WO2007002557A1 - Niacin receptor agonists, compositions containing such compounds and methods of treatment - Google Patents
Niacin receptor agonists, compositions containing such compounds and methods of treatment Download PDFInfo
- Publication number
- WO2007002557A1 WO2007002557A1 PCT/US2006/024740 US2006024740W WO2007002557A1 WO 2007002557 A1 WO2007002557 A1 WO 2007002557A1 US 2006024740 W US2006024740 W US 2006024740W WO 2007002557 A1 WO2007002557 A1 WO 2007002557A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- groups
- halo
- optionally substituted
- group
- Prior art date
Links
- 0 *c(cc1)cnc1C#N Chemical compound *c(cc1)cnc1C#N 0.000 description 5
- GPHOYYDXVWHBPG-UHFFFAOYSA-N CC(/N=C1/Sc(cccc2)c2N1)=C Chemical compound CC(/N=C1/Sc(cccc2)c2N1)=C GPHOYYDXVWHBPG-UHFFFAOYSA-N 0.000 description 1
- XLPGAEWMBGEKLS-UHFFFAOYSA-N CC(C)(CC1)CC(NC(CCc2nc(-c(cc3)ncc3O)n[o]2)=O)=C1C(O)=O Chemical compound CC(C)(CC1)CC(NC(CCc2nc(-c(cc3)ncc3O)n[o]2)=O)=C1C(O)=O XLPGAEWMBGEKLS-UHFFFAOYSA-N 0.000 description 1
- HQBVWNXKGVHWGP-UHFFFAOYSA-N CC(CCC1C(O)=O)CC1NC(CCc1nc(-c(cc2)ncc2O)n[o]1)=O Chemical compound CC(CCC1C(O)=O)CC1NC(CCc1nc(-c(cc2)ncc2O)n[o]1)=O HQBVWNXKGVHWGP-UHFFFAOYSA-N 0.000 description 1
- NRMUKJXMGAGWOY-UHFFFAOYSA-N CCCC(CC1C(O)=O)C(C)CC1NC(CCc(ccc1c2)cc1ccc2O)=O Chemical compound CCCC(CC1C(O)=O)C(C)CC1NC(CCc(ccc1c2)cc1ccc2O)=O NRMUKJXMGAGWOY-UHFFFAOYSA-N 0.000 description 1
- OMWPNORWFNGMCJ-UHFFFAOYSA-N COC(C(CCC(C1)C(F)(F)F)=C1N)=O Chemical compound COC(C(CCC(C1)C(F)(F)F)=C1N)=O OMWPNORWFNGMCJ-UHFFFAOYSA-N 0.000 description 1
- RMXRUKCQJHLQFA-UHFFFAOYSA-N COC(C(CCCC1)=C1N)=O Chemical compound COC(C(CCCC1)=C1N)=O RMXRUKCQJHLQFA-UHFFFAOYSA-N 0.000 description 1
- BJNFEVNETHSCLN-UHFFFAOYSA-N COC(c1cnc(-[n]2ncc(O)c2)[s]1)=O Chemical compound COC(c1cnc(-[n]2ncc(O)c2)[s]1)=O BJNFEVNETHSCLN-UHFFFAOYSA-N 0.000 description 1
- OCWZDGIDMPPHBM-UHFFFAOYSA-N Cc([nH]nc1-c2c3cccc2)c1S3(=O)=O Chemical compound Cc([nH]nc1-c2c3cccc2)c1S3(=O)=O OCWZDGIDMPPHBM-UHFFFAOYSA-N 0.000 description 1
- ZWALEOSSKIASEA-UHFFFAOYSA-N Cc(cc1)cc(cc2)c1c1c2NN=[I]1 Chemical compound Cc(cc1)cc(cc2)c1c1c2NN=[I]1 ZWALEOSSKIASEA-UHFFFAOYSA-N 0.000 description 1
- GUTIDBNICULQBN-UHFFFAOYSA-N Cc1c(CCC2=C3[I]=CC=C2)c3n[o]1 Chemical compound Cc1c(CCC2=C3[I]=CC=C2)c3n[o]1 GUTIDBNICULQBN-UHFFFAOYSA-N 0.000 description 1
- CEQWQJPIFHYJQO-UHFFFAOYSA-N Cc1c(c(OC)nc2c3cccc2)[n]3nn1 Chemical compound Cc1c(c(OC)nc2c3cccc2)[n]3nn1 CEQWQJPIFHYJQO-UHFFFAOYSA-N 0.000 description 1
- LZHLZVFMEZBISD-UHFFFAOYSA-N Cc1n[nH]c2c1CCC1=C2[I]=CC=C1 Chemical compound Cc1n[nH]c2c1CCC1=C2[I]=CC=C1 LZHLZVFMEZBISD-UHFFFAOYSA-N 0.000 description 1
- HQHTXMMHXWMLEY-UHFFFAOYSA-N Cc1n[nH]c2c1cc1[s]cnc1c2 Chemical compound Cc1n[nH]c2c1cc1[s]cnc1c2 HQHTXMMHXWMLEY-UHFFFAOYSA-N 0.000 description 1
- ZKLBYOFSSKCQRQ-UHFFFAOYSA-N Cc1n[nH]c2c3[I]=CC=Cc3cnc12 Chemical compound Cc1n[nH]c2c3[I]=CC=Cc3cnc12 ZKLBYOFSSKCQRQ-UHFFFAOYSA-N 0.000 description 1
- RPUWRDHXNPLYFH-UHFFFAOYSA-N Cc1n[nH]c2c3[s]cnc3ccc12 Chemical compound Cc1n[nH]c2c3[s]cnc3ccc12 RPUWRDHXNPLYFH-UHFFFAOYSA-N 0.000 description 1
- FQRIHARWLRXNRT-UHFFFAOYSA-N Cc1n[o]c2c3nc[s]c3ccc12 Chemical compound Cc1n[o]c2c3nc[s]c3ccc12 FQRIHARWLRXNRT-UHFFFAOYSA-N 0.000 description 1
- YHADNJOEQWBXAR-UHFFFAOYSA-N O=C(CCC1)CC1C(F)(F)F Chemical compound O=C(CCC1)CC1C(F)(F)F YHADNJOEQWBXAR-UHFFFAOYSA-N 0.000 description 1
- PPVADDJJLWKJLE-UHFFFAOYSA-N OC(C(CCCC1)=C1NC(CCC1=CN(c(cc2)ncc2O)[I]=C1)=O)=O Chemical compound OC(C(CCCC1)=C1NC(CCC1=CN(c(cc2)ncc2O)[I]=C1)=O)=O PPVADDJJLWKJLE-UHFFFAOYSA-N 0.000 description 1
- CEEDKNHIBNHBQW-UHFFFAOYSA-N OC(C(CCCC1)=C1NC(CCc1n[o]c-2c1CCc1c-2ccc(O)c1)=O)=O Chemical compound OC(C(CCCC1)=C1NC(CCc1n[o]c-2c1CCc1c-2ccc(O)c1)=O)=O CEEDKNHIBNHBQW-UHFFFAOYSA-N 0.000 description 1
- CXQPATCGBJNQBH-UHFFFAOYSA-N OC(c1ccc(C(F)(F)F)cc1NC(CCc1nc(-c(cc2)ncc2O)n[o]1)=O)=O Chemical compound OC(c1ccc(C(F)(F)F)cc1NC(CCc1nc(-c(cc2)ncc2O)n[o]1)=O)=O CXQPATCGBJNQBH-UHFFFAOYSA-N 0.000 description 1
- UGEJOEBBMPOJMT-UHFFFAOYSA-N Oc1cccc(C(F)(F)F)c1 Chemical compound Oc1cccc(C(F)(F)F)c1 UGEJOEBBMPOJMT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/20—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/45—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/196—Carboxylic acids, e.g. valproic acid having an amino group the amino group being directly attached to a ring, e.g. anthranilic acid, mefenamic acid, diclofenac, chlorambucil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/45—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups
- C07C233/46—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/45—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups
- C07C233/52—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/45—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups
- C07C233/53—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a six-membered aromatic ring
- C07C233/55—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a six-membered aromatic ring having the carbon atom of the carboxamide group bound to a carbon atom of an unsaturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/32—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton containing six-membered aromatic rings
- C07C235/36—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton containing six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/20—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/04—1,2,3-Triazoles; Hydrogenated 1,2,3-triazoles
- C07D249/06—1,2,3-Triazoles; Hydrogenated 1,2,3-triazoles with aryl radicals directly attached to ring atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D257/00—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms
- C07D257/02—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D257/04—Five-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/06—1,2,4-Oxadiazoles; Hydrogenated 1,2,4-oxadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/22—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D277/30—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/78—Benzo [b] furans; Hydrogenated benzo [b] furans
- C07D307/79—Benzo [b] furans; Hydrogenated benzo [b] furans with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/14—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/16—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D309/28—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/10—Systems containing only non-condensed rings with a five-membered ring the ring being unsaturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/16—Systems containing only non-condensed rings with a six-membered ring the ring being unsaturated
Definitions
- the present invention relates to cycloalkene compounds, their derivatives, compositions containing such compounds and methods of treatment or prevention in a mammal relating to dyslipidemias.
- Dyslipidemia is a condition wherein serum lipids are abnormal. Elevated cholesterol and low levels of high density lipoprotein (HDL) are independent risk factors for atherosclerosis associated with a greater-than-normal risk of atherosclerosis and cardiovascular disease. Factors known to affect serum cholesterol include genetic predisposition, diet, body weight, degree of physical activity, age and gender. While cholesterol in normal amounts is a vital building block for cell membranes and essential organic molecules such as steroids and bile acids, cholesterol in excess is known to contribute to cardiovascular disease. For example, cholesterol, through its relationship with foam cells, is a primary component of plaque which collects in coronary arteries, resulting in the cardiovascular disease termed atherosclerosis.
- Niacin or nicotinic acid is a drug that reduces coronary events in clinical trials. It is commonly known for its effect in elevating serum levels of high density lipoproteins (HDL). Importantly, niacin also has a beneficial effect on other lipid profiles. Specifically, it reduces low density lipoproteins (LDL), very low density lipoproteins (VLDL), and triglycerides (TG).
- LDL low density lipoproteins
- VLDL very low density lipoproteins
- TG triglycerides
- the clinical use of nicotinic acid is limited by a number of adverse side-effects including cutaneous vasodilation, sometimes called flushing.
- the present invention relates to compounds that have been discovered to have effects in modifying serum lipid levels.
- the invention thus provides compositions for effecting reduction in total cholesterol and triglyceride concentrations and raising HDL, in accordance with the methods described. Consequently one object of the present invention is to provide a nicotinic acid receptor agonist that can be used to treat dyslipidemias, atherosclerosis, diabetes, metabolic syndrome and related conditions while minimizing the adverse effects that are associated with niacin treatment.
- Yet another object is to provide a pharmaceutical composition for oral use.
- X represents CH 2 , O, S, S(O), SO 2 or NH, such that when X represents NH, the nitrogen atom may be optionally substituted with R 6 , C(O)R 6 , or SO 2 R 6 , wherein:
- R 6 represents Ci -3 alkyl optionally substituted with 1-3 groups, 0-3 of which are halo, and 0-1 of which are selected from the group consisting of: OCi -3 alkyl, OH, NH 2 , NHCi. 3 alkyl, N(Ci -3 alkyl) 2 , CN, Hetcy, Aryl and HAR, said Aryl and HAR being further optionally substituted with 1-3 groups, 1-3 of which are halo, and 0-1 of which are selected from the group consisting of: OH, NH 2 , Ci -3 alkyl, Ci -3 alkoxy, haloQ, 3 alkyl and haloCi -3 alkoxy groups;
- a and b are each integers 1, 2 or 3, such that the sum of a and b is 2, 3 or 4;
- ring A represents a 6-10 membered aryl, a 5-13 membered heteroaryl or a partially aromatic heterocyclic group, said heteroaryl and partially aromatic heterocyclic group containing at least one heteroatom selected from O, S, S(O), S(O) 2 and N, and optionally containing 1 other heteroatom selected from O and S, and optionally containing 1-3 additional N atoms, with up to 5 heteroatoms being present;
- each R 2 and R 3 is independently H, Ci -3 alkyl, haloCi -3 alkyl, OCi -3 alkyl, haloCi -3 alkoxy, OH or F;
- n represents an integer of from 1 to 5;
- each R 4 is H or is independently selected from halo and R 6 ;
- R 5 represents -CO 2 H, — i N" H N or -C(O)NHSO 2 R 6 wherein R c represents Ci -4 alkyl or phenyl, said Ci -4 alkyl and
- each R 1 is H or is independently selected from the group consisting of: a) halo, OH, CO 2 H, CN, NH 2, S(O) 0-2 R 6 , C(O)R 6 , OC(O)R 6 and CO 2 R e , wherein R e is as previously defined; b) Ci- 6 alkyl and OCi- ⁇ alkyl, said C 1-6 alkyl and alkyl portion of OCi -6 alkyl being optionally substituted with 1-3 groups, 1-3 of which are halo and 1-2 of which are selected from: OH, CO 2 H, CO 2 C 1-4 alkyl, CO 2 C ]-4 haloalkyl, OCO 2 C 1-4 alkyl, NH 2 , NHC M alkyl, N(C M alkyl) 2 , Hetcy and CN; c) NHCi -4 alkyl and the alkyl portions of which are optionally substituted as set forth in (b) above; d) C
- R 55 represents (a) Ci -8 alkyl optionally substituted with 1-4 groups, 0-4 of which are halo, and 0-1 of which are selected from the group consisting of: OC ⁇ alkyl, OH, CO 2 H, CO 2 Ci_ 4 alkyl, CO 2 C 1-4 haloalkyl, NH 2 , NHC 1-4 alkyl, N(C 1-4 alkyl) 2 , CN, Hetcy, Aryl and HAR, said Hetcy, Aryl and HAR being further optionally substituted with 1-3 halo, C 1- 4 alkyl, Q ⁇ alkoxy, haloCi -4 alkyl or haloC ⁇ alkoxy groups;
- Hetcy, Aryl or HAR each being optionally substituted with 1-3 members selected from the group consisting of: halo, Q ⁇ alkoxy, haloQ ⁇ alkyl and haloCi. 4 alkoxy groups; and R 555 representing H or R";
- Alkyl as well as other groups having the prefix "alk”, such as alkoxy, alkanoyl and the like, means carbon chains which may be linear, branched, or cyclic, or combinations thereof, containing the indicated number of carbon atoms. If no number is specified, 1-6 carbon atoms are intended for linear and 3-7 carbon atoms for branched alkyl groups. Examples of alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, sec- and tert-butyl, pentyl, hexyl, heptyl, octyl, nonyl and the like.
- Cycloalkyl is a subset of alkyl; if no number of atoms is specified, 3-7 carbon atoms are intended, forming 1-3 carbocyclic rings that are fused. "Cycloalkyl” also includes monocyclic rings fused to an aryl group in which the point of attachment is on the non-aromatic portion. Examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, tetrahydronaphthyl, decahydronaphthyl, indanyl and the like.
- Haloalkoxy and haloOalkyl are used interchangeably and refer to halo substituted alkyl groups linked through the oxygen atom.
- Haloalkyl and haloalkoxy include mono- substituted as well as multiple substituted alkyl and alkoxy groups, up to perhalo substituted alkyl and alkoxy. For example, trifluoromethyl and trifluoromethoxy are included.
- alkenyl means carbon chains which contain at least one carbon-carbon double bond, and which may be linear or branched or combinations thereof. Examples of alkenyl include vinyl, allyl, isopropenyl, pentenyl, hexenyl, heptenyl, 1-propenyl, 2-butenyl, 2-methyl-2-butenyl, and the like.
- Alkynyl means carbon chains which contain at least one carbon-carbon triple bond, and which may be linear or branched or combinations thereof. Examples of alkynyl include ethynyl, propargyl, 3 -methyl- 1-pentynyl, 2-heptynyl and the like.
- Aryl (Ar) means mono- andbicyclic aromatic rings containing 6-10 carbon atoms. Examples of aryl include phenyl, naphthyl, indenyl and the like.
- Heteroaryl (HAR) unless otherwise specified, means mono-, bicyclic and tricyclic aromatic ring systems containing at least one heteroatom selected from O, S, S(O), SO 2 and N, with each ring containing 5 to 6 atoms.
- HAR groups may contain from 5-14, preferably 5-13 atoms.
- Examples include, but are not limited to, pyrrolyl, isoxazolyl, isothiazolyl, pyrazolyl, pyridyl, oxazolyl, oxadiazolyl, thiadiazolyl, thiazolyl, imidazolyl, triazolyl, tetrazolyl, furanyl, triazinyl, thienyl, pyrimidyl, pyridazinyl, pyrazinyl, benzoxazolyl, benzothiazolyl, benzimidazolyl, benzofuranyl, benzothiophenyl, benzopyrazolyl, benzotriazolyl, fbro(2,3-b)pyridyl, benzoxazinyl, tetrahydrohydroquinolinyl, tetrahydroisoquinolinyl., quinolyl, isoquinolyl, indolyl, dihydroindoly
- Heteroaryl also includes aromatic carbocyclic or heterocyclic groups fused to heterocycles that are non-aromatic or partially aromatic, and optionally containing a carbonyl.
- additional heteroaryl groups include indolinyl, dihydrobenzofuranyl, dihydrobenzothiophenyl, dihydrobenzoxazolyl, and aromatic heterocyclic groups fused to cycloalkyl rings. Examples also include the following:
- Heteroaryl also includes such groups in charged form, e.g., pyridinium.
- Heterocyclyl (Hetcy) unless otherwise specified, means mono- and bicyclic saturated and partially saturated rings and ring systems containing at least one heteroatom selected from N, S and O, each of said ring having from 3 to 10 atoms in which the point of attachment may be carbon or nitrogen.
- heterocyclyl include, but are not limited to, azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, imidazolidinyl, tetrahydrofuranyl, 1,4-dioxanyl, morpholinyl, thiomorpholinyl, tetrahydrothienyl and the like.
- Heterocycles can also exist in tautomeric forms, e.g., 2- and 4-pyridones. Heterocycles moreover includes such moieties in charged form, e.g., piperidinium.
- Hydroogen includes fluorine, chlorine, bromine and iodine.
- the phrase "in the absence of substantial flushing" refers to the side effect that is often seen when nicotinic acid is administered in therapeutic amounts. The flushing effect of nicotinic acid usually becomes less frequent and less severe as the patient develops tolerance to the drug at therapeutic doses, but the flushing effect still occurs to some extent and can be transient.
- flushing refers to the reduced severity of flushing when it occurs, or fewer flushing events than would otherwise occur.
- the incidence of flushing is reduced by at least about a third, more preferably the incidence is reduced by half, and most preferably, the flushing incidence is reduced by about two thirds or more.
- the severity is preferably reduced by at least about a third, more preferably by at least half, and most preferably by at least about two thirds.
- a one hundred percent reduction in flushing incidence and severity is most preferable, but is not required.
- One aspect of the invention relates to a compound represented by formula I:
- X represents CH 2 , O, S, S(O), SO 2 or NH, such that when X represents NH, the nitrogen atom may be optionally substituted with R 6 , C(O)R 6 , or SO 2 R 6 , wherein: R 6 represents Ci -3 alkyl optionally substituted with 1-3 groups, 0-3 of which are halo, and
- a and b are each integers 1, 2 or 3, such that the sum of a and b is 2, 3 or 4;
- ring A represents a 6-10 membered aryl, a 5-13 membered heteroaryl or a partially aromatic heterocyclic group, said heteroaryl and partially aromatic heterocyclic group containing at least one heteroatom selected from O, S, S(O), S(O) 2 and N, and optionally containing 1 other heteroatom selected from O and S, and optionally containing 1-3 additional N atoms, with up to 5 heteroatoms being present;
- each R 2 and R 3 is independently H, d. 3 alkyl, haloCi. 3 alkyl, OC ]-3 alkyl, haloCi -3 alkoxy, OH or F;
- n represents an integer of from 1 to 5;
- each R 4 is H or is independently selected from halo and R 6 ;
- R e represents C )-4 alkyl or phenyl, said Q ⁇ alkyl and phenyl each being optionally substituted with 1-3 groups, 1-3 of which are selected from halo and Ci -3 alkyl, and 1-2 of which are selected from the group consisting of: OCi -3 alkyl, haloCi -3 alkyl, haloC 1-3 alkoxy, OH, NH 2 and NHC 1-3 alkyl;
- each R 1 is H or is independently selected from the group consisting of: a) halo, OH, CO 2 H, CN, NH 2 , S(O) 0-2 R 6 , C(O)R 6 , OC(O)R 6 and CO 2 R 6 , wherein R e is as previously defined; b) C 1-5 alkyl and OCi -6 alkyl, said Ci -6 alkyl and alkyl portion of OCi -5 alkyl being optionally substituted with 1-3 groups, 1-3 of which are halo and 1-2 of which are selected from: OH, CO 2 H, COaCi- t alkyl, CO 2 C 1-4 haloalkyl, OCO 2 C 1-4 alkyl, NH 2 , NHC 1-4 alkyl, N(Ci -4 alkyl) 2 , Hetcy and CN; c) and N(Ci- 4 alkyl) 2 , the alkyl portions of which are optionally substituted as set forth
- R'" representing H or R"
- f phenyl or a 5-6 membered heteroaryl or a Hetcy group attached at any available ring atom and each being optionally substituted with 1-3 groups, 1-3 of which are selected from halo, C 1- 3 alkyl and haloC ⁇ alkyl groups, and 1-2 of which are selected from OCi -3 alkyl and haloOCi -3 alkyl groups, and 0-1 of which is selected from the group consisting of: i) OH; CO 2 H; CN; NH 2 and S(O) 0-2 R 6 wherein R e is as described above; ii) and N(Ci.
- R 2 and R 3 moieties are selected from the group consisting of: Ci -3 alkyl, haloCi_ 3 alkyl, OCi -3 alkyl, haloC 1-3 alkoxy, OH and F, and any remaining R 2 and R 3 moieties represent H.
- ring A is a phenyl or naphthyl group, a 5-6 membered monocyclic heteroaryl group or a 9-13 membered bicyclic or tricyclic heteroaryl group.
- ring A is a phenyl or naphthyl group, a 5-6 membered monocyclic heteroaryl group or a 9-13 membered bicyclic or tricyclic heteroaryl group.
- a subset of compounds that is of interest relates to compounds of formula I wherein ring A is selected from the group consisting of: phenyl; naphthyl;
- HAR which is a member selected from the group consisting of: pyrrolyl, isoxazolyl, isothiazolyl, pyrazolyl, pyridyl, oxazolyl, oxadiazolyl, thiadiazolyl, thiazolyl, imidazolyl, triazolyl, tetrazolyl, furanyl, triazinyl, thienyl, pyrimidyl, pyridazinyl, pyrazinyl, benzoxazolyl, benzothiazolyl, benzimidazolyl, benzofuranyl, benzothiophenyl, benzopyrazolyl, benzotriazolyl, furo(2,3-b)pyridyl, benzoxazinyl, tetrahydrohydroquinolinyl, tetrahydroisoquinolinyl., quinolyl, isoquinolyl, indolyl, dihydr
- an aspect of the invention that is of interest relates to a compound of formula I wherein ring A is selected from the group consisting of: phenyl; naphthyl;
- HAR which is a member selected from the group consisting of: isoxazolyl, pyrazolyl, oxazolyl, oxadiazolyl, thiazolyl, triazolyl, thienyl, benzothiazolyl, or a member selected from the group consisting of:
- each R 1 is H or is selected from the group consisting of: a) halo, OH, CN, NH 2 and S(O) 0-2 R 6 wherein R e is methyl or phenyl optionally substituted with 1-3 halo groups; b) Ci- 3 alkyl and OCi -3 alkyl, each being optionally substituted with 1-3 groups, 1-3 of which are halo and 1-2 of which are selected from: OH, NH 2 , NHCi. 4 alkyl and CN; c) NR 5 SO 2 R" and NR'C(0)NR"R'" wherein: R' represents H, Ci -3 alkyl or haloCi -3 alkyl,
- R" represents (a) Ci -8 alkyl optionally substituted with 1-4 groups, 0-4 of which are halo, and 0-1 of which are selected from the group consisting of: OCi -6 alkyl, OH, CO 2 H, CO 2 Ci -4 alkyl, CO 2 C 1-4 haloalkyl, OCO 2 C 1-4 alkyl, NH 2 , NHC 1-4 alkyl, N(C I-4 alkyl) 2 , CN, Hetcy, Aryl and HAR, said Hetcy, Aryl and HAR being further optionally substituted with 1-3 groups selected from: halo,
- alkyl groups and 1 of which is selected from the group consisting of: i) OH; CO 2 H; CN; NH 2 and S(O) 0-2 R' wherein R e is as described above; ii) NHC ⁇ alkyl, the alkyl portion of which is optionally substituted with 1-3 groups, 1-3 of which are halo and 1 of which is selected from: OH, CO 2 H, CO 2 Ci -4 alkyl, CO 2 C 1- 4 haloalkyl, NH 2 , NHCi.
- each R 1 is H or is selected from the group consisting of: a) halo, OH, CN and NH 2 ; b) C ]-3 alkyl and OCi -3 alkyl, each being optionally substituted with 1-3 groups, 1-3 of which are halo and 1-2 of which are selected from: OH, NH 2 , NHCi- 4 alkyl and CN; c) phenyl or a 5-6 membered heteroaryl or a heterocyclic group attached at any available point and being optionally substituted with 1-3 groups, 1-3 of which are halo, C 1-3 alkyl or haloCi -3 alkyl groups, 1-2 of which are OC 1-3 alkyl or haloOCi -3 alkyl groups, and 1 of which is selected from the group consisting of: i) OH, CN and NH 2 .
- all other variables are as
- Another subset of compounds that is of interest relates to compounds of formula I wherein a and b are 1 or 2 such that the sum of a and b is 2 or 3. Within this subset of compounds, all other variables are as defined with respect to formula I.
- Another subset of compounds that is of interest relates to compounds of formula I wherein X represents O, S, N or CH 2 . Within this subset of compounds, all other variables are as defined with respect to formula I. More particularly, another subset of compounds that is of interest relates to compounds of formula I wherein X represents O or CH 2 . Within this subset of compounds, all other variables are as defined with respect to formula I.
- Another subset of compounds that is of interest relates to compounds of formula I wherein R 2 and R 3 are independently H, Ci -3 alkyl, OH or haloCi -3 alkyl. Within this subset of compounds, all other variables are as defined with respect to formula I. More particularly, another subset of compounds that is of interest relates to compounds of formula I wherein R 2 and R 3 are independently H, Ci. 3 alkyl or haloC 1-3 alkyl. Within this subset of compounds, all other variables are as defined with respect to formula I.
- a subset of compounds that is of interest relates to compounds of formula I wherein R 2 and R 3 are independently H or methyl. Within this subset of compounds, all other variables are as defined with respect to formula I.
- Another subset of compounds that is of interest relates to compounds of formula I wherein n represents an integer of from 2 to 4. Within this subset of compounds, all other variables are as defined with respect to formula I. More particularly, a subset of compounds that is of interest relates to compounds of formula I wherein n is 2. Within this subset of compounds, all other variables are as defined with respect to formula I.
- each R 4 is H or is independently selected from the group consisting of: halo, C 1-3 alkyl optionally substituted with 1-3 halo groups and 0-1 OC 1-3 alkyl groups.
- all other variables are as defined with respect to formula I.
- a particular subset of compounds that is of interest relates to compounds of formula I or a pharmaceutically acceptable salt or solvate thereof wherein: ring A is a phenyl or naphthyl group, a 5-6 membered monocyclic heteroaryl group or a 9-13 membered bicyclic or tricyclic heteroaryl group; each R 1 is H or is selected from the group consisting of: a) halo, OH, CN, NH 2 and S(O) 0-2 R 6 wherein R e is methyl or phenyl optionally substituted with 1-3 halo groups; b) Ci -3 alkyl and OCi -3 alkyl, each being optionally substituted with 1-3 groups, 1-3 of which are halo and 1-2 of which are selected from: OH, NH 2 , NHC M alkyl and CN; c) NR 5 SO 2 R" and NR'C(O)NR"R'" wherein: R' represents H, C 1-3 alkyl or
- R 2 and R 3 are independently H, OH, Ci -3 alkyl or haloCi -3 alkyl; n represents 2;
- R 4 is H or is independently selected from the group consisting of: halo, Ci -3 alkyl optionally substituted with 1-3 halo groups or 0-1 OCi -3 alkyl groups; and R 5 represents -CO 2 H.
- ring A is selected from the group consisting of:
- each R 1 is independently H, CH 3 , phenyl, 4-hydroxy-phenyl, OH, 2-hydroxy-phenyl, 3- hydroxy-phenyl, 3 -amino-phenyl, 2,3-dihydro-benzofuran-6-yl, 2-chloro-4-hydroxy-phenyl, lH-pyrazol- 4-yl, 5-hydroxy-pyridin-2-yl, 4-hydroxy-pyrazol-l-yl, lH-[l,2,3]triazol-4-yl, or 5-fluoro-pyridin-2-yl; a and b are 1 or 2 such that the sum of a and b is 2 or 3; X represents CH 2 ; each R 2 and R 3 is independently H, OH or CH 3 ; n represents 2;
- R 4 is H, CH 3 , CH 2 CH 3 , CF 3 or CH 2 OCH 3 ; and R 5 represents -CO 2 H.
- racemic mixtures of compounds may be separated so that individual enantiomers are isolated.
- the separation can be carried out by methods well known in the art, such as the coupling of a racemic mixture of compounds of Formula I to an enantiomerically pure compound to form a diastereomeric mixture, which is then separated into individual diastereomers by standard methods, such as fractional crystallization or chromatography.
- the coupling reaction is often the formation of salts using an enantiomerically pure acid or base.
- the diasteromeric derivatives may then be converted to substantially pure enantiomers by cleaving the added chiral residue from the diastereomeric compound.
- racemic mixture of the compounds of Formula I can also be separated directly by chromatographic methods utilizing chiral stationary phases, which methods are well known in the art.
- enantiomers of compounds of the general Formula I may be obtained by stereoselective synthesis using optically pure starting materials or reagents.
- tautomers which have different points of attachment for hydrogen accompanied by one or more double bond shifts.
- a ketone and its enol form are keto-enol tautomers.
- a 2-hydroxyquinoline can reside in the tautomeric 2-quinolone form. The individual tautomers as well as mixtures thereof are included.
- the dosages of compounds of formula I or a pharmaceutically acceptable salt or solvate thereof vary within wide limits.
- the specific dosage regimen and levels for any particular patient will depend upon a variety of factors including the age, body weight, general health, sex, diet, time of administration, route of administration, rate of excretion, drug combination and the severity of the patient's condition. Consideration of these factors is well within the purview of the ordinarily skilled clinician for the purpose of determining the therapeutically effective orprophylactically effective dosage amount needed to prevent, counter, or arrest the progress of the condition.
- the compounds will be administered in amounts ranging from as low as about 0.01 mg/day to as high as about 2000 mg/day, in single or divided doses.
- a representative dosage is about 0.1 mg/day to about 1 g/day.
- additional active agents may be administered with the compounds described herein.
- the additional active agent or agents can be lipid modifying compounds or agents having other pharmaceutical activities, or agents that have both lipid-modifying effects and other pharmaceutical activities.
- additional active agents which may be employed include but are not limited to HMG-CoA reductase inhibitors, which include statins in their lactonized or dihydroxy open acid forms and pharmaceutically acceptable salts and esters thereof, including but not limited to lovastatin (see US Patent No. 4,342,767), simvastatin (see US Patent No. 4,444,784), dihydroxy open-acid simvastatin, particularly the ammonium or calcium salts thereof, pravastatin, particularly the sodium salt thereof (see US Patent No.
- HMG-CoA synthase inhibitors include squalene epoxidase inhibitors; squalene synthetase inhibitors (also known as squalene synthase inhibitors), acyl-coenzyme A: cholesterol acyltransferase (ACAT) inhibitors including selective inhibitors of ACAT-I or ACAT-2 as well as dual inhibitors of ACAT-I and -2; microsomal triglyceride transfer protein (MTP) inhibitors; endothelial lipase inhibitors; bile acid sequestrants; LDL receptor inducers; platelet aggregation inhibitors, for example glycoprotein Eb/ ⁇ ia fibrinogen receptor antagonists and aspirin; human peroxisome proliferator activated receptor gamma (PPAR-gamma) agonists including the compounds commonly referred to as glitazones for example pioglitazone and rosiglitazone and, including those compounds included within the structural
- Cholesterol absorption inhibitors can also be used in the present invention. Such compounds block the movement of cholesterol from the intestinal lumen into enterocytes of the small intestinal wall, thus reducing serum cholesterol levels.
- Examples of cholesterol absorption inhibitors are described in U.S. Patent Nos. 5,846,966, 5,631,365, 5,767,115, 6,133,001, 5,886,171, 5,856,473, 5,756,470, 5,739,321, 5,919,672, and in PCT application Nos. WO 00/63703, WO 00/60107, WO 00/38725, WO 00/34240, WO 00/20623, WO 97/45406, WO 97/16424, WO 97/16455, and WO 95/08532.
- ezetimibe also known as l-(4- fluorophenyl)-3(R)-[3(S)-(4-fluorophenyl)-3-hydroxypropyl)]-4(S)-(4-hydroxyphenyl)-2-azetidinone, described in U.S. Patent Nos. 5,767,115 and 5,846,966.
- Therapeutically effective amounts of cholesterol absorption inhibitors include dosages of from about 0.01 tng/kg to about 30 mg/kg of body weight per day, preferably about 0.1 mg/kg to about 15 mg/kg.
- the compounds used in the present invention can be administered with conventional diabetic medications.
- a diabetic patient receiving treatment as described herein may also be taking insulin or an oral antidiabetic medication.
- an oral antidiabetic medication useful herein is metformin.
- niacin receptor agonists induce some degree of vasodilation
- the compounds of formula I may be co-dosed with a vasodilation suppressing agent.
- one aspect of the methods described herein relates to the use of a compound of formula I or a pharmaceutically acceptable salt or solvate thereof in combination with a compound that reduces flushing.
- Conventional compounds such as aspirin, ibuprofen, naproxen, indomethacin, other NSAIDs, COX-2 selective inhibitors and the like are useful in this regard, at conventional doses.
- DP antagonists are useful as well. Doses of the DP receptor antagonist and selectivity are such that the DP antagonist selectively modulates the DP receptor without substantially modulating the CRTH2 receptor.
- the DP receptor antagonist ideally has an affinity at the DP receptor (i.e., Ki) that is at least about 10 times higher (a numerically lower K,- value) than the affinity at the CRTH2 receptor.
- Ki affinity at the DP receptor
- Any compound that selectively interacts with DP according to these guidelines is deemed "DP selective". This is in accordance with US Published Application No. 2004/0229844A1 published on November 18, 2004, incorporated herein by reference.
- Dosages for DP antagonists as described herein, that are useful for reducing or preventing the flushing effect in mammalian patients, particularly humans, include dosages ranging from as low as about 0.01 mg/day to as high as about 100 mg/day, administered in single or divided daily doses. Preferably the dosages are from about 0.1 mg/day to as high as about 1.0 g/day, in single or divided daily doses.
- the compound of formula I or a pharmaceutically acceptable salt or solvate thereof and the DP antagonist can be administered together or sequentially in single or multiple daily doses, e.g., bid, tid or qid, without departing from the invention.
- sustained release such as a sustained release product showing a release profile that extends beyond 24 hours, dosages may be administered every other day.
- single daily doses are preferred.
- morning or evening dosages can be utilized.
- Salts and solvates of the compounds of formula I are also included in the present invention, and numerous pharmaceutically acceptable salts and solvates of nicotinic acid are useful in this regard.
- Alkali metal salts in particular, sodium and potassium, form salts that are useful as described herein.
- alkaline earth metals in particular, calcium and magnesium, form salts that are useful as described herein.
- Various salts of amines, such as ammonium and substituted ammonium compounds also form salts that are useful as described herein.
- solvated forms of the compounds of formula I are useful within the present invention. Examples include the hemihydrate, mono-, di-, tri- and sesquihydrate.
- the compounds of the invention also include esters that are pharmaceutically acceptable, as well as those that are metabolically labile.
- Metabolically labile esters include Q -4 alkyl esters , preferably the ethyl ester.
- Many prodrug strategies are known to those skilled in the art. One such strategy involves engineered amino acid anhydrides possessing pendant nucleophiles, such as lysine, which can cyclize upon themselves, liberating the free acid. Similarly, acetone-ketal diesters, which can break down to acetone, an acid and the active acid, can be used.
- the compounds used in the present invention can be administered via any conventional route of administration.
- the preferred route of administration is oral.
- compositions described herein are generally comprised of a compound of formula I or a pharmaceutically acceptable salt or solvate thereof, in combination with a pharmaceutically acceptable carrier.
- suitable oral compositions include tablets, capsules, troches, lozenges, suspensions, dispersible powders or granules, emulsions, syrups and elixirs.
- carrier ingredients include diluents, binders, disintegrants, lubricants, sweeteners, flavors, colorants, preservatives, and the like.
- diluents include, for example, calcium carbonate, sodium carbonate, lactose, calcium phosphate and sodium phosphate.
- granulating and disintegrants include corn starch and alginic acid.
- binding agents include starch, gelatin and acacia.
- lubricants include magnesium stearate, calcium stearate, stearic acid and talc.
- the tablets may be uncoated or coated by known techniques. Such coatings may delay disintegration and thus, absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- One embodiment of the invention that is of interest is a tablet or capsule that is comprised of a compound of formula I or a pharmaceutically acceptable salt or solvate thereof in an amount ranging from about O.lmg to about lOOOmg, in combination with a pharmaceutically acceptable carrier.
- a compound of formula I or a pharmaceutically acceptable salt or solvate thereof is combined with another therapeutic agent and the carrier to form a fixed combination product.
- This fixed combination product may be a tablet or capsule for oral use.
- a compound of formula I or a pharmaceutically acceptable salt or solvate thereof (about 0.1 to about 1000 mg) and the second therapeutic agent (about 0.1 to about 500 mg) are combined with the pharmaceutically acceptable carrier, providing a tablet or capsule for oral use.
- Sustained release over a longer period of time may be particularly important in the formulation.
- a time delay material such as glyceryl monostearate or glyceryl distearate may be employed.
- the dosage form may also be coated by the techniques described in the U.S. Patent Nos. 4,256,108; 4,166,452 and 4,265,874 to form osmotic therapeutic tablets for controlled release.
- Typical ingredients that are useful to slow the release of nicotinic acid in sustained release tablets include various cellulosic compounds, such as methylcellulose, ethylcellulose, propylcellulose, hydroxypropylcellulose, hydroxyethylcellulose, hydroxypropylmethylcellulose, microcrystalline cellulose, starch and the like.
- Various natural and synthetic materials are also of use in sustained release formulations. Examples include alginic acid and various alginates, polyvinyl pyrrolidone, tragacanth, locust bean gum, guar gum, gelatin, various long chain alcohols, such as cetyl alcohol and beeswax.
- a tablet as described above, comprised of a compound of formula I or a pharmaceutically acceptable salt or solvate thereof, and further containing an HMG Co-A reductase inhibitor, such as simvastatin or atorvastatin.
- This particular embodiment optionally contains the DP antagonist as well.
- Typical release time frames for sustained release tablets in accordance with the present invention range from about 1 to as long as about 48 hours, preferably about 4 to about 24 hours, and more preferably about 8 to about 16 hours.
- Hard gelatin capsules constitute another solid dosage form for oral use. Such capsules similarly include the active ingredients mixed with carrier materials as described above.
- Soft gelatin capsules include the active ingredients mixed with water-miscible solvents such as propylene glycol, PEG and ethanol, or an oil such as peanut oil, liquid paraffin or olive oil.
- Aqueous suspensions are also contemplated as containing the active material in admixture with excipients suitable for the manufacture of aqueous suspensions.
- excipients include suspending agents, for example sodium carboxymethylcellulose, methylcellulose, hydroxypropylmethylcellulose, sodium alginate, polyvinylpyrrolidone, tragacanth and acacia; dispersing or wetting agents,e.g., lecithin; preservatives, e.g., ethyl, or n-propyl para-hydroxybenzoate, colorants, flavors, sweeteners and the like.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredients in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives.
- a dispersing or wetting agent e.g., kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, ka
- Syrups and elixirs may also be formulated.
- a pharmaceutical composition that is of interest is a sustained release tablet that is comprised of a compound of formula I or a pharmaceutically acceptable salt or solvate thereof, and a DP receptor antagonist that is selected from the group consisting of compounds A through AJ in combination with a pharmaceutically acceptable carrier.
- compositions that is of more interest are comprised of a compound of formula I or a pharmaceutically acceptable salt or solvate thereof and a DP antagonist compound selected from the group consisting of compounds A, B, D, E, X, AA, AF, AG, AH, AI and AJ, in combination with a pharmaceutically acceptable carrier.
- a DP antagonist compound selected from the group consisting of compounds A, B, D, E, X, AA, AF, AG, AH, AI and AJ, in combination with a pharmaceutically acceptable carrier.
- compositions that is of more particular interest relate to a sustained release tablet that is comprised of a compound of formula I or a pharmaceutically acceptable salt or solvate thereof, a DP receptor antagonist selected from the group consisting of compounds A, B, D, E, X, AA, AF, AG, AH, AI and AJ, and simvastatin or atorvastatin in combination with a pharmaceutically acceptable carrier.
- a DP receptor antagonist selected from the group consisting of compounds A, B, D, E, X, AA, AF, AG, AH, AI and AJ
- simvastatin or atorvastatin in combination with a pharmaceutically acceptable carrier.
- composition in addition to encompassing the pharmaceutical compositions described above, also encompasses any product which results, directly or indirectly, from the combination, complexation or aggregation of any two or more of the ingredients, active or excipient, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients. Accordingly, the pharmaceutical composition of the present invention encompasses any composition made by admixing or otherwise combining the compounds, any additional active ingredient(s), and the pharmaceutically acceptable excipients.
- Another aspect of the invention relates to the use of a compound of formula I or a pharmaceutically acceptable salt or solvate thereof and a DP antagonist in the manufacture of a medicament.
- This medicament has the uses described herein.
- another aspect of the invention relates to the use of a compound of formula I or a pharmaceutically acceptable salt or solvate thereof, a DP antagonist and an HMG Co-A reductase inhibitor, such as simvastatin, in the manufacture of a medicament.
- This medicament has the uses described herein.
- Compounds of the present invention have anti-hyperlipidemic activity, causing reductions in LDL-C, triglycerides, lipoprotein (a), free fatty acids and total cholesterol, and increases in HDL-C. Consequently, the compounds of the present invention are useful in treating dyslipidemias.
- the present invention thus relates to the treatment, prevention or reversal of atherosclerosis and the other diseases and conditions described herein, by administering a compound of formula I or a pharmaceutically acceptable salt or solvate in an amount that is effective for treating, preventing or reversing said condition.
- a compound of formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective to treat or prevent said condition, while preventing, reducing or minimizing flushing effects in terms of frequency and/or severity.
- One aspect of the invention that is of interest relates to a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof for use in a method of treatment of the human or animal body by therapy.
- Another aspect of the invention that is of interest relates to a compound in accordance with formula I or a pharmaceutically acceptable salt or solvate thereof for use in a method for the treatment of atherosclerosis, dyslipidemia, diabetes, metabolic syndrome or a related condition in the human or animal body by therapy. More particularly, an aspect of the invention that is of interest is a method of treating atherosclerosis in a human patient in need of such treatment comprising administering to the patient a compound of formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective for treating atherosclerosis in the absence of substantial flushing.
- Another aspect of the invention that is of interest relates to a method of raising serum HDL levels in a human patient in need of such treatment, comprising administering to the patient a compound of formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective for raising serum HDL levels.
- Another aspect of the invention that is of interest relates to a method of treating dyslipidemia in a human patient in need of such treatment comprising administering to the patient a compound of formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective for treating dyslipidemia.
- Another aspect of the invention that is of interest relates to a method of reducing serum VLDL or LDL levels in a human patient in need of such treatment, comprising administering to the patient a compound of formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective for reducing serum VLDL or LDL levels in the patient in the absence of substantial flushing.
- Another aspect of the invention that is of interest relates to a method of reducing serum triglyceride levels in a human patient in need of such treatment, comprising administering to the patient a compound of formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective for reducing serum triglyceride levels.
- Another aspect of the invention that is of interest relates to a method of reducing serum Lp(a) levels in a human patient in need of such treatment, comprising administering to the patient a compound of formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective for reducing serum Lp(a) levels.
- Lp(a) refers to lipoprotein (a).
- Another aspect of the invention that is of interest relates to a method of treating diabetes, and in particular, type 2 diabetes, in a human patient in need of such treatment comprising administering to the patient a compound of formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective for treating diabetes.
- Another aspect of the invention that is of interest relates to a method of treating metabolic syndrome in a human patient in need of such treatment comprising administering to the patient a compound of formula I or a pharmaceutically acceptable salt or solvate thereof in an amount that is effective for treating metabolic syndrome.
- Another aspect of the invention that is of particular interest relates to a method of treating atherosclerosis, dyslipidemias, diabetes, metabolic syndrome or a related condition in a human patient in need of such treatment, comprising administering to the patient a compound of formula I or a pharmaceutically acceptable salt or solvate thereof and a DP receptor antagonist, said combination being administered in an amount that is effective to treat atherosclerosis, dyslipidemia, diabetes or a related condition in the absence of substantial flushing.
- Another aspect of the invention that is of particular interest relates to the methods described above wherein the DP receptor antagonist is selected from the group consisting of compounds A through AJ and the pharmaceutically acceptable salts and solvates thereof.
- Scheme 3 Shown in Scheme 3 is the preparation of acid of the structure 10 from commercially available material 9 by methods known to one skilled in the art, such as hydrogenation in a polar solvent, such as methanol or ethanol, using Pd/C as a catalyst.
- a polar solvent such as methanol or ethanol
- 6-methoxy-2-naphthaldehyde 11 can be treated with a suitable ylide such as (tert-Butoxycarbonyl- methylene)triphenyl-phospharane in a non-polar solvent such as toluene or xylenes under refluxing conditions to give the desired olefin 12.
- a suitable ylide such as (tert-Butoxycarbonyl- methylene)triphenyl-phospharane in a non-polar solvent such as toluene or xylenes under refluxing conditions to give the desired olefin 12.
- Hydrogenation of the double bond can be accomplished using standard conditions such as H 2 (g), Pd/C in a suitable polar solvent like methanol or ethanol to give 13.
- Removal of the methyl group in the methoxynaphthyl moiety can be accomplished with boron tribromide at low temperature, followed by a careful quenching of the reaction with methanol to give the trans- esterified product 14. Saponification of the ester was accomplished using conditions described earlier.
- the naphthol can be protected as the TBS ether using TBSOTf or TBS-Cl in the presence of a suitable base such as triethylamine or imidazole in dichloromethane.
- the TBS ester can be hydrolyzed with a mild acid such as acetic acid in THF-H 2 O to give the desired acid 17.
- Compounds with the structure 28 can be prepared by the chemistry outlined in Scheme 5.
- a suitable tetralone such as 20 with LDA at low temperature followed by the addition of a suitable acylating agent such as 4-chloro-4-oxobutyrate provides the desired diketo-ester 21.
- the ester can be saponified using standard conditions known to one skilled in the art to give the acid 22.
- the di- ketone 22 can be converted to the fused isoxazole of the structure 23 by refluxing with hydroxylamine hydrochloride in the presence of a base such as triethylamine in an alcoholic solvent such as methanol or ethanol.
- De-protection of the methyl ether can be done with boron tribromide in a suitable solvent such as dichloromethane to give the desired alcohol 24.
- a silylating agent such as TBS-Cl in the presence of a base such as imidazole or triethylamine in a chlorinated solvent like DCM gives the bis-silyl protected ester 25.
- the silyl ester 25 can be treated with oxalyl chloride in a solvent such as DCM under anhydrous conditions followed by coupling with methyl 2- aminocyclo-pent-1-ene-l-carboxylate to give the desired amide 26.
- the TBS group can be removed using aqueous TBAF.
- the methyl ester can be saponified using standard conditions to give compounds of the structure 28.
- Scheme 6 outlines the strategy used to synthesize compounds of the structure 32.
- Coupling commercially available methyl or ethyl 2-aminocyclo-hex-l-ene-l-carboxylate 5 or 6 with 3-(3- bromophenyl) propionic acid 29 in the presence of methanesulfonyl chloride and DMAP gives the desired amide 30.
- the bromide 30 can be converted to 31 via a Suzuki reaction with a suitable boronic acid such as 4-hydroxy phenyl boronic acid in the presence of a catalyst such as Bis-tert-butyl-ferrocene palladium dichloride.
- the ester can be saponified by methods known to those skilled in the art providing compounds of the structure 32.
- Scheme 7 outlines the strategy used to synthesize compounds of the structure 37. Homologating aldehyde 33, followed by reduction provides 34 which may be resolved into its enantiomers via chiral BDPLC . One enantiomer is shown in Scheme 7 for illustrative purposes. Hydrolysis of the ethyl esters provides the acid 35, followed by demethylation and silylation to give 36. This intermediate can be acylated with the cyclopentene fragment, and saponified to provide biaryl products such as 37. Scheme 8
- Scheme 8 outlines the strategy used to synthesize compounds of the structure 43.
- the aminobenzothiazole 38 may be N-alkylated and cyclized to form intermediate 39.
- the ester can be reduced to the aldehyde, and homologated to the enoate 40.
- This intermediate can then be reduced and saponified to provide the acid 41.
- Demethylation and silylation affords intermediate 42, which can be acylated and saponified once again as in Scheme 7 above, to provide products such as 43.
- Scheme 9 outlines the strategy used to synthesize compounds of the structure 50.
- Adiponitrile can be converted to the amino-nitrile 45 via a Thorpe-Ziegler reaction using a suitable base such as LDA in a solvent such as THF. Coupling the amino nitrile 45 to 3-(4-bromophenyl) propionic acid 46 in the presence of methanesulfonyl chloride and DMAP gives the desired amide 47.
- the bromide 47 can be converted to 49 via a Suzuki reaction with a suitable boronic acid such as phenyl boronic acid in the presence of a catalyst such as 1,1 bis(di-ter ⁇ butylphosphino)-ferrocene palladium dichloride.
- Scheme 10 outlines the strategy used to synthesize compounds of the structure 54.
- the heterocyclic bromo aldehyde 51 can be homologated to the intermediate 52 via several transformations including the displacement of an activated bromide with a malonate anion.
- the bromide 52 can be arylated and demethylated to provide acid 53, which in turn may be acylated and deprotected to provide compounds such as 54.
- Scheme 11 displays a method for generating compounds of the structure 55.
- the acid intermediate 52 from Scheme 10 above may be acylated, the bromide coupled with a heterocyclic boronate ester, and this intermediate deprotected to provide compounds such as 55.
- Scheme 12
- Scheme 12 demonstrates a synthetic strategy to access compounds of the structure 59.
- a pyridyl bromo nitrile such as 56
- the bromide may be displaced, the nitrile transformed to an N-hydroxy amidine, this intermediate acylated and then cyclized to provide the intermediate 57.
- Saponification of 57 can give acid 58, which may be acylated, and after deprotection provide compounds such as 59.
- the pyrazole 60 may be N-arylated, this intermediate homologated to nitro 61, which in turn may be transformed to hydroxy acid 62. Upon acylation and deprotection, compounds such as 63 may be obtained.
- Scheme 14 displays a method to access compounds of the structure 66.
- the intermediate 64 generated in Scheme 12 may be elaborated into 65 via an intermediate oxime cycloaddition with an alkyne.
- the hydroxy acid 65 can be protected, acylated and deprotected to provide compounds such as 66.
- Scheme 15 shows a synthetic route used to generate compounds of the structure 68.
- the intermediate 67 may be accessed from an oxidative cleavage of the requisite olefin. This alpha-methyl acid 67 may then be condensed with an N-hydroxy amidine, and this intermediate elaborated into compounds such as 68 using methods illustrated in the Schemes above.
- Scheme 16 outlines the strategy used to synthesize compounds of the structure 70. This methodology follows closely to that illustrated in Scheme 15 above, where intermediate 69 can now be doubly deprotected in one step to provide compounds such as 70, containing a methyl group alpha to the amide moiety.
- Scheme 17 displays methodology to synthesize compounds of the structure 74.
- An amino heterocycle such as 71 can be converted to its halide and displaced with a nitrogen anionic heterocycle, followed by hydroxyl introduction to provide intermediate 72.
- This ester 72 may be homologated to 73, and upon acylation and protecting group manipulation, converted to compounds such as 74.
- Scheme 18 displays a method for generating compounds of the structure 77.
- the intermediate 75 can be accessed from 4-methoxyaniline via dipolar cycloaddition, and then homologated into 76. Following some of the methods illustrated in the Schemes above, 76 can be converted into compounds such as 77.
- Scheme 19 displays a method to access compounds of the structure 80.
- Alkyne 78 may be homologated and undergo a cycloaddition reaction to generate intermediate 79.
- the enoate 79 may then be converted into compounds such as 80 using methods illustrated in the Schemes above.
- Scheme 20
- Scheme 20 illustrates a strategy used to synthesize compounds of the structure 83.
- Malic acid 81 can be orthogonally protected, condensed with an N-hydroxy amidine, and deprotected to generate 82.
- the bis-hydroxyacid 82 may be globally silylated, and then acylated and deprotected to provide alpha-hydroxy compounds such as 83.
- Scheme 21 displays a strategy used to generate compounds of the structure 86.
- Orthogonally protected acid ester intermediate 84 can be obtained via oxidative degradation of the requisite olefinic starting material.
- the acid 84 may then be condensed with an N-hydroxy amidine, and manipulated to provide a primary carboxamide 85.
- a primary carboxamide intermediate such as 85 may undergo a coupling reaction with an enol triflate, and upon further deprotection reactions, provide geminal dimethyl compounds such as 86.
- Scheme 22 outlines a methodology to access compounds of the structure 88.
- Scheme 23 displays a strategy to access compounds of the structure 89.
- Commercially available symmetrical ketones such as 4-ethylcyclohexanone, can be acylated with Mander's reagent, followed by enol triflate formation with Comins' reagent.
- Using similar metal catalyzed coupling methodology illustrated in Scheme 21, different regioisomerically substituted cyclohexene compounds such as 89 may be accessed.
- Scheme 24 illustrates a methodology to access compounds of the structure 90.
- Commercially available 3-(trifluoromethyl)phenol can be reduced to a hydroxy cyclohexane, oxidized with Dess-Martin reagent to the ketone, acylated with Mander's reagent, and followed by enamine formation.
- compounds such as 90 may be obtained that possess a trifluoromethyl substituted cyclohexene.
- Scheme 25 displays a strategy to access compounds of the structure 91.
- Commercially available 3-methyl-2-cyclohexen-l-one can be substituted to generate a 3-geminal-dialkyl cyclohexanone.
- enamine formation Upon acylation with Mander's reagent, enamine formation, and following similar methodologies illustrated above, compounds such as 91 may be obtained that possess a geminal dialkyl substituted cyclohexene.
- Scheme 26 shows a method to access compounds of the structure 94.
- Commercially available pyridine 92 can be fluorinated and incorporated into a fluoro biaryl intermediate such as 93. Subsequent hydrolysis, acylation, and saponification following similar methodologies illustrated above, may provide fluoropyridyl compounds such as 94.
- Scheme 27 illustrates a method to generate compounds of the structure 97.
- Scheme 28 displays methodology to access compounds of the structure 100.
- Commercially available tetrahydro-4-H-pyran-4-one can be converted into the dihydropyran triflate intermediate 98 via similar methodologies described above.
- commercially available 6- methoxy-2-naphthaldehyde can be converted into the primary carboxamide intermediate 99, also via similar methodologies described above.
- Intermediates 98 and 99 may be coupled under similar metal catalyzed methodology illustrated in Scheme 21, to generate compounds such as 100 that possess a dihydropyran carboxylic acid moiety.
- Scheme 29 illustrates a methodology to access compounds of the structure 105.
- the ketone 101 (for preparation see Danishefsky, et alJ. Am. Chem. Soc. 2004, 126, 14358) can be converted to the olefin 102, followed by reduction of the double bond using standard hydrogenation conditions, and acid catalyzed removal of the ketal protecting group to provide the ketone 103.
- This material can be acylated using Mander's reagent to give the desired ketoester that is converted to the enol trifate 104 with Comins' reagent.
- Intermediates 104 and 99 may be coupled using similar metal catalyzed methodology illustrated in Scheme 21. Saponification of the methyl ester using standard conditions can generate vicinal disubstituted cyclohexene compounds such as 105.
- nM nanomolar
- the aqueous layer was further extracted with 30% of isopropanol in chloroform (2 x 30 mL). The organic fractions were combined, dried with sodium sulfate and concentrated in vacuo to give the tricycle as a pale yellow solid.
- This intermediate was dissolved in dichloromethane (20 mL) and borontribromide (10 mL, 1 M in dichloromethane) was added at 0 0 C. The resulting dark solution was stirred at room temperature for 4 h before it was quenched with 10O mL of water at O 0 C. The mixture was extracted with 30% isopropanol in chloroform. The aqueous layer contained a lot of product as a yellow solid, which was collected by filtration.
- Example 6 was stirred at room temperature for 3 h. The mixture was concentrated and dissolved in DMSO. The mixture was purified by reverse phase HPLC (Gilson) to provide Example 6 as a light brown solid.
- 1 H NMR acetone-d 6 , 500 MHz) ⁇ 10.4 (IH, s), 7.44 (IH, d), 6.85 (s, IH), 6.80 (IH, dd), 3.13 (2H, t), 2.98 (4H, q), 2.83 (2H, t), 2.73 (2H, t), 2.49 (2H, t), 1.87 (2H, t); LCMS m/z 369 (M+l).
- Example 15 utilized triethylamine as base instead of imidazole/DMAP described for the TBSCl silylation step in Example 6.
- This enoate intermediate (1.74 g, 5.8 mmol) and Pd/C (10%, 170 mg) in 200 mL of methanol was stirred under 1 atm of hydrogen gas (balloon) for 12 h.
- the slurry was filtered and concentrated in vacuo.
- the residue was dissolved in ethanol/methanol (1:1) and purified by chiral OJ-H (9 mL/min, 28% isopropanol/heptane, isocratic, 40 min/run) to give the enantiomers as white solids. Elution times of these enantiomeric intermediates were 18 min and 22 min using analytical Chiralcel-OJ, (25% isopropanol in heptane, isocratic).
- the ethyl ester enantiomer (400 mg, 1.32 mmoL) was combined with concentrated HCl (2 mL) and 4 mL of acetic acid, and was heated at 80 0 C for 3 h. The mixture was concentrated in vacuo, and to it was added 15 mL of water. The mixture was extracted with 30% isopropanol/chloroform. The organic layer was dried with sodium sulfate and concentrated in vacuo to give the acid product as a white solid.
- Example 17 The enantiomers of Example 17 were prepared under similar conditions as described in the Examples above.
- Example 18 was obtained.
- Example 19 was prepared under similar conditions as described in the Examples above. 1 H NMR (methanol ⁇ , 500 MHz) ⁇ 8.04 (IH, s), 7.83 (IH, d), 7.37 (IH, d), 7.08 (IH, dd), 3.12 (2H, t), 2.93 (2H, m), 2.81 (2H, t), 2.34 (2H, m), 1.65 (4H, m); LCMS m/z 386 (M+l).
- This material was purified by flash chromatography using 15 % ethyl acetate-hexanes as the eluant to give the cyclopentene aminonitrile as an off white solid.
- This intermediate cyclopentene aminonitrile was coupled with 3-(4-bromophenyl) propionic acid, using similar procedures as described in the Examples above, to provide the amide bromide.
- Example 23 was collected by filtration to give the desired Example 23 as a light brown solid.
- methylpyrazole 820 mg in one portion. After 2 h, to this mixture was added the pyridyl nitrobromide (1.83 g). The mixture was then heated at 130 0 C overnight. To the resulting mixture was added 100 mL of water and 100 mL of ethyl acetate. The aqueous layer was extracted with 100 mL of dichloromethane. The combined organic layers were dried with sodium sulfate and concentrated in vacuo to give a green slurry. To the slurry was added hexane to remove the mineral oil. The mixture was then filtered to give the pyridylpyrazole as a green solid.
- Example 24 1 H NMR (Acetone-d 6 , 500 MHz) ⁇ 11.7 (IH, s), 8.32 (IH, s), 8.01 (IH, s), 7.78 (IH, d), 7.56 (IH, s), 7.40 (IH, d), 2.93 (2H, m), 2.87 (2H, t), 2.63 (2H, t), 2.32 (2H, m), 1.60 (4H, m); LCMS m/z 357 (M+l).
- the cyanopyridine prepared above was reduced with DIBAL-H under standard conditions, and the aldehyde (173 mg), in 5 mL of THF and 2 mL of water was combined with hydroxylamine hydrochloride (99 mg). The mixture was stirred for 5 h and concentrated in vacuo. The residue was purified by Biotage eluting with 5%-20% of ethyl acetate in 1:1 mixture of dichloromethane and hexane to give the oxime.
- Example 26 To this acid intermediate (1 g) in 10 mL of toluene was added thionyl chloride (2 mL) at room temperature. The mixture was heated at 80 0 C for 1 h, and the volatiles were removed and azetroped with toluene. The residue was then dissolved in pyridine (10 mL), and to the mixture was added the N-hydroxy amidine (1.0 g) described in the above Examples. The resulting mixture was heated at 120 0 C for 2 h and then purified by Biotage (5-40% ethyl acetate in hexane) to give the oxadiazole as a brown oil. Following similar procedures described in the Examples above, acylation and deprotection gave the desired Example 26 as a white solid.
- This oxadiazole intermediate (5 g) was purified by chrial AD-H to give two enantiomers.
- LiOH 15 mL, 1 N
- the residue was extracted with 30% isopropanol in chloroform.
- the organic layer was dried with sodium sulfate. The removal of solvent in vacuo gave the acid containing some inorganic salt.
- Example 27 These enantiomers of Example 27 were subsequently repurified and resolved by preparative SFC chiral chromatography (ChiralPak AD, 35% methanol(TFA) - CO 2 ) to obtain each enantiomer at 98-99% ee.
- Enantiomer A 1 H NMR (DMSO-d 6 , 500 MHz) ⁇ 11.7 (IH, s), 10.7 (IH 5 bs), 8.27 (IH, d), 7.88 (IH, d), 7.31 (IH, dd), 3.24 (IH 5 dd), 3.07 (IH 5 dd) 5 3.02 (IH 5 m), 2.75 (2H, m), 2.21 (2H, m), 1.51 (4H 5 m), 1.27 (3H, d); LCMS m/z 373 (M+l).
- Enantiomer B 1 H NMR (CD 3 OD-d 6 , 500 MHz) ⁇ 8.25 (IH 5 d), 8.05 (IH, d), 7.42 (IH, dd), 3.34 (IH, dd), 3.12 (2H, m), 2.87 (2H 5 m), 2.34 (2H, m), 1.62 (4H 5 m), 1.37 (3H, d); LCMS m/z 373 (M+l).
- Example 28 was obtained as a white solid.
- 1 H NMR (Acetone-d 6 , 500 MHz) ⁇ 11.8 (IH, s), 7.84 (IH, s), 7.42 (IH, s), 7.27 (IH, s), 3.13 (2H, t), 2.95 (2H, t), 2.71 (2H, t), 2.34 (2H, m), 1.62 (4H, m); LCMS m/z 363 (M+l).
- Example 29 was obtained as a white solid.
- Example 31 was obtained after silylation, amide formation and hydrolysis.
- Example 32 The precipitate was collected by filtration, washed with water and diethyl ether to give the product as a crude material. This material was dissolved in 30% isopropanol in chloroform and filtered. The filtrate was concentrated to a small volume and the homogeneous mixture was kept at 0 0 C overnight. The precipitate was collected, washed with methanol, then diethyl ether and dried under vacuum to provide Example 32 as a white solid.
- Example 27 As before, the enantiomeric ester intermediates from Example 27 above were each converted to their respective acids. One enantiomeric acid was then submitted directly for the next amidation step with the methyl (R)-cyclohexene aminoester as described in the Examples above. Following the similar hydrolysis procedures as described in the Examples above, and subsequent purification by reverse phase HPLC, provided the third diastereomer of Example 33 containing minor epimerization.
- Example 34 The enantiomers of Example 34 were generated from the respective methyl (R)- cyclohexene aminoester and methyl (S)-cyclohexene aminoester intermediates prepared in Example 33 above. These enantiomeric methyl cyclohexene aminoesters were acylated with the requisite carboxylic acid intermediate from Example 23, and the amides converted to the desired products following the procedures described in the Examples above.
- the methyl ester intermediate from Example 23 can be directly converted to its primary carboxamide.
- a solution of this methyl ester in dioxane in a pressure tube was added 7N ammonia in methanol.
- the resulting solution was heated at 70 0 C for 16 hours.
- the reaction mixture was cooled to room temperature and concentrated to give the desired primary carboxamide intermediate as a white solid.
- the residue was purified by flash chromatography using 10% ethyl acetate-hexanes to give the ketoester as a colorless oil.
- This ketoester intermediate (860 mg, 3.83 mmol) in methanol was added ammonium acetate (1.48 g, 19.2 mmol). After stirring the mixture at room temperature for 16 h, it was concentrated. The residue was diluted with ethyl acetate and washed with water. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated. A white solid of the trifluorocyclohexene aminoester was obtained.
- the carboxylic acid intermediate from Example 23 can be used to acylate this trifluorocyclohexene aminoester.
- anhydrous dichloromethane 5 mL
- oxalyl chloride 0.562 mL, 2.0 M solution in DCM
- the reaction mixture was warmed to 23 °C and stirred for 30 min.
- the reaction mixture was concentrated, and the residue was dissolved in anhydrous dichloromethane.
- the carboxylic acid intermediate from Example 23 can be used to acylate this cyclohexene aminoester.
- oxalyl chloride 0.281 mL, 0.563 mmol
- the solution was stirred at RT for 30 min, then concentrated.
- methylene chloride 3 mL
- a solution of the cyclohexene aminoester intermediate 129 mg, 0.703 mmol
- methylene chloride 2 mL
- Example 39 was obtained after amide formation and hydrolysis. The residue was purified by reverse phase HPLC to provide Example 39.
- 1 H NMR 500 MHz, DMSO-d 6 ) ⁇ 11.66 (s, IH), 8.76 (d, IH), 8.14 (dd, IH), 7.94 (m, 1), 3.24 - 3.20 (m, 7H), 3.01 - 2.89 (m, 3H), 2.43 (t, IH), 2.37 (d, IH), 2.16 (m, IH) 5 1.75 (m, IH), 1.68 (br d, IH), 1.14 (m, IH); LCMS m/z 403 (M-I).
- Potassium hexamethyldisilazide 35 mL of a 0.5 M solution in THF, 17.5 mmol was added to propyl triphenylphosphonium bromide (7.1g, 18.5 mmol) in toluene (75 mL) at 0 0 C. The solution was stirred for 15 min, and the ketone (2.1 g, 12.3 mmol) in toluene (50 mL) was added. The solution was stirred at 0 0 C for 1 h and then heated at 100 0 C overnight. Solvent was removed, and the product was purified by flash chromatography (Biotage, Horizon) 0 to 10% ethyl acetate/hexanes.
- the product was dissolved in methanol (150 mL) and stirred over palladium on carbon (5%, Ig) under an atmosphere of hydrogen overnight. The solution was filtered through celite and solvent was removed. The product was dissolved in THF/MeOH/3N HCl (50mL/20mL/10mL) for 36 h. The mixture was neutralized with saturated sodium bicarbonate, and the solvent was removed. The solution was washed with ethyl acetate, and the resulting organic layer was washed with brine and dried over Na 2 SO 4 . The product was purified by flash chromatography (Biotage, Horizon) 0 to 20% ethyl acetate/hexanes.
- Example 41 1 HNMR(SOO MHz, DMSO-d 6 ) ⁇ 7.65 (d, IH), 7.58 (d, IH), 7.56 (s, IH), 7.28 (d, IH), 7.06-7.02 (m, 2H), 2.97-2.88 (m, 3H), 2.65-2.63 (m, 3H), 2.45-2.33 (m, 2H), 1.84-1.71 (m, IH), 1.65-1.62(m, IH), 1.51-1.15(m, 4H),0.901 (d, 3H), 0.86 (t, 3H); LCMS m/z 396 (M+l).
- niacin receptor affinity and function The activity of the compounds of the present invention regarding niacin receptor affinity and function can be evaluated using the following assays:
- Membrane preps are stored in liquid nitrogen in:
- the compounds of the invention generally have an IC 5 O in the 3 H-nicotinic acid competition binding assay within the range of 1 nM to about 25 ⁇ M.
- Membranes prepared from Chinese Hamster Ovary (CHO)-Kl cells stably expressing the niacin receptor or vector control (7 ⁇ g/assay) were diluted in assay buffer (100 mM HEPES, 100 mM NaCl and 10 mM MgCl 2 , pH 7.4 ) in Wallac Scintistrip plates and pre-incubated with test compounds diluted in assay buffer containing 40 ⁇ M GDP (final [GDP] was 10 ⁇ M) for ⁇ 10 minutes before addition Of 35 S-GTPyS to 0.3 nM. To avoid potential compound precipitation, all compounds were first prepared in 100% DMSO and then diluted with assay buffer resulting in a final concentration of 3% DMSO in the assay.
- assay buffer 100 mM HEPES, 100 mM NaCl and 10 mM MgCl 2 , pH 7.4
- Binding was allowed to proceed for one hour before centrifuging the plates at 4000 rpm for 15 minutes at room temperature and subsequent counting in a TopCount scintillation counter. Non-linear regression analysis of the binding curves was performed in GraphPad Prism.
- CHO-Kl cell culture medium F-12 Kaighn's Modified Cell Culture Medium with 10% FBS, 2 mM L- Glutamine, 1 mM Sodium Pyruvate and 400 ⁇ g/ml G418
- the pellet may be frozen at -80°C for later use or it can be used immediately.
- Guanosine 5 '-diphosphate sodium salt (GDP, Sigma-Aldrich Catalog #87127)
- the compounds of the invention generally have an EC 50 in the functional in vitro GTP7S binding assay within the range of about less than 1 uM to as high as about 100 ⁇ M.
- mice Male C57B16 mice ( ⁇ 25g) are anesthetized using 10mg/ml/kg Nembutal sodium. When antagonists are to be administered they are co-injected with the Nembutal anesthesia. After ten minutes the animal is placed under the laser and the ear is folded back to expose the ventral side. The laser is positioned in the center of the ear and focused to an intensity of 8.4-9.0 V (with is generally ⁇ 4.5cm above the ear). Data acquisition is initiated with a 15 by 15 image format, auto interval, 60 images and a 20sec time delay with a medium resolution. Test compounds are administered following the 10th image via injection into the peritoneal space. Images 1-10 are considered the animal's baseline and data is normalized to an average of the baseline mean intensities.
Abstract
Description


























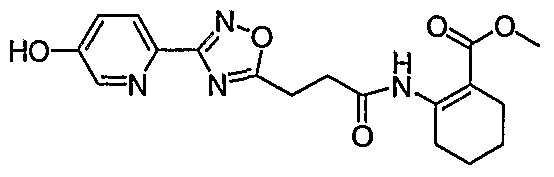



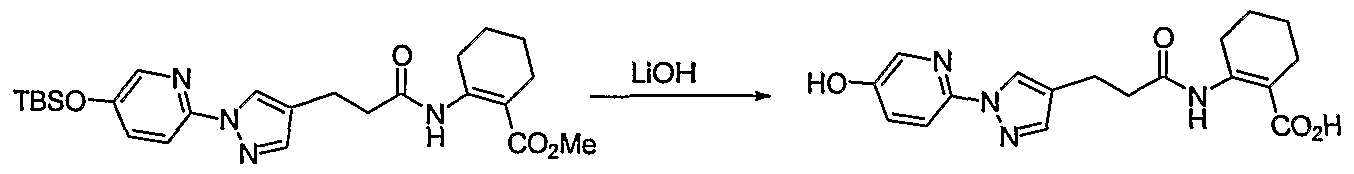

















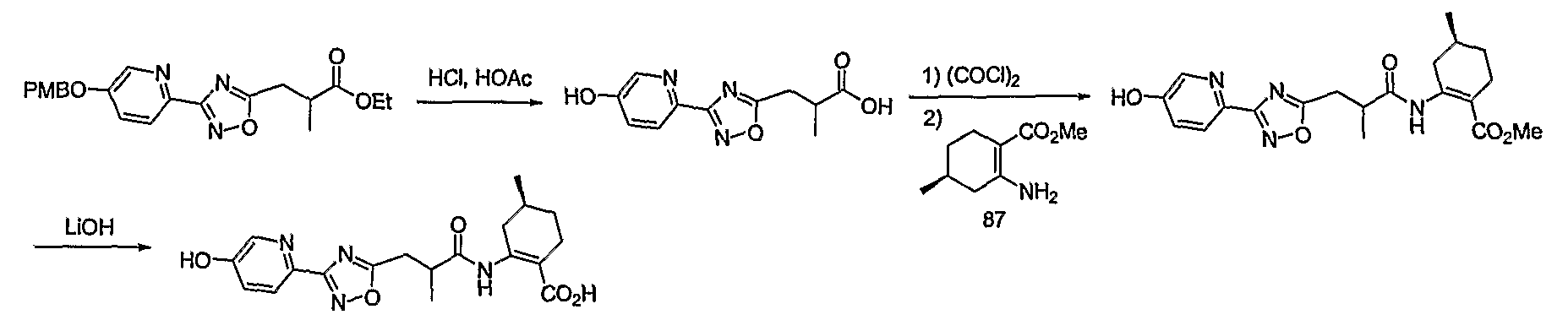










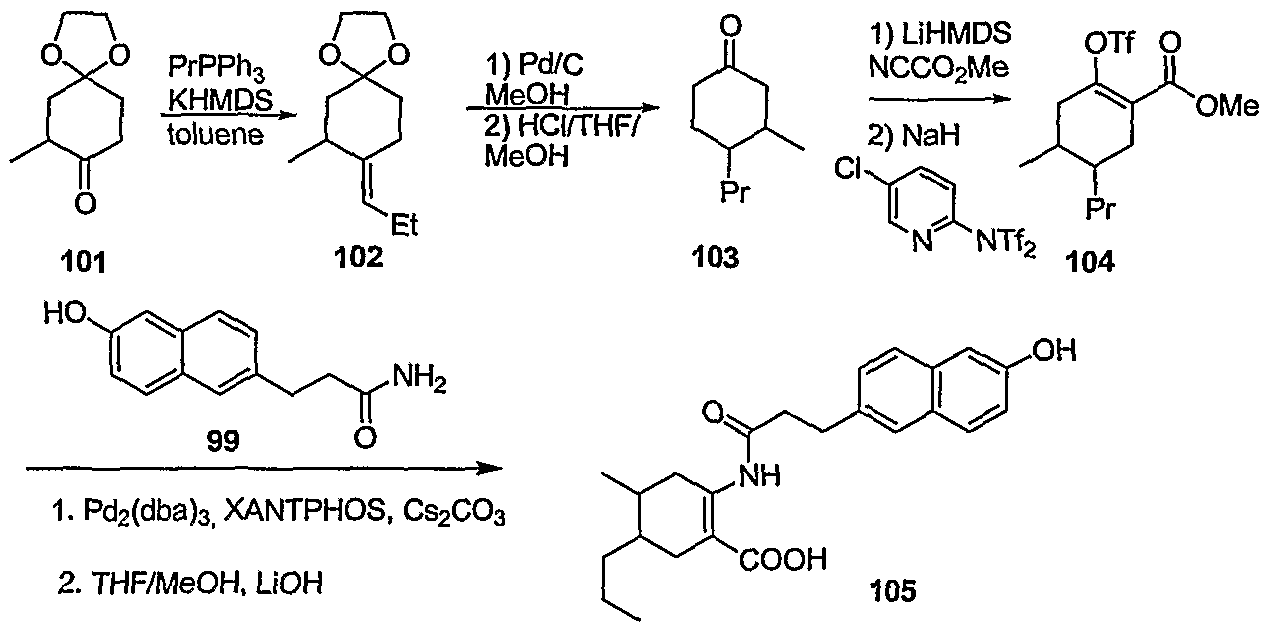










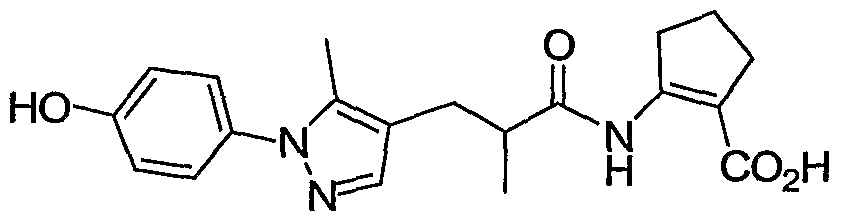



















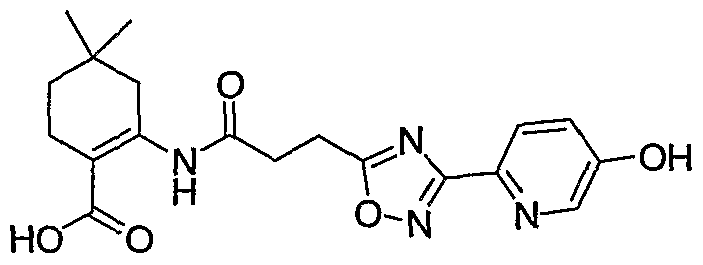




Claims









Priority Applications (16)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT06785553T ATE499932T1 (en) | 2005-06-28 | 2006-06-26 | NIACIN RECEPTOR AGONISTS, COMPOSITIONS CONTAINING SUCH COMPOUNDS AND TREATMENT METHODS |
| BRPI0612117-9A BRPI0612117A2 (en) | 2005-06-28 | 2006-06-26 | compound or a pharmaceutically acceptable salt or solvate thereof, pharmaceutical composition, and use of a compound |
| US11/922,629 US8168649B2 (en) | 2005-06-28 | 2006-06-26 | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| JP2008519441A JP5113046B2 (en) | 2005-06-28 | 2006-06-26 | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| PL06785553T PL1901731T3 (en) | 2005-06-28 | 2006-06-26 | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| CA002611552A CA2611552A1 (en) | 2005-06-28 | 2006-06-26 | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| DK06785553.6T DK1901731T3 (en) | 2005-06-28 | 2006-06-26 | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| MX2008000113A MX2008000113A (en) | 2005-06-28 | 2006-06-26 | Niacin receptor agonists, compositions containing such compounds and methods of treatment. |
| AU2006261839A AU2006261839B2 (en) | 2005-06-28 | 2006-06-26 | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| SI200631019T SI1901731T1 (en) | 2005-06-28 | 2006-06-26 | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| EP06785553A EP1901731B1 (en) | 2005-06-28 | 2006-06-26 | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| DE602006020447T DE602006020447D1 (en) | 2005-06-28 | 2006-06-26 | NIACIN RECEPTOR AGONISTS, COMPOSITIONS WITH DE |
| IL188329A IL188329A0 (en) | 2005-06-28 | 2007-12-23 | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| NO20080484A NO20080484L (en) | 2005-06-28 | 2008-01-25 | Niacin Receptor Antagonists, Preparations Containing Such Compounds, and Treatment Methods |
| HR20110295T HRP20110295T1 (en) | 2005-06-28 | 2011-04-21 | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| US13/423,618 US20120178750A1 (en) | 2005-06-28 | 2012-03-19 | Niacin Receptor Agonists, Compositions Containing Such Compounds and Methods of Treatment |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US69471105P | 2005-06-28 | 2005-06-28 | |
| US60/694,711 | 2005-06-28 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/423,618 Division US20120178750A1 (en) | 2005-06-28 | 2012-03-19 | Niacin Receptor Agonists, Compositions Containing Such Compounds and Methods of Treatment |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007002557A1 true WO2007002557A1 (en) | 2007-01-04 |
Family
ID=37134234
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/024740 WO2007002557A1 (en) | 2005-06-28 | 2006-06-26 | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
Country Status (31)
| Country | Link |
|---|---|
| US (3) | US8168649B2 (en) |
| EP (1) | EP1901731B1 (en) |
| JP (1) | JP5113046B2 (en) |
| KR (1) | KR20080019653A (en) |
| CN (1) | CN101208082A (en) |
| AR (1) | AR057408A1 (en) |
| AT (1) | ATE499932T1 (en) |
| AU (1) | AU2006261839B2 (en) |
| BR (1) | BRPI0612117A2 (en) |
| CA (1) | CA2611552A1 (en) |
| CR (1) | CR9589A (en) |
| CY (1) | CY1112019T1 (en) |
| DE (1) | DE602006020447D1 (en) |
| DK (1) | DK1901731T3 (en) |
| EC (1) | ECSP078018A (en) |
| ES (1) | ES2360726T3 (en) |
| HR (1) | HRP20110295T1 (en) |
| IL (1) | IL188329A0 (en) |
| MA (1) | MA29647B1 (en) |
| MX (1) | MX2008000113A (en) |
| MY (1) | MY141024A (en) |
| NI (1) | NI200700319A (en) |
| NO (1) | NO20080484L (en) |
| PE (2) | PE20070097A1 (en) |
| PL (1) | PL1901731T3 (en) |
| PT (1) | PT1901731E (en) |
| RU (1) | RU2008102936A (en) |
| SI (1) | SI1901731T1 (en) |
| TW (1) | TWI313258B (en) |
| WO (1) | WO2007002557A1 (en) |
| ZA (1) | ZA200710645B (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1983993A2 (en) * | 2006-02-07 | 2008-10-29 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2010004972A1 (en) | 2008-07-08 | 2010-01-14 | 第一三共株式会社 | Nitrogen-containing aromatic heterocyclyl compound |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| US8168649B2 (en) | 2005-06-28 | 2012-05-01 | Merk Sharp & Dohme Corp. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| US8354418B2 (en) | 2006-04-06 | 2013-01-15 | Boehringer Ingelheim International Gmbh | Thiazolyl-dihydro-quinazolines |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US9574117B2 (en) | 2008-07-02 | 2017-02-21 | 3M Innovative Properties Company | Low surface energy adhesive |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7094572B2 (en) * | 2003-03-14 | 2006-08-22 | Bristol-Myers Squibb | Polynucleotide encoding a novel human G-protein coupled receptor variant of HM74, HGPRBMY74 |
| WO2007027532A2 (en) * | 2005-08-29 | 2007-03-08 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| EP2640366A2 (en) | 2010-11-15 | 2013-09-25 | Exelixis, Inc. | Benzoxazepines as inhibitors of pi3k/mtor and methods of their use and manufacture |
| US20140113824A1 (en) | 2011-05-10 | 2014-04-24 | Bayer Intellectual Property Gmbh | Bicyclic (thio)carbonylamidines |
| KR20150047609A (en) | 2012-08-30 | 2015-05-04 | 니뽄 신야쿠 가부시키가이샤 | Pyridine derivative and medicine |
| CN108290839A (en) | 2015-12-01 | 2018-07-17 | 巴斯夫欧洲公司 | Pyridine compounds as fungicide |
| BR112018010316A2 (en) | 2015-12-01 | 2018-12-04 | Basf Se | compounds of formula, composition, use of a compound of formula, method for combating phytopathogenic fungi and seed |
| CR20190580A (en) | 2017-05-30 | 2020-02-10 | Basf Se | Pyridine and pyrazine compounds |
| CN113563166B (en) * | 2019-04-24 | 2023-08-01 | 珠海市柏瑞医药科技有限公司 | Synthesis method of vanillyl alcohol ether |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005016870A1 (en) * | 2003-08-14 | 2005-02-24 | Smithkline Beecham Corporation | 2-substituted benzoic acid derivatives as hm74a receptor agonists |
| WO2006052555A2 (en) * | 2004-11-04 | 2006-05-18 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2006057922A2 (en) * | 2004-11-23 | 2006-06-01 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
Family Cites Families (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU6087194A (en) | 1993-01-15 | 1994-08-15 | Agouron Pharmaceuticals, Inc. | Hiv protease inhibitors |
| AU7966098A (en) | 1997-06-12 | 1998-12-30 | Smithkline Beecham Corporation | Hm74a receptor |
| WO1999026615A1 (en) * | 1997-11-24 | 1999-06-03 | Merck & Co., Inc. | Cyclic amino acid derivatives as cell adhesion inhibitors |
| IT1309593B1 (en) | 1999-03-05 | 2002-01-24 | Novuspharma Spa | HETEROCYCLIC COMPOUNDS WITH ANTI-TUMOR ACTIVITY. |
| EP1242448A2 (en) | 1999-11-17 | 2002-09-25 | Arena Pharmaceuticals, Inc. | Endogenous and non-endogenous versions of human g protein-coupled receptors |
| WO2001077320A2 (en) | 2000-04-05 | 2001-10-18 | Bayer Aktiengesellschaft | Regulation of human hm74-like g protein coupled receptor |
| AU2002242910A1 (en) | 2001-04-11 | 2002-10-28 | Glaxo Group Limited | Medicaments which are modulators of hm74 and/or hm74a activity |
| AUPR738301A0 (en) | 2001-08-30 | 2001-09-20 | Starpharma Limited | Chemotherapeutic agents |
| US6902902B2 (en) | 2001-11-27 | 2005-06-07 | Arena Pharmaceuticals, Inc. | Human G protein-coupled receptors and modulators thereof for the treatment of metabolic-related disorders |
| JP2005170790A (en) * | 2002-01-09 | 2005-06-30 | Ajinomoto Co Inc | N-alkylsulfonyl-substituted amide derivative |
| WO2004004665A2 (en) * | 2002-07-09 | 2004-01-15 | Bristol-Myers Squibb Company | Substituted heterocyclic derivatives useful as antidiabetic and antiobesity agents and method |
| GB0319126D0 (en) | 2003-08-14 | 2003-09-17 | Smithkline Beecham Corp | Chemical compounds |
| PE20050483A1 (en) | 2003-10-31 | 2005-08-25 | Arena Pharm Inc | TETRAZOLE DERIVATIVES OF FORMULA (I), ITS PHARMACEUTICAL COMPOSITIONS AND PROCESSES TO PRODUCE PHARMACEUTICAL COMPOSITIONS |
| KR101100601B1 (en) | 2004-02-14 | 2011-12-29 | 글락소스미스클라인 엘엘씨 | Novel compounds |
| WO2005082887A1 (en) * | 2004-02-26 | 2005-09-09 | Aska Pharmaceutical Co., Ltd. | Pyrimidine derivative |
| WO2006045565A1 (en) | 2004-10-22 | 2006-05-04 | Smithkline Beecham Corporation | Xanthine derivatives with hm74a receptor activity |
| PE20060949A1 (en) | 2004-12-23 | 2006-10-11 | Arena Pharm Inc | FUSED DERIVATIVES OF PIRAZOLE AS NIACIN RECEPTOR AGONISTS |
| GB0503056D0 (en) | 2005-02-14 | 2005-03-23 | Smithkline Beecham Corp | Chemical compounds |
| GB0503053D0 (en) | 2005-02-14 | 2005-03-23 | Smithkline Beecham Corp | Chemical compounds |
| US20080221108A1 (en) | 2005-02-14 | 2008-09-11 | Richard Hatley | Anthranilic Acid Derivatives As Hm74A Receptor Agonists |
| GB0503054D0 (en) | 2005-02-14 | 2005-03-23 | Smithkline Beecham Corp | Chemical compounds |
| BRPI0610133A2 (en) | 2005-05-17 | 2010-06-01 | Schering Corp | heterocycles as nicotinic acid receptor agonists for the treatment of dyslipidemia |
| AU2006259137B2 (en) | 2005-06-14 | 2010-04-01 | F. Hoffmann-La Roche Ag | Anthranilic acid derivatives |
| PL1901731T3 (en) | 2005-06-28 | 2011-08-31 | Merck Sharp & Dohme | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
-
2006
- 2006-06-26 PL PL06785553T patent/PL1901731T3/en unknown
- 2006-06-26 WO PCT/US2006/024740 patent/WO2007002557A1/en active Application Filing
- 2006-06-26 KR KR1020077030918A patent/KR20080019653A/en not_active Application Discontinuation
- 2006-06-26 CA CA002611552A patent/CA2611552A1/en not_active Abandoned
- 2006-06-26 ES ES06785553T patent/ES2360726T3/en active Active
- 2006-06-26 EP EP06785553A patent/EP1901731B1/en active Active
- 2006-06-26 JP JP2008519441A patent/JP5113046B2/en not_active Expired - Fee Related
- 2006-06-26 AR ARP060102740A patent/AR057408A1/en unknown
- 2006-06-26 SI SI200631019T patent/SI1901731T1/en unknown
- 2006-06-26 AT AT06785553T patent/ATE499932T1/en active
- 2006-06-26 RU RU2008102936/04A patent/RU2008102936A/en not_active Application Discontinuation
- 2006-06-26 CN CNA2006800231512A patent/CN101208082A/en active Pending
- 2006-06-26 DE DE602006020447T patent/DE602006020447D1/en active Active
- 2006-06-26 BR BRPI0612117-9A patent/BRPI0612117A2/en not_active IP Right Cessation
- 2006-06-26 US US11/922,629 patent/US8168649B2/en active Active
- 2006-06-26 US US11/474,646 patent/US20060293364A1/en not_active Abandoned
- 2006-06-26 PE PE2006000734A patent/PE20070097A1/en not_active Application Discontinuation
- 2006-06-26 DK DK06785553.6T patent/DK1901731T3/en active
- 2006-06-26 AU AU2006261839A patent/AU2006261839B2/en not_active Ceased
- 2006-06-26 PT PT06785553T patent/PT1901731E/en unknown
- 2006-06-26 MX MX2008000113A patent/MX2008000113A/en active IP Right Grant
- 2006-06-27 MY MYPI20063030A patent/MY141024A/en unknown
- 2006-06-27 TW TW095123164A patent/TWI313258B/en not_active IP Right Cessation
-
2007
- 2007-12-06 NI NI200700319A patent/NI200700319A/en unknown
- 2007-12-06 ZA ZA200710645A patent/ZA200710645B/en unknown
- 2007-12-13 CR CR9589A patent/CR9589A/en unknown
- 2007-12-14 EC EC2007008018A patent/ECSP078018A/en unknown
- 2007-12-23 IL IL188329A patent/IL188329A0/en unknown
-
2008
- 2008-01-25 NO NO20080484A patent/NO20080484L/en not_active Application Discontinuation
- 2008-01-28 MA MA30599A patent/MA29647B1/en unknown
-
2009
- 2009-12-15 PE PE2009001309A patent/PE20110541A1/en not_active Application Discontinuation
-
2011
- 2011-04-21 HR HR20110295T patent/HRP20110295T1/en unknown
- 2011-06-01 CY CY20111100527T patent/CY1112019T1/en unknown
-
2012
- 2012-03-19 US US13/423,618 patent/US20120178750A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005016870A1 (en) * | 2003-08-14 | 2005-02-24 | Smithkline Beecham Corporation | 2-substituted benzoic acid derivatives as hm74a receptor agonists |
| WO2006052555A2 (en) * | 2004-11-04 | 2006-05-18 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| WO2006057922A2 (en) * | 2004-11-23 | 2006-06-01 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8168649B2 (en) | 2005-06-28 | 2012-05-01 | Merk Sharp & Dohme Corp. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| EP1983993A4 (en) * | 2006-02-07 | 2010-09-22 | Merck Sharp & Dohme | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| EP1983993A2 (en) * | 2006-02-07 | 2008-10-29 | Merck & Co., Inc. | Niacin receptor agonists, compositions containing such compounds and methods of treatment |
| US8354418B2 (en) | 2006-04-06 | 2013-01-15 | Boehringer Ingelheim International Gmbh | Thiazolyl-dihydro-quinazolines |
| US9574117B2 (en) | 2008-07-02 | 2017-02-21 | 3M Innovative Properties Company | Low surface energy adhesive |
| RU2481330C2 (en) * | 2008-07-08 | 2013-05-10 | Дайити Санкио Компани, Лимитед | Nitrogen-containing aromatic heterocyclic compound |
| CN102149680A (en) * | 2008-07-08 | 2011-08-10 | 第一三共株式会社 | Nitrogen-containing aromatic heterocyclyl compound |
| US8648103B2 (en) | 2008-07-08 | 2014-02-11 | Daiichi Sankyo Company, Limited | Nitrogen-containing aromatic heterocyclyl compound |
| JP5461398B2 (en) * | 2008-07-08 | 2014-04-02 | 第一三共株式会社 | Nitrogen-containing aromatic heterocyclyl compounds |
| CN102149680B (en) * | 2008-07-08 | 2014-12-10 | 第一三共株式会社 | Nitrogen-containing aromatic heterocyclyl compound |
| US9150563B2 (en) | 2008-07-08 | 2015-10-06 | Daiichi Sankyo Company, Limited | Nitrogen-containing aromatic heterocyclyl compound |
| WO2010004972A1 (en) | 2008-07-08 | 2010-01-14 | 第一三共株式会社 | Nitrogen-containing aromatic heterocyclyl compound |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1901731B1 (en) | Niacin receptor agonists, compositions containing such compounds and methods of treatment | |
| US20100204278A1 (en) | Niacin Receptor Agonists, Compositions Containing Such Compounds and Methods of Treatment | |
| US20090062269A1 (en) | Niacin Receptor Agonists, Compositions Containing Such Compounds and Methods of Treatment | |
| US20090170891A1 (en) | Niacin Receptor Agonists, Compositions Containing Such Compounds and Methods of Treatment | |
| US20090042926A1 (en) | Niacin Receptor Agonists, Compositions Containing Such Compounds and Methods of Treatment | |
| EP1942905A2 (en) | Niacin receptor agonists, compositions containing such compounds and methods of treatment | |
| WO2006057922A2 (en) | Niacin receptor agonists, compositions containing such compounds and methods of treatment | |
| US20110028462A1 (en) | Niacin Receptor Agonists, compositions Containing Such Compounds and Methods of Treatment |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680023151.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 564042 Country of ref document: NZ |
|
| ENP | Entry into the national phase |
Ref document number: 2611552 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2007000769 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12007502824 Country of ref document: PH Ref document number: 2006261839 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2007-009589 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/000113 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11922629 Country of ref document: US Ref document number: 07134572 Country of ref document: CO Ref document number: 2006785553 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 188329 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2008519441 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020077030918 Country of ref document: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2006261839 Country of ref document: AU Date of ref document: 20060626 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200800223 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008102936 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0612117 Country of ref document: BR Kind code of ref document: A2 Effective date: 20071227 |