WO2006044503A2 - Compounds for nonsense suppression, use of these compounds for the manufacture of a medicament for treating somatic mutation-related diseases - Google Patents
Compounds for nonsense suppression, use of these compounds for the manufacture of a medicament for treating somatic mutation-related diseases Download PDFInfo
- Publication number
- WO2006044503A2 WO2006044503A2 PCT/US2005/036762 US2005036762W WO2006044503A2 WO 2006044503 A2 WO2006044503 A2 WO 2006044503A2 US 2005036762 W US2005036762 W US 2005036762W WO 2006044503 A2 WO2006044503 A2 WO 2006044503A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- optionally substituted
- alkyl
- groups
- independently selected
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 Cc1c(C=CC=C)[o]*(*)*1* Chemical compound Cc1c(C=CC=C)[o]*(*)*1* 0.000 description 6
- LERMECNLIVEERL-UHFFFAOYSA-N CC(C1)OC(C)CN1N Chemical compound CC(C1)OC(C)CN1N LERMECNLIVEERL-UHFFFAOYSA-N 0.000 description 2
- MCTWTZJPVLRJOU-UHFFFAOYSA-N C[n]1cncc1 Chemical compound C[n]1cncc1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 2
- MWZDIEIXRBWPLG-UHFFFAOYSA-N C[n]1ncnc1 Chemical compound C[n]1ncnc1 MWZDIEIXRBWPLG-UHFFFAOYSA-N 0.000 description 2
- UOKZUTXLHRTLFH-UHFFFAOYSA-N NOc1ccccc1 Chemical compound NOc1ccccc1 UOKZUTXLHRTLFH-UHFFFAOYSA-N 0.000 description 2
- WQZAHVGNMHBOKQ-UHFFFAOYSA-N CN(CC1)/C1=[O]\c1ccc(CCN2N)c2c1 Chemical compound CN(CC1)/C1=[O]\c1ccc(CCN2N)c2c1 WQZAHVGNMHBOKQ-UHFFFAOYSA-N 0.000 description 1
- YLPBUMOJYDPQJB-UHFFFAOYSA-N CN(CC1)CCC11OCCO1 Chemical compound CN(CC1)CCC11OCCO1 YLPBUMOJYDPQJB-UHFFFAOYSA-N 0.000 description 1
- XYOIIDRRMQCAIX-UHFFFAOYSA-N CN(CC1)CCC1NN1CCN(C)CC1 Chemical compound CN(CC1)CCC1NN1CCN(C)CC1 XYOIIDRRMQCAIX-UHFFFAOYSA-N 0.000 description 1
- RJWLLQWLBMJCFD-UHFFFAOYSA-N CN(CC1)CCN1N Chemical compound CN(CC1)CCN1N RJWLLQWLBMJCFD-UHFFFAOYSA-N 0.000 description 1
- SJRJJKPEHAURKC-UHFFFAOYSA-N CN1CCOCC1 Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 1
- IBZUHUCXTVBZCU-UHFFFAOYSA-N COC(c1cc(-c2nc(cc(cc3)Br)c3[o]2)ccc1)=O Chemical compound COC(c1cc(-c2nc(cc(cc3)Br)c3[o]2)ccc1)=O IBZUHUCXTVBZCU-UHFFFAOYSA-N 0.000 description 1
- UQFQONCQIQEYPJ-UHFFFAOYSA-N C[n]1[n]ccc1 Chemical compound C[n]1[n]ccc1 UQFQONCQIQEYPJ-UHFFFAOYSA-N 0.000 description 1
- GASCFHSWYPJXSN-UHFFFAOYSA-N NC[n]1cncc1 Chemical compound NC[n]1cncc1 GASCFHSWYPJXSN-UHFFFAOYSA-N 0.000 description 1
- SBMSLRMNBSMKQC-UHFFFAOYSA-N NN1CCCC1 Chemical compound NN1CCCC1 SBMSLRMNBSMKQC-UHFFFAOYSA-N 0.000 description 1
- LWMPFIOTEAXAGV-UHFFFAOYSA-N NN1CCCCC1 Chemical compound NN1CCCCC1 LWMPFIOTEAXAGV-UHFFFAOYSA-N 0.000 description 1
- MKQLBNJQQZRQJU-UHFFFAOYSA-N NN1CCOCC1 Chemical compound NN1CCOCC1 MKQLBNJQQZRQJU-UHFFFAOYSA-N 0.000 description 1
- NYIGEYYREVRXES-UHFFFAOYSA-N N[n]1nccc1 Chemical compound N[n]1nccc1 NYIGEYYREVRXES-UHFFFAOYSA-N 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N O=C1NCCC1 Chemical compound O=C1NCCC1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- NAGAIICDOFCRFU-UHFFFAOYSA-N OC(c1cccc(-c2nc3cc(N(CCC4)C4=O)ccc3[o]2)c1)=O Chemical compound OC(c1cccc(-c2nc3cc(N(CCC4)C4=O)ccc3[o]2)c1)=O NAGAIICDOFCRFU-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/06—1,2,4-Oxadiazoles; Hydrogenated 1,2,4-oxadiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4155—1,2-Diazoles non condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/64—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms, e.g. histidine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D251/00—Heterocyclic compounds containing 1,3,5-triazine rings
- C07D251/02—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings
- C07D251/12—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D251/14—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hydrogen or carbon atoms directly attached to at least one ring carbon atom
- C07D251/24—Heterocyclic compounds containing 1,3,5-triazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hydrogen or carbon atoms directly attached to at least one ring carbon atom to three ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/08—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/32—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/10—1,3,4-Oxadiazoles; Hydrogenated 1,3,4-oxadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/10—1,3,4-Oxadiazoles; Hydrogenated 1,3,4-oxadiazoles
- C07D271/107—1,3,4-Oxadiazoles; Hydrogenated 1,3,4-oxadiazoles with two aryl or substituted aryl radicals attached in positions 2 and 5
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/22—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D277/30—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D285/00—Heterocyclic compounds containing rings having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by groups C07D275/00 - C07D283/00
- C07D285/01—Five-membered rings
- C07D285/02—Thiadiazoles; Hydrogenated thiadiazoles
- C07D285/04—Thiadiazoles; Hydrogenated thiadiazoles not condensed with other rings
- C07D285/12—1,3,4-Thiadiazoles; Hydrogenated 1,3,4-thiadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/56—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/68—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/24—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6527—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having nitrogen and oxygen atoms as the only ring hetero atoms
- C07F9/653—Five-membered rings
- C07F9/65306—Five-membered rings containing two nitrogen atoms
Definitions
- the present invention relates to methods, compounds, and compositions for treating or preventing diseases associated with nonsense mutations in an mRNA by administering the compounds or compositions of the present invention. More particularly, the present invention relates to methods, compounds, and compositions for suppressing premature translation termination associated with a nonsense mutation in an mRNA.
- Gene expression in cells depends upon the sequential processes of transcription and translation. Together, these processes produce a protein from the nucleotide sequence of its corresponding gene. Transcription involves the synthesis of mRNA from DNA by RNA polymerase. Transcription begins at a promoter region of the gene and continues until termination is induced, such as by the formation of a stem-loop structure in the nascent RNA or the binding of the rho gene product. Protein is then produced from mRNA by the process of translation, occurring on the ribosome with the aid of tRNA, tRNA synthetases and various other protein and RNA species. Translation comprises the three phases of initiation, elongation and termination.
- Translation is initiated by the formation of an initiation complex consisting of protein factors, mRNA, tRNA, cofactors and the ribosomal subunits that recognize signals on the mRNA that direct the translation machinery to begin translation on the mRNA.
- initiation complex consisting of protein factors, mRNA, tRNA, cofactors and the ribosomal subunits that recognize signals on the mRNA that direct the translation machinery to begin translation on the mRNA.
- growth of the polypeptide chain occurs by the repetitive addition of amino acids by the peptidyl transferase activity of the ribosome as well as tRNA and tRNA synthetases.
- the presence of one of the three termination codons (UAA, UAG, UGA) in the A site of the ribosome signals the polypeptide chain release factors (RFs) to bind and recognize the termination signal.
- RFs polypeptide chain release factors
- Mutations of the DNA sequence in which the number of bases is altered are categorized as insertion or deletion mutations (frameshift mutations) and can result in major disruptions of the genome. Mutations of the DNA that change one base into another and result in an amino acid substitution are labeled missense mutations. Base substitutions are subdivided into the classes of transitions (one purine to another purine, or one pyrimidine to another pyrimidine) and transversions (a purine to a pyrimidine, or a pyrimidine to a purine).
- Transition and transversion mutations can result in a nonsense mutation changing an amino acid codon into one of the three stop codons. These premature stop codons can produce aberrant proteins in cells as a result of premature translation termination.
- a nonsense mutation in an essential gene can be lethal and can also result in a number of human diseases, such as, cancers, lysosomal storage disorders, the muscular dystrophies, cystic fibrosis and hemophilia, to name a few.
- the human p53 gene is the most commonly mutated gene in human cancer (Zambetti, G.P. and Levine, A., FASEB 7:855-865 (1993)). Found in both genetic and spontaneous cancers, over 50 different types of human cancers contain p53 mutations and mutations of this gene occur in 50-55% of all human cancers (Hollstein, M., et al, Nucleic Acids Res. 22:3551-55 (1994); International Agency for Research on Cancer (IARC) database). Approximately 70% of colorectal cancer, 50% of lung cancer and 40% of breast cancers contain mutant p53 (Koshland, D., Science 262:1953 (1993)).
- p53 Aberrant forms of p53 are associated with poor prognosis, more aggressive tumors, metastasis, and lower 5 year survival rates (Id). p53's role in the induction of cell growth arrest and/or apoptosis upon DNA damage is believed to be essential for the destruction of mutated cells that would have otherwise gained a growth advantage. In addition, p53 sensitizes rapidly dividing cells to apoptotic signals. Of greater than 15,000 reported mutations in the p53 gene, approximately 7% are nonsense mutations. Accordingly, there is a need for a safe and effective treatment directed to p53 nonsense mutations.
- suppression of the nonsense mutation can arise as a result of a mutation in one of the tRNA molecules so that the mutant tRNA can recognize the nonsense codon, as a result of mutations in proteins that are involved in the translation process, as a result of mutations in the ribosome (either the ribosomal RNA or ribosomal proteins), or by the addition of compounds known to alter the translation process (for example, cycloheximide or the aminoglycoside antibiotics).
- the result is that an amino acid will be incorporated into the polypeptide chain, at the site of the nonsense mutation, and translation will not prematurely terminate at the nonsense codon.
- the inserted amino acid will not necessarily be identical to the original amino acid of the wild-type protein, however, many amino acid substitutions do not have a gross effect on protein structure or function.
- a protein produced by the suppression of a nonsense mutation would be likely to possess activity close to that of the wild-type protein. This scenario provides an opportunity to treat diseases associated with nonsense mutations by avoiding premature termination of translation through suppression of the nonsense mutation.
- small molecule therapeutics or prophylactics that suppress premature translation termination by mediating the misreading of the nonsense codon would be useful for the treatment of a number of diseases.
- the discovery of small molecule drugs, particularly orally bioavailable drugs, can lead to the introduction of a broad spectrum of selective therapeutics or prophylactics to the public which can be used against disease caused by nonsense mutations is just beginning.
- Clitocine (6-Amino-5-nitro-4-( ⁇ -D-ribo-furanosylamino)pyrimidine) is a naturally occurring exocyclic amino nucleoside that was first isolated from the mushroom CUtocybe inversa (Kubo et al, Tet. Lett. 27: 4277 (1986)).
- the present invention provides compounds of Formula (1) which are useful for suppressing premature translation termination associated with a nonsense mutation in mRNA, and for treating diseases associated with nonsense mutations in mRNA:
- Y and Z are independently selected from N or C; W is N or CH; n is 0, 1, 2 or 3;
- R 1 is hydrogen, a C 6 to C 8 aryl which is optionally substituted with a carboxy group, or R 1 is absent when Z is N;
- R 2 is hydrogen; a C 6 to C 8 aryl which is optionally substituted with one, two, or three independently selected R a groups; a four to seven membered heterocycle which is optionally substituted with one or more independently selected C 1 -C 6 alkyl groups or a three to seven membered heterocycle; or R 2 is absent when Y is N;
- R is independently selected from a halogen; a carboxy group; a C 1 -C 6 alkyl group optionally substituted with a four to seven membered heterocycle, a C 6 -C 8 aryloxy group, or an amino group, wherein the four to seven membered heterocycle, C 6 -C 8 aryloxy group, and amino group are optionally substituted with one or two independently selected
- C 1 -C 6 alkyl or C 6 -C 8 aryl groups which C 6 -C 8 aryl groups are optionally and independently substituted with one or more C 1 -C 6 alkyl groups; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 -C 8 aryl optionally substituted with one or more independently selected halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, oxy, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; an amino group optionally substituted with one or two independently selected C 6 -C 8 aryl or
- C 1 -C 6 alkyl groups which are optionally substituted with a hydroxy, a C 6 -C 8 aryl, or a nine to ten membered heterocycle having two ring structures; a carbonyl group substituted with a five to six membered heterocycle group; a four to seven membered heterocycle group optionally substituted with one more C 1 -C 4 alkyl or oxo groups; a nine to ten membered heterocycle having two ring structures; or two R groups, wherein R may also include an oxy group, together with the hetero-bicycle to which they are attached form a twelve to thirteen membered heterocycle having three ring structures; wherein R a is a halogen; a C 1 -C 6 alkyl; a C 1 -C 6 alkoxy which is optionally substituted with one or more independently selected halogen groups; a C 6 -C 8 aryl; a four to seven membered heterocycle which is optionally substituted with one or more
- compounds of Formula (2) are provided which are useful for suppressing premature translation termination associated with a nonsense mutation in mRNA, and for treating diseases associated with nonsense mutations in mRNA:
- Y and Z are independently selected from N or C; W is N or CH; n is 0, 1, 2 or 3;
- R 1 is hydrogen, a C 6 to C 8 aryl which is optionally substituted with a carboxy group, or R 1 is absent when Z is N;
- R 2 is hydrogen; a C 6 to C 8 aryl which is optionally substituted with one, two, or three independently selected R a groups; a four to seven membered heterocycle which is optionally substituted with one or more independently selected C 1 -C 6 alkyl groups or a three to seven membered heterocycle; or R 2 is absent when Y is N;
- R is independently selected from a halogen; a carboxy group; a C 1 -C 6 alkyl group optionally substituted with a four to seven membered heterocycle, a C 6 -C 8 aryloxy group, or an amino group, wherein the four to seven membered heterocycle, C 6 -C 8 aryloxy group, and amino group are optionally substituted with one or two independently selected
- C 1 -C 6 alkyl or C 6 -C 8 aryl groups which C 6 -C 8 aryl groups are optionally and independently substituted with one or more C 1 -C 6 alkyl groups; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 -C 8 aryl optionally substituted with one or more independently selected halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, oxy, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; an amino group optionally substituted with one or two independently selected C 6 -Cs aryl or
- C 1 -C 6 alkyl groups which are optionally substituted with a hydroxy, a C 6 -C 8 aryl, or a nine to ten membered heterocycle having two ring structures; a carbonyl group substituted with a five to six membered heterocycle group; a four to seven membered heterocycle group optionally substituted with one more Cj-C 4 alkyl or oxo groups; a nine to ten membered heterocycle having two ring structures; or two R groups, wherein R may also include an oxy group, together with the hetero-bicycle to which they are attached form a twelve to thirteen membered heterocycle having three ring structures; wherein R a is a hydroxy group; a halogen; a C 1 -C 6 alkyl which is optionally substituted with one or more independently selected halogen or hydroxy groups; a C 1 -C 6 alkoxy which is optionally substituted with one or more independently selected halogen or phenyl groups; a C
- compounds of Formula (3) are provided which are useful for suppressing premature translation termination associated with a nonsense mutation in mRNA, and for treating diseases associated with nonsense mutations in mRNA:
- Y and Z are independently selected from N or C;
- W is N or CH; n is O, 1, 2 or 3;
- R 1 is absent or a C 6 to C 8 aryl which is optionally substituted with a carboxy group
- R 2 is absent; a C 6 to C 8 aryl which is optionally substituted with one, two, or three independently selected R a groups; or a four to seven membered heterocycle which is optionally substituted with one or more C 1 -C 4 alkyl groups, or a four to six membered heterocycle;
- R is independently selected from a halogen; a carboxy group; a C 1 -C 6 alkyl group optionally substituted with a C 6 -C 8 aryloxy group, an imidazole group, or an amino group which is optionally substituted with one or two independently selected C 1 -C 6 alkyl or C 6 - C 8 aryl groups; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 -C 8 aryl optionally substituted with one or more halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; an amino group optionally substituted with one or two independently selected C 6 - C 8 aryl or C 1 -C 6 alkyl groups, which are optionally substituted with a hydroxy, a phenyl, or a benzo[1,3]diox
- diseases include, but are not limited to, genetic diseases caused by premature translation termination associated with a nonsense mutation, such as a CNS disease, an inflammatory disease, a neurodegenerative disease, an autoimmune disease, a cardiovascular disease, or a pulmonary disease; more preferably the disease is cancer (or other proliferative diseases), amyloidosis, Alzheimer's disease, atherosclerosis, giantism, dwarfism, hypothyroidism, hyperthyroidism, cystic fibrosis, aging, obesity, Parkinson's disease, Niemann Pick's disease, familial hypercholesterolemia, retinitis pigmentosa, Marfan syndrome, lysosomal storage disorders, the muscular dystrophies, cystic fibrosis, hemophilia, or classical late infantile neuronal ceroid lipofuscinosis (LINCL).
- the invention is directed to methods for suppressing premature translation termination associated with a nonsense mutation in mRNA comprising administering a nonsense-suppressing amount of at least one compound of the invention to a subject in need thereof.
- methods for treating cancer, lysosomal storage disorders, a muscular dystrophy, cystic fibrosis, hemophilia, or classical late infantile neuronal ceroid lipofuscinosis comprising administering a therapeutically effective amount of at least one compound of the invention to a subject in need thereof.
- a method of treating or preventing a disease resulting from a somatic mutation comprising administering to a patient in need thereof an effective amount of a compound of Formula 3:
- Y and Z are independently selected from N or C;
- W is N or CH; n is 0, 1, 2 or 3;
- R 1 is absent or a C 6 to C 8 aryl which is optionally substituted with a carboxy group
- R 2 is absent; a C 6 to C 8 aryl which is optionally substituted with one, two, or three independently selected R a groups; or a four to seven membered heterocycle which is optionally substituted with one or more C 1 -C 4 alkyl groups, or a four to six membered heterocycle;
- R is independently selected from a halogen; a carboxy group; a C 1 -C 6 alkyl group optionally substituted with a C 6 -C 8 aryloxy group, an imidazole group, or an amino group which is optionally substituted with one or two independently selected C 1 -C 6 alkyl or C 6 - C 8 aryl groups; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 -C 8 aryl optionally substituted with one or more halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; an amino group optionally substituted with one or two independently selected C 6 - C 8 aryl or C 1 -C 6 alkyl groups, which are optionally substituted with a hydroxy, a phenyl, or a benzo[1,3]diox
- R 3 is independently selected from: a halogen; a C 1 to C 6 alkyl; a C 1 to C 6 haloalkyl; a Cj to C 6 alkoxy; a C 1 to C 6 haloalkoxy; a C 6 to C 8 aryl group; a carboxy group; a carbamoyl group; an amino group which is optionally substituted with one or two independently selected hydroxy groups, halogens, C 1 to C 6 alkyls, or C 1 to C 6 haloalkyls; or a four to six membered heterocycle optionally substituted with an oxo group. 5.
- the four to seven membered heterocycle is selected from an azetidine group, a pyrrolidine group, a piperidine group, a piperazine group, a morpholine group, a [1,4]diazepane group, a pyrazole group, an imidazole group, a [1,2,4] triazole group, a pyridine group, a furan group, and a thiophene group. 6.
- the nine to ten membered heterocycle having two ring structures is selected from a benzofuran group, a 2,3-dihydro-benzofuran group, a benzo[1,3]dioxole group, a 2,3-dihydro-isoindole group, a 2,3-dihydro-indole group, a 1,2,3,4-tetrahydro-isoquinoline group, and a l,4-dioxa-8-aza-spiro[4.5]decane group.
- R 2 is selected from the following, wherein the * indicates the bond of attachment:
- R 2 is a C 6 to Cs aryl, optionally substituted with one, two, or three -R a groups, wherein R a is independently selected from: a halogen; a C 1 to C 6 alkyl; a C 1 to C 6 haloalkyl; a C 1 to C 6 alkoxy; a C 1 to C 6 haloalkoxy; a C 6 to C 8 aryl group; a carboxy group; a carbamoyl group; an amino group which is optionally substituted with one or two independently selected hydroxy groups, halogens, C 1 to C 6 alkyls, or C 1 to C 6 haloalkyls; or a four to six membered heterocycle optionally substituted with an oxo group.
- R 2 is a phenyl group optionally substituted with a carboxy group.
- R 1 is a phenyl group optionally substituted with a carboxy group.
- R is a C 1 -C 6 alkyl; a C 1 -C 6 alkoxy; a C 6 -C 8 aryl optionally substituted with one or more halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; or two R groups together with the hetero- bicycle to which they are attached form a twelve to thirteen membered heterocycle having three ring structures.
- a method of treating or preventing a disease resulting from a somatic mutation comprising administering to a patient in need thereof an effective amount of a compound of Formula 4 :
- Y and Z are independently selected from N or C; W is N or CH;
- R 1 is absent or a C 6 to C 8 aryl which is optionally substituted with a carboxy group
- R 2 is absent; a C 6 to C 8 aryl which is optionally substituted with one, two, or three independently selected -R a groups; or a four to seven membered heterocycle which is optionally substituted with one or more independently selected C 1 -C 4 alkyl groups, or a four to six membered heterocycle;
- R 3 is hydrogen, a halogen, a carboxy group, or R 3 together with R 4 and the heterocycle to which they are attached preferably form a twelve to thirteen membered heterocycle with three ring structures;
- R 4 is hydrogen, a halogen; a carboxy group; a C 1 -C 6 alkyl group; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 -C 8 aryl optionally substituted with one or more halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; an amino group optionally substituted with one or two independently selected C 6 -C 8 aryl or C 1 -C 6 alkyl groups, which are optionally substituted with a hydroxy, a phenyl, or a benzo[1,3]dioxole group; a carbony
- R 4 is selected from:
- R 5 is selected from the following, wherein the * indicates the bond of attachment:
- R 2 is a C 6 to C 8 aryl, optionally substituted with one, two, or three -R a groups, wherein R a is independently selected from: a halogen; a C 1 to C 6 alkyl; a C 1 to C 6 haloalkyl; a C 1 to C 6 alkoxy; a C 1 to C 6 haloalkoxy; a C 6 to C 8 aryl group; a carboxy group; a carbamoyl group; an amino group which is optionally substituted with one or two independently selected hydroxy groups, halogens, C 1 to C 6 alkyls, or C 1 to C 6 haloalkyls; or a four to six membered heterocycle optionally substituted with an oxo group.
- R a is independently selected from: a halogen; a C 1 to C 6 alkyl; a C 1 to C 6 haloalkyl; a C 1 to C 6 alkoxy; a C 1 to C
- a method of treating or preventing an autoimmune disease, a blood disease, a collagen disease, diabetes, a neurodegenerative disease, a cardiovascular disease, a pulmonary disease, or an inflammatory disease or central nervous system disease comprising administering to a patient in need thereof an effective amount of a compound of Formula 3 or 4, or a pharmaceutically acceptable salt, hydrate, solvate, clathrate, racemate, stereoisomer, or polymorph thereof
- central nervous system disease is multiple sclerosis, muscular dystrophy, Duchenne muscular dystrophy, Alzheimer's disease, a neurodegenerative disease or Parkinson's disease.
- a method of treating or preventing cancer in a human comprising administering to a human in need thereof an effective amount of a compound of Formula 3, or a pharmaceutically acceptable salt, hydrate, solvate, clathrate, racemate, stereoisomer, or polymorph thereof.
- the method of embodiment 34, wherein the administration is intravenous.
- the cancer is of the head and neck, eye, skin, mouth, throat, esophagus, chest, bone, blood, lung, colon, sigmoid, rectum, stomach, prostate, breast, ovaries, kidney, liver, pancreas, brain, intestine, heart or adrenals.
- the compound, or a pharmaceutically acceptable salt, hydrate, solvate, clathrate, racemate, stereoisomer, or polymorph thereof comprises a pharmaceutically acceptable carrier or diluent.
- cancer is sarcoma, carcinoma, fibrosarcoma, myxosarcoma, liposarcoma, chondrosarcoma, osteogenic sarcoma, chordoma, angiosarcoma, endotheliosarcoma, lymphangiosarcoma, lymphangioendotheliosarcoma, synovioma, mesothelioma, Ewing's tumor, leiomyosarcoma, rhabdomyosarcoma, colon carcinoma, pancreatic cancer, breast cancer, ovarian cancer, prostate cancer, squamous cell carcinoma, basal cell carcinoma, adenocarcinoma, sweat gland carcinoma, sebaceous gland carcinoma, papillary carcinoma, papillary adenocarcinomas, cystadenocarcinoma, medullary carcinoma, bronchogenic carcinoma, renal cell carcinoma, hepatoma, bile duct carcinoma, choriocarcinom
- the cancer is acute lymphoblastic leukemia, acute lymphoblastic B-cell leukemia, acute lymphoblastic T-cell leukemia, acute myeloblastic leukemia, acute promyelocyte leukemia, acute monoblastic leukemia, acute erythroleukemic leukemia, acute megakaryoblastic leukemia, acute myelomonocytic leukemia, acute nonlymphocyctic leukemia, acute undifferentiated leukemia, chronic myelocytic leukemia, chronic lymphocytic leukemia, hairy cell leukemia, or multiple myeloma. 41.
- a method of treating or preventing a disease associated with a mutation of the p53 gene comprising administering to a patient in need thereof an effective amount of a compound of Formula 3 or 4, or a pharmaceutically acceptable salt, hydrate, solvate, clathrate, racemate, stereoisomer, or polymorph thereof. 42. The method of embodiment 41 , wherein the administration is intravenous.
- a method of inhibiting the growth of a cancer cell comprising contacting the cancer cell with an effective amount of a compound of Formula 3 or 4, or a pharmaceutically acceptable salt, hydrate, solvate, clathrate, racemate, stereoisomer, or polymorph thereof.
- a method for selectively producing a protein in a mammal comprising transcribing a gene containing a nonsense mutation in the mammal; and providing an effective amount of a compound of the present invention to said mammal, wherein said protein is produced by said mammal.
- Figure 1 provides schematic representations of constructs for luciferase based assays to evaluate the suppression of a nonsense mutation.
- Figure 2 provides schematic representations of the luciferase constructs engineered to harbor one or more epitope tags in the N-terminus of the luciferase protein.
- Figure 3 provides schematic representations of constructs for luciferase based assays to evaluate readthrough efficiency.
- Premature translation termination can produce aberrant proteins which can be lethal or can cause a number of diseases, including as non-limiting examples, cancers, lysosomal storage disorders, the muscular dystrophies, cystic fibrosis and hemophilia.
- diseases including as non-limiting examples, cancers, lysosomal storage disorders, the muscular dystrophies, cystic fibrosis and hemophilia.
- compounds that suppress nonsense mutations have been identified, and methods for their use provided.
- compounds of the invention are provided which are useful in suppression of a nonsense mutation.
- Compounds of the present invention are also useful for increasing the expression of a protein.
- the compounds of the invention specifically suppresses a nonsense mutation, while in other embodiments, the compounds of the invention suppress a nonsense mutation as well as treat a disease, including as non-limiting examples, cancers, lysosomal storage disorders, the muscular dystrophies, cystic fibrosis and hemophilia.
- the present invention provides compounds of Formula (1) which are useful for suppressing premature translation termination associated with a nonsense mutation in mRNA, and for treating diseases associated with nonsense mutations in mRNA:
- Y and Z are independently selected from N or C; W is N or CH; n is 0, 1, 2 or 3;
- R 1 is hydrogen, a C 6 to C 8 aryl which is optionally substituted with a carboxy group, or R 1 is absent when Z is N;
- R 2 is hydrogen; a C 6 to C 8 aryl which is optionally substituted with one, two, or three independently selected R a groups; a four to seven membered heterocycle which is optionally substituted with one or more independently selected C 1 -C 6 alkyl groups or a three to seven membered heterocycle; or R 2 is absent when Y is N;
- R is independently selected from a halogen; a carboxy group; a C 1 -C 6 alkyl group optionally substituted with a four to seven membered heterocycle, a C 6 -C 8 aryloxy group, or an amino group, wherein the four to seven membered heterocycle, C 6 -C 8 aryloxy group, and amino group are optionally substituted with one or two independently selected
- C 1 -C 6 alkyl or C 6 -C 8 aryl groups which C 6 -C 8 aryl groups are optionally and independently substituted with one or more C 1 -C 6 alkyl groups; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 -C 8 aryl optionally substituted with one or more independently selected halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, oxy, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; an amino group optionally substituted with one or two independently selected C 6 -C 8 aryl or
- C 1 -C 6 alkyl groups which are optionally substituted with a hydroxy, a C 6 -C 8 aryl, or a nine to ten membered heterocycle having two ring structures; a carbonyl group substituted with a five to six membered heterocycle group; a four to seven membered heterocycle group optionally substituted with one more C 1 -C 4 alkyl or oxo groups; a nine to ten membered heterocycle having two ring structures; or two R groups, wherein R may also include an oxy group, together with the hetero-bicycle to which they are attached form a twelve to thirteen membered heterocycle having three ring structures; wherein R a is a halogen; a C 1 -C 6 alkyl; a C 1 -C 6 alkoxy which is optionally substituted with one or more independently selected halogen groups; a C 6 -C 8 aryl; a four to seven membered heterocycle which is optionally substituted with one or more
- compounds of Formula (2) are provided which are useful for suppressing premature translation termination associated with a nonsense mutation in mRNA, and for treating diseases associated with nonsense mutations in mRNA:
- Y and Z are independently selected from N or C; W is N or CH; n is 0, 1, 2 or 3;
- R 1 is hydrogen, a C 6 to C 8 aryl which is optionally substituted with a carboxy group, or R 1 is absent when Z is N;
- R 2 is hydrogen; a C 6 to C 8 aryl which is optionally substituted with one, two, or three independently selected R a groups; a four to seven membered heterocycle which is optionally substituted with one or more independently selected C 1 -C 6 alkyl groups or a three to seven membered heterocycle; or R 2 is absent when Y is N;
- R is independently selected from a halogen; a carboxy group; a C 1 -C 6 alkyl group optionally substituted with a four to seven membered heterocycle, a C 6 -C 8 aryloxy group, or an amino group, wherein the four to seven membered heterocycle, C 6 -C 8 aryloxy group, and amino group are optionally substituted with one or two independently selected
- C 1 -C 6 alkyl or C 6 -C 8 aryl groups which C 6 -C 8 aryl groups are optionally and independently substituted with one or more C 1 -C 6 alkyl groups; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 -C 8 aryl optionally substituted with one or more independently selected halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, oxy, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; an amino group optionally substituted with one or two independently selected C 6 -C 8 aryl or
- C 1 -C 6 alkyl groups which are optionally substituted with a hydroxy, a C 6 -C 8 aryl, or a nine to ten membered heterocycle having two ring structures; a carbonyl group substituted with a five to six membered heterocycle group; a four to seven membered heterocycle group optionally substituted with one more C 1 -C 4 alkyl or oxo groups; a nine to ten membered heterocycle having two ring structures; or two R groups, wherein R may also include an oxy group, together with the hetero-bicycle to which they are attached form a twelve to thirteen membered heterocycle having three ring structures; wherein R 3 is a hydroxy group; a halogen; a C 1 -C 6 alkyl which is optionally substituted with one or more independently selected halogen or hydroxy groups; a C 1 -C 6 alkoxy which is optionally substituted with one or more independently selected halogen or phenyl groups; a C
- compounds of Formula (3) are provided which are useful for suppressing premature translation tennination associated with a nonsense mutation in mRNA, and for treating diseases associated with nonsense mutations in mRNA:
- Y and Z are independently selected from N or C;
- W is N or CH; n is O, 1, 2 or 3;
- R 1 is absent or a C 6 to C 8 aryl which is optionally substituted with a carboxy group
- R 2 is absent; a C 6 to C 8 aryl which is optionally substituted with one, two, or three independently selected R a groups; or a four to seven membered heterocycle which is optionally substituted with one or more C 1 -C 4 alkyl groups, or a four to six membered heterocycle;
- R is independently selected from a halogen; a carboxy group; a C 1 -C 6 alkyl group optionally substituted with a C 6 -C 8 aryloxy group, an imidazole group, or an amino group which is optionally substituted with one or two independently selected C 1 -C 6 alkyl or C 6 - C 8 aryl groups; a Ci-C ⁇ alkoxy; a C 6 -C 8 aryloxy; a C ⁇ -C$ aryl optionally substituted with one or more halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; an amino group optionally substituted with one or two independently selected C 6 - C 8 aryl or C 1 -C 6 alkyl groups, which are optionally substituted with a hydroxy, a phenyl, or a benzo[1,3]dioxole group
- a compound of Formula 1 is provided with the proviso that at least one Of R 1 and R 2 is not hydrogen.
- compounds of Formulas IA and IB are provided with the proviso that at least one OfR 1 and R 2 is not hydrogen.
- a compound of Formula 2 is provided with the proviso that at least one of R 1 and R 2 is not hydrogen.
- compounds of Formulas 2A and 2B are provided with the proviso that at least one Of R 1 and R 2 is not hydrogen.
- a compound of Formula 4 is provided with the proviso that at least one Of R 1 and R 2 is not hydrogen.
- a compound of Formula 4A is provided with the proviso that at least one of R 1 and R 2 is not hydrogen.
- Y is N.
- Z is N.
- both Y and Z are N.
- neither Y nor Z is N.
- Y is C.
- Z is C.
- both Y and Z are C.
- neither Y nor Z is C.
- W is CH.
- W is N.
- n is 0. In another embodiment of Formulas 1, 2, and 3, n is 1. In another embodiment of Formulas 1, 2, and 3, n is 2. In a further embodiment of Formulas 1, 2, and 3, n is 3. In a preferred embodiment of Formulas 1, 2, and 3, n is 1. In an embodiment of Formulas 1, 2, and 3, R 1 is absent. In another embodiment of Formulas 1, 2, and 3, Z is N and R 1 is absent. In a further embodiment of Formulas 1, 2, and 3, Y is C, Z is N, and R 1 is absent.
- R 1 is C 6 to C 8 aryl optionally substituted with a carboxy group. In another embodiment of Formulas 1, 2, and 3, R 1 is a phenyl group optionally substituted with a carboxy group. In another embodiment of Formulas 1, 2, and 3, R 1 is a phenyl group substituted with a carboxy group, wherein the carboxy group is in the ortho, meta or para position.
- R 1 is a phenyl group substituted with a carboxy group, wherein the carboxy group is in the ortho, meta or para position, hi a further embodiment of Formulas 1, 2, and 3, Y is N, n is 1 or 2, and R 1 is C 6 to C 8 aryl optionally substituted with a carboxy group. In a further embodiment of Formulas 1, 2, and 3, Y is N, n is 1 or 2, and R 1 is a phenyl group optionally substituted with a carboxy group. In another embodiment of Formulas 1, 2, and 3, Y is N, n is 1, and R 1 is phenyl group substituted with a carboxy group, wherein the carboxy group is in the ortho, meta or para, and preferably in the meta or para, position.
- R 2 is absent. In another embodiment of Formulas 1, 2, and 3, R 2 is absent and R 1 is C 6 to C 8 aryl optionally substituted with a carboxy group. In another embodiment of Formulas 1, 2, and 3, R 2 is absent and R 1 is a phenyl group optionally substituted with a carboxy group. In another embodiment of Formulas 1, 2, and 3, R 2 is absent and R 1 is a phenyl group substituted with a carboxy group, wherein the carboxy group is in the ortho, meta or para position, hi another embodiment of Formulas 1 , 2, and 3, R 2 is absent and R 1 is a phenyl group substituted with a carboxy group, wherein the carboxy group is in the meta or para position. In a further embodiment of Formulas 1, 2, and 3, Y is N, n is 1 or 2, R 2 is absent and R 1 is C 6 to C 8 aryl optionally substituted with a carboxy group.
- R 2 is C 6 to C 8 aryl optionally substituted with one, two, or three independently selected R a groups. In an embodiment of Formulas 1, 2, and 3, R 2 is C 6 to C 8 aryl optionally substituted with a carboxy group. In another embodiment of Formulas 1, 2, and 3, R 2 is a phenyl group optionally substituted with a carboxy group. In another embodiment of Formulas 1, 2, and 3, R 2 is a phenyl group substituted with a carboxy group, wherein the carboxy group is in the ortho, meta or para position. In a further embodiment of Formulas 1, 2, and 3, Z is N and R 2 is C 6 to C 8 aryl optionally substituted with a carboxy group.
- Z is N and R 2 is a phenyl group optionally substituted with a carboxy group.
- Z is N and R 2 is a phenyl group substituted with a carboxy group, wherein the carboxy group is in the ortho, meta or para position.
- Z is N and R 2 is a phenyl group substituted with a carboxy group, wherein the carboxy group is in the meta or para position.
- R 2 is C 6 -C 8 aryl optionally substituted with one, two or three independently selected R a groups.
- R 2 is an unsubstituted C 6 -C 8 aryl.
- R 2 is a phenyl group optionally substituted with one, two or three independently selected R 3 groups.
- R 2 is an unsubstituted phenyl group.
- R 2 is C 6 -C 8 aryl substituted with one C 1 - C 6 alkyl group.
- R 2 is C 6 -C 8 aryl substituted with one C 1 -C 4 alkyl group.
- R 2 is C 6 -C 8 aryl substituted with one methyl group.
- R 2 is C 6 -C 8 aryl substituted with one propyl group. In an embodiment of Formulas 1, 2, and 3, R 2 is C 6 -C 8 aryl substituted with one isopropyl group. In an embodiment of Formulas 1,
- R 2 is a phenyl group substituted with one C 1 -C 4 alkyl group. In an embodiment of Formulas 1, 2, and 3, R 2 is phenyl group substituted with one methyl group. In an embodiment of Formulas 1, 2, and 3, R 2 is phenyl group substituted with one propyl group. In another embodiment of Formulas 1, 2, and 3, R 2 is phenyl group substituted with one isopropyl group.
- R 2 is C 6 -C 8 aryl substituted with one C 6 -C 8 aryl group. In another embodiment of Formulas 1, 2, and 3, R 2 is C 6 -C 8 aryl substituted with one phenyl group. In an embodiment of Formulas 1, 2, and 3, R 2 is a phenyl group substituted with one C 6 to C 8 aryl group. In a further embodiment of Formulas 1, 2, and 3, R 2 is phenyl group substituted with one phenyl group.
- R 2 is a C 6 -C 8 aryl group substituted with one four to seven membered heterocycle, wherein the four to seven membered heterocycle is optionally substituted with an oxo group.
- R 2 is a C 6 -C 8 aryl group substituted with one four to six membered heterocycle, wherein the four to six membered heterocycle is optionally substituted with an oxo group.
- R 2 is C 6 -C 8 aryl substituted with one pyrrolidine group, wherein the pyrrolidine is optionally substituted with an oxo group.
- R 2 is C 6 -C 8 aryl substituted with one morpholine group. In another embodiment of Formulas 1, 2, and 3, R 2 is C 6 -C 8 aryl substituted with one pyrazole group. In another embodiment of Formulas 1, 2, and 3, R 2 is C 6 -C 8 aryl substituted with one azetidine group. In an embodiment of Formulas 1 and 2, R 2 is phenyl group substituted with one one four to seven membered heterocycle, which four to seven membered heterocycle is optionally substituted with an oxo group.
- R 2 is phenyl group substituted with one one four to six membered heterocycle, which four to six membered heterocycle is optionally substituted with an oxo group
- R 2 is a phenyl group substituted with one pyrrolidine group, which pyrrolidine is optionally substituted with an oxo group.
- R 2 is a phenyl group substituted with one morpholine group.
- R 2 is phenyl group substituted with one pyrazole group.
- R 2 is phenyl group substituted with one azetidine group.
- R 2 is a C 6 -C 8 aryl group substituted with one halogen. In an embodiment of Formulas 1, 2, and 3, R 2 is a C 6 -C 8 aryl group substituted with one bromine. In an embodiment of Formulas 1, 2, and 3, R 2 is a C 6 -C 8 aryl group substituted with one fluorine. In an embodiment of Formulas 1, 2, and 3, R 2 is a phenyl group substituted with one halogen. In another embodiment of Formulas 1, 2, and 3, R 2 is a phenyl group substituted with one bromine. In another embodiment of Formulas 1, 2, and 3, R 2 is a phenyl group substituted with one fluorine.
- R 2 is a C 6 -C 8 aryl group substituted with one, two or three C 1 -C 6 alkoxy groups, each of which is optionally substituted with one or more independently selected halogens.
- R 2 is a C 6 -C 8 aryl group substituted with one, two or three C 1 -C 6 alkoxy groups, each of which is optionally substituted with one or more independently selected halogen or phenyl groups.
- R 2 is a C 6 -C 8 aryl group substituted with one, two or three C 1 -C 4 alkoxy groups, each of which is optionally substituted with one or more independently selected halogen or phenyl groups.
- R 2 is a C 6 -C 8 aryl group substituted with one methoxy group. In an embodiment of Formulas 1, 2, and 3, R 2 is a C 6 -C 8 aryl group substituted with one methoxy group, wherein the methoxy group is substituted with up to three independently selected halogens. In an embodiment of Formulas 1, 2, and 3, R 2 is a C 6 -C 8 aryl group substituted with a trifluoromethoxy group. In an embodiment of Formulas 1, 2, and 3, R 2 is a C 6 -C 8 aryl group substituted with two methoxy groups. In an embodiment of Formulas 1, 2, and 3, R 2 is a C 6 -C 8 aryl group substituted with two trifluoromethoxy groups.
- R 2 is a phenyl group substituted with one, two or three C 1 -C 4 alkoxy groups.
- R 2 is a phenyl group substituted with one methoxy group.
- R 2 is a phenyl group substituted with one, two or three C 1 -C 4 alkoxy groups, each of which is optionally substituted with one or more independently selected halogens.
- R 2 is a phenyl group substituted with one methoxy group.
- R 2 is a phenyl group substituted with one methoxy group, which is substituted with three independently selected halogens. In an embodiment of Formulas 1, 2, and 3, R 2 is a C 6 -C 8 aryl group substituted with one trifluoromethoxy group. In an embodiment of Formulas 1, 2, and 3, R 2 is a phenyl group substituted with two methoxy groups.
- R 2 is a C 6 -C 8 aryl group substituted with an amino group, wherein the amino group is optionally substituted with a C 1 -C 6 alkyl, which is optionally substituted with one or more independently selected halogens.
- R 2 is a C 6 -C 8 aryl group substituted with an amino group, wherein the amino group is optionally substituted with a C 1 -C 6 alkyl, which is optionally substituted with one or more hydroxyl groups.
- R 2 is a C 6 -C 8 aryl group substituted with an amino group, wherein the amino group is substituted with a propyl group, which is optionally substituted with one or more independently selected halogens.
- R 2 is a C 6 -C 8 aryl group substituted with an amino group, wherein the amino group is substituted with a propyl group, which is optionally substituted with one chlorine.
- R 2 is a C 6 -C 8 aryl group substituted with an amino group, wherein the amino group is substituted with a propyl group, which is optionally substituted with one hydroxyl group.
- R 2 is a C 6 -C 8 aryl group substituted with an amino group, wherein the amino group is substituted with a pentyl group, which is optionally substituted with one hydroxyl group.
- R 2 is a phenyl group substituted with an amino group, wherein the amino group is optionally substituted with a C 1 -C 6 alkyl, which is optionally substituted with one or more independently selected halogens.
- R 2 is a phenyl group substituted with an amino group, wherein the amino group is optionally substituted with a C 1 -C 6 alkyl, which is optionally substituted with one or more hydroxyl groups.
- R 2 is a phenyl group substituted with an amino group, wherein the amino group is substituted with a propyl group, which is optionally substituted with one or more independently selected halogens.
- R 2 is a phenyl group substituted with an amino group, wherein the amino group is substituted with a propyl group, which is optionally substituted with one chlorine.
- R 2 is a phenyl group substituted with an amino group, wherein the amino group is substituted with a propyl group, which is optionally substituted with one hydroxyl group.
- R 2 is a phenyl group substituted with an amino group, wherein the amino group is substituted with a pentyl group, which is optionally substituted with one hydroxyl group.
- R 2 is C 6 to C 8 aryl optionally substituted with a carbamoyl group. In another embodiment of Formulas 1, 2, and 3, R 2 is phenyl group optionally substituted with a carbamoyl group.
- R 2 is C 6 to C 8 aryl optionally substituted with two independently selected R a groups.
- R 2 is C 6 to C 8 aryl substituted with two R 3 groups, wherein the two R 3 groups together with the C 6 to C 8 aryl to which they are attached form a nine to ten membered heterocycle having two ring structures, wherein the nine to ten membered heterocycle having two ring structures is optionally substituted with one or more independently selected halogens.
- R 2 is C 6 to C 8 aryl substituted with two R 3 groups, wherein the two R 3 groups together with the C 6 to C 8 aryl form a benzo[1,3]dioxole, which benzo[1,3]dioxole is optionally substituted with one or more independently selected halogens.
- R 2 is C 6 to C 8 aryl substituted with two R 3 groups, wherein the two R 3 groups together with the C 6 to C 8 aryl to which they are attached form a benzo[1,3]dioxole, wherein the benzo[1,3]dioxole is optionally substituted with two fluorines.
- R 2 is C 6 to C 8 aryl substituted with two R 3 groups, wherein the two R 3 groups together with the C 6 to C 8 aryl form a 2,3-dihydro-benzofuran.
- R 2 is phenyl optionally substituted with two independently selected R 3 groups.
- R 2 is phenyl substituted with two R 3 groups, wherein the two R 3 groups together with the phenyl to which they are attached form a nine to ten membered heterocycle having two ring structures, wherein the nine to ten membered heterocycle having two ring structures is optionally substituted with one or more independently selected halogens.
- R 2 is phenyl substituted with two R 3 groups, wherein the two R 3 groups together with the phenyl form a benzo[1,3]dioxole, which benzo[1,3]dioxole is optionally substituted with one or more independently selected halogens.
- R 2 is phenyl substituted with two R 3 groups, wherein the two R a groups together with the phenyl to which they are attached form a benzo[1,3]dioxole, wherein the benzo[1,3]dioxole is optionally substituted with two fluorines.
- R 2 is phenyl substituted with two R a groups, wherein the two R a groups together with the phenyl form a 2,3-dihydro- benzofuran.
- R 2 is a four to seven membered heterocycle, which is optionally substituted with one or more independently selected C 1 to C 6 alkyl groups or a three to seven membered heterocycle.
- R 2 is a four to seven membered heterocycle, which is optionally substituted with one or more independently selected C 1 to C 4 alkyl groups or a four to six membered heterocycle.
- R 2 is a five membered heterocycle optionally substituted with one or more independently selected alkyl groups.
- R 2 is a furyl group.
- R 2 is a furyl group substituted with one or more independently selected C 1 to C 6 alkyl groups.
- R 2 is a furyl group substituted with one or more independently selected C 1 to C 4 alkyl groups.
- R 2 is a thiophenyl group substituted with one or more independently selected C 1 to C 6 alkyl groups.
- R 2 is a thiophenyl group substituted with one or more independently selected C 1 to C 4 alkyl groups.
- R 2 is a furyl group substituted with two methyl groups.
- R 2 is a thiophenyl group substituted with a methyl group.
- R 2 is a six membered heterocycle, which is optionally substituted with a three to seven membered heterocycle.
- R 2 is a six membered heterocycle, which is optionally substituted with a four to six membered heterocycle.
- R 2 is a six membered heterocycle, which is optionally substituted with a four membered heterocycle.
- R 2 is a six membered heterocycle, which is optionally substituted with a five membered heterocycle.
- R 2 is a six membered heterocycle, which is optionally substituted with a six membered heterocycle.
- R 2 is a pyridine group, optionally substituted with a four to six membered heterocycle. In another embodiment of Formulas 1, 2, and 3, R 2 is a six membered heterocycle, which is optionally substituted with a pyrrolidine, mophiline, piperidine, azetidine, or piperazine group. In another embodiment of Formulas 1, 2, and 3, R 2 is a pyridine group, optionally substituted with a pyrrolidine group. In another embodiment of Formulas 1, 2, and 3, R 2 is a pyridine group, optionally substituted with a morpholine group. In another embodiment of Formulas 1, 2, and 3, R 2 is a pyridine group, optionally substituted with a piperidine group.
- R 2 is a pyridine group, optionally substituted with an azetidine group. In another embodiment of Formulas 1, 2, and 3, R 2 is a pyridine group, optionally substituted with a piperazine group.
- R is a C 1 -C 6 alkyl group optionally substituted with a C 6 -C 8 aryloxy group, wherein the C 6 -C 8 aryloxy group is optionally substituted with one or two independently selected C 1 -C 6 alkyl groups.
- R is an unsubstituted C 1 -C 6 alkyl group.
- R is a methyl group.
- R is a propyl group.
- R is an isopropyl group.
- R is a butyl group.
- R is a tert-butyl group. In an embodiment of Formulas 1, 2, and 3, R is a pentyl group. In an embodiment of Formulas 1 and 2, R is a C 1 -C 6 alkyl group optionally substituted with an amino group, wherein the amino group is optionally substituted with one C 6 -C 8 aryl group wherein the C 6 -C 8 aryl group is optionally and independently substituted with one or more C 1 -C 6 alkyl groups.
- R is a methyl group substituted with an amino group, wherein the amino group is optionally substituted with one C 6 -C 8 aryl group wherein the C 6 -C 8 aryl group is optionally and independently substituted with one or more C 1 -C 6 alkyl groups.
- R is a methyl group substituted with an amino group, wherein the amino group is substituted with one C 6 -C 8 aryl group wherein the C 6 -C 8 aryl group is optionally and independently substituted with one or more C 1 -C 6 alkyl groups.
- R is a methyl group substituted with an amino group, wherein the amino group is substituted with one C 6 -C 8 aryl group wherein the C 6 -C 8 aryl group is substituted with one or more independently selected C 1 -C 6 alkyl groups.
- R is a methyl group substituted with an amino group, which is substituted with a phenyl group, which is optionally substituted with one or more independently selected C 1 -C 6 alkyl groups.
- R is a methyl group, which is substituted with an amino group, which is substituted with a phenyl group, which is optionally substituted with one propyl group.
- R is a methyl group, which is substituted with an amino group, which is substituted with a phenyl group, which is optionally substituted with one isopropyl group.
- R is a C 1 -C 6 alkyl group optionally substituted with a four to seven membered heterocycle. In an embodiment of Formulas 1 and 2, R is a C 1 -C 6 alkyl group substituted with a four to seven membered heterocycle that comprises nitrogen. In another embodiment of Formulas 1 and 2, R is a methyl group substituted with a four to seven membered heterocycle. In another embodiment of Formulas 1 and 2, R is a methyl group substituted with a four to seven membered heterocycle that comprises nitrogen. In a further embodiment of Formulas 1 and 2, R is a methyl group substituted with an imidazolidine group.
- R is a Ci-Ce alkoxy group. In another embodiment of Formulas 1, 2, and 3, R is methoxy.
- R is a C 6 -C 8 aryloxy group. In an embodiment of Formulas 1, 2, and 3, R is a phenoxy group.
- R is a C 6 -C 8 aryl group optionally substituted with one or more independently selected C 1 -C 4 alkyl groups.
- R is an unsubstituted C 6 -C 8 aryl group.
- R is a C 6 -C 8 aryl group optionally substituted with one or more methyl groups.
- R is a C 6 -C 8 aryl group optionally substituted with one or more propyl groups.
- R is a C 6 -C 8 aryl group substituted with two methyl groups.
- R is a C 6 -C 8 aryl group substituted with two propyl groups. In an embodiment of Formulas 1, 2, and 3, R is a C 6 -C 8 aryl group optionally substituted with one or more isopropyl groups. In an embodiment of Formulas 1, 2, and 3, R is a C 6 -C 8 aryl group substituted with two isopropyl groups. In an embodiment of Formulas 1, 2, and 3, R is a phenyl group optionally substituted with one or more methyl groups. In an embodiment of Formulas 1, 2, and 3, R is a phenyl group optionally substituted with one or more propyl groups.
- R is a phenyl group optionally substituted with one or more isopropyl groups. In an embodiment of Formulas 1, 2, and 3, R is a phenyl group substituted with two methyl groups. In an embodiment of Formulas 1, 2, and 3, R is a phenyl group substituted with two propyl groups. In an embodiment of Formulas 1, 2, and 3, R is a phenyl group substituted with two isopropyl groups.
- R is a C 6 -C 8 aryl group optionally substituted with one or more independently selected C 1 -C 4 haloalkyl groups. In another embodiment of Formulas 1, 2, and 3, R is a C 6 -C 8 aryl group optionally substituted with one halomethyl group. In another embodiment of Formulas 1, 2, and 3, R is a C 6 -C 8 aryl group optionally substituted with one trifluoromethyl group. In an embodiment of Formulas 1, 2, and 3, R is a phenyl group. In an embodiment of Formulas 1, 2, and 3, R is a phenyl group optionally substituted with one or more independently selected C 1 -C 4 haloalkyl groups. In an embodiment of Formulas 1, 2, and 3, R is a phenyl group optionally substituted with one halomethyl group, hi an embodiment of Formulas 1, 2, and 3, R is a phenyl group substituted with one trifluoromethyl group.
- R is a C 6 -C 8 aryl group optionally substituted with one or more independently selected C 1 -C 4 haloalkoxy groups. In another embodiment of Formulas 1, 2, and 3, R is a C 6 -C 8 aryl group optionally substituted with one halomethoxy group. In another embodiment of Formulas 1, 2, and 3, R is a C 6 -C 8 aryl group optionally substituted with one trifluoromethoxy group. In an embodiment of Formulas 1, 2, and 3, R is a phenyl group optionally substituted with one or more independently selected C 1 -C 4 haloalkoxy groups. In an embodiment of Formulas 1, 2, and 3, R is a phenyl group optionally substituted with one halomethoxy group. In an embodiment of Formulas 1, 2, and 3, R is a phenyl group substituted with one trifluoromethoxy group.
- R is a C 6 -C 8 aryl group optionally substituted with one or more independently selected halogens. In an embodiment of Formulas 1, 2, and 3, R is a C 6 -C 8 aryl group optionally substituted with one halogen. In an embodiment of Formulas 1, 2, and 3, R is a C 6 -C 8 aryl group substituted with two halogens. In an embodiment of Formulas 1, 2, and 3, R is a C 6 -C 8 aryl group substituted with one or more fluorines. In an embodiment of Formulas 1, 2, and 3, R is a C 6 -C 8 aryl group substituted with one fluorine.
- R is a C 6 - C 8 aryl group substituted with two fluorines.
- R is a phenyl group substituted with one halogen.
- R is a phenyl group substituted with two halogens.
- R is a phenyl group substituted with one or more fluorines.
- R is a phenyl group substituted with one fluorine.
- R is a phenyl group substituted with two fluorines.
- R is a four to seven membered heterocycle group optionally substituted with one or more oxo groups.
- R is a four membered heterocycle.
- R is a four membered heterocycle optionally substituted with an oxo group.
- R is a five membered heterocycle.
- R is a five membered heterocycle optionally substituted with an oxo group.
- R is a six membered heterocycle.
- R is a six membered heterocycle optionally substituted with an oxo group.
- R is a four to seven membered heterocycle group that comprises nitrogen. In an embodiment of Formulas 1, 2, and 3, R is a four to seven membered heterocycle group that comprises two or more nitrogens. In an embodiment of Formulas 1, 2, and 3, R is a four to seven membered heterocycle group that comprises at least one of both nitrogen and oxygen. In a further embodiment of Formulas 1, 2, and 3, R is a piperidine group. In another embodiment of Formulas 1, 2, and 3, R is a morpholine group. In an embodiment of Formulas 1, 2, and 3, R is a pyrrolidine group. In a further embodiment of Formulas 1, 2, and 3, R is an imidazolidine group.
- R is an azetidine group optionally substituted with one or more oxo groups. In another embodiment of Formulas 1, 2, and 3, R is an unsubstituted azetidine group. In another embodiment of Formulas 1, 2, and 3, R is an azetidine group substituted with one oxo group.
- R is a four to seven membered heterocycle group optionally substituted with one or more independently selected C 1 -C 4 alkyl groups.
- R is an unsubstituted four to seven membered heterocycle group.
- R is an unsubstituted four to seven membered heterocycle group that comprises one or more nitrogens.
- R is an unsubstituted four to seven membered heterocycle group that comprises one nitrogen.
- R is an unsubstituted four to seven membered heterocycle group that comprises two nitrogens.
- R is an unsubstituted four to seven, or five to seven, membered heterocycle group that comprises three nitrogens.
- R is a pyrrolidine group.
- R is a triazole group.
- R is a four to seven membered heterocycle group substituted with one C 1 -C 4 alkyl group.
- R is a four to seven membered heterocycle group substituted with one methyl group.
- R is a four to seven membered heterocycle group substituted with two C 1 -C 4 alkyl groups.
- R is a four to seven membered heterocycle group substituted with two methyl groups.
- R is a four to seven membered heterocycle group that comprises one or more oxygen and is optionally substituted with one or more independently selected C 1 -C 4 alkyl groups.
- R is a four to seven membered heterocycle group that comprises one or more nitrogen and is optionally substituted with one or more independently selected C 1 -C 4 alkyl groups.
- R is a four to seven membered heterocycle group that comprises one or more oxygen and one or more nitrogen and is optionally substituted with one or more independently selected C 1 -C 4 alkyl groups.
- R is a morpholine group that is optionally substituted with one or more independently selected C 1 -C 4 alkyl groups.
- R is a piperazine group that is optionally substituted with one or more independently selected C 1 -C 4 alkyl groups.
- R is a four to seven membered heterocycle group that comprises oxygen and nitrogen and is optionally substituted with two methyl groups.
- R is a four to seven membered heterocycle group that comprises two nitrogens and is optionally substituted with one methyl group.
- R is a morpholinyl group that is substituted with two methyl groups.
- R is a piperazine group that is substituted with one methyl group.
- R is a diazepane group that is substituted with one methyl group.
- R is a four to seven membered heterocycle optionally substituted with one or more oxo groups. In an embodiment of Formulas 1, 2, and 3, R is a four to seven membered heterocycle that comprises nitrogen and is optionally substituted with one or more oxo groups. In an embodiment of Formulas I 5 2, and 3, R is a pyrrolidine group optionally substituted with an oxo group. In an embodiment of Formulas 1, 2, and 3, R is a pyrrolidine group substituted with an oxo group.
- R is a nine to ten membered heterocycle having two ring structures.
- Li an embodiment of Formulas 1, 2, and 3, R is a nine to ten membered heterocycle comprising at least one nitrogen and having two ring structures.
- R is a nine to ten membered heterocycle comprising at least one oxygen and having two ring structures.
- R is a nine to ten membered heterocycle comprising at least two oxygens and having two ring structures.
- R is a nine to ten membered heterocycle comprising at least two oxygens and at least one nitrogen and having two ring structures.
- R is a nine to ten membered heterocycle having two ring structures, one of which is a furan. In an embodiment of Formulas 1, 2, and 3, R is 2,3-dihydro-benzofuran. In an embodiment of Formulas 1, 2, and 3, R is a nine to ten membered heterocycle having two ring structures, one of which is a dioxolane. In an embodiment of Formulas 1, 2, and 3, R is a nine to ten membered heterocycle having two ring structures, one of which is a piperidine. In an embodiment of Formulas 1, 2, and 3, R is a nine to ten membered heterocycle having two ring structures, one of which is a pyrrolidine.
- R is a nine to ten membered heterocycle having two ring structures, one of which is a dioxolane.
- R is l,4-dioxa-8-aza-spiro-[4.5]decane.
- R is a nine to ten membered heterocycle having two ring structures, one of which is a pyrrolidine.
- R is an indane.
- R is an amino group optionally substituted with one or two independently selected C 1 -C 6 alkyl groups, each of which is optionally and independently substituted with a hydroxy group, a C 6 -C 8 aryl group, or a nine to ten membered heterocycle having two ring structures.
- R is an amino group optionally substituted with one or two independently selected C 1 -C 6 alkyl groups, each of which is optionally and independently substituted with a hydroxy group, a phenyl group, or a benzo[1,3]-dixoxole group.
- R is an amino group substituted with an ethyl group, wherein the ethyl group is optionally substituted with a hydroxy group.
- R is an ammo group substituted with a butyl group, wherein the butyl group is optionally substituted with a hydroxy group.
- R is an amino group substituted with a hexyl group, wherein the hexyl group is optionally substituted with a hydroxy group.
- R is an amino group substituted with an ethyl group, wherein the ethyl group is substituted with a hydroxy group.
- R is an amino group substituted with a butyl group, wherein the butyl group is substituted with a hydroxy group.
- R is an amino group substituted with a hexyl group, wherein the hexyl group is substituted with a hydroxy group.
- R is an amino group substituted with an ethyl group, wherein the ethyl group is optionally substituted with a phenyl group.
- R is an amino group substituted with a propyl group, wherein the propyl group is optionally substituted with a phenyl group.
- R is an amino group substituted with an ethyl group, wherein the ethyl group is substituted with a phenyl group.
- R is an amino group substituted with a propyl group, wherein the propyl group is substituted with a phenyl group.
- R is an amino group substituted with a pentyl group, wherein the pentyl group is optionally substituted with a hydroxy group.
- R is an amino group substituted with a pentyl group, wherein the pentyl group is substituted with a hydroxy group.
- R is an amino group optionally substituted with two methyl groups, both of which are optionally and independently substituted with a phenyl group.
- R is an amino group optionally substituted with two methyl groups, one of which is substituted with a phenyl group.
- R is an amino group optionally substituted with one or two independently selected C 6 -C 8 aryl groups. In an embodiment of Formulas 1, 2, and 3, R is an amino group optionally substituted with a phenyl group. In an embodiment of Formulas 1, 2, and 3, R is a carboxy group. In an embodiment of Formulas 1, 2, and 3, R is a carbonyl group substituted with a five to six membered heterocycle group. In an embodiment of Formulas 1, 2, and 3, R is a carbonyl group substituted with a five membered heterocycle group. In another embodiment of Formulas 1, 2, and 3, R is a carbonyl group substituted with a six membered heterocycle group.
- R is a carbonyl group substituted with a five to six membered heterocycle group that comprises nitrogen. In an embodiment of Formulas 1, 2, and 3, R is a carbonyl group substituted with a pyrrolidine group.
- each R is an independently selected halogen.
- R is chlorine.
- R is bromine.
- R is fluorine
- R is a carbonyl group substituted with a five to six membered heterocycle group.
- R is a carbonyl group substituted with a heterocycle that comprises oxygen.
- R is a carbonyl group substituted with a heterocycle that comprises nitrogen.
- R is a carbonyl group substituted with a heterocycle that comprises oxygen and nitrogen.
- R is a carbonyl group substituted with a morpholine group.
- two R groups together with the hetero-bicycle to which they are attached form a twelve to thirteen membered heterocycle having three ring structures.
- two alkoxy groups together with the hetero-bicycle to which they are attached form a twelve to thirteen membered ring.
- two methoxy groups together with the hetero-bicycle to which they are attached form a twelve to thirteen membered ring.
- one alkoxy and one oxy group together with the hetero-bicycle to which they are attached form a twelve to thirteen membered ring.
- a methoxy and an oxy group together with the hetero-bicycle to which they are attached form a twelve to thirteen membered ring.
- W is N, Y is C, and Z is N. In a further embodiment of Formulas 1, 2, and 3, W is N, Y is C, Z is N, and n is 0. In a further embodiment of Formulas 1, 2, and 3, W is N, Y is C, Z is N, n is 0, and R 2 is C 6 to C 8 aryl. In another embodiment of Formulas 1, 2, and 3, W is N 3 Y is C, Z is N, n is 0, and R 2 is a phenyl group. In a further embodiment of Formulas 1, 2, and 3, W is N, Y is C, Z is N, and n is
- W is N, Y is C, Z is N, n is 1, and R is a halogen.
- W is N, Y is C, Z is N, n is 1,
- R is a halogen
- R 2 is C 6 to C 8 aryl.
- W is N
- Y is C
- Z is N 5 n is 1
- R is a halogen
- R 2 is a phenyl group.
- certain compounds of the invention may include at least one chiral center, and as such may exist as racemic mixtures or as enantiomerically pure compositions.
- enantiomerically pure refers to compositions consisting substantially of a single isomer, preferably consisting of 90%, 92%, 95%, 98%, 99%, or 100% of a single isomer.
- alkyl generally refers to saturated hydrocarbyl radicals of straight, branched or cyclic configuration including methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, n-hexyl, cyclohexyl, n-heptyl, octyl, n- octyl, and the like.
- alkyl substituents may be C 1 to C 8 , C 1 to C 6 , or C 1 to C 4 alkyl groups.
- the alkyl group may be optionally substituted with one or more independently selected halogen or alkoxy groups.
- the alkyl group may be a haloalkyl, including monohaloalkyl, dihaloalkyl, and trihaloalkyl.
- alkenyl generally refers to linear, branched or cyclic alkene radicals having one or more carbon-carbon double bonds, such as C 2 to C 6 alkylene groups including 3-propenyl.
- aryl refers to a carbocyclic aromatic ring structure. Included in the scope of aryl groups are aromatic rings having from five to twenty carbon atoms.
- Aryl ring structures include compounds having one or more ring structures, such as mono-, bi-, or tricyclic compounds. Examples of aryl groups that include phenyl, tolyl, anthracenyl, fluorenyl, indenyl, azulenyl, phenanthrenyl (i.e., phenanthrene), and napthyl ⁇ i.e., napthalene) ring structures. In certain embodiments, the aryl group may be optionally substituted.
- heterocycle refers to cyclic ring structures in which one or more atoms in the ring, the heteroatom(s), is an element other than carbon. Heteroatoms are typically O, S or N atoms. Included within the scope of heterocycle, and independently selectable, are O, N, and S heterocycle ring structures.
- the ring structure may include compounds having one or more ring structures, such as mono-, bi-, or tricyclic compounds, and may be aromatic, i.e., the ring structure may be a heteroaryl.
- heterocyclo groups include morpholinyl, pyrrolidinonyl, pyrrolidinyl, piperidinyl, piperazinyl, hydantoinyl, valerolactamyl, oxiranyl, oxetanyl, tetrahydrofuranyl, tetrahydropyranyl, tetrahydropyridinyl, tetrahydroprimidinyl, tetrahydrothiophenyl or tetrahydrothiopyranyl and the like.
- the heterocycle may optionally be substituted.
- heteroaryl refers to cyclic aromatic ring structures in which one or more atoms in the ring, the heteroatom(s), is an element other than carbon. Heteroatoms are typically O, S or N atoms. Included within the scope of heteroaryl, and independently selectable, are O, N, and S heteroaryl ring structures.
- the ring structure may include compounds having one or more ring structures, such as mono-, bi-, or tricyclic compounds.
- the heteroaryl groups may be selected from heteroaryl groups that contain two or more heteroatoms, three or more heteroatoms, or four or more heteroatoms.
- Heteroaryl ring structures may be selected from those that contain five or more atoms, six or more atoms, or eight or more atoms. In a preferred embodiment, the heteroaryl including five to ten atoms.
- heteroaryl ring structures include: acridine, benzimidazole, benzoxazole, benzodioxole, benzofuran, 1,3- diazine, 1,2-diazine, 1,2-diazole, 1,4-diazanaphthalene, furan, furazan, imidazole, indole, isoxazole, isoquinoline, isothiazole, oxazole, purine, pyridazine, pyrazole, pyridine, pyrazine, pyrimidine, pyrrole, quinoline, quinoxaline, thiazole, thiophene, 1,3,5-triazine, 1,2,4-triazine, 1,2,3-tria
- alkoxy generally refers to a group with the structure -O-R.
- R may be an alkyl group, such as a C 1 to C 8 , C 1 to C 6 alkyl group, or C 1 to C 4 alkyl group, hi certain embodiments, the R group of the alkoxy may optionally be substituted with at least one halogen.
- the R group of the alkoxy may be a haloalkyl, i.e., haloalkoxy.
- Halogen substituents may be independently selected from the halogens such as fluorine, chlorine, bromine, iodine, and astatine.
- each functionality or substituent appearing at any location within the disclosed compounds may be independently selected, and as appropriate, independently substituted.
- a more generic substituent is set forth for any position in the molecules of the present invention, it is understood that the generic substituent may be replaced with more specific substituents, and the resulting molecules are within the scope of the molecules of the present invention.
- R a is preferably independently selected from: a halogen; a C 1 to C 6 alkyl; a C 1 to C 6 haloalkyl; a C 1 to C 6 alkoxy; a C 1 to C 6 haloalkoxy; a C 6 to C 8 aryl group; a carboxy group; a carbamoyl group; an amino group which is optionally substituted.
- the four to seven membered heterocycles of Formulas 1, 2, and 3 are preferably selected from: an azetidine group, a pyrrolidine group, a piperidine group, a piperazine group, a morpholine group, a [1,4]diazepane group, a pyrazole group, an imidazole group, a [1,2,4] triazole group, a pyridine group, a furan group, and a thiophene group.
- the four to seven membered heterocycles may be optionally substituted as illustrated in Formulas 1, 2, and 3.
- the nine to ten membered heterocycle having two ring structures is preferably selected from the group consisting of a benzofuran group, a 2,3-dihydro-benzofuran group, a benzo[1,3]dioxole group, a 2,3- dihydro-isoindole group, a 2,3-dihydro-indole group, a 1,2,3,4-tetrahydro-isoquinoline group, and a l,4-dioxa-8-aza-spiro[4.5]decane group.
- R 2 is preferably selected from the following, wherein the * indicates the bond of attachment:
- R is preferably selected from the following, wherein the * indicates the bond of attachment:
- Y is C 5 Z is N, and R 1 is absent (Formula 1/ 2/3- A):
- n is preferably 1 or 2, and R group is in the 5 and/or 6 position.
- R is preferably a carboxy group.
- R 2 is a C 6 to C 8 aryl, optionally substituted with one, two, or three - R a groups, wherein R a is preferably independently selected from: a halogen; a C 1 to C 6 alkyl; a C 1 to C 6 haloalkyl; a C 1 to C 6 alkoxy; a C 1 to C 6 haloalkoxy; a carboxy group; a carbamoyl group; an amino group which is optionally substituted; or a four to six membered heterocycle optionally substituted with an oxo group.
- R 2 is a C 6 to C 8 aryl, optionally substituted with one, two, or three -R a groups, wherein R a is preferably independently selected from: a halogen; a C 1 to C 6 alkyl; a C 1 to C 6 alkoxy; a C 1 to C 6 haloalkoxy; a carboxy group; a carbamoyl group; an amino group which is optionally substituted; or a four to six membered heterocycle optionally substituted with an oxo group.
- R 2 is a phenyl group optionally substituted with a carboxy group.
- W is CH
- n is 1
- R is a carboxy group and is located in the 5 -position, as follows (Formula 1/2/3 -A-I):
- W is CH
- n is 1
- R is a carboxy group and is located in the 6-position, as follows (Formula 1/2/3-A-2):
- R 2 is a phenyl group substituted with a carboxy group
- W is CH
- n is 1
- R is located in the 5 -position, as follows (Formula 1/2/3-A-3):
- the carboxy group is preferably in a meta or para position.
- R is a halogen; a carboxy group; a C 1 -C 6 alkyl group; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 - C 8 aryl optionally substituted with one or more independently selected halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; an amino group optionally substituted with one or two independently selected C 6 -C 8 aryl or C 1 -C 6 alkyl groups, which are optionally substituted with a hydroxy, a phenyl, or a benzo[1,3]dioxole group; a carbonyl group substituted with a five to
- R is a halogen; a carboxy group; a C 1 -C 6 alkyl group; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 -C 8 aryl optionally substituted with one or more independently selected halogen, C 1 -C 4 alkyl or C 1 -C 4 haloalkoxy groups; an amino group optionally substituted with one or two independently selected C 1 -C 6 alkyl groups, which are optionally substituted with a hydroxy, a phenyl, or a benzo[1,3]dioxole group; a carbonyl group substituted with a five to six membered heterocycle group; or a four to seven membered heterocycle group optionally substituted with one more oxo groups; or a nine to ten membered heterocycle having two ring structures.
- R is selected from a halogen; a carboxy group; a C 1 -C 6 alkyl
- R 2 is a phenyl group substituted with a carboxy group, W is CH, n is 1, and R is located in the 6-position, as follows (Formula 1/2/3-A-4):
- the carboxy group is preferably in a meta or para positions.
- R is a halogen; a carboxy group; a C 1 -C 6 alkyl group optionally substituted with a C 6 -C 8 aryloxy group, an imidazole group, or an amino group which is optionally substituted with one or two independently selected C 1 -C 6 alkyl or C 6 - C 8 aryl groups; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 -C 8 aryl optionally substituted with one or more independently selected halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; a carbonyl group substituted with a five to six membered heterocycle group; a four to seven membered heterocycle group optionally substitute
- R is a halogen; a carboxy group; a C 1 -C 6 alkyl group; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 -C 8 aryl optionally substituted with one or more independently selected halogen, Cj-C 4 alkyl or C 1 -C 4 haloalkoxy groups; an amino group optionally substituted with one or two independently selected C 1 -C 6 alkyl groups, which are optionally and independently substituted with a hydroxy, a phenyl, or a benzo[1,3]dioxole group; a carbonyl group substituted with a five to six membered heterocycle group; or a four to seven membered heterocycle group optionally substituted with one more oxo groups; or a nine to ten membered heterocycle having two ring structures.
- R is selected from
- R 2 is a phenyl group substituted with a carbamoyl group, W is CH, n is 1, and R is located in the 5- position, as follows (Formula 1/2/3-A-5):
- the carbamoyl group is preferably in a meta or para positions.
- R is a halogen; a carboxy group; a C 1 -C 6 alkyl group; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 -C 8 aryl optionally substituted with one or more independently selected halogen, C 1 -C 4 alkyl, Q- C 4 haloalkyl, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; an amino group optionally substituted with one or two independently selected C 6 -C 8 aryl or C 1 -C 6 alkyl groups, which are optionally substituted with a hydroxy, a phenyl, or a benzo[1,3]dioxole group; a carbonyl group substituted with a five to six membered heterocycle group
- R is a halogen; a C 1 -C 6 alkyl group; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; an amino group optionally substituted with one or two independently selected C 6 -C 8 aryl or C 1 -C 6 alkyl groups, which are optionally substituted with a phenyl; or a four to seven membered heterocycle group.
- R 2 is a phenyl group substituted with a carbamoyl group, W is CH, n is 1, and R is located in the 6-position, as follows (Formula 1/2/3-A-6):
- the carbamoyl group is preferably in a meta or para positions.
- R is selected from the group consisting of a halogen; a carboxy group; a C 1 -C 6 alkyl group optionally substituted with a C 6 -C 8 aryloxy group, an imidazole group, or an amino group which is optionally substituted with one or two independently selected C 1 -C 6 alkyl or C 6 -C 8 aryl groups; a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 -C 8 aryl optionally substituted with one or more independently selected halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; a carbonyl group substituted with a five to six membered heterocycle group; a four to seven membered hetero
- R is a halogen; a C 1 -C 6 alkyl group; a Cj-C 6 alkoxy; a C 6 -C 8 aryloxy; an amino group optionally substituted with one or two independently selected C 6 -C 8 aryl or C 1 -C 6 alkyl groups, which are optionally substituted with a phenyl; or a four to seven membered heterocycle group.
- Y is N
- Z is C
- W is CH
- R 2 is absent
- R 1 is preferably a phenyl group optionally substituted with a carboxy group.
- R is a C 1 -C 6 alkyl; a C 1 -C 6 alkoxy; a C 6 -C 8 aryl optionally substituted with one or more independently selected halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; or two R groups together with the hetero-bicycle to which they are attached form a twelve to thirteen membered heterocycle having three ring structures.
- the twelve to thirteen membered heterocycle is selected from the following, wherein the * indicates the bond of attachment to R 1 :
- preferred compounds of the invention also include the compounds of Formula 4:
- Y and Z are independently selected from N or C; W is N or CH;
- R 1 is hydrogen, a C 6 to C 8 aryl which is optionally substituted with a carboxy group, or R 1 is absent when Z is N;
- R 2 is hydrogen; a C 6 to C 8 aryl which is optionally substituted with one, two, or three independently selected R 3 groups; a four to seven membered heterocycle which is optionally substituted with one or more independently selected C 1 -C 6 alkyl groups or a three to seven membered heterocycle; or R 2 is absent when Y is N;
- R 3 is absent; a halogen; a carboxy group; an alkoxy group; or R 3 , wherein R 3 may also include an oxy group, together with R 4 and the heterocycle to which they are attached preferably form a twelve to thirteen membered heterocycle with three ring structures;
- R 4 is absent, a halogen; a carboxy group; a C 1 -C 6 alkyl group; a C 1 -C 6 alkoxy; a
- C 6 -C 8 aryloxy a C 6 -C 8 aryl optionally substituted with one or more independently selected halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; an amino group optionally substituted with one or two independently selected C 6 -
- C 8 aryl or C 1 -C 6 alkyl groups which are optionally substituted with a hydroxy, a phenyl, or a benzo[1,3]dioxole group; a carbonyl group substituted with a five to six membered heterocycle group; or a four to seven membered heterocycle group optionally substituted with one more C 1 -C 4 alkyl or oxo groups; a nine to ten membered heterocycle having two ring structures; or R 3 together with R 4 and the heterocycle to which they are attached preferably form a twelve to thirteen membered heterocycle with three ring structures;
- R 5 is independently selected from: absent, a halogen; a carboxy group; a C 1 -C 6 alkyl group optionally substituted with a C 6 -C 8 aryloxy group, an imidazole group, or an amino group which is optionally substituted with one or two independently selected C 1 -
- C 6 alkyl or C 6 -C 8 aryl groups a C 1 -C 6 alkoxy; a C 6 -C 8 aryloxy; a C 6 -C 8 aryl optionally substituted with one or more independently selected halogen, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, C 1 -C 4 alkoxy, or C 1 -C 4 haloalkoxy groups; a carbonyl group substituted with a five to six membered heterocycle group; a four to seven membered heterocycle group optionally substituted with one more C 1 -C 4 alkyl or oxo groups; or a nine to ten membered heterocycle having two ring structures; or a pharmaceutically acceptable salt, hydrate, solvate, clathrate, racemate, stereoisomer, or polymorph of said compound of Formula 4.
- R 3 is hydrogen, halogen, or a carboxy group.
- R 3 together with R 4 and the heterocycle to which they are attached preferably form a twelve to thirteen membered heterocycle with three ring structures.
- R 4 is selected from:
- R 5 is selected from the following, wherein the * indicates the bond of attachment:
- R 2 is preferably a C 6 to C 8 aryl, optionally substituted with one, two, or three -R a groups, wherein R 3 is independently selected from: a halogen; a C 1 to C 6 alkyl; a C 1 to C 6 haloalkyl; a C 1 to C 6 alkoxy; a C 1 to C 6 haloalkoxy; a C 6 to C 8 aryl group; a carboxy group; a carbamoyl group; an amino group which is optionally substituted with one or two independently selected hydroxy groups, halogens, C 1 to C 6 alkyls, or C 1 to C 6 haloalkyls; or a four to six membered heterocycle optionally substituted with an oxo group.
- R 2 is a phenyl group optionally substituted with a carboxy group.
- Y is N
- Z is C
- W is CH and R 2 is
- Rj is a phenyl group optionally substituted with a carboxy group.
- R 3 is preferably hydrogen, a halogen, or a carboxy group.
- R 4 and R 5 are preferably independently selected from: hydrogen, a C 1 -C 6 alkyl group, a C 1 -
- R 3 together with R 4 and the heterocycle to which they are attached preferably form a twelve to thirteen membered heterocycle with three ring structures.
- the twelve to thirteen membered heterocycle is selected from the following, wherein the * indicates the bond of attachment to R 1 :
- Preferred compounds of the invention include the following.
- Particularly preferred compounds are Compound NOs: 19, 20, 42, 49, 53, 56, 58, 59, 85, 91, 94, 96, 100, 101, 102.
- Compounds of the invention may be produced in any manner known in the art.
- certain benzooxazole compounds of Formulas 1-A and 2- A may be prepared in the manner shown in Scheme A.
- m (or p)-cyanato methyl benzoate (I) may be synthesized as follows. A solution of m (or p)-cyanobenzoic acid (1.0 equiv.) and K 2 CO 3 (1.5 equiv.) in DMF is treated with iodomethane (1.5 equiv.) and stirred at room temperature for 5 h until the complete consumption of the starting material. The reaction mixture is poured into ice-water to afford a white crystalline solid which is collected by filtration and washed with water and hexanes in sequence.
- Compound (I) is then used in the synthesis of m (or p)-methoxycarboniumidoyl- benzoic acid methyl ester hydrochloride (II) as follows.
- Compound (I) (1.0 equiv.) is dissolved in methanol, acetyl chloride (> 20.0 equiv.) is added to the solution at room temperature under stirring. After 5 ⁇ 6 h, the mixture is dried under reduced pressure to give a crude product which was washed with ethyl ether to obtain a pure product.
- compound (III) is used in the synthesis of m (or p) benzooxazol-2-yl- benzoic acid (IV) as follows.
- Compound (III) 1.0 equiv.
- LiI > 10.0 equiv.
- the mixture is refluxed for 5 h ⁇ overnight until invisible starting material by TLC.
- IN HCl is added to the reaction mixture until PH ⁇ 7.
- the resulting acidic mixture is extracted with ethyl acetate (2-3 times).
- the combined ethyl estate is dried over Mg 2 SO 4 , and evaporated under reduced pressure to give the crude product followed by washing with ethyl ether or recrystallization for further purification to obtain the product, compound (IV)
- benzooxazole compounds of Formula 1-A and 2-A may be synthesized using a palladium catalyzed Suzuki coupling reaction, as shown in Scheme B. See, e.g., Nicholas E. Leadbeater and Maria Marco. Organic Letters. 2002, 4(17), 2973-2976.
- compounds of the invention may generally be synthesized as follows.
- 3-(5-bromo-benzooxazole-2-yl) benzoic acid methyl ester (l.Oequiv.) substituted phenylboronic acid (1.0 equiv.), Na 2 CO 3 (3.0 equiv.), t-butylammonia iodide (TBAI, as a phase-transfer catalyst, 1.0 equiv.), 6-8 ml of H 2 O, and a stirring bar.
- the vessel is sealed and placed into the microwave cavity. 6Ow of power and 150 °C is used for the reaction condition. The reaction is held for 5-10 min.
- reaction mixture is acidified using IN HCl until PH ⁇ 7, and partitioned between ethyl acetate and water. Water layer is extracted with ethyl acetate 3 times. Ethyl acetate is combined and washed with Brine (in most of the cases, solid is crushed out in organic phase), then dried over Mg 2 SO 4 and evaporated. The residue is washed with ethyl ether to afford the desired product as a crystalline solid (in some cases, recrystallization is needed to improve the purity).
- benzooxazole compounds of Formula 1-A and 2- A may be synthesized using a copper catalyzed amidation reaction, as shown in Scheme C. See, e.g., Artis Klapars, Xiaohua Huang, and Stephen L. Buchwald. J. Am. Chem. Soc. 2002, 124, 7421-7428.
- compounds of the invention may generally be synthesized as follows.
- a 50ml culture tube is placed with 3-(5-bromo-benzooxazole-2- yl) benzoic acid methyl ester (1.0 equiv.) , CuI (5.0% equiv.), Cyclic amide (1.25 equiv.), and K 2 CO 3 (2.0 equiv.), evacuated, and backed charged with N 2 .
- Trans-diamino- cyclohexane (10.0% equiv.) and toluene (2.0-3.OmI) are added under N 2 .
- the tube is sealed with a Teflon cap and the reaction mixture is stirred at 110 °C for 15-20 h, or until no further reaction proceeding.
- the resulting reaction mixture is added water, and extracted with ethyl acetate.
- the organic layer is washed with H 2 O to afford a colorful solid which is purified by flash chromatography using a mixture of ethyl acetate - heaxnes followed by methanol - methylene chloride in different ratio to provide desired product as a solid.
- benzooxazole compounds of Formula 1-A and 2-A may be synthesized using a commercial unavailable amino phenols and aromatic nuclephilic substitution seaction, as shown in Scheme D.
- compounds of the invention may generally be synthesized as follows.
- a mixture of 5-fluoro-2-nitrophenol (1.0 equiv.), amines (1.5-3.0 equiv.) in DMSO (4.0-10.0ml) is charged in a 50 ml culture tube and stirred at room temperature to 98 °C for 2 h-overnight. Yellow solid is precipitated by addition of water. Desired product is collected by filtration and washed with water, hexanes in sequence.
- the obtained product from the above reaction is dissolved or suspended in methanol (20- 30ml), and is added with 20% (by weight) of Pd/C.
- Schemes E, F, G, and H may generally be used to synthesize the compounds of Formula 1-B and 2-B, as follows.
- compounds of the invention may be resolved to enantiomerically pure compositions or synthesized as enantiomerically pure compositions using any method known in art.
- compounds of the invention may be resolved by direct crystallization of enantiomer mixtures, by diastereomer salt formation of enantiomers, by the formation and separation of diasteriomers or by enzymatic resolution of a racemic mixture.
- methods are provided for the suppression of premature translation termination, which may be associated with a nonsense mutation, and for the prevention or treatment of diseases.
- diseases are associated with mutations of mRNA, especially nonsense mutations.
- Exemplary diseases include, but are not limited to, cancer, lysosomal storage disorders, the muscular dystrophies, cystic fibrosis, hemophilia, epidermolysis bullosa and classical late infantile neuronal ceroid lipofuscinosis.
- methods for treating cancer, lysosomal storage disorders, a muscular dystrophy, cystic fibrosis, hemophilia, or classical late infantile neuronal ceroid lipofuscinosis comprising administering a therapeutically effective amount of at least one compound of the invention to a subject in need thereof.
- the present invention is directed to methods for increasing the expression of one or more specific, functional proteins. Any compound of the invention can be used to specifically increase expression of functional protein.
- a specific increase in expression of functional protein occurs when premature translation termination is suppressed by administering a therapeutically effective amount of at least one compound of the invention to a subject in need thereof.
- premature translation termination is associated with a nonsense mutation in mRNA.
- a specific increase in expression of functional protein occurs when mRNA decay is reduced in a patient.
- the abnormality in a patient is caused by mutation-mediated mRNA decay.
- mutation-mediated mRNA decay is the result of a nonsense mutation.
- the methods of the present invention are not limited by any particular theory.
- the invention encompasses methods of treating and preventing diseases or disorders ameliorated by the suppression of premature translation termination, nonsense- mediated mRNA decay, or premature translation termination and nonsense-mediated mRNA decay in a patient which comprise administering to a patient in need of such treatment or prevention a therapeutically effective amount of a compound of the invention.
- the present invention encompasses the treatment or prevention of any disease that is associated with a gene exhibiting premature translation termination, nonsense-mediated mRNA decay, or premature translation termination and nonsense-mediated mRNA decay.
- the disease is due, in part, to the lack of or reduced expression of the gene resulting from a premature stop codon.
- Diseases ameliorated by the suppression of premature translation termination, nonsense-mediated mRNA decay, or premature translation termination and nonsense- mediated mRNA decay include, but are not limited to: genetic diseases, somatic diseases, cancers, autoimmune diseases, blood diseases, collagen diseases, diabetes, neurodegenerative diseases, proliferative diseases, cardiovascular diseases, pulmonary diseases, inflammatory diseases or central nervous system diseases.
- diseases to be treated or prevented by administering to a patient in need thereof an effective amount of a compound of the invention include, but are not limited to, amyloidosis, hemophilia, Alzheimer's disease, Tay Sachs disease, Niemann Pick disease, atherosclerosis, giantism, dwarfism, hypothyroidism, hyperthyroidism, aging, obesity, Parkinson's disease, cystic fibrosis, muscular dystrophy, heart disease, kidney stones, ataxia-telangiectasia, familial hypercholesterolemia, retinitis pigmentosa, Duchenne muscular dystrophy, epidermolysis bullosa and Marfan syndrome.
- the diseases are associated with a nonsense mutation.
- the compounds of the invention are useful for treating or preventing an autoimmune disease.
- the autoimmune disease is associated with a nonsense mutation.
- the autoimmune disease is rheumatoid arthritis or graft versus host disease.
- the compounds of the invention are useful for treating or preventing a blood disease.
- the blood disease is associated with a nonsense mutation.
- the blood disease is hemophilia, Von Willebrand disease, ataxia-telangiectasia, ⁇ -thalassemia or kidney stones.
- the compounds of the invention are useful for treating or preventing a collagen disease.
- the collagen disease is associated with a nonsense mutation.
- the collagen disease is osteogenesis imperfecta or cirrhosis.
- the compounds of the invention are useful for treating or preventing diabetes.
- the diabetes is associated with a nonsense mutation.
- the compounds of the invention are useful for treating or preventing an inflammatory disease.
- the inflammatory disease is associated with a nonsense mutation.
- the inflammatory disease is arthritis, rheumatoid arthritis or osteoarthritis.
- the compounds of the invention are useful for treating or preventing a central nervous system disease.
- the central nervous system disease is associated with a nonsense mutation.
- the central nervous system disease is a neurodegenerative disease.
- the central nervous system disease is multiple sclerosis, muscular dystrophy, Duchenne muscular dystrophy, Alzheimer's disease, Tay Sachs disease, Niemann Pick disease, late infantile neuronal ceroid lipofuscinosis (LINCL) or Parkinson's disease.
- the compounds of the invention are useful for treating or preventing cancer, particularly in humans.
- the cancer is of the head and neck, eye, skin, mouth, throat, esophagus, chest, bone, blood, lung, colon, sigmoid, rectum, stomach, prostate, breast, ovaries, kidney, liver, pancreas, brain, intestine, heart or adrenals.
- the cancer is a solid tumor.
- the cancer is associated with a nonsense mutation.
- the cancer is associated with a genetic nonsense mutation.
- the cancer is associated with a somatic mutation.
- the use of the compounds of the invention against cancer may relate to its action against mutations of the p53 gene.
- the cancer is not a blood cancer. In another embodiment, the cancer is not leukemia. In another embodiment, the cancer is not multiple myeloma. In another embodiment, the cancer is not prostate cancer.
- the compounds of the invention are useful for treating or preventing cancer associated with a mutation of tumor suppressor gene.
- genes include, but are not limited to PTEN, BRCAl, BRCA2, Rb, and the p53 gene.
- the mutation is a genetic mutation.
- the mutation is a somatic mutation.
- the methods of the invention are particularly useful for treating or preventing a cancer associated with a nonsense mutation in the in a tumor suppressor gene.
- the methods of the invention are particularly useful for treating or preventing a cancer associated with a p53 gene due to the role of p53 in apoptosis.
- apoptosis can be induced by contacting a cell with an effective amount of a compound of the invention resulting in suppression of the nonsense mutation, which, in turn, allows the production of full-length p53 to occur.
- Nonsense mutations have been identified in the p53 gene and have been implicated in cancer.
- Several nonsense mutations in the p53 gene have been identified (see, e.g., Masuda et al., 2000, Tokai J Exp Clin Med. 25(2):69-77; Oh et al., 2000, MoI Cells 10(3):275-80; Li et al., 2000, Lab Invest.
- diseases to be treated or prevented by administering to a patient in need thereof an effective amount of a compound of the invention include, but are not limited to, solid tumors such as sarcoma, carcinomas, fibrosarcoma, myxosarcoma, liposarcoma, chondrosarcoma, osteogenic sarcoma, chordoma, angiosarcoma, endotheliosarcoma, lymphangiosarcoma, lymphangioendotheliosarcoma, synovioma, mesothelioma, Ewing's tumor, leiomyosarcoma, rhabdomyosarcoma, colon carcinoma, pancreatic cancer, breast cancer, ovarian cancer, prostate cancer, squamous cell carcinoma, basal cell carcinoma, adenocarcinoma, sweat gland carcinoma, sebaceous gland carcinoma, papillary carcinoma, papillary adenocarcinomas, cystadenocarcinoma, medull
- diseases to be treated or prevented by administering to a patient in need thereof an effective amount of a compound of the invention include, but are not limited to, a blood-born tumor such as acute lymphoblastic leukemia, acute lymphoblastic B-cell leukemia, acute lymphoblastic T-cell leukemia, acute myeloblastic leukemia, acute promyelocytic leukemia, acute monoblastic leukemia, acute erythroleukemic leukemia, acute megakaryoblastic leukemia, acute myelomonocytic leukemia, acute nonlymphocyctic leukemia, acute undifferentiated leukemia, chronic myelocytic leukemia, chronic lymphocytic leukemia, hairy cell leukemia, or multiple myeloma.
- a blood-born tumor such as acute lymphoblastic leukemia, acute lymphoblastic B-cell leukemia, acute lymphoblastic T-cell leukemia, acute myeloblastic leukemia, acute promy
- the invention encompasses the treatment of a human afflicted with a solid tumor or a blood tumor.
- the invention encompasses a method of treating or preventing a disease ameliorated by modulation of premature translation termination, nonsense-mediated mRNA decay, or premature translation termination and nonsense- mediated mRNA decay, or ameliorating one or more symptoms associated therewith comprising contacting a cell with a therapeutically effective amount of a compound of the invention.
- Cells encompassed by the present methods include animal cells, mammalian cells, bacterial cells, and virally infected cells.
- the nonsense mutation is a genetic mutation (i.e., the nonsense codon was present in the progenitor DNA).
- the nonsense mutation is a somatic mutation (i.e., the nonsense codon arose spontaneously or from mutagenesis).
- a compound of the invention is administered to a subject, including but not limited to a plant, reptile, avian, amphibian or preferably a mammal, more preferably a human, as a preventative measure against a disease associated with premature translation termination, nonsense-mediated mRNA decay, or premature translation termination and nonsense-mediated mRNA decay.
- the patient is suffering from a disease associated with premature translation termination and/or nonsense-mediated mRNA decay.
- the patient has undergone a screening process to determine the presence of a nonsense mutation comprising the steps of screening a subject, or cells extracted therefrom, by an acceptable nonsense mutation screening assay.
- the DNA of the patient can be sequenced or subjected to Southern Blot, polymerase chain reaction (PCR), use of the Short Tandem Repeat (STR), or polymorphic length restriction fragments (RFLP) analysis to determine if a nonsense mutation is present in the DNA of the patient.
- PCR polymerase chain reaction
- STR Short Tandem Repeat
- RFLP polymorphic length restriction fragments
- the patient is an unborn child who has undergone screening in utero for the presence of a nonsense mutation.
- Administration of a compound of the invention can occur either before or after birth.
- the therapy is personalized in that the patient is screened for a nonsense mutation screening assay and treated by the administration of one or more compounds of the invention; particularly, the patient may be treated with a compound particularly suited for the mutations in question; e.g., depending upon the disease type, cell type, and the gene in question.
- Such methods are well known to one of skill in the art.
- the cells are screened for premature translation termination and/or nonsense-mediated mRNA decay with a method such as that described above (i.e., the DNA of the cell can be sequenced or subjected to Southern Blot, polymerase chain reaction (PCR), use of the Short Tandem Repeat (STR), or polymorphic length restriction fragments (RFLP) analysis to determine if a nonsense mutation is present in the DNA of the cell; the RNA of the cell can be subjected to quantitative real time PCR to determine transcript abundance).
- Specific methods of the invention further comprise the administration of an additional therapeutic agent (i.e., a therapeutic agent other than a compound of the invention).
- the compounds of the invention can be used in combination with at least one other therapeutic agent.
- Therapeutic agents include, but are not limited to non-opioid analgesics; non-steroid anti- inflammatory agents; steroids, antiemetics; ⁇ -adrenergic blockers; anticonvulsants; antidepressants; Ca 2+ -channel blockers; anticancer agent(s) and antibiotics and mixtures thereof.
- the compounds of the invention can be administered or formulated in combination with anticancer agents.
- Suitable anticancer agents include, but are not limited to: alkylating agents; nitrogen mustards; folate antagonists; purine antagonists; pyrimidine antagonists; spindle poisons; topoisomerase inhibitors; apoptosis inducing agents; angiogenesis inhibitors; podophyllotoxins; nitrosoureas; cisplatin; carboplatin; interferon; asparginase; tamoxifen; leuprolide; flutamide; megestrol; mitomycin; bleomycin; doxorubicin; irinotecan and taxol.
- the compounds of the invention can be administered or formulated in combination with antibiotics.
- the antibiotic is an aminoglycoside (e.g., tobramycin), a cephalosporin (e.g., cephalexin, cephradine, cefuroxime, cefprozil, cefaclor, cefixime or cefadroxil), a clarithromycin (e.g., clarithromycin), a macrolide (e.g., erythromycin), a penicillin (e.g., penicillin V) or a quinolone (e.g., ofloxacin, ciprofloxacin or norfloxacin).
- the antibiotic is active against Pseudomonas aeruginosa.
- the methods of the present invention act through a combination of mechanisms that suppress nonsense mutations.
- the methods of the invention comprise administering a therapeutically effective amount of at least one compound of the invention, e.g., a compound of Formula 1.
- Relative activity of the compounds of the invention may be determined by any method known in the art, including the assay described in Example 2 herein.
- Compounds of the invention can be characterized with an in vitro luciferase nonsense suppression assay. Luciferase assays are included in the methods of the present invention.
- Luciferase can be used as a functional reporter gene assay (light is only produced if the protein is functional), and luciferase is extremely sensitive (Light intensity is proportional to luciferase concentration in the nM range).
- an assay of the present invention is a cell-based luciferase reporter assay.
- a luciferase reporter construct containing a premature termination codon (UGA, UAA, or UAG) is stably transfected in 293 Human Embryonic Kidney cells.
- a preferred assay is a biochemical assay consisting of rabbit reticulocyte lysate and a nonsense-containing luciferase reporter mRNA.
- the assay is a biochemical assay consisting of prepared and optimized cell extract (Lie & Macdonald, 1999, Development 126(22) :4989-4996 and Lie & Macdonald, 2000, Biochem. Biophys. Res. Commun. 270(2):473-481).
- mRNA containing a premature termination codon (UGA, UAA, or UAG) is used as a reporter in an in vitro translation reaction using rabbit reticulocyte lysate supplemented with tRNA, hemin, creatine kinase, amino acids, KOAc, Mg(O Ac)2, and creatine phosphate.
- Translation of the mRNA is initiated within a virus derived leader sequence, which significantly reduces the cost of the assay because capped RNA is not required.
- Synthetic mRNA is prepared in vitro using the T7 promoter and the MegaScript in vitro transcription kit (Ambion, Inc.; Austin, Texas).
- assays of the present invention addition of gentamicin, an aminoglycoside known to allow readthrough of premature termination codons, results in increased luciferase activity and can be used as an internal standard.
- Assays of the present invention can be used in high- throughput screens. Hundreds of thousands of compounds can be screened in cell-based and biochemical assays of the present invention. In a preferred aspect, a functional cell- based assay similar to the one described.
- Compounds of the present invention include compounds capable of increasing specific, functional protein expression from mRNA molecules comprising premature termination codons.
- compounds of the present invention can preferentially suppress premature translation termination.
- a compound of the present invention can be capable of suppressing a nonsense mutation if the mutation results in UAA, but not capable of suppressing a nonsense mutation if the mutation results in UAG.
- Another non-limiting example can occur when a compound of the present invention can be capable of suppressing a nonsense mutation if the mutation results in UAA and is followed, in-frame by a cytosine at the +1 position, but not capable of suppressing a nonsense mutation if the mutation results in UAA and is followed, in- frame by an adenine at the +1 position.
- a stable cell line harboring the UGA nonsense-containing luciferase gene can be treated with a test compound.
- cells can be grown in standard medium supplemented with 1% penicillin- streptomycin (P/S) and 10% fetal bovine serum (FBS) to 70% confluency and split 1:1 the day before treatment. The next day, cells are trypsinized and 40,000 cells are added to each well of a 96-well tissue culture dish. Serial dilutions of each compound are prepared to generate a six-point dose response curve spanning 2 logs (30 ⁇ M to 0.3 ⁇ M). The final concentration of the DMSO solvent remains constant at 1% in each well. Cells treated with 1% DMSO serve as the background standard, and cells treated with gentamicin serve as a positive control.
- P/S penicillin- streptomycin
- FBS fetal bovine serum
- a bronchial epithelial cell line harboring a nonsense codon at amino acid 1282 (W1282X) can be treated with a compound of the invention and CFTR function is monitored as a cAMP-activated chloride channel using the SPQ assay (Yang et al, Hum. MoI. Genet. 2(8):1253-1261 (1993) and Howard et al, Nat. Med. 2(4):467-469(1996)).
- the increase in SPQ fluorescence in cells treated with a compound of the invention is compared to those treated with cAMP and untreated cells.
- the invention includes compounds produced by a process comprising contacting a compound of this invention with a mammalian tissue or a mammal for a period of time sufficient to yield a metabolic product thereof.
- Such products typically are identified by preparing a radio-labeled (e.g.
- C ⁇ or H ⁇ ) compound of the invention administering it in a detectable dose (e.g., greater than about 0.5 mg/kg) to a mammal such as rat, mouse, guinea pig, monkey, or to man, allowing sufficient time for metabolism to occur (typically about 30 seconds to 30 hours), and isolating its conversion products from urine, blood or other biological samples.
- a detectable dose e.g., greater than about 0.5 mg/kg
- a mammal such as rat, mouse, guinea pig, monkey, or to man
- sufficient time for metabolism to occur typically about 30 seconds to 30 hours
- the metabolite structures are determined in conventional fashion, e.g., by MS or NMR analysis. In general, analysis of metabolites may be done in the same way as conventional drug metabolism studies well-known to those skilled in the art.
- the conversion products so long as they are not otherwise found in vivo, are useful in diagnostic assays for therapeutic dosing of the compounds of the invention even if they
- compositions useful in the methods of the invention are provided.
- the pharmaceutical compositions of the invention may be formulated with pharmaceutically acceptable excipients such as carriers, solvents, stabilizers, adjuvants, diluents, etc., depending upon the particular mode of administration and dosage form.
- the pharmaceutical compositions should generally be formulated to achieve a physiologically compatible pH, and may range from a pH of about 3 to a pH of about 11, preferably about pH 3 to about pH 7, depending on the formulation and route of administration.
- the pharmaceutical compositions of the present invention may be formulated from about pH 4 to about pH 7.
- it may be preferred that the pH is adjusted to a range from about pH 5 to about pH 8.
- compositions of the invention comprise a therapeutically or prophylactically effective amount of at least one compound of the present invention, together with one or more pharmaceutically acceptable excipients.
- the pharmaceutical compositions of the invention may comprise a combination of compounds of the present invention, or may include a second active ingredient useful in the treatment of cancer, diabetic retinopathy, or exudative macular degeneration.
- Formulations of the present invention are most typically solids, liquid solutions, emulsions or suspensions, while inhaleable formulations for pulmonary administration are generally liquids or powders, with powder formulations being generally preferred.
- a preferred pharmaceutical composition of the invention may also be formulated as a lyophilized solid that is reconstituted with a physiologically compatible solvent prior to administration.
- Alternative pharmaceutical compositions of the invention may be formulated as syrups, creams, ointments, tablets, and the like.
- compositions of the invention can be administered to the subject via any drug delivery route known in the art.
- Specific exemplary administration routes include oral, ocular, rectal, buccal, topical, nasal, ophthalmic, subcutaneous, intramuscular, intraveneous (bolus and infusion), intracerebral, transdermal, and pulmonary.
- pharmaceutically acceptable excipient refers to an excipient for administration of a pharmaceutical agent, such as the compounds of the present invention.
- the term refers to any pharmaceutical excipient that may be administered without undue toxicity.
- Pharmaceutically acceptable excipients are determined in part by the particular composition being administered, as well as by the particular method used to administer the composition. Accordingly, there exists a wide variety of suitable formulations of pharmaceutical compositions of the present invention ⁇ see, e.g., Remington's Pharmaceutical Sciences, 18 th Ed., Mack Publishing Co., 1990).
- Suitable excipients may be carrier molecules that include large, slowly metabolized macromolecules such as proteins, polysaccharides, polylactic acids, polyglycolic acids, polymeric amino acids, amino acid copolymers, and inactive virus particles.
- Other exemplary excipients include antioxidants such as ascorbic acid; chelating agents such as EDTA; carbohydrates such as dextrin, hydroxyalkylcellulose, hydroxyalkylmethylcellulose, stearic acid; liquids such as oils, water, saline, glycerol and ethanol; wetting or emulsifying agents; pH buffering substances; and the like. Liposomes are also included within the definition of pharmaceutically acceptable excipients.
- compositions of the invention may be formulated in any form suitable for the intended method of administration.
- tablets, troches, lozenges, aqueous or oil suspensions, non-aqueous solutions, dispersible powders or granules (including micronized particles or nanoparticles), emulsions, hard or soft capsules, syrups or elixirs may be prepared.
- Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions, and such compositions may contain one or more agents including sweetening agents, flavoring agents, coloring agents and preserving agents, in order to provide a palatable preparation.
- compositions particularly suitable for use in conjunction with tablets include, for example, inert diluents, such as celluloses, calcium or sodium carbonate, lactose, calcium or sodium phosphate; disintegrating agents, such as croscarmellose sodium, cross-linked povidone, maize starch, or alginic acid; binding agents, such as povidone, starch, gelatin or acacia; and lubricating agents, such as magnesium stearate, stearic acid or talc. Tablets may be uncoated or may be coated by known techniques including microencapsulation to delay disintegration and adsorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- inert diluents such as celluloses, calcium or sodium carbonate, lactose, calcium or sodium phosphate
- disintegrating agents such as croscarmellose sodium, cross-linked povidone, maize starch, or alginic acid
- binding agents such as povidone, starch
- a time delay material such as glyceryl monostearate or glyceryl distearate alone or with a wax may be employed.
- Formulations for oral use may be also presented as hard gelatin capsules where the active ingredient is mixed with an inert solid diluent, for example celluloses, lactose, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with non-aqueous or oil medium, such as glycerin, propylene glycol, polyethylene glycol, peanut oil, liquid paraffin or olive oil.
- compositions of the invention may be formulated as suspensions comprising a compound of the present invention in admixture with at least one pharmaceutically acceptable excipient suitable for the manufacture of a suspension.
- pharmaceutical compositions of the invention may be formulated as dispersible powders and granules suitable for preparation of a suspension by the addition of suitable excipients.
- Excipients suitable for use in connection with suspensions include suspending agents, such as sodium carboxymethylcellulose, methylcellulose, hydroxypropyl methylcelluose, sodium alginate, polyvinylpyrrolidone, gum tragacanth, gum acacia, dispersing or wetting agents such as a naturally occurring phosphatide (e.g., lecithin), a condensation product of an alkylene oxide with a fatty acid (e.g., polyoxyethylene stearate), a condensation product of ethylene oxide with a long chain aliphatic alcohol (e.g., heptadecaethyleneoxycethanol), a condensation product of ethylene oxide with a partial ester derived from a fatty acid and a hexitol anhydride (e.g., polyoxyethylene sorbitan monooleate); and thickening agents, such as carbomer, beeswax, hard paraffin or cetyl alcohol.
- suspending agents such as sodium carboxymethylcellulose
- the suspensions may also contain one or more preservatives such as acetic acid, methyl and/or n-propyl p-hydroxy-benzoate; one or more coloring agents; one or more flavoring agents; and one or more sweetening agents such as sucrose or saccharin.
- preservatives such as acetic acid, methyl and/or n-propyl p-hydroxy-benzoate
- coloring agents such as acetic acid, methyl and/or n-propyl p-hydroxy-benzoate
- flavoring agents such as sucrose or saccharin.
- sweetening agents such as sucrose or saccharin.
- the pharmaceutical compositions of the invention may also be in the form of oil- in-water emulsions.
- the oily phase may be a vegetable oil, such as olive oil or arachis oil, a mineral oil, such as liquid paraffin, or a mixture of these.
- Suitable emulsifying agents include naturally-occurring gums, such as gum acacia and gum tragacanth; naturally occurring phosphatides, such as soybean lecithin, esters or partial esters derived from fatty acids; hexitol anhydrides, such as sorbitan monooleate; and condensation products of these partial esters with ethylene oxide, such as polyoxyethylene sorbitan monooleate.
- the emulsion may also contain sweetening and flavoring agents.
- Syrups and elixirs may be formulated with sweetening agents, such as glycerol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative, a flavoring or a coloring agent.
- the pharmaceutical compositions of the invention may be in the form of a sterile injectable preparation, such as a sterile injectable aqueous emulsion or oleaginous suspension. This emulsion or suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, such as a solution in 1 ,2-propane-diol.
- the sterile injectable preparation may also be prepared as a lyophilized powder.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution, and isotonic sodium chloride solution.
- sterile fixed oils may be employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid may likewise be used in the preparation of injectables.
- the compounds of the present invention useful in the methods of the present invention are substantially insoluble in water and are sparingly soluble in most pharmaceutically acceptable protic solvents and in vegetable oils.
- the compounds are generally soluble in medium chain fatty acids (e.g., caprylic and capric acids) or triglycerides and have high solubility in propylene glycol esters of medium chain fatty acids.
- medium chain fatty acids e.g., caprylic and capric acids
- triglycerides e.g., triglycerides
- propylene glycol esters of medium chain fatty acids e.g., propylene glycol esters of medium chain fatty acids.
- compounds which have been modified by substitutions or additions of chemical or biochemical moieties which make them more suitable for delivery e.g., increase solubility, bioactivity, palatability, decrease adverse reactions, etc.
- esterification glycosylation, PEGylation, etc.
- the compounds of the present invention may be formulated for oral administration in a lipid-based formulation suitable for low solubility compounds.
- Lipid-based formulations can generally enhance the oral bioavailability of such compounds.
- a preferred pharmaceutical composition of the invention comprises a therapeutically or prophylactically effective amount of a compound of the present invention, together with at least one pharmaceutically acceptable excipient selected from the group consisting of: medium chain fatty acids or propylene glycol esters thereof (e.g., propylene glycol esters of edible fatty acids such as caprylic and capric fatty acids) and pharmaceutically acceptable surfactants such as polyoxyl 40 hydrogenated castor oil.
- cyclodextrins may be added as aqueous solubility enhancers.
- Preferred cyclodextrins include hydroxypropyl, hydroxyethyl, glucosyl, maltosyl and maltotriosyl derivatives of ⁇ -, ⁇ -, and ⁇ -cyclodextrin.
- a particularly preferred cyclodextrin solubility enhancer is hydroxypropyl- ⁇ -cyclodextrin (HPBC), which may be added to any of the above-described compositions to further improve the aqueous solubility characteristics of the compounds of the present invention.
- the composition comprises 0.1% to 20% hydroxypropyl- ⁇ - cyclodextrin, more preferably 1% to 15% hydroxypropyl- ⁇ -cyclodextrin, and even more preferably from 2.5% to 10% hydroxypropyl- ⁇ -cyclodextrin.
- solubility enhancer employed will depend on the amount of the compound of the present invention in the composition.
- the therapeutically effective amount refers to an amount of a pharmaceutical composition of the invention to treat, ameliorate, or modulate an identified disease or condition, or to exhibit a detectable therapeutic or inhibitory effect.
- the effect can be detected by, for example, assays of the present invention.
- the effect can also be the prevention of a disease or condition where the disease or condition is predicted for an individual or a high percentage of a population.
- the precise effective amount for a subject will depend upon the subject's body weight, size, and health; the nature and extent of the condition; the therapeutic or combination of therapeutics selected for administration, the protein half-life, the mRNA half-life and the protein localization.
- Therapeutically effective amounts for a given situation can be determined by routine experimentation that is within the skill and judgment of the clinician.
- the therapeutically effective amount can be estimated initially either in cell culture assays, e.g., of neoplastic cells, or in animal models, usually rats, mice, rabbits, dogs, or pigs.
- the animal model may also be used to determine the appropriate concentration range and route of administration. Such information can then be used to determine useful doses and routes for administration in humans.
- Therapeutic/prophylactic efficacy and toxicity may be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e.g., ED 50 (the dose therapeutically effective in 50% of the population) and LD 50 (the dose lethal to 50% of the population).
- the dose ratio between toxic and therapeutic effects is the therapeutic index, and it can be expressed as the ratio, LD 50 / ED 50 .
- compositions that exhibit large therapeutic indices are preferred.
- the data obtained from cell culture assays and animal studies may be used in fo ⁇ nulating a range of dosage for human use.
- the dosage contained in such compositions is preferably within a range of circulating concentrations that include an ED 50 with little or no toxicity.
- the dosage may vary within this range depending upon the dosage form employed, sensitivity of the patient, and the route of administration.
- the concentration-biological effect relationships observed with regard to the compound(s) of the present invention indicate an initial target plasma concentration ranging from approximately 5 ⁇ g/mL to approximately 100 ⁇ g/mL, preferably from approximately 10 ⁇ g/mL to approximately 50 ⁇ g/mL , more preferably from approximately 10 ⁇ g/mL to approximately 25 ⁇ g/mL.
- the compounds of the invention may be administered at doses that vary from 1 mg/kg to 150 mg/kg, depending upon the route of administration.
- Guidance as to particular dosages and methods of delivery is provided in the literature and is generally available to practitioners in the art.
- the dose will be in the range of about lmg/day to about 10g/day, or about O.lg to about 3g/day, or about 0.3g to about 3g/day, or about 0.5g to about 2g/day, in single, divided, or continuous doses for a patient weighing between about 40 to about 100 kg (which dose may be adjusted for patients above or below this weight range, particularly children under 40 kg).
- the magnitude of a prophylactic or therapeutic dose of a particular active ingredient of the invention in the acute or chronic management of a disease or condition will vary, however, with the nature and severity of the disease or condition, and the route by which the active ingredient is administered.
- the dose, and perhaps the dose frequency will also vary according to the age, body weight, and response of the individual patient.
- Suitable dosing regimens can be readily selected by those skilled in the art with due consideration of such factors.
- the recommended daily dose range for the conditions described herein lie within the range of from about 1 mg/kg to about 150 mg/kg per day.
- the compound of the invention is given as a single once-a-day dose.
- the compound of the invention is given as divided doses throughout a day. More specifically, the daily dose is administered in a single dose or in equally divided doses.
- a daily dose range should be from about 5 mg/kg to about 100 mg/kg per day, more preferably, between about 10 mg/kg and about 90mg/kg per day, even more preferably 20 mg/kg to 60 mg/kg per day.
- the therapy should be initiated at a lower dose, perhaps about 200 mg to about 300 mg , and increased if necessary up to about 600 mg to about 4000 mg per day as either a single dose or divided doses, depending on the patient's global response. It may be necessary to use dosages of the active ingredient outside the ranges disclosed herein in some cases, as will be apparent to those of ordinary skill in the art. Furthermore, it is noted that the clinician or treating physician will know how and when to interrupt, adjust, or terminate therapy in conjunction with individual patient response.
- Dosage and administration are adjusted to provide sufficient levels of the active agent(s) or to maintain the desired effect. Factors which may be taken into account include the severity of the disease state, general health of the subject, age, weight, and gender of the subject, diet, time, protein of interest half-life, RNA of interest half-life, frequency of administration, drug combination(s), reaction sensitivities, and tolerance/response to therapy.
- Long-acting pharmaceutical compositions may be administered every 3 to 4 days, every week, or once every two weeks depending on half-life and clearance rate of the particular formulation.
- any compound of the present invention with one or more other active ingredients useful in the treatment of diseases associated with nonsense mutations of mRNA as described herein, including compounds in a unitary dosage form, or in separate dosage forms intended for simultaneous or sequential administration to a patient in need of treatment.
- the combination may be administered in two or more administrations.
- the combination of active ingredients may be: (1) co-formulated and administered or delivered simultaneously in a combined formulation; (2) delivered by alternation or in parallel as separate formulations; or (3) by any other combination therapy regimen known in the art.
- the methods of the invention may comprise administering or delivering the active ingredients sequentially, e.g., in separate solution, emulsion, suspension, tablets, pills or capsules, or by different injections in separate syringes.
- an effective dosage of each active ingredient is administered sequentially, i.e., serially, whereas in simultaneous therapy, effective dosages of two or more active ingredients are administered together.
- Various sequences of intermittent combination therapy may also be used.
- the compounds of the present invention or other nonsense compounds can be utilized in combination with gene therapy.
- a gene can be introduced or provided to a mammal, preferably a human that contains a specified nonsense mutation in the desired gene.
- the desired gene is selected from the group consisting of IGFl, EPO, p53, pi 9ARF, p21, PTEN, EI 24 and ApoAI.
- the patient or mammal would be provided with an effective amount of a compound of the present invention or other nonsense suppression compound when such polypeptide is desired.
- nucleic acid that contain a nonsense mutation (optionally contained in a vector) into the patient's cells
- in vivo and ex vivo the nucleic acid is injected directly into the patient, usually at the sites where the polypeptide is required, i.e., the site of synthesis of the polypeptide, if known, and the site (e.g. solid tumor) where biological activity of the polypeptide is needed.
- site e.g. solid tumor
- the patient's cells are removed, the nucleic acid is introduced into these isolated cells, and the modified cells are administered to the patient either directly or, for example, encapsulated within porous membranes that are implanted into the patient (see e.g., U.S. Pat.
- nucleic acids there are a variety of techniques available for introducing nucleic acids into viable cells. The techniques vary depending upon whether the nucleic acid is transferred into cultured cells in vitro, or transferred in vivo in the cells of the intended host. Techniques suitable for the transfer of nucleic acid into mammalian cells in vitro include the use of liposomes, electroporation, microinjection, transduction, cell fusion, DEAE-dextran, the calcium phosphate precipitation method, etc. Transduction involves the association of a replication-defective, recombinant viral (preferably retroviral) particle with a cellular receptor, followed by introduction of the nucleic acids contained by the particle into the cell. A commonly used vector for ex vivo delivery of the gene is a retrovirus.
- the currently preferred in vivo nucleic and transfer techniques include transfection with viral or non-viral vectors (such as adenovirus, lentivirus, Herpes simplex I virus, or adeno-associated virus (AAV)) and lipid-based systems (useful lipids for lipid-mediated transfer of the gene are, for example, DOTMA, DOPE, and DC-Choi; see, e.g., Tonkinson et al., Cancer Investigation, 14 (1): 54-65 (1996)).
- the most preferred vectors for use in gene therapy are viruses, most preferably adenoviruses, AAV, lentiviruses, or retroviruses.
- a viral vector such as a retroviral vector includes at least one transcriptional promoter/enhancer or locus-defining element(s), or other elements that control gene expression by other means such as alternate splicing, nuclear RNA export, or post-translational modification of messenger.
- a viral vector such as a retroviral vector includes a nucleic acid molecule that, when transcribed with a gene encoding a polypeptide, is operably linked to the coding sequence and acts as a translation initiation sequence.
- Such vector constructs also include a packaging signal, long terminal repeats (LTRs) or portions thereof, and positive and negative strand primer binding sites appropriate to the virus used (if these are not already present in the viral vector).
- such vector typically includes a signal sequence for secretion of the polypeptide from a host cell in which it is placed.
- the signal sequence for this purpose is a mammalian signal sequence, most preferably the native signal sequence for the polypeptide.
- the vector construct may also include a signal that directs polyadenylation, as well as one or more restriction sites and a translation termination sequences.
- such vectors will typically include a 5' LTR, a tRNA binding site, a packaging signal, a origin of second-strand DNA synthesis, and a 3' LTR or a portion thereof.
- Other vectors can be used that are non-viral, such as cationic lipids, polylysine, and dendrimers.
- the nucleic acid source with an agent that targets the target cells, such as an antibody specific for a cell-surface membrane protein or the target cell, a ligand for a receptor on the target cell, etc.
- an agent that targets the target cells such as an antibody specific for a cell-surface membrane protein or the target cell, a ligand for a receptor on the target cell, etc.
- proteins that bind to a cell-surface membrane protein associated with endocytosis may be used for targeting and/or to facilitate uptake, e.g., capsid proteins or fragments thereof tropic for a particular cell type, antibodies for proteins that undergo internalization in cycling, and proteins that target intracellular localization and enchance intracellular half-life.
- the technique of recpto-mediated endocytosis is described, for example, by Wu et al., J Biol. Chern.
- Suitable gene therapy and methods for making retroviral particles and structural proteins can be found in, e.g. U.S. Pat. Nos. 5.681, 746; 6,800, 604 and 6,800,731.
- 3-Cyano-benzoic acid methyl ester (0.5 Ig, 3.17mmol) is dissolved in 10 ml of methanol, acetyl chloride (6.0ml, 84.38mmol) is added to the solution at room temperature under stirring. After 5-6 h, the mixture is dried under reduced pressure to give a crude product which is washed with ethyl ether to obtain a pure product as white crystal flakes. The product is used immediately for the next step.
- the reaction mixture is acidified using IN HCl until PH ⁇ 7, and partitioned between ethyl acetate and water. Water layer is extracted with ethyl acetate (3x15ml). Ethyl acetate is combined and washed with Brine, solid is crushed out in organic phase. Ethyl acetate is removed by reduce pressure. The residue is washed with water followed by ethyl ether to afford the desired product as a white crystalline solid. The obtained compound is >95% pure as determined by LC-MS. Mp: 305-308 °C.
- a 50ml culture tube is placed with 3-(5-bromo-benzooxazole-2-yl) benzoic acid methyl ester (478.5 mg, 1.44 mmol), CuI (13.7 mg, 0.72 mmol.), pyrrolidin-2-one (153.3 mg, 1.80 mmol.), and K 2 CO 3 (398.2 mg, 2.88 mmol), evacuated, and backed charged with N 2 .
- Trans-diamino-cyclohexane (16.4 mg, 0,14 mmol) and toluene (3.0 ml) are added under N 2 .
- the tube is sealed with a Teflon cap and the reaction mixture is stirred at 110 °C for 15 h.
- 1,2,3- Trimethoxybenzene (1.01 g, 6 mmol), and aluminum chloride (798 mg, 6 mmol) are added sequentially. This is stirred at rt overnight, and then at 60 °C for 2 h. The reaction mixture is poured into icy aqueous HCl, and this is then EtO Ac-extracted. Purification by column (6:4 CH 2 Cl 2 : Hex) yields product as a yellow semisolid (1.733 g, 75%). This material has 7% 1,2,3-trimethoxybenzene impurity that is carried through the next reaction.
- Trifluoro-methanesulfonic acid 3-(4-iodo-phenyl)-benzo[ ⁇ T]isoxazol-6-yl ester (390 mg, 0.83 mmol) is converted into white solid product (261 mg, 81%) at 85% purity using a procedure similar to Scheme F, step 5. The crude material is used as is in the next step.
- a functional, cell-based translation assay based on luciferase-mediated chemoluminescence permits quantitative assessment of the level of nonsense suppression.
- Human embryonic kidney cells (293 cells) are grown in medium containing fetal bovine serum (FBS). These cells can be stably transfected with the luciferase gene containing a premature termination codon at amino acid position 190.
- FBS fetal bovine serum
- each of the 3 possible nonsense codons (TAA, TAG, or TGA) and each of the 4 possible nucleotides (adenine, thymine, cytosine, or guanine) at the contextually important downstream +1 position following the nonsense codon are introduced by site- directed mutagenesis.
- amino acid 190 in the luciferase gene containing a premature termination codon is TAA, TAG, or TGA.
- nucleotide following amino acid 190 of luciferase gene containing a premature termination codon can be replaced with an adenine, thymine, cytosine, or guanine (A, T, C, G) such that these mutations do not change the reading frame of the luciferase gene.
- Figure 1 Schematics of these constructs are depicted in Figure 1.
- Activity measurements in Table 2 are determined in a cell-based luciferase reporter assay of the present invention construct containing a UGA premature termination codon.
- Gentamicin an aminoglycoside antibiotic known to allow readthrough of premature termination codons, is used as an internal standard.
- Activity measurements are based on the qualitative ratio between the minimum concentration of compound required to produce a given protein in a cell versus the amount of protein produced by the cell at that concentration.
- Compounds which are found to have either or both very high potency and very high efficacy of protein synthesis are classified as "***** » .
- Compounds which are found to have intermediate potency and/or efficacy of protein synthesis are classified as » **** » . « *** » . or « ** »
- compounds which are found to have lower potency and/or efficacy of protein synthesis are classified as "*".
- a functional, cell-based translation assay based on luciferase-mediated chemoluminescence permits assessment of translation- readthough of the normal stop codon in a mRNA.
- Human embryonic kidney cells (293 cells) are grown in medium containing fetal bovine serum (FBS). These cells are stably transfected with the luciferase gene containing a premature termination codon at amino acid position 190.
- FBS fetal bovine serum
- each of the 3 possible nonsense codons (TAA, TAG, or TGA) and each of the 4 possible nucleotides (adenine, thymine, cytosine, or guanine) at the contextually important downstream +1 position following the nonsense codon are introduced by site-directed mutagenesis.
- amino acid 190 in the luciferase gene containing a premature termination codon is either TAA, TAG, or TGA.
- Another assay of the present invention can evaluate compounds that promote nonsense mutation suppression.
- the luciferase constructs described above in Figure 1 are engineered to harbor two epitope tags in the N-terminus of the luciferase protein. Based on luciferase protein production, these constructs qualitatively assess the level of translation-readthrough. The presence of the full-length luciferase protein produced by suppression of the premature termination codon is measured by immunoprecipitation of the suppressed luciferase protein (using an antibody against a His tag) followed by western blotting using an antibody against the second epitope (the XpressTM epitope; Invitrogen ® ; Carlsbad, California). These constructs are depicted in Figure 2.
- Cells that harbor the constructs of Figure 2 show increased full-length protein production when treated with a compound of the present invention.
- cells containing the constructs of Figure 2 are collected and an antibody recognizing the His epitope is used to immunoprecipitate the luciferase protein.
- western blotting is performed using the antibody to the XpressTM epitope (Invitrogen ® ; Carlsbad, California) to detect the truncated luciferase (produced when no nonsense suppression occurs) and to detect the full-length protein (produced by suppression of the nonsense codon).
- Treatment of cells with a test compound produces full-length protein and not a readthrough protein (See e.g., Figure 3).
- the readthrough protein is produced if suppression of the normal termination codon occurs.
- Compounds of the present invention suppress the premature, i.e. nonsense mutation, but not the normal termination codon in the luciferase mRNA.
- Compounds of the present invention selectively act on premature termination codons but not normal termination codons in mammals.
- Rats and dogs are administered high doses of compound (up to 1800 mg/kg) by gavage (oral) once daily for 14 days. After the treatment, tissues are collected, lysates are prepared, and Western blot analysis is performed. Selection of the proteins for evaluation of normal termination codon readthrough is based primarily on the corresponding mRNA having a second stop codon in the 3'-UTR that is in-frame with the normal termination codon. Between these 2 stop codons, each selected protein has an intervening sequence of nucleotides that codes for an extension of the protein in the event of ribosomal readthrough of the first termination codon.
- an elongated protein is differentiated from the wild- type protein using Western blot.
- Tissues are collected from rats and are analyzed for suppression of the normal termination codon (UAA) in the vimentin mRNA. No evidence of suppression is apparent.
- Tissues are collected from dogs treated with compounds of the present invention. There is no evidence of suppression of the normal termination codon of beta actin, which harbors a UAG stop codon.
- a single dose of a compound of the present invention (200 mg/kg) is administered orally. Blood samples are collected, plasma is prepared, and a Western blot is conducted using plasma samples from female and male subjects. C-reactive protein (CRP), which harbors a UGA termination codon, is used to determine if treatment of subjects with compounds of the present invention result in suppression of the normal termination codon in the CRP mRNA. A luciferase assay in combination with a premature termination assay demonstrates selective suppression of premature termination codons but not normal termination codons.
- Example 4 Animal Models Animal model systems can also be used to demonstrate the safety and efficacy of a compound of the present invention. The compounds of the present invention are tested for biological activity using animal models for a disease, condition, or syndrome of interest. These include animals engineered to contain the target RNA element coupled to a functional readout system, such as a transgenic mouse. Cystic Fibrosis
- Examples of animal models for cystic fibrosis include, but are not limited to, cftr(-A-) mice (see, e.g., Freedman et al., 2001, Gastroenterology 121(4):950-7), cftr(tmlHGU/tmlHGU) mice (see, e.g., Bernhard et al, 2001, Exp Lung Res 27(4):349- 66), CFTR-deficient mice with defective cAMP -mediated Cl(-) conductance (see, e.g., Stotland et al, 2000, Pediatr Pulmonol 30(5):413-24), and C57BL/6- Cftr(mlUNC)/Cftr(m IUNC) knockout mice (see, e.g., Stotland et al, 2000, Pediatr Pulmonol 30(5):413-24).
- Muscular Dystrophy examples include, but are not limited to, mouse, hamster, cat, dog, and C. elegans.
- mouse models for muscular dystrophy include, but are not limited to, the dy-/- mouse (see, e.g., Connolly et ah, 2002, J Neiiroimmiinol 127(1 -2): 80-7), a muscular dystrophy with myositis (mdm) mouse mutation (see, e.g., Garvey et ah, 2002, Genomics 79(2):146-9), the mdx mouse (see, e.g., Nakamura et ah, 2001, Neuromuscul Disord l l(3):251-9), the utrophin-dystrophin knockout (dko) mouse (see, e.g., Nakamura et ah, 2001, Neuromuscul Disord 11(3):25
- Examples of hamster models for muscular dystrophy include, but are not limited to, sarcoglycan-deficient hamsters (see, e.g., Nakamura et ah, 2001, Am J Physiol Cell Physiol 281(2):C690-9) and the BIO 14.6 dystrophic hamster (see, e.g., Schlenker & Burbach, 1991, JAppI Physiol 71(5): 1655-62).
- An example of a feline model for muscular dystrophy includes, but is not limited to, the hypertrophic feline muscular dystrophy model (see, e.g., Gaschen & Burgunder, 2001, Acta Neuropathol (Berl) 101(6):591-600).
- Canine models for muscular dystrophy include, but are not limited to, golden retriever muscular dystrophy (see, e.g., Fletcher et ah, 2001, Neuromuscul Disord l l(3):239-43) and canine X-linked muscular dystrophy (see, e.g., Valentine et ah, 1992, Am J Med Genet 42(3):352-6). Examples of C. elegans models for muscular dystrophy are described in Chamberlain & Benian, 2000, Curr Biol 10(21):R795-7 and Culette & Sattelle, 2000, Hum MoI Genet 9(6):869-77.
- mice lacking functional LDL receptor genes see, e.g., Aji et ah, 1997, Circulation 95(2):430-7
- Yoshida rats see, e.g., Fantappie et ah, 1992, Life Sci 50(24):1913-24
- the JCR:LA-cp rat see, e.g., Richardson et ah, 1998, Atherosclerosis 138(l):135-46
- swine see, e.g., Hasler-Rapacz et ah, 1998, Am J Med Genet 76(5):379- 86
- Watanabe heritable hyperlipidaemic rabbit see, e.g., Tsutsumi et ah, 2000, Arzneistoffforschung 50(2):118-21; Harsch et ah, 1998, Br J Pharmacol 124(2):
- an animal model for human cancer in general includes, but is not limited to, spontaneously occurring tumors of companion animals (see, e.g., Vail & MacEwen, 2000, Cancer Invest 18(8):781-92).
- animal models for lung cancer include, but are not limited to, lung cancer animal models described by Zhang & Roth (1994, In Vivo 8(5):755-69) and a transgenic mouse model with disrupted p53 function (see, e.g., Morris et al, 1998, JLa State Med Soc 150(4): 179-85).
- An example of an animal model for breast cancer includes, but is not limited to, a transgenic mouse that overexpresses cyclin Dl (see, e.g., Hosokawa et al., 2001, Transgenic Res 10(5):471-8).
- An example of an animal model for colon cancer includes, but is not limited to, a TCRbeta and p53 double knockout mouse (see, e.g., Kado et al., 2001, Cancer Res 61(6):2395-8).
- animal models for pancreatic cancer include, but are not limited to, a metastatic model of PancO2 murine pancreatic adenocarcinoma (see, e.g., Wang et al., 2001, Int J Pancreatol 29(l):37-46) and nu-nu mice generated in subcutaneous pancreatic tumours (see, e.g., Ghaneh et al., 2001, Gene Ther 8(3): 199- 208).
- animal models for non-Hodgkin's lymphoma include, but are not limited to, a severe combined immunodeficiency ("SCID") mouse (see, e.g., Bryant et al, 2000, Lab Invest 80(4):553-73) and an IgHmu-HOXll transgenic mouse (see, e.g., Hough et al, 1998, Proc Natl Acad Sci USA 95(23): 13853-8).
- An example of an animal model for esophageal cancer includes, but is not limited to, a mouse transgenic for the human papillomavirus type 16 E7 oncogene (see, e.g., Herber et al, 1996, J Virol 70(3): 1873-81).
- animal models for colorectal carcinomas include, but are not limited to, Ape mouse models (see, e.g., Fodde & Smits, 2001, Trends MoI Med 7(8):369-73 and Kuraguchi et al, 2000, Oncogene 19(50):5755-63).
- An example of an animal model for neurofibromatosis includes, but is not limited to, mutant NFl mice (see, e.g., Cichowski et al, 1996, Semin Cancer Biol 7(5):291-8).
- animal models for retinoblastoma include, but are not limited to, transgenic mice that expression the simian virus 40 T antigen in the retina (see, e.g., Howes et al, 1994, Invest Ophthalmol Vis Sci 35(2):342-51 and Windle et al, 1990, Nature 343(6259):665-9) and inbred rats (see, e.g., Nishida et al, 1981, Curr Eye Res l(l):53-5 and Kobayashi et al, 1982, Acta Neuropathol (Berl) 57(2-3):203-8).
- Examples of animal models for Wilm's tumor include, but are not limited to, a WTl knockout mice (see, e.g., Scharnhorst et al, 1997, Cell Growth Differ 8(2): 133-43), a rat subline with a high incidence of nephroblastoma (see, e.g., Mesf ⁇ n & Breech, 1996, Lab Anim Sci 46(3):321-6), and a Wistar/Furth rat with Wilms' tumor (see, e.g., Murphy et al., 1987, Anticancer Res 7(4B):717-9).
- Examples of animal models for retinitis pigmentosa include, but are not limited to, the Royal College of Surgeons ("RCS") rat (see, e.g., Vollrath et al, 2001, Proc Natl Acad Sci USA 98(22);12584-9 and Hanitzsch et al, 1998, Acta Anat (Basel) 162(2- 3): 119-26), a rhodopsin knockout mouse (see, e.g., J aissle et al, 2001, Invest Ophthalmol Vis Sci 42(2):506-13), and Wag/Rij rats (see, e.g., Lai et al, 1980, Am J Pathol 98(l):281-4).
- RCS Royal College of Surgeons
- animal models for cirrhosis include, but are not limited to, CCl 4 - exposed rats (see, e.g., Kloehn et al, 2001, Horm Metab Res 33(7):394-401) and rodent models instigated by bacterial cell components or colitis (see, e.g., Vierling, 2001, Best PractRes Clin Gastroenterol 15(4):591-610).
- animal models for hemophilia include, but are not limited to, rodent models for hemophilia A (see, e.g., Reipert et al, 2000, Thromb Haemost 84(5):826-32; Jarvis et al,.
- von Willebrand Disease examples include, but are not limited to, an inbred mouse strain RIIIS/J (see, e.g., Nichols et al, 1994, 83(11):3225-31 and Sweeney et al, 1990, 76(l l):2258-65), rats injected with botrocetin (see, e.g., Sanders et al, 1988, Lab Invest 59(4):443-52), and porcine models for von Willebrand disease (see, e.g.
- animal models for ⁇ -thalassemia include, but are not limited to, murine models with mutations in globin genes (see, e.g., Lewis et al, 1998, Blood 91(6):2152-6; R ⁇ et al, 1994, Br J Haematol 86(l):156-62; Popp et al, 1985, 445:432- 44; and Skow et al, 1983, Cell 34(3):1043-52).
- kidney stones examples include, but are not limited to, genetic hypercalciuric rats (see, e.g., Bushinsky et al, 1999, Kidney Int 55(l):234-43 and
- animal models for ataxia-telangiectasia include, but are not limited to, murine models of ataxia-telangiectasia (see, e.g., Barlow et al, 1999, Proc Natl Acad Sci USA 96(17):9915-9 and Inoue et al, 1986, Cancer Res 46(8):3979-82).
- animal models for lysosomal storage diseases include, but are not limited to, mouse models for mucopolysaccharidosis type VII (see, e.g., Brooks et al, 2002, Proc Natl Acad Sci USA. 99(9):6216-21; Monroy et al, 2002, Bone 30(2):352-9; Vogler et al., 2001, Pediatr Dev Pathol. 4(5):421-33; Vogler et al, 2001, Pediatr Res. 49(3):342-8; and Wolfe et al, 2000, MoI Ther.
- mice model for metachromatic leukodystrophy see, e.g., Matzner et al, 2002, Gene Ther. 9(l):53-63
- a mouse model of Sandhoff disease see, e.g., Sango et al, 2002, Neuropathol Appl Neurobiol.
- mice 28(l):23-34 mouse models for mucopolysaccharidosis type III A (see, e.g., Bhattacharyya et al, 2001, Glycobiology l l(l):99-10 and Bhaumik et al, 1999, Glycobiology 9(12):1389-96.), arylsulfatase A (ASA)-deficient mice (see, e.g., D'Hooge et al, 1999, Brain Res. 847(2):352-6 and D'Hooge et al, 1999, Neurosci Lett.
- ASA arylsulfatase A
- mice with an aspartylglucosaminuria mutation see, e.g., Jalanko et al., 1998, Hum MoI Genet. 7(2):265-72
- feline models of mucopolysaccharidosis type VI see, e.g., Crawley et al, 1998, J Clin Invest. 101(l):109-19 and Norrdin et al., 1995, Bone 17(5):485-9
- a feline model of Niemann-Pick disease type C see, e.g., March et al, 1997, Acta Neuropathol (Berl).
- TSC tuberous sclerosis
- a mouse model of TSCl see, e.g., Kwiatkowski et al, 2002, Hum MoI Genet. l l(5):525-34
- Tscl TSCl homologue
- a TSC2 gene mutant(Eker) rat model see, e.g., Hino 2000, Nippon Rinsho 58(6): 1255-61; Mizuguchi et al, 2000, J Neuropathol Exp Neurol.
- Example 5 mdx mouse, an animal model study
- mice The mutation in the mdx mouse that causes premature translation termination of the 427 kDa dystrophin polypeptide has been shown to be a C to T transition at position 3185 in exon 23 (Sicinski et al, Science 244(4912):1578-1580(1989)).
- Mouse primary skeletal muscle cultures derived from 1-day old mdx mice are prepared as described previously (Barton-Davis et al, J. Clin. Invest. 104(4):375-381 (1999)). Cells are cultured for 10 days in the presence of a compound of the invention. Culture medium is replaced every four days and the presence of dystrophin in myoblast cultures is detected by immunostaining as described previously (Barton-Davis et al, J. Clin.
- a primary monoclonal antibody to the C-terminus of the dystrophin protein is used undiluted and rhodamine conjugated anti-mouse IgG is used as the secondary antibody.
- the antibody detects the full-length protein produced by suppression of the nonsense codon. Staining is viewed using a Leica DMR microscope, digital camera, and associated imaging software.
- compound is delivered by Alzet osmotic pumps implanted under the skin of anesthetized mice. Two doses of a compound of the invention are administered. Gentamicin serves as a positive control and pumps filled with solvent only serve as the negative control. Pumps are loaded with appropriate compound such that the calculated doses to which tissue is exposed are 10 mM and 20 niM. The gentamicin concentration is calculated to achieve tissue exposure of approximately 200 mM. In the initial experiment, mice are treated for 14 days, after which animals are anesthetized with ketamine and exsanguinated.
- TA tibialis anterior
- the tibialis anterior (TA) muscle of the experimental animals is then excised, frozen, and used for immunofluorescence analysis of dystrophin incorporation into striated muscle.
- the presence of dystrophin in TA muscles is detected by immunostaining, as described previously (Barton-Davis et al, J. CHn. Invest. 104(4):375-381(1999).
- Quadricep muscles from an mdx mouse treated with a compound of the present invention for 4 weeks are analyzed by western blot using a commercially available antibody to dystrophin. Protein extracted from the quadriceps of a wild-type mouse serve as a positive control. Production of full-length dystrophin is observed in the treated animal. The amount of full-length dystrophin produced, as a result of nonsense suppression, but not limited by this theory, is approximately 10% of wild-type levels of expression.
- mice After 2 weeks of treatment, mice are sacrificed for the removal of muscles to determine dystrophin readthrough efficiency. Immunofluorescence (IF) is performed on 10 ⁇ m cryosections using a dystrophin antibody. The antibody recognizes an epitope C-terminal to the premature stop mutation found in mdx mice. Image analysis is performed in an identical manner in all sections. Images from treated and untreated mice are analyzed and a signal greater than the signal on the untreated control is deemed positive and indicates that suppression of the premature termination codon in the dystrophin mRNA occurred. Muscle mechanics
- Optimum muscle length is defined as the length that produced maximum twitch tension.
- Maximum tetanic force at Lo is measured using a 120Hz, 500 msec pulse at supramaximal voltage. Protection against mechanical injury, induced by a series of 5 eccentric tetanic contractions, is monitored. These measurements are performed using a 700 msec stimulation period during which the muscle is held in an isometric contraction for the first 500 msec followed by a stretch of 8 or 10% Lo at a rate of 0.5Lo/sec. Protection against mechanical injury is evaluated at 80Hz stimulation frequency. Damage is determined as the loss in force between the first and last eccentric contraction.
- Example 6 Suppression of a nonsense mutation in the p53 gene
- CAOV-3 cells (1 x 10 7 ) are injected into the flanks of nude/nude mice. After 12 days, mice are randomized (10 mice per group) and treated subcutaneously (5 days per week) with 3 mg/kg of a compound of the present invention or intraperitonealy (1 day per week) with 30 mg/kg of a compound of the present invention. Tumor volumes are measured weekly. Suppression of nonsense mutations in the p53 gene by a compound of the present invention can inhibit cancer growth in vivo.
- Example 7 Access to specific nucleotides of the 28S rRNA can be modified by compounds of the present invention
- gentamicin and other members of the aminoglycoside family that decrease the fidelity of translation bind to the A site of the 16S rRNA.
- gentamicin has been shown to bind at the A site (comprised of nucleotides 1400-1410 and 1490-1500, E. coli numbering) of the rRNA at nucleotides 1406, 1407, 1494, and 1496 (Moazed & Noller, Nature 327(6121):389-394 (1978); Woodcock et al, EMBO J. 10(10):3099-3103 (1991); and Schroeder et al.
- Ribosomes prepared from HeLa cells are incubated with the small molecules (at a concentration of 100 mM), followed by treatment with chemical modifying agents (dimethyl sulfate [DMS] and kethoxal [KE]).
- DMS dimethyl sulfate
- KE kethoxal
- rRNA is phenol-chloroform extracted, ethanol precipitated, analyzed in primer extension reactions using end-labeled oligonucleotides hybridizing to different regions of the three rRNAs and resolved on 6% polyacrylamide gels.
- Probes for primer extension cover the entire 18S (7 oligonucleotide primers), 28S (24 oligonucleotide primers), and 5 S (one primer) rRNAs.
- Controls in these experiments include DMSO (a control for changes in rRNA accessibility induced by DMSO), paromomycin (a marker for 18S rRNA binding), and anisomycin (a marker for 28S rRNA binding). All publications and patent applications cited herein are incorporated by reference to the same extent as if each individual publication or patent application was specifically and individually indicated to be incorporated by reference.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Diabetes (AREA)
- Immunology (AREA)
- Neurology (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Obesity (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Rheumatology (AREA)
- Epidemiology (AREA)
- Urology & Nephrology (AREA)
- Endocrinology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Emergency Medicine (AREA)
- Transplantation (AREA)
- Psychiatry (AREA)
- Child & Adolescent Psychology (AREA)
- Psychology (AREA)
- Ophthalmology & Optometry (AREA)
- Vascular Medicine (AREA)
- Hospice & Palliative Care (AREA)
Abstract
Description
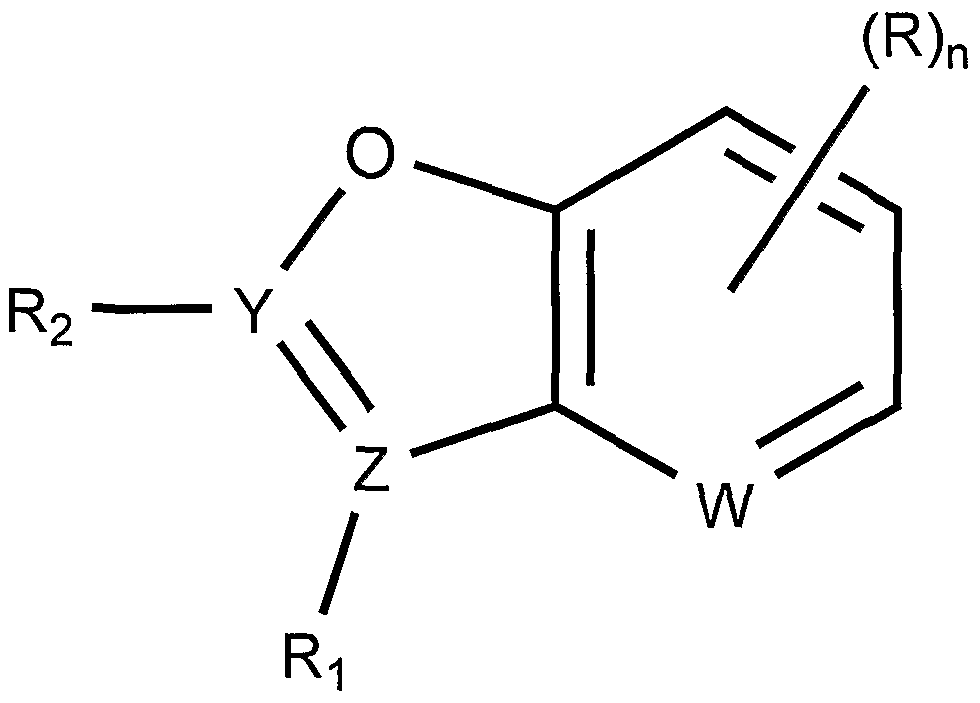



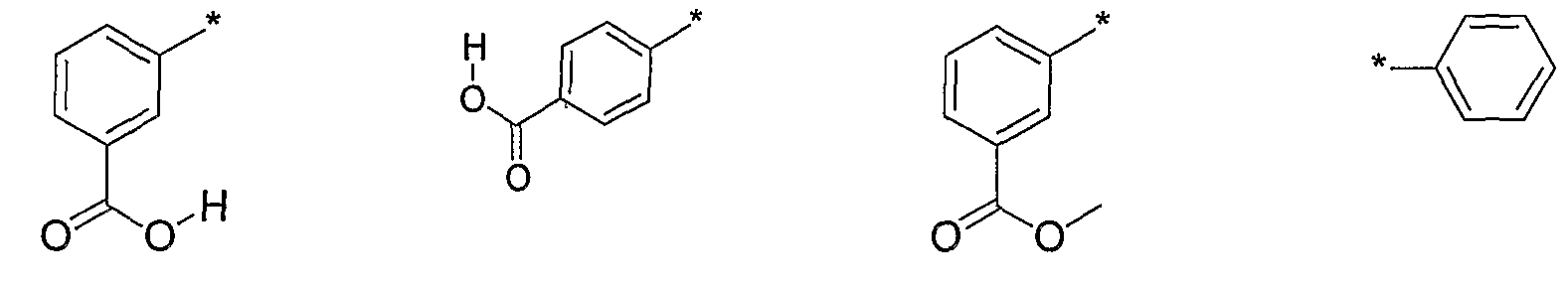






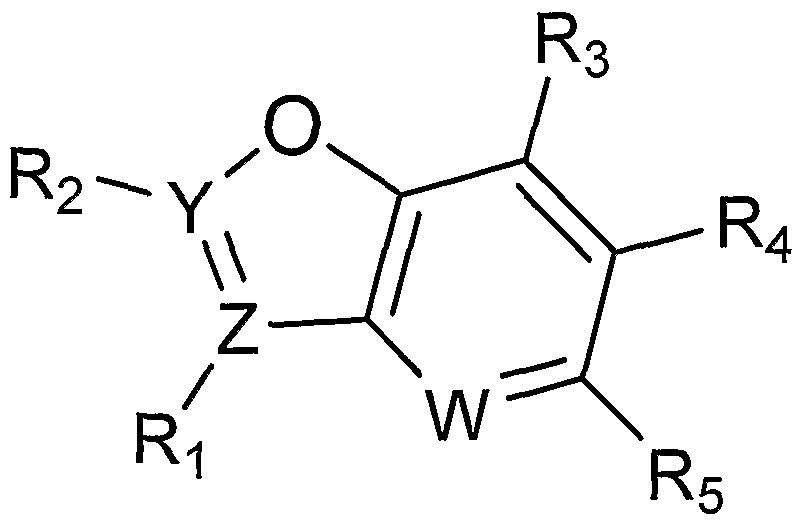


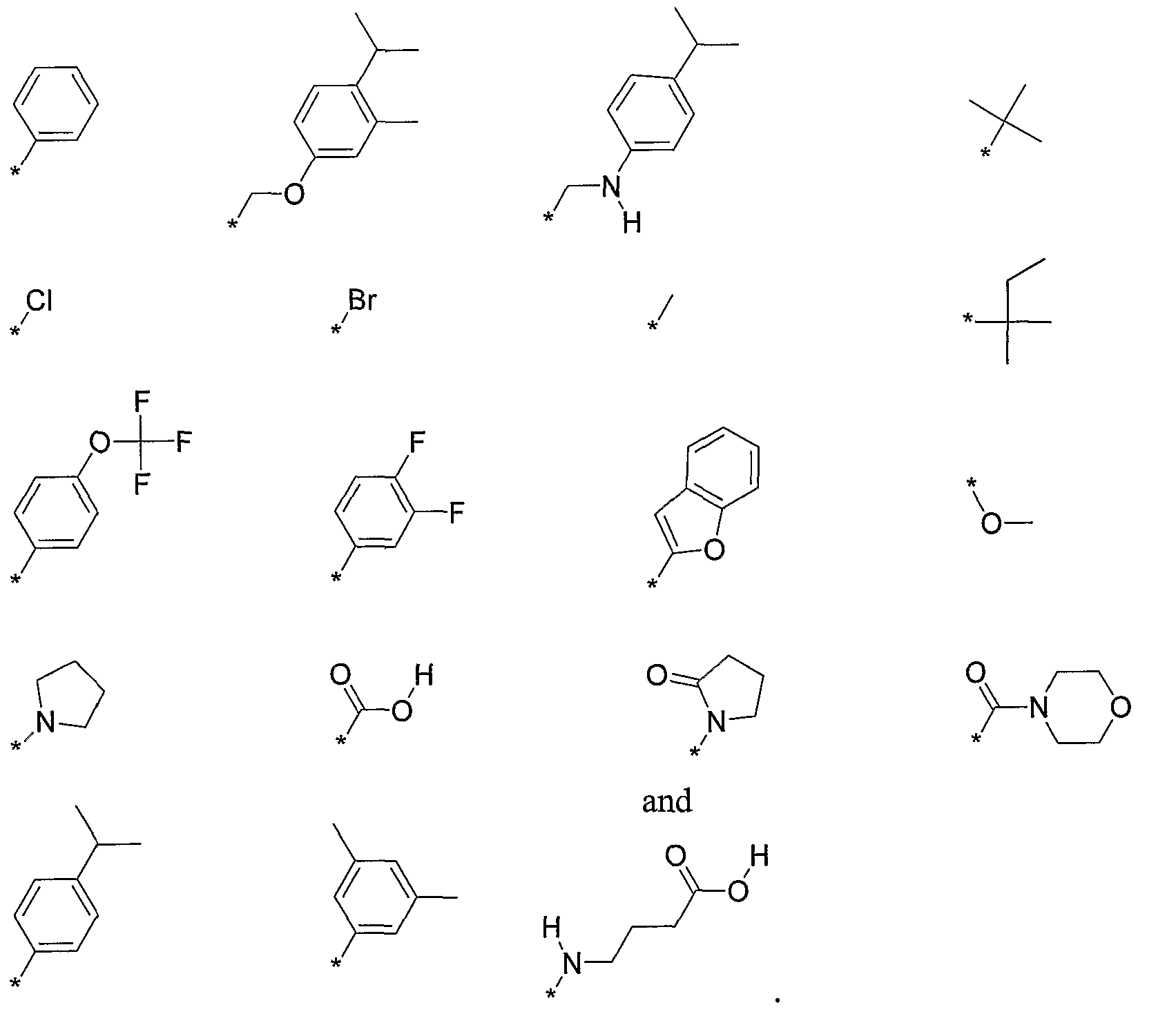

























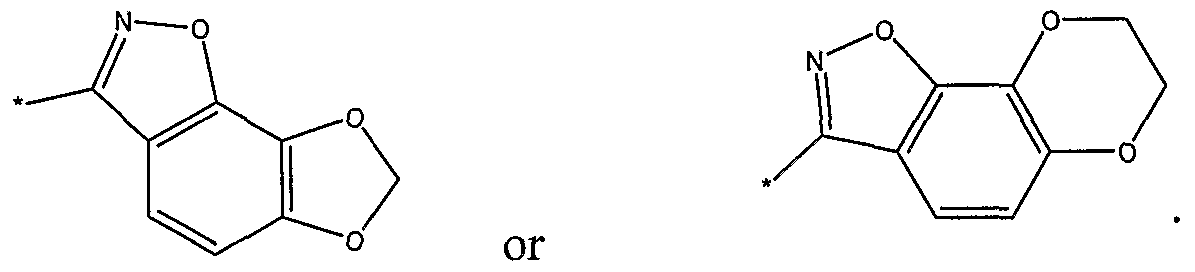












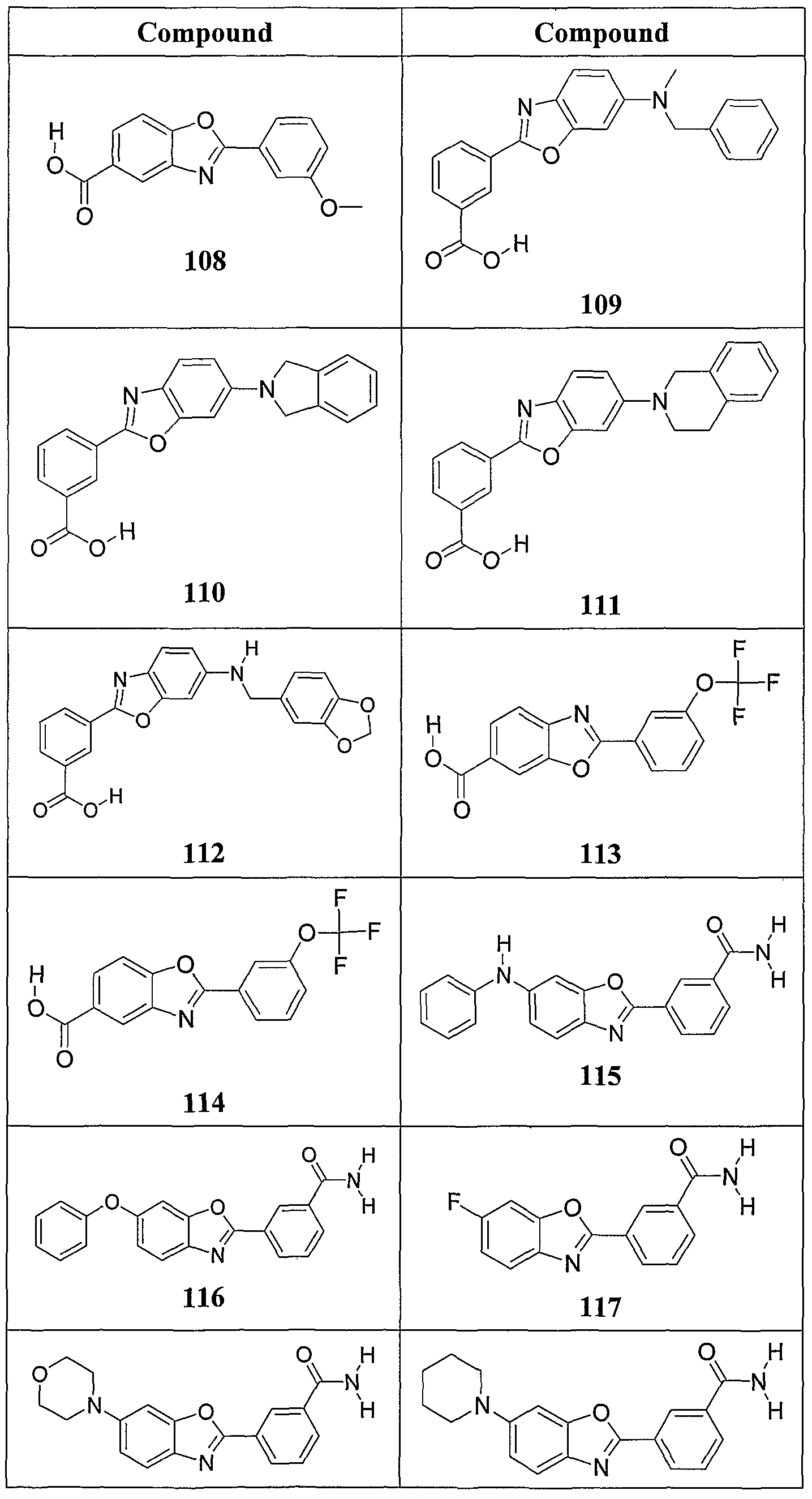





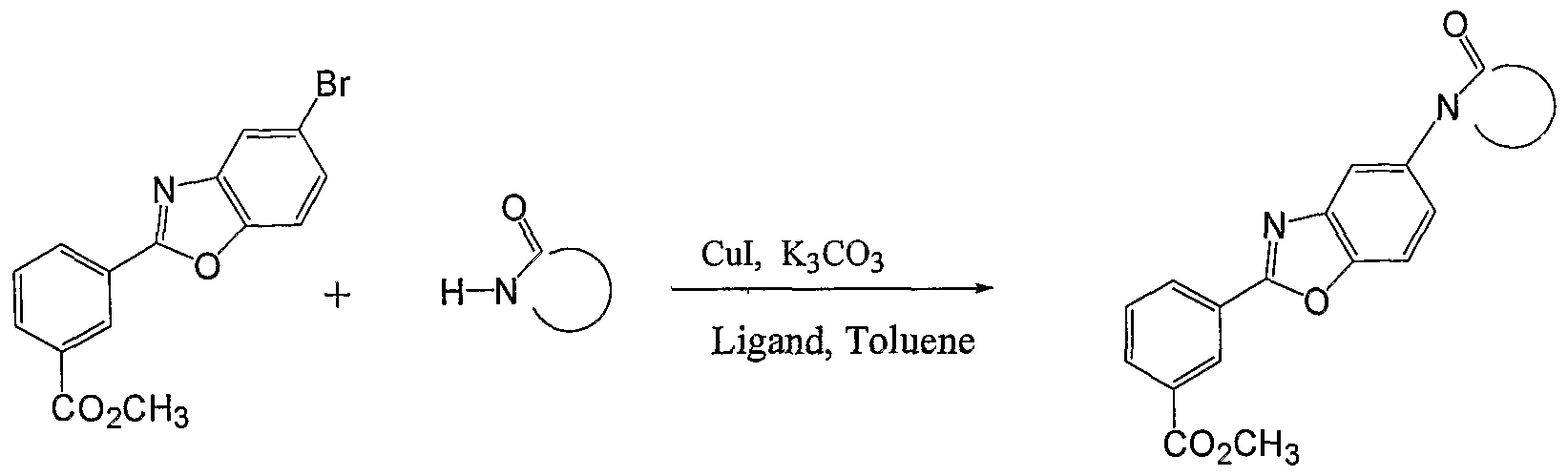













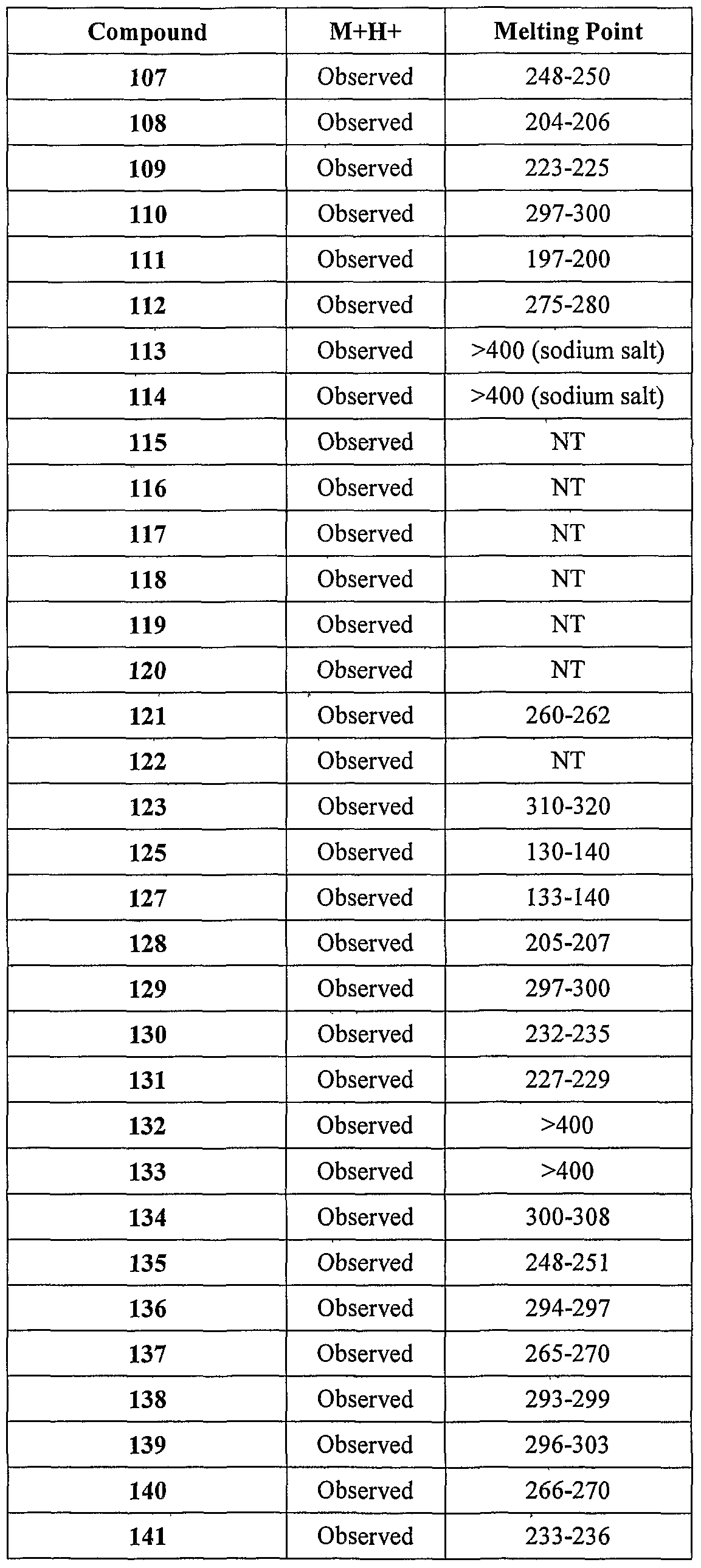






Claims







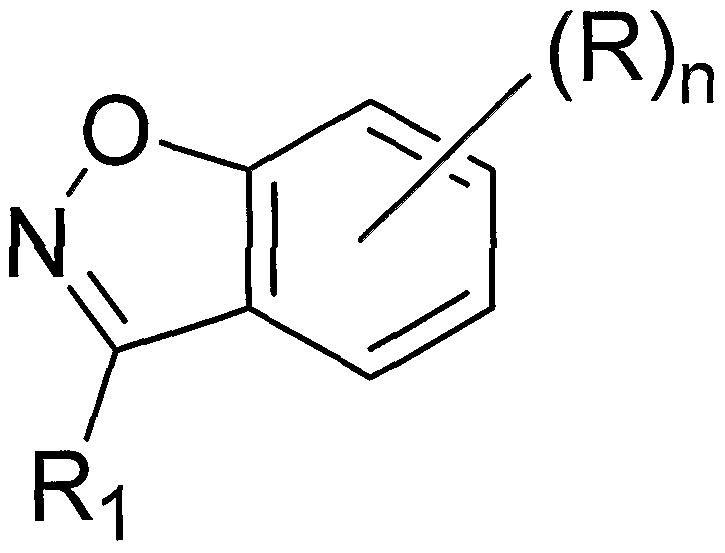





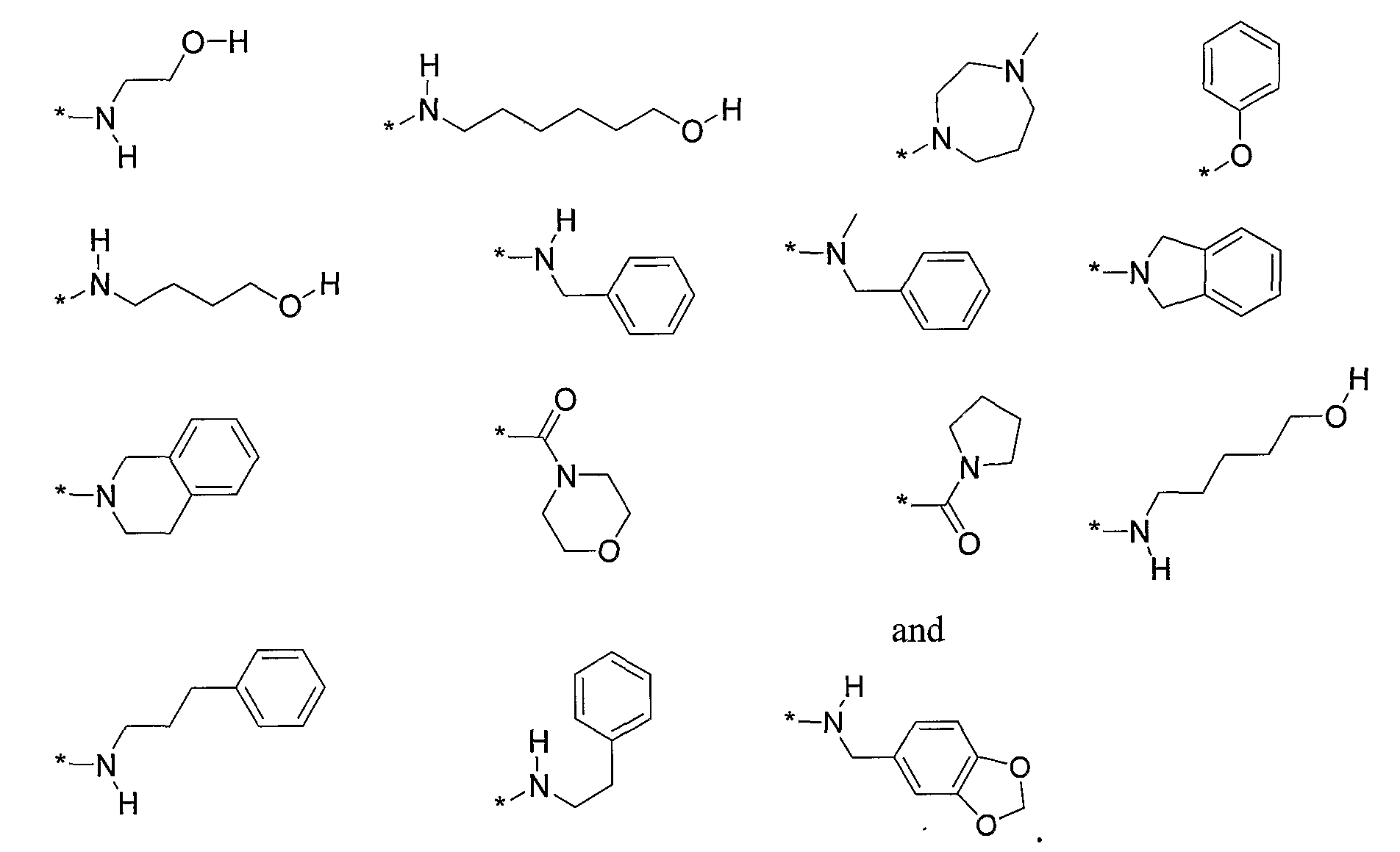
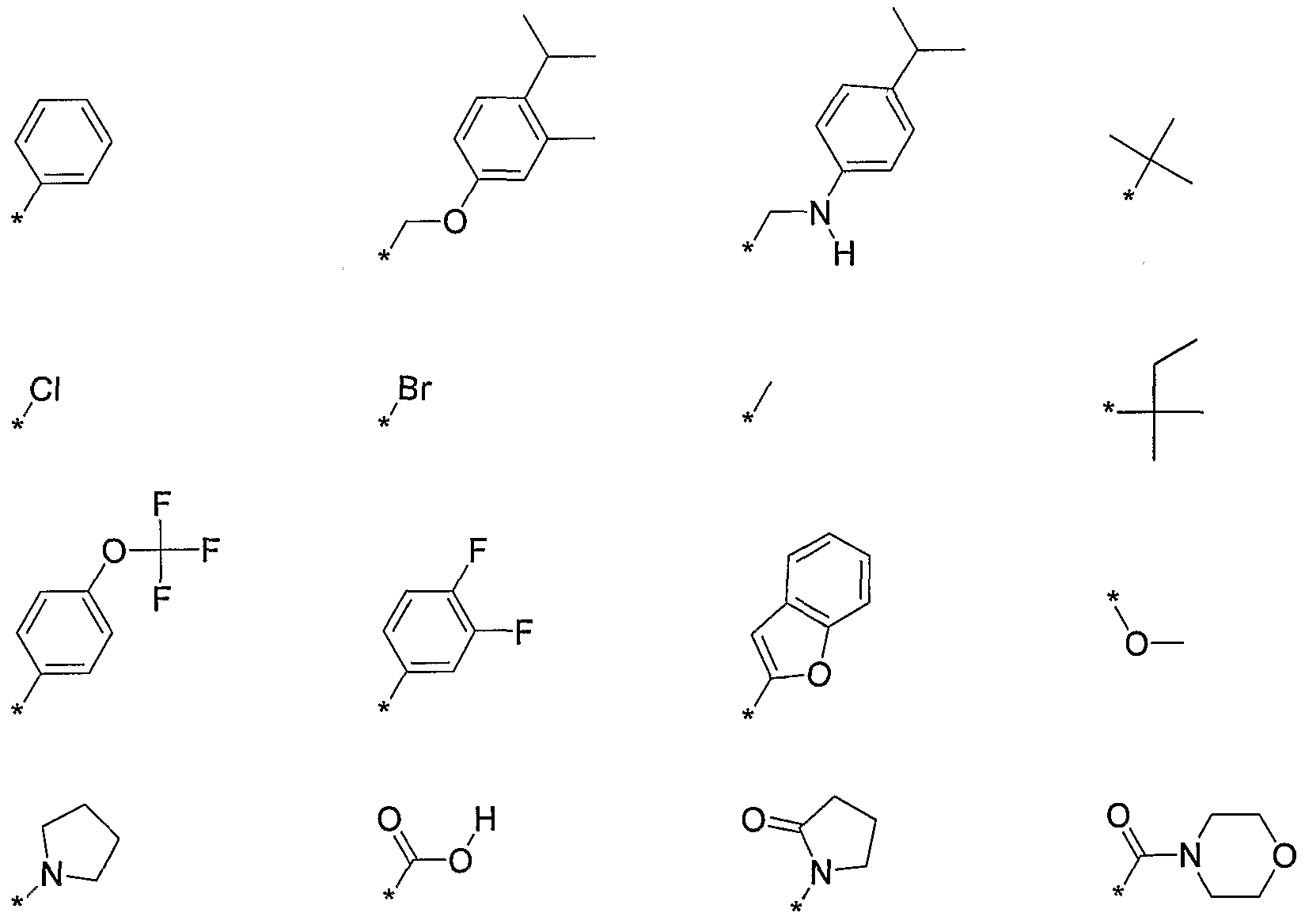





Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/577,176 US20080269191A1 (en) | 2004-10-13 | 2005-10-13 | Compounds for Nonsense Suppression, Use of These Compounds for the Manufacture of a Medicament for Treating Somatic Mutation-Related Diseases |
| CA002582885A CA2582885A1 (en) | 2004-10-13 | 2005-10-13 | Compounds for nonsense suppression, use of these compounds for the manufacture of a medicament for treating somatic mutation-related diseases |
| EP05804171A EP1812071A2 (en) | 2004-10-13 | 2005-10-13 | Compounds for nonsense suppression, use of these compounds for the manufacture of a medicament for treating somatic mutation-related diseases |
Applications Claiming Priority (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US61763304P | 2004-10-13 | 2004-10-13 | |
| US61765504P | 2004-10-13 | 2004-10-13 | |
| US61763404P | 2004-10-13 | 2004-10-13 | |
| US61767004P | 2004-10-13 | 2004-10-13 | |
| US61765304P | 2004-10-13 | 2004-10-13 | |
| US60/617,633 | 2004-10-13 | ||
| US60/617,670 | 2004-10-13 | ||
| US60/617,655 | 2004-10-13 | ||
| US60/617,653 | 2004-10-13 | ||
| US60/617,634 | 2004-10-13 | ||
| US62417004P | 2004-11-03 | 2004-11-03 | |
| US60/624,170 | 2004-11-03 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2006044503A2 true WO2006044503A2 (en) | 2006-04-27 |
| WO2006044503A3 WO2006044503A3 (en) | 2006-07-06 |
Family
ID=35517967
Family Applications (5)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/036673 Ceased WO2006044456A1 (en) | 2004-10-13 | 2005-10-13 | Compounds for nonsense suppression, and methods for their use |
| PCT/US2005/036762 Ceased WO2006044503A2 (en) | 2004-10-13 | 2005-10-13 | Compounds for nonsense suppression, use of these compounds for the manufacture of a medicament for treating somatic mutation-related diseases |
| PCT/US2005/037052 Ceased WO2006044682A1 (en) | 2004-10-13 | 2005-10-13 | Compounds for nonsense suppression, and methods for their use |
| PCT/US2005/036764 Ceased WO2006044505A2 (en) | 2004-10-13 | 2005-10-13 | Compounds for nonsense suppression, and methods for their use |
| PCT/US2005/036761 Ceased WO2006044502A2 (en) | 2004-10-13 | 2005-10-13 | Pyrazole or triazole compounds and their use for the manufacture of a medicament for treating somatic mutation-related diseases |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/036673 Ceased WO2006044456A1 (en) | 2004-10-13 | 2005-10-13 | Compounds for nonsense suppression, and methods for their use |
Family Applications After (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/037052 Ceased WO2006044682A1 (en) | 2004-10-13 | 2005-10-13 | Compounds for nonsense suppression, and methods for their use |
| PCT/US2005/036764 Ceased WO2006044505A2 (en) | 2004-10-13 | 2005-10-13 | Compounds for nonsense suppression, and methods for their use |
| PCT/US2005/036761 Ceased WO2006044502A2 (en) | 2004-10-13 | 2005-10-13 | Pyrazole or triazole compounds and their use for the manufacture of a medicament for treating somatic mutation-related diseases |
Country Status (11)
| Country | Link |
|---|---|
| US (10) | US9315467B2 (en) |
| EP (13) | EP1799659A1 (en) |
| JP (3) | JP2008515992A (en) |
| KR (3) | KR20070067201A (en) |
| AU (3) | AU2005295778A1 (en) |
| BR (2) | BRPI0515995A (en) |
| CA (5) | CA2583177A1 (en) |
| IL (3) | IL182459A0 (en) |
| MX (3) | MX2007004484A (en) |
| SG (2) | SG156640A1 (en) |
| WO (5) | WO2006044456A1 (en) |
Cited By (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007091106A2 (en) | 2006-02-10 | 2007-08-16 | Summit Corporation Plc | Treatment of duchenne muscular dystrophy |
| JP2008515985A (en) * | 2004-10-13 | 2008-05-15 | ピーティーシー セラピューティクス,インコーポレーテッド | Nonsense-inhibiting compounds and methods of use |
| WO2009019504A1 (en) | 2007-08-03 | 2009-02-12 | Summit Corporation Plc | Drug combinations for the treatment of duchenne muscular dystrophy |
| US7842685B2 (en) | 2008-06-20 | 2010-11-30 | Amgen Inc. | S1P1 receptor agonists and use thereof |
| JP2010540407A (en) * | 2006-10-02 | 2010-12-24 | デジタル バイオテック カンパニー リミテッド | Composition for the treatment and prevention of cognitive impairment including a novel benzofuran derivative and use thereof |
| WO2011119984A1 (en) | 2010-03-25 | 2011-09-29 | Vertex Pharmaceuticals Incorporated | Solid forms of (r)-1(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihyderoxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide |
| WO2011127241A2 (en) | 2010-04-07 | 2011-10-13 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyriodin-2-yl)benzoic acid and administration thereof |
| WO2011127290A2 (en) | 2010-04-07 | 2011-10-13 | Vertex Pharmaceuticals Incorporated | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| WO2011133751A2 (en) | 2010-04-22 | 2011-10-27 | Vertex Pharmaceuticals Incorporated | Process of producing cycloalkylcarboxamido-indole compounds |
| WO2012027247A2 (en) | 2010-08-23 | 2012-03-01 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition of (r)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihydroxy propyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide and administration therof |
| US8242133B2 (en) | 2008-04-21 | 2012-08-14 | Sumitomo Chemical Company, Limited | Arthropod pest control compositions comprising substituted oxazolo [5,4-b ] pyridines |
| JP2012524715A (en) * | 2009-04-02 | 2012-10-18 | メルク セローノ ソシエテ アノニム | Dihydroorotate dehydrogenase inhibitor |
| WO2012170061A1 (en) | 2011-06-08 | 2012-12-13 | Vertex Pharmaceuticals Incorporated | Formulations of (r)-1-(2,2-diflurobenzo[d][1,3] dioxol-5-yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methyl propan-2-yl)-1h-indol-5-yl)cyclopropanecarboxamide |
| EP2578571A1 (en) | 2007-11-16 | 2013-04-10 | Vertex Pharmaceuticals Incorporated | Isoquinoline modulators of ATP-binding cassette transporters |
| WO2013070961A1 (en) | 2011-11-08 | 2013-05-16 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| EP2615085A1 (en) | 2008-03-31 | 2013-07-17 | Vertex Pharmaceuticals Incorporated | Pyridyl derivatives as CFTR modulators |
| WO2013112804A1 (en) | 2012-01-25 | 2013-08-01 | Vertex Pharmaceuticals Incorporated | Formulations of 3-(6-(1-(2.2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| US8530483B2 (en) | 2008-05-30 | 2013-09-10 | Merck Sharp & Dohme Corp. | Substituted azabenzoxazoles |
| WO2013151666A2 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | Modified polynucleotides for the production of biologics and proteins associated with human disease |
| WO2013151668A2 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | Modified polynucleotides for the production of secreted proteins |
| WO2014071122A1 (en) | 2012-11-02 | 2014-05-08 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cftr mediated diseases |
| US8980864B2 (en) | 2013-03-15 | 2015-03-17 | Moderna Therapeutics, Inc. | Compositions and methods of altering cholesterol levels |
| WO2015051214A1 (en) | 2013-10-03 | 2015-04-09 | Moderna Therapeutics, Inc. | Polynucleotides encoding low density lipoprotein receptor |
| WO2015160787A1 (en) | 2014-04-15 | 2015-10-22 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| US9181319B2 (en) | 2010-08-06 | 2015-11-10 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| WO2016014846A1 (en) | 2014-07-23 | 2016-01-28 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of intrabodies |
| US9255129B2 (en) | 2012-04-02 | 2016-02-09 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding SIAH E3 ubiquitin protein ligase 1 |
| WO2016057572A1 (en) | 2014-10-06 | 2016-04-14 | Mark Thomas Miller | Modulators of cystic fibrosis transmembrane conductance regulator |
| US9376448B2 (en) | 2012-04-24 | 2016-06-28 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors |
| US9572897B2 (en) | 2012-04-02 | 2017-02-21 | Modernatx, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| US9587003B2 (en) | 2012-04-02 | 2017-03-07 | Modernatx, Inc. | Modified polynucleotides for the production of oncology-related proteins and peptides |
| US9657295B2 (en) | 2010-10-01 | 2017-05-23 | Modernatx, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| EP3170818A1 (en) | 2007-12-07 | 2017-05-24 | Vertex Pharmaceuticals Incorporated | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid |
| US9732080B2 (en) | 2006-11-03 | 2017-08-15 | Vertex Pharmaceuticals Incorporated | Azaindole derivatives as CFTR modulators |
| US9758510B2 (en) | 2006-04-07 | 2017-09-12 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US9950068B2 (en) | 2011-03-31 | 2018-04-24 | Modernatx, Inc. | Delivery and formulation of engineered nucleic acids |
| US9974781B2 (en) | 2006-04-07 | 2018-05-22 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US9987284B2 (en) | 2013-03-12 | 2018-06-05 | Vertex Pharmaceuticals Incorporated | Substituted benzooxadiazole DNA-PK inhibitors |
| US10022352B2 (en) | 2006-04-07 | 2018-07-17 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US10022425B2 (en) | 2011-09-12 | 2018-07-17 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US10039761B2 (en) | 2013-10-17 | 2018-08-07 | Vertex Pharmaceuticals Incorporated | Co-crystals and pharmaceutical compositions comprising the same |
| US10058546B2 (en) | 2012-07-16 | 2018-08-28 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of (R)-1-(2,2-difluorobenzo[D][1,3]dioxo1-5-y1)-N-(1-(2,3-dihydroxypropy1)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-y1)-1H-indol-5-y1) cyclopropanecarbox-amide and administration thereof |
| US10081621B2 (en) | 2010-03-25 | 2018-09-25 | Vertex Pharmaceuticals Incorporated | Solid forms of (R)-1(2,2-difluorobenzo[D][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| WO2019101709A1 (en) | 2017-11-23 | 2019-05-31 | Universita' Degli Studi Di Palermo | Oxadiazole derivatives for the treatment of genetic diseases due to nonsense mutations |
| JP2020504766A (en) * | 2016-12-20 | 2020-02-13 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Compounds useful as immunomodulators |
| US10815291B2 (en) | 2013-09-30 | 2020-10-27 | Modernatx, Inc. | Polynucleotides encoding immune modulating polypeptides |
| US11110108B2 (en) | 2016-09-27 | 2021-09-07 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of DNA-damaging agents and DNA-PK inhibitors |
| US11744801B2 (en) | 2017-08-31 | 2023-09-05 | Modernatx, Inc. | Methods of making lipid nanoparticles |
| US11786607B2 (en) | 2017-06-15 | 2023-10-17 | Modernatx, Inc. | RNA formulations |
| US12090235B2 (en) | 2018-09-20 | 2024-09-17 | Modernatx, Inc. | Preparation of lipid nanoparticles and methods of administration thereof |
| US12304901B2 (en) | 2022-11-03 | 2025-05-20 | Sentonix, Inc. | Selective ligands for tau aggregates |
| USRE50453E1 (en) | 2006-04-07 | 2025-06-10 | Vertex Pharmaceuticals Incorporated | Indole derivatives as CFTR modulators |
Families Citing this family (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7927791B2 (en) | 2002-07-24 | 2011-04-19 | Ptc Therapeutics, Inc. | Methods for identifying small molecules that modulate premature translation termination and nonsense mediated mRNA decay |
| EP4101846B1 (en) | 2003-04-11 | 2023-08-02 | PTC Therapeutics, Inc. | 1,2,4-oxadiazole benzoic acid compounds and their use for nonsense suppression and the treatment of disease |
| EP1940801A2 (en) * | 2005-10-14 | 2008-07-09 | NeuroSearch A/S | Imidazole derivatives and their use for modulating the gaba-a receptor complex |
| WO2007047706A2 (en) * | 2005-10-17 | 2007-04-26 | Children's Hospital | Methods and compositions for regulating gene expression |
| EP2387995A1 (en) | 2006-03-30 | 2011-11-23 | PTC Therapeutics, Inc. | Methods for the production of functional protein from DNA having a nonsense mutation and the treatment of disorders associated therewith |
| NZ575795A (en) * | 2006-09-25 | 2012-03-30 | Ptc Therapeutics Inc | Crystalline forms of 3-[5-(2-fluorophenyl)-[1,2,4]oxadiazol-3-yl]-benzoic acid |
| WO2008157407A2 (en) * | 2007-06-13 | 2008-12-24 | University Of Virginia Patent Foundation | Thiadiazole, oxadiazole and triazole derivatives for treating leukemia |
| CA2702094C (en) | 2007-10-10 | 2018-05-01 | Parion Sciences, Inc. | Delivering osmolytes by nasal cannula |
| WO2010008831A2 (en) * | 2008-06-24 | 2010-01-21 | Irm Llc | Compounds and methods for modulating g protein-coupled receptors |
| WO2011022393A2 (en) * | 2009-08-17 | 2011-02-24 | The Brigham And Women's Hospital, Inc. | Phosphatidylcholine transfer protein inhibitors |
| WO2011045224A1 (en) | 2009-10-12 | 2011-04-21 | Bayer Cropscience Ag | 1- (pyrid-3-yl) -pyrazole and 1- (pyrimid-5-yl) -pyrazole as pesticide |
| WO2011085269A1 (en) * | 2010-01-08 | 2011-07-14 | Selexagen Therapeutics, Inc. | Raf kinase inhibitors |
| KR101799429B1 (en) * | 2010-05-03 | 2017-11-21 | 에스케이바이오팜 주식회사 | Pharmaceutical composition for inhibiting apoptosis of neuron or neurodegeneration |
| US8945605B2 (en) | 2011-06-07 | 2015-02-03 | Parion Sciences, Inc. | Aerosol delivery systems, compositions and methods |
| EP2717855A4 (en) | 2011-06-07 | 2014-11-12 | Parion Sciences Inc | PROCESSING METHODS |
| AR086745A1 (en) | 2011-06-27 | 2014-01-22 | Parion Sciences Inc | 3,5-DIAMINO-6-CHLORINE-N- (N- (4- (4- (2- (HEXIL (2,3,4,5,6-PENTAHYDROXIHEXIL)) AMINO) ETOXI) PHENYL) BUTIL) CARBAMIMIDOIL) PIRAZINA -2-CARBOXAMIDE |
| EP2545964A1 (en) | 2011-07-13 | 2013-01-16 | Phenex Pharmaceuticals AG | Novel FXR (NR1H4) binding and activity modulating compounds |
| RU2648950C2 (en) | 2011-10-03 | 2018-04-02 | Модерна Терапьютикс, Инк. | Modified nucleosides, nucleotides and nucleic acids and their application |
| CN104114572A (en) | 2011-12-16 | 2014-10-22 | 现代治疗公司 | Modified nucleosides, nucleotides and nucleic acid compositions |
| KR101452235B1 (en) * | 2012-02-03 | 2014-10-22 | 서울대학교산학협력단 | Novel pyrimidine derivatives or pharmaceutically acceptable salts thereof, process for the preparation thereof and pharmaceutical composition for prevention or treatment of RAGE receptor related diseases containing the same as an active ingredient |
| HUE039734T2 (en) | 2012-05-29 | 2019-01-28 | Parion Sciences Inc | Dendrimer like amino amides possessing sodium channel blocker activity for the treatment of dry eye and other mucosal diseases |
| WO2014081507A1 (en) | 2012-11-26 | 2014-05-30 | Moderna Therapeutics, Inc. | Terminally modified rna |
| EP2928471B1 (en) | 2012-12-06 | 2020-10-14 | Celgene Quanticel Research, Inc. | Histone demethylase inhibitors |
| ES2674665T3 (en) | 2012-12-17 | 2018-07-03 | Parion Sciences, Inc. | 3,5-Diamino-6-Chloro-N- (N- (4-phenylbutyl) carbamimidoyl) -pyrazine-2-carboxamide compounds |
| CN108658876A (en) | 2012-12-17 | 2018-10-16 | 帕里昂科学公司 | The chloro- N- of 3,5- diamino -6- (N- (4- phenyl butyls) carbonamidine base) pyrazine -2- benzamide compounds |
| CA2895512C (en) | 2012-12-17 | 2021-10-19 | Parion Sciences, Inc. | Chloro-pyrazine carboxamide derivatives useful for the treatment of diseases favoured by insufficient mucosal hydration |
| WO2014163934A1 (en) * | 2013-03-13 | 2014-10-09 | Dow Agrosciences Llc | Preparation of 1,3-(substituted-diaryl)-1,2,4-triazoles |
| CA2903103C (en) | 2013-03-15 | 2020-06-09 | Discoverybiomed, Inc. | Coumarin derivatives and methods of use in treating cystic fibrosis, chronic obstructive pulmonary disease, and misfolded protein disorders |
| JP6348568B2 (en) * | 2013-03-15 | 2018-06-27 | モンサント テクノロジー エルエルシー | N-, C-disubstituted azoles for nematode pest control |
| KR20150132483A (en) | 2013-03-15 | 2015-11-25 | 디스커버리바이오메드 인코포레이티드 | Coumarin derivatives and methods of use in treating hyperproliferative diseases |
| FR3011468B1 (en) * | 2013-10-04 | 2015-12-04 | Inventiva | USE OF ODIPARCIL IN THE TREATMENT OF MUCOPOLYSACCHARIDOSE |
| US9611263B2 (en) | 2013-10-08 | 2017-04-04 | Calcimedica, Inc. | Compounds that modulate intracellular calcium |
| TWI695717B (en) | 2014-03-06 | 2020-06-11 | 美商 Ptc 治療公司 | Pharmaceutical compositions and salts of a 1,2,4-oxadiazole benzoic acid |
| EP3227281A4 (en) | 2014-12-01 | 2018-05-30 | Zenith Epigenetics Ltd. | Substituted pyridinones as bromodomain inhibitors |
| CN107207488B (en) * | 2014-12-17 | 2020-11-17 | 伦敦皇家学院 | Bicyclic heteroaryl-benzoic acid compounds as retinoic acid receptor beta (RAR) agonists |
| US10647688B2 (en) * | 2014-12-19 | 2020-05-12 | Galderma Research & Development | Compounds, synthesis method thereof and use of same in medicine and in cosmetics |
| WO2016105491A1 (en) | 2014-12-23 | 2016-06-30 | Fgh Biotech | Compositions of fatostatin based heterocyclic compounds and uses thereof |
| BR112018001414B1 (en) * | 2015-07-24 | 2022-02-22 | Syngenta Participations Ag | COMPOUNDS DERIVED FROM 1, 2, 4-TRIAZOLE FROM THE PESTICIDE POINT OF VIEW WITH SUBSTITUENTS CONTAINING SULFUR, PESTICIDE COMPOSITION, METHOD FOR PEST CONTROL AND METHOD FOR PROTECTING PLANT PROPAGATION MATERIAL FROM PEST ATTACK |
| CN108348527A (en) | 2015-10-30 | 2018-07-31 | Ptc医疗公司 | Method for treating epilepsy |
| JP6943857B2 (en) | 2015-12-22 | 2021-10-06 | シンジェンタ パーティシペーションズ アーゲー | Pest control active pyrazole derivative |
| JP2019502753A (en) | 2015-12-23 | 2019-01-31 | ムーンショット ファーマ エルエルシー | Induction of immune response by suppression of nonsense mutation-dependent mRNA degradation mechanism |
| KR102498741B1 (en) | 2016-04-29 | 2023-02-10 | 에프지에이치 바이오테크 인코포레이티드 | Di-substituted pyrazole compounds for the treatment of disease |
| CA3252823A1 (en) | 2016-06-13 | 2025-02-25 | Gilead Sciences, Inc. | Substituted tert-butyl-3-(2-chloro-phenyl)-3-hydroxyazetidine-1-carboxylate compounds |
| MX385718B (en) | 2016-06-13 | 2025-03-18 | Gilead Sciences Inc | FXR MODULATING COMPOUNDS (NR1H4). |
| GB201610867D0 (en) | 2016-06-22 | 2016-08-03 | King S College London | Crystalline forms of a therapeutic compound and processes for their preparation |
| JP7071981B2 (en) | 2016-09-07 | 2022-05-19 | エフジーエイチ バイオテック,インコーポレーテッド | Disubstituted pyrazole compounds for the treatment of diseases |
| PT3600309T (en) | 2017-03-28 | 2022-10-03 | Gilead Sciences Inc | THERAPEUTIC COMBINATIONS FOR THE TREATMENT OF LIVER DISEASES |
| WO2018187479A1 (en) * | 2017-04-04 | 2018-10-11 | Case Western Reserve University | Method of modulating ribonucleotide reductase |
| IL271149B2 (en) * | 2017-06-14 | 2024-05-01 | Trevena Inc | Compounds for modulating S1P1 activity and methods of using them |
| EP3641752A4 (en) | 2017-06-22 | 2021-03-17 | Moonshot Pharma LLC | CANCER TREATMENT METHODS USING COMPOSITIONS INCLUDING AMLEXANOX AND IMMUNOMODULATORS |
| WO2019040724A1 (en) * | 2017-08-23 | 2019-02-28 | The Regents Of The University Of Michigan | Small molecule inhibitors of myc and uses thereof |
| WO2019124577A1 (en) * | 2017-12-20 | 2019-06-27 | 재단법인 경기도경제과학진흥원 | Triazole derivative and use thereof |
| CA3094256A1 (en) * | 2018-03-29 | 2019-10-03 | Explo Engineering Ag | Device and method for generating high amplitude pressure waves |
| US12152242B2 (en) | 2018-04-23 | 2024-11-26 | The Curators Of The University Of Missouri | CRISPR therapy |
| EP3911647B1 (en) | 2019-01-15 | 2023-12-13 | Gilead Sciences, Inc. | Isoxazole compound as fxr agonist and pharmaceutical compositions comprising same |
| CA3233305A1 (en) | 2019-02-19 | 2020-08-27 | Gilead Sciences, Inc. | Solid forms of fxr agonists |
| JP2022527294A (en) * | 2019-03-26 | 2022-06-01 | ニューロポア セラピーズ インコーポレイテッド | Compounds and compositions as regulators of TLR signaling |
| KR102663365B1 (en) * | 2019-09-25 | 2024-05-08 | 한국원자력의학원 | Composition for preventing or treating cancer comprising novel trifluoromethylphenylpyrazol derivative |
| JP2022550109A (en) * | 2019-09-25 | 2022-11-30 | コリア インスティチュート オブ レディオロジカル アンド メディカル サイエンシズ | Cancer preventive or therapeutic composition containing a novel trifluoromethylphenylpyrazole derivative as an active ingredient |
| EP4061352A4 (en) | 2019-11-19 | 2024-02-28 | Trevena, Inc. | Compounds and methods of preparing compounds s1p1 modulators |
| JP7677646B2 (en) | 2019-12-10 | 2025-05-15 | ザ・トラスティーズ・オブ・インディアナ・ユニバーシティー | Replication protein A (RPA)-DNA interaction inhibitors |
| KR102847693B1 (en) * | 2019-12-27 | 2025-08-18 | 서울대학교산학협력단 | Composition for Preventing or Treating Obesity Comprising Oxadiazole Derivatives |
| EP3912628A1 (en) | 2020-05-20 | 2021-11-24 | Institut de Recherche en Semiochimie et Ethologie Appliquée | Nucleoside analogues to inhibit the main protease of a coronavirus |
| CN117042759A (en) * | 2021-02-04 | 2023-11-10 | 国立大学法人九州大学 | IL-31 production inhibitors and pharmaceutical compositions containing the same |
| CN115594642B (en) * | 2021-06-28 | 2024-12-27 | 广西医科大学 | A Ji Ruige triazole derivative and application thereof in resisting breast cancer |
| EP4173485A1 (en) * | 2021-10-27 | 2023-05-03 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Protein aggregation inhibiting compounds for plant disease control |
| AU2023238460A1 (en) * | 2022-03-25 | 2024-10-31 | Vasthera Co., Ltd. | 3-phenylisoxazole derivative, and pharmaceutical composition for preventing or treating eye disease, containing same as active ingredient |
| GB202300021D0 (en) * | 2023-01-03 | 2023-02-15 | Thirtyfivebio Ltd | Compounds |
| WO2025116596A1 (en) * | 2023-11-29 | 2025-06-05 | 가천대학교 산학협력단 | Novel compound, and composition for preventing or treating eye diseases, comprising same |
| CN119144004B (en) * | 2024-08-12 | 2025-10-28 | 兰州大学 | M-HOF material, preparation method thereof and application thereof in hydroxide ion identification |
Family Cites Families (98)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3153017A (en) * | 1961-06-29 | 1964-10-13 | Eastman Kodak Co | Nitrogen-containing linear polyesters from amino methyl cyclohexanol and dicarboxylic acids |
| CH489510A (en) * | 1967-02-07 | 1970-04-30 | Geigy Ag J R | Process for the preparation of substituted γ-triazoles |
| DE1938904A1 (en) | 1968-08-02 | 1970-02-05 | Innothera Lab Sa | 1-phenylpyrroles |
| GB1248070A (en) * | 1968-12-16 | 1971-09-29 | Science Union & Cie | Thiazolyl-benzoic acid derivatives and process for preparing them |
| US3803162A (en) | 1969-10-03 | 1974-04-09 | Sandoz Ltd | 7-triazolyl-3-phenylcoumarins |
| US3679669A (en) * | 1970-09-23 | 1972-07-25 | Ciba Geigy Ag | Phenyl-1,3,4-oxdiazole compounds |
| US3964896A (en) * | 1971-08-09 | 1976-06-22 | Uniroyal, Inc. | Oxadiazole benzoic acid derivatives as herbicides |
| US3882138A (en) * | 1971-08-09 | 1975-05-06 | Uniroyal Inc | Oxazole and oxadiazole benzoic acid derivatives as herbicides |
| BE789948A (en) | 1971-10-13 | 1973-04-11 | Sandoz Sa | NEW DERIVATIVES OF PYRAZOLE, THEIR PREPARATION AND THEIR APPLICATION AS MEDICINAL PRODUCTS |
| US3948937A (en) * | 1972-02-29 | 1976-04-06 | E. I. Du Pont De Nemours And Company | Pyrazole plant growth regulants |
| US4035175A (en) * | 1974-07-29 | 1977-07-12 | Uniroyal Inc. | Method of retarding the growth of plants using certain oxadiazolyl, oxazolyl or thiadiazolyl benzoic acid derivatives |
| US3947263A (en) * | 1974-07-29 | 1976-03-30 | Uniroyal, Inc. | Plant growth regulants |
| AU497898B2 (en) * | 1975-01-10 | 1979-01-18 | Commonwealth Scientific And Industrial Research Organisation | Plant growth regulating method and composition for use therein |
| US4038285A (en) * | 1975-06-25 | 1977-07-26 | E. I. Du Pont De Nemours And Company | 1-(2-carboxyaryl)-4-arylimidazoles and 1-(2-carboxyaryl)-3-aryl-1,2,4-triazoles |
| US4032644A (en) * | 1975-12-29 | 1977-06-28 | Sandoz, Inc. | Isoxazolyl benzoic acids |
| DE2722331A1 (en) * | 1976-05-18 | 1977-11-24 | Ricoh Kk | (5)-Para-diethylaminophenyl-(1,3,4)-oxadiazole derivs. - prepd. from diethylaminophenyl-tetrazole and used in electrophotography |
| US4135910A (en) * | 1977-05-12 | 1979-01-23 | Monsanto Company | Oxadiazol-3-yl-benzoates as plant growth regulants |
| US4140515A (en) * | 1977-05-12 | 1979-02-20 | Monsanto Company | Aryl-3-isoxazole benzoates as plant growth regulants and herbicides |
| US4166732A (en) * | 1977-05-12 | 1979-09-04 | Monsanto Company | Oxadiazol-5-yl-benzoates |
| US4210762A (en) * | 1978-08-17 | 1980-07-01 | Monsanto Company | 2[5-(3-Trifluoromethylphenyl)-1,3,4-oxadiazol-2-yl] benzoates |
| US4268299A (en) * | 1978-08-17 | 1981-05-19 | Monsanto Company | Method and composition for plant growth regulation containing trifluoromethyl 1,3,4 oxadiazol benzoates |
| CA1125768A (en) * | 1978-10-31 | 1982-06-15 | Hoffmann-La Roche Limited | Benzoxazole derivatives |
| US4229204A (en) * | 1978-12-04 | 1980-10-21 | Monsanto Company | Trifluoromethylphenyl isoxazolyl benzoates |
| JPS58177977A (en) | 1982-04-09 | 1983-10-18 | Grelan Pharmaceut Co Ltd | 4-phenylpyrazoles |
| SU1063100A1 (en) * | 1982-04-22 | 1985-06-30 | Предприятие П/Я А-7850 | Liquid crystalline material for electrooptical devices |
| US4760066A (en) * | 1982-09-21 | 1988-07-26 | Wayne State University | Method for treating human tumor cell metastasis |
| US4546113A (en) * | 1983-04-14 | 1985-10-08 | Pfizer Inc. | Antiprotozoal diamidines |
| DD226883A1 (en) * | 1984-07-30 | 1985-09-04 | Neubauer T Paedagog Hochschule | PROCESS FOR THE PREPARATION OF 1,3-DISUBSTITUTED 5-ALKOXY-1,2,4-TRIAZOLEN AND 1,2,4-TRIAZOLIN-5-ONEN |
| US5436252A (en) * | 1986-12-19 | 1995-07-25 | Merrell Dow Pharmaceuticals Inc. | 5-aryl-3H-1,2,4-triazol-3-ones and their use in the treatment of neurodegenerative disorders |
| US4892538A (en) | 1987-11-17 | 1990-01-09 | Brown University Research Foundation | In vivo delivery of neurotransmitters by implanted, encapsulated cells |
| US5283187A (en) | 1987-11-17 | 1994-02-01 | Brown University Research Foundation | Cell culture-containing tubular capsule produced by co-extrusion |
| DE3819037A1 (en) | 1988-06-04 | 1989-12-14 | Hoechst Ag | 2,4-disubstituted oxazole derivatives, process for their preparation, and their use as agents for the therapy of diseases caused by rhinoviruses |
| US5260451A (en) | 1989-05-11 | 1993-11-09 | Merckle Gmbh | Substituted pyrrole compounds and use thereof in pharmaceutical compositions |
| US5022915A (en) * | 1990-03-01 | 1991-06-11 | Ici Americas Inc. | Substituted 2,4-diarylpyrimidines and their use as herbicides |
| DK0513379T3 (en) * | 1990-11-30 | 1996-09-30 | Teijin Ltd | 2-Arylthiazole derivatives and pharmaceutical compositions containing them |
| FR2672050A1 (en) | 1991-01-30 | 1992-07-31 | Atochem | DIARYL-2,5-OXADIAZOLES 1,3,4-HYDROXYESTER, HYDROXYACIDE AND ACETOXYACIDE, PROCESS FOR THEIR SYNTHESIS. |
| EP0646178A1 (en) | 1992-06-04 | 1995-04-05 | The Regents Of The University Of California | expression cassette with regularoty regions functional in the mammmlian host |
| US5466823A (en) * | 1993-11-30 | 1995-11-14 | G.D. Searle & Co. | Substituted pyrazolyl benzenesulfonamides |
| IL112290A (en) * | 1994-01-12 | 1999-01-26 | Novartis Ag | Substituted aryl and heteroaryl pyrimidines and herbicidal compositions containing them |
| US5521189A (en) | 1994-05-06 | 1996-05-28 | The University Of Nc At Ch | Methods of treating pneumocystis carinii pneumonia |
| US5486534A (en) * | 1994-07-21 | 1996-01-23 | G. D. Searle & Co. | 3,4-substituted pyrazoles for the treatment of inflammation |
| RU2165423C2 (en) * | 1994-10-07 | 2001-04-20 | Фудзисава Фармасьютикал Ко., Лтд. | Polypeptide compound, method of its synthesis and pharmaceutical composition |
| US5681746A (en) | 1994-12-30 | 1997-10-28 | Chiron Viagene, Inc. | Retroviral delivery of full length factor VIII |
| JP3964478B2 (en) * | 1995-06-30 | 2007-08-22 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | Heterocycle-containing carboxylic acid derivative and pharmaceutical containing the same |
| DE19536811A1 (en) * | 1995-10-02 | 1997-04-03 | Basf Ag | Intermediates and processes for the production of substituted salicylic acid derivatives as crop protection agents |
| KR100485642B1 (en) * | 1996-03-18 | 2005-09-30 | 에자이 가부시키가이샤 | Fused-ring carboxylic acid derivatives |
| DE19620041A1 (en) | 1996-05-17 | 1998-01-29 | Merck Patent Gmbh | Adhesion receptor antagonists |
| TW467902B (en) * | 1996-07-31 | 2001-12-11 | Bristol Myers Squibb Co | Diphenyl heterocycles as potassium channel modulators |
| MY128323A (en) | 1996-09-30 | 2007-01-31 | Otsuka Pharma Co Ltd | Thiazole derivatives for inhibition of cytokine production and of cell adhesion |
| AU4945697A (en) | 1996-10-09 | 1998-05-05 | Novartis Ag | Solid phase synthesis of heterocyclic compounds |
| KR20000069128A (en) * | 1996-11-25 | 2000-11-25 | 데이비드 엠 모이어 | 2-imidazolinylaminobenzoxazole compounds useful as alpha-2 adrenoceptor agonists |
| WO1998028421A1 (en) | 1996-12-20 | 1998-07-02 | Human Genome Sciences, Inc. | Human oncogene induced secreted protein i |
| JP2001518501A (en) | 1997-10-06 | 2001-10-16 | ビーエーエスエフ アクチェンゲゼルシャフト | Indeno [1,2-c]-, naphtho [1,2-c]-and benzo [6,7] cyclohepta [1,2-c] pyrazole derivatives |
| SI1023063T1 (en) * | 1997-10-06 | 2004-02-29 | Abbott Gmbh & Co. Kg | INDENO(1,2-c), NAPHTHO(1,2-c)- AND BENZO(6,7)CYCLOHEPTA(1,2-c)PYRAZOLE DERIVATIVES |
| DE19816880A1 (en) | 1998-04-17 | 1999-10-21 | Boehringer Ingelheim Pharma | New diaryl substituted heterocyclic compounds are AMPA receptor antagonists useful in treatment of neurodegenerative disorders and cerebral ischemia |
| CN1240713C (en) * | 1998-02-09 | 2006-02-08 | 藤泽药品工业株式会社 | New compound |
| WO1999059586A1 (en) * | 1998-05-19 | 1999-11-25 | Regents Of The University Of California | Thiazolidine and oxazolidine derivatives for the treatment of acute myocardial infarction and inhibition of cardiomyocyte apoptosis |
| EP1102755B1 (en) * | 1998-08-07 | 2006-01-04 | Chiron Corporation | Substituted isoxazole derivatives as estrogen receptor modulators |
| DE19904389A1 (en) * | 1999-02-04 | 2000-08-10 | Bayer Ag | Use of substituted isoxazolecarboxylic acids and derivatives and new substances |
| DE19904406A1 (en) * | 1999-02-04 | 2000-08-10 | Bayer Ag | Substituted pyrazole carboxylic acids |
| DE60022386T2 (en) | 1999-12-16 | 2006-06-22 | Schering Corp. | SUBSTITUTED IMIDAZOLE AS NEUROPEPTIDE Y Y5 RECEPTOR ANTAGONISTS |
| US7729756B2 (en) | 2000-01-18 | 2010-06-01 | Siemens Aktiengesellschaft | Measurement system for examining a section of tissue on a patient and the use of a measurement system of this type |
| DK1255829T3 (en) | 2000-02-11 | 2009-12-14 | Genentech Inc | Hepatocyte growth factor activator inhibitor for use in modulating angiogenesis and cardiovascularization |
| US6605615B2 (en) * | 2000-03-01 | 2003-08-12 | Tularik Inc. | Hydrazones and analogs as cholesterol lowering agents |
| DZ3347A1 (en) * | 2000-05-22 | 2001-11-29 | Aventis Pharma Inc | ARYLMETHYLAMINE DERIVATIVES FOR THEIR USE AS TRYPTASE INHIBITORS |
| GB0012362D0 (en) * | 2000-05-22 | 2000-07-12 | Aventis Pharma Ltd | Chemical compounds |
| WO2002036580A2 (en) * | 2000-10-31 | 2002-05-10 | Lynn Bonham | Benzoxazole lpaat- beta inhibitors and uses thereof |
| CN1853630A (en) * | 2001-02-21 | 2006-11-01 | Nps制药公司 | Heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists |
| AUPR362001A0 (en) | 2001-03-08 | 2001-04-05 | Fujisawa Pharmaceutical Co., Ltd. | New compound |
| JP2003026663A (en) | 2001-05-09 | 2003-01-29 | Takeda Chem Ind Ltd | Azole compound, method for producing the same and application of the same |
| IL158693A0 (en) | 2001-05-09 | 2004-05-12 | Sumitomo Chem Takeda Agro Co | Azole compounds, process for preparation of the same and use thereof |
| EP1405636A4 (en) * | 2001-06-26 | 2009-04-15 | Takeda Pharmaceutical | REGULATOR OF RECTINTOR FUNCTION RELATING TO RETINOIDS |
| JP2003081832A (en) | 2001-06-26 | 2003-03-19 | Takeda Chem Ind Ltd | Function regulator for retinoid relative receptor |
| DE10136066A1 (en) * | 2001-07-25 | 2003-02-13 | Bayer Cropscience Ag | Tetrahydropyridazine derivatives |
| US20070010562A1 (en) * | 2001-08-13 | 2007-01-11 | Ulrike Bauer | Nr1h4 nuclear receptor binding compounds |
| CA2461363A1 (en) | 2001-09-26 | 2003-04-03 | Bayer Pharmaceuticals Corporation | Substituted 3-pyridyl indoles and indazoles as c17,20 lyase inhibitors |
| CA2469821C (en) | 2001-12-18 | 2009-10-20 | Merck & Co., Inc. | Heteroaryl substituted triazole modulators of metabotropic glutamate receptor-5 |
| WO2003051833A2 (en) * | 2001-12-18 | 2003-06-26 | Merck & Co., Inc. | Heteroaryl substituted pyrazole modulators of metabotropic glutamate receptor-5 |
| CA2470519C (en) * | 2001-12-21 | 2009-11-17 | Merck & Co., Inc. | Heteroaryl substituted pyrrole modulators of metabotropic glutamate receptor-5 |
| CA2484233A1 (en) | 2002-05-13 | 2003-11-27 | Eli Lilly And Company | Multicyclic compounds for use as melanin concentrating hormone antagonists in the treatment of obesity and diabetes |
| AU2003234567A1 (en) | 2002-05-15 | 2003-12-02 | Janssen Pharmaceutica N.V. | N-substituted tricyclic 3-aminopyrazoles as pdfg receptor inhibitors |
| AU2003256755A1 (en) * | 2002-07-24 | 2004-02-09 | Ptc Therapeutics, Inc. | Ureido substituted benzoic acid compounds, their use for nonsense suppression and the treatment of diseases caused by such mutations |
| AU2003254157A1 (en) * | 2002-07-24 | 2004-02-09 | Ptc Therapeutics, Inc. | Acetylamino benzoic acid compounds and their use for nonsense suppression and the treatment of disease |
| CA2493816A1 (en) | 2002-07-24 | 2004-01-29 | Ptc Therapeutics, Inc. | Use of nucleoside compounds for nonsense suppression and the treatment of genetic diseases |
| PA8578101A1 (en) * | 2002-08-13 | 2004-05-07 | Warner Lambert Co | HETEROBIARILO DERIVATIVES AS METALOPROTEINASE IN MATRIX INHIBITORS |
| CA2495285A1 (en) * | 2002-08-13 | 2004-02-19 | Merck Sharp & Dohme Limited | Phenylpyridazine derivatives as ligands for gaba receptors |
| PL379544A1 (en) * | 2002-09-09 | 2006-10-02 | Amgen Inc. | 1,4,5-substituted 1,2-dihydro-pyrazol-3-one and 3-alkoxy-1h-pyrazole derivatives s tnf-alpha and interleukin lowering agents for the treatment of inflammations |
| WO2004046122A2 (en) * | 2002-11-16 | 2004-06-03 | Oxford Glycosciences (Uk) Ltd | Benzoxazole, benzthiazole and benzimidazole acid derivatives and their use as heparanase inhibitors |
| US20040176264A1 (en) * | 2002-12-30 | 2004-09-09 | The Procter & Gamble Company | Rinse aid composition containing water-soluble metal salt for use in automatic dishwashing for glassware corrosion protection |
| GB0303503D0 (en) * | 2003-02-14 | 2003-03-19 | Novartis Ag | Organic compounds |
| ES2325440T3 (en) * | 2003-02-20 | 2009-09-04 | Smithkline Beecham Corporation | PIRIMIDINE COMPOUNDS. |
| US7320986B2 (en) | 2003-03-07 | 2008-01-22 | Abbott Labortories | Fused tri and tetra-cyclic pyrazole kinase inhibitors |
| EP4101846B1 (en) | 2003-04-11 | 2023-08-02 | PTC Therapeutics, Inc. | 1,2,4-oxadiazole benzoic acid compounds and their use for nonsense suppression and the treatment of disease |
| AU2004232973A1 (en) | 2003-04-18 | 2004-11-04 | Memory Pharmaceuticals Corporation | Pyrazole derivatives as phosphodiesterase 4 inhibitors |
| ES2534605T3 (en) | 2004-08-23 | 2015-04-24 | Eli Lilly And Company | Histamine H3 receptor agents, preparation and therapeutic uses |
| EP1799659A1 (en) * | 2004-10-13 | 2007-06-27 | PTC Therapeutics, Inc. | Compounds for nonsense suppression, and methods for their use |
| WO2007059356A2 (en) | 2005-11-19 | 2007-05-24 | Government Of The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Furfurylidene- pyrazolidines and their use as inhibitors of ubiquitin e1 |
| EP2284154A1 (en) | 2009-07-29 | 2011-02-16 | Argon Pharma S.L. | Antitumor 1,2-diphenylpyrrole compounds and their preparation process |
-
2005
- 2005-10-13 EP EP05807667A patent/EP1799659A1/en not_active Withdrawn
- 2005-10-13 JP JP2007536867A patent/JP2008515992A/en active Pending
- 2005-10-13 MX MX2007004484A patent/MX2007004484A/en unknown
- 2005-10-13 US US11/577,189 patent/US9315467B2/en active Active
- 2005-10-13 KR KR1020077010767A patent/KR20070067201A/en not_active Withdrawn
- 2005-10-13 BR BRPI0515995-4A patent/BRPI0515995A/en not_active IP Right Cessation
- 2005-10-13 US US11/577,177 patent/US9051342B2/en not_active Expired - Fee Related
- 2005-10-13 AU AU2005295778A patent/AU2005295778A1/en not_active Abandoned
- 2005-10-13 EP EP10185133A patent/EP2363129A1/en not_active Withdrawn
- 2005-10-13 US US11/577,192 patent/US7902235B2/en not_active Expired - Fee Related
- 2005-10-13 SG SG200906828-9A patent/SG156640A1/en unknown
- 2005-10-13 EP EP10185130.1A patent/EP2311455B1/en not_active Expired - Lifetime
- 2005-10-13 AU AU2005295730A patent/AU2005295730A1/en not_active Abandoned
- 2005-10-13 EP EP05807462A patent/EP1799212A2/en not_active Withdrawn
- 2005-10-13 MX MX2007004487A patent/MX2007004487A/en unknown
- 2005-10-13 EP EP05804194A patent/EP1799207A2/en not_active Withdrawn
- 2005-10-13 SG SG200906829-7A patent/SG156641A1/en unknown
- 2005-10-13 WO PCT/US2005/036673 patent/WO2006044456A1/en not_active Ceased
- 2005-10-13 WO PCT/US2005/036762 patent/WO2006044503A2/en not_active Ceased
- 2005-10-13 WO PCT/US2005/037052 patent/WO2006044682A1/en not_active Ceased
- 2005-10-13 EP EP10185137A patent/EP2316452A1/en not_active Withdrawn
- 2005-10-13 EP EP10185128.5A patent/EP2301536B1/en not_active Expired - Lifetime
- 2005-10-13 CA CA002583177A patent/CA2583177A1/en not_active Abandoned
- 2005-10-13 EP EP05804171A patent/EP1812071A2/en not_active Withdrawn
- 2005-10-13 AU AU2005295727A patent/AU2005295727A1/en not_active Abandoned
- 2005-10-13 EP EP16163831.7A patent/EP3067053A1/en not_active Withdrawn
- 2005-10-13 EP EP10185139A patent/EP2402323A3/en not_active Withdrawn
- 2005-10-13 CA CA002583971A patent/CA2583971A1/en not_active Abandoned
- 2005-10-13 CA CA002582885A patent/CA2582885A1/en not_active Abandoned
- 2005-10-13 KR KR1020077009154A patent/KR20070067165A/en not_active Withdrawn
- 2005-10-13 MX MX2007004479A patent/MX2007004479A/en unknown
- 2005-10-13 JP JP2007536837A patent/JP2008515985A/en active Pending
- 2005-10-13 JP JP2007536865A patent/JP2008515990A/en active Pending
- 2005-10-13 US US11/577,191 patent/US9611230B2/en not_active Expired - Fee Related
- 2005-10-13 BR BRPI0516110-0A patent/BRPI0516110A/en not_active IP Right Cessation
- 2005-10-13 CA CA002583159A patent/CA2583159A1/en not_active Abandoned
- 2005-10-13 KR KR1020077010794A patent/KR20070065429A/en not_active Withdrawn
- 2005-10-13 EP EP10185126.9A patent/EP2301538B1/en not_active Expired - Lifetime
- 2005-10-13 EP EP15168072.5A patent/EP2939674A1/en not_active Withdrawn
- 2005-10-13 US US11/577,176 patent/US20080269191A1/en not_active Abandoned
- 2005-10-13 CA CA002583976A patent/CA2583976A1/en not_active Abandoned
- 2005-10-13 WO PCT/US2005/036764 patent/WO2006044505A2/en not_active Ceased
- 2005-10-13 WO PCT/US2005/036761 patent/WO2006044502A2/en not_active Ceased
- 2005-10-13 EP EP05815159.8A patent/EP1815206B1/en not_active Expired - Lifetime
-
2007
- 2007-04-11 IL IL182459A patent/IL182459A0/en unknown
- 2007-04-11 IL IL182461A patent/IL182461A0/en unknown
- 2007-04-11 IL IL182464A patent/IL182464A0/en unknown
-
2011
- 2011-03-07 US US13/042,456 patent/US8710031B2/en not_active Expired - Fee Related
-
2014
- 2014-04-28 US US14/263,478 patent/US9273076B2/en not_active Expired - Lifetime
-
2015
- 2015-06-08 US US14/733,615 patent/US20150274674A1/en not_active Abandoned
-
2016
- 2016-04-15 US US15/130,318 patent/US20160229818A1/en not_active Abandoned
-
2017
- 2017-03-31 US US15/475,240 patent/US20170204073A1/en not_active Abandoned
Cited By (115)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008515985A (en) * | 2004-10-13 | 2008-05-15 | ピーティーシー セラピューティクス,インコーポレーテッド | Nonsense-inhibiting compounds and methods of use |
| EP1986633B1 (en) * | 2006-02-10 | 2014-07-30 | Summit Corporation Plc | Treatment of duchenne muscular dystrophy |
| US8518980B2 (en) | 2006-02-10 | 2013-08-27 | Summit Corporation Plc | Treatment of Duchenne muscular dystrophy |
| WO2007091106A2 (en) | 2006-02-10 | 2007-08-16 | Summit Corporation Plc | Treatment of duchenne muscular dystrophy |
| USRE50453E1 (en) | 2006-04-07 | 2025-06-10 | Vertex Pharmaceuticals Incorporated | Indole derivatives as CFTR modulators |
| US11639347B2 (en) | 2006-04-07 | 2023-05-02 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US10022352B2 (en) | 2006-04-07 | 2018-07-17 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US9758510B2 (en) | 2006-04-07 | 2017-09-12 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US10975061B2 (en) | 2006-04-07 | 2021-04-13 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US10987348B2 (en) | 2006-04-07 | 2021-04-27 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US9974781B2 (en) | 2006-04-07 | 2018-05-22 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US10239867B2 (en) | 2006-04-07 | 2019-03-26 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| JP2010540407A (en) * | 2006-10-02 | 2010-12-24 | デジタル バイオテック カンパニー リミテッド | Composition for the treatment and prevention of cognitive impairment including a novel benzofuran derivative and use thereof |
| US9732080B2 (en) | 2006-11-03 | 2017-08-15 | Vertex Pharmaceuticals Incorporated | Azaindole derivatives as CFTR modulators |
| EP3251694A1 (en) | 2007-08-03 | 2017-12-06 | Summit (Oxford) Limited | Drug combinations for the treatment of duchenne muscular dystrophy |
| US8501713B2 (en) | 2007-08-03 | 2013-08-06 | Summit Corporation Plc | Drug combinations for the treatment of duchenne muscular dystrophy |
| WO2009019504A1 (en) | 2007-08-03 | 2009-02-12 | Summit Corporation Plc | Drug combinations for the treatment of duchenne muscular dystrophy |
| EP3012250A1 (en) | 2007-11-16 | 2016-04-27 | Vertex Pharmaceuticals Incorporated | Isoquinoline modulators of atp-binding cassette transporters |
| EP2578571A1 (en) | 2007-11-16 | 2013-04-10 | Vertex Pharmaceuticals Incorporated | Isoquinoline modulators of ATP-binding cassette transporters |
| EP3683218A1 (en) | 2007-12-07 | 2020-07-22 | Vertex Pharmaceuticals Incorporated | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid |
| EP3170818A1 (en) | 2007-12-07 | 2017-05-24 | Vertex Pharmaceuticals Incorporated | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl) benzoic acid |
| EP2980077A1 (en) | 2008-03-31 | 2016-02-03 | Vertex Pharmaceuticals Incorporated | Pyridyl derivatives as cftr modulators |
| EP2615085A1 (en) | 2008-03-31 | 2013-07-17 | Vertex Pharmaceuticals Incorporated | Pyridyl derivatives as CFTR modulators |
| US8609705B2 (en) | 2008-04-21 | 2013-12-17 | Sumitomo Chemical Company, Limited | Fused heterocyclic compounds, and compositions thereof, for use in arthropod pest control |
| US8242133B2 (en) | 2008-04-21 | 2012-08-14 | Sumitomo Chemical Company, Limited | Arthropod pest control compositions comprising substituted oxazolo [5,4-b ] pyridines |
| US8324387B2 (en) | 2008-04-21 | 2012-12-04 | Sumitomo Chemical Company, Limited | Substituted Oxazolo[5,4-b]pyridines |
| US8445522B2 (en) | 2008-04-21 | 2013-05-21 | Sumitomo Chemical Company, Limited | Substituted 2-(4-pyridyl)benzoxazoles, and compositions thereof, for use in arthropod pest control |
| US8530483B2 (en) | 2008-05-30 | 2013-09-10 | Merck Sharp & Dohme Corp. | Substituted azabenzoxazoles |
| US7842685B2 (en) | 2008-06-20 | 2010-11-30 | Amgen Inc. | S1P1 receptor agonists and use thereof |
| AU2010233917B2 (en) * | 2009-04-02 | 2015-07-02 | Merck Serono S.A. | Dihydroorotate dehydrogenase inhibitors |
| JP2012524715A (en) * | 2009-04-02 | 2012-10-18 | メルク セローノ ソシエテ アノニム | Dihydroorotate dehydrogenase inhibitor |
| US10081621B2 (en) | 2010-03-25 | 2018-09-25 | Vertex Pharmaceuticals Incorporated | Solid forms of (R)-1(2,2-difluorobenzo[D][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| WO2011119984A1 (en) | 2010-03-25 | 2011-09-29 | Vertex Pharmaceuticals Incorporated | Solid forms of (r)-1(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihyderoxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide |
| US10906891B2 (en) | 2010-03-25 | 2021-02-02 | Vertex Pharmaceuticals Incoporated | Solid forms of (R)-1(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| EP4253381A2 (en) | 2010-03-25 | 2023-10-04 | Vertex Pharmaceuticals Incorporated | Solid forms of (r)-1-(2,2-difluorobenzo[d][1,3]d ioxol-5-yl)-n-(1-(2,3-dihydroxyp ropyl)-6-fluoro-2-(1-hydroxy- 2-methylpropan-2-yl)-1h-indol- 5-yl) cyclopropanecarboxamide |
| EP2826776A1 (en) | 2010-03-25 | 2015-01-21 | Vertex Pharmaceuticals Incorporated | Solid amorphous form of (R)-1(2,2-difluorobenzo(D)(1,3)dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)-cyclopropanecarboxamide |
| EP3181561A1 (en) | 2010-03-25 | 2017-06-21 | Vertex Pharmaceuticals Incorporated | Synthetic intermediate of (r)-1(2,2 -difluorobenzo[d][1,3]dioxol-5yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2yl)-1h-indol-5yl)cyclopropanecarboxamide |
| US11578062B2 (en) | 2010-03-25 | 2023-02-14 | Vertex Pharmaceuticals Incorporated | Solid forms of (R)-1(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| EP3835297A1 (en) | 2010-03-25 | 2021-06-16 | Vertex Pharmaceuticals Incorporated | Synthesis and intermediates of (r)-1(2,2 -difluorobenzo[d][1,3]dioxol-5yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2yl)-1h-indol-5yl)cyclopropanecarboxamide |
| WO2011127290A2 (en) | 2010-04-07 | 2011-10-13 | Vertex Pharmaceuticals Incorporated | Solid forms of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| WO2011127241A2 (en) | 2010-04-07 | 2011-10-13 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyriodin-2-yl)benzoic acid and administration thereof |
| EP3150198A1 (en) | 2010-04-07 | 2017-04-05 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyriodin-2-yl)benzoic acid and administration thereof |
| EP4005559A1 (en) | 2010-04-07 | 2022-06-01 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyriodin-2-yl)benzoic acid and administration thereof |
| EP3381899A1 (en) | 2010-04-22 | 2018-10-03 | Vertex Pharmaceuticals Incorporated | Intermediate compound for process of producing cycloalkylcarboxamido-indole compounds |
| US10071979B2 (en) | 2010-04-22 | 2018-09-11 | Vertex Pharmaceuticals Incorporated | Process of producing cycloalkylcarboxamido-indole compounds |
| WO2011133751A2 (en) | 2010-04-22 | 2011-10-27 | Vertex Pharmaceuticals Incorporated | Process of producing cycloalkylcarboxamido-indole compounds |
| EP3045452A1 (en) | 2010-04-22 | 2016-07-20 | Vertex Pharmaceuticals Inc. | Process of producing cycloalkylcarboxamido-indole compounds |
| US9937233B2 (en) | 2010-08-06 | 2018-04-10 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US9181319B2 (en) | 2010-08-06 | 2015-11-10 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| WO2012027247A2 (en) | 2010-08-23 | 2012-03-01 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition of (r)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihydroxy propyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide and administration therof |
| US10064959B2 (en) | 2010-10-01 | 2018-09-04 | Modernatx, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US9657295B2 (en) | 2010-10-01 | 2017-05-23 | Modernatx, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US9950068B2 (en) | 2011-03-31 | 2018-04-24 | Modernatx, Inc. | Delivery and formulation of engineered nucleic acids |
| WO2012170061A1 (en) | 2011-06-08 | 2012-12-13 | Vertex Pharmaceuticals Incorporated | Formulations of (r)-1-(2,2-diflurobenzo[d][1,3] dioxol-5-yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methyl propan-2-yl)-1h-indol-5-yl)cyclopropanecarboxamide |
| US10751386B2 (en) | 2011-09-12 | 2020-08-25 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US10022425B2 (en) | 2011-09-12 | 2018-07-17 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| WO2013070961A1 (en) | 2011-11-08 | 2013-05-16 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| WO2013112804A1 (en) | 2012-01-25 | 2013-08-01 | Vertex Pharmaceuticals Incorporated | Formulations of 3-(6-(1-(2.2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| US9814760B2 (en) | 2012-04-02 | 2017-11-14 | Modernatx, Inc. | Modified polynucleotides for the production of biologics and proteins associated with human disease |
| US9828416B2 (en) | 2012-04-02 | 2017-11-28 | Modernatx, Inc. | Modified polynucleotides for the production of secreted proteins |
| WO2013151736A2 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | In vivo production of proteins |
| US9827332B2 (en) | 2012-04-02 | 2017-11-28 | Modernatx, Inc. | Modified polynucleotides for the production of proteins |
| US9255129B2 (en) | 2012-04-02 | 2016-02-09 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding SIAH E3 ubiquitin protein ligase 1 |
| US9878056B2 (en) | 2012-04-02 | 2018-01-30 | Modernatx, Inc. | Modified polynucleotides for the production of cosmetic proteins and peptides |
| WO2013151668A2 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | Modified polynucleotides for the production of secreted proteins |
| US9782462B2 (en) | 2012-04-02 | 2017-10-10 | Modernatx, Inc. | Modified polynucleotides for the production of proteins associated with human disease |
| US9587003B2 (en) | 2012-04-02 | 2017-03-07 | Modernatx, Inc. | Modified polynucleotides for the production of oncology-related proteins and peptides |
| US10501512B2 (en) | 2012-04-02 | 2019-12-10 | Modernatx, Inc. | Modified polynucleotides |
| US9572897B2 (en) | 2012-04-02 | 2017-02-21 | Modernatx, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| WO2013151666A2 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | Modified polynucleotides for the production of biologics and proteins associated with human disease |
| US10501439B2 (en) | 2012-04-24 | 2019-12-10 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors |
| US10076521B2 (en) | 2012-04-24 | 2018-09-18 | Vertex Pharamceuticals Incorporated | DNA-PK inhibitors |
| US11008305B2 (en) | 2012-04-24 | 2021-05-18 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors |
| US11021465B2 (en) | 2012-04-24 | 2021-06-01 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors |
| US9925188B2 (en) | 2012-04-24 | 2018-03-27 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors and uses thereof |
| US9878993B2 (en) | 2012-04-24 | 2018-01-30 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors for treatment of cancer |
| US9376448B2 (en) | 2012-04-24 | 2016-06-28 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors |
| US10391095B2 (en) | 2012-04-24 | 2019-08-27 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors |
| US10442791B2 (en) | 2012-04-24 | 2019-10-15 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors |
| US10058546B2 (en) | 2012-07-16 | 2018-08-28 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of (R)-1-(2,2-difluorobenzo[D][1,3]dioxo1-5-y1)-N-(1-(2,3-dihydroxypropy1)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-y1)-1H-indol-5-y1) cyclopropanecarbox-amide and administration thereof |
| EP4556008A2 (en) | 2012-11-02 | 2025-05-21 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cftr mediated diseases |
| WO2014071122A1 (en) | 2012-11-02 | 2014-05-08 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cftr mediated diseases |
| EP3470063A1 (en) | 2012-11-02 | 2019-04-17 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cftr mediated diseases |
| US9987284B2 (en) | 2013-03-12 | 2018-06-05 | Vertex Pharmaceuticals Incorporated | Substituted benzooxadiazole DNA-PK inhibitors |
| US12251387B2 (en) | 2013-03-12 | 2025-03-18 | Vertex Pharmaceuticals Incorporated | Substituted quinoxalines and benzo[c][1,2,5]oxadiazoles as DNA-PK inhibitors |
| US10786512B2 (en) | 2013-03-12 | 2020-09-29 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors |
| US10258627B2 (en) | 2013-03-12 | 2019-04-16 | Vertex Pharmaceutical Incorporated | DNA-PK inhibitors |
| US11813267B2 (en) | 2013-03-12 | 2023-11-14 | Vertex Pharmaceuticals Incorporated | DNA-PK inhibitors |
| US10973830B2 (en) | 2013-03-12 | 2021-04-13 | Vertex Pharmaceuticals Incorporated | Substituted quinoxaline DNA-PK inhibitors |
| US8980864B2 (en) | 2013-03-15 | 2015-03-17 | Moderna Therapeutics, Inc. | Compositions and methods of altering cholesterol levels |
| US10815291B2 (en) | 2013-09-30 | 2020-10-27 | Modernatx, Inc. | Polynucleotides encoding immune modulating polypeptides |
| WO2015051214A1 (en) | 2013-10-03 | 2015-04-09 | Moderna Therapeutics, Inc. | Polynucleotides encoding low density lipoprotein receptor |
| US10323076B2 (en) | 2013-10-03 | 2019-06-18 | Modernatx, Inc. | Polynucleotides encoding low density lipoprotein receptor |
| US10039761B2 (en) | 2013-10-17 | 2018-08-07 | Vertex Pharmaceuticals Incorporated | Co-crystals and pharmaceutical compositions comprising the same |
| US10716789B2 (en) | 2013-10-17 | 2020-07-21 | Vertex Pharmaceuticals Incorporated | Co-crystals and pharmaceutical compositions comprising the same |
| EP4223294A1 (en) | 2014-04-15 | 2023-08-09 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| WO2015160787A1 (en) | 2014-04-15 | 2015-10-22 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| EP3424534A1 (en) | 2014-04-15 | 2019-01-09 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| US10206877B2 (en) | 2014-04-15 | 2019-02-19 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| EP3925607A1 (en) | 2014-04-15 | 2021-12-22 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| US11951212B2 (en) | 2014-04-15 | 2024-04-09 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| US10980746B2 (en) | 2014-04-15 | 2021-04-20 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of cystic fibrosis transmembrane conductance regulator mediated diseases |
| WO2016014846A1 (en) | 2014-07-23 | 2016-01-28 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of intrabodies |
| WO2016057572A1 (en) | 2014-10-06 | 2016-04-14 | Mark Thomas Miller | Modulators of cystic fibrosis transmembrane conductance regulator |
| US11110108B2 (en) | 2016-09-27 | 2021-09-07 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of DNA-damaging agents and DNA-PK inhibitors |
| US11980633B2 (en) | 2016-09-27 | 2024-05-14 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of DNA-damaging agents and DNA-PK inhibitors |
| JP2020504766A (en) * | 2016-12-20 | 2020-02-13 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Compounds useful as immunomodulators |
| JP7106572B2 (en) | 2016-12-20 | 2022-07-26 | ブリストル-マイヤーズ スクイブ カンパニー | Compounds Useful as Immunomodulators |
| US11786607B2 (en) | 2017-06-15 | 2023-10-17 | Modernatx, Inc. | RNA formulations |
| US11744801B2 (en) | 2017-08-31 | 2023-09-05 | Modernatx, Inc. | Methods of making lipid nanoparticles |
| US12357575B2 (en) | 2017-08-31 | 2025-07-15 | Modernatx, Inc. | Methods of making lipid nanoparticles |
| WO2019101709A1 (en) | 2017-11-23 | 2019-05-31 | Universita' Degli Studi Di Palermo | Oxadiazole derivatives for the treatment of genetic diseases due to nonsense mutations |
| US11203578B2 (en) | 2017-11-23 | 2021-12-21 | Universita'degli Studi Dipalermo | Oxadiazole derivatives for the treatment of genetic diseases due to nonsense mutations |
| US12090235B2 (en) | 2018-09-20 | 2024-09-17 | Modernatx, Inc. | Preparation of lipid nanoparticles and methods of administration thereof |
| US12304901B2 (en) | 2022-11-03 | 2025-05-20 | Sentonix, Inc. | Selective ligands for tau aggregates |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2301536B1 (en) | Compounds and their use for treating nonsense-related diseases | |
| NO332843B1 (en) | 1,2,4-oxadiazole benzoic acid compounds, their use and pharmaceutical composition containing such a compound. | |
| US8101641B2 (en) | Hydroxylated 1,2,4-oxadiazole benzoic acid compounds and compositions thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV LY MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2582885 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005804171 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWP | Wipo information: published in national office |
Ref document number: 2005804171 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11577176 Country of ref document: US |