WO2007022015A1 - Methods to evaluate amyloid beta-lowering agents using wild-type mice - Google Patents
Methods to evaluate amyloid beta-lowering agents using wild-type mice Download PDFInfo
- Publication number
- WO2007022015A1 WO2007022015A1 PCT/US2006/031517 US2006031517W WO2007022015A1 WO 2007022015 A1 WO2007022015 A1 WO 2007022015A1 US 2006031517 W US2006031517 W US 2006031517W WO 2007022015 A1 WO2007022015 A1 WO 2007022015A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- peptide
- binding substance
- amount
- antibody
- mouse
- Prior art date
Links
Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5082—Supracellular entities, e.g. tissue, organisms
- G01N33/5088—Supracellular entities, e.g. tissue, organisms of vertebrates
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6893—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids related to diseases not provided for elsewhere
- G01N33/6896—Neurological disorders, e.g. Alzheimer's disease
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/46—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans from vertebrates
- G01N2333/47—Assays involving proteins of known structure or function as defined in the subgroups
- G01N2333/4701—Details
- G01N2333/4709—Amyloid plaque core protein
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/28—Neurological disorders
- G01N2800/2814—Dementia; Cognitive disorders
- G01N2800/2821—Alzheimer
Definitions
- the present invention relates generally to methods and compositions for detecting endogenous brain A ⁇ , and more particularly to assays, such as immunoassays, for screening for compounds that specifically alter the level of A ⁇ .
- Amyloid beta is a major component of plaques in Alzheimer's disease (AD) and Down syndrome.
- a ⁇ is produced from the amyloid precursor protein (APP) by sequential proteolytic processing at the N- and C-termini of the A ⁇ domain by beta- and gamma- secretases, respectively.
- APP amyloid precursor protein
- a ⁇ occurs principally in two forms consisting of 40 and 42 amino acids, A ⁇ l-40 and 1-42 (Selkoe DJ. 1993 Trends in Neurosciences 16:403-409).
- a ⁇ lowering is a promising therapeutic approach for AD.
- the first transgenic mouse model to form A ⁇ deposits overexpressed a mutant (V717) form of APP at high levels, generated elevated levels of A ⁇ in general, and A ⁇ 42 in particular, and formed A ⁇ plaques at approximately 6 months of age (Games D. et al. 1995 Nature 373:523-527).
- a second mouse model overexpressed the "Swedish" mutant APP (APPK 67ON , M67IL ), showed elevated A ⁇ 40 and 42 levels, and developed amyloid deposits between 10 and 14 months of age (Hsiao K. et al. 1996 Science 274:99-102).
- PS1 MI46L Mutant presenilin 1 transgenic mice have subtly elevated levels of the A ⁇ 42 peptide, but they lack pathological changes (Duff K. et al. 1996 Nature 383:710- 713).
- Transgenic mice derived from a cross between the APP line, Tg2576 and Duffs mutant PSl mice (known as PS/APP) show markedly accelerated accumulation of A ⁇ into visible deposits (deposition is initiated at approximately 10 weeks of age) compared with APP singly transgenic mice (Holcomb L. et al. 1998 Nat Med 4:97-100).
- transgenic models of amyloidosis has significantly aided progress in the development of therapeutic approaches designed to lower brain A ⁇ load.
- all transgenic mice express their transgenes under the control of an autologous promoter.
- testing using non-genetically manipulated animal is required.
- Three amino acids were different between rodent and primate A ⁇ , and human-specific enzyme linked immunosorbent assay (ELISA) is not useful for detection of rodent A ⁇ .
- the invention relates to a method for determining whether a compound alters the amount of amyloid beta (A ⁇ ) peptide present in a wildtype animal, comprising the steps of administering the compound to a wildtype animal, measuring the amount of the A ⁇ peptide in a sample from the animal and determining whether the measured amount in the animal is different from the amount expected in a sample from a wildtype animal to which no compound has been administered, whereby a difference between the measured amount in the animal and the amount expected in the animal to which no compound has been administered indicates that the compound alters the amount of an A ⁇ peptide present in the animal, wherein the amount of the A ⁇ peptide is measured by immunoassay, wherein the immunoassay is a sandwich immunoassay using a capture binding substance bound to a solid phase and a labeled detection binding substance, and wherein the binding substances are specific for A ⁇ peptide. Also included are kits with means to perform the method of the invention, compounds identified by the method of the invention, and methods of making compositions that comprise
- FIG. 1 Schematic diagram of the amyloid precursor protein (APP) and its principal metabolic derivatives, hi the second line, the sequence within amyloid precursor protein that contains the ⁇ -amyloid protein and TM regions is expanded (SEQ ID NO: 49).
- Figure 2. A) Amino acid sequence of mouse amyloid precursor protein (APP)
- C A ⁇ burden (A ⁇ 42 positive plaque area) of the cortex (p ⁇ 0.01) and hippocampus (p ⁇ 0.05) was significantly lower in the 82El group than in the control group.
- D Insoluble A ⁇ l-42 and A ⁇ x-42 were significantly reduced in the 82El group based on ELISA measurements. *p ⁇ 0.05, **p ⁇ 0.01.
- FIG. 4 Characterization of ⁇ -cleavage site-specific antibody, clone 82El.
- S Fibril-free soluble
- S/F soluble and fibril-containing
- B APP transgenic mouse (Tg2576) brain extract was run on a gel and probed with 82El.
- 82El detects A ⁇ , but not with full-length APP.
- C ⁇ -cleavage site specificity was further confirmed using the transfected cells.
- 82El does not react with full-length APP (lane C), although non- ⁇ site-specific antibody 6E10 detects (lane C).
- ⁇ CTF is strongly detectable after treatment with a ⁇ -secretase inhibitor, N-[N-(3,5-difluorophenacetyl-l- alanyl)]-S-phenylglycine t-butylester (DAPT) (Dovey, H.F. et al. 2001 JNeurochem 76:173- 181) (lane D).
- FIG. 1 Characterization of A ⁇ 40-specific antibody, clone IAlO.
- a ⁇ 40 and A ⁇ 42 were run on a gel and probed with IAlO.
- IAlO specifically detected A ⁇ 40 and did not show cross-reactivity to A ⁇ 42 (A).
- a ⁇ was also detected in brain A ⁇ extract from an APP transgenic mouse, Tg2576 (B).
- FIG. 7 A ⁇ reduction after treatment with A ⁇ -lowering agents in non-transgenic mice.
- HRP horseradish peroxidase
- FIG. 1 Immunoblot detection of A ⁇ with 14Fl monoclonal antibody.
- FIG. 9 A ⁇ reduction after treatment with A ⁇ -lowering agents in non-transgenic mice.
- HRP-coupled 14Fl was used for detection by ELISA.
- Figure 10 Characterization of newly developed mouse cross-reactive N terminus end-specific Abeta antibody, clone 14Fl, and ELISA development. Cross reactivity of clone 14Fl with human and mouse Abeta peptide was examined by immunoblotting (A).
- FIG 11. Newly developed ELISA determined BACEl and gamma secretase inhibitors-mediated Abeta-lowering effects.
- Primary cultured neurons prepared from mouse embryos were treated with a gamma secretase inhibitor, DAPT (Dovey et al. 2001) (A) or a BACEl inhibitor, Inhibitor IV (Stachel et al. 2004) (B) at various doses for 24 hours.
- Levels of endogenous full-length 1-40 and x-40 Abeta in the culture medium were determined using Abeta ELISAs, 14F1/1A10 and 12B2/1A10, respectively.
- sAPPbeta Ba and Ca
- sAPPalpha Bb and Cb
- AD Alzheimer's disease
- a ⁇ amyloid beta
- APP amyloid precursor protein
- Lowering A ⁇ is widely considered to be a primary AD therapeutic goal, and A ⁇ -lowering strategies are usually evaluated by applying ELISA methods to detect human A ⁇ to transgenic mice expressing mutant (typically Swedish) APP.
- Transgenic mice are now widely available, but substantial effort in colony maintenance as well as complex intellectual property issues limit flexibility with this approach. Further, all transgenic mice express their transgenes under the control of an autologous promoter; testing in a more physiologically relevant environment is desirable.
- non-transgenic mice can be considered, but have not yet been widely used in the assessment of A ⁇ lowering interventions; a primary reason is the limited sensitivity of mouse A ⁇ ELISAs.
- a ⁇ -lowering agents for example, secretase inhibitors
- Non-transgenic mice lack A ⁇ plaque pathology and so the disease- modifying impact of A ⁇ -lowering agents must be confirmed in plaque-bearing APP transgenic mice.
- the study of A ⁇ reduction in non-transgenic mice using this sensitive mouse A ⁇ ELISA provides a valuable tool in the development of therapeutic agents.
- Amyloid Precursor Protein and Proteolytic Fragments ⁇ -Amyloid protein is derived from amyloid precursor protein by sequential cleavages by proteases referred to as ⁇ -secretase and ⁇ -secretase ( Figure 1).
- the amyloid precursor protein comprises ubiquitously expressed proteins whose heterogeneity comes from the "alternative splicing" together of different protein coding regions (exons) within the amyloid precursor protein gene and also from post-translational modifications, such as the addition of sugar or phosphate groups to the protein backbone.
- spliced forms of amyloid precursor protein containing 751 or 770 amino acids are widely expressed in cells throughout the body and also occur in neurons. However, neurons express much higher levels of a 695-amino acid splice form. The difference between the 751-, 770-, and 695-residue forms is the retention in the former of an exon that encodes an amino acid sequence that is homologous to certain inhibitors of serine proteases.
- amyloid precursor protein 751 that is in human platelets has been shown to inhibit factor XIa (a serine protease) in the clotting cascade.
- amyloid precursor protein APP
- the first line depicts the largest of the known (amyloid precursor protein alternate splice forms, comprising 770 amino acids. Regions of interest are indicated at their correct relative positions. A 17-residue signal peptide occurs at the N- terminus (box with vertical lines). Two alternatively spliced exons of 56 and 19 amino acids are inserted at residue 289; the first contains a serine protease inhibitor domain of the Kunitz type (KPl). A single membrane-spanning domain (transmembrane, TM) at amino acids 700 through 723 is indicated (dotted lines).
- the ⁇ -amyloid protein (A ⁇ ) fragment includes 28 residues just outside the membrane plus the first 12 to 14 residues of the TM domain.
- the sequence within amyloid precursor protein that contains the ⁇ -amyloid protein and TM regions is expanded (SEQ ID NO: 49).
- the underlined residues represent the ⁇ -amyloid proteins 1 to 42 peptide.
- the green letters below the wildtype sequence indicate the currently known missense mutations identified in certain families with Alzheimer disease or hereditary cerebral hemorrhage with amyloidosis.
- the 3 -digit numbers are codon numbers (amyloid precursor protein 770 isoform).
- the first arrow indicates the site (after residue 687) of a cleavage by ⁇ -secretase that enables secretion of the large, soluble ectodomain of amyloid precursor protein (APP s - ⁇ ) into the medium and retention of the 83-residue C-terminal fragment (C83) in the membrane.
- the C83 fragment can undergo cleavage by the protease called ⁇ -secretase at residue 711 or residue 713 to release the p3 peptides.
- the fourth line depicts the alternative proteolytic cleavage after residue 671 by ⁇ -secretase that causes the secretion of the slightly truncated APP s - ⁇ molecule and the retention of a 99 residue C-terminal fragment (C99).
- the C99 fragment can also undergo cleavage by ⁇ -secretase to release the ⁇ amyloid peptides.
- Cleavage of both C83 and C99 by ⁇ -secretase releases the ⁇ amyloid precursor protein intracellular domain (AICD) into the cytoplasm.
- AICD ⁇ amyloid precursor protein intracellular domain
- All three members of the amyloid precursor protein family are single transmembrane proteins that resemble receptors, with the large amino-terminal region (the ectodomain) projecting from the cell surface or into the lumens of intracellular vesicles (for example, the endoplasmic reticulum, Golgi, trans-Golgi network, and endosomes) and the short carboxy-terminal region projecting into the cytoplasm (Figure 1, first line).
- the 3 secretase enzymes In the normal processing of amyloid precursor protein by the 3 secretase enzymes
- ⁇ -Secretases such as tumor necrosis factor-converting enzyme, are membrane-anchored proteases able to cleave diverse single- transmembrane proteins.
- ⁇ -Secretase also called ⁇ -amyloid cleaving enzyme 1
- ⁇ -Secretase is a membrane-anchored aspartyl protease with its active site in its ectodomain (Vassar, R. et al. 1999 Science 286:735-741; Sinha, S. et al. 1999 Nature 402:537-540; Yan, R. et al. 1999 Nature 402:533-537).
- a slightly shorter form of the amyloid precursor protein ectodomain amyloid precursor proteinp is released from the cell surface, and a C-terminal fragment of 99 amino acids (C99) embedded in the membrane is left.
- the C99 fragment can subsequently be cleaved by the unusual protease referred to as ⁇ -secretase to create ⁇ - amyloid protein.
- the C83 fragment made by ⁇ -secretase can undergo cleavage by the same ⁇ -secretase to generate a small peptide that makes up the latter two thirds of the ⁇ - amyloid protein region (designated as p3).
- C99 and C83 are the substrates of ⁇ -secretase, which create ⁇ -amyloid protein and p3, respectively.
- ⁇ -secretase which create ⁇ -amyloid protein and p3, respectively.
- a substantially larger portion of total cellular amyloid precursor protein molecules is cleaved by ⁇ -secretase than by ⁇ -secretase.
- a ⁇ l-40 and A ⁇ l-42 peptides are constitutive products of cellular metabolism and occur in normal biological fluids throughout life. Genetic defects that cause autosomal dominant AD, such as mutations in amyloid precursor protein (APP) or the presenilin (PS) genes PSl and PSl augment the amyloidogenic pathway of APP processing in all cells in a way that favors production of the highly self-aggregating A ⁇ l-42 variant over the slightly shorter and less hydrophobic A ⁇ l-40 form.
- a ⁇ l-42 normally comprises only about 5-10% of total secreted A ⁇ peptides, but this fraction rises to about 15-40% when either APP or PS is mutant.
- a ⁇ peptides particularly A ⁇ l-42
- a ⁇ peptides are overproduced or insufficiently cleared, they become prone to aggregation into stable oligomers and larger polymers, apparently culminating in mature amyloid fibrils.
- the clinical diagnosis of Alzheimer disease is confirmed by observing numerous neuritic (amyloid) plaques and neurofibrillary tangles in the hippocampus, amygdala and association neocortex.
- the plaques (extracellular) are composed of the A ⁇ l-40 and A ⁇ l-42 proteins, whereas the tangles (intraneuronal) are composed of modified forms of the microtubule-associated protein, tau.
- the Alzheimer's disease-linked amyloid- ⁇ precursor protein belongs to a superfamily of proteins, which also comprises the amyloid- ⁇ precursor-like proteins APLPl (Wasco, W. et al. 1992 Proc Natl Acad Sd USA 89:10758-10762) and APLP2 (Slunt, H.H. et al. 1994 J Biol Chem 269:2637-2644).
- APLPl and APLP2 are highly homologous to APP, but not within the A ⁇ region.
- the A ⁇ region of APP is highly conserved across divergent species as shown by the sequence alignment in Fig. 2. Antibodies
- Antibodies come in a variety of classes, affinities, and idiotypes. They can be polyclonal or monoclonal, engineered or natural, and raised in animals or generated in vitro. Antibodies can be divided into three groups according to how they react with their target proteins. One class of antibodies reacts with the target protein independently of its protein conformation. Antibodies of this type are particularly useful for measuring the total amount of the target protein in crude preparations or in cell extracts. They are usually polyclonal in nature and are prepared by immunizing animals with partially denatured protein or with a peptide whose sequence corresponds to part of the intact protein. However, monoclonal antibodies that are pan-specific are not uncommon.
- Antibodies of this type are typically monoclonal, have been raised against native protein, and recognize a given sequence of amino acids only when it occurs in its native three-dimensional configuration. These antibodies are useful for testing whether mutated forms of proteins that have been generated by in vitro mutagenesis are folded correctly or whether a wildtype protein expressed in heterologous cells is assembled into a correct three-dimensional configuration.
- a third class of antibodies reacts only with denatured forms of the target protein. These antibodies are raised against fully denatured antigens and can be either monoclonal or polyclonal. Antibodies of this type are useful for western blotting.
- Hybridomas producing monoclonal antibodies are generated by the somatic cell fusion of two cell types: antibody-producing cells from an immunized animal, which by themselves die in tissue culture in a relatively short time, and myeloma cells, which contribute their immortality in tissue culture to the hybrid cell.
- the myeloma cells are variants carrying drug selection markers, so that only those myeloma cells that have fused with spleen cells providing the missing enzyme will survive under selective conditions.
- Successful hybridoma production is influenced by the characteristics of each of the cell populations, the fusion conditions, and the subsequent selection and screening of the hybrids. Immunization of Donor Animals
- Immunization protocols for fusion purposes have been developed empirically.
- a wide variety of standard routes and schedules of immunization can be used, the main distinguishing feature being the use of a final intravenous boost with antigen 2 to 4 days before fusion.
- the importance and required timing of the intravenous boost are thought to be related to the type of cell that preferentially fuses: Peak hybridoma production was found to precede the peak plaque-forming cell and the peak of serum antibody but corresponded to the peak of proliferation.
- HAT hypoxanthine, aminopterin, and thymidine
- HGPRT hypoxanthine-guanine-phosphoribosyltransferase
- the myeloma cells mutagenized and selected to be HGPRT-negative, are killed by the HAT-containing medium unless they have fused and therefore contain the enzymes of the spleen cell. Thus, for several days after a fusion, there is extensive cell death; subsequently, the culture should contain only cells resulting from spleen-myeloma fusion. Other drug markers are occasionally used, for example, ouabain resistance.
- the myeloma cells should be cycled through selective medium such as 8-azaguanine, to assure that they have not reverted to a drug-resistant phenotype, although this would be unnecessary for cell lines in which the mutation responsible for drug sensitivity is a deletion. Incorporation of 8-azaguanine into DNA is dependent on the salvage pathway, so it selectively kills HGPRT-positive cells. Fusion Methods
- Methods for testing supernatants for desired antibodies can include the same range of methods used for studying such antibodies. Fusions have been successfully screened using RIA, ELISA, or other binding assays; visual immunofluorescence or flow cytometry; cytotoxicity assays; and assays for activation or blocking of biologic effects such as cell- mediated lympholysis (CML), receptor activation, and lymphokine activity. Fusions can also be screened by hybridization to detect mRNA for immunoglobulin of certain types in the cells, rather than antibody in the supernatant.
- CML cell- mediated lympholysis
- screening assay The major issue in choosing a screening assay is that the assay not be subject to fluctuation, which would lead to many false-positive identifications and a large investment of effort in maintaining and cloning hybrid cells of no interest. Thus, clear-cut discrimination between positives and negatives is often more important than 17, sensitivity. Most supernatants contain at least several milligrams per milliliter of antibody, which is enough to detect by numerous methods. A good screening assay should be convenient to use with hundreds of samples and should give results quickly, so that cells of greatest interest are still healthy when identified. If a parameter of interest is more difficult or time consuming to measure, it is often practical to use another assay for primary screening and then evaluate likely candidates in the more demanding assay.
- a simple binding assay can be used for primary screening, and the positives then tested by immune precipitation to determine which bind to a particular component.
- Multiple-pass screening is useful in many other situations in which two or more antibody characteristics are important in the choice of clones to keep.
- a screening assay difficult to perform on very large numbers of samples can also be applied by testing supernatants pooled from groups of hybrids, provided the assay sensitivity would allow detection of one positive supernatant diluted in a pool of negative ones. Components of positive pools are then screened individually. Post-fusion Processing of Hybridomas
- hybridomas After identification of positive cultures, hybridomas must be cloned to assure production of only one antibody, and cells must be frozen for future use. Since a great deal of labor and material are consumed by processing of candidate hybridomas, an efficient strategy must be used.
- Cloning can be performed by limiting dilution or by colony selection from soft agar, hi either case, it is best to clone promptly, before possible nonproducing cells in the same well, including variants of the positive cell, can overgrow the antibody producers.
- Newly derived hybridomas are often unstable in their antibody production, perhaps because somatic cell hybrids are aneuploid and throw off chromosomes. The uncloned lines are maintained until active clones have been well established. Clones producing desired antibody must then be expanded, supernatant is collected for antibody preparation, cells are frozen, and frozen cells are thawed for verification of viability and antibody secretion.
- mice are the most common species immunized for hybridoma production, but for a variety of reasons, other animal species often have advantages. If an antigen of interest is nonpolymorphic in the mouse, the mouse component might be immunogenic in other species, while mice would be tolerant to it. In the case of hybridomas for clinical use, mouse antibodies have the drawback of inducing anti-mouse immunoglobulin immune responses with possible deleterious effects, so derivation of human hybridomas would become desirable.
- hybridomas in species other than mouse.
- interspecies hybridization can be performed using mouse myeloma fusion partners.
- the resulting hybrids are often unstable and throw off chromosomes but clones can sometimes be selected that produce antibody in a stable fashion. Examples of this would be rat-mouse fusion to produce antibody to the mouse Fc receptor, and hamster- mouse fusion to produce antibody to mouse CD3.
- Rabbit-mouse hybridomas have also been described.
- a second approach is the use of fusion partner cells from the desired species.
- Myeloma variants carrying drug selection markers are available in a number of species.
- a rat myeloma line adapted for this purpose, IR983F, was described in the literature. This approach avoids some of the instability in interspecies hybrids and allows ascites production in homologous hosts.
- Monoclonal antibodies produced by hybridoma technology are derived from B cells of immunized animals.
- An alternative technology uses gene libraries and expression systems instead. This approach has the advantages of avoiding labor-intensive immunizations of animals and the screening of antibody-containing supernatants. Another advantage of the approach is circumventing tolerance.
- V H and V K libraries involved preparation of V H and V K libraries and expression of the libraries in bacteria.
- a VH domain alone binds antigen with reasonable affinity, and so the expressed products of the gene library could be screened directly.
- Further development of the system led to use of V H and V L libraries made separately, and then preparation of a combinatorial library by cleaving, mixing, and religating the libraries at a restriction site.
- a linker can be used so that V H and V L can both be expressed on one covalent polypeptide; the flexibility of the linker allows association of the VH and V L in a normal three dimensional configuration and thus formation of an antigen-binding site.
- V H and VK genes are expressed on the surface of bacteriophage as fusion proteins with a phage protein, to permit rapid screening of large numbers of sequences. Adsorption of antibody-bearing phage on antigen-coated surfaces allows positive selection of phage containing DNA encoding the desired Fv.
- V-region variable region gene
- Human antibody gene sequences can be recovered by polymerase chain reaction (PCR) from peripheral blood cells, bone marrow, or human cells reimmunized in SCID-hu mice.
- PCR polymerase chain reaction
- the phage display technique can then be used to select antigen-binding clones and derive reagents of desired specificity.
- the antibody sequence may be mutated.
- a single bacterium will harbor both wildtype and mutant phage, leading to mixed sequence protein displayed on each phage.
- the phage must be grown in nonmutator bacteria, so that each bacterium contains only one antibody sequence and antibody expressed on a phage matches its sequence.
- Multiple rounds of mutation followed by growth in nonmulator bacteria and then selection for high-affinity binding led to an overall 100-fold increase in affinity.
- Improved affinity has also been achieved by use of site-directed mutagenesis to alter residues in hypervariable regions affecting dissociation rates.
- monoclonal antibodies in clinical use are due to the foreign immunoglobulin constant regions. Recognition of foreign epitopes can lead to sensitization and so preclude subsequent use in the same individual of different monoclonal antibodies. Thus, monoclonal antibodies with some or all structure derived from human immunoglobulins have advantages.
- mice Production of fully human mAbs in transgenic mice has now been achieved by multiple laboratories.
- the strategy has involved insertion of constructs containing clusters of human immunoglobulin V, D, J, and C genes into the mouse germ line to generate one transgenic line, and targeted disruption of the mouse heavy-chain and ⁇ -chain loci to generate another transgenic line. From these two lines, mice are then bred that express only human antibodies. Specificity of Monoclonal Antibodies
- each different antibody has a distinct range of reactivity, and the only common feature would be detectable reactivity with the antigen used for immunization or testing.
- the serum as a whole may show only a low-titered cross-reaction with any particular other antigen, and that cross-reaction can be removed by absorption, leaving substantial activity against the immunizing antigen.
- a polyclonal serum may be "more specific" than anyone of its clonal parts and may be more useful.
- Polyclonal sera also have advantages in certain technical situations, such as immunoprecipitation in which multivalency is important. Many antigens are univalent with respect to monoclonal antibody binding but display multiple distinct sites that can be recognized by different components of polyclonal sera. Thus a greater degree of cross- linking can be achieved.
- the ultimate serologic reagent in many cases may well be a mixture of monoclonal antibodies that have been chosen according to their cross-reactions. The mixture would be better defined and more reproducible than a polyclonal antiserum and would have the same advantage of overlapping specificities.
- Antibodies are Specific Binding Substances
- Antibodies whether monoclonal or polyclonal, provide a unique type of reagent that can be made with high specificity for almost any desired organic or biochemical structure, often with extremely high affinity. These can be naturally divalent (e.g., in the case of IgG) or multivalent (e.g., in the case of IgM) or can be made as monovalent molecules, such as Fab or recombinant Fv fragments. They serve not only as a major arm of host defense, playing a major role in the protective efficacy of most existing antiviral and antibacterial vaccines, but also as very versatile tools for research and clinical use.
- Radioimmunoassay (RLA) and ELISA have revolutionized the detection of minute quantities of biologic molecules, such as hormones and cytokines, and thus have become indispensable for clinical diagnosis and monitoring of patients, as well as for basic and applied research.
- Current solid-phase versions of these take advantage not only of the intrinsic affinity and specificity of the antibodies, but also of the implicit multivalency and local high concentration on a solid surface. Binding Assays
- Solid phase immunological binding assays have the advantages of ease of processing large numbers of samples and potentially increased affinity (compared to the intrinsic affinity of the antibody due to the effects at the solid-liquid interface. 1. Radioimmunoassay (RIA)
- RIA The central concept of RIA is that the binding of an infinitesimal concentration of highly radioactive tracer antigen to low concentrations of a high-affinity specific antibody is very sensitive to competition by unlabeled antigen and is also very specific for that antigen. Thus concentrations of antigen in unknown samples can be determined by their ability to compete with tracer for binding to antibody.
- the method can be used to measure very low concentrations of a molecule, even in the presence of the many impurities in biologic fluids.
- Enzyme-linked Immunosorbent Assay An alternative solid-phase readout system for the detection of antigen-antibody reactions is the ELISA assay. In principle, the only difference from RIAs is that antibodies antigen are covalently coupled to an enzyme instead of a radioisotope, so that bound enzyme activity is measured instead of bound counts per minute.
- a basic strategy for using ELISA assays to detect antigen involves the sandwich technique for detecting antigen.
- Specific antibody is used to coat the micro titer wells. Antigen is then bound to the solid-phase antibody. Finally, a second antibody, linked to enzyme, is added. This binds to the solid-phase antigen-antibody complex, carrying enzyme along with it. Excess second antibody is washed off and substrate is added. The absorbance produced is a function of the antigen concentration of the test solution, which can be determined from a standard curve. Specificity of the assay depends on the specificity of the antibodies used to coat the plate and detect antigen.
- Sensitivity depends on affinities as well as the amount of the first antibody bound to the well, which can be increased by using affinity-purified antibodies in the coating step.
- the binding of both antibodies of the sandwich depends on divalency of the antigen, or else the two antibodies must be specific for different antigenic determinants on the same antigen molecule.
- sequence of a protein is known or can be deduced from the nucleic acid sequence, specific antisera can be raised by immunizing animals with a synthetic peptide corresponding in sequence to a segment of the native protein. If information about the primary sequence of the target protein is limited, there may be little or no choice in the peptide sequence used as an immunogen. However, there is a good chance that peptides chosen at random will be at least partially buried in the native protein and they may be too hydrophobic to be efficient immunogens. Antibodies directed against these peptides may be of low titer and/or may react only with denatured protein.
- Hydrophilic peptides that contain charged residues are much better immunogens and also have a high probability of occupying a surface location on the native protein.
- Antipeptide antibodies raised against conformationally flexible surface features of proteins such as turns and ⁇ -loops are likely to be of high titer and may react efficiently with the native protein.
- Most of the computing packages that are commonly used to analyze DNA sequences contain programs to search protein sequences for surface peptides that are likely to be good antigens. The goal of these programs is to choose a sequence of amino acids that contains a preponderance of polar residues and no more than four adjacent hydrophobic residues. Such peptides are likely to be soluble in aqueous solvents and therefore easy to couple to carrier proteins.
- binding substance refers to a polypeptide substantially encoded by an immunoglobulin gene or immunoglobulin genes, or fragments thereof, which specifically bind and recognize an analyte (antigen).
- the recognized immunoglobulin genes include the kappa, lambda, alpha, gamma, delta, epsilon and mu constant region genes, as well as the myriad immunoglobulin variable region genes.
- Antibodies exist, e.g., as intact immunoglobulins or as a number of well characterized fragments produced by digestion with various peptidases. This includes, e.g., Fab' and F(ab)'2 fragments.
- binding substance also includes antibody fragments either produced by the modification of whole antibodies or those synthesized de novo using recombinant DNA methodologies.
- immunoassay is an assay that utilizes a binding substance to specifically bind an analyte.
- the immunoassay is characterized by the use of specific binding properties of a particular antibody to isolate, target, and/or quantify the analyte.
- a binding substance "specifically binds to” or “is specifically immunoreactive with” a protein when the binding substance functions in a binding reaction which is determinative of the presence of the protein in the presence of a heterogeneous population of proteins and other biologies.
- the specified binding substances bind preferentially to a particular protein and do not bind in a significant amount to other proteins present in the sample.
- Specific binding to a protein under such conditions requires an antibody that is selected for its specificity for a particular protein.
- a variety of immunoassay formats may be used to select binding substances specifically immunoreactive with a particular protein. For example, solid-phase ELISA immunoassays are routinely used to select monoclonal antibodies specifically immunoreactive with a protein.
- label is a composition detectable by spectroscopic, photochemical, biochemical, immunochemical, or chemical means.
- useful labels include P, fluorescent dyes, electron-dense reagents, enzymes (e.g., as commonly used in an ELISA), biotin, digoxigenin, or haptens and proteins for which antisera or monoclonal antibodies are available.
- Binding substances can be made detectible, e.g., by incorporating a radiolabel into the peptide, and used to detect antibodies specifically reactive with the peptide.
- a label often generates a measurable signal, such as radioactivity, fluorescent light or enzyme activity, which can be used to quantify the amount of bound label.
- a ⁇ Monoclonal Antibodies means an immunoglobulin molecule or a fragment of an immunoglobulin molecule having the ability to specifically bind to a particular antigen. Antibodies are well known to those of ordinary skill in the science of immunology. As used herein, the term “antibody” means not only full-length antibody molecules but also fragments of antibody molecules retaining antigen binding ability. Such fragments are also well known in the art and are regularly employed both in vitro and in vivo. In particular, as used herein, the term “antibody” means not only full-length immunoglobulin molecules but also antigen binding active fragments such as the well- known active fragments F(ab')2, Fab, Fv, and Fd.
- substantially pure means that the polypeptides are essentially free of other substances with which they may be found in nature or in vivo systems to an extent practical and appropriate for their intended use.
- the polypeptides are sufficiently pure and are sufficiently free from other biological constituents of their host cells so as to be useful in, for example, generating antibodies, sequencing, or producing pharmaceutical preparations.
- substantially pure polypeptides may be produced in light of the nucleic acid and amino acid sequences disclosed herein. Because a substantially purified polypeptide of the invention may be admixed with a pharmaceutically acceptable carrier in a pharmaceutical preparation, the polypeptide may comprise only a certain percentage by weight of the preparation. The polypeptide is nonetheless substantially pure in that it has been substantially separated from the substances with which it may be associated in living systems.
- isolated means: (i) amplified in vitro by, for example, polymerase chain reaction (PCR); (ii) recombinantly produced by cloning; (iii) purified, as by cleavage and gel separation; or (iv) synthesized by, for example, chemical synthesis.
- An isolated nucleic acid is one which is readily manipulable by recombinant DNA techniques well known in the art.
- PCR polymerase chain reaction
- An isolated nucleic acid may be substantially purified, but need not be.
- a nucleic acid that is isolated within a cloning or expression vector is not pure in that it may comprise only a tiny percentage of the material in the cell in which it resides.
- Such a nucleic acid is isolated, however, as the term is used herein because it is readily manipulable by standard techniques known to those of ordinary skill in the art.
- a coding sequence and regulatory sequences are said to be "operably joined” when they are covalently linked in such a way as to place the expression or transcription of the coding sequence under the influence or control of the regulatory sequences. If it is desired that the coding sequences be translated into a functional protein, two DNA sequences are said to be operably joined if induction of a promoter in the 5' regulatory sequences results in the transcription of the coding sequence and if the nature of the linkage between the two DNA sequences does not (1) result in the introduction of a frame-shift mutation, (2) interfere with the ability of the promoter region to direct the transcription of the coding sequences, or (3) interfere with the ability of the corresponding RNA transcript to be translated into a protein.
- a promoter region would be operably joined to a coding sequence if the promoter region were capable of effecting transcription of that DNA sequence such that the resulting transcript might be translated into the desired protein or polypeptide.
- the precise nature of the regulatory sequences needed for gene expression may vary between species or cell types, but shall in general include, as necessary, 5' non-transcribing and 5' non-translating sequences involved with initiation of transcription and translation respectively, such as a TATA box, capping sequence, CAAT sequence, and the like.
- 5' non-transcribing regulatory sequences will include a promoter region which includes a promoter sequence for transcriptional control of the operably joined gene. Regulatory sequences may also include enhancer sequences or upstream activator sequences, as desired.
- a "vector" may be any of a number of nucleic acids into which a desired sequence may be inserted by restriction and ligation for transport between different genetic environments or for expression in a host cell.
- Vectors are typically composed of DNA although RNA vectors are also available.
- Vectors include, but are not limited to, plasmids and phagemids.
- a cloning vector is one which is able to replicate in a host cell, and which is further characterized by one or more endonuclease restriction sites at which the vector may be cut in a determinable fashion and into which a desired DNA sequence may be ligated such that the new recombinant vector retains its ability to replicate in the host cell, hi the case of plasmids, replication of the desired sequence may occur many times as the plasmid increases in copy number within the host bacterium or just a single time per host before the host reproduces by mitosis. In the case of phage, replication may occur actively during a lytic phase or passively during a lysogenic phase.
- An expression vector is one into which a desired DNA sequence may be inserted by restriction and ligation such that it is operably joined to regulatory sequences and may be expressed as an RNA transcript.
- Vectors may further contain one or more marker sequences suitable for use in the identification and selection of cells which have been transformed or transfected with the vector. Markers include, for example, genes encoding proteins which increase or decrease either resistance or sensitivity to antibiotics or other compounds, genes which encode enzymes whose activities are detectable by standard assays known in the art (e.g., ⁇ - galactosidase or alkaline phosphatase), and genes which visibly affect the phenotype of transformed or transfected cells, hosts, colonies or plaques.
- Preferred vectors are those capable of autonomous replication and expression of the structural gene products present in the DNA segments to which they are operably joined.
- an antibody from which the pFc' region has been enzymatically cleaved, or which has been produced without the pFc' region designated an F(ab')2 fragment, retains both of the antigen binding sites of a full- length antibody.
- an antibody from which the Fc region has been enzymatically cleaved, or which has been produced without the Fc region designated an Fab fragment, retains one of the antigen binding sites of a full-length antibody molecule.
- Fab fragments consist of a covalently bound antibody light chain and a portion of the antibody heavy chain denoted Fd.
- the Fd fragments are the major determinant of antibody specificity (a single Fd fragment may be associated with up to ten different light chains without altering antibody specificity) and Fd fragments retain epitope-binding ability in isolation.
- CDRs complementarity determining regions
- FRs framework regions
- CDRl through CDR3 complementarity determining regions
- SEQ ID NOs: 1, 17 and 33 disclose the amino acid sequences of the Fd fragment of the A ⁇ monoclonal antibodies.
- amino acid sequences of the heavy chain FRl, CDRl, FR2, CDR2, FR3, CDR3 and FR4 regions are disclosed as (FRl, SEQ ID NOs: 2, 18 and 34); (CDRl, SEQ ID NOs: 3, 19 and 35); (FR2, SEQ ID NOs: 4, 20 and 36); (CDR2, SEQ ID NOs: 5, 21 and 37); (FR3, SEQ ID NOs: 6, 22 and 38); (CDR3, SEQ ID NOs: 7, 23 and 39); and (FR4, SEQ ID NOs: 8, 24 and 40).
- SEQ ID NOs: 9, 25 and 41 disclose the amino acid sequences of the light chains of the A ⁇ monoclonal antibodies.
- amino acid sequences of the light chain FRl, CDRl, FR2, CDR2, FR3, CDR3 and FR4 regions are disclosed as (FRl, SEQ ID NOs: 10, 26 and 42); (CDRl, SEQ ID NOs: 11, 27 and 43); (FR2, SEQ ID NOs: 12, 28 and 44); (CDR2, SEQ ID NOs: 13, 29 and 45); (FR3, SEQ ID NOs: 14, 30 and 46); (CDR3, SEQ ID NOs: 15, 31 and 47); and (FR4, SEQ ID NOs: 16, 32 and 48).
- the present invention includes substantially pure polypeptides comprising an antibody selectively binding an A ⁇ antigenic determinant, wherein said antibody includes a heavy chain CDR3 region having the amino acid sequence selected from the group consisting of SEQ ID NO: 7, SEQ ID NO: 23 and SEQ ID NO: 39 and substantially pure polypeptides of the same wherein said antibody comprises an Fd fragment or an Fab fragment.
- the invention also includes substantially pure polypeptides comprising an antibody selectively binding an A ⁇ epitope, wherein said antibody includes a heavy chain CDR2 region having the amino acid sequence of SEQ ID NO: 5 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 21 (when heavy chain CDR3 region is SEQ ID NO: 23), or SEQ ID NO: 37 (when heavy chain CDR3 region is SEQ ID NO: 39).
- the invention also includes substantially pure polypeptides comprising an antibody selectively binding an A ⁇ epitope, wherein said antibody includes a heavy chain CDRl region having the amino acid sequence of SEQ ID NO: 3 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 19 (when heavy chain CDR3 region is SEQ ID NO: 23), or SEQ ID NO: 35 (when heavy chain CDR3 region is SEQ ID NO: 39).
- the invention also includes substantially pure polypeptides comprising an antibody selectively binding an A ⁇ epitope, wherein said antibody includes a heavy chain Fd region including the amino acid sequence of SEQ ID NO: 1 (when heavy chain CDR3 region is
- SEQ ID NO: 7 SEQ ID NO: 17 (when heavy chain CDR3 region is SEQ ID NO: 23), or
- SEQ ID NO: 33 when heavy chain CDR3 region is SEQ ID NO: 39.
- the invention also includes substantially pure polypeptides comprising an antibody selectively binding an A ⁇ epitope, wherein said antibody includes a light chain CDR3 region having the amino acid sequence of SEQ ID NO: 15 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 31 (when heavy chain CDR3 region is SEQ ID NO: 15 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 31 (when heavy chain CDR3 region is SEQ ID
- SEQ ID NO: 23 when heavy chain CDR3 region is SEQ ID NO: 39.
- the invention also includes substantially pure polypeptides comprising an antibody selectively binding an A ⁇ epitope, wherein said antibody includes a light chain CDR2 region having the amino acid sequence of SEQ ID NO: 13 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 29 (when heavy chain CDR3 region is SEQ ID NO:
- SEQ ID NO: 23 when heavy chain CDR3 region is SEQ ID NO: 39.
- the invention also includes substantially pure polypeptides comprising an antibody selectively binding an A ⁇ epitope, wherein said antibody includes a light chain CDRl region having the amino acid sequence of SEQ ID NO: 11 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 27 (when heavy chain CDR3 region is SEQ ID NO:
- the invention also includes substantially pure polypeptides comprising an antibody selectively binding an A ⁇ epitope, wherein said antibody includes a light chain region including the amino acid sequence of SEQ ID NO: 9 (when heavy chain CDR3 region is
- SEQ ID NO: 7 SEQ ID NO: 25 (when heavy chain CDR3 region is SEQ ID NO: 23), or SEQ ID NO: 41 (when heavy chain CDR3 region is SEQ ID NO: 39).
- the invention also includes substantially pure polypeptides comprising an antibody selectively binding an A ⁇ epitope, wherein said antibody includes a heavy chain Fd region including the CDR amnio acid sequences of SEQ ID NO: 1 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 17 (when heavy chain CDR3 region is SEQ ID NO: 23), or SEQ ID NO: 33 (when heavy chain CDR3 region is SEQ ID NO: 39).
- the invention also includes substantially pure polypeptides comprising an antibody selectively binding an A ⁇ epitope, wherein said antibody includes a light chain region including the CDR amino acid sequences of SEQ ID NO: 9 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 25 (when heavy chain CDR3 region is SEQ ID NO: 23), or SEQ ID NO: 41 (when heavy chain CDR3 region is SEQ ID NO: 39).
- the present invention also contemplates isolated nucleic acids that encode the polypeptides described herein that comprise an antibody that selectively binds an A ⁇ antigenic determinant, isolated nucleic acids that comprise a vector including a regulatory sequence operably joined to the nucleic acids that encode the polypeptides that comprise an antibody that selectively binds an A ⁇ antigenic determinant as described herein, and host cells that include a vector comprising a nucleic acid that encodes a polypeptide described herein that comprising an antibody selectively binding an A ⁇ antigenic determinant.
- Alzheimer's disease is characterized by the initial deposition of A ⁇ peptide in the form of amyloid plaques in the brain. Therefore, effective treatments for AD are expected to decrease the production of these peptides, whereas agents that hasten progress of the disease are expected to increase production of the peptide. Because A ⁇ peptides are the major constituent of neuritic plaques, it is useful to identify compounds that specifically inhibit the amount of these peptides. Accordingly, this invention provides methods for screening compounds that specifically elevate or decrease the the amount of A ⁇ peptide in an animal. Compounds that decrease the amount of A ⁇ peptide are candidates for use in treating the disease, while compounds that increase the amount may hasten the disease and are to be avoided by humans.
- Screening methods of this invention for determining whether a test compound specifically alters the amount of A ⁇ peptide present in a wildtype animal involve administering the compound to the animal, measuring the amount of A ⁇ peptide present in a sample from the animal, and determining whether this amount is greater than, less than or the same as an amount expected in a sample from a wildtype animal to which no compound has been administered. If the amounts are different, then the compound affects the presence of A ⁇ peptide in the animal. This amount can be measured, for example, from brain extracts.
- the expected amount generally will be a control amount determined by measuring A ⁇ peptide present in the animal in the absence of the compound. However, one also may determine the expected amount by extrapolation; measuring the amount of A ⁇ peptide present in the animal upon administration of different amounts of the compound to the animal, and using these figures to calculate the expected amount. In certain instances measuring a control amount for the purposes of comparison may not be necessary because the effect of the compound on A ⁇ peptide presence is clear. For example, a compound may render A ⁇ peptide undetectable in an animal that normally posesses detectable amounts, indicating that the compound decreases A ⁇ peptide from the amount expected in its absence.
- Drugs are often initially screened by a cell-free assay. For example, enzyme activity may be assayed to investigate ⁇ - and ⁇ -secretase inhibitors. Then, compounds are screened using in vitro cultured cells. Primary cultured neurons, which are the most physiologically relevant system, can be assayed using the methods of this invention. Amyloid- ⁇ Peptide and Related Proteins and Peptides
- amyloid- ⁇ peptide refers to an approximately 4.2 kD protein which, in the brains of AD, Down's Syndrome, Hereditary Cerebral Hemorrhage with Amyloidosis Dutch type (HCHWA-D) and some normal aged subjects, forms the subunit of the amyloid filaments comprising the senile (amyloid) plaques and the amyloid deposits in small cerebral and meningeal blood vessels (amyloid angiopathy).
- a ⁇ can occur in a filamentous polymeric form (in this form, it exhibits the Congo-red and thioflavin-S dye-binding characteristics of amyloid described in connection therewith).
- a ⁇ can also occur in a non-filamentous form ("preamyloid” or "amorphous” or “diffuse” deposits) in brain tissue, in which form no birefringent staining by Congo red occurs.
- a ⁇ is an approximately 40-42 amino acid fragment of a large membrane-spanning glycoprotein, referred to as the amyloid precursor protein (APP) , encoded by a gene on the long arm of human chromosome 21. Forms of A ⁇ longer than 42 amino acids are also contemplated herein.
- a ⁇ is further characterized by its relative mobility in SDS- polyacrylamide gel electrophoresis or in high perfo ⁇ nance liquid chromatography (HPLC).
- HPLC high perfo ⁇ nance liquid chromatography
- a ⁇ also refers to related polymorphic forms of A ⁇ , including those that result from mutations in the A ⁇ region of the APP gene.
- a ⁇ fragment refers to fragments and degradation products of A ⁇ which are generated at low concentrations by mammalian cells. Particular A ⁇ fragments have a molecular weight of approximately 3 kD and are presently believed to include peptides with, for example, amino acid residues 3-34, 6-27, 6-34, 6-35, 6-42, 11-34, 11-40, 11-43, 12-43, 17-40 a ⁇ d 17-42 of Ab (Vigo-Pelfrey et al. 1993 J Neurochem 61:1965-1968).
- a ⁇ peptide refers to A ⁇ or an A ⁇ fragment whose amino-terminus begins at amino acid number 1 of A ⁇ or which is amino-terminally truncated, and whose carboxy-terminus extends no further than amino acid number 40 or 42. These peptides and fragments also comprise a heterogenous group.
- the term “A ⁇ (40)” or “A ⁇ 40” or “A ⁇ (l-40)” or “A ⁇ l-40” refers to A ⁇ or an A ⁇ fragment whose C-terminal amino acid is #40 of A ⁇ .
- a ⁇ (42)" or “A ⁇ 42” or “A ⁇ (l-42)" or "A ⁇ l-42” refers to A ⁇ or an A ⁇ fragment whose C-terminal amino acid is #42 of A ⁇ .
- N terminus of the A ⁇ peptide as used herein refers to a region of A ⁇ which is located approximately between amino acid residues 1 and 16 (e.g., Aspl and Lysl6).
- C terminus of the A ⁇ peptide as used herein refers to a region of A ⁇ which is located approximately between amino acid residues 35 and 40 (e.g., Met35 and VaWO) or between amino acids 38 and 42 (e.g., Gly38 and Ala42), referring to an A ⁇ fragment where the C-terminal amino acid is #40 of A ⁇ and an A ⁇ fragment where the C- terminal amino acid is #42 of A ⁇ , respectively.
- middle region refers to a region of A ⁇ which is located approximately between amino acid residues 11 and 28 (e.g. Glul 1 and Lys28).
- Alzheimer's disease which includes familial Alzheimer's disease
- Down's Syndrome Down's Syndrome
- HCHWA-D Advanced aging of the brain
- p3 refers to a peptide whose amino acid sequence is substantially similar to A ⁇ , but whose amino-terminal amino acid begins at amino acid 17 of A ⁇ .
- p3 fragment refers to fragments and degradation products of p3. Whereas p3 is produced through a different processing pathway than A ⁇ , for the purposes of the detection methods of this invention, p3 and p3 fragments are considered to be a subset of A ⁇ peptides, because certain detection techniques that recognize A ⁇ solely from the carboxy terminus generally also will recognize p3. Also it is shown that the same apparent mechanisms generate the p3 and A ⁇ carboxy-termini.
- APP amyloid- ⁇ precursor protein
- APP is defined as a polypeptide that is encoded by a gene of the same name localized in humans on the long arm of chromosome 21 and that includes the A ⁇ region within the carboxyl third of its coding region.
- APP is a glycosylated, single-membrane spanning protein expressed in a wide variety of cells in many mammalian tissues. Examples of specific isotypes of APP which are currently known to exist in humans are the 695-amino acid polypeptide described by Kang et al. 1987 Nature 325:733-736; the 751-amino acid polypeptide described by Ponte et al.
- a ⁇ peptides can be detected by any method known in the art.
- the method involves an immunoassay employing binding substances specific for the peptides.
- binding substances specific for the peptides.
- One step of the screening methods of this invention involves measuring the amount of at least one A ⁇ peptide, specifically, in a sample.
- Measuring A ⁇ peptides specifically means measuring A ⁇ peptides so as to distinguish them from APP.
- Specific measurement of A ⁇ peptide preferably is performed by the use of binding substances that specifically recognize A ⁇ peptides.
- Another method of this invention involves screening compounds to determine their ability to alter the amount of A ⁇ peptides. Such methods can involve the use of binding substances that can distinguish A ⁇ peptides from APP.
- immunological detection techniques i.e., immunoassays employing binding substances
- detection techniques include ELISA, Western blotting, radioimmunoassay, and the like.
- Suitable immunological methods employing a single antibody are also contemplated, for example, radioimmunoassay using an antibody specific for A ⁇ peptide, or single antibody ELISA methods. It will be clear that the particular forms of A ⁇ detected by such methods depend upon the particular binding substances employed. For example, binding substances directed to the middle portion of A ⁇ may detect A ⁇ peptides whose amino termini do not extend to amino acid no. 1 of A ⁇ .
- binding substances directed to the carboxy-terminal end of A ⁇ peptide may detect peptides ending at amino acids 40-42. Therefore, determining the specificity of the binding substances will assist in determining exactly which A ⁇ peptides the assay is detecting.
- the method to detect A ⁇ peptides is an immunoassay involving two antibodies.
- One antibody is specific for an epitope containing amino acids approximately between 35 and 40 or amino acids approximately between 38 and 42.
- a preferred immunoassay technique is a two-site or "sandwich" assay. This assay involves a capture binding substance, usually bound to a solid phase, and a labelled detection binding substance, hi this method, A ⁇ peptides are captured from the sample using a first binding substance specific for A ⁇ peptides (usually bound to a solid phase). The capture of A ⁇ peptides is detected using a labeled second binding substance specific for A ⁇ against a different antigenic determinant on the same A ⁇ peptide.
- labeled binding substances include those directed to the middle region (amino acids ⁇ 11 to ⁇ 28) or binding substances specific for amino-terminal amino acids (amino acids ⁇ 1 to -16).
- a sandwich assay using an antibody against the middle region of A ⁇ can be used to specifically measure A ⁇ and A ⁇ fragments whose amino-terminus begins at approximately amino acid 17 of A ⁇ .
- Such assays may recognize p3 or p3 fragments, since those peptides begin at amino acid number 17 of A ⁇ .
- binding substances that recognize A ⁇ peptides and do not recognize p3 are useful in the methods of this invention.
- Antibodies specific for A ⁇ can be prepared, e.g., by immunizing an animal with a peptide whose amino acid sequence corresponds to amino acids from the N-terminus, C- terminus or middle portion of A ⁇ .
- human A ⁇ peptides consisting of amino acid residues 1-16, 11-28, 35-40 and 38-42 may be used as imrnunogens for A ⁇ N- terminus, middle region and C-terminus-specific antibodies respectively.
- Antibodies specific for A ⁇ (l-40) or A ⁇ (l-42) can be prepared, e.g., by immunizing an animal with a peptide whose amino acid sequence corresponds with amino acids 35-40 or 38-42 of A ⁇ .
- Synthetic polypeptide haptens may be produced by the well-known Merrifield solid- phase synthesis technique in which amino acids are sequentially added to a growing chain (Merrifield (1963) J Am Chem Soc 85:2149-2156). Suitable peptide haptens will usually comprise at least five contiguous residues within A ⁇ and may include more than six residues. The amino acid sequences may be based on the sequence of A ⁇ set forth herein.
- polypeptide hapten may be conjugated to a suitable immunogenic carrier, such as serum albumin, keyhole limpet hemocyanin, or other suitable protein carriers, as generally described in Hudson and Hay, Practical Immunology, Blackwell Scientific Publications, Oxford, Chapter 1.3, 1980.
- a suitable immunogenic carrier such as serum albumin, keyhole limpet hemocyanin, or other suitable protein carriers, as generally described in Hudson and Hay, Practical Immunology, Blackwell Scientific Publications, Oxford, Chapter 1.3, 1980.
- Antibodies specific for the desired epitope may be produced by in vitro or in vivo techniques. In vitro techniques involve exposure of lymphocytes to the immunogens, while in vivo techniques require the injection of the immunogens into a suitable vertebrate host. Suitable vertebrate hosts are non-human, including mice, rats, rabbits, sheep, goats, and the like.
- Immunogens are injected into the animal according to a predetermined schedule, and the animals are periodically bled, with successive bleeds having improved titer and specificity.
- the injections may be made intramuscularly, intraperitoneally, subcutaneously, or the like, and an adjuvant, such as incomplete Freund's adjuvant, may be employed.
- monoclonal antibodies can be obtained by preparing immortalized cell lines capable of producing antibodies having desired specificity.
- immortalized cell lines may be produced in a variety of ways. Conveniently, a small vertebrate, such as a mouse, is hyperimmunized with the desired immunogen by the method just described. The vertebrate is then killed, usually several days after the final immunization, the spleen cells removed, and the spleen cells immortalized. The manner of immortalization is not critical. Presently, the most common technique is fusion with a myeloma cell fusion partner, as first described by Kohler and Milstein 1975 Nature 256:495-497.
- EBV transformation transformation with bare DNA, e.g., oncogenes, retroviruses, etc., or any other method which provides for stable maintenance of the cell line and production of monoclonal antibodies.
- Specific techniques for preparing monoclonal antibodies are described in Antibodies: A Laboratory Manual, Harlow and Lane, eds., Cold Spring Harbor Laboratory, 1988.
- the detection techniques of the present invention will also be able to use antibody fragments, such as F(ab), Fv, VL, VH, and other fragments.
- kits In the use of polyclonal antibodies, however, it may be necessary to adsorb the anti-sera against the target epitopes in order to produce a monospecific antibody population. It will also be possible to employ recombinantly produced antibodies (immunoglobulins) and variations thereof. It would also be possible to prepare other recombinant proteins which would mimic the binding specificity of antibodies prepared as just described. Kits
- kits for performing assays of the invention include means for detecting specifically A ⁇ peptides.
- the means can include any means known or described above, e.g., binding substances.
- the kit is useful for immunoassays including two antibodies for each antigen.
- the kit can comprise a binding substance specific for the amino terminal region of A ⁇ , the carboxy terminal region of A ⁇ , or the middle region of A ⁇ . Such antibodies are useful for the capture or detection of A ⁇ peptides.
- the binding substance specific for the carboxy terminal region of A ⁇ is bound to a solid phase, and the binding substances specific for a different antigenic determinant on the same A ⁇ peptide are detectably labeled.
- the detectable labels can be any known and used in the art including, e.g., a biotinylation label, a radioactive label, a light scattering label, an enzymatic label, a fluorescent label and the like.
- the kit can further comprise a substrate for the enzyme.
- test compounds can be any molecule, compound, or other substance which can be administered to the test animal without substantially interfering with animal viability.
- Suitable test compounds may be small molecules (i.e., molecules whose molecular mass is no more than 1000 Daltons), biological polymers, such as polypeptides, polysaccharides, polynucleotides, and the like.
- the test compounds will typically be administered at a dosage of from 1 ng/kg to lg/kg, usually from 10 ⁇ g/kg to 100 mg/kg.
- Test compounds which are able to inhibit amounts of A ⁇ peptides are considered as candidates for further determinations of the ability to block ⁇ -amyloid amounts in animals and humans. Such compounds can be tested in in vivo studies, as described below. Inhibition of amounts of A ⁇ peptides indicates that levels of A ⁇ peptides are unavailable for forming ⁇ -amyloid plaques.
- wildtype animal models can be used to detect quantitative differences in A ⁇ peptide. Wildtype animals are useful for screening compounds that alter the amount of A ⁇ peptide in the assays of this invention, indicating their ability to affect the course of Alzheimer's disease, both to ameliorate and aggravate the condition. Rodent models, and in particular murine models, are suitable for this use.
- a common approach to examine the role of a particular gene in the APP pathway is to first create a transgenic or knockout mouse model with respect to the gene of interest. Typically, the knockout or transgenic mouse is subsequently crossed with transgenic mice expressing human APP so that human A ⁇ level may be measured using existing human A ⁇ assay techniques. This additional step in creating an animal model is time consuming. Using the methods of this invention, wildtype mouse A ⁇ level can be measured, eliminating the need for the secondary cross with a transgenic mouse expressing human APP and saving time and resources.
- test compounds can be administered to additional test animals, in which deviation from the average control value would indicate that the test compound had an effect on the amount of A ⁇ peptide in the animal.
- Test substances which are considered positive, i.e., likely to be beneficial in the treatment of Alzheimer's disease or other ⁇ -amyloid-related conditions will be those which are able to reduce the level of A ⁇ peptide amount, preferably by at least 5%, more preferably by at least 10%, and most preferably by at least 20, 30, 40, 50, 60, 70, 80, 90, 95 or 100%.
- the present invention further comprises pharmaceutical compositions incorporating a compound selected by methods described herein, including a pharmaceutically acceptable carrier.
- Such pharmaceutical compositions should contain a therapeutic or prophylactic amount of at least one compound identified by the method of the present invention.
- the pharmaceutically acceptable carrier can be any compatible non-toxic substance suitable to deliver the compounds to an intended host. Sterile water, alcohol, fats, waxes and inert solids may be used as the carrier. Pharmaceutically acceptable adjuvants, buffering agents, dispersing agents and the like may also be incorporated into the pharmaceutical compositions. Preparation of pharmaceutical conditions incorporating active agents is well described in the medical and scientific literature. See for example, Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, 16th Ed., 1982.
- compositions just described are suitable for systemic administration to the host, including parenteral, topical and oral administration.
- the pharmaceutical compositions may be administered parenterally, i.e., subcutaneously, intramuscularly, or intravenously.
- the present invention provides compositions for administration to a host, where the compositions comprise a pharmaceutically acceptable solution of the identified compound in an acceptable carrier, as described above.
- the N-terminal peptide of amyloid ⁇ used as antigen comprises the amino acid sequence starting from the N terminus and continuing toward the C terminus of the amino acid sequence of amyloid ⁇ (1-40) or amyloid ⁇ (1-42), comprising the sequence of amino acids 1 through 28 of amyloid ⁇ being preferable, and a peptide comprising the sequence of amino acids 1 through 16 of amyloid ⁇ , illustrated in the following amino acid sequence being especially preferable.
- DAEFRHDSGYEVHHQK SEQ ID NO: 63
- a peptide comprising the above amino acid sequence can be obtained by various methods, without any particular limitations. For example, if may be synthesized by a method publicly known in the industry, or else a synthesized product can be purchased from Synpep Corporation, TANA Laboratories (USA), or the like.
- a peptide comprising the above amino acid sequence is next conjugated with a biopolymer compound to make the antigen. In this case, it is preferable to perform conjugation by attaching a cysteine (C) to the C-terminal side amino acid of the aforementioned peptide.
- C cysteine
- KLH keyhole limpet hemocyanin
- OVA egg white albumin
- BSA bovine serum albumin
- RSA rabbit serum albumin
- thyroglobulin thyroglobulin
- the conjugation of the aforementioned peptide and biopolymer can be performed by a publicly known method such as the mixed acid anhydride method (Erlanger, B.F. et al.1959 J Biol Chem 234:1090-1094) or the activated ester method (Kara, A.E et al. 1994 J AgHc Food Chem 42 301-309).
- a publicly known method such as the mixed acid anhydride method (Erlanger, B.F. et al.1959 J Biol Chem 234:1090-1094) or the activated ester method (Kara, A.E et al. 1994 J AgHc Food Chem 42 301-309).
- the mixed acid anhydride used in the mixed acid anhydride method is obtained by subjecting the aforementioned peptide to a conventional Schotten-Baumann reaction, which is then reacted with a biopolymer compound to prepare the target conjugate of peptide and biopolymer.
- halofumaric acid esters which may be used in this mixed acid anhydride method include methyl chlorofumarate, methyl bromofumarate, ethyl chlorofumarate, ethyl bromofumarate, isobutyl chlorofumarate and the like.
- the proportions of peptide, halofumaric acid ester, and biopolymer used in this method can be selected as appropriate from a broad range.
- the Schotten-Baumann reaction is conducted in the presence of a basic compound.
- the basic compounds which can be used for this reaction include compounds commonly used for Schotten-Baumann reactions, such as triethylamine, trimethylamine, pyridine, dimethylaniline, N-methylmorpholine, diazabicyclononene (DBN), diazabicycloundecene (DBU), diazabicyclooctane (DABCO) and other organic bases, potassium carbonate, sodium carbonate, potassium hydrogen carbonate, sodium hydrogen carbonate and other inorganic bases, and the like.
- the aforementioned reaction is normally conducted at -2O 0 C to 100°C, preferably O 0 C to 5O 0 C, and the reaction time is about 5 minutes to 10 hours, preferably 5 minutes to 2 hours.
- the reaction of the obtained mixed acid anhydride and biopolymer compound is normally conducted at -2O 0 C to 15O 0 C, preferably O 0 C to 100 0 C, with the reaction time being about 5 minutes to 10 hours, preferably 5 minutes to 5 hours.
- the mixed acid anhydride method is commonly conducted in a solvent.
- any solvent commonly used in the mixed acid anhydride method include halogenated hydrocarbons such as dichloromethane, chloroform and dichloroethane, aromatic hydrocarbons such as benzene, toluene, and xylene, ethers such as diethyl ether, dioxane, tetrahydrofuran, and dimethoxyethane, esters such as methyl acetate and ethyl acetate, and aprotic polar solvents such as N,N-dimethyl formamide, dimethyl sulfoxide, and hexamethylphosphoric triamide.
- halogenated hydrocarbons such as dichloromethane, chloroform and dichloroethane
- aromatic hydrocarbons such as benzene, toluene, and xylene
- ethers such as diethyl ether, dioxane, tetrahydrofuran, and dimethoxyethane
- esters such as methyl
- the activated ester method can be generally carried out as follows. First, the aforementioned peptide is dissolved in organic solvent and is reacted with N-hydroxysuccinic acid imide in the presence of a coupling agent to produce N-hydroxysuccinic acid imide activated ester.
- the coupling agent used in this reaction one can employ coupling agents commonly used in condensation reactions, such as dicyclohexyl carbodiimide, carbonyl diimidazole, and water-soluble carbodiimide. Furthermore, for the organic solvent, N,N-dimethyl formamide (DMF), dimethyl sulfoxide, dioxane, or the like can be used.
- the molar ratio of peptide to coupling agent such as N-hydroxysuccinic acid imide used in the reaction is preferably between 1:10 and 10:1, with 1 :1 being most preferable.
- the reaction temperature is 0 to 50 0 C, preferably 22 to 27°C, and the reaction time is 5 minutes to 24 hours, preferably 1 to 2 hours. With regard to the reaction temperature, the reaction can be conducted at a temperature above the melting points but below the boiling points.
- the reaction liquid is added to a solution in which the biopolymer compound has been dissolved and is reacted, whereupon, for example if the biopolymer compound has free amino groups, an acid-amide bond will be formed between those amino groups and the carboxyl groups of the aforementioned peptide.
- the reaction temperature is 0 to 60°C, preferably 5 to 40°C, more preferably 22 to 27 0 C, and the reaction time is 5 minutes to 24 hours, preferably 1 to 16 hours, more preferably 1 to 2 hours.
- conjugate 1 A conjugate of the aforementioned peptide and biopolymer compound (hereinafter referred to simply as "Conjugate 1”) can be obtained by purifying the reaction product obtained by either of the above methods by means of dialysis, a desalting column or the like.
- N-terminal antibody of the present invention using the aforementioned conjugate, an animal is given an immunization using the conjugated antigen, followed by booster immunization(s) using the conjugated antigen, and antibodies are collected from that animal.
- the antigen conjugate is dissolved in sodium phosphate buffer solution (hereinafter referred to as "PBS"), which is mixed with an auxiliary such as Freund's complete adjuvant or incomplete adjuvant or alum and used as the antigen to immunize the animals.
- PBS sodium phosphate buffer solution
- any type of animal commonly used in this field can be used for the immunized animals: examples include mammals such as mice, rats, rabbits, goats, and cows.
- the method of administration of the immunogen for the immunization subcutaneous injection, intraperitoneal injection, intravenous injection, intracutaneous injection, or intramuscular injection may be used, but subcutaneous injection or intraperitoneal injection is preferable.
- the immunization can be performed a single time or multiple times at a suitable interval, preferably, multiple times at an interval of one to five weeks.
- Animals, which have been immunized with the antigen, as described above are then boosted with antigen in the same manner.
- the immunization can be performed multiple times at a suitable interval, preferably, multiple times at an interval of one to five weeks.
- myeloma cells and immune cells obtained from animals immunized as described above are fused to obtain hybridomas, and antibodies are collected from a culture of said hybridomas to obtain monoclonal antibodies against the N terminus of amyloid ⁇ .
- N-terminal antibodies of the present invention obtained in this manner can be used as immunological assay reagents or as a treatment drugs for Alzheimer's disease and the like.
- labeling or immobilization is preferably performed if necessary.
- Labeling is performed by conjugating a labeling substance, for instance an enzyme such as horseradish peroxidase (HRP) or alkaline phosphatase, a fluorescent substance such as fluorescein isocyanate or rhodamine, a radioactive substance such as 32 P or 125 I, or a chemoluminescent substance, with the N-terminal antibodies.
- a labeling substance for instance an enzyme such as horseradish peroxidase (HRP) or alkaline phosphatase, a fluorescent substance such as fluorescein isocyanate or rhodamine, a radioactive substance such as 32 P or 125 I, or a chemoluminescent substance
- immobilization is performed by binding the N-terminal antibodies to a suitable solid phase. Any solid phase commonly used in immunological assays can be used for the solid phase, for instance, a plate such as a polystyrene 96-well microtiter plate or an amino bonded microtiter plate,
- N-terminal antibodies of the present invention can be used as a reagent, which al-lows accurate determination of amyloid ⁇ by combining with antibodies which recognize amyloid ⁇ (1-40) or amyloid ⁇ (1-42). Antibodies against the middle portion of A ⁇
- a peptide corresponding to the middle portion of amyloid ⁇ used as antigen may comprise, for example, the sequence of amino acids ⁇ 11 through ⁇ 28 of amyloid ⁇ , illustrated in the following amino acid sequence.
- a peptide comprising the above amino acid sequence can be obtained by various methods, without any particular limitations. For example, it may be synthesized by a method publicly known in the industry, or else a synthesized product can be purchased from Synpep Corporation, TANA Laboratories (USA), or the like.
- a peptide comprising the above amino acid sequence is next conjugated with a biopolymer compound to make the antigen.
- a biopolymer compound to make the antigen.
- conjugation by attaching a cysteine (C) to the C-terminal side amino acid of the aforementioned peptide.
- the antibodies which recognize amyloid ⁇ (1-40) or amyloid ⁇ (1-42) which are combined with the N-terminal antibodies of the present invention one can use for instance polyclonal antibodies or monoclonal antibodies which can be obtained by conventional methods using part or all of the amino acid sequence of amyloid ⁇ (1-40) or amyloid ⁇ (1- 42) as an antigen, hi the present invention, among these antibodies, it is especially preferable, from the standpoint of accuracy of determination, to use antibodies, which recognize a C-terminal peptide of amyloid ⁇ (hereinafter referred to as "C-terminal antibodies").
- C-terminal antibodies there is no particular limitation on the aforementioned C-terminal antibodies so long as they are antibodies which distinguish the C-terminal peptide of the amyloid ⁇ (1-40) or amyloid ⁇ (1-42) that is to be measured, but antibodies which do not recognize amyloid ⁇ (1-43) besides the amyloid ⁇ (1-40) or amyloid ⁇ (1-42) which is to be measured are preferable.
- a C-terminal antibody which recognizes amyloid ⁇ (1-40) (hereinafter referred to as "C-Terminal Antibody 1”) can be obtained by conventional methods using a peptide (C-terminal peptide) comprising an amino acid sequence starting from the C terminus and continuing toward the N terminus of amyloid ⁇ (1-40) as an antigen.
- the C-terminal peptide used as antigen is preferably a peptide comprising the sequence of amino acids 18 through 40 of amyloid ⁇ (1-40), with a peptide comprising 35 through 40 of amyloid ⁇ (1-40), shown in the following amino acid sequence being especially preferable.
- C-Terminal Antibody 2 a C-terminal antibody which recognizes amyloid ⁇ (1-42)
- C-Terminal Antibody 2 a C-terminal antibody which recognizes amyloid ⁇ (1-42)
- the peptide used as antigen is preferably a peptide comprising the sequence of amino acids 18 through 42 of amyloid ⁇ (1-42), with a peptide comprising 38 through 42 of amyloid ⁇ (1-42), shown in the following amino acid sequence being especially preferable.
- GWIA SEQ ID NO: 66
- conjugate of the aforementioned peptide and a biopolymer compound is preferably conjugated by attaching lysine and cysteine (KC), for example, to the N-terminal amino acid of the aforementioned peptide.
- KC lysine and cysteine
- N-terminal antibody (or middle region antibody) of the present invention Utilizing the N-terminal antibody (or middle region antibody) of the present invention and an antibody which recognizes amyloid ⁇ (1-40) or amyloid ⁇ (1-42) makes it possible to accurately measure amyloid ⁇ (1-40) or amyloid ⁇ (1-42).
- Specific measurement methods include the various methods commonly used in immunological assays ("The hybridoma method and monoclonal antibodies," published by R&D Planning Co., Ltd., pages 30-53, 5 March 1982), such as radioimmunoassay (RIA), ELISA ( Engvall, E. et al. 1980: Methods in Enzymol 70:419-439), fluorescent antibody technique, plaque technique, spot technique, aggregation technique, ouchterlony, immunochromatography, etc.
- RIA radioimmunoassay
- ELISA Engvall, E. et al. 1980: Methods in Enzymol 70:419-439
- fluorescent antibody technique fluorescent antibody technique
- an antibody which recognizes amyloid ⁇ (1-40) or amyloid ⁇ (1-42) is immobilized on a carrier. Then, in Process (B), the carrier surface on which no antibodies are immobilized is blocked for instance by a protein unrelated to amyloid ⁇ .
- Process (C) specimens containing various concentrations of amyloid ⁇ are added to form complexes of amyloid ⁇ and antibody, after which, in Process (D), labeled N- terminal antibody (or middle region antibody) of the present invention is added and the amyloid ⁇ -antibody complex is bound thereto, and finally, in Process (E), by measuring the quantity of label, the quantity of amyloid ⁇ in a specimen can be determined based on a calibration curve prepared in advance. Measurement is possible also if the antibodies used in Process (A) and Process (D) are reversed, but immobilizing the antibody which recognizes amyloid ⁇ (1-40) or amyloid ⁇ (1-42) is preferable from the standpoint of detection sensitivity.
- the carrier used for immobilizing the antibody which recognizes amyloid ⁇ (1-40), or amyloid ⁇ (1-42)
- any carrier commonly used in immunological assays can be used. Possible examples include a polystyrene 96-well microtiter plate or an amino bonded microtiter plate.
- the aforementioned antibody can be immobilized for example by placing a buffer solution containing the aforementioned antibody onto the carrier and incubating.
- a publicly known buffer solution can be used, for example, 10 mM PBS.
- the concentration of the aforementioned antibody in the buffer solution can be selected from a wide range, but normally, about 0.01 to 100 ⁇ g/ml, preferably 0.1 to 20 mg/ml is appropriate. Furthermore, when using a 96-well microtiter plate as a carrier, one would use 300 ⁇ l/well or less, with about 20 to 150 ⁇ l/well being preferable. Moreover, while there are no particular limitations as to the incubation conditions, normally, overnight incubation at about 4°C is appropriate.
- the blocking of Process (B) is carried out with the aim of preventing this.
- a blocking agent one can for instance use BSA or skim milk solution, or a commercial blocking agent such as Block-Ace (made by Dainippon Pharmaceutical (Code No. UK-25B)). While there are no limitations as to the specifics of the blocking, it can be performed for instance by adding a suitable quantity of Block- Ace to the areas where antigen was immobilized, incubating overnight at approximately 4°C, and then washing with buffer solution.
- specimen containing amyloid ⁇ is brought into contact with immobilized antibodies, capturing the amyloid ⁇ on the immobilized antibodies and producing a complex of immobilized antibodies and amyloid ⁇ .
- immobilized antibodies While there are no limitations as to the conditions of producing this complex, one can conduct the reaction for approximately 1 hour to overnight at about 4 0 C to 37 0 C. After completion of reaction, it is preferable to wash the carrier with buffer solution and remove unreacted proteins and the like.
- the buffer solution used for this reaction preferably has a composition of 10 mM PBS (pH 7.2) and 0.05% (v/v) Tween 20.
- a labeled antibody which recognizes a different epitope of amyloid ⁇ captured by the immobilized antibody is added to form an immobilized antibody-amyloid ⁇ -labeled antibody complex.
- the buffer solution described above can be used for this reaction.
- the quantity of labeled antibody used in Process (D) is approximately 5,000 to 10,000 times the quantity of immobilized antibody, and is preferably a quantity diluted such that the final absorbance will be 1.5 to 2.0.
- Buffer solution can be used for the dilution, and although there are no particular limitations as to the reaction conditions, it is preferable to conduct the reaction for approximately 1 hour at about 4 0 C to 37 0 C and wash with buffer solution after the reaction.
- an immobilized antibody-amyloid ⁇ -labeled antibody complex can be formed.
- a chromogenic substrate solution which reacts with the labeling substance of the immobilized antibody-amyloid ⁇ -labeled antibody complex is added and absorbance is measured, whereby the quantity of amyloid ⁇ can be computed based on a calibration curve.
- a chromogenic substrate solution containing hydrogen peroxide and 3,3',5,5'-tetramethyl benzene hereinafter referred to as "TMB"
- the chromogenic reaction can be conducted by adding the chromogenic substrate solution, reacting for approximately 30 minutes at approximately 25°C, and then adding 1 to 2N sulfuric acid to stop the enzymatic reaction.
- TMB color development can be measured based on absorbance at 450 nm.
- an appropriate method would be to induce color development with p-nitrophenyl phosphoric acid as the substrate, add 2N NaOH to stop the enzymatic reaction and measure absorbance at 415 nm.
- concentration of amyloid ⁇ in a specimen can be computed using a calibration curve prepared in advance based on the absorbance of reaction liquids to which a known concentration of amyloid ⁇ has been added.
- amyloid ⁇ assay kit characterized in that it includes a first reagent containing the N-terminal antibody (or middle region antibody) of the present invention and a second reagent containing an antibody which recognizes amyloid ⁇ (1-40) or amyloid ⁇ (1-42) (hereinafter referred to as "the kit of the present invention") is favorably used to practice the method of measuring amyloid ⁇ described above.
- the kit of the present invention can be fabricated by conventional methods. Specifically, the N-terminal antibody (or middle region antibody) of the present invention and a labeled antibody which recognizes either amyloid ⁇ (1-40) or amyloid ⁇ (1-42) can be used as standard antibodies, and combined furthermore with a dilution buffer solution, standard substance, substrate buffer solution, stop solution, wash solution and the like.
- the measurement method and assay kit obtained in this manner make it possible to accurately measure amyloid ⁇ (1-40) or amyloid ⁇ (1-42) completely retaining its full length in a specimen such as blood plasma or blood serum.
- Example 1 A ⁇ N-terminal-end specific antibody reduced ⁇ -amyloid in Alzheimer-model mice
- AD Alzheimer's disease
- a ⁇ amyloid ⁇ protein
- 82El an A ⁇ N-terminal-end specific monoclonal antibody named 82El was developed, which does not cross-react with full-length A ⁇ precursor.
- Passive intraperitoneal administration of 82El markedly reduced total plaque area (A ⁇ burden) in the Tg2576 mouse brains. This was confirmed by the ELISA measurement of insoluble A ⁇ in the brain homogenates.
- mice After 12 times administrations for over 3 months, blood sample was collected transcardially for the ELISA measurement of soluble A ⁇ in the plasma level under the deep anesthesia by sodium pentobarbital (25 mg/kg), and mice were euthanized by transcardial perfusion of 100 ml of 10 U/ml heparin in saline. The left cerebral hemisphere was fixed in 4% buffered formaldehyde solution and embedded in paraffin.
- a ⁇ 1 -40 and A ⁇ 1 -42 were measured by combinations of 82El with A ⁇ 40 or A ⁇ 42 C-terminal end-specific antibodies IAlO and 1C3, respectively, as described in Example 2.
- a ⁇ x-40 and A ⁇ x-42 were also measured by combinations of 12B2, A ⁇ middle portion antibody, with IAlO or 1C3 as described in Example 2. Results and discussion
- the A ⁇ burden was significantly reduced in the 82El group compared with that in the control group for both A ⁇ 40 (30% reduction, p ⁇ 0.05, in cortex; 40% reduction, p ⁇ 0.05, in hippocampus) and A ⁇ 42 (43% reduction, p ⁇ 0.01 in cortex; 40% reduction, p ⁇ 0.05, in hippocampus) (Fig. 3C).
- a ⁇ ELISA analysis of TBS-insoluble fraction was consistent with the findings on immunohistochemical studies.
- Significant reduction of A ⁇ 42 was found in the 82El group.
- the 82El group showed about 50% less A ⁇ l-42 and A ⁇ x-42 than the control group (Fig. 3D).
- Fig. 3D was measured by ELISA.
- the unit of measurement in Fig. 3D is ⁇ g/g brain tissue.
- a measurement of 20 ⁇ g/ml is equivalent to approximately 500 fmol/ml.
- AD Alzheimer's disease
- senile plaques are a pathological hallmark of AD, and amyloid ⁇ (A ⁇ ) peptides are a major component of these plaques.
- a ⁇ peptides are derived from proteolytic cleavage of the A ⁇ protein precursor (APP) by ⁇ - and ⁇ -secretases to generate two principal species, A ⁇ l-40 and A ⁇ l-42. Antibodies against the N- and C-termini of these peptides and an ELISA for accurate and sensitive quantitative assessment were developed.
- Sandwich ELISA composed of N-terminus (A ⁇ l) end-specific antibody, clone 82El, and C- termini end-specific antibodies, and clones IAlO and 1C3 for A ⁇ 40 and A ⁇ 42, respectively, detects full-length A ⁇ l -40 and 1-42 with a sensitivity in the sub single digit fmol/ml (equivalent to single digit pg/ml) range with no cross-reactivity to APP.
- a combination of C-termini antibodies and an antibody against the middle region of A ⁇ detects mouse A ⁇ in non-transgenic mouse brains.
- mice (BALB/c, Charles River, Japan) were immunized weekly with thyroglobulin conjugated A ⁇ peptides (50 ⁇ g/mouse).
- Partial human A ⁇ peptide consisting of amino acid residues of 1-16 (DAEFRHD S GYEVHHQK) (SEQ ID NO: 63), 11-28 (EVHHQKLVFFAEDVGSNK) (SEQ ID NO: 64), 35-40 (MVGGW) (SEQ ID NO: 65), and 38-42 (GWIA) (SEQ ID NO: 66) was used as immunogens for A ⁇ N-terminus, middle region, and C-terminus-specific antibodies, respectively.
- cysteine residue was combined in each immunogen beforehand for binding to the carrier protein, bovine thyroglobulin. After four to six immunizations, the spleen was isolated and fused with X63 Ag8 myeloma cells. Epitopes and cross-reactivity to human and rodent A ⁇ of selected clones were determined by a microplate assay using various A ⁇ fragments (American Peptides, Sunnyvale, CA).
- a 96-well plate (Maxisorp, Nunc, Denmark) was coated with various A ⁇ fragments (human A ⁇ sequence unless otherwise indicated as "r"), such as -11 to -1, -4-8, -3-5, 1-5, 1-7, rl-9, 1-11, rl-15, 1-17, 1-40, rl-40, 1-42, rl-42, 2-40, 3-7, 3-13, 3- 40, 7-17, 10-20, 11-28, 13-19, 17-28, 20-29, 35-40, and 38-42 (100 ng/well) and blocked with Block Ace (Serotec) overnight at 4 0 C.
- Block Ace Block Ace
- the plates were incubated with selected antibodies for 4 h, then with an HRP-coupled anti-mouse IgG antibody (Southern Biotechnology, Birmingham, AL), and visualized by TMB substrate (Pierce, Rockford, IL).
- HRP-coupled anti-mouse IgG antibody Southern Biotechnology, Birmingham, AL
- TMB substrate Pieris, Rockford, IL
- various A ⁇ containing samples were prepared, such as fibril-free soluble and mixture of soluble and fibril A ⁇ from synthetic peptides, culture medium from APP overexpressing HEK cells, and brain homogenate from APP overexpressing transgenic mice. Lyophilized A ⁇ (American Peptide) was sequentially dissolved in trifluoroacetic acid and l,l,l,3,3,3-hexafiuoro-2-propanol followed by complete elimination by exposure to nitrogen gas.
- a ⁇ peptide was then re-suspended in dimethyl sulfoxide and used as fibril-free A ⁇ .
- a ⁇ was aggregated at room temperature for 10 days with mixing at 500rpm and used as fibril-containing A ⁇ .
- HEK293 cells expressing Swedish mutant APP were cultured with or without a gamma-secretase inhibitor, N-[N-(3,5-difluorophenacetyl-l -alanyl)]-S-phenylglycine t-butylester (DAPT) (Dovey, H.F. et al.
- 96-well plate (Maxisorp, Nunc, Denmark) was coated with 100 ⁇ l of either A ⁇ 40 or A ⁇ 42-specific antibody (5.0 ⁇ g IgG/ml each), clones IAlO and 1C3, respectively, overnight at 4 0 C in 100 mM carbonate buffer, pH 9.6, containing 0.05% sodium azide. After blocking with 1 % Block Ace in PBS overnight at 4 0 C, standards (human or mouse synthetic A ⁇ peptides 1-40 and 1-42) and samples were loaded and incubated overnight at 4 0 C. Human plasma specimens were obtained from the Mount Sinai Alzheimer's Disease Research Center.
- Mouse brain A ⁇ was extracted in 0.4% diacetylamine containing buffer, centrifuged, and the resultant supernatant was used.
- HRP- coupled detection antibody either 82El or 12B2 was incubated for 4 h at room temperature and visualized using a TMB substrate. Results and discussion
- Clones 12B2, IAlO, and 1C3 reacted with both human and rodent A ⁇ to a similar degree because their epitopes do not include a human-specific residue.
- Table 2 summarizes epitopes, specificity, cross-reactivity to human and rodent A ⁇ , and applications of these antibodies.
- Clone 82El reacted with both soluble and fibrillar A ⁇ to a similar degree (Fig. 4A).
- Clone 82El detected A ⁇ in the SDS extract from APP-expressing transgenic mice (Tg2576), but did not detect non-cleaved APP (Fig. 4B). N-terminus specificity was further confirmed using HEK cells expressing Swedish mutant APP. 82El did not react with full-length APP (Fig.
- Clone 12B2 was raised against the middle region of A ⁇ peptide using A ⁇ l 1-28 as an imrnunogen.
- This antibody is cross-reactive to human and rodent A ⁇ in a plate assay, and a sandwich ELISA made with a combination of C-terminus-specific antibodies, IAlO and 1C3, detects mouse endogenous brain and cerebrospinal fluid (CSF) A ⁇ (see below).
- a sandwich ELISA was developed by a combination of these antibodies.
- C- terminus antibodies were selected, either A ⁇ 40 (IAlO) or 42 specific (1C3), as the capturing antibody, and another antibody, either 82El or 12B2, as a detection antibody (reporter).
- Other combinations were not considered (82E1/12B2 for capturing and 1A10/1C3 for detection) because A ⁇ 40 is much more abundant than A ⁇ 42 in biological specimens and A ⁇ 40 might monopolize the capturing antibody and reduce the sensitivity of A ⁇ 42 detection.
- the epitope of clone 12B2 is 17-28 which is an identical sequence in human and rodent A ⁇ , and 12B2 reacted to human and rodent A ⁇ to a similar degree.
- This antibody was used as a reporter antibody in an ELISA developed to detect endogenous mouse brain A ⁇ x-40 and x-42.
- a combination of 1A10/12B2 and 1C3/12B2 gave a linear standard curve in the range of 1-500 pg/ml (typical correlation factor >0.96).
- N-terminal peptide and C-terminal peptide of amyloid ⁇ were purchased as HPLC purified products from Synpep Corporation and TANA Laboratories, USA. The amino acid sequences of these peptides are as indicated below.
- the first peptide is a peptide made by attaching cysteine (C) to the sequence of amino acids 1 through 16 of amyloid ⁇ .
- the next peptide is a peptide made by attaching KC to the sequence of amino acids 35 through 40 of amyloid , /? (1-40) and the last peptide is a peptide made by attaching lysine and cysteine (KC) to the sequence of amino acids 38 through 42 of amyloid ⁇ (1-42).
- DAEFRHDSGYEVHHQKC SEQ ID NO: 67
- KCMVGGW (SEQ ID NO: 68)
- KCGWIA (SEQ ID NO: 69)
- EMCS N-(6-maleimidocaproyloxy)-succinimide
- Example 3 4 mg of each peptide of Example 3 was dissolved in approximately 1 ml of distilled water. Furthermore, 5 mg of thyroglobulin dissolved in 1 ml of 0.01 M phosphate buffer (pH 7.0) and 80 ⁇ g/ ⁇ l EMCS dissolved in dimethyl formamide were mixed at the aforementioned molar equivalent to prepare a thyroglobulin-EMCS complex solution. This complex solution was divided into four portions and a peptide solution was added to each portion at the aforementioned molar equivalent to prepare a conjugate solution of peptide and thyroglobulin cross-linked with EMCS.
- phosphate buffer pH 7.0
- conjugate solution was dialyzed using PBS, and the concentration was adjusted to 10 ⁇ g/ ⁇ l in terms of conjugate.
- the conjugates of each aforementioned peptide and thyroglobulin obtained in this manner were used as immunization antigens in the following examples.
- mice were immunized using the conjugate of the peptide having amino acids 35-40 of A ⁇ and thyroglobulin obtained in Example 4 as an immunization antigen, by administering 50 ⁇ (50 ⁇ g) of a conjugate solution at one week or two week intervals.
- the antigen was mixed with Freund's complete adjuvant only for the initial immunization, and with Freund's incomplete adjuvant for the second and subsequent immunizations.
- Splenic monocytes of the immunized mice and fusion partner X63-Ag8-653 were subjected to polyethylene glycol mediated cell fusion, and hybridomas were selected by a method described in the literature ⁇ J Immunol 146:3721-3728). The selection was performed by selecting cells, which appeared to react to fixed peptide having amino acids 35-40 of A ⁇ and to not react to the peptide having amino acids 38-42 of A ⁇ .
- Example 6 Verification of specificity of IAlO by Western blotting
- Western blotting was performed by conventional methods (e.g. "Fundamental Experimental Methods of Molecular Biology," Nankodo) on the peptides amyloid ⁇ (1-40), amyloid ⁇ (1-42) and amyloid ⁇ (1-43) using IAlO antibodies. It was verified that monoclonal antibodies IAlO did not react to amyloid,/? (1- 42) or amyloid ⁇ (1-43), and recognized only amyloid ⁇ (1-40).
- mice were immunized by the same technique as in Example 5, using the conjugate of thyroglobulin and the peptide having amino acids 38-42 of A ⁇ obtained in Example 4 as an immunization antigen.
- Splenic monocytes of the immunized mice and fusion partner X63-Ag8-653 were subjected to polyethylene glycol mediated cell fusion, and hybridomas were selected by a method described in the literature (J. Immunol. 146: 3721—3728). The selection was performed by selecting cells, which appeared to react to fixed peptide having amino acids 38-42 of A ⁇ and to not react to the peptide having amino acids 35-40 of A ⁇ .
- the cells selected as described above were purified in the same manner as in Example 5 to obtain antibodies, which recognize the C-terminal peptide of amyloid/ (1- 42). These antibodies were named 1C3.
- 6E10 antibodies (made by Signet Laboratories, Inc.) were used for the final blot detection.
- mice were immunized using the conjugate of thyroglobulin and the peptide having amino acids 1-16 of A ⁇ obtained in Example 4 as an immunization antigen. After four immunizations, another two immunizations were performed using a conjugate of a peptide having amino acids 1-5 of A ⁇ and thyroglobulin. Splenic monocytes of the immunized mice and fusion partner X63-Ag8-653 were subjected to polyethylene glycol mediated cell fusion, and hybridomas were selected by a method described in the literature
- the cells selected as described above were purified in the same manner as in Example 5 to obtain antibodies, which recognize the N-terminal peptide of amyloid /. These antibodies were named 82El.
- Example 9 Western blotting was performed by conventional methods (e.g. "Fundamental Experimental Methods of Molecular Biology," Nankodo) on the peptides amyloid , /? (1-40), amyloid,/? (2-40) and amyloid./? (3-40) using 82El antibodies. Furthermore, for comparison, Chinese hamster cells forced to express amyloid./? precursor protein (APP) were homogenized in a buffer solution containing 1% Triton and centrifuged, after which the supernatant was used as a sample and subjected to Western blotting in the same manner.
- APP amyloid./? precursor protein
- this supernatant contained / C-terminal fragments (/SCTF) cleaved at the / site form APP and amyloid ⁇ .
- /SCTF C-terminal fragments
- Western blotting was also performed on 6E10 antibodies (made by Signet Laboratories, Inc.), which are said to recognize an N-terminal peptide of amyloid/, using the same samples as above. It was verified that 82El antibodies do not react with amyloid/ (2-40) or amyloid/
- a conjugate of HRP and the 82El antibody obtained in Example 9 was prepared as follows. A necessary quantity of HRP was dissolved in distilled water and oxidized with NaIO 4 , and was then dialyzed overnight in pH 4.4 1 mM acetate buffer solution. Furthermore, 2 mg of 82El antibodies were also dialyzed overnight in pH 9.5 0.1 M carbonate buffer solution. The dialyzed 82El antibodies and HRP were mixed at 0.4 mg HRP per 1 mg of antibodies and reacted for two hours at room temperature. Subsequently, NaBH 4 was added thereto, reacting for two hours in ice, followed by overnight dialysis in PBS. Moreover, this reaction substance was gel-filtered to prepare a conjugate of 82El antibodies and HRP.
- Example 12 Creation of sandwich ELISA technique Creation of a sandwich ELISA technique using the antibodies obtained in the above examples was conducted as follows. First, 20 ⁇ g/wl IAlO antibodies or 1C3 antibodies were added at 100 ⁇ each to a 96-well ELISA plate. This was then reacted overnight at 4°C, after which blocking was performed with a 1% BSA/PBS/NaN 3 solution to make a sandwich ELISA plate. Furthermore, a conjugate of 82El antibody prepared in Example 8 and HRP was used as the labeled antibody.
- mice given 82El the number of senile plaques was lower in the brains of mice given 82El as compared to control. Furthermore, in mice given 82El, the number of senile plaques was clearly lower as compared to control in the basal nucleus, where the accumulation of amyloid ⁇ is lower as compared to the cortex and hippocampus. This reduction was also confirmed by the image analyzer.
- amyloid ⁇ was significantly reduced in mice given 82El both in the case of amyloid ⁇ (1- 40) (30% reduction in the cortex, 40% reduction in the hippocampus (for both, p ⁇ 0.05)) and amyloid ⁇ (1-42) (43% reduction in the cortex (p ⁇ 0.01), 40% reduction in the hippocampus (p ⁇ 0.05)).
- amyloid ⁇ (1-42) and amyloid ⁇ (x-42) in the brain using ELISA showed a significant decrease (a 50% decrease in both cases (for both, p ⁇ 0.05)) in mice given 82El.
- DAPT a gamma secretase inhibitor which reduced brain A ⁇ load in transgenic mice
- mice were treated with vehicle (5% ethanol in corn oil).
- mice were killed and the brain was quickly isolated.
- the brain was homogenized in Tris-HCl buffer, pH 7.4, containing protease inhibitor cocktail.
- a ⁇ was extracted in 4% DEA followed by ultracentrifugation at 100,000 g for 45 min. Supernatants were used for A ⁇ quantification.
- Brain A ⁇ level in the mice receiving DAPT was significantly reduced approximately 10% (from 24 to 21.4 fmol A ⁇ /ml brain homogenate) compared to vehicle-received controls. Discussion
- Transgenic mice have been commonly used for evaluation of A ⁇ -lowering strategies; however, transgenic mice are not the optimal system to test A ⁇ -lowering strategies.
- Transgenic mice carry mutant genes, such as APP and presenilin; but genetic mutation in these genes is involved in less than 5% of AD patients. Majority of AD patients (over 95%) are free from genetic mutation. Therefore, more physiological testing system is desired.
- mice ELISA is sensitive enough to measure endogenous brain A ⁇ levels.
- mice ELISA can evaluate A ⁇ lowering strategies and therapeutic agents in non-transgenic mice.
- Genes and mutations used in transgenic mice are commonly covered by someone's patent; but this approach only uses non-transgenic mice which are free from patent protection.
- mice (BALB/c, Charles River, Japan) were immunized weekly with thyroglobulin conjugated A ⁇ peptides (50 meg/mouse).
- a ⁇ peptide consisting of amino acid residues of 1-16 (DAEFRHDSGYEVHHQK) as an immunogen.
- the cysteine residue was combined in each immunogen beforehand for binding to the carrier protein, bovine thyroglobulin.
- the spleen was isolated and fused with X63Ag8 myeloma cells.
- Epitope mapping using various A ⁇ peptide fragments identified that the epitope of clone 14Fl locates between l-9 th amino acid residues of A ⁇ . 14Fl detects A ⁇ in the immunoblotting (see Figure 8).
- Example 16 Example 16
- HRP-coupled 14Fl was used by the ELISA methods described in Example 14 (see Figure 9). Brain A ⁇ level in the mice receiving DAPT was significantly reduced approximately 15% (from 24 to 20.2 fmol A ⁇ /ml brain homogenate) compared to vehicle- received controls.
- Example 17
- AD amyloid beta peptide
- APP beta-site amyloid precursor protein
- BACEl beta-site amyloid precursor protein
- AD cases which represent the majority of AD patients, are free from mutation and do not necessarily have overproduction of APP.
- commonly used Swedish mutant APP alters APP cleavage. Therefore, testing Abeta-lowering strategies in transgenic mice may not be optimal.
- BACEl inhibition in non- transgenic mice with physiologically relevant APP expression.
- Existing Abeta ELISAs are either relatively insensitive to mouse Abeta or not specific to full-length Abeta.
- AD Alzheimer's disease
- Abeta amyloid beta
- APP beta-site amyloid precursor protein
- BACEl BACEl
- BACEl knockout mice have been crossed with transgenic mice that overexpress human mutant APP.
- mice obtained by crossing BACEl-/- mice and APP transgenic mice showed virtually no Abeta production (Luo Y. et al. 2003 Neurobiol Dis 14:81-88).
- double transgenic mice obtained by crossing transgenic mice that overexpress BACEl and APP showed enhanced Abeta generation and exacerbated Abeta pathology (Willem M. et al. 2004 Am J Pathol 165:1621-1631).
- Abeta-lowering strategies such as pharmacologic BACEl inhibition, are commonly investigated in APP transgenic mice.
- APP expression is not necessarily increased in sporadic AD, which represents the majority of AD patients. Consequently, testing Abeta- lowering approaches in transgenic mice may not be optimal.
- the amount of substrate (APP) and the efficiency of APP cleavage are important factors that may determine Abeta production.
- Commonly used APP transgenic mice carry Swedish mutation APP, which is cleaved by BACEl much more efficiently (>50 fold) than wildtype (Tomasselli A. G. et al. 2003 J Neurochem 84:1006-1017).
- DAEFRHDSGYEVHHQK SEQ ID NO: 63
- This peptide was conjugated to the carrier protein, bovine thyroglobulin, through an additional cysteine residue at the C-terminus.
- Mice BALB/c, Charles River, Yokohama, Japan
- this antigen 50 ⁇ g/mouse
- the spleen was isolated and fused with X63Ag8 myeloma cells.
- Epitopes and cross reactivity to mouse and human Abeta of selected clones were determined by a microplate assay using various human and mouse Abeta fragments including fragments starting with 2nd and 3rd amino acid residue of Abeta.
- a 96-well plate (Maxisorp, Nunc, Rochester, NY, USA) was coated with Abeta fragments and non-specific binding was blocked.
- the plates were incubated with selected antibodies for 30 minutes at 37 °C, then with an HRP-coupled anti-mouse IgG antibody (500 ng/ml, Immuno-Biological Laboratories (IBL), Takasaki, Japan), and visualized by OPD substrate (Sigma, St Louis, MO, USA).
- HRP-coupled anti-mouse IgG antibody 500 ng/ml, Immuno-Biological Laboratories (IBL), Takasaki, Japan
- OPD substrate Sigma, St Louis, MO, USA.
- APP transgenic mice Tg2576 (Hsiao K. et al. 1996 Science 274:99-102), were perfused with 10 mM phosphate-buffered saline, pH 7.4 (PBS) followed by fixative consisting of 4% paraformaldehyde. After dehydration with 20% sucrose in 0.1 M phosphate buffer, brains were cut into 4 ⁇ m-thick sections. Serial sections were incubated with the anti-Abeta antibody, clone 14Fl (2 ⁇ g IgG/ml) along with other N terminus end (specific to the beta cleavage site)- and N terminus region-specific antibodies, clones 82El (Horikoshi Y.
- a 96-well plate (Maxisorp) was coated with an anti- Abeta 40 antibody, clone IAlO (Horikoshi Y. et al. 2004 Biochem Biophys Res Commun 319:733-737), overnight at 4 0 C. After blocking overnight at 4 0 C, standards (synthetic mouse Abeta peptide 1-40) and samples were loaded and incubated overnight at 4 °C. The plate was incubated with HRP- coupled detection antibody, 14Fl, and visualized using a TMB substrate. To determine the specificity to full-length Abeta 1-40, we incubated Abeta fragments, 2-40 and 3-40, and developed the plate as described above. We also used human Abeta 1-40 and mouse Abeta 1 -40 to determine the mouse/human cross reactivity.
- Mouse primary neurons were prepared from the cerebral cortex and hippocampal formation of mouse embryos (ICR, Charles River) at embryonic days 17-18, and cultured in NeuroBasal medium supplemented with B-27 (Invitrogen, Carlsbad, CA, USA). Cells were studied on the 6th day after preparation.
- a gamma secretase inhibitor N-[N-(3,5- difluorophenacetyl-L-alanyl)]-S-phenylglycine t-butyl ester (DAPT, Calbiochem, San Diego, CA, USA) (Dovey H. F. et al. 2001 J Neurochem 76:173-181) and a BACEl inhibitor, N-(I S,2R)- 1 -benzyl-3-(cyclopropylamino)-2-hydroxypropyl)-5-
- the brain was isolated from BACEl knockout mice (Cai H. et al. 2001 Nat Neurosci 4:233-234) carrying wildtype, heterozygous and homozygous (BACE+/+, BACE+/- and BACE-/-, respectively) genotypes (3 female mice at 11 weeks of age in each genotype,).
- the brain was homogenized in 10-fold volume of 50 mM Tris-HCl buffer, pH 7.6, containing 250 mM sucrose and protease inhibitor cocktail (Sigma).
- full-length APP, sAPPalpha, and sAPPbeta were prepared.
- the full-length cDNA of human APP was amplified from human brain cDNA (Clontech, Mountain View, CA, USA) using a forward primer, 5'- CGGTCGACTCGCGATGCTGCCCGGTTTGGC-S' (SEQ ID NO: 70) and a reverse primer, 5'-GGGCGGCCGCGTCTAGTTCTGCATCTGCTC-S' (SEQ ID NO: 71).
- the amplified products were digested with Sail and Notl, ligated into pGEX-6P-l vector (Amersham, Piscataway, NJ, USA) and transformed into E. coli JMl 09.
- sAPPalpha and sAPPbeta cDNA were amplified using APP695 cDNA as a template.
- forward primer 5'- CGGTCGACTCGCGATGCTGCCCGGTTTGGC-3' (SEQ ID NO: 72) for both sAPPalpha and sAPPbeta
- specific reverse primer for sAPPalpha and sAPPbeta; i.e., 5'-GCGCGGCCGCCTATTTTTGATGATGAACTT-S' (SEQ ID NO: 73) for sAPPalpha and 5'-GCGCGGCCGCCTACATCTTCACTTCAGAGAT-S' (SEQ ID NO: 74) for sAPPbeta.
- the amplified products were digested with Sail and Notl, ligated into pGEX- 6P-1 vector and used to transform E. coli JMl 09 cells.
- APP695, sAPPalpha and sAPPbeta cDNAs in pGEX-6P- 1 vector were transferred into pcDNA3.1(+) (Invitrogen), and transfected into COS-I cells by using FuGENE ⁇ (Roche Diagnostics, Basel, Switzerland). After 2 days, cells were harvested in 10 mM Tris, pH 8.0, consisting of 1% Nonident P-40, 150 mM NaCl andl mM EDTA 5 and used as positive controls for immunoblotting.
- sAPPbeta and sAPPalpha detection crude homogenates were mixed in diethylamine to yield 0.4% concentration, and centrifuged at 100,000 g for 45 minutes. The resultant supernatants were mixed with SDS sample buffer and run on a gel. Proteins were transferred to a PVDF membrane and the membrane was incubated with non-fat skim milk to minimize non-specific binding. The membranes were probed with a primary antibody overnight at 4 °C.
- Non-transgenic wildtype male mice ICR, Charles River
- a BACEl inhibitor, Inhibitor IV (Stachel S. J. et al. 2004 JMeJ Chem 47:6447-6450) (10 ⁇ g Inhibitor IV in 2 ⁇ l 40% DMSO/mouse) was stereotaxicially administered into the cerebral ventricle (Bregma -0.7 mm,1.8 mm lateral, 2.5 mm depth).
- 40% DMSO in saline vehicle
- the brain was isolated and the samples were prepared as described above. Antibody characterization and development of a full-length Abeta ELISA sensitive enough to detect endogenous mouse Abeta in wildtype mtiuse brain
- the epitope of our newly developed clone 14Fl is located between amino acid residues 1-4 of Abeta, and the ELISA combining 14Fl with the Abeta 40-specific IAlO antibody measures full-length Abeta 1-40.
- Our new 14F1/1A10 ELISA achieved sensitivity to single digit fmol/ml (equivalent to sub pg/ml) and detected endogenous full-length Abeta 1-40 in non-transgenic mouse brain homogenate (Fig. 12) and cell culture medium from primary cultured neurons (Fig. 11).
- IAlO 2 IgGl C-terminus end-specific (Epitope: Abeta 35-40)
- Inhibitor IV Inhibitor IV, there was significant reduction in Abeta 1-40 level (30% reduction compare to vehicle-administered mice, P ⁇ 0.01, Fig. 12Ba). However, the Abeta x-40 level did not change significantly (13% reduction, no significance, Fig. 12Bb). With systemic administration, Inhibitor IV did not reduce brain Abeta, although it reduced plasma Abeta (36% reduction compared to vehicle-treated mice, 100 mg/kg subcutaneous injection). Inhibitor IV is a substrate of P-glycoprotein brain efflux transporter (Stachel S. J. et al. 2006 Bioorg Med Chem Lett 16:641-644); thus we suspect that the lack of efficacy following systemic administration might be due to insufficient drug concentration in the brain.
- Beta and gamma secretases are promising therapeutic targets in AD, and transgenic mice that overexpress mutant human APP have been commonly used for evaluation of potential treatments.
- early-onset AD cases with increased generation of Abeta caused by genetic mutation represent a small percentage of the overall population suffering from AD (Rocca W. A. et al. 1991 Ann Neurol 30:381-390).
- APP expression is upregulated in sporadic AD cases. Therefore, testing therapeutic agents under physiologically relevant conditions, i.e. in mice with physiological APP expression, is important.
- 82El includes the human-unique 5th amino acid residue within the epitope, and it was not sensitive enough to detect endogenous Abeta in non-transgenic (wildtype) mouse brain.
- clone 14Fl N terminus end specific antibody, clone 14Fl, with an epitope within amino acid residues 1-4 of the Abeta peptide.
- amino acid residues 1-4 of Abeta are identical in humans and mice
- clone 14Fl showed preference to mouse Abeta.
- conformational differences between human and mouse Abeta determined this species preference of clone 14Fl.
- the ELISA which is composed of 14Fl and the Abeta 40-specific clone IAlO (Horikoshi Y. et al. 2004 Biochem Biophys Res Commun 319:733-737), detects full-length endogenous Abeta 1-40 in non-transgenic mouse brain homogenate. This is the first full-length Abeta ELISA which detects Abeta change in wildtype mice.
- Beta-secretase cleaves APP and generates sAPPbeta and the C99 fragment, and then gamma-secretase cleaves C99 and generates Abeta.
- Another cascade is known: alpha-secretase cleaves the APP molecule in the middle of the Abeta domain and generates sAPPalpha and the C83 fragment, and then gamma-secretase cleaves C83 and generates P3.
- additional cleavage sites are known.
- cells overexpressing BACEl generate C89 (the secondary beta-cleavage product starting with the Glul 1 amino acid of Abeta domain) in addition to C99 (the primary beta-cleavage product starting with Aspl of Abeta domain) (Haass C. et al. 1992 Nature 359:322-325; Gouras G. K. et al. 1998 JNeurochem 71:1920-1925; Vassar R. et al. 1999 Science 286:735-741).
- There are some mouse cross- reactive antibodies and they typically have epitopes within amino acid residues 14-28, a region that is identical in human and mouse Abeta.
- a commonly used mouse cross-reactive Abeta ELISA includes clone BNT77 (Suzuki N. et al. 1994 Science 264:1336-1340), which has an epitope within amino acid residue 11-16 of Abeta (Fukumoto H. et al. 1999 Neuroreport 10:2965-2969).
- the BNT77 ELISA is not cross-reactive to P3, but still detects the Abeta 11-40 fragment (generated by cleavage at the beta 1 (Glul l)-site), which is undesirable. Determination of full-length Abeta is essential for the accurate evaluation of Abeta changes, and our newly developed 14F1/1A10 ELISA provides the most accurate and useful results currently available.
- Full-length Abeta ELISA provides the optimal measure for the evaluation of Abeta- lowering strategies
- Abeta is generated by sequential proteolytic processing at the N- and C-termini of the Abeta domain by beta- and gamma-secretases, respectively (Hardy J. and Selkoe D. J. 2002 Science 297:353-356).
- a gamma secretase inhibitor, DAPT Dovey H. F. et al. 2001 J Neurochem 76:173-181), reduced Abeta production in a dose-dependent manner in primary cultured neurons, and diminished Abeta production at 1 ⁇ M.
- DAPT gamma secretase inhibitor
- a P3 cross- reactive x-40 ELISA detected much less significant Abeta reduction, probably due to an increase in P3 generation through compensatory upregulation of alpha secretase.
- BACEl as a therapeutic target Abeta-lowering approaches, including secretase inhibition, are being extensively pursued as potential disease-modifying therapies for AD (Aisen P. S.
- BACEl initiates Abeta generation and BACEl enzyme activity and/or BACEl protein level were elevated in sporadic AD brains (Fukumoto H. et al. 2002 Arch Neurol 59:1381-1389; Holsinger R. M. et al. 2002 Ann Neurol 51:783-786; Yang L. B. et al. 2003 Nat Med 9:3-4).
- BACEl is among the most aggressively pursued pharmacological targets in AD.
- a gamma secretase inhibitor 2-[(lR)-l-[[(4-chlorophenyl)sulfony](2,5- difluorophenyl)amino]ethyl]-5-fluorobenzenepropanoic acid (BMS-299897), showed greater efficacy in APP transgenic mice with excess APP, compared to APP-YAC mice (Lamb B. T. et al. 1993 Nat Genet 5:22-30) and guinea pigs with more physiologically- relevant APP expression (Anderson J. J. et al.
- BACEl inhibition elevated sAPPalpha levels in vivo as suggested by cell culture study (Skovronsky D. M. et al. 2000 J Biol Chem 275:2568-2575). sAPPalpha is not only non-amyloidogenic, but also neurotrophic and neuroprotective (Schubert D. and Behl C. 1993 Brain Res 629:275-282; Mattson M. P. et al. 1993 Neuron 10:243-254; Mattson M. P. et al. 1997 Brain Res Rev 23:47-61). BACEl inhibition reduces harmful Abeta, and the resulting compensatory increase of sAPPalpha has a favorable effect; this reconfirms BACEl as a promising therapeutic target.
- testing candidate interventions in wildtype mice is rapid and cost- effective; thus this approach is valuable as first-line in vivo testing of secretase inhibitors and other Abeta-lowering strategies. While non-transgenic mice do not recapitulate Abeta plaques, testing lead drug candidates in wildtype mice is advantageous to explore disease- modifying effects. Appendix 1
- FRL(SEQ ID NO: 2) EVQLHQSGAELVKPGASVKLSCTASGFNIK
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Immunology (AREA)
- Hematology (AREA)
- Chemical & Material Sciences (AREA)
- Urology & Nephrology (AREA)
- Molecular Biology (AREA)
- Cell Biology (AREA)
- Analytical Chemistry (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
- Food Science & Technology (AREA)
- Medicinal Chemistry (AREA)
- Physics & Mathematics (AREA)
- General Health & Medical Sciences (AREA)
- Microbiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Toxicology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Peptides Or Proteins (AREA)
Abstract
The invention relates to a method for determining whether a compound alters the amount of amyloid beta (A )? peptide present in a wildtype animal, comprising the steps of administering the compound to a wildtype animal, measuring the amount of the A peptide in a sample from the animal and determining whether the measured amount in the animal is different from the amount expected in a sample from a wildtype animal to which no compound has been administered, whereby a difference between the measured amount in the animal and the amount expected in the animal to which no compound has been administered indicates that the compound alters the amount of an A peptide present in the animal, wherein the amount of the A peptide is measured by immunoassay, wherein the immunoassay is a sandwich immunoassay using a capture binding substance bound to a solid phase and a labeled detection binding substance, and wherein the binding substances are specific for A peptide. Also included are kits with means to perform the method of the invention, compounds identified by the method of the invention, and methods of making compositions that comprise the compounds identified by the method of the invention.
Description
METHODS TO EVALUATE AMYLOID BETA-LOWERING AGENTS USING WILD-TYPE MICE
Related Applications
This application claims benefit of US Provisional Application No. 60/763,648 filed January 31, 2006, US Provisional Application No. 60/737,191 filed November 16, 2005 and US Provisional Application No. 60/707,713 filed August 12, 2005, all of which are hereby incorporated by reference in their entireties.
Statement Regarding Federally Sponsored Research or Development The U.S. Government has a paid-up license in this invention and the right in limited circumstances to require the patent owner to license others on reasonable terms as provided for by the terms of Grant No. AG022455 awarded by National Institutes of Health.
Field of the Invention
The present invention relates generally to methods and compositions for detecting endogenous brain Aβ, and more particularly to assays, such as immunoassays, for screening for compounds that specifically alter the level of Aβ .
Description of the Related Art
Amyloid beta (Aβ) is a major component of plaques in Alzheimer's disease (AD) and Down syndrome. Aβ is produced from the amyloid precursor protein (APP) by sequential proteolytic processing at the N- and C-termini of the Aβ domain by beta- and gamma- secretases, respectively. Aβ occurs principally in two forms consisting of 40 and 42 amino acids, Aβl-40 and 1-42 (Selkoe DJ. 1993 Trends in Neurosciences 16:403-409). Aβ lowering is a promising therapeutic approach for AD.
The first transgenic mouse model to form Aβ deposits overexpressed a mutant (V717) form of APP at high levels, generated elevated levels of Aβ in general, and Aβ42 in particular, and formed Aβ plaques at approximately 6 months of age (Games D. et al. 1995 Nature 373:523-527). A second mouse model (known as Tg2576) overexpressed the "Swedish" mutant APP (APPK67ON, M67IL), showed elevated Aβ40 and 42 levels, and developed amyloid deposits between 10 and 14 months of age (Hsiao K. et al. 1996 Science 274:99-102). Mutant presenilin 1 (PS1MI46L) transgenic mice have subtly elevated levels of
the Aβ42 peptide, but they lack pathological changes (Duff K. et al. 1996 Nature 383:710- 713). Transgenic mice derived from a cross between the APP line, Tg2576 and Duffs mutant PSl mice (known as PS/APP) show markedly accelerated accumulation of Aβ into visible deposits (deposition is initiated at approximately 10 weeks of age) compared with APP singly transgenic mice (Holcomb L. et al. 1998 Nat Med 4:97-100).
The generation of transgenic models of amyloidosis has significantly aided progress in the development of therapeutic approaches designed to lower brain Aβ load. However, all transgenic mice express their transgenes under the control of an autologous promoter. To investigate Aβ lowering strategies in a more physiologically relevant environment, testing using non-genetically manipulated animal is required. Three amino acids were different between rodent and primate Aβ, and human-specific enzyme linked immunosorbent assay (ELISA) is not useful for detection of rodent Aβ.
Segue to the Invention In this study, we developed a new ELISA using Aβ-specific antibodies and used it to monitor an Aβ-lowering strategy using non-transgenic mice.
Summary of the Invention
The invention relates to a method for determining whether a compound alters the amount of amyloid beta (Aβ) peptide present in a wildtype animal, comprising the steps of administering the compound to a wildtype animal, measuring the amount of the Aβ peptide in a sample from the animal and determining whether the measured amount in the animal is different from the amount expected in a sample from a wildtype animal to which no compound has been administered, whereby a difference between the measured amount in the animal and the amount expected in the animal to which no compound has been administered indicates that the compound alters the amount of an Aβ peptide present in the animal, wherein the amount of the Aβ peptide is measured by immunoassay, wherein the immunoassay is a sandwich immunoassay using a capture binding substance bound to a solid phase and a labeled detection binding substance, and wherein the binding substances are specific for Aβ peptide. Also included are kits with means to perform the method of the invention, compounds identified by the method of the invention, and methods of making compositions that comprise the compounds identified by the method of the invention.
Brief Description of the Drawings
Figure 1. Schematic diagram of the amyloid precursor protein (APP) and its principal metabolic derivatives, hi the second line, the sequence within amyloid precursor protein that contains the β-amyloid protein and TM regions is expanded (SEQ ID NO: 49).. Figure 2. A) Amino acid sequence of mouse amyloid precursor protein (APP)
(Ml 8373) (SEQ ID NO: 50). B) Sequence alignment of the APP Aβ region from human (Y00264) (SEQ ID NO: 51); mouse (Ml 8373) (SEQ ID NO: 52); Guinea Pig (X97631) (SEQ ID NO: 53); Macaque (M58727) (SEQ ID NO: 54); Chicken (AF289218) (SEQ ID NO: 55); Rat (NM_019288) (SEQ ID NO: 56); Xenopus (AJ298151) (SEQ ID NO: 57); Japanese sleeper ray (AB005544) (SEQ ID NO: 58); Zebra fish (NM_152886) (SEQ ID NO: 59); Japanese Puffer Fish (AF090120) (SEQ ID NO: 60); and Green spotted puffer fish (AF018165) (SEQ ID NO: 61). Shaded amino acid residues differ from human sequence within the Aβ region. Positions cleaved by α, β and γ secretases are indicated.
Figure 3. (A) Aβ burden was reduced by administration of 82El antibody when compared to that in the control group; Aβ42 staining. Bar = 500 μm. (B) Senile plaques in basal ganglia (in the circle) were absent in the 82El group; Aβ42 staining. Bar = 500 μm. (C) Aβ burden (Aβ42 positive plaque area) of the cortex (p<0.01) and hippocampus (p<0.05) was significantly lower in the 82El group than in the control group. (D) Insoluble Aβl-42 and Aβx-42 were significantly reduced in the 82El group based on ELISA measurements. *p<0.05, **p<0.01.
Figure 4. Characterization of β-cleavage site-specific antibody, clone 82El. (A) Fibril-free soluble (S) and soluble and fibril-containing (S/F) Aβ were prepared from synthetic Aβ peptide, run on a gel, and probed with 82El. Clone 82El detects both soluble and fibril Aβ. (B) APP transgenic mouse (Tg2576) brain extract was run on a gel and probed with 82El. 82El detects Aβ, but not with full-length APP. (C) β-cleavage site specificity was further confirmed using the transfected cells. 82El does not react with full-length APP (lane C), although non-β site-specific antibody 6E10 detects (lane C). βCTF is strongly detectable after treatment with a γ-secretase inhibitor, N-[N-(3,5-difluorophenacetyl-l-
alanyl)]-S-phenylglycine t-butylester (DAPT) (Dovey, H.F. et al. 2001 JNeurochem 76:173- 181) (lane D). (D) Human AD brain was stained with 82El. Bar = 200 μm.
Figure 5. Characterization of Aβ40-specific antibody, clone IAlO. Aβ40 and Aβ42 were run on a gel and probed with IAlO. IAlO specifically detected Aβ40 and did not show cross-reactivity to Aβ42 (A). Aβ was also detected in brain Aβ extract from an APP transgenic mouse, Tg2576 (B). Immunohistochemistry stained Aβ plaques in AD brain (C).
Bar = 200 μm.
Figure 6. Immunostaining of AD brains. Serial sections (A-D) from AD patient were stained with antibodies generated in this study. An asterisk indicates an identical Aβ plaque. Bar= 100 μm.
Figure 7. Aβ reduction after treatment with Aβ-lowering agents in non-transgenic mice. For detection by ELISA, horseradish peroxidase (HRP)-coupled 12B2 was used.
Figure 8. Immunoblot detection of Aβ with 14Fl monoclonal antibody.
Figure 9. Aβ reduction after treatment with Aβ-lowering agents in non-transgenic mice. For detection by ELISA, HRP-coupled 14Fl was used.
Figure 10. Characterization of newly developed mouse cross-reactive N terminus end- specific Abeta antibody, clone 14Fl, and ELISA development. Cross reactivity of clone 14Fl with human and mouse Abeta peptide was examined by immunoblotting (A). Synthetic human (lane H) and mouse (lane M) Abeta, and APP Tg2576 transgenic mouse brain homogenate (lane Tg) were run on a gel, transferred, and probed with anti-Abeta antibodies against N terminus end with strong preference to human (clone 82El, Aa), N terminus region (uncleaved APP cross-reactive) with preference to human (clone 6E10, Ab) and N terminus end with preference to mouse (clone 14Fl, Ac). Transgenic mouse brain serial sections containing the hippocampal formation were stained using these antibodies (B). Bar = 200 μm. Epitopes and detectable Abeta fragments by ELISA are summarized (C). Aβ peptide and surrounding sequences (SEQ ID NO: 62). Three amino acids, 5th, 10th and 13th amino acid residues, are different between human and mouse, and indicated with underlined bold. Please refer to Table 2 for more details.
Figure 11. Newly developed ELISA determined BACEl and gamma secretase inhibitors-mediated Abeta-lowering effects. Primary cultured neurons prepared from mouse
embryos were treated with a gamma secretase inhibitor, DAPT (Dovey et al. 2001) (A) or a BACEl inhibitor, Inhibitor IV (Stachel et al. 2004) (B) at various doses for 24 hours. Levels of endogenous full-length 1-40 and x-40 Abeta in the culture medium were determined using Abeta ELISAs, 14F1/1A10 and 12B2/1A10, respectively. Figure 12. BACEl inhibitor-induced Abeta reduction in non-transgenic mouse brain in vivo. Levels of brain Abeta 1-40 (a) and x-40 (b) are determined in BACEl knockout mice (A) and non-transgenic mice received BACEl inhibitor, Inhibitor IV (B) using 14F1/1A10 and 12B2/1A10 ELISAs, respectively. l)Abeta level in BACEl-/- mice was at the background level. **P<0.01, t-test. Figure 13. BACEl inhibition shifted APP cleavage to non-amyloidogenic alpha cleavage site. Specificity of antibodies against sAPPbeta (Aa) and sAPPalpha (Ab) are confirmed using recombinant sAPPalpha, sAPPbeta and uncleaved APP. Levels of sAPPbeta (Ba and Ca) and sAPPalpha (Bb and Cb), were determined in the brain homogenates from BACEl knockout mice and mice received intracerebroventricular injection of BACEl inhibitor, Inhibitor IV. Brain homogenates were run on a gel and probed with sAPPalpha- and sAPPbeta-specific antibodies.
Table 1. Brief Description of mAb SEO ID NOs.
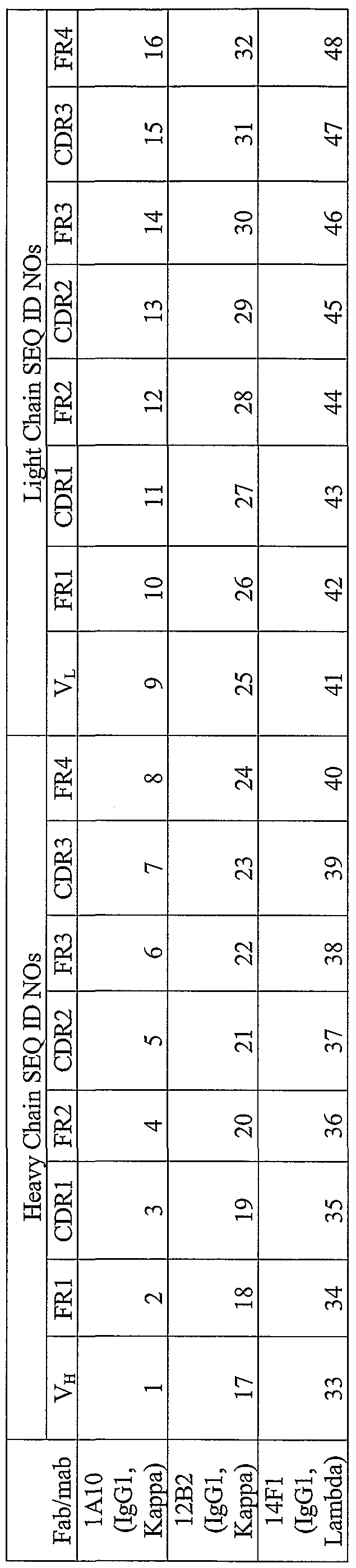
Detailed Description of the Preferred Embodiment
Alzheimer's disease (AD) is a neurodegenerative affliction associated with memory dysfunction. Accumulation and deposition of amyloid beta (Aβ) is the pathological hallmark of AD. Aβ is generated from a parent molecule, amyloid precursor protein (APP), by sequential proteolytic processing. Lowering Aβ is widely considered to be a primary AD therapeutic goal, and Aβ-lowering strategies are usually evaluated by applying ELISA methods to detect human Aβ to transgenic mice expressing mutant (typically Swedish) APP. Transgenic mice are now widely available, but substantial effort in colony maintenance as well as complex intellectual property issues limit flexibility with this approach. Further, all transgenic mice express their transgenes under the control of an autologous promoter; testing in a more physiologically relevant environment is desirable. Alternatively, non-transgenic mice can be considered, but have not yet been widely used in the assessment of Aβ lowering interventions; a primary reason is the limited sensitivity of mouse Aβ ELISAs. We developed a new Aβ ELISA detecting mouse endogenous Aβ 40 and 42 at the sub nM range, which is below 1/10th of endogenous Aβ levels in non- transgenic mouse brain. This ELISA also successfully quantified endogenous Aβ in the plasma and cerebrospinal fluid. To test the feasibility of evaluation of Aβ-lowering strategies in non-transgenic mice, we administered Aβ-lowering agents (for example, secretase inhibitors) and quantified brain Aβ levels. As expected, brain Aβ was reduced with treatment. Non-transgenic mice lack Aβ plaque pathology and so the disease- modifying impact of Aβ-lowering agents must be confirmed in plaque-bearing APP transgenic mice. However, the study of Aβ reduction in non-transgenic mice using this sensitive mouse Aβ ELISA provides a valuable tool in the development of therapeutic agents. Definitions
Unless defined otherwise, technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. See, e.g., Singleton P and Sainsbury D., Dictionary of Microbiology and Molecular Biology 3rd ed., J. Wiley & Sons, Chichester, New York, 2001. Amyloid Precursor Protein and Proteolytic Fragments β -Amyloid protein is derived from amyloid precursor protein by sequential cleavages by proteases referred to as β-secretase and γ-secretase (Figure 1). The amyloid
precursor protein comprises ubiquitously expressed proteins whose heterogeneity comes from the "alternative splicing" together of different protein coding regions (exons) within the amyloid precursor protein gene and also from post-translational modifications, such as the addition of sugar or phosphate groups to the protein backbone. Alternatively spliced forms of amyloid precursor protein containing 751 or 770 amino acids are widely expressed in cells throughout the body and also occur in neurons. However, neurons express much higher levels of a 695-amino acid splice form. The difference between the 751-, 770-, and 695-residue forms is the retention in the former of an exon that encodes an amino acid sequence that is homologous to certain inhibitors of serine proteases. The existence of this form suggests one normal function for these longer amyloid precursor protein isoforms; indeed, the amyloid precursor protein 751 that is in human platelets has been shown to inhibit factor XIa (a serine protease) in the clotting cascade.
In Figure 1, the amyloid precursor protein (APP) and its principal metabolic derivatives are depicted. The first line depicts the largest of the known (amyloid precursor protein alternate splice forms, comprising 770 amino acids. Regions of interest are indicated at their correct relative positions. A 17-residue signal peptide occurs at the N- terminus (box with vertical lines). Two alternatively spliced exons of 56 and 19 amino acids are inserted at residue 289; the first contains a serine protease inhibitor domain of the Kunitz type (KPl). A single membrane-spanning domain (transmembrane, TM) at amino acids 700 through 723 is indicated (dotted lines). The β-amyloid protein (Aβ) fragment includes 28 residues just outside the membrane plus the first 12 to 14 residues of the TM domain. In the second line, the sequence within amyloid precursor protein that contains the β-amyloid protein and TM regions is expanded (SEQ ID NO: 49). The underlined residues represent the β-amyloid proteins 1 to 42 peptide. The green letters below the wildtype sequence indicate the currently known missense mutations identified in certain families with Alzheimer disease or hereditary cerebral hemorrhage with amyloidosis. The 3 -digit numbers are codon numbers (amyloid precursor protein 770 isoform). In the third line, the first arrow indicates the site (after residue 687) of a cleavage by α-secretase that enables secretion of the large, soluble ectodomain of amyloid precursor protein (APPs-α) into the medium and retention of the 83-residue C-terminal fragment (C83) in the membrane. The C83 fragment can undergo cleavage by the protease called γ-secretase at residue 711 or residue 713 to release the p3 peptides. The fourth line depicts the alternative proteolytic cleavage after residue 671 by β-secretase that causes the secretion of the slightly
truncated APPs-β molecule and the retention of a 99 residue C-terminal fragment (C99). The C99 fragment can also undergo cleavage by γ-secretase to release the β amyloid peptides. Cleavage of both C83 and C99 by γ-secretase releases the β amyloid precursor protein intracellular domain (AICD) into the cytoplasm. Completely deleting the amyloid precursor protein gene in mice results in only minor deficits (Zheng, H. et al. 1995 Cell 81:525-531), suggesting that its 2 close homologs- amyloid precursor-like proteins 1 and 2 (Wasco, W. et al. 1992 Proc Natl Acad Sd USA 89:10758-10762; Slunt, H.H. et al. 1994 J Biol Chem 269:2637-2644)- have functions similar to amyloid precursor protein. All three members of the amyloid precursor protein family are single transmembrane proteins that resemble receptors, with the large amino-terminal region (the ectodomain) projecting from the cell surface or into the lumens of intracellular vesicles (for example, the endoplasmic reticulum, Golgi, trans-Golgi network, and endosomes) and the short carboxy-terminal region projecting into the cytoplasm (Figure 1, first line). In the normal processing of amyloid precursor protein by the 3 secretase enzymes
(designated as α-, β-, and γ-secretases), the most common cut occurs between amino acids 16 and 17 of the β -amyloid protein region, that is, 12 residues N-terminal to the transmembrane region (Figure 1, third line) and is carried out by α-secretase. This scission creates a large, soluble ectodomain fragment (β amyloid precursor proteinα) that is released from the cell surface and leaves a C-terminal fragment of 83 amino acids (C83) still embedded in the membrane (Figure 1, third line). α-Secretases, such as tumor necrosis factor-converting enzyme, are membrane-anchored proteases able to cleave diverse single- transmembrane proteins. Some amyloid precursor protein holoproteins that are not subjected to the α-secretase cleavage can instead be cut by β-secretase (Figure 1, fourth line). β-Secretase (also called β-amyloid cleaving enzyme 1) is a membrane-anchored aspartyl protease with its active site in its ectodomain (Vassar, R. et al. 1999 Science 286:735-741; Sinha, S. et al. 1999 Nature 402:537-540; Yan, R. et al. 1999 Nature 402:533-537). A slightly shorter form of the amyloid precursor protein ectodomain amyloid precursor proteinp) is released from the cell surface, and a C-terminal fragment of 99 amino acids (C99) embedded in the membrane is left. The C99 fragment can subsequently be cleaved by the unusual protease referred to as γ-secretase to create β- amyloid protein. Also, the C83 fragment made by α-secretase can undergo cleavage by the
same γ-secretase to generate a small peptide that makes up the latter two thirds of the β- amyloid protein region (designated as p3). In summary, C99 and C83 are the substrates of γ-secretase, which create β-amyloid protein and p3, respectively. In general, a substantially larger portion of total cellular amyloid precursor protein molecules is cleaved by α-secretase than by β-secretase.
Both Aβl-40 and Aβl-42 peptides are constitutive products of cellular metabolism and occur in normal biological fluids throughout life. Genetic defects that cause autosomal dominant AD, such as mutations in amyloid precursor protein (APP) or the presenilin (PS) genes PSl and PSl augment the amyloidogenic pathway of APP processing in all cells in a way that favors production of the highly self-aggregating Aβl-42 variant over the slightly shorter and less hydrophobic Aβl-40 form. Aβl-42 normally comprises only about 5-10% of total secreted Aβ peptides, but this fraction rises to about 15-40% when either APP or PS is mutant.
If Aβ peptides, particularly Aβl-42, are overproduced or insufficiently cleared, they become prone to aggregation into stable oligomers and larger polymers, apparently culminating in mature amyloid fibrils. The clinical diagnosis of Alzheimer disease is confirmed by observing numerous neuritic (amyloid) plaques and neurofibrillary tangles in the hippocampus, amygdala and association neocortex. The plaques (extracellular) are composed of the Aβl-40 and Aβl-42 proteins, whereas the tangles (intraneuronal) are composed of modified forms of the microtubule-associated protein, tau.
The Alzheimer's disease-linked amyloid-β precursor protein (APP) belongs to a superfamily of proteins, which also comprises the amyloid-β precursor-like proteins APLPl (Wasco, W. et al. 1992 Proc Natl Acad Sd USA 89:10758-10762) and APLP2 (Slunt, H.H. et al. 1994 J Biol Chem 269:2637-2644). APLPl and APLP2 are highly homologous to APP, but not within the Aβ region. The Aβ region of APP is highly conserved across divergent species as shown by the sequence alignment in Fig. 2. Antibodies
Antibodies come in a variety of classes, affinities, and idiotypes. They can be polyclonal or monoclonal, engineered or natural, and raised in animals or generated in vitro. Antibodies can be divided into three groups according to how they react with their target proteins.
One class of antibodies reacts with the target protein independently of its protein conformation. Antibodies of this type are particularly useful for measuring the total amount of the target protein in crude preparations or in cell extracts. They are usually polyclonal in nature and are prepared by immunizing animals with partially denatured protein or with a peptide whose sequence corresponds to part of the intact protein. However, monoclonal antibodies that are pan-specific are not uncommon.
Another class of antibodies reacts only with epitopes specific to the native form of the target protein. Antibodies of this type are typically monoclonal, have been raised against native protein, and recognize a given sequence of amino acids only when it occurs in its native three-dimensional configuration. These antibodies are useful for testing whether mutated forms of proteins that have been generated by in vitro mutagenesis are folded correctly or whether a wildtype protein expressed in heterologous cells is assembled into a correct three-dimensional configuration.
A third class of antibodies reacts only with denatured forms of the target protein. These antibodies are raised against fully denatured antigens and can be either monoclonal or polyclonal. Antibodies of this type are useful for western blotting.
Although there is no way to guarantee the production of particular types of antibodies, it is nevertheless possible to choose an immunization regimen that will favor the production of antibodies with the desired characteristics. However, it is always necessary to screen several independent antisera or a series of monoclonal antibodies to identify those suited to the tasks at hand. Derivation of Hybridomas
Hybridomas producing monoclonal antibodies are generated by the somatic cell fusion of two cell types: antibody-producing cells from an immunized animal, which by themselves die in tissue culture in a relatively short time, and myeloma cells, which contribute their immortality in tissue culture to the hybrid cell. The myeloma cells are variants carrying drug selection markers, so that only those myeloma cells that have fused with spleen cells providing the missing enzyme will survive under selective conditions. Successful hybridoma production is influenced by the characteristics of each of the cell populations, the fusion conditions, and the subsequent selection and screening of the hybrids.
Immunization of Donor Animals
Immunization protocols for fusion purposes have been developed empirically. A wide variety of standard routes and schedules of immunization can be used, the main distinguishing feature being the use of a final intravenous boost with antigen 2 to 4 days before fusion. The importance and required timing of the intravenous boost are thought to be related to the type of cell that preferentially fuses: Peak hybridoma production was found to precede the peak plaque-forming cell and the peak of serum antibody but corresponded to the peak of proliferation. Some investigators feel that animals should not be given the final boost and fused when they are at peak antibody titers, but rather should be rested until antibody levels decline, and then boosted for fusion. Myeloma Cell Lines Used as Fusion Partners
One technical advance necessary for successful production of hybridomas was the development of drug-sensitive variants of myeloma cell lines. A commonly used selective marker is sensitivity to medium containing hypoxanthine, aminopterin, and thymidine (HAT). Aminopterin poisons the de no vo synthesis of purines. Spleen cells expressing the enzyme hypoxanthine-guanine-phosphoribosyltransferase (HGPRT) necessary for the salvage pathway to recycle purines are able to survive in HAT medium, but die after a short time unless immortalized by fusion with the myeloma cell. The myeloma cells, mutagenized and selected to be HGPRT-negative, are killed by the HAT-containing medium unless they have fused and therefore contain the enzymes of the spleen cell. Thus, for several days after a fusion, there is extensive cell death; subsequently, the culture should contain only cells resulting from spleen-myeloma fusion. Other drug markers are occasionally used, for example, ouabain resistance.
Initial work used myeloma cells, which retained the capacity to secrete their own immunoglobulin products. Later, such fusion partners were replaced by myeloma variants that express only one endogenous chain or that fail to express immunoglobulin, so that the fused cell secretes primarily or exclusively antibody of the desired specificity. This confers a large advantage since, assuming random association, a cell making two heavy chains and two light chains would make immunoglobulin of which only one-sixteenth was of the desired type.
Periodically, the myeloma cells should be cycled through selective medium such as 8-azaguanine, to assure that they have not reverted to a drug-resistant phenotype, although this would be unnecessary for cell lines in which the mutation responsible for drug
sensitivity is a deletion. Incorporation of 8-azaguanine into DNA is dependent on the salvage pathway, so it selectively kills HGPRT-positive cells. Fusion Methods
Several different agents have been used to cause cell-cell fusion. Early somatic cell fusion work used Sendai virus. That approach has been replaced for routine fusions by use of polyethylene glycol (PEG). Both of these methods cause random fusions; many of the donor cells fusing will not be B cells in the right state to allow antibody production by the hybrid cell, and of those producing antibody, many will not make antibody of the desired specificity. Fusion efficiency is influenced by reagents in poorly understood ways. PEG or fetal bovine serum or other sera used in the medium must be tested in actual fusions and batches screened to select those able to support the fusion step. Ability of reagents to support cell growth is not an adequate test. Mycoplasma contamination of cell lines can also interfere, and monitoring for mycoplasma should be carried out.
Of growth-positive wells, the proportion secreting antibody of the desired specificity is quite variable. The original descriptions of fusions producing anti-SRBC antibodies reported 10% specific antibody among viable hybrids, but many antigens give far lower yields than that. The poor yield of desired hybridomas from random fusion methods led some investigators to attempt selective fusion of those cells expressing receptors specific for a particular antigen. Selective fusion methods involve attaching antigen to the myeloma cell, directly or indirectly, for example via an avidin biotin bridge. Immune spleen cells are then mixed with the antigen-coated myeloma cells and cell aggregates are allowed to form. The aggregates are fused by addition of PEG or by application of a strong electric field. Methods of Screening Fusions Methods for testing supernatants for desired antibodies can include the same range of methods used for studying such antibodies. Fusions have been successfully screened using RIA, ELISA, or other binding assays; visual immunofluorescence or flow cytometry; cytotoxicity assays; and assays for activation or blocking of biologic effects such as cell- mediated lympholysis (CML), receptor activation, and lymphokine activity. Fusions can also be screened by hybridization to detect mRNA for immunoglobulin of certain types in the cells, rather than antibody in the supernatant.
The major issue in choosing a screening assay is that the assay not be subject to fluctuation, which would lead to many false-positive identifications and a large investment
of effort in maintaining and cloning hybrid cells of no interest. Thus, clear-cut discrimination between positives and negatives is often more important than exquisite sensitivity. Most supernatants contain at least several milligrams per milliliter of antibody, which is enough to detect by numerous methods. A good screening assay should be convenient to use with hundreds of samples and should give results quickly, so that cells of greatest interest are still healthy when identified. If a parameter of interest is more difficult or time consuming to measure, it is often practical to use another assay for primary screening and then evaluate likely candidates in the more demanding assay. For example, a simple binding assay can be used for primary screening, and the positives then tested by immune precipitation to determine which bind to a particular component. Multiple-pass screening is useful in many other situations in which two or more antibody characteristics are important in the choice of clones to keep.
A screening assay difficult to perform on very large numbers of samples can also be applied by testing supernatants pooled from groups of hybrids, provided the assay sensitivity would allow detection of one positive supernatant diluted in a pool of negative ones. Components of positive pools are then screened individually. Post-fusion Processing of Hybridomas
After identification of positive cultures, hybridomas must be cloned to assure production of only one antibody, and cells must be frozen for future use. Since a great deal of labor and material are consumed by processing of candidate hybridomas, an efficient strategy must be used.
It is often best to retest all hybrids before cloning any of them. Only those hybrids producing specific antibody again at the second screening are then cloned. Cloning can be performed by limiting dilution or by colony selection from soft agar, hi either case, it is best to clone promptly, before possible nonproducing cells in the same well, including variants of the positive cell, can overgrow the antibody producers. Newly derived hybridomas are often unstable in their antibody production, perhaps because somatic cell hybrids are aneuploid and throw off chromosomes. The uncloned lines are maintained until active clones have been well established. Clones producing desired antibody must then be expanded, supernatant is collected for antibody preparation, cells are frozen, and frozen cells are thawed for verification of viability and antibody secretion.
Hybridomas Derived from Species Other than Mice
Laboratory mice are the most common species immunized for hybridoma production, but for a variety of reasons, other animal species often have advantages. If an antigen of interest is nonpolymorphic in the mouse, the mouse component might be immunogenic in other species, while mice would be tolerant to it. In the case of hybridomas for clinical use, mouse antibodies have the drawback of inducing anti-mouse immunoglobulin immune responses with possible deleterious effects, so derivation of human hybridomas would become desirable.
Several approaches have been taken to the derivation of hybridomas in species other than mouse. First, interspecies hybridization can be performed using mouse myeloma fusion partners. The resulting hybrids are often unstable and throw off chromosomes but clones can sometimes be selected that produce antibody in a stable fashion. Examples of this would be rat-mouse fusion to produce antibody to the mouse Fc receptor, and hamster- mouse fusion to produce antibody to mouse CD3. Rabbit-mouse hybridomas have also been described.
A second approach is the use of fusion partner cells from the desired species. Myeloma variants carrying drug selection markers are available in a number of species. A rat myeloma line adapted for this purpose, IR983F, was described in the literature. This approach avoids some of the instability in interspecies hybrids and allows ascites production in homologous hosts.
Production of human hybridomas is of special interest, because their use in therapies would avoid the problem of human immune responses to immunoglobulin derived from other animal species. Use of Gene Libraries to Derive Monoclonal Antibodies Monoclonal antibodies produced by hybridoma technology are derived from B cells of immunized animals. An alternative technology uses gene libraries and expression systems instead. This approach has the advantages of avoiding labor-intensive immunizations of animals and the screening of antibody-containing supernatants. Another advantage of the approach is circumventing tolerance. One can derive mAbs to antigens expressed in the responding animal species, including highly conserved antigens for which there may be no available responder that does not express the antigen.
The first version of such an approach involved preparation of VH and VK libraries and expression of the libraries in bacteria. In some cases, a VH domain alone binds antigen
with reasonable affinity, and so the expressed products of the gene library could be screened directly. Further development of the system led to use of VH and VL libraries made separately, and then preparation of a combinatorial library by cleaving, mixing, and religating the libraries at a restriction site. A linker can be used so that VH and VL can both be expressed on one covalent polypeptide; the flexibility of the linker allows association of the VH and VL in a normal three dimensional configuration and thus formation of an antigen-binding site.
Another technical innovation involves expression of VH and VK genes on the surface of bacteriophage as fusion proteins with a phage protein, to permit rapid screening of large numbers of sequences. Adsorption of antibody-bearing phage on antigen-coated surfaces allows positive selection of phage containing DNA encoding the desired Fv.
The phage technique has been applied to combinatorial variable region gene (V-region) libraries. In one study, rearranged VH and VK genes from immunized mice were used and the resulting VH-linker-Vκ products were screened for antigen binding. By using either VH or VK genes that gave activity, new sets, termed hierarchic libraries, were developed by pairing the known active gene segment with all elements of the library for the other chain from immunized mice. New active combinations were found by this approach.
Human antibody gene sequences can be recovered by polymerase chain reaction (PCR) from peripheral blood cells, bone marrow, or human cells reimmunized in SCID-hu mice. The phage display technique can then be used to select antigen-binding clones and derive reagents of desired specificity.
One limitation in the phage library technique initially was low affinity of the mAbs derived. Since they were generated by a random process and not subject to further somatic mutation, they did not achieve the exquisite fit of antibodies produced in vivo. Several approaches have now been used to improve affinities. Hypermutation and selection has now been achieved in vitro by a strategy using a bacterial mutator strain. The mutD5 strain of E. coli has an error-prone DNA polymerase III, leading to a high mutation rate, up to 105-fold enhanced over wildtype.
If a phage encoding antibody V regions infects such bacteria, the antibody sequence may be mutated. A single bacterium will harbor both wildtype and mutant phage, leading to mixed sequence protein displayed on each phage. Thus, before stringent selection for high-affinity mutants, the phage must be grown in nonmutator bacteria, so that each bacterium contains only one antibody sequence and antibody expressed on a phage matches
its sequence. Multiple rounds of mutation followed by growth in nonmulator bacteria and then selection for high-affinity binding led to an overall 100-fold increase in affinity. Improved affinity has also been achieved by use of site-directed mutagenesis to alter residues in hypervariable regions affecting dissociation rates. Since arbitrary combinatorial possibilities of VH and VL can occur in the various libraries discussed, the antibodies generated do not reflect the combinations actually selected and expressed in immune responses. It has been suggested that one could recover a "natural library" by recovering V-region genes from individual cells by PCR. However, recovering genes from a large enough number of representative cells does not seem a reasonable or efficient approach to repertoire studies. Thus, the combinatorial library technology does not replace hybridoma technology for many immunologic studies, including studies of the immune repertoire and patterns of its expression in immune responses. What combinatorial gene libraries do provide is a powerful way to derive antibody reagents of desired specificity, including some that would not occur naturally and so could not be derived by other means.
Production of Human or Humanized Monoclonal Antibodies
Many of the side effects of monoclonal antibodies in clinical use are due to the foreign immunoglobulin constant regions. Recognition of foreign epitopes can lead to sensitization and so preclude subsequent use in the same individual of different monoclonal antibodies. Thus, monoclonal antibodies with some or all structure derived from human immunoglobulins have advantages.
Several approaches have been taken employing fusion of human cells with animal myelomas or with human tumor cells of various kinds, and use of Epstein-Barr virus to immortalize antibody-producing cells. Production of populations of sensitized human cells to be fused presents another special problem, since the donors cannot be immunized at will. In one example, in vitro stimulation of lymphocytes with antigen followed by fusion with mouse myeloma cells has been used to generate a series of antibodies to varicella zoster.
Another approach to production of monoclonal antibodies with human characteristics involves application of genetic engineering. When mouse monoclonal antibodies are used clinically, many of the complications are due to reactions to mouse immunoglobulin as a foreign protein, and thus there could be an advantage to minimizing the part of the antibody structure recognized as foreign by humans. Human constant regions can be combined with mouse V regions or even just mouse hypervariable segments
by molecular genetic techniques. Antigen-binding specificity is retained in some cases, and the "humanized" chimeric molecules may have many of the advantages of human hybridomas.
Production of fully human mAbs in transgenic mice has now been achieved by multiple laboratories. The strategy has involved insertion of constructs containing clusters of human immunoglobulin V, D, J, and C genes into the mouse germ line to generate one transgenic line, and targeted disruption of the mouse heavy-chain and κ-chain loci to generate another transgenic line. From these two lines, mice are then bred that express only human antibodies. Specificity of Monoclonal Antibodies
Since all of the molecules in a sample of monoclonal antibody have the same V- region structure, barring variants arising after cloning, they all have the same specificity. This uniformity has the advantage that batches of monoclonal antibody do not vary in specificity as polyclonal sera often do. The most obvious fact about cross-reactions of monoclonal antibodies is that they are characteristic of all molecules and cannot be removed by absorption without removing all activity. An exception would be an apparent cross-reaction due to a subset of denatured antibody molecules, which could be removed on the basis of that binding. The homogeneity of monoclonal antibodies allows refinement of specificity analysis that was not possible with polyclonal sera. Polyclonal Versus Monoclonal Antibodies
When monoclonal antibodies first became available, some people expected that they would be exquisitely specific and would be superior to polyclonal sera for essentially all purposes. Further thought about the issues discussed above, however, suggests that this is not always the case and depends on the intended use of the antibodies. Not only do monoclonal antibodies cross-react, but when they do, the cross-reaction is not minor and cannot be removed by absorption. A large panel of monoclonal antibodies may be needed before one is identified with the precise range of reactivity desired for a study.
In polyclonal sera, on the other hand, each different antibody has a distinct range of reactivity, and the only common feature would be detectable reactivity with the antigen used for immunization or testing. Thus the serum as a whole may show only a low-titered cross-reaction with any particular other antigen, and that cross-reaction can be removed by absorption, leaving substantial activity against the immunizing antigen. For the purposes of
an experiment, a polyclonal serum may be "more specific" than anyone of its clonal parts and may be more useful.
Polyclonal sera also have advantages in certain technical situations, such as immunoprecipitation in which multivalency is important. Many antigens are univalent with respect to monoclonal antibody binding but display multiple distinct sites that can be recognized by different components of polyclonal sera. Thus a greater degree of cross- linking can be achieved.
The ultimate serologic reagent in many cases may well be a mixture of monoclonal antibodies that have been chosen according to their cross-reactions. The mixture would be better defined and more reproducible than a polyclonal antiserum and would have the same advantage of overlapping specificities. Antibodies are Specific Binding Substances
Antibodies, whether monoclonal or polyclonal, provide a unique type of reagent that can be made with high specificity for almost any desired organic or biochemical structure, often with extremely high affinity. These can be naturally divalent (e.g., in the case of IgG) or multivalent (e.g., in the case of IgM) or can be made as monovalent molecules, such as Fab or recombinant Fv fragments. They serve not only as a major arm of host defense, playing a major role in the protective efficacy of most existing antiviral and antibacterial vaccines, but also as very versatile tools for research and clinical use. Radioimmunoassay (RLA) and ELISA have revolutionized the detection of minute quantities of biologic molecules, such as hormones and cytokines, and thus have become indispensable for clinical diagnosis and monitoring of patients, as well as for basic and applied research. Current solid-phase versions of these take advantage not only of the intrinsic affinity and specificity of the antibodies, but also of the implicit multivalency and local high concentration on a solid surface. Binding Assays
Solid phase immunological binding assays have the advantages of ease of processing large numbers of samples and potentially increased affinity (compared to the intrinsic affinity of the antibody due to the effects at the solid-liquid interface. 1. Radioimmunoassay (RIA)
The central concept of RIA is that the binding of an infinitesimal concentration of highly radioactive tracer antigen to low concentrations of a high-affinity specific antibody is very sensitive to competition by unlabeled antigen and is also very specific for that antigen.
Thus concentrations of antigen in unknown samples can be determined by their ability to compete with tracer for binding to antibody. The method can be used to measure very low concentrations of a molecule, even in the presence of the many impurities in biologic fluids. To do this, one must prepare the appropriate high-affinity antibody and radiolabeled antigen, develop a method to distinguish bound from free labeled antigen, determine the optimal concentrations of antibody and tracer-labeled antigen to maximize sensitivity, and generate a standard curve, using known concentrations of competing unlabeled antigen, from which to read off the concentrations in unknown samples. 2. Enzyme-linked Immunosorbent Assay (ELISA) An alternative solid-phase readout system for the detection of antigen-antibody reactions is the ELISA assay. In principle, the only difference from RIAs is that antibodies antigen are covalently coupled to an enzyme instead of a radioisotope, so that bound enzyme activity is measured instead of bound counts per minute. In practice, the safety and convenience of nonradioactive materials and the commercial availability of plate readers that can measure the absorbance of wells in a few seconds account for ELISA' s popularity. Since both ELISA and RIA are governed by the same thermodynamic constraints, and the enzyme can be detected in the same range of molarity as commonly used radioisotopes, the sensitivity and specificity are comparable.
A basic strategy for using ELISA assays to detect antigen involves the sandwich technique for detecting antigen. Specific antibody is used to coat the micro titer wells. Antigen is then bound to the solid-phase antibody. Finally, a second antibody, linked to enzyme, is added. This binds to the solid-phase antigen-antibody complex, carrying enzyme along with it. Excess second antibody is washed off and substrate is added. The absorbance produced is a function of the antigen concentration of the test solution, which can be determined from a standard curve. Specificity of the assay depends on the specificity of the antibodies used to coat the plate and detect antigen. Sensitivity depends on affinities as well as the amount of the first antibody bound to the well, which can be increased by using affinity-purified antibodies in the coating step. The binding of both antibodies of the sandwich depends on divalency of the antigen, or else the two antibodies must be specific for different antigenic determinants on the same antigen molecule. Antipeptide Antibodies
If the sequence of a protein is known or can be deduced from the nucleic acid sequence, specific antisera can be raised by immunizing animals with a synthetic peptide
corresponding in sequence to a segment of the native protein. If information about the primary sequence of the target protein is limited, there may be little or no choice in the peptide sequence used as an immunogen. However, there is a good chance that peptides chosen at random will be at least partially buried in the native protein and they may be too hydrophobic to be efficient immunogens. Antibodies directed against these peptides may be of low titer and/or may react only with denatured protein.
Hydrophilic peptides that contain charged residues are much better immunogens and also have a high probability of occupying a surface location on the native protein. Antipeptide antibodies raised against conformationally flexible surface features of proteins such as turns and β-loops are likely to be of high titer and may react efficiently with the native protein. Most of the computing packages that are commonly used to analyze DNA sequences contain programs to search protein sequences for surface peptides that are likely to be good antigens. The goal of these programs is to choose a sequence of amino acids that contains a preponderance of polar residues and no more than four adjacent hydrophobic residues. Such peptides are likely to be soluble in aqueous solvents and therefore easy to couple to carrier proteins.
Several scales of hydrophobicity and hydrophilicity have been proposed for amino acids. Although these scales are in good general agreement, they differ slightly in the order of amino acids and in the relative hydrophobicity values assigned to them. This variability arises because the hydrophobicity of an amino acid residue is the product of several different factors, including electrostatic charge, hydrogen-bonding capability, and surface area. In addition, the hydrophobicity of an amino acid can be assessed experimentally by partitioning into solvents of various types. The variation in hydrophobicity scales of amino acids reflects the particular weightings that different investigators have attached to these and other factors.
Computer programs to predict strongly antigenic sites in proteins rely on hydrophobicity scales alone or in combination with programs that predict secondary structure. The strongest antigenic sites are predicted in regions of the protein surface that are high in charge and low in hydrophobicity. Rarely found in ordered structures such as helices or sheets, such regions usually map to turns and loops that are rich in residues with H-bonding potential.
Highly charged regions also often occur at the amino and carboxyl termini of proteins, which tend to be regions of high flexibility. However, if the protein of interest is a
secretory or transmembrane protein, or is located in organelles such as mitochondria or chloroplasts, it is better not to use peptides derived from the terminal regions. The amino- terminal peptide is likely to be part of a signal or leader sequence that will be cleaved during posttranslational processing. The carboxy-terminal region of transmembrane proteins may be located on the cytoplasmic side of the membrane and may only be accessible to antibodies after cells are permeabilized, fixed, or lysed. Screening Compounds
The term "binding substance" refers to a polypeptide substantially encoded by an immunoglobulin gene or immunoglobulin genes, or fragments thereof, which specifically bind and recognize an analyte (antigen). The recognized immunoglobulin genes include the kappa, lambda, alpha, gamma, delta, epsilon and mu constant region genes, as well as the myriad immunoglobulin variable region genes. Antibodies exist, e.g., as intact immunoglobulins or as a number of well characterized fragments produced by digestion with various peptidases. This includes, e.g., Fab' and F(ab)'2 fragments. The term "binding substance," as used herein, also includes antibody fragments either produced by the modification of whole antibodies or those synthesized de novo using recombinant DNA methodologies.
The term "immunoassay" is an assay that utilizes a binding substance to specifically bind an analyte. The immunoassay is characterized by the use of specific binding properties of a particular antibody to isolate, target, and/or quantify the analyte.
A binding substance "specifically binds to" or "is specifically immunoreactive with" a protein when the binding substance functions in a binding reaction which is determinative of the presence of the protein in the presence of a heterogeneous population of proteins and other biologies. Thus, under designated immunoassay conditions, the specified binding substances bind preferentially to a particular protein and do not bind in a significant amount to other proteins present in the sample. Specific binding to a protein under such conditions requires an antibody that is selected for its specificity for a particular protein. A variety of immunoassay formats may be used to select binding substances specifically immunoreactive with a particular protein. For example, solid-phase ELISA immunoassays are routinely used to select monoclonal antibodies specifically immunoreactive with a protein. See Harlow and Lane (1988) Antibodies, A Laboratory Manual, Cold Spring Harbor Publications, New York, for a description of immunoassay formats and conditions that can be used to determine specific immunoreactivity.
A "label" is a composition detectable by spectroscopic, photochemical, biochemical, immunochemical, or chemical means. For example, useful labels include P, fluorescent dyes, electron-dense reagents, enzymes (e.g., as commonly used in an ELISA), biotin, digoxigenin, or haptens and proteins for which antisera or monoclonal antibodies are available. Binding substances can be made detectible, e.g., by incorporating a radiolabel into the peptide, and used to detect antibodies specifically reactive with the peptide. A label often generates a measurable signal, such as radioactivity, fluorescent light or enzyme activity, which can be used to quantify the amount of bound label. Aβ Monoclonal Antibodies As used herein, the term "antibody" means an immunoglobulin molecule or a fragment of an immunoglobulin molecule having the ability to specifically bind to a particular antigen. Antibodies are well known to those of ordinary skill in the science of immunology. As used herein, the term "antibody" means not only full-length antibody molecules but also fragments of antibody molecules retaining antigen binding ability. Such fragments are also well known in the art and are regularly employed both in vitro and in vivo. In particular, as used herein, the term "antibody" means not only full-length immunoglobulin molecules but also antigen binding active fragments such as the well- known active fragments F(ab')2, Fab, Fv, and Fd.
As used herein with respect to polypeptides, the term "substantially pure" means that the polypeptides are essentially free of other substances with which they may be found in nature or in vivo systems to an extent practical and appropriate for their intended use. In particular, the polypeptides are sufficiently pure and are sufficiently free from other biological constituents of their host cells so as to be useful in, for example, generating antibodies, sequencing, or producing pharmaceutical preparations. By techniques well known in the art, substantially pure polypeptides may be produced in light of the nucleic acid and amino acid sequences disclosed herein. Because a substantially purified polypeptide of the invention may be admixed with a pharmaceutically acceptable carrier in a pharmaceutical preparation, the polypeptide may comprise only a certain percentage by weight of the preparation. The polypeptide is nonetheless substantially pure in that it has been substantially separated from the substances with which it may be associated in living systems.
As used herein with respect to nucleic acids, the term "isolated" means: (i) amplified in vitro by, for example, polymerase chain reaction (PCR); (ii) recombinantly
produced by cloning; (iii) purified, as by cleavage and gel separation; or (iv) synthesized by, for example, chemical synthesis. An isolated nucleic acid is one which is readily manipulable by recombinant DNA techniques well known in the art. Thus, a nucleotide sequence contained in a vector in which 5' and 3' restriction sites are known or for which polymerase chain reaction (PCR) primer sequences have been disclosed is considered isolated but a nucleic acid sequence existing in its native state in its natural host is not. An isolated nucleic acid may be substantially purified, but need not be. For example, a nucleic acid that is isolated within a cloning or expression vector is not pure in that it may comprise only a tiny percentage of the material in the cell in which it resides. Such a nucleic acid is isolated, however, as the term is used herein because it is readily manipulable by standard techniques known to those of ordinary skill in the art.
As used herein, a coding sequence and regulatory sequences are said to be "operably joined" when they are covalently linked in such a way as to place the expression or transcription of the coding sequence under the influence or control of the regulatory sequences. If it is desired that the coding sequences be translated into a functional protein, two DNA sequences are said to be operably joined if induction of a promoter in the 5' regulatory sequences results in the transcription of the coding sequence and if the nature of the linkage between the two DNA sequences does not (1) result in the introduction of a frame-shift mutation, (2) interfere with the ability of the promoter region to direct the transcription of the coding sequences, or (3) interfere with the ability of the corresponding RNA transcript to be translated into a protein. Thus, a promoter region would be operably joined to a coding sequence if the promoter region were capable of effecting transcription of that DNA sequence such that the resulting transcript might be translated into the desired protein or polypeptide. The precise nature of the regulatory sequences needed for gene expression may vary between species or cell types, but shall in general include, as necessary, 5' non-transcribing and 5' non-translating sequences involved with initiation of transcription and translation respectively, such as a TATA box, capping sequence, CAAT sequence, and the like. Especially, such 5' non-transcribing regulatory sequences will include a promoter region which includes a promoter sequence for transcriptional control of the operably joined gene. Regulatory sequences may also include enhancer sequences or upstream activator sequences, as desired.
As used herein, a "vector" may be any of a number of nucleic acids into which a desired sequence may be inserted by restriction and ligation for transport between different genetic environments or for expression in a host cell. Vectors are typically composed of DNA although RNA vectors are also available. Vectors include, but are not limited to, plasmids and phagemids. A cloning vector is one which is able to replicate in a host cell, and which is further characterized by one or more endonuclease restriction sites at which the vector may be cut in a determinable fashion and into which a desired DNA sequence may be ligated such that the new recombinant vector retains its ability to replicate in the host cell, hi the case of plasmids, replication of the desired sequence may occur many times as the plasmid increases in copy number within the host bacterium or just a single time per host before the host reproduces by mitosis. In the case of phage, replication may occur actively during a lytic phase or passively during a lysogenic phase. An expression vector is one into which a desired DNA sequence may be inserted by restriction and ligation such that it is operably joined to regulatory sequences and may be expressed as an RNA transcript. Vectors may further contain one or more marker sequences suitable for use in the identification and selection of cells which have been transformed or transfected with the vector. Markers include, for example, genes encoding proteins which increase or decrease either resistance or sensitivity to antibiotics or other compounds, genes which encode enzymes whose activities are detectable by standard assays known in the art (e.g., β- galactosidase or alkaline phosphatase), and genes which visibly affect the phenotype of transformed or transfected cells, hosts, colonies or plaques. Preferred vectors are those capable of autonomous replication and expression of the structural gene products present in the DNA segments to which they are operably joined.
Significantly, as is well-known in the art, only a small portion of an antibody molecule, the paratope, is involved in the binding of the antibody to its epitope (see, in general, Clark, W.R. 1986 in The Experimental Foundations of Modern Immunology Wiley & Sons, Inc., New York; Roitt, I. 1991 in Essential Immunology. 7th Ed., Blackwell Scientific Publications, Oxford). The pFc' and Fc regions, for example, are effectors of the complement cascade but are not involved in antigen binding. An antibody from which the pFc' region has been enzymatically cleaved, or which has been produced without the pFc' region, designated an F(ab')2 fragment, retains both of the antigen binding sites of a full- length antibody. Similarly, an antibody from which the Fc region has been enzymatically cleaved, or which has been produced without the Fc region, designated an Fab fragment,
retains one of the antigen binding sites of a full-length antibody molecule. Proceeding further, Fab fragments consist of a covalently bound antibody light chain and a portion of the antibody heavy chain denoted Fd. The Fd fragments are the major determinant of antibody specificity (a single Fd fragment may be associated with up to ten different light chains without altering antibody specificity) and Fd fragments retain epitope-binding ability in isolation.
Within the antigen-binding portion of an antibody, as is well-known in the art, there are complementarity determining regions (CDRs), which directly interact with the epitope of the antigen, and framework regions (FRs), which maintain the tertiary structure of the paratope (see, in general, Clark, 1986, supra; Roitt, 1991, supra). In both the heavy chain Fd fragment and the light chain of IgG immunoglobulins, there are four framework regions (FRl through FR4) separated respectively by three complementarity determining regions (CDRl through CDR3). The CDRs, and in particular the CDR3 regions, and more particularly the heavy chain CDR3, are largely responsible for antibody specificity. The complete amino acid sequences of the antigen-binding Fab portions of the Aβ monoclonal antibodies as well as the relevant FR and CDR regions are disclosed herein. SEQ ID NOs: 1, 17 and 33 disclose the amino acid sequences of the Fd fragment of the Aβ monoclonal antibodies. The amino acid sequences of the heavy chain FRl, CDRl, FR2, CDR2, FR3, CDR3 and FR4 regions are disclosed as (FRl, SEQ ID NOs: 2, 18 and 34); (CDRl, SEQ ID NOs: 3, 19 and 35); (FR2, SEQ ID NOs: 4, 20 and 36); (CDR2, SEQ ID NOs: 5, 21 and 37); (FR3, SEQ ID NOs: 6, 22 and 38); (CDR3, SEQ ID NOs: 7, 23 and 39); and (FR4, SEQ ID NOs: 8, 24 and 40). SEQ ID NOs: 9, 25 and 41 disclose the amino acid sequences of the light chains of the Aβ monoclonal antibodies. The amino acid sequences of the light chain FRl, CDRl, FR2, CDR2, FR3, CDR3 and FR4 regions are disclosed as (FRl, SEQ ID NOs: 10, 26 and 42); (CDRl, SEQ ID NOs: 11, 27 and 43); (FR2, SEQ ID NOs: 12, 28 and 44); (CDR2, SEQ ID NOs: 13, 29 and 45); (FR3, SEQ ID NOs: 14, 30 and 46); (CDR3, SEQ ID NOs: 15, 31 and 47); and (FR4, SEQ ID NOs: 16, 32 and 48).
The present invention includes substantially pure polypeptides comprising an antibody selectively binding an Aβ antigenic determinant, wherein said antibody includes a heavy chain CDR3 region having the amino acid sequence selected from the group consisting of SEQ ID NO: 7, SEQ ID NO: 23 and SEQ ID NO: 39 and substantially pure
polypeptides of the same wherein said antibody comprises an Fd fragment or an Fab fragment.
The invention also includes substantially pure polypeptides comprising an antibody selectively binding an Aβ epitope, wherein said antibody includes a heavy chain CDR2 region having the amino acid sequence of SEQ ID NO: 5 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 21 (when heavy chain CDR3 region is SEQ ID NO: 23), or SEQ ID NO: 37 (when heavy chain CDR3 region is SEQ ID NO: 39).
The invention also includes substantially pure polypeptides comprising an antibody selectively binding an Aβ epitope, wherein said antibody includes a heavy chain CDRl region having the amino acid sequence of SEQ ID NO: 3 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 19 (when heavy chain CDR3 region is SEQ ID NO: 23), or SEQ ID NO: 35 (when heavy chain CDR3 region is SEQ ID NO: 39).
The invention also includes substantially pure polypeptides comprising an antibody selectively binding an Aβ epitope, wherein said antibody includes a heavy chain Fd region including the amino acid sequence of SEQ ID NO: 1 (when heavy chain CDR3 region is
SEQ ID NO: 7), SEQ ID NO: 17 (when heavy chain CDR3 region is SEQ ID NO: 23), or
SEQ ID NO: 33 (when heavy chain CDR3 region is SEQ ID NO: 39).
The invention also includes substantially pure polypeptides comprising an antibody selectively binding an Aβ epitope, wherein said antibody includes a light chain CDR3 region having the amino acid sequence of SEQ ID NO: 15 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 31 (when heavy chain CDR3 region is SEQ ID
NO: 23), or SEQ ID NO: 47 (when heavy chain CDR3 region is SEQ ID NO: 39).
The invention also includes substantially pure polypeptides comprising an antibody selectively binding an Aβ epitope, wherein said antibody includes a light chain CDR2 region having the amino acid sequence of SEQ ID NO: 13 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 29 (when heavy chain CDR3 region is SEQ ID
NO: 23), or SEQ ID NO: 45 (when heavy chain CDR3 region is SEQ ID NO: 39).
The invention also includes substantially pure polypeptides comprising an antibody selectively binding an Aβ epitope, wherein said antibody includes a light chain CDRl region having the amino acid sequence of SEQ ID NO: 11 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 27 (when heavy chain CDR3 region is SEQ ID
NO: 23), or SEQ ID NO: 43 (when heavy chain CDR3 region is SEQ ID NO: 39).
The invention also includes substantially pure polypeptides comprising an antibody selectively binding an Aβ epitope, wherein said antibody includes a light chain region including the amino acid sequence of SEQ ID NO: 9 (when heavy chain CDR3 region is
SEQ ID NO: 7), SEQ ID NO: 25 (when heavy chain CDR3 region is SEQ ID NO: 23), or SEQ ID NO: 41 (when heavy chain CDR3 region is SEQ ID NO: 39).
The invention also includes substantially pure polypeptides comprising an antibody selectively binding an Aβ epitope, wherein said antibody includes a heavy chain Fd region including the CDR amnio acid sequences of SEQ ID NO: 1 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 17 (when heavy chain CDR3 region is SEQ ID NO: 23), or SEQ ID NO: 33 (when heavy chain CDR3 region is SEQ ID NO: 39).
The invention also includes substantially pure polypeptides comprising an antibody selectively binding an Aβ epitope, wherein said antibody includes a light chain region including the CDR amino acid sequences of SEQ ID NO: 9 (when heavy chain CDR3 region is SEQ ID NO: 7), SEQ ID NO: 25 (when heavy chain CDR3 region is SEQ ID NO: 23), or SEQ ID NO: 41 (when heavy chain CDR3 region is SEQ ID NO: 39).
The present invention also contemplates isolated nucleic acids that encode the polypeptides described herein that comprise an antibody that selectively binds an Aβ antigenic determinant, isolated nucleic acids that comprise a vector including a regulatory sequence operably joined to the nucleic acids that encode the polypeptides that comprise an antibody that selectively binds an Aβ antigenic determinant as described herein, and host cells that include a vector comprising a nucleic acid that encodes a polypeptide described herein that comprising an antibody selectively binding an Aβ antigenic determinant. In vitro Screening
Alzheimer's disease is characterized by the initial deposition of Aβ peptide in the form of amyloid plaques in the brain. Therefore, effective treatments for AD are expected to decrease the production of these peptides, whereas agents that hasten progress of the disease are expected to increase production of the peptide. Because Aβ peptides are the major constituent of neuritic plaques, it is useful to identify compounds that specifically inhibit the amount of these peptides. Accordingly, this invention provides methods for screening compounds that specifically elevate or decrease the the amount of Aβ peptide in an animal. Compounds that decrease the amount of Aβ peptide are candidates for use in
treating the disease, while compounds that increase the amount may hasten the disease and are to be avoided by humans.
Screening methods of this invention for determining whether a test compound specifically alters the amount of Aβ peptide present in a wildtype animal involve administering the compound to the animal, measuring the amount of Aβ peptide present in a sample from the animal, and determining whether this amount is greater than, less than or the same as an amount expected in a sample from a wildtype animal to which no compound has been administered. If the amounts are different, then the compound affects the presence of Aβ peptide in the animal. This amount can be measured, for example, from brain extracts.
The expected amount generally will be a control amount determined by measuring Aβ peptide present in the animal in the absence of the compound. However, one also may determine the expected amount by extrapolation; measuring the amount of Aβ peptide present in the animal upon administration of different amounts of the compound to the animal, and using these figures to calculate the expected amount. In certain instances measuring a control amount for the purposes of comparison may not be necessary because the effect of the compound on Aβ peptide presence is clear. For example, a compound may render Aβ peptide undetectable in an animal that normally posesses detectable amounts, indicating that the compound decreases Aβ peptide from the amount expected in its absence.
Drugs are often initially screened by a cell-free assay. For example, enzyme activity may be assayed to investigate β- and γ-secretase inhibitors. Then, compounds are screened using in vitro cultured cells. Primary cultured neurons, which are the most physiologically relevant system, can be assayed using the methods of this invention. Amyloid-β Peptide and Related Proteins and Peptides
The terms "amyloid-β peptide", "Aβ" or "βAP" as used herein refer to an approximately 4.2 kD protein which, in the brains of AD, Down's Syndrome, Hereditary Cerebral Hemorrhage with Amyloidosis Dutch type (HCHWA-D) and some normal aged subjects, forms the subunit of the amyloid filaments comprising the senile (amyloid) plaques and the amyloid deposits in small cerebral and meningeal blood vessels (amyloid angiopathy). Aβ can occur in a filamentous polymeric form (in this form, it exhibits the Congo-red and thioflavin-S dye-binding characteristics of amyloid described in connection
therewith). Aβ can also occur in a non-filamentous form ("preamyloid" or "amorphous" or "diffuse" deposits) in brain tissue, in which form no birefringent staining by Congo red occurs.
Aβ is an approximately 40-42 amino acid fragment of a large membrane-spanning glycoprotein, referred to as the amyloid precursor protein (APP) , encoded by a gene on the long arm of human chromosome 21. Forms of Aβ longer than 42 amino acids are also contemplated herein. Aβ is further characterized by its relative mobility in SDS- polyacrylamide gel electrophoresis or in high perfoπnance liquid chromatography (HPLC). As used herein, Aβ also refers to related polymorphic forms of Aβ, including those that result from mutations in the Aβ region of the APP gene.
The term "Aβ fragment" as used herein refers to fragments and degradation products of Aβ which are generated at low concentrations by mammalian cells. Particular Aβ fragments have a molecular weight of approximately 3 kD and are presently believed to include peptides with, for example, amino acid residues 3-34, 6-27, 6-34, 6-35, 6-42, 11-34, 11-40, 11-43, 12-43, 17-40 aηd 17-42 of Ab (Vigo-Pelfrey et al. 1993 J Neurochem 61:1965-1968).
As used herein, the term "Aβ peptide" refers to Aβ or an Aβ fragment whose amino-terminus begins at amino acid number 1 of Aβ or which is amino-terminally truncated, and whose carboxy-terminus extends no further than amino acid number 40 or 42. These peptides and fragments also comprise a heterogenous group. The term "Aβ(40)" or "Aβ40" or "Aβ(l-40)" or "Aβl-40" refers to Aβ or an Aβ fragment whose C-terminal amino acid is #40 of Aβ. The term "Aβ(42)" or "Aβ42" or "Aβ(l-42)" or "Aβl-42" refers to Aβ or an Aβ fragment whose C-terminal amino acid is #42 of Aβ.
The term "N terminus" of the Aβ peptide as used herein refers to a region of Aβ which is located approximately between amino acid residues 1 and 16 (e.g., Aspl and Lysl6).
The term "C terminus" of the Aβ peptide as used herein refers to a region of Aβ which is located approximately between amino acid residues 35 and 40 (e.g., Met35 and VaWO) or between amino acids 38 and 42 (e.g., Gly38 and Ala42), referring to an Aβ fragment where the C-terminal amino acid is #40 of Aβ and an Aβ fragment where the C- terminal amino acid is #42 of Aβ, respectively.
The term "middle region" as used herein refers to a region of Aβ which is located approximately between amino acid residues 11 and 28 (e.g. Glul 1 and Lys28).
The term "Aβ-related condition" as used herein is defined as including Alzheimer's disease (which includes familial Alzheimer's disease), Down's Syndrome, HCHWA-D, and advanced aging of the brain.
As used herein, the term "p3" refers to a peptide whose amino acid sequence is substantially similar to Aβ, but whose amino-terminal amino acid begins at amino acid 17 of Aβ. The term "p3 fragment" as used herein refers to fragments and degradation products of p3. Whereas p3 is produced through a different processing pathway than Aβ, for the purposes of the detection methods of this invention, p3 and p3 fragments are considered to be a subset of Aβ peptides, because certain detection techniques that recognize Aβ solely from the carboxy terminus generally also will recognize p3. Also it is shown that the same apparent mechanisms generate the p3 and Aβ carboxy-termini.
The term "amyloid-β precursor protein" (APP) as used herein is defined as a polypeptide that is encoded by a gene of the same name localized in humans on the long arm of chromosome 21 and that includes the Aβ region within the carboxyl third of its coding region. APP is a glycosylated, single-membrane spanning protein expressed in a wide variety of cells in many mammalian tissues. Examples of specific isotypes of APP which are currently known to exist in humans are the 695-amino acid polypeptide described by Kang et al. 1987 Nature 325:733-736; the 751-amino acid polypeptide described by Ponte et al. 1988 Nature 331:525-527 (1988) and Tanzi et al. 1988 Nature 331:528-530; and the 770-amino acid polypeptide described by Kitaguchi et al. 1988 Nature 331:530- 532. Examples of specific variants of APP include point mutations which can differ in both position and resultant neuropathological phenotype (for a review of known variant mutations see Hardy 1992 Nature Genet 1:233-234). Measuring Aβ Peptide
Aβ peptides can be detected by any method known in the art. Preferably, the method involves an immunoassay employing binding substances specific for the peptides. Optionally, one can detect Aβ peptides by determining their size, e.g., by HPLC or by mass spectrometry.
A. Binding Substances
One step of the screening methods of this invention involves measuring the amount of at least one Aβ peptide, specifically, in a sample. Measuring Aβ peptides specifically means measuring Aβ peptides so as to distinguish them from APP. Specific measurement of Aβ peptide preferably is performed by the use of binding substances that specifically recognize Aβ peptides.
Another method of this invention involves screening compounds to determine their ability to alter the amount of Aβ peptides. Such methods can involve the use of binding substances that can distinguish Aβ peptides from APP. B. Immunoassays
The use of immunological detection techniques, i.e., immunoassays employing binding substances, is preferred. Particularly suitable detection techniques include ELISA, Western blotting, radioimmunoassay, and the like. Suitable immunological methods employing a single antibody are also contemplated, for example, radioimmunoassay using an antibody specific for Aβ peptide, or single antibody ELISA methods. It will be clear that the particular forms of Aβ detected by such methods depend upon the particular binding substances employed. For example, binding substances directed to the middle portion of Aβ may detect Aβ peptides whose amino termini do not extend to amino acid no. 1 of Aβ. Also, binding substances directed to the carboxy-terminal end of Aβ peptide may detect peptides ending at amino acids 40-42. Therefore, determining the specificity of the binding substances will assist in determining exactly which Aβ peptides the assay is detecting.
In one embodiment, the method to detect Aβ peptides is an immunoassay involving two antibodies. One antibody is specific for an epitope containing amino acids approximately between 35 and 40 or amino acids approximately between 38 and 42. A preferred immunoassay technique is a two-site or "sandwich" assay. This assay involves a capture binding substance, usually bound to a solid phase, and a labelled detection binding substance, hi this method, Aβ peptides are captured from the sample using a first binding substance specific for Aβ peptides (usually bound to a solid phase). The capture of Aβ peptides is detected using a labeled second binding substance specific for Aβ against a different antigenic determinant on the same Aβ peptide. For example, labeled binding substances include those directed to the middle region (amino acids ~11 to
~28) or binding substances specific for amino-terminal amino acids (amino acids ~1 to -16).
Particular methods for preparing such antibodies and utilizing such antibodies in an exemplary ELISA are set forth in the Experimental section hereinafter. A sandwich assay using an antibody against the middle region of Aβ can be used to specifically measure Aβ and Aβ fragments whose amino-terminus begins at approximately amino acid 17 of Aβ. Such assays may recognize p3 or p3 fragments, since those peptides begin at amino acid number 17 of Aβ. hi certain embodiments, binding substances that recognize Aβ peptides and do not recognize p3 are useful in the methods of this invention. Antibodies specific for Aβ(l-40), i.e., which do not cross react with Aβ(=39) and antibodies specific for Aβ(l-42), i.e., which do not cross react with Aβ(=41) are also useful in the methods of this invention. These antibodies can be made by immunizing animals with synthetic peptides that include amino acids from the C-terminus of Aβ(l-40) or Aβ(l- 42). For example, the synthetic peptide can include amino acids Aβ(35-40) or Aβ(38-42). C. Preparation of Antibodies
Antibodies specific for Aβ can be prepared, e.g., by immunizing an animal with a peptide whose amino acid sequence corresponds to amino acids from the N-terminus, C- terminus or middle portion of Aβ. For example, human Aβ peptides consisting of amino acid residues 1-16, 11-28, 35-40 and 38-42 may be used as imrnunogens for Aβ N- terminus, middle region and C-terminus-specific antibodies respectively. Antibodies specific for Aβ(l-40) or Aβ(l-42) can be prepared, e.g., by immunizing an animal with a peptide whose amino acid sequence corresponds with amino acids 35-40 or 38-42 of Aβ.
Synthetic polypeptide haptens may be produced by the well-known Merrifield solid- phase synthesis technique in which amino acids are sequentially added to a growing chain (Merrifield (1963) J Am Chem Soc 85:2149-2156). Suitable peptide haptens will usually comprise at least five contiguous residues within Aβ and may include more than six residues. The amino acid sequences may be based on the sequence of Aβ set forth herein.
Once a sufficient quantity of polypeptide hapten has been obtained, it may be conjugated to a suitable immunogenic carrier, such as serum albumin, keyhole limpet hemocyanin, or other suitable protein carriers, as generally described in Hudson and Hay, Practical Immunology, Blackwell Scientific Publications, Oxford, Chapter 1.3, 1980.
Antibodies specific for the desired epitope may be produced by in vitro or in vivo techniques. In vitro techniques involve exposure of lymphocytes to the immunogens, while in vivo techniques require the injection of the immunogens into a suitable vertebrate host. Suitable vertebrate hosts are non-human, including mice, rats, rabbits, sheep, goats, and the like. Immunogens are injected into the animal according to a predetermined schedule, and the animals are periodically bled, with successive bleeds having improved titer and specificity. The injections may be made intramuscularly, intraperitoneally, subcutaneously, or the like, and an adjuvant, such as incomplete Freund's adjuvant, may be employed.
If desired, monoclonal antibodies can be obtained by preparing immortalized cell lines capable of producing antibodies having desired specificity. Such immortalized cell lines may be produced in a variety of ways. Conveniently, a small vertebrate, such as a mouse, is hyperimmunized with the desired immunogen by the method just described. The vertebrate is then killed, usually several days after the final immunization, the spleen cells removed, and the spleen cells immortalized. The manner of immortalization is not critical. Presently, the most common technique is fusion with a myeloma cell fusion partner, as first described by Kohler and Milstein 1975 Nature 256:495-497. Other techniques include EBV transformation, transformation with bare DNA, e.g., oncogenes, retroviruses, etc., or any other method which provides for stable maintenance of the cell line and production of monoclonal antibodies. Specific techniques for preparing monoclonal antibodies are described in Antibodies: A Laboratory Manual, Harlow and Lane, eds., Cold Spring Harbor Laboratory, 1988. hi addition to monoclonal antibodies and polyclonal antibodies (antisera), the detection techniques of the present invention will also be able to use antibody fragments, such as F(ab), Fv, VL, VH, and other fragments. In the use of polyclonal antibodies, however, it may be necessary to adsorb the anti-sera against the target epitopes in order to produce a monospecific antibody population. It will also be possible to employ recombinantly produced antibodies (immunoglobulins) and variations thereof. It would also be possible to prepare other recombinant proteins which would mimic the binding specificity of antibodies prepared as just described. Kits
This invention also provides kits for performing assays of the invention. The kits include means for detecting specifically Aβ peptides. The means can include any means known or described above, e.g., binding substances.
In another embodiment, the kit is useful for immunoassays including two antibodies for each antigen. For example, the kit can comprise a binding substance specific for the amino terminal region of Aβ, the carboxy terminal region of Aβ, or the middle region of Aβ. Such antibodies are useful for the capture or detection of Aβ peptides. In one kit useful for a sandwich ELISA, the binding substance specific for the carboxy terminal region of Aβ is bound to a solid phase, and the binding substances specific for a different antigenic determinant on the same Aβ peptide are detectably labeled.
The detectable labels can be any known and used in the art including, e.g., a biotinylation label, a radioactive label, a light scattering label, an enzymatic label, a fluorescent label and the like. When the label is enzymatic, the kit can further comprise a substrate for the enzyme. Test Compounds
The test compounds can be any molecule, compound, or other substance which can be administered to the test animal without substantially interfering with animal viability. Suitable test compounds may be small molecules (i.e., molecules whose molecular mass is no more than 1000 Daltons), biological polymers, such as polypeptides, polysaccharides, polynucleotides, and the like. The test compounds will typically be administered at a dosage of from 1 ng/kg to lg/kg, usually from 10 μg/kg to 100 mg/kg.
Test compounds which are able to inhibit amounts of Aβ peptides are considered as candidates for further determinations of the ability to block β-amyloid amounts in animals and humans. Such compounds can be tested in in vivo studies, as described below. Inhibition of amounts of Aβ peptides indicates that levels of Aβ peptides are unavailable for forming β-amyloid plaques. In Vivo Screening The inventors have found that wildtype animal models can be used to detect quantitative differences in Aβ peptide. Wildtype animals are useful for screening compounds that alter the amount of Aβ peptide in the assays of this invention, indicating their ability to affect the course of Alzheimer's disease, both to ameliorate and aggravate the condition. Rodent models, and in particular murine models, are suitable for this use. Many genes are known to be involved in APP cascade. A common approach to examine the role of a particular gene in the APP pathway is to first create a transgenic or knockout mouse model with respect to the gene of interest. Typically, the knockout or transgenic mouse is subsequently crossed with transgenic mice expressing human APP so
that human Aβ level may be measured using existing human Aβ assay techniques. This additional step in creating an animal model is time consuming. Using the methods of this invention, wildtype mouse Aβ level can be measured, eliminating the need for the secondary cross with a transgenic mouse expressing human APP and saving time and resources.
In all cases, it will be necessary to obtain a control value which is characteristic of the level of Aβ peptide amount in the test animal in the absence of test compound(s). In cases where the animal is sacrificed, it will be necessary to base the value on an average or a typical value from other wildtype test animals, but which have not received the administration of any test compounds or any other substances expected to affect the amount of Aβ peptide. Once such control level is determined, test compounds can be administered to additional test animals, in which deviation from the average control value would indicate that the test compound had an effect on the amount of Aβ peptide in the animal. Test substances which are considered positive, i.e., likely to be beneficial in the treatment of Alzheimer's disease or other β -amyloid-related conditions, will be those which are able to reduce the level of Aβ peptide amount, preferably by at least 5%, more preferably by at least 10%, and most preferably by at least 20, 30, 40, 50, 60, 70, 80, 90, 95 or 100%.
The present invention further comprises pharmaceutical compositions incorporating a compound selected by methods described herein, including a pharmaceutically acceptable carrier. Such pharmaceutical compositions should contain a therapeutic or prophylactic amount of at least one compound identified by the method of the present invention. The pharmaceutically acceptable carrier can be any compatible non-toxic substance suitable to deliver the compounds to an intended host. Sterile water, alcohol, fats, waxes and inert solids may be used as the carrier. Pharmaceutically acceptable adjuvants, buffering agents, dispersing agents and the like may also be incorporated into the pharmaceutical compositions. Preparation of pharmaceutical conditions incorporating active agents is well described in the medical and scientific literature. See for example, Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, 16th Ed., 1982.
The pharmaceutical compositions just described are suitable for systemic administration to the host, including parenteral, topical and oral administration. The pharmaceutical compositions may be administered parenterally, i.e., subcutaneously, intramuscularly, or intravenously. Thus, the present invention provides compositions for
administration to a host, where the compositions comprise a pharmaceutically acceptable solution of the identified compound in an acceptable carrier, as described above. Antibodies against the Amino terminus of Aβ
The N-terminal peptide of amyloid β used as antigen comprises the amino acid sequence starting from the N terminus and continuing toward the C terminus of the amino acid sequence of amyloid β (1-40) or amyloid β (1-42), comprising the sequence of amino acids 1 through 28 of amyloid β being preferable, and a peptide comprising the sequence of amino acids 1 through 16 of amyloid β, illustrated in the following amino acid sequence being especially preferable. DAEFRHDSGYEVHHQK (SEQ ID NO: 63)
A peptide comprising the above amino acid sequence can be obtained by various methods, without any particular limitations. For example, if may be synthesized by a method publicly known in the industry, or else a synthesized product can be purchased from Synpep Corporation, TANA Laboratories (USA), or the like. A peptide comprising the above amino acid sequence is next conjugated with a biopolymer compound to make the antigen. In this case, it is preferable to perform conjugation by attaching a cysteine (C) to the C-terminal side amino acid of the aforementioned peptide.
Examples of the biopolymer conjugated with the aforementioned peptide include keyhole limpet hemocyanin (hereinafter referred to as "KLH"), egg white albumin (hereinafter referred to as "OVA"), bovine serum albumin (hereinafter referred to as "BSA"), rabbit serum albumin (hereinafter referred to as "RSA") and thyroglobulin, with KLH and thyroglobulin being more preferable.
The conjugation of the aforementioned peptide and biopolymer can be performed by a publicly known method such as the mixed acid anhydride method (Erlanger, B.F. et al.1959 J Biol Chem 234:1090-1094) or the activated ester method (Kara, A.E et al. 1994 J AgHc Food Chem 42 301-309).
The mixed acid anhydride used in the mixed acid anhydride method is obtained by subjecting the aforementioned peptide to a conventional Schotten-Baumann reaction, which is then reacted with a biopolymer compound to prepare the target conjugate of peptide and biopolymer. Examples of halofumaric acid esters which may be used in this mixed acid anhydride method include methyl chlorofumarate, methyl bromofumarate, ethyl chlorofumarate, ethyl bromofumarate, isobutyl chlorofumarate and the like. The
proportions of peptide, halofumaric acid ester, and biopolymer used in this method can be selected as appropriate from a broad range.
The Schotten-Baumann reaction is conducted in the presence of a basic compound. The basic compounds which can be used for this reaction include compounds commonly used for Schotten-Baumann reactions, such as triethylamine, trimethylamine, pyridine, dimethylaniline, N-methylmorpholine, diazabicyclononene (DBN), diazabicycloundecene (DBU), diazabicyclooctane (DABCO) and other organic bases, potassium carbonate, sodium carbonate, potassium hydrogen carbonate, sodium hydrogen carbonate and other inorganic bases, and the like. Furthermore, the aforementioned reaction is normally conducted at -2O0C to 100°C, preferably O0C to 5O0C, and the reaction time is about 5 minutes to 10 hours, preferably 5 minutes to 2 hours.
The reaction of the obtained mixed acid anhydride and biopolymer compound is normally conducted at -2O0C to 15O0C, preferably O0C to 1000C, with the reaction time being about 5 minutes to 10 hours, preferably 5 minutes to 5 hours. The mixed acid anhydride method is commonly conducted in a solvent. For the solvent, one can employ any solvent commonly used in the mixed acid anhydride method; specific examples include halogenated hydrocarbons such as dichloromethane, chloroform and dichloroethane, aromatic hydrocarbons such as benzene, toluene, and xylene, ethers such as diethyl ether, dioxane, tetrahydrofuran, and dimethoxyethane, esters such as methyl acetate and ethyl acetate, and aprotic polar solvents such as N,N-dimethyl formamide, dimethyl sulfoxide, and hexamethylphosphoric triamide.
Furthermore, the activated ester method can be generally carried out as follows. First, the aforementioned peptide is dissolved in organic solvent and is reacted with N-hydroxysuccinic acid imide in the presence of a coupling agent to produce N-hydroxysuccinic acid imide activated ester.
As the coupling agent used in this reaction, one can employ coupling agents commonly used in condensation reactions, such as dicyclohexyl carbodiimide, carbonyl diimidazole, and water-soluble carbodiimide. Furthermore, for the organic solvent, N,N-dimethyl formamide (DMF), dimethyl sulfoxide, dioxane, or the like can be used. The molar ratio of peptide to coupling agent such as N-hydroxysuccinic acid imide used in the reaction is preferably between 1:10 and 10:1, with 1 :1 being most preferable. The reaction temperature is 0 to 500C, preferably 22 to 27°C, and the reaction time is 5 minutes to
24 hours, preferably 1 to 2 hours. With regard to the reaction temperature, the reaction can be conducted at a temperature above the melting points but below the boiling points.
After the coupling reaction, the reaction liquid is added to a solution in which the biopolymer compound has been dissolved and is reacted, whereupon, for example if the biopolymer compound has free amino groups, an acid-amide bond will be formed between those amino groups and the carboxyl groups of the aforementioned peptide. The reaction temperature is 0 to 60°C, preferably 5 to 40°C, more preferably 22 to 270C, and the reaction time is 5 minutes to 24 hours, preferably 1 to 16 hours, more preferably 1 to 2 hours.
A conjugate of the aforementioned peptide and biopolymer compound (hereinafter referred to simply as "Conjugate 1") can be obtained by purifying the reaction product obtained by either of the above methods by means of dialysis, a desalting column or the like.
To prepare the N-terminal antibody of the present invention using the aforementioned conjugate, an animal is given an immunization using the conjugated antigen, followed by booster immunization(s) using the conjugated antigen, and antibodies are collected from that animal.
Regarding the specific method of immunization, the antigen conjugate is dissolved in sodium phosphate buffer solution (hereinafter referred to as "PBS"), which is mixed with an auxiliary such as Freund's complete adjuvant or incomplete adjuvant or alum and used as the antigen to immunize the animals.
Any type of animal commonly used in this field can be used for the immunized animals: examples include mammals such as mice, rats, rabbits, goats, and cows. Furthermore, as the method of administration of the immunogen for the immunization, subcutaneous injection, intraperitoneal injection, intravenous injection, intracutaneous injection, or intramuscular injection may be used, but subcutaneous injection or intraperitoneal injection is preferable. The immunization can be performed a single time or multiple times at a suitable interval, preferably, multiple times at an interval of one to five weeks.
Animals, which have been immunized with the antigen, as described above are then boosted with antigen in the same manner. The immunization can be performed multiple times at a suitable interval, preferably, multiple times at an interval of one to five weeks.
Finally, following conventional methods, myeloma cells and immune cells obtained from animals immunized as described above are fused to obtain hybridomas, and antibodies
are collected from a culture of said hybridomas to obtain monoclonal antibodies against the N terminus of amyloid β.
The N-terminal antibodies of the present invention obtained in this manner can be used as immunological assay reagents or as a treatment drugs for Alzheimer's disease and the like.
For use as immunological assay reagents, labeling or immobilization is preferably performed if necessary.
Labeling is performed by conjugating a labeling substance, for instance an enzyme such as horseradish peroxidase (HRP) or alkaline phosphatase, a fluorescent substance such as fluorescein isocyanate or rhodamine, a radioactive substance such as 32P or 125I, or a chemoluminescent substance, with the N-terminal antibodies. Furthermore, immobilization is performed by binding the N-terminal antibodies to a suitable solid phase. Any solid phase commonly used in immunological assays can be used for the solid phase, for instance, a plate such as a polystyrene 96-well microtiter plate or an amino bonded microtiter plate, or various types of beads. N-terminal antibodies can be immobilized for instance by placing buffer solution containing the antibodies onto a carrier and incubating.
The N-terminal antibodies of the present invention can be used as a reagent, which al-lows accurate determination of amyloid β by combining with antibodies which recognize amyloid β (1-40) or amyloid β (1-42). Antibodies against the middle portion of Aβ
A peptide corresponding to the middle portion of amyloid β used as antigen may comprise, for example, the sequence of amino acids ~11 through ~28 of amyloid β, illustrated in the following amino acid sequence.
EVHHQKLVFFAEDVGSNK (SEQ ID NO: 64) A peptide comprising the above amino acid sequence can be obtained by various methods, without any particular limitations. For example, it may be synthesized by a method publicly known in the industry, or else a synthesized product can be purchased from Synpep Corporation, TANA Laboratories (USA), or the like.
A peptide comprising the above amino acid sequence is next conjugated with a biopolymer compound to make the antigen. In this case, it is preferable to perform conjugation by attaching a cysteine (C) to the C-terminal side amino acid of the aforementioned peptide. Methods of immunizing animals and deriving monoclonal antibodies are described above.
Antibodies against the C-terminus of Aβ
As the antibodies which recognize amyloid β (1-40) or amyloid β (1-42) which are combined with the N-terminal antibodies of the present invention, one can use for instance polyclonal antibodies or monoclonal antibodies which can be obtained by conventional methods using part or all of the amino acid sequence of amyloid β (1-40) or amyloid β (1- 42) as an antigen, hi the present invention, among these antibodies, it is especially preferable, from the standpoint of accuracy of determination, to use antibodies, which recognize a C-terminal peptide of amyloid β (hereinafter referred to as "C-terminal antibodies"). There is no particular limitation on the aforementioned C-terminal antibodies so long as they are antibodies which distinguish the C-terminal peptide of the amyloid β (1-40) or amyloid β (1-42) that is to be measured, but antibodies which do not recognize amyloid β (1-43) besides the amyloid β (1-40) or amyloid β (1-42) which is to be measured are preferable. Specifically, a C-terminal antibody which recognizes amyloid β (1-40) (hereinafter referred to as "C-Terminal Antibody 1") can be obtained by conventional methods using a peptide (C-terminal peptide) comprising an amino acid sequence starting from the C terminus and continuing toward the N terminus of amyloid β (1-40) as an antigen. More specifically, the C-terminal peptide used as antigen is preferably a peptide comprising the sequence of amino acids 18 through 40 of amyloid β (1-40), with a peptide comprising 35 through 40 of amyloid β (1-40), shown in the following amino acid sequence being especially preferable.
MVGGW (SEQ ID NO: 65)
Similarly, a C-terminal antibody which recognizes amyloid β (1-42) (hereinafter referred to as "C-Terminal Antibody 2") can be obtained by conventional methods using a peptide comprising an amino acid sequence starting from the C terminus and continuing toward the N terminus of amyloid β (1-42) as an antigen. More specifically, the peptide used as antigen is preferably a peptide comprising the sequence of amino acids 18 through 42 of amyloid β (1-42), with a peptide comprising 38 through 42 of amyloid β (1-42), shown in the following amino acid sequence being especially preferable. GWIA (SEQ ID NO: 66)
Furthermore, to prepare C-terminal antibodies using the aforementioned peptide, one can use a conjugate of the aforementioned peptide and a biopolymer compound as an
antigen, just as in the method of preparing the aforementioned N-terminal antibodies. The conjugate of aforementioned peptide and biopolymer compound is preferably conjugated by attaching lysine and cysteine (KC), for example, to the N-terminal amino acid of the aforementioned peptide. For the antibody that recognizes amyloid β (1-40) or amyloid β (1-42) as described above, one can utilize Anti-Amyloid β (35-40); (IAlO) Mouse IgG MoAb (product number: 10047), sold by Immuno-Biological Laboratories Co., Ltd.
Utilizing the N-terminal antibody (or middle region antibody) of the present invention and an antibody which recognizes amyloid β (1-40) or amyloid β (1-42) makes it possible to accurately measure amyloid β (1-40) or amyloid β (1-42). Specific measurement methods include the various methods commonly used in immunological assays ("The hybridoma method and monoclonal antibodies," published by R&D Planning Co., Ltd., pages 30-53, 5 March 1982), such as radioimmunoassay (RIA), ELISA ( Engvall, E. et al. 1980: Methods in Enzymol 70:419-439), fluorescent antibody technique, plaque technique, spot technique, aggregation technique, ouchterlony, immunochromatography, etc.
These assay methods can be selected among as appropriate based on various perspectives, but ELISA is preferable from the standpoint of sensitivity, convenience, and the like. More specifically, taking for example the sandwich technique, which is one type of ELISA, the procedure of the amyloid β assay can be described as follows.
First, in Process (A), an antibody, which recognizes amyloid β (1-40) or amyloid β (1-42) is immobilized on a carrier. Then, in Process (B), the carrier surface on which no antibodies are immobilized is blocked for instance by a protein unrelated to amyloid β. Furthermore, in Process (C), specimens containing various concentrations of amyloid β are added to form complexes of amyloid β and antibody, after which, in Process (D), labeled N- terminal antibody (or middle region antibody) of the present invention is added and the amyloid β-antibody complex is bound thereto, and finally, in Process (E), by measuring the quantity of label, the quantity of amyloid β in a specimen can be determined based on a calibration curve prepared in advance. Measurement is possible also if the antibodies used in Process (A) and Process (D) are reversed, but immobilizing the antibody which recognizes amyloid β (1-40) or amyloid β (1-42) is preferable from the standpoint of detection sensitivity.
Specifically, in Process (A), there are no particular limitations as to the carrier used for immobilizing the antibody, which recognizes amyloid β (1-40), or amyloid β (1-42), and any carrier commonly used in immunological assays can be used. Possible examples include a polystyrene 96-well microtiter plate or an amino bonded microtiter plate. Furthermore, the aforementioned antibody can be immobilized for example by placing a buffer solution containing the aforementioned antibody onto the carrier and incubating. A publicly known buffer solution can be used, for example, 10 mM PBS. The concentration of the aforementioned antibody in the buffer solution can be selected from a wide range, but normally, about 0.01 to 100 μg/ml, preferably 0.1 to 20 mg/ml is appropriate. Furthermore, when using a 96-well microtiter plate as a carrier, one would use 300 μl/well or less, with about 20 to 150 μl/well being preferable. Moreover, while there are no particular limitations as to the incubation conditions, normally, overnight incubation at about 4°C is appropriate.
Furthermore, since areas may exist where the amyloid β in the subsequently added specimen will be adsorbed independently of the antigen-antibody reaction in carriers that immobilize the antibody in Process (A), the blocking of Process (B) is carried out with the aim of preventing this. As a blocking agent, one can for instance use BSA or skim milk solution, or a commercial blocking agent such as Block-Ace (made by Dainippon Pharmaceutical (Code No. UK-25B)). While there are no limitations as to the specifics of the blocking, it can be performed for instance by adding a suitable quantity of Block- Ace to the areas where antigen was immobilized, incubating overnight at approximately 4°C, and then washing with buffer solution.
Moreover, in Process (C), specimen containing amyloid β is brought into contact with immobilized antibodies, capturing the amyloid β on the immobilized antibodies and producing a complex of immobilized antibodies and amyloid β. While there are no limitations as to the conditions of producing this complex, one can conduct the reaction for approximately 1 hour to overnight at about 40C to 370C. After completion of reaction, it is preferable to wash the carrier with buffer solution and remove unreacted proteins and the like. The buffer solution used for this reaction preferably has a composition of 10 mM PBS (pH 7.2) and 0.05% (v/v) Tween 20.
Moreover, in Process (D), a labeled antibody which recognizes a different epitope of amyloid β captured by the immobilized antibody is added to form an immobilized antibody-amyloid β-labeled antibody complex. After completion of this reaction, it is
preferable to wash the carrier with buffer solution and remove unreacted proteins and the like. The buffer solution described above can be used for this reaction. The quantity of labeled antibody used in Process (D) is approximately 5,000 to 10,000 times the quantity of immobilized antibody, and is preferably a quantity diluted such that the final absorbance will be 1.5 to 2.0. Buffer solution can be used for the dilution, and although there are no particular limitations as to the reaction conditions, it is preferable to conduct the reaction for approximately 1 hour at about 40C to 370C and wash with buffer solution after the reaction. By means of the above reaction, an immobilized antibody-amyloid β-labeled antibody complex can be formed. Finally, in Process (E), a chromogenic substrate solution which reacts with the labeling substance of the immobilized antibody-amyloid β-labeled antibody complex is added and absorbance is measured, whereby the quantity of amyloid β can be computed based on a calibration curve.
When using the enzyme peroxidase as the labeling substance of the labeled antibody, one can for instance use a chromogenic substrate solution containing hydrogen peroxide and 3,3',5,5'-tetramethyl benzene (hereinafter referred to as "TMB"). Although not being limited hereto, the chromogenic reaction can be conducted by adding the chromogenic substrate solution, reacting for approximately 30 minutes at approximately 25°C, and then adding 1 to 2N sulfuric acid to stop the enzymatic reaction. When using TMB, color development can be measured based on absorbance at 450 nm. Furthermore, when using the enzyme alkaline phosphatase as the labeling substance, an appropriate method would be to induce color development with p-nitrophenyl phosphoric acid as the substrate, add 2N NaOH to stop the enzymatic reaction and measure absorbance at 415 nm. The concentration of amyloid β in a specimen can be computed using a calibration curve prepared in advance based on the absorbance of reaction liquids to which a known concentration of amyloid β has been added.
An amyloid β assay kit characterized in that it includes a first reagent containing the N-terminal antibody (or middle region antibody) of the present invention and a second reagent containing an antibody which recognizes amyloid β (1-40) or amyloid β (1-42) (hereinafter referred to as "the kit of the present invention") is favorably used to practice the method of measuring amyloid β described above.
The kit of the present invention can be fabricated by conventional methods. Specifically, the N-terminal antibody (or middle region antibody) of the present invention
and a labeled antibody which recognizes either amyloid β (1-40) or amyloid β (1-42) can be used as standard antibodies, and combined furthermore with a dilution buffer solution, standard substance, substrate buffer solution, stop solution, wash solution and the like.
The measurement method and assay kit obtained in this manner make it possible to accurately measure amyloid β (1-40) or amyloid β (1-42) completely retaining its full length in a specimen such as blood plasma or blood serum.
Example 1 Aβ N-terminal-end specific antibody reduced β-amyloid in Alzheimer-model mice
Alzheimer's disease (AD) is a neurodegenerative disease with memory dysfunction that is causing serious medical problems in modern society. For the fundamental treatment of AD, an amyloid β protein (Aβ) vaccine is considered to be the most potent candidate. To cure AD, an Aβ N-terminal-end specific monoclonal antibody named 82El was developed, which does not cross-react with full-length Aβ precursor. Passive intraperitoneal administration of 82El markedly reduced total plaque area (Aβ burden) in the Tg2576 mouse brains. This was confirmed by the ELISA measurement of insoluble Aβ in the brain homogenates. The density of diffuse plaques, which increases in the late stage, was markedly reduced by the administration of 82El, suggesting that the reduction of the Aβ burden was due to the prevention of newly developed diffuse plaques. Materials and methods AU animal protocols were in accordance with the guidelines of the Animal Use
Ethics Committee of the Saitama Medical Center/School and NIH guidelines [DHHS publication No. (NIH) 85-23, revised 1985].
Purified Aβ N-terminal-end specific monoclonal antibody 82El (1 mg/ml in PBS) was administered once a week by intraperitoneal injection at a dose of 10 mg/kg to 16- month-old Tg2576 mice (82El group, n = 3), while PBS was administered for the control group of Tg2576 mice (control group, n = 2) (Bard, F. et al. 2000 Nat Med 6:916-919). After 12 times administrations for over 3 months, blood sample was collected transcardially for the ELISA measurement of soluble Aβ in the plasma level under the deep anesthesia by sodium pentobarbital (25 mg/kg), and mice were euthanized by transcardial perfusion of 100 ml of 10 U/ml heparin in saline. The left cerebral hemisphere was fixed in 4% buffered formaldehyde solution and embedded in paraffin. Serial sections (5-μm in thickness) of the left cerebral hemisphere at 10 predetermined coronal planes separated by 1-mm intervals were sequentially cut and immunostained with Aβ40 and Aβ42 end specific
polyclonal antibodies (2 μg/ml; IBL., Japan) after brief formic acid pretreatment using an ABC Elite kit (Vector, USA). Additionally, to examine the distribution of 82El into the brain parenchyma, one mouse in the 82El group was intraperitoneally administered by the biotinylated 82El 1 day before sacrifice. The distribution of biotinylated 82El was visualized using an avidin-horseradish peroxidase in 4% buffered formaldehyde-fixed vibratome sections. Microglia in paraffin sections were visualized using Iba-1 antibody (2 μg/ml; Wako, Japan).
Images of 10 selected sections at fixed intervals from the cortex and five from hippocampus of each mouse were acquired using an Olympus BX60 microscope with an attached digital camera system (DP-50, Olympus, Japan), and the digital image was routed into a Windows PC for quantitative analysis using SimplePCI software (Compix, Imaging Systems, USA). Aβ40 and Aβ42 burdens were presented as the percentage of immunolabeled area captured (positive pixels) divided by the full area captured (total pixels). TB S -insoluble Aβ was extracted using 6 M-guanidine-HCl (Morishima-
Kawashima, M. 2000 Am J Pathol 157:2093-2099) from the right frontal quarter of brains and then used for the ELISA measurement together with plasma. Aβ 1 -40 and Aβ 1 -42 were measured by combinations of 82El with Aβ40 or Aβ42 C-terminal end-specific antibodies IAlO and 1C3, respectively, as described in Example 2. Aβx-40 and Aβx-42 were also measured by combinations of 12B2, Aβ middle portion antibody, with IAlO or 1C3 as described in Example 2. Results and discussion
The effect of 82El administration was observed. Immunohistochemistry for Aβ42 showed a smaller number of plaques in the 82El group than in the control group (Figs. 3 A and B). In the basal ganglia, where Aβ deposit occurred later than that in the cortex and hippocampus, senile plaques were markedly less in the 82El group than in the control group (Fig. 3B). This reduction was confirmed by image analysis (Fig. 3C). The Aβ burden was significantly reduced in the 82El group compared with that in the control group for both Aβ40 (30% reduction, p < 0.05, in cortex; 40% reduction, p < 0.05, in hippocampus) and Aβ42 (43% reduction, p < 0.01 in cortex; 40% reduction, p < 0.05, in hippocampus) (Fig. 3C).
Aβ ELISA analysis of TBS-insoluble fraction was consistent with the findings on immunohistochemical studies. Significant reduction of Aβ42 was found in the 82El group. The 82El group showed about 50% less Aβl-42 and Aβx-42 than the control group (Fig. 3D). Aβ40 tended to reduce in the 82El group compared to that in the control group, although its reduction did not reach a significant level (data not shown). The Aβ level in plasma was not significantly different between the 82El and control groups (data not shown). Fig. 3D was measured by ELISA. The unit of measurement in Fig. 3D is μg/g brain tissue. A measurement of 20 μg/ml is equivalent to approximately 500 fmol/ml.
Example 2 Development of Aβ terminal end-specific antibodies and sensitive ELISA for Aβ variant
Alzheimer's disease (AD) is a neurodegenerative affliction associated with memory dysfunction. Senile plaques are a pathological hallmark of AD, and amyloid β (Aβ) peptides are a major component of these plaques. Aβ peptides are derived from proteolytic cleavage of the Aβ protein precursor (APP) by β- and γ-secretases to generate two principal species, Aβl-40 and Aβl-42. Antibodies against the N- and C-termini of these peptides and an ELISA for accurate and sensitive quantitative assessment were developed. Sandwich ELISA composed of N-terminus (Aβl) end-specific antibody, clone 82El, and C- termini end-specific antibodies, and clones IAlO and 1C3 for Aβ40 and Aβ42, respectively, detects full-length Aβl -40 and 1-42 with a sensitivity in the sub single digit fmol/ml (equivalent to single digit pg/ml) range with no cross-reactivity to APP. A combination of C-termini antibodies and an antibody against the middle region of Aβ detects mouse Aβ in non-transgenic mouse brains. Materials and methods Mice (BALB/c, Charles River, Japan) were immunized weekly with thyroglobulin conjugated Aβ peptides (50 μg/mouse). Partial human Aβ peptide consisting of amino acid residues of 1-16 (DAEFRHD S GYEVHHQK) (SEQ ID NO: 63), 11-28 (EVHHQKLVFFAEDVGSNK) (SEQ ID NO: 64), 35-40 (MVGGW) (SEQ ID NO: 65), and 38-42 (GWIA) (SEQ ID NO: 66) was used as immunogens for Aβ N-terminus, middle region, and C-terminus-specific antibodies, respectively. The cysteine residue was combined in each immunogen beforehand for binding to the carrier protein, bovine thyroglobulin. After four to six immunizations, the spleen was isolated and fused with
X63 Ag8 myeloma cells. Epitopes and cross-reactivity to human and rodent Aβ of selected clones were determined by a microplate assay using various Aβ fragments (American Peptides, Sunnyvale, CA). A 96-well plate (Maxisorp, Nunc, Denmark) was coated with various Aβ fragments (human Aβ sequence unless otherwise indicated as "r"), such as -11 to -1, -4-8, -3-5, 1-5, 1-7, rl-9, 1-11, rl-15, 1-17, 1-40, rl-40, 1-42, rl-42, 2-40, 3-7, 3-13, 3- 40, 7-17, 10-20, 11-28, 13-19, 17-28, 20-29, 35-40, and 38-42 (100 ng/well) and blocked with Block Ace (Serotec) overnight at 40C. The plates were incubated with selected antibodies for 4 h, then with an HRP-coupled anti-mouse IgG antibody (Southern Biotechnology, Birmingham, AL), and visualized by TMB substrate (Pierce, Rockford, IL). For Western blotting analysis, various Aβ containing samples were prepared, such as fibril-free soluble and mixture of soluble and fibril Aβ from synthetic peptides, culture medium from APP overexpressing HEK cells, and brain homogenate from APP overexpressing transgenic mice. Lyophilized Aβ (American Peptide) was sequentially dissolved in trifluoroacetic acid and l,l,l,3,3,3-hexafiuoro-2-propanol followed by complete elimination by exposure to nitrogen gas. Aβ peptide was then re-suspended in dimethyl sulfoxide and used as fibril-free Aβ. Aβ was aggregated at room temperature for 10 days with mixing at 500rpm and used as fibril-containing Aβ. To assess the N-terminus specificity, HEK293 cells expressing Swedish mutant APP were cultured with or without a gamma-secretase inhibitor, N-[N-(3,5-difluorophenacetyl-l -alanyl)]-S-phenylglycine t-butylester (DAPT) (Dovey, H.F. et al. 2001 J Neurochem 76:173-181), for 24h and cell lysates were analyzed on a 10-20% NuPAGE gel (Invitrogen, Carlsbad, CA). Also, overexpressing APP transgenic mice (Hsiao, K. et al. 1996 Science 274:99-102) known as Tg2576 were analyzed. At 18 months of age, the brains were homogenized in PBS containing 2% SDS. All samples, except cell lysate, were run on a 16.5% Tris-Tricine gel (Bio-Rad, Hercules, CA) and transferred to a PVDF membrane. After blocking with 5% non-fat dried milk, the membrane was probed with anti-Aβ antibody followed by HRP- coupled anti-mouse IgG (Southern Biotechnology, Birmingham, AL). For a comparison, N-terminus region antibody, 6E10 (Signet, Dedham, MA) was used. Bands were visualized with a kit (Super Signal West Pico, Pierce). For immunohistochemistry, 4 μm-thickness paraffin sections of the temporal cortex from human autopsied AD brain, were incubated with 82El, 12B2, IAlO, and 1C3 (2, 1, 2, and 2 μg IgG/ml, respectively) overnight after brief pretreatment with formic acid. Immunoreactivity was visualized using an ABC elite
kit (ABC elite, Vector Laboratories, Burlingame, CA). Sections were briefly counterstained with hematoxylin.
For sandwich ELISA, 96-well plate (Maxisorp, Nunc, Denmark) was coated with 100 μl of either Aβ40 or Aβ42-specific antibody (5.0 μg IgG/ml each), clones IAlO and 1C3, respectively, overnight at 40C in 100 mM carbonate buffer, pH 9.6, containing 0.05% sodium azide. After blocking with 1 % Block Ace in PBS overnight at 40C, standards (human or mouse synthetic Aβ peptides 1-40 and 1-42) and samples were loaded and incubated overnight at 40C. Human plasma specimens were obtained from the Mount Sinai Alzheimer's Disease Research Center. Mouse brain Aβ was extracted in 0.4% diacetylamine containing buffer, centrifuged, and the resultant supernatant was used. HRP- coupled detection antibody, either 82El or 12B2, was incubated for 4 h at room temperature and visualized using a TMB substrate. Results and discussion
We selected clones that showed high and selective affinity to the Aβ peptide used for immunization. Four potential clones, 82El, 12B2, IAlO, and 1C3, which react with different regions of Aβ peptide were identified. The specific epitope for each of these antibodies was determined by plate assays using various Aβ fragments. Human and rodent cross-reactivity was examined using human and rodent Aβl-40 and 1-42. Clone 82El showed higher selectivity for human Aβ (approximately 10% cross-reactivity to rodent Aβ), probably because the human-specific 5th amino acid is a part of the epitope. Clones 12B2, IAlO, and 1C3 reacted with both human and rodent Aβ to a similar degree because their epitopes do not include a human-specific residue. Table 2 summarizes epitopes, specificity, cross-reactivity to human and rodent Aβ, and applications of these antibodies. Clone 82El reacted with both soluble and fibrillar Aβ to a similar degree (Fig. 4A). Clone 82El detected Aβ in the SDS extract from APP-expressing transgenic mice (Tg2576), but did not detect non-cleaved APP (Fig. 4B). N-terminus specificity was further confirmed using HEK cells expressing Swedish mutant APP. 82El did not react with full-length APP (Fig. 4C, lane C), although a control antibody, 6E10 (epitope within 3-10), detected full- length APP (Fig. 4C, lane C). β-secretase (BACE) generates β-cleaved C-terminus fragment (βCTF) from the full-length APP, but βCTF was not detectable in this transfected cell probably due to high γ-secretase activity. After treatment with a γ-secretase inhibitor, DAPT, for 24h, βCTF is strongly detectable with 82El (Fig. 4C, lane D). 82El stained
senile plaques in AD patients with brief formic acid treatment (Figs. 4D and 6C) Thus, 82El specifically detected fragments generated by β-secretase cleavage.
Clone 12B2 was raised against the middle region of Aβ peptide using Aβl 1-28 as an imrnunogen. This antibody is cross-reactive to human and rodent Aβ in a plate assay, and a sandwich ELISA made with a combination of C-terminus-specific antibodies, IAlO and 1C3, detects mouse endogenous brain and cerebrospinal fluid (CSF) Aβ (see below).
C-terminus-specific antibodies, clones IAlO and 1C3 (Aβ40 and Aβ42 specific, respectively), showed virtually no cross-reactivity to each other (<1.8 and <0.1%, respectively). The Aβ40-specific antibody, IAlO, worked well in immunoblotting (Figs. 5 A and B), immunohistochemistry (Figs. 5C and 6B), immunoprecipitation, and ELISA, although Aβ42-specific antibody, 1C3, worked well only in immunohistochemistry (Fig. 6D) and ELISA.
A sandwich ELISA was developed by a combination of these antibodies. C- terminus antibodies were selected, either Aβ40 (IAlO) or 42 specific (1C3), as the capturing antibody, and another antibody, either 82El or 12B2, as a detection antibody (reporter). Other combinations were not considered (82E1/12B2 for capturing and 1A10/1C3 for detection) because Aβ40 is much more abundant than Aβ42 in biological specimens and Aβ40 might monopolize the capturing antibody and reduce the sensitivity of Aβ42 detection. A combination of 1A10/82E1 and 1C3/82E1 gave a linear standard curve in the range of 1-1000 pg/ml (typical correlation factor >0.98) and detected human Aβ as low as 1.5 pg/ml (equivalent to sub fmol/ml range). Human Aβ in the plasma of AD patients and non-AD controls was measured (Table 3). Previous publications showed that human plasma has a wide range of Aβ, and there is no significant difference between levels in AD and non-AD controls (Kuo, Y.M. et al. 1999 Biochem Biophys Res Commun 257:787-791; Mehta, P.D. 2001 Neurosci Lett 304:102-106; Olsson, A. et al. 2003 Dement Geriatr Cogn Disord 16:93-97; Farrer, L.A. et al. 1997 JAMA 278:1349-1356; Naslund, J. et al. 2000 JAMA 283:1571-1577). The sandwich ELISAs using 1A10/82E1 and 1C3/82E1 similarly revealed no significant difference between AD and non-AD control cases (p = 0.442).
The epitope of clone 12B2 is 17-28 which is an identical sequence in human and rodent Aβ, and 12B2 reacted to human and rodent Aβ to a similar degree. This antibody was used as a reporter antibody in an ELISA developed to detect endogenous mouse brain Aβx-40 and x-42. A combination of 1A10/12B2 and 1C3/12B2 gave a linear standard curve
in the range of 1-500 pg/ml (typical correlation factor >0.96). Endogenous brain and CSF Aβ was measured in wildtype (non-transgenic) mice, finding 25.2 ± 4.8 fmol Aβ40/ml (means ± SE, n = 4) and 817 ± 73 fmol Aβ40/ml (means ± SE, n = 2), respectively.
In conclusion, a series of anti-Aβ antibodies were generated, including those that are specific both for the N-terminus β-cleavage site (clone 82El) and for C-terminus γ- cleavage sites (clones IAlO and 1C3). Combinations of these antibodies were used in
ELISA to quantify full-length Aβl-40 and 1-42, and rodent endogenous Aβx-40 and x-42.
The question is whether these assays can detect changes in Aβ level in response to compounds, allowing for very sensitive and accurate evaluation of anti-Aβ therapeutic approaches.
Table 2. Antibodies and applications
Applications
Clone Selectivity, human/rodent cross-reactivity WB IHC ELISA IP
82El (IgGl) N-terminus end specific (Aβ 1-5) +++ +++ +++ +++
No cross-reactivity to non-cleaved APP fragments Preference to human Aβ (~ 10% cross-reactivity to mouse Aβ)
12B2 (IgGl) Middle region-specific (Aβ 17-28) +a +++ +++ +b
Cross-reactive human and rodent Aβ at similar degree
IAlO (IgGl) C-terminus end specific (Aβ40) +++ +++ +++ -m-
Cross-reactive human and rodent Ab at similar degree <1.8% cross-reactivity to Aβ42
1C3 (IgGl) C-terminus end specific (Aβ42) - +++ +++ +b
Cross-reactive human and rodent Ab at similar degree <0.1% cross-reactivity to Aβ40
The utility of the antibodies for various applications is indicated as follows: +++, very useful; +, useful with limitations; and -, not useful under the testing conditions described. a12B2 did not work in tissue homogenates, but worked with synthetic Ab peptides (>50 ng Ab). b12B2 and 1C3 worked in IP but not as well as 82El and IAlO. WB, Western blotting; IHC, immunohistochemistry; IP, immunoprecipitation.
Table 3. Levels of Aβ40 in human plasma
Case No. Gender Age (year) NMSE ApoEl/E2 Aβ40 (finol/ml)
(A) Non-AD controls
052 M 71.8 29 3,3 52
2065 F 69.2 30 3,4 4
9415 F 74.4 29 3,3 798
4019 M 67.4 30 3,3 n.d.
5487 M 70.5 28 3,4 33
0505 M 74.5 30 3,3 224
3138 M 70.9 30 3,3 182 71.2 ±2.4 29 ±1 184 ±264
B)AD
8640 M 73.7 27 3,3 27
7564 F 84.3 8 3,4 60
6270 F 85.7 8 3,3 n.d.
9029 M 74.4 27 4,4 120
5207 F 76.3 8 4,4 6
2502 F 75.5 24 3,3 19
6625 M 83.2 9 3,4 475
4379 F 78.8 9 3,3 66
6003 F 79.3 13 3,4 33 79.0 ±4.2 15±8 89 ±141
Levels of Aβ in the human plasma were compared between non-AD controls (A) and AD patients (B). AD patients met NINDS- ADRDA criteria (McKhann, G. et al.1984 Neurology 34:939-944) for probable AD, and had lower MMSE scores (Folstein, M.F. et al.1975 J Psychiatr Res 12:189-198) than controls, p = 0.001. No significant difference in Aβ levels between AD patients and controls was detected (p = 0.442).
Example 3 Acquisition of N-terminal peptide and C-terminal peptide of amyloid/
N-terminal peptide and C-terminal peptide of amyloid β were purchased as HPLC purified products from Synpep Corporation and TANA Laboratories, USA. The amino acid sequences of these peptides are as indicated below. The first peptide is a peptide made by attaching cysteine (C) to the sequence of amino acids 1 through 16 of amyloid β. The next peptide is a peptide made by attaching KC to the sequence of amino acids 35 through 40 of amyloid,/? (1-40) and the last peptide is a peptide made by attaching lysine and cysteine (KC) to the sequence of amino acids 38 through 42 of amyloid β (1-42). DAEFRHDSGYEVHHQKC (SEQ ID NO: 67)
KCMVGGW (SEQ ID NO: 68) KCGWIA (SEQ ID NO: 69)
Example 4
Preparation of immunization antigen Conjugates of each of the above peptides and thyroglobulin were prepared by the
EMCS (N-(6-maleimidocaproyloxy)-succinimide) method as follows. For making the conjugate, the molar ratio of thyroglobulin, peptide, and EMCS was set at 1 :300:400.
First, 4 mg of each peptide of Example 3 was dissolved in approximately 1 ml of distilled water. Furthermore, 5 mg of thyroglobulin dissolved in 1 ml of 0.01 M phosphate buffer (pH 7.0) and 80 μg/μl EMCS dissolved in dimethyl formamide were mixed at the aforementioned molar equivalent to prepare a thyroglobulin-EMCS complex solution. This complex solution was divided into four portions and a peptide solution was added to each portion at the aforementioned molar equivalent to prepare a conjugate solution of peptide and thyroglobulin cross-linked with EMCS. This conjugate solution was dialyzed using PBS, and the concentration was adjusted to 10 μg/μl in terms of conjugate. The conjugates of each aforementioned peptide and thyroglobulin obtained in this manner were used as immunization antigens in the following examples.
Example 5 Preparation of antibody, which recognizes the C-terminal peptide of amyloid/ (1-40)
Mice were immunized using the conjugate of the peptide having amino acids 35-40 of Aβ and thyroglobulin obtained in Example 4 as an immunization antigen, by administering 50 μ\ (50 μg) of a conjugate solution at one week or two week intervals. The
antigen was mixed with Freund's complete adjuvant only for the initial immunization, and with Freund's incomplete adjuvant for the second and subsequent immunizations. Splenic monocytes of the immunized mice and fusion partner X63-Ag8-653 were subjected to polyethylene glycol mediated cell fusion, and hybridomas were selected by a method described in the literature {J Immunol 146:3721-3728). The selection was performed by selecting cells, which appeared to react to fixed peptide having amino acids 35-40 of Aβ and to not react to the peptide having amino acids 38-42 of Aβ.
Cells selected as above were made to produce antibodies in a GIT medium (made by Wako Pure Chemical Industries Ltd.), which is a serum free medium, until 80% of the cells had died. Subsequently, cells were removed from this medium by centrifuging (1,000 rpm, 15 min), and then ammonium sulfate was added to a 50% saturated state, leaving overnight at 40C, and the sediment was collected by centrifuging (1,000 rpm, 30 min). Furthermore, this sediment was dissolved in 2-fold diluted binding buffer (Protein A MAPS II kit), after which IgG was adsorbed to a Protein A column (made by Pharmacia- Amersham). Subsequently, PBS dialysis was conduced overnight to purify the antibodies, and antibodies, which recognize the C-terminal peptide of amyloid ./? (1-40) were obtained. These antibodies were named IAlO.
Example 6 Verification of specificity of IAlO by Western blotting Next, to verify that the IAlO antibodies obtained in Example 5 recognize the C- terminal peptide of amyloid β (1-40), Western blotting was performed by conventional methods (e.g. "Fundamental Experimental Methods of Molecular Biology," Nankodo) on the peptides amyloid β (1-40), amyloid β (1-42) and amyloid β (1-43) using IAlO antibodies. It was verified that monoclonal antibodies IAlO did not react to amyloid,/? (1- 42) or amyloid^ (1-43), and recognized only amyloid^ (1-40).
Example 7 Preparation of antibody, which recognizes the C-terminal peptide of amyloid β (1-42)
Mice were immunized by the same technique as in Example 5, using the conjugate of thyroglobulin and the peptide having amino acids 38-42 of Aβ obtained in Example 4 as an immunization antigen. Splenic monocytes of the immunized mice and fusion partner X63-Ag8-653 were subjected to polyethylene glycol mediated cell fusion, and hybridomas were selected by a method described in the literature (J. Immunol. 146: 3721—3728). The
selection was performed by selecting cells, which appeared to react to fixed peptide having amino acids 38-42 of Aβ and to not react to the peptide having amino acids 35-40 of Aβ.
The cells selected as described above were purified in the same manner as in Example 5 to obtain antibodies, which recognize the C-terminal peptide of amyloid/ (1- 42). These antibodies were named 1C3.
Example 8 Verification of reactivity of 1C3 antibodies by immunoprecipitation
Next, to verify that the 1C3 antibodies obtained in Example 7 recognize the C- terminal peptide of amyloid β (1-42), immunoprecipitation was performed by conventional methods (e.g. "Fundamental Experimental Methods of Molecular Biology," Nankodo) on the peptides amyloid/ (1-40), amyloid,/? (1-41) and amyloid/ (1-42) using 1C3 antibodies.
6E10 antibodies (made by Signet Laboratories, Inc.) were used for the final blot detection.
It was verified that 1C3 antibodies specifically recognize amyloid/ (1-42) and do not recognize amyloid/ (1-40), amyloid/ (1-41) or amyloid/ (1-43). Example 9
Preparation of antibody, which recognizes the N-terminal peptide of amyloid/
BALB/c mice were immunized using the conjugate of thyroglobulin and the peptide having amino acids 1-16 of Aβ obtained in Example 4 as an immunization antigen. After four immunizations, another two immunizations were performed using a conjugate of a peptide having amino acids 1-5 of Aβ and thyroglobulin. Splenic monocytes of the immunized mice and fusion partner X63-Ag8-653 were subjected to polyethylene glycol mediated cell fusion, and hybridomas were selected by a method described in the literature
(J. Immunol. 146: 3721-3728). The selection was performed by selecting cells which reacted to fixed peptide having amino acids 1-16 of Aβ and a peptide having amino acids I- 5 of Aβ.
The cells selected as described above were purified in the same manner as in Example 5 to obtain antibodies, which recognize the N-terminal peptide of amyloid /. These antibodies were named 82El.
Example 10 Verification of specificity of 82El antibodies by Western blotting
Next, to verify that the 82El antibodies obtained in Example 9 recognize the N- terminal peptide of amyloid /, Western blotting was performed by conventional methods (e.g. "Fundamental Experimental Methods of Molecular Biology," Nankodo) on the
peptides amyloid,/? (1-40), amyloid,/? (2-40) and amyloid./? (3-40) using 82El antibodies. Furthermore, for comparison, Chinese hamster cells forced to express amyloid./? precursor protein (APP) were homogenized in a buffer solution containing 1% Triton and centrifuged, after which the supernatant was used as a sample and subjected to Western blotting in the same manner. In addition to APP, this supernatant contained / C-terminal fragments (/SCTF) cleaved at the / site form APP and amyloid β. Moreover, for comparison to a conventional antibody, Western blotting was also performed on 6E10 antibodies (made by Signet Laboratories, Inc.), which are said to recognize an N-terminal peptide of amyloid/, using the same samples as above. It was verified that 82El antibodies do not react with amyloid/ (2-40) or amyloid/
(3-40) and only recognize amyloid / (1-40). Furthermore, it was verified that 6E10 antibodies do not react to amyloid/ (1-40) and recognize amyloid/ (2-40) and amyloid/ (3-40). Furthermore, it was verified that the monoclonal antibodies 82El do not react to APP and recognize only amyloid / and /CTF. Moreover, it was verified that 6E10 antibodies recognize APP.
Example 11
Preparation of conjugate of HRP and antibody, which recognizes the N-terminal peptide of amyloid/ (82El antibody)
A conjugate of HRP and the 82El antibody obtained in Example 9 was prepared as follows. A necessary quantity of HRP was dissolved in distilled water and oxidized with NaIO4, and was then dialyzed overnight in pH 4.4 1 mM acetate buffer solution. Furthermore, 2 mg of 82El antibodies were also dialyzed overnight in pH 9.5 0.1 M carbonate buffer solution. The dialyzed 82El antibodies and HRP were mixed at 0.4 mg HRP per 1 mg of antibodies and reacted for two hours at room temperature. Subsequently, NaBH4 was added thereto, reacting for two hours in ice, followed by overnight dialysis in PBS. Moreover, this reaction substance was gel-filtered to prepare a conjugate of 82El antibodies and HRP.
Example 12 Creation of sandwich ELISA technique Creation of a sandwich ELISA technique using the antibodies obtained in the above examples was conducted as follows. First, 20 μg/wl IAlO antibodies or 1C3 antibodies were added at 100 μ\ each to a 96-well ELISA plate. This was then reacted overnight at 4°C, after which blocking was performed with a 1% BSA/PBS/NaN3 solution to make a
sandwich ELISA plate. Furthermore, a conjugate of 82El antibody prepared in Example 8 and HRP was used as the labeled antibody.
The standard curve results obtained from measurements using the peptides synthetic amyloid / (1-40) and synthetic amyloid ,/? (1-42), using the aforementioned ELISA plate and standard antibodies were obtained. A measurement of synthetic amyloid / (1-40) with a combination of IAlO antibodies and HRP labeled 82El antibodies was generated. A measurement of synthetic amyloid / (1-42) with a combination of 1C3 antibodies and HRP labeled 82El antibodies was produced. Both measurements showed good concentration- dependent linearity. Example 13
Amyloid/ accumulation inhibition test
16-month old Tg2576 recombinant mice (n = 3) expressing amyloid precursor protein (APP) were intraperitoneally administered 82El antibodies obtained in Example 9 which had been adjusted to 1 mg/ml using PBS, once per week at the rate of 10 mg/kg (mouse body weight). Furthermore, PBS alone was administered in the same manner for the control (n = 2). Mice which had been given 12 doses (over approximately three months) of 82El antibodies or PBS were sacrificed, and the left cerebral hemisphere was fixed in 4% buffered formaldehyde solution and then embedded in paraffin. Subsequently, 5 μm continuous slices were prepared from the paraffin-embedded specimen, and were immune stained using amyloid/ (1-40) and amyloid/ (1-42) polyclonal antibodies (both made by Immuno-Biological Laboratories Co., Ltd.: product numbers 18580 and 18582).
Furthermore, 10 slices were selected from the cortex and 5 from the hippocampus and subjected to image analysis using a microscope digital camera and Simple PCI software (Compix, Inc. Imaging Systems, USA). Accumulation of amyloid/ (1-40) and amyloid/ (1-42) was expressed as the proportion (%) of immune stain positive area in the total area.
Furthermore, pH 7.6, 0.05 M tris buffered saline (TBS) insoluble amyloid/ was extracted with 6 M guanidine hydrochloride from the right frontal part of the head (1/4 of the brain), and amyloid/ (x-42) was measured by the sandwich ELISA system prepared in Example 12 using an amyloid/ (1-42) and amyloid/ (1-42) assay kit (made by Imrnuno- Biological Laboratories Co., Ltd.; product number 17711). The method of extraction followed a method described in the literature (M. Morishima-Kawashima, et al., Am. J. Pathol., 157 (2000) 2093-2099).
Based on the immune stain image of amyloid,/? (1-42), the number of senile plaques was lower in the brains of mice given 82El as compared to control. Furthermore, in mice given 82El, the number of senile plaques was clearly lower as compared to control in the basal nucleus, where the accumulation of amyloid^ is lower as compared to the cortex and hippocampus. This reduction was also confirmed by the image analyzer. Accumulation of amyloid^ was significantly reduced in mice given 82El both in the case of amyloid^ (1- 40) (30% reduction in the cortex, 40% reduction in the hippocampus (for both, p < 0.05)) and amyloid β (1-42) (43% reduction in the cortex (p < 0.01), 40% reduction in the hippocampus (p < 0.05)). Moreover, the results of measuring amyloid β (1-42) and amyloid β (x-42) in the brain using ELISA showed a significant decrease (a 50% decrease in both cases (for both, p < 0.05)) in mice given 82El.
Example 14
Method to evaluate Aβ-lowering agents using wildtype mice ELISA development Maxisorp plate (Nunc) was coated with Aβ 40-specific capture antibody (clone
IAlO, IBL) overnight at 4°C. After blocking with blockace (Serotec) for 4 hours at room temperature, brain homogenates were loaded. For standard, synthetic rodent Aβl-40 peptide (American Peptide) was used. Samples were incubated overnight at 40C, and then detected by HRP-coupled anti-Aβ antibody (clone 12B2, IBL). Signal was visualized using TMB kit (Pierce) and read by a plate reader. This ELISA can detect mouse endogenous Aβ as low as single digit fmol Aβ/ml sample. ELISA Use
Conventional (non-transgenic) ICR mice (9 weeks of age, male, n=6) were treated with DAPT (37.5 mg/kg, ip), a gamma secretase inhibitor which reduced brain Aβ load in transgenic mice (Dovey H.F. et al. 2001 J Neurochem 76:173-181). For control, mice were treated with vehicle (5% ethanol in corn oil). Four hours after the injection, mice were killed and the brain was quickly isolated. The brain was homogenized in Tris-HCl buffer, pH 7.4, containing protease inhibitor cocktail. Aβ was extracted in 4% DEA followed by ultracentrifugation at 100,000 g for 45 min. Supernatants were used for Aβ quantification. Brain Aβ level in the mice receiving DAPT was significantly reduced approximately 10% (from 24 to 21.4 fmol Aβ/ml brain homogenate) compared to vehicle-received controls.
Discussion
Transgenic mice have been commonly used for evaluation of Aβ-lowering strategies; however, transgenic mice are not the optimal system to test Aβ-lowering strategies. Transgenic mice carry mutant genes, such as APP and presenilin; but genetic mutation in these genes is involved in less than 5% of AD patients. Majority of AD patients (over 95%) are free from genetic mutation. Therefore, more physiological testing system is desired.
Newly developed mouse ELISA is sensitive enough to measure endogenous brain Aβ levels. We treated non-transgenic mice with a gamma secretase inhibitor (Dovey H.F. et al. 2001 J Neurochem 76:173-181) and evaluated Aβ-lowering effect by this ELISA. Brain Aβ level was statistically significant reduced.
Overall, newly developed mouse ELISA can evaluate Aβ lowering strategies and therapeutic agents in non-transgenic mice. Genes and mutations used in transgenic mice are commonly covered by someone's patent; but this approach only uses non-transgenic mice which are free from patent protection.
Example 15
Mice (BALB/c, Charles River, Japan) were immunized weekly with thyroglobulin conjugated Aβ peptides (50 meg/mouse). We used partial Aβ peptide consisting of amino acid residues of 1-16 (DAEFRHDSGYEVHHQK) as an immunogen. The cysteine residue was combined in each immunogen beforehand for binding to the carrier protein, bovine thyroglobulin. After four to six immunizations, the spleen was isolated and fused with X63Ag8 myeloma cells. Epitope mapping using various Aβ peptide fragments identified that the epitope of clone 14Fl locates between l-9th amino acid residues of Aβ. 14Fl detects Aβ in the immunoblotting (see Figure 8). Example 16
For detection of Aβ reduction after treatment with Aβ-lowering agents in non- transgenic mice, HRP-coupled 14Fl was used by the ELISA methods described in Example 14 (see Figure 9). Brain Aβ level in the mice receiving DAPT was significantly reduced approximately 15% (from 24 to 20.2 fmol Aβ/ml brain homogenate) compared to vehicle- received controls.
Example 17
BACEl inhibition reduces endogenous Abeta and alters APP processing in wildtype mice
Accumulation of amyloid beta peptide (Abeta) in brain is a hallmark of Alzheimer's disease (AD). Inhibition of beta-site amyloid precursor protein (APP)-cleaving enzyme- 1 (BACEl), the enzyme that initiates Abeta production, and other Abeta-lowering strategies are commonly tested in transgenic mice overexpressing mutant APP. However, sporadic
AD cases, which represent the majority of AD patients, are free from mutation and do not necessarily have overproduction of APP. In addition, commonly used Swedish mutant APP alters APP cleavage. Therefore, testing Abeta-lowering strategies in transgenic mice may not be optimal. In this study, we investigated the impact of BACEl inhibition in non- transgenic mice with physiologically relevant APP expression. Existing Abeta ELISAs are either relatively insensitive to mouse Abeta or not specific to full-length Abeta. A newly developed ELISA detected significant reduction of full-length soluble Abeta 1-40 in mice with BACEl homozygous gene deletion or BACEl inhibitor treatment, while the level of x-40 Abeta was moderately reduced due to detection of non-full-length Abeta and compensatory activation of alpha-secretase. These results confirmed the feasibility of Abeta reduction through BACEl inhibition under physiological conditions. Studies using our new ELISA in nontransgenic mice provide more accurate evaluation of Abeta reducing strategies than was previously feasible. Introduction
Alzheimer's disease (AD) is a neurodegenerative affliction associated with cognitive dysfunction. Accumulation and deposition of amyloid beta (Abeta) peptides are the hallmarks of AD pathophysiology. Neurotoxic Abeta is generated by sequential proteolytic processing at the N and C termini of the Abeta domain by beta- and gamma- secretases, respectively (Hardy J. and Selkoe D. J. 2002 Science 297:353-356). Beta-site amyloid precursor protein (APP)-cleaving enzyme 1 (BACEl), the primary beta-secretase (Sinha S. et al. 1999 Nature 402:537-540; Vassar R. et al. 1999 Science 286:735-741; Yan R. et al. 1999 Nature 402:533-537) that initiates Abeta production, plays an essential and pivotal role in AD pathogenesis. The role of BACEl in Abeta production has been investigated. In primary cultured neurons prepared from homozygous BACEl knockout (BACEl-/-) mice, Abeta was not detectable (Cai H. et al. 2001 Nat Neurosci 4:233-234; Luo Y. et al. 2001 Nat Neurosci 4:231-232). Due to the limited sensitivity of mouse Abeta ELISAs, the brain Abeta levels
in BACEl knockout mice were unquantifiable. In order to assess the role of BACEl in
Abeta production, BACEl knockout mice have been crossed with transgenic mice that overexpress human mutant APP. As expected, mice obtained by crossing BACEl-/- mice and APP transgenic mice showed virtually no Abeta production (Luo Y. et al. 2003 Neurobiol Dis 14:81-88). Conversely, double transgenic mice obtained by crossing transgenic mice that overexpress BACEl and APP showed enhanced Abeta generation and exacerbated Abeta pathology (Willem M. et al. 2004 Am J Pathol 165:1621-1631). These results suggest that BACEl is a strong candidate for anti-AD pharmacological intervention.
Abeta-lowering strategies, such as pharmacologic BACEl inhibition, are commonly investigated in APP transgenic mice. However, APP expression is not necessarily increased in sporadic AD, which represents the majority of AD patients. Consequently, testing Abeta- lowering approaches in transgenic mice may not be optimal. Particularly in studies of secretase inhibitors, the amount of substrate (APP) and the efficiency of APP cleavage are important factors that may determine Abeta production. Commonly used APP transgenic mice carry Swedish mutation APP, which is cleaved by BACEl much more efficiently (>50 fold) than wildtype (Tomasselli A. G. et al. 2003 J Neurochem 84:1006-1017). These factors may compromise the outcome of therapeutic studies using APP transgenic mice. Thus, testing BACEl inhibitors in wildtype mice with physiologically relevant APP expression, may provide more meaningful results. However, commonly available ELISAs have a preference for human Abeta peptide, and do not have sufficient sensitivity to measure endogenous mouse Abeta. In addition, there are multiple cleavage sites in the Abeta domain, and various Abeta fragments are generated; thus measurement of full-length Abeta is essential for accurate evaluation of Abeta-lowering strategies. However, currently available ELISAs use N terminus region (but not end-specific) antibodies, and the results may reflect changes in various Abeta fragments. Our recently developed ELISA utilizing the N terminus end-specific antibody 82El measures only full-length Abeta, but cross- reactivity to mouse Abeta is limited (Horikoshi Y. et al. 2004 Biochem Biophys Res Commun 319:733-737).
In this study, we developed a full-length Abeta ELISA detecting endogenous Abeta in non-transgenic (wildtype) mouse brain. We used this tool to investigate how BACEl activity affects Abeta levels and APP processing using BACEl knockout mice and BACEl inhibitor treatment in mice with physiologically-relevant levels of non-mutant wildtype APP expression.
Development and characterization of mouse/human cross-reactive N terminus end- specific anti-Abeta antibody, clone 14Fl
We used a partial Abeta peptide consisting of amino acid residues of Abeta 1—16
(DAEFRHDSGYEVHHQK) (SEQ ID NO: 63) as an immunogen. This peptide was conjugated to the carrier protein, bovine thyroglobulin, through an additional cysteine residue at the C-terminus. Mice (BALB/c, Charles River, Yokohama, Japan) were immunized weekly with this antigen (50 μg/mouse).
After four to six immunizations, the spleen was isolated and fused with X63Ag8 myeloma cells. Epitopes and cross reactivity to mouse and human Abeta of selected clones were determined by a microplate assay using various human and mouse Abeta fragments including fragments starting with 2nd and 3rd amino acid residue of Abeta. A 96-well plate (Maxisorp, Nunc, Rochester, NY, USA) was coated with Abeta fragments and non-specific binding was blocked. The plates were incubated with selected antibodies for 30 minutes at 37 °C, then with an HRP-coupled anti-mouse IgG antibody (500 ng/ml, Immuno-Biological Laboratories (IBL), Takasaki, Japan), and visualized by OPD substrate (Sigma, St Louis, MO, USA).
For Western blotting analysis, we used synthetic Abeta peptides and brain homogenate from APP overexpressing transgenic mice, Tg2576 (Hsiao K. et al. 1996 Science 274:99-102) (Taconic, Hudson, NY, USA). All samples were run on a 15 % SDS PAGE gel and transferred to a PVDF membrane. After blocking with 3% non-fat dried milk, the membrane was probed with an anti-Abeta antibody, clones 14Fl, 82El and 6E10 (1.5, 1.0 and 1.0 μg IgG/ml, respectively), followed by HRP-coupled anti-mouse IgG.
For histochemical analysis, APP transgenic mice, Tg2576 (Hsiao K. et al. 1996 Science 274:99-102), were perfused with 10 mM phosphate-buffered saline, pH 7.4 (PBS) followed by fixative consisting of 4% paraformaldehyde. After dehydration with 20% sucrose in 0.1 M phosphate buffer, brains were cut into 4 μm-thick sections. Serial sections were incubated with the anti-Abeta antibody, clone 14Fl (2 μg IgG/ml) along with other N terminus end (specific to the beta cleavage site)- and N terminus region-specific antibodies, clones 82El (Horikoshi Y. et al. 2004 Biochem Biophys Res Commun 319:733-737) and 6E10 (1 and 2 μg IgG/ml), respectively, in 1 % BSA and 0.05% sodium azide in PBS, pH7.4, overnight at 4 °C. Then sections were with biotinylated anti-mouse IgG for 30 minutes, and visualized using a kit (Vectastain ABC kit, Vector Laboratories, Burlingame, CA, USA).
Development of Abeta ELISA detecting endogenous mouse Abeta in non-transgenic mice
A 96-well plate (Maxisorp) was coated with an anti- Abeta 40 antibody, clone IAlO (Horikoshi Y. et al. 2004 Biochem Biophys Res Commun 319:733-737), overnight at 4 0C. After blocking overnight at 4 0C, standards (synthetic mouse Abeta peptide 1-40) and samples were loaded and incubated overnight at 4 °C. The plate was incubated with HRP- coupled detection antibody, 14Fl, and visualized using a TMB substrate. To determine the specificity to full-length Abeta 1-40, we incubated Abeta fragments, 2-40 and 3-40, and developed the plate as described above. We also used human Abeta 1-40 and mouse Abeta 1 -40 to determine the mouse/human cross reactivity.
In vitro studies using primary cultured neurons from mice
Mouse primary neurons were prepared from the cerebral cortex and hippocampal formation of mouse embryos (ICR, Charles River) at embryonic days 17-18, and cultured in NeuroBasal medium supplemented with B-27 (Invitrogen, Carlsbad, CA, USA). Cells were studied on the 6th day after preparation. A gamma secretase inhibitor, N-[N-(3,5- difluorophenacetyl-L-alanyl)]-S-phenylglycine t-butyl ester (DAPT, Calbiochem, San Diego, CA, USA) (Dovey H. F. et al. 2001 J Neurochem 76:173-181) and a BACEl inhibitor, N-(I S,2R)- 1 -benzyl-3-(cyclopropylamino)-2-hydroxypropyl)-5-
(methyl(methylsulfonyl)amino-N'-((lR)-l-phenylethyl)isophthalamide (Inhibitor IV, Calbiochem) (Stachel S. J. et al. 2004 J Med Chem 47:6447-6450), were dissolved in DMSO and added to culture medium at a 1:100 dilution. For the control, we added DMSO to yield 1% concentration in the culture medium. We treated cells for 24 hours and determined Abeta levels in the culture medium by ELISA. After collecting the culture medium, cells were further incubated for 2 hours with a tetrazolium salt, 2-(2-methoxy-4- nitrophenyl)-3-(4-nitrophenyl)- 5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt, and the cell toxicity was determined using MTT (Mosmann 1983)-based cell assay kit (WST-8 kit, Kishida Chemical, Osaka, Japan). In vivo studies using BACEl knockout mice
The brain was isolated from BACEl knockout mice (Cai H. et al. 2001 Nat Neurosci 4:233-234) carrying wildtype, heterozygous and homozygous (BACE+/+, BACE+/- and BACE-/-, respectively) genotypes (3 female mice at 11 weeks of age in each genotype,). The brain was homogenized in 10-fold volume of 50 mM Tris-HCl buffer, pH 7.6, containing 250 mM sucrose and protease inhibitor cocktail (Sigma).
For Abeta ELISA, crude homogenate was mixed with 3-[(3- cholamidopropyl)dimethylammonio]-l-propanesulfonate (CHAPS, Sigma) to yield the final concentration of 1%, ultra centrifuged at 100,000 g for 45 minutes at 4 °C, and then the Abeta level was determined by ELISA. Mouse Abeta standards were prepared in 1% CHAP S -containing buffer because CHAPS slightly compromises the immobilized C- terminus capture antibody on the ELISA plate.
For positive controls, full-length APP, sAPPalpha, and sAPPbeta were prepared. In brief, the full-length cDNA of human APP was amplified from human brain cDNA (Clontech, Mountain View, CA, USA) using a forward primer, 5'- CGGTCGACTCGCGATGCTGCCCGGTTTGGC-S' (SEQ ID NO: 70) and a reverse primer, 5'-GGGCGGCCGCGTCTAGTTCTGCATCTGCTC-S' (SEQ ID NO: 71). The amplified products were digested with Sail and Notl, ligated into pGEX-6P-l vector (Amersham, Piscataway, NJ, USA) and transformed into E. coli JMl 09. After the sequence of cloned APP (APP695) was confirmed, sAPPalpha and sAPPbeta cDNA were amplified using APP695 cDNA as a template. We used forward primer, 5'- CGGTCGACTCGCGATGCTGCCCGGTTTGGC-3' (SEQ ID NO: 72) for both sAPPalpha and sAPPbeta, and specific reverse primer for sAPPalpha and sAPPbeta; i.e., 5'-GCGCGGCCGCCTATTTTTGATGATGAACTT-S' (SEQ ID NO: 73) for sAPPalpha and 5'-GCGCGGCCGCCTACATCTTCACTTCAGAGAT-S' (SEQ ID NO: 74) for sAPPbeta. The amplified products were digested with Sail and Notl, ligated into pGEX- 6P-1 vector and used to transform E. coli JMl 09 cells. We confirmed the sequence of cloned sAPPalpha and sAPPbeta. APP695, sAPPalpha and sAPPbeta cDNAs in pGEX-6P- 1 vector were transferred into pcDNA3.1(+) (Invitrogen), and transfected into COS-I cells by using FuGENEό (Roche Diagnostics, Basel, Switzerland). After 2 days, cells were harvested in 10 mM Tris, pH 8.0, consisting of 1% Nonident P-40, 150 mM NaCl andl mM EDTA5 and used as positive controls for immunoblotting.
For sAPPbeta and sAPPalpha detection, crude homogenates were mixed in diethylamine to yield 0.4% concentration, and centrifuged at 100,000 g for 45 minutes. The resultant supernatants were mixed with SDS sample buffer and run on a gel. Proteins were transferred to a PVDF membrane and the membrane was incubated with non-fat skim milk to minimize non-specific binding. The membranes were probed with a primary antibody overnight at 4 °C. We used a commercially avairable antibodies, which are mouse monoclonal anti-sAPPalpha antibody (clone 2B3, 5 μg IgG/ml, IBL, Gunma, Japan) and a
rabbit polyclonal anti-wildtype sAPPbeta antibody (2 μg IgG/ml, IBL). The membranes were then incubated with an HRP-coupled secondary antibody for 2 hours at room temperature, and protein bands were visualized using a chemiluminescence kit (Pierce, Rockford, IL, USA). Protein bands were densitometrically analyzed (Quantity One, BioRad, Hercules, CA, USA), and statistical significance was determined by t-test. In vivo studies using BACEl inhibitor
Non-transgenic wildtype male mice (ICR, Charles River) at 8 weeks of age (body weight 34.8 ± 1.5 g) were used (n=10 in each vehicle- and drug-treated group). A BACEl inhibitor, Inhibitor IV (Stachel S. J. et al. 2004 JMeJ Chem 47:6447-6450) (10 μg Inhibitor IV in 2 μl 40% DMSO/mouse) was stereotaxicially administered into the cerebral ventricle (Bregma -0.7 mm,1.8 mm lateral, 2.5 mm depth). For control, we injected 40% DMSO in saline (vehicle). After 4 hours, the brain was isolated and the samples were prepared as described above. Antibody characterization and development of a full-length Abeta ELISA sensitive enough to detect endogenous mouse Abeta in wildtype mtiuse brain
We screened numerous clones and selected 14Fl, which is highly sensitive to mouse Abeta. The plate assay using various Abeta fragments revealed that the epitope of clone 14Fl locates amino acid residues 1-4 of Abeta. Clone 14Fl detects both human and mouse Abeta synthetic peptides, but has a preference for mouse Abeta (Fig. lOAc, lanes H vs M). Since amino acid residues 1-4 of Abeta are identical in humans and mice, conformational differences between human and mouse Abeta presumably determine the mouse preference of 14Fl. Our N terminus end specific antibody, clone 82El (epitope: 1- 5), and a commercially available N terminus region antibody, clone 6E10 (epitope: 3-8), show a preference for human Abeta because the 5th amino acid residue, which is specific to humans, is within their epitopes (Fig. 10Aa and Ab, lanes H vs M). These antibodies detected Abeta in APP Tg2576 transgenic mouse brain homogenate (Fig. 1OA, lanes Tg). Because clone 14Fl has a preference for mouse Abeta, the 14Fl -detected Abeta band in transgenic mouse brain homogenate was weaker than the 82El - and 6E10-detected bands. An ELISA study using human and mouse Abeta 1-40 determined that 14Fl /IAlO ELISA has approximately 50% cross reactivity to human Abeta. Clone 6E10 detected uncleaved APP in addition to Abeta (Fig. 10Ab), while clones 82El and 14Fl did not (Fig. 10Aa and IAc). Clone 14Fl stained Abeta plaques in APP Tg2576 mouse brain sections in a manner
similar to other Abeta antibodies, clones 82El and 6E10 (Fig. 10B). We also found that clone 14Fl can be utilized for immunoprecipitation.
We used two Abeta ELISAs, 14F1/1A10 and 12B2/1A10, in this study (Fig 1OC and Table 4B). An ELISA composed of 12B2/1A10 detected human and mouse Abeta equally and detected endogenous Abeta in non-transgenic mouse brain homogenate. However, the epitope of 12B2 (epitope: within 17-28) is located between the alpha and gamma secretase cleavage sites. Thus the 12B2/1A10 ELISA measures the alpha and gamma secretase-cleaved fragment, P3, in addition to Abeta (Horikoshi Y. et al. 2004 Biochem Biophys Res Commun 319:733-737). The epitope of our newly developed clone 14Fl is located between amino acid residues 1-4 of Abeta, and the ELISA combining 14Fl with the Abeta 40-specific IAlO antibody measures full-length Abeta 1-40. To confirm the specificity to full-length Abeta 1-40, we examined cross reactivity to Abeta 2-40 and 3-40 using Abeta fragments, and determined this to be 0.78% and 0.08%, respectively. Our new 14F1/1A10 ELISA achieved sensitivity to single digit fmol/ml (equivalent to sub pg/ml) and detected endogenous full-length Abeta 1-40 in non-transgenic mouse brain homogenate (Fig. 12) and cell culture medium from primary cultured neurons (Fig. 11).
Table 4. Antibodies and ELISAs we used in this study.
A. Antibodies used in this study
Clone Subclass Specificity, human/rodent cross reactivity
14Fl 1} IgGl N terminus end region-specific (Epitope: Abeta 1-4)
Strong preference to mouse Abeta
82El 2) IgGl N terminus end-specific (Epitope: Abeta 1-5)
Strong preference to human Abeta
12B2 2) IgGl Middle region-specific (Epitope: Abeta 17-28)
Fully cross-reactive to human and rodent Abeta
IAlO 2) IgGl C-terminus end-specific (Epitope: Abeta 35-40)
Fully cross-reactive to human and rodent Abeta
B. ELISAs used in this study (See also Figure 10C)
Antibody combination Specificity
14F1/1 AlO Single digit fmol/ml sensitivity (equivalent to sub single digit pg/ml)
Specific to full-length Abeta 1-40
12B2/1 AlO Single digit fmol/ml sensitivity (equivalent to sub single digit pg/ml)
Detect Abeta x-40 including P340 fragment
1^ See Figure 10 for characterization.
2) Clones IAlO, 12B2, and 82El were previously published (Horikoshi Y. et al. 2004
Biochem Biophys Res Commun 319:733-737). Full-length Abeta ELISA provides accurate assessment of secretase inhibitor- mediated Abeta reduction in primary cultured neurons
Primary cultured neurons prepared from mouse embryos were treated with a beta and gamma secretase inhibitor, Inhibitor IV (Stachel S. J. et al. 2004 J Med Chem 47:6447- 6450) and DAPT (Dovey H. F. et al. 2001 J Neurochem 76:173-181), respectively, and Abeta levels in the culture medium were determined by 14F1/1A10 and 12B2/1A10 ELISAs. In the primary cultured neurons treated with the gamma secretase inhibitor, both full-length 1-40 (14F1/1A10) and P3 cross-reactive x-40 (12B2/1A10) Abeta ELISAs provided similar results (Fig. HA). In the case of the BACEl inhibitor (Fig. HB), Abeta levels determined by P3 cross-reactive Abeta x-40 ELISA (12B2/1A10) were always higher than full-length Abeta determined by 14F1/1A10 ELISA. At the highest tested concentration, 1 μM, full-length Abeta ELISA indicated virtually complete inhibition of
Abeta production, while P3 cross-reactive Abeta x-40 ELISA detected only 67% Abeta reduction (Fig. HB, the difference is indicated by bilateral arrow). At all testing concentrations, MTT determined cell toxicity to be <10% of vehicle-treated cells. Genetic and pharmacological BACEl inhibition lowered brain Abeta levels in wildtype (non-trasngenic) mice
We determined brain Abeta levels using 1-40 and x-40 Abeta ELISAs in BACEl knockout mice and mice treated with a BACEl inhibitor, Inhibitor IV. Full-length 1-40 Abeta ELISA (14F1/1A10) determined that levels in BACEl-/- mice were at the background level (OD=0.067-0.073, compared to the background level, OD=0.055-0.071). Full-length Abeta 1-40 level in BACEl+/- mice was virtually the same as in BACE1+/+ controls (Fig. 12Aa). While full-length Abeta 1-40 is diminished in BACEl-/- mice, P3 cross-reactive Abeta x-40 ELISA (12B2/1A10) detected much less significant Abeta reduction (only 33% reduction compared to BACE1+/+ control, PO.01, Fig. 12Ab).
In mice receiving the BACEl inhibitor, Inhibitor IV, there was significant reduction in Abeta 1-40 level (30% reduction compare to vehicle-administered mice, P<0.01, Fig. 12Ba). However, the Abeta x-40 level did not change significantly (13% reduction, no significance, Fig. 12Bb). With systemic administration, Inhibitor IV did not reduce brain Abeta, although it reduced plasma Abeta (36% reduction compared to vehicle-treated mice, 100 mg/kg subcutaneous injection). Inhibitor IV is a substrate of P-glycoprotein brain efflux transporter (Stachel S. J. et al. 2006 Bioorg Med Chem Lett 16:641-644); thus we suspect that the lack of efficacy following systemic administration might be due to insufficient drug concentration in the brain.
Upregulation of alpha secretase cleavage in response to genetic and pharmacological BACEl inhibition We used commercially avairable sAPPalpha- and sAPPbeta-specific antibodies to determine how BACEl inhibition alters APP processing. Specificities of these antibodies were confirmed by western blotting, that sAPPalpha and sAPPbeta specific antibodies did not cross react with each other or uncleaved APP (Fig. 13A). hi BACEl-/- mice, the levels of sAPPbeta were diminished (protein bands were undetectable) (Fig. 13Ba), and sAPPalpha was significantly elevated (250% of control, P<0.001) (Fig. 13Bb). Intracerebroventricular injection with a BACEl inhibitor, Inhibitor IV, significantly reduced sAPPbeta level (59% reduction, P<0.01, Fig. 13Ca), and significantly elevated sAPPalpha level (60% elevation, P<0.05, Fig. 13Cb).
Determination of Abeta change under physiological condition is more relevant to sporadic AD
Beta and gamma secretases are promising therapeutic targets in AD, and transgenic mice that overexpress mutant human APP have been commonly used for evaluation of potential treatments. However, early-onset AD cases with increased generation of Abeta caused by genetic mutation represent a small percentage of the overall population suffering from AD (Rocca W. A. et al. 1991 Ann Neurol 30:381-390). There is no clear evidence that APP expression is upregulated in sporadic AD cases. Therefore, testing therapeutic agents under physiologically relevant conditions, i.e. in mice with physiological APP expression, is important. We previously developed an N terminus end-specific antibody, clone 82El, which detects full-length Abeta in combination with C terminus antibodies. However, 82El includes the human-unique 5th amino acid residue within the epitope, and it was not sensitive enough to detect endogenous Abeta in non-transgenic (wildtype) mouse brain. In this study, we developed an N terminus end specific antibody, clone 14Fl, with an epitope within amino acid residues 1-4 of the Abeta peptide. Although amino acid residues 1-4 of Abeta are identical in humans and mice, clone 14Fl showed preference to mouse Abeta. We suspect that conformational differences between human and mouse Abeta determined this species preference of clone 14Fl. The ELISA, which is composed of 14Fl and the Abeta 40-specific clone IAlO (Horikoshi Y. et al. 2004 Biochem Biophys Res Commun 319:733-737), detects full-length endogenous Abeta 1-40 in non-transgenic mouse brain homogenate. This is the first full-length Abeta ELISA which detects Abeta change in wildtype mice.
Multiple secretases are involved in Abeta generation. Beta-secretase cleaves APP and generates sAPPbeta and the C99 fragment, and then gamma-secretase cleaves C99 and generates Abeta. Another cascade is known: alpha-secretase cleaves the APP molecule in the middle of the Abeta domain and generates sAPPalpha and the C83 fragment, and then gamma-secretase cleaves C83 and generates P3. hi addition to these primary cleavage sites, additional cleavage sites are known. For example, cells overexpressing BACEl generate C89 (the secondary beta-cleavage product starting with the Glul 1 amino acid of Abeta domain) in addition to C99 (the primary beta-cleavage product starting with Aspl of Abeta domain) (Haass C. et al. 1992 Nature 359:322-325; Gouras G. K. et al. 1998 JNeurochem 71:1920-1925; Vassar R. et al. 1999 Science 286:735-741). There are some mouse cross- reactive antibodies, and they typically have epitopes within amino acid residues 14-28, a
region that is identical in human and mouse Abeta. A commonly used mouse cross-reactive Abeta ELISA includes clone BNT77 (Suzuki N. et al. 1994 Science 264:1336-1340), which has an epitope within amino acid residue 11-16 of Abeta (Fukumoto H. et al. 1999 Neuroreport 10:2965-2969). The BNT77 ELISA is not cross-reactive to P3, but still detects the Abeta 11-40 fragment (generated by cleavage at the beta1 (Glul l)-site), which is undesirable. Determination of full-length Abeta is essential for the accurate evaluation of Abeta changes, and our newly developed 14F1/1A10 ELISA provides the most accurate and useful results currently available. Full-length Abeta ELISA provides the optimal measure for the evaluation of Abeta- lowering strategies
Abeta is generated by sequential proteolytic processing at the N- and C-termini of the Abeta domain by beta- and gamma-secretases, respectively (Hardy J. and Selkoe D. J. 2002 Science 297:353-356). A gamma secretase inhibitor, DAPT (Dovey H. F. et al. 2001 J Neurochem 76:173-181), reduced Abeta production in a dose-dependent manner in primary cultured neurons, and diminished Abeta production at 1 μM. Both full-length and P3 cross-reactive x-40 ELISA (14F1/1A10 and 12B2/1A10, respectively) provided similar results in experiments using the gamma secretase inhibitor (DAPT). A BACEl inhibitor significantly reduced full-length Abeta in a dose-dependent manner. However, a P3 cross- reactive x-40 ELISA detected much less significant Abeta reduction, probably due to an increase in P3 generation through compensatory upregulation of alpha secretase. Similar results were found in vitro; i.e. alpha and beta secretases compete in cultured cells (Skovronsky D. M. et al. 2000 J Biol Chem 275:2568-2575). Measurement of full-length Abeta is thus important, particularly in the case of BACEl inhibition. BACEl as a therapeutic target Abeta-lowering approaches, including secretase inhibition, are being extensively pursued as potential disease-modifying therapies for AD (Aisen P. S. 2005 Drugs 19:989- 996). BACEl initiates Abeta generation and BACEl enzyme activity and/or BACEl protein level were elevated in sporadic AD brains (Fukumoto H. et al. 2002 Arch Neurol 59:1381-1389; Holsinger R. M. et al. 2002 Ann Neurol 51:783-786; Yang L. B. et al. 2003 Nat Med 9:3-4). Thus, BACEl is among the most aggressively pursued pharmacological targets in AD.
In our study, we found that endogenous full-length mouse Abeta is diminished in BACEl-/- mice, confirming that BACEl is primarily responsible for Abeta generation.
While complete BACEl gene deletion yielded expected results, the effect of partial gene deletion seems complicated. Heterozygous BACEl knockout mice were crossed with Swedish APP transgenic mice, and Abeta levels were investigated. At a pre-pathological stage, Abeta was slightly reduced (-20%) (Luo Y. et al. 2003 Neiirobiol Dis 14:81-88). Abeta plaque load was significantly reduced (37%) at the moderate pathological stage, but the efficacy was lost at the severe pathological stage (Laird F. M. et al. 2005 J Neurosci 25:11693-11709). Transient BACEl gene silencing through intrahippocampal injection with small interfering RNA (siRNA) reduced Abeta load in APP transgenic mice (Laird F. M. et al. 2005 J Neurosci 25:11693-11709; Singer O. et al. 2005 Nat Neurosci 8:1343- 1349). While partial gene deletion reduced Abeta level in mice overexpressing Swedish APP, we found no change in endogenous mouse Abeta in non-transgenic mice with physiological APP expression. Under conditions of excess substrate (APP), BACEl is more sensitive to inhibitory actions, and the impact on Abeta levels may be exaggerated. In the case of gamma secretase inhibition, the amount of the substrate affected the Abeta reduction in vivo. A gamma secretase inhibitor, 2-[(lR)-l-[[(4-chlorophenyl)sulfony](2,5- difluorophenyl)amino]ethyl]-5-fluorobenzenepropanoic acid (BMS-299897), showed greater efficacy in APP transgenic mice with excess APP, compared to APP-YAC mice (Lamb B. T. et al. 1993 Nat Genet 5:22-30) and guinea pigs with more physiologically- relevant APP expression (Anderson J. J. et al. 2005 Biochem Pharmacol 69:689-698; Barten D. M. et al. 2005 J Pharmacol Exp Ther 312:635-643). This suggests that testing secretase modulating approaches in APP overexpressing transgenic mice may yield artificially amplified results, resulting in overly optimistic efficacy projections. It should also be noted that Swedish mutation APP is a highly efficient BACEl substrate (>50 fold compared to wildtype) (Tomasselli A. G. et al. 2003 J Neurochem 84:1006-1017). It is advantageous to use plaque-forming, transgenic mice to test disease-modifying effects, but the effect of the APP mutation on outcome should be carefully considered.
A potential pharmacological agent and complete gene deletion significantly reduced endogenous brain Abeta levels in non-transgenic mice. This suggests that both approaches, enzyme inhibition and protein expression inhibition, may have therapeutic value. Since heterozygous gene deletion did not reduce endogenous Abeta levels, substantial reduction (beyond 50%) of BACEl protein level is presumably necessary. A potential enzyme inhibitor (IC50=6 nM in primary cultured neurons) showed significant but moderate efficacy (30% reduction). It is unknown how much Abeta reduction will be required for
clinical efficacy in AD; simultaneous inhibition of additional targets such as gamma secretase may be required. Along with the benefit of Abeta reduction, BACEl inhibition elevated sAPPalpha levels in vivo as suggested by cell culture study (Skovronsky D. M. et al. 2000 J Biol Chem 275:2568-2575). sAPPalpha is not only non-amyloidogenic, but also neurotrophic and neuroprotective (Schubert D. and Behl C. 1993 Brain Res 629:275-282; Mattson M. P. et al. 1993 Neuron 10:243-254; Mattson M. P. et al. 1997 Brain Res Rev 23:47-61). BACEl inhibition reduces harmful Abeta, and the resulting compensatory increase of sAPPalpha has a favorable effect; this reconfirms BACEl as a promising therapeutic target. In conclusion, we developed a full-length Abeta ELISA that facilitates the evaluation of endogenous Abeta in non-transgenic mice. We previously developed a full- length human Abeta ELISA using N terminus end specific antibody, clone 82El (Horikoshi Y. et al. 2004 Biochem Biophys Res Commun 319:733-737). However, clone 82El has strong preference to human Abeta, and the ELISA using 82El does not detect endogenous Abeta in wildtype mouse brain. Our new antibody, 14Fl, is also speficic to the N terminus end, and the ELISA composed of 14Fl with an anti-Abeta 40 antibody detects endogenous full-length mouse Abeta. This new ELISA facilitates in vivo investigations of Abeta- lowering strategies using wildtype (non-transgenic) mice with physiologically relevant APP expression. With this tool, we confirmed the feasibility of Abeta reduction through modulation of BACEl using BACEl knockout mice and a pharmacological agent. Testing in non-transgenic mice may be essential, because excess APP in transgenic mice may exaggerate the therapeutic impact of BACEl inhibition and other Abeta-lowering strategies. In addition, testing candidate interventions in wildtype mice is rapid and cost- effective; thus this approach is valuable as first-line in vivo testing of secretase inhibitors and other Abeta-lowering strategies. While non-transgenic mice do not recapitulate Abeta plaques, testing lead drug candidates in wildtype mice is advantageous to explore disease- modifying effects.
Appendix 1
IAlO VH (SEQ ID NO: 1)
EVQLHQSGAELVIaOASVKLSCTASGFNIICHTYFHWVRQRPEQGLEWIGRIDPANLNT KYDPICFQDI'CATITADTSSNTAYLHLSSLTSEDTAVYYCSNLYSVMTYWGQGTSVTVSS
FRL(SEQ ID NO: 2) EVQLHQSGAELVKPGASVKLSCTASGFNIK
CDRl (SEO ID NO: 3)
HTYFH
FR2_(SEQ ID NO: 4)
WVRQRPEQGLEWIG
CDR2 (SEO ID NO: 5)
RIDPANLNTKYDPKFQD
FRl(SEQ ID NO: 6) KATITADTSSNTAYLHLSSLTSEDTAVYYCSN
CDR3_(SEQ ID NO: 7) LYSVMTY
FR4 (SEO ID NO: 8)
WGQGTSVTVSS
YL(SEQ ID NO: 9)
DWMTQTPLILSVTIGQPASISCKSSQSLLFSDGRTYLNWLLQRPGQSPKRLI YLVSKLDSGVPDRFTGSGSGTDFTLKISRVEAEDLGHYYCWQGTHFPLTFG AGTKLELK
FRL(SEQ ID NO: 10)
DVVMTQTPLILSVTIGQPASISC
CDRl (SEO ID NO: 11)
KSSQSLLFSDGRTYLN
FR2_(SEQ ID NO: 12) WLLQRPGQSPKRLIY
CDR2 (SEO ID NO: 13) LVSKLDS
FRl(SEQ ID NO: 14) GVPDRFTGSGSGTDFTLKISRVEAEDLGHYYC
CDRl(SEQ ID NO: 15) WQGTHFPL
FRl(SEQ ID NO: 16) TFGAGTICLELK
B2
VH.(SEQ ID NO: 17) EVQLQQSGPELVIOOASVKISCKTSGYTFTEYTMHWVKQSHGKSLEWIGGINPNNGVT
NYNQKΪKGICATLTVDKSSSTAYMELRSLTSDDSAVYYCARSPYYFNYGDCWGQGTT LTVSS
FJRL(SEQ ID NO: 18) EVQLQQSGPELVKPGASVKISCKTSGYTFT
CDRl (SEO ID NO: 19)
EYTMH FR2_(SEQ ID NO: 20)
WVKQSHGKSLEWIG
CDR2 (SEO ID NO: 21)
GINPNNGVTNYNQKFKG
FR3 (SEO ID NO: 22) KATLTVDKSSSTAYMELRSLTSDDSAVYΥCAR
CDR3 (SEO ID NO: 23) SPYYFNYGDC
FRl(SEQ ID NO: 24) WGQGTTLTVSS
YLISEQ ID NO: 25)
DIVLTQSPASLAVFLGQRATISCRASKSVSTSGYTYMHWYQQKPGQPPKLLI
YLASNLESGVPARFSGSGSGTDFTLNIHPVEEEDAATYYCQHSRELPFTFGS
GTKLEIK
FRl (SEO ID NO: 26)
DΓVLTQSP ASLAVFLGQRATISC
CDRl (SEO ID NO: 27)
RASKSVSTSGYTYMH
FR2 (SEQ ID NO: 28) WYQQKPGQPPKLLIY
CDR2 (SEO ID NO: 29) LASNLES
FR3_(SEQ ID NO: 30) GVPARFSGSGSGTDFTLNIHPVEEEDAATYYC
CDR3 (SEO ID NO: 31)
QHSRELPF
FR4 (SEO ID NO: 32)
TFGSGTKLEIK
Fl
VHXSEQ ID NO: 33)
DVHLQASGPDLVRPSQSLSLTCTVTDYSITSDYSWHWIRQFPGNKLEWMGYILFSGSSN FNPSLKSRISITRDTSKNQFFLHLNSVTTEDTATYYCARRAYDGNFSWFAYWGQGTLV
TVSA
FRL(SEQ ID NO: 34) DVHLQASGPDLVRPSQSLSLTCTVTDYSITS
CDRl (SEO ID NO: 35)
DYSWH
FR2_(SEQ ID NO: 36) WIRQFPGNKLEWMG
CDR2 (SEO ID NO: 37)
YILFSGSSNFNPSLKS FR3_(SEQ ID NO: 38)
RISITRDTSKNQFFLHLNSVTTEDTATYYCAR
CDR-L(SEQ ID NO: 39) RAYDGNFSWFAY
FR4 (SEQ ID NO: 40)
WGQGTLVTVSA
V1^(SEQ ID NO: 41) QAWTQESALTTSPGETVTLTCRSSAGAVTISNYANWVQEKPDHLFTGLIGG
TNNRAPGVPARFSGSLIGDKAALTITGAQTEDEAIYFCALWYGNYWVFGGG TKLTVLGQ
FRl (SEO ID NO: 42) QAWTQESALTTSPGETVTLTC
CDRl (SEO ID NO: 43)
RSSAGAVTISNYAN
FR2_(SEQ ID NO: 44) WVQEICPDHLFTGLIG
CDR2 (SEO ID NO: 45) GTNNRAP
FR3 (SEO ID NO: 46)
GVPARFSGSLIGDKAALTITGAQTEDEAIYFC
CDR3 (SEO ID NO: 47)
ALWYGNYW
FRl(SEQ ID NO: 48) VFGGGTKLTVL
***
While the present invention has been described in some detail for purposes of clarity and understanding, one skilled in the art will appreciate that various changes in form and detail can be made without departing from the true scope of the invention. All figures, tables, and appendices, as well as patents, applications, and publications, referred to above, are hereby incorporated by reference.
Claims
1. A method for determining whether a compound alters the amount of amyloid beta (Aβ) peptide present in a wildtype animal, comprising the steps of: administering the compound to a wildtype animal; measuring the amount of the Aβ peptide in a sample from the animal; and determining whether the measured amount in the animal is different from the amount expected in a sample from a wildtype animal to which no compound has been administered, whereby a difference between the measured amount in the animal and the amount expected in the animal to which no compound has been administered indicates that the compound alters the amount of an Aβ peptide present in the animal, wherein the amount of the Aβ peptide is measured by immunoassay, wherein the immunoassay is a sandwich immunoassay using a capture binding substance bound to a solid phase and a labeled detection binding substance, and wherein the binding substances are specific for Aβ peptide.
2. The method of claim 1, wherein the capture binding substance or labeled detection binding substance is specific to the C-terminus of the Aβ peptide.
3. The method of claim 2, wherein the capture binding substance or labeled detection binding substance is raised against peptide MVGGW (SEQ ID NO: 65).
4. The method of claim 3, wherein the capture binding substance or labeled detection binding substance is IAlO.
5. The method of claim 1, wherein the capture binding substance or labeled detection binding substance is specific to the N-terminus of the Aβ peptide.
6. The method of claim 5, wherein the capture binding substance or labeled detection binding substance is raised against peptide DAEFRHDSGYEVHHQK (SEQ ID NO: 63).
7. The method of claim 6, wherein the capture binding substance or labeled detection binding substance is 14Fl .
8. The method of claim 1, wherein the capture binding substance or labeled detection binding substance is specific to the middle region of the Aβ peptide.
9. The method of claim 8, wherein the capture binding substance or labeled detection binding substance is raised against peptide EVHHQKLVFF AEDVGSNK (SEQ ID NO: 64).
10. The method of claim 9, wherein the capture binding substance or labeled detection binding substance is 12B2.
11. The method of any one of Claims 1-10, wherein the animal is a rodent.
12. The method of Claim 11 , wherein the rodent is a mouse.
13. A kit for determining whether a compound alters the amount of amyloid beta
(Aβ) peptide present in a wildtype animal comprising: means for measuring by a sandwich immunoassay an amount of Aβ peptide in a wildtype animal to which a compound has been administered in comparison to an amount of Aβ peptide expressed from a wildtype animal to which no compound has been administered, and a capture binding substance and a labeled detection binding substance, wherein the binding substances are specific for Aβ peptide.
14. The kit of claim 13, wherein the capture binding substance is specific to the C-terminus of the Aβ peptide.
15. The kit of claim 14, wherein the capture binding substance is raised against peptide MVGGW (SEQ ID NO: 65).
16. The kit of claim 15, wherein the capture binding substance is IAlO.
17. The kit of claim 13, wherein the capture binding substance is specific to the N-terminus of the Aβ peptide.
18. The kit of claim 17, wherein the capture binding substance is raised against peptide DAEFRHD S GYEVHHQK (SEQ ID NO: 63).
19. The kit of claim 18, wherein the capture binding substance is 14F 1.
20. The kit of claim 13, wherein the capture binding substance is specific to the middle region of the Aβ peptide.
21. The kit of claim 20, wherein the capture binding substance is raised against peptide EVHHQKLVFFAED VGSNK (SEQ ID NO : 64).
22. The kit of claim 21 , wherein the capture binding substance is 12B2.
23. A compound that alters the amount of Aβ peptide present in a wildtype animal identified by the method of any one of Claims 1-12.
24. A method of making a composition comprised of a compound that alters the amount of Aβ peptide present in a wildtype animal comprising combining the compound identified by the method of any one of Claims 1-12 and a physiologically acceptable carrier.
25. A method for determining whether a compound alters the amount of amyloid beta (Aβ) peptide present in a mouse, comprising the steps of: administering the compound to a mouse; measuring the amount of the Aβ peptide in a sample from the mouse; and determining whether the measured amount in the mouse is different from the amount expected in a sample from a mouse to which no compound has been administered, whereby a difference between the measured amount in the mouse and the amount expected in the mouse to which no compound has been administered indicates that the compound alters the amount of an Aβ peptide present in the mouse, wherein the amount of the Aβ peptide is measured by immunoassay, wherein the immunoassay is a sandwich immunoassay using a capture binding substance bound to a solid phase and a labeled detection binding substance, and wherein the binding substances are specific for Aβ peptide.
26. A method for determining whether a compound alters the amount of amyloid beta (Aβ) peptide present in a neuron of a mouse, comprising the steps of: administering the compound to a neuron of a mouse; measuring the amount of the Aβ peptide in the neuron from the mouse; and determining whether the measured amount in the neuron is different from the amount expected in a neuron from a mouse to which no compound has been administered, whereby a difference between the measured amount in the neuron and the amount expected in the neuron to which no compound has been administered indicates that the compound alters the amount of an Aβ peptide present in the neuron, wherein the amount of the Aβ peptide is measured by immunoassay, wherein the immunoassay is a sandwich immunoassay using a capture binding substance bound to a solid phase and a labeled detection binding substance, and wherein the binding substances are specific for Aβ peptide.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US70771305P | 2005-08-12 | 2005-08-12 | |
| US60/707,713 | 2005-08-12 | ||
| US73719105P | 2005-11-16 | 2005-11-16 | |
| US60/737,191 | 2005-11-16 | ||
| US76364806P | 2006-01-31 | 2006-01-31 | |
| US60/763,648 | 2006-01-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007022015A1 true WO2007022015A1 (en) | 2007-02-22 |
Family
ID=37488482
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/031517 WO2007022015A1 (en) | 2005-08-12 | 2006-08-11 | Methods to evaluate amyloid beta-lowering agents using wild-type mice |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2007022015A1 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009033743A1 (en) * | 2007-09-13 | 2009-03-19 | University Of Zurich Prorektorat Forschung | Monoclonal amyloid beta (abeta)-specific antibody and uses thereof |
| WO2009067272A1 (en) * | 2007-11-19 | 2009-05-28 | Abbott Laboratories | Immunoassay for detection and quantification of amyloid-b peptides |
| WO2011070174A1 (en) | 2009-12-11 | 2011-06-16 | Araclon Biotech, S.L. | Methods and reagents for improved detection of amyloid beta peptides |
| EP2511296A1 (en) | 2011-04-12 | 2012-10-17 | Araclón Biotech, S. L. | Antibody, kit and method for determination of amyloid peptides |
| US8323647B2 (en) | 2007-09-13 | 2012-12-04 | Delenex Therapeutics Ag | Humanized antibodies against the β-amyloid peptide |
| WO2015077473A1 (en) * | 2013-11-20 | 2015-05-28 | University Of Iowa Research Foundation | Methods and compositions for treating amyloid deposits |
| WO2015099931A1 (en) * | 2013-12-23 | 2015-07-02 | Arizona Board Of Regents On Behalf Of Arizona State University | Method of distinguishing between different neurodegenerative diseases |
| CN110950960A (en) * | 2019-11-26 | 2020-04-03 | 中国农业大学 | Preparation method of small molecule compound antibody based on high-throughput sequencing and hybrid hybridoma technology |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020107173A1 (en) * | 1999-11-04 | 2002-08-08 | Lawrence Friedhoff | Method of treating amyloid beta precursor disorders |
| EP1717250A1 (en) * | 2004-02-20 | 2006-11-02 | Immuno-Biological Laboratories Co., Ltd. | Monoclonal antibody and use thereof |
-
2006
- 2006-08-11 WO PCT/US2006/031517 patent/WO2007022015A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020107173A1 (en) * | 1999-11-04 | 2002-08-08 | Lawrence Friedhoff | Method of treating amyloid beta precursor disorders |
| EP1717250A1 (en) * | 2004-02-20 | 2006-11-02 | Immuno-Biological Laboratories Co., Ltd. | Monoclonal antibody and use thereof |
Non-Patent Citations (2)
| Title |
|---|
| HORIKOSHI Y ET AL: "Development of Abeta terminal end-specific antibodies and sensitive ELISA for Abeta variant", BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, ACADEMIC PRESS INC. ORLANDO, FL, US, vol. 319, no. 3, 2 July 2004 (2004-07-02), pages 733 - 737, XP004512500, ISSN: 0006-291X * |
| KOUHEI NISHITOMI ET AL.: "BACE1 inhibition reduces endogenous Abeta and alters APP processing in wild-type mice", JOURNAL OF NEUROCHEMISTRY, vol. 99, no. 6, December 2006 (2006-12-01), England, pages 1555 - 1563, XP002411184 * |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8323647B2 (en) | 2007-09-13 | 2012-12-04 | Delenex Therapeutics Ag | Humanized antibodies against the β-amyloid peptide |
| WO2009033743A1 (en) * | 2007-09-13 | 2009-03-19 | University Of Zurich Prorektorat Forschung | Monoclonal amyloid beta (abeta)-specific antibody and uses thereof |
| US9273126B2 (en) | 2007-09-13 | 2016-03-01 | Delenex Therapeutics Ag | Humanized antibodies against the beta-amyloid peptide |
| WO2009067272A1 (en) * | 2007-11-19 | 2009-05-28 | Abbott Laboratories | Immunoassay for detection and quantification of amyloid-b peptides |
| WO2011070174A1 (en) | 2009-12-11 | 2011-06-16 | Araclon Biotech, S.L. | Methods and reagents for improved detection of amyloid beta peptides |
| EP2511296A1 (en) | 2011-04-12 | 2012-10-17 | Araclón Biotech, S. L. | Antibody, kit and method for determination of amyloid peptides |
| US9255932B2 (en) | 2011-04-12 | 2016-02-09 | Aracion Biotech, S.L. | Antibody, kit and method for determining amyloid peptides |
| WO2012140296A1 (en) | 2011-04-12 | 2012-10-18 | Araclon Biotech, S.L. | Antibody, kit and method for determining amyloid peptides |
| US9863961B2 (en) | 2011-04-12 | 2018-01-09 | Araclon Biotech, S.L. | Method for determination of amyloid peptides with anti-amyloid antibody |
| WO2015077473A1 (en) * | 2013-11-20 | 2015-05-28 | University Of Iowa Research Foundation | Methods and compositions for treating amyloid deposits |
| WO2015099931A1 (en) * | 2013-12-23 | 2015-07-02 | Arizona Board Of Regents On Behalf Of Arizona State University | Method of distinguishing between different neurodegenerative diseases |
| US20160320412A1 (en) * | 2013-12-23 | 2016-11-03 | Arizona Board Of Regents On Behalf Of Arizona State University | Method of distinguishing between different neurodegenerative diseases |
| CN110950960A (en) * | 2019-11-26 | 2020-04-03 | 中国农业大学 | Preparation method of small molecule compound antibody based on high-throughput sequencing and hybrid hybridoma technology |
| CN110950960B (en) * | 2019-11-26 | 2021-05-14 | 中国农业大学 | Preparation method of small molecule compound antibody based on high-throughput sequencing and hybrid hybridoma technology |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9939452B2 (en) | N-11 truncated amyloid-beta monoclonal antibodies, compositions, methods and uses | |
| US10208111B2 (en) | Alpha-synuclein antibodies and uses thereof | |
| EP0906577B1 (en) | Screening compounds for the ability to alter the production of amyloid-beta peptide | |
| US6114133A (en) | Methods for aiding in the diagnosis of Alzheimer's disease by measuring amyloid-β peptide (x-≧41) | |
| US8673593B2 (en) | Antibodies to alpha-synuclein | |
| JP3634375B2 (en) | Method for assisting diagnosis of Alzheimer's disease by measuring amyloid β peptide (X− ≧ 41) and tau | |
| WO2007022015A1 (en) | Methods to evaluate amyloid beta-lowering agents using wild-type mice | |
| WO1997048983A9 (en) | SCREENING COMPOUNDS FOR THE ABILITY TO ALTER THE PRODUCTION OF AMYLOID-β PEPTIDE (x-≥41) | |
| WO2007021255A1 (en) | Antibodies to alpha-synuclein | |
| JPWO2005080435A1 (en) | Monoclonal antibodies and uses thereof | |
| US7682795B2 (en) | Method of diagnosing Alzheimer's Disease | |
| JP6742590B2 (en) | Antibody or antigen-binding fragment thereof and hybridoma | |
| DK2289909T3 (en) | The screening method, method of purification of non-diffusing alpha-beta oligomers selective antibodies to said non-diffunderingsdygtige alpha-beta oligomers and a method of producing said antibodies | |
| JP7481733B2 (en) | Monoclonal antibody against brain-derived neurotrophic factor and hybridoma producing the antibody | |
| WO2024032823A1 (en) | Diagnostic use of highly toxic amyloid oligomer |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06801343 Country of ref document: EP Kind code of ref document: A1 |