WO2007051846A1 - Tricyclo substituted amides - Google Patents
Tricyclo substituted amides Download PDFInfo
- Publication number
- WO2007051846A1 WO2007051846A1 PCT/EP2006/068088 EP2006068088W WO2007051846A1 WO 2007051846 A1 WO2007051846 A1 WO 2007051846A1 EP 2006068088 W EP2006068088 W EP 2006068088W WO 2007051846 A1 WO2007051846 A1 WO 2007051846A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pharmaceutically acceptable
- acceptable salt
- compound according
- compound
- formula
- Prior art date
Links
- 0 C*c1ncc(C)cc1 Chemical compound C*c1ncc(C)cc1 0.000 description 3
- IOFLMISZYNEXKT-UHFFFAOYSA-N OC(C(CC1CCOCC1)c(cc1)cc(Cl)c1S(C1CC1)(=O)=O)=O Chemical compound OC(C(CC1CCOCC1)c(cc1)cc(Cl)c1S(C1CC1)(=O)=O)=O IOFLMISZYNEXKT-UHFFFAOYSA-N 0.000 description 1
- ZSOAGUHLSCFQFY-UHFFFAOYSA-N OC(C(CC1CCOCC1)c(cc1)cc(Cl)c1SC1CC1)=O Chemical compound OC(C(CC1CCOCC1)c(cc1)cc(Cl)c1SC1CC1)=O ZSOAGUHLSCFQFY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/04—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D309/06—Radicals substituted by oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention is directed to tri(cyclo) substituted amide compounds.
- the present invention is directed to amide compounds substituted i) at the carbonyl carbon with an ethyl attached to a phenyl ring and a heterocyclic ring, and ii) at the amino with a nitrogen bearing heteroaryl ring, which are modulators of glucokinase and are useful in the prophylactic or therapeutic treatment of hyperglycemia and diabetes, particularly type II diabetes.
- Glucokinase (“GK”) is believed to be important in the body's regulation of its plasma glucose level. GK, found principally in the liver and pancreas, is one of four hexokinases that catalyze the initial metabolism of glucose.
- GK pathway is saturated at higher glucose levels than the other hexokinase pathways (see R.L. Printz et al., Annu. Rev. Nutr., 13:463- 496 (1993)).
- GK is critical to maintaining the glucose balance in mammals. Animals that do not express GK die soon after birth with diabetes, while animals that overexpress GK have improved glucose tolerance. Activation of GK can lead to hyperinsulinemic hypoglycemia (see, for example, H.B.T. Christesen et al., Diabetes, 51:1240-1246 (2002)). Additionally, type II maturity-onset diabetes of the young is caused by the loss of function mutations in the GK gene, suggesting that GK operates as a glucose sensor in humans (Y.
- International Patent Publication No. WO2003/015774 describes compounds as GK modulators.
- International Patent Publication No. WO2003047626 describes the use of a GK activator in combination with a glucagon antagonist for treating type II diabetes.
- International Patent Publication No. WO2003/055482 describes amide derivatives as GK activators.
- International Patent Publication No. WO2003/080585 describes aminobenzamide derivatives with GK activity for the treatment of diabetes and obesity.
- International Patent Publication No. WO2003/097824 describes human liver GK crystals and their used for structure-based drug design.
- International Patent Publication No. WO2004/002481 discloses arylcarbonyl derivatives as GK activators.
- International Patent Publication Nos. WO2004/072031 and WO2004/072066 disclose tri(cyclo) substituted amide compounds as GK activators.
- the present invention provides novel GK activators which may demonstrate improved properties desirable for pharmaceutical products compared to known GK activators, such as increased potency.
- (I) or pharmaceutically acceptable salts thereof are useful in the prophylactic or therapeutic treatment of hyperglycemia and diabetes, particularly type II diabetes.
- A is a nitrogen containing heteroaryl ring selected from 5-methylpyrazin-2- yl, 5-methylpyrid-2-yl, 5-chloropyrid-2-yl, pyrid-2-yl, 5-methylisoxazol-3-yl, isoxazol-3-yl, 5-methylthiazol-2-yl, 6-methylpyridazin-3-yl, l-methylpyrazol-3-yl, pyrazin-2-yl and pyrimidin-4-yl; and pharmaceutically acceptable salts thereof.
- A is preferably 5-methylpyrazin-2-yl or pyrazin-2-yl.
- A represents 5-methylpyrazin-2-yl
- A represents 5-methylpyrid-2-yl
- A represents 5-chloropyrid-2-yl
- A represents pyrid-2-yl
- A represents 5-methylisoxazol-3-yl
- A represents isoxazol-3-yl
- A represents 5-methylthiazol-2-yl
- A represents 6-methylpyridazin-3- yl
- A represents l-methylpyrazol-3-yl:
- A represents pyrazin-2-yl
- A represents pyrimidin-4-yl
- the carbon atom linking the phenyl ring and the tetrahydropyran containing sidechain to the amide carbonyl carbon is a chiral centre. Accordingly, at this centre, the compound may be present either as a racemate or as a single enantiomer in the (R)- or ( ⁇ -configuration.
- salts includes salts prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- Such acids include, for example, acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid and the like.
- Particularly preferred are citric, hydrobromic, hydrochloric, maleic, phosphoric, sulfuric, methanesulfonic, and tartaric acids.
- the present invention includes any possible solvates and polymorphic forms.
- the type of solvent that forms the solvate is not particularly limited so long as the solvent is pharmacologically acceptable.
- water, ethanol, propanol, acetone or the like can be used.
- the compounds of Formula (I) are intended for pharmaceutical use they are preferably provided in substantially pure form, for example at least 60% pure, more suitably at least 75% pure, at least 95% pure and especially at least 98% pure (% are on a weight for weight basis).
- the invention also encompasses a pharmaceutical composition that is comprised of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, in combination with a pharmaceutically acceptable carrier.
- a pharmaceutically acceptable carrier Preferably the composition is comprised of a pharmaceutically acceptable carrier and a non-toxic therapeutically effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof.
- the invention encompasses a pharmaceutical composition for the prophylaxis or treatment of hyperglycemia and diabetes, particularly type II diabetes, by the activation of GK, comprising a pharmaceutically acceptable carrier and a non-toxic therapeutically effective amount of compound of Formula (I), or a pharmaceutically acceptable salt thereof.
- a pharmaceutical composition for the prophylaxis or treatment of hyperglycemia and diabetes, particularly type II diabetes, by the activation of GK comprising a pharmaceutically acceptable carrier and a non-toxic therapeutically effective amount of compound of Formula (I), or a pharmaceutically acceptable salt thereof.
- the invention also provides the use of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, as a pharmaceutical.
- the compounds and compositions of the present invention are effective for treating hyperglycemia and diabetes, particularly type II diabetes, in mammals such as, for example, humans.
- the invention also provides a method of prophylactic or therapeutic treatment of a condition where activation of GK is desirable comprising a step of administering an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof.
- the invention also provides a method of prophylactic or therapeutic treatment of hyperglycemia or diabetes, particularly type II diabetes, comprising a step of administering an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof.
- the invention also provides a method for the prevention of diabetes, particularly type II diabetes, in a human demonstrating pre-diabetic hyperglycemia or impaired glucose tolerance comprising a step of administering an effective prophylactic amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof.
- the invention also provides the use of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, as a GK activator.
- the invention also provides the use of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, for the prophylactic or therapeutic treatment of hyperglycemia or diabetes, particularly type II diabetes.
- the invention also provides the use of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, for the prevention of diabetes, particularly type II diabetes, in a human demonstrating pre-diabetic hyperglycemia or impaired glucose tolerance.
- the invention also provides the use of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the activation of GK.
- the invention also provides the use of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the prophylactic or therapeutic treatment of hyperglycemia or diabetes, particularly type II diabetes.
- the invention also provides the use of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the prevention of diabetes, particularly type II diabetes, in a human demonstrating pre-diabetic hyperglycemia or impaired glucose tolerance.
- the compounds and compositions of the present invention may be optionally employed in combination with one or more other anti-diabetic agents or anti-hyperglycemic agents, which include, for example, sulfonylureas (e.g.
- glyburide glimepiride, glipyride, glipizide, chlorpropamide, gliclazide, glisoxepid, acetohexamide, glibornuride, tolbutamide, tolazamide, carbutamide, gliquidone, glyhexamide, phenbutamide, tolcyclamide, etc.), biguanides (e.g. metformin, phenformin, buformin, etc.), glucagon antagonists (e.g. a peptide or non-peptide glucagon antagonist), glucosidase inhibitors (e.g. acarbose, miglitol, etc.), insulin secetagogues, insulin sensitizers (e.g.
- compositions of the present invention comprise a compound of Formula (I), or a pharmaceutically acceptable salt thereof, as an active ingredient, a pharmaceutically acceptable carrier and optionally other therapeutic ingredients or adjuvants.
- compositions include compositions suitable for oral, rectal, topical, and parenteral (including subcutaneous, intramuscular, and intravenous) administration, as well as administration through inhaling, although the most suitable route in any given case will depend on the particular host, and nature and severity of the conditions for which the active ingredient is being administered.
- the pharmaceutical compositions may be conveniently presented in unit dosage form and prepared by any of the methods well known in the art of pharmacy.
- compositions according to the invention are preferably adapted for oral administration.
- the compounds of Formula (I), or pharmaceutically acceptable salts thereof can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques.
- the carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g. oral or parenteral (including intravenous).
- the pharmaceutical compositions of the present invention can be presented as discrete units suitable for oral administration such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient.
- compositions can be presented as a powder, as granules, as a solution, as a suspension in an aqueous liquid, as a non-aqueous liquid, as an oil- in- water emulsion, or as a water-in-oil liquid emulsion.
- the compounds of Formula (I), or a pharmaceutically acceptable salt thereof may also be administered by controlled release means and/or delivery devices.
- the compositions may be prepared by any of the methods of pharmacy. In general, such methods include a step of bringing into association the active ingredient with the carrier that constitutes one or more necessary ingredients. In general, the compositions are prepared by uniformly and intimately admixing the active ingredient with liquid carriers or finely divided solid carriers or both.
- the product can then be conveniently shaped into the desired presentation.
- compositions of this invention may include a pharmaceutically acceptable carrier and a compound of Formula (I), or a pharmaceutically acceptable salt thereof.
- the compounds of Formula (I), or pharmaceutically acceptable salts thereof, can also be included in pharmaceutical compositions in combination with one or more other therapeutically active compounds.
- compositions of this invention include pharmaceutically acceptable liposomal formulations containing a compound of Formula (I), or a pharmaceutically acceptable salt thereof.
- the pharmaceutical carrier employed can be, for example, a solid, liquid, or gas.
- solid carriers include lactose, terra alba, sucrose, talc, gelatin, agar, pectin, acacia, magnesium stearate, and stearic acid.
- liquid carriers are sugar syrup, peanut oil, olive oil, and water.
- gaseous carriers include carbon dioxide and nitrogen.
- oral liquid preparations such as suspensions, elixirs and solutions
- carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents, and the like
- oral solid preparations such as powders, capsules, and tablets.
- tablets and capsules are the preferred oral dosage units whereby solid pharmaceutical carriers are employed.
- tablets may be coated by standard aqueous or nonaqueous techniques.
- a tablet containing the composition of this invention may be prepared by compression or molding, optionally with one or more accessory ingredients or adjuvants.
- Compressed tablets may be prepared by compressing, in a suitable machine, the active ingredient in a free-flowing form such as powder or granules, optionally mixed with a binder, lubricant, inert diluent, surface active or dispersing agent or other such excipient.
- excipients may be, for example, inert diluents such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example, starch, gelatin, or acacia; and lubricating agents, for example, magnesium stearate, stearic acid, or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer time.
- a time delay material such as glyceryl monostearate, or glyceryl distearate may be used.
- the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate, or kaolin.
- the active ingredient is mixed with water or an oil medium, for example, peanut oil, liquid paraffin, or olive oil.
- Molded tablets may be made by molding in a suitable machine, a mixture of the powdered compound moistened with an inert liquid diluent.
- Each tablet preferably contains from about 0.05mg to about 5g of the active ingredient and each cachet or capsule preferably contains from about 0.05mg to about 5g of the active ingredient.
- a formulation intended for the oral administration to humans may contain from about 0.5mg to about 5g of active agent, compounded with an appropriate and convenient amount of carrier material which may vary from about 5 to about 95% of the total composition.
- Unit dosage forms will generally contain between from about lmg to about 2g of the active ingredient, typically 25mg, 50mg, lOOmg, 200mg, 300mg, 400mg, 500mg, 600mg, 800mg, or lOOOmg.
- compositions of the present invention suitable for parenteral administration may be prepared as solutions or suspensions of the active compounds in water.
- a suitable surfactant can be included such as, for example, hydroxypropylcellulose.
- Dispersions can also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof in oils. Further, a preservative can be included to prevent the detrimental growth of microorganisms .
- compositions of the present invention suitable for injectable use include sterile aqueous solutions or dispersions.
- the compositions can be in the form of sterile powders for the extemporaneous preparation of such sterile injectable solutions or dispersions.
- the final injectable form must be sterile and must be effectively fluid for easy syringability.
- the pharmaceutical compositions must be stable under the conditions of manufacture and storage and thus, preferably should be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g. glycerol, propylene glycol, and liquid polyethylene glycol), vegetable oils, and suitable mixtures thereof.
- compositions of the present invention can be in a form suitable for topical use such as, for example, an aerosol, cream, ointment, lotion, dusting powder, or the like. Further, the compositions can be in a form suitable for use in transdermal devices.
- formulations may be prepared, utilizing a compound of Formula (I), or a pharmaceutically acceptable salt thereof, via conventional processing methods.
- a cream or ointment is prepared by admixing hydrophilic material and water, together with about 5wt% to about 10wt% of the compound of Formula (I), to produce a cream or ointment having a desired consistency.
- compositions of this invention can be in a form suitable for rectal administration wherein the carrier is a solid. It is preferable that the mixture forms unit dose suppositories. Suitable carriers include cocoa butter and other materials commonly used in the art. The suppositories may be conveniently formed by first admixing the composition with the softened or melted carrier(s) followed by chilling and shaping in molds.
- compositions of this invention can be in a form suitable for inhaled administration.
- Such administration can be in forms and utilizing carriers described in, for example, 1) Particulate Interactions in Dry Powder Formulations for Inhalation, Xian Zeng et al, 2000, Taylor and Francis, 2) Pharmaceutical Inhalation Aerosol Technology, Anthony Hickey, 1992, Marcel Dekker, 3) Respiratory Drug Delivery, 1990, Editor: P.R. Byron, CRC
- compositions described above may include, as appropriate, one or more additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface-active agents, thickeners, lubricants, preservatives (including anti- oxidants) and the like.
- additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface-active agents, thickeners, lubricants, preservatives (including anti- oxidants) and the like.
- additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface-active agents, thickeners, lubricants, preservatives (including anti- oxidants) and the like.
- additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface-active agents, thickeners, lubricants, preservatives (including anti- oxidants) and the like.
- other adjuvants can be included to render the formulation isotonic with the blood of the
- dosage levels of the order of from about O.Olmg/kg to about 150mg/kg of body weight per day are useful in the treatment of the above- indicated conditions, or alternatively about 0.5mg to about 1Og per patient per day.
- type II diabetes may be effectively treated by the administration of from about 0.01 to lOOmg of the compound per kilogram of body weight per day, or alternatively about 0.5mg to about 7g per patient per day. It is understood, however, that the specific dose level for any particular patient will depend upon a variety of factors including the age, body weight, general health, sex, diet, time of administration, route of administration, rate of excretion, drug combination and the severity of the disease in the particular diabetic patient undergoing therapy.
- the compounds and salts thereof of this invention can be administered at subtherapeutic levels prophylactically in anticipation of a hyperglycemic condition.
- the compounds of Formula (I) may exhibit advantageous properties compared to known glucokinase activators, such properties may be illustrated in the assays described herein or in other assays known to those skilled in the art.
- the compounds of the invention may also demonstrate one or more of the following properties compared to known compounds: reduced neurotoxicity, longer duration of action (e.g. improved half-life/higher plasma protein binding), improved bioavailability, and /or increased potency (e.g. in vitro or in vivo).
- the carboxylic acid II, or an activated derivative thereof may be condensed with the amine III, or a salt thereof e.g. the hydrochloride salt, using a variety of coupling conditions known to those skilled in the art.
- a reagent that causes negligible racemisation e.g. benzotriazol-l-yloxytris(pyrrolidino)phosphonium hexafluorophosphate (J. Coste et al., Tetrahedron Lett., 1990, 31, 205-208), to furnish enantiopure amides of Formula (I).
- the carboxylic acid carboxylic acid II may be treated with (COC1) 2 and DMF in dichloromethane e.g. at -45°C, followed by the addition of the amine III and pyridine.
- a racemic mixture of amides can be prepared from racemic carboxylic acid II and then separated by means of chiral high performance liquid chromatography employing a chiral stationary phase (which can be purchased from, for example, Daicel Chemical Industries, Ltd, Tokyo, Japan) to provide the desired compound of Formula (I).
- a chiral stationary phase which can be purchased from, for example, Daicel Chemical Industries, Ltd, Tokyo, Japan
- the amines III are commercially available or are readily prepared using known techniques.
- the carboxylic acid II can be prepared following the protocol illustrated in Scheme 2 below: SCHEME 2
- the compound of formula IV will typically be converted to the enantiomerically pure carboxylic acid V (illustrated as the (R)-isomer) by catalytic hydrogenation (i), followed by the reaction with an enantiomerically pure chiral agent, separation of diasteriomers using conventional techniques, and finally the removal of the chiral resolving agent (ii).
- Racemic compounds V can be condensed, for example, with a chiral oxazolidinone derivative (see, for instance, F. T. Bizzarro et al. WO 00/58293) to generate a mixture of diastereoisomeric imides that are separable by any conventional method, e.g. column chromatography. Hydrolysis of the pure imides affords the stereopure (R)- and (S)-carboxylic acids that can then be condensed with heteroaryl amines III.
- the compound of formula IV can be converted to the enantiomerically pure carboxylic acid II directly by means of an asymmetric reduction (iii).
- Cyclopropylation of compound VI may be performed by means such as those described in J. Am. Chem. Soc. ⁇ 911 99:3080-3087 to provide the compound VII. Briefly, this involves treatment with Br(CH 2 ) 3 Cl in the presence of potassium hydroxide, followed by treatment with KNH 2 in the presence of FeN ⁇ 3. Subsequently compound VII may be converted into the acid of formula IV by a four step (Friedel-Crafts, oxidation, Wittig, saponification) process.
- the initial Friedel-Crafts step may be carried out in an analogous manner to that described in WO03/95438, namely compound VII may be treated with ethyl chlorooxoacetate in the presence of AICI3 in a solvent such as chloroform to yield (3-chloro-4-cyclopropylsulfanyl-phenyl)oxoacetic acid ethyl ester.
- the subsequent steps may be performed by analogous means to those described in WO2004/072031 for the preparation of the non- chlorinated equivalent of compound IV (Preparations 22 and 23 therein).
- labile functional groups in the intermediate compounds e.g. hydroxy, oxo, carboxy and amino groups
- the protecting groups may be removed at any stage in the synthesis of the compounds of Formula (I) or may be present on the final compound of Formula (I).
- a comprehensive discussion of the ways in which various labile functional groups may be protected and methods for cleaving the resulting protected derivatives is given in, for example, Protective Groups in Organic Chemistry, T. W. Greene and P.G.M. Wuts, (1991) Wiley-Interscience, New York, 2 nd edition.
- the invention also provides the novel intermediate of formula (II) and protected or activated derivatives thereof and the use of such compound in the synthesis of novel GK activators.
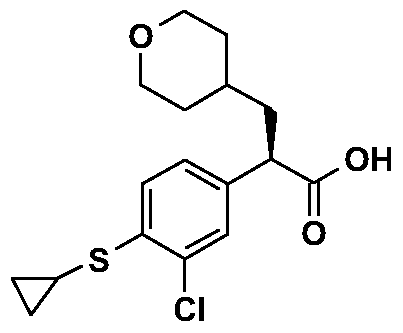
- the invention provides the compound (2R)-2-(3-chloro-4- cyclopropanesulfonylphenyl)-3-(tetrahydropyran-4-yl)propionic acid and protected or activated derivatives thereof.
- Tetrabutylammoniumchloride (Ig, 0.0035mol, 0.002eq) was added to a mixture of 2- bromo-3-chloropropane (262g, 1.660mol, 1.2eq), KOH (508g, 3.680mol, 2.66eq) and tBME (3.0L).
- a solution of 2-chlorothiophenol (20Og, 1.383mol, leq.) in tBME (0.2L) was added at rt followed by a first portion Of H 2 O (0.003L) which afforded gas evolution and an increase of the reaction temperature.
- a further amount Of H 2 O (0.027L) was carefully added keeping the temperature below 30 0 C.
- the crude product was dissolved in EtOAc (0.4L) and washed with an aqueous solution of citric acid (IM, 0.3L), H 2 O (2 x 10OmL), aqueous NaHC ⁇ 3 (10OmL), H 2 O (5OmL) and brine (5OmL).
- IM citric acid
- H 2 O 2 x 10OmL
- aqueous NaHC ⁇ 3 10OmL
- H 2 O H 2 O
- brine 5OmL
- the organic phase was dried (MgSOzi) and concentrated under reduced pressure to yield a foam.
- the crude product was used in the following step without further purification.
- Preparation 6 (52.Og, 0.133mol, leq) and THF (0.5L) were cooled to -50 0 C and a solution of lithium diisopropylamide (20OmL, 2M, 0.400mol, 3eq) added. The reaction was stirred at -40 0 C and a solution of 4-iodomethyltetrahydropyran (WO2004/072031, 30.2g, 0.133mol, leq) in THF (0.15L) was added. The cooling bath was removed and the reaction stirred overnight. The THF was evaporated under reduced pressure and the crude product triturated with aqueous citric acid (IL, 0.2M) and then extracted with tBME (0.5L).
- IL aqueous citric acid
- (2R)-2-(3-Chloro-4-cyclopropanesulfonylphenyl)-3-(tetrahydropyran-4-yl)propionic acid (Preparation 9) was coupled with amines selected from 2-amino-5-methylpyrazine, 3- amino-5-methylisoxazole, 3-aminoisoxazole, 2-amino-5-methylthiazole, 3-amino-6- methylpyridazine, l-methyl-3-aminopyrazole, 2-aminopyrazine and 4-aminopyrimidine using the following procedure to provide Examples 1-8.
- (2R)-2-(3-Chloro-4-cyclopropanesulfonylphenyl)-3-(tetrahydropyran-4-yl)propionic acid may also be coupled with amines selected from 2-amino-5- methylpyridine, 2-amino-5-chloropyridine and 2-aminopyridine using the procedure described above to provide Examples 9-11.
- GK activity may be measured by coupling the production of G6P by GST-GK to the generation of NADH with G6PDH as the coupling enzyme.
- the assay is performed at room temperature (23 0 C) in clear flat bottom 96-well plates in a total volume of 100 ⁇ l consisting of 25 mM Hepes (pH 7.4), 25 mM KCl, 5 mM D- glucose, 1 mM ATP, 1 mM NADP, 2 mM MgCl 2 , 1 mM dithiothreitol, 0.2 ⁇ g purified GST- GK derived from human liver GK and a range of activator concentrations in a final concentration of 5 % DMSO.
- the incubation time is 15 minutes at which time the reaction has been shown to be linear.
- the generation of NADH, as an indirect determination of GK activity, is measured at OD 340 in a SpectraMAX 190 microplate spectrophotometer (Molecular Devices Corp).
- a pre-dose blood GIc reading is acquired by snipping off a small portion of the animals' tails ( ⁇ lmm) and collecting 20 ⁇ L blood for analysis. After GK activator treatment, further blood GIc readings are taken at 0.5, 1.0, and 2.5h post-dose from the same tail wound. Results are interpreted by comparing the mean blood GIc values of the vehicle treated mice with the the GK activator treated mice over the study duration. Compounds are considered active when they exhibited a statistically significant decrease in blood GIc compared to vehicle for 2 consecutive assay time points following compound administration.
- GK activators typically reduce the area under the glucose curve by at least 20% in the 2h following administration of glucose.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Diabetes (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description






















Claims




Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BRPI0618067-1A BRPI0618067A2 (en) | 2005-11-03 | 2006-11-03 | compound or a pharmaceutically acceptable salt thereof, pharmaceutical composition, and use and process for the preparation of a compound or a pharmaceutically acceptable salt thereof |
| EP06819251A EP1948645A1 (en) | 2005-11-03 | 2006-11-03 | Tricyclo substituted amides |
| CA002628486A CA2628486A1 (en) | 2005-11-03 | 2006-11-03 | Tricyclo substituted amides |
| AU2006310475A AU2006310475A1 (en) | 2005-11-03 | 2006-11-03 | Tricyclo substituted amides |
| JP2008538365A JP2009514836A (en) | 2005-11-03 | 2006-11-03 | Tricyclo-substituted amide |
| US12/091,376 US20080293730A1 (en) | 2005-11-03 | 2006-11-11 | Tricyclo Substituted Amides |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0522456.3 | 2005-11-03 | ||
| GB0522456A GB0522456D0 (en) | 2005-11-03 | 2005-11-03 | Compounds |
| GB0603132A GB0603132D0 (en) | 2006-02-16 | 2006-02-16 | Compounds |
| GB0603132.2 | 2006-02-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007051846A1 true WO2007051846A1 (en) | 2007-05-10 |
Family
ID=37635686
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/068088 WO2007051846A1 (en) | 2005-11-03 | 2006-11-03 | Tricyclo substituted amides |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20080293730A1 (en) |
| EP (1) | EP1948645A1 (en) |
| JP (1) | JP2009514836A (en) |
| AU (1) | AU2006310475A1 (en) |
| BR (1) | BRPI0618067A2 (en) |
| CA (1) | CA2628486A1 (en) |
| WO (1) | WO2007051846A1 (en) |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2008012227A3 (en) * | 2006-07-24 | 2008-05-15 | Hoffmann La Roche | Pyrazoles as glucokinase activators |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2009091014A1 (en) | 2008-01-18 | 2009-07-23 | Astellas Pharma Inc. | Phenyl acetamide derivative |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| US7741327B2 (en) | 2008-04-16 | 2010-06-22 | Hoffmann-La Roche Inc. | Pyrrolidinone glucokinase activators |
| JP2010540667A (en) * | 2007-10-09 | 2010-12-24 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Pyridine derivatives useful as glucokinase activators |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| CN102146078A (en) * | 2010-02-09 | 2011-08-10 | 中国科学院上海药物研究所 | Substituted propanamide compounds and preparation method and application thereof |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| US8063079B2 (en) | 2008-12-19 | 2011-11-22 | Eli Lilly And Company | Cyclopropyl compounds |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| US8222416B2 (en) | 2009-12-14 | 2012-07-17 | Hoffmann-La Roche Inc. | Azaindole glucokinase activators |
| US8258134B2 (en) | 2008-04-16 | 2012-09-04 | Hoffmann-La Roche Inc. | Pyridazinone glucokinase activators |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8946440B2 (en) | 2008-04-28 | 2015-02-03 | Kyorin Pharmaceutical Co., Ltd. | Cyclopentylacrylamide derivative |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BRPI0514147A (en) | 2004-08-12 | 2008-05-27 | Prosidion Ltd | compound or a pharmaceutically acceptable salt thereof, pharmaceutical composition, methods of prophylactic or therapeutic treatment of a condition where gk activation is desirable, prophylactic or therapeutic treatment of hyperglycemia or diabetes, and prevention of diabetes in a human demonstrating hyperglycemia. pre-diabetic or impaired glucose tolerance, and process for preparing the compound |
| BRPI0618062A2 (en) * | 2005-11-03 | 2011-08-16 | Prosidion Ltd | compound or a pharmaceutically acceptable salt thereof, pharmaceutical composition, use of a compound or a pharmaceutically acceptable salt thereof, and process for the preparation of a compound |
| TW200831081A (en) * | 2006-12-25 | 2008-08-01 | Kyorin Seiyaku Kk | Glucokinase activator |
| EP2116533B1 (en) * | 2007-03-07 | 2013-07-10 | Kyorin Pharmaceutical Co., Ltd. | Glucokinase activator |
| AR070107A1 (en) * | 2008-01-15 | 2010-03-17 | Lilly Co Eli | R-2- (4-CYCLOPROPANSULFONYL-PHENYL) -N-PIRAZIN-2-IL-3- (TETRAHIDROPIRAN-4-IL) -PROPIONAMIDE IN CRYSTALINE FORM, PHARMACEUTICAL COMPOSITION THAT UNDERSTANDS AND USES IT FOR THE MANUFACTURE OF A USEFUL MEDICINAL PRODUCT FOR THE PREVENTION OR TREATMENT OF HYPERGLYCEMIA |
| BRPI0912802A2 (en) * | 2008-05-16 | 2015-10-13 | Takeda San Diego Inc | glycokinase activators |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004072031A2 (en) * | 2003-02-11 | 2004-08-26 | Prosidion Limited | Phenylacetamides and their use as glucokinase modulators |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080293741A1 (en) * | 2005-11-03 | 2008-11-27 | Matthew Colin Thor Fyfe | Tricyclo Substituted Amides as Glucokinase Modulators |
| BRPI0618062A2 (en) * | 2005-11-03 | 2011-08-16 | Prosidion Ltd | compound or a pharmaceutically acceptable salt thereof, pharmaceutical composition, use of a compound or a pharmaceutically acceptable salt thereof, and process for the preparation of a compound |
-
2006
- 2006-11-03 WO PCT/EP2006/068088 patent/WO2007051846A1/en active Application Filing
- 2006-11-03 JP JP2008538365A patent/JP2009514836A/en not_active Withdrawn
- 2006-11-03 EP EP06819251A patent/EP1948645A1/en not_active Withdrawn
- 2006-11-03 CA CA002628486A patent/CA2628486A1/en not_active Abandoned
- 2006-11-03 BR BRPI0618067-1A patent/BRPI0618067A2/en not_active IP Right Cessation
- 2006-11-03 AU AU2006310475A patent/AU2006310475A1/en not_active Abandoned
- 2006-11-11 US US12/091,376 patent/US20080293730A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004072031A2 (en) * | 2003-02-11 | 2004-08-26 | Prosidion Limited | Phenylacetamides and their use as glucokinase modulators |
Cited By (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7935699B2 (en) | 2006-07-24 | 2011-05-03 | Hoffmann-La Roche Inc. | Pyrazole glucokinase activators |
| WO2008012227A3 (en) * | 2006-07-24 | 2008-05-15 | Hoffmann La Roche | Pyrazoles as glucokinase activators |
| EP2261216A3 (en) * | 2006-07-24 | 2011-12-14 | F. Hoffmann-La Roche AG | Pyrazoles as glucokinase activators |
| EP2261216A2 (en) * | 2006-07-24 | 2010-12-15 | F. Hoffmann-La Roche AG | Pyrazoles as glucokinase activators |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| JP2010540667A (en) * | 2007-10-09 | 2010-12-24 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Pyridine derivatives useful as glucokinase activators |
| WO2009091014A1 (en) | 2008-01-18 | 2009-07-23 | Astellas Pharma Inc. | Phenyl acetamide derivative |
| US8329707B2 (en) | 2008-01-18 | 2012-12-11 | Astellas Pharma Inc. | Substituted pyrazine compounds |
| JP5287730B2 (en) * | 2008-01-18 | 2013-09-11 | アステラス製薬株式会社 | Phenylacetamide derivatives |
| US7741327B2 (en) | 2008-04-16 | 2010-06-22 | Hoffmann-La Roche Inc. | Pyrrolidinone glucokinase activators |
| US8258134B2 (en) | 2008-04-16 | 2012-09-04 | Hoffmann-La Roche Inc. | Pyridazinone glucokinase activators |
| US8946440B2 (en) | 2008-04-28 | 2015-02-03 | Kyorin Pharmaceutical Co., Ltd. | Cyclopentylacrylamide derivative |
| US9452977B2 (en) | 2008-04-28 | 2016-09-27 | Kyorin Pharmaceutical Co., Ltd. | Cyclopentylacrylamide derivative |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| US8063079B2 (en) | 2008-12-19 | 2011-11-22 | Eli Lilly And Company | Cyclopropyl compounds |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| US8222416B2 (en) | 2009-12-14 | 2012-07-17 | Hoffmann-La Roche Inc. | Azaindole glucokinase activators |
| CN102146078A (en) * | 2010-02-09 | 2011-08-10 | 中国科学院上海药物研究所 | Substituted propanamide compounds and preparation method and application thereof |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2006310475A1 (en) | 2007-05-10 |
| US20080293730A1 (en) | 2008-11-27 |
| JP2009514836A (en) | 2009-04-09 |
| BRPI0618067A2 (en) | 2011-08-16 |
| CA2628486A1 (en) | 2007-05-10 |
| EP1948645A1 (en) | 2008-07-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1948645A1 (en) | Tricyclo substituted amides | |
| US20080293741A1 (en) | Tricyclo Substituted Amides as Glucokinase Modulators | |
| EP1594867B1 (en) | Phenylacetamides and their use as glucokinase modulators | |
| US20090005391A1 (en) | Tricyclo Substituted Amides | |
| US7745491B2 (en) | Substituted phenylacetamides and their use as glucokinase activators | |
| US7262196B2 (en) | Tri(cyclo) substituted amide glucokinase activator compounds | |
| US20080242869A1 (en) | Tri(Cyclo) Substituted Amide Compounds | |
| NO330139B1 (en) | 5-substituted pyrazine or pyridine derivatives, the preparation of such, pharmaceutical preparations containing such, such compounds as active substances and the use of such compounds for the preparation of drugs for the treatment or prevention of disease | |
| CN101291930A (en) | Tricyclo substituted amides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680039164.9 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006310475 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2816/DELNP/2008 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2006310475 Country of ref document: AU Date of ref document: 20061103 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12091376 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006310475 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2008538365 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2628486 Country of ref document: CA Ref document number: MX/a/2008/005745 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006819251 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006819251 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0618067 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080429 |