WO2007057775A1 - Spiropiperidine derivatives - Google Patents
Spiropiperidine derivatives Download PDFInfo
- Publication number
- WO2007057775A1 WO2007057775A1 PCT/IB2006/003320 IB2006003320W WO2007057775A1 WO 2007057775 A1 WO2007057775 A1 WO 2007057775A1 IB 2006003320 W IB2006003320 W IB 2006003320W WO 2007057775 A1 WO2007057775 A1 WO 2007057775A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- aminopyridin
- piperidine
- spiro
- carboxamide
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- ZRRKIBBHARIWBI-KRWDZBQOSA-N C([C@@H]1c2nnc(-c3ccccc3)[o]2)C2(CCNCC2)c2c1cccc2 Chemical compound C([C@@H]1c2nnc(-c3ccccc3)[o]2)C2(CCNCC2)c2c1cccc2 ZRRKIBBHARIWBI-KRWDZBQOSA-N 0.000 description 1
- KGSIOJZCEWLTCF-UHFFFAOYSA-N C1C2(CCNCC2)c2ccccc2C1Nc1ncccc1 Chemical compound C1C2(CCNCC2)c2ccccc2C1Nc1ncccc1 KGSIOJZCEWLTCF-UHFFFAOYSA-N 0.000 description 1
- VWILPRXTTLMHQV-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1(C1)c2ccccc2C1(C)C(N(C)C)=O)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1(C1)c2ccccc2C1(C)C(N(C)C)=O)=O VWILPRXTTLMHQV-UHFFFAOYSA-N 0.000 description 1
- KQOQFYKSAOQUMZ-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC11OC(C(O)=O)c2ccccc12)=O Chemical compound CC(C)(C)OC(N(CC1)CCC11OC(C(O)=O)c2ccccc12)=O KQOQFYKSAOQUMZ-UHFFFAOYSA-N 0.000 description 1
- ODVJVEBQBXOGJF-INIZCTEOSA-N CC(C)(C)OC(N(CC1)CCC11c2ccccc2O[C@H]1C(N(C)C)=O)=O Chemical compound CC(C)(C)OC(N(CC1)CCC11c2ccccc2O[C@H]1C(N(C)C)=O)=O ODVJVEBQBXOGJF-INIZCTEOSA-N 0.000 description 1
- UFYSVGNJNSBYCA-UHFFFAOYSA-N CCN(CC1)CCN1C(c1c2OCC(CC3)(CCN3C(C3)N=CC=C3N)c2ccc1)=O Chemical compound CCN(CC1)CCN1C(c1c2OCC(CC3)(CCN3C(C3)N=CC=C3N)c2ccc1)=O UFYSVGNJNSBYCA-UHFFFAOYSA-N 0.000 description 1
- YFIANJUHVWWFDC-UHFFFAOYSA-N CN(C)C(C1OC2(CCNCC2)c2ccccc12)=O Chemical compound CN(C)C(C1OC2(CCNCC2)c2ccccc12)=O YFIANJUHVWWFDC-UHFFFAOYSA-N 0.000 description 1
- PTIVTVMZNHOPRD-UHFFFAOYSA-N COc1ccc(CN(C2(C=CNCC2)c2ccccc22)C2=O)cc1 Chemical compound COc1ccc(CN(C2(C=CNCC2)c2ccccc22)C2=O)cc1 PTIVTVMZNHOPRD-UHFFFAOYSA-N 0.000 description 1
- VIIKVBZZBYRWSF-UHFFFAOYSA-N CS(NC(CC1(CC2)CCN2c2cc(N)ccn2)c2c1cccc2)(=O)=O Chemical compound CS(NC(CC1(CC2)CCN2c2cc(N)ccn2)c2c1cccc2)(=O)=O VIIKVBZZBYRWSF-UHFFFAOYSA-N 0.000 description 1
- XQWVFQJURPACMK-UHFFFAOYSA-N N#Cc1cccc(Br)c1OCC1=CCN(Cc2ccccc2)CC1 Chemical compound N#Cc1cccc(Br)c1OCC1=CCN(Cc2ccccc2)CC1 XQWVFQJURPACMK-UHFFFAOYSA-N 0.000 description 1
- VICQPIDPFCIFDE-CQSZACIVSA-N Nc1cc(N(CC2)CCC2(C2)c3ccccc3[C@H]2C#N)ncc1 Chemical compound Nc1cc(N(CC2)CCC2(C2)c3ccccc3[C@H]2C#N)ncc1 VICQPIDPFCIFDE-CQSZACIVSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
Definitions
- This invention relates to spiropiperidine derivatives. More particularly, this invention relates to (aminopyridyl)spiropiperidine derivatives and to processes for the preparation of, intermediates used in the preparation of, compositions containing and the uses of, such derivatives.
- the spiropiperidine derivatives of the present invention are delta opioid receptor agonists and have a number of therapeutic applications, particularly in the treatment of pain, especially neuropathic pain.
- Delta opioid receptors DORs
- MORs mu opioid receptors
- KORs kappa opioid receptors
- GPCRs G-protein coupled receptors
- Stimulation of DOR induces analgesia in a wide variety of animal models especially inflammatory models (Quock et al, 1999, above).
- Mouse KO studies have shown that the abuse liability and other CNS risks associated with opiates, such as morphine, are mediated through the mu opioid receptor (Kieffer and Gaveriaux-Ruff 2002 Prog. Neurobio. 66: 285-306).
- a DOR agonist approach may be devoid of the side effect profiles of mu and kappa opioids (Quock et al., 1999, above). Generally there is good correlation in DOR expression between species with few exceptions e.g. in the spinal cord where, in humans expression is restricted to laminae Ml, an important region for nociception (Mennicken et al., 2003 J. Comparative Neurol. 465: 349-360).
- Systemic administration of the selective agonist SB-235863 reverses thermal hyperalgesia resulting from sciatic nerve ligation without any effect on gastrointestinal transit or motor coordination (Petrillo et al, above).
- Systemic administration of DOR agonists has little effect on acute pain but results in potent analgesia following morphine pre-treatment or following inflammatory stimuli (Morinville et al., above). This seems to occur as a consequence of increased trafficking and membrane insertion of DORs located in cytoplasmic vesicles (Cahill et al., above).
- WO 2004/074273 discloses compounds of formula:
- WO 2004/004715 describes a method for modulation of conditions associated with the delta opioid receptor, comprising administration of a compound of formula:
- the present invention provides a compound of formula (I):
- R 1 represents H, (Ci -10 )alkyl (optionally substituted by one or more substituents ⁇ ), -(CR 2 ) n -
- (C 3-8 )cycloalkyl (wherein the cycloalkyl part is optionally substituted by one or more substituents ⁇ ), -(CR 2 ) n -Ar, -(CR 2 ) n -Het 1 , -(CR 2 ) n -Het 2 , or -CN, n is O or an integer from 1 to 6;
- R represents H or (C 1-10 )alkyl; or, when a group R and a group R 1 substitute a nitrogen atom, the groups together with the nitrogen atom form a ring Het 3 ;
- R 2 and R 3 are the same or different and each represent R or -(CR 2 )-Ar, or R 2 and R 3 , together with the nitrogen atom to which they are attached, form a ring Het 3 ;
- R 4 represents (C 1-10 )alkyl; a is O, 1 or 2;
- R 5 represents (C ⁇ iojalkyl, (C 1-10 )haloalkyl, hydroxy-(C 1-10 )alkyl, (C 1-10 )alkoxy(C 1-10 )alkyl, halogen, OH, (C 1-10 )alkoxy, (C ⁇ n ⁇ haloalkoxy, phenoxy, benzyloxy, (C 3-8 )cycloalkyIoxy, (C 3-
- R 6 , R 7 , R 8 and R 9 are each independently H or (C 1- i 0 )alkyI, or two groups selected from (a) R 6 or R 7 and (b) R 8 or R 9 together form a bond or a (C 1-3 )alkylene group;
- R 10 represents H, (Ci -10 )alky! (optionally substituted by one or more substituents ⁇ ), -(CR 2 ) q -
- (C 3-8 )cycloaikyl (wherein the cycloalkyl part is optionally substituted by one or more substituents ⁇ ), -(CR 2 ) q -Ar, -(CR 2 ) q -Het 1 , or -(CR 2 ) q -Het 2 ; q is 0 or an integer from 1 to 6;
- R 11 represents H, (Ci.i O )alkyl or -(CR 2 )-Ar, or two groups R 11 , together with the nitrogen atom to which they are attached, form a ring Het 3 ;
- Ar is a phenyl or naphthyl group optionally substituted by one or more substituents ⁇ ;
- Het 1 is a group selected from (1) a 5- or 6-membered aromatic heterocyclic group having as ring heteroatoms (a) 1 to 4 nitrogen atoms, (b) 1 oxygen or 1 sulphur atom or (c) 1 oxygen atom or 1 sulphur atom and 1 or 2 nitrogen atoms, is optionally substituted (i) on carbon by one or more substituents ⁇ and/or (ii) on nitrogen by one or more substituents ⁇ , (2) a bicyclic group comprising an optionally substituted 5- or 6-membered aromatic heterocyclic group, as defined in (1 ) above, fused with a benzene ring (the benzene ring being optionally substituted with one or more substituents ⁇ ) or (3) a bicyclic group comprising two optionally substituted 5- or 6-membered aromatic heterocyclic group, as defined in (1 ) above, fused together
- alkyl' means a straight or branched monovalent saturated hydrocarbon chain containing from 1 to 10 carbon atoms.
- alkyl groups include methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, t-butyl, n-pentyl, i-pentyl, neopentyl, n-hexyl, i-hexyl, n-heptyl, n-octyl, n-nonyl and n-decyl.
- 'alkyl' is (Ci -S )alkyl, more preferably (C 1-4 )alkyl, and most preferably methyl or ethyl.
- the alkyl group may optionally be substituted by one or more substituents ⁇ , defined below, the number of substituents being limited by the number of substitutable positions (eg a methyl group is limited to 3 substituents).
- substituents ⁇ there are preferably 1 to 3 substituents ⁇ , more preferably 1 or 2 substituents ⁇ , and most preferably one substituent ⁇ .
- 'Cycloalkyl' and 'cycloalkane' means a saturated carbocyclic ring having from 3 to 8 carbon atoms, the term 'cycloalkyl' referring to a monovalent group and the term 'cycloalkane ring' referring to a ring completed by two substituents together with the carbon atom to which they are attached, or a ring fused to one of the heterocyclic groups defined below.
- cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
- 'cycloalkyl' is (C 3-6 )cycloalkyl.
- the cycloalkyl group or cycloalkane ring may optionally be substituted by one or more substituents ⁇ , defined below, the number of substituents being limited by the number of substitutable positions. Where the cycloalkyl group or cycloalkane ring is substituted, there are preferably 1 to 3 substituents ⁇ , more preferably 1 or 2 substituents ⁇ , and most preferably one substituent ⁇ .
- the cycioalkyl group or cycloalkane ring may also be fused with a benzene ring (the benzene ring being optionally substituted with one or more substituents ⁇ ). Examples of such fused groups include indanyl and tetralinyl, of which indanyl is preferred.
- 'Alkylene' means a straight or branched divalent saturated hydrocarbon chain containing from 1 to 3 carbon atoms.
- alkyl groups include methylene, ethylene, methylmethylene and propylene.
- 'alkylene' is methylene or ethylene, more preferably methylene.
- 'Alkyleneoxy' means a divalent group -alkylene-O-, wherein the alkylene moiety is a straight or branched saturated divalent hydrocarbon chain containing 1 to 4 carbon atoms. The oxygen atom may be attached to the remainder of the molecule at either position.
- Preferred aikyleneoxy groups include methyleneoxy and ethyleneoxy.
- alkylenedioxy' means a divalent group -O-alkylene-0-, wherein the alkylene moiety is a straight or branched saturated divalent hydrocarbon chain containing 1 to 4 carbon atoms.
- Preferred alkylenedioxy groups include methylenedioxy and ethylenedioxy.
- 'Halogen' means fluoro, chloro, bromo or iodo.
- 'Oxo' means a doubly bonded oxygen atom.
- 'Alkoxy' means 'alkyl-O-', wherein 'alkyl' is as defined above (either in its broadest aspect or a preferred aspect).
- alkoxy groups include methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, i-butoxy, sec-butoxy, t-butoxy, n-pentyloxy, i-pentyloxy, neopentyloxy, n-hexyloxy, i- hexyloxy, n-heptyloxy, n-octyloxy, n-nonyloxy and n-decyloxy.
- 'alkoxy' is (C 1-6 )alkoxy, more preferably (C ⁇ alkoxy, and most preferably methoxy or ethoxy.
- 'Phenoxy' means 'phenyl-O-' and 'benzyloxy' means 'phenyl-CH 2 -O-'.
- 'Cycloalkyloxy' means 'cycloalkyl-O-', wherein 'cycloalkyl' is as defined above (either in its broadest aspect or a preferred aspect).
- cycloalkoxy groups include cyclopropyloxy, cyclobutyloxy, cyclopentyloxy, cyclohexyloxy, cycloheptyloxy, and cyclooctyloxy.
- 'cycloalkyloxy' is (C 3 . 6 )cycloalkyloxy.
- 'Cycloalkylalkoxy means alkoxy substituted with cycloalkyl, wherein 'alkoxy' and 'cycloalkyl' are as defined above (either in the broadest aspect or a preferred aspect).
- Examples of cycloalkylalkoxy groups include cyclopropylmethoxy, 1- or 2-cyclopropylethoxy, 1-, 2- or 3- cyclopropylpropoxy, 1-, 2-, 3- or 4-cyclopropylbutoxy, cyclobutylmethoxy, 1- or 2- cyclobutylethoxy, 1-, 2- or 3- cyclobutylpropoxy, 1-, 2-, 3- or 4-cyclobutylbutoxy, cyclopentylmethoxy, 1- or 2-cyclopentylethoxy, 1-, 2- or 3- cyclopentylpropoxy, 1-, 2-, 3- or 4- cyclopentylbutoxy, cyclohexylmethoxy, 1- or 2-cyclohexylethoxy, 1-, 2- or
- Haloalkyl means an alkyl group, as defined above (either in its broadest aspect or a preferred aspect) substituted by one or more halogen atoms (preferably fluorine or chlorine atoms, more preferably fluorine atoms), the number of halogen atoms being limited by the number of substitutable positions.
- halogen atoms preferably fluorine or chlorine atoms, more preferably fluorine atoms
- 'haloalkyl' examples include mono-, di- or trifluoromethyl, mono-, di- or trichloromethyl, bromomethyl, 2-fluoroethyl, 2,2-difluoroethyl, 2,2,2-trifluoroethyl, 2-chloroethyl, 2,2-dichloroethyl, 2,2,2-trichloroethyl, perfluoroethyl, perfluoropropyl and perfluorobutyl.
- 'haloaikyi' is (C 1-6 )haloalkyl, more preferably (C 1-4 )haloalkyl, and most preferably mono-, di- or trifluoromethyl, mono-, di- or trichloromethyl, or perfluoroethyl.
- 'Haloalkoxy' means 'haloalkyl-O-', where 'haloaikyi' is as defined above (either in its broadest aspect or a preferred aspect), the number of halogen atoms being limited by the number of substitutable positions.
- 'haloalkoxy' include mono-, di- or trifluoromethoxy, mono-, di- or trichloromethoxy, bromomethoxy, 2-fluoroethoxy, 2,2-difluoroethoxy, 2,2,2- trifluoroethoxy, 2-chloroethoxy, perfluoroethoxy, perfluoropropoxy and perfluorobutoxy.
- 'haloalkoxy' is (C ⁇ haloalkoxy, more preferably (C ⁇ Jhaloalkoxy, and most preferably mono-, di- or trifluoromethoxy.
- 'Hydroxyalkyl' means alkyl, as defined above (either in its broadest aspect or a preferred aspect) substituted by a hydroxy (-OH) group.
- hydroxyalkyl groups include hydroxymethyl, 1- or 2-hydroxyethyl, 1-, 2- or 3-hydroxypropyl, and 1-, 2-, 3- or 4- hydroxybutyl.
- 'hydroxyalkyl' is (C ⁇ alkyl substituted with hydroxy, more preferably (C 1-4 )alkyl substituted with hydroxy. Hydroxymethyl and 1- or 2-hydroxyethyl groups are especially preferred.
- alkoxyalkyl means alkyl, as defined above (either in its broadest aspect or a preferred aspect) substituted by an alkoxy group, as defined above (either in its broadest aspect or a preferred aspect).
- alkoxyalkyl groups include methoxymethyl, ethoxymethyl, propoxymethyl, butoxymethyl, 1- or 2-methoxyethyl, 1- or 2-ethoxyethyl, 1- or 2-propoxyethyl, 1- or 2-butoxyethyl, 1-, 2- or 3-methoxypropyl, 1-, 2- or 3-ethoxypropyl, 1-, 2- or 3- propoxypropyl, 1-, 2- or 3-butoxypropyl, 1-, 2-, 3- or 4-methoxybutyl, 1-, 2-, 3- or 4- ethoxybutyl, 1-, 2-, 3- or 4-propoxybutyl and 1-, 2-, 3- or 4-butoxybutyl.
- 'alkoxyalkyl means (C 1-6 )alkyl substituted with (C 1-4 )alkoxy, more preferably (C 1-4 )alkyl substituted with (C 1 ⁇ aIkOXy.
- Methoxymethyl, ethoxymethyl and 2-methoxyethyl groups are especially preferred.
- R is hydrogen or (Ci -10 )alkyl, as defined above.
- R is hydrogen or (C 1- 6 )alkyl.
- R is hydrogen or (C ⁇ alkyl.
- R 1 represents H, (C 1-10 )alkyl (optionally substituted by one or more substituents ⁇ ), -(CR2)n-(C3-8)cycloalkyl (wherein the cycloalkyl part is optionally substituted by one or more substituents ⁇ ), -(CR 2 ) n -Het 1 , -(CR 2 ) n -Het 2 , -(CR 2 ) n -Ar, or -CN.
- R 1 represents H, (Ci. 10 )alkyl (optionally substituted by one substituent ⁇ ), -(CR 2 ) n -
- (C 3 . 8 )cycloalkyl (wherein the cycloalkyl part is optionally substituted by one substituent ⁇ ), - (CR 2 ) n -Het ⁇ -(CR 2 ) n -Het 2 , -(CR 2 ) n -Ar, or -CN.
- R 1 represents H, (Ci. 10 )alkyl, phenyl, benzyl (wherein the phenyl part is optionally substituted with (C 1-4 )alkyl or (C 1-4 )BIkOXy), -Het 1 , -(CH 2 )-Het 1 or -CN.
- R 1 is R 1a wherein R 1a represents H, C 1-4 alkyl, phenyl, benzyl (wherein the phenyl part is optionally substituted with (C 1-4 )alkyl or (C 1-4 )alkoxy), pyridyl, -(CH 2 )- pyridyl, oxadiazolyl (optionally substituted with phenyl) or oxazolyl (optionally substituted with C 1-4 alkyl).
- R 1 is R 1z wherein R 1z represents H, C 1-6 alkyl, -(CR 2 )n-(C 3 . 8 )cycloalkyl, -(CR 2 ) ⁇ -Ar, or -(CR 2 ) n -Het 2 .
- R 1 is R 1y wherein R 1y represents H, C 1-4 alkyl, C 3-8 cycloalkyl or phenyl.
- each R may be the same or different.
- each R is H or methyl. More preferably, each R is H.
- n is 0, 1 or 2; more preferably 0 or 1.
- the group Ar is an aryl group selected from phenyl and naphthyl.
- the group Ar is optionally substituted by one or more substituents ⁇ , defined below, the number of substituents being limited by the number of substitutable positions. Where the group Ar is substituted, there are preferably 1 to 5 substituents ⁇ , more preferably 1 to 3 substituents ⁇ , even more preferably 1 or 2 substituents ⁇ , and most preferably one substituent ⁇ .
- Ar is phenyl optionally substituted by one substituent ⁇ .
- Ar is unsubstituted phenyl or phenyl substituted with one substituent selected from (C ⁇ alkyl and (C ⁇ alkoxy.
- the group Het 1 is selected from (1 ) a 5- or 6-membered aromatic heterocyclic group having as ring heteroatoms (a) 1 to 4 nitrogen atoms, (b) 1 oxygen or 1 sulphur atom or (c) 1 oxygen atom or 1 sulphur atom and 1 or 2 nitrogen atoms, is optionally substituted (i) on carbon by one or more substituents ⁇ and/or (ii) on nitrogen by one or more substituents ⁇ , (2) a bicyclic group comprising an optionally substituted 5- or 6-membered aromatic heterocyclic group, as defined in (1 ) above, fused with a benzene ring (the benzene ring being optionally substituted with one or more substituents ⁇ ) or (3) a bicyclic group comprising two optionally substituted 5- or 6-membered aromatic heterocyclic group, as defined in (1) above, fused together.
- Het 1 examples include thienyl, furanyl, pyrrolyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, triazolyl, oxadiazolyl, thiadiazolyl, tetrazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, indolyl, benzofuranyl, benzothienyl, benzpyrazolyl, benzimidazolyl, indazolyl, benzothiazolyl, benzoxazolyl, benzotriazolyl, quinolyl, isoquinolinyl, cinnolinyl, quinazolinyl, quinoxalinyl, phthalazinyl, naphthyridinyl, pteridinyl and pyrazolopyrimidiny
- Het 1 is a group selected from (1 ) and (2) above. Where the group Het 1 is substituted on carbon, there are preferably 1 or 2 substituents ⁇ , and most preferably one substituent ⁇ . In addition, when the group Het 1 is optionally substituted on the one or more ring nitrogen atoms, if present, there is preferably only one substituent ⁇ .
- Het 1 is thienyl, furanyl, pyrrolyl, imidazolyl, oxadiazolyl, oxazolyl, thiazolyl or pyridyl (these groups being optionally substituted on carbon with one substituent ⁇ , and/or on nitrogen with one substituent ⁇ ).
- Het 1 is pyridyl, oxadiazolyl or oxazolyl (these groups being optionally substituted with a phenyl group or a (C ⁇ alkyl group).
- Het 2 include oxiranyl, thiiranyl, aziridinyl, oxetanyl, thietanyl, azetidinyl, tetrahydrofuranyl, tetrahydrothiophenyl, pyrrolidinyl, pyrazolidinyl, imidazolidinyl, dioxolanyl, oxazolidinyl, thiazolidinyi, isoxazolidinyl, isothiazolidinyl, tetrahydropyranyl, tetrahydrothiopyranyl, piperidinyl, morpholinyl, thiomorpholinyl, dioxanyl, oxathianyl, dithianyl, piperazinyl, azepanyl, oxepanyl, oxazepanyl, diazepanyl and azocinyl.
- Het 2 is a group (4) above. Where the group Het 2 is substituted on carbon, there are preferably 1 or 2 substituents ⁇ , and most preferably one substituent ⁇ . In addition, when the group is optionally substituted on the one or more ring nitrogen atoms, if present, there is preferably only one substituent ⁇ .
- Het 2 is azetidinyl, pyrrolidinyl, piperidinyl, morpholinyl, thiomorpholinyl or piperazinyl, these groups being optionally substituted on carbon by one substituent ⁇ and/or on nitrogen by one substituent ⁇ .
- Het 2 is azetidinyl, pyrrolidinyl, piperidinyl or piperazinyl, these groups being optionally substituted on carbon and/or nitrogen by a (C M )alkyl group.
- the ring is formed by two groups described above together with the nitrogen atom to which they are attached.
- Specific examples of Het 3 include aziridine, azetidine, pyrrolidine, piperidine, morpholine, thiomorpholine, piperazine, azepane, oxazepane, diazepane and azocine.
- the ring Het 3 is a ring (7) above. Where the ring Het 3 is substituted on carbon, there are preferably 1 or 2 substituents ⁇ , and most preferably one substituent ⁇ . In addition, when the ring is substituted on the one or more ring nitrogen atoms, there is preferably only one substituent ⁇ .
- Het 3 is azetidine, pyrrolidine, piperidine, morpholine, thiomorpholine or piperazine, these groups being optionally substituted on carbon by one substituent ⁇ and/or on nitrogen by one substituent ⁇ .
- Het 3 is azetidine, pyrrolidine, piperidine or piperazine, these groups being optionally substituted on carbon and/or nitrogen by a (C 1-4 )alkyl group.
- substituents ⁇ are selected from substituents ⁇ 1 comprising halogen, (C 1-6 )alkoxy, phenoxy, (C 1-4 )alkylenedioxy, NR 2 , pyridyl, CN, CONR 2 and NRCOR.
- substituents a are selected from substituents ⁇ 2 comprising halogen, (C 1-4 )alkoxy, phenoxy, (C 1-2 )alkylenedioxy, CONR 2 and pyridyl.
- substituents ⁇ are selected from substituents ⁇ 1 comprising (C 1-6 )alkyl, (C 1- 6 )haloalkyl, phenyl, halogen, (C 1-6 )alkoxy, phenoxy, CN, CONR 2 , NRCOR, and COOR. More preferably, substituents ⁇ are selected from substituents ⁇ 2 comprising (C 1-4 )alkyl, halogen, (C 1-4 )haloaikyl (especially CF 3 ) and (C ⁇ alkoxy.
- substituents ⁇ are selected from substituents ⁇ 1 comprising (C 1-6 )alkyl, phenyl and pyridyl.
- substituents ⁇ are selected from substituents ⁇ 2 comprising (C 1-4 )alkyl.
- the group NR 2 R 3 is present at the 4-position of the pyridine ring (the pyridine ring nitrogen being the 1 -position).
- the bond to the left of the groups is connected to the spiro ring and the bond to the right is connected to the group R 1 .
- the bond between X and Y is a single bond.
- X represents C(L-R 1 ) 2
- preferably one group (L-R 1 ) is H and the other group (L-R 1 ) is other than H.
- both groups (L-R 1 ) are H.
- R, R 1 and Het 1 are as defined above, either in its broadest aspect or in a preferred aspect; or a group R and a group R 1 , together with the nitrogen atom to which they are attached, together form a group Het 3 wherein Het 3 is as defined above, either in its broadest aspect or in a preferred aspect.
- X represents C(L C -R 1a ) 2> N(L b '-R 1a ) or O, wherein
- R 1a represents H, Ci -4 alkyl, phenyl, benzyl, pyridyl, oxadiazolyl (optionally substituted with phenyl) or oxazolyl (optionally substituted with Ci -4 alkyl).
- Y represents C(L-R 1 ) 2
- preferably one group (L-R 1 ) is H and the other group (L-R 1 ) is other than H.
- both groups (L-R 1 ) are H.
- R and R 1 are as defined above, either in its broadest aspect or in a preferred aspect; or a group R and a group R 1 , together with the nitrogen atom to which they are attached, together form a group Het 3 wherein Het 3 is as defined above, either in its broadest aspect or in a preferred aspect.
- R and Het 2 are as defined above either in its broadest aspect or in a preferred aspect; or a group R and a group R 1 , together with the nitrogen atom to which they are attached, together form a piperidine, piperazine or azepine group which is optionally fused with a benzene ring.
- m may be 0 or 1.
- Z represents C(L-R 1 ) 2 (wherein L and R 1 are as defined above, either in its broadest aspect or in a preferred aspect) or O.
- Z represents CH 2 or O, even more preferably CH 2 .
- R 2 and R 3 are the same or different and each represent R (as defined above, either in its broadest aspect or in a preferred aspect) or benzyl, or R 2 and R 3 , together with the nitrogen atom to which they are attached, form a group Het 3 (as defined above, either in its broadest aspect or in a preferred aspect).
- R 2 and R 3 are the same or different and each represent H, C 1-4 alkyl or benzyl.
- R 2 and R 3 each represent H.
- a is 0 or 1 , suitably 0.
- b is 0, 1 or 2, suitably 0 or 1.
- n is 0 or 1 , suitably 0.
- R 5 represents (C 1-6 )alkyl, F, Cl, Br, OH, (C 1-6 JaIkOXy, CN,
- R 5 represents methyl, OH or methoxy.
- p is O or 1 , more preferably 1.
- R 6 , R 7 , R 8 and R 9 are each independently H or (C 1-4 )alkyl. More preferably, R 6 , R 7 , R 8 and R 9 are all H.
- R 10 represents H, (C 1-6 )alkyl (optionally substituted by one or more substituents ⁇ ), (C 3 . 6 )cycloalkyl (wherein the cycloalkyl part is optionally substituted by one or more substituents ⁇ ), benzyl (wherein the phenyl part is optionally substituted by one or more substituents ⁇ 1 ), or -Het 1 .
- R 10 represents H, (C 1-4 )alkyl, benzyl (wherein the phenyl part is optionally substituted by one or more substituents ⁇ 2 ), or pyridyl.
- q is O, 1 or 2, more preferably 0 or 1.
- R 11 represents H or (C 1-6 )alkyl.
- the compounds of formula (I) are preferably compounds of formulae (1-1), (I-2), (I-3), (I-4), (I- 5), (I-6), (I-7) or (I-8):
- L, L', R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , a and b are as defined above, either in the broadest aspect or a preferred aspect.
- Particularly preferred compounds of the invention include those in which each variable in Formula (I) is selected from the suitable and/or preferred groups for each variable. Even more preferred compounds of the invention include those where each variable in Formula (I) is selected from the more preferred or most preferred groups for each variable.
- R 1 represents H, (C 1-10 )alkyl (optionally substituted by one or more substituents ⁇ ), -(CR 2 ) n - (C 3-8 )cycloalkyl (wherein the cycloalkyl part is optionally substituted by one or more substituents ⁇ ), -(CR 2 ) n -Ar, -(CR 2 ) n -Het 1 , -(CR 2 ) n -Het 2 , or -CN, n is O or an integer of from 1 to 6;
- R represents H or (C 1-10 )alkyl; or, when a group R and a group R 1 substitute a nitrogen atom, the groups together with the nitrogen atom form a ring Het 3 ;
- R 2 and R 3 are the same or different and each represent R or -(CR 2 )-Ar, or R 2 and R 3 , together with the nitrogen atom to which they are attached, form a ring Het 3 ;
- R 4 represents (C 1-10 )alkyl; a is O, 1 or 2;
- b is O or an integer from 1 to 4;
- R 6 , R 7 , R 8 and R 9 are hydrogen;
- Ar is a phenyl or naphthyl group optionally substituted by one or more substituents ⁇ ;
- Het 1 is a 5- or 6-membered aromatic heterocyclic group having as ring heteroatoms (a) 1 to 4
- Het 3 is a 3- to 8-membered saturated heterocyclic ring containing 1 or 2 heteroatoms of which one heteroatom is a nitrogen atom and the other heteroatom, if present, is selected from nitrogen, oxygen and sulphur, is optionally substituted on carbon by one or more substituents ⁇ and/or on nitrogen by one or more substituents ⁇ and/or on sulphur by one or two oxo groups and is optionally fused with a benzene ring (the benzene ring being optionally substituted by one or more substituents ⁇ ) or a (C 3 .
- substituents ⁇ are selected from (C 1-10 )alkyI, phenyl, halogen, CF 3 , OR, O-phenyl,
- substituents ⁇ are selected from (C 1-1o )alkyl, phenyl, pyridyl, CONR 2 and COOR; or a pharmaceutically acceptable salt or solvate thereof.
- the compounds of the invention are delta opioid receptor agonists.
- they show an affinity for the delta opioid receptor which is greater than their affinity for the mu and kappa opioid receptors.
- Preferred compounds of the invention show at least a 10-fold selectivity for the delta opioid receptor as compared with the mu opioid receptor.
- the compounds of formula (I), being delta opioid receptor agonists, are potentially useful in the treatment of a range of disorders.
- the present invention also comprises a compound of formula (I), as defined above, or a pharmaceutically acceptable salt or solvate thereof, for use as a medicament.
- the present invention also comprises use of a compound of formula (I), as defined above, or a pharmaceutically acceptable salt or solvate thereof, in the manufacture of a medicament for the treatment of diseases or conditions mediated by the delta opioid receptor.
- the present invention also comprises a method for the treatment of a disease mediated by the delta opioid receptor, comprising administering to a mammal an effective amount of a compound of formula (I), as defined above, or a pharmaceutically acceptable salt or solvate thereof.
- Physiological pain is an important protective mechanism designed to warn of danger from potentially injurious stimuli from the external environment.
- the system operates through a specific set of primary sensory neurones and is activated by noxious stimuli via peripheral transducing mechanisms (see Millan, 1999, Prog. Neurobiol., 57, 1-164 for a review).
- These sensory fibres are known as nociceptors and are characteristically small diameter axons with slow conduction velocities. Nociceptors encode the intensity, duration and quality of noxious stimulus and by virtue of their topographically organised projection to the spinal cord, the location of the stimulus.
- nociceptive nerve fibres of which there are two main types, A-delta fibres (myelinated) and C fibres (non-myelinated).
- A-delta fibres myelinated
- C fibres non-myelinated
- the activity generated by nociceptor input is transferred, after complex processing in the dorsal horn, either directly, or via brain stem relay nuclei, to the ventrobasal thalamus and then on to the cortex, where the sensation of pain is generated. Pain may generally be classified as acute or chronic. Acute pain begins suddenly and is short-lived (usually twelve weeks or less). It is usually associated with a specific cause such as a specific injury and is often sharp and severe. It is the kind of pain that can occur after specific injuries resulting from surgery, dental work, a strain or a sprain.
- Acute pain does not generally result in any persistent psychological response.
- chronic pain is long- term pain, typically persisting for more than three months and leading to significant psychological and emotional problems.
- Common examples of chronic pain are neuropathic pain (e.g. painful diabetic neuropathy, postherpetic neuralgia), carpal tunnel syndrome, back pain, headache, cancer pain, arthritic pain and chronic post-surgical pain.
- Clinical pain is present when discomfort and abnormal sensitivity feature among the patient's symptoms. Patients tend to be quite heterogeneous and may present with various pain symptoms. Such symptoms include: 1) spontaneous pain which may be dull, burning, or stabbing; 2) exaggerated pain responses to noxious stimuli (hyperalgesia); and 3) pain produced by normally innocuous stimuli (allodynia - Meyer et al., 1994, Textbook of Pain, 13- 44). Although patients suffering from various forms of acute and chronic pain may have similar symptoms, the underlying mechanisms may be different and may, therefore, require different treatment strategies. Pain can also therefore be divided into a number of different subtypes according to differing pathophysiology, including nociceptive, inflammatory and neuropathic pain.
- Nociceptive pain is induced by tissue injury or by intense stimuli with the potential to cause injury. Pain afferents are activated by transduction of stimuli by nociceptors at the site of injury and activate neurons in the spinal cord at the level of their termination. This is then relayed up the spinal tracts to the brain where pain is perceived (Meyer et al., 1994, Textbook of Pain, 13-44).
- the activation of nociceptors activates two types of afferent nerve fibres. Myelinated A-deita fibres transmit rapidly and are responsible for sharp and stabbing pain sensations, whilst unmyelinated C fibres transmit at a slower rate and convey a dull or aching pain.
- Moderate to severe acute nociceptive pain is a prominent feature of pain from central nervous system trauma, strains/sprains, burns, myocardial infarction and acute pancreatitis, postoperative pain (pain following any type of surgical procedure), posttraumatic pain, renal colic, cancer pain and back pain.
- Cancer pain may be chronic pain such as tumour related pain (e.g. bone pain, headache, facial pain or visceral pain) or pain associated with cancer therapy (e.g. postchemotherapy syndrome, chronic postsurgical pain syndrome or post radiation syndrome). Cancer pain may also occur in response to chemotherapy, immunotherapy, hormonal therapy or radiotherapy.
- Back pain may be due to herniated or ruptured intervertabral discs or abnormalities of the lumber facet joints, sacroiliac joints, paraspinal • muscles or the posterior longitudinal ligament. Back pain may resolve naturally but in some patients, where it lasts over 12 weeks, it becomes a chronic condition which can be particularly debilitating.
- Neuropathic pain is currently defined as pain initiated or caused by a primary lesion or dysfunction in the nervous system. Nerve damage can be caused by trauma and disease and thus the term 'neuropathic pain' encompasses many disorders with diverse aetiologies. These include, but are not limited to, peripheral neuropathy, diabetic neuropathy, post herpetic neuralgia, trigeminal neuralgia, back pain, cancer neuropathy, HIV neuropathy, phantom limb pain, carpal tunnel syndrome, central post-stroke pain and pain associated with chronic alcoholism, hypothyroidism, uremia, multiple sclerosis, spinal cord injury, Parkinson's disease, epilepsy and vitamin deficiency. Neuropathic pain is pathological as it has no protective role.
- neuropathic pain are difficult to treat, as they are often heterogeneous even between patients with the same disease (Woolf & Decosterd, 1999, Pain Supp., 6, S141-S147; Woolf and Mannion, 1999, Lancet, 353, 1959-1964). They include spontaneous pain, which can be continuous, and paroxysmal or abnormal evoked pain, such as hyperalgesia (increased sensitivity to a noxious stimulus) and allodynia (sensitivity to a normally innocuous stimulus).
- the inflammatory process is a complex series of biochemical and cellular events, activated in response to tissue injury or the presence of foreign substances, which results in swelling and pain (Levine and Taiwo, 1994, Textbook of Pain, 45-56).
- Arthritic pain is the most common inflammatory pain.
- Rheumatoid disease is one of the commonest chronic inflammatory conditions in developed countries and rheumatoid arthritis is a common cause of disability. The exact aetiology of rheumatoid arthritis is unknown, but current hypotheses suggest that both genetic and microbiological factors may be important (Grennan & Jayson, 1994, Textbook of Pain, 397-407).
- Pain associated with osteoarthritis or rheumatoid arthritis includes joint pain associated with osteoarthritis or rheumatoid arthritis and soft tissue (e.g., muscle) pain due to change in gait or other body movement in response to joint pain.
- Joint pain associated with rheumatoid arthritis may be due to inflammation in the joint or degradation of cartilage of the joint.
- Joint pain associated with osteoarthritis is usually due to degradation of cartilage of the joint, although such pain may be due to episodic flares of inflammation in osteoarthritic joints.
- Preferred joints are knee, hip, and joints of hands.
- Visceral pain is pain associated with the viscera, which encompass the organs of the abdominal cavity. These organs include the sex organs, spleen and part of the digestive system. Pain associated with the viscera can be divided into digestive visceral pain and non-digestive visceral pain.
- Gl gastrointestinal
- FBD functional bowel disorder
- IBD inflammatory bowel disease
- Gl disorders include a wide range of disease states that are currently only moderately controlled, including, in respect of FBD, gastro-esophageal reflux, dyspepsia, irritable bowel syndrome (IBS) and functional abdominal pain syndrome (FAPS), and, in respect of IBD, Crohn's disease, ileitis and ulcerative colitis, all of which regularly produce visceral pain.
- Other types of visceral pain include the pain associated with dysmenorrhea, cystitis and pancreatitis and pelvic pain.
- musculoskeletal disorders including myalgia, fibromyalgia, spondylitis, sero-negative (non-rheumatoid) arthropathies, non-articular rheumatism, dystrophinopathy, glycogenosis, polymyositis and pyomyositis; • heart and vascular pain, including pain caused by angina, myocardical infarction, mitral stenosis, pericarditis, Raynaud's phenomenon, scleredoma and skeletal muscle ischemia;
- head pain such as migraine (including migraine with aura and migraine without aura), cluster headache, tension-type headache mixed headache and headache associated with vascular disorders; and
- orofacial pain including dental pain, otic pain, burning mouth syndrome and temporomandibular myofascial pain.
- the compounds of formula (I) are also useful in the treatment of conditions other than pain.
- the compounds of formula (I) are useful in the treatment of conditions of lower urinary tract dysfunction including but not exclusively restricted to overactive bladder, increased daytime frequency, nocturia, urgency, urinary incontinence (any condition in which there is an involuntary leakage of urine), including stress urinary incontinence, urge urinary incontinence and mixed urinary incontinence, overactive bladder with associated urinary incontinence, enuresis, nocturnal enuresis, continuous urinary incontinence, situational urinary incontinence such as incontinence during sexual intercourse, and lower urinary tract symptoms (LUTS) associated with benign prostatic hyperplasia (BPH).
- LUTS benign prostatic hyperplasia
- PE premature ejaculation
- PE is a relatively common sexual dysfunction in men. It has been defined in several different ways but the most widely accepted is the Diagnostic and Statistical Manual of Mental Disorders IV one which states: "PE is a lifelong persistent or recurrent ejaculation with minimal sexual stimulation before, upon or shortly after penetration and before the patient wishes it. The clinician must take into account factors that affect duration of the excitement phase, such as age, novelty of the sexual partner or stimulation, and frequency of sexual activity. The disturbance causes marked distress of interpersonal difficulty.”
- the International Classification of Diseases 10 definition states: "There is an inability to delay ejaculation sufficiently to enjoy lovemaking, manifest as either of the following: (1) occurrence of ejaculation before or very soon after the beginning of intercourse (if a time limit is required: before or within 15 seconds of the beginning of intercourse); (2) ejaculation occurs in the absence of sufficient erection to make intercourse possible. The problem is not the result of prolonged abstinence from sexual activity"
- Ejaculation is dependent on the sympathetic and parasympathetic nervous systems. Efferent impulses via the sympathetic nervous system to the vas deferens and the epididymis produce smooth muscle contraction, moving sperm into the posterior urethra. Similar contractions of the seminal vesicles, prostatic glands and the bulbouretheral glands increase the volume and fluid content of semen.
- Expulsion of semen is mediated by efferent impulses originating from a population of lumber spinothalamic cells in the lumbosacral spinal cord (Coolen & Truitt, Science, 2002, 297, 1566) which pass via the parasympathetic nervous system and cause rhythmic contractions of the bulbocavernous, ischiocavernous and pelvic floor muscles.
- Cortical control of ejaculation is still under debate in humans.
- the medial pre-optic area and the paraventricular nucleus of the hypothalamus seem to be involved in ejaculation.
- Ejaculation comprises two separate components - emission and ejaculation.
- Emission is the deposition of seminal fluid and sperm from the distal epididymis, vas deferens, seminal vesicles and prostrate into the prostatic urethra. Subsequent to this deposition is the forcible expulsion of the seminal contents from the urethral meatus.
- Ejaculation is distinct from orgasm, which is purely a cerebral event. Often the two processes are coincidental.
- Premature female orgasm may be defined as: a) Persistent or recurrent orgasm with minimal sexual stimulation, occurring before, upon or shortly after penetration and generally before the person/couple wishes it. b) Interpersonal difficulties arise from female lack of motivation for/discomfort with continued sexual stimulation, c) The disturbance gives rise to negative psychosocial consequences where arousal occurs in inappropriate situations, d) The disturbance itself causes marked distress or interpersonal difficulty.
- WSCDI means ⁇ S-dimethylaminopropyO-S-ethylcarbodiimide hydrochloride
- DCC means N.N'-dicyclohexylcarbodiimide
- HOBT means 1 -hydroxybenzotriazole hydrate
- HOAT means 1-hydroxy-7-azabenzotriazole
- PyBOP® means benzotriazol-1-yloxytrispyrrolidinophosphonium hexafluorophosphate
- PyBrOP® means bromotrispyrrolidinophosphonium hexafluorophosphate
- CDI means 1 ,1 '-carbonyldiimidazole
- HBTU means 0-benzotriazol-1-yl-N,N,N',N'-tetramethyluronium hexafluorophosphate
- DIAD diisopropyl azodicarboxylate
- AIBN 2,2'-azobis-(2-methylpropionitrile)
- Pd(dppf) 2 means 1 ,1 '-bis[(diphenylphosphino)ferrocene]palladium;
- Pd 2 (dba) 3 means tris(dibenzylideneacetone)dipailadium (0)
- NMM 4-methylmorpholine
- H ⁇ nig's base means N-ethyldiisopropylamine
- Et 3 N means triethylamine
- BOC means terf-butoxycarbonyl
- CBz means benzyloxycarbonyl
- MeOH means methanol
- EtOH means ethanol
- EtOAc means ethyl acetate
- Et 2 O means diethyl ether
- THF means tetrahydrofuran
- DMSO means dimethyl sulfoxide
- DCM dichloromethane
- AcOH acetic acid
- TFA means trifluoroacetic acid
- STAB means sodium triacetoxyborohydride
- NaHMDS means sodium hexamethyldisilazane
- DMF means dimethylformamide
- TLC means thin layer chromatography
- DMA means dimethylacetamide
- Bu 3 SnH means tributyltin hydride
- (Me 3 Si) 3 SiH means tris(trimethylsilyl)si!yl hydride
- Pd(OAc) 2 means palladium diacetate; and TfO means trifluoromethanesulfonyloxy.
- R a represents (N-R)-R 1 or 0-R 1 and PG represents a protecting group, suitable examples of which are described in "Protecting Groups in Organic Synthesis” by T. W. Greene and P. Wuts, Wiley and Sons, 1991.
- PG is preferably BOC.
- Compounds of formula (II) may be obtained commercially or from published methods (eg as described in WO 97/36873, step E) unless their synthesis is described in this specification.
- Single enantiomers of (II) (e.e. > 95%) may be obtained by conventional chiral separation methods, preferably by repeated crystallisation in the presence of chiral amines, preferably (S)- or (R)- ⁇ -methyl- benzylamine, in toluene.
- Step a): Compounds of formula (III) may be synthesised by coupling an amine, HNRR 1 or alcohol R 1 OH with an acid (II), in the presence of a conventional coupling agent, optionally in the presence of a catalyst, with an excess of base in a suitable solvent.
- the preferred conditions are: 1-hydroxybenzotriazole monohydrate (1 eq.), 1-(3- dimethylaminopropyl)-3-ethylcarbodiimide HCI (1.2 eq.), and N-methylmorpholine (1.5 eq.) and the appropriate amine/aicohol (1.1 eq.) in dichloromethane at room temperature for up to
- Step b): Deprotection of compounds of formula (III) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is BOC are TFA in DCM for about 4 hrs at room temperature.
- an alcohol solvent preferably 2-methyl-2-butanol
- Step d): Compounds of formula (Ia) may be prepared by reduction of the crude reaction mixture, or the isolated product, of step c). Typically, reduction is carried out in the presence of a suitable catalyst, e.g. palladium on carbon or palladium hydroxide on carbon, under an atmosphere of hydrogen or with a suitable hydrogen transfer reagent (e.g. cyclohexene or ammonium formate), or in the presence of a reducing agent, typically iron powder, in organic acid at room temperature to 65 0 C for 2 to 18 hours.
- a suitable catalyst e.g. palladium on carbon or palladium hydroxide on carbon
- a suitable hydrogen transfer reagent e.g. cyclohexene or ammonium formate
- a reducing agent typically iron powder
- Preferred conditions are ammonium formate (6 eq.) and 20% palladium hydroxide on carbon (10% by weight) at 65 0 C until conventional methods of detection, preferably TLC, indicate complete conversion.
- the isolated product from step c) may be dissolved in an organic acid, preferably acetic acid, and stirred in the presence of iron powder at room temperature for 2-10 hours, preferably 2-4 hours.
- Compounds of formula (Vl) may be obtained from published methods (eg as described in WO 2004/028459, example 21 , compound xxii). Steps a) to c): Compounds of formula (Ic) may be obtained from compounds of formula (Vl) using the conditions described in steps c), d) and b) for compounds of formula (Ia) depicted in Scheme 1.
- Step d): Compounds of formula (Ib) may be synthesised by coupling a sulphonyl chloride, R 1 SO 2 CI, with an amine (Ic), in the presence of an excess of base in a suitable solvent.
- the preferred conditions are: 1 eq. of R 1 SO 2 CI, 1.5 eq. triethylamine in dichloromethane at room temperature for 4-16 hours.
- R b represents (C 1-10 )alkyl or Het 1 (as defined above)
- R c represents (C ⁇ ojalkyl
- PG represents a suitable protecting group, suitable examples of which are described in "Protecting Groups in Organic Synthesis” (referred to above), preferably BOC.
- Compounds of formula (VII) may be obtained from published methods (eg as described in US 5578593, compound 21 , scheme 10).
- Step a): Deprotection of compounds of formula (VII) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis” (referred to above).
- PG is BOC 1
- preferred conditions are 4M hydrochloric acid in dioxane at room temperature.
- the imine is hydrolysed during work up.
- Preferred conditions are 1 eq. diamine (VIII), 3.5 eq. sodium carbonate in methyl isobutyl ketone at 110 0 C for 3 hours, then (V) at 8O 0 C overnight and finally 2-propanol at room temperature for 4 hours.
- the intermediate nitro compound can then be reduced according to the conditions described in the synthesis of compounds of formula (Ia), step d), Scheme 1.
- Step d): Compounds of formula (Ie) may be prepared by coupling the amine of formula (If) with the appropriate sulphonyl chloride R 1 SO 2 CI optionally in the presence of a base.
- the conditions are amine and sulphonyl chloride R 1 SO 2 CI, optionally with an excess of tertiary amine such as triethylamine, H ⁇ nig's base or NMM, in DCM or THF, at room temperature for 1 to 24 hours.
- the preferred conditions are amine and 1.1 eq. of R 1 SO 2 CI, 3 eq. triethylamine in dichioromethane at room temperature for up to 18 hours.
- Step e): Compounds of formula (IX) may be obtained by coupling the amine of formula (VII) with the appropriate acid or acid derivative. The reaction may be undertaken using either:
- amine and anhydride (R 1 CO) 2 O optionally with an excess of tertiary amine such as Et 3 N, H ⁇ nig's base or NMM, in DCM or THF, without heating for 1 to 24 hours.
- Preferred conditions are: Amine and 1eq. acid R 1 COOH, 1.1 eq. 1-hydroxybenzotriazole monohydrate, 1.1 eq. 1-(3- dimethylaminopropyl)-3-ethylcarbodiimide HCI, and 3 eq. triethylamine in dichloromethane at room temperature for up to 18 hrs.
- a base e.g. sodium tert-butoxide or sodium methoxide
- a suitable catalyst e.g. palladium (II) acetate or Pd 2 (dba) 3
- a ligand e.g.
- 2,2'-bis(diphenylphosphino)-1 ,1 '-binaphthyl in a suitable solvent, typically toluene, 1 ,4- dioxane or DMF ' with heating.
- Preferred conditions are: 1 eq. of amine, 1 eq. of 2- chloropyridine, 1.4 eq. of sodium tert-butoxide in the presence of 0.1 eq. palladium (II) acetate and 0.1 eq. 2,2'-bis(diphenylphosphino)-1 ,1'-binaphthyl in toluene at 80 0 C for up to 4 hours.
- a base e.g. sodium hydride or NaHMDS
- a suitable solvent e.g. tetrahydrofuran or DMF at room temperature
- Preferred conditions are: 1 eq. of compound (VII), 3.7 eq. of iodomethane, 1.2 eq. of sodium hydride in tetrahydrofuran at room temperature for up to 3 hours.
- Step h): Deprotection of compounds of formula (IX) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis” (referred to above).
- PG is BOC
- preferred conditions are 4M hydrochloric acid in dioxane at room temperature or TFA in DCM at room temperature.
- Step k): Reaction of compounds of formula (Ie) wherein L is -NH-C( O)- and R 2 and R 3 are H with a compound of formula R C -LG, wherein LG is a suitable leaving group, eg Cl, Br, I, OTf, in the presence of a base, eg sodium hydride or NaHMDS in a suitable solvent, eg THF or DMF.
- a base eg sodium hydride or NaHMDS in a suitable solvent, eg THF or DMF.
- the preferred conditions are sodium hydride and R c -I in THF at room temperature for 18 hours.
- Steps c) and d): Reaction of the ketone of formula (Ik) with a homochiral sulfinamide of formula R d S( O)N H 2 (wherein R d represents (C 1-10 )alkyl), preferably in the presence of titanium ethoxide in a suitable organic solvent, e.g. THF or toluene at reflux followed by reduction at low temperature with a suitable reducing agent, e.g sodium borohydride or sodium triacetoxyborohydride, may provide compounds of formula (Ij).
- Preferred conditions are: 1 eq. of compound (Ik), 1.5 eq. of sulfinamide, 3.5 eq. of titanium ethoxide in THF at reflux for 18 hours, followed by 4 eq. of sodium borohydride at -48 0 C to room temperature over 18 hours.
- Step e): The sulfinamide may be cleaved to give compounds of formula (Ii) under acidic conditions, typically HCI or trifluoromethanesulfonic acid in an appropriate solvent, e.g. methanol, ethanol, DCM.
- acidic conditions typically HCI or trifluoromethanesulfonic acid in an appropriate solvent, e.g. methanol, ethanol, DCM.
- the preferred conditions are: 1 eq. of compound (Ij) in methanol with an excess of a 4M solution of hydrogen chloride in dioxane at room temperature for about 30 minutes.
- Step f): Compounds of formula (Ih) may be prepared by coupling amines of formula (Ii) with the appropriate acid or acid derivative using conditions described in step e) for compounds of formula (Ie) depicted in Scheme 3.
- the preferred conditions are amine (Ii), 1eq. acid R 1 COOH, 1.1 eq. 1-hydroxybenzotriazole monohydrate, 1.1 eq. 1-(3-dimethylaminopropyl)-3- ethylcarbodiimide HCI, and 3 eq. triethylamine in dichloromethane at room temperature for up to i ⁇ hrs.
- Step a): Compounds of formula (XIV) may be prepared by methods analogous to those described in "Comprehensive Heterocyclic Chemistry", A.R. Katritzky, CW. Rees, Pergamon Press, Oxford, 1984.
- compounds of formula (XIV) where Het 1 is oxadiazolyl may be prepared by coupling an acid with the appropriate hydrazide of formula R 6 CONHNH 2 , followed by ring closure with an appropriate dehydrating reagent, e.g. phosphorus oxychloride, thionyl chloride or 2-chloro-1 ,3-dimethyl-2-imidazolinium tetrafluoroborate.
- dehydrating reagent e.g. phosphorus oxychloride, thionyl chloride or 2-chloro-1 ,3-dimethyl-2-imidazolinium tetrafluoroborate.
- the preferred conditions are: 1 eq.
- Step b): Deprotection of compounds of formula (XIV) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is BOC are TFA in DCM for about 3 hrs at room temperature.
- Steps c) and d): Compounds of formula (I/) may be obtained from compounds of formula (XV) using conditions described in steps c) and d) for compounds of formula (Ia) depicted in Scheme 1.
- PG represents a protecting group, suitable examples of which are described in "Protecting Groups in Organic Synthesis” (referred to above).
- PG is preferably BOC.
- Step a): Compounds of formula (XVII) may be synthesised by coupling the appropriate amine of formula NH 2 CHR 1 CH 2 OH, with acid (XVI), in the presence of a conventional coupling agent, optionally in the presence of a catalyst, with an excess of base in a suitable solvent.
- Preferred conditions are 1eq. acid of formula (XVI), 2 eq. triethylamine, 1.1 eq NH 2 CHR f CH 2 OH, 1 eq. 2-chloro-1 ,3-dimethyl-2-imidazolinium tetrafluoroborate in DCM at room temperature for 72 hours.
- Step b): Compounds of formula (XVIII) may be prepared by methods analogous to those described in "Comprehensive Heterocyclic Chemistry" (referred to above). For example, oxidation of the alcohol of formula (XVII) with a suitable oxidising reagent, e.g. 1 ,1 ,1- triacetoxy-1 ,1-dihydro-1 ,2-benziodoxol-3(1 H)-one, standard Swern conditions (typically oxalyl chloride, DMSO and triethylamine) or pyridinium chlorochromate, followed by cyclisation of the intermediate aldehyde with a suitable dehydrating reagent e.g.
- a suitable dehydrating reagent e.g.
- Step c): Deprotection of compounds of formula (XVIII) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is BOC are TFA in DCM for about 3 hrs at room temperature.
- a suitable catalyst and solvent typically platinum oxide or palladium on carbon in methanol or ethanol at 1-4 atmospheres of hydrogen, preferably platinum oxide in ethanol at room temperature under 3 atmospheres of hydrogen.
- Step b): Deprotection of compounds of formula (XXI) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis” (referred to above). Preferred conditions are 1 eq. methyl piperidine (XXI), 3 eq. 1-chloroethylchloroformate, 1 eq. 1 ,8-bis-(dimethylamino)naphthalene in 1 ,2-dichIoroethane at reflux for 3 hours.
- Step c): Protection of compounds of formula (XXII) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis” (referred to above). Preferred conditions when PG is BOC are 1 eq. piperidine (XXII) 1 1.1 eq. triethylamine, 1 ,1 eq. di-'butyl-dicarbonate in dichloromethane at room temperature for 18 hours.
- Step d): Debenzylation of compounds of formula (XXIV) followed by protection may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is BOC are 1 eq. (XXIV), palladium on carbon, in ethanol at 4O 0 C under hydrogen for 6 hours, followed by 1 eq. triethylamine, 1eq. di-'butyl dicarbonate in dichloromethane at room temperature for 3 hours.
- Step e): Hydrolysis of esters of formula (XXIII) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions are 1 eq. ester (XXIII), 1.5 eq. 1 M aqueous sodium hydroxide in methanol at room temperature for 18 hours.
- Single enantiomers of acids of formula (XXV) (e.e. > 95%) may be obtained by conventional chiral separation methods, preferably by repeated crystallisation in the presence of chiral amines, preferably (S)- or (R)- ⁇ -methyl- benzylamine, in toluene.
- Step f): Compounds of formula (XXVI) may be prepared by coupling the acid of formula (XXV) with the appropriate amine of formula NHRR 1 in the presence of a conventional coupling agent, optionally in the presence of a catalyst, with an excess of base in a suitable solvent.
- Preferred conditions are 1 eq. acid (XXIV), 1 eq. HOBT, 1.1 eq. WSCDI, 2- 10 eq. NHRR 1 , in dimethylformamide or dichloromethane at room temperature for 18 hours.
- Steps h) and i) and I) and m) Compounds of formula (In) and (lo) may be obtained from compounds of formula (XXVII) and (XXIX) respectively using conditions described in steps c) and d) for compounds of formula (Ia) depicted in Scheme 1.
- Step j): Esters of general formula (XXVIII) may be obtained using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions are 1 eq. acid (XXV), 1 eq. HOBT, 1 eq. WSCDI, in dichloromethane with excess R 1 OH.
- Step n): Hydrolysis of esters of formula (lo) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions are 1 eq. ester (lo), 1.2 eq. lithium hydroxide in tetrahydrofuran at room temperature for 7 hours.
- Step o): Compounds of formula (In) may be prepared by coupling the acid of formula (Ip) with the appropriate amine of formula NHRR 1 using conditions described in step f) for compounds of formula (In) above.
- the preferred conditions are 1 eq. acid (Ip), 1.5 eq. HBTU, 1.2 eq. NHRR 1 , 14 eq. triethylamine in N-methylpyrrolidine at room temperature for 18 hours.
- Scheme 8 In Scheme 8, PG represents a suitable protecting group, preferably benzyl.
- PG represents a suitable protecting group, preferably benzyl.
- Compounds of formula (XXX) may be obtained by methods described in this specification (e.g Preparation 52).
- Step a): Compounds of formula (XXXII) may be obtained from alcohols of formula (XXXI) by a Mitsunobu reaction with the appropriate phenol, using standard methodology.
- a suitable phosphine such as tri- "butylphosphine or triphenylphosphine
- a suitable dehydrating agent typically an azo compound such as diisopropyl azodicarboxylate or di-terf- butyl azodicarboxylate, in a solvent such as dichloromethane, tetrahydrofuran or N 1 N- dimethylformamide, at temperatures between 25-115 0 C, for 1-48 hours, optionally in the absence of light.
- Step b): Compounds of formula (XXXIII) may be prepared by a radical initiated cyclisation of the compound of formula (XXXII), in the presence of a suitable radical initiator (e.g. AIBN) and radical carrier source (e.g. Bu 3 SnH, (Me 3 Si) 3 SiH) in a suitable solvent (e.g. toluene) at 60- 8O 0 C for 4 hours.
- a suitable radical initiator e.g. AIBN
- radical carrier source e.g. Bu 3 SnH, (Me 3 Si) 3 SiH
- solvent e.g. toluene
- Step c): The compound of formula (XXXIV) may be obtained by removal of the N-protecting group using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above).
- PG represents benzyl
- this may be achieved by catalytic hydrogenation in the presence of a suitable catalyst e.g. Pd/C or Pd(OH) 2 in a suitable alcoholic solvent, e.g. H 2 O, MeOH, or EtOH at between room temperature and 6O 0 C under an atmosphere of H 2 .
- a suitable catalyst e.g. Pd/C and Pd(OH) 2 and a hydrogen donor, e.g.
- a suitable solvent e.g. EtOH or MeOH
- deprotection can be achieved using 1-chloroethylchloroformate in an appropriate organic solvent, e.g 1 ,2- dichloroethane followed by hydrolysis with, for example, sodium hydroxide or methanol.
- Preferred conditions 1eq. compound (XXXIII), 5 eq. NH 4 CO 2 H, 10% Pd/C in EtOH at the reflux temperature of the reaction solvent for about 1.5 hrs, Or, 1 eq. compound (XXXIII), 1 eq. 1-chloroethylchloroformate, 1eq. triethylamine in 1 ,2- dichloroethane at O 0 C to reflux for 2 hours followed by methanol at reflux for 45 minutes.
- Steps d) and e): Compounds of formula (Iq) may be obtained from compounds of formula (XXXIV) using conditions described in steps c) to d) for compounds of formula (Ia) depicted in Scheme 1.
- Steps f) and g): Hydrolysis of esters of formula (Iq) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions are 1 eq. ester (Iq) with 2M hydrochloric acid in THF at 35 0 C for 20 hours.
- Compounds of formula (Ir) may be prepared by coupling the intermediate acid with the appropriate amine using conditions described in step f) for compounds of formula (In) depicted in Scheme 7.
- Preferred conditions are 1 eq. acid, 1 eq. HOBT, 1.2 eq. WSCDI 1 3 eq. amine NHRR 1 in DMF at room temperature for 16 hours.
- PG represents a suitable protecting group, suitable examples of which are described in "Protecting Groups in Organic Synthesis” (referred to above).
- PG is preferably BOC.
- Compounds of formula (XXXV) may be obtained by reacting an N-protected 4-piperidone with an amine of formula R 1 NH 2 in an appropriate organic solvent, e.g. trimethylorthoformate followed by reaction with an appropriately substituted 2-bromobenzoyl chloride in the presence of a base e.g. triethylamine or H ⁇ nig's base in an appropriate organic solvent e.g. dichioromethane, THF.
- Preferred conditions are 1 eq. N-protected 4-piperidone, 1 eq. R 1 NH 2 in trimethylorthoformate at room temperature for 72 hours followed by 1 eq. 2- bromobenzoyl chloride, 2 eq. triethylamine in dichloromethane at O 0 C to room temperature for 18 hours.
- Step c): Compounds of formula (XXXVI) may be obtained by cyclising alkenes of formula (XXXV) under standard Heck conditions.
- compounds of formula (XXXV) are cyclised in the presence of a suitable catalyst, e.g. PdCI 2 or Pd(OAc) 2 , a phosphine ligand, e.g. triphenylphosphine and a base, e.g. triethylamine in a suitable organic solvent, e.g. dimethylformamide or acetonitrile at room temperature to 100 0 C.
- Preferred conditions are 1 eq. (XXXIV), 1 eq.
- Step d): Deprotection of compounds of formula (XXXVI) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is BOC, are TFA in DCM for about 18 hours at room temperature.
- a suitable catalyst e.g. palladium on carbon or palladium hydroxide
- a solvent e.g. ethyl acetate, methanol or ethanol
- Preferred conditions are 10% palladium on carbon in ethyl acetate and acetic acid at room temperature under 4 atmospheres of hydrogen for 18 hours.
- an appropriate reducing reagent eg sodium borohydride or lithium aluminium hydride in a suitable organic solvent, eg methanol or tetrahydrofuran.
- Preferred conditions are 1 eq. ketone (Ik), 1.3 eq. sodium borohydride or lithium aluminium hydride in tetrahydrofuran at room temperature for 18 hours.
- a base e.g. sodium hydride
- Preferred conditions are 1 eq. alcohol (Iu), 1.1 eq. sodium hydride, 1.1 eq. R 1 I in tetrahydrofuran at room temperature for 18 hours. .
- Step b): Compounds of general formula (Iv) may be obtained from the reaction of compounds of formulae (XXXIX) and (XL) under Buchwald reaction conditions. These are typically the reaction of a compound of formula (XXXIX) with an amine of formula (XL) in the presence of a metal catalyst e.g. Pd(OAc) 2 , Pd(dppf) 2 , and a base, e.g. sodium tert-butoxide, in an appropriate solvent, e.g. dioxane or toluene. Preferred conditions are 1 eq. (XL), 1 eq.
- acidic conditions e.g. hydrochloric acid or sulphuric acid
- an appropriate solvent e.g. dioxane or water.
- the preferred conditions are 1 eq. nitrile (Iy) in dioxane and 6N hydrochloric acid at 15O 0 C for 15 hours.
- Step b): Compounds of formula (Iw) may be prepared by coupling the acid of formula (Ix) with the appropriate amine of formula RNHR using conditions described in step f) for compounds of formula (In) depicted in Scheme 7.
- Preferred conditions are 1 eq. acid (Ix), 1.1 eq. WSCDI, 1.3 eq. HOBT, 11 eq. RNHR in dimethylformamide at room temperature for 72 hours.
- Step a): Compounds of formula (XLII) may be obtained from acid (XLI) and R 9 -LG wherein R 9 is (C ⁇ ojalkyl and LG is a suitable leaving group, e.g. Br, I, with a base, eg lithium diisopropylamide or sodium hydride, in an appropriate organic solvent, eg THF.
- Preferred conditions are 1 eq. acid (XLI), 3 eq. lithium diisopropylamide, 3 eq. R 9 -LG in THF at O 0 C to room temperature for 18 hours.
- Step b): Compounds of formula (XLIII) may be prepared by coupling the acid of formula (XLII) with the appropriate amine using conditions described in step e) for compounds of formula (Ie) depicted in scheme 3. Preferred conditions are 1 eq. acid (XL), excess NHRR 1 , 1.5 eq. HBTU in THF at 5O 0 C for 18 hours.
- Step c): Deprotection of compounds of formula (XLIII) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is BOC are 4M hydrochloric acid in dioxane for about 2 hours at room temperature.
- PG represents a suitable protecting group, suitable examples of which are described in "Protecting Groups in Organic Synthesis” (referred to above).
- PG is preferably benzyloxycarbonyl.
- Compounds of formula (XLV) may be obtained from published methods (eg as described in Xie et al, Tetrahedron, 2004, 60, 4875-4878).
- Step b): Deprotection of compounds of formula (XLVI) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is benzyloxycarbonyl are 1-methyl-1 ,4-cyclohexadiene and catalytic Pd(OH) 2 on carbon in ethanol at 5O 0 C for 30 minutes.
- Step c) Compounds of formula (laa) may be obtained from compounds of formula (XLVII) using the conditions described in steps c), d) for compounds of formula (Ia) depicted in Scheme 1.
- the present invention also comprises a pharmaceutical composition
- a pharmaceutical composition comprising an effective amount of a compound of formula (I) as defined above, or a pharmaceutically acceptable salt or solvate thereof, together with a pharmaceutically acceptable carrier or diluent.
- Pharmaceutically acceptable salts of the compounds of formula (I) include the acid addition and base salts thereof.
- Suitable acid addition salts are formed from acids which form non-toxic salts. Examples include the acetate, adipate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, cyclamate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, pyroglutamate, saccharate, ste
- Suitable base salts are formed from bases which form non-toxic salts. Examples include the aluminium, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and zinc salts.
- Hemisalts of acids and bases may also be formed, for example, hemisulphate and hemicalcium salts.
- the resulting salt may precipitate out and be collected by filtration or may be recovered by evaporation of the solvent.
- the degree of ionisation in the resulting salt may vary from completely ionised to almost non-ionised.
- the compounds of the invention may exist in a continuum of solid states ranging from fully amorphous to fully crystalline.
- the term 'amorphous' refers to a state in which the material lacks long range order at the molecular level and, depending upon temperature, may exhibit the physical properties of a solid or a liquid. Typically such materials do not give distinctive X- ray diffraction patterns and, while exhibiting the properties of a solid, are more formally described as a liquid.
- a change from solid to liquid properties occurs which is characterised by a change of state, typically second order ('glass transition')-
- 'crystalline' refers to a solid phase in which the material has a regular ordered internal structure at the molecular level and gives a distinctive X-ray diffraction pattern with defined peaks.
- Such materials when heated sufficiently will also exhibit the properties of a liquid, but the change from solid to liquid is characterised by a phase change, typically first order ('melting point').
- the compounds of the invention may also exist in unsolvated and solvated forms.
- 'solvate' is used herein to describe a molecular complex comprising the compound of the invention and one or more pharmaceutically acceptable solvent molecules, for example, ethanol.
- solvent molecules for example, ethanol.
- 'hydrate' is employed when said solvent is water.
- Isolated site hydrates are ones in which the water molecules are isolated from direct contact with each other by intervening organic molecules.
- channel hydrates the water molecules lie in lattice channels where they are next to other water molecules.
- metal-ion coordinated hydrates the water molecules are bonded to the metal ion.
- the complex When the solvent or water is tightly bound, the complex will have a well-defined stoichiometry independent of humidity. When, however, the solvent or water is weakly bound, as in channel solvates and hygroscopic compounds, the water/solvent content will be dependent on humidity and drying conditions. In such cases, non-stoichiometry will be the norm.
- references to compounds of formula (I) include references to pharmaceutically acceptable salts and solvates thereof.
- the compounds of the invention include compounds of formula (I) as hereinbefore defined, including all polymorphs and crystal habits thereof, prodrugs and isomers thereof (including optical, geometric and tautomeric isomers) as hereinafter defined and isotopically-labeled compounds of formula (I).
- Compounds of formula (I) containing one or more asymmetric carbon atoms can exist as two or more stereoisomers. Where a compound of formula (I) contains an alkenyl or alkenylene group, geometric cisltrans (or Z/E) isomers are possible. Where structural isomers are interconvertible via a low energy barrier, tautomeric isomerism ('tautomerism') can occur. This can take the form of proton tautomerism in compounds of formula (I) containing, for example, an imino, keto, or oxime group, or so-called valence tautomerism in compounds which contain an aromatic moiety. It follows that a single compound may exhibit more than one type of isomerism.
- Cis/trans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallisation.
- the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of formula (I) contains an acidic or basic moiety, a base or acid such as 1-phenylethylamine or tartaric acid.
- a suitable optically active compound for example, an alcohol, or, in the case where the compound of formula (I) contains an acidic or basic moiety, a base or acid such as 1-phenylethylamine or tartaric acid.
- the resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereo isomers converted to the corresponding pure enantiomer(s) by means well known to a skilled person.
- Chiral compounds of the invention may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine. Concentration of the eluate affords the enriched mixture.
- chromatography typically HPLC
- a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine.
- the first type is the racemic compound (true racemate) referred to above wherein one homogeneous form of crystal is produced containing both enantiomers in equimolar amounts.
- the second type is the racemic mixture or conglomerate wherein two forms of crystal are produced in equimolar amounts each comprising a single enantiomer.
- Racemic mixtures may be separated by conventional techniques known to those skilled in the art - see, for example, Stereochemistry of Organic Compounds by E. L. Eliel and S. H. Wilen (Wiley, 1994).
- the present invention includes all pharmaceutically acceptable isotopically-Iabelled compounds of formula (I) wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number which predominates in nature.
- isotopes suitable for inclusion in the compounds of the invention include isotopes of hydrogen, such as 2 H and 3 H, carbon, such as 11 C, 13 C and 14 C, chlorine, such as 36 CI, fluorine, such as 18 F, iodine, such as 123 I and 125 I, nitrogen, such as 13 N and 15 N, oxygen, such as 15 0, 17 O and 18 O, phosphorus, such as 32 P, and sulphur, such as 35 S.
- isotopically-labelled compounds of formula (I), for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies.
- the radioactive isotopes tritium, i.e. 3 H, and carbon-14, i.e. 14 C, are particularly useful for this purpose in view of their ease of incorporation and ready means of detection.
- substitution with heavier isotopes such as deuterium, i.e. 2 H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances.
- Isotopically-labeled compounds of formula (I) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagent in place of the non-labeled reagent previously employed.
- solvates in accordance with the invention include those wherein the solvent of crystallization may be isotopically substituted, e.g. D 2 O, d 6 -acetone, d 6 -DMSO.
- intermediate compounds of formula (I)I as hereinbefore defined, all salts, solvates and complexes thereof and all solvates and complexes of salts thereof as defined hereinbefore for compounds of formula (I).
- the invention includes all polymorphs of the aforementioned species and crystal habits thereof.
- Compounds of the invention intended for pharmaceutical use may be administered as crystalline or amorphous products. They may be obtained, for example, as solid plugs, powders, or films by methods such as precipitation, crystallization, freeze drying, spray drying, or evaporative drying. Microwave or radio frequency drying may be used for this purpose.
- excipients may be administered alone or in combination with one or more other compounds of the invention or in combination with one or more other drugs (or as any combination thereof). Generally, they will be administered as a formulation in association with one or more pharmaceutically acceptable excipients.
- excipient' is used herein to describe any ingredient other than the compound(s) of the invention. The choice of excipient will to a large extent depend on factors such as the particular mode of administration, the effect of the excipient on solubility and stability, and the nature of the dosage form.
- compositions suitable for the delivery of compounds of the present invention and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation may be found, for example, in Remington's Pharmaceutical Sciences. 19th Edition (Mack Publishing Company, 1995).
- the compounds of the invention may be administered orally.
- Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, and/or buccal, lingual, or sublingual administration by which the compound enters the blood stream directly from the mouth.
- Formulations suitable for oral administration include solid, semi-solid and liquid systems such as tablets; soft or hard capsules containing multi- or nano-particulates, liquids, or powders; lozenges (including liquid-filled); chews; gels; fast dispersing dosage forms; films; ovules; sprays; and buccal/mucoadhesive patches.
- Liquid formulations include suspensions, solutions, syrups and elixirs. Such formulations may be employed as fillers in soft or hard capsules (made, for example, from gelatin or hydroxypropylmethylcellulose) and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet.
- the compounds of the invention may also be used in fast-dissolving, fast-disintegrating dosage forms such as those described in Expert Opinion in Therapeutic Patents, H (6), 981- 986, by Liang and Chen (2001).
- the drug may make up from 1 weight % to 80 weight % of the dosage form, more typically from 5 weight % to 60 weight % of the dosage form.
- tablets generally contain a disintegrant.
- disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl-substituted hydroxypropyl cellulose, starch, pregelatinised starch and sodium alginate.
- the disintegrant will comprise from 1 weight % to 25 weight %, preferably from 5 weight % to 20 weight % of the dosage form.
- Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate.
- lactose monohydrate, spray-dried monohydrate, anhydrous and the like
- mannitol xylitol
- dextrose sucrose
- sorbitol microcrystalline cellulose
- starch dibasic calcium phosphate dihydrate
- Tablets may also optionally comprise surface active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc.
- surface active agents such as sodium lauryl sulfate and polysorbate 80
- glidants such as silicon dioxide and talc.
- surface active agents may comprise from 0.2 weight % to 5 weight % of the tablet, and glidants may comprise from 0.2 weight % to 1 weight % of the tablet.
- Tablets also generally contain lubricants such as magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate.
- Lubricants generally comprise from 0.25 weight % to 10 weight %, preferably from 0.5 weight % to 3 weight % of the tablet.
- ingredients include anti-oxidants, colourants, flavouring agents, preservatives and taste-masking agents.
- Exemplary tablets contain up to about 80% drug, from about 10 weight % to about 90 weight % binder, from about 0 weight % to about 85 weight % diluent, from about 2 weight % to about 10 weight % disintegrant, and from about 0.25 weight % to about 10 weight % lubricant.
- Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting.
- the final formulation may comprise one or more layers and may be coated or uncoated; it may even be encapsulated.
- Consumable oral films for human or veterinary use are typically pliable water-soluble or water-swellable thin film dosage forms which may be rapidly dissolving or mucoadhesive and typically comprise a compound of formula (I), a film-forming polymer, a binder, a solvent, a humectant, a plasticiser, a stabiliser or emulsifier, a viscosity-modifying agent and a solvent.
- Some components of the formulation may perform more than one function.
- the compound of formula (I) may be water-soluble or insoluble.
- a water-soluble compound typically comprises from 1 weight % to 80 weight %, more typically from 20 weight % to 50 weight %, of the solutes. Less soluble compounds may comprise a greater proportion of the composition, typically up to 88 weight % of the solutes.
- the compound of formula (I) may be in the form of multiparticulate beads.
- the film-forming polymer may be selected from natural polysaccharides, proteins, or synthetic hydrocolloids and is typically present in the range 0.01 to 99 weight %, more typically in the range 30 to 80 weight %.
- ingredients include anti-oxidants, colorants, flavourings and flavour enhancers, preservatives, salivary stimulating agents, cooling agents, co-solvents (including oils), emollients, bulking agents, anti-foaming agents, surfactants and taste-masking agents.
- Films in accordance with the invention are typically prepared by evaporative drying of thin aqueous films coated onto a peelable backing support or paper. This may be done in a drying oven or tunnel, typically a combined coater dryer, or by freeze-drying or vacuuming.
- Solid formulations for oral administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- Suitable modified release formulations for the purposes of the invention are described in US Patent No. 6,106,864. Details of other suitable release technologies such as high energy dispersions and osmotic and coated particles are to be found in Pharmaceutical Technology On-line. 25(2), 1-14, by Verma ef a/ (2001 ). The use of chewing gum to achieve controlled release is described in WO 00/35298.
- the compounds of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ.
- Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular, intrasynovial and subcutaneous.
- Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
- Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile non-aqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
- excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9)
- a suitable vehicle such as sterile, pyrogen-free water.
- parenteral formulations under sterile conditions may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
- solubility of compounds of formula (I) used in the preparation of parenteral solutions may be increased by the use of appropriate formulation techniques, such as the incorporation of solubility-enhancing agents.
- Formulations for parenteral administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- compounds of the invention may be formulated as a suspension or as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound.
- examples of such formulations include drug-coated stents and semi-solids and suspensions comprising drug- loaded poly( ⁇ 7-lactic-coglyco!ic)acid (PGLA) microspheres.
- PGLA poly( ⁇ 7-lactic-coglyco!ic)acid
- the compounds of the invention may also be administered topically, (intra)dermally, or transdermal ⁇ to the skin or mucosa.
- Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibres, bandages and microemulsions. Liposomes may also be used.
- Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol. Penetration enhancers may be incorporated - see, for example, J Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999).
- topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and microneedle or needle-free (e.g. PowderjectTM, BiojectTM, etc.) injection.
- Formulations for topical administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- the compounds of the invention can also be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler, as an aerosol spray from a pressurised container, pump, spray, atomiser (preferably an atomiser using electrohydrodynamics to produce a fine mist), or nebuliser, with or without the use of a suitable propellent, such as 1 ,1 ,1 ,2-tetrafluoroethane or 1 ,1 ,1 ,2,3,3,3-heptafluoropropane, or as nasal drops.
- the powder may comprise a bioadhesive agent, for example, chitosan or cyclodextrin.
- the pressurised container, pump, spray, atomizer, or nebuliser contains a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- the drug product Prior to use in a dry powder or suspension formulation, the drug product is micronised to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenisation, or spray drying.
- comminuting method such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenisation, or spray drying.
- Capsules made, for example, from gelatin or hydroxypropylmethylcellulose
- blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the compound of the invention, a suitable powder base such as lactose or starch and a performance modifier such as /-leucine, mannitol, or magnesium stearate.
- the lactose may be anhydrous or in the form of the monohydrate, preferably the latter.
- Other suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
- a suitable solution formulation for use in an atomiser using electrohydrodynamics to produce a fine mist may contain from 1 ⁇ g to 20mg of the compound of the invention per actuation and the actuation volume may vary from 1 ⁇ l to 100 ⁇ l.
- a typical formulation may comprise a compound of formula (I), propylene glycol, sterile water, ethanol and sodium chloride.
- Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
- Suitable flavours such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations of the invention intended for inhaled/intranasal administration.
- Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, PGLA.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- the compounds of the invention may be administered rectally or vaginally, for example, in the form of a suppository, pessary, or enema.
- Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate.
- Formulations for rectal/vaginal administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- the compounds of the invention may also be administered directly to the eye or ear, typically in the form of drops of a micronised suspension or solution in isotonic, pH-adjusted, sterile saline.
- Other formulations suitable for ocular and aural administration include ointments, gels, biodegradable (e.g. absorbable gel sponges, collagen) and non-biodegradable (e.g. silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes.
- a polymer such as crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose, or a heteropolysaccharide polymer, for example, gelan gum, may be incorporated together with a preservative, such as benzalkonium chloride.
- a preservative such as benzalkonium chloride.
- Such formulations may also be delivered by iontophoresis.
- Formulations for ocular/aural administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted, or programmed release.
- the compounds of the invention may be combined with soluble macromolecular entities, such as cyclodextrin and suitable derivatives thereof or polyethylene glycol-containing polymers, in order to improve their solubility, dissolution rate, taste-masking, bioavailability and/or stability for use in any of the aforementioned modes of administration.
- soluble macromolecular entities such as cyclodextrin and suitable derivatives thereof or polyethylene glycol-containing polymers
- Drug-cyclodextrin complexes are found to be generally useful for most dosage forms and administration routes. Both inclusion and non-inclusion complexes may be used.
- the cyclodextrin may be used as an auxiliary additive, i.e. as a carrier, diluent, or solubiliser. Most commonly used for these purposes are alpha-, beta- and gamma-cyclodextrins, examples of which may be found in International Patent Publication Nos. WO 91/11172, WO 94/02518 and WO 98/55148.
- two or more pharmaceutical compositions may conveniently be combined in the form of a kit suitable for coadministration of the compositions. .
- the kit of the invention comprises two or more separate pharmaceutical compositions, at least one of which contains a compound of formula (I) in accordance with the invention, and means for separately retaining said compositions, such as a container, divided bottle, or divided foil packet.
- a container, divided bottle, or divided foil packet An example of such a kit is the familiar blister pack used for the packaging of tablets, capsules and the like.
- the kit of the invention is particularly suitable for administering different dosage forms, for example, oral and parenteral, for administering the separate compositions at different dosage intervals, or for titrating the separate compositions against one another.
- the kit typically comprises directions for administration and may be provided with a so-called memory aid.
- the total daily dose of the compounds of the invention is typically in the range 1 mg to 5000 mg, preferably 10 mg to 2000 mg depending, of course, on the mode of administration.
- oral administration may require a total daily dose of from 50 mg to 1000 mg; an intravenous dose may require from 50 mg to 1000 mg.
- the total daily dose may be administered in single or divided doses and may, at the physician's discretion, fall outside of the typical range given herein.
- These dosages are based on an average human subject having a weight of about 60kg to 70kg. The physician will readily be able to determine doses for subjects whose weight falls outside this range, such as infants and the elderly.
- references herein to "treatment” include references to curative, palliative and prophylactic treatment.
- the ability of the compounds of the formula (I) to bind to the delta opioid receptor may be measured using the assay described below.
- Stimulation buffer was made at 2x concentrated to allow for 1 in 2 dilution with compound as follows:
- Stimulation buffer Final concentration: 5mM HEPES pH 7.4, HBSS, 0.1 % BSA, 1OmM MgCI 2 .6H 2 O and 0.5mM IBMX in purified H 2 O:
- Detection buffer 5mM HEPES pH 7.4, HBSS, 0.1 % BSA and 0.3% Tween-20 in purified H 2 O: 50ml 10x HBSS, 600mg HEPES, 500mg BSA, 1.5ml Tween-20 and 450ml purified H 2 O.
- Acceptor beads 3.75mg/ml stock.
- Cell suspension 20 ⁇ l acceptor beads/ml cells
- cAMP curve 1 ⁇ l acceptor beads/500 ⁇ l stimulation buffer
- Detection mix 2 ⁇ l biotin-cAMP + 800 ⁇ l detection buffer.
- the assay protocol dictates that compounds will be diluted by a factor of 2 when the final assay mix is assembled.
- Compounds were made as 4mM stocks in 100% DMSO and 10 ⁇ l transferred to a v-bottom 96-well plate for further dilutions to generate 11 pt concentration effect curves with Vz log dilutions. Test compounds were evaluated in the range of 0.03nM - 10 ⁇ M (final assay concentration).
- DOR cells were cultured in T225 flasks in DMEM/nutrient mix F12 supplemented with 2mM L- glutamine, 500 ⁇ g/ml hygromycin and 10% heat inactivated foetal bovine serum. Media was changed every 3 days. Cells were kept in an incubator at 37 0 C, with 95% O 2 and 5% CO 2 . Cell culture took place under sterile conditions in a Class Il Microbiological Safety Cabinet. Cells were split by removing the media, washing twice with 1OmIs PBS and adding 3mls of cell dissociation solution. The usual split ratio is 1 :5 to 1 :30. Cell dissociation solution was inactivated after approximately 5 minutes with supplemented media.
- DOR cells are harvested and a viable cell count performed (Trypan Blue exclusion to identify non-viable cells). The cells were then centrifuged and resuspended in 2x stimulation buffer to the required cell density (1.6E06 cells/ml equates to 8000/well in 5 ⁇ l).
- the plate was then read on a Fusion reader and EC 50 values calculated for the test compounds.
- the delta opioid receptor agonists of the present invention may be usefully combined with another pharmacologically active compound, or with two or more other pharmacologically active compounds, particularly in the treatment of pain.
- a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, as defined above may be administered simultaneously, sequentially or separately in combination with one or more agents selected from:
- an opioid analgesic e.g. morphine, heroin, hydromorphone, oxymorphone, levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine, dihydrocodeine, oxycodone, hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, nalbuphine or pentazocine;
- NSAID nonsteroidal antiinflammatory drug
- NSAID nonsteroidal antiinflammatory drug
- diclofenac diflusinal, etodolac
- fenbufen fenoprofen
- flufenisal flurbiprofen
- ibuprofen indomethacin
- ketoprofen ketorolac
- meclofenamic acid mefenamic acid
- meloxicam nabumetone, naproxen, nimesulide, nitroflurbiprofen, olsalazine, oxaprozin, phenylbutazone, piroxicam, sulfasalazine, sulindac, tolmetin or zomepirac
- NSAID nonsteroidal antiinflammatory drug
- a barbiturate, sedative e.g. amobarbital, aprobarbital, butabarbital, butabital, mephobarbital, metharbital, methohexital, pentobarbital, phenobartital, secobarbital, talbutal, theamylal or thiopental;
- a benzodiazepine having a sedative action e.g. chlordiazepoxide, clorazepate, diazepam, flurazepam, lorazepam, oxazepam, temazepam or triazolam;
- an Hi antagonist having a sedative action e.g. diphenhydramine, pyrilamine, promethazine, chlorpheniramine or chlorcyciizine; • a sedative such as glutethimide, meprobamate, methaqualone or dichloralphenazone;
- a skeletal muscle relaxant e.g. baclofen, carisoprodol, chlorzoxazone, cyclobenzaprine, methocarbamol or orphrenadine;
- an NMDA receptor antagonist e.g. dextromethorphan ((+)-3-hydroxy-N- methylmorphinan) or its metabolite dextrorphan ((+)-3-hydroxy-N-methylmorphinan), ketamine, memantine, pyrroloquinoline quinine, cis-4-(phosphonomethyl)-2- piperidinecarboxylic acid, budipine, EN-3231 (MorphiDex®, a combination formulation of morphine and dextromethorphan), topiramate, neramexane or perzinfotel including an NR2B antagonist, e.g.
- an NMDA receptor antagonist e.g. dextromethorphan ((+)-3-hydroxy-N- methylmorphinan) or its metabolite dextrorphan ((+)-3-hydroxy-N-methylmorphinan), ketamine, memantine, pyrroloquinoline quinine, cis-4-(phosphonomethyl
- an alpha-adrenergic e.g. doxazosin, tamsulosin, clonidine, guanfacine, dexmetatomidine, modafinil, or 4-amino-6,7-dimethoxy-2-(5-methane-sulfonamido- 1 ,2,3,4-tetrahydroisoquinol-2-yl)-5-(2-pyridyl)quinazoIine;
- a tricyclic antidepressant e.g. desipramine, imipramine, amitriptyline or nortriptyline
- an anticonvulsant e.g. carbamazepine, lamotrigine, topiratmate or valproate
- a tachykinin (NK) antagonist particularly an NK-3, NK-2 or NK-1 antagonist, e.g. ( ⁇ R,9R)-7-[3,5-bis(trifluoromethyl)benzyl]-8,9,10,11-tetrahydro-9-methyl-5-(4- methylphenyl)-7H-[1 ,4]diazocino[2,1-g][1 ,7]-naphthyridine-6-13-dione (TAK-637), 5- [[(2R,3S)-2-[(1 R)-1-[3,5-bis(trifluoromethyl)phenyl]ethoxy-3-(4-fluorophenyl)-4- morpholinyl]-methyl]-1,2-dihydro-3H-1,2,4-triazol-3-one (MK-869), aprepitant, lanepitant, dapitant or 3-[[2-methoxy-5-(trifluoromethoxy)phenyl]
- a muscarinic antagonist e.g oxybutynin, tolterodine, propiverine, tropsium chloride, darifenacin, solifenacin, temiverine and ipratropium
- a COX-2 selective inhibitor e.g. celecoxib, rofecoxib, parecoxib, valdecoxib, deracoxib, etoricoxib, or iumiracoxib;
- a neuroleptic such as droperidol, chlorpromazine, haloperidol, perphenazine, thioridazine, mesoridazine, trifluoperazine, fluphenazine, clozapine, olanzapine, risperidone, ziprasidone, quetiapine, sertindole, aripiprazole, sonepiprazole, blonanserin, iloperidone, perospirone, raclopride, zotepine, bifeprunox, asenapine, lurasidone, amisulpride, balaperidone, palindore, eplivanserin, osanetant, rimonabant, meclinertant, Miraxion® or sarizotan; • a vanilloid receptor agonist (e.g. resinferatoxin) or antagonist (e.g. capsazepine);
- a beta-adrenergic such as propranolol
- a corticosteroid such as dexamethasone
- a 5-HT receptor agonist or antagonist particularly a 5-HTI B /ID agonist such as eletriptan, sumatriptan, naratriptan, zolmitriptan or rizatriptan;
- a 5-HT 2A receptor antagonist such as (R)-(+)-alpha-(2,3-dimethoxy-phenyl)-1-[2-(4- fluorophenylethyl)]-4-piperidinemethanol (MDL-100907);
- a cholinergic (nicotinic) analgesic such as ispronicline (TC-1734), (E)-N-methyl-4-(3- pyridinyl)-3-buten-1 -amine (RJR-2403), (R)-5-(2-azetidinylmethoxy)-2-chloropyridine (ABT-594) or nicotine;
- a PDEV inhibitor such as 5-[2-ethoxy-5-(4-methyl-1-piperazinyl-sulphonyl)phenyl]-1- methyl-3-n-propyl-1 ,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil),
- an alpha-2-delta ligand such as gabapentin, pregabalin, 3-methylgabapentin, (1 ⁇ ,3 ⁇ ,5 ⁇ )(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid, (3S.5R)- 3-aminomethyl-5-methyl-heptanoic acid, (3S,5R)-3-amino-5-methyl-heptanoic acid, (3S,5R)-3-amino-5-methyl-octanoic acid, (2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3-f!uorobenzyl)-proline, [(1 R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6- yl]acetic acid, 3-(1-aminomethyl-cyclohexylmethyl)-4H-[1 ,2,4]oxadiazol-5-one,
- mGluRI metabotropic glutamate subtype 1 receptor
- a serotonin reuptake inhibitor such as sertraline, sertraline metabolite demethylsertraline, fluoxetine, norfluoxetine (fluoxetine desmethyl metabolite), fluvoxamine, paroxetine, citalopram, citalopram metabolite desmethylcitalopram, escitalopram, d,l-fenfluramine, femoxetine, ifoxetine, cyanodothiepin, litoxetine, dapoxetine, nefazodone, cericlamine and trazodone;
- a noradrenaline (norepinephrine) reuptake inhibitor such as maprotiline, lofepramine, mirtazepine, oxaprotiline, fezolamine, tomoxetine, mianserin, buproprion, buproprion metabolite hydroxybuproprion, nomifensine and viloxazine (Vivalan®), especially a selective noradrenaline reuptake inhibitor such as reboxetine, in particular (S 1 S)- reboxetine;
- a dual serotonin-noradrenaline reuptake inhibitor such as venlafaxine, venlafaxine metabolite O-desmethylvenlafaxine, clomipramine, clomipramine metabolite desmethylclomipramine, duloxetine, milnacipran and imipramine;
- an inducible nitric oxide synthase (iNOS) inhibitor such as S-[2-[(1- iminoethyl)amino]ethyl]-L-homocysteine, S-[2-[(1-iminoethyl)-amino]ethyl]-4,4-dioxo- L-cysteine, S-[2-[(1 -iminoethyl)amino]ethyl]-2-methyl-L-cysteine, (2S,5Z)-2-amino-2- methyl-7-[(1-iminoethyl)amino]-5-heptenoic acid, 2-[[(1 R,3S)-3-amino-4-hydroxy-1-(5- thiazolyl)-butyl]thio]-5-chloro-3-pyridinecarbonitri!e; 2-[[(1 R,3S)-3-amino-4-hydroxy-1- (5-thiazo
- an acetylcholinesterase inhibitor such as donepezil
- a prostaglandin E 2 subtype 4 (EP4) antagonist such as ⁇ /-[( ⁇ 2-[4-(2-ethyl-4,6- dimethyl-I H-imidazo ⁇ . ⁇ -clpyridin-i-yOpheny ⁇ ethylJaminoJ-carbonyll ⁇ -methyl- benzenesulfonamide or 4-[(1S)-1-( ⁇ [5-chloro-2-(3-fluorophenoxy)pyridin-3-yl]- carbonyl ⁇ amino)ethyl]benzoic acid;
- a leukotriene B4 antagonist such as 1-(3-biphenyl-4-ylmethyl-4-hydroxy-chroman-7- yl)-cyclopentanecarboxylic acid (CP-105696), 5-[2-(2-Carboxyethyl)-3-[6-(4- methoxyphenyl)-5E- hexenyl]oxyphenoxy]-valeric acid (ONO-4057) or DPC-11870, • a 5-lipoxygenase inhibitor, such as zileuton, 6-[(3-fluoro-5-[4-methoxy-3,4,5,6- tetrahydro-2H-pyran-4-yl])phenoxy-methyl]-1-methyl-2-quinolone (ZD-2138), or 2,3,5- trimethyl-6-(3-pyridylmethyl),1 ,4-benzoquinone (CV-6504);
- a leukotriene B4 antagonist such as 1-(3-biphenyl-4-yl
- a sodium channel blocker such as lidocaine
- a 5-HT3 antagonist such as ondansetron
- Optical Rotation is measured on a Perkin Elmer 341 Polarimeter. This high-resolution instrument has an accuracy of ⁇ 0.0020°. The rotation can be measured at 5 wavelengths, 589nm, 578nm, 546nm, 436nm and 365nm and the Polarimeter cell is temperature controlled with a Peltier unit, normally operated at 25°C.
- Example 30 The ketone of Example 30 (400mg, 1.36mmol) was dissolved in tetrahydrofuran.
- the reaction mixture was cooled to -48 0 C then added via a cannula to a suspension of sodium borohydride (205mg, 5.4mmol) in tetrahydrofuran (5ml) stirring at -48 0 C.
- the reaction mixture was allowed to slowly warm to room temperature overnight.
- reaction mixture was quenched with water then extracted from water into dichloromethane, dried over magnesium sulphate filtered and evaporated in vacuo.
- residue was purified by column chromatography using an ISCO ® silica cartridge eluting a linear gradient from dichloromethane - 90/10/1 dichloromethane/ methanoi/0.880 ammonia.
- the product was collected as a white solid (320mg, 0.8mmol,
- step (a) The sulfinamide of step (a) (384mg, 0.96mmol) was dissolved in methanol (600 ⁇ L). 4M Hydrogen chloride in dioxane (560 ⁇ L) was added and the reaction mixture stirred at room temperature for 30 minutes. The reaction mixture was diluted with ether and the resultant precipitate collected by filtration. The product was collected as a pale pink solid (340mg, 0.96mmol, 100%).
- step (b) The title compound was obtained as a cream solid in 80% yield from the sulfanamide of step (a), following the procedure described in Example 1 , step (b).
- Example 9 The alcohol of Example 9 (70mg, 0.24mmol) was dissolved in tetrahydrofuran (5ml). Sodium hydride (10mg, 0.26mmol) was added and the reaction mixture stirred for 15 minutes before the addition of iodomethane (16 ⁇ l_, 0.26mmol, 1.1eq). The reaction mixture was allowed to stir at room temperature for 18 hours. The reaction mixture was quenched with water then extracted from water into dichloromethane, dried over magnesium sulphate, filtered and evaporated in vacuo. The residue was purified by column chromatography using an ISCO ® silica cartridge eluting with a linear gradient from dichloromethane - 90:10:1 dichloromethane/methanol/0.880 ammonia.
- Example 13 1'-[4-(Dimethylamino)pyridin-2-yl1-N,N-dimethyl-2,3-dihvdrospirorindene-1.4'-piperidine1-3- carboxamide
- the crude material was purified by column chromatography using an ISCO ® silica cartridge eluting with dichloromethane/ methanol/0.880 ammonia (95:5:0.5). The product was obtained as a white solid (42mg, 0.09mmol, 39%).
- step (a) The ester of step (a) (336mg, 0.985mmol) was dissolved in water: dioxane (4ml:5ml). 2M sodium hydroxide (0.6ml, 1.2mmol) was added and the reaction mixture stirred at room temperature for 1 hour. The solution was neutralised with 2M hydrochloric acid. The reaction was concentrated in vacuo to yield the title compound in quantitative yield.
- Example 19 The nitrile of Example 19 (35mg, 0.113mmol) was dissolved in 1,4-dioxane (0.5ml) and 6N aqueous hydrochloric acid (5.0ml) added. The reaction mixture was stirred at 15O 0 C for 15 hours. The reaction mixture was concentrated in vacuo to yield the title compound as a yellow gum.
- step (a) The acid of step (a) (50mg, 0.138mmol), 1-hydroxybenzotriazole monohydrate (26mg, 0.180mmol), 1-(3-dimethylaminopropyl)-3-ethyIcarbodiimide HCI (31 mg, 0.160mmol) and ethyipiperazine (200 ⁇ L, 1.57mmol) were combined in dimethylformamide (3ml) and stirred at room temperature for 72 hours. The reaction mixture was concentrated in vacuo then redissolved in ethyl acetate (20ml). The solution was washed with 3% aqueous sodium hydrogen carbonate (2x1 OmI), dried over sodium sulphate, filtered and evaporated in vacuo.
- the crude product was purified by column chromatography using an ISCO ® silica cartridge eluting with dichloromethane - 90:10:1 dichloromethane/methanol/0.880 ammonia. (7mg, 0.015mmol, 12%).
- Example 132 The amine of Example 132 (110mg, 0.373mmol), benzenesulphonyl chloride (73mg, 0.411mmol) and triethylamine (156 ⁇ l_, 1.12mmol) were combined in dichloromethane and stirred at room temperature overnight. Benzenesulphonyl chloride (2.0 ⁇ L, 15mmol) was added and the reaction mixture stirred for a further 20 minutes. The reaction mixture was concentrated in vacuo and the residue purified by column chromatography using an ISCO ® silica cartridge eluting with dichloromethane - 90:10:1 dichloromethane/methanol/0.880 ammonia.
- Example 132 and methanesulphonyl chloride, following a similar procedure to that described in Example 27.
- the nitro-pyridine N-oxide of Preparation 103 (100mg, 0.3mmol) was dissolved in ethyl acetate (20ml), 10% palladium on carbon (10mg, 10% by weight) was added and the reaction mixture stirred at room temperature under 3.4 atmospheres of hydrogen overnight. The reaction mixture was concentrated in vacuo and the residue purified by column chromatography using an ISCO ® silica cartridge. Dichloromethane/methanol/0.880 ammonia (90:10:1 ) was used as the eiuent. The title compound was obtained as a white solid (10mg, 0.03mmol, 11%).
- the nitro-pyridine N-oxide of Preparation 115 (500mg, 1.5mmol) was dissolved in acetic acid (5ml), iron powder (491 mg, 8.8mmol) was added and the reaction mixture stirred at room temperature for 3 hours. The reaction mixture was concentrated in vacuo and the residue extracted from 2M sodium hydroxide into dichloromethane. The combined organic extracts were dried over magnesium sulphate, filtered and evaporated to yield the crude amino pyridine. The material was purified by column chromatography on silica gel using dichloromethane/methanoI/0.880 ammonia as eluent. The title compound was obtained as a white solid (260mg, 0.88mmol, 59%).
- the nitro-pyridine N-oxide of Preparation 105 (11mg, 0.023mmoi) was dissolved in acetic acid (2ml), iron powder (8mg, 0.143mmol) was added and the reaction mixture stirred at room temperature overnight. The reaction mixture was concentrated in vacuo and the residue extracted from 2M sodium hydroxide into dichloromethane. The combined organic extracts were dried over sodium sulphate, filtered and evaporated to yield the crude amino pyridine. The material was purified by column chromatography using an ISCO ® silica cartridge, eluting with dichloromethane/methanol/0.880 ammonia. The title compound was obtained as a colourless gum (5mg, 0.012mmol, 52%).
- Example 110 following a similar procedure to that described in Example 31.
- the product was purified by crystallisation from dich!oromethane:methanol.
- the crude product was purified by column chromatography on silica gel using dichloromethane/ methanol/0.880 ammonia (90:10:1) as eluent to afford the title compound as a brown solid (560mg, 1.65mmol, 72%).
- step (a) The ester of step (a) was dissolved in tetrahydrofuran (7ml). A solution of lithium hydroxide (47mg, 1.95mmol) in water (20ml) was added and the reaction mixture was stirred at room temperature for 7 hours. The reaction mixture was acidified to pH 3 with 2N hydrochloric acid and then extracted into ethyl acetate, dried over magnesium sulphate, filtered and evaporated to yield the title compound as a powder (442mg, 1.3mmol, 78%).
- LCMS was performed using a Phenomenex Luna C18, 5 ⁇ m, 30 x 4.6 mm id column with a flow rate of 2.5 ml/min.
- the solvent was evaporated and the residue dissolved in dimethylsulphoxide: water (3:1 ).
- The-material was purified by HPLC on a Phenomenex Luna C18, 10 ⁇ m, 150 x 10 mm id column using acetonitrile: 0.05% aqueous diethylamine as the mobile phase.
- LCMS was performed using a Phenomenex Luna C18, 5 ⁇ m, 30 x 4.6 mm id column with a flow rate of 2.5 ml/min.
- Detection is performed at 225nm. Retention times are quoted for ELSD detection.
- the reaction mixture was decanted, discarding the catalyst and the methanol evaporated.
- the material was purified by HPLC on a Phenomenex Luna C18, 10 ⁇ m, 150 x 10 mm id column using acetonitrile: 0.05% aqueous diethylamine as the mobile phase.
- LCMS was performed using a Phenomenex Luna C18, 5 ⁇ m, 30 x 4.6 mm id column with a flow rate of 2.5 ml/min.
- the amide was redissolved in dichloromethane (2ml) and treated with trifluoroacetic acid (1ml). The solution was stirred at room temperature for 4 hours and then concentrated in vacuo. The residue was extracted from 2M sodium hydroxide (2ml) into dichloromethane (3x2ml) and the combined organic extracts dried over sodium sulphate, filtered and evaporated.
- the nitro-pyridine N-oxide of Preparation 106 (65mg, 0.16mmol) was dissolved in ethanol (3ml), cyclohexene (0.4ml, 3.2mmol) and 10% palladium on carbon (15mg) were added and the reaction mixture heated at 7O 0 C for 8 hours. 10% Palladium on carbon (10mg) and cyclohexene (0.3ml) were added and the reaction heated for a further 2.5 hours. The catalyst was removed by filtration through Arbocel ® and the filtrates concentrated in vacuo. The crude product was purified by column chromatography on silica gel using dichloromethane: methanol as the eluent (18mg, O. ⁇ mmol, 30%).
- step (a) The ester of step (a) (23mg, 0.068mmol) and lithium hydroxide (2.5mg, 0.102mmol) were dissolved in tetrahydrofuran (0.2ml). Water (0.2ml) was added and the reaction mixture stirred at room temperature overnight. 2N hydrochloric acid (0.034ml, 0.102mmol) was added, then the reaction mixture evaporated in vacuo. The residue was triturated with ether. to yield the title compound as a beige solid (25mg, 0.07mmol).
- Example 131 The ester of Example 131 (70mg, 0.2mmol) was dissolved in tetrahydrofuran (2ml) and treated with 2M hydrochloric acid (2mi). The reaction mixture was stirred at 35 0 C for 20 hours. The solvent was evaporated in vacuo. The intermediate acid was dissolved in dimethylformamide (3ml) and treated with 1-hydroxybenzotriazole monohydrate (31 mg,
- the reaction mixture was concentrated in vacuo and the residue extracted from 2M sodium hydroxide into dichloromethane.
- the combined organic extracts were dried over sodium sulphate, filtered and evaporated to yield the crude aminopyridine.
- the material was purified by column chromatography over silica gel eluting with dichloromethane: methanol: 0.88 ammonia (95:5:0.5) to yield the title compound (155mg, 0.52mmol).
- nitro-pyridine N-oxide was dissolved in methanol. Palladium hydroxide (10% by weight) and ammonium formate (6eq) were added and the reaction mixture heated at reflux for 2-18 hours. The reaction mixture was allowed to cool to room temperature and the catalyst removed by filtration through Arbocel ® . The combined filtrates were concentrated in vacuo and the residue extracted from saturated sodium hydrogen carbonate into dichloromethane, dried over magnesium sulphate, filtered and evaporated to yield the crude amino pyridine. The material was purified by column chromatography on silica gel using dichIoromethane/methanol/0.880 ammonia as eluent to afford the title compounds. ⁇
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pain & Pain Management (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention provides compounds of formula (I): wherein the dotted line represents an optional covalent bond between X and Y, and X, Y, Z, m, R2, R3, R4, R5, R6, R7, R8, R9, a and b have the meanings given in the specification, and pharmaceutically acceptable salts and solvates thereof. The compounds are delta opioid receptor agonists and have a number of therapeutic applications, particularly in the treatment of pain.
Description
Spiropiperidine derivatives
Field of the Invention
This invention relates to spiropiperidine derivatives. More particularly, this invention relates to (aminopyridyl)spiropiperidine derivatives and to processes for the preparation of, intermediates used in the preparation of, compositions containing and the uses of, such derivatives.
The spiropiperidine derivatives of the present invention are delta opioid receptor agonists and have a number of therapeutic applications, particularly in the treatment of pain, especially neuropathic pain.
Description of the Prior Art
Delta opioid receptors (DORs), mu opioid receptors (MORs) and kappa opioid receptors (KORs) belong to a family of G-protein coupled receptors (GPCRs) that share extensive structural and sequence homology (Quock et al., 1999 Pharmacological Reviews 51 : 503) and are widely expressed in the central and peripheral nervous system of many species including man. Stimulation of DOR induces analgesia in a wide variety of animal models especially inflammatory models (Quock et al, 1999, above). Mouse KO studies have shown that the abuse liability and other CNS risks associated with opiates, such as morphine, are mediated through the mu opioid receptor (Kieffer and Gaveriaux-Ruff 2002 Prog. Neurobio. 66: 285-306). A DOR agonist approach may be devoid of the side effect profiles of mu and kappa opioids (Quock et al., 1999, above). Generally there is good correlation in DOR expression between species with few exceptions e.g. in the spinal cord where, in humans expression is restricted to laminae Ml, an important region for nociception (Mennicken et al., 2003 J. Comparative Neurol. 465: 349-360). Systemic administration of DOR agonists have little effect on acute pain but results in potent analgesia following certain inflammatory stimuli (Morinville, 2004 Pain 109(3):266-273) via a MOR-mediated increase in DOR trafficking and membrane insertion (Cahill et al., 2003 Pain 101 : 199-208; Morinville et al., 2004 J. Neurosci. 24(24):5549-5559). Systemic administration of the selective agonist SB-235863 reverses thermal hyperalgesia resulting from sciatic nerve ligation without any effect on gastrointestinal transit or motor coordination (Petrillo et al., 2003 JPET 307:1079-1089). Systemic administration of the selective agonist SB-235863 reverses thermal hyperalgesia resulting from sciatic nerve ligation without any effect on gastrointestinal transit or motor coordination (Petrillo et al, above). Systemic administration of DOR agonists has little effect on acute pain but results in potent analgesia following morphine pre-treatment or following inflammatory stimuli (Morinville et al., above). This seems to occur as a consequence of increased
trafficking and membrane insertion of DORs located in cytoplasmic vesicles (Cahill et al., above).
WO 2004/074273 discloses compounds of formula:

These compounds are stated to be useful in the treatment of a number of conditions, particularly the modulation of autoimmune disease.
WO 2004/004715 describes a method for modulation of conditions associated with the delta opioid receptor, comprising administration of a compound of formula:

These compounds are stated to be useful in the treatment of a number of conditions, including pain and allergic and inflammatory diseases.
Summary of the Invention
The present invention provides a compound of formula (I):

(I)
wherein: the dotted line represents an optional covalent bond between X and Y; when the groups X and Y are connected by a single bond, X and Y may be the same or different and each represent C(=O), C(L-R1)2, N(L-R1), O or S; when the groups X and Y are connected by a double bond, X and Y may be the same or different and each represent C(L-R1) or N;
Z represents C(=O), C(L-R1 )2, N(U-R1), O or S; m is 0 or 1 ; L represents a single bond, -O-, -S-, -(C=O)-, -(C=O)-O-, -(C=O)-NR-, -SO-,
-SO-NR-, -SO2-, -SO2-NR-, -NR-, -NR-SO-, -NR-SO2-, -NR(C=O)- -N(Het1)(C=O)-,
-NR(C=O)-O-, Or1-NR(C=O)-NR-;
L' represents a single bond, -(C=O)- -(C=O)-O-, -(C=O)-NR-, or -SO2-; with the provisos that not more than one of X, Y and Z represents O or S, and not more than two of X, Y and Z represents C(=0) or N(L-R1);
R1 represents H, (Ci-10)alkyl (optionally substituted by one or more substituents α), -(CR2)n-
(C3-8)cycloalkyl (wherein the cycloalkyl part is optionally substituted by one or more substituents α), -(CR2)n-Ar, -(CR2)n-Het1, -(CR2)n-Het2, or -CN, n is O or an integer from 1 to 6; R represents H or (C1-10)alkyl; or, when a group R and a group R1 substitute a nitrogen atom, the groups together with the nitrogen atom form a ring Het3;
R2 and R3 are the same or different and each represent R or -(CR2)-Ar, or R2 and R3, together with the nitrogen atom to which they are attached, form a ring Het3; R4 represents (C1-10)alkyl; a is O, 1 or 2;
R5 represents (C^iojalkyl, (C1-10)haloalkyl, hydroxy-(C1-10)alkyl, (C1-10)alkoxy(C1-10)alkyl, halogen, OH, (C1-10)alkoxy, (C^n^haloalkoxy, phenoxy, benzyloxy, (C3-8)cycloalkyIoxy, (C3-
8)cycloalkyl-(C1-10)alkoxy, -(CR2)P-CN, -(CR2)P-C(=O)R10, -(CR2)p-C(=O)Het2, -(CR2)P- C(=O)OR10, -(CR2)P-C(=O)N(R11)2, -(CR2)p-C(=O)NR-(CR2)n-N(R11)2, -(CR2)P-N(R11)2,
-(CR2)P-NRC(=O)R10, -(CR2)p-NRS(=O)R10, -(CR2)P-NRS(=O)2R10, SR, S(=O)R, S(=O)2R,
S(=O)N(R11)2 or pyridyl (optionally substituted by a (Ci-io)alkyl group), or two groups R5 together form a (C1-3)alkylene group, a (C1-3)alkyleneoxy group or a (C1-3)alkylenedioxy group; b is O or an integer from 1 to 4; p is O or an integer from 1 to 6;
R6, R7, R8 and R9 are each independently H or (C1-i0)alkyI, or two groups selected from (a) R6 or R7 and (b) R8 or R9 together form a bond or a (C1-3)alkylene group;
R10 represents H, (Ci-10)alky! (optionally substituted by one or more substituents α), -(CR2)q-
(C3-8)cycloaikyl (wherein the cycloalkyl part is optionally substituted by one or more substituents α), -(CR2)q-Ar, -(CR2)q-Het1, or -(CR2)q-Het2;
q is 0 or an integer from 1 to 6;
R11 represents H, (Ci.iO)alkyl or -(CR2)-Ar, or two groups R11, together with the nitrogen atom to which they are attached, form a ring Het3;
Ar is a phenyl or naphthyl group optionally substituted by one or more substituents β; Het1 is a group selected from (1) a 5- or 6-membered aromatic heterocyclic group having as ring heteroatoms (a) 1 to 4 nitrogen atoms, (b) 1 oxygen or 1 sulphur atom or (c) 1 oxygen atom or 1 sulphur atom and 1 or 2 nitrogen atoms, is optionally substituted (i) on carbon by one or more substituents α and/or (ii) on nitrogen by one or more substituents γ, (2) a bicyclic group comprising an optionally substituted 5- or 6-membered aromatic heterocyclic group, as defined in (1 ) above, fused with a benzene ring (the benzene ring being optionally substituted with one or more substituents β) or (3) a bicyclic group comprising two optionally substituted 5- or 6-membered aromatic heterocyclic group, as defined in (1 ) above, fused together; Het2 is a group selected from (4) a 3- to 8-membered saturated heterocyclic group containing 1 or 2 heteroatoms selected from nitrogen, oxygen and sulphur, is optionally substituted (i) on carbon by one or more substituents α and/or (ii) on nitrogen by one or more substituents γ and/or (iii) on sulphur by one or two =0 groups, (5) a bicyclic group comprising an optionally substituted 3- to 8-membered saturated heterocyclic group, as defined in (4) above, fused with a benzene ring (the benzene ring being optionally substituted by one or more substituents β) or (6) a bicyclic group comprising an optionally substituted 3- to 8-membered saturated heterocyclic group, as defined in (4) above, fused with a (C3-8)cycloalkane ring (the cycloalkane ring being optionally substituted by one or more substituents α); Het3 is a group selected from (7) a 3- to 8-membered saturated heterocyclic ring containing 1 or 2 heteroatoms of which one heteroatom is a nitrogen atom and the other heteroatom, if present, is selected from nitrogen, oxygen and sulphur, is optionally substituted (i) on carbon by one or more substituents α and/or (ii) on nitrogen by one or more substituents γ and/or (iii) on sulphur by one or two =0 groups, (8) a bicyclic ring comprising an optionally substituted 3- to 8-membered saturated heterocyclic ring, as defined in (7) above, fused with a benzene ring (the benzene ring being optionally substituted by one or more substituents β), or (9) a bicyclic ring comprising an optionally substituted 3- to 8-membered saturated heterocyclic ring, as defined in (7) above, fused with a (C3-8)cycloalkane ring (the cycloalkane ring being optionally substituted by one or more substituents α); substituents α are selected from (C1-10)alkyl, (C1-10)haloalkyl, hydroxy-(Ci.10)alkyl, (C1- 10)alkoxy(C1-io)alkyl, phenyl, pyridyl, halogen, OH, (C1-10)alkoxy, (C1-i0)haloalkoxy, phenoxy, benzyloxy, (C3-8)cycloalkyloxy, (C3-8)cycloalkyl-(C1-10)alkoxy, C(=O)R, CN, C(=O)OR, C(=O)NR2, NR2, NRC(=O)R, NRS(=O)R, NRS(=O)2R, SR, S(=O)R, S(=O)2R, S(=O)NR2 and =0, or two substituents α together form a (C1-4)alkylenedioxy group; substituents β are selected from (C1-10)alkyl, (C1-10)haloalkyl, hydroxy-(Ci.i0)alkyl, (C1- 10)alkoxy(C1-10)alkyl, phenyl, halogen, OH, (C1-10)alkoxy, (C1-10)haloalkoxy, phenoxy, benzyloxy, (C3-8)cycloalkyloxy,
 C(=O)R, CN, C(=O)OR, C(O)NR2, NR2, NRC(=O)R, NRS(=O)R, NRS(O)2R, SR, S(O)R, S(O)2R, S(O)NR2 and
pyridyl (optionally substituted by a (C^iojalkyl group), or two substituents β together form a (C^alkyleneoxy group or a (C^alkylenedioxy group; substituents γ are selected from (C1.10)alkyl, (CMoJhaloalkyl, phenyl, pyridyl, C(=O)R, CN, C(=O)OR, C(=O)NR2, S(=O)R and S(=O)2R; or a pharmaceutically acceptable salt or solvate thereof.
C(=O)R, CN, C(=O)OR, C(O)NR2, NR2, NRC(=O)R, NRS(=O)R, NRS(O)2R, SR, S(O)R, S(O)2R, S(O)NR2 and
pyridyl (optionally substituted by a (C^iojalkyl group), or two substituents β together form a (C^alkyleneoxy group or a (C^alkylenedioxy group; substituents γ are selected from (C1.10)alkyl, (CMoJhaloalkyl, phenyl, pyridyl, C(=O)R, CN, C(=O)OR, C(=O)NR2, S(=O)R and S(=O)2R; or a pharmaceutically acceptable salt or solvate thereof.
Detailed Description of Preferred Embodiments
In the present specification, 'alkyl' means a straight or branched monovalent saturated hydrocarbon chain containing from 1 to 10 carbon atoms. Examples of alkyl groups include methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, t-butyl, n-pentyl, i-pentyl, neopentyl, n-hexyl, i-hexyl, n-heptyl, n-octyl, n-nonyl and n-decyl. Preferably, 'alkyl' is (Ci-S)alkyl, more preferably (C1-4)alkyl, and most preferably methyl or ethyl. Where stated, the alkyl group may optionally be substituted by one or more substituents α, defined below, the number of substituents being limited by the number of substitutable positions (eg a methyl group is limited to 3 substituents). Where the alkyl group is substituted, there are preferably 1 to 3 substituents α, more preferably 1 or 2 substituents α, and most preferably one substituent α.
'Cycloalkyl' and 'cycloalkane' means a saturated carbocyclic ring having from 3 to 8 carbon atoms, the term 'cycloalkyl' referring to a monovalent group and the term 'cycloalkane ring' referring to a ring completed by two substituents together with the carbon atom to which they are attached, or a ring fused to one of the heterocyclic groups defined below. Examples of cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl. Preferably, 'cycloalkyl' is (C3-6)cycloalkyl. The cycloalkyl group or cycloalkane ring may optionally be substituted by one or more substituents α, defined below, the number of substituents being limited by the number of substitutable positions. Where the cycloalkyl group or cycloalkane ring is substituted, there are preferably 1 to 3 substituents α, more preferably 1 or 2 substituents α, and most preferably one substituent α. The cycioalkyl group or cycloalkane ring may also be fused with a benzene ring (the benzene ring being optionally substituted with one or more substituents β). Examples of such fused groups include indanyl and tetralinyl, of which indanyl is preferred.
'Alkylene' means a straight or branched divalent saturated hydrocarbon chain containing from 1 to 3 carbon atoms. Examples of alkyl groups include methylene, ethylene, methylmethylene and propylene. Preferably, 'alkylene' is methylene or ethylene, more preferably methylene.
'Alkyleneoxy' means a divalent group -alkylene-O-, wherein the alkylene moiety is a straight or branched saturated divalent hydrocarbon chain containing 1 to 4 carbon atoms. The
oxygen atom may be attached to the remainder of the molecule at either position. Preferred aikyleneoxy groups include methyleneoxy and ethyleneoxy.
'Alkylenedioxy' means a divalent group -O-alkylene-0-, wherein the alkylene moiety is a straight or branched saturated divalent hydrocarbon chain containing 1 to 4 carbon atoms. Preferred alkylenedioxy groups include methylenedioxy and ethylenedioxy.
'Halogen' means fluoro, chloro, bromo or iodo.
'Oxo' means a doubly bonded oxygen atom.
'Alkoxy' means 'alkyl-O-', wherein 'alkyl' is as defined above (either in its broadest aspect or a preferred aspect). Examples of alkoxy groups include methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, i-butoxy, sec-butoxy, t-butoxy, n-pentyloxy, i-pentyloxy, neopentyloxy, n-hexyloxy, i- hexyloxy, n-heptyloxy, n-octyloxy, n-nonyloxy and n-decyloxy. Preferably, 'alkoxy' is (C1-6)alkoxy, more preferably (C^alkoxy, and most preferably methoxy or ethoxy.
'Phenoxy' means 'phenyl-O-' and 'benzyloxy' means 'phenyl-CH2-O-'.
'Cycloalkyloxy' means 'cycloalkyl-O-', wherein 'cycloalkyl' is as defined above (either in its broadest aspect or a preferred aspect). Examples of cycloalkoxy groups include cyclopropyloxy, cyclobutyloxy, cyclopentyloxy, cyclohexyloxy, cycloheptyloxy, and cyclooctyloxy. Preferably, 'cycloalkyloxy' is (C3.6)cycloalkyloxy.
'Cycloalkylalkoxy' means alkoxy substituted with cycloalkyl, wherein 'alkoxy' and 'cycloalkyl' are as defined above (either in the broadest aspect or a preferred aspect). Examples of cycloalkylalkoxy groups include cyclopropylmethoxy, 1- or 2-cyclopropylethoxy, 1-, 2- or 3- cyclopropylpropoxy, 1-, 2-, 3- or 4-cyclopropylbutoxy, cyclobutylmethoxy, 1- or 2- cyclobutylethoxy, 1-, 2- or 3- cyclobutylpropoxy, 1-, 2-, 3- or 4-cyclobutylbutoxy, cyclopentylmethoxy, 1- or 2-cyclopentylethoxy, 1-, 2- or 3- cyclopentylpropoxy, 1-, 2-, 3- or 4- cyclopentylbutoxy, cyclohexylmethoxy, 1- or 2-cyclohexylethoxy, 1-, 2- or 3- cyclohexylpropoxy, 1-, 2-, 3- or 4-cyclohexylbutoxy, cycloheptylmethoxy, 1- or 2- cycloheptyiethoxy, 1-, 2- or 3- cycloheptylpropoxy, 1-, 2-, 3- or 4-cycloheptylbutoxy, cyclooctylmethoxy, 1- or 2-cyclooctyl ethoxy, 1-, 2- or 3- cyclooctylpropoxy and 1-, 2-, 3- or 4- cyclooctylbutoxy. Preferably, 'cycloalkylalkoxy' is (C3.6)cycloalkyl-(C1-6)alkoxy.
'Haloalkyl' means an alkyl group, as defined above (either in its broadest aspect or a preferred aspect) substituted by one or more halogen atoms (preferably fluorine or chlorine atoms, more preferably fluorine atoms), the number of halogen atoms being limited by the number of substitutable positions. Examples of 'haloalkyl' include mono-, di- or
trifluoromethyl, mono-, di- or trichloromethyl, bromomethyl, 2-fluoroethyl, 2,2-difluoroethyl, 2,2,2-trifluoroethyl, 2-chloroethyl, 2,2-dichloroethyl, 2,2,2-trichloroethyl, perfluoroethyl, perfluoropropyl and perfluorobutyl. Preferably, 'haloaikyi' is (C1-6)haloalkyl, more preferably (C1-4)haloalkyl, and most preferably mono-, di- or trifluoromethyl, mono-, di- or trichloromethyl, or perfluoroethyl.
'Haloalkoxy' means 'haloalkyl-O-', where 'haloaikyi' is as defined above (either in its broadest aspect or a preferred aspect), the number of halogen atoms being limited by the number of substitutable positions. Examples of 'haloalkoxy' include mono-, di- or trifluoromethoxy, mono-, di- or trichloromethoxy, bromomethoxy, 2-fluoroethoxy, 2,2-difluoroethoxy, 2,2,2- trifluoroethoxy, 2-chloroethoxy, perfluoroethoxy, perfluoropropoxy and perfluorobutoxy. Preferably, 'haloalkoxy' is (C^haloalkoxy, more preferably (C^Jhaloalkoxy, and most preferably mono-, di- or trifluoromethoxy.
'Hydroxyalkyl' means alkyl, as defined above (either in its broadest aspect or a preferred aspect) substituted by a hydroxy (-OH) group. Examples of hydroxyalkyl groups include hydroxymethyl, 1- or 2-hydroxyethyl, 1-, 2- or 3-hydroxypropyl, and 1-, 2-, 3- or 4- hydroxybutyl. Preferably, 'hydroxyalkyl' is (C^alkyl substituted with hydroxy, more preferably (C1-4)alkyl substituted with hydroxy. Hydroxymethyl and 1- or 2-hydroxyethyl groups are especially preferred.
'Alkoxyalkyl' means alkyl, as defined above (either in its broadest aspect or a preferred aspect) substituted by an alkoxy group, as defined above (either in its broadest aspect or a preferred aspect). Examples of alkoxyalkyl groups include methoxymethyl, ethoxymethyl, propoxymethyl, butoxymethyl, 1- or 2-methoxyethyl, 1- or 2-ethoxyethyl, 1- or 2-propoxyethyl, 1- or 2-butoxyethyl, 1-, 2- or 3-methoxypropyl, 1-, 2- or 3-ethoxypropyl, 1-, 2- or 3- propoxypropyl, 1-, 2- or 3-butoxypropyl, 1-, 2-, 3- or 4-methoxybutyl, 1-, 2-, 3- or 4- ethoxybutyl, 1-, 2-, 3- or 4-propoxybutyl and 1-, 2-, 3- or 4-butoxybutyl. Preferably, 'alkoxyalkyl' means (C1-6)alkyl substituted with (C1-4)alkoxy, more preferably (C1-4)alkyl substituted with (C1^aIkOXy. Methoxymethyl, ethoxymethyl and 2-methoxyethyl groups are especially preferred.
The group R is hydrogen or (Ci-10)alkyl, as defined above. Suitably, R is hydrogen or (C1- 6)alkyl. Preferably, R is hydrogen or (C^alkyl.
The group R1 represents H, (C1-10)alkyl (optionally substituted by one or more substituents α), -(CR2)n-(C3-8)cycloalkyl (wherein the cycloalkyl part is optionally substituted by one or more substituents α), -(CR2)n-Het1, -(CR2)n-Het2, -(CR2)n-Ar, or -CN.
Preferably, R1 represents H, (Ci.10)alkyl (optionally substituted by one substituent α), -(CR2)n-
(C3.8)cycloalkyl (wherein the cycloalkyl part is optionally substituted by one substituent α), - (CR2)n-Het\ -(CR2)n-Het2, -(CR2)n-Ar, or -CN.
More preferably, R1 represents H, (Ci.10)alkyl, phenyl, benzyl (wherein the phenyl part is optionally substituted with (C1-4)alkyl or (C1-4)BIkOXy), -Het1, -(CH2)-Het1 or -CN.
In one embodiment, R1 is R1a wherein R1a represents H, C1-4 alkyl, phenyl, benzyl (wherein the phenyl part is optionally substituted with (C1-4)alkyl or (C1-4)alkoxy), pyridyl, -(CH2)- pyridyl, oxadiazolyl (optionally substituted with phenyl) or oxazolyl (optionally substituted with C1-4 alkyl).
In other embodiments, R1 is R1z wherein R1z represents H, C1-6 alkyl, -(CR2)n-(C3.8)cycloalkyl, -(CR2)π-Ar, or -(CR2)n-Het2.
More preferably, R1 is R1y wherein R1y represents H, C1-4 alkyl, C3-8 cycloalkyl or phenyl.
In the definition of the linking group -(CR2)n-, each R may be the same or different. Preferably each R is H or methyl. More preferably, each R is H.
Preferably, n is 0, 1 or 2; more preferably 0 or 1.
The group Ar is an aryl group selected from phenyl and naphthyl. The group Ar is optionally substituted by one or more substituents β, defined below, the number of substituents being limited by the number of substitutable positions. Where the group Ar is substituted, there are preferably 1 to 5 substituents β, more preferably 1 to 3 substituents β, even more preferably 1 or 2 substituents β, and most preferably one substituent β.
Preferably, Ar is phenyl optionally substituted by one substituent β.
More preferably, Ar is unsubstituted phenyl or phenyl substituted with one substituent selected from (C^alkyl and (C^alkoxy.
The group Het1 is selected from (1 ) a 5- or 6-membered aromatic heterocyclic group having as ring heteroatoms (a) 1 to 4 nitrogen atoms, (b) 1 oxygen or 1 sulphur atom or (c) 1 oxygen atom or 1 sulphur atom and 1 or 2 nitrogen atoms, is optionally substituted (i) on carbon by one or more substituents α and/or (ii) on nitrogen by one or more substituents γ, (2) a bicyclic group comprising an optionally substituted 5- or 6-membered aromatic heterocyclic group, as defined in (1 ) above, fused with a benzene ring (the benzene ring being optionally substituted with one or more substituents β) or (3) a bicyclic group comprising two optionally substituted
5- or 6-membered aromatic heterocyclic group, as defined in (1) above, fused together. Examples of Het1 include thienyl, furanyl, pyrrolyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, triazolyl, oxadiazolyl, thiadiazolyl, tetrazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, indolyl, benzofuranyl, benzothienyl, benzpyrazolyl, benzimidazolyl, indazolyl, benzothiazolyl, benzoxazolyl, benzotriazolyl, quinolyl, isoquinolinyl, cinnolinyl, quinazolinyl, quinoxalinyl, phthalazinyl, naphthyridinyl, pteridinyl and pyrazolopyrimidinyl. Preferably, Het1 is a group selected from (1 ) and (2) above. Where the group Het1 is substituted on carbon, there are preferably 1 or 2 substituents α, and most preferably one substituent α. In addition, when the group Het1 is optionally substituted on the one or more ring nitrogen atoms, if present, there is preferably only one substituent γ.
Preferably, Het1 is thienyl, furanyl, pyrrolyl, imidazolyl, oxadiazolyl, oxazolyl, thiazolyl or pyridyl (these groups being optionally substituted on carbon with one substituent α, and/or on nitrogen with one substituent γ).
More preferably, Het1 is pyridyl, oxadiazolyl or oxazolyl (these groups being optionally substituted with a phenyl group or a (C^alkyl group).
The group Het2 is a group selected from (4) a 3- to 8-membered saturated heterocyclic group containing 1 or 2 heteroatoms selected from nitrogen, oxygen and sulphur, is optionally substituted (i) on carbon by one or more substituents α and/or (ii) on nitrogen by one or more substituents γ and/or (iii) on sulphur by one or two =0 groups, (5) a bicyclic group comprising an optionally substituted 3- to 8-membered saturated heterocyclic group, as defined in (4) above, fused with a benzene ring (the benzene ring being optionally substituted by one or more substituents β) or (6) a bicyclic group comprising an optionally substituted 5- or 6- membered aromatic heterocyclic group, as defined in (1) above, fused with a (C3- 8)cycloalkane ring (the cycloalkane ring being optionally substituted by one or more substituents α). Specific examples of Het2 include oxiranyl, thiiranyl, aziridinyl, oxetanyl, thietanyl, azetidinyl, tetrahydrofuranyl, tetrahydrothiophenyl, pyrrolidinyl, pyrazolidinyl, imidazolidinyl, dioxolanyl, oxazolidinyl, thiazolidinyi, isoxazolidinyl, isothiazolidinyl, tetrahydropyranyl, tetrahydrothiopyranyl, piperidinyl, morpholinyl, thiomorpholinyl, dioxanyl, oxathianyl, dithianyl, piperazinyl, azepanyl, oxepanyl, oxazepanyl, diazepanyl and azocinyl. Preferably, Het2 is a group (4) above. Where the group Het2 is substituted on carbon, there are preferably 1 or 2 substituents α, and most preferably one substituent α. In addition, when the group is optionally substituted on the one or more ring nitrogen atoms, if present, there is preferably only one substituent γ.
Preferably, Het2 is azetidinyl, pyrrolidinyl, piperidinyl, morpholinyl, thiomorpholinyl or piperazinyl, these groups being optionally substituted on carbon by one substituent α and/or on nitrogen by one substituent γ.
More preferably, Het2 is azetidinyl, pyrrolidinyl, piperidinyl or piperazinyl, these groups being optionally substituted on carbon and/or nitrogen by a (CM)alkyl group.
The ring Het3 is selected from (7) a 3- to 8-membered saturated heterocyclic ring containing 1 or 2 heteroatoms of which one heteroatom is a nitrogen atom and the other heteroatom, if present, is selected from nitrogen, oxygen and sulphur, is optionally substituted (i) on carbon by one or more substituents α and/or (ii) on nitrogen by one or more substituents γ and/or (iii) on sulphur by one or two =0 groups, (8) a bicyclic ring comprising an optionally substituted 3- to 8-membered saturated heterocyclic ring, as defined in (7) above, fused with a benzene ring (the benzene ring being optionally substituted by one or more substituents β), or (9) a bicyclic ring comprising an optionally substituted 3- to 8-membered saturated heterocyclic ring, as defined in (7) above, fused with a (C3.8)cycloalkane ring (the cycloalkane ring being optionally substituted by one or more substituents α). The ring is formed by two groups described above together with the nitrogen atom to which they are attached. Specific examples of Het3 include aziridine, azetidine, pyrrolidine, piperidine, morpholine, thiomorpholine, piperazine, azepane, oxazepane, diazepane and azocine. Preferably, the ring Het3 is a ring (7) above. Where the ring Het3 is substituted on carbon, there are preferably 1 or 2 substituents α, and most preferably one substituent α. In addition, when the ring is substituted on the one or more ring nitrogen atoms, there is preferably only one substituent γ.
Preferably, Het3 is azetidine, pyrrolidine, piperidine, morpholine, thiomorpholine or piperazine, these groups being optionally substituted on carbon by one substituent α and/or on nitrogen by one substituent γ.
More preferably, Het3 is azetidine, pyrrolidine, piperidine or piperazine, these groups being optionally substituted on carbon and/or nitrogen by a (C1-4)alkyl group.
Preferably, substituents α are selected from substituents α1 comprising halogen, (C1-6)alkoxy, phenoxy, (C1-4)alkylenedioxy, NR2, pyridyl, CN, CONR2 and NRCOR.
More preferably, substituents a are selected from substituents α2 comprising halogen, (C1-4)alkoxy, phenoxy, (C1-2)alkylenedioxy, CONR2 and pyridyl.
Preferably, substituents β are selected from substituents β1 comprising (C1-6)alkyl, (C1- 6)haloalkyl, phenyl, halogen, (C1-6)alkoxy, phenoxy, CN, CONR2, NRCOR, and COOR.
More preferably, substituents β are selected from substituents β2 comprising (C1-4)alkyl, halogen, (C1-4)haloaikyl (especially CF3) and (C^alkoxy.
Preferably substituents γ are selected from substituents γ1 comprising (C1-6)alkyl, phenyl and pyridyl.
More preferably substituents γ are selected from substituents γ2 comprising (C1-4)alkyl.
Preferably, the group NR2R3 is present at the 4-position of the pyridine ring (the pyridine ring nitrogen being the 1 -position).
In the definitions of the divalent linking groups L and L', the bond to the left of the groups is connected to the spiro ring and the bond to the right is connected to the group R1.
In one embodiment, the group L is La wherein La represents a single bond, -O-, -(C=O)-O-, -(C=O)-NR-, -SO2-, -NR-, -NR-SO2- -NR(C=O)-, or -N(Het1)(C=O)-.
More preferably, L is Lb wherein Lb represents a single bond, -0-, -(C=O)-NR-, -NR-SO2-, -NR(C=O)-, -or N(pyridyl)(C=O)-.
Even more preferably, L is Lc wherein Lc represents a single bond, -0-, -(C=O)-NR-, -NR-SO2-, Or -NR(C=O)-.
In other embodiments, the group L is Lz wherein Lz represents a single bond, -(C=O)-O-, or -(C=O)-NR-.
Preferably, L is Ly wherein Ly represents a single bond, -(C=O)-O-, or -(C=O)-NR-.
Preferably, the group L' is La wherein La represents a single bond, -(C=O)-, -(C=O)-O-, or -SO2-.
More preferably, L' is Lb wherein Lb' represents a single bond, -(C=O)-O-, or -SO2-.
In other embodiments, the group L' is Lz wherein Lz represents a single bond or -(C=O)-.
Preferably, the bond between X and Y is a single bond.
When X represents C(L-R1)2, preferably one group (L-R1) is H and the other group (L-R1) is other than H. In other embodiments, both groups (L-R1) are H.
Preferably, the group X represents C(=O), C(La-R1)2, N(La'-R1) or O1 wherein La represents a single bond, -O-, -(C=O)-O-, -(C=O)-NR-, -SO2-, -NR-, -NR-SO2-, -NR(C=O)-, or
-N(Het1 )(C=0)-; La> represents a single bond, -(C=O)- -(C=O)-O- or -SO2-; and
R, R1 and Het1 are as defined above, either in its broadest aspect or in a preferred aspect; or a group R and a group R1, together with the nitrogen atom to which they are attached, together form a group Het3 wherein Het3 is as defined above, either in its broadest aspect or in a preferred aspect.
More preferably, X represents C(=0), C(Lb-R1)2, N(Lb'-R1) or O, wherein
Lb represents a single bond, -O- -(C=O)-NR-, -NR-SO2-, -NR(C=O)-, or
-N(pyridyl)(C=O)-;
Lb> represents a single bond, -(C=O)-O- Or -SO2-; and R and R1 are as defined above, either in its broadest aspect or in a preferred aspect; or a group R and a group R1, together with the nitrogen atom to which they are attached, together form an azetidine, pyrrolidine, piperidine or piperazine group which is optionally substituted by a Ci-4 alkyl group.
Even more preferably, X represents C(LC-R1a)2> N(Lb'-R1a) or O, wherein
Lc represents a single bond, -O- -(C=O)-NR-, -NR-SO2-, or -NR(C=O)-;
Lb' represents a single bond, -(C=O)-O-, -SO2-; and
R1a represents H, Ci-4 alkyl, phenyl, benzyl, pyridyl, oxadiazolyl (optionally substituted with phenyl) or oxazolyl (optionally substituted with Ci-4 alkyl).
When Y represents C(L-R1)2, preferably one group (L-R1) is H and the other group (L-R1) is other than H. In other embodiments, both groups (L-R1) are H.
Preferably, the group Y represents C(=0), C(LZ-R1)2, N(LZ'-R1) or O, wherein Lz represents a single bond, -(C=O)-O-, or -(C=O)-NR-;
Lz' represents a single bond or -(C=O)-; and
R and R1 are as defined above, either in its broadest aspect or in a preferred aspect; or a group R and a group R1, together with the nitrogen atom to which they are attached, together form a group Het3 wherein Het3 is as defined above, either in its broadest aspect or in a preferred aspect.
More preferably, Y represents C(=O), C(Ly-R1z)2, N(Ly>- R1z) or O, wherein Ly represents a single bond, -(C=O)-O-, Or -(C=O)-NR-; Ly' represents a single bond or -(C=O)-; R1z represents H, C1-6 alkyl, -(CR2)n-(C3-8)cycloalkyl, -(CR2)n-Ar, or -(CR2)n-Het2;
n' is 0, 1 or 2; and
R and Het2 are as defined above either in its broadest aspect or in a preferred aspect; or a group R and a group R1, together with the nitrogen atom to which they are attached, together form a piperidine, piperazine or azepine group which is optionally fused with a benzene ring.
Even more preferably, Y represents C(=O), C(LX-R1y)2 or O, wherein L* represents a single bond or -(C=O)-Q-; and R1y represents H, Ci-4 alkyl, or phenyl.
m may be 0 or 1.
When m is 1 , Z represents C(=O), C(L-R1)2, N(L'- R1), O or S.
Preferably, Z represents C(L-R1)2 (wherein L and R1 are as defined above, either in its broadest aspect or in a preferred aspect) or O.
More preferably, Z represents CH2 or O, even more preferably CH2.
Preferably, R2 and R3 are the same or different and each represent R (as defined above, either in its broadest aspect or in a preferred aspect) or benzyl, or R2 and R3, together with the nitrogen atom to which they are attached, form a group Het3 (as defined above, either in its broadest aspect or in a preferred aspect).
More preferably, R2 and R3 are the same or different and each represent H, C1-4 alkyl or benzyl.
Even more preferably, R2 and R3 each represent H.
Preferably, a is 0 or 1 , suitably 0.
Preferably, b is 0, 1 or 2, suitably 0 or 1.
Preferably, n is 0 or 1 , suitably 0.
Preferably, R5 represents (C1-6)alkyl, F, Cl, Br, OH, (C1-6JaIkOXy, CN,
-(CR2)P-C(=O)OR10, -(CR2)p-C(=O)Het2, -(CR2)p-NRC(=O)R10,
-(CR2)p-C(=O)N(R11)2, or -(CR2)p-C(=0)NR-(CR2)n-N(R11)2, wherein R, n and Het3 are as defined above, either in its broadest aspect or in a preferred aspect, and p, R10 and R11 are as defined below, either in its broadest aspect or a preferred aspect.
More preferably, R5 represents (C1-4)alkyl, F, OH, (C^alkoxy, CN, C(=O)NR2, -CH2- NRC(=O)R10 or C(=O)NR-(CR2)n-NR2, where R is as defined above, either in its broadest aspect or in a preferred aspect, and R10 is as defined below, either in its broadest aspect or a preferred aspect.
Even more preferably, R5 represents methyl, OH or methoxy.
Preferably, p is O or 1 , more preferably 1.
Preferably, R6, R7, R8 and R9 are each independently H or (C1-4)alkyl. More preferably, R6, R7, R8 and R9 are all H.
Preferably, R10 represents H, (C1-6)alkyl (optionally substituted by one or more substituents α), (C3.6)cycloalkyl (wherein the cycloalkyl part is optionally substituted by one or more substituents α), benzyl (wherein the phenyl part is optionally substituted by one or more substituents β1), or -Het1.
Preferably, R10 represents H, (C1-4)alkyl, benzyl (wherein the phenyl part is optionally substituted by one or more substituents β2), or pyridyl.
Preferably q is O, 1 or 2, more preferably 0 or 1.
Preferably, R11 represents H or (C1-6)alkyl.
The compounds of formula (I) are preferably compounds of formulae (1-1), (I-2), (I-3), (I-4), (I- 5), (I-6), (I-7) or (I-8):

(I-5) (I-6) (I-7) (I-8)
In the formulae (1-1) to (I-8), L, L', R1, R2, R3, R4, R5, R6, R7, R8, R9, a and b are as defined above, either in the broadest aspect or a preferred aspect.
In the formulae (1-1), (I-2), (I-3), (I-7) and (I-8), which contain groups (L-R1), in one embodiment one group (L-R1) is other than H and the other group or groups (L-R1) are H; in another embodiment all of the groups (L-R1) are H.
Particularly preferred compounds of the invention include those in which each variable in Formula (I) is selected from the suitable and/or preferred groups for each variable. Even more preferred compounds of the invention include those where each variable in Formula (I) is selected from the more preferred or most preferred groups for each variable.
In one embodiment, the present invention provides a compound of formula (I) wherein: the dotted line represents an optional covalent bond between X and Y; when the groups X and Y are connected by a single bond, X and Y may be the same or different and each represent C(=O), C(L-R1)2, N(L-R1), O or S; when the groups X and Y are connected by a double bond, X and Y may be the same or different and each represent C(L-R1) or N;
Z represents C(=O), C(L-R1)2, N(U-R1), O or S; m is 0 or 1 ;
L represents a single bond, -O- -S-, -(C=O)-, -(C=O)-O-, -(C=O)-NR-, -SO- -SO-NR-, -SO2-, -SO2-NR-, -NR-, -NR-SO-, -NR-SO2-, -NR(C=O)- -N(Het1)(C=O)-, -N R(C=O)-O-, or -N R(C=O)-N R-;
L' represents a single bond ,'-(C=O)-, -(C=O)-O-, -(C=O)-NR-, or -SO2-; with the provisos that not more than one of X, Y and Z represents O or S, and not more than two of X, Y and Z represents C(=0) or N(L -R1);
R1 represents H, (C1-10)alkyl (optionally substituted by one or more substituents α), -(CR2)n- (C3-8)cycloalkyl (wherein the cycloalkyl part is optionally substituted by one or more substituents α), -(CR2)n-Ar, -(CR2)n-Het1, -(CR2)n-Het2, or -CN, n is O or an integer of from 1 to 6; R represents H or (C1-10)alkyl; or, when a group R and a group R1 substitute a nitrogen atom, the groups together with the nitrogen atom form a ring Het3;
R2 and R3 are the same or different and each represent R or -(CR2)-Ar, or R2 and R3, together with the nitrogen atom to which they are attached, form a ring Het3; R4 represents (C1-10)alkyl; a is O, 1 or 2; R5 represents C1-10 alkyl, halogen, CF3, OR, CN, C(=0)R, C(=0)NR2, C(=O)NR-(CR2)n-NR2, C(=O)Het2 or C(=0)0R; b is O or an integer from 1 to 4; R6, R7, R8 and R9 are hydrogen; Ar is a phenyl or naphthyl group optionally substituted by one or more substituents β; Het1 is a 5- or 6-membered aromatic heterocyclic group having as ring heteroatoms (a) 1 to 4 nitrogen atoms, (b) one oxygen or one sulphur atom or (c) 1 oxygen atom or 1 sulphur atom and 1 or 2 nitrogen atoms, the group being optionally substituted on carbon by one or more substituents α and/or on nitrogen by one or more substituents γ and is optionally fused with a benzene ring (the benzene ring being optionally substituted by one or more substituents β); Het2 is a 3- to 8-membered saturated heterocyclic group containing 1 or 2 heteroatoms selected from nitrogen, oxygen and sulphur, the group being optionally substituted on carbon by one or more substituents α and/or on nitrogen by one or more substituents γ and/or on sulphur by one or two oxo groups, is optionally fused with a benzene ring (the benzene ring being optionally substituted by one or more substituents β) or a (C3-8)cycloalkane ring (the cycloalkane ring being optionally substituted by one or more substituents α);
Het3 is a 3- to 8-membered saturated heterocyclic ring containing 1 or 2 heteroatoms of which one heteroatom is a nitrogen atom and the other heteroatom, if present, is selected from nitrogen, oxygen and sulphur, is optionally substituted on carbon by one or more substituents α and/or on nitrogen by one or more substituents γ and/or on sulphur by one or two oxo groups and is optionally fused with a benzene ring (the benzene ring being optionally
substituted by one or more substituents β) or a (C3.8)cycloalkane ring (the cycloalkane ring being optionally substituted by one or more substituents α); substituents α are selected from (C1-10)alkyI, phenyl, halogen, CF3, OR, O-phenyl,
(C^alkylenedioxy, NR2, pyridyl, CN, CONR2, NRCOR, NRSOR1 NRSO2R, COOR and =0; substituents β are selected from (C1-10)alkyl, phenyl, halogen, CF3, OR,
O-phenyl, (C1-4)alkylenedioxy, NR2, pyridyl, CN, CONR2, NRCOR, NRSOR, NRSO2R and
COOR; substituents γ are selected from (C1-1o)alkyl, phenyl, pyridyl, CONR2 and COOR; or a pharmaceutically acceptable salt or solvate thereof.
The following compounds are preferred:
(3S)-1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-amine;
(3R)- 1 '-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-amine;
N-[(3S)-1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]-acetamide; N-[(3S)-1 '-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]-pyridine-2- carboxamide;
N-[(3S)-1 '-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]- cyclopropanecarboxamide;
N-[(3S)-1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]- cyclohexanecarboxamide;
N-[(3S)-1l-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]-2,2,2- trifluoroacetamide;
N-[(3f?)-1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]-pyridine-2- carboxamide; 1 '-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-ol;
2-(3-methoxy-2,3-dihydro-1 Η-spirotindene-1 ,4'-piperidin]-1 '-yl)pyridin-4-amine;
N,N-dimethyl-1'-(4-piperidin-1-ylpyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
1'-{4-[benzyl(methyI)amino]pyridin-2-yl}-N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4l-piperidine]- 3-carboxamide;
^-^-(dimethylaminoJpyridin^-yO-N.N-dimethyl^.S-dihydrospirofindene-i ^'-piperidinel-S- carboxamide;
1'-{4-[ethyl(methyI)amino]pyridin-2-yl}-N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide; N,N-dimethyl-1'-(4-pyrrolidin-1-ylpyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(2R)-1'-(4-aminopyridin-2-yl)-N-(3-chlorobenzyl)spiro[1-benzofuran-3,4'-piperidine]-2- carboxamide; methyl (2R)-1'-(4-aminopyridin-2-yl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxylate;
(2S)-r-(4-aminopyridin-2-yl)-N-(3-chlorobenzyl)spiro[1-benzofuran-3,4'-piperidine]-2- carboxamide;
1'-(4-aminopyridin-2-yl)spiro[1-benzofuran-3,4'-piperidine]-7-carbonitrile; methyl 1 '-(4-aminopyridin-2-yl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3-carboxylate; 1 '-(4-aminopyridin-2-yl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3-carboxylic acid;
2-{7-[(4-ethylpiperazin-1-yl)carbonyl]-1Η-spiro[1-benzofuran-3,4I-piperidin]-1l-yl}pyridin-4- amine;
1'-(4-aminopyridin-2-yl)-N-(1-ethylpropyl)spiro[1-benzofuran-3,4'-piperidine]-7-carboxamide;
1'-(4-aminopyridin-2-yl)-N,N-dimethylspiro[1-benzofuran-3,4'-piperidine]-7-carboxamide; 1'-(4-aminopyridin-2-y!)-N-[3-(dimethylamino)-2,2-dimethylpropyl]spiro[1-benzofuran-3,4'- piperidine]-7-carboxamide; r-(4-aminopyridin-2-yl)spiro[1-benzofuran-3,4'-piperidine]-5-carbonitrile;
N-{r-[4-(dimethylamino)pyridin-2-yI]-2,3-dihydrospiro[indene-1,4'-piperidin]-3-yl}-N- methylpyridine-2-carboxamide; N-[1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1,4'-piperidin]-3-yl]-benzenesulfonamide;
N-[1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4I-piperidin]-3-yl]-methanesulfonamide;
1 '-(4-aminopyridin-2-yl)spiro[indole-3,4'-piperidin]-2(1 H)-one;
1'-(4-aminopyridin-2-yl)spiro[indene-1 ,4'-piperidin]-3(2H)-one;
1'-(4-aminopyridin-2-yl)-2-(4-methoxybenzyl)spirotisoindole-1 ,4'-piperidin]-3(2H)-one; 1 '-(4-aminopyridin-2-yl)-2,3-dihydrospirotindene-1 ,4'-piperidine]-3-carbonitrile;
2-(5-fluoro-1 Η,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1 '-yl)pyridin-4-amine;
2-(6-methyl-1 Η,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1 '-yl)pyridin-4-amine;
1'-(4-aminopyridin-2-yl)-4-methoxy-3H-spiro[2-benzofuran-1 ,4'-piperidin]-3-one;
2-(3-phenyl-1 Η,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1 '-yl)pyridin-4-amine; 2-(1 'H-spiro[1 -benzofuran-3,4'-piperidin]-1 '-yl)pyridin-4-amine;
2-(1H,1'H-spiro[isochromene-4,4'-piperidin]-1'-yl)pyridin-4-amine; methyl 1 '-(4-aminopyridin-2-yl)spiro[1 -benzofuran-3,4'-piperidine]-2-carboxylate;
1'-(4-aminopyridin-2-yi)spiro[1-benzofuran-3,4'-piperidine]-2-carboxylic acid;
1'-(4-aminopyridiπ-2-yl)-N-isopropylspiro[1-benzofuran-3,4'-piperidine]-2-carboxamide; 1'-(4-aminopyridin-2-yl)-N-(3-fluorobenzyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
2-[2-(1 ,2,4,5-tetrahydro-3H-3-benzazepin-3-yIcarbonyl)-1 'H-spiro[1-benzofuran-3,4'-piperidin]-
1 '-yl]pyridin-4-amine;
1'-(4-aminopyridin-2-yl)-N-(cyclohexylmethyl)spiro[1-benzofuran-3,4'-pipeπdine]-2- carboxamide; 2-[2-(1 ,4-dioxa-8-azaspiro[4.5]dec-8-ylcarbonyI)-1 Η-spiro[1 -benzofuran-3,4'-piperidin]-1 '- - yl]pyridin-4-amine; r-(4-aminopyridin-2-yl)-N-methyl-N-propylspiro[1-benzofuran-3,4I-piperidine3-2-carboxamide;
1l-(4-aminopyridin-2-yl)-N-(2-fluorobenzyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
1'-(4-aminopyridin-2-yl)-N-(2-chlorobenzyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxamide; 1 l-(4-aminopyridin-2-yl)-N-ethyl-N-methylspiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
1l-(4-aminopyridin-2-yl)-N-(3-chlorobenzyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
I'-C^aminopyridin^-yO-N-ethylspiroti-benzofuran-S^'-pipericiinel^-carboxannide;
1'-(4-aminopyridin-2-yl)-N-(2-methoxybenzyl)spiro[1-benzofuran-3,4l-piperidine]-2- carboxamide; 2-[2-(piperidin-1 -ylcarbonyl)-1 'H-spiro[1 -benzofuran-3,4'-piperidin]-1 '-yl]pyridin-4-amine;
1'-(4-aminopyridin-2-yl)-N-(4-methoxybenzyl)spiro[1-benzofuran-3,4'-piperidine]-2- carboxamide;
1'-(4-aminopyridin-2-yl)-N-(3-ethoxypropyl)spiro[1-benzofuran-3,4l-piperidine]-2-carboxamide;
1'-(4-aminopyridin-2-yl)-N-propylspiro[1-benzofuran-3,4I-piperidine]-2-carboxamide; 1'-(4-aminopyridin-2-yl)-N-(4-phenylbutyl)spiro[1-ben2ofuran-3,4'-piperidine]-2-carboxamide;
1l-(4-aminopyridin-2-y))-N-[2-(trifluoromethyl)benzyl]spiro[1-benzofuran-3,4'-piperidine]-2- carboxamide;
1'-(4-aminopyridin-2-yl)-N-(3-methoxypropyl)spiro[1-benzofuran-3,4'-piperidine]-2- carboxamide; 1 '-(4-aminopyridin-2-yl)-N-(2-phenoxyethyl)spiro[1 -benzofuran-3,4'-piperidine]-2-carboxamide;
1'-(4-aminopyridin-2-yl)-N-[4-(trifluoromethyl)benzyl]spiro[1-benzofuran-3,4'-piperidine]-2- carboxamide;
2-{2-[(4-methoxypiperidin-1 -yl)carbony!]-1 'H-spiro[1 -benzofuran-3,4'-piperidin]-1 '-yl}pyridin-4- amine; 2-{2-[(4-pyridin-2-ylpiperazin-1 -yl)carbonyl]-1 Η-spiro[1 -benzofuran-3,4'-piperidin]-1 '-yljpyridin-
4-amine;
1l-(4-aminopyridin-2-yl)-N-methyl-N-(2-phenylethyl)spiro[1-benzofuran-3,4'-piperidine]-2- carboxamide;
1 '-(4-aminopyridin-2-yl)-N-(4-fluorobenzyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxamide; 1'-(4-aminopyridin-2-yl)-N-(4-chlorobenzyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
1'-(4-aminopyridin-2-yl)-N-pentylspiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
1'-(4-aminopyridin-2-yl)-N-(3-chlorobenzyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide; r-(4-aminopyridin-2-yl)-N-(4-fluorobenzyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N-(2-methoxyethyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N-(3-methoxybenzyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide; 1'-(4-aminopyridin-2-yl)-N-(1-ethylpropyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide; r-(4-aminopyridin-2-yl)-N-(4-fluorobenzyl)-N-methyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide; 2-[3-(piperidin-1-ylcarbonyl)-1'H,3H-spiro[2-benzofuran-1 ,4l-piperidin]-r-yl]pyridin-4-amine;
1I-(4-aminopyridin-2-yl)-N-[3-(dimethylamino)-2,2-dimethylpropyl]-3H-spiro[2-benzofuran-1 ,4I- piperidine]-3-carboxamide;
1'-(4-aminopyridin-2-yl)-N-[2-(3-fluorophenyl)ethyl]-3H-spiro[2-benzofuran-1 ,4'-pipendine]-3- carboxamide; 2-{3-[(4-ethylpiperazin-1 -yl)carbonyl]-1 'H,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1 '-yl}pyridin-4- amine;
1I-(4-aminopyridin-2-yl)-N-(3,5-dimethylbenzyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N-butyl-N-methyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N-(3-isopropoxypropyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
2-[3-(pyrrolidin-1-ylcarbonyl)-1Η,3H-spiro[2-benzofuran-1,4'-piperidin]-1 '-yl]pyridin-4-amine; r-(4-aminopyridin-2-yl)-N-(2-methoxy-2-methylpropyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]- 3-carboxamide;
1'-(4-aminopyridin-2-yl)-N-ethyl-N-methyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yI)-N,N-dimethyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3-carboxamide; r-(4-aminopyridin-2-yl)-N-(2-phenoxyethyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N-butyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3-carboxamide;
1'-(4-aminopyridin-2-yl)-N-isobutyl-N-methyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
2-[3-(morpholin-4-ylcarbonyl)-1Η,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1'-yl]pyridin-4-amine; 1 '-(4-aminopyridin-2-yl)-N-(2-chlorobenzyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N-(2-fluorobenzyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N-cyclopentyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3-carboxamide; 1 '-(4-aminopyridin-2-yl)-N-(4-methoxybenzyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N-(2-phenylethyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N-(4-methylbenzyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1 '-(4-aminopyridin-2-yl)-1 -methylspiro[indole-3,4'-piperidin]-2(1 H)-one;
2-(1 -acetyl-1 ,2-dihydro-1 'H-spiro[indole-3,4'-piperidin]-1 '-yl)pyridin-4-amine;
2-(2,3-dihydro-1'H-spiro[indene-1 ,4'-piperidin]-1'-yl)pyridin-4-amine;
1'-(4-aminopyridin-2-yl)-5-fluoro-1-methylspiro[indole-3,4'-piperidin]-2(1 H)-one; (3S)-1'-(4-aminopyridin-2-yl)-N-ethyl-2,3-dihydrospiro[indene-1 ,4I-piperidine]-3-carboxamide;
(SSVI'-C^aminopyridin-Σ-ylJ-N-isopropyl-N-nnethyl^.S-clihycirospiropndene-i ^'-pipericlinel-S- carboxamide;
(3S)-r-(4-aminopyridin-2-yl)-N-(cyclopropylmethyl)-N-methy)-2,3-dihydro-spiro[indene-1 ,4'- piperidine]-3-carboxamide; 2-[(3S)-3-(azetidin-1 -ylcarbonyl)-2,3-dihydro-1 'H-spiro[indene-1 ,4'-piperidin]-1 '-yI]pyridin-4- amine;
2-{(3S)-3-[(4,4-difiuoropiperidin-1-yl)carbonyl]-2,3-dihydro-1'H-spiro[indene-1 ,4'-piperidin]-1'- yl}pyridin-4-amine;
2-[(3S)-3-(morpholin-4-ylcarbonyl)-2,3-dihydro-1lH-spiro[indene-1l4'-piperidin]-1'-yl]pyridin-4- amine;
(3S)-1'-(4-aminopyridin-2-yl)-N-(2-methoxyethyl)-N-methyl-2,3-dihydro-spiro[indene-1 ,4'- piperidine]-3-carboxamide;
2-[(3S)-3-(pyrrolidin-1-ylcarbonyl)-2,3-dihydro-1'H-spiro[indene-1 ,4'-piperidin]-1'-yl]pyridin-4- amine; (3S)-1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxamide;
(3S)-1'-(4-aminopyridin-2-yl)-N-cyclobutyl-2,3-dihydrospiro[indene-1 ,4l-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxamide;
1 '-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxylic acid; 2-[1-(methylsulfonyl)-1 ,2-dihydro-1'H-spiro[indole-3,4'-piperidin]-r-yl]pyridin-4-amine;
2-[1-(ethylsulfonyI)-1 ,2-dihydro-1Η-spiro[indole-3)4'-piperidin]-r-yl]pyridin-4-amine;
2-[1-(propylsulfonyl)-1 ,2-dihydro-1 'H-spiro[indole-3,4'-piperidin]-1 '-yl]pyridin-4-amine;
2-[1-(isopropylsulfonyl)-1 ,2-dihydro-1'H-spiro[indole-3,4l-piperidin]-1'-yl]pyridin-4-amine;
2-[1-(butylsulfonyl)-1 ,2τdihydro-1'H-spiro[indole-3,4'-piperidin]-1'-yl]pyridin-4-amine; 2-{1-[(2,2,2-trifluoroethyl)sulfonyl]-1 ,2-dihydro-1Η-spiro[indole-3,4'-piperidin]-1'-yl}pyridin-4- amine;
2-[1-(phenylsuIfonyl)-1 ,2-dihydro-1'H-spiro[indole-3,4'-piperidin]-r-yl]pyridin-4-amine;
2-(1 -{[4-(trifluoromethyl)phenyl]sulfonyl}-1 ,2-dihydro-1 Η-spiro[indole-3,4'-piperidin]-1 '- yl)pyridin-4-amine; 2-[1-(benzylsulfonyl)-1,2-dihydro-1'H-spiro[indole-3,4'-piperidin]-1'-yl]pyridin-4-amine;
1'-(4-aminopyridin-2-yl)-N,N-dimethyl-2,3-dihydrospiro[chromene-4,4'-piperidine]-2- carboxamide;
2-(2,3-dihydro-1lH-spiro[chromene-4,4'-piperidin]-1'-yl)pyridin-4-amine;
(3RS)-N-I '-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]benzamide; (3R)-N-[I '-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4l-piperidin]-3-yl]benzamide;
(3S)-N-[I I-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]benzamide;
2-[(3S)-3-(5-phenyl-1 ,3,4-oxadiazol-2-yl)-2,3-dihydro-1 Η-spiro[indene-1 ,4'-piperidin]-1 '- yl]pyridin-4-amine;
2-[(3S)-3-(5-methyl-1 ,3,4-oxadiazol-2-yl)-2,3-dihydro-1 Η-spiro[indene-1 ,4'-piperidin]-1 '- yl]pyridin-4-amine;
2-[(3S)-3-(4-methyl-1 ,3-oxazol-2-yl)-2,3-dihydro-1 Η-spiro[indene-1 ,4'-piperidin]-1 '-yl] pyridine- amine;
N-II'-CΦaminopyridin^-yl^.S-dihydrospiroIindene-i ^'-piperidinJ-S-yllpyridine^- carboxamide; 2-(1'H,3H-spiro[2-benzofuran-1 ,4l-piperidin]-1I-yl)pyridin-4-amine;
1'-(4-aminopyridin-2-yl)-N,N,3-trimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N-pyridin-2-yl-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-amine;
2-(6-fluoro-1'H-spiro[1-benzofuran-3,4'-piperidin]-1'-yl)pyridin-4-amine; N-[1'-(4-aminopyridin-2-yl)-2,3-dihydrospirotindene-1 ,4'-piperidin]-3-yl]-2-methylpropanamide; methyl 1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[chromene-4,4'-piperidine]-2-carboxylate;
1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-amine;
(2f?)-r-(4-aminopyridin-2-yI)-N-benzylspiro[1-benzofuran-3,4l-piperidine]-2-carboxamide;
(2R)-1'-(4-aminopyridin-2-yl)-N,N-dimethylspiro[1-benzofuran-3,4'-piperidine]-2-carboxamide; (2S)-1 '-(4-aminopyridin-2-yl)-N-benzylspiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
(2S)-r-(4-aminopyridin-2-yl)-N,N-dimethylspiro[1-benzofuran-3,4l-piperidine]-2-carboxamide; methyl (2S)- 1 '-(4-aminopyridin-2-yl)spiro[1 -benzofuran-3,4'-piperidine]-2-carboxylate;
(3S)-r-(4-aminopyπdin-2-yl)-N-methyl-N-propyl-2,3-dihydrospiropndene-1 ,4'-piperidine]-3- carboxamide; (3S)-1'-(4-aminopyridin-2-yl)-N-ethyl-N-methyl-2,3-dihydrospiro[indene-1 ,4I-piperidine]-3- carboxamide;
N-[1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]-N-pyridin-2- ylacetamide;
N-[1'-(4-aminopyridin-2-yl)-2,3-dihydrospirotiηdene-1 ,4'-piperidin]-3-yl]-N-methylpyridine-2- carboxamide;
1'-(4-aminopyridin-2-yl)-2-methylspiro[isoindole-1 ,4'-piperidin]-3(2H)-one;
(3S)-1'-(4-aminopyridin-2-yl)-N-pyridin-2-yl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-1'-(4-aminopyridin-2-yl)-N-benzyl-2,3-dihydrospiro[indene-1 l4I-piperidine]-3- carboxamide;
(3S)-1'-(4-aminopyridin-2-yl)-N-{5-[(diethylamino)carbonyl]pyridin-2-yl}-2,3- dihydrospiro[indene-1 ,4'-piperidine]-3-carboxamide; r-(4-aminopyridin-2-yl)-2-benzylspiro[isoindole-1 ,4'-piperidin]-3(2H)-one;
(3R)-1 '-(4-aminopyridin-2-yl)-N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-1I-(4-aminopyridin-2-yl)-N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide; methyl T-(4-aminopyridin-2-yl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxylate;
(3S)-1'-(4-aminopyridin-2-yl)-N-cyclopropyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(SSJ-i'-C^aminopyridin^-yO-N-propyl^.S-dihydrospirotindene-i ^'-piperidinel-S-carboxamide;
(3S)-1'-(4-aminopyridin-2-yl)-N,N-diethyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
(3f?)-1'-(4-aminopyridin-2-yl)-NlN-diethyl-3H-spiro[2-benzofuran-1,4'-piperidine]-3- carboxamide;
2-(1 ,2-dihydro-1 Η-spiro[indole-3,4'-piperidin3-1 '-yl)pyridin-4-amine;
(3S)-1'-(4-aminopyridin-2-yl)-N,N-diethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(+)-1'-(4-aminopyridin-2-yl)-N,N-dimethyl-5-methoxy-3H-spiro[2-benzo-furan-1,4'-piperidine]- 3-carboxamide;
(+)-1'-(4-aminopyridin-2-yl)-N,N-diethyl-5-methoxy-3H-spiro[2-benzofuran-1 I4'-piperidine]-3- carboxamide; r-(4-aminopyridin-2-yl)-N,N-dimethyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3-carboxamide;
1'-(4-aminopyridin-2-yl)-N-ethyl-N-methyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
2-[7-(aminomethyl)-1 'H-spiro[1 -benzofuran-3,4'-piperidin]-1 '-yl]pyridine-4-amine;
N-{[1'-(4-aminopyridin-2-yl)spiro[1-benzofuran-3,4'-piperidin]-7-yl]methyl}-acetamide;
N-{[1'-(4-aminopyridin-2-yl)spiro[1-benzofuran-3,4'-piperidin]-7-yl]methyl}-pyridine-2- carboxamide; methyl 1 '-(4-aminopyridin-2-yl)spiro[indole-3,4'-piperidine]-1 (2H)-carboxylate; ethyl 1 '-(4-aminopyridin-2-yl)spiro[indole-3,4'-piperidine]-1 (2H)-carboxylate; isopropyl 1 '-(4-aminopyridin-2-yl)spiro[indole-3,4'-piperidine]-1 (2H)-carboxylate; and pharmaceutically acceptable salts and solvates thereof.
The following compounds are more preferred:
N-[(3S)-1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4I-piperidin]-3-yl]-acetamide;
N-[(3S)-r-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1,4'-piperidin]-3-yl]-pyridine-2- carboxamide;
N-[(3S)-r-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]- cyclopropanecarboxamide;
N-t(3S)-1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]- cyclohexanecarboxamide;
N-[(3S)-1l-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4l-piperidin]-3-yl]-2,2,2- trifluoroacetamide; N-[(3R)-1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1,4'-piperidin]-3-yl]pyridine-2- carboxamide;
1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1,4'-piperidin]-3-ol;
2-(3-methoxy-2,3-dihydro-1 Η-spiro[indene-1 ,4'-piperidin]-1 '-yl)pyridin-4-amine;
I'-^-lbenzyKmethylJaminolpyridin^-ylϊ-N.N-dimethyl^.S-dihydrospiroIindene-i ^'-piperidine]- 3-carboxamide;
(2R)-1l-(4-aminopyridin-2-yl)-N-(3-chlorobenzyl)spiro[1-benzofuran-3l4l-piperidine]-2- carboxamide; methyl (2R)-1'-(4-aminopyridin-2-yl)spirot1-benzofuran-3,4'-piperidine]-2-carboxylate;
(2S)-1'-(4-aminopyridin-2-yl)-N-(3-chlorobenzyl)spiro[1-benzofuran-3,4'-piperidine]-2- carboxamide; r-(4-aminopyridin-2-yl)spiro[1-benzofuran-3,4'-piperidine]-7-carbonitrile;
1'-(4-aminopyridin-2-yl)-N-(1-ethylpropyl)spiro[1-benzofuran-3,4'-piperidine]-7-carboxamide; 1 '-(4-aminopyridin-2-yl)-N-[3-(dimethylamino)-2,2-dimethylpropyl]spiro[1 -benzofuran-3,4'- piperidine]-7-carboxamide; N-{1'-[4-(dimethylamino)pyridin-2-yI]-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl}-N- methylpyridine-2-carboxamide;
N-[r-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]benzenesulfonamide;
N-[1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]methanesulfonamide;
1'-(4-aminopyridin-2-yl)spiro[indole-3,4'-piperidin]-2(1 H)-one; 1'-(4-aminopyridin-2-yl)spiro[indene-1 ,4'-piperidin]-3(2H)-one;
1'-(4-aminopyridin-2-yl)-2-(4-methoxybenzyl)spiro[isoindole-1 ,4'-piperidin]-3(2H)-one; r-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carbonitrile;
2-(5-fluoro-1 Η,3H-spirot2-beπzofuran-1 ,4'-piperidin]-1 '-yl)pyridin-4-amine;
2-(6-methyl-1 Η,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1 '-yl)pyridin-4-amine; 1 '-(4-aminopyridin-2-yl)-4-methoxy-3H-spiro[2-benzofuran-1 ,4'-piperidin]-3-one;
2-(3-phenyl-1 'H,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1 '-yl)pyridin-4-amine;
2-(rH-spiro[1-benzofuran-3,4'-piperidin]-1'-yl)pyridin-4-amine;
2-(1 H,1'H-spiro[isochromene-4,4'-piperidin]-1'-yl)pyridin-4-amine; r-(4-aminopyridin-2-yl)-N-(3-fluorobenzyI)spiro[1-benzofuran-3,4'-piperidine]-2-carboxamide; 2-[2-(1 ,2,4,5-tetrahydro-3H-3-benzazepin-3-ylcarbonyl)-1 'H-spiro[1 -benzofuran-3,4'-piperidin]-
1 '-yl]pyridin-4-amine;
1'-(4-aminopyridin-2-yl)-N-(cyclohexylmethyl)spiro[1-benzofuran-3,4'-piperidine]-2- carboxamide;
1I-(4-aminopyridin-2-yl)-N-methyl-N-propylspiro[1-benzofuran-3,4'-piperidine]-2-carboxamide; 1'-(4-aminopyridin-2-yl)-N-(2-fluorobenzyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxamide; r-(4-aminopyridin-2-yl)-N-(2-chlorobenzyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
1l-(4-aminopyridin-2-yl)-N-ethyl-N-methylspiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
1l-(4-aminopyridin-2-yl)-N-(3-chlorobenzyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
1'-(4-aminopyridin-2-yl)-N-(2-methoxybenzyl)spiro[1-benzofuran-3,4l-piperidine]-2- carboxamide;
2-[2-(piperidin-1-ylcarbonyl)-rH-spiro[1-benzofuran-3,4'-piperidin]-1'-yl]pyridin-4-amine; r-(4-aminopyridin-2-yl)-N-(3-ethoxypropyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
1'-(4-aminopyridin-2-yl)-N-(4-phenylbutyl)spiro[1-benzofuran-3,4l-piperidine]-2-carboxamide; r-(4-aminopyndin-2-yl)-N-(2-phenoxyethyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
r-(4-aminopyridin-2-yl)-N-[4-(trifluoromethyl)benzyl]spiro[1-benzofuran-3,4l-piperidine]-2- carboxamide;
2-{2-[(4-methoxypiperidin-1 -yl)carbonyl]-1 'H-spiro[1 -benzofuran-3,4'-piperidin]-1 '-yl}pyridin-4- amine; 2-{2-[(4-pyridin-2-ylpiperazin-1-yl)carbonyl]-1'H-spiro[1-benzofuran-3,4'-piperidin]-1'-yl}pyridin-
4-amine;
1'-(4-aminopyridin-2-yl)-N-methyl-N-(2-phenylethyl)spiro[1-benzofuran-3,4'-piperidine]-2- carboxamide; r-(4-aminopyridin-2-yl)-N-(4-fluorobenzyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxamide; 1'-(4-aminopyridin-2-yl)-N-(4-chlorobenzyl)spiro[1-benzofuraπ-3,4'-piperidine]-2-carboxamide;
1 '-(4-aminopyridin-2-yl)-N-pentylspiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
1'-(4-aminopyridin-2-yl)-N-(3-chlorobenzyl)-3H-spiro[2-benzofuran-1 ,4I-piperidine]-3- carboxamide;
1 '-(4-aminopyridin-2-yl)-N-(4-fluorobenzyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- • carboxamide;
1'-(4-aminopyridin-2-yl)-N-(2-methoxyethyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1 '-(4-aminopyridin-2-yl)-N-(3-methoxybenzyl)-3H-spirot2-benzofuran-1 ,4'-piperidine]-3- carboxamide; 1'-(4-aminopyridin-2-yl)-N-(1-ethylpropyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide; r-(4-aminopyridin-2-yl)-N-(4-fluorobenzyl)-N-methyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
2-[3-(piperidin-1-ylcarbonyl)-1IH,3H-spiro[2-benzofuran-1 ,4I-piperidin]-1'-yl]pyridin-4-amine; 1'-(4-aminopyridin-2-yl)-N-[3-(dimethylamino)-2,2-dimethylpropyl]-3H-spiro[2-benzofuran-1 ,4'- piperidine]-3-carboxamide;
1'-(4-aminopyridin-2-yl)-N-[2-(3-fluorophenyl)ethyI]-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
2-{3-[(4-ethylpiperazin-1 -yl)carbonyl]-1 Η,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1 '-ylJpyridin-4- amine;
(3RS)-1l-(4-aminopyridin-2-yl)-N-(3,5-dimethylbenzyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-
3-carboxamide;
(3R)-1'-(4-aminopyridin-2-yl)-N-butyl-N-methyI-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide; (3S)-1'-(4-aminopyridin-2-yl)-N-(3-isopropoxypropyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
2-[3-(pyrrolidin-1 -ylcarbonyl)-1 'H,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1 '-yl]pyridin-4-amine;
1'-(4-aminopyridin-2-yl)-N-(2-methoxy-2-methyIpropyl)-3H-spiro[2-benzofuran-1 ,4l-piperidine]-
3-carboxamide;
1'-(4-aminopyridin-2-yl)-N-ethyl-N-methyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N,N-climethyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3-carboxamide;
11-(4-aminopyridin-2-yl)-N-(2-phenoxyethyI)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-y!)-N-isobuty!-N-methyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide; r-(4-aminopyridin-2-yl)-N-(2-chlorobenzyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide; 1 '-(4-aminopyridin-2-yl)-N-(2-fluorobenzyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N-cyclopentyl-3H-spiro[2-benzofuran-1,4'-piperidine]-3-carboxamide;
1'-(4-aminopyridin-2-yl)-N-(4-methoxybenzyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide; 1 '-(4-aminopyridin-2-yl)-N-(2-phenylethyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide; r-(4-aminopyridin-2-yl)-1-methylspiro[indole-3,4'-piperidin]-2(1 H)-one; r-(4-aminopyridin-2-yl)-5-fluoro-1-methylspiro[indole-3,4'-piperidin]-2(1 H)-one;
(3S)-1l-(4-aminopyridin-2-yl)-N-ethyl-2,3-dihydrospiro[indene-1 l4'-piperidine]-3-carboxamide; (3S)-1'-(4-aminopyridin-2-yl)-N-isopropyl-N-methyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-1'-(4-aminopyridin-2-yl)-N-(cyclopropyImethyl)-N-methyl-2,3-dihydrospiro[indene-1 ,4'- piperidine]-3-carboxamide;
2-[(3S)-3-(azetidin-1-ylcarbonyl)-2,3-dihydro-1'H-spiro[indene-1 ,4'-piperidin]-1'-yl]pyridin-4- amine;
2-{(3S)-3-[(4)4-difluoropiperidin-1-yl)carbonyl]-2,3-dihydro-1'H-spiro[indene-1 ,4l-piperidin]-11- yl}pyridin-4-amine;
2-[(3S)-3-(morpholin-4-yIcarbonyl)-2,3-dihydro-1'H-spiro[indene-1,4'-piperidin]-1 '-yl]pyridin-4- amine; (3S)-1'-(4-aminopyridin-2-yl)-N-(2-methoxyethyl)-N-methyl-2,3-dihydrospiro[indene-1 ,4'- piperidine]-3-carboxamide;
2-[(3S)-3-(pyrrolidin-1 -ylcarbonyl)-2,3-dihydro-1 'H-spirofindene-1 ,4'-piperidin]-1 '-yl]pyridin-4- amine;
(3S)-1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxamide; (3S)-1 '-(4-aminopyridin-2-yl)-N-cyclobutyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxamide;
2-[1-(methylsulfonyl)-1 )2-dihydro-1'H-spiro[indole-3,4'-piperidin]-1'-yl]pyridin-4-amine;
2-[1-(ethylsulfonyI)-1 ,2-dihydro-1'H-spiro[indole-3,4'-piperidin]-1'-yl]pyridin-4-amine; 2-[1 -(propylsulfonyl)-i ,2-dihydro-1 'H-spiro[indole-3,4'-piperidin]-1 '-yl]pyridin-4-amine;
2-t1-(isopropylsulfonyl)-1 ,2-dihydro-1'H-spiro[indole-3,4I-piperidin]-1l-yl]pyridin-4-amine;
2-[1 -(butylsulfonyl)-i ,2-dihydro-1 'H-spiro[indole-3,4'-piperidin]-1 '-yI]pyridin-4-amine;
2-{1-[(2,2,2-trifluoroethyl)sulfonyl]-1 ,2-dihydro-rH-spiro[indole-3,4'-piperidin]-1 '-yl}pyridin-4- amine; 2-[1 -(phenylsulfonyl)-i ,2-dihydro-1 'H-spiropndole-S^'-piperidinH '-yl]pyridin-4-amine;
2-(1 -{[4-(trifluoromethyl)phenyl]sulfonyl}-1 ,2-dihydro-1 'H-spiro[indole-3,4'-piperidin]-1 '- yl)pyridin-4-amine;
2-[1 -(benzylsulfonyl)-i ,2-dihydro-1 'H-spiro[indole-3,4'-piperidin]-1 '-yI]pyridin-4-amine;
2-(2,3-dihydro-1'H-spiro[chromene-4,4'-piperidin]-1'-yl)pyridin-4-amine; N-[1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]-benzamide;
N-[1l-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]-benzamide;
N-[1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro{indene-1 ,4'-piperidin]-3-yl]-benzamide;
2-[(3S)-3-(5-phenyl-1 ,3,4-oxadiazol-2-yl)-2,3-dihydro-1 Η-spiro[indene-1 ,4'-pipeπdin]-1 '- yl]pyridin-4-amine; 2-[(3S)-3-(5-methyl-1 ,3,4-oxadiazol-2-yl)-2I3-dihydro-1'H-spiro[indene-1 ,4'-piperidin]-1'- yl]pyridin-4-amine;
2-[(3S)-3-(4-methyl-1 ,3-oxazol-2-yI)-2,3-dihydro-1 'H-spiro[indene-1 ,4'-piperidin]-1 '-yl]pyridin-4- amine;
N-[1I-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]pyridine-2- carboxamide;
2-(1 'H,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1 '-yl)pyridin-4-amine; r-(4-aminopyridin-2-yl)-N-pyridin-2-yl-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-amine;
2-(6-fluoro-1 'H-spiro[1 -benzofuran-3,4'-piperidin]-1 '-yl)pyridin-4-amine;
N-[r-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]-2-tnethylpropanamide; methyl 1 '-(4-aminopyridin-2-yl)-2,3-dihydrospiro[chromene-4,4'-piperidine]-2-carboxylate;
(2R)-1'-(4-aminopyridin-2-yl)-N-benzylspiro[1-benzofuran-3,4'-piperidine]-2-carboxamide;
(2S)-r-(4-aminopyridin-2-yl)-N-benzylspiro[1-benzofuran-3,4'-piperidine]-2-carboxamide; methyl (2S)-1 '-(4-aminopyridin-2-yl)spiro[1 -benzofuran-3,4'-piperidine]-2-carboxylate;
(3S)-1'-(4-aminopyridin-2-yl)-N-methyl-N-propyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-1 '-(4-aminopyridin-2-yl)-N-ethyl-N-methyl-2,3-dihydrospiro[iπdene-1 ,4'-piperidine]-3- carboxamide;
N-[1I-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]-N-pyridin-2- ylacetamide; N-[1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]-N-methylpyridine-2- carboxamide;
1'-(4-aminopyridin-2-yl)-2-methyIspiro[isoindole-1 ,4'-piperidin]-3(2H)-one;
(3S)-1'-(4-aminopyridin-2-yl)-N-pyridin-2-yl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-144-aminopyridin-2-yl)-N-benzyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-1'-(4-aminopyridin-2-yl)-N-{5-[(diethylamino)carbonyl]pyridin-2-yl}-2,3- dihydrospiro[indene-1 ,4'-piperidine]-3-carboxamide; 1 '-(4-aminopyridin-2-yl)-2-benzylspiro[isoindole-1 ,4'-piperidin]-3(2H)-one;
(3R)-1'-(4-aminopyridin-2-yl)-N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-1'-(4-aminopyridin-2-yl)-N,N-dimethy!-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide; (3S)-1 '-(4-aminopyridin-2-yl)-N-cyclopropyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-1l-(4-aminopyridin-2-yl)-N-propyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxamide;
(3S)-r-(4-amiπopyridin-2-yl)-N,N-diethyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide; (3R)-1'-(4-aminopyridin-2-yl)-N,N-diethyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
(3S)-1 l-(4-aminopyridin-2-yl)-N,N-diethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(+)-1'-(4-aminopyridin-2-yl)-N,N-dimethyl-5-methoxy-3H-spiro[2-benzo-furan-1 ,4I-piperidine]- 3-carboxamide;
(+)-1'-(4-aminopyridin-2-yl)-N,N-diethyl-5-methoxy-3H-spiro[2-benzofuran-1,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N,N-dimethyl-3H-spiro[2-benzofuran-1 ,4I-piperidine]-3-carboxamide;
1 '-(4-aminopyridin-2-yl)-N-ethyl-N-methyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
N-{[1'-(4-aminopyridin-2-yl)spiro[1-benzofuran-3,4'-piperidin]-7-yl]methyl}-acetamide;
N-{[1'-(4-aminopyridin-2-yl)spiro[1-benzofuran-3,4'-piperidin]-7-yl]methyl}-pyridine-2- carboxamide; methyl 1'-(4-aminopyridin-2-yl)spiro[indole-3,4'-piperidine]-1(2H)-carboxylate; ethyl 1I-(4-aminopyridin-2-yl)spiro[indoIe-3,4'-piperidine]-1 (2H)-carboxylate; isopropyl 1'-(4-aminopyridin-2-yl)spiro[indole-3,4'-piperidine]-1(2H)-carboxylate; and pharmaceutically acceptable salts and solvates thereof.
The following compounds are most preferred: N-[(3S)-1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]-ρyridine-2- carboxamide;
2-(3-methoxy-2,3-dihydro-1'H-spiro[indene-1 ,4'-piperidin]-1'-yl)pyridin-4-amine;
I'^-tbenzyKmethyOaminolpyridin^-y^-N.N-dimethyl^.S-dihydrospirolindene-i ^'-piperidine]-
3-carboxamide; N-[r-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4I-piperidin]-3-yl]benzenesulfonamide;
I'-C^aminopyridin^-yO^.S-dihydrospiroOndeπe-i ^'-piperidineJ-S-carbonitrile;
2-(6-methyl-1 Η,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1 '-yl)pyridin-4-amine;
2-(3-phenyl-1 Η,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1 '-yl)pyridin-4-amine;
2-(1 H,rH-spiro[isochromene-4,4l-piperidin]-1 '-yl)pyridin-4-amine; 1'-(4-aminopyridin-2-yl)-N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxamide;
2-[1 -(methylsulfonyl)-i ,2-dihydro-i 'H-spirotindoIe-3,4'-piperidin]-1 '-yl]pyridin-4-amine;
2-[1 -(ethylsulfonyl)-i ,2-dihydro-1 Η-spiro[indole-3,4'-piperidin]-1 '-yl]pyridin-4-amine;
2-(2,3-dihydro-1'H-spiro[chromene-4,4'-piperidin]-1'-yl)pyridin-4-amine;
(3f?)-N-[r-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]benzamide; (3S)-N-[I '-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]benzamide;
2-[(3S)-3-(5-phenyl-1 ,3,4-oxadiazo!-2-yl)-2,3-dihydro-1 'H-spiro[indene-1 ,4'-piperidin]-1 '- yl]pyridin-4-amine;
2-[(3S)-3-(4-methyl-1 ,3-oxazol-2-yl)-2,3-dihydro-1 Η-spiro[indene-1 ,4'-piperidin]-1 '-yl]pyridin-4- amine; N-[1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4I-piperidin]-3-yl]pyridine-2- carboxamide; r-(4-aminopyridin-2-yl)-N-pyridin-2-yl-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-amine; methyl 1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[chromene-4,4'-piperidine]-2-carboxylate;
(3S)-1'-(4-aminopyridin-2-yl)-N-methyl-N-propyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-1 '-(4-aminopyridin-2-yl)-N-ethyl-N-methyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-1 l-(4-aminopyridin-2-yl)-N-pyridin-2-yl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide; (3S)-1l-(4-aminopyridin-2-yl)-N-benzyl-2l3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-1'-(4-aminopyridin-2-yl)-N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-r-(4-aminopyridin-2-yl)-N,N-diethyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
(3f?)-1'-(4-aminopyridin-2-yl)-N,N-diethyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
(3S)-r-(4-aminopyridin-2-yl)-N,N-diethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide; (+)-1I-(4-aminopyridin-2-yl)-N,N-dimethyl-5-methoxy-3H-spiro[2-benzo-furan-1 ,4'-piperidine]-
3-carboxamide;
(+)-1'-(4-aminopyridin-2-yl)-N,N-diethyl-5-methoxy-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide
1'-(4-aminopyridin-2-yl)-N,N-dimethyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3-carboxamide;
r-(4-aminopyridin-2-yl)-N-ethyl-N-methyl-3H-spiro[2-benzofuran-1 ,4l-piperidine]-3- carboxamide; and methyl 1 '-(4-aminopyridin-2-yl)spiro[indole-3,4'-piperidine]-1 (2H)-carboxylate; and pharmaceutically acceptable salts and solvates thereof.
The compounds of the invention are delta opioid receptor agonists. In particular, they show an affinity for the delta opioid receptor which is greater than their affinity for the mu and kappa opioid receptors. Preferred compounds of the invention show at least a 10-fold selectivity for the delta opioid receptor as compared with the mu opioid receptor.
The compounds of formula (I), being delta opioid receptor agonists, are potentially useful in the treatment of a range of disorders.
Therefore, the present invention also comprises a compound of formula (I), as defined above, or a pharmaceutically acceptable salt or solvate thereof, for use as a medicament.
In particular, the present invention also comprises use of a compound of formula (I), as defined above, or a pharmaceutically acceptable salt or solvate thereof, in the manufacture of a medicament for the treatment of diseases or conditions mediated by the delta opioid receptor.
Furthermore, the present invention also comprises a method for the treatment of a disease mediated by the delta opioid receptor, comprising administering to a mammal an effective amount of a compound of formula (I), as defined above, or a pharmaceutically acceptable salt or solvate thereof.
The treatment of pain, particularly neuropathic pain or inflammatory pain, is a preferred use. Physiological pain is an important protective mechanism designed to warn of danger from potentially injurious stimuli from the external environment. The system operates through a specific set of primary sensory neurones and is activated by noxious stimuli via peripheral transducing mechanisms (see Millan, 1999, Prog. Neurobiol., 57, 1-164 for a review). These sensory fibres are known as nociceptors and are characteristically small diameter axons with slow conduction velocities. Nociceptors encode the intensity, duration and quality of noxious stimulus and by virtue of their topographically organised projection to the spinal cord, the location of the stimulus. The nociceptors are found on nociceptive nerve fibres of which there are two main types, A-delta fibres (myelinated) and C fibres (non-myelinated). The activity generated by nociceptor input is transferred, after complex processing in the dorsal horn, either directly, or via brain stem relay nuclei, to the ventrobasal thalamus and then on to the cortex, where the sensation of pain is generated.
Pain may generally be classified as acute or chronic. Acute pain begins suddenly and is short-lived (usually twelve weeks or less). It is usually associated with a specific cause such as a specific injury and is often sharp and severe. It is the kind of pain that can occur after specific injuries resulting from surgery, dental work, a strain or a sprain. Acute pain does not generally result in any persistent psychological response. In contrast, chronic pain is long- term pain, typically persisting for more than three months and leading to significant psychological and emotional problems. Common examples of chronic pain are neuropathic pain (e.g. painful diabetic neuropathy, postherpetic neuralgia), carpal tunnel syndrome, back pain, headache, cancer pain, arthritic pain and chronic post-surgical pain.
When a substantial injury occurs to body tissue, via disease or trauma, the characteristics of nociceptor activation are altered and there is sensitisation in the periphery, locally around the injury and centrally where the nociceptors terminate. These effects lead to a hightened sensation of pain. In acute pain these mechanisms can be useful, in promoting protective behaviours which may better enable repair processes to take place. The normal expectation would be that sensitivity returns to normal once the injury has healed. However, in many chronic pain states, the hypersensitivity far outlasts the healing process and is often due to nervous system injury. This injury often leads to abnormalities in sensory nerve fibres associated with maladaptation and aberrant activity (Woolf & Salter, 2000, Science, 288, 1765-1768).
Clinical pain is present when discomfort and abnormal sensitivity feature among the patient's symptoms. Patients tend to be quite heterogeneous and may present with various pain symptoms. Such symptoms include: 1) spontaneous pain which may be dull, burning, or stabbing; 2) exaggerated pain responses to noxious stimuli (hyperalgesia); and 3) pain produced by normally innocuous stimuli (allodynia - Meyer et al., 1994, Textbook of Pain, 13- 44). Although patients suffering from various forms of acute and chronic pain may have similar symptoms, the underlying mechanisms may be different and may, therefore, require different treatment strategies. Pain can also therefore be divided into a number of different subtypes according to differing pathophysiology, including nociceptive, inflammatory and neuropathic pain.
Nociceptive pain is induced by tissue injury or by intense stimuli with the potential to cause injury. Pain afferents are activated by transduction of stimuli by nociceptors at the site of injury and activate neurons in the spinal cord at the level of their termination. This is then relayed up the spinal tracts to the brain where pain is perceived (Meyer et al., 1994, Textbook of Pain, 13-44). The activation of nociceptors activates two types of afferent nerve fibres. Myelinated A-deita fibres transmit rapidly and are responsible for sharp and stabbing pain sensations, whilst unmyelinated C fibres transmit at a slower rate and convey a dull or aching pain. Moderate to severe acute nociceptive pain is a prominent feature of pain from central nervous
system trauma, strains/sprains, burns, myocardial infarction and acute pancreatitis, postoperative pain (pain following any type of surgical procedure), posttraumatic pain, renal colic, cancer pain and back pain. Cancer pain may be chronic pain such as tumour related pain (e.g. bone pain, headache, facial pain or visceral pain) or pain associated with cancer therapy (e.g. postchemotherapy syndrome, chronic postsurgical pain syndrome or post radiation syndrome). Cancer pain may also occur in response to chemotherapy, immunotherapy, hormonal therapy or radiotherapy. Back pain may be due to herniated or ruptured intervertabral discs or abnormalities of the lumber facet joints, sacroiliac joints, paraspinal • muscles or the posterior longitudinal ligament. Back pain may resolve naturally but in some patients, where it lasts over 12 weeks, it becomes a chronic condition which can be particularly debilitating.
Neuropathic pain is currently defined as pain initiated or caused by a primary lesion or dysfunction in the nervous system. Nerve damage can be caused by trauma and disease and thus the term 'neuropathic pain' encompasses many disorders with diverse aetiologies. These include, but are not limited to, peripheral neuropathy, diabetic neuropathy, post herpetic neuralgia, trigeminal neuralgia, back pain, cancer neuropathy, HIV neuropathy, phantom limb pain, carpal tunnel syndrome, central post-stroke pain and pain associated with chronic alcoholism, hypothyroidism, uremia, multiple sclerosis, spinal cord injury, Parkinson's disease, epilepsy and vitamin deficiency. Neuropathic pain is pathological as it has no protective role. It is often present well after the original cause has dissipated, commonly lasting for years, significantly decreasing a patient's quality of life (Woolf and Mannion, 1999, Lancet, 353, 1959-1964). The symptoms of neuropathic pain are difficult to treat, as they are often heterogeneous even between patients with the same disease (Woolf & Decosterd, 1999, Pain Supp., 6, S141-S147; Woolf and Mannion, 1999, Lancet, 353, 1959-1964). They include spontaneous pain, which can be continuous, and paroxysmal or abnormal evoked pain, such as hyperalgesia (increased sensitivity to a noxious stimulus) and allodynia (sensitivity to a normally innocuous stimulus).
The inflammatory process is a complex series of biochemical and cellular events, activated in response to tissue injury or the presence of foreign substances, which results in swelling and pain (Levine and Taiwo, 1994, Textbook of Pain, 45-56). Arthritic pain is the most common inflammatory pain. Rheumatoid disease is one of the commonest chronic inflammatory conditions in developed countries and rheumatoid arthritis is a common cause of disability. The exact aetiology of rheumatoid arthritis is unknown, but current hypotheses suggest that both genetic and microbiological factors may be important (Grennan & Jayson, 1994, Textbook of Pain, 397-407). It has been estimated that almost 16 million Americans have symptomatic osteoarthritis (OA) or degenerative joint disease, most of whom are over 60 years of age, and this is expected to increase to 40 million as the age of the population increases, making this a public health problem of enormous magnitude (Houge & Mersfelder,
2002, Ann Pharmacother., 36, 679-686; McCarthy et al., 1994, Textbook of Pain, 387-395). Most patients with osteoarthritis seek medical attention because of the associated pain. Arthritis has a significant impact on psychosocial and physical function and is known to be the leading cause of disability in later life. Ankylosing spondylitis is also a rheumatic disease that causes arthritis of the spine and sacroiliac joints. It varies from intermittent episodes of back pain that occur throughout life to a severe chronic disease that attacks the spine, peripheral joints and other body organs.
Pain associated with osteoarthritis or rheumatoid arthritis includes joint pain associated with osteoarthritis or rheumatoid arthritis and soft tissue (e.g., muscle) pain due to change in gait or other body movement in response to joint pain. Joint pain associated with rheumatoid arthritis may be due to inflammation in the joint or degradation of cartilage of the joint. Joint pain associated with osteoarthritis is usually due to degradation of cartilage of the joint, although such pain may be due to episodic flares of inflammation in osteoarthritic joints. Preferred joints are knee, hip, and joints of hands.
Another type of inflammatory pain is visceral pain which includes pain associated with inflammatory bowel disease (IBD). Visceral pain is pain associated with the viscera, which encompass the organs of the abdominal cavity. These organs include the sex organs, spleen and part of the digestive system. Pain associated with the viscera can be divided into digestive visceral pain and non-digestive visceral pain. Commonly encountered gastrointestinal (Gl) disorders that cause pain include functional bowel disorder (FBD) and inflammatory bowel disease (IBD). These Gl disorders include a wide range of disease states that are currently only moderately controlled, including, in respect of FBD, gastro-esophageal reflux, dyspepsia, irritable bowel syndrome (IBS) and functional abdominal pain syndrome (FAPS), and, in respect of IBD, Crohn's disease, ileitis and ulcerative colitis, all of which regularly produce visceral pain. Other types of visceral pain include the pain associated with dysmenorrhea, cystitis and pancreatitis and pelvic pain.
It should be noted that some types of pain have multiple aetiologies and thus can be classified in more than one area, e.g. back pain and cancer pain have both nociceptive and neuropathic components.
Other types of pain include:
• pain resulting from musculoskeletal disorders, including myalgia, fibromyalgia, spondylitis, sero-negative (non-rheumatoid) arthropathies, non-articular rheumatism, dystrophinopathy, glycogenosis, polymyositis and pyomyositis;
• heart and vascular pain, including pain caused by angina, myocardical infarction, mitral stenosis, pericarditis, Raynaud's phenomenon, scleredoma and skeletal muscle ischemia;
• head pain, such as migraine (including migraine with aura and migraine without aura), cluster headache, tension-type headache mixed headache and headache associated with vascular disorders; and
• orofacial pain, including dental pain, otic pain, burning mouth syndrome and temporomandibular myofascial pain.
The compounds of formula (I) are also useful in the treatment of conditions other than pain. For example, the compounds of formula (I) are useful in the treatment of conditions of lower urinary tract dysfunction including but not exclusively restricted to overactive bladder, increased daytime frequency, nocturia, urgency, urinary incontinence (any condition in which there is an involuntary leakage of urine), including stress urinary incontinence, urge urinary incontinence and mixed urinary incontinence, overactive bladder with associated urinary incontinence, enuresis, nocturnal enuresis, continuous urinary incontinence, situational urinary incontinence such as incontinence during sexual intercourse, and lower urinary tract symptoms (LUTS) associated with benign prostatic hyperplasia (BPH). Activity of such compounds on lower urinary tract function, and thus their potential usefulness in treating conditions involving lower urinary tract dysfunction, can be investigated and assessed utilising a number of standard in vivo models known to those skilled in the art and frequently described in the literature (Morrison, J., et al., Neurophysiology and Neuropharmacology. In: Incontinence, Ed. Abrams, P., Cardozo, C, Khoury, S. and Wein, A. Report of the World Health Organisation Consensus Conference. Paris, France: Health Publications Ltd., 2002: 83-163; Brune ME et al. Comparison of alpha 1 -adrenoceptor agonists in canine urethral pressure profilometry and abdominal leak point pressure models. J Urol. (2001), 166:1555-9; Schroder et al. (2003) J.Urol. 170, 1017-1021 ).
The compounds of formula (I) are also useful in the treatment of premature ejaculation (PE). PE is a relatively common sexual dysfunction in men. It has been defined in several different ways but the most widely accepted is the Diagnostic and Statistical Manual of Mental Disorders IV one which states: "PE is a lifelong persistent or recurrent ejaculation with minimal sexual stimulation before, upon or shortly after penetration and before the patient wishes it. The clinician must take into account factors that affect duration of the excitement phase, such as age, novelty of the sexual partner or stimulation, and frequency of sexual activity. The disturbance causes marked distress of interpersonal difficulty."
The International Classification of Diseases 10 definition states: "There is an inability to delay ejaculation sufficiently to enjoy lovemaking, manifest as either of the following: (1) occurrence of ejaculation before or very soon after the beginning of intercourse (if a time limit is required:
before or within 15 seconds of the beginning of intercourse); (2) ejaculation occurs in the absence of sufficient erection to make intercourse possible. The problem is not the result of prolonged abstinence from sexual activity"
Other definitions which have been used include classification on the following criteria: related to partner's orgasm; duration between penetration and ejaculation; and number of thrust and capacity for voluntary control.
Psychological factors may be involved in PE, with relationship problems, anxiety, depression, prior sexual failure all playing a role.
Ejaculation is dependent on the sympathetic and parasympathetic nervous systems. Efferent impulses via the sympathetic nervous system to the vas deferens and the epididymis produce smooth muscle contraction, moving sperm into the posterior urethra. Similar contractions of the seminal vesicles, prostatic glands and the bulbouretheral glands increase the volume and fluid content of semen. Expulsion of semen is mediated by efferent impulses originating from a population of lumber spinothalamic cells in the lumbosacral spinal cord (Coolen & Truitt, Science, 2002, 297, 1566) which pass via the parasympathetic nervous system and cause rhythmic contractions of the bulbocavernous, ischiocavernous and pelvic floor muscles. Cortical control of ejaculation is still under debate in humans. In the rat the medial pre-optic area and the paraventricular nucleus of the hypothalamus seem to be involved in ejaculation.
Ejaculation comprises two separate components - emission and ejaculation. Emission is the deposition of seminal fluid and sperm from the distal epididymis, vas deferens, seminal vesicles and prostrate into the prostatic urethra. Subsequent to this deposition is the forcible expulsion of the seminal contents from the urethral meatus. Ejaculation is distinct from orgasm, which is purely a cerebral event. Often the two processes are coincidental.
Furthermore, the compounds of formula (I) are useful in the treatment of premature female orgasm. Premature female orgasm may be defined as: a) Persistent or recurrent orgasm with minimal sexual stimulation, occurring before, upon or shortly after penetration and generally before the person/couple wishes it. b) Interpersonal difficulties arise from female lack of motivation for/discomfort with continued sexual stimulation, c) The disturbance gives rise to negative psychosocial consequences where arousal occurs in inappropriate situations, d) The disturbance itself causes marked distress or interpersonal difficulty.
All of the compounds of the formula (I) can be prepared by the procedures described in the General Methods presented below or by the specific methods described in the Examples section and the Preparations section, or by routine modifications thereof. The present
invention also encompasses any one or more of these processes for preparing the compounds of formula (I), in addition to any novel intermediates used therein.
General Methods Unless otherwise provided herein:
WSCDI means ΗS-dimethylaminopropyO-S-ethylcarbodiimide hydrochloride;
DCC means N.N'-dicyclohexylcarbodiimide;
HOBT means 1 -hydroxybenzotriazole hydrate;
HOAT means 1-hydroxy-7-azabenzotriazole; PyBOP® means benzotriazol-1-yloxytrispyrrolidinophosphonium hexafluorophosphate;
PyBrOP® means bromotrispyrrolidinophosphonium hexafluorophosphate;
CDI means 1 ,1 '-carbonyldiimidazole;
HBTU means 0-benzotriazol-1-yl-N,N,N',N'-tetramethyluronium hexafluorophosphate;
DIAD means diisopropyl azodicarboxylate; AIBN means 2,2'-azobis-(2-methylpropionitrile);
Pd(dppf)2 means 1 ,1 '-bis[(diphenylphosphino)ferrocene]palladium;
Pd2(dba)3 means tris(dibenzylideneacetone)dipailadium (0);
NMM means 4-methylmorpholine;
Hϋnig's base means N-ethyldiisopropylamine; Et3N means triethylamine;
BOC means terf-butoxycarbonyl;
CBz means benzyloxycarbonyl;
MeOH means methanol;
EtOH means ethanol; EtOAc means ethyl acetate;
Et2O means diethyl ether;
THF means tetrahydrofuran;
DMSO means dimethyl sulfoxide;
DCM means dichloromethane; AcOH means acetic acid;
TFA means trifluoroacetic acid;
STAB means sodium triacetoxyborohydride;
NaHMDS means sodium hexamethyldisilazane;
DMF means dimethylformamide; TLC means thin layer chromatography;
DMA means dimethylacetamide;
Bu3SnH means tributyltin hydride;
(Me3Si)3SiH means tris(trimethylsilyl)si!yl hydride;
Pd(OAc)2 means palladium diacetate; and
TfO means trifluoromethanesulfonyloxy.
Compounds of formula (Ia), which are compounds of formula (I) when X represents C(L-R1)2 or O, Y represents C(L-R1)2 or O, Z represents C(L-R1)2 or O, m represents O or 1 , at least one L represents -(C=O)-N-R- or -(C=O)-O, and R, R1, R2, R3, R4, R5, R6, R7, R8, R9, a and b are as defined above, may be prepared as shown in Scheme 1.

Scheme 1
In Scheme 1 , Ra represents (N-R)-R1 or 0-R1 and PG represents a protecting group, suitable examples of which are described in "Protecting Groups in Organic Synthesis" by T. W. Greene and P. Wuts, Wiley and Sons, 1991. PG is preferably BOC. Compounds of formula (II) may be obtained commercially or from published methods (eg as described in WO 97/36873, step E) unless their synthesis is described in this specification. Single enantiomers of (II) (e.e. > 95%) may be obtained by conventional chiral separation methods, preferably by repeated crystallisation in the presence of chiral amines, preferably (S)- or (R)-α-methyl- benzylamine, in toluene.
Step a): Compounds of formula (III) may be synthesised by coupling an amine, HNRR1 or alcohol R1OH with an acid (II), in the presence of a conventional coupling agent, optionally in the presence of a catalyst, with an excess of base in a suitable solvent.
Typically the conditions are as follows: Acid (II), WSCDI/DCC, in the presence or absence of
HOBT/HOAT, the amine/alcohol, with an excess of NMM1 Et3N, or Hϋnig's base in THF,
DCM, DMA or EtOAc, at room temperature for 4 to 48 hours; or acid (II), PyBOP®/PyBrOP®/HBTU/CDI, an excess of amine/alcohol, with an excess of NMM, Et3N, or
Hϋnig's base in THF, ■ DCM, DMA, N-methylpyrrolidine or EtOAc, at between room temperature and about 6O0C for 4 to 24 hours.
The preferred conditions are: 1-hydroxybenzotriazole monohydrate (1 eq.), 1-(3- dimethylaminopropyl)-3-ethylcarbodiimide HCI (1.2 eq.), and N-methylmorpholine (1.5 eq.) and the appropriate amine/aicohol (1.1 eq.) in dichloromethane at room temperature for up to
18 hrs.
Step b): Deprotection of compounds of formula (III) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is BOC are TFA in DCM for about 4 hrs at room temperature.
Step c): Typically, amine (IV), a heterocycle of formula (V)1 (wherein R4 and a are defined above and LG is a suitable leaving group, preferably chloride or bromide) (0.95 eq.), and base, preferably sodium hydrogen carbonate (1.1 eq.) may be stirred in an alcohol solvent, preferably 2-methyl-2-butanol, at temperatures between 48 and 12O0C, preferably at 480C, for 15-24 hours.
Step d): Compounds of formula (Ia) may be prepared by reduction of the crude reaction mixture, or the isolated product, of step c). Typically, reduction is carried out in the presence of a suitable catalyst, e.g. palladium on carbon or palladium hydroxide on carbon, under an atmosphere of hydrogen or with a suitable hydrogen transfer reagent (e.g. cyclohexene or ammonium formate), or in the presence of a reducing agent, typically iron powder, in organic acid at room temperature to 650C for 2 to 18 hours.
Preferred conditions are ammonium formate (6 eq.) and 20% palladium hydroxide on carbon (10% by weight) at 650C until conventional methods of detection, preferably TLC, indicate complete conversion. Or: The isolated product from step c) may be dissolved in an organic acid, preferably acetic acid, and stirred in the presence of iron powder at room temperature for 2-10 hours, preferably 2-4 hours.
Compounds of formula (Ib), which are compounds of formula (I) wherein X represents N(L-R1), Y represents C(L-R1 )2 wherein each (L-R1) represents H, m represents 0, L' represents -SO2- and R, R1, R2, R3, R4, R5, R6, R7, R8, R9, a and b are as defined above, may be prepared as shown in Scheme 2.

(Ib)
Scheme 2
Compounds of formula (Vl) may be obtained from published methods (eg as described in WO 2004/028459, example 21 , compound xxii).
Steps a) to c): Compounds of formula (Ic) may be obtained from compounds of formula (Vl) using the conditions described in steps c), d) and b) for compounds of formula (Ia) depicted in Scheme 1.
Step d): Compounds of formula (Ib) may be synthesised by coupling a sulphonyl chloride, R1SO2CI, with an amine (Ic), in the presence of an excess of base in a suitable solvent. The preferred conditions are: 1 eq. of R1SO2CI, 1.5 eq. triethylamine in dichloromethane at room temperature for 4-16 hours.
Compounds of formulae (Ie) and (Ig) which are compounds of formula (I) wherein X and Y represent C(L-R1)2, m represents 0, in the case of X, one group L represents -NR-(C=O)- or -NH-(SO2)- and the other group (L-R1) represents H, for Y both (L-R1) are H and R, R1, R2, R3, R4, R5, R6, R7, R8, R9, a and b are as defined above, may be prepared as shown in Scheme 3.

Scheme 3
In Scheme 3, Rb represents (C1-10)alkyl or Het1 (as defined above), Rc represents (C^ojalkyl and PG represents a suitable protecting group, suitable examples of which are described in "Protecting Groups in Organic Synthesis" (referred to above), preferably BOC. Compounds of formula (VII) may be obtained from published methods (eg as described in US 5578593, compound 21 , scheme 10).
Step a): Deprotection of compounds of formula (VII) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above).
When PG is BOC1 preferred conditions are 4M hydrochloric acid in dioxane at room temperature.
Steps b) and c): Monofunctionalisation of compounds of formula (VIII) via in situ protection of the primary amine, typically as an imine, using a suitable ketone and base. The imine is hydrolysed during work up. Preferred conditions are 1 eq. diamine (VIII), 3.5 eq. sodium carbonate in methyl isobutyl ketone at 1100C for 3 hours, then (V) at 8O0C overnight and finally 2-propanol at room temperature for 4 hours. The intermediate nitro compound can then be reduced according to the conditions described in the synthesis of compounds of formula (Ia), step d), Scheme 1.
Step d): Compounds of formula (Ie) may be prepared by coupling the amine of formula (If) with the appropriate sulphonyl chloride R1SO2CI optionally in the presence of a base. Typically, the conditions are amine and sulphonyl chloride R1SO2CI, optionally with an excess of tertiary amine such as triethylamine, Hϋnig's base or NMM, in DCM or THF, at room temperature for 1 to 24 hours. The preferred conditions are amine and 1.1 eq. of R1SO2CI, 3 eq. triethylamine in dichioromethane at room temperature for up to 18 hours.
Step e): Compounds of formula (IX) may be obtained by coupling the amine of formula (VII) with the appropriate acid or acid derivative. The reaction may be undertaken using either:
(i) the amine + anhydride (R1CO)2O or acid chloride R1COCI, which may be generated in-situ, with an excess of base in a suitable solvent; or
(ii) the amine of formula (VII) with a conventional coupling agent + acid R1COOH, optionally in the presence of a catalyst, with an excess of base in a suitable solvent.
Typically conditions are as follows:
(i) amine and acid chloride R1COCI, optionally with an excess of tertiary amine such as Et3N,
Hϋnig's base or NMM, in DCM or THF, at room temperature for 1 to 24 hours; or
(ii) amine and acid R1COOH, WSCDI /DCC and HOBT /HOAT, the amine, with an excess of NMM, Et3N, Hϋnig's base in THF, DCM, DMA or EtOAc, at room temperature for 4 to 48 hours; or
(iii) amine and acid R1COOH, PyBOP®/PyBrOP®/O-benzotriazol-1-yl-N,N,N',N'- tetramethyluronium hexafluorophosphate/ CDI, an excess of amine, with an excess of NMM, Et3N, or Hϋnig's base in THF, DCM, DMA or EtOAc, at between room temperature and 60°C for 4 to 24 hours.
(iv) amine and anhydride (R1CO)2O, optionally with an excess of tertiary amine such as Et3N, Hϋnig's base or NMM, in DCM or THF, without heating for 1 to 24 hours. Preferred conditions are:
Amine and 1eq. acid R1COOH, 1.1 eq. 1-hydroxybenzotriazole monohydrate, 1.1 eq. 1-(3- dimethylaminopropyl)-3-ethylcarbodiimide HCI, and 3 eq. triethylamine in dichloromethane at room temperature for up to 18 hrs.
Step f): Cross coupling reaction of amine of formula (VII) with a heteroaryl halide Het1-LG (wherein LG is a suitable leaving group e.g. Cl, Br, I), e.g. 2-chloropyridine, using standard Buchwald-Hartwig conditions, which typically utilise a base e.g. sodium tert-butoxide or sodium methoxide, a suitable catalyst, e.g. palladium (II) acetate or Pd2(dba)3 and a ligand e.g. 2,2'-bis(diphenylphosphino)-1 ,1 '-binaphthyl, in a suitable solvent, typically toluene, 1 ,4- dioxane or DMF'with heating. Preferred conditions are: 1 eq. of amine, 1 eq. of 2- chloropyridine, 1.4 eq. of sodium tert-butoxide in the presence of 0.1 eq. palladium (II) acetate and 0.1 eq. 2,2'-bis(diphenylphosphino)-1 ,1'-binaphthyl in toluene at 800C for up to 4 hours.
Step g): Reaction of amine of formula (VII) with a compound of formula Rb-LG (wherein LG is a suitable leaving group, e.g Cl, Br, I, OTf) in the presence of a base, e.g. sodium hydride or NaHMDS in a suitable solvent, e.g. tetrahydrofuran or DMF at room temperature provides compounds of formula (IX). Preferred conditions are: 1 eq. of compound (VII), 3.7 eq. of iodomethane, 1.2 eq. of sodium hydride in tetrahydrofuran at room temperature for up to 3 hours.
Step h): Deprotection of compounds of formula (IX) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). When PG is BOC, preferred conditions are 4M hydrochloric acid in dioxane at room temperature or TFA in DCM at room temperature.
Steps i) and j): Compounds of formula (Ie) may be obtained from compounds of formula (Xl) using conditions described in steps c) and d) for compounds of formula (Ia) depicted in Scheme 1.
Step k): Reaction of compounds of formula (Ie) wherein L is -NH-C(=O)- and R2 and R3 are H with a compound of formula RC-LG, wherein LG is a suitable leaving group, eg Cl, Br, I, OTf, in the presence of a base, eg sodium hydride or NaHMDS in a suitable solvent, eg THF or DMF. The preferred conditions are sodium hydride and Rc-I in THF at room temperature for 18 hours.
Compounds of formula (Ih), which are compounds of formula (I) wherein X represents C(L- R1)2, where one group L represents -NR-(C=O)- and the other group (L-R1) represents H, Y indicates C(L-R1J2 where each L represents a bond and each R1 is H, m is 0 and wherein R1, R2, R3, R4, R5, R6, R7, R8, R9, a and b are as defined above, may be prepared as single
enantiomers as shown in Scheme 4. Compounds of formula (Xl) may be obtained by published methods (eg as described in Bioorg. Med. Chem. Lett. 1997, 7 (2), 213-218).

Scheme 4
Steps a) and b): Compounds of formula (Ik) may be obtained from compounds of formula (Xl) using conditions described in steps c) and d) for compounds of formula (Ia) depicted in Scheme 1.
Steps c) and d): Reaction of the ketone of formula (Ik) with a homochiral sulfinamide of formula RdS(=O)N H2 (wherein Rd represents (C1-10)alkyl), preferably in the presence of titanium ethoxide in a suitable organic solvent, e.g. THF or toluene at reflux followed by reduction at low temperature with a suitable reducing agent, e.g sodium borohydride or sodium triacetoxyborohydride, may provide compounds of formula (Ij). Preferred conditions are: 1 eq. of compound (Ik), 1.5 eq. of sulfinamide, 3.5 eq. of titanium ethoxide in THF at reflux for 18 hours, followed by 4 eq. of sodium borohydride at -480C to room temperature over 18 hours.
Step e): The sulfinamide may be cleaved to give compounds of formula (Ii) under acidic conditions, typically HCI or trifluoromethanesulfonic acid in an appropriate solvent, e.g. methanol, ethanol, DCM. The preferred conditions are: 1 eq. of compound (Ij) in methanol with an excess of a 4M solution of hydrogen chloride in dioxane at room temperature for about 30 minutes.
Step f): Compounds of formula (Ih) may be prepared by coupling amines of formula (Ii) with the appropriate acid or acid derivative using conditions described in step e) for compounds of formula (Ie) depicted in Scheme 3. The preferred conditions are amine (Ii), 1eq. acid
R1COOH, 1.1 eq. 1-hydroxybenzotriazole monohydrate, 1.1 eq. 1-(3-dimethylaminopropyl)-3- ethylcarbodiimide HCI, and 3 eq. triethylamine in dichloromethane at room temperature for up to iδ hrs.
Or, amine (Ii) and 1 eq. of (R1CO)2O, 3 eq. triethylamine in dichloromethane at room temperature for up to 18 hours.
Compounds of formula (I/), which are compounds of formula (I) wherein X represents C(l_-R1)2 , where one group (L-R1) represents (CR2)n-Het1-Re, wherein Rθ is selected from substituents α and the other group (L-R1) represents H, Y represents C(L-R1J2 in which each group L represents a bond and each R1 is H, m represents O, otherwise
 R, R1, R2, R3, R4, R5, R6, R7, R8, R9, a, b and n are as defined above, may be prepared as single enantiomers as shown in Scheme 5, wherein PG represents a suitable protecting group, preferably BOC, suitable examples of which are described in "Protecting Groups in Organic Synthesis" (referred to above). Compounds of formula (XIII) may be obtained by published methods (eg as described in WO 97/36873, step E).
R, R1, R2, R3, R4, R5, R6, R7, R8, R9, a, b and n are as defined above, may be prepared as single enantiomers as shown in Scheme 5, wherein PG represents a suitable protecting group, preferably BOC, suitable examples of which are described in "Protecting Groups in Organic Synthesis" (referred to above). Compounds of formula (XIII) may be obtained by published methods (eg as described in WO 97/36873, step E).

Scheme 5
Step a): Compounds of formula (XIV) may be prepared by methods analogous to those described in "Comprehensive Heterocyclic Chemistry", A.R. Katritzky, CW. Rees, Pergamon Press, Oxford, 1984. For example, compounds of formula (XIV) where Het1 is oxadiazolyl may be prepared by coupling an acid with the appropriate hydrazide of formula R6CONHNH2, followed by ring closure with an appropriate dehydrating reagent, e.g. phosphorus oxychloride, thionyl chloride or 2-chloro-1 ,3-dimethyl-2-imidazolinium tetrafluoroborate. The preferred conditions are: 1 eq. acid of formula (XIII), 4 eq. triethylamine, 1 eq. R6CONHNH2, 3 eq. 2-chloro-1 ,3-dimethyl-2-imidazoIinium tetrafluoroborate in dichloromethane at room temperature to 5O0C for 18 hours.
Step b): Deprotection of compounds of formula (XIV) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is BOC are TFA in DCM for about 3 hrs at room temperature.
Steps c) and d): Compounds of formula (I/) may be obtained from compounds of formula (XV) using conditions described in steps c) and d) for compounds of formula (Ia) depicted in Scheme 1.
Compounds of formula (Im), which are compounds of formula (I) wherein X represents C(L- R1)2, where one group (L-R1) rePresents (CR2)n-Het1-Rf and the other group (L-R1) represents H, Y represents C(L-R1)2 in which each group L represents a bond and each R1 is H, m represents 0, Het1 is oxazolyl, Rf is selected from substituents α, and R, R1, R2, R3, R4, R5, R6, R7, R8, R9, a and b are as defined above, may be prepared as single enantiomers as shown in Scheme 6. Compounds of formula (XVI) may be obtained by published methods (eg as described in WO 97/36873, step E).

(XIX) (Im)
Scheme 6
In Scheme 6, PG represents a protecting group, suitable examples of which are described in "Protecting Groups in Organic Synthesis" (referred to above). PG is preferably BOC.
Step a): Compounds of formula (XVII) may be synthesised by coupling the appropriate amine of formula NH2CHR1CH2OH, with acid (XVI), in the presence of a conventional coupling agent, optionally in the presence of a catalyst, with an excess of base in a suitable solvent.
Typically the conditions are as follows: Acid (XVI), WSCDI/ DCC, in the presence or absence of HOBT/HOAT, the amine of formula NH2CHRfCH2OH, with an excess of NMM, Et3N, or Hϋnig's base in THF, DCM, DMA or EtOAc, at room temperature for 4 to 48 hours; or acid (XVI), PyBOP®/PyBrOP®/0-benzotriazo!-1-yI-N,N,N',N'-tetramethyluronium hexafluoro- phosphate/ CDI, an excess of amine NH2CHRfCH2OH, with an excess of NMM, Et3N, or
Hϋnig's base in THF, DCM, DMA, N-methylpyrrolidine or EtOAc, at between room temperature and 600C for 4 to 24 hours.
Preferred conditions are 1eq. acid of formula (XVI), 2 eq. triethylamine, 1.1 eq NH2CHRfCH2OH, 1 eq. 2-chloro-1 ,3-dimethyl-2-imidazolinium tetrafluoroborate in DCM at room temperature for 72 hours.
Step b): Compounds of formula (XVIII) may be prepared by methods analogous to those described in "Comprehensive Heterocyclic Chemistry" (referred to above). For example, oxidation of the alcohol of formula (XVII) with a suitable oxidising reagent, e.g. 1 ,1 ,1- triacetoxy-1 ,1-dihydro-1 ,2-benziodoxol-3(1 H)-one, standard Swern conditions (typically oxalyl chloride, DMSO and triethylamine) or pyridinium chlorochromate, followed by cyclisation of the intermediate aldehyde with a suitable dehydrating reagent e.g. thionyl chloride or triphenylphosphine and iodine. The preferred conditions are similar to the method described by P. Wipf and Chris Miller, J. Org. Chem, 1993, 58, 3604; 1 eq. alcohol of formula (XVII), 1.5 eq. 1 ,1 ,1-triacetoxy-1 ,1-dihydro-1 ,2-benziodoxol-3(1 H)-one in dichloromethane at room temperature for 18 hours followed by 1.4 eq. 2,6-ditbutylpyridine, 1.4eq. dibromotetrachloroethane, 1.4 eq. triphenylphosphine, 1.4eq. 1 ,8-diazabicyclo(5.4.0)undec-7- ene in dichloromethane at room temperature for 18 hours.
Step c): Deprotection of compounds of formula (XVIII) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is BOC are TFA in DCM for about 3 hrs at room temperature.
Steps d) and e): Compounds of formula (Im) may be obtained from compounds of formula (XIX) using conditions described in steps c) to d) for compounds of formula (Ia) depicted in Scheme 1.
Compounds of formulae (In), (lo) and (Ip), which are compounds of formula (I) wherein m represents 0, X and Y can represent O or C(L-R1 )2 wherein, in the case of compounds of formulae (lo) and (Ip) one group L represents -(C=O)-O- and the other group (L-R1) represents H, in the case of compounds of formula (In) one group L represents -(C=O)-NR- and the other group (L-R1) represents H, otherwise R, R1, R2, R3, R4, R5, R6, R7, R8, R9, a and b are as defined above, and PG represents a suitable protecting group, preferably BOC, may be prepared as shown in Scheme 7. Compounds of formulae (XX) and (XXIV) may be obtained by published methods (eg as described in J. Org. Chem. 1983, 48 (18), 3061 , or J. Med. Chem. 2002, 45, 438).
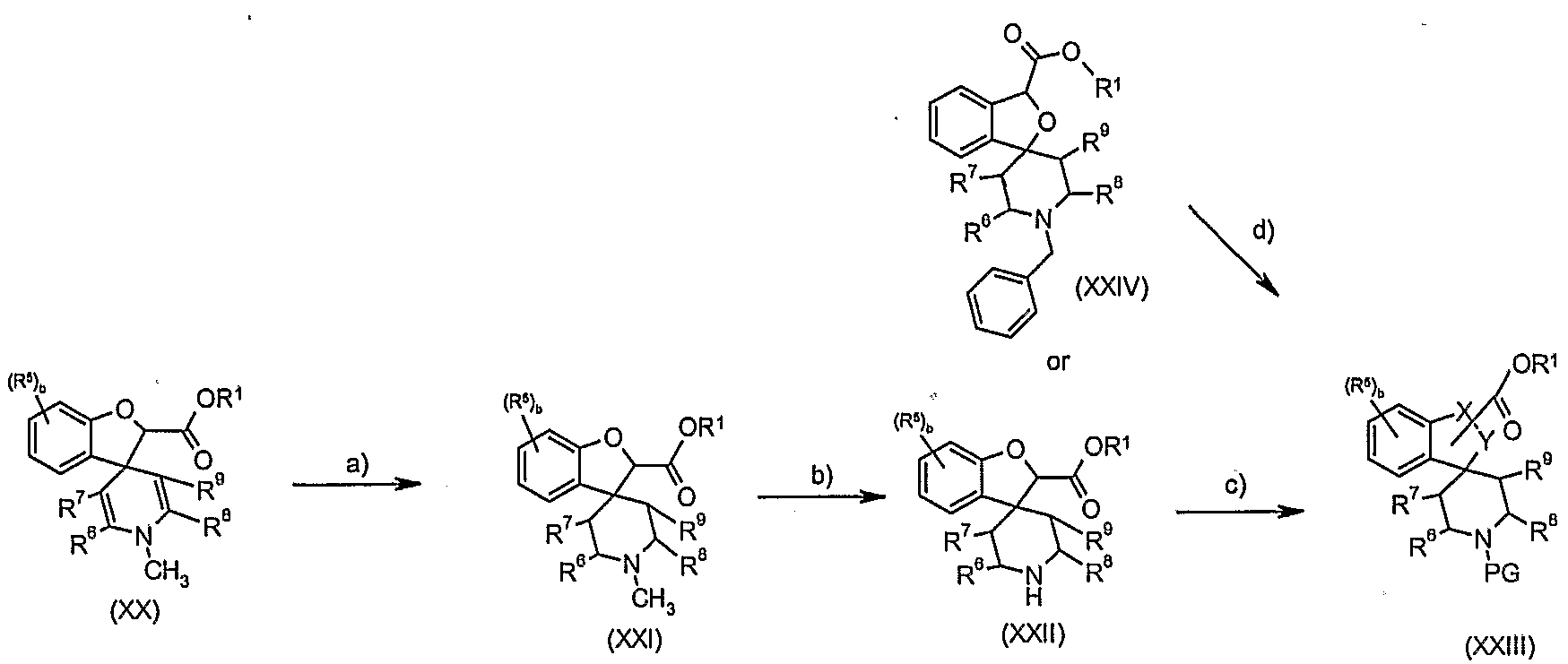 (XXIII)
(XXIII)

Scheme 7
Step a): Reduction of compound (XX) using catalytic hydrogenation, with a suitable catalyst and solvent, typically platinum oxide or palladium on carbon in methanol or ethanol at 1-4 atmospheres of hydrogen, preferably platinum oxide in ethanol at room temperature under 3 atmospheres of hydrogen.
Step b): Deprotection of compounds of formula (XXI) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions are 1 eq. methyl piperidine (XXI), 3 eq. 1-chloroethylchloroformate, 1 eq. 1 ,8-bis-(dimethylamino)naphthalene in 1 ,2-dichIoroethane at reflux for 3 hours.
Step c): Protection of compounds of formula (XXII) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is BOC are 1 eq. piperidine (XXII)1 1.1 eq. triethylamine, 1 ,1 eq. di-'butyl-dicarbonate in dichloromethane at room temperature for 18 hours.
Step d): Debenzylation of compounds of formula (XXIV) followed by protection may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is BOC are 1 eq. (XXIV), palladium on carbon, in ethanol at 4O0C under hydrogen for 6 hours, followed by 1 eq. triethylamine, 1eq. di-'butyl dicarbonate in dichloromethane at room temperature for 3 hours.
Step e): Hydrolysis of esters of formula (XXIII) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions are 1 eq. ester (XXIII), 1.5 eq. 1 M aqueous sodium hydroxide in methanol at room temperature for 18 hours. Single enantiomers of acids of formula (XXV) (e.e. > 95%) may be obtained by conventional chiral separation methods, preferably by repeated crystallisation in the presence of chiral amines, preferably (S)- or (R)-α-methyl- benzylamine, in toluene.
Step f): Compounds of formula (XXVI) may be prepared by coupling the acid of formula (XXV) with the appropriate amine of formula NHRR1 in the presence of a conventional coupling agent, optionally in the presence of a catalyst, with an excess of base in a suitable solvent. Typically the conditions are as follows: Acid (XXV), WSCDI/DCC, in the presence or absence of HOBT/HOAT, the amine NHRR1, with an excess of NMM, Et3N, or Hϋnig's base in THF, DCM, DMA or EtOAc, at room temperature for 4 to 48 hours; or acid (XXV), PyBOP®/PyBrOP®/0-benzotriazol-1-yl-N,N,N\N'-tetramethyluronium hexafluorophosphate/ CDI, an excess of amine NHRR1, with an excess of NMM, Et3N, or Hϋnig's base in THF, DCM, DMA, N-methyl pyrrolidine or EtOAc, at between room temperature and 600C for 4 to 24 hours. Preferred conditions are 1 eq. acid (XXIV), 1 eq. HOBT, 1.1 eq. WSCDI, 2- 10 eq. NHRR1, in dimethylformamide or dichloromethane at room temperature for 18 hours.
Steps g) and k): Deprotection of compounds of formula (XXVI) and (XXVIII) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis"(referred to above). Preferred conditions when PG is BOC are TFA in DCM for about 1-4 hrs at room temperature.
Steps h) and i) and I) and m): Compounds of formula (In) and (lo) may be obtained from compounds of formula (XXVII) and (XXIX) respectively using conditions described in steps c) and d) for compounds of formula (Ia) depicted in Scheme 1.
Step j): Esters of general formula (XXVIII) may be obtained using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions are 1 eq. acid (XXV), 1 eq. HOBT, 1 eq. WSCDI, in dichloromethane with excess R1OH.
Step n): Hydrolysis of esters of formula (lo) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions are 1 eq. ester (lo), 1.2 eq. lithium hydroxide in tetrahydrofuran at room temperature for 7 hours.
Step o): Compounds of formula (In) may be prepared by coupling the acid of formula (Ip) with the appropriate amine of formula NHRR1 using conditions described in step f) for compounds of formula (In) above. The preferred conditions are 1 eq. acid (Ip), 1.5 eq. HBTU, 1.2 eq. NHRR1, 14 eq. triethylamine in N-methylpyrrolidine at room temperature for 18 hours.
Compounds of formulae (Iq) and (Ir) may be prepared as shown in Scheme 8 wherein X represents O, m can be either 0 or 1 and Y indicates C(L-R1)2 where one group L can represent either -C(=O)-O-, -C(=O)NR- or a bond in which case R1 indicates H and the other group (L-R1) represents H, where m is 1 , Z represents C(L-R1 )2 where each L represents a bond and each R1 is H, otherwise R, R1, R2, R3, R4, R5, R6, R7, R8, R9, a and b are as defined above.

(XXXIII) (XXXIV)

Scheme 8
In Scheme 8, PG represents a suitable protecting group, preferably benzyl. Compounds of formula (XXX) may be obtained by methods described in this specification (e.g Preparation 52).
Step a): Compounds of formula (XXXII) may be obtained from alcohols of formula (XXXI) by a Mitsunobu reaction with the appropriate phenol, using standard methodology. In a typical procedure the compounds of formulae (XXX) and (XXXI) are treated with a suitable phosphine such as tri- "butylphosphine or triphenylphosphine, followed by a suitable dehydrating agent, typically an azo compound such as diisopropyl azodicarboxylate or di-terf- butyl azodicarboxylate, in a solvent such as dichloromethane, tetrahydrofuran or N1N- dimethylformamide, at temperatures between 25-1150C, for 1-48 hours, optionally in the absence of light.
Preferred conditions: 1 eq. (XXX), 1.2 eq. (XXXI), 1.2 eq. PPh3, 1.0 eq. DIAD, in THF between O0C and room temperature for up to 24 hrs.
Step b): Compounds of formula (XXXIII) may be prepared by a radical initiated cyclisation of the compound of formula (XXXII), in the presence of a suitable radical initiator (e.g. AIBN) and radical carrier source (e.g. Bu3SnH, (Me3Si)3SiH) in a suitable solvent (e.g. toluene) at 60- 8O0C for 4 hours. Preferred conditions: 1eq. (XXXII), cat. AIBN, 4 eq. Bu3SnH, in toluene at reflux for about 3 hrs.
Step c): The compound of formula (XXXIV) may be obtained by removal of the N-protecting group using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). When PG represents benzyl, typically this may be achieved by catalytic hydrogenation in the presence of a suitable catalyst e.g. Pd/C or Pd(OH)2 in a suitable alcoholic solvent, e.g. H2O, MeOH, or EtOH at between room temperature and 6O0C under an atmosphere of H2. This may alternatively be achieved by transfer hydrogenation, in the presence of a suitable catalyst, e.g. Pd/C and Pd(OH)2 and a hydrogen donor, e.g. formic acid or NH4CO2H, in a suitable solvent, e.g. EtOH or MeOH, at 60-800C. Alternatively deprotection can be achieved using 1-chloroethylchloroformate in an appropriate organic solvent, e.g 1 ,2- dichloroethane followed by hydrolysis with, for example, sodium hydroxide or methanol. Preferred conditions: 1eq. compound (XXXIII), 5 eq. NH4CO2H, 10% Pd/C in EtOH at the reflux temperature of the reaction solvent for about 1.5 hrs, Or, 1 eq. compound (XXXIII), 1 eq. 1-chloroethylchloroformate, 1eq. triethylamine in 1 ,2- dichloroethane at O0C to reflux for 2 hours followed by methanol at reflux for 45 minutes.
Steps d) and e): Compounds of formula (Iq) may be obtained from compounds of formula (XXXIV) using conditions described in steps c) to d) for compounds of formula (Ia) depicted in Scheme 1.
(optional) Steps f) and g): Hydrolysis of esters of formula (Iq) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions are 1 eq. ester (Iq) with 2M hydrochloric acid in THF at 35 0C for 20 hours.
Compounds of formula (Ir) may be prepared by coupling the intermediate acid with the appropriate amine using conditions described in step f) for compounds of formula (In) depicted in Scheme 7. Preferred conditions are 1 eq. acid, 1 eq. HOBT, 1.2 eq. WSCDI1 3 eq. amine NHRR1 in DMF at room temperature for 16 hours.
Compounds of formula (Is) which are compounds of formula (I) wherein X represents C(=O), Y represents N(L'-R1)where L' represents a bond, m is 0 and otherwise R, R1, R2, R3, R4, R5, R6, R7, R8, R9, a and b are as defined above, may be prepared as shown in Scheme 9.
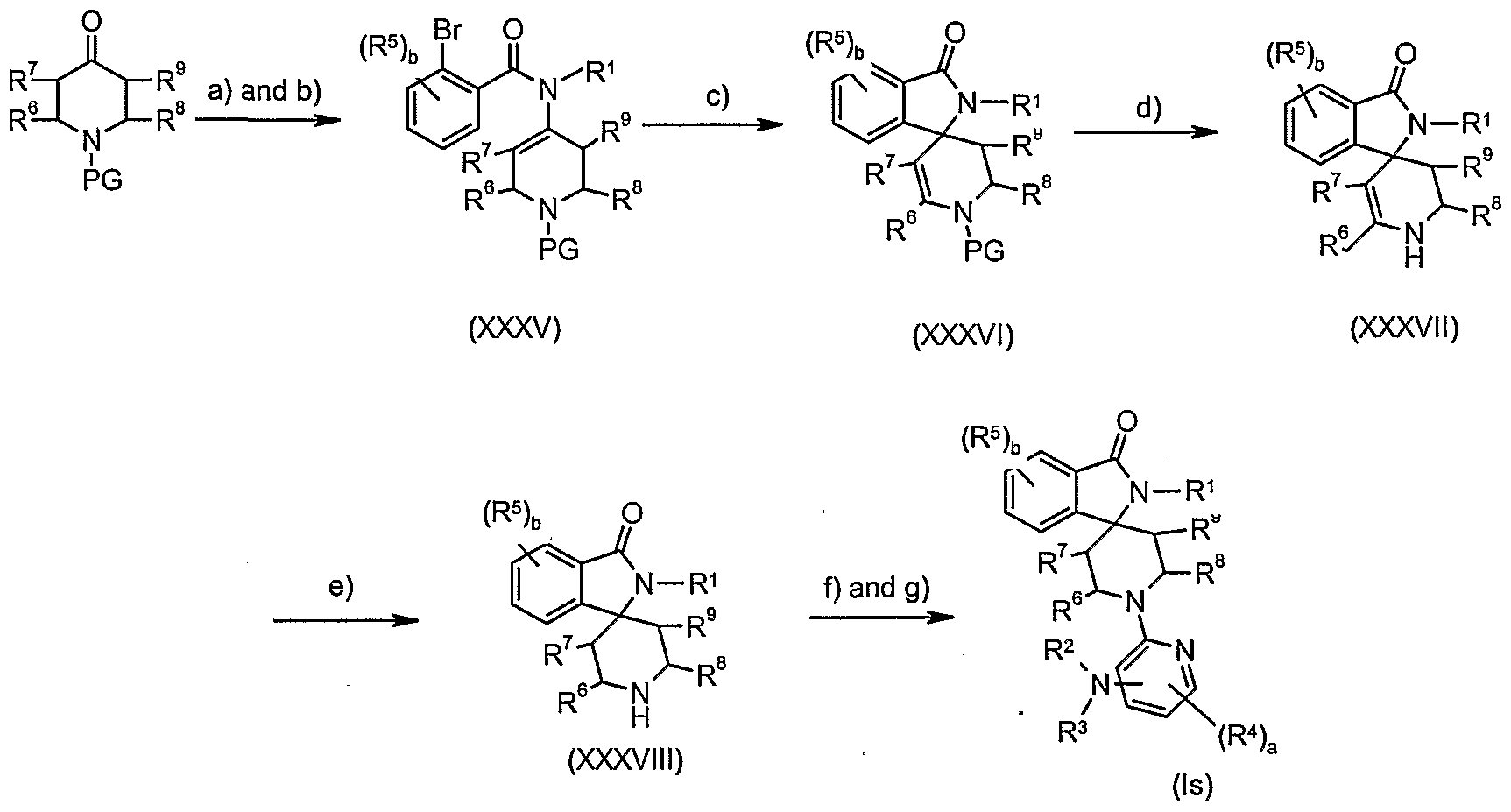
Scheme 9
In Scheme 9, PG represents a suitable protecting group, suitable examples of which are described in "Protecting Groups in Organic Synthesis" (referred to above). PG is preferably BOC.
Steps a) and b): Compounds of formula (XXXV) may be obtained by reacting an N-protected 4-piperidone with an amine of formula R1NH2 in an appropriate organic solvent, e.g. trimethylorthoformate followed by reaction with an appropriately substituted 2-bromobenzoyl chloride in the presence of a base e.g. triethylamine or Hϋnig's base in an appropriate organic solvent e.g. dichioromethane, THF. Preferred conditions are 1 eq. N-protected 4-piperidone, 1 eq. R1NH2 in trimethylorthoformate at room temperature for 72 hours followed by 1 eq. 2-
bromobenzoyl chloride, 2 eq. triethylamine in dichloromethane at O0C to room temperature for 18 hours.
Step c): Compounds of formula (XXXVI) may be obtained by cyclising alkenes of formula (XXXV) under standard Heck conditions. Typically, compounds of formula (XXXV) are cyclised in the presence of a suitable catalyst, e.g. PdCI2 or Pd(OAc)2, a phosphine ligand, e.g. triphenylphosphine and a base, e.g. triethylamine in a suitable organic solvent, e.g. dimethylformamide or acetonitrile at room temperature to 1000C. Preferred conditions are 1 eq. (XXXIV), 1 eq. tetraethylammonium chloride, 0.07eq. triphenylphosphine, 2 eq. potassium carbonate, 0.07 eq. palladium acetate in acetonitrile at 8O0C for 18 hours.
Step d): Deprotection of compounds of formula (XXXVI) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is BOC, are TFA in DCM for about 18 hours at room temperature.
Step e): Reduction of compounds of formula (XXXVII) using catalytic hydrogenation, with a suitable catalyst, e.g. palladium on carbon or palladium hydroxide in a solvent, e.g. ethyl acetate, methanol or ethanol, under between 3-4 atmospheres of hydrogen. Preferred conditions are 10% palladium on carbon in ethyl acetate and acetic acid at room temperature under 4 atmospheres of hydrogen for 18 hours.
Steps f) and g): Compounds of formula (Is) may be obtained from compounds of formula (XXXVIII) using conditions described in steps c) and d) for compounds of formula (Ia) depicted in Scheme 1.
Compounds of formula (It) which are compounds of formula (I) wherein X represents C(L-R1)2 where one L group represents O and the other group (L-R1) represents H, Y represents C(L- R1)2 in which each L represents a bond and each R1 is H, m is 0 and otherwise R, R1, R2, R3, R4, R5, R6, R7, R8, R9, a and b are as defined above, may be prepared as shown in Scheme 10. Ketones of formula (Ik) may be prepared as shown in Scheme 4.
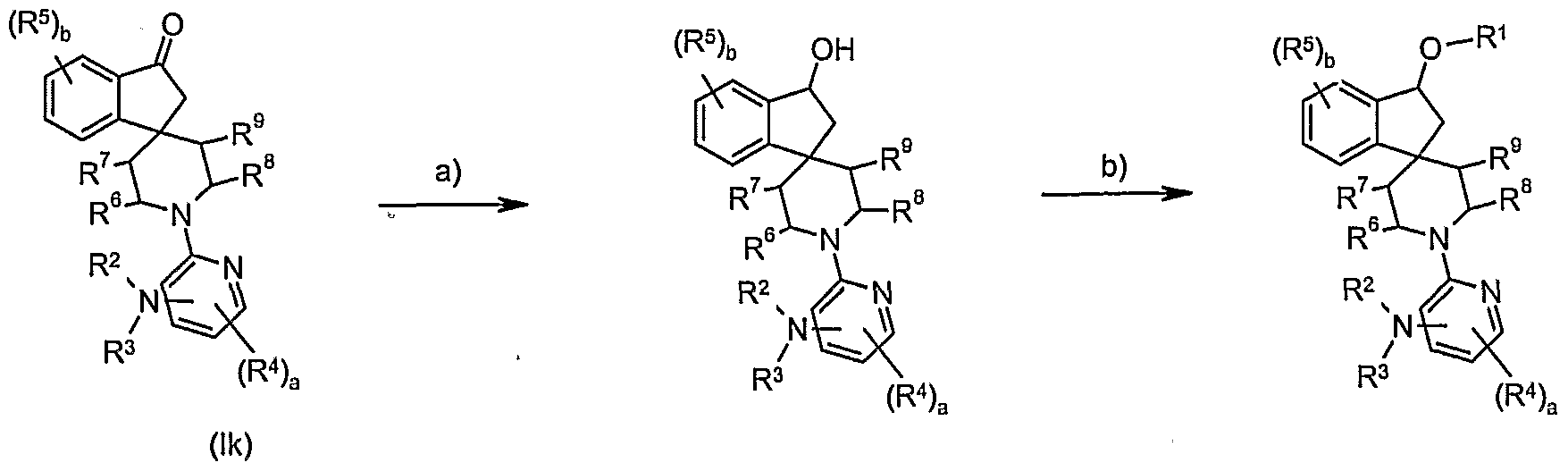
(Iu)
Scheme 10
Step a): Reduction of ketones of general formula (Ik) using an appropriate reducing reagent, eg sodium borohydride or lithium aluminium hydride in a suitable organic solvent, eg methanol or tetrahydrofuran. Preferred conditions are 1 eq. ketone (Ik), 1.3 eq. sodium borohydride or lithium aluminium hydride in tetrahydrofuran at room temperature for 18 hours.
Step b): Alkylation of alcohols of general formula (Iu) using a compound of formula R1-LG where R1 represents (C1-10)alkyl and LG represents a suitable leaving group, e.g. Cl, Br, I, OTf, in the presence of a base, e.g. sodium hydride, in an appropriate organic solvent, e.g. tetrahydrofuran or dimethylformamide. Preferred conditions are 1 eq. alcohol (Iu), 1.1 eq. sodium hydride, 1.1 eq. R1I in tetrahydrofuran at room temperature for 18 hours. .
Compounds of general formula (Iv), which are compounds of formula (I) wherein X represents C(L-R1)2 where one L group represents -C(=O)-NR- and the other group (L-R1) represents H, Y represents C(L-R1 )2 in which each L represents a bond and R1 is H, m is 0 and otherwise R, R1, R2, R3, R4, R5, R6, R7, R8, R9, a and b are as defined above, may be prepared as shown in Scheme 11. Compounds of general formula (XL) may be prepared by methods published in the literature (eg as described in WO 01/04119).
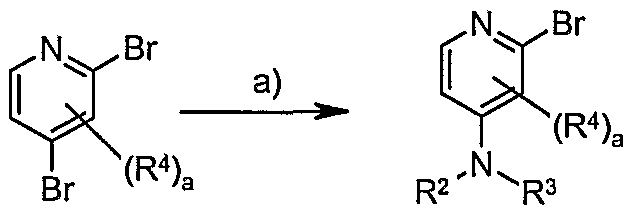
(XXXIX)
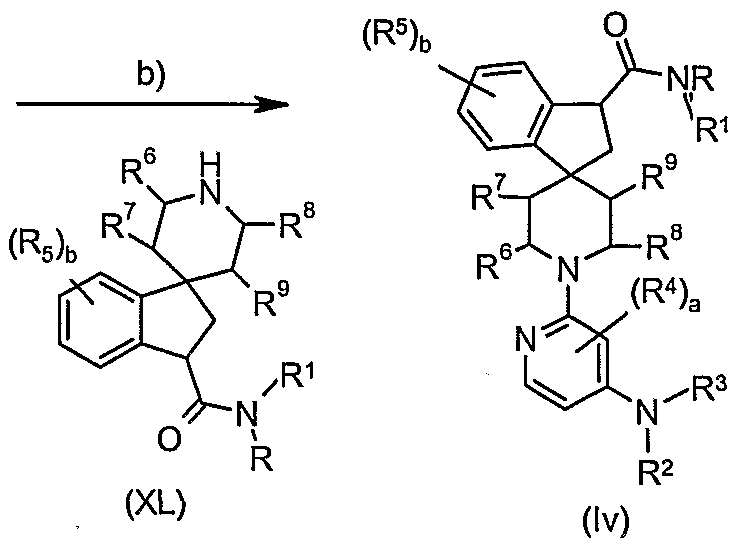
Scheme 11
Step a): Compounds of general formula (XXXIX) can be obtained by reaction of an appropriately substituted 2,4-dibromopyridine with an amine of formula NHR2R3 in an appropriate organic solvent, e.g. isopropanol or ethanol at 50 - 750C for 5 - 72 hours. The preferred conditions are 1eq. 2,4-dibromopyridine, 2 - 10 eq. amine NHR2R3, in ethanol or isopropanol at 50 - 750C for up to 72 hours.
Step b): Compounds of general formula (Iv) may be obtained from the reaction of compounds of formulae (XXXIX) and (XL) under Buchwald reaction conditions. These are typically the reaction of a compound of formula (XXXIX) with an amine of formula (XL) in the presence of a metal catalyst e.g. Pd(OAc)2, Pd(dppf)2, and a base, e.g. sodium tert-butoxide, in an appropriate solvent, e.g. dioxane or toluene. Preferred conditions are 1 eq. (XL), 1 eq. (XXXIX), catalytic dipalladium-tris(dibenzylidene acetone) chloroform complex, 1.5 eq. sodium tert-butoxide in toluene, irradiated in a microwave at 15O0C for 15 minutes.
Compounds of formula (Iw), which are compounds of formula (I) wherein X represents O, Y represents C(L-R1) in which each (L-R1) represents H, m is 0, b is 1 , R5 is CONRR where each R may be the same or different and otherwise R, R1, R2, R3, R4, R6, R7, R8, R9 and a are as defined above, may be prepared as shown in Scheme 12.

Scheme 12
Compounds of formula (Iy) may be obtained by the method indicated in Scheme 8.
Step a): Hydrolysis of the nitrile of formula (Iy) under acidic conditions e.g. hydrochloric acid or sulphuric acid in an appropriate solvent, e.g. dioxane or water. The preferred conditions are 1 eq. nitrile (Iy) in dioxane and 6N hydrochloric acid at 15O0C for 15 hours.
Step b): Compounds of formula (Iw) may be prepared by coupling the acid of formula (Ix) with the appropriate amine of formula RNHR using conditions described in step f) for compounds of formula (In) depicted in Scheme 7. Preferred conditions are 1 eq. acid (Ix), 1.1 eq. WSCDI, 1.3 eq. HOBT, 11 eq. RNHR in dimethylformamide at room temperature for 72 hours.
Compounds of formula (Iz), which are compounds of formula (I) wherein X represents C(L- R1)2 where one group (L-R1) represents -C(=O)-OH and the other (L-R1) group represents (C1- 10)alkyl, Y represents C(L-R1)2in which each (L-R1) group is H, m is 0, R, R1, R2, R3, R4, R5, R6, R7, R8, R9, a and b are as defined above and PG represents a suitable protecting group, preferably BOC, maybe prepared as shown in Scheme 13.. Compounds of formula (XLI) may be obtained by published methods (eg as described in WO 97/36873, step E).

Scheme 13
Step a): Compounds of formula (XLII) may be obtained from acid (XLI) and R9-LG wherein R9 is (C^ojalkyl and LG is a suitable leaving group, e.g. Br, I, with a base, eg lithium diisopropylamide or sodium hydride, in an appropriate organic solvent, eg THF. Preferred conditions are 1 eq. acid (XLI), 3 eq. lithium diisopropylamide, 3 eq. R9-LG in THF at O0C to room temperature for 18 hours.
Step b): Compounds of formula (XLIII) may be prepared by coupling the acid of formula (XLII) with the appropriate amine using conditions described in step e) for compounds of formula (Ie) depicted in scheme 3. Preferred conditions are 1 eq. acid (XL), excess NHRR1, 1.5 eq. HBTU in THF at 5O0C for 18 hours.
Step c): Deprotection of compounds of formula (XLIII) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is BOC are 4M hydrochloric acid in dioxane for about 2 hours at room temperature.
Steps d) and e): Compounds of formula (Iz) may be obtained from compounds of formula (XLIV) using conditions described in steps c) and d) for compounds of formula (Ia) depicted in Scheme 1.
Compounds of formula (laa), which are compounds of formula (I) wherein X represents N(L'- Rh) wherein L' is -(C=O)-O- and Rh is (C1.10)alkyl (optionally substituted by one or more substituents α) or Ar, Y represents C(L-R1) in which each (L-R1) represents H, m is 0, otherwise R, R2, R3, R4, R5, R6, R7, R8, R9, Ar, a and b are as defined above, may be prepared as shown in Scheme 14.

(XLV) (XLVI) (XLVII)

Scheme 14
In Scheme 14, PG represents a suitable protecting group, suitable examples of which are described in "Protecting Groups in Organic Synthesis" (referred to above). PG is preferably benzyloxycarbonyl. Compounds of formula (XLV) may be obtained from published methods (eg as described in Xie et al, Tetrahedron, 2004, 60, 4875-4878).
Step a): Compounds of formula (XLVI) may be synthesised by coupling a chloroformate, RhO(C=O)CI, with an amine (XLV), in the presence of an excess of base in a suitable solvent. Preferred conditions are: 1 eq. of RhO(C=O)CI, 1.25 eq. triethylamine in dichloromethane at room temperature for 4-18 hours.
Step b): Deprotection of compounds of formula (XLVI) may be undertaken using standard methodology, as described in "Protecting Groups in Organic Synthesis" (referred to above). Preferred conditions when PG is benzyloxycarbonyl are 1-methyl-1 ,4-cyclohexadiene and catalytic Pd(OH)2 on carbon in ethanol at 5O0C for 30 minutes.
Step c) Compounds of formula (laa) may be obtained from compounds of formula (XLVII) using the conditions described in steps c), d) for compounds of formula (Ia) depicted in Scheme 1.
The present invention also comprises a pharmaceutical composition comprising an effective amount of a compound of formula (I) as defined above, or a pharmaceutically acceptable salt or solvate thereof, together with a pharmaceutically acceptable carrier or diluent.
Pharmaceutically acceptable salts of the compounds of formula (I) include the acid addition and base salts thereof.
Suitable acid addition salts are formed from acids which form non-toxic salts. Examples include the acetate, adipate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, cyclamate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate,
malate, maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, pyroglutamate, saccharate, stearate, succinate, tannate, tartrate, tosylate, trifluoroacetate and xinofoate salts.
Suitable base salts are formed from bases which form non-toxic salts. Examples include the aluminium, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and zinc salts.
Hemisalts of acids and bases may also be formed, for example, hemisulphate and hemicalcium salts.
For a review on suitable salts, see Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, 2002).
Pharmaceutically acceptable salts of compounds of formula (I) may be prepared by one or more of three methods: ■
(i) by reacting the compound of formula (I) with the desired acid or base; (ii) by removing an acid- or base-labile protecting group from a suitable precursor of the compound of formula (I) or by ring-opening a suitable cyclic precursor, for example, a lactone or lactam, using the desired acid or base; or (iii) by converting one salt of the compound of formula (I) to another by reaction with an appropriate acid or base or by means of a suitable ion exchange column. ' '
All three reactions are typically carried out in solution. The resulting salt may precipitate out and be collected by filtration or may be recovered by evaporation of the solvent. The degree of ionisation in the resulting salt may vary from completely ionised to almost non-ionised.
The compounds of the invention may exist in a continuum of solid states ranging from fully amorphous to fully crystalline. The term 'amorphous' refers to a state in which the material lacks long range order at the molecular level and, depending upon temperature, may exhibit the physical properties of a solid or a liquid. Typically such materials do not give distinctive X- ray diffraction patterns and, while exhibiting the properties of a solid, are more formally described as a liquid. Upon heating, a change from solid to liquid properties occurs which is characterised by a change of state, typically second order ('glass transition')- The term 'crystalline' refers to a solid phase in which the material has a regular ordered internal structure at the molecular level and gives a distinctive X-ray diffraction pattern with defined peaks. Such materials when heated sufficiently will also exhibit the properties of a liquid, but
the change from solid to liquid is characterised by a phase change, typically first order ('melting point').
The compounds of the invention may also exist in unsolvated and solvated forms. The term 'solvate' is used herein to describe a molecular complex comprising the compound of the invention and one or more pharmaceutically acceptable solvent molecules, for example, ethanol. The term 'hydrate' is employed when said solvent is water.
A currently accepted classification system for organic hydrates is one that defines isolated site, channel, or metal-ion coordinated hydrates - see Polymorphism in Pharmaceutical Solids by K. R. Morris (Ed. H. G. Brittain, Marcel Dekker, 1995). Isolated site hydrates are ones in which the water molecules are isolated from direct contact with each other by intervening organic molecules. In channel hydrates, the water molecules lie in lattice channels where they are next to other water molecules. In metal-ion coordinated hydrates, the water molecules are bonded to the metal ion.
When the solvent or water is tightly bound, the complex will have a well-defined stoichiometry independent of humidity. When, however, the solvent or water is weakly bound, as in channel solvates and hygroscopic compounds, the water/solvent content will be dependent on humidity and drying conditions. In such cases, non-stoichiometry will be the norm.
In this specification all references to compounds of formula (I) include references to pharmaceutically acceptable salts and solvates thereof.
The compounds of the invention include compounds of formula (I) as hereinbefore defined, including all polymorphs and crystal habits thereof, prodrugs and isomers thereof (including optical, geometric and tautomeric isomers) as hereinafter defined and isotopically-labeled compounds of formula (I).
Compounds of formula (I) containing one or more asymmetric carbon atoms can exist as two or more stereoisomers. Where a compound of formula (I) contains an alkenyl or alkenylene group, geometric cisltrans (or Z/E) isomers are possible. Where structural isomers are interconvertible via a low energy barrier, tautomeric isomerism ('tautomerism') can occur. This can take the form of proton tautomerism in compounds of formula (I) containing, for example, an imino, keto, or oxime group, or so-called valence tautomerism in compounds which contain an aromatic moiety. It follows that a single compound may exhibit more than one type of isomerism.
Included within the scope of the present invention are all stereoisomers, geometric isomers and tautomeric forms of the compounds of formula (I), including compounds exhibiting more
than one type of isomerism, and mixtures of one or more thereof. Also included are acid addition or base salts wherein the counterion is optically active, for example, d-lactate or /- lysine, or racemic, for example, (//-tartrate or d/-arginine.
Cis/trans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallisation.
Conventional techniques for the preparation/isolation of individual enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate (or the racemate of a salt or derivative) using, for example, chiral high pressure liquid chromatography (HPLC).
Alternatively, the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of formula (I) contains an acidic or basic moiety, a base or acid such as 1-phenylethylamine or tartaric acid. The resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereo isomers converted to the corresponding pure enantiomer(s) by means well known to a skilled person.
Chiral compounds of the invention (and chiral precursors thereof) may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine. Concentration of the eluate affords the enriched mixture.
When any racemate crystallises, crystals of two different types are possible. The first type is the racemic compound (true racemate) referred to above wherein one homogeneous form of crystal is produced containing both enantiomers in equimolar amounts. The second type is the racemic mixture or conglomerate wherein two forms of crystal are produced in equimolar amounts each comprising a single enantiomer.
While both of the crystal forms present in a racemic mixture have identical physical properties, they may have different physical properties compared to the true racemate. Racemic mixtures may be separated by conventional techniques known to those skilled in the art - see, for example, Stereochemistry of Organic Compounds by E. L. Eliel and S. H. Wilen (Wiley, 1994).
The present invention includes all pharmaceutically acceptable isotopically-Iabelled compounds of formula (I) wherein one or more atoms are replaced by atoms having the same
atomic number, but an atomic mass or mass number different from the atomic mass or mass number which predominates in nature.
Examples of isotopes suitable for inclusion in the compounds of the invention include isotopes of hydrogen, such as 2H and 3H, carbon, such as 11C, 13C and 14C, chlorine, such as 36CI, fluorine, such as 18F, iodine, such as 123I and 125I, nitrogen, such as 13N and 15N, oxygen, such as 150, 17O and 18O, phosphorus, such as 32P, and sulphur, such as 35S.
Certain isotopically-labelled compounds of formula (I), for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies. The radioactive isotopes tritium, i.e. 3H, and carbon-14, i.e. 14C, are particularly useful for this purpose in view of their ease of incorporation and ready means of detection.
Substitution with heavier isotopes such as deuterium, i.e. 2H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances.
Substitution with positron emitting isotopes, such as 11C, 18F, 15O and 13N, can be useful in Positron Emission Topography (PET) studies for examining substrate receptor occupancy.
Isotopically-labeled compounds of formula (I) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagent in place of the non-labeled reagent previously employed.
Pharmaceutically acceptable solvates in accordance with the invention include those wherein the solvent of crystallization may be isotopically substituted, e.g. D2O, d6-acetone, d6-DMSO.
Also within the scope of the invention are intermediate compounds of formula (I)I as hereinbefore defined, all salts, solvates and complexes thereof and all solvates and complexes of salts thereof as defined hereinbefore for compounds of formula (I). The invention includes all polymorphs of the aforementioned species and crystal habits thereof.
When preparing compounds of formula (I) in accordance with the invention, it is open to a person skilled in the art to routinely select the form of compound of formula (I) which provides the best combination of features for this purpose. Such features include the melting point, solubility, processability and yield of the intermediate form and the resulting ease with which the product may be purified on isolation.
The compounds of formula (I) should be assessed for their biopharmaceutical properties, such as solubility and solution stability (across pH), permeability, efα, in order to select the most appropriate dosage form and route of administration for treatment of the proposed indication.
Compounds of the invention intended for pharmaceutical use may be administered as crystalline or amorphous products. They may be obtained, for example, as solid plugs, powders, or films by methods such as precipitation, crystallization, freeze drying, spray drying, or evaporative drying. Microwave or radio frequency drying may be used for this purpose.
They may be administered alone or in combination with one or more other compounds of the invention or in combination with one or more other drugs (or as any combination thereof). Generally, they will be administered as a formulation in association with one or more pharmaceutically acceptable excipients. The term 'excipient' is used herein to describe any ingredient other than the compound(s) of the invention. The choice of excipient will to a large extent depend on factors such as the particular mode of administration, the effect of the excipient on solubility and stability, and the nature of the dosage form.
Pharmaceutical compositions suitable for the delivery of compounds of the present invention and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation may be found, for example, in Remington's Pharmaceutical Sciences. 19th Edition (Mack Publishing Company, 1995).
ORAL ADMINISTRATION
The compounds of the invention may be administered orally. Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, and/or buccal, lingual, or sublingual administration by which the compound enters the blood stream directly from the mouth.
Formulations suitable for oral administration include solid, semi-solid and liquid systems such as tablets; soft or hard capsules containing multi- or nano-particulates, liquids, or powders; lozenges (including liquid-filled); chews; gels; fast dispersing dosage forms; films; ovules; sprays; and buccal/mucoadhesive patches.
Liquid formulations include suspensions, solutions, syrups and elixirs. Such formulations may be employed as fillers in soft or hard capsules (made, for example, from gelatin or hydroxypropylmethylcellulose) and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more
emulsifying agents and/or suspending agents. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet.
The compounds of the invention may also be used in fast-dissolving, fast-disintegrating dosage forms such as those described in Expert Opinion in Therapeutic Patents, H (6), 981- 986, by Liang and Chen (2001).
For tablet dosage forms, depending on dose, the drug may make up from 1 weight % to 80 weight % of the dosage form, more typically from 5 weight % to 60 weight % of the dosage form. In addition to the drug, tablets generally contain a disintegrant. Examples of disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl-substituted hydroxypropyl cellulose, starch, pregelatinised starch and sodium alginate. Generally, the disintegrant will comprise from 1 weight % to 25 weight %, preferably from 5 weight % to 20 weight % of the dosage form.
Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate.
Tablets may also optionally comprise surface active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc. When present, surface active agents may comprise from 0.2 weight % to 5 weight % of the tablet, and glidants may comprise from 0.2 weight % to 1 weight % of the tablet.
Tablets also generally contain lubricants such as magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate. Lubricants generally comprise from 0.25 weight % to 10 weight %, preferably from 0.5 weight % to 3 weight % of the tablet.
Other possible ingredients include anti-oxidants, colourants, flavouring agents, preservatives and taste-masking agents.
Exemplary tablets contain up to about 80% drug, from about 10 weight % to about 90 weight % binder, from about 0 weight % to about 85 weight % diluent, from about 2 weight % to about 10 weight % disintegrant, and from about 0.25 weight % to about 10 weight % lubricant.
Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting. The final formulation may comprise one or more layers and may be coated or uncoated; it may even be encapsulated.
The formulation of tablets is discussed in Pharmaceutical Dosage Forms: Tablets. Vol. 1 , by H. Lieberman and L. Lachman (Marcel Dekker, New York, 1980).
Consumable oral films for human or veterinary use are typically pliable water-soluble or water-swellable thin film dosage forms which may be rapidly dissolving or mucoadhesive and typically comprise a compound of formula (I), a film-forming polymer, a binder, a solvent, a humectant, a plasticiser, a stabiliser or emulsifier, a viscosity-modifying agent and a solvent. Some components of the formulation may perform more than one function.
The compound of formula (I) may be water-soluble or insoluble. A water-soluble compound typically comprises from 1 weight % to 80 weight %, more typically from 20 weight % to 50 weight %, of the solutes. Less soluble compounds may comprise a greater proportion of the composition, typically up to 88 weight % of the solutes. Alternatively, the compound of formula (I) may be in the form of multiparticulate beads.
The film-forming polymer may be selected from natural polysaccharides, proteins, or synthetic hydrocolloids and is typically present in the range 0.01 to 99 weight %, more typically in the range 30 to 80 weight %.
Other possible ingredients include anti-oxidants, colorants, flavourings and flavour enhancers, preservatives, salivary stimulating agents, cooling agents, co-solvents (including oils), emollients, bulking agents, anti-foaming agents, surfactants and taste-masking agents.
Films in accordance with the invention are typically prepared by evaporative drying of thin aqueous films coated onto a peelable backing support or paper. This may be done in a drying oven or tunnel, typically a combined coater dryer, or by freeze-drying or vacuuming.
Solid formulations for oral administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
Suitable modified release formulations for the purposes of the invention are described in US Patent No. 6,106,864. Details of other suitable release technologies such as high energy dispersions and osmotic and coated particles are to be found in Pharmaceutical Technology
On-line. 25(2), 1-14, by Verma ef a/ (2001 ). The use of chewing gum to achieve controlled release is described in WO 00/35298.
PARENTERAL ADMINISTRATION
The compounds of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ. Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular, intrasynovial and subcutaneous. Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile non-aqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
The preparation of parenteral formulations under sterile conditions, for example, by lyophilisation, may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
The solubility of compounds of formula (I) used in the preparation of parenteral solutions may be increased by the use of appropriate formulation techniques, such as the incorporation of solubility-enhancing agents.
Formulations for parenteral administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release. Thus compounds of the invention may be formulated as a suspension or as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound. Examples of such formulations include drug-coated stents and semi-solids and suspensions comprising drug- loaded poly(α7-lactic-coglyco!ic)acid (PGLA) microspheres.
TOPICAL ADMINISTRATION
The compounds of the invention may also be administered topically, (intra)dermally, or transdermal^ to the skin or mucosa. Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibres, bandages and microemulsions. Liposomes
may also be used. Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol. Penetration enhancers may be incorporated - see, for example, J Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999).
Other means of topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and microneedle or needle-free (e.g. Powderject™, Bioject™, etc.) injection.
Formulations for topical administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
INHALED/INTRANASAL ADMINISTRATION
The compounds of the invention can also be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler, as an aerosol spray from a pressurised container, pump, spray, atomiser (preferably an atomiser using electrohydrodynamics to produce a fine mist), or nebuliser, with or without the use of a suitable propellent, such as 1 ,1 ,1 ,2-tetrafluoroethane or 1 ,1 ,1 ,2,3,3,3-heptafluoropropane, or as nasal drops. For intranasal use, the powder may comprise a bioadhesive agent, for example, chitosan or cyclodextrin.
The pressurised container, pump, spray, atomizer, or nebuliser contains a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
Prior to use in a dry powder or suspension formulation, the drug product is micronised to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenisation, or spray drying.
Capsules (made, for example, from gelatin or hydroxypropylmethylcellulose), blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the compound of the invention, a suitable powder base such as lactose or starch and a
performance modifier such as /-leucine, mannitol, or magnesium stearate. The lactose may be anhydrous or in the form of the monohydrate, preferably the latter. Other suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
A suitable solution formulation for use in an atomiser using electrohydrodynamics to produce a fine mist may contain from 1μg to 20mg of the compound of the invention per actuation and the actuation volume may vary from 1 μl to 100μl. A typical formulation may comprise a compound of formula (I), propylene glycol, sterile water, ethanol and sodium chloride. Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
Suitable flavours, such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations of the invention intended for inhaled/intranasal administration.
Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, PGLA. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
RECTAL/INTRAVAGINAL ADMINISTRATION
The compounds of the invention may be administered rectally or vaginally, for example, in the form of a suppository, pessary, or enema. Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate.
Formulations for rectal/vaginal administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
OCULAR/AURAL ADMINISTRATION
The compounds of the invention may also be administered directly to the eye or ear, typically in the form of drops of a micronised suspension or solution in isotonic, pH-adjusted, sterile saline. Other formulations suitable for ocular and aural administration include ointments, gels, biodegradable (e.g. absorbable gel sponges, collagen) and non-biodegradable (e.g. silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes. A polymer such as crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose, or a heteropolysaccharide polymer, for example, gelan gum, may be
incorporated together with a preservative, such as benzalkonium chloride. Such formulations may also be delivered by iontophoresis.
Formulations for ocular/aural administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted, or programmed release.
OTHER TECHNOLOGIES
The compounds of the invention may be combined with soluble macromolecular entities, such as cyclodextrin and suitable derivatives thereof or polyethylene glycol-containing polymers, in order to improve their solubility, dissolution rate, taste-masking, bioavailability and/or stability for use in any of the aforementioned modes of administration.
Drug-cyclodextrin complexes, for example, are found to be generally useful for most dosage forms and administration routes. Both inclusion and non-inclusion complexes may be used. As an alternative to direct complexation with the drug, the cyclodextrin may be used as an auxiliary additive, i.e. as a carrier, diluent, or solubiliser. Most commonly used for these purposes are alpha-, beta- and gamma-cyclodextrins, examples of which may be found in International Patent Publication Nos. WO 91/11172, WO 94/02518 and WO 98/55148.
KIT-OF-PARTS
Inasmuch as it may desirable to administer a combination of active compounds, for example, for the purpose of treating a particular disease or condition, it is within the scope of the present invention that two or more pharmaceutical compositions, at least one of which contains a compound in accordance with the invention, may conveniently be combined in the form of a kit suitable for coadministration of the compositions. .
Thus the kit of the invention comprises two or more separate pharmaceutical compositions, at least one of which contains a compound of formula (I) in accordance with the invention, and means for separately retaining said compositions, such as a container, divided bottle, or divided foil packet. An example of such a kit is the familiar blister pack used for the packaging of tablets, capsules and the like.
The kit of the invention is particularly suitable for administering different dosage forms, for example, oral and parenteral, for administering the separate compositions at different dosage intervals, or for titrating the separate compositions against one another. To assist compliance, the kit typically comprises directions for administration and may be provided with a so-called memory aid.
DOSAGE
For administration to human patients, the total daily dose of the compounds of the invention is typically in the range 1 mg to 5000 mg, preferably 10 mg to 2000 mg depending, of course, on the mode of administration. For example, oral administration may require a total daily dose of from 50 mg to 1000 mg; an intravenous dose may require from 50 mg to 1000 mg. The total daily dose may be administered in single or divided doses and may, at the physician's discretion, fall outside of the typical range given herein.
These dosages are based on an average human subject having a weight of about 60kg to 70kg. The physician will readily be able to determine doses for subjects whose weight falls outside this range, such as infants and the elderly.
For the avoidance of doubt, references herein to "treatment" include references to curative, palliative and prophylactic treatment.
The ability of the compounds of the formula (I) to bind to the delta opioid receptor may be measured using the assay described below.
Preparation of Working Solutions
Stimulation buffer was made at 2x concentrated to allow for 1 in 2 dilution with compound as follows:
Stimulation buffer: Final concentration: 5mM HEPES pH 7.4, HBSS, 0.1 % BSA, 1OmM MgCI2.6H2O and 0.5mM IBMX in purified H2O:
2x: 200ml 10x HBSS, 2.4g HEPES, 2g BSA, 4.08ml MgCI2.6H2O, 2ml 50OmM IBMX and 900ml purified H2O.
Detection buffer: 5mM HEPES pH 7.4, HBSS, 0.1 % BSA and 0.3% Tween-20 in purified H2O: 50ml 10x HBSS, 600mg HEPES, 500mg BSA, 1.5ml Tween-20 and 450ml purified H2O.
Acceptor beads: 3.75mg/ml stock. Cell suspension: 20μl acceptor beads/ml cells cAMP curve:1θμl acceptor beads/500μl stimulation buffer
Detection mix: 5OnM = 2μl biotin-cAMP + 800μl detection buffer.
50μl streptavidin donor beads (5mg/ml) 6.5ml detection buffer
Preparation of Compounds
The assay protocol dictates that compounds will be diluted by a factor of 2 when the final assay mix is assembled. Compounds were made as 4mM stocks in 100% DMSO and 10μl transferred to a v-bottom 96-well plate for further dilutions to generate 11 pt concentration effect curves with Vz log dilutions. Test compounds were evaluated in the range of 0.03nM - 10μM (final assay concentration).
Following dilution, 5μl of compound, max (full effect using standard compound) and min (0.5% DMSO in deionised H2O) were dispensed into white 384-well assay plates. Compounds were tested in the presence of a final concentration of 20μM forskolin, which was added to the cell suspension as detailed below.
Culture of human CHO delta opioid receptor FLP-IN (DOR) cell line
DOR cells were cultured in T225 flasks in DMEM/nutrient mix F12 supplemented with 2mM L- glutamine, 500μg/ml hygromycin and 10% heat inactivated foetal bovine serum. Media was changed every 3 days. Cells were kept in an incubator at 370C, with 95% O2 and 5% CO2. Cell culture took place under sterile conditions in a Class Il Microbiological Safety Cabinet. Cells were split by removing the media, washing twice with 1OmIs PBS and adding 3mls of cell dissociation solution. The usual split ratio is 1 :5 to 1 :30. Cell dissociation solution was inactivated after approximately 5 minutes with supplemented media.
Assay procedure
Note: Assay components are light and temperature sensitive
1. All compound dose responses curves were prepared in advance of the experiment.
2. DOR cells are harvested and a viable cell count performed (Trypan Blue exclusion to identify non-viable cells). The cells were then centrifuged and resuspended in 2x stimulation buffer to the required cell density (1.6E06 cells/ml equates to 8000/well in 5μl).
3. 50μl acceptor beads (3.75mg/ml stock) was added to 2.5ml of cell suspension (final assay concentration is 15μg/ml) per assay plate.
4. Just prior to adding cells and acceptor beads to the plate, 5μl of 2OmM forskolin was added per 2.5m! of cell suspension, giving a final assay concentration of 20μM.
5. 5μl of cells containing acceptor beads and forskolin were added to all wells, and these were covered with a foil seal.
6. These were then centrifuged for 30 seconds at 1200rpm and incubated for 30 minutes in the dark, at room temperature.
7. Detection mix was prepared 30 minutes prior to addition.
8- 15μl detection mix was added to all wells, they were then covered with a foil seal, centrifuged for 30 seconds at 1200rpm and incubated in the dark, overnight at room temperature.
9. The plate was then read on a Fusion reader and EC50 values calculated for the test compounds.
The compounds described in the Examples below may be tested according to the above assay procedure. The EC50 values for the Examples are as follows:



NT - Not tested
The delta opioid receptor agonists of the present invention may be usefully combined with another pharmacologically active compound, or with two or more other pharmacologically active compounds, particularly in the treatment of pain. For example, a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, as defined above, may be administered simultaneously, sequentially or separately in combination with one or more agents selected from:
• an opioid analgesic, e.g. morphine, heroin, hydromorphone, oxymorphone, levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine,
dihydrocodeine, oxycodone, hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, nalbuphine or pentazocine;
• a nonsteroidal antiinflammatory drug (NSAID), e.g. aspirin, diclofenac, diflusinal, etodolac, fenbufen, fenoprofen, flufenisal, flurbiprofen, ibuprofen, indomethacin, ketoprofen, ketorolac, meclofenamic acid, mefenamic acid, meloxicam, nabumetone, naproxen, nimesulide, nitroflurbiprofen, olsalazine, oxaprozin, phenylbutazone, piroxicam, sulfasalazine, sulindac, tolmetin or zomepirac;
• a barbiturate, sedative, e.g. amobarbital, aprobarbital, butabarbital, butabital, mephobarbital, metharbital, methohexital, pentobarbital, phenobartital, secobarbital, talbutal, theamylal or thiopental;
• a benzodiazepine having a sedative action, e.g. chlordiazepoxide, clorazepate, diazepam, flurazepam, lorazepam, oxazepam, temazepam or triazolam;
• an Hi antagonist having a sedative action, e.g. diphenhydramine, pyrilamine, promethazine, chlorpheniramine or chlorcyciizine; • a sedative such as glutethimide, meprobamate, methaqualone or dichloralphenazone;
• a skeletal muscle relaxant, e.g. baclofen, carisoprodol, chlorzoxazone, cyclobenzaprine, methocarbamol or orphrenadine;
• an NMDA receptor antagonist, e.g. dextromethorphan ((+)-3-hydroxy-N- methylmorphinan) or its metabolite dextrorphan ((+)-3-hydroxy-N-methylmorphinan), ketamine, memantine, pyrroloquinoline quinine, cis-4-(phosphonomethyl)-2- piperidinecarboxylic acid, budipine, EN-3231 (MorphiDex®, a combination formulation of morphine and dextromethorphan), topiramate, neramexane or perzinfotel including an NR2B antagonist, e.g. ifenprodil, traxoprodil or (-)-(R)-6-{2-[4-(3-fluorophenyl)-4- hydroxy-1 -piperidinyl]-1 -hydroxyethyl-3,4-dihydro-2(1 H)-quinolinone;
• an alpha-adrenergic, e.g. doxazosin, tamsulosin, clonidine, guanfacine, dexmetatomidine, modafinil, or 4-amino-6,7-dimethoxy-2-(5-methane-sulfonamido- 1 ,2,3,4-tetrahydroisoquinol-2-yl)-5-(2-pyridyl)quinazoIine;
• a tricyclic antidepressant, e.g. desipramine, imipramine, amitriptyline or nortriptyline; • an anticonvulsant, e.g. carbamazepine, lamotrigine, topiratmate or valproate;
• a tachykinin (NK) antagonist, particularly an NK-3, NK-2 or NK-1 antagonist, e.g. (αR,9R)-7-[3,5-bis(trifluoromethyl)benzyl]-8,9,10,11-tetrahydro-9-methyl-5-(4- methylphenyl)-7H-[1 ,4]diazocino[2,1-g][1 ,7]-naphthyridine-6-13-dione (TAK-637), 5- [[(2R,3S)-2-[(1 R)-1-[3,5-bis(trifluoromethyl)phenyl]ethoxy-3-(4-fluorophenyl)-4- morpholinyl]-methyl]-1,2-dihydro-3H-1,2,4-triazol-3-one (MK-869), aprepitant, lanepitant, dapitant or 3-[[2-methoxy-5-(trifluoromethoxy)phenyl]-methylamino]-2- phenylpiperidine (2S.3S);
• a muscarinic antagonist, e.g oxybutynin, tolterodine, propiverine, tropsium chloride, darifenacin, solifenacin, temiverine and ipratropium;
• a COX-2 selective inhibitor, e.g. celecoxib, rofecoxib, parecoxib, valdecoxib, deracoxib, etoricoxib, or iumiracoxib;
• a coal-tar analgesic, in particular paracetamol;
• a neuroleptic such as droperidol, chlorpromazine, haloperidol, perphenazine, thioridazine, mesoridazine, trifluoperazine, fluphenazine, clozapine, olanzapine, risperidone, ziprasidone, quetiapine, sertindole, aripiprazole, sonepiprazole, blonanserin, iloperidone, perospirone, raclopride, zotepine, bifeprunox, asenapine, lurasidone, amisulpride, balaperidone, palindore, eplivanserin, osanetant, rimonabant, meclinertant, Miraxion® or sarizotan; • a vanilloid receptor agonist (e.g. resinferatoxin) or antagonist (e.g. capsazepine);
• a beta-adrenergic such as propranolol;
• a local anaesthetic such as mexiletine;
• a corticosteroid such as dexamethasone;
• a 5-HT receptor agonist or antagonist, particularly a 5-HTIB/ID agonist such as eletriptan, sumatriptan, naratriptan, zolmitriptan or rizatriptan;
• a 5-HT2A receptor antagonist such as (R)-(+)-alpha-(2,3-dimethoxy-phenyl)-1-[2-(4- fluorophenylethyl)]-4-piperidinemethanol (MDL-100907);
• a cholinergic (nicotinic) analgesic, such as ispronicline (TC-1734), (E)-N-methyl-4-(3- pyridinyl)-3-buten-1 -amine (RJR-2403), (R)-5-(2-azetidinylmethoxy)-2-chloropyridine (ABT-594) or nicotine;
• Tramadol®;
• a PDEV inhibitor, such as 5-[2-ethoxy-5-(4-methyl-1-piperazinyl-sulphonyl)phenyl]-1- methyl-3-n-propyl-1 ,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil),
(6R, 12aR)-2,3, 6,7, 12,12a-hexahydro-2-methyl-6-(3,4-methylenedioxyphenyl)- pyrazino[2',r:6,1]-pyrido[3,4-b]indole-1 ,4-dione (IC-351 or tadalafil), 2-[2-ethoxy-5-(4- ethyl-piperazin-1-yl-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1- f][1 ,2,4]triazin-4-one (vardenafil), 5-(5-acetyl-2-butoxy-3-pyridinyl)-3-ethyl-2-(1-ethyl-3- azetidinyl)-2,6-dihydro-7H-pyrazolo[4,3-tf|pyrimidin-7-one, 5-(5-acetyl-2-propoxy-3- pyridinyl)-3-ethyl-2-(1-isopropyl-3-azetidinyl)-2,6-dihydro-7H-pyrazolo[4,3-c(lpyrimidin- • 7-one, 5-[2-ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2- methoxyethyl]-2,6-dihydro-7H-pyrazoIo[4,3-d]pyrimidin-7-one, 4-[(3-chloro-4- methoxybenzyl)amino]-2-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-N-(pyrimidin-2- ylmethyl)pyrimidine-5-carboxamide, 3-(1 -methyl-7-oxo-3-propyl-6,7-dihydro-1 H- pyrazolo[4,3-d]pyrimidin-5-yI)-N-[2-(1-methylpyrrolidin-2-yl)ethyl]-4- propoxybenzenesu.lfonamide;
• an alpha-2-delta ligand such as gabapentin, pregabalin, 3-methylgabapentin, (1 α,3α,5α)(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid, (3S.5R)- 3-aminomethyl-5-methyl-heptanoic acid, (3S,5R)-3-amino-5-methyl-heptanoic acid, (3S,5R)-3-amino-5-methyl-octanoic acid, (2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3-f!uorobenzyl)-proline, [(1 R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-
yl]acetic acid, 3-(1-aminomethyl-cyclohexylmethyl)-4H-[1 ,2,4]oxadiazol-5-one, C-[1- (1 H-tetrazol-5-ylmethyl)-cycloheptyl]-methylamine, (3S,4S)-(1-aminomethyl-3,4- dimethyl-cyclopentyl)-acetic acid, (SS.δR^S-aminomethyl-S-methyl-octanoic acid, (3S,5R)-3-amino-5-methyl-nonanoic acid, (3S,5R)-3-amino-5-methyl-octanoic acid, (3R,4R,5R)-3-amino-4,5-dimethyl-heptanoic acid and (3R,4R,5R)-3-amino-4,5- dimethyl-octanoic acid;
• a cannabinoid;
• metabotropic glutamate subtype 1 receptor (mGluRI ) antagonist;
• a serotonin reuptake inhibitor such as sertraline, sertraline metabolite demethylsertraline, fluoxetine, norfluoxetine (fluoxetine desmethyl metabolite), fluvoxamine, paroxetine, citalopram, citalopram metabolite desmethylcitalopram, escitalopram, d,l-fenfluramine, femoxetine, ifoxetine, cyanodothiepin, litoxetine, dapoxetine, nefazodone, cericlamine and trazodone;
• a noradrenaline (norepinephrine) reuptake inhibitor, such as maprotiline, lofepramine, mirtazepine, oxaprotiline, fezolamine, tomoxetine, mianserin, buproprion, buproprion metabolite hydroxybuproprion, nomifensine and viloxazine (Vivalan®), especially a selective noradrenaline reuptake inhibitor such as reboxetine, in particular (S1S)- reboxetine;
• a dual serotonin-noradrenaline reuptake inhibitor, such as venlafaxine, venlafaxine metabolite O-desmethylvenlafaxine, clomipramine, clomipramine metabolite desmethylclomipramine, duloxetine, milnacipran and imipramine;
• an inducible nitric oxide synthase (iNOS) inhibitor such as S-[2-[(1- iminoethyl)amino]ethyl]-L-homocysteine, S-[2-[(1-iminoethyl)-amino]ethyl]-4,4-dioxo- L-cysteine, S-[2-[(1 -iminoethyl)amino]ethyl]-2-methyl-L-cysteine, (2S,5Z)-2-amino-2- methyl-7-[(1-iminoethyl)amino]-5-heptenoic acid, 2-[[(1 R,3S)-3-amino-4-hydroxy-1-(5- thiazolyl)-butyl]thio]-5-chloro-3-pyridinecarbonitri!e; 2-[[(1 R,3S)-3-amino-4-hydroxy-1- (5-thiazoIyl)butyl]thio]-4-chlorobenzonitrile, (2S,4R)-2-amino-4-[[2-chloro-5- (trifluoromethyl)phenyl]thio]-5-thiazolebutanol, 2-[[(1 R,3S)-3-amino-4-hydroxy-1-(5- thiazolyl)butyl]thio]-6-(trifluoromethyl)-3-pyridinecarbonitrile, 2-[[(1 R,3S)-3- amino-4- hydroxy-1-(5-thiazolyl)butyl]thio]-5-chlorobenzonitrile, N-[4-[2-(3-chlorobenzylamino)- ethyl]phenyl]thiophene-2-carboxamidine, or guanidinoethyldisulfide;
• an acetylcholinesterase inhibitor such as donepezil;
• a prostaglandin E2 subtype 4 (EP4) antagonist such as Λ/-[({2-[4-(2-ethyl-4,6- dimethyl-I H-imidazoμ.δ-clpyridin-i-yOphenyπethylJaminoJ-carbonyll^-methyl- benzenesulfonamide or 4-[(1S)-1-({[5-chloro-2-(3-fluorophenoxy)pyridin-3-yl]- carbonyl}amino)ethyl]benzoic acid;
• a leukotriene B4 antagonist; such as 1-(3-biphenyl-4-ylmethyl-4-hydroxy-chroman-7- yl)-cyclopentanecarboxylic acid (CP-105696), 5-[2-(2-Carboxyethyl)-3-[6-(4- methoxyphenyl)-5E- hexenyl]oxyphenoxy]-valeric acid (ONO-4057) or DPC-11870,
• a 5-lipoxygenase inhibitor, such as zileuton, 6-[(3-fluoro-5-[4-methoxy-3,4,5,6- tetrahydro-2H-pyran-4-yl])phenoxy-methyl]-1-methyl-2-quinolone (ZD-2138), or 2,3,5- trimethyl-6-(3-pyridylmethyl),1 ,4-benzoquinone (CV-6504);
• a sodium channel blocker, such as lidocaine; • a 5-HT3 antagonist, such as ondansetron;
and the pharmaceutically acceptable salts and solvates thereof.
Such combinations offer significant advantages, including synergistic activity, in therapy.
The following Examples illustrate the preparation of compounds of the formula (I). The Preparations illustrate the preparation of suitable intermediates.
1H Nuclear magnetic resonance (NMR) spectra were in all cases consistent with the proposed structures. Characteristic chemical shifts (δ) are given in parts per million downfield from tetramethylsilane using conventional abbreviations for designation of major peaks: e.g. s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; b, broad. The low resolution mass spectra (LRMS m/z) were recorded using either electrospray (ES), electrospray ionisation (ESI) or atmospheric pressure chemical ionisation (APCI). The following abbreviations have been used for common solvents: CDCI3, deuterochloroform; D6-DMSO, deuterodimethylsulphoxide; CD3OD, deuteromethanol. 'Ammonia' refers to a concentrated solution of ammonia in water possessing a specific gravity of 0.88. Where thin layer chromatography (TLC) has been used it refers to silica gel TLC using silica gel 60 F254 plates, Rf is the distance travelled by a compound divided by the distance travelled by the solvent front on a TLC plate. Optical Rotation is measured on a Perkin Elmer 341 Polarimeter. This high-resolution instrument has an accuracy of ±0.0020°. The rotation can be measured at 5 wavelengths, 589nm, 578nm, 546nm, 436nm and 365nm and the Polarimeter cell is temperature controlled with a Peltier unit, normally operated at 25°C.
Examples
Example 1 (3S)-1'-(4-Aminopyridin-2-yl)-2,3-dihvdrospironndene-1 ,4'-piperidinl-3-amine
(a) N-FQSV-I '-(4-Aminopyridin-2-vO-2.3-dihvdrospiroNndene-1 ,4'-piperidinl-3-yll-2- methvlpropane-2-sulfinamide

The ketone of Example 30 (400mg, 1.36mmol) was dissolved in tetrahydrofuran. (S)-2- methyl-2-propanesulfinamide (246mg, 2.0mmol, 1.5eq.) and titanium ethoxide (990μL, 4.8mmol) were added and the reaction mixture heated to reflux for 18 hours. The reaction mixture was cooled to -480C then added via a cannula to a suspension of sodium borohydride (205mg, 5.4mmol) in tetrahydrofuran (5ml) stirring at -480C. The reaction mixture was allowed to slowly warm to room temperature overnight. The reaction mixture was quenched with water then extracted from water into dichloromethane, dried over magnesium sulphate filtered and evaporated in vacuo. The residue was purified by column chromatography using an ISCO® silica cartridge eluting a linear gradient from dichloromethane - 90/10/1 dichloromethane/ methanoi/0.880 ammonia. The product was collected as a white solid (320mg, 0.8mmol,
1H-NMR (CD3OD, 400MHz): δ 1.3 (s, 9H), 1.5-1.8 (m, 3H), 1.9 (dd, 1 H), 2.2 (t, 1 H), 2.75 (dd, 1 H), 2.9 (t, 1 H), 3.05 (t, 1 H), 4.1 (dd, 2H), 4.9 (t, 1 H), 6.1 (s, 2H), 7.2 (m, 3H), 7.55 (d, 1 H), 7.65 (d, 1 H). LRMS m/z (ESI) 399 [MH]+. [α]D 25 -19.88 (c = 2.05, methanol).
(b) (3S)-1'-(4-Aminopyridin-2-vO-2.3-dihvdrospirorindene-1 ,4'-piperidinl-3-amine

The sulfinamide of step (a) (384mg, 0.96mmol) was dissolved in methanol (600μL). 4M Hydrogen chloride in dioxane (560μL) was added and the reaction mixture stirred at room temperature for 30 minutes. The reaction mixture was diluted with ether and the resultant precipitate collected by filtration. The product was collected as a pale pink solid (340mg, 0.96mmol, 100%).
1H-NMR (CD3OD, 400MHz): δ 1.7 (m, 1H), 2.9 (m, 2H), 2.0 (dd, 1 H), 2.3 (t, 1H), 2.95 (dd, 1 H), 3.3 (m, 2H), 4.9 (dd, 2H), 5.9 (t, 1 H), 6.15 (s, 1 H), 6.3 (d, 1 H), 7.4 (m, 3H), 7.55 (m, 2H). LRMS m/z (APCI) 295 [MH]+. [α]D 25 -33.79 (c = 1.65, methanol).
Example 2
(3f?)-1'-(4-Aminopyridin-2-yl)-2,3-dihydrospiroπndene-1 ,4'-piperidinel-3-amine
(a) N-lϊ3f?)-1 '-(4-Aminopyridin-2-yl)-2,3-dihvdrospirorindene-1 ,4'-piperidinl-3-vn-2- methvlpropane-2-sulfinamide

The title compound was obtained as a white solid in 55% yield from the ketone of Example 30 and (R)-2-methyl-2-propanesulfinamide, following the procedure described in Example 1 , step (a).
1H-NMR (CD3OD, 400MHz): δ 1.3 (s, 9H), 1.6 (m, 1 H), 1.6-1.8 (m, 2H), 1.9 (t, 1 H), 2.2 (t, 1 H), 2.75 (t, 1H), 3.0 (m, 2H), 4.0 (m, 2H), 4.9 (m, 1H), 6.05 (s, 2H), 7.2 (m, 3H), 7.55 (s, 1 H), 7.6 (s, 1 H). LRMS m/z (APCI) 399 [MH]+. [α]D 25 +13.42 (c = 1.9, methanol).
(b) (3f?V1'-(4-Aminopyridin-2-yl)-2,3-dihvdrospiroflndene-1 ,4'-piperidine1-3-amine

The title compound was obtained as a cream solid in 80% yield from the sulfanamide of step (a), following the procedure described in Example 1 , step (b).
1H-NMR (CD3OD, 400MHz): S 1.7 (d, 1 H), 1.85 (d, 2H), 2.0 (q, 1 H), 2.3 (t, 1 H), 3.0 (q, 1 H), 3.3 (m, 2H), 4.0 (q, 2H), 4.9 (t, 1 H), 6.1 (s, 1 H), 6.35 (d, 1 H), 7.4 (m, 3H), 7.6 (d, 2H). LRMS m/z (APCI) 295 [MH]+. [α]D 25 +36.36 (c = 1.65, methanol).
Example 3
N-r(3S)-1 '-(4-Aminopyridin-2-yl)-2,3-dihvdrospirorindene-1 ,4'-piperidinl-3-yllacetamide

The amine of Example 1 (85mg, 0.26mmol), acetic acid (16μl_, 0.27mmol), triethylamine (108μl_, 0.75mmol), 1 -hydroxybenzotriazole monohydrate (36mg, 0.27mmol) and 1-(3- dimethylaminopropyl)-3-ethylcarbodiimide HCI (42mg, 0.27mmol) were combined in dichloromethane (5ml). The reaction was allowed to stir at room temperature for 18 hours. The reaction mixture was diluted with dichloromethane and washed with water, dried over magnesium sulphate, filtered and evaporated in vacuo. The residue was purified by column chromatography using an ISCO® silica cartridge using dichloromethane/methanol/0.880 ammonia as the eluent.
1H-NMR (CD3OD, 400MHz): δ 1.6-1.8 (m, 4H), 2.0 (s, 3H), 2.2 (m, 1 H), 2.75 (q, 1 H), 2.9 (t, 1 H), 3.1 (t, 1 H), 4.0 (d, 1 H), 4.1 (d, 1 H), 5.45 (t, 1 H), 6.1 (s, 2H), 7.25 (m, 4H), 7.65 (d, 1 H). LRMS m/z (APCI) 337 [MH]+. [α]D 25 -69.26 (c = 1.7, methanol).
Example 4
N-r(3S)-1'-(4-Aminopyridin-2-yl)-2,3-dihvdrospiro[indene-1 ,4'-piperidin"|-3-vnpyridine-2- carboxamide

The title cpimpound was obtained as a white solid in 45% yield from the amine of Example 1 and picolinic acid, following the procedure described in Example 3.
1H-NMR (CD3OD, 400MHz): δ 1.65 (m, 2H), 1.8 (m, 1 H), 1.9 (q, 1 H), 2.25 (t, 1 H), 2.9 (q, 1 H), 2.95 (t, 1 H), 3.05 (t, 1 H), 4.1 (m, 2H), 5.7 (t, 1 H), 6.05 (s, 2H), 7.3 (m, 4H), 7.55 (q, 1 H), 7.65 (d, 1 H), 8.0 (t, 1 H), 8.2 (d, 1 H), 8.6 (d, 1 H). LRMS m/z (APCI) 400 [MH]+. [α]D 25 -103.13 (c = 1.6, methanol).
Example 5 N-r(3S)-1'-(4-Aminopyridin-2-yl)-2,3-dihydrospiroπndene-1 ,4'-piperidin1-3- yllcvclopropanecarboxamide

The title compound was obtained as a white solid in 36% yield from the amine of Example 1 and cyclopropanecarboxylic acid, following the procedure described in Example 3. 1H-NMR (CD3OD, 400MHz): δ 0.8 (m, 2H), 0.9 (m, 2H), 1.6 (m, 3H), 1.8 (m, 2H), 2.2 (t, 1 H), 2.75 (q, 1 H), 2.9 (t, 1 H), 3.05 (t, 1 H), 4.0 (d, 1 H), 4.1 (d, 1 H),
5.5 (t, 1 H), 6.05 (s, 2H), 7.2 (m, 4H), 7.65 (d, 1 H). LRMS m/z (APCI) 363 [MH]+. [α]D 25 -59.06 (c = 1.6, methanol).
Example 6
N-r(3S)-1'-(4-Aminopyridin-2-vn-2,3-dihvdrospirorindene-1 ,4'-piperidin1-3- ylicyclohexanecarboxamide

The title compound was obtained as a white solid in 28% yield from the amine of Example 1 and cyclohexanecarboxylic acid, following the procedure described in Example 3. 1H-NMR (CD3OD, 400MHz): δ 1.2-1.9 (m, 14H), 2.2 (m, 2H), 1.75 (q, 1 H), 2.95 (t, 1 H), 3.05 (t, 1 H), 3.95 (d, 1 H), 4.05 (d, 1 H), 5.45 (t, 1 H), 6.05 (s, 1 H), 6.1 (d, 1 H), 7.2 (m, 4H), 7.6 (d, 1 H). LRMS m/z (APCI) 404 [MH]+. [α]D 25 -61.00 (c = 1.5, methanol).
Example 7
N-r(3SV1'-(4-Aminopyridin-2-vπ-2.3-dihvdrospiroπndene-1 ,4'-piperidin1-3-vn-212,2- trifluoroacetamide

The amine of Example 1 (85mg, 0.26mmol), triethylamine (108μL, 0.75mmol), and trifluoroacetic anhydride (36μL, 0.26mmol) were combined in dichloromethane (5ml) and allowed to stir at room temperature overnight. The reaction mixture was diluted with dichloromethane and washed with water, dried over magnesium sulphate, filtered and evaporated in vacuo. The residue was purified by column chromatography using an ISCO® silica cartridge eluting with a linear gradient from dichloromethane - 80:20:5 dichloromethane/ methanol/0.880 ammonia. The product was obtained as a white solid (5mg, 0.0128mmol, 5%).
1H-NMR (CD3OD, 400MHz): δ 1.6-2.0 (m, 4H), 2.3 (m, 1 H), 2.85 (m, 1 H), 2.95 (t, 1 H), 3.05 (t, 1 H), 4.1 (m, 2H), 5.6 (t, 1 H), 6.1 (m, 2H), 7.3 (m, 4H), 7.6 (d, 1 H). LRMS m/z (APCI) 391 [MH]+.
Example 8
N-r(3f?V1'-(4-Aminopyridin-2-vπ-2,3-dihvdrospiro[indene-1 ,4'-piperidin]-3-vnpyridine-2- carboxamide

The title compound was obtained as a white solid in 56% yield from the amine of Example 2 and picolinic acid, following the procedure described in Example 3.
1H-NMR (CD3OD, 400MHz): δ 1.6-1.7 (m, 2H), 1.8 (t, 1 H), 1.95 (q, 1 H), 2.25 (t, 1 H), 2.9 (q, 1 H), 2.95 (t, 1 H), 3.05 (t, 1 H), 4.05 (m, 2H), 5.7 (t, 1 H), 6.05 (s, 2H), 7.2-7.3 (m, 4H), 7.55 (m, 1 H), 7.65 (d, 1 H), 8.0 (t, 1 H), 8.2 (d, 1 H), 8.6 (d, 1 H). LRMS m/z APCI (400) [MH]+. [α]D 25 +66.57 (c = 2.7, methanol).
Example 9 1'-(4-Aminopyridin-2-vO-2.3-dihvdrospirorindene-1 ,4'-piperidinl-3-ol

The ketone of Example 30 (120mg, 0.41 mmol) was dissolved in tetrahydrofuran (5ml).
Sodium borohydride (20mg, 0.53mmol) was added and the reaction mixture stirred at room temperature for 18 hours. The reaction mixture was quenched with water and extracted into dichloromethane, dried over magnesium sulphate, filtered and evaporated in vacuo. The product was obtained as a white solid (70mg, 0.236mmol, 57%).
1H-NMR (CD3OD, 400MHz): δ 1.5 (d, 1 H), 1.7 (d, 1 H), 1.8-2.0 (m, 2H), 2.1 (t, 1 H), 2.6 (dd,
1 H), 3.0 (m, 2H), 4.1 (dd, 2H), 5.25 (t, 1 H), 6.1 (s, 2H), 7.2-7.3 (m, 3H), 7.4 (d, 1 H), 7.6 (d, 1 H). LRMS m/z (APCI) 296 [MH]+
Example 10
2-(3-Methoxy-2,3-dihvdro-1 'H-spiroπndene-1 ,4'-piperidinl-r-vπpyridin-4-amine

The alcohol of Example 9 (70mg, 0.24mmol) was dissolved in tetrahydrofuran (5ml). Sodium hydride (10mg, 0.26mmol) was added and the reaction mixture stirred for 15 minutes before the addition of iodomethane (16μl_, 0.26mmol, 1.1eq). The reaction mixture was allowed to stir at room temperature for 18 hours. The reaction mixture was quenched with water then extracted from water into dichloromethane, dried over magnesium sulphate, filtered and evaporated in vacuo. The residue was purified by column chromatography using an ISCO® silica cartridge eluting with a linear gradient from dichloromethane - 90:10:1 dichloromethane/methanol/0.880 ammonia. The product was obtained as a colourless gum (15mg, 0.048mmol, 20%). 1H-NMR (CD3OD, 400MHz): δ 1.45 (d, 1 H), 1.8 (d, 1 H), 2.7 (d, 1 H), 2.0 (m, 2H), 2.2 (d, 1 H), 2.4 (dd, 1H), 3.0 (t, 2H), 3.45 (t, 3H), 4.05 (t, 2H), 4.2 (d, 1H), 4.3 (d, 1 H), 6.05 (s, 2H), 7.2 (m, 2H), 7.3 (m, 1 H), 7.4 (d, 1 H), 7.65 (d, 1 H). LRMS m/z (APCI) 310 [MH]+
Example 11 N.N-Dimethyl-1'-(4-piperidin-1-ylpyridin-2-vπ-2,3-dihvdrospiroπndene-1.4'-piperidine1-3- carboxamide

The bromopyridine of Preparation 70 (25mg, O.immol), N,N-dimethyl-2,3- dihydrospiro[indene-1 ,4'-piperidine]-3-carboxamide (26mg, O.immol), dipalladium- tris(dibenzylidene acetone) chloroform complex (4mg) and sodium tert-butoxide (14mg) were combined in toluene and sealed in a microwave tube and irradiated at 15O0C for 15 minutes. The solvent was evaporated in vacuo and the residue purified by column chromatography
over silica gel using dichloromethane/methanol/0.880 ammonia as the eluent. (4mg, 0.01 mmol, 10%).
1H-NMR (CD3OD, 400MHz): δ 1.6 (m, 8H)1 1.8 (m, 2H), 2.2 (m, 2H), 2.6 (m, 1 H), 3.0 (s, 6H), 3.4 (m, 5H), 4.1 (d, 2H), 4.6 (t, 1 H), 6.1 (s, 1 H), 6.3 (d, 1 H), 7.1 (d, 1 H), 7.2 (m, 3H), 7.7 (d, 1 H). LRMS m/z (ESI) 419 [MH]+.
Example 12
1'-(4-rBenzyl(methyl)aminolPyridin-2-yl>-N.N-dimethyl-2,3-dihvdrospiroπndene-1 ,4'-piperidinel-
3-carboxamide

The title compound was obtained in 36% yield from the bromopyridine of Preparation 71 and N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxamide following a similar procedure to that described in Example 11. The reaction time was 20 minutes. 1H-NMR (CDCI3, 400MHz): δ 1.6 (m, 1 H), 1.8 (m, 2H), 2.1 (m, 1 H), 2.3 (m, 1 H), 2.5 (m, 1 H), 3.0 (s, 6H), 3.3 (s, 3H), 4.1 (m, 1 H), 4.3 (m, 1 H), 4.4 (t, 3H), 4.6 (s, 2H), 5.9 (s, 1 H), 6.1 (d, 1H), 7.1-7.4 (m, 9H), 7.9 (d, 1 H). LRMS m/z (APCI) 455 [MH]+.
Example 13 1'-[4-(Dimethylamino)pyridin-2-yl1-N,N-dimethyl-2,3-dihvdrospirorindene-1.4'-piperidine1-3- carboxamide

The title compound was obtained in 10% yield from the bromopyridine of Preparation 72 and NrN-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxamide following a similar procedure to that described in Example 11. The reaction time was 20 minutes.
1H-NMR (CD3OD, 400MHz): δ 1.6-1.8 (m, 3H), 2.2 (m, 2H), 2.6 (m, 1 H), 3.0 (m, 11 H), 3.3 (s, 3H), 4.1 (m, 2H), 4.6 (t, 1 H), 5.9 (s, 1 H), 6.2 (m, 1 H), 7.1 (d, 1 H), 7.2 (m, 3H), 7.8 (d, 1 H). LRMS m/z (APCI) 379 [MH]+.
Example 14
I '^-rEthylfmethvπamino'lpyridin^-yll-N.N-dimethyl^.S-dihvdrospirorinclene-I Λ'-piperidinel-
3-carboxamide

The title compound was obtained in 5% yield from the bromopyridine of Preparation 73 and N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxamide following a similar procedure to that described in Example 11. The reaction time was 20 minutes. 1H-NMR (CD3OD, 400MHz): δ 1.2 (t, 3H), 1.7-1.9 (m, 3H), 2.2 (m, 2H), 2.6 (m, 1 H), 3.0 (s, 3H), 3.05 (m, 5H), 3.3 (s, 3H), 3.5 (q, 2H), 4.1 (m, 2H), 4.6 (t, 1 H), 5.9 (s, 1 H), 6.2 (m, 1 H), 7.1 (d, 1 H), 7.2 (t, 3H), 7.7 (d, 1 H). LRMS m/z (APCI) 393 [MH]+.
Example 15 N,N-Dimethyl-1 '-(4-pyrrolidin-1 -ylpyridin-2-yl)-2,3-dihvdrospirorindene-1 ,4'-piperidine1-3-

The title compound was obtained in 18% yield from the bromopyridine of Preparation 74 and N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxamide following a similar procedure to that described in Example 11. The reaction time was 20 minutes. 1H-NMR (CD3OD, 400MHz): δ 1.6 (m, 1 H), 1.8 (m, 2H), 2.0 (m, 4H), 2.2 (m, 1 H), 2.4 (m, 1 H), 2.5 (m, 1 H), 3.0 (m, 2H), 3.05 (s, 3H), 3.25 (s, 3H), 3.3 (m, 4H), 4.1 (m, 1 H), 4.35 (m, 1 H), 4.4 (t, 1 H), 5.7 (s, 1 H), 5.95 (d, 1 H), 7.1 (d, 1 H), 7.2 (m, 3H), 7.9 (d, 1H). LRMS m/z (APCI) 405 [MH]+.
Example 16
(2f?)-r-(4-Aminopyridiπ-2-yl)-N-(3-chlorobenzvπspiroπ-benzofuran-3,4'-piperidinel-2- carboxamide
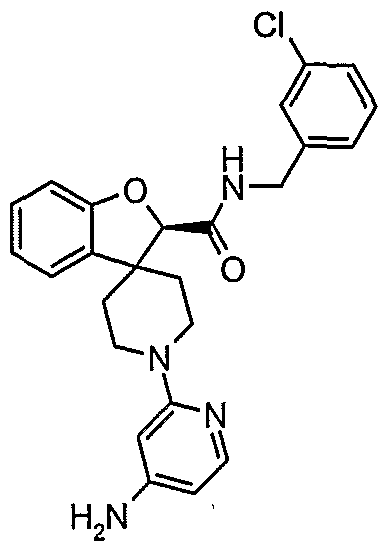
The piperidine of Preparation 21 (85mg, 0.237mmol), 2-chloro-4-nitropyridine N-oxide (42mg, 0.241 mmol) and sodium hydrogen carbonate (37mg, 0.443mmol) were combined in 2-methyl- 2-butanol (4ml). The reaction mixture was heated at 500C overnight. The solvent was evaporated in vacuo and the residue redissolved in acetic acid (4ml), iron powder (122mg, 2.1 mmol) was added and the reaction mixture stirred at room temperature for 1 hour. The reaction mixture was extracted from 2M sodium hydroxide into ethyl acetate. The combined organic extracts were dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane/ methanol/0.880 ammonia (95:5:0.5). The product was obtained as a white solid (42mg, 0.09mmol, 39%).
1H-NMR (CD3OD, 400MHz): δ 1.8 (m, 1 H), 1.9 (m, 1 H), 2.0 (m, 1 H), 2.1 (m, 1 H), 2.5 (m, 2H), 3.7 (m, 2H), 4.4 (d, 2H), 4.85 (s, 1 H), 6.0 (s, 1 H), 6.1 (d, 1 H), 6.9 (t, 2H), 7.2-7.4 (m, 6H), 7.6 (d, 1 H). LRMS m/z (APCI) 449 [MH]+.
Example 17 Methyl (2R)-1 '-(4-aminopyridin-2-v0spirori -benzofuran-3.4'-piperidinel-2-carboxvlate

The title compound was obtained in 8% yield from the piperidine of Preparation 25, following a similar procedure to that described in Example 16 using methanol as the reaction solvent.
1H-NMR (CDCI3, 400MHz): δ 1.8 (m, 1 H), 1.9-2.1 (m, 3H), 3.5 (m, 1 H), 3.6 (m, 1 H), 3.8 (s, 3H), 3.8 (m, 1 H), 3.9 (m, 1 H), 4.0 (s, 1 H), 5.9 (s, 1 H), 6.05 (d, 1 H), 6.9 (t, 2H), 7.15-7.3 (m, 2H), 7.9 (d, 1 H). LRMS m/z (APCI) 340 [MH]+.
Example 18
(2S)-1^4-Aminopyridin-2-yl)-N-(3-chlorobenzvflspiroH-benzofuran-3.4'-piperidine1-2- carboxamide

The title compound was obtained as a white solid in 17% yield from the piperidine of Preparation 27, following a similar procedure to that described in Example 16. 1H-NMR (CD3OD, 400MHz): δ 1.8 (m, 1 H), 1.9 (m, 1 H), 2.0 (m, 1 H), 2.1 (m, 1 H), 3.5 (m, 2H), 3.7 (m, 2H), 4.4 (d, 2H), 4.8 (s, 1 H), 6.0 (s, 1 H), 6.1 (s, 1 H), 6.9 (t, 2H), 7.15-7.35 (m, 6H), 7.6 (d, 1 H). LRMS m/z (APCI) 449 [MH]+.
Example 19 1'-(4-Aminopyridin-2-yl)spirori-benzofuran-3.4'-piperidinel-7-carbonithle

The title compound was obtained as a white solid in 30% yield from the piperidine of Preparation 55, following a similar procedure to that described in Example 16. 1H-NMR (CD3OD, 400MHz): S 1.8 (d, 2H), 2.0 (t, 2H), 2.9 (t, 2H), 4.1 (m, 2H), 4.7 (s, 2H), 6.05 (s, 1 H), 6.1 (d, 1 H), 7.0 (t, 1 H), 7.4 (d, 1 H), 7.45 (d, 1 H), 7.65 (d, 1 H). LRMS m/z (APCI) 307 [MH]+.
Example 20 1 '-(4-Aminopyridin-2-yl)-3H-spiror2-benzofuran-1 Λ'-piperidinel-S-carboxylic acid
(a) Methyl 1 '-(4-aminopyridin-2-vπ-3H-spiror2-benzofuran-1.4'-piperidine1-3-carboxylate

The title compound was obtained as a white solid in 58% yield from the piperidine of Preparation 61 , following a similar procedure to that described in Example 16. 1H-NMR (CDCI3, 400MHz): S 1.8 (m, 2H), 2.0 (m, 3H)1 3.4 (m, 2H), 3.8 (s, 3H), 4.2 (m, 1 H), 4.3 (m, 3H), 5.8 (s, 1H), 6.0 (s, 1H), 6.05 (d, 1H), 7.1 (d, 1H), 7.3 (m, 2H), 7.4 (m, 1 H). LRMS m/z (APCI) 340 [MH]+.
(b) 1 '-(4-Aminopyridin-2-yl)-3H-spiro[2-benzofuran-1 ,4'-piperidine1-3-carboxylic acid

The ester of step (a) (336mg, 0.985mmol) was dissolved in water: dioxane (4ml:5ml). 2M sodium hydroxide (0.6ml, 1.2mmol) was added and the reaction mixture stirred at room temperature for 1 hour. The solution was neutralised with 2M hydrochloric acid. The reaction was concentrated in vacuo to yield the title compound in quantitative yield. 1H-NMR (CD3OD, 400MHz): δ 1.8 (m, 1 H), 2.0 (m, 2H), 2.2 (m, 1 H), 3.6 (m, 2H), 3.7 (m, 1 H), 3.9 (m, 2H), 6.2 (s, 1 H), 6.3 (d, 1 H), 7.2 (m, 2H), 7.3 (m, 2H), 7.6 (m, 2H). LRMS m/z (APCI) 326 [MH]+.
Example 21
1 '-(4-Aminopyridin-2-yl V3H-spirof2-benzofuran-1 ,4'-piperidinel-3-carboxylic acid (a) 1 '-(4-Aminopyridin-2-yl)spirori -benzofuran-3,4'-piperidinel-7-carboxylic acid

The nitrile of Example 19 (35mg, 0.113mmol) was dissolved in 1,4-dioxane (0.5ml) and 6N aqueous hydrochloric acid (5.0ml) added. The reaction mixture was stirred at 15O0C for 15 hours. The reaction mixture was concentrated in vacuo to yield the title compound as a yellow gum.
1H-NMR (CD3OD, 400MHz): δ 0.8 (m, 2H), 2.1 (m, 2H), 3.2 (m, 2H), 3.9 (m, 2H), 4.6 (s, 2H), 6.1 (s, 1 H), 6.3 (d, 1 H), 6.9 (t, 1 H), 7.4 (d, 1 H), 7.6 (d, 1 H), 7.8 (d, 1 H). LRMS m/z (APCI) 326 [MH]+.
(b) {7-r(4-Ethylpiperazin-1-vπcarbonyll-1'H-spiroF1-benzofuran-3,4'-piperidin1-1'-yllpyridin-4- amine

The acid of step (a) (50mg, 0.138mmol), 1-hydroxybenzotriazole monohydrate (26mg, 0.180mmol), 1-(3-dimethylaminopropyl)-3-ethyIcarbodiimide HCI (31 mg, 0.160mmol) and ethyipiperazine (200μL, 1.57mmol) were combined in dimethylformamide (3ml) and stirred at room temperature for 72 hours. The reaction mixture was concentrated in vacuo then redissolved in ethyl acetate (20ml). The solution was washed with 3% aqueous sodium hydrogen carbonate (2x1 OmI), dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography using an ISCO® silica cartridge, eluting with dichloromethane/methanol/0.880 ammonia (90:10:1) as eluent. The title compound was obtained as a yellow solid (10mg, 0.023mmol, 17%).
1H-NMR (CDCI3, 400MHz): δ 1.1 (t, 3H), 1.8 (m, 2H), 2.0 (m, 2H), 2.4-2.6 (m, 6H), 2.9 (m,
2H), 3.4 (bs, 2H), 3.8 (bs, 2H), 4.1 (bs, 2H), 4.2 (m, 2H), 4.5 (s, 2H), 5.9 (s, 1 H), 6.0 (d, 1 H), 6.9 (t, 1H), 7.15 (d, 1H), 7.2 (d, 1H), 7.9 (d, 1H). LRMS m/z (APCI) 422 [MH]+.
Example 22
1'-(4-Aminopyridiπ-2-vπ-N-(1-θthylpropyl)spirori-benzofuran-3.4'-piperidinel-7-carboxamide

The title compound was obtained in 25% yield from the acid of Example 21 , step (a), and 1- ethylpropylamine, following a similar procedure to that described in Example 21 , step (b).
1H-NMR (CDCI3, 400MHz): δ 1.0 (s, 3H), 0.8 (m, 2H), 2.0 (m, 2H), 2.2 (m, 3H), 2.3 (s, 3H), 2.9 (m, 2H), 3.4 (d, 2H), 4.0 (bs, 2H), 4.2 (m, 2H), 4.6 (s, 2H), 5.9 (s, 1 H), 6.0 (m, 1 H), 7.0 (t, 1 H), 7.2 (d, 1 H), 7.9 (d, 1 H), 8.0 (d, 1 H), 8.4 (bs, 1 H). LRMS m/z (APCI) 395 [MH]+.
Example 23
1'-(4-Aminopyridin-2-yl)-N,N-dimethylspiro|'1-benzofuran-3,4'-piperidine]-7-carboxamide

The title compound was obtained in 25% yield from the acid of Example 21 , step (a), and 2M dimethylamine in tetrahydrofuran, following a similar procedure to that described in Example
21 , step (b).
1H-NMR (CDCI3, 400MHz): δ 0.8 (m, 2H), 2.0 (m, 2H), 3.0 (s, 3H), 3.0 (m, 2H), 3.1 (s, 3H), 4.1
(bs, 2H), 4.2 (m, 2H), 4.5 (s, 2H), 5.3 (s, 2H), 5.9 (s, 1 H), 6.0 (d, 1 H), 6.9 (t, 1 H), 7.1 (d, 1 H),
7.2 (d, 1H), 7.9 (d, 1H). LRMS m/z (APCI) 353 [MH]+.
Example 24
1'-(4-Aminopyridin-2-yl)-N-r3-(dimethylamino)-2,2-dimethylpropynspirori-benzofuraπ-3.4'- piperidiπel-7-carboxamide

The title compound was obtained in 13% yield from the acid of Example 21 , step (a) and dimethylaminoneopentylamine, following a similar procedure to that described in Example 21 , step (b).
1H-NMR (CDCI3, 400MHz): δ 0.9 (t, 6H), 1.2 (m, 3H), 1.5 (m, 2H), 1.6 (m, 2H), 1.8 (m, 2H), 2.0 (m, 2H), 2.9 (m, 2H), 4.1 (m, 3H), 4.2 (m, 2H), 4.6 (s, 2H), 5.9 (s, 1 H), 6.05 (d, 1 H), 7.0 (t, 3H), 7.2 (d, 1 H), 7.3 (m, 1 H), 7.9 (d, 1 H), 8.0 (d, 1 H). LRMS m/z (APCI) 438 [MH]+.
Example 25 1'-(4-Aminopyridin-2-yl)spirori-benzofuran-3,4'-piperidine1-5-carbonitrile
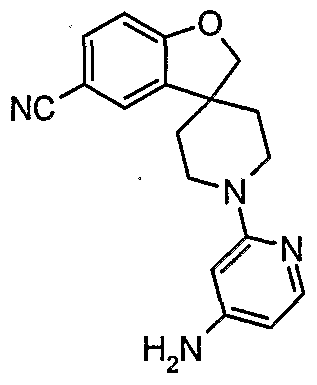
The title compound was obtained as a white solid in 45% yield from the piperidine in Preparation 58, following a similar procedure to that described in Example 16. 1H-NMR (CDCI3, 400MHz): δ 1.8 (m, 2H), 2.0 (m, 2H), 3.0 (t, 2H), 4.1 (m, 2H), 4.2 (m, 2H), 4.6 (s, 2H), 5.95 (s, 1 H), 6.1 (d, 1 H), 6.8 (d, 1 H), 7.4 (s, 1 H), 7.5 (d, 1 H), 7.9 (d, 1 H). LRMS m/z (APCI) 307 [MH]+.
Example 26
N-(1'-r4-(Dimethylamino)pyridin-2-vn-2,3-dihvdrospiroπndene-1.4'-piperidin1-3-yl)-N- methvlpvridine-2-carboxamide

Sodium hydride (6mg, 0.15mmol) was added to a solution of the amino pyridine of Example 125 in tetrahydrofuran. The reaction mixture was allowed to stir for 5 minutes at room temperature before the addition of iodomethane (21 mg, 0.15mmol). The reaction mixture was stirred at room temperature overnight. Sodium hydride (6mg, 0.15mmol) and iodomethane (21 mg, 0.15mmol) were added and the reaction mixture stirred at room temperature for a further 24 hours. The reaction mixture was partitioned between water and dichloromethane. The organic phase was washed with brine, dried over sodium sulphate, filtered and evaporated. The crude product was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane - 90:10:1 dichloromethane/methanol/0.880 ammonia. (7mg, 0.015mmol, 12%).
1H-NMR (CD3OD, 400MHz): δ 1.8 (m, 2H), 2.0 (m, 2H), 2.5 (m, 1 H), 2.9 (m, 1 H), 3.1 (m, 1 H), 3.2 (m, 1 H), 3.2 (s, 6H), 3.4 (m, 2H), 3.9 (s, 3H), 5.7 (t, 1 H), 6.4 (s, 1 H), 6.8 (d, 1 H), 7.3 (m, 2H), 7.4 (m, 2H), 7.6 (m, 1 H), 7.95 (d, 1 H), 8.0 (t, 1 H), 8.2 (d, 1 H), 8.6 (d, 1 H). LRMS m/z (ESI) 442 [MH]!.
Example 27 N-ri'-(4-Aminopyridin-2-vπ-2,3-dihvdrospirorindene-1.4'-piperidinl-3-vnbenzenesulfonamide

The amine of Example 132 (110mg, 0.373mmol), benzenesulphonyl chloride (73mg, 0.411mmol) and triethylamine (156μl_, 1.12mmol) were combined in dichloromethane and stirred at room temperature overnight. Benzenesulphonyl chloride (2.0μL, 15mmol) was added and the reaction mixture stirred for a further 20 minutes. The reaction mixture was concentrated in vacuo and the residue purified by column chromatography using an ISCO®
silica cartridge eluting with dichloromethane - 90:10:1 dichloromethane/methanol/0.880 ammonia. This product was isolated as a clear gum (25mg, O.OΘmmol, 15%). 1H-NMR (CD3OD, 400MHz): δ 1.4 (m, 1 H), 1.6 (m, 1 H), 1.7 (m, 2H), 2.1 (m, 1 H), 2.4 (m, 1 H), 2.8 (m, 2H), 4.0 (t, 2H), 4.85 (t, 1H), 6.0 (s, 1H), 6.05 (d, 1H), 7.2 (d, 1H), 7.2 (m, 2H), 7.25 (t, 1 H), 7.6 (m, 5H), 8.0 (d, 1 H). LRMS m/z (ESI) 435 [MH]+.
Example 28 N-Ii '-(4-Aminopvridin-2-vl)-2,3-dihvdrospirorindene-1.4'-Piperidin1-3-vllmethanesulfonamide

Example 132 and methanesulphonyl chloride, following a similar procedure to that described in Example 27.
1H-NMR (CD3OD, 400MHz): δ 1.55 (m, 1 H), 1.65 (m, 1H), 1.8 (m, 1H), 1.85 (m, 1H), 2.2 (m, 1 H), 2.85 (m, 1 H), 3.0 (m, 2H), 3.1 (s, 3H), 4.1 (m, 3H), 5.0 (t, 1 H), 6.1 (m, 2H), 7.2 (m, 3H), 7.4 (d, 1 H), 7.7 (d, 1 H). LRMS m/z (ESI) 373 [MH]+.
Example 29 1'-(4-Aminopyridin-2-yl)spiroπndole-3.4'-piperidin1-2(1 H')-one

The nitro-pyridine N-oxide of Preparation 103 (100mg, 0.3mmol) was dissolved in ethyl acetate (20ml), 10% palladium on carbon (10mg, 10% by weight) was added and the reaction mixture stirred at room temperature under 3.4 atmospheres of hydrogen overnight. The reaction mixture was concentrated in vacuo and the residue purified by column chromatography using an ISCO® silica cartridge. Dichloromethane/methanol/0.880 ammonia (90:10:1 ) was used as the eiuent. The title compound was obtained as a white solid (10mg, 0.03mmol, 11%).
1H-NMR (CD3OD, 400MHz): δ 1.8-2.0 (m, 4H), 3.7 (m, 2H), 3.9 (m, 2H), 6.1 (m, 2H), 6.9 (d, 1H), 7.05 (t, 1H), 7.2 (t, 1 H), 7.4 (d, 1H), 7.6 (d, 1H). LRMS m/z (APCI) 295 [MH]+.
Example 30
1'-(4-Aminopyridin-2-yl)spirorindene-1 ,4'-piperidinl-3(2H)-one

The nitro-pyridine N-oxide of Preparation 115 (500mg, 1.5mmol) was dissolved in acetic acid (5ml), iron powder (491 mg, 8.8mmol) was added and the reaction mixture stirred at room temperature for 3 hours. The reaction mixture was concentrated in vacuo and the residue extracted from 2M sodium hydroxide into dichloromethane. The combined organic extracts were dried over magnesium sulphate, filtered and evaporated to yield the crude amino pyridine. The material was purified by column chromatography on silica gel using dichloromethane/methanoI/0.880 ammonia as eluent. The title compound was obtained as a white solid (260mg, 0.88mmol, 59%).
1H-NMR (CDCI3, 400MHz): δ 1.6 (d, 2H), 2.1 (t, 2H), 2.7 (s, 2H), 2.9 (t, 2H), 4.0 (s, 2H), 4.3 (d, 2H), 5.9 (s, 1 H), 6.0 (d, 1 H), 7.4 (t, 1 H), 7.5 (d, 1 H), 7.6 (t, 1 H), 7.7 (d, 1 H), 7.9 (d, 1 H). LRMS m/z (APCI) 294 [MH]+.
Example 31
1'-(4-Aminopyridin-2-yl)-2-(4-methoxybenzvπspiroπsoindole-1 ,4'-piperidinl-3(2H)-one

The nitro-pyridine N-oxide of Preparation 105 (11mg, 0.023mmoi) was dissolved in acetic acid (2ml), iron powder (8mg, 0.143mmol) was added and the reaction mixture stirred at room temperature overnight. The reaction mixture was concentrated in vacuo and the residue extracted from 2M sodium hydroxide into dichloromethane. The combined organic extracts were dried over sodium sulphate, filtered and evaporated to yield the crude amino pyridine.
The material was purified by column chromatography using an ISCO® silica cartridge, eluting with dichloromethane/methanol/0.880 ammonia. The title compound was obtained as a colourless gum (5mg, 0.012mmol, 52%).
1H-NMR (CD3OD1 400MHz): δ 1.4 (m, 2H)1 2.3 (m, 2H), 3.5 (m, 2H)1 3.8 (s, 3H)1 4.1 (m, 2H)1 4.8 (s, 2H)1 6.0 (s, 1 H)1 6.1 (m, 1 H)1 6.8 (d, 2H), 7.2 (d, 2H), 7.6 (m, 3H), 7.9 (d, 1 H), 8.0 (d, 1 H). LRMS m/z (APCI) 415 [MH]+.
Example 32
1'-(4-Aminopyridin-2-vπ-2.3-dihvdrospiroπndene-1 ,4'-piperidine1-3-carbonitrile

The title compound was obtained as a white foam in 86% yield from the nitro-pyridine-N-oxide of Preparation 107, following a similar procedure to that described in Example 31. 1H-NMR (CDCI3, 400MHz): δ 1.6 (m, 1 H)1 1.8 (m, 2H)1 2.1 (m, 1 H)1 2.35 (m, 1 H), 2.7 (m, 1 H), 3.0 (m, 2H), 4.1 (m, 5H)1 5.9 (s, 1 H), 6.05 (m, 1 H), 7.2 (m, 3H), 7.4 (m, 1 H), 7.9 (d, 1 H). LRMS m/z 305 [MH]+.
Example 33 2-(5-Fluoro-1 'H,3H-spiror2-benzofuran-1 ,4'-piperidin1-1 '-yl)pyridin-4-amine

The title compound was obtained in 60% yield from the nitro-pyridine-N-oxide of Preparation 108, following a similar procedure to that described in Example 31.
1H-NMR (CDCI3, 400MHz): δ 1.8 (m, 2H)1 1.9 (m, 2H)1 3.3 (m, 2H), 4.0 (bs, 2H), 4.2 (m, 2H), 5.05 (s, 2H)1 5.95 (s, 1 H), 6.0 (d, 1 H)1 7.0 (m, 3H)1 7.9 (d, 1 H). LRMS m/z 300 [MH]+.
Example 34 2-(6-Methyl-1 'H.3H-spiror2-benzofuran-1 ,4'-piperidin1-1 '-yl)Pyridin-4-amine

The title compound was obtained in 33% yield from the nitro-pyridine-N-oxide of Preparation 109, following a similar procedure to that described in Example 31.
1H-NMR (CDCI3, 400MHz): δ 1.8 (m, 2H), 1.9 (m, 2H), 2.4 (s, 3H), 3.3 (m, 2H), 4.0 (bs, 2H), 4.2 (m, 2H), 5.05 (s, 2H), 5.95 (s, 1 H), 6.0 (d, 1 H), 6.9 (s, 1 H), 7.1 (m, 2H), 7.9 (d, 1 H). LRMS m/z 296 [MH]+.
Example 35 1'-(4-Aminopyridin-2-vQ-4-methoxy-3H-spiror2-benzofuran-1 ,4'-piperidinl-3-one

The title compound was obtained in 27% yield from the nitro-pyridine-N-oxide of Preparation
110, following a similar procedure to that described in Example 31. The product was purified by crystallisation from dich!oromethane:methanol.
1H-NMR (CD3OD, 400MHz): δ 1.7 (m, 2H), 2.2 (m, 2H), 3.2 (m, 2H), 4.0 (s, 3H), 4.2 (m, 2H), 6.1 (m, 2H), 7.1 (m, 2H), 7.7 (m, 2H). LRMS m/z 326 [MH]+.
Example 36 2-(3-Phenyl-1 'H.3H-spiror2-benzofuran-1.4'-pioeridin1-1 '-yl)pyridin-4-amine

The title compound was obtained in 48% yield from the nitro-pyridine-N-oxide of Preparation 111 , following a similar procedure to that described in Example 31.
1H-NMR (CDCI3, 400MHz): δ 1.9 (m, 2H), 2.2 (m, 2H), 3.4 (m, 2H), 4.1 (bs, 2H), 4.2 (m, 2H), 5.95 (s, 1 H), 6.0 (d, 1H), 6.2 (s, 1 H), 7.0 (d, 1H), 7.2 (d, 1 H), 7.3 (m, 7H), 7.9 (d, 1 H). LRMS m/z 358 [MH]+.
Example 37 2-(1'H-Spirori-benzofuran-3,4'-piperidinl-1'-yl)pyridin-4-amine

The title compound was obtained in 73% yield from the nitro-pyridine-N-oxide of Preparation
112, following a similar procedure to that described in Example 31.
1H-NMR (CDCI3, 400MHz): δ 1.8 (m, 2H), 2.0 (m, 2H), 3.0 (m, 2H), 4.1 (bs, 2H), 4.2 (m, 2H),
4.4 (s, 2H), 5.95 (s, 1 H), 6.05 (m, 1 H), 6.8 (d, 1 H), 6.9 (t, 1 H), 7.1 (m, 2H), 7.9 (d, 1 H). LRMS m/z 282 [MH]+
Example 38
2-(1 H,1'H-Spiroπsochromene-4,4'-piperidin1-1'-yl)pyridin-4-amine
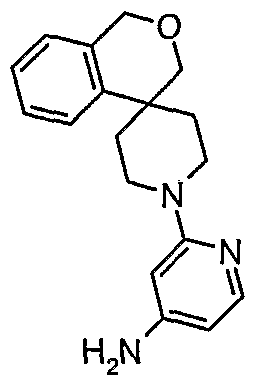
Example 39
(a) Methyl 1 '-(4-aminopyridin-2-yl)spirori-benzofuran-3.4'-piperidine1-2-carboxylate

The piperidine of Preparation 19 (569mg,,2.3mmol), 2-chloro-4-nitropyridine N-oxide (384mg, 2.2mmol) and sodium hydrogen carbonate (285mg, 3.44mmol) were combined in 2-methyl-2- butanol (10ml) and the reaction mixture was heated at 480C for 72 hours. After this time ammonium formate (1.48g, 23.6mmol) and palladium hydroxide (152mg) were added and the reaction mixture heated for a further 2 hours at 650C. This was repeated until complete reduction was seen. The reaction mixture was filtered through Arbocel® and the combined filtrates concentrated in vacuo. The crude product was purified by column chromatography on silica gel using dichloromethane/ methanol/0.880 ammonia (90:10:1) as eluent to afford the title compound as a brown solid (560mg, 1.65mmol, 72%).
1H-NMR (CDCI3, 400MHz): δ 1.8 (m, 1 H), 2.0 (m, 3H), 3.5 (m, 2H), 3.6 (m, 2H), 3.8 (m, 1 H), 3.8 (s, 3H), 4.0 (m, 4H), 5.0 (s, 1 H), 5.9 (s, 1H), 6.0 (d, 1 H), 6.9 (m, 2H), 7.2 (m, 2H), 7.9 (d, 1 H). LRMS m/z (APCI) 340 [MH]+.
(b) 1'-(4-Aminopyridin-2-yl)spirori-benzofuran-3,4'-piperidine1-2-carboxylic acid

The ester of step (a) (560mg, 1.65mmol) was dissolved in tetrahydrofuran (7ml). A solution of lithium hydroxide (47mg, 1.95mmol) in water (20ml) was added and the reaction mixture was stirred at room temperature for 7 hours. The reaction mixture was acidified to pH 3 with 2N
hydrochloric acid and then extracted into ethyl acetate, dried over magnesium sulphate, filtered and evaporated to yield the title compound as a powder (442mg, 1.3mmol, 78%). 1H-NMR (DMSO, 400MHz): δ 1.6-1.9 (m, 4H), 3.6-3.8 (m, 4H), 4.9 (s, 1 H), 5.9 (s, 1 H), 6.0 (m, 1 H), 6.8 (m, 2H), 7.1 (m, 1 H), 7.2 (d, 1 H), 7.6 (s, 1 H). LRMS m/z 296 [MH]+.
Examples 40-66

A solution of the acid of Example 39, step (b), (25μmol, 1eq) in N-methylpyrrolidine (0.2M) was added to a solution of the appropriate amine (30μmol, 1.2eq) in N-methylpyrrolidine
(0.1M). A solution of O-benzotriazol-1-yl-N,N,N',N'-tetramethyluronium hexafluorophosphate (35.7 μmol, 1.5eq) in N-methylpyrrolidine (0.375M) was added followed by triethylamine (50μL, 0.36mmol, 14eq). The reaction mixture was shaken at 6O0C overnight. The solvent was evaporated and the residue dissolved in dimethylsulphoxide/ water (3:1). The material was purified by HPLC on a Phenomenex Luna C18, 10μm, 150 x 10 mm id column using acetonitrile/0.05% aqueous diethylamine as the mobile phase.



LCMS was performed using a Phenomenex Luna C18, 5μm, 30 x 4.6 mm id column with a flow rate of 2.5 ml/min. The LC pump gradient timetable is shown beiow where solvent A = an aqueous solution of 0.05% ammonium acetate and 5% acetonitrile and B = acetonitrile.

Detection is performed at 225nm. Retention times are quoted for ELSD detection.
Examples 67-93

A solution of the acid of Example 20, step (b) (25μmol, 1eq) in N-methylpyrroIidine (0.25M) was added to a solution of the appropriate amine (30μmol, 1.2eq) in N-methylpyrrolidine (0.2M). A solution of O-benzotriazol-1-yl-N,N,N',N'-tetramethyluronium hexafluorophosphate (35.7 μmol, 1.5eq) in N-methylpyrrolidine (0.375M) was added followed by triethylamine (50μL, 0.36mmol, 14eq). The reaction mixture was shaken at 6O0C for 48 hours. The solvent was evaporated and the residue dissolved in dimethylsulphoxide: water (3:1 ). The-material was purified by HPLC on a Phenomenex Luna C18, 10μm, 150 x 10 mm id column using acetonitrile: 0.05% aqueous diethylamine as the mobile phase.



LCMS was performed using a Phenomenex Luna C18, 5μm, 30 x 4.6 mm id column with a flow rate of 2.5 ml/min. The LC pump gradient time table is shown below where solvent A = an aqueous solution of 0.05% ammonium acetate and 5% acetonitriie and B = acetonitrile.
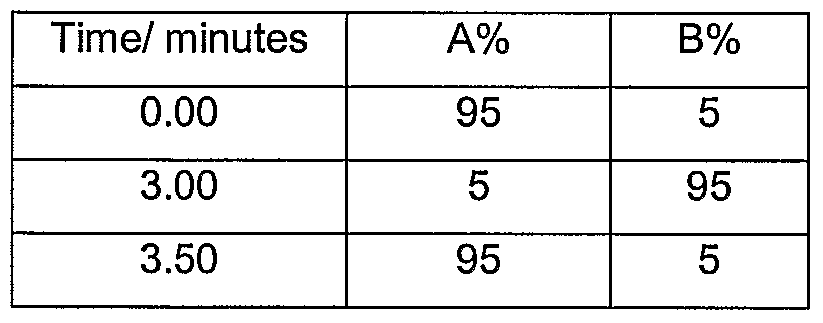
Detection is performed at 225nm. Retention times are quoted for ELSD detection.
Examples 94-97
H2N ■M
The appropriate piperidine (150μmol, 5eq) was dissolved in N-methylpyrrolidine (100μL), N1N- diisopropylethylamine (150μmol, 5eq) was added followed by a solution of 2-chloro-4- nitropyridine N-oxide (30μmol, 1eq) in N-methylpyrrolidine (200μL). The reaction mixture was sealed and heated at 480C for 48 hours. The reaction mixture was concentrated in vacuo then redissolved in methanol (100μL). A solution 1 M solution of ammonium formate in methanol (400μL) was added followed by 10% palladium on carbon (approx 10% by weight). The
reaction mixture was sealed and shaken at room temperature overnight. The reaction mixture was decanted, discarding the catalyst and the methanol evaporated. The material was purified by HPLC on a Phenomenex Luna C18, 10μm, 150 x 10 mm id column using acetonitrile: 0.05% aqueous diethylamine as the mobile phase.

LCMS was performed using a Phenomenex Luna C18, 5μm, 30 x 4.6 mm id column with a flow rate of 2.5 ml/min. The LC pump gradient time table is shown below where solvent A = an aqueous solution of 0.05% ammonium acetate and 5% acetonitrile and B = acetonitrile.

Detection is performed at 225nm. Retention times are quoted for ELSD detection.
Examples 98-107

(3S)-1'-(Tert-butoxycarbonyl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxylic acid
(150mg, 0.45mmol), [described in WO97/36873, step E] 1-hydroxybenzotriazole monohydrate (71 mg, 0.466mmol) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide HCI (104mg, 0.543mmol), N-methylmorpholine (0.075ml, 0.679mmol) and the appropriate amine (1.1 eq) were combined in dichloromethane (3ml) and stirred at room temperature overnight. The reaction mixture was washed with 3% aqueous sodium hydrogen carbonate (3ml), dried over sodium sulphate, filterered and evaporated in vacuo. The crude material was purified by column chromatography on silica gel using ethyl acetate: pentane as the eluent.
The amide was redissolved in dichloromethane (2ml) and treated with trifluoroacetic acid (1ml). The solution was stirred at room temperature for 4 hours and then concentrated in vacuo. The residue was extracted from 2M sodium hydroxide (2ml) into dichloromethane (3x2ml) and the combined organic extracts dried over sodium sulphate, filtered and evaporated.
The resultant piperidine (1eq), 2-chloro-4-nitropyridine N-oxide (0.95eq) and sodium hydrogen carbonate (1.1 eq) were combined in 2-methyl-2-butanoI. The reaction mixture was heated at 480C for 18 hours. After this time ammonium formate (6eq) and palladium hydroxide (10% by weight) were added and the reaction mixture heated for a further 2 hours at 650C. This was repeated until complete reduction was seen. The reaction mixture was filtered through Arbocel® and the combined filtrates concentrated in vacuo. The crude product was purified by column chromatography on silica gel using dichloromethane: methanol: 0.880 ammonia as eluent to afford the title compounds as racemates.



A= Dimethylformamide was used as the reaction solvent during the amide formation.
Example 108 r-(4-Aminopyridin-2-yl)-N,N-dimethyl-2,3-dihvdrospiroπndene-1.4'-piperidinel-3-carboxamide

The nitro-pyridine N-oxide of Preparation 106 (65mg, 0.16mmol) was dissolved in ethanol (3ml), cyclohexene (0.4ml, 3.2mmol) and 10% palladium on carbon (15mg) were added and the reaction mixture heated at 7O0C for 8 hours. 10% Palladium on carbon (10mg) and cyclohexene (0.3ml) were added and the reaction heated for a further 2.5 hours. The catalyst was removed by filtration through Arbocel® and the filtrates concentrated in vacuo. The crude
product was purified by column chromatography on silica gel using dichloromethane: methanol as the eluent (18mg, O.δmmol, 30%).
1H-NMR (CDCI3, 400MHz): δ 1.6 (m, 2H), 1.8 (m, 2H), 2.1 (m, 1 H), 2.3 (m, 1 H), 2.5 (m, 1H), 3.0 (m, 2H), 3.05 (s, 3H), 4.1 (m, 2H), 4.35 (m, 1H), 4.4 (t, 1 H), 5.95 (s, 1 H), 6.0 (d, 1H), 7.2 (m, 4H), 7.9 (d, 1 H). LRMS m/z (APCI) 351 [MH]+.
Example 109 I'-^-Aminopyridin^-vD^^-dihvdrospiroπndene-I Λ'-piperidinel-S-carboxylic acid
(a) Methyl (3S)- 1 '-(4-aminopyridin-2-yl')-2.3-dihvdrospirofindene-1 ,4'-piperidinel-3-carboxylate
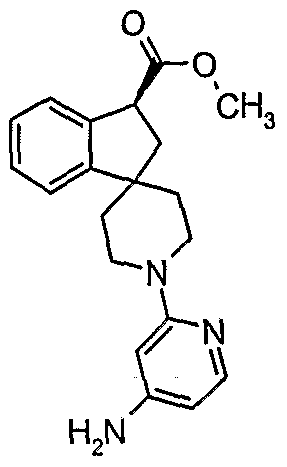
The piperidine of Preparation 47 (100mg, 0.41 mmol), 2-chloro-4-nitropyridirie N-oxide (68mg, 0.389mmo!) and sodium hydrogen carbonate (68mg, 0.82mmol) were combined in 2-methyl- 2-butanol (2.5ml) and the reaction mixture was heated at 480C for 18 hours. After this time ammonium formate (154mg, 2.46mmol) and palladium hydroxide (10mg) were added and the reaction mixture heated for a further 2 hours at 650C. This was repeated until complete reduction was seen by thin layer chromatography. The reaction mixture was filtered through Arbocel® and the combined filtrates concentrated in vacuo. The crude product was purified by column chromatography on silica gel using dichloromethane/methanol/0.880 ammonia (95:5:0.5) as eluent to afford the title compound as a white foam (90mg, 0.26mmol, 63%).
1H-NMR (CDCI3, 400MHz): δ 1.6 (m, 1H), 1.8 (m, 1 H), 1.9 (m, 1 H), 2.0 (m, 1 H), 2.4 (d, 2H), 3.0 (m, 2H), 3.8 (s, 3H), 4.0 (bs, 2H), 4.2 (m, 2H), 4.3 (m, 1 H), 5.9 (s, 1 H), 6.0 (d, 1H), 7.2 (m, 3H), 7.4 (d, 1 H), 7.9 (d, 1 H). Rf: 0.35 dichloromethane/methanol/0.880 ammonia (94.5: 5: 0.5).
(b) 1'-(4-Aminopyridin-2-yl)-2,3-dihvdrospiroπndene-1 ,4'-piperidine1-3-carboxvlic acid

1H-NMR (CD3OD, 400MHz): δ 1.6 (m, 1 H), 1.8 (m, 2H), 2.1 (m, 1 H), 2.3 (m, 1 H), 2.5 (m, 1 H), 3.1 (m, 2H), 4.0 (m, 4H), 6.05 (s, 1 H), 6.1 (d, 1 H), 7.1 (m, 3H), 7.4 (d, 1 H), 7.6 (d, 1 H). LRMS m/z (APCI) 324 [MH]+.
Examples 110-118

The appropriate sulphonyl chloride (1eq) was added to a stirred solution of the amine from Example 154 (1eq) and triethylamine (1.5eq) in dichloromethane. The reaction mixture was stirred at room temperature for 16 hours. The solvent was evaporated in vacuo and the residue partitioned between 3% aqueous sodium hydrogen carbonate and ethyl acetate. The organic layer was dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography on silica gel using dichloromethane/methanol as the eluent to afford the title compounds.


Example 119 r-(4-Aminopyridin-2-yl)-N,N-dimethyl-2,3-dihvdrospirorchromene-4,4'-piperidinel-2- carboxamide

The ester of Example 131 (70mg, 0.2mmol) was dissolved in tetrahydrofuran (2ml) and treated with 2M hydrochloric acid (2mi). The reaction mixture was stirred at 350C for 20 hours. The solvent was evaporated in vacuo. The intermediate acid was dissolved in dimethylformamide (3ml) and treated with 1-hydroxybenzotriazole monohydrate (31 mg,
0.204mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide HCI (46mg, 0.24mmol) and 2M dimethylamine in tetrahydrofuran (0.3ml, O.δmmol). The reaction mixture was stirred at room temperature for 16 hours. The reaction mixture was partitioned between 3% sodium hydrogen carbonate (12ml) and ethyl acetate (12ml). The organic layer was dried over sodium sulphate, filtered and evaporated. The crude material was purified by column chromatography over silica gel using dichloromethane/methanol/ammonia (93:7:0.5) as the eluent. (12mg, 0.03mmol, 14%).
1H-NMR (CDCI3, 400MHz): δ 1.4 (m, 2H), 1.8-2.1 (m, 3H), 2.4 (m, 2H), 3.6 (d, 1 H), 3.0 (s, 3H), 3.2 (s, 3H), 4.0 (bs, 2H), 4.2 (m, 2H), 4.8 (d, 1 H), 5.9 (s, 1 H), 6.0 (d, 1 H), 6.9 (m, 2H), 7.1 (m, 1 H), 7.3 (d, 1 H), 7.9 (d, 1 H). LRMS m/z (ESI) 367 [MH]+.
Example 120
2-(2.3-Dihvdro-1Η-spirofchromene-4Λ'-piperidin]-1 '-yl)pyridin-4-amine

2,3-Dihydrospiro[chromene-4,4'-piperidine] [described in J. Med. Chem. 1995, 38, (11), 2009- 2017] (0.8g, 1.36mmol), 2-chloro-4-nitropyridine N-oxide (226mg, 1.29mmol) and sodium hydrogen carbonate (171 mg, 2.0mmol) were combined in 2-methyl-2-butanol (4ml). The reaction mixture was heated at 480C for 16 hours. The reaction mixture was concentrated in vacuo. The residue was extracted from water into dichloromethane. The crude material was redissolved in acetic acid (5ml). Iron powder (143mg, 2.55mmol) was added and the reaction mixture stirred at room temperature overnight. The reaction mixture was concentrated in vacuo and the residue extracted from 2M sodium hydroxide into dichloromethane. The combined organic extracts were dried over sodium sulphate, filtered and evaporated to yield the crude aminopyridine. The material was purified by column chromatography over silica gel eluting with dichloromethane: methanol: 0.88 ammonia (95:5:0.5) to yield the title compound (155mg, 0.52mmol).
1H-NMR (CDCI3, 400MHz): δ 1.6 (m, 1 H), 1.7 (m, 2H), 2.1 (m, 2H), 2.2 (m, 2H), 3.0 (m, 2H), 4.0 (m, 2H), 4.2 (m, 3H), 5.9 (s, 1 H), 6.0 (d, 1 H), 6.8-7.2 (m, 3H), 7.3 (m, 1 H), 7.9 (m, 1 H). LRMS m/z (APCI) 296 [MH]+.
Examples 121-132
The nitro-pyridine N-oxide was dissolved in methanol. Palladium hydroxide (10% by weight) and ammonium formate (6eq) were added and the reaction mixture heated at reflux for 2-18 hours. The reaction mixture was allowed to cool to room temperature and the catalyst removed by filtration through Arbocel®. The combined filtrates were concentrated in vacuo and the residue extracted from saturated sodium hydrogen carbonate into dichloromethane, dried over magnesium sulphate, filtered and evaporated to yield the crude amino pyridine. The material was purified by column chromatography on silica gel using dichIoromethane/methanol/0.880 ammonia as eluent to afford the title compounds.
δ
(t,
(s,

126 Preparation 1H-NMR (CDCI3, 400MHZ): 110 δ1.8(m, 2H), 1.95 (m, 2H)13.3 (t, 2H), 4.0 (s, 2H), 4.2 (m, 2H), 5.1 (s, 2H), 6.0 (s, 1H), 6.05 (d, 1H), 7.1 (d, 1H), 7.2
 (m, 3H), 7.9 (d, 1H). LRMS m/z (APCI) 282 [MH]+
(m, 3H), 7.9 (d, 1H). LRMS m/z (APCI) 282 [MH]+
127 Preparation 1H-NMR (CDCI3, 400MHz): 101 δ 1.6 (s, 3H), 2.65 (d, 1H), 1.8 (d, 1H), 1.9 (m, 1H), 2.1 (m, 1H), 2.35 (q, 2H), 2.8 (m, 6H), 3.0 (t, 1H), 3.1 (t, 1H), 4.15 (d,
 1H), 4.25 (d, 1H), 4.9 (m, 2H), 6.0(s, 1H), 6.1 (d, 1H), 7.1 (m, 2H), 7.2 (m, 2H), 7.8 (d, 1H). LRMS m/z (APCI) 365 [MH]+
1H), 4.25 (d, 1H), 4.9 (m, 2H), 6.0(s, 1H), 6.1 (d, 1H), 7.1 (m, 2H), 7.2 (m, 2H), 7.8 (d, 1H). LRMS m/z (APCI) 365 [MH]+
128 Preparation 1H-NMR (CD3OD, 400MHz): 102 δ 1.6 (d, 2H), 1.8 (m, 2H), 2.2 (m, 1H), 2.85 (m, 1H), 2.95 (t, 1H), 3.1 (t, 1H), 4.05 (t, 2H), 5.5 (m, 1H), 6.1 (m, 2H), 6.6 (m, 2H), 7.2 (m, 4H), 7.4 (t,
 1H), 7.6 (d, 1H), 8.0 (d, 1H). LRMS m/z (APCI) 372 [MH]+
1H), 7.6 (d, 1H), 8.0 (d, 1H). LRMS m/z (APCI) 372 [MH]+
129L Preparation 1H-NMR (CD3OD, 400MHz): 116 δ 1.8 (m, 2H), 2.0 (m, 2H), 3.0 (m, 2H), 4.0 (m, 2H), 4.6 (s, 2H), 6.05 (s, 1H), 6.1 (d, 1H), 6.5 (d, 1H), 6.6 (t, 1H), 7.1 (m,
 1H), 7.6 (d, 1H). LRMS m/z (APCI) 300 [MH]+
1H), 7.6 (d, 1H). LRMS m/z (APCI) 300 [MH]+

B = Racemic product was separated via chiral HPLC to provide both enantiomers.
C = Ethyl acetate was used as the column eluent.
D = Ethanol was used as the reaction solvent.
E = 2-Methyl-2-butanol was used as the reaction solvent.
Examples 133-152
The piperidine (1eq), 2-chloro-4-nitropyridine N-oxide (1eq) and sodium hydrogen carbonate (1.1 eq) were combined in 2-methyl-2-butanol and the reaction mixture was heated at 480C for 18 hours. After this time ammonium formate (6eq) and palladium hydroxide (10% by weight) were added and the reaction mixture heated for a further 2 hours at 650C. This was repeated until complete reduction was seen. The reaction mixture was filtered through Arbocel® and the combined filtrates concentrated in vacuo. The crude product was purified by column
chromatography on silica gel using dichioromethane/ methanol/0.880 ammonia as eluent to afford the title compounds.
(d,

3.7
(s,
1.2
(m, 3.0
(m, 6.0

4.3
4.2


146 Preparation 94 1H-NMR (CDCI3, 400MHz): 51.5 (m, 2H), 2.3 (m, 2H), 3.5 (m, 2H), 4.0 (bs, 2H), 4.2 (m, 2H), 4.8 (s, 2H), 5.9 (S, 2H), 6.1 (d, 1H), 7.2 (m, 5H), 7.55 (m, 1H), 7.8 (m, 1H), 7.9 (d, 1H), 8.0 (m, 1H). LRMS m/z (APCI) 385 [MH]+
147 Preparation 93 1H-NMR (CDCI3, 400MHz): 51.6 (m, 1H), 1.8 (m, 2H), 2.1 (m, 1H), 2.3 (m, 1H), 2.5 (m, 1H), 3.0 (m, 2H), 3.0 (s, 3H), 3.3 (s, 3H), 4.1 (m, 3H), 4.3 (m, 1H), 4.4 (t, 1H), 5.9 (s, 1H), 6.0 (d, 1H), 7.1-7.3
 (m, 4H), 7.9 (d, 1H). LRMS m/z (APCI) 351 [MH]+
(m, 4H), 7.9 (d, 1H). LRMS m/z (APCI) 351 [MH]+
148" Preparation 91 1H-NMR (CDCI3, 400MHz): δ 1.6 (m, 1H), 1.8 (m, 2H), 2.1 (m, 1H), 2.3 (m, 1H), 2.5 (m, 1H), 3.0 (m, 2H), 3.05 (s, 3H), 3.2 (s, 3H), 4.2 (bs, 2H), 4.1 (m, 1H), 4.3 (m, 1H), 4.4 (t, 1H), 5.95(s, 1H), 6.0
 (d, 1H), 7.1-7.3 (m, 4H), 7.9 (d, 1 H). LRMS m/z (APCI) 351 [MH]+
(d, 1H), 7.1-7.3 (m, 4H), 7.9 (d, 1 H). LRMS m/z (APCI) 351 [MH]+
149 Preparation 19 1H-NMR (CDCI3, 400MHz): 51.8 (m, 1H), 2.0 (m, 3H), 3.5 (m, 2H), 3.6 (m, 2H), 3.8 (m, 1H), 3.8 (s, 3H), 4.0 (m, 4H), 5.0 (s, 1H), 5.9 (s, 1H)16.0 (d, 1H), 6.9 (m, 2H),
 7.2 (m, 2H), 7.9 (d, 1H). LRMS m/z (APCI) 340 [MH]+
7.2 (m, 2H), 7.9 (d, 1H). LRMS m/z (APCI) 340 [MH]+
150 Preparation 50 1H-NMR (CDCI3, 400MHz): δθ.4 (m, 2H)10.8 (m, 2H)11.2 (m, 1H)1
1.6 (m, 1H), 1.9 (m, 1H), 2.0 (m, 1H), 2.3 (m, 1H), 2.5 (m, 1 H), 2.7 (m, 1H), 3.0 (m, 2H), 3.9 (m, 1H), 4.0 (bs, 2H), 4.2 (m, 2H), 5.7 (bs,
 1H), 5.9 (s, 1H), 6.0 (d, 1H), 7.3 (m, 4H), 7.9 (d, 1H). LRMS m/z (APCI) 363 [MH]+
1H), 5.9 (s, 1H), 6.0 (d, 1H), 7.3 (m, 4H), 7.9 (d, 1H). LRMS m/z (APCI) 363 [MH]+
1511 Preparation 51 1H-NMR (CDCI3, 400MHz): δ 0.9 (t, 3H), 1.4-1.7 (m, 4H), 1.9 (m, 1H), 2.0 (m, 1H), 2.4 (m, 1H), 2.6 (m, 1H), 3.0 (m, 2H), 3.3 (m, 2H), 4.0 (t, 1H), 4.05 (bs, 2H), 4.2 (m, 2H), 5.6 (bs, 1H), 5.9 (s, 1H), 6.0
 (d, 1H), 7.2 (d, 1H), 7.3 (m, 3H), 7.9 (d, 1H). LRMS m/z (APCI) 365 [MH]+
(d, 1H), 7.2 (d, 1H), 7.3 (m, 3H), 7.9 (d, 1H). LRMS m/z (APCI) 365 [MH]+
152H Preparation 62 1H-NMR (CDCI3, 400MHz): δ 1.2 (derived from (t, 3H), 1.3 (t, 3H), 1.8 (m, 1H), resolution with 2.0 (m, 3H), 3.3 (m, 2H), 3.4 (m, (R)-(+)-α- 2H), 3.6 (m, 2H), 3.9 (bs, 2H), methylbenzyl- 4.1 (m, 1H), 4.3 (m, 1H), 5.9 (s, amine) 1H), 5.95 (s, 1H), 6.0 (d, 1H), 7.1
 (m, 1H), 7.3 (m, 3H), 7.9 (d, 1H). chiral LRMS m/z (ESI) 381 [MH]+
(m, 1H), 7.3 (m, 3H), 7.9 (d, 1H). chiral LRMS m/z (ESI) 381 [MH]+
153H Preparation 63 1H-NMR (CDCI3, 400MHz): δ 1.2 (derived from (t, 3H), 1.3 (t, 3H), 1.8 (m, 1H), resolution with 2.0 (m, 3H), 3.3 (m, 2H), 3.4 (m, (S)-(-)-α- 2H), 3.6 (m, 2H), 3.9 (bs, 2H), methylbenzyl- 4.1 (m, 1H), 4.3 (m, 1H), 5.9 (s, amine) 1H), 5.95 (s, 1H), 6.0 (d, 1H), 7.1
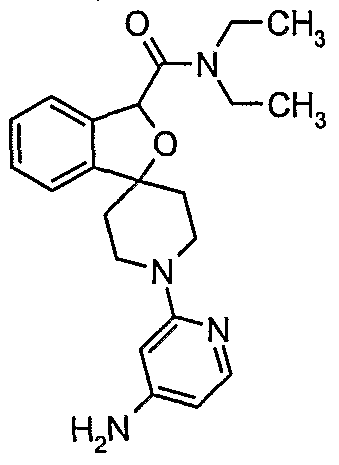 (m, 1H), 7.3 (m, 3H), 7.9 (d, 1H). chiral LRMS m/z (APCI) 381 [MH]+
(m, 1H), 7.3 (m, 3H), 7.9 (d, 1H). chiral LRMS m/z (APCI) 381 [MH]+
E = Methanol was used as the reaction solvent.
F = Triethylamine was used as the base.
G = Ethyl acetate/methanol/0.880 ammonia (90:10:1) was used as the column eluent.
H = Diethyl ether/methanol/0.880 ammonia (90:10:1) was used as the column eluent.
Example 154 2-M .2-Dihvdro-i 'H-spiroπndole-3,4'-piperidin1-1 '-yl)pyridin-4-amine
(a) Tert-butyl r-(4-aminopyridin-2-yl)spirorindole-3,4'-piperidine1-1 (2H)-carboxylate

Tert-butyl spiro[indole-3,4'-piperidine]-1(2H)-carboxylate. HCI (1g, 3.1 mmol) [described in WO2004/028459, example 21 , compound xxii], 2-chloro-4-nitropyridine N-oxide (538mg, 2.94mmol), sodium hydrogen carbonate (310mg, 3.72mmol) and triethylamine (0.472ml, 3.4mmol) were combined in 2-methyl-2-butanol (15ml) and the reaction mixture heated at 480C for 18 hours. After this time ammonium formate (1.16g, 18.6mmol) and palladium hydroxide (100mg) were added and the reaction mixture heated for a further 2 hours at 650C. This was repeated until complete reduction was seen. The reaction mixture was filtered through Arbocel® and the combined filtrates concentrated in vacuo. The crude product was purified by column chromatography on silica gel using dichloromethane: methanol: 0.880 ammonia (90:10:1) as eluent to afford the title compound as a beige foam (1.1g, 2.89mmol,
1H-NMR (CDCI3, 400MHz): δ 1.6 (s, 10H), 1.8 (m, 2H), 2.0 (m, 2H), 2.9 (m, 2H), 3.9 (bs, 2H), 4.1 (bs, 1 H), 4.2 (m, 2H), 5.9 (s, 1 H), 6.05 (d, 1 H), 7.0 (t, 1 H), 7.1 (d, 1 H), 7.2 (t, 1 H), 7.9 (d, 1 H). LRMS m/z (APCI) 381 [MH]+.
(b) 2-(1 ,2-Dihvdro-1 'H-spiroπndole-3.4'-piperidin1-1 '-ylbyridin-4-amine

The protected piperidine of step (a) (1g, 2.6mmol) was dissolved in dichloromethane: trifluoroacetic acid (8ml:2ml) and the reaction mixture stirred at room temperature for 3 hours. The solvent was evaporated and the residue partitioned between 2N sodium hydroxide and
dichloromethane. The organic phase was dried over sodium sulphate, filtered and evaporated to yield the title compound (680mg, 2.4mmol, 93%).
1H-NMR (CDCI3, 400MHz): δ 1.8 (m, 2H), 1.9 (m, 2H), 3.0 (t, 2H), 3.5 (s, 2H), 4.0 (bs, 2H), 4.2 (m, 2H)1 5.9 (s, 1 H), 6.0 (d, 1 H), 6.7 (d, 1 H), 6.8 (t, 1 H), 7.0 (m, 2H), 7.9 (d, 1 H). LRMS m/z (APCI) 281 [MH]+.
Example 155
(3S)-1'-(4-aminopyridin-2-yl)-N,N-diethyl-2,3-dihvdrospiro[indene-1 ,4'-piperidinel-3- carboxamide

The piperidine of Preparation 118 (92mg. 0.3mmol), 2-chloro-4-nitropyridine N-oxide (50mg, 0.28mmol) and sodium hydrogen carbonate (28mg, 0.33mmol) were combined in 2-methyl-2- butanol (2ml) and the reaction mixture was heated at 480C for 18 hours. After this time ammonium formate (113mg, 1.8mmol) and palladium hydroxide (11 mg) were added and the reaction mixture heated for a further 2 hours at 650C. This was repeated until complete reduction was seen. The reaction mixture was filtered through Arbocel® and the combined filtrates concentrated in vacuo. The crude product was purified by column chromatography on silica gel using dichloromethane/ methanol/0.880 ammonia (95:5:0.5) as eluent to afford the title compound as a gum (67mg, 0.18mmol, 56%).
1H-NMR (CDCI3, 400MHz): δ 1.2 (m, 3H), 1.3 (m, 3H), 1.6 (m, 1H), 1.8 (m, 2H), 2.2 (m, 1 H)1 2.4 (m, 1 H), 2.5 (m, 1 H), 3.0 (m, 2H), 3.4 (m, 1 H), 3.5 (m, 1 H), 3.6 (m, 2H), 3.9 (s, 2H), 4.1 (m, 1H), 4.3 (m, 2H), 5.9 (s, 1H), 6.0 (d, 1H), 7.1 (m, 1H), 7.2 (m, 3H), 7.9 (d, 1H). LRMS m/z (ESI) 379 [MH]+.
Example 156
(a) Ethyl r-(4-aminopyridin-2-yl)-5-methoxy-3H-spiro[2-benzofuran-1 ,4'-piperidine1-3- carboxylate

1H-NMR (CDCI3, 400MHz): δ 1.3 (t, 3H), 1.80 (m, 1 H), 1.95 (m, 3H), 3.4 (m, 2H), 3.8 (s, 3H), 4.0 (S, 2H), 4.2 (m, 4H), 5.65 (s, 1H), 5.95 (d, 1H), 6.0 (m, 1 H), 6.85 (m, 1 H), 6.95 (d, 1 H), 7.0 (m, 1 H), 7.9 (d, 1 H). LRMS m/z (ES) 384 [MH]+
(b) (+)-1'-(4-aminopyridin-2-yl)-N.N-dimethyl-5-methoxy-3H-spirof2-benzo-furan-1 ,4'- piperidinel-3-carboxamide

The chiral resolution of the racemic title compound was achieved by chiral HPLC using a Chiralpak AD-H column at ambient temperature, eluting with hexane:isopropanol:diethylamine (70:30:0.1). The first isomer to elute had a positive rotation [α]D 25 (c.1.01mg/ml methanol) +20.3°
Example 157
(+)-1l-(4-aminopyridin-2-yl)-N.N-diethyl-5-methoxy-3H-spiro[2-benzofuran-1.4'-piperidine1-3- carboxamide

The title compound was obtained as a white foam in 29% yield from the ester of Example 156, step (a) and diethylamine, following a similar procedure to that described in Example 156, step (b).
1H-NMR (CDCI3, 400MHz): δ 1.15 (t, 3H), 1.25(t, 3H), 1.80 (m, 1 H), 1.95 (m, 3H), 3.5 (m, 6H), 3.8 (s, 3H), 4.0 (s, 2H), 4.15 (m, 1H), 4.25 (m, 1 H), 5.85 (s, 1 H), 5.95 (d, 1 H), 6.0 (m, 1H), 6.85 (m, 3H), 7.0 (m, 1 H), 7.9 (d, 1 H). LRMS m/z (APCI) 411 [MH+].
The chiral resolution of the racemic title compound was achieved by chiral HPLC using a Chiralpak AD-H column at ambient temperature, eluting with hexane:isopropanol:diethylamine (70:30:0.1). The first isomer to elute had a positive rotation [α]D 25 (c.1.01 mg/ml methanol) +14.6°
Example 158 1'-(4-aminopvridin-2-vl)-N,N-dimethvl-3H-SDirof2-benzofuran-1 ,4'-piperidine1-3-carboxamide

The title compound, as a single enantiomer, was obtained as a white foam in 48% yield from the amine of Preparation 121, following a similar procedure to that described in Example 155.
1H-NMR (CDCI3, 400MHz): δ 1.8 (m, 1 H), 2.0 (m, 3H), 3.0 (m, 3H), 3.3 (m, 5H), 3.95 (s, 2H), 4.15 (m, 1 H), 4.3 (m, 1 H), 5.9 (s, 1 H), 5.95 (d, 1 H), 6.0 (m, 1 H), 7.1 (m, 1 H), 7.3 (m, 3H), 7.9 (d, 1 H). LRMS m/z (ESI) 353 [MH]+.
Example 159
1'-(4-aminopyridin-2-vπ-N-ethyl-N-methyl-3H-spiro[2-benzofuran-1.4'-piperidine1-3- carboxamide

The title compound, as a single enantiomer, was obtained as a white foam in 40% yield from the amine of Preparation 122, following a similar procedure to that described in Example 155.
1H-NMR (CDCI3, 400MHz): δ 1.2 (m, 3H), 1.8 (m, 1 H), 2.0 (m, 3H), 3.4 (m, 7H), 3.95 (s, 2H), 4.15 (m, 1 H), 4.3 (m, 1H), 5.9 (s, 1H), 5.95 (d, 1 H), 6.0 (m, 1 H), 7.1 (m, 1H), 7.3 (m, 3H), 7.9 (d, 1 H). LRMS m/z (APCI) 367 [MH]+.
Example 160 2-[7-(Aminomethvπ-1 'H-spirori-benzofuran-3,4'-piperidinl-1'-yllpyridine-4-amine

The piperidine of Preparation 123 (250 mg, 0.708 mmol) was dissolved in ammonia (25 ml of a 2M solution in ethanol) and the reaction was hydrogenated at 4 atmospheres over Raney Nickel (100 mg) at room temperature for 3 hours. The reaction mixture was filtered through Arbocel® and after evaporation, the crude material was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane - dichloromethane/methanol/ 0.880 ammonia (90:10:1 ) to yield the title compound as a white solid (172 mg, 0.555 mmol, 78%).
1H-NMR (CDCI3, 400MHz): δ 1.8-1.9 (d, 2H), 1.95-2.05 (m, 2H), 2.9-3.05 (m, 2H), 3.85 (s, 2H), 4.0 (s, 2H), 4.15-4.25 (m, 2H), 4.5 (s, 2H), 5.95 (s, 1 H), 6.0 (s, 1 H), 6.85 (t, 1 H), 7.05 (d, 1 H), 7.15 (d, 1 H), 7.9 (d, 1 H). LRMS m/z (APCI) 311 [MH]+.
Example 161 N-{ri '-(4-aminopyridin-2-yl)spirori-benzofuran-3,4'-piperidin1-7-yllmethyl)-acetamide

Acetic acid (2.5 μl, 0.044 mmol), HOBT (7mg, 0.052 mmol) and WSCDI (10mg, 0.052) were dissolved in dichloromethane (3ml). The reaction mixture was stirred at room temperature under nitrogen for 30 minutes. The amine of Example 160 (14mg, 0.0463 mmol) was added and the reaction mixture stirred at room temperature under nitrogen overnight. The reaction mixture was partitioned between sodium hydroxide solution (2N) and ethyl acetate. The organic phase was dried over sodium sulfate, filtered and evaporated. The crude product was
purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane - dichloromethane/methanol/ 0.880 ammonia (90:10:1 ) to yield the title compound as a white solid (6mg, 0.017 mmol, 39%).
1H-NMR (CD3OD, 400MHz): δ 1.75-1.8 (d, 2H), 1.90-2.05 (m, 2H), 1.95 (s, 3H), 2.9-3.0 (m, 2H), 4.0-4.1 (m, 2H), 4.3 (s, 2H), 4.55 (s, 2H), 6.05 (s, 1 H), 6.10 (s, 1 H), 6.85 (t, 1 H), 7.05 (m, 2H), 7.65 (d, 1 H). LRMS m/z (ESI) 353 [MH]+.
Example 162 N-iri'-f4-aminoDyridin-2-vπspirori-benzofuran-3.4'-piperidinl-7-yllmethyll-pyridine-2- carboxamide

Picolinic acid (5mg, 0.0414 mmol), HOBT (7mg, 0.048 mmol) and WSCDI (10mg, 0.047 mmol) were dissolved in dichloromethane (3ml). The reaction mixture was stirred at room temperature under nitrogen for 30 minutes. The amine of Example 160 (11mg, 0.036 mmol) amine was added and the reaction mixture stirred at room temperature under nitrogen overnight. The reaction mixture was partitioned between sodium hydroxide solution (2N) and ethyl acetate. The organic phase was dried over sodium sulfate, filtered and evaporated. The crude product was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane - dichloromethane/methanol/0.880 ammonia (90:10:1) to yield the title compound as a yellow solid (5mg, 0.017 mmol, 30 %).
1H-NMR (CD3OD, 400MHz): δ 1.75-1.8 (d, 2H), 1.90-2.05 (m, 2H), 2.9-3.0 (m, 2H), 4.0-4.1 (m, 2H), 4.55 (s, 2H), 4.60 (s, 2H), 6.05 (s, 1 H), 6.10 (d, 1 H), 6.85 (t, 1 H), 7.05-7.10 (m, 2H), 7.65 (m, 1 H), 7.75 (d, 1 H), 7.95 (m, 1 H), 8.10 (d, 1 H), 8.60 (m, 1 H). LRMS m/z (ESI) 416 [MH]+.
Example 163 Methyl 1'-(4-aminopyridin-2-yl)spirorindole-3,4'-piperidinel-1 (2H)-carboxylate

The compound of Preparation 130 (420 mg, 1.09 mmol), ammonium formate (340 mg, 5.46 mmol) and Pd(OH)2 on carbon (40 mg) were stirred in ethanol at reflux for 2 hours. The mixture was allowed to cool and the catalyst removed by filtration. The filtrate was concentrated and loaded onto a silica gel column, eluting with ethyl acetate:cyclohexane (50:50 - 100:0) to yield the title compound as a white foam (260 mg, 0.76 mmol, 70%).
1H-NMR (CDCI3, 400MHz): δ 1.67 (m, 2H), 1.91 (m, 2H)1 2.88 (m, 2H), 3.75-4.00 (m, 5H), 4.17 (m, 2H), 5.87 (m, 1 H), 5.96 (m, 1 H), 6.92 (t, 1 H), 7.05 (d, 1 H), 7.15 (t, 1 H), 7.45 (m, 1 H), 7.63 (d, 1 H).
MS m/z (APCI) 339 [MH]+.
Example 164 Ethyl 1 '-(4-aminopyridin-2-yl)spirorindole-3.4'-piperidinel-1 (2H)-carboxvlate

The compound of Preparation 131 (465 mg, 1.17 mmol), ammonium formate (368 mg, 5.84 mmol) and Pd(OH)2 on carbon (40 mg) were stirred in ethanol at reflux for 2 hours. The mixture was allowed to cool and the catalyst removed by filtration. The filtrate was concentrated and loaded onto a silica gel column, eluting with ethyl acetate:cyclohexane (50:50 - 100:0) to yield the title compound as a white foam (290 mg, 0.82 mmol, 70%).
1H-NMR (CDCI3, 400MHz): δ 1.30 (m, 3H), 1.67 (m, 2H), 1.91 (m, 2H), 2.88 (m, 2H), 3.80- 4.00 (m, 4H), 4.15-4.35 (m, 3H), 5.88 (m, 1 H), 5.96 (m, 1 H), 6.92 (t, 1 H), 7.05 (d, 1 H), 7.15 (t, 1 H), 7.45 (m, 1 H), 7.63 (d, 1 H). MS m/z (APCI) 353 [MH]+.
Example 165 Isopropyl 1 '-(4-aminopyridin-2-yl)spiroπndole-3,4'-piperidinel-1 (2H)-carboxvlate

The compound of Preparation 132 (260 mg, 0.63 mmol), ammonium formate (199 mg, 3.15 mmol) and Pd(OH)2 on carbon (30 mg) were stirred in ethanol at reflux for 2 hours. The mixture was allowed to cool and the catalyst removed by filtration. The filtrate was concentrated and loaded onto a silica gel column, eluting with ethyl acetate:cyclohexane (50:50 - 100:0) to yield the title compound as a white foam (160 mg, 0.44 mmol, 69%).
1H-NMR (CDCI3, 400MHz): δ 1.36 (m, 6H), 1.74 (m, 2H), 1.97 (m, 2H), 2.96 (m, 2H), 3.85- 4.04 (m, 4H), 4.24 (m, 2H), 5.10 (m, 1 H), 5,95 (m, 1 H), 6.02 (d, 1 H), 6.98 (t, 1 H), 7.11 (d, 1 H), 7.20 (t, 1 H), 7.53 (m, 1 H), 7.90 (d, 1 H). MS m/z (APCI) 367 [MH]+.
Preparations
Preparation 1 Tert-butyl 3-(benzoylamino)-2,3-dihvdro-1 'H-spiroHndene-i ,4'-piperidine1-1 '-carboxvlate

Tert-butyl S-amino^.S-dihydro-i'H-spirotindene-i ^'-piperidineJ-i'-carboxylate [described in US5578593, compound 21 , scheme 10] (300mg, 0.88mmol, 1eq), benzoic acid (107mg,
0.88mmol), triθthylamine (367μL, 2.64mmol), 1-hydroxybenzotriazole monohydrate (135mg,
1.0 mmol) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide HCI (155mg, I .Ommol, 1.2mmol) were combined in dichloromethane (10ml). The reaction was allowed to stir at room temperature for 48 hours. The reaction mixture was diluted with dichloromethane and washed with saturated sodium hydrogen carbonate, 2M hydrochloric acid and water. The combined organic layers were dried over magnesium sulphate, filtered and evaporated in vacuo. The residue was purified by column chromatography using an ISCO® silica cartridge eluting with a linear gradient from 0-50% ethyl acetate/pentane. The product was obtained as a white foam (200mg, 0.49mmol, 56%). 1H-NMR (CD3OD, 400MHz): δ 1.5 (s, 9H)1 1.6 (m, 3H), 1.9 (dd, 1 H), 2.1 (t, 1 H), 2.9 (dd, 1 H), 3.0 (bs, 1 H), 3.1 (bs, 1 H), 4.1 (m, 2H), 5.7 (t, 1 H), 7.3 (m, 4H), 7.4 (t, 2H), 7.55 (t, 1 H), 7.9 (d, 2H). LRMS m/z (APCI) 351 [M- WyIH]+.
Preparation 2 N-(2.3-dihvdrospirorindene-1 ,4'-piperidinl-3-v0benzamide

The protected piperidine of Preparation 1 (200mg, 0.49mmol, 1eq) was dissolved in dichloromethane (5ml), trifluoroacetic acid was added (1ml) and the reaction mixture stirred at room temperature for 4 hours. The reaction mixture was concentrated in vacuo and residual trifluoroacetic acid azeotroped with toluene, then ether. The residue was extracted from saturated sodium hydrogen carbonate into dichloromethane, dried over magnesium sulphate, filtered and evaporated in vacuo. The product was isolated as a white foam (130mg, 0.42mmol, 85%). 1H-NMR (CD3OD, 400MHz): δ 1.55-1.7 (m, 3H), 1.85 (dd, 1 H), 2.2 (t, 1 H), 2.8-2.95 (m, 3H), 3.1 (d, 2H), 5.7 (t, 1 H), 7.3 (m, 4H), 7.45 (t, 2H), 7.55 (t, 1 H), 7.9 (d, 2H). LRMS m/z (APCI) 307 [MHf.
Preparation 3
Tert-butyl 3-r(pyridin-2-ylcarbonv0amino1-2,3-dihvdro-1 'H-spirorindene-1 ,4'-piperidine1-1 '-

The title compound was obtained in 78% yield from tert-butyl 3-amino-2,3-dihydro-1'H- spiro[indene-1 ,4'-piperidine]-1'-carboxylate and picolinic acid, following a similar procedure to that described in Preparation 1. Dichloromethane/methanoI/0.880 ammonia was used as the column eluent.
1H-NMR (CD3OD, 400MHz): δ 1.5 (s, 9H), 1.6 (m, 3H), 1.9 (m, 1 H), 2.1 (m, 1 H), 2.8-3.2 (m, 3H), 4.1 (m, 2H), 5.6 (m, 1H), 7.2 (m, 4H), 7.5 (m, 1 H), 8.0 (m, 1 H), 8.2 (m, 1 H), 8.6 (m, 1 H).
Preparation 4 N-(2,3-Dihvdrospirorindene-1 ,4'-piperidinl-3-yl)pyridine-2-carboxamide

The protected amine of Preparation 3 (470mg, 1.15mmol) was dissolved in dichloromethane, 4M hydrochloric acid in dioxane (4ml) was added and the reaction mixture stirred at room temperature overnight. The solvent was concentrated in vacuo and the residue partitioned between 1 N sodium carbonate and dichloromethane. The organic phase was washed with brine, dried over sodium sulphate, filtered and evaporated in vacuo. The crude product was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane - 90:10:1 dichloromethane/ methanol/0.880 ammonia. The product was isolated as a white solid (290mg, 0.94mmol, 82%).
1H-NMR (CD3OD, 400MHz): δ 1.6 (m, 3H), 1.9 (m, 1 H), 2.1 (m, 1 H), 2.8 (m, 3H), 3.0 (m, 2H), 5.7 (t, 1 H), 7.2 (m, 2H), 7.3 (m, 2H), 7.5 (m, 1 H), 7.95 (t, 1 H), 8.2 (d, 1 H), 8.6 (d, 1 H). LRMS m/z (APCI) 308 [MH]+.
Preparation 5 , Tert-butvl 3-(isobutvrylamino)-2,3-dihvdro-1 'H-spiroHndene-i .4'-piperidine1-1 '-carboxvlate

The title compound was obtained in 60% yield from tert-butyl 3-amino-2,3-dihydro-1'H- spiro[indene-1 ,4'-piperidine]-r-carboxylate and isobutyric acid, following a similar procedure to that described in Preparation 1.
1H-NMR (CDCI3, 400MHz): δ 1.1 (t, 6H), 1.4 (s, 9H), 1.5 (m, 2H), 1.9 (m, 2H), 2.4 (m, 1 H), 1.7 (m, 1 H), 2.8 (m, 2H), 4.0 (m, 2H), 5.4 (q, 1 H), 6.2 (d, 1 H), 7.1 (d, 1 H), 7.2 (m, 3H). LRMS m/z (APCI) 273 [M-BOCH]+
Preparation 6, N-(213-dihvdrospiroπndene-1 ,4'-piperidin1-3-yl)-2-methylpropanamide

The title compound was obtained in 36% yield from the protected piperidine of Preparation 5 following a similar procedure to that described in Preparation 2.
1H-NMR (CDCI3, 400MHz): δ 1.2 (m, 6H), 1.5 (m, 2H), 1.6 (m, 2H), 2.05 (m, 1 H), 2.4 (m, 1 H), 2.8 (m, 2H), 3.0 (m, 2H), 5.5 (q, 1 H), 5.7 (bd, 1 H), 7.3 (m, 4H). LRMS m/z (APCI) 273 [MH]+
Preparation 7
Tert-butyl (3S)-3-(5-phenyl-1 ,3,4-oxadiazol-2-vπ-2,3-dihvdro-1'H-spiro[indene-1 ,4'-piperidine1-
1 '-carboxvlate

(3S)-1 '-(Tert-butoxycarbonyl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxylic acid
(300mg, 0.9mmol), was dissolved in dichloromethane (5ml), benzoylhydrazine (123mg, 0.9mmol), triethylamine (0.5ml, 3.6mmol) and 2-chloro-1 ,3-dimethyI-2-imidazolinium
tetrafluoroborate (598mg, 2.7mmol) were added and the reaction mixture stirred at room temperature for 18 hours. The reaction mixture was diluted with dichloromethane and washed with saturated sodium hydrogen carbonate, 2M hydrochloric acid, water, dried over magnesium sulphate, filtered and concentrated in vacuo. The crude material was purified by column chromatography using an ISCO® silica cartridge, using ethyl acetate/pentane as the eluent. The title compound was obtained as a white foam (172mg, 0.4mmol, 44%). 1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 9H), 1.55-1.8 (m, 4H), 2.0 (m, 1 H), 2.5 (q, 1 H), 2.75 (q, 1H), 3.0 (m, 2H), 4.2 (bs, 2H), 7.2-7.3 (m, 4H), 7.5 (m, 3H), 8.0 (d, 2H). LRMS m/z (APCI) 432 [MH]+.
Preparation 8 (3S)-3-(5-phenyl-1 ,3,4-oxadiazol-2-yl)-2,3-dihvdrospirorindene-1 ,4'-piperidinel

The protected amine of Preparation 7 (160mg, 0.37mmol, 1eq) was dissolved in dichloromethane (5ml), trifluoroacetic acid (0.5ml) was added and the reaction mixture stirred at room temperature for 2 hours. The reaction mixture was concentrated in vacuo, azeotroped with toluene then ether. The residue was extracted from saturated sodium hydrogen carbonate into dichloromethane, dried over magnesium sulphate, filtered and concentrated in vacuo. The crude material was purified by column chromatography using an ISCO® silica cartridge, eluting a linear gradient from dichloromethane - 90:10:1 dichloromethane/methanol/0.880 ammonia. The product was isolated as a white foam (90mg, 0.27mmol, 74%). 1H-NMR (CDCI3, 400MHz): δ 1.6-1.8 (m, 3H), 2.1 (m, 1 H), 2.5 (m, 1 H), 2.8 (q, 1 H), 2.9 (m, 2H), 3.2 (m, 2H), 4.8 (t, 1 H), 7.2-7.3 (m, 4H), 7.5 (m, 3H), 8.0 (d, 2H). LRMS m/z (APCI) 332 [MH]+.
Preparation 9
Tert-butyl (3S)-3-(5-methyl-1 ,3,4-oxadiazol-2-yl V2.3-dihvdro-1 'H-spiroOndene-i ,4'-piperidinel-
1 '-carboxylate

(3S)-1 '-(Tert-butoxycarbonyl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxylic acid (300mg, 0.91 mmol), triethylamine (506μL, 3.64mmol), acethydrazide (67mg, 0.91 mmol) and 2-chloro-1 ,3-dimethyl-2-imidazolinium tetrafluoroborate (598mg, 2.71 mmol) were combined in dichloromethane and stirred at room temperature for three days. 2-Chloro-1 ,3-dimethyl-2- imidazolinium tetrafluoroborate (200mg, 0.91 mmol) was added and the reaction mixture heated at 5O0C overnight. The reaction mixture was washed with water, dried over magnesium sulphate, filtered and evaporated in vacuo. The material was purified by column chromatography using an ISCO® silica cartridge eluting a linear gradient from 0-70% ethyl acetate/pentane. The product was isolated as an oily solid (80mg, 0.2mmoi, 24%). 1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 9H), 1.5-1.8 (m, 3H), 1.9-2.0 (m, 1 H), 2.4 (m, 1 H), 2.5 (s, 3H), 2.7 (dd, 1 H), 2.9 (m, 2H), 4.1 (m, 2H), 4.7 (t, 1 H), 7.2 (m, 3H), 7.3 (m, 1 H). LRMS m/z (APCI) 370 [MH]+.
Preparation 10 (3S)-3-(5-methyl-1.3,4-oxadiazol-2-vD-2,3-dihvdrospirorindene-1 ,4'-piperidinel

The protected amine of Preparation 9 (80mg, 0.2mmol) was dissolved in dichloromethane (5ml), trifluoroacetic acid (300μL) was added and the reaction mixture stirred at room temperature for 3 hours. The solvent was evaporated in vacuo and the residue extracted from saturated sodium hydrogen carbonate into dichloromethane. The combined organics were dried over magnesium sulphate, filtered and evaporated in vacuo (50mg, 0.185mmol, 93%). 1H-NMR (CD3OD, 400MHz): δ 1.65 (t, 2H), 1.75 (t, 1 H), 2.1 (t, 1 H), 2.4 (dd, 1 H), 2.5 (s, 3H), 2.8 (dd, 1 H), 3.9 (q, 2H), 3.1 (m, 2H), 4.75 (t, 1 H), 7.2 (m, 2H), 7.3 (s, 2H). LRMS m/z (APCI) 270 [MHf.
Preparation 11
Tert-butyl OSVS-drd ^-Σ-hvdroxy-i-methylethvnaminolcarbonvD-Z.S-dihvdro-IΗ- spirgJindene-1.4'-Diperidine1-1 '-carboxylate

Preparation 12 Tert-butyl (3S)-3-(4-methyl-1.3-oxazol-2-ylV2.3-dihvdro-1 'H-spiroHndene-i .4'-piperidinel-1 '- carboxylate

The alcohol of Preparation 11 (200mg, 0.51 mmol) was dissolved in dichloromethane (5ml), 1 ,1 ,1-triacetoxy-1 ,1-dihydro-1 ,2-benziodoxol-3(1 H)-one (327mg, 0.77mmo!) was added and the reaction mixture stirred at room temperature overnight. The reaction mixture was quenched with saturated aqueous sodium thiosulphate solution (2ml), saturated sodium hydrogen carbonate (2ml) and ether (5ml) then left to stir at room temperature for 30 minutes. The reaction mixture was extracted from water into dichloromethane, dried over magnesium
sulphate, filtered and evaporated to yield the crude aldehyde as a white solid (200mg). The material was dissolved in dichloromethane (10ml). 2,6-Ditbutylpyridine (169μL, 0.73mmol), dibromotetrachloroethane (238mg, 0.73mmol) and triphenylphosphine (191mg, 0.73mmol) were added and the reaction mixture stirred at room temperature for 1 hour before the addition of 1 ,8-diazabicyclo(5.4.0)undec-7-ene (108μL, 0.73mmol). The reaction mixture was stirred at room temperature overnight. The mixture was extracted from water into dichloromethane, dried over magnesium sulphate, filtered and evaporated in vacuo. The crude product was purified by column chromatography using an ISCO® silica cartridge eluting with a linear gradient from 0-100% ethyl acetate / pentane. The product was isolated as a colourless oil (80mg, 0.21 mmol, 36%).
1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 9H), 1.5-1.7 (m, 3H), 2.0 (m, 1 H), 2.15 (s, 3H), 2.4 (dd, 1 H), 2.7 (dd, 1 H), 3.0 (m, 2H), 4.1 (bs, 2H), 4.6 (t, 1 H), 7.2 (m, 4H), 7.3 (s, 1 H). LRMS m/z (APCI) SIS [M- WyIH]+.
Preparation 13
(3S)-3-(4-Methyl-1 ,3-oxazol-2-vD-2,3-dihvdrospirorindene-1 ,4'-piperidinel

The protected amine of Preparation 12 (80mg, 0.21 mmol) was dissolved in dichloromethane (5ml), trifluoroacetic acid (0.5ml) was added and the reaction mixture stirred at room temperature overnight. The reaction mixture was extracted from saturated sodium hydrogen carbonate into dichloromethane, dried over magnesium sulphate, filtered and evaporated in vacuo. The product was isolated as a colourless gum (40mg, 0.15mmol, 71 %). 1H-NMR (CD3OD, 400MHz): δ 1.6-1.8 (m, 3H), 2.05 (dd, 1 H), 2.1 (s, 3H), 2.3 (dd, 1 H), 2.7-2.9 (m, 3H), 3.1 (m, 2H), 4.6 (t, 1 H), 7.1 (d, 1 H), 7.2 (m, 1 H), 7.3 (s, 2H), 7.55 (s, 1 H). LRMS m/z (APCI) 269 [MH]+.
Preparation 14 Ethyl 1'-methylspirori-benzofuran-3,4'-piperidinel-2-carboxylate

Ethyl 1 '-methyl-1'H-spiro[1-benzofuran-3,4'-pyridine]-2-carboxylate [described in J. Org. Chem. 1983, 48 (18), 3061] (9.4g, 34.7mmol) was dissolved in ethanol (250ml), platinum
oxide (1g) was added and the reaction mixture stirred under 3 atmospheres of hydrogen at room temperature overnight. The reaction mixture was filtered through Arbocel® washing with ethanol then dichloromethane. The combined filtrates were evaporated in vacuo to yield an orange oil (8.9g, 32.3mmol, 94%).
1H-NMR (CDCI3, 400MHz): δ 1.3 (t, 3H), 1.8 (m, 1 H), 2.0-2.2 (m, 3H), 2.4-2.6 (m, 5H), 2.9 (m, 2H), 4.2 (m, 2H), 4.95 (s, 1 H), 6.9 (q,.2H), 7.2 (t, 2H). LRMS m/z (APCI) 276 [MH]+.
Preparation 15
Ethyl spirori-benzofuran-3.4'-piperidinel-2-carboxylate

The methyl piperidine of Preparation 14 (53mg, 0.19mmol) was dissolved in 1 ,2- dichloroethane (2ml) and 1-chloroethylchloroformate (41 μL, 0.38mmol) was added at O0C. The reaction mixture was heated to reflux for 10 minutes before the addition of 1 ,8-bis- (dimethylamino)naphthalene (40mg, 0.19mmol). The reaction mixture was heated at reflux for 1 hour, 1-chloroethylchloroformate (20μL) was added and the reaction mixture heated for a further 2 hours. The reaction mixture was diluted with dichloromethane (5ml) and washed with 2N hydrochloric acid (1 OmI), dried over magnesium sulphate, filtered and evaporated in vacuo. The residue was dissolved in methanol (5ml) and the solution heated at reflux for 30 minutes. The solvent was evaporated in vacuo to yield the title compound as a pale beige foam (67mg).
1H-NMR (dβ-DMSO, 400MHz): δ 1.2 (t, 3H), 1.7 (m, 1 H), 1.9 (m, 2H), 2.2 (m, 1 H), 3.0 (m, 1 H), 3.3 (m, 3H), 4.2 (q, 2H), 5.3 (s, 1 H), 6.9 (m, 2H), 7.2 (m, 2H). LRMS m/z (APCI) 262 [MH]+.
Preparation 16
1 '-Tert-butyl 2-ethyl 1 'H-spiroM -benzofuran-3,4'-piperidinel-1 ',2-dicarboxylate
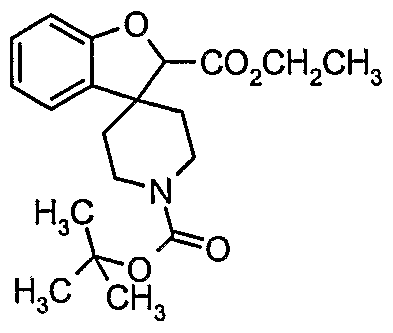
A solution of the piperidine of Preparation 15 (9.1 g, 30.6mmol) in dichloromethane (200ml) was treated with triethylamine (4.7ml, 33.8mmol) and the solution stirred at room temperature for 5 minutes before the addition of a solution of di-'butyl-dicarbonate (7.4g, 33.9mmol) in dichloromethane (20ml). The reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated in vacuo and the residue purified by column
chromatography over silica gel eluting with dichloromethane to 99:1 dichloromethane: methanol. The title compound was obtained as a pale yellow oil (8.2g, 22.7mmol, 74%). 1H-NMR (CDCI3, 400MHz): δ 1.2 (t, 3H), 1.4 (s, 9H), 1.6 (m, 1 H), 1.8 (m, 3H), 3.4 (m, 2H), 3.6 (m, 1 H), 3.8 (m, 1 H), 4.2 (m, 2H), 4.9 (s, 1 H), 6.9 (m, 2H), 7.2 (m, 2H). LRMS m/z (APCI) 262 [M-BOCH]+.
Preparation 17
1 '-(Tert-butoxycarbonyDspiroH -benzofuran-3.4'-piperidinel-2-carboxylic acid
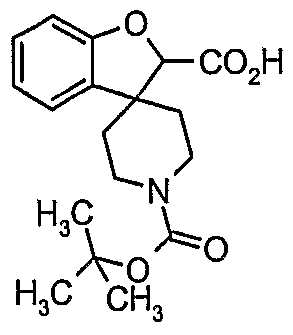
The ester of Preparation 16 (198mg, 0.57mmol) was dissolved in methanol (5ml), 1M aqueous sodium hydroxide (0.86ml, 0.86mmol) was added and the reaction mixture stirred at room temperature overnight. The reaction mixture was concentrated then diluted with water (10ml) then acidified to pH 1 with 2N hydrochloric acid. The acidic solution was extracted into ethyl acetate (2x15ml) and the combined organic phases dried over magnesium sulphate, filtered and evaporated in vacuo to yield a white foam (152mg, 0.45mmol, 80%). 1H-NMR (CDCI3, 400MHz): δ 1.45 (s, 9H), 1.8 (m, 3H), 2.0 (m, 1 H), 3.6 (m, 3H), 3.8 (m, 1 H), 4.95 (s, 1 H), 7.0 (m, 2H), 7.2 (m, 2H). LRMS m/z (APCI) 234 [M-BOCH]+ The racemic acid was recrystallised with (S)-methylbenzylamine from toluene to yield (2S)-1'- (tert-butoxycarbonyl)spiro[1 -benzofuran-3,4'-piperidine]-2-carboxylic acid.
The racemic acid was recrystallised with (R)-methylbenzylamine from toluene to yield (2R)-I1- (tert-butoxycarbonyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxylic acid.
Preparation 18 1 '-Tert-butyl 2-methyl 1 'H-spiroH -benzofuran-3,4'-piperidinel-1 ',2-dicarboxylate

The racemic acid of Preparation 17 (1.13g, 3.41mmol), 1-hydroxybenzotriazole monohydrate (453mg, 3.37mmol) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide HCI (654mg, 3.4mmol) were combined in dichloromethane (12ml) and stirred at room temperature for 30 minutes before the addition of methanol (10ml). The reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated in vacuo and the residue
redissolved in ethyl acetate (50ml) and washed with saturated aqueous sodium hydrogen carbonate (3x40ml). The organic phase was dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane: methanol: 0.880 ammonia (95:5:0.5). The title compound was obtained as an oil (809mg, 3.2mmol, 68%).
1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 9H), 1.6-1.9 (m, 4H), 3.4 (m, 2H), 3.6 (m, 1 H), 3.75 (s, 3H), 3.8 (m, 1 H), 4.9 (s, 1 H), 6.9 (m, 2H), 7.2 (m, 2H). LRMS m/z (APCI) 348 [MH]+.
Preparation 19 Methyl spiroH -benzofuran-3.4'-piperidinel-2-carboxylate

The title compound was obtained in quantitative yield from the protected piperidine of Preparation 18, following a similar procedure to that described in Preparation 21. 1H-NMR (CDCI3, 400MHz): δ 2.0 (m, 1 H), 2.3 (m, 3H), 3.2-3.6 (m, 4H), 3.8 (s, 3H), 4.9 (d, 1 H), 6.9 (m, 2H), 7.2 (m, 2H), 9.9 (bs, 1 H). LRMS m/z (APCI) 248 [MH]+.
Preparation 20 ■
Tert-butyl (2R)-2-(r(3-chlorobenzyl)amino1carbonyl|-1 'H-spiroH -benzofuran-3,4'-piperidine1-1 '- carboxylate

(2f?)-1 '-(Tert-butoxycarbonyl)spiro[1 -benzofuran-3,4'-piperidine]-2-carboxylic acid of Preparation 17 (107mg, 0.323mmol), 1-hydroxybenzotriazole monohydrate (45mg, 0.323mmoi) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide HCI (67mg, 0.366mmol) and 3-chlorobenzylamine (400μL, 3.33mmol) were combined in dimethylformamide (3ml) and stirred at room temperature overnight. The reaction mixture was concentrated in vacuo and the residue extracted from saturated aqueous sodium hydrogen carbonate (20ml) into dichloromethane (3x5ml). The combined organic phases were dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography
on silica gel using ethyl acetate: pentane as the eluent. The title compound was obtained as an oil.
1H-NMR (CDCI3, 400MHz): δ 1.4 (s, 9H), 1.5 (m, 1 H), 1.75 (m, 2H), 2.0 (m, 1 H), 3.3 (m, 1 H), 3.6 (m, 2H), 3.7 (m, 1 H), 4.4 (m, 2H), 4.7 (s, 1 H), 6.75 (d, 1 H), 6.85 (t, 1 H), 7.1-7.3 (m, 6H). LRMS m/z (APCl) 455 [M]".
Preparation 21 (2f?VN-(3-chlorobenzyl)spirori-benzofuran-3.4'-piperidinel-2-carboxamide

The protected piperidine of Preparation 20 (163mg, 0.35mmol) was dissolved in dichloromethane (4ml), trifluoroacetic acid (1ml) was added and the reaction mixture stirred at room temperature for 1 hour. The reaction mixture was concentrated in vacuo and the residue azeotroped with toluene. The crude product was extracted from 2M sodium hydroxide into dichloromethane, dried over magnesium sulphate, filtered and evaporated. The material was purified by column chromatography using an ISCO® silica cartridge, eluting with dichloromethane/methanol/0.880 ammonia (80:20:5). The title compound was obtained as an oil (85mg, 0.24mmol, 73%). 1H-NMR (CDCI3, 400MHz): δ 1.6 (m, 1 H), 1.75 (m, 1 H), 1.85 (m, 1 H), 2.1 (m, 3H), 2.9 (m, 1 H), 3.0 (m, 1 H), 3.1-3.2 (m, 2H), 4.4 (d, 2H), 4.75 (s, 1 H), 6.8 (d, 1 H), 7.0 (t, 2H), 7.2 (m, 4H), 7.4 (d, 1 H). LRMS m/z (APCI) 357 [MH]+.
Preparation 22
Tert-butyl (2fl)-2-f(dimethylamino)carbonyr|-1 'H-spiroH -benzofuran-3,4'-piperidinel-1 '- carboxylate

The title compound was obtained as an oil in 81 % yield from (2R)-1 '-(tert- butoxycarbonyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxylic acid of Preparation 17 and
dimethylamine (2M in tetrahydrofuran), following a similar procedure to that described in Preparation 20. Dichloromethane was used as the reaction solvent.
1H-NMR (CDCI3, 400MHz): δ 1.4(s, 9H), 1.8 (m, 4H), 2.95 (s, 3H), 3.05 (s, 3H), 3.4 (m, 1 H), 3.6 (m, 3H), 5.2 (s, 1 H), 6.8 (d, 1 H), 6.85 (t, 1 H), 7.1 (t, 1 H), 7.2 (d, 1 H). LRMS m/z (APCI) 361 [MH]+.
Preparation 23 (2f?)-N,N-Dimethvlspirori-benzofuran-3.4'-piperidine1-2-carboxamide

The title compound was obtained as an oil in 91 % yield from the protected piperidine of Preparation 22, following a similar procedure to that described in Preparation 21. 1H-NMR (CDCI3, 400MHz): δ 1.9 (m, 4H), 2.2 (bs, 1 H), 2.8 (m, 1 H), 3.0 (s, 3H), 3.0 (m, 1 H), 3.1 (s, 3H), 3.1 (m, 2H), 5.2 (s, 1 H), 6.8 (d, 1 H), 6.9 (t, 1H), 7.2 (t, 1 H), 7.4 (d, 1H). LRMS m/z (APCI) 261 [MHf.
Preparation 24 1 '-Tert-butyl 2-methyl (2R)- 1 'H-spirof 1 -benzofuran-3.4'-piperidinel-1 ',2-dicarboxvlate

The title compound was obtained as an oil in 100% yield from (2R)-1 '-(tert- butoxycarbonyOspiroti-benzofuran-S^'-piperidine^-carboxylic acid of Preparation 17 and methanol, following a similar procedure to that described in Preparation 20. Dichloromethane/methanol/0.880 ammonia was used as the column solvent. 1H-NMR (CDCI3, 400MHz): δ 1.4 (s, 9H), 1.6 (m, 1 H), 1.7-1.9 (m, 3H), 3.4 (m, 1 H), 3.5 (m, 1 H), 3.65 (m, 1H), 3.7 (s, 3H), 3.8 (m, 1H), 4.9 (s, 1H), 6.9 (t, 2H), 7.2 (q, 2H). LRMS m/z (APCI) 292 [MH]+.
Preparation 25 Methyl (2f?)-spiroπ -benzofuran-3,4'-piperidine1-2-carboxylate

The title compound was obtained as an oil in 90% yield from the protected piperidine of Preparation 24, following a similar procedure to that described in Preparation 21. 1H-NMR (CDCI3, 400MHz): δ 1.65 (m, 1 H), 1.8 (m, 1 H), 1.9 (m, 2H), 2.2 (bs, 1 H), 2.9 (m, 1 H), 3.0 (m, 2H), 3.2 (m, 1 H), 3.8 (s, 3H), 4.9 (s, 1 H), 6.9 (t, 2H), 7.2 (t, 1 H), 7.3 (t, 1 H). LRMS m/z (APCI) 248 [MH]+.
Preparation 26
Tert-butyl (2S)-2-{r(3-chlorobenzyl)aminolcarbonyl)-1'H-spirori-benzofuran-3.4'-piperidinel-1'- carboxylate

The title compound was obtained as an oil in 96% yield from (2S)-1 '-(tert- butoxycarbonyOspiroti-benzofuran-S^'-piperidinel^-carboxylic acid of Preparation 17 and 3- chlorobenzylamine following the procedure described in Preparation 20. Dichloromethane was used as the reaction solvent.
1H-NMR (CDCI3, 400MHz): δ 1.4 (s, 9H), 1.6 (m, 1 H), 1.8 (m, 2H), 2.1 (m, 1 H), 3.4 (m, 1 H), 3.6-3.8 (m, 3H), 4.4 (m, 2H), 4.8 (s, 1 H), 6.8 (d, 1 H), 6.9 (t, 1 H), 7.0 (t,' 1 H), 7.1-7.3 (m, 5H). LRMS m/z (APCI) 455 [M]".
Preparation 27 (2S)-N-(3-Chlorobenzvπsoirori-benzofuran-3,4'-piperidine1-2-carboxamide

The title compound was obtained as an oil in 99% yield from the protected piperidine of Preparation 26, following a similar procedure to that described in Preparation 21. 1H-NMR (CDCI3, 400MHz): δ 1.6 (m, 1 H), 1.75 (m, 1 H), 1.85 (m, 1 H), 2.1 (m, 1 H), 2.9 (m, 1 H), 3.0 (m, 1 H), 3.15 (m, 2H), 4.45 (d, 2H), 4.75 (s, 1 H), 6.8 (d, 1 H), 7.0 (m, 2H), 7.1-7.3 (m, 4H), 7.4 (d, 1 H). LRMS m/z (APCI) 357 [MH]+.
Preparation 28
Tert-butyl (2SV2-f(dimethylamino)carbonyll-1 'H-spiroH -benzofuran-3.4'-piperidine1-1 '- carboxylate

The title compound was obtained as an oil in 80% yield from (2S)-1 '-(tert- butoxycarbonyOspiroti-benzofuran-S^'-piperidineJ^-carboxylic acid of Preparation 17 and dimethylamine (2M in tetrahydrofuran), following a similar procedure to that described in Preparation 20. Dichloromethane was used as the reaction solvent.
1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 9H), 1.9 (m, 4H), 3.0 (s, 3H), 3.1 (s, 3H), 3.45 (m, 1 H), 3.6 (m, 2H), 3.7 (m, 1 H), 5.2 (s, 1 H), 6.9 (m, 2H), 7.2 (t, 1 H), 7.3 (d, 1 H). LRMS m/z (APCI) 305 [M-*butylH]+.
Preparation 29 (2SVN,N-dimethylspirof1-benzofuran-3,4'-pipericline1-2-carboxamide

The title compound was obtained as an oil in 100% yield from the protected piperidine of Preparation 28, following a similar procedure to that described in Preparation 21. 1H-NMR (CDCI3, 400MHz): δ 1.9 (m, 3H), 2.8 (m, 1H), 2.95 (m, 1 H), 3.0 (s, 3H), 3.1 (m, 1 H), 3.15 (s, 3H), 5.2 (s, 1 H), 6.85 (d, 1H), 6.9 (t, 1 H), 7.15 (t, 3H), 7.35 (d, 1 H). LRMS m/z (APCI) 261 [MH]+.
Preparation 30 1'-Tert-butyl 2-methyl (2S)-1'H-spirof1-benzofuran-3,4'-piperidine1-1',2-dicarboxylate

The title compound was obtained as an oil in 90% yield from (2S)-1'-(tert- butoxycarbonyl)spiro[1-benzofuran-3,4'-piperidine]-2-carboxyiic acid of Preparation 17 and methanol, following a similar procedure to that described in Preparation 20. 1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 9H), 1.7 (m, 1 H), 1.75-2.0 (m, 3H), 3.45 (m, 1 H), 3.5 (m, 1 H), 3.7 (m, 1 H), 3.8 (s, 3H), 3.85 (m, 1 H), 4.9 (s, 1 H), 6.9 (t, 2H), 7.2 (m, 2H). LRMS m/z (APCI) 292 [M-WyIH]+.
Preparation 31 Methyl (2S)-spirof1-benzofuran-3,4'-piperidine1-2-carboxylate

The title compound was obtained as an oil in 43% yield from the protected piperidine of Preparation 30, following a similar procedure to that described in Preparation 21.
1H-NMR (CDCI3, 400MHz): δ 1.6 (m, 1H), 1.8 (m, 1 H), 1.9 (m, 2H), 2.9 (m, 1 H), 3.0 (m, 2H), 3.2 (m, 1 H), 3.8 (s, 3H), 4.95 (s, 1 H), 6.9 (q, 2H), 7.2 (t, 1H), 7.3 (t, 1 H). LRMS m/z (APCI) 248 [MH]+.
Preparation 32 Ethyl 3-pyridin-4-yloxirane-2-carboxylate

4-Pyridinecarboxaldehyde (50.5g, 0.47mol) was added to a 21 % solution of sodium ethoxide in ethanol (305ml). Ethyl chloroacetate (87g, 0.7mol) was added dropwise over 30 minutes while cooling in ice. The reaction mixture was stirred at room temperature for 3 hours. The reaction mixture was poured into ice (1200ml) then extracted into ethyl acetate (2x800ml).
The solvent was evaporated in vacuo. The crude material was purified by column chromatography over silica gel using ethyl acetate/pentane as the eluent. (12.3g, O.OΘmol, 13%).
1H-NMR (CDCI3, 400MHz): δ 1.4 (t, 1H), 3.4 (s, 1H), 4.1 (s, 1H), 4.3 (m, 2H), 7.2 (d, 2H), 8.6
(m, 2H).
Preparation 33 Ethyl 2-hydroxy-3-pyridin-4-ylpropanoate

The epoxide of Preparation 32 (12.3g, 63.7mmol) was dissolved in ethyl acetate (70ml), 20% palladium hydroxide on charcoal (1.2g) was added and the reaction mixture stirred under 3.4 atmospheres of hydrogen at room temperature for 16 hours. The reaction mixture was filtered through Arbocel® and the solvent evaporated in vacuo to yield the title compound (9.9g, O.Oδmol, 80%).
1H-NMR (CDCi3, 400MHz): δ 1.3 (t, 3H), 2.9 (m, 1 H), 3.1 (m, 1 H), 4.2 (q, 2H), 4.4 (m, 1 H), 7.2 (d, 2H), 8.5 (m, 2H).
Preparation 34
Methyl 3-(1 -benzyl-1 ,2,3,6-tetrahvdropyridin-4-vD-2-hvdroxypropanoate

The alcohol of Preparation 33 (9.9g, 50.7mmol) and benzyl chloride (6.42ml, 55.7mmol) were combined in methanol (50ml) and heated at reflux for 20 hours. The solvent was evaporated in vacuo. The residue was redissolved in methanol (90ml) and cooled in ice. Sodium borohydride (3.07g, 81.1mmol) was added portionwise over 15 minutes. The reaction mixture was stirred at room temperature for 3 hours, water (20ml) was added and the methanol evaporated in vacuo. The remaining aqueous slurry was treated with 3% aqueous sodium hydrogen carbonate (30ml) then extracted into ethyl acetate (2x60ml). The organic phase was dried over sodium sulphate, filtered and evaporated. The crude material was purified by column chromatography over silica gel using dichloromethane : methanol as the eluent. The title compound was obtained as an oil (7.6g, 0.27mol, 55%).
1H-NMR (CDCI3, 400MHz): δ 2.2 (m, 2H), 2.4 (m, 1 H)1 2.5 (m, 1 H), 2.6 (bs, 2H), 2.7 (bs, 1 H), 3.0 (bs, 2H), 3.6 (bs, 2H), 3.8 (s, 3H), 4.4 (m, 1 H), 5.6 (s, 1 H), 7.3 (m, 5H). LRMS m/z (APCI) 275 [MH]+.
Preparation 35
Methyl 3-(1 -benzyl-1 ,2,3,6-tetrahvdropyridin-4-vD-2-(2-bromophenoxy)propanoate

A solution of diisopropyl azodicarboxylate (6.15ml, 31.74mmol) in toluene (15ml) was added to a solution of the alcohol of Preparation 34 (7.6g, 27.6mmol), triphenylphosphine (8.3g, 31.74mmol) and 2-bromophenol (5.01 g, 29mmol) in toluene (85ml) at O0C. The reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated in vacuo. Heptane: ether (1 :1 , 250ml) was added and the resultant precipitate removed by filtration. The filtrate was evaporated in vacuo to yield the crude product. The material was
purified by column chromatography over silica gel using ether/heptane as the eluent. (9.Og, 0.02mol, 76%).
1H-NMR (CDCi3, 400MHz): δ 2.3 (m, 2H), 2.5-2.7 (m, 4H), 3.0 (m, 2H), 3.6 (s, 2H), 3.7 (s, 3H), 4.8 (m, 1 H), 5.6 (s, 1 H), 6.75 (m, 1 H), 6.8 (m, 1 H), 7.3 (m, 6H), 7.6 (m, 1 H).
Preparation 36
Methyl I'-benzyl^.S-dihvdrospirorchromene^'-piperidinel-Σ-carboxylate

Tributyltin hydride (2Og, 62.7mmol) was added to a solution of the alkene of Preparation 35 (9g, 20.9mmol) in toluene (350ml). The reaction mixture was heated to reflux and 2,2'-azobis- (2-methylpropionitrile) (666mg, 3.97mmol) added. The reaction mixture was heated at reflux for 4.5 hours. The reaction mixture was concentrated in vacuo. Ether (300ml) and saturated aqueous potassium fluoride (300ml) were added and the mixture stirred overnight. The phases were separated and the organic phase evaporated in vacuo. The crude material was purified by column chromatography over silica gel eluting with 1-3% dichloromethane: methanol as the eluent. (550mg, 0.001 mol, 7%).
1H-NMR (CDCI3, 400MHz): δ 1.4 (m, 1 H), 1.8 (m, 2H), 2.0 (m, 1 H), 2.2-2.4 (m, 3H), 2.6 (m, 1 H), 2.8 (m, 2H), 3.6 (s, 2H), 3.8 (s, 3H), 4.6 (d, 1 H), 7.0 (m, 2H), 7.1 (m, 1 H), 7.4 (m, 6H). LRMS m/z (APCI) 352 [MH]+.
Preparation 37 Methyl 2,3-dihvdrospirorchromene-4,4'-piperidine1-2-carboxylate

The benzylamine of Preparation 36 (0.15g, 0.43mmol) was dissolved in ethanol (3ml), palladium hydroxide (15mg) and ammonium formate (134mg, 2.15mmol) were added and the reaction mixture heated at reflux for 2 hours. The reaction mixture was filtered through Arbocel® and the solvent evaporated in vacuo. The crude material was purified by column chromatography over silica gel using dichloromethane/methanol/0.880 ammonia (90:10:1) as the eluent (100mg, 0.38mmol, 89%).
1H-NMR (CDCI3, 400MHz): δ 1.4 (m, 1 H), 1.8 (m, 3H)1 2.2 (m, 1 H), 2.7 (d, 1 H), 2.8 (m, 1 H), 3.0 (m, 3H), 3.8 (s, 3H), 4.6 (d, 1 H), 7.0 (m, 2H), 7.1 (m, 1 H), 7.4 (d, 1 H).
Preparations 38-43

(3S)-1'-(Tert-butoxycarbonyl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxylic acid (1eq), 1-hydroxybenzotriazole monohydrate (1.03eq) and 1-(3-dimethylaminopropyl)-3- ethylcarbodiimide HCI (1.2eq), N-methylmorpholine (1.5eq) and the appropriate amine/alcohol (1-2 eq) were combined in dichloromethane and stirred at room temperature overnight. The reaction mixture was washed with 3% aqueous sodium hydrogen carbonate, dried over sodium sulphate, filterered and evaporated in vacuo. The crude material was purified by column chromatography on silica gel using 25-75% ethyl acetate/pentane as the eluent.


I = N-methylmorpholine was omitted from the reaction.
J = An excess of methanol was used.
K = Dichloromethane was used as the column eluent.
L = Dichloromethane/methanol/0.880 ammonia (95:5:0.5) was used as the column eluent.
Preparations 44-51



M = Ethyl acetate and 10% aqueous sodium carbonate were used for the work up. N = The crude material was purified by column chromatography over silica gel using dichloromethane/methanol/0.880 ammonia as the eluent.
Preparation 52 (1-Benzyl-1 ,2,3,6-tetrahvdropvridin-4-vPmethanol

4-(Hydroxymethyl)pyridine (517mg, 4.74mmol) was dissolved in methanol (5ml), benzyl chloride (550μL, 4.78mmol) was added and the reaction mixture heated at reflux for 24 hours. The solvent was evaporated and the residue redissolved in methanol (4ml). The solution was cooled in ice and sodium borohydride (260mg, 6.70mmol) added portionwise. The reaction mixture was stirred at room temperature for 3 hours. The solvent was evaporated in vacuo and the residue dissolved in ethyl acetate (30ml) and washed with 2N sodium hydroxide (3x 30ml). The organic phase was dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography using an ISCO® silica cartridge eluting with ethyl acetate/methanol. The product was obtained as a yellow oil (778mg, 3.8mmol, 81%). 1H-NMR (CDCI3, 400MHz): δ 2.2 (bs, 2H), 2.6 (t, 2H), 3.0 (s, 2H), 3.60 (s, 2H), 4.05 (s, 2H), 5.65 (s, 1 H), 7.2-7.4 (m, 5H). LRMS m/z (APCI) 204 [MH]+.
Preparation 53 2-r(1-Benzvl-1 ,2.3,6-tetrahvdropvridin-4-v0methoxvl-3-bromobenzonitrile

The alcohol of Preparation 52 (389mg, 1.92mmol) and triphenylphosphine (532mg, 2.3mmol) were dissolved in tetrahydrofuran (5ml) and cooled in ice. 3-Bromo-2-hydroxy-benzonitrile
[described in J. Org. Chem. 1997, 62, 4504-4506] (330mg, 1.67mmol) was added followed by diisopropyl azodicarboxylate (360μl_, 1.8mmol). The reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated in vacuo. The crude material was purified by column chromatography over silica gel using dichloromethane: methanol (95:5) as the eluent. The title compound was obtained as a yellow, oil (377mg, 0.98mmol,
1H-NMR (CDCI3, 400MHz): δ 2.4 (bs, 2H), 2.65 (t, 2H), 3.0 (s, 2H), 3.6 (s, 2H)1 4.6 (s, 2H), 5.8 (s, 1 H), 7.05 (t, 1 H), 7.2-7.4 (m, 5H), 7.5 (d, 1 H), 7.75 (d, 1 H). LRMS m/z (APCI) 383 [MH]+.
Preparation 54
1'-Benzylspirof1-benzofuran-3,4'-piperidinel-7-carbonitrile

Tributyltin hydride (1.1ml, 4.09mmol) was added to a solution of the alkene of Preparation 53 (377mg, 0.984mmol) in toluene (6ml). The reaction mixture was heated to reflux and 2,2'- azobis-(2-methylpropionitriIe) (32mg, 0.197mmol) added. The reaction mixture was heated at reflux for 4 hours. The reaction mixture was concentrated in vacuo. The residue was redissolved in ether and saturated aqueous potassium fluoride (1 :1) and the mixture stirred overnight. The mixture was filtered and the phases separated The organic phase was evaporated in vacuo. The crude material was purified by column chromatography using an ISCO® silica cartridge eluting with ethyl acetate/ pentane (1 :1). The title compound was obtained as an oil (206mg, 0.68mmol, 69%).
1H-NMR (CDCI3, 400MHz): δ 1.75 (d, 2H), 1.9-2.1 (m, 4H), 2.9 (d, 2H), 3.55 (s, 2H), 4.5 (s, 2H), 6.9 (t, 1 H), 7.2-7.4 (m, 7H). LRMS m/z (APCI) 305 [MH]+.
Preparation 55 Spirori-benzofuran-3,4'-piperidinel-7-carbonitrile

The benzyl piperidine of Preparation 54 (204mg, 0.671 mmol) was dissolved in 1 ,2- dichloroethane (2ml) and a solution of 1-chloroethylchIoroformate (73μL, 0.677mmol) in 1 ,2- dichloroethane (2ml) was added at O0C. The reaction mixture was stirred for 30 minutes
before the addition of triethylamine (93μL, 0.671 mmol). The reaction mixture was then heated at reflux for 2 hours. The solvent was evaporated in vacuo and the residue redissolved in methanol (3ml) and the solution heated at reflux for 45minutes. The solvent was evaporated in vacuo and the residue dissolved in dichloromethane. The remaining solid removed by filtration. The filtrate was evaporated to yield the title compound as a hydrochloride salt (63mg, 0.25mmol, 49%).
1H-NMR (CD3OD, 400MHz): δ 2.0 (d, 2H), 2.2 (m, 2H), 3.2 (m, 2H), 3.5 (m, 2H), 4.7 (s, 2H), 7.05 (t, 1 H), 7.45 (d, 1 H), 7.55 (d, 1 H). LRMS m/z (APCI) 215 [MH]+.
Preparation 56
4-f(1-Benzyl-1.2,3.6-tetrahvdropyridin-4-v0methoxyl-3-bromobenzonitrile

The title compound was obtained as a brown oil in 31% yield from 3-bromo-4- hydroxybenzonitrile and the alcohol of Preparation 52 following a similar procedure to that described in Preparation 53.
1H-NMR (CDCI3, 400MHz): δ 2.29 (bm, 2H), 2.69 (bm, 2H), 3.09 (bm, 2H), 3.66 (bs, 2H), 4.54 (bs, 2H), 5.82 (bm, 1 H), 6.91 (d, 1 H), 7.24-7.41 (m, 5H), 7.55 (m, 1 H), 7.81 (s, 1 H). LRMS ' m/z (ESI) 385 [MH]+
Preparation 57
1 '-Benzylspiroπ -benzofuran-3,4'-piperidinel-5-carbonitrile
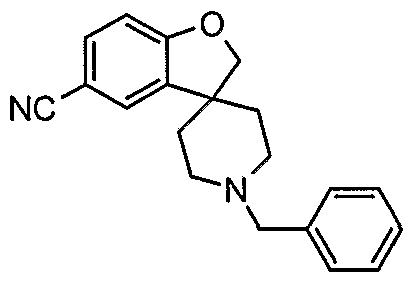
The title compound was obtained as a white solid in 90% yield from the alkene of Preparation 56 following a similar procedure to that described in Preparation 54.
1H-NMR (CDCI3, 400MHz): δ 1.69-1.74 (m, 2H), 1.91-2.10 (m, 4H), 2.89 (bm, 2H), 3.54 (bs, 2H), 4.44 (s, 2H), 6.81 (d, 1 H), 7.23-7.42 (m, 6H), 7.44 (dd, 1 H). LRMS m/z (APCI) 305 [MH]+.
Preparation 58 Spirori-benzofuran-3.4'-piperidine1-5-carbonitrile

The title compound was obtained in 65% yield from the benzyl piperidine of Preparation 57 following a similar procedure to that described in Preparation 55. The compound was isolated by trituration with ether.
1H-NMR (CD3OD, 400MHz): 52.0 (m, 2H), 2.1 (m, 2H), 3.2 (t, 2H), 3.5 (m, 4H), 6.95 (d, 1 H), 7.6 (d, 1 H), 7.65 (s, 1 H). LRMS m/z (APCI) 215 [MH]+.
Preparation 59 1 '-Tert-butyl 3-ethyl 1 H3H-spiror2-benzofuran-1 ,4'-piperidinel-1 ',3-dicarboxylate

Ethyl 1'-benzyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3-carboxylate [described in J. Med.
Chem. 2002, 45, 438] (20.Og, 0.056mol) was dissolved in ethanol (350ml) and 10% palladium on carbon (1.5g) added. The reaction mixture was stirred at 4O0C under hydrogen for 6 hours. 10% Palladium on carbon (1.5g) was added and the reaction mixture returned to the reaction conditions for 18 hours. The reaction mixture was filtered through Celite® and the combined filtrates concentrated in vacuo. The crude material was redissolved in dichloromethane
(350ml). Triethylamine (7.2ml, 0.0519mol) was added followed by di-'butyl dicarbonate and the reaction mixture stirred at room temperature for 3 hours. The reaction mixture was concentrated and the residue purified by column chromatography over silica gel using 10- 20% ethyl acetate: pentane as the eluent. The title compound was obtained as an oil (14.08g, 0.039mol, 69%).
1H-NMR (CDCI3, 400MHz): δ 1.3 (t, 3H), 1.5 (s, 9H), 1.7-2.0 (m, 4H), 3.2-3.4 (m, 2H), 4.1 (m, 2H), 4.2 (q, 2H), 5.7 (s, 1 H), 7.1 (d, 1H), 7.3 (m, 2H), 7.4 (d, 1 H). LRMS m/z (APCI) 362 [MH]+.
Preparation 60 1 '-(Tert-butoxvcarbonyl)-3H-spiror2-benzofuran-1 Λ'-Piperidinel-S-carboxvlic acid

The ester of Preparation 59 (11.2g, 0.031 mmol) was dissolved in ethanol (10ml) and a solution of sodium hydroxide (2.48g, 0.062mmol) in water (40ml) added. The reaction mixture was heated at reflux for two hours. 1 N hydrochloric acid was added until pH 3-4 was reached. The mixture was extracted into ethyl acetate and the combined extracts dried over magnesium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography over silica gel eluting with 40-50% ethyl acetate:pentane. The title compound was obtained as a white solid (7.58g, 0.02mmol, 77%).
1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 9H), 1.7 (m, 1 H), 1.8-2.0 (m, 3H), 3.1-3.3 (m, 2H), 4.1 (m, 2H), 5.6 (s, 1 H), 7.1 (d, 1 H), 7.35 (m, 2H), 7.5 (d, 1 H). LRMS m/z (APCI) 334 [MH]+ Recrystallisation of the racemic acid with (f?)-(+)-α-methylbenzylamine from toluene yielded one enantiomer of the title compound.
Recrystallisation of the racemic acid with (S)-(-)-α-methylbenzylamine from toluene yielded the other enantiomer of the title compound.
Preparation 61
Methyl 3H-spiror2-benzofuran-1 ,4'-piperidine1-3-carboxylate

The acid of Preparation 60 (1.51g, 4.55mmol), 1-hydroxybenzotriazole monohydrate (619mg, 4.58mmol) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide HCI (885mg, 4.62mmol) were combined in dichloromethane (10ml). Methanol (10ml) was added and stirred at room temperature for 72 hours. The reaction mixture was concentrated in vacuo then redissolved in ethyl acetate (20ml). The solution was washed with 3% aqueous sodium hydrogen carbonate (2x1 OmI), dried over sodium sulphate, filtered and evaporated in vacuo. The crude material
was redissolved in dichloromethane (8ml) and trifluoroacetic acid (2ml) added. The solution was stirred at room temperature for 45 minutes. The reaction mixture was concentrated in vacuo and the residue redissolved in dichloromethane, washed with 2N sodium hydroxide, dried over sodium sulphate, filtered and evaporated. The crude material was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane: methanol: ammonia (90:10:1 ). The title compound was obtained as a colourless oil (396mg, 1.2mmol, 34%).
1H-NMR (CDCI3, 400MHz): δ 1.5 (m, 1 H), 1.8 (m, 2H), 2.8-3.1 (m, 3H), 3.2 (bs, 2H), 3.6 (s, 3H), 5.5 (s, 1 H), 7.0 (d, 1 H), 7.2 (m, 2H), 7.25 (m, 1 H). LRMS m/z (ESI) 248 [MH]+.
Preparation 62 N.N-Diethyl-3H-spiror2-benzofuran-1 ,4'-piperidine1-3-carboxamide

The enantiomer of Preparation 60 (derived from resolution with with (R)-(+)-α-methyl- benzylamine) (0.1g, 0.3mmol), 1-hydroxybenzotriazole monohydrate (47mg, 0.309mmol), 1- (3-dimethylaminopropyl)-3-ethylcarbodiimide HCI (69mg, 0.36mmol) and diethylamine (44mg, O.θmmol) were combined in dichloromethane (3ml) and stirred at room temperature for 16 hours. The reaction mixture was concentrated in vacuo then extracted from 3% aqueous sodium hydrogen carbonate (20ml) into ethyl acetate (2x20ml), dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography over silica gel eluting with ethyl acetate/pentane (1 :1 ). The amide was redissolved in dichloromethane (1.5ml). Trifluoroacetic acid (0.5ml) added and the solution was stirred at room temperature for 4 hours. The reaction mixture was concentrated in vacuo and the residue extracted from 2N sodium hydroxide (15ml) into dichloromethane (3x15ml), dried over sodium sulphate, filtered and evaporated. The crude material was purified by column chromatography over silica gel eluting with dichloromethane/methanol/0.880 ammonia (90:10:1). The title compound, as a single enantiomer, was obtained as a foam in quantitative yield. 1H-NMR (CDCI3, 400MHz): δ 1.1 (t, 3H), 1.3 (t, 3H), 1.8 (m, 1 H), 1.9 (m, 3H), 2.6 (bs, 1 H), 3.1 (m, 4H), 3.4 (m, 2H), 3.6 (m, 2H), 5.8 (s, 1 H), 7.2 (m, 1 H), 7.3 (m, 3H). LRMS m/z (ESI) 289 [MH]+.
Preparation 63 (3f?)-N,N-Diethyl-3H-spiror2-benzofuran-1 ,4'-piperidine1-3-carboxamide

The title compound was obtained, as a single enantiomer, in quantitative yield from the enantiomer of Preparation 60 (derived from resolution with (S)-(-)-α-methylbenzylamine) and diethylamine following a similar procedure to that described in Preparation 62. 1H-NMR (CDCI3, 400MHz): δ 1.1 (t, 3H), 1.3 (t, 3H), 1.8 (m, 1 H), 1.9 (m, 3H), 2.6 (bs, 1 H), 3.1 (m, 4H), 3.4 (m, 2H), 3.6 (m, 2H), 5.8 (s, 1 H), 7.2 (m, 1 H), 7.3 (m, 3H). LRMS m/z (APCI) 289 [MH]+.
Preparation 64
1-Benzyl-4-r(2-bromo-5-fluorophenoxy)methyl]-1.2,3,6-tetrahvdropyridine

The title compound was obtained as a brown oil in 74% yield from 5-fluoro-2-bromophenol and the alcohol of Preparation 52 following a similar procedure to that described in Preparation 53. 1H-NMR (CDCI3, 400MHz): δ 2.27 (bm, 2H), 2.65 (t, 2H), 3.05 (bm, 2H), 3.61 (s, 2H), 4.44 (s, 2H), 5.82 (m, 1 H), 6.56 (m, 1 H), 6.62 (dd, 1 H), 7.23-7.38 (m, 5H), 7.45 (dd, 1 H). LRMS m/z (ESI) 378 [MH]+.
Preparation 65 1 '-Benzyl-6-f luorospiroH -benzofuran-3.4'-piperidinel

The title compound was obtained as a yellow oil in 48% yield from the alkene of Preparation 64 following a similar procedure to that described in Preparation 54.
1H-NMR (CDCI3, 400MHz): δ 1.65-1.74 (m, 2H), 1.88-2.08 (m, 4H), 2.89-2.93 (m, 2H), 3.46- 3.58 (m, 2H), 4.39 (s, 2H), 6.48 (m, 1 H), 6.55 (m, 1 H), 7.03 (m, 1 H), 7.24-7.39 (m, 5H). LRMS m/z (APCI) 298 [MH]+.
Preparation 66 6-Fluorospirori-benzofuran-3.4'-piperidinel

The benzyl piperidine of Preparation 65 (1.09g, 3.67mmol) was dissolved in ethanol (25ml). 10% Palladium on carbon (750mg) was added followed by ammonium formate (1.15g,
18.3mmol). The reaction mixture was heated at reflux for 1.25 hours. The reaction mixture was allowed to cool to room temperature and the catalyst removed by filtration through Arbocel®. The solids were washed with ethanol and the combined filtrates evaporated in vacuo. The crude material was purified by column chromatography over silica gel using dichloromethane: methanol: 0.880 ammonia (90:10:1) as the eluent. The title compound was obtained as a white solid (584mg, 2.82mmol, 77%).
1H-NMR (CDCI3, 400MHz): δ (m, 2H), 1.75-1.88 (m, 3H), 2.68 (m, 2H), 3.10 (m, 2H), 4.43 (s, 2H), 6.50 (m, 1 H), 6.56 (m, 1 H), 7.03 (m, 1 H). LRMS m/z (APCI) 208 [MH]+
Preparation 67
1 '-(Tert-butoxycarbonyl)-3-methyl-2,3-dihvdrospirorindene-1 ,4'-piperidinel-3-carboxylic acid

1'-(Tert-butoxycarbonyl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxylic acid (500mg, 1.5mmol) was dissolved in tetrahydrofuran (5ml) and cooled to O0C, lithium diisopropylamide (1.5M in hexanes, 3ml, 4.5mmol) was added and the reaction mixture allowed to warm to room temperature and stirred for 90 minutes before the addition of iodomethane (638mg, 4.5mmol). The reaction mixture was stirred at room temperature overnight. The reaction was quenched with water, acidified with 2M hydrochloric acid and extracted into dichloromethane. The organic phase was washed with brine, dried over magnesium sulphate, filtered and evaporated in vacuo. The crude product was purified by column chromatography using an lSCO® silica cartridge eluting with dichloromethane - 90:10 dichloromethane/methanol. The product was isolated as a foam (439mg, 1.2mmol, 84%).
1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 10H), 1.6 (m, 5H), 1.8 (d, 1 H), 1.9 (m, 1 H), 2.9 (m, 3H), 4.1 (m, 2H), 7.1 (d, 1 H), 7.3 (m, 2H), 7.4 (d, 1 H).
Preparation 68 Tert-butyl 3-Kdimethylamino)carbonvπ-3-methyl-2,3-dihydro-1'H-spiro)'indene-1.4'-piperidiπe1-
1 '-carboxylate

The acid of Preparation 67 (380mg, 1.09mmol), dimethylamine (33% in ethanol (5ml) and O- benzotriazol-1-yl-N,N,N',N'-tetramethyluronium hexafluorophosphate (625mg, 1.64mmol) were combined in tetrahydrofuran. The reaction mixture was heated at 5O0C overnight. The reaction mixture was concentrated in vacuo and the residue partitioned between dichloromethane and water. The organic phase was washed with brine, dried over magnesium sulphate, filtered and evaporated in vacuo. The crude product was purified by column chromatography using an ISCO® silica cartridge eluting with 20-100% pentane/ethyl acetate. The product was isolated as a cream solid (320mg, 0.86mmol, 79%). 1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 10H), 1.55 (s, 3H), 1.65 (m, 1 H), 1.8 (m, 1 H), 1.9 (m, 1 H), 2.3 (q, 2H), 1.7 (m 7H), 3.0 (m, 1 H), 4.1 (m, 2H), 7.1 (m, 2H), 7.25 (m, 2H).
Preparation 69
N,N,3-Trimethyl-2,3-dihvdrospiroπndene-1 ,4'-piperidine1-3-carboxamide

The protected piperidine of Preparation 68 (320mg, 0.859mmol) was dissolved in dichloromethane (5ml), 4M hydrochloric acid in dioxane (5ml) was added and the reaction mixture stirred at room temperature for 2 hours. The solvent was concentrated in vacuo and the residue partitioned between 1 N sodium carbonate and dichloromethane. The organic phase was washed with brine, dried over sodium sulphate, filtered and evaporated in vacuo. The crude product was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane - 90:10:1 dichloromethane/methanol/ 0.880 ammonia. The product was isolated as a clear gum (140mg, 0.51 mmol, 60%).
1H-NMR (CD3OD, 400MHz): δ 1.5 (s, 3H), 1.6 (d, 1 H), 1.65 (d, 1 H), 1.8 (m, 2H), 2.8 (m, 8H), 3.0 (d, 2H), 7.0 (d, 1H), 7.3 (m, 3H). LRMS m/z (APCI) 273 [MH]+.
Preparation 70 2-Bromo-4-piperidin-1-ylpyridine

2,4-Dibromopyridine (50mg, 0.211mmol) and piperidine (90mg, 1.05mmol) were combined in isopropyl alcohol and heated at 750C for 72 hours. The reaction mixture was concentrated in vacuo and the residue purified by column chromatography over silica gel using ethyl acetate: pentane as the eluent. (45mg, 0.18mmol, 88%).
1H-NMR (CDCI3, 400MHz): δ 1.6 (m, 6H), 3.3 (m, 4H), 6.6 (d, 1 H), 6.8 (s,1H), 7.9 (d, 1H). LRMS m/z (ESI) 241 [MH]+.
Preparation 71
N-Benzyl-2-bromo-N-methylpyridin-4-amine
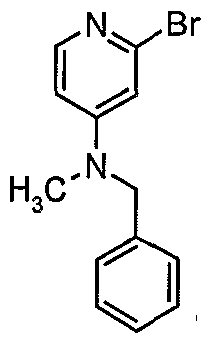
2,4-Dibromopyridine (400mg, 1.25mmol) and benzylmethylamine (760mg, 2.7mmol) were combined in ethanol and heated at reflux for 18 hours. The reaction mixture was concentrated in vacuo and the residue purified by column chromatography using an ISCO® silica cartridge eluting with 0-100% ethyl acetate/pentane. The title compound was obtained as a colourless gum (240mg, 0.86mmol, 67%).
1H-NMR (CDCI3, 400MHz): δ 3.0 (s, 3H), 4.6 (s, 2H), 6.5 (m, 1 H), 6.7 (s, 1 H), 7.1 (d, 2H), 7.3 (m, 3H), 7.9 (d, 1 H). LRMS m/z (APCI) 277 [MH]+.
Preparation 72 2-Bromo-N.N-dimethylpyridin-4-amine

1H-NMR (CDCI3, 400MHz): δ 3.0 (s, 6H), 6.4 (m, 1H), 6.6 (s, 1 H), 7.9 (d, 1H). LRMS m/z (APCI) 201 [MH]+.
Preparation 73 2-Bromo-N-ethyl-N-methylpyridin-4-amine

2,4-Dibromopyridine (300mg, 0.94mmol) and N-ethylmethylamine (550mg, 9.4mmol) were combined in ethanol and heated at 7O0C overnight. The reaction mixture was concentrated in vacuo and the residue purified by column chromatography using an ISCO® silica cartridge eluting with 0-80% ethyl acetate/pentane. (144mg, 0.0.66mmol, 70%).
1H-NMR (CDCI3, 400MHz): δ 1.1 (t, 3H), 2.9 (s, 3H), 3.4 (q, 2H), 6.4 (m, 1 H), 6.6 (s, 1 H), 5.9 (d, 1 H). LRMS m/z (APCI) 215 [MH]+.
Preparation 74
2-Bromo-4-pyrrolidin-1-ylpyridine

2,4-Dibromopyridine (300mg, 0.94mmol) and pyrrolidine (335mg, 4.7mmol) were combined in ethanol and heated at 700C overnight. The reaction mixture was concentrated in vacuo and the residue purified by column chromatography using an ISCO® silica cartridge eluting with 0- 70% ethyl acetate: pentane. (208mg, 0.91 mmol, 97%).
1H-NMR (CDCI3, 400MHz): δ 2.0 (m, 4H), 3.3 (m, 4H), 6.3 (m, 1 H), 6.6 (s, 1 H)1 7.9 (d, 1 H). LRMS m/z (APCI) 227 [MH]+.
Preparation 75 Tert-butyl 3-(pyridin-2-ylamino)-2,3-dihydro-1 'H-spiroFindene-i ,4'-piperidine1-1 '-carboxylate

Tert-butyl 3-amino-2,3-dihydro-1'H-spiro[indene-1 ,4'-piperidine]-1'-carboxylate (840mg, 2.77mmol), 2-chIoropyridine (315mg, 2.77mmol), sodium tert-butoxide (373mg, 3.88mmol), palladium (II) acetate (62mg, 0.27mmol) and 2,2'-bis(diphenylphosphino)-1 ,1 >-binaphthyl (172mg, 0.27mmol) were combined in toluene and heated at 8O0C for 4 hours. The reaction mixture was partitioned between ethyl acetate and water. The organic phase was washed with water and brine, dried over magnesium sulphate, filtered and evaporated in vacuo. The material was purified by column chromatography using an ISCO® silica cartridge eluting with 0-80% ethyl acetate/pentane. The product was obtained as a pale brown gum (450mg, 1.18mmol, 43%). 1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 9H), 1.55 (m, 2H), 1.7 (m, 2H), 2.0 (m, 1 H), 2.8 (m, 1 H), 2.9 (m, 1 H), 3.0 (m, 1 H), 4.1 (m, 2H), 4.8 (d, 1 H), 5.5 (q, 1 H), 6.5 (d, 1 H), 6.6 (m, 1 H), 7.3 (m, 5H), 7.5 (t, 1H), 8.1 (m, 1H). LRMS m/z (APCI) 380 [MH]+.
Preparation 76 N-Pyridin-2-yl-2.3-dihydrospiroπndene-1.4'-piperidinl-3-amine

The protected piperidine of Preparation 75 (180mg, 0.474mmol) was dissolved in dichloromethane, 4M hydrochloric acid was added and the reaction mixture stirred at room temperature overnight. The solvent was evaporated in vacuo and the residue partitioned between 1 M sodium carbonate and dichloromethane. The organic phase was washed with brine, dried over magnesium sulphate, filtered and evaporated in vacuo. The crude product was purified by column chromatography using an ISCO® silica cartridge eluting with
dichloromethane - 90:10:1 dichloromethane/methanol/ 0.880 ammonia. The product was isolated as a clear gum (105mg, 0.37mmol, 79%).
1H-NMR (CD3OD, 400MHz): δ 1.6 (m, 2H), 1.7 (m, 2H), 2.1 (m, 1 H), 2.8 (m, 2H), 2.9 (m, 1 H), 3.0 (m, 2H), 5.45 (t, 1 H), 6.6 (m, 2H), 7.2 (m, 1 H), 7.3 (m, 3H), 7.4 (t, 1 H), 7.95 (d, 1 H). LRMS m/z (APCI) 280 [MH]+.
Preparation 77
Tert-butyl 3-facetyl(pyridin-2-yl)aminol-2,3-dihvdro-1 'H-spiroNndene-1.4'-piperidinel-1 '- carboxylate

The amino-pyridine of Preparation 75 (200mg, 0.527mmol), acetyl chloride (206mg, 2.63mmol) and triethylamine (2ml) were combined in tetrahydrofuran and stirred at room temperature over the weekend. The reaction mixture was partitioned between dichloromethane and water. The organic phase was washed with water and brine then dried over magnesium sulphate, filtered and evaporated in vacuo. The material was purified by column chromatography using an ISCO® silica cartridge eluting with 0-100% ethylacetate/pentane. (180mg, 0.42mmol, 81 %). 1H-NMR (CD3OD, 400MHz): δ 0.8 (m, 1 H), 1.2 (m, 2H), 1.4 (s, 9H), 1.6 (m, 1 H), 1.8 (m, 1 H), 1.95 (s, 2H), 2.15 (s, 1 H), 2.6 (m, 1 H), 2.7 (m, 1 H), 2.9 (m, 1 H), 3.4 (m, 1 H), 4.0 (m, 2H), 6.4 (m, 1 H), 6.8 (m, 1 H), 7.1 (m, 1 H), 7.2 (m, 2H), 7.3 (m, 1 H), 7.6 (m, 1 H), 8.5 (m, 1 H).
Preparation 78 N-(2.3-Dihvdrospirorindene-1.4'-piperidin1-3-yl)-N-pyridin-2-vlacetamide

The protected amine of Preparation 77 (180mg, 0.42mmol) was dissolved in dichloromethane, trifluoroacetic acid (0.5ml) was added and the reaction mixture stirred at room temperature for 4 hours. The reaction mixture was diluted with water then basified with
1 M sodium carbonate. The material was extracted into dichloromethane and the organic phase washed with brine, dried over sodium sulphate, filtered and evaporated in vacuo. The material was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane - 80:20:5 dichIoromethane/methanol/0.880 ammonia. The product was isolated as a clear gum (30mg, 0.093mmol, 22%).
1H-NMR (CD3OD, 400MHz): δ 0.9 (m, 1 H), 1.4 (m, 1 H), 1.6 (m, 1 H), 1.8 (m, 1 H), 2.9 (s, 3H), 2.95 (m, 1 H), 2.6 (m, 2H)1 2.8 (m, 2H), 3.0 (m, 1 H), 6.4 (m, 1 H), 7.0 (d, 1 H), 7.2 (m, 3H), 7.3 (m, 2H), 7.7 (m, 1 H), 8.5 (d, 1 H). LRMS m/z (APCI) 322 [MH]+.
Preparation 79
Tert-butyl 3-rmethyl(pyridin-2-ylcarbonyl)aminol-2,3-dihydro-1 'H-spirorindene-1 ,4'-piperidine1-
1 '-carboxylate
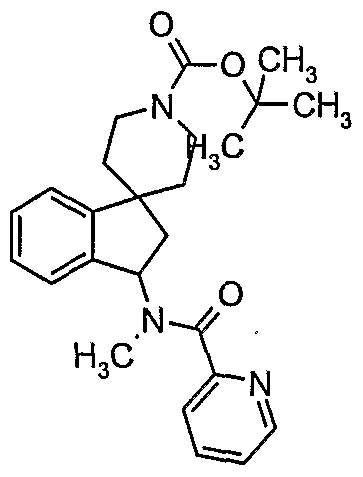
A solution of the pyridyl amide of Preparation 3 (100mg, 0.245mmol) in tetrahydrofuran was added dropwise to a suspension of sodium hydride (12mg, 0.294mmol) in tetrahydrofuran (2ml). The reaction mixture was allowed to stir at room temperature for 10 minutes before the addition of a solution of iodomethane (138mg, 0.9mmol) in tetrahydrofuran. The reaction mixture was allowed to stir at room temperature for 3 hours. The reaction mixture was quenched with water, then extracted from water into dichloromethane. The organic phase was washed with brine, dried over magnesium sulphate, filtered and concentrated in vacuo. The material was purified by column chromatography using an ISCO® silica cartridge eluting with 0-100% ethyl acetate/pentane. The product was isolated as a clear gum (83mg, 0.2mmol, 80%). 1H-NMR (CD3OD, 400MHz): δ 1.45 (d, 9H), 1.6 (m, 2H), 2.0 (m, 2H), 2.6 (m, 0.5H), 2.65 (s, 1.5H), 2.75 (m, 0.5H), 2.8 (s, 1.5H), 3.0 (m, 2H), 4.0 (m, 0.5H), 4.1 (m, 2.5H), 5.4 (t, 0.5H), 6.4 (t, 0.5H), 7.3 (m, 4H), 7.5 (m, 1 H), 7.7 (t, 1 H), 8.0 (q, 1 H), 8.6 (t, 1 H).
Preparation 80 N-(2.3-Dihvdrospiroπndene-1 ^'-piperidini-S-vh-N-methvlpyridine^-carboxamide

The protected piperidine of Preparation 79 (84mg, 0.2mmol) was dissolved in dichloromethane, 4M hydrochloric acid in dioxane (1 ml) was added and the reaction mixture stirred at room temperature overnight. The reaction mixture was concentrated in vacuo. The residue was partitioned between dichloromethane and water then basified with sodium carbonate. The mixture was extracted into dichioromethane, then dried over sodium sulphate, filtered and evaporated in vacuo. The material was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane - 90:10:1 dichloromethane/methanol/0.880 ammonia (55mg, 0.17mmol, 85%). 1H-NMR (CD3OD, 400MHz): δ 1.6 (m, 2H), 1.9 (m, 1 H), 2.1 (m, 1 H), 2.6 (m, 2H), 2.8 (m, 4H), 2.9 (m, 1 H), 3.0 (m, 2H), 5.4 (t, 0.5H), 6.3 (t, 0.5H), 7.3 (m, 4H), 7.5 (m, 1 H), 7.7 (t, 1 H), 8.0 (q, 1 H), 8.6 (t, 1 H). LRMS m/z (APCI) 322 [MH]+.
Preparation 81
2,3-Dihvdrospirorindene-1 ,4'-piperidin1-3-amine

Tert-butyl 3-amino-2,3-dihydro-1'H-spiro[indene-1 ,4'-piperidine]-1'-carboxylate (2g, 5.9mmol) was suspended in dichloromethane and 4M hydrochloric acid in dioxane (5ml) was added dropwise. The solvent was evaporated in vacuo and the resultant salt recrystallised from methanol and acetone. The product was isolated as a white solid (1.2g, 3.9mmol, 66%).
1H-NMR (CD3OD, 400MHz): δ 1.8 (m, 1 H), 2.0 (m, 3H), 2.4 (m, 1 H), 2.9 (m, 1 H), 3.2 (m, 2H), 3.5 (m, 2H), 4.9 (t, 1 H), 7.4 (m, 3H), 7.6 (d, 1 H).
Preparation 82 1'-(4-Nitro-1-oxidopyridin-2-ylV2,3-dihvdrospirorindene-1 ,4'-piperidin1-3-amine

The amine of Preparation 81 (260mg, 0.95mmol) and sodium carbonate (352mg, 3.32mmol) were combined in methyl isobutyl ketone (10ml) and heated at 11O0C for 3 hours. The reaction mixture was cooled to 8O0C before the addition of 2-chIoro-4-nitropyridine N-oxide. The reaction mixture was heated at 8O0C overnight. The reaction mixture was washed with water and the solvent evaporated in vacuo. The residue was redissolved in water / 2-propanol and stirred at room temperature for 4 hours. The reaction mixture was concentrated in vacuo then partitioned between dichloromethane and water. The organic phase was washed with brine, dried over sodium sulphate, filtered and evaporated in vacuo. The crude product was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane - 90:10:1 dichloromethane/methanol/0.880 ammonia. The product was isolated as an orange gum (230mg, 0.67mmol, 70%).
1H-NMR (CD3OD, 400MHz): δ 1.6 (m, 3H), 2.0 (t, 1 H), 2.4 (t, 1 H), 2.8 (m, 1 H), 3.1 (m, 2H), 4.0 (m, 2H), 4.4 (m, 1 H), 7.2 (m, 3H), 7.4 (s, 1 H), 7.8 (s, 1 H), 7.9 (s, 1 H), 8.4 (s, 1 H).
Preparation 83
Tert-butyl 4-r(2-bromobenzov0(4-methoxybenzv0amino1-3.6-dihvdropyridine-1 (2H)- carboxylate

4-Methoxybenzylamine (1.37g, 0.01 mol) was dissolved in trimethylorthoformate (20ml). 1- Tert-butoxycarbonyl-4-piperidone (2g, 0.01 mol) was added portionwise and the reaction mixture stirred at room temperature for three days. The solvent was evaporated and the residue redissolved in dichloromethane (100ml). A solution of 2-bromobenzoyI chloride (2.19g, 0.01 mol) in dichloromethane (20ml) was added at O0C followed by triethylamine (2.78ml, 0.02mol). The reaction mixture was allowed to stir at room temperature overnight.
The reaction mixture was washed with saturated aqueous citric acid, saturated aqueous sodium hydrogen carbonate and brine. The organic phase was dried over sodium sulphate, filtered and evaporated. The residue was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane/methanol (95:5). The product was collected as an impure yellow foam (1.6g).
1H-NMR (CDCI3, 400MHz): δ 1.4 (s, 9H), 2.2 (t, 2H), 3.2 (bs, 2H), 3.6 (bs, 2H), 3.7 (m, 2H), 3.8 (s, 3H), 5.4 (bs, 1 H)1 6.85 (d, 2H), 7.2 (m, 3H), 7.3 (m, 2H), 7.5 (d, 1 H). LRMS m/z (APCI) 501 [MH]+.
Preparation 84
Tert-butyl 2-(4-methoxybenzyl)-3-oxo-2,2'.3.3'-tetrahydro-1 'H-spiroFisoindole-i ,4'-pyridine1-1 '- carboxylate

The alkene of Preparation 83 (1.6g, 3mmol), tetraethylammonium chloride (390mg, 3mmol), triphenylphosphine (59mg, 0.22mmol) and potassium carbonate (830mg, 6mmol) were combined in acetonitrile (40ml), palladium acetate (50mg, 0.22mmol) was added and the reaction mixture heated at 8O0C overnight. The reaction mixture was concentrated in vacuo and the residue extracted from water into ethyl acetate. The combined organic layer were washed with brine, dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography using an ISCO® silica cartridge eluting with 0-50% ethyl acetate/pentane. The title compound was obtained as a white foam (300mg, OJmmol, 24%).
1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 9H), 1.7 (m, 1 H), 2.15 (m, 2H), 2.4 (t, 1 H), 3.7 (m, 2H), 3.8 (s, 3H), 4.2 (d, 1 H), 4.9 (d, 1 H), 6.8 (d, 2H), 7.2 (m, 2H), 7.4 (d, 1 H), 7.5 (m, 2H), 7.9 (d, 1 H).
Preparation 85 2-(4-Methoxybenzvn-2'.3'-dihvdro-1'H-SDirorisoindole-1 ,4'-Pyridinl-3(2H)-one

The protected enamine of Preparation 84 (400mg, 0.95mmol) was dissolved in dichloromethane (10ml), trifluoroacetic acid (2ml) was added and the reaction mixture stirred at room temperature overnight. The solvent was evaporated in vacuo and the residue azeotroped with toluene, then ether.
1H-NMR (CDCI3, 400MHz): δ 1.8 (m, 1 H), 2.5 (m, 1 H), 2.8 (m, 1 H), 3.2 (m, 1 H), 3.8 (s, 3H), 4.2 (m, 2H), 4.6 (d, 1 H), 5.0 (d, 1 H), 6.8 (m, 3H), 7.6 (m, 3H), 8.1 (d, 1 H), 8.8 (s, 1 H).
Preparation 86
2-(4-Methoxybenzyl)spirorisoindole-1 ,4'-piperidinl-3(2H)-one

The enamine of Preparation 85 (250mg, 0.78mmol) was dissolved in ethyl acetate (5ml). Acetic acid (0.5ml) was added followed by 10% palladium on carbon (25mg). The reaction mixture was stirred at room temperature under 4 atmospheres of hydrogen overnight. The reaction mixture was filtered through Arbocel® and the solvent evaporated in vacuo. The residue was extracted from saturated sodium hydrogen carbonate into ethyl acetate, dried over sodium sulphate, filtered and evaporated. The crude material was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane/methanoI/0.880 ammonia (90:10:1). The title compound was obtained as a colourless gum (40mg, 0.12mmol, 16%).
1H-NMR (CDCI3, 400MHz): δ 1.4 (m, 2H), 2.2 (m, 2H), 3.2 (m, 2H), 3.3 (m, 2H), 3.8 (s, 3H), 4.8 (s, 2H), 6.8 (d, 2H), 7.3 (m, 2H), 7.5 (m, 2H), 7.9 (d, 1 H), 8.0 (d, 1 H). LRMS m/z (APCl) 323 [MH]+
Preparation 87
Tert-butyl (3S)-3-r(pyridin-2-ylamino)carbonvπ-2,3-dihvdro-1 'H-spiroFindene-i ,4'-piperidine1-1 ' carboxylate

(3S)-1 '-(Tert-butoxycarbonyl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxylic acid
(1636mg, O.δmmol) and 1 ,1-carbonyldiimidazole (97mg, O.θmmol) were combined in dichloromethane (0.5ml) and stirred at room temperature for 35 minutes before the addition of 2-aminopyridine (56mg, O.θmmol). The reaction mixture was stirred at room temperature overnight. The reaction mixture was extracted from 3% aqueous sodium hydrogen carbonate (5ml) into ethyl acetate (2 x 5ml), dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography over silica gel eluting with ethyl acetate/pentane (1 :2). The title compound was obtained as a foam (186mg, 0.45mmol, 91 %).
1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 9H), 1.55 (m, 1 H), 1.7 (m, 2H), 1.9 (m, 1 H), 2.4 (m, 1 H), 2.6 (m, 1 H), 3.0 (m, 2H), 4.2 (m, 3H), 7.1 (m, 1 H), 7.3 (m, 4H), 7.8 (m, 1 H), 8.3 (d, 1 H), 8.4 (d, 1 H).
Preparation 88
Tert-butyl (3S)-3-r((5-r(diethylamino)carbonyllpyridin-2-yl)amino)carbonyll-2,3-dihvdro-1'H- spirorindene-1 ,4'-piperidine1-1 '-carboxylate

The title compound was obtained in 56% yield from (3S)-1'-(tert-butoxycarbonyl)-2,3- dihydrospiro[indene-1 ,4'-piperidine]-3-carboxylic acid and 6-amino-N,N-diethylnicotinamide
[Aminoethenes, Ger. Offen, 1984] following a similar procedure to that described in Preparation 87.
1H-NMR (CDCl3, 400MHz): δ 1.2 (m, 6H)1 1.5 (s, 10H), 1.7 (m, 1 H), 1.75 (m, 1 H), 1.9 (m, 1 H), 2.4 (m, 1 H)1 2.6 (m, 1 H), 2.9 (m, 2H), 3.4 (m, 4H), 4.2 (m, 3H), 7.3 (m, 4H), 7.8 (d, 1 H), 8.3 (m, 2H), 8.5 (bs, 1 H).
Preparation 89 (3S)-2,3-dihvdrospiro[indene-1.4'-piperidine1-3-carbonitrile

The title compound was obtained 90% pure from tert-butyl (3S)-3-cyano-2,3-dihydro-1 'H- spiro[indene-1 ,4'-piperidine]-1'-carboxylate [described in US5578593, compound 46, scheme 19] following a procedure similar to that described in Preparation 2. The reaction time was 3 hours.
1H-NMR (CDCI3, 400MHz): δ 1.3 (m, 1 H), 1.5 (m, 1 H), 1.8 (m, 1 H), 2.0 (m, 1 H), 2.3 (m, 1 H), 2.7 (m, 1 H), 2.8 (m, 2H), 3.1 (m, 2H), 4.2 (m, 1 H), 7.3 (m, 4H).
Preparation 90
Tert-butyl (3S)-3-r(dimethylamino)carbonyll-2,3-dihvdro-1 'H-spiroHndene-i ,4'-piperidinel-1 '- carboxvlate

(3S)-1'-(Tert-butoxycarbonyl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxylic acid
(732mg, 2.21 mmol), 1-hydroxybenzotriazole monohydrate (346mg, 2.25mmol) and 1-(3- dimethylaminopropyl)-3-ethylcarbodiimide HCI (508mg, 2.65mmol) and 33% dimethylamine in ethanol (0.895ml, 6.63mmol) were combined in dichloromethane and stirred at room temperature overnight. The reaction mixture was concentrated in vacuo, then extracted from 3% aqueous sodium hydrogen carbonate (20ml) into ethyl acetate (2x 20ml). The combined organic extracts were dried over sodium sulphate, filterered and evaporated in vacuo. The crude material was purified by column chromatography on silica gel using ethyl acetate/pentane as the eluent. 1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 10H), 1.7 (m, 2H), 2.0 (m, 1 H), 2.3 (m, 1 H), 2.4 (m, 1 H), 2.8 (bs, 1 H), 2.9 (m, 1 H), 3.05 (s, 3H), 3.25 (s, 3H), 4.1 (m, 2H), 4.4 (t, 1 H), 7.1-7.3 (m, 4H).
Preparation 91
(3S)-N,N-Dimethyl-2,3-dihvdrospirorindene-1.4'-piperidine1-3-carboxamide

The protected piperidine of Preparation 90 was dissolved in dichloromethane: trifluoroacetic acid (8ml:2ml) and the reaction mixture stirred at room temperature for 4 hours. The solvent was evaporated and the residue extracted from 2N sodium hydroxide (15ml) into dichloromethane (3x15ml). The combined organic extracts were dried over sodium sulphate, filtered and evaporated to yield the title compound as a gum (0.36g, 63%). 1H-NMR (CDCI3, 400MHz): δ 1.6 (m, 1 H), 1.8 (m, 2H), 2.1 (m, 1 H), 2.3 (m, 1 H), 2.4 (m, 1 H), 2.9 (q, 2H), 3.1 (s, 3H), 3.2 (m, 2H), 3.3 (s, 3H), 4.4 (t, 1 H), 7.1 (d, 1 H), 7.2 (m, 3H). LRMS m/z (APCI) 259 [MH]+.
Preparation 92 Tert-butyl (3f?)-3-r(dimethylamino)carbonyll-2,3-dihvdro-1 'H-spiroFindene-i ,4'-piperidinel-1 '- carboxylate

The title compound was obtained as a gum in 80% yield from (3R)-1'-(tert-butoxycarbonyl)- 2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxylic acid following the procedure described in Preparation 90.
1H-NMR (CDCI3, 400MHz): δ 1.5 (s, 10H), 1.7 (m, 2H), 2.0 (m, 1H), 2.3 (m, 1H), 2.4 (m, 1 H), 2.8 (bs, 1 H), 2.9 (m, 1 H), 3.1 (s, 3H), 3.3 (s, 3H), 4.1 (m, 2H), 4.4 (t, 1 H), 7.1-7.3 (m, 4H).
Preparation 93
(3f?)-N.N-Dimethyl-2.3-dihvdrospiroπndene-1.4'-piperidine1-3-carboxamide

Preparation 94 2-Benzylspirofisoindole-1 ,4'-piperidin1-3(2H)-one
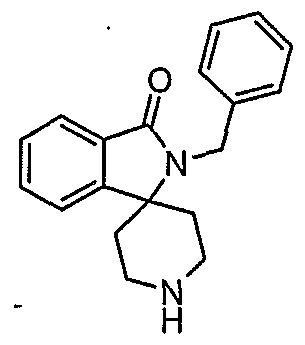
Tert-butyl 2-benzyl-3-oxo-2,2',3,3'-tetrahydro-1 Η-spiro[isoindole-1 ,4'-pyridine]-1 '-carboxylate [described in US2002/188124, compound 7, production process B] (3.25g, 8mmol) was dissolved in methanol (30ml). 4M hydrochloric acid in dioxane (5ml, 20mmol) was added and the reaction mixture was stirred at room temperature for 72 hours. 4M hydrochloric acid in dioxane (5ml) was added and the reaction mixture heated to 5O0C for 1 hour. The reaction mixture was concentrated in vacuo. The alkene was dissolved in ethanol and a slurry of 10% palladium on carbon in ethanol added. The mixture was stirred at room temperature under 2.72 atmospheres of hydrogen for 72 hours. Water was added and the reaction mixture was filtered through Celite®. The combined filtrates were concentrated in vacuo and the precipitated solid collected by filtration.
1H-NMR (CD3OD400MHz): δ 1.7 (m, 2H), 2.5 (m, 2H), 3.6 (m, 4H), 4.9 (m, 2H), 7.3 (m, 5H), 7.7 (m, 2H), 8.0 (m, 2H). LRMS m/z (ES) 293 [MH]+.
Preparations 95-115
The piperidine (1eq), 2-chloro-4-nitropyridine N-oxide (1eq) and sodium hydrogen carbonate (1.5eq) were combined in 2-methyl-2-butanol. The reaction mixture was heated at 48-12O0C for 18 hours. The reaction mixture was concentrated in vacuo. The residue was extracted from water into dichloromethane, dried over magnesium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography on silica gel using ethyl acetate/pentane as the eluent to afford the title compounds.







O = Ethyl acetate, then dichloromethane was used as the column solvent.
P = The compound was purified by crystallisation from dichloromethane and ethyl acetate.
Q = Dichloromethane/methanol was used as the column solvent.
R = Triethylamine was used as the base.
S = Dichloromethane/methanol/0.880 ammonia was used as the column solvent.
T= Ethyl acetate/dichloromethane was used as the column solvent.
Preparation 116
6-Fluoro-1'-(4-nitro-1-oxidopyridin-2-yl)spirori-benzofuran-3,4'-piperidinel

The piperidine of Preparation 66 (167mg, O.δmmol), 2-chloro-4-nitropyridine N-oxide (150mg, 0.89mmol), N,N-diisopropylethylamine (150μL, 0.86mmol), and pyridine (5mg) were combined in 2-methyl-2-butanol (8ml) and heated at reflux for 1 hour. The reaction mixture was diluted with pentane (25ml) and filtered through a silica plug. The title compound was eluted with ether. The material was collected as an orange solid (117mg, 0.34mmol, 42%). 1H-NMR (CD3OD, 400MHz): δ 1.85 (m, 2H), 2.2 (m, 2H), 3.0 (t, 2H), 3.95 (m, 2H), 4.6 (s, 2H), 6.5 (d, 1H), 6.6 (m, 1 H), 7.2 (m, 1H), 7.8 (m, 1H), 7.9 (s, 1 H), 8.4 (d, 1 H). LRMS m/z (APCI) 346 [MH]+.
Preparation 117
Tert-butyl (3S)-3-r(diethylamino)carbonvπ-2,3-dihvdro-1 'H-spiroNndene-i ,4'-piperidine]-1 '- carboxylate

(3S)-1 '-(Tert-butoxycarbonyl)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carboxylic acid
(150mg, 0.45mmol), 1-hydroxybenzotriazole monohydrate (71 mg, 0.466) and 1-(3- dimethylaminopropyl)-3-ethylcarbodiimide HCI (104mg, 0.54mmol) and diethylamine (66mg, 0.906mmol) were combined in dichloromethane (3ml) and stirred at room temperature for 16 hours. The reaction mixture was washed with 3% aqueous sodium hydrogen carbonate, dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography on silica gel using ethyl acetate/pentane (1 :1) as the eluent to give the title compound as a gum (115mg, 0.3mmol, 66%). 1H-NMR (CDCI3, 400MHz): δ 1.2 (t, 3H), 1.3 (t, 3H), 1.5 (s, 9H), 1.7 (m, 3H), 2.0 (m, 1H), 2.4 (m, 2H), 2.9 (m, 2H), 3.3 (m, 1 H), 3.5 (m, 1 H), 3.7 (m, 2H), 4.1 (m, 2H), 4.3 (t, 1 H), 7.05 (d, 1 H), 7.2 (m, 3H).
Preparation 118 (3S)-N,N-diethyl-2,3-dihvdrospiroπndene-1 ,4'-piperidine1-3-carboxamide

The protected piperidine of Preparation 117 (115mg, 0.3mmol) was dissolved in dichloromethane^rifluoroacetic acid (4ml:1ml) and the reaction mixture stirred at room temperature for 4 hours. The solvent was evaporated and the residue partitioned between 2N sodium hydroxide and dichloromethane. The organic phase was dried over sodium sulphate, filtered and evaporated to yield the title compound.
1H-NMR (CDCI3, 400MHz): δ 1.2 (t, 3H), 1.3 (t, 3H), 1.6 (m, 1 H), 1.8 (m, 2H), 2.1 (m, 1 H), 2.4 (m, 2H), 2.9 (m, 2H), 3.2 (m, 2H), 3.3 (m, 1 H), 3.5 (m, 1 H), 3.6 (m, 2H), 4.3 (t, 1 H), 7.1 (d, 1 H), 7.2 (m, 3H).
Preparation 119 Ethvl 1'-benzvl-5-methoxv-3H-spirof2-benzofuran-1.4'-piperidine1-3-carboxvlate

The title compound was synthesised from 2-bromo-5-methoxybenzaldehyde dimethyl acetal (described in Org. Lett, 2002, 4, 2711) using methods similar to those outlined for the des- methoxy analogue Preparation 59 (methods described in J. Med. Chem., 2002, 45, 438 and J. Med. Chem., 2002, 45, 4923).
1H-NMR (CDCI3, 400MHz): δ 1.25 (t, 3H), 1.7 (m, 1 H), 1.85-2.05 (m, 3H), 2.45 (m, 1 H), 2.55 (m, 1 H), 2.85 (m, 2H), 3.6 (s, 2H), 3.75 (s, 3H), 5.2 (q, 2H), 5.6 (s, 1 H), 6.85 (m, 1 H), 6.9 (d, 1H), 7.0 (m, 1 H), 7.2-7.35 (m, 5H). LRMS m/z (APCI) 382 [MH]+
Preparation 120
Ethyl-5-methoxy-3H-spiro[2-benzofuran-1,4'-piperidine1-3-carboxvlate
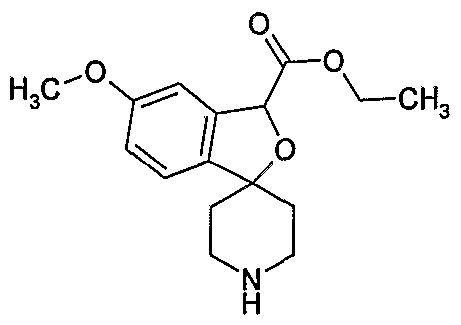
The amine of Preparation 119 (25g, 65.6mmol) was dissolved in ethanol (350ml) and stirred under 2.7 atmospheres of hydrogen at 6O0C for 16 hours. TLC indicated that no debenzylation had occurred. The catalyst was removed by filtration (using Arbocel®) and the solvent evaporated. The reaction was repeated using fresh catalyst and solvent. After 16 hours, TLC indicated that only a small amount of debenzylation had occurred. The reaction was continued at 3.5 atmospheres of hydrogen and 7O0C for 65 hours. LCMS indicated that the reaction was 40% complete. The catalyst was removed by filtration (using Arbocel®) and the solvent evaporated. The reagents and solvents were refreshed and the reaction was stirred at 7O0C under 3.5 atmospheres of hydrogen for 48 hours. The catalyst was removed by filtration (using Arbocel®) and the solvent evaporated to give the product as a pale yellow gum (15.8g, 82%).
1H-NMR (CDCI3, 400MHz): 5 1.25 (t, 3H), 1.65 (m, 1 H), 1.85 (m, 3H), 3.1 (m, 4H), 3.75 (s, 3H), 4.2 (q, 2H), 5.6 (s, 1 H), 6.85 (m, 2H)1 7.0 (m, 2H).
Preparation 121 N,N-dimethyl-3H-spiror2-benzofuran-1.4'-piperidinel-3-carboxamide

The title compound was obtained as a colourless gum in 64% yield from 2M dimethylamine solution (in THF) and the chiral compound 1'-(tert-butoxy-carbonyl)-3H-spiro[2-benzofuran- 1 ,4'-piperidine]-3-carboxylic acid (derived from resolution with (f?)-(+)-α-methylbenzylamine) of Preparation 60, following a similar procedure to that described in Preparation 62.
1H-NMR (CDCI3, 400MHz): δ 1.65-1.95 (m, 5H), 2.0 (m, 3H), 2.95-3.15 (m, 7H), 3.25 (s, 3H), 5.85 (s, 1 H), 7.15 (m, 1 H), 7.25 (m, 3H). LRMS m/z (ESI) 261 [MH]+.
Preparation 122 N-ethyl-N-methyl-3H-spirof2-benzofuran-1 ,4'-piperidine1-3-carboxamide

The title compound was obtained as a colourless gum in 84% yield from ethylmethylamine and the chiral compound 1'-(tert-butoxycarbonyl)-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxylic acid (derived from resolution with (R)-(+)-α-methylbenzylamine) of Preparation 60, following a similar procedure to that described in Preparation 62.
1H-NMR (CDCI3, 400MHz): δ 1.2 (m, "3H), 1.6-1.9 (m, 5H), 2.9-3.25 (m, 7H), 3.3-3.7 (m, 2H), 5.85 (s, 1 H), 7.15 (m, 1H), 7.25 (m, 1H). LRMS m/z (ESI) 275 [MH]+.
Preparation 123
1 '-(4-nitro-1 -oxidopyridin-2-yl)spirori -benzofuran-3,4'-piperidinel-7-carbonitrile

The piperidine of Preparation 55 (565 mg, 2.28 mmol) was dissolved in 2-methyl-2-butanol (10ml). 2-chloro-4-nitropyridine N-oxide (398 mg, 2.28 mmol) and sodium hydrogen carbonate (300 mg 3.57 mmol) were added and the reaction was heated at 5O0C for 48 hours. The reaction mixture was concentrated in vacuo and the residue taken up in ethyl acetate (15ml). The organic phase was washed with water (3 x 10ml) and then dried over sodium sulfate. The material was purified by column chromatography using an ISCO® silica cartridge eluting with a gradient of heptane to 70:30 heptane/ethyl acetate to yield the title compound as a yellow solid (459 mg, 1.30 mmol, 58%).
1H-NMR (CDCI3, 400MHz): δ 1.9-1.95 (d, 2H), 2.10-2.15 (m, 2H), 3.0-3.05 (m, 2H), 3.95-4.05 (m, 2H), 5.75 (s, 2H), 7.05 (t, 1 H), 7.40 (d, 1 H), 7.55 (d, 1 H), 7.8 (m, 1 H), 7.90 (m, 1 H), 8.4 (d, 1H).
LRMS m/z (APCI) 353 [MH]+.
Preparation 124
1'-benzyl 1-methyl 1'H-spiroπndole-3,4'-piperidine1-1 ,1'(2H)-dicarboxylate

Benzyl i ^-dihydro-i'H-spiroIindole-S^'-piperidineJ-i'-carboxylate [described in J. Xie et al., Tetrahedron, 2004, 60, 4875-4878] (251 mg, 0.78 mmol), methyl chloroformate (81 mg, 0.86 mmol) and triethylamine (0.15 ml, 0.97 mmol) were stirred in dichloromethane (5 ml) at room
temperature for 18 hours. The organic phase was washed with saturated aqueous sodium hydrogen carbonate (5 ml), dried (MgSO4), and loaded onto a silica gel column. Gradient elution with cyclohexane:Et2O (90:10 - 50:50) yielded the title compound as a colourless gum (85 mg, 0.22 mmol, 29%).
MS m/z (APCI) 381 [MH]+
Preparation 125
1 '-benzyl 1-ethyl 1'H-spiroπndole-3.4'-piperidinel-1 ,1'(2H)-dicarboxylate

Benzyl 1 ,2-dihydro-1Η-spiro[indole-3,4'-piperidine]-r-carboxylate [described in J. Xie et al., Tetrahedron, 2004, 60, 4875-4878] (251 mg, 0.78 mmol), ethyl chloroformate (93 mg, 0.86 mmol) and triethylamine (0.15 ml, 0.97 mmol) were stirred in dichloromethane (5 ml) at room temperature for 18 hours. The organic phase was washed with saturated aqueous sodium hydrogen carbonate (5 ml), dried (MgSO4), and loaded onto a silica gel column. Gradient elution with cydohexane:Et2O (90:10 - 50:50) yielded the title compound as a colourless gum (95 mg, 0.24 mmol, 31 %). MS m/z (APCI) 395 [MH]+
Preparation 126 1 '-Benzyl 1-isopropyl 1 'H-spirorindole-3,4'-piperidinel-1.1 '(2H)-dicarboxylate

Preparation 127 Methyl spirorindole-3.4'-pir->eridine1-1 (2H)-carboxvlate

The compound of Preparation 124 (515 mg, 1.35 mmol) was stirred with 1-methyl-1 ,4- cyclohexadiene (1.52 ml, 13.5 mmol) and Pd(OH)2 on carbon (50 mg) in ethanol at 500C for 30 minutes. The catalyst was removed by filtration and the filtrate concentrated to yield the title compound as a gum (340 mg, 1.35 mmol, quantitative).
1H-NMR (CDCI3, 400MHz): δ 1.65 (m, 2H), 1.85 (m, 2H), 2.77 (m, 2H), 3.11 (m, 2H), 3.88 (m, 4H), 7.01 (t, 1 H), 7.17 (d, 1 H), 7.21 (t, 1 H), 7.86 (m, 1 H). MS m/z (APCI) 247 [MH]+.
Preparation 128 Ethyl spirorindole-3.4'-piperidinel-1 (2H)-carboxylate

The compound of Preparation 125 (555 mg, 1.41 mmol) was stirred with 1-methyl-1 ,4- cyclohexadiene (1.58 ml, 14.1 mmol) and Pd(OH)2 on carbon (60 mg) in ethanol at 500C for
30 minutes. The catalyst was removed by filtration and the filtrate concentrated to yield the title compound as a gum (350 mg, 1.35 mmol, 96%).
1H-NMR (CDCI3, 400MHz): δ 1.38 (m, 3H), 1.66 (m, 2H), 1.85 (m, 2H), 2.76 (m, 2H), 3.09 (m, 2H), 3.90 (m, 2H), 4.32 (m, 2H)1 7.00 (t, 1 H), 7.15 (d, 1 H), 7.20 (t, 1 H), 7.86 (m, 1 H). MS m/z (APCI) 261 [MH]+.
Preparation 129
Isopropyl spirofindole-3,4'-piperidinel-1 (2H)-carboxylate

The compound of Preparation 126 (365 mg, 0.89 mmol) was stirred with 1-methyl-1 ,4- cyclohexadiene (1.00 ml, 8.94 mmol) and Pd(OH)2 on carbon (40 mg) in ethanol at 500C for 30 minutes. The catalyst was removed by filtration and the filtrate concentrated to yield the title compound as a gum (230 mg, 0.84 mmol, 94%).
1H-NMR (CDCI3, 400MHz): δ 1.37 (d, 6H), 1.65 (m, 2H), 1.86 (m, 2H), 2.79 (t, 2H), 3.11 (m, 2H), 3.88 (m, 2H), 5.10 (m, 1 H), 6.99 (t, 1 H), 7.16 (d, 1 H), 7.19 (t, 1 H), 7.86 (m, 1 H). MS m/z (APCI) 275 [MH]+.
Preparation 130
Methyl 1 '-(4-nitro-1 -oxidopyridin-2-v0spirorindole-3,4'-piperidinel-1 (2H)-carboxylate

The compound of Preparation 127 (340 mg, 1.38 mmol) was stirred with 2-chloro-4- nitropyridine-N-oxide (361 mg, 2.07 mmol) and triethylamine (0.29 ml, 2.07 mmol) at 500C in
t-amyl alcohol (5 ml) overnight. The mixture was cooled and the volatiles removed in vacuo. The residue was resuspended in toluene, dried (MgSO4) and filtered. The filtrate was loaded onto a silica gel column and eluted with cyclohexane:ethyl acetate (100:0 - 0:100) to yield the title compound as a yellow foam (420 mg, 1.09 mmol, 79%).
1H-NMR (CDCI3, 400MHz): δ 1.65 (m, 2H), 2.24 (m, 2H), 2.96 (m, 2H), 3.85-4.06 (m, 6H), 7.03 (t, 1 H), 7.20 (d, 1 H), 7.25 (t, 1 H), 7.68-7.74 (m, 2H), 7.90 (m, 1 H), 8.26 (d, 1 H). MS m/z (APCI) 385 [MH]+.
Preparation 131
Ethyl 1 '-(4-nitro-1 -oxidopyridin-2-v0spirorindole-3,4'-piperidinel-1 (2H)-carboxylate

The compound of Preparation 128 (350 mg, 1.34 mmol) was stirred with 2-chloro-4- nitropyridine-N-oxide (352 mg, 2.02 mmol) and triethylamine (0.28 ml, 2.02 mmol) at 500C in t-amyl alcohol (5 ml) overnight. The mixture was cooled and the volatiles removed in vacuo.
The residue was resuspended in toluene, dried (MgSO4) and filtered. The filtrate was loaded onto a silica gel column and eluted with cyclohexane:ethyl acetate (100:0 - 0:100) to yield the title compound as a yellow foam (465 mg, 1.17 mmol, 87%).
1H-NMR (CDCI3, 400MHz): δ 1.39 (m, 3H), 1.65 (m, 2H), 2.25 (m, 2H), 2.97 (m, 2H), 3.94-
4.06 (m, 4H), 3.32 (m, 2H), 7.03 (t, 1 H), 7.20 (d, 1H), 7.24 (t, 1 H), 7.68-7.74 (m, 2H), 8.25 (m,
1 H). MS m/z (APCI) 399 [MH]+.
Preparation 132 Isopropyl 1 '-(4-nitro-1 -oxidopyridin-2-v0spirorindole-3,4'-piperidinel-1 (2H Vcarboxylate
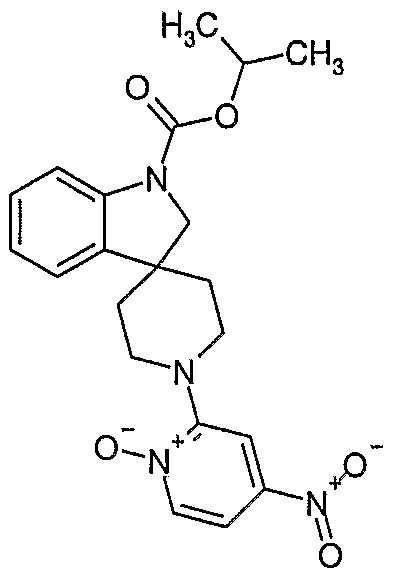
The compound of Preparation 129 (230 mg, 0.84 mmάl) was stirred with 2-chloro-4- nitropyridine-N-oxide (219 mg, 1.26 mmol) and triethylamine (0.18 ml, 1.26 mmol) at 500C in t-amyl alcohol (5 ml) overnight. The mixture was cooled and the volatiles were removed in vacuo. The residue was resuspended in toluene, dried (MgSO4) and filtered. The filtrate was loaded onto a silica gel column and eluted with cyclohexane:ethyl acetate (100:0 - 0:100) to yield the title compound as a yellow glass (260 mg, 0.63 mmol, 75%).
1H-NMR (CDCI3, 400MHz): δ 1.39 (m, 6H), 1.65 (m, 2H), 2.23 (m, 2H), 2.97 (m, 2H), 3.93- 4.07 (m, 4H), 5.12 (m, 1 H), 7.02 (t, 1 H), 7.20 (d, 1 H), 7.23 (t, 1 H), 7.71 (d, 1 H), 7.74 (m, 1 H), 7.90 (m, 1 H), 8.26 (d, 1 H). MS m/z (APCI) 413 [MH]+.
Claims
1. A compound of formula (I):

(I) '
wherein: the dotted line represents an optional covalent bond between X and Y; when the groups X and Y are connected by a single bond, X and Y may be the same or different and each represent C(=O), C(L-R1)2, N(L -R1), O or S; when the groups X and Y are connected by a double bond, X and Y may be the same or different and each represent C(L-R1) or N;
Z represents C(=O), C(L-R1)2> N(L'-R1), O or S; m is 0 or 1 ;
L represents a single bond, -O-, -S-, -(C=O)- -(C=O)-O-, -(C=O)-NR-, -SO-,
-SO-NR-, -SO2-, -SO2-NR- -NR-, -NR-SO-, -NR-SO2-, -NR(C=O)- -N(Het1)(C=O)-,
-NR(C=O)-O-, Or -NR(C=O)-NR-;
L' represents a single bond, -(C=O)-, -(C=O)-O-, -(C=O)-NR-, Or -SO2-; with the provisos that not more than one of X, Y and Z represents O or S, and not more than two of X, Y and Z represents C(=0) or N(L-R1);
R1 represents H, (C1-i0)alkyl (optionally substituted by one or more substituents α), -(CR2)n-
(C3.8)cycloalkyl (wherein the cycloalkyl part is optionally substituted by one or more substituents α), -(CR2)n-Ar, -(CR2)n-Het1, -(CR2)n-Het2, or -CN, n is O or an integer from 1 to 6;
R represents H or (C1-10)alkyl; or, when a group R and a group R1 substitute a nitrogen atom, the groups together with the nitrogen atom form a ring Het3;
R2 and R3 are the same or different and each represent R or -(CR2)-Ar, or R2 and R3, together with the nitrogen atom to which they are attached, form a ring Het3;
R4 represents (C1-10)alkyl; a is O, 1 or 2; R5 represents (C1-10)a!kyl, (C1-10)haloalkyl, hydroxy-(C1-10)alkyl, (C1.10)alkoxy(C1-10)alky], halogen, OH, (C1-10)alkoxy, (C1-10)haloalkoxy, phenoxy, benzyloxy, (C3.8)cycloalkyloxy, (C3-8)cycloalkyl-(C1.10)alkoxy, -(CR2)P-CN, -(CR2)P-C(=O)R10, -(CR2)p-C(=O)Het2, . -(CR2)P-C(=O)OR10, -(CR2)p-C(=O)N(R11)2, -(CR2)p-C(=O)NR-(CR2)n-N(R11)2, -(CR2)P-N(R11)2, -(CR2)P-NRC(=O)R10, -(CR2)P-NRS(=O)R10, -(CR2)P-NRS(=O)2R10, SR, S(=O)R, S(=O)2R, S(=O)N(R11)2 or pyridyl (optionally substituted by a (C1-10)alkyl group), or two groups R5 together form a (C-^alkylene group, a (C^alkyleneoxy group or a (Ci- 3)alkylenedioxy group; b is 0 or an integer from 1 to 4; p is 0 or an integer from 1 to 6;
R6, R7, R8 and R9 are each independently H or (C1-10)alkyl, or two groups selected from (a) R6 or R7 and (b) R8 or R9 together form a bond or a (C1-3)alkylene group; R10 represents H, (C1-10)alkyl (optionally substituted by one or more substituents α), -(CR2)q-(C3-8)cycloalkyl (wherein the cycloalkyl part is optionally substituted by one or more substituents o), -(CR2)q-Ar, -(CR2)q-Het1, or -(CR2)q-Het2; q is 0 or an integer from 1 to 6;
R11 represents H, (C1-10)afkyl or -(CR2)-Ar, or two groups R11, together with the nitrogen atom to which they are attached, form a ring Het3; Ar is a phenyl or naphthyl group optionally substituted by one or more substituents β; Het1 is a group selected from (1 ) a 5- or 6-membered aromatic heterocyclic group having as ring heteroatoms (a) 1 to 4 nitrogen atoms, (b) 1 oxygen or 1 sulphur atom or (c) 1 oxygen atom or 1 sulphur atom and 1 or 2 nitrogen atoms, is optionally substituted (i) on carbon by one or more substituents α and/or (ii) on nitrogen by one or more substituents γ, (2) a bicyclic group comprising an optionally substituted 5- or 6-membered aromatic heterocyclic group, as defined in (1) above, fused with a benzene ring (the benzene ring being optionally substituted with one or more substituents β) or (3) a bicyclic group comprising two optionally substituted 5- or 6-membered aromatic heterocyclic group, as defined in (1) above, fused together; Het2 is a group selected from (4) a 3- to 8-membered saturated heterocyclic group containing 1 or 2 heteroatoms selected from nitrogen, oxygen and sulphur, is optionally substituted (i) on carbon by one or more substituents α and/or (ii) on nitrogen by one or more substituents γ and/or (iii) on sulphur by one or two =0 groups, (5) a bicyclic group comprising an optionally substituted 3- to 8-membered saturated heterocyclic group, as defined in (4) above, fused with a benzene ring (the benzene ring being optionally substituted by one or more substituents β) or (6) a bicyclic group comprising an optionally substituted 3- to 8-membered saturated heterocyclic group, as defined in (4) above, fused with a (C3.8)cycloalkane ring (the cycloalkane ring being optionally substituted by one or more substituents α); Het3 is a group selected from (7) a 3- to 8-membered saturated heterocyclic ring containing 1 or 2 heteroatoms of which one heteroatom is a nitrogen atom and the other heteroatom, if present, is selected from nitrogen, oxygen and sulphur, is optionally substituted (i) on carbon by one or more substituents α and/or (ii) on nitrogen by one or more substituents γ and/or (iii) on sulphur by one or two =0 groups, (8) a bicyclic ring comprising an optionally substituted 3- to 8-membered saturated heterocyclic ring, as defined in (7) above, fused with a benzene ring (the benzene ring being optionally substituted by one or more substituents β), or (9) a bicyclic ring comprising an optionally substituted 3- to 8-membered saturated heterocyclic ring, as defined in (7) above, fused with a (C3.8)cycloalkane ring (the cycloalkane ring being optionally substituted by one or more substituents α); substituents α are selected from (C1-10)alkyl, (C1-10)haloalkyl, hydroxy-(C1-i0)alkyl, (C1- 10)alkoxy(Ci-10)alkyl, phenyl, pyridyl, halogen, OH1 (C^ojalkoxy, (Ci-i0)haloalkoxy, phenoxy, benzyloxy, (C3-8)cycloalkyloxy, (C3.8)cycloalkyl-(C1-10)alkoxy, C(=O)R, CN, C(=O)OR, C(O)NR2, NR2, NRC(=O)R, NRS(O)R, NRS(O)2R, SR, S(O)R, S(O)2R, S(O)NR2 and O, or two substituents α together form a (C1-4)alkylenedioxy group; substituents β are selected from (C1-10)alkyI, (C1-10)haloalkyl, hydroxy-(C1-i0)alkyl, (C1- 10)alkoxy(Ci,10)alkyl, phenyl, halogen, OH, (C1-10)alkoxy,  phenoxy, benzyloxy, (C3.8)cycloalkyloxy, (C3-8)cycloalkyl-(C1-10)alkoxy, C(O)R, CN, C(O)OR, C(O)NR2, NR2, NRC(O)R, NRS(O)R, NRS(O)2R, SR, S(O)R, S(O)2R, S(O)NR2 and pyridyl (optionally substituted by a (C1-10)alkyl group), or two substituents β together form a (C1-4)alkyleneoxy group or a (C1-4)alkylenedioxy group; substituents γ are selected from (C^alkyl, (C1-10)haloalkyl, phenyl, pyridyl, C(O)R, CN, C(O)OR, C(O)NR2, S(O)R and S(O)2R; or a pharmaceutically acceptable salt or solvate thereof.
phenoxy, benzyloxy, (C3.8)cycloalkyloxy, (C3-8)cycloalkyl-(C1-10)alkoxy, C(O)R, CN, C(O)OR, C(O)NR2, NR2, NRC(O)R, NRS(O)R, NRS(O)2R, SR, S(O)R, S(O)2R, S(O)NR2 and pyridyl (optionally substituted by a (C1-10)alkyl group), or two substituents β together form a (C1-4)alkyleneoxy group or a (C1-4)alkylenedioxy group; substituents γ are selected from (C^alkyl, (C1-10)haloalkyl, phenyl, pyridyl, C(O)R, CN, C(O)OR, C(O)NR2, S(O)R and S(O)2R; or a pharmaceutically acceptable salt or solvate thereof.
2. A compound according to Claim 1 , wherein the group NR2R3 is present at the 4- position of the pyridine ring (the pyridine ring nitrogen being the 1-position).
3. A compound according to Claim 1 or Claim 2, wherein X represents C(O), C(La-
R1)2, N(Lai-R1) or O, wherein
La represents a single bond, -0-, -(CO)-O-, -(CO)-NR-, -SO2-, -NR-, -NR-SO2-,
-NR(CO)-, or -N(Het1)(CO)-;
La> represents a single bond, -(CO)-, -(CO)-O- or -SO2-; and R, R1 and Het1 are as defined above, either in its broadest aspect or in a preferred aspect; or a group R and a group R1, together with the nitrogen atom to which they are attached, together form a group Het3 wherein Het3 is as defined in Claim 1.
4. A compound according to any one of Claims 1 to 3, wherein Y represents C(O), C(LZ-R1)2, N(LZ'-R1) or O, wherein
Lz represents a single bond, -(C=O)-O-, or -(CO)-NR-;
LZl represents a single bond or -(CO)-; and
R and R1 are as defined in claim 1 ; or a group R and a group R1, together with the nitrogen atom to which they are attached, together form a group Het3 wherein Het3 is as defined in Claim 1.
5. A compound according to any one of Claims 1 to 4, wherein m is 0.
6. A compound according to any one of Claims 1 to 4, wherein m is 1 and Z represents O or C(L-R1 )2, wherein L and R1 are as defined in Claim 1.
7. A compound according to any one of Claims 1 to 6, wherein R2 and R3 are the same or different and each represent R or benzyl, or R2 and R3, together with the nitrogen atom to which they are attached, form a group Het3, wherein R and Het3 are as defined in Claim 1.
8. A compound according to any one of Claims 1 to 7, wherein a is 0.
9. A compound according to any one of Claims 1 to 8, wherein b is 0.
10. A compound according to any one of Claims 1 to 8, wherein b is 1 and R5 represents (C^alkyl, F, Cl, Br, OH, (C1-6)alkoxy, CN, -(CR2)P-C(=O)OR10, -(CR2)p-C(=O)Het2, -(CR2)p-NRC(=O)R10, -(CR2)p-C(=O)N(R11)2, or -(CR2)p-C(=O)NR-(CR2)n-N(R11)2, wherein R, n, p, Het2, R10 and R11 are as defined in claim 1.
11. A compound according to any one of Claims 1 to 10, wherein R6, R7, R8 and R9 are each independently H or (C1-4)alkyl.
12. A compound according to Claim 1 , selected from:
N-[(3S)-1 '-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]-pyridine-2- carboxamide;
2-(3-methoxy-2,3-dihydro-rH-spiro[indene-1,4'-piperidin]-1'-yl)pyridin-4-amine;
1'-{4-[ben2yl(methyl)amino]pyridin-2-yl}-N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-
3-carboxamide;
N-[1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]benzenesulfonamide; 1'-(4-aminopyridin-2-yI)-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3-carbonitrile;
2-(6-methyl-1 'H,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1 '-yl)pyridin-4-amine;
2-(3-phenyl-1 Η,3H-spiro[2-benzofuran-1 ,4'-piperidin]-1 '-yl)pyridin-4-amine;
2-(1 H,1 Η-spiro[isochromene-4,4'-piperidin]-1 '-yl)pyridin-4-amine;
1'-(4-aminopyridin-2-yl)-N,N-dimethyl-2,3-dihydrospiro[indene-1,4'-piperidine]-3-carboxamide; ^-ti-CmethylsulfonyO-i ^-dihydro-VH-spirofindole-S^'-piperidinl-i'-yOpyridin^-amine;
2-[1-(ethylsulfonyl)-1 ,2-dihydro-1'H-spiro[indole-3,4l-piperidin]-1'-yl]pyridin-4-amine;
2-(2,3-dihydro-1'H-spiro[chromene-4,4'-piperidin]-1'-yl)pyridin-4-amine;
(3R)-N-[I ■-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1,4'-piperidin]-3-yl]benzamide;
(3S)-N-[r-(4-aminopyridin-2-yl)-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-yl]benzamide; 2-[(3S)-3-(5-phenyl-1 ,3,4-oxadiazol-2-yl)-2,3-dihydro-1Η-spiro[indene-1 ,4l-pipericiin]-1l-yl]- pyridin-4-amine;
2-[(3S)-3-(4-methyl-1 ,3-oxazol-2-yl)-2,3-dihydro-1 Η-spiro[indene-1 ,4'-piperidin]-1 '-yl]pyridin-4- amine; N-[1 '-(4-aminopyridin-2-yl)-2,3:dihydrospiro[indene-1 ,4'-piperidin]-3-yl]pyridine-2- carboxamide;
1'-(4-aminopyridin-2-yl)-N-pyridin-2-yl-2,3-dihydrospiro[indene-1 ,4'-piperidin]-3-amine; methyl 1'-(4-aminopyridin-2-yl)-2,3-dihydrospiro[chromene-4,4'-piperidine]-2-carboxylate;
(3S)-1'-(4-aminopyridin-2-yl)-N-methyI-N-propyl-2,3-dihydrospiro[indene-1,4'-piperidine]-3- carboxamide;
(3S)-1'-(4-aminopyridin-2-yl)-N-ethyl-N-methyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-1'-(4-aminopyridin-2-yl)-N-pyridin-2-yl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide; (3S)-1'-(4-aminopyridin-2-yl)-N-benzyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-1'-(4-aminopyridin-2-yl)-N,N-dimethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide;
(3S)-1'-(4-aminopyridin-2-yi)-N,N-diethyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
(3R)-1'-(4-aminopyridin-2-yl)-N,N-diethyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
(3S)-1'-(4-aminopyridin-2-yl)-N,N-diethyl-2,3-dihydrospiro[indene-1 ,4'-piperidine]-3- carboxamide; (+)-1'-(4-aminopyridin-2-yl)-N,N-dimethyl-5-methoxy-3H-spiro[2-benzo-furan-1,4'-piperidine]-
3-carboxamide;
(+)-r-(4-aminopyridin-2-yl)-N,N-diethyl-5-methoxy-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide;
1'-(4-aminopyridin-2-yl)-N,N-dimethyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3-carboxamide; 1 '-(4-aminopyridin-2-yl)-N-ethyl-N-methyl-3H-spiro[2-benzofuran-1 ,4'-piperidine]-3- carboxamide; and methyl 1 '-(4-aminopyridin-2-yl)spiro[indole-3,4'-piperidine]-1 (2H)-carboxylate; or a pharmaceutically acceptable salt or solvate thereof.
13. A pharmaceutical composition comprising an effective amount of a compound according to any one of claims 1 to 12, or a pharmaceutically acceptable salt or solvate thereof, together with a pharmaceutically acceptable carrier or diluent.
14. A compound according to any one of claims 1 to 12, or a pharmaceutically acceptable salt or solvate thereof, for use as a medicament.
15. Use of a compound according to any one of claims 1 to 12, or a pharmaceutically acceptable salt or solvate thereof, in the manufacture of a medicament for the treatment of diseases or conditions mediated by the delta opioid receptor.
16. Use according to claim 15, wherein the disease or condition is pain.
17. Use according to claim 16, wherein the pain is neuropathic pain.
18. A method for the treatment of a disease or condition mediated by the delta opioid receptor, comprising administering to a mammal an effective amount of a compound according to any one of claims 1 to 12, or a pharmaceutically acceptable salt or solvate thereof.
19. A method according to claim 18, wherein the disease or condition is pain.
20. A method according to claim 19, wherein the pain is neuropathic pain.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US73881005P | 2005-11-21 | 2005-11-21 | |
| US60/738,810 | 2005-11-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007057775A1 true WO2007057775A1 (en) | 2007-05-24 |
Family
ID=37762568
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2006/003320 Ceased WO2007057775A1 (en) | 2005-11-21 | 2006-11-16 | Spiropiperidine derivatives |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2007057775A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012004378A1 (en) | 2010-07-09 | 2012-01-12 | Abbott Healthcare Products B.V. | Spiro-cyclic amine derivatives as s1p modulators |
| CN102464663A (en) * | 2010-11-11 | 2012-05-23 | 上海药明康德新药开发有限公司 | Synthesis method of 1 '-tert-butyloxycarbonyl-spiro [ chroman-4, 4' -piperidine ] -2-formic acid |
| WO2012168162A1 (en) | 2011-06-06 | 2012-12-13 | F. Hoffmann-La Roche Ag | Benzocycloheptene acetic acids |
| WO2012172043A1 (en) | 2011-06-15 | 2012-12-20 | Laboratoire Biodim | Purine derivatives and their use as pharmaceuticals for prevention or treatment of bacterial infections |
| WO2014072903A1 (en) | 2012-11-06 | 2014-05-15 | Actelion Pharmaceuticals Ltd | Novel aminomethyl-phenol derivatives as antimalarial agents |
| EP3601239A4 (en) * | 2017-03-23 | 2020-05-13 | Jacobio Pharmaceuticals Co., Ltd. | NOVEL HETEROCYCLIC DERIVATIVES USEFUL AS SHP2 INHIBITORS |
| US11535632B2 (en) | 2019-10-31 | 2022-12-27 | ESCAPE Bio, Inc. | Solid forms of an S1P-receptor modulator |
| WO2023284833A1 (en) * | 2021-07-14 | 2023-01-19 | 宜昌人福药业有限责任公司 | Spirocyclic piperidine derivative, pharmaceutical composition thereof, preparation method therefor, and use thereof |
| EP4192957A4 (en) * | 2020-08-07 | 2024-09-04 | Casma Therapeutics, Inc. | TRPML MODULATORS |
| US12233062B2 (en) | 2018-09-26 | 2025-02-25 | Jacobio Pharmaceuticals Co., Ltd. | Heterocyclic derivatives useful as SHP2 inhibitors |
| WO2025201477A1 (en) * | 2024-03-28 | 2025-10-02 | Pyrotech (Beijing) Biotechnology Co.,Ltd. | Spirocyclic gsdme inhibitors |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004004715A2 (en) * | 2002-07-09 | 2004-01-15 | Glaxosmithkline Spa | Novel delta opioid receptor ligands |
| US20050038060A1 (en) * | 2001-06-26 | 2005-02-17 | Koji Ando | Spiropoperidine compounds as ligands for orl-1 receptor |
| WO2005092858A2 (en) * | 2004-03-29 | 2005-10-06 | Pfizer Japan Inc. | Alpha aryl or heteroaryl methyl beta piperidino propanamide compounds as orl1-receptor antagonist |
-
2006
- 2006-11-16 WO PCT/IB2006/003320 patent/WO2007057775A1/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050038060A1 (en) * | 2001-06-26 | 2005-02-17 | Koji Ando | Spiropoperidine compounds as ligands for orl-1 receptor |
| WO2004004715A2 (en) * | 2002-07-09 | 2004-01-15 | Glaxosmithkline Spa | Novel delta opioid receptor ligands |
| WO2005092858A2 (en) * | 2004-03-29 | 2005-10-06 | Pfizer Japan Inc. | Alpha aryl or heteroaryl methyl beta piperidino propanamide compounds as orl1-receptor antagonist |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10807991B2 (en) | 2010-07-09 | 2020-10-20 | Abbvie B.V. | Spiro-cyclic amine derivatives as S1P modulators |
| US11427598B2 (en) | 2010-07-09 | 2022-08-30 | AbbVie Deutschland GmbH & Co. KG | Spiro-cyclic amine derivatives as S1P modulators |
| EP3144312A1 (en) | 2010-07-09 | 2017-03-22 | AbbVie B.V. | Spiro-cyclic amine derivatives as s1p modulators |
| WO2012004378A1 (en) | 2010-07-09 | 2012-01-12 | Abbott Healthcare Products B.V. | Spiro-cyclic amine derivatives as s1p modulators |
| JP2013535412A (en) * | 2010-07-09 | 2013-09-12 | アブビー・ベー・ブイ | Spirocyclic amine derivatives as S1P modulators |
| CN102464663A (en) * | 2010-11-11 | 2012-05-23 | 上海药明康德新药开发有限公司 | Synthesis method of 1 '-tert-butyloxycarbonyl-spiro [ chroman-4, 4' -piperidine ] -2-formic acid |
| WO2012168162A1 (en) | 2011-06-06 | 2012-12-13 | F. Hoffmann-La Roche Ag | Benzocycloheptene acetic acids |
| WO2012172043A1 (en) | 2011-06-15 | 2012-12-20 | Laboratoire Biodim | Purine derivatives and their use as pharmaceuticals for prevention or treatment of bacterial infections |
| WO2014072903A1 (en) | 2012-11-06 | 2014-05-15 | Actelion Pharmaceuticals Ltd | Novel aminomethyl-phenol derivatives as antimalarial agents |
| US10988466B2 (en) | 2017-03-23 | 2021-04-27 | Jacobio Pharmaceuticals Co., Ltd. | Heterocyclic derivatives useful as SHP2 inhibitors |
| EP3601239A4 (en) * | 2017-03-23 | 2020-05-13 | Jacobio Pharmaceuticals Co., Ltd. | NOVEL HETEROCYCLIC DERIVATIVES USEFUL AS SHP2 INHIBITORS |
| US12233062B2 (en) | 2018-09-26 | 2025-02-25 | Jacobio Pharmaceuticals Co., Ltd. | Heterocyclic derivatives useful as SHP2 inhibitors |
| US11535632B2 (en) | 2019-10-31 | 2022-12-27 | ESCAPE Bio, Inc. | Solid forms of an S1P-receptor modulator |
| EP4192957A4 (en) * | 2020-08-07 | 2024-09-04 | Casma Therapeutics, Inc. | TRPML MODULATORS |
| WO2023284833A1 (en) * | 2021-07-14 | 2023-01-19 | 宜昌人福药业有限责任公司 | Spirocyclic piperidine derivative, pharmaceutical composition thereof, preparation method therefor, and use thereof |
| WO2025201477A1 (en) * | 2024-03-28 | 2025-10-02 | Pyrotech (Beijing) Biotechnology Co.,Ltd. | Spirocyclic gsdme inhibitors |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN104968647B (en) | Acid amides as sodium channel modulators | |
| US9051296B2 (en) | Aryl carboxamide derivatives as TTX-S blockers | |
| CA2574600C (en) | Pyridine derivatives | |
| US8969383B2 (en) | Picolinamide derivatives as TTX-S blockers | |
| KR102533094B1 (en) | Oxadiazole transient receptor potential channel inhibitor | |
| TWI769266B (en) | AMIDE DERIVATIVES AS Nav1.7 AND Nav1.8 BLOCKERS | |
| US12503439B2 (en) | Heteroaryl compounds for the treatment of pain | |
| KR102067235B1 (en) | Amide derivatives as ttx-s blockers | |
| JP7055528B1 (en) | Ketoamide derivative as a protease inhibitor | |
| US12110287B2 (en) | Heterocyclic derivatives as Nav1.7 and Nav1.8 blockers | |
| WO2012020567A1 (en) | Acyl piperazine derivatives as ttx-s blockers | |
| CN114874209A (en) | Oxadiazole transient receptor potential channel inhibitors | |
| WO2007057775A1 (en) | Spiropiperidine derivatives | |
| JP2019521163A (en) | Bicyclic proline compounds | |
| US20150250785A1 (en) | Tropomyosin-Related Kinase Inhibitors | |
| CA3059687C (en) | Amide derivatives as nav1.7 and nav1.8 blockers | |
| HK40070400A (en) | Oxadiazole transient receptor potential channel inhibitors | |
| WO2016020784A1 (en) | N-acylpyrrolidine ether tropomyosin-related kinase inhibitors | |
| HK40053076A (en) | Heterocyclic derivatives as nav1.7 and nav1.8 blockers | |
| HK40009776A (en) | Oxadiazolones as transient receptor potential channel inhibitors | |
| BR112019021736B1 (en) | AMIDE-DERIVED COMPOUND AS A NAV 1.7 AND NAV 1.8 BLOCKER, PHARMACEUTICAL COMPOSITION, USE OF THE COMPOUND AND PROCESS FOR PREPARING A PHARMACEUTICAL COMPOSITION | |
| HK1223611A1 (en) | Fused piperidine amides as modulators of ion channels |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06809246 Country of ref document: EP Kind code of ref document: A1 |