WO2006102759A1 - Novel substituted 2-nitroimidazoles useful for hypoxic cell therapy and imaging - Google Patents
Novel substituted 2-nitroimidazoles useful for hypoxic cell therapy and imaging Download PDFInfo
- Publication number
- WO2006102759A1 WO2006102759A1 PCT/CA2006/000482 CA2006000482W WO2006102759A1 WO 2006102759 A1 WO2006102759 A1 WO 2006102759A1 CA 2006000482 W CA2006000482 W CA 2006000482W WO 2006102759 A1 WO2006102759 A1 WO 2006102759A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- nitroimidazole
- deoxy
- compound
- cells
- present
- Prior art date
Links
- 0 C[C@]1OC(CO*)CC1* Chemical compound C[C@]1OC(CO*)CC1* 0.000 description 3
- HBMINVNVQUDERA-UHFFFAOYSA-N C[n]1c([N+]([O-])=O)ncc1 Chemical compound C[n]1c([N+]([O-])=O)ncc1 HBMINVNVQUDERA-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/052—Imidazole radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/041—Heterocyclic compounds
- A61K51/044—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins
- A61K51/0453—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/0491—Sugars, nucleosides, nucleotides, oligonucleotides, nucleic acids, e.g. DNA, RNA, nucleic acid aptamers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Abstract
The present invention provides for substituted 2-nitroimidazoles suitable for the therapeutic treatment of hypoxic tissues, particularly for application in radiotherapy, chemosensitization and radiosensitization The present invention further provides substituted 2-nitroimidazoles derivatives suitable for radioimaging of hypoxic cells.
Description
NOVEL COMPOUNDS FOR HYPOXIC CELL THERAPY AND IMAGING
RELATED APPLICATION
This application claims the benefit of United States Provisional Application Serial Number 60/665,876, filed March 29, 2005, under 35 U.S.C. 119(e). The entire disclosure of the prior application is hereby incorporated by reference.
FIELD OF THE INVENTION
The present invention relates to the fields of human therapeutics, diagnostics, radioimaging and chemotherapy.
BACKGROUND OF THE INVENTION
All of the publications, patents and patent applications cited within this application are herein incorporated by reference in their entirety to the same extent as if the disclosure of each individual publication, patent application or patent was specifically and individually indicated to be incorporated by reference in its entirety.
Decreased oxygen levels in tumor cells increases their resistance to the damaging effects of ionizing radiations (Tomlinson R.H., et al Br J Cancer 9:539 (1955)), an effect that is thought to greatly reduce the efficacy of conventional low linear energy transfer radiation (e.g. X-ray) therapies (Brown J.M. Cancer Res 59:5863 (1999)). 2- nitroimidaole (azomycin) nucleosides are highly diffusible radiosensitizers that readily permeate hypoxic tissues, where they are bioreductively activated by single electron transfer and subsequently selectively bound as molecular adducts within
viable hypoxic cells. The reversibility of this single electron reduction in the presence of oxygen limits adduct formation to cells that are pathologically hypoic (Bigalow, J.E. et al Biochem Pharmac 35:77 (1986)).
This oxygen-dependent selectivity forms the basis for non-invasive (imaging) diagnosis of a hypoxic region with radiolabeled nitroimidazoles. (Chapman J.D. et al Cancer 43:456 (1981); Adams, G.E. Radia Res 67:9 (1976)). In the past, a number of radioiodinated azomycin α-nucleosides have been synthesized and explored to detect and monitor regional hypoxia (Jette D.C. et al Radiat Res 105:169 (1986); Wiebe L.I in "Nuclear Medicine in Clinical Oncology" 402 (1986)). Of these, l-α-D-(5-dexoy- 5-iodorabinofuranosyl)-2-nitroimidazole (IAZA) has been widely studied and clinically used in a variety of pathologies involving tissue hypoxia (Parliament M.B. et al Br J Radiol 65:90 (1991); Groshar D. Nucl Med 34:885 (1993); Urtasun R.C. et al Br J Cancer 74:S209 (1996); Al-Arafaj A. et al Europ J Nucl Med 21:1338 (1994); McEwan A.J.B. et al J Nucl Med 38:300 (1997); Vinjamuri S. Clin Nucl Med 24:8912 (1999)).
Nitroimidazole radiosensitizers were used to overcome the Oxygen effect' through an oxygen mimicking processes that results in radiosensitization through selective bioactivation and consequent binding (adduct formation) to tissue components (Adams G.E. et al Int J Radiat Biol 15:457 (1969)). In this process, the first-electron reduction is reversible in the presence of oxygen, therefore, the ultimate degree of binding is dependent on the absence (low concentration) of oxygen. Reducing equivalents (electrons) for this process are metabolically-derived (Bigalow J.E. et al Biochem Pharmac ?>5:11 (1986)), and therefore the adduct-based accumulation of
SUBSTITUH SHEET (RULE 26)
azomycins is restricted to viable tissue that is O2-deficient, with no accumulation in necrotic cells and little accumulation and low toxicity in most normally-oxygenated cells.
Flavin-dependent cytochrome P450 reductase and related enzymes, including xanthine and aldehyde oxidases, and quinone oxidase are thought capable of carrying out this reductive bioactivation. Electron affinity of the substrate (e.g. azomycins) dictates both sensitivity to O2 and toxicity of the tracer. If the first, single-electron, reduction potential (first electron reduction potential, ES) approaches that of O2 (-155 mV), then selectivity for hypoxia will be diminished; if it is not sufficiently electron- affinic (E1 7 < -450 mV), then sensitivity will be lost. This step is critical, since it is reversible by O2 and is therefore responsible for selective binding to only those tissues that are O2 deficient. The EV s of most 2-nitroimidazoles lie around -390 mV, an electron affinity considered to be optimal for selectivity and sensitivity (Adams G.E. et al Radiat Res 67:9 (1976)). A schematic representation of these processes is depected in Figure 1.
Thus, hypoxia-sensitive radiopharmaceuticals are reduced by electrons produced during glycolysis and by the Krebs Cycle. Flavin-dependent cytochrome P450 reductase, and xanthine-, aldehyde- and quinone- oxidases are among the activating (i.e. reductive) enzymes. (Biaglow J.E. et al Biochem Pharmac 35:77 (1986)). The cell must be viable, even if oxidatively quiescent, to carry out this function, a property which discriminates between dead and stunned but salvageable tissue.
Hypoxic tissue is also ischemic. It is therefore equally important that the radiosensitizer is a facile tissue permeant, meaning that the molecules must be
SUBSTITUTE SHEET (RULE 28)
moderately lipophilic. The ability of radiosensitizers (and any compound that is not actively transported) to move freely across cell membranes is based on their lipophilicity (Brown J.M. et al Radiat Res 82:171 (1980)). However, if lipophilicity is too high, they will dissolve in lipoidal tissues and exhibit selective toxicities (e.g. neuropathies). If they are too hydrophilic, they will not diffuse readily through cell membranes. Moreover, hydrophilic compounds tend to be cleared very rapidly via the kidney, severely reducing the amount of tracer available for bioreductive activation and hypoxia-dependent binding.
The limitations of the halogenated azomycin compounds, in being transported into the cell and in establishing therapeutic and diagnosticly relevant residence time are known in the art. It is therefore an object of the present invention to describe a class of compounds capable of transport into a cell through equilabrative and/or concentrative means.
It is a further object of the present invention to describe a class of compounds capable of increased cellular residence time.
It is a further object of the present invention to describe a class of compounds capable of increased tumor specificity.
It is a further object of the present invention to describe a class of comopunds capable of increased therapeutic effect.
SUMMARY OF THE INVENTION
SUBSTiTUTE SHEET (RJLE #)
In another embodiment, the present invention provides for compounds suitable for diagnostics, radiotherapy, chemotherapy, radiosensitization and chemosensitization of hypoxic cells; said compounds selected from the group comprising (together "Compound(s) of the Present Invention")
l-β-D-(Substituted pentosyl/hexosyl)-2-nitroimidazoles and 1 -α-D-(Substituted furanosyl/hexopyranosyl)-2-nitroimidazoles, more particularly described as:

R = H, Ac, Bz, Piv, any halogen, TIPS, TBDPS, TBDMS, SO2R1; Ri = CH3, toluyl, CF3, p-nitrobenzene and any other Leaving

; and X= -CH2, or -CHCH2OH
SUBSTiTUTE SHEET (RUE 26)

l-β-D-[(2/3-Substituted) or (2,3-disusbstituted) or (2,2-disubstituted) or (3/3-disubstituted) furanosyl/hexopyranosyl)-2-nitroimidazoles; Wherein
-I (except at 2'-arabinose position),



S(JBSTiTUTE SHEET (WLE 26)

l-β-D-[(2/3-Epoxy)-5-susbstituted furanosyI/hexopyranosyl)-2- nitroimidazoles;
Wherein
R = OH, OAc, OBz, OPiv, any halogen or OSO2R1 substituents at these positions;
R] = CH3, toluyl, CF3, p-nitrobenzene and any other Leaving
Group.

X= -CH2, or -CHCH2OH
SUBSTITU7Ε SHEET (RULE 26)

l-α-D-[(2/3-Substituted) or (2,3-disusbstituted) or (2,2- disubstituted) or (3/3-disubstituted) furanosyl/hexopyranosyl)-2- nitroimidazoles. Wherein 5I,


SUBSTITUTE SHEET (RULE 26)

l-α-D-[(2/3-Epoxy)-5-susbstituted furanosyl/hexopyranosyl)-2- nitroimidazoles.
Wherein
R = OH, OAc, OBz, OPiv, every halogen and OSO2Ri substituents;
Ri = CH3, toluyl, CF3, p-nitrobenzene and any other Leaving
Group;

X= -CH2, or -CHCH2OH
The present invention further provides for said Compound of the Present Invention to contain a radionuclide suitable for radiotherapy, said radionuclide selected from the group consisting of 211At, 1251, 131I, 76Br, 77Br, 82Br, 34mCl and 24CI;
The present invention further provides for use of said compound in association with radiotherapy so as to render a hypoxic cell more susceptible to radiotherapy.
The present invention further provides for use of said compound in association with chemotherapy so as to render a hypoxic cell more susceptible to chemotherapy.
The present invention further provides for the use of said compound for radioimaging hypoxic cells wherein the halogen in said compound is replaced with a radionuclide suitable for radioimaging.
The present invention further provides for compounds suitable for radiotherapy, chemotherapy, radiosensitization and chmeosensitization of hypoxic cells; said compounds selected from the group consisting of: l-β-D-[3-deoxy-3-fluoroxylofuranosyl]-2-nitroimidazole;
l-β-D-[3-deoxy-3chloroxylofuranosyl]-2-nitroimidazole;
l-β-D-[3-deoxy-3-bromoroxylofuranosyl]-2-nitroimidazole;
1 -β-D-[3-deoxy-3-fluororibofuranosyl]-2-nitroimidazole; l-β-D-[3-deoxy-3-chlororibofuranosyl]-2-nitroimidazole; l-β-D-[3-deoxy-3-bromoribofuranosyl]-2-nitroimidazole; l-β-D-[2-deoxy-2-fluororibofuranosyl]-2-nitroimidazole;
l-β-D-[2-deoxy-2-chlororibofuranosyl]-2-nitroimidazole;
1 -β-D-[2-deoxy-2-bromoribofuranosyl]-2-nitroimidazole;
l-β-D-[3-deoxy-3-fluoroarabinofuranosyl]-2-nitroimidazole; l-β-D-[3-deoxy-3-chloroarabinofuranosyl]-2-nitroimidazole;
l-β-D-[3-deoxy-3-bromoarabinofuranosyl]-2-nitroimidazole; l-β-D-[2-deoxy-2-fluoroarabinofuranosyl]-2-nitroimidazole; 1 -β-D-[2-deoxy-2-chloroarabinofuranosyl]-2-nitroimidazole; l-β-D-[2-deoxy-2-bromoarabinofuranosyl]-2-nitroimidazole;
1 -β-D-[3 ,2-dideoxy-3 -fluoroarabinofuranosy 1] -2-nitroimidazole ;
srørruTE SHEET (RULE W)
l-β-D-[3,2-dideoxy-3-chloroarabinofuranosyl]-2-nitroimidazole;
l-β-D-[3,2-dideoxy-3-bromoarabinofuranosyl]-2-nitroimidazole;
l-β-D-[3,2-dideoxy-3-fluoroarabinofuranosyl]-2-nitroimidazole; l-β-D-[3,2-dideoxy-3-chloroarabinofuranosyl]-2-nitroimidazole; 1 -β-D-[3,2-dideoxy-3-bromoarabinofuranosyl]-2-nitroimidazole; l-β-D-[2,3-dideoxy-2-fluoroarabinofuranosyl]-2-nitroimidazole; l-β-D-[2,3-dideoxy-2-chloroarabinofuranosyl]-2-nitroimidazole; , l-β-D-[2,3-dideoxy-2-bromoarabinofuranosyl]-2-nitroimidazole;
l-β-D-[2,3-dideoxy-2-fluororibofuranosyl]-2-nitroimidazole;
l-β-D-[2,3-dideoxy-2-chlororibofuranosyl]-2-nitroimidazole;
l-β-D-[2,3-dideoxy-2-bromoribofuranosyl]-2-nitroimidazole; l-β-D-[2-deoxy-2-keto-arabinofuranosyl]-2-nitroimidazole;
l-β-D-[3-deoxy-3-keto-arabinofuranosyl]-2-nitroimidazole; and l-β-D-[2,3-dideoxy-2,3-epoxyarabinofuranosyl]-2-nitroimidazole.
In another aspect, the present invention provides for methods of treating a patient suffering from cancer comprising administration of an effective amount of at least one Compound of the Present Invention followed by radiotherapy.
In the case of diagnostic applications, increased localization of a Compound of the Present Invention in hypoxic cells as compared to less hypoxic or oxic cells, the Compound of the Present Invention labelled with a radioisotope capable of being imaged, facilitates the detection of the hypoxic cells.
In the case of radiotherapy applications, increased localization of a Compound of the Present Invention in hypoxic cells as compared to less hypoxic or oxic cells, the Compound of the Present Invention radioactive as a result of the compound being radiolabeled, permits the product to preferentially accumulate in hypoxic cells, thus facilitating radiotherapeutic effects directed specifically at hypoxic cells.
In a first embodiment relating to diagnostic applications of the invention, the invention comprises a method for monitoring hypoxic cells within a population of less hypoxic or oxic cells, comprising the steps of administering to the cells an effective amount of a labelled Compound of the Present Invention so that the labelled compound accumulates preferentially in hypoxic tissues and then detecting labelled compound. In this first embodiment, the invention also comprises the use of labelled Compounds of the Present Invention in performing this diagnostic method.
In a preferred diagnostic method, the method for monitoring hypoxic cells throughout the population of cells is comprised of the following steps:
(a) administering to the cells an effective amount of at least one labelled
Compound of the Present Invention;
(b) waiting a period of time such that a substantial amount of the at least one labelled Compound of the Present has been expelled from less hypoxic or oxic cells and such that a detectable amount of the at least one labelled Compound of the Present Invention remains within hypoxic cells; and
SUBSTiTUTE SHEET (WlE 26)
(c) determining the extent and location of hypoxic cells throughout the population of cells by detecting the at least one labelled Compound of the Present Invention.
The labelled Compound of the Present Invention is preferably radiolabeled, but any other form of labelling which facilitates detection of the labelled Compound of the Present Invention may also be suitable. One non-limiting example is the inclusion of isotopes in the Compound of the Present Invention which are identifiable using Nuclear Magnetic Resonance or Magnetic Resonance Imaging.
In the case of diagnostic applications, preferential localization of the Compound of the Present Invention in hypoxic cells as compared less hypoxic or oxic cells permits the compound of the Present Invention to accumulate in hypoxic cells in order to facilitate the detection of the labelled product in those cells.
Accordingly, following the administration of an effective amount of the labelled Compound of the Present Invention to a patient such that the labelled Compound of the Present Invention accumulates preferentially in hypoxic tissues, the diagnostic method includes waiting a period of time such that a substantial amount of the labelled Compound of the Present Invention has been expelled from the less hypoxic or oxic cells and such that a detectable amount of the labelled Compound of the Present Invention remains within hypoxic cells. As there is a preferential accumulation of the Compound of the Present Invention in cells experiencing more hypoxic conditions than those of lesser hypoxic or oxic conditions, this is a matter of waiting a period of time allowing the preferred amount of clearance from lesser hypoxic cells or oxic cells as compared to hypoxic cells. One skilled in the art will be
SUBST1TUTE ShEET (RULE 26)
capable of determining the appropriate amount of time with observation and as a function of administered dose, patient weight, patient age, patient sex, and suspected hypoxic cell location in the body.
The period of time for waiting, prior to performing the step of determining the extent and location of hypoxic cells throughout the population of cells by detecting the labelled Compound of the Present Invention, will be determined or selected depending upon a number of various factors including the properties of each of the labelled Compound of the Present Invention. For instance, the rate of expulsion or clearance of each of the labelled Compound of the Present Invention in hypoxic cells compared to cells of lesser hypoxia or cells in oxic conditions. The time period is selected to achieve a balance between the amount of the labelled Compound of the Present Invention present in the hypoxic cells and the amount of the Compound of the Present Invention present in the cells of lesser hypoxia or oxic conditions at the time of detecting the labelled Compound of the Present Invention. First, the amount of the labelled Compound of the Present Invention is preferably minimized in order to enhance or increase the accuracy of the diagnostic method as the presence of significant or substantial amounts of the labelled Compound of the Present Invention may interfere with the detection of the labelled Compound of the Present Invention localized in hypoxic cells. For instance, in radiolabelling of the Compound of the Present Invention, radioimaging may be unable to distinguish between the presence of the labelled Compound of the Present Invention in hypoxic cells as compared with lesser hypoxic or oxic cells if too much labelled Compound of the Present Invention is administered. Second, the amount of the labelled Compound of the Present Invention within the hypoxic cells is preferably maximized to facilitate the detection of the
SUBSTiTUTE SHEET (RUE 26)
labelled Compound of the Present Invention in hypoxic cells as compared to cells of lesser hypoxia or cells in oxic conditions and to also enhance or increase the accuracy of the diagnostic method.
Most preferably, the labelled Compound of the Present Invention rate of clearance in hypoxic cells compared to cells of lesser hypoxia or cells in oxic conditions is such that following the passage of a determined or selected period of time, all or substantially all of the labelled Compound of the Present Invention has been expelled from the cells of lesser hypoxia or cells in oxic conditions while all or substantially all of the labelled Compound of the Present Invention remains within the hypoxic cells. In other words, the expulsion of the labelled Compound of the Present Invention from cells of lesser hypoxia or cells in oxic conditions and the expulsion of the labelled Compound of the Present Invention from hypoxic cells do not overlap such that the expulsion of the labelled Compound of the Present Invention from cells of lesser hypoxia or cells in oxic conditions is complete or substantially complete prior to the commencement of any expulsion or any substantial expulsion of the labelled Compound of the Present Invention from hypoxic cells.
However, the relative rates of clearance may provide for an overlap of the expulsion of the labelled Compound of the Present Invention in hypoxic cells compared to cells of lesser hypoxia or cells in oxic conditions. In this case, the period of time is selected or determined according to the desired degree of accuracy or the desired statistical significance of the diagnostic test results as discussed above. As indicated, the period of time is selected so that preferably a substantial amount of the labelled Compound of the Present Invention has been expelled. For a substantial amount to be expelled,
SUBSTtTUTE SHEET (BULE 26)
any remaining labelled Compound of the Present Invention in cells of lesser hypoxia or cells in oxic conditions is not enough to significantly interfere with the detection of the labelled Compound of the Present Invention in hypoxic cells and is such that the diagnostic test results achieve the desired degree of accuracy or statistical significance. The period of time is also selected so that a detectable amount of the labelled Compound of the Present Invention remains within the hypoxic cells. A detectable amount is present if there is a sufficient amount to permit effective detection according to the selected detection method or process and such that the diagnostic test results achieve the desired degree of accuracy or statistical significance. For instance, where radiolabelling and radioimaging are used, a sufficient amount of the labelled Compound of the Present Invention must remain in the hypoxic cells to provide adequate signal measurement.
Once this period of time has passed, the extent and location of the hypoxic cells throughout the population of lesser hypoxic or oxic cells is determined by detecting the labelled Compound of the Present Invention. The determination of the extent and location of the Compound of the Present Invention in the cells provides for or permits the monitoring of regions of hypoxia. These regions of hypoxia correlate with the presence of a collection of cancerous cells or tumors
The method of detection is selected according to the type or manner of the labelling of the Compound of the Present Invention. However, in the preferred embodiment, the labelled Compound of the Present Invention is radiolabeled and the detection is performed using nuclear medicine imaging techniques.
'8UBSTITUTE SHEET (RUlE 26)
In a second embodiment relating to radiotherapy applications of the invention, the invention comprises a method of radiotherapy for use with a population of hypoxic cells, comprising the step of administering to the cells an effective radiotherapeutic dose of a radiolabeled Compound of the Present Invention so that the radiolabeled Compound of the Present Invention becomes preferentially localized within hypoxic cells. In this second embodiment, the invention also comprises the use of radiolabeled Compound of the Present Invention in performing this radiotherapy method. The substituents for the specific preferred radiolabeled Compound of the Present Invention for use with this radiotherapy applications are the same as for the diagnostic method of the invention, except that the radiolabeled compounds are selected from the group consisting of, but not limited to, 123I, 125I and 131I.
In a third embodiment relating to chemotherapy applications of the invention, the invention comprises a method of chemotherapy for use with a population of cells suspected of containing hypoxic cells, comprising the step of administering to the cells an effective chemotherapeutic amount of a Compound of the Present Invention wherein the Compound of the Present Invention is cytotoxic or cytostatic. In this embodiment, the invention also comprises the use of Compound of the Present Invention in performing this chemotherapy method.
The appropriate time interval or period of time between injection of the radiolabeled Compound of the Present Invention and imaging depends on, amongst other factors, the half-life of the radiolabeled Compound of the Present Invention, the rate of clearance of the radiolabeled Compound of the Present Invention in hypoxic cells compared to and the rate of clearance of the radiolabeled Compound of the Present
Invention in cells of lesser hypoxia or cells in oxic conditions. Thus, the time period must be particularly determined or selected for each specific labelling and dosing paradigm. A time period of 1.5-24 h is most common, with the shorter periods used for 18F imaging and the longer times for radiolabels like 123I. After the appropriate time period, retained radioactivity will be due to the Compound of the Present Invention in hypoxic cells. Optimal times are selected to provide best image contrast, that is, the time when excretion of the radiolabeled Compound of the Present Invention in cells of lesser hypoxia or cells in oxic conditions is complete or substanially complete, and sufficient radiolabeled Compound of the Present Invention in hypoxic cells remains for adequate signal measurement. A positive image will show uptake of radioactivity in a region, which reflects proof of cellular hypoxia (i.e. measurement by imaging). Nuclear medicine imaging techniques, including planar (2-dimensional), positron emission tomography (PET) and single photon emission tomography (SPECT) imaging, and their interpretations, are known to practitioners versed in the art.
Those skilled in medical radiotherapeutic methods and uses will be able to calculate a suitable effective dose of the radiolabeled Compound of the Present Invention for human or other uses based on their experience with other compounds carrying similar radiolabels. However, as indicated previously, when the radiolabeled Compound of the Present Invention is used for diagnostic purposes, as small a dosage as possible should be used in order to minimize any toxicity to the population of cells or surrounding tissue. When using the compound for radiotherapeutic purposes, an effective radiotherapeutic dose of the radiolabeled Compound of the Present Invention must be used. Typically, the dosage of the radiolabeled Compound of the
Present Invention for therapeutic purposes will be greater than that used for diagnostic purposes in order to achieve the desired radiotherapeutic effect. When used on cancerous cells, the desired radiotherapeutic effect will be a cytotoxic or cytostatic effect on the cells in which the radiolabelled Compound of the Present Invention is present. For use as a radiosensitizer, one skilled in the art will recognize that a dosage of Compound of the Present Invention administered will be that which achieves an increase in therapeutic effect of the radiation when the patient is administered with a Compound of the Present Invention, as compared to a patient in which a Compound of the Present Invention is not administered. One skilled in the art will recognize that administration of at least one Compound of the Present Invention can result in an increased therapeutic kill of hypoxic cells, including cancerous or tumor cells, with a given radiation dose, or alternatively reduce the radiation dose utilized to effect a therapeutic kill of hypoxic cells, including cancerous or tumor cells.
The accompanying description illustrates preferred embodiments of the present invention and serves to explain the principles of the present invention.
BRIEF DESCRIPTION OF THE DRAWNINGS
FIGURE 1 shows a schematic of the reduction of azomycins under hypoxic conditions; FIGURE 2 shows the sensitization of the human colorectal carcinoma cell line HCTl 16 to radiotherapy with select Compounds of the Present Invention; and FIGURE 3 shows cytotoxicity of β-IAZA in various cell lines as a function of concentration.
DETAILED DESCRIPTION QF THE INVENTION Symbols used in this description are explained below.
Symbols Chemical Name α-AZA l-α-D-(arabinofuranosyl)-2-nitroimidazole
At Astatine β-AZA l-β-D-(arabinofuranosyl)-2-nitroimidazole
Ac Acetyl
Ac2O Acetic anhydride
Br Bromine
Bz Benzoyl
CH2C12 Dichloromethane
CH3CN Acetonitrile
Cl Chlorine
CrO3 Chromium trioxide
D Deuterium
DAST Diethylaminosulfurtrifluoride
DMAP N,N-Dimethylaminopyridine
DME Dimethoxyethane
EtOH Ethyl alcohol
EtOAc Ethyl acetate
F Fluorine/Fluoride
HRMS Igh resolution mass spectroscopy
K2CO3 Potassium carbonate
KF Potassium fluoride
MeCN Acetonitrile
MeOH Methanol
N2H2 Hydrazine
NaBDO4 Sodium tetraborodeuteride
NaBTO4 Sodium tetraborotritide
Na2SO4 Sodium sulphate
NH3 Ammonia
NMR Nuclear magnetic resonance
Nosyl p-Nitrobenzenesulfonyl
Piv. Pivaloyl
R4NF Teraalkylammonium fluoride
T Tritium
THF Tetrahydrofuran
TLC Thin layer chromatography
Tosyl Toluenesulfonyl
Triflyl Trifluoromethanesulfonyl v/v Volume/volume
- 20 -
SUBSTiTUTE SHEET (BtIE 26)
As used herein "Leaving Group" means the Sulfonyl related leaving groups Methanesulfonyl, substituted methane sulfonyl, trifluoromethanesulfonyl, benzenesulfonyl and all substituted benzenesulfonyl (including but not limited to toluenesulfonyl, nitrobenzenesulfonyl and related compounds); OH, C=O and Halogen related leaving groups including but not limited to Cl, Br and iodine.
An "effective amount" is an amount of a Compound of the Present Invention sufficient to achieve the intended purpose. For example, an effective amount of a Compound of the Present Invention to kill hypoxic or cancerous cells comprising a tumor is an amount sufficient, in vivo to result in an increased killing of hypoxic or cancerous cells as compared to non-hypoxic cells. An effective amount of a Compound of the Present Invention to image hypoxic or cancerous cells comprising a tumor is an amount sufficient, to identify an increased localization of hypoxic or cancerous cells as compared to lesser hypoxic or oxic cells. An effective amount of a Compound of the Present Invention to treat or ameliorate a cancerous disease or condition is an amount of the Compound of the Present Invention sufficient to reduce or remove the symptoms of the cancerous disease or condition. The effective amount of a given Compound of the Present Invention will vary with factors such as the nature of the agent, the route of administration, the size and species of the animal or patient to receive the therapeutic agent, and the purpose of the administration. The effective amount in each individual case may be determined empirically by a skilled artisan according to established methods in the art and the teachings herein.
Chapman postulated that scintigraphic imaging of tumor hypoxia using gamma-
SUBSTITUTE SfEET (RULE 26)
emitting nitroimidazole radiosensitizers could form the basis of a useful predictive assay for radiation therapy planning. (Chapman J.D. et al Brit J Cancer 43:546 (1981)). For nitroimidazole-based radiopharmaceuticals, this means that the sensitizer characteristics already identified have to be adjusted to accommodate design limitations imposed by the radionuclide.
Preliminary in vivo biodistribution studies in a murine tumor model, and pharmacokinetic studies in rats indicated that [3H] 1-α-D-FAZA has biodistribution, tumor uptake and pharmacokinetic properties similar to those of 123I-IAZA, a clinically-proven radiopharmaceutical for SPECT-imaging of hypoxic tissues (Kumar P. et al J Label Comp Radiopharm 42:3 (1999)). In vitro and in vivo comparisons of [18F] 1-α-D-FAZA and [18F]FMISO indicated that hypoxia-selective uptake similar,
with faster clearance of [18F] 1-α-D-FAZA from blood, viscera and muscle tissue, via the renal system of rats (Sorger D. et al Nucl Med Biol 30:317 (2003)). In three different murine tumor models, tumoπblood ratios were 2-4 times greater for [18F] 1- α-D-FAZA than [18F]FMISO, an effect attributable to rapid blood clearance of [18F] 1- α-D-FAZA, since tumor uptake, as a fraction of dose, was similar between these tracers. In a nude mouse model bearing a subcutaneous A431 tumor, tumor- background was 9.3:1 for animals breathing room air, compared to 5.3:1 for animals breathing 100 % O2, demonstrating the oxygen-sensitivity of [18F] 1 -α-D-FAZA binding (Piert M. J Nucl Med 43:278P (2002)).
Initial clinical studies complement animal data, reflecting strong uptake by hypoxic tumor and rapid clearance from the vascular compartment, to provide strong target (tumor) to background contrast, and with little hepatic and gastrointestinal signal. In
SUBSTiTUTE SHEET (BUlE 26)
patients studied sequentially with [18F] 1-α-D-FAZA, [18F]FMISO and [18F]FDG, virtually identical images were obtained. One major difference was the absence of [18F] 1-α-D-FAZA uptake in normal brain tissue, compared to [18F]FMISO, which is taken up non-specifically by brain, and [18F]FDG, which is taken up as a reflection of glucose metabolism in healthy brain (Wiebe L.I. in press).
Previous studies on the synthesis of l-α-D-[5-deoxy-5-iodoarabinofuranosyl]-2- aminoimidazole (iodo aminoimidazole arabinoside; IAIA)5 a potential nitroreducrase reduction metabolite of IAZA (Mannan R.H. et al J Nucl Med 32:1764 (1991); Edwards D.I. J Antimicrob Chemother 31 :9 (1993)), revealed that IAZA, which had previously been assigned the β-configuration, was actually the α-anomer (Lee H.C. et al Nucleosides & Necleorides 18:1995 (1999)). Furthermore, in vitro studies indicated that IAZA was not transported by the NBMPR (nitrobenzylthioinosine)- sensitive equilibrative nucleoside transporter in erythrocytes (Wange L. et al Unpublished) which was not unexpected given that these transporters handle physiological nucleosides that have the β-nucleoside configuration (Cass CE. in "Drug Transport in Antimicrobial and Anticancer Chemotherapy" 403 (1995)). Therefore a class of compounds useful for selectively residing in hypoxic cells and actively transported into cells, in the β-nucleoside configuration are defined and disclosed herein.
Nucleoside kinases bioactivate nucleosides for incorporation into DNA and RNA by 5 '-phosphorylation. In mammalian cells, four deoxyribonucleoside kinases have been characterized, two of which (thymidine kinase; TKl) and deoxycytidine kinase (dCK) are found in the cytoplasm, whereas thymidine kinase (TK2) and the deoxyguanosine
SUBSTITUTE SHEET (RULE 28)
kinase (dGK) are predominantly localized in mitochondria (Arner, E.SJ. et al Pharmacol Ther 67:155 (1995)). These kinases are generally substrate-specific, with specificities governed by the base (pyrimidine or purine), the sugar (deoxyribose and ribose), and the configuration of the glycoside bond at the anomeric carbon (Cl'; only β anomer) of the sugar. There are important exceptions for each nucleoside kinase; important examples include phosphorylation of 2'-/3'-fluoro-2-73'- ribo/arabinofyranosyl pyrimidine nucleosides (altered sugar) (De Clercq E. Mini Rev Med Chem 2:163 (2002)), and imidocarboxamide ribosides (altered base) like EICAR (Balzarini, J. et al Adv Exp Med Biol 431:723 (1998)). Nucleoside kinases play a crucial role in the chemotherapy of cancer and viral infections. These enzymes catalyze the rate-limiting phosphorylation of the nucleoside-analogue pro-drugs into their cytotoxic phosphorylated forms. Interestingly, elevated levels of deoxynucleoside kinases are detected in proliferating cells such as cancer cells.
Importantly for viral chemotherapy and some 'suicide' gene therapy paradigms, kinases from viruses are found to have broad substrate specificity, phosphorylating a variety of nucleosides analogues. Although no specific information of nucleoside kinase activity in hypoxia is known in the art, reports of over-replication of DNA
(Young, D.S. et al Proc Nat Acad Sci USA 85:95533 (1988)) and signalling endothelial cell proliferation (Schafer M. et al FASEB J 17:449 (2003)) in hypoxia implies active DNA synthesis, which likely includes nucleoside phosphorylation as a first metabolic step.
Tables 1-4 disclose a series of new compounds with potential applications in radiosensitization, chemosensitization and chemotherapy of cancer. They are selected
SUBST1TUTE ShSET (RULE 26)
so as to undergo selective 'nucleoside-type' transport and, most importantly, bioactivation (e.g. phosphorylation) to enhance selective accumulation and promote selective toxicity to hypoxic cells, thereby producing enhanced concentrations in viable but hypoxic cells. Their advantage lies in their improved concentration, and residence half-life, in target cells. Though an exact understanding of the mechanism of action of the present invention is not needed to practise the invention and the present invention is not intended to be limited by any proposed mechanism of action; these characteristics could result because of metabolic trapping as a result of phosphorylation. The compounds disclosed in Tables 1-4 represent azomycin nucleosides that are phosphorylated by nucleoside kinases, and thereby transported by equilibrative, high capacity nucleoside transporters and/or by concentrative nucleoside transporters. Furthermore the halogenated azomycin nucleosides offer optimal lipophilicity (Mannan, R.H. et al J Nucl Med 32:1764 (1991)).
SUBSTrrUTΕ SHEET (FRJLE 20)

SUBSΠTUTE SHEET (RULE 26)

Table 2; 5, 3 and 2- Halo β-Arabinofuranosyl Azomycin derivatives

SUBSTITUTE SHEET (RULE 26)

SUBSTITUTE SHEET (RULE 26)

SUBSTITUTE SHEET RULE 26),

 l-β-D-[2/3/5-
l-β-D-[2/3/5-
TsO 1.A11 leaving groups at 5 O- (Ts, Ns5 TFMs, Ms, X with TrisubstitutedfUranosyl]-2- protective groups at T and nitroimidazoles 3' when the molecule is
CH3COO OCOCH3 ribose, arabinose, 3 '-xylo, Threo
2. All leaving groups at 3'0~ (Ts, Ns, TFMs, Ms, X with protective groups at 2' and 5' when the molecule is ribose, arabinose, 3'-xylo, threo
3. All leaving groups at 2'O- (Ts, Ns, TFMs, Ms, X with protective groups at 5' and 3' when the molecule is ribose, arabinose, 3'-xylo, Threo
SUBSTiTUTE SHEET (RlE 26)
The increased transport of the compounds disclosed in Tables 1-4 results in a class of compounds capable of increased residence time, concentration and .therefore bioavailability in hypoxic cells, such as tumor cells. The presence of halogens within the compounds allows for inclusion of radioisotopes, the selection of which would be within the ability of one skilled in the art, for radioimaging of hypoxic cells and tissues.
Through inclusion of appropriate radionuclides, the compounds will be appropriate for use in a therapeutic capacity. Examples of appropriate therapeutic radionuclides include the radioiodines 125I and 131I; the radiobromines 76Br, 77Br, and 82Br; the radiochlorines 34mCl and 24Cl; and astatine 211At.
Furthermore, the compounds disclosed in Tables 1-4 are capable of acting as radiosensitizers and chemosensitizers, when administered in association with radio- or chemo- therapy respectively, under conditions known or determinable by those skilled in the art.
Pharmaceutical compositions are also provided, comprising at least one Compound of the Present Invention, and a pharmaceutically acceptable excipient and/or carrier.
The pharmaceutical compositions can be prepared by mixing the desired Compound(s) of the Present Invention with an appropriate vehicle suitable for the intended route of administration. In making the pharmaceutical compositions of this invention, the Compound(s) of the Present Invention are usually mixed with an excipient, diluted by an excipient or enclosed within such a carrier which can be in the
form of a capsule, sachet, paper or other container. When the pharmaceutically acceptable excipient serves as a diluent, it can be a solid, semi-solid, or liquid material, which acts as a vehicle, carrier or medium for the therapeutic agent. Thus, the compositions can be in the form of tablets, pills, powders, lozenges, sachets, cachets, elixirs, suspensions, emulsions, solutions, syrups, aerosols (as a solid or in a liquid medium), ointments containing, for example, up to 10% by weight of the Compounds of the Present Invention, soft and hard gelatin capsules, suppositories, sterile injectable solutions, and sterile packaged powders.
Some examples of suitable excipients include lactose, dextrose, sucrose, sorbitol, mannitol, starches, gum acacia, calcium phosphate, alginates, tragacanth, gelatin, calcium silicate, microcrystalline cellulose, polyvinylpyrrolidone, cellulose, sterile water, syrup, and methyl cellulose. The formulations can additionally include: lubricating agents such as talc, magnesium stearate, and mineral oil; wetting agents; emulsifying and suspending agents; preserving agents such as methyl- and propylhydroxy-benzoates; sweetening agents; and flavoring agents. The compositions of the invention can be formulated so as to provide quick, sustained or delayed release of the Compound(s) of the Present Invention after administration to the patient by employing procedures known in the art.
For preparing solid compositions such as tablets, the Compound(s) of the Present Invention is mixed with a pharmaceutical excipient to form a solid preformulation composition containing a homogeneous mixture of a compound of the present invention. When referring to these preformulation compositions as homogeneous, it is meant that the Compound(s) of the Present Invention are dispersed evenly
throughout the composition so that the composition may be readily subdivided into equally effective unit dosage forms such as tablets, pills and capsules.
The tablets or pills of the present invention may be coated or otherwise compounded to provide a dosage form affording the advantage of prolonged action. For example, the tablet or pill can comprise an inner dosage and an outer dosage component, the latter being in the form of an envelope over the former. The two components can be separated by an enteric layer which serves to resist disintegration in the stomach and permit the inner component to pass intact into the duodenum or to be delayed in release. A variety of materials can be used for such enteric layers or coatings, such materials including a number of polymeric acids and mixtures of polymeric acids with such materials as shellac, cetyl alcohol, and cellulose acetate.
The liquid forms in which the Compound(s) of the Present Invention may be incorporated for administration orally or by injection include aqueous solutions, suitably flavored syrups, aqueous or oil suspensions, and flavored emulsions with edible oils such as corn oil, cottonseed oil, sesame oil, coconut oil, or peanut oil, as well as elixirs and similar pharmaceutical vehicles.
Compositions for inhalation or insufflation include solutions and suspensions in pharmaceutically acceptable, aqueous or organic solvents, or mixtures thereof, and powders. The liquid or solid compositions may contain suitable pharmaceutically acceptable excipients as described herein. The compositions are administered by the oral or nasal respiratory route for local or systemic effect. Compositions in preferably pharmaceutically acceptable solvents may be nebulized by use of inert gases. Nebulized solutions may be inhaled directly from the nebulizing device or the
SUBSTiTUTE SHEET (fi)LE 2β)
nebulizing device may be attached to a face mask tent, or intermittent positive pressure breathing machine. Solution, suspension, or powder compositions may be administered, preferably orally or nasally, from devices which deliver the formulation in an appropriate manner.
Another formulation employed in the methods of the present invention employs transdermal delivery devices ("patches"). Such transdermal patches may be used to provide continuous or discontinuous infusion of the therapeutic agent of the present invention in controlled amounts. The construction and use of transdermal patches for the delivery of pharmaceutical agents is well known in the art. See, for example, U.S. Pat. No. 5,023,252, herein incorporated by reference. Such patches may be constructed for continuous, pulsatile, or on demand delivery of pharmaceutical agents.
Other suitable formulations for use in the present invention can be found in Remington's Pharmaceutical Sciences.
The following examples are offered to illustrate this invention and are not to be construed in any way as limiting the scope of the present invention.
EXAMPLE 1 : Preparation of l-β-D-(Substituted furanosyl/hexopyranosyl)-2- nitroimidazoles
Main synthons l-β-D-(3,5-O-Tetraisopropyldisilyloxy ribofuranosyl)-2-
nitroimidazole and l -β-D-(2,3-Di-O-acetyl/benzoyl arabinofuranosyl)-2- nitroimidazoles were prepared by the methods described in the literature (Kumar, P. et al Chem Pharm Bull 51 :399 (2003); Kumar, P. et al. Tetrahedron Lett 43:4427-4429
SU8STJTUTE SHEET {WIE 26)
(2002)) and were derivatized to develop the compounds claimed in Genus 1. Few compounds under this sub-category are described below.
l-β-D-(2-0-MethyIthiomethyl-3,5-0-tetraisopropyIdisiIyIoxyribofuranosyl)-2- nitroimidazole
A solution of l-β-D-(3,5-O-tetraisopropyldisilyloxyribofuranosyl)-2-nitroimidazole (24 mg, 0.05 mmol) in DMSO (0.2 ml) was treated with Ac2O (0.125 ml) and the mixture was stirred at 220C for 2 days. Then 1 ml water was added and extracted with EtOAc and the organic phase was washed with water and dried (Na2SO4). After evaporation the residue was chromatographed on silica gel column, eluting with hexanes-ethyl acetate (10:1) to give l-β-D-(2-O-methylthiomethyl-3,5-0- tetraisopropyldisilyloxy ribofuranosyl)-2-nitroimidazole (lOmg, 37%) as a viscous oil. 1H-NMR, MS.
l-β-D-(5-0-AcetylribofuranosyI)-2-nitroimidazoIe,
A solution of l-β-D-(2,3,5-tri-O-acetylribofuranosyl)-2-nitroimidazole (37.1 mg, 0.1 mmol) (Naimi, E. et al Nucleosides Nucleotides Nucleic Acids 24:173 (2005)) and N2H2 (12.8 mg, 0.4 mmol) in glacial acetic acid-pyridine (1:4, 1 ml) was heated at 800C for 3 h. After quenching with acetone (0.5 ml) and stirring at 22°C for 1 h, the solvents were evaporated and the residue was purified on silica gel column, using ethyl acetate-hexanes (80:20, v/v) to give l-β-D-(5-<9-acetylribofuranosyl)-2- nitroimidazole (20 mg, 70%); 1H-NMR, HRMS.
SUBSTiTUTE SHEET (RtIE 28)
l-β-D-(3,5-Di-0-acetyl ribofuranosyl)-2-nitroimidazole and l-β-D-(2,5-Di-0- acetyl ribofuranosyl)-2-nitroimidazole
A solution of l-β-D-(2,3,5-tri-O-acetylribofuranosyl)-2-nitroimidazole (9.3 mg, 0.025 mmol) and N2H2 (1.2 mg, 0.037 mmol) in glacial acetic acid-pyridine (1:4, 0.25 ml) was stored at 22°C for 9h. After quenching with acetone, the solvents were evaporated and the residue was purified on preparative TLC, CHCl3-MeOH (9:5, v/v) to give mixture of l-β-D-(3,5-di-O-acetylribofuranosyl)-2-nitroimidazole and 1-β-D- (2,5-di-0-acetylribofuranosyl)-2-nitroimidazole; 1H-NMR, and trace of l-β-D-(5-0- acetylribofuranosyl)-2-nitroimidazole.
l-β-D-(3-0-p-Toluenesulfonyl ribofuranosyl)-2-nitroimidazole and l-β-D~(2~O-p- Toluenesulfonyl ribofuranosyl)~2-nitroimidazole
To a stirred suspension of l-β-D-(ribofuranosyl)-2-nitroimidazole (49 mg, 0.20 mmol) in CH3CN (6 ml) was added Bu2SnO (56 mg, 0.225 mmol), p-toluenesulfonyl chloride (64 mg, 0.335 mmol) and TBAF in THF (1.0 M solution, 0.2 ml, 0.20 mmol) at 220C After 24 h stirring, another portion of/?-toluenesulfonyl chloride (38 mg, 0.20 mmol) was added and stirring continued overnight. The solvent was evaporated and the residue was chromatographed on silica gel column, using dichloromethane-ethyl acetate (70:30) to give l-β-D-(3-0-p-toluenesulfonyl ribofuranosyl)-2-nitroimidazole (19 mg, 24%); m.p. 172-173°C; 1H-NMR, HRMS, and l-β-D-(2-<9-/?-toluenesulfonyl ribofuranosyl)-2-nitroimidazole (31 mg, 39%); m.p. 165-166°C, 1H-NMR, HRMS.
SUBSTITUTΕ SHEET (RULE 26
l-β-D-(3,5-6>-Tetraisopropyldisilyloxy-2-0-/;-toluenesulfonylribofuranosyl)-2- nitroimidazole
A mixture of l-β-D-(3,5-<9-Tetraisopropyldisilyloxyribofuranosyl)-2-nitroimidazole (48.7 mg, 0.1 mmol), p-toluenesulfonyl chloride (95.3 mg, 0.5 mmol) and DMAP (6.1 mg, 0.05 mmol) in dry pyridine (1 ml) was stirred at 50-550C overnight. Another portion of jc-toluenesulfonyl chloride (78.6 mg, 0.4 mmol) and DMAP (4.9 mg, 0.04 mmol) was added and stirring at 50-550C continued for 12-14 h. After evaporation of solvent and purification on silica gel column, using hexanes-ethyl acetate (87.5:12.5, v/v), gave l-β-D-(3,5-(9-tetraisopropyldisiIyloxy-2-<9-/?- toluenesulfonylribofuranosyl)-2-nitroimidazole (49 mg, 75%); m.p. 146-1470C, 1H- NMR, mass, HRMS.
l-β-D-(3,5-Di-O-acetyl-2-O-/?-toIuenesulfonylribofuranosyI)-2-nitroimidazole
Ac2O (0.045 ml) was added to a solution of l-(2-0-/>-toluenesulfonyl-β-D- ribofuranosyl)-2-nitroimidazole (24 mg, 0.06 mmol) in anhydrous pyridine and the mixture was stirred at 220C overnight. The solvent was removed and the residue was purified on silica gel column, using hexanes-ethyl acetate (50:50) to give 1-(3,5-Di-O- acetyl-2-O-p-toluenesulfonyl-β-D-ribofuranosyl)-2-nitroimidazole (26 mg, 90%).
l-β-D-(5-0-tert-Butyldiphenylsilyl-2,3-di-0-acetyl ribofuranosyl)-2- nitroimidazole and l-β-D-(3,5-Di-0-tert-butyIdiphenyIsiIyl-2-0-acetyl ribofuranosyl)-2-nitroimidazole
SUBSTITUTE SHEET (RULE 26 )
l-β-D-(ribofuranosyl)-2-nitroimidazole (270 mg, 1.1 mmol) was dissolved in dry pyridine (1.25 ml), and tert -butyldiphenylsilyl chloride (316 mg, 1.15 mmol) was added. The reaction mixture was stirred at 22°C overnight. Additional tert- butyldiphenylsilyl chloride (32mg) was added and after completion, Ac2O (0.415 ml, 4.0 mmol) was added and stirred overnight. After solvent evaporation, the residue was purified on column, eluting with hexanes-ethyl acetate (70 : 30) to give l-β~D-(5-O - tert-Butyldiphenylsilyl-2,3-di- O-acetylribofuranosyl)-2-nitroimidazole (478 mg, 77%); m.p 52-53°C, 1H-NMR, 13C-NMR, HRMS, and 1-(3, 5 -Di-O-tert- butyldiphenylsilyl-2-O -acetyl-β-D-ribofuranosyl)-2-nitroimidazole (33 mg, 4%) as a viscous oil; 1H-NMR, HRMS.
l-β-D-(2,3-Di~O-acetyl ribofuranosyl)-2-nitroimidazole
A suspension of l-β-D-(5-O -tert-Butyldiphenylsilyl-2,3-di-O-acetyl ribofuranosyl)-2- nitroimidazole (454 mg, 0.8 mmol), benzoic acid (683 mg, 5.6 mmol) and KF (325 mg, 5.6 mmol) in MeCN (20 ml) was heated at 75-80°C for 16 h. After cooling and filtration, the filtrate was evaporated and the residue was chromatographed on silica gel column, eluted with ethyl acetate-hexanes (70:30) to afford l-β-D-(2,3-Di-O- acetylribofuranosyl)-2-nitroimidazole (246mg, 93%); m.p. 122-123°C; 1H-NMR, 13C-NMR, HRMS.
l-β-D-(2,3-Di-0-acetyl-5-O-toluenesulfonyl arabinofuranosyI)-2-nitroimidazole A solution of toluenesulfonyl chloride (0.23 g, 1.2 mmol) in anhydrous pyridine (5 mL) was added to a pre-cooled solution of l-β-D-(2-O-acetylarabinofuranosyl)-2-
nitroimidazole (0.23 g, 0.8 mmol) in anhydrous pyridine (20 mL) and stirred at 0°C for 18h. Once the starting precursor was completely consumed (determined by tic examination), acetic anhydride (1.2 mmol) was added to this mixture and the stirring was continued for an additional 3h. The solvent was evaporated after the reaction was complete and the mixture was purified on a silica gel column using hexanes/ethyl acetate (1:1, v/v) as eluent, m.p. 117-119°C, 1H-NMR, 13C-NMR.
EXAMPLE 2: Preparation of l-β-D-[(2/3/5-Substituted) or (2,3-disusbstituted) or (2,2-disubstituted) or (3/3-disubstituted) or (2,5-disubstituted) or (3,5-disubstituted) furanosyl/hexoρyranosyl)-2-nitroimidazoles.
l-β-D-(2-Deuterio-3,5-0-tetraisopropyldisilyloxyarabinofuranosyl)-2- nitroimidazole
A stirred suspension of CrO3 (15 mg) in CH2Cl2 (1 ml) was cooled to 0°C and Ac2O
(0.015 ml) and pyridine (0.025 ml) were added. After 3 min, l-β-D-(3,5-O- tetraisopropyldisilyloxy ribofuranosyl)-2-nitroimidazole (24mg, 0.05 mmol) was added and then allowed to warm to 5-10°C over a period of 2 h. Volatile materials
were evaporated and the residue was cooled to 0°C and dissolved in absolute EtOH (1.0 ml). The stirred mixture was treated by addition ofNaBD4 (3 mg). After 30 min a second portion OfNaBD4 (3 mg) was added and a 2 mg portion was added at 1 h and allowed to warm to 10-12°C and stirred for 30 min. After evaporation, the residue was chromatographed on a silica column, eluting with hexanes-ethyl acetate (80:20) to afford 1 -β-D-(2-deuterio-3,5-O-tetraisopropyldisilyloxy arabinofuranosyl)-2-
nitroimidazole (15 mg, 61%); m.p. 160-161°C, 1H-NMR, 13C-NMR, mass, HRMS.
SUBSTΓΠJTΈ SHEET RULE 26
l-β-D-(2-Deuterio -arabinofuranosyl)-2-nitroimidazoIe
A suspension of KF (41 mg, 0.7 mmol), benzoic acid (85 mg, 0.7 mmol) and 1-β-D- (2-Deuterio-3,5-0-tetraisopropyIdisilyloxy arabinofuranosyl)-2-nitroimidazole (58 mg, 0.12 mmol) in MeCN (8 ml) was heated at 75°C for 3.5 h. After cooling and filtration, the filtrate was evaporated and the residue was purified by column chromatography, using ethyl acetate as a eluent to give l-β-D-(2-Deuterio arabinofuranosyl)-2-nitroimidazole (23 mg, 78%); m.p. 163-164°C, 1H-NMR, 13C- NMR, HRMS.
l-β-D-(2-Deuterio-5-deoxy-5-fluoroarabinofuranosyl)-2-nitroimidazole l-β-D-(2-Deuterio arabinofuranosyl)-2-nitroimidazole (20 mg, 0.08 mmol) was dissolved in anhydrous DME (6 ml) and cooled to -10°C and DAST (14 mg, 0.0.09 mmol) was added. After 1 h, the reaction mixture was allowed to warm to 0°C and after 2.5 h additional DAST (20 mg, 0.12 mmol) was added in three portions every 1 h and stirred for additional 5 h at 0°C. Additional DAST (10 mg, 0.06 mmol) was added until starting material disappeared on TLC. After quenching with MeOH and solvent evaporation, the products were separated by preparative TLC gave l-β-D-(2- deuterio-5-deoxy-5-fluoroarabinofuranosyl)-2-nitroimidazole (2 mg); 1H-NMR, 19F- NMR, MS
l-β-D-(2,3-Di-0-acetyl-5-deoxy-5-fluoroarabinofuranosyI)-2-nitroimidazole
SUBSΠTUTE SHEET (RULE 26)
DAST (0.613 g, 3.8 mmol) was added to a solution of l-β-D-(2,3-Di-<3- acetylarabinofuranosyl)-2-nitroimidazole in CH2Cl2 (9 mL) and pyridine (1 mL) at - 78°C and stirred at this temperature for 8h. The temperature was warmed up to 22°C and the stirring was continued for an additional 16h. Afterwards, the reaction mixture was cooled down to °C and quenched by adding ice water to it. Column chromatographic purification of the mixture, after removal of the solvents, using hexanes/ethyl acetate (1:1, v/v) afforded pure l-β-D-(2,3-Di-O-acetyl-5-deoxy-5- fluoroarabinofuranosyl)-2-nitroimidazole. m.p. 112-114°C, 1H NMR, 13C NMR, 19F- NMR.
l-β-D-(5-Deoxy-5-fluoroarabinofuranosyl)-2-nitroimidazole Treatment of 1 -β-D-(2,3-Di-O-acetyl-5-deoxy-5-fluoroarabinofuranosyl)-2- nitroimidazole with 2M. NH3/MeOH solution at 22°C afforded l-(-β-D-5-Deoxy-5- fluoroarabinofuranosyl)-2-nitroimidazole which was purified by column chromatography using ethyl acetate as an eluent, 1H NMR, 13C NMR3 19F NMR.
l-β-D-(2,3-Di-0-acetyl-5-deoxy-5-chloroarabinofuranosyI)-2-nitroimidazole
Trifluoromethanesulfonyl fluoride (0.53 g, 3.19 mmol) was added to a pre-cooled stirred solution (-80°C) of l-β-D-(2,3-Di-O-acetyl arabinofuranosyl)-2-nitroimidazole (0.55 g, 1.67 mmol) and dimethylamino pyridine (0.62g, 5 mmol) in anhydrous dichloromethane (40 mL) and the reaction was continued for 2h. Afterwards the temperature of the reaction was raised to 22°C and, then, quenched with water. The solvent was removed from the eraction mixture and the product was chromatographed
SUBSTiTUTE SHEET (RULE SM)
on a silica gel column using hexanes/ethyl acetate (40/60, v/v) to afford this product, m.p. 128°C, 1H NMR, HRMS, CHN.
l-β-D-(3,5-Di-0-benzoyl-2-deoxy-2-fluoroarabinofuranosyI)-2-nitroimidazole and l-β-D-(2,5-di-0-benzoyl-3-deoxy-3-fluorolyxofuranosyl)-2-nitroimidazole DAST (280 mg, 1.8 mmol) was added to a solution of l-β-D-(3,5-Di-O - benzoylarabinofuranosyl)-2-nitroimidazole (32) and l-β-D-(2,5-di-O - benzoylribofuranosyl)-2-nitroimidazole mixture (163 mg, 0.36 mmol) in pyridine (0.24 ml) and CH2Cl2 (10 ml) under ice bath, and stirred at 0-5°C for Ih and then allowed to warm to 22°C . After 7h, second portion of DAST (140 mg, 0.9 mmol) was added and stirring continued at same temperature for an additional 14h. Then, the reaction mixture was quenched by addition of MeOH, the solvents were evaporated and the residue was purified by preparative thin layer chromatography, using CHCl3 as a solvent to afford l-β-D-(3,5-di-0-benzoyl-2-deoxy-2-fluoroarabinofuranosyl)-2- nitroimidazole (67 mg, 41%); m.p 162-1630C, 1H-NMR, 13C-NMR, 19F-NMR,. MS, HR-mass, and 1 -β-D-(2,5-di-0-benzoyl-3~deoxy-3-fluorolyxofuranosyl)-2- nitroimidazole (35 mg, 21%) as a viscous oil; 1H-NMR, 13C-NMR, 19F-NMR, MS.
l-β-D-(2-Deoxy-2-fluoroarabinofuranosyl)-2-nitroimidazoIe A solution of l-β-D-(3,5-di-0-benzoy-l2-deoxy-2-fluoroarabinofuranosyl)-2- nitroimidazole (55 mg, 0.12 mmol) in methanolic ammonia (4 ml, 2.0 M) was stirred at 22°C for 9.5 h. After evaporation of solvent, the residue was chromatographed on silica gel column, eluting with CHCl3-MeOH (92.5:7.5) to give l-β-D-(2-Deoxy-2-
SUBSTiJUTE SHEET (RULE 26)
fluoroarabinofuranosyl)-2-nitroimidazole (27 mg, 92%); m.p 183-185°C, 1H-NMR, 13C-NMR5 19F-NMR.
l-β-D-(3-Deoxy-3-fluorolyxofuranosyl)-2-nitroimidazole
A solution of l-β-D-(2,5-di-O-benzoyl-3-deoxy-3-fluorolyxofuranosyl)-2-
nitroimidazole (30 mg) in methanolic ammonia (3 ml, 2.0 M) was stirred at 22°C for 14 h. After evaporation of solvent, the residue was chromatographed on silica gel column to give l-β-D-(3-deoxy-3-fluorolyxofuranosyl)-2-nitroimidazole (16 mg) with minor impurity; 1H-NMR, 19F-NMR.
l-β-D-(5-Deoxy-5-fluororibofuranosyl)-2-nitroimidazole
To a solution of l-β-D-(2,3-Di-O-acetylribofuranosyl)-2-nitroimidazole (228 mg,
0.69 mmol) and pyridine (0.51 ml) in CH2Cl2 at -20°C, DAST was added and then
allowed to warm to 0°C Over a period of 3 h. The reaction solution was stirred at 0°C
for 12 h. After addition of DAST (0.09 ml, 0.69 mmol), stirring was continued at 0°C for 12 h but not completed so additional DAST (0.09 ml, 0.69 mmol) was added and stirred at 5 °C for 12 h to completion. Then, the reaction solution was quenched with MeOH and evaporated. NH3/MeOH (2.9 M, 15 ml) was added and stirred at 5°C overnight. After evaporation of solvent, the residue was chromatographed on silica gel column, using CH2Cl2-MeOH (96:4, v/v) to afford l-β-D-(5-deoxy-5- fluororibofuranosyl)-2-nitroimidazole (75 mg, 44%); m.p 151-153°C; 1H-NMR, 13C- NMR, 19F-NMR, mass, HRMS.
SUBSTITUTE- SHEET (RULE 26)
EXAMPLE 3: Preparation of l-β-D-[(2/3-Eρoxy)-5- susbstitutedfuranosyl/hexopyranosylsyl)-2-nitroimidazoles
l-β-D-[(2/3-Epoxy)-5-deoxy-5-fluorofuranosyl/hexosyl)-2-nitroimidazoIe
This product was obtained as a side product during the synthesis of l-β-D-(5-deoxy- 5-fluoroarabinofuranosyl)-2-nitroimidazole. It was isolated and characterized by 1H- NMR, 19F-NMR, MS.
l-β-D-[(2/3-Epoxy)-2-deutero-5-deoxy~5-fluorofuranosyI/hexosyl)-2- nitroimidazole This product was obtained as a side product during the synthesis of,l-β-D-(2- deuterio-5-deoxy-5-fluoroarabinofuranosyl)-2-nitroimidazole. It was isolated and characterized by 1H-NMR, 19F-NMR, MS.
EXAMPLE 4: Preparation of l-α-D-[(2/3-Substituted) or (2,3-disusbstituted) or (2,2- disubstituted) or (3/3-disubstituted) furanosyl/hexopyranosyl)-2-nitroimidazoles
l-α-D-(3,5-Di-0-benzoyl-arabinofuranosyl)-2-nitroimidazo!e l-α-D-(2,3,5-Tri-O-benzoylarabinofuranosyl)-2-nitroimidazole (REF) was dissolved in anhydrous terahydrofuran (THF, 6 mL) and chilled to -56°C. IM solution of potassium tert-butoxide in THF (6.1 mL) was added to this solution under sitrring in an inert atmosphere. After the reaction was complete (15 min), the mixture was quenched by adding DOWEX-50™ until the pH was 7. The resin was filtered and the filtrate was subjected to solvent removal by evaporation. Column chromatography of
SUBSTiTUTE SHEET (BULE 26)
this mixture on a silica gel column using hexanes/ethyl acetate (1:3, v/v) and, then recrystallization of purified product in hexanes/ethylacetate (3:1, /v/v, 6 mL) afforded pure l-α-D-(3,5-Di-0-benzoylarabinofuranosyl)-2-nitroimidazole. m.p. 194-196°C, 1H NMR5 13C NMR.
l-α-D-(3,5-Di-0-benzoyl-2-0-[toluene/p-nitrobenzene]suIfonyIribofuranosyl)-2- nitroimidazole
A mixture of l-α-D-(3,5-di-0-benzoylribofuranosyl)-2-nitroimidazole (0.2 g, 0.44 mmol), toluenesulfonyl chloride or p-nitrobenzenesulfonyl chloride (1.32 mmol) an DMAP (0.16 g, 1.32 mmol) was taken in anhydrous pyridine (15 mL) and stirred at 45-50°C under argon for 16h and, then, the solvent was removed by evaporation. Purification of this mixture on a silica gel column using hexanes/ethyl acetate (2:1, v/v) gave this product, m.p. 60-61°C, 1H NMR, 13C NMR, CHN.
1-α-D -(3,5-Di-0-benzoyl-2-deoxy-2-fluororibofuranosyI)-2-nitroimidazole l-α-D-(3,5-Di-0-benzoyl-arabinofuranosyl)-2-nitroimidazole (0.6 g, 1.32 mmol) was treated with DAST (1.07 g, 6.6 mmol) in anhydrous dichloromethane (10 mL) and pyridine (0.2 mmol) at 0°C for 4h. Work up and purification of this product was done as described for other fluorinated compounds in this patent application, m.p. 56-57°C, 1H NMR, 13C NMR, 19F NMR, CHN.
l-α-D-(2-Deoxy-2-Fluororibofuranosyl)-2-nitroimidazole
SUBSTfTUTE SHEET (RDlE 20)
1 -(3 ,5-Di-0-benzoyl-2-deoxy-2-fluoro-α-D-ribofu ranosyl)-2-nitroimidazole (50 mg) was dissolved in NH3MeOH (2.0 M, 8 ml) and stirred overnight at 5°C. then the solvent was removed and the residue was purified on a silica gel column using MeOH-CHCl3 (7 : 93) to give l-α-D-(2-deoxy-2-fluororibofuranosyI)-2- nitroimidazole (20 mg).m.p. 155-157°C, 1H NMR, 13C NMR, 19F NMR, CHN.
l-(2-Deuterio-3,5-0-tetraisopropyldisilyloxy-α-D-ribofuranosyl)-2- nitroimidazole
A stirred suspension of CrO3 (15 mg) in CH2Cl2 (1 ml) was cooled to 0°C and Ac2O (0.015 ml) and pyridine (0.025 ml) were added. After 3 min, l-α-D-(3,5-O-O- tetraisopropyldisilyloxyarabinofuranosyl)-2-nitroimidazole (3,5-TIPS-α-AZA, (24mg,
0.05 mmol) was added to this solution and then allowed to warm to 5-10°C over a period of 2 h. Volatile materials were evaporated and the residue was cooled to 0°C and dissolved in absolute EtOH (1.0 ml). The stirred mixture was treated by addition OfNaBD4 (3 mg). After 30 min a second portion OfNaBD4 (3 mg) was added and a 2 mg portion was added at 1 h and allowed to warm to 10-12°C and stirred for 30 min.
After evaporation, the residue was chromatographed on a silica column, eluting with hexanes-ethyl acetate (80:20, v/v) to afford l-α-D-(2-deuterio-3,5-O,O- tetraisopropyldisilyloxy ribofuranosyl)-2 -nitroimidazole (15 mg, 61%); m.p 83-84°C, 1H-NMR, 13C-NMR, HRMS.
l-α-D-(2-Deuterio ribofuranosyl)-2-nitroimidazoIe
SUBSTITUTE SHEET PlE 20)
A stirred suspension of potassium fluoride (29 mg, 0.5 mmol), benzoic acid (61 mg, 0.5 mmol) and l-α-D-(2-deuterio-3,5-O,O-tetraisopropyldisilyloxyribofuranosyl)-2- nitroimidazole (41 mg, 0.084 mmol) in CH3CN (5 ml) was heated to 750C. After the reaction was over, the mixture was cooled, filtered and the filtrate was evaporated over a rotavapor. The residue was purified by column chromatography using MeOH:CHCl3 (8:92, v/v) to afford pure product. 1H-NMR, 13C-NMR.
EXAMPLE 5: Preparation of Epoxy)-5-susbstituted
 furanosyl/hexopyranosyl)-2-nitroimidazoles
furanosyl/hexopyranosyl)-2-nitroimidazoles
l-α-D-(2,3-Epoxy-5-deoxy-5-fluororibofuranosyl)-2-nitroimidazoIe
DAST (0.5 mmol) was added to a cooled (00C) suspension of α-AZA (0.1 mmol) in anhydrous DME and the stirring was continued at this temperature for 8h. After quenching with MeOH, the solvents were removed and l-α-D-(2,3-epoxy-5-fluoro- ribofuranosyl)-2-nitroimidazole was isolated by chromatography. 1H-NMR, 19F-NMR
EXAMPLE 6: Radiochemical Synthesis
^ ^^ analogs Qf ^
 products described under two genii and their labeling procedures are claimed under this patent. The details of general synthesis procedures for isotopic labeling of the Compounds of the Present Invention are described below.
products described under two genii and their labeling procedures are claimed under this patent. The details of general synthesis procedures for isotopic labeling of the Compounds of the Present Invention are described below.
Tritiation/Deuteration :
SUBSTITU SHEET (RULE 26)
A stirred suspension of CrO3 (15 mg) in CH2Cl2 (1 ml) was cooled to 0°C and Ac2O
(0.015 ml) and pyridine (0.025 ml) were added. After 3 min, l-β-D-(3,5-O- tetraisopropyldisilyloxy arabinofuranosyl)-2-nitroimidazole (24 mg, 0.05 mmol) is added and then allowed to warm to 5-10°C over a period of 2 h. Volatile materials were evaporated and the residue was cooled to 0°C and dissolved in absolute EtOH
(1.0 ml). The stirred mixture is treated by addition Of NaBT4 (3 mg). After 30 min a second portion of NaBT4 (3 mg) was added and a 2 mg portion was added at 1 h and allowed to warm to 10-12°C and stirred for 30 min. After evaporation, the residue is chromatographed on a silica column, eluting with hexanes-ethyl acetate (80:20) to afford corresponding tritiated product.

Deute rated and tritiated products
Similarly, the syntheses of other tritiated Compounds of the Present Invention may be undertaken.
SUBSTITUTE SHEET (WlE 28}
Radiohalogenation: abeled
 analogs of the Compounds of the Present Invention. Radiofluorination process with 18F isotope is provided as an illustrative example of radiohalogenation, though one skilled in the art would recognize that other radiohalogens could be utilised.
analogs of the Compounds of the Present Invention. Radiofluorination process with 18F isotope is provided as an illustrative example of radiohalogenation, though one skilled in the art would recognize that other radiohalogens could be utilised.
Radiofluorination of the Compounds of the Present Invention is carried out by using three radiofluorinated reagents namely

R4N[18F]F.
The precursors for radio(fluorin)halogention, pre-dissolved in appropriate solvent are allowed to react with appropriate radiofluorination reagen
 complex,
complex,
 an inert atmosphere. This process is temperature and time specific for radiohalogenation of every precursor. This is followed by removal of the protective groups and purification (automated or designed HPLC chromatography) to afford pure radiofluorinated product. The radiofluorination process is outlined below:
an inert atmosphere. This process is temperature and time specific for radiohalogenation of every precursor. This is followed by removal of the protective groups and purification (automated or designed HPLC chromatography) to afford pure radiofluorinated product. The radiofluorination process is outlined below:
2-NI
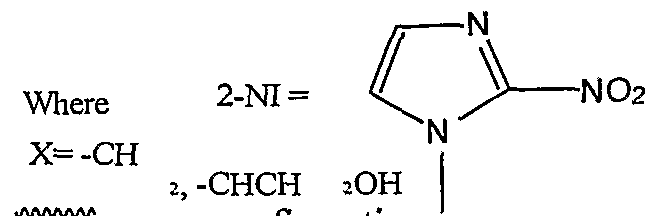
 * — = any configuration
* — = any configuration
Radiofluorination using K-2.2.2/ 18FZK2CO3 complexor
18F-DASTOr R4N[ 18F]F
2-NI

One of the -OR is leaving group and is replaced by radiofluorine Other -ORs represent the substituents claimed in the patent
The products radiofluorinated using this process include l-α-D-(2-deoxy-2-fluoro - ribofuranosyl)-2-nitroimidazole, l-β-D-(5-deoxy-5-fIuoroarabinofuranosyl)-2- nitroimidazole and l-β-D-(2-deoxy-2-fluororibofuranosyl)-2-nitroimidazole
Although the disclosure describes and illustrates various embodiments of the invention, it is to be understood that the invention is not limited to these particular embodiments. Many variations and modifications will now occurr to those skilled in the art.
EXAMPLE 7: Use of Compounds as Sensitizers in Cells Under Hypoxic Conditions The human colorectal carcinoma cell line HCTl 16 (WT) was used to observe the effects of selected Compounds of the Present Invention in sensitizing the cells, under
- 52 -
SUBSTfITUTE SHEET (M.E 26)
conditions of hypoxia, to radiotherapy. All hypoxia sensitizers with the exception of β-3 -FTAZR, which was dissolved in DMSO, were dissolved in 95% ethanol to the concentration of 10 mM. Cell treatment was performed for 30 min. prior to degassing and irradiation at 100 μM concentration of the tested sensitizer. Cells were irradiated in 60Co γ-irradiator at doses: 4 - 8 - 12 - 16 & 20 Gy
Approximately 300,000 cells were seeded per T60 glass dish with 3 ml DMEM/ F12 media added per dish. Dishes were incubated in 5% CO2 at 370C overnight. On the second day media in each dish was replaced with 2 ml fresh DMEM/F12. The plates were degassed in nitrogen in 6 groups of 2 dishes per chamber. Dishes were incubated for 30 min. on oscillating shaker at R/T X 60 cycles per min and irradiated as follows:
N2 chamber/2 dishes at 0 Gy (Control)
N2 chamber/2 dishes at 4 Gy
N2 chamber/2 dishes at 8 Gy N2 chamber/2 dishes at 12 Gy
N2 chamber/2 dishes at 16 Gy
N2 chamber/2 dishes at 20 Gy
Air chamber/2 dishes at 0 Gy (Control)
Air chamber/2 dishes at 4 Gy Air chamber/2 dishes at 8 Gy
Air chamber/2 dishes at 12 Gy
Air chamber/2 dishes at 16 Gy
Air chamber/2 dishes at 20 Gy
SUeSTJTUTE SHEET (RULE 26)
Cells were trypsinized from each dish and plate them at density, from 100 to 15000 cells/5 ml media for oxic conditions and 100 & 5000 cells/5 ml media for hypoxic conditions. Media was decanted from the dish, washed twice with PBS and 500 μl of Trypsin added. Trypsinization was quenched with 4.5 ml fresh media and serial dilutions of cell cells were made as follows: (A) 1:10; (B) 1 :100; (C) 1 : 1000. To each dish was added 1000 μl of dilution (C) for 100 cells per dish or 100 μl of dilution (B) for 100 cells per dish. Cells were incubated 10 to 14 days at 5% CO2 at 370C. On Day 10 to 14 colonies were stained with Methylene Blue or Crystal Violet stain in EtOH. Colonies were counted and plotted accordingly. As can be seen in Figure 2, use of the selected compound of the present invention decreased survival of cells under hypoxic conditions, compared to those cells under hypoxic conditions not similarly treated.
As shown in Figure 3 Compounds of the Present Invention, in particular IAZA compounds, can be utilized to effect a cytotoxicity in a cell population.
While particular embodiments of the present invention have been described in the foregoing, it is to be understood that other embodiments are possible within the scope of the invention and are intended to be included herein. It will be clear to any person skilled in the art that modifications of and adjustments to this invention, not shown, are possible without departing from the spirit of the invention as demonstrated through the exemplary embodiments. The invention is therefore to be considered limited solely by the scope of the appended claims. ,
SUBSTiTUTE SHEET (RULE M)
Claims
1. A compound useful for the therapeutic killing of hypoxic cells in a patient, said compound comprising the following structure:

wherein
R = H, Ac, Bz, Piv, TIPS, TBDPS, TBDMS, SO2Ri ;
Ri = CH3, toluyl, CF3, p-nitrobenzene and any other Leaving

; and X= -CH2, or -CHCH2OH.
2. A compound useful for the therapeutic killing of hypoxic cells in a patient, said compound comprising the following structure:
SUBSTITUTE SHEET (RULE 26) 
Wherein
-I (except at T -arabinose position),  t and 21 1At,;
t and 21 1At,; 


3. A compound useful for the therapeutic killing of hypoxic cells in a patient, said compound comprising the following structure:

Wherein
R = OH, OAc, OBz, OPiv, any halogen or OSO2Ri substituents at these positions;
Ri = CH3, toluyl, CF3, p-nitrobenzene and any other Leaving
Group.

X= -CH2, or -CHCH2OH
4. A compound useful for the therapeutic killing of hypoxic cells in a patient, said compound comprising the following structure:
SUSSTiTUTE SHEET (RULE 26) 
Wherein
Y = H, D, T, OH, F, ' 8F, Br, 75/76/77Br, Cl, 34m/34/36ci, I, 125I, 1231, 1311, 124I, At and 21 1At ; R = H ;

5. A compound useful for the therapeutic killing of hypoxic cells in a patient, said compound comprising the following structure:

SUBSTITUTE SHEET (PULE 26) 1 -α-D-[(2/3-Epoxy)-5-susbstituted furanosyl/hexopyranosyl)-2- nitroimidazoles.
Wherein
R = OH, OAc, OBz, OPiv, every halogen and OSO2Ri substituents;
Ri = CH3, toluyl, CF3, p-nitrobenzene and any other Leaving
Group;

X= -CH2, or -CHCH2OH
6. A compound useful for therapeutic treatment of hypoxic cells in a patient, said compound selected from the group consisting of : l-β-D-[3-deoxy-3-fluoroxylofuranosyl]-2-nitroimidazole, l-β-D-[3-deoxy-3chloroxylofuranosyl]-2-nitroimidazole,
l-β-D-[3-deoxy-3-brornoroxylofuranosyl]-2-nitroimidazole, l-β-D-[3-deoxy-3-fluororibofuranosyl]-2-nitroimidazole,
l-β-D-[3-deoxy-3-chlororibofuranosyl]-2-nitroimidazole,
l-β-D-[3-deoxy-3-bromoribofuranosyl]-2-nitroimidazole, l-β-D-[2-deoxy-2-fluororibofuranosyl]-2-nitroimidazole,
l-β-D-[2-deoxy-2-chlororibofuranosyl]-2-nitroimidazole,
SUBSTiTUTE SB€ET (RiIE 26) l-β-D-[2-deoxy-2-bromoribofuranosyl]-2-nitiOimidazole, l-β-D-[3-deoxy-3-fluoroarabinofuranosyl]-2-nitroimidazole, l-β-D-[3-deoxy-3-chloroarabinofuranosyl]-2-nitroimidazole, l-β-D-[3-deoxy-3-bromoarabinofuranosyl]-2-nitroimidazole,
l-β-D-[2-deoxy-2-fluoroarabinofuranosyl]-2-nitroimidazole,
l-β-D-[2-deoxy-2-chloroarabinofuranosyl]-2-nitroimidazole, l-β-D-[2-deoxy-2-bromoarabinofuranosyl]-2-nitroimidazole,
l-β-D-[3,2-dideoxy-3-fluoroarabinofuranosyl]-2-nitroimidazole,
l-β-D-[3,2-dideoxy-3-chloroarabinofuranosyl]-2-nitroimidazole, l-β-D-[3,2-dideoxy-3-bromoarabinofuranosyl]-2-nitroimidazole, l-β-D-[3,2-dideoxy-3-fluoroarabinofuranosyl]-2-nitroimidazole, l-β-D-[3,2-dideoxy-3-chloroarabinofuranosyl]-2-nitroimidazole,
l-β-D-[3,2-dideoxy-3-bromoarabinofuranosyl]-2-nitroimidazole,
l-β-D-[2,3-dideoxy-2-fluoroarabinofuranosyl]-2-nitroimidazole,
l-β-D-[2,3-dideoxy-2-chloroarabinofuranosyl]-2-nitroimidazole, l-β-D-[2,3-dideoxy-2-bromoarabinofuranosyl]-2-nitroimidazole, l-β-D-[2,3-dideoxy-2-fluororibofuranosyl]-2-nitroimidazole,
l-β-D-[2,3-dideoxy-2-chlororibofuranosyl]-2-nitroimidazole, l-β-D-[2,3-dideoxy-2-bromoribofuranosyl]-2-nitroimidazole, l-β-D-[2-deoxy-2-keto-arabinofuranosyl]-2-nitroimidazole,
1 -β-D-[3-deoxy-3-keto-arabinofuranosyl]-2-nitroimidazole, and
l-β-D-[2,3-dideoxy-2,3-epoxyarabinofuranosyl]-2-nitroimidazole.
SϋBSTJTUTΕ SHEET (FUlE 26)
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US66587505P | 2005-03-29 | 2005-03-29 | |
| US60/665,875 | 2005-03-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006102759A1 true WO2006102759A1 (en) | 2006-10-05 |
Family
ID=37052917
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CA2006/000482 WO2006102759A1 (en) | 2005-03-29 | 2006-03-29 | Novel substituted 2-nitroimidazoles useful for hypoxic cell therapy and imaging |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2006102759A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007079902A1 (en) * | 2005-12-21 | 2007-07-19 | Eberhard-Karls-Universitaet Tuebingen Universitaetsklinikum | Derivatives of 2-nitro-1,3-imidazole coupled to amino acids and deoxyribose useful for the detection of hypoxic biological tissue |
| WO2012087115A1 (en) | 2010-12-24 | 2012-06-28 | Stichting Maastricht Radiation Oncology "Maastro-Clinic" | Cancer targeting using carbonic anhydrase isoform ix inhibitors |
| WO2015025283A2 (en) | 2013-08-20 | 2015-02-26 | Stichting Maastricht Radiation Oncology "Maastro-Clinic" | Dual action carbonic anhydrase inhibitors |
| CN114344736A (en) * | 2021-12-09 | 2022-04-15 | 吉林大学 | Tumor treatment system and method involving in vivo photonuclear reaction |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1339130C (en) * | 1989-08-18 | 1997-07-29 | Leonard Irving Wiebe | Markers of tissue hypoxia |
| CA2400092A1 (en) * | 2000-02-14 | 2001-08-16 | Virexx Research, Inc. | Diagnostic and therapeutic compositions and methods for affecting tumor growth using oxygen mimetic agents |
-
2006
- 2006-03-29 WO PCT/CA2006/000482 patent/WO2006102759A1/en not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1339130C (en) * | 1989-08-18 | 1997-07-29 | Leonard Irving Wiebe | Markers of tissue hypoxia |
| CA2400092A1 (en) * | 2000-02-14 | 2001-08-16 | Virexx Research, Inc. | Diagnostic and therapeutic compositions and methods for affecting tumor growth using oxygen mimetic agents |
Non-Patent Citations (5)
| Title |
|---|
| KUMAR ET AL.: "SYNTHESIS OF BETA-AZOMYCIN NUCLEOSIDES: 1-(beta-D-2-IODO-2-DEOXYARABINOFURANOSYL)-2-NITROIMIDAZOLE (beta-2-IAZA), A NOVEL MARKER OF TISSUE HYPOXIA", TETRAHEDRON LETTERS, vol. 43, no. 25, 2002, pages 4427 - 4429, XP004361168, DOI: doi:10.1016/S0040-4039(02)00845-6 * |
| PATT ET AL.: "ADDUCT OF 2-[18F]FDG AND 2-NITROIMIDAZOLE AS A PUTATIVE RADIOTRACER FOR THE DETECTION OF HYPOXIA WITH PET: SYNTHESIS IN VITRO AND IN VIVO-CHARACTERIZATION", APPLIED RADIATION AND ISOTOPES, vol. 57, no. 5, 2002, pages 705 - 712, XP004381725, DOI: doi:10.1016/S0969-8043(02)00186-0 * |
| PEDERSE ET AL.: "AZOMYCIN RIBOSE: A NEW RADIOSENSITIZER", INT. J. RADIATION ONCOLOGY BIOL. PHYS., vol. 8, no. 3 AND 4, 1982, pages 415 - 418, XP026841598 * |
| SAKAGUCHI ET AL.: "POTENTIAL RADIOSENSITIZING AGENTS. 4. 2-NITROIMIDAZOLE NUCLEOSIDES", J. MED. CHEM., vol. 25, no. 11, 1982, pages 1339 - 1342, XP002979334, DOI: doi:10.1021/jm00353a013 * |
| SCHNEIDER ET AL.: "THE SYNTHESIS AND RADIOLABELING OF NOVEL MARKERS OF TISSUE HYPOXIA OF THE IODINATED AZOMYCIN NUCLEOSIDE CLASS", JOURNAL OF LABELLED COMPOUNDS AND RADIOPHARMACEUTICALS, vol. 39, no. 7, 1996, pages 541 - 557, XP008040431, DOI: doi:10.1002/(SICI)1099-1344(199707)39:7<541::AID-JLCR5>3.0.CO;2-B * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007079902A1 (en) * | 2005-12-21 | 2007-07-19 | Eberhard-Karls-Universitaet Tuebingen Universitaetsklinikum | Derivatives of 2-nitro-1,3-imidazole coupled to amino acids and deoxyribose useful for the detection of hypoxic biological tissue |
| WO2012087115A1 (en) | 2010-12-24 | 2012-06-28 | Stichting Maastricht Radiation Oncology "Maastro-Clinic" | Cancer targeting using carbonic anhydrase isoform ix inhibitors |
| US8980932B2 (en) | 2010-12-24 | 2015-03-17 | Stichting Maastricht Radiation Oncology “Maastro-Clinic” | Cancer targeting using carbonic anhydrase isoform IX inhibitors |
| WO2015025283A2 (en) | 2013-08-20 | 2015-02-26 | Stichting Maastricht Radiation Oncology "Maastro-Clinic" | Dual action carbonic anhydrase inhibitors |
| CN114344736A (en) * | 2021-12-09 | 2022-04-15 | 吉林大学 | Tumor treatment system and method involving in vivo photonuclear reaction |
| CN114344736B (en) * | 2021-12-09 | 2022-09-13 | 吉林大学 | Tumor treatment system and method involving in vivo photonuclear reaction |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Mannan et al. | Radioiodinated 1-(5-iodo-5-deoxy-β-D-arabinofuranosyl)-2-nitroimidazole (iodoazomycin arabinoside: IAZA): a novel marker of tissue hypoxia | |
| US7928210B2 (en) | Nucleoside based proliferation imaging markers | |
| US7045115B2 (en) | Drugs for the diagnosis of tissue-reproductive activity or the treatment of proliferative diseases | |
| US8845999B2 (en) | Tracers for monitoring the activity of sodium/glucose cotransporters in health and disease | |
| US11865194B2 (en) | Benzene ring-containing glucose derivative and use thereof | |
| WO2006102759A1 (en) | Novel substituted 2-nitroimidazoles useful for hypoxic cell therapy and imaging | |
| Yang et al. | Synthesis and bioevaluation of radioiodinated nitroimidazole hypoxia imaging agents by one-pot click reaction | |
| Ilem‐Ozdemir et al. | Radiolabeling and in vitro evaluation of a new 5‐fluorouracil derivative with cell culture studies | |
| ES2397255T3 (en) | Nucleoside analogs useful as positron emission tomography imaging (PET) agents | |
| US20060270610A1 (en) | Novel compounds for hypoxic cell therapy and imaging | |
| Gonzalez et al. | The 4-N-acyl and 4-N-alkyl gemcitabine analogues with silicon-fluoride-acceptor: application to 18F-radiolabeling | |
| US5401490A (en) | Markers of tissue hypoxia | |
| EP1916003A1 (en) | Thymidine analogs for imaging | |
| US8420049B2 (en) | Labeled adenosine for use in positron emission tomography | |
| Schweifer et al. | [18F] Fluoro-azomycin-2´-deoxy-β-d-ribofuranoside—A new imaging agent for tumor hypoxia in comparison with [18F] FAZA | |
| Singh et al. | Synthesis and preliminary evaluation of a 99m Tc labelled deoxyglucose complex {[99m Tc] DTPA-bis (DG)} as a potential SPECT based probe for tumor imaging | |
| Mannan et al. | Radioiodinated 1-(2-fluoro-4-iodo-2, 4-dideoxy-β-L-xylopyranosyl)-2-nitroimidazole: a novel probe for the noninvasive assessment of tumor hypoxia | |
| Misra et al. | Synthesis of 131I, 125I, 123I and 82Br labelled 5-halo-1-(2-deoxy-2-fluoro-β-d-arabinofuranosyl) uracils | |
| US8969546B2 (en) | Compounds useful in imaging and therapy | |
| Celen et al. | Synthesis and biological evaluation of 11C-labeled β-galactosyl triazoles as potential PET tracers for in vivo LacZ reporter gene imaging | |
| Ishiwata et al. | Brain tumor accumulation and plasma pharmacokinetic parameters of 2′-deoxy-5-18 F-fluorouridine | |
| US20150133777A1 (en) | Positron emission tomography probe to monitor selected sugar metabolism in vivo | |
| TWI394587B (en) | A radiolabeled nucleoside analogue, a method for preparing the same and the use thereof | |
| Nascimento et al. | Synthesis and quality control of [18 F] fluorothymidine | |
| GUNDOGDU et al. | Radiolabeling and in vitro evaluation of a new 5-Fluorouracil derivative with cell culture studies |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: RU |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06721741 Country of ref document: EP Kind code of ref document: A1 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 6721741 Country of ref document: EP |