WO2007092751A2 - Compounds and methods for modulating fx-receptors - Google Patents
Compounds and methods for modulating fx-receptors Download PDFInfo
- Publication number
- WO2007092751A2 WO2007092751A2 PCT/US2007/061515 US2007061515W WO2007092751A2 WO 2007092751 A2 WO2007092751 A2 WO 2007092751A2 US 2007061515 W US2007061515 W US 2007061515W WO 2007092751 A2 WO2007092751 A2 WO 2007092751A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
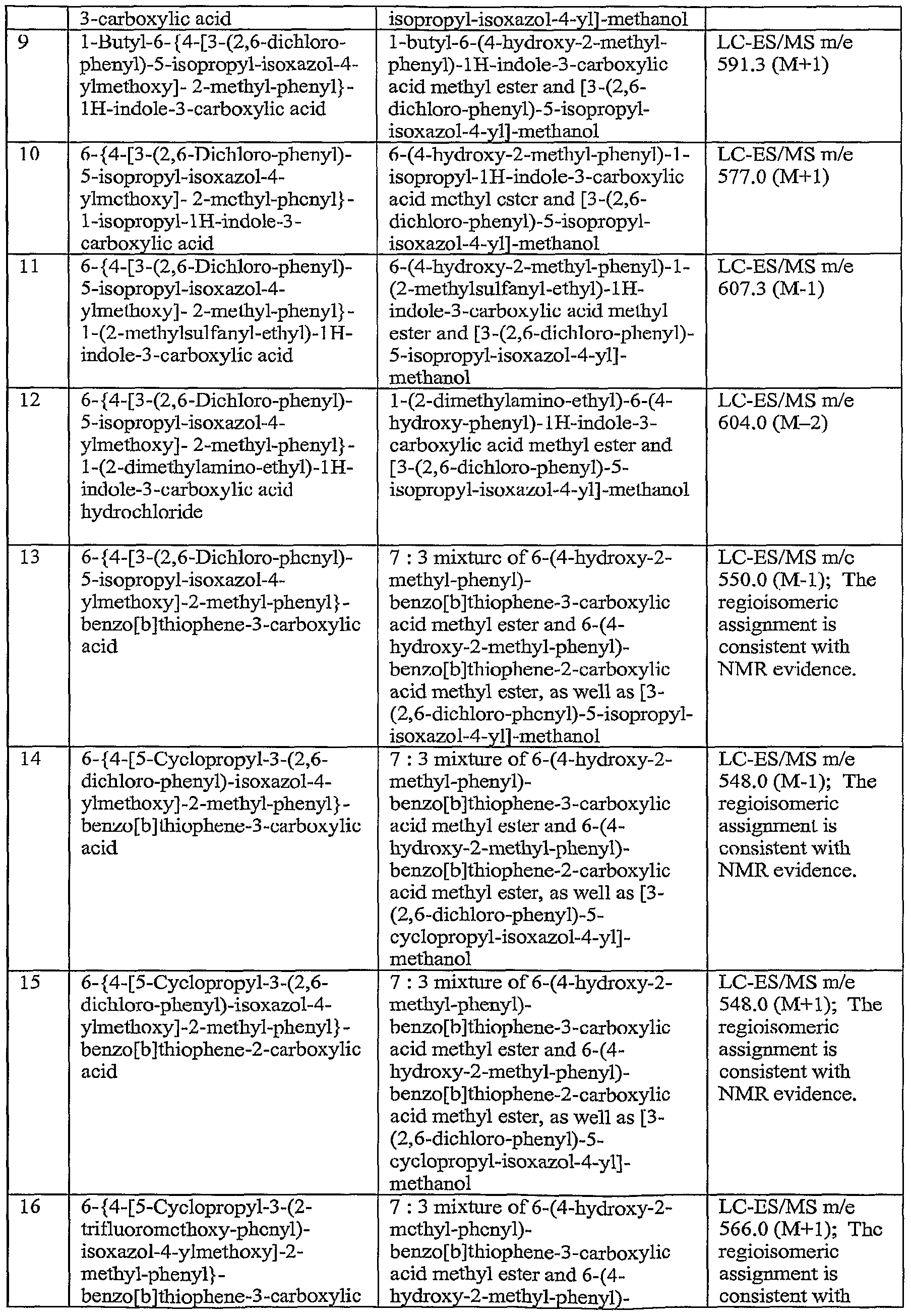
- carboxylic acid
- methyl
- isoxazol
- isopropyl
- Prior art date
Links
- LKXUYGICYUDILG-UHFFFAOYSA-N CC(C)c1c(COc(cc2C)ccc2-c(cc2)cc3c2c(C(O)=O)c[nH]3)c(-c(c(Cl)ccc2)c2Cl)n[o]1 Chemical compound CC(C)c1c(COc(cc2C)ccc2-c(cc2)cc3c2c(C(O)=O)c[nH]3)c(-c(c(Cl)ccc2)c2Cl)n[o]1 LKXUYGICYUDILG-UHFFFAOYSA-N 0.000 description 1
- UICHUVJHVIHQHD-UHFFFAOYSA-N Cc1c(C(O)=O)c(ccc(-c(c(C)c2)ccc2OCc2c(C3CC3)[o]nc2-c2ccccc2OC(F)(F)F)c2)c2[s]1 Chemical compound Cc1c(C(O)=O)c(ccc(-c(c(C)c2)ccc2OCc2c(C3CC3)[o]nc2-c2ccccc2OC(F)(F)F)c2)c2[s]1 UICHUVJHVIHQHD-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
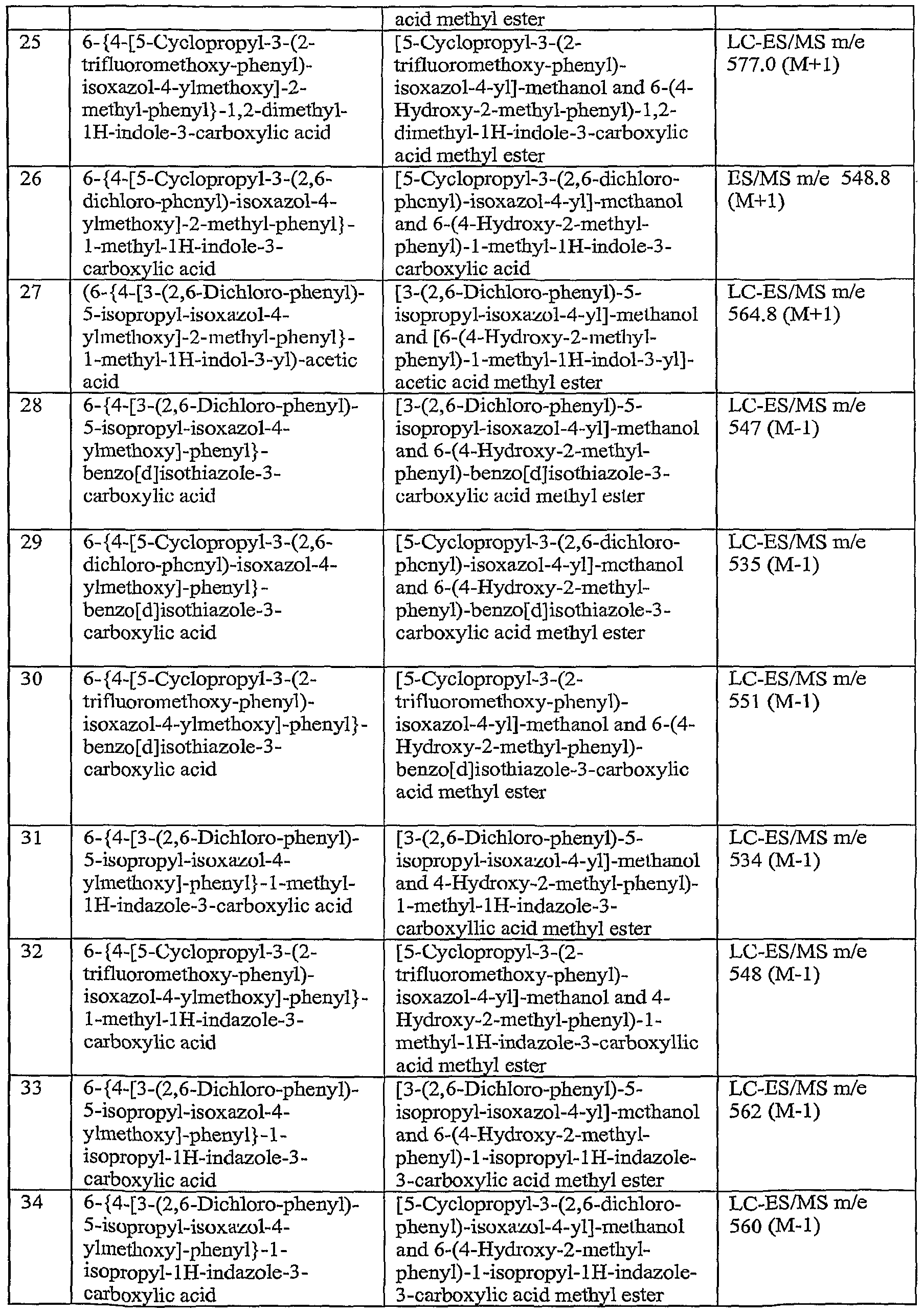
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
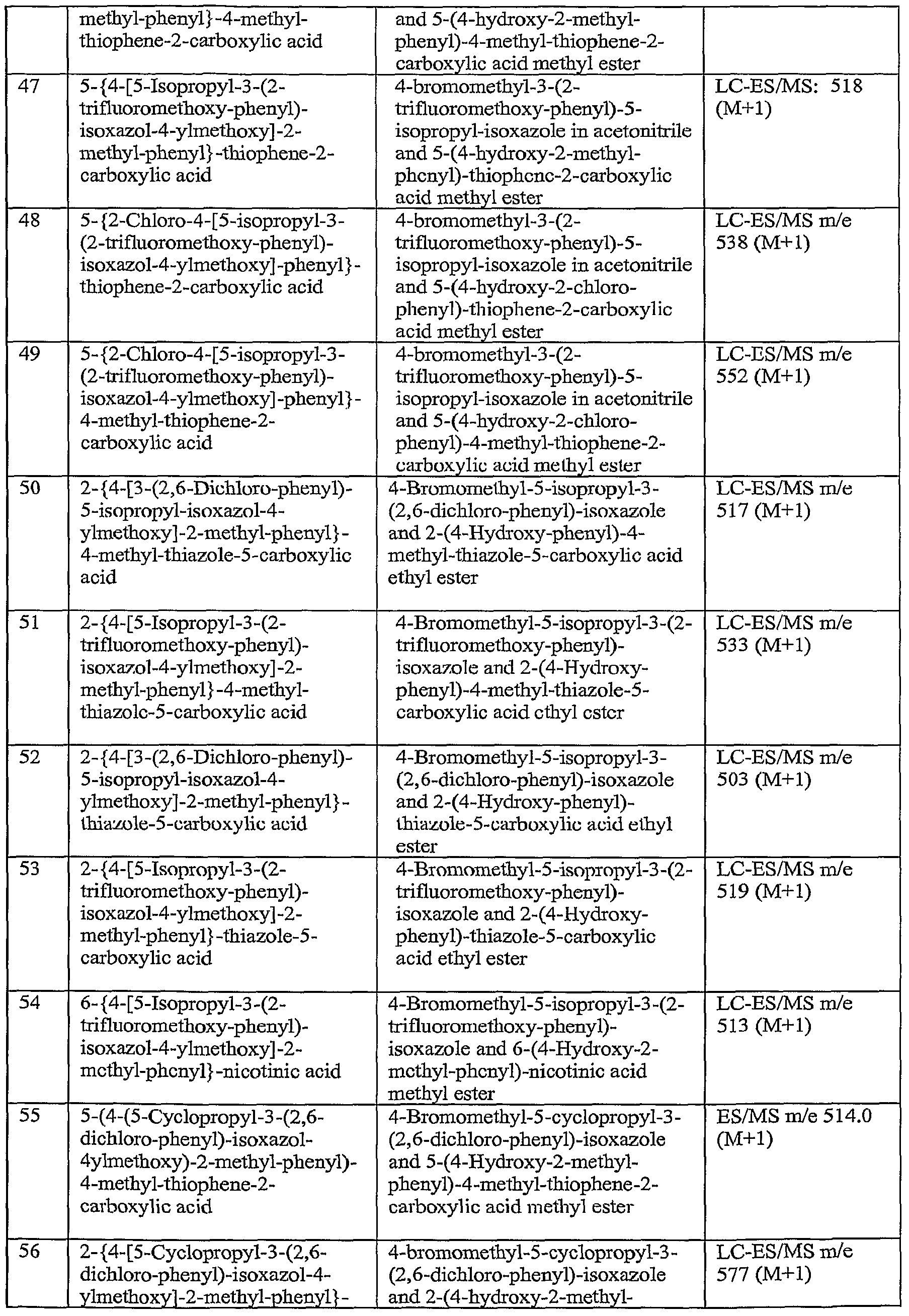
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
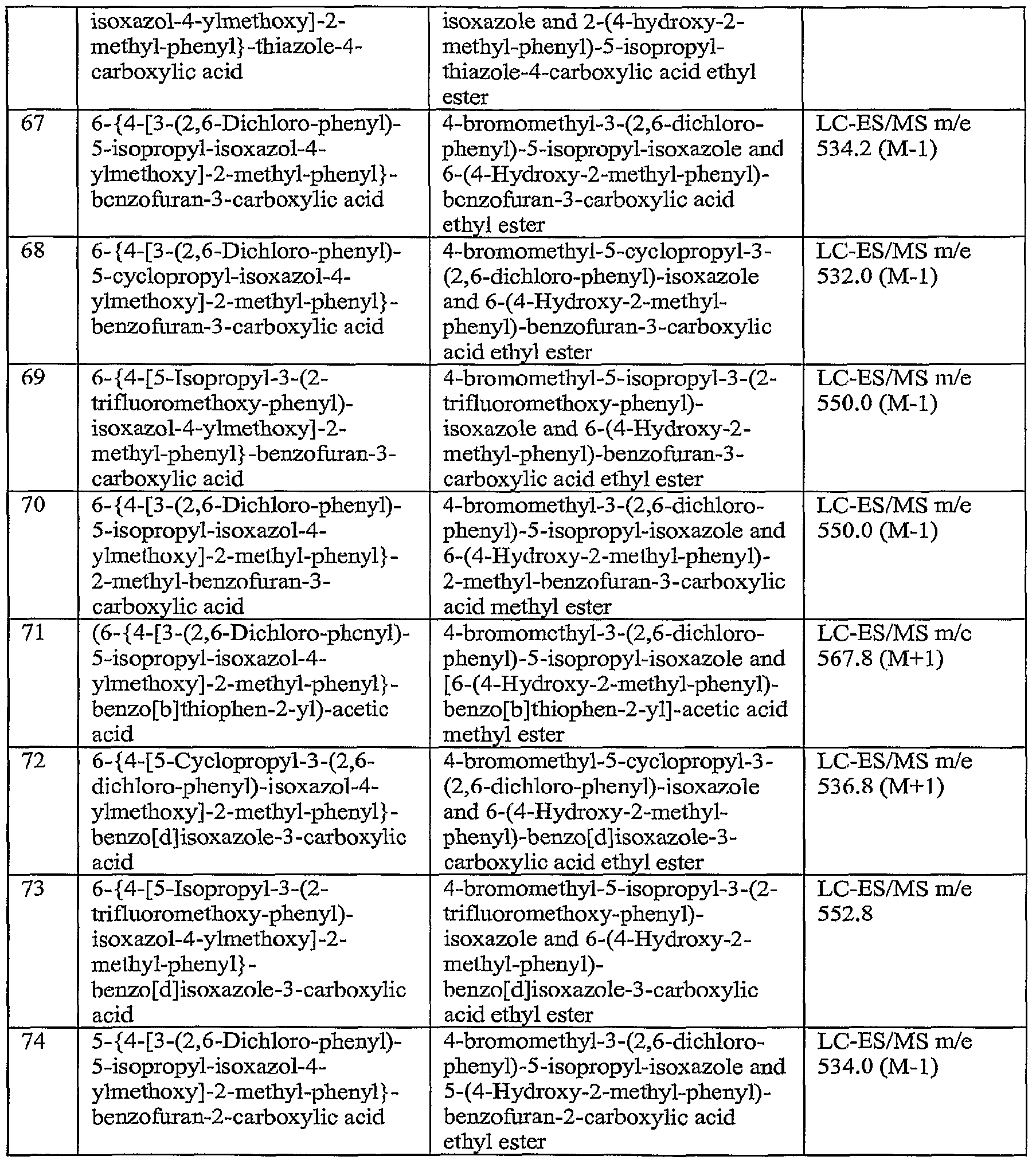
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
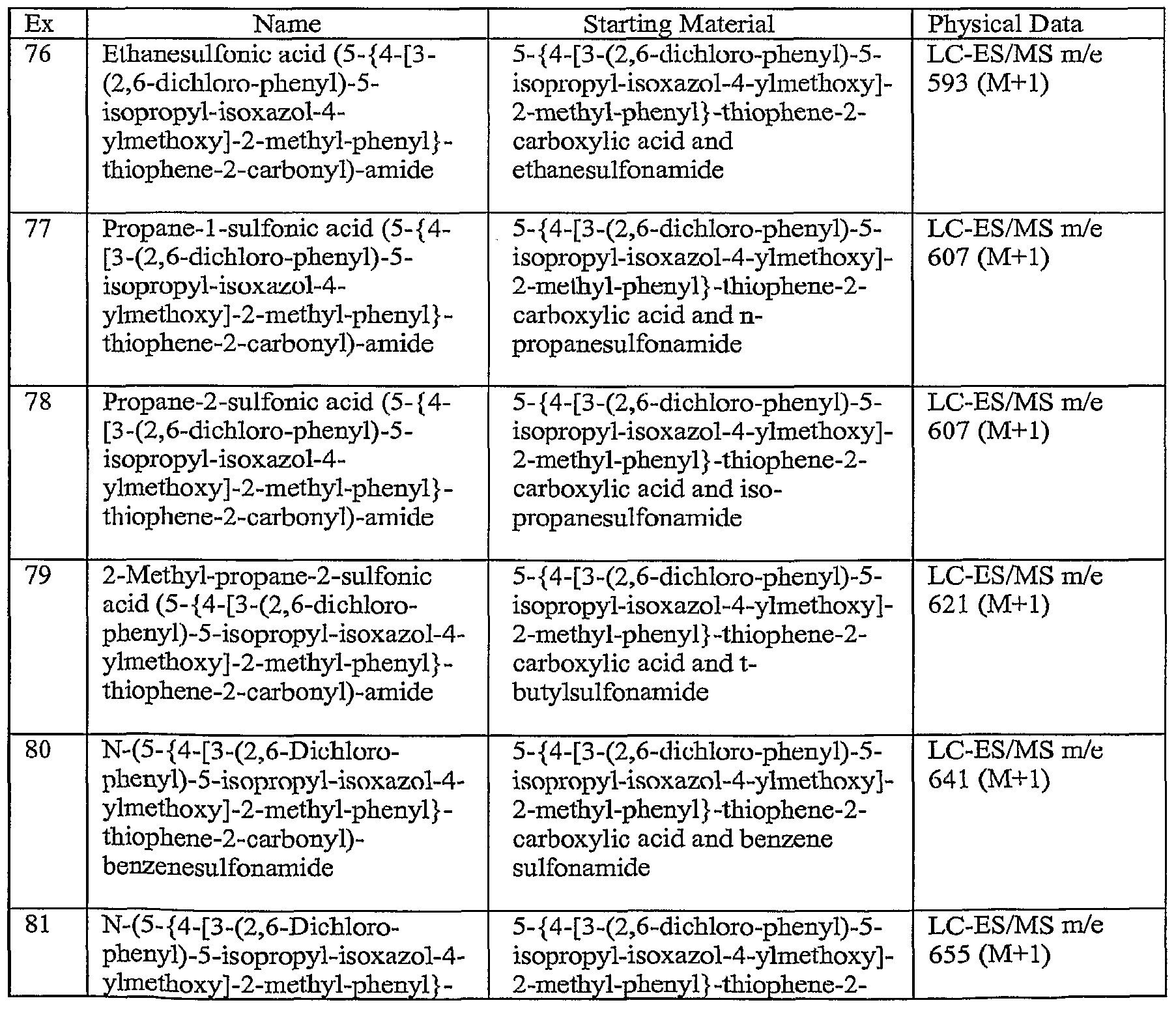
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the current invention relates to the fields of medicinal organic chemistry, pharmacology and medicine.
- Favorable modulation of plasma lipid such as triglycerides, HDL cholesterol, and LDL cholesterol is desirable.
- International application WO 03/015771 Al discloses certain isoxazoles for use in treating diseases mediated by the FXR NR1H4 receptor.
- International application WO 00/37077 discloses certain isoxazoles that bind to the farncsoid X receptor (FXR).
- International application WO 2004/048349 Al discloses compounds useful as farnesoid X receptor agonists.
- International application WO 98/28269 discloses compounds useful as factor Xa inhibitors.
- the nuclear hormone receptors, FXRs regulate the metabolism of plasma cholesterol and HDL.
- FXRs The nuclear hormone receptors, FXRs, regulate the metabolism of plasma cholesterol and HDL.
- compounds which modulate the FXRs would enhance the profile of lipid regulation, particularly increased HDL levels.
- Such compounds are desirable and would be useful for treatment of disorders characterized by or resulting from an undesirable lipid profile including dyslipidemia, atherosclerosis, diabetes and related diseases.
- the present invention provides novel, selective and potent FXR agonists for beneficial regulation of lipid profiles including raising HDL levels.
- the present invention provides a compound of formula
- p is 0 or 1;
- R 1 and R 2 are independently selected from the group consisting of hydrogen, -Ci-C 6 alkyl, -Ci-Ce haloalkyl, -Ci-C 6 alkoxy-, -Ci-C 6 haloalkoxy-, halo, -SR 11 , and -S-Ci-C 3 haloalkyl; each R 3 is independently selected from the group consisting of -Cj-Cc alkyl, -Ci-Cc haloalkyl, -Ci-Cg alkoxy-, -Ci-C 6 haloalkoxy-, and halo;
- R 4 is selected from the group consisting of hydrogen, -Ci-C 6 alkyl, -Ci-C 6 haloalkyl,
- R 5 and R 5a are independently selected from the group consisting of hydrogen, and -Ci-C 3 alkyl;
- R 6 is selected from the group consisting of hydrogen, -Ci-C 6 alkyl, -Ci-C 6 haloalkyl, and halo;
- Ar 1 is selected from the group consisting of indolyl, pyridinyl, thienyl, benzothienyl, indazolyl, benzothiazolyl, benzisoxazolyl, benzofuranyl and thiazolyl, each optionally substituted with one or two groups independently selected from the group consisting of hydroxy, -Cj-C 6 alkyl, C 3 -C 8 cyeloalkyl, -Ci-C 4 alkylSOzd-Ca alkyl, -C 1 -C 4 alkylSQ-Cz alkyl, -C 1 -C 4 alkylNR 1 ⁇ 11 , phenyl, -C 1 -C 4 alkyl-O-C r C 4 alkyl, and -NHC(
- R 7 is selected from the group consisting Of-CH 2 COOR 10 , -COOR 10 , -CONR 11 R 11 , -C(O)NHSO 2 C 1 -C 4 alkyl, -C(O)NHSO 2 R 12 , oxadiazolethione, and oxadiazolone; each R 10 is independently selected from the group consisting of hydrogen, -C 1 -C 4 alkyl, and phenyl; each R 11 is independently hydrogen, or -Cy-Ce alkyl;
- R 12 is -Q-C 6 alkyl or phenyl optionally substituted with -C 1 -Cs alkyl, or a pharmaceutically acceptable salt thereof.
- the compounds of the invention are modulators of FXRs.
- the compounds of the invention are useful for beneficially altering lipid profiles, including but not limited to lowering total cholesterol, lowering LDL cholesterol, lowering VLDL cholesterol levels, raising HDL levels, lowering triglyceride levels and beneficially sensitizing the effects of insulin.
- the present invention provides a method for treating FXR mediated conditions such as dyslipidemia and diseases related to dyslipidemia comprising administering a therapeutically effective amount of a compound of the invention to a patient in need thereof.
- the present invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the invention and a pharmaceutically acceptable carrier.
- the present invention also relates to the use of a compound of the invention for the manufacture of a medicament.
- the present invention also provides for the use of a compound of the invention for the manufacture of a medicament for treating FXR mediated conditions described herein.
- modulation and modulator as used herein xefer to beneficial regulation of genes and enzymatic processes resulting in or from agonism of the FXR receptor.
- FXR modulates key genes in multiple metabolic pathways, including cholesterol, triglyceride, bile acid and glucose metabolism.
- dyslipidemia refers to abnormality in, or abnormal amounts of lipids and lipoproteins in the blood and the disease states resulting, caused by, exacerbated by, or adjunct to such abnormality (see Dorland's Illustrated Medical Dictionary, 29th edition, W.B Saunders publishing Company, New York, NY).
- Disease states encompassed within the definition of dyslipidemia as used herein include hyperlipidemia, hypertriglyceremia, low plasma HDL, high plasma LDL, high plasmaVLDL, liver cholestasis, and hypercholesterolemia.
- diabetes related to dyslipidemia refers to cardiovascular diseases including but not limited to atherosclerosis, thrombosis, coronary artery disease, stroke, and hypertension.
- Diseases related to dyslipidemia also include diabetes, insulin resistance, and complications thereof. Complications of diabetes include but are not limited to diabetic retinopathy and obesity.
- the term “patient” includes human and non-human animals such as companion animals (dogs and cats and the like) and livestock animals.
- treatment include inhibiting, ameliorating, halting, slowing, and reversing the progression of, or reducing the severity of, pathological symptoms of dyslipidemia and diseases related to dyslipidemia.
- terapéuticaally effective amount means an amount of a compound of the invention that is part of an approved therapeutic regimen, or is determined by a qualified prescriber to be sufficient taken as directed, for treating a condition, or detrimental effects thereof herein described.
- pharmaceutically acceptable is used herein as an adjective and means substantially non-deleterious to the recipient patient.
- Ci-C 6 alkyl or the like (e.g. -C r C 2 atkyl, -Ci-C 3 alkyl, -Ci-C 4 alkyl,
- -C1-C 5 alkyl represents a straight or branched hydrocarbon moiety having from 1 to 6 (or as indicated) carbon atoms, including but not limited to methyl, ethyl, w-propyl, isopropyl, M-butyl, isobutyl, sec-butyl, t- butyl, and the like.
- An optionally substituted akyl group is divalent when connected to the substrate or molecular backbone.
- C 3 -Cg cycloalkyl or similar terms refer to a saturated carbocyclic ring having from 3 to 8 carbon atoms (or as indicated), including but not limited to cyclopropyl, cyclopentyl and cyclohexyl.
- C 4 -C 8 alkylcycloalkyl and the like (depending on indicated number of carbon atoms) as used herein refer to the combination of an alkyl and a cycloalkyl group such that the total number of carbon atoms is 4 to 8 or as indicated and the entire group is bonded to the substrate via the alkyl portion.
- C 4 -Cs alkylcycloalkyl includes cycloalkyl rings (e.g. C 3 -C 7 cycloalkyl) bonded to at least one carbon atom, such that the total number of carbon atoms is anywhere from 4 to 8 as in for example, -CH 2 cyclopropyl.
- halo means halogens including iodo, chloro, bromo and fluoro.
- C 2 -C 6 haloalkyl refers to a Ci-C 6 alkyl (or as indicated) group substituted with one, two, three or more halogen atoms as indicated or chemically appropriate.
- Ci -C 6 haloalkyl include but are not limited to trifluoromethyl, chloroethyl, and 2- chloropropyl.
- Ci-C 6 alkoxy e.g. Ci-C 3 alkoxy, C 2 -C 6 alkoxy, etc
- Ci-Ce alkyl or as indicated moiety connected through an oxy linkage.
- alkoxy groups include but are not limited to methoxy (-OMe), ethoxy(-OEt), propoxy (-OPr), isopropoxy (-OiPr), butoxy (-OBu), etc.
- Ci-C 6 haloalkoxy or the like (e.g. Ci-C 3 haloalkoxy) encompasses Ci-C 6 alkoxy wherein one or more of the hydrogen have been replaced with halogens.
- haloalkoxy groups include difluoromethoxy, trifluoromethoxy, 2-haloethoxy, 2,2,2-trifluoroethoxy, 4,4,4-trifluorobutoxy, up to and including like groups having the indicated number of carbon atoms.
- a compound of the invention as illustrated by the invention may occur as any one of its isomers all of which are objects of the invention. Certain compounds of the invention may possess one or more chiral centers, and thus, may exist in optically active forms. All such isomers as well as the mixtures thereof are within the ambit of the present invention. If a particular stereoisomer is desired, it can be prepared by methods well known in the art. Prcfcrrcd Embodiments of the Invention
- p is 0 or 1. More preferably p is 0
- R 1 and R 2 are each independently selected from the group consisting of hydrogen, Q- C 3 alkyl, -C r C 3 haloalkyl, Ci-C 3 alkoxy-, C]-C 3 haloalkoxy-, -SC r C 3 alkyl, -SCi-C 3 haloalkyl, and halo. More preferred R 1 and R 2 groups arc independently selected from the group consisting of hydrogen, chloro, fluoro, trifluoromethyl, thiotrifluoromethyl, and trifluoromethoxy.
- a preferred R 3 group is selected from the group consisting OfCi-C 4 alkyl, -Ci-C 3 haloalkyl, Ci-C 3 alkoxy-, Ci-C 3 haloalkoxy-, and halo. More preferred is an R 3 group selected from the group consisting of chloro, fluoro, trifluoromethoxy, thiotrifluoromethyl, and trifluoromethyl. Most preferably, R 3 is absent (p is O).
- R 4 is selected from H, methyl, ethyl, isopropyl, cyclopropyl, and methylcyclopropyl. Most preferably R 4 is isopropyl or cyclopropyl.
- R 5 and R 5a are each independently selected from the group consisting of hydrogen, methyl and ethyl. More preferably, R 5 and R 5a are both hydrogen.
- a preferred R 6 group is selected from the group consisting of hydrogen, halo, and Ci-C 3 alkyl. More preferably, R 6 is hydrogen or methyl.
- a preferred Ar 1 group is selected from lhe group consisting of optionally subslituted indolyl, thienyl, pyridinyl, benzothienyl, indazolyl, benzothiazolyl, benzisoxazolyl, benzofuranyl and thiazolyl each attached to the chain of the compound of the invention at any available carbon atom.
- Ar 1 is indolyl, thienyl, benzothienyl, and thiazolyl each optionally substituted with one or two groups independently selected from the group consisting of halo, C r C 5 alkyl, -Ci-C 3 alkylSO 2 Ci-C 3 alkyl, -C r C 3 alkyl-O-d-Ca alkyl, -Cj-C 3 alkyl-S-Ci-C 3 alkyl, -Cj-C 3 alkylNH(Ci-C 3 alkyl), -Cj-C 3 alkylN(C r C 3 alkyl) 2 , phenyl, and -NHC(O)Ci-C 3 alkyl wherein said substitution may be on carbon and/or nitrogen. More preferably, Ar 1 is substituted once at the nitrogen atom of a nitrogen containing group.
- a preferred R 7 substituent is selected from the group consisting of -COOH,
- R 7 group is -COOH.
- Each R 10 is preferably hydrogen, or Ci-C 6 alkyl.
- Each R 11 is preferably Ci-Ce alkyl.
- R 12 is preferably phenyl optionally substituted with Ci-C 3 alkyl.
- p is O, or 1 ;
- R 1 and R 2 are independently selected from the group consisting of hydrogen, fluoro, chloro, CF 3 , and- OCF 3 ,
- R 3 is fluoro, chloro, CF 3 , SCF 3 , or OCF 3 ;
- R 4 is H, isopropyl or cyclopropyl;
- R 5 and R 5a are each independently selected from H or methyl;
- Ar 1 is indolyl, pyridinyl, thienyl, thiazolyl and benzothienyl each optionally substituted with one group selected from the group consisting of Ci-C 4 . alkyl, CF 3 , -CH 2 CH 2 SCH 2 , -CH 2 CH 2 OCH 3 , -CH 2 CH 2 SO 2 CH 3 ,
- R 6 is hydrogen, or methyl
- E 7 is -COOH, -COOC 1 -C 2 alkyl, -CONHSO 2 C 1 -C 4 alkyl, -CONHSO 2 phenyl, CONHSO 2 phenylmethyl, oxadiazolone, and thiadiazolone; each R 10 is independently hydrogen or Cj-Cc alkyl; and each R u is independently hydrogen or Ci-C 6 alkyl.
- each p is O;
- R 1 and R 2 are independently selected from the group consisting of chloro, fluoro, Irifluoromelhyl, and Irifiuoromelhoxy;
- R 4 is isopropyl or cyclopropyl;
- R 5 and R 5a are both hydrogen;
- R 6 is hydrogen, methyl, ethyl or chloro;
- Ar 1 is thienyl, benzothienyl, indolyl or thiazolyl, each bound at any available carbon atom and each optionally substituted with a group selected from methyl, ethyl, propyl, butyl, isopropyl, cyclopropyl, -CH 2 CH 2 SO 2 CH 3 , - CH 2 CH 2 N(CH 3 ) 2 , -CH 2 CH 2 SCH 2 , -CH 2 CH 2 OCH 2 , and phenyl; and R 7 is -COOH or -COOMe.
- the compounds of the invention may be prepared by a variety of procedures known in the art and those described below.
- the products of each step in the Scheme below can be recovered by conventional methods including extraction, evaporation, precipitation, chromatography, filtration, trituration, crystallization, and the like.
- substituents unless otherwise indicated, are as previously defined and suitable reagents are well known and appreciated in the art.
- Scheme 1 depicts the reaction of an appropriate compound of formula appropriate compound of formula (2) to give a compound of formula (I).
- the reaction in Scheme 1 can be carried out by at least two variants discussed below.
- an appropriate compound of formula (1) is one in which R 1 , R 2 , R 3 , p, R 4 , R 5 , and R 5a are defined for formula I, and Y is —OH and an appropriate compound of formula (2) is one in which R 6 , R 7 , and Ar 1 are as defined in formula (I) or a group which gives rise to R 7 as defined in formula (I), for example, by formation of an ester, amide, sulfonamide, or acid.
- a compound of formula (1) is reacted with a compound of formula (2) in a Mitsunobu reaction using a suitable diazo reagent, such as DEAD or ADDP, and the like, and a suitable phosphine reagent such as triphenyl phosphine or tributylphosphine, and the like.
- a suitable solvent such as toluene, tetrahydrofuran, and the like.
- the reactions are carried out at temperatures of from about 0 0 C to 50 0 C.
- Typical stoichiometry for this reaction based on the compound of formula (1) is about 1 to 2 equivalents of a compound of formula (2) and about 1 to 2 equivalents each of the dia ⁇ o and phosphine reagents.
- an appropriate compound of formula (1) in which R 1 , R 2 , R 3 , p, R 4 , R 5 , and R 5a are defined for formula 1 and Y is a leaving group and an appropriate compound of formula (2) is as defined above are reacted to form the compound of formula (I) with appropriate protections and/or deprotections or other processing steps known to one of skill in the art or disclosed herein.
- Suitable leaving groups are well-known in the art and include halides, particularly chloro, bromo, and iodo; and sulfonate esters, such as brosyl, tosyl, methanesulfonyl, and trifluromethanesulfonyl.
- a compound of formula (1) is reacted with a compound of formula (2) in a suitable solvent, such as acetonitrile, dimethylformamide, tetrahydrofuran, pyridine, methylethyl ketone and the luce.
- a suitable solvent such as acetonitrile, dimethylformamide, tetrahydrofuran, pyridine, methylethyl ketone and the luce.
- a suitable base including sodium hydride, potassium carbonate, sodium carbonate, cesium carbonate, sodium bicarbonate, triethylamine, diisopropyethylamine.
- Such reactions generally are carried out at temperatures of about room temperature to about the reflux temperature of the chosen solvent and typically use from about 1 to 2 equivalents of the compound of formula (2).
- compounds of formula (T) wherein R 7 is an ester can be converted to compounds of formula (I) wherein R 7 is an acid via methods well known to one of ordinary skill in the art.
- the microwave may be used as an energy/heat source, especially when the ester is sterically hindered.
- a laboratory microwave utilizing the lowest power setting at about 125 0 C for about 20 minutes in solvent mixtures described above is useful.
- R 7 is a t-butyl ester the acid can be formed under acidic conditions well known to those skilled in the art.
- compounds of formula (I) wherein R 7 is a carboxylic acid can be converted to compounds of formula (I) wherein R 7 is an amide or sulfonamide by coupling procedures well known in the art.
- a compound of formula (I) wherein R 7 is an acid is reacted with an amine or sulfonamide compound in the presence of a coupling agent such as dicyclohexylcarbodiimide, l-[3- (dimethylamino) propyl] -3 -ethylcarbodiimide hydrochloride, and the like, and optionally N 5 N- dimethylaminopyridine and/or an amine base, such as triethylamine, diisopropylethylamine, and the like, in a suitable solvent, such as DMF, THF, and the like.
- a suitable solvent such as DMF, THF, and the like.
- a pharmaceutically acceptable salt of a compound of formula (I) is formed.
- the formation of such salts is well known and appreciated in the art.
- compounds of formula (1) and (2) can be readily prepared by methods that arc well-known and established in the art including methods and procedures similar to those described herein.
- compounds of formula (1) are prepared by the reaction of optionally substituted benzaldehydes with hydroxylamine followed by chlorination with a suitable chlorinating agent, such as N-Chloro succinimide, to afford chloroximes (see for example J. Med. Chem. 2000, 43 (16), 2971- 2974).
- a suitable base such as triethylamine or sodium methoxide
- the esters can be reduced to the alcohol compounds of formula (1) with well known methods (e.g. DIBAL-H, LAH) and subsequently converted to a leaving group.
- Compounds of formula (2) are prepared by carbon-carbon bond formation/coupling reactions. Also, it is recognized that the steps required Io prepare a compound of formula (I) can be carried out in any order. For example, including reaction of a partial compound of formula (2) with a compound of formula (1), such that the later carried out carbon-carbon bond formation/coupling reaction provide a compound of formula I. More specifically, a compound of formula (3) can be reacted with a compound of formula (1) as described above to afford compounds of formula (4) which can be converted to compounds of formula (I) via carbon-carbon bond forming reactions with compounds of formula (5) (Scheme 2).
- Certain compounds of the invention exist as solid amorphous or crystalline forms.
- a compound of the invention may also exist in multiple crystalline forms wherein one or more of the crystalline forms are preferred over others on account of having more desirable properties such as, for example, improved solubility, improved bioavailability and/or improved stability. All such crystalline forms are within the ambit of the present invention.
- the compound of example 101 has been found to exist in two forms (forms I and II).
- LDLR-/- mice from Jackson Laboratories (Stock number 002207, Bar Harbor, Maine, USA). Acclimate animals for one week prior to study initiation. House mice individually in polycarbonate cages with filter tops, and maintain mice on a 12:12 hour light-dark cycle (lights on at 6:00 AM) at 21 0 C. Provide deionized water ad libitum and maintain for two weeks on 'western diet' TD 88137 Diet (42 % fat, 0.15 % cholesterol, Harlan Teklad) ad libitum. Optimize groups of five ten-week-old male LDLR-/- mice based on serum triglyceride and cholesterol levels.
- VLDL, LDL, HDL lipoprotein cholesterol fraction values
- tested compounds of the invention reduce total cholesterol up to 80% and triglycerides up to 90% when dosed at 10 mg/kg. More specifically the compound of Example 7 lowers total cholesterol 63% and triglycerides 61% when dosed at 10 mg/kg.
- a typical daily dose will contain a nontoxic dosage level of from about 0.1 mg to about 500 mg/day of a compound of lhe present invention.
- Preferred daily doses generally will be from about 1 mg to about 250 mg/day.
- the compounds of this invention may be administered by a variety of routes including oral, xectal, transdermal, subcutaneous, intravenous, intramuscular, and intranasal. These compounds preferably are formulated prior to administration. The selection of appropriate dose and route of administration will be decided by the attending physician.
- a pharmaceutical composition comprising an effective amount of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier, diluent, or excipient.
- compositions of the present invention may be adapted for these various routes and may be administered to the patient, for example, in the form of tablets, capsules, cachets, papers, lozenges, wafers, elixirs, ointments, transdermal patches, aerosols, inhalants, suppositories, solutions, and suspensions.
- the total active ingredients in such composition comprises from 0.1 % to 99.9 % by weight of the formulation.
- Compounds of the invention may be formulated as elixirs or solutions for convenient oral administration or as solutions appropriate for parenteral administration, for example, by intramuscular, subcutaneous or intravenous routes. Additionally, the compounds may be formulated as sustained release dosage forms and the like. The formulations can be constituted such that they release the active ingredient only or preferably in a particular physiological location, possibly over a period of time.
- the coatings, envelopes, and protective matrices may be made, for example, from polymeric substances or waxes.
- 2,6-Dichloro-benzaldelryde (7.0 g,40 mmol) is added to 10 mL of water and 30 mL of methanol.
- Sodium hydroxide (4.0 g, 100 mmol) is dissolved in 8 mL of water slowly.
- the sodium hydroxide solution is added to the benzaldel ⁇ yde solution.
- the reaction is stirred overnight.
- the reaction mixture is partitioned between ethyl acetate and water.
- the organic layer is washed with brine and dried over solid sodium sulfate.
- the organic layer is filtered and the solvent is removed under reduced pressure to yield the title compound.
- Step 4 (5-CvclopropyI-3-(2.6-dichloro-phcnyl ' )-isoxazoI-4-yl ' )-mcthanol
- Preparation 2B r5-Cvclopropyl-3-r2-fluoro-6-trifluoromethyl-phenyl')-isoxazol-4-yll-methanol. utilizing 2-fluoro-6-trifluoromethyl-benzaldehyde, 1 H NMR (400M Hz, CDC13) ⁇ 7.67-7.55 (m, 2H), 7.37 (m, IH), 4.34(s, 2H), 2.13 (m, IH), 1.22 (m, 2H), 1.10 (m, 2H);
- Preparation 2C r5-Isopropyl-3-f2-isopropyl-phenvD-isoxazol-4-y ⁇ -methanol utilizing 2-isopropyl- benzaldehyde, ES/MS m/e 260.0 (M+l), 258.0 (M-I).
- the title compound is prepared essentially as described in the preparation of 6-bromo-l-mcthyl- lH-indole-3-carboxylic acid methyl ester utilizing 6-bromo-lH-indole-2-carboxylic acid methyl ester, ES/MS m/e 270.0 (M+2).
- Preparation 15E 6-( " 4-Hvdroxy-2-methyl-phenylVl-methyl-lH-indole-3-carboxylic acid methyl ester. utilizing ⁇ -bromo-l-methyl-lH-indole-S-carboxylic acid methyl ester, LC-ES/MS m/e 296.0 (M+l); Preparation 15F: 6-C4-Hvdroxy-2-methyl-phenyr)-l -methyl- lH-indole-2-carhoxyric acid methyl ester.
- Preparation 15G 5-(4-Hvdroxy-2-methyl-phenylVbenzorblthiophene-3-carboxylic acid methyl ester. utilizing S-bromo-benzotbJthiophene-S-carboxylic acid methyl ester, ES/MS m/e 299.1 (M+l); Preparation 15H: 6-(4-Hv ⁇ joxy-2-methyl-phemylVbenzorb1thiophene-2-carboxylic acid methyl ester. utilizing 6-bromo-benzo[b]tliiophene-2-carboxylic acid metliyl ester, ES/MS m/e 297.3 (M-I);
- Preparation 16B 2-(4-Hvdroxy-2-methyl-phenyl)-4-trifluoromethyl-thiazole-5-carboxylic acid ethyl ester. utilizing 2-amino-4-trifluoromethyl-thiazole-5-carboxylic acid ethyl ester, LC-ES/MS m/e 332 (M+l), 330 (M-I), 91.2%;
- Preparation 16C 2-(4-Hvdroxy-2-mcthyl-phcnyl)-4-phcnyl-thiazolc-5-carboxylic acid ethyl ester, utilizing 2-amrno-4-phenyl-thia7:ole-5-carboxylic acid ethyl ester, 1 H NMR (DMSO-d 6 ) ⁇ 10.06 (s, 1 H), 7.76, 7.79 (m, 3H), 7.44, 7.46 (m, 3H), 6.74, 6.77 (m, 2H), 4.23 (q, 2H), 2.56 (s, 3H), 1.21 (t, 3H);
- Preparation 16D 2-(4-Hvdroxy-2-methyl-phenyl)-thiazole-4-carboxylic acid ethyl ester, utilizing 2- bromo-thiazole-4-carboxylic acid ethyl ester, LC-ES/MS m/e 264 (M+l
- a solution of «-butyllithium (2.5M in hexanes, 2.866 L, 7.17 mol) is added dropwise to a stirred solution of diisopropylamine (745.7 g, 7.37 mol) in tetrahydrofuran (1.630L) such that the temperature is maintained hi the range -60 to -78 0 C.
- the resulting suspension is stirred for 1.5 h at -75 to -78 °C.
- a solution of l-bromo-3-fluorobenzene (1.228 Kg, 7.02 mol) in tetrahydrofuran (2.40 L) is added slowly to the reaction mixture over 1.5 h. Stirring is continued for 30 min at -70 to -71°C.
- the combined extracts are washed with hydrochloric acid (1 M, 2x 5.00 L) and water (4.0 L), dried over magnesium sulfate, filtered, and concentrated under reduced pressure to afford the crude product (640 g) as a brown oil which solidifies on standing overnight.
- the residue is slurried in methanol (500 mL, 0.5 vol) at -10 to 0 °C for 1.5 h, the observed solid material is collected by filtration and pulled dry on the filter.
- the methanolic mother liquors are concentrated by rotary evaporation.
- the residue is combined with the isolated solid material, slurried in TBME (2.0 L) and the collected solids are washed with TBME (660 L).
- the title compound (12Og, 86%) is prepared essentially as described in the preparation of 4- carboxy-benzo[b]thiophen-2- boronic acid utilizing benzotfelthiophene- ⁇ -carboxylic acid, ES/MS m/e 221 M-I).
- Acetyl chloride (14.8 mmol; 1.05 mL) is added to a solution of benzo[ ⁇ ]thiophene-7-carboxylic acid (4.94 mmol; 880 mg) in methanol (20 rnL). The reaction mixture is stirred at reflux for 24 h. The solvent is removed under reduced pressure. The residue is taken up in ethyl acetate washed with water, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to yield the title compound (880 mg, 92%) as colorless oil.
- Cesium carbonate (9.70 mmol; 3.19 g) is dried in a resealable tube at 150 0 C in vacuo for 2 h and cooled to room temperature.
- Copper(I) iodide (9.70 mmol; 1.86 g)
- Pd(OAc) 2 (0.24 mmol; 55 mg)
- triphcnylphosphinc 0.85 mmol; 128.50 mg
- 2-bromo-5-mcthoxytolucnc (9.70 mmol; 2.14 mL)
- benzo[ ⁇ ]thiophene-5-carboxylic acid ethyl ester (4.85 mmol; Ig) and anhydrous dimethylforrnarnide (24 mL) are added under nitrogen atmosphere and the mixture is stirred at 140 0 C.
- the title compound (85 mg, 69%) is prepared essentially as described in the synthesis of 2-(4- Hyd ⁇ oxy-2-methyl-pheny1)-ben7:o[ ⁇ ]thiophene-5-ca ⁇ boxylic acid ethyl ester, utilizing 2-(4-Methoxy-2- methyl-phenyl)-benzo[ ⁇ ]thiophene-7-carboxylic acid methyl ester and methanol.
- Neat 3-cyclopropylamino-but-2-enoie acid ethyl esler (5.63 g, 33.2 mmol) is added to a mixture of p-benzoquinone (7.19 g, 66.5 mmol) and acetic acid (120 mL). The mixture is stirred at room temperature for 5 h and a dark solid is precipitated. The solid is washed with acetic acid and water, dried, adsorbed onto silica gel, and purified via flash chromatography eluting with dichloromethane. The product is triturated in dichloromethane-hexane to afford the title compound (440 mg, 21%).
- a mixture of sodium hydride (60% in mineral oil, 2.60 g, 65.0 mmol) and DMF (52 mL) is stirred in an ice bath and methylacetoacetate (6.46 mL, 59.9 mmol) is added via a syringe over 10 minutes. The mixture is stirred for an additional 10 minutes and the ice bath is removed. The solution is stirred at ambient temperature for 30 minutes and transferred via cannula into a flask containing 4-chloro-l-fluoro-2- nitrobenzene (5.00 g, 28.5 mmol) cooled to 0°Cvia an ice bath. The reaction is allowed to warm slowly and is stirred for two days at room temperature.
- the black mixture is acidified with 2 N HCl, turning it yellow.
- the resulting solution is diluted with water and extracted with ether.
- the combined ether layers are washed with water and brine and dried over MgSO 4 to give the crude title compound (8.26 g), which contains a small amount of mineral oil.
- the material is used without purification in the next step.
- the title compound is prepared essentially according to the preparation of 6-(4-hydroxy-2-rnethyl ⁇ phenyl)-l-isopropyl-2-methyl-lH-indole-3-carboxylic acid methyl ester utilizing 6-Chloro-l,2-dimethyl- lH-indolc-3-carboxylic acid methyl ester.
- ES/MS m/c 310.3 (M + 1).
- Preparation 32 6-C4-Hvdroxy-2-methyl-phenylVbenzordlisoxa2ole-3-carboxylic acid ethyl ester
- Step 1 (4-bromo-2-nitro-phenyl ' )-acetic acid methyl ester
- Step 1 4-(3-methoxy- ⁇ henylsulfanyl)-3-oxo-butyric acid ethyl ester
- Step 3 f ⁇ -h y droxy-benzorbithiophen-S-vD-acetic acid ethyl ester
- the residual aqueous solution is extracted with EtOAc (50 mL x2).
- the combined organic layers are dried over Na 2 SO 4 , filtered, and concentrated under reduced pressure.
- the crude product is purified via flash chromatography eluting with 10% EtOAc/Hexanes. The appropriate fractions arc combined and concentrated under reduced pressure to a residue.
- the residue is dissolved in THF (50 mL) and treated with HCl (5 N, 5 mL). The mixture is stirred at 70 0 C for 60 minutes and neutalized with NaOH (5 N, 5 mL). The mixture is concentrated to a residue under reduced pressure.
- the residual aqueous mixure is extracted with EtOAc (50 mL x2).
- Step 5 (6-bromo-benzorb1thiophen-2-vD-acetic acid methyl ester
- 6-bromo-benzorb1thiophen-2-vD-acetic acid methyl ester To a 0 0 C solution of (6-bromo-benzo[b]thioplien-2-yl)-acetaldeliyde (2.21 g; 8.66 mmol) in t- butyl alcohol (50 mL; 526.36 mmoles; 50.00 mL; 39.015 g) and 2-methyl-2-butene (20 mL; 188 mmol) is added a solution of sodium chlorite (6.27 g; 69.3 mr ⁇ l) in water (20 mL) and a solution of sodium phosphate monobasic (4.20 g; 34.6 mmol) in water (10 mL).
- the reaction mixture is stirred at 0 0 C for 12 hours while warming to room temperature.
- the mixture is extracted with EtOAc (50 mL x2).
- the combined organic layers are dried over Na 2 SO 4 , filtered, and concentrated under reduced pressure.
- the crude product is dissolved in methanol (30 mL).
- Sulfuric acid (1.0 mL; 18.8 mmol) is added and the mixture is stirred at 95 0 C for 4 hours.
- the mixture is neutralized with aqeous NaHCU3 and concentrated under reduced pressure.
- the residual aqueous mixture is extracted with EtOAc (50 mL x2).
- the combined organic layers are dried over Na 2 SO 4 , filtered, and concentrated under reduced pressure.
- Step 4 (l-Methyl- ⁇ -trifluoromethanesulfonyloxy-lH-indol-S-vD-acetic acid methyl ester
- Preparation 42A 6-(4-Hvdroxy-2-methyl-phenyl')-benzord1isoxazole-3-carboxylic acid ethyl ester (0.065 g, 47%) utilizing 2-methyl-6-trifluoromethanesulfonyloxy-benzofuran-3-carboxylic acid methyl ester stirring at 100 0 C for 16 hours.
- ES/MS m/e 297.0 (M+l), 295.0 (M-I).
- Triphenylphosphine (1.1 equiv., 51.5 mmoles, 13.5 g) is added in small portions to a 0 "C solution of [5-Cyclopropyl-3-(2-tiifluoromethoxy-plienyl)-isoxazol-4-yl]-methanol and carbon tetrabromide (1.1 equiv, 51.5 mmoles, 17.1 g) in dichloromethane (187.1 mL). The reaction mixture is stirred at ambient temperature for 15 h and concentrated under reduced pressure.
- reaction mixture Upon completion of the reaction, as evidenced by thin layer chromatography, the reaction mixture is cooled to 0-20 0 C. The reaction mixture is filtered and the filter cake is washed with acetonitrile (2 x 100 mL). The reaction mixture is concentrated under reduced pressure to a solid which is co-evaporated with 1,4-dioxane (500 mL). The title compound is used without further purification.
- Oxalyl chloride (717.2 g, 5.65 mol, 3.5 eq) is added to a 0-5 0 C suspension of dichloromethane (3.44 L) and aluminum chloride (753.4 g, 5.65 mol, 3.5 eq). The resulting suspension is stirred for 30-60 minutes at 0-5 0 C and cooled -20 to -25 °C.
- a solution of 6-bromobenzo[b]thiophene (344 g, 1.614 mol, 1 eq) in dichloromethane (1.72 L) is added over 1 h while maintaining the temperature at -20 to -25 0 C.
- the reaction mixture is stirred for 30 minutes at -20 to -25 0 C and warmed to 18 to 20 0 C using a warm water bath.
- the reaction mixture is stirred for 1.5 h at this temperature.
- the reaction mixture is filtered and the filter cake is washed with dichloromethane (3 x 300 mL).
- the combined filtrate is concentrated to yield a thick black oil in the flask (600 g). This residue is dissolved in dichloromethane (1 L) and added to ethanol (3.5 L) at -10 to 0 0 C in portions at such rate to maintain temperature at 10 to 20 0 C. Once the addition is complete, the reaction mixture is partially concentrated to remove the dichloromethane only and then the vacuum is released.
- the reaction mixture is heated to 60-70 0 C and stirred at this temperature for 1 h. Upon completion of the reaction, the solution is decanted from the resulting tars. The tars are discarded. The ethanol solution is evaporated to a residue. The residue is diluted with EtOAc (2 L).
- the current reaction mixture is combined with another reaction mixture for further work up (started with 330 g of 6-Bromobenzo[b]thiophene, 1.549 mol).
- the combined reaction mixture is poured into a stirred mixture of EtOAc (1 L) and brine solution (10 L). The layers are separated and the organic layer is washed with brine solution (2 L). The combined aqueous layer is extracted with EtOAc (4 L). The organic layer is washed ilh brine solution (1 L). The combined organic layers are dried over magnesium sulfate and charcoal, filtered, and concentrated under reduced pressure. The resulting oil is further concentrated in a vacuum oven for 15 h at room temperature to afford waxy solids after drying (750 g).
- the solids are suspended in heptane (5 L) with stirring and the suspension is heated to 70 0 C.
- Magnesium sulfate (300 g) is added and the resulting suspension is stirred for 10 minutes at 70 0 C.
- the suspension is filtered.
- the solids are suspended in heptane (5 L) and heated to 70 0 C.
- the suspension is stirred for 10-20 minutes at this temperature and filtered.
- the filter cake is washed with heptane (1 L).
- the heptane filtrates are collected and concentrated under reduced pressure to give light brown solids (550 g).
- the solids are dissolved in heptane (4 L) at 60 0 C.
- the resulting solution is cooled to 35 to 50 0 C.
- 6-(4-r3-C2.6-Dichloro-phenyl)-5-isopropyl-isoxazol-4-ylmethoxyl-2-methyl-phenvU-lH-indole-3- carboxylic acid A mixture of 6- ⁇ 4-[3 -(2, 6-dichloro-plienyl)-5-isopropyl-isoxazol-4-ylme1iioxy] -2-methyl-phenyl ⁇ - lH-indole-3-carboxylic acid methyl ester (80 mg, 0.15 mmol), 5 N sodium hydroxide (150 ⁇ L, 0.750 mmol), methanol (2 mL) and THF (1 mL) is heated at 85 0 C for five hours.
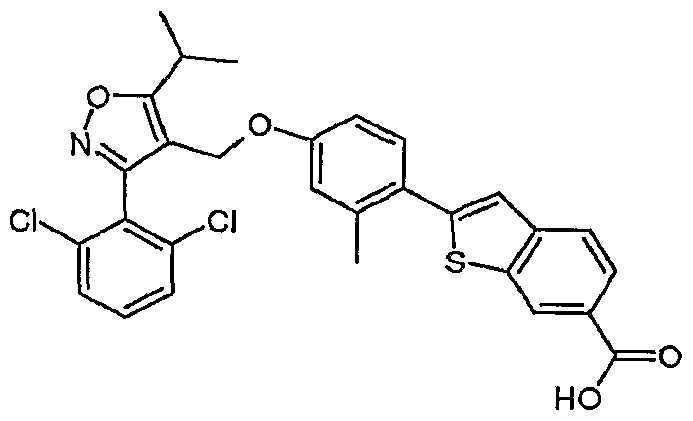
- the compounds in Table 1 are prepared essentially according to the preparation of 6- ⁇ 4-[3-(2,6- dichloro-phenyl)-5-isopropyl-isoxazol-4-ylmethoxy]-2-methyl-phenyl ⁇ -lH-indole-3-carboxylic acid.
- the compounds in Table 3 are prepared essentially according to the preparation of 2-(4-(5- Cyclopropyl-3-(2-fluoro-6-trifluoromethyl-phenyl)-isoxazol-4-yhnethoxy)-2-methyl-phenyl)-4-methyl- thiazole-5-carboxylic acid.
- the compounds in Table 4 are prepared essentially according to the preparation of N-(5- ⁇ 4-[3- (2,6-dichloro- ⁇ henyl)-5-isopropyl-isoxazol-4-ylmethoxy]-2-methyl-phenyl ⁇ -thio ⁇ hene-2-carbonyl)- methanesulfonamide.
- the organic solvent is removed under reduced pressure and the residue is acidified with IN HCl to pH 4.
- the resulting solution is extracted with dichloromethane.
- the organic layers are combined, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure.
- the residue is chromatographed using a gradient from 100% dichloromethane to 100% ethyl acetate to obtain a solid which is washed with acetonitrile to yield the title compound (135 mg, 27%) as a white solid.
- the title compound is prepared essentially according to the preparation of 2- ⁇ 4-[3-(2,6-dichloro- phenyl)-5-isopropyl-isoxazol-4-ylmethoxy] -2-methyl-phenyl ⁇ -benzo[ ⁇ ]thiophene-4-carboxylic acid utilizing 6-carboxy-benzo[b]thiophen-2-boronic acid.
- the mixture is diluted with water and ethyl acetate.
- the ethyl acetate layer is washed with brine, dried over MgSO 4 , and concentrated under reduced pressure.
- the residue is purified via radial chromatography eluting with 2% MeOH-CH 2 Cl 2 .
- the purification is repeated for impure fractions containing product.
- the title compound (44 mg, 21%) is obtained as a gray solid.
- the compounds in Table 5 are prepared essentially according to the preparation of 6- ⁇ 4-[3-(2,6- Dichloro-phenyl)-5-isopropyl-isoxazol-4-ylmethoxy]-2-methyl-phenyl ⁇ -l-isopropyl-2-methyl-lH-indole- 3-carboxylic acid.
- Step 1 4-(4-bromo-3-methyl-phenoxymethyl')-5-cvclopropyl-3-(2.6-dicnloro- ⁇ henylVisoxazole
- the compounds in Table 6 are prepared essentially according to the preparation of 6- ⁇ 4-[3-(2,6- Dichloro-phenyl)-5-cyclopropyl-isoxazol-4-ylmethoxy]-2-methyl-phenyl ⁇ -2-methyl-benzofuran-3- carboxylic acid.
- a solution water (850 mL), potassium carbonate (212.08 g, 1.5345 mol, 3 eq), dioxane (500 mL), ⁇ -bromo-benzojVlthiophene-S-carboxylic acid ethyl ester (175 g, 0.6138 mol, 1.2 eq) and tetrakis(triphenylphosphine)palladium (35.47 g, 0.03 mol,0.06 eq) is heated to reflux (87-90 °C).
- the oil (737 g) is dissolved in EtOAc (500 mL). Heptane is added until the solution becomes hazy and the oil is separated ( ⁇ 3 L). The resulting hazy solution is stirred for 1 h. The suspension is filtered and the filter cake is washed with heptane (200 mL). The combined filtrate is purified via column chromatograpy on silica gel columns (2 X 1.5 Kg) eluting with EtOAc (5 to 20 %) in heptane. The appropriate fractions are combined and concentrated under reduced pressure to afford a pale yellow oil (401 g). The impure product fractions are concentrated (-200 g of oil) and the silica gel purification is repeated using 2 Kg of silica gel. The appropriate fractions are combined and concentrated to afford the title compound as a thick pale yellow oil (496 g, 99 %).
- Solution A A solution of 6- ⁇ 4-[5-cyclopropyl-3-(2-trifluoxomethoxy-phenyl)-isoxazol-4- y]memoxy]-2-methyl-phenyl ⁇ -benzo[b]tmophene-3-carboxylic acid ethyl ester (470 g) in EtOH (1.8 L) is heated to 40 °C.
- Solution B In a separate flask is added 50 % NaOH (158.6 g, 1.9825 mol, 2.5 eq) in water (125 mL) and EtOH (600 niL).
- Solution B is added via addition fiinnel to Solution A at 40-50 0 C at a such a rate to prevent formation of significant amount of solids.
- the reaction mixture is heated to 65-75 0 C and stirred at this temperature for 1 h.
- the reaction mixture is cooled to room temperature.
- the reaction mixture is diluted with water (3 L), EtOAc (2.5 L), and 10 % aqueous citric acid solution (3 L). The layers are separated and lhe aqueous layer is extracted with EtOAc (2 L). The combined organic layer is washed with brine (3 L), dried over magnesium sulfate, filtered, and concentrated to afford a pale yellow oil (480 g).
- the oil is co-evapotated with EtOH (1 L) and dissolved (51O g) in MeOH (1.2 L).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Emergency Medicine (AREA)
- Vascular Medicine (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Child & Adolescent Psychology (AREA)
- Ophthalmology & Optometry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
Description

































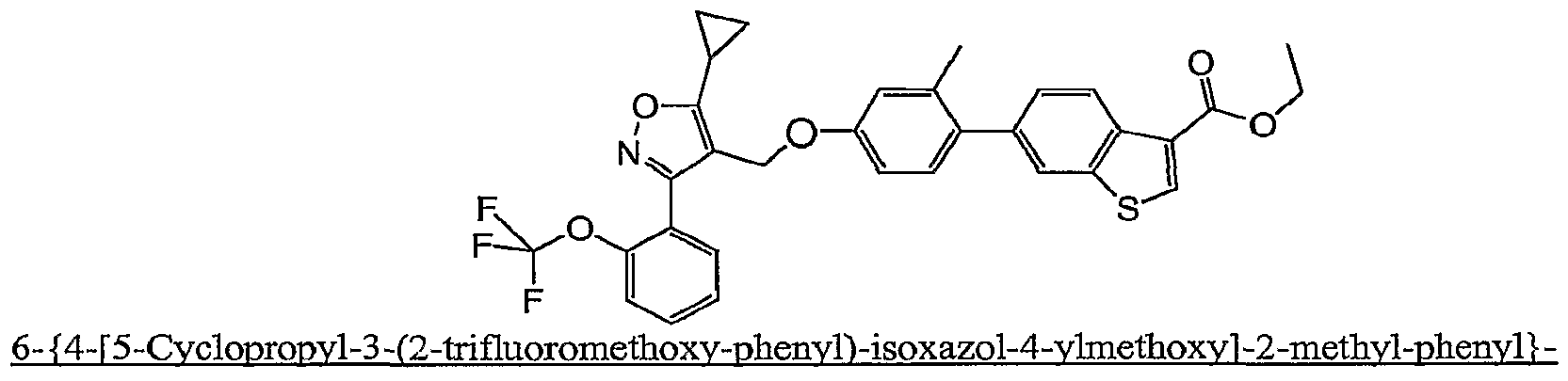
Claims





Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002640476A CA2640476A1 (en) | 2006-02-03 | 2007-02-02 | Compounds and methods for modulating fx-receptors |
| BRPI0707427-1A BRPI0707427A2 (en) | 2006-02-03 | 2007-02-02 | a compound or a pharmaceutically acceptable salt thereof, a method for treating dyslipidemia, atherosclerosis, and diabetes and complications thereof, to elevate plasma levels of hdl, and to decrease plasma triglycerides, pharmaceutical composition, and use of a compound or salt. pharmaceutically acceptable thereof |
| US12/159,224 US7863302B2 (en) | 2006-02-03 | 2007-02-02 | Compounds and methods for modulating FX-receptors |
| ES07763142.2T ES2452031T3 (en) | 2006-02-03 | 2007-02-02 | Compounds and procedures to modulate FX receivers |
| AU2007212126A AU2007212126B2 (en) | 2006-02-03 | 2007-02-02 | Compounds and methods for modulating FX-receptors |
| CN2007800038562A CN101374834B (en) | 2006-02-03 | 2007-02-02 | Compounds and methods for modulating FXR |
| EP07763142.2A EP1984360B1 (en) | 2006-02-03 | 2007-02-02 | Compounds and methods for modulating FX-receptors |
| JP2008553518A JP5301286B2 (en) | 2006-02-03 | 2007-02-02 | Compounds and methods for modulating FX receptors |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US76540706P | 2006-02-03 | 2006-02-03 | |
| US60/765,407 | 2006-02-03 | ||
| US80631006P | 2006-06-30 | 2006-06-30 | |
| US60/806,310 | 2006-06-30 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007092751A2 true WO2007092751A2 (en) | 2007-08-16 |
| WO2007092751A3 WO2007092751A3 (en) | 2007-12-27 |
Family
ID=38229980
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/061515 WO2007092751A2 (en) | 2006-02-03 | 2007-02-02 | Compounds and methods for modulating fx-receptors |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US7863302B2 (en) |
| EP (1) | EP1984360B1 (en) |
| JP (1) | JP5301286B2 (en) |
| CN (1) | CN101374834B (en) |
| AU (1) | AU2007212126B2 (en) |
| BR (1) | BRPI0707427A2 (en) |
| CA (1) | CA2640476A1 (en) |
| ES (1) | ES2452031T3 (en) |
| WO (1) | WO2007092751A2 (en) |
Cited By (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1962838A2 (en) * | 2005-12-19 | 2008-09-03 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| WO2009012125A1 (en) | 2007-07-16 | 2009-01-22 | Eli Lilly And Company | Compounds and methods for modulating fxr |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| US7618973B2 (en) | 2007-12-04 | 2009-11-17 | Hoffmann-La Roche Inc. | Isoxazolo-pyrazine derivatives |
| EP2128158A1 (en) | 2008-05-26 | 2009-12-02 | Phenex Pharmaceuticals AG | Heterocyclic cyclopropyl-substituted FXR binding compounds |
| EP2170072A1 (en) * | 2007-06-13 | 2010-04-07 | GlaxoSmithKline LLC | Farnesoid x receptor agonists |
| WO2010079443A1 (en) | 2009-01-12 | 2010-07-15 | Pfizer Limited | Sulfonamide derivatives |
| WO2011020615A1 (en) | 2009-08-19 | 2011-02-24 | Phenex Pharmaceuticals Ag | Novel fxr (nr1h4 ) binding and activity modulating compounds |
| US7902201B2 (en) | 2007-12-04 | 2011-03-08 | Hoffmann-La Roche Inc. | Isoxazolo-pyrazine derivatives |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| US8163728B2 (en) | 2009-05-05 | 2012-04-24 | Hoffmann-La Roche Inc. | Pyrazoles |
| US8173652B2 (en) | 2009-02-19 | 2012-05-08 | Hoffmann-La Roche Inc. | Isoxazole-isoxazoles and isoxazole-isothiazoles |
| US8178522B2 (en) | 2009-05-05 | 2012-05-15 | Hoffmann-La Roche Inc. | Thiazoles |
| WO2012087521A1 (en) * | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| WO2012087520A1 (en) * | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| US8222246B2 (en) | 2009-04-02 | 2012-07-17 | Hoffmann-La Roche Inc. | Substituted isoxazoles |
| US8227461B2 (en) | 2009-04-30 | 2012-07-24 | Hoffmann-La Roche Inc. | Isoxazoles |
| WO2012114285A1 (en) | 2011-02-23 | 2012-08-30 | Lupin Limited | Heteroaryl derivatives as alpha7 nachr modulators |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| US8293738B2 (en) | 2010-05-12 | 2012-10-23 | Abbott Laboratories | Indazole inhibitors of kinase |
| WO2013005153A1 (en) | 2011-07-05 | 2013-01-10 | Lupin Limited | Biaryl derivatives as nachr modulators |
| EP2545964A1 (en) | 2011-07-13 | 2013-01-16 | Phenex Pharmaceuticals AG | Novel FXR (NR1H4) binding and activity modulating compounds |
| US8357703B2 (en) | 2009-05-07 | 2013-01-22 | Hoffmann-La Roche Inc. | Pyridines |
| US8389550B2 (en) | 2009-02-25 | 2013-03-05 | Hoffmann-La Roche Inc. | Isoxazoles / O-pyridines with ethyl and ethenyl linker |
| WO2013037482A1 (en) | 2011-09-15 | 2013-03-21 | Phenex Pharmaceuticals Ag | Farnesoid x receptor agonists for cancer treatment and prevention |
| US8410104B2 (en) | 2009-05-05 | 2013-04-02 | Hoffmann-La Roche Inc. | Pyridazines |
| US8415379B2 (en) | 2009-05-05 | 2013-04-09 | Hoffmann-La Roche Inc. | Pyridines |
| WO2013125732A1 (en) | 2012-02-24 | 2013-08-29 | Takeda Pharmaceutical Company Limited | Aromatic ring compound |
| US8785435B2 (en) | 2011-10-20 | 2014-07-22 | Hoffmann-La Roche Inc. | Solid forms |
| US9150568B2 (en) | 2010-12-20 | 2015-10-06 | Novartis Ag | Compositions and methods for modulating FXR |
| EP3006939A1 (en) | 2014-10-06 | 2016-04-13 | Gilead Sciences, Inc. | Histidine-rich Glycoprotein as a marker for hepatic Farnesoid X receptor activation |
| WO2016086218A1 (en) | 2014-11-26 | 2016-06-02 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as fxr/tgr5 agonists and methods of use thereof |
| EP3034501A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Hydroxy containing FXR (NR1H4) modulating compounds |
| EP3034499A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Novel FXR (NR1H4) modulating compounds |
| US9388196B2 (en) | 2012-03-06 | 2016-07-12 | Lupin Limited | Thiazole derivatives as alpha 7 nAChR modulators |
| WO2016127924A1 (en) | 2015-02-13 | 2016-08-18 | Sunshine Lake Pharma Co., Ltd. | Tricyclic compounds and uses thereof in medicine |
| WO2018178260A1 (en) | 2017-03-30 | 2018-10-04 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for reducing persistence and expression of episomal viruses |
| US10329286B2 (en) | 2016-06-13 | 2019-06-25 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| WO2019149158A1 (en) | 2018-02-02 | 2019-08-08 | Sunshine Lake Pharma Co., Ltd. | Nitrogenous tricyclic compounds and uses thereof in medicine |
| US10421730B2 (en) | 2016-06-13 | 2019-09-24 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| EP3711762A1 (en) | 2013-09-11 | 2020-09-23 | INSERM (Institut National de la Santé et de la Recherche Médicale) | A farnesoid x receptor agonsits foruse and pharmaceutical compositions for the treatment of chronic hepatitis b virus infection |
| WO2021009332A1 (en) | 2019-07-18 | 2021-01-21 | Enyo Pharma | Method for decreasing adverse-effects of interferon |
| US10988449B2 (en) | 2017-04-12 | 2021-04-27 | Il Dong Pharmaceutical Co., Ltd. | Isoxazole derivatives as nuclear receptor agonists and uses thereof |
| WO2021144330A1 (en) | 2020-01-15 | 2021-07-22 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of fxr agonists for treating an infection by hepatitis d virus |
| US11225473B2 (en) | 2019-01-15 | 2022-01-18 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| WO2022152770A1 (en) | 2021-01-14 | 2022-07-21 | Enyo Pharma | Synergistic effect of a fxr agonist and ifn for the treatment of hbv infection |
| WO2022229302A1 (en) | 2021-04-28 | 2022-11-03 | Enyo Pharma | Strong potentiation of tlr3 agonists effects using fxr agonists as a combined treatment |
| US11524005B2 (en) | 2019-02-19 | 2022-12-13 | Gilead Sciences, Inc. | Solid forms of FXR agonists |
| US11833150B2 (en) | 2017-03-28 | 2023-12-05 | Gilead Sciences, Inc. | Methods of treating liver disease |
Families Citing this family (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2030974B1 (en) * | 2006-06-13 | 2016-02-17 | Shanghai Institute of Materia Medica, Chinese Academy of Sciences | Thiazole non-nucleoside compounds, their preparation, pharmaceutical composition and their use as antiviral agents |
| US8889730B2 (en) | 2012-04-10 | 2014-11-18 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| JP6064062B2 (en) | 2013-03-15 | 2017-01-18 | ファイザー・インク | Indazole compounds that activate AMPK |
| MA41094A (en) * | 2014-12-02 | 2017-10-10 | Lilly Co Eli | KIDNEY DISORDERS TREATMENT METHODS |
| CN107427527B (en) | 2015-03-31 | 2021-01-26 | 英安塔制药有限公司 | Bile acid derivatives as FXR/TGR5 agonists and methods of use thereof |
| CN106995416A (en) * | 2016-01-26 | 2017-08-01 | 上海翰森生物医药科技有限公司 | FXR activators and its preparation method and application |
| WO2017128896A1 (en) * | 2016-01-26 | 2017-08-03 | 江苏豪森药业集团有限公司 | Fxr agonist and preparation method and use thereof |
| WO2017189663A1 (en) | 2016-04-26 | 2017-11-02 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as fxr agonists and methods of use thereof |
| US10080741B2 (en) | 2016-04-26 | 2018-09-25 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| US10080742B2 (en) | 2016-04-26 | 2018-09-25 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| US10144729B2 (en) | 2016-05-18 | 2018-12-04 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as FXR agonists and methods of use thereof |
| US10149835B2 (en) | 2016-05-18 | 2018-12-11 | Elmore Patent Law Group, P.C. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| US10138228B2 (en) | 2016-05-18 | 2018-11-27 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use therof |
| TW201808283A (en) | 2016-08-05 | 2018-03-16 | 廣東東陽光藥業有限公司 | Nitrogen-containing tricyclic compounds and uses thereof in medicine |
| RU2019113066A (en) | 2016-10-04 | 2020-11-09 | Энанта Фармасьютикалс, Инк. | ISOXAZOLE ANALOGUES AS FXR AGONISTS AND METHODS OF THEIR APPLICATION |
| US10597391B2 (en) | 2016-10-26 | 2020-03-24 | Enanta Pharmaceuticals, Inc. | Urea-containing isoxazole derivatives as FXR agonists and methods of use thereof |
| CN108017636A (en) * | 2016-11-04 | 2018-05-11 | 合帕吉恩治疗公司 | Nitrogen-containing heterocycle compound as FXR conditioning agents |
| CN108218852A (en) * | 2016-12-15 | 2018-06-29 | 宁波百纳西药业有限公司 | A kind of spiro-compound, preparation method, composition and purposes |
| CN109071468B (en) * | 2017-01-20 | 2022-09-02 | 四川科伦博泰生物医药股份有限公司 | Heterocyclic compound and preparation method and application thereof |
| CN106977535A (en) * | 2017-04-28 | 2017-07-25 | 大连联化化学有限公司 | One kind synthesis fluorobenzoic boric acid technique of 2 cyano group 3 |
| CN109575008B (en) * | 2017-09-29 | 2020-11-17 | 轩竹生物科技有限公司 | FXR receptor agonists |
| KR20200081435A (en) | 2017-11-01 | 2020-07-07 | 브리스톨-마이어스 스큅 컴퍼니 | Multicyclic compounds as farnesoid X receptor modulators |
| JP7264906B2 (en) | 2017-11-01 | 2023-04-25 | ブリストル-マイヤーズ スクイブ カンパニー | Alkene compounds as farnesoid X receptor modulators |
| CA3080893A1 (en) | 2017-11-01 | 2019-05-09 | Bristol-Myers Squibb Company | Alkene spirocyclic compounds as farnesoid x receptor modulators |
| US10689391B2 (en) | 2017-12-12 | 2020-06-23 | Enanta Pharmaceuticals, Inc. | Isoxazole analogs as FXR agonists and methods of use thereof |
| WO2019160813A1 (en) | 2018-02-14 | 2019-08-22 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as fxr agonists and methods of use thereof |
| CN111825667B (en) * | 2019-04-19 | 2023-07-25 | 中国科学院上海药物研究所 | FXR small molecule agonist and preparation method and application thereof |
| US11555032B2 (en) | 2019-05-13 | 2023-01-17 | Enanta Pharmaceuticals, Inc. | Isoxazole derivatives as FXR agonists and methods of use thereof |
| CN111004211B (en) * | 2019-12-29 | 2021-04-02 | 苏州诚和医药化学有限公司 | Synthetic method of brexpiprazole intermediate 4-bromobenzo [ b ] thiophene |
| WO2021145543A1 (en) * | 2020-01-15 | 2021-07-22 | 재단법인 대구경북과학기술원 | Conjugate consisting of extracellular matrix and anticancer drug, and medical use thereof |
| CN112876516B (en) * | 2021-02-05 | 2022-02-15 | 昆明理工大学 | N- (4-indolyl) N-heterocyclic carbene palladium complex and application thereof |
| CN112898247B (en) * | 2021-03-09 | 2023-03-14 | 中国科学院兰州化学物理研究所 | Synthetic method of furan acetate compound |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9914977D0 (en) * | 1999-06-25 | 1999-08-25 | Glaxo Group Ltd | Chemical compounds |
| ATE381542T1 (en) * | 2001-08-13 | 2008-01-15 | Phenex Pharmaceuticals Ag | NR1H4 CORE RECEPTOR BINDING COMPOUNDS |
| EP1562915A1 (en) * | 2002-11-22 | 2005-08-17 | SmithKline Beecham Corporation | Farnesoid x receptor agonists |
| JP5081161B2 (en) * | 2005-12-19 | 2012-11-21 | スミスクライン ビーチャム コーポレーション | Farnesoid X receptor agonist |
-
2007
- 2007-02-02 JP JP2008553518A patent/JP5301286B2/en active Active
- 2007-02-02 WO PCT/US2007/061515 patent/WO2007092751A2/en active Application Filing
- 2007-02-02 US US12/159,224 patent/US7863302B2/en active Active
- 2007-02-02 AU AU2007212126A patent/AU2007212126B2/en not_active Ceased
- 2007-02-02 ES ES07763142.2T patent/ES2452031T3/en active Active
- 2007-02-02 CA CA002640476A patent/CA2640476A1/en not_active Abandoned
- 2007-02-02 BR BRPI0707427-1A patent/BRPI0707427A2/en not_active IP Right Cessation
- 2007-02-02 CN CN2007800038562A patent/CN101374834B/en not_active Expired - Fee Related
- 2007-02-02 EP EP07763142.2A patent/EP1984360B1/en active Active
Non-Patent Citations (6)
| Title |
|---|
| "Dorland's Illustrated Medical Dictionary", WB SAUNDERS PUBLISHING COMPANY |
| "Remington's Pharmaceutical Sciences", 1990, MACK PUBLISHING CO. |
| ALDRICHIMICA ACTA, vol. 17, no. 1, 1984 |
| J. MED. CHEM., vol. 43, no. 16, 2000, pages 2971 |
| MED. CHEM., vol. 43, no. 16, 2000, pages 2971 - 2974 |
| THEODORA GREENE: "Protecting Groups in Organic Synthesis", WILEY-INTERSCIENCE |
Cited By (79)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1962838A4 (en) * | 2005-12-19 | 2009-11-18 | Smithkline Beecham Corp | Farnesoid x receptor agonists |
| EP1962838A2 (en) * | 2005-12-19 | 2008-09-03 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| EP2170072A4 (en) * | 2007-06-13 | 2010-10-27 | Glaxosmithkline Llc | Farnesoid x receptor agonists |
| EP2170072A1 (en) * | 2007-06-13 | 2010-04-07 | GlaxoSmithKline LLC | Farnesoid x receptor agonists |
| JP2010533722A (en) * | 2007-07-16 | 2010-10-28 | イーライ リリー アンド カンパニー | Compounds and methods for modulating FXR |
| WO2009012125A1 (en) | 2007-07-16 | 2009-01-22 | Eli Lilly And Company | Compounds and methods for modulating fxr |
| US8153624B2 (en) | 2007-07-16 | 2012-04-10 | Eli Lilly And Company | Compounds and methods for modulating FXR |
| EA016475B1 (en) * | 2007-07-16 | 2012-05-30 | Эли Лилли Энд Компани | Azol compounds for use as fxr modulators |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| US7618973B2 (en) | 2007-12-04 | 2009-11-17 | Hoffmann-La Roche Inc. | Isoxazolo-pyrazine derivatives |
| US7902201B2 (en) | 2007-12-04 | 2011-03-08 | Hoffmann-La Roche Inc. | Isoxazolo-pyrazine derivatives |
| EP2128158A1 (en) | 2008-05-26 | 2009-12-02 | Phenex Pharmaceuticals AG | Heterocyclic cyclopropyl-substituted FXR binding compounds |
| WO2010079443A1 (en) | 2009-01-12 | 2010-07-15 | Pfizer Limited | Sulfonamide derivatives |
| US8173652B2 (en) | 2009-02-19 | 2012-05-08 | Hoffmann-La Roche Inc. | Isoxazole-isoxazoles and isoxazole-isothiazoles |
| US8389550B2 (en) | 2009-02-25 | 2013-03-05 | Hoffmann-La Roche Inc. | Isoxazoles / O-pyridines with ethyl and ethenyl linker |
| US8222246B2 (en) | 2009-04-02 | 2012-07-17 | Hoffmann-La Roche Inc. | Substituted isoxazoles |
| US8227461B2 (en) | 2009-04-30 | 2012-07-24 | Hoffmann-La Roche Inc. | Isoxazoles |
| US8415379B2 (en) | 2009-05-05 | 2013-04-09 | Hoffmann-La Roche Inc. | Pyridines |
| US8178522B2 (en) | 2009-05-05 | 2012-05-15 | Hoffmann-La Roche Inc. | Thiazoles |
| US8163728B2 (en) | 2009-05-05 | 2012-04-24 | Hoffmann-La Roche Inc. | Pyrazoles |
| US8410104B2 (en) | 2009-05-05 | 2013-04-02 | Hoffmann-La Roche Inc. | Pyridazines |
| US8357703B2 (en) | 2009-05-07 | 2013-01-22 | Hoffmann-La Roche Inc. | Pyridines |
| WO2011020615A1 (en) | 2009-08-19 | 2011-02-24 | Phenex Pharmaceuticals Ag | Novel fxr (nr1h4 ) binding and activity modulating compounds |
| CN102548974A (en) * | 2009-08-19 | 2012-07-04 | 菲尼克斯药品股份公司 | Novel FXR (NR1H4 ) binding and activity modulating compounds |
| CN102548974B (en) * | 2009-08-19 | 2015-11-25 | 菲尼克斯药品股份公司 | Novel FXR (NR1H4) combines and activity modulating compounds |
| US8952042B2 (en) | 2009-08-19 | 2015-02-10 | Phenex Pharmaceuticals Ag | FXR (NR1H4) binding and activity modulating compounds |
| EP2289883A1 (en) | 2009-08-19 | 2011-03-02 | Phenex Pharmaceuticals AG | Novel FXR (NR1H4) binding and activity modulating compounds |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| US8293738B2 (en) | 2010-05-12 | 2012-10-23 | Abbott Laboratories | Indazole inhibitors of kinase |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2012087520A1 (en) * | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| US9150568B2 (en) | 2010-12-20 | 2015-10-06 | Novartis Ag | Compositions and methods for modulating FXR |
| WO2012087521A1 (en) * | 2010-12-20 | 2012-06-28 | Irm Llc | Compositions and methods for modulating farnesoid x receptors |
| US9393247B2 (en) | 2011-02-23 | 2016-07-19 | Lupin Limited | Heteroaryl derivatives as alpha7 nAChR modulators |
| WO2012114285A1 (en) | 2011-02-23 | 2012-08-30 | Lupin Limited | Heteroaryl derivatives as alpha7 nachr modulators |
| US9072731B2 (en) | 2011-02-23 | 2015-07-07 | Lupin Limited | Heteroaryl derivatives as alpha7 nAChR modulators |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2013005153A1 (en) | 2011-07-05 | 2013-01-10 | Lupin Limited | Biaryl derivatives as nachr modulators |
| US8946432B2 (en) | 2011-07-05 | 2015-02-03 | Lupin Limited | Biaryl derivatives as nAChR modulators |
| EP2987532A1 (en) | 2011-07-13 | 2016-02-24 | Gilead Sciences, Inc. | Novel fxr (nr1h4) binding and activity modulating compounds |
| US9539244B2 (en) | 2011-07-13 | 2017-01-10 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| US9139539B2 (en) | 2011-07-13 | 2015-09-22 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| US10485795B2 (en) | 2011-07-13 | 2019-11-26 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| EP3246070A1 (en) | 2011-07-13 | 2017-11-22 | Gilead Sciences, Inc. | Novel fxr (nr1h4) binding and activity modulating azoles |
| WO2013007387A1 (en) | 2011-07-13 | 2013-01-17 | Phenex Pharmaceuticals Ag | Novel fxr (nr1h4) binding and activity modulating compounds |
| US9820979B2 (en) | 2011-07-13 | 2017-11-21 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| US10220027B2 (en) | 2011-07-13 | 2019-03-05 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| EP4400503A2 (en) | 2011-07-13 | 2024-07-17 | Gilead Sciences, Inc. | Novel fxr (nr1h4) binding and activity modulating compounds |
| EP2545964A1 (en) | 2011-07-13 | 2013-01-16 | Phenex Pharmaceuticals AG | Novel FXR (NR1H4) binding and activity modulating compounds |
| WO2013037482A1 (en) | 2011-09-15 | 2013-03-21 | Phenex Pharmaceuticals Ag | Farnesoid x receptor agonists for cancer treatment and prevention |
| US8785435B2 (en) | 2011-10-20 | 2014-07-22 | Hoffmann-La Roche Inc. | Solid forms |
| WO2013125732A1 (en) | 2012-02-24 | 2013-08-29 | Takeda Pharmaceutical Company Limited | Aromatic ring compound |
| US9388196B2 (en) | 2012-03-06 | 2016-07-12 | Lupin Limited | Thiazole derivatives as alpha 7 nAChR modulators |
| EP3711762A1 (en) | 2013-09-11 | 2020-09-23 | INSERM (Institut National de la Santé et de la Recherche Médicale) | A farnesoid x receptor agonsits foruse and pharmaceutical compositions for the treatment of chronic hepatitis b virus infection |
| EP3006939A1 (en) | 2014-10-06 | 2016-04-13 | Gilead Sciences, Inc. | Histidine-rich Glycoprotein as a marker for hepatic Farnesoid X receptor activation |
| WO2016086218A1 (en) | 2014-11-26 | 2016-06-02 | Enanta Pharmaceuticals, Inc. | Bile acid derivatives as fxr/tgr5 agonists and methods of use thereof |
| EP3034499A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Novel FXR (NR1H4) modulating compounds |
| EP3034501A1 (en) | 2014-12-17 | 2016-06-22 | Gilead Sciences, Inc. | Hydroxy containing FXR (NR1H4) modulating compounds |
| WO2016127924A1 (en) | 2015-02-13 | 2016-08-18 | Sunshine Lake Pharma Co., Ltd. | Tricyclic compounds and uses thereof in medicine |
| US11247986B2 (en) | 2016-06-13 | 2022-02-15 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10421730B2 (en) | 2016-06-13 | 2019-09-24 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10774054B2 (en) | 2016-06-13 | 2020-09-15 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10981881B2 (en) | 2016-06-13 | 2021-04-20 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10329286B2 (en) | 2016-06-13 | 2019-06-25 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US11739065B2 (en) | 2016-06-13 | 2023-08-29 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US11833150B2 (en) | 2017-03-28 | 2023-12-05 | Gilead Sciences, Inc. | Methods of treating liver disease |
| WO2018178260A1 (en) | 2017-03-30 | 2018-10-04 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for reducing persistence and expression of episomal viruses |
| US10988449B2 (en) | 2017-04-12 | 2021-04-27 | Il Dong Pharmaceutical Co., Ltd. | Isoxazole derivatives as nuclear receptor agonists and uses thereof |
| WO2019149158A1 (en) | 2018-02-02 | 2019-08-08 | Sunshine Lake Pharma Co., Ltd. | Nitrogenous tricyclic compounds and uses thereof in medicine |
| US11225473B2 (en) | 2019-01-15 | 2022-01-18 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US11524005B2 (en) | 2019-02-19 | 2022-12-13 | Gilead Sciences, Inc. | Solid forms of FXR agonists |
| WO2021009332A1 (en) | 2019-07-18 | 2021-01-21 | Enyo Pharma | Method for decreasing adverse-effects of interferon |
| WO2021144330A1 (en) | 2020-01-15 | 2021-07-22 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of fxr agonists for treating an infection by hepatitis d virus |
| WO2022152770A1 (en) | 2021-01-14 | 2022-07-21 | Enyo Pharma | Synergistic effect of a fxr agonist and ifn for the treatment of hbv infection |
| WO2022229302A1 (en) | 2021-04-28 | 2022-11-03 | Enyo Pharma | Strong potentiation of tlr3 agonists effects using fxr agonists as a combined treatment |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2007092751A3 (en) | 2007-12-27 |
| US20080306125A1 (en) | 2008-12-11 |
| JP2009525984A (en) | 2009-07-16 |
| CA2640476A1 (en) | 2007-08-16 |
| EP1984360A2 (en) | 2008-10-29 |
| JP5301286B2 (en) | 2013-09-25 |
| BRPI0707427A2 (en) | 2011-05-03 |
| ES2452031T3 (en) | 2014-03-31 |
| AU2007212126A1 (en) | 2007-08-16 |
| CN101374834B (en) | 2011-12-14 |
| CN101374834A (en) | 2009-02-25 |
| AU2007212126B2 (en) | 2012-07-19 |
| EP1984360B1 (en) | 2014-01-15 |
| US7863302B2 (en) | 2011-01-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1984360B1 (en) | Compounds and methods for modulating FX-receptors | |
| US8093278B2 (en) | Substituted indoles | |
| EP1513812B1 (en) | Substituted indoles | |
| JP4894517B2 (en) | 2-Phenylpyridine derivative | |
| CA2771445C (en) | Novel fxr (nr1h4) binding and activity modulating compounds | |
| CN101180290B (en) | Hydogrenated benzo[c] thiophene derivatives as immunomodulators | |
| KR20100044810A (en) | Farnesoid x receptor agonists | |
| TW200536832A (en) | Process for producing 5-hydroxy-4-thiomethylpyrazole compound | |
| PT1852433E (en) | Carbazole derivative, solvate thereof, or pharmaceutically acceptable salt thereof | |
| EA015632B1 (en) | Fxr agonists | |
| TW201021798A (en) | Amide acetate derivative having inhibitory activity on endothelial lipase | |
| JPWO2006022375A1 (en) | 2-Phenylthiophene derivatives | |
| KR20100031725A (en) | Pyridone compound | |
| JPWO2007097403A1 (en) | Gastrointestinal ulcer treatment or prevention | |
| KR20120080183A (en) | Novel phenol derivative | |
| JPWO2008016175A1 (en) | Peroxisome proliferator-activated receptor activator | |
| MX2011000044A (en) | Nitrogen-containing aromatic heterocyclyl compound. | |
| JP4986927B2 (en) | Medicine | |
| TW201925211A (en) | Process for preparing benzothiophen-2yl boronate | |
| JPH0673012A (en) | 4-iminoquinoline, its preparation and its use | |
| CN101094827B (en) | Benzene compound having two or more substituents | |
| MX2008010004A (en) | Compounds and methods for modulating fx-receptors | |
| EP2475642A1 (en) | Use of indole derivatives as nurr-1 activators for the application thereof as a medicament for the treatment of parkinson's disease | |
| JP2003267870A (en) | Pulmonary hypertension-preventing or treating agent | |
| JPH01104060A (en) | Haloalkylthiazole and manufacture |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 12159224 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007212126 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2788/KOLNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007763142 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2640476 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780003856.2 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2007212126 Country of ref document: AU Date of ref document: 20070202 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/010004 Country of ref document: MX Ref document number: 2008553518 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: PI0707427 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080801 |