WO2007039125A2 - An optically active thyroid receptor agonist and optically active key intermediates in its production - Google Patents
An optically active thyroid receptor agonist and optically active key intermediates in its production Download PDFInfo
- Publication number
- WO2007039125A2 WO2007039125A2 PCT/EP2006/009184 EP2006009184W WO2007039125A2 WO 2007039125 A2 WO2007039125 A2 WO 2007039125A2 EP 2006009184 W EP2006009184 W EP 2006009184W WO 2007039125 A2 WO2007039125 A2 WO 2007039125A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- ester
- formula
- amide
- salt
- Prior art date
Links
- 0 *c(cc(C[C@@](C(OCC1CCCC1)=O)F)cc1Br)c1O Chemical compound *c(cc(C[C@@](C(OCC1CCCC1)=O)F)cc1Br)c1O 0.000 description 8
- RSFXMOLIDUZAGZ-TWGQIWQCSA-N COC(/C(/F)=C/c(cc1)ccc1O)=O Chemical compound COC(/C(/F)=C/c(cc1)ccc1O)=O RSFXMOLIDUZAGZ-TWGQIWQCSA-N 0.000 description 1
- PDFUCDWETSQSSU-UHFFFAOYSA-N COC(C(Cc(cc1)ccc1O)O)=O Chemical compound COC(C(Cc(cc1)ccc1O)O)=O PDFUCDWETSQSSU-UHFFFAOYSA-N 0.000 description 1
- DPWHKCSGAXUYGL-UHFFFAOYSA-N COC(C(Cc(cc1Br)cc([Br]=C)c1O)O)=O Chemical compound COC(C(Cc(cc1Br)cc([Br]=C)c1O)O)=O DPWHKCSGAXUYGL-UHFFFAOYSA-N 0.000 description 1
- PDFUCDWETSQSSU-SECBINFHSA-N COC([C@@H](Cc(cc1)ccc1O)O)=O Chemical compound COC([C@@H](Cc(cc1)ccc1O)O)=O PDFUCDWETSQSSU-SECBINFHSA-N 0.000 description 1
- WOTHQHCCFZSVFW-UHFFFAOYSA-N COc1ccc(CC(/C(/O)=[O]/C2CCCC2)O)cc1 Chemical compound COc1ccc(CC(/C(/O)=[O]/C2CCCC2)O)cc1 WOTHQHCCFZSVFW-UHFFFAOYSA-N 0.000 description 1
- FTVDGXRTMDRADG-UHFFFAOYSA-N COc1ccc(CC(CO)O)cc1 Chemical compound COc1ccc(CC(CO)O)cc1 FTVDGXRTMDRADG-UHFFFAOYSA-N 0.000 description 1
- AKUZGLJFNSWSAV-UHFFFAOYSA-N CS(Nc1cccc(COc(c(Br)cc(CC(C(O)=O)F)c2)c2Br)c1Cl)(=O)=O Chemical compound CS(Nc1cccc(COc(c(Br)cc(CC(C(O)=O)F)c2)c2Br)c1Cl)(=O)=O AKUZGLJFNSWSAV-UHFFFAOYSA-N 0.000 description 1
- RGHHSNMVTDWUBI-UHFFFAOYSA-N Oc1ccc(C=O)cc1 Chemical compound Oc1ccc(C=O)cc1 RGHHSNMVTDWUBI-UHFFFAOYSA-N 0.000 description 1
- SGWSNMOWHZXGTE-AWEZNQCLSA-N Oc1ccc(C[C@@H](C(OCC2CCCC2)=O)F)cc1 Chemical compound Oc1ccc(C[C@@H](C(OCC2CCCC2)=O)F)cc1 SGWSNMOWHZXGTE-AWEZNQCLSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/42—Unsaturated compounds containing hydroxy or O-metal groups
- C07C59/56—Unsaturated compounds containing hydroxy or O-metal groups containing halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/07—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by halogen atoms
- C07C205/11—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by halogen atoms having nitro groups bound to carbon atoms of six-membered aromatic rings
- C07C205/12—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by halogen atoms having nitro groups bound to carbon atoms of six-membered aromatic rings the six-membered aromatic ring or a condensed ring system containing that ring being substituted by halogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/13—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by hydroxy groups
- C07C205/26—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by hydroxy groups and being further substituted by halogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/27—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by etherified hydroxy groups
- C07C205/35—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by etherified hydroxy groups having nitro groups and etherified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/49—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by carboxyl groups
- C07C205/56—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by carboxyl groups having nitro groups bound to carbon atoms of six-membered aromatic rings and carboxyl groups bound to acyclic carbon atoms of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/76—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings and etherified hydroxy groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/01—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms
- C07C311/02—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C311/08—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/48—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups having nitrogen atoms of sulfonamide groups further bound to another hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/73—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids
- C07C69/732—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids of unsaturated hydroxy carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
Definitions
- the present invention relates to optically active compounds which are agonists or partial agonists of the thyroid receptor and the use of such compounds for therapeutic purposes.
- thyroid agonists and antagonists for treatment of other important clinical indications, such as hypercholesterolemia, dyslipidemia, obesity, diabetes, atherosclerosis and cardiac diseases.
- Thyroid hormones affect the metabolism of virtually every cell of the body. At normal levels, these hormones maintain body weight, metabolic rate, body temperature and mood, and influence blood levels of serum lipoproteins. Thus, in hypothyroidism there is weight gain, high levels of LDL cholesterol, and depression. In hyperthyroidism, these hormones lead to weight loss, hypermetabolism, lowering of serum LDL cholesterol levels, cardiac arrhythmias, heart failure, muscle weakness, bone loss in postmenopausal women, and anxiety.
- Thyroid hormones are currently used primarily as replacement therapy for patients with hypothyroidism. Therapy with L-thyroxine returns metabolic functions to normal and can easily be monitored with routine serum measurements of levels of thyroid-stimulating hormone (TSH), thyroxine (3,5,3',5'-tetraiodo-L-thyronine, or T 4 ) and triiodothyronine (3,5,3'-triiodo-L-thyronine, or T 3 ).
- TSH thyroid-stimulating hormone
- thyroxine 3,5,3',5'-tetraiodo-L-thyronine, or T 4
- triiodothyronine 3,5,3'-triiodo-L-thyronine, or T 3
- replacement therapy particularly in older individuals, may be restricted by certain detrimental effects from thyroid hormones.
- thyroid hormones may be therapeutically useful in non-thyroid disorders if adverse effects can be minimized or eliminated.
- these potentially useful influences include for example, lowering of serum LDL levels, weight reduction, amelioration of depression and stimulation of bone formation.
- Prior attempts to utilize thyroid hormones pharmacologically to treat these disorders have been limited by manifestations of hyperthyroidism, and in particular by cardiovascular toxicity.
- useful thyroid agonist drugs should minimize the potential for undesired consequences due to locally induced hypothyroidism, i.e. sub-normal levels of thyroid hormone activity in certain tissues or organs. This can arise because increased circulating thyroid hormone agonist concentrations may cause the pituitary to suppress the secretion of thyroid stimulating hormone (TSH), thereby reducing thyroid hormone synthesis by the thyroid gland (negative feedback control). Since endogenous thyroid hormone levels are reduced, localized hypothyroidism can result wherever the administered thyroid agonist drug fails to compensate for the reduction in endogenous hormone levels in specific tissues.
- TSH thyroid stimulating hormone
- Tissue-selective thyroid hormone agonists may be obtained by selective tissue uptake or extrusion, topical or local delivery, targeting to cells through other ligands attached to the agonist and targeting receptor subtypes. Tissue selectivity can also be achieved by selective regulation of thyroid hormone responsive genes in a tissue specific manner.
- the compounds that are thyroid hormone receptor ligands, particularly selective agonists of the thyroid hormone receptor are expected to demonstrate a utility for the treatment or prevention of diseases or disorders associated with thyroid hormone activity, for example: (1) hypercholesterolemia, dyslipidemia or any other lipid disorder manifested by an unbalance of blood or tissue lipid levels; (2) atherosclerosis; (3) replacement therapy in elderly subjects with hypothyroidism who are at risk for cardiovascular complications; (4) replacement therapy in elderly subjects with subclinical hypothyroidism who are at risk for cardiovascular complications; (5) obesity; (6) diabetes (7) depression; (8) osteoporosis (especially in combination with a bone resorption inhibitor); (9) goiter; (10) thyroid cancer; (11) cardiovascular disease or congestive heart failure; (12) glaucoma; and (13) skin disorders.
- diseases or disorders associated with thyroid hormone activity for example: (1) hypercholesterolemia, dyslipidemia or any other lipid disorder manifested by an unbalance of blood or tissue lipid levels; (2) atherosclerosis; (3) replacement therapy in elderly subjects with
- the present invention provides the optically active (iS)-form of the compound of formula (I) 5
- ester, amide, solvate or salt thereof including a salt of such an ester or amide, and a solvate of such an ester, amide or salt.
- the compound is the (-) enantiomer.
- the (-) form of the compound is believed on the basis of X-ray crystal structure analysis to be the (S) isomer.
- the compound is (-)-3-[3,5- dibromo-4-( ⁇ 2-chloro-3- [(methylsulfonyl)amino]benzyl ⁇ oxy)phenyl] -2-fluoropropanoic acid, or (5)-3-[3,5-dibrom ⁇ -4-( ⁇ 2-chloro-3-[(methylsulfonyl)amino]benzyl ⁇ oxy)phenyl]-2- fluoropropanoic acid, or a pharmaceutically acceptable ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt.
- the optically active compound of the invention has surprisingly been found to be a particularly effective ligand of the thyroid receptor, in particular agonist or partial agonist of the thyroid receptor.
- the compounds accordingly have use in the treatment or prophylaxis of conditions associated with thyroid receptor activity.
- the (S) enantiomer ((-)-enantiomer) of the compound of the invention has advantages over the racemic mixture or the (+)-enantiomer in terms of various parameters.
- the compound is more effective in lowering lipid levels in vivo and it is more effective at binding to the thyroid receptor than the racemic mixture or the (+)-enantiomer.
- Optically active compounds stated to be the (+)-form or (-)-form are the compounds which show respectively (+)-or (-)- rotatory power with respect to the sodium D line.
- References to optically active compounds should be taken to include compounds in a mixture of enantiomers, but having an enantiomeric excess of at least 75%, preferably at least 85%, more preferably at least 95%, most preferably at least 97%, especially at least 99%. Accordingly, references to optically active compounds should be taken to include compounds in a mixture of enantiomers, but having a molar ratio of enantiomers of at least 7:1, preferably at least 37:3, more preferably at least 39: 1, most preferably at least 197:3, especially at least 199:1.
- Salts and solvates of compounds of formula (I) which are suitable for use in medicine are those wherein a counterion or associated solvent is pharmaceutically acceptable.
- salts and solvates having non-pharmaceutically acceptable counterions or associated solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of the compounds of formula (I) and their pharmaceutically acceptable salts, solvates and physiologically functional derivatives.
- physiologically functional derivatives include esters and amides.
- Suitable salts according to the invention include those formed with organic or inorganic acids or bases.
- Pharmaceutically acceptable acid addition salts include those formed from hydrochloric, hydrobromic, sulphuric, nitric, citric, tartaric, acetic, phosphoric, lactic, pyruvic, acetic, trifluoroacetic, succinic, perchloric, fumaric, maleic, glycollic, lactic, salicylic, oxaloacetic, methanesulfonic, ethanesulfonic, p-toluenesulfonic, formic, benzoic, malonic, naphthalene-2-sulfonic, benzenesulfonic, and isethionic acids.
- compositions of formula (I) may have an appropriate group, for example an acid group, converted to a Ci -6 alkyl, Cs -I0 aryl, C 5 . 10 aryl-Ci -6 alkyl, or amino acid ester or amide.
- a compound which, upon administration to the recipient, is capable of being converted into a compound of formula (I) as described above or an active metabolite or residue thereof, is known as a "prodrug".
- a prodrug may, for example, be converted within the body, e. g. by hydrolysis in the blood, into its active form that has medical effects.
- Pharmaceutically acceptable prodrugs are described in T. Higuchi and V. Stella, Prodrugs as Novel Delivery Systems, Vol. 14 of the A. C. S. Symposium Series (1976); and in Edward B. Roche, ed.,
- alkyl means both straight and branched chain saturated hydrocarbon groups.
- alkyl groups include methyl, ethyl, n-propyl, iso-propyl, n-butyl, t-butyl, i-butyl, sec-butyl pentyl, hexyl, heptyl, octyl, nonyl and decyl groups.
- unbranched alkyl groups there are preferred methyl, ethyl, n-propyl, iso-propyl, n-butyl groups.
- branched alkyl groups there may be mentioned t-butyl, i-butyl,
- aryl means a monocyclic or bicyclic aromatic carbocyclic group.
- aryl groups include phenyl and naphthyl.
- a naphthyl group may be attached through the 1 or the 2 position.
- one of the rings may, for example, be partially saturated.
- examples of such groups include indanyl and tetrahydronaphthyl.
- C 5-I0 aryl is used herein to mean a group comprising from 5 to 10 carbon atoms in a monocyclic or bicyclic aromatic group.
- a particularly preferred C 5-I0 aryl group is phenyl.
- the compounds of the invention have activity as thyroid receptor ligands.
- the compounds of the invention are preferably selective agonists or partial agonists of the thyroid receptor.
- compounds of the present invention possess activity as agonists of the thyroid receptor, preferably selective agonists of the thyroid receptor-beta. They may thus be used in the treatment of diseases or disorders associated with thyroid receptor activity, particularly diseases or disorders for which selective agonists of the thyroid receptor-beta are indicated. In particular, compounds of the present invention may be used in the treatment of diseases or disorders associated with metabolism dysfunction or which are dependent upon the expression of a T 3 regulated gene.
- Clinical conditions for which an agonist or partial agonist is indicated include, but are not limited to, hypothyroidism; subclinical hyperthyroidism; non-toxic goiter; atherosclerosis; thyroid hormone replacement therapy (e.g., in the elderly); malignant tumor cells containing the thyroid receptor; papillary or follicular cancer; maintenance of muscle strength and function (e.g., in the elderly); reversal or prevention of frailty or age-related functional decline ("ARPD”) in the elderly (e.g., sarcopenia); treatment of catabolic side effects of glucocorticoids; prevention and/or treatment of reduced bone mass, density or growth (e.g., osteoporosis and osteopenia); treatment of chronic fatigue syndrome (CFS); accelerating healing of complicated fractures (e.g.
- distraction osteogenesis in joint replacement; eating disorders (e.g., anorexia); treatment of obesity and growth retardation associated with obesity; treatment of depression, nervousness, irritability and stress; treatment of reduced mental energy and low self-esteem (e.g., motivation/assertiveness); improvement of cognitive function (e.g., the treatment of dementia, including Alzheimer's disease and short term memory loss); treatment of catabolism in connection with pulmonary dysfunction and ventilator dependency; treatment of cardiac dysfunction (e.g., associated with valvular disease, myocardial infarction, cardiac hypertrophy or congestive heart failure); lowering blood pressure; protection against ventricular dysfunction or prevention of reperfusion events; treatment of hyperinsulinemia; stimulation of osteoblasts, bone remodeling and cartilage growth; regulation of food intake; treatment of insulin resistance, including NIDDM, in mammals (e.g., humans); treatment of insulin resistance in the heart; treatment of congestive heart failure; treatment of musculoskeletal impairment (e.g., in the elderly); improvement of the overall pulmonary function; skin disorders
- the compounds of the invention find particular application in the treatment or prophylaxis of the following: (1) hypercholesterolemia, dyslipidemia or any other lipid disorder manifested by an unbalance of blood or tissue lipid levels ; (2) atherosclerosis; (3) replacement therapy in elderly subjects with hypothyroidism who are at risk for cardiovascular complications; (4) replacement therapy in elderly subjects with subclinical hypothyroidism who are at risk for cardiovascular complications; (5) obesity; (6) diabetes (7) depression; (8) osteoporosis (especially in combination with a bone resorption inhibitor); (9) goiter; (10) thyroid cancer; (11) cardiovascular disease or congestive heart failure; (12) glaucoma; and (13) skin disorders.
- the compounds of the invention find especial application in the treatment or prophylaxis of the following: (1) hypercholesterolemia, dyslipidemia or any other lipid disorder manifested by an unbalance of blood or tissue lipid levels; (2) atherosclerosis; (3) obesity; (4) diabetes.
- the invention also provides a method for the treatment or prophylaxis of a condition in a mammal mediated by a thyroid receptor, which comprises administering to the mammal a therapeutically effective amount of a compound of formula (I) as defined above or a pharmaceutically acceptable ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt.
- a thyroid receptor that may be treated by the method of the invention are those described above.
- the invention also provides the use of a compound of formula (I) as defined above or a pharmaceutically acceptable ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt, for the manufacture of a medicament for the treatment or prophylaxis of a condition mediated by a thyroid receptor.
- Clinical conditions mediated by a thyroid receptor that may be treated by the method of the invention are those described above.
- active ingredient means a compound of formula (I) as defined above, or a pharmaceutically acceptable ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt.
- a compound of the invention may be administered orally or via injection at a dose of from 0.001 to 1500 mg/kg per day, preferably from 0.01 to 1500 mg/kg per day, more preferably from 0.1 to 1500 mg/kg per day, most preferably from 0.1 to 500 mg/kg per day.
- the dose range for adult humans is generally from 5 mg to 35 g per day and preferably 5 mg to 2 g per day. Tablets or other forms of presentation provided in discrete units may conveniently contain an amount of compound of the invention which is effective at such dosage or as a multiple of the same, for example units containing 5 mg to 500 mg, usually around 10 mg to 200 mg.
- the invention provides a pharmaceutical formulation comprising a compound of formula (I) as defined above or a pharmaceutically acceptable ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt, and a pharmaceutically acceptable excipient.
- the pharmaceutical formulations according to the invention include those suitable for oral, parenteral (including subcutaneous, intradermal, intramuscular, intravenous, and intraarticular), inhalation (including fine particle dusts or mists which may be generated by means of various types of metered does pressurized aerosols), nebulizers or insufflators, rectal and topical (including dermal, buccal, sublingual, and intraocular) administration, although the most suitable route may depend upon, for example, the condition and disorder of the recipient.
- parenteral including subcutaneous, intradermal, intramuscular, intravenous, and intraarticular
- inhalation including fine particle dusts or mists which may be generated by means of various types of metered does pressurized aerosols
- nebulizers or insufflators rectal and topical (including dermal, buccal, sublingual, and intraocular) administration, although the most suitable route may depend upon, for example, the condition and disorder of the recipient.
- the formulations may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy. All methods include the step of bringing the active ingredient into association with the carrier which constitutes one or more accessory ingredients. In general the formulations are prepared by uniformly and intimately bringing into association the active ingredient with liquid carriers or finely divided solid carriers or both and then, if necessary, shaping the product into the desired formulation.
- Formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous liquid or a non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in- oil liquid emulsion.
- the active ingredient may also be presented as a bolus, electuary or paste.
- a tablet may be made by compression or moulding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free-flowing form such as a powder or granules, optionally mixed with a binder, lubricant, inert diluent, lubricating, surface active or dispersing agent.
- Moulded tablets may be made by moulding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
- the tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredient therein.
- the present compounds can, for example, be administered in a form suitable for immediate release or extended release.
- Immediate release or extended release can be achieved by the use of suitable pharmaceutical compositions comprising the present compounds, or, particularly in the case of extended release, by the use of devices such as subcutaneous implants or osmotic pumps.
- the present compounds can also be administered liposomally.
- compositions for oral administration include suspensions which can contain, for example, microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners or flavoring agents such as those known in the art; and immediate release tablets which can contain, for example, microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and/or lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants such as those known in the art.
- a compound of formula (I) can also be delivered through the oral cavity by sublingual and/or buccal administration.
- Molded tablets, compressed tablets or freeze-dried tablets are exemplary forms which may be used.
- Exemplary compositions include those formulating the present compound(s) with fast dissolving diluents such as mannitol, lactose, sucrose and/or cyclodextrins. Also included in such formulations may be high molecular weight excipients such as celluloses (avicel) or polyethylene glycols (PEG).
- Such formulations can also include an excipient to aid mucosal adhesion such as hydroxy propyl cellulose (HPC), hydroxy propyl methyl cellulose (HPMC), sodium carboxy methyl cellulose (SCMC), maleic anhydride copolymer (e.g., Gantrez), and agents to control release such as polyacrylic copolymer (e.g. Carbopol 934).
- HPC hydroxy propyl cellulose
- HPMC hydroxy propyl methyl cellulose
- SCMC sodium carboxy methyl cellulose
- maleic anhydride copolymer e.g., Gantrez
- agents to control release such as polyacrylic copolymer (e.g. Carbopol 934).
- Lubricants, glidants, flavors, coloring agents and stabilizers may also be added for ease of fabrication and use.
- Formulations for parenteral administration include aqueous and non-aqueous sterile injection solutions which may contain anit-oxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- the formulations may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilised) condition requiring only the addition of the sterile liquid carrier, for example saline or water- for-injection, immediately prior to use.
- compositions for parenteral administration include injectable solutions or suspensions which can contain, for example, suitable non-toxic, parenterally acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid, or Cremaphor.
- suitable non-toxic, parenterally acceptable diluents or solvents such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid, or Cremaphor.
- compositions for nasal aerosol or inhalation administration include solutions in saline, which can contain, for example, benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, and/or other solubilizing or dispersing agents such as those known in the art.
- Formulations for rectal administration may be presented as a suppository with the usual carriers such as cocoa butter, synthetic glyceride esters or polyethylene glycol. Such carriers are typically solid at ordinary temperatures, but liquify and/or dissolve in the rectal cavity to release the drug.
- Formulations for topical administration in the mouth include lozenges comprising the active ingredient in a flavoured basis such as sucrose and acacia or tragacanth, and pastilles comprising the active ingredient in a basis such as gelatin and glycerine or sucrose and acacia.
- exemplary compositions for topical administration include a topical carrier such as Plastibase (mineral oil gelled with polyethylene).
- Preferred unit dosage formulations are those containing an effective dose, as hereinbefore recited, or an appropriate fraction thereof, of the active ingredient.
- formulations of this invention may include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavouring agents.
- a compound of the invention may be used as the sole active ingredient in a medicament, it is also possible for the compound to be used in combination with one or more further active agents.
- Such further active agents may be a further compound according to the invention, or they may be different therapeutic agents, for example an anti- dyslipidemic agent or other pharmaceutically active material.
- a compound of the present invention may be employed in combination with one or more other modulators and/or ligands of the thyroid receptor or one or more other suitable therapeutic agents selected from the group consisting of cholesterol/lipid lowering agents, hypolipidemic agents, anti-atherosclerotic agents, anti-diabetic agents, anti-osteoporosis agents, anti-obesity agents, growth promoting agents, anti-inflammatory agents, antianxiety agents, anti-depressants, anti-hypertensive agents, cardiac glycosides, appetite supressants, bone resorption inhibitors, thyroid mimetics, anabolic agents, anti-tumor agents and retinoids.
- suitable therapeutic agents selected from the group consisting of cholesterol/lipid lowering agents, hypolipidemic agents, anti-atherosclerotic agents, anti-diabetic agents, anti-osteoporosis agents, anti-obesity agents, growth promoting agents, anti-inflammatory agents, antianxiety agents, anti-depressants, anti-hyper
- hypolipidemic agents for use in combination with a compound of the present invention also include ezetimibe, simvastatin, atorvastatin, rosuvastatin, cerivastatin, fluvastatin, lovastatin, pravastatin, fenofibrate, gemfibrozil and bezafibrate.
- Suitable anti-diabetic agents for use in combination with a compound of the present invention include biguanides (e.g., metformin or phenformin), glucosidase inhibitors (e.g,. acarbose or miglitol), insulins (including insulin secretagogues or insulin sensitizers), meglitinides (e.g., repaglinide), sulfonylureas (e.g., glimepiride, glyburide, glipyride, gliclazide, chlorpropamide and glipizide), biguanide/glyburide combinations (e.g., Glucovance®), thiazolidinediones (e.g., troglitazone, rosiglitazone, englitazone, darglitazone and pioglitazone), PPAR-alpha agonists, PPAR-gamma agonists, PPAR alpha/gamma dual
- Suitable anti-osteoporosis agents for use in combination with a compound of the present invention include alendronate, risedronate, PTH, PTH fragment, raloxifene, calcitonin, RANK ligand antagonists, calcium sensing receptor antagonists, TRAP inhibitors, selective estrogen receptor modulators (SERM) and AP-I inhibitors.
- Suitable anti-obesity agents for use in combination with a compound of the present invention include aP2 inhibitors, PPAR gamma antagonists, PPAR delta agonists, beta 3 adrenergic agonists, such as AJ9677 (Takeda/Dainippon), L750355 (Merck), or CP331648 (Pfizer) or other known beta 3 agonists as disclosed in U.S. Patent Nos.
- a lipase inhibitor such as orlistat or ATL-962 (Alizyme)
- a serotonin (and dopamine) reuptake inhibitor such as sibutramine, topiramate (Johnson & Johnson) or axokine (Regeneron)
- other thyroid receptor beta drugs such as a thyroid receptor ligand as disclosed in WO 97/21993 (U. CaI SF), WO 99/00353 (KaroBio) and GB98/284425 (KaroBio), CB-I (cannabinoid receptor) antagonists (see G. Colombo et al, "Appetite Suppression and Weight Loss After the
- Cannabionid Antagonist SR 141716 Life Sciences, VoI 63, PL 113-117 (1998)) and/or an anorectic agent, such as dexamphetamine, phentermine, phenylpropanolamine or mazindol.
- anorectic agent such as dexamphetamine, phentermine, phenylpropanolamine or mazindol.
- a compound of the present invention may be combined with growth promoting agents, such as, but not limited to, TRH, diethylstilbesterol, theophylline, enkephalins, E series prostaglandins, compounds disclosed in U.S. Patent No. 3,239,345, e.g., zeranol, and compounds disclosed in U.S. Patent No. 4,036,979, e.g., sulbenox or peptides disclosed in U.S. Patent No. 4,411,890.
- growth promoting agents such as, but not limited to, TRH, diethylstilbesterol, theophylline, enkephalins, E series prostaglandins, compounds disclosed in U.S. Patent No. 3,239,345, e.g., zeranol, and compounds disclosed in U.S. Patent No. 4,036,979, e.g., sulbenox or peptides disclosed in U.S. Patent No. 4,411,890
- a compound of the invention may also be used in combination with growth hormone secretagogues such as GHRP-6, GHRP-I (as described in U.S. Patent No. 4,411,890 and publications WO 89/07110 and WO 89/07111), GHRP-2 (as described in WO 93/04081), NN703 (Novo Nordisk), LY444711 (Lilly), MK-677 (Merck), CP424391 (Pfizer) and B- HT920, or with growth hormone releasing factor and its analogs or growth hormone and its analogs or somatomedins including IGF-I and IGF-2, or with alpha-adrenergic agonists, such as clonidine or serotinin 5-HT D agonists, such as sumatriptan, or agents which inhibit somatostatin or its release, such as physostigmine and pyridostigmine.
- growth hormone secretagogues such as GHRP-6, GHRP-I (as
- a still further use of a disclosed compound of the invention is in combination with parathyroid hormone, PTH(I -34) or bisphosphonates, such as MK-217 (alendronate).
- suitable anti-inflammatory agents include prednisone, dexamethasone, Enbrel®, cyclooxygenase inhibitors (i.e., COX-I and/or COX-2 inhibitors such as NSAIDs, aspirin, indomethacin, ibuprofen, piroxicam, Naproxen®, Celebrex®, Vioxx®), CTLA4-Ig agonists/antagonists, CD40 ligand antagonists, IMPDH inhibitors, such as mycophenolate (CellCept®), integrin antagonists, alpha-4 beta-7 integrin antagonists, cell adhesion inhibitors, interferon gamma antagonists, ICAM-I, tumor necrosis factor (TNF) antagonists (e.g., inf
- Suitable anti-anxiety agents for use in combination with a compound of the present invention include diazepam, lorazepam, buspirone, oxazepam, and hydroxyzine pamoate.
- Suitable anti-depressants for use in combination with a compound of the present invention include citalopram, fluoxetine, nefazodone, sertraline, and paroxetine.
- Suitable anti-hypertensive agents for use in combination with a compound of the present invention include beta adrenergic blockers, calcium channel blockers (L-type and T-type; e.g. diltiazem, verapamil, nifedipine, amlodipine and mybefradil), diuretics (e.g., chlorothiazide, hydrochlorothiazide, flumethiazide, hydroflumethiazide, bendroflumethiazide, methylchlorothiazide, trichloromethiazide, polythiazide, benzthiazide, ethacrynic acid tricrynafen, chlorthalidone, furosemide, musolimine, bumetanide, triamtrenene, amiloride, spironolactone), renin inhibitors, ACE inhibitors (e.g., captopril, zofenopril, fosin
- Dual ET/AII antagonist e.g., compounds disclosed in WO 00/01389
- neutral endopeptidase (NEP) inhibitors neutral endopeptidase (NEP) inhibitors
- vasopepsidase inhibitors dual NEP- ACE inhibitors
- omapatrilat and gemopatrilat examples include digitalis and ouabain.
- Suitable cholesterol/lipid lowering agents for use in combination with a compound of the present invention include HMG-CoA reductase inhibitors, squalene synthetase inhibitors, fibrates, bile acid sequestrants, ACAT inhibitors, MTP inhibitors, lipooxygenase inhibitors, an ileal Na ⁇ 7bile acid cotransporter inhibitor, cholesterol absorption inhibitors, and cholesterol ester transfer protein inhibitors (e.g., CP-529414).
- HMG-CoA reductase inhibitors e.g., squalene synthetase inhibitors, fibrates, bile acid sequestrants, ACAT inhibitors, MTP inhibitors, lipooxygenase inhibitors, an ileal Na ⁇ 7bile acid cotransporter inhibitor, cholesterol absorption inhibitors, and cholesterol ester transfer protein inhibitors (e.g., CP-529414).
- MTP inhibitors which may be employed herein in combination with a compound of formula (I) include MTP inhibitors as disclosed in U.S. Patent No. 5,595,872, U.S. Patent No. 5,739,135, U.S. Patent No. 5,712,279, U.S. Patent No. 5,760,246, U.S. Patent No. 5,827,875, U.S. Patent No. 5,885,983 and U.S. Patent No. 5,962,440 all incorporated herein by reference.
- the HMG CoA reductase inhibitors which may be employed in combination with a compound of formula (I) include mevastatin and related compounds as disclosed in U.S. Patent No. 3,983,140, lovastatin (mevinolin) and related compounds as disclosed in U.S. Patent No. 4,231 ,938, pravastatin and related compounds such as disclosed in U.S. Patent No. 4,346,227, simvastatin and related compounds as disclosed in U.S. Patent Nos. 4,448,784 and 4,450,171.
- Further HMG CoA reductase inhibitors which may be employed herein include fluvastatin, disclosed in U.S. Patent No. 5,354,772, cerivastatin disclosed in U.S. Patent Nos.
- the squalene synthetase inhibitors which may be used in combination with a compound of the present invention include, but are not limited to, ⁇ -phosphono-sulfonates disclosed in U.S. Patent No. 5,712,396, those disclosed by Biller et al, J. Med. Chem., 1988, Vol. 31, No. 10, pp 1869-1871, including isoprenoid (phosphinylmethyl)phosphonates, terpenoid pyrophosphates disclosed by P. Ortiz de Montellano et al, J. Med.
- Bile acid sequestrants which may be used in combination with a compound of the present invention include cholestyramine, colestipol and DEAE-Sephadex (Secholex®, policexide®), as well as lipostabil (Rhone-Poulenc), Eisai E-5050 (an N-substituted ethanolamine derivative), imanixil (HOE-402), tetrahydrolipstatin (THL), istigmastanylphos-phorylcholine (SPC, Roche), aminocyclodextrin (Tanabe Seiyoku), Ajinomoto AJ-814 (azulene derivative), melinamide (Sumitomo), Sandoz 58-035, American Cyanamid CL-277,082 and CL-283,546 (disubstituted urea derivatives), nicotinic acid, acipimox, acifran, neomycin, p-aminosalicylic acid, aspirin, poly(
- ACAT inhibitors suitable for use in combination with a compound of the invention include ACAT inhibitors as described in, Drugs of the Future 24, 9-15 (1999), (Avasimibe); "The ACAT inhibitor, Cl-1011 is effective in the prevention and regression of aortic fatty streak area in hamsters", Nicolosi et al, Atherosclerosis (Shannon, Irel). (1998), 137(1), 77-85; "The pharmacological profile of FCE 27677: a novel ACAT inhibitor with potent hypolipidemic activity mediated by selective suppression of the hepatic secretion of ApoB 100-containing lipoprotein", Ghiselli, Giancarlo, Cardiovasc. Drug Rev.
- Suitable cholesterol absorption inhibitor for use in combination with a compound of the invention include SCH48461 (Schering-Plough), as well as those disclosed in Atherosclerosis 115, 45-63 (1995) and J. Med. Chem. 41, 973 (1998).
- Suitable thyroid mimetics for use in combination with a compound of the present invention include thyrotropin, polythyroid, KB-130015, and dronedarone.
- a compound of the present invention may be used alone or optionally in combination with a retinoid, such as tretinoin, or a vitamin D analog.
- a still further use of a compound of the invention is in combination with estrogen, testosterone, a selective estrogen receptor modulator, such as tamoxifen or raloxifene, or other androgen receptor modulators, such as those disclosed in Edwards, J. P. et al., Bio. Med. Chem. Let., 9, 1003-1008 (1999) and Hamann, L. G. et al., J. Med. Chem., 42, 210-212 (1999).
- a further use of a compound of this invention is in combination with steriodal or non-steroidal progesterone receptor agonists ("PRA”), such as levonorgestrel, medroxyprogesterone acetate (MPA).
- PRA steriodal or non-steroidal progesterone receptor agonists
- MPA medroxyprogesterone acetate
- a compound of formula (I) When combined with a hypolypidemic agent, an antidepressant, a bone resorption inhibitor and/or an appetite suppressant, a compound of formula (I) may be employed in a weight ratio to the additional agent within the range from about 500: 1 to about 0.005 : 1 , preferably from about 300: 1 to about 0.01:1.
- a compound of formula (I) may be employed in a weight ratio to biguanide within the range from about 0.01:1 to about 100:1, preferably from about 0.5:1 to about 2:1.
- a compound of formula (I) may be employed in a weight ratio to a glucosidase inhibitor within the range from about 0.01:1 to about 100:1, preferably from about 0.5:1 to about 50:1.
- a compound of formula (I) may be employed in a weight ratio to a sulfonylurea in the range from about 0.01:1 to about 100:1, preferably from about 0.2:1 to about 10:1.
- a compound of formula (I) may be employed in a weight ratio to a thiazolidinedione in an amount within the range from about 0.01:1 to about 100:1, preferably from about 0.5:1 to about 5:1.
- the thiazolidinedione may be employed in amounts within the range from about 0.01 to about 2000 mg/day, which may optionally be administered in single or divided doses of one to four times per day.
- these additional agents may be incorporated into a combined single tablet with a therapeutically effective amount of a compound of formula (I).
- Metformin, or salt thereof may be employed with a compound of formula (I) in amounts within the range from about 500 to about 2000 mg per day, which may be administered in single or divided doses one to four times daily.
- a compound of formula (I) may be employed in a weight ratio to a PPAR-alpha agonist, a PPAR-gamma agonist, a PPAR-alpha/gamma dual agonist, an SGLT2 inhibitor and/or an aP2 inhibitor within the range from about 0.01:1 to about 100:1, preferably from about 0.5:1 to about 5:1.
- An MTP inhibitor may be administered orally with a compound of formula (I) in an amount within the range of from about 0.01 mg/kg to about 100 mg/kg and preferably from about 0.1 mg/kg to about 75 mg/kg, one to four times daily.
- a preferred oral dosage form such as tablets or capsules, may contain the MTP inhibitor in an amount of from about 1 to about 500 mg, preferably from about 2 to about 400 mg, and more preferably from about 5 to about 250 mg, administered on a regimen of one to four times daily.
- the MTP inhibitor may be employed in an amount within the range of from about 0.005 mg/kg to about 10 mg/kg and preferably from about 0.005 mg/kg to about 8 mg/kg, administered on a regimen of one to four times daily.
- a HMG CoA reductase inhibitor may be administered orally with a compound of formula (I) within the range of from about 1 to 2000 mg, and preferably from about 4 to about 200 mg.
- a preferred oral dosage form, such as tablets or capsules, will contain the HMG CoA reductase inhibitor in an amount from about 0.1 to about 100 mg, preferably from about 5 to about 80 mg, and more preferably from about 10 to about 40 mg.
- a squalene synthetase inhibitor may be administered with a compound of formula (I) within the range of from about 10 mg to about 2000 mg and preferably from about 25 mg to about 200 mg.
- a preferred oral dosage form, such as tablets or capsules, will contain the squalene synthetase inhibitor in an amount of from about 10 to about 500 mg, preferably from about 25 to about 200 mg.
- a compound of formula (I) as described above also finds use, optionally in labelled form, as a diagnostic agent for the diagnosis of conditions associated with malfunction of the thyroid receptor.
- a diagnostic agent for the diagnosis of conditions associated with malfunction of the thyroid receptor.
- such a compound may be radioactively labelled.
- a compound of formula (I) as described above, optionally in labelled form, also finds use as a reference compound in methods of discovering other antagonists or partial antagonists of the thyroid receptor.
- the invention provides a method of discovering a ligand of the thyroid receptor which comprises use of a compound of the invention or a compound of the invention in labelled form, as a reference compound.
- a method may involve a competitive binding experiment in which binding of a compound of formula (I) to the thyroid receptor is reduced by the presence of a further compound which has thyroid receptor-binding characteristics, for example stronger thyroid receptor-binding characteristics than the compound of formula (I) in question.
- the invention also provides a method for preparing a compound of formula (I) in accordance with the invention as described above comprising a step of reacting - a compound of formula (II)
- R groups include methyl, ethyl and propyl, particularly methyl.
- Methanesulphonylating reagents include methanesulphonyl chloride and methanesulphonyl bromide.
- Suitable bases include pyridine or alkylamines, for example diisopropylamine and triethylamine. Other bases may be employed, as is known by the person skilled in the art.
- one or more coupling reagents may be employed.
- the hydrolysis step may be carried out by reaction with an alkali metal hydroxide. Suitable alkali metal hydroxides include lithium hydroxide and sodium hydroxide.
- the reaction mixture is stirred at room temperature, or heated until the starting materials have been consumed.
- the reaction may be carried out with protecting groups present and those protecting groups may be removed after the reaction. Suitable protecting groups are known to the person skilled in the art (see T. W. Greene, "Protective Groups in Organic Synthesis", 3 rd Edition, New York, 1999).
- a compound of formula (H) can be synthesized by reacting: a compound of formula (III)
- Suitable reducing agents include tin(II)chloride in ethanol.
- the reaction mixture is stirred at room temperature, or heated until the starting materials have been consumed.
- the reaction may be carried out with protecting groups present and those protecting groups may be removed after the reaction. Suitable protecting groups are known to the person skilled in the art (see T. W. Greene, "Protective Groups in Organic Synthesis", 3 rd Edition, New York, 1999).
- a compound of formula (III) can be synthesized by reacting: the compound of formula (IVa)
- L is a suitable leaving group, in the presence of a suitable base.
- a compound of formula (III) can be synthesized by reacting: - a compound of formula (IVb)
- L is a suitable leaving group, in the presence of a suitable base, followed by conversion of the R 3 group to a fluoro group with (S) stereochemistry.
- the conversion of the R 3 group to a fluoro group with (S) stereochemistry may proceed with either retention or inversion of stereochemistry depending on the nature of the R 3 group and the reaction conditons.
- R amino
- a convenient method of converting the group to a fluoro group uses a diazotisation reaction.
- R is H, the reaction typically proceeds with retention of configuration.
- R 3 is hydroxy
- a convenient method of converting the group to a fluoro group makes use of a reaction that first converts the hydroxyl group into an improved leaving group (for example methanesulfonyl, trifluroacetyl or toluensulfonyl) followed by substitution of the leaving group.
- the reaction typically proceeds with inversion of configuration.
- Suitable leaving groups L are for example halogens, hydroxyl and mesylate.
- Suitable bases include inorganic bases and organic bases (for example amines or pyridines).
- the base may be selected from hydroxide salts, carbonate salts and bicarbonate salts or from triethylamine and pyridine.
- a particularly suitable base is potassium carbonate.
- the reaction mixture is stirred at room temperature, or heated until the starting materials have been consumed.
- the reaction may be carried out with protecting groups present and those protecting groups may be removed after the reaction. Suitable protecting groups are known to the person skilled in the art (see T. W. Greene, "Protective Groups in Organic Synthesis", 3 rd Edition, New York, 1999).
- Suitable leaving groups L include halogen, OMs and OTs.
- Suitable bases can be included in the coupling reaction. Suitable bases include inorganic bases and organic bases (for example amines or pyridines). For example, the base may be selected from hydroxide salts, carbonate salts and bicarbonate salts or from triethylamine and pyridine in the presence of a nucleophilic catalyst, for example iodide. A particularly suitable base is potassium carbonate.
- Suitable leaving groups Y include Ms and Ts.
- a further alternative method for the preparation of the compound of formula (I) is provided by deprotecting a compound of formula (Ia) or an ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt:
- Compound (Ia) can, in turn, be prepared by coupling the intermediate of formula (IVa) with a compound of formula (IX), wherein L is a suitable leaving group, optionally followed by hydrolysis to the free acid:
- Suitable leaving groups L include halogen, OMs and OTs.
- Suitable bases can be included in the coupling reaction. Suitable bases include inorganic bases and organic bases (for example amines or pyridines). For example, the base may be selected from hydroxide salts, carbonate salts and bicarbonate salts or from triethylamine and pyridine. A particularly suitable base is potassium carbonate.
- a compound of formula (VII) or (IX), wherein L is a suitable leaving group (for example halogen, OMs or OTs) and Y is H, Ts or Ms, can be obtained from (3-aminophenyl)- methanol compounds (VIII) using standard literature procedures.
- the method may, in particular make use of a compound of formula (VII) which is the compound of formula (IX): MeO 2
- the intermediate of formula (IVa) or (IVb) can be prepared as a single enantiomer.
- the (S) form of 2-fluoro-3-(3,5-dibromo-4-hydroxy-phenyl)-propanoic acid methyl ester can be obtained from the corresponding (S) form of 2-fluoro-3-(4-hydroxy-phenyl)- propanoic acid using a brominating agent such as bromine.
- the (S) form of 2-fluoro-3-(4- hydroxy-phenyl)-propanoic acid can be obtained from diazotization of (S) form of 2- fluoro-3-(4-amino-phenyl)-propanoic acid.
- the (S) form of 2-fluoro-3-(4-amino-phenyl)- propanoic acid can be obtained from 4-nitro-phenyl alanine as set ou t in the general references: Tet, 55, 36, 1999, 10971; Arzneim. Forsch, 30, 5, 1980, 751; Chem. Pharm. Bull, 31 (10), 1983, 3424; J. Chem. Soc, 1947, 1571; HeIv. Chim. Acta; 64 (8), 1981, 2526; Tet; 52 (46), 1996 14501-14506.
- the (R) and (S) forms of 2-fluoro-3-(4-hydroxy-phenyl)-propanoic acid can be obtained by separation of a racemic mixture of 2-fluoro-3-(4-hydroxy-phenyl)-propanoic acid, by enzymatic kinetic resolution as set out in the general references: ref. Organic Lett. 2 (8), 2000, 1037-1040, Tet Asymmetry. 11, 2000, 889-896, J. Org. Chem. 55, 1990, 812-815, or by forming diasteromers of the 2-fluoro-3-(4-hydroxy-phenyl)-propanoic acid with an optical pure compound, for example an amine.
- the (S) form of 2-fluoro-3-(4-hydroxy-phenyl)-propanoic acid methyl ester can be obtained from the corresponding 2-O-substituted-3-(4-hydroxy-phenyl)-propanoic acid methyl ester wherein X is a group such that OX is a leaving group, by reaction with a suitable fluoride source.
- the reaction typically proceeds with inversion of configuration.
- X may be Ms, Ts or Tf.
- the reaction may also be carried out on the free acid.
- the S form of 2-fluoro-3-(4-methoxy-phenyl)-propanoic acid can be obtained from by the treatment of (S)- 2-fiuoro-3-(4-methoxy-phenyl)-propanoic acid-(i?)- 2-oxo-5-phenyl-pyrolidin-l-yl-ester with base and H 2 O 2 .
- (S)- 2-fluoro-3-(4-methoxy- phenyl)-propanoic acid-(i?)-2-oxo-5-phenyl-pyrolidin-l-yl-ester can be obtained from 3-(4- methoxy-phenyl)-propanoic acid (i?)-2-oxo-5-phenyl-pyrolidin-l-yl-ester for example by reaction with an electrophilic fluorinating agent (eg Ts 2 NF or (PHSO 2 ) 2 NF) in the presence of a base (eg Lithium bis (trimethyl silyl)amide (LiHMDS) or Na HMDS).
- an electrophilic fluorinating agent eg Ts 2 NF or (PHSO 2 ) 2 NF
- a base eg Lithium bis (trimethyl silyl)amide (LiHMDS) or Na HMDS.
- 3-(4-methoxy- phenyl)-propanoic acid (i?)-2-oxo-5-phenyl-pyrolidin-l-yl-ester can be obtained from 3-(4- methoxy-phenyl)-propanoic acid.
- a compound of formula (IVa) or (IVb) can be synthesized in its enantiomerically pure form from a single enantiomer of a compound of formula (VI) (p- hydroxyphenyllactic acid, HPLA):
- R and S forms of 2-hydroxy-3-(4-hydroxy-phenyl)-propanoic acid can be obtained by separation of the commercially available racemic mixture by forming diastereomers of the acid with an optically pure amine (ref. L. Pretlow, R. Williams and M. Elliot; Chirality, 2003, 15, 674-679) and by enzymatic separation (ref. Y. Momose, T. Maekawa, T. Yamano, M. Kawada, H. Odaka, H. Ikeda and T. Sohda; J. Med. Chem, 2002, 45, 1518-1534).
- the single enantiomers can be obtained by enzymatic reduction of methyl p- hydroxyphenylpyruvate by the enzyme lactate dehydrogenase (LDH) (ref. L. Pretlow, R. Williams and M. Elliot; Chirality, 2003, 75, 674-679).
- LDH lactate dehydrogenase
- the single isomer compounds of the invention may be obtained by use of an enantiospecific enzymic reaction.
- many lipase enzymes will treat one enantiomer of an ⁇ - hydroxyester as a substrate, but not the other.
- Lipase PS-C "amino" will acetylate the (S) isomer (Fernando F. Huerta, Y. R. Santosh Laxmi, and Jan-E. Backvall Org. Lett., Vol. 2, No. 8, 2000, 1037-1040).
- a racemic substrate (X) in which R is Cl-6 alkyl and Z is H, 3-amino-2-chlorobenzyl, 3-nitro-2-chlorobenzyl, 2- chloro-3-[(methylsulfonyl)amino]benzyl or 2-chloro-3-[bis(methylsulfonyl)amino]benzyl
- XI a racemic substrate
- R is Cl-6 alkyl and Z is H
- Some other enzymes convert the racemic starting materials into the (R) acylated product (XIII), leaving the (S) isomer as unreacted starting material (XIV).
- the two isomers can be readily separated on the basis of the different physico-chemical properties of the acylated and unacylated compounds.
- the invention thus provides a method of obtaining an optically active compound (XI), (XII), (XIII) or (XIV) comprising the step of treating a less optically active compound of formula (X) (in which R is Q -6 alkyl and Z is H, 3-amino-2-chlorobenzyl, 3-nitro-2- chlorobenzyl, 2-chloro-3-[(methylsulfonyl)amino]benzyl or 2-chloro-3- [bis(methylsulfonyl)amino]benzyl) with a suitable enzyme, for example a lipase enzyme.
- the invention also provides a synthesis of a compound of formula (I), (Ia), (II), (III), (IVa) or (IVb) comprising such a step.
- the present invention further provides the optically active (S) form of the compound of formula (Ha),
- the compound is useful as an advanced intermediate for the synthesis of a compound of formula (I).
- the methyl ester is a particular example of an ester of the compound.
- the present invention also provides the optically active (S) form of the compound of formula (Ilia),
- the compound is useful as an advanced intermediate for the synthesis of a compound of formula (I).
- the methyl ester is a particular example of an ester of the compound.
- the present invention further provides the optically active (S) form of the compound of formula (Ia), or an ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt.
- the compound is useful as an advanced intermediate for the synthesis of a compound of formula (I).
- the methyl ester is a particular example of an ester of the compound.
- the present invention also provides the optically active (S) form of the compound of formula (IVaa),
- the present invention also provides a compound of formula (VII):
- L is as defined above.
- Preferred L groups are Cl and OMs.
- LCMS analyzed on HPLC-MS with alternating +/- API and equipped with different brands of 50 mm*2.1mm, 5 ⁇ C8 columns. Eluted with 0.05% formic acid/ACN or 0.05% ammonium acetate/ ACN
- MW calc. (molecular weight) is an isotopic average and the "found mass” is referring to the most abundant isotope detected in the LC-MS.
- the “found mass” refers to M+l, M, M-I or M-2 as stated.
- the crude product was purified by silica flash chromatography column (ethyl acetate/heptane; 1 :9 to 2:8) to afford 10.9 g of methyl 2-hydroxy-3-(4-benzyloxy-phenyl)- propanoate as yellow solid.
- the total yield was 30% for four steps.
- reaction mixture was diluted with brine and extracted with ethyl acetate (3 x 20 mL).
- the reaction mixture was quenched with sodium hydrogen carbonate aqueous solution (saturated).
- the aqueous phase was extracted with ethyl acetate (3 x 40 mL) and the combined organic phases were washed with water and brine and dried over magnesium sulphate.
- the pH of reaction mixture was approximately 2 to 3.
- the reaction mixture was quenched with 2N aqueous NaOH (105 mL) and H 2 O (45 mL).
- the reaction temperature was increased from 22° C to 25.5° C during the addition of 2N aqueous NaOH. After the addition of 2N aqueous NaOH, the pH of the reaction mixture was approximately 10 to 11.
- the aqueous layer was washed with tert-butylmethylether (2x200 mL) and the combined organic layers were washed with H 2 O (2x300 mL).
- the combined aqueous phases were acidified with 2N HCl (25 mL) from pH ⁇ 10-11 to pH ⁇ 1 and extracted with EtOAc (4x300 mL).
- End B 100 (isocratic, only B pump used)
- the retention-time of the (R) form of 2-hydroxy-3-(4-hydroxy-phenyl)-propionic acid is approximately 26 min and the retention-time of (S) form is approximately 31 min.
- reaction mixture was monitored by HPLC, TLC (heptane: EtOAc 65:35), LC/MS and shows that all starting material has been consumed.
- the aqueous was extracted with ethyl acetate ( 3 x 600 ml) and the extracts combined and stirred with water (1000 ml) and saturated K 2 CO 3 (1000 ml).
- the aqueous was adjusted to pH 7-8 with K 2 CO 3 (317.7 g) and the organics removed.
- the aqueous was extracted with ethyl acetate (500 ml), the organics combined, dried and evaporated.
- the crude product was slurried in methanol (200 ml) at 45°C for 45 mins then left at room temperature overnight and filtered.
- Methyl 3-[3,5-dibromo-4-( ⁇ 2-chloro-3-[(methylsulfonyl)amino]benzyl ⁇ oxy)phenyl]-2- fluoropropanoate (8.6 g, 15 mmol) was dissolved in dioxane (150 mL), and sodium hydroxide (lithium hydroxide has also been used) (1 N in water, 150 mL) was added and the mixture was stirred at 5O 0 C for 1 hour. After neutralization with hydrochloric acid (1 N), the product was extracted into ethyl acetate. The combined organic phases were dried over sodium sulphate and concentrated under vacuum.
- sodium hydroxide lithium hydroxide has also been used
- Example 1-Fl 98 min.
- Example 1-F2 109 min.
- the (S) isomer compound of the invention can also be separated from the (R) isomer by an enzymatic method. Such a method may be carried out as follows:
- the recovered reaction products comprise unreacted starting material (the non-substrate enantiomer) and acetylated product of the substrate starting material (methyl 3-[3,5-dibromo-4-( ⁇ 2-chloro-3-[(methylsulfonyl)amino]benzyl ⁇ oxy)phenyl]-2-acetoxypropanoate)
- the stereochemistry of the acetylated compound is (S) (Fernando F. Huerta, Y. R. Santosh Laxmi, and Jan-E. Backvall Org. Lett., Vol. 2, No. 8, 2000, 1037-1040).
- the (R) isomer can be isolated by separating it from the acetylated (S) compound.
- the aqueous pH was adjusted to 1 with saturated citric acid (ca. 20 ml) and the suspension extracted into dichloromethane (3 x 50 ml). Some solid was seen at the phase boundary and this was left with the aqueous layer.
- End B 100 (isocratic, only B pump used)
- the retention time of the (R) form of -2-fluoro-3-[3, 5-dibromo-4-(2-chloro-3- methanesulphonyl amino-benzyloxy)-phenyl]-propionic acid is 120-134 min and Retention time of (S) form of -2-fluoro-3-[3, 5-dibromo-4-(2-chloro-3-methanesulphonyl amino- benzyloxy)-phenyl] -propionic acid is 128-143 min
- Pharmacodynamic parameters were measured for the compound of Example 2. Such parameters are for example plasma levels of LDL-C (lipid parameters), which are known to those skilled in the art.
- Example 1 the compound of Example 1 was analysed in a cholesterol-fed rat model:
- mice Male Sprague Dawley rats 4 weeks old are placed on Paigan diet (D 12336 from Research Diets, US) for 2 weeks before transfer to the laboratory. Upon arrival, animals are adapted to the new environment for 6 days. Rats are given free access to paigan diet and water, and placed on a 12:12h light dark cycle. Before the start of the experiment, body weights are measured, animals are marked for individual tracking and divided into dose groups (usually six animals per group). Animals are dosed p.o. with compound once daily for seven days. At the end of the experiment animals are fasted for at least 12h prior to sacrifice. In the study below, animals were dosed with 0.1; 0.3 or 0.9 mg/kg body weight of the (S) enantiomer of Example 1 or with the (R) enantiomer of Example 1.
- Paigan diet D 12336 from Research Diets, US
- mice are adapted to the new environment for 6 days. Rats are given free access to paigan diet and water, and placed on a 12:12h
- the serum LDL cholesterol and the serum total cholesterol were measured before the experiment and after seven days. The changes in serum LDL cholesterol and the serum total cholesterol levels over the timecourse of the experiments are shown in Figures 1 and 2.
- the animals treated with the (S) isomer of Example 2 have a signifantly lower LDL serum cholesterol level at all doses tested (p ⁇ 0.05).
- the animals treated with the (S) isomer of Example 1 have a signifantly lower total serum cholesterol level at all doses tested (p ⁇ 0.05).
- a compound of the present invention to bind with high affinity to the thyroid hormone receptor and to activate the thyroid hormone receptor in genetically engineered cells (in vitro assays) is appreciated by those skilled in the art as being beneficial in terms of therapeutic use. For example, it diminishes the possibility for cross-reactivity with other receptors.
- Barkhem T. et al. : High level expression of functional full-length human thyroid hormone receptor ⁇ 1 in insect cells using a recombinant baculovirus. J. Steroid Biochem. MoI. Biol, 1991, 38, 667-75.
- Chinese hamster ovary cells have been stably transfected with the human thyroid hormone receptor beta and with a reporter gene driven by thyroid hormone receptor response elements.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The invention provides an optically active form of the compound of formula (I), or a pharmaceutically acceptable ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt; uses of the compound; and methods of preparation of the compound.
Description
Compounds
Field of the invention
The present invention relates to optically active compounds which are agonists or partial agonists of the thyroid receptor and the use of such compounds for therapeutic purposes.
Background of the invention
While the extensive role of thyroid hormones in regulating metabolism in humans is well recognized, the discovery and development of new specific drugs for improving the treatment of hyperthyroidism and hypothyroidism has been slow. This has also limited the development of thyroid agonists and antagonists for treatment of other important clinical indications, such as hypercholesterolemia, dyslipidemia, obesity, diabetes, atherosclerosis and cardiac diseases.
Thyroid hormones affect the metabolism of virtually every cell of the body. At normal levels, these hormones maintain body weight, metabolic rate, body temperature and mood, and influence blood levels of serum lipoproteins. Thus, in hypothyroidism there is weight gain, high levels of LDL cholesterol, and depression. In hyperthyroidism, these hormones lead to weight loss, hypermetabolism, lowering of serum LDL cholesterol levels, cardiac arrhythmias, heart failure, muscle weakness, bone loss in postmenopausal women, and anxiety.
Thyroid hormones are currently used primarily as replacement therapy for patients with hypothyroidism. Therapy with L-thyroxine returns metabolic functions to normal and can easily be monitored with routine serum measurements of levels of thyroid-stimulating hormone (TSH), thyroxine (3,5,3',5'-tetraiodo-L-thyronine, or T4) and triiodothyronine (3,5,3'-triiodo-L-thyronine, or T3). However, replacement therapy, particularly in older individuals, may be restricted by certain detrimental effects from thyroid hormones.
In addition, some effects of thyroid hormones may be therapeutically useful in non-thyroid disorders if adverse effects can be minimized or eliminated. These potentially useful influences include for example, lowering of serum LDL levels, weight reduction, amelioration of depression and stimulation of bone formation. Prior attempts to utilize
thyroid hormones pharmacologically to treat these disorders have been limited by manifestations of hyperthyroidism, and in particular by cardiovascular toxicity.
Furthermore, useful thyroid agonist drugs should minimize the potential for undesired consequences due to locally induced hypothyroidism, i.e. sub-normal levels of thyroid hormone activity in certain tissues or organs. This can arise because increased circulating thyroid hormone agonist concentrations may cause the pituitary to suppress the secretion of thyroid stimulating hormone (TSH), thereby reducing thyroid hormone synthesis by the thyroid gland (negative feedback control). Since endogenous thyroid hormone levels are reduced, localized hypothyroidism can result wherever the administered thyroid agonist drug fails to compensate for the reduction in endogenous hormone levels in specific tissues.
Development of specific and selective thyroid hormone receptor ligands, particularly agonists of the thyroid hormone receptor, is expected to lead to specific therapies for these common disorders, while avoiding the cardiovascular and other toxicity of native thyroid hormones. Tissue-selective thyroid hormone agonists may be obtained by selective tissue uptake or extrusion, topical or local delivery, targeting to cells through other ligands attached to the agonist and targeting receptor subtypes. Tissue selectivity can also be achieved by selective regulation of thyroid hormone responsive genes in a tissue specific manner.
Accordingly, the compounds that are thyroid hormone receptor ligands, particularly selective agonists of the thyroid hormone receptor, are expected to demonstrate a utility for the treatment or prevention of diseases or disorders associated with thyroid hormone activity, for example: (1) hypercholesterolemia, dyslipidemia or any other lipid disorder manifested by an unbalance of blood or tissue lipid levels; (2) atherosclerosis; (3) replacement therapy in elderly subjects with hypothyroidism who are at risk for cardiovascular complications; (4) replacement therapy in elderly subjects with subclinical hypothyroidism who are at risk for cardiovascular complications; (5) obesity; (6) diabetes (7) depression; (8) osteoporosis (especially in combination with a bone resorption inhibitor); (9) goiter; (10) thyroid cancer; (11) cardiovascular disease or congestive heart failure; (12) glaucoma; and (13) skin disorders.
Summary of the invention
The present invention provides the optically active (iS)-form of the compound of formula (I)5
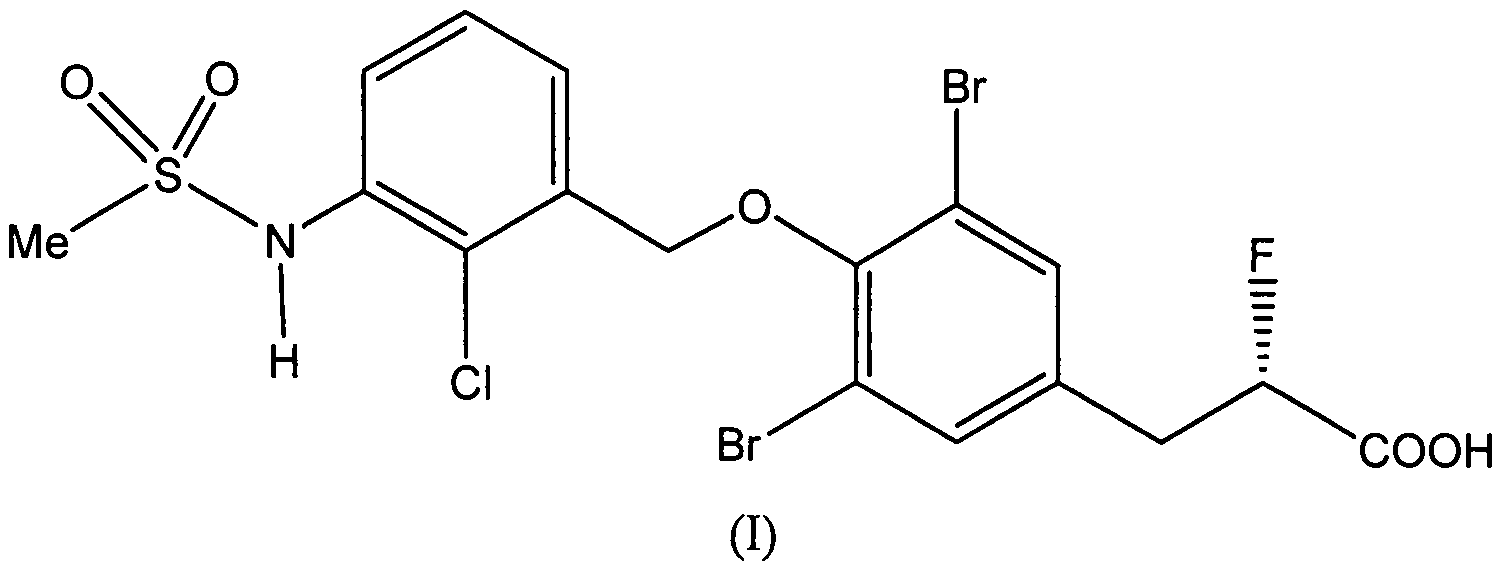
The compound is the (-) enantiomer. The (-) form of the compound is believed on the basis of X-ray crystal structure analysis to be the (S) isomer. Thus the compound is (-)-3-[3,5- dibromo-4-( { 2-chloro-3- [(methylsulfonyl)amino]benzyl } oxy)phenyl] -2-fluoropropanoic acid, or (5)-3-[3,5-dibromό-4-({2-chloro-3-[(methylsulfonyl)amino]benzyl}oxy)phenyl]-2- fluoropropanoic acid, or a pharmaceutically acceptable ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt.
The optically active compound of the invention has surprisingly been found to be a particularly effective ligand of the thyroid receptor, in particular agonist or partial agonist of the thyroid receptor. The compounds accordingly have use in the treatment or prophylaxis of conditions associated with thyroid receptor activity.
Surpririsingly, it has further been found that the the (S) enantiomer ((-)-enantiomer) of the compound of the invention has advantages over the racemic mixture or the (+)-enantiomer in terms of various parameters. The compound is more effective in lowering lipid levels in vivo and it is more effective at binding to the thyroid receptor than the racemic mixture or the (+)-enantiomer.
Detailed description of the invention
Optically active compounds stated to be the (+)-form or (-)-form are the compounds which show respectively (+)-or (-)- rotatory power with respect to the sodium D line. References
to optically active compounds should be taken to include compounds in a mixture of enantiomers, but having an enantiomeric excess of at least 75%, preferably at least 85%, more preferably at least 95%, most preferably at least 97%, especially at least 99%. Accordingly, references to optically active compounds should be taken to include compounds in a mixture of enantiomers, but having a molar ratio of enantiomers of at least 7:1, preferably at least 37:3, more preferably at least 39: 1, most preferably at least 197:3, especially at least 199:1.
The compound names given herein were generated in accordance with IUPAC by the ACD Labs/Name program, version 7.08 build 21 and with ISIS DRAW Autonom 2000.
Salts and solvates of compounds of formula (I) which are suitable for use in medicine are those wherein a counterion or associated solvent is pharmaceutically acceptable. However, salts and solvates having non-pharmaceutically acceptable counterions or associated solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of the compounds of formula (I) and their pharmaceutically acceptable salts, solvates and physiologically functional derivatives. According to the present invention, examples of physiologically functional derivatives include esters and amides.
Suitable salts according to the invention include those formed with organic or inorganic acids or bases. Pharmaceutically acceptable acid addition salts include those formed from hydrochloric, hydrobromic, sulphuric, nitric, citric, tartaric, acetic, phosphoric, lactic, pyruvic, acetic, trifluoroacetic, succinic, perchloric, fumaric, maleic, glycollic, lactic, salicylic, oxaloacetic, methanesulfonic, ethanesulfonic, p-toluenesulfonic, formic, benzoic, malonic, naphthalene-2-sulfonic, benzenesulfonic, and isethionic acids. Other acids such as oxalic, while not in themselves pharmaceutically acceptable, may be useful as intermediates in obtaining the compounds of the invention and their pharmaceutical acceptable acid addition salts. Pharmaceutically acceptable base salts include ammonium salts, alkali metal salts, for example those of potassium and sodium, alkaline earth metal salts, for example those of calcium and magnesium, and salts with organic bases e.g. primary, secondary or tertiary organic amines, for example dicyclohexylamine, and N-methyl-D-glucomine.
Pharmaceutically acceptable esters and amides of a compound of formula (I) may have an appropriate group, for example an acid group, converted to a Ci-6 alkyl, Cs-I0 aryl, C5.10 aryl-Ci-6 alkyl, or amino acid ester or amide.
Those skilled in the art of organic chemistry will appreciate that many organic compounds can form complexes with solvents in which they are reacted or from which they are precipitated or crystallized. These complexes are known as "solvates". For example, a complex with water is known as a "hydrate".
A compound which, upon administration to the recipient, is capable of being converted into a compound of formula (I) as described above or an active metabolite or residue thereof, is known as a "prodrug". A prodrug may, for example, be converted within the body, e. g. by hydrolysis in the blood, into its active form that has medical effects. Pharmaceutically acceptable prodrugs are described in T. Higuchi and V. Stella, Prodrugs as Novel Delivery Systems, Vol. 14 of the A. C. S. Symposium Series (1976); and in Edward B. Roche, ed.,
Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, both of which are incorporated herein by reference.
As used herein, the term "alkyl" means both straight and branched chain saturated hydrocarbon groups. Examples of alkyl groups include methyl, ethyl, n-propyl, iso-propyl, n-butyl, t-butyl, i-butyl, sec-butyl pentyl, hexyl, heptyl, octyl, nonyl and decyl groups.
Among unbranched alkyl groups, there are preferred methyl, ethyl, n-propyl, iso-propyl, n-butyl groups. Among branched alkyl groups, there may be mentioned t-butyl, i-butyl,
1-ethylpropyl, 1-ethylbutyl and 1-ethylpentyl groups.
As used herein, the term "aryl" means a monocyclic or bicyclic aromatic carbocyclic group.
Examples of aryl groups include phenyl and naphthyl. A naphthyl group may be attached through the 1 or the 2 position. In a bicyclic aromatic group, one of the rings may, for example, be partially saturated. Examples of such groups include indanyl and tetrahydronaphthyl. Specifically, the term C5-I0 aryl is used herein to mean a group comprising from 5 to 10 carbon atoms in a monocyclic or bicyclic aromatic group. A particularly preferred C5-I0 aryl group is phenyl.
As mentioned above, the compounds of the invention have activity as thyroid receptor ligands. The compounds of the invention are preferably selective agonists or partial agonists of the thyroid receptor. Preferably compounds of the present invention possess activity as agonists of the thyroid receptor, preferably selective agonists of the thyroid receptor-beta. They may thus be used in the treatment of diseases or disorders associated with thyroid receptor activity, particularly diseases or disorders for which selective agonists of the thyroid receptor-beta are indicated. In particular, compounds of the present invention may be used in the treatment of diseases or disorders associated with metabolism dysfunction or which are dependent upon the expression of a T3 regulated gene.
Clinical conditions for which an agonist or partial agonist is indicated include, but are not limited to, hypothyroidism; subclinical hyperthyroidism; non-toxic goiter; atherosclerosis; thyroid hormone replacement therapy (e.g., in the elderly); malignant tumor cells containing the thyroid receptor; papillary or follicular cancer; maintenance of muscle strength and function (e.g., in the elderly); reversal or prevention of frailty or age-related functional decline ("ARPD") in the elderly (e.g., sarcopenia); treatment of catabolic side effects of glucocorticoids; prevention and/or treatment of reduced bone mass, density or growth (e.g., osteoporosis and osteopenia); treatment of chronic fatigue syndrome (CFS); accelerating healing of complicated fractures (e.g. distraction osteogenesis); in joint replacement; eating disorders (e.g., anorexia); treatment of obesity and growth retardation associated with obesity; treatment of depression, nervousness, irritability and stress; treatment of reduced mental energy and low self-esteem (e.g., motivation/assertiveness); improvement of cognitive function (e.g., the treatment of dementia, including Alzheimer's disease and short term memory loss); treatment of catabolism in connection with pulmonary dysfunction and ventilator dependency; treatment of cardiac dysfunction (e.g., associated with valvular disease, myocardial infarction, cardiac hypertrophy or congestive heart failure); lowering blood pressure; protection against ventricular dysfunction or prevention of reperfusion events; treatment of hyperinsulinemia; stimulation of osteoblasts, bone remodeling and cartilage growth; regulation of food intake; treatment of insulin resistance, including NIDDM, in mammals (e.g., humans); treatment of insulin resistance in the heart; treatment of congestive heart failure; treatment of musculoskeletal impairment (e.g., in the elderly); improvement of the overall pulmonary function; skin disorders or diseases, such as dermal atrophy, glucocorticoid induced dermal atrophy, including restoration of dermal atrophy induced by topical glucocorticoids, and the prevention of dermal atrophy induced
by topical glucocorticoids (such as the simultaneous treatment with topical glucocorticoid or a pharmacological product including both glucocorticoid and a compound of the invention), the restoration/prevention of dermal atrophy induced by systemic treatment with glucocorticoids, restoration/prevention of atrophy in the respiratory system induced by local treatment with glucocorticoids, UV -induced dermal atrophy, dermal atrophy induced by aging (wrinkles, etc.), wound healing, post surgical bruising caused by laser resurfacing, keloids, stria, cellulite, roughened skin, actinic skin damage, lichen planus, ichtyosis, acne, psoriasis, Dernier's disease, eczema, atopic dermatitis, chloracne, pityriasis and skin scarring. In addition, the conditions, diseases, and maladies collectively referenced to as "Syndrome X" or Metabolic Syndrome as detailed in Johannsson J. Clin. Endocrinol. Metab., 82, 727-34 (1997), may be treated employing a compound of the invention. The term treatment includes, where appropriate, prophylactic treatment.
The compounds of the invention find particular application in the treatment or prophylaxis of the following: (1) hypercholesterolemia, dyslipidemia or any other lipid disorder manifested by an unbalance of blood or tissue lipid levels ; (2) atherosclerosis; (3) replacement therapy in elderly subjects with hypothyroidism who are at risk for cardiovascular complications; (4) replacement therapy in elderly subjects with subclinical hypothyroidism who are at risk for cardiovascular complications; (5) obesity; (6) diabetes (7) depression; (8) osteoporosis (especially in combination with a bone resorption inhibitor); (9) goiter; (10) thyroid cancer; (11) cardiovascular disease or congestive heart failure; (12) glaucoma; and (13) skin disorders.
The compounds of the invention find especial application in the treatment or prophylaxis of the following: (1) hypercholesterolemia, dyslipidemia or any other lipid disorder manifested by an unbalance of blood or tissue lipid levels; (2) atherosclerosis; (3) obesity; (4) diabetes.
The invention also provides a method for the treatment or prophylaxis of a condition in a mammal mediated by a thyroid receptor, which comprises administering to the mammal a therapeutically effective amount of a compound of formula (I) as defined above or a pharmaceutically acceptable ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt. Clinical conditions mediated by a thyroid receptor that may be treated by the method of the invention are those described above.
The invention also provides the use of a compound of formula (I) as defined above or a pharmaceutically acceptable ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt, for the manufacture of a medicament for the treatment or prophylaxis of a condition mediated by a thyroid receptor. Clinical conditions mediated by a thyroid receptor that may be treated by the method of the invention are those described above.
Hereinafter, the term "active ingredient" means a compound of formula (I) as defined above, or a pharmaceutically acceptable ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt.
The amount of active ingredient which is required to achieve a therapeutic effect will, of course, vary with the particular compound, the route of administration, the subject under treatment, and the particular disorder or disease being treated. A compound of the invention may be administered orally or via injection at a dose of from 0.001 to 1500 mg/kg per day, preferably from 0.01 to 1500 mg/kg per day, more preferably from 0.1 to 1500 mg/kg per day, most preferably from 0.1 to 500 mg/kg per day. The dose range for adult humans is generally from 5 mg to 35 g per day and preferably 5 mg to 2 g per day. Tablets or other forms of presentation provided in discrete units may conveniently contain an amount of compound of the invention which is effective at such dosage or as a multiple of the same, for example units containing 5 mg to 500 mg, usually around 10 mg to 200 mg.
While it is possible for the active ingredient to be administered alone, it is preferable for it to be present in a pharmaceutical formulation. Accordingly, the invention provides a pharmaceutical formulation comprising a compound of formula (I) as defined above or a pharmaceutically acceptable ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt, and a pharmaceutically acceptable excipient.
The pharmaceutical formulations according to the invention include those suitable for oral, parenteral (including subcutaneous, intradermal, intramuscular, intravenous, and intraarticular), inhalation (including fine particle dusts or mists which may be generated by
means of various types of metered does pressurized aerosols), nebulizers or insufflators, rectal and topical (including dermal, buccal, sublingual, and intraocular) administration, although the most suitable route may depend upon, for example, the condition and disorder of the recipient.
The formulations may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy. All methods include the step of bringing the active ingredient into association with the carrier which constitutes one or more accessory ingredients. In general the formulations are prepared by uniformly and intimately bringing into association the active ingredient with liquid carriers or finely divided solid carriers or both and then, if necessary, shaping the product into the desired formulation.
Formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous liquid or a non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in- oil liquid emulsion. The active ingredient may also be presented as a bolus, electuary or paste.
A tablet may be made by compression or moulding, optionally with one or more accessory ingredients. Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free-flowing form such as a powder or granules, optionally mixed with a binder, lubricant, inert diluent, lubricating, surface active or dispersing agent. Moulded tablets may be made by moulding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent. The tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredient therein. The present compounds can, for example, be administered in a form suitable for immediate release or extended release. Immediate release or extended release can be achieved by the use of suitable pharmaceutical compositions comprising the present compounds, or, particularly in the case of extended release, by the use of devices such as subcutaneous implants or osmotic pumps. The present compounds can also be administered liposomally.
Exemplary compositions for oral administration include suspensions which can contain, for example, microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners or flavoring agents such as those known in the art; and immediate release tablets which can contain, for example, microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and/or lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants such as those known in the art. A compound of formula (I) can also be delivered through the oral cavity by sublingual and/or buccal administration. Molded tablets, compressed tablets or freeze-dried tablets are exemplary forms which may be used. Exemplary compositions include those formulating the present compound(s) with fast dissolving diluents such as mannitol, lactose, sucrose and/or cyclodextrins. Also included in such formulations may be high molecular weight excipients such as celluloses (avicel) or polyethylene glycols (PEG). Such formulations can also include an excipient to aid mucosal adhesion such as hydroxy propyl cellulose (HPC), hydroxy propyl methyl cellulose (HPMC), sodium carboxy methyl cellulose (SCMC), maleic anhydride copolymer (e.g., Gantrez), and agents to control release such as polyacrylic copolymer (e.g. Carbopol 934). Lubricants, glidants, flavors, coloring agents and stabilizers may also be added for ease of fabrication and use.
Formulations for parenteral administration include aqueous and non-aqueous sterile injection solutions which may contain anit-oxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents. The formulations may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (lyophilised) condition requiring only the addition of the sterile liquid carrier, for example saline or water- for-injection, immediately prior to use. Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described. Exemplary compositions for parenteral administration include injectable solutions or suspensions which can contain, for example, suitable non-toxic, parenterally acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid, or Cremaphor.
Exemplary compositions for nasal aerosol or inhalation administration include solutions in saline, which can contain, for example, benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, and/or other solubilizing or dispersing agents such as those known in the art.
Formulations for rectal administration may be presented as a suppository with the usual carriers such as cocoa butter, synthetic glyceride esters or polyethylene glycol. Such carriers are typically solid at ordinary temperatures, but liquify and/or dissolve in the rectal cavity to release the drug.
Formulations for topical administration in the mouth, for example buccally or sublingually, include lozenges comprising the active ingredient in a flavoured basis such as sucrose and acacia or tragacanth, and pastilles comprising the active ingredient in a basis such as gelatin and glycerine or sucrose and acacia. Exemplary compositions for topical administration include a topical carrier such as Plastibase (mineral oil gelled with polyethylene).
Preferred unit dosage formulations are those containing an effective dose, as hereinbefore recited, or an appropriate fraction thereof, of the active ingredient.
It should be understood that in addition to the ingredients particularly mentioned above, the formulations of this invention may include other agents conventional in the art having regard to the type of formulation in question, for example those suitable for oral administration may include flavouring agents.
Whilst a compound of the invention may be used as the sole active ingredient in a medicament, it is also possible for the compound to be used in combination with one or more further active agents. Such further active agents may be a further compound according to the invention, or they may be different therapeutic agents, for example an anti- dyslipidemic agent or other pharmaceutically active material.
A compound of the present invention may be employed in combination with one or more other modulators and/or ligands of the thyroid receptor or one or more other suitable therapeutic agents selected from the group consisting of cholesterol/lipid lowering agents,
hypolipidemic agents, anti-atherosclerotic agents, anti-diabetic agents, anti-osteoporosis agents, anti-obesity agents, growth promoting agents, anti-inflammatory agents, antianxiety agents, anti-depressants, anti-hypertensive agents, cardiac glycosides, appetite supressants, bone resorption inhibitors, thyroid mimetics, anabolic agents, anti-tumor agents and retinoids..
Examples of suitable hypolipidemic agents for use in combination with a compound of the present invention include an acyl coenzyme A cholesterol acyltransferase (ACAT) inhibitor, a microsomal triglyceride transfer protein (MTP) inhibitor, a cholesterol ester transfer protein (CETP) inhibitor, a ileal bile acid transporter (IBAT) inhibitor, any cholesterol absorption inhibitor, a 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, a squalene synthetase inhibitor, a bile acid sequestrant, a peroxisome proliferator-activator receptor (PPAR)-alpha agonist, a peroxisome proliferator-activator receptor (PPAR)-delta agonist, any peroxisome proliferator-activator receptor (PPAR)- gamma/delta dual agonist, any peroxisome proliferator-activator receptor (PPAR)- alpha/delta dual agonist, a nicotinic acid or a derivative thereof, and a thiazolidinedione or a derivative thereof.
Examples of suitable hypolipidemic agents for use in combination with a compound of the present invention also include ezetimibe, simvastatin, atorvastatin, rosuvastatin, cerivastatin, fluvastatin, lovastatin, pravastatin, fenofibrate, gemfibrozil and bezafibrate.
Examples of suitable anti-diabetic agents for use in combination with a compound of the present invention include biguanides (e.g., metformin or phenformin), glucosidase inhibitors (e.g,. acarbose or miglitol), insulins (including insulin secretagogues or insulin sensitizers), meglitinides (e.g., repaglinide), sulfonylureas (e.g., glimepiride, glyburide, glipyride, gliclazide, chlorpropamide and glipizide), biguanide/glyburide combinations (e.g., Glucovance®), thiazolidinediones (e.g., troglitazone, rosiglitazone, englitazone, darglitazone and pioglitazone), PPAR-alpha agonists, PPAR-gamma agonists, PPAR alpha/gamma dual agonists, PPAR alpha/delta dual agonists, SGLT 1, 2 or 3 inhibitors, glycogen phosphorylase inhibitors, inhibitors of fatty acid binding protein (aP2), glucagon- like peptide- 1 (GLP-I), glucocorticoid (GR) antagonist and dipeptidyl peptidase IV (DP4) inhibitors.
Examples of suitable anti-osteoporosis agents for use in combination with a compound of the present invention include alendronate, risedronate, PTH, PTH fragment, raloxifene, calcitonin, RANK ligand antagonists, calcium sensing receptor antagonists, TRAP inhibitors, selective estrogen receptor modulators (SERM) and AP-I inhibitors.
Examples of suitable anti-obesity agents for use in combination with a compound of the present invention include aP2 inhibitors, PPAR gamma antagonists, PPAR delta agonists, beta 3 adrenergic agonists, such as AJ9677 (Takeda/Dainippon), L750355 (Merck), or CP331648 (Pfizer) or other known beta 3 agonists as disclosed in U.S. Patent Nos. 5,541,204, 5,770,615, 5,491,134, 5,776,983 and 5,488,064, a lipase inhibitor, such as orlistat or ATL-962 (Alizyme), a serotonin (and dopamine) reuptake inhibitor, such as sibutramine, topiramate (Johnson & Johnson) or axokine (Regeneron), other thyroid receptor beta drugs, such as a thyroid receptor ligand as disclosed in WO 97/21993 (U. CaI SF), WO 99/00353 (KaroBio) and GB98/284425 (KaroBio), CB-I (cannabinoid receptor) antagonists (see G. Colombo et al, "Appetite Suppression and Weight Loss After the
Cannabionid Antagonist SR 141716", Life Sciences, VoI 63, PL 113-117 (1998)) and/or an anorectic agent, such as dexamphetamine, phentermine, phenylpropanolamine or mazindol.
A compound of the present invention may be combined with growth promoting agents, such as, but not limited to, TRH, diethylstilbesterol, theophylline, enkephalins, E series prostaglandins, compounds disclosed in U.S. Patent No. 3,239,345, e.g., zeranol, and compounds disclosed in U.S. Patent No. 4,036,979, e.g., sulbenox or peptides disclosed in U.S. Patent No. 4,411,890.
A compound of the invention may also be used in combination with growth hormone secretagogues such as GHRP-6, GHRP-I (as described in U.S. Patent No. 4,411,890 and publications WO 89/07110 and WO 89/07111), GHRP-2 (as described in WO 93/04081), NN703 (Novo Nordisk), LY444711 (Lilly), MK-677 (Merck), CP424391 (Pfizer) and B- HT920, or with growth hormone releasing factor and its analogs or growth hormone and its analogs or somatomedins including IGF-I and IGF-2, or with alpha-adrenergic agonists, such as clonidine or serotinin 5-HTD agonists, such as sumatriptan, or agents which inhibit somatostatin or its release, such as physostigmine and pyridostigmine. A still further use of a disclosed compound of the invention is in combination with parathyroid hormone, PTH(I -34) or bisphosphonates, such as MK-217 (alendronate).
Examples of suitable anti-inflammatory agents for use in combination with a compound of the present invention include prednisone, dexamethasone, Enbrel®, cyclooxygenase inhibitors (i.e., COX-I and/or COX-2 inhibitors such as NSAIDs, aspirin, indomethacin, ibuprofen, piroxicam, Naproxen®, Celebrex®, Vioxx®), CTLA4-Ig agonists/antagonists, CD40 ligand antagonists, IMPDH inhibitors, such as mycophenolate (CellCept®), integrin antagonists, alpha-4 beta-7 integrin antagonists, cell adhesion inhibitors, interferon gamma antagonists, ICAM-I, tumor necrosis factor (TNF) antagonists (e.g., infliximab, OR1384), prostaglandin synthesis inhibitors, budesonide, clofazimine, CNI- 1493, CD4 antagonists (e.g., priliximab), p38 mitogen-activated protein kinase inhibitors, protein tyrosine kinase (PTK) inhibitors, IKK inhibitors, and therapies for the treatment of irritable bowel syndrome (e.g., Zelmac® and Maxi-K® openers such as those disclosed in U.S. Patent No. 6,184,231 Bl).
Examples of suitable anti-anxiety agents for use in combination with a compound of the present invention include diazepam, lorazepam, buspirone, oxazepam, and hydroxyzine pamoate.
Examples of suitable anti-depressants for use in combination with a compound of the present invention include citalopram, fluoxetine, nefazodone, sertraline, and paroxetine.
Examples of suitable anti-hypertensive agents for use in combination with a compound of the present invention include beta adrenergic blockers, calcium channel blockers (L-type and T-type; e.g. diltiazem, verapamil, nifedipine, amlodipine and mybefradil), diuretics (e.g., chlorothiazide, hydrochlorothiazide, flumethiazide, hydroflumethiazide, bendroflumethiazide, methylchlorothiazide, trichloromethiazide, polythiazide, benzthiazide, ethacrynic acid tricrynafen, chlorthalidone, furosemide, musolimine, bumetanide, triamtrenene, amiloride, spironolactone), renin inhibitors, ACE inhibitors (e.g., captopril, zofenopril, fosinopril, enalapril, ceranopril, cilazopril, delapril, pentopril, quinapril, ramipril, lisinopril), AT-I receptor antagonists (e.g., losartan, irbesartan, valsartan), ET receptor antagonists (e.g., sitaxsentan, atrsentan and compounds disclosed in U.S. Patent Nos. 5,612,359 and 6,043,265), Dual ET/AII antagonist (e.g., compounds disclosed in WO 00/01389), neutral endopeptidase (NEP) inhibitors, vasopepsidase inhibitors (dual NEP- ACE inhibitors) (e.g., omapatrilat and gemopatrilat), and nitrates.
Examples of suitable cardiac glycosides for use in combination with a compound of the present invention include digitalis and ouabain.
Examples of suitable cholesterol/lipid lowering agents for use in combination with a compound of the present invention include HMG-CoA reductase inhibitors, squalene synthetase inhibitors, fibrates, bile acid sequestrants, ACAT inhibitors, MTP inhibitors, lipooxygenase inhibitors, an ileal Na~7bile acid cotransporter inhibitor, cholesterol absorption inhibitors, and cholesterol ester transfer protein inhibitors (e.g., CP-529414).
MTP inhibitors which may be employed herein in combination with a compound of formula (I) include MTP inhibitors as disclosed in U.S. Patent No. 5,595,872, U.S. Patent No. 5,739,135, U.S. Patent No. 5,712,279, U.S. Patent No. 5,760,246, U.S. Patent No. 5,827,875, U.S. Patent No. 5,885,983 and U.S. Patent No. 5,962,440 all incorporated herein by reference.
The HMG CoA reductase inhibitors which may be employed in combination with a compound of formula (I) include mevastatin and related compounds as disclosed in U.S. Patent No. 3,983,140, lovastatin (mevinolin) and related compounds as disclosed in U.S. Patent No. 4,231 ,938, pravastatin and related compounds such as disclosed in U.S. Patent No. 4,346,227, simvastatin and related compounds as disclosed in U.S. Patent Nos. 4,448,784 and 4,450,171. Further HMG CoA reductase inhibitors which may be employed herein include fluvastatin, disclosed in U.S. Patent No. 5,354,772, cerivastatin disclosed in U.S. Patent Nos. 5,006,530 and 5,177,080, atorvastatin disclosed in U.S. Patent Nos. 4,681,893, 5,273,995, 5,385,929 and 5,686,104, pyrazole analogs of mevalonolactone derivatives as disclosed in U.S. Patent No. 4,613,610, indene analogs of mevalonolactone derivatives, as disclosed in PCT application WO 86/03488, 6-[2-(substituted-pyrrol-l-yl)- alkyl)pyran-2-ones and derivatives thereof, as disclosed in U.S. Patent No. 4,647,576, Searle's SC-45355 (a 3-substituted pentanedioic acid derivative) dichloroacetate, imidazole analogs of mevalonolactone, as disclosed in PCT application WO 86/07054, 3-carboxy-2- hydroxy-propane-phosphonic acid derivatives, as disclosed in French Patent No. 2,596,393, 2,3-disubstituted pyrrole, furan and thiophene derivatives, as disclosed in European Patent Application No. 0221025, naphthyl analogs of mevalonolactone, as disclosed in U.S. Patent No. 4,686,237, octahydronaphthalenes, such as disclosed in U.S. Patent No. 4,499,289, keto
analogs of mevinolin (lovastatin), as disclosed in European Patent Application No.0,142,146 A2, as well as other known HMG CoA reductase inhibitors.
The squalene synthetase inhibitors which may be used in combination with a compound of the present invention include, but are not limited to, α-phosphono-sulfonates disclosed in U.S. Patent No. 5,712,396, those disclosed by Biller et al, J. Med. Chem., 1988, Vol. 31, No. 10, pp 1869-1871, including isoprenoid (phosphinylmethyl)phosphonates, terpenoid pyrophosphates disclosed by P. Ortiz de Montellano et al, J. Med. Chem., 1977, 20, 243- 249, the farnesyl diphosphate analog A and presqualene pyrophosphate (PSQ-PP) analogs as disclosed by Corey and Volante, J. Am. Chem. Soc, 1976, 98, 1291-1293, phosphinylphosphonates reported by McClard, R.W. et al, J.A.C.S., 1987, 109, 5544 and cyclopropanes reported by Capson, T.L., PhD dissertation, June, 1987, Dept. Med. Chem. U of Utah, Abstract, Table of Contents, pp 16, 17, 40-43, 48-51, as well as other squalene synthetase inhibitors as disclosed in U.S. Patent No. 4,871,721 and 4,924,024 and in Biller, S. A., Neuenschwander, K., Ponpipom, M.M., and Poulter, CD., Current Pharmaceutical Design, 2, 1-40 (1996).
Bile acid sequestrants which may be used in combination with a compound of the present invention include cholestyramine, colestipol and DEAE-Sephadex (Secholex®, Policexide®), as well as lipostabil (Rhone-Poulenc), Eisai E-5050 (an N-substituted ethanolamine derivative), imanixil (HOE-402), tetrahydrolipstatin (THL), istigmastanylphos-phorylcholine (SPC, Roche), aminocyclodextrin (Tanabe Seiyoku), Ajinomoto AJ-814 (azulene derivative), melinamide (Sumitomo), Sandoz 58-035, American Cyanamid CL-277,082 and CL-283,546 (disubstituted urea derivatives), nicotinic acid, acipimox, acifran, neomycin, p-aminosalicylic acid, aspirin, poly(diallylmethylamine) derivatives such as disclosed in U.S. Patent No. 4,759,923, quaternary amine poly(diallyldimethylammonium chloride) and ionenes such as disclosed in U.S. Patent No. 4,027,009, and other known serum cholesterol lowering agents.
ACAT inhibitors suitable for use in combination with a compound of the invention include ACAT inhibitors as described in, Drugs of the Future 24, 9-15 (1999), (Avasimibe); "The ACAT inhibitor, Cl-1011 is effective in the prevention and regression of aortic fatty streak area in hamsters", Nicolosi et al, Atherosclerosis (Shannon, Irel). (1998), 137(1), 77-85; "The pharmacological profile of FCE 27677: a novel ACAT inhibitor with potent
hypolipidemic activity mediated by selective suppression of the hepatic secretion of ApoB 100-containing lipoprotein", Ghiselli, Giancarlo, Cardiovasc. Drug Rev. (1998), 16(1), 16-30; "RP 73163: a bioavailable alkylsulfinyl-diphenylimidazole ACAT inhibitor", Smith, C, et al, Bioorg. Med. Chem. Lett. (1996), 6(1), 47-50; "ACAT inhibitors: physiologic mechanisms for hypolipidemic and anti-atherosclerotic activities in experimental animals", Krause et al, Editor(s): Ruffolo, Robert R., Jr.; Hollinger, Mannfred A., Inflammation: Mediators Pathways (1995), 173-98, Publisher: CRC, Boca Raton, FIa.; "ACAT inhibitors: potential anti-atherosclerotic agents", Sliskovic et al, Curr. Med. Chem. (1994), 1(3), 204-25; "Inhibitors of acyl-CoA:cholesterol O-acyl transferase (ACAT) as hypocholesterolemic agents. 6. The first water-soluble ACAT inhibitor with lipid-regulating activity. Inhibitors of acyl-CoA:cholesterol acyltransferase (ACAT). 7. Development of a series of substituted N-phenyl-N'-[(l-phenylcyclopentyl)methyl]ureas with enhanced hypocholesterolemic activity", Stout et al, Chemtracts: Org. Chem. (1995), 8(6), 359-62.
Examples of suitable cholesterol absorption inhibitor for use in combination with a compound of the invention include SCH48461 (Schering-Plough), as well as those disclosed in Atherosclerosis 115, 45-63 (1995) and J. Med. Chem. 41, 973 (1998).
Examples of suitable ileal Na+/bile acid cotransporter inhibitors for use in combination with a compound of the invention include compounds as disclosed in Drugs of the Future, 24, 425-430 (1999).
Examples of suitable thyroid mimetics for use in combination with a compound of the present invention include thyrotropin, polythyroid, KB-130015, and dronedarone.
Examples of suitable anabolic agents for use in combination with a compound of the present invention include testosterone, TRH diethylstilbesterol, estrogens, β-agonists, theophylline, anabolic steroids, dehydroepiandrosterone, enkephalins, E-series prostagladins, retinoic acid and compounds as disclosed in U.S. Pat. No. 3,239,345, e.g., Zeranol®; U.S. Patent No. 4,036,979, e.g., Sulbenox® or peptides as disclosed in U.S. Pat. No. 4,411,890.
For the treatment of skin disorders or diseases as described above, a compound of the present invention may be used alone or optionally in combination with a retinoid, such as tretinoin, or a vitamin D analog.
A still further use of a compound of the invention is in combination with estrogen, testosterone, a selective estrogen receptor modulator, such as tamoxifen or raloxifene, or other androgen receptor modulators, such as those disclosed in Edwards, J. P. et al., Bio. Med. Chem. Let., 9, 1003-1008 (1999) and Hamann, L. G. et al., J. Med. Chem., 42, 210-212 (1999).
A further use of a compound of this invention is in combination with steriodal or non-steroidal progesterone receptor agonists ("PRA"), such as levonorgestrel, medroxyprogesterone acetate (MPA).
The above other therapeutic agents, when employed in combination with a compound of the present invention, may be used, for example, in those amounts indicated in the Physicians' Desk Reference (PDR) or as otherwise determined by one of ordinary skill in the art.
Where a compound of the invention is utilized in combination with one or more other therapeutic agent(s), either concurrently or sequentially, the following combination ratios and dosage ranges are preferred:
When combined with a hypolypidemic agent, an antidepressant, a bone resorption inhibitor and/or an appetite suppressant, a compound of formula (I) may be employed in a weight ratio to the additional agent within the range from about 500: 1 to about 0.005 : 1 , preferably from about 300: 1 to about 0.01:1.
Where the antidiabetic agent is a biguanide, a compound of formula (I) may be employed in a weight ratio to biguanide within the range from about 0.01:1 to about 100:1, preferably from about 0.5:1 to about 2:1.
A compound of formula (I) may be employed in a weight ratio to a glucosidase inhibitor within the range from about 0.01:1 to about 100:1, preferably from about 0.5:1 to about 50:1.
A compound of formula (I) may be employed in a weight ratio to a sulfonylurea in the range from about 0.01:1 to about 100:1, preferably from about 0.2:1 to about 10:1.
A compound of formula (I) may be employed in a weight ratio to a thiazolidinedione in an amount within the range from about 0.01:1 to about 100:1, preferably from about 0.5:1 to about 5:1. The thiazolidinedione may be employed in amounts within the range from about 0.01 to about 2000 mg/day, which may optionally be administered in single or divided doses of one to four times per day. Further, where the sulfonylurea and thiazolidinedione are to be administered orally in an amount of less than about 150 mg, these additional agents may be incorporated into a combined single tablet with a therapeutically effective amount of a compound of formula (I).
Metformin, or salt thereof, may be employed with a compound of formula (I) in amounts within the range from about 500 to about 2000 mg per day, which may be administered in single or divided doses one to four times daily.
A compound of formula (I) may be employed in a weight ratio to a PPAR-alpha agonist, a PPAR-gamma agonist, a PPAR-alpha/gamma dual agonist, an SGLT2 inhibitor and/or an aP2 inhibitor within the range from about 0.01:1 to about 100:1, preferably from about 0.5:1 to about 5:1.
An MTP inhibitor may be administered orally with a compound of formula (I) in an amount within the range of from about 0.01 mg/kg to about 100 mg/kg and preferably from about 0.1 mg/kg to about 75 mg/kg, one to four times daily. A preferred oral dosage form, such as tablets or capsules, may contain the MTP inhibitor in an amount of from about 1 to about 500 mg, preferably from about 2 to about 400 mg, and more preferably from about 5 to about 250 mg, administered on a regimen of one to four times daily. For parenteral administration, the MTP inhibitor may be employed in an amount within the range of from about 0.005 mg/kg to about 10 mg/kg and preferably from about 0.005 mg/kg to about 8 mg/kg, administered on a regimen of one to four times daily.
A HMG CoA reductase inhibitor may be administered orally with a compound of formula (I) within the range of from about 1 to 2000 mg, and preferably from about 4 to about 200 mg. A preferred oral dosage form, such as tablets or capsules, will contain the HMG CoA
reductase inhibitor in an amount from about 0.1 to about 100 mg, preferably from about 5 to about 80 mg, and more preferably from about 10 to about 40 mg.
A squalene synthetase inhibitor may be administered with a compound of formula (I) within the range of from about 10 mg to about 2000 mg and preferably from about 25 mg to about 200 mg. A preferred oral dosage form, such as tablets or capsules, will contain the squalene synthetase inhibitor in an amount of from about 10 to about 500 mg, preferably from about 25 to about 200 mg.
A compound of formula (I) as described above also finds use, optionally in labelled form, as a diagnostic agent for the diagnosis of conditions associated with malfunction of the thyroid receptor. For example, such a compound may be radioactively labelled.
A compound of formula (I) as described above, optionally in labelled form, also finds use as a reference compound in methods of discovering other antagonists or partial antagonists of the thyroid receptor. Thus, the invention provides a method of discovering a ligand of the thyroid receptor which comprises use of a compound of the invention or a compound of the invention in labelled form, as a reference compound. For example, such a method may involve a competitive binding experiment in which binding of a compound of formula (I) to the thyroid receptor is reduced by the presence of a further compound which has thyroid receptor-binding characteristics, for example stronger thyroid receptor-binding characteristics than the compound of formula (I) in question.
The invention also provides a method for preparing a compound of formula (I) in accordance with the invention as described above comprising a step of reacting - a compound of formula (II)

(H)
wherein R is a C1-6alkyl group
- with a methanesulphonylating reagent, in the presence of a suitable base;
- optionally followed by hydrolysis of the ester to give the free acid
Exemplary R groups include methyl, ethyl and propyl, particularly methyl.
Methanesulphonylating reagents include methanesulphonyl chloride and methanesulphonyl bromide. Suitable bases include pyridine or alkylamines, for example diisopropylamine and triethylamine. Other bases may be employed, as is known by the person skilled in the art. Optionally, one or more coupling reagents may be employed. The hydrolysis step may be carried out by reaction with an alkali metal hydroxide. Suitable alkali metal hydroxides include lithium hydroxide and sodium hydroxide. The reaction mixture is stirred at room temperature, or heated until the starting materials have been consumed. The reaction may be carried out with protecting groups present and those protecting groups may be removed after the reaction. Suitable protecting groups are known to the person skilled in the art (see T. W. Greene, "Protective Groups in Organic Synthesis", 3rd Edition, New York, 1999).
A compound of formula (H) can be synthesized by reacting: a compound of formula (III)

(III) wherein R is as defined above
- with a suitable reducing agent.
Suitable reducing agents include tin(II)chloride in ethanol. The reaction mixture is stirred at room temperature, or heated until the starting materials have been consumed. The reaction may be carried out with protecting groups present and those protecting groups may be removed after the reaction. Suitable protecting groups are known to the person skilled in the art (see T. W. Greene, "Protective Groups in Organic Synthesis", 3rd Edition, New York, 1999).
A compound of formula (III) can be synthesized by reacting: the compound of formula (IVa)

(IVa) wherein R is as defined above
with a compound of formula (V)
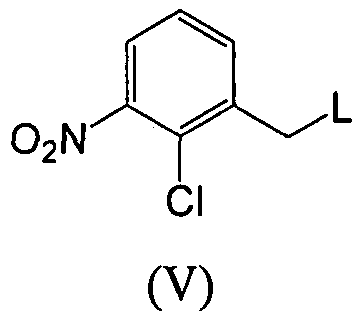
Alternatively, a compound of formula (III) can be synthesized by reacting: - a compound of formula (IVb)

(IVb) wherein R3 is a masked nucleofugal group (for example amino or hydroxyl, optionally in protected form), and R is as defined above, - with a compound of formula (V)

Suitable leaving groups L are for example halogens, hydroxyl and mesylate. Suitable bases include inorganic bases and organic bases (for example amines or pyridines). For example, the base may be selected from hydroxide salts, carbonate salts and bicarbonate salts or from triethylamine and pyridine. A particularly suitable base is potassium carbonate. The reaction mixture is stirred at room temperature, or heated until the starting materials have been consumed. The reaction may be carried out with protecting groups present and those protecting groups may be removed after the reaction. Suitable protecting groups are known to the person skilled in the art (see T. W. Greene, "Protective Groups in Organic Synthesis", 3rd Edition, New York, 1999).
An alternative method for the preparation of the compound of formula (I) is provided by coupling the intermediate of formula (IVa) with a compound of formula (VII) wherein L is a suitable leaving group and Y is H or a suitable leaving group, optionally followed by hydrolysis to the free acid:

Suitable leaving groups L include halogen, OMs and OTs. Suitable bases can be included in the coupling reaction. Suitable bases include inorganic bases and organic bases (for
example amines or pyridines). For example, the base may be selected from hydroxide salts, carbonate salts and bicarbonate salts or from triethylamine and pyridine in the presence of a nucleophilic catalyst, for example iodide. A particularly suitable base is potassium carbonate. Suitable leaving groups Y include Ms and Ts.
A further alternative method for the preparation of the compound of formula (I) is provided by deprotecting a compound of formula (Ia) or an ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt:

(Ia)

Compound (Ia) can, in turn, be prepared by coupling the intermediate of formula (IVa) with a compound of formula (IX), wherein L is a suitable leaving group, optionally followed by hydrolysis to the free acid:

Suitable leaving groups L include halogen, OMs and OTs. Suitable bases can be included in the coupling reaction. Suitable bases include inorganic bases and organic bases (for example amines or pyridines). For example, the base may be selected from hydroxide salts, carbonate salts and bicarbonate salts or from triethylamine and pyridine. A particularly suitable base is potassium carbonate.
A compound of formula (VII) or (IX), wherein L is a suitable leaving group (for example halogen, OMs or OTs) and Y is H, Ts or Ms, can be obtained from (3-aminophenyl)- methanol compounds (VIII) using standard literature procedures.

(VIII) (VII) or (IX)
The method may, in particular make use of a compound of formula (VII) which is the compound of formula (IX):
MeO2

(IX) in which L is as defined above.
The intermediate of formula (IVa) or (IVb) can be prepared as a single enantiomer.
Many methods exist in the literature for the chiral synthesis of the intermediate of formula (IVa). In particular, the intermediate of formula (IVa) can be synthesized by any of the following described synthetic procedures.
a) The (S) form of 2-fluoro-3-(3,5-dibromo-4-hydroxy-phenyl)-propanoic acid methyl ester can be obtained from the corresponding (S) form of 2-fluoro-3-(4-hydroxy-phenyl)- propanoic acid using a brominating agent such as bromine. The (S) form of 2-fluoro-3-(4- hydroxy-phenyl)-propanoic acid can be obtained from diazotization of (S) form of 2- fluoro-3-(4-amino-phenyl)-propanoic acid. The (S) form of 2-fluoro-3-(4-amino-phenyl)- propanoic acid can be obtained from 4-nitro-phenyl alanine as set ou t in the general references: Tet, 55, 36, 1999, 10971; Arzneim. Forsch, 30, 5, 1980, 751; Chem. Pharm. Bull, 31 (10), 1983, 3424; J. Chem. Soc, 1947, 1571; HeIv. Chim. Acta; 64 (8), 1981, 2526; Tet; 52 (46), 1996 14501-14506.


b) The (S) form of 2-fluoro-3-(3, 5-dibromo-4-hydroxy-phenyl)-propanoic acid methyl ester can be obtained from 4-hydroxy-benzaldehyde by hydrogenation using a chiral catalyst as
set out in the general references: J. Chem. Soc. Perkin Trans 1, 1996, 2895-2990; Tet Lett, 33, 1992, 7877; G. K. Surya Parkash, 2006.
Hydrogenation

c) The (R) and (S) forms of 2-fluoro-3-(4-hydroxy-phenyl)-propanoic acid can be obtained by separation of a racemic mixture of 2-fluoro-3-(4-hydroxy-phenyl)-propanoic acid, by enzymatic kinetic resolution as set out in the general references: ref. Organic Lett. 2 (8), 2000, 1037-1040, Tet Asymmetry. 11, 2000, 889-896, J. Org. Chem. 55, 1990, 812-815, or by forming diasteromers of the 2-fluoro-3-(4-hydroxy-phenyl)-propanoic acid with an optical pure compound, for example an amine.
Resolution


d) The (S) form of 2-fluoro-3-(4-hydroxy-phenyl)-propanoic acid methyl ester can be obtained from the corresponding 2-O-substituted-3-(4-hydroxy-phenyl)-propanoic acid methyl ester wherein X is a group such that OX is a leaving group, by reaction with a suitable fluoride source. The reaction typically proceeds with inversion of configuration. For example X may be Ms, Ts or Tf. The reaction may also be carried out on the free acid.

Many methods exist in the literature for the chiral synthesis of intermediates of formula (IVb). For example, the intermediate of formula (IVb) wherein R3 is hydroxy can be synthesized by the following described synthetic procedures.

(IVb)
a) The (S) and (R) forms of 2-hydroxy-3-(3, 5-dibromo-4-hydroxy-phenyl)-propanoic acid methyl ester can be obtained from 4-allyl anisole. [ref. KoIb et al., Chemical Reviews, 94 (1994) 8; Pinard et al.,Bioorg and Med Chem. Lett. 11, 2001, 2173-2176; Liu et al., Org Lett 6 (13), 2004, 2269-2272; Guo et al., Tet Asymmetry. 11, 2000, 4105-4111; Moitessier et al., J. Carbohydrate Chemistry. 22 (1), 2003, 25-34; Alessio et al., Tet Asymmetry 7 (4), 1996, 1101-1104] by chiral oxidation of the allyl double bond followed by oxidation of the primary alcohol, esterification of the acid group and subsequent bromination of the phenyl ring.

Many other methods exist in the literature for the chiral synthesis of alpha-fluoro propanoic acids. For example, the S form of 2-fluoro-3-(4-methoxy-phenyl)-propanoic acid can be obtained from by the treatment of (S)- 2-fiuoro-3-(4-methoxy-phenyl)-propanoic acid-(i?)- 2-oxo-5-phenyl-pyrolidin-l-yl-ester with base and H2O2. (S)- 2-fluoro-3-(4-methoxy- phenyl)-propanoic acid-(i?)-2-oxo-5-phenyl-pyrolidin-l-yl-ester can be obtained from 3-(4- methoxy-phenyl)-propanoic acid (i?)-2-oxo-5-phenyl-pyrolidin-l-yl-ester for example by reaction with an electrophilic fluorinating agent (eg Ts2NF or (PHSO2)2NF) in the presence of a base (eg Lithium bis (trimethyl silyl)amide (LiHMDS) or Na HMDS). 3-(4-methoxy- phenyl)-propanoic acid (i?)-2-oxo-5-phenyl-pyrolidin-l-yl-ester can be obtained from 3-(4- methoxy-phenyl)-propanoic acid. [ref. Shi Z-D, Liu H, Zhang M, Yang D, Burke Jr T R. Synthetic Communications, 34 (21) 2004, 3883-3889; Gao Y, Wei C-Q, Burke Jr T R, Org. Lett. 3 (11), 2001, 1617-1620; Kalaritis P, Regenye R W, Partridge J J, Coffen D L. J. Org. Chem. 55, 1990, 812-815.


a) For use in comparative experiments, the (S) form of 2-hydroxy-3-(4-hydroxy- phenyl)-propanoic acid is commercially available and it can be transformed into the corresponding fluoro derivative with inversion of configuration (to the (R) form) according to literature (ref. M. Shiozaki, Y. Kobayashi and M. Arai; Tetrahedron.
1991, 67, 7021-7028 and S. De Jonghe; I. Van Overmeire; S. Van Calenbergh; C.
Hendrix; R. Busson; D. De Keukeleire; and P. Herdewijn Eur. J. Org. Chem. 2000, 3177-3183).

b) For use in comparative experiments, the (R) form of 2-hydroxy-3-(4-hydroxy- phenyl)-propanoic acid can be obtained from the corresponding amino acid with retention of configuration following the described procedure (ref. N. Vails, M. Vallribera, M. Lopez-Canet and J. Bonjoch; J. Org. Chem. 2002, 67, 4945-4950).
c) The R and S forms of 2-hydroxy-3-(4-hydroxy-phenyl)-propanoic acid can be obtained by separation of the commercially available racemic mixture by forming diastereomers of the acid with an optically pure amine (ref. L. Pretlow, R. Williams and M. Elliot; Chirality, 2003, 15, 674-679) and by enzymatic separation (ref. Y. Momose, T. Maekawa, T. Yamano, M. Kawada, H. Odaka, H. Ikeda and T. Sohda; J. Med. Chem, 2002, 45, 1518-1534).
d) The single enantiomers can be obtained by enzymatic reduction of methyl p- hydroxyphenylpyruvate by the enzyme lactate dehydrogenase (LDH) (ref. L. Pretlow, R. Williams and M. Elliot; Chirality, 2003, 75, 674-679).

The single isomer compounds of the invention may be obtained by use of an enantiospecific enzymic reaction. For example, many lipase enzymes will treat one enantiomer of an α- hydroxyester as a substrate, but not the other. For example, Lipase PS-C "amino" will acetylate the (S) isomer (Fernando F. Huerta, Y. R. Santosh Laxmi, and Jan-E. Backvall Org. Lett., Vol. 2, No. 8, 2000, 1037-1040). That is to say that a racemic substrate (X) (in which R is Cl-6 alkyl and Z is H, 3-amino-2-chlorobenzyl, 3-nitro-2-chlorobenzyl, 2- chloro-3-[(methylsulfonyl)amino]benzyl or 2-chloro-3-[bis(methylsulfonyl)amino]benzyl) is typically converted into the (S) acylated product (XI), leaving the (R) as unreacted starting material (XII). Some other enzymes convert the racemic starting materials into the (R) acylated product (XIII), leaving the (S) isomer as unreacted starting material (XIV). The two isomers can be readily separated on the basis of the different physico-chemical properties of the acylated and unacylated compounds.

(XIII) (XIV)
The invention thus provides a method of obtaining an optically active compound (XI), (XII), (XIII) or (XIV) comprising the step of treating a less optically active compound of formula (X) (in which R is Q-6 alkyl and Z is H, 3-amino-2-chlorobenzyl, 3-nitro-2- chlorobenzyl, 2-chloro-3-[(methylsulfonyl)amino]benzyl or 2-chloro-3-
[bis(methylsulfonyl)amino]benzyl) with a suitable enzyme, for example a lipase enzyme. The invention also provides a synthesis of a compound of formula (I), (Ia), (II), (III), (IVa) or (IVb) comprising such a step.
The present invention further provides the optically active (S) form of the compound of formula (Ha),

(Ha) or an ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt. The compound is useful as an advanced intermediate for the synthesis of a compound of formula (I). The methyl ester is a particular example of an ester of the compound.
The present invention also provides the optically active (S) form of the compound of formula (Ilia),

(Ilia) or an ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt. The compound is useful as an advanced intermediate for the synthesis of a compound of formula (I). The methyl ester is a particular example of an ester of the compound.
The present invention further provides the optically active (S) form of the compound of formula (Ia),
 or an ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt. The compound is useful as an advanced intermediate for the synthesis of a compound of formula (I). The methyl ester is a particular example of an ester of the compound.
or an ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt. The compound is useful as an advanced intermediate for the synthesis of a compound of formula (I). The methyl ester is a particular example of an ester of the compound.
The present invention also provides the optically active (S) form of the compound of formula (IVaa),

(IVaa) or an ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt. The compound is useful as an intermediate for the synthesis of a compound of formula (I).
The present invention also provides a compound of formula (VII):

(VII) in which L is as defined above and Y is H, Ts or Ms. A particular example of a compound of formula (VII) is the compound of formula (IX):
MeO2

(IX) in which L is as defined above. Preferred L groups are Cl and OMs.
The invention will now be illustrated by the following Examples, which do not in any way limit the scope of the invention.
EXAMPLES
The following compounds illustrate compounds of the invention or, where appropriate, compounds for use in the invention.
General Experimental Procedures LCMS: analyzed on HPLC-MS with alternating +/- API and equipped with different brands of 50 mm*2.1mm, 5μ C8 columns. Eluted with 0.05% formic acid/ACN or 0.05% ammonium acetate/ ACN
MW calc. (molecular weight) is an isotopic average and the "found mass" is referring to the most abundant isotope detected in the LC-MS. The "found mass" refers to M+l, M, M-I or M-2 as stated.
Intermediate 1 2-Chloro-3-nitrobenzyIbromide
2-Chloro-3-nitrobenzoic acid (0.30 g, 1.5 mmol) was added to BH3 (IM, 4.0 mL, 4.0 mmol). The reaction was refluxed for 3 hours and then cooled to room temperature. The reaction was quenched with water and extracted with diethyl ether (3x15 mL). Purification on silica (100% dichloromethane) gave 0.28g, (99% yield) of the 2-chloro-3- nitrobenzylalcohol.
To a solution of 2-chloro-3-nitrobenzylalcohol (0.45 g, 2.3 mmol) in toluene (7 mL) was added PBr3 (0.22 mL, 2.3 mmol). The reaction was stirred over night at room temperature.
Filtration through silica and evaporation gave a crude product (2-chloro-3- nitrobenzylbromide) which was used in the next step without purification.
Intermediate 2 Methyl 2-hvdroxy-3-(4-hvdroxy-phenvD-propanoate
To a mixture of 2-hydroxy-3-(4-hydroxy-phenyl)-propanoic acid (4 g, 22 mmol) in dry methanol (150 mL) was added concentrated hydrochloric acid (2 rnL). The reaction mixture was heated at reflux for 3 hours, cooled to room temperature and concentrated under vacuum. The residue was dissolved in ethyl acetate (200 mL) and washed with sodium hydrogen carbonate aqueous solution (saturated) and brine, then dried over magnesium sulphate. The pure compound methyl 2-hydroxy-3-(4-hydroxy-phenyl)- propanoate (4.10 g) was obtained in 95% of yield.
Intermediate 2 Methyl 2-hvdroxy-3-(4-hydroxy-phenvD-propanoate (alternative method)
To a solution of D,L-tyrosine (27.1 g, 0.15 mol) in sodium hydroxide solution (2N aq., 170 mL), copper sulphate solution (12 g Of CuSO4 in 8OmL of water) was added. The mixture was heated at 6O0C for 1.5 h. The reaction mixture was cooled to room temperature and methanol (550 mL) and sodium hydroxide (aq. solution, 2N, 20 mL) were added, followed by dropwise addition of benzylbromide (18 mL, 0.15 mol). The mixture was stirred overnight at room temperature. The precipitate was filtered and washed with water to give 2-amino-3-(4-benzyloxy-phenyl)-propanoic acid as pale solid (36.2g).
To a solution of 2-amino-3-(4-benzyloxy-phenyl)-propanoic acid (36.2g, 0.133mol) in glacial acetic acid (600 mL) and chloroform (150 mL) at between 0 and -50C, isoamyl nitrite (49 mL, 0.36 mol) was added slowly. The reaction mixture was stirred overnight at room temperature, then diluted with water and extracted with chloroform (3 x 250 mL). The combined organic layers were washed with water, dried over sodium sulphate and concentrated to give crude 2-acetoxy-3-(4-benzyloxy-phenyl)-propanoic acid.
The crude 2-acetoxy-3-(4-benzyloxy-phenyl)-propanoic acid and sodium hydroxide solution (10%, 250 mL) were stirred at room temperature overnight. The reaction mixture was poured into water (300 mL) and acidified with concentrated hydrochloric acid to pH=3. The mixture was extracted with chloroform (3 x 250 mL), washed with brine and the
solvent removed in under vacuum to give the crude 2-hydroxy-3-(4-benzyloxy-phenyl)- propanoic acid as yellow solid (25.6 g).
To a solution of the crude 2-hydroxy-3-(4-benzyloxy-phenyl)-propanoic acid (25.6 g) in methanol (400 mL), concentrated hydrochloric acid (1 mL) was added. The reaction mixture was heated at 7O0C for 5 h. After evaporation of the solvent, the residue was dissolved in ethyl acetate (500 mL). The organic layers were washed with sodium hydrogen carbonate aqueous solution (saturated), brine and dried over sodium sulphate. The crude product was purified by silica flash chromatography column (ethyl acetate/heptane; 1 :9 to 2:8) to afford 10.9 g of methyl 2-hydroxy-3-(4-benzyloxy-phenyl)- propanoate as yellow solid. The total yield was 30% for four steps.
PdVC (10%, 0.21 g) was added to a solution of methyl 2-hydroxy-3-(4-benzyloxy-phenyl)- propanoate (10.9g, 38.07mmol) in methanol (300 mL), and then the reaction mixture was stirred under hydrogen at room temperature until the starting material disappeared. The mixture was filtered through celite and the solvent removed under vacuum. The residue methyl 2-hydroxy-3-(4-hydroxy-phenyl)-propanoate (7.1 g. 95% of yield) was pure enough to be used in the next step without further purification.
Intermediate 3
Methyl 2-hvdroxy-3-(4-hvdroxy-3,5-dibromophenvD propanoate
To a solution of methyl 2-hydroxy-3-(4-hydroxy-phenyl)-propanoate (intermediate 2, 2.1 g,
10.7 mmol) in acetonitrile (130 mL) was added bromine (3.4 g, 21.4 mmol) in acetonitrile (20 mL) dropwise at room temperature, and the reaction mixture was stirred overnight. After evaporation of the solvent, the residue was dissolved in ethyl acetate (150 mL) and washed with washed with NaHSO3 (aq.) and brine, dried over magnesium sulphate and concentrated in vacuo to give methyl 2-hydroxy-3-(4-hydroxy-3,5-dibromophenyl) propanoate (3.14 g, 83%).
Intermediate 4
Methyl 2-fluoro-3-(4-hvdroxy-3,5-dibromophenyl) propanoate
A mixture of methyl 2-hydroxy-3-(4-hydroxy-3,5-dibromophenyl) propanoate (intermediate
3, 3.4 g, 9.5 mmol) and potassium carbonate (1.45 g, 1051 mmol) in acetone (85 mL) was
added to methyl iodide (1.5 g, 10.5 mmol). The mixture was stirred at 60°C overnight. After cooling to room temperature, acetone was removed and hydrochloric acid aqueous solution (1 M) was added and extracted with ethyl acetate (3 x 150 mL). The organic layer was washed with sodium hydrogen carbonate aqueous solution (saturated) and brine and then dried over magnesium sulphate. The reaction mixture was evaporated to get crude methyl 2-hydroxy-3-(4-methoxy-3,5-dibromophenyl) propanoate and used directly in next step without additional purification.
A solution of crude methyl 2-hydroxy-3-(4-methoxy-3,5-dibromophenyi) propanoate (3.7 g, 10.2 mmol) in dry dichloromethane (30 mL) was added slowly to a solution of DAST (Et2NSF3) (1.76 g, 10.9 mmol) in dry dichloromethane (10 mL) at 0°C under nitrogen atmosphere. The mixture was stirred for 15 min and allowed to reach room temperature and poured into a mixture of water and ice. The organic layer was separated and the water was extracted with dichloromethane (2 x 30 mL). The combined organic layers were washed with water and dried over magnesium sulphate. The obtained residue was purified by flash chromatography (ethyl acetate/heptane 5:95). Methyl 2-fluoro-3-(4-methoxy-3,5- dibromophenyl) propanoate (2.4 g) was obtained in 65% yield.
To a dichloromethane (30 mL) solution of methyl 2-fluoro-3-(4-methoxy-3,5- dibromophenyl) propanoate (2.4 g, 6.5 mmol) at -780C was added BF3-SMe2 (40 mL) very slowly. The mixture was allowed to warm up to room temperature and stirred overnight.
The reaction mixture was diluted with brine and extracted with ethyl acetate (3 x 20 mL).
The combined organic phases were dried (magnesium sulphate), filtered and concentrated.
The obtained residue was purified by flash chromatography to give the title compound in 70% yield (1.6 g).
Intermediate 5 (Method Al)
Methyl 2-fluoro-3-(4-[(3-amino-2-chlorobenzyl)oxy]-3,5-dibromophenyl| propanoate

A mixture of the nitro derivative (e.g. methyl 2-fluoro-3-{4-[(3-nitro-2-chlorobenzyl)oxy]- 3, 5 -dibromophenyl} propanoate) and tin(II)chloride dihydrate (5 eq.) in absolute ethanol (40 mL/mmol ester) was heated to 75°C for 4 h. The reaction mixture was quenched with sodium hydrogen carbonate aqueous solution (saturated). The aqueous phase was extracted with ethyl acetate (3 x 40 mL) and the combined organic phases were washed with water and brine and dried over magnesium sulphate. After evaporation of the solvent, the residue was purified by flash chromatography (dichloromethane/diethylether 90:10) to yield the desired amino derivative (e.g. methyl 2-fluoro-3-{4-[(3-amino-2-chlorobenzyl)oxy]-3,5- dibromophenyl} propanoate [intermediate 5]).
Intermediate 5 (alternative method) METHOD A2
Methyl 2-fluoro-3-(4-[(3-amino-2-chlorobenzyI)oxy1-3,5-dibromophenvU propanoate

A mixture of the appropriate alphahydroxy-phenol (methyl 2-hydroxy-3-(4-hydroxy-3,5- dibromophenyl) propanoate [intermediate 3]) (1 eq.), the appropriate 3-nitrobenzylbromide (2-chloro-3-nitrobenzylbromide [intermediate I]) (1 eq.) and potassium carbonate (4 eq.) in dry acetone (12 mL/mmol phenol) was heated to reflux for 3 h. The reaction mixture was concentrated, diluted with ethyl acetate and washed with with hydrochloric acid (IM) and brine. The combined organic phases were dried over sodium sulphate, concentrated under vacuum and purified on a column (silica, ethyl acetate:heptane; 1 :9) to give the alphahydroxy-nitro derivative (methyl 2-hydroxy-3-{4-[(3-nitro-2-chlorobenzyl)oxy]-3,5- dibromophenyl} propanoate)

A solution of the appropriate alpha-hydroxy-nitro derivative (methyl 2-hydroxy-3-{4-[(3- nitro-2-chlorobenzyl)oxy]-3,5-dibromophenyl} propanoate) (1 eq.) in dry dichloromethane (10 mL/mmol of alphahydroxy derivative) was added slowly to the solution of DAST (Et2NSF3) (1 eq.) in dry dichloromethane (3 mL/mmol of DAST) at 0°C under nitrogen atmosphere. The mixture was stirred 15 min at O0C and allowed to come to room temperature and stirred for 1 h more. The reaction mixture was poured into a mixture of water and ice. The organic layer was separated and the water was extracted with dichloromethane. The combined organic layers were washed with water and dried over magnesium sulphate. The obtained residue was purified by flash chromatography (ethyl acetate/heptane 5:95 to 1 :9) to afford the appropriate alphafluoro compound (methyl 2- fluoro-3 - { 4- [(3 -nitro-2 -chlorobenzy l)oxy] -3,5 -dibromophenyl } propanoate) .
A mixture of the nitro derivative (methyl 2-fluoro-3-{4-[(3-nitro-2-chlorobenzyl)oxy]-3,5- dibromophenyl} propanoate and tin(II)chloride dihydrate (5 eq.) in absolute ethanol (40
mL/mmol ester) was heated to 8O0C for 16 h. After evaporation of the solvent, NaOH aqueous solution (25%) was added. The aqueous phase was extracted with ethyl acetate and the combined organic phases were washed with water and brine and dried over potassium carbonate. After evaporation of the solvent, the residue was recrystallized from ethyl acetate/heptane to yield the wanted amino derivative (methyl 2-fluoro-3-{4-[(3- amino-2-chlorobenzyl)oxy]-3,5-dibromophenyl} propanoate [intermediate 5]).

Yields and Mass spectrometry analysis for Intermediate 5:

Intermediate 6 (R)-2-hvdroxy-3-(4-hvdroxy-phenvD-propionic acid

A solution of 4-hydroxyphenyl pyruvic acid (14.8 g, 82.15 mmol) in dry THF (200 mL) was treated with Et3N at -2O0C. After 10 min. at this temperature, a solution of (-)-β- chlorodiisopinocamphyl borane in THF (80 mL) was added in approximately 65 minutes. The inside temperature of the reaction mixture was -220C to -2O0C under the addition of (-)- β -chlorodiisopinocamphyl borane. The reaction mixture was further stirred at -20° C for 20
minutes. The cooling bath was removed and the reaction mixture was stirred for 24 hours at room temperature.
The reaction mixture was monitored by HPLC, TLC (MeOH: EtOAc 1 :1, Rf= 0.42), LC/MS and shows that all starting material have been consumed. The pH of reaction mixture was approximately 2 to 3. The reaction mixture was quenched with 2N aqueous NaOH (105 mL) and H2O (45 mL). The reaction temperature was increased from 22° C to 25.5° C during the addition of 2N aqueous NaOH. After the addition of 2N aqueous NaOH, the pH of the reaction mixture was approximately 10 to 11.
The aqueous layer was washed with tert-butylmethylether (2x200 mL) and the combined organic layers were washed with H2O (2x300 mL). The combined aqueous phases were acidified with 2N HCl (25 mL) from pH ~ 10-11 to pH ~ 1 and extracted with EtOAc (4x300 mL).
The organic layer was dried over Na2SO4 and concentrated to give the title compound (R)- 2-hydroxy-3-(4-hydroxy-phenyl)-propionic acid (14.93 g, 99 %). The purity of the (R)-2- hydroxy-3-(4-hydroxy-phenyl)-propionic acid was 88.8% and ee 89.5%. The product was used for next step without any purification.
Chiral analysis of C/?)-2-hvdroxy-3-(4-hvdroxy-phenyr)-propionic acid:
Column: Chiral Reprosil-NR, 4,6x250 mm, 8μm Max: 400bar
Mobile phase: n-heptane/IPA/TFA 95/5/0.1% Software: UniPoint, Version 2.00
Sample compound was dissolved in MeOH
Flow: 1.4 mL/min Inj.volume: 5 and 10 μL (conc.= 9mg/mL)
Start B: 100 (isocratic, only B pump used)
End B: 100 (isocratic, only B pump used)
Grad. time: 45 min
Coll. wave: 225 nm
OBS wave: 274 nm
The retention-time of the (R) form of 2-hydroxy-3-(4-hydroxy-phenyl)-propionic acid is approximately 26 min and the retention-time of (S) form is approximately 31 min.
Intermediate 7
(J?)-2-hvdroxy-3-(4-hvdroxy-phenyl)-propionie acid methyl ester

(7?)-2-hydroxy-3-(4-hydroxy-phenyl)-propionic acid (intermediate 6, 14.34 g, 78.71 mmol) was dissolved in MeOH (55 mL) and 1-4 drops Of H2SO4 (95%-97%) was added. The reaction mixture was refluxed for 1 hour. The reaction mixture was monitored by HPLC, TLC (heptane: EtOAc 65:35), LC/MS and shows that all starting material has been consumed. The solvent was evaporated and EtOAc (300 mL) was added to the mixture. The organic layer was washed with saturated aqueous solution OfNaHCO3 (2*30 mL, pH ~ 8), and saturated aqueous solution of NaCl (2*25 mL, pH ~ 7). The organic layer was dried over Na2SO4 and concentrated to give 14.1 g (J?)-2-hydroxy-3-(4-hydroxy-phenyl)- propionic acid methyl ester in 91% yield. The HPLC purity of (/?)-2-hydroxy-3-(4-hydroxy- phenyl)-propionic acid methyl ester is 80.7% and ee 87%. The product was used for next step without any purification.
Chiral analysis of compound (/0-2-hvdroxy-3-(4-hydroxy-phenyl>propionic acid methyl ester:
Column: Chiral Reprosil-NR, 4,6x250 mm, 8μm Max: 400bar Mobile phase: n-heptane/IPA/TFA 90/10/0.1% Software: UniPoint, Version 2.00
Sample dissolved in MeOH
Flow: 0.8 mL/min Inj .volume: 5 and 10 μL (conc.= 9mg/mL)
Start B: 100 (isocratic, only B pump used)
End B: 100 (isocratic, only B pump used)
Grad. time: 45 min Coll. wave: 225 nm
OBS wave: 274 nm
Retention-time enantiomer (R): ca 21.3 min [(Λ)-2-hydroxy-3-(4-hydroxy-phenyl)- propionic acid methyl ester] Retention-time enantiomer (S): ca 24.7 min
The two peaks were not baseline separated but sufficiently separated for adequate chiral separation. Small injections with higher concentrations were found to give better peak shapes.
Chiral GCMS analysis of compound (/?V2-hvdroxy-3-(4-hvdroxy-phenyl)-propionic acid methyl ester:
Instrument: GC Agilent 8690 series with Autosampler and MS detector,
Jeol GC-mate
Chromatography Software: GCmate ver. 1.9.00 Column: Astec, Beta Cyclodextrin Permethyl, B-PM, 30m x 0.25mm
Ionisation: EI
Injection volume: 1 μl
Gas He
GC -method: Flow lmL/min Split 1 :100
Temp, program:
170oC to 230°C at l0°C/min
23O0C 4 minutes
Total run time 30 min MS -Method Scan interval: 56-413 amu
Scan speed: 0.8 sec
Scan delay 0.1 sec
Analysis time 30 min
Intermediate 8
(7?)-2-hydroxy-3-(3, 5-dibromo-4-hvdroxy-phenvD-propionic acid methyl ester

(/?)-2-hydroxy-3-(4-hydroxy-phenyl)-propionic acid methyl ester (intermediate 7, 13.14 g, 66.97 mmol) was dissolved in acetic acid (145 mL) and cooled with ice bath (inside temperature of the reaction mixture was +1O0C) and NaOAc (27.5 g, 335 mmol) was added to the reaction mixture (inside temperature increased to +120C). The reaction mixture was further stirred for 5 minutes. A solution of bromine (26.8 g, 167.4 mmol, 8577μl) in acetic acid (30 mL) was added to the reaction mixture at 10°- 12° C temperature over 50 minutes. The reaction mixture was stirred for a further 120 minutes at this temperature (inside temperature was +1O0C, and outside O0C).
The reaction mixture was monitored by HPLC, TLC (heptane: EtOAc 65:35), LC/MS and shows that all starting material has been consumed.
To the reaction mixture was added saturated aqueous solution OfNaHSO3 (300 mL, pH~4- 5) and it was extracted with EtOAc (600 mL). The organic layer was washed with saturated aqueous solution OfNaHCO3 (5x300 mL, pH ~ 6-7) and saturated aqueous solution of NaCl (4x300 mL, pH ~ 7). The organic layer was dried over Na2SO4 and filtered through celite and evaporated to give 21.8 g of (i?)-2-hydroxy-3-(3, 5-dibromo-4-hydroxy-phenyl)- propionic acid methyl ester in 92%. The HPLC purity of the (Λ)-2-hydroxy-3-(3, 5- dibromo-4-hydroxy-phenyl)-propionic acid methyl ester was 88% and ee 81%..
Crystallization of CflV2-hydroxy-3-(3, 5-dibromo-4-hydroxy-phenyl')-propionic acid methyl ester:
Re-crystallisation has been completed by dissolving under stirring 21.8 g of crude mixture in MeOH (6OmL), and adding H2O (100 mL) drop-wise over two hours at room temperature. The yellow solid product (12 g) was filtered. The HPLC purity of compound (i?)-2-hydroxy-3-(3, 5-dibromo-4-hydroxy-phenyl)-propionic acid methyl ester was 88% and ee 85%.
The filtrate was evaporated to give 9.8 g which was again crystallised by dissolving in MeOH (50 mL) and adding H2O (80 mL) drop-wise over two hours. The yellow solid product was filtered to give 4.1 g. The HPLC purity of second crystallisation of compound (/?)-2-hydroxy-3-(3, 5-dibromo-4-hydroxy-phenyl)-propionic acid methyl ester was 88% and ee 81%
Chiral analysis of (i?*)-2-hydroxy-3-(3, S-dibromo^-hvdroxy-phenvQ-propionic acid methyl ester:
Column: Chiral Reprosil-NR, 4,6x250 mm, 8μm Max: 400bar
Mobile phase: n-heptane/IPA/TFA 90/10/0.1%
Software: UniPoint, Version 2.00
Sample dissolved in MeOH
Flow: 1.0 mL/min
Inj.volume: 5 and 10 μL (conc.= 9mg/mL)
Start B: 100 (isocratic, only B pump used)
End B: 100 (isocratic, only B pump used) Grad. time: 30 min
Coll. wave: 225 nm
OBS wave: 288 nm
The retention-time of the (R) form of [2-hydroxy-3-(3, 5-dibromo-4-hydroxy-phenyl)- propionic acid methyl ester] is approximately 14 min and the retention-time of the (S) form is approximately 16.5 min.
Intermediate 9
(R)-2-hydroxy-3-[3, 5-dibromo-4-(2-chloro-3-nitro-benzyloxy)-phenyri-propionic acid methyl ester

(/?)-2-hydroxy-3-(3, 5-dibromo-4-hydroxy-phenyl)-propionic acid methyl ester (intermediate 8, 16.12 g, 45.54 mmol) (mixture of re-crystallisation 1 and 2 from previous step) was dissolved in acetone (100 mL) and K2CO3 (25.17g, 182.2 mmol), 1- bromomethyl-2-chloro-3-nitro-benzene (11.4Ig, 45.54 mmol) were added. The reaction mixture was refluxed for 3 hours. The reaction mixture was monitored by HPLC, TLC (heptane: EtOAc 65:35), LC/MS and shows that all starting material has been consumed.
To the reaction mixture was added EtOAc (500 mL) and IM HCl (350 mL) and extracted (pH ~ 1). The organic layer was washed with brine (2*300 mL pH ~ 7) and dried over Na2SO4 and filtered through celite and evaporated to give 23.51 g (98.6%). The HPLC purity of the (i?)-2-hydroxy-3-[3, 5-dibromo-4-(2-chloro-3-nitro-benzyloxy)-phenyl]- propionic acid methyl ester was 91.2% and ee 89%.
The product was used for the next step without any purification.
Intermediate 10
(S)-2-fluoro-3-[3, 5-dibromo-4-(2-chloro-3-nitro-benzyloxy)-phenyll-propionie acid methyl ester

To a stirred solution of (/?)-2-hydroxy-3-[3, 5-dibromo-4-(2-chloro-3-nitro-benzyloxy)- phenyl] -propionic acid methyl ester (intermediate 9, 118.6 g) in THF (3000 ml) at 0-20C
was added a solution of DAST (60 ml) in THF (600 ml) over 10 mins with the temperature below 2°C. The solution was then stirred at room temperature overnight. TLC (30% EtOAc/hexanes) showed no starting material remained and so the solution was added to 5% KHCO3 (11600 ml). The organics were removed, the aqueous extracted with DCM (3 x 3000 ml) and the combined organics dried and evaporated to give a tan solid of (5)-2- fluoro-3-[3, 5-dibromo-4-(2-chloro-3-nitro-benzyloxy)-phenyl]-propionic acid methyl ester.
Yield = 203.2 g (>100%). 1H NMR and MS correspond to the desired structure.
The large scale of the DAST reaction has been completed and the material purified by column chromatography. This gave two crops of material 50.0 g (42% yield; 95.8% @ 230 nm) and 59.6 g (50%; 94.2% @ 230 nm). These were then reduced as two separate batches. The two next step reactions were then carried out and combined for purification by chromatography to give a total of 76.6 g (74% isolated yield; 92.2% @ 254 nm).
Intermediate 11
(S)-2-fluoro-3-[3, 5-dibromo-4-(3-amino-2-chloro-benzyloxy)-phenyll-propionic acid methyl ester

Intermediate 12
(S)-2-fluoro-3-f3, 5-dibromo-4-(2-chloro-3-[bis(methanesuIphonyl)aminol-benzyloxy)- phenyll-propionic acid methyl ester

To a stirred solution of («S)-2-fluoro-3-[3, 5-dibromo-4-(3-amino-2-chloro-benzyloxy)- phenyl] -propionic acid methyl ester (intermediate 11, 75.7 g) in dichloromethane (750 ml) was added, under nitrogen, Et3N (106 ml) followed by methane sulphonyl chloride (47.5 ml) with the temperature being kept below 15°C and the resultant suspension stirred at 0- 10°C for 1 h then room temperature for Ih. LCMS shows only bis-sulfonated product so the suspension was poured onto water (750 ml) and the dichloromethane removed. The aqueous layer was extracted with DCM (2 x 250 ml) and the combined organics were washed with water (500 ml), saturated citric acid (500 ml), saturated K2CO3, water (500 ml) and dried.
Yield = 115.4% (116%) sticky orange solid.
The crude product was slurried in methanol (200 ml) at 45°C for 45 mins then left at room temperature overnight and filtered.
Yield = 86.1 g (86%). Purity = 93.8% (230 nm)
This solid was absorbed onto silica (96.3 g) and the product chromatographed (dry flash) on silica (400 g) eluting with 30% EtOAc in hexanes followed by DCM.
Yield = 83.1 g (97% recovery from column; 83% overall yield). Purity = 92.7% (230 nm)
Example 1
3-[3,5-dibromo-4-({2-chloro-3-[(methylsulfonyl)aminolbenzvUoxy)phenyll-2- fluoropropanoic acid

Methyl 3-[3,5-dibromo-4-({2-chloro-3-[(methylsulfonyl)amino]benzyl}oxy)phenyl]-2- fluoropropanoate (8.6 g, 15 mmol) was dissolved in dioxane (150 mL), and sodium hydroxide (lithium hydroxide has also been used) (1 N in water, 150 mL) was added and the mixture was stirred at 5O0C for 1 hour. After neutralization with hydrochloric acid (1 N), the product was extracted into ethyl acetate. The combined organic phases were dried over sodium sulphate and concentrated under vacuum. The residue was recrystallized from ethyl acetate/heptane to yield 3-[3,5-dibromo-4-({2-chloro-3-[(methylsulfonyl)- amino]benzyl}oxy)phenyl]-2-fluoropropanoic acid as a white solid (7.5 g, 89% yield) (MW=559.6). LC/MS (ESI): m/z 557.9 (M-2).
Chiral HPLC separation HPLC Analyses ReproSil Chiral-NR (4.6 mm id x 250 mm, 8 μm (Dr.Maisch GmbH, Ammerbuch, Germany)) was used for HPLC analyses. Analyses were carried out using M-heptane: 2-propanol:trifluoroacetic acid (90:10:0.1%) as a mobile phase at a flow rate of 1,0 mL/min and room temperature. Detection was carried out at UV 254 run. Under these conditions, the retention times were as follows:
Example 1-Fl = 51 min. Example 1-F2 = 55 min.
HPLC Preparative separation ReproSil Chiral-NR (20 mm id x 250 mm, 8 μm (Dr.Maisch GmbH, Ammerbuch,
Germany)) was used for HPLC preparative separation. The separations were carried out using n-heptane:2-propanol:trifluoroacetic acid (90:10:0.1%) as a mobile phase at a flow rate of 10.0 mL/min and room temperature. Detection was carried out at UV 254 nm. Under these conditions, the retention times were as follows:
Example 1-Fl = 98 min. Example 1-F2 = 109 min.
Optical rotation Optical rotations were obtained on a Perkin-elmer 241 Polarimeter and are reported as follows:
[α]temperature Waveiength, c = concentration in g/100mL and solvent.
[α]23CD=589nm = α times 100 divided by c
Example 1 : (3-[3,5-dibromo-4-({2-chloro-3-[(methylsulfonyl)amino]benzyl}oxy)phenyl]-2- fluoropropanoic acid) fraction 1 : c (Ex-2-Fl) = 2.08mg in l.20mL MeOH = 0.17 α = +0.019 plus positive M23D = +11.2 (c = 0.17, MeOH)
Example 1 : (3-[3,5-dibromo-4-({2-chloro-3-[(methylsulfonyl)amino]benzyl}oxy)phenyl]-2- fluoropropanoic acid) fraction 2: c (Ex-2-F2) = 2.21mg in l.28mL MeOH = 0.17 α = -0.019 minus negative [(X]23 D = -11.2 (c = 0.17, MeOH)
The absolute stereochemistry of each of the two fractions (Fl = + isomer, F2 = - isomer) was determined by single crystal X-ray diffraction analysis. That analysis gaves absolute
structure parameters Flack x=0.00001, esd=0.078 and Flack x=0.064, esd=0.092 for Fraction 1 (+ isomer) and Fraction 2 (- isomer) respectively with the enantiomorphs containing the R-enantiomer and S-enantiomer configuration respectively. The absolute structure parameters are consistent with Fraction 1 (+ isomer) being the (Λ)-enantiomer and Fraction 2 (- isomer) being the (5)-enantiomer.
Enzymatic separation
The (S) isomer compound of the invention can also be separated from the (R) isomer by an enzymatic method. Such a method may be carried out as follows:
To methyl 2-hydroxy-3 - [3 ,5-dibromo-4-( { 2-chloro-3- [(methylsulfonyl)amino]benzyl } oxy)- phenyl] propanoate (synthesable by a method analogous to Intermediate 5, Method A2 with omission of the fluorination step, 1 equivalent) in THF is added vinylacetate (3 equivalents) and enzyme (Lipase PS-C "amano"). The reaction is stirred at room temperature over night and then at 50°C for three more days. The reaction products are then filtered through cotton and purified using preparative HPLC. The recovered reaction products comprise unreacted starting material (the non-substrate enantiomer) and acetylated product of the substrate starting material (methyl 3-[3,5-dibromo-4-({2-chloro-3-[(methylsulfonyl)amino]benzyl} oxy)phenyl]-2-acetoxypropanoate) According to the mechanism of the enzymatic reaction, the stereochemistry of the acetylated compound is (S) (Fernando F. Huerta, Y. R. Santosh Laxmi, and Jan-E. Backvall Org. Lett., Vol. 2, No. 8, 2000, 1037-1040). The (R) isomer can be isolated by separating it from the acetylated (S) compound.
To recover the (S) deacetylated product, the methyl 3-[3,5-dibromo-4-({2-chloro-3- [(methylsulfonyl)amino]benzyl}oxy)phenyl]-2-acetoxypropanoate is dissolved in dioxane and KOH (2M) added. The mixture is stirred for 1.5 h. The reaction mixture is then acidified, extracted into dichloromethane and concentrated. The crude product is dissolved in methanol (3 mL) and some drops of concentrated sulphuric acid are added. The reaction is then refluxed for 2 h. The product is concentrated, extracted into dichloromethane and analysed on chiral HPLC. The methyl 3-[3,5-dibromo-4-({3-chloro-5-[(methylsulfonyl) amino]benzyl}oxy)phenyl]-2-hydroxypropanoate product is isolated with high ee. No racemization is observed; the compounds are enantiostable in acidic and basic media.
Methyl 3 -[3 ,5 -dibromo-4-( { 2-chloro-3- [(methylsulfonyl)amino] benzyl } oxy)phenyl]- 2-hydroxypropanoate can be transformed into the 2-fluoro derivative by treatment with DAST using the already described conditions, which leads to an inversion of configuration.
Example 2
(S)-2-fluoro-3-f3, 5-dibromo-4-(2-chloro-3-methanesulphonyI amino-benzyloxy)- phenyll -propionic acid

(intermediate 12, 1054 mg) in methanol (10.5 ml) was added an aqueous solution of lithium hydroxide (2M; 8.4 ml). The suspension was then left to stir at room temperature for 2 h where it became a hazy solution. The reaction mixture was stirred at room temperature overnight where TLC (30% EtOAc/hexanes) indicated no starting material.
The methanol was evaporated off (bath temperature = 40°C) and the aqueous diluted with water (20 ml) before being extracted with dichloromethane (20 ml). The aqueous pH was adjusted to 1 with saturated citric acid (ca. 20 ml) and the suspension extracted into dichloromethane (3 x 50 ml). Some solid was seen at the phase boundary and this was left with the aqueous layer.
The combined organics were dried, filtered and evaporated.
Yield = 751 mg (83%); purity = 96.5 % (230 nm)
The white solid was slurried in 30% EtOAc/hexanes (8 ml) for 30 mins then filtered to give (ιS)-2-fluoro-3-[3, 5-dibromo-4-(2-chloro-3-methanesulphonyl amino-benzyloxy)-phenyl]- propionic acid.
Yield = 520 mg (69% recovery, 57% yield overall); purity = 97.3% (230 nm)
Chiral analysis of rS)-2-fluoro-3-[3, 5-dibromo-4-(2-chloro-3-methanesulphonyl amino- benzyloxy Vphenyl] -propionic acid :
Column: Reprosil, Chiral-NR, 4,6x250 mm, 8μm Max: 400bar Mobile phase: n-heptan/IPA/TFA 90/10/0.1% Software: UniPoint, Version 2.00
Sample dissolved in IPA:n-heptane 1 : 1
Inj.volume: 20μL (l mg/mL) Flow: 0.8 mL/min
Start B: 100 (isocratic, only B pump used)
End B: 100 (isocratic, only B pump used)
Grad timers): ca 90 min
Coll wave: 225 nm OBS wave: 254 nm
Retention time of (7?)-2-fluoro-3-[3, 5-dibromo-4-(2-chloro-3-methanesulphonyl amino- benzyloxy)-phenyl] -propionic acid: ca 74 min and Retention time of (S)-2-fluoro-3-[3, 5- dibromo-4-(2-chloro-3-methanesulphonyl amino-benzyloxy)-phenyl]-propionic acid ca 80 min
Chiral separation of ("5)-2-fluoro-3-[3, 5-dibromo-4-('2-chloro-3-methanesulphonyl amino- benzyloxyVphenyl] -propionic acid:
Column: Reprosil, Chiral-NR, 20x250 mm, 8μm Max: 400bar
Precolumn: Reprosil, 100 Chiral-NR, 20x30 mm, 8μm
Mobile phase: n-heptane/IPA/TFA, 90/10/0.1%
Software: UniPoint, Version 2.00
Sample dissolved in THF:n-heptane 1 : 1
Inj .amount: ca 4 mg
Flow: 10 mL/min
Start B: 100 (isocratic, only B pump used)
End B 100 (isocratic, only B pump used)
Grad time(495): ca 180 min
Coll wave: 225 run
OBS wave: 254 run
The retention time of the (R) form of -2-fluoro-3-[3, 5-dibromo-4-(2-chloro-3- methanesulphonyl amino-benzyloxy)-phenyl]-propionic acid is 120-134 min and Retention time of (S) form of -2-fluoro-3-[3, 5-dibromo-4-(2-chloro-3-methanesulphonyl amino- benzyloxy)-phenyl] -propionic acid is 128-143 min
Intermediate 13
(S)-2-fluoro-3-[3, 5-dibromo-4-(2-chloro-3-methanesulphonyl amino-benzyloxy)- phenyll -propionic acid methyl ester

To a stirred solution of (S)-2-fluoro-3-[3, 5-dibromo-4-(2-chloro-3-methanesulphonyl amino-benzyloxy)-phenyl]-propionic acid (Example 2; 53.7 g; 91.2% pure) in methanol (537 ml) was added cone. H2SO4 (6 drops) and the solution was heated at reflux for a total of 6 h. HPLC showed trace starting material remaining (0.7%) so the solution was cooled. The solid was filtered off, washed well with cold methanol (50 ml) and dried at 40°C overnight.
Yield = 46.2 g (84%); purity (HPLC) = 97.5 % (230 ran)
Example 3
(S)-2-fluoro-3-[3, 5-dibromo-4-(2-chloro-3-inethanesulphonyl amino-benzyloxy)- phenyll-nropionic acid

To a stirred suspension of (iS)-2-fluoro-3-[3, 5-dibromo-4-(2-chloro-3-methanesulphonyl amino-benzyloxy)-phenyl] -propionic acid methyl ester (Intermediate 13, 46.1 g) in methanol (460 ml) was added an aqueous solution of lithium hydroxide (2M; 420 ml). The suspension was then left to stir at room temperature for 2 h where TLC (30% EtOAc/hexanes), NMR and HPLC indicated no starting material. The solution was adjusted to pH 4 with saturated citric acid (200 ml), then the methanol was evaporated off (bath temperature < 40°C). The slurry was extracted with EtOAc (500 ml then 2x 250ml), the organics were combined, washed with water (250 ml), dried and evaporated. Yield (batch B) = 40.5 g (90%); purity (HPLC) = 96.7 % (230 nm)
On standing more solid was precipitated out of the aqueous liquors. This was extracted into EtOAc (3x150ml), dried and evaporated. Yield (batch E) = 8.0 g (18%, EtOAc wet); purity (HPLC) = 98.4% (230 nm)
The white solid from batch B was slurried in 30% EtO Ac/heptane (12 volumes) at 45°C for
30 min., cooled and filtered.
Yield (batch F) = 38.8 g (86%; 96% recovery); purity (HPLC)= 95.9% (230 nm)
The liquors were combined with the solid (batch F) and re-evaporated. This solid was then blended with the solid material from batch E and slurried in 30% EtO Ac/heptane (200 ml) at 40°C for 30 min., cooled and filtered.
Yield (final batch G) = 45.0 g (100%); purity (HPLC) = 97.3% (230 nm); e.e. 89%, m.p. 167°C.
The enantiomeric excess (e.e.) may be improved through recrystallisation using alcohol, water, acetic acid, ethyl acetate or combinations hereof.
Biological Assays
a) Pharmacodynamics
The ability of the compounds of the present invention in terms of demonstrating favorable pharmacodynamics in animals can be demonstrated and evaluated by those skilled in the art, using for example the protocol found in the following scientific literature:
1) Liu Ye et al. : Thyroid Receptor Ligands. 1. Agonist Ligands Selective for the Thyroid Receptor βi. J. Med. Chem., 2003, 45, 1580-1588.
2) Liu Ye et al. : Selective Thyroid Hormone Receptor-β Activation: A Strategy for
Reduction of Weight, Cholesterol, and Lp(a) with Reduced Cardiovascular Liability. PNAS, 2003, 100, 10067-10072.
Pharmacodynamic parameters were measured for the compound of Example 2. Such parameters are for example plasma levels of LDL-C (lipid parameters), which are known to those skilled in the art.
It was found that (-)-(3-[3,5-dibromo-4-({2-chloro-3-[(methylsulfonyl)amino]benzyl}oxy)- phenyl]-2-fluoropropanoic acid) ("fraction 2" (Exl-F2)) gives a greater reduction of LDL-C compared with "the racemic mixture" of (3-[3,5-dibromo-4-({2-chloro-3-
[(methylsulfonyl)amino] -benzyl }oxy)phenyl]-2-fluoropropanoic acid) or the (+) enantiomer ((+)-(3- [3 , 5 -dibromo-4-( { 2-chloro-3 -[(methylsulfonyl)amino]benzyl } oxy)-phenyl]-2- fluoropropanoic acid), "fraction 1" (ExI-Fl))).
In particular, the compound of Example 1 was analysed in a cholesterol-fed rat model:
Cholesterol fed rat model
Male Sprague Dawley rats 4 weeks old are placed on Paigan diet (D 12336 from Research Diets, US) for 2 weeks before transfer to the laboratory. Upon arrival, animals are adapted
to the new environment for 6 days. Rats are given free access to paigan diet and water, and placed on a 12:12h light dark cycle. Before the start of the experiment, body weights are measured, animals are marked for individual tracking and divided into dose groups (usually six animals per group). Animals are dosed p.o. with compound once daily for seven days. At the end of the experiment animals are fasted for at least 12h prior to sacrifice. In the study below, animals were dosed with 0.1; 0.3 or 0.9 mg/kg body weight of the (S) enantiomer of Example 1 or with the (R) enantiomer of Example 1.
The serum LDL cholesterol and the serum total cholesterol were measured before the experiment and after seven days. The changes in serum LDL cholesterol and the serum total cholesterol levels over the timecourse of the experiments are shown in Figures 1 and 2.
As is seen in Figure 1 , the animals treated with the (S) isomer of Example 2 have a signifantly lower LDL serum cholesterol level at all doses tested (p<0.05). As is seen in Figure 2, the animals treated with the (S) isomer of Example 1 have a signifantly lower total serum cholesterol level at all doses tested (p<0.05).
b) Receptor binding
The ability of a compound of the present invention to bind with high affinity to the thyroid hormone receptor and to activate the thyroid hormone receptor in genetically engineered cells (in vitro assays) is appreciated by those skilled in the art as being beneficial in terms of therapeutic use. For example, it diminishes the possibility for cross-reactivity with other receptors.
1) Barkhem, T. et al. : High level expression of functional full-length human thyroid hormone receptor β 1 in insect cells using a recombinant baculovirus. J. Steroid Biochem. MoI. Biol, 1991, 38, 667-75.
2) Carlsson, B., et al: Synthesis and preliminary characterization of a novel antiarrhythmic compound (KB 130015) with an improved toxicity profile compared with amiodarone. J. Med. Chem., 2002, 45, 623-630.
3) Liu Ye et al: Thyroid Receptor Ligands. 1. Agonist Ligands Selective for the Thyroid Receptor βi. J Med. Chem., 2003, 45, 1580-1588.
The literature above contain not only protocols for binding experiments to the TR-receptor, but also vector constructs, generation of reporter cell lines and the corresponding assay procedures. It has been found that compounds of the invention display binding at the TR- receptor with affinity < 500 nM. In particular it has been found that the (S) isomer of Example 1 (the (-) enantiomer) binds to the TR-receptor with a binding affinity of more than double that of the (R) isomer of Example 1 (the (+) enantiomer):

The following particular cell-based binding experiment was carried out:
Chinese hamster ovary cells have been stably transfected with the human thyroid hormone receptor beta and with a reporter gene driven by thyroid hormone receptor response elements.
Cells are grown under standard conditions using stripped serum. Cells are treated with increasing concentrations of test compound for 48h under serum-free conditions after which reporter gene expression is quantified. Efficacy is quantified relative to the natural hormone, T3. It was found that the the (S) isomer of Example 1 (the (-) enantiomer) has a higher potency (EC50 = 57OnM) than the (R) isomer of Example 1 (the (+) enantiomer) (EC50=1700nM).
Claims
1. An optically active (S) form of the compound of formula (I),

2. A compound as claimed in claim 1 which is the optically active (S) form of 3-[3,5- dibromo-4-({2-chloro-3-[(methylsulfonyl)amino]benzyl}-oxy)phenyl]-2-fluoropropanoic acid or a pharmaceutically acceptable ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt.
3. A compound as claimed in claim 1 or claim 2 for use as a medicament.
4. A compound as claimed in claim 3 for use in the treatment or prophylaxis of a condition associated with a disease or disorder associated with thyroid receptor activity.
5. A method for the treatment or prophylaxis of a disease or disorder associated with thyroid receptor activity in a mammal, which comprises administering to the mammal a therapeutically effective amount of a compound as claimed in claim 1 or claim 2.
6. Use of a compound as claimed in claim 1 or claim 2 for the manufacture of a medicament for the treatment or prophylaxis of a disease or disorder associated with thyroid receptor activity.
7. A pharmaceutical formulation comprising a compound as claimed in claim 1 or claim 2 and a pharmaceutically acceptable excipient.
8. A pharmaceutical formulation as claimed in claim 7 further comprising an additional therapeutic agent selected from cholesterol/lipid lowering agents, hypolipidemic agents, anti-atherosclerotic agents, anti-diabetic agents, anti-osteoporosis agents, anti-obesity agents, growth promoting agents, anti-inflammatory agents, anti-anxiety agents, antidepressants, anti-hypertensive agents, cardiac glycosides, appetite supressants, bone resorption inhibitors, thyroid mimetics, anabolic agents, anti-tumor agents and retinoids.
9. A method of discovering a ligand of the thyroid hormone receptor comprising use of a compound as claimed in claim 1 or claim 2 or a compound as claimed in claim 1 or claim 2 in labelled form, as a reference compound.
10. A compound as claimed in claim 4, a method as claimed in claim 5, a use as claimed in claim 6, or a pharmaceutical formulation as claimed in claim 7 or claim 8 wherein the condition associated with a disease or disorder associated with thyroid receptor activity is selected from (1) hypercholesterolemia, dyslipidemia or any other lipid disorder manifested by an unbalance of blood or tissue lipid levels; (2) atherosclerosis; (3) replacement therapy in elderly subjects with hypothyroidism who are at risk for cardiovascular complications; (4) replacement therapy in elderly subjects with subclinical hypothyroidism who are at risk for cardiovascular complications; (5) obesity; (6) diabetes (7) depression; (8) osteoporosis (especially in combination with a bone resorption inhibitor); (9) goiter; (10) thyroid cancer; (11) cardiovascular disease or congestive heart failure; (12) glaucoma; and (13) skin disorders.
11. A method for preparing a compound of formula (I) as claimed in claim 1 comprising a step of reacting
- a compound of formula (II)

(H) wherein R is a Ci-6alkyl group with a methanesulphonylating reagent, in the presence of a suitable base; - optionally followed by hydrolysis of the ester to give the free acid.
12. A method for preparing a compound of formula (I) as claimed in claim 1 comprising a step of reacting a compound of formula (IVa) with a compound of formula (VII):

wherein L is a suitable leaving group and Y is H or a suitable leaving group; optionally followed by hydrolysis to the free acid.
13. A method for preparing a compound of formula (I) as claimed in claim 1 comprising deprotecting a compound of formula (Ia) or an ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt:

(Ia)

14. The optically active (S) form of the compound of formula (Ha) 
(Ha) or an ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt.
15. The optically active (S) form) of the compound of the compound of formula (Ia),

16. The optically active (S) form of the compound of formula (Ilia),

(Ilia) or an ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt.
17. The optically active (S) form of the compound of formula (IVaa),  (IVaa) or an ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt.
(IVaa) or an ester, amide, solvate or salt thereof, including a salt of such an ester or amide, and a solvate of such an ester, amide or salt.
18. A compound of formula (VII):

(VII) in which L is a suitable leaving group and Y is H, Ts or Ms
19. A compound as claimed in claim 18 which is a compound of formula (IX):
MeO5

(IX) in which L is Cl or OMs.
20. A method of preparing a compound of formula (III) which comprises reacting: - a compound of formula (IVb)

(IVb) wherein R3 is a masked nucleofugal group (for example amino or hydroxyl, optionally in protected form), and R is a Ci-6alkyl group, with a compound of formula (V)

(V) wherein L is a suitable leaving group, in the presence of a suitable base, followed by conversion of the R3 group to a fluoro group with (S) stereochemistry.
21. A method of preparing a compound of formula (III) which comprises reacting a compound of formula (IVa)

(IVa) wherein R is a Ci-6alkyl group
with a compound of formula (V)

(V) wherein L is a suitable leaving group, in the presence of a suitable base.
22. A method of preparing an optically active compound (XI), (XII), (XIII) or (XIV) comprising the step of treating a less optically active compound of formula (X) (in which R is Ci-6 alkyl and Z is H, 3-amino-2-chlorobenzyl, 3-nitro-2-chlorobenzyl, 2-chloro-3- [(methylsulfonyl)amino]benzyl or 2-chloro-3-[bis(methylsulfonyl)amino] benzyl) with a suitable enzyme: 
(XIII) (XIV)
23. A method of preparing a compound of formula (I) as claimed in claim 1 comprising a step as claimed in claim 22.
24. A pharmaceutical formulation as claimed in claim 7 wherein the additional therapeutic agent is a hypolipidemic agent selected from the group consisting of an acyl coenzyme A cholesterol acyltransferase (ACAT) inhibitor, a microsomal triglyceride transfer protein (MTP) inhibitor, a cholesterol ester transfer protein (CETP) inhibitor, a ileal bile acid transporter (IBAT) inhibitor, any cholesterol absorption inhibitor, a 3-hydroxy-3- methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, a squalene synthetase inhibitor, a bile acid sequestrant, a peroxisome proliferator-activator receptor (PPAR)-alpha agonist, a peroxisome proliferator-activator receptor (PPAR)-delta agonist, any peroxisome proliferator-activator receptor (PPAR)-gamma/delta dual agonist, a nicotinic acid or a derivative thereof, and a thiazolidinedione or a derivative thereof.
25. A pharmaceutical formulation as claimed in claim 7 wherein the additional therapeutic agent is a hypolipidemic agent selected from the group consisting of ezetimibe, simvastatin, atorvastatin, rosuvastatin, cerivastatin, fluvastatin, lovastatin, pravastatin, fenofibrate, gemfibrozil and bezafibrate.
26. A pharmaceutical formulation as claimed in claim 7 wherein the additional therapeutic agent is an antidiabetic agent selected from the group consisting of a biguanide, a glucosidase inhibitor, a meglitinide, a sulfonylurea, a thiazolidinedione, a peroxisome proliferator-activator receptor (PPAR)-alpha agonist, a peroxisome proliferator-activator receptor (PPAR)-gamma agonist, a peroxisome proliferator-activator receptor (PPAR) alpha/gamma dual agonist, a sodium glucose co-transporter (SGLT) 1, 2 or 3 inhibitor, a glycogen phosphorylase inhibitor, an aP2 inhibitor, a glucagon-like peptide- 1 (GLP-I), a dipeptidyl peptidase IV inhibitor, a glucocorticoid (GR) antagonist and insulin.
27. A pharmaceutical formulation as claimed in claim 7 wherein the additional therapeutic agent is an antidiabetic agent selected from the group consisting of metformin, glyburide, glimepiride, glipyride, glipizide, chlorpropamide, gliclazide, acarbose, miglitol, troglitazone, pioglitazone, englitazone, darglitazone, rosiglitazone and insulin.
28. A pharmaceutical formulation as claimed in claim 7 wherein the additional therapeutic agent is an anti-obesity agent is selected from the group consisting of an aP2 inhibitor, a peroxisome proliferator-activator receptor (PPAR) gamma antagonist, a peroxisome proliferator-activator receptor (PPAR) delta agonist, a beta-3 adrenergic agonist, a lipase inhibitor, a serotonin reuptake inhibitor and an anorectic agent.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0519294A GB0519294D0 (en) | 2005-09-21 | 2005-09-21 | Compounds |
| GB0519294.3 | 2005-09-21 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007039125A2 true WO2007039125A2 (en) | 2007-04-12 |
| WO2007039125A3 WO2007039125A3 (en) | 2007-06-28 |
Family
ID=35335228
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/009184 WO2007039125A2 (en) | 2005-09-21 | 2006-09-21 | An optically active thyroid receptor agonist and optically active key intermediates in its production |
Country Status (2)
| Country | Link |
|---|---|
| GB (1) | GB0519294D0 (en) |
| WO (1) | WO2007039125A2 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| US10064850B2 (en) | 2007-04-11 | 2018-09-04 | Omeros Corporation | Compositions and methods for prophylaxis and treatment of addictions |
| US11241420B2 (en) | 2007-04-11 | 2022-02-08 | Omeros Corporation | Compositions and methods for prophylaxis and treatment of addictions |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0887346A2 (en) * | 1997-06-23 | 1998-12-30 | F. Hoffmann-La Roche Ag | Phenyl-and aminophenyl-alkylsulfonamide and urea derivatives, their preparation and their use as alpha1A/1L adrenoceptor agonists |
| WO2001036365A2 (en) * | 1999-11-17 | 2001-05-25 | Karo Bio Ab | Thyroid receptor antagonists for the treatment of cardiac and metabolic disorders |
| WO2005092317A1 (en) * | 2004-03-22 | 2005-10-06 | Karo Bio Ab | Thyroid receptor agonists |
| WO2005092316A1 (en) * | 2004-03-22 | 2005-10-06 | Karo Bio Ab | Novel pharmaceutical compositions comprising agonists of the thyroid receptor |
-
2005
- 2005-09-21 GB GB0519294A patent/GB0519294D0/en not_active Ceased
-
2006
- 2006-09-21 WO PCT/EP2006/009184 patent/WO2007039125A2/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0887346A2 (en) * | 1997-06-23 | 1998-12-30 | F. Hoffmann-La Roche Ag | Phenyl-and aminophenyl-alkylsulfonamide and urea derivatives, their preparation and their use as alpha1A/1L adrenoceptor agonists |
| WO2001036365A2 (en) * | 1999-11-17 | 2001-05-25 | Karo Bio Ab | Thyroid receptor antagonists for the treatment of cardiac and metabolic disorders |
| WO2005092317A1 (en) * | 2004-03-22 | 2005-10-06 | Karo Bio Ab | Thyroid receptor agonists |
| WO2005092316A1 (en) * | 2004-03-22 | 2005-10-06 | Karo Bio Ab | Novel pharmaceutical compositions comprising agonists of the thyroid receptor |
Non-Patent Citations (2)
| Title |
|---|
| F.F.HUERTA ET AL.: "Dynamic kinetic resolution of alpha-hydroxy acid esters" ORGANIC LETTERS, vol. 2, no. 8, 2000, pages 1037-1040, XP002422791 cited in the application * |
| HAIGH D ET AL: "Anomalous fluorination of 3-aryl-2-hydroxypropanoic esters by diethylaminosulfur trifluoride (DAST)" JOURNAL OF THE CHEMICAL SOCIETY, PERKIN TRANSACTIONS 1, CHEMICAL SOCIETY. LETCHWORTH, GB, no. 24, 1996, pages 2895-2900, XP002153922 ISSN: 0300-922X cited in the application * |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| US10064850B2 (en) | 2007-04-11 | 2018-09-04 | Omeros Corporation | Compositions and methods for prophylaxis and treatment of addictions |
| US11241420B2 (en) | 2007-04-11 | 2022-02-08 | Omeros Corporation | Compositions and methods for prophylaxis and treatment of addictions |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| GB0519294D0 (en) | 2005-11-02 |
| WO2007039125A3 (en) | 2007-06-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007039125A2 (en) | An optically active thyroid receptor agonist and optically active key intermediates in its production | |
| US6747048B2 (en) | Pyridine-based thyroid receptor ligands | |
| US8263659B2 (en) | Thyroid receptor ligands | |
| US6831102B2 (en) | Phenyl naphthol ligands for thyroid hormone receptor | |
| US20080004251A1 (en) | Novel Pharmaceutical Compositions Comprising Agonists of the Thyroid Receptor | |
| US20100004271A1 (en) | Heterocyclic compounds as aganist for the thyroid receptor | |
| US20080221210A1 (en) | Thyroid Receptor Agonists | |
| ZA200500167B (en) | Benzamide or phenylacetamide derivatives useful as thyroid receptor ligands | |
| WO2004018421A1 (en) | Idol derivatives as new thyroid receptor agonist ligands and methods | |
| US20090233979A1 (en) | Indole Derivatives and Their Use as Thyroid Receptor Ligands | |
| US20090318514A1 (en) | Novel Pharmaceutical Compositions | |
| US7342127B2 (en) | Substituted anilide ligands for the thyroid receptor | |
| US20040176425A1 (en) | Cycloalkyl containing anilide ligands for the thyroid receptor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06805797 Country of ref document: EP Kind code of ref document: A2 |