WO2007116092A1 - Enzyme inhibitors - Google Patents
Enzyme inhibitors Download PDFInfo
- Publication number
- WO2007116092A1 WO2007116092A1 PCT/EP2007/053560 EP2007053560W WO2007116092A1 WO 2007116092 A1 WO2007116092 A1 WO 2007116092A1 EP 2007053560 W EP2007053560 W EP 2007053560W WO 2007116092 A1 WO2007116092 A1 WO 2007116092A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound according
- aryl
- groups
- heteroaryl
- Prior art date
Links
- BRXUZDMVZBBWOL-UHFFFAOYSA-N NCc1c(-c(ccc(F)c2)c2F)nc[n]1Cc(cccc1)c1OC(F)(F)F Chemical compound NCc1c(-c(ccc(F)c2)c2F)nc[n]1Cc(cccc1)c1OC(F)(F)F BRXUZDMVZBBWOL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/02—Nutrients, e.g. vitamins, minerals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/64—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms, e.g. histidine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Definitions
- the present invention relates to the area of dipeptidyl peptidase IV (DP IV) inhibition and, particularly, relates to novel specific DP IV-inhibitors, which are able to cross the blood- brain-barrier in mammals, pharmaceutical compositions containing said compounds, and the use of said compounds for specifically inhibiting DP IV with low or no activity against related enzymes (e.g. DP IV-like enzymes).
- DP IV dipeptidyl peptidase IV
- DP IV is a serine protease, which cleaves N-terminal dipeptides from a peptide chain containing, preferably, a proline residue in the penultimate position.
- GLP-1 glucose-dependent insulinotropic peptide
- GIP gastric- inhibitory peptide
- DP IV inhibitors may be useful for the treatment of impaired glucose tolerance and diabetes mellitus (WO 99/61431 ; Pederson RA et al, Diabetes 1998 47(8):1253-1258 and Pauly RP et al, Metabolism 1999 48(3):385-389).
- WO 99/61431 discloses DP IV inhibitors comprising an amino acid residue and a thiazolidine or pyrrolidine group, especially L-f/?reo-isoleucyl thiazolidine, L-a//o-isoleucyl thiazolidine, L- f/?reo-isoleucyl pyrrolidine, L-a//o-isoleucyl thiazolidine, L-a//o-isoleucyl pyrrolidine, and salts thereof.
- WO 03/72556 discloses DP IV inhibitors comprising a glutaminyl residue and a thiazolidine or pyrrolidine group, especially glutaminyl thiazolidine and glutaminyl pyrrolidine, and salts thereof.
- low molecular weight DP IV inhibitors are agents such as tetrahydroisoquinolin-3-carboxamide derivatives, N-substituted 2-cyanopyroles and - pyrrolidines, N-(N'-substituted glycyl)-2-cyanopyrrolidines, N-(substituted glycyl)- thiazolidines, N-(substituted glycylH-cyanothiazolidines, boronyl inhibitors and cyclopropyl-fused pyrrolidines.
- agents such as tetrahydroisoquinolin-3-carboxamide derivatives, N-substituted 2-cyanopyroles and - pyrrolidines, N-(N'-substituted glycyl)-2-cyanopyrrolidines, N-(substituted glycyl)- thiazolidines, N-(substit
- Inhibitors of DP IV are described in US 6,01 1 ,155; US 6,107,317; US 6,1 10,949; US 6,124,305; US 6,172,081 ; WO 99/61431 , WO 99/67278, WO 99/67279, DE 198 34 591 , WO 97/40832, DE 196 16 486 C 2, WO 95/15309, WO 98/19998, WO 00/07617, WO 99/38501 , WO 99/46272, WO 99/38501 , WO 01/68603, WO 01/40180, WO 01/81337, WO 01/81304, WO 01/55105, WO 02/02560, WO 01/34594, WO 02/38541 , WO 02/083128, WO 03/072556, WO 03/002593, WO 03/000250, WO 03/000180, WO 03/000181 , EP 1 258 476, WO 03
- DP IV-inhibitors for the treatment of neurological diseases.
- WO 02/34242 and WO 02/34242 disclose the medical use of DP IV-inhibitors for maintenance or potentiation of endogenous neurological and neuropsychological effects of brain neuropeptide Y (NPY) systems via a potentiation of NPY Y1 receptor mediated effects within the central nervous system (CNS).
- NPY brain neuropeptide Y
- CNS central nervous system
- WO 01/34594 discloses DP IV-inhibitors comprising a proline mimetic, and a method of treating a patient having a disorder selected from the group consisting of strokes, tumors, ischemia, Parkinson's disease, memory loss, hearing loss, vision loss, migraines, brain injury, spinal cord injury, Alzheimer's disease, amyotrophic lateral, multiple sclerosis, diabetic neuropathy and prostate abnormalities.
- WO 05/079795 relates to the use of a DP IV inhibitor for the prevention, delay of progression or the treatment of neurodegenerative disorders, cognitive disorders and for improving memory (both short term and long term) and learning ability.
- the DPP8/9 inhibitor was also shown to attenuate T-cell activation in human in vitro models; a selective DP IV inhibitor was inactive in these assays. Moreover, it was found that DP IV inhibitors which were previously reported to be active in models of immune function are more potent inhibitors of DPP8/9. These results suggest that assessment of selectivity of potential clinical candidates may be important to an optimal safety profile for this class of agents.
- the term "pharmaceutically acceptable” embraces both human and veterinary use: for example the term “pharmaceutically acceptable” embraces a veterinarily acceptable compound or a compound acceptable in human medicine and health care.
- both the E and Z configuration are comprised within the scope of the present invention.
- the processes for the preparation of the compounds according to the invention give rise to a mixture of stereoisomers
- these isomers may be separated by conventional techniques such as preparative chromatography.
- the compounds may be prepared in racemic form, or individual enantiomers may be prepared either by enantiospecific synthesis or by resolution.
- the compounds may, for example, be resolved into their components enantiomers by standard techniques, such as the formation of diastereomeric pairs by salt formation with an optically active acid, such as (-)-di-p-toluoyl-d-tartaric acid and/or (+)-di-p-toluoyl-l-tartaric acid followed by fractional crystallization and regeneration of the free base.
- the compounds may also be resolved by formation of diastereomeric esters or amides, followed by chromatographic separation and removal of the chiral auxiliary. Alternatively, the compounds may be resolved using a chiral HPLC column.
- the pharmaceutically acceptable salt generally takes a form in which one or more basic moieties is protonated with an inorganic or organic acid.
- organic or inorganic acids include hydrochloric, hydrobromic, perchloric, sulfuric, nitric, phosphoric, acetic, propionic, glycolic, lactic, succinic, maleic, fumaric, malic, tartaric, citric, benzoic, mandelic, methanesulfonic, hydroxyethanesulfonic, benzenesulfonic, oxalic, pamoic, 2- naphthalenesulfonic, p-toulenesulfonic, cyclohexanesulfamic, salicylic, saccharinic or trifluoroacetic acid. All pharmaceutically acceptable acid addition salt forms of the compounds of the present invention are intended to be embraced by the scope of this invention.
- Non- pharmaceutically acceptable salts forms of the compounds of the invention may be of use in the preparation of pharmaceutically
- Compounds of the invention may form solvates with water (i.e. hydrate) or common organic solvents which are embraced as an aspect of the invention.
- Pharmaceutically acceptable solvents e.g. hydrates are of particular interest.
- compounds of the invention may exist as crystalline solids and all polymorphic forms thereof are included within the scope of the present invention.
- the present invention further includes within its scope prodrugs of the compounds of this invention.
- prodrugs will be functional derivatives of the compounds which are readily convertible in vivo into the desired therapeutically active compound.
- the term "administering" shall encompass the treatment of the various disorders described with prodrug versions of one or more of the claimed compounds of the invention, but which convert to one or more of the compounds of the invention in vivo after administration to the subject.
- DP IV-inhibitor or "dipeptidyl peptidase IV inhibitor” is generally known to a person skilled in the art and means enzyme inhibitors, which inhibit the catalytic activity of DP IV. DP IV-activity
- DP IV-activity is defined as the catalytic activity of dipeptidyl peptidase IV (DP IV).
- DP IV dipeptidyl peptidase IV
- These enzymes are post-proline (to a lesser extent post-alanine, post-serine or post- glycine) cleaving serine proteases found in various tissues of the body of a mammal including kidney, liver, and intestine, where they remove dipeptides from the N-terminus of biologically active peptides with a high specificity when proline or alanine form the residues that are adjacent to the N-terminal amino acid in their sequence.
- DP IV was originally believed to be the only membrane-bound enzyme specific for proline as the penultimate residue at the amino-terminus of the polypeptide chain.
- DP IV-like enzymes which are identified so far, are e.g.
- fibroblast activation protein ⁇ dipeptidyl peptidase IV ⁇ , dipeptidyl aminopeptidase-like protein, N- acetylated ⁇ -linked acidic dipeptidase, quiescent cell proline dipeptidase, dipeptidyl peptidase II, attractin and dipeptidyl peptidase IV related protein (DPP 8), and are described in the review article by Sedo & Malik (Sedo & Malik, Dipeptidyl peptidase IV-like molecules: homologous proteins or homologous activities? Biochimica et Biophysica Acta 2001 36506: 1 -10).
- DPIV-like enzymes are disclosed in WO 01/19866, WO 02/04610, WO 02/34900 and WO02/31 134.
- WO 01/19866 discloses novel human dipeptidyl aminopeptidase (DPP8) with structural und functional similarities to DPIV and fibroblast activation protein (FAP).
- WO 02/04610 provides reagents, which regulate human dipeptidyl peptidase IV- like enzyme and reagents which bind to human dipeptidyl peptidase IV-like enzyme gene product. These reagents can play a role in preventing, ameliorating, or correcting dysfunctions or diseases including, but not limited to, tumors and peripheral and central nervous system disorders including pain and neurodegenerative disorders.
- the dipeptidyl peptidase IV-like enzyme of WO 02/04610 is well known in the art. In the Gene Bank data base, this enzyme is registered as KIAA1492 (registration in February 2001 , submitted on April 04, 2000, AB040925).
- WO 02/34900 discloses a dipeptidyl peptidase 9 (DPP9) with significant homology with the amino acid sequences of DP IV and DPP8.
- WO 02/31 134 discloses three DP IV-like enzymes, DPRP1 , DPRP2 and DPRP3. Sequence analysis revealed, that DPRP1 is identical to DPP8, as disclosed in WO 01/19866, that DPRP2 is identical to DPP9 and that
- DPRP3 is identical to KIAA1492 as disclosed in WO 02/04610.
- subject refers to an animal, such as a mammal, in particular a human, who has been the object of treatment, observation or experiment.
- terapéuticaally effective amount means that amount of a compound of the invention compound or pharmaceutical agent that is sufficient to elicit a biological or medicinal response in a tissue system, animal or human, being sought by a researcher, veterinarian, medical doctor or other clinician, which includes alleviation of the symptoms of the disease or disorder being treated.
- composition is intended to encompass a product comprising the claimed compounds in the therapeutically effective amounts, as well as any product which results, directly or indirectly, from combinations of the claimed compounds.
- Co-administration includes administration of a formulation, which includes at least one DP IV-inhibitor of the present invention and at least one further drug agent (for example as listed in the section "Pharmaceutical combinations") or the essentially simultaneous administration of separate formulations of each agent.
- the DP IV-inhibitor and the other drug agent may be administered concomitantly or sequentially, as may be required by the appropriate treatment regime, via the same or different routes of administration.
- prevention means prophylactic administration of the combination to healthy patients to prevent the outbreak of the conditions mentioned herein. Moreover, the term “prevention” means prophylactic administration of such combination to patients being in a pre-stage of the conditions, to be treated. Delay
- delay of progression means administration of the combination, such as a combined preparation or pharmaceutical composition, to patients being in a pre- stage of the condition to be treated in which patients a pre-form of the corresponding condition is diagnosed.
- treatment is understood the management and care of a patient for the purpose of combating the disease, condition, or disorder.
- ementia includes Alzheimer type dementia, Parkinson type dementia, Huntington type dementia, Pick's type dementia, Creutzfeldt-Jakob type dementia, senile dementia, pre-senile dementia, idiopathic-related dementia, trauma- related dementia, stroke-related dementia, cranial bleed-related dementia, vascular dementia, and includes acute, chronic or recurring forms.
- alkyl denotes a d. 12 alkyl group, such as a Ci -6 alkyl group (for example a Ci -4 alkyl group).
- Alkyl groups may be straight chain or branched. Suitable alkyl groups include, for example, methyl, ethyl, propyl (e.g. n-propyl and isopropyl), butyl (e.g. n-butyl, tert-butyl and sec-butyl), pentyl (e.g. n-pentyl, hexyl (e.g. n-hexyl heptyl (e.g. n-heptyl) and octyl
- alkyl groups include 1 -ethyl-propyl, 3-methyl-butyl and 2,2-dimethyl-propyl), and 3,3-dimethyl-butyl), nonyl (e.g. n-nonyl and 7-methyl-octyl) and decyl (e.g. n-decyl), in particular methyl and ethyl.
- Further suitable alkyl groups include isobutyl.
- “Lower alkyl” refers to an alkyl group having 1 -4 carbon atoms e.g. methyl or ethyl.
- alk for example in the expression “alkoxy”, should be interpreted in accordance with the definition of "alkyl”.
- alkoxy groups include methoxy, ethoxy, propoxy, butoxy, pentyloxy, hexyloxy, heptyloxy and octyloxy.
- Other examples include nonyloxy and decyloxy.
- Further examples include n-propoxy, n-butoxy, n-pentoxy, n-hexoxy, n-heptoxyand n-octoxy).
- alk in the expression “thioalkyl” should be interpreted in accordance with the definition of "alkyl”.
- exemplary thioalkyl groups include methylthio-.
- alkan for example in the expression “alkanoyl” should be interpreted in accordance with the definition of "alkyl”.
- alkanoyl i.e. acyl groups
- ethanoyl i.e. acetyl
- propionyl i.e. acetyl
- butyryl i.e. acetyl
- acyl denotes a d-i 2 acyl residue, such as a Ci- 8 acyl residue e.g. a Ci -6 acyl residue and in particular a d- 4 acyl residue.
- acyl include the alkanoyl groups mentioned previously.
- alk in the expressions “haloalkyl” and “haloalkoxy” should be interpreted in accordance with the definition of “alkyl”.
- Ci- 6 haloalkyl is meant a Ci- 6 alkyl group which is substituted by at least one halo atom (for example fluoro, chloro or bromo).
- CWIuoroalkyl represents a Ci_ 6 alkyl group (such as those specifically recited above) which is substituted by at least one fluoro atom, including for example, fluoromethyl, difluoromethyl and trifluoromethyl (in particular trifluoromethyl).
- the expressions d-ehaloalkoxy and CWIuoroalkoxy can be interpreted accordingly.
- alkenyl denotes a C 2 -i 2 alkenyl group, such as a C 2 -6alkenyl group (for example a C 2 . 4 alkenyl group), which contains at least one double bond at any desired location and which does not contain any triple bonds.
- Alkenyl groups may be straight chain or branched.
- Exemplary alkenyl groups include propenyl, butenyl. Other examples include ethenyl, pentenyl and hexenyl.
- Exemplary alkenyl groups including two double bonds include pentadienyl, e.g. (1 E, 3E)-pentadienyl.
- alkynyl denotes a C 2 _i 2 alkynyl group, such as a C 2 - 6 alkynyl group (for example a C 2 . 4 alkynyl group), which contains at least one triple bond at any desired location.
- Alkynyl groups may be straight chain or branched. Exemplary alkynyl groups include ethynyl, propynyl and butynyl. Further examples include ethynyl, pentynyl and hexynyl.
- alkynyl group comprises also compounds having double bonds as well as triple bonds, i.e.
- alkeninyl groups for example having one double bond and additionally, one triple bond.
- amino means a primary, secondary or tertiary amine group.
- amino is represented by formula -NR a R b wherein R a and R b are selected from hydrogen or alkyl (e.g. Ci- 4 alkyl) or R a and R b may be joined to form a 4-7 membered ring optionally containing a further N or O atom.
- Examples of amino include NH 2 , NHMe, NMe 2 , NHEt, NEt 2 , NMeEt, azetidine, pyrrolidine, piperidine, morpholine, piperazine and N- methylpiperazine.
- amine unless qualified as “secondary amine” or “tertiary amine” means NH 2 .
- Exemplary cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
- Carbocycle denotes any ring system in which all the ring atoms are carbon and which typically contain between three and twelve ring carbon atoms, suitably between three and ten carbon atoms and more suitably between three and eight carbon atoms.
- Carbocycle groups may be saturated or partially unsaturated, but do not include aromatic rings. Examples of carbocycle groups include monocyclic, bicyclic, and tricyclic ring systems, in particular monocyclic and bicyclic ring systems. Other carbocylcyl groups include bridged ring systems (e.g. bicyclo[2.2.1]heptenyl).
- a specific example of a carbocycle group is a cycloalkyl group. A further example of a carbocycle group is a cycloalkenyl group. A further example of a carbocycle group is a cycloalkynyl group.
- cycloalkyl denotes a C 3 -i 2 cycloalkyl group, such as a C 3 -i 0 cycloalkyl (for example a C 3 . 8 cycloalkyl group).
- exemplary cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
- a most suitable number of ring carbon atoms is three to six.
- cycloalkenyl denotes a C 5 -i 2 cycloalkenyl group, for example a C 5 -i 0 cycloalkenyl group such as a C 5 . 8 cycloalkenyl (for example a C 6 - 8 cycloalkenyl group or a C 5 . 6 cycloalkenyl group).
- a specific example is cyclohexenyl. Further specific examples include cyclopropenyl, cycloheptenyl and cyclooctenyl. A most suitable number of ring carbon atoms is five to six.
- exemplary carbocycle groups include bridged ring systems (e.g. bicyclo[2.2.1]heptanyl bicyclo[2.2.1 ]heptenyl and adamantane, which are considered to be examples of cycloalkenyl and cycloalkyl groups respectively).
- bridged ring systems e.g. bicyclo[2.2.1]heptanyl bicyclo[2.2.1 ]heptenyl and adamantane, which are considered to be examples of cycloalkenyl and cycloalkyl groups respectively.
- C 3 - 6 cycloalkylimine is meant C 3 - 6 cycloalkyl group in which one of the ring carbon atoms is replaced by a nitrogen atom.
- exemplary C 3 - 6 cycloalkylimine groups include azetidine (also known as trimethylene imine, which may be 1 -azetidine, 2- azetidine or 3-azetidine, in particular 3-azetidine), pyrrolidine (including pyrrolidin-1 -yl, pyrrolidin-2-yl and pyrrolidin-3-yl) and piperidine (including piperidin-1 -yl, piperidin-2-yl, piperidin-3-yl and piperidin-4-yl).
- azetidine also known as trimethylene imine, which may be 1 -azetidine, 2- azetidine or 3-azetidine, in particular 3-azetidine
- pyrrolidine including pyrrolidin-1 -yl, pyrroli
- heterocyclic or “heterocycle”, unless specifically limited, denotes a carbocyclic residue (for example a cycloalkyl group, e.g. cyclopentyl or more particularly cyclohexyl), wherein one or more (e.g. 1 , 2, 3 or 4, such as 1 , 2 or 3, in particular 1 or 2, especially 1 ) ring atoms are replaced by heteroatoms selected from N, S or O.
- C 3 -i 2 heterocycle is meant a C 3 .i 2 carbocyclyl group in which at least one of the ring carbon atoms is replaced by a heteroatom.
- a heterocyclic group could therefore be monocylic or could alternatively be bicyclic.
- heterocyclic groups containing one hetero atom include: three membered rings (e.g. oxirane aziridine, thiirane); four membered rings (e.g. oxetane, azetidine, thietane); five membered rings (e.g. pyrrolidine and tetrahydrofuran, but also pyrroline and tetrahydrothiophene); and six membered rings (e.g. piperidine or tetrahydropyran).
- Exemplary heterocyclic groups containing two hetero atoms include five membered rings (e.g.
- a heterocycle group is a cycloalkenyl group (e.g. a cyclohexenyl group) wherein one or more (e.g. 1 , 2 or 3, particularly 1 or 2, especially 1 ) ring atoms are replaced by heteroatoms selected from N, S and O.
- a group is dihydropyranyl (e.g. 3,4-dihydro-2H-pyran-2-yl-).
- An example of a bridged heterocyclic group is utropine.
- aryl denotes a C 6- i 2 aryl group, suitably a C 6 - io aryl group, more suitably a C 6 - 8 aryl group.
- Aryl groups will contain at least one aromatic ring (e.g. one, two or three rings), but may also comprise partially or fully unsaturated rings.
- An example of a typical aryl group with one aromatic ring is phenyl.
- aromatic groups with two aromatic rings include naphthyl.
- naphthyl include naphth-1 -yl- and naphth-2-yl-.
- aryl groups which contain partially or fully unsaturated rings include pentalene, indene and indane.
- Other aryl groups include tricyclic rings such as anthracene.
- heteroaryl denotes as an aryl residue, wherein one or more (e.g. 1 , 2, 3, or 4, such as 1 , 2 or 3) ring atoms are replaced by heteroatoms selected from N, S and O or else a 5-membered aromatic ring containing one or more (e.g. 1 , 2, 3, or 4, such as 1 , 2 or 3) ring atoms selected from N, S and O.
- exemplary monocyclic heteroaryl groups having one heteroatom include: five membered rings (e.g. pyrrole, furan, thiophene); and six membered rings (e.g.
- pyridine such as pyridin-2-yl, pyridin-3-yl and pyridin-4-yl
- exemplary monocyclic heteroaryl groups having two heteroatoms include: five membered rings (e.g. pyrazole (e.g. pyrazol-3-yl), oxazole, isooxazole, thiazole, isothiazole, imidazole, such as imidazol-1 -yl, imidazol-2-yl imidazol- 4-yl); six membered rings (e.g. pyridazine, pyrimidine, pyrazine).
- Exemplary monocyclic heteroaryl groups having three heteroatoms include: triazole e.g. 1 ,2,3-triazole and 1 ,2,4- triazole.
- Exemplary monocyclic heteroaryl groups having four heteroatoms include tetrazole.
- Exemplary bicyclic heteroaryl groups include quinoline, benzothiophene, indazole, indole and purine. Exemplary bicyclic heteroaryl groups also include isoquinoline, quinolizine, benzodioxolane, benzodioxane, benzodioxepine (e.g. indol-6-yl), indoline, benzimidazole Further examples of bicyclic heteroaryl groups include (e.g. 1 H-indol-6-yl), benzimidazole, chromene, benzodioxolane, benzodioxane (e.g. 2,3-dihydro-benzo[1 ,4]dioxin-6-yl) and benzodioxepine.
- Exemplary tricyclic heteroaryl groups include carbazole and acridine groups.
- -alkylaryl denotes an aryl residue which is connected via an alkylene moiety such as a Ci- 6 alkylene moiety e.g. a Ci. 4 alkylene moiety.
- alkylaryl examples include: -methylaryl and -ethylaryl (e.g. 1 -arylethyl- or 2- arylethyl-); or phenylalkyl-, which may be optionally substituted.
- Specific examples of - alkylaryl functions include: phenylmethyl- (i.e. benzyl), phenylethyl- (e.g.
- -alkylheteroaryl denotes a heteroaryl residue which is connected via an alkylene moiety such as a Ci- 6 alkylene moiety e.g. a Ci- 4 alkylene moiety.
- alkylene moiety such as a Ci- 6 alkylene moiety e.g. a Ci- 4 alkylene moiety.
- -alkylheteroaryl include -methylheteroaryl and -ethylheteroaryl (e.g. 1 -heteroarylethyl- and 2-heteroarylethyl-).
- - alkylheteroaryl groups include pyridinylmethyl-, N-methyl-pyrrol-2-methyl- N-methyl-pyrrol- 2-ethyl-, N-methyl-pyrrol-3-methyl-, N-methyl-pyrrol-3-ethyl-, 2-methyl-pyrrol-1 -methyl-, 2- methyl-pyrrol-1 -ethyl-, 3-methyl-pyrrol-1 -methyl-, 3-methyl-pyrrol-1 -ethyl-, 4-pyridino- methyl-, 4-pyridino-ethyl-, 2-(thiazol-2-yl)-ethyl-, 2-ethyl-indol-1 -methyl-, 2-ethyl-indol-1 - ethyl-, 3-ethyl-indol-1 -methyl-, 3-ethyl-indol-1 -ethyl-, 4-methyl-pyridin-2-methyl-, 4-methyl-pyridin-2-yl-y
- halogen and “halo” include fluorine, chlorine, bromine and iodine, especially fluorine, chlorine and bromine, in particular fluorine and chlorine (e.g. fluorine).
- R 1 represents a group selected from the list consisting of:
- R 2 represents a group selected for the list consisting of -d- 6 alkylNR 10 R 11 and -C 3 .
- ecycloalkylimine optionally N substituted by R 12 ; and R 9 , R 10 , R 11 and R 12 independently represents hydrogen or lower alkyl.
- R 3 represents H; halogen; C 1-4 alkyl; d_ 4 haloalkyl; d_ 4 alkoxy or d_ 4 haloalkoxy;
- R 4 represents H; halogen; d_ 4 alkyl; d_ 4 haloalkyl; d_ 4 alkoxy or d_ 4 haloalkoxy;
- R 5 represents H; halogen; C 1-4 alkyl; d_ 4 haloalkyl; d_ 4 alkoxy or d_ 4 haloalkoxy; and wherein any of the aforesaid carbocycle and heterocycle groups may optionally be substituted by one or more groups (for example 1 , 2, or 3, in particular one or two groups) selected from the list consisting of:
- d- 6 haloalkyl e.g. d- 6 fluoroalkyl, such as -CF 3 );
- halogen e.g. fluoro, chloro and bromo
- oxo e.g. oxo
- amino e.g. -NH 2 , -NHCi- 6 alkyl (e.g. -NHMe), -N(d_ 6 alkyl) 2 (e.g. dimethylamino-)
- -OR 13 wherein R 13 may represent hydrogen, d- 6 alkyl, d. 6 alkenyl, d_ 6 alkynyl or d- 6 haloalkyl (e.g. hydrogen or d- 6 alkyl (e.g.
- any of the aforesaid aryl and heteroaryl groups may optionally be substituted by one or more groups (for example 1 , 2, or 3, in particular one or two groups) selected from the list consisting of:
- d- 6 haloalkyl e.g. d. 6 fluoroalkyl, such as -CF 3 );
- halogen e.g. fluoro, chloro and bromo
- amino e.g. -NH 2 , -NHCi- 6 alkyl (e.g. -NHMe), -N(d- 6 alkyl) 2 (e.g. dimethylamino-)
- -OR 13 wherein R 13 may represent hydrogen, d. 6 alkyl, d. 6 alkenyl, d. 6 alkynyl or Ci-ehaloalkyl (e.g. hydrogen or d- 6 alkyl (e.g.
- FIG. 1 Tetradic encounter test: Total distance travelled (upper panel) and distance travelled during first and during last 5 minutes of the test (lower panel) for animals treated with vehicle and with the different doses of Example 130b.
- Example 130b Mean values ⁇ SEM.
- Figure 7 Tetradic encounter test: Behavioural synchronisation of animals treated with vehicle and with the different doses of Example 130b. Synchronisation was tested according to the four patterns non-social activity, friendly contact, aggression, defense. Mean values ⁇ SEM.
- Figure 8 Tetradic encounter test: Behavioural synchronisation of animals treated with vehicle and with the different doses of Example 130b. Synchronisation was tested according to the three patterns concerning social behaviour friendly contact, aggression, defense. Mean values ⁇ SEM.
- Figure 9 Tetradic encounter test: Behavioural synchronisation of animals treated with vehicle and with the different doses of Example 130b. Synchronisation was tested according to friendly contact. Mean values ⁇ SEM.
- FIG. 10 Tetradic encounter test: Behavioural synchronisation of animals treated with vehicle and with the different doses of Example 130b. Synchronisation was tested according to aggression. Mean values ⁇ SEM.
- Figure 1 Development of body weight and food intake for animals treated with vehicle and with the different doses of Example 130b. Days of circadian measurement are indicated by a square, time points of forced swimming are indicated by arrows. Mean values ⁇ SEM.
- Figure 12 Development of fluid intake and Example 130b doses for animals treated with vehicle and with the different doses of Example 130b. Days of circadian measurement are indicated by a square, time points of forced swimming are indicated by arrows. Mean values ⁇ SEM.
- Figure 13 Total distance travelled during day 2 of circadian measurement for animals treated with vehicle and with the different doses of Example 130b. Mean values ⁇ SEM. The dark phase is indicated by a square. Figure 14 Rapid locomotion during day 2 of circadian measurement for animals treated with vehicle and with the different doses of Example 130b. Mean values ⁇
- Example 130b Mean values ⁇ SEM. Figure 19 Portion of time spent with rearing during day 2 of circadian measurement for animals treated with vehicle and with the different doses of Example 130b.
- Example 130b Mean values ⁇ SEM. Figure 21 Portion of time spent with rearing at fluid place during day 2 of circadian measurement for animals treated with vehicle and with the different doses of
- Example 130b Mean values ⁇ SEM. Figure 22 Cumulated number of drinking bouts on day 2 of circadian measurement for animals treated with vehicle and with the different doses of Example 130b.
- carbocycle and heterocycle are not substituted or are substituted by at most one substituent (for example they are not substituted).
- aryl and heteroaryl are not substituted or are substituted by at most one substituent (for example they are substituted by one substituent).
- substituted aryl groups include 4-fluoro-phenyl-, 3-fluoro-phenyl-, pentafluoro-phenyl-, 4-hydroxyphenyl-, 3-nitro-phenyl-, 4-(trifluoromethyl)-phenyl- and A- anilinyl-, 2-chlorophenyl, 3-fluoro-5-trifluoromethylphenyl, 2-chloro-6-fluorophenyl, 4- bromophenyl, 3-fluorophenyl, 2-methylphenyl, 2,3-dichlorophenyl, 3-chlorophenyl, 2,4- difluorophenyl, 3,4-difluorophenyl, 4-fluorophenyl, 2,4-dimethoxyphenyl, 3,4- dimethoxyphenyl, 2-fluoro-5-bromophenyl, 2,4-dichlorophenyl, 4-methoxyphenyl, 2,6- difluorophenyl, 4-chlor
- substituted heteroaryl groups include N-methyl-2-pyrrolyl, 2-methyl-1 -pyrrolyl and 3-methyl-2-pyrrolyl, 6F-(1 H)-indol-3-yl-, 5-nitro-pyridin-2-yl, 5-methyl-furan-2-yl, 5- methyl-1 H-pyrazol-4-yl, 5-methyl-pyrazin-2-yl.
- substituted carbocycle groups include methylcyclohexyl. Generally carbocycle is not substituted.
- substituted heterocycle groups include pyrrolidinone. Generally heterocycle is not substituted.
- R 1 represents d-i 2 alkyl (e.g. Ci- 6 alkyl).
- Particular R 1 Ci-i 2 alkyl (e.g. d- 6 alkyl) groups of interest include ethyl and pentyl.
- Other R 1 d-i 2 alkyl groups of interest include propyl, hexyl and nonyl for example ethyl-, n-propyl-, n-pentyl-, 3-methyl-butyl-, 2,2-dimethyl-propyl-, n-hexyl-, 3, 3, -dimethyl-butyl- or 7-methyl-octyl-.
- R 1 represents Ci -6 alkyl
- further examples include methyl, n-propyl, isopropyl, butyl (e.g. n-butyl, iso-butyl, sec-butyl, tert-butyl), (e.g. n-pentyl), 2,2-dimethylpropyl and hexyl (e.g. n-hexyl).
- substituted alkyl examples include ethan-2-ol-, propan-2-ol-, propan-3-ol-, pentan-5-ol- and hexan-6-ol-.
- R 1 represents Particular R 1 C 1 . 12 alkenyl groups of interest include ethenyl and propenyl.
- R 1 represents other examples include propen-2-yl, 2-methyl-propen-2- yl, buten-2-yl and buten-3-yl.
- R 1 represents C 2 -i 2 alkynyl.
- Particular R 1 Ci_ i 2 alkynyl groups of interest include ethynyl and propynyl.
- R 1 represents C 2 - i 2 alkynyl, other examples include propyn-2-yl, butyn-2-yl and butyn-3-yl.
- R 1 represents aryl.
- a particular R 1 aryl groups of interest is phenyl which may optionally be substituted.
- R 1 represents heteroaryl.
- Particular R 1 heteroaryl groups of interest include pyridinyl (e.g. 2-pyridinyl) which may optionally be substituted.
- R 1 represents Ci- 6 alkyl0Ci- 6 alkyl, for example Ci- 6 alkylOMe such as 2-(methoxy)-ethyl-.
- Ci- 6 alkylOMe such as 2-(methoxy)-ethyl-.
- alkyl is substituted is 1 - (ethoxy)-ethan-2-ol-.
- R 1 represents Examples include 2-(ethylamine)-ethyl-, 2-(cyclohexylamine)-ethyl-, 2-(diisopropylamine)-ethyl- and 3- (cyclohexylamine)-propyl-.
- R 1 represents -aryl-aryl, for example biphenyl.
- R 1 represents -Ci_ 6 alkylaryl (e.g. -Ci -4 alkyl-aryl).
- Particular d- 6 alkylaryl groups of interest include methylaryl, ethylaryl, propylaryl and butylaryl especially methylaryl and ethylaryl.
- the aryl group is phenyl which may be optionally substituted.
- the phenyl group is substituted by one or two substituents (in particular one substituent).
- substituents may be selected from Ci -6 alkyl (e.g. methyl), halo (e.g. Br, Cl, F) Ci-ehaloalkoxyl (e.g.
- Ci- 6 alkoxy e.g. methoxy
- Other exemplary substituents include hydroxyl, and Ci- ehaloalkyl (e.g. trifluoromethyl). Further exemplary substituents are selected from, ethyl, ethoxy, thiomethyl, thioethyl and trifluoromethyl.
- R 1 d- 6 alkylaryl group is: benzyl-.
- -alkyl-aryl include 2-fluoro-benzyl-, (3-fluoro-benzyl)-, (2- chloro-benzyl)-, (4-chloro-benzyl)-, (2-methyl-benzyl)-, (4-methyl-benzyl)-, (3-methoxy- benzyl)-, (2-ethoxy-benzyl)-, (3-trifluoromethyl-benzyl)-, (2-trifluoromethyl-benzyl)-, (2,4- difluoro-benzyl)-, (3,4-difluoro-benzyl)-, (2,5-difluoro-benzyl)-, (2,6-difluoro-benzyl)-, (2,3- dichloro-benzyl)-, (2,4-dichloro-benzyl)-, (3,4-dichlor
- Examples include 2-phenyl-ethyl-, 3-phenyl-propyl- and 4-phenyl-butyl-. Further specific examples of include: ((4-bromo-benzyl)-, (2-methoxy-benzyl)-, (4- methoxy-benzyl)-, (4-trifluoromethyl-benzyl)-, (2-trifluoromethoxy-benzyl)-, (4- trifluoromethoxy-benzyl)-, (5-bromo-2-fluoro-benzyl)-, (2-fluoro-3-trifluoromethyl-benzyl)-, (3-fluoro-5-trifluoromethyl-benzyl)-, (2,4-dimethoxy-benzyl)-, (3,4-dimethoxy-benzyl)-, (4- hydroxy-phenyl)-ethyl-, (1 -phenyl)-ethan-2-ol-, 2-(2-fluoro-phenyl)-ethyl
- R 1 represents d- 6 alkylaryl-aryl, for example (biphen-4-yl)-methyl-.
- R 1 represents Ci -6 alkyl(aryl) 2 .
- Examples include 2,2-diphenyl-ethyl- and 3,3-diphenyl-propyl-.
- R 1 represents C ⁇ -ealkyKheteroaryO ⁇ e.g. ethyl(2,2- heteroaryl) 2 .
- examples include 2,2-dipyridinyl-ethyl-, 3,3-dipyridinyl-propyl- and 2,2- bifuranyl-ethyl.
- R 1 represents C 2 . 6 alkenylaryl.
- examples include ethenylaryl e.g. ethenylphenyl which phenyl may optionally be substituted.
- R 1 represents C 2 -6alkynylaryl.
- examples include ethynylaryl e.g. ethynylphenyl which phenyl may optionally be substituted.
- R 1 represents Ci. 6 alkylaryl-heteroaryl, for example (2-pyridinyl)-4-phenyl-2-ethyl.
- R 1 represents d- 6 alkylheteroaryl-aryl, for example
- R 1 represents d-ealkylheteroaryl-heteroaryl, for example 4-(2-pyridinyl)-2-pyridinyl-2-ethyl.
- R 1 represents Ci. 6 alkyl(heteroaryl)(aryl), for example 2-phenyl-2-pyridinyl-ethyl.
- R 1 represents d- 6 alkylheteroaryl (e.g. -Ci- 4 alkyl- heteroaryl).
- Particular d- 6 alkylheteroaryl groups of interest include methylheteroaryl and ethylheteroaryl.
- Another d- 6 alkylheteroaryl groups of interest is propylheteroaryl. For example, d.
- 6 alkylheteroaryl groups of interest include d- 6 alkylbenzodioxolyl-, d- 6 alkylfuryl-, d- 6 alkylthiophenyl-, d- 6 alkylpyridinyl-, d- 6 alkylpyrazinyl-, d- 6 alkylbenzodioxolyl-, d- 6 alkylfuryl-, d- 6 alkylthiophenyl-, d- 6 alkylpyridinyl-, d- 6 alkylpyrazinyl-, d-
- R 1 represents d. 6 alkylheteroaryl
- the heteroaryl group is substituted by one or two substituents (in particular one substituent), for example selected from halo, d- 6 alkyl, d- 6 alkoxy, d-ethioalkyl, d- 6 haloalkyl and d- 6 haloalkoxy, particularly halo and d. 6 alkyl.
- the Ci_ 6 alkyl group is methyl or ethyl, particularly methyl.
- the d_ 6 alkoxy suitably the d_ 6 alkoxygroup is methoxy or ethoxy, particularly methoxy.
- the d-ethioalkyl suitably the d-ethioalkyl group is thiomethyl or thioethyl particularly thiomethyl.
- the d- 6 haloalkyl group is a d-efluoroalkyl group e.g. trifluoromethyl.
- the d-ehaloalkoxygroup is a d- efluoroalkoxy group e.g. trifluoromethoxy.
- the halo group is fluoro, chloro or bromo, particularly fluoro or chloro.
- R 1 d. 6 alkylheteroaryl groups include: (thiophen-3-yl)- methyl-, (pyridin-2-yl)-methyl-, (pyridin-3-yl)-methyl-, (pyridin-4-yl)-methyk
- Other specific examples of d. 6 alkylheteroaryl include 3-(imidazol-1 -yl)-propyl- 2-(pyridin-2-yl)-ethyl-, 2- (pyridin-3-yl)-ethyl-, (benzo[1 ,3]dioxol-5-yl)-methyl-, 2-(1 H-indol-3-yl)-ethyl.
- substituted R 1 d. 6 alkylheteroaryl groups include: (5-methyl-furan-2-yl)- methyl-, (1 -methyl-1 H-pyrazol-4-yl)-methyl-, 3-(5-methyl-1 H-pyrazol-4-yl)-propyl-, (5- methyl-isoxazol-3-yl)-methyl-, (5-methyl-pyrazin-2-yl)-methyl-, 2-(6-fluoro-1 H-indol-3-yl)- ethyl- 2-(6-chloro-1 H-indol-3-yl)-ethyl- and (6-methyl-1 H-indol-3-yl)-ethyl.
- Further examples include (1 -methyl-pyrrol-2-yl)-methyl-, (5-methyl-furan-2-yl)-methyl-, (benzo-
- R 1 represents C 2 - 6 alkenylheteroaryl, for example ethenylheteroaryl.
- R 1 represents C 2 . 6 alkynylheteroaryl, for example ethynylheteroaryl.
- R 1 represents C 3 -i 2 carbocycle, for example cyclohexyl.
- R 1 represents carbocycle
- other examples include cyclopentyl, and cyclohexenyl (e.g. cyclohexen-2-yl, cyclohexen-3-yl).
- substituted carbocycle include methylcyclopentyl-, methylcyclohexyl- (e.g. 2-methyl-cyclohexyl-, 3-methyl- cyclohexyl, 4-methyl-cyclohexyl) and methylcyclohexenyl.
- R 1 represents C 3 -i 2 heterocycle, for example piperidinyl.
- R 1 represents d-ealkylCs. ⁇ carbocycle (e.g. -C ⁇ alkylCa- ⁇ carbocycle).
- R 1 represents d-ealkylCa. ⁇ carbocycle
- the C 3 . 12 carbocycle group is unsubstituted or substituted by one or two substituents (in particular one substituent).
- Particular d-ealkylCa. ⁇ carbocycle groups of interest include methylC 3 . 12 carbocycle and ethylC 3 . 12 carbocycle (e.g. -methyl-cyclopentyl,
- -methyl-cyclohexyl which carbocycles may be optionally substituted.
- substituted -alkyl- C 3 .i 2 carbocycle include methylcyclopentyl-methyl- and methylcyclohexyl-methyl-.
- d- 6 alkylC 3 -i 2 carbocycle groups of interest include Ci- 6 alkylC 3 -i 2 cycloalkenyl and Ci- 6 alkylC 3 .i 2 cycloalkynyl, which cycloalkyl, cycloalkenyl and cycloalkynyl groups may be optionally substituted.
- Ci- 6 alkylC 3 -i 2 cycloalkenyl is -methyl-cyclohexenyl.
- R 1 d- 6 alkylC 3 -i 2 carbocycle groups include (adamantan-i -yl)-methyl- and 2- (cyclohex-1 -enyl)-ethyl-.
- R 1 represents C 2 . 6 alkenylC 3 -i 2 carbocycle, for example ethenylC 3 -i 2 carbocycle, C 2 - 6 alkenylcyclohexyl or d-ealkenylcyclohexenyl.
- R 1 represents C 2 -6alkynylC 3 -i 2 carbocycle, for example ethynylC 3 . 12 carbocycle, C 2 - 6 alkynylcyclohexyl or C ⁇ ealkynylcyclohexenyl.
- R 1 represents C 1 . 6 alkylNR 9 aryl, for example ethylNR 9 aryl or d. 6 alkylNR 9 phenyl which phenyl may optionally be substituted.
- a specific example is 2-phenylamine-ethyl.
- R 1 represents Ci. 6 alkylNR 9 heteroaryl, for example ethylNR 9 heteroaryl or d- 6 alkylNR 9 pyridinyl which pyridinyl may optionally be substituted.
- a specific example is 2-(nitropyridinylamine)-ethyl e.g. 2-(5-nitro-pyridin2-yl)-ethyl or 2- (pyridin-2-yl-amine)-ethyl-.
- R 1 represents Ci- 6 alkylOC 3 -i 2 carbocycle, for example ethylOd- i 2 carbocycle or d-ealkylOd-sCycloalkyl.
- R 1 represents Ci- 6 alkylNR 9 C 3 -i 2 carbocycle, for example ethylNR 9 C 3 -i 2 carbocycle or Ci- 6 alkylNR 9 C 3 - 8 cycloalkyl.
- a particular example is ethyl- NHcyclohexyl.
- R 1 represents d-ealkylOaryl, for example C 1 . 6 alkyl0phenyl which phenyl may optionally be substituted or methyl-O-aryl. Specific examples include 2-phenoxyethyl.
- R 1 represents d-ealkylOheteroaryl, for example Ci- 6 alkyl ⁇ pyridinyl which pyridinyl may optionally be substituted or methyl-O-heteroaryl. Specific examples include 4-pyridinyloxyethyl.
- R 1 represents Ci- 6 alkylC 3 .i 2 heterocycle (e.g. d- 4 alkylC 3 -i 2 heterocycle), which heterocycle may be optionally substituted.
- Particular d- 6 alkylC 3 -i 2 heterocycle groups of interest include methylC 3 -i 2 heterocycle, ethylC 3 . i 2 heterocycle and propyld- ⁇ heterocycle.
- Further specific Ci- 6 alkylC 3 -i 2 heterocycle groups of interest include Ci. 6 alkyltetrahydrofuranyl (e.g. -methyl-tetrahydrofuranyl) and d- 6 alkylpyrrolidinyl (e.g.
- R 1 represents d- 6 alkylC 3 -i 2 heterocycle
- the d-ealkyld- ⁇ heterocycle group may be substituted by one or two substituents (in particular one substituent), for example selected from oxo and lower alkyl e.g. methyl.
- substituents in particular one substituent
- Specific examples of unsubstituted d- ealkylCa- ⁇ heterocycle include (tetrahydrofuran-2-yl)-methyl-, 2-(pyrrolidin-1 -yl)-ethyl-, 2-
- R 1 represents C 2 - 6 alkenylC 3 . 12 heterocycle, for example ethenylCa. ⁇ heterocycle.
- R 1 represents C 2 -6alkynylC 3 -i 2 heterocycle, for example ethynylCa- ⁇ heterocycle.
- R 1 represents alkyl substituted by halogen (i.e. haloalkyl)
- examples include fluoromethyl, trifluoromethyl, fluoroethyl and fluoropropyl.
- R 2 represents -Ci- 6 alkyl-NR 10 R 11
- examples include amino-methyl-, 1 -amino-ethyl-, 2-amino-ethyl-, 1 -amino-propyl-, 2-amino-propyl- and 3-amino-propyl-.
- R 2 represents Ca-ecycloalkylimine optionally N-substituted by R 12
- examples include azetidinyl (e.g. 3-azetidinyl) and pyrrolidinyl.
- R 3 represents Hal
- examples include F, Cl and Br.
- R 3 represents C 1 4 alkyl
- examples include methyl, ethyl, n-propyl, iso-propyl and butyl.
- R 3 represents C ⁇ haloalkyl
- examples include fluoromethyl and trifluoromethyl.
- R 3 represents C ⁇ alkoxy
- examples include methoxy, ethoxy and propoxy.
- R 3 represents Ci. 4 haloalkoxy
- examples include fluoromethoxy and trifluoromethoxy.
- R 4 represents Hal
- examples include F, Cl and Br.
- R 4 represents d- 4 alkyl
- examples include methyl, ethyl, n-propyl, iso-propyl and butyl.
- R 4 represents Ci. 4 haloalkyl
- examples include fluoromethyl and trifluoromethyl.
- R 4 represents Ci. 4 alkoxy
- examples include methoxy, ethoxy and propoxy.
- R 4 represents Ci_ 4 haloalkoxy
- examples include fluoromethoxy and trifluoromethoxy.
- R 5 represents Hal
- examples include F, Cl and Br.
- R 5 represents d- 4 alkyl
- examples include methyl, ethyl, n-propyl, iso-propyl and butyl.
- R 5 represents d. 4 haloalkyl
- examples include fluoromethyl and trifluoromethyl.
- R 5 represents d. 4 alkoxy
- examples include methoxy, ethoxy and propoxy.
- R 5 represents d. 4 haloalkoxy
- examples include fluoromethoxy and trifluoromethoxy.
- suitable compounds are compounds of formula (I) wherein:
- R 1 represents d-i 2 alkyl; C 2 -i 2 alkenyl, wherein the double bond is not at the C-1 position; C 2 -i 2 alkynyl, wherein the triple bond is not at the C-1 position; C 3 -i 2 carbocycle; - which may optionally be substituted by one or more methyl groups; d- 6 alkyl-C 3 - i 2 carbocycle, in which the carbocycle ring may optionally be substituted by one or more methyl groups; Ci-ehaloalkyl; -Ci_ 6 alkyl-aryl; in which the heterocycle ring may optionally be substituted by one or more methyl groups; or -Ci- 6 alkyl-heteroaryl;
- R 2 represents -Ci- 4 alkyl-NH 2 ; azetidin-2-yl; azetidin-3-yl; pyrrolidin-2-yl or pyrrolidin-3-yl;
- R 3 represents H; halogen (e.g. F, Cl, Br); d. 4 alkyl; d. 4 haloalkyl (e.g. d_ 4 fluoroalkyl); C 1 . 4 alkoxy or d_ 4 haloalkoxy (e.g. d_ 4 fluoroalkoxy);
- R 4 represents H; halogen (e.g. F, Cl, Br); d_ 4 alkyl; C 1 _ 4 haloalkyl (e.g. d_ 4 fluoroalkyl) ; C 1 . 4 alkoxy or d_ 4 haloalkoxy (e.g. d_ 4 fluoroalkoxy);
- R 5 represents H; halogen (e.g. F, Cl, Br); d_ 4 alkyl; d_ 4 haloalkyl (e.g. d_ 4 fluoroalkyl); C 1 . 4 alkoxy or d- 4 haloalkoxy (e.g. d- 4 fluoroalkoxy); wherein any of the forementioned aryl and heteroaryl groups may optionally be substituted by one or more (e.g. 1 , 2 or 3, suitably 1 or 2) substituent groups selected from C 1 . 12 alkyl, C 2 _i 2 alkenyl, C 2 _i 2 alkynyl, d- 6 haloalkyl (e.g.
- d-efluoroalkyl d-efluoroalkyl
- -thiod_ 6 alkyl e.g. -thiomethyl
- -S0 2 d- 6 alkyl e.g. SO 2 Me
- d- 6 alkoxy- e.g. OMe
- C 3- i 2 cycloalkyl e.g. -S0 2 C 3 -i 2 cycloalkyl
- d- 6 alkoxy d- 6 alkyl- nitro, halogen (e.g.
- substituents will be selected from Ci-i 2 alkyl (e.g. Me), Ci_ 6 fluoroalkyl (e.g. CF 3 ), Ci- 6 alkoxy (e.g. OMe), halogen (e.g. F, Cl, Br) and hydroxy; or a pharmaceutically acceptable salt, polymorph or solvate thereof, including all tautomers and stereoisomers thereof.
- substituted aryl groups include fluorophenyl- (e.g. 4-fluoro-phenyl- or 3- fluoro-phenyl-), pentafluoro-phenyl-, 4-hydroxyphenyl-, 3-nitro-phenyl-, 4-(trifluoromethyl)- phenyl- and 4-anilinyl- groups.
- exemplary substituted monocyclic heteroaryl groups include methylfuranyl-.
- Exemplary substituted bicyclic heteroaryl groups include chromen-4-one, chromen-2-one and methylbenzothiophenyl.
- R 1 represents -Ci- 6 alkyl-aryl
- aryl is suitably optionally substituted phenyl.
- alkyl is suitably -CH 2 .
- R 1 represents -CH 2 -phenyl wherein phenyl is optionally substituted by one or more e.g. one or two substituents.
- Suitable optional substituents of aryl are selected from F, Cl, Br, methyl, methoxy, ethoxy, trifluoromethyl and trifluoromethoxy.
- Examples include 4-fluoro-benzyl, 3-methoxy-benzyl, benzyl, 3,4-difluoro-benzyl, 2-ethoxy-benzyl, 2-trifluoromethoxy-benzyl, 2-fluoro-benzyl, 2- chloro-benzyl, 3-trifluoromethyl-benzyl, 2,4-difluoro-benzyl, 3-fluoro-benzyl and 2-methyl- benzyl.
- R 1 represents -C ⁇ -ealkyl-heteroaryl
- heteroaryl is suitably monocyclic.
- alkyl is suitably -CH 2 .
- Heteroaryl is suitably an optionally substituted five or six-membered ring containing one atom selected from N, S and O and is optionally substituted. Most suitably, heteroaryl is unsubstituted or is substituted by methyl.
- R 1 represents Ci-i 2 alkyl, -Ci- 6 alkyl-aryl or -Ci-ealkyl-heteroaryl, particularly - Ci- 6 alkyl-aryl or -Ci-ealkyl-heteroaryl, wherein any of aryl and heteroaryl are optionally substituted.
- R 2 represents -Ci- 4 alkyl-NH 2
- R 2 suitably represents amino-methyl-, 1 -amino-ethyl-, 2-amino-ethyl or 3-amino propyl.
- R 2 represents amino-methyl-, 1 -amino-ethyl-, 2-amino-ethyl-, 3-amino- propyl- or azetidin-3-yl, especially amino-methyl-.
- R 3 represents Ci- 4 alkyl
- R 3 suitably represents methyl.
- R 3 represents alkoxy
- R 3 suitably represents methoxy.
- R 3 represents halogen or methyl, particularly halogen. More suitably, R 3 represents Cl or F.
- R 3 represents Cl
- R 4 represents C 1-4 alkyl
- R 4 suitably represents methyl
- R 4 represents alkoxy
- R 4 suitably represents methoxy
- R 4 represents halogen, methyl or methoxy, particularly halogen. More suitably, R 4 represents Cl or F. Most suitably R 4 represents Cl.
- R 5 represents d- 4 alkyl
- R 5 suitably represents methyl
- R 5 represents alkoxy
- R 5 suitably represents methoxy
- R 5 represents Hal
- R 5 suitably represents Cl or F. More suitably R 5 represents Cl.
- R 5 represents H.
- suitable compounds are compounds of formula formula (I) wherein:
- R 1 represents a group selected from the list consisting of:
- R 2 represents a group selected from the list consisting of: d- 6 alkylNR 10 R 11 and C 3 - 6 cycloalkylimine optionally N substituted by R 12 ; and R 9 , R 10 , R 11 and R 12 independently represents hydrogen or d- 4 alkyl.
- R 3 represents Cl
- R 4 represents Cl
- R 5 represents H; wherein any of the aforementioned aryl, heteroaryl, carbocycle and heterocycle groups may optionally be substituted by one or more (for example 1 , 2, or 3, in particular one or two) groups selected from the list consisting of:
- Ci_ 6 alkyl Ci -6 alkenyl, Ci -6 alkynyl (e.g. C 1-6 alkyl);
- haloalkyl e.g. C ⁇ efluoroalkyl, such as -CF 3 ;
- halogen e.g. fluoro, chloro and bromo
- R 13 may represent hydrogen, d- 6 alkyl, d- 6 alkenyl, d- 6 alkynyl or Ci- 6 haloalkyl (e.g. hydrogen or d- 6 alkyl (e.g. Me));
- substituent groups are selected from Ci -6 alkyl, Ci -6 fluoroalkyl, halogen, nitro, amino, hydroxyl and d- 6 alkoxy.
- substituent groups are selected from d. 6 alkyl, d- 6 fluoroalkyl, halogen, nitro, amino, hydroxyl, d. 6 alkoxy and oxo.
- carbocycle and heterocyle are unsubstituted.
- carbocycle and heterocycle are substituted by one or more methyl groups.
- R 1 represents a group selected from -Ci-i 2 alkyl, -d- 6 alkyl ⁇ d- 6 alkyl, C 2 -i 2 alkenyl or C 2 -i 2 alkynyl, any of which alkyl, alkenyl or alkynyl groups may optionally be substituted by one or more halogen (e.g. fluorine) groups or a hydroxyl group; -C 1 .
- halogen e.g. fluorine
- R 1 represents a group selected from Ci-i 2 alkyl optionally substituted by one or more halogen groups or by a hydroxyl group, -Ci_ 6 alkyl (optionally substituted by hydroxy)-OCi- 6 alkyl, -Ci-i 2 alkylamino, aryl, -Ci. 6 alkylaryl, -Ci.
- R 1 represents a group selected from Ci-i 2 alkyl optionally substituted by hydroxy; -Ci. 6 alkylaryl (e.g. Ci_ 2 alkylaryl); -Ci. 6 alkylheteroaryl (e.g. Ci_ 2 alkylheteroaryl); -d- 6 alkylcarbocycle (e.g. d ⁇ alkylcarbocycle), -d ⁇ alkylamino, -C 1-6 alkyl(aryl) 2 (e.g. C 1 . 3 alkyl(aryl) 2 ), -d. 6 alkylNR 9 heteroaryl, -C 1 . 6 alkylNR 9 C 3 .
- aryl represents optionally subtituted phenyl
- heteroaryl represents optionally substituted pyridinyl, furanyl, pyrazolyl, pyrazinyl, indolyl or imidazolyl
- carbocycle represents cycloalkenyl especially cyclohexenyl or cycloalkyl especially cyclohexyl
- heterocycle represents tetrahydrofuran or morpholinyl.
- Preferred substituents for aryl and heteroaryl are one or two groups selected from nitro, hydroxyl, C h alky!
- R 1 represents a group selected from d- 12 alkyl optionally substituted by hydroxy; -d. 6 alkylaryl (e.g. d_ 2 alkylaryl); and d. 6 alkylheteroaryl (e.g. d_ 2 alkylheteroaryl) especially in which aryl represents phenyl optionally substituted by substituents (e.g.
- R 9 represents hydrogen, methyl or ethyl, more suitably hydrogen or methyl, most suitably hydrogen.
- R 10 represents hydrogen, methyl or ethyl, more suitably hydrogen or methyl, most suitably hydrogen.
- R 11 represents hydrogen, methyl or ethyl, more suitably hydrogen or methyl, most suitably hydrogen.
- R 12 represents hydrogen, methyl or ethyl, more suitably hydrogen or methyl, most suitably hydrogen.
- R 2 represents Ci- 6 alkylNR 10 R 11 .
- Specific examples include methylamine, ethylamine (e.g. 1 -ethylamine and 2-ethylamine) and propylamine (e.g. 2-(1 -methyl)-ethylamine and 3-propylamine, in particular 3-propylamine).
- R 2 represents Ci- 6 alkylNR 10 R 11
- R 2 is methylamine, ethylamine or propylamine.
- One R 2 group of interest is methylamine.
- Another R 2 group of interest is 1 -ethylamine.
- a further R 2 group of interest is 2-ethylamine.
- An additional R 2 group of interest is 3- propylamine.
- R 2 represents C 3 - 6 cycloalkylimine optionally N substituted by R 11 and suitably represents C ⁇ ecycloalkylimine, in particular azetidine (for example 1 -azetidine, 2-azetidine or 3-azetidine, especially 3-azetidine).
- azetidine for example 1 -azetidine, 2-azetidine or 3-azetidine, especially 3-azetidine.
- R 2 represents a group selected from:
- the R 2 group of most particular interest is methylamine.
- the compounds of the present invention may have an improved ability to cross the blood- brain-barrier in mammals, preferably in humans, and may thus be especially suitable for the treatment of neurological disorders and/or CNS diseases.
- the ability of the compounds of the present invention to cross the blood-brain-barrier is demonstrated in the Biological Example 7.
- the compounds of the present invention may be highly specific against DP IV, i.e. may have less or no effectivity against DP IV-like enzymes, e.g. DP9, compared to conventional DP IV inhibitors.
- the high specificity of the compounds of the present invention is demonstrated in the Biological Example 1 .
- the compounds of the present invention may be less cytotoxic than conventional DP IV inhibitors.
- the low cytotoxicity of the compounds of the present invention is demonstrated in the Biological Example 2.
- the compounds of the present invention are well transported through Caco-2 monolayers.
- the transportability of the compounds of the present invention through Caco-2 monolayers is demonstrated in the Biological Example 3.
- the compounds of the present invention may be suitable for the treatment of anxiety.
- the suitability of the compounds of the present invention for the treatment of anxiety is demonstrated in the Biological Example 4.
- the compounds of the present invention may be suitable for the improvement of the social behaviour and treatment of schizophrenia.
- the suitability of the compounds of the present invention for the improvement of the social behaviour and treatment of schizophrenia may be demonstrated in the Tetradic Encounter Test, as shown in the Biological Example 5.
- the compounds of the present invention have a high bioavailability in vivo.
- the bioavailability of the compounds of the present invention is demonstrated in the Biological Example 6.
- the present invention provides a composition, preferably a pharmaceutical composition, comprising at least one DP IV inhibitor optionally in combination with at least one other agent e.g. selected from the group consisting of nootropic agents, neuroprotectants, antiparkinsonian drugs, amyloid protein deposition inhibitors, beta amyloid synthesis inhibitors, antidepressants, anxiolytic drugs, antipsychotic drugs and anti-multiple sclerosis drugs.
- at least one DP IV inhibitor optionally in combination with at least one other agent e.g. selected from the group consisting of nootropic agents, neuroprotectants, antiparkinsonian drugs, amyloid protein deposition inhibitors, beta amyloid synthesis inhibitors, antidepressants, anxiolytic drugs, antipsychotic drugs and anti-multiple sclerosis drugs.
- said DP IV inhibitor is a compound of of formula (I) of the present invention.
- the aforementioned other agent is selected from the group consisting PEP-inhibitors, LiCI, inhibitors of glutaminyl cyclase (QC), other inhibitors of DP IV or DP IV-like enzymes; acetylcholinesterase (AChE) inhibitors, PIMT enhancers, inhibitors of beta secretases, inhibitors of gamma secretases, inhibitors of neutral endopeptidase, inhibitors of Phosphodiesterase-4 (PDE-4), TNFalpha inhibitors, muscarinic M1 receptor antagonists, NMDA receptor antagonists, sigma-1 receptor inhibitors, histamine H3 antagonists, immunomodulatory agents, immunosuppressive agents or an agent selected from the group consisting of antegren (natalizumab), Neurelan (fampridine-SR), campath (alemtuzumab), IR 208, NBI 5788/MSP 771 (tiplimotide), paclitaxel, Anergix.
- the further drug agent may be selected from the group consisting of glutaminyl cyclase (QC) inhibitors, prolyl endopeptidase (PEP) inhibitors, LiCI, inhibitors of dipeptidyl aminopeptidases, other inhibitors of DP IV or DP IV-like enzymes, ACE inhibitors, PIMT enhancers, inhibitors of beta secretases, inhibitors of gamma secretases, inhibitors of neutral endopeptidase, inhibitors of PDE-4, TNFalpha inhibitors, amyloid protein or amyloid peptide deposition inhibitors, sigma-1 receptor inhibitors and histamine H3 antagonists.
- the other agent may be, for example, an anti-anxiety drug or antidepressant selected from the group consisting of
- Benzodiazepines e.g. alprazolam, chlordiazepoxide, clobazam, clonazepam, clorazepate, diazepam, fludiazepam, loflazepate, lorazepam, methaqualone, oxazepam, prazepam, tranxene,
- alprazolam chlordiazepoxide
- clobazam clonazepam
- clorazepate diazepam
- fludiazepam fludiazepam
- loflazepate lorazepam
- methaqualone oxazepam
- prazepam tranxene
- SSRI's Selective serotonin re-uptake inhibitors
- SSRI's e.g. citalopram, fluoxetine, fluvoxamine, escitalopram, sertraline, paroxetine
- Tricyclic antidepressants e.g. amitryptiline, clomipramine, desipramine, doxepin, imipramine
- Azapirones e.g. buspirone, tandopsirone
- SNRI's Serotonin-norepinephrine reuptake inhibitors
- NRI's Norepinephrine reuptake inhibitors (NRI's), e.g. reboxetine,
- NPY-receptor ligands NPY agonists or antagonists.
- the other agent may be, for example, an anti-multiple sclerosis drug selected from the group consisting of
- dihydroorotate dehydrogenase inhibitors e.g. SC-12267, teriflunomide, MNA-715, HMR-1279 (syn. to HMR-1715, MNA-279), b) autoimmune suppressant, e.g. laquinimod, c) paclitaxel, d) antibodies, e.g. AGT-1 , anti-granulocyte-macrophage colony-stimulating factor (GM-CSF) monoclonal antibody, Nogo receptor modulators, ABT-874, alemtuzumab (CAMPATH), anti-OX40 antibody, CNTO-1275, DN-1921 , natalizumab (syn.
- dihydroorotate dehydrogenase inhibitors e.g. SC-12267, teriflunomide, MNA-715, HMR-1279 (syn. to HMR-1715, MNA-279), b) autoimmune suppressant, e.g. la
- PNA peptide nucleic acid
- interferon beta-1 a like Avonex, Betron (Rebif), interferon beta analogs, interferon beta-transferrin fusion protein, recombinant interferon beta-1 b like Betaseron, h) interferon tau, i) peptides, e.g. AT-008, AnergiX.MS, lmmunokine (alpha-lmmunokine-NNS03), cyclic peptides like ZD-7349, j) therapeutic enzymes, e.g. soluble CD8 (sCD8), k) multiple sclerosis-specific autoantigen-encoding plasmid and cytokine-encoding plasmid, e.g. BHT-3009;
- peptides e.g. AT-008, AnergiX.MS, lmmunokine (alpha-lmmunokine-NNS03), cyclic peptides like ZD-7349
- therapeutic enzymes e.g.
- TNF-alpha e.g. BLX- 1002, thalidomide, SH-636, m) TNF antagonists, e.g. solimastat, lenercept (syn. to RO-45-2081 , Tenefuse), onercept (sTNFRI ), CC-1069, n) TNF alpha, e.g. etanercept (syn. to Enbrel, TNR-001 ) o) CD28 antagonists, e.g.
- TNF antagonists e.g. solimastat, lenercept (syn. to RO-45-2081 , Tenefuse), onercept (sTNFRI ), CC-1069
- TNF alpha e.g. etanercept (syn. to Enbrel, TNR-001 )
- CD28 antagonists e.g.
- chemokine receptor-1 (CCR1 ) antagonist e.g. BX-471 , t) CCR2 antagonists, u) AMPA receptor antagonists, e.g. ER- 167288-01 and ER-099487, E-2007, talampanel, v) potassium channel blockers, e.g.
- tosyl-proline-phenylalanine small-molecule antagonists of the VLA-4/VCAM interaction e.g. TBC-3342, x) cell adhesion molecule inhibitors, e.g. TBC-772, y) antisense oligonucleotides, e.g. EN-101 , z) antagonists of free immunoglobulin light chain (IgLC) binding to mast cell receptors, e.g. F-991 , aa) apoptosis inducing antigenes, e.g. Apogen MS, bb) alpha-2 adrenoceptor agonist, e.g. tizanidine (syn. to Zanaflex, Ternelin, Sirdalvo,
- IgLC free immunoglobulin light chain
- Sirdalud, Mionidine cc) copolymer of L-tyrosine, L-lysine, L-glutamic acid and L-alanine, e.g. glatiramer acetate (syn. to Copaxone, COP-1 , copolymer-1 ), dd) topoisomerase Il modulators, e.g. mitoxantrone hydrochloride, ee) adenosine deaminase inhibitor, e.g. cladribine (syn. to Leustatin, Mylinax, RWJ-
- interleukin-10 e.g. ilodecakin (syn. to Tenovil, Sch-52000, CSIF), gg) interleukin-12 antagonists, e.g. lisofylline (syn. to CT-1501 R, LSF, lysofylline), hh) Ethanaminum, e.g. SRI-62-834 (syn. to CRC-8605, NSC-614383), ii) immunomodulators, e.g. SAIK-MS, PNU-156804, alpha-fetoprotein peptide (AFP),
- interleukin-10 e.g. ilodecakin (syn. to Tenovil, Sch-52000, CSIF)
- gg) interleukin-12 antagonists e.g. lisofylline (syn. to CT-1501 R, LSF, lysofylline), hh) Ethanaminum, e.g. SRI-
- IPDS, jj) retinoid receptor agonists e.g. adapalene (syn. to Differin, CD-271 ), kk) TGF-beta, e.g. GDF-1 (growth and differentiation factor 1 ), II) TGF-beta-2, e.g. BetaKine, mm) MMP inhibitors, e.g. glycomed, nn) phosphodiesterase 4 (PDE4) inhibitors, e.g. RPR-122818, oo) purine nucleoside phosphorylase inhibitors, e.g. 9-(3-pyridylmethyl)-9- deazaguanine, peldesine (syn.
- alpha-4/beta-1 integrin antagonists e.g. ISIS-104278, qq) antisense alpha4 integrin (CD49d), e.g. ISIS-17044, ISIS-27104, rr) cytokine-inducing agents, e.g. nucleosides, ICN-17261 , ss) cytokine inhibitors, tt) heat shock protein vaccines, e.g. HSPPC-96, uu) neuregulin growth factors, e.g. GGF-2 (syn.
- the present invention provides a composition, preferably a pharmaceutical composition, comprising at least one compound of formula 1 , optionally in combination with at least one anti-multiple sclerosis drug selected from the groups mentioned above.
- PIMT enhancers are 10-aminoaliphatyl-dibenz[b, f] oxepines described in WO 98/15647 and WO 03/057204, respectively. Further useful according to the present invention are modulators of PIMT activity described in WO 2004/039773.
- PIMT enhancers are 10-aminoaliphatyl-dibenz[b, f]oxepines of the general formula:
- alk is a divalent aliphatic radical
- R is an amino group that is unsubstituted or mono- or di-substituted by monovalent aliphatic and/or araliphatic radicals or disubstituted by divalent aliphatic radicals
- Ri, R 2 , R3 and R 4 are each, independently of the others, hydrogen, lower alkyl, lower alkoxy, halogen or trifluoromethyl.
- WO 98/15647 discloses the compounds above for use in methods for modulating the activity of PIMT in order to specifically enhance or prevent apoptotic processes in a cell.
- WO 03/057204 discloses the compounds above for use in methods for preventing or alleviating an autoimmune response in a mammal, which may act through the activity of PIMT.
- modulators of PIMT activity are compounds of the general formulae I - IV:
- WO 2004/039773 discloses the compounds above for use in methods for preventing or alleviating diabetes, autoimmune diseases and neurodegenerative diseases which may act through the activity of PIMT and/or glyceraldehyde-3-phosphate dehydrogenase.
- WO 98/15647, WO 03/057204 and WO 2004/039773 are incorporated herein in their entirety and are part of this invention with regard to the synthesis and use of the compounds described therein in pharmaceutical combinations comprising a compound of the present invention.
- Inhibitors of beta secretase and compositions containing such inhibitors are described, e.g. in WO03/059346, WO2006/099352, WO2006/078576, WO2006/060109,
- beta secretase inhibitors for the purpose of the present invention are WY-25105 (Wyeth); Posiphen, (+)-phenserine (TorreyPines / NIH); LSN-2434074, LY- 2070275, LY-2070273, LY-2070102 (EIi Lilly & Co.); PNU-159775A, PNU-178025A, PNU- 17820A, PNU-33312, PNU-38773, PNU-90530 (Elan / Pfizer); KMI-370, KMI-358, kmi-008 (Kyoto University); OM-99-2, OM-003 (Athenagen Inc.); AZ- 12304146 (AstraZeneca / Astex); GW-840736X (GlaxoSmithKline pic.) and DNP-004089 (De Novo Pharmaceuticals Ltd.).
- Inhibitors of gamma secretase and compositions containing such inhibitors are described, e.g. in WO2005/008250, WO2006/004880, US 7,122,675, US 7,030,239, US 6,992,081 , US 6,982,264, WO2005/097768, WO2005/028440, WO2004/101562, US 6,756,51 1 , US 6,683,091 , WO03/066592, WO03/014075, WO03/013527, WO02/36555, WO01 /53255, US 7,109,217, US 7,101 ,895, US 7,049,296, US 7,034,182, US 6,984,626, WO2005/040126, WO2005/030731 , WO2005/014553, US 6,890,956, EP 1334085, EP 1263774, WO2004/101538, WO2004/00958, WO2004/08991 1 , WO2004/07
- Suitable gamma secretase inhibitors for the purpose of the present invention are GSI-953, WAY-GSI-A, WAY-GSI-B (Wyeth); MK-0752, MRK-560, L-852505, L-685-458, L-852631 , L-852646 (Merck & Co. Inc.); LY-450139, LY-41 1575, AN-37124 (EIi Lilly & Co.); BMS- 299897, BMS-433796 (Bristol-Myers Squibb Co.); E-2012 (Eisai Co. Ltd.); EHT-0206, EHT-206 (ExonHit Therapeutics SA); and NGX-555 (TorreyPines Therapeutics Inc.).
- Suitable inhibitors of beta and/or gamma secretases and compositions containing such inhibitors are described, e.g. in GB 2 385 124, GB 2 389 1 13, US 2002-1 15616, WO 01/87293, WO 03/057165, WO 2004/052348 and WO 2004/062652. These references are incorporated herein in their entirety and are part of this invention with regard to the synthesis, manufacture and use of the compounds and compositions described therein in pharmaceutical combinations comprising a compound of the present invention.
- a potent selective and cell permeable gamma secretase inhibitor is (5S)-(t- Butoxycarbonylamino)-6-phenyl-(4R)hydroxy-(2R)benzylhexanoyl)-L-leu-L-phe-amide with the formula:
- a potent beta secretase inhibitor is PNU-33312 of the formula:
- Suitable beta amyloid synthesis inhibitors for the purpose of the present invention are for example Bisnorcymserine (Axonyx Inc.); (R)-flurbiprofen (MCP-7869; Flurizan) (Myriad Genetics); nitroflurbiprofen (NicOx); BGC-20-0406 (Sankyo Co. Ltd.) and BGC-20-0466 (BTG pic).
- Suitable amyloid protein deposition inhibitors for the purpose of the present invention are for example SP-233 (Samaritan Pharmaceuticals); AZD-103 (Ellipsis Neurotherapeutics Inc.); AAB-001 (Bapineuzumab), AAB-002, ACC-001 (Elan Corp pic); Colostrinin (ReGen Therapeutics pic); AdPEDI-(amyloid-beta1 -6)1 1 ) (Vaxin Inc.); MPI-127585, MPI-423948 (Mayo Foundation); SP-08 (Georgetown University); ACU-5A5 (Acumen / Merck); Transthyretin (State University of New York); PTI-777, DP-74, DP 68, Exebryl (ProteoTech Inc.); m266 (EIi Lilly & Co.); EGb-761 (Dr.
- Suitable PDE-4 inhibitors for the purpose of the present invention are for example Doxofylline (Instituto Biologico Chemioterapica ABC SpA.); idudilast eye drops, tipelukast, ibudilast (Kyorin Pharmaceutical Co.
- Suitable PDE-4 inhibitors are, e.g. shown in the table below:
- a more suitable PDE-4-inhibitor is rolipram.
- MAO inhibitors and compositions containing such inhibitors are described, e.g. in WO2006/091988, WO2005/007614, WO2004/089351 , WO01/26656, WO01/12176, WO99/57120, WO99/571 19, WO99/13878, WO98/40102, WO98/01 157, WO96/20946, WO94/07890 and WO92/21333.
- Suitable MAO-inhibitors for the purpose of the present invention are for example Linezolid (Pharmacia Corp.); RWJ-416457 (RW Johnson Pharmaceutical Research Institute); budipine (Altana AG); GPX-325 (BioResearch Ireland); isocarboxazid; phenelzine; tranylcypromine; indantadol (Chiesi Farmaceutici SpA.); moclobemide (Roche Holding AG); SL-25.1 131 (Sanofi-Synthelabo); CX-1370 (Burroughs Wellcome Co.); CX-157 (Krenitsky Pharmaceuticals Inc.); desoxypeganine (HF Arzneiffenforschung GmbH & Co.
- Linezolid Pharmacia Corp.
- RWJ-416457 RW Johnson Pharmaceutical Research Institute
- budipine Altana AG
- GPX-325 BioResearch Ireland
- isocarboxazid phenelzine
- a suitable MAO-inhibitor is the compound ladostigil of the formula
- Suitable histamine H3 antagonists are, e.g. shown in the table below:
- PEP inhibitors and compositions containing such inhibitors are described, e.g. in JP 01042465, JP 03031298, JP 04208299, WO 00/71 144, US 5847155; JP 09040693, JP 10077300, JP 05331072, JP 05015314, WO 95/15310, WO 93/00361 , EP 0556482, JP 06234693, JP 01068396, EP 0709373, US 5,965,556, US 5,756,763, US 6,121 ,31 1 , JP 63264454, JP 64000069, JP 63162672, EP 0268190, EP 0277588, EP 0275482, US 4,977,180, US 5,091 ,406, US 4,983,624, US 5,1 12,847, US 5,100,904, US 5,254,550, US 5,262,431 , US 5,340,832, US 4,956,380, EP 0303434, JP 03056486, J
- Suitable prolyl endopeptidase inhibitors for the purpose of the present invention are, e.g. Fmoc-Ala-Pyrr-CN, Z-Phe-Pro-Benzothiazole (Probiodrug), Z-321 (Zeria Pharmaceutical Co Ltd.); ONO-1603 (Ono Pharmaceutical Co Ltd); JTP-4819 (Japan Tobacco Inc.) and S-17092 (Servier).
- Suitable inhibitors of prolyl endopeptidase are, e.g. chemical derivatives of proline or small peptides containing terminal prolines.
- Benzyloxycarbonyl-prolyl-prolinal has been shown to be a specific transition state inhibitor of the enzyme (WiIk, S. and Orloeski, M., J. Neurochem., 41 , 69 (1983), Friedman, et al., Neurochem., 42, 237 (1984)).
- N-terminal substitutions of L-proline or L-prolylpyrrolidine (Atack, et al., Eur. J.
- EP-A-O 286 928 discloses 2-acylpyrrolidine derivatives useful as propyl endopeptidase inhibitors.
- prolyl endopeptidase inhibitors are, e.g. Fmoc-Ala-Pyrr-CN and those listed below:
- PEP-inhibitor of the formula:
- NPY neuropeptide Y
- NPY mimetic NPY mimetic
- NPY agonist or antagonist NPY ligand of the NPY receptors.
- Preferred according to the present invention are antagonists of the NPY receptors.
- Suitable ligands or antagonists of the NPY receptors are 3a,4,5,9b-tetrahydro-1 h- benz[e]indol-2-yl amine-derived compounds as disclosed in WO 00/68197.
- NPY receptor antagonists which may be mentioned include those disclosed in European patent applications EP 0 614 91 1 , EP 0 747 357, EP 0 747 356 and EP 0 747 378; international patent applications WO 9417035, WO 971991 1 , WO 9719913, WO 9612489, WO 9719914, WO 9622305, WO 9640660, WO 9612490, WO 9709308, WO 9720820, WO 9720821 , WO 9720822, WO 9720823, WO 9719682, WO 9725041 , WO 9734843, WO 9746250, WO 9803492, WO 9803493, WO 9803494 and WO 9807420; WO 0030674, US patents Nos.
- Preferred NPY antagonists include those compounds that are specifically disclosed in these patent documents. More preferred compounds include amino acid and non-peptide-based NPY antagonists.
- Amino acid and non-peptide-based NPY antagonists which may be mentioned include those disclosed in European patent applications EP O 614 91 1 , EP O 747 357, EP O 747 356 and EP O 747 378; international patent applications WO 9417035, WO 971991 1 , WO 9719913, WO 9612489, WO 9719914, WO 9622305, WO 9640660, WO 9612490, WO 9709308, WO 9720820, WO 9720821 , WO 9720822, WO 9720823, WO 9719682, WO 9725041 , WO 9734843, WO 9746250, WO 9803492, WO 9803493, WO 9803494, WO 9807420 and WO 9915498 ; US patents Nos.
- Preferred amino acid and non-peptide-based NPY antagonists include those compounds that are specifically disclosed in these patent documents. Particularly preferred compounds include amino acid-based NPY antagonists. Amino acid- based compounds which may be mentioned include those disclosed in international patent applications WO 9417035, WO 971991 1 , WO 9719913, WO 9719914 or, preferably, WO 9915498.
- Preferred amino acid-based NPY antagonists include those that are specifically disclosed in these patent documents, for example BIBP3226 and, especially, (R)-N2-(diphenylacetyl)-(R)-N-[1 -(4-hydroxy- phenyl) ethyl] arginine amide (Example 4 of international patent application WO 9915498).
- M1 receptor agonists and compositions containing such inhibitors are described, e.g. in WO2004/087158, WO91/10664.
- Suitable M1 receptor antagonists for the purpose of the present invention are for example CDD-0102 (Cognitive Pharmaceuticals); Cevimeline (Evoxac) (Snow Brand Milk Products Co. Ltd.); NGX-267 (TorreyPines Therapeutics); sabcomeline (GlaxoSmithKline); alvameline (H Lundbeck A/S); LY-593093 (EIi Lilly & Co.); VRTX-3 (Vertex Pharmaceuticals Inc.); WAY-132983 (Wyeth) and CI-101 / (PD-151832) (Pfizer Inc.).
- Acetylcholinesterase inhibitors and compositions containing such inhibitors are described, e.g. in WO2006/071274, WO2006/070394, WO2006/040688, WO2005/092009, WO2005/079789, WO2005/039580, WO2005/027975, WO2004/084884,
- WO2004/037234 WO2004/032929, WO03/101458, WO03/091220, WO03/082820, WO03/020289, WO02/32412, WO01/85145, WO01/78728, WO01/66096, WO00/02549, WO01/00215, WO00/15205, WO00/23057, WO00/33840, WO00/30446, WO00/23057, WO00/15205, WO00/09483, WO00/07600, WO00/02549, WO99/47131 , WO99/07359, WO98/30243, WO97/38993, WO97/13754, WO94/29255, WO94/20476, WO94/19356, WO93/03034 and WO92/19238.
- Suitable acetylcholinesterase inhibitors for the purpose of the present invention are for example Donepezil (Eisai Co. Ltd.); rivastigmine (Novartis AG); (-)-phenserine (TorreyPines Therapeutics); ladostigil (Hebrew University of Jerusalem); huperzine A (Mayo Foundation); galantamine (Johnson & Johnson); Memoquin (Universita di Bologna); SP-004 (Samaritan Pharmaceuticals Inc.); BGC-20-1259 (Sankyo Co.
- NMDA receptor antagonists and compositions containing such inhibitors are described, e.g. in WO2006/094674, WO2006/058236, WO2006/058059, WO2006/010965, WO2005/000216, WO2005/102390, WO2005/079779, WO2005/079756,
- Suitable NMDA receptor antagonists for the purpose of the present invention are for example Memantine (Merz & Co. GmbH); topiramate (Johnson & Johnson); AVP-923 (Neurodex) (Center for Neurologic Study); EN-3231 (Endo Pharmaceuticals Holdings Inc.); neramexane (MRZ-2/579) (Merz and Forest); CNS-5161 (CeNeS Pharmaceuticals Inc.); dexanabinol (HU-21 1 ; Sinnabidol; PA-5021 1 ) (Pharmos); EpiCept NP-1 (Dalhousie University); indantadol (V-3381 ; CNP-3381 ) (Vernalis); perzinfotel (EAA-090, WAY- 126090, EAA-129) (Wyeth); RGH-896 (Gedeon Richter Ltd.); traxoprodil (CP-101606), besonprodil (PD-196860, CI-1041 ) (Pf
- DP IV-inhibitors and compositions containing such inhibitors are described, e.g. in US 6,01 1 ,155; US 6,107,317; US 6,1 10,949; US 6,124,305; US 6,172,081 ; WO99/61431 , WO99/67278, WO99/67279, DE19834591 , WO97/40832, WO95/15309, WO98/19998, WO00/07617, WO99/38501 , WO99/46272, WO99/38501 , WO01/68603, WO01/40180, WO01/81337, WO01/81304, WO01/55105, WO02/02560, WO01/34594, WO02/38541 , WO02/083128, WO03/072556, WO03/002593, WO03/000250, WO03/000180, WO03/000181 , EP1258476, WO03
- Suitable DP IV-inhibitors for the purpose of the present invention are for example Sitagliptin, des-fluoro-sitagliptin (Merck & Co. Inc.); vildagliptin, DPP-728, SDZ-272-070 (Novartis) ; ABT-279, ABT-341 (Abbott Laboratories); denagliptin, TA-6666 (GlaxoSmithKline pic); SYR-322 (Takeda San Diego Inc.); talabostat (Point Therapeutics Inc.); Ro-0730699, R-1499, R-1438 (Roche Holding AG); FE-99901 1 (Ferring Pharmaceuticals); TS-021 (Taisho Pharmaceutical Co.
- GRC-8200 (Glenmark Pharmaceuticals Ltd.); ALS-2-0426 (Alantos Pharmaceuticals Holding Inc.); ARI-2243 (Arisaph Pharmaceuticals Inc.); SSR-162369 (Sanofi-Synthelabo); MP-513 (Mitsubishi Pharma Corp.); DP-893, CP-867534-01 (Pfizer Inc.); TSL-225, TMC-2A (Tanabe Seiyaku Co.
- PHX-1 149 Phenomenix Corp.
- saxagliptin Bristol-Myers Squibb Co.
- PSN- 9301 ((OSI) Prosidion), S-40755 (Servier)
- KRP-104 ActivX Biosciences Inc.
- sulphostin Zaidan Hojin
- KR-62436 Korea Research Institute of Chemical Technology
- P32/98 Bl- A, Bl-B (Boehringer lngelheim Corp.); SK-0403 (Sanwa Kagaku Kenkyusho Co. Ltd.); and
- dipeptide-like compounds disclosed in WO 99/61431 , e.g. N-valyl prolyl, O-benzoyl hydroxylamine, alanyl pyrrolidine, isoleucyl thiazolidine like L-allo-isoleucyl thiazolidine, L- threo-isoleucyl pyrrolidine and salts thereof, especially the fumaric salts, and L-allo- isoleucyl pyrrolidine and salts thereof;
- peptide structures disclosed in WO 03/002593, e.g. tripeptides; (iii) peptidylketones, disclosed in WO 03/033524; (vi) substituted aminoketones, disclosed in WO 03/040174; (v) topically active DP IV-inhibitors, disclosed in WO 01/14318; (vi) prodrugs of DP IV-inhibitors, disclosed in WO 99/67278 and WO 99/67279; and (v) glutaminyl based DP IV-inhibitors, disclosed in WO 03/072556 and WO 2004/099134.
- Inhibitors of QC are described in WO 2004/098625, WO 2004/098591 , WO 2005/039548 and WO 2005/075436.
- the present invention provides a composition, preferably a pharmaceutical composition, comprising at least one compound of the invention, optionally in combination with at least one anti-diabetic drug selected from the group consisting of
- insulin sensitizers selected from the group consisting of
- glucagon receptor agonists e.g. glucagon receptor agonists
- GLP-1 GLP-1 mimetics, e.g. NN-221 1 (liraglutide from Novo Nordisk), and
- GLP-1 receptor agonists (h) GLP-2; GLP-2 mimetics, e.g. ALX-0600 (teduglutide from NPS Allelix Corp.) and GLP-2 receptor agonists; (i) exendin-4 and exendin-4 mimetics, e.g. exenatide (AC-2993, synthetic exendin-
- antioxidants include antioxidants; (m) PPAR ⁇ agonists; (n) antiobesity compounds; (o) an ileal bile acid transporter inhibitor; and (p) anti-inflammatory agents.
- compositions or pharmaceutical compositions according to any one of the embodiments described above optionally comprise additionally at least one carrier or excipient.
- compositions e.g. for parenteral, enteral or oral administration, comprising at least one DP IV inhibitor of formula (I) optionally in combination with at least one of the other aforementioned agents.
- the invention provides a method for the treatment of these conditions.
- the method comprises either co-administration of at least one DP IV inhibitor of formula (I) and at least one of the other agents or the sequential administration thereof.
- Co-administration includes administration of a formulation, which comprises at least one DP IV inhibitor of formula (I) and at least one of the other agents or the essentially simultaneous administration of separate formulations of each agent.
- compositions or pharmaceutical compositions according to any one of the embodiments described above comprise additionally at least one carrier or excipient.
- compositions which comprise a compound of the invention together with one or more pharmaceutically acceptable diluents or carriers.
- compositions are in unit dosage forms from such as tablets, pills, capsules, powders, granules, sterile parenteral solutions or suspensions, metered aerosol or liquid sprays, drops, ampoules, autoinjector devices or suppositories.
- the composition may be formulated for administration to a patient by any conventional route, including, but not limited to, intravenous, oral, subcutaneous, intramuscular, intradermal, parenteral, intranasal, sublingual or rectal administration, or for administration by inhalation or insufflation.
- compositions of this invention To prepare the pharmaceutical compositions of this invention, one or more compounds of the present invention, especially the DP IV inhibitors according to formula 1 , as well as optionally, other agents as described for the "pharmaceutical combinations", and their corresponding pharmaceutically acceptable acid addition salt forms, as the active ingredients, are intimately admixed with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques, which carrier may take a wide variety of forms depending of the form of preparation desired for administration.
- a pharmaceutical carrier may take a wide variety of forms depending of the form of preparation desired for administration.
- Compounds of the present invention may also be coupled with soluble polymers as targetable drug carriers.
- Homogeneous preparation For preparing solid compositions such as tablets, the principal active ingredient is ideally mixed with a pharmaceutical carrier, e.g. conventional tableting ingredients such as corn starch, lactose, sucrose, sorbitol, talc, stearic acid, magnesium stearate, dicalcium phosphate or gums, and other pharmaceutical diluents, e.g. water, to form a solid preformulation composition containing a homogeneous mixture of a compound of the present invention, or a pharmaceutically acceptable salt thereof.
- a pharmaceutical carrier e.g. conventional tableting ingredients such as corn starch, lactose, sucrose, sorbitol, talc, stearic acid, magnesium stearate, dicalcium phosphate or gums, and other pharmaceutical diluents, e.g. water, to form a solid preformulation composition containing a homogeneous mixture of a compound of the present invention, or a pharmaceutically acceptable salt thereof.
- preformulation compositions as homogeneous, it is meant that the active ingredient is ideally dispersed evenly throughout the composition so that the composition may be readily subdivided into equally effective dosage forms such as tablets, pills and capsules.
- This solid preformulation composition may then be subdivided into unit dosage forms of the type described above containing from about 0.1 to about 1000 mg, preferably from about 5 to about 500 mg of the active ingredient of the present invention.
- compositions herein will contain, per dosage unit, e.g., tablet, capsule, powder, injection, suppository, teaspoonful and the like, of from about 0.01 mg to about 1000 mg (preferably about 5 to about 500 mg) and may be given at a dosage of from about 0.1 to about 300 mg/kg bodyweight per day (preferably 1 to 50 mg/kg per day).
- compositions suitable for oral administration include solid forms, such as pills, tablets, caplets, capsules (each including immediate release, timed release and sustained release formulations), granules, and powders.
- suitable carriers and additives may advantageously include starches, sugars, diluents, granulating agents, lubricants, binders, disintegrating agents and the like.
- the active drug component can be combined with an oral, non-toxic pharmaceutically acceptable inert carrier such as ethanol, glycerol, water and the like.
- tablets, pills and capsules represent the most advantageous oral dosage unit form, in which case solid pharmaceutical carriers are employed.
- the tablets, pills or capsules of the novel composition can be advantageously sugar coated or enteric coated by standard techniques or otherwise compounded to provide a dosage form affording the advantage of prolonged action.
- the tablet or pill can comprise an inner dosage and an outer dosage component, the latter being in the form of an envelope over the former.
- the two components can be separated by an enteric layer which serves to resist disintegration in the stomach and permits the inner component to pass intact into the duodenum or to be delayed in release.
- compositions of the present invention can be advantageously incorporated for administration orally or by injection, include aqueous solutions, suitably flavoured syrups, elixirs, aqueous or oil suspensions, and flavoured emulsions with edible oils such as cottonseed oil, sesame oil, coconut oil or peanut oil, as well as elixirs and similar pharmaceutical vehicles.
- Suitable dispersing or suspending agents for aqueous suspensions include synthetic and natural gums such as tragacanth, acacia, alginate, dextran, sodium carboxymethylcellulose, methylcellulose, polyvinylpyrrolidone or gelatin.
- the liquid forms are suitable in flavored suspending or dispersing agents such as the synthetic and natural gums, for example, tragacanth, acacia, methyl-cellulose and the like.
- Isotonic preparations which generally contain suitable preservatives are employed when intravenous administration is desired.
- suitable carriers and additives may advantageously include water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents and the like.
- Forms useful for parenteral administration include sterile solutions, emulsions and suspensions.
- the carrier will usually comprise sterile water, through other ingredients, for example, for purposes such as aiding solubility or for preservation, may be included.
- injectable suspensions may also be prepared, in which case appropriate liquid carriers, suspending agents and the like may be employed.
- sterile suspensions and solutions are desired.
- the pharmaceutical compositions herein will contain, per dosage unit, e.g. solution, suspension, emulsion, injection, teaspoonful and the like, an amount of the active ingredient necessary to deliver an effective dose as described above.
- compositions for intramuscular injection may be presented in a form suitable for once-weekly or once-monthly administration; for example, an insoluble salt of the active compound, such as the decanoate salt, may be adapted to provide a depot preparation for intramuscular injection.
- compounds for the present invention can be administered in intranasal form via topical use of suitable intranasal vehicles, or via transdermal skin patches well known to those of ordinary skill in that art.
- the dosage administration will, of course, be continuous rather than intermittent throughout the dosage regimen and dosage strength will need to be accordingly modified to obtain the desired therapeutic effects.
- the compound of the present invention can also be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles, and multilamellar vesicles.
- Liposomes can be formed from a variety of phospholipids, such as cholesterol, stearylamine or phosphatidylcholines using processes well described in the art.
- Compounds of this invention may be administered in any of the foregoing compositions and according to dosage regimens established in the art whenever treatment of the addressed disorders is required.
- compounds of the present invention may be administered in a single daily dose, or the total daily dosage may be administered in divided doses of two, three or four times daily.
- the daily dosage of the products may be varied over a wide range from 0.01 to 1.000 mg per adult human per day.
- the compositions are preferably provided in the form of tablets containing, 0.01 , 0.05, 0.1 , 0.5, 1 .0, 2.5, 5.0, 10.0, 15.0, 25.0, 50.0, 100, 150, 200, 250, 500 and 1000 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- An effective amount of the drug is ordinarily supplied at a dosage level of from about 0.1 mg/kg to about 300 mg/kg of body weight per day.
- the range is from about 1 to about 50 mg/kg of body weight per day.
- the compounds may be administered on a regimen of 1 to 4 times per day.
- Optimal dosages to be administered may be readily determined by those skilled in the art, and will vary with the particular compound used, the mode of administration, the strength of the preparation, bioavailability due to the mode of administration, and the advancement of disease condition. In addition, factors associated with the particular patient being treated, including patient age, weight, diet and time of administration, should generally be considered in adjusting dosages.
- the dosages may be varied depending upon the requirement of the patients, the severity of the condition being treated and the compound being employed. The use of either daily administration or post-periodic dosing may be employed. Typically the dosage will be regulated by the physician based on the characteristics of the patient, his/her condition and the therapeutic effect desired.
- the compounds or compositions of the present invention may be taken before a meal, while taking a meal or after a meal. When taken before a meal the compounds or composition of the present invention an be taken 1 hour, preferably 30 or even 15 or 5 minutes before eating. When taken while eating, the compounds or compositions of the present invention can be mixed into the meal or taken in a separate dosage form as described above. When taken after a meal, the compounds or compositions of the present invention can be taken 5, 15 or 30 minutes or even 1 hour after finishing a meal.
- the inhibitors according to the present invention will typically possess a K,-value of 100 nM or less, for example 10 nM or less, especially 5 nM or less.
- Compounds of the invention of particular interest possess a K,-value of 1 nM or less, for example 100 pM or less (such as 10 pM).
- the compounds of the invention molecular weights of 1000 Da or less, for example 800 Da or less, in particular 500 Da or less (such as 400 Da or less) e.g. 350Da or less or 300 Da or less.
- the compounds of the invention are reversible non-covalent inhibitors.
- Certain compounds of the present invention typically show a selectivity for DP IV over DPII (i.e. as defined by the ratio of K,-values) which is at least 10 fold, such as at least 100 fold, especially at least 1 ,000 fold.
- Compounds of the present invention which are of particular interest for their selectivity over DPII will typically have a selectivity of at least 10,000 fold, such as at least 20,000, especially at least 30,000 fold (for example at least 40,000 fold or 50,000 fold).
- Certain compounds of the present invention typically show a selectivity for DP IV over DP9 (i.e. as defined by the ratio of K,-values) which is at least 10 fold, such as at least 100 fold, especially at least 1 ,000 fold.
- Compounds of the present invention which are of particular interest for their selectivity over DP9 will typically have a selectivity of at least 10,000 fold, such as at least 20,000, especially at least 30,000 fold (for example at least 40,000 fold or 50,000 fold).
- Preferred compounds of the present invention are not cytotoxic or may have less cytotoxicity than prior art compounds.
- the compounds of the invention are to be used to treat disorders involving the central nervous system
- the compounds will suitably be sufficiently neutral and non-polar such that they can cross the blood-brain barrier via passive diffusion.
- compounds that cannot cross by passive diffusion instead cross by active transport.
- administration approaches also can be employed when treating the central nervous system to avoid adverse interference from the blood-brain barrier.
- the present invention provides specific DP IV-inhibitors which are expected to have efficacy for the prophylaxis and treatment of neurological diseases, especially of the central nervous system (CNS), compared with DP IV-inhibitors of the prior art.
- the compounds of the present invention are believed to be especially useful for the prevention or treatment of a disease selected from anxiety and depression.
- a disease selected from anxiety and depression.
- important class specific disadvantages and side effects have been observed, which are, for instance sedative effects, withdrawal effects and risk for substance abuse in case of benzodiazepines, and slow onset of effect, nausea, restlessness, dizziness, weight loss and insomnia for the other classes. Consequently new treatment concepts with lower side effects and an acute action are needed for treatment of anxiety and panic attacks.
- An important problem of current anti-anxiety or anti-depressive medications is the time until onset of action and the speed of action.
- the sedative drugs work fast - in hours or days - whereas the onset of action of antidepressants and 5-HT1 A acting drugs is delayed - and usually takes weeks or even months.
- Another clinically important problem with current treatments is that the antidepressants and selective serotonin reuptake inhibitors (SSRIs) as well as 5-HT1 A agonist drugs may actually worsen anxiety at the start of therapy.
- SSRIs selective serotonin reuptake inhibitors
- DP IV-inhibitors of the present invention are preferred, which are effective after acute and chronic dosing.
- the present invention provides the use of a compound of the invention as a medicament. Also provided is the use of a compound of the invention in the manufacture of a medicament for the prevention, delay or treatment of a disease listed in section "Diseases" below.
- a method of prevention, delay or treatment of diseases selected from the group consisting of the diseases listed in section "diseases" below, comprising the step of administering a safe and therapeutically effective amount of a compound of the invention to a subject in need thereof.
- Neuronal disorders as well as psychosomatic, neuropsychiatric and depressive illnesses, such as anxiety, depression, sleep disorders, chronic fatigue, schizophrenia, epilepsy, nutritional disorders, spasm and chronic pain.
- the indications above refer each to both acute and chronic form of the disease. It may be expected that the compounds of the invention are highly effective in the treatment of the acute form of the diseases above.
- Metabolic diseases like impaired glucose tolerance, glucosuria, hyperlipidemia, metabolic acidosis, diabetes mellitus, non-insulin dependent diabetes mellitus, diabetic neuropathy and nephropathy and of sequelae caused by diabetes mellitus; high blood pressure and disturbance of signal action at the cells of the islets of Langerhans and insulin sensitivity in the peripheral tissue in the postprandial phase, metabolism-related hypertension and cardiovascular sequelae caused by hypertension in mammals.
- pathological states selected from the group consisting of impaired glucose tolerance (IGT), impaired fasting glucose (IFG) and impaired glucose metabolism (IGM);
- the present invention can be used for treatment and/or prophylaxis of cancer and tumors and the prophylaxis and inhibition of metastasis and tumor colonization including, but not limited to, adenocarcinomas, melanomas, lymphomas, sarcomas, leukemias, and different organ tumors like lung, breast, ovarian, head and/or neck, prostate, cervical, endometrial, colorectal, gastric, liver, fallopian tubes, esophagus, small intestine, pancreas, kidney, adrenal, vaginal, vulvar, brain and testicular tumors.
- Dermal diseases like skin diseases and diseases of the mucosae, for example psoriasis, neurodermitis, acne;
- Immune and autoimmune disorders Immune and autoimmune disorders, multiple sclerosis, and inflammatory conditions, arthritis, obesity, allograft transplantation.
- Neurodegenerative disorders a cognitive disorders and for improving memory (both short term and long term) and learning ability.
- the neurodegenerative disorder is selected from conditions and diseases like dementia (e.g. senile dementia, pre-senile dementia (also known as mild cognitive impairment), Alzheimer related dementia (Alzheimer type dementia)), Huntington's chorea, tardive dyskinesia, hyperkinesias, mania, Morbus Parkinson, steel-Richard syndrome, Down's syndrome, myasthenia gravis, nerve and brain trauma, vascular amyloidosis, cerebral haemorrhage with amyloidosis, brain inflammation, Friedrich's ataxia, acute confusion disorders and especially those in which apoptotic necrocytosis plays a part, such as amyotrophic lateral sclerosis, glaucoma and especially Alzheimer's disease.
- dementia e.g. senile dementia, pre-senile dementia (also known as mild cognitive impairment), Alzheimer related dementia (Alzheimer type dementia)
- Huntington's chorea e.g. senile dementia, pre-senile dementia
- the neurodegenerative disorder is selected from Alzheimer's disease and dementia, preferably senile dementia, mild cognitive impairment or Alzheimer type dementia (for example the neurodegenerative disorder is Alzheimer's disease).
- the cognitive disorder is selected from conditions and diseases like cognitive deficits associated with schizophrenia, age-induced memory impairment, cognitive deficits associated with psychosis, cognitive impairment associated with diabetes, cognitive deficits associated with post-stroke, memory defects associated with hypoxia, cognitive and attention deficits associated with senile dementia, attention-deficit disorders, memory problems associated with mild cognitive impairment, impaired cognitive function associated with dementias, impaired cognitive function associated with Alzheimer's disease, impaired cognitive function associated with Parkinson's disease, impaired cognitive function associated with vascular dementia, cognitive problems associated with brain tumors, Pick's disease, cognitive deficits due to autism, cognitive deficits post electroconvulsive therapy, cognitive deficits associated with traumatic brain injury, amnesic disorders, delirium, dementias.
- schizophrenia age-induced memory impairment
- cognitive deficits associated with psychosis cognitive impairment associated with diabetes
- cognitive deficits associated with post-stroke memory defects associated with hypoxia
- cognitive and attention deficits associated with senile dementia attention-deficit disorders
- memory problems associated with mild cognitive impairment impaired cognitive function associated with dementias
- Cognitive disorder also include, but are not limited to, disorders of learning acquisition (learning disorders), memory consolidation, retrieval memory and retention disorders.
- the cognitive disorder is selected from cognitive impairment associated with diabetes, impaired cognitive function associated with Alzheimer's disease, impaired cognitive function associated with Parkinson's disease, cognitive deficits associated with post-stroke, cognitive and attention deficits associated with senile dementia, memory problems associated with mild cognitive impairment (for example, cognitive impairment associated with diabetes and impaired cognitive function associated with Alzheimer's disease, cognitive deficits associated with post-stroke).
- the compounds of the present invention are useful for the prophylaxis or treatment of psychosomatic, neuropsychiatric and depressive illness, and neurodegenerative diseases such as anxiety, depression, sleep disorders, chronic fatigue, schizophrenia, epilepsy, nutritional disorders, spasm, and chronic pain, and a simple method for the treatment of those disorders.
- the DP IV-inhibitors of the present invention are of particular interest for the prophylaxis or the treatment of neurological diseases, especially for the treatment of a disease selected from anxiety, depression and schizophrenia.
- Anxiety is a vital reaction and experience, which similar to pain, constitutes a biological alarm system to protect a living organism from threats coming from the environment, or alternatively, being represented internally.
- Pathological anxiety is an escalating reaction of the body becoming independent, negatively influencing the decision making process and the development of strategies to accomplish survival. This state differs from normal anxiety through its intensity, duration and inadequate timing to the triggering event and may need treatment, if this anxiety causes a significant disability, which cannot be explained by external cause and which cannot be solved by the patient.
- GAD Generalized anxiety disorder
- Panic disorder PD
- Phobia is the fear of certain objects or situations
- SAD plays a specific role in this later form.
- Secondary anxiety forms can be caused by a series of psychotic or physical disorders like depression, schizophrenia, obsessive-compulsive disorder (OCD), cardiovascular and metabolic diseases, epilepsy as well as posttraumatic stress disorder (PTSD) So far the reasons for pathological anxiety are not fully understood. Besides the individual digestion of events and experiences, a genetical predisposition as well as congenital and acquired changes in the brain are being discussed. A major role is attributed to the hypothalamic-pituitary-adrenal axis. Besides the Corticotropin-Releasing-Hormone (CRH), Neurotransmitters, like gamma-aminobutyric acid (GABA), Serotonin and Noradrenalin, play an important role.
- CRCH Corticotropin-Releasing-Hormone
- GABA gamma-aminobutyric acid
- Serotonin Noradrenalin
- the primary or secondary amine group of R 2 is protected e.g. by Boc.
- This protecting group can be removed at the end of the process e.g. by treatment with HCI.
- the reaction is typically carried out in a polar, protic organic solvent such as an alcohol (e.g. methanol) and may be carried out at elevated temperature.
- a polar, protic organic solvent such as an alcohol (e.g. methanol)
- Exemplary anion stabilising leaving groups L include sulfonic acid derivatives such as - SO 2 X where X represents aryl or alkyl or -alkylaryl e.g. tosyl, mesyl, especially tosyl.
- Novel intermediate compounds are provided as an aspect of the invention.
- compounds of the invention may generally be prepared by the following route:
- an R 2 containing compound of formula (IV), or a protected derivative thereof is mixed with the R 1 containing compound of formula (V), or a protected derivative thereof, in a suitable solvent (such as dry methanol).
- a suitable solvent such as dry methanol.
- the amine function of the compound of formula (IV) will be protected, for example by a Boc protecting group.
- the compound of formula (II), wherein L indicates a leaving group may be added. Reaction is suitably facilitated at elevated temperature (for example around 45 0 C) for a sufficient period of time (for example around
- L can be any suitable leaving group known in organic chemistry.
- L is: a halogen atom, (such as a bromine atom); arylsulfonyl optionally substituted by alkyl (such as toluenesulfonyl); or optionally halogenated alkylsulfonyl (such as mesyl or CF 3 -SO 2 -).
- R 5 can, for example, represent H.
- R 3 and R 4 can, for example, represent Cl.
- Boc-protecting group was cleaved away with an addition of 200 ⁇ l of a 4 M solution of HCI in 1 ,4 Dioxan, 20 ⁇ l H 2 O, and shaking for 3 hours at room temperature. Afterwards the solvent was evaporated.
- IC 50 values were found to be in the range of 1 to 50 ⁇ M when tested directly following synthesis (i.e. without purification). All compounds were detected via HPLC-MS either as mass plus proton ([M+H]) or mass plus sodium ([M+Na]) or both when calculated (CaIc.) and experimentally determined (Det.) masses did not differ by more than 0.1 Dalton.
- Solvent A methanol, 0,3% acetic acid
- Solvent B water, 0,3 % acetic acid Time table for gradient:
- Solvent A acetonitrile,0.5% acetic acid
- Solvent B 90% water, 10% acetonitrile, 0,5 % acetic acid Time table for gradient:
- Substances are screened for inhibition of dipeptidyl peptidase (DP IV) by their ability to inhibit the hydrolysis of the DP IV specific substrate Glycyl-Prolyl-4-Nitroanilide (Gly-Pro- pNA). All compounds are diluted in DMSO and IC 50 will be determined with 1 1 serial dilutions (1 :2) starting with an final concentration of 100 ⁇ M. The final DMSO concentration in the assay is 5 % (v/v). The method was automated using a Tecan Genesis Freedom liquid handler and a Genius Pro (Tecan) microplate reader.
- DP IV from porcine kidney (pDP IV) or recombinant human DP IV (rhDP IV) is diluted in HEPES buffer to a final activity of approximately 40-50 ⁇ U * ml "1 .
- the DP IV is stored in a cooling rack (4 °C) on the liquid handler.
- the assay may becarried out at room temperature using Freedom liquid handler connected with a Genios Pro microplate reader. Each sample is measured in duplicate. Each sample stock solution is prediluted with DMSO in a ratio of 1 :5 in a polypropylen 96- well plate. From this predilution 10 serial (1 :2) dilutions in DMSO are prepared. 13.5 ⁇ l of the 1 1 DMSO dilutions and DMSO alone are transferred to the measurement microplate (duplicates) and 86.5 ⁇ l water, 100 ⁇ l HEPES buffer and 50 ⁇ l substrate solution are added to each well.
- the enzymatic reaction is started by addition of 20 ⁇ l enzyme solution to each well, and then the microplate is immediately transferred to the microplate reader. After short mixing (20 s) the release of 4-Nitroanilin (pNA) is monitored at 405 nm for 30 cycles (approx. 18 min).
- pNA 4-Nitroanilin
- Post assay calculations are done by the Magellan (Tecan) and Graphit Software. The linear slopes are calculated for each well and averaged for the duplicates. The residue activities are calculated in relation to a sample containing only solvent instead of inhibitor and expressed in %. Residue activities are plotted against the log of inhibitor concentrations and IC 50 is calculated by an 4-parameter fitting ranging from 0 to 100 %.
- Measurement of DP IV activities for K determination is carried out as described for IC 50 .
- K 1 determination samples were analysed in triplicates. 7 serial dilutions of the inhibitor (range calculated from the IC 50 determination) in water are analyzed with 4 serial dilutions of the substrate.
- K,-value and inhibition type are determined using the inhibitor equation (competitive, pell competitive, non-competitive) which gives the best fitting results.
- DP9 GIy Pro-AMC is used as substrate in a final concentration of 0.25 mM.
- DPII DPII instead of the HEPES buffer a 0.2 M sodium acetate buffer pH 5.5 is used.
- As substrate Gly-Pro-AMC is used in a final concentration of 1 mM .
- Cytotoxicity of the compounds may be screened using the liver cell carcinoma cell line Hep-G2.
- LDH activity in the cell culture media after 24 h of incubation are analysed.
- the untreated control is set to 0 % cytotoxicity.
- LDH activity of lysed untreated cells are analysed.
- liver carcinoma cell line HEP-G2 and the neuroblastoma cell line SY5Y may be used.
- the effect of 8 serial dilutions of the compounds (highest concentration 1 mM) is investigated as described above.
- cytotoxicity of certain example compounds was screened using Hep-G2 cells. Compounds were tested in concentrations of 100 ⁇ M, 2 ⁇ M and 40 nM.
- the concentration of compounds in the Caco experiments may be determined from their potency to inhibit recombinant human DP IV.
- 100 ⁇ l sample, 100 ⁇ l HEPES buffer and 50 ⁇ l substrate (GIy Pro-pNA, 0.15 mM final concentration, corresponding to 1.5 Km) are mixed. Reaction is started by addition of 20 ⁇ l rhDP IV and release of pNA was monitored at 405 nm. Samples are analyzed in several dilutions (undiluted, 1 :10, 1 :100, 1 :000). Dilutions are done in the Caco assay buffer. Activity measured with Caco-assay buffer alone is set to 100 % and inhibitor concentration is calculated from the dilutions resulting in 20 to 80 % inhibition according to the equation for competitive inhibition.
- GIy Pro-pNA 0.15 mM final concentration, corresponding to 1.5 Km
- V 0 velocity of the reaction without inhibitor (Caco assay buffer alone)
- v, velocity of the reaction with inhibitor sample
- the human colon adenocarcinome cell line Caco-2 are grown in 6- or 24- transwell microplates (Costar) for 21 days with MEM-alpha containing 10 % FBS and 0.5% Gentamycin (Invitrogen). After 21 days of cultivation integrity of the cell layer are assessed by measurement of the transepitelial electrical resistance.
- Transport measurements are carried out in triplicates at a pH of 7.5.
- a-b anterior to basal
- b-a basal to apical
- Samples are collected from the donor and acceptor compartments at the beginning of the experiment and after 60 min (for 6-well plates, Donor at 0 and 120 min, Acceptor at 15, 30, 60 and 120 min). Samples are analyzed in several dilutions for their ability to inhibit human recombinant DP
- IV and compound concentrations may be calculated using the prior determined K,-value (see above).
- the permeability coefficient P app is calculated according to the following equation:
- VA Volume of the acceptor compartment
- CAt concentration in the acceptor compartment at sampling time
- Papp-values >1 e-5 cm/s are assessed as good permeable, value ⁇ 1 e-6 cm/s as not permeable.
- P app -values in between as medium permeable.
- Compounds where no significant difference were found between a-b and b-a transport are assessed to be passive transported.
- P app -values above 1 e-5 cm/s for all compounds indicate a good transport through the cell layer. These determined transport rates are in the same range as the predicted transport rates. Comparable transport rates from apical to basal and from basal to apical indicate that there is a pure passive transport and the compounds are not substrates of P- Glycoprotein (PgP).
- PgP P- Glycoprotein
- the compounds of the invention may be expected to be of use for the prevention or treatment of anxiety or depression.
- the efficacy of the compounds of the invention can be tested in the following in vivo models:
- Test compound C Example 130b as hydrochloride salt
- Example 130b was dissolved in 0.5% Natrosol and administered acutely p.o. (gavage) 40 minutes before behavioural testing.
- Example 130b was administered in doses of 0.3, 1.0 and 3.0 mg/kg in a volume of 5 ml/kg, each.
- Example 130b as hydrochloride salt was dissolved in tap water in concentrations of 0.03,
- solution offer was limited to 40 ml per day for each animal.
- Chlordiazepoxide was dissolved in 0.9 % NaCI and administered acutely i.p. 30 minutes before behavioural testing with a dose of 3 mg/kg in a volume of 1 ml/kg (test group TG 2, see below). The solution was prepared freshly every day.
- Vehicle 0.9 % NaCI (Saline) lmipramine was dissolved 0.5 % Natrosol and administered once daily i.p. for 8 days (test group TG 1 1 , see below) with a dose of 15 mg/kg/day in a volume of 1 ml/kg.
- test group TG 1 In the control group (test group TG 1 , see below) 0.5 % Natrosol was administered acutely p.o. (gavage) 40 minutes before behavioural testing in a volume of 5 ml/kg.
- test group TG 10 see below
- mice 216 male Sprague Dawley rats.
- the animals were 6 weeks old at the start of the study. They were housed in groups of 4 animals ("acute animals") or in single cages ("chronic animals") at 12h/12h light/dark (start of the light phase at 2 a.m.), 20 ⁇ O, 60 % rH. Animals were provided by Elevage Janvier, France.
- mice Depending on the test group ("acute animals” or “chronic animals”), animals were housed in groups of four animals per cage in Makrolon type IV group housing cages ("acute animals”) or in single cages Makrolon type III (“chronic animals”), each with heightened lids (5 cm). During the whole phase, standard diet (Altromin 1324) and tap water were provided ad libitum, except for the period of food restriction prior to the H maze test and for the phase of chronic compound administration (see below). Animals' cages were changed twice weekly. At this time, the animals' weight was recorded on protocol sheets.
- the animals were assigned to the following test groups (TG) by chance.
- Infrared activity monitoring cages TSE Scientific Equipment GmbH, Bad Homburg. Cages are 46 x 46 x 26 cm 3 . Each cage is equipped with 2 infrared frames in 4.5 and 13.5 cm height respectively. 16 beams per axis allow a spatial resolution of 1.4 cm and a temporal resolution of 20 ms
- Animals were kept in group cages until compound administration. Each two animals derived from the same home cage and were treated the same way. During the time from compound administration to the beginning of the test, animals were kept in single cages for 40 minutes.
- the tetradic encounter test was conducted according to the protocol of Wolffgramm (1990). Four rats were placed for 15 minutes in an open field arena of 1 m x 1 m; all four animals of an encounter group were treated in the same way, they received either test compound, reference compound, or vehicle. Each two animals were from the same home cage. The open field was illuminated by dim red light (3-4 Lux). Rats were familiar with the open field and their encounter partners by means of three prior training sessions (no compound administration). Testing (compound administration) started at 2:20 p.m. (dark phase). All animals were marked individually by means of black symbols on their back. Behaviour was recorded on videotapes. Each animal was tested once.
- the reference compound lmipramine was given i.p. (one administration per day, one hour before the respective tests). Fluid supply was restricted to 40 ml per day for all test groups.
- Raw data were provided as videotapes and protocol sheets of application and test time.
- Raw data were provided as videotapes and protocol sheets of application and test time. From the videotapes the social behaviour of each rat was rated by an experienced ethologist using a list of predefined behavioural patterns. The observer was blind to the animal's treatment. The resulting file containing the time series of behavioural patterns was transferred to a computer program for further analysis.
- coefficients of synchrony were calculated that base on rank correlation coefficients.
- the time series of behavioural patterns was subdivided into equidistant interval sections (here: 1 s intervals).
- the user To calculate synchrony coefficients, the user must define "pattern sets of interest". Each set contained one or more behavioural patterns that were taken together for the analysis (e.g. "friendly contact” or "all kinds of social interaction").
- a computerized program that is part of the program system first calculated the relative portion of time for each interval and each set of patterns, then the data was submitted to a Spearman rank transformation (portion parameters are converted to ranks within each time series). The rank numbers were subsequently correlated with each other among the participants of an encounter. If, for instance, the encounter mates were well synchronized according to non-agonistic ("friendly") contacts, there were high contacting ranks concomitantly for all rats in the same intervals and correspondingly, low ranks coincided in the time periods with a low frequency of contacting. A Spearman rank correlation coefficient was calculated for each animal versus the three other ones.
- Synchrony may be extended over more than a single time interval.
- cross-correlation calculations were used. For this purpose, the two time series of the encounter mates (each one containing the rank numbers) were shifted against each other. The temporal dislocation was done stepwise: 1 interval, 2 intervals and so on. In the present study, all participants of an encounter had received the same acute treatment. Therefore, the cross-correlation curves were necessarily symmetric and it was sufficient to shift them only into one direction. In the present study, cross-synchrony coefficients were calculated for different sets of behavioural parameters:
- Defensive behaviour Active rejection of body contact from the partner, withdrawal from the partner, which tries to contact, flight
- Raw data were provided as computer files accompanied by written protocol sheets.
- Meal pattern parameters reflecting e.g. the duration and recurrence of feeding and drinking bouts
- Feeding and drinking bouts and bout-related analysis On the basis of high-resolution infrared beam recordings, feeding and drinking is identified by means of beam interruption in the upper frame near to the positions of food or water. Feeding or drinking acts are then defined as temporally contingent behavioural pattern separated from each other by intervals not longer than 0.5 seconds. Normally, several acts form a feeding or drinking bout. The intervals within a bout are definitely shorter than between subsequent bouts. A threshold interval can be set to distinguish between intra- bout and inter-bout intervals.
- the curve is exponential provided that the end of an interval (the start of the next feeding act) is occurring by chance. If the probability of surviving is shown in a diagram against time and the y-axis (probability) is logarithmic, the curve will be linear. This log survivor plot enables a good assessment whether intervals are really distributed by chance or whether there exist accumulations of short intervals indicating bouts.
- Stereotyped locomotion was assessed by means of statistical comparisons between the observed locomotor sequences (as described by the movement from one of the 4 x 4 subsquares to another) and a "random walk". The comparison is performed at 5 different levels:
- Level 1 Number of transitions from one subsquare to a specified other one
- Level 2 Number of 2nd order transitions (second order: S1 ⁇ S2 ⁇ S3)
- Level 3 Number of 3rd order transitions (third order: S1 ⁇ S2 ⁇ S3 ⁇ S4)
- Level 4 Number of 4th order transitions (fourth order: S1 -» S2 ⁇ S3 -» S4 ⁇ S5 )
- the maximum value of SC 100% indicates that the animal always uses the same pathway.
- Protocol sheets were entered into a computer program. In case of missing or obviously wrong measurements (e.g. empty drinking bottles due to activities of the rat) missing values were introduced (less than 0.1 % of the data). Missing values were interpolated by the program. After verification of the data, relevant parameters were calculated. The resulting time courses were the basis for subsequent statistical analysis.
- GLM general linear model
- GLM general linear model
- a GLM general linear model
- Chlordiazepoxide affected inter-individual synchrony of behaviour. In all analyses, a statistically significant interaction of treatment and displacement appeared compared with vehicle-treated animals. Chlordiazepoxide tended to reduce immediate inter-individual synchrony of behaviour, especially concerning aggressive behaviour, whereas it also prolonged synchrony. Effects of chronic administration of lmipramine
- Body weight, food and fluid consumption lmipramine significantly reduced body weight gain, food and fluid intake during the period of administration.
- Example 130b led to a statistically significant increase of the number of defensive acts and of the distance travelled during the first five minutes of the test compared with vehicle-treated animals.
- the high dose of Example 130b significantly increased the time spent in the centre of the open field and the usage of available space (Fig. 1 to 4).
- the medium dose tended to increase the time spent with friendly (non- agonistic) social behaviour.
- Example 130b led to a significantly higher distance to the encounter mates compared with vehicle treatment. This effect was also seen in the distribution of spatial distances to the encounter mates, resulting in a significantly lower amount of time spent in a distance of less than 25 cm (high dose) and a distance of 25-50 cm (low dose) and a higher amount in a distance of 50-75 cm (high dose), 75-100 cm (low and medium dose) and more than 100 cm (low dose, Fig. 5). The effect was also seen in the cross-distance analyses (Fig. 6).
- Example 130b revealed strong effects on the inter-individual synchrony of behaviour. Statistical analysis yielded a significant interaction between treatment and displacement in the synchrony analyses including all behavioural types, social behaviour, and friendly (non-agonistic) social behaviour and an effect of treatment on the synchrony of aggressive behaviour. Especially the high dose of Example 130b increased synchrony, with the strongest effect on aggressive behaviour (Fig. 7 to 10).
- Example 130b led to a statistically significant effect of treatment and an interaction between time and treatment on fluid intake compared with vehicle controls whereas body weight and food intake were not affected.
- Example 130b dose-dependently led to a reduction of fluid intake with the highest dose having the strongest effect. Fluid intake remained slightly lower during ongoing treatment (Fig. 1 1 , 12).
- Example 130b dose ranged between 2.8 - 3.1 mg/kg/day (low dose), 9.1 - 9.9 mg/kg/day (medium dose) and 26.0 - 29.0 mg/kg/day (high dose, Fig. 12).
- Example 130b led to a significantly slight decrease of fluid intake, measured during the last day of circadian recording compared with vehicle-treated animals. Body weight and food intake were not affected.
- Example 130b There were no statistically significant effects of Example 130b on horizontal or vertical locomotor activity compared with vehicle-treated animals. There were no statistically significant effects of Example 130b on usage of available space or stereotypy index. Access times to food and fluid did not differ from vehicle-treated animals whereas there was a statistically significant time x treatment interaction effect on the cumulated number of drinking bouts compared with vehicle treated animals. Example 130b-treated animals dose-dependently reached a lower total number of drinking bouts during the 22 hours of measurement (Fig. 13 to 23).
- Chlordiazepoxide had no effects on locomotor activity and led to weak effects on behavioural patterns. It decreased defensive and increased non-agonistic social behaviour. There were indications of a weakly increasing effect on the distance between the encounter mates. Chlordiazepoxide had clear effects on inter-individual synchrony of behaviour in reducing immediate and prolonging synchronisation especially for aggressive behaviour. This course indicates a unidirectional way of interaction (no mutual stimulation).
- Example 130b altered locomotor activity.
- the low and high dose increased the activity at the beginning of the tests. Only the high dose increased the time spent in the centre of the open field and the use of available space. Low and high dose increased the distance to encounter mates.
- Example 130b had strong effects on inter-individual synchrony of behaviour. Effects were strongest for the high dose and to a lower degree visible for the low dose.
- Example 130b increased immediate synchrony in all cases. The effects were strongest for aggressive behaviour indicating bidirectional (mutual) inter-individual interactions.
- Body weight, food and fluid consumption during chronic compound administration lmipramine reduced body weight gain, food and fluid consumption throughout the time of administration compared with vehicle-treated animals.
- Example 130b dose-dependently led to a reduction in fluid intake especially during the first days of administration compared with vehicle-treated animals. The effect remained visible for the whole period of treatment. It was most probably due to the fact that the animals had to adapt to the changed taste of their drinking solution, resulting in a strong reduction of fluid intake at the first day of administration that did not completely recover. Food intake and body weight were not affected.
- Circadian courses of activity, feeding and drinking behaviour lmipramine had strong effects on almost all parameters of the circadian registration compared with vehicle-treated animals. It clearly reduced horizontal and vertical activity, feeding and drinking especially during the first and the last three hours of the dark phase. It also reduced total food and fluid intake.
- Example 130b had no effects on time courses of activity and feeding compared with vehicle-treated animals. While the reduction of fluid intake did not lead to differences in time course of fluid access, it could be seen in a dose dependent reduction of cumulated drinking bouts.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Immunology (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Rheumatology (AREA)
- Anesthesiology (AREA)
- Nutrition Science (AREA)
- Endocrinology (AREA)
- Transplantation (AREA)
- Hospice & Palliative Care (AREA)
- Emergency Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description
















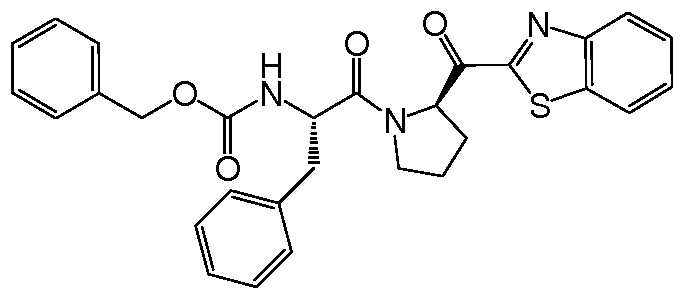








































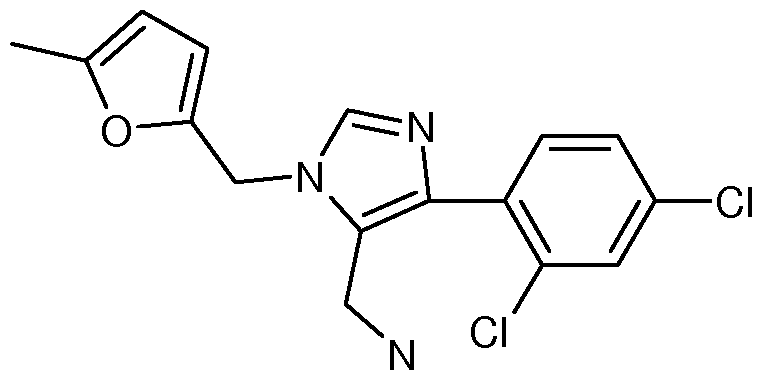





















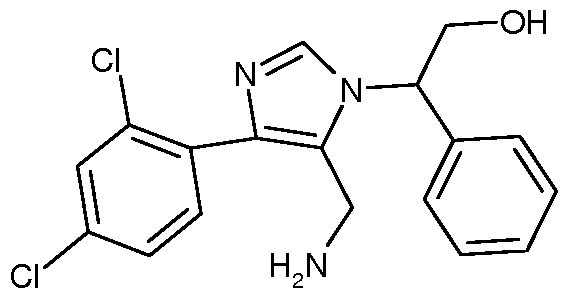













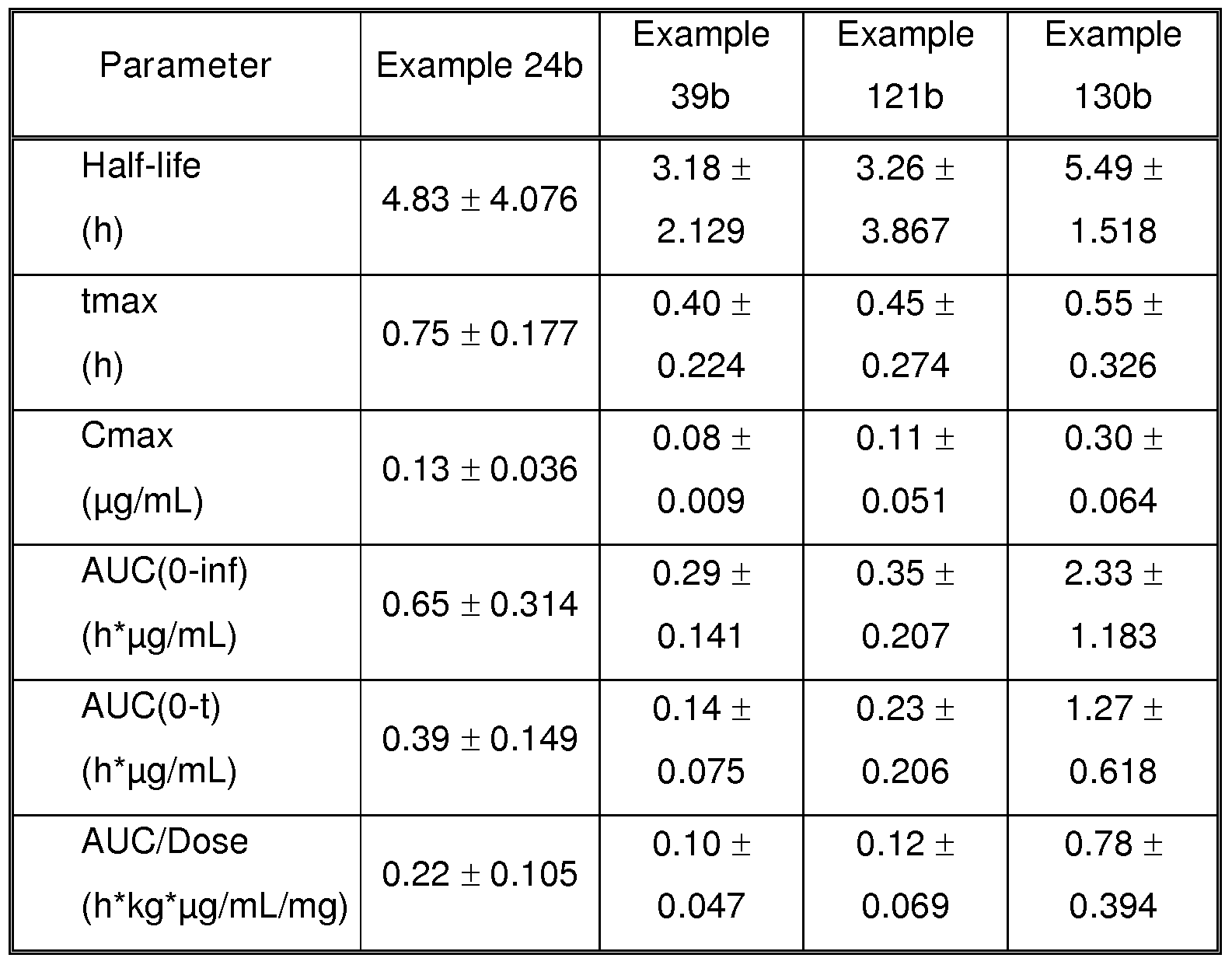



Claims


Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07728027A EP2007734A1 (en) | 2006-04-12 | 2007-04-12 | Enzyme inhibitors |
| JP2009504750A JP2009533393A (en) | 2006-04-12 | 2007-04-12 | Enzyme inhibitor |
| NZ571761A NZ571761A (en) | 2006-04-12 | 2007-04-12 | 5-phenylimidazoles |
| EA200802054A EA200802054A1 (en) | 2006-04-12 | 2007-04-12 | ENZYME INHIBITORS |
| MX2008013130A MX2008013130A (en) | 2006-04-12 | 2007-04-12 | Enzyme inhibitors. |
| AU2007235876A AU2007235876A1 (en) | 2006-04-12 | 2007-04-12 | Enzyme inhibitors |
| CA002649209A CA2649209A1 (en) | 2006-04-12 | 2007-04-12 | Enzyme inhibitors |
| BRPI0709984-3A BRPI0709984A2 (en) | 2006-04-12 | 2007-04-12 | enzyme inhibitors |
| IL194339A IL194339A0 (en) | 2006-04-12 | 2008-09-25 | Enzyme inhibitors |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US79130206P | 2006-04-12 | 2006-04-12 | |
| US60/791,302 | 2006-04-12 | ||
| US87016506P | 2006-12-15 | 2006-12-15 | |
| US60/870,165 | 2006-12-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007116092A1 true WO2007116092A1 (en) | 2007-10-18 |
Family
ID=38256669
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/053560 WO2007116092A1 (en) | 2006-04-12 | 2007-04-12 | Enzyme inhibitors |
Country Status (12)
| Country | Link |
|---|---|
| US (2) | US7728146B2 (en) |
| EP (1) | EP2007734A1 (en) |
| JP (1) | JP2009533393A (en) |
| KR (1) | KR20090004950A (en) |
| AU (1) | AU2007235876A1 (en) |
| BR (1) | BRPI0709984A2 (en) |
| CA (1) | CA2649209A1 (en) |
| EA (1) | EA200802054A1 (en) |
| IL (1) | IL194339A0 (en) |
| MX (1) | MX2008013130A (en) |
| NZ (1) | NZ571761A (en) |
| WO (1) | WO2007116092A1 (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| RU2614737C1 (en) * | 2016-04-12 | 2017-03-28 | Общество с ограниченной ответственностью "Нормофарм" | Method for memory improvement |
| WO2017164173A1 (en) * | 2016-03-22 | 2017-09-28 | 国立研究開発法人科学技術振興機構 | Imidazole compound and medicine comprising same |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112933073A (en) | 2013-01-22 | 2021-06-11 | 维斯塔津治疗公司 | Dosage forms and therapeutic uses of L-4-chlorokynurenine |
| EA201792571A1 (en) * | 2015-05-22 | 2018-06-29 | Вистаджен Терапьютикс, Инк. | THERAPEUTIC APPLICATION OF L-4-CHLOROKINURENIN |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996040143A1 (en) | 1995-06-07 | 1996-12-19 | Smithkline Beecham Corporation | Imidazole compounds |
| WO2004018460A1 (en) | 2002-08-21 | 2004-03-04 | Neurogen Corporation | Amino methyl imidazoles as c5a receptor modulators |
| WO2004098591A2 (en) | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Inhibitors of glutaminyl cyclase and their use in the treatment of neurological diseases |
| WO2005079795A2 (en) | 2004-02-20 | 2005-09-01 | Novartis Ag | Dpp-iv inhibitors for treating neurodegeneration and cognitive disorders |
Family Cites Families (104)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5668136A (en) * | 1990-09-25 | 1997-09-16 | Eisai Co., Ltd. | Trisubstituted benzene derivatives, composition and methods of treatment |
| IL111785A0 (en) | 1993-12-03 | 1995-01-24 | Ferring Bv | Dp-iv inhibitors and pharmaceutical compositions containing them |
| DE122010000020I1 (en) | 1996-04-25 | 2010-07-08 | Prosidion Ltd | Method for lowering the blood glucose level in mammals |
| TW492957B (en) | 1996-11-07 | 2002-07-01 | Novartis Ag | N-substituted 2-cyanopyrrolidnes |
| US6011155A (en) | 1996-11-07 | 2000-01-04 | Novartis Ag | N-(substituted glycyl)-2-cyanopyrrolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| AU766219B2 (en) | 1998-02-02 | 2003-10-09 | 1149336 Ontario Inc. | Method of regulating glucose metabolism, and reagents related thereto |
| JP2002506075A (en) | 1998-03-09 | 2002-02-26 | フォンダテッヒ・ベネルクス・ナムローゼ・フェンノートシャップ | Serine peptidase modulator |
| DE19823831A1 (en) | 1998-05-28 | 1999-12-02 | Probiodrug Ges Fuer Arzneim | New pharmaceutical use of isoleucyl thiazolidide and its salts |
| DE19827387A1 (en) | 1998-06-19 | 1999-12-23 | Bayer Ag | Use of ligands of the high affinity binding site of diisopropyl-fluorophosphate in treatment of central nervous system disorders |
| DE19828113A1 (en) | 1998-06-24 | 2000-01-05 | Probiodrug Ges Fuer Arzneim | Prodrugs of Dipeptidyl Peptidase IV Inhibitors |
| CN1185259C (en) | 1998-06-24 | 2005-01-19 | 阿温蒂斯药物公司 | Fluorophenyl resin compounds |
| DE19828114A1 (en) | 1998-06-24 | 2000-01-27 | Probiodrug Ges Fuer Arzneim | Produgs of unstable inhibitors of dipeptidyl peptidase IV |
| AU5027299A (en) | 1998-07-31 | 2000-02-28 | Novo Nordisk A/S | Use of glp-1 and analogues for preventing type ii diabetes |
| DE19834591A1 (en) | 1998-07-31 | 2000-02-03 | Probiodrug Ges Fuer Arzneim | Use of substances that decrease the activity of dipeptidyl peptidase IV to increase blood sugar levels, e.g. for treating hypoglycemia |
| US6110949A (en) | 1999-06-24 | 2000-08-29 | Novartis Ag | N-(substituted glycyl)-4-cyanothiazolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| US6107317A (en) | 1999-06-24 | 2000-08-22 | Novartis Ag | N-(substituted glycyl)-thiazolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| DE60029230T2 (en) | 1999-09-10 | 2007-06-06 | The University Of Sydney, Sydney | Dipeptidyl peptidase |
| WO2001034594A1 (en) | 1999-11-12 | 2001-05-17 | Guilford Pharmaceuticals, Inc. | Dipeptidyl peptidase iv inhibitors and methods of making and using dipeptidyl peptidase iv inhibitors |
| GB9928330D0 (en) | 1999-11-30 | 2000-01-26 | Ferring Bv | Novel antidiabetic agents |
| EP1254113A1 (en) | 2000-01-24 | 2002-11-06 | Novo Nordisk A/S | N-substituted 2-cyanopyroles and -pyrrolines which are inhibitors of the enzyme dpp-iv |
| US6395767B2 (en) | 2000-03-10 | 2002-05-28 | Bristol-Myers Squibb Company | Cyclopropyl-fused pyrrolidine-based inhibitors of dipeptidyl peptidase IV and method |
| GB0010183D0 (en) | 2000-04-26 | 2000-06-14 | Ferring Bv | Inhibitors of dipeptidyl peptidase IV |
| GB0010188D0 (en) | 2000-04-26 | 2000-06-14 | Ferring Bv | Inhibitors of dipeptidyl peptidase IV |
| MXPA02012272A (en) | 2000-07-04 | 2003-04-25 | Novo Nordisk As | Heterocyclic compounds, which are inhibitors of the enzyme dpp-iv. |
| AU2001269123A1 (en) | 2000-07-10 | 2002-01-21 | Bayer Aktiengesellschaft | Regulation of human dipeptidyl-peptidase iv-like enzyme |
| CA2425001A1 (en) | 2000-10-12 | 2002-04-18 | Ferring Bv | Novel serine protease genes related to dppiv |
| CN101143217A (en) | 2000-10-27 | 2008-03-19 | 前体生物药物股份有限公司 | Treatment of neurological and neuropsychological disorders |
| AUPR107800A0 (en) | 2000-10-27 | 2000-11-23 | University Of Sydney, The | Peptide and nucleic acid molecule ii |
| TWI243162B (en) | 2000-11-10 | 2005-11-11 | Taisho Pharmaceutical Co Ltd | Cyanopyrrolidine derivatives |
| US6573287B2 (en) | 2001-04-12 | 2003-06-03 | Bristo-Myers Squibb Company | 2,1-oxazoline and 1,2-pyrazoline-based inhibitors of dipeptidyl peptidase IV and method |
| FR2824825B1 (en) | 2001-05-15 | 2005-05-06 | Servier Lab | NOVEL ALPHA-AMINOACID DERIVATIVES, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| JP2005500308A (en) | 2001-06-20 | 2005-01-06 | メルク エンド カムパニー インコーポレーテッド | Dipeptidyl peptidase inhibitors for the treatment of diabetes |
| CA2450579A1 (en) | 2001-06-20 | 2003-01-03 | Merck & Co., Inc. | Dipeptidyl peptidase inhibitors for the treatment of diabetes |
| GB0115517D0 (en) | 2001-06-25 | 2001-08-15 | Ferring Bv | Novel antidiabetic agents |
| DE10150203A1 (en) | 2001-10-12 | 2003-04-17 | Probiodrug Ag | Use of dipeptidyl peptidase IV inhibitor in treatment of cancer |
| IL158923A0 (en) | 2001-06-27 | 2004-05-12 | Smithkline Beecham Corp | Fluoropyrrolidines as dipeptidyl peptidase inhibitors |
| DE10154689A1 (en) | 2001-11-09 | 2003-05-22 | Probiodrug Ag | Substituted amino ketone compounds |
| CA2419888A1 (en) | 2001-06-27 | 2003-01-09 | Probiodrug Ag | Peptide structures useful for competitive modulation of dipeptidyl peptidase iv catalysis |
| JP4300108B2 (en) | 2001-06-27 | 2009-07-22 | スミスクライン ビーチャム コーポレーション | Pyrrolidines as dipeptidyl peptidase inhibitors |
| ATE370943T1 (en) | 2001-06-27 | 2007-09-15 | Smithkline Beecham Corp | FLUOROPYRROLIDINE AS DIPEPTIDYL-PEPTIDASE INHIBITORS |
| EP1404675B1 (en) | 2001-07-03 | 2008-03-12 | Novo Nordisk A/S | Dpp-iv-inhibiting purine derivatives for the treatment of diabetes |
| TWI246510B (en) | 2001-09-14 | 2006-01-01 | Mitsubishi Pharma Corp | Thiazolidine derivatives and pharmaceutical uses thereof |
| WO2003024965A2 (en) | 2001-09-19 | 2003-03-27 | Novo Nordisk A/S | Heterocyclic compounds that are inhibitors of the enzyme dpp-iv |
| GB0125445D0 (en) | 2001-10-23 | 2001-12-12 | Ferring Bv | Protease Inhibitors |
| GB0125446D0 (en) | 2001-10-23 | 2001-12-12 | Ferring Bv | Novel anti-diabetic agents |
| US6861440B2 (en) | 2001-10-26 | 2005-03-01 | Hoffmann-La Roche Inc. | DPP IV inhibitors |
| JP4771661B2 (en) | 2001-11-26 | 2011-09-14 | トラスティーズ オブ タフツ カレッジ | Post-proline cleavage enzyme pseudopeptide inhibitor |
| AU2002360732A1 (en) | 2001-12-26 | 2003-07-24 | Guilford Pharmaceuticals | Change inhibitors of dipeptidyl peptidase iv |
| US6727261B2 (en) | 2001-12-27 | 2004-04-27 | Hoffman-La Roche Inc. | Pyrido[2,1-A]Isoquinoline derivatives |
| WO2004004498A2 (en) | 2002-07-05 | 2004-01-15 | Bernd Elbert | Watertight leg garment |
| KR101072236B1 (en) | 2002-09-19 | 2011-10-12 | 아보트 러보러터리즈 | -- Pharmaceutical compositions as inhibitors of dipeptidyl peptidase-IVDPP-IV |
| AU2003269850A1 (en) | 2002-10-08 | 2004-05-04 | Novo Nordisk A/S | Hemisuccinate salts of heterocyclic dpp-iv inhibitors |
| JP4352001B2 (en) | 2002-10-18 | 2009-10-28 | メルク エンド カムパニー インコーポレーテッド | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| RU2005117383A (en) | 2002-11-07 | 2006-01-20 | Мерк энд Ко., Инк. (US) | Phenylalanine derivatives as dipepididyl peptidase inhibitors for the treatment or prevention of diabetes |
| AU2002952946A0 (en) | 2002-11-27 | 2002-12-12 | Fujisawa Pharmaceutical Co., Ltd. | Dpp-iv inhibitor |
| CA2508487A1 (en) | 2002-12-04 | 2004-06-17 | Merck & Co., Inc. | Phenylalanine derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7420079B2 (en) | 2002-12-09 | 2008-09-02 | Bristol-Myers Squibb Company | Methods and compounds for producing dipeptidyl peptidase IV inhibitors and intermediates thereof |
| JP4504924B2 (en) | 2002-12-20 | 2010-07-14 | メルク・シャープ・エンド・ドーム・コーポレイション | 3-Amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment and prevention of diabetes |
| US7265128B2 (en) | 2003-01-17 | 2007-09-04 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| US7388019B2 (en) | 2003-01-31 | 2008-06-17 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| WO2004071454A2 (en) | 2003-02-13 | 2004-08-26 | Guilford Pharmaceuticals Inc. | Substituted azetidine compounds as inhibitors of dipeptidyl peptidase iv |
| WO2004076434A1 (en) | 2003-02-28 | 2004-09-10 | Aic | Dipeptidyl peptidase inhibitors |
| AU2003207881A1 (en) | 2003-02-28 | 2004-09-17 | Aic | Dipeptidyl peptidase inhibitors |
| CN1894234A (en) | 2003-03-25 | 2007-01-10 | 武田药品工业株式会社 | Dipeptidyl peptidase inhibitors |
| WO2004089362A1 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | 2-cyanopyrroles and their analogues as ddp-iv inhibitors |
| AU2003902260A0 (en) | 2003-05-09 | 2003-05-29 | Fujisawa Pharmaceutical Co., Ltd. | Dpp-iv inhibitor |
| CA2524531A1 (en) | 2003-05-14 | 2004-12-02 | Merck And Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| EP1625122A1 (en) | 2003-05-14 | 2006-02-15 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| AU2003902828A0 (en) | 2003-06-05 | 2003-06-26 | Fujisawa Pharmaceutical Co., Ltd. | Dpp-iv inhibitor |
| CA2526770A1 (en) | 2003-06-06 | 2004-12-23 | Merck & Co., Inc. | Fused indoles as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| AU2003902946A0 (en) | 2003-06-12 | 2003-06-26 | Fujisawa Pharmaceutical Co., Ltd. | Dpp-iv inhibitor |
| CA2527806A1 (en) | 2003-06-17 | 2004-12-29 | Merck & Co., Inc. | Cyclohexylglycine derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| MXPA05013734A (en) | 2003-06-20 | 2006-03-08 | Hoffmann La Roche | Hexahydropyridoisoqinolines as dpp-iv inhibitors. |
| DE602004030244D1 (en) | 2003-06-20 | 2011-01-05 | Hoffmann La Roche | ITOREN |
| DE602004018503D1 (en) | 2003-07-31 | 2009-01-29 | Merck & Co Inc | HEXAHYDRODIAZEPINONE AS INHIBITORS OF DIPEPTIDYLPEPTIDASE-IV FOR TREATMENT OR PREVENTION OF DIABETES |
| MXPA06001601A (en) | 2003-08-13 | 2006-08-25 | Takeda Pharmaceutical | 4-pyrimidone derivatives and their use as peptidyl peptidase inhibitors. |
| WO2005023762A1 (en) | 2003-09-04 | 2005-03-17 | Abbott Laboratories | Pyrrolidine-2-carbonitrile derivatives and their use as inhibitors of dipeptidyl peptidase-iv (dpp-iv) |
| WO2005026148A1 (en) | 2003-09-08 | 2005-03-24 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| US20050065144A1 (en) | 2003-09-08 | 2005-03-24 | Syrrx, Inc. | Dipeptidyl peptidase inhibitors |
| WO2005025554A2 (en) | 2003-09-09 | 2005-03-24 | Japan Tobacco Inc. | Dipeptidyl peptidase iv inhibitor |
| GB0324236D0 (en) | 2003-10-16 | 2003-11-19 | Astrazeneca Ab | Chemical compounds |
| TW200519105A (en) | 2003-10-20 | 2005-06-16 | Lg Life Science Ltd | Novel inhibitors of DPP-IV, methods of preparing the same, and pharmaceutical compositions containing the same as an active agent |
| US7238683B2 (en) | 2003-11-04 | 2007-07-03 | Merck & Co., Inc. | Fused phenylalanine derivatives as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| EP1997533B8 (en) | 2003-11-12 | 2014-10-29 | Sino-Med International Alliance, Inc. | Heterocyclic boronic acid compounds, dipeptidyl peptidase IV inhibitors |
| DE10355304A1 (en) | 2003-11-27 | 2005-06-23 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Novel 8- (piperazin-1-yl) and 8 - ([1,4] diazepan-1-yl) xanthines, their preparation and their use as pharmaceuticals |
| EP1541148A1 (en) | 2003-12-09 | 2005-06-15 | Graffinity Pharmaceuticals Aktiengesellschaft | Dpp-iv inhibitors |
| EP1541143A1 (en) | 2003-12-09 | 2005-06-15 | Graffinity Pharmaceuticals Aktiengesellschaft | Dpp-iv inhibitors |
| WO2005058849A1 (en) | 2003-12-15 | 2005-06-30 | Glenmark Pharmaceuticals Ltd. | New dipeptidyl peptidase in inhibitors; process for their preparation and compositions containing them |
| WO2005075426A1 (en) | 2004-02-03 | 2005-08-18 | Glenmark Pharmaceuticals Ltd. | Novel dipeptidyl peptidase iv inhibitors; processes for their preparation and compositions thereof |
| PT1758905E (en) | 2004-02-18 | 2009-07-16 | Boehringer Ingelheim Int | 8-[3-amino-piperidin-1-yl]-xanthine, the production thereof and the use in the form of a ddp-iv inhibitor |
| BRPI0507972A (en) | 2004-02-23 | 2007-07-24 | Tufts College | compound, pharmaceutical composition, use of a compound, method for inhibiting the proteolytic activity of a postproline cleavage enzyme and packaged pharmaceutical composition |
| EP1593671A1 (en) | 2004-03-05 | 2005-11-09 | Graffinity Pharmaceuticals AG | DPP-IV inhibitors |
| CN1942186B (en) | 2004-03-09 | 2010-10-06 | 国家卫生研究院 | Pyrrolidine compounds |
| CN102079743B (en) | 2004-03-15 | 2020-08-25 | 武田药品工业株式会社 | Dipeptidyl peptidase inhibitors |
| WO2005095339A1 (en) | 2004-03-31 | 2005-10-13 | Pfizer Products Inc. | Dicyanopyrrolidines as dipeptidyl peptidase iv inhibitors |
| WO2005108382A1 (en) | 2004-05-04 | 2005-11-17 | Merck & Co., Inc. | 1,2,4-oxadiazole derivatives as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| GEP20084421B (en) | 2004-05-12 | 2008-07-10 | Pfizer Prod Inc | Proline derivatives and their use as dipeptidyl peptidase iv inhibitors |
| WO2005116029A1 (en) | 2004-05-18 | 2005-12-08 | Merck & Co., Inc. | Cyclohexylalanine derivatives as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| EP1598341A1 (en) | 2004-05-21 | 2005-11-23 | Santhera Pharmaceuticals (Deutschland) Aktiengesellschaft | DPP-IV inhibitors |
| WO2005118555A1 (en) | 2004-06-04 | 2005-12-15 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| EP1604989A1 (en) | 2004-06-08 | 2005-12-14 | Santhera Pharmaceuticals (Deutschland) Aktiengesellschaft | DPP-IV inhibitors |
| EP1604980A1 (en) | 2004-06-08 | 2005-12-14 | Santhera Pharmaceuticals (Deutschland) Aktiengesellschaft | DPP-IV inhibitors |
| EP1604662A1 (en) | 2004-06-08 | 2005-12-14 | Santhera Pharmaceuticals (Deutschland) Aktiengesellschaft | 1-[(3R)-Amino-4-(2-fluoro-phenyl)-butyl]-pyrrolidine-(2R)-carboxylic acid benzyl amine derivatives and related compounds as dipeptidyl peptidase IV (DPP-IV) inhibitors for the treatment of type 2 diabetes mellitus |
| GB0413389D0 (en) | 2004-06-16 | 2004-07-21 | Astrazeneca Ab | Chemical compounds |
-
2007
- 2007-04-12 JP JP2009504750A patent/JP2009533393A/en not_active Abandoned
- 2007-04-12 EA EA200802054A patent/EA200802054A1/en unknown
- 2007-04-12 AU AU2007235876A patent/AU2007235876A1/en not_active Abandoned
- 2007-04-12 KR KR1020087024915A patent/KR20090004950A/en not_active Application Discontinuation
- 2007-04-12 MX MX2008013130A patent/MX2008013130A/en not_active Application Discontinuation
- 2007-04-12 US US11/734,369 patent/US7728146B2/en not_active Expired - Fee Related
- 2007-04-12 BR BRPI0709984-3A patent/BRPI0709984A2/en not_active IP Right Cessation
- 2007-04-12 CA CA002649209A patent/CA2649209A1/en not_active Abandoned
- 2007-04-12 EP EP07728027A patent/EP2007734A1/en not_active Withdrawn
- 2007-04-12 NZ NZ571761A patent/NZ571761A/en unknown
- 2007-04-12 WO PCT/EP2007/053560 patent/WO2007116092A1/en active Application Filing
-
2008
- 2008-09-25 IL IL194339A patent/IL194339A0/en unknown
-
2010
- 2010-03-31 US US12/750,930 patent/US20100234397A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996040143A1 (en) | 1995-06-07 | 1996-12-19 | Smithkline Beecham Corporation | Imidazole compounds |
| WO2004018460A1 (en) | 2002-08-21 | 2004-03-04 | Neurogen Corporation | Amino methyl imidazoles as c5a receptor modulators |
| WO2004098591A2 (en) | 2003-05-05 | 2004-11-18 | Probiodrug Ag | Inhibitors of glutaminyl cyclase and their use in the treatment of neurological diseases |
| WO2005079795A2 (en) | 2004-02-20 | 2005-09-01 | Novartis Ag | Dpp-iv inhibitors for treating neurodegeneration and cognitive disorders |
Non-Patent Citations (2)
| Title |
|---|
| LANKAS GR ET AL., DIABETES, vol. 54, no. 10, 2005, pages 2988 - 2994 |
| XU J, BIOORG MED CHEM LETT, vol. 16, no. 5, 1 March 2006 (2006-03-01), pages 1346 - 9 |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| WO2017164173A1 (en) * | 2016-03-22 | 2017-09-28 | 国立研究開発法人科学技術振興機構 | Imidazole compound and medicine comprising same |
| RU2614737C1 (en) * | 2016-04-12 | 2017-03-28 | Общество с ограниченной ответственностью "Нормофарм" | Method for memory improvement |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2007734A1 (en) | 2008-12-31 |
| KR20090004950A (en) | 2009-01-12 |
| NZ571761A (en) | 2010-07-30 |
| EA200802054A1 (en) | 2009-04-28 |
| AU2007235876A1 (en) | 2007-10-18 |
| IL194339A0 (en) | 2009-08-03 |
| BRPI0709984A2 (en) | 2011-08-02 |
| US20070244177A1 (en) | 2007-10-18 |
| US7728146B2 (en) | 2010-06-01 |
| US20100234397A1 (en) | 2010-09-16 |
| MX2008013130A (en) | 2008-11-19 |
| CA2649209A1 (en) | 2007-10-18 |
| JP2009533393A (en) | 2009-09-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7728146B2 (en) | Enzyme inhibitors | |
| EP2091945B1 (en) | Novel inhibitors of glutaminyl cyclase | |
| EP2086960B1 (en) | Novel inhibitors of glutaminyl cyclase | |
| EP2089383B1 (en) | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases | |
| JP5891278B2 (en) | Thiourea derivatives as glutaminyl cyclase inhibitors | |
| CN102186475B (en) | novel inhibitors | |
| JP5676249B2 (en) | Aminopyridine derivatives as glutaminyl cyclase inhibitors | |
| EP2142513B1 (en) | Nitrovinyl-diamine derivatives as glutaminyl cyclase inhibitors | |
| CN102695546B (en) | Heterocylcic derivatives as inhibitors of glutaminyl cyclase | |
| US8420684B2 (en) | Inhibitors of glutaminyl cyclase | |
| EP2160380A1 (en) | Cyano-guanidine derivatives as glutaminyl cyclase inhibitors | |
| EP2146968A1 (en) | Urea derivatives as glutaminyl cyclase inhibitors | |
| EP2142515A1 (en) | Novel inhibitors | |
| AZ et al. | substituted by one or more groups selected from alkyl, hydroxy and oxo;-heteroaryl; or-hydroxyalkylaryl; R represents H or C,. alkyl; R rcpresents H or C., alkyl; and X rcprescnts O or S. | |
| Niestroj | i, Patent Application Publication to, Pub. No.: US 2009/0269301 A1 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780021894.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07728027 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007728027 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 194339 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 571761 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/013130 Country of ref document: MX Ref document number: 2009504750 Country of ref document: JP Ref document number: 2007235876 Country of ref document: AU Ref document number: 2649209 Country of ref document: CA Ref document number: 4117/KOLNP/2008 Country of ref document: IN Ref document number: 1020087024915 Country of ref document: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2007235876 Country of ref document: AU Date of ref document: 20070412 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200802054 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: PI0709984 Country of ref document: BR Kind code of ref document: A2 Effective date: 20081010 |