-10 | 2014-06-10 | Hickory Springs Manufacturing Company | Flotation device |
| KR101241148B1 (en) * | 2012-09-26 | 2013-03-11 | 곽승민 | Flame proofing agents comprising an aromatic phosphate ester compound and methods for manufacturing the same |
| US20150266908A1 (en) * | 2012-09-26 | 2015-09-24 | Seung Min Kwak | Flame retardant comprising aromatic phosphate ester-based compound, and method for preparing same |
| KR101302555B1 (en) * | 2012-09-26 | 2013-09-02 | 곽승민 | Flame proofing agents comprising an aromatic phosphate ester compound and methods for manufacturing the same |
| KR101354039B1 (en) * | 2013-03-15 | 2014-01-23 | 곽승민 | Double cooling apparatus and apparatus for synthesizing an aromatic phosphate ester compound |
| JP6432935B2 (en) * | 2014-11-20 | 2018-12-05 | アキレス株式会社 | Vinyl chloride resin sheet |
| CN105040154B (en) * | 2015-07-22 | 2017-10-24 | 东华大学 | A kind of composite fibre of Flameproof polyamide 66 and preparation method thereof |
| EP3208276A1 (en) | 2016-02-18 | 2017-08-23 | PCC Rokita SA | Process for the preparation of a triaryl phosphate ester composition |
| CN111372991B (en) * | 2017-09-15 | 2023-11-17 | 吉昂功能材料(东莞)有限公司 | Flame retardant poly (vinyl chloride) composites |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2118662A5 (en) | 1970-12-16 | 1972-07-28 | Monsanto Co | Alkyl aryl phosphate esters - for hydraulic fluids with high thermal stability and wide viscosity range |
| US3859395A (en) | 1973-10-09 | 1975-01-07 | Fmc Corp | Triaryl phosphate esters |
| US4069279A (en) | 1972-03-23 | 1978-01-17 | Albright & Wilson Limited | Dialkylphenol phosphorylation |
| US4093680A (en) | 1966-06-18 | 1978-06-06 | Ciba-Geigy Ag | Phosphorylated tertiary butylated phenol/phenol ester reaction mixtures |
| US4139487A (en) | 1965-12-01 | 1979-02-13 | Albright & Wilson Limited | Mixed tri-aryl (phenyl and alkylphenyl) phosphate esters |
| US4370281A (en) | 1978-07-21 | 1983-01-25 | Ciba-Geigy Ag | Triaryl phosphates |
| EP0324716A2 (en) | 1988-01-12 | 1989-07-19 | Fmc Corporation (Uk) Limited | Triaryl phosphates |
| US5206404A (en) | 1992-04-27 | 1993-04-27 | Fmc Corporation | Triaryl phosphate ester composition and process for its preparation |
| US5624968A (en) | 1993-07-26 | 1997-04-29 | Monsanto Company | Flexible water-blown polyurethane foams |
| WO2000017210A1 (en) | 1998-09-21 | 2000-03-30 | Akzo Nobel N.V. | Triaryl phosphate ester composition |
| US6232485B1 (en) | 1997-10-29 | 2001-05-15 | Great Lakes Chemical Corporation | Production of phosphate esters |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1146173A (en) * | 1966-06-18 | 1969-03-19 | Geigy Uk Ltd | Production of triaryl phosphates |
| US4232359A (en) | 1979-04-09 | 1980-11-04 | Berkey-Colortran, Inc. | Spotlight or other illuminator |
| US4559184A (en) * | 1983-11-14 | 1985-12-17 | Stauffer Chemical Company | Phosphate ester synthesis without phosphorylation catalyst |
| JP2999036B2 (en) * | 1991-11-11 | 2000-01-17 | 本州化学工業株式会社 | Method for producing aryl phosphoric chlorides |
| US5302303A (en) * | 1993-08-24 | 1994-04-12 | Miles Inc. | Storage stable isocyanate-reactive compositions for use in flame-retardent systems |
| US6242631B1 (en) * | 1998-09-21 | 2001-06-05 | Akzo Nobel Nv | Triaryl phosphate ester composition |
| US8143433B2 (en) * | 2006-04-24 | 2012-03-27 | Albemarle Corporation | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation |
-
2007
- 2007-04-23 US US12/297,273 patent/US8143433B2/en not_active Expired - Fee Related
- 2007-04-23 AU AU2007244958A patent/AU2007244958A1/en not_active Abandoned
- 2007-04-23 CA CA002649249A patent/CA2649249A1/en not_active Abandoned
- 2007-04-23 KR KR1020087025850A patent/KR20080112323A/en not_active Application Discontinuation
- 2007-04-23 WO PCT/US2007/067188 patent/WO2007127691A2/en active Application Filing
- 2007-04-23 TW TW096114184A patent/TW200745148A/en unknown
- 2007-04-23 EP EP07797267A patent/EP2021347A2/en not_active Withdrawn
- 2007-04-23 JP JP2009507914A patent/JP2009534469A/en active Pending
- 2007-04-23 BR BRPI0711409-5A patent/BRPI0711409A2/en not_active IP Right Cessation
- 2007-04-23 MX MX2008013263A patent/MX2008013263A/en active IP Right Grant
-
2008
- 2008-10-05 IL IL194529A patent/IL194529A0/en unknown
-
2011
- 2011-09-14 US US13/232,388 patent/US8350074B2/en not_active Expired - Fee Related
-
2013
- 2013-04-12 JP JP2013083876A patent/JP2013151552A/en active Pending
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4139487A (en) | 1965-12-01 | 1979-02-13 | Albright & Wilson Limited | Mixed tri-aryl (phenyl and alkylphenyl) phosphate esters |
| US4093680A (en) | 1966-06-18 | 1978-06-06 | Ciba-Geigy Ag | Phosphorylated tertiary butylated phenol/phenol ester reaction mixtures |
| FR2118662A5 (en) | 1970-12-16 | 1972-07-28 | Monsanto Co | Alkyl aryl phosphate esters - for hydraulic fluids with high thermal stability and wide viscosity range |
| US4069279A (en) | 1972-03-23 | 1978-01-17 | Albright & Wilson Limited | Dialkylphenol phosphorylation |
| US3859395A (en) | 1973-10-09 | 1975-01-07 | Fmc Corp | Triaryl phosphate esters |
| US4370281A (en) | 1978-07-21 | 1983-01-25 | Ciba-Geigy Ag | Triaryl phosphates |
| EP0324716A2 (en) | 1988-01-12 | 1989-07-19 | Fmc Corporation (Uk) Limited | Triaryl phosphates |
| US5206404A (en) | 1992-04-27 | 1993-04-27 | Fmc Corporation | Triaryl phosphate ester composition and process for its preparation |
| US5624968A (en) | 1993-07-26 | 1997-04-29 | Monsanto Company | Flexible water-blown polyurethane foams |
| US6232485B1 (en) | 1997-10-29 | 2001-05-15 | Great Lakes Chemical Corporation | Production of phosphate esters |
| WO2000017210A1 (en) | 1998-09-21 | 2000-03-30 | Akzo Nobel N.V. | Triaryl phosphate ester composition |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012511089A (en) * | 2008-12-08 | 2012-05-17 | アルベマール・コーポレーシヨン | Phosphorus flame retardant and its use |
| RU2751888C1 (en) * | 2020-10-29 | 2021-07-19 | Общество с ограниченной ответственностью Научно-производственное предприятие КВАЛИТЕТ ООО НПП КВАЛИТЕТ | Method for obtaining fire-resistant hydraulic fluid base |
| EP4431560A1 (en) * | 2023-03-15 | 2024-09-18 | LANXESS Deutschland GmbH | Phosphoric acid ester compositions for flame retardant soft pvc with high colour stability |
| WO2024188705A1 (en) * | 2023-03-15 | 2024-09-19 | Lanxess Deutschland Gmbh | Phosphoric acid ester compositions for flame-retardant soft pvc with high colour stability |
Also Published As
| Publication number | Publication date |
|---|---|
| US8350074B2 (en) | 2013-01-08 |
| CA2649249A1 (en) | 2007-11-08 |
| WO2007127691A3 (en) | 2008-01-03 |
| IL194529A0 (en) | 2009-08-03 |
| JP2009534469A (en) | 2009-09-24 |
| US20120004438A1 (en) | 2012-01-05 |
| US20100012906A1 (en) | 2010-01-21 |
| EP2021347A2 (en) | 2009-02-11 |
| BRPI0711409A2 (en) | 2011-11-01 |
| MX2008013263A (en) | 2008-10-27 |
| AU2007244958A1 (en) | 2007-11-08 |
| KR20080112323A (en) | 2008-12-24 |
| US8143433B2 (en) | 2012-03-27 |
| TW200745148A (en) | 2007-12-16 |
| JP2013151552A (en) | 2013-08-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8143433B2 (en) | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation | |
| US8575225B2 (en) | Use of low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates as flame retardants in polyurethane or polyisocyanurate foams | |
| US8119711B2 (en) | Oligomeric bisphosphate flame retardants and compositions containing the same | |
| US20100130654A1 (en) | Use of low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates as flame retardants in resins | |
| CN101426801B (en) | Low triphenylphosphate, high phosphorous content isopropyl phenyl phosphates with high ortho alkylation | |
| TWI448470B (en) | Water miscible solvent based process | |
| WO2007057900A2 (en) | Flame retardant bromobenzyl systems | |
| EP1199310B1 (en) | Process for the preparation of condensed phosphoric esters | |
| RU2008146092A (en) | ISOPROPYLPHENYL PHOSPHATES OF A HIGH DEGREE OF ORTHO-ALKYLATION WITH A LOW CONTENT OF TRIPHENYL PHOSPHATE, A HIGH CONTENT OF PHOSPHORUS | |
| PL219211B1 (en) | Composition of isopropylphenyl phosphates with reduced content of triphenyl phosphate (TPP) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007244958 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 194529 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12008502247 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2649249 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/013263 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009507914 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087025850 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780014574.2 Country of ref document: CN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2007244958 Country of ref document: AU Date of ref document: 20070423 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007797267 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008146092 Country of ref document: RU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12297273 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0711409 Country of ref document: BR Kind code of ref document: A2 Effective date: 20081112 |