WO2007024536A2 - Process for preparing maytansinoid antibody conjugates - Google Patents
Process for preparing maytansinoid antibody conjugates Download PDFInfo
- Publication number
- WO2007024536A2 WO2007024536A2 PCT/US2006/031653 US2006031653W WO2007024536A2 WO 2007024536 A2 WO2007024536 A2 WO 2007024536A2 US 2006031653 W US2006031653 W US 2006031653W WO 2007024536 A2 WO2007024536 A2 WO 2007024536A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cell
- drug
- antibody
- mixture
- chromatography
- Prior art date
Links
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 12
- 229940127121 immunoconjugate Drugs 0.000 title description 2
- 239000003814 drug Substances 0.000 claims abstract description 108
- 229940079593 drug Drugs 0.000 claims abstract description 107
- 239000011230 binding agent Substances 0.000 claims abstract description 90
- 238000000034 method Methods 0.000 claims abstract description 85
- 230000008569 process Effects 0.000 claims abstract description 52
- 230000001268 conjugating effect Effects 0.000 claims abstract description 12
- 239000000203 mixture Substances 0.000 claims description 53
- 230000000274 adsorptive effect Effects 0.000 claims description 37
- 238000009295 crossflow filtration Methods 0.000 claims description 32
- 238000006243 chemical reaction Methods 0.000 claims description 21
- 238000004587 chromatography analysis Methods 0.000 claims description 21
- 239000012539 chromatography resin Substances 0.000 claims description 19
- 230000027455 binding Effects 0.000 claims description 17
- 229940127089 cytotoxic agent Drugs 0.000 claims description 16
- 239000002254 cytotoxic agent Substances 0.000 claims description 16
- 231100000599 cytotoxic agent Toxicity 0.000 claims description 16
- 239000003431 cross linking reagent Substances 0.000 claims description 15
- UOWVMDUEMSNCAV-WYENRQIDSA-N rachelmycin Chemical compound C1([C@]23C[C@@H]2CN1C(=O)C=1NC=2C(OC)=C(O)C4=C(C=2C=1)CCN4C(=O)C1=CC=2C=4CCN(C=4C(O)=C(C=2N1)OC)C(N)=O)=CC(=O)C1=C3C(C)=CN1 UOWVMDUEMSNCAV-WYENRQIDSA-N 0.000 claims description 11
- 239000000243 solution Substances 0.000 claims description 11
- 229940123237 Taxane Drugs 0.000 claims description 10
- 230000001588 bifunctional effect Effects 0.000 claims description 10
- 238000001914 filtration Methods 0.000 claims description 10
- 238000001556 precipitation Methods 0.000 claims description 10
- 229930006000 Sucrose Natural products 0.000 claims description 9
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 claims description 9
- 239000005720 sucrose Substances 0.000 claims description 9
- 239000002253 acid Substances 0.000 claims description 8
- 239000006227 byproduct Substances 0.000 claims description 8
- 229910052588 hydroxylapatite Inorganic materials 0.000 claims description 8
- 238000001597 immobilized metal affinity chromatography Methods 0.000 claims description 8
- XYJRXVWERLGGKC-UHFFFAOYSA-D pentacalcium;hydroxide;triphosphate Chemical compound [OH-].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O XYJRXVWERLGGKC-UHFFFAOYSA-D 0.000 claims description 8
- 108010002350 Interleukin-2 Proteins 0.000 claims description 7
- 102000000588 Interleukin-2 Human genes 0.000 claims description 7
- 238000004191 hydrophobic interaction chromatography Methods 0.000 claims description 7
- 125000003396 thiol group Chemical group [H]S* 0.000 claims description 7
- 108091005804 Peptidases Proteins 0.000 claims description 6
- 102000035195 Peptidases Human genes 0.000 claims description 6
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 6
- 238000004255 ion exchange chromatography Methods 0.000 claims description 6
- 235000019833 protease Nutrition 0.000 claims description 6
- 108090000371 Esterases Proteins 0.000 claims description 5
- 102000000646 Interleukin-3 Human genes 0.000 claims description 5
- 108010002386 Interleukin-3 Proteins 0.000 claims description 5
- 102000004388 Interleukin-4 Human genes 0.000 claims description 5
- 108090000978 Interleukin-4 Proteins 0.000 claims description 5
- 108090001005 Interleukin-6 Proteins 0.000 claims description 5
- 239000006172 buffering agent Substances 0.000 claims description 5
- 239000000126 substance Substances 0.000 claims description 5
- 102000004338 Transferrin Human genes 0.000 claims description 4
- 108090000901 Transferrin Proteins 0.000 claims description 4
- 239000003446 ligand Substances 0.000 claims description 4
- 239000008363 phosphate buffer Substances 0.000 claims description 4
- 239000012581 transferrin Substances 0.000 claims description 4
- 229960000575 trastuzumab Drugs 0.000 claims description 4
- 101150021185 FGF gene Proteins 0.000 claims description 3
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 claims description 3
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 claims description 3
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 claims description 3
- 102000004877 Insulin Human genes 0.000 claims description 3
- 108090001061 Insulin Proteins 0.000 claims description 3
- 102000014150 Interferons Human genes 0.000 claims description 3
- 108010050904 Interferons Proteins 0.000 claims description 3
- 101800004564 Transforming growth factor alpha Proteins 0.000 claims description 3
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 claims description 3
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 claims description 3
- 239000008351 acetate buffer Substances 0.000 claims description 3
- 238000001042 affinity chromatography Methods 0.000 claims description 3
- 229950002903 bivatuzumab Drugs 0.000 claims description 3
- 239000007979 citrate buffer Substances 0.000 claims description 3
- 230000002209 hydrophobic effect Effects 0.000 claims description 3
- 230000006698 induction Effects 0.000 claims description 3
- 229940125396 insulin Drugs 0.000 claims description 3
- 229940047124 interferons Drugs 0.000 claims description 3
- 239000003456 ion exchange resin Substances 0.000 claims description 3
- 229920003303 ion-exchange polymer Polymers 0.000 claims description 3
- 238000004366 reverse phase liquid chromatography Methods 0.000 claims description 3
- 229960004641 rituximab Drugs 0.000 claims description 3
- 229950008684 sibrotuzumab Drugs 0.000 claims description 3
- 239000008362 succinate buffer Substances 0.000 claims description 3
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 claims description 2
- 102000004889 Interleukin-6 Human genes 0.000 claims 4
- 229940076264 interleukin-3 Drugs 0.000 claims 4
- 229940028885 interleukin-4 Drugs 0.000 claims 4
- 229940100601 interleukin-6 Drugs 0.000 claims 4
- 102000009024 Epidermal Growth Factor Human genes 0.000 claims 2
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 claims 2
- 102100028123 Macrophage colony-stimulating factor 1 Human genes 0.000 claims 2
- 101710127797 Macrophage colony-stimulating factor 1 Proteins 0.000 claims 2
- 101710098940 Pro-epidermal growth factor Proteins 0.000 claims 2
- 102000006747 Transforming Growth Factor alpha Human genes 0.000 claims 2
- 238000000746 purification Methods 0.000 abstract description 31
- 210000004027 cell Anatomy 0.000 description 103
- 125000005647 linker group Chemical group 0.000 description 82
- 239000000562 conjugate Substances 0.000 description 61
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 54
- 239000011347 resin Substances 0.000 description 44
- 229920005989 resin Polymers 0.000 description 44
- 125000004432 carbon atom Chemical group C* 0.000 description 40
- 125000003342 alkenyl group Chemical group 0.000 description 33
- 230000021615 conjugation Effects 0.000 description 32
- 239000000427 antigen Substances 0.000 description 28
- 102000036639 antigens Human genes 0.000 description 27
- 239000011780 sodium chloride Substances 0.000 description 27
- 108091007433 antigens Proteins 0.000 description 26
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 25
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 22
- 206010028980 Neoplasm Diseases 0.000 description 21
- -1 4-methyl-4-mercapto-l- oxopentyl Chemical group 0.000 description 20
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 18
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 18
- 125000000217 alkyl group Chemical group 0.000 description 16
- 239000008057 potassium phosphate buffer Substances 0.000 description 16
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 16
- 150000001875 compounds Chemical class 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 14
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 14
- 201000011510 cancer Diseases 0.000 description 14
- 239000012507 Sephadex™ Substances 0.000 description 13
- 102220589754 YjeF N-terminal domain-containing protein 3_G25F_mutation Human genes 0.000 description 12
- JSHOVKSMJRQOGY-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-(pyridin-2-yldisulfanyl)butanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCSSC1=CC=CC=N1 JSHOVKSMJRQOGY-UHFFFAOYSA-N 0.000 description 11
- GTBCXYYVWHFQRS-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-(pyridin-2-yldisulfanyl)pentanoate Chemical compound C=1C=CC=NC=1SSC(C)CCC(=O)ON1C(=O)CCC1=O GTBCXYYVWHFQRS-UHFFFAOYSA-N 0.000 description 11
- 239000012634 fragment Substances 0.000 description 11
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 10
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 10
- 229920002684 Sepharose Polymers 0.000 description 10
- 239000000872 buffer Substances 0.000 description 10
- 239000003153 chemical reaction reagent Substances 0.000 description 10
- 239000011541 reaction mixture Substances 0.000 description 10
- QWPXBEHQFHACTK-KZVYIGENSA-N (10e,12e)-86-chloro-12,14,4-trihydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-15,16-dihydro-14h-7-aza-1(6,4)-oxazina-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-6-one Chemical compound CN1C(=O)CC(O)C2(C)OC2C(C)C(OC(=O)N2)CC2(O)C(OC)\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 QWPXBEHQFHACTK-KZVYIGENSA-N 0.000 description 9
- 238000012986 modification Methods 0.000 description 9
- 230000004048 modification Effects 0.000 description 9
- 210000004408 hybridoma Anatomy 0.000 description 8
- 239000000178 monomer Substances 0.000 description 8
- 229910000160 potassium phosphate Inorganic materials 0.000 description 8
- 235000011009 potassium phosphates Nutrition 0.000 description 8
- 239000000047 product Substances 0.000 description 7
- 125000003107 substituted aryl group Chemical group 0.000 description 7
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 6
- 210000003719 b-lymphocyte Anatomy 0.000 description 6
- 230000001472 cytotoxic effect Effects 0.000 description 6
- 238000005516 engineering process Methods 0.000 description 6
- 210000004881 tumor cell Anatomy 0.000 description 6
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 5
- QWPXBEHQFHACTK-UHFFFAOYSA-N Maytansinol Natural products CN1C(=O)CC(O)C2(C)OC2C(C)C(OC(=O)N2)CC2(O)C(OC)C=CC=C(C)CC2=CC(OC)=C(Cl)C1=C2 QWPXBEHQFHACTK-UHFFFAOYSA-N 0.000 description 5
- 241001529936 Murinae Species 0.000 description 5
- 241000699670 Mus sp. Species 0.000 description 5
- 239000000919 ceramic Substances 0.000 description 5
- 238000003776 cleavage reaction Methods 0.000 description 5
- 231100000433 cytotoxic Toxicity 0.000 description 5
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 5
- 229940126619 mouse monoclonal antibody Drugs 0.000 description 5
- 230000007017 scission Effects 0.000 description 5
- 239000001509 sodium citrate Substances 0.000 description 5
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 5
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- 108060003951 Immunoglobulin Proteins 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 125000003118 aryl group Chemical group 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 230000008033 biological extinction Effects 0.000 description 4
- 125000002228 disulfide group Chemical group 0.000 description 4
- AMRJKAQTDDKMCE-UHFFFAOYSA-N dolastatin Chemical compound CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)C)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 AMRJKAQTDDKMCE-UHFFFAOYSA-N 0.000 description 4
- 102000018358 immunoglobulin Human genes 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 239000002953 phosphate buffered saline Substances 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- WHMDPDGBKYUEMW-UHFFFAOYSA-N pyridine-2-thiol Chemical compound SC1=CC=CC=N1 WHMDPDGBKYUEMW-UHFFFAOYSA-N 0.000 description 4
- 239000001488 sodium phosphate Substances 0.000 description 4
- 229910000162 sodium phosphate Inorganic materials 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 229910052717 sulfur Inorganic materials 0.000 description 4
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 230000001988 toxicity Effects 0.000 description 4
- 231100000419 toxicity Toxicity 0.000 description 4
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 4
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 3
- 238000002965 ELISA Methods 0.000 description 3
- 229930126263 Maytansine Natural products 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 3
- 229930012538 Paclitaxel Natural products 0.000 description 3
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 3
- 108020004511 Recombinant DNA Proteins 0.000 description 3
- 241000187747 Streptomyces Species 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- 239000000611 antibody drug conjugate Substances 0.000 description 3
- 229940049595 antibody-drug conjugate Drugs 0.000 description 3
- 230000030833 cell death Effects 0.000 description 3
- 125000003636 chemical group Chemical group 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 125000000392 cycloalkenyl group Chemical group 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 229960003668 docetaxel Drugs 0.000 description 3
- 239000013628 high molecular weight specie Substances 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 229960001592 paclitaxel Drugs 0.000 description 3
- 239000001103 potassium chloride Substances 0.000 description 3
- 235000011164 potassium chloride Nutrition 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 238000003127 radioimmunoassay Methods 0.000 description 3
- 239000000376 reactant Substances 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000003998 size exclusion chromatography high performance liquid chromatography Methods 0.000 description 3
- 239000012064 sodium phosphate buffer Substances 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 125000004434 sulfur atom Chemical group 0.000 description 3
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 3
- NKUZQMZWTZAPSN-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-bromoacetate Chemical compound BrCC(=O)ON1C(=O)CCC1=O NKUZQMZWTZAPSN-UHFFFAOYSA-N 0.000 description 2
- VRDGQQTWSGDXCU-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-iodoacetate Chemical compound ICC(=O)ON1C(=O)CCC1=O VRDGQQTWSGDXCU-UHFFFAOYSA-N 0.000 description 2
- LLXVXPPXELIDGQ-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(2,5-dioxopyrrol-1-yl)benzoate Chemical compound C=1C=CC(N2C(C=CC2=O)=O)=CC=1C(=O)ON1C(=O)CCC1=O LLXVXPPXELIDGQ-UHFFFAOYSA-N 0.000 description 2
- JWDFQMWEFLOOED-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(pyridin-2-yldisulfanyl)propanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCSSC1=CC=CC=N1 JWDFQMWEFLOOED-UHFFFAOYSA-N 0.000 description 2
- WGMMKWFUXPMTRW-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-[(2-bromoacetyl)amino]propanoate Chemical compound BrCC(=O)NCCC(=O)ON1C(=O)CCC1=O WGMMKWFUXPMTRW-UHFFFAOYSA-N 0.000 description 2
- PVGATNRYUYNBHO-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-(2,5-dioxopyrrol-1-yl)butanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCN1C(=O)C=CC1=O PVGATNRYUYNBHO-UHFFFAOYSA-N 0.000 description 2
- PMJWDPGOWBRILU-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-[4-(2,5-dioxopyrrol-1-yl)phenyl]butanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCC(C=C1)=CC=C1N1C(=O)C=CC1=O PMJWDPGOWBRILU-UHFFFAOYSA-N 0.000 description 2
- WCMOHMXWOOBVMZ-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-[3-(2,5-dioxopyrrol-1-yl)propanoylamino]hexanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCCCNC(=O)CCN1C(=O)C=CC1=O WCMOHMXWOOBVMZ-UHFFFAOYSA-N 0.000 description 2
- DLKUYSQUHXBYPB-NSSHGSRYSA-N (2s,4r)-4-[[2-[(1r,3r)-1-acetyloxy-4-methyl-3-[3-methylbutanoyloxymethyl-[(2s,3s)-3-methyl-2-[[(2r)-1-methylpiperidine-2-carbonyl]amino]pentanoyl]amino]pentyl]-1,3-thiazole-4-carbonyl]amino]-2-methyl-5-(4-methylphenyl)pentanoic acid Chemical compound N([C@@H]([C@@H](C)CC)C(=O)N(COC(=O)CC(C)C)[C@H](C[C@@H](OC(C)=O)C=1SC=C(N=1)C(=O)N[C@H](C[C@H](C)C(O)=O)CC=1C=CC(C)=CC=1)C(C)C)C(=O)[C@H]1CCCCN1C DLKUYSQUHXBYPB-NSSHGSRYSA-N 0.000 description 2
- KTLUMOZOWZSJFC-UHFFFAOYSA-N 3-azatetracyclo[7.5.0.02,6.012,14]tetradeca-1,3,5,7,9,11,13-heptaene Chemical compound C1=C2C=CN=C2C2=C3C=C3C=CC2=C1 KTLUMOZOWZSJFC-UHFFFAOYSA-N 0.000 description 2
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 2
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 description 2
- 102000000844 Cell Surface Receptors Human genes 0.000 description 2
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 2
- 230000004568 DNA-binding Effects 0.000 description 2
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 2
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 2
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- 108090001090 Lectins Proteins 0.000 description 2
- 102000004856 Lectins Human genes 0.000 description 2
- 102000008072 Lymphokines Human genes 0.000 description 2
- 108010074338 Lymphokines Proteins 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- 206010035226 Plasma cell myeloma Diseases 0.000 description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 2
- 206010060862 Prostate cancer Diseases 0.000 description 2
- 102000005157 Somatostatin Human genes 0.000 description 2
- 108010056088 Somatostatin Proteins 0.000 description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 description 2
- 230000002152 alkylating effect Effects 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 230000003013 cytotoxicity Effects 0.000 description 2
- 231100000135 cytotoxicity Toxicity 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 230000017858 demethylation Effects 0.000 description 2
- 238000010520 demethylation reaction Methods 0.000 description 2
- 229930188854 dolastatin Natural products 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- VQNATVDKACXKTF-XELLLNAOSA-N duocarmycin Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C([C@@]64C[C@@H]6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-XELLLNAOSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 229910052587 fluorapatite Inorganic materials 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 125000005179 haloacetyl group Chemical group 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 2
- 229940072221 immunoglobulins Drugs 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 239000002523 lectin Substances 0.000 description 2
- 239000013627 low molecular weight specie Substances 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- WKPWGQKGSOKKOO-RSFHAFMBSA-N maytansine Chemical compound CO[C@@H]([C@@]1(O)C[C@](OC(=O)N1)([C@H]([C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(C)=O)CC(=O)N1C)C)[H])\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 WKPWGQKGSOKKOO-RSFHAFMBSA-N 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- 238000006011 modification reaction Methods 0.000 description 2
- 201000000050 myeloid neoplasm Diseases 0.000 description 2
- 230000000955 neuroendocrine Effects 0.000 description 2
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 238000002823 phage display Methods 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 238000001542 size-exclusion chromatography Methods 0.000 description 2
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 2
- 229960000553 somatostatin Drugs 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 2
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- 238000001262 western blot Methods 0.000 description 2
- TYKASZBHFXBROF-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-(2,5-dioxopyrrol-1-yl)acetate Chemical compound O=C1CCC(=O)N1OC(=O)CN1C(=O)C=CC1=O TYKASZBHFXBROF-UHFFFAOYSA-N 0.000 description 1
- BQWBEDSJTMWJAE-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-[(2-iodoacetyl)amino]benzoate Chemical compound C1=CC(NC(=O)CI)=CC=C1C(=O)ON1C(=O)CCC1=O BQWBEDSJTMWJAE-UHFFFAOYSA-N 0.000 description 1
- VLARLSIGSPVYHX-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-(2,5-dioxopyrrol-1-yl)hexanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCCCN1C(=O)C=CC1=O VLARLSIGSPVYHX-UHFFFAOYSA-N 0.000 description 1
- OJQSISYVGFJJBY-UHFFFAOYSA-N 1-(4-isocyanatophenyl)pyrrole-2,5-dione Chemical compound C1=CC(N=C=O)=CC=C1N1C(=O)C=CC1=O OJQSISYVGFJJBY-UHFFFAOYSA-N 0.000 description 1
- OWDQCSBZQVISPN-UHFFFAOYSA-N 2-[(2,5-dioxopyrrolidin-1-yl)amino]-4-(2-iodoacetyl)benzoic acid Chemical compound OC(=O)C1=CC=C(C(=O)CI)C=C1NN1C(=O)CCC1=O OWDQCSBZQVISPN-UHFFFAOYSA-N 0.000 description 1
- GUPXYSSGJWIURR-UHFFFAOYSA-N 3-octoxypropane-1,2-diol Chemical compound CCCCCCCCOCC(O)CO GUPXYSSGJWIURR-UHFFFAOYSA-N 0.000 description 1
- HBEDKBRARKFPIC-UHFFFAOYSA-N 6-(2,5-dioxopyrrol-1-yl)hexanoic acid;1-hydroxypyrrolidine-2,5-dione Chemical compound ON1C(=O)CCC1=O.OC(=O)CCCCCN1C(=O)C=CC1=O HBEDKBRARKFPIC-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- 241000186046 Actinomyces Species 0.000 description 1
- 108010083359 Antigen Receptors Proteins 0.000 description 1
- 102000006306 Antigen Receptors Human genes 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 239000012619 Butyl Sepharose® Substances 0.000 description 1
- 0 C[C@@](C(OC(CC(N(C)c(cc(CC(C)=CC=CC(C(C1)(N2)O)OC)cc3OC)c3Cl)=O)C3(C)OC3C(C)C1OC2=O)=O)N(C)C(*)=O Chemical compound C[C@@](C(OC(CC(N(C)c(cc(CC(C)=CC=CC(C(C1)(N2)O)OC)cc3OC)c3Cl)=O)C3(C)OC3C(C)C1OC2=O)=O)N(C)C(*)=O 0.000 description 1
- 241000251730 Chondrichthyes Species 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 1
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 101100421450 Drosophila melanogaster Shark gene Proteins 0.000 description 1
- 101150029707 ERBB2 gene Proteins 0.000 description 1
- 101710181478 Envelope glycoprotein GP350 Proteins 0.000 description 1
- 102400001368 Epidermal growth factor Human genes 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 1
- 208000009329 Graft vs Host Disease Diseases 0.000 description 1
- 102000004457 Granulocyte-Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 241001622557 Hesperia Species 0.000 description 1
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 1
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 1
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 description 1
- 101001099381 Homo sapiens Peroxisomal biogenesis factor 19 Proteins 0.000 description 1
- 102000009786 Immunoglobulin Constant Regions Human genes 0.000 description 1
- 108010009817 Immunoglobulin Constant Regions Proteins 0.000 description 1
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 1
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 1
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 102000003996 Interferon-beta Human genes 0.000 description 1
- 108090000467 Interferon-beta Proteins 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102000007651 Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 240000001427 Mallotus nudiflorus Species 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 102000029749 Microtubule Human genes 0.000 description 1
- 108091022875 Microtubule Proteins 0.000 description 1
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 1
- 108010069196 Neural Cell Adhesion Molecules Proteins 0.000 description 1
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 description 1
- 102100023616 Neural cell adhesion molecule L1-like protein Human genes 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- 241000187654 Nocardia Species 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- 241000047703 Nonion Species 0.000 description 1
- 101710160107 Outer membrane protein A Proteins 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 102000057297 Pepsin A Human genes 0.000 description 1
- 108090000284 Pepsin A Proteins 0.000 description 1
- 102100038883 Peroxisomal biogenesis factor 19 Human genes 0.000 description 1
- 231100000742 Plant toxin Toxicity 0.000 description 1
- 208000009052 Precursor T-Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 239000012614 Q-Sepharose Substances 0.000 description 1
- JQYMGXZJTCOARG-UHFFFAOYSA-N Reactive blue 2 Chemical compound C1=2C(=O)C3=CC=CC=C3C(=O)C=2C(N)=C(S(O)(=O)=O)C=C1NC(C=C1S(O)(=O)=O)=CC=C1NC(N=1)=NC(Cl)=NC=1NC1=CC=CC(S(O)(=O)=O)=C1 JQYMGXZJTCOARG-UHFFFAOYSA-N 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- 108010029180 Sialic Acid Binding Ig-like Lectin 3 Proteins 0.000 description 1
- 102000001555 Sialic Acid Binding Ig-like Lectin 3 Human genes 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 102000004584 Somatomedin Receptors Human genes 0.000 description 1
- 108010017622 Somatomedin Receptors Proteins 0.000 description 1
- 231100000632 Spindle poison Toxicity 0.000 description 1
- 241000133426 Streptomyces zelensis Species 0.000 description 1
- 208000029052 T-cell acute lymphoblastic leukemia Diseases 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 102400001320 Transforming growth factor alpha Human genes 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- 235000019897 UltracelTM Nutrition 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 201000011186 acute T cell leukemia Diseases 0.000 description 1
- 150000001263 acyl chlorides Chemical class 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 125000005042 acyloxymethyl group Chemical group 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 150000001413 amino acids Chemical group 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 230000005875 antibody response Effects 0.000 description 1
- 239000003972 antineoplastic antibiotic Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 239000013602 bacteriophage vector Substances 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 238000010504 bond cleavage reaction Methods 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 229930195731 calicheamicin Natural products 0.000 description 1
- HXCHCVDVKSCDHU-LULTVBGHSA-N calicheamicin Chemical compound C1[C@H](OC)[C@@H](NCC)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@](C/3=C/CSSSC)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HXCHCVDVKSCDHU-LULTVBGHSA-N 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 230000022534 cell killing Effects 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 229940044683 chemotherapy drug Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- 238000011210 chromatographic step Methods 0.000 description 1
- 229940047120 colony stimulating factors Drugs 0.000 description 1
- 230000006957 competitive inhibition Effects 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- UFULAYFCSOUIOV-UHFFFAOYSA-N cysteamine Chemical compound NCCS UFULAYFCSOUIOV-UHFFFAOYSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 238000006298 dechlorination reaction Methods 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 125000005414 dithiopyridyl group Chemical group 0.000 description 1
- 229960005501 duocarmycin Drugs 0.000 description 1
- 229930184221 duocarmycin Natural products 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 210000001163 endosome Anatomy 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 229940044627 gamma-interferon Drugs 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 208000024908 graft versus host disease Diseases 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 238000013415 human tumor xenograft model Methods 0.000 description 1
- 210000004754 hybrid cell Anatomy 0.000 description 1
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 238000001114 immunoprecipitation Methods 0.000 description 1
- 229940051026 immunotoxin Drugs 0.000 description 1
- 239000002596 immunotoxin Substances 0.000 description 1
- 231100000608 immunotoxin Toxicity 0.000 description 1
- 230000002637 immunotoxin Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000009830 intercalation Methods 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 108010034897 lentil lectin Proteins 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000037841 lung tumor Diseases 0.000 description 1
- 210000003712 lysosome Anatomy 0.000 description 1
- 230000001868 lysosomic effect Effects 0.000 description 1
- 239000012516 mab select resin Substances 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- SVVGCFZPFZGWRG-OTKBOCOUSA-N maytansinoid dm4 Chemical compound CO[C@@H]([C@@]1(O)C[C@H](OC(=O)N1)C(C)(C)[C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(=O)CCC(C)(C)S)CC(=O)N1C)\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 SVVGCFZPFZGWRG-OTKBOCOUSA-N 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 229960003151 mercaptamine Drugs 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 230000000394 mitotic effect Effects 0.000 description 1
- 108091005573 modified proteins Proteins 0.000 description 1
- 102000035118 modified proteins Human genes 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 210000000066 myeloid cell Anatomy 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 150000007523 nucleic acids Chemical group 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 239000003123 plant toxin Substances 0.000 description 1
- 229920000889 poly(m-phenylene isophthalamide) Polymers 0.000 description 1
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 230000006337 proteolytic cleavage Effects 0.000 description 1
- ZCOSUVZGFDGWFV-UHFFFAOYSA-N pyrrolo[2,3-e]indole Chemical compound C1=CC2=NC=CC2=C2N=CC=C21 ZCOSUVZGFDGWFV-UHFFFAOYSA-N 0.000 description 1
- 229940051022 radioimmunoconjugate Drugs 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 239000004627 regenerated cellulose Substances 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 231100000057 systemic toxicity Toxicity 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- RCINICONZNJXQF-XAZOAEDWSA-N taxol® Chemical compound O([C@@H]1[C@@]2(CC(C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3(C21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-XAZOAEDWSA-N 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- YONPGGFAJWQGJC-UHFFFAOYSA-K titanium(iii) chloride Chemical compound Cl[Ti](Cl)Cl YONPGGFAJWQGJC-UHFFFAOYSA-K 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229930184737 tubulysin Natural products 0.000 description 1
- 238000013060 ultrafiltration and diafiltration Methods 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/20—Interleukins [IL]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6867—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from a cell of a blood cancer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2896—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against molecules with a "CD"-designation, not provided for elsewhere
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6807—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug or compound being a sugar, nucleoside, nucleotide, nucleic acid, e.g. RNA antisense
- A61K47/6809—Antibiotics, e.g. antitumor antibiotics anthracyclins, adriamycin, doxorubicin or daunomycin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2839—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the integrin superfamily
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2884—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against CD44
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/40—Immunoglobulins specific features characterized by post-translational modification
Definitions
- This invention pertains to a process for preparing conjugates of substantially high purity and stability, wherein the conjugates comprise a cell-binding agent chemically coupled to a drug.
- Such compounds are typically referred to as toxin, radionuclide, and drag "conjugates," respectively. Often they also are referred to as immunoconjugates, radioimmunoconjugates, and immunotoxins.
- Tumor cell killing occurs upon binding of the drag conjugate to a tumor cell and release or/and activation of the cytotoxic activity of the drug.
- the selectivity afforded by drug conjugates minimizes toxicity to normal cells, thereby enhancing tolerability of the drag in the patient.
- Patents 5,208,020 and 5,416,064 further describe conjugation of a modified antibody with an excess of a sulfhydryl-containing cytotoxic agent at pH 7, followed by purification on SephadexTM G25 chromatography columns. Purification of antibody-drug conjugates by size exclusion chromatography (SEC) also has been described (see, e.g., Liu et al., Proc. Natl. Acad. Set (USA), 93: 8618-8623 (1996), and Chari et al., Cancer Research, 52: 127-131 (1992)).
- SEC size exclusion chromatography
- the invention provides a process for preparing a conjugate of substantially high purity and stability comprising a cell-binding agent chemically coupled to a drag.
- the process comprises (a) contacting a cell-binding agent with a bifunctional crosslinking reagent to covalently attach a linker to the cell-binding agent and thereby prepare a first mixture comprising cell-binding agents having linkers bound thereto, (b) subjecting the first mixture to tangential flow filtration, adsorptive chromatography, adsorptive filtration, selective precipitation, or combination thereof, and thereby prepare a purified first mixture of cell- binding agents having linkers bound thereto, (c) conjugating a drug to the cell-binding agents having linkers bound thereto in the purified first mixture by reacting the cell-binding agents having linkers bound thereto with a drug in a solution having a pH of about 4 to about 9 to prepare a second mixture comprising (i) cell-binding agent chemically coupled through the linker to the drug, (ii
- the invention provides a process for preparing cell-binding agent-drug conjugates of substantially high purity and stability.
- Such compositions can be used for treating diseases because of the high purity and stability of the conjugates.
- Compositions comprising a cell- binding agent, such as an antibody, chemically coupled to a drug, such as a maytansinoid, are described in, for example, U.S. Patent Application Publication No. 2004/0241174 Al.
- substantially high purity is considered to be: (a) greater than 90%, preferably greater than 95%, of conjugate species are monomeric, and/or (b) free drug level in the conjugate preparation is less than 2% (relative to total drug).
- the inventive process comprises (a) modifying the cell-binding agent with a bifunctional crosslinking reagent to covalently attach a linker to the cell-binding agent and thereby prepare a first mixture comprising cell-binding agents having linkers bound thereto, (b) subjecting the first mixture to tangential flow filtration, adsorptive chromatography, adsorptive filtration, selective precipitation, or combinations thereof, to purify the cell-binding agents having linkers bound thereto from other components of the first mixture and thereby prepare a purified first mixture of cell-binding agents having linkers bound thereto, (c) conjugating a drug to the cell-binding agents having linkers bound thereto in the purified first mixture by reacting the cell-binding agents having linkers bound thereto with the drug in a solution having a pH of about 4 to about 9 to prepare a second mixture comprising (i) cell-binding agent chemically coupled through the linker to the drug, (ii) free drug, and (iii)
- tangential flow filtration also known as cross flow filtration, ultrafiltration and diafiltration
- adsorptive chromatography resins are utilized in the purification steps.
- TFF tangential flow filtration
- step c tangential flow filtration
- step d adsorptive chromatography resin
- the adsorptive chromatography resin is a non-ion exchange resin.
- TFF is utilized in both purification steps, or adsorptive chromatography resins are utilized in both purification steps.
- an adsorptive chromatography resin is utilized in the first purification step, and TFF is utilized in the second purification step.
- TFF is utilized in the first and/or second purification step as well.
- Any suitable TFF systems may be utilized, including a Pellicon type system (Millipore, Billerica, MA), a Sartocon Cassette system (Sartorius AG, Edgewood, NY), and a Centrasette type system (Pall Corp., East Hills, NY).
- any suitable adsorptive chromatography resin may be utilized.
- Preferred adsorptive chromatography resins include resins for hydroxyapatite chromatography, hydrophobic charge induction chromatography (HCIC), hydrophobic interaction chromatography (HIC), ion exchange chromatography, mixed mode ion exchange chromatography, immobilized metal affinity chromatography (IMAC), dye ligand chromatography, affinity chromatography, reversed phase chromatography, and combinations thereof.
- Suitable hydroxyapatite resins include ceramic hydroxyapatite (CHT Type I and Type II, Bio-Rad Laboratories, Hercules, CA), HA Ultrogel hydroxyapatite (Pall Corp., East Hills, NY), and ceramic fluoroapatite (CFT Type I and Type II, Bio-Rad Laboratories, Hercules, CA).
- CHT Type I and Type II Ceramic hydroxyapatite
- HA Ultrogel hydroxyapatite Pall Corp., East Hills, NY
- CFT Type I and Type II Bio-Rad Laboratories, Hercules, CA
- An example of a suitable HCIC resin is MEP Hypercel resin (Pall Corp., East Hills, NY).
- HIC resins examples include Butyl-Sepharose, Hexyl-Sepaharose, Phenyl-Sepharose, and Octyl Sepharose resins (all from GE Healthcare, Piscataway, NJ), as well as Macro-prep Methyl and Macro-Prep t-Butyl resins (Biorad Laboratories, Hercules, CA).
- suitable ion exchange resins include SP- Sepharose, CM-Sepharose, and Q-Sepharose resins (all from GE Healthcare, Piscataway, NJ), and Unosphere S resin (Bio-Rad Laboratories, Hercules, CA).
- suitable mixed mode ion exchangers include Bakerbond ABx resin (JT Baker, Phillipsburg NJ).
- suitable IMAC resins include Chelating Sepharose resin (GE Healthcare, Piscataway, NJ) and Profinity IMAC resin (Bio-Rad Laboratories, Hercules, CA).
- suitable dye ligand resins include Blue Sepharose resin (GE Healthcare, Piscataway, NJ) and Affi-gel Blue resin (Bio-Rad Laboratories, Hercules, CA).
- suitable affinity resins include Protein A Sepharose resin (e.g., MabSelect, GE Healthcare, Piscataway, NJ), where the cell binding agent is an antibody, and lectin affinity resins, e.g.
- Lentil Lectin Sepharose resin (GE Healthcare, Piscataway, NJ), where the cell binding agent bears appropriate lectin binding sites.
- an antibody specific to the cell binding agent may be used.
- Such an antibody can be immobilized to, for instance, Sepharose 4 Fast Flow resin (GE Healthcare, Piscataway, NJ).
- suitable reversed phase resins include C4, C8, and Cl 8 resins (Grace Vydac, Hesperia, CA).
- a first mixture is produced comprising the cell-binding agent having linkers bound thereto, as well as reactants and other byproducts.
- Purification of the modified cell-binding agent from reactants and by-products is carried out by subjecting the first mixture to a purification process.
- the first mixture can be purified using tangential flow filtration (TFF), e.g., a membrane-based tangential flow filtration process, adsorptive chromatography, adsorptive filtration, or selective precipitation, or any other suitable purification process, as well as combinations thereof.
- TMF tangential flow filtration
- This first purification step provides a purified first mixture, i.e., an increased concentration of the cell-binding agents having linkers bound thereto and a decreased amount of unbound bifunctional crosslinking reagent, as compared to the first mixture prior to purification in accordance with the invention.
- a drug is conjugated to the cell-binding agents having linkers bound thereto in the first purified mixture by reacting the cell-binding agents having linkers bound thereto with a drug in a solution having a pH from about 4 to about 9, whereupon a second mixture comprising (i) the cell-binding agent chemically coupled through the linker to the drug, (ii) free drug, and (iii) reaction by-products is produced.
- the conjugation reaction is performed at a pH of about 4 to about pH 9
- the reaction is preferably performed at a pH of about 6 or below or at a pH of about 6.5 or greater, most preferably at a pH of about 4 to about 6 or at a pH of about 6.5 to about 9, and especially at a pH of 4 to less than 6 or at a pH of greater than 6.5 to 9.
- the conjugation step is performed at a pH of about 6.5 or greater, some sulhydryl-containing drugs may be prone to dimerize by disulfide-bond formation. Removal of trace metals and/or oxygen from the reaction mixture, as well as optional addition of antioxidants or the use of linkers with more reactive leaving groups, or addition of drug in more than one aliquot, may be required to allow for efficient reaction in such a situation.
- purification of the modified cell binding agent may be omitted.
- the drug may be added simultaneously with the crosslinking reagent or at some later point, e.g., 1, 2, 3, or more hours after addition of the crosslinking reagent to the cell binding agent.
- the inventive method may optionally include the addition of sucrose to the conjugation step used in the inventive method to increase solubility and recovery of the cell- binding agent-drug conjugates.
- sucrose is added at a concentration of about 0.1% (w/v) to about 20% (w/v) (e.g., about 0.1% (w/v), 1% (w/v), 5% (w/v), 10% (w/v), 15% (w/v), or 20% (w/v)).
- sucrose is added at a concentration of about 1% (w/v) to 10% (w/v) (e.g., about 2% (w/v), about 4% (w/v), about 6% (w/v), or about 8% (w/v)).
- the conjugation reaction also can comprise the addition of a buffering agent.
- a buffering agent Any suitable buffering agent known in the art can be used. Suitable buffering agents include, for example, a citrate buffer, an acetate buffer, a succinate buffer, and a phosphate buffer.
- the second mixture is subjected to a purification step.
- the second mixture can be purified using tangential flow filtration (TFF), e.g., a membrane-based tangential flow filtration process, adsorptive chromatography, absorptive filtration, selective precipitation, or any other suitable purification process, as well as combinations thereof, which are set-forth herein.
- TMF tangential flow filtration
- This second purification step provides a purified second mixture, i.e., an increased concentration of the cell-binding agents chemically coupled through the linkers to the drug and a decreased amount of one or more other components of the second mixture, as compared to the second mixture prior to purification in accordance with the invention.
- the cell-binding agent can be any suitable agent that binds to a cell, typically and preferably an animal cell (e.g., a human cell).
- the cell-binding agent preferably is a peptide or a polypeptide.
- Suitable cell-binding agents include, for example, antibodies (e.g., monoclonal antibodies and fragments thereof), lymphokines, hormones, growth factors, nutrient-transport molecules (e.g., transferrin), and any other agent or molecule that specifically binds a target molecule on the surface of a cell.
- antibody refers to any immunoglobulin, any immunoglobulin fragment, such as Fab, F(ab') 2 , dsFv, sFv, diabodies, and triabodies, or immunoglobulin chimera, which can bind to an antigen on the surface of a cell (e.g., which contains a complementarity determining region (CDR)).
- CDR complementarity determining region
- Any suitable antibody can be used as the cell-binding agent.
- One of ordinary skill in the art will appreciate that the selection of an appropriate antibody will depend upon the cell population to be targeted.
- cell surface molecules i.e., antigens
- a particular cell population typically and preferably a diseased cell population
- Cell surface expression profiles are known for a wide variety of cell types, including tumor cell types, or, if unknown, can be determined using routine molecular biology and histochemistry techniques.
- the antibody can be polyclonal or monoclonal, but is most preferably a monoclonal antibody.
- polyclonal antibodies refer to heterogeneous populations of antibody molecules, typically contained in the sera of immunized animals.
- Monoclonal antibodies refer to homogenous populations of antibody molecules that are specific to a particular antigen.
- Monoclonal antibodies are typically produced by a single clone of B lymphocytes ("B cells").
- B cells B cells
- Monoclonal antibodies may be obtained using a variety of techniques known to those skilled in the art, including standard hybridoma technology (see, e.g., K ⁇ hler and Milstein, Eur. J.
- the hybridoma method of producing monoclonal antibodies typically involves injecting any suitable animal, typically and preferably a mouse, with an antigen (i.e., an "immunogen"). The animal is subsequently sacrificed, and B cells isolated from its spleen are fused with human myeloma cells.
- an antigen i.e., an "immunogen”
- hybrid cell is produced (i.e., a "hybridoma"), which proliferates indefinitely and continuously secretes high titers of an antibody with the desired specificity in vitro.
- Any appropriate method known in the art can be used to identify hybridoma cells that produce an , antibody with the desired specificity. Such methods include, for example, enzyme-linked immunosorbent assay (ELISA), Western blot analysis, and radioimmunoassay.
- ELISA enzyme-linked immunosorbent assay
- the population of hybridomas is screened to isolate individual clones, each of which secretes a single antibody species to the antigen.
- each hybridoma is a clone derived from fusion with a single B cell, all the antibody molecules it produces are identical in structure, including their antigen binding site and isotype.
- Monoclonal antibodies also may be generated using other suitable techniques including EBV-hybridoma technology (see, e.g., Haskard and Archer, J Immunol.
- the monoclonal antibody can be isolated from or produced in any suitable animal, but is preferably produced in a mammal, more preferably a mouse or human, and most preferably a human. Methods for producing an antibody in mice are well known to those skilled in the art and are described herein. With respect to human antibodies, one of ordinary skill in the art will appreciate that polyclonal antibodies can be isolated from the sera of human subjects vaccinated or immunized with an appropriate antigen. Alternatively, human antibodies can be generated by adapting known techniques for producing human antibodies in non-human animals such as mice (see, e.g., U.S. Patents 5,545,806, 5,569,825, and 5,714,352, and U.S. Patent Application Publication No.
- human antibodies While being the ideal choice for therapeutic applications in humans, human antibodies, particularly human monoclonal antibodies, typically are more difficult to generate than mouse monoclonal antibodies. Mouse monoclonal antibodies, however, induce a rapid host antibody response when administered to humans, which can reduce the therapeutic or diagnostic potential of the antibody-drug conjugate. To circumvent these complications, a monoclonal antibody preferably is not recognized as "foreign" by the human immune system.
- phage display can be used to generate the antibody.
- phage libraries encoding antigen-binding variable (V) domains of antibodies can be generated using standard molecular biology and recombinant DNA techniques (see, e.g., Sambrook et al. (eds.), Molecular Cloning, A Laboratory Manual, 3 rd Edition, Cold Spring Harbor Laboratory Press, New York (2001)). Phage encoding a variable region with the desired specificity are selected for specific binding to the desired antigen, and a complete human antibody is reconstituted comprising the selected variable domain.
- Nucleic acid sequences encoding the reconstituted antibody are introduced into a suitable cell line, such as a myeloma cell used for hybridoma production, such that human antibodies having the characteristics of monoclonal antibodies are secreted by the cell (see, e.g., Janeway et al., supra, Huse et al., supra, and U.S. Patent 6,265,150).
- a suitable cell line such as a myeloma cell used for hybridoma production
- monoclonal antibodies can be generated from mice that are transgenic for specific human heavy and light chain immunoglobulin genes.
- Such methods are known in the art and described in, for example, U.S. Patents 5,545,806 and 5,569,825, and Janeway et al., supra.
- the antibody is a humanized antibody.
- a "humanized” antibody is one in which the complementarity-determining regions (CDR) of a mouse monoclonal antibody, which form the antigen binding loops of the antibody, are grafted onto the framework of a human antibody molecule. Owing to the similarity of the frameworks of mouse and human antibodies, it is generally accepted in the art that this approach produces a monoclonal antibody that is antigenically identical to a human antibody but binds the same antigen as the mouse monoclonal antibody from which the CDR sequences were derived. Methods for generating humanized antibodies are well known in the art and are described in detail in, for example, Janeway et al., supra, U.S.
- Humanized antibodies can also be generated using the antibody resurfacing technology described in U.S. Patent 5,639,641 and Pedersen et al., J MoI Biol, 235: 959- 973 (1994). While the antibody employed in the conjugate of the inventive composition most preferably is a humanized monoclonal antibody, a human monoclonal antibody and a mouse monoclonal antibody, as described above, are also within the scope of the invention.
- Antibody fragments that have at least one antigen binding site, and thus recognize and bind to at least one antigen or receptor present on the surface of a target cell also are within the scope of the invention.
- proteolytic cleavage of an intact antibody molecule can produce a variety of antibody fragments that retain the ability to recognize and bind antigens.
- limited digestion of an antibody molecule with the protease papain typically produces three fragments, two of which are identical and are referred to as the Fab fragments, as they retain the antigen binding activity of the parent antibody molecule.
- F(ab') 2 fragment Cleavage of an antibody molecule with the enzyme pepsin normally produces two antibody fragments, one of which retains both antigen-binding arms of the antibody molecule, and is thus referred to as the F(ab') 2 fragment.
- Reduction of a F(ab') 2 fragment with dithiothreitol or mercaptoethylamine produces a fragment referred to as a Fab' fragment.
- a single-chain variable region fragment (sFv) antibody fragment which consists of a truncated Fab fragment comprising the variable (V) domain of an antibody heavy chain linked to a V domain of a light antibody chain via a synthetic peptide, can be generated using routine recombinant DNA technology techniques (see, e.g., Janeway et al., supra).
- disulfide-stabilized variable region fragments (dsFv) can be prepared by recombinant DNA technology (see, e.g., Reiter et al., Protein Engineering, 7: 697-704 (1994)).
- Antibody fragments in the context of the invention are not limited to these exemplary types of antibody fragments.
- Antibody fragments that recognizes and binds to a desired cell surface receptor or antigen can be employed.
- Antibody fragments are further described in, for example, Parham, J Immunol, 131: 2895-2902 (1983), Spring et al, J Immunol, 113: 470-478 (1974), and Nisonoff et A., Arch. Biochem. Biophys., 89: 230-244 (1960).
- Antibody-antigen binding can be assayed using any suitable method known in the art, such as, for example, radioimmunoassay (RIA), ELISA, Western blot, immunoprecipitation, and competitive inhibition assays (see, e.g., Janeway et al., supra, and U.S. Patent Application Publication No. 2002/0197266 Al).
- the antibody can be a chimeric antibody or an antigen binding fragment thereof.
- chimeric it is meant that the antibody comprises at least two immunoglobulins, or fragments thereof, obtained or derived from at least two different species (e.g., two different immunoglobulins, such as a human immunoglobulin constant region combined with a murine immunoglobulin variable region).
- the antibody also can be a domain antibody (dAb) or an antigen binding fragment thereof, such as, for example, a camelid antibody (see, e.g., Desmyter et al., Nature Struct.
- the monoclonal antibody J5 is a murine IgG2a antibody that is specific for Common Acute Lymphoblastic Leukemia Antigen (CALLA) (Rite et al., Nature, 283: 583-585 (1980)), and can be used to target cells that express CALLA (e.g., acute lymphoblastic leukemia cells).
- CALLA Common Acute Lymphoblastic Leukemia Antigen
- the monoclonal antibody MY9 is a murine IgGl antibody that binds specifically to the CD33 antigen (Griffin et al., Leukemia Res., 8: 521 (1984)), and can be used to target cells that express CD33 (e.g., acute myelogenous leukemia (AML) cells).
- AML acute myelogenous leukemia
- the monoclonal antibody anti-B4 (also referred to as B4) is a murine IgGl antibody that binds to the CD19 antigen on B cells (Nadler et al., J. Immunol., 131: 244-250 (1983)), and can be used to target B cells or diseased cells that express CD19 (e.g., non-Hodgkin's lymphoma cells and chronic lymphoblastic leukemia cells).
- N901 is a murine monoclonal antibody that binds to the CD56 (neural cell adhesion molecule) antigen found on cells of neuroendocrine origin, including small cell lung tumor, which can be used in the conjugate to target drugs to cells of neuroendocrine origin.
- the J5, MY9, and B4 antibodies preferably are resurfaced or humanized prior to their use as part of the conjugate. Resurfacing or humanization of antibodies is described in, for example, Roguska et al., Proc. Natl. Acad. Sci. USA, 91: 969-73 (1994).
- the monoclonal antibody C242 binds to the CanAg antigen (see, e.g., U.S. Patent 5,552,293), and can be used to target the conjugate to CanAg expressing tumors, such as colorectal, pancreatic, non-small cell lung, and gastric cancers.
- HuC242 is a humanized form of the monoclonal antibody C242 (see, e.g., U.S. Patent 5,552,293).
- the hybridoma from which HuC242 is produced is deposited with ECACC identification Number 90012601.
- HuC242 can be prepared using CDR-grafting methodology (see, e.g., U.S.
- HuC242 can be used to target the conjugate to tumor cells expressing the CanAg antigen, such as, for example, colorectal, pancreatic, non-small cell lung, and gastric cancer cells.
- an anti-MUCl antibody can be used as the cell-binding agent in the conjugate.
- Anti-MUCl antibodies include, for example, anti-HMFG-2 (see, e.g., Taylor-Papadimitriou et al., Int. J. Cancer, 28: 17-21 (1981)), hCTMOl (see, e.g., van Hof et al., Cancer Res., 56: 5179-5185 (1996)), and DS6.
- Prostate cancer cells also can be targeted with the conjugate by using an anti-prostate-specific membrane antigen (PSMA) as the cell-binding agent, such as J591 (see, e.g., Liu et al., Cancer Res., 57: 3629-3634 (1997)).
- PSMA anti-prostate-specific membrane antigen
- cancer cells that express the Her2 antigen such as breast, prostate, and ovarian cancers, can be targeted using the antibody trastuzumab.
- Anti-IGF-IR antibodies that bind to insulin-like growth factor receptor also can be used in the conjugate.
- Particularly preferred antibodies are humanized monoclonal antibodies, examples of which include huN901, huMy9-6, huB4, huC242, trastuzumab, bivatuzumab, sibrotuzumab, and rituximab (see, e.g., U.S. Patents 5,639,641 and 5,665,357, U.S. Provisional Patent Application No. 60/424,332 (which is related to U.S. Patent Application Publication No.
- the antibody is the huN901 humanized monoclonal antibody or the huMy9-6 humanized monoclonal antibody.
- Other preferred antibodies include CNTO95, huDS6, huB4, and huC242.
- Other humanized monoclonal antibodies are known in the art and can be used in connection with the invention.
- the cell-binding agent preferably is an antibody
- the cell-binding agent also can be a non-antibody molecule.
- suitable non-antibody molecules include, for example, interferons (e.g., alpha, beta, or gamma interferon), lymphokines (e.g., interleukin 2 (IL-2), IL-3, IL-4, or IL-6), hormones (e.g., insulin), growth factors (e.g., EGF, TGF-alpha, FGF, and VEGF), colony-stimulating factors (e.g., G-CSF, M-CSF, and GM-CSF (see, e.g., Burgess, Immunology Today, 5: 155-158 (1984)), somatostatin, and transferrin (see, e.g., O'Keefe et al, J Biol.
- interferons e.g., alpha, beta, or gamma interferon
- GM-CSF 3 which binds to myeloid cells, can be used as a cell-binding agent to target acute myelogenous leukemia cells.
- IL-2 which binds to activated T-cells, can be used for prevention of transplant graft rejection, for therapy and prevention of graft- versus-host disease, and for treatment of acute T-cell leukemia.
- Epidermal growth factor (EGF) can be used to target squamous cancers such as lung cancer and head and neck cancer.
- Somatostatin can be used to target neuroblastoma cells and other tumor cell types.
- the conjugate can comprise any suitable drug, typically a cytotoxic agent.
- Suitable cytotoxic agents include, for example, maytansinoids and maytansinoid analogs, taxoids, CC- 1065 and CC-1065 analogs, and dolastatin and dolastatin analogs.
- the cytotoxic agent is a maytansinoid, including maytansinol and maytansinol analogs. Maytansinoids are compounds that inhibit microtubule formation and are highly toxic to mammalian cells.
- Suitable maytansinol analogues include those having a modified aromatic ring and those having modifications at other positions. Such maytansinoids are described in, for example, U.S. Patents 4,256,746, 4,294,757, 4,307,016, 4,313,946, 4,315,929, 4,322,348, 4,331,598, 4,361,650, 4,362,663, 4,364,866, 4,424,219, 4,371,533, 4,450,254, 5,475,092, 5,585,499, 5,846,545, and 6,333,410. [0034] Examples of maytansinol analogs having a modified aromatic ring include:
- Patent 4,424,219) (prepared by the reaction of maytansinol with H 2 S or P 2 S 5 ), (2) C-14-alkoxymethyl (demethoxy/CH 2 OR) (U.S. Patent 4,331,598), (3) C-14-hydroxymethyl or acyloxymethyl (CH 2 OH or CH 2 OAc) (U.S. Patent 4,450,254) (prepared from Nocardia), (4) C-15-hydroxy/acyloxy (U.S. Patent 4,364,866) (prepared by the conversion of maytansinol by Streptomyces), (5) C-15-methoxy (U.S.
- Patents 4,313,946 and 4,315,929) isolated from Trewia nudiflora
- (6) C-18-N-demethyl U.S. Patents 4,362,663 and 4,322,348
- 4,5-deoxy U.S. Patent 4,371,533
- the conjugate utilizes the thiol- containing maytansinoid DMl, also known as N 2 -deacetyl-N 2 -(3-mercapto-l-oxopropyl)- maytansine, as the cytotoxic agent.
- DMl also known as N 2 -deacetyl-N 2 -(3-mercapto-l-oxopropyl)- maytansine
- the conjugate utilizes the thiol- containing maytansinoid DM4, also known as N -deacetyl-N ⁇ (4-methyl-4-mercapto-l- oxopentyl)-maytansine, as the cytotoxic agent.
- DM4 is represented by formula (II):
- maytansines may be used in the context of the invention, including, for example, thiol and disulfide-containing maytansinoids bearing a mono or di-alkyl substitution on the carbon atom bearing the sulfur atom.
- Particularly preferred is a maytansinoid having at the C-3 position (a) C- 14 hydroxymethyl, C- 15 hydroxy, or C-20 desmethyl functionality, and (b) an acylated amino acid side chain with an acyl group bearing a hindered sulfhydryl group, wherein the carbon atom of the acyl group bearing the thiol functionality has one or two substituents, said substituents being CH 3 , C 2 H 5 , linear or branched alkyl or alkenyl having from 1 to 10 carbon atoms, cyclic alkyl or alkenyl having from 3 to 10 carbon atoms, phenyl, substituted phenyl, or heterocyclic aromatic or heterocycloalkyl radical, and further wherein one of
- R 1 and R 2 are each independently CH 3 , C 2 H 5 , linear alkyl or alkenyl having from 1 to 10 carbon atoms, branched or cyclic alkyl or alkenyl having from 3 to 10 carbon atoms, phenyl, substituted phenyl or heterocyclic aromatic or heterocycloalkyl radical, and wherein
- R 2 also can be H, wherein A, B, D are cycloalkyl or cycloalkenyl having 3-10 carbon atoms, simple or substituted aryl, or heterocyclic aromatic, or heterocycloalkyl radical, wherein R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 11 , and R 12 are each independently H, CH 3 , C 2 H 5 , linear alkyl or alkenyl having from 1 to 10 carbon atoms, branched or cyclic alkyl or alkenyl having from 3 to 10 carbon atoms, phenyl, substituted phenyl or heterocyclic aromatic, or heterocycloalkyl radical, wherein 1, m, n, o, p, q, r, s, and t are each independently zero or an integer from 1 to 5, provided that at least two of 1, m, n, o, p, q, r, s and t are not zero at
- Preferred embodiments of formula (III) include compounds of formula (III) wherein (a) R 1 is H, R 2 is methyl and Z is H, (b) R 1 and R 2 are methyl and Z is H, (c) R 1 is H 5
- Such additional maytansines also include compounds represented by formula (IV- L) 5 (IV-D), or (IV-D 3 L):
- Y represents (CR 7 R 8 )i(CR 5 R 6 ) m (CR 3 R 4 ) n CR 1 R 2 SZ, wherein R 1 and R 2 are each independently CH 3 , C 2 H 5 , linear alkyl, or alkenyl having from 1 to 10 carbon atoms, branched or cyclic alkyl or alkenyl having from 3 to 10 carbon atoms, phenyl, substituted phenyl, or heterocyclic aromatic or heterocycloalkyl radical, and wherein
- R 2 also can be H 3 wherein R 3 , R 4 , R 5 , R 6 , R 7 , and R 8 are each independently H, CH 3 , C 2 H 5 , linear alkyl or alkenyl having from 1 to 10 carbon atoms, branched or cyclic alkyl or alkenyl having from 3 to 10 carbon atoms, phenyl, substituted phenyl, or heterocyclic aromatic or heterocycloalkyl radical, wherein 1, m, and n are each independently an integer of from 1 to 5, and in addition n can be zero, wherein Z is H, SR, or COR wherein R is linear or branched alkyl or alkenyl having from 1 to 10 carbon atoms, cyclic alkyl or alkenyl having from 3 to 10 carbon atoms, or simple or substituted aryl or heterocyclic aromatic or heterocycloalkyl radical, and wherein May represents a maytansinoid which bears the side chain at C-3, C- 14
- Preferred embodiments of formulas (IV-L), (IV-D) and (IV-D 5 L) include compounds of formulas (IV-L), (IV-D) and (IV-D 5 L) wherein (a) R 1 is H 5 R 2 is methyl, R 5 ,
- R 6 , R 7 , and R 8 are each H, 1 and m are each 1 , n is O 5 and Z is H, (b) R 1 and R 2 are methyl,
- R 5 , R 6 , R 7 , Rg are each H, 1 and m are I 5 n is O 5 and Z is H 5 (c) R 1 is H, R 2 is methyl, R 5 , R 6 ,
- R 7 , and R 8 are each H, 1 and m are each 1, n is 0, and Z is -SCH 3 , or (d) Ri and R 2 are methyl, R 5 , R 6 , R 7 , R 8 are each H 5 1 and m are I 5 n is O 5 and Z is -SCH 3 .
- cytotoxic agent is represented by formula (IV-L).
- Additional preferred maytansines also include compounds represented by formula (V):
- Y represents (CR 7 R 8 )i(CR 5 R 6 ) m (CR 3 R 4 ) n CR 1 R 2 SZ, wherein R 1 and R 2 are each independently CH 3 , C 2 H 5 , linear alkyl, or alkenyl having from 1 to 10 carbon atoms, branched or cyclic alkyl or alkenyl having from 3 to 10 carbon atoms, phenyl, substituted phenyl or heterocyclic aromatic or heterocycloalkyl radical, and wherein
- R 2 also can be H, wherein R 3 , R 4 , R 5 , R 6 , R 7 , and R 8 are each independently H, CH 3 , C 2 H 5 , linear alkyl or alkenyl having from 1 to 10 carbon atoms, branched or cyclic alkyl or alkenyl having from 3 to 10 carbon atoms, phenyl, substituted phenyl, or heterocyclic aromatic or heterocycloalkyl radical, wherein 1, m, and n are each independently an integer of from 1 to 5, and in addition n can be zero, and wherein Z is H, SR or COR, wherein R is linear alkyl or alkenyl having from 1 to 10 carbon atoms, branched or cyclic alkyl or alkenyl having from 3 to 10 carbon atoms, or simple or substituted aryl or heterocyclic aromatic or heterocycloalkyl radical.
- Preferred embodiments of formula (V) include compounds of formula (V) wherein (a) R 1 is H, R 2 is methyl, R 5 , R 6 , R 7 , and R 8 are each H; 1 and m are each 1; n is 0; and Z is H, (b) R 1 and R 2 are methyl; R 5 , R 6 , R 7 , R 8 are each H, 1 and m are 1; n is 0; and Z is H, (c) R 1 is H, R 2 is methyl, R 5 , R 6 , R 7 , and R 8 are each H 5 1 and m are each 1, n is 0, and Z is -SCH 3 , or (d) R 1 and R 2 are methyl, R 5 , R 6 , R 7 , R 8 are each H, 1 and m are 1, n is 0, and Z is -SCH 3 .
- Still further preferred maytansines include compounds represented by formula (VI-L), (VI-D), or (VI-D 3 L):
- VI-L VI-D
- VI-D VI-D, L
- Y 2 represents (CR 7 R 8 ) 1 (CR 5 R 6 ) m (CR 3 R 4 ) n CR 1 R 2 SZ 2 , wherein R 1 and R 2 are each independently CH 3 , C 2 H 5 , linear alkyl or alkenyl having from 1 to 10 carbon atoms, branched or cyclic alkyl or alkenyl having from 3 to 10 carbon atoms, phenyl, substituted phenyl or heterocyclic aromatic or heterocycloalkyl radical, and wherein
- R 2 also can be H, wherein R 3 , R 4 , R 5 , R 6 , R 7 , and R 8 are each independently H, CH 3 , C 2 H 5 , linear cyclic alkyl or alkenyl having from 1 to 10 carbon atoms, branched or cyclic alkyl or alkenyl having from
- n 3 to 10 carbon atoms, phenyl, substituted phenyl or heterocyclic aromatic or heterocycloalkyl radical, wherein 1, m, and n are each independently an integer of from 1 to 5, and in addition n can be zero, wherein Z 2 is SR or COR, wherein R is linear alkyl or alkenyl having from 1 to 10 carbon atoms, branched or cyclic alkyl or alkenyl having from 3 to 10 carbon atoms, or simple or substituted aryl or heterocyclic aromatic or heterocycloalkyl radical, and wherein May is a maytansinoid.
- Additional preferred maytansines include compounds represented by formula
- R 1 and R 2 are each independently CH 3 , C 2 H 5 , linear branched or alkyl or alkenyl having from 1 to 10 carbon atoms, cyclic alkyl or alkenyl having from 3 to 10 carbon atoms, phenyl, substituted phenyl or heterocyclic aromatic or heterocycloalkyl radical, and in addition R 2 can be H, wherein A, B, and D each independently is cycloalkyl or cycloalkenyl having 3 to 10 carbon atoms, simple or substituted aryl, or heterocyclic aromatic or heterocycloalkyl radical, wherein R 3 , R 4 , R 5 , R 6 , R 7 , Rg, R
- Preferred embodiments of formula (VII) include compounds of formula (VII), wherein R 1 is H and R 2 is methyl.
- the cytotoxic agent used in the conjugate can be a taxane or derivative thereof.
- Taxanes are a family of compounds that includes paclitaxel (Taxol®), a cytotoxic natural product, and docetaxel (Taxotere®), a semi-synthetic derivative, which are both widely used in the treatment of cancer. Taxanes are mitotic spindle poisons that inhibit the depolymerization of tubulin, resulting in cell death.
- docetaxel and paclitaxel are useful agents in the treatment of cancer, their antitumor activity is limited because of their non-specific toxicity towards normal cells. Further, compounds like paclitaxel and docetaxel themselves are not sufficiently potent to be used in conjugates of cell-binding agents.
- a preferred taxane for use in the preparation of a cytotoxic conjugate is the taxane of formula (VIII):
- the cytotoxic also can be CC- 1065 or a derivative thereof.
- CC-1065 is a potent anti-tumor antibiotic isolated from the culture broth of Streptomyces zelensis.
- CC- 1065 is about 1000-fold more potent in vitro than commonly used anti-cancer drugs, such as doxorubicin, methotrexate, and vincristine (Bhuyan et al. 5 Cancer Res., 42: 3532-3537 (1982)).
- CC-1065 and its analogs are disclosed in U.S. Patents 5,585,499, 5,846,545, 6,340,701, and 6,372,738.
- the cytotoxic potency of CC-1065 has been correlated with its alkylating activity and its DNA-binding or DNA-intercalating activity. These two activities reside in separate parts of the molecule.
- the alkylating activity is contained in the cyclopropapyrroloindole (CPI) subunit and the DNA-binding activity resides in the two pyrroloindole subunits of CC-1065.
- CPI cyclopropapyrroloindole
- CC- 1065 analogs are known in the art and also can be used as the cytotoxic agent in the conjugate (see, e.g., Warpehoski et al., J Med. Chem., 31: 590-603 (1988)).
- a series of CC-1065 analogs has been developed in which the CPI moiety is replaced by a cyclopropabenzindole (CBI) moiety (Boger et al., J Org. Chem., 55: 5823- 5833 (1990), and Boger et al., Bioorg. Med. Chem. Lett, 1: 115-120 (1991)).
- CBI cyclopropabenzindole
- These CC- 1065 analogs maintain the high in vitro potency of the parental drug, without causing delayed toxicity in mice.
- these compounds are alkylating agents that covalently bind to the minor groove of DNA to cause cell death.
- CC-1065 analogs can be greatly improved by changing the in vivo distribution through targeted delivery to a tumor site, resulting in lower toxicity to non-targeted tissues, and thus, lower systemic toxicity.
- conjugates of analogs and derivatives of CC-1065 with cell-binding agents that specifically target tumor cells have been generated (see, e.g., U.S. Patents 5,475,092, 5,585,499, and 5,846,545). These conjugates typically display high target-specific cytotoxicity in vitro, and anti-tumor activity in human tumor xenograft models in mice (see, e.g., Chari et al., Cancer Res., 55: 4079-4084 (1995)).
- Drugs such as methotrexate, daunorubicin, doxorubicin, vincristine, vinblastine, melphalan, mitomycin C, chlorambucil, calicheamicin, tubulysin and tubulysin analogs, duocarmycin and duocarmycin analogs, dolastatin and dolastatin analogs also can be used in the context of the invention.
- Doxarubicin and daunorubicin compounds can also be used as the drug.
- the drug conjugates may be prepared by in vitro methods.
- a linking group is used.
- Suitable linking groups are well known in the art and include disulfide groups, acid labile groups, photolabile groups, peptidase labile groups, and esterase labile groups.
- Preferred linking groups are disulfide groups.
- conjugates can be constructed using a disulfide exchange reaction between the antibody and the drug or prodrug.
- the drug molecules also can be linked to a cell-binding agent through an intermediary carrier molecule such as serum albumin.
- the cell-binding agent is modified by reacting a bifunctional crosslinking reagent with the cell-binding agent, thereby resulting in the covalent attachment of a linker molecule to the cell-binding agent.
- a "bifunctional crosslinking reagent” is any chemical moiety that covalently links a cell-binding agent to a drug, such as the drugs described herein.
- a portion of the linking moiety is provided by the drug.
- the drug comprises a linking moiety that is part of a larger linker molecule that is used to join the cell-binding agent to the drug.
- the side chain at the C-3 hydroxyl group of maytansine is modified to have a free sulfhydryl group (SH).
- This thiolated form of maytansine can react with a modified cell-binding agent to form a conjugate. Therefore, the final linker is assembled from two components, one of which is provided by the crosslinking reagent, while the other is provided by the side chain from DMl.
- any suitable bifunctional crosslinking reagent can be used in connection with the invention, so long as the linker reagent provides for retention of the therapeutic, e.g., cytotoxicity, and targeting characteristics of the drug and the cell-binding agent, respectively.
- the linker molecule joins the drug to the cell-binding agent through chemical bonds (as described above), such that the drug and the cell-binding agent are chemically coupled (e.g., covalently bonded) to each other.
- the linking reagent is a cleavable linker. More preferably, the linker is cleaved under mild conditions, i.e., conditions within a cell under which the activity of the drug is not affected.
- cleavable linkers examples include disulfide linkers, acid labile linkers, photolabile linkers, peptidase labile linkers, and esterase labile linkers.
- Disulfide containing linkers are linkers cleavable through disulfide exchange, which can occur under physiological conditions.
- Acid labile linkers are linkers cleavable at acid pH. For example, certain intracellular compartments, such as endosomes and lysosomes, have an acidic pH (pH 4-5), and provide conditions suitable to cleave acid labile linkers.
- Photo labile linkers are useful at the body surface and in many body cavities that are accessible to light. Furthermore, infrared light can penetrate tissue.
- Peptidase labile linkers can be used to cleave certain peptides inside or outside cells (see e.g., Trouet et al, Proc. Natl. Acad. Sci. USA, 79: 626-629 (1982), and Umemoto et al., Int. J. Cancer, 43: 677-684 (1989)).
- the drug is linked to a cell-binding agent through a disulfide bond.
- the linker molecule comprises a reactive chemical group that can react with the cell-binding agent.
- Preferred reactive chemical groups for reaction with the cell-binding agent are N- succinimidyl esters and JV-sulfosuccinimidyl esters.
- linker molecule comprises a reactive chemical group, preferably a dithiopyridyl group, that can react with the drug to form a disulfide bond.
- linker molecules include, for example, N-succinimidyl 3-(2-pyridyldithio)propionate (SPDP) (see, e.g., Carlsson et al., Biochem. J, 173: 723-737 (1978)), N-succinimidyl 4-(2-pyridyldithio)butanoate (SPDB) (see, e.g., U.S.
- SPDP N-succinimidyl 3-(2-pyridyldithio)propionate
- SPDB N-succinimidyl 4-(2-pyridyldithio)butanoate
- Patent 4,563,304 N-succinimidyl 4-(2-pyridyldithio)pentanoate (SPP) (see, e.g., CAS Registry number 341498-08-6), and other reactive cross-linkers which are described in U.S. Patent 6,913,748, which is incorporated herein in its entirety by reference.
- SPP N-succinimidyl 4-(2-pyridyldithio)pentanoate
- other reactive cross-linkers which are described in U.S. Patent 6,913,748, which is incorporated herein in its entirety by reference.
- cleavable linkers preferably are used in the inventive method, a non- cleavable linker also can be used to generate the above-described conjugate.
- a non-cleavable linker is any chemical moiety that is capable of linking a drug, such as a maytansinoid, a taxane, or a CC- 1065 analog, to a cell-binding agent in a stable, covalent manner.
- non- cleavable linkers are substantially resistant to acid-induced cleavage, light-induced cleavage, peptidase-induced cleavage, esterase-induced cleavage, and disulfide bond cleavage, at conditions under which the drug or the cell-binding agent remains active.
- Suitable crosslinking reagents that form non-cleavable linkers between a drug and the cell-binding agent are well known in the art.
- non-cleavable linkers include linkers having an N-succinimidyl ester or N-sulfosuccinimidyl ester moiety for reaction with the cell-binding agent, as well as a maleimido- or haloacetyl-based moiety for reaction with the drug.
- Crosslinking reagents comprising a maleimido-based moiety include N- succinimidyl 4-(maleimidomethyl)cyclohexanecarboxylate (SMCC), N-succinimidyl-4-(N- maleimidomethyl)-cyclohexane-l-carboxy-(6-amidocaproate), which is a "long chain" analog of SMCC (LC-SMCC), ⁇ -maleimidoundecanoic acid N-succinimidyl ester (KMUA), ⁇ -maleimidobutyric acid N-succinimidyl ester (GMBS), ⁇ -maleimidocaproic acid N- hydroxysuccinimide ester (EMCS), m-maleimidobenzoyl-N-hydroxysuccinimide ester (MBS), N-( ⁇ -maleimidoacetoxy)-succinimide ester (AMAS), succinimidyl-6-( ⁇ - maleimid
- Cross-linking reagents comprising a haloacetyl-based moiety include N-succinimidyl-4-(iodoacetyl)-aminobenzoate (SIAB) 5 N-succinimidyl iodoacetate (SIA), N-succinimidyl bromoacetate (SBA), and N- succinimidyl 3-(bromoacetamido)propionate (SBAP).
- SIAB N-succinimidyl-4-(iodoacetyl)-aminobenzoate
- SIA N-succinimidyl iodoacetate
- SBA N-succinimidyl bromoacetate
- SBAP N- succinimidyl 3-(bromoacetamido)propionate
- linkers can be derived from dicarboxylic acid based moieties.
- Suitable dicarboxylic acid based moieties include, but are not limited to, ⁇ , ⁇ - dicarboxylic acids of the general formula (IX):
- X is a linear or branched alkyl, alkenyl, or alkynyl group having 2 to 20 carbon atoms
- Y is a cycloalkyl or cycloalkenyl group bearing 3 to 10 carbon atoms
- Z is a substituted or unsubstituted aromatic group bearing 6 to 10 carbon atoms, or a substituted or unsubstituted heterocyclic group wherein the hetero atom is selected from N, O or S, and wherein 1, m, and n are each 0 or 1, provided that 1, m, and n are all not zero at the same time.
- Patent Application No. 10/960,602 which corresponds to U.S. Patent Application Publication
- the drug can be first modified to introduce a reactive ester suitable to react with a cell-binding agent. Reaction of these maytansinoids containing an activated linker moiety with a cell-binding agent provides another method of producing a cleavable or non-cleavable cell-binding agent maytansinoid conjugate.
- This example demonstrates the purification of an antibody modified with a heterobifunctional modification reagent using TFF.
- the huN901 monoclonal antibody (final concentration of 8 mg/ml) was incubated with N-succinimidyl 4-(2-pyridyldithio)pentanoate (SPP, 5.6-fold molar excess) for approximately 180 minutes at 20° C in 50 mM potassium phosphate buffer (pH 7.5) containing 50 mM NaCl, 2 mM EDTA, and 5% ethanol.
- SPP N-succinimidyl 4-(2-pyridyldithio)pentanoate
- the reaction mixture was purified using a Pellicon XL TFF system (Millipore, Billerica, MA), and the antibody was diafiltered (5 volumes) into 50 mM potassium phosphate, 50 mM NaCl (pH 6.5), and 2 mM EDTA using a 10,000 molecular weight cutoff membrane (UltracelTM regenerated cellulose membrane, Millipore, Billerica, MA). Both samples were conjugated with DMl (1.7 fold molar excess over the unbound linker) for 18 hours at pH 6.5 in potassium phosphate buffer containing 50 mM NaCl and a final concentration of 3% DMA.
- TFF drug conjugate product of at least equivalent quality to the nonadsorptive chromatography (G25) process while being more convenient and scaleable.
- This example demonstrates the purification of an antibody modified with a heterobifunctional modification reagent using adsorptive chromatography.
- the huB4 antibody was modified with N-succinimidyl 4-(2- pyridyldithio)butanoate (SPDB, 5.4 fold molar excess) for 120 minutes at room temperature in 50 mM potassium phosphate buffer (pH 6.5) containing 50 mM NaCl, 2 mM EDTA, and 5% ethanol.
- SPDB N-succinimidyl 4-(2- pyridyldithio)butanoate
- the reaction mixture was purified using the SephadexTM G25F resin as described in Example 1.
- reaction mixture was loaded onto a column of ceramic hydroxyapatite (CHT, Bio-Rad Laboratories, Hercules, CA), which was equilibrated in 12.5 mM potassium phosphate buffer (pH 6.5) and eluted with 80 mM potassium phosphate buffer (pH 6.5).
- CHT ceramic hydroxyapatite
- the CNTO95 antibody (final concentration of 10 mg/ml) was modified with N- succinimidyl 4-(2-pyridyldithio)butanoate (SPDB, 4.5 fold molar excess) for 120 minutes at 20° C in 10 mM sodium phosphate buffer (pH 7.5) containing 2.7 % sucrose and 5% ethanol.
- SPDB N- succinimidyl 4-(2-pyridyldithio)butanoate
- the reaction mixture was purified using SephadexTM G25F resin in 12.5 mM potassium phosphate buffer (pH 6.6) containing 12.5 mM NaCl and 0.5 mM EDTA.
- This example demonstrates the beneficial effects of conjugating a modified antibody with a drug at a pH of above 6.5.
- Example 2 The modified antibody was then divided into two groups. In the first group, conjugation was performed in 12.5 mM potassium phosphate at pH 6.5 containing 12.5 mM
- the conjugation reaction was at pH 7.5.
- the conjugated antibody was purified over NAP-IO columns.
- huB4 humanized monoclonal antibody was modified with either (a) a 4.9-fold molar excess of SPDB relative to antibody, or (b) a 4.8-fold molar excess of SPDB relative to antibody.
- reaction was in 50 mM potassium phosphate, 50 mM potassium chloride, and 2 mM EDTA (pH 6.5) in 5% ethanol for a total of 120 minutes at room temperature.
- Sample (a) was purified over a column of SephadexTM G25F resin equilibrated in 50 mM potassium phosphate, 50 mM sodium chloride, and 2 mM EDTA at pH 6.5.
- Sample (b) was purified equivalently except that the chromatography buffer was adjusted to pH 7.5. Both samples were conjugated with DM4 (1.7 fold molar excess over bound linker) for 18 hours at room temperature in a final concentration of dimethylacetamide (DMA) of 3%.
- DMA dimethylacetamide
- sample (a) was conjugated at pH 6.5
- sample (b) was conjugated at pH 7.5.
- the samples were then purified over a column of SephadexTM G25F resin equilibrated in 9.6 mM potassium phosphate and 4.2 mM sodium chloride at pH 6.5. Both samples were incubated at 4° C for up to 7 months and subjected to analysis of released free drug at intervals. The resulting data are set forth in Table 3.
- release of free drug is substantially slower from sample (b) that had been conjugated at pH 7.5 relative to sample (a) that had been conjugated at pH 6.5. Accordingly, drug conjugate product prepared at pH 7.5 is shown to be more stable with respect to release of free drug over time as compared to the drug conjugate product prepared at pH 6.5. The conjugation at pH 7.5 also shows a better drug incorporation than at pH 6.5, thereby requiring less drug to be used.
- This example demonstrates the beneficial effects of conjugating a modified antibody with a drug at a pH of below 6.0.
- the huN901 monoclonal antibody (final concentration of 8 mg/ml) was incubated with N-succinimidyl 4-(2-pyridyldithio)pentanoate (SPP, 5.6-fold molar excess) for approximately 180 minutes at 20° C in 50 mM potassium phosphate buffer (pH 7.5) containing 50 mM NaCl, 2 mM EDTA, and 5% ethanol.
- SPP N-succinimidyl 4-(2-pyridyldithio)pentanoate
- the reaction mixture was purified using a column of SephadexTM G25F resin equilibrated and eluted in 50 mM sodium citrate buffer (pH 5.0) containing 50 mM NaCl and 2 mM EDTA.
- reaction mixture was purified using a column of SephadexTM G25F resin equilibrated and eluted in 50 mM potassium phosphate buffer (pH 6.5) containing 50 mM NaCl and 2 mM
- linker/antibody ratios were determined by treatment with dithiothreitol to release pyridine-2-thione, which has an extinction coefficient of 8,080
- the first group had a 4.3 linker/antibody ratio.
- the second group had a 4.2 linker/antibody ratio.
- conjugate that is made by conjugating the modified antibody with the drug at a pH of 5.0 reaches a higher and more stable level of bound drug during the course of the conjugation reaction than conjugate made at a conjugation pH of 6.5.
- the results indicate that a higher drug/antibody level is achieved upon conjugation at pH 5.0 than, when using the same amount of drug at a conjugation pH of 6.5, thereby indicating more efficient usage of drug at pH 5.0.
- the conjugate monomer amounts were determined over time. The resulting data are set forth in Table 5.
- conjugate that is made by conjugating the modified antibody with the drug at a pH of 5.0 has a higher level of conjugate monomer than conjugate made at a conjugation pH of 6.5.
- This example further demonstrates benefits of conjugating a drug to a modified antibody at a pH of less than 6.
- BIWA 4 antibody was modified with SPP (molar excess of SPP as shown in Table 6) for 120-140 minutes at room temperature in 50 mM potassium phosphate buffer (pH 6.5), 50 mM NaCl, 2 mM EDTA, and 5% ethanol. Aliquots of modified antibody were purified on separate NAP 25 columns equilibrated in buffers having various pH values (pH 4.6 — 6.5). The pH 4.6 - 5.9 buffers were composed of 35 mM sodium citrate, 150 mM sodium chloride, and 2 mM EDTA. The pH 6.5 buffer was PBS with 2 mM EDTA.
- Modified antibody at each pH was conjugated with DMl (1.7 fold molar excess over linker) in dimethylacetamide (DMA, final concentration of 3%). After incubation for 17-18 hours at room temperature, the conjugated antibody samples were purified by chromatography on NAP 25 columns equilibrated in PBS (pH 6.5). Linker/antibody ratios (L/A in Table 6) were determined by treatment with dithiothreitol to release pyridine-2- thione, which has an extinction coefficient of 8,080 M -1 Cm "1 at 343 nM. Drug/antibody ratios were determined spectrophotometrically (wavelengths of 280 nm and 252 nm) for the conjugation step.
- DMA dimethylacetamide
- Conjugate monomer, high molecular weight species, and low molecular weight species were determined by SEC-HPLC using a TSKG3000SWXL column equilibrated and developed in 0.2 M potassium phosphate buffer (pH 7.0) containing 0.2 M potassium chloride and 20% isopropanol. [0098] The results of this analysis are set forth in Table 6.
- the humanized monoclonal antibody CNTO95 was modified at a concentration of 20 mg/mL with the bifunctional modifying reagent SPDB at a molar excess of SPDB over antibody of 4.6 for 120 min at 20° C.
- the modification buffer was 44 mM phosphate buffer (pH 7.5) containing 5.3% sucrose and 5% ethanol.
- One aliquot of the modified antibody was purified over SephadexTM G25F resin (standard four-step process), equilibrated and eluted in 12.5 mM potassium phosphate buffer (pH 7.5) containing 12.5 mM NaCl, and was subsequently conjugated with DM4 (1.7 fold molar excess of drug over bound linker) at a final modified antibody concentration of 10 mg/mL in 12.5 mM potassium phosphate buffer (pH 7.5) containing 12.5 mM NaCl and 10% DMA for 20 hours at room temperature.
- a second aliquot of the modified antibody was conjugated immediately at the end of the 120 minute modification reaction (three-step process), without being further purified.
- Linker/antibody ratios were determined by treatment with dithiothreitol to release pyridine-2-thione, which has an extinction coefficient of 8,080 M -1 Cm "1 at 343 nM.
- Drug/antibody (D/ A) ratios and yield were determined spectrophotometrically (wavelengths of 280 nm and 252 nm) for the conjugation step. Percentages of monomer were assayed by SEC-HPLC. Percentages of free drug were assayed by HPLC on a Hisep column. The results of these analyses are set forth in Table 7.
- This example demonstrates an improved means of purifying antibody that has been modified with a heterobifunctional modification reagent and then conjugated with a maytansinoid.
- a second conjugate sample was purified by a Pellicon XL TFF system (Millipore,
- a third conjugate sample was purified using a column of MEP Hypercell resin equilibrated in 50 mM Tris (pH 8.0), and eluted with 50 mM sodium acetate (pH 4.0).
- a fourth conjugate sample was purified using a column of UNOsphere S resin equilibrated in 50 mM sodium phosphate (pH 6.5) and eluted with 0.2 M NaCl and 50 mM sodium phosphate (pH 6.5).
- a fifth conjugate sample was purified using a column of CHT resin (Bio-Rad
- a sixth conjugate sample was purified using a column of SP Sepharose resin equilibrated in 35 mM sodium citrate, 10 mM sodium chloride (pH 5.0), and eluted with 0.25
- Conjugate monomer was determined by SEC-HPLC using a column of
- the conjugation step yield was determined by dividing the yield of conjugated antibody by the amount of modified antibody that was conjugated (determined spectrophotometrically at a wavelength of 280 nm).
- CHT ceramic hydroxyapatite
- CFT ceramic fluoroapatite
- both the CHT and CFT resins may be used in non-adsorptive mode so that the desired product (substantially monomeric conjugate) is not retained by the resins, whereas high molecular weight species are retained and thereby separated from the desired product.
- a standard buffer/solvent composition for conjugation comprises 3% DMA, 50 mM potassium phosphate, 50 mM NaCl, and 2 mM EDTA at pH 6.5 (as utilized in Example 1)
- other compositions are more compatible with some of the chromatographic steps described herein and provide other benefits relative to the standard process.
- conjugation may be performed in 3% DMA, 12.5 mM potassium phosphate, 12.5 mM NaCl, and 0.5 mM EDTA at pH 6.5.
- the amount of DM4 incorporated relative to the amount of linker incorporated in huB4 antibody was about 10% higher than for the standard conditions.
- these conditions are more compatible with loading onto resins such as cation exchange and CHT resins.
Abstract
Description













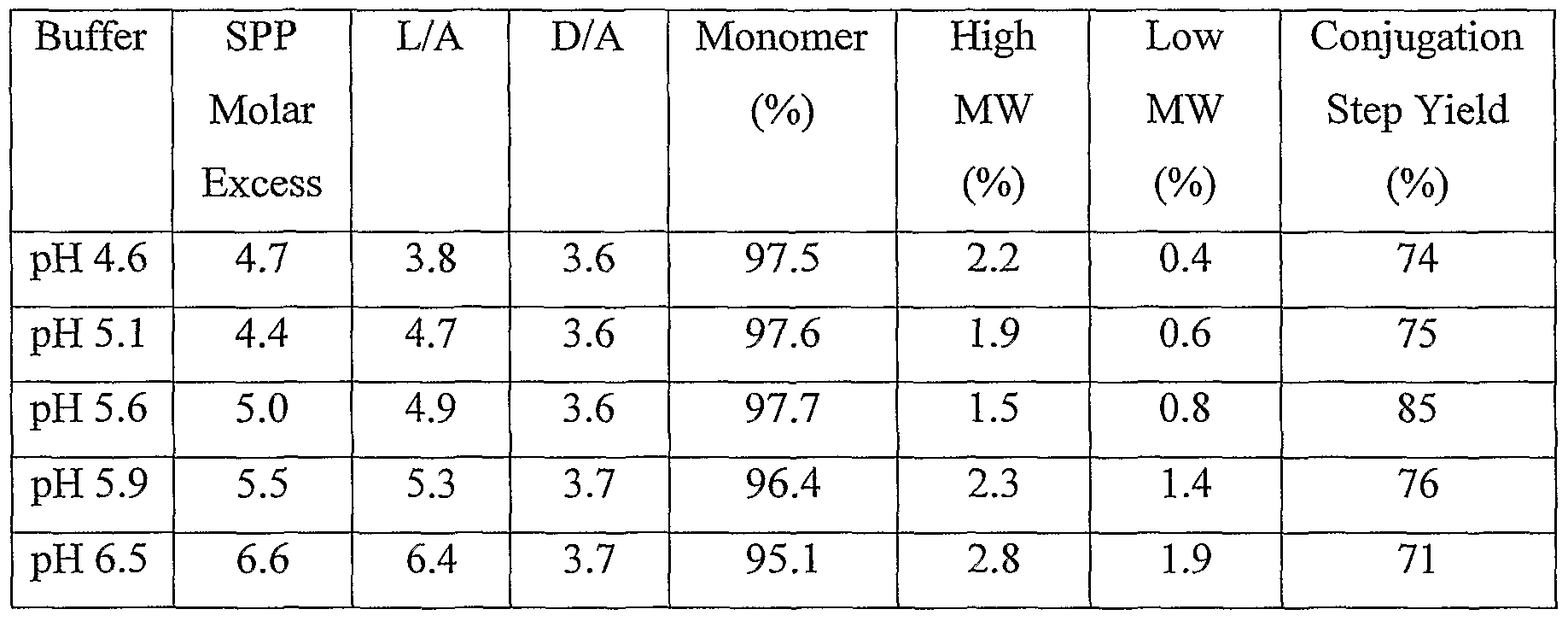


Claims
Priority Applications (27)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MX2008002597A MX2008002597A (en) | 2005-08-24 | 2006-08-14 | Process for preparing purified drug conjugates. |
| EP06801436A EP1928503B1 (en) | 2005-08-24 | 2006-08-14 | Process for preparing maytansinoid antibody conjugates |
| RS20120425A RS52470B (en) | 2005-08-24 | 2006-08-14 | Process for preparing maytansinoid antibody conjugates |
| AU2006283726A AU2006283726C1 (en) | 2005-08-24 | 2006-08-14 | Process for preparing maytansinoid antibody conjugates |
| DK06801436.4T DK1928503T3 (en) | 2005-08-24 | 2006-08-14 | Process for Preparation of Purified Drug Conjugates |
| BRPI0615049-7A BRPI0615049B1 (en) | 2005-08-24 | 2006-08-14 | PROCESS FOR THE PREPARATION OF AN ANTIBODY- MAITANSINOID CONJUGATE |
| IL282138A IL282138B2 (en) | 2005-08-24 | 2006-08-14 | Process for preparing purified drug conjugates |
| EP19156303.0A EP3539572A1 (en) | 2005-08-24 | 2006-08-14 | Process for preparing maytansinoid antibody conjugates |
| KR1020087004084A KR101301011B1 (en) | 2005-08-24 | 2006-08-14 | Process for preparing maytansinoid antibody conjugates |
| IL305084A IL305084A (en) | 2005-08-24 | 2006-08-14 | Process for preparing purified drug conjugates |
| PL06801436T PL1928503T3 (en) | 2005-08-24 | 2006-08-14 | Process for preparing maytansinoid antibody conjugates |
| SI200631435T SI1928503T1 (en) | 2005-08-24 | 2006-08-14 | Process for preparing maytansinoid antibody conjugates |
| CA2620343A CA2620343C (en) | 2005-08-24 | 2006-08-14 | Process for preparing antibody maytansinoid conjugates |
| EA200800657A EA013327B1 (en) | 2005-08-24 | 2006-08-14 | Process for preparing purified drug conjugate |
| JP2008527970A JP5350792B2 (en) | 2005-08-24 | 2006-08-14 | Method for preparing maytansinoid antibody complex |
| NZ565964A NZ565964A (en) | 2005-08-24 | 2006-08-14 | Process for preparing maytansinoid antibody conjugates by purifying with tangential flow filtration |
| NZ595430A NZ595430A (en) | 2005-08-24 | 2006-08-14 | Process for preparing maytansinoid antibody conjugates |
| ES06801436T ES2390826T3 (en) | 2005-08-24 | 2006-08-14 | Procedure for preparing purified drug conjugates |
| CN2006800342426A CN101267841B (en) | 2005-08-24 | 2006-08-14 | Process for preparing maytansinoid antibody conjugates |
| IL189461A IL189461A (en) | 2005-08-24 | 2008-02-12 | Process for preparing cell-binding agent-cytotoxic agent conjugates |
| ZA2008/01564A ZA200801564B (en) | 2005-08-24 | 2008-02-15 | Process for preparaing maytansinoid antibody conjugates |
| HK08112150.3A HK1120407A1 (en) | 2005-08-24 | 2008-11-06 | Process for preparing maytansinoid antibody conjugates |
| IL220816A IL220816A (en) | 2005-08-24 | 2012-07-08 | Process for preparing cell-binding agent-cytotoxic agent conjugates |
| HRP20120794TT HRP20120794T1 (en) | 2005-08-24 | 2012-10-04 | Process for preparing maytansinoid antibody conjugates |
| IL226985A IL226985A (en) | 2005-08-24 | 2013-06-17 | Process for preparing purified drug conjugates |
| CR20150350A CR20150350A (en) | 2005-08-24 | 2015-07-03 | PROCESS TO PREPARE ANTIBODY AND MAYTANSINOID CONJUGATES |
| IL248076A IL248076B (en) | 2005-08-24 | 2016-09-27 | Process for preparing purified drug conjugates |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US71085805P | 2005-08-24 | 2005-08-24 | |
| US60/710,858 | 2005-08-24 | ||
| US79771306P | 2006-05-04 | 2006-05-04 | |
| US60/797,713 | 2006-05-04 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP11004940.0A Previously-Filed-Application EP2399609B1 (en) | 2005-08-24 | 2006-08-14 | Process for preparing maytansinoid antibody conjugates |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007024536A2 true WO2007024536A2 (en) | 2007-03-01 |
| WO2007024536A3 WO2007024536A3 (en) | 2007-07-05 |
Family
ID=37666423
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/031653 WO2007024536A2 (en) | 2005-08-24 | 2006-08-14 | Process for preparing maytansinoid antibody conjugates |
Country Status (25)
| Country | Link |
|---|---|
| US (6) | US7811572B2 (en) |
| EP (4) | EP3539572A1 (en) |
| JP (3) | JP5350792B2 (en) |
| KR (1) | KR101301011B1 (en) |
| CN (2) | CN101267841B (en) |
| AU (1) | AU2006283726C1 (en) |
| BR (1) | BRPI0615049B1 (en) |
| CA (5) | CA2620343C (en) |
| CR (2) | CR9742A (en) |
| CY (1) | CY1113206T1 (en) |
| DK (1) | DK1928503T3 (en) |
| EA (1) | EA013327B1 (en) |
| EC (1) | ECSP088212A (en) |
| ES (2) | ES2533992T3 (en) |
| HK (2) | HK1120407A1 (en) |
| HR (1) | HRP20120794T1 (en) |
| IL (6) | IL282138B2 (en) |
| MX (1) | MX2008002597A (en) |
| NZ (4) | NZ609752A (en) |
| PL (1) | PL1928503T3 (en) |
| PT (1) | PT1928503E (en) |
| RS (1) | RS52470B (en) |
| SI (1) | SI1928503T1 (en) |
| WO (1) | WO2007024536A2 (en) |
| ZA (1) | ZA200801564B (en) |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010126979A1 (en) | 2009-04-29 | 2010-11-04 | Bio-Rad Laboratories, Inc. | Purification of immunoconjugates |
| KR20120088549A (en) * | 2009-06-03 | 2012-08-08 | 이뮤노젠 아이엔씨 | Conjugation methods |
| WO2012143496A2 (en) | 2011-04-21 | 2012-10-26 | Bayer Intellectual Property Gmbh | Novel binder-drug conjugates (adcs) and their use |
| WO2012135517A3 (en) * | 2011-03-29 | 2013-02-21 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| WO2014055842A1 (en) * | 2012-10-04 | 2014-04-10 | Immunogen, Inc. | Process for preparing stable antibody maytansinoid conjugates |
| US8795673B2 (en) | 2011-03-29 | 2014-08-05 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| US8933205B2 (en) | 2005-08-24 | 2015-01-13 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| WO2015009740A2 (en) | 2013-07-15 | 2015-01-22 | Cell Signaling Technology, Inc. | Anti-mucin 1 binding agents and uses thereof |
| CN104524592A (en) * | 2008-04-30 | 2015-04-22 | 伊缪诺金公司 | Cross-linkers and their uses |
| EP2903450A4 (en) * | 2012-10-04 | 2016-05-11 | Immunogen Inc | Use of an ion exchange membrane to remove impurities from cell-binding agent cytotoxic agent conjugates |
| WO2016207089A1 (en) | 2015-06-22 | 2016-12-29 | Bayer Pharma Aktiengesellschaft | Antibody drug conjugates (adcs) and antibody prodrug conjugates (apdcs) with enzymatically cleavable groups |
| WO2017060322A2 (en) | 2015-10-10 | 2017-04-13 | Bayer Pharma Aktiengesellschaft | Ptefb-inhibitor-adc |
| WO2017162663A1 (en) | 2016-03-24 | 2017-09-28 | Bayer Pharma Aktiengesellschaft | Prodrugs of cytotoxic active agents having enzymatically cleavable groups |
| WO2018114798A1 (en) | 2016-12-21 | 2018-06-28 | Bayer Aktiengesellschaft | Prodrugs of cytotoxic active agents having enzymatically cleavable groups |
| WO2018114578A1 (en) | 2016-12-21 | 2018-06-28 | Bayer Pharma Aktiengesellschaft | Antibody drug conjugates (adcs) having enzymatically cleavable groups |
| US10022453B2 (en) | 2013-12-23 | 2018-07-17 | Bayer Pharma Aktiengesellschaft | Antibody drug conjugates (ADCs) with kinesin spindel protein (KSP) |
| US10035817B2 (en) | 2012-10-04 | 2018-07-31 | Immunogen, Inc. | Method of purifying cell-binding agent-cytotoxic agent conjugates with a PVDF membrane |
| US20180311375A1 (en) * | 2017-04-27 | 2018-11-01 | Cadila Healthcare Limited | Process of preparing antibody-drug conjugate |
| WO2019105835A1 (en) | 2017-11-29 | 2019-06-06 | Bayer Consumer Care Ag | Combinations of copanlisib and anetumab ravtansine |
| WO2020022363A1 (en) | 2018-07-25 | 2020-01-30 | 第一三共株式会社 | Effective method for manufacturing antibody-drug conjugate |
| WO2020084115A1 (en) | 2018-10-25 | 2020-04-30 | Pharma Mar, S.A. | Antibody drug conjugates comprising ecteinascidin derivatives |
| US10640508B2 (en) | 2017-10-13 | 2020-05-05 | Massachusetts Institute Of Technology | Diazene directed modular synthesis of compounds with quaternary carbon centers |
| WO2020234114A1 (en) | 2019-05-21 | 2020-11-26 | Bayer Aktiengesellschaft | A novel stable high concentration formulation for anetumab ravtansine |
| US10918735B2 (en) | 2012-12-04 | 2021-02-16 | Massachusetts Institute Of Technology | Substituted pyrazino[1′,2′:1,5]pyrrolo[2,3-b]indole-1,4-diones for cancer treatment |
| US10918627B2 (en) | 2016-05-11 | 2021-02-16 | Massachusetts Institute Of Technology | Convergent and enantioselective total synthesis of Communesin analogs |
| WO2021043951A1 (en) | 2019-09-05 | 2021-03-11 | Pharma Mar, S.A. | Drug antibody conjugates |
| US11001636B2 (en) | 2016-06-15 | 2021-05-11 | Bayer Pharma Aktiengesellschaft | Specific antibody-drug-conjugates (ADCs) with KSP inhibitors and anti-CD123-antibodies |
| WO2021214126A1 (en) | 2020-04-21 | 2021-10-28 | Pharma Mar, S.A. | Drug antibody conjugates |
| US11160871B2 (en) | 2015-10-28 | 2021-11-02 | Tarveda Therapeutics, Inc. | SSTR-targeted conjugates and particles and formulations thereof |
| US11274121B2 (en) | 2018-01-12 | 2022-03-15 | Immunogen, Inc. | Methods for antibody drug conjugation, purification, and formulation |
| US11433140B2 (en) | 2016-12-21 | 2022-09-06 | Bayer Pharma Aktiengesellschaft | Specific antibody drug conjugates (ADCs) having KSP inhibitors |
| US11535634B2 (en) | 2019-06-05 | 2022-12-27 | Massachusetts Institute Of Technology | Compounds, conjugates, and compositions of epipolythiodiketopiperazines and polythiodiketopiperazines and uses thereof |
| US11634508B2 (en) | 2019-07-10 | 2023-04-25 | Cybrexa 2, Inc. | Peptide conjugates of cytotoxins as therapeutics |
| US11932650B2 (en) | 2017-05-11 | 2024-03-19 | Massachusetts Institute Of Technology | Potent agelastatin derivatives as modulators for cancer invasion and metastasis |
Families Citing this family (62)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002092771A2 (en) | 2001-05-11 | 2002-11-21 | Ludwig Institute For Cancer Research | Specific binding proteins and uses thereof |
| US20100056762A1 (en) | 2001-05-11 | 2010-03-04 | Old Lloyd J | Specific binding proteins and uses thereof |
| JP5149620B2 (en) | 2004-07-23 | 2013-02-20 | エンドサイト,インコーポレイテッド | Bivalent linker and conjugate thereof |
| US20110166319A1 (en) * | 2005-02-11 | 2011-07-07 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| MX2009007987A (en) * | 2007-01-25 | 2010-03-01 | Dana Farber Cancer Inst Inc | Use of anti-egfr antibodies in treatment of egfr mutant mediated disease. |
| NZ599239A (en) | 2007-03-14 | 2013-10-25 | Endocyte Inc | Binding ligand linked drug delivery conjugates of tubulysins |
| US9023356B2 (en) | 2007-03-15 | 2015-05-05 | Ludwig Institute For Cancer Research Ltd | Treatment method using EGFR antibodies and SRC inhibitors and related formulations |
| US9877965B2 (en) | 2007-06-25 | 2018-01-30 | Endocyte, Inc. | Vitamin receptor drug delivery conjugates for treating inflammation |
| WO2009002993A1 (en) | 2007-06-25 | 2008-12-31 | Endocyte, Inc. | Conjugates containing hydrophilic spacer linkers |
| JP5532486B2 (en) | 2007-08-14 | 2014-06-25 | ルードヴィッヒ インスティテュート フォー キャンサー リサーチ | Monoclonal antibody 175 targeting EGF receptor and derivatives and uses thereof |
| US20100092495A1 (en) * | 2008-04-30 | 2010-04-15 | Immunogen Inc. | Potent cell-binding agent drug conjugates |
| US20110076232A1 (en) * | 2009-09-29 | 2011-03-31 | Ludwig Institute For Cancer Research | Specific binding proteins and uses thereof |
| AR078471A1 (en) * | 2009-10-02 | 2011-11-09 | Sanofi Aventis | MAITANSINOID COMPOUNDS AND THE USE OF THESE TO PREPARE CONJUGATES WITH AN ANTIBODY WHICH ARE USED AS ANTI-BANKING AGENTS AND THE PREPARATION PROCEDURE OF THESE CONJUGATES |
| AU2011215900A1 (en) | 2010-02-10 | 2012-07-26 | Immunogen, Inc. | CD20 antibodies and uses thereof |
| KR102099462B1 (en) | 2010-11-30 | 2020-04-10 | 제넨테크, 인크. | Low affinity blood brain barrier receptor antibodies and uses therefor |
| RU2748733C2 (en) | 2011-02-15 | 2021-05-31 | Иммуноджен, Инк. | Cytotoxic benzodiazepine derivatives |
| MX2013011201A (en) * | 2011-03-29 | 2013-12-16 | Immunogen Inc | Process for manufacturing conjugates of improved homogeneity. |
| US9289512B2 (en) | 2011-06-21 | 2016-03-22 | Immunogen, Inc. | Maytansinoid derivatives with peptide linker and conjugates thereof |
| AU2012352210A1 (en) * | 2011-12-13 | 2014-07-24 | Immunogen, Inc. | Use of N-hydroxysuccinimide to improve conjugate stability |
| US10080805B2 (en) | 2012-02-24 | 2018-09-25 | Purdue Research Foundation | Cholecystokinin B receptor targeting for imaging and therapy |
| US20140080175A1 (en) | 2012-03-29 | 2014-03-20 | Endocyte, Inc. | Processes for preparing tubulysin derivatives and conjugates thereof |
| CA2869704A1 (en) * | 2012-04-04 | 2013-10-10 | Perseus Proteomics Inc. | Drug conjugate comprising anti-cdh3 (pcadherin) antibody |
| WO2013151649A1 (en) * | 2012-04-04 | 2013-10-10 | Sialix Inc | Glycan-interacting compounds |
| EP2887965A1 (en) | 2012-08-22 | 2015-07-01 | ImmunoGen, Inc. | Cytotoxic benzodiazepine derivatives |
| WO2014058947A1 (en) | 2012-10-12 | 2014-04-17 | Sanofi | Compositions and methods for treating cancer using pi3k inhibitor and anti-cd19 maytansinoid immunoconjugate |
| IN2015DN04147A (en) | 2012-10-16 | 2015-10-16 | Endocyte Inc | |
| EP2961434A2 (en) | 2013-02-28 | 2016-01-06 | ImmunoGen, Inc. | Conjugates comprising cell-binding agents and cytotoxic agents |
| JP6423804B2 (en) | 2013-02-28 | 2018-11-14 | イミュノジェン・インコーポレーテッド | Complex containing cell binding agent and cytotoxic agent |
| US9498532B2 (en) | 2013-03-13 | 2016-11-22 | Novartis Ag | Antibody drug conjugates |
| US10570151B2 (en) | 2013-03-15 | 2020-02-25 | Regeneron Pharmaceuticals, Inc. | Biologically active molecules, conjugates thereof, and therapeutic uses |
| MA38396B1 (en) | 2013-03-15 | 2019-05-31 | Novartis Ag | Conjugated drug antibodies and their pharmaceutical compositions for treating a ckit positive cancer |
| WO2014182970A1 (en) * | 2013-05-08 | 2014-11-13 | Zymeworks Inc. | Bispecific her2 and her3 antigen binding constructs |
| WO2014194030A2 (en) | 2013-05-31 | 2014-12-04 | Immunogen, Inc. | Conjugates comprising cell-binding agents and cytotoxic agents |
| SG11201601770YA (en) | 2013-09-12 | 2016-04-28 | Halozyme Inc | Modified anti-epidermal growth factor receptor antibodies and methods of use thereof |
| TWI541022B (en) | 2013-12-18 | 2016-07-11 | 應克隆公司 | Compounds to fibroblast growth factor receptor-3 (fgfr3) and methods of treatment |
| ES2785551T3 (en) | 2014-06-30 | 2020-10-07 | Glykos Finland Oy | Saccharide derivative of a toxic payload and its conjugates with antibodies |
| EP3160518A4 (en) | 2014-06-30 | 2018-05-23 | Tarveda Therapeutics, Inc. | Targeted conjugates and particles and formulations thereof |
| WO2016024195A1 (en) | 2014-08-12 | 2016-02-18 | Novartis Ag | Anti-cdh6 antibody drug conjugates |
| WO2016036861A1 (en) * | 2014-09-02 | 2016-03-10 | Immunogen, Inc. | Methods for formulating antibody drug conjugate compositions |
| TWI697493B (en) | 2014-09-03 | 2020-07-01 | 美商免疫原公司 | Cytotoxic benzodiazepine derivatives |
| WO2016036804A1 (en) | 2014-09-03 | 2016-03-10 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives |
| CN104208719B (en) * | 2014-09-24 | 2017-05-03 | 北京天广实生物技术股份有限公司 | ADC (antibody-drug conjugate) cation exchange chromatographic purification method |
| EP4183806A3 (en) | 2014-11-12 | 2023-08-02 | Seagen Inc. | Glycan-interacting compounds and methods of use |
| WO2016077739A1 (en) * | 2014-11-13 | 2016-05-19 | The Curators Of The University Of Missouri | Multiple human antibody-nanoparticle conjugates and methods of formation |
| TW201711702A (en) | 2015-06-04 | 2017-04-01 | 應克隆公司 | Therapies utilizing compounds to fibroblast growth factor receptor-3 (FGFR3) |
| US20190194315A1 (en) | 2015-06-17 | 2019-06-27 | Novartis Ag | Antibody drug conjugates |
| IL302822A (en) | 2015-11-12 | 2023-07-01 | Seagen Inc | Glycan-interacting compounds and methods of use |
| FR3045058B1 (en) * | 2015-12-11 | 2017-12-29 | Ifp Energies Now | NOVEL POLYAMINES, PROCESS FOR THEIR SYNTHESIS AND THEIR USE FOR THE SELECTIVE REMOVAL OF H2S FROM A GASEOUS EFFLUENT COMPRISING CO2 |
| WO2017161206A1 (en) | 2016-03-16 | 2017-09-21 | Halozyme, Inc. | Conjugates containing conditionally active antibodies or antigen-binding fragments thereof, and methods of use |
| EP3541847A4 (en) | 2016-11-17 | 2020-07-08 | Seattle Genetics, Inc. | Glycan-interacting compounds and methods of use |
| JOP20190187A1 (en) | 2017-02-03 | 2019-08-01 | Novartis Ag | Anti-ccr7 antibody drug conjugates |
| KR102653141B1 (en) | 2017-03-03 | 2024-04-01 | 씨젠 인크. | Glycan-interacting compounds and methods of use |
| AU2018241654A1 (en) * | 2017-03-30 | 2019-11-07 | Jiangsu Hengrui Medicine Co., Ltd. | Method for preparing antibody-drug conjugate |
| WO2018185618A1 (en) | 2017-04-03 | 2018-10-11 | Novartis Ag | Anti-cdh6 antibody drug conjugates and anti-gitr antibody combinations and methods of treatment |
| TW201839001A (en) | 2017-04-20 | 2018-11-01 | 美商伊繆諾金公司 | Cytotoxic benzodiazepine derivatives and conjugates thereof |
| WO2019133652A1 (en) | 2017-12-28 | 2019-07-04 | Immunogen, Inc. | Benzodiazepine derivatives |
| EP3552631A1 (en) * | 2018-04-10 | 2019-10-16 | Inatherys | Antibody-drug conjugates and their uses for the treatment of cancer |
| JP2022529583A (en) | 2019-03-29 | 2022-06-23 | イミュノジェン・インコーポレーテッド | Its complex with cytotoxic bis-benzodiazepine derivatives and cell binding agents for inhibiting aberrant cell proliferation or treating proliferative disorders |
| FI3958977T3 (en) | 2019-04-26 | 2023-12-12 | Immunogen Inc | Camptothecin derivatives |
| BR112022000297A2 (en) | 2019-07-10 | 2022-03-15 | Cybrexa 3 Inc | Peptide conjugates of microtubule targeting agents as therapeutics |
| CN113125732B (en) * | 2019-12-31 | 2023-08-08 | 科美博阳诊断技术(上海)有限公司 | Homogeneous detection kit for interleukin 6 and application thereof |
| US20230181756A1 (en) | 2020-04-30 | 2023-06-15 | Novartis Ag | Ccr7 antibody drug conjugates for treating cancer |
Family Cites Families (163)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US585499A (en) * | 1897-06-29 | Oil-can | ||
| US585089A (en) * | 1897-06-22 | durey | ||
| US552293A (en) * | 1895-12-31 | ktjhn | ||
| SE430062B (en) | 1977-03-04 | 1983-10-17 | Pharmacia Fine Chemicals Ab | COUPLING OR TIOLATION REAGENTS |
| US4137230A (en) | 1977-11-14 | 1979-01-30 | Takeda Chemical Industries, Ltd. | Method for the production of maytansinoids |
| US4307016A (en) | 1978-03-24 | 1981-12-22 | Takeda Chemical Industries, Ltd. | Demethyl maytansinoids |
| US4256746A (en) | 1978-11-14 | 1981-03-17 | Takeda Chemical Industries | Dechloromaytansinoids, their pharmaceutical compositions and method of use |
| JPS55102583A (en) | 1979-01-31 | 1980-08-05 | Takeda Chem Ind Ltd | 20-acyloxy-20-demethylmaytansinoid compound |
| JPS55162791A (en) | 1979-06-05 | 1980-12-18 | Takeda Chem Ind Ltd | Antibiotic c-15003pnd and its preparation |
| JPS5645483A (en) | 1979-09-19 | 1981-04-25 | Takeda Chem Ind Ltd | C-15003phm and its preparation |
| JPS5645485A (en) | 1979-09-21 | 1981-04-25 | Takeda Chem Ind Ltd | Production of c-15003pnd |
| EP0028683A1 (en) | 1979-09-21 | 1981-05-20 | Takeda Chemical Industries, Ltd. | Antibiotic C-15003 PHO and production thereof |
| WO1982001188A1 (en) | 1980-10-08 | 1982-04-15 | Takeda Chemical Industries Ltd | 4,5-deoxymaytansinoide compounds and process for preparing same |
| US4450254A (en) | 1980-11-03 | 1984-05-22 | Standard Oil Company | Impact improvement of high nitrile resins |
| US4313946A (en) | 1981-01-27 | 1982-02-02 | The United States Of America As Represented By The Secretary Of Agriculture | Chemotherapeutically active maytansinoids from Trewia nudiflora |
| US4315929A (en) | 1981-01-27 | 1982-02-16 | The United States Of America As Represented By The Secretary Of Agriculture | Method of controlling the European corn borer with trewiasine |
| US4563304A (en) | 1981-02-27 | 1986-01-07 | Pharmacia Fine Chemicals Ab | Pyridine compounds modifying proteins, polypeptides or polysaccharides |
| JPS57192389A (en) | 1981-05-20 | 1982-11-26 | Takeda Chem Ind Ltd | Novel maytansinoid |
| US4664911A (en) | 1983-06-21 | 1987-05-12 | Board Of Regents, University Of Texas System | Immunotoxin conjugates employing toxin B chain moieties |
| EP0397292B1 (en) | 1985-01-22 | 1993-04-21 | Saint-Gobain Vitrage International | Process for producing a thin metal oxide layer on a substrate, especially glass, and its use as glazing |
| GB8600582D0 (en) * | 1986-01-10 | 1986-02-19 | Ca Minister Nat Defence | Purifying biological materials |
| GB8607679D0 (en) | 1986-03-27 | 1986-04-30 | Winter G P | Recombinant dna product |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| US4859449A (en) | 1987-09-14 | 1989-08-22 | Center For Molecular Medicine And Immunology | Modified antibodies for enhanced hepatocyte clearance |
| US5241078A (en) | 1988-06-14 | 1993-08-31 | Cetus Oncology | Coupling agents and sterically hindered disulfide linked conjugates prepared therefrom |
| US5024834A (en) | 1988-07-12 | 1991-06-18 | Cetus Corporation | Thioether linked immunotoxin conjugates |
| CA2006408A1 (en) | 1988-12-27 | 1990-06-27 | Susumu Iwasa | Bispecific monoclonal antibody, its production and use |
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US5714149A (en) | 1989-02-10 | 1998-02-03 | Celltech Therapeutics Limited | Crosslinked antibodies and processes for their preparation |
| CA2026147C (en) | 1989-10-25 | 2006-02-07 | Ravi J. Chari | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US6316003B1 (en) | 1989-12-21 | 2001-11-13 | Whitehead Institute For Biomedical Research | Tat-derived transport polypeptides |
| US5137877B1 (en) | 1990-05-14 | 1996-01-30 | Bristol Myers Squibb Co | Bifunctional linking compounds conjugates and methods for their production |
| GB9015198D0 (en) | 1990-07-10 | 1990-08-29 | Brien Caroline J O | Binding substance |
| US5545806A (en) | 1990-08-29 | 1996-08-13 | Genpharm International, Inc. | Ransgenic non-human animals for producing heterologous antibodies |
| ES2108048T3 (en) | 1990-08-29 | 1997-12-16 | Genpharm Int | PRODUCTION AND USE OF LOWER TRANSGENIC ANIMALS CAPABLE OF PRODUCING HETEROLOGICAL ANTIBODIES. |
| US5256294A (en) | 1990-09-17 | 1993-10-26 | Genentech, Inc. | Tangential flow filtration process and apparatus |
| CA2048078A1 (en) | 1990-11-15 | 1992-05-16 | Wolfgang A. Wrasidlo | Chemical modification of antibodies for creation of immunoconjugates |
| US5252714A (en) | 1990-11-28 | 1993-10-12 | The University Of Alabama In Huntsville | Preparation and use of polyethylene glycol propionaldehyde |
| SE9102074D0 (en) | 1991-07-03 | 1991-07-03 | Kabi Pharmacia Ab | TOMOUR ANTIGEN SPECIFIC ANTIBODY |
| SE470006B (en) | 1991-09-26 | 1993-10-25 | Corline Systems Ab | New conjugate, its preparation and use, and substrates prepared with the conjugate |
| ES2227512T3 (en) | 1991-12-02 | 2005-04-01 | Medical Research Council | PRODUCTION OF ANTIBODIES AGAINST SELF-ANTIGENS FROM REPERTORIES OF ANTIBODY SEGMENTS FIXED IN A PHOTO. |
| CA2076465C (en) | 1992-03-25 | 2002-11-26 | Ravi V. J. Chari | Cell binding agent conjugates of analogues and derivatives of cc-1065 |
| US5639641A (en) | 1992-09-09 | 1997-06-17 | Immunogen Inc. | Resurfacing of rodent antibodies |
| US5556623A (en) | 1993-03-30 | 1996-09-17 | Eli Lilly And Company | Antibody-drug conjugates |
| GB9320575D0 (en) | 1993-10-06 | 1993-11-24 | Amp Gmbh | Coaxial connector having improved locking mechanism |
| IL111748A0 (en) | 1993-12-03 | 1995-01-24 | Zeneca Ltd | Proteins |
| DE19503164A1 (en) * | 1994-02-04 | 1995-08-10 | Siemens Comp Inc | Optically coupled data switching system and handset cradle switch |
| US5919758A (en) | 1994-03-22 | 1999-07-06 | Beth Israel Deaconess Medical Center | Modified polypeptides with altered biological activity |
| US5580853A (en) | 1994-03-22 | 1996-12-03 | New England Deaconess Hospital | Modified polypeptides with increased biological activity |
| US5747446A (en) | 1994-03-22 | 1998-05-05 | Beth Israel Deaconess Medical Center | Modified polypeptides with increased biological activity |
| US5612474A (en) | 1994-06-30 | 1997-03-18 | Eli Lilly And Company | Acid labile immunoconjugate intermediates |
| AU5908296A (en) | 1995-05-31 | 1996-12-24 | Fred Hutchinson Cancer Research Center | Compositions and methods for targeted delivery of effector m olecules |
| US6265150B1 (en) | 1995-06-07 | 2001-07-24 | Becton Dickinson & Company | Phage antibodies |
| US5714352A (en) | 1996-03-20 | 1998-02-03 | Xenotech Incorporated | Directed switch-mediated DNA recombination |
| GB9610992D0 (en) | 1996-05-24 | 1996-07-31 | Glaxo Group Ltd | Concentrated antibody preparation |
| US6340461B1 (en) | 1996-12-17 | 2002-01-22 | David Stephen Terman | Superantigen based methods and compositions for treatment of diseases |
| US6462070B1 (en) | 1997-03-06 | 2002-10-08 | The General Hospital Corporation | Photosensitizer conjugates for pathogen targeting |
| US6371975B2 (en) | 1998-11-06 | 2002-04-16 | Neomend, Inc. | Compositions, systems, and methods for creating in situ, chemically cross-linked, mechanical barriers |
| CA2304254C (en) | 1997-06-11 | 2012-05-22 | Hans Christian Thogersen | Trimerising module |
| US6171586B1 (en) | 1997-06-13 | 2001-01-09 | Genentech, Inc. | Antibody formulation |
| US5958677A (en) | 1997-07-28 | 1999-09-28 | The New York Blood Center, Inc. | Method for purifying viral nucleic acids |
| CA2297070A1 (en) | 1997-08-01 | 1999-02-11 | Morphosys Ag | Novel method and phage for the identification of nucleic acid sequences encoding members of a multimeric (poly)peptide complex |
| US5965714A (en) * | 1997-10-02 | 1999-10-12 | Connaught Laboratories, Inc. | Method for the covalent attachment of polysaccharides to protein molecules |
| IL135148A0 (en) | 1997-10-03 | 2001-05-20 | Galenica Pharmaceuticals Inc | A polysaccharide conjugate and pharmaceutical compositions containing the same |
| US6121236A (en) | 1998-03-24 | 2000-09-19 | The Children's Medical Center Corporation | Multivalent ligands which modulate angiogenesis |
| US5963761A (en) * | 1998-04-15 | 1999-10-05 | Xerox Corporation | Area coverage sensor calibration and algorithm for seam detection noise eliminator on a seamed photoreceptor |
| US6573245B1 (en) | 1998-04-28 | 2003-06-03 | Galenica Pharmaceuticals, Inc. | Modified polysaccharide adjuvant-protein antigen conjugates, the preparation thereof and the use thereof |
| US5981564A (en) | 1998-07-01 | 1999-11-09 | Universite Laval | Water-soluble derivatives of paclitaxel, method for producing same and uses thereof |
| BR9912053A (en) | 1998-07-13 | 2001-04-03 | Univ Texas | Cancer treatment processes using therapeutic conjugates that bind to aminophospholipids |
| AUPQ014799A0 (en) | 1999-05-04 | 1999-05-27 | Access Pharmaceuticals Australia Pty Limited | Amplification of folate-mediated targeting to tumor cells using polymers |
| EP2266607A3 (en) | 1999-10-01 | 2011-04-20 | Immunogen, Inc. | Immunoconjugates for treating cancer |
| ATE349438T1 (en) | 1999-11-24 | 2007-01-15 | Immunogen Inc | CYTOTOXIC ACTIVE INGREDIENTS CONTAINING TAXANES AND THEIR THERAPEUTIC USE |
| WO2001049698A1 (en) | 1999-12-29 | 2001-07-12 | Immunogen, Inc. | Cytotoxic agents comprising modified doxorubicins and daunorubicins and their therapeutic use |
| WO2001058479A1 (en) | 2000-02-08 | 2001-08-16 | The Penn State Research Foundation | Immunotherapy using interleukin 13 receptor subunit alpha 2 |
| US6632979B2 (en) | 2000-03-16 | 2003-10-14 | Genentech, Inc. | Rodent HER2 tumor model |
| US7097840B2 (en) | 2000-03-16 | 2006-08-29 | Genentech, Inc. | Methods of treatment using anti-ErbB antibody-maytansinoid conjugates |
| AU8295301A (en) | 2000-07-24 | 2002-02-05 | Microcell Corp | Microcell electrochemical devices and assemblies, and method of making and usingthe same |
| US6333410B1 (en) | 2000-08-18 | 2001-12-25 | Immunogen, Inc. | Process for the preparation and purification of thiol-containing maytansinoids |
| US6596503B1 (en) | 2000-08-18 | 2003-07-22 | East Carolina University | Monoclonal antibody DS6, tumor-associated antigen CA6, and methods of use thereof |
| US20060062786A1 (en) | 2000-11-08 | 2006-03-23 | Human Genome Sciences, Inc. | Antibodies that immunospecifically bind to TRAIL receptors |
| CA2436092A1 (en) | 2001-01-29 | 2002-08-08 | Idec Pharmaceutical Corporation | Modified antibodies and methods of use |
| MXPA03007037A (en) | 2001-02-07 | 2003-11-18 | Beth Israel Hospital | Modified psma ligands and uses related thereto. |
| EP1389090A2 (en) | 2001-04-26 | 2004-02-18 | Board of Regents, The University of Texas System | Diagnostic imaging compositions, their methods of synthesis and use |
| WO2002092127A1 (en) | 2001-05-11 | 2002-11-21 | Board Of Regents, The University Of Texas System | Anti-cd26 monoclonal antibodies as therapy for diseases associated with cells expressing cd26 |
| EP1258255A1 (en) | 2001-05-18 | 2002-11-20 | Boehringer Ingelheim International GmbH | Conjugates of an antibody to CD44 and a maytansinoid |
| US6441163B1 (en) * | 2001-05-31 | 2002-08-27 | Immunogen, Inc. | Methods for preparation of cytotoxic conjugates of maytansinoids and cell binding agents |
| JP4619651B2 (en) | 2001-06-01 | 2011-01-26 | コーネル・リサーチ・ファンデーション・インコーポレイテッド | Modified antibodies against prostate-specific membrane antigen and uses thereof |
| TW593239B (en) | 2001-06-04 | 2004-06-21 | Kevin Dale Allen | One-step production of 1,3-propanediol from ethylene oxide and syngas with a catalyst with a phospholanoalkane ligand |
| ATE419005T1 (en) | 2001-12-11 | 2009-01-15 | Merck & Co Inc | STAPHYLOCOCCUS AUREUS EXOPOLYSACCHARIDE AND METHOD |
| US6716821B2 (en) | 2001-12-21 | 2004-04-06 | Immunogen Inc. | Cytotoxic agents bearing a reactive polyethylene glycol moiety, cytotoxic conjugates comprising polyethylene glycol linking groups, and methods of making and using the same |
| EP1467758A4 (en) | 2002-01-03 | 2007-11-14 | Smithkline Beecham Corp | Methods for preparing immunoconjugates |
| US8877901B2 (en) | 2002-12-13 | 2014-11-04 | Immunomedics, Inc. | Camptothecin-binding moiety conjugates |
| US7591994B2 (en) | 2002-12-13 | 2009-09-22 | Immunomedics, Inc. | Camptothecin-binding moiety conjugates |
| US6756397B2 (en) | 2002-04-05 | 2004-06-29 | Immunogen, Inc. | Prodrugs of CC-1065 analogs |
| US6534660B1 (en) | 2002-04-05 | 2003-03-18 | Immunogen, Inc. | CC-1065 analog synthesis |
| US7323553B2 (en) * | 2002-04-26 | 2008-01-29 | Genentech, Inc. | Non-affinity purification of proteins |
| LT1507556T (en) | 2002-05-02 | 2016-10-10 | Wyeth Holdings Llc | Calicheamicin derivative-carrier conjugates |
| US20050276812A1 (en) * | 2004-06-01 | 2005-12-15 | Genentech, Inc. | Antibody-drug conjugates and methods |
| US20090068178A1 (en) | 2002-05-08 | 2009-03-12 | Genentech, Inc. | Compositions and Methods for the Treatment of Tumor of Hematopoietic Origin |
| US6596757B1 (en) | 2002-05-14 | 2003-07-22 | Immunogen Inc. | Cytotoxic agents comprising polyethylene glycol-containing taxanes and their therapeutic use |
| EP1551875A4 (en) | 2002-06-21 | 2006-06-28 | Biogen Idec Inc | Buffered formulations for concentrating antibodies and methods of use thereof |
| US7390898B2 (en) | 2002-08-02 | 2008-06-24 | Immunogen Inc. | Cytotoxic agents containing novel potent taxanes and their therapeutic use |
| ATE499116T1 (en) | 2002-08-16 | 2011-03-15 | Immunogen Inc | HIGH REACTIVITY AND SOLUBILITY CROSS-LINKERS AND THEIR USE IN THE PRODUCTION OF CONJUGATES FOR THE TARGETED DELIVERY OF SMALL MOLECULAR DRUGS |
| AU2003273413A1 (en) | 2002-10-08 | 2004-05-04 | Fresenius Kabi Deutschland Gmbh | Pharmaceutically active oligosaccharide conjugates |
| PT1558744E (en) | 2002-10-30 | 2011-09-22 | Nuevolution As | Enzymatic encoding |
| AU2003285878B2 (en) | 2002-11-07 | 2011-04-28 | Immunogen, Inc. | Anti-CD33 antibodies and method for treatment of acute myeloid leukemia using the same |
| CN102940889A (en) | 2003-05-14 | 2013-02-27 | 伊缪诺金公司 | Drug conjugate composition |
| US8088387B2 (en) | 2003-10-10 | 2012-01-03 | Immunogen Inc. | Method of targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via a non-cleavable linker, said conjugates, and methods of making said conjugates |
| EP3851126A1 (en) * | 2003-05-20 | 2021-07-21 | ImmunoGen, Inc. | Maytansinoid-cell-binding agent conjugates |
| US7276497B2 (en) | 2003-05-20 | 2007-10-02 | Immunogen Inc. | Cytotoxic agents comprising new maytansinoids |
| AU2004259398A1 (en) | 2003-06-27 | 2005-02-03 | Amgen Fremont Inc. | Antibodies directed to the deletion mutants of epidermal growth factor receptor and uses thereof |
| WO2005002597A1 (en) | 2003-07-02 | 2005-01-13 | Polycord, Inc. | Method for delivering polymerized therapeutic agent compositions and compositions thereof |
| MXPA06005104A (en) | 2003-11-05 | 2007-01-25 | Palingen Inc | Enhanced b cell cytotoxicity of cdim binding antibody. |
| US20050175619A1 (en) * | 2004-02-05 | 2005-08-11 | Robert Duffy | Methods of producing antibody conjugates |
| US20110064754A1 (en) | 2005-03-03 | 2011-03-17 | Center For Molecular Medicine And Immunology | Immunoconjugates Comprising Poxvirus-Derived Peptides and Antibodies Against Antigen-Presenting Cells for Subunit-Based Poxvirus Vaccines |
| EP1610818A4 (en) * | 2004-03-03 | 2007-09-19 | Millennium Pharm Inc | Modified antibodies to prostate-specific membrane antigen and uses thereof |
| JP2008505853A (en) | 2004-04-13 | 2008-02-28 | クインテセンス バイオサイエンシーズ インコーポレーティッド | Non-natural ribonuclease complex as a cytotoxic agent |
| US20060073528A1 (en) | 2004-05-14 | 2006-04-06 | Jean-Michel Lecerf | Measurement methods |
| CA2564076C (en) | 2004-05-19 | 2014-02-18 | Medarex, Inc. | Chemical linkers and conjugates thereof |
| US7691962B2 (en) | 2004-05-19 | 2010-04-06 | Medarex, Inc. | Chemical linkers and conjugates thereof |
| US7313946B2 (en) * | 2004-07-15 | 2008-01-01 | Matsuo Electric Co., Ltd. | Moisture detector |
| AR052774A1 (en) | 2004-10-08 | 2007-04-04 | Wyeth Corp | IMMUNOTHERAPY FOR AUTOIMMUNE DISORDERS |
| EP1817341A2 (en) | 2004-11-29 | 2007-08-15 | Seattle Genetics, Inc. | Engineered antibodies and immunoconjugates |
| US7408030B2 (en) * | 2005-01-13 | 2008-08-05 | North Carolina State University | Purification of immunoglobulins using affinity chromatography and peptide ligands |
| US20110166319A1 (en) | 2005-02-11 | 2011-07-07 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| AU2006213662B2 (en) | 2005-02-11 | 2010-08-05 | Immunogen, Inc. | Process for preparing stable drug conjugates |
| EP1868649A4 (en) | 2005-04-15 | 2011-06-29 | Immunogen Inc | Elimination of heterogeneous or mixed cell population in tumors |
| GB2427360A (en) | 2005-06-22 | 2006-12-27 | Complex Biosystems Gmbh | Aliphatic prodrug linker |
| CN101262890B (en) | 2005-07-15 | 2015-08-26 | 安吉奥化学公司 | Aprotinin polypeptides is as the purposes of carrier in drug conjugate |
| CA2615122A1 (en) | 2005-08-03 | 2007-02-15 | Immunogen, Inc. | Immunoconjugate formulations |
| ES2533992T3 (en) * | 2005-08-24 | 2015-04-16 | Immunogen, Inc. | Procedure for preparing conjugates of maitansinoid antibody |
| CN101443044A (en) | 2005-09-22 | 2009-05-27 | 哈达斯特医学研究服务与开发有限公司 | Dextran and arabinogalactan conjugates of therapeutically active compounds |
| BRPI0617546A2 (en) | 2005-09-26 | 2011-07-26 | Medarex Inc | drug-antibody conjugate, pharmaceutical formulation, method for killing a tumor cell, method for retarding or arresting tumor growth in a mammalian subject and compound |
| EP2298308B1 (en) | 2005-11-14 | 2013-01-16 | University Of Southern California | Integrin-binding small molecules |
| US7964415B2 (en) | 2006-04-26 | 2011-06-21 | Cardiogenics Inc. | Stable water-soluble polyethylenimine conjugates and methods of use thereof |
| US7307016B1 (en) * | 2006-06-07 | 2007-12-11 | Grace Semiconductor Manufacturing Corporation | Method of processing metal surface in dual damascene manufacturing |
| KR101528939B1 (en) | 2006-07-18 | 2015-06-15 | 사노피 | Antagonist antibody against epha2 for the treatment of cancer |
| US20080213349A1 (en) | 2006-09-11 | 2008-09-04 | Deepak Ramesh Thakker | Liposome Complexes Containing Pharmaceutical Agents and Methods |
| WO2008057683A2 (en) | 2006-10-03 | 2008-05-15 | Novo Nordisk A/S | Methods for the purification of polypeptide conjugates |
| SI2019104T1 (en) | 2007-07-19 | 2013-12-31 | Sanofi | Cytotoxic agents comprising new tomaymycin derivatives and their therapeutic use |
| EP2276506A4 (en) | 2008-04-30 | 2014-05-07 | Immunogen Inc | Potent conjugates and hydrophilic linkers |
| KR20230003298A (en) | 2008-04-30 | 2023-01-05 | 이뮤노젠 아이엔씨 | Cross-linkers and their uses |
| GB0811743D0 (en) | 2008-06-26 | 2008-07-30 | Hemosol Biopharma Inc | Composition |
| TW201021852A (en) | 2008-11-17 | 2010-06-16 | Enzon Pharmaceuticals Inc | Releasable fusogenic lipids for nucleic acids delivery systems |
| IL271761B (en) | 2009-02-05 | 2022-09-01 | Immunogen Inc | (12as)-8-methoxy-9-benzyloxy-11,12,12a,13-tetrahydro-6h-indolo[2,1-c][1,4]benzodiazepin-6-one, 4-benzyloxy-5-methoxy-2-nitrobenzoic acid and processes for preparing the same |
| KR102444399B1 (en) | 2009-06-03 | 2022-09-16 | 이뮤노젠 아이엔씨 | Methods for preparing a maytansinoid |
| FR2947269B1 (en) | 2009-06-29 | 2013-01-18 | Sanofi Aventis | NEW ANTICANCER COMPOUNDS |
| AR078470A1 (en) | 2009-10-02 | 2011-11-09 | Sanofi Aventis | ANTIBODIES THAT SPECIFICALLY JOIN THE EPHA2 RECEIVER |
| SG10201501342UA (en) | 2010-02-24 | 2015-04-29 | Immunogen Inc | Folate receptor 1 antibodies and immunoconjugates and uses thereof |
| KR20110103182A (en) | 2010-03-12 | 2011-09-20 | 삼성전자주식회사 | 3 dimensional image display device |
| US20120149732A1 (en) | 2010-12-14 | 2012-06-14 | Alexander Chucholowski | Multifunctional linkers and methods for the use thereof |
| RU2748733C2 (en) | 2011-02-15 | 2021-05-31 | Иммуноджен, Инк. | Cytotoxic benzodiazepine derivatives |
| MY171008A (en) | 2011-03-29 | 2019-09-23 | Immunogen Inc | Preparation of maytansinoid antibody conjugates by a one-step process |
| UA116524C2 (en) | 2011-03-29 | 2018-04-10 | Іммуноджен, Інк. | Preparation of maytansinoid antibody conjugates by a one-step process |
| MX2013011201A (en) | 2011-03-29 | 2013-12-16 | Immunogen Inc | Process for manufacturing conjugates of improved homogeneity. |
| SG193514A1 (en) | 2011-04-01 | 2013-10-30 | Immunogen Inc | Methods for increasing efficacy of folr1 cancer therapy |
| US20130071482A1 (en) | 2011-09-20 | 2013-03-21 | The University Of Kentucky Research Foundation | Block copolymer cross-linked nanoassemblies as modular delivery vehicles |
| AU2012352210A1 (en) | 2011-12-13 | 2014-07-24 | Immunogen, Inc. | Use of N-hydroxysuccinimide to improve conjugate stability |
| US20150307596A1 (en) | 2012-10-04 | 2015-10-29 | Immunogen, Inc. | Use of an ion exchange membrane to remove impurities from cell-binding agent cytotoxic agent conjugates |
| RU2661083C2 (en) | 2012-10-04 | 2018-07-11 | Иммуноджен, Инк. | Use of pvdf membrane to purify cell-binding agent - cytotoxic agent conjugates |
| WO2014055842A1 (en) | 2012-10-04 | 2014-04-10 | Immunogen, Inc. | Process for preparing stable antibody maytansinoid conjugates |
| TR201905612T4 (en) | 2012-12-05 | 2019-05-21 | Univ Heidelberg Ruprecht Karls | Conjugates of proteins and polyvalent cell-penetrating peptides and their uses. |
-
2006
- 2006-08-14 ES ES11004940.0T patent/ES2533992T3/en active Active
- 2006-08-14 CN CN2006800342426A patent/CN101267841B/en active Active
- 2006-08-14 PT PT06801436T patent/PT1928503E/en unknown
- 2006-08-14 RS RS20120425A patent/RS52470B/en unknown
- 2006-08-14 EP EP19156303.0A patent/EP3539572A1/en active Pending
- 2006-08-14 DK DK06801436.4T patent/DK1928503T3/en active
- 2006-08-14 EA EA200800657A patent/EA013327B1/en active IP Right Revival
- 2006-08-14 JP JP2008527970A patent/JP5350792B2/en active Active
- 2006-08-14 EP EP20130178039 patent/EP2662096A1/en not_active Withdrawn
- 2006-08-14 NZ NZ609752A patent/NZ609752A/en unknown
- 2006-08-14 US US11/503,781 patent/US7811572B2/en active Active
- 2006-08-14 IL IL282138A patent/IL282138B2/en unknown
- 2006-08-14 CA CA2620343A patent/CA2620343C/en active Active
- 2006-08-14 MX MX2008002597A patent/MX2008002597A/en active IP Right Grant
- 2006-08-14 CA CA3190867A patent/CA3190867A1/en active Pending
- 2006-08-14 NZ NZ595430A patent/NZ595430A/en unknown
- 2006-08-14 CN CN201210308200.4A patent/CN102989000B/en active Active
- 2006-08-14 EP EP06801436A patent/EP1928503B1/en active Active
- 2006-08-14 CA CA2893252A patent/CA2893252C/en active Active
- 2006-08-14 NZ NZ716641A patent/NZ716641A/en unknown
- 2006-08-14 KR KR1020087004084A patent/KR101301011B1/en active IP Right Grant
- 2006-08-14 BR BRPI0615049-7A patent/BRPI0615049B1/en active IP Right Grant
- 2006-08-14 CA CA3000520A patent/CA3000520C/en active Active
- 2006-08-14 ES ES06801436T patent/ES2390826T3/en active Active
- 2006-08-14 EP EP11004940.0A patent/EP2399609B1/en active Active
- 2006-08-14 PL PL06801436T patent/PL1928503T3/en unknown
- 2006-08-14 AU AU2006283726A patent/AU2006283726C1/en active Active
- 2006-08-14 SI SI200631435T patent/SI1928503T1/en unknown
- 2006-08-14 NZ NZ565964A patent/NZ565964A/en unknown
- 2006-08-14 IL IL305084A patent/IL305084A/en unknown
- 2006-08-14 WO PCT/US2006/031653 patent/WO2007024536A2/en active Application Filing
- 2006-08-14 CA CA2794554A patent/CA2794554C/en active Active
-
2008
- 2008-02-12 IL IL189461A patent/IL189461A/en active IP Right Grant
- 2008-02-15 ZA ZA2008/01564A patent/ZA200801564B/en unknown
- 2008-02-18 CR CR9742A patent/CR9742A/en unknown
- 2008-02-22 EC EC2008008212A patent/ECSP088212A/en unknown
- 2008-11-06 HK HK08112150.3A patent/HK1120407A1/en unknown
-
2010
- 2010-10-08 US US12/901,039 patent/US8383122B2/en active Active
-
2012
- 2012-06-27 HK HK12106280.2A patent/HK1165329A1/en unknown
- 2012-07-08 IL IL220816A patent/IL220816A/en active IP Right Grant
- 2012-09-11 JP JP2012199467A patent/JP5738248B2/en active Active
- 2012-10-04 HR HRP20120794TT patent/HRP20120794T1/en unknown
- 2012-10-17 CY CY20121100972T patent/CY1113206T1/en unknown
-
2013
- 2013-02-25 US US13/776,097 patent/US8933205B2/en active Active
- 2013-06-17 IL IL226985A patent/IL226985A/en active IP Right Grant
-
2014
- 2014-05-12 JP JP2014098796A patent/JP6028001B2/en active Active
-
2015
- 2015-01-05 US US14/589,541 patent/US9789204B2/en active Active
- 2015-07-03 CR CR20150350A patent/CR20150350A/en unknown
-
2016
- 2016-09-27 IL IL248076A patent/IL248076B/en active IP Right Grant
-
2018
- 2018-04-27 US US15/964,418 patent/US11471536B2/en active Active
-
2022
- 2022-09-08 US US17/940,297 patent/US20230158168A1/en active Pending
Non-Patent Citations (5)
| Title |
|---|
| "Antibodies: A Laboratory Manual", 1988, CSH PRESS |
| "Immunobiology", 2001, GARLAND PUBLISHING |
| HASKARD; ARCHER, J. IMMUNOL. METHODS, vol. 74, no. 2, 1984, pages 361 - 67 |
| KOHLER; MILSTEIN, EUR. J. IMMUNOL., vol. 5, 1976, pages 511 - 519 |
| REITER ET AL., PROTEIN ENGINEERING, vol. 7, 1994, pages 697 - 704 |
Cited By (68)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9789204B2 (en) | 2005-08-24 | 2017-10-17 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| US11471536B2 (en) | 2005-08-24 | 2022-10-18 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| US8933205B2 (en) | 2005-08-24 | 2015-01-13 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| US11046762B2 (en) | 2008-04-30 | 2021-06-29 | Immunogen, Inc. | Cross-linkers and their uses |
| CN104524592A (en) * | 2008-04-30 | 2015-04-22 | 伊缪诺金公司 | Cross-linkers and their uses |
| US10494431B2 (en) | 2008-04-30 | 2019-12-03 | Immunogen, Inc. | Cross-linkers and their uses |
| EP2424875A4 (en) * | 2009-04-29 | 2012-10-31 | Bio Rad Laboratories | Purification of immunoconjugates |
| US8481694B2 (en) | 2009-04-29 | 2013-07-09 | Bio-Rad Laboratories, Inc. | Purification of immunoconjugates |
| EP2424875A1 (en) * | 2009-04-29 | 2012-03-07 | Bio-Rad Laboratories, Inc. | Purification of immunoconjugates |
| WO2010126979A1 (en) | 2009-04-29 | 2010-11-04 | Bio-Rad Laboratories, Inc. | Purification of immunoconjugates |
| EP2424875B1 (en) | 2009-04-29 | 2016-01-06 | Bio-Rad Laboratories, Inc. | Purification of immunoconjugates |
| EP2437790A4 (en) * | 2009-06-03 | 2015-06-24 | Immunogen Inc | Conjugation methods |
| KR20120088549A (en) * | 2009-06-03 | 2012-08-08 | 이뮤노젠 아이엔씨 | Conjugation methods |
| US11498979B2 (en) | 2009-06-03 | 2022-11-15 | Immunogen, Inc. | Methods for preparing a purified maytansinoid conjugate in a solution |
| US9771432B2 (en) | 2009-06-03 | 2017-09-26 | Immunogen, Inc. | Conjugation methods |
| KR102139019B1 (en) * | 2009-06-03 | 2020-07-28 | 이뮤노젠 아이엔씨 | Conjugation methods |
| US10233257B2 (en) | 2009-06-03 | 2019-03-19 | Immunogen, Inc. | Methods for preparing antibody-drug conjugates |
| US9376500B2 (en) | 2009-06-03 | 2016-06-28 | Immunogen, Inc. | Conjugation methods |
| KR20190015590A (en) * | 2009-06-03 | 2019-02-13 | 이뮤노젠 아이엔씨 | Conjugation methods |
| KR101947176B1 (en) * | 2009-06-03 | 2019-02-12 | 이뮤노젠 아이엔씨 | Conjugation methods |
| US10815309B2 (en) | 2009-06-03 | 2020-10-27 | Immunogen, Inc. | Methods for preparing antibody-drug conjugates |
| EP3545977A1 (en) * | 2011-03-29 | 2019-10-02 | ImmunoGen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| AU2012236398B2 (en) * | 2011-03-29 | 2016-02-11 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| US11744900B2 (en) | 2011-03-29 | 2023-09-05 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| US9914748B2 (en) | 2011-03-29 | 2018-03-13 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| AU2016202832B2 (en) * | 2011-03-29 | 2018-05-10 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| US11090390B2 (en) | 2011-03-29 | 2021-08-17 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| WO2012135517A3 (en) * | 2011-03-29 | 2013-02-21 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| US8795673B2 (en) | 2011-03-29 | 2014-08-05 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| EA033468B1 (en) * | 2011-03-29 | 2019-10-31 | Immunogen Inc | Processes for preparing an "antibody-maytansinoid" conjugate |
| US9428543B2 (en) | 2011-03-29 | 2016-08-30 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| US10435432B2 (en) | 2011-03-29 | 2019-10-08 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| WO2012143496A2 (en) | 2011-04-21 | 2012-10-26 | Bayer Intellectual Property Gmbh | Novel binder-drug conjugates (adcs) and their use |
| WO2012143499A2 (en) | 2011-04-21 | 2012-10-26 | Bayer Intellectual Property Gmbh | Novel binder-drug conjugates (adcs) and their use |
| WO2014055842A1 (en) * | 2012-10-04 | 2014-04-10 | Immunogen, Inc. | Process for preparing stable antibody maytansinoid conjugates |
| EP2903450A4 (en) * | 2012-10-04 | 2016-05-11 | Immunogen Inc | Use of an ion exchange membrane to remove impurities from cell-binding agent cytotoxic agent conjugates |
| US10035817B2 (en) | 2012-10-04 | 2018-07-31 | Immunogen, Inc. | Method of purifying cell-binding agent-cytotoxic agent conjugates with a PVDF membrane |
| US10918735B2 (en) | 2012-12-04 | 2021-02-16 | Massachusetts Institute Of Technology | Substituted pyrazino[1′,2′:1,5]pyrrolo[2,3-b]indole-1,4-diones for cancer treatment |
| EP3699200A1 (en) | 2013-07-15 | 2020-08-26 | Cell Signaling Technology, Inc. | Anti-mucin 1 binding agents and uses thereof |
| WO2015009740A2 (en) | 2013-07-15 | 2015-01-22 | Cell Signaling Technology, Inc. | Anti-mucin 1 binding agents and uses thereof |
| US10022453B2 (en) | 2013-12-23 | 2018-07-17 | Bayer Pharma Aktiengesellschaft | Antibody drug conjugates (ADCs) with kinesin spindel protein (KSP) |
| US11123439B2 (en) | 2015-06-22 | 2021-09-21 | Bayer Pharma Aktiengesellschaft | Antibody drug conjugates (ADCS) and antibody prodrug conjugates (APDCS) with enzymatically cleavable groups |
| WO2016207089A1 (en) | 2015-06-22 | 2016-12-29 | Bayer Pharma Aktiengesellschaft | Antibody drug conjugates (adcs) and antibody prodrug conjugates (apdcs) with enzymatically cleavable groups |
| WO2017060322A2 (en) | 2015-10-10 | 2017-04-13 | Bayer Pharma Aktiengesellschaft | Ptefb-inhibitor-adc |
| US11160871B2 (en) | 2015-10-28 | 2021-11-02 | Tarveda Therapeutics, Inc. | SSTR-targeted conjugates and particles and formulations thereof |
| US11685714B2 (en) | 2016-03-24 | 2023-06-27 | Bayer Pharma Aktiengesellschaft | Prodrugs of cytotoxic active agents having enzymatically cleavable groups |
| WO2017162663A1 (en) | 2016-03-24 | 2017-09-28 | Bayer Pharma Aktiengesellschaft | Prodrugs of cytotoxic active agents having enzymatically cleavable groups |
| US10918627B2 (en) | 2016-05-11 | 2021-02-16 | Massachusetts Institute Of Technology | Convergent and enantioselective total synthesis of Communesin analogs |
| US11001636B2 (en) | 2016-06-15 | 2021-05-11 | Bayer Pharma Aktiengesellschaft | Specific antibody-drug-conjugates (ADCs) with KSP inhibitors and anti-CD123-antibodies |
| US11643469B2 (en) | 2016-06-15 | 2023-05-09 | Bayer Pharma Aktiengesellschaft | Specific antibody-drug-conjugates (ADCs) with KSP inhibitors and anti-CD123-antibodies |
| WO2018114798A1 (en) | 2016-12-21 | 2018-06-28 | Bayer Aktiengesellschaft | Prodrugs of cytotoxic active agents having enzymatically cleavable groups |
| US11433140B2 (en) | 2016-12-21 | 2022-09-06 | Bayer Pharma Aktiengesellschaft | Specific antibody drug conjugates (ADCs) having KSP inhibitors |
| WO2018114578A1 (en) | 2016-12-21 | 2018-06-28 | Bayer Pharma Aktiengesellschaft | Antibody drug conjugates (adcs) having enzymatically cleavable groups |
| US11478554B2 (en) | 2016-12-21 | 2022-10-25 | Bayer Pharma Aktiengesellschaft | Antibody drug conjugates (ADCS) having enzymatically cleavable groups |
| US11660351B2 (en) | 2016-12-21 | 2023-05-30 | Bayer Aktiengesellschaft | Antibody drug conjugates (ADCs) having enzymatically cleavable groups |
| US20180311375A1 (en) * | 2017-04-27 | 2018-11-01 | Cadila Healthcare Limited | Process of preparing antibody-drug conjugate |
| US11932650B2 (en) | 2017-05-11 | 2024-03-19 | Massachusetts Institute Of Technology | Potent agelastatin derivatives as modulators for cancer invasion and metastasis |
| US10640508B2 (en) | 2017-10-13 | 2020-05-05 | Massachusetts Institute Of Technology | Diazene directed modular synthesis of compounds with quaternary carbon centers |
| WO2019105835A1 (en) | 2017-11-29 | 2019-06-06 | Bayer Consumer Care Ag | Combinations of copanlisib and anetumab ravtansine |
| US11274121B2 (en) | 2018-01-12 | 2022-03-15 | Immunogen, Inc. | Methods for antibody drug conjugation, purification, and formulation |
| KR20210038904A (en) | 2018-07-25 | 2021-04-08 | 다이이찌 산쿄 가부시키가이샤 | Effective method for preparing antibody-drug conjugates |
| WO2020022363A1 (en) | 2018-07-25 | 2020-01-30 | 第一三共株式会社 | Effective method for manufacturing antibody-drug conjugate |
| WO2020084115A1 (en) | 2018-10-25 | 2020-04-30 | Pharma Mar, S.A. | Antibody drug conjugates comprising ecteinascidin derivatives |
| WO2020234114A1 (en) | 2019-05-21 | 2020-11-26 | Bayer Aktiengesellschaft | A novel stable high concentration formulation for anetumab ravtansine |
| US11535634B2 (en) | 2019-06-05 | 2022-12-27 | Massachusetts Institute Of Technology | Compounds, conjugates, and compositions of epipolythiodiketopiperazines and polythiodiketopiperazines and uses thereof |
| US11634508B2 (en) | 2019-07-10 | 2023-04-25 | Cybrexa 2, Inc. | Peptide conjugates of cytotoxins as therapeutics |
| WO2021043951A1 (en) | 2019-09-05 | 2021-03-11 | Pharma Mar, S.A. | Drug antibody conjugates |
| WO2021214126A1 (en) | 2020-04-21 | 2021-10-28 | Pharma Mar, S.A. | Drug antibody conjugates |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11471536B2 (en) | Process for preparing purified drug conjugates | |
| US20160045616A1 (en) | Process for preparing purified drug conjugates | |
| EP1853322A2 (en) | Process for preparing maytansinoid antibody conjugates | |
| AU2017218969B2 (en) | Process for preparing maytansinoid antibody conjugates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680034242.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 189461 Country of ref document: IL Ref document number: 2006283726 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 565964 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2008-009742 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087004084 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 885/CHENP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/002597 Country of ref document: MX Ref document number: 2008527970 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 2620343 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08019768 Country of ref document: CO Ref document number: 08019768B Country of ref document: CO |
|
| ENP | Entry into the national phase |
Ref document number: 2006283726 Country of ref document: AU Date of ref document: 20060814 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006801436 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200800657 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: PI0615049 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080221 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 220816 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2012/0425 Country of ref document: RS |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2015-000350 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 248076 Country of ref document: IL |