WO2007003964A1 - G-protein coupled receptor agonists - Google Patents
G-protein coupled receptor agonists Download PDFInfo
- Publication number
- WO2007003964A1 WO2007003964A1 PCT/GB2006/050182 GB2006050182W WO2007003964A1 WO 2007003964 A1 WO2007003964 A1 WO 2007003964A1 GB 2006050182 W GB2006050182 W GB 2006050182W WO 2007003964 A1 WO2007003964 A1 WO 2007003964A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pharmaceutically acceptable
- alkyl
- acceptable salt
- substituted
- compound according
- Prior art date
Links
- QCXZDZGTWWKNNT-UHFFFAOYSA-N CC(C)(C)OC(N1CCC(CCN(CC2)CCN2c(cc2)ccc2-c2n[o]c(C)n2)CC1)=O Chemical compound CC(C)(C)OC(N1CCC(CCN(CC2)CCN2c(cc2)ccc2-c2n[o]c(C)n2)CC1)=O QCXZDZGTWWKNNT-UHFFFAOYSA-N 0.000 description 1
- BJAXWGSHKDWPMQ-UHFFFAOYSA-N CS(c(cc1)ccc1N1CCNCC1)=O Chemical compound CS(c(cc1)ccc1N1CCNCC1)=O BJAXWGSHKDWPMQ-UHFFFAOYSA-N 0.000 description 1
- CGQXXPSFURARGQ-IBGZPJMESA-N C[C@@H](C1)N(CCC(CC2)CCN2C(OC(C)(C)C)=O)CCN1c(cc1)ccc1S(C)(=O)=O Chemical compound C[C@@H](C1)N(CCC(CC2)CCN2C(OC(C)(C)C)=O)CCN1c(cc1)ccc1S(C)(=O)=O CGQXXPSFURARGQ-IBGZPJMESA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/20—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by singly bound oxygen or sulphur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/26—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
Definitions
- the present invention is directed to G-protein coupled receptor (GPCR) agonists.
- GPCR G-protein coupled receptor
- the present invention is directed to GPCR agonists that are useful for the treatment of obesity, e.g. as regulators of satiety, and for the treatment of diabetes.
- Obesity is characterized by an excessive adipose tissue mass relative to body size.
- body fat mass is estimated by the body mass index (BMI; weight(kg)/height(m) 2 ), or waist circumference.
- BMI body mass index
- Individuals are considered obese when the BMI is greater than 30 and there are established medical consequences of being overweight. It has been an accepted medical view for some time that an increased body weight, especially as a result of abdominal body fat, is associated with an increased risk for diabetes, hypertension, heart disease, and numerous other health complications, such as arthritis, stroke, gallbladder disease, muscular and respiratory problems, back pain and even certain cancers.
- Drugs aimed at the pathophysiology associated with insulin dependent Type I diabetes and non-insulin dependent Type II diabetes have many potential side effects and do not adequately address the dyslipidaemia and hyperglycaemia in a high proportion of patients. Treatment is often focused at individual patient needs using diet, exercise, hypoglycaemic agents and insulin, but there is a continuing need for novel antidiabetic agents, particularly ones that may be better tolerated with fewer adverse effects.
- metabolic syndrome which is characterized by hypertension and its associated pathologies including atherosclerosis, lipidemia, hyperlipidemia and hypercholesterolemia have been associated with decreased insulin sensitivity which can lead to abnormal blood sugar levels when challenged.
- Myocardial ischemia and microvascular disease is an established morbidity associated with untreated or poorly controlled metabolic syndrome.
- GPRl 19 (previously referred to as GPRl 16) is a GPCR identified as SNORF25 in WO00/50562 which discloses both the human and rat receptors, US 6,468,756 also discloses the mouse receptor (accession numbers: AAN95194 (human), AAN95195 (rat) and ANN95196 (mouse)).
- GPRl 19 is expressed in the pancreas, small intestine, colon and adipose tissue.
- the expression profile of the human GPRl 19 receptor indicates its potential utility as a target for the treatment of obesity and diabetes.
- (I) or pharmaceutically acceptable salts thereof are agonists of GPRl 19 and are useful for the prophylactic or therapeutic treatment of obesity and diabetes.
- the present invention is directed to a compound of formula (I):
- Z represents an aryl, heteroaryl, -Ci -4 alkylaryl or -Ci -4 alkylheteroaryl group, any of which may optionally be substituted by one or more groups selected from halogen, Ci -4 alkyl, OR 9 , NR 3 R 4 , S(O) n R 9 , S(O) 2 NR 9 R 99 , C(O)NR 9 R 99 , NR 10 C(O)R 9 , NR 10 C(O)NR 9 R 99 , NR 10 SO 2 R 9 , C(O)R 9 , C(O)OR 9 , -P(O)(CH 3 ) 2 , NO 2 , cyano or -(CH 2 )J-C 3-7 cycloalkyl, -(CH 2 ) r aryl, -(CH 2 ) r heterocyclyl, -(CH 2 ) r heteroaryl, any of which cycloalkyl, aryl, heterocycl
- Ci -4 alkylene chain represents a branched or unbranched Ci -4 alkylene chain or Ci -4 alkenylene chain, either of which may optionally be substituted by one or more groups selected from halogen, hydroxy or oxo, and wherein one CH 2 group may be replaced by O or NR 8 , provided that the group >A 2 -B- does not contain any direct N-O, N-C-O, N-N, N-C-N or N-C-halogen bonds;
- G represents CHR 2 or NR 1 ;
- R 1 is C(O)OR 5 , C(O)R 5 , S(O) 2 R 5 , C(O)NR 5 R 8 , Ci -4 alkylene-C(O)OR 5 , C(O)C(O)OR 5 , or P(O)(O-Ph) 2 ; or heterocyclyl or heteroaryl, either of which may optionally be substituted by one or two groups selected from Ci -4 alkyl, Ci -4 alkoxy or halogen; R 2 is C 3 _ 6 alkyl;
- R 3 and R 4 are independently hydrogen, methoxy, Ci -4 alkyl, which may optionally be substituted by halo (e.g. fluoro), hydroxy, Ci -4 alkyloxy-, aryloxy-, arylCi -4 alkyloxy-, Ci -4 alkylS(O) n -, C 3-7 heterocyclyl, -C(O)OR 14 or N(R 10 ) 2 ; or may be C 3-7 cycloalkyl, aryl, heterocyclyl or heteroaryl, wherein the cyclic groups may be substituted with one or more substituents selected from halo, Ci -4 alkyl, Ci -4 fluoroalkyl, OR 13 , CN, SO 2 CH 3 , N(R 10 ) 2 and NO 2 ; or taken together R 3 and R 4 may form a 5- or 6-membered heterocyclic ring optionally substituted by hydroxy, Ci -4 alkyl or Ci -4 hydroxyalkyl and optionally
- R 5 and R 55 are independently Ci -8 alkyl, C 2-8 alkenyl or C 2-8 alkynyl, any of which may be optionally substituted by one or more halo atoms, NR 6 R 66 , OR 6 , C(O)OR 6 , OC(O)R 6 or cyano, and may contain a CH 2 group that is replaced by O or S; or a C 3-7 cycloalkyl, aryl, heterocyclyl, heteroaryl, Ci -4 alkylenearyl, or Ci -4 alkyleneheteroaryl, any of which may be substituted with one or more substituents selected from halo, Ci -4 alkyl, Ci -4 fluoroalkyl, OR 7 , CN, NR 7 R 77 , SO 2 Me, NO 2 or C(O)OR 7 ;
- R 6 , R 66 , R 7 , and R 77 each independently are hydrogen or or, taken together, R 6 and R 66 or R 7 and R 77 may independently form a 5- or 6-membered heterocyclic ring;
- R 8 hydrogen or
- R 9 and R 99 are independently hydrogen, methoxy, Ci -4 alkyl, which may optionally be substituted by halo (e.g. fluoro), hydroxy, Ci -4 alkoxy-, -aryloxy-, arylCi -4 alkyloxy-, C 3-7 heterocyclyl, -C(O)OR 14 or N(R 10 ) 2 ; or may be C 3-7 cycloalkyl, aryl, heterocyclyl or heteroaryl, wherein the cyclic groups may be substituted with one or more substituents selected from halo, Ci -4 alkyl, Ci -4 fluoroalkyl, OR 13 , CN, SO 2 CH 3 , N(R 10 ) 2 and NO 2 ; or taken together R 9 and R 99 may form a 5- or 6-membered heterocyclic ring optionally substituted by hydroxy, Ci -4 alkyl or Ci -4 hydroxyalkyl and optionally containing a further heteroatom selected from O and NR 10 ;
- R 10 is hydrogen, Ci -4 alkyl; or a group N(R 10 ) 2 may form a 4- to 7-membered heterocyclic ring optionally containing a further heteroatom selected from O and NR 10 ;
- R 11 is hydrogen or hydroxy, or when B represents and there is a point of unsaturation adjacent to CR 11 then R 11 is absent;
- R 12 is each independently hydroxy, oxo, methyl; or two R 12 groups may form a bridging methylene;
- R 13 is hydrogen, Ci -2 alkyl or Ci -2 fluoroalkyl
- R 14 is hydrogen or Ci -4 alkyl; x is 0, 1, 2 or 3; and y is 1, 2, 3, 4 or 5; with the proviso that x + y is 2, 3, 4 or 5.
- the molecular weight of the compounds of formula (I) is preferably less than 800, more preferably less than 600, even more preferably less than 500.
- Z represents an aryl or heteroaryl group, either of which may optionally be substituted by one or more groups selected from halogen, NR 3 R 4 , S(O) 1n R 9 , S(O) 2 NR 9 R 99 C(O)NR 9 R 99 , C(O)R 9 , C(O)OR 9 , aryl, heterocyclyl, heteroaryl or cyano; or C ⁇ alkyl, C 2 - 4 alkenyl, or C 2 ⁇ alkynyl any of which three may optionally be substituted by one or more halogen, hydroxy, NR 3 R 4 , oxo or one of Ai and A 2 is N, and the other is CH or N; d is 0, 1, 2, or 3; e is 1 or 2; with the proviso that d + e is 2, 3, 4 or 5, and that if Ai and A 2 are both N, d is 2 or 3 and e is 2; m is 1, 2 or 3;
- G represents CHR 2 or NR 1 ;
- R 1 is C(O)OR 5 , C(O)R 5 , S(O) 2 R 5 , C(O)NR 5 R 8 , Ci -4 alkylene-C(O)OR 5 , C(O)C(O)OR 5 , S(O) 2 R 5 , C(O)R 5 or P(O)(O-Ph) 2 ; or heterocyclyl or heteroaryl, either of which may optionally be substituted by one or two groups selected from Ci -4 alkyl, Ci -4 alkoxy or halogen;
- R 2 is C 3 _ 6 alkyl
- R 3 and R 4 are independently hydrogen, C 3 _ 7 cycloalkyl, or aryl, which may optionally be substituted with 1 or 2 substituents selected from halo, cyano, and S(O) 2 Me; or, taken together, R 4 and R 44 may form a 5- or 6-membered heterocyclic ring;
- R 5 and R 55 are independently Ci -8 alkyl, C 2-8 alkenyl or C 2-8 alkynyl, any of which may be optionally substituted by one or more halo atoms, NR 6 R 66 , OR 6 , C(O)OR 6 , OC(O)R 6 or cyano, and may contain a CH 2 group that is replaced by O or S; or a C 3-7 cycloalkyl, aryl, heterocyclyl, heteroaryl, Ci -4 alkylenearyl, or Ci -4 alkyleneheteroaryl, any of which may be substituted with one or more substituents selected from halo, Ci -4 alkyl, Ci -4 fluoroalkyl, OR 7 , CN, NR 7 R 77 , SO 2 Me, NO 2 or C(O)OR 7 ;
- R 6 , R 66 , R 7 , and R 77 each independently are hydrogen or or, taken together, R 6 and R 66 or R 7 and R 77 may independently form a 5- or 6-membered heterocyclic ring;
- R 8 hydrogen or
- R 9 and R 99 are independently hydrogen, Ci -4 alkyl, which may optionally be substituted by halo (e.g. fluoro), hydroxy, Ci -4 alkyloxy-, Ci -4 alkylthio-, C 3-7 heterocyclyl or N(R 10 ) 2 ; or may be C 3-7 cycloalkyl, aryl, heterocyclyl or heteroaryl, wherein the cyclic groups may be substituted with one or more substituents selected from halo, Ci -4 alkyl, Ci -4 fluoroalkyl, OR 9 , CN, SO 2 CH 3 , N(R 10 ) 2 and NO 2 ; x is 0, 1, 2 or 3; and y is 1, 2, 3, 4 or 5; with the proviso that x + y is 2, 3, 4 or 5.
- halo e.g. fluoro
- Exemplary aryl groups which Z may represent include phenyl and naphthalenyl (either of which may be optionally substituted as described above), in particular phenyl.
- Exemplary heteroaryl groups which Z may represent include 5 membered monocyclic rings, 6 membered monocyclic rings, 8 membered bicyclic rings, 9 membered bicyclic rings and 10 membered bicyclic rings (any of which may be optionally substituted as described above), in particular 6 membered monocyclic rings (such as those containing one or two nitrogen atoms).
- Z is a heteroaryl or -Ci -4 alkylheteroaryl group it will typically contain up to four heteroatoms selected from O, N and S.
- Z is preferably phenyl or 6-membered heteroaryl preferably containing one nitrogen atom.
- Z is a substituted phenyl or a 6-membered heteroaryl group containing one nitrogen atom it is preferably substituted by up to 3 substituents preferably in the meta and para positions.
- Preferred groups by which Z may be substituted include S(O) n R 9 e.g. SOMe or SO 2 Me, C(O)NR 9 R", NR 10 C(O)NR 9 R 99 , 5- or 6-membered heteroaryl, halogen e.g. fluoro or chloro, Ci -4 alkyl e.g. methyl and cyano.
- G is preferably NR 1 .
- R 1 is preferably C(O)OR 5 , C(O)NR 5 R 8 , Ci -4 alkylene-C(O)OR 5 , C(O)C(O)OR 5 , heterocyclyl, heteroaryl, S(O) 2 R 5 , C(O)R 5 or P(O)(O-Ph) 2 ; especially C(O)OR 5 , C(O)NR 5 R 8 , Ci- 4 alkyl-C(O)OR 5 , heteroaryl, S(O) 2 R 5 or C(O)R 5 ; in particular C(O)OR 5 , C(O)NR 5 R 8 , heteroaryl, S(O) 2 R 5 or C(O)R 5 .
- R 1 is C(O)OR 5 , C(O)NR 5 R 8 or heteroaryl.
- R 1 is most preferably COOR 5 .
- the heteroaryl ring is preferably a 5- or 6-membered heteroaryl ring, for example pyrimidinyl, especially pyrimidin-2-yl.
- R 5 represents Ci -8 alkyl, C 2-8 alkenyl or C 2-8 alkynyl optionally substituted by one or more halo atoms or cyano, and which may contain a CH 2 group that is replaced by O or S; or a C 3-7 cycloalkyl, aryl or cycloalkyl, any of which may be substituted with one or more substituents selected from halo, Ci -4 alkyl, Ci -4 fluoroalkyl, OR 7 , CN, NR 7 R 77 , NO 2 and C(O)OCi -4 alkyl.
- R 5 represents Ci -8 alkyl, C 2-8 alkenyl or C 2-8 alkynyl optionally substituted by one or more halo atoms or cyano, and which may contain a CH 2 group that is replaced by O or S; or a C 3-7 cycloalkyl or aryl, either of which may be substituted with one or more substituents selected from halo, Ci -4 alkyl, Ci -4 fluoroalkyl, OR 7 , CN, NR 7 R 77 , NO 2 and C(O)OCi -4 alkyl.
- R 5 groups are C 3-5 alkyl optionally substituted by one or more halo atoms or cyano, and may contain a CH 2 group that is replaced by O or S; or C 3-5 cycloalkyl optionally substituted by Ci -4 alkyl. In one embodiment of the invention the group represented by R 5 is unsubstituted.
- x + y is 2, 3, or 4. In a preferred embodiment of the invention x and y each represent 1. In a more preferred embodiment of the invention x and y each represent 2.
- B represents a branched or unbranched Ci -4 alkylene which may optionally be substituted by one or more groups selected from halogen, hydroxy or oxo.
- B represents a branched or unbranched Ci -4 alkenylene which may optionally be substituted by one or more groups selected from halogen, hydroxy or oxo.
- substituent groups e.g. 1 or 2.
- Ai and A 2 represent N.
- Ai represents N and and A 2 represents CH.
- Ai represents CH and and A 2 represents N.
- a subgroup of compounds of formula (I) of are those of formula (Ib):
- a 2 is N or CH; when A 2 is N, Y is CH 2 ; when A 2 is CH, Y is O or NR 8 ;
- W is a branched or unbranched Ci -3 alkylene chain or Ci -3 alkenylene chain, either of which may optionally be substituted by one or more groups selected from halogen, hydroxy or oxo; one of R a , R b and R c is selected from S(O) n R 9 , S(O) 2 NR 9 R 99 , C(O)NR 9 R 99 , NR 10 C(O)NR 9 R 99 and 5- or 6-membered heteroaryl, and the other two of R a , R b and R c are selected from hydrogen, halogen, Ci -4 alkyl and cyano; and
- R 1 is C(O)OR 5 , C(O)NR 5 R 8 or 5- or 6-membered heteroaryl.
- the H may be replaced by one of the substituents listed above for R a , R b and R c .
- one of E 1 or E 2 is preferably N.
- preferred compounds of this invention include those in which several or each variable in formulae (I), (Ia) and (Ib) is selected from the preferred, more preferred or particularly listed groups for each variable. Therefore, this invention is intended to include all combinations of preferred, more preferred and particularly listed groups.
- provisos may optionally be used (individually or in any combination) to exclude certain compounds from the scope of the invention: i) when G represents N-C(O)O-tert-butyl; B represents an ethylene group; Ai and A 2 each represent N; d, e, x, and y each represent 2; R 11 represents H; k represents O; suitably Z does not represent:
- G represents N-(naphthylen-l-ylsulphonyl-); B represents an ethylene group; Ai and A 2 each represent N, or Ai represents CH and A 2 represents N; d and e each represent 2; x represents 0; y represents 4; R 11 represents H; k represents 0; suitably Z does not represent phenyl, pyridine-2-yl-, 2-methylphenyl-, 4-trifluoromethylphenyl- or 3-trifluoromethylphenyl.
- G when G represents N-(4-trifluoromethylphenylsulphonyl-); B represents a methylene group; Ai represents CH and A 2 represents N; d, e, x and y each represent 2; R 11 represents H; k represents 0; suitably Z does not represent pyridine-5-yl-.
- Z when Z represents 2-methoxyphenyl-; B represents a methylene group; Ai and A 2 represent N; d and e each represent 2; x represents 1 and y represents 3, or x represents 2 and y represents 2 ; R 11 represents H; k represents 0; suitably G does not represent N-C(O)-phenyl or N-C(O)-cyclohexyl.
- B represents a methylene group
- Ai and A 2 represent N
- d and e each represent 2
- x represents 1 and y represents 3, or x represents 2 and y represents 2
- R 11 represents H
- k represents 0
- Z represents 3-(dimethylamino)phenyl-, 3-(acetamido)phenyl-, 2- methoxyphenyl-, pyrid-2-yl, suitably G does not represent N-(4-methylphenylsulphonyl-), N-(4- fluorophenylsulphonyl-) or N-(cyclohexylmethanesulphonyl-).
- alkyl as well as other groups having the prefix “alk” such as, for example, alkenyl, alkynyl, and the like, means carbon chains which may be linear or branched or combinations thereof. Examples of alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, sec- and tert-b ⁇ tyl, pentyl, hexyl, heptyl and the like. "Alkenyl”, “alkynyl” and other like terms include carbon chains having at least one unsaturated carbon- carbon bond.
- fluoroalkyl includes alkyl groups substituted by one or more fluorine atoms, e.g. CH 2 F, CHF 2 and CF 3 .
- cycloalkyl means carbocycles containing no heteroatoms, and includes monocyclic and bicyclic saturated and partially saturated carbocycles.
- examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl.
- Examples of partially saturated cycloalkyl groups include cyclohexene and indane. Cycloalkyl groups will typically contain 3 to 10 ring carbon atoms in total (e.g. 3 to 6, or 8 to 10).
- halo includes fluorine, chlorine, bromine, and iodine atoms (in particular fluorine or chlorine).
- aryl includes phenyl and naphthyl, in particular phenyl.
- heterocyclyl and “heterocyclic ring” includes 4- to 10-membered monocyclic and bicyclic saturated rings, e.g. 4- to 7-membered monocyclic saturated rings, containing up to three heteroatoms selected from N, O and S.
- heterocyclic rings examples include oxetane, tetrahydrofuran, tetrahydropyran, oxepane, oxocane, thietane, tetrahydrothiophene, tetrahydrothiopyran, thiepane, thiocane, azetidine, pyrrolidine, piperidine, azepane, azocane, [l,3]dioxane, oxazolidine, piperazine, and the like.
- Other examples of heterocyclic rings include the oxidised forms of the sulfur-containing rings.
- tetrahydrothiophene 1 -oxide tetrahydrothiophene 1,1 -dioxide
- tetrahydrothiopyran 1 -oxide tetrahydrothiopyran 1,1 -dioxide
- tetrahydrothiopyran 1,1 -dioxide tetrahydrothiophene 1,1 -dioxide
- heteroaryl includes mono- and bicyclic 5- to 10- membered, e.g. monocyclic 5- or 6-membered, heteroaryl rings containing up to 4 heteroatoms selected from N, O and S.
- heteroaryl rings are furyl, thienyl, pyrrolyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, triazolyl, oxadiazolyl, thiadiazolyl, tetrazolyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl and triazinyl.
- Bicyclic heteroaryl groups include bicyclic heteroaromatic groups where a 5- or 6-membered heteroaryl ring is fused to a phenyl or another heteroaromatic group.
- bicyclic heteroaromatic rings are benzofuran, benzothiophene, indole, benzoxazole, benzothiazole, indazole, benzimidazole, benzotriazole, quinoline, isoquinoline, quinazoline, quinoxaline and purine.
- Preferred heteroaryl groups are monocyclic 5- or 6-membered, heteroaryl rings containing up to 4 heteroatoms selected from N, O and S.
- Compounds described herein may contain one or more asymmetric centers and may thus give rise to diastereomers and optical isomers.
- the present invention includes all such possible diastereomers as well as their racemic mixtures, their substantially pure resolved enantiomers, all possible geometric isomers, and pharmaceutically acceptable salts thereof.
- the above formula (I) is shown without a definitive stereochemistry at certain positions.
- the present invention includes all stereoisomers of formula (I) and pharmaceutically acceptable salts thereof. Further, mixtures of stereoisomers as well as isolated specific stereoisomers are also included. During the course of the synthetic procedures used to prepare such compounds, or in using racemization or epimerization procedures known to those skilled in the art, the products of such procedures can be a mixture of stereoisomers.
- the present invention includes any possible tautomers and pharmaceutically acceptable salts thereof, and mixtures thereof, except where specifically drawn or stated otherwise.
- the invention includes all keto and enol forms which may be encompassed by the definition of B.
- the compound of formula (I) and pharmaceutically acceptable salts thereof exist in the form of solvates or polymorphic forms
- the present invention includes any possible solvates and polymorphic forms.
- a type of a solvent that forms the solvate is not particularly limited so long as the solvent is pharmacologically acceptable. For example, water, ethanol, propanol, acetone or the like can be used.
- salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids.
- pharmaceutically acceptable non-toxic bases including inorganic bases and organic bases.
- Salts derived from such inorganic bases include aluminum, ammonium, calcium, copper (ic and ous), ferric, ferrous, lithium, magnesium, potassium, sodium, zinc and the like salts. Particularly preferred are the ammonium, calcium, magnesium, potassium and sodium salts.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, as well as cyclic amines and substituted amines such as naturally occurring and synthesized substituted amines.
- organic non-toxic bases from which salts can be formed include arginine, betaine, caffeine, choline, N'JF- dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
- the compound of the present invention When the compound of the present invention is basic, its corresponding salt can be conveniently prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include, for example, acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid and the like
- the compounds of formula (I) are intended for pharmaceutical use they are preferably provided in substantially pure form, for example at least 60% pure, more suitably at least 75% pure, especially at least 98% pure (% are on a weight for weight basis).
- the compounds of formula (I) can be prepared as described below, wherein the groups Z, A 1 , A 2 , R 11 , R 12 , d, e, m, x, y, and G are as defined above.
- Compounds of formula (I) may be prepared from amine 4 and compound 5 where L is a leaving group such as mesylate/tosylate/halide with triethylamine, DIPEA or potassium carbonate. Where R 12 is oxo and adjacent to A 2 , sodium hydride is used as the base.
- compounds of formula 17 may be reacted with sulfonyl chlorides, carboxylic acid chlorides, and carbamyl chlorides Cl-R 1 , which are generally commercially available or can readily be synthesised, in a suitable solvent, such as CH 2 Cl 2 , in the presence of a suitable base, such as triethylamine, to afford compounds of formula (I) where R 1 is S(O) 2 R 5 , C(O)R 5 , and C(O)NR 5 R 8 , respectively.
- compounds of formula (I) in which R 1 is heteroaryl may be prepared by reacting the amine 12 with the appropriate heteroaryl chloride or bromide under Pd(O) catalysis in the presence of a suitable ligand and base (Urgaonkar, S.; Hu, J.-H.; Verkade, J. G. J. Org. Chent. 2003, 68, 8416-8423).
- compounds of the formula (I) where R 1 is heteroaryl may be prepared by condensation of amine 17 with a heteroaryl chloride in the presence of base (Barillari, C. et al. Eur. J. Org. Chem. 2001, 4737-4741; Birch, A. M. et al. J. Med. Chem. 1999, 42, 3342-3355).
- Z is cyanoaryl further modification may be carried out by treatment with hydroxylamine to give the amidoxime which can be condensed with acids to give oxadiazole examples.
- Z is the methylthioaryl further modification may be carried out by oxidation of the sulfide to the sulfoxide and sulfone, N-oxides can be isolated as a by-product of the sulfone oxidation.
- the compounds of formula (I) may be prepared singly or as compound libraries comprising at least 2, for example 5 to 1,000, compounds and more preferably 10 to 100 compounds of formula (I).
- Compound libraries may be prepared by a combinatorial "split and mix” approach or by multiple parallel synthesis using either solution or solid phase chemistry, using procedures known to those skilled in the art.
- labile functional groups in the intermediate compounds e.g. hydroxy, carboxy and amino groups
- the protecting groups may be removed at any stage in the synthesis of the compounds of formula (I) or may be present on the final compound of formula (I).
- a comprehensive discussion of the ways in which various labile functional groups may be protected and methods for cleaving the resulting protected derivatives is given in, for example, Protective Groups in Organic Chemistry, T.W. Greene and P.G.M. Wuts, (1991) Wiley-Interscience, New York, 2 nd edition. Any novel intermediates, such as those defined above, may be of use in the synthesis of formula (I) and are therefore also included within the scope of the invention, for example compounds of formula 12:
- the compounds of formula (I) are useful as GPRl 19 agonists, e.g. for the treatment and/or prophylaxis of obesity and diabetes.
- the compounds of formula (I) will generally be administered in the form of a pharmaceutical composition.
- the invention also provides a compound of formula (I), or a pharmaceutically acceptable salt thereof, for use as a pharmaceutical.
- the invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I), in combination with a pharmaceutically acceptable carrier.
- composition is comprised of a pharmaceutically acceptable carrier and a non-toxic therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- the invention also provides a pharmaceutical composition for the treatment of disease by modulating GPRl 19, resulting in the prophylactic or therapeutic treatment of obesity, e.g. by regulating satiety, or for the treatment of diabetes, comprising a pharmaceutically acceptable carrier and a non-toxic therapeutically effective amount of compound of formula (I), or a pharmaceutically acceptable salt thereof.
- compositions may optionally comprise other therapeutic ingredients or adjuvants.
- the compositions include compositions suitable for oral, rectal, topical, and parenteral (including subcutaneous, intramuscular, and intravenous) administration, although the most suitable route in any given case will depend on the particular host, and nature and severity of the conditions for which the active ingredient is being administered.
- the pharmaceutical compositions may be conveniently presented in unit dosage form and prepared by any of the methods well known in the art of pharmacy.
- the compounds of formula (I), or pharmaceutically acceptable salts thereof can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques.
- the carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g. oral or parenteral (including intravenous).
- compositions can be presented as discrete units suitable for oral administration such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient. Further, the compositions can be presented as a powder, as granules, as a solution, as a suspension in an aqueous liquid, as a non-aqueous liquid, as an oil-in- water emulsion, or as a water-in-oil liquid emulsion.
- the compound of formula (I), or a pharmaceutically acceptable salt thereof may also be administered by controlled release means and/or delivery devices.
- the compositions may be prepared by any of the methods of pharmacy.
- such methods include a step of bringing into association the active ingredient with the carrier that constitutes one or more necessary ingredients.
- the compositions are prepared by uniformly and intimately admixing the active ingredient with liquid carriers or finely divided solid carriers or both. The product can then be conveniently shaped into the desired presentation.
- the compounds of formula (I), or pharmaceutically acceptable salts thereof, can also be included in pharmaceutical compositions in combination with one or more other therapeutically active compounds.
- the pharmaceutical carrier employed can be, for example, a solid, liquid, or gas.
- solid carriers include lactose, terra alba, sucrose, talc, gelatin, agar, pectin, acacia, magnesium stearate, and stearic acid.
- liquid carriers are sugar syrup, peanut oil, olive oil, and water.
- gaseous carriers include carbon dioxide and nitrogen.
- oral liquid preparations such as suspensions, elixirs and solutions
- carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents, and the like may be used to form oral solid preparations such as powders, capsules and tablets.
- oral solid preparations such as powders, capsules and tablets.
- tablets and capsules are the preferred oral dosage units whereby solid pharmaceutical carriers are employed.
- tablets may be coated by standard aqueous or nonaqueous techniques.
- a tablet containing the composition of this invention may be prepared by compression or molding, optionally with one or more accessory ingredients or adjuvants.
- Compressed tablets may be prepared by compressing, in a suitable machine, the active ingredient in a free-flowing form such as powder or granules, optionally mixed with a binder, lubricant, inert diluent, surface active or dispersing agent. Molded tablets may be made by molding in a suitable machine, a mixture of the powdered compound moistened with an inert liquid diluent.
- Each tablet preferably contains from about 0.05mg to about 5g of the active ingredient and each cachet or capsule preferably containing from about 0.05mg to about 5g of the active ingredient.
- a formulation intended for the oral administration to humans may contain from about 0.5mg to about 5g of active agent, compounded with an appropriate and convenient amount of carrier material which may vary from about 5 to about 95 percent of the total composition.
- Unit dosage forms will generally contain between from about lmg to about 2g of the active ingredient, typically 25mg, 50mg, lOOmg, 200mg, 300mg, 400mg, 500mg, 600mg, 800mg, or lOOOmg.
- compositions of the present invention suitable for parenteral administration may be prepared as solutions or suspensions of the active compounds in water.
- a suitable surfactant can be included such as, for example, hydroxypropylcellulose.
- Dispersions can also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof in oils. Further, a preservative can be included to prevent the detrimental growth of microorganisms.
- compositions of the present invention suitable for injectable use include sterile aqueous solutions or dispersions.
- the compositions can be in the form of sterile powders for the extemporaneous preparation of such sterile injectable solutions or dispersions.
- the final injectable form must be sterile and must be effectively fluid for easy syringability.
- the pharmaceutical compositions must be stable under the conditions of manufacture and storage; thus, preferably should be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g. glycerol, propylene glycol and liquid polyethylene glycol), vegetable oils, and suitable mixtures thereof.
- compositions of the present invention can be in a form suitable for topical use such as, for example, an aerosol, cream, ointment, lotion, dusting powder, or the like. Further, the compositions can be in a form suitable for use in transdermal devices. These formulations may be prepared, using a compound of formula (I), or a pharmaceutically acceptable salt thereof, via conventional processing methods. As an example, a cream or ointment is prepared by admixing hydrophilic material and water, together with about 5wt% to about 10wt% of the compound, to produce a cream or ointment having a desired consistency. Pharmaceutical compositions of this invention can be in a form suitable for rectal administration wherein the carrier is a solid.
- the mixture forms unit dose suppositories.
- Suitable carriers include cocoa butter and other materials commonly used in the art.
- the suppositories may be conveniently formed by first admixing the composition with the softened or melted carrier(s) followed by chilling and shaping in molds.
- the pharmaceutical formulations described above may include, as appropriate, one or more additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface-active agents, thickeners, lubricants, preservatives (including anti-oxidants) and the like.
- additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface-active agents, thickeners, lubricants, preservatives (including anti-oxidants) and the like.
- additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface-active agents, thickeners, lubricants, preservatives (including anti-oxidants) and the like.
- additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface-active agents, thickeners, lubricants, preservatives (including anti-oxidants) and the like.
- other adjuvants can be included to render the formulation isotonic with the blood of the intended recipient
- dosage levels on the order of 0.01mg/kg to about 150mg/kg of body weight per day are useful in the treatment of the above-indicated conditions, or alternatively about 0.5mg to about 7g per patient per day.
- obesity may be effectively treated by the administration of from about 0.01 to 50mg of the compound per kilogram of body weight per day, or alternatively about 0.5mg to about 3.5g per patient per day.
- the compounds of formula (I) may be used in the treatment of diseases or conditions in which GPRl 19 plays a role.
- the invention also provides a method for the treatment of a disease or condition in which GPRl 19 plays a role comprising a step of administering to a subject in need thereof an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- Diseases or conditions in which GPRl 19 plays a role include obesity and diabetes.
- the treatment of obesity is intended to encompass the treatment of diseases or conditions such as obesity and other eating disorders associated with excessive food intake e.g. by reduction of appetite and body weight, maintenance of weight reduction and prevention of rebound and diabetes (including Type 1 and Type 2 diabetes, impaired glucose tolerance, insulin resistance and diabetic complications such as neuropathy, nephropathy, retinopathy, cataracts, cardiovascular complications and dyslipidaemia).
- the compounds of the invention may also be used for treating metabolic diseases such as metabolic syndrome (syndrome X), impaired glucose tolerance, hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels and hypertension.
- the compounds of the invention may offer advantages over compounds acting via different mechanisms for the treatment of the above mentioned disorders in that they may offer beta-cell protection, increased cAMP and insulin secretion and also slow gastric emptying.
- the invention also provides a method for the regulation of satiety comprising a step of administering to a subject in need thereof an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- the invention also provides a method for the treatment of obesity comprising a step of administering to a subject in need thereof an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- the invention also provides a method for the treatment of diabetes, including Type 1 and Type 2 diabetes, particularly type 2 diabetes, comprising a step of administering to a patient in need thereof an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- the invention also provides a method for the treatment of metabolic syndrome (syndrome X), impaired glucose tolerance, hyperlipidemia, hypertriglyceridemia, hypercholesterolemia, low HDL levels or hypertension comprising a step of administering to a patient in need thereof an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- metabolic syndrome sekunder X
- impaired glucose tolerance hyperlipidemia
- hypertriglyceridemia hypercholesterolemia
- low HDL levels or hypertension comprising a step of administering to a patient in need thereof an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof.
- the invention also provides a compound of formula (I), or a pharmaceutically acceptable salt thereof, for use in the treatment of a condition as defined above.
- the invention also provides the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of a condition as defined above.
- treatment includes both therapeutic and prophylactic treatment.
- the compounds of formula (I), or pharmaceutically acceptable salts thereof, may be administered alone or in combination with one or more other therapeutically active compounds.
- the other therapeutically active compounds may be for the treatment of the same disease or condition as the compounds of formula (I) or a different disease or condition.
- the therapeutically active compounds may be administered simultaneously, sequentially or separately.
- the compounds of formula (I) may be administered with other active compounds for the treatment of obesity and/or diabetes, for example insulin and insulin analogs, gastric lipase inhibitors, pancreatic lipase inhibitors, sulfonyl ureas and analogs, biguanides, ⁇ 2 agonists, glitazones, PPAR- ⁇ agonists, mixed PPAR- ⁇ / ⁇ agonists, RXR agonists, fatty acid oxidation inhibitors, ⁇ -glucosidase inhibitors, dipeptidyl peptidase IV inhibitors, GLP-I agonists e.g.
- active compounds for the treatment of obesity and/or diabetes for example insulin and insulin analogs, gastric lipase inhibitors, pancreatic lipase inhibitors, sulfonyl ureas and analogs, biguanides, ⁇ 2 agonists, glitazones, PPAR- ⁇ agonists, mixed PPAR- ⁇ / ⁇ agonists,
- GLP-I analogues and mimetics ⁇ -agonists, phosphodiesterase inhibitors, lipid lowering agents, glycogen phosphorylase inhibitors, antiobesity agents e.g. pancreatic lipase inhibitors, MCH-I antagonists and CB-I antagonists (or inverse agonists), amylin antagonists, lipoxygenase inhibitors, somostatin analogs, glucokinase activators, glucagon antagonists, insulin signalling agonists, PTPlB inhibitors, gluconeogenesis inhibitors, antilypolitic agents, GSK inhibitors, galanin receptor agonists, anorectic agents, CCK receptor agonists, leptin, serotonergic/dopaminergic antiobesity drugs, reuptake inhibitors e.g.
- sibutramine CRF antagonists, CRF binding proteins, thyromimetic compounds, aldose reductase inhibitors, glucocorticoid receptor antagonists, NHE-I inhibitors or sorbitol dehydrogenase inhibitors.
- Combination therapy comprising the administration of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and at least one other antiobesity agent represents a further aspect of the invention.
- the present invention also provides a method for the treatment of obesity in a mammal, such as a human, which method comprises administering an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and another antiobesity agent, to a mammal in need thereof.
- the invention also provides the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and another antiobesity agent for the treatment of obesity.
- the invention also provides the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for use in combination with another antiobesity agent, for the treatment of obesity.
- the compound of formula (I), or a pharmaceutically acceptable salt thereof, and the other antiobesity agent(s) may be co-administered or administered sequentially or separately.
- Co-administration includes administration of a formulation which includes both the compound of formula (I), or a pharmaceutically acceptable salt thereof, and the other antiobesity agent(s), or the simultaneous or separate administration of different formulations of each agent. Where the pharmacological profiles of the compound of formula (I), or a pharmaceutically acceptable salt thereof, and the other antiobesity agent(s) allow it, coadministration of the two agents may be preferred.
- the invention also provides the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and another antiobesity agent in the manufacture of a medicament for the treatment of obesity.
- the invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I), or a pharmaceutically acceptable salt thereof, and another antiobesity agent, and a pharmaceutically acceptable carrier.
- the invention also encompasses the use of such compositions in the methods described above.
- GPRl 19 agonists are of particular use in combination with centrally acting antiobesity agents.
- the other antiobesity agent for use in the combination therapies according to this aspect of the invention is preferably a CB-I modulator, e.g. a CB-I antagonist or inverse agonist.
- CB-I modulators include SR141716 (rimonabant) and SLV-319 ((4S)-(-)-3-(4- chlorophenyl)-N-methyl-N-[(4-chlorophenyl)sulfonyl]-4-phenyl-4,5-dihydro-lH-pyrazole-l- carboxamide); as well as those compounds disclosed in EP576357, EP656354, WO 03/018060, WO 03/020217, WO 03/020314, WO 03/026647, WO 03/026648, WO 03/027076, WO 03/040105, WO 03/051850, WO 03/051851, WO 03/053431, WO 03/063781, WO 03
- GPRl 19 diseases or conditions in which GPRl 19 has been suggested to play a role include those described in WO 00/50562 and US 6,468,756, for example cardiovascular disorders, hypertension, respiratory disorders, gestational abnormalities, gastrointestinal disorders, immune disorders, musculoskeletal disorders, depression, phobias, anxiety, mood disorders and Alzheimer's disease.
- All publications, including, but not limited to, patents and patent application cited in this specification, are herein incorporated by reference as if each individual publication were specifically and individually indicated to be incorporated by reference herein as fully set forth.
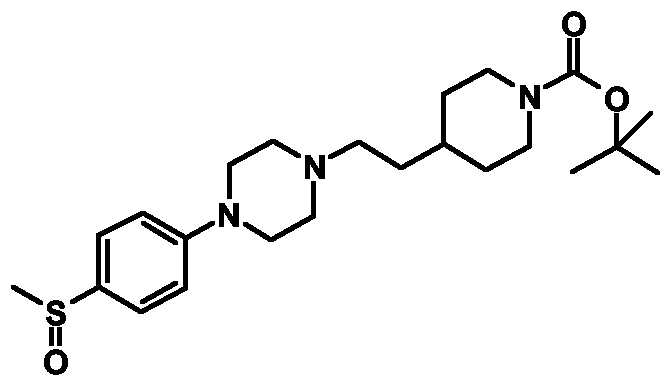
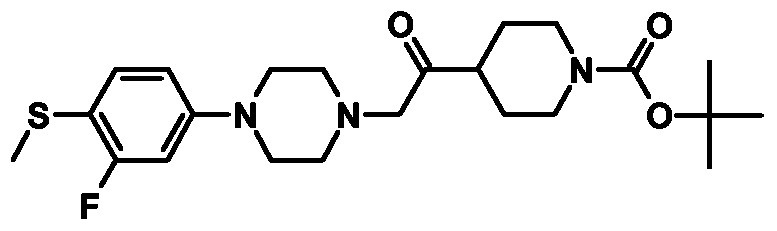
- Example 1 4-[4-(4-Mcthancsulfonylphcnyl)pipcrazin-l -ylmcthyljpipcridinc-l -carboxylic acid tert-butyl ester
- Example 12 4- ⁇ 2-[4-(5-Fluoro-2-methanesulfonylphenyl)piperazin-l-yl]ethyl ⁇ piperidinc- 1-carboxylic acid tert-butyl ester
- Example 13 4- ⁇ 2-[4-(4-Carboxyphenyl)piperazin-l-yl]ethyl ⁇ piperidine-l-carboxylic acid terf-butyl ester
- Example 14 4- ⁇ 2-[4-(4-Carbamoylphcnyl)pipcrazin-l-yl]cthyl ⁇ pipcridinc-l-carboxylic acid terf-butyl ester
- the adsorbed sample was purified by flash chromatography eluting with EtOAc to give 4- ⁇ 2-[4-(4- nitrophenyl)piperazin-l-yl]ethyl ⁇ cyclohexanecarboxylic acid tert-butyl ester (0.54 g, 1.30 mmol), which was taken up in EtOH (25 mL), 10% palladium on carbon was added and the mixture was stirred under an hydrogen atmosphere at rt for 2Oh.
- Example 26 4- ⁇ 2-[4-(4-Propionylaminophcnyl)pipcrazin-l-yl]cthyl ⁇ cyclo hcxanccarboxylic acid tert-butyl ester
- Example 32 4-[2-(4- ⁇ 4-[2-(2-Mcthoxycthoxy)acctylamino]phcnyl ⁇ pipcrazin-l- yl)ethyl]piperidine-l-carboxylic acid tert-butyl ester
- Example 37 4-(2- ⁇ 4-[4-(5-Methyl[l,2,4]oxadiazol-3-yl)phenyl]piperazin-l- yl ⁇ ethyl)piperidine-l-carboxylic acid tert-butyl ester
- Example 40 4-(2- ⁇ 4-[4-(3- ⁇ sopropyl[l,2,4]oxadiazol-5-yl)phcnyl]pipcrazin-l- yl ⁇ ethyl)piperidine-l-carboxylic acid terf-butyl ester
- Example 41 4-(2- ⁇ 4-[4-(3-Isopropyl[l,2,4]oxadiazol-5-yl)phenyl]piperazin-l- yl ⁇ ethyl)piperidine-l-carboxylic acid tert-butyl ester
- the adsorbed sample was purified by flash chromatography eluting with 5:95 MeOH:DCM to give 4-(2- ⁇ 4-[4-(N'-acetylhydrazino carbonyl)phenyl]piperazin-l-yl ⁇ ethyl)piperidine-l -carboxylic acid tert-butyl ester.
- Example 42 4-[l-(4-Methanesulfonylphenyl)piperidin-4-yloxymethyl] pipcridinc-1- carboxylic acid tert-butyl ester
- Example 43 4- ⁇ [l-(4-Mcthancsulfonylphcnyl)pipcridin-4-ylamino]mcthyl ⁇ pipcridinc-l- carboxylic acid tert-butyl ester
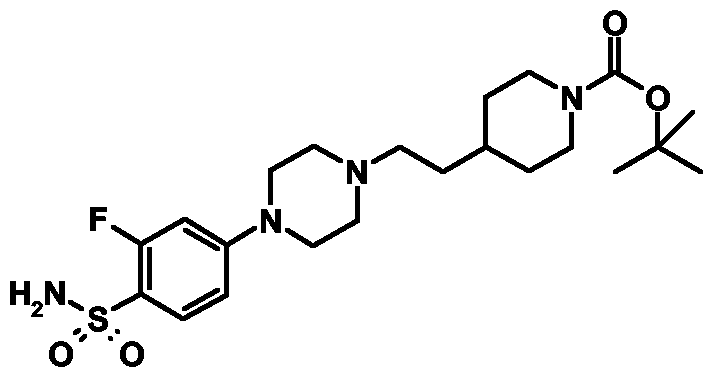
- Example 44 4- ⁇ 2-[4-(4-Sulfamoylphcnyl)pipcrazin-l-yl]cthyl ⁇ pipcridinc-l-carboxylic acid tert-butyl ester hydrochloride
- Example 45 4- ⁇ 2-[4-(3-Fluoro-4-sulfamoylphenyl)piperazin-l-yl]ethyl ⁇ piperidine-l- carboxylic acid tert-butyl ester
- Example 46 4- ⁇ 2-[4-(4-(Pyrrolidine-l-sulfonyl)phenyl)piperazin-l-yl]ethyl ⁇ piperidine-l- carboxylic acid tert-butyl ester
- Example 51 4- ⁇ 2-[4-(3-Fluoro-4-mcthancsulfonylphcnyl)pipcrazin-l-yl] cthyljpipcridinc- 1-carboxylic acid ferf-butyl ester
- Example 52 4- ⁇ 2-[4-(3-Fluoro-4-mcthancsulfonylphcnyl)pipcrazin-l-yl]cthyl ⁇ pipcridinc- 1-carboxylic acid terf-butyl ester
- Example 53 4- ⁇ 2-[4-(3-Fluoro-4-methanesulfonylphenyl)-l-oxypiperazin-l- yl]ethyl ⁇ piperidine-l-carboxylic acid ferf-butyl ester
- Example 54 4- ⁇ 2-[4-(4-Ethancsulfonylphcnyl)pipcrazin-l-yl]cthyl ⁇ pipcridinc-l- carboxylic acid ferf-butyl ester
- Example 55 4- ⁇ 2-[4-(4-Methanesulfonylphenyl)piperidin-l-yl]ethyl ⁇ piperidine-l- carboxylic acid tert-butyl ester
- Example 56 4- ⁇ 2-[4-(4-Methylsulfanylphenyl)piperidin-l-yl]ethyl ⁇ piperidine-l-carboxylic acid tert-butyl ester
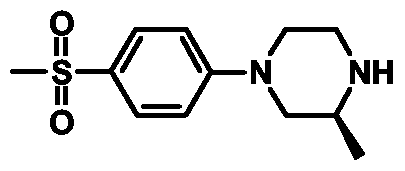
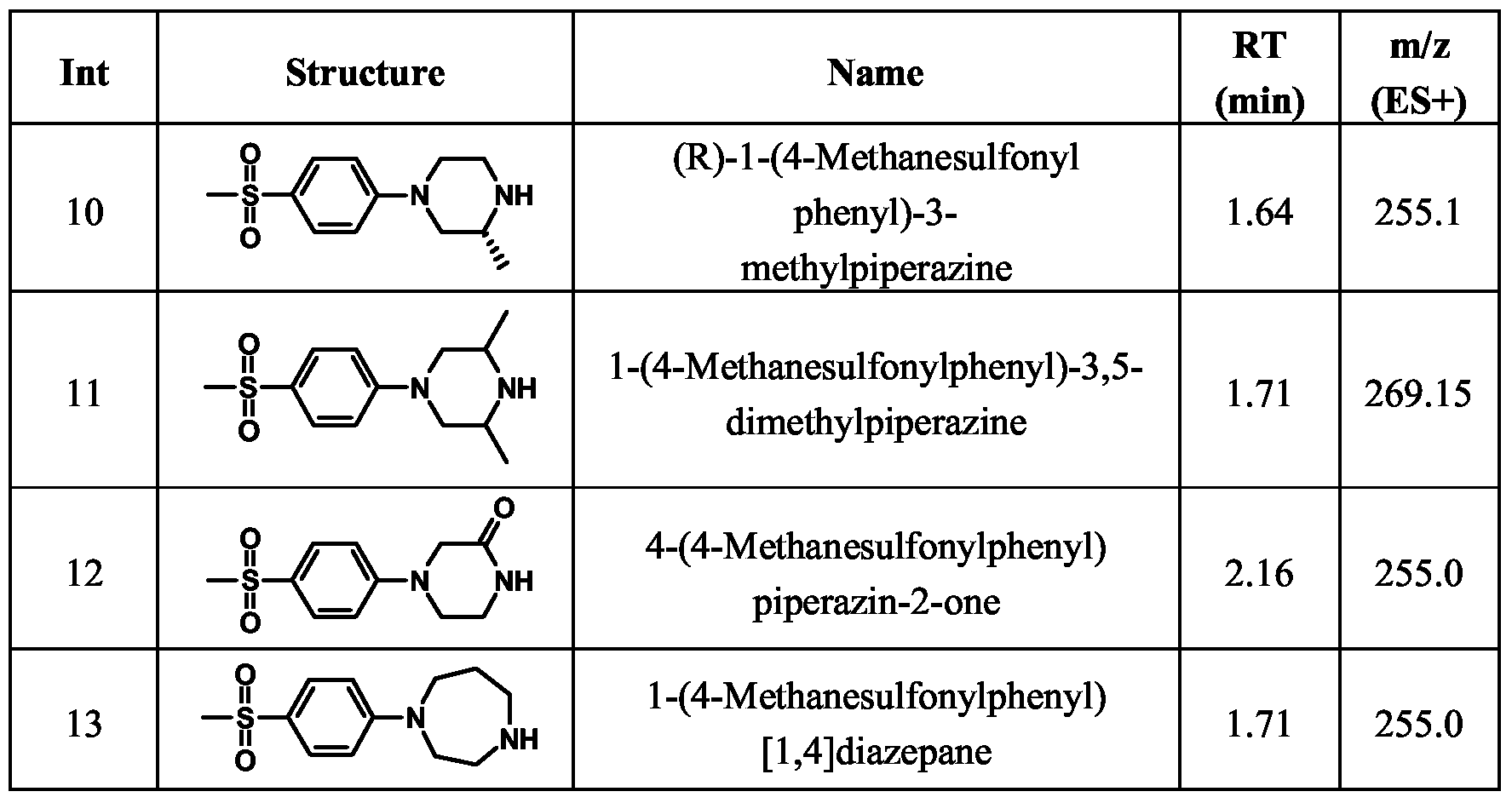
- Example 57 4- ⁇ 2-[(S)-4-(4-Methanesulfonylphenyl)-2-methylpiperazin-l-yl] cthyl ⁇ pipcridinc-l-carboxylic acid tert-butyl ester
- Example 62 4- ⁇ 2-[4-(4-Mcthancsulfonylphcnyl)-2,6-dimcthylpipcrazin-l- yl]ethyl ⁇ piperidine-l-carboxylic acid tert-butyl ester
- Example 63 2-[4-(4-mcthancsulfonylphcnyl)-2-oxopipcrazin-l-yl]cthyl ⁇ pipcridinc-l- carboxylic acid terf-butyl ester
- Example 64 4- ⁇ 2-[4-(4-Methylsulfanylphenyl)-3-oxopiperazin-l-yl]ethyl ⁇ piperidine-l- carboxylic acid tert-butyl ester
- Example 65 4- ⁇ 2-[4-(4-Methanesulfonylphenyl)-3-oxopiperazin-l-yl]ethyl ⁇ pipcridinc-1- carboxylic acid tert-butyl ester
- Example 66 4- ⁇ 2-[4-(4-Mcthancsulfinylphcnyl)-3-oxopipcrazin-l-yl]cthyl ⁇ piperidine-1-carboxylic acid ferf-butyl ester
- Example 68 4- ⁇ 2-[4-(3-Fluoro-4-methylsulfanylphenyl)piperazin-l-yl] acctyl ⁇ pipcridinc-l - carboxylic acid tert-butyl ester
- Example 69 4- ⁇ 2-[4-(3-Fluoro-4-methanesulfonylphenyl)piperazin-l-yl] acctyl ⁇ pipcridinc-l -carboxylic acid tert-butyl ester
- Example 71 4- ⁇ l,l-Difluoro-2-[4-(4-mcthancsulfonylphcnyl)pipcrazin-l-yl] ethyljpiperidine-l-carboxylic acid tert-butyl ester
- Example 72 4- ⁇ l,l-Difluoro-2-[4-(3-fluoro-4-methanesulfonylphenyl)piperazin-l- yl]ethyl ⁇ piperidine-l-carboxylic acid terf-butyl ester
- Example 73 4- ⁇ l-Hydroxy-2-[4-(4-methanesulfonylphenyl)piperaziii-l-yl] ethyl ⁇ piperidine-l-carboxylic acid terf-butyl ester
- Example 74 4- ⁇ l-Chloro-2-[4-(4-mcthancsulfonylphcnyl)pipcrazin-l-yl] ethyljpiperidine-l-carboxylic acid ferf-butyl ester
- Example 75 4- ⁇ l-Fluoro-2-[4-(4-methanesulfonylphenyl)piperazin-l-yl]ethyl ⁇ piperidine- 1-carboxylic acid ferf-butyl ester
- Example 76 4- ⁇ 2-Fluoro-l-[4-(4-methanesulfonylphenyl)piperazin-l-yl]ethyl ⁇ piperidine-l- carboxylic acid tert-butyl ester
- Example 77 4- ⁇ 2-[4-(4-Methanesulfonylphenyl)piperazin-l-yl]ethylidene ⁇ piperidine-l- carboxylic acid tert-butyl ester
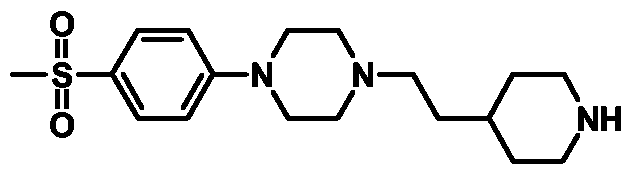
- This suspension was thus added to a preformed mixture of l-(4-methanesulfonylphenyl)piperazine (0.487g, 2.03mmol) and sodium hydride (0.1 Ig of a 60% dispersion in mineral oil, 2.77mmol) in DMF (5mL) at rt.
- the mixture was allowed to stir for 2h then treated with water.
- the aqueous was extracted with EtOAc and the combined organic layers washed with water, brine, dried (MgSO 4 ) and the solvent removed in vacuo.
- Example 78 4- ⁇ 2-[4-Hydroxy-4-(4-methylsulfanylphenyl)piperidin-l-yl] cthyljpipcridinc- 1-carboxylic acid terf-butyl ester
- Example 79 4- ⁇ 2-[4-Hydroxy-4-(4-methanesulfonylphenyl)piperidin-l-yl] ethyl ⁇ piperidine-l-carboxylic acid terf-butyl ester
- Example 80 4- ⁇ 2-[4-(4-Mcthancsulfonylphcnyl)pipcrazin-l-yl]-2-oxocthyl ⁇ pipcridinc-l- carboxylic acid ferf-butyl ester
- Example 81 4- ⁇ 2-[4-(4-Mcthancsulfonylphcnyl)pipcrazin-l-yl]-2-oxocthyl ⁇ pipcridinc-l- carboxylic acid tert-butyl ester
- Example 82 4- ⁇ 2-[4-(6-Chloropyridin-3-yl)piperazin-l-yl]ethyl ⁇ piperidine -1-carboxylic acid terf-butyl ester
- Example 83 4- ⁇ 2-[4-(6-Methanesulfonylpyridin-3-yl)piperazin-l-yl]ethyl ⁇ piperidine-l- carboxylic acid terf-butyl ester
- the solid was dissolved in DCM (8mL) and treated with N-boc-piperidinyl-4-acetaldehyde (0.088g, 0.39 mmol) and 4A molecular sieves (O.lg) at room temperature. The solution was allowed to stir for 3h then treated with NaHB(OAc) 3 (0.106g, 0.5 mmol). The resulting solution was stirred at rt for 2Oh. DCM (2OmL) was added and the organic layer washed with saturated Na 2 CO 3 solution, brine, dried (MgSO 4 ) and the solvent removed in vacuo.
- Example 84 4- ⁇ 2-[4-(5-Methanesulfonylpyridin-2-yl)piperazin-l-yl]ethyl ⁇ piperidine-l- carboxylic acid tert-butyl ester
- Example 86 4- ⁇ 2-[4-(4-Methanesulfonylphenyl)piperazin-l-yl]ethyl ⁇ piperidine-l- carboxylic acid isopropyl ester
- Example 88 2-(4- ⁇ 2-[4-(4-Mcthancsulfonylphcnyl)pipcrazin-l-yl]cthyl ⁇ pipcridin-l-yl)-5- mcthylpyrimidinc
- Example 85 To a degassed solution of Example 85 (0.2g, 0.57mmol), 2-chloro-5-fluoro pyrimidine (0.076g, 0.57mmol), sodium tert-butoxide (0.082g, 0.86mmol), 9,9-dimethyl-4,5- bis(diphenylphosphino)xanthene (0.02g, 0.032mmol) in toluene was added Pd 2 (dba) 3 (0.01 Ig, 0.01 mmol). The mixture was heated at reflux for 2h, cooled and the solvent removed in vacuo.
- the biological activity of the compounds of the invention may be tested in the following assay systems: Yeast Reporter Assay
- yeast cell-based reporter assays have previously been described in the literature (e.g. see Miret J. J. et al, 2002, J. Biol. Chem., 277:6881-6887; Campbell R.M. et al, 1999, Bioorg. Med. Chem. Lett., 9:2413-2418; King K. et al, 1990, Science, 250:121-123); WO 99/14344; WO 00/12704; and US 6,100,042).
- yeast cells have been engineered such that the endogenous yeast G-alpha (GPAl) has been deleted and replaced with G-protein chimeras constructed using multiple techniques.
- yeast GPCR Ste3 has been deleted to allow for heterologous expression of a mammalian GPCR of choice.
- elements of the pheromone signaling transduction pathway which are conserved in eukaryotic cells (for example, the mitogen-activated protein kinase pathway), drive the expression of Fusl.
- ⁇ -galactosidase LacZ
- Fuslp Fusl promoter
- Yeast cells were transformed by an adaptation of the lithium acetate method described by Agatep et al, (Agatep, R. et al, 1998, Transformation of Saccharomyces cerevisiae by the lithium acetate/single-stranded carrier DNA/polyethylene glycol (LiAc/ss-DNA/PEG) protocol. Technical Tips Online, Trends Journals, Elsevier). Briefly, yeast cells were grown overnight on yeast tryptone plates (YT).
- Carrier single-stranded DNA (lO ⁇ g), 2 ⁇ g of each of two Fuslp- LacZ reporter plasmids (one with URA selection marker and one with TRP), 2 ⁇ g of GPRl 16 (human or mouse receptor) in yeast expression vector (2 ⁇ g origin of replication) and a lithium acetate/ polyethylene glycol/ TE buffer was pipetted into an Eppendorf tube.
- the yeast expression plasmid containing the receptor/ no receptor control has a LEU marker.
- Yeast cells were inoculated into this mixture and the reaction proceeds at 3O 0 C for 60min.
- the yeast cells were then heat-shocked at 42 0 C for 15min.
- the cells were then washed and spread on selection plates.
- the selection plates are synthetic defined yeast media minus LEU, URA and TRP (SD- LUT). After incubating at 3O 0 C for 2-3 days, colonies that grow on the selection plates were then tested in the LacZ assay.
- yeast cells carrying the human or mouse GPRl 16 receptor were grown overnight in liquid SD-LUT medium to an unsaturated concentration (i.e. the cells were still dividing and had not yet reached stationary phase). They were diluted in fresh medium to an optimal assay concentration and 90 ⁇ l of yeast cells added to 96-well black polystyrene plates (Costar). Compounds, dissolved in DMSO and diluted in a 10% DMSO solution to 1OX concentration, were added to the plates and the plates placed at 3O 0 C for 4h. After 4h, the substrate for the ⁇ -galactosidase was added to each well.
- Fluorescein di ⁇ -D-galactopyranoside
- FDG Fluorescein di
- a substrate for the enzyme that releases fluorescein allowing a fluorimetric read-out.
- 20 ⁇ l per well of 500 ⁇ M FDG/2.5% Triton XlOO was added (the detergent was necessary to render the cells permeable).
- 20 ⁇ l per well of IM sodium carbonate was added to terminate the reaction and enhance the fluorescent signal.
- the plates were then read in a fluorimeter at 485/535nm.
- the compounds of the invention give an increase in fluorescent signal of at least ⁇ 1.5- fold that of the background signal (i.e. the signal obtained in the presence of 1% DMSO without compound).
- Compounds of the invention which give a increase of at least 5-fold that of the background signal may be preferred.
- cyclic AMP cyclic AMP
- the cell monolayers were washed with phosphate buffered saline and stimulated at 37°C for 30min with various concentrations of compound in stimulation buffer plus 1% DMSO. Cells were then lysed and cAMP content determined using the Perkin Elmer AlphaScreenTM (Amplified Luminescent Proximity Homogeneous Assay) cAMP kit. Buffers and assay conditions were as described in the manufacturer's protocol. Compounds of the invention showed a concentration-dependant increase in intracellular cAMP level.
- Compounds of the invention produced a concentration-dependent increase in intracellular cAMP level and generally had an EC 50 of ⁇ 10 ⁇ M. Compounds showing an EC 50 of less than lum in the cAMP assay may be preferred.
- Test compounds and reference compounds are dosed by appropriate routes of administration (e.g. intraperitoneally or orally) and measurements made over the following 24 h.
- Rats are individually housed in polypropylene cages with metal grid floors at a temperature of 21 ⁇ 4°C and 55 ⁇ 20% humidity. Polypropylene trays with cage pads are placed beneath each cage to detect any food spillage. Animals are maintained on a reverse phase light-dark cycle (lights off for 8 h from 09.30-17.30 h) during which time the room was illuminated by red light.
- Animals have free access to a standard powdered rat diet and tap water during a two week acclimatization period.
- the diet is contained in glass feeding jars with aluminum lids. Each lid has a 3-4 cm hole in it to allow access to the food.
- Animals, feeding jars and water bottles are weighed (to the nearest 0.1 g) at the onset of the dark period. The feeding jars and water bottles are subsequently measured 1, 2, 4, 6 and 24 h after animals are dosed with a compound of the invention and any significant differences between the treatment groups at baseline compared to vehicle-treated controls.
- HIT-T15 cells (passage 60) were obtained from ATCC, and were cultured in RPMI1640 medium supplemented with 10% fetal calf serum and 3OnM sodium selenite. All experiments were done with cells at less than passage 70, in accordance with the literature, which describes altered properties of this cell line at passage numbers above 81 (Zhang HJ, Walseth TF, Robertson RP. Insulin secretion and cAMP metabolism in HIT cells. Reciprocal and serial passage-dependent relationships. Diabetes. 1989 Jan;38(l):44-8).
- cAMP assay HIT-T 15 cells were plated in standard culture medium in 96-well plates at 100,000 cells/ 0.1ml/ well and cultured for 24 hr and the medium was then discarded. Cells were incubated for 15min at room temperature with lOO ⁇ l stimulation buffer (Hanks buffered salt solution, 5mM HEPES, 0.5mM IBMX, 0.1% BSA, pH 7.4). This was discarded and replaced with compound dilutions over the range 0.001, 0.003, 0.01, 0.03, 0.1, 0.3, 1, 3, 10, 30 ⁇ M in stimulation buffer in the presence of 0.5% DMSO. Cells were incubated at room temperature for 30min.
- lOO ⁇ l stimulation buffer Hors buffered salt solution, 5mM HEPES, 0.5mM IBMX, 0.1% BSA, pH 7.4
- 75ul lysis buffer (5mM HEPES, 0.3% Tween-20, 0.1% BSA, pH 7.4) was added per well and the plate was shaken at 900 rpm for 20 min. Particulate matter was removed by centrifugation at 3000rpm for 5min, then the samples were transferred in duplicate to 384-well plates, and processed following the Perkin Elmer AlphaScreen cAMP assay kit instructions. Briefly 25 ⁇ l reactions were set up containing 8 ⁇ l sample, 5 ⁇ l acceptor bead mix and 12 ⁇ l detection mix, such that the concentration of the final reaction components is the same as stated in the kit instructions. Reactions were incubated at room temperature for 150min, and the plate was read using a Packard Fusion instrument.
- Measurements for cAMP were compared to a standard curve of known cAMP amounts (0.01, 0.03, 0.1, 0.3, 1, 3, 10, 30, 100, 300, 1000 nM) to convert the readings to absolute cAMP amounts. Data was analysed using XLfit 3 software.
- Representative compounds of the invention were found to increase cAMP at an EC 50 of less than 10 ⁇ M. Compounds showing an EC 50 of less than 1 ⁇ M in the cAMP assay may be preferred.
- HIT-T 15 cells were plated in standard culture medium in 12- well plates at 106 cells/ 1 ml/ well and cultured for 3 days and the medium was then discarded. Cells were washed x 2 with supplemented Krebs-Ringer buffer (KRB) containing 119 mM NaCl, 4.74 mM KCl, 2.54 mM CaCl 2 , 1.19 mM MgSO 4 , 1.19 mM KH2PO4, 25 mM NaHCO 3 , 1OmM HEPES at pH 7.4 and 0.1% bovine serum albumin. Cells were incubated with ImI KRB at 37°C for 30 min which was then discarded.
- KRB Krebs-Ringer buffer
- mice The effects of compounds of the invention on oral glucose (GIc) tolerance may be evaluated in male C57B1/6 or male oblob mice.
- Food is withdrawn 5 h before administration of GIc and remains withdrawn throughout the study. Mice have free access to water during the study.
- a cut is made to the animals' tails, then blood (20 ⁇ L) is removed for measurement of basal GIc levels 45 min before administration of the GIc load.
- the mice are weighed and dosed orally with test compound or vehicle (20% aqueous hydroxypropyl- ⁇ -cyclodextrin or 25% aqueous Gelucire 44/14) 30 min before the removal of an additional blood sample (20 ⁇ L) and treatment with the GIc load (2-5 g kg "1 p.o.).
- Blood samples (20 ⁇ L) are taken 25, 50, 80, 120, and 180 min after GIc administration.
- the 20 ⁇ L blood samples for measurement of GIc levels are taken from the cut tip of the tail into disposable micro-pipettes (Dade Diagnostics Inc., Puerto Rico) and the sample added to 480 ⁇ L of haemolysis reagent.
- Duplicate 20 ⁇ L aliquots of the diluted haemolysed blood are added to 180 ⁇ L of Trinders glucose reagent (Sigma enzymatic (Trinder) colorimetric method) in a 96-well assay plate. After mixing, the samples are left at rt for 30 min before being read against GIc standards (Sigma glucose/urea nitrogen combined standard set).
Abstract
Description























































































Claims



Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2006264651A AU2006264651A1 (en) | 2005-06-30 | 2006-06-30 | G-protein coupled receptor agonists |
| JP2008520009A JP2008545010A (en) | 2005-06-30 | 2006-06-30 | G protein-coupled receptor agonist |
| MX2007016545A MX2007016545A (en) | 2005-06-30 | 2006-06-30 | G-protein coupled receptor agonists. |
| EP06744362A EP1909791A1 (en) | 2005-06-30 | 2006-06-30 | G-protein coupled receptor agonists |
| BRPI0612599-9A BRPI0612599A2 (en) | 2005-06-30 | 2006-06-30 | compound, pharmaceutical composition comprising the same, method of treatment and use thereof |
| US11/922,826 US20090203676A1 (en) | 2005-06-30 | 2006-06-30 | G-protein Coupled Receptor Agonists |
| CA002613236A CA2613236A1 (en) | 2005-06-30 | 2006-06-30 | G-protein coupled receptor agonists |
| NZ564758A NZ564758A (en) | 2005-06-30 | 2006-06-30 | G-protein coupled receptor agonists |
| NO20080052A NO20080052L (en) | 2005-06-30 | 2008-01-04 | G-Protein Coupled Receptor Agonists |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0513276A GB0513276D0 (en) | 2005-06-30 | 2005-06-30 | Compounds |
| GB0513276.6 | 2005-06-30 | ||
| GB0612897A GB0612897D0 (en) | 2006-06-29 | 2006-06-29 | GPCR agonists |
| GB0612897.9 | 2006-06-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007003964A1 true WO2007003964A1 (en) | 2007-01-11 |
Family
ID=37057179
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2006/050182 WO2007003964A1 (en) | 2005-06-30 | 2006-06-30 | G-protein coupled receptor agonists |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US20090203676A1 (en) |
| EP (1) | EP1909791A1 (en) |
| JP (1) | JP2008545010A (en) |
| KR (1) | KR20080025190A (en) |
| AU (1) | AU2006264651A1 (en) |
| BR (1) | BRPI0612599A2 (en) |
| CA (1) | CA2613236A1 (en) |
| MX (1) | MX2007016545A (en) |
| NO (1) | NO20080052L (en) |
| NZ (1) | NZ564758A (en) |
| WO (1) | WO2007003964A1 (en) |
Cited By (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007120702A2 (en) | 2006-04-11 | 2007-10-25 | Arena Pharmaceuticals, Inc. | Use of gpr119 receptor agonists for increasing bone mass and for treating osteoporosis, and combination therapy relating thereto |
| WO2007130712A1 (en) * | 2006-01-31 | 2007-11-15 | Janssen Pharmaceutica, Nv | Substituted dipiperidine as ccr2 antagonists for the treatment of inflammatory diseases |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2008081205A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081208A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081204A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081207A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081206A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008085316A1 (en) * | 2006-12-20 | 2008-07-17 | Merck & Co., Inc. | Bipiperidinyl compounds, compositions containing such compounds and methods of treatment |
| JP2008239623A (en) * | 2005-01-10 | 2008-10-09 | Arena Pharmaceuticals Inc | Combination therapy for treating diabetes and condition related thereto and for treating condition ameliorated by increasing blood glp-1 level |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2009034388A1 (en) | 2007-09-10 | 2009-03-19 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| WO2009038974A1 (en) | 2007-09-20 | 2009-03-26 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| WO2009050523A1 (en) | 2007-10-18 | 2009-04-23 | Prosidion Limited | Azetidinyl g-protein coupled receptor agonists |
| WO2009050522A1 (en) | 2007-10-18 | 2009-04-23 | Prosidion Limited | Azetidinyl g-protein coupled receptor agonists |
| WO2009105722A1 (en) * | 2008-02-22 | 2009-08-27 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| JP2009532453A (en) * | 2006-04-06 | 2009-09-10 | プロシディオン・リミテッド | Heterocyclic GPCR agonist |
| EP2108960A1 (en) | 2008-04-07 | 2009-10-14 | Arena Pharmaceuticals, Inc. | Methods of using A G protein-coupled receptor to identify peptide YY (PYY) secretagogues and compounds useful in the treatment of conditons modulated by PYY |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2010084512A1 (en) | 2008-12-24 | 2010-07-29 | Cadila Healthcare Limited | Novel oxime derivatives |
| WO2010103333A1 (en) | 2009-03-12 | 2010-09-16 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| WO2010103334A1 (en) | 2009-03-12 | 2010-09-16 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| WO2010103335A1 (en) | 2009-03-12 | 2010-09-16 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| WO2011002067A1 (en) * | 2009-07-02 | 2011-01-06 | 武田薬品工業株式会社 | Heterocyclic compound and use thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| JP2011513232A (en) * | 2008-02-22 | 2011-04-28 | アイアールエム・リミテッド・ライアビリティ・カンパニー | Compounds and compositions as modulators of GPR119 activity |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011113947A1 (en) | 2010-03-18 | 2011-09-22 | Boehringer Ingelheim International Gmbh | Combination of a gpr119 agonist and the dpp-iv inhibitor linagliptin for use in the treatment of diabetes and related conditions |
| WO2011128394A1 (en) | 2010-04-14 | 2011-10-20 | Prosidion Limited | 3-substituted 5-(pyrrolidine-1-carbonyl) pyrrolidine and its derivatives for use in the treatment of metabolic disorders |
| WO2011128395A1 (en) | 2010-04-14 | 2011-10-20 | Prosidion Limited | N- substituted 3-amino 4 - ( pyrrolidine - 1 - carbonyl) pyrrolidine and its derivatives for use in the treatment of metabolic disorders |
| WO2011147951A1 (en) | 2010-05-28 | 2011-12-01 | Prosidion Limited | Cycloamino derivatives as gpr119 antagonists |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| EP2402750A1 (en) | 2006-04-11 | 2012-01-04 | Arena Pharmaceuticals, Inc. | Methods of using GPR119 receptor to identify compounds useful for increasing bone mass in an individual |
| US8093248B2 (en) | 2007-12-05 | 2012-01-10 | Astrazeneca Ab (Publ) | Compounds useful for the treatment of conditions associated with weight gain |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012046249A1 (en) | 2010-10-08 | 2012-04-12 | Cadila Healthcare Limited | Novel gpr 119 agonists |
| WO2012066077A1 (en) | 2010-11-18 | 2012-05-24 | Prosidion Limited | 1,4 di substituted pyrrolidine - 3 - yl -amine derivatives and their use for the treatment of metabolic disorders |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| CN102686579A (en) * | 2009-10-09 | 2012-09-19 | Irm责任有限公司 | Compounds and compositions as modulators of GPR119 activity |
| US8293729B2 (en) | 2009-06-24 | 2012-10-23 | Boehringer Ingelheim International Gmbh | Compounds, pharmaceutical composition and methods relating thereto |
| WO2012170702A1 (en) * | 2011-06-08 | 2012-12-13 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2012170867A1 (en) | 2011-06-09 | 2012-12-13 | Rhizen Pharmaceuticals Sa | Novel compounds as modulators of gpr-119 |
| US8343990B2 (en) | 2008-04-14 | 2013-01-01 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment |
| WO2013026587A1 (en) | 2011-08-22 | 2013-02-28 | Prosidion Limited | 1,4 disubstituted pyrrolidine - 3 - yl -amine derivatives and their use for the treatment of metabolic disorders |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013066869A1 (en) * | 2011-11-03 | 2013-05-10 | Array Biopharma Inc. | Piperidinyl-substituted lactams as gpr119 modulators |
| US8481731B2 (en) | 2009-06-24 | 2013-07-09 | Boehringer Ingelheim International Gmbh | Compounds, pharmaceutical composition and methods relating thereto |
| US8754226B2 (en) | 2010-05-17 | 2014-06-17 | Array Biopharma Inc. | Piperidinyl-substituted lactams as GPR119 modulators |
| US8933083B2 (en) | 2003-01-14 | 2015-01-13 | Arena Pharmaceuticals, Inc. | 1,2,3-trisubstituted aryl and heteroaryl derivatives as modulators of metabolism and the prophylaxis and treatment of disorders related thereto such as diabetes and hyperglycemia |
| US8957062B2 (en) | 2011-04-08 | 2015-02-17 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment |
| US9006228B2 (en) | 2011-06-16 | 2015-04-14 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds, and methods of treatment |
| US9018200B2 (en) | 2011-10-24 | 2015-04-28 | Merck Sharp & Dohme Corp. | Substituted piperidinyl compounds useful as GPR119 agonists |
| US9018224B2 (en) | 2011-11-15 | 2015-04-28 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds useful as GPR119 agonists |
| US9422266B2 (en) | 2011-09-30 | 2016-08-23 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment |
| US9452990B2 (en) | 2012-06-20 | 2016-09-27 | Novartis Ag | Complement pathway modulators and uses thereof |
| US10894787B2 (en) | 2010-09-22 | 2021-01-19 | Arena Pharmaceuticals, Inc. | Modulators of the GPR119 receptor and the treatment of disorders related thereto |
| US11007175B2 (en) | 2015-01-06 | 2021-05-18 | Arena Pharmaceuticals, Inc. | Methods of treating conditions related to the S1P1 receptor |
| US11534424B2 (en) | 2017-02-16 | 2022-12-27 | Arena Pharmaceuticals, Inc. | Compounds and methods for treatment of primary biliary cholangitis |
| US11884626B2 (en) | 2015-06-22 | 2024-01-30 | Arena Pharmaceuticals, Inc. | Crystalline L-arginine salt of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclo-penta [b]indol-3-yl)acetic acid(Compound1) for use in S1P1 receptor-associated disorders |
| WO2024072930A1 (en) * | 2022-09-30 | 2024-04-04 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Dopamine d3/d2 receptor partial agonists for the treatment of neuropsychiatric disorders |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG10201500639TA (en) | 2010-01-27 | 2015-03-30 | Arena Pharm Inc | Processes for the preparation of (r)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid and salts thereof |
| WO2012145361A1 (en) * | 2011-04-19 | 2012-10-26 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2013062837A1 (en) * | 2011-10-24 | 2013-05-02 | Merck Sharp & Dohme Corp. | Piperidine derivatives useful as gpr119 agonists |
| WO2016040554A1 (en) * | 2014-09-10 | 2016-03-17 | Temple University-Of The Commonwealth System Of Higher Education | Novel 5-hydroxytryptamine receptor 7 activity modulators and their method of use |
| US10568882B2 (en) | 2015-08-21 | 2020-02-25 | Srx Cardio, Llc | Phenylpiperazine proprotein convertase subtilisin/kexin type 9 (PCSK9) modulators and their use |
| EP3337564A4 (en) | 2015-08-21 | 2019-01-23 | Portola Pharmaceuticals, Inc. | Composition and methods of use of tetrahydroisoquinoline small molecules to bind and modulate pcsk9 protein activity |
| US10821106B2 (en) | 2015-08-21 | 2020-11-03 | Srx Cardio, Llc | Composition and methods of use of novel phenylalanine small organic compounds to directly modulate PCSK9 protein activity |
| WO2017147328A1 (en) | 2016-02-23 | 2017-08-31 | Portola Pharmaceuticals, Inc. | Compounds for binding proprotein convertase subtilisin/kexin type 9 (pcsk9) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005007647A1 (en) * | 2003-07-11 | 2005-01-27 | Arena Pharmaceuticals, Inc. | Trisubstituted aryl and heteroaryl derivatives as modulators of metabolism and the prophylaxis and treatment of disorders related thereto |
| WO2006067531A1 (en) * | 2004-12-24 | 2006-06-29 | Prosidion Ltd | G-protein coupled receptor (gpr116) agonists and use thereof for treating obesity and diabetes |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3332949A (en) * | 1962-11-13 | 1967-07-25 | Sterling Drug Inc | 1-phenyl-4-[2-(2-pyridyl)-ethyl] piperazines |
| US3133061A (en) * | 1962-11-13 | 1964-05-12 | Sterling Drug Inc | Piperidine carboxamides and derivatives thereof |
| FR2285883A1 (en) * | 1974-09-30 | 1976-04-23 | Cerm Cent Europ Rech Mauvernay | NEW CHEMICAL COMPOUNDS WITH BRONCHOSPASMOLYTIC AND ANTITUSSIVE ACTIVITY, AND PROCESS FOR OBTAINING THEM |
| US5229398A (en) * | 1990-02-27 | 1993-07-20 | Adir Et Compagnie | Aminomethylpiperidine compounds |
| GB9516709D0 (en) * | 1995-08-15 | 1995-10-18 | Zeneca Ltd | Medicament |
| DE19614204A1 (en) * | 1996-04-10 | 1997-10-16 | Thomae Gmbh Dr K | Carboxylic acid derivatives, medicaments containing these compounds, their use and processes for their preparation |
| ATE230399T1 (en) * | 1996-08-14 | 2003-01-15 | Astrazeneca Ab | SUBSTITUTED PYRIMIDINE DERIVATIVES AND THEIR PHARMACEUTICAL USE |
| PT966462E (en) * | 1997-02-13 | 2003-10-31 | Astrazeneca Ab | UTERIC HETEROCYCLICAL COMPOUNDS AS INHIBITORS OF THE OXIDE-ESKALENE CYCLASE |
| AU5999698A (en) * | 1997-02-13 | 1998-09-08 | Zeneca Limited | Heterocyclic compounds useful as oxido-squalene cyclase inhibitors |
| CA2294344A1 (en) * | 1997-06-18 | 1998-12-23 | Michael A. Patane | Alpha 1a adrenergic receptor antagonists |
| US6255302B1 (en) * | 1998-12-17 | 2001-07-03 | American Home Products Corporation | 1,4-piperazine derivatives |
| KR20030024799A (en) * | 2000-07-20 | 2003-03-26 | 뉴로젠 코포레이션 | Capsaicin receptor ligands |
| PE20030385A1 (en) * | 2001-07-26 | 2003-05-08 | Schering Corp | UREA COMPOUNDS AS ANTAGONISTS OF NEUROPEPTIDE Y5 RECEPTORS AND |
| ES2197003B1 (en) * | 2002-04-08 | 2005-03-16 | J. URIACH & CIA S.A. | NEW ANTAGONIST COMPOUNDS OF INTEGRINAS ALFA. |
| AR047759A1 (en) * | 2003-09-26 | 2006-02-22 | Vertex Pharma | FENIL DERIVATIVES - PIPERAZINE AS MODULATORS OF MUSCARNIC RECEPTORS |
| DK2607362T3 (en) * | 2005-02-17 | 2015-01-19 | Astellas Pharma Inc | Piperidine and piperazine carboxylates as FAAH inhibitors |
-
2006
- 2006-06-30 NZ NZ564758A patent/NZ564758A/en not_active IP Right Cessation
- 2006-06-30 JP JP2008520009A patent/JP2008545010A/en active Pending
- 2006-06-30 CA CA002613236A patent/CA2613236A1/en not_active Abandoned
- 2006-06-30 EP EP06744362A patent/EP1909791A1/en not_active Withdrawn
- 2006-06-30 KR KR1020087002531A patent/KR20080025190A/en not_active Application Discontinuation
- 2006-06-30 MX MX2007016545A patent/MX2007016545A/en unknown
- 2006-06-30 WO PCT/GB2006/050182 patent/WO2007003964A1/en active Application Filing
- 2006-06-30 BR BRPI0612599-9A patent/BRPI0612599A2/en not_active IP Right Cessation
- 2006-06-30 US US11/922,826 patent/US20090203676A1/en not_active Abandoned
- 2006-06-30 AU AU2006264651A patent/AU2006264651A1/en not_active Abandoned
-
2008
- 2008-01-04 NO NO20080052A patent/NO20080052L/en not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005007647A1 (en) * | 2003-07-11 | 2005-01-27 | Arena Pharmaceuticals, Inc. | Trisubstituted aryl and heteroaryl derivatives as modulators of metabolism and the prophylaxis and treatment of disorders related thereto |
| WO2006067531A1 (en) * | 2004-12-24 | 2006-06-29 | Prosidion Ltd | G-protein coupled receptor (gpr116) agonists and use thereof for treating obesity and diabetes |
Cited By (77)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8933083B2 (en) | 2003-01-14 | 2015-01-13 | Arena Pharmaceuticals, Inc. | 1,2,3-trisubstituted aryl and heteroaryl derivatives as modulators of metabolism and the prophylaxis and treatment of disorders related thereto such as diabetes and hyperglycemia |
| JP2008239623A (en) * | 2005-01-10 | 2008-10-09 | Arena Pharmaceuticals Inc | Combination therapy for treating diabetes and condition related thereto and for treating condition ameliorated by increasing blood glp-1 level |
| WO2007130712A1 (en) * | 2006-01-31 | 2007-11-15 | Janssen Pharmaceutica, Nv | Substituted dipiperidine as ccr2 antagonists for the treatment of inflammatory diseases |
| JP2009532453A (en) * | 2006-04-06 | 2009-09-10 | プロシディオン・リミテッド | Heterocyclic GPCR agonist |
| EP2402750A1 (en) | 2006-04-11 | 2012-01-04 | Arena Pharmaceuticals, Inc. | Methods of using GPR119 receptor to identify compounds useful for increasing bone mass in an individual |
| WO2007120702A2 (en) | 2006-04-11 | 2007-10-25 | Arena Pharmaceuticals, Inc. | Use of gpr119 receptor agonists for increasing bone mass and for treating osteoporosis, and combination therapy relating thereto |
| EP2253311A2 (en) | 2006-04-11 | 2010-11-24 | Arena Pharmaceuticals, Inc. | Use of GPR119 receptor agonists for increasing bone mass and for treating osteoporosis, as well as combination therapy relating thereto |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2008085316A1 (en) * | 2006-12-20 | 2008-07-17 | Merck & Co., Inc. | Bipiperidinyl compounds, compositions containing such compounds and methods of treatment |
| WO2008081205A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081206A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081207A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081204A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| WO2008081208A1 (en) | 2007-01-04 | 2008-07-10 | Prosidion Limited | Piperidine gpcr agonists |
| EP2383270A1 (en) | 2007-01-04 | 2011-11-02 | Prosidion Limited | Piperidine GPCR agonists |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2009034388A1 (en) | 2007-09-10 | 2009-03-19 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| RU2443699C2 (en) * | 2007-09-20 | 2012-02-27 | Айрм Ллк | Compounds and compositions as gpr119 activity modulators |
| US8153635B2 (en) | 2007-09-20 | 2012-04-10 | Irm Llc | Compounds and compositions as modulators of GPR119 activity |
| JP2010540441A (en) * | 2007-09-20 | 2010-12-24 | アイアールエム・リミテッド・ライアビリティ・カンパニー | Compounds and compositions as modulators of GPR119 activity |
| WO2009038974A1 (en) | 2007-09-20 | 2009-03-26 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| US8258156B2 (en) | 2007-09-20 | 2012-09-04 | Irm Llc | Compounds and compositions as modulators of GPR119 activity |
| WO2009050522A1 (en) | 2007-10-18 | 2009-04-23 | Prosidion Limited | Azetidinyl g-protein coupled receptor agonists |
| WO2009050523A1 (en) | 2007-10-18 | 2009-04-23 | Prosidion Limited | Azetidinyl g-protein coupled receptor agonists |
| US8093248B2 (en) | 2007-12-05 | 2012-01-10 | Astrazeneca Ab (Publ) | Compounds useful for the treatment of conditions associated with weight gain |
| WO2009105722A1 (en) * | 2008-02-22 | 2009-08-27 | Irm Llc | Compounds and compositions as modulators of gpr119 activity |
| JP2011513232A (en) * | 2008-02-22 | 2011-04-28 | アイアールエム・リミテッド・ライアビリティ・カンパニー | Compounds and compositions as modulators of GPR119 activity |
| US8883714B2 (en) | 2008-04-07 | 2014-11-11 | Arena Pharmaceuticals, Inc. | Pharmaceutical compositions comprising GPR119 agonists which act as peptide YY (PYY) secretagogues |
| EP2108960A1 (en) | 2008-04-07 | 2009-10-14 | Arena Pharmaceuticals, Inc. | Methods of using A G protein-coupled receptor to identify peptide YY (PYY) secretagogues and compounds useful in the treatment of conditons modulated by PYY |
| US8343990B2 (en) | 2008-04-14 | 2013-01-01 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2010084512A1 (en) | 2008-12-24 | 2010-07-29 | Cadila Healthcare Limited | Novel oxime derivatives |
| US8742117B2 (en) | 2008-12-24 | 2014-06-03 | Cadila Healthcare Limited | Oxime derivatives |
| WO2010103335A1 (en) | 2009-03-12 | 2010-09-16 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| WO2010103333A1 (en) | 2009-03-12 | 2010-09-16 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| WO2010103334A1 (en) | 2009-03-12 | 2010-09-16 | Prosidion Limited | Compounds for the treatment of metabolic disorders |
| US8481731B2 (en) | 2009-06-24 | 2013-07-09 | Boehringer Ingelheim International Gmbh | Compounds, pharmaceutical composition and methods relating thereto |
| US8293729B2 (en) | 2009-06-24 | 2012-10-23 | Boehringer Ingelheim International Gmbh | Compounds, pharmaceutical composition and methods relating thereto |
| WO2011002067A1 (en) * | 2009-07-02 | 2011-01-06 | 武田薬品工業株式会社 | Heterocyclic compound and use thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| CN102686579A (en) * | 2009-10-09 | 2012-09-19 | Irm责任有限公司 | Compounds and compositions as modulators of GPR119 activity |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011113947A1 (en) | 2010-03-18 | 2011-09-22 | Boehringer Ingelheim International Gmbh | Combination of a gpr119 agonist and the dpp-iv inhibitor linagliptin for use in the treatment of diabetes and related conditions |
| WO2011128395A1 (en) | 2010-04-14 | 2011-10-20 | Prosidion Limited | N- substituted 3-amino 4 - ( pyrrolidine - 1 - carbonyl) pyrrolidine and its derivatives for use in the treatment of metabolic disorders |
| WO2011128394A1 (en) | 2010-04-14 | 2011-10-20 | Prosidion Limited | 3-substituted 5-(pyrrolidine-1-carbonyl) pyrrolidine and its derivatives for use in the treatment of metabolic disorders |
| US8754226B2 (en) | 2010-05-17 | 2014-06-17 | Array Biopharma Inc. | Piperidinyl-substituted lactams as GPR119 modulators |
| WO2011147951A1 (en) | 2010-05-28 | 2011-12-01 | Prosidion Limited | Cycloamino derivatives as gpr119 antagonists |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| US10894787B2 (en) | 2010-09-22 | 2021-01-19 | Arena Pharmaceuticals, Inc. | Modulators of the GPR119 receptor and the treatment of disorders related thereto |
| WO2012046249A1 (en) | 2010-10-08 | 2012-04-12 | Cadila Healthcare Limited | Novel gpr 119 agonists |
| WO2012066077A1 (en) | 2010-11-18 | 2012-05-24 | Prosidion Limited | 1,4 di substituted pyrrolidine - 3 - yl -amine derivatives and their use for the treatment of metabolic disorders |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| US8957062B2 (en) | 2011-04-08 | 2015-02-17 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment |
| WO2012170702A1 (en) * | 2011-06-08 | 2012-12-13 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| WO2012170867A1 (en) | 2011-06-09 | 2012-12-13 | Rhizen Pharmaceuticals Sa | Novel compounds as modulators of gpr-119 |
| US9006228B2 (en) | 2011-06-16 | 2015-04-14 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds, and methods of treatment |
| WO2013026587A1 (en) | 2011-08-22 | 2013-02-28 | Prosidion Limited | 1,4 disubstituted pyrrolidine - 3 - yl -amine derivatives and their use for the treatment of metabolic disorders |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US9422266B2 (en) | 2011-09-30 | 2016-08-23 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment |
| US9018200B2 (en) | 2011-10-24 | 2015-04-28 | Merck Sharp & Dohme Corp. | Substituted piperidinyl compounds useful as GPR119 agonists |
| WO2013066869A1 (en) * | 2011-11-03 | 2013-05-10 | Array Biopharma Inc. | Piperidinyl-substituted lactams as gpr119 modulators |
| US9018224B2 (en) | 2011-11-15 | 2015-04-28 | Merck Sharp & Dohme Corp. | Substituted cyclopropyl compounds useful as GPR119 agonists |
| US9452990B2 (en) | 2012-06-20 | 2016-09-27 | Novartis Ag | Complement pathway modulators and uses thereof |
| US11007175B2 (en) | 2015-01-06 | 2021-05-18 | Arena Pharmaceuticals, Inc. | Methods of treating conditions related to the S1P1 receptor |
| US11884626B2 (en) | 2015-06-22 | 2024-01-30 | Arena Pharmaceuticals, Inc. | Crystalline L-arginine salt of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclo-penta [b]indol-3-yl)acetic acid(Compound1) for use in S1P1 receptor-associated disorders |
| US11534424B2 (en) | 2017-02-16 | 2022-12-27 | Arena Pharmaceuticals, Inc. | Compounds and methods for treatment of primary biliary cholangitis |
| WO2024072930A1 (en) * | 2022-09-30 | 2024-04-04 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Dopamine d3/d2 receptor partial agonists for the treatment of neuropsychiatric disorders |
Also Published As
| Publication number | Publication date |
|---|---|
| NO20080052L (en) | 2008-01-23 |
| BRPI0612599A2 (en) | 2010-11-23 |
| AU2006264651A1 (en) | 2007-01-11 |
| NZ564758A (en) | 2011-03-31 |
| EP1909791A1 (en) | 2008-04-16 |
| MX2007016545A (en) | 2008-02-21 |
| JP2008545010A (en) | 2008-12-11 |
| CA2613236A1 (en) | 2007-01-11 |
| KR20080025190A (en) | 2008-03-19 |
| US20090203676A1 (en) | 2009-08-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007003964A1 (en) | G-protein coupled receptor agonists | |
| EP1838698B1 (en) | Pyrimidine derivatives as gpcr agonists | |
| EP2321308B1 (en) | Piperidine gpcr agonists | |
| EP2013201B1 (en) | Heterocyclic gpcr agonists | |
| EP2114931B1 (en) | Piperidine gpcr agonists | |
| EP2114935B1 (en) | Piperidine gpcr agonists | |
| EP2114933B1 (en) | Piperidine gpcr agonists | |
| EP2318399B1 (en) | Piperidinyl gpcr agonists | |
| US20110212939A1 (en) | Heterocyclic GPCR Agonists | |
| US20110178054A1 (en) | Heterocyclic GPCR Agonists | |
| EP1838311A1 (en) | G-protein coupled receptor (gpr116) agonists and use thereof for treating obesity and diabetes | |
| WO2007003961A2 (en) | Gpcr agonists | |
| WO2008081206A1 (en) | Piperidine gpcr agonists | |
| US20100298299A1 (en) | non-peptide derivatives as bradykinin b1 antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/016545 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4960/KOLNP/2007 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006264651 Country of ref document: AU Ref document number: 2613236 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 564758 Country of ref document: NZ |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008520009 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006744362 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087002531 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006264651 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006264651 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680032073.2 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006744362 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11922826 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0612599 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080102 |