WO2006069798A1 - Process for the preparation of chiral amines - Google Patents
Process for the preparation of chiral amines Download PDFInfo
- Publication number
- WO2006069798A1 WO2006069798A1 PCT/EP2005/014170 EP2005014170W WO2006069798A1 WO 2006069798 A1 WO2006069798 A1 WO 2006069798A1 EP 2005014170 W EP2005014170 W EP 2005014170W WO 2006069798 A1 WO2006069798 A1 WO 2006069798A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- group
- process according
- cyclo
- compound according
- Prior art date
Links
- 0 CC(C)(C(N)=O)N=C(*)* Chemical compound CC(C)(C(N)=O)N=C(*)* 0.000 description 2
- WYVWLIXZTPLZCL-ISKYKNHCSA-N C=C[C@H](C(N)=O)/N=C/c(cc1)cc2c1OCO2 Chemical compound C=C[C@H](C(N)=O)/N=C/c(cc1)cc2c1OCO2 WYVWLIXZTPLZCL-ISKYKNHCSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/12—Preparation of carboxylic acid amides by reactions not involving the formation of carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/30—Preparation of optical isomers
- C07C227/32—Preparation of optical isomers by stereospecific synthesis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C249/00—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C249/02—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton of compounds containing imino groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/44—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D317/46—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D317/48—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring
- C07D317/50—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to atoms of the carbocyclic ring
Definitions
- the present invention relates to a process for the preparation of chiral amines, which amines are defined in this application as chiral beta-amino acid esters or amides, or derivatives hereof e.g. acids, or chiral beta amino ketones or chiral beta amino nitriles. These compounds are used e.g. in the manufacture of pharmaceutical or agrochemically active compounds.
- a process for the preparation of chiral amines is known from EP-A-
- the BrZnCH 2 CO 2 -Su reagent, in US 5,840,961 referred to as a Reformatsky agent, is prepared separately by activation of zinc with dibromoethane in tetrahydrofuran, followed by reaction with tert-butyl bromoacetate and isolation by filtration. Given this Reformatsky agent's low stability it has to be stored at low temperature (i.e. -10 0 C, as mentioned in Example 2 of US 5,840,961). Disadvantage of the process as disclosed in US 5,840,961 is that said process comprises many reaction steps resulting in a laborious process for the preparation of chiral beta-amino acids.
- Object of the present invention is to provide a process for the preparation of chiral amines, which process has less process steps than the process as disclosed in US 5,840,961.
- R 1, R 2 H, a substituted or unsubstituted: (cyclo)alkyl group, (cyclo)alkenyl group, aryl group, cyclic of acyclic heteroalkyl group or heteroaryl group with one or more N, O, S atoms whereby Ri ⁇ R 2 is contacted with an enantiomerically enriched phenylglycine amide according to formula (II)
- R 4 a substituted or unsubstituted phenyl or naphtyl group
- R 5 H or alkyl with 1-6 carbon atoms
- R 3 COOR 8 , CONR 8 R 9 , COSR 8 , COR 8 or CN R 6 , R 7 independent of each other, H, halogen (for example Cl, Br, I or F), a substituted or unsubstituted: (cyclo)alkyl group, (cyclo)alkenyl group, aryl group, cyclic or acyclic heteroalkyl group or a heteroaryl group with one or more N, O, S atoms.
- halogen for example Cl, Br, I or F
- R 8 , R 9 independent of each other, a substituted or unsubstituted: (cyclo)alkyl group, (cyclo)alkenyl group, aryl group, cyclic or acyclic heteroalkyl group or a heteroaryl group with one or more N, O, S atoms.
- the process according to the invention has less process steps than the process as disclosed in US 5,840,961.
- process according to the invention refrains from using environmentally undesirable compounds such as e.g. lead tetra acetate as used in US 5,840,961. Furthermore the process according to the invention is not restricted to the use of t-butylesters, as in US 5,840,961.
- Suitable compounds according to formula (I) are aldehydes or ketones for example, benzaldehyde, 4-methoxybenzaldehyde, 3,4-dimethoxybenzaldehyde, 4- fluorobenzaldehyde, 4-trifluoromethylbenzaldehyde, piperonal, acetaldehyde, cyclopropylaldehyde or isopropylaldehyde.
- the R 1 , R 2 group may be substituted with for example alkoxy, halo, trifluoromethyl, alkyl or alkylamino groups or aryl or arylamino groups
- R 1 should not equal R 2 in order to obtain chiral amines with the process according to the invention.
- R 1 is a (cyclo)alkyl or (hetero)aryl group with 3-10 carbon atoms. In the process according to the invention this gives a high yield of the compound according to formula (V).
- R 2 comprises H or from 3 to 6 carbon atoms.
- Compounds according to formula (II) are chiral compounds wherein R 4 is a substituted or unsubstituted phenyl- or naphthyl-group, and R 5 is H or an alkyl group
- the phenyl- or naphthyl-group of R 4 may be monosubstituted or polysubstituted with for example halogen, in particular chlorine or bromine, a hydroxy group, an alkyl or (hetero)aryl group with for example 1-10 carbon atoms and/or an alkoxy group or acyloxy group with for example 1-10 carbon atoms.
- R 5 is H or an alkyl group with 1 to 6 carbon atoms, more preferably R 5 is H. In the process according to the invention this gives a high enantiomeric excess, as defined below, of a compound according to formula (V).
- a particular preferred compound according to formula (II) is a compound where R 4 is a phenyl-group and R 5 is a H, hereinafter referred to as phenyl glycine amide (PGA).
- PGA phenyl glycine amide
- An advantage of PGA is that it gives compounds according to formula (V) that are generally solids. This means that compounds of formula (IV), which are not completely diastereomerically pure, can be purified to diastereomerically pure compounds in one crystallization step.
- Examples of possible substituents on R 6 , R 7 , Rs or R 9 are: alkoxy, halo, trifluoromethyl, alkyl, aryl, alkylamino and arylamino.
- the enantiomeric excess (ee) is defined as the difference between the amounts of enantiomers divided by the sum of the amounts of the enantiomers, which quotient can be expressed as a percentage after multiplication by 100.
- ee the enantiomeric excess
- the compound according to formula (II) is an enantiomerically enriched compound, which means that the compound according to formula (II) has an enantiomeric excess of at least 80%, more preferably of at least 90%, most preferably of at least 98%.
- the compounds according to formula (I) and (II) are contacted, preferably in a solvent.
- a solvent Preferably water, alcohols, esters, aliphatic or aromatic hydrocarbons or haloalkanes are used as solvents. More preferred solvents include methanol, ethanol, toluene, dichloromethane, ethylacetate or isopropylacetate. In these solvents the compounds according to formula (I) and (II) can be dissolved easily.
- a catalyst may be used upon contacting the compounds according to formula (I) and (II).
- Preferred catalysts include acids, such as for example p- toluenesulphonic acid, pyridinium-p-toluenesulphonate or Lewis acids such as for example titaniumtetrachloride, titaniumtetra-isopropoxide, BF 3 .O(CH 2 CH 3 ) 2 or Si(OCH 2 CH 3 ) 4 ..
- the temperature at which the compounds according to formula (I) and (II) are contacted preferably is between 0-140°C, more preferably between 20-120 0 C.
- a reaction mixture comprising compound (III) is formed.
- Compound (III) is an imine, and in this application also referred to as a 'Schiff base'.
- the reaction mixture comprising compound (III) may be purified before the subsequent contacting with the Reformatsky reagent XZnCR 6 R 7 R 3 .
- a beta amino acid ester is produced with the process according to the invention. If required the beta amino acid ester may be hydrolyzed to the beta amino acid with techniques well known to the skilled man.
- R 3 is CONR 8 R 9 , or COSR 8 , a beta amino acid amide respectively a beta amino acid thio ester is produced with the process according to the invention.
- R 3 is COR 8 , or CN a beta amino ketone respectively a beta amino nitril is produced with the process according to the invention.
- R 3 is -COOR 8 , because the chiral beta amino acid esters produced thereof can be incorporated directly into pharmaceutical or agrochemically active compounds.
- the Reformatsky reagent XZnCR 6 R 7 R 3 may be prepared separately or may be prepared in situ in the process according to the invention.
- X Br.
- the advantage hereof is that this gives the fastest reaction with the compound according to formula (III).
- these Reformatsky reagents BrZnCR 6 R 7 R 3 give the highest yield upon reacting with the compound according to formula (III).
- the Reformatsky reagent XZnCR 6 R 7 R 3 is prepared separately this can suitably done with methods as disclosed in "Organozinc reagents in Organic Synthesis", Erdik, E, Ed., CRC Press, 1996, p 55-61.
- the Reformatsky reagent XZnCR 6 R 7 R 3 Js preferably prepared in situ in the process according to the invention by heating Zn and XCR 6 R 7 R 3 preferably at a temperature between 40 and 120 0 C, which in situ formed Reformatsky reagent then subsequently reacts with the compound according to formula (III).
- Zinc may be used in any form, for example as dust, granules, foil or wire.
- the zinc may be used without prior activation, however if the reactivity of the zinc would not be sufficient and consequently resulting in a low yield, the zinc may be activated in situ by methods known to the skilled man, for example by treatment of the zinc with dibromoethane, iodine, trimethylsilylchloride, HCI or by application of ultrasound.
- Zinc preferably is used in 0.8-10 equivalents related to compound (III), more preferably in 1.0-6 equivalents and most preferred in 1.2-4 equivalents. This results in a good balance between high speed of reaction and limited amounts of zinc to be removed.
- a second method of in situ preparation of the Reformatsky reagent XZnCR 6 R 7 R 3 is by contacting XCR 6 R 7 R 3 with wherein Ri 0 is alkyl, alkenyl or aryl, in the presence of an additive comprising an element from group 4-12 of the periodic table with the compound according to formula III.
- 0 ) 2 Zn are (C 2 H 5 ) 2 Zn and (CH 3 ) 2 Zn because these reagents give fast formation of the Reformatsky reagent XZnCR 6 R 7 R 3 .
- Preferred additives are Cr(lll)(acetylacetonatate) 3 , Fe(lll)(acetylacetonate) 3 , Ni(ll)(acetylacetonate) 2 , PdCI 2 , RhCI(PPh 3 ), IrCI 2 and CeCI 3 . More preferably, Fe(lll)(acetylacetonate) 3 or Ni(ll)(acetylacetonate) 2 are used, because a higher yield is obtained when these additives are present during contacting the Reformatsky reagent with the compound according to formula (III).
- the additive is preferably added in an amount of less than 20 mol% based on the total amount of compound according to formula (III), more preferably in an amount of less than 10 mol% and most preferred in an amount of less than 5 mol%.
- XZnCR 6 R 7 R 3 is that the process according to the invention comprises even less process steps than the process as disclosed in US 5,840,961.
- An additional advantage of the second method of in situ preparation is that a high yield in the compound according to formula (V) is obtained.
- the reaction mixture comprising compound (III) is subsequently contacted with the Reformatsky reagent XZnCR 6 R 7 R 3 in the event that this reagent is produced separately.
- the temperature at which the compound according to formula (III) and the Reformatsky reagent are contacted preferably is between 0 and 140 0 C, more preferably between 20-120 0 C.
- a reaction mixture comprising compound (IV) is formed.
- this compound may be recrystallized, with known techniques, as such or as a salt from a suitable solvent.
- Suitable salts include those formed with HCI, HBr, acetic acid, p-toluenesulphonic acid, benzenesulphonic acid, tartaric acid.
- Suitable solvents include heptane, methylisobutylketone, acetone, methyltertbutylether, isopropylacetate, ethylacetate, isopropanol, ethanol or methanol.
- Hydrogenolysis may suitably be done with H 2 using for example Pd as a catalyst.
- the chiral centre comprising R 4 and R 5 is split off from the compound according to formula (IV), resulting in the corresponding chiral amine according to formula (V).
- Temperature during hydrogenolysis is chosen preferably between 0 and 40 0 C, more preferably between 20 and 30 0 C. This results in a high yield of the chiral amine according to formula (V)
- Another method of converting the compound according to formula (IV) into the corresponding chiral amine is by means of a so-called retro Strecker reaction.
- This retro Strecker reaction is a one step reaction, in which the amide group of the compound according to formula (IV) is first converted to a nitril, by a dehydrating agent, for example by thionylchloride, phosphoroxy trichloride, phosphorpentachloride, p-toluenesulphonic acid/pyridine, Vilsmeyer reagent/Et 3 N or pyridine, cyanuric chloride, acetic acid anhydride, trifluoroacetic acid anhydride and other dehydrating agents, for example as described in March, Advanced Organic Chemistry, 5 th Edition, Wiley Interscience, Eds.M.B. Smith and J.
- a dehydrating agent for example by thionylchloride, phosphoroxy trichloride, phosphorpentachloride, p-toluenesulphonic acid/pyridine, Vilsmeyer reagent/Et 3 N or pyridine, cyanuric chloride,
- a retro Strecker reaction i.e. elimination of HCN, for example by addition of a base or by heating.
- suitable bases are (earth) alkali metal hydroxides, (earth) alkalimetal carbonates, (earth) alkalimetal phospates and organic bases.
- Suitable bases are for instance KOH, Na 2 CO 3 or K 2 CO 3 ,followed by hydrolysis with methods known to the skilled man of the obtained imine to the chiral amine.
- Hydrolysis may suitable be done by e.g. heating the imine to temperatures up to 120 0 C, addition of acids, or transimination by for example NH 2 OH. HCI or NaHSO 3 .
- This retro Strecker reaction is carried out in one vessel without isolation of intermediates thereby making it a one step reaction.
- a further method of converting the compound according to formula (IV) into the corresponding chiral amine is by means of oxidation followed by hydrolysis of the imine to the chiral amine according to formula (V).
- oxidation in this application is understood treatment with peracids, O 3 , metal-based oxidative agents such as e.g. KMnO 4 and hypochlorites such as e.g. NaOCI.
- metal-based oxidative agents such as e.g. KMnO 4
- hypochlorites such as e.g. NaOCI.
- Second step Synthesis of (ff)-3-(Tfffl-2-amino-2-oxo-1-phenylethv ⁇ amino)-3-phenyl- propionic acid ethyl ester: Reformatsky reaction of compound from Example Ia with Zn, BrCH 2 COOEt to form: Hy CONH 2 ⁇ COOEt
- Example Ia The Schiff base of Example Ia (4.8 g, 20 mmol) was dissolved in CH 2 CI 2 (50 ml_) and Zn dust was added (5.2, 80 mmol). The reaction mixture was heated until reflux and BrCH 2 COOEt (10.4 g, 62 mmol) was dosed in ca 200 min. The mixture was then stirred under reflux (ca 42°C) for 2 h. After cooling the reaction mixture was filtered and added to a solution of 4N aqueous HCI (75 ml_). After stirring for approximately 15 h, the precipitated solid was isolated by filtration to give the product as HCI salt (5.2 g, 72%, cfe 96%).
- second step lb-2 as given above may be employed:
- Second step Synthesis of (fi)-3-(r(f?)-2-amino-2-oxo-1-phenylethyl1amino)-3-phenyl- propionic acid ethyl ester: Reformatsky reaction of compound from Example Ia with Et 2 Zn, BrCH 2 COOEt in CH 2 CI 2 as solvent.
- Example Ia The Schiff base of Example Ia (8.6 g, 36 mmol) was dissolved in CH 2 CI 2 (100 mL) and cooled to ca. -30 0 C. The following reagents were added in succession: ZnEt 2 , 1 M in hexane (91 mL, 2.5 eq), BrCH 2 COOEt (4.9 mL, 1.2 eq) and Ni(acetylacetonatate) 2 (0.5 g, 5 mol%), either drop wise or in portions. The reaction mixture was allowed to warm to room temperature in a controlled fashion and stirred for 3 h, then poured onto about 150 g of crushed ice.
- Example Ib The compound of Example Ib (1.54 g, 4.7 mmol) was dissolved in THF (40 ml_). Et 3 N (7.2 mL, 12 eq) was added and the mixture was cooled on ice. POCI 3 (0.9 ml_, 2.2 eq) was added dropwise, then the mixture was allowed to warm to room temperature, stirred for 1 h and subsequently refluxed for 5 h. At room temperature, a solution of NH 2 OH-HCI (0.91 g, 3 eq) in H 2 O (50 mL) was added and the resulting mixture was stirred for 16 h. The pH was adjusted to 1 with 30% HCI and THF was removed in vacuo.
- third step lc-2 instead of third step lc-1 , third step lc-2 as given above may be employed: lc-2: Third step: Synthesis of (f?)-3-amino-3-phenyl-propionic acid ethyl ester via oxidation
- Example Ib The product of Example Ib (0.84 g, 2.37 mmol) was dissolved in acetone (40 mL). KMnO 4 (0.94 g, 2.5 eq) was added and the mixture was stirred for 16 h, then 6 mL of 1 M aqueous NaOH (2.5 eq) was added and the mixture was filtered over Celite. Acetone was removed in vacuo and the residue was partitioned between CH 2 CI 2 (40 mL) and H 2 O (40 mL). The layers were separated, the organic layer was dried on Na 2 SO 4 and volatiles were removed in vacuo, yielding a yellow oil (1.03 g).
- Phenylglycine amide (50 g, 0.31 mol, assay 94%) and piperonal (50 g, 0.30 mol) were added to 400 mL of CH 3 OH. After heating to 30 0 C a clear solution was obtained. The mixture was stirred at room temperature for 18 h and a precipitate was observed. After filtration, washing with 2 x 25 mL of CH 3 OH, a white solid was obtained (73 g, 86 %).
- Second step Synthesis of (fi)-3-(r(R)-2-amino-2-oxo-1-phenylethv ⁇ amino)-3- r(benzo ⁇ .31dioxol-5-yl)-3-propionic acid ethyl ester: Reformatsky reaction of compound from Example Ha with Et 2 Zn, BrCH 2 COOEt to form:
- Hc Third step: Synthesis of (ffl-3-amino-3-benzori ,31dioxol-5-y
- Phenylglycine amide (30 g, 0.19 mol, assay 94%) and 5 g Na 2 SO 4 were added to 250 mL of CH 2 CI 2 .
- acetaldehyde (8.8 g, 0.20 mol).
- the mixture was stirred at room temperature for 1 h and the solids were removed by filtration.
- the CH 2 CI 2 was removed by evaporation and the residu stirred in 150 mL EtOAc.
- the solid was filtrated and washed with 2 x 25 ml_ of EtOAc. After drying the imine was obtained as a white solid (26.2 g, 78.4%).
- Step IVa Synthesis of Schiff base: see example Ia
- Step IVb Synthesis of (f?)-1-(r(f?V2-amino-2-oxo-1-phenylethyllamino)-4,4-dimethyl-1- phenyl-pentane-3-one: Reformatsky reaction of compound from Example IVa with Et 2 Zn, BrCH 2 COOEt i n CH 2 CI 2 as solvent to form :
- Example. IVa The Schiff base of Example. IVa (0.87 g, 3.65 mmol) was dissolved in CH 2 CI 2 (40 mL). The following reagents were added in succession: Et 2 Zn, (1 M in hexane, (9.1 mL, 2.5 eq), BrCH 2 COf-Bu (0.59 mL, 1.2 eq), Ni(acetylacetonatate) 2 (0.05 g, 5 mol%) and PPh 3 (0.1O g, 10 mol%). The resulting orange solution was stirred for 2 h, then saturated NH 4 CI (50 mL) was added.
- Step IVc Synthesis of (R)-1-Amino-4.4-dimethyl-1-phenyl-pentane-3-one via retro Strecker method to form:
Abstract
The invention relates to a process for the preparation of a chiral amine, whereby a ketone or aldehyde is contacted with an enantiomerically enriched phenylglycine amide to give an imine, whereby the imine is subsequently contacted with a Reformatsky reagent and the formed compound is subsequently converted into a chiral amine by means of hydrogenolysis, oxidation or a retro Strecker method.
Description
PROCESS FOR THE PREPARATION OF CHIRAL AMINES
The present invention relates to a process for the preparation of chiral amines, which amines are defined in this application as chiral beta-amino acid esters or amides, or derivatives hereof e.g. acids, or chiral beta amino ketones or chiral beta amino nitriles. These compounds are used e.g. in the manufacture of pharmaceutical or agrochemically active compounds. A process for the preparation of chiral amines is known from EP-A-
0355819 which publication relates specifically to the preparation of chiral beta-amino acid esters. In this disclosure a process is given comprising the steps of 1) reacting an aldehyde with an amine to produce a Schiff base, 2) reacting said Schiff base with a methyl haloacetate and a metal such as Zn to produce a diastereomeric mixture of a beta- lactam, 3) hydrolysing said beta-lactam to produce a diastereomeric mixture of a first beta- amino acid, 4) esterifying said first beta-amino acid, 5) isolating one isomer of the ester of said diastereomeric mixture of said first beta-amino acid, 6) hydrogenolysing said ester to produce one stereoisomer of a second beta amino acid. This process however is known to give very low diastereoselectivities. A process for the preparation of chiral amines is furthermore known from
US 5,840,961. In this disclosure, a 4-step process is described in which process in a first step an aldehyde is reacted with (R)- or (S)-phenylglycinol as a chiral auxiliary to form an imino-alcohol; in a second step this imino-alcohol is reacted with BrZnCH2CO2-Su to form an amino-alcohol; in a third step the chiral auxiliary is removed from the amino alcohol by reaction with sodium periodate or lead tetraacetate to form an imine and finally in a fourth step, the imine is hydrolyzed in the presence of para toluene sulphonic acid to obtain an ester of an (R)-or (S)-beta amino acid.
The BrZnCH2CO2-Su reagent, in US 5,840,961 referred to as a Reformatsky agent, is prepared separately by activation of zinc with dibromoethane in tetrahydrofuran, followed by reaction with tert-butyl bromoacetate and isolation by filtration. Given this Reformatsky agent's low stability it has to be stored at low temperature (i.e. -10 0C, as mentioned in Example 2 of US 5,840,961).
Disadvantage of the process as disclosed in US 5,840,961 is that said process comprises many reaction steps resulting in a laborious process for the preparation of chiral beta-amino acids.
Object of the present invention is to provide a process for the preparation of chiral amines, which process has less process steps than the process as disclosed in US 5,840,961.
This object is achieved with a process according to the invention in which process a compound according to formula (I)
O
R-i R2 (i)
in which formula
R1, R2= H, a substituted or unsubstituted: (cyclo)alkyl group, (cyclo)alkenyl group, aryl group, cyclic of acyclic heteroalkyl group or heteroaryl group with one or more N, O, S atoms whereby Ri ≠ R2 is contacted with an enantiomerically enriched phenylglycine amide according to formula (II)

in which formula
R4 = a substituted or unsubstituted phenyl or naphtyl group
R5 = H or alkyl with 1-6 carbon atoms
* = a chiral center, which may have the (R) or (S) configuration to give an imine according to formula (III), in this application also referred to as a 'Schiff base',

The Schiff base of formula (III) is converted into a compound according to formula (IV)

R3 = COOR8, CONR8R9, COSR8, COR8 or CN R6, R7 independent of each other, H, halogen (for example Cl, Br, I or F), a substituted or unsubstituted: (cyclo)alkyl group, (cyclo)alkenyl group, aryl group, cyclic or acyclic heteroalkyl group or a heteroaryl group with one or more N, O, S atoms.
R8, R9 = independent of each other, a substituted or unsubstituted: (cyclo)alkyl group, (cyclo)alkenyl group, aryl group, cyclic or acyclic heteroalkyl group or a heteroaryl group with one or more N, O, S atoms.
Finally the compound according to formula (IV) is then converted into the chiral amine according to formula (V) or an amine salt thereof by hydrogenolysis, oxidation or a retro Strecker method.

The process according to the invention has less process steps than the process as disclosed in US 5,840,961.
An additional advantage is that the process according to the invention uses less costly chiral auxiliaries than those disclosed in US 5,840,961.
Moreover the process according to the invention refrains from using environmentally undesirable compounds such as e.g. lead tetra acetate as used in US 5,840,961. Furthermore the process according to the invention is not restricted to the use of t-butylesters, as in US 5,840,961.
Suitable compounds according to formula (I) are aldehydes or ketones for example, benzaldehyde, 4-methoxybenzaldehyde, 3,4-dimethoxybenzaldehyde, 4- fluorobenzaldehyde, 4-trifluoromethylbenzaldehyde, piperonal, acetaldehyde, cyclopropylaldehyde or isopropylaldehyde. Optionally the R1, R2 group may be substituted with for example alkoxy, halo, trifluoromethyl, alkyl or alkylamino groups or aryl or arylamino groups
Furthermore R1 should not equal R2 in order to obtain chiral amines with the process according to the invention. Preferably R1 is a (cyclo)alkyl or (hetero)aryl group with 3-10 carbon atoms. In the process according to the invention this gives a high yield of the compound according to formula (V).
Preferably R2 comprises H or from 3 to 6 carbon atoms. In the process according to the invention this gives a high yield of the compound according to formula (V). Compounds according to formula (II) are chiral compounds wherein R4 is a substituted or unsubstituted phenyl- or naphthyl-group, and R5 is H or an alkyl group
If so desired, the phenyl- or naphthyl-group of R4 may be monosubstituted or polysubstituted with for example halogen, in particular chlorine or bromine, a hydroxy group, an alkyl or (hetero)aryl group with for example 1-10 carbon atoms and/or an alkoxy group or acyloxy group with for example 1-10 carbon atoms.
Preferably R5 is H or an alkyl group with 1 to 6 carbon atoms, more preferably R5 is H. In the process according to the invention this gives a high enantiomeric excess, as defined below, of a compound according to formula (V).
A particular preferred compound according to formula (II) is a compound where R4 is a phenyl-group and R5 is a H, hereinafter referred to as phenyl glycine amide (PGA). An advantage of PGA is that it gives compounds according to formula (V) that are generally solids. This means that compounds of formula (IV), which are not completely diastereomerically pure, can be purified to diastereomerically pure compounds in one crystallization step. Examples of possible substituents on R6, R7, Rs or R9 are: alkoxy, halo, trifluoromethyl, alkyl, aryl, alkylamino and arylamino.
The enantiomeric excess (ee) is defined as the difference between the amounts of enantiomers divided by the sum of the amounts of the enantiomers, which quotient can be expressed as a percentage after multiplication by 100. Depending on the desired chirality of the compound according to formula
(V), either an (R)- or (S)-configuration of the compound according to formula (II) may be chosen.
In the present invention the compound according to formula (II) is an enantiomerically enriched compound, which means that the compound according to formula (II) has an enantiomeric excess of at least 80%, more preferably of at least 90%, most preferably of at least 98%.
In the process according to the invention the compounds according to formula (I) and (II) are contacted, preferably in a solvent. Preferably water, alcohols, esters, aliphatic or aromatic hydrocarbons or haloalkanes are used as solvents. More preferred solvents include methanol, ethanol, toluene, dichloromethane, ethylacetate or isopropylacetate. In these solvents the compounds according to formula (I) and (II) can be dissolved easily. Optionally a catalyst may be used upon contacting the compounds according to formula (I) and (II). Preferred catalysts include acids, such as for example p- toluenesulphonic acid, pyridinium-p-toluenesulphonate or Lewis acids such as for example titaniumtetrachloride, titaniumtetra-isopropoxide, BF3.O(CH2CH3)2 or Si(OCH2CH3)4..
The temperature at which the compounds according to formula (I) and (II) are contacted preferably is between 0-140°C, more preferably between 20-1200C.
Upon contacting the compounds according to formula (I) and formula (II) in the process according to the invention a reaction mixture comprising compound (III) is formed. Compound (III) is an imine, and in this application also referred to as a 'Schiff base'. The reaction mixture comprising compound (III) may be purified before the subsequent contacting with the Reformatsky reagent XZnCR6R7R3.
In the next step, the Reformatsky reagent XZnCR6R7Rs, is contacted with the compound according to formula (III).
In the event that R3 is -COOR8, a beta amino acid ester is produced with the process according to the invention. If required the beta amino acid ester may be hydrolyzed to the beta amino acid with techniques well known to the skilled man.
In the event that R3 is CONR8R9, or COSR8, a beta amino acid amide respectively a beta amino acid thio ester is produced with the process according to the invention.
In the event that R3 is COR8, or CN a beta amino ketone respectively a beta amino nitril is produced with the process according to the invention.
Preferably R3 is -COOR8, because the chiral beta amino acid esters produced thereof can be incorporated directly into pharmaceutical or agrochemically active compounds.
The Reformatsky reagent XZnCR6R7R3, may be prepared separately or may be prepared in situ in the process according to the invention. In the Reformatsky reagent XZnCR6R7R3, preferably X = Br. The advantage hereof is that this gives the fastest reaction with the compound according to formula (III). Moreover these Reformatsky reagents BrZnCR6R7R3 give the highest yield upon reacting with the compound according to formula (III). In the case that the Reformatsky reagent XZnCR6R7R3 is prepared separately this can suitably done with methods as disclosed in "Organozinc reagents in Organic Synthesis", Erdik, E, Ed., CRC Press, 1996, p 55-61.
The Reformatsky reagent XZnCR6R7R3Js preferably prepared in situ in the process according to the invention by heating Zn and XCR6R7R3 preferably at a temperature between 40 and 120 0C, which in situ formed Reformatsky reagent then subsequently reacts with the compound according to formula (III). Zinc may be used in any form, for example as dust, granules, foil or wire. The zinc may be used without prior activation, however if the reactivity of the zinc would not be sufficient and consequently
resulting in a low yield, the zinc may be activated in situ by methods known to the skilled man, for example by treatment of the zinc with dibromoethane, iodine, trimethylsilylchloride, HCI or by application of ultrasound. Zinc preferably is used in 0.8-10 equivalents related to compound (III), more preferably in 1.0-6 equivalents and most preferred in 1.2-4 equivalents. This results in a good balance between high speed of reaction and limited amounts of zinc to be removed.
A second method of in situ preparation of the Reformatsky reagent XZnCR6R7R3, is by contacting XCR6R7R3 with
 wherein Ri0 is alkyl, alkenyl or aryl, in the presence of an additive comprising an element from group 4-12 of the periodic table with the compound according to formula III. Most preferred compounds of (R-|0)2Zn are (C2H5)2Zn and (CH3)2Zn because these reagents give fast formation of the Reformatsky reagent XZnCR6R7R3. Preferred additives are Cr(lll)(acetylacetonatate)3, Fe(lll)(acetylacetonate)3, Ni(ll)(acetylacetonate)2, PdCI2, RhCI(PPh3), IrCI2 and CeCI3. More preferably, Fe(lll)(acetylacetonate)3 or Ni(ll)(acetylacetonate)2 are used, because a higher yield is obtained when these additives are present during contacting the Reformatsky reagent with the compound according to formula (III). The additive is preferably added in an amount of less than 20 mol% based on the total amount of compound according to formula (III), more preferably in an amount of less than 10 mol% and most preferred in an amount of less than 5 mol%. An additional advantage of in situ preparation of the Reformatsky reagent
wherein Ri0 is alkyl, alkenyl or aryl, in the presence of an additive comprising an element from group 4-12 of the periodic table with the compound according to formula III. Most preferred compounds of (R-|0)2Zn are (C2H5)2Zn and (CH3)2Zn because these reagents give fast formation of the Reformatsky reagent XZnCR6R7R3. Preferred additives are Cr(lll)(acetylacetonatate)3, Fe(lll)(acetylacetonate)3, Ni(ll)(acetylacetonate)2, PdCI2, RhCI(PPh3), IrCI2 and CeCI3. More preferably, Fe(lll)(acetylacetonate)3 or Ni(ll)(acetylacetonate)2 are used, because a higher yield is obtained when these additives are present during contacting the Reformatsky reagent with the compound according to formula (III). The additive is preferably added in an amount of less than 20 mol% based on the total amount of compound according to formula (III), more preferably in an amount of less than 10 mol% and most preferred in an amount of less than 5 mol%. An additional advantage of in situ preparation of the Reformatsky reagent
XZnCR6R7R3 is that the process according to the invention comprises even less process steps than the process as disclosed in US 5,840,961.
An additional advantage of the second method of in situ preparation is that a high yield in the compound according to formula (V) is obtained. The reaction mixture comprising compound (III) is subsequently contacted with the Reformatsky reagent XZnCR6R7R3 in the event that this reagent is produced separately. The temperature at which the compound according to formula (III) and the Reformatsky reagent are contacted preferably is between 0 and 140 0C, more preferably between 20-1200C. Upon contacting the compound according to formula (III) and the
Reformatsky reagent XZnCR6R7R3 in the process according to the invention a reaction mixture comprising compound (IV) is formed.
In order to increase the diastereoselectivity or purity of the compound according to formula (IV), this compound may be recrystallized, with known techniques, as such or as a salt from a suitable solvent. Suitable salts include those formed with HCI, HBr, acetic acid, p-toluenesulphonic acid, benzenesulphonic acid, tartaric acid. Suitable solvents include heptane, methylisobutylketone, acetone, methyltertbutylether, isopropylacetate, ethylacetate, isopropanol, ethanol or methanol.
Compound (IV) is finally converted through hydrogenolysis, a retro Strecker method or oxidation into the chiral amine according to formula (V).
Hydrogenolysis may suitably be done with H2 using for example Pd as a catalyst. Through hydrogenolysis the chiral centre comprising R4 and R5 is split off from the compound according to formula (IV), resulting in the corresponding chiral amine according to formula (V). Temperature during hydrogenolysis is chosen preferably between 0 and 40 0C, more preferably between 20 and 30 0C. This results in a high yield of the chiral amine according to formula (V) Another method of converting the compound according to formula (IV) into the corresponding chiral amine is by means of a so-called retro Strecker reaction. This retro Strecker reaction is a one step reaction, in which the amide group of the compound according to formula (IV) is first converted to a nitril, by a dehydrating agent, for example by thionylchloride, phosphoroxy trichloride, phosphorpentachloride, p-toluenesulphonic acid/pyridine, Vilsmeyer reagent/Et3N or pyridine, cyanuric chloride, acetic acid anhydride, trifluoroacetic acid anhydride and other dehydrating agents, for example as described in March, Advanced Organic Chemistry, 5th Edition, Wiley Interscience, Eds.M.B. Smith and J. March, 2001 , p1350 followed by a retro Strecker reaction, i.e. elimination of HCN, for example by addition of a base or by heating. Examples of suitable bases are (earth) alkali metal hydroxides, (earth) alkalimetal carbonates, (earth) alkalimetal phospates and organic bases. Suitable bases are for instance KOH, Na2CO3 or K2CO3,followed by hydrolysis with methods known to the skilled man of the obtained imine to the chiral amine. Hydrolysis may suitable be done by e.g. heating the imine to temperatures up to 1200C, addition of acids, or transimination by for example NH2OH. HCI or NaHSO3. This retro Strecker reaction is carried out in one vessel without isolation of intermediates thereby making it a one step reaction.
A further method of converting the compound according to formula (IV) into the corresponding chiral amine is by means of oxidation followed by hydrolysis of the
imine to the chiral amine according to formula (V). With oxidation in this application is understood treatment with peracids, O3, metal-based oxidative agents such as e.g. KMnO4 and hypochlorites such as e.g. NaOCI. These oxidation methods as such are known and are described e.g. in March, Advanced Organic Chemistry, 5th Edition, Wiley lnterscience Eds.M.B. Smith and J. March, 2001 , Chapter! 9..
Preferably compound (IV) converted through hydrogenolysis into the chiral amine according to formula (V). The advantage hereof is that this method is less prone to side reactions.
The invention will now be further elucidated with the following examples, without being limited hereto.
Examples
Example I: Synthesis of (f?)-3-amino-3-phenyl-propionic acid ethyl ester
Ia. First step: Synthesis of (R)-2-[(phenylmethylene)-aminol-2-phenylacetamide according to formula:

Phenylglycine amide (50 g, 0.31 mol, assay 94%) and benzaldehyde
(34.8 g, 0.33 mol were added to 250 mL of CH3OH. The mixture was stirred at room temperature. After 15 min crystallization was observed. The mixture was stirred for 18 h.
After filtration, washing with 2 x 25 mL of CH3OH, a white solid was obtained (63.6 g, 81%). 1H NMR (CDCI3): δ 4.99 (s, 1 H), 5.8 (bs, 1 H), 7.0 (bs, 1 H), 7.4 (m, 8H), 7.8 (m, 2H),
8.31 (s, 1 H),
lb-1. Second step: Synthesis of (ff)-3-(Tfffl-2-amino-2-oxo-1-phenylethvπamino)-3-phenyl- propionic acid ethyl ester: Reformatsky reaction of compound from Example Ia with Zn, BrCH2COOEt to form:
Hy CONH2 ^COOEt
The Schiff base of Example Ia (4.8 g, 20 mmol) was dissolved in CH2CI2 (50 ml_) and Zn dust was added (5.2, 80 mmol). The reaction mixture was heated until reflux and BrCH2COOEt (10.4 g, 62 mmol) was dosed in ca 200 min. The mixture was then stirred under reflux (ca 42°C) for 2 h. After cooling the reaction mixture was filtered and added to a solution of 4N aqueous HCI (75 ml_). After stirring for approximately 15 h, the precipitated solid was isolated by filtration to give the product as HCI salt (5.2 g, 72%, cfe 96%).1H NMR (CDCI3): δ 1.3 (m), 2.1 (s), 2.8 (m), 4.0 (s), 4.3 (m), 6.1 (br s), 7.2-7.5 (m), 7.9 (s).
Instead of second step lb-1, second step lb-2 as given above may be employed:
lb-2. Second step: Synthesis of (fi)-3-(r(f?)-2-amino-2-oxo-1-phenylethyl1amino)-3-phenyl- propionic acid ethyl ester: Reformatsky reaction of compound from Example Ia with Et2Zn, BrCH2COOEt in CH2CI2 as solvent.
The Schiff base of Example Ia (8.6 g, 36 mmol) was dissolved in CH2CI2 (100 mL) and cooled to ca. -30 0C. The following reagents were added in succession: ZnEt2, 1 M in hexane (91 mL, 2.5 eq), BrCH2COOEt (4.9 mL, 1.2 eq) and Ni(acetylacetonatate)2 (0.5 g, 5 mol%), either drop wise or in portions. The reaction mixture was allowed to warm to room temperature in a controlled fashion and stirred for 3 h, then poured onto about 150 g of crushed ice. The mixture was filtered over Celite, the layers were separated, the organic layer was dried on Na2SO4 and the volatiles were removed in vacuo, yielding a yellow oil. This oil was dissolved in methyl-f-butylether (250 mL) and CH3OH (25 mL), to which about 4.6 mL of 30% aqueous HCI (1.5 eq) was added then. CH3OH (50 mL) was added and the mixture was stirred for 16 h, after which a white suspension had formed. The solid was filtered off, washed with methyl-f-butylether (100 mL) and dried in vacuo, affording the product as HCI salt as white solid (8.10 g, 62%). HPLC-MS (Discovery C18): 97 assay % at 216 nm, 97 assay % at 286 nm, 98% de.
lc-1 : Third step: Synthesis of (ffl-S-amino-S-phenyl-propionic acid ethyl ester via retro- Strecker method to form:
NH2
XOOEt
The compound of Example Ib (1.54 g, 4.7 mmol) was dissolved in THF (40 ml_). Et3N (7.2 mL, 12 eq) was added and the mixture was cooled on ice. POCI3 (0.9 ml_, 2.2 eq) was added dropwise, then the mixture was allowed to warm to room temperature, stirred for 1 h and subsequently refluxed for 5 h. At room temperature, a solution of NH2OH-HCI (0.91 g, 3 eq) in H2O (50 mL) was added and the resulting mixture was stirred for 16 h. The pH was adjusted to 1 with 30% HCI and THF was removed in vacuo. The aqueous layer was washed with methyl-f-butylether (40 mL), then CH2CI2 was added (50 mL) and the pH was adjusted to 10 with 10% aqueous NaOH. The layers were separated, the organic layer was dried on Na2SO4 and the volatiles were removed in vacuo, affording a brown oil (0.51 g, 54%). 1H NMR (CDCI3): δ 1.5 (q, 3H), 2.6 (d, 2H), 3.9 (q, 2H), 4.4 (t, 1 H), 7.4 (m, 5H). HPLC (Chiralcel OD): >99% ee.
Instead of third step lc-1 , third step lc-2 as given above may be employed: lc-2: Third step: Synthesis of (f?)-3-amino-3-phenyl-propionic acid ethyl ester via oxidation
The product of Example Ib (0.84 g, 2.37 mmol) was dissolved in acetone (40 mL). KMnO4 (0.94 g, 2.5 eq) was added and the mixture was stirred for 16 h, then 6 mL of 1 M aqueous NaOH (2.5 eq) was added and the mixture was filtered over Celite. Acetone was removed in vacuo and the residue was partitioned between CH2CI2 (40 mL) and H2O (40 mL). The layers were separated, the organic layer was dried on Na2SO4 and volatiles were removed in vacuo, yielding a yellow oil (1.03 g). This was dissolved in tetrahydrofuran (20 mL) and H2O (20 mL) and NH2OH HCI (0.50 g, 3 eq) was added. The mixture was stirred for 16 h, then THF was removed in vacuo. The pH was adjusted to 1 with 1 M HCI and the aqueous layer was washed with EtOAc (30 mL). The pH was adjusted to 10 with 10% aqueous NaOH and the aqueous layer was extracted with CH2CI2 (50 mL). The organic layer was dried on Na2SO4 and the volatiles were removed in vacuo,
affording a yellow oil (0.23 g, 44%). 1H NMR (CDCI3): vide supra. HPLC (Chiralcel OD): 99% ee.
Example II: Synthesis of (ffl-3-amino-3-benzoH ,31dioxol-5-yl-propionic acid ethyl ester
Na. First step: Synthesis of (ffl-2-[(benzo[1 ,3ldioxol-5-ylmethylene)-aminol-2- phenylacetamide
Phenylglycine amide (50 g, 0.31 mol, assay 94%) and piperonal (50 g, 0.30 mol) were added to 400 mL of CH3OH. After heating to 300C a clear solution was obtained. The mixture was stirred at room temperature for 18 h and a precipitate was observed. After filtration, washing with 2 x 25 mL of CH3OH, a white solid was obtained (73 g, 86 %). 1H NMR (CDCI3): δ 4.9 (s, 1H), 6.0, (m, 3H), 6.8 (m, 1 H), 7.01 (br s, 1H), 7.3 (m, 7H)1 8.2 (s, 1H)
lib. Second step: Synthesis of (fi)-3-(r(R)-2-amino-2-oxo-1-phenylethvπamino)-3- r(benzoπ .31dioxol-5-yl)-3-propionic acid ethyl ester: Reformatsky reaction of compound from Example Ha with Et2Zn, BrCH2COOEt to form:

The Schiff base of example Ha (10.3 g, 36 mmol) was dissolved in CH2CI2 (100 mL) and cooled to about -30 0C. The following reagents were added in succession: Et2Zn (1 M in hexane. 91 mL, 2.5 eq), BrCH2COOEt (4.9 mL, 1.2 eq) and Ni(acetylacetonatate)2 (0.5 g, 5 mol%), either dropwise or in portions. The reaction mixture was allowed to warm to room temperature in ca 30 min and stirred for 3 h, then poured onto ca. 150 g of crushed ice. The mixture was filtered over Celite, the layers were separated, the organic layer was dried on Na2SO4 and the volatiles were removed in vacuo, yielding yellow oil. This was dissolved in methyl-f-butylether (250 mL) and CH3OH (25 mL), to which ca. 4.6 mL of 30% aqueous HCI (1.5 eq) was added. A precipitate formed initially, but rapidly oiled out. CH3OH (25 mL) was added and the mixture was
stirred for 16 h, after which a white suspension had formed. The solid was filtered off, washed with methyl-f-butylether (100 mL) and dried in vacuo, affording a white solid (7.89 g, 59%). HPLC-MS (Discovery C18): 97 assay % at 216 nm, 97 assay % at 286 nm, 98% de.
Hc: Third step: Synthesis of (ffl-3-amino-3-benzori ,31dioxol-5-y|-propionic acid ethyl ester via hvdrogenolysis to form:
^H2
^COOEt
The compound of example lib (6 g, 0.13 mol) was dissolved in dry EtOH (100 mL) and 5% Pd/C (1.9 g, 50 % H2O) was added. The mixture was shaken at 9 bar H2 for 16h. The Pd/C was removed by filtration over Celite and after addition of 5 mL of concentrated aqueous HCI the solvent was removed by evaporation. To the residu EtOAc (150 mL) was added and the solvent removed by evaporation. Then again EtOAc was added, stirred for 15 h to give the product as HCI salt (3.1 g, 87%, ee 96% containing a small amount (8%) of phenylacetamide. 1H NMR (CDCI3): δ 1.3 (t, 3H), 1.8 (br s, 2H), 2.6 (d, 2H), 4.2 (q, 2H), 4.4 (t, 1 H), 6.0 (s, 2H), 6.8-7.0 (m, 3H).
Example III: Synthesis of (S)-3-amino-butyric acid ethyl ester.
Ilia. First step: Synthesis of (ffl-2-f(ethylidene)-aminol-2-phenylacetamide:
N CONH2 H3C J^
Phenylglycine amide (30 g, 0.19 mol, assay 94%) and 5 g Na2SO4 were added to 250 mL of CH2CI2. To the stirred mixture was added acetaldehyde (8.8 g, 0.20 mol). The mixture was stirred at room temperature for 1 h and the solids were removed by filtration. The CH2CI2 was removed by evaporation and the residu stirred in 150 mL EtOAc.
The solid was filtrated and washed with 2 x 25 ml_ of EtOAc. After drying the imine was obtained as a white solid (26.2 g, 78.4%). 1H NMR (CDCI3): δ 2.1 (d, 3H), 4.7 (s, 1 H), 5.9 (br s, 1 H), 6.9 (br s, 1 H), 7.4 (m, 5H), 7.8 (q, 1 H).
HIb; Second step: (S)-3-(r(fl)-2-amino-2-oxo-1-phenylethyl1amino)-butyric acid ethyl ester; Reformatsky reaction of compound from Example Ilia with Et2Zn, BrCH2COOEt and CH2CI2 to form:
HN CONH2
^COOEt
The Schiff base of Example Ilia (67.3 g, 0.38 mol) was dissolved in
CH2CI2 (1500 mL) and cooled on ice-NaCI. The following reagents were added in succession: Et2Zn (1 M in hexane, 955 mL, 2.5 eq), BrCH2COOEt (63.5 mL, 1.2 eq) and Ni(acetylacetonatate)2 (4.91 g, 5 mol%), either dropwise or in portions. The deep-orange solution was allowed to warm to room temperature and stirred for 2 h, then poured onto about 2000 g of crushed ice. The mixture was stirred to room temperature over 16 h and then filtered over Celite. The layers were separated, the organic layer was dried on Na2SO4 and the volatiles were removed in vacuo, yielding a brown oil (81.5 g, 81%). 1H NMR (CDCI3): δ 1.2 (d, 3H), 1.3 (t, 3H), 1.7 (br, 1H), 2.3-2.6 (m, 2H), 3.2 (m, 1 H), 4.2 (m, 3H)1 4.4 (s, 1 H), 6.1 (br s, 1 H), 7.2-7.4 (m, 5H), 7.6 (br s, 1 H). HPLC-MS (Discovery C18): 73 assay % at 218 nm, >99% de.
HIc: Third step: Synthesis of (S)-3-amino-butyric acid ethyl ester via hydrogenolvsis
NH2 H3C^C00B
The compound of example IHb (33.3 g, 0.13 mol) was dissolved in dry EtOH (160 mL). AcOH (36.5 mL, 5 eq) and dry reduced 10% Pd/C (3.33 g, 10% w/w) were added with care. The mixture was shaken at 5 bar H2 for 24 h, then filtered over Celite. The residue was extracted with H2O (250 mL) and EtOH was removed in vacuo until a crystalline white precipitate formed (PhCH2CONH2). This was filtered off and washed with
H2O (50 ml_). CH2CI2 (400 mL) was added to the combined filtrates, then the pH was adjusted to 9 with 10% aqueous NaOH. The layers were separated, the organic layer was dried on Na2SO4 and the volatiles were removed in vacuo without external heating. The residue was extracted with pentane (2 x 125 mL) and the pentane removed in vacuo without heating. This afforded a yellow oil (8.81 g, 53%). 1H NMR (CDCI3): δ 1.1 (d, 3H), 1.2 (t, 3H), 1.6 (s, 2H), 2.2-2.4 (m, 2H), 3.4 (m, 1 H), 4.1 (q, 2H). HPLC (Crownpak): 92% ee.
Example IV: Synthesis of (f?)-1-Amino-4,4-dimethyl-1-phenyl-pentane-3-one
Step IVa: Synthesis of Schiff base: see example Ia
Step IVb: Synthesis of (f?)-1-(r(f?V2-amino-2-oxo-1-phenylethyllamino)-4,4-dimethyl-1- phenyl-pentane-3-one: Reformatsky reaction of compound from Example IVa with Et2Zn, BrCH2COOEt i n CH2CI2 as solvent to form :

The Schiff base of Example. IVa (0.87 g, 3.65 mmol) was dissolved in CH2CI2 (40 mL). The following reagents were added in succession: Et2Zn, (1 M in hexane, (9.1 mL, 2.5 eq), BrCH2COf-Bu (0.59 mL, 1.2 eq), Ni(acetylacetonatate)2 (0.05 g, 5 mol%) and PPh3 (0.1O g, 10 mol%). The resulting orange solution was stirred for 2 h, then saturated NH4CI (50 mL) was added. The layers were separated, the organic layer was dried on Na2SO4 and the volatiles were removed in vacuo, affording a yellow oil (1.52 g), HPLC-MS (Discovery C18): 58 assay %, 99% de. 1H NMR (CDCI3): δ 1.3 (s, 9H), 2.2 (bm, 1 H) 2.6-3.4 (m, 2H), 4.1 (s,1 H), 4.3-4.4 (m, 1 H), 6.2 (br s, 1 H), 7.2-7.9 (m, 10H), 8.1 (bs, 1H).
Step IVc: Synthesis of (R)-1-Amino-4.4-dimethyl-1-phenyl-pentane-3-one via retro Strecker method to form:

Procedure as described in Example Ic gives 0.34 g brown oil (54 %, assay 90%). 1H NMR (CDCI3): δ 1.2 (s, 9H), 1.9 (bm, 2H) 2.8-3.2 (m, 2H), 4.5-4.6 (m,1H), 7.3-7.6 (m, 5H).
Claims
1. Process for the preparation of a chiral amine whereby a compound according to formula (I)

in which formula Ri1 R2 = H, a substituted or unsubstituted: (cyclo)alkyl group, (cyclo)alkenyl group, aryl group, cyclic of acyclic heteroalkyl group or heteroaryl group with one or more N, O, S atoms, whereby R1 ≠ R2 is contacted with an enantiomerically enriched phenylglycine amide according to formula (II)
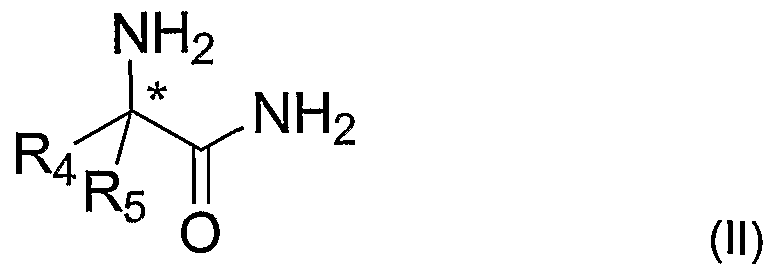
in which formula
R4 = a substituted or unsubstituted phenyl or naphthyl group R5 = H or alkyl with 1-6 carbon atoms
* = a chiral center, which may have the (R) or (S) configuration
to give an imine according to formula (III),


by contacting with a Reformatsky reagent BrZnCR6R7Rs, in which formulas
X = Cl, Br or I R3 = COOR8, CONR8R9, COSR8, COR8Or CN
R6, R7 independent of each other, H, halogen, a substituted or unsubstituted: (cyclo)alkyl group, (cyclo)alkenyl group, aryl group, cyclic of acyclic heteroalkyl group or a heteroaryl group with one or more N, O, S atoms Rβ.Rθ = independent of each other, a substituted or unsubstituted:
(cyclo)alkyl group, (cyclo)alkenyl group, aryl group, cyclic of acyclic heteroalkyl group or a heteroaryl group with one or more N, O, S atoms
and finally the compound according to formula (IV) is converted into a chiral amine according to formula (V) by means of hydrogenolysis, oxidation or a retro Strecker method

2. Process according to claim 1 , where, in the compound according to formula (II), R4 is a phenyl-group and R5 is a H.
3. Process according to claim 1 or 2, where the compound according to formula (II) has an enantiomeric excess of at least 80%.
4 Process according to any one of claims 1-3, whereby said Reformatsky reagent
XZnCR6R7R3 is prepared in situ by contacting XCR6R7R3 with (R10)2Zn, wherein R10 is alkyl, alkenyl or aryl and additive comprising an element from group 4-12 of the periodic table is present
5. Process according to claim 4, wherein (RiO)2Zn is (C2H5)2Zn.
6. Process according to claim 4 or 5 wherein the additive is
Cr(lll)(acetylacetonatate)3, Fe(III)(acetylacetonate)3 or Ni(ll)(acetylacetonate)2.
7. Process according to any one of claims 1-6, where, in the Reformatsky reagent XZnCR6R7R3, X is Br and R3 is COOR8.
8. Process according to claim 7 whereby the obtained compound according to formula (V) is subsequently hydrolysed.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP04078541.2 | 2004-12-28 | ||
| EP04078541 | 2004-12-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006069798A1 true WO2006069798A1 (en) | 2006-07-06 |
Family
ID=34928784
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2005/014170 WO2006069798A1 (en) | 2004-12-28 | 2005-12-22 | Process for the preparation of chiral amines |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2006069798A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107698484A (en) * | 2017-11-13 | 2018-02-16 | 广东中科药物研究有限公司 | A kind of preparation method of derivative of lenalidomide and application |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998002410A1 (en) * | 1996-07-12 | 1998-01-22 | G.D. Searle & Co. | Asymetric synthesis of chiral beta-amino acids |
| WO2001042173A2 (en) * | 1999-12-08 | 2001-06-14 | Dsm N.V. | Method for the preparation of enantiomerically enriched compounds |
-
2005
- 2005-12-22 WO PCT/EP2005/014170 patent/WO2006069798A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998002410A1 (en) * | 1996-07-12 | 1998-01-22 | G.D. Searle & Co. | Asymetric synthesis of chiral beta-amino acids |
| US5840961A (en) * | 1996-07-12 | 1998-11-24 | G. D. Searle & Co. | Asymmetric synthesis of chiral beta-amiNo acids |
| WO2001042173A2 (en) * | 1999-12-08 | 2001-06-14 | Dsm N.V. | Method for the preparation of enantiomerically enriched compounds |
Non-Patent Citations (3)
| Title |
|---|
| COMPTES RENDUS DES SEANCES DE L'ACADEMIE DES SCIENCES, SERIE C: SCIENCES CHIMIQUES , 268(25), 2228-30 CODEN: CHDCAQ; ISSN: 0567-6541, 1969 * |
| DATABASE CA [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; DARDOIZE, FRANCOIS ET AL: "Reactivity of metallic Reformatsky intermediates toward imines", XP002328137, retrieved from STN Database accession no. 1969:460644 * |
| SOLOSHONOK V A ET AL: "Convenient, large-scale asymmetric synthesis of beta-aryl-substituted alpha,alpha-difluoro-beta-amino acids", TETRAHEDRON LETTERS, ELSEVIER SCIENCE PUBLISHERS, AMSTERDAM, NL, vol. 43, no. 31, 29 July 2002 (2002-07-29), pages 5445 - 5448, XP004370281, ISSN: 0040-4039 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107698484A (en) * | 2017-11-13 | 2018-02-16 | 广东中科药物研究有限公司 | A kind of preparation method of derivative of lenalidomide and application |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6046353A (en) | Method of making (S)-3-(aminomethyl)-5-methylhexanoic acid | |
| KR101699095B1 (en) | Process for manufacture and resolution of 2-acylamino-3-diphenylpropanoic acid | |
| CZ374297A3 (en) | Process for preparing (s)-3-(aminomethyl)-5-methylhexanoic acid | |
| CA2610776C (en) | Process for production of mono-substituted alkylated compound using aldimine or derivative thereof | |
| IL150633A (en) | Asymmetric synthesis of pregabalin and some such novel intermediates | |
| JP5008553B2 (en) | Optically active quaternary ammonium salt having axial asymmetry and method for producing α-amino acid and derivatives thereof using the same | |
| JP2024023340A (en) | Preparation method of glufosinate ammonium | |
| CA2450545A1 (en) | Process for the production of 3-phenylisoserine | |
| TW201718462A (en) | Process for the manufacture of organic compounds and intermediates | |
| EP1235786B1 (en) | Method for the preparation of enantiomerically enriched compounds | |
| WO2006069798A1 (en) | Process for the preparation of chiral amines | |
| JP2011057665A (en) | Method for producing optically active 1-amino-2-vinylcyclopropanecarboxylic acid ester | |
| CA2166061C (en) | Novel processes for preparing (s)-4-amino-hepta-5,6-dienoic acid and intermediates thereof | |
| JP5153631B2 (en) | Novel imidazolidinone derivative, method for producing the same, and method for producing optically active amino acids | |
| US20040006225A1 (en) | Preparation of enantiomerically enriched amine-functionalized compounds | |
| JP4524841B2 (en) | Optically active amino acid ester tartaric acid amide and process for producing the same | |
| NL1013789C2 (en) | New diastereomerically enriched phenylglycine amide derivatives useful in the preparation of enantiomerically enriched compounds such as alpha- and beta-amino acids and amines | |
| US4188343A (en) | Process for preparing anthranylaldehyde derivatives | |
| JP4212351B2 (en) | Process for producing optically active 1,2-diamine derivatives and optically active imidazolidinone derivatives | |
| KR20070030488A (en) | New aminosalicylic acid analogs, it's pharmaceutically acceptable salts and their preparations | |
| JPH0987259A (en) | Optically active oxazolines and production of asymmetric cyclopropanecarboxylic acids using the same | |
| JPS5835994B2 (en) | Quinolinone imine derivative and method for producing the same | |
| JP2004155696A (en) | Method for producing optically active 2-amino-2-phenylethanols and their intermediate | |
| JP2002105071A (en) | Method for producing cyclic acetal compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 05847777 Country of ref document: EP Kind code of ref document: A1 |