WO2006090272A1 - Isoquinoline [1,8]naphthyridin-2-ones and related compounds for treatment of schizophrenia - Google Patents
Isoquinoline [1,8]naphthyridin-2-ones and related compounds for treatment of schizophrenia Download PDFInfo
- Publication number
- WO2006090272A1 WO2006090272A1 PCT/IB2006/000476 IB2006000476W WO2006090272A1 WO 2006090272 A1 WO2006090272 A1 WO 2006090272A1 IB 2006000476 W IB2006000476 W IB 2006000476W WO 2006090272 A1 WO2006090272 A1 WO 2006090272A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- disorder
- naphthyridin
- butoxy
- piperazin
- disorders
- Prior art date
Links
- 0 CC(C1NCCC[U]CC1C*C1)C1=C Chemical compound CC(C1NCCC[U]CC1C*C1)C1=C 0.000 description 4
- BXLACVBTKVVTOT-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCN1c1c(cccc2)c2cc(C)n1)=O Chemical compound CC(C)(C)OC(N(CC1)CCN1c1c(cccc2)c2cc(C)n1)=O BXLACVBTKVVTOT-UHFFFAOYSA-N 0.000 description 1
- VTBOTOBFGSVRMA-UHFFFAOYSA-N CC1(CCCCC1)O Chemical compound CC1(CCCCC1)O VTBOTOBFGSVRMA-UHFFFAOYSA-N 0.000 description 1
- UNRGNIQJHIOPFN-UHFFFAOYSA-N Cc1c(C=CC(N2)=O)c2nc(OCCCCOC2OCCCC2)n1 Chemical compound Cc1c(C=CC(N2)=O)c2nc(OCCCCOC2OCCCC2)n1 UNRGNIQJHIOPFN-UHFFFAOYSA-N 0.000 description 1
- BMLWCDIRNIPFBY-UHFFFAOYSA-N Cc1nc(OCCCC=O)nc(N2)c1C=CC2=O Chemical compound Cc1nc(OCCCC=O)nc(N2)c1C=CC2=O BMLWCDIRNIPFBY-UHFFFAOYSA-N 0.000 description 1
- DVTUBNBXWLJYTE-UHFFFAOYSA-N O=C(C=C1)Nc2c1cnc(OCCCCOC1OCCCC1)n2 Chemical compound O=C(C=C1)Nc2c1cnc(OCCCCOC1OCCCC1)n2 DVTUBNBXWLJYTE-UHFFFAOYSA-N 0.000 description 1
- QLZMRYMVGWSNCR-UHFFFAOYSA-N OCCCCOc1nc(NC(C=C2)=O)c2cn1 Chemical compound OCCCCOc1nc(NC(C=C2)=O)c2cn1 QLZMRYMVGWSNCR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
Definitions
- This invention relates to [l,8]naphthyridin-2-ones and related compounds, methods of making such compounds, pharmaceutical compositions containing them, and their use for the treatment of schizophrenia and other central nervous system (CNS) disorders.
- CNS central nervous system
- the [l,8]naphthyridin-2-ones and related compounds of this invention bind to dopamine D 2 receptors. Some exhibit activity as partial agonists of D 2 receptors, while others exhibit activity as antagonists of such receptors. Other heterocyclic derivatives that are useful for the treatment of schizophrenia are referred to in United States patent 5,350,747, which issued on September 27, 1994; in United States patent 6,127,357, which issued on October 3, 2000; in WO 93/04684, which published on March 18, 1993; and European patent application EP 402644A, which was published on
- A is -(CH 2 ) n O- wherein n is 4 or 5, and one or two of the carbon atoms of - (CH 2 ) n O- can be substituted, optionally and independently, with one or two substituents that are selected, independently, from fluoro and methyl, or with two substituents attached to the same carbon atom that form, together with the carbon to which they are attached, a spirocyclopropyl or spirocyclobutyl ring;
- D is N, C, or CH, provided that when D is N, each carbon atom attached to D is attached through a single bond;
- J and K are independently selected from N, CH, and C, provided that at least one of J and K is N;
- Z and Q are independently selected from N and CH, provided that at least one of Z and Q is N;
- R and R are selected, independently, from hydrogen and methyl;
- ring AA is a saturated or unsaturated 5-, 6-, or 7-membered carbocyclic ring wherein one, two or three of the carbon atoms of ring AA that are not shared with the other fused ring of formula (i) can be replaced, optionally and independently, by a nitrogen, oxygen or sulfur atom; and the pharmaceutically acceptable salts of such compounds
- This invention also relates to a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of the formula 1, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the compounds of formula 1 have useful pharmaceutical and medicinal properties.
- the invention also relates to a pharmaceutical composition for treating a disorder or condition selected from single episodic or recurrent major depressive disorders, dysthymic disorders, depressive neurosis and neurotic depression, melancholic depression; atypical depression; bipolar disorder; cyclothymic disorder; conduct disorder; disruptive behavior disorder; attention deficit hyperactivity disorder; behavioral disturbances associated with mental retardation, autistic disorder, and conduct disorder; anxiety disorders; borderline personality disorder; schizophrenia and other psychotic disorders; delirium, dementia, and amnestic and other cognitive or neurodegenerative disorders; movement disorders, dyskinesias; extra-pyramidal movement disorders; chemical dependencies and additions; behavioral addictions; and ocular disorders, comprising: an amount of a compound of formula 1, or a pharmaceutically acceptable salt thereof that is effective in treating the disorder or condition, and a pharmaceutically acceptable carrier.
- the invention further relates to a pharmaceutical composition for treating a disorder or condition selected from those listed above, comprising: (a) a compound of formula 1, or a pharmaceutically acceptable salt thereof; and (b) an antidepressant or an anti-anxiety agent; and (c) a pharmaceutically acceptable carrier; wherein active agents (a) and (b) are not the same and are present in amounts that render the combination of them effective in treating said disorder or condition.
- This invention also relates to a method of treating a disorder or condition selected from those listed above, comprising administering to a mammal in need of such treatment an amount of a compound according to formula 1, or a pharmaceutically acceptable salt thereof, that is effective in treating the disorder or condition.
- the invention also relates to a method of treating a disorder or condition selected from those listed above, comprising administering to a mammal in need of such treatment (a) a compound of formula 1, or a pharmaceutically acceptable salt thereof; and (b) an antidepressant or an anti-anxiety agent; wherein the active agents (a) and (b) are not the same and are present in amounts that render the combination of them effective in treating the disorder or condition.
- alkyl as used herein, unless otherwise indicated, includes saturated monovalent hydrocarbon radicals having straight, branched or cyclic moieties or combinations thereof.
- alkyl groups include, but are not limited to, methyl, ethyl, propyl, isopropyl, butyl, iso- sec- and tert-butyl, pentyl, hexyl, heptyl, 3- ethylbutyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, norbornyl, and the like.
- alkoxy as used herein, unless otherwise indicated, means “alkyl-O- ", wherein “alkyl” is as defined above.
- alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy, butoxy and pentoxy.
- aryl as used herein, unless otherwise indicated, includes an aromatic ring system with no heteroatoms as ring members, which can be either unsubstituted or substituted with one, two or three substituents selected from the group consisting of halo, (C 1 -C 4 ) ⁇ yI optionally substituted with from one to three fluorine atoms and (Q-GOalkoxy optionally substituted with from one to three fluorine atoms.
- aryloxy as used herein, unless otherwise indicated, means "aryl- O-", wherein “aryl” is as defined above.
- one or more substituents refers to a number of substituents that equals from one to the maximum number of substituents possible based on the number of available bonding sites.
- halo and halogen, as used herein, unless otherwise indicated, include, fluoro, chloro, bromo and iodo.
- therapeutically effective amount refers to a quantity of active agent sufficient to treat one or more of the disorders or conditions referred to above, when one or more doses of a pharmaceutical composition of the invention are administered to a subject with one or more of the disorders or conditions.
- a number of factors will generally be considered, including the experience of the medical practitioner or veterinarian administering the composition, published clinical studies, the subject's age, sex, weight and general condition, as well as the type and extent of the disorder or condition being treated, and the use of other medications, if any, by the subject. Determination of a proper dose for a particular situation, and preparation of a pharmaceutical composition containing a suitable dose of active agent for that situation, is within the skill of the medical or veterinary arts.
- treating refers to reversing, alleviating, inhibiting the progress of, or preventing the disorder or condition to which such term applies, or preventing one or more symptoms of such condition or disorder.
- treatment refers to the act of treating, as "treating” is defined immediately above.
- Examples of preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein AA is a 6-membered unsaturated carbocyclic ring.
- Other preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein A is -(CH 2 ) 4 O-.
- Other preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein Q is N.
- Other preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein -X — Y- is -CH 2 -NH-.
- R 10 and R 11 are compounds of formula 1 and their pharmaceutically acceptable salts, wherein each of R 10 and R 11 is, independently, hydrogen or fluoro .
- K' and J' are independently selected from N, CH, and C, provided that at least one of K' and J' is N;
- R 8' is H, (C 1 -C 4 ) alkyl, or (C 1 -C 4 ) alkoxy;
- R 10' is H, (C 1 -C 4 ) alkyl, (C 1 -C 4 ) alkoxy, or halo;
- U, V, and W are independently selected from N, CH, and C, provided that no more than two of U, V, and W are N.
- R 8 is preferably attached to K'.
- R 8 is preferably H or methyl.
- R 10 is preferably attached to U or V.
- R 10 is preferably H, methyl, or fluoro.
- G is preferably an isoquinoline of formula (ii), more preferably an isoquinoline selected from the group consisting of 1-isoquinolinyl, 7-fluoro-l-isoquinolinyl, 6- fluoro-1-isoquinolinyl, 3-methyl-l-isoquinolinyl, and 7-fluoro-3 -methyl- 1- isoquinolinyl.
- Compounds of the formula 1 may contain chiral centers and therefore may exist in different enantiomeric and diastereomeric forms.
- This invention relates to all optical isomers and all stereoisomers of compounds of the formula 1, both as racemic mixtures and as individual enantiomers and diastereoisomers of such compounds, and mixtures thereof, and to all pharmaceutical compositions and methods of treatment defined above that contain or employ them, respectively.
- Individual isomers can be obtained by known methods, such as optical resolution, fractional crystallization, optically selective reaction, or chromatographic separation in the preparation of the final product or its intermediate.
- Individual enantiomers of the compounds of formula 1 may have advantages, as compared with the racemic mixtures of these compounds, in the treatment of various disorders or conditions.
- the compounds of formula 1 are basic compounds, they are all capable of forming a wide variety of different salts with various inorganic and organic acids. Although such salts must be pharmaceutically acceptable for administration to animals, it is often desirable in practice to initially isolate the base compound from the reaction mixture as a pharmaceutically unacceptable salt and then simply convert to the free base compound by treatment with an alkaline reagent and thereafter convert the free base to a pharmaceutically acceptable acid addition salt.
- the acid addition salts of the base compounds of this invention are readily prepared by treating the base compound with a substantially equivalent amount of the chosen mineral or organic acid in an aqueous solvent or in a suitable organic solvent, such as methanol or ethanol. Upon careful evaporation of the solvent, the desired solid salt is readily obtained.
- the acids which are used to prepare the pharmaceutically acceptable acid addition salts of the aforementioned base compounds of this invention are those which form non-toxic acid addition salts, Le., salts containing pharmaceutically acceptable anions, such as the hydrochloride, hydrobromide, hydroiodide, nitrate, sulfate or bisulfate, phosphate or acid phosphate, acetate, lactate, citrate or acid citrate, tartrate or bi-tartrate, succinate, maleate, fumarate, gluconate, saccharate, benzoate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate and pamoate (i.e., l,l'-methylene-bis-(2-hydroxy- 3-naphthoate)) salts.
- pharmaceutically acceptable anions such as the hydrochloride, hydrobromide, hydroiodide, nitrate, s
- the present invention also includes isotopically labeled compounds, which are identical to those of formula 1, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be incorporated into compounds of the present invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, sulfur, fluorine and chlorine, such as 2 H, 3 H, 13 C, 11 C, 14 C, 15 N, 18 0, 17 0, 31 P, 32 P, 35 S, 18 F, and 36 Cl, respectively.
- Compounds of the present invention, prodrugs thereof, and pharmaceutically acceptable salts of said compounds or of said prodrugs which contain the aforementioned isotopes and/or other isotopes of other atoms are within the scope of this invention.
- Certain isotopically labeled compounds of the present invention, for example those into which radioactive isotopes such as 3 H and 14 C are incorporated, are useful in drug and/or substrate tissue distribution assays. Tritiated, i.e., 3 H, and carbon-14, Le., 14 C, isotopes are particularly preferred for their ease of preparation and detectability.
- isotopically labeled compounds of formula 1 and prodrugs thereof can generally be prepared by carrying out the procedures disclosed in the Schemes and/or in the Examples below, by substituting a readily available isotopically labeled reagent for a non-isotopically labeled reagent.
- compositions and methods of the present invention are suitable for use in treatment of a disease or disorder selected from single episodic or recurrent major depressive disorders, dysthymic disorders, depressive neurosis and neurotic depression, melancholic depression including anorexia, weight loss, insomnia, early morning waking or psychomotor retardation; atypical depression (or reactive depression) including increased appetite, hypersomnia, psychomotor agitation or irritability, seasonal affective disorder and pediatric depression; bipolar disorders or manic depression, for example, bipolar I disorder, bipolar II disorder and cyclothymic disorder; conduct disorder; disruptive behavior disorder; attention deficit hyperactivity disorder (ADHD); behavioral disturbances associated with mental retardation, autistic disorder, and conduct disorder; anxiety disorders such as panic disorder with or without agoraphobia, agoraphobia without history of panic disorder, specific phobias, for example, specific animal phobias, social anxiety, social phobia, obsessive-compulsive disorder, stress disorders including post-traumatic stress disorder and acute stress disorder, and generalized anxiety disorders; border
- the disorder or condition treated is selected from major depression, single episode depression, recurrent depression, child abuse induced depression, postpartum depression, dysthymia, cyclothymia and bipolar disorder.
- the disorder or condition treated is selected from schizophrenia, schizoaffective disorder, delusional disorder, substance-induced psychotic disorder, brief psychotic disorder, shared psychotic disorder, psychotic disorder due to a general medical condition, and schizophreniform disorder.
- the disorder or condition that is being treated is selected from autism, pervasive development disorder, speech impediments such as stuttering, and attention deficit hyperactivity disorder.
- the disorder or condition that is being treated is selected from generalized anxiety disorder, panic disorder, obsessive-compulsive disorder, posttraumatic stress disorder, and phobias, including social phobia, agoraphobia, and specific phobias.
- the disorder or condition that is being treated is selected from movement disorders such as akinesias, dyskinesias, including familial paroxysmal dyskinesias, spasticities, Tourette's syndrome, Scott syndrome, PALSYS and akinetic- rigid syndrome; and extra-pyramidal movement disorders such as medication-induced movement disorders, for example, neuroleptic-induced Parkinsonism, neuroleptic malignant syndrome, neuroleptic-induced acute dystonia, neuroleptic-induced acute akathisia, neuroleptic-induced tardive dyskinesia and medication-induced postural tremor.
- movement disorders such as akinesias, dyskinesias, including familial paroxysmal dyskinesias, spasticities, Tourette's syndrome, Scott syndrome, PALSYS and akinetic- rigid syndrome
- extra-pyramidal movement disorders such as medication-induced movement disorders, for example, neuroleptic-induced Parkinsonism, neuroleptic malignant syndrome, neuroleptic-induced acute dystonia, neuro
- the disorder or condition that is being treated is selected from delirium, dementia, and amnestic and other cognitive or neurodegenerative disorders, such as Parkinson's disease (PD), Huntington's disease (HD), Alzheimer's disease, senile dementia, dementia of the Alzheimer's type, memory disorder, vascular dementia, and other dementias, for example, due to HIV disease, head trauma, Parkinson's disease, Huntington's disease, Pick's disease, Creutzfeldt- Jakob disease, or due to multiple etiologies.
- Parkinson's disease PD
- Huntington's disease HD
- Alzheimer's disease senile dementia
- dementia of the Alzheimer's type dementia of the Alzheimer's type
- memory disorder vascular dementia
- other dementias for example, due to HIV disease, head trauma, Parkinson's disease, Huntington's disease, Pick's disease, Creutzfeldt- Jakob disease, or due to multiple etiologies.
- the compound of formula 1 is administered to a human for the treatment of any two or more comorbid disorders or conditions selected from those disorders and conditions referred to in any of the above methods.
- compositions of the present invention comprising (a) a compound of formula 1, or a pharmaceutically acceptable salt thereof, and (b) an antidepressant or an anti-anxiety agent, as described above, or in methods of the present invention comprising administering active agents (a) and (b)
- classes antidepressants suitable for use in the invention include norepinephrine reuptake inhibitors, selective serotonin reuptake inhibitors (SSRIs), NK-I receptor antagonists, monoamine oxidase inhibitors (MAOIs), reversible inhibitors of monoamine oxidase (RIMAs), serotonin and noradrenaline reuptake inhibitors (SNRIs), corticotropin releasing factor (CRF) antagonists, ⁇ -adrenoreceptor antagonists, and atypical antidepressants.
- norepinephrine reuptake inhibitors selective serotonin reuptake inhibitors (SSRIs), NK-I receptor antagonists, monoamine oxid
- Suitable norepinephrine reuptake inhibitors include tertiary amine tricyclics and secondary amine tricyclics.
- Suitable tertiary amine tricyclics and secondary amine tricyclics include amitriptyline, clomipramine, doxepin, imipramine, trimipramine, dothiepin, butripyline, iprindole, lofepramine, nortriptyline, protriptyline, amoxapine, desipramine and maprotiline.
- Suitable selective serotonin reuptake inhibitors include fluoxetine, fluvoxamine, paroxetine and sertraline.
- Examples of monoamine oxidase inhibitors include isocarboxazid, phenelzine, and tranylcyclopramine.
- Suitable reversible inhibitors of monoamine oxidase include moclobemide.
- Suitable serotonin and noradrenaline reuptake inhibitors of use in the present invention include venlafaxine.
- Suitable CRF antagonists include those compounds described in International Patent Application Nos. WO 94/13643, WO 94/13644, WO 94/13661, WO 94/13676 and WO 94/13677.
- Suitable atypical antidepressants include bupropion, lithium, nefazodone, trazodone and viloxazine.
- Suitable NK-I receptor antagonists include those referred to in World Patent Publication WO 01/77100.
- Suitable classes of anti-anxiety agents that can be used in combination with the compounds of formula 1 in the pharmaceutical compositions and methods of this invention include benzodiazepines and serotonin IA (5-HT ⁇ A ) agonists or antagonists, especially 5-HT 1A partial agonists, and corticotropin releasing factor (CRF) antagonists.
- Suitable benzodiazepines include alprazolam, chlordiazepoxide, clonazepam, chlorazepate, diazepam, halazepam, lorazepam, oxazepam, and prazepam.
- Suitable 5- HT 1A receptor agonists or antagonists include buspirone, flesinoxan, gepirone and ipsapirone.
- the active compounds of this invention may be prepared as described below. Unless otherwise indicated, in the reaction schemes and discussion that follow, A, Z, D, Q, ring AA, G, X, Y, R 1 through R 11 , formula 1, the dotted line connecting X and Y, and groups of formulas (i) and (ii) are defined as above, m is 3 or 4 in each of the reaction schemes, below.
- Scheme A illustrates a method for preparing compounds of the formula 1, optionally substituted as indicated in the definition of formula 1 above (also referred to as compounds of the formula IA).
- This method involves oxidation of a compound of the formula 2 with Dess-Martin Periodinane or another suitable oxidizing agent such as EBX (o-iodoxybenzoic acid), oxalyl chloride in dimethyl sulfoxide (DMSO) (Swern oxidation) or PCC (pyridinium chlorochromate) to form the corresponding aldehyde of formula 3.
- This reaction may be carried out in dichloromethane (CH 2 Cl 2 ), tetrahydrofuran (THF), DMSO, or a combination of two or more of these solvents.
- the reductive amination can be performed, for example, utilizing catalytic hydrogenation methods or using a hydride reducing agent such as sodium triacetoxyborohydride (NaBH(OAc) 3 ) or sodium cyanoborohydride.
- the reaction solvent can be 1,2-dichloroethane, tetrahydrofuran, acetonitrile, dimethylformamide or a combination of two or more of these solvents, with the optional addition of 1-10 equivalents of acetic acid.
- a base such as triethylamine is typically added.
- compounds of the formula IA can be prepared according to Scheme B.
- the hydroxy group of the compound of formula 2 is converted into a leaving group (L) using conventional methods to provide the corresponding compound of formula 4 wherein L is mesylate (OMs), tosylate (OTs) or a halogen such as bromide, iodide or chloride.
- L is mesylate (OMs), tosylate (OTs) or a halogen such as bromide, iodide or chloride.
- OMs mesylate
- OTs tosylate
- a halogen such as bromide, iodide or chloride.
- the resulting compound of formula 4 is then reacted with a G-substituted piperazine or piperidine, as depicted in Scheme B, to yield the desired compound of formula IA.
- This reaction is preferably run in the presence of a base such as potassium carbonate, sodium carbonate, cesium carbonate, triethy
- the solvent used may be acetonitrile, water, tetrahydrofuran, dioxane, acetone, methyl isobutyl ketone, benzene or toluene, or a combination of two or more of these solvents.
- Inorganic salts such as sodium or potassium iodide may be employed as catalysts in the reaction.
- the temperature of the reaction may vary from about ambient temperature to about the reflux temperature of the solvent used.
- the reaction may also be conducted under microwave irradiation.
- Scheme C illustrates a method for preparing compounds of the formula 2A (wherein X is double bonded to Y) and 2B (wherein X is single bonded to Y).
- R 12 is most preferably fluoro.
- This reaction is conducted in the presence of a base such as potassium tert-butoxide, sodium tert-butoxide, sodium hydride, potassium hydride, lithium diisopropylamide, lithium bis(trimethylsilyl)amide, potassium bis(trimethylsilyl)amide, or sodium bis(trimethylsilyl)amide.
- a base such as potassium tert-butoxide, sodium tert-butoxide, sodium hydride, potassium hydride, lithium diisopropylamide, lithium bis(trimethylsilyl)amide, potassium bis(trimethylsilyl)amide, or sodium bis(trimethylsilyl)amide.
- Suitable solvents for this reaction include THF, dioxane, ethylene glycol dimethylether, DMF, N-methylpyrrolidinone (NMP), or DMSO, or a combination of two or more of these solvents.
- the temperature of the reaction may vary from about ambient temperature to about the reflux temperature of the
- the reaction of the compound of formula 5 with the mono-protected diol of the formula POCH 2 (CH 2 ) m OH to yield the compound of formula 6, as described above, is preferably conducted in the presence of a catalytic amount of a phase transfer catalyst, such as tetrabutyl ammonium chloride or bromide.
- a phase transfer catalyst accelerates the rate of the coupling, and allows one to carry out the reaction at a lower temperature than would be possible without the catalyst.
- Use of the phase transfer catalyst also significantly reduces the formation of dimeric by-products.
- Compounds of the formula 2A or compounds of the formula 6 wherein P is H or benzyl can be reduced using catalytic hydrogenation methods to provide the corresponding compounds of formula 2B.
- the hydrogenation can be conducted using 5 to 20 % palladium on activated carbon in a solvent such as methanol, ethanol, tetrahydrofuran, acetic acid, dimethylformamide, or a combination of two or more of these solvents for a period of about 5 hours to about 48 hours, preferably for about 24 hours, under a hydrogen pressure from about 1 to about 5 atmosphere, preferably about 1 atmosphere.
- R 12 Cl, F, Br, S(O)Me, SO 2 Me
- Suitable solvents for this reaction include THF, dioxane, ethylene glycol dimethylether, DMF, NMP, and DMSO, or a combination of two or more of these solvents.
- the reaction temperature can range from about -78 °C to about ambient temperature, and is preferably from about -20 ° to about 0°C.
- Catalytic hydrogenation of compounds of the formula IA-I (when G is compatible with the hydrogenation conditions) provides compounds of formula 1A-2.
- the hydrogenation can be conducted using 5 to 20% palladium on activated carbon in a solvent such as methanol, ethanol, THF, acetic acid, or DMF, or a combination of two or more of these solvents, for a period of about 5 hours to about 48 hours, preferably for about 12 to 24 hours.
- a solvent such as methanol, ethanol, THF, acetic acid, or DMF, or a combination of two or more of these solvents, for a period of about 5 hours to about 48 hours, preferably for about 12 to 24 hours.
- Scheme E illustrates an alternative method for preparing compounds of the formula 5A-1.
- Compounds of the formula 5A-1 are compounds of the formula 5 wherein X is CR 3 , Y is CR 2 and there is a double bond between X and Y).
- R 3 is (C 1 -C 4 ) alkyl.
- Ortho metalation of compounds of the formula 8 and subsequent treatment with electrophiles having the formula shown in Scheme E results in compounds of the formula 9.
- Condensation of compounds of the formula 9 with the enolates of the alkyl esters having the formula shown in Scheme E provides the corresponding compounds of formula 10.
- Refluxing compounds of the formula 10 in aqueous acid such as 3N hydrochloric acid with the optional use of a co- solvent such as dioxane, generates the corresponding compounds of formula 5A-1.
- Scheme F illustrates an alternative method for preparing compounds of the formula 5A-1.
- Scheme F illustrates another method for preparing compounds of the formula 2A-1.
- R 3 is (C 1 -C 4 ) alkyl.
- Addition of a suitably mono-protected diol, where n is 3 or 4 and P is tetrahydropyranyl (THP), benzyl, or te/t-butyldimethysilyl, to compounds of the formula 9 provides compounds of the formula 11.
- the reaction is conducted in the presence of a base such as potassium tert-butoxide, sodium tert- butoxide, sodium hydride, potassium hydride, lithium diisopropylamide, lithium bis(trimethylsilyl)amide, potassium bis(trimethylsilyl)amide, or sodium bis(trimethylsilyl)amide.
- a base such as potassium tert-butoxide, sodium tert- butoxide, sodium hydride, potassium hydride, lithium diisopropylamide, lithium bis(trimethylsilyl)amide, potassium bis(trimethylsilyl)amide, or sodium bis(trimethylsilyl)amide.
- the solvents used may be THF, dioxane, ethylene glycol dimethylether, DMF, NMP, or DMSO or a combination of two or more of these solvents.
- the temperature of the reaction may vary from about O 0 C to about the reflux temperature of the solvent.
- compounds of the formula 12 can be prepared from compounds of the formula 11 according to the method described in Scheme E for the preparation of compounds of the formula 9.
- This reaction can also be conducted using lithium chloride and a base, such as DBU (l,8-diazabicyclo[5.4.0]undec-7-ene) or triethylamine, in a solvent such as acetonitrile or THF.
- a base such as DBU (l,8-diazabicyclo[5.4.0]undec-7-ene) or triethylamine
- a solvent such as acetonitrile or THF.
- the intermediate ⁇ , ⁇ - unsaturated esters are then treated with aqueous hydrochloric acid with the optional use of a co-solvent such as dioxane to provide the desired compounds of the formula 2A-1.
- the temperature of this reaction may vary from about ambient temperature to about the reflux temperature of the solvent.
- Scheme G illustrates a method for preparing compounds of the formula 5B-1, 5B-2 and 5B-3.
- Ortho metalation of compounds of the formula 8, as described in Scheme E, and subsequent treatment with 3-oxopropionic acid esters of the formula shown in Scheme G above provides the corresponding compounds of formula 13.
- the reaction can be conducted in a solvent such as tetrahydrofuran at temperatures ranging from about -78°C to about ambient temperature, preferably from about -78 0 C to about -O 0 C.
- Refluxing compounds of the formula 13 in an aqueous acid such as 3N hydrochloric acid with the optional use of a co-solvent such as dioxane, generates the corresponding compounds of formula 5B-1.
- Compounds of the formula 5B-2 can be prepared by treating the corresponding compounds of the formula 5B-1 with triethylsilane in trifluoroacetic acid at a temperature from about room temperature to the reflux temperature of the solvent.
- Compounds of the formula 5B-3 can be prepared by treating compounds of the formula 5B-1 with an oxidizing agent such as Dess Martin periodinane, IBX or PCC at about ambient temperature in a solvent such as dichloromethane, dichloroethane, TEDF or DMSO, or a combination of two or more of these solvents.
- nitro group of compounds of the formula 15 can be reduced with iron powder and acetic acid, with or without the addition of a solvent such methanol or water, at temperatures from about room temperature to about the reflux temperature of the solvent mixture used. These conditions also result in ring closure to yield compounds of the formula 5B-4.
- Scheme I illustrates a method for preparing compounds of the formula 5B-5.
- Compounds of the formula 16 can be heated with liquid ammonia in a sealed reaction vessel at temperatures of about 40°C to about 100 0 C, in a solvent such as THF, to yield compounds of the formula 17.
- Scheme J illustrates an alternative method for preparing compounds of the formula 2B-2.
- Addition of a suitably mono-protected diol, where n is 3 and P is THP, benzyl, or TBS, to compounds of the formula 20 provides the corresponding compounds of formula 21.
- This reaction is typically conducted in the presence of a base such as potassium tert-butoxide, sodium tert-butoxide, sodium hydride, lithium diisopropylamide, lithium bis(trimethylsilyl)amide, potassium bis(trimethylsilyl)amide, or sodium bis(trimethylsilyl)amide, in a solvent such as THF, dioxane, or ethylene glycol dimethylether, preferably THF.
- a base such as potassium tert-butoxide, sodium tert-butoxide, sodium hydride, lithium diisopropylamide, lithium bis(trimethylsilyl)amide, potassium bis(trimethylsilyl)amide, or sodium bis(trimethylsilyl)amide
- the temperature of the reaction may vary from about -78 °C to about room temperature.
- Compounds of the formula 21 can be reacted with sodium azide in solvents such as DMF, NMP or DMSO, or a combination of two or more of these solvents, to provide compounds of the formula 22.
- the temperature of the reaction may vary from about room temperature to about the reflux temperature of the solvent, and is preferably about 7O 0 C.
- the azide of compounds of the formula 22 can be reduced to an amine using conventional reducing agents known to those skilled in the art, preferably using hexamethyldisilthiane [(Me 3 Si) 2 S] in a solvent such as methanol or ethanol.
- Compounds of the formula 24 can be hydrogenated using methods known to those skilled in the art, using, for example, palladium on activated carbon, palladium on barium sulfate, or Raney- nickel, in a solvent such as methanol, ethanol, THF, or a combination of two of the formerly mentioned solvents.
- the resulting amino esters can be cyclized to give the corresponding compounds of the formula 2B-2 by heating at a temperature from about 50°C to about the reflux temperature of the solvent, in a solvent such as ethanol, methanol or isopropanol, or a mixture of two or more of these solvents.
- a catalytic amount of acid for example, toluene sulphonic acid
- base for example, l,8-diazabicyclo[5.4.0]undec-7-ene
- pressure is not critical unless otherwise indicated. Pressures from about 0.5 atmospheres to about 5 atmospheres are generally acceptable, and ambient pressure, i.e., about 1 atmosphere, is preferred as a matter of convenience.
- pressure is not critical unless otherwise indicated. Pressures from about 0.5 atmospheres to about 5 atmospheres are generally acceptable, and ambient pressure, i.e., about 1 atmosphere, is preferred as a matter of convenience.
- the compounds of the formula 1 and their pharmaceutically acceptable salts can be administered to mammals via either the oral, parenteral (such as subcutaneous, intravenous, intramuscular, intrasternal and infusion techniques), rectal, buccal or intranasal routes.
- these compounds are most desirably administered in doses ranging from about 3 mg to about 600 mg per day, in single or divided doses (i.e., from 1 to 4 doses per day), although variations will necessarily occur depending upon the species, weight and condition of the patient being treated and the patient's individual response to said medicament, as well as on the type of pharmaceutical formulation chosen and the time period and interval at which such administration is carried out.
- a dosage level that is in the range of about 10 mg to about 100 mg per day is most desirably employed. In some instances, dosage levels below the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effects, provided that such higher dose levels are first divided into several small doses for administration throughout the day.
- novel compounds of the present invention may be administered alone or in combination with pharmaceutically acceptable carriers or diluents by any of the routes previously indicated, and such administration may be carried out in single or multiple doses. More particularly, the novel therapeutic agents of this invention can be administered in a wide variety of different dosage forms, i.e., they may be combined with various pharmaceutically acceptable inert carriers in the form of tablets, capsules, lozenges, troches, hard candies, suppositories, jellies, gels, pastes, ointments, aqueous suspensions, injectable solutions, elixirs, syrups, and the like. Such carriers include solid diluents or fillers, sterile aqueous media and various non-toxic organic solvents, etc.
- oral pharmaceutical compositions can be suitably sweetened and/or flavored.
- the weight ratio of the novel compounds of this invention to the pharmaceutically acceptable carrier will be in the range from about 1:6 to about 2:1, and preferably from about 1:4 to about 1:1.
- tablets containing various excipients such as microcrystalline cellulose, sodium citrate, calcium carbonate, dicalcium phosphate and glycine may be employed along with various disintegrants such as starch (and preferably corn, potato or tapioca starch), alginic acid and certain complex silicates, together with granulation binders like polyvinylpyrrolidone, sucrose, gelatin and acacia.
- lubricating agents such as magnesium stearate, sodium lauryl sulfate and talc are often very useful for tabletting purposes.
- Solid compositions of a similar type may also be employed as fillers in gelatin capsules; preferred materials in this connection also include lactose or milk sugar as well as high molecular weight polyethylene glycols.
- the active ingredient may be combined with various sweetening or flavoring agents, coloring matter or dyes, and, if so desired, emulsifying and/or suspending agents as well, together with such diluents as water, ethanol, propylene glycol, glycerin and various like combinations thereof.
- solutions of a compound of the present invention in either sesame or peanut oil or in aqueous propylene glycol may be employed.
- the aqueous solutions should be suitably buffered (preferably pH greater than 8) if necessary and the liquid diluent first rendered isotonic.
- These aqueous solutions are suitable for intravenous injection purposes.
- the oily solutions are suitable for intraarticular, intra-muscular and subcutaneous injection purposes. The preparation of all these solutions under sterile conditions is readily accomplished by standard pharmaceutical techniques well known to those skilled in the art.
- This invention relates to methods of treating anxiety, depression, schizophrenia and the other disorders referred to in the description of the methods of the present invention, wherein a novel compound of this invention and one or more of the other active agents referred to above (e.g., an NKl receptor antagonist, tricyclic antidepressant, 5HT ID receptor antagonist, or serotonin reuptake inhibitor) are administered together, as part of the same pharmaceutical composition, as well as to methods in which such active agents are administered separately as part of an appropriate dose regimen designed to obtain the benefits of the combination therapy.
- the appropriate dose regimen, the amount of each dose of an active agent administered, and the specific intervals between doses of each active agent will depend upon the subject being treated, the specific active agent being administered and the nature and severity of the specific disorder or condition being treated.
- novel compounds of this invention when used as a single active agent or in combination with another active agent, will be administered to an adult human in an amount from about 3 mg to about 300 mg per day, in single or divided doses, preferably from about 10 to about 100 mg per day.
- Such compounds may be administered on a regimen of up to 6 times per day, preferably 1 to 4 times per day, especially 2 times per day and most especially once daily. Variations may nevertheless occur depending upon the species of animal being treated and its individual response to said medicament, as well as on the type of pharmaceutical formulation chosen and the time period and interval at which such administration is carried out.
- dosage levels below the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effect, provided that such larger doses are first divided into several small doses for administration throughout the day.
- a proposed daily dose of a 5HT reuptake inhibitor, preferably sertraline, in the combination methods and compositions of this invention, for oral, parenteral or buccal administration to the average adult human for the treatment of the conditions referred to above, is from about 0.1 mg to about 2000 mg, preferably from about 1 mg to about 200 mg of the 5HT reuptake inhibitor per unit dose, which could be administered, for example, 1 to 4 times per day.
- a proposed daily dose of a 5HT1D receptor antagonist in the combination methods and compositions of this invention, for oral, parenteral, rectal or buccal administration to the average adult human for the treatment of the conditions referred to above, is from about 0.01 mg to about 2000 mg, preferably from about 0.1 mg to about 200 mg of the 5HT1D receptor antagonist per unit dose, which could be administered, for example, 1 to 4 times per day.
- the novel compounds of the invention are conveniently delivered in the form of a solution or suspension from a pump spray container that is squeezed or pumped by the patient or as an aerosol spray presentation from a pressurized container or a nebulizer, with the use of a suitable propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- a suitable propellant e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- the dosage unit may be determined by providing a valve to deliver a metered amount.
- the pressurized container or nebulizer may contain a solution or suspension of the active compound.
- Capsules and cartridges for use in an inhaler or insufflator may be formulated containing a powder mix of a compound of the invention and a suitable powder base such as lactose or starch.
- Formulations of the active compounds of this invention for treatment of the conditions referred to above in the average adult human are preferably arranged so that each metered dose or "puff' of aerosol contains 20 ⁇ g to 1000 ⁇ g of active compound.
- the overall daily dose with an aerosol will be within the range 100 ⁇ g to 10 mg.
- Administration may be several times daily, for example 2, 3, 4 or 8 times, giving for example, 1, 2 or 3 doses each time.
- the ability of the novel compounds of this invention to bind to the dopamine D 2 receptor can be determined using conventional radioligand receptor binding assays. All receptors can be heterologously expressed in cell lines and binding assays can be conducted in membrane preparations from the cell lines using procedures outlined below. IC 50 concentrations can be determined by nonlinear regression of concentration- dependent reduction in specific binding. The Cheng-Prussoff equation can be used to convert the IC 50 to Ki concentrations. See Example 21, below, for a description of the assay used to determine the binding of the compounds of this invention to the dopamine D 2 receptor, and the binding data obtained for the assayed compounds. Compounds of the present invention preferably exhibit Ki values of no more than 100 nM, more preferably no more than 50 nM, even more preferably no more than 25 nM, most preferably no more than 10 nM.
- the yellow mixture was diluted with saturated NaCl solution and extracted with ethyl acetate.
- the extracts were dried over anhydrous Na 2 SO 4 , filtered and concentrated to a yellow oil.
- the oil was purified by column chromatography (chloroform/ethyl acetate, 2:3), to afford 4-(3 -Methyl- isoquinolin-l-yl)-piperazine-l-carboxylic acid tert-butyl ester (0.305 g, 37 %), as a yellow oil.
- the concentrate was diluted with a mixture of 1 liter of ethyl acetate and 200ml THF, and filtered again through Celite. The filtrate was washed sequentially with 20% potassium bicarbonate (800ml), brine, and dried over sodium sulfate. The suspension was filtered, concentrated to a crystalline solid and washed with a 50:50 mixture of ethyl acetate and hexane (100ml). The solid obtained from drying at 50 0 C under vacuum was 7-(4-Hydroxy-butoxy)-3,4-dihydro-lH- [l,8]naphthyridin-2-one, 75.8g, 87% yield, MP 103-106 0 C.
- 7-Fluoro-4,4-dimethyl-3,4-dihydro-lH-[l,8]naphthyridin-2-one was produced as follows: HF-pyridine (100 mL) was cooled to -42 0 C in a 1000 mL HDPE bottle using an CH 3 CN dry ice bath. While stirring vigorously, 7-amino-4,4-dimethyl-3,4- dihydro-lH-[l,8]naphthyridin-2-one (24.6 g, 0.129 mol) was added portionwise to control the exotherm. After the addition, NaNO 2 (8.9 g, 0.1291 mol) was added portionwise. Significant exotherms were observed for both additions.
- the reaction mixture was then allowed to warm to 0 0 C and stir for 2 h.
- the reaction mixture was quenched into a 4 L high density polyethylene (HDPE) bottle full of ice.
- the aqueous slurry was then neutralized using 2 N KOH.
- the resulting aqueous solution was extracted 3 times with CH 2 Cl 2 .
- the organic layers were dried over Na 2 SO 4 , filtered and concentrated to dryness. Excess pyridine was azeotroped with heptane.
- the product was dried under vacuum (2 mm Hg) for 3 h.
- the orange red colored reaction mixture is stirred at RT for 1.5 h, cooled and quenched with saturated NH 4 Cl solution (150 mL) and water (2 L). The mixture is extracted with ethyl acetate (2 x 0.75 L) and the organic layer is washed with brine (300 mL), dried over anhydrous sodium sulfate, filtered through a small bed of silica gel eluting with 5% methanol in ethyl acetate (750 mL) and concentrated.
- 6-Fluoro-l-piperazin-l-yl- isoquinoline trifluoroacetate salt (WO 98/42712), (520 mg, 1.29 mmol) and 4-(7-Oxo- 7,8-dihydro-[l,8]naphthyridin-2-yloxy)-butyraldehyde (300 mg, 1.29 mmol) (Example 1) were dissolved in dichloroethane (10 mL). Triethylamine (392 mg, 3.87 mmol) was added and the mixture was stirred for 5 minutes. Sodium triacetoxyborohydride (383 mg, 1.81 mmol) was added and the mixture was stirred for 1.5 hours.
- Example 2 4-(7-oxo-7,8-dihydro-[l,8]naphthyridin-2-yloxy)-butyraldehyde (0.21g, 0.90 mmol) (Example 1) and the hydrochloride salt of 3 -Methyl- 1-piperazin-l-yl- isoquinoline (260mg, 0.99 mmol) (Example 2), were suspended in dichloroethane (20 mL). Triethylamine (0.25ml, 1.79 mmol) was added followed by sodium triacetoxyborohydride (0.266 g, 1.26 mmol). The yellow solution was stirred at room temperature for 1 hour, then quenched with water and extracted with dichloromethane.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Psychology (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Compounds of formula 1 (1) are disclosed, wherein G is as shown in formula (i): (i). A of formula 1 is -(CH2)nCO-, wherein n is 4 or 5, and one or two of the carbon atoms of A can be substituted as described in the specification. D, Z, Q, X, Y, K, J, R1, R4 through R7, and R9 through R11 of formulae 1 and (i) are defined in the specification. Also provided are descriptions of processes for preparing compounds of formula 1, intermediates used in making the same, and pharmaceutical compositions containing such compounds and their use in the treatment of central nervous system disorders and other disorders.
Description
ISOOUINOLIKE rUlNAPHTHYRIDIN-2-ONES AND RELATED COMPOUNDS FOR THE TREATMENT OF SCHIZOPHRENIA
BACKGROUND OF THE INVENTION This invention relates to [l,8]naphthyridin-2-ones and related compounds, methods of making such compounds, pharmaceutical compositions containing them, and their use for the treatment of schizophrenia and other central nervous system (CNS) disorders.
The [l,8]naphthyridin-2-ones and related compounds of this invention bind to dopamine D2 receptors. Some exhibit activity as partial agonists of D2 receptors, while others exhibit activity as antagonists of such receptors. Other heterocyclic derivatives that are useful for the treatment of schizophrenia are referred to in United States patent 5,350,747, which issued on September 27, 1994; in United States patent 6,127,357, which issued on October 3, 2000; in WO 93/04684, which published on March 18, 1993; and European patent application EP 402644A, which was published on
December 19, 1990. The foregoing patents and patent applications are incorporated herein by reference in their entireties.
SUMMARY OF THE INVENTION The present invention relates to compounds of formula 1, below:


A is -(CH2)nO- wherein n is 4 or 5, and one or two of the carbon atoms of - (CH2)nO- can be substituted, optionally and independently, with one or two substituents that are selected, independently, from fluoro and methyl, or with two substituents attached to the same carbon atom that form, together with the carbon to which they are attached, a spirocyclopropyl or spirocyclobutyl ring;
D is N, C, or CH, provided that when D is N, each carbon atom attached to D is attached through a single bond;
J and K are independently selected from N, CH, and C, provided that at least one of J and K is N;
Z and Q are independently selected from N and CH, provided that at least one of Z and Q is N;
R1 is hydrogen, -C(=O)CH3, or (C1-C3) alkyl;
-X=Y- is -CH2-CH2-, -CH=CH-, -CH2-NH-, -NH-CH2-, -N=CH-, -CH=N-, -0-CH2-, or -CH2-O-, wherein -X- — Y- can optionally be substituted, at any available bonding site, by one to four substituents R2, R2 , R3 and R3 ;
R2, R2 , R3 and R3 are independently selected from hydrogen, halo, cyano, oxo, hydroxy, -C(=O)CH3, (C1-C4) alkyl, and (C1-C4) alkoxy, wherein the alkyl moieties of the (C1-C4) alkyl, (C1-C4) alkoxy, and -C(=O)CH3 groups can be optionally substituted with from one to three fluoro atoms and can also be optionally substituted with an amino or hydroxy substituent;
R4 and R5 are independently selected from hydrogen, halo, cyano, hydroxy, -C(=O)CH3, (C1-C4) alkyl, and (C1-C4) alkoxy, wherein the alkyl moieties of the (C1- C4) alkyl, (C1-C4) alkoxy, and -C(=O)CH3 groups can be optionally substituted with from one to three fluoro atoms and can also be optionally substituted with an amino or hydroxy substituent;
R and R are selected, independently, from hydrogen and methyl;
R8, R9, R10 and Rπare independently selected from hydrogen, halo, -C(=O)CH3, (C1-C4) alkyl, and (C1-C4) alkoxy, wherein the alkyl moieties of the (C1-C4) alkyl, (C1- C4) alkoxy, and -C(=O)CH3 groups can be optionally substituted with from one to three fluoro atoms and can also be optionally substituted with an amino or hydroxy substituent; ring AA is a saturated or unsaturated 5-, 6-, or 7-membered carbocyclic ring wherein one, two or three of the carbon atoms of ring AA that are not shared with the other fused ring of formula (i) can be replaced, optionally and independently, by a nitrogen, oxygen or sulfur atom; and the pharmaceutically acceptable salts of such compounds.
This invention also relates to a pharmaceutical composition comprising a therapeutically effective amount of a compound of the formula 1, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
The compounds of formula 1 have useful pharmaceutical and medicinal properties.
The invention also relates to a pharmaceutical composition for treating a disorder or condition selected from single episodic or recurrent major depressive disorders, dysthymic disorders, depressive neurosis and neurotic depression, melancholic depression; atypical depression; bipolar disorder; cyclothymic disorder; conduct disorder; disruptive behavior disorder; attention deficit hyperactivity disorder; behavioral disturbances associated with mental retardation, autistic disorder, and conduct disorder; anxiety disorders; borderline personality disorder; schizophrenia and other psychotic disorders; delirium, dementia, and amnestic and other cognitive or neurodegenerative disorders; movement disorders, dyskinesias; extra-pyramidal movement disorders; chemical dependencies and additions; behavioral addictions; and ocular disorders, comprising: an amount of a compound of formula 1, or a pharmaceutically acceptable salt thereof that is effective in treating the disorder or condition, and a pharmaceutically acceptable carrier.
The invention further relates to a pharmaceutical composition for treating a disorder or condition selected from those listed above, comprising: (a) a compound of formula 1, or a pharmaceutically acceptable salt thereof; and (b) an antidepressant or an anti-anxiety agent; and (c) a pharmaceutically acceptable carrier; wherein active agents
(a) and (b) are not the same and are present in amounts that render the combination of them effective in treating said disorder or condition.
This invention also relates to a method of treating a disorder or condition selected from those listed above, comprising administering to a mammal in need of such treatment an amount of a compound according to formula 1, or a pharmaceutically acceptable salt thereof, that is effective in treating the disorder or condition.
The invention also relates to a method of treating a disorder or condition selected from those listed above, comprising administering to a mammal in need of such treatment (a) a compound of formula 1, or a pharmaceutically acceptable salt thereof; and (b) an antidepressant or an anti-anxiety agent; wherein the active agents (a) and (b) are not the same and are present in amounts that render the combination of them effective in treating the disorder or condition.
The term "alkyl", as used herein, unless otherwise indicated, includes saturated monovalent hydrocarbon radicals having straight, branched or cyclic moieties or combinations thereof. Examples of "alkyl" groups include, but are not limited to, methyl, ethyl, propyl, isopropyl, butyl, iso- sec- and tert-butyl, pentyl, hexyl, heptyl, 3- ethylbutyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, norbornyl, and the like.
The term "alkoxy", as used herein, unless otherwise indicated, means "alkyl-O- ", wherein "alkyl" is as defined above. Examples of "alkoxy" groups include, but are not limited to, methoxy, ethoxy, propoxy, butoxy and pentoxy.
The term "aryl", as used herein, unless otherwise indicated, includes an aromatic ring system with no heteroatoms as ring members, which can be either unsubstituted or substituted with one, two or three substituents selected from the group consisting of halo, (C1-C4)^yI optionally substituted with from one to three fluorine atoms and (Q-GOalkoxy optionally substituted with from one to three fluorine atoms.
The term "aryloxy", as used herein, unless otherwise indicated, means "aryl- O-", wherein "aryl" is as defined above.
The term "one or more substituents", as used herein, refers to a number of substituents that equals from one to the maximum number of substituents possible based on the number of available bonding sites.
The terms "halo" and "halogen", as used herein, unless otherwise indicated, include, fluoro, chloro, bromo and iodo.
The term "therapeutically effective amount," as used herein, refers to a quantity of active agent sufficient to treat one or more of the disorders or conditions referred to above, when one or more doses of a pharmaceutical composition of the invention are administered to a subject with one or more of the disorders or conditions. In determining what constitutes a therapeutically effective amount of an active agent in a composition or delivered in a method of the present invention, a number of factors will generally be considered, including the experience of the medical practitioner or veterinarian administering the composition, published clinical studies, the subject's age, sex, weight and general condition, as well as the type and extent of the disorder or condition being treated, and the use of other medications, if any, by the subject. Determination of a proper dose for a particular situation, and preparation of a pharmaceutical composition containing a suitable dose of active agent for that situation, is within the skill of the medical or veterinary arts.
The term "treating", as used herein, refers to reversing, alleviating, inhibiting the progress of, or preventing the disorder or condition to which such term applies, or preventing one or more symptoms of such condition or disorder.
The term "treatment", as used herein, refers to the act of treating, as "treating" is defined immediately above.
The compounds of formula 1, and the pharmaceutically acceptable salts of these compounds are referred to herein, collectively, as the "novel compounds of this invention" and the "active compounds of this invention".
DETAILED DESCRIPTION OF THE INVENTION Examples of preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein AA is a 6-membered unsaturated carbocyclic ring.
Other preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein J is N.
Other preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein A is -(CH2)4O-. Other preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein Q is N.
Other preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein -X — Y- is -CH2-NH-.
Other preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein -X—Y- is -CH2-CH2- or -CH=CH-.
Other preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein R1 is hydrogen.
Other preferred embodiments of this invention are compounds of formula 1 and their pharmaceutically acceptable salts, wherein each of R10 and R11 is, independently, hydrogen or fluoro .
Other preferred embodiments of this invention are compounds of formula 1 and their pharmaceutically acceptable salts, wherein D is N, Q is N, Z is CH, -X — Y- is -CH2-CH2- or -CH=CH-. and R1, R4, and R5 are hydrogen.
Other preferred embodiments of this invention are compounds of formula 1 and their pharmaceutically acceptable salts, wherein Q and Z are both N.
Other preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein R4 and R5 are hydrogen.
Other preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein D is N. Other preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein Q is N and Z is C or CH.
Other preferred embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein Q is N and Z is N.
Other embodiments of this invention are compounds of the formula 1, and their pharmaceutically acceptable salts, wherein Q is C or CH, and Z is N.
Other embodiments of this invention relates to compounds of the formula 1, and their pharmaceutically acceptable salts, wherein -X=Y- is -0-CH2-.
Other embodiments of this invention relates to compounds of the formula 1, and their pharmaceutically acceptable salts, wherein -X1^771Y- is -CH2-NH-. In another preferred embodiment, the invention relates to compounds of formula
1, and their pharmaceutically acceptable salts, wherein G has a structure shown in formula (ii), below:

(ϋ) wherein K' and J' are independently selected from N, CH, and C, provided that at least one of K' and J' is N;
R8' is H, (C1-C4) alkyl, or (C1-C4) alkoxy;
R10' is H, (C1-C4) alkyl, (C1-C4) alkoxy, or halo; and
U, V, and W are independently selected from N, CH, and C, provided that no more than two of U, V, and W are N.
R8 is preferably attached to K'.
R8 is preferably H or methyl.
R10 is preferably attached to U or V.
R10 is preferably H, methyl, or fluoro.
G is preferably an isoquinoline of formula (ii), more preferably an isoquinoline selected from the group consisting of 1-isoquinolinyl, 7-fluoro-l-isoquinolinyl, 6- fluoro-1-isoquinolinyl, 3-methyl-l-isoquinolinyl, and 7-fluoro-3 -methyl- 1- isoquinolinyl.
Specific preferred embodiments of this invention are the following compounds and their pharmaceutically acceptable salts:
7-[4-(4-Isoquinolin- 1 -yl-piperazin- 1 -yl)-butoxy] -3 ,4-dihydro- IH- [l,8]naphthyridin-2-one;
7-[4-(4-Isoquinolin-4-yl-piperazin-l-yl)-butoxy]-3,4-dihydro-lH- [ 1 ,8]naphthyridin-2-one;
7-[4-(4-Isoquinolin-l-yl-piperazin-l-yl)-butoxy]-lH-[l,8]naphthyridin-2-one;
7- { 4- [4-(6-Fluoro-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } -3 ,4-dihydro- IH- [1 ,8]naphthyridin-2-one;
7- { 4-[4-(6-Fluoro-isoquinolin- l-yl)-piperazin- 1 -yl] -butoxy } - IH- [l,8]naphthyridin-2-one;
7- { 4-[4-(7-Fluoro-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } -3 ,4-dihydro- IH- [1 ,8]naphthyridin-2-one;
7- { 4- [4-(7-Fluoro-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } - IH- [ 1 ,8]naphthyridin-2-one; 7- { 4- [4-(7-Fluoro-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } -4,4-dimethyl-3 ,4- dihydro-lH-[ 1 ,8]naphthyridin-2-one;
2- { 4- [4-(7-Fluoro-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } -8H-pyrido [2,3- d]pyrimidin-7-one;
2- { 4- [4-(7-Fluoro-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } -4-methyl-8H- pyrido[2,3-d]pyrimidin-7-one;
7-[4-(4-[2,7]Naphthyridin-l-yl-piperazin-l-yl)-butoxy]-lH-[l,8]naphthyridin-2- one;
7-[4-(4-[2,7]Naphthyridin- 1 -yl-piperazin- 1 -yl)-butoxy]-3 ,4-dihydro- IH- [l,8]naphthyridin-2-one; 7-{4-[4-(3-Methyl-isoquinolin-l-yl)-piperazin-l-yl]-butoxy}-3,4-dihydro-lH-
[ 1 ,8]naphthyridin-2-one;
7- { 4- [4-(3 -Methyl-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } - IH- [ 1 , 8]naphthyridin-2-one.
Specific embodiments of this invention further relate to the following compounds and their pharmaceutically acceptable salts:
7-[4-(4-[l,6]Naphthyridin-8-yl-piperazin-l-yl)-butoxy]-3,4-dihydro-lH- [1 ,8]naphthyridin-2-one;
7-[4-(4-[l,6]Naphthyridin-8-yl-piperazin-l-yl)-butoxy]-lH-[l,8]naphthyridin-2- one; 7- { 4- [4-(7-Huoro-3-methyl-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } -3 ,4- dihydro- IH- [ 1 , 8]naphthyridin-2-one;
7-{4-[4-(7-Fluoro-3-methyl-isoquinolin-l-yl)-piperazin-l-yl]-butoxy}-lH- [1 ,8]naphthyridin-2-one;
7- { 4-[4-(6-Fluoro-3-methyl-isoquinolin- 1 -yl)-piperazin- 1 -yl]-butoxy } -3 ,4- dihydro-lH-[l ,8]naρhthyridin-2-one;
7- { 4-[4-(6-Fluoro-3-methyl-isoquinolin- 1 -yl)-piperazin- 1 -yl]-butoxy } - IH- [l,8]naphthyridin-2-one;
7-[4-(4-[l,7]Naphthyridin-8-yl-piperazin-l-yl)-butoxy]-3,4-dihydro-lH- [ 1 , 8]naphthyridin-2-one
7-[4-(4-[l,7]Naphthyridin-8-yl-piperazin-l-yl)-butoxy]-lH-[l,8]naphthyridin-2- one; 7-[4-(4-Thieno[2,3-c]pyridin-7-yl-piperazin-l-yl)-butoxy]-3,4-dihydro-lH-
[l,8]naphthyridin-2-one; and
7-[4-(4-Thieno[2,3-c]pyridin-7-yl-piperazin-l-yl)-butoxy]-lH- [ 1 ,8]naphthyiidin-2-one.
Compounds of the formula 1 may contain chiral centers and therefore may exist in different enantiomeric and diastereomeric forms. This invention relates to all optical isomers and all stereoisomers of compounds of the formula 1, both as racemic mixtures and as individual enantiomers and diastereoisomers of such compounds, and mixtures thereof, and to all pharmaceutical compositions and methods of treatment defined above that contain or employ them, respectively. Individual isomers can be obtained by known methods, such as optical resolution, fractional crystallization, optically selective reaction, or chromatographic separation in the preparation of the final product or its intermediate. Individual enantiomers of the compounds of formula 1 may have advantages, as compared with the racemic mixtures of these compounds, in the treatment of various disorders or conditions. In so far as the compounds of formula 1 are basic compounds, they are all capable of forming a wide variety of different salts with various inorganic and organic acids. Although such salts must be pharmaceutically acceptable for administration to animals, it is often desirable in practice to initially isolate the base compound from the reaction mixture as a pharmaceutically unacceptable salt and then simply convert to the free base compound by treatment with an alkaline reagent and thereafter convert the free base to a pharmaceutically acceptable acid addition salt. The acid addition salts of the base compounds of this invention are readily prepared by treating the base compound with a substantially equivalent amount of the chosen mineral or organic acid in an aqueous solvent or in a suitable organic solvent, such as methanol or ethanol. Upon careful evaporation of the solvent, the desired solid salt is readily obtained. The acids which are used to prepare the pharmaceutically acceptable acid addition salts of the aforementioned base compounds of this invention are those which form non-toxic acid addition salts, Le., salts containing pharmaceutically acceptable anions, such as the
hydrochloride, hydrobromide, hydroiodide, nitrate, sulfate or bisulfate, phosphate or acid phosphate, acetate, lactate, citrate or acid citrate, tartrate or bi-tartrate, succinate, maleate, fumarate, gluconate, saccharate, benzoate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate and pamoate (i.e., l,l'-methylene-bis-(2-hydroxy- 3-naphthoate)) salts.
The present invention also includes isotopically labeled compounds, which are identical to those of formula 1, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes that can be incorporated into compounds of the present invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, sulfur, fluorine and chlorine, such as 2H, 3H, 13C, 11C, 14C, 15N, 180, 170, 31P, 32P, 35S, 18F, and 36Cl, respectively. Compounds of the present invention, prodrugs thereof, and pharmaceutically acceptable salts of said compounds or of said prodrugs which contain the aforementioned isotopes and/or other isotopes of other atoms are within the scope of this invention. Certain isotopically labeled compounds of the present invention, for example those into which radioactive isotopes such as 3H and 14C are incorporated, are useful in drug and/or substrate tissue distribution assays. Tritiated, i.e., 3H, and carbon-14, Le., 14C, isotopes are particularly preferred for their ease of preparation and detectability. Further, substitution with heavier isotopes such as deuterium, i.e., 2H, can afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements and, hence, may be preferred in some circumstances. Isotopically labeled compounds of formula 1 and prodrugs thereof can generally be prepared by carrying out the procedures disclosed in the Schemes and/or in the Examples below, by substituting a readily available isotopically labeled reagent for a non-isotopically labeled reagent.
The pharmaceutical compositions and methods of the present invention are suitable for use in treatment of a disease or disorder selected from single episodic or recurrent major depressive disorders, dysthymic disorders, depressive neurosis and neurotic depression, melancholic depression including anorexia, weight loss, insomnia, early morning waking or psychomotor retardation; atypical depression (or reactive depression) including increased appetite, hypersomnia, psychomotor agitation or irritability, seasonal affective disorder and pediatric depression; bipolar disorders or
manic depression, for example, bipolar I disorder, bipolar II disorder and cyclothymic disorder; conduct disorder; disruptive behavior disorder; attention deficit hyperactivity disorder (ADHD); behavioral disturbances associated with mental retardation, autistic disorder, and conduct disorder; anxiety disorders such as panic disorder with or without agoraphobia, agoraphobia without history of panic disorder, specific phobias, for example, specific animal phobias, social anxiety, social phobia, obsessive-compulsive disorder, stress disorders including post-traumatic stress disorder and acute stress disorder, and generalized anxiety disorders; borderline personality disorder; schizophrenia and other psychotic disorders, for example, schizophreniform disorders, schizoaffective disorders, delusional disorders brief psychotic disorders, shared psychotic disorders, psychotic disorders with delusions or hallucinations, psychotic episodes of anxiety, anxiety associated with psychosis, psychotic mood disorders such as severe major depressive disorder; mood disorders associated with psychotic disorders such as acute mania and depression associated with bipolar disorder; mood disorders associated with schizophrenia; delirium, dementia, and amnestic and other cognitive or neurodegenerative disorders, such as Parkinson's disease (PD), Huntington's disease (HD), Alzheimer's disease, senile dementia, dementia of the Alzheimer's type, memory disorders, loss of executive function, vascular dementia, and other dementias, for example, due to HTV disease, head trauma, Parkinson's disease, Huntington's disease, Pick's disease, Creutzfeldt- Jakob disease, or due to multiple etiologies; movement disorders such as akinesias, dyskinesias, including familial paroxysmal dyskinesias, spasticities, Tourette's syndrome, Scott syndrome, PALSYS and akinetic-rigid syndrome; extra-pyramidal movement disorders such as medication-induced movement disorders, for example, neuroleptic-induced Parkinsonism, neuroleptic malignant syndrome, neuroleptic-induced acute dystonia, neuroleptic-induced acute akathisia, neuroleptic-induced tardive dyskinesia and medication-induced postural tremor; chemical dependencies and addictions (e.g., dependencies on, or addictions to, alcohol, heroin, cocaine, benzodiazepines, nicotine, or phenobarbitol) and behavioral addictions such as an addiction to gambling; and ocular disorders such as glaucoma and ischemic retinopathy in a mammal in need of such treatment, including a human, comprising an amount of a compound of the formula 1, or a pharmaceutically acceptable salt thereof, that is effective in treating such disorder or condition, and a pharmaceutically acceptable carrier.
In a specific embodiment of the methods or pharmaceutical compositions of the present invention, the disorder or condition treated is selected from major depression, single episode depression, recurrent depression, child abuse induced depression, postpartum depression, dysthymia, cyclothymia and bipolar disorder. In another, more specific, embodiment of the methods or pharmaceutical compositions of the present invention, the disorder or condition treated is selected from schizophrenia, schizoaffective disorder, delusional disorder, substance-induced psychotic disorder, brief psychotic disorder, shared psychotic disorder, psychotic disorder due to a general medical condition, and schizophreniform disorder. In another more specific embodiment of the methods or pharmaceutical compositions of the present invention, the disorder or condition that is being treated is selected from autism, pervasive development disorder, speech impediments such as stuttering, and attention deficit hyperactivity disorder.
In another more specific embodiment of the methods or pharmaceutical compositions of this invention, the disorder or condition that is being treated is selected from generalized anxiety disorder, panic disorder, obsessive-compulsive disorder, posttraumatic stress disorder, and phobias, including social phobia, agoraphobia, and specific phobias.
In another more specific embodiment of the methods or pharmaceutical compositions of this invention, the disorder or condition that is being treated is selected from movement disorders such as akinesias, dyskinesias, including familial paroxysmal dyskinesias, spasticities, Tourette's syndrome, Scott syndrome, PALSYS and akinetic- rigid syndrome; and extra-pyramidal movement disorders such as medication-induced movement disorders, for example, neuroleptic-induced Parkinsonism, neuroleptic malignant syndrome, neuroleptic-induced acute dystonia, neuroleptic-induced acute akathisia, neuroleptic-induced tardive dyskinesia and medication-induced postural tremor.
In another more specific embodiment of the methods or pharmaceutical compositions of this invention, the disorder or condition that is being treated is selected from delirium, dementia, and amnestic and other cognitive or neurodegenerative disorders, such as Parkinson's disease (PD), Huntington's disease (HD), Alzheimer's disease, senile dementia, dementia of the Alzheimer's type, memory disorder, vascular dementia, and other dementias, for example, due to HIV disease, head trauma,
Parkinson's disease, Huntington's disease, Pick's disease, Creutzfeldt- Jakob disease, or due to multiple etiologies.
In another more specific embodiment of the methods of the present invention, the compound of formula 1 is administered to a human for the treatment of any two or more comorbid disorders or conditions selected from those disorders and conditions referred to in any of the above methods.
In embodiments of the pharmaceutical compositions of the present invention, comprising (a) a compound of formula 1, or a pharmaceutically acceptable salt thereof, and (b) an antidepressant or an anti-anxiety agent, as described above, or in methods of the present invention comprising administering active agents (a) and (b), examples of classes antidepressants suitable for use in the invention include norepinephrine reuptake inhibitors, selective serotonin reuptake inhibitors (SSRIs), NK-I receptor antagonists, monoamine oxidase inhibitors (MAOIs), reversible inhibitors of monoamine oxidase (RIMAs), serotonin and noradrenaline reuptake inhibitors (SNRIs), corticotropin releasing factor (CRF) antagonists, α-adrenoreceptor antagonists, and atypical antidepressants. Suitable norepinephrine reuptake inhibitors include tertiary amine tricyclics and secondary amine tricyclics. Suitable tertiary amine tricyclics and secondary amine tricyclics include amitriptyline, clomipramine, doxepin, imipramine, trimipramine, dothiepin, butripyline, iprindole, lofepramine, nortriptyline, protriptyline, amoxapine, desipramine and maprotiline. Suitable selective serotonin reuptake inhibitors include fluoxetine, fluvoxamine, paroxetine and sertraline. Examples of monoamine oxidase inhibitors include isocarboxazid, phenelzine, and tranylcyclopramine. Suitable reversible inhibitors of monoamine oxidase include moclobemide. Suitable serotonin and noradrenaline reuptake inhibitors of use in the present invention include venlafaxine. Suitable CRF antagonists include those compounds described in International Patent Application Nos. WO 94/13643, WO 94/13644, WO 94/13661, WO 94/13676 and WO 94/13677. Suitable atypical antidepressants include bupropion, lithium, nefazodone, trazodone and viloxazine. Suitable NK-I receptor antagonists include those referred to in World Patent Publication WO 01/77100.
Suitable classes of anti-anxiety agents that can be used in combination with the compounds of formula 1 in the pharmaceutical compositions and methods of this invention include benzodiazepines and serotonin IA (5-HTΪA) agonists or antagonists,
especially 5-HT1A partial agonists, and corticotropin releasing factor (CRF) antagonists. Suitable benzodiazepines include alprazolam, chlordiazepoxide, clonazepam, chlorazepate, diazepam, halazepam, lorazepam, oxazepam, and prazepam. Suitable 5- HT1A receptor agonists or antagonists include buspirone, flesinoxan, gepirone and ipsapirone.
The active compounds of this invention may be prepared as described below. Unless otherwise indicated, in the reaction schemes and discussion that follow, A, Z, D, Q, ring AA, G, X, Y, R1 through R11, formula 1, the dotted line connecting X and Y, and groups of formulas (i) and (ii) are defined as above, m is 3 or 4 in each of the reaction schemes, below.
Scheme A

Scheme A illustrates a method for preparing compounds of the formula 1, optionally substituted as indicated in the definition of formula 1 above (also referred to as compounds of the formula IA). This method involves oxidation of a compound of the formula 2 with Dess-Martin Periodinane or another suitable oxidizing agent such as EBX (o-iodoxybenzoic acid), oxalyl chloride in dimethyl sulfoxide (DMSO) (Swern oxidation) or PCC (pyridinium chlorochromate) to form the corresponding aldehyde of formula 3. This reaction may be carried out in dichloromethane (CH2Cl2), tetrahydrofuran (THF), DMSO, or a combination of two or more of these solvents.
Reductive amination of a G-substituted piperidine or piperizine, as shown in Scheme A, using methods well known to those of skill in the art, with a compound of formula 3 yields the corresponding compound of formula IA. The reductive amination can be performed, for example, utilizing catalytic hydrogenation methods or using a hydride reducing agent such as sodium triacetoxyborohydride (NaBH(OAc)3) or sodium cyanoborohydride. The reaction solvent can be 1,2-dichloroethane, tetrahydrofuran,
acetonitrile, dimethylformamide or a combination of two or more of these solvents, with the optional addition of 1-10 equivalents of acetic acid. When the piperazine or piperidine hydrochloride or hydrobromide salt is used, a base such as triethylamine is typically added.
Scheme B

L = OMs, OTs, Br, Cl

Alternatively, compounds of the formula IA can be prepared according to Scheme B. The hydroxy group of the compound of formula 2 is converted into a leaving group (L) using conventional methods to provide the corresponding compound of formula 4 wherein L is mesylate (OMs), tosylate (OTs) or a halogen such as bromide, iodide or chloride. The resulting compound of formula 4 is then reacted with a G-substituted piperazine or piperidine, as depicted in Scheme B, to yield the desired compound of formula IA. This reaction is preferably run in the presence of a base such as potassium carbonate, sodium carbonate, cesium carbonate, triethylamine or diisopropylethylamine. The solvent used may be acetonitrile, water, tetrahydrofuran, dioxane, acetone, methyl isobutyl ketone, benzene or toluene, or a combination of two or more of these solvents. Inorganic salts such as sodium or potassium iodide may be employed as catalysts in the reaction. The temperature of the reaction may vary from about ambient temperature to about the reflux temperature of the solvent used. The reaction may also be conducted under microwave irradiation.
Scheme C

deprotection

Scheme C illustrates a method for preparing compounds of the formula 2A (wherein X is double bonded to Y) and 2B (wherein X is single bonded to Y). Addition of 2 to 20 equivalents of a diol of the formula HOCH2(CH2)mOH, where m is 3 or 4, or one to four equivalents of a suitably mono-protected diol of the formula POCH2(CH2)mOH, wherein P is tetrahydropyranyl (THP), benzyl (Bn), p- methoxybenzyl, tert-butyldimethysilyl (TBS), or tert-butyldiphenylsilyl (TBDPS), to a compound of the formula 5, wherein R is chloro, fluoro, bromo, S(O)CH3 or SO2CH3, provides the corresponding compound of formula 6 (or 2A when the unprotected diol reactant is used). R12 is most preferably fluoro. This reaction is conducted in the presence of a base such as potassium tert-butoxide, sodium tert-butoxide, sodium hydride, potassium hydride, lithium diisopropylamide, lithium bis(trimethylsilyl)amide, potassium bis(trimethylsilyl)amide, or sodium bis(trimethylsilyl)amide. Suitable solvents for this reaction include THF, dioxane, ethylene glycol dimethylether, DMF, N-methylpyrrolidinone (NMP), or DMSO, or a combination of two or more of these solvents. The temperature of the reaction may vary from about ambient temperature to about the reflux temperature of the solvent used.
The reaction of the compound of formula 5 with the mono-protected diol of the formula POCH2(CH2)mOH to yield the compound of formula 6, as described above, is preferably conducted in the presence of a catalytic amount of a phase transfer catalyst, such as tetrabutyl ammonium chloride or bromide. The use of a phase transfer catalyst accelerates the rate of the coupling, and allows one to carry out the reaction at a lower
temperature than would be possible without the catalyst. Use of the phase transfer catalyst also significantly reduces the formation of dimeric by-products.
Compounds of the formula 6 where, P is tetrahydropyranyl (THP), can be deprotected using conventional methods such as treatment with PPTS (pyridinium p- tolunesulfonate) orp-toluenesulfonic acid in ethanol to give the corresponding compounds of formula 2A. Compounds of the formula 6 wherein P is tert- butyldimethysilyl or tert-butyldiphenylsilyl can be deprotected using conventional methods such as treatment with tetrabutylammonium fluoride in tetrahydrofuran to yield compounds of the formula 2A. Compounds of the formula 2A or compounds of the formula 6 wherein P is H or benzyl can be reduced using catalytic hydrogenation methods to provide the corresponding compounds of formula 2B. For example, the hydrogenation can be conducted using 5 to 20 % palladium on activated carbon in a solvent such as methanol, ethanol, tetrahydrofuran, acetic acid, dimethylformamide, or a combination of two or more of these solvents for a period of about 5 hours to about 48 hours, preferably for about 24 hours, under a hydrogen pressure from about 1 to about 5 atmosphere, preferably about 1 atmosphere.
Scheme D
R1

R12 = Cl, F, Br, S(O)Me, SO2Me

IA. Addition of a compound of the formula 5 to a compound of the formula 7 provides the corresponding compound of formula IA-I. Compounds of the formula (7) obtained from arylpiperazines and piperidines prepared per the general procedure of Acta Physica et Chemica, 28(3-4), 225-44; 1982, can be reacted in the presence of a base such as potassium tert-butoxide, sodium te?t-butoxide, sodium hydride, potassium hydride, lithium diisopropylamide, lithium bis(trimethylsilyl)amide, potassium bis(trimethylsilyl)amide, or sodium bis(trimethylsilyl)amide. Suitable solvents for this
reaction include THF, dioxane, ethylene glycol dimethylether, DMF, NMP, and DMSO, or a combination of two or more of these solvents. The reaction temperature can range from about -78 °C to about ambient temperature, and is preferably from about -20 ° to about 0°C. Catalytic hydrogenation of compounds of the formula IA-I (when G is compatible with the hydrogenation conditions) provides compounds of formula 1A-2. For example, the hydrogenation can be conducted using 5 to 20% palladium on activated carbon in a solvent such as methanol, ethanol, THF, acetic acid, or DMF, or a combination of two or more of these solvents, for a period of about 5 hours to about 48 hours, preferably for about 12 to 24 hours.
Scheme E

Scheme E illustrates an alternative method for preparing compounds of the formula 5A-1. (/. Org. Chem. 1990, 55, 4744). (Compounds of the formula 5A-1 are compounds of the formula 5 wherein X is CR3, Y is CR2 and there is a double bond between X and Y). In Scheme E, R3 is (C1-C4) alkyl. Ortho metalation of compounds of the formula 8 and subsequent treatment with electrophiles having the formula shown in Scheme E results in compounds of the formula 9. Condensation of compounds of the formula 9 with the enolates of the alkyl esters having the formula shown in Scheme E provides the corresponding compounds of formula 10. Refluxing compounds of the formula 10 in aqueous acid such as 3N hydrochloric acid, with the optional use of a co- solvent such as dioxane, generates the corresponding compounds of formula 5A-1.
Scheme F

Scheme F illustrates another method for preparing compounds of the formula 2A-1. In Scheme F, R3 is (C1-C4) alkyl. Addition of a suitably mono-protected diol, where n is 3 or 4 and P is tetrahydropyranyl (THP), benzyl, or te/t-butyldimethysilyl, to compounds of the formula 9 provides compounds of the formula 11. The reaction is conducted in the presence of a base such as potassium tert-butoxide, sodium tert- butoxide, sodium hydride, potassium hydride, lithium diisopropylamide, lithium bis(trimethylsilyl)amide, potassium bis(trimethylsilyl)amide, or sodium bis(trimethylsilyl)amide. The solvents used may be THF, dioxane, ethylene glycol dimethylether, DMF, NMP, or DMSO or a combination of two or more of these solvents. The temperature of the reaction may vary from about O0C to about the reflux temperature of the solvent.
Alternatively, compounds of the formula 12 can be prepared from compounds of the formula 11 according to the method described in Scheme E for the preparation of compounds of the formula 9.
Condensation of compounds of the formula 12 with the enolates of the esters having the formula shown in Scheme F provides α-hydroxy ester intermediates, which are treated with an aqueous acid such as 3N hydrochloric acid, with the optional use of a co-solvent such as dioxane, at temperatures varying from about ambient temperature
to about the reflux temperature of the solvent, to generate compounds of the formula 2A-1.
Alternatively, Horner-Wadsworth-Emmons reaction of compounds of the formula 12 with ketophosphonates having the formula shown in Scheme F, in the presence of a base such as sodium hydride, sodium ethoxide, or butyl lithium, in a solvent such as TBDF, DMSO, dioxane, ethylene glycol dimethylether, ethanol, or benzene, or a combination of two or more of these solvents, to give the corresponding intermediate α,β-unsaturated esters. This reaction can also be conducted using lithium chloride and a base, such as DBU (l,8-diazabicyclo[5.4.0]undec-7-ene) or triethylamine, in a solvent such as acetonitrile or THF. The intermediate α,β- unsaturated esters are then treated with aqueous hydrochloric acid with the optional use of a co-solvent such as dioxane to provide the desired compounds of the formula 2A-1. The temperature of this reaction may vary from about ambient temperature to about the reflux temperature of the solvent. Scheme G

Scheme G illustrates a method for preparing compounds of the formula 5B-1, 5B-2 and 5B-3. Ortho metalation of compounds of the formula 8, as described in Scheme E, and subsequent treatment with 3-oxopropionic acid esters of the formula shown in Scheme G above provides the corresponding compounds of formula 13. The reaction can be conducted in a solvent such as tetrahydrofuran at temperatures ranging from about -78°C to about ambient temperature, preferably from about -780C to about -O0C. Refluxing compounds of the formula 13 in an aqueous acid such as 3N hydrochloric acid, with the optional use of a co-solvent such as dioxane, generates the corresponding compounds of formula 5B-1. Compounds of the formula 5B-2 can be
prepared by treating the corresponding compounds of the formula 5B-1 with triethylsilane in trifluoroacetic acid at a temperature from about room temperature to the reflux temperature of the solvent. Compounds of the formula 5B-3 can be prepared by treating compounds of the formula 5B-1 with an oxidizing agent such as Dess Martin periodinane, IBX or PCC at about ambient temperature in a solvent such as dichloromethane, dichloroethane, TEDF or DMSO, or a combination of two or more of these solvents.
Scheme H

14 L = Br1 I1 CI1 OMs1 OTs 15
X = O, S1 NH 5B-4 Scheme H illustrates a method for preparing compounds of the formula 5B-4
(see PCT Patent Application WO 02/056882). Alkylation of compounds of the formula 14 with an ester of the formula shown in Scheme I (L = Br, I, Cl, OMs, OTs) yields the corresponding compounds of formula 15. This reaction is typically run in the presence of a base such as potassium carbonate or sodium hydride, in a solvent such as acetonitrile, TBOF, dioxane, acetone, methyl isobutyl ketone, benzene, toluene or DMF, or a combination of two or more of these solvents. The temperature of the reaction may vary from about ambient temperature to about the reflux temperature of the solvent. The nitro group of compounds of the formula 15 can be reduced with iron powder and acetic acid, with or without the addition of a solvent such methanol or water, at temperatures from about room temperature to about the reflux temperature of the solvent mixture used. These conditions also result in ring closure to yield compounds of the formula 5B-4.
Scheme I

Ph3PCHCO2Et

Scheme I illustrates a method for preparing compounds of the formula 5B-5. Compounds of the formula 16 can be heated with liquid ammonia in a sealed reaction vessel at temperatures of about 40°C to about 1000C, in a solvent such as THF, to yield compounds of the formula 17. Reduction of the ester of compound 17 to the corresponding alcohol, using lithium aluminum hydride, under conventional conditions well known to those of skill in the art, followed by oxidation of the alcohol with an oxidizing agent such as barium manganate, manganese dioxide, EBX, Dess Martin periodinane, or PCC, in a solvent such as dichloromethane, THF, or DMSO, or a combination of two or more of these solvents, yields the corresponding aldehydes of formula 18. Compounds of the formula 18 can then be reacted with (carbethoxymethylene)triphenylphosphorane or a similar Wittig reagent in a solvent such as dichloromethane, chloroform, THF, benzene or toluene, at a temperature from about room temperature to about the reflux temperature of the solvent, to give the corresponding compounds of formula 19. In the case of barium manganate, the oxidation and the Wittig reaction can be carried out using a one-pot procedure (/. Org. Chern. 1998, 63, 4489). Hydrogenation of compounds of the formula 19, using methods known to those skilled in the art, for example, as described above, preferably using palladium on barium sulfate in a solvent such as THF, provides the corresponding amino esters. The resulting amino esters can be cyclized to give compounds of the formula 5B-5 by heating at a temperature from about 5O0C to about the reflux temperature of the solvent, in a solvent such as ethanol, methanol or isopropanol, preferably with a catalytic amount of acid (i.e., TsOH) or base (Le., DBU).
Scheme J

20 21
,i); r reedαuuccee a azziidαee
 a 2) DDllBBAALLHH *"
a 2) DDllBBAALLHH *"
 22 23
22 23

Scheme J illustrates an alternative method for preparing compounds of the formula 2B-2. Addition of a suitably mono-protected diol, where n is 3 and P is THP, benzyl, or TBS, to compounds of the formula 20 provides the corresponding compounds of formula 21. This reaction is typically conducted in the presence of a base such as potassium tert-butoxide, sodium tert-butoxide, sodium hydride, lithium diisopropylamide, lithium bis(trimethylsilyl)amide, potassium bis(trimethylsilyl)amide, or sodium bis(trimethylsilyl)amide, in a solvent such as THF, dioxane, or ethylene glycol dimethylether, preferably THF. The temperature of the reaction may vary from about -78 °C to about room temperature. Compounds of the formula 21 can be reacted with sodium azide in solvents such as DMF, NMP or DMSO, or a combination of two or more of these solvents, to provide compounds of the formula 22. The temperature of the reaction may vary from about room temperature to about the reflux temperature of the solvent, and is preferably about 7O0C. The azide of compounds of the formula 22 can be reduced to an amine using conventional reducing agents known to those skilled in the art, preferably using hexamethyldisilthiane [(Me3Si)2S] in a solvent such as methanol or ethanol. Subsequent reduction of the cyano group to an aldehyde using diisobutylaluminum hydride (DIB ALH) in a solvent such as THF at about 0 0C provides compounds of the formula 23. Compounds of the formula 23 can be reacted with (carbethoxymethylene)triphenylphosphorane or a similar Wittig reagent in a solvent such as dichloromethane, chloroform, THF, benzene or toluene, or a mixture of two or more of these solvents, at about room temperature to about the reflux
temperature of the solvent, to give compounds of the formula 24. Compounds of the formula 24 can be hydrogenated using methods known to those skilled in the art, using, for example, palladium on activated carbon, palladium on barium sulfate, or Raney- nickel, in a solvent such as methanol, ethanol, THF, or a combination of two of the formerly mentioned solvents. The resulting amino esters can be cyclized to give the corresponding compounds of the formula 2B-2 by heating at a temperature from about 50°C to about the reflux temperature of the solvent, in a solvent such as ethanol, methanol or isopropanol, or a mixture of two or more of these solvents. Preferably, a catalytic amount of acid (for example, toluene sulphonic acid) or base (for example, l,8-diazabicyclo[5.4.0]undec-7-ene) is used.
The preparation of other compounds of the formula 1 not specifically described in the foregoing experimental section can be accomplished using combinations of the reactions described above that will be apparent to those skilled in the art.
In each of the reactions discussed or illustrated above, pressure is not critical unless otherwise indicated. Pressures from about 0.5 atmospheres to about 5 atmospheres are generally acceptable, and ambient pressure, i.e., about 1 atmosphere, is preferred as a matter of convenience.
The compounds of the formula 1 and the intermediates shown in the above reaction schemes can be isolated and purified by conventional procedures, such as recrystallization or chromatographic separation.
The preparation of other compounds of the formula 1 not specifically described in the foregoing experimental section can be accomplished using combinations of the reactions described above that will be apparent to those skilled in the art.
In each of the reactions discussed or illustrated above, pressure is not critical unless otherwise indicated. Pressures from about 0.5 atmospheres to about 5 atmospheres are generally acceptable, and ambient pressure, i.e., about 1 atmosphere, is preferred as a matter of convenience.
The compounds of the formula 1 and the intermediates shown in the above reaction schemes can be isolated and purified by conventional procedures, such as recrystallization or chromatographic separation.
The compounds of the formula 1 and their pharmaceutically acceptable salts, can be administered to mammals via either the oral, parenteral (such as subcutaneous, intravenous, intramuscular, intrasternal and infusion techniques), rectal, buccal or
intranasal routes. In general, these compounds are most desirably administered in doses ranging from about 3 mg to about 600 mg per day, in single or divided doses (i.e., from 1 to 4 doses per day), although variations will necessarily occur depending upon the species, weight and condition of the patient being treated and the patient's individual response to said medicament, as well as on the type of pharmaceutical formulation chosen and the time period and interval at which such administration is carried out. However, a dosage level that is in the range of about 10 mg to about 100 mg per day is most desirably employed. In some instances, dosage levels below the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effects, provided that such higher dose levels are first divided into several small doses for administration throughout the day.
The novel compounds of the present invention may be administered alone or in combination with pharmaceutically acceptable carriers or diluents by any of the routes previously indicated, and such administration may be carried out in single or multiple doses. More particularly, the novel therapeutic agents of this invention can be administered in a wide variety of different dosage forms, i.e., they may be combined with various pharmaceutically acceptable inert carriers in the form of tablets, capsules, lozenges, troches, hard candies, suppositories, jellies, gels, pastes, ointments, aqueous suspensions, injectable solutions, elixirs, syrups, and the like. Such carriers include solid diluents or fillers, sterile aqueous media and various non-toxic organic solvents, etc. Moreover, oral pharmaceutical compositions can be suitably sweetened and/or flavored. In general, the weight ratio of the novel compounds of this invention to the pharmaceutically acceptable carrier will be in the range from about 1:6 to about 2:1, and preferably from about 1:4 to about 1:1. For oral administration, tablets containing various excipients such as microcrystalline cellulose, sodium citrate, calcium carbonate, dicalcium phosphate and glycine may be employed along with various disintegrants such as starch (and preferably corn, potato or tapioca starch), alginic acid and certain complex silicates, together with granulation binders like polyvinylpyrrolidone, sucrose, gelatin and acacia. Additionally, lubricating agents such as magnesium stearate, sodium lauryl sulfate and talc are often very useful for tabletting purposes. Solid compositions of a similar type may also be employed as fillers in gelatin capsules; preferred materials in this connection also include lactose or milk sugar as well as high molecular weight
polyethylene glycols. When aqueous suspensions and/or elixirs are desired for oral administration, the active ingredient may be combined with various sweetening or flavoring agents, coloring matter or dyes, and, if so desired, emulsifying and/or suspending agents as well, together with such diluents as water, ethanol, propylene glycol, glycerin and various like combinations thereof.
For parenteral administration, solutions of a compound of the present invention in either sesame or peanut oil or in aqueous propylene glycol may be employed. The aqueous solutions should be suitably buffered (preferably pH greater than 8) if necessary and the liquid diluent first rendered isotonic. These aqueous solutions are suitable for intravenous injection purposes. The oily solutions are suitable for intraarticular, intra-muscular and subcutaneous injection purposes. The preparation of all these solutions under sterile conditions is readily accomplished by standard pharmaceutical techniques well known to those skilled in the art.
This invention relates to methods of treating anxiety, depression, schizophrenia and the other disorders referred to in the description of the methods of the present invention, wherein a novel compound of this invention and one or more of the other active agents referred to above (e.g., an NKl receptor antagonist, tricyclic antidepressant, 5HT ID receptor antagonist, or serotonin reuptake inhibitor) are administered together, as part of the same pharmaceutical composition, as well as to methods in which such active agents are administered separately as part of an appropriate dose regimen designed to obtain the benefits of the combination therapy. The appropriate dose regimen, the amount of each dose of an active agent administered, and the specific intervals between doses of each active agent will depend upon the subject being treated, the specific active agent being administered and the nature and severity of the specific disorder or condition being treated. In general, the novel compounds of this invention, when used as a single active agent or in combination with another active agent, will be administered to an adult human in an amount from about 3 mg to about 300 mg per day, in single or divided doses, preferably from about 10 to about 100 mg per day. Such compounds may be administered on a regimen of up to 6 times per day, preferably 1 to 4 times per day, especially 2 times per day and most especially once daily. Variations may nevertheless occur depending upon the species of animal being treated and its individual response to said medicament, as well as on the type of pharmaceutical formulation chosen and the time period and interval at which
such administration is carried out. In some instances, dosage levels below the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effect, provided that such larger doses are first divided into several small doses for administration throughout the day.
A proposed daily dose of a 5HT reuptake inhibitor, preferably sertraline, in the combination methods and compositions of this invention, for oral, parenteral or buccal administration to the average adult human for the treatment of the conditions referred to above, is from about 0.1 mg to about 2000 mg, preferably from about 1 mg to about 200 mg of the 5HT reuptake inhibitor per unit dose, which could be administered, for example, 1 to 4 times per day. A proposed daily dose of a 5HT1D receptor antagonist in the combination methods and compositions of this invention, for oral, parenteral, rectal or buccal administration to the average adult human for the treatment of the conditions referred to above, is from about 0.01 mg to about 2000 mg, preferably from about 0.1 mg to about 200 mg of the 5HT1D receptor antagonist per unit dose, which could be administered, for example, 1 to 4 times per day.
For intranasal administration or administration by inhalation, the novel compounds of the invention are conveniently delivered in the form of a solution or suspension from a pump spray container that is squeezed or pumped by the patient or as an aerosol spray presentation from a pressurized container or a nebulizer, with the use of a suitable propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas. In the case of a pressurized aerosol, the dosage unit may be determined by providing a valve to deliver a metered amount. The pressurized container or nebulizer may contain a solution or suspension of the active compound. Capsules and cartridges (made, for example, from gelatin) for use in an inhaler or insufflator may be formulated containing a powder mix of a compound of the invention and a suitable powder base such as lactose or starch. Formulations of the active compounds of this invention for treatment of the conditions referred to above in the average adult human are preferably arranged so that each metered dose or "puff' of aerosol contains 20 μg to 1000 μg of active compound. The overall daily dose with an aerosol will be within the range 100 μg to 10 mg. Administration may be several times daily, for example 2, 3, 4 or 8 times, giving for example, 1, 2 or 3 doses each time.
The ability of the novel compounds of this invention to bind to the dopamine D2 receptor can be determined using conventional radioligand receptor binding assays. All receptors can be heterologously expressed in cell lines and binding assays can be conducted in membrane preparations from the cell lines using procedures outlined below. IC50 concentrations can be determined by nonlinear regression of concentration- dependent reduction in specific binding. The Cheng-Prussoff equation can be used to convert the IC50 to Ki concentrations. See Example 21, below, for a description of the assay used to determine the binding of the compounds of this invention to the dopamine D2 receptor, and the binding data obtained for the assayed compounds. Compounds of the present invention preferably exhibit Ki values of no more than 100 nM, more preferably no more than 50 nM, even more preferably no more than 25 nM, most preferably no more than 10 nM.
The following Examples illustrate the preparation of several compounds of the present invention. Melting points are uncorrected. NMR data are reported in parts per million and are referenced to the deuterium lock signal from the sample solvent. Any reference to a "title compound" in an example, below, refers to the compound named in the title of that particular example.
EXAMPLES
EXAMPLE l

7-(4-Hydroxy-butoxy)-lH-[l,8]naphthyridin-2-one
To 1 liter (L) of n-methylpyrrolidinone was added 60% sodium hydride suspension (83.6g, 2.09moles). With external cooling to maintain 50°C, 1,4-butanediol (3.39 moles) was added dropwise, causing offgassing. The mixture was stirred for 15 minutes at 60°C, followed by addition of 7-Chloro-lH-[l,8]naphthyridin-2-one (Journal of Organic Chemistry, 55(15), 4744-50; 1990, 146g, 0.813 moles) while stirring at 680C for 20 hours (h). To the mixture was added 5 liters of acetonitrile followed by filtration of a solid which was washed with a 50:50 mixture of acetonitrile and tetrahydrofuran. The solid was resuspended in 3 liters of tetrahydrofuran, to which was
added 3N HCl in methanol (290ml, 0.870 moles). After heating for 1 hour at 6O0C, the mixture was filtered thru celite, washed with 1 liter of tetrahydrofuran and concentrated to 500ml in volume. To the residue was added 1.5 liters of tetrahydrofuran, 1Og Darco activated charcoal, and 100ml Magnesol. After stirring at 40°C for 30 minutes (min.), the mixture was filtered and washed with tetrahydrofuran and concentrated to 500ml in volume. To this residue was added 1 liter of acetonitrile followed by concentration of the mixture to 1 liter in volume. The resulting solid precipitate was filtered, washed with acetonitrile and ether, and dried at 50°C giving 7-(4-Hydroxy-butoxy)-lH- [l,8]naphthyridin-2-one, 101g, (53% yield).

4-(7-Oxo-7,8-dihydro-[lj8]naphthyridin-2-yloxy)-butyraIdehyde
7-(4-Hydroxy-butoxy)-lH-[l,8]naphthyridin-2-one (69.44g, 296mmol) was dissolved in ethyl acetate (2.1L). IBX (232.42g, 828mmol) was added to make a slurry, and the reaction was heated to 75°C for three 8 hour periods over the course of two days. The reaction was cooled, filtered and the solids washed wtih dichloromethane. The filtrate was stripped to give a waxy solid which was triturated with methyl-t-butyl ether, from which was obtained 4-(7-Oxo-7,8-dihydro-[l,8]naphthyridin-2-yloxy)- butyraldehyde, as a solid (58.61g, 85% yield).
EXAMPLE 2

4-(3-Methyl-isoquinolin-l-yl)-piperazine-l-carboxylic acid tert-butyl ester
A solution of 1-boc-piperazine (1.179 g, 6.33 mmol) in tetrahydrofuran (15 mL) was cooled to - 78 0C, then treated with n-butyllithium (2.5 M in hexanes, 2.5 mL, 6.25 mmol) dropwise. The colourless solution was stirred at -780C for 30 minutes, then treated with l-Chloro-3-methyl-isoquinoline (US 6,403,608 Bl, 0.45g, 2.53 mmol) in tetrahydrofuran (5 mL) dropwise. The purple mixture was stirred at -78 0C for 2 hours,
then quenched with saturated NH4Cl solution. The yellow mixture was diluted with saturated NaCl solution and extracted with ethyl acetate. The extracts were dried over anhydrous Na2SO4, filtered and concentrated to a yellow oil. The oil was purified by column chromatography (chloroform/ethyl acetate, 2:3), to afford 4-(3 -Methyl- isoquinolin-l-yl)-piperazine-l-carboxylic acid tert-butyl ester (0.305 g, 37 %), as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.03 (d, IH), 7.66 (d, IH), 7.56 (t, IH), 7.43 (t, IH), 7.10 (s, IH), 3.71-3.67 (m, 4H), 3.38-3.33 (m, 4H), 2.54 (s, 3H), 1.50 (s, 9H).

An ice-cold solution of methanol (10 mL) was treated with acetyl chloride (0.8 mL, 11.3 mmol). After 15 minutes, this mixture was added to 4-(3-Methyl- isoquinolin-l-yl)-piperazine-l-carboxylic acid tert-butyl ester (0.340 g, 1.04 mmol) and the resulting solution was stirred at room temperature for 2 hours. The mixture was concentrated under vacuum, to afford 3-Methyl-l-piperazin-l-yl-isoquinoline Hydrochloride (0.274 g, quantitative), as a yellow solid. 1H NMR (400 MHz, DMSO- d6) δ 9.51 (br s, 2H), 8.15 (d, IH), 7.86 (d, IH), 7.77 (t, IH), 7.60 (t, IH), 7.36 (s, IH), 3.74-3.62 (m, 4H), 3.43-3.31 (m, 4H), 2.50 (s, 3H).
EXAMPLE 3

7-(4-Hydroxy-butoxy)-3,4-dihydro-lH-[l,8]naphthyridin-2-one
To 1.5 liters refluxing methanol was added 7-(4-Hydroxy-butoxy)-lH- [l,8]naphthyridin-2-one (87 g). After 30 minutes, the mixture was allowed to cool to 220C for 16 hours, followed by filtration through Celite to remove insoluble impurities. The pH of the filtrate was adjusted to 3 by addition of 2N HCl (2ml). This solution was added to a stainless steel hydrogenation vessel containing 10% palladium on carbon (PMC #1940C, 54% water, 3Og) and hydrogenated at 34 psi for 48 hours. The catalyst
was removed by filtration through Celite, and the filtrate was concentrated to 100ml in volume under vacuum. The concentrate was diluted with a mixture of 1 liter of ethyl acetate and 200ml THF, and filtered again through Celite. The filtrate was washed sequentially with 20% potassium bicarbonate (800ml), brine, and dried over sodium sulfate. The suspension was filtered, concentrated to a crystalline solid and washed with a 50:50 mixture of ethyl acetate and hexane (100ml). The solid obtained from drying at 500C under vacuum was 7-(4-Hydroxy-butoxy)-3,4-dihydro-lH- [l,8]naphthyridin-2-one, 75.8g, 87% yield, MP 103-1060C.

4-(7-Oxo-5,6,7,8-tetrahydro-[l,8]naphthyridin-2-yIoxy)-butyraldehyde
4-(7-Oxo-5,6,7,8-tetrahydro-[l,8]naphthyridin-2-yloxy)-butyraldehyde, was produced using either a Dess-Martin oxidation reaction or a Swern oxidation reaction, as described below. Dess-Martin oxidation: To a cloudy solution of the Dess-Martin periodinane
(2.80 g, 6.60 mmol, 1.5 equiv) in CH2Cl2 (13 mL) was added a solution of 7-(4- hydroxy-butoxy)-3,4-dihydro-lH-[l,8]naphthyridin-2-one (1.04 g, 4.40 mmol) in CH2Cl2 (25 mL) via cannula. The reaction was stirred at room temperature for 5 h and stored in the freezer overnight. A 1:1 mixture of saturated Na2S2O3 and saturated NaHCO3 (50 mL) was added followed by ethyl ether (Et2O). The mixture was stirred for 10 min and then extracted with Et2O/EtOAc (2: 1). The organic layer was washed with saturated NaHCO3 and brine, dried over Na2SO4 and concentrated to give the title compound as a pale yellow oil (1.06 g, used crude in next reaction). MS: APCI: M+l: 235.1 (Exact Mass: 234.10). Swern oxidation: A solution of oxalyl chloride (9.97 mL, 112 mmol) in CH2Cl2 was cooled to -70 °C and DMSO (15.6 mL, 220 mmol) was carefully added. The solution was stirred at -60 °C for 10 min and then a solution of 7-(4-hydroxy-butoxy)~ 3,4-dihydro-lH-[l,8]naphthyridin-2-one (23 g, 97.5 mmol) in DMSO (70 mL) was added dropwise at -50—60 0C. The reaction mixture was stirred at -60 0C for 20 min and then triethylamine (72 mL, 0.513 mol) was added dropwise. The reaction was warmed to room temp and stirred for 30 min. The mixture was poured into ice-water
and the organic phase was separated. The aqueous phase was extracted with CH2Cl2, combined with the organic phase, washed with brine, dried over Na2SO4, and concentrated under vacuum to give the crude product. Purification by column chromatography (hexane:ethyl acetate 2:1) followed by recrystallization provided 4-(7- Oxo-5,6,7,8-tetrahydro-[l,8]naphthyridin-2-yloxy)-butyraldehyde (12.7 g, 54.3 mmol, 56%).
EXAMPLE 4

7-Amino-4,4-dimethyl-3,4-dihydro-lH-[l,8]naphthyridin-2-one, was produced as follows: 3-Methyl-but-3-enoic acid (6-amino-pyridin-2-yl)-amide (49.2 g, 0.26 mol) was dissolved in 500 mL CH2Cl2 in a 1000 mL 3-neck flask equipped with mechanical stirring, a 125 mL addition funnel and a thermal couple. While stirring, MeSO3H (50 mL, 0.78 mol) was added to the flask dropwise. The exotherm upon addition was controlled to maintain a temperature <20 0C by an ice/water bath. The mixture was allowed to stir for 15 minutes. AlCl3 (274 g, 2.08 mol) was suspended in 1500 mL CH2Cl2 in a 5 L 4-neck flask equipped with mechanical stirring, 1000 mL addition funnel, N2 line and a thermal couple. To this suspension, the amide solution was added dropwise. The exotherm from the addition was again controlled to maintain a temperature <200C with an ice/water bath. The reaction was allowed to warm to room temperature and stir overnight. The reaction had consumed all the beta gamma unsaturated isomer and was deemed complete. The reaction mixture was slowly added to ice as an inverse quench. The quenched mixture was brought to pH 8-10 with 2 N KOH. The salts precipitated out of solution and saturated the aqueous phase. The suspension was transferred to a separatory funnel and extracted twice with 100:8:1 CH2C^EtORNH4OH. The organic layers were combined, dried over Na2SO4, filtered and concentrated to a crude solid. The solid was triturated with EtOAc and filtered. The resulting solid was 7-Amino-4,4-dimethyl-3,4-dihydro-lH-[l,8]naphthyridin-2-one (22.4 g, 0.117 mol, 46%). MS: APCI: M+l: 192.2 (Exact Mass: 191.11).

7-Fluoro-4,4-dimethyI-3,4-dihydro-lH-[l,8]naphthyridin-2-one
7-Fluoro-4,4-dimethyl-3,4-dihydro-lH-[l,8]naphthyridin-2-one, was produced as follows: HF-pyridine (100 mL) was cooled to -42 0C in a 1000 mL HDPE bottle using an CH3CN dry ice bath. While stirring vigorously, 7-amino-4,4-dimethyl-3,4- dihydro-lH-[l,8]naphthyridin-2-one (24.6 g, 0.129 mol) was added portionwise to control the exotherm. After the addition, NaNO2 (8.9 g, 0.1291 mol) was added portionwise. Significant exotherms were observed for both additions. The reaction mixture was then allowed to warm to 0 0C and stir for 2 h. The reaction mixture was quenched into a 4 L high density polyethylene (HDPE) bottle full of ice. The aqueous slurry was then neutralized using 2 N KOH. The resulting aqueous solution was extracted 3 times with CH2Cl2. The organic layers were dried over Na2SO4, filtered and concentrated to dryness. Excess pyridine was azeotroped with heptane. The product was dried under vacuum (2 mm Hg) for 3 h. The resulting product, 7-fluoro-4,4- dimethyl-3,4-dihydro-lH-[l,8]naphthyridin-2-one, was in the form of a white powder (23.06 g, 0.119 mol, 92%). MS: APCI: M+l: 195.1 (Exact Mass: 194.09).

7-(4-Hydroxy-butoxy)-4,4-dimethyI-3,4-dihydro-lH-[l,8]naphthyridin-2-one 7-(4-Hydroxy-butoxy)-4,4-dimethyl-3,4-dihydro-lH-[l,8]naphthyridin-2-one, was produced as follows: The 7-fluoro-4,4-dimethyl-3,4-dihydro-lH- [l,8]naphthyridin-2-one (5.09 g, 26.2 mmol) and butane-l,4-diol (11.81 g, 131.0 mmol) were combined in a dried 2-necked flask under N2. NMP (50 mL) was added and the solution was heated in an oil bath to 700C overnight. The reaction was cooled to room temperature and poured into ice water. The solid that formed was collected and triturated in acetonitrile to give the title compound as a tan powder (1.72 g). The mother liquor was extracted with CH2Cl2, dried over Na2SO4, filtered and purified by medium pressure liquid chromatography (MPLC) (gradient of 100% CH2Cl2 to 100% ethyl acetate). The compound was isolated as a mixture with diol byproducts. 7-(4-
Hydroxy-butoxy)-4,4-dimethyl-3,4-dihydro-lH-[l,8]naphthyridin-2-one was formed as clear crystals for a total of 3.15 g (11.9 rnmol, 45.5%). MS: APCI: M+l: 265.1 (Exact Mass: 264.15).

4-(5,5-Dimethyl-7-oxo-5,6,7,8-tetrahydro-[l,8]naphthyridin-2-yloxy)- butyraldehyde
4-(5,5-Dimethyl-7-oxo-5,6,7,8-tetrahydro-[l,8]naphthyridin-2-yloxy)- butyraldehyde, was produced as follows: 7-(4-Hydroxy-butoxy)-4,4-dimethyl-3,4- dihydro-lH-[l,8]naphthyridin-2-one (1.72 g, 6.51 mmol) was dissolved in ethyl acetate (50 mL, 0.14 M solution) and IBX (13 g, 46.4 mmol) was added. The suspension was immersed in an oil bath set at 80 °C and stirred vigorously with a condenser. After 1.5 h, the reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated to give 4-(5,5-Dimethyl-7-oxo-5,6,7,8-tetrahydro-[l,8]naphthyridin-2- yloxy)-butyraldehyde as a tan solid (1.62 g, 6.18 mmol, 95%). MS: APCI: M+l: 263.1 (Exact Mass: 262.13).
EXAMPLE 5

To a suspension of 2-methylsulfanyl-8H-pyrido[2,3-d]pyrimidin-7-one (WO 9833798 A2, 5.0 g, 25.9 mmol) in CH2Cl2 (100 mL), CHCl3 (50 mL) and MeOH (10 mL, the starting material still did not dissolve) was added the oxaziridine (8.11 g, 31.05 mmol, 1.2 equiv) as a solid. The reaction became homogenous after 3 h and was stirred overnight at RT. The reaction was concentrated and CH2Cl2ZMeOH was added to dissolve the residue. Much of the solid did not dissolve so the mixture was filtered to give 2-Methanesulfinyl-8H-pyrido[2,3-d]pyrimidin-7-one, as an off-white solid (2.31 g, 11.04 mmol, 43%). MS: APCI: M+l: 210.1 (Exact Mass: 209.03).
 2-[4-(Tetrahydro-pyran-2-yloxy)-butoxy]-8H-pyrido[2,3-d] pyrimidin-7-one
2-[4-(Tetrahydro-pyran-2-yloxy)-butoxy]-8H-pyrido[2,3-d] pyrimidin-7-one
To a solution of 4-(Tetrahydro-pyran-2-yloxy)-butan-l-ol (4.45 g, 25.3 mmol, 2.5 equiv) in THF (20 mL) cooled to 0 °C was added IM potassium terf-butoxide (KO1Bu) in THF (25 mL, 25 mmol). The solution was stirred at 0 °C for 20 min and then added to a suspension of 2-methanesulfinyl-8H-pyrido[2,3-d]pyrimidin-7-one (2.12 g, 10.13 mmol) in DMF (30 mL) at RT. The reaction became homogenous and was stirred at RT for 1 h. Saturated NH4Cl and H2O were added to quench the reaction. The mixture was extracted with EtOAc. The organic layer was washed with H2O and brine, dried over Na2SO4 and concentrated. Purification by liquid chromatography
(70% EtOAc/Hexanes to 100% EtOAc) gave 2-[4-(Tetrahydro-pyran-2-yloxy)-butoxy]- 8H-pyrido[2,3-d]pyrimidin-7-one, as a white solid (1.95 g, 6.11 mmol, 60%). MS: APCI: M+l: 320.2 (Exact Mass: 319.15).

2-(4-Hydroxy-butoxy)-8H-pyrido[2,3-d]pyrimidin-7-one
To a suspension of 2-[4-(tetrahydro-pyran-2-yloxy)-butoxy]-8H-pyrido[2,3- d]pyrimidin-7-one (1.95 g, 6.11 mmol) in EtOH (30 mL) and CH2Cl2 (2 mL, added to help dissolve the starting material) was added PPTS (151 mg, 0.6 mmol). The solution was stirred overnight at RT and then heated at 60 °C for 5 h. The reaction was concentrated to give a white solid. Purification by liquid chromatography (6% MeOHZCH2Cl2) gave 2-(4-Hydroxy-butoxy)-8H-pyrido[2,3-d]pyrimidin-7-one, as a white solid (1.22 g, 5.19 mmol, 85%). MS: APCI: M+l: 236.1 (Exact Mass: 235.10).

4-(7-Oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-yIoxy)-butyraldehyde
To a solution of 2-(4-hydroxy-butoxy)-8H-pyrido[2,3-d]pyrimidin-7-one (251 mg, 1.07 mmol) in DMSO (3 mL) was added a solution of IBX (597 mg, 2.13 mmol) in DMSO (7 mL, 0.3 M). The reaction was stirred at RT for 90 min, cooled to 0 0C and
quenched with 5% NaHCO3. The mixture was extracted with CH2Cl2 (4x). The organic layer was washed with 5% NaHCO3 and brine, dried over Na2SO4 and concentrated to give 4-(7-Oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-yloxy)- butyraldehyde, as a white solid (171 mg, 0.733 mmol, 69%). MS: APCI: M+l: 234.1 (Exact Mass: 233.08).
EXAMPLE 6

2-MethanesuIfonyl-4-methyl-8H-pyrido[2,3-d]pyrimidin-7-one A solution of 4-methyl-2-methylsulfanyl-8H-pyrido[2,3-d]pyrimidin-7-one (24 g, 0.1158 mol, US 6,498,163) in a mixture of CH2Cl2 (1.9 L) and methanol (300 mL) is treated with m-chloroperbenzoic acid (103 g, 60%, 0.345 mo)) in portions at room temperature. The mixture is stirred for 24 h, cooled to ~5 0C and quenched with saturated sodium bicarbonate solution. The solids are filtered, washed thoroughly with water followed by ether and dried in vacuum to give 2-Methanesulfonyl-4-methyl-8H- pyrido[2,3-d]pyrimidin-7-one, as a solid (10 g, 0.042 mol, 36%). MS: APCI: M+l: 240.0 (Exact Mass: 239.04).

To an ice bath cooled solution of 4-(tetrahydro-pyran-2-yloxy)-butan-l-ol (27.3 g, 0.1567 mol) in dry THF (125 mL) is added drop wise a solution of KOtBu (IM, 155 mL, 0.155 mol) in THF within 15 min. The mixture is then stirred at 00C for 2 h. To this mixture is added a suspension of 2-methanesulfonyl-4-methyl-8H-pyrido[2,3- d]pyrimidin-7-one (15 g, 0.0627 mol) in DMF (225 mL) at RT within 15 min. The orange red colored reaction mixture is stirred at RT for 1.5 h, cooled and quenched with saturated NH4Cl solution (150 mL) and water (2 L). The mixture is extracted with ethyl
acetate (2 x 0.75 L) and the organic layer is washed with brine (300 mL), dried over anhydrous sodium sulfate, filtered through a small bed of silica gel eluting with 5% methanol in ethyl acetate (750 mL) and concentrated. The residue is then triturated with hexane, filtered and dried to give 4-Methyl-2-[4-(tetrahydro-pyran-2-yloxy)- butoxy]-8H-pyrido[2,3-d]pyrimidin-7-one, as a white solid (16.5 g, 0.0495 mol 78%). MS: APCI: M+l: 334.0 (Exact Mass: 333.17).

2-(4-Hydroxy-butoxy)-4-methyI-8H-pyrido[2,3-d]pyrimidin-7-one A mixture of 4-methyl-2-[4-(tetrahydro-pyran-2-yloxy)-butoxy]-8H-pyrido[2,3- d]pyrimidin-7-one (16.5 g, 0.049 mol) and PPTS (1.24 g, 0.0049 mol) in ethanol (250 mL) and CH2Cl2 (20 mL) is stirred at room temperature for 16 h, followed by heating at reflux (-900C) for 3 h. The cloudy reaction mixture is evaporated under vacuum and the residue is triturated in hexane-ethyl acetate (150 mL, 1:1) and dried to give 2-(4- Hydroxy-butoxy)-4-methyl-8H-pyrido[2,3-d]pyrimidin-7-one, as a yellow powder (12.5 g, 0.049 mol, 100%). MS: APCI: M+l: 250.0 (Exact Mass: 249.11).

4-(4-Methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-yIoxy)-butyraldehyde A stirred solution of IBX (26 g, 0.092 mol) in DMSO (220 ml) is treated with 2-
(4-hydroxy-butoxy)-4-methyl-8H-pyrido[2,3-d]pyrimidin-7-one (11 g, 0.0467 mol) portion wise while stirring at RT during 30 min and the reaction is stirred at RT for an additional 2 h. The mixture is cooled and treated with saturated NaHCO3 (150 mL) and extracted with chloroform (4 x 0.5 L). The combined organic layer is washed with brine/ice (2 x), dried over Na2SO4, filtered and concentrated. The residue is stirred with ether, filtered, washed with ether and dried to give 6 g of the crude, which shows it to be a mixture. The ether filtrate residue also shows some product, but mostly starting material. The residue from the filtrate and the crude (11 g) are subjected to re-oxidation as above using fresh BX (15.5 g, 0.055 mol) in DMSO (150 mL), but stirred at 300C
for 3 h. Workup as above yielded 4-(4-Methyl-7-oxo-7,8-dihydro-pyrido[2,3- d]pyrimidin-2-yloxy)-butyraldehyde, as an off-white powder (8.3 g, 0.057 mol, 66.8%). MS: APCI: M+l: 248.0 (Exact Mass: 247.10).
EXAMPLE 7

7-[4-(4-IsoquinoIin-l-yl-piperazin-l-yl)-butoxy]-3,4-dihydro-lH- [l,8]naphthyridin-2-one
The dihydrochloride salt of 1-Piperazin-l-yl-isoquinoline (WO 98/42712), (550mg, 1.92 mmol) was suspended in dichloroethane (10 mL). Triethylamine (1.04g, 10.3 mmol) was added and the mixture was stirred for 5 minutes. 4-(7-oxo-7,8-dihydro- [l,8]naphthyridin-2-yloxy)-butyraldehyde (665 mg, 2.84 mmol), produced as described in Example 1, above, was added, followed by addition of 0.3g 3 Angstrom molecular sieves. After stirring for 2 hours, Sodium triacetoxyborohydride (765mg, 3.61 mmol) was added and the mixture was stirred for 18 hours. The mixture was quenched with water (20 mL), filtered, and the aqueous phase was adjusted to pH 6 by addition of IN citric acid, agitated and decanted. The dichloromethane was again extracted with water adjusted to pH 3 by addition of IN citric acid. The aqueous phase was adjusted to pH 13 by addition of 50% NaOH solution and extracted with dichloromethane. The organic layer was washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and evaporated. The crude solid was purified by column chromatography (dichloromethane / methanol / ethyl acetate) to yield the title compound (Yield: 520 mg, 46%) as a white foam, MS ES: m/z 432.3 (M+H)+ (Exact mass: 431.23).
EXAMPLE 8

7-[4-(4-Isoquinolin-4-yl-piperazin-l-yl)-butoxy]-3,4-dihydro-lH- [l,8]naphthyridin-2-one In a manner similar to Example 7, above, the dihydrochloride salt of 4-
Piperazin-1-yl-isoquinoline (Chem. Pharm. Bull. 49(10) 1313-1320 (2001)), and 4-(7- oxo-7,8-dihydro-[l,8]naphthyridin-2-yloxy)-butyraldehyde (Example 1, above) gave a crude solid that was purified by column chromatography (dichloromethane / methanol / ethyl acetate) to yield the title compound (Yield: 447mg, 64%) as a white solid, MS ES: m/z 432.3 (M+H)+ (Exact mass: 431.23).
EXAMPLE 9

7-[4-(4-Isoquinolin-l-yl-piperazin-l-yI)-butoxy]-lH-[l,8]naphthyridin-2-one In a similar manner, the dihydrochloride salt of 1-Piperazin-l-yl-isoquinoline
(WO 98/42712), (700 mg, 2.4 mmol) and 4-(7-Oxo-7,8-dihydro-[l,8]naphthyridin-2- yloxy)-butyraldehyde (520 mg, 2.2 mmol) (Example 1, above) were dissolved in dichloroethane (10 mL). Triethylamine (l.Omg, 13 mmol) was added and the mixture was stirred for 5 minutes. Sodium triacetoxyborohydride (660 mg, 3.1 mmol) was added and the mixture was stirred overnight. The mixture was quenched with water (20 mL) and extracted with dichloromethane (20 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and evaporated. The residue was purified by column chromatography. The crude residue was purified by column chromatography (dichloromethane/rnethanol/ethyl acetate) to yield the title compound (Yield: 443 mg, 46%) as a white solid, MS: APCI: M+l: 430.3 (Exact Mass: 429.22).
EXAMPLE 10

7-{4-[4-(6-Fluoro-isoquinoIin-l-yI)-piperazin-l-yI]-butoxy}-3,4-dihydro-lH-
[l,8]naphthyridin-2-one 6-Fluoro-l-piperazin-l-yl-isoquinoline trifluoroacetate salt (WO 98/42712),
(515 mg, 1.28 mmol) and 4-(7-oxo-7,8-dihydro-[l,8]naphthyridin-2-yloxy)- butyraldehyde (300 mg, 1.28 mmol) were dissolved in dichloroethane (10 mL). Triethylamine (389 mg, 3.84 mmol) was added and the mixture was stirred for 5 minutes. Sodium triacetoxyborohydride (380 mg, 1.79 mmol) was added and the mixture was stirred for 1.5 hours. The mixture was quenched with water (20 mL) and extracted with dichloromethane (20 mL). The organic layer was washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and evaporated. The crude solid was purified by column chromatography (10:90 methanol / ethyl acetate) to yield the title compound (Yield: 410 mg, 71%) as a white solid, mp 57-59 0C, MS ES+ 450.22 (M+l) + Exact Mass: 449.22.
EXAMPLE Il

7-{4-[4-(6-Fluoro-isoquinoIin-l-yl)-piperazin-l-yl]-butoxy}-lH-[l,8]naphthyridin- 2-one
In a process similar to Example 10, above, 6-Fluoro-l-piperazin-l-yl- isoquinoline trifluoroacetate salt (WO 98/42712), (520 mg, 1.29 mmol) and 4-(7-Oxo- 7,8-dihydro-[l,8]naphthyridin-2-yloxy)-butyraldehyde (300 mg, 1.29 mmol) (Example 1) were dissolved in dichloroethane (10 mL). Triethylamine (392 mg, 3.87 mmol) was
added and the mixture was stirred for 5 minutes. Sodium triacetoxyborohydride (383 mg, 1.81 mmol) was added and the mixture was stirred for 1.5 hours. The mixture was quenched with water (20 mL) and extracted with dichloromethane (20 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and evaporated. The residue was purified by column chromatography (10:90 methanol / ethyl acetate) to yield the title compound (Yield: 408 mg, 71%) as a beige solid, mp 79-181 0C MS ES+ 448.16 (M+l)+ExactMass: 447.2.
EXAMPLE 12

7-{4-[4-(7-Fluoro-isoquinolin-l-yl)-piperazin-l-yl]-butoxy}-3,4-dihydro-lH-
[l,8]naphthyridin-2-one
7-Fluoro-l-piperazin-l-yl-isoquinoline trifluoroacetate salt (WO 98/42712), (250 mg, 1.08 mmol) and 4-(7-oxo-7,8-dihydro-[l,8]naphthyridin-2-yloxy)- butyraldehyde (253mg, 1.08 mmol) (Example 1) were dissolved in dichloroethane (10 mL). Triethylamine (219 mg, 2.16 mmol) was added and the mixture was stirred for 5 minutes. Sodium triacetoxyborohydride (321 mg, 1.51 mmol) was added and the mixture was stirred for 1.5 hours. The mixture was quenched with water (20 mL) and extracted with dichloromethane (20 mL). The organic layer was washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and evaporated. The crude solid was purified by column chromatography (10:90 methanol / ethyl acetate) to yield the title compound (Yield: 402 mg, 83%) as a white solid, mp 63-65 0C, MS ES+ 450.19 (M+l)+, Exact Mass: 449.22.
EXAMPLE 13

7-{4-[4-(7-Fluoro-isoquinolin-l-yl)-piperazin-l-yl]-butoxy}-lH-[l,8]πaphthyridin-
2-one In a similar manner, 7-Fluoro-l-piperazin-l-yl-isoquinoline trifluoroacetate salt
(WO 98/42712), (250 mg, 1.08 mmol) and 4-(7-Oxo-7,8-dihydro-[l,8]naphthyridin-2- yloxy)-butyraldehyde (251 mg, 1.08 mmol) (Example 1) were dissolved in dichloroethane (10 mL). Triethylamine (219 mg, 2.16 mmol) was added and the mixture was stirred for 5 minutes. Sodium triacetoxyborohydride (321 mg, 1.51 mmol) was added and the mixture was stirred for 1.5 hours. The mixture was quenched with water (20 mL) and extracted with dichloromethane (20 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and evaporated. The residue was purified by column chromatography (10:90 methanol/ethyl acetate) to yield the title compound (Yield: 410 mg, 85%) as a white solid, mp 74-76 0C, MS ES+ 448.20 (M+l)+, Exact Mass: 447.21.
EXAMPLE 14

7-{4-[4-(7-Fluoro-isoquinolin-l-yl)-piperazin-l-yl]-butoxy}-4,4-dimethyI-3,4- dihydro-lH-[l,8]naphthyridin-2-one
7-Fluoro-l-piperazin-l-yl-isoquinoline trifluoroacetate salt (WO 98/42712), (263 mg, 0.82 mmol) and 4-(5,5-Dimethyl-7-oxo-5,6,7,8-tetrahydro-[l,8]naphthyridin- 2-yloxy)-butyraldehyde (210mg, 0.80mmol) (Example 4, above) were dissolved in dichloroethane (10 mL). Triethylamine (170 mg, 1.68 mmol) was added and the mixture was stirred for 5 minutes. Sodium triacetoxyborohydride (237 mg, 1.12 mmol) was added and the mixture was stirred for 1.5 hours. The mixture was quenched with
water (20 mL) and extracted with dichloromethane (20 mL). The organic layer was washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and evaporated. The crude solid was purified by column chromatography eluted with a gradient of dichloromethane to 10:90 methanol in ethyl acetate to yield the title compound (Yield: 148 mg, 39%, as a solid, mp 151 0C, MS: APCI: M+l: 478.4 (Exact mass: 477.25).
EXAMPLE 15

7-Fluoro-l-piperazin-l-yl-isoquinoline trifluoroacetate salt (WO 98/42712), (263 mg, 0.82 mmol) and 4-(7-Oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-yloxy)- butyraldehyde (187mg, 0.80mmol) (Example 5, above) were dissolved in dichloroethane (10 mL). Triethylamine (170 mg, 1.68 mmol) was added and the mixture was stirred for 5 minutes. Sodium triacetoxyborohydride (237 mg, 1.12 mmol) was added and the mixture was stirred for 1.5 hours. The mixture was quenched with water (20 mL) and extracted with dichloromethane (20 mL). The organic layer was washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and evaporated. The crude solid was purified by column chromatography eluted with a gradient of dichloromethane to 100:8:1 dichloromethane:ethanol:concentrated ammonium hydroxide to yield the title compound (Yield: 280 mg, 78%, as a solid, mp 500C, MS: APCI: M+l: 449.2 (Exact mass: 448.20).
EXAMPLE 16

In a similar manner, 7-Fluoro-l-piperazin-l-yl-isoquinoline trifluoroacetate salt (WO 98/42712), (263 mg, 0.82 mmol) and 4-(4-Methyl-7-oxo-7,8-dihydro-pyrido[2,3- d]pyrimidin-2-yloxy)-butyraldehyde (197mg, 0.80mmol) (Example 6) were dissolved in dichloroethane (10 mL). Triethylamine (170 mg, 1.68 mmol) was added and the mixture was stirred for 5 minutes. Sodium triacetoxyborohydride (237 mg, 1.12 mmol) was added and the mixture was stirred for 1.5 hours. The mixture was quenched with water (20 mL) and extracted with dichloromethane (20 mL). The organic layer was washed with brine (20 mL), dried over anhydrous sodium sulfate, filtered and evaporated. The crude solid was purified by column chromatography eluted with a gradient of dichloromethane to 100:8:1 dichloromethane: ethanol: concentrated ammonium hydroxide to yield the title compound (Yield: 261 mg, 71%, as a solid, MS: APCI: M+l: 463.1 (Exact mass: 462.22).
EXAMPLE 17

7-[4-(4.[2,7]Naphthyridin-l-yl-piperazin-l-yl)-butoxy]-lH-[l,8]naphthyridin-2- one To a mixture of l-Piperazin-l-yl-[2,7]naphthyiidine (U.S. Pat No 4,859,671,
0.29 g, 1.36 mmol), 4-(7-Oxo-7,8-dihydro-[l,8]naphthyridin-2-yloxy)-butyraldehyde (0.314 g, 1.36 mmol) (Example 1), and l-methyl-2-pyrrolidinone (15 mL) was added NaBH(OAc)3 (0.35 g, 1.63 mmol,) in portions, over 20 mim. After the addition was over, the mixture was left stirring overnight. After quenching with H2O (50 mL), the reaction mixture was extracted with CH2Cl2 (3 x 100 mL). The combined organic phases were washed with brine (100 mL), dried and concentrated. The residue was purified by chromatography on silica gel to give the title compound (310 mg) as a white solid. MS: 431 (M++!), Exact Mass: 430.21.
EXAMPLE 18

7.[4-(4-[2,7]Naphthyridin-l-yl-piperazm-l-yl)-butoxy]-3,4-dihydro-lH-
[l,8]naphthyridin-2-one
In a manner similar to that above, l-Piperazin-l-yl-[2,7]naphthyridine (U.S.4,859,671, 0.36 g) and 4-(7-Oxo-5,6,7,8-tetrahydro-[l,8]naphthyridin-2-yloxy)- butyraldehyde (Example 3) gave the title compound, 0.6Og.
EXAMPLE 19

7-{4-[4-(3-Methyl-isoquinolin-l-yI)-piperazin-l-yl]-butoxy}-3,4-dihydro-lH-
[l,8]naphthyridin-2-one
4-(7-oxo-7,8-dihydro-[l,8]naphthyridin-2-yloxy)-butyraldehyde (0.21g, 0.90 mmol) (Example 1) and the hydrochloride salt of 3 -Methyl- 1-piperazin-l-yl- isoquinoline (260mg, 0.99 mmol) (Example 2), were suspended in dichloroethane (20 mL). Triethylamine (0.25ml, 1.79 mmol) was added followed by sodium triacetoxyborohydride (0.266 g, 1.26 mmol). The yellow solution was stirred at room temperature for 1 hour, then quenched with water and extracted with dichloromethane. The extracts were dried over anhydrous Na2SO4, filtered and concentrated in vacuo to give a yellow oil. The oil was purified by column chromatography (methanol :ethyl acetate, 3:97) to yield the title compound (0.351 g, 88%), as a light yellow solid, mp 65- 68 0C. ES MS: 446.42 (M+).
EXAMPLE 20

7-{4-[4-(3-Methyl-isoquinolin-l-yl)-piperazin-l-yl]-butoxy}-lH-[l,8]naphthyridin-
2-one In a similar manner, the hydrochloride salt of 3-Methyl-l-piperazin-l-yl- isoquinoline, (0.250 g, 0.95 mmol) (Example 2) and 4-(7-Oxo-7,8-dihydro- [l,8]naphthyridin-2-yloxy)-butyraldehyde (0.20Og, 0.86 mmol) (Example 1) were dissolved in dichloroethane (10 mL). Triethylamine (0.25mL, 1.79mmol) was added, followed by sodium triacetoxyborohydride (0.255g, 1.20mmol). The yellow mixture was stirred at room temperature for 1 hour, then quenched with water and extracted with dichloromethane. The extracts were dried over anhydrous Na2SO4, filtered and concentrated in vacuo to give a yellow oil. The oil was purified by column chromatography (1:9 methanokethyl acetate) to yield the title compound (0.235 g, 62 %), as a light yellow foam, mp 85-87 0C. ES MS: 444.37 (M+).
EXAMPLE 21
Dopamine D2 Receptor Binding Assay:
The compounds produced as described in Examples 7-21, above were each tested in a Dopamine D2 Receptor Binding Assay, as follows. [3H] Spiperone binding to a membrane preparation from CHO-hD2L cells was carried out in 250 μl of 50 mM Tris-HCl buffer containing 100 niM NaCl, 1 mM MgCl2 and 1% DMSO at pH 7.4. Duplicate samples containing (in order of addition) the test compounds, 0.4 nM [3H]spiperone and approximately 12 μg protein were incubated for 120 minutes at room temperature. Bound radioligand was separated by rapid filtration under reduced pressure through Whatman GF/B glass fiber filters previously treated with 0.3% polyethyleneimine. Radioactivity retained on the filter is determined by liquid scintillation spectrophotometry.
Specific binding was determined in the presence of 1 mM haloperidol was 95%. See Table 1, below, for results obtained from each of the compounds tested.
TABLE 1

Claims
1. A compound of formula 1


A is -(CH2)nO- wherein n is 4 or 5, and one or two of the carbon atoms of -(CH2)nO- can be substituted, optionally and independently, with one or two substituents that are selected, independently, from fluoro and methyl, or with two substituents attached to the same carbon atom that form, together with the carbon to which they are attached, a spirocyclopropyl or spirocyclobutyl ring;
D is N, C, or CH, provided that when D is N, each carbon atom covalently attached to D is attached through a single bond;
J and K are independently selected from N, CH, or C, provided that at least one of J and K is N;
Z and Q are independently selected from N or CH, provided that at least one of Z and Q is N;
R1 is hydrogen, -C(=O)CH3, or (Ci-C3) alkyl;
-X7^Y- is -CH2-CH2-, -CH=CH-, -CH2-NH-, -NH-CH2-, -N=CH-, -CH=N-, -0-CH2-, or -CH2-O-, wherein -X=^7Y- can optionally be substituted, at any available bonding site, by one to four substituents R2, R2 , R3 and R3 ; R , R , R and R are independently selected from hydrogen, halo, cyano, oxo, hydroxy, -C(=O)CH3, (C1-C4) alkyl, and (C1-C4) alkoxy, wherein the alkyl moieties of the (C1-C4) alkyl, (C1-C4) alkoxy, and -C(=O)CH3 groups can be optionally substituted with from one to three fluoro atoms and can also be optionally substituted with an amino or hydroxy substituent;
R4 and R5 are independently selected from hydrogen, halo, cyano, hydroxy, -C(=O)CH3, (C1-C4) alkyl, and (C1-C4) alkoxy, wherein the alkyl moieties of the (C1- C4) alkyl, (C1-C4) alkoxy, and -C(=O)CH3 groups can be optionally substituted with from one to three fluoro atoms and can also be optionally substituted with an amino or hydroxy substituent;
R6 and R7 are selected, independently, from hydrogen and methyl;
R8, R9, R10 and R11 are independently selected from hydrogen, halo, -C(=O)CH3, (C1-C4) alkyl, and (C1-C4) alkoxy, wherein the alkyl moieties of the (Ci-C4) alkyl, (C1- C4) alkoxy, and -C(=O)CH3 groups can be optionally substituted with from one to three fluoro atoms and can also be optionally substituted with an amino or hydroxy substituent; ring AA is a saturated or unsaturated 5- 6- or 7-membered carbocyclic ring wherein one, two or three of the carbon atoms of ring AA thai are not shared with the other fused ring of formula (i) can be replaced, optionally and independently, by a nitrogen, oxygen or sulfur atom; and the pharmaceutically acceptable salts thereof.
2. The compound or salt according to claim 1, wherein ring AA is a 6-membered unsaturated carbocyclic ring.
3. The compound or salt according to claim 1, wherein A is -(CH2)4O-.
4. The compound or salt according to claim 1, wherein -X=^Y- is -CH2NH-, -CH2-CH2- or -CH=CH-.
5. The compound or salt according to claim 1, wherein R4, R5, R10, and R11 are independently hydrogen or fluoro.
6. The compound or salt according to claim 1, wherein G is as shown formula (ii):
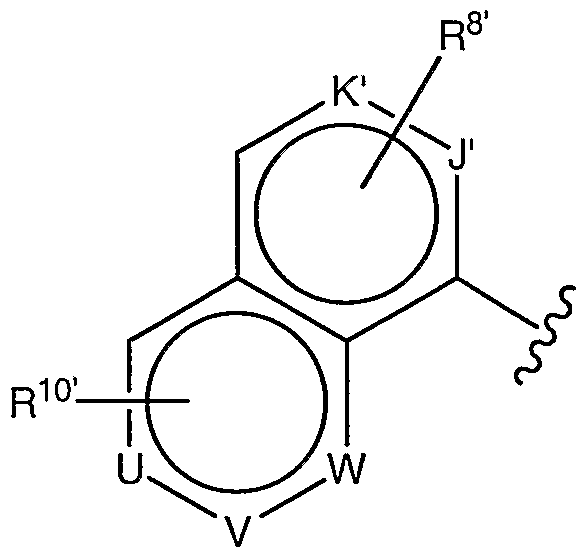
(ii) where K' and J' are independently N, CH, or C, provided that at least one of K' or J' is N; R8' is H, (C1-C4) alkyl, or (C1-C4) alkoxy;
R10' is H, (C1-C4) alkyl, or (C1-C4) alkoxy, or halo; and
U, V, and W are independently N, CH, or C, provided that no more than two of U, V, and W are N.
7. The compound or salt according to claim 1, selected from the group consisting of:
7-[4-(4-Isoquinolin-l-yl-piperazin-l-yl)-butoxy]-3,4-dihydro-lH- [1 ,8]naphthyridin-2-one;
7- [4-(4-Isoquinolin-4-yl-piperazin- 1 -yl)-butoxy] -3 ,4-dihydro- IH- [l,8]naphthyridin-2-one;
7- [4-(4-Isoquinolin- 1 -yl-piperazin- 1 -yl)-butoxy] -IH-[1 , 8]naphthyridin-2-one; 7- { 4- [4-(6-Fluoro-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } -3 ,4-dihydro- IH- [l,8]naphthyridin-2-one;
7- { 4- [4-(6-Fluoro-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } - IH- [l,8]naphthyridin-2-one;
7- { 4- [4-(7-Fluoro-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } -3 ,4-dihydro- IH- [l,8]naphthyridin-2-one;
7- { 4- [4-(7-Fluoro-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } - IH- [l,8]naphthyridin-2-one; 7-{4-[4-(7-Fluoro-isoquinolin-l-yl)-piperazin-l-yl]-butoxy}-4,4-dimethyl-3,4- dihydro-lH-[l,8]naphthyridin-2-one; 2-{4-[4-(7-Fluoro-isoquinolin-l-yl)-piperazin-l-yl]-butoxy}-8H-pyrido[2,3- d]pyrimidin-7-one;
2- { 4- [4-(7-Fluoro-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } -4-methyl-8H- pyrido[2,3-d]pyrimidin-7-one; 7-[4-(4-[2,7]Naphthyridin-l-yl-piperazin-l-yl)-butoxy]-lH-[l,8]naphthyridin-2- one;
7-[4-(4-[2,7]Naphthyridin-l-yl-piperazin-l-yl)-butoxy]-3,4-dihydro-lH- [1 ,8]naphthyridin-2-one;
7- { 4- [4-(3-Methyl-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } -3 ,4-dihydro- 1 H- [l,8]naphthyridm-2-one;
7- { 4- [4-(3 -Methyl-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } - IH- [1 ,8]naphthyridin-2-one;
7-[4-(4-[l,6]Naphthyridin-8-yl-piperazin-l-yl)-butoxy]-3,4-dihydro-lH- [l,8]naphthyridin-2-one; 7-[4-(4-[l,6]Naphthyridin-8-yl-piperazin-l-yl)-butoxy]-lH-[l,8]naphthyridin-2- one;
7-[4-(4-[l,6]Naphthyridin-8-yl-piperazin-l-yl)-butoxy]-lH-[l,8]naphthyridin-2- one;
7- { 4- [4-(7-Fluoro-3-methyl-isoquinolin- 1 -yl)-piperazin- 1 -yl]-butoxy } -3 ,4- dihydro-lH-[l,8]naphthyridin-2-one;
7- { 4- [4-(7-Ruoro-3-methyl-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } - IH- [1 ,8]naphthyridin-2-one;
7- { A- [4-(6-Fluoro-3-methyl-isoquinolin- 1 -yl)-piperazin- 1-yl] -butoxy } -3 ,4- dihydro-lH-[l,8]naphthyridin-2-one; 7- { 4- [4-(6-Fluoro-3-methyl-isoquinolin- 1 -yl)-piperazin- 1 -yl] -butoxy } - IH-
[ 1 ,8]naphthyridin-2-one;
7-[4-(4-[l,7]Naphthyridin-8-yl-piperazin-l-yl)-butoxy]-3,4-dihydro-lH- [ 1 ,8]naphthyridin-2-one
7-[4-(4-[l,7]Naphthyridin-8-yl-piperazin-l-yl)-butoxy]-lH-[l,8]naphthyridin-2- one;
7-[4-(4-Thieno[2,3-c]pyridin-7-yl-piperazin-l-yl)-butoxy]-3,4-dihydro-lH- [l,8]naphthyridin-2-one; and 7-[4-(4-Thieno[2,3-c]pyridin-7-yl-piperazin-l-yl)-butoxy]-lH- [ 1 ,8]naphthyridin-2-one.
8. A pharmaceutical composition, comprising: (a) a compound according to claim 1, or a pharmaceutically acceptable salt thereof, and
(b) a pharmaceutically acceptable carrier.
9. A pharmaceutical composition useful for treating a disorder or condition in a mammal, comprising:
(a) a compound according to claim 1, or a pharmaceutically acceptable salt thereof; and
(b) an antidepressant or an anti-anxiety agent, or a pharmaceutically acceptable salt thereof; wherein the disorder or condition is selected from the group consisting of single episodic or recurrent major depressive disorders, dysthymic disorders, depressive neurosis and neurotic depression, melancholic depression; atypical depression; bipolar disorder; cyclothymic disorder; conduct disorder; disruptive behavior disorder; attention deficit hyperactivity disorder; behavioral disturbances associated with mental retardation, autistic disorder, and conduct disorder; anxiety disorders; borderline personality disorder; schizophrenia and other psychotic disorders; delirium, dementia, and amnestic and other cognitive or neurodegenerative disorders; movement disorders, dyskinesias; extra-pyramidal movement disorders; chemical dependencies and additions; behavioral addictions; and ocular disorders, and wherein the compound and the antidepressant or anti-anxiety agent, or pharmaceutically acceptable salts thereof, are present in amounts that render the combination effective in treating such disorder or condition.
10. A method for treating a disorder or condition in a mammal, comprising administering to the mammal an effective amount of a compound according to claim 1, or a pharmaceutically acceptable salt thereof, wherein the disorder or condition is selected from the group consisting of single episodic or recurrent major depressive disorders, dysthymic disorders, depressive neurosis and neurotic depression, melancholic depression; atypical depression; bipolar disorder; cyclothymic disorder; conduct disorder; disruptive behavior disorder; attention deficit hyperactivity disorder; behavioral disturbances associated with mental retardation, autistic disorder, and conduct disorder; anxiety disorders; borderline personality disorder; schizophrenia and other psychotic disorders; delirium, dementia, and amnestic and other cognitive or neurodegenerative disorders; movement disorders, dyskinesias; extra-pyramidal movement disorders; chemical dependencies and additions; behavioral addictions; and ocular disorders.
11. The method according to claim 10, wherein the disorder or condition is selected from the group consisting of schizophrenia, schizoaffective disorder, delusional disorder, substance-induced psychotic disorder, brief psychotic disorder, shared psychotic disorder, psychotic disorder due to a general medical condition, and schizophreniform disorder.
12. A compound according to claim 1 for use as a medicament.
13, A compound according to claim 1 for use in the treatment of a disorder or condition selected from the group consisting of single episodic or recurrent major depressive disorders, dysthymic disorders, depressive neurosis and neurotic depression, melancholic depression; atypical depression; bipolar disorder; cyclothymic disorder; conduct disorder; disruptive behavior disorder; attention deficit hyperactivity disorder; behavioral disturbances associated with mental retardation, autistic disorder, and conduct disorder; anxiety disorders; borderline personality disorder; schizophrenia and other psychotic disorders; delirium, dementia, and amnestic and other cognitive or neurodegenerative disorders; movement disorders, dyskinesias; extra-pyramidal movement disorders; chemical dependencies and additions; behavioral addictions; and ocular disorders.
14. A composition comprising a compound according to claim 1 for use as a medicament.
15. A composition comprising a compound according to claim 1 for use in the treatment of a disorder or condition selected from the group consisting of single episodic or recurrent major depressive disorders, dysthymic disorders, depressive neurosis and neurotic depression, melancholic depression; atypical depression; bipolar disorder; cyclothymic disorder; conduct disorder; disruptive behavior disorder; attention deficit hyperactivity disorder; behavioral disturbances associated with mental retardation, autistic disorder, and conduct disorder; anxiety disorders; borderline personality disorder; schizophrenia and other psychotic disorders; delirium, dementia, and amnestic and other cognitive or neurodegenerative disorders; movement disorders, dyskinesias; extra-pyramidal movement disorders; chemical dependencies and additions; behavioral addictions; and ocular disorders.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US65504005P | 2005-02-22 | 2005-02-22 | |
| US60/655,040 | 2005-02-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006090272A1 true WO2006090272A1 (en) | 2006-08-31 |
Family
ID=36658633
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2006/000476 WO2006090272A1 (en) | 2005-02-22 | 2006-02-14 | Isoquinoline [1,8]naphthyridin-2-ones and related compounds for treatment of schizophrenia |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2006090272A1 (en) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007099828A1 (en) * | 2006-02-23 | 2007-09-07 | Shionogi & Co., Ltd. | Nirogenous heterocyclic derivatives substituted with cyclic groups |
| US7887784B2 (en) * | 2007-03-21 | 2011-02-15 | University Of Montana | 1-[(2′-substituted)-piperazin-1′ -yl]-isoquinolines as norepinephrine transporter inhibitor therapeutics and positron emission tomography imaging agents |
| CN102134240A (en) * | 2010-06-29 | 2011-07-27 | 嘉兴宜博生物医药科技有限公司 | Synthesis method of 6-fluorine-7-azaindole |
| US8507478B2 (en) | 2008-10-10 | 2013-08-13 | Actelion Pharmaceuticals Ltd. | Oxazolidinyl antibiotics |
| US20150072974A1 (en) * | 2012-05-09 | 2015-03-12 | Sunovion Pharmaceuticals Inc. | Heteroaryl compounds and methods of use thereof |
| US9156822B2 (en) | 2010-07-02 | 2015-10-13 | The University Of North Carolina At Chapel Hill | Functionally selective ligands of dopamine D2 receptors |
| JP2017537938A (en) * | 2014-12-17 | 2017-12-21 | アジェンデ・キミケ・リウニテ・アンジェリニ・フランチェスコ・ア・チ・エレ・ア・エフェ・ソシエタ・ペル・アチオニAziende Chimiche Riunite Angelini Francesco A.C.R.A.F.Societa Per Azioni | New antibacterial compounds |
| US9902703B2 (en) | 2015-07-01 | 2018-02-27 | Crinetics Pharmaceuticals, Inc. | Somatostatin modulators and uses thereof |
| US10154988B2 (en) | 2012-11-14 | 2018-12-18 | The Johns Hopkins University | Methods and compositions for treating schizophrenia |
| US11028068B2 (en) | 2017-07-25 | 2021-06-08 | Crinetics Pharmaceuticals, Inc. | Somatostatin modulators and uses thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0367141A2 (en) * | 1988-10-31 | 1990-05-09 | Otsuka Pharmaceutical Co., Ltd. | Carbostyril derivatives |
| WO2005019215A1 (en) * | 2003-08-22 | 2005-03-03 | Warner-Lambert Company Llc | [1,8]naphthyridin-2-ones and related compounds for the treatment of schizophrenia |
-
2006
- 2006-02-14 WO PCT/IB2006/000476 patent/WO2006090272A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0367141A2 (en) * | 1988-10-31 | 1990-05-09 | Otsuka Pharmaceutical Co., Ltd. | Carbostyril derivatives |
| WO2005019215A1 (en) * | 2003-08-22 | 2005-03-03 | Warner-Lambert Company Llc | [1,8]naphthyridin-2-ones and related compounds for the treatment of schizophrenia |
Non-Patent Citations (3)
| Title |
|---|
| KIKUCHI T ET AL: "PHARMACOLOGICAL PROFILE OF CPC-14597, A NOVEL ANTI-PSYCHOTIC DRUG (1): PRESYNAPTIC DOPAMINE AUTORECEPTOR AGONISTIC ACTIVITY AND POSTSYNAPTIC DOPAMINE D2 RECEPTOR ANTAGONISTIC ACTIVITY", JAPANESE JOURNAL OF PHARMACOLOGY, THE JAPANESE PHARMACOLOGICAL SOCIETY, KYOTO, JP, vol. 67, no. SUPPL 1, 1995, pages 144P,ANP1 - 235, XP008005977, ISSN: 0021-5198 * |
| MCCORT G ET AL: "Synthesis and SAR of 3- and 4-substituted quinolin-2-ones: discovery of mixed 5-HT(1B)/5-HT(2A) receptor antagonists.", BIOORGANIC & MEDICINAL CHEMISTRY. AUG 2001, vol. 9, no. 8, August 2001 (2001-08-01), pages 2129 - 2137, XP002391169, ISSN: 0968-0896 * |
| ORJALES A ET AL: "New 3-benzisothiazolyl and 3-benzisoxazolylpiperazine derivatives with atypical antipsychotic binding profile", EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, EDITIONS SCIENTIFIQUE ELSEVIER, PARIS, FR, vol. 37, no. 9, September 2002 (2002-09-01), pages 721 - 730, XP004383155, ISSN: 0223-5234 * |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2007099828A1 (en) * | 2006-02-23 | 2009-07-16 | 塩野義製薬株式会社 | Nitrogen-containing heterocyclic derivatives substituted with cyclic groups |
| US7935706B2 (en) | 2006-02-23 | 2011-05-03 | Shionogi & Co., Ltd. | Nitrogen-containing heterocycle derivatives substituted with cyclic group |
| WO2007099828A1 (en) * | 2006-02-23 | 2007-09-07 | Shionogi & Co., Ltd. | Nirogenous heterocyclic derivatives substituted with cyclic groups |
| US7887784B2 (en) * | 2007-03-21 | 2011-02-15 | University Of Montana | 1-[(2′-substituted)-piperazin-1′ -yl]-isoquinolines as norepinephrine transporter inhibitor therapeutics and positron emission tomography imaging agents |
| US8507478B2 (en) | 2008-10-10 | 2013-08-13 | Actelion Pharmaceuticals Ltd. | Oxazolidinyl antibiotics |
| CN102134240A (en) * | 2010-06-29 | 2011-07-27 | 嘉兴宜博生物医药科技有限公司 | Synthesis method of 6-fluorine-7-azaindole |
| US9156822B2 (en) | 2010-07-02 | 2015-10-13 | The University Of North Carolina At Chapel Hill | Functionally selective ligands of dopamine D2 receptors |
| US20190031685A1 (en) * | 2012-05-09 | 2019-01-31 | Sunovion Pharmaceuticals Inc. | Heteroaryl compounds and methods of use thereof |
| US9951088B2 (en) * | 2012-05-09 | 2018-04-24 | Sunovion Pharmaceuticals Inc. | D2 receptor modulators and methods of use thereof in the treatment of diseases and disorders |
| US20150072974A1 (en) * | 2012-05-09 | 2015-03-12 | Sunovion Pharmaceuticals Inc. | Heteroaryl compounds and methods of use thereof |
| US10154988B2 (en) | 2012-11-14 | 2018-12-18 | The Johns Hopkins University | Methods and compositions for treating schizophrenia |
| EP3610890A1 (en) | 2012-11-14 | 2020-02-19 | The Johns Hopkins University | Methods and compositions for treating schizophrenia |
| US10624875B2 (en) | 2012-11-14 | 2020-04-21 | The Johns Hopkins University | Methods and compositions for treating schizophrenia |
| JP2017537938A (en) * | 2014-12-17 | 2017-12-21 | アジェンデ・キミケ・リウニテ・アンジェリニ・フランチェスコ・ア・チ・エレ・ア・エフェ・ソシエタ・ペル・アチオニAziende Chimiche Riunite Angelini Francesco A.C.R.A.F.Societa Per Azioni | New antibacterial compounds |
| US10369130B2 (en) * | 2014-12-17 | 2019-08-06 | AZIEN DE CHIMICHE RIUNITE ANGELINI FRANCESCO A.C.R.A.F. S.p.A. | Antibacterial compounds |
| US9902703B2 (en) | 2015-07-01 | 2018-02-27 | Crinetics Pharmaceuticals, Inc. | Somatostatin modulators and uses thereof |
| US11028068B2 (en) | 2017-07-25 | 2021-06-08 | Crinetics Pharmaceuticals, Inc. | Somatostatin modulators and uses thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006090272A1 (en) | Isoquinoline [1,8]naphthyridin-2-ones and related compounds for treatment of schizophrenia | |
| WO2006090273A2 (en) | [1,8]naphthyridin-2-ones and related compounds with keto or hydroxyl linkers for the treatment of schizophrenia | |
| US20040138230A1 (en) | Heterocyclic substituted piperazines for the treatment of schizophrenia | |
| AU2009335019A1 (en) | Substituted 1H-pyrazolo[3,4-d]pyrimidine-6-amine compounds | |
| JP2007503386A (en) | [1,8] Naphthyridin-2-one and related compounds for the treatment of schizophrenia | |
| US20030120072A1 (en) | Decahydro-isoquinolines | |
| AU2017208119B2 (en) | 6,7,8,9-tetrahydro-5H-pyrido[2,3-d]azepine dopamine D3 ligands | |
| IE842476L (en) | Antidepressant 1, 2, 4 - triazolone compounds | |
| US20060234997A1 (en) | Tetrahydro-pyridoazepin-8-ones and related compounds for the treatment of schizophrenia | |
| US20170224713A1 (en) | Heterocyclic compounds for treating or preventing disorers caused by reduced neurotransmission of serotonin, norephnephrine or dopamine | |
| TW555758B (en) | Azabicyclic 5HT1 receptor ligands | |
| JPH0641095A (en) | Antidepressant 3-halophenylpiperazinyl-propyl derivative of substituted triazolone and triazoledione | |
| JPH04234860A (en) | Thiazine (or oxazine) derivative, its production and synthetic intermediate therefor | |
| TW210338B (en) | ||
| US20040067960A1 (en) | Heterocyclic substituted piperazines for the treatment of schizophrenia | |
| WO2018139471A1 (en) | Dibenzodiazepine derivative | |
| US20040142933A1 (en) | Oxindole substituted piperazine derivatives | |
| JP2021104931A (en) | Dibenzoazepine derivative having nitrogen-containing heterocycle | |
| TW201348215A (en) | Heterocyclic compound | |
| KR20240035517A (en) | Hydrogenated Quinoxaline | |
| PL183618B1 (en) | Derivatives of tetracyclic substituted oxazepin and thiazepin exhibiting affinity in respect to 5-ht2 receptors | |
| CN117062815A (en) | VMAT2 inhibitors and methods of use | |
| JP2020055752A (en) | Dibenzo diazepine derivative | |
| JPS62277356A (en) | Novel imide derivative and production thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06710499 Country of ref document: EP Kind code of ref document: A1 |