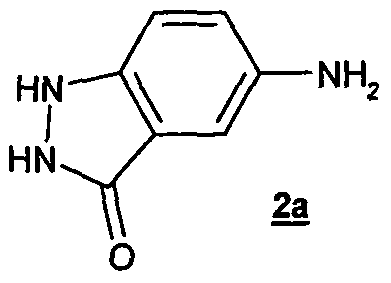
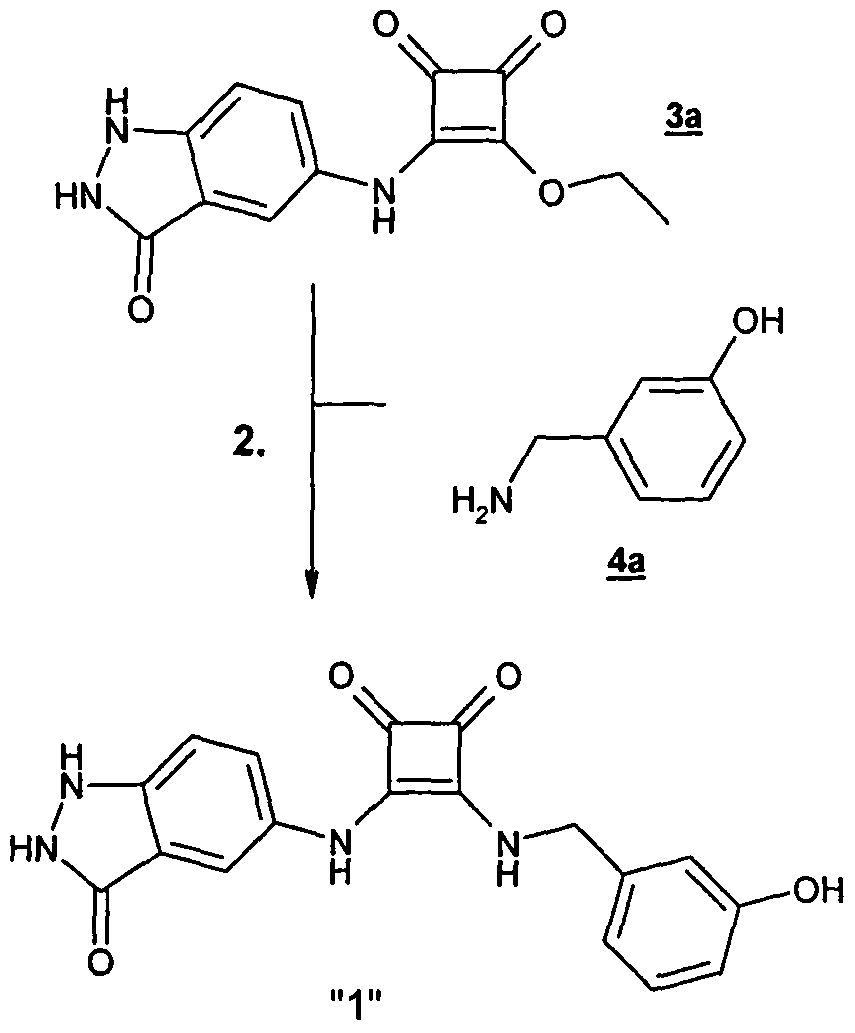
3-Oxo-indazol-quadratsäurederivate
HINTERGRUND DER ERFINDUNG
5 Die vorliegende Erfindung betrifft Verbindungen und die Verwendung von
Verbindungen, bei denen die Hemmung, Regulierung und/oder Modulation der Signaltransduktion von Kinasen, insbesondere der Tyrosinkinasen und/oder Serin/Threonin-Kinasen eine Rolle spielt, ferner pharmazeutische
.Q Zusammensetzungen, die diese Verbindungen enthalten, sowie die Verwendung der Verbindungen zur Behandlung kinasebedingter Krankheiten.
Die vorliegende Erfindung betrifft Verbindungen, bei denen die Hemmung,
15 Regulierung und/oder Modulation insbesondere der zellvolumenregulierten humanen Kinase h-sgk (human serum and glucocorticoid dependent kinase oder SGK), sowie der CHK1- und CHK2 - Kinase, eine Rolle spielt, ferner pharmazeutische Zusammensetzungen, die diese Verbindungen 20 enthalten, sowie die Verwendung der Verbindungen zur Behandlung CHK1-, CHK2- und SGK-bedingter Krankheiten.
Zellzyklus-Kontrollpunkte sind Regulationswege, die die Reihenfolge und je den Zeitpunkt von Zellzyklusübergängen steuern. Sie gewährleisten, daß wichtige Ereignisse, wie DNA-Replikation und Chromosomensegregation, mit hoher Zuverlässigkeit abgeschlossen werden. Die Steuerung dieser Zellzyklus-Kontrollpunkte ist eine wichtige Determinante der Art und
Weise, wie Tumorzellen auf viele Chemotherapien und Bestrahlung ant-
30 worten. Viele effiziente Krebstherapien wirken, indem sie eine DNA- Schädigung hervorrufen; Resistenz gegen diese Mittel bleibt jedoch eine erhebliche Einschränkung bei der Behandlung von Krebs. Es gibt verschiedene Mechanismen der Arzneistoffresistenz; einen wichtigen führt
35 man auf die Verhinderung der Zellzyklus-Progression aufgrund der
Steuerung der kritischen Aktivierung eines Kontrollpunkt-Weges zurück,
der den Zellzyklus arretiert, um Zeit für die Reparatur bereitzustellen, und die Transkription von Genen induziert, so daß die Reparatur erleichtert und sofortiger Zelltod verhindert wird.
Im Zellzyklus gibt es zwei dieser Kontrollpunkte - den G1/S-Kontrollpunkt, 5 der durch p53 gesteuert wird, und den G2/M-Kontrollpunkt, der durch die Ser/Thr-Kinase Checkpoint-Kinase 1 (CHK1) überwacht wird. Gelänge es, Kontrollpunkt-Arretierungen, beispielsweise am G2-Kontroll- punkt, aufzuheben, ließe sich möglicherweise synergistisch der durch
10 DNA-Schädigung induzierte Tumorzelltod verbessern und die Resistenz umgehen. (Shyjan et al., U.S.-Patent 6,723,498 (2004)). Humane CHK1 spielt eine Rolle bei der Steuerung der Zellzyklus-Arretierung, indem sie die Phosphatase cdc25 an Serin 216 phosphoryliert, was möglicherweise
«lg dazu beiträgt, die Aktivierung von cdc2/Cyclin B zu verhindern und die Mitose einzuleiten. (Sanchez et al., Science, 277:1497 (1997)). Daher sollte die Hemmung von CHK1 die Wirkung DNA-schädigender Substanzen verstärken, indem die Mitose eingeleitet wird, bevor die DNA- Reparatur abgeschlossen ist, und dadurch den Tumorzelltod hervorrufen.
20
Ein Ansatz für das Design von Chemosensibilisatoren, die den G2/M-
Kontrollpunkt aufheben, besteht darin, Inhibitoren der regulatorischen Schlüssel-G2/M-Kinase CHK1 zu entwickeln. In einer Reihe von Konzeptüberprüfungsstudien wurde gezeigt, daß dieser Ansatz funktioniert 25 (Koniaras et al., Oncogene, 2001, 20:7453; Luo et al., Neoplasie, 2001 , 3:411 ; Busby et al., Cancer Res., 2000, 60:2108; Jackson et al., Cancer Res., 2000, 60:566).
3Q Eine weitere essentielle Checkpoint Kinase, die eine kritische Rolle bei der p53-abhängigen Apoptosis spielt, ist CHK2 zu nennen. Die Inhibierung von CHK2 kann normales empfindliches Gewebe gegen chemotherapeutische Agenzien schützen (B. -B S. Zhou et al., Progress in Cell Cycle Research,
Vol. 5, 413-421 , 2003). 35
Für Verbindungen der Formel I kann gezeigt werden, daß sie die Checkpoint-Kinase-Aktivität hemmen. Für Inhibitoren der Checkpoint- Kinase kann gezeigt werden, daß sie es den Zellen ermöglichen, unangebracht zur Metaphase der Mitose voranzuschreiten, was zur Apoptose betroffener Zellen führt, und deshalb antiproliferative Wirkungen besitzen. Die Verbindungen der Formel I können zur Behandlung von neoplastischer Erkrankung verwendet werden können. Die Verbindungen der Formel I und ihre Salze können gegen neoplastische Erkrankungen,
10 wie Karzinom des Hirns, der Brust, der Eierstöcke, der Lunge, des
Dickdarms, der Prostata, der Haut oder anderer Gewebe sowie gegen Leukämien und Lymphome, Tumoren des zentralen und peripheren Nervensystems und andere Tumortypen, wie Melanom, Sarkom,
^c Fibrosarkom und Osteosarkom verwendet werden. Die Verbindungen der Formel I sind auch zur Behandlung anderer proliferativer Erkrankungen geeignet. Die Verbindungen der Formel I können auch in Kombination mit einem breiten Spektrum von DNA-schädigenden Mitteln verwendet werden, können aber auch als einzelne Substanz verwendet werden. 20
Die vorliegende Erfindung betrifft daher die Verwendung der Verbindungen der Formel I zur Behandlung von Krankheiten oder Zuständen, bei denen eine Hemmung der CHK1- und/oder CHK2 - Aktivität vorteilhaft ist. 25
Wie CHK1 und CHK2 gehört SGK zu den Serin/Threonin-Kinasen.
Die vorliegende Erfindung betrifft die Verwendung der Verbindungen der 3Q Formel I, wobei insbesondere die Hemmung, Regulierung und/oder
Modulation der Signaltransduktion der zellvolumenregulierten humanen Kinase h-sgk (human serum and glucocorticoid dependent kinase oder SGK) eine Rolle spielt, zur Behandlung SGK-bedingter Krankheiten.
35
Die SGK mit den Isoformen SGK-1 , SGK-2 und SGK-3 sind eine
Serin/Threonin-Proteinkinase Familie (WO 02/17893).
Die erfindungsgemäßen Verbindungen sind Inhibitoren der SGK-1. Ferner können sie Inhibitoren der SGK-2 und/oder SGK-3 sein.
Die vorliegende Erfindung betrifft somit die Verwendung der Verbindungen der Formel I1 die die Signaltransduktion der SGK hemmen, regulieren und/oder modulieren, Zusammensetzungen, die diese Verbindungen enthalten, sowie Verfahren zu ihrer Verwendung zur Behandlung von SGK-bedingten Krankheiten und Leiden wie Diabetes (z.B. Diabetes mellitus, diabetische Nephropathie, diabetische Neuropathie, diabetische Angiopathie und Mikroangiopathie), Fettsucht, metabolisches Syndrom (Dyslipidämie), systemische und pulmonale Hypertonie, Herzkreislauferkrankungen (z.B. kardiale Fibrosen nach Myokardinfarkt, Herzhypertrophie und Herzinsuffizienz, Arteriosklerose) und Nierenerkrankungen (z.B. Glomerulosklerose, Nephrosklerose, Nephritis, Nephropathie,
Störung der Elektrolytausscheidung), allgemein bei jeglicher Art von
Fibrosen und entzündlichen Prozessen (z.B. Leberzirrhose, Lungenfibrose, fibrosierende Pankreatitis, Rheumatismus und Arthrosen, Morbus Crohn, chronische Bronchitis, Strahlenfibrose, Sklerodermitis, zystische Fibrose, Narbenbildung, Morbus Alzheimer). Die erfindungsgemäßen Verbindungen können auch das Wachstum von Tumorzellen und Tumormetastasen hemmen und sind deshalb für die Tumortherapie geeignet. Die erfindungsgemäßen Verbindungen finden weiterhin Verwendung zur Behandlung von Koagulopathien, wie z.B. Dysfibrinogenämie, Hypopro- konvertinämie, Hämophilie B, Stuart-Prower-Defekt, Prothrombin- Komplex-Mangel, Verbrauchskoagulopathie, Hyperfibrinolyse, Immuno- koagulopathie oder komplexer Koagulopathien, wie auch bei neuronaler
Erregbarkeit, z.B. Epilepsie. Die erfindungsgemäßen Verbindungen können auch bei der Behandlung eines Glaukoms oder Katarakt therapeutisch eingesetzt werden.
Die erfindungsgemäßen Verbindungen finden ferner Verwendung bei der Behandlung bakterieller Infektionen sowie in einer antiinfektiösen Therapie. Die erfindungsgemäßen Verbindungen können auch zur Steigerung der Lernfähigkeit und Aufmerksamkeit therapeutisch eingesetzt werden.
Darüberhinaus wirken die erfindungsgemäßen Verbindungen der Zellalterung und Stress entgegen und steigern somit die Lebenserwartung und die Fitness im Alter.
Die erfindungsgemäßen Verbindungen finden ferner Verwendung bei der Behandlung von Tinitus.
Die Identifikation von kleinen Verbindungen, die die Signaltransduktion der SGK hemmen, regulieren und/oder modulieren, ist daher wünschenswert und ein Ziel der vorliegenden Erfindung.
Es wurde gefunden, daß die erfindungsgemäßen Verbindungen und ihre Salze bei guter Verträglichkeit sehr wertvolle pharmakologische
Eigenschaften besitzen.
So zeigen sie insbesondere inhibierende Eigenschaften der SGK.
Gegenstand der vorliegenden Erfindung sind deshalb erfindungsgemäße Verbindungen als Arzneimittel und/oder Arzneimittelwirkstoffe bei der Behandlung und/oder Prophylaxe der genannten Erkrankungen und die
Verwendung von erfindungsgemäßen Verbindungen zur Herstellung eines Pharmazeutikums für die Behandlung und/oder Prophylaxe der genannten Erkrankungen wie auch ein Verfahren zur Behandlung der genannten Erkrankungen umfassend die Verabreichung eines oder mehrerer erfindungsgemäßer Verbindungen an einen Patienten mit Bedarf an einer derartigen Verabreichung.
Der Wirt oder Patient kann jeglicher Säugerspezies angehören, z. B. einer
Primatenspezies, besonders Menschen; Nagetieren, einschließlich
Mäusen, Ratten und Hamstern; Kaninchen; Pferden, Rindern, Hunden, Katzen usw. Tiermodelle sind für experimentelle Untersuchungen von Interesse, wobei sie ein Modell zur Behandlung einer Krankheit des
Menschen zur Verfügung stellen.
Zur Identifizierung eines Signalübertragungswegs und zum Nachweis von Wechselwirkungen zwischen verschiedenen Signalübertragungswegen wurden von verschiedenen Wissenschaftlern geeignete Modelle oder Modellsysteme entwickelt, z.B. Zellkulturmodelle (z.B. Khwaja et al.,
EMBO, 1997, 16, 2783-93) und Modelle transgener Tiere (z.B. White et al., Oncogene, 2001, 20, 7064-7072). Zur Bestimmung bestimmter Stufen in der Signalübertragungskaskade können wechselwirkende Verbindungen genutzt werden, um das Signal zu modulieren (z.B. Stephens et al., Biochemical J., 2000, 351 , 95-105). Die erfindungsgemäßen Verbindungen können auch als Reagenzien zur Testung kinaseabhängiger Signalübertragungswege in Tieren und/oder Zellkulturmodellen oder in den in dieser Anmeldung genannten klinischen Erkrankungen verwendet werden.
Die Messung der Kinaseaktivität ist eine dem Fachmann wohlbekannte Technik. Generische Testsysteme zur Bestimmung der Kinaseaktivität mit Substraten, z.B. Histon (z.B. Alessi et al., FEBS Lett. 1996, 399, 3, Seiten 333-338) oder dem basischen Myelinprotein sind in der Literatur beschrieben (z.B. Campos-Gonzälez, R. und Glenney, Jr., J. R. 1992, J. Biol. Chem. 267, Seite 14535).
Zur Identifikation von Kinase-Inhibitoren stehen verschiedene Assay- Systeme zur Verfügung. Beim Scintillation-Proximity-Assay (Sorg et al., J. of. Biomolecular Screening, 2002, 7, 11-19) und dem FlashPlate-Assay wird die radioaktive Phosphorylierung eines Proteins oder Peptids als
Substrat mit γATP gemessen. Bei Vorliegen einer inhibitorischen Verbin-
dung ist kein oder ein vermindertes radioaktives Signal nachweisbar. Ferner sind die Homogeneous Time-resolved Fluorescence Resonance Energy Transfer- (HTR-FRET-) und Fluoreszenzpolarisations- (FP-) Technologien als Assay-Verfahren nützlich (SiIIs et al., J. of Biomolecular Screening, 2002, 191-214).
Andere nicht radioaktive ELISA-Assay-Verfahren verwenden spezifische Phospho-Antikörper (Phospho-AK). Der Phospho-AK bindet nur das phosphorylierte Substrat. Diese Bindung ist mit einem zweiten Peroxidase- konjugierten Anti-Schaf-Antikörper durch Chemolumineszenz nachweisbar (Ross et alM Biochem. J., 2002, 366, 977-981).
STAND DER TECHNIK
In der US 5,466,712 und US 5,605,909 sind andere N-Aryl- und N-
Heteroaryl-1 ,2-diaminocyclobuten-3,4-dione als Relaxantien der glatten
Muskulatur beschrieben.
Quadratsäure-amide als Stabilisatoren von synthetischen Harzen sind in
US 4,170,588 und DE 1669798 beschrieben.
In der WO 02/083624, WO 02/076926, US 2003/0204085 und WO
03/080053 sind 3,4-substituierte Cyclobuten-1 ,2-dione als CXC-Chemokin-
Rezeptorliganden zur Behandlung von Chemokin-induzierten Krankheiten, wie Entzündungen oder Krebs, beschrieben.
Andere 3,4-substituierte Cyclobuten-1,2-dione zur Behandlung von Chemokin-(insbes. IL-8) induzierten Krankheiten, kennt man als IL-8 Rezeptorantagonisten aus WO 01/92202 und WO 01/64208.
In der WO 00/62781 ist die Verwendung von Arzneimitteln enthaltend Hemmstoffe der zellvolumenregulierten humanen Kinase H-SGK beschrieben.
Die Verwendung von Kinase-Inhibitoren in der antiinfektiösen Therapie ist von C.Doerig in Cell. Mol. Biol. Lett. Vol.8, No. 2A, 2003, 524-525 beschrieben.
Die Verwendung von Kinase-Inhibitoren bei Fettsucht ist von N.Perrotti in J. Biol. Chem. 2001 , März 23; 276(12):9406-9412 beschrieben.
In nachstehenden Literaturstellen wird die Verwendung von SGK- Hemmem bei der Behandlung von Krankheiten nahegelegt und/oder beschrieben:
1: Chung EJ, Sung YK, Farooq M, Kim Y, Im S, Tak WY, Hwang YJ, Kim Yl1 Han HS, Kim JC, Kim MK. Gene expression profile analysis in human hepatocellular Carcinoma by cDNA microarray. Mol CeIIs. 2002; 14:382-7.
2: Brickley DR, Mikosz CA, Hagan CR, Conzen SD. Ubiquitin modification of serum and glucocorticoid-induced protein kinase-1(SGK-1). J Biol
Chem. 2002;277:43064-70.
3: Fillon S, Klingel K, Warntges S, Sauter M, Gabrysch S, Pestel S, Tanneur V, Waldegger S, Zipfel A, Viebahn R, Haussinger D, Broer S, Kandolf R, Lang F. Expression of the serine/threonine kinase hSGK1 in chronic viral hepatitis. Cell Physiol Biochem. 2002; 12:47-54.
4: Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival Signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol 2001 ;21 :952-65
5: Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD. Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem. 2001 ;276: 16649-54.
6: Zuo Z1 Urban G, Scammell JG, Dean NM, McLean TK, Aragon I1 Honkanen RE. Ser/Thr protein Phosphatase type 5 (PP5) is a negative regulator of glucocorticoid receptor-mediated growth arrest. Biochemistry. 1999;38.8849-57.
7: Buse P, Tran SH1 Luther E, Phu PT, Aponte GW1 Firestone GL. Cell cycle and hormonal control of nuclear-cytoplasmic localization of the serum- and glucocorticoid-inducible protein kinase, Sgk, in mammary tumor cells. A novel convergence point of anti-proliferative and proliferative cell signalling pathways. J Biol Chem. 1999;274:7253-63.
8: M. Hertweck, C. Göbel, R. Baumeister: C.elegans SGK-1 is the critical component in the Akt/PKB Kinase complex to control stress response and life span. Developmental Cell, Vol. 6, 577-588, April, 2004.
ZUSAMMENFASSUNG DER ERFINDUNG
Die Erfindung betrifft Verbindungen der Formel
R1 H, A, HaI, CN, NO2, C(=O)A, CHO, CH(OH)A1 NH2, NH(C=O)A, COOH, COOA, SO2NH2, CONH2, CONA2, (CH2JmAr oder Het,
R^ OH1 OA1 HaI, CF3, SO2NH2, NHAc oder NHSO2A,
R2 , R2 jeweils unabhängig voneinander H oder HaI1 Ac Acetyl,
Ar unsubstituiertes oder ein-, zwei- oder dreifach durch HaI1 A, OH, OA, NH2, NO2, CN, COOH, COOA, CONH2, NHCOA, NHCONH2, NHSO2A, SO2NH2 und/oder S(O)111A substituiertes Phenyl,
Het unsubstituiertes oder ein-, zwei- oder dreifach durch A, HaI, OH und/oder OA substituiertes Furyl, Thienyl, Pyrrolyl, Imidazolyl, Pyridyl, Pyrimidinyl, Pyrazolyl, Thiazolyl oder Indolyl, unverzweigtes oder verzweigtes Alkyl mit 1-10 C-Atomen, worin 1- 7 H-Atome durch F ersetzt sein können,
CH '2, fehlt, CH2, CHA1 CA2 oder c' / \
(CH2)n .
HaI F, Cl, Br oder I, m O, 1 oder 2, n 1 , 2, 3 oder 4, bedeuten, sowie ihre pharmazeutisch verwendbaren Derivate, Tautomere, Salze,
Solvate und Stereoisomere, einschließlich deren Mischungen in allen
Verhältnissen.
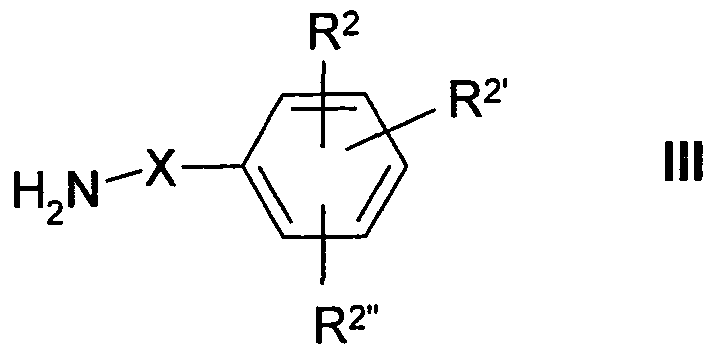
Gegenstand der Erfindung sind die Verbindungen der Formel I und ihre Salze sowie ein Verfahren zur Herstellung von Verbindungen der Formel I sowie ihrer pharmazeutisch verwendbaren Derivate, Tautomere, Solvate, Salze und Stereoisomere, dadurch gekennzeichnet, daß man
a) eine Verbindung der Formel
A Alkyl mit 1 , 2, 3 oder 4 C-Atomen bedeutet und
R1 die in Anspruch 1 angegebene Bedeutung hat,
mit einer Verbindung der Formel III
worin X
1 R
2, R
2 und R
2 die in Anspruch 1 angegebenen Bedeutungen haben,
umsetzt,
oder
b) in einer Verbindung der Formel I einen Rest R2 in einen anderen
Rest R2 umwandelt, indem man einen Ether spaltet,
und/oder eine Base oder Säure der Formel I in eines ihrer Salze umwandelt.
Gegenstand der Erfindung sind auch die Stereoisomeren, Tautomeren sowie die Hydrate und Solvate dieser Verbindungen. Unter Solvate der Verbindungen werden Anlagerungen von inerten Lösungsmittelmolekülen an die Verbindungen verstanden, die sich aufgrund ihrer gegenseitigen
Anziehungskraft ausbilden. Solvate sind z.B. Mono- oder Dihydrate oder Alkoholate.
Unter pharmazeutisch verwendbaren Derivaten versteht man z.B. die
Salze der erfindungsgemäßen Verbindungen als auch sogenannte
Prodrug-Verbindungen.
Unter Prodrug-Derivaten versteht man mit z. B. Alkyl- oder Acylgruppen,
Zuckern oder Oligopeptiden abgewandelte Verbindungen der Formel I1 die im Organismus rasch zu den wirksamen erfindungsgemäßen Verbindungen gespalten werden.
Hierzu gehören auch bioabbaubare Polymerderivate der erfindungsgemäßen Verbindungen, wie dies z. B. in Int. J. Pharm. IJjj, 61-67 (1995) beschrieben ist.
Der Ausdruck "wirksame Menge" bedeutet die Menge eines Arzneimittels oder eines pharmazeutischen Wirkstoffes, die eine biologische oder medizinische Antwort in einem Gewebe, System, Tier oder Menschen hervorruft, die z.B. von einem Forscher oder Mediziner gesucht oder erstrebt wird.
Darüberhinaus bedeutet der Ausdruck "therapeutisch wirksame Menge" eine Menge, die, verglichen zu einem entsprechenden Subjekt, das diese Menge nicht erhalten hat, folgendes zur Folge hat: verbesserte Heilbehandlung, Heilung, Prävention oder Beseitigung einer Krankheit, eines Krankheitsbildes, eines Krankheitszustandes, eines Leidens, einer Störung oder von Nebenwirkungen oder auch die Verminderung des Fortschreitens einer Krankheit, eines Leidens oder einer Störung.
Die Bezeichnung "therapeutisch wirksame Menge" umfaßt auch die Mengen, die wirkungsvoll sind, die normale physiologische Funktion zu erhöhen.
Gegenstand der Erfindung sind auch Mischungen der erfindungsgemäßen Verbindungen der Formel I, z.B. Gemische zweier Diastereomerer z.B. im Verhältnis 1 :1 , 1 :2, 1:3, 1 :4, 1 :5, 1 :10, 1 :100 oder 1 :1000. Besonders bevorzugt handelt es sich dabei um Mischungen stereoisomerer Verbindungen.
Für alle Reste, die mehrfach auftreten, gilt, daß deren Bedeutungen unabhängig voneinander sind. Vor- und nachstehend haben die Reste bzw. Parameter R1, R2, R2, R2 und X die bei der Formel I angegebenen Bedeutungen, falls nicht ausdrücklich etwas anderes angegeben ist.
A bedeutet Alkyl, ist unverzweigt (linear) oder verzweigt, und hat 1 , 2, 3, 4, 5 oder 6 C-Atome. A bedeutet vorzugsweise Methyl, weiterhin Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, sek.-Butyl oder tert.-Butyl, ferner auch Pentyl, 1-, 2- oder 3-Methylbutyl, 1 ,1- , 1 ,2- oder 2,2-Dimethylpropyl, 1-
Ethylpropyl, Hexyl, 1- , 2- , 3- oder 4-Methylpentyl, 1 ,1- , 1 ,2- , 1 ,3- ,
2,2- , 2,3- oder 3,3-Dimethylbutyl, 1- oder 2-Ethylbutyl, 1-Ethyl-1-methyl- propyl, 1-Ethyl-2-methylpropyl, 1 ,1 ,2- oder 1 ,2,2-Trimethylpropyl, weiter bevorzugt z.B. Trifluormethyl.
A bedeutet ganz besonders bevorzugt Alkyl mit 1 , 2, 3, 4, 5 oder 6 C- Atomen, vorzugsweise Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, sek.-Butyl, tert.-Butyl, Pentyl, Hexyl, Trifluormethyl, Pentafluorethyl oder 1 ,1 ,1-Trifluorethyl. Ac bedeutet Acetyl.
Ar bedeutet z.B. Phenyl, o-, m- oder p-Tolyl, o-, m- oder p-Ethylphenyl, o-, m- oder p-Propylphenyl, o-, m- oder p-lsopropylphenyl, o-, m- oder p-tert.- Butylphenyl, o-, m- oder p-Hydroxyphenyl, o-, m- oder p-Nitrophenyl, o-, m- oder p-Aminophenyl, o-, m- oder p-Acetamidophenyl, o-, m- oder p-
Methoxyphenyl, o-, m- oder p-Ethoxyphenyl, o-, m- oder p-Ethoxycarbonyl- phenyl, o-, m- oder p-Aminocarbonyl-phenyl, o-, m- oder p-Fluorphenyl, o-,
m- oder p-Bromphenyl, o-, m- oder p- Chlorphenyl, o-, m- oder p-(Methyl- sulfonamido)-phenyl, o-, m- oder p-(Methylsulfonyl)-phenyl, o-, m- oder p- Cyanphenyl, o-, m- oder p-Ureidophenyl, o-, m- oder p-Aminosulfonyl- phenyl, o-, m- oder p-Carboxyphenyl, weiter bevorzugt 2,3-, 2,4-, 2,5-, 2,6-,
5 3,4- oder 3,5-Difluorphenyl, 2,3-, 2,4-, 2,5-, 2,6-, 3,4- oder 3,5-Dichlor- phenyl, 2,3-, 2,4-, 2,5-, 2,6-, 3,4- oder 3,5-Dibromphenyl, 2,4- oder 2,5- Dinitrophenyl, 2,5- oder 3,4-Dimethoxyphenyl, 3-Nitro-4-chlorphenyl, 3- Amino-4-chlor-, 2-Amino-3-chlor-, 2-Amino-4-chlor-, 2-Amino-5-chlor- oder
10 2-Amino-6-chlorphenyl, 2,3-Diaminophenyl, 2,3,4-, 2,3,5-, 2,3,6-, 2,4,6- oder 3,4,5-Trichlorphenyl, 2,4,6-Trimethoxyphenyl, 2-Hydroxy-3,5-dichlor- phenyl, p-lodphenyl, 3,6-Dichlor-4-aminophenyl, 4-Fluor-3-chlorphenyl, 2- Fluor-4-bromphenyl, 2,5-Difluor-4-bromphenyl, 3-Brom-6-methoxyphenyl,
^ 3-Chlor-6-methoxyphenyl, 3-Chlor-4-acetamidophenyl, 3-Fluor-4-methoxy- phenyl, 3-Amino-6-methylphenyl, 3-Chlor-4-acetamidophenyl oder 2,5- Dimethyl-4-chlorphenyl.
Ar bedeutet besonders bevorzugt unsubstituiertes oder ein-, zwei- oder
20 dreifach durch HaI substituiertes Phenyl. Ar bedeutet ganz besonders bevorzugt Phenyl.
Het bedeutet vorzugsweise unsubstituiertes oder ein-, zwei- oder dreifach 25 durch A, HaI, OH und/oder OA substituiertes Furyl, Thienyl, Pyrrolyl, Imidazolyl, Pyridyl, Pyrimidinyl, Pyrazolyl, Thiazolyl oder Indolyl. Het bedeutet ganz besonders bevorzugt Pyridyl.
O0 X bedeutet besonders bevorzugt CH2 oder CHA, wobei A vorzugsweise Alkyl mit 1 , 2, 3 oder 4 C-Atomen bedeutet.
R1 bedeutet vorzugsweise H, A, HaI, CN, NO2, CH(OH)A, C(=O)A, COOH, COOA, SO2NH2, Benzyl, Phenyl oder Pyridyl; besonders bevorzugt H oder 35 A.
In einer weiteren Ausführungsform bedeutet R1 vorzugsweise H, A, Ar oder Het.
R2 bedeutet vorzugsweise OH1 OCH3, HaI, CF3, SO2NH2, NHAc oder NHSO2A, wie z.B. NHSO2CH3.
Die Verbindungen der Formel I können ein oder mehrere chirale Zentren besitzen und daher in verschiedenen stereoisomeren Formen vorkommen. 0 Die Formel I umschließt alle diese Formen.
Dementsprechend sind Gegenstand der Erfindung insbesondere diejenigen Verbindungen der Formel I1 in denen mindestens einer der genannten e Reste eine der vorstehend angegebenen bevorzugten Bedeutungen hat.
Einige bevorzugte Gruppen von Verbindungen können durch die folgenden Teilformeln Ia bis Ii ausgedrückt werden, die der Formel I entsprechen und worin die nicht näher bezeichneten Reste die bei der Formel I angegebene
Bedeutung haben, worin jedoch 0 in Ia R1 H, A, Ar oder Het bedeutet;
5 in Ib A unverzweigtes oder verzweigtes Alkyl mit 1-6 C-Atomen, worin 1-5 H-Atome durch F ersetzt sein können, bedeutet;
Q in Ic Ar unsubstituiertes oder ein-, zwei- oder dreifach durch HaI substituiertes Phenyl, bedeutet;
in Id Het unsubstituiertes oder ein-, zwei- oder dreifach durch A, 5
HaI, OH und/oder OA substituiertes Furyl, Thienyl,
006/007650
- 16 -
Pyrrolyl, Imidazolyl, Pyridyl, Pyrimidinyl, Pyrazolyl, Thiazolyl oder Indolyl, bedeutet;
in Ie R1 H bedeutet;
in If Ar Phenyl bedeutet;
in Ig Het Pyridyl bedeutet;
in Ih R1 H, A, Ar oder Het,
OH1 OA1 HaI1 CF3, SO2NH2, NHAc oder NHSO2A,
R2', R2" jeweils unabhängig voneinander H oder HaI, Ar unsubstituiertes oder ein-, zwei- oder dreifach durch HaI substituiertes Phenyl,
Het unsubstituiertes oder ein-, zwei- oder dreifach durch A, HaI, OH und/oder OA substituiertes Furyl,
Thienyl, Pyrrolyl, Imidazolyl, Pyridyl, Pyrimidinyl,
Pyrazolyl, Thiazolyl oder Indolyl, unverzweigtes oder verzweigtes Alkyl mit 1-6 C-
Atomen, worin 1-5 H-Atome durch F ersetzt sein können,
X fehlt, CH2, CHA oder CA2,
HaI F, Cl, Br oder I bedeuten;
in R1 H, R2 OH, OA, HaI, CF3, SO2NH2, NHAc oder NHSO2A, R2', R2" jeweils unabhängig voneinander H oder HaI,
A unverzweigtes oder verzweigtes Alkyl mit 1 -6 C-
Atomen, worin 1-5 H-Atome durch F ersetzt sein können,
X CH2, CHA oder CA2, HaI F1 Cl, Br oder I bedeuten; sowie ihre pharmazeutisch verwendbaren Derivate, Tautomere, Salze, Solvate und Stereoisomere, einschließlich deren Mischungen in allen Verhältnissen.
Die erfindungsgemäßen Verbindungen und auch die Ausgangsstoffe zu ihrer Herstellung werden im übrigen nach an sich bekannten Methoden hergestellt, wie sie in der Literatur (z.B. in den Standardwerken wie
Houben-Weyl, Methoden der organischen Chemie, Georg-Thieme-Verlag, Stuttgart) beschrieben sind, und zwar unter Reaktionsbedingungen, die für die genannten Umsetzungen bekannt und geeignet sind. Dabei kann man auch von an sich bekannten, hier nicht näher erwähnten Varianten
Gebrauch machen.
Die Ausgangsstoffe können, falls erwünscht, auch in situ gebildet werden, so daß man sie aus dem Reaktionsgemisch nicht isoliert, sondern sofort weiter zu den erfindungsgemäßen Verbindungen umsetzt.
Die Ausgangsverbindungen sind in der Regel bekannt. Sind sie neu, so können sie aber nach an sich bekannten Methoden hergestellt werden.
Verbindungen der Formel I können vorzugsweise erhalten werden, indem man eine Verbindung der Formel Il mit einer Verbindung der Formel III umsetzt.
Die Umsetzung erfolgt nach Methoden, die dem Fachmann bekannt sind.
Die Umsetzung erfolgt in der Regel in einem inerten Lösungsmittel.
AIs inerte Lösungsmittel eignen sich z.B. Kohlenwasserstoffe wie Hexan, Petrolether, Benzol, Toluol oder XyIoI; chlorierte Kohlenwasserstoffe wie
Trichlorethylen, 1 ,2-Dichlorethan,Tetrachlorkohlenstoff, Chlorform oder
Dichlormethan; Alkohole wie Methanol, Ethanol, Isopropanol, n-Propanol, n-Butanol oder tert.-Butanol; Ether wie Diethylether, Diisopropylether, Tetrahydrofuran (THF) oder Dioxan; Glykolether wie Ethylenglykol- monomethyl- oder -monoethylether (Methylglykol oder Ethylglykol), Ethylenglykoldimethylether (Diglyme); Ketone wie Aceton oder Butanon; Amide wie Acetamid, Dimethylacetamid oder Dimethylformamid (DMF); Nitrile wie Acetonitril; Sulfoxide wie Dimethylsulfoxid (DMSO); Schwefelkohlenstoff; Carbonsäuren wie Ameisensäure oder Essigsäure; Nitrover- bindungen wie Nitromethan oder Nitrobenzol; Ester wie Ethylacetat oder Gemische der genannten Lösungsmittel.
Die Reaktionszeit liegt je nach den angewendeten Bedingungen zwischen einigen Minuten und 14 Tagen, die Reaktionstemperatur zwischen etwa
-30° und 140°, normalerweise zwischen -10° und 110°, insbesondere zwischen etwa 20° und etwa 100°.
Die Spaltung eines Ethers erfolgt unter Methoden, wie sie dem Fachmann bekannt sind.
Eine Standardmethode zur Etherspaltung ist die Verwendung von Bortribromid.
Pharmazeutische Salze und andere Formen
Die genannten erfindungsgemäßen Verbindungen lassen sich in ihrer endgültigen Nichtsalzform verwenden. Andererseits umfaßt die vorliegende Erfindung auch die Verwendung dieser Verbindungen in Form ihrer pharmazeutisch unbedenklichen Salze, die von verschiedenen organi- sehen und anorganischen Säuren und Basen nach fachbekannten Vorgehensweisen abgeleitet werden können. Pharmazeutisch unbedenkliche
Salzformen der Verbindungen der Formel I werden größtenteils konventionell hergestellt. Sofern die Verbindung der Formel I eine Carbonsäuregruppe enthält, läßt sich eines ihrer geeigneten Salze dadurch bilden, daß man die Verbindung mit einer geeigneten Base zum entsprechenden Basenadditionssalz umsetzt. Solche Basen sind zum Beispiel Alkalimetall- hydroxide, darunter Kaliumhydroxid, Natriumhydroxid und Lithiumhydroxid; Erdalkalimetallhydroxide wie Bariumhydroxid und Calciumhydroxid; Alkali- metallalkoholate, z.B. Kaliumethanolat und Natriumpropanolat; sowie verschiedene organische Basen wie Piperidin, Diethanolamin und
N-Methylglutamin. Die Aluminiumsalze der Verbindungen der Formel I zählen ebenfalls dazu. Bei bestimmten Verbindungen der Formel I lassen sich Säureadditionssalze dadurch bilden, daß man diese Verbindungen mit pharmazeutisch unbedenklichen organischen und anorganischen
Säuren, z.B. Halogenwasserstoffen wie Chlorwasserstoff, Bromwasserstoff oder Jodwasserstoff, anderen Mineralsäuren und ihren entsprechenden Salzen wie Sulfat, Nitrat oder Phosphat und dergleichen sowie Alkyl- und
Monoarylsulfonaten wie Ethansulfonat, Toluolsulfonat und Benzolsulfonat, sowie anderen organischen Säuren und ihren entsprechenden Salzen wie
Acetat, Trifluoracetat, Tartrat, Maleat, Succinat, Citrat, Benzoat, Salicylat, Ascorbat und dergleichen behandelt. Dementsprechend zählen zu pharmazeutisch unbedenklichen Säureadditionssalzen der Verbindungen der Formel I die folgenden: Acetat, Adipat, Alginat, Arginat, Aspartat, Benzoat, Benzolsulfonat (Besylat), Bisulfat, Bisulfit, Bromid, Butyrat, Campherat, Camphersulfonat, Caprylat, Chlorid, Chlorbenzoat, Citrat, Cyclopentanpropionat, Digluconat, Dihydrogenphosphat, Dinitrobenzoat, Dodecylsulfat, Ethansulfonat, Fumarat, Galacterat (aus Schleimsäure), Galacturonat, Glucoheptanoat, Gluconat, Glutamat, Glycerophosphat, Hemisuccinat, Hemisulfat, Heptanoat, Hexanoat, Hippurat, Hydrochlorid, Hydrobromid, Hydroiodid, 2-Hydroxyethansulfonat, lodid, Isethionat,
Isobutyrat, Lactat, Lactobionat, Malat, Maleat, Malonat, Mandelat,
Metaphosphat, Methansulfonat, Methylbenzoat, Monohydrogenphosphat,
2-Naphthalinsulfonat, Nicotinat, Nitrat, Oxalat, Oleat, Pamoat, Pectinat,
Persulfat, Phenylacetat, 3-Phenylpropionat, Phosphat, Phosphonat, Phthalat, was jedoch keine Einschränkung darstellt.
Weiterhin zählen zu den Basensalzen der erfindungsgemäßen 5
Verbindungen Aluminium-, Ammonium-, Calcium-, Kupfer-, Eisen(lll)-, Eisen(ll)-, Lithium-, Magnesium-, Mangan(lll)-, Mangan(ll), Kalium-, Natrium- und Zinksalze, was jedoch keine Einschränkung darstellen soll. Bevorzugt unter den oben genannten Salzen sind Ammonium; die
10 Alkalimetallsalze Natrium und Kalium .sowie die Erdalkalimetalsalze
Calcium und Magnesium. Zu Salzen der Verbindungen der Formel I, die sich von pharmazeutisch unbedenklichen organischen nicht-toxischen Basen ableiten, zählen Salze primärer, sekundärer und tertiärer Amine,
A c substituierter Amine, darunter auch natürlich vorkommender substituierter Amine, cyclischer Amine sowie basischer lonenaustauscherharze, z.B. Arginin, Betain, Koffein, Chlorprocain, Cholin, N.N'-Dibenzylethylendiamin (Benzathin), Dicyclohexylamin, Diethanolamin, Diethylamin, 2-Diethyl- aminoethanol, 2-Dimethylaminoethanol, Ethanolamin, Ethylendiamin, N-
20
Ethylmorpholin, N-Ethylpiperidin, Glucamin, Glucosamin, Histidin,
Hydrabamin, Iso-propylamin, Lidocain, Lysin, Meglumin, N-Methyl-D- glucamin, Morpholin, Piperazin, Piperidin, Polyaminharze, Procain, Purine, Theobromin, Triethanolamin, Triethylamin, Trimethylamin, Tripropylamin 25 sowie Tris-(hydroxymethyl)-methylamin (Tromethamin), was jedoch keine Einschränkung darstellen soll.
Verbindungen der vorliegenden Erfindung, die basische stickstoffhaltige OQ Gruppen enthalten, lassen sich mit Mitteln wie (C1-C4) Alkylhalogeniden, z.B. Methyl-, Ethyl-, Isopropyl- und tert.-Butylchlorid, -bromid und -iodid; Di(C1 -C4)Alkylsulfaten, z.B. Dimethyl-, Diethyl- und Diamylsulfat; (C10- C18)Alkylhalogeniden, z.B. Decyl-, Dodecyl-, Lauryl-, Myristyl- und
Stearylchlorid, -bromid und -iodid; sowie Aryl-(CrC4)Alkylhalogeniden, z.B.
35
Benzylchlorid und Phenethylbromid, quartemisieren. Mit solchen Salzen
können sowohl wasser- als auch öllösliche erfindungsgemäße Verbindungen hergestellt werden.
Zu den oben genannten pharmazeutischen Salzen, die bevorzugt sind, zählen Acetat, Trifluoracetat, Besylat, Citrat, Fumarat, Gluconat, Hemisuccinat, Hippurat, Hydrochlorid, Hydrobromid, Isethionat, Mandelat, Meglumin, Nitrat, Oleat, Phosphonat, Pivalat, Natriumphosphat, Stearat, Sulfat, Sulfosalicylat, Tartrat, Thiomalat, Tosylat und Tromethamin, was jedoch keine Einschränkung darstellen soll.
Die Säureadditionssalze basischer Verbindungen der Formel I werden dadurch hergestellt, daß man die freie Basenform mit einer ausreichenden Menge der gewünschten Säure in Kontakt bringt, wodurch man auf übliche Weise das Salz darstellt. Die freie Base läßt sich durch In-Kontakt-Bringen der Salzform mit einer Base und Isolieren der freien Base auf übliche Weise regenerieren. Die freien Basenformen unterscheiden sich in gewissem Sinn von ihren entsprechenden Salzformen in bezug auf bestimmte physikalische Eigenschaften wie Löslichkeit in polaren Lösungsmitteln; im
Rahmen der Erfindung entsprechen die Salze jedoch sonst ihren jeweiligen freien Basenformen.
Wie erwähnt werden die pharmazeutisch unbedenklichen Basenadditionssalze der Verbindungen der Formel I mit Metallen oder Aminen wie Alkalimetallen und Erdalkalimetallen oder organischen Aminen gebildet. Bevorzugte Metalle sind Natrium, Kalium, Magnesium und Calcium. Bevor- zugte organische Amine sind N.N'-Dibenzylethylendiamin, Chlorprocain, Cholin, Diethanolamin, Ethylendiamin, N-Methyl-D-glucamin und Procain.
Die Basenadditionssalze von erfindungsgemäßen sauren Verbindungen werden dadurch hergestellt, daß man die freie Säureform mit einer ausreichenden Menge der gewünschten Base in Kontakt bringt, wodurch man das Salz auf übliche Weise darstellt. Die freie Säure läßt sich durch
In-Kontakt-Bringen der Salzform mit einer Säure und Isolieren der freien Säure auf übliche Weise regenerieren. Die freien Säureformen unterscheiden sich in gewissem Sinn von ihren entsprechenden Salzformen in bezug auf bestimmte physikalische Eigenschaften wie Löslichkeit in polaren Lösungsmitteln; im Rahmen der Erfindung entsprechen die Salze jedoch sonst ihren jeweiligen freien Säureformen.
Enthält eine erfindungsgemäße Verbindung mehr als eine Gruppe, die solche pharmazeutisch unbedenklichen Salze bilden kann, so umfaßt die Erfindung auch mehrfache Salze. Zu typischen mehrfachen Salzformen zählen zum Beispiel Bitartrat, Diacetat, Difumarat, Dimeglumin, Diphosphat, Dinatrium und Trihydrochlorid, was jedoch keine Ein- schränkung darstellen soll.
Im Hinblick auf das oben Gesagte sieht man, daß unter dem Ausdruck "pharmazeutisch unbedenkliches Salz" im vorliegenden Zusammenhang ein Wirkstoff zu verstehen ist, der eine Verbindung der Formel I in der
Form eines ihrer Salze enthält, insbesondere dann, wenn diese Salzform dem Wirkstoff im Vergleich zu der freien Form des Wirkstoffs oder irgendeiner anderen Salzform des Wirkstoffs, die früher verwendet wurde, verbesserte pharmakokinetische Eigenschaften verleiht. Die pharma- zeutisch unbedenkliche Salzform des Wirkstoffs kann auch diesem
Wirkstoff erst eine gewünschte pharmakokinetische Eigenschaft verleihen, über die er früher nicht verfügt hat, und kann sogar die Pharmakodynamik dieses Wirkstoffs in bezug auf seine therapeutische Wirksamkeit im Körper positiv beeinflussen.
Erfindungsgemäße Verbindungen der Formel I können aufgrund ihrer Molekülstruktur chiral sein und können dementsprechend in verschiedenen enantiomeren Formen auftreten. Sie können daher in racemischer oder in optisch aktiver Form vorliegen.
Da sich die pharmazeutische Wirksamkeit der Racemate bzw. der Stereoisomeren der erfindungsgemäßen Verbindungen unterscheiden kann, kann es wünschenswert sein, die Enantiomere zu verwenden. In diesen Fällen kann das Endprodukt oder aber bereits die Zwischenprodukte in enantiomere Verbindungen, durch dem Fachmann bekannte chemische oder physikalische Maßnahmen, aufgetrennt oder bereits als solche bei der Synthese eingesetzt werden.
Im Falle racemischer Amine werden aus dem Gemisch durch Umsetzung mit einem optisch aktiven Trennmittel Diastereomere gebildet. Als Trennmittel eignen sich z.B. optisch aktiven Säuren, wie die R- und S-Formen von Weinsäure, Diacetylweinsäure, Dibenzoylweinsäure, Mandelsäure, Äpfelsäure, Milchsäure, geeignet N-geschützte Aminosäuren (z.B. N-Ben- zoylprolin oder N-Benzolsulfonylprolin) oder die verschiedenen optisch aktiven Camphersulfonsäuren. Vorteilhaft ist auch eine chromatographische Enantiomerentrennung mit Hilfe eines optisch aktiven Trennmittels (z.B. Dinitrobenzoylphenylglycin, Cellulosetriacetat oder andere Derivate von Kohlenhydraten oder auf Kieselgel fixierte chiral derivatisierte Methacrylatpolymere). Als Laufmittel eignen sich hierfür wäßrige oder alkoholische Lösungsmittelgemische wie z.B. Hexan/Isopropanol/ Acetonitril z.B. im Verhältnis 82:15:3.
Gegenstand der Erfindung ist ferner die Verwendung der Verbindungen und/oder ihrer physiologisch unbedenklichen Salze zur Herstellung eines Arzneimittels (pharmazeutische Zubereitung), insbesondere auf nicht- chemischem Wege. Hierbei können sie zusammen mit mindestens einem festen, flüssigen und/oder halbflüssigen Träger- oder Hilfsstoff und gegebenenfalls in Kombination mit einem oder mehreren weiteren Wirkstoffen in eine geeignete Dosierungsform gebracht werden.
Gegenstand der Erfindung sind ferner Arzneimittel, enthaltend mindestens eine erfindungsgemäße Verbindung und/oder ihre pharmazeutisch verwendbaren Derivate, Tautomere, Solvate und Stereoisomere, einschließlich deren Mischungen in allen Verhältnissen, sowie gegebenenfalls Träger- und/oder Hilfsstoffe.
Pharmazeutische Formulierungen können in Form von Dosiseinheiten, die eine vorbestimmte Menge an Wirkstoff pro Dosiseinheit enthalten, dargereicht werden. Eine solche Einheit kann beispielsweise 0,5 mg bis
1 g, vorzugsweise 1 mg bis 700 mg, besonders bevorzugt 5 mg bis 100 mg einer erfindungsgemäßen Verbindung enthalten, je nach dem behandelten Krankheitszustand, dem Verabreichungsweg und dem Alter, Gewicht und Zustand des Patienten, oder pharmazeutische Formulierungen können in Form von Dosiseinheiten, die eine vorbestimmte Menge an Wirkstoff pro Dosiseinheit enthalten, dargereicht werden. Bevorzugte Dosierungs- einheitsformulierungen sind solche, die eine Tagesdosis oder Teildosis, wie oben angegeben, oder einen entsprechenden Bruchteil davon eines
Wirkstoffs enthalten. Weiterhin lassen sich solche pharmazeutischen
Formulierungen mit einem der im pharmazeutischen Fachgebiet allgemein bekannten Verfahren herstellen.
Pharmazeutische Formulierungen lassen sich zur Verabreichung über einen beliebigen geeigneten Weg, beispielsweise auf oralem (einschließlich buccalem bzw. sublingualem), rektalem, nasalem, topischem (einschließlich buccalem, sublingualem oder transdermalem), vaginalem oder parenteralem (einschließlich subkutanem, intramuskulärem, intravenösem oder intradermalem) Wege, anpassen. Solche Formulierungen können mit allen im pharmazeutischen Fachgebiet bekannten Verfahren hergestellt werden, indem beispielsweise der Wirkstoff mit dem bzw. den Trägerstoff(en) oder Hilfsstoff(en) zusammengebracht wird.
An die orale Verabreichung angepaßte pharmazeutische Formulierungen können als separate Einheiten, wie z.B. Kapseln oder Tabletten; Pulver oder Granulate; Lösungen oder Suspensionen in wäßrigen oder nichtwäßrigen Flüssigkeiten; eßbare Schäume oder Schaumspeisen; oder
ÖI-in-Wasser-Flüssigemulsionen oder Wasser-in-öl-Flüssigemulsionen dargereicht werden.
So läßt sich beispielsweise bei der oralen Verabreichung in Form einer Tablette oder Kapsel die Wirkstoffkomponente mit einem oralen, nichttoxischen und pharmazeutisch unbedenklichen inerten Trägerstoff, wie z.B. Ethanol, Glycerin, Wasser u.a. kombinieren. Pulver werden hergestellt, indem die Verbindung auf eine geeignete feine Größe zerkleinert und mit einem in ähnlicher Weise zerkleinerten pharmazeutischen
Trägerstoff, wie z.B. einem eßbaren Kohlenhydrat wie beispielsweise Stärke oder Mannit vermischt wird. Ein Geschmacksstoff, Konservierungsmittel, Dispersionsmittel und Farbstoff können ebenfalls vorhanden sein.
Kapseln werden hergestellt, indem ein Pulvergemisch wie oben beschrieben hergestellt und geformte Gelatinehüllen damit gefüllt werden. Gleit- und Schmiermittel wie z.B. hochdisperse Kieselsäure, Talkum, Magnesiumstearat, Calciumstearat oder Polyethylenglykol in Festform können dem Pulvergemisch vor dem Füllvorgang zugesetzt werden. Ein
Sprengmittel oder Lösungsvermittler, wie z.B. Agar-Agar, Calciumcarbonat oder Natriumcarbonat, kann ebenfalls zugesetzt werden, um die Verfügbarkeit des Medikaments nach Einnahme der Kapsel zu verbessern.
Außerdem können, falls gewünscht oder notwendig, geeignete Bindungs-,
Schmier- und Sprengmittel sowie Farbstoffe ebenfalls in das Gemisch eingearbeitet werden. Zu den geeigneten Bindemitteln gehören Stärke,
Gelatine, natürliche Zucker, wie z.B. Glukose oder Beta-Lactose, Süß-
Stoffe aus Mais, natürliche und synthetische Gummi, wie z.B. Akazia,
Traganth oder Natriumalginat, Carboxymethylzellulose, Polyethylenglykol,
Wachse, u.a. Zu den in diesen Dosierungsformen verwendeten Schmiermitteln gehören Natriumoleat, Natriumstearat, Magnesiumstearat, Natrium- benzoat, Natriumacetat, Natriumchlorid u.a. Zu den Sprengmitteln gehören, ohne darauf beschränkt zu sein, Stärke, Methylzellulose, Agar, 5
Bentonit, Xanthangummi u.a. Die Tabletten werden formuliert, indem beispielsweise ein Pulvergemisch hergestellt, granuliert oder trocken- verpreßt wird, ein Schmiermittel und ein Sprengmittel zugegeben werden und das Ganze zu Tabletten verpreßt wird. Ein Pulvergemisch wird
10 hergestellt, indem die in geeigneter Weise zerkleinerte Verbindung mit einem Verdünnungsmittel oder einer Base, wie oben beschrieben, und gegebenenfalls mit einem Bindemittel, wie z.B. Carboxymethylzellulose, einem Alginat, Gelatine oder Polyvinylpyrrolidon, einem Lösungsverlang-
-j c samer, wie z.B. Paraffin, einem Resorptionsbeschleuniger, wie z.B. einem quaternären Salz und/oder einem Absorptionsmittel, wie z.B. Bentonit, Kaolin oder Dicalciumphosphat, vermischt wird. Das Pulvergemisch läßt sich granulieren, indem es mit einem Bindemittel, wie z.B. Sirup, Stärkepaste, Acadia-Schleim oder Lösungen aus Zellulose- oder Polymer-
20 materialen benetzt und durch ein Sieb gepreßt wird. Als Alternative zur
Granulierung kann man das Pulvergemisch durch eine Tablettiermaschine laufen lassen, wobei ungleichmäßig geformte Klumpen entstehen, die in Granulate aufgebrochen werden. Die Granulate können mittels Zugabe
25 von Stearinsäure, einem Stearatsalz, Talkum oder Mineralöl gefettet werden, um ein Kleben an den Tablettengußformen zu verhindern. Das gefettete Gemisch wird dann zu Tabletten verpreßt. Die erfindungsgemäßen Verbindungen können auch mit einem freifließenden inerten
O0 Trägerstoff kombiniert und dann ohne Durchführung der Granulierungs- oder Trockenverpressungsschritte direkt zu Tabletten verpreßt werden. Eine durchsichtige oder undurchsichtige Schutzschicht, bestehend aus einer Versiegelung aus Schellack, einer Schicht aus Zucker oder Polymermaterial und einer Glanzschicht aus Wachs, kann vorhanden sein. Diesen
35
Beschichtungen können Farbstoffe zugesetzt werden, um zwischen unterschiedlichen Dosierungseinheiten unterscheiden zu können.
Orale Flüssigkeiten, wie z.B. Lösung, Sirupe und Elixiere, können in Form von Dosierungseinheiten hergestellt werden, so daß eine gegebene Quantität eine vorgegebene Menge der Verbindung enthält. Sirupe lassen sich herstellen, indem die Verbindung in einer wäßrigen Lösung mit geeignetem Geschmack gelöst wird, während Elixiere unter Verwendung eines nichttoxischen alkoholischen Vehikels hergestellt werden. Suspensionen können durch Dispersion der Verbindung in einem nicht-
10 toxischen Vehikel formuliert werden. Lösungsvermittler und Emulgiermittel, wie z.B. ethoxylierte Isostearylalkohole und Polyoxyethylensorbitolether, Konservierungsmittel, Geschmackszusätze, wie z.B. Pfefferminzöl oder natürliche Süßstoffe oder Saccharin oder andere künstliche Süßstoffe, u.a.
,.C können ebenfalls zugegeben werden.
Die Dosierungseinheitsformulierungen für die orale Verabreichung können gegebenenfalls in Mikrokapseln eingeschlossen werden. Die Formulierung läßt sich auch so herstellen, daß die Freisetzung verlängert oder retardiert
20 wird, wie beispielsweise durch Beschichtung oder Einbettung von partikulärem Material in Polymere, Wachs u.a.
Die erfindungsgemäßen Verbindungen sowie Salze, Solvate und 25 physiologisch funktionelle Derivate davon lassen sich auch in Form von
Liposomenzuführsystemen, wie z.B. kleinen unilamellaren Vesikeln, großen unilamellaren Vesikeln und multilamellaren Vesikeln, verabreichen.
Liposomen können aus verschiedenen Phospholipiden, wie z.B. 30 Cholesterin, Stearylamin oder Phosphatidylcholinen, gebildet werden.
Die erfindungsgemäßen Verbindungen sowie die Salze, Solvate und physiologisch funktionellen Derivate davon können auch unter
Verwendung monoklonaler Antikörper als individuelle Träger, an die die
35
Verbindungsmoleküle gekoppelt werden, zugeführt werden. Die
Verbindungen können auch mit löslichen Polymeren als zielgerichtete
Arzneistoffträger gekoppelt werden. Solche Polymere können Polyvinyl- pyrrolidon, Pyran-Copolymer, Polyhydroxypropylmethacrylamidphenol, Polyhydroxyethylaspartamidphenol oder Polyethylenoxidpolylysin, substituiert mit Palmitoylresten, umfassen. Weiterhin können die 5
Verbindungen an eine Klasse von biologisch abbaubaren Polymeren, die zur Erzielung einer kontrollierten Freisetzung eines Arzneistoffs geeignet sind, z.B. Polymilchsäure, Polyepsilon-Caprolacton, Polyhydroxybutter- säure, Polyorthoester, Polyacetale, Polydihydroxypyrane, Polycyano- 10 acrylate und quervernetzte oder amphipatische Blockcopolymere von Hydrogelen, gekoppelt sein.
An die transdermale Verabreichung angepaßte pharmazeutische Aß Formulierungen können als eigenständige Pflaster für längeren, engen Kontakt mit der Epidermis des Empfängers dargereicht werden. So kann beispielsweise der Wirkstoff aus dem Pflaster mittels lontophorese zugeführt werden, wie in Pharmaceutical Research, 3(6), 318 (1986) allgemein beschrieben. 20
An die topische Verabreichung angepaßte pharmazeutische Verbindungen können als Salben, Cremes, Suspensionen, Lotionen, Pulver, Lösungen, Pasten, Gele, Sprays, Aerosole oder öle formuliert sein.
25
Für Behandlungen des Auges oder anderer äußerer Gewebe, z.B. Mund und Haut, werden die Formulierungen vorzugsweise als topische Salbe oder Creme appliziert. Bei Formulierung zu einer Salbe kann der Wirkstoff o0 entweder mit einer paraffinischen oder einer mit Wasser mischbaren Cremebasis eingesetzt werden. Alternativ kann der Wirkstoff zu einer Creme mit einer Öl-in-Wasser-Cremebasis oder einer Wasser-in-ÖI-Basis formuliert werden.
35
Zu den an die topische Applikation am Auge angepaßten pharmazeutischen Formulierungen gehören Augentropfen, wobei der Wirkstoff in
einem geeigneten Träger, insbesondere einem wäßrigen Lösungsmittel, gelöst oder suspendiert ist.
An die topische Applikation im Mund angepaßte pharmazeutische
Formulierungen umfassen Lutschtabletten, Pastillen und Mundspülmittel.
An die rektale Verabreichung angepaßte pharmazeutische Formulierungen können in Form von Zäpfchen oder Einlaufen dargereicht werden.
An die nasale Verabreichung angepaßte pharmazeutische
Formulierungen, in denen die Trägersubstanz ein Feststoff ist, enthalten ein grobes Pulver mit einer Teilchengröße beispielsweise im Bereich von 20-500 Mikrometern, das in der Art und Weise, wie Schnupftabak aufgenommen wird, verabreicht wird, d.h. durch Schnellinhalation über die Nasenwege aus einem dicht an die Nase gehaltenen Behälter mit dem Pulver. Geeignete Formulierungen zur Verabreichung als Nasenspray oder Nasentropfen mit einer Flüssigkeit als Trägersubstanz umfassen Wirkstofflösungen in Wasser oder öl.
An die Verabreichung durch Inhalation angepaßte pharmazeutische Formulierungen umfassen feinpartikuläre Stäube oder Nebel, die mittels verschiedener Arten von unter Druck stehenden Dosierspendern mit Aerosolen, Verneblern oder Insufflatoren erzeugt werden können.
An die vaginale Verabreichung angepaßte pharmazeutische
Formulierungen können als Pessare, Tampons, Cremes, Gele, Pasten,
Schäume oder Sprayformulierungen dargereicht werden.
Zu den an die parenterale Verabreichung angepaßten pharmazeutischen Formulierungen gehören wäßrige und nichtwäßrige sterile Injektions- lösungen, die Antioxidantien, Puffer, Bakteriostatika und Solute, durch die die Formulierung isotonisch mit dem Blut des zu behandelnden
Empfängers gemacht wird, enthalten; sowie wäßrige und nichtwäßrige sterile Suspensionen, die Suspensionsmittel und Verdicker enthalten können. Die Formulierungen können in Einzeldosis- oder Mehrfachdosisbehältern, z.B. versiegelten Ampullen und Fläschchen, dargereicht und in gefriergetrocknetem (lyophilisiertem) Zustand gelagert werden, so daß nur die Zugabe der sterilen Trägerflüssigkeit, z.B. Wasser für Injektionszwecke, unmittelbar vor Gebrauch erforderlich ist. Rezepturmäßig hergestellte Injektionslösungen und Suspensionen können aus sterilen Pulvern, Granulaten und Tabletten hergestellt werden.
Es versteht sich, daß die Formulierungen neben den obigen besonders erwähnten Bestandteilen andere im Fachgebiet übliche Mittel mit Bezug auf die jeweilige Art der Formulierung enthalten können; so können beispielsweise für die orale Verabreichung geeignete Formulierungen Geschmacksstoffe enthalten.
Eine therapeutisch wirksame Menge einer Verbindung der vorliegenden Erfindung hängt von einer Reihe von Faktoren ab, einschließlich z.B. dem Alter und Gewicht des Menschen oder Tiers, dem exakten Krankheitszustand, der der Behandlung bedarf, sowie seines Schweregrads, der Beschaffenheit der Formulierung sowie dem Verabreichungsweg, und wird letztendlich von dem behandelnden Arzt bzw. Tierarzt festgelegt. Jedoch liegt eine wirksame Menge einer erfindungsgemäßen Verbindung für die Behandlung im allgemeinen im Bereich von 0,1 bis 100 mg/kg Körpergewicht des Empfängers (Säugers) pro Tag und besonders typisch im
Bereich von 1 bis 10 mg/kg Körpergewicht pro Tag. Somit läge für einen
70 kg schweren erwachsenen Säuger die tatsächliche Menge pro Tag für gewöhnlich zwischen 70 und 700 mg, wobei diese Menge als Einzeldosis pro Tag oder üblicher in einer Reihe von Teildosen (wie z.B. zwei, drei, vier, fünf oder sechs) pro Tag gegeben werden kann, so daß die Gesamt- tagesdosis die gleiche ist. Eine wirksame Menge eines Salzes oder
Solvats oder eines physiologisch funktionellen Derivats davon kann als
Anteil der wirksamen Menge der erfindungsgemäßen Verbindung perse bestimmt werden. Es läßt sich annehmen, daß ähnliche Dosierungen für die Behandlung der anderen, obenerwähnten Krankheitszustände geeignet sind.
Gegenstand der Erfindung sind ferner Arzneimittel enthaltend mindestens eine erfindungsgemäße Verbindung und/oder ihre pharmazeutisch verwendbaren Derivate, Tautomere, Solvate und Stereoisomere, 0 einschließlich deren Mischungen in allen Verhältnissen, und mindestens einen weiteren Arzneimittelwirkstoff.
Gegenstand der Erfindung ist auch ein Set (Kit), bestehend aus getrennten e Packungen von
(a) einer wirksamen Menge an einer erfindungsgemäßen Verbindung und/oder ihrer pharmazeutisch verwendbaren Derivate, Tautomere, Solvate und Stereoisomere, einschließlich deren Mischungen in allen Verhältnissen, und
(b) einer wirksamen Menge eines weiteren Arzneimittelwirkstoffs.
Das Set enthält geeignete Behälter, wie Schachteln oder Kartons, 5 individuelle Flaschen, Beutel oder Ampullen. Das Set kann z.B. separate Ampullen enthalten, in denen jeweils eine wirksame Menge an einer erfindungsgemäßen Verbindung und/oder ihrer pharmazeutisch verwendbaren Derivate, Tautomere, Solvate und Stereoisomere, Q einschließlich deren Mischungen in allen Verhältnissen, und einer wirksamen Menge eines weiteren Arzneimittelwirkstoffs gelöst oder in lyophilisierter Form vorliegt.
5
VERWENDUNG
1. Die offenbarten Verbindungen der Formel I sind bei therapeu- tischen Anwendungen in Verbindung mit einer durch CHK1 vermittelten
Störung geeignet. Wie hier verwendet, umfasst der Begriff "durch CHK1 vermittelte Störung" jede Störung, jede Erkrankung oder jeden Zustand, die/der durch einen Anstieg der CHK1 -Expression oder -Aktivität verur- sacht wird oder gekennzeichnet ist oder der CHK1 -Aktivität erfordert. Der Begriff "durch CHK1 vermittelte Störung" umfasst ferner jede Störung, jede Erkrankung oder jeden Zustand, bei der/dem eine Hemmung der CHK1- Aktivität vorteilhaft ist.
CHK1-Hemmung kann dazu verwendet werden, eine günstige therapeutische oder prophylaktische Wirkung, beispielsweise bei Patienten mit einer proliferativen Störung, zu erzielen. Nichtbeschränkende Beispiele für proliferative Störungen sind u.a. chronische entzündliche proliferative Störungen, z.B. Psoriasis und rheumatoide Arthritis, proliferative Augenstörungen, z.B. diabetische Retinopathie, gutartige proliferative Störungen, z.B. Hämangiome, sowie Krebs. Wie hier verwendet, betrifft der Begriff "Krebs" eine zelluläre Störung, die durch eine unkontrollierte oder falsch regulierte Zellproliferation, verringerte Zelldifferenzierung, die unange- messene Fähigkeit, in umgebendes Gewebe einzudringen, und/oder die Fähigkeit, neues Wachstum an ektopischen Stellen zu etablieren, gekennzeichnet ist. Der Begriff "Krebs" umfasst, ist aber nicht beschränkt auf, solide Tumoren und im Blut entstehende Tumoren. Der Begriff "Krebs" umfasst Erkrankungen von Haut, Geweben, Organen, Knochen, Knorpel,
Blut und Gefäßen. Der Begriff "Krebs" umfasst ferner primäre und metastasierende Krebserkrankungen.
Nichtbeschränkende Beispiele für solide Tumoren, die mit den offenbarten CHK1 -Inhibitoren behandelt werden können, sind u.a. Pankreaskrebs,
Blasenkrebs, Kolorektalkrebs, Brustkrebs, einschließlich metastasieren- dem Brustkrebs, Prostatakrebs, einschließlich androgenabhängigem und androgenunabhängigem Prostatakrebs, Nierenkrebs, einschließlich z.B. metastasierendem Nierenzellkarzinom, Leberzellkrebs, Lungenkrebs, 5 einschließlich z.B. nicht-kleinzelligem Lungenkrebs (NSCLC),
Bronchioloalveolarkarzinom (BAC) und Adenokarzinom der Lunge, Ovarkrebs, einschließlich z.B. progressivem epithelialem oder primären Peritonealkrebs, Gebärmutterhalskrebs, Magenkrebs, Speiseröhrenkrebs,
10 Kopf- und Halskrebs, einschließlich z.B. Schuppenzellkarzinom des
Kopfes und des Halses, Melanom, neuroendokriner Krebs, einschließlich metastasierender neuroendokriner Tumoren, Hirntumoren, einschließlich z.B. Gliom, anaplastischem Oligodendrogliom, Glioblastoma multiforme
Λ c bei Erwachsenen und anaplastischem Astrozytom bei Erwachsenen, Knochenkrebs und Weichgewebesarkom.
Nichtbeschränkende Beispiele für hämatologische Malignitäten, die mit den offenbarten CHK1 -Inhibitoren behandelt werden können, sind u.a.
20 akute myeloische Leukämie (AML), chronische myeologene Leukämie
(CML), einschließlich beschleunigter CML und CML-Blastenphase (CML- BP)1 akute lymphoblastische Leukämie (ALL), chronische lymphozytische Leukämie (CLL), Hodgkin-Erkrankung (HD), Non-Hodgkin-Lymphom
25 (NHL), einschließlich follikulärem Lymphom und Mantelzelllymphom, B- Zell-Lymphom, T-Zell-Lymphom, multiples Myelom (MM), Waldenström- Makroglobulinämie, myelodysplastische Syndrome (MDS), einschließlich refraktärer Anämie (RA), refraktärer Anämie mit Ringsideroblasten
O0 (RARS), refraktärer Anämie mit Blastenüberschuss (RAEB) und RAEB in Transformation (RAEB-T), sowie myeloproliferative Syndrome.
Die offenbarten Verbindungen der Formel I eignen sich zur Behandlung von Krebsarten oder Zelltypen, bei denen CHK1 -Protein oder -Aktivität
35 hochreguliert ist, einschließlich, ohne Beschränkung, schnell proliferierender Zellen und arzneistoffresistenter Zellen (Shyjan et al., U.S.-
Patent Nr. 6,723,498 (2004)) sowie Retinoblastomen, wie Rb-negative oder inaktivierte Zellen (Gottifredi et al., Mol. Cell Biol., 21 :1066 (2001)), oder bei denen der ARFp14/p19-Locus inaktiviert oder falsch reguliert ist. Die offenbarten CHK1-Inhibitoren eignen sich auch besonders zur Behandlung von Krebsarten oder Zelltypen, bei denen ein anderer Kontrollpunkt-Weg mutiert oder aufgehoben ist, einschließlich, ohne Beschränkung, Krebsarten oder Zelltypen, bei denen p53 oder der p53-Weg inaktiviert oder aufgehoben ist.
Die offenbarten Verbindungen der Formel I können in Verbindung mit anderen Therapeutika, einschließlich Antikrebsmitteln, verabreicht werden. Wie hier verwendet, betrifft der Begriff "Antikrebsmittel" jedes Mittel, das einem Patienten mit Krebs zum Zweck der Behandlung des Krebses verabreicht wird.
Die hier definierte Antikrebsbehandlung kann als alleinige Therapie angewendet werden oder zusätzlich zu der erfindungsgemäßen Verbindung herkömmliche Operation oder Strahlungstherapie oder Chemotherapie umfassen. Eine derartige Chemotherapie kann eine oder mehrere der folgenden Kategorien von Antitumormitteln umfassen:
(i) antiproliferative/antineoplastische/DNA schädigende Mittel und Kombinationen davon, wie in der medizinischen Onkologie verwendet, wie Alkylierungsmittel (zum Beispiel Cisplatin, Carboplatin, Cyclophosphamid, Nitrogen Mustard, Melphalan, Chlorambucil, Busulphan und Nitroso- harnstoffe); Antimetaboliten (z.B. Antifolate, wie Fluorpyrimidine, wie 5- Fluoruracil und Tegafur, Raltitrexed, Methotrexat, Cytosinarabinosid, Hydroxyharnstoff und Gemcitabin); Antitumor-Antibiotika (z.B. Anthra- cycline, wie Adriamycin, Bleomycin, Doxorubicin, Daunomycin, Epirubicin, Idarubicin, Mitomycin-C, Dactinomycin und Mithramycin); antimitotische
Mittel (zum Beispiel Vinca-Alkaloide, wie Vincristin, Vinblastin, Vindesin und Vinorelbin, und Taxoide, wie Taxol und Taxoter); Topoisomerase-
Inhibitoren (zum Beispiel Epipodophyllotoxine, wie Etoposid und
Teniposid, Amsacrin, Topotecan, Irinotecan und Camptothecin) und zeildifferenzierende Mittel (zum Beispiel all-trans-Retinsäure, 13-cis- Retinsäure und Fenretinid);
(ii) zytostatische Mittel, wie Anti-Östrogene (z.B. Tamoxifen,
Toremifen, Raloxifen, Droloxifen und lodoxyfen), den Östrogenrezeptor nach unten regulierende Mittel (zum Beispiel Fulvestrant), Anti-Androgene (z.B. Bicalutamid, Flutamid, Nilutamid und Cyproteronacetat), LHRH- Antagonisten oder LHRH-Agonisten (zum Beispiel Goserelin, Leuprorelin und Buserelin), Progesterone (zum Beispiel Megestrolacetat), Aromatase- Inhibitoren (zum Beispiel Anastrozol, Letrozol, Vorazol und Exemestan) und Inhibitoren der 5α-Reduktase, wie Finasterid; (iii) Mittel, die die Invasion von Krebszellen hemmen (zum Beispiel Metalloproteinase-Inhibitoren, wie Marimastat und Inhibitoren der Urokinase-Plasminogenaktivator-Rezeptor-Funktion); (iv) Inhibitoren der Wachstumsfaktor-Funktion, zum Beispiel umfassen solche Inhibitoren Wachstumsfaktor-Antikörper, Wachstumsfaktor- Rezeptor-Antikörper (zum Beispiel den Anti-erbb2-Antikörper Trastuzumab
[Herceptin™] und den Anti-erbb1 -Antikörper Cetuximab [C225]), Famesyl- transferase-lnhibitoren, Tyrosinkinase-Inhibitoren und Serin / Threonin- Kinase-Inhibitoren, zum Beispiel Inhibitoren der epidermalen Wachstumsfaktor-Familie (zum Beispiel Inhibitoren der Tyrosinkinasen der EGFR- Familie, wie N-(3-Chlor-4-fluorphenyl)-7-methoxy-6-(3-morpholinopropoxy)- chinazolin-4-amin (Gefitinib, AZD1839), N-(3-Ethinylphenyl)-6,7-bis(2- methoxyethoxy)chinazolin-4-amin (Erlotinib, OSI-774) und 6-Acrylamido-N- (3-chlor-4-fluorphenyl)-7-(3-morpholinopropoxy)chinazolin-4-amin (Cl 1033)), zum Beispiel Inhibitoren der von Plättchen abstammenden Wachstumsfaktor-Familie und zum Beispiel Inhibitoren der Hepatozytenwachs- tumsfaktor-Familie; (v) antiangiogene Mittel, wie solche, die die Wirkungen des vaskulären endothelialen Wachstumsfaktors hemmen (zum Beispiel der
Antikörper gegen den vaskulären Endothelzell-Wachstumsfaktor
Bevacizumab [Avastin™], Verbindungen, wie die in den veröffentlichten internationalen Patentanmeldungen WO 97/22596, WO 97/30035, WO 97/32856 und WO 98/13354 offenbarten) und Verbindungen, die durch andere Mechanismen wirken (zum Beispiel Linomid, Inhibitoren der lntegrin-αvß3-Funktion und Angiostatin);
(vi) gefäßschädigende Mittel, wie Combretastatin A4 und in den internationalen Patentanmeldungen WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 und WO 02/08213 offenbarte Verbindungen;
(vii) Antisense-Therapien, zum Beispiel diejenigen, die gegen die vorstehend aufgelisteten Ziele gerichtet sind, wie ISIS 2503, ein anti-Ras- Antisense; (viii) Genetherapieansätze, einschließlich beispielsweise Ansätze zum Ersetzen von veränderten Genen, wie verändertem p53 oder verändertem BRCA1 oder BRCA2, GDEPT- (gene-directed enzyme pro-drug-Therapie-) Ansätze, die diejenigen, die Cytosindesaminase, Thymidinkinase oder ein bakterielles Nitroreduktase-Enzym verwenden, sowie Ansätze zur
Erhöhung der Patiententoleranz gegenüber Chemotherapie oder
Strahlungstherapie, wie Multi-Drug-Resistence-Gen-Therapie; und
(ix) Immuntherapieansätze, einschließlich beispielsweise Ex-vivo- und
In-vivo-Ansätzen zur Erhöhung der Immunogenität von Patiententumor- zellen, wie Transfektion mit Cytokinen, wie Interleukin 2, Interleukin 4 oder Granulozyten-Makrophagen-Kolonie-stimulierendem Faktor, Ansätze zur Verringerung der T-Zell-Anergie, Ansätze unter Verwendung transfizierter Immunzellen, wie mit Cytokin transfizierter dendritischer Zellen, Ansätze unter Verwendung mit Cytokin transfizierter Tumorzelllinien und Ansätze unter Verwendung anti-idiotypischer Antikörper.
Bevorzugt aber nicht ausschliesslich werden die Arzneimittel der nachstehenden Tabelle 1 mit den Verbindungen der Formel I kombiniert.
Tabelle !
Alkylierungsmittel Cyclophosphamid Lomustin
Busulfan Procarbazin
Ifosfamid Altretamin
Melphalan Estramustinphosphat
Hexamethylmelamin Mechlorethamin
Thiotepa Streptozocin
Chlorambucil Temozolomid
Dacarbazin Semustin
Carmustin
Platinmittel Cisplatin Carboplatin
Oxaliplatin ZD-0473 (AnorMED)
Spiroplatin Lobaplatin (Aetema)
Carboxyphthalatoplatinum Satraplatin (Johnson
Tetraplatin Matthey)
Ormiplatin BBR-3464 (Hoffrnann-La
Iproplatin Roche)
SM-11355 (Sumitomo)
AP-5280 (Access)
Antimetabolite Azacytidin Tomudex
Gemcitabin Trimetrexate
Capecitabin Deoxycoformycin
5-Fluoruracil Fludarabin
Floxuridin Pentostatin
2-Chlordesoxyadenosin Raltitrexed
6-Mercaptopurin Hydroxyharnstoff
6-Thioguanin Decitabin (SuperGen)
Cytarabin Clofarabin (Bioenvision)
2-Fluordesoxycytidin Irofulven (MGI Pharma)
Methotrexat DMDC (Hoffmann-La
Idatrexate Roche)
Ethinylcytidin (Taiho )
Topoisomerase- Amsacrin Rubitecan (SuperGen)
Inhibitoren Epirubicin Exatecanmesylat (Daiichi)
Etoposid Quinamed (ChemGenex)
Teniposid oder Gimatecan (Sigma- Tau)
Mitoxantron Diflomotecan (Beaufour- lrinotecan (CPT-11) Ipsen)
7-Ethyl-10- TAS-103 (Taiho) hydroxycamptothecin Elsamitrucin (Spectrum)
Topotecan J-107088 (Merck & Co)
Dexrazoxanet BNP-1350 (BioNumerik)
(TopoTarget) CKD-602 (Chong Kun
Pixantron (Novuspharrna) Dang)
Rebeccamycin-Analogon KW-2170 (Kyowa Hakko)
(Exelixis)
BBR-3576 (Novuspharrna)
Antitumor- Dactinomycin (Actinomycin Amonafid
Antibiotika D) Azonafid
Doxorubicin (Adriamycin) Anthrapyrazol
Deoxyrubicin Oxantrazol
Valrubicin Losoxantron
Daunorubicin Bleomycinsulfat
(Daunomycin) (Blenoxan)
Epirubicin Bleomycinsäure
Therarubicin Bleomycin A
Idarubicin Bleomycin B
Rubidazon Mitomycin C
Plicamycinp MEN-10755 (Menarini)
Porfiromycin GPX-100 (Gem
Cyanomorpholino- Pharmaceuticals) doxorubicin
Mitoxantron (Novantron)
Antimitotische Paclitaxel SB 408075
Mittel Docetaxel (GlaxoSmithKline)
Colchicin E7010 (Abbott)
Vinblastin PG-TXL (Cell
Vincristin Therapeutics)
Vinorelbin IDN 5109 (Bayer)
Vindesin A 105972 (Abbott)
Dolastatin 10 (NCI) A 204197 (Abbott)
Rhizoxin (Fujisawa) LU 223651 (BASF)
Mivobulin (Warner- D 24851 (ASTA Medica)
Lambert) ER-86526 (Eisai)
Cemadotin (BASF) Combretastatin A4 (BMS)
RPR 109881A (Aventis) Isohomohalichondrin-B
TXD 258 (Aventis) (PharmaMar)
Epothilon B (Novartis) ZD 6126 (AstraZeneca)
T 900607 (Tularik) PEG-Paclitaxel (Enzon)
T 138067 (Tularik) AZ10992 (Asahi)
Cryptophycin 52 (EIi Lilly) !DN-5109 (lndena)
Vinflunin (Fabre) AVLB (Prescient
Auristatin PE (Teikoku NeuroPharma)
Hormone) Azaepothilon B (BMS)
BMS 247550 (BMS) BNP- 7787 (BioNumerik)
BMS 184476 (BMS) CA-4-Prodrug (OXiGENE)
BMS 188797 (BMS) Dolastatin-10 (NrH)
Taxoprexin (Protarga) CA-4 (OXiGENE)
Retinsäure- Fenretinid (Johnson & Alitretinoin (Ligand) rezeptor- Johnson)
Agonisten LGD-1550 (Ligand)
ImmunInterferon Dexosom-Therapie modulatoren Oncophage (Antigenics) (Anosys)
GMK (Progenics) Pentrix (Australian Cancer
Adenokarzinom-Impfstoff Technology)
(Biomira) JSF-154 (Tragen)
CTP-37 (AVI BioPharma) Krebsimpfstoff (Intercell)
JRX-2 (Immuno-Rx) Norelin (Biostar)
PEP-005 (Peplin Biotech) BLP-25 (Biomira)
Synchrovax-Impfstoffe MGV (Progenics)
(CTL Immuno) !3-Alethin (Dovetail)
Melanom-Impfstoff (CTL CLL-Thera (Vasogen)
Immuno) p21-RAS-lmpfstoff
(GemVax)
Hormonelle und Östrogene Prednison antihormonelle konjugierte Östrogene Methylprednisolon
Mittel Ethinylestradiol Prednisolon
Chlortrianisen Aminoglutethimid
Idenestrol Leuprolid
Hydroxyprogesteron- Goserelin caproat Leuporelin
Medroxyprogesteron Bicalutamid
Testosteron Flutamid
Testosteronpropionat Octreotid
Fluoxymesteron Nilutamid
Methyltestosteron Mitotan
Diethylstilbestrol P-04 (Novogen)
Megestrol 2-Methoxyöstradiol
Tamoxifen (EntreMed)
Toremofin Arzoxifen (EIi Lilly)
Dexamethason
Photodynamische Talaporfin (Light Sciences) Pd-Bacteriopheophorbid
Mittel Theralux (Yeda)
(Theratechnologies) Lutetium-Texaphyrin
Motexafin-Gadolinium (Pharmacyclics)
(Pharmacyclics) Hypericin
Tyrosinkinase- Imatinib (Novartis) Kahalid F (PharmaMar)
Inhibitoren Leflunomid CEP- 701 (Cephalon)
(Sugen/Pharmacia) CEP-751 (Cephalon)
ZDI839 (AstraZeneca) MLN518 (Millenium)
Erlotinib (Oncogene P KC412 (Novartis)
Science) Phenoxodiol O
Canertjnib (Pfizer) Trastuzumab (Genentech)
Squalamin (Genaera) C225 (ImCIone)
SU5416 (Pharmacia) rhu-Mab (Genentech)
SU6668 (Pharmacia) MDX-H210 (Medarex)
ZD4190 (AstraZeneca) 2C4 (Genentech)
ZD6474 (AstraZeneca) MDX-447 (Medarex)
Vatalanib (Novartis) ABX-EGF (Abgenix)
PKI166 (Novartis) IMC-1C11 (ImCIone)
GW2016
(GlaxoSmithKline)
EKB-509 (Wyeth)
EKB-569 (Wyeth)
Verschiedene SR-27897 (CCK-A- BCX-1777 (PNP-lnhibitor,
Mittel Inhibitor, Sanofi- BioCryst)
Synthelabo) Ranpirnase
Tocladesin (cyclisches- (Ribonuclease-Stimulans,
AMP-Agonist, Ribapharm) Alfacell)
Alvocidib (CDK-Inhibitor, Galarubicin (RNA-
Aventis) Synthese-Inhibitor, Dong-
CV-247 (COX-2-lnhibitor, A)
Ivy Medical) Tirapazamin
P54 (COX-2-lnhibitor, (Reduktionsmittel, SRI
Phytopharm) International)
CapCell™ (CYP450- N-Acetylcystein
Stimulans, Bavarian (Reduktionsmittel,
Nordic) Zambon)
GCS-IOO (gal3- R-Flurbiprofen (NF-
Antagonist, kappaB-lnhibitor, Encore)
GlycoGenesys) 3CPA (NF-kappaB-
G17DT-Immunogen Inhibitor, Active Biotech)
(Gastrin-Inhibitor, Aphton) Seocalcitol (Vitamin-D-
Efaproxiral (Oxygenator, Rezeptor-Agonist, Leo)
Allos Therapeutics) 131-I-TM-601 (DNA-
PI-88 (Heparanase- Antagonist,
Inhibitor, Progen) TransMolecular)
Tesmilifen (Histamin- Eflornithin (ODC-Inhibitor,
Antagonist, YM ILEX Oncology)
BioSciences) Minodronsäure
Histamin (Histamin-H2- (Osteoclasten-Inhibitor,
Rezeptor- Agonist, Maxim) Yamanouchi)
Tiazofurin (IMPDH- Indisulam (p53-Stimulans,
Inhibitor, Ribapharm) Eisai)
Cilengitid (Integrin- Aplidin (PPT-Inhibitor,
Antagonist, Merck KGaA) PharmaMar)
SR-31747 (IL-1- Rituximab (CD20-
Antagonist, Sanofi- Antikörper, Genentech)
Synthelabo) Gemtuzumab (CD33-
CCI-779 (mTOR-Kinase- Antikörper, Wyeth Ayerst)
Inhibitor, Wyeth) PG2 (Hämatopoese-
Exisulind (PDE-V-Inhibitor, Verstärker,
Cell Pathways) Pharmagenesis)
CP-461 (PDE-V-Inhibitor, Immunol™ (Triclosan-
Cell Pathways) Oralspülung, Endo)
AG-2037 (GART-Inhibitor, Triacetyluridin (Uridin-
Pfizer) Prodrug, Wellstat)
WX-UK1 SN-4071 (Sarkom-Mittel,
(Plasminogenaktivator- Signature BioScience)
Inhibitor, Wilex) TransMID-107™
PBI-1402 (PMN-Stimulans, (Immunotoxin, KS
ProMetic LifeSciences) Biomedix)
Bortezomib (Proteasom- PCK-3145 (Apoptose-
Inhibitor, Millennium) Förderer, Procyon)
SRL-172 (T-ZeII- Doranidazol (Apoptose-
Stimulans, SR Pharma) Förderer, PoIa)
TLK-286 (Glutathion-S- CHS-828 (cytotoxisches
Transferase-Inhibitor, Mittel, Leo)
Telik) trans-Retinsäure
PT-100 (Wachstumsfaktor- (Differentiator, NIH)
Agonist, Point MX6 (Apoptose-Förderer,
Therapeutics) MAXIA)
Midostaurin (PKC-Inhibitor, Apomin (Apoptose-
Novartis) Förderer, ILEX Oncology)
Bryostatin-1 (PKC- Urocidin (Apoptose-
Stimulans, GPC Biotech) Förderer, Bioniche)
CDA-II (Apoptose- Ro-31-7453 (Apoptose-
Förderer, Everlife) Förderer, La Roche)
SDX-101 (Apoptose- Brostallicin (Apoptose-
Förderer, Salmedix) Förderer, Pharmacia)
Ceflatonin (Apoptose-
Förderer, ChemGenex)
Alkylierungsmittel Cyclophosphamid Lomustin
Busulfan Procarbazin
Ifosfamid Altretamin
Melphalan Estramustinphosphat
Hexamethylmelamin Mechlorethamin
Thiotepa Streptozocin
Chlorambucil Temozolomid
Dacarbazin Semustin
Carmustin
Platinmittel Cisplatin Carboplatin
Oxaliplatin ZD-0473 (AnorMED)
Spiroplatin Lobaplatin (Aetema)
Carboxyphthalatoplatinum Satraplatin (Johnson
Tetraplatin Matthey)
Ormiplatin BBR-3464 (Hoffrnann-La
Iproplatin Roche)
SM-11355 (Sumitomo)
AP-5280 (Access)
Antimetabolite Azacytidin Tomudex
Gemcitabin Trimetrexate
Capecitabin Deoxycoformycin
5-Fluoruracil Fludarabin
Floxuridin Pentostatin
2-Chlordesoxyadenosin Raltitrexed
6-Mercaptopurin Hydroxyhamstoff
6-Thioguanin Decitabin (SuperGen)
Cytarabin Clofarabin (Bioenvision)
2-Fluordesoxycytidin Irofulven (MGI Pharma)
Methotrexat DMDC (Hoffmann-La
Idatrexate Roche)
Ethinylcytidin (Taiho )
Topoisomerase- Amsacrin Rubitecan (SuperGen)
Inhibitoren Epirubicin Exatecanmesylat (Daiichi)
Etoposid Quinamed (ChemGenex)
Teniposid oder Gimatecan (Sigma- Tau)
Mitoxantron Diflomotecan (Beaufour- lrinotecan (CPT-11) Ipsen)
7-Ethyl-10- TAS-103 (Taiho) hydroxycamptothecin Elsamitrucin (Spectrum)
Topotecan J-107088 (Merck & Co)
Dexrazoxanet BNP-1350 (BioNumerik)
(TopoTarget) CKD-602 (Chong Kun
Pixantron (Novuspharrna) Dang)
Rebeccamycin-Analogon KW-2170 (Kyowa Hakko)
(Exelixis)
BBR-3576 (Novuspharrna)
Antitumor- Dactinomycin (Actinomycin Amonafid
Antibiotika D) Azonafid
Doxorubicin (Adriamycin) Anthrapyrazol
Deoxyrubicin Oxantrazol
Valrubicin Losoxantron
Daunorubicin Bleomycinsulfat
(Daunomycin) (Blenoxan)
Epirubicin Bleomycinsäure
Therarubicin Bleomycin A
Idarubicin Bleomycin B
Rubidazon Mitomycin C
Plicamycinp MEN-10755 (Menarini)
Porfiromycin GPX-100 (Gem
Cyanomorpholinodoxorubi Pharmaceuticals) ein
Mitoxantron (Novantron)
Antimitotische Paclitaxel SB 408075
Mittel Docetaxel (GlaxoSmithKline)
Colchicin E7010 (Abbott)
Vinblastin PG-TXL (Cell
Vincristin Therapeutics)
Vinorelbin IDN 5109 (Bayer)
Vindesin A 105972 (Abbott)
Dolastatin 10 (NCI) A 204197 (Abbott)
Rhizoxin (Fujisawa) LU 223651 (BASF)
Mivobulin (Warner- D 24851 (ASTA Medica)
Lambert) ER-86526 (Eisai)
Cemadotin (BASF) Combretastatin A4 (BMS)
RPR 109881A (Aventis) Isohomohalichondrin-B
TXD 258 (Aventis) (PharmaMar)
Epothilon B (Novartis) ZD 6126 (AstraZeneca)
T 900607 (Tularik) PEG-Paclitaxel (Enzon)
T 138067 (Tularik) AZ10992 (Asahi)
Cryptophycin 52 (EIi Lilly) IDN-5109 (Indena)
Vinflunin (Fabre) AVLB (Prescient
Auristatin PE (Teikoku NeuroPharma)
Hormone) Azaepothilon B (BMS)
BMS 247550 (BMS) BNP- 7787 (BioNumerik)
BMS 184476 (BMS) CA-4-Prodrug (OXiGENE)
BMS 188797 (BMS) Dolastatin-10 (NrH)
Taxoprexin (Protarga) CA-4 (OXiGENE)
Aromatase- Aminoglutethimid Exemestan
Inhibitoren Letrozol Atamestan (BioMedicines)
Anastrazol YM-511 (Yamanouchi)
Formestan
ImmunInterferon Dexosom-Therapie modulatoren Oncophage (Antigenics) (Anosys)
GMK (Progenics) Pentrix (Australian Cancer
Adenokarzinom-Impfstoff Technology)
(Biomira) JSF-154 (Tragen)
CTP-37 (AVI BioPharma) Krebsimpfstoff (Intercell)
JRX-2 (Immuno-Rx) Norelin (Biostar)
PEP-005 (Peplin Biotech) BLP-25 (Biomira)
Synchrovax-Impfstoffe MGV (Progenics)
(CTL Immuno) !3-Alethin (Dovetail)
Melanom-Impfstoff (CTL CLL-Thera (Vasogen)
Immuno) p21-RAS-lmpfstoff
(GemVax)
Hormonelle und Östrogene Prednison antihormonelle konjugierte Östrogene Methylprednisolon
Mittel Ethinylöstradiol Prednisolon
Chlort rianisen Aminoglutethimid ldenestrol Leuprolid
Hydroxyprogesteroncaproa Goserelin t Leuporelin
Medroxyprogesteron Bicalutamid
Testosteron Flutamid
Testosteronpropionat Octreotid
Fluoxymesteron Nilutamid
Methyltestosteron Mitotan
Diethylstilbestrol P-04 (Novogen)
Megestrol 2-Methoxyöstrad iol
Tamoxifen (EntreMed)
Toremofm Arzoxifen (EIi Lilly)
Dexamethason
Photodynamische Talaporfin (Light Sciences) Pd-Bacteriopheophorbid
Mittel Theralux (Yeda)
(Theratechnologies) Lutetium-Texaphyrin
Motexafin-Gadolinium (Pharmacyclics)
(Pharmacyclics) Hypericin
Tiazofurin (IMPDH- Rituximab (CD20-
Inhibitor, Ribapharm) Antikörper, Genentech)
Cilengitid (Integrin- Gemtuzumab (CD33-
Antagonist, Merck KGaA) Antikörper, Wyeth Ayerst)
SR-31747 (IL-1- PG2 (Hämatopoese-
Antagonist, Sanofi- Verstärker,
Synthelabo) Pharmagenesis)
CCI-779 (mTOR-Kinase- Immunol™ (Triclosan-
Inhibitor, Wyeth) Oralspülung, Endo)
Exisulind (PDE-V- Triacetyluridin (Uridin-
Inhibitor, Cell Pathways) Prodrug, Wellstat)
10 CP-461 (PDE-V-Inhibitor, SN-4071 (Sarkom-Mittel,
Cell Pathways) Signature BioScience)
AG-2037 (GART-Inhibitor, TransMID-107™
Pfizer) (Immunotoxin, KS
WX-UK1 Biomedix)
(Plasminogenaktivator- PCK-3145 (Apoptose-
Inhibitor, Wilex) Förderer, Procyon)
15 PBI-1402 (PMN- Doranidazol (Apoptose-
Stimulans, ProMetic Förderer, PoIa)
LifeSciences) CHS-828 (cytotoxisches
Bortezomib (Proteasom- Mittel, Leo)
Inhibitor, Millennium) trans-Retinsäure
SRL-172 (T-ZeII- (Differentiator, NIH)
20 Stimulans, SR Pharma) MX6 (Apoptose-Förderer,
TLK-286 (Glutathion-S- MAXIA)
Transferase-Inhibitor, Apomin (Apoptose-
Telik) Förderer, ILEX Oncology)
PT-100 Urocidin (Apoptose-
(Wachstumsfaktor- Förderer, Bioniche)
Agonist, Point Ro-31-7453 (Apoptose-
25 Therapeutics) Förderer, La Roche)
Midostaurin (PKC- Brostallicin (Apoptose-
Inhibitor, Novartis) Förderer, Pharmacia)
Bryostatin-1 (PKC-
Stimulans, GPC Biotech)
CDA-II (Apoptose-
30 Förderer, Everlife)
SDX-101 (Apoptose-
Förderer, Salmedix)
Ceflatonin (Apoptose-
Förderer, ChemGenex)
35
Eine derartige gemeinsame Behandlung kann mithilfe gleichzeitiger, aufeinander folgender oder getrennter Dosierung der einzelnen Komponenten der Behandlung erzielt werden. Solche Kombinationsprodukte setzen die erfindungsgemäßen Verbindungen ein. 5
2. Die vorliegenden Verbindungen der Formel I eignen sich insbesondere als pharmazeutische Wirkstoffe für Säugetiere, insbesondere für den Menschen, bei der Behandlung von SGK-bedingten 10 Krankheiten.
Gegenstand der Erfindung ist somit die Verwendung von Verbindungen nach Anspruch 1 , sowie ihrer pharmazeutisch verwendbaren Derivate, ^g Solvate und Stereoisomere, einschließlich deren Mischungen in allen Verhältnissen, zur Herstellung eines Arzneimittels zur Behandlung von Krankheiten, bei denen die Hemmung, Regulierung und/oder Modulation der Signaltransduktion von Kinasen eine Rolle spielt.
Bevorzugt ist die Verwendung von Verbindungen gemäß Anspruch 1 ,
20 sowie ihrer pharmazeutisch verwendbaren Derivate, Solvate und Stereoisomere, einschließlich deren Mischungen in allen Verhältnissen, zur Herstellung eines Arzneimittels zur Behandlung von Krankheiten, die durch Inhibierung der SGK durch die Verbindungen nach Anspruch 1 5 beeinflußt werden.
Die vorliegende Erfindung umfasst die Verwendung der erfindungsgemäßen Verbindungen nach Anspruch 1 und/oder ihre physiologisch Q unbedenklichen Salze und Solvate zur Herstellung eines Arzneimittels zur Behandlung oder Vorbeugung von Diabetes (z.B. Diabetes mellitus, diabetische Nephropathie, diabetische Neuropathie, diabetische Angiopathie und Mikroangiopathie), Fettsucht, metabolisches Syndrom
(Dyslipidämie), systemische und pulmonale Hypertonie, Herzkreis- 5 lauferkrankungen (z.B. kardiale Fibrosen nach Myokardinfarkt, Herzhypertrophie und Herzinsuffizienz, Arteriosklerose) und Nierener-
krankungen (z.B. Glomerulosklerose, Nephrosklerose, Nephritis, Nephropathie, Störung der Elektrolytausscheidung), allgemein bei jeglicher Art von Fibrosen und entzündlichen Prozessen (z.B. Leberzirrhose,
Lungenfibrose, fibrosierende Pankreatitis, Rheumatismus und Arthrosen, 5
Morbus Crohn, chronische Bronchitis, Strahlenfibrose, Sklerodermitis, zystische Fibrose, Narbenbildung, Morbus Alzheimer). Die erfindungsgemäßen Verbindungen können auch das Wachstum von Krebs, Tumorzellen und Tumormetastasen hemmen und sind deshalb für
10 die Tumortherapie geeignet.
Die erfindungsgemäßen Verbindungen finden weiterhin Verwendung zur Behandlung von Koagulopathien, wie z.B. Dysfibrinogenämie, Hypopro- konvertinämie, Hämophilie B, Stuart-Prower-Defekt, Prothrombin-
<j c Komplex-Mangel, Verbrauchskoagulopathie, Hyperfibrinolyse, Immuno- koagulopathie oder komplexer Koagulopathien, wie auch bei neuronaler Erregbarkeit, z.B. Epilepsie. Die erfindungsgemäßen Verbindungen können auch bei der Behandlung eines Glaukoms oder Katarakt therapeutisch eingesetzt werden.
20
Die erfindungsgemäßen Verbindungen finden ferner Verwendung bei der
Behandlung bakterieller Infektionen sowie in einer antiinfektiösen Therapie. Die erfindungsgemäßen Verbindungen können auch zur Steigerung der Lernfähigkeit und Aufmerksamkeit therapeutisch eingesetzt 5 werden.
Bevorzugt ist die Verwendung von Verbindungen gemäß Anspruch 1, sowie ihrer pharmazeutisch verwendbaren Derivate, Solvate und Stereo- Q isomere, einschließlich deren Mischungen in allen Verhältnissen, zur Herstellung eines Arzneimittels zur Behandlung oder Vorbeugung von Diabetes, Fettsucht, metabolischem Syndrom (Dyslipidämie), systemischer und pulmonaler Hypertonie, Herzkreislauferkrankungen und Nierenerkrankungen, allgemein bei jeglicher Art von Fibrosen und entzündlichen 5
Prozessen, Krebs, Tumorzellen, Tumormetastasen, Koagulopathien, neuronaler Erregbarkeit, Glaukom, Katarakt, bakteriellen Infektionen sowie
in einer antiinfektiösen Therapie, zur Steigerung der Lernfähigkeit und Aufmerksamkeit, sowie zur Behandlung und Prophylaxe von Zellalterung und Stress.
Bei Diabetes handelt es sich vorzugsweise um Diabetes mellitus, diabetische Nephropathie, diabetische Neuropathie, diabetische Angiopathie und Mikroangiopathie.
Bei Herzkreislauferkrankungen handelt es sich vorzugsweise um kardiale Fibrosen nach Myokardinfarkt, Herzhypertrophie, Herzinsuffizienz und Arteriosklerose.
Bei Nierenerkrankungen handelt es sich vorzugsweise um Glomerulo-
Sklerose, Nephrosklerose, Nephritis, Nephropathie und Störung der
Elektrolytausscheidung.
Bei Fibrosen und entzündlichen Prozessen handelt es sich vorzugsweise um Leberzirrhose, Lungenfibrose, fibrosierende Pankreatitis, Rheumatismus und Arthrosen, Morbus Crohn, chronische Bronchitis, Strahlenfibrose, Sklerodermitis, zystische Fibrose, Narbenbildung, Morbus Alzheimer.
ASSAYS
Die in den Beispielen beschriebenen erfindungsgemäßen Verbindungen wurden in den unten beschriebenen Assays geprüft, und es wurde gefunden, dass sie eine kinasehemmende Wirkung aufweisen. Weitere
Assays sind aus der Literatur bekannt und könnten vom Fachmann leicht durchgeführt werden (siehe z.B. Dhanabal et al., Cancer Res. 59:189-197; Xin et al., J. Biol. Chem. 274:9116-9121 ; Sheu et al., Anticancer Res. 18:4435-4441 ; Ausprunk et al., Dev. Biol. 38:237-248; Gimbrone et al., J. Natl. Cancer Inst. 52:413-427; Nicosia et al., In Vitro 18:538- 549).
Messung der CHK1 Kinaseaktivität
5 Die CHK1 Kinase wird zum Zweck der Proteinproduktion in Insektenzellen
(Sf21 ; S. frugiperda) und der anschließenden affinitätschromato- graphischen Aufreinigung als Fusionsprotein mit Glutathion S-Transferase in einem Baculovirus-Expressionsvektor exprimiert. Die Kultivierung,
1 f. Infektion und der Aufschluss der Zellen, sowie die säulenchromato- graphische Aufreinigung des Fusionsproteins erfolgen entsprechend Hersteller-orientierter generischer Arbeitsanweisungen.
Zur Messung der Kinase-Aktivität wird auf verschiedene zur Verfügung stehender Meßsysteme zurückgegriffen. Beim Scintillation-Proximity- (Sorg et al., J. of. Biomolecular Screening, 2002, 7, 11-19), dem FlashPlate- Verfahren oder dem Filterbindungstest wird die radioaktive Phosphorylierung eines Proteins oder Peptids als Substrat mit radioaktiv markiertem
ATP (32P-ATP, (33P-ATP) gemessen. Bei Vorliegen einer inhibitorischen 90.
Verbindung ist kein oder ein vermindertes radioaktives Signal nachweisbar. Ferner sind die Homogeneous Time-resolved Fluorescence Resonance Energy Transfer- (HTR-FRET-) und Fluoreszenzpolarisations- (FP-) Technologien als Assay-Verfahren nützlich (SiIIs et al., J. of 25 Biomolecular Screening, 2002, 191-214).
Andere nicht radioaktive ELISA-Assay-Verfahren verwenden spezifische Phospho-Antikörper (Phospho-AK). Der Phospho-Antikörper bindet nur das phosphorylierte Substrat. Diese Bindung ist mit einem zweiten 30 Peroxidase-konjugierten Antikörper durch Chemilumineszenz nachweisbar (Ross et al., 2002, Biochem. J.).
Flashplate-Verfahren (CHKD:
Als Testplatten dienen 384-well Streptavidin-beschichtete Flashplates
PlusR der Firma Perkin Eimer (Cat.No. SMP410A001PK). Die Assay Platte
wird 30 min vor Versuchsbeginn mit je 75 μl Assay-Puffer pro well equilibriert. Der Puffer wird vor Versuchsbeginn abgesaugt und die Komponenten der unten beschriebenen Kinasereaktion werden auf die
Platte pipettiert.
Die CHK1 Kinase, ein biotinyliertes Substratpeptid (z. Bsp. CHKtide: KKKVSRSGLYRSPSMPENLNRPR) wird mit radioaktiv markiertem ATP in An- und Abwesenheit von Testsubstanzen bei 30° Celsius und einem Gesamtvolumen von 50 μl inkubiert. Die Reaktion wird mit 25μl einer 0,2 M EDTA-Lösung abgestoppt. Nach Inkubation für 30 min bei Raumtemperatur werden die Überstände abgesaugt und die wells dreimal mit je 100 μl 0,9% NaCI-Lösung gewaschen. Die Messung der gebundenen Radioaktivität erfolgt mittels eines Szintillationsmessgerätes (Topcount NXT, Fa. Perkin-Elmer).
Als Vollwert wird die Inhibitor-freie Kinasereaktion verwendet. Dieser sollte ca. im Bereich von 3000-4000 cpm liegen. Als pharmakologischer Nullwert wird Staurosporin in einer Endkonzentration von 0,1 μM verwendet. Eine
Bestimmung der Hemmwerte (IC50) erfolgt unter Verwendung des
Programms RS1_MTS ().
Kinase-Reaktionsbedingungen pro well: 5-20 mU CHK1 Kinase 0, 15 μg CHKtide (KKKVSRSGLYRSPSMPENLNRPR) 8 μM ATP, kalt 0,2 μCi 33P-ATP 50 μl Gesamtvolumen (1-fach Assaypuffer-Reaktionsbedingungen)
Verwendete Lösungen:
- Assay-Puffer: 50 mM Tris
0,1 mM Titriplex VI (EGTA
10 mM Magnesiumacetat
0,1 % Mercaptoethanol
0,02% Brij35 pH= 7,5 (einzustellen mit Salzsäure)
Die Zugabe von Rinderserumalbumin (Endkonzentration 0,1%) erfolgt erst kurz vor Verwendung. 5
- Stopp-Lösung:
0,2 M Titriplexlll (EDTA)
10 - 33P-ATP (Perkin-Elmer)
- CHK1 Kinasepräparationen: spezifische Aktivität > 50 U/mg
- CHKtide-Lösung: biotinyliert.es Peptidsubstrat (Firma Biotrend) als Stocklösung (Konzentration 0,15 mg/ml) aufbewahrt.
15
Filterbindunqs- Verfahren (CHK1 ):
5-20 mU CHK1 Kinase (verdünnt in 20 mM MOPS pH7.5, 1 mM EDTA, 0,1% ß-Mercaptoethanol, 0,01% Brij-35, 5% Glyzerin, 1 mg/ml BSA) werden in Gegenwart von 30-200 μM CHKtide in 25,5 μl in 1-fach
20 Reaktionspuffer (8 mM MOPS pH7, 0,2 mM EDTA1 10 mM
Magnesiumacetat, 0,02 mM 33P-ATP [500-1000 cpm/pmol]) für 30 min bei Raumtemperatur inkubiert. Die Reaktion wird mit 5 μl 0,5 M Orthophosphorsäure gestoppt und durch P81 Filterplatten filtriert. Nach
25 mehrmaligem Waschen der Filterplatten erfolgt die Bestimmung der gebundenen Radioaktivität im Szintillationszähler.
Messung der CHK2 Kinaseaktivität
•an
0 Filterbindungs-Verfahren (CHK2):
5-20 mU CHK2 Kinase (verdünnt in 20 mM MOPS pH7.5, 1 mM EDTA, 0,1% ß-Mercaptoethanol, 0,01% Brij-35, 5% Glyzerin, 1 mg/ml BSA) werden in Gegenwart von 30-200 μM CHKtide
35 (KKKVSRSGLYRSPSMPENLNRPR) in 25,5 μl in 1-fach Reaktionspuffer (8 mM MOPS pH7, 0,2 mM EDTA, 10 mM Magnesiumacetat, 0,02 mM
33P-ATP [500-1000 cpm/pmol]) für 30 min bei Raumtemperatur inkubiert. Die Reaktion wird mit 5 μl 0,5 M ortho-Phosphorsäure gestoppt und durch P81 Filterplatten filtriert. Nach mehrmaligem Waschen der Filterplatten erfolgt die Bestimmung der gebundenen Radioaktivität im Szintillations-
Zähler.
Die Hemmung der SGK1 Proteinkinase kann im Filterbindungsverfahren (analog zu CHK1 , CHK2) bestimmt werden.
Vor- und nachstehend sind alle Temperaturen in 0C angegeben. In den nachfolgenden Beispielen bedeutet "übliche Aufarbeitung": Man gibt, falls erforderlich, Wasser hinzu, stellt, falls erforderlich, je nach Konstitution des Endprodukts auf pH-Werte zwischen 2 und 10 ein, extrahiert mit
Ethylacetat oder Dichlormethan, trennt ab, trocknet die organische Phase über Natriumsulfat, dampft ein und reinigt durch Chromatographie an Kieselgel und /oder durch Kristallisation. Rf-Werte an Kieselgel; Laufmittel:
Ethylacetat/Methanol 9:1.
Massenspektrometrie (MS): El (Elektronenstoß-Ionisation) M+
FAB (Fast Atom Bombardment) (M+H)+ ESI (Electrospray lonization) (M+H)+ (wenn nichts anderes angegeben) Beispiel 1
Die Herstellung von 3-(3-Oxo-2,3-dihydro-1W-indazol-5-ylamino)-4-(3- hydroxy-benzylamino)-cyclobut-3-en-1 ,2-dion ("1") erfolgt analog nachstehendem Schema:
1. Die Darstellung des 5-Amino-1 ,2-dihydro-indazol-3-ons 2a ist von Kenner und Witham in J. Chem. Soc. 1921, 119, 1057 und von Pfannstiel und Janecke in Chem. Ber. 1942, 75, 1096 und 1104 beschrieben.
2. 1 ,597 g (9,39 mmol) 3,4-Diethoxy-3-cyclobuten-1 ,2-dion Ia werden in 20 ml_ Ethanol gelöst, mit 1 ,4 g (9,39 mmol) 2a versetzt und 20 h bei 750C gerührt. Danach arbeitet man wie üblich auf und so erhält man 1 ,77 g (69%) 3-Ethoxy-4-(3-oxo-2,3-dihydro-1H-indazol-5-ylamino)-cyclobut-3-en- 1 ,2-dion 3a; MS-FAB (M+H+) = 274.
200 mg (0,732 mmol) 3a werden in 2 ml_ Ethanol gelöst, mit 112,7 mg (0,915 mmol) 3-Aminomethyl-phenol 4a versetzt und 48 h bei 75°C gerührt. Danach arbeitet man wie üblich auf und so erhält man 188 mg "1"; MS-FAB (M+H+) = 351.
Analog erhält man
3-(3-Oxo-2,3-dihydro-1H-indazol-5-ylamino)-4-[(R)-1-(3-methoxy- phenyl)-ethylamino]-cyclobut-3-en-1 ,2-dion ("2"), MS-FAB (M+H+) = 379
S-CS-Oxo^.S-dihydro-IH-indazol-δ-ylaminoH-^RJ-i-CS-hydroxy- phenyl)-ethylamino]-cyclobut-3-en-1 ,2-dion ("3"), MS-FAB (M+H+) = 365;
3-(3-Oxo-2,3-dihydro-1H-indazol-5-ylamino)-4-[(R)-1-(3-fluor-phenyl)- ethylamino]-cyclobut-3-en-1 ,2-dion ("4"),
3-(3-Oxo-2,3-dihydro-1/7-indazol-5-ylamino)-4-(3-fluor-benzylamino)- cyclobut-3-en-1 ,2-dion ("5"),
3-(3-Oxo-2,3-dihydro-1H-indazol-5-ylamino)-4-(3-acetamido- benzylamino)-cyclobut-3-en-1 ,2-dion ("6"),
3-(3-Oxo-2,3-dihydro-1/-/-indazol-5-ylamino)-4-(3-methylsulfonamido- benzylamino)-cyclobut-3-en-1 ,2-dion ("7"),
3-(3-Oxo-2,3-dihydro-1H-indazol-5-ylamino)-4-(2,3-difluor- benzylamino)-cyclobut-3-en-1 ,2-dion ("8"),
3-(3-Oxo-2,3-dihydro-1H-indazol-5-ylamino)-4-(3-chlor-benzylamino)- cyclobut-3-en-1 ,2-dion ("9").
Die nachfolgenden Beispiele betreffen pharmazeutische Zubereitungen:
Beispiel A: Injektionsgläser
Eine Lösung von 100 g eines erfindungsgemäßen Wirkstoffes und 5 g
Dinatriumhydrogenphosphat wird in 3 I zweifach destilliertem Wasser mit 2 n Salzsäure auf pH 6,5 eingestellt, steril filtriert, in Injektionsgläser abgefüllt, unter sterilen Bedingungen lyophilisiert und steril verschlossen. Jedes Injektionsglas enthält 5 mg Wirkstoff.
Beispiel B: Suppositorien
Man schmilzt ein Gemisch von 20 g eines erfindungsgemäßen Wirkstoffes mit 100 g Sojalecithin und 1400 g Kakaobutter, gießt in Formen und läßt erkalten. Jedes Suppositorium enthält 20 mg Wirkstoff.
Beispiel C: Lösung
Man bereitet eine Lösung aus 1 g eines erfindungsgemäßen Wirkstoffes,
9,38 g NaH2PO4 • 2 H2O, 28,48 g Na2HPO4 • 12 H2O und 0,1 g
Benzalkoniumchlorid in 940 ml zweifach destilliertem Wasser. Man stellt auf pH 6,8 ein, füllt auf 1 I auf und sterilisiert durch Bestrahlung. Diese
Lösung kann in Form von Augentropfen verwendet werden.
Beispiel D: Salbe
Man mischt 500 mg eines erfindungsgemäßen Wirkstoffes mit 99,5 g Vaseline unter aseptischen Bedingungen.
Beispiel E: Tabletten
Ein Gemisch von 1 kg Wirkstoff, 4 kg Lactose, 1 ,2 kg Kartoffelstärke, 0,2 kg Talk und 0,1 kg Magnesiumstearat wird in üblicher weise zu
Tabletten verpreßt, derart, daß jede Tablette 10 mg Wirkstoff enthält.
Beispiel F: Dragees
Analog Beispiel E werden Tabletten gepreßt, die anschließend in üblicher Weise mit einem Überzug aus Saccharose, Kartoffelstärke, Talk, Tragant und Farbstoff überzogen werden.
Beispiel G: Kapseln
2 kg Wirkstoff werden in üblicher Weise in Hartgelatinekapseln gefüllt, so daß jede Kapsel 20 mg des Wirkstoffs enthält.
Beispiel H: Ampullen
Eine Lösung von 1 kg eines erfindungsgemäßen Wirkstoffes in 60 I zweifach destilliertem Wasser wird steril filtriert, in Ampullen abgefüllt, unter sterilen Bedingungen lyophilisiert und steril verschlossen. Jede Ampulle enthält 10 mg Wirkstoff.