SUBSTITUTED N-ALKYLPYRIMIDINONES
CROSS REFERENCE TO RELATED APPLICATIONS
This application claims priority from U.S. Provisional Application Serial Number 60/637949, filed December 21, 2004, which claims priority to U.S. Provisional Application Serial Number 60/618856, filed October 13, 2004, the disclosure of each of which is incorporated herein by reference in its entirety.
FIELD OF THE INVENTION
[0001] This invention is directed to compounds that inhibit p38 kinase (particularly p38α kinase), TNF (particularly TNF-α), and/or cyclooxygenase (particularly cyclooxygenase-2 or "COX-2") activity. This invention also is directed to compositions of such compounds, methods for making such compounds, and methods for treating (including preventing) conditions (typically pathological conditions) associated with p38 kinase activity, TNF activity, and/or cyclooxygenase-2 activity.
BACKGROUND OF THE INVENTION
[0002] Mitogen- activated protein kinases (MAP) constitute a family of proline- directed serine/threonine kinases that activate their substrates by dual phosphorylation. The kinases are activated by a variety of signals, including nutritional and osmotic stress, UV light, growth factors, endotoxin, and inflammatory cytokines. The p38 MAP kinase group is a MAP family of various isoforms, including p38α, p38β, and p38γ. These kinases are responsible for phosphorylating and activating transcription factors (e.g., ATF2, CHOP, and MEF2C), as well as other kinases (e.g., MAPKAP-2 and MAPKAP- 3). The p38 isoforms are activated by bacterial lipopolysaccharide, physical and chemical stress, and pro- inflammatory cytokines, including tumor necrosis factor ("TNF') and interleukin-1 ("IL-I"). The products of the p38 phosphorylation mediate the production of inflammatory cytokines, including TNF, IL-I, and cyclooxygenase-2. [0003] It is believed that p38α kinase can cause or contribute to the effects of, for example, inflammation generally; arthritis; neuroinflammation; pain; fever; pulmonary disorders; cardiovascular diseases; cardiomyopathy; stroke; ischemia; reperfusion injury;
renal reperfusion injury; brain edema; neurotrauma and brain trauma; neurodegenerative disorders; central nervous system disorders; liver disease and nephritis; gastrointestinal conditions; ulcerative diseases; ophthalmic diseases; ophthalmological conditions; glaucoma; acute injury to the eye tissue and ocular traumas; diabetes; diabetic nephropathy; skin-related conditions; viral and bacterial infections; myalgias due to infection; influenza; endotoxic shock; toxic shock syndrome; autoimmune disease; bone resorption diseases; multiple sclerosis; disorders of the female reproductive system; pathological (but non-malignant) conditions, such as hemaginomas, angiofibroma of the nasopharynx, and avascular necrosis of bone; benign and malignant tumors/neoplasia including cancer; leukemia; lymphoma; systemic lupus erthrematosis (SLE); angiogenesis including neoplasia; and metastasis.
[0004] TNF is a cytokine produced primarily by activated monocytes and ' macrophages. Excessive or unregulated TNF production (particularly TNF-α) has been implicated in mediating a number of diseases. It is believed, for example, that TNF can cause or contribute to the effects of inflammation (e.g., rheumatoid arthritis and inflammatory bowel disease), asthma, autoimmune disease, graft rejection, multiple sclerosis, fibrotic diseases, cancer, fever, psoriasis, cardiovascular diseases (e.g., post-ischemic reperfusion injury and congestive heart failure), pulmonary diseases (e.g., hyperoxic alveolar injury), hemorrhage, coagulation, radiation damage, and acute phase responses like those seen with infections and sepsis and during shock (e.g., septic shock and hemodynamic shock). Chronic release of active TNF can cause cachexia and anorexia. And TNF can be lethal.
[0005] TNF also has been implicated in infectious diseases. These include, for example, malaria, mycobacterial infection and meningitis. These also include viral infections, such as HIV, influenza virus, and herpes virus, including herpes simplex virus type-1 (HSV-I), herpes simplex virus type-2 (HS V-2), cytomegalovirus (CMV), varicella-zoster virus (VZV), Epstein-Barr virus, human herpesvirus-6 (HHV-6), human herpesvirus-7 (HHV -7), human herpesvirus-8 (HHV-8), pseudorabies and rhinotracheitis, among others.
[0006] IL- 8 is another pro-inflammatory cytokine, which is produced by mononuclear cells, fibroblasts, endothelial cells, and keratinocytes. This cytokine is associated with conditions including inflammation.
[0007] IL-I is produced by activated monocytes and macrophages, and is involved in inflammatory responses. IL-I plays a role in many pathophysiological responses, including rheumatoid arthritis, fever, and reduction of bone resorption.
[0008] TNF, IL-I, and IL-8 affect a wide variety of cells and tissues, and are important inflammatory mediators of a wide variety of conditions. The inhibition of these cytokines by inhibition of the p38 kinase is beneficial in controlling, reducing, and alleviating many of these disease states.
[0009] Various substituted pyrimidinones have previously been described:
[0010] US patent application serial number 10/808,146 (filed March 24, 2004) refers to certain substituted pyrimidinones.
[0011] In view of the importance of substituted pyrimidinones in the treatment of several pathological conditions (particularly those associated with p38 kinase activity,
TNF activity, and/or cyclooxygenase-2 activity), there continues to be a need for substituted pyrimidinones compounds exhibiting an improved safety profile, solubility, and/or potency. The following disclosure describes substituted pyrimidinones compounds that exhibit one or more such desirable qualities.
SUMMARY OF THE INVENTION
[0012] This invention is directed to substituted pyrimidinone compounds that inhibit p38 kinase activity, TNF activity, and/or cyclooxygenase-2 activity. This invention also is directed to, for example, a method for inhibiting p38 kinase, TNF, and/or cyclooxygenase-2 activity, and particularly to a method for treating a condition (typically a pathological condition) mediated by p38 kinase activity, TNF activity, and/or cyclooxygenase-2 activity. Such a method is typically suitable for use with mammals in need of such treatment.
[0013] Briefly, therefore, this invention is directed, in part, to compounds that generally fall within structure of Formula I:
or a pharmaceutically acceptable salt, enantiomer or racemate thereof, wherein R
1 is selected from the group consisting of alkenyl, alkoxycarbonylalkylamino, alkoxycarbonylaminoalkoxy, alkoxycarbonylaminoheterocyclo, alkoxycarbonylaryl, alkoxycarbonylarylalkylamino, alkoxycarbonylheterocyclo, alkyl, alkylamino, alkylaminocarbonylalkyl, alkylaminocarbonylalkylamino, alkylaminocarbonylaminoalkoxy, alkylaminoheterocyclo, alkylcarbonylaminoalkoxy, alkylcarbonylaminoalkyl, alkylcarbonylaminoalkylamino, alkylcarbonylaminoheterocyclo, alkylcarbonylheterocycloamino, alkylcarbonyloxyalkylcarbonylaminoalkoxy, alkylcarbonyloxyalkylcarbonylaminoheterocyclo, alkylsulfonyl, alkylsulfonylaminoalkoxy, alkylsulfonylaminoalkyl, alkylsulfonylaminoalkylamino, alkylthio, aminoalkoxy, aminoalkyl, aminoalkylamino, aminoalkylcarbonylaminoheterocyclo, aminoalkylcarbonylheterocyclo, aminocarbonylalkoxy, aminocarbonylalkyl, aminocarbonylalkylamino, aminocarbonylalkylheterocyclo, aminocarbonylaminoalkoxy, aminocarbonylaminoalkylamino, aminocarbonylaryl, aminocarbonyldialkylamino, aminocarbonylheterocyclo, aminoheterocyclo, aryl, carboxyalkoxy, carboxyalkyl, carboxyaryl, carboxydialkylamino, cycloalkyl, dialkylaminoalkylamino, dihydroxyalkylamino, halo, haloalkylsulfonyloxy, haloarylalkylamino, heteroarylalkoxycarbonylaminoheterocyclo, heterocyclocarbonylalkylamino, heterocyclo, hydrogen, hydroxy, hydroxyalkoxy, hydroxyalkyl, hydroxyalkylamino, hydroxyalkylaminocarbonylalkoxy, hydroxyalkylaminocarbonylalkyl, hydroxyalkylaminocarbonylalkylamino, hydroxyalkylaminocarbonylaminoalkoxy, hydroxyalkylaminoheterocyclo, hydroxyalkylcarbonylaminoalkylamino, hydroxyalkylcarbonylaminoheterocyclo, hydroxyalkylcarbonylheterocyclo, hydroxyalkylheterocyclo, and hydroxyheterocyclo; R
2 is selected from the group consisting of alkyl, cycloalkyl and hydrogen; R
3 is selected from the group consisting of hydrogen, alkyl, alkoxy and halo; wherein each alky wherever they occur, are independently and optionally substituted with alkoxy, amino, carboxy, halo and hydroxyl; and R
4A, R
4B, R
4C, R
4D and R
4E are each independently selected form the group consisting of alkylaminocarbonyl aminoalkyl,
alkylarylheteroarylaminocarbonylaminoalkyl, aminoalkyl, arylcycloakylaminocarbonyldialkylaminoalkyl, arylcycloalkylaminocarbonylaminoalkyl, c'yano, cycloalkylaminocarbonylaminoalkyl, cycloalkylaminocarbonyldialkylaminoalkyl, halo, and hydrogen; wherein each aryl and heteroaryl where ever they occur, are independently and optionally substituted with alkyl.
[0014] This invention also is directed, in part, to pharmaceutical compositions comprising a therapeutically-effective amount of an above-described compound or pharmaceutically acceptable salt thereof.
[0015] This invention also is directed, in part, to a method for treating an inflammatory condition in a mammal. The method comprises administering an above- described compound or pharmaceutically acceptable salt thereof, to the mammal in an amount that is therapeutically-effective to treat the condition.
[0016] Further benefits of Applicants' invention will be apparent to one skilled in the art from reading this specification.
DETAILED DESCRIPTION
[0017] This detailed description of embodiments is intended only to acquaint others skilled in the art with Applicants' invention, its principles, and its practical application so that others skilled in the art may adapt and apply the invention in its numerous forms, as they may be best suited to the requirements of a particular use. This detailed description and its specific examples, while indicating embodiments of this invention, are intended for purposes of illustration only. This invention, therefore, is not limited to the embodiments described in this specification, and may be variously modified.
Compounds of This Invention
[0018] In accordance with this invention, it has been found that certain substituted pyrimidinone compounds are effective for inhibiting the activity (particularly pathological activity) of p38 kinase, TNF, and/or cyclooxygenase-2. [0019] Among its many embodiments the present invention provides a compound of Formula I:
or a pharmaceutically acceptable salt, enantiomer or racemate thereof, wherein R1 is selected from the group consisting of alkenyl, alkoxycarbonylalkylamino, alkoxycarbonylaminoalkoxy, alkoxycarbonylaminoheterocyclo, alkoxycarbonylaryl, alkoxycarbonylarylalkylamino, alkoxycarbonylheterocyclo, alkyl, alkylamino, alkylaminocarbonylalkyl, alkylaminocarbonylalkylamino, alkylaminocarbonylaminoalkoxy, alkylaminoheterocyclo, alkylcarbonylaminoalkoxy, alkylcarbonylaminoalkyl, alkylcarbonylaminoalkylamino, alkylcarbonylaminoheterocyclo, alkylcarbonylheterocycloamino, alkylcarbonyloxyalkylcarbonylaminoalkoxy, alkylcarbonyloxyalkylcarbonylaminoheterocyclo, alkylsulfonyl, alkylsulfonylaminoalkoxy, alkylsulfonylaminoalkyl, alkylsulfonylaminoalkylamino, alkylthio, aminoalkoxy, aminoalkyl, aminoalkyl amino, aminoalkylcarbonylaminoheterocyclo, aminoalkylcarbonylheterocyclo, aminocarbonylalkoxy, aminocarbonylalkyl, aminocarbonylalkylamino, aminocarbonylalkylheterocyclo, aminocarbonyl aminoalkoxy, aminocarbonylaminoalkylamino, aminocarbonylaryl, aminocarbonyldialkylamino, aminocarbonylheterocyclo, aminoheterocyclo, aryl, carboxyalkoxy, carboxyalkyl, carboxyaryl, carboxydialkylamino, cycloalkyl, dialkylaminoalkylamino, dihydroxyalkylamino, halo, haloalkylsulfonyloxy, haloarylalkylamino, heteroarylalkoxycarbonylaminoheterocyclo, heterocyclocarbonylalkylamino, heterocyclo, hydrogen, hydroxy, hydroxyalkoxy, hydroxyalkyl, hydroxyalkylamino, hydroxyalkylaminocarbonylalkoxy, hydroxyalkylaminocarbonylalkyl, hydroxyalkylaminocarbonylalkylamino, hydroxyalkylaminocarbonylaminoalkoxy, hydroxyalkylaminoheterocyclo, hydroxyalkylcarbonylaminoalkylamino, hydroxyalkylcarbonylaminoheterocyclo, hydroxyalkylcarbonylheterocyclo, hydroxyalkylheterocyclo, and hydroxyheterocyclo; R2 is selected from the group
consisting of alkyl, cycloalkyl and hydrogen; R is selected from the group consisting of hydrogen, alkyl, alkoxy and halo; wherein each alky wherever they occur, are independently and optionally substituted with alkoxy, amino, carboxy, halo and h'ydroxyl; and R4A, R4B, R4C, R4D and R4E are each independently selected form the group consisting of alkylaminocarbonylaminoalkyl, alkyl aryϊheteroarylaminocarbonylaminoalkyl , aminoalkyl , arylcycloakylaminocarbonyldialkylaminoalkyl, arylcycloalkylaminocarbonylaminoalkyl, cyano, cycloalkylaminocarbonylaminoalkyl, cycloalkylaminocarbonyldialkylaminoalkyl, halo, and hydrogen; wherein each aryl and heteroaryl where ever they occur, are independently and optionally substituted with alkyl.
[0020] In another embodiment, R1 is selected from the group consisting of (C2-C1O)- alkenyl, (C1-Ci 0)-alkoxycarbonyl-(C i -C i o)-alkylamino, (Ci-Ci o)-alkoxycarbonylamino- (C i -C i o)- alkoxy, (C i -C i o)- alkoxycarbonyl aminoheterocyclo, (C i -C i o)- alkoxycarbonylaryl, (Ci-Cio)-alkoxycarbonylaryl-(Ci-Cio)-alkylamino, (CI-CJO)- alkoxycarbonylheterocyclo, (Ci-Cio)-alkyl, (Ci-Cio)-alkylamino, (Ci-Cio)- alkylaminocarbonyl-(Ci-Cio)-alkyl, (Ci-Cio)-alkylaminocarbonyl-(Ci-Cio)-alkylamino, (Ci-Ci o)-alkylaminocarbonylamino-(C i -C i o)-alkoxy, (C i -Ci o)-alkylaminoheterocyclo, (Ci-Ci o)-alkylcarbonyl amino-(C i -C 10)-alkoxy, (C i -C io)-alkylcarbonylamino-(C ] -C io)- alkyl, (Ci-Ci o)-alkylcarbonylamino-(C i -C i o)-alkylamino, (Ci-Ci o)- alkylcarbonylaminoheterocyclo, (Ci-Cio)-alkylcarbonylheterocycloamino, (Ci-Cio)- alkylcarbonyl oxy-(C i -C 10)-alkylcarbonylamino-(Ci -C io)-alkoxy, (C i -C io)- alkylcarbonyloxy-(Ci-Cio)-alkylcarbonylaminoheterocyclo, (Ci-Cio)-alkylsulfonyl, (Ci- Cjo)-alkylsulfonylamino-(Ci-Cio)-alkoxy, (Ci-Cio)-alkylsulfonylamino-(Ci-Cio)-alkyl, (Ci-Cio)-alkylsulfonylamino-(Ci-Cio)-alkylamino, (Ci-Cio)-alkylthio, amino-(Ci-Cio)- alkoxy, amino-(Ci-Cio)-alkyl, amino-(Ci-Cio)-alkylamino, amino-(Ci-Cio)- alkylcarbonylaminoheterocyclo, amino-(C i -C i o)-alkylcarbonylheterocyclo, aminocarbonyl-(Ci-Cio)-alkoxy, aminocarbonyl-(Ci-Cio)-alkyl, aminocarbonyl-(Ci-Cio)- alkylamino, aminocarbonyl-(Ci-Cio)-alkylheterocyclo, aminocarbonylamino-(Ci-Cio)- alkoxy, aminocarbonylamino-(Ci-Cio)-alkylamino, aminocarbonylaryl, aminocarbonyl- (Ci-Cio)-dialkylamino, aminocarbonylheterocyclo, aminoheterocyclo, aryl, carboxy-(d- Cio)-alkoxy, carboxy-(Ci-Cio)-alkyl, carboxyaryl, carboxy-(Ci-Cio)-dialkylamino, (Cj- Cio)-cycloalkyl, (Ci-C]0)-dialkylamino-(Ci-Cio)-alkylamino, dihydroxy-(Ci-Cio)-
alkylamino, halo, halo-(Ci-Cio)-alkylsulfonyloxy, haloaryl-(C1-C10)-alkylamino, heteroaryl-(C1-C1o)-alkoxycarbonylaminoheterocyclo, heterocyclocarbonyl-(Ci-Cio)- alkylamino, heterocyclo, hydrogen, hydroxy, hydroxy-(C1-C1o)-alkoxy, hydroxy-(Cp C10)-alkylamino, hydroxy-(C1-Cio)-alkylaminocarbonyl-(Ci-C10)-alkoxy, hydroxy-(Ci- Cio)-alkylaminocarbonyl-(C1-Cio)-alkyl, hydroxy-(Ci-C10)-alkylaminocarbonyl-(C1- C10)-alkylamino, hydroxy-(C1-C10)-alkylaminocarbonylamino-(Ci-Cjo)-alkoxy, hydroxy- (C i -C i o)- alkylaminoheterocyclo, hydroxy- (C i -C i o)- alkylcarbonylamino-(C i -C i o)- alkylamino, hydroxy-(Ci-C1o)-alkylcarbonylaminoheterocyclo, hydroxy-(Ci-C!o)- alkylcarbonylheterocyclo, hydroxy-(Ci-C]o)-alkylheterocyclo, and hydroxyheterocyclo; R2 is selected from the group consisting of (Ci-Cio)-alkyl, (C1-C1o)-cycloalkyl and hydrogen; R3 is selected from the group consisting of hydrogen, (Ci-Cio)-alkyl, (C1-C1O)- alkoxy and halo; wherein each alky wherever they occur, are independently and optionally substituted with (C1-Cio)-alkoxy, amino, carboxy, halo and hydroxyl; and R4A, R4B, R4C, R4D and R4E are each independently selected form the group consisting of (C1- C10)-alkylaminocarbonylamino-(Ci-C10)-alkyl, (C1-C10)- alkylarylheteroarylaminocarbonylamino-(C1 -C ] o)-alkyl, amino-(C i -C 10)-alkyl, aryl-(C i - Cio)-cycloakylaminocarbonyl-(Ci-Cio)-dialkylamino-(Ci-Cio)-alkyl, aryl-(Ci-Cio)- cycloalkylaminocarbonylamino-(Ci-C1o)-alkyl, cyano, (C1-CiO)- cycloalkylaminocarbonylamino-(C]-Cio)-alkyl, (C1-C1o)-cycloalkylaminocarbonyl-(Ci- Cio)-dialkylamino-(Ci-Cio)-alkyl, halo, and hydrogen; wherein each aryl and heteroaryl where ever they occur, are independently and optionally substituted with (C1-C1o)-alkyl. [0021] In another embodiment, R1 is selected from the group consisting of (C2-C8)- alkenyl, (Ci-C8)-alkoxycarbonyl-(Ci-C8)-alkylamino, (Ci-C8)-alkoxycarbonylamino-(C1- C8)-alkoxy, (C]-C8)-alkoxycarbonylaminoheterocyclo, (Cj-C8)-alkoxycarbonylaryl, (Ci- C8)-alkoxycarbonylaryl-(C \ -C8)-alkylamino, (C i -C8)-alkoxycarbonylheterocyclo, (C i - C8)-alkyl, (Ci-C8)-alkylamino, (Ci-C8)-alkylaminocarbonyl-(CrC8)-alkyl, (Ci-C8)- alkylaminocarbonyl-(Ci-C8)-alkylamino, (Ci-C8)-alkylaminocarbonylamino-(Ci-C8)- alkoxy, (Ci-C8)-alkylaminoheterocyclo, (Ci-C8)-alkylcarbonylamino-(Ci-C8)-alkoxy, (Ci-C8)-alkylcarbonylamino-(Ci-C8)-alkyl, (Ci-C8)-alkylcarbonylamino-(Ci-C8)- alkylamino, (Ci-C8)-alkylcarbonylaminoheterocyclo, (Ci-C8)- alkylcarbonylheterocycloamino, (Ci-C8)-alkylcarbonyloxy-(Ci-C8)-alkylcarbonylamino- (Ci-Cg)-alkoxy, (Ci-C8)-alkylcarbonyloxy-(Ci-C8)-alkylcarbonylaminoheterocyclo, (Cr
C8)-alkylsulfonyl, (C1-C8)-alkylsulfonylamino-(Ci-C8)-alkoxy, (C1-C8)- alkylsulfonylamino-(Ci-C8)-alkyl, (C1-C8)-alkylsulfonylamino-(C1-C8)-alkylamino, (C1- C8)-alkylthio, amino-tCrC^-alkoxy, amino-(Ci-C8)-alkyl, amino-(Ci-C8)-alkylamino, amino-tC] -C8)-alkylcarbonylaminoheterocyclo, amino-(C \ -C8)- alkylcarbonylheterocyclo, aminocarbonyl-(Ci-C8)-alkoxy, aminocarbonyl-(C1-C8)-alkyl, aminocarbonyl-(Ci-C8)-alkylamino, aminocarbonyl-(Ci-C8)-alkylheterocyclo, aminocarbonylamino-(C1-C8)-alkoxy, aminocarbonylamino-(C1-C8)-alkylamino, aminocarbonylaryl, aminocarbonyl-(C1-C8)-dialkylamino, aminocarbonylheterocyclo, aminoheterocyclo, aryl, carboxy-(Ci-C8)-alkoxy, carboxy-(C1-C8)-alkyl, carboxyaryl, carboxy-(C1-C8)-dialkylamino, (Ci-C8)-cycloalkyl, (C1-C8)-dialkylamino-(C1-C8)- alkylamino, dihydroxy-(C1-C8)-alkylamino, halo, HaIo-(C1 -C8)-alkylsulfonyloxy, haloaryl-(Ci-C8)-alkylamino, heteroaryl-(Ci-C8)-alkoxycarbonylaminoheterocyclo, heterocyclocarbonyl-(C1-C8)-alkylamino, heterocyclo, hydrogen, hydroxy, hydroxy-(Cr C8)-alkoxy, hydroxy-(Ci-C8)-alkylamino, hydroxy-(C1-C8)-alkylaminocarbonyl-(Ci-C8)- alkoxy, hydroxy-(C)-C8)-alkylaminocarbonyl-(C1-C8)-alkyl, hydroxy-(Ci-C8)- alkylaminocarbonyl-(Ci-C8)-alkylamino, hydroxy-(C1-C8)-alkylaminocarbonylamino- (Ci-C8)-alkoxy, hydroxy-(Ci-C8)-alkylaminoheterocyclo, hydroxy-(Ci-C8)- alkylcarbonylamino-(C i -C8)-alkylamino, hydroxy-(C ] -C8)- alkylcarbonylaminoheterocyclo, hydroxy-^ -C8)-alkylcarbonylheterocyclo, hydroxy- (Ci-C8)-alkylheterocyclo, and hydroxyheterocyclo; R2 is selected from the group consisting of (Ci-C8)-alkyl, (Ci-C8)-cycloalkyl and hydrogen; R3 is selected from the group consisting of hydrogen, (Q-C^-alkyl, (Cj-C8)-alkoxy and halo; wherein each alky wherever they occur, are independently and optionally substituted with (C1-C8)-alkoxy, amino, carboxy, halo and hydroxyl; and R4A, R4B, R4C, R4D and R4E are each independently selected form the group consisting of (Ci-C8)-alkylaminocarbonylamino- (C ] -C8)-alkyl, (C i -C8)-alkylarylheteroarylaminocarbonylamino-(C \ -C8)-alkyl, amino- (C \ -C8)-alkyl, aryl-(C i -C8)-cycloakylaminocarbonyl-(C i -C8)-dialkylamino-(C i -C8)-alkyl, aryl-(Ci-C8)-cycloalkylaminocarbonylamino-(C1-C8)-alkyl, cyano, (C1-C8)- cycloalkylaminocarbonylamino-(C i -C8)-alkyl, (Cj -C8)-cycloalkylaminocarbonyl-(C i - C8)-dialkylamino-(Ci-C8)-alkyl, halo, and hydrogen; wherein each aryl and heteroaryl where ever they occur, are independently and optionally substituted with (Ci-C8)-alkyl.
[0022] In yet another embodiment, R1 is selected from the group consisting of (C2-C6)- alkenyl, (Ci-C6)-alkoxycarbonyl-(Ci-C6)-alkylamino, (C1-C6)-alkoxycarbonylamino-(C1- C6)-alkoxy, (C1-C6)-alkoxycarbonylaminoheterocyclo, (Ci-C6)-alkoxycarbonylaryl, (Ci- C6)-alkoxycarbonylaryl-(Ci-C6)-alkylamino, (CrC^-alkoxycarbonylheterocyclo, (Ci- C6)-alkyl, (CrC6)-alkylamino, (C1-C6)-alkylaminocarbonyl-(Ci-C6)-alkyl, (Ci-C6)- alkylaminocarbonyl-(C]-C6)-alkylamino, (CrC^-alkylaminocarbonylamino-^i-Cδ)- alkoxy, (Ci-C6)-alkylaminoheterocyclo, (Ci-C6)-alkylcarbonylamino-(C1-C6)-alkoxy, (Ci-C6)-alkylcarbonylamino-(C1-C6)-alkyl, (Ci-C6)-alkylcarbonylamino-(Ci-C6)- alkylamino, (Ci-C6)-alkylcarbonylaminoheterocyclo, (Ci-C6)- alkylcarbonylheterocycloamino, (C i -C6)-alkylcarbonyloxy-(C i -C6)-alkylcarbonylamino- (C i -C6)-alkoxy, (C i -C6)-alkylcarbonyloxy-(C i -C6)-alkylcarbonylaminoheterocyclo, (C i - C6)-alkylsulfonyl, (Ci-C6)-alkylsulfonylamino-(C1-C6)-alkoxy, (C1-C6)- alkylsulfonylamino-(C)-C6)-alkyl, (Ci-C6)-alkylsulfonylamino-(C1-C6)-alkylamino, (C1- C6)-alkylthio, amino-(C1-C6)-alkoxy, amino-(Ci-C6)-alkyl, amino-(C!-C6)-alkylamino, amino-(C i -C6)-alkylcarbonylaminoheterocyclo, amino-(C \ -C6)- alkylcarbonylheterocyclo, aminocarbonyl-(C i -C6)-alkoxy, aminocarbonyl-(C i -C6)-alkyl, aminocarbonyHCj-C^-alkylamino, aminocarbonyl-^i-C^-alkylheterocyclo, aminocarbonylamino-(C1-C6)-alkoxy, aminocarbonylamino-(C1-C6)-alkylamino, aminocarbonylaryl, aminocarbonyl-(d-C6)-dialkylamino, aminocarbonylheterocyclo, aminoheterocyclo, aryl, carboxy-(Ci-C6)-alkoxy, carboxy-(Ci-C6)-alkyl, carboxyaryl, carboxy-(C]-C6)-dialkylamino, (CrC6)-cycloalkyl, (C1-Cό)-dialkylamino-(Ci-C6)- alkylamino, dihydroxy-(Ci-C6)-alkylamino, halo, halo-^rC^-alkylsulfonyloxy, haloaryl-(Ci-C6)-alkylamino, heteroaryl-^j-C^-alkoxycarbonylaminoheterocyclo, heterocyclocarbonyl-(C1-C6)-alkylamino, heterocyclo, hydrogen, hydroxy, hydroxy-(Ci- C6)-alkoxy, hydroxy-(Ci-C6)-alkylamino, hydroxy-(C]-C6)-alkylaminocarbonyl-(Ci-C6)- alkoxy, hydroxy-(Ci-C6)-alkylaminocarbonyl-(Ci-C6)-alkyl, hydroxy- (Ci -C6)- alkylaminocarbonyl-(Ci-C6)-alkylamino, hydroxy-(Ci-C6)-alkylaminocarbonylamino- (Ci-C6)-alkoxy, hydroxy-(Ci-C6)-alkylaminoheterocyclo, hydroxy-(Ci-C6)- alkylcarbonylamino-tC] -C6)-alkylamino, hydroxy-(d -C6)- alkylcarbonylaminoheterocyclo, hydroxy-(Ci-C6)-alkylcarbonylheterocyclo, hydroxy- (Ci-C6)-alkylheterocyclo, and hydroxyheterocyclo; R2 is selected from the group consisting of (Ci-C6)-alkyl, (Ci-C6)-cycloalkyl and hydrogen; R3 is selected from the
group consisting of hydrogen, (Ci-C6)-alkyl, (d-C6)-alkoxy and halo; wherein each alky wherever they occur, are independently and optionally substituted with (Ci-C6)-alkoxy, amino, carboxy, halo and hydroxyl; and R4A, R4B, R4C, R4D and R4E are each independently and optionally selected form the group consisting of (C1-C6)- alkylaminocarbonylamino-(C \ -C6)-alkyl, (Ci-C6)- alkylarylheteroarylaminocarbonylamino-(Ci-C6)-alkyl, amino-(Ci-C6)-alkyl, 3TyI-(C1- C^-cycloakylaminocarbonyl-^i-C^-dialkylamino-fCrC^-alkyl, 3TyI-(C1-C6)- cycloalkylaminocarbonylamino-(C1-C6)-alkyl, cyano, (C1-C6)- cycloalkylaminocarbonylamino-(d-C6)-alkyl, (C1-C6)-cycloalkylaminocarbonyl-(C1- C6)-dialkylamino-(CrC6)-alkyl, halo, and hydrogen; wherein each aryl and heteroaryl where ever they occur, are independently and optionally substituted with (d-C6)-alkyl. [0023] In another embodiment, R1 is selected from the group consisting of (C2-C6)-alkenyl, (d-C6)-alkyl, (d-C6)-alkylamino, (d-C6)-alkylaminocarbonyl-(d- C6)-alkylamino, (d-C^-alkylaminoheterocyclo, (C1-C6)-alkylcarbonylamino-(Cj-C6)- alkoxy, (d-C^-alkylcarbonylamino^d-C^-alkylamino, (C1-C6)- alkylcarbonylheterocycloamino, (C i -C6)-alkylsulfonylamino-(C i -C6)-alkoxy, (C1-C6)- alkylsulfonylamino-(C1-C6)-alkyl, (C1-C6)-alkylsulfonylamino-(Ci-C6)-alkylamino, (C1- C6)-alkylsulphonylamino-(C]-C6)-alkylamino, amino-(Ci-C6)-alkylamino, amino-(d- C6)-alkylcarbonylheterocyclo, aminocarbonyl-(C i -C6)-alkoxy, aminocarbonyl-(C i -C6)- alkylamino, aminocarbonylamino-(C1-C6)-alkoxy, aminocarbonylamino-(C1-C6)- alkylamino, aminocarbonylaryl,aminocarbonylheterocyclo, aminoheterocyclo, aryl, dihydroxy-(C1-C6)-alkylamino, haloaryl-(Ci-C6)-alkylamino, hydrogen, hydroxyl- (d-C6)-alkoxy, hydroxyl-(C!-C6)-alkyl, hydroxyl-(d-C6)-alkylamino, hydroxyl-(Ci- C6)-alkylcarbonylamino-(Ci-C6)-alkylamino, hydroxyl-(CrC6)-alkylheterocyclo and hydroxyl-(C i -C6)-alkylcarbonylamino-(C i -C6)-alkylamino. [0024] In another embodiment, R2 is (d-C6)-alkyl. [0025] In another embodiment, R3 is halo.
[0026] In another embodiment, R4A, R4B, R4C, R4D and R4E are each independently selected from the group consisting of hydrogen, halo and (C1-C6)- alkylaminocarbonylamino-(Ci-C6)-alkyl.
[0027] In another embodiment, R2 is (CrC6)-alkyl; R3 is halo; and R4A, R4B, R4C, R4D and R4E are each independently selected from the group consisting of hydrogen, halo and
(C1-C6)-alkylaminocarbonylamino-(Ci-C6)-alkyl.
[0028] In another embodiment, R1 is selected from the group consisting of
(Ci-QO-alkylaminoheterocyclo, (C1-C6)-alkylcarbonylamino-(C1-C6)-alkylamino, (C1-
C6)-alkylsulfonylamino-(C1-C6)-alkoxy, (C1-C6)-alkylsulfonylamino-(Ci-C6)- alkylamino, (C1-C6)-alkylsulphonylamino-(C1-C6)-alkylamino, aminocarbonyl-(C J-C6)- alkylamino^minocarbonylamino-CCt-C^-alkoxy, aminocarbonylamino-(C!-C6)- alkylamino, aminocarbonylheterocyclo, dihydroxy-(C1-C6)-alkylamino, hydroxyl-(Ci-
C6)-alkylamino and hydroxy-(Ci-C6)-alkylcarbonylamino-(Cj-C6)-alkylamino.
[0029] In another embodiment, R2 is (Ci-C6)-alkyl.
[0030] In another embodiment, R3 is halo.
[0031] In another embodiment, R4A, R4B, R4C, R4D and R4E are each independently selected from the group consisting of hydrogen and halo.
[0032] In another embodiment, R2 is (Ci-C6)-alkyl; R3 is halo; and R4A, R4B, R4C, R4D and R4E are each independently and optionally selected from the group consisting of hydrogen and halo.
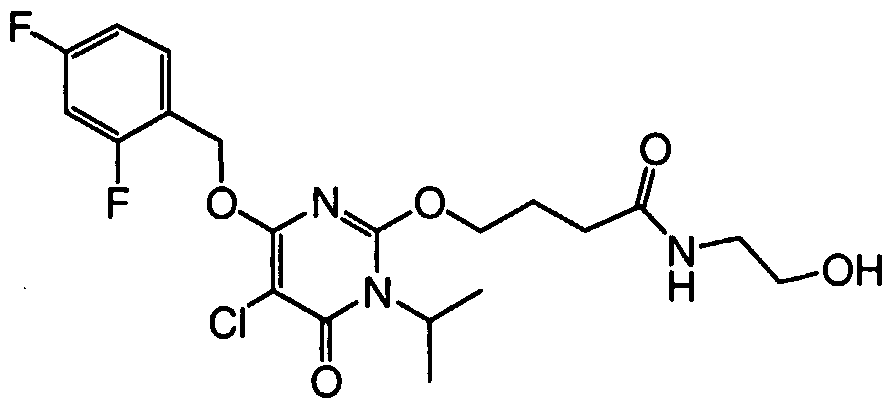
[0033] In another embodiment, the compound is selected from the group consisting of
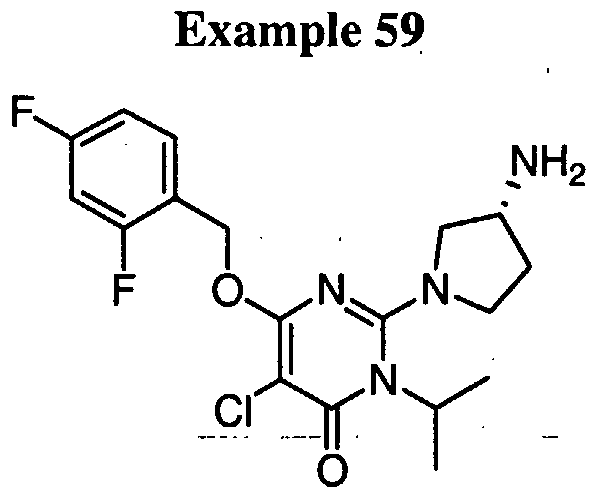
N- [2-( { 5-bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } amino)ethyl]methanesulfonamide,
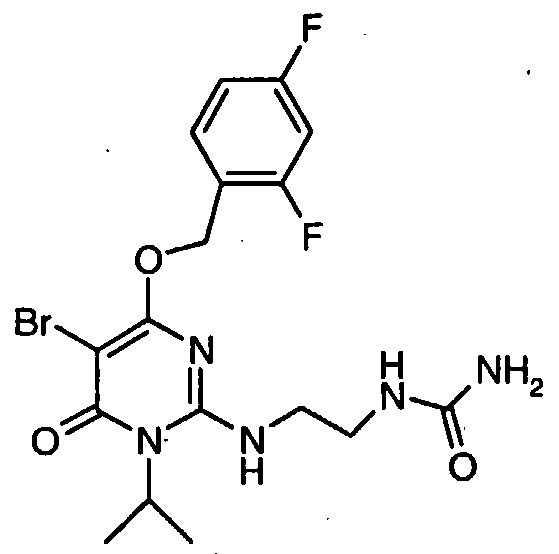
N-[2-( { 5-bromo-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } oxy)ethyl]urea,
N~2~- { 5-bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } -L-alaninamide,
N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)propyl]urea, iV-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)propyl]methane sulfonamide,
N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)propyl]-2-hydroxyacetamide,
_ 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-[(3R)-3-
(methylamino)pyrrolidin-l-yl]pyrimidin-4(3H)-one,
N~3~-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } -beta-alaninamide,
; N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-
I dihydropyrimidin-2-yl } amino)ethyl]-2-hydroxyacetamide,
N- [3-( { 5-bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } amino)propyl]acetamide,
N~3~-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } -beta-alaninamide,
N~2~-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } glycinamide,
N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)ethyl]urea,
N-[2-( { 5-bromo-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl}amino)ethyl]-2-hydroxy-2-methylpropanamide,
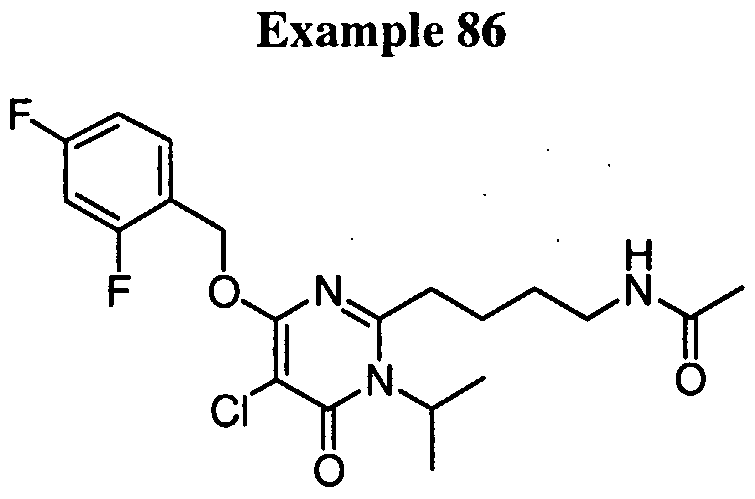
4-( { 5-bromo-4- [(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } amino)butanamide,
5-Bromo-6-[(2,4-difluorobenzyl)oxy]-2-{ [(2S)-2,3- dihydroxypropyl]amino } -3-isopropylpyrimidin-4(3H)-one,
N-[2-( { 5-bromo-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } oxy)ethyl]methanesulfonamide,
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)ethyl]urea,
N-[2-( { 5-chloro-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } amino)ethyl]-2-hydroxyacetamide,
N- [2-( { 5-chloro-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } amino)ethyl]methanesulfonamide,
5-Bromo-6-[(2,4-difluorobenzyl)oxy]-2-{[(2R)-2,3- dihydroxypropyl]amino } -3-isopropylpyrimidin-4(3H)-one, l-{5-Bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}prolinamide,
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } oxy)ethyl]urea,
N- [3-( { 5-bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxy-2-methylpropanamide,
5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(4-fluorobenzyl)amino]-3- isopropylpyrimidin-4(3H)-one,
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)ethyl]acetamide,
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } oxy)ethyl]methanesulfonamide,
5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(2S)-2- (hydroxymethyl)pyrrolidin-l-yl]-3-isopropylpyrimidin-4(3H)-one trifluoroacetate,
5-bromo-6- [(2,4-difluorobenzyl)oxy] -2- [(2-hydroxy-2- methylpropyl)amino]-3-isopropylpyrimidin-4(3H)-one,
1 - { 5-Bromo-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } prolinamide,
7V-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)ethyl]-2-hydroxy-2-methylpropanamide,
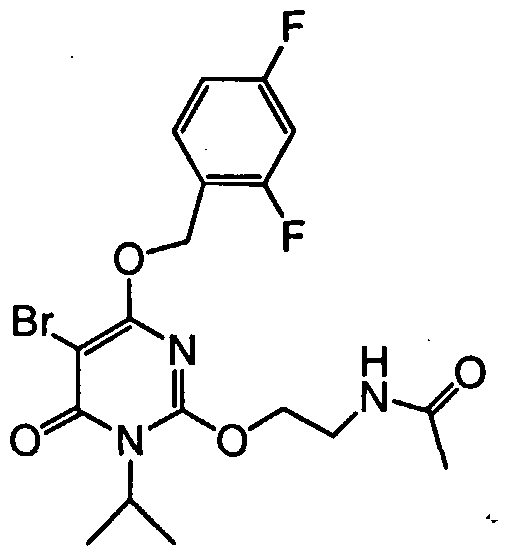
7V-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } oxy)ethyl]acetamide,
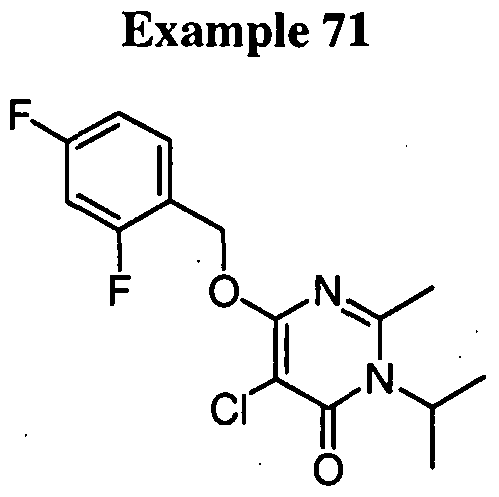
5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one,
N-( { 5-bromo-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } methyl)methanesulfonamide,
2-[(3R)-3-aminopyrrolidin-l-yl]-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3- isopropylpyrimidin-4(3H)-one trifluoroacetate,
2-[(l-acetylpiperidin-4-yl)amino]-5-bromo-6-[(2,4-difluorobenzyl)oxy]- 3-isopropylpyrimidin-4(3H)-one,
N~2~- { 5-bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } -N- 1 —methylglycinamide,
N~2—{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } glycinamide,
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } oxy)ethyl]acetamide,
5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-phenylpyrimidin- 4(3H)-one,
; 4-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-
I dihydropyrimidin-2-yl Jbenzamide, l-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } piperidine-3-carboxamide,
N-(2- { [(5-bromo- 1 -isopropyl^-methyl-ό-oxo- 1 ,6-dihydropyrimidin-4- yl)oxy]methyl } -5-fluorobenzyl)-N'-ethylurea,
2-(3-Aminopyrrolidin-l-yl)-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3- isopropylpyrimidin-4(3H)-one trifluoroacetate,
5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(3,3-dimethylbutyl)amino]-3- isopropylpyrimidin-4(3H)-one,
5-chloro-6-[(2,4-difluorobenzyl)oxy]-2-(4-hydroxybutoxy)-3- isopropylpyrimidin-4(3H)-one,
5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(3R)-3-(ethylamino)pyrrolidin-l- yl] -3 -isopropylpyrimidin-4(3H)-one,
4-( { 5-chloro-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } oxy)butanamide,
2-[(2-aminoethyl)amino]-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3- isopropylpyrimidin-4(3H)-one trifluoroacetate, and
2-but-3-enyl-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin- 4(3H)-one, or a pharmaceutically acceptable salt thereof. In another embodiment, the compound is selected from the group consisting of
N- [2-( { 5-bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } amino)ethyl]methanesulfonamide,
N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } oxy)ethyl]urea,
N~2~- { 5-bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } -L-alaninamide,
N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)propyl]urea,
N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyriπαidin-2-yl } amino)propyl] methane sulfonamide,
N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)propyl]-2-hydroxyacetamide,
5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-[(3R)-3- (methylamino)pyrrolidin- 1 -yl]pyrimidin-4(3H)-one,
N~3~-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } -beta-alaninamide,
N- [2-( { 5-bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } amino)ethyl]-2-hydroxyacetamide,
N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)propyl] acetamide,
N~3~- { 5-bromo-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } -beta-alaninamide,
N~2—{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } glycinamide,
N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)ethyl]urea,
N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)ethyl]-2-hydroxy-2-methylpropanamide,
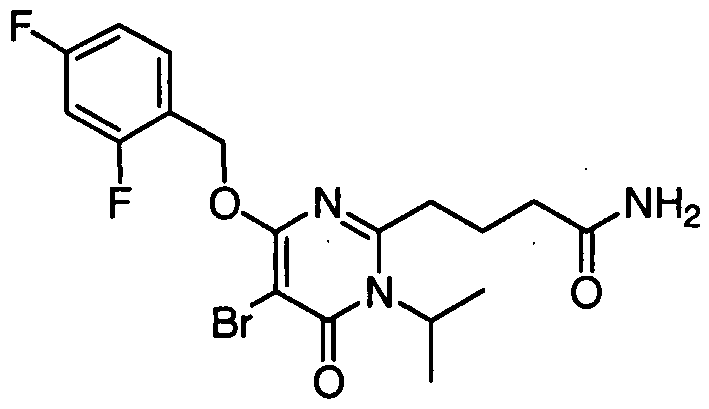
4-( { 5-bromo-4- [(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } amino)butanamide,
5-Bromo-6-[(2,4-difluorobenzyl)oxy]-2-{[(2S)-2,3- dihydroxypropyl]amino } -3-isopropylpyrimidin-4(3H)-one,
N- [2-( { 5-bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } oxy)ethyl]methanesulfonamide,
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)ethyl]urea,
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)ethyl]-2-hydroxyacetamide,
- - N-[2-({ 5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)ethyl]methanesulfonamide,
5-Bromo-6-[(2,4-difluorobenzyl)oxy]-2-{[(2R)-2,3- dihydroxypropyl]amino}-3-isopropylpyrimidin-4(3H)-one, : 1- { 5-Bromo-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-
I dihydropyrimidin-2-yl Jprolinamide,
N- [2-( { 5-chloro-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } oxy)ethyl]urea,
N- [3 -( { 5-bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } amino)ρropyl]-2-hydroxy-2-methylpropanamide, and
5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(4-fluorobenzyl)amino]-3- isopropylpyrimidin-4(3H)-one, or a pharmaceutically acceptable salt thereof. [0034] In one embodiment, a pharmacaeutical composition comprising a compound of Formula I and a pharmaceutically acceptable excipient.
[0035] In one embodiment, a method for the treatment or prevention of an inflammatory disorder in a subject in need of such treatment or prevention, wherein the method comprises administering to the subject an amount of a compound of Formula I wherein the amount of the compound is effective for the treatment or prevention of the inflammatory disorder.
[0036] In one embodiment, the inflammatory disorder is arthritis. [0037] In one embodiment, the inflammatory disorder is osteoarthritis. [0038] In one embodiment, the inflammatory disorder is rheumatoid arthritis. [0039] In one embodiment, the inflammatory disorder is asthma. [0040] This invention also is directed to tautomers of such compounds, as well as salts (particularly pharmaceutically-acceptable salts) of such compounds and tautomers. [0041] This invention also is directed, in part, to a method for treating a condition mediated by pathological p38 kinase activity (particularly p38α activity) in a mammal. The method comprises administering an above-described compound or pharmaceutically acceptable salt thereof, to the mammal in an amount that is therapeutically-effective to treat the condition.
[0042] This invention also is directed, in part, to a method for treating a condition mediated by pathological TNF activity (particularly TNF-α activity) in a mammal. The method comprises administering an above-described compound or pharmaceutically
acceptable salt thereof, to the mammal in an amount that is therapeutically-effective to treat the condition.
[0043] This invention also is directed, in part, to a method for treating a condition mediated by pathological cyclooxygenase-2 activity in a mammal. The method comprises administering an above-described compound or pharmaceutically acceptable salt thereof, to the mammal in an amount that is therapeutically-effective to treat the condition.
Compounds of this Invention Having One or More Asymmetric Carbons [0044] The present invention also comprises compounds of Formulas I having one or more asymmetric carbons. It is known to those skilled in the art that the compounds of the present invention having asymmetric carbon atoms may exist in diastereomeric, racemic, or optically active forms. All of these forms are contemplated within the scope of this invention. More specifically, the present invention includes enantiomers, diastereomers, racemic mixtures, and other mixtures thereof.
Salts of the Compounds of this Invention
[0045] The compounds of this invention may be used in the form of salts derived from inorganic or organic acids. Depending on the particular compound, a salt of the compound may be advantageous due to one or more of the salt's physical properties, such as enhanced pharmaceutical stability in differing temperatures and humidities, or a desirable solubility in water or oil. In some instances, a salt of a compound also may be used as an aid in the isolation, purification, and/or resolution of the compound. [0046] Where a salt is intended to be administered to a patient (as opposed to, for example, being used in an in vitro context), the salt preferably is pharmaceutically acceptable. Pharmaceutically acceptable salts include salts commonly used to form alkali metal salts and to form addition salts of free acids or free bases. In general, these salts typically may be prepared by conventional means with a compound of this invention by reacting, for example, the appropriate acid or base with the compound. [0047] Pharmaceutically- acceptable acid addition salts of the compounds of this invention may be prepared from an inorganic or organic acid. Examples of suitable inorganic acids include hydrochloric, hydrobromic acid, hydroionic, nitric, carbonic,
sulfuric, and phosphoric acid. Suitable organic acids generally include, for example, aliphatic, cycloaliphatic, aromatic, araliphatic, heterocyclyl, carboxyic, and sulfonic classes of organic acids. Specific examples of suitable organic acids include acetate, trifluoroacetate, formate, propionate, succinate, glycolate, gluconate, digluconate, lactate, malate, tartaric acid, citrate, ascorbate, glucuronate, maleate, fumarate, pyruvate, aspartate, glutamate, benzoate, anthranilic acid, mesylate, stearate, salicylate, p-hydroxybenzoate, phenyl acetate, mandelate, embonate (pamoate), methanesulfonate, ethanesulfonate, benzenesulfonate, pantothenate, toluenesulfonate, 2-hydroxyethanesulfonate, sufanilate, cyclohexylaminosulfonate, algenic acid, b-hydroxybutyric acid, galactarate, galacturonate, adipate, alginate, bisulfate, butyrate, camphorate, camphorsulfonate, cyclopentanepropionate, dodecylsulfate, glycoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, nicotinate, 2-naphthalesulfonate, oxalate, palmoate, pectinate, persulfate, 3-phenylpropionate, picrate, pivalate, thiocyanate, tosylate, and undecanoate.
[0048] Pharmaceutically-acceptable base addition salts of the compounds of this invention include, for example, metallic salts and organic salts. Preferred metallic salts include alkali metal (group Ia) salts, alkaline earth metal (group Ha) salts, and other physiological acceptable metal salts. Such salts may be made from aluminum, calcium, lithium, magnesium, potassium, sodium, and zinc. Preferred organic salts may be made from tertiary amines and quaternary amine salts, such as tromethamine, diethylamine, N,N'-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethyl enediamine, meglumine (N-methylglucamine), and procaine. Basic nitrogen-containing groups may be quaternized with agents such as lower alkyl (C1-C6) halides (e.g., methyl, ethyl, propyl, and butyl chlorides, bromides, and iodides), dialkyl sulfates (e.g., dimethyl, diethyl, dibuytl, and diamyl sulfates), long chain halides (e.g., decyl, lauryl, myristyl, and stearyl chlorides, bromides, and iodides), arylalkyl halides (e.g., benzyl and phenethyl bromides), and others.
Treating Conditions Using the Compounds of this Invention [0049] This invention is directed, in part, to a method for treating a condition (typically a pathological condition) in mammals, such as humans, other primates {e.g., monkeys, chimpanzees, etc.), companion animals (e.g., dogs, cats, horses, etc.), farm
animals (e.g., goats, sheep, pigs, cattle, etc.), laboratory animals (e.g., mice, rats, etc.), and wild and zoo animals (e.g., wolves, bears, deer, etc.) having or disposed to having such a condition.
[0050] In this specification, the phrase "treating a condition" means ameliorating, suppressing, eradicating, reducing the severity of, decreasing the frequency of incidence of, preventing, reducing the risk of, or delaying the onset of the condition. [0051] Some embodiments of this invention are directed to a method for treating a p38-mediated condition. As used herein, the term "p38-mediated condition" refers to any condition (particularly pathological conditions, i.e., diseases and disorders) in which p38 kinase (particularly p38α kinase) plays a role, either by control of p38 kinase itself, or by p38 kinase causing another factor to be released, such as, for example, DL-I, lL-6, or IL-8. A disease state in which, for instance, IL-I is a major component, and whose production or action is exacerbated or secreted in response to p38, would therefore be considered a disorder mediated by p38.
[0052] The compounds of this invention generally are useful for treating pathological conditions that include, but are not limited to:
(a) inflammation;
(b) arthritis, such as rheumatoid arthritis, spondyloarthropathies, gouty arthritis, osteoarthritis, systemic lupus erythematosus arthritis, juvenile arthritis, osteoarthritis, and gouty arthritis;
(c) neuroinflammation;
(d) pain (i.e., use of the compounds as analgesics), such as neuropathic pain;
(e) fever (i.e., use of the compounds as antipyretics);
(f) pulmonary disorders or lung inflammation, such as adult respiratory distress syndrome, pulmonary sarcoisosis, asthma, silicosis, and chronic pulmonary inflammatory disease;
(g) cardiovascular diseases, such as atherosclerosis, myocardial infarction (such as post-myocardial infarction indications), thrombosis, congestive heart failure, cardiac reperfusion injury, and complications associated with hypertension and/or heart failure such as vascular organ damage;
_(h) cardiomyopathy;
(i) stroke, such as ischemic and hemorrhagic stroke;
(J) ischemia, such as brain ischemia and ischemia resulting from cardiac/coronary bypass; ' (k) reperfusion injury; J (1) renal reperfusion injury;
(m) brain edema;
(n) neurotrauma and brain trauma, such as closed head injury;
(o) neurodegenerative disorders;
(p) central nervous system disorders (these include, for example, disorders having an inflammatory or apoptotic component), such as Alzheimer's disease, Parkinson's disease, Huntington's Disease, amyotrophic lateral sclerosis, spinal cord injury, and peripheral neuropathy;
(q) liver disease and nephritis;
(r) gastrointestinal conditions, such as inflammatory bowel disease, Crohn's disease, gastritis, irritable bowel syndrome, and ulcerative colitis;
(s) ulcerative diseases, such as gastric ulcer;
(t) ophthalmic diseases, such as retinitis, retinopathies (such as diabetic retinopathy), uveitis, ocular photophobia, nonglaucomatous optic nerve atrophy, and age- related macular degeneration (ARMD) (such as ARMD-atrophic form);
(u) ophthalmological conditions, such as corneal graft rejection, ocular neovascularization, retinal neovascularization (such as neovascularization following injury or infection), and retrolental fibroplasia;
(v) glaucoma, such as primary open angle glaucoma (POAG), juvenile onset primary open-angle glaucoma, angle-closure glaucoma, pseudoexfoliative glaucoma, anterior ischemic optic neuropathy (AION), ocular hypertension, Reiger's syndrome, normal tension glaucoma, neovascular glaucoma, ocular inflammation, and corticosteroid-induced glaucoma;
(w) acute injury to the eye tissue and ocular traumas, such as post- traumatic glaucoma, traumatic optic neuropathy, and central retinal artery occlusion (CRAO);
(x) diabetes;
(y) diabetic nephropathy;
(z) skin-related conditions, such as psoriasis, eczema, burns, dermatitis, keloid formation, scar tissue formation, and angiogenic disorders;
(aa) viral and bacterial infections, such as sepsis, septic shock, gram negative sepsis, malaria, meningitis, opportunistic infections, cachexia secondary to infection or malignancy, cachexia secondary to acquired immune deficiency syndrome (AIDS), AE)S, ARC (AIDS related complex), pneumonia, and herpes virus;
(bb) myalgias due to infection;
(cc) influenza;
(dd) endotoxic shock;
(ee) toxic shock syndrome;
(ff) autoimmune disease, such as graft vs. host reaction and allograft rejections;
(gg) bone resorption diseases, such as osteoporosis;
(hh) multiple sclerosis;
(ii) disorders of the female reproductive system, such as endometriosis;1
(Jj) pathological, but non-malignant, conditions, such as hemaginomas (such as infantile hemaginomas), angiofibroma of the nasopharynx, and avascular necrosis of bone;
(kk) benign and malignant tumors/neoplasia including cancer, such as colorectal cancer, brain cancer, bone cancer, epithelial cell-derived neoplasia (epithelial carcinoma) such as basal cell carcinoma, adenocarcinoma, gastrointestinal cancer such as Hp cancer, mouth cancer, esophageal cancer, small bowel cancer and stomach cancer, colon cancer, liver cancer, bladder cancer, pancreas cancer, ovarian cancer, cervical cancer, lung cancer, breast cancer, skin cancer such as squamus cell and basal cell cancers, prostate cancer, renal cell carcinoma, and other known cancers that affect epithelial cells throughout the body;
(11) leukemia;
(mm) lymphoma, such as B cell lymphoma;
(nn) systemic lupus erthrematosis (SLE);
(oo) angiogenesis including neoplasia; and
(pp) metastasis.
[0053] The compounds of this invention generally are also useful for treating pathological conditions that include, but are not limited to:
(a) asthma of whatever type, etiology, or pathogenesis, in particular asthma that is a member selected from the group consisting of atopic asthma, non-atopic asthma, allergic asthma, atopic bronchial lgE-mediated asthma, bronchial asthma, essential asthma, true asthma, intrinsic asthma caused by pathophysiologic disturbances, extrinsic asthma caused by environmental factors, essential asthma of unknown or inapparent cause, non-atopic asthma, bronchitic asthma, emphysematous asthma, exercise-induced asthma, allergen induced asthma, cold air induced asthma, occupational asthma, infective asthma caused by bacterial, fungal, protozoal, or viral infection, non-allergic asthma, incipient asthma, wheezy infant syndrome and bronchiolytis;
(b) chronic or acute bronchoconstriction, chronic bronchitis, small airways obstruction, and emphysema;
(c) obstructive or inflammatory airways diseases of whatever type, etiology, or pathogenesis, in particular an obstructive or inflammatory airways disease that is a member selected from the group consisting of chronic eosinophilic pneumonia, chronic obstructive pulmonary disease (COPD), COPD that includes chronic bronchitis, pulmonary emphysema or dyspnea associated or not associated with COPD, COPD that is characterized by irreversible, progressive airways obstruction, adult respiratory distress syndrome (ARDS), exacerbation of airways hyper-reactivity consequent to other drug therapy and airways disease that is associated with pulmonary hypertension;
(d) bronchitis of whatever type, etiology, or pathogenesis, in particular bronchitis that is a member selected from the group consisting of acute bronchitis, acute laryngotracheal bronchitis, arachidic bronchitis, catarrhal bronchitis, croupus bronchitis, dry bronchitis, infectious asthmatic bronchitis, productive bronchitis, staphylococcus or streptococcal bronchitis and vesicular bronchitis;
(e) acute lung injury; and
(f) bronchiectasis of whatever type, etiology, or pathogenesis, in particular bronchiectasis that is a member selected from the group consisting of cylindric bronchiectasis, sacculated bronchiectasis, fusiform bronchiectasis, capillary bronchiectasis, cystic bronchiectasis, dry bronchiectasis and follicular bronchiectasis.
[0054] The compounds of this invention generally are also useful in treating obstructive or inflammatory airways diseases of whatever type, etiology, or pathogenesis, in particular an obstructive or inflammatory airways disease that is a member selected from the group consisting of chronic eosinophilic pneumonia, chronic obstructive pulmonary disease (COPD), COPD that includes chronic bronchitis, pulmonary emphysema or dyspnea associated or not associated with COPD, COPD that is characterized by irreversible, progressive airways obstruction, adult respiratory distress syndrome (ARDS), exacerbation of airways hyper-reactivity consequent to other drug therapy and airways disease that is associated with pulmonary hypertension. [0055] Some embodiments of this invention are alternatively (or additionally) directed to a method for treating a TNF-mediated condition. As used herein, the term "TNF-mediated condition" refers to any condition (particularly any pathological conditions, i.e., diseases or disorders) in which TNF plays a role, either by control of TNF itself, or by TNF causing another monokine to be released, such as, for example, IL-I, IL-6, and/or IL-8. A disease state in which, for instance, IL-I is a major component and whose production or action is exacerbated or secreted in response to TNF, would therefore be considered a disorder mediated by TNF.
[0056] Examples of TNF-mediated conditions include inflammation (e.g., rheumatoid arthritis), autoimmune disease, graft rejection, multiple sclerosis, a fibrotic disease, cancer, an infectious disease (e.g., malaria, mycobacterial infection, meningitis, etc.), fever, psoriasis, a cardiovascular disease (e.g., post-ischemic reperfusion injury and congestive heart failure), a pulmonary disease, hemorrhage, coagulation, hyperoxic alveolar injury, radiation damage, acute phase responses like those seen with infections and sepsis and during shock (e.g., septic shock, hemodynamic shock, etc.), cachexia, and anorexia. Such conditions also include infectious diseases. Such infectious diseases include, for example, malaria, mycobacterial infection and meningitis. Such infectious diseases also include viral infections, such as HIV, influenza virus, and herpes virus,
including herpes simplex virus type-1 (HSV-I), herpes simplex virus type-2 (HS V-2), cytomegalovirus (CMV), varicella-zoster virus (VZV), Epstein-Barr virus, human herpesvirus-6 (HHV-6), human herpesvirus-7 (HHV-7), human herpesvirus-8 (HHV-8), p'seudorabies and rhinotracheitis, among others.
[0057] As TNF-β has close structural homology with TNF-α (also known as cachectin), and because each induces similar biologic responses and binds to the same cellular receptor, the synthesis of both TNF-α and TNF-β are inhibited by the compounds of this invention and thus are herein referred to collectively as "TNF" unless specifically delineated otherwise.
[0058] Some embodiments of this invention are alternatively (or additionally) directed to a method for treating a cyclooxygenase-2-mediated condition. As used herein, the term "cyclooxygenase-2-mediated condition" refers to any condition (particularly pathological conditions, i.e., diseases and disorders) in which cyclooxygenase-2 plays a role, either by control of cyclooxygenase-2 itself, or by cyclooxygenase-2 causing another factor to be released. Many cyclooxygenase-2- mediated conditions are known in the art, and include, for example, inflammation and other cyclooxygenase-mediated disorders listed by Carter et al. in U.S. Patent No. 6,271,253.
[0059] In some embodiments of particular interest, the condition treated by the methods of this invention comprises inflammation.
[0060] In some embodiments of particular interest, the condition treated by the methods of this invention comprises arthritis.
[0061] In some embodiments of particular interest, the condition treated by the methods of this invention comprises rheumatoid arthritis.
[0062] In some embodiments of particular interest, the condition treated by the methods of this invention comprises asthma.
[0063] In some embodiments of particular interest, the condition treated by the methods of this invention comprises a coronary condition.
[0064] In some embodiments of particular interest, the condition treated by the methods of this invention comprises bone loss.
[0065] In some embodiments of particular interest, the condition treated by the methods of this invention comprises B cell lymphoma.
[0066] In some embodiments of particular interest, the condition treated by the methods of this invention comprises COPD.
[0067] The compounds of the invention can also be used in the treatment of a TNF- mediated disease such as smoke-induced airway inflammation, inflammation enhanced cough, for the control of myogenesis, for treating mucin overproduction, and/or for treating mucus hypersecretion.
[0068] In another embodiment of the invention, the compounds of the invention are preferably administered by inhalation.
[0069] In one embodiment the obstructive or inflammatory airways disease is COPD. [0070] According to another embodiment of the present invention, the compounds of the invention can also be used as a combination with one or more additional therapeutic agents to be co- administered to a patient to obtain some particularly desired therapeutic end result such as the treatment of pathophysiological^- relevant disease processes including, but not limited to (i) bronchoconstriction, (ii) inflammation, (iii) allergy, (iv) tissue destruction, (v) signs and symptoms such as breathlessness, cough. The second and more additional therapeutic agents may also be a compound of the invention, or one or more P38 and/or TNF inhibitors known in the art. More typically, the second and more therapeutic agents will be selected from a different class of therapeutic agents. [0071] As used herein, the terms "co- administration", "co-administered" and "in combination with", referring to the compounds of the invention and one or more other therapeutic agents, is intended to mean, and does refer to and include the following:
(a) simultaneous administration of such combination of compound(s) of the invention) and therapeutic agent(s) to a patient in need of treatment, when such components are formulated together into a single dosage form which releases said components at substantially the same time to said patient,
(b) substantially simultaneous administration of such combination of compound(s) of the invention and therapeutic agent(s) to a patient in need of treatment, when such components are formulated apart from each other into separate dosage forms which are taken at substantially the same time by said patient, whereupon said components are released at substantially the same time to said patient,
(c) sequential administration of such combination compound(s) of the invention and therapeutic agent(s) to a patient in need of treatment, when such components are
formulated apart from each other into separate dosage forms which are taken at consecutive times by said patient with a significant time interval between each administration, whereupon said components are released at substantially different times to said patient; and
(d) sequential administration of such combination of compound(s) of the invention and therapeutic agent(s) to a patient in need of treatment, when such components are formulated together into a single dosage form which releases said components in a controlled manner whereupon they are concurrently, consecutively, and/or overlappingly administered at the same and/or different times by said patient, where each part may be administered by either the same or different route.
[0072] Suitable examples of other therapeutic agents which may be used in combination with the compound(s) of the invention, or pharmaceutically acceptable salts, solvates or compositions thereof, include, but are by no means limited to:
(a) 5-Lipoxygenase (5-LO) inhibitors or 5-liρoxygenase activating protein (FLAP) antagonists,
(b) Leukotriene antagonists (LTRAs) including antagonists of LTB4, LTC4, LTD4, and LTE4,
(c) Histamine receptor antagonists including Hl and H3 antagonists,
(d) (X1- and α2-adrenoceptor agonist vasoconstrictor sympathomimetic agents for decongestant use,
(θ) muscarinic M3 receptor antagonists or anticholinergic agents,
(f) PDE inhibitors, e.g. PDE3, PDE4 and PDE5 inhibitors,
(g) Theophylline,
(h) Sodium cromoglycate,
(i) COX inhibitors both non-selective and selective COX-I or COX-2 inhibitors
(NSAIDs), (j) Oral and inhaled glucocorticosteroids, such as DAGR (dissociated agonists of the corticoid receptor)
(k) Monoclonal antibodies active against endogenous inflammatory entities, (I) β2 agonists
(m)Adhesion molecule inhibitors including VLA-4 antagonists, (n) Kinin-B] - and B2 -receptor antagonists,
(o) Immunosuppressive agents,
(p) Inhibitors of matrix metalloproteases (MMPs),
(q) Tachykinin NKj, NK2 and NK3 receptor antagonists,
(r) Elastase inhibitors,
(s) Adenosine A2a receptor agonists,
(t) Inhibitors of urokinase,
(u) Compounds that act on dopamine receptors, e.g. D2 agonists,
(v) Modulators of the NFiφ pathway, e.g. IKK inhibitors,
(w) modulators of cytokine signalling pathways such as syk kinase, or JAK kinase inhibitors,
(x) Agents that can be classed as mucolytics or anti-tussive, (y) Antibiotics,
(z) HDAC (histone deacetylase) inhibitors, and (aa) PI3 kinase inhibitors.
[0073] According to one embodiment of the present invention, combination of the compounds of the invention with:
- H3 antagonists,
- Muscarinic M3 receptor antagonists,
- PDE4 inhibitors,
- glucocorticosteroids,
- Adenosine A2a receptor agonists,
- β2 agonists
- Modulators of cytokine signalling pathyways such as syk kinase, or,
- Leukotriene antagonists (LTRAs) including antagonists of LTB4, LTC4, LTD4, and LTE4, can be used.
[0074] According to one embodiment of the present invention, combination of the compounds of the invention with:
-glucocorticosteroids, in particular inhaled glucocorticosteroids with reduced systemic side effects, including prednisone, prednisolone, flunisolide, triamcinolone acetonide, beclomethasone dipropionate, budesonide, fluticasone propionate, ciclesonide, and mometasone furoate,
-muscarinic M3 receptor antagonists or anticholinergic agents including in particular ipratropium salts, namely bromide, tiotropium salts, namely bromide, oxitropium salts, namely bromide, perenzepine, and telenzepine, -or β2 agonists can be used.
[0075] A wide variety of methods may be used alone or in combination to administer the compounds described above. For example, the compounds may be administered orally, intravascularly (IV), intraperitoneally, subcutaneously, intramuscularly (IM), by inhalation spray, rectally, or topically.
[0076] Typically, a compound described in this specification is administered in an amount effective to inhibit p38 kinase (particularly p38α kinase), TNF (particularly TNF-α), and/or cyclooxygenase (particularly cyclooxygenase-2). The preferred total daily dose of the compound (administered in single or divided doses) is typically from about 0. 01 to about 100 mg/kg, more preferably from about 0.1 to about 50 mg/kg, and even more preferably from about 0.5 to about 30 mg/kg {i.e., mg compound per kg body weight). Dosage unit compositions may contain such amounts or submultiples thereof to make up the daily dose. In many instances, the administration of the compound will be repeated a plurality of times in a day (typically no greater than 4 times). Multiple doses per day typically may be used to increase the total daily dose, if desired. [0077] Factors affecting the preferred dosage regimen include the type, age, weight, sex, diet, and condition of the patient; the severity of the pathological condition; the route of administration; pharmacological considerations, such as the activity, efficacy, pharmacokinetic, and toxicology profiles of the particular compound employed; whether a drug delivery system is utilized; and whether the compound is administered as part of a drug combination. Thus, the dosage regimen actually employed can vary widely, and, therefore, can deviate from the preferred dosage regimen set forth above. [0078] The present compounds may be used in co-therapies, partially or completely, in place of other conventional anti-inflammatory, such as together with steroids, cyclooxygenase-2 inhibitors, non-steroidal anti-inflammatory drugs ("NSAIDs"), disease-modifying anti-rheumatic drugs ("DMARDs"), immunosuppressive agents, 5- lipoxygenase inhibitors, leukotriene B4 ("LTB4") antagonists, and leukotriene A4 ("LTA4") hydrolase inhibitors.
Pharmaceutical Compositions Containing the Compounds of this Invention [0079] This invention also is directed to pharmaceutical compositions (or "medicaments") comprising the compounds described above (including tautomers of the compounds, and pharmaceutically-acceptable salts of the compounds and tautomers), and to methods for making pharmaceutical compositions comprising those compounds in combination with one or more conventional non-toxic, pharmaceutically-acceptable carriers, diluents, wetting or suspending agents, vehicles, and/or adjuvants (the carriers, diluents, wetting or suspending agents, vehicles, and adjuvants sometimes being collectively referred to in this specification as "carrier materials"); and/or other active ingredients. The preferred composition depends on the method of administration. Formulation of drugs is generally discussed in, for example, Hoover, John E., Remington's Pharmaceutical Sciences (Mack Publishing Co., Easton, PA: 1975) (incorporated by reference into this specification). See also, Liberman, H.A., Lachman, L., eds., Pharmaceutical Dosage Forms (Marcel Decker, New York, N. Y., 1980) (incorporated by reference into this specification). In many preferred embodiments, the pharmaceutical composition is made in the form of a dosage unit containing a particular amount of the active ingredient. Typically, the pharmaceutical composition contains from about 0.1 to 1000 mg (and more typically, 7.0 to 350 mg) of the compound. [0080] Solid dosage forms for oral administration include, for example, hard or soft capsules, tablets, pills, powders, and granules. In such solid dosage forms, the compounds are ordinarily combined with one or more adjuvants. If administered per os, the compounds may be mixed with lactose, sucrose, starch powder, cellulose esters of alkanoic acids, cellulose alkyl esters, talc, stearic acid, magnesium stearate, magnesium oxide, sodium and calcium salts of phosphoric and sulfuric acids, gelatin, acacia gum, sodium alginate, polyvinylpyrrolidone, and/or polyvinyl alcohol, and then tableted or encapsulated for convenient administration. Such capsules or tablets may contain a controlled-release formulation, as may be provided in a dispersion of the compound of this invention in hydroxypropylmethyl cellulose, hi the case of capsules, tablets, and pills, the dosage forms also may comprise buffering agents, such as sodium citrate, or magnesium or calcium carbonate or bicarbonate. Tablets and pills additionally may be prepared with enteric coatings. ._ ._
[0081] Liquid dosage forms for oral administration include, for example, pharmaceutically acceptable emulsions, solutions, suspensions, syrups, and elixirs containing inert diluents commonly used in the art (e.g., water). Such compositions also may comprise adjuvants, such as wetting, emulsifying, suspending, flavoring (e.g., sweetening), and/or perfuming agents.
[0082] "Parenteral administration" includes subcutaneous injections, intravenous injections, intramuscular injections, intrasternal injections, and infusion. Injectable preparations (e.g., sterile injectable aqueous or oleaginous suspensions) may be formulated according to the known art using suitable dispersing, wetting agents, and/or suspending agents. Acceptable carrier materials include, for example, water, 1,3-butanediol, Ringer's solution, isotonic sodium chloride solution, bland fixed oils (e.g., synthetic mono- or diglycerides), dextrose, mannitol, fatty acids (e.g., oleic acid), dimethyl acetamide, surfactants (e.g., ionic and non-ionic detergents), and/or polyethylene glycols (e.g., PEG 400).
[0083] Formulations for parenteral administration may, for example, be prepared from sterile powders or granules having one or more of the carriers materials mentioned for use in the formulations for oral administration. The compounds may be dissolved in water, polyethylene glycol, propylene glycol, ethanol, com oil, cottonseed oil, peanut oil, sesame oil, benzyl alcohol, sodium chloride, and/or various buffers. The pH may be adjusted, if necessary, with a suitable acid, base, or buffer.
[0084] The compounds of this invention preferably make up from about 0.075 to about 30% (w/w) (more preferably 0.2 to 20% (w/w), and even more preferably 0.4 to 15% (w/w)) of a pharmaceutical composition used for topical or rectal administration. [0085] The compounds of the invention can also be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an aerosol spray from a pressurised container, pump, spray, atomiser (preferably an atomiser using electrohydrodynamics to produce a fine mist), or nebuliser, with or without the use of a suitable propellant, such as 1,1,1,2-tetrafluoroethane or 1,1,1,2,3,3,3-heptafluoropropane. For intranasal use, the powder may comprise a bioadhesive agent, for example, chitosan or cyclodextrin.
[0086] The pressurised container, pump, spray, atomizer, or nebuliser contains a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
[0087] Prior to use in a dry powder or suspension formulation, the drug product is micronised to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenisation, or spray drying.
[0088] Capsules (made, for example, from gelatin or hydroxypropylmethylcellulose), blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the compound of the invention, a suitable powder base such as lactose or starch and a performance modifier such as 1-leucine, mannitol, or magnesium stearate. The lactose may be anhydrous or in the form of the monohydrate, preferably the latter. Other suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
[0089] A suitable solution formulation for use in an atomiser using electrohydrodynamics to produce a fine mist may contain from lμg to 20mg of the compound of the invention per actuation and the actuation volume may vary from lμl to lOOμl. A typical formulation may comprise a compound of the invention, propylene glycol, sterile water, ethanol and sodium chloride. Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol. [0090] Suitable flavours, such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations of the invention intended for inhaled/intranasal administration.
[0091] Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, PGLA. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
_-[0092] In the case of dry powder inhalers and aerosols, the dosage unit is determined by means of a valve which delivers a metered amount. Units in accordance with the
invention are typically arranged to administer a metered dose or "puff containing from O.OOlmg to lOmg of the compound of the invention. The overall daily dose will typically be in the range O.OOlmg to 40mg which may be administered in a single dose or, more usually, as divided doses throughout the day.
[0093] Suppositories for rectal administration may be prepared by, for example, mixing a compound of this invention with a suitable nonirritating excipient that is solid at ordinary temperatures, but liquid at the rectal temperature and will therefore melt in the rectum to release the drug. Suitable excipients include, for example, such as cocoa butter; synthetic mono-, di-, or triglycerides; fatty acids; and/or polyethylene glycols. [0094] "Topical administration" includes transdermal administration, such as via transdermal patches or iontophoresis devices. Compositions for topical administration also include, for example, topical gels, sprays, ointments, and creams. [0095] When formulated in an ointment, the compounds of this invention may be employed with, for example, either a paraffinic or a water-miscible ointment base. When formulated in a cream, the active ingredient(s) may be formulated with, for example, an oil-in- water cream base. If desired, the aqueous phase of the cream base may include, for example at least about 30% (w/w) of a polyhydric alcohol, such as propylene glycol, butane- 1,3-diol, mannitol, sorbitol, glycerol, polyethylene glycol, and mixtures thereof. [0096] A topical formulation may include a compound which enhances absorption or penetration of the active ingredient through the skin or other affected areas. Examples of such dermal penetration enhancers include dimethylsulfoxide and related analogs. [0097] When the compounds of this invention are administered by a transdermal device, administration will be accomplished using a patch either of the reservoir and porous membrane type or of a solid matrix variety. In either case, the active agent is delivered continuously from the reservoir or microcapsules through a membrane into the active agent permeable adhesive, which is in contact with the skin or mucosa of the recipient. If the active agent is absorbed through the skin, a controlled and predetermined flow of the active agent is administered to the recipient. In the case of microcapsules, the encapsulating agent may also function as the membrane. The transdermal patch may include the compound in a suitable solvent system with an adhesive system, such as an acrylic emulsion, and a polyesterpatch. The oily phase of the emulsions of this invention may be constituted from known ingredients in a known
manner. While the phase may comprise merely an emulsifier, it may comprise, for example, a mixture of at least one emulsifier with a fat or an oil or with both a fat and an oil. Preferably, a hydrophilic emulsifier is included together with a lipophilic emulsifier which acts as a stabilizer. It is also preferable to include both an oil and a fat. Together, the emulsifier(s) with or without stabilizer(s) make-up the so-called emulsifying wax, and the wax together with the oil and fat make up the so-called emulsifying ointment base which forms the oily dispersed phase of the cream formulations. Emulsifiers and emulsion stabilizers suitable for use in the formulation of the present invention include Tween 60, Span 80, cetostearyl alcohol, myristyl alcohol, glyceryl monostearate, and sodium lauryl sulfate, among others. The choice of suitable oils or fats for the formulation is based on achieving the desired cosmetic properties, given that the solubility of the active compound in most oils likely to be used in pharmaceutical emulsion formulations is very low. Thus, the cream should preferably be a non-greasy, non-staining and washable product with suitable consistency to avoid leakage from tubes or other containers. Straight or branched chain, mono- or dibasic alkyl esters such as di- isoadipate, isocetyl stearate, propylene glycol diester of coconut fatty acids, isopropyl myristate, decyl oleate, isopropyl palmitate, butyl stearate, 2-ethylhexyl palmitate or a blend of branched chain esters, for example, may be used. These may be used alone or in combination depending on the properties required. Alternatively, high melting point lipids such as white soft paraffin and/or liquid paraffin or other mineral oils may be used. Formulations suitable for topical administration to the eye also include eye drops wherein the compound of this invention is dissolved or suspended in suitable carrier, typically comprising an aqueous solvent. The compounds of this invention are preferably present in such formulations in a concentration of from about 0.5 to about 20% (w/w) (more preferably 0.5 to 10% (w/w), and often even more preferably about 1.5% (w/w)). [0098] Other carrier materials and modes of administration known in the pharmaceutical art may also be used.
Definitions
[0099] The term "alkyl" (alone or in combination with another term(s)) means a _.straight-or branched-.chain saturated hydrocarbyl substituent (i.e., a substituent containing only carbon and hydrogen) typically containing from 1 to about 20 carbon
atoms, more typically from 1 to about 12 carbon atoms, even more typically from 1 to about 8 carbon atoms, and still even more typically from 1 to about 6 carbon atoms. Examples of such substituents include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, pentyl, iso-amyl, hexyl, and octyl. [00100] The term "alkenyl" (alone or in combination with another term(s)) means a straight- or branched-chain hydrocarbyl substituent containing one or more double bonds and typically from 2 to about 20 carbon atoms, more typically from 2 to about 12 carbon atoms, even more typically from 2 to about 8 carbon atoms, and still even more typically from 2 to about 6 carbon atoms. Examples of such substituents include ethenyl (vinyl); 2-propenyl; 3-propenyl; 1,4-pentadienyl; 1,4-butadienyl; 1-butenyl; 2-butenyl; 3-butenyl; and decenyl.
[0100] The term "alkynyl" (alone or in combination with another term(s)) means a straight- or branched-chain hydrocarbyl substituent containing one or more triple bonds and typically from 2 to about 20 carbon atoms, more typically from 2 to about 12 carbon atoms, even more typically from 2 to about 8 carbon atoms, and still even more typically from 2 to about 6 carbon atoms. Examples of such substituents include ethynyl, 1-propynyl, 2-propynyl, decynyl, 1-butynyl, 2-butynyl, 3-butynyl, and 1-pentynyl. [0101] The term "cycloalkyl" (alone or in combination with another term(s)) means a saturated carbocyclyl substituent containing from 3 to about 14 carbon ring atoms, more typically from 3 to about 12 carbon ring atoms, and even more typically from 3 to about 8 carbon ring atoms. A cycloalkyl may be a single carbon ring, which typically contains from 3 to 6 carbon ring atoms. Examples of single-ring cycloalkyls include cyclopropyl (or "cyclopropanyl"), cyclobutyl (or "cyclobutanyl"), cyclopentyl (or "cyclopentanyl"), and cyclohexyl (or "cyclohexanyl"). A cycloalkyl alternatively may be 2 or 3 carbon rings fused together, such as, for example, decalinyl or norpinanyl. [0102] The term "cycloalkylalkyl" (alone or in combination with another term(s)) means alkyl substituted with cycloalkyl. Examples of such substituents include cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, and cyclohexylmethyl. [0103] The term "aryl" (alone or in combination with another term(s)) means an aromatic carbocyclyl containing from 6 to 14 carbon ring atoms. Examples of aryls include phenyl, naphthalenyl, and indenyl.
[0104] In some instances, the number of carbon atoms in a hydrocarbyl substituent
(e.g., alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, aryl, etc.) is indicated by the prefix "Cx-Cy", wherein x is the minimum and y is the maximum number of carbon atoms in the substituent. Thus, for example, "Q-Cδ-alkyl" refers to an alkyl substituent containing from 1 to 6 carbon atoms. Illustrating further, Cs-Cβ-cycloalkyl means a saturated carbocyclyl containing from 3 to 6 carbon ring atoms.
[0105] The term "arylalkyl" (alone or in combination with another term(s)) means alkyl substituted with aryl.
[0106] The term "benzyl" (alone or in combination with another term(s)) means a methyl radical substituted with phenyl, i.e., the following structure:
[0107] The term "benzene" means the following structure:
[0108] The term "hydrogen" (alone or in combination with another term(s)) means a hydrogen radical, and may be depicted as -H.
[0109] The term "hydroxy" or "hydroxyl" (alone or in combination with another term(s)) means -OH.
[0110] The term "hydroxyalkyl" (alone or in combination with another term(s)) means alkyl substituted with one more hydroxy.
[0111] The term "nitro" (alone or in combination with another term(s)) means -NO2.
[0112] The term "cyano" (alone or in combination with another term(s)) means -CN, which also may be depicted:
[0113] The term "keto" (alone or in combination with another term(s)) means an oxo radical, and may be depicted as =0.
[0114] The term "carboxy" or "carboxyl" (alone or in combination with another term(s)) means -C(O)-OH, which also may be depicted as:
[0115] The term "amino" (alone or in combination with another term(s)) means -NH2. The term "monosubstituted amino" (alone or in combination with another term(s)) means an amino substituent wherein one of the hydrogen radicals is replaced by a non-hydrogen substituent. The term "disubstituted amino" (alone or in combination with another term(s)) means an amino substituent wherein both of the hydrogen atoms are replaced by non-hydrogen substituents, which may be identical or different. [0116] The term "halogen" (alone or in combination with another term(s)) means a fluorine radical (which may be depicted as -F), chlorine radical (which may be depicted as -Cl), bromine radical (which may be depicted as -Br), or iodine radical (which may be depicted as -I). Typically, a fluorine radical or chlorine radical is preferred, with a fluorine radical often being particularly preferred.
[0117] The prefix "halo" indicates that the substituent to which the prefix is attached is substituted with one or more independently selected halogen radicals. For example, haloalkyl means an alkyl substituent wherein at least one hydrogen radical is replaced with a halogen radical. Where there are more than one hydrogens replaced with halogens, the halogens may be the identical or different. Examples of haloalkyls include chloromethyl, dichloromethyl, difluorochloromethyl, dichlorofluoromethyl, trichloromethyl, 1-bromoethyl, fluoromethyl, difluoromethyl, trifluoromethyl, 1,1,1-trifluoroethyl, difluoroethyl, pentafluoroethyl, difluoropropyl, dichloropropyl, and heptafluoropropyl. Illustrating further, "haloalkoxy" means an alkoxy substituent wherein at least one hydrogen radical is replaced by a halogen radical. Examples of haloalkoxy substituents include chloromethoxy, 1-bromoethoxy, fluoromethoxy, difluoromethoxy, trifluoromethoxy (also known as "perfluoromethyloxy"), and 1,1,1,-trifluoroethoxy. It should be recognized that if a substituent is substituted by
more than one halogen radical, those halogen radicals may be identical or different (unless otherwise stated).
[0118] The prefix "perhalo" indicates that each hydrogen radical on the substituent to which the prefix is attached is replaced with an independently selected halogen radical. If all the halogen radicals are identical, the prefix may identify the halogen radical. Thus, for example, the term "perfluoro" means that every hydrogen radical on the substituent to which the prefix is attached is substituted with a fluorine radical. To illustrate, the term "perfluoroalkyl" means an alkyl substituent wherein a fluorine radical is in the place of each hydrogen radical. Examples of perfluoroalkyl substituents include trifluoromethyl (-CF3), perfluorobutyl, perfluoroisopropyl, perfluorododecyl, and perfluorodecyl. To illustrate further, the term "perfluoroalkoxy" means an alkoxy substituent wherein each hydrogen radical is replaced with a fluorine radical. Examples of perfluoroalkoxy substituents include trifluoromethoxy (-0-CF3), perfluorobutoxy, perfluoroisopropoxy, perfluorododecoxy, and perfluorodecoxy. [0119] The term "carbonyl" (alone or in combination with another term(s)) means -C(O)-, which also may be depicted as:
This term also is intended to encompass a hydrated carbonyl substituent, i.e., -C(OH)2-. [0120] The term "aminocarbonyl" (alone or in combination with another term(s)) means -C(O)-NH2, which also may be depicted as:
[0121] The term "oxy" (alone or in combination with another term(s)) means an ether substituent, and may be depicted as -O-.
[0122] The term "alkoxy" (alone or in combination with another term(s)) means an alkylether substituent, i.e., -O-alkyl. Examples of such a substituent include methoxy
(-0-CH3), ethoxy, n-propoxy, isopropoxy, n-butoxy, iso-butoxy, sec-butoxy, and tert-butoxy.
[0123] The term "alkylthio" (alone or in combination with another term(s)) means -S-alkyl. For example, "methylthio" is -S-CH3. Other examples of alkylthio substituents include ethylthio, propylthio, butylthio, and hexylthio.
[0124] The term "alkylcarbonyl" or "alkanoyl" (alone or in combination with another term(s)) means -C(O)-alkyl. For example, "ethylcarbonyl" may be depicted as:
Examples of other often preferred alkylcarbonyl substituents include methylcarbonyl, propylcarbonyl, butylcarbonyl, pentylcarbonyl, and hexylcarbonyl. [0125] The term "aminoalkylcarbonyl" (alone or in combination with another term(s)) means -C(O)-alkyl-NH2. For example, "aminomethylcarbonyl" may be depicted as:
[0126] The term "alkoxycarbonyl" (alone or in combination with another term(s)) means -C(O)-O-alkyl. For example, "ethoxycarbonyl" may be depicted as:
[0127] Examples of other often preferred alkoxycarbonyl substituents include methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, butoxycarbonyl, pentoxycarbonyl, and hexyloxycarbonyl. [0128] The term "carbocyclylcarbonyl" (alone or in combination with another term(s)) means -C(O)-carbocyclyl. For example, "phenylcarbonyl" may be depicted as:
Similarly, the term "heterocyclylcarbonyl" (alone or in combination with another term(s)) means -C(O)-heterocyclyl.
[0129] The term "carbocyclylalkylcarbonyl" (alone or in combination with another term(s)) means -C(O)-alkyl-carbocyclyl. For example, "phenylethylcarbonyl" may be depicted as:
Similarly, the term "heterocyclylalkylcarbonyl" (alone or in combination with another term(s)) means -C(O)-alkyl-heterocyclyl.
[0130] The term "carbocyclyloxycarbonyl" (alone or in combination with another term(s)) means -C(O)-O-carbocyclyl. For example, "phenyloxycarbonyl" may be depicted as:
[0131] The term "carbocyclylalkoxycarbonyl" (alone or in combination with another term(s)) means -C(O)-O-alkyl-carbocyclyl. For example, "phenylethoxycarbonyl" may be depicted as:
[0132] The term "thio" or "thia" (alone or in combination with another term(s)) means a thiaether substituent, i.e., an ether substituent wherein a divalent sulfur atom is in the place of the ether oxygen atom. Such a substituent may be depicted as -S-. This, for example, "alkyl-thio-alkyl" means alkyl-S-alkyl.
[0133] The term "thiol" (alone or in combination with another term(s)) means a sulfhydryl substituent, and may be depicted as -SH.
[0134] The term "sulfonyl" (alone or in combination with another term(s)) means
-S(O)
2-, which also may be depicted as:
Thus, for example, "alkyl-sulfonyl-alkyl" means alkyl-S(O)2-alkyl. Examples of typically preferred alkylsulfonyl substituents include methylsulfonyl, ethylsulfonyl, and propylsulfonyl.
[0135] The term "aminosulfonyl" (alone or in combination with another term(s)) means -S(O)2-NH2, which also may be depicted as:
[0136] The term "sulfinyl" or "sulfoxido" (alone or in combination with another term(s)) means -S(O)-, which also may be depicted as:
Thus, for example, "alkylsulfinylalkyl" or "alkylsulfoxidoalkyl" means alkyl-S(O)-alkyl. Typically preferred alkylsulfinyl groups include methylsulfinyl, ethylsulfinyl, butyl sulfinyl, and hexylsulfinyl.
[0137] The term "heterocyclyl" (alone or in combination with another term(s)) means a saturated (i.e., "heterocycloalkyl"), partially saturated (i.e., "heterocycloalkenyl"), or completely unsaturated (i.e., "heteroaryl") ring structure containing a total of 3 to 14 ring atoms. At least one of the ring atoms is a heteroatom (i.e., oxygen, nitrogen, or sulfur), with the remaining ring atoms being independently selected from the group consisting of carbon, oxygen, nitrogen, and sulfur. [0138] A heterocyclyl may be a single ring, which typically contains from 3 to 7 ring atoms, more typically from 3 to 6 ring atoms, and even more typically 5 to 6 ring atoms. Examples of single-ring heterocyclyls include furanyl, dihydrofurnayl, tetradydrofurnayl, thiophenyl (also known as "thiofuranyl"), dihydrothiophenyl, tetrahydrothiophenyl, pyrrolyl, isopyrrolyl, pyrrolinyl, pyrrolidinyl, imidazolyl, isoimidazolyl, imidazolinyl,
imidazolidinyl, pyrazolyl, pyrazolinyl, pyrazolidinyl, triazolyl, tetrazolyl, dithiolyl, oxathiolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, thiazolinyl, isothiazolinyl, thiazolidinyl, isothiazolidinyl, thiodiazolyl, oxathiazolyl, oxadiazolyl (including l;,2,3-oxadiazolyl, 1,2,4-oxadiazolyl (also known as "azoximyl"), 1,2,5-oxadiazolyl (also known as "furazanyl"), or 1,3,4-oxadiazolyl), oxatriazolyl (including 1,2,3,4-oxatriazolyl or 1,2,3,5-oxatriazolyl), dioxazolyl (including 1,2,3-dioxazolyl, 1,2,4-dioxazolyl, 1,3,2-dioxazolyl, or 1,3,4-dioxazolyl), oxathiazolyl, oxathiolyl, oxathiolanyl, pyranyl (including 1,2-pyranyl or 1,4-pyranyl), dihydropyranyl, pyridinyl (also known as "azinyl"), piperidinyl, diazinyl (including pyridazinyl (also known as "1,2-diazinyl"), pyrimidinyl (also known as "1,3 -diazinyl" or "pyrimidyl"), or pyrazinyl (also known as "1,4-diazinyl")), piperazinyl, triazinyl (including s-triazinyl (also known as "1,3,5-triazinyl"), as-triazinyl (also known 1,2,4-triazinyl), and v-triazinyl (also known as "1,2,3-triazinyl")), oxazinyl (including 1,2,3-oxazinyl, 1,3,2-oxazinyl, 1,3,6-oxazinyl (also known as "pentoxazolyl"), 1,2,6-oxazinyl, or 1,4-oxazinyl), isoxazinyl (including o-isoxazinyl or p-isoxazinyl), oxazolidinyl, isoxazolidinyl, oxathiazinyl (including 1,2,5-oxathiazinyl or 1,2,6-oxathiazinyl), oxadiazinyl (including 1,4,2-oxadiazinyl or 1,3,5,2-oxadiazinyl), morpholinyl, azepinyl, oxepinyl, thiepinyl, and diazepinyl. [0139] A heterocyclyl alternatively may be 2 or 3 rings fused together, wherein at least one such ring contains a heteroatom as a ring atom (i.e., nitrogen, oxygen, or sulfur). Such substituents include, for example, indolizinyl, pyrindinyl, pyranopyrrolyl, 4H-quinolizinyl, purinyl, naphthyridinyl, pyridopyridinyl (including pyrido[3,4-b]-pyridinyl, pyrido[3,2-b] -pyridinyl, or pyrido[4,3-b]-pyridinyl), and pteridinyl. Other examples of fused-ring heterocyclyls include benzo-fused heterocyclyls, such as indolyl, isoindolyl (also known as "isobenzazolyl" or "pseudoisoindolyl"), indoleninyl (also known as "pseudoindolyl"), isoindazolyl (also known as "benzpyrazolyl"), benzazinyl (including quinolinyl (also known as "1-benzazinyl") or isoquinolinyl (also known as "2-benzazinyl")), phthalazinyl, quinoxalinyl, quinazolinyl, benzodiazinyl (including cinnolinyl (also known as "1,2-benzodiazinyl") or quinazolinyl (also known as "1,3-benzodiazinyl")), benzopyranyl (including "chromanyl" or "isochromanyl"), benzothiopyranyl (also known as "thiochromanyl"), benzoxazolyl, indoxazinyl (also known as "benzisoxazolyl"), anthranilyl, benzodioxolyl, benzodioxanyl, benzoxadiazolyl,
benzofuranyl (also known as "coumaronyl"), isobenzofuranyl, benzothienyl (also known as "benzothiophenyl", "thionaphthenyl", or "benzothiofuranyl"), isobenzothienyl (also known as "isobenzothiophenyl", "isothionaphthenyl", or "isobenzothiofuranyl"), benzothiazolyl, benzothiadiazolyl, benzimidazolyl, benzotriazolyl, benzoxazinyl (including 1,3,2-benzoxazinyl , 1,4,2-benzoxazinyl , 2,3,1 -benzoxazinyl , or
3,1,4-benzoxazinyl ), benzisoxazinyl (including 1,2-benzisoxazinyl or 1,4-benzisoxazinyl), tetrahydroisoquinolinyl , carbazolyl, xanthenyl, and acridinyl. [0140] The term "2-fused'ring" heterocyclyl (alone or in combination with another term(s)) means a saturated, partially saturated, or aryl heterocyclyl containing 2 fused rings. Examples of 2-fused-ring heterocyclyls include indolizinyl, pyrindinyl, pyranopyrrolyl, 4H-quinolizinyl, purinyl, naphthyridinyl, pyridopyridinyl, pteridinyl, indolyl, isoindolyl, indoleninyl, isoindazolyl, benzazinyl, phthalazinyl, quinoxalinyl, quinazolinyl, benzodiazinyl, benzopyranyl, benzothiopyranyl, benzoxazolyl, indoxazinyl, anthranilyl, benzodioxolyl, benzodioxanyl, benzoxadiazolyl, benzofuranyl, isobenzofuranyl, benzothienyl, isobenzothienyl, benzothiazolyl, benzothiadiazolyl, benzimidazolyl, benzotriazolyl, benzoxazinyl, benzisoxazinyl, and tetrahydroisoquinolinyl.
[0141] The term "heteroaryl" (alone or in combination with another term(s)) means an aromatic heterocyclyl containing from 5 to 14 ring atoms. A heteroaryl may be a single ring or 2 or 3 fused rings. Examples of heteroaryl substituents include 6-membered ring substituents such as pyridyl, pyrazyl, pyrimidinyl, and pyridazinyl; 5-membered ring substituents such as 1,3,5-, 1,2,4- or 1,2,3-tiiazinyl, imidazyl, furanyl, thiophenyl, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, 1,2,3-, 1,2,4-, 1,2,5-, or 1,3,4-oxadiazolyl and isothiazolyl; 6/5-membered fused ring substituents such as benzothiofuranyl, isobenzothiofuranyl, benzisoxazolyl, benzoxazolyl, purinyl, and anthranilyl; and 6/6-membered fused rings such as 1,2-, 1,4-, 2,3- and 2, 1-benzopyronyl, quinolinyl, isoquinolinyl, cinnolinyl, quinazolinyl, and 1,4-benzoxazinyl. [0142] The term "heterocyclylalkyl" (alone or in combination with another term(s)) means alkyl substituted with a heterocyclyl.
[0143] The term "heterocycloalkyl" (alone or in combination with another term(s)) means a fully saturated heterocyclyl.
[0144] This specification uses the terms "substituent" and "radical" interchangeably.
[0145] A prefix attached to a multi-component substituent only applies to the first component. To illustrate, the term "alkylcycloalkyl" contains two components: alkyl and cycloalkyl. Thus, the C1-C6- prefix on C1 -C6- alkylcycloalkyl means that the alkyl component of the alkylcycloalkyl contains from 1 to 6 carbon atoms; the C1-C6- prefix does not describe the cycloalkyl component. To illustrate further, the prefix "halo" on haloalkoxyalkyl indicates that only the alkoxy component of the alkoxyalkyl substituent is substituted with one or more halogen radicals. If halogen substitution may alternatively or additionally occur on the alkyl component, the substituent would instead be described as "halogen-substituted alkoxyalkyl" rather than "haloalkoxyalkyl." And finally, if the halogen substitution may only occur on the alkyl component, the substituent would instead be described as "alkoxyhaloalkyl."
[0146] If substituents are described as being "independently selected" from a group, each substituent is selected independent of the other. Each substituent therefore may be identical to or different from the other substituent(s).
[0147] When words are used to describe a substituent, the rightmost-described component of the substituent is the component that has the free valence. To illustrate, benzene substituted with methoxyethyl has the following structure:
As can be seen, the ethyl is bound to the benzene, and the methoxy is the component of the substituent that is the component furthest from the benzene. As further illustration, benzene substituted with cyclohexanylthiobutoxy has the following structure:
[0148] When words are used to describe a linking element between two other elements of a depicted chemical structure, the rightmost-described component of the substituent is the component that is bound to the left element in the depicted structure. To illustrate, if the chemical structure is X-L-Y and L is described as methylcyclohexanyl ethyl, then the chemical would be X-ethyl-cyclohexanyl-methyl-Y.
[0149] When a chemical formula is used to describe a substituent, the dash on the left side of the formula indicates the portion of the substituent that has the free valence. To illustrate, benzene substituted with -C(O)-OH has the following structure:
[0150] When a chemical formula is used to describe a linking element between two other elements of a depicted chemical structure, the leftmost dash of the substituent indicates the portion of the substituent that is bound to the left element in the depicted structure. The rightmost dash, on the other hand, indicates the portion of the substituent that is bound to the right element in the depicted structure. To illustrate, if the depicted chemical structure is X-L-Y and L is described as -C(O)-N(H)-, then the chemical would be:
[0151] The term "pharmaceutically acceptable" is used adjectivally in this specification to mean that the modified noun is appropriate for use as a pharmaceutical product or as a part of a pharmaceutical product.
[0152] With reference to the use of the words "comprise" or "comprises" or "comprising" in this patent (including the claims), Applicants note that unless the context requires otherwise, those words are used on the basis and clear understanding that they are to be interpreted inclusively, rather than exclusively, and that Applicants intend each of those words to be so interpreted in construing this patent, including the claims below.
General Synthetic Procedures
[0153] Representative procedures for the preparation of compounds of the invention are outlined below in the Schemes. The starting materials can be purchased or prepared using methods known to those skilled in the art. Similarly, the preparation of the various intermediates can be achieved using methods known in the art. The starting materials may be varied and additional steps employed to produce compounds encompassed by the
invention, as demonstrated by the examples below. In addition, different solvents and reagents can typically be used to achieve the above transformations. Furthermore, in certain situations, it may be advantageous to alter the order in which the reactions are performed. Protection of reactive groups may also be necessary to achieve the above transformations. In general, the need for protecting groups, as well as the conditions necessary to attach and remove such groups, will be apparent to those skilled in the art of organic synthesis. When a protecting group is employed, deprotection will generally be required. Suitable protecting groups and methodology for protection and deprotection such as those described in Protecting Groups in Organic Synthesis by Greene and Wuts are known and appreciated in the art.
The following schemes are representative of the methods that can be used to prepare these compounds.
SCHEME 1
R2 = alkyl, cycloalkyl, substituted alkyl
[0154] Scheme 1 depicts the general manner by which the C-5 alkyl pyrmidinone scaffold was assembled. In these procedures a substituted amine is condensed with potassium thiocyanate in the presence of acid. The generated thiourea is then condensed with a substituted malonate derivative. The thiol group can then be alkylated with an alkyl halide and the hydroxyl group is alkylated using a substituted benzyl halide or
subjected to a Mitsunobu reaction with a substituted benzyl alcohol. The sulfide is then oxidized to the sulfone using standard reagents.
SCHEME 2
mCPBA or Oxone
O R1
[0155] Scheme 2 depicts the general manner by which the C-5 halo pyrmidinone scaffold was assembled. In these procedures a substituted amine is condensed with potassium thiocyanate in the presence of acid. The generated thiourea is then condensed with a substituted malonate derivative. The thiol group can then be alkylated with an alkyl halide and the hydroxyl group is alkylated using a substituted benzyl halide or subjected to a Mitsunobu reaction with a substituted benzyl alcohol. The C-5 halogen is
introduced using N-bromosuccinimide or ΛT-chlorosuccinimide. The sulfide is then oxidized to the sulfone using standard reagents.
' SCHEME 3
X = Cl, Br, alkyl
Yn = anything
R, R1 = H1 alkyl, cycloalkyl, independent of one another
Z = O1 N1 S
R2 = alkyl cycloalkyl, substituted alkyl
[0156] Scheme 3 depicts the manner that the pyrimidinone sulfone is further elaborated to provide a number of C-2 heteroatom substituted derivatives. The sulfone is reacted with a substituted amine, alcohol or thiol in the presence of a base.
SCHEME 4
X = Cl, Br, alkyl
Yn = anything
R1 R1 = H, alkyl, cycloalkyl, independent of one another
R2 = alkyl cycloalkyl, substituted alkyl
[0157] Scheme 4 depicts the manner that the pyimidinone sulfone is further elaborated to provide a number of C-2 alkyl substituted derivatives. The sulfone is condensed with a functionalized or non-functionalized organometallic reagent, which can then be further elaborated using standard conditions.
SCHEME 5
X = Cl, Br, alkyl
Yn = anything
R, Ri = H, alkyl, cycloalkyl, independent of one another
[0158] Scheme 5 depicts the manner that the pyrimidinone sulfone is further elaborated by reaction with potassium cyanide. The cyanide function can then be elaborated to a number of derivatives.
SCHEME 6
Yn = anything
R, R1 = H, alkyl, cycloalkyl, independent of one another
Z = aromatic
[0159] Scheme 6 depicts the manner that the pyrimidinone sulfide is further elaborated to provide C-2 aryl substituted derivatives. The sulfide is reacted with a substituted arylboronic acid in the presence of a copper and palladium catalyst.
Detailed Preparative Method
[0160] The detailed examples below illustrate preparation of compounds of this invention. Other compounds of this invention may be prepared using the methods illustrated in these examples, either alone or in combination with techniques generally known in the art. The following examples are merely illustrative, and not limiting to the remainder of this disclosure in any way. [0161] The following abbreviations are used: g - gram mg - milligram mmol - millimole 0C - degrees celcius M - molar ml - milliliter
NMR - nuclear magnetic resonance 1H - proton MHz - megahertz s - singlet dd - doublet of doublets d - doublet t - triplet q - quartet br - broad m - multiplet app - apparent J - coupling constant Hz - hertz
LC/MS - liquid chromatograph/mass spectrometer tr - time of retention min - minute nm - nanometers
ES-MS - electrospray mass spectrometer m/z - mass to charge ratio
ES-HRMS - electrospray high resolution mass spectrometer
calcd - calculated
N - normal
L - liter dq - doublet of quartets dt - doublet of triplets ddd - doublet of doublet of doublets it - room temperature h - hour ddt - doublet of doublet of triplets w/w - weight to weight psi - pounds per square inch
M+H - exact mass + 1
HPLC - high performance liquid chromatography
DCM - dichloromethane
TFA - trifluoroacetic acid
DMF - dimethylformamide
DBU - l,8-Diazabicylo[5.4.0]-undec-7-ene
NBS - N-Bromosuccinimide
NCS - N-Chlorosuccinimide
ES-HRMS - Electrospray high-resolution mass spectrometry t-BOC - tert-butyloxycarbonyl
DMAP - dimethylaminopyridine
DCM - dichloromethane
EtOAc - ethyl acetate
MCPBA - meta-Chloroperbenzoic acid
Example 1
Step 1: Preparation of 6-hvdroxy-3-isopropyl-2-(methylthio)pyrimidin-4(3H)-one.
[0162] To a mechanically stirred mixture of N-isopropyl thiourea (124.03 g, 1.05 mol) and diethyl malonate (168 g, 1.05 mol) was added in one portion, commercial (Aldrich) sodium methoxide (25% wt) in methanol (468 mL) at ambient temperature. The thick white suspension formed initially was heated to 68°C (gentle reflux) for 4.5 hrs. At this point, the reaction mixture turns clear (hazy). Cooling was initiated and upon attaining 50°C, methyl iodide (149 g, 1.05 mol) was added in a steady stream over 10 minutes while maintaining 50°C. An exotherm is noted and moderate cooling is applied to maintain this temperature, (methyl iodide is volatile, hence the reflux condenser has to remain operational at this point as well). After 30 minutes, the reaction is cooled to 35°C and quenched with 120 mL of glacial acetic acid, added rapidly with good stirring. The temperature increases to 42°C and a thick white suspension was formed. This prevents the sudden precipitation of solids, allowing smooth stirring leading to the formation of a uniform and rapidly filterable crystalline mass. After a further 30 minutes of stirring to allow cooling to ambient temperature, (water bath) the contents were filtered and washed four times with 500 mL of water. Filtrations were rapid and the white semi-crystalline solid obtained was dried to constant weight at 50
0C and 20mm vacuum. The weight was 190 g (90%) (purity >98%)
1H NMR (400 MHz, DMSO) δ 11.2 (br s, IH), 5.0 (s, IH) 4.4 (m, IH), 2.42 (s, 3H), 1.42 (d, J = 6.6 Hz, 6H).
Step 2: Preparation of 6-(2,4-difluorobenzyloxy)-3-isopropyl-2-(methvthio)pyrimidin- 4(3H)-one.
[0163] A suspension of 6-hydroxy-3-isopropyl-2-(methylthio)pyrimidin-4(3H)-one_(190 g, 0.95 mol) from Step 1 and 325 mesh potassium carbonate (16O g, 1.15 mol) in 600 mL of N-methyl pyrrolidinone was warmed to 500C. An additional 100 rnL of solvent was added to ease the stirring and prevent lump formation or caking of the salt that is formed. 2,4-difluorobenzyl bromide (197 g, 0.96 mol) is added without dilution over 20 minutes, to allow an exotherm of 1O0C (final temp is 600C). The suspension now becomes thinner and easier to stir in about 10 minutes, indicating significant completion. The temperature was lowered to 5O0C and held for a further 3 hours. The reaction was tested for completion by withdrawing a small aliquot. The mixture was cooled to ambient temperature and poured into a large flask containing 3 L of water with good stirring. The precipitated solids were filtered and washed three times with 600 mL of water and dried at 500C and 20mm vacuum. The NMR purity at this stage was about 75-80% and weighed about 300 g. The crude product was dissolved in 2 L of ethyl acetate and concentrated to 750 mL (distillation of 1250 mL) on a large rotary evaporator at 35-400C at 100 mm vacuum. The heat and vacuum was turned off as the crystallization had begun but the rotation was continued and the bath was cooled slowly over 30 min to a final temperature of 15°C. The suspension was filtered (occurs rapidly) and washed twice with 200 mL of Hexanes. The fine white crystals of dried at 5O0C and 20 mm to obtain 160 g (52%). Additional quantities of product are obtainable from the mother liquors by crystallization or chromatography. 1H NMR (400 MHz, DMSO) δ 7.55 (m, IH), 7.27
(m, IH), 7.11 (m, IH), 5.37 (s, IH), 5..21 (s, 2H), 4.47-4.38 (m, IH), 2.48 (s, 3H), 1.47 (d, J = 6.7 Hz, 6H).
Step 3: Preparation of 6-(2,4-difluorobenzyloxy)-5-bromo-3-isopropyl-2- (methylthio)pyrimidin-4(3H)-one.
[0164] Substrate 6-(2,4-difluorobenzyloxy)-3-isopropyl-2-(methythio)pyrimidin-4(3H)- one (137 g, 0.42 mol) was dissolved in methylene chloride and N-bromosuccinimide (178 g, 0.42 mol) was added in two equal portions with good stirring at ambient temperature. A moderate rise in temperature (3 to 50C) was noted. After 2 hours of additional stirring, the solvent was distilled to near dryness on a rotary evaporator. 750 mL of water was subsequently added and stirred well for 10 minutes. The solids were filtered (occurs rapidly) and washed thrice with 200 mL of water. The off-white solid was dried at 500C and 20 mm vacuum. The yield of product was 170 g (100%). 1H NMR (400 MHz, DMSO) 67.55 (m, IH), 7.27 (m, IH), 7.15 (m, IH), 5.47 (s, 2H), 4.51-4.40 (m, IH), 2.58 (s, 3H), 1.52 (d, J = 6.7 Hz, 6H).
Step 4: Preparation of 6-(2,4-difluorobenzyloxy)-5-bromo-3-isopropyl-2- (methylsulfonyl)pyrimidin-4(3H)-one.
[0165] Oxone (600 g, 0.98 mol) was added to a stirred solution of 6-(2,4- difluorobenzyloxy)-5-bromo-3-isopropyl-2-(methylthio)pyrimidin-4(3H)-one (170 g, 0.42 mol) in 3 L of THF and 300 mL of water held at ambient temperature. The exotherm was minimal (2-30C) and the reaction remained heterogeneous, as the oxone had a low solubility in aqueous THF. The mixture was stirred for an additional 5 days at ambient temperature, (heating to speed up the reaction is not recommended as decomposition is observed). The reaction mixture was filtered and the residue washed twice with 500 mL of ethyl acetate. The filtrates were concentrated to approx 1/3 of the original volume (2800 mL of distillate) followed by the addition of 1 L of ethyl acetate. After washing thrice with 200 mL of water, the organic layer was dried over sodium sulfate and concentrated on a rotary evaporator to a semi-pasty solid. 500 mL of ether was then added and stirred well. A crystalline solid separates out rapidly, which was subsequently filtered and washed once with 100 mL of cold (100C) ether and once with 100 mL of hexane. The product was obtained as a colorless solid was dried in a vacuum oven at 500C and 20 mm to constant weight of 100 g (51%). 1H NMR (400 MHz, CDCl3) δ 7.40 (m, IH), 6.92 (m, IH), 6.82 (m, IH), 5.41 (s, 2H), 4.21-4.28 (m, IH), 3.38 (s, 3H), 1.60 (d, J = 6.7 Hz, 6H).
Example 2
CF3COOH
5-Bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-piperazin-l-ylpyrimidin-4(3H)- one trifluoroacetate
Step 1: Preparation of t-Butyl 4-f 5-bromo-4-[(2,4-difluorobenzyl)oxy1-l-isoDropyl-6- oxo- 1 ,6-dihvdropyrimidin-2-yl 1 piperazine- 1-carbox ylate.
[0166] A mixture of t-butyl-1 -piperazine carboxylate (1.0 g, 0.0054 mol), 5-Bromo-6- [(2,4-difluorobenzyl)oxy]-3-isopropyl-2-(methylsulfonyl)pyrimidin-4(3H)-one (1.8 g, 0.004 mol) obtained from step 3, diisopropylethyl amine (0.79g, 0.006 mol) and dimethylaminopyridine (0.15 g, 0.0012 mol) in dioxane (20.0 mL) was heated at 70 °C for 16 h under argon. After removal of the solvents in vacuo, the residue was partitioned cold 5% citric acid (10 mL) and dichloromethane (60.0 mL). The organic extract was washed with water, dried (Na2SO4), and concentrated to dryness. The resulting material was purified by reverse-phase HPLC using 10-90% CH3CN/Water gradient (40 min) containing 0.5% trifluoroacetic acid at a flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 543) were combined and concentrated to a small volume (~25 mL), added 5% sodium bicarbonate (5 mL) and extracted with dichloromethane (2 x 20 mL) dried (Na2SO4), and concentrated to dryness under reduced pressure. The combined organic extracts were washed with water, dried (Na2SO4), and concentrated to dryness in vacuo to afford the title compound as an amorphous powder (0.85 g, 36%): 1H NMR (CD3OD/ 400 MHz) δ 7.51 (m, IH), 6.98 (m, 2H), 5.44 (s, 2H), 4.61 (m, IH), 3.55 (br, 4H), 3.24 (m, 4H), 1.57 (d, 6H, 7 = 6.8 Hz, and 1.46 (s, 9H); ES-HRMS m/z 543.1441 (M+H calcd for C23H30N4O4F2Br requires 543.1413).
Step 2: Preparation of the title compound.
[0167] A solution of f-butyl 4-{ 5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- 1,6 dihydropyrimidin-2-yl}piperazine-l-carboxylate (0.26 g, 4.6 mmol) obtained from step 4 in a mixture of trifluoroacetic acid (0.3 mL) and dichloromethane (0.3 mL) was stirred at room temperature for 30 min and the product was isolated by reverse-phase HPLC using 10-90% CH3CN/Water gradient (40 min) containing 0.5% trifluoroacetic acid at a flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 443) were combined and lyophilized to afford the title compound as a white powder (0.18g, 67%): 1H NMR (CD3OD/ 400 MHz) 67.50 (m, IH), 6.99 (m, 2H), 5.46 (s, 2H), 4.61(m, IH), 3.49 (m, 4H), 3.39 (m, 4H), 1.58 (d, 6H, J = 6.8 Hz); ES-HRMS m/z 443.0859 (M+H calcd for C18H22N4O4F2Br requires 443.0889).
Example 3
5-Bromo-6-[(2,4-difluorobenzyl)oxy]-2-(4-glycylpiperazin-l-yl)-3- isopropylpyrimidin-4(3H)-one trifluoroacetate
[0168] To a solution of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-piperazin-l- ylpyrimidin-4(3H)-one trifluoroacetate (0.055 g, 0.1 mmol)in DMF (0.25 mL), N- methylmorpholine (0.015 g, 0.15mmol) and BOC-glycine-N-hydroxysuccimide (0.04 g, 0.15 mmol) were added and stirred at room temperature for 2 h. The reaction mixture was concentrated in vacuo and the residue was stirred with trifluoroacetic acid (0.125 mL) and dichloromethane (0,125 mL) at room temperature for 15 min. The solution was diluted with acetonitrile (3.0 mL) and the product was isolated by reverse-phase HPLC using 10-90% CH3CN/Water gradient (40 min) containing 0.5% trifluoroacetic acid at a
flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 500) were combined and lyophilized to afford the title compound as a white powder (0.04g): 1H NMR (CD3OD/ 400 MHz) δ 7.45 (m, IH), 6.96 (m, 2H), 5.45 (s, 2H), 4.64(m, IH), 3.97 (s, 2H), 3.68 (br, 2H), 3.58 (m, 2H), 3.33 (m, 4H), 1.59 (d, 6H, J = 6.8 Hz); ES-HRMS m/z 500.1112 (M+H calcd for C20H25N5O3F2Br requires 500.1103).
Example 4
5-Bromo-6-[(2,4-difluorobenzyl)oxy]-2-(4-glycoIoyIpiperazin-l-yl)-3- isopropylpyrimidin-4(3H)-one
[0169] To a solution of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-piperazin-l- ylpyrimidin-4(3H)-one trifluoroacetate (0.1 g, 0.18 mmol) in dichloromethane (3.0 mL) at 0 °C, N-methylmorpholine (0.037 g, 0.36 mmol) and acetoxyacetyl chloride (0.05 g, 0.37 mmol) were added and stirred at 0 0C for 30 min. The reaction mixture was concentrated in vacuo and the product was isolated by reverse-phase HPLC using 10- 90% CH3CN/Water gradient (40 min) containing 0.5% trifluoroacetic acid at a flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 500) were combined and lyophilized to afford 0.065 g (MH+ m/z = 543) as a white powder. This was stirred with a mixture of 1.5 N NaOH (0.3 mL) and dioxane (0.3 mL) at room temperature for 1 h This mixture was diluted with water (5.0 mL) acidified with trifluoroacetic acid and the product was purified by reverse-phase HPLC as described above to afford the title compound (0.025 g) as a white powder: 1H NMR (CD3OD/ 400 MHz) δ 7.51 (m, IH), 6.97 (m, 2H), 5.47 (s, 2H), 4.63(m, IH), 4.25 (s, 2H), 3.67 (br, 2H), 3.59 (br, 2H), 3.33
(m, 4H), 1.59 (d, 6H, J = 6.8 Hz); ES-HR MS m/z 501.0954 (M+H calcd for C20H24N4O4F2Br requires 501.0943).
j Example 5
5-Bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-[4-(2-methylaIanyl)piperazin-l- yl]pyrimidin-4(3H)-one trifluoroacetate
[0170] A mixture of di-t-butyl dicarbonate (0.2 g, 0.8 mmol), N-(tert-butoxycarbonyl)-2- methylalanine (0.15 g, 0.74 mmol) 3-hydroxy-3,4-dihydrobenzotriazine-4-one (0.12 g), and triethylamine (0.1 mL) in acetonitrile was stirred at room temperature for 1 h. A solution of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-piperazin-l-ylpyrimidin- 4(3H)-one (0.22 g, 0.5 mmol) in DMF (2.0 mL) was then added and the resulting mixture was stirred at room temperature for 3 h under argon. After the removal of the solvents in vacuo, the residue was partitioned between 5% citric acid (5 mL) and dichloromethane (20 mL). The organic phase was washed with dried (Na2SO4), and concentrated to dryness. The resulting material was stirred with a mixture of trifluoroacetic acid (0.5 mL) and dichloromethane (0.5 mL) for 30 min and the product was purified by purified by reverse-phase HPLC using 10-90% CH3CN/Water gradient (40 min) containing 0.5% trifluoroacetic acid at a flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 500) were combined and lyophilized to afford the title compound (0.155g) as a white powder: 1H NMR (CD3OD/ 400 MHz) δ 7.45 (m, IH), 6.95 (m, 2H), 5.43 (s, 2H), 4.62 (m, IH), 3.79 (br, 4H), 3.30 (m, 4H), 1.67 (s, 6H), and
1.59 (d, 6H, J = 6.8 Hz); ES-HRMS m/z 528.1435 (M+H calcd for C22H29N5O3F2Br requires 528.1416).
Example 6
2-(4-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin- 2-yl}piperazin-l-yl)acetamide trifluoroacetate
[0171] A mixture of chloroacetamide (0.1 g, 1.1 mmol), 5-bromo-6-[(2,4- difluorobenzyl)oxy]-3-isopropyl-2-piperazin-l-ylpyrimidin-4(3H)-one (0.30 g, 068 mmol), diisopropylethyl amine (0.081 g, 0.63 mmol) and DMAP (0.01 g) in dioxane (6.0 mL) was heated at 70 °C under argon for 16 h. After the removal of solvents under reduced pressure the residue was purified by HPLC using 10-90% CH3CN/Water gradient (40 min) containing 0.5% trifluoroacetic acid at a flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 500) were combined and lyophilized to afford the title compound (0.085g) as a white powder: 1H NMR (CD3OD/ 400 MHz) δ 7.50 (m, IH), 6.98 (m, 2H), 5.44 (s, 2H), 4.59 (m, IH), 3.80 (br, 2H), 3.52 (m, 4H), 3.29 (m, 4H), and 1.57 (d, 6H, J = 6.8 Hz); ES-HRMS m/z 500.1092 (M+H calcd for C20H25N5O3F2Br requires 500.1103).
2-(2-aminoethoxy)-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyIpyrimidin-
4(3H)-one trifluoroacetate
Step 1: Preparation of t-butyl 2-({5-bromo-4-r(2,4-difluorobenzyl)oxyl-l-isopropyl-6- oxo- 1 ,6-dihydrop yrimidin-2- yl ) oxy)ethyl carbamate.
[0172] A mixture of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)-pyrimidin-4(3H)-one (0.25 g, 0.57 mmol), t-butyl 2- hydroxyethylcarbamate (0.138g, 0.86 mmol), and DBU (0.1 g, 0.66 mmol) in THF (5.0 mL) containing DMAP (0.01 g) was stirred at room temperature under argon. After 4 h, the mixture was concentrated under reduced pressure and the residue was partitioned between dichloromethane (15.0 mL) and 5% citric acid (5.0 mL). The organic phase was washed with water, dried (Na2SO4) and concentrated. The resulting syrup was dissolved in acetonitrile and added water to turbidity when solids separated out. These were filtered, washed with acetonorile/water (1:1 v/v) and dried in vacuo to furnish the title compound as a white powder: ES-HRMS mJz 518.1134 (M+H calcd for C21H27N3O5F2Br requires 518.1097).
Step 2: Preparation of the title compound.
[0173] A solution of t-butyl 2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6- oxo-l,6-dihydropyrimidin-2-yl}oxy)ethylcarbamate (0.1 g, 0.19 mmol) in dichloromethane (0.3 mL) and trifluoroacetic acid (0.2 mL) was stirred at room temperature for 30 min. The product was isolated by reverse-phase HPLC using 10-90% CH3CN/Water gradient (40 min) containing 0.5% trifluoroacetic acid at a flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 418) were combined and lyophilized to afford the title compound (0.07g) as a white powder: 1H NMR (CD3OD/ 400 MHz) δ 7.51 (m, IH), 6.98 (m, 2H), 5.45 (s, 2H), 5.21 (br, IH), 4.68 (m, 2H), 3.43 (br, 2H), and 1.47 (d, 6H, J= 7.2 Hz); ES-HRMS m/z 418.0580 (M+H calcd for C16H]9N3O3F2Br requires 418.0572).
Example 8
l-{5-Bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yljprolinamide
[0174] A mixture of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)-pyrimidin-4(3H)-one (0.43 g, 0.99 mmol), S -prolineamide (0.23 g, 2.0 mmol), diisopropylethylamine (0.25 g, 1.95 mmol) in THF (5.0 mL) containing DMAP (0.015 g) was heated at 70 °C for 16 h under argon. The reaction mixture was concentrated under reduced pressure and the residue was partitioned between 5% citric acid (10.0 mL and dichloromethane (15.0 mL). The organic phase was washed with water, dried (Na2SO4) and concentrated to dryness under reduced pressure. The resulting material was purified by silica gel flash chromatography using EtOAc containing 1%
methanol as the eluent. The appropriate fractions (MH+ m/z = 471) were combined, concentrated to dryness, and the residue was further purified by reverse-phase HPLC using 10-90% CH3CN/Water gradient (40 min) containing 0.5% trifluoroacetic acid at a flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 471) were combined, concentrated to a small volume (~ 25 mL), added sodium bicarbonate (1.0 g) and extracted with dichloromethane (2 x 20 mL). The combined organic extracts were washed with water (2 x 10 mL), dried (Na2SO4) and concentrated to dryness in vacuo to afford the title compound (0.13 g) as a white powder: 1H NMR (CD3OD/ 400 MHz) δ 7.50 (m, IH), 6.97 (m. 2H), 5.38 (q, 2H, 7 = 12.8 Hz), 4.58 (m, 2H), 3.81 (m, IH), 3.61 (m, IH), 2.35 (m, IH), 2.11 (m, IH), 1.89 (m, 2H), 1.70 (d, 3H, J = 6.8 Hz), and 1.45 (d, 3H, J = 6.8 Hz); ES-HRMS m/z 471.0820 (M+H calcd for C19H22N4O3F2Br requires 471.0838).
Example 9
l-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl }piperidine-3-carboxamide
[0175] A mixture of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)-pyrimidin-4(3H)-one (0.50 g, 1.15 mmol), nipectomide(0.22 g, 1.7 mmol), diisopropylethylamine (0.22 g, 1.7mmol) in dioxane (5.0 mL) containing DMAP (0.02 g) was heated at 70 °C for 3 h under argon. It was concentrated under reduced pressure and the residue was partitioned between 5% citric acid (10.0 mL) and dichloromethane (15.0 mL). The organic phase was washed with water, dried (Na2SO4) and concentrated to dryness under reduced pressure. The resulting material was purified by reverse-phase HPLC using 10-90% CH3CN/Water gradient (40 min) containing 0.5%
trifluoroacetic acid at a flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 485) were combined, concentrated to a small volume, added sodium bicarbonate (0.5 g) and extracted with dichloromethane (2 x 20 mL). The combined organic extracts were washed with water (2 x 10 mL), dried (Na2SO4) and concentrated to dryness in vacuo to afford the title compound (0.13 g) as a white powder: 1H NMR (CD3OD/ 400 MHz) δ 7.49 (m~ IH), 6.95 (m, 2H), 5.42 (s, 2H), 4.52 (m, IH), 3.57 (m, IH), 3.47 (m, IH), 3.1 (m IH), 2.98 (m, IH), 2.62 (m, IH), 1.98 (m, IH), 1.82 (m IH), 1.69 (m, 2H), and 1.55 (m, 6H); ES-HRMS m/z 485.1016 (M+H calcd for C20H24N4O3F2Br requires 485.0994).
Example 10
N~2~-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}glycinamide
[0176] A mixture of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)-pyrimidin-4(3H)-one (0.50 g, 1.15 mmol), glycineamide hydrochloride (0.19 g, 1.72 mmol), diisopropylethylamine (0.44 g, 3.4 mmol) in dioxane (5.0 mL) containing DMAP (0.02 g) was heated at 70 °C for 2 h under argon. The reaction mixture was concentrated under reduced pressure and the residue purified by reverse- phase HPLC using 10-90% CH3CN/Water gradient (40 min) containing 0.5% trifluoroacetic acid at a flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 431) were combined, concentrated to a small volume, added sodium bicarbonate (0.5 g) and extracted with dichloromethane (2 x 20 mL). The combined organic extracts were washed with water (2 x 10 mL), dried (Na2SO4) and concentrated to dryness in vacuo to afford the title compound (0.13 g) as a white powder: 1H NMR (CD3OD/ 400 MHz) δ
7.49 (m, IH), 6.95 (m, 2H), 5.36 (s, 2H), 4.55 (br, 2H), 4.00 (s, 2H), and 1.52 (d, 6H, J -- 6.8 Hz); ES-HRMS m/z 431.0527 (M+H calcd for C16H]8N4O3F2Br requires 431.0525).
2-(3-Aminopyrrolidin-l-yl)-5-bromo-6-[(2,4-difIuorobenzyl)oxy]-3- isopropylpyrimidin-4(3H)-one trifluoroacetate
Step 1 : Preparation of t-butyl l-(5-bromo-4-r(2,4-difluorobenzyl)oxyl-l-isopropyl-6- oxo-l,6-dihvdropyrimidin-2-yl)pyrrolidin-3-ylcarbamate.
[0177] To a solution of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)-pyrimidin-4(3H)-one (0.50 g, 1.15 mmol) in THF (5.0 mL),3- t-butoxycarbonylaminopyrrolodine (0.27 g, 1.45 mmol), DBU (0.25 g, 1.6 mL) and DMAP (0.02 g) were added. The resulting mixture was stirred at room temperature for 2 h under argon and concentrated under reduced pressure. The residue was partitioned between 5% citric acid (20.0 mL and dichloromethane (15.0 mL). The organic phase
was washed with water, dried (Na2SO4) and concentrated to dryness under reduced pressure. The resulting material was purified by silica gel flash chromatography using 50% EtOAc in hexane to give 0.46(MH+ m/z = 543) as a white powder: ES-HRMS m/z 543.1405(M+H calcd for C23H30N4O4F2Br requires 543.1413).
Step 2: Preparation of the title compound.
[0178] A solution of t-butyl l-{5-brorno-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}pyrrolidin-3-ylcarbamate (0.4 g, 0.74 mmol) in dichloromethane (0.5 mL) and trifluoroacetic acid (0.5 mL) was stirred at room temperature for 30 min. The product was isolated by reverse-phase HPLC using 10-90% CH3CN/Water gradient (40 min) containing 0.5% trifluoroacetic acid at a flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 443) were combined and lyophilized to afford the title compound (0.34g, 82%) as a white powder: 1H NMR (CD3OD/ 400 MHz) 67.51 (m, IH), 7.00 (m, 2H), 5.44 (s, 2H), 4.49 (m, IH), 3.95 (m, 2H), 3.77 (m, IH), 3.68 (m, IH), 3.62 (m,lH), 2.42(m, IH), 2.14 (m, IH), 1.62 (d, 3H, J = 6.8 Hz), and 1.58 (d, 3H, J = 6.8 Hz); ES-HR MS m/z 443.0882 (M+H calcd for C18H22N4O2F2Br requires 443.0889).
Example 12
2-[(3R)-3-aminopyrrolidin-l-yl]-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3- isopropylpyrimidin-4(3H)-one trifluoroacetate
[0179] The title compound was prepared by a similar procedure described for 2-(3- Aminopyrrolidin-l-yl)-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin- 4(3H)-one trifluoroacetate using 3i?-3-t-butoxycarbonylaminopyrrolidine in place of 3-t- b'utoxycarbonylamino-pyrrolidine: 1H NMR (CD3OD/ 400 MHz) δ 7.50 (m, IH), 6.96 (m, 2H), 5.41 (s, 2H), 4.45 (m, IH), 3.95 (m, 2H), 3.74 (m, IH), 3.68 (m, IH), 3.60 (m,lH), 2.41(m, IH), 2.15 (m, IH), 1.60 (d, 3H, J = 6.8 Hz), and 1.55 (d, 3H, J = 6.8 Hz); ES-HR MS m/z 443.0831 (M+H calcd for Ci8H22N4O2F2Br requires 443.0889).
Example 13
NH2. CF3COOH
2-[(3S)-3-aminopyrrolidin-l-yl]-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3- isopropylpyrimidin-4(3H)-one trifluoroacetate
[0180] The title compound was prepared by a similar procedure described for 2-(3- Aminopyrrolidin-l-yl)-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin- 4(3H)-one trifluoroacetate using 35-3-t-butoxycarbonylaminopyrrolidine in place of 3-t- butoxycarbonylamino-pyrrolidine : 1H NMR (CD3OD/ 400 MHz) δ 7.50 (m, IH), 6.96 (m, 2H), 5.41 (s, 2H), 4.46 (m, IH), 3.94 (m, 2H), 3.75 (m, IH), 3.61 (m, IH), 3.60 (m,lH), 2.41(m, IH), 2.15 (m, IH), 1.59 (d, 3H, J = 6.8 Hz), and 1.55 (d, 3H, J = 6.8 Hz); ES-HRMS m/z 443.0914 (M+H calcd for C8H22N4O2F2Br requires 443.0889).
CF3COOH
5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(2S)-2-(hydroxymethyI)pyrroIidin-l-yl]-3- isopropylpyrimidin-4(3H)-one trifluoroacetate
[0181] A mixture of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)-pyrimidin-4(3H)-one (0.43 g, 0.99 mmol), S-prolinol(0.12 g, 1.2 mmol) in dioxane (3.0 mL) containing DMAP (0.025 g, 0.2 mmol) was stirred at room temperature for 2 h under argon. The reaction mixture was then concentrated under reduced pressure and the residue was partitioned between water (10.0 mL ) and dichloromethane (15.0 mL). The organic phase was washed with water, dried (Na2SO4) and concentrated to dryness under reduced pressure and the residue was purified by silica gel flash chromatography using 50% EtOAc in hexane as the eluent . The appropriate fractions (MH+ m/z = 458) were combined and concentrated and the resulting residue was further purified by reverse-phase HPLC using 10-90% CH3CN/Water gradient (40 min) containing 0.5% trifluoroacetic acid at a flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 458) were combined, and lyophilized to afford the title compound (0.02 g) as a white powder: 1H NMR (CD3OD/ 400 MHz) δ 7.49 (m, IH), 6.95 (m, 2H), 5.38 (q, 2H, J = 10.8 Hz), 4.48 (m, 2H), 3.61 (m, 3H), 3.45 (m, IH), 2.18 (m, IH), 2.01 (m, IH), 1.85 (m, 2H), 1.73 (d, 3H, J = 6.8 Hz), and 1.39(d, 3H, J = 6.8 Hz); ES-HRMS m/z 458.0835 (M+H calcd for C19H23N3O3F2Br requires 458.0885).
Example 15
N~3~-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}-beta-alaninamide
[0182] A mixture of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)-pyrimidin-4(3H)-one (0.50 g, 1.15 mmol), β-alanineamide hydrochloride (0.19 g, 1.75 mmol), diisopropylethylamine (0.33 g, 2.6 mmol) in dioxane (5.0 mL) containing DMAP (0.025 g, 0.2 mmol) was heated at 65 °C for 2 h under argon. The reaction mixture was concentrated under reduced pressure and the residue was partitioned between 5% citric acid (10.0 mL) and dichloromethane (20.0 mL). The organic phase was washed with water, dried (Na2SO4) and concentrated to dryness under reduced pressure and the residue was purified by silica gel flash chromatography using EtOAc containing 1% methanol as the eluent. The appropriate fractions (MH+ m/z - 445) were combined and concentrated to dryness under reduced pressure and the resulting solid was crystallized from EtOAc/hexane to afford the title compound (0.16 g) as a white powder: 1H NMR (CD3OD/ 400 MHz) δ 7.49 (m, IH), 6.95 (m, 2H), 5.40 (s, 2H), 4.55 (br, IH), 3.67 (t, 2H, J = 6.8 Hz), and 2.51 (t, 2H, J = 6.8 Hz), and 1.46 (d, 6H, J = 6.8 Hz); ES-HR MS m/z 445.0645 (M+H calcd for CnH20N4O3F2Br requires 445.0681).
Example 16
N~2~-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimJdin-2-yl}-L-alaninamide
[0183] A mixture of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)-pyrimidin-4(3H)-one (0.50 g, 1.15 mmol), S-alanineamide • hydrochloride (0.18 g, 1.44 mmol), diisopropylethylamine (0.39 g, 3.0 mmol) in dioxane (5.0 mL) containing DMAP (0.020 g, 0.16 mmol) was heated at 65 0C for 3 h under argon. The reaction mixture was then concentrated under reduced pressure and the residue was partitioned between 5% citric acid (5.0 mL ) and dichloromethane (20.0 mL). The organic phase was washed with water, dried (Na2SO4) and concentrated to dryness under reduced pressure and the residue was purified by silica gel flash chromatography using EtOAc as the eluent. The appropriate fractions (MH+ m/z = 445) were combined and concentrated to dryness under reduced pressure. The resulting substance was further purified by reverse-phase HPLC using 10-90% CH3CN/Water gradient (40 min) containing 0.5% trifluoroacetic acid at a flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 445) were combined, sodium bicarbonate (0.5 g) was added and extracted with dichloromethane (2 x 20 mL). The combined organic extracts were washed with water (2 x 10 mL), dried (Na2SO4) and concentrated to dryness under reduced pressure to afford the title compound (0.13 g) as a white powder: 1H NMR (CD3OD/ 400 MHz) δ 7.49 (m, IH), 6.95 (m, 2H), 5.37 (q, 2H, J = 4.0 Hz), 4.46 (m, IH), and 1.53 (m, 9H); ES-HRMS m/z 445.0672 (M+H calcd for CnH20N4O3F2Br requires 445.0681).
N~l~-(l-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyI-6-oxo-l,6- dihydropyrimidin-2-yl}pyrrolidin-3-yl)glycinamide trifluoroacetate
[0184] To a solution of 2-(3-Aminopyrrolidin-l-yl)-5-bromo-6-[(2,4- difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one trifluoroacetate (0.15 g, 0.27 mmol) in DMF (2.0 mL), N-methylmorpholine (0.04 g, 0.4 mmol), N-t-BOC-glycine-N- succinimide ester (0.1 g, 0.37 mmol) and DMAP (0.05 g) were added. The reaction mixture was stirred at room temperature for 16 h under argon. The solvents were distilled in vacuo, the residue was dissolved in dichloromethane (0.5 mL) and trifluoroacetic acid (0.5 mL), and the mixture was stirred at room temperature for 1 h. The product was isolated by reverse-phase HPLC using 10-90% CH3CN/Water gradient (40 min) containing 0.5% trifluoroacetic acid at a flow rate of 80 mL/min. The appropriate fractions (MH+, m/z = 500) were combined and lyophilized to afford the title compound (0.09 g) asa white powder: 1H NMR (CD3OD/ 400 MHz) δ 7.48 (m, IH), 6.95 (m, 2H), 5.39 (s, 2H), 4.44 (m, 2H), 3.89 (dd, IH, J = 6.0 Hz, 11.2 Hz), 3.76 (m, IH), 3.62 (m, 3H), 3.42 (2d, IH, J = 4.0 Hz), 2.21 (m, IH), 1.97 (m, 1H)1.59 (d, 3H, J = 6.4 Hz), and 1.50 (d, 3H, J = 6.4 Hz); ES-HRMS m/z 500.1079 (M+H calcd for C20H25N5O3F2Br requires 500.1103).
Example 18
N~2~-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}-N~l~-methylglycinamide
[0185] A mixture of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)-pyrimidin-4(3H)-one (0.50 g, 1.15 mmol), N-methylglycineamide hydrochloride (0.21 g, 1.7 mmol), diisopropylethylamine (0.46 g, 3.6 mmol) in dioxane (5.0 mL) containing DMAP (0.02 g, 0.16 mmol) was stirred at room temperature for 2 h under argon. The residue was partitioned between 5% citric acid (5.0 mL ) and ethylacetate (20.0 mL). The organic phase was washed with water, dried (Na2SO4) and concentrated to dryness under reduced pressure and the residue was crystallized from dichloromethane/hexane to afford the title compound (0.16 g, 31%) as a white powder: 1H NMR (CD3OD/ 400 MHz) δ 7.49 (m, IH), 6.93 (m, 2H), 5.32 (s, 2H), 3.96 (s, 2H), 2.69 (s, 3H), and 1.53 (d, 6H, J = 7.2 Hz); ES-HRMS mJz 445.0682 (M+H calcd for Ci7H20N4O3F2Br requires 445.0681).
Example 19
2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}oxy)acetamide
[0186] To a solution of 2-hydroxyacetamide (0.08 g, 1.1 mmol) and 5-bromo-6-[(2,4- difluorobenzyl)oxy]-3-isopropyl-2-(methylsulfonyl)-pyrimidin-4(3H)-one (0.30 g, 1.67 mmol), at 10 0C in dioxane (5.0 mL), sodium hydride (0.04 g, 1.67 mmol) was added and stirred at room temperature under argon. After 30 min, DMAP (0.01 g) was added and the mixture was heated at 65 °C for an additional 30 min. The reaction mixture was cooled, added acetic acid (0.1 mL) and concentrated under reduced pressure. The resulting material was partitioned between 5% citric acid (5.0 mL) and ethyl acetate (20.0 mL). The organic phase was washed with water, dried (Na2SO4), concentrated to dryness under reduced pressure, and the residue was crystallized from ethyl acetate to afford the title compound (0.12 g, 40%) as a white powder: 1H NMR (CD3OD/ 400 MHz) δ 7.49 (m, IH), 6.96 (m, 2H), 5.39 (s, 2H), 5.24 (br, IH), 4.94 (s, 2H), and 1.46 (d, 6H, J = 6.8 Hz); ES-HRMS m/z 432.0354 (M+H calcd for C16H17N3O4F2Br requires 432.0365).
Example 20
5-Bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(3R)-3-hydroxypyrrolidin-l-yl]-3- isopropylpyrimidin-4(3H)-one
[0187] A mixture of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)-pyrimidin-4(3H)-one (0.40 g, 0.92 mmol), (37?)-pyrrolidin-3-ol (0.08 g, 0.93 mmol), and diisopropylethylamine (0.15 g, 1.15 mmol) in dioxane (5.0 mL) containing DMAP (0.02 g, 0.16 mmol) was stirred at room temperature for 1 h under argon. The reaction mixture was concentrated under reduced pressure and the residue was partitioned between 5% citric acid (5.0 mL) and dichloromethane (15.0 mL). The
organic phase was washed with water, dried (Na2SO4) and concentrated to dryness under reduced pressure and the residue was purified by silica gel flash chromatography using ethyl acetate as the eluent to afford the title compound (0.27 g, 67%) as an amorphous material: 1H NMR (CD3OD/ 400 MHz) δ 7.49 (m, IH), 6.95 (m, 2H), 5.39 (s, 2H), 4.43 (m, 2H), 3.89 (m, IH), 3.79 (m, IH), 3.55 (m, IH), 3.38 (m, IH), 1.98 (m, 2H), 1.65 (d, 3H, J = 7.2 Hz), and 1.43 (d, 3H, J = 7.2 Hz); ES-HRMS m/z 444.0740 (M+H calcd for C18H2]N3O3F2Br requires 444.0729).
Example 21
5-Bromo-6-[(2,4-difluorobenzyl)oxy]-2-(4-hydroxypiperidin-l-yl)-3- isopropylpyrimidin-4(3H)-one
[0188] The title compound was prepared by a similar procedure as described for 5- Bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(3R)-3-hydroxypyrrolidin-l-yl]-3- isopropylpyrimidin-4(3H)-one, substituting 4-hydroxypiperidine for (3R)-pyrrolidin-3- ol: 1H NMR (CD3OD/ 400 MHz) δ 7.49 (m, IH), 6.95 (m, 2H), 5.42 (s, 2H), 4.53 (m,lH), 3.81 (m, IH), 3.52 (m, 2H), 3.12 (m, 2H), 1.96 (m, 2H), 1.61 (m, 2H), and 1.58 (d, 6H, J = 7.2 Hz); ES-HRMS m/z 458.0842 (M+H calcd for C9H23N3O3F2Br requires 458.0885).
5-Bromo-6-[(2,4-difluorobenzyl)oxy]-2-{[(2S)-2,3-dihydroxypropyl]amino}-3- isopropylpyrimidin-4(3H)-one
[0189] A mixture of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)-pyrimidin-4(3H)-one (0.50 g, 1.15 mmol), S-l-amino-2-propanol (0.13 g, 1.43 mmol), and diisopropylethylamine (0.22 g, 1.7 mmol) in dioxane (5.0 mL) containing DMAP (0.02 g, 0.16 mmol) was stirred at room temperature for 16 h under argon. The reaction mixture was concentrated under reduced pressure and the residue was partitioned between 5% citric acid (5.0 mL ) and dichloromethane (15.0 mL). The organic phase was washed with water, dried (Na2SO4) and concentrated to dryness under reduced pressure and the residue was purified by silica gel flash chromatography using ethyl acetate containing l%methanol as the eluent to afford the title compound (0.19 g, 37%) as an amorphous material: 1H NMR (CD3OD/ 400 MHz) 57.49 (m, IH), 6.95 (m, 2H), 5.40 (s, 2H), 4.85 (br, IH), 3.86 (m, IH), 3.65 (m, IH), 3.54 (m, 2H), 3.38 (m, IH), and 1.48 (2d, 6H, J = 1.6 Hz); ES-HRMS mJz 448.0659 (M+H calcd for C17H2IN3O4F2Br requires 448.0678).
Example 23
5-Bromo-6-[(2,4-difluorobenzyl)oxy]-2-{[(2R)-2,3-dihydroxypropyl]amino}-3- isopropylpyrimidin-4(3H)-one
[0190] The title compound was prepared by a similar procedure as described for 5- Bromo-6-[(2,4-difluorobenzyl)oxy]-2-{[(2S)-2,3-dihydroxypropyl]amino}-3- isopropylpyrimidin-4(3H)-one, substituting 5-l-amino-2-propanol for /?-l-amino-2- propanol: 1H NMR (CD3OD/ 400 MHz) 67.49 (m, IH), 6.95 (m, 2H), 5.40 (s, 2H), 3.83 (m, IH), 3.62 (m, 1H), 3.54 (m, 2H), 3.38 (m, IH), and 1.48 (2d, 6H, J = 1.6 Hz); ES- HRMS m/z 448.0705 (M+H calcd for C17H2]N3O4F2Br requires 448.0678).
Example 24
5-Bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-moφholin-4-ylpyrimidin-4(3H)-one [0191] A mixture of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-
(methylsulfonyl)-pyrimidin-4(3H)-one (0.44 g, 1.0 mmol), morpholine (0.14 g, 1.6 mmol), and DMAP (0.016 g) in dioxane (5.0 mL) containing diisopropylethyl amine (0.13 g, 1.0 mmol) was stirred at room temperature for 16 h under nitrogen atmosphere. The reaction mixture was diluted with water (15.0 mL) and extracted with dichloromethane (2 x 15 mL). The combined organic extracts were washed with water, dried (Na2SO4), concentrated and the residue was purified by reverse-phase HPLC using 10-90% CH3CN/Water gradient (45 min) containing 0.5% trifluoroacetic acid at a flow rate of 80 mL/min.
[0192] The appropriate fractions (MH+, m/z = 444/446) were combined, and concentrated to -25 mL, added 5% sodium bicarbonate (10 mL) and extracted the product with dichloromethane (2 x 15 mL). The combined organic extracts were washed with water (2 x 10 mL), dried (Na2SO4) and concentrated to dryness under reduced pressure to afford the title compound (0.21 g, 47%) as a pale powder: 1H NMR (CD3OD/
400 MHz) 57.48 (m, IH), 6.96 (m, 2H), 5.45 (s, 2H), 4.62 (m, IH), 3.79 (m, 4H), 3.27 (m, 4H), and 1.57 (d, 6H, J = 6.4 Hz); ES-HRMS m/z 444.0744 (M+H calcd for C18H21N3O3F2Br requires 444.0729).
Example 25
5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one
Step 1: Preparation of 6-[(2,4-difluorobenzyl)oxyl-3-isopropylpyrimidin-4(3H)-one.
[0193] To a mixture of 6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylthio)pyrimidin-4(3H)-one (0.53 g, 1.62 mmol) in ethanol (5.0 mL) was added a suspension Raney nickel (1.0 mL). The reaction mixture was heated at reflux for 2 h. Added additional Raney nickel (0.5 mL) and ethanol (2.5 mL) and heating was continued for an additional 2 h. The catalyst was allowed to settle and pipetted off the supernatent. The catalyst was washed with ethanol (2 x 25 mL), combined the superenatent and the organic washings, concentrated, and dried in vacuo. The resulting residue was used without further purification in the next step.
Step 2: Preparation of the title compound.
[0194] To a cooled (0°C) solution of 6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin- 4(3H)-one (0.34 g, 1.2 mmol) in DCM (3 mL), JV-bromosuccinimide (0.19 g, 1.1 mmol) was added. The reaction mixture was allowed to warm to ambient temperature. After 4
h the reaction mixture was concentrated and the residue was purified by preparatory HPLC using a 10-90% CH3CNfH2O (30 min) gradient containing 0.5% TFA at a flow rate of 80 mL/min. Appropriate fractions (M+H m/z =360) were combined and concentrated to approximately to 20 mL under reduced pressure. Added 5% NaHCO3 (20 mL) and extracted with dichloromethane (3 x 15 mL). The combined organic extracts were dried over Na2SO4, filtered, concentrated under reduced pressure, and dried in vacuo to give the desired product as a white solid (0.32 g, 55% ): 1H NMR (CD3OD/ 400MHz) 58.33 (s, IH), 7.53 (q, IH, J = 8.0 Hz), 6.95 (m, 2H), 5.48 (s, 2H), 4.93 (quintet, IH, J = 6.8 Hz), 1.43 (d, 6H, J = 6.8 Hz). ES-HRMS m/z 359.0202 (M+H calculated for C14H14BrF2N2O2 requires 359.0201).
Example 26
5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-(methylthio)pyrimidin-4(3H)- one
[0195] To a cooled (0 °C) solution of 6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylthio)pyrimidin-4(3H)-one (0.20 g, 0.61 mmol) in DCM (2 mL) was added N- bromosuccinimide (0.11 g, 0.61 mmol). Stirred at 00C for 30 min, ambient temperature for 4h. Concentrated and purified crude residue by preparatory HPLC using a 10-90% CH3CN/H2O (30 min) gradient containing 0.5% TFA at a flow rate of 80 mL/min. Appropriate fractions (M+H m/z =406) were combined and concentrated to approximately 25 mL under reduced pressure. Added 5% NaHCO3 (20 mL) and extracted with DCM (3 x 15 mL). The organic extracts were dried over Na2SO4, filtered, concentrated under reduced pressure, and dried in vacuo to give the desired product as a white solid (0.13 g, 53%). 1H NMR (CD3OD/ 400MHz) 67.49 (q, IH, J = 8.4 Hz), 6.97 (m, 2H), 5.51 (s, 2H), 4.63 (m, IH), 2.57 (s, 3H), 1.55 (d, 6H, 7 = 6.8 Hz). ES-HRMS m/z 405.0089 (M+H calculated for Ci5Hi6BrF2N2O2S requires 405.0078).
5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(3R)-3-(ethylamino)pyrroIidin-l-yI]-3- isopropyIpyrimidin-4(3H)-one
Step 1. Preparation of tert-butyl (3R)-l-(5-bromo-4-r(2,4-difluorobenzyl)oxyl-l- isopropyI-6-oxo-l,6-dihvdropyrimidin-2-yl)pyiτolidm-3-yl(ethyl)carbarnate.
[0196] To a cold solution of tert-butyl (3R)-l-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-ylcarbamate (0.4 g, 0.74 mmol) in anhydrous tetrahydrofuran (4 mL) was added NaH (0.026 g 1.1 mmol) followed by the addition of iodoethane (0.12 mL, 1.5 mmol). The reaction mixture was stirred at room temperature overnight under nitrogen. Subsequent addition of iodoethane (0.05 mL, 0.5 equiv) was needed. The reaction mixture was heated for 3 h in an oil bath at 69 °C. It was cooled, diluted with a cold solution of citric acid (5%) and extracted with ethyl acetate. The organic extracts were concentrated, the residue was treated with
acetonitrile/water (2: 1 v/v) and purified by reverse phase HPLC using a 10-90% acetonitrile in water containing 0.5% TFA (30 min) gradient at a flow rate of 80 mL/min. T; he appropriate fractions (M+H m/z = 571) were combined and freeze-dried to yield 0.180 g (43 %) of tert-butyl (3R)-l-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6- oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-yl(ethyl)carbamate as a white solid: 1H- NMR-(CD3OD, 400 MHz) 57.51 (q, IH, J= 8.4 Hz), 6.96 (m, 2H), 5.43 (m, 2H), 4.43 (m, 2H), 3.60 (m, 4H), 3.27 (m, IH), 2.12 (m, 2H), 1.67 (d, 3HJ= 6.8 Hz), 1.46 (m, 12H), 1.13 (t, 3H, J= 6.8 Hz). ES-HRMS m/z 571.1721 (M+H C25H34BrF2N4O4 requires 571.1726).
Step 2. Preparation of title compound.
[0197] To a solution of tert-butyl (3R)-l-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-yl(ethyl)carbamate (0.162 g, 0.28 mmol) from Step 1 in dichloromethane (3 mL) was added trifluoroacetic acid (0.5 mL). The reaction mixture stirred at room temperature overnight. The reaction mixture was concentrated to remove solvent, the residue was treated with acetonitrile/water (2: 1 v/v) and purified by reverse phase HPLC using a 10-90% acetonitrile in water containing 0.5% TFA (30 min) gradient at a flow rate of 80 mL/min. The appropriate fractions (M+H m/z = 471) were combined and freeze-dried to yield 0.110 g (82 %) of 5-bromo-6- [(2,4-difluorobenzyl)oxy]-2-[(3R)-3-(ethylamino)pyrrolidin-l-yl]-3-isopropylpyrimidin- 4(3H)-one as a white solid: 1H-NMR (CD3OD, 400 MHz) δ 7.51 (q, IH, J= 8.4 Hz), 6.93 (m, 2H), 5.43 (s, 2H), 4.48 (m, IH), 3.96 (m, 2H), 3.69 (m, 3H), 3.17 (m, 2H), 2.43 (m, IH), 2.19 (m, IH), 1.61 (dd, 6H, J= 2.4 Hz), 1.37 (t, 3H, J= 12 Hz). ES-HRMS m/z 471.1169 (M+H C20H26BrF2N4O4 requires 471.1202).
5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-[(3R)-3- (methylamino)pyrrolidin-l-yI]pyrimidin-4(3H)-one
Step 1. Preparation of tert-butyl (3R)-l-(5-bromo-4-r(2,4-difluorobenzyl)oxyl-l- isopropyl-6-oxo-l,6-dihvdropyrimidin-2-yl)pyrrolidin-3-yl(methyl)carbamate.
[0198] To a cold solution of tert-butyl (3R)-l-{ 5-bromo-4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-ylcarbamate (0.4 g, 0.74 mmol) in anhydrous tetrahydrofuran (4 mL) was added NaH (0.026 g 1.1 mmol) followed by the addition iodomethane (0.09 mL, 1.5 mmol). The reaction mixture stirred at room temperature for 3 h under nitrogen. Subsequent addition of iodomethane (0.06 mL, 1.0 equiv) and NaH (0.013g, 0.5 equiv) was needed. The reaction mixture continued to stir at room temperature for 3 h and then cooled, treated with a cold solution of citric acid (5%) and extracted with ethyl acetate. The organic extracts were concentrated, the residue was treated with acetonitrile/water (2:1 v/v) and purified by reverse phase HPLC
using a 10-90% acetonitrile in water containing 0.5% TFA (30 min) gradient at a flow rate of 80 mL/min. The appropriate fractions (M+H m/z = 557) were combined and freeze-dried to yield 0.33 g (80 %) of tert-butyl (3R)-l-{5-bromo-4-[(2,4- difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3- yl(methyl)carbamate as a white solid: 1H-NMR (CD3OD, 400 MHz) δ 7.51 (q, IH, /= 8.4 Hz), 6.97 (m, 2H), 5.43 (q, 2H), 4.65 (m, IH), 4.45 (m, IH) 3.61 (m, 4H), 2.83 (s, 3H), 2.10 (m, 2H), 1.65 (d, 3H, J= 6.4 Hz), 1.50 (d, 3H, J= 6.4 Hz), 1.13 (s, 9H). ES- HRMS m/z 557.1522 (M+H C24H32BrF2N4O4 requires 557.1570).
Step 2. Preparation of title compound.
[0199] To a solution tert-butyl (3R)-l-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl }pyrrolidin-3-yl(methyl)carbamate (0.30 g, 0.53 mmol) from Step 2 in dichloromethane (3 mL) was added trifluoroacetic acid (1.0 mL). The reaction mixture stirred at room temperature for 30 min. It was concentrated to remove solvent and the residue was treated with acetonitrile/water (2:1 v/v) and purified by reverse phase HPLC using a 10-90% acetonitrile in water containing 0.5% TFA (30 min) gradient at a flow rate of 80 mL/min. The appropriate fractions (M+H m/z = 457) were combined and freeze-dried to yield 0.20 g (81 %) of 5-bromo-6-[(2,4- difluorobenzyl)oxy]-3-isopropyl-2-[(3R)-3-(methylamino)pyrrolidin-l-yl]pyrimidin- 4(3H)-one as a white solid: 1H-NMR (CD3OD, 400 MHz) δ 7.51 (q, IH, J= 8.4 Hz), 6.99 (m, 2H), 5.43 (s, 2H), 4.48 (m, IH), 3.96 (m, 2H), 3.69 (m, 3H), 2.97 (s, 3H), 2.43 (m, IH), 2.19 (m, IH), 1.62 (d, 3H, J= 6.4 Hz), 1.59 (d, 3H, J= 6.4 Hz). ES-HRMS m/z 457.1169 (M+H C19H23BrF2N4O4 requires 457.1202).
Example 29
pyridin-3-yImethyl (3R)-l-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}pyrrolidin-3-ylcarbamate
[0200] To a solution of 3-hydroxy methyl pyridine (0.044 mL, 0.45 mmol) in anhydrous tetrahydrofuran (3 mL) was added 4-nitrophenyl chloroformate (0.109 g, 0.54 mmol) and triethylamine (0.075 mL, 0.54 mmol). The reaction mixture stirred at room temperature for 1 h under nitrogen at which time 2-[(3R)-3-aminopyrrolidin-l-yl]-5-bromo-6-[(2,4- difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one (0.20 g, 0.45 mmol) was added. The reaction mixture stirred at room temperature overnight. The solvent was concentrated and the residue was treated with acetonitrile/water (2:1 v/v) and purified by reverse phase HPLC using a 10-90% acetonitrile in water containing 0.5% TFA (30 min) gradient at a flow rate of 80 mL/min. The appropriate fractions (M+H m/z - 578) were combined and freeze-dried to yield 0.13 g (42 %) of pyridin-3-ylmethyl (3R)-l-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-ylcarbamate as a white solid: 1H- NMR (CD3OD, 400 MHz) δ 8.76 (s, IH), 8.68 (d, IH, J= 5.6 Hz), 8.34 (d, IH, /= 8 Hz) 7.86 (dd, IH, J= 5.6 Hz), 7.51 (q, IH, J= 8.4 Hz), 6.96 (m, 2H), 5.40 (s, 2H), 5.25 (s, 2H), 4.43 (m, IH), 4.20 (m, IH), 3.80 (m, 2H), 3.68 (m, IH), 3.44 (m, IH), 2.19 (m, IH), 2.02 (m, IH), 1.61 (d, 3H, J= 6.4 Hz), 1.54 (d, 3H, J= 6.4 Hz). ES-HRMS m/z 578.1234 (M+H C25H27BrF2N5O4 requires 578.1209).
Example 30
pyridin-2-ylmethyl (3R)-l-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}pyrrolidin-3-yIcarbamate
[0201] The title compound was prepared by a procedure similar to the one described for pyridin-3-ylmethyl (3R)-l-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}pyrrolidin-3-ylcarbamate using 2-hydroxy methyl pyridine (0.051 mL, 0.62 mmol). After reverse phase HPLC purification, the appropriate fractions were combined and freeze-dried to afford 0.20 g (47%) of pyridin-2-ylmethyl (3R)-l-{5- bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}pyrrolidin-3-ylcarbamate as a white solid: 1H-NMR (CD3OD, 400 MHz) δ 8.60 (d, IH, J= 5.2 Hz), 8.08 (m, IH), 7.67 (d, IH, J= 8 Hz), 7.52 (m, 2H), 6.96 (m, 2H), 5.40 (s, 2H), 5.25 (s, 2H), 4.42 (m, IH), 4.20 (m, IH), 3.80 (m, 2H), 3.68 (m, IH), 3.49 (m, IH), 2.19 (m, IH), 2.02 (m, IH), 1.61 (d, 3H, J= 6.8 Hz), 1.54 (d, 3H, J= 6.8 Hz). ES-HRMS m/z 578.1234 (M+H C25H27BrF2N5O4 requires 578.1209).
5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-{(3R)-3-[(2-hydroxyethyl)amino]pyrrolidin- l-yl}-3-isopropylpyrimidin-4(3H)-one
Step 1. Preparation of 5-bromo-6-r(2,4-difluorobenzyl)oxy"|-3-isopropyl-2-((3R)-3-{r2- (tetrahvdro-2H-pyran-2-yloxy)ethyl1amino)pyrrolidm-l-yl)pyrimidin-4(3HVone.
[0202] To a cold solution of tert-butyl (3R)-l-{ 5-bromo-4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-ylcarbamate (0.7 g, 1.29 mmol) in anhydrous tetrahydrofuran (7 mL) was added NaH (0.046 g 1.96 mmol) followed by the addition of 2-(2-bromoethoxy)tetrahydro-2H-pyran (0.29 mL, 1.96 mmol). The reaction mixture was heated in an oil bath at 69 0C overnight under nitrogen. Subsequent addition of 2-(2-bromoethoxy)tetrahydro-2H-pyran (0.13 mL, 0.5 eq) and NaH (0.02g, 0.5 eq) was needed. The reaction mixture continued to heat overnight. It was cooled, diluted with a cold solution of citric acid (5%) and extracted with ethyl
acetate. The organic extracts were concentrated, the residue was treated with acetonitrile/water (2:1 v/v) and purified by reverse phase HPLC using a 10-90% acetonitrile in water containing 0.5% TFA (30 min) gradient at a flow rate of 80 mL/min. the appropriate fractions (M+H m/z = 671) were combined and freeze-dried to yield 0.29 g (29 %)of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-((3R)-3-{[2- (tetrahydro-2H-pyran-2-yloxy)ethyl]amino}pyrrolidin-l-yl)pyrimidin-4(3H)-one.
Step 2. Preparation of title compound.
[0203] To a solution of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-((3R)-3-{ [2- (tetrahydro-2H-pyran-2-yloxy)ethyl]amino}pyrrolidin-l-yl)pyrimidin-4(3H)-one (0.29 g, 0.43 mmol) from Step 1 in dichloromethane was added trifluoroacetic acid (1.0 mL). The reaction mixture stirred at room temperature for 2.5 h. It was treated with acetonitrile/water (2: 1 v/v) and purified by reverse phase HPLC using a 10-90% acetonitrile in water containing 0.5% TFA (30 min) gradient at a flow rate of 80 mL/min. The appropriate fractions (M+H m/z = 487) were combined and freeze-dried to yield 0.040 g (19 %) 5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-{(3R)-3-[(2- hydroxyethyl)amino]pyrrolidin-l-yl}-3-isopropylpyrimidin-4(3H)-one as a white solid: 1H-NMR (CD3OD, 400 MHz) δ 7.53 (q, IH, J= 5.2 Hz), 7.01 (m, 2H), 5.44 (s, 2H), 4.48 (m, IH), 3.97 (m, 2H), 3.83 (t, 2H, J= 5.2 Hz ), 3.74 (m, 3H), 3.25 (m, 2H), 2.45 (m, IH), 2.19 (m, IH), 1.61 (d, 3H, /= 6.8 Hz), 1.54 (d, 3H, J= 6.8 Hz). ES-HRMS m/z 487.1187 (M+H C20H26BrF2N4O3 requires 487.1151).
Example 32
N2-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-methyl-6-oxo-l,6-dihydropyrimidin-2- yl}glycinamide
Step 1: Preparation of 6-hvdroxy-3-methyl-2-(methylthio')pyrimidin-4GH')-one as described in US patent 4.152.426 (1979).
Step 2: Preparation of 6-(2,4-difluorobenzyloxy)-3-methyl-2-(methvthio)pyrimidin- 4(3HVone.
[0204] A suspension of 6-hydroxy-3-methyl-2-(methylthio)pyrimidin-4(3H)-one (64 g, 0.371 mol) from Step 1 and 325 mesh potassium carbonate (52 g, 0.372 mol) in 170 mL of N-methyl pyrolidinone was warmed to 50 0C to obtain a pasty suspension. 2,4- difluorobenzyl bromide (77 g, 0.372 mol) was added without dilution over 20 minutes, to allow an exotherm of 10 °C (final temp is 55-60 0C). The suspension became thinner and easier to stir in about 10 min, indicating significant completion. The temperature was lowered to 50 0C and held for a further 2 h. The mixture was cooled to ambient temperature and 400 mL of water was added with vigorous stirring. The precipitated solids were filtered and washed four times with 250 mL of water and dried at 50 0C and 20mm vacuum. The crude product was purified by flash chromatography on a Biotage 75L column using 10% ethyl acetate in hexanes to obtain 50 g (46%) of the 6-(2,4- difluorobenzyloxy)-3-methyl-2-(methythio)pyrimidin-4(3H)-one: 1H NMR (400 MHz, CDCl3) δ 7.40-7.32 (m, IH), 6.75-6.9 (m, 2H), 7.11 (m, IH), 5.57 (s, IH), 5.29 (s, 2H),
3.42(s, 3H), 2.53 (s, 3H).
Step 3: Preparation of 6-(2.4-difluorobenzyloxy)-5-bromo-3-methyl-2- (methylsulfonyl)pyrimidin-4(3HVone.
[0205] N-bromosucinimide (29.2 g, 0.16 mol) was added in portions to a stirred solution of 6-(2,4-difluorobenzyloxy)-3-methyl-2-(methythio)pyrimidin-4(3H)-one (49 g, 0.16 mol) from Step 2 in 400 mL of methylene chloride held at ambient temperature. The exotherm was minimal (2-3 0C) and the reaction was stirred for two hours. MCPBA (meta chloro perbenzoic acid) (73 g, 0.32 mol based on 77% assay) was added in three portions at ambient temperature over a period of one hour. The exotherm was mild as before and this suspension stirred vigorously for 12 hours. The reaction mixture was filtered to remove most of the reduced by products and the filtrate was concentrated and passed through a pad of neutral Alumina (150 g) using methylene chloride as the solvent. 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-methyl-2-(methylsulfonyl)pyrimidin-4(3H)-one (29 g, 55%) was obtained after drying as a colorless semi-crystalline solid. 1H NMR (400 MHz, CDCl3) δ 7.39-7.48 (m, IH), 6.78-6.92 (m, 2H), 5.42 (s, 2H), 3.83 (s, 3H), 3.39 (s, 3H).
Step 4: Preparation of title compound.
[0206] To a solution of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-methyl-2- (methylsulfonyl)pyrimidin-4(3H)-one (0.5 g, 1.22 mmol) from Step 3 in anhydrous 1,4- dioxane (5 mL) was added diisopropylethyl amine (0.43 mL, 2.44 mmol), glycinamide hydrochloride (0.135 g, 1.22 mmol) and DMAP (0.03 g). The reaction mixture was heated at 70 0C in an oil bath for 2.5 h under nitrogen. The reaction mixture was treated with acetonitrile/water (2:1 v/v) and purified by reverse phase HPLC using a 10-90%
acetonitrile in water containing 0.5% TFA (30 min) gradient at a flow rate of 80 rnL/min. The appropriate fractions (M+H mJz - 403) were combined and freeze-dried to yield 0.066 g (13 %) of N2-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-methyl-6-oxo-l,6- dihydropyrimidin-2-yl}glycinamide as a white solid: 1H-NMR (CD3OD, 400 MHz) 5 7.53 (q, IH, J= 5.2 Hz), 6.96 (m, 2H), 5.39 (s, 2H), 4.04 (s, 2H), 3.44 (s, 3H). ES- HRMS m/z 403.0198 (M+H Ci4H14BrF2N4O3 requires 403.0212).
Example 33
N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]acetamide
Step 1. Preparation of 6-(2,4-difluorobenzyloxy)-2-(3-aminopropylamino)-5-bromo-3- isopropylpyrimidin-4(3H)-one.
[0207] 6-(2,4-difluorobenzyloxy)-5-bromo-3-isopropyl-2-(methylsulfonyl)pyrimidin- 4(3H)-one (3.0 g, 6.9 mmol) was dissolved in dichloromethane (30 mL). Propane-1,3- diamine (0.65 mL, 7.6 mmol) was added followed by triethylamine (1.9 mL, 13.8 mmol). The reaction was stirred at room temperature for 5 minutes. Then the reaction was concentrated and the residue was washed with ethyl acetate/water. The aqueous layer was extracted into ethyl acetate. The combined organics were dried over MgSO
4, filtered, and concentrated. The residue was dissolved in methanol to approximately 1.0 M and trifluoroacetic acid (0.4 mL, 5.5 mmol) was added to afford a white solid (2.9 g, 80%).
1H NMR (400 MHz, CD
3OD) δ ppm 1.52 (d, J = 6.98 Hz, 6H) 1.97 (m, 2H) 2.94 (m, 2H) 3.54 (t, J = 6.71 Hz, 2H) 4.73 (m, IH) 5.42 (s, 2H) 6.99 (m, 2H) 7.53 (m, IH). LC/MS, t
r = 1.99 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O
0C), ES-MS m/z 433 (M+l).
Step 2. Preparation of N-[3-({5-bromo-4-r(2,4-difluorobenzyl)oxyl-l-isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl I amino)propyll acetamide. [0208] 6-(2,4-difluorobenzyloxy)-2-(3-aminopropylamino)-5-bromo-3- isopropylpyrimidin-4(3H)-one trifluoroacetic acid salt (from step 1) (0.4 g, 0.76 mmol) was dissolved in dichloromethane (4 mL) and cooled in an ice bath. Triethylamine (0.3 mL, 2.3 mmol) was added. A solution of acetyl chloride (0.07 mL, 0.84 mmol) in dichloromethane (1 mL), pre-cooled in an ice bath, was added to the reaction mixture. The cooling was removed and the reaction mixture was warmed to room temperature over 1 hour. The reaction was quenched by the addition of solution of NaHCO3. The aqueous layer was extracted into dichloromethane. The combined organics were dried over MgSO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, hexanes/ethyl acetate with 10% methanol) to afford a white solid (0.092 g, 26%). 1H NMR (400 MHz, CD3OD) 6 ppm 1.53 (d, J = 6.98 Hz, 6H) 1.93 (s, 3H) 3.20 (t, J = 6.71 Hz, 2H) 3.46 (t, J = 6.71 Hz, 2H) 4.76 (m, IH) 5.42 (s, 2H) 6.98 (m, 2H) 7.49 (m, IH), ES-HRMS m/z 473.0986 (M+H calcd for C19H24BrF2N4O3 requires 473.0904).
iV-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]urea
[0209] Preparation of N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } amino)propyl]urea. 6-(2,4-difluorobenzyloxy)-2-(3- aminopropylamino)-5-bromo-3-isopropylpyrimidin-4(3H)-one trifluoroacetic acid salt (from Example 33, step 1) (0.40 g, 0.76 mmol) was dissolved in dichloromethane (4 mL) followed by triethylamine (0.3 mL, 2.3 mmol) addition at room temperature. Trimethylsilyl isocyanate (0.1 mL, 0.84 mmol) in dichloromethane (1 mL) was added to the reaction mixture. The reaction mixture was stirred overnight at room temperature. The resulting precipitate was filtered then washed with ether and dried on a vacuum pump to afford a white solid (0.175 g, 49%). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.43 (d, J = 6.71 Hz, 6H) 1.57 (m, 2H) 3.00 (m, 2H) 3.35 (m, 2H) 4.78 (m, IH) 5.35 (s, 2H) 7.12 (m, IH) 7.28 (m, IH) 7.52 (m, IH), ES-HRMS 474.0918 (M+H calcd for Ci8H23BrF2N5O3 requires 474.0947).
iV-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyI]methane sulfonamide
[0210] Preparation of N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6- oxo- 1 ,6-dihydropyrimidin-2-yl } amino)propyl]methane sulfonamid. Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]acetamide (step 2) utilizing 6-(2,4- difluorobenzyloxy)-2-(3-aminopropylamino)-5-bromo-3-isopropylpyrimidin-4(3H)-one trifluoroacetic acid salt (N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}amino)propyl]acetamide, step 1) (0.399 g, 0.76 mmol) and methanesulfonyl chloride (0.065 mL, 0.83 mmol) to afford a white solid (0.123 g, 32%). 1H NMR (400 MHz, CD3OD) δ ppm 1.51 (d, J = 6.98 Hz, 6H) 1.83 (m, 2H) 2.91 (s, 3H) 3.11 (t, J = 6.71 Hz, 2H) 3.54 (t, J = 6.71 Hz, 2H) 4.74 (m, IH) 5.43 (s, 2H) 6.98 (m, 2H) 7.50 (m, IH), ES-HRMS 509.0655 (M+H calcd for C18H24BrF2N4O4S requires 509.0664).
Example 36
N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxy-2-methyIpropanamide
Step 1. Preparation of 2-(3-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihvdro-l- isopropyl-6-oxopyrimidin-2-ylamino)propylcarbamoyl)propan-2-yl acetate.
[0211] Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]acetamide (step 2) utilizing 6-(2,4- difluorobenzyloxy)-2-(3-aminopropylamino)-5-bromo-3-isopropylpyrimidin-4(3H)-one trifluoroacetic acid salt (N-[3-({5-bromo-4-[(2,4-difIuorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}amino)propyl]acetamide, step 1) (0.399 g, 0.76 mmol) and 2- (chlorocarbonyl)propan-2-yl acetate (0.14 mL, 0.84 mmol) to afford a white solid (0.234
g, 55%). LC/MS, tr = 2.89 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mlVmin, at 254 nm, at 5O0C), ES-MS m/z 561 (M+l).
Step 2. Preparation of N-r3-({5-bromo-4-r(2,4-difluorobenzyl)oxyl-l-isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl ) amino)propyπ-2-hvdroxy-2-methylpropanamide. [0212] 2-(3-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihydro-l-isopropyl-6- oxopyrimidin-2-ylamino)propylcarbamoyl)propan-2-yl acetate (from step 1) (0.234 g, 0.42 mmol) was dissolved in methanol /water (3 mL) and K2CO3 (0.087 g, 0.63 mmol) was added. The reaction mixture was stirred at room temperature overnight. The solvent was removed and the residue was dissolved in ethyl acetate and washed with water.
The aqueous layer was neutralized (pH 7) with 1.0 M HCl. The aqueous layer was extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated to afford an off white solid (0.193 g, 89%). 1H ΝMR (400 MHz, CD3OD) 5 ppm 1.35 (s, 6H) 1.53 (d, J = 6.98 Hz, 6H) 1.76 (m, 2H) 3.25 (t, J = 6.71 Hz, 2H) 3.46 (t, J = 6.58 Hz, 2H) 4.85 (m, IH) 5.42 (s, 2H) 6.98 (m, 2H) 7.49 (m, IH), ES- HRMS 517.1256 (M+H calcd for C2JH28BrF2N4O4 requires 517.1256).
Example 37
N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxyacetainide
Step 1. Preparation of (3-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihvdro-l-isopropyl- 6-oxopyrimidin-2-ylamino)propylcarbamoyl)methyl acetate.
[0213] Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}amino)propyl]acetamide (step 2) utilizing 6-(2,4- difluorobenzyloxy)-2-(3-aminopropylamino)-5-bromo-3-isopropylpyrimidin-4(3H)-one trifluoroacetic acid salt (N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}amino)propyl]acetamide, step 1) (0.405 g, 0.77 mmol) and (chlorocarbonyl)methyl acetate (0.091 mL, 0.85 mmol) to afford a white solid (0.112 g, 27%). LC/MS, tr = 2.60 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mlVmin, at 254 run, at 5O0C), ES-MS m/z 533 (M+l).
Step 2. Preparation of N-[3-({5-bromo-4-f(2,4-difluorobenzyl)oxy1-l-isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl ) amino)propyll-2-hvdroxyacetamide.
[0214] Prepared as iV-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}amino)propyl]-2-hydroxy-2-methylpropanamide (step 3) utilizing (3-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihydro-l-isopropyl-6- oxopyrimidin-2-ylamino)propylcarbamoyl)methyl acetate (from step 1) (0.112 g, 0.21 mmol) and K2CO3 (0.045 g, 0.32 mmol) to afford a white solid (0.022 g, 21%). 1H NMR (400 MHz, CD3OD) δ ppm 1.53 (d, J = 6.98 Hz, 6H) 1.77 (m, 2H) 3.30 (t, J = 6.58 Hz, 2H) 3.47 (t, J = 6.58 Hz, 2H) 3.96 (s, 2H) 4.89 (m, IH) 5.42 (s, 2H) 6.97 (m, 2H) 7.49 (m, IH), ES-HRMS 489.0934 (M+H calcd for C19H24BrF2N4O4 requires 489.0943).
Example 38
6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-phenylpyrimidin-4(3H)-one
[0215] Preparation of 6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-phenylpyrimidin- 4(3H)-one. 6-(2,4-difluorobenzyloxy)-3-isopropyl-2-(methythio)pyrimidin-4(3H)-one (0.10 g, 0.31 mmol), phenyl bornic acid (0.063 g, 0.52 mmol), 2-hydroxy-3- methylbenzoic acid copper salt (0.16 g, 0.68 mmol), and palladium(O) tetrakis(triphenylphosphine) (0.036 g, 0.031 mmol) were added to a dry flask and evacuated followed by purging with N2. This process was repeated three times. Dioxane (2 mL) was added and the reaction mixture was heated to 80°C for 15 hours. The reaction mixture was cooled and the residue was dissolved in ethyl acetate, washed with brine. The aqueous layer was extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, hexanes/ethyl acetate) to afford a clear oil (0.096 g, 86%). 1H NMR (400 MHz, CD3OD) δ ppm 1.51 (d, J = 6.98 Hz, 6H) 4.35 (m, IH) 5.27 (s, 2H) 5.72 (s, IH) 6.98 (s, 2H) 7.53 (m, 6H). LC/MS, tr = 3.09 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 500C), ES-MS m/z 357 (M+l).
5-bromo-6-[(2,4-difluorobenzyI)oxy]-3-isopropyl-2-phenylpyrimidin-4(3H)-one
[0216] Preparation of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- phenylpyrimidin-4(3H)-one. 6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- phenylpyrimidin-4(3H)-one (0.040 g, 0.112 mmol) was dissolved in acetonitrile (1 mL) and cooled in an ice bath. N-Bromosuccinimide (0.022 g, 0.123 mmol) was added slowly and the reaction mixture was stirred for 1 hour with cooling. The reaction was concentrated and dissolved in ethyl acetate then washed with water. The aqueous layer was extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated. The residue was dissolved in 30% ethyl acetate/hex anes and passed through a pad of silica gel to afford a yellow colored oil (0.045 g, 92%). 1H NMR (400 MHz, CD3OD) δ ppm 1.53 (d, J = 6.71 Hz, 6H) 4.40 (m, IH) 5.43 (s, 2H) 6.94 (m, 2H) 7.55 (m, 6H), ES-HRMS 435.0530 (M+H calcd for C20H18BrF2N2O2 requires 435.0514).
Example 40
methyl 3-{4-[(2,4-difluorobenzyI)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- : yljbenzoate
[0217] Preparation of methyl 3-{4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}benzoate. Prepared as 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3- isopropyl-2-phenylpyrimidin-4(3H)-one utilizing 3-(methoxycarbonyl)phenylboronic acid (0.936 g, 5.2 mmol) in place of phenyl bornic acid, to afford an off white solid (0.923 g, 72%). 1H NMR (400 MHz, CD3OD) 5 ppm 1.52 (d, J = 6.71 Hz, 6H) 4.30 (m, IH) 4.84 (s, 3H) 5.28 (s, 2H) 5.74 (s, IH) 6.94 (m, 2H) 7.49 (m, IH) 7.68 (t, J = 8.06 Hz, IH) 7.78 (ddd, J = 1.34, 1.48, 7.92 Hz, IH) 8.15 (m, IH) 8.20 (dt, J = 1.48, 7.79 Hz, IH), ES-HRMS 415.1470 (M+H calcd for C22H2IF2N2O4 requires 415.1464).
4-{4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yljbenzamide
[0218] Preparation of 4-{4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}benzamide. Prepared as 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3- isopropyl-2-phenylpyrimidin-4(3H)-one utilizing 4-carbamoylphenylboronic acid (0.264 g, 1.6 mmol) in place of phenyl bornic acid, to afford a light green solid (0.254 g, 69%). 1H NMR (400 MHz, CD3OD) 5 ppm 1.52 (d, J = 6.71 Hz, 6H) 4.29 (m, IH) 5.28 (s, 2H)
5.74 (s, IH) 6.96 (m, 2H) 7.49 (m, IH) 7.63 (d, J = 8.59 Hz, 2H) 8.03 (d, J = 8.59 Hz, 2H), ES-HRMS 400.1480 (M+H calcd for C2iH20F2N3O3 requires 400.1467).
5-chloro-6-[(2,4-difluorobenzyI)oxy]-3-isopropyl-2-phenylpyrimidin-4(3H)-one
[0219] Preparation of 5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- phenylpyrimidin-4(3H)-one. 6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- phenylpyrimidin-4(3H)-one (0.318 g, 0.89 mmol) was dissolved in acetonitrile (3 mL) and cooled in an ice bath. JV-Chlorosuccinimide (0.13 g, 0.98 mmol) was added slowly and the reaction mixture was stirred for 1 hour with cooling. Dichloroacetic acid (cat.) was added and the reaction mixture was warmed to room temperature overnight. The reaction mixture was concentrated and the residue was dissolved in ethyl acetate then washed with water. The aqueous layer was extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, ethyl acetate/hexanes) to afford a white solid (0.204 g, 59%). 1H NMR (400 MHz, CD3OD) δ ppm 1.53 (d, J = 6.71 Hz, 6H) 4.40 (m, IH) 5.44 (s, 2H) 6.95 (m, 2H) 7.55 (m, 6H), ES-HRMS 391.1034 (M+H calcd for C20Hi8ClF2N2O2 requires 391.1019).
4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yljbenzamide
[0220] Preparation of 4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}benzamide. Prepared as 5-chloro-6-[(2,4-difluorobenzyl)oxy]-3- isopropyl-2-phenylpyrimidin-4(3H)-one utilizing 4-{4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}benzamide (0.102 g, 0.26 mmol) and N- chlorosuccinimide (0.039 g, 0.29 mmol) to afford a white solid (0.051 g, 46%). 1H NMR (400 MHz, CD3OD) δ ppm 1.54 (d, J = 6.71 Hz, 6H) 4.34 (m, IH) 5.44 (s, 2H) 6.94 (m, 2H) 7.49 (m, IH) 7.64 (d, J = 8.86 Hz, 2H) 8.04 (d, J = 8.59 Hz, 2H), ES-HRMS 434.1059 (M+H calcd for C21H19ClF2N3O3 requires 434.1078).
4-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yljbenzamide
[0221] Preparation of 4- {5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- 1,6- dihydropyrimidin-2-yl}benzamide. Prepared as 5-bromo-6-[(2,4-difluorobenzyl)oxy]-3- isopropyl-2-phenylpyrimidin-4(3H)-one utilizing 4-{4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}benzamide (0.076 g, 0.19 mmol) and N- bromosuccinimide (0.038 g, 0.21 mmol) to afford a light brown solid (0.065 g, 74%). 1H NMR (400 MHz, CD3OD) δ ppm 1.54 (d, J = 6.71 Hz, 6H) 4.34 (m, IH) 5.44 (s, 2H) 6.96 (m, 2H) 7.49 (m, IH) 7.65 (d, J = 8.59 Hz, 2H) 8.05 (d, J = 8.59 Hz, 2H), ES- HRMS 478.0541 (M+H calcd for C21H19BrF2N3O3 requires 478.0572).
3-{4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}benzoic acid
[0222] Preparation of 3-{4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}benzoic acid. Methyl 3-{4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}benzoate (0.87 g, 2.1 mmol) was dissolved in tetrahydrofuran (20 mL) and 2.5 N NaOH (8 mL, 20.0 mmol) was added. The reaction mixture was stirred at room temperature overnight. The solvent was removed and the aqueous layer was neutralized (pH 7) with 1.0 M HCl. The aqueous layer was extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated to afford an off white solid (0.84 g, 100%). 1H NMR (400 MHz,
CD3OD) δ ppm 1.53 (d, J = 6.44 Hz, 6H) 4.32 (m, IH) 5.29 (s, 2H) 5.73 (s, IH) 6.96 (m, 2H) 7.57 (m, 2H) 7.72 (m, IH) 8.14 (m, 2H). LC/MS, tr = 2.60 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS m/z 401 (M+l).
3-{4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yljbenzamide
[0223] Preparation of 3-{4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } benzamide. 3- { 4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl} benzoic acid (0.840 g, 2.1 mmol) was dissolved in tetrahydrofuran (7 mL). 2-chloro-4,6-dimethoxy-l,3,5-triazine (0.444 g, 2.5 mmol) was added followed by 4-methylmorpholine (0.7 mL, 6.1 mmol). The reaction was stirred at room temperature for 1.5 hours. An excess of aqueous ammonium hydroxide was added and the resulting mixture was stirred at room temperature overnight. The reaction was concentrated and the residue was dissolved in ethyl acetate then washed with water. The aqueous layer was extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, ethyl acetate with 10% methanol/hexanes to afford a white solid (0.56 g, 67%). 1H NMR (400 MHz, CDCl3) δ ppm 1.53 (d, J = 6.71 Hz, 6H) 4.26 (m, IH) 5.17 (s, 2H) 5.73 (s, IH) 6.82 (m, 2H) 7.39 (m, IH) 7.58 (m, 2H) 7.96 (m, 2H), ES-HRMS 400.1444 (M+H calcd for C2]H20F2N3O3 requires 400.1467).
3-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yljbenzamide
[0224] Preparation of 3-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6- oxo-l,6-dihydropyrimidin-2-yl}benzamide. Prepared as 5-bromo-6-[(2,4- difluorobenzyl)oxy]-3-isopropyl-2-phenylpyrimidin-4(3H)-one (step 2) utilizing 3-{4- [(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}benzamide (0.200 g, 0.50 mmol) and N-bromosuccinimide (0.098 g, 0.55 mmol) to afford a white solid (0.188 g, 79%). 1H NMR (400 MHz, CD3OD) 6 ppm 1.55 (d, J = 6.98 Hz, 6H) 4.36 (m, IH) 5.44 (s, 2H) 6.96 (m, 2H) 7.52 (m, IH) 7.67 (m, IH) 7.74 (m, IH) 8.04 (m, IH) 8.09 (m, IH), ES-HRMS 478.0573 (M+H calcd for C21Hi9BrF2N3O3 requires 478.0572).
3-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yljbenzamide
[0225] Preparation of 3-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}benzamide. Prepared as 5-chloro-6-[(2,4-difluorobenzyl)oxy]-3- isopropyl-2-phenylpyrimidin-4(3H)-one utilizing 3- { 4-[(2,4-difluorobenzyl)oxy]- 1 - isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}benzamide (0.200 g, 0.50 mmol) and N- chlorosuccinimide (0.077 g, 0.55 mmol) to afford a white solid (0.133 g, 61%). 1H NMR (400 MHz, CD3OD) δ ppm 1.55 (d, J = 6.98 Hz, 6H) 4.36 (m, IH) 5.45 (s, 2H) 6.96 (m, 2H) 7.51 (m, IH) 7.67 (m, 2H) 8.04 (m, 2H), ES-HRMS 434.1081 (M+H calcd for C21H19ClF2N3O3 requires 434.1078).
Example 48
N~3~-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yI}-beta-alaninamide
Step 1. Preparation of ethyl 3-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihydro-l- isopropyl-6-oxopyrimidin-2-ylamino)propanoate.
[0226] Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]acetamide (step 1) utilizing ethyl 3- aminopropanoate (0.388 g, 2.52 mmol) in place of propane- 1,3 -diamine to afford a clear oil (0.47 g, 43%). 1H NMR (400 MHz, CD3OD) δ ppm 1.21 (t, J = 7.12 Hz, 3H) 1.50 (d, J = 6.98 Hz, 6H) 2.59 (t, J = 6.71 Hz, 2H) 3.69 (t, J = 6.85 Hz, 2H) 4.11 (q, J = 7.25 Hz, 2H) 4.79 (m, IH) 5.42 (s, 2H) 6.97 (m, 2H) 7.50 (m, IH). LC/MS, tr = 3.07 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 ran, at 5O0C), ES-MS m/z AlA (M+l).
Step 2. Preparation of 3-(4-(2,4-difIuorobenzyIoxy)-5-bromo-l,6-dihydro-l- isopropyl-6-oxopyrimidin-2-ylamino)propanoic acid.
[0227] Prepared as 3-{4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl} benzoic acid utilizing ethyl 3-(4-(2,4-difluorobenzyloxy)-5- bromo-l,6-dihydro-l-isopropyl-6-oxopyrimidin-2-ylamino)propanoate (from step 1)
(0.47 g, 0.99 mmol) and 2.5 N NaOH (2.0 mL, 4.95 mmol) to afford an off white solid
(0.13 g, 30%). 1H NMR (400 MHz, CD3OD) 5 ppm 1.50 (d, J = 6.98 Hz, 6H) 2.57 (t, J = 6.71 Hz, 2H) 3.69 (t, J = 6.71 Hz, 2H) 4.90 (m, IH) 5.43 (s, 2H) 6.97 (m, 2H) 7.51 (m, IH). LC/MS, tr = 2.49 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nra, at 5O0C), ES-MS m/z 446 (M+l).
Step 3. Preparation of iV~3~- {5-bromo-4-r(2,4-difluorobenzv0oxyl-l-isopropyl-6-oxo- L6-dihvdropyrimidin-2-yl)-beta-alaninamide.
[0228] 3-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihydro-l-isopropyl-6-oxopyrimidin- 2-ylamino)propanoic acid (from step 2) (0.13 g, 0.29 mmol) was dissolved in tetrahydrofuran (1 mL). 2-chloro-4,6-dimethoxy-l,3,5-triazine (0.062 g, 0.35 mmol) was added followed by 4-methylmorpholine (0.1 mL, 0.87 mmol). The reaction was stirred at room temperature for 1.5 hours. An excess of aqueous ammonium hydroxide was added and the resulting mixture was stirred at room temperature overnight. The reaction was concentrated and the residue was dissolved in ethyl acetate then washed with water. The aqueous layer was extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, ethyl acetate with 10% methanol /hexanes) to afford a white solid (0.046 g, 36%). 1H NMR (400 MHz, CD3OD) δ ppm 1.50 (d, J = 6.98 Hz, 6H) 2.54 (t, J = 6.58 Hz, 2H) 3.70 (t, J = 6.58 Hz, 2H) 4.79 (m, IH) 5.43 (s, 2H) 6.98 (m, 2H) 7.54 (m, IH), ES-HRMS 445.0651 (M+H calcd for C17H20BrF2N4O3 requires 445.0681).
Example 49
4-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isoprop
yl-6-oxo-l,6-dihydropyrimidin-2- yl}amino)butanamide
Step 1. Preparation of ethyl 4-(4-(2.4-difluorobenzyloxy)-5-bromo-l,6-dihydro-l- isopropyl-6-oxopyrimidin-2-ylamino')butanoate.
[0229] Prepared as N~3~-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}-beta-alaninamide (step 1) utilizing ethyl 4-aminobutanoate (0.436 g, 2.59 mmol) in place of propane- 1,3 -diamine to afford a clear oil (0.291 g, 25%). 1H NMR (400 MHz, CD3OD) δ ppm 1.21 (t, J = 7.12 Hz, 3H) 1.51 (d, J = 6.98 Hz, 6H) 1.88 (m, 2H) 2.35 (t, J = 7.12 Hz, 2H) 3.46 (t, J = 6.98 Hz, 2H) 4.08 (q, J = 7.25 Hz, 2H) 4.70 (m, IH) 5.43 (s, 2H) 6.98 (m, 2H) 7.50 (m, IH). LC/MS, tr = 3.15 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS m/z 488 (M+ 1).
Step 2. Preparation of 4-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihydro-l-isopropyl-6- oxopyrimidin-2-ylamino)butanoic acid.
Prepared as N~3~-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}-beta-alaninamide (step 2) utilizing ethyl 4-(4-(2,4- difluorobenzyloxy)-5 -bromo- 1 ,6-dihydro- 1 -isopropyl-6-oxopyrimidin-2- ylamino)butanoate (from step 1) (0.291 g, 0.60 mmol) and 2.5 N NaOH (2.0 mL, 4.8 mmol) to afford a clear oil (0.160 g, 25%). 1H NMR (400 MHz, CD3OD) 8 ppm 1.51 (d, J = 6.98 Hz, 6H) 1.88 (m, 2H) 2.33 (t, J = 7.12 Hz, 2H) 3.47 (t, J = 6.98 Hz, 2H) 4.72 (m, IH) 5.43 (s, 2H) 6.97 (m, 2H) 7.49 (m, IH). LC/MS, t, = 2.56 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS mJz 460 (M+l).
Step 3. Preparation of 4-((5-bromo-4-[(2,4-difluorobenzyl)oxyl-l-isopropyl-6-oxo-l,6- dihvdropyrimidin-2-yl ) amino)butanamide.
[0230] Prepared as _V~3~- {5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}-beta-alaninamide (step 3) utilizing 4-(4-(2,4-difluorobenzyloxy)- 5-bromo-l,6-dihydro-l-isopropyl-6-oxopyrimidin-2-ylamino)butanoic acid (from step 2) (0.160 g, 0.35 mmol), 2-chloro-4,6-dimethoxy-l,3,5-triazine (0.075 g, 0.42 mmol), 4- methylmorpholine (0.115 mL, 1.05 mmol) and an excess ammonium hydroxide to afford a white solid (0.042 g, 26%). 1H NMR (400 MHz, CD3OD) δ ppm 1.52 (d, J = 6.98 Hz, 6H) 1.90 (m, 2H) 2.27 (t, J = 7.25 Hz, 2H) 3.47 (t, J = 6.85 Hz, 2H) 4.72 (m, IH) 5.43 (s, 2H) 6.98 (m, 2H) 7.51 (m, IH), ES-HRMS 459.0841 (M+H calcd for Ci8H22BrF2N4O3 requires 459.0838).
Example 50
tert-butyl 2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)ethyIcarbamate
[0231] Preparation of tert-butyl 2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6- oxo-l,6-dihydropyrimidin-2-yl}oxy)ethylcarbamate. t-Butyl 2-hydroxyethylcarbamate (2.2 mL, 14.0 mmol) was dissolved in dioxane (100 mL) and cooled in an ice bath. Sodium hydride (0.770 g, 19.1 mmol) was added portion wise to the reaction mixture. The reaction mixture was warmed to room temperature over 1 hour then re-cooled in an ice bath. A solution of 5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)pyrimidin-4(3H)-one (5.0 g, 12.8 mmol) in dioxane (30 mL) was slowly added to the reaction mixture. The reaction mixture was allowed to warm to room temperature overnight. The reaction was quenched by the addition of water and the solvent was removed. The aqueous layer was extracted into ethyl acetate, dried over MgSO4, filtered, and concentrated. The residue was dissolved in 50% ethyl acetate/hexanes and passed through a pad of silica gel to afford a white solid (4.92g, 81%). 1H NMR (400 MHz, CD3OD) δ ppm 1.41 (s, 9H) 1.45 (d, J = 6.71 Hz, 6H) 3.49 (m, 2H) 4.51 (t, J = 5.10 Hz, 2H) 5.28 (m, IH) 5.47 (s, 2H) 6.98 (m, 2H) 7.53 (m, IH). LC/MS, tr = 3.30 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS m/z 474 (M+l).
Example 51
2-{[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)ethyl]amino}-2-oxoethyl acetate
Step 1. Preparation of 6-(2,4-difluorobenzyloxy)-2-(3-aminopropoxy)-5-chloro-3- isopropylpyrimidin-4(3H)-one trifluoroacetic acid.
[0232] Tert-butyl 2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)ethylcarbamate (9.4 g, 10.4 mmol) was dissolved in dichloromethane (10 mL) and trifluoroacetic acid (10 mL) was added. The reaction mixture was stirred at room temperature for 1 hour. The reaction was then concentrated and the residue was dissolved in ethyl acetate then washed with a solution of NaHCO3. The combined organics were dried over MgSO4, filtered and concentrated to afford yellow colored foam (4.58 g, 90%). 1H NMR (400 MHz, CD3OD) δ ppm 1.49 (d, J = 6.98 Hz, 6H) 3.45 (m, 2H) 4.72 (m, 2H) 5.22 (m, IH) 5.48 (s, 2H) 7.01 (m, 2H) 7.54 (m,
IH). LC/MS, tr = 1.94 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 ran, at 5O0C), ES-MS m/z 374 (M+l).
Step 2. Preparation of 2-{ r2-((5-chloro-4-r(2,4-difluorobenzyl)oxy1-l-isoρropyl-6-oxo- 1 ,6-dihvdropyrimidin-2-yl |oxy)ethyllamino ) -2-oxoethyl acetate.
[0233] Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6- oxo-l,6-dihydropyrimidin-2-yl}amino)propyl]-2-hydroxyacetamide (step 2) utilizing 2- { [2-( { 5-chloro-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}oxy)ethyl] amino }-2-oxoethyl acetate and (chlorocarbonyl)methyl acetate (0.121 mL, 1.13 mmol) to afford a white solid (0.326 g, 67%). 1H NMR (400 MHz, CDCl3) δ ppm 1.42 (d, J = 6.98 Hz, 6H) 2.12 (s, 3H) 3.74 (m, 2H) 4.51 (t, J = 5.50 Hz, 2H) 4.57 (s, 2H) 5.36 (m, IH) 5.39 (s, 2H) 6.83 (m, 2H) 7.44 (m, IH). LC/MS, tr = 2.58 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS m/z 474 (M+l).
Example 52
N-[2-({5-chloro-4-[(2,4-dIfluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)ethyl]acetainide
[0234] Preparation of N-[2-({ 5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}oxy)ethyl]acetamide. Prepared as Λf-[3-({5-bromo-4-[(2,4- difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}amino)propyl]acetamide (step 2) utilizing 6-(2,4-difluorobenzyloxy)-2-(3- aminopropoxy)-5-chloro-3-isopropylpyrimidin-4(3H)-one trifluoroacetic acid (from 2-
{ [2-( { 5-chloro-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}oxy)ethyl] amino }-2-oxoethyl acetate, step 1) (0.497 g, 1.02 mmol) and acetyl chloride (0.119 mL, 1.53 mmol) to afford a light red solid (0.312 g, 74%). 1H NMR (400 tøHz, CDCl3) δ ppm 1.40 (d, J = 6.98 Hz, 6H) 2.02 (s, 3H) 3.68 (m, 2H) 4.50 (t, / = 5.50 Hz, 2H) 5.32 (m, IH) 5.38 (s, 2H) 6.84 (m, 2H) 7.44 (m, IH), ES-HRMS 416.1175 (M+H calcd for C18H2IClF2N3O4 requires 416.1183).
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)ethyl]methanesulfonamide
[0235] Preparation of N-[2-({ 5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}oxy)ethyl]methanesulfonamide. Prepared as iV-[3-({5- bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}amino)propyl]acetamide (step 2) utilizing 6-(2,4-difluorobenzyloxy)-2-(3- aminopropoxy)-5-chloro-3-isopropylpyrimidin-4(3H)-one trifluoroacetic acid (from 2- { [2-( { 5-chloro-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}oxy)ethyl]amino}-2-oxoethyl acetate, step 1) (0.510 g, 1.05 mmol) and methanesulfonyl chloride (0.089 mL, 1.16 mmol) to afford a white solid (0.369 g, 78%). 1H NMR (400 MHz, CDCl3) δ ppm 1.43 (d, J = 6.98 Hz, 6H) 2.97 (s, 3H) 3.54 (m, 2H) 4.54 (t, J = 5.50 Hz, 2H) 5.31 (m, IH) 5.39 (s, 2H) 6.82 (m, 2H) 7.45 (m, IH), ES- HRMS 452.0846 (M+H calcd for Ci7H2iClF2N3O5S requires 452.0853).
Example 54
2-{[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)ethyl]amino}-l,l-dimethyl-2-oxoethyl acetate
[0236] Preparation of 2-{ [2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-
6-oxo- 1 ,6-dihydropyrimidin-2-yl } oxy)ethyl] amino } - 1 , 1 -dimethyl-2-oxoethyl acetate. Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxy-2-methylpropanamide (step 2) utilizing 6-(2,4-difluorobenzyloxy)-2-(3-aminopropoxy)-5-chloro-3-isopropylpyrimidin-4(3H)- one trifluoroacetic acid (from 2-{ [2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl- 6-oxo-l,6-dihydropyrimidin-2-yl}oxy)ethyl] amino }-2-oxoethyl acetate, step 1) (0.502 g, 1.03 mmol) and 2-(chlorocarbonyl)propan-2-yl acetate (0.185 mL, 1.13 mmol) to afford a white solid (0.364 g, 71%). 1H NMR (400 MHz, CDCl3) δ ppm 1.42 (d, J = 6.98 Hz, 6H) 1.61 (s, 6H) 2.03 (s, 3H) 3.69 (m, 2H) 4.50 (t, / = 5.37 Hz, 2H) 5.34 (m, IH) 5.39 (s, 2H) 6.82 (m, 2H) 7.44 (m, IH). LC/MS, tr = 2.87 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS m/z 502 (M+l).
Example 55
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)ethyl]urea
[0237] Preparation of N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-
6-oxo- 1 ,6-dihydropyrimidin-2-yl } oxy)ethyl]urea. Prepared as N-[3-( { 5-bromo-4-[(2,4- difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } amino)propyl]urea utilizing 6-(2,4-difluorobenzyloxy)-2-(3-aminopropoxy)-5-chloro-3-isopropylpyrimidin- 4(3H)-one trifluoroacetic acid (from 2-{[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } oxy)ethyl] amino } -2-oxoethyl acetate, step 1) (0.510 g, 1.05 mmol) and trimethylsilyl isocyanate (0.132 mL, 1.15 mmol) to afford a white solid (0.105 g, 24%). 1H NMR (400 MHz, CD3OD) δ ppm 1.46 (d, J = 6.98 Hz, 6H) 3.55 (t, J = 5.37 Hz, 2H) 4.53 (t, J = 5.37 Hz, 2H) 5.29 (m, IH) 5.47 (s, 2H) 6.99 (m, 2H) 7.54 (m, IH), ES-HRMS 417.1145 (M+H calcd for C17H20ClF2N4O4 requires 417.1136).
Example 56
(3S)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}pyrrolidin-3-ylcarbamate
[0238] Preparation of (35)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-
6-oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-ylcarbamate. 5-chloro-6-[(2,4- difluorobenzyl)oxy]-3-isopropyl-2-(methylsulfonyl)pyrimidin-4(3H)-one (1.0 g, 2.55 mmol) was dissolved in dioxane (25 mL). t-Butyl (S)-pyrrolidin-3-ylcarbamate (0.57 g, 3.06 mmol) was added followed by triethylamine (0.7 mL, 5.1 mmol). The resulting mixture was stirred at room temperature for 2 hours. The reaction was concentrated and the residue was washed with ethyl acetate/water. The aqueous layer was extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, ethyl acetate/hexanes) to afford an off white solid (0.810 g, 64%). 1H NMR (300 MHz, CD3OD) δ ppm 1.44 (m, 9H) 1.59 (dd, J = 6.65, 12.89 Hz, 6H) 1.85 (m, 2H) 3.21 (m, IH) 3.41 (m, IH) 3.80 (m, IH) 4.12 (m, 2H) 4.44 (m, IH) 5.43 (m, 2H) 6.99 (m, 2H) 7.55 (m, IH). LC/MS, tr = 3.36 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mlVmin, at 254 ran, at 5O0C), ES-MS m/z 499 (M+l).
Example 57
(3R)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}pyrrolidin-3-ylcarbamate
[0239] Preparation of (3/?)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-
6-oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-ylcarbamate. Prepared as (3S)-I- {5-
chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}pyrrolidin-3-ylcarbamate utilizing t-butyl (/?)-pyrrolidin-3-ylcarbamate (2.84 g, 15.3 mmol) to afford an off white solid (4.10 g, 64%). 1H NMR (400 MHz, CD3OD) δ ppm lj.44 (s, 9H) 1.59 (dd, J = 6.71, 17.45 Hz, 6H) 2.14 (m, 2H) 3.42 (m, IH) 3.60 (m, IH) 3.76 (m, 2H) 4.14 (m, IH) 4.43 (m, IH) 5.42 (s, 2H) 6.97 (m, 2H) 7.50 (m, IH). LC/MS, tr = 3.36 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS m/z 499 (M+l).
Example 58
2-[(35)-3-aminopyrrolidin-l-yl]-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3- isopropyIpyrimidin-4(3H)-one
[0240] Preparation of 2-[(3S)-3-aminopyrrolidin-l-yl]-5-chloro-6-[(2,4- difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one. (3S)-l-{5-chloro-4-[(2,4- difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3- ylcarbamate (3.42 g, 6.87 mmol) was dissolved in dichloromethane (14 mL) and trifluoroacetic acid (14 mL) was added and the resulting mixture was stirred at room temperature for 1 hour. The reaction was then concentrated and placed in a vacuum oven overnight to afford a brown solid (3.65 g, > 100%). 1H NMR (400 MHz, CD3OD) 5 ppm 1.60 (dd, J = 23.36, 6.71 Hz, 6H) 2.16 (m, IH) 2.42 (m, IH) 3.66 (m, 2H) 3.92 (m, 3H) 4.50 (m, IH) 5.43 (s, 2H) 6.98 (m, 2H) 7.51 (m, IH), ES-HRMS 399.1390 (M+H calcd for Ci8H22ClF2N4O2 requires 390.1394).
2-[(3/?)-3-aminopyrrolidin-l-yI]-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3- isopropylpyrimidin-4(3H)-one
[0241] Preparation of 2-[(3/?)-3-aminopyrrolidin-l-yl]-5-chloro-6-[(2,4- difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one. Prepared as 2-[(35)-3- ' aminopyrrolidin-l-yl]-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin- 4(3H)-one utilizing (3R)- 1 - { 5-chloro-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl}pyrrolidin-3-ylcarbamate (3.87 g, 7.77 mmol) and trifluoroacetic acid (15mL) to afford a red solid (3.82 g, 96%). 1H NMR (400 MHz, CD3OD) δ ppm 1.60 (dd, J = 22.42, 6.58 Hz, 6H) 2.15 (m, IH) 2.44 (m, IH) 3.67 (m, 2H) 3.82 (m, IH) 3.97 (m, 2H) 4.50 (s, IH) 5.43 (s, 2H) 6.97 (m, 2H) 7.51 (m, IH), ES-HRMS 399.1390 (M+H calcd for C]8H22ClF2N4O2 requires 390.1394).
N-((3S)-l-{5-chIoro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}pyrrolidin-3-yl)acetamide
[0242] Preparation of N-((3S)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6- oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-yl)acetamide. Prepared as N-[3-({5-bromo- 4,- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}amino)propyl]acetamide (step 2) utilizing 2-[(35)-3-aminopyrrolidin-l-yl]-5-chloro- 6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one (0.50 g, 0.98 mmol) and acetyl chloride (0.085 mL, 1.08 mmol) to afford a tan/red solid (0.273 g, 63%). 1H NMR (400 MHz, CD3OD) δ ppm 1.59 (dd, J = 6.58, 26.45 Hz, 6H) 1.94 (m, 4H) 2.19 (m, IH) 3.42 (dd, J = 3.76, 10.74 Hz, IH) 3.63 (m, IH) 3.75 (m, IH) 3.85 (dd, J = 5.91, 11.01 Hz, IH) 4.37 (m, IH) 4.44 (m, IH) 5.42 (s, 2H) 6.96 (m, 2H) 7.50 (m, IH), ES-HRMS 441.1475 (M+H calcd for C20H24ClF2N4O3 requires 441.1500).
N-((3R)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}pyrrolidin-3-yl)acetamide
[0243] Preparation of N-((3R)-l-{ 5-chloro-4-[(2,4-difluorobenzyl)oxy]- l-isopropyl-6- oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-yl)acetamide. Prepared as Λf-[3-({5-bromo- 4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}amino)propyl]acetamide (step 2) utilizing 2-[(3/?)-3-aminopyrrolidin-l-yl]-5-chloro- 6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one (0.50 g, 0.98 mmol) and acetyl chloride (0.085 mL, 1.08 mmol) to afford a tan solid (0.300 g, 70 %). 1H NMR (400 MHz, CD3OD) δ ppm 1.59 (dd, J = 6.71, 26.58 Hz, 6H) 1.94 (m, 4H) 2.19 (m, IH) 3.42 (dd, J = 3.76, 10.74 Hz, IH) 3.63 (m, IH) 3.75 (m, IH) 3.85 (dd, J = 5.91, 11.01 Hz, IH) 4.37 (m, IH) 4.43 (m, IH) 5.42 (s, 2H) 6.96 (m, 2H) 7.50 (m, IH), ES-HRMS 441.1464 (M+H calcd for C20H24ClF2N4O3 requires 441.1500).
2-[((3S)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yI}pyrrolidin-3-yl)amino]-2-oxoethyl acetate
[0244] Preparation of 2-[((3S)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-yl)amino]-2-oxoethyl acetate. Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxyacetamide (step 2) utilizing 2-[(35)-3- aminopyrrolidin-l-yl]-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin- 4(3H)-one (0.50 g, 0.98 mmol) and (chlorocarbonyl)methyl acetate (0.115 mL, 1.07 mmol) to afford a light yellow solid (0.292 g, 60 %). 1H NMR (400 MHz, CD3OD) δ ppm 1.59 (dd, J = 6.71, 20.68 Hz, 6H) 2.03 (m, IH) 2.11 (s, 3H) 2.21 (m, IH) 3.49 (dd, J = 4.16, 10.88 Hz, IH) 3.65 (m, IH) 3.75 (m, IH) 3.86 (dd, J = 5.91, 11.01 Hz, IH) 4.43 (m, 2H) 4.53 (s, 2H) 5.42 (s, 2H) 6.96 (m, 2H) 7.50 (m, IH). LC/MS, tr = 2.63 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS m/z 499 (M+l).
2-[((3R)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- ; dihydropyrimidin-2-yl}pyrrolidin-3-yI)amino]-2-oxoethyl acetate
[0245] Preparation of 2-[((3R)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-yl)amino]-2-oxoethyl acetate. Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxyacetamide (step 2) utilizing 2-[(3R)-3- aminopyrrolidin-l-yl]-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin- 4(3H)-one (0.50 g, 0.98 mmol) and (chlorocarbonyl)methyl acetate (0.115 mL, 1.07 mmol) to afford a light yellow solid (0.279 g, 57%). 1H NMR (400 MHz, CD3OD) δ ppm 1.59 (dd, J = 6.58, 20.81 Hz, 6H) 2.02 (m, IH) 2.11 (s, 3H) 2.21 (m, IH) 3.49 (dd, J = 4.16, 10.88 Hz, IH) 3.65 (m, IH) 3.75 (m, IH) 3.86 (dd, J = 5.91, 11.01 Hz, IH) 4.44 (m, 2H) 4.53 (s, 2H) 5.42 (s, 2H) 6.96 (m, 2H) 7.50 (m, IH). LC/MS, tr = 2.63 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mlVmin, at 254 nm, at 5O0C), ES-MS m/z 499 (M+l).
2-[((35)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}pyrrolidin-3-yl)amino]-l,l-dimethyl-2-oxoethyI acetate
[0246] Preparation of 2-[((3S)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } pyrrolidin-3-yl)amino]- 1 , 1 -dimethyl-2- oxoethyl acetate. Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-
6-oxo- 1 ,6-dihydropyrimidin-2-yl } amino)propyl]-2-hydroxy-2-methylpropanamide (step
2) utilizing 2-[(35)-3-aminopyrrolidin-l-yl]-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3- isopropylpyrimidin-4(3H)-one (0.50 g, 0.98 mmol) and 2-(chlorocarbonyl)propan-2-yl acetate (0.18 mL, 1.07 mmol) to afford a white solid (0.354 g, 69%). 1H NMR (400 MHz, CD3OD) δ ppm 1.59 (dd, J = 6.71, 30.88 Hz, 6H) 1.63 (d, J = 6.71 Hz, 3H) 1.98 (s, 3H) 2.10 (m, 2H) 3.51 (m, 2H) 3.78 (m, 2H) 4.38 (m, 2H) 5.42 (s, 2H) 6.98 (m, 2H) 7.51 (m, IH). LC/MS, tr = 2.87 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS m/z 527 (M+l).
Example 65
2-[((3R)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}pyrrolidin-3-yl)amino]-l,l-dimethyl-2-oxoethyl acetate
[0247] Preparation of 2-[((3/?)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6- oxo- 1 ,6-dihydropyrimidin-2-yl } pyrrolidin-3-yl)amino] -1,1 -dimethyl-2-oxoethyl acetate. Prepared as N- [3 -( { 5-bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxy-2-methylpropanamide (step 2) utilizing 2-[(3/?)-3-aminopyrrolidin-l-yl]-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3- isopropylpyrimidin-4(3H)-one (0.50 g, 0.98 mmol) and 2-(chlorocarbonyl)propan-2-yl acetate (0.18 mL, 1.07 mmol) to afford a white solid (0.405 g, 79%). 1H NMR (400 MHz, CD3OD) 6 ppm 1.59 (dd, J = 6.58, 31.02 Hz, 6H) 1.63 (d, J = 6.71 Hz, 3H) 1.98 (s, 3H) 2.00 (m, 2H) 3.53 (m, 2H) 3.77 (m, 2H) 4.41 (m, 2H) 5.42 (s, 2H) 6.97 (m, 2H) 7.51 (m, IH). LC/MS, tr = 2.86 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS m/z 527 (M+l).
Example 66
N-((3S)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}pyrrolidin-3-yl)-2-hydroxyacetamide
[0248] Preparation of N-((3S)- 1- { 5-chloro-4-[(2,4-difluorobenzyl)oxy]- l-isopropyl-6- oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-yl)-2-hydroxyacetamide. Prepared as N-[3- ({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}amino)propyl]-2-hydroxyacetamide (step 3) utilizing (0.228 g, 0.46 mmol) and K2CO3 (0.095 g, 0.69 mmol) to afford a white solid (0.183 g, 87%). 1H NMR (300 MHz, CD3OD) δ ppm 1.59 (d, J = 6.65 Hz, 6H) 2.10 (m, 2H) 3.69 (m, 4H) 3.99 (d, J = 1.21 Hz, 2H) 4.46 (m, 2H) 5.43 (s, 2H) 6.97 (m, 2H) 7.52 (m, IH), ES-HRMS 457.1404 (M+H calcd for C20H24ClF2N4O4 requires 457.1449).
Example 67
N-((3R)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}pyrrolidin-3-yl)-2-hydroxyacetamide
[0249] Preparation of N-((3/?)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6- oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-yl)-2-hydroxyacetamide. Prepared as iV-[3-
({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}amino)propyl]-2-hydroxyacetamide (step 3) utilizing 2-[((3/?)-l-{5-chloro-4-[(2,4- difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3- yl)amino]-2-oxoethyl acetate (0.217 g, 0.44 mmol) and K2CO3 (0.090 g, 0.65 mmol) to afford a white solid (0.194 g, 97%). 1H NMR (300 MHz, CD3OD) δ ppm 1.59 (d, / = 6.65 Hz, 6H) 2.22 (s, 2H) 3.73 (m, 4H) 3.98 (s, 2H) 4.46 (m, 2H) 5.43 (s, 2H) 6.98 (m, 2H) 7.51 (m, IH), ES-HRMS 457.1454 (M+H calcd for C20H24ClF2N4O4 requires 457.1449).
N-((3S)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}pyrrolidin-3-yl)-2-hydroxy-2-methylpropanamide
[0250] Preparation of N-((35)- 1 - { 5-chloro-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6- oxo-l,6-dihydropyrimidin-2-yl}pyrrolidin-3-yl)-2-hydroxy-2-methylpropanamide. Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxyacetamide (step 3) utilizing 2-[((3S)-I- { 5-chloro-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}pyrrolidin-3-yl)amino]-l,l-dimethyl-2-oxoethyl acetate (0.304 g, 0.58 mmol) and K2CO3 (0.120 g, 0.87 mmol) to afford a white solid (0.206 g, 73%). 1H NMR (300 MHz, CD3OD) δ ppm 1.35 (s, 3H) 1.36(s, 3H) 1.59 (dd, J = 3.52, 6.54 Hz, 6H) 2.00 (m, 2H) 3.69 (m, 4H) 4.47 (m, 2H) 5.44 (s, 2H) 6.98 (m, 2H) 7.51 (m, IH), ES-HRMS 485.1723 (M+H calcd for C22H28ClF2N4O4 requires 485.1762).
Example 69
iV-((3R)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}pyrrolidin-3-yl)-2-hydroxy-2-methylpropanamide
[0251] Preparation of N-((3/?)-l-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6- oxo-l,6-dihydropyrimidin-2-yl}pyπOlidin-3-yl)-2-hydroxy-2-methylpropanamide. Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxyacetamide (step 3) utilizing 2-[((3/?)-l- { 5-chloro-4-[(2,4-difluorobenzyl)oxy]- l-isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}pyrrolidin-3-yl)amino]-l,l-dimethyl-2-oxoethyl acetate (0.363 g, 0.69 mmol) and K2CO3 (0.143 g, 1.04 mmol) to afford a white solid (0.313 g, 94%). 1H NMR (300 MHz, CD3OD) δ ppm 1.35 (s, 3H) 1.36 (s, 3H) 1.59 (dd, J = 3.42, 6.65Hz 6H) 2.00 (m, 2H) 3.69 (m, 4H) 4.47 (m, 2H) 5.44 (s, 2H) 6.97 (m, 2H) 7.51 (m, IH), ES-HRMS 485.1766 (M+H calcd for C22H28ClF2N4O4 requires 485.1762).
Example 70
5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one
[0252] Preparation of 5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)- one. 5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-(methylsulfonyl)pyrimidin- 4(3H)-one (0.390 g, 1.0 mmol) was dissolved in methanol (3.0 mL) and cooled in an ice bath. Sodium borohydride (0.076 g, 3.0 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. The reaction was quenched with water and the methanol was removed. The aqueous layer was extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered and concentrated. The residue was purified by chromatography (silica gel, ethyl acetate/hexanes) to afford a white solid (0.140 g, 44%). 1H NMR (300 MHz, CD3OD) δ ppm 1.46 (d, J = 6.85 Hz, 6H) 4.96 (m, IH) 5.51 (s, 2H) 6.99 (m, 2H) 7.55 (m, IH) 8.35 (s, IH), ES-HRMS 315.0696 (M+H calcd for Ci4H14ClF2N2O2 requires 315.0706).
5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropyI-2-methylpyrimidin-4(3H)-one
Step 1. Preparation of 4-(2,4-difluorobenzyloxy)-5-chloro-l,6-dihvdro-l-isopropyl-6- oxopyrimidine-2-carbonitrile.
[0253] 5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-(methylsulfonyl)pyrimidin-
4(3H)-one (10.0 g, 25.4 mmol) was dissolved in dimethylformamide (56 mL).
Potassium cyanide (3.31 g, 50.9 mmol) was added. The resulting mixture was stirred at room temperature for 3 hours. The reaction was diluted with ethyl acetate (100 mL) and poured into ethyl acetate (100 mL)/ice water (300 mL). The aqueous layer was further extracted into ethyl acetate. The combined organics were dried over NaSO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, ethyl " acetate/hexanes) to afford a light yellow solid (3.34 g, 39 %). 1H NMR (300 MHz, CDCl3) δ ppm 1.67 (m, 6H) 5.16 (m, IH) 5.45 (s, 2H) 6.88 (m, 2H) 7.46 (m, IH). LC/MS, tr = 3.14 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS m/z 340 (M+l).
Step 2. Preparation of 5-chloro-6-[(2,4-difluorobenzyl)oxyl-3-isopropyl-2- methylpyrirnidin-4(3H)-one.
[0254] 4-(2,4-difluorobenzyloxy)-5-chloro-l,6-dihydro-l-isopropyl-6-oxopyrimidine-2- carbonitrile (from step 1) (0.170 g, 0.50 mmol) was dissolved in tetrahydrofuran (17 mL) and cooled to -78°C. Methyl magnesium bromide (3.0 M in THF, 0.40 mL) was added slowly to the reaction mixture. The reaction was then warmed to 0°C, quenched by the addition of water. The aqueous layer was extracted into ethyl acetate. The combined organics were washed with brine, dried over MgSO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, ethyl acetate/hexanes) to afford an off white solid (0.091 g, 56%). 1H NMR (400 MHz, CDCl3) δ ppm 1.59 (d, / = 6.71 Hz, 6H) 2.52 (s, 3H) 4.49 (m, IH) 5.41 (s, 2H) 6.83 (m, 2H) 7.45 (m, IH), ES-HRMS 329.0898 (M+H calcd for C15H16ClF2N2O2 requires 329.0863).
2-but-3-enyl-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one
[0255] Preparation of 2-but-3-enyl-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3- isopropylpyrimidin-4(3H)-one. 5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)pyrimidin-4(3H)-one (0.100 g, 0.25 mmol) was dissolved in tetrahydrofuran (5 mL) and cooled to -780C. But-l-ene-4-magnesium bromide (0.5 M, 0.900 mL) was added slowly. The reaction mixture was allowed to warm to room temperature overnight. The reaction mixture was quenched by the addition of a solution of NH4Cl then extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated. The crude solid was purified by chromatography (silica gel, ethyl acetate/hexanes) to afford a white solid (0.048 g, 52%). 1H NMR (300 MHz, CD3OD) 6 ppm 1.58 (d, J = 6.85 Hz, 6H) 2.52 (m, 2H) 2.95 (t, J = 7.45 Hz, 2H) 4.68 (m, IH) 5.04 (m, 2H) 5.48 (s, 2H) 5.88 (m, IH) 6.96 (m, 2H) 7.50 (m, IH), ES- HRMS 369.1156 (M+H calcd for Ci8H20ClF2N2O2 requires 369.1176).
Example 73
5-chloro-6-[(2,4-difluorobenzyl)oxy]-2-(4-hydroxybutyl)-3-isopropylpyrimidin-
4(3H)-one
[0256] Preparation of 5-chloro-6-[(2,4-difluorobenzyl)oxy]-2-(4-hydroxybutyl)-3- isopropylpyrimidin-4(3H)-one. 2-but-3-enyl-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3- isopropylpyrimidin-4(3H)-one (0.310 g, 0.84 mmol) was dissolved in tetrahydrofuran (3 mL). BH3-THF (1.0 M, 0.53 mL) was then added at room temperature. After the evolution of gas ceased, sodium hydroxide (2.5 N, 1 mL) was added to the reaction mixture followed by hydrogen peroxide (30%, 4 mL). The resulting reaction was stirred overnight. The reaction mixture was then extracted into ethyl acetate and washed with brine. The combined organics were dried over MgSO4, filtered and concentrated. The
residue was purified by chromatography (silica gel, ethyl acetate/hexanes) to afford an off white solid (0.118 g, 36%). 1H NMR (300 MHz, CD3OD) δ ppm 1.59 (d, J = 6.65 Hz, 6H) 1.64 (m, 2H) 1.83 (m, 2H) 2.89 (m, 2H) 3.61 (t, J = 6.24 Hz, 2H) 4.67 (m, IH) 5.50 (s, 2H) 6.99 (m, 2H) 7.53 (m, IH), ES-HRMS 387.1237 (M+H calcd for C18H22ClF2N2O3 requires 387.1282).
Example 74
4-{5-chIoro-4-[(2,4-difluorobenzyI)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}butanoic acid
[0257] Preparation of 4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl }butanoic acid. 5-chloro-6-[(2,4-difluorobenzyl)oxy]-2-(4- hydroxybutyl)-3-isopropylpyrimidin-4(3H)-one (1.32 g, 3.42 mmol) was dissolved in dimethylformamide (17 niL). Pyridinium dichromate (4.5 g, 12.0 mmol) was added and the reaction was stirred overnight at room temperature. The reaction was quenched by the addition of water (125 mL). The aqueous layer was extracted into ethyl acetate. The combined organics were dried over MgSO
4, filtered, and concentrated. The crude solid was washed with small amounts of dichloromethane and filtered to afford a tan solid (0.750 g, 55%).
1H NMR (300 MHz, CD
3OD) 6 ppm 1.59 (d, J = 6.65 Hz, 6H) 2.06 (m, 2H) 2.45 (t, J = 7.05 Hz, 2H) 2.92 (t, J = 7.35 Hz, 2H) 4.70 (m, IH) 5.49 (m, 2H) 6.98 (m, 2H) 7.55 (m, IH), ES-HRMS 401.1040 (M+H calcd for Ci
8H
20ClF
2N
2O
4 requires 401.1074).
4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}butanamide
[0258] Preparation of 4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}butanamide. Prepared as iV~3~-{5-bromo-4-[(2,4- difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } -beta-alaninamide (step 3) utilizing 4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}butanoic acid (0.50 g, 1.25 mmol), 2-chloro-4,6-dimethoxy- 1,3,5-triazine (0.33 g, 1.88 mmol), 4-methylmorpholine (0.41 mL, 3.75 mmol), and an excess of aqueous ammonium hydroxide to afford a tan solid (0.153 g, 31%). 1H NMR (300 MHz, CD3OD) δ ppm 1.59 (d, J = 6.85 Hz, 6H) 2.07 (m, 2H) 2.35 (t, J = 7.15 Hz, 2H) 2.90 (t, J = 7.45 Hz, 2H) 4.69 (m, IH) 5.50 (s, 2H) 6.98 (m, 2H) 7.54 (m, IH), ES- HRMS 400.1265 (M+H calcd for C18H21ClF2N3O3 requires 400.1234).
4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yI}-N-[(2R)-2-hydroxypropyl]butanamide
[0259] Preparation of 4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}-N-[(2R)-2-hydroxypropyl]butanamide. 4-{5-chloro-4-[(2,4- difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl Jbutanoic acid (0.210 g, 0.53 mmol) was dissolved in dioxane (8 mL). N-Hydroxybenzotriazole (0.072 g, 0.53 mmol), and carbodiimide resin (1.31 mmol/g, 1.39 g) were added and the reaction was agitated for 15 minutes. (R)-l-Aminopropan-2-ol (0.080 g, 1.06 mmol) was added and the resulting reaction mixture was agitated overnight at room temperature. Dioxane (20 mL), polyamine resin (2.87 mmol/g, 1.44 g), and PS-isocyanate resin (1.47 mmol/g, 2.67 g) were added and the reaction mixture was agitated for 4 hours. The reaction mixture was filtered and concentrated to afford a clear oil (0.145 g, 60 %). 1H NMR (400 MHz, CD3OD) δ ppm 1.13 (d, J = 6.44 Hz, 3H) 1.59 (d, J = 6.71 Hz, 6H) 2.08 (m, 2H) 2.36 (t, J = 7.12 Hz, 2H) 2.89 (t, J = 7.38 Hz, 2H) 3.19 (m, 2H) 3.81 (m, IH) 4.66 (m, IH) 5.49 (s, 2H) 6.98 (m, 2H) 7.54 (m, IH), ES-HRMS 458.1634 (M+H calcd for C2IH27ClF2N3O4 requires 458.1653).
4-{5-ch]oro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}-7V-(2-hydroxyethyl)butanamide
[0260] Preparation of 4- { 5-chloro-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6- dihydropyrimidin-2-yl } -N-(2-hydroxyethyl)butanamide. Prepared as 4- { 5-chloro-4- [(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}-N-[(2R)-2- hydroxypropyl]butanamide utilizing 4- { 5-chloro-4-[(2,4-difluorobenzyl)oxy]- 1- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}butanoic acid (0.40 g, 1.0 mmol) and ethanolamine (0.12 mL, 2.0 mmol) to afford an off white solid (0.244 g, 55%). 1H NMR
(400 MHz, CD3OD) δ ppm 1.59 (d, J = 6.71 Hz, 6H) 2.08 (m, 2H) 2.35 (t, J = 7.12 Hz,
2H) 2.89 (t, J = 7.52 Hz, 2H) 3.30 (m, 2H) 3.58 (t, J = 5.77 Hz, 2H) 4.67 (m, IH) 5.50 (s, 2H) 6.98 (m, 2H) 7.54 (m, IH), ES-HRMS 444.1488 (M+H calcd for C20H25ClF2N3O4 requires 444.1496).
Example 78
4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}-N-methylbutanamide
[0261] Preparation of 4-{ 5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}-N-methylbutanamide. Prepared as 4-{5-chloro-4-[(2,4- difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } -iV-[(2R)-2- hydroxypropyl]butanamide utilizing 4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}butanoic acid (0.40 g, 1.0 mmol) and methylamine (2.0 M in dioxane, 1.0 mL, 2.0 mmol) to afford a light brown-red solid (0.239 g, 58%). 1H NMR (400 MHz, CD3OD) δ ppm 1.59 (d, / = 6.71 Hz, 6H) 2.06 (m, 2H) 2.31 (t, J = 7.25 Hz, 2H) 2.71 (s, 3H) 2.88 (t, J = 7.38 Hz, 2H) 4.67 (m, IH) 5.49 (s, 2H) 6.98 (m, 2H) 7.53 (m, IH), ES-HRMS 414.1373 (M+H calcd for C19H23ClF2N3O3 requires 414.1391).
S-chloro-θ-tCl^-difluorobenzy^oxyl^-hydroxy-a-isopropylpyrimidin^Ca^-one
[0262] Preparation of 5-chloro-6-[(2,4-difluorobenzyl)oxy]-2-hydroxy-3- isopropylpyrimidin-4(3H)-one. 5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)pyrimidin-4(3H)-one (5.00 g, 12.7 mmol) was dissolved in dioxane (130 mL) and 2.5 N NaOH (25.5 mL, 63.7 mmol)) was added to the reaction mixture at room temperature. After 15 minutes, the dioxane was removed and the aqueous layer was made acidic (pH 4.5) with 1.0 M HCl. The precipitate was collected by filtration and washed with ether to afford a white solid (3.99 g, 95%). 1H NMR (300 MHz, CDCl3) δ ppm 1.42 (d, J = 7.05 Hz, 6H) 5.15 (m, IH) 5.45 (s, 2H) 6.91 (m, 2H) 7.49 (m, IH), ES- HRMS 331.0622 (M+H calcd for C14Hi4ClF2N2O3 requires 331.0656).
Example 80
5-chIoro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl trifluoromethanesulfonate
[0263] Preparation of 5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl trifluoromethanesulfonate. 5-chloro-6-[(2,4-difluorobenzyl)oxy]- 2-hydroxy-3-isopropylpyrimidin-4(3H)-one (2.00 g, 6.1 mmol) was dissolved in dichloromethane (20 mL) and cooled in an ice bath. Pyridine (1.5 mL, 18.3 mmol) was added and the reaction was stirred, with cooling, for 30 minutes. Trifluoromethanesulfonic anhydride (2.1 mL, 12.2 mmol) was added and the reaction was allowed to warm to room temperature over 2.5 hours. The reaction was quenched by the addition of water and extracted into dichloromethane. The combined organics were dried over MgSO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, ethyl acetate/hexanes) to afford white solid (1.14 g, 41 %).
1H NMR (400 MHz, CD3OD) δ ppm 1.51 (d, J = 6.98 Hz, 6H) 5.26 (m, IH) 5.43 (s, 2H) 7,.Ol (m, 2H) 7.54 (m, IH). LC/MS, tr = 3.64 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mlVmin, at 254 nm, at 5O0C), ES-MS m/z 463 (M+l).
Example 81
2-allyl-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one
[0264] Preparation of 2-allyl-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3- isopropylpyrimidin-4(3H)-one. Prepared as 2-but-3-enyl-5-chloro-6-[(2,4- difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one utilizing prop-l-ene-3-magnesium bromide (1.0 M, 3.75 mL, 3.75 mmol) to afford a yellow oil (0.620 g, 70 %). 1H NMR (400 MHz, CD3OD) δ ppm 1.56 (d, J = 6.98 Hz, 6H) 3.66 (d, J = 5.91 Hz, 2H) 4.61 (m, IH) 5.15 (dd, J = 1.34, 17.19 Hz, IH) 5.25 (dd, J = 1.34, 10.20 Hz, IH) 5.46 (m, 2H) 6.03 (m, IH) 6.96 (m, 2H) 7.54 (d, J = 6.44 Hz, IH), ES-HRMS 355.1019 (M+H calcd for C17Hi8ClF2N2O2 requires 355.1019).
Example 82
5-chloro-6-[(2,4-difluorobenzyl)oxy]-2-(3-hydroxypropyl)-3-isopropylpyrimidin-
4(3H)-one
[0265] Preparation of 5-chloro-6-[(2,4-difluorobenzyl)oxy]-2-(3-hydroxypropyl)-3- isopropylpyrimidin-4(3H)-one. Prepared as 5-chloro-6-[(2,4-difluorobenzyl)oxy]-2-(4- hydroxybutyl)-3-isopropylpyrimidin-4(3H)-one utilizing 2-allyl-5-chloro-6-[(2,4- difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one (0.516, 1.46 mmol), BH3-THF (1.0 M in tetrahydrofuran, 0.9 mL, 0.92 mmol), hydrogen peroxide (30%, 1.7 mL, 0.51 mmolX and 2.5 N NaOH (0.25 mL, 0.09 mmol) to afford an off white solid (0.110 g, 20 %). 1H NMR (400 MHz, CD3OD) δ ppm 1.58 (d, J = 6.98 Hz, 6H) 1.99 (m, 2H) 2.95 (m, 2H) 3.67 (t, J = 6.18 Hz, 2H) 4.68 (m, IH) 5.47 (s, 2H) 6.95 (m, 2H) 7.51 (m, IH), ES-HRMS 373.1111 (M+H calcd for C17H20ClF2N2O3 requires 373.1125).
3-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yljpropanoic acid
[0266] Preparation of 3-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl} propanoic acid. Prepared as 4-{5-chloro-4-[(2,4- difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } butanoic acid utilizing 5-chloro-6-[(2,4-difluorobenzyl)oxy]-2-(3-hydroxypropyl)-3- isopropylpyrimidin-4(3H)-one (0.910 g, 2.3 mmol) and pyridinium dichromate (3.03 g, 8.1 mmol) to afford an off white solid (0.410 g, 46%). 1H NMR (400 MHz, CD3OD) 6 ppm 1.60 (d, J = 6.71 Hz, 6H) 2.82 (m, 2H) 3.17 (m, 2H) 4.74 (m, IH) 5.47 (s, 2H) 6.98 (m, 2H) 7.52 (m, IH), ES-HRMS 387.0897 (M+H calcd for C17H18ClF2N2O4 requires 387.0918).
Example 84
S-IS-chloro^-tCl^-difluorobenzyOoxyl-l-isopropyl-ό-oxo-l.θ-dihydropyrimidin-Z- yljpropanamide
[0267] Preparation of 3-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}propanamide. Prepared as 4-{5-chloro-4-[(2,4- difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}butanamide utilizing 3-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yljpropanoic acid (0.180 g, 0.47 mmol) and ammonia (0.5 M in dioxane, 1.86 mL, 0.93 mmol) to afford a light yellow solid (0.156 g, 86%). 1H NMR (400 MHz, CD3OD) 6 ppm 1.61 (d, J = 6.71 Hz, 6H) 2.77 (t, J = 6.31 Hz, 2H) 3.17 (t, J = 6.44 Hz, 2H) 4.75 (m, IH) 5.49 (s, 2H) 6.99 (m, 2H) 7.53 (m, IH), ES-HRMS 386.1055 (M+H calcd for C17H19ClF2N3O3 requires 386.1078).
Example 85
3-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}-N-methylpropanamide
[0268] Preparation of 3-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}-N-methylpropanamide. Prepared as 4-{5-chloro-4-[(2,4-
difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } -N- methylbutanamide utilizing 3-{ 5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl} propanoic acid (0.180 g, 0.47 mmol) and methylamine (2.0 M in dioxane, 1.0 mL, 2.0 mmol) to afford an off white solid (0.053 g, 28%). 1H NMR (400 MHz, CD3OD) δ ppm 1.61 (d, J = 6.98 Hz, 6H) 2.70 (s, 3H) 2.71 (m, 2H) 3.18 (t, J = 6.31 Hz~ 2H) 4.75 (m, IH) 5.44 (s, 2H) 7.00 (m, 2H) 7.52 (m, IH), ES-HRMS 400.1232 (M+H calcd for C18H2]ClF2N3O3 requires 400.1234).
3-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}-N-methylpropanamide
Step 1. Preparation of 6-(2,4-difluorobenzyloxy)-2-(4-aminobutyl)-5-chloro-3- isopropylpyrimidin-4(3H)-one.
[0269] To a dry flask of 4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}butanamide (0.400 g, 1.0 mmol), 1.0 M BH3-THF (1.5 mL, 1.5 mmol) was added and the resulting reaction was brought to reflux for 1 hour. The reaction mixture was the removed from the heat and cooled in an ice bath. 1.0 M
Hydrochloric acid (11 mL) was slowly added and the reaction mixture was warmed to
room temperature overnight. The reaction mixture was made alkaline (pH 9) with 2.5 N NaOH the extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated. The residue was dissolved in methanol and 1.0 M hydrochloric acid (1 mL) was added. The residue was concentrated to afford a white solid (0.300 g, 71 %). 1H NMR (300 MHz, CD3OD) δ ppm 1.59 (d, J = 6.65 Hz, 6H) 1.83 (m, 4H) 3.00 (m, 4H) 4.74 (m, IH) 5.50 (s, 2H) 6.98 (m, 2H) 7.53 (m, IH). LC/MS, tr = 3.94 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS m/z 413 (M+l).
Step 2. Preparation of 3-{5-chloro-4-r(2,4-difluorobenzyl)oxyl-l-isopropyl-6-oxo-l,6- dihvdropyrimidin-2-yl I -■/V-methylpropanamide.
[0270] Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]acetamide (step 2) utilizing 6-(2,4- difluorobenzyloxy)-2-(4-aminobutyl)-5-chloro-3-isopropylpyrimidin-4(3H)-one (from step 1) (0.300 g, 0.71 mmol) and acetyl chloride (0.060 mL, 0.85 mmol) to afford a clear oil (0.104 g, 34%). 1H NMR (300 MHz, CD3OD) δ ppm 1.58 (d, J = 6.85 Hz, 6H) 1.60 (m, 2H) 1.79 (m, 2H) 1.91 (s, 3H) 2.88 (t, J = 7.45 Hz, 2H) 3.21 (m, 2H) 4.66 (m, IH) 5.49 (s, 2H) 6.99 (m, 2H) 7.52 (m, IH), ES-HRMS 428.1509 (M+H calcd for C20H25ClF2N3O3 requires 428.1547).
5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-(4-hydroxybutyl)-3-isopropylpyrimidin-
4(3H)-one
Step 1. Preparation of 6-(2,4-difluorobenzyloxy)-5-bromo-2-(but-3-enyl)-3- isopropylpyrimidin-4(3H)-one.
[0271] Prepared as 2-but-3-enyl-5-chloro-6-[(2,4-difluorobenzyl)oxy]-3- isopropylpyrimidin-4(3H)-one utilizing 6-(2,4-difluorobenzyloxy)-5-bromo-3-isopropyl- 2-(methylsulfonyl)pyrimidin-4(3H)-one (5.00 g, 11.4 mmol) and but-l-ene-4-magnesium bromide (0.5 M in tetrahydrofuran, 34.0 mL, 17.2 mmol) to afford a light yellow solid (4.18 g, 89%). 1H NMR (300 MHz, CD3OD) δ ppm 1.58 (d, J = 6.65 Hz, 6H) 2.53 (m, 2H) 2.95 (t, J = 7.35 Hz, 2H) 4.68 (m, IH) 5.05 (m, 2H) 5.49 (s, 2H) 5.87 (m, IH) 6.98 (m, 2H) 7.52 (m, IH). LC/MS, tr = 3.94 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS mJz 413 (M+l).
Step 2. Preparation of 5-bromo-6-r(2,4-difluorobenzvI)oxyl-2-(4-hvdroxybutyl)-3- isopropylpyrimidin-4(3H)-one.
[0272] Prepared as 5-chloro-6-[(2,4-difluorobenzyl)oxy]-2-(4-hydroxybutyl)-3- isopropylpyrimidin-4(3H)-one utilizing 6-(2,4-difluorobenzyloxy)-5-bromo-2-(but-3- enyl)-3-isopropylpyrimidin-4(3H)-one (from step 1) (4.00 g, 9.7 mmol) and BH3-THF (1.0 M in tetrahydrofuran, 5.8 mL, 5.8 mmol), hydrogen peroxide (35%, 9.7 mL, 3.4 mmol), and 2.5 NNaOH (8.5 mL, 21.3 mmol) to afford a white solid (2.34 g, 56 %). 1H NMR (300 MHz, CD3OD) δ ppm 1.58 (d, J = 6.85 Hz, 6H) 1.63 (m, 2H) 1.84 (m, 2H) 2.89 (m, 2H) 3.61 (t, J = 6.24 Hz, 2H) 4.68 (m, IH) 5.49 (s, 2H) 6.98 (m, 2H) 7.53 (m, IH), ES-HRMS 431.0756 (M+H calcd for C18H22BrF2N2O3 requires 431.0776).
Example 88
4-{5-bromo-4-[(2,4-difluorobenzyI)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- y 1 }-N- methy lbutanamide
Step 1. Preparation of 4-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihvdro-l-isopropyl-6- oxopyrimidin-2-yl)butanoic acid.
[0273] Prepared as 4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}butanoic acid utilizing 5-bromo-6-[(2,4-difluorobenzyl)oxy]-2- (4-hydroxybutyl)-3-isopropylpyrimidin-4(3H)-one (2.34 g, 5.4 mmol) and pyridinium dichromate (7.11 g, 18.9 mmol) to afford a white solid (1.15 g, 48%). 1H NMR (300 MHz, CD3OD) 5 ppm 1.58 (d, J = 6.85 Hz, 6H) 2.04 (m, 2H) 2.45 (t, J = 6.95 Hz, 2H) 2.92 (t, J = 7.35 Hz, 2H) 4.71 (m, IH) 5.49 (s, 2H) 6.98 (m, 2H) 7.54 (m, IH). LC/MS, tr = 2.66 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS m/z 445 (M+l).
Step 2. Preparation of 4-{5-bromo-4-r(2,4-difluorobenzyl)oxyl-l-isopropyl-6-oxo-l,6- dihvdropyrimidin-2-yl I -N-methylbutanamide.
[0274] Prepared as 4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-γl } -./V-methylbutanamide utilizing 4-(4-(2,4-difluorobenzyloxy)-5-
bromo-l,6-dihydro-l-isopropyl-6-oxopyrimidin-2-yl)butanoic acid (from step 1) (0.30 g, 0.68 mmol) and methyl amine (2.0 M in dioxane, 0.68 mL, 1.35 mmol) to afford a white solid (0.068 g, 22%). 1H NMR (400 MHz, CD3OD) δ ppm 1.58 (d, J = 6.71 Hz, 6H) 2.07 (m, 2H) 2.31 (t, J = 7.25 Hz, 2H) 2.70 (s, 3H) 2.88 (t, J = 7.52 Hz, 2H) 4.68 (m, IH) 5.49 (s, 2H) 6.97 (m, 2H) 7.53 (m, IH), ES-HRMS 458.0923 (M+H calcd for C19H23BrF2N3O3 requires 458.0885).
Example 89
4-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yljbutanamide
[0275] Preparation of 4-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}butanamide. Prepared as 4-{5-chloro-4-[(2,4- difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } butanamide utilizing 4-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihydro-l-isopropyl-6-oxopyrimidin-2- yl)butanoic acid (step 1) (0.30 g, 0.68 mmol) and ammonia (0.5 M in dioxane, 2.7 mL, 1.35 mmol) to afford a white solid (0.148 g, 49 %). 1H NMR (400 MHz, CD3OD) δ ppm 1.58 (d, J = 6.71 Hz, 6H) 2.06 (m, 2H) 2.35 (t, J = 7.12 Hz, 2H) 2.89 (t, J = 7.38 Hz, 2H) 4.68 (m, IH) 5.49 (s, 2H) 6.98 (m, 2H) 7.55 (m, IH), ES-HRMS 444.0740 (M+H calcd for C I8H21BrF2N3O3 requires 444.0729).
Example 90
4-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}-N-(2-hydroxyethyl)butanamide
[0276] Preparation of 4-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}-N-(2-hydroxyethyl)butanamide. Prepared as 4-{5-chloro-4- [(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}-iV-(2- hydroxyethyl)butanamide utilizing 4-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihydro-l- isopropyl-6-oxopyrimidin-2-yl)butanoic acid (step 1) (0.30 g, 0.68 mmol) and ethanolamine (0.081 mL, 1.35 mmol) to afford a white solid (0.131 g, 39%). 1H NMR (400 MHz, CD3OD) δ ppm 1.59 (d, J = 6.71 Hz, 6H) 2.08 (m, 2H) 2.36 (t, J = 7.25 Hz, 2H) 2.89 (t, J = 7.52 Hz, 2H) 3.30 (m, 2H) 3.59 (t, / = 5.77 Hz, 2H) 4.69 (m, IH) 5.49 (s, 2H) 6.98 (m, 2H) 7.54 (m, IH), ES-HRMS 488.0976 (M+H calcd for C20H25BrF2N3O4 requires 488.0991).
Example 91
2-[(l-acetyIpiperidin-4-yl)amino]-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3- ~ ~isopropylpyririiidin-4(3H)-one
Step 1. Preparation of tert-butyl 4-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihvdro-l- isopropyl-6-oxoρyrimidin-2-ylamino)piperidine-l-carboxylate.
[0277] 6-(2,4-difluorobenzyloxy)-5-bromo-3-isopropyl-2-(methylsulfonyl)pyrimidin- 4(3H)-one (1.0 g, 2.29 mmol) was dissolved in dichloromethane (10 mL). Triethylamine (0.64 mL, 2.0 mmol) and t-butyl 4-aminopiperidine-l-carboxylate (0.504 g, 2.52 mmol) were added and the reaction mixture was stirred at room temperature overnight. The reaction was quenched by the addition of a solution Of NaHCO3, then extraction into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, ethyl acetate/hexanes) to afford a yellow solid (0.285 g, 22%). 1H NMR (400 MHz, CD3OD) δ ppm 1.45 (s, 9H) 1.51 (d, J = 6.98 Hz, 6H) 1.85 (m, 2H) 2.83 (m, 2H) 4.08 (m, 4H) 4.57 (m, IH) 5.21 (m, IH) 5.43 (s, 2H) 7.00 (m, 2H) 7.44 (m, IH). LC/MS, tr = 3.52 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS mJz 557 (M+l).
Step 2. Preparation of 6-(2,4-difluorobenzyloxy)-5-brorno-3-isopropyl-2-(piperidin-4- ylamino)pyrimidin-4(3H)-one di-trifluoroacetic acid salt.
[0278] Prepared as 2-[(3S)-3-aminopyrrolidin-l-yl]-5-chloro-6-[(2,4- difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one utilizing t-butyl 4-(4-(2,4- difluorobenzyloxy)-5-bromo- 1 ,6-dihydro- 1 -isopropyl-6-oxopyrimidin-2- ylamino)piperidine-l-carboxylate (from step 1) (0.270 g, 0.49 mmol) and trifluoroacetic acid (1 mL) to afford a brown solid (0.374 g, > 100%). 1H NMR (400 MHz, CD3OD) δ ppm 1.53 (d, J = 6.71 Hz, 6H) 1.80 (m, 2H) 2.15 (m, 2H) 3.09 (m, 2H) 3.45 (m, 2H) 4.19 (m, IH) 4.55 (m, IH) 5.44 (s, 2H) 7.00 (m, 2H) 7.49 (m, IH). LC/MS, tr = 2.10 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 ran, at 5O0C), ES-MS m/z 457 (M+l).
Step 3. Preparation of 2-r(l-acetylpiperidin-4-yl)amino1-5-bromo-6-[(2,4- difluorobenzyl)oxy1-3-isopropylpyrimidin-4(3H)-one.
[0279] Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]acetamide (step 2) utilizing 6-(2,4- difluorobenzyloxy)-5-bromo-3-isopropyl-2-(piperidin-4-ylamino)pyrimidin-4(3H)-one di-trifluoroacetic acid salt (from step 2) (0.374 g, 0.5 mmol) and acetyl chloride (0.043 mL, 0.55 mmol) to afford a white solid (0.115 g, 46%). 1H NMR (300 MHz, CDCl3) δ ppm 1.41 (m, 2H) 1.45 (d, J = 7.25 Hz, 6H) 2.11 (m, 2H) 2.11 (s, 3H) 2.77 (m, IH) 3.19 (m, IH) 3.80 (m, IH) 4.07 (m, IH) 4.51 (m, IH) 4.83 (m, IH) 5.39 (s, 2H) 6.85 (m, 2H) 7.44 (m, IH), ES-HRMS 499.1143 (M+H calcd for C2]H26BrF2N4O3 requires 499.1151).
Example 92
methyl 4-[({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)methyl]benzoate
[0280] Preparation of methyl 4-[({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6- oxo- 1 ,6-dihydropyrimidin-2-yl } amino)methyl]benzoate. 6-(2,4-difluorobenzylόxy)-5- bromo-3-isopropyl-2-(methylsulfonyl)pyrimidin-4(3H)-one (2.00 g, 4.58 mmol) was dissolved in dichloromethane (23 mL). Triethylamine (1.9 mL, 13.74 mmol) and methyl 4-(aminomethyl)benzoate (1.01 g, 5.04 mmol) were added and stirred at room temperature overnight. The reaction was quenched by the addition of a solution of NaHCO3 then extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered and concentration. The residue was purified by chromatography (silica gel, ethyl acetate/hexanes) to afford a white solid (0.644 g, 27%). 1H NMR (400 MHz, CDCl3) 6 ppm 1.49 (d, J = 7.25 Hz, 6H) 3.90 (s, 3H) 4.68 (d, J = 5.10 Hz, 2H) 5.25 (s, 2H) 5.78 (m, IH) 6.70 (m, IH) 6.80 (m, IH) 7.29 (m, 3H) 7.96 (d, J = 8.32 Hz, 2H), ES- HRMS 522.0853 (M+H calcd for C23H23BrF2N3O4 requires 522.0835).
Example 93
5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(4-fluorobenzyl)amino]-3- isopropylpyrimidin-4(3H)-one
[0281] Preparation of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(4-fluorobenzyl)amino]- 3risopropylpyrimidin-4(3H)-one. Prepared as methyl 4-[({5-bromo-4-[(2,4- difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl } amino)methyl]benzoate utilizing (4-fluorophenyl)methanamine (0.50 g, 1.14 mmol) to afford a light pink solid (0.054 g, 11%). 1H NMR (300 MHz, CDCl3) 5 ppm 1.46 (d, J = 7.25 Hz, 6H) 4.59 (d, J = 5.03 Hz, 2H) 5.33 (s, 2H) 5.33 (m, IH) 6.75 (m, IH) 6.85 (m, IH) 7.02 (m, 2H) 7.22 (m, 2H) 7.37 (m, IH), ES-HRMS 482.0645 (M+H calcd for C2IH20BrF3N3O2 requires 482.0685).
Example 94
5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(3,3-dimethylbutyl)amino]-3- isopropylpyrimidin-4(3H)-one
[0282] Preparation of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(3,3- dimethylbutyl)amino]-3-isopropylpyrimidin-4(3H)-one. Prepared as methyl 4-[({5- bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}amino)methyl]benzoate utilizing 3,3-dimethylbutan-l-amine (0.50 g, 1.14 mmol) to afford a white solid (0.084 g, 16%). 1H NMR (400 MHz, CDCl3) δ ppm 0.96 (s, 9H) 1.47 (d, J = 7.25 Hz, 6H) 1.49 (m, 2H) 3.45 (m, 2H) 4.78 (m, IH) 5.42 (s, 2H) 6.80 (m, IH) 6.89 (m, IH) 7.48 (m, IH), ES-HRMS 458.1102 (M+H calcd for C20H27BrF2N3O2 requires 458.1255).
Example 95
N~2~-{5-chloro-4-[(2,4-difluorobenzyI)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}glycinamide
[0283] Preparation of N~2~-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } glycinamide. 5-chloro-6-[(2,4-difluorobenzyl)oxy]-3- isopropyl-2-(methylsulfonyl)pyrimidin-4(3H)-one (1.73 g, 3.9 mmol) was dissolved in dimethylformamide (10 mL) and added slowly to a suspension of triethylamine (1.95 mL, 14.0 mmol) and 2-aminoacetamide hydrochloride salt (1.54 g, 14.0 mmol) in dimethylformamide (20 mL) at room temperature. After 10 minutes, the reaction was quenched by the addition of water then extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated. The solids were washed with a small amount of ethyl acetate and filtered to afford a white solid (0.164 g, 33%). 1H NMR (400 MHz, CD3OD) 5 ppm 1.56 (d, J = 6.98 Hz, 6H) 4.03 (s, 2H) 4.82 (m, IH) 5.39 (s, 2H) 6.98 (m, 2H) 7.51 (m, IH), ES-HRMS 387.0995 (M+H calcd for C16H18ClF2N4O3 requires 387.1030).
Example 96
N~2~-{5-bromo-4-[(2,4-difluorobenzyI)oxy]-l-isopropyI-6-oxo-l,6- dihydropyrimidin-2-yl}-N~l~-(2-hydroxyethyl)glycinamide
Step 1. Preparation of ethyl 2-(4-(2.4-difluorobenzyloxy)-5-bromo-l,6-dihvdro-l- isopropyl-6-oxopyrimidin-2-ylamino)acetate.
[0284] Prepared as N~3— {5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}-beta-alaninamide (step 1) utilizing 6-(2,4-difluorobenzyloxy)-5- bromo-3-isopropyl-2-(methylsulfonyl)pyrimidin-4(3H)-one (1.75 g, 12.6 mmol) and ethyl 2-aminoacetate hydrochloric acid salt (1.75 g, 12.6 mmol) to afford a clear oil (1.8 g, 34%). 1H NMR (400 MHz, CDCl3) δ ppm 1.31 (t, J = 7.12 Hz, 3H) 1.51 (d, J = 7.25 Hz, 6H) 4.15 (d, J = 4.57 Hz, 2H) 4.26 (q, J = 7.25 Hz, 2H) 5.36 (s, 2H) 5.67 (m, IH) 6.83 (m, 2H) 7.46 (m, IH). LC/MS, tr = 2.96 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mlVmin, at 254 nm, at 5O0C), ES-MS m/z 460 (M+l).
Step 2. Preparation of 2-(4-(2,4-difluorobenzyloxy)-5-bromo-h6-dihvdro-l-isopropyl-6- oxopyrimidin-2-ylamino')acetic acid.
[0285] Prepared as W~3~-{5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}-beta-alaninamide (step 2) utilizing ethyl 2-(4-(2,4- difluorobenzyloxy)-5-bromo-l,6-dihydro-l-isopropyl-6-oxopyrimidin-2-ylamino)acetate
(from step 1) (1.73 g, 3.9 mmol) and 2.5 N NaOH (16.0 mL, 39.0 mmol) to afford a
white solid (1.19 g, 71%). 1H NMR (400 MHz, CD3OD) δ ppm 1.55 (d, J = 6.98 Hz, 6H) 4.09 (s, 2H) 4.89 (m, IH) 5.36 (s, 2H) 6.96 (m, 2H) 7.49 (m, IH). LC/MS, tr = 2.57 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 rtiL/min, at 254 nm, at 5O0C), ES-MS m/z 432 (M+l).
Step 3. Preparation of N~2~-(5-bromo-4-r(2,4-difluorobenzyl)oxy1-l-isopropyl-6-oxo- l,6-dihvdropyrimidin-2-yl)-N~l~-(2-hvdroxyethyl)glvcinamide.
[0286] Prepared as 4-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } -N-[(2R)-2-hydroxypropyl]butanamide utilizing 2-(4-(2,4- difluorobenzyloxy)-5-bromo-l,6-dihydro-l-isopropyl-6-oxopyrimidin-2-ylamino)acetic acid (from step 2) (0.39 g, 0.90 mmol) and ethanolamine (0.11 mL, 1.81 mmol)'to afford a light yellow solid (0.269 g, 63%). 1H NMR (400 MHz, CD3OD) δ ppm 1.56 (d, / = 6.98 Hz, 6H) 3.31 (m, 2H) 3.57 (t, J = 5.77 Hz, 2H) 4.04 (s, 2H) 4.85 (m, IH) 5.35 (s, 2H) 6.98 (m, 2H) 7.50 (m, IH), ES-HRMS 475.0822 (M+H calcd for C8H22BrF2N4O4 requires 475.0787).
6-(2,4-difluorobenzyloxy)-2-(2-oxo-2-(pyrrolidin-l-yl)ethylamino)-5-brorno-3- isopropylpyrimidin-4(3H)-one
[0287] Preparation of 6-(2,4-difluorobenzyloxy)-2-(2-oxo-2-(pyrrolidin- 1 - yl)ethylamino)-5-bromo-3-isopropylpyrimidin-4(3H)-one. Prepared as 4-{5-chloro-4- [(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } -N-[(2R)-2- hydroxypropyl]butanamide utilizing 2-(4-(2,4-difluorobenzyloxy)-5-bromo- 1 ,6-dihydro- l-isopropyl-6-oxopyrimidin-2-ylamino)acetic acid (from N~2~-{5-bromo-4-[(2,4-
difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } -N- 1 ~-(2- hydroxyethyl)glycinamide step 2) (0.39 g, 0.9 mmol) and pyrrolidine (0.15 mL, 1.81 rnmol) to afford a light yellow solid (0.385 g, 89%). 1H NMR (400 MHz, CD3OD) 5 ppm 1.56 (d, J = 6.98 Hz, 6H) 1.86 (m, 2H) 2.00 (m, 2H) 3.42 (t, J = 6.98 Hz, 2H) 3.53 (t, J = 6.71 Hz, 2H) 4.15 (s, 2H) 5.00 (m, IH) 5.35 (s, 2H) 6.99 (m, 2H) 7.48 (m, IH), ES-HRMS 485.0985 (M+H calcd for C20H24BrF2N4O3 requires 485.0994).
Example 98
5-chloro-6-[(2,4-difluorobenzyl)oxy]-2-(4-hydroxybutoxy)-3-isopropyIpyrimidin-
4(3H)-one
[0288] Preparation of 5-chloro-6-[(2,4-difluorobenzyl)oxy]-2-(4-hydroxybutoxy)-3- isopropylpyrimidin-4(3H)-one. Prepared as Tert-butyl 2-({5-chloro-4-[(2,4- difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } oxy)ethylcarbamate utilizing 5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-(methylsulfonyl)pyrimidin- 4(3H)-one (5.0 g, 12.8 mmol) and butane- 1,4-diol (1.24 mL, 14.0 mmol) to afford a white solid (2.98 g, 58%). 1H NMR (400 MHz, CD3OD) δ ppm 1.45 (d, J = 6.98 Hz, 6H) 1.70 (m, 2H) 1.88 (m, 2H) 3.62 (t, J = 6.44 Hz, 2H) 4.52 (t, J = 6.58 Hz, 2H) 5.30 (m, IH) 5.48 (s, 2H) 6.98 (m, 2H) 7.52 (m, IH), ES-HRMS 403.1212 (M+H calcd for C18H22ClF2N2O4 requires 403.1231).
Example 99
4-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}oxy)butanoic acid
[0289] Preparation of 4-({ 5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)butanoic acid. Prepared as 4-{5-chloro-4-[(2,4- difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } butanoic acid utilizing 5-chloro-6-[(2,4-difluorobenzyl)oxy]-2-(4-hydroxybutoxy)-3- isopropylpyrimidin-4(3H)-one (2.98 g, 7.4 mmol) and pyridinium dichromate (9.74 g, 25.9 mmol) to afford a white solid (0.615 g, 20 %). 1H NMR (300 MHz, CD3OD) 5 ppm 1.45 (d, J = 7.05 Hz, 6H) 2.12 (m, 2H) 2.48 (t, J = 7.15 Hz, 2H) 4.54 (t, J = 6.44 Hz, 2H) 5.28 (m, IH) 5.48 (s, 2H) 7.00 (m, 2H) 7.53 (m, IH), ES-HRMS 417.1028 (M+H calcd for C18H20ClF2N2O5 requires 417.1023).
Example 100
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)ethyl]-iV'-methylurea
[0290] Preparation of N-[2-({ 5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl }oxy)ethyl]-ΛT-methylurea. 6-(2,4-difluorobenzyloxy)-2-(3-
aminopropoxy)-5-chloro-3-isopropylpyrimidin-4(3H)-one trifluoroacetic acid (from 2- { [2-( { 5-chloro-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl} ox y)ethyl] amino }-2-oxoethyl acetate, step 1) (0.50 g, 1.03 mmol) was dissolved in dimethylacetamide (15 mL) and cooled in an ice bath. Diisopropylethylamine (0.480 mL, 2.7 mmol) and 4-nitrophenyl chloroformate (0.310 g, 1.54 mmol) were added and the resulting mixture was stirred with cooling for 10 minutes. Methylamine (2.0 M in tetrahydrofuran, 1.5 mL) was added and the reaction mixture was warmed to room temperature overnight. The reaction was quenched by the addition of water then extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, ethyl acetate/hexanes) to afford a red-brown solid (0.093 g, 21%). 1H NMR (400 MHz, CD3OD) δ ppm 1.45 (d, J = 6.71 Hz, 6H) 2.67 (s, 3H) '3.56 (t, J = 4.97 Hz, 2H) 4.52 (t, J = 5.10 Hz, 2H) 5.28 (m, IH) 5.47 (s, 2H) 7.00 (m, 2H) 7.54 (m, IH), ES-HRMS 431.1297 (M+H calcd for Ci8H22ClF2N4O4 requires 431.1292).
Example 101
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)ethyl]-iVl-(2-hydroxyethyl)urea
[0291] Preparation of N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } oxy)ethyl]-ΛT-(2-hydroxyethyl)urea. Prepared as N-[2-( { 5- chloro-4- [(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}oxy)ethyl]-iV-methylurea utilizing ethanolamine (0.19 mL, 3.09 mmol) to afford a brown solid (0.101 g, 21 %). 1H NMR (400 MHz, CD3OD) 6 ppm 1.45 (d, J = 6.98 Hz, 6H) 3.22 (t, J = 5.77 Hz, 2H) 3.56 (q, J = 5.64 Hz, 4H) 4.52 (t, J = 5.37 Hz, 2H) 5.28
(m, IH) 5.47 (s, 2H) 7.00 (m, 2H) 7.54 (m, IH), ES-HRMS 461.1428 (M+H calcd for C19H24ClF2N4O5 requires 1398).
Example 102
4-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}oxy)butanamide
[0292] Preparation of 4-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } oxy)butanamide. Prepared as N~3~- { 5-bromo-4-[(2,4- difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } -beta-alaninamide utilizing 4-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)butanoic acid (0.30 g, 0.72 mmol) and ammonia (0.5 M in dioxane, 2.8 mL, 1.4 mmol) to afford a white solid (0.220 g, 74%). 1H NMR (400 MHz, CD3OD) δ ppm 1.46 (d, J = 6.98 Hz, 6H) 2.13 (m, 2H) 2.39 (t, J = 7.38 Hz, 2H) 4.53 (t, J = 6.44 Hz, 2H) 5.28 (m, IH) 5.47 (s, 2H) 6.98 (m, 2H) 7.53 (m, IH), ES-HRMS 416.1171 (M+H calcd for C18H21ClF2N3O4 requires 416.1183).
Example 103
4-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}oxy)-iV-(2-hydroxyethyl)butanamide
[0293] Preparation of 4-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)-//-(2-hydroxyethyl)butanamide. Prepared as 4-{5-chloro-4- [(2,4-difluorobenzyl)oxy]- l-isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } -N-(2- hydroxyethyl)butanamide utilizing 4-( { 5-chloro-4- [(2,4-difluorobenzyl)oxy] - 1 - isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}oxy)butanoic acid (0.30 g, 0.72 mmol) and ethanolamine (0.085 mL, 1.4 mmol) to afford a light red oil (0.131 g, 40%). 1H NMR (300 MHz, CD3OD) δ ppm 1.46 (d, J = 7.05 Hz, 6H) 2.13 (m, 2H) 2.39 (t, J = 7.35 Hz, 2H) 3.30 (m, 2H) 3.58 (t, J = 5.74 Hz, 2H) 4.52 (t, / = 6.44 Hz, 2H) 5.29 (m, IH) 5.47 (s, 2H) 6.99 (m, 2H) 7.53 (m, IH), ES-HRMS 460.1457 (M+H calcd for C20H25ClF2N3O5 requires 460.1445).
2-[(2-aminoethyl)amino]-5-bromo-6-[(2,4-difluorobenzyI)oxy]-3- isopropylpyrimidin-4(3H)-one trifluoroacetate
Step 1. Preparation of tert-butyl 2-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihydro-l- isopropyl-6-oxopyrimidin-2-ylamino)ethylcarbamate.
[0294] 6-(2,4-difluorobenzyloxy)-5-bromo-3-isopropyl-2-(methylsulfonyl)pyrimidin- 4(3H)-one (1.77 g, 4.1 mmol) was dissolved in dioxane (40 mL) and t-butyl 2- aminoethylcarbamate (1.3 mL, 8.2 mmol) was added. After 2 hours, the solvent was removed and then the residue was dissolved in ethyl acetate and washed with water. The aqueous layer was extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated. The residue was dissolved in ethyl acetate and passed through a pad of silica gel to afford the desired product (1.95 g, 90 %). LC/MS, tr = 3.16 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 ran, at 5O0C), ES-MS m/z 517 (M+l).
Step 2. Preparation of 2-r(2-aminoethyl)amino"l-5-brorno-6-r(2,4-difluorobenzyl)oxyl-3- isopropylpyrimidin-4(3H)-one trifluoroacetate.
[0295] Prepared as 2-{ [2-({ 5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)ethyl]amino}-2-oxoethyl acetate (step 1) utilizing t-butyl 2- (4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihydro-l-isopropyl-6-oxopyrimidin-2- ylamino)ethylcarbamate (from step 1) (1.95 g, 3.8 mmol) and trifluoroacetic acid (1 mL) to afford a yellowish solid (1.96 g, 87%). 1H NMR (400 MHz, CD3OD) 5 ppm 1.51 (d, J = 6.71 Hz, 6H) 3.20 (t, J = 5.91 Hz, 2H) 3.74 (t, J = 6.04 Hz, 2H) 4.61 (m, IH) 5.43 (s, 2H) 6.98 (m, 2H) 7.51 (m, IH), ES-HRMS 417.0749 (M+H calcd for C16H20BrF2N4O2 requires 417.0732).
N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)ethyl]urea
[0296] Preparation of N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}amino)ethyl]urea. 2-[(2-aminoethyl)amino]-5-bromo-6- [(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one trifluoroacetate (0.3 g, 0.7 mmol) was dissolved in dichloromethane (4 mL) and triethylamine (0.2 mL, 1.44 mmol) was added. Trimethylsilylisocyanate (0.13 mL, 1.08 mmol) was added to the mixture and stirred at room temperature for 45 minutes. The precipitate that formed was collected by filtration, washed with water and ether to afford a white solid (0.225 g, 68%). 1H NMR (400 MHz, CD3OD) 6 ppm 1.53 (d, J = 6.98 Hz, 6H) 3.35 (t, J = 5.77 Hz, 2H) 3.49 (t, J = 5.77 Hz, 2H) 4.74 (m, IH) 5.43 (s, 2H) 6.98 (m, 2H) 7.51 (m, IH), ES-HRMS m/z 460.0803 (M+H calcd for C17H21BrF2N5O3 requires 460.0790).
Example 106
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)ethyl]acetamide
[0297] Preparation of N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}amino)ethyl]acetamide. Prepared as N-[3-({5-bromo-4-[(2,4- difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl } amino)propyl]acetamide utilizing 2-[(2-aminoethyl)amino]-5-bromo-6-[(2,4- difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one trifluoroacetate (0.301 g, 0.72 mmol) and acetyl chloride (0.090 mL, 1.08 mmol). The crude residue was purified by chromatography (silica gel, ethyl acetate/hexanes) to afford a white solid (0.068 g, 20 %). 1H NMR (400 MHz, CD3OD) δ ppm 1.52 (d, J = 6.98 Hz, 6H) 1.93 (s, 3H) 3.39 (t, J = 5.91 Hz, 2H) 3.52 (t, J = 5.91 Hz, 2H) 4.74 (m, IH) 5.44 (s, 2H) 6.98 (m, 2H) 7.51 (m, IH), ES-HRMS 415.1321 (M+H calcd for C18H22ClF2N4O3 requires 415.1343). After chromatography, LCMS shows mass corresponding to chloride isotope signature instead of bromide. High-resolution MS confirms chloride exchange.
Example 107
iV-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)ethyI]methanesulfonamide
[0298] Preparation of N-[2-({ 5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } amino)ethyl]methanesulfonamide. Prepared as N-[3-( { 5- bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl}amino)propyl]methane sulfonamide utilizing 2-[(2-aminoethyl)amino]-5-bromo-6-
[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one trifluoroacetate (0.299 g, 0.72 mmol) and methanesulfonyl chloride (0.090, 1.08 mmol) to afford a light yellow solid (0.094 g, 29%). 1H NMR (400 MHz, CD3OD) δ ppm 1.53 (d, J = 6.98 Hz, 6H) 2.90 (s, 3H) 3.26 (t, J = 6.18 Hz, 2H) 3.55 (m, 2H) 4.74 (m, IH) 5.44 (s, 2H) 6.96 (m, 2H) 7.50 (m, IH), ES-HRMS 451.0989 (M+H calcd for C17H22ClF2N4O4S requires 451.1013). Same bromide to chloride exchange as seen with iV-[2-({5-chloro-4-[(2,4- difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl } amino)ethyl]acetamide.
Example 108
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)ethyl]-2-hydroxyacetamide
Step 1. Preparation of (2-(4-(2,4-difluorobenzyloxy)-5-chloro-l,6-dihvdro-l-isopropyl- 6-oxopyrimidin-2-ylamino')diethylcarbamoyl) methyl acetate.
[0299] Prepared as N-[3-({ 5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxyacetamide (step 1) utilizing 2-[(2- aminoethyl)amino]-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)- one trifluoroacetate (0.300 g, 0.72 mmol) and (chlorocarbonyl)methyl acetate (0.118 mL, 1.08 mmol) to afford an off white solid (0.181 g, 44%).
1H ΝMR (400 MHz, CD
3OD) δ ppm 1.51 (d, J = 6.98 Hz, 6H) 2.07 (s, 6H) 3.69 (t, J = 6.04 Hz, 2H) 3.91 (t, J = 5.91 Hz, 2H) 4.62 (m, IH) 4.95 (s, 4H) 5.44 (s, 2H) 6.98 (m, 2H) 7.51 (m, IH). LC/MS, t
r = 2.85 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O
0C), ES-MS mJz 573 (M+l). Same bromide to chloride exchange seen as in N-[2-({5- chloro-4-[(2,4-difluorobenzyl)oxy]- l-isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl } amino)ethyl]acetamide.
Step 2. Preparation of N-f2-({5-chloro-4-[(2,4-difluorobenzyl)oxyl-l-isopropyl-6-oxo- 1 ,6-dihvdropyrimidin-2-yl ] amino)ethyll-2-hvdroxyacetamide.
[0300] Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxyacetamide (step 2) utilizing (2-(4-(2,4- difluorobenzyloxy)-5-chloro-l,6-dihydro-l-isopropyl-6-oxopyrimidin-2- ylamino)ethylcarbamoyl)methyl acetate (from step 1) (0.181 g, 0.32 mmol) and K2CO3 (0.065 g, 0.47 mmol) to afford an off white solid (0.100 g, 73%). 1H ΝMR (400 MHz, CD3OD) 5 ppm 1.51 (d, J = 6.71 Hz, 6H) 3.48 (t, J = 6.58 Hz, 2H) 3.57 (t, J = 5.64 Hz, 2H) 3.95 (s, 2H) 4.74 (m, IH) 5.45 (s, 2H) 6.97 (m, J = 8.59 Hz, 2H) 7.51 (m, IH), ES- HRMS 431.1284 (M+H calcd for C18H22ClF2N4O4 requires 431.1292).
Example 109
N-[2-({5-chIoro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)ethyl]-2-hydroxy-2-methylpropanamide
Step 1. Preparation of 2-(2-(4-(2,4-difluorobenzyloxy)-5-chloro-l,6-dihvdro-l- isopropvl-6-oxopvrimidin-2-vlamino)ethvlcarbamoyl)propan-2-vl acetate.
[0301] Prepared as iV-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxy-2-methylpropanamide (step 1) utilizing 2-[(2-aminoethyl)amino]-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin- 4(3H)-one trifluoroacetate (0.307 g, 0.74 mmol) and 2-(chlorocarbonyl)propan-2-yl acetate (0.18 mL, 1.11 mmol) to afford an off white solid (0.127 g, 34%). LC/MS, tr = 2.72 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 nm, at 5O0C), ES-MS m/z 501 (M+l). Same bromide to chloride exchange seen as in N-[2-({5- chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-2- yl } amino)ethyl] acetamide.
Step 2. Preparation of Λ^r2-(f 5-chloro-4-f(2,4-difIuorobenzyl)oxyl-l-isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl 1 amino)ethyl"|-2-hvdroxy-2-methylpropanamide. [0302] Prepared as iv*-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxy-2-methylpropanamide (step 2) utilizing (2-(4-(2,4-difluorobenzyloxy)-5-chloro-l,6-dihydro-l-isopropyl-6-oxopyrimidin-2- ylamino)ethylcarbamoyl)methyl acetate (from step 1) (0.127 g, 0.25 mmol) and K2CO3 (0.053 g, 0.38 mmol) to afford an off white solid (0.044 g, 38%). 1H NMR (400 MHz, CD3OD) δ ppm 1.31 (s, 6H) 1.52 (d, J = 6.98 Hz, 6H) 3.45 (m, 2H) 3.57 (m, 2H) 4.75
(m, IH) 5.46 (s, 2H) 6.98 (m, 2H) 7.52 (m, IH), ES-HRMS 459.1593 (M+H calcd for C20H26ClF2N4O4 requires 459.1603).
Example 110
N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)ethyl]methanesulfonamide
[0303] Preparation of N-[2-({ 5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}amino)ethyl]methanesulfonamide. Prepared as iV-[3-({5- bromo-4- [(2,4-difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}amino)propyl]methane sulfonamide utilizing 2-[(2-aminoethyl)amino]-5-bromo-6- [(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one trifluoroacetate (0.303 g, 0.57 mmol) and methanesulfonyl chloride (0.050 mL, 0.63 mmol) to afford a white solid (0.100 g, 35%). 1H NMR (400 MHz, CD3OD) δ ppm 1.52 (d, J = 6.98 Hz, 6H) 2.90 (s, 3H) 3.26 (t, J = 6.18 Hz, 2H) 3.55 (t, J = 6.04 Hz, 2H) 4.74 (m, IH) 5.43 (s, 2H) 6.97 (m, 2H) 7.50 (m, IH), ES-HRMS 495.0540 (M+H calcd for CnH22BrF2N4O4S requires 495.0508).
N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)ethyl]-2-hydroxyacetamide
Step 1. Preparation of (2-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihydro-l-isopropyl- 6-oxopyrimidin-2-ylamino)ethylcarbamoyl)methyl acetate.
[0304] Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxyacetamide (step 1) utilizing 2-[(2- aminoethyl)amino]-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)- one trifluoroacetate (0.404 g, 0.97 mmol) and (chlorocarbonyl)methyl acetate (0.113 mL, 1.06 mmol) to afford a white solid (0.292 g, 64%). 1H NMR (400 MHz, CD3OD) δ ppm 1.51 (d, J = 6.98 Hz, 6H) 2.09 (s, 3H) 3.46 (t, / = 5.77 Hz, 2H) 3.56 (t, J = 5.77 Hz, 2H) 4.50 (s, 2H) 4.71 (m, IH) 5.43 (s, 2H) 6.97 (m, J = 8.86 Hz, 2H) 7.51 (m, IH). UZIMS, tr = 2.48 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mlVmin, at 254 nm, at 5O0C), ES-MS m/z 517 (M+l).
Step 2. Preparation of N-[2-({5-bromo-4-r(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihvdropyrimidin-2-yl)amino")ethyll-2-hydroxyacetamide.
[0305] Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } amino)propyl]-2-hydroxyacetamide utilizing (2-(4-(2,4- difluorobenzyloxy)-5-bromo-l,6-dihydro-l-isopropyl-6-oxopyrimidin-2- ylamino)ethylcarbamoyl)methyl acetate (from step 1) (0.292 g, 0.56 mmol) and K2CO3 (0.116 g, 0.84 mmol) to afford a white solid (0.264 g, 100 %). 1H NMR (400 MHz, CD3OD) δ ppm 1.51 (d, J = 6.98 Hz, 6H) 3.48 (t, / = 5.77 Hz, 2H) 3.58 (t, J = 5.77 Hz, 2H) 3.95 (s, 2H) 4.73 (m, IH) 5.45 (s, 2H) 6.98 (m, 2H) 7.52 (m, IH), ES-HRMS 475.0795 (M+H calcd for C8H22BrF2N4O4 requires 475.0787).
Example 112
N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)ethyl]-2-hydroxy-2-methylpropanamide
Step 1. Preparation of 2-(2-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihvdro-l- isopropyl-6-oxopyrimidin-2-ylamino)ethylcarbamoyl)propan-2-yl acetate.
[0306] Prepared as Λ43-({5-bromo-4-[(2,4-difIuorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxy-2-methylpropanamide (step 1) utilizing 2-[(2-aminoethyl)amino]-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin- 4(3H)-one trifluoroacetate (0.403 g, 0.97 mmol) and 2-(chlorocarbonyl)propan-2-yl acetate (0.17 mL, 1.06 mmol) to afford a light pink colored solid (0.335 g, 63%). 1H NMR (400 MHz, CD3OD) δ ppm 1.51 (s, 6H) 1.52 (m, 6H) 2.02 (s, 3H) 3.45 (t, J = 5.37 Hz, 2H) 3.55 (t, J = 5.64 Hz, 2H) 4.68 (m, IH) 5.43 (s, 2H) 6.98 (m, 2H) 7.53 (m, IH). LC/MS, tr = 2.77 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mL/min, at 254 run, at 5O0C), ES-MS mJz 545 (M+l).
Step 2. Preparation of 7V-[2-({5-bromo-4-r(2,4-difluorobenzyl)oxyl-l-isopropyl-6-oxo- 1 ,6-dihvdropyrimidin-2-yl ) amino)ethvπ-2-hvdroxy-2-rnethylpropanamide. [0307] Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]-2-hydroxy-2-methylpropanamide (step 2) utilizing 2-(2-(4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihydro-l-isopropyl-6-oxopyrimidin-2- ylamino)ethylcarbamoyl)propan-2-yl acetate (from step 1) (0.335 g, 0.61 mmol) and K2CO3 (0.127 g, 0.92 mmol) to afford a white solid (0.171 g, 56%). 1H NMR (400 MHz, CD3OD) δ ppm 1.31 (s, 6H) 1.51 (d, J = 6.71 Hz, 6H) 3.45 (t, J = 5.77 Hz, 2H) 3.57 (t, / = 5.77 Hz, 2H) 4.75 (m, IH) 5.45 (s, 2H) 6.98 (s, 2H) 7.52 (m, IH), ES-HRMS 503.1071 (M+H calcd for C20H26BrF2N4O4 requires 503.1100).
Example 113
N-[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)ethyl]urea
[0308] Preparation of Λ^[2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}amino)ethyl]urea. N-[2-({5-bromo-4-[(2,4- difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } amino)ethyl]urea (0.283 g, 0.62 mmol) was dissolved in dioxane (1.5 mL) and tetrabutylammonium chloride (0.690 g, 2.84 mmol) was added. The reaction was heated to 100°C for 48 hours. The reaction was concentrated and the residue was dissolved in ethyl acetate and washed with water. The aqueous layer was extracted into ethyl acetate. The combined organics were dried over MgSO4, filtered, and concentrated. The residue was purified by chromatography (silica gel, ethyl acetate with 10% methanol/hexanes) to afford a light yellow solid (0.094 g, 37%). 1H NMR (400 MHz, CD3OD) δ ppm 1.52 (dd, J = 1.21, 6.85 Hz, 6H) 3.35 (t, J = 5.77 Hz, 2H) 3.49 (t, J = 5.77 Hz, 2H) 4.74 (m, IH) 5.44 (s, 2H) 6.97 (m, 2H) 7.51 (m, IH), ES-HRMS 416.1325 (M+H calcd for C17H21ClF2N5O3 requires 416.1296).
Example 114
N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)ethyl]acetamide
Step 1. Preparation of tert-butyl 2-(4-(2,4-difluorobenzyloxy)-5-bromo-1.6-dihydro-l- isopropyl-6-oxopyrimidin-2-yloxy)ethylcarbamate.
[0309] Prepared as tert-butyl 2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6- oxo-l,6-dihydropyrimidin-2-yl}oxy)ethylcarbamate utilizing 6-(2,4-difluorobenzyloxy)- 5-bromo-3-isopropyl-2-(methylsulfonyl)pyrimidin-4(3H)-one (3.0 g, 6.9 mmol) and t- butyl 2-hydroxyethylcarbamate (1.18 mL, 7.6 mmol) to afford a white solid (0.440 g, 37%). 1H NMR (400 MHz, CD3OD) δ ppm 1.41 (s, 9H) 1.45 (d, J = 6.98 Hz, 6H) 3.49 (t, J = 5.24 Hz, 2H) 4.51 (t, J = 5.24 Hz, 2H) 5.31 (m, IH) 5.47 (s, 2H) 7.00 (m, 2H) 7.54 (m, IH). LC/MS, tr = 3.30 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mlVmin, at 254 nm, at 5O0C), ES-MS m/z 518 (M+l).
Step 2. Preparation of 6-(2,4-difluorobenzyloxyy2-(2-aminoethoxy)-5-bromo-3- isopropvlpvrimidin-4(3H)-one trifluoroacetic acid.
[0310] Prepared as 2-{ [2-({5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}oxy)ethyl] amino }-2-oxoethyl acetate (stepl) utilizing t-butyl 2- (4-(2,4-difluorobenzyloxy)-5-bromo-l,6-dihydro-l-isopropyl-6-oxopyrimidin-2- yloxy)ethylcarbamate (from step 1) (2.36 g, 4.57 mmol) and trifluoroacetic acid (5 mL) to afford a light brown oil (0.489 g, >100 %). 1H NMR (400 MHz, CD3OD) δ ppm 1.49 (d, J = 6.98 Hz, 6H) 3.46 (m, 2H) 4.72 (m, 2H) 5.22 (m, IH) 5.48 (s, 2H) 7.01 (m, 2H) 7.54 (m, IH). LC/MS, tr = 1.86 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 mLVmin, at 254 nm, at 5O0C), ES-MS m/z 418 (M+l).
Step 3. Preparation of N-r2-({5-brorno-4-r(2,4-difluorobenzyl)oxyl-l-isopropyl-6-oxo- 1 ,6-dihvdropyrimidin-2-yl ) oxytethyliacetamide.
[0311] Prepared as N-[3-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}amino)propyl]acetamide (step 2) utilizing 6-(2,4- difluorobenzyloxy)-2-(2-aminoethoxy)-5-bromo-3-isopropylpyrimidin-4(3H)-one trifluoroacetic acid (from step 1) (0.400 g, 0.75 mmol) and acetyl chloride (0.064 mL, 0.83 mmol) to afford a pink solid (0.200 g, 58%). 1H NMR (400 MHz, CD3OD) δ ppm 1.44 (d, J = 6.98 Hz, 6H) 1.94 (s, 3H) 3.61 (t, J = 5.37 Hz, 2H) 4.54 (t, J = 5.37 Hz, 2H) 5.28 (m, IH) 5.47 (m, 2H) 7.00 (m, 2H) 7.55 (m, IH), ES-HRMS 460.0697 (M+H calcd for Ci8H2IBrF2N3O4 requires 460.0678).
Example 115
N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yI}oxy)ethyl]urea
[0312] Preparation of N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}oxy)ethyl]urea. Prepared as N-[3-({5-bromo-4-[(2,4- difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl } amino)propyl]urea utilizing 6-(2,4-difluorobenzyloxy)-2-(2-aminoethoxy)-5-bromo-3-isopropylpyrimidin- 4(3H)-one trifluoroacetic acid (from 7V-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}oxy)ethyl]acetamide step 2) (0.406 g, 0.76 mmol) and trimethylsilyl isocyanate (0.96 mL, 0.84 mmol) to afford a white solid (0.146 g, 42%). 1H NMR (400 MHz, CD3OD) δ ppm 1.45 (d, J = 6.98 Hz, 6H) 3.55 (t, J = 5.37 Hz, 2H) 4.53 (t, J = 5.37 Hz, 2H) 5.28 (m, IH) 5.47 (s, 2H) 6.98 (m, 2H) 7.54 (m, IH), ES-HRMS 461.0613 (M+H calcd for CnH20BrF2N4O4 requires 0630).
Example 116
N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yI}oxy)ethyl]methanesulfonamide
[0313] Preparation of N-[2-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo- l,6-dihydropyrimidin-2-yl}oxy)ethyl]methanesulfonamide. Prepared as iV-[3-({5- bromo-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}amino)propyl]methane sulfonamide utilizing 6-(2,4-difluorobenzyloxy)-2-(2- aminoethoxy)-5-bromo-3-isopropylpyrimidin-4(3H)-one trifluoroacetic acid (from N-[2- ( { 5-bromo-4-[(2,4-difluorobenzyl)oxy]- 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}oxy)ethyl]acetamide, step 2) (0.489 g, 0.92 mmol) and methanesulfonyl chloride (0.077 mL, 1.0 mmol) to afford a yellow-orange solid (0.352 g, 77%). 1H NMR (400 MHz, CD3OD) δ ppm 1.47 (d, J = 6.71 Hz, 6H) 2.95 (s, 3H) 3.49 (t, J = 5.37 Hz, 2H) 4.57 (t, J = 5.50 Hz, 2H) 5.30 (m, IH) 5.47 (s, 2H) 7.00 (m, 2H) 7.54 (m, IH), ES- HRMS 496.0322 (M+H calcd for C17H21BrF2N3O5S requires 496.0348).
5-chloro-6-[(2,4-difluorobenzyI)oxy]-3-isopropyl-2-(methylsuIfonyl)pyriinidin-
4(3H)-one
Step 1. Preparation of 5-chloro-6-r(2,4-difluorobenzyl)oxyl-3-isopropyl-2- (methylthio)pyrimidin-4(3H)-one.
[0314] 6-(2,4-difluorobenzyloxy)-3-isopropyl-2-(methythio)pyrimidin-4(3H)-one (5.00 g, 15.32 mmol) was dissolved in dichloromethane (50 mL) and cooled in an ice-bath. N- Chlorosuccinimide (2.25 g, 16.85 mmol) was added and the reaction was allowed to stir, with warming, for 24 hours. The reaction mixture was then washed with H2O, dried over Na2SO4, filtered and concentrated to provide a white solid (5.61g, >100%). 1H NMR (400 MHz, CDCl3) δ 7.43 (app q, J = 7.7 Hz, IH), 6.91-6.87 (m, IH), 6.84-6.79 (m, IH), 5.47 (s, 2H), 4.58-4.51 (m, IH), 2.51 (s, 3H), 1.59 (d, J = 6.7 Hz, 6H). LC/MS, tr = 3.25 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 ml/min, at 254 run, at 5O0C), ES-MS m/z 361 (M + H).
Step 2. Preparation of 5-chloro-6-[(2,4-difluorobenzyl)oxyl-3-isopropyl-2- (methylsulfonyl)pyrimidin-4(3H)-one.
[0315] 5-Chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2-(methylthio)pyrimidin- 4(3H)-one (crude from Stepl) (5.61 g, 15.55 mmol) was dissolved in tetrahydrofuran (50 mL) and H2O (5 mL). Oxone (38.3g, 62.2 mmol) was added and the resulting mixture was stirred at room temperature for 4 days. The reaction mixture was then filtered washing the solids with ethyl acetate. The filtrate was diluted with H2O and then extracted with ethyl acetate. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated. The resulting solids were washed with diethyl ether to afford a white solid (4.02 g, 66%). 1H NMR (400 MHz, CDCl3) δ 7.42 (app q , / = 8.5 Hz, IH), 6.93 (dt, J = 8.6, 1.5 Hz, IH), 6.84 (dt, J = 9.3, 2.4 Hz, IH), 5.40 (s, 2H),
5.23 (septet, J = 6.6 Hz, IH), 3.38 (s, 3H), 1.64 (d, J = 6.6 Hz, 6H). LC/MS, tr = 2.60
minutes (5 to 95% acetonitrile/water over 5 minutes at 1 ml/min, at 254 nm, at 5O0C), ES-MS m/z 415 (M + Na).
N~2~-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyI-6-oxo-l,6- dihydropyrimidin-2-yl}-N~2~-methylglycinamide
Step 1. Preparation of tert-butyl N-|5-chloro-4-r(2,4-difluorobenzyl)oxyl-l-isopropyl-6- oxo-l,6-dihydropyrimidin-2-yl)glvcinate.
[0316] Glycine tert-butyl ester hydrochloride (1.71 g, 10.18 mmol) was suspended in tetrahydrofuran (20 mL). Dimethylamine bound to silica gel (6.79 g, Loading = 1.50 mmol/g) was added and the resulting mixture was agitated at room temperature for 30 minutes. 5-chloro-6-[(2,4-difluorobenzyl)oxy]-3-isopropyl-2- (methylsulfonyl)pyrimidin-4(3H)-one (2.00 g, 5.09 mmol) was added and the mixture was agitated at room temperature for 2 days. The reaction mixture was filtered and concentrated. The residue was treated with methanol and the precipitate was collected by filtration to provide a white solid (0.79 g, 35%). 1H NMR (300 MHz, CDCl3) δ 7.46 (app q, J = 7.7 Hz, IH), 6.88 (dt, J = 8.4, 2.2 Hz, IH), 6.82 (m, IH), 5.69 (br s, IH), 5.48 (br m IH), 5.38 (s, 2H), 4.05 (d, J = 4.2 Hz, 2H), 1.52 (d, J = 7.3 Hz, 6H) 1.50 (s, 9H).
LC/MS, tr = 3.26 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 ml/min, at 254 nm, at 5O0C), ES-MS m/z 444 (M + H).
Step 2. Preparation of tert-butyl N-|5-chloro-4-[(2,4-difluorobenzyl)oxyl-l-isopropyl-6- oxo- 1 ,6-dihydropyrimidin-2-vl ) -N-methvlgl vcinate.
[0317] Tert-Butyl N-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}glycinate (N~2~-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l- isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}-N~2~-methylglycinamide, step 1) was suspended in tetrahydrofuran (10 mL) and cooled in an ice-bath. Sodium hydride (60% in mineral oil, 0.106 g, 2.64 mmol) was added and the ice-bath was removed. After 30 minute the ice-bath was replaced and iodomethane (0.151 mL, 2.42 mmol) was added. After stirring, with warming, overnight additional sodium hydride (60% in mineral oil, 0.026 g, 0.66 mmol) was added followed by iodomethane (0.068 mL, 1.10 mmol). The resulting mixture was stirred at room temperature for 1.5 hours then quenched by the addition of saturated NH4Cl and extracted into ethyl acetate. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated. Chromatography (silica gel, hexanes/ethyl acetate) afforded a pale yellow solid (0.920 g, 91%). 1H NMR (400 MHz, CDCl3) δ 7.45 (app q, J = 7.8 Hz, IH), 6.87 (m, IH), 6.81 (m, IH), 5.34 (s, 2H), 4.47 (septet, J = 6.7 Hz, IH), 3.83 (s, 2H), 3.01 (s, 3H), 1.63 (d, J = 6.7Hz, 6H) 1.44 (s, 9H). LC/MS, tr = 3.47 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 ml/min, at 254 nm, at 5O0C), ES-MS m/z 458 (M + H).
Step 3. Preparation of N-{5-chloro-4-[(2,4-difluorobenzyl)oxyl-l-isopropyl-6-oxo-1.6- dihvdropyrimidin-2-vU-N-methyl glycine.
[0318] Tert-Butyl N-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl}-N-methylglycinate (Step 2) (0.920 g, 2.01 mmol) was treated with formic acid (4.0 mL) and stirred at room temperature for 24 hours. The reaction mixture was partially concentrated and portioned between H2O and ethyl acetate. The aqueous layer was further extracted with ethyl acetate. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated to provide a pale yellow solid (0.690 g, 85%). 1H NMR (300 MHz, CDCl3) 67.42 (app q, J = 7.8 Hz, IH), 6.86 (m, IH), 6.79 (m, IH), 5.32 (s, 2H), 4.47 (septet, 7 = 6.8 Hz, IH), 3.90 (s, 2H), 3.04 (s, 3H), 1.61 (d, J = 6.7 Hz, 6H). LC/MS, tr = 2.51 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 ml/min, at 254 nm, at 500C), ES-MS tn/z 402 (M + H).
Step 4. Preparation of N~2~-{5-chloro-4-r(2,4-difluorobenzyl)oxy1-l-isopropyl-6-oxo- l,6-dihvdropyrimidin-2-yl}-N~2~-methylglvcinamide. [0319] N-{5-chloro-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } -N-methylglycine (N~2~- { 5-chloro-4-[(2,4-difluorobenzyl)oxy]- l-isopropyl-6-oxo-l,6-dihydropyrimidin-2-yl}-N~2~-methylglycinamide, Step 3) (0.300g, 0.747 mmol) was dissolved in dioxane (7.0 mL). 1-Hydroxybenzotriazole (0.101 g, 0.747 mmol) was added followed by polymer bound carbodiimide resin (1.0Og, loading = 1.8 mmol/g). The mixture was then agitated at room temperature for 15 minutes. Ammonia in dioxane (2.62 mL, 1.31 mmol, 0.5 M) was added and the reaction mixture was agitated at room temperature overnight. At this time the reaction was diluted with dioxane (10 mL) and treated with polyamine resin (1.30 g, loading = 2.87 mmol/g) followed by methylisocyanate fucntionalized polystyrene (2.54 g, loading = 1.47 mmol/g). The resulting reaction was agitated at room temperature for 3 hours and the filtered and concentrated to provide the title compound as a pale yellow solid (0.220
g, 74%). 1H NMR (300 MHz, CDCl3) δ 7.40 (app q, J = 7.8 Hz, IH), 6.87-6.74 (m, 2H), 6.55 (br s, IH), 5.91 (br s, IH), 5.33 (s, 2H), 4.45 (septet, J = 6.6 Hz, IH), 3.85 (s, 2H), 2.98 (s, 3H), 1.58 (d, J = 6.7Hz, 6H). LC/MS, tr = 2.28 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 ml/min, at 254 nm, at 500C), ES-MS m/z 401 (M + H). ES-HRMS m/z 401.1202 (M+H calcd for C17H20ClF2N4O3 requires 401.1187).
Example 119
5-bromo-6-[(2,4-difluorobenzyI)oxy]-2-[(2-hydroxy-2-methylpropyl)amino]-3- isopropyIpyrimidin-4(3H)-one
[0320] Preparation of 5-bromo-6-[(2,4-difluorobenzyl)oxy]-2-[(2-hydroxy-2- methylpropyl)amino]-3-isopropylpyrimidin-4(3H)-one. l-Amino-2-methyl-2-propanol hydrochloride (0.15 g, 1.19 mmol) was dissolved in dioxane (10 mL). Carbonate bound to silica gel (3.53 g, loading = 0.7 mmol/g) was added and the mixture was stirred at room temperature for 5 minutes. 6-(2,4-difluorobenzyloxy)-5-bromo-3-isopropyl-2- (methylsulfonyl)pyrimidin-4(3H)-one (0.300 g, 0.686 mmol) was added and the resulting mixture was stirred at room temperature for 18 hours. The reaction mixture was then filtered and concentrated. The filtrate was subjected to chromatography (silica gel, hexanes/ethyl acetate) to provide an off-white oily solid (0.143 g, 47%). 1H NMR (400 MHz, CDCl3) δ 7.44 (app q , J = 7.8 Hz, IH), 6.93 (app t, J = 8.3 Hz, IH), 6.84 (dt, J = 9.1, 2.3 Hz, IH), 5.54 (br s, IH), 5.36 (s, 2H), 3.42 (d, J = 5.0 Hz, 2H), 1.46 (d, J = 7.3 Hz, 6H) 1.26 (s, 6H). LC/MS, tr = 2.77 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 ml/min, at 254 nm, at 5O0C), ES-MS m/z 446 (M + H). ES-HRMS m/z 446.0862 (M+H calcd for C]8H23BrF2N3O3 requires 446.0885).
N-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-
2-yl}methyl)methanesulfonamide
Step 1. Preparation of 5-bromo-4-r(2,4-difluorobenzyl)oxyl-l-isopropyl-6-oxo-l,6- dihvdropyrimidine-2-carbonitrile.
[0321] 6-(2,4-difluorobenzyloxy)-5-bromo-3-isopropyl-2-(methylsulfonyl)pyrimidin- 4(3H)-one (9.85 g, 22.53 mmol) was dissolved in N,iV-dimethylacetamide (50 mL). Potassium cyanide (2.93 g, 45.06 mmol) was added and the resulting mixture was stirred at room temperature for 3 hours. Ethyl acetate (100 mL) was added to the reaction mixture, which was then poured into ice/water (100 mL). The reaction was then extracted into ethyl acetate. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated. The residue was subjected to chromatography (silica gel, hexanes/ethyl acetate) to provide a bright yellow solid (5.05 g, 58%). 1H NMR (400 MHz, CDCl3) δ 7.45 (app q, J = 7.7 Hz, IH), 6.89 (m, IH), 6.84 (m, IH), 5.44 (s, 2H), 5.14 (m, IH), 1.66 (d, J = 7.0 Hz, 6H). LC/MS, tr = 3.13 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 ml/min, at 254 nm, at 500C), ES-MS mJz 384 (M + H).
Step 2. Preparation'of 2-(aminomethylV5-bromo-6-[(2,4-difluorobenzyl')oxyl-3- isopropvlpvrimidin-4(3H)-one.
[0322] 5-Bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidine- 2-carbonitrile (Stepl) (1.30 g, 3.38 mmol) was dissolved in ethyl acetate (4.0 mL) and acetic acid (4.0 mL). 10% Pd/C (0.676 g) was added and the flask was fitted with a balloon containing H2. The resulting mixture was stirred at room temperature for 1 hour. The reaction was filtered through a pad of Celite® washing with ethyl acetate. The reaction mixture was made alkaline (pH 10-11) with 2.5 N NaOH. The aqueous layer was removed and the organic layer was washed with brine, dried over Na2SO4, filtered and concentrated. The residue was dissolved in dichloromethane (10 mL). N- Bromosuccinimide (0.141 g, 0.792 mmol) was added and the mixture was stirred at room temperature for 4 hours. Additional N-bromosuccinimide (0.028 g, 0.159 mmol) was added and the resulting mixture was stirred at room temperature overnight. The reaction was concentrated and then ethyl acetate and H2O were added. The reaction was made alkaline (pH 10) with 2.5 Ν NaOH. The aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to afford an orange/brown foam (1.26 g, >100%). LC/MS, tr = 1.76 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 ml/min, at 254 nm, at 5O0C), ES-MS m/z 388 (M + H).
Step 3. Preparation of N-({5-bromo-4-[Y2,4-difluorobenzyl)oxy1-l-isopropyl-6-oxo-l,6- dihvdropyrimidin-2-yl I methvPmethanesulfonamide.
[0323] 2-(Aminomethyl)-5-bromo-6-[(2,4-difluorobenzyl)oxy]-3-isopropylpyrimidin- 4(3H)-one (crude from Step 2) (0.300 g, 0.773 mmol) was dissolved in dichloromethane
(2.0 mL) and cooled in an ice bath. Triethylamine (0.162 mL, 1.16 mmol) was added followed be methanesulfonyl chloride (0.066 mL, 0.850 mmol). The resulting mixture was stirred for 1.5 hours with cooling. The reaction was quenched by the addition of saturated NaHCO3 and extracted in dichloromethane. The combined organic layers were dried over Na2SO4, filtered and concentrated. Chromatography (silica gel, hexanes/ethyl acetate with 10% methanol) provided the title compound as a pale pink solid (0.250 g, 69%). 1H NMR (400 MHz, CDCl3) δ 7.45 (app q , J = 7.8 Hz, IH), 6.88 (app t, J = 8.3 Hz, IH), 6.83 (dt, J = 9.5, 2.3 Hz, IH), 5.64 (m, IH), 5.44 (s, 2H), 4.35 (d, J = 5.2 Hz, 2H), 2.97 (s, 3H), 1.58 (d, J = 7.3 Hz, 6H). LC/MS, tr = 2.52 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 ml/min, at 254 nm, at 5O0C), ES-MS mJz 466 (M + H). ES-HRMS mJz 466.0219 (M+H calcd for Ci6H19BrF2N3O4S requires 446.0242).
N-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6-dihydropyrimidin-
2-yl}methyI)acetamide
[0324] Preparation of N-({5-bromo-4-[(2,4-difluorobenzyl)oxy]-l-isopropyl-6-oxo-l,6- dihydropyrimidin-2-yl } methyl)acetamide. 2-(aminomethyl)-5-bromo-6-[(2,4- difluorobenzyl)oxy]-3-isopropylpyrimidin-4(3H)-one (N-({5-bromo-4-[(2,4- difluorobenzyl)oxy] - 1 -isopropyl-6-oxo- 1 ,6-dihydropyrimidin-2- yl}methyl)methanesulfonamide, step 2) (0.300 g, 0.773 mmol) was dissolved in dichloromethane (2.0 mL) and cooled in an ice bath. Triethylamine (0.162 mL, 1.16 mmol) was added followed by acetyl chloride (0.060 mL, 0.850 mmmol). The resulting mixture was stirred, with cooling, for 2 hours. The reaction was quenched by the addition of saturated NaHCO3 and extracted in dichloromethane. The combined organic layers were dried over Na2SO4, filtered and concentrated. Chromatography (silica gel, hexanes/ethyl acetate with 10% methanol) provided the title compound as an off-white
solid (0.170 g, 51%). 1H NMR (400 MHz, CDCl3) δ 7.46 (app q , J = 8.5 Hz, IH), 6.88 (app t, J = 7.9 Hz, IH), 6.81 (dt, J = 9.5, 2.3 Hz, IH), 6.68 (br s, IH), 5.45 (s, 2H), 4.45 (d, J = 4.4Hz, 2H), 4.35 (m, IH), 2.08 (s, 3H), 1.56 (d, J = 6.7 Hz, 6H). LC/MS, tr = 2.34 minutes (5 to 95% acetonitrile/water over 5 minutes at 1 ml/min, at 254 nm, at 5O0C), ES-MS m/z 430 (M + H). ES-HRMS m/z 430.0532 (M+H calcd for CnH19BrF2N3O3 requires 430.0572).
Example 122
N-(2-{[(5-bromo-l-isopropyl-2-methyl-6-oxo-l,6-dihydropyrimidin-4- yl)oxy]methyl}-5-fluorobenzyl)-N'-ethylurea
Step 1: Preparation of 6-hvdroxy-2-methyl-3-(methylethyl)-3-hvdropyrimidin-4-one.
[0325] A mixture of (iminoethyl)(methylethyl)amine (6.08g, 60.8 mmol) and sodium methoxide (25% in methanol, 26.3 mL) was stirred at room temperature for 15 min, and heated in an oil bath to remove methanol. Diethyl malonate (9.69 ml, 63.84 mmol) was added and the mixture was heated to 110 0C with stirring for 15 min. The resulting yellow solid was purified by silica gel flash chromatography using 7% methanol in dichloromethane as eluant to give 4.65g (45.5%) of the desired compound as a beige
solid: 1H NMR (CD3OD/400MHz) δ 5.20 (s,lH), 4.60 (br,lH), 2.59 (s, 3H), 1.56 (d, 6H, 7=6.6 Hz); ES-MS m/z=169.09.
Step 2: Preparation of 5-bromo-6-hvdroxy-2-methyl-3-(methylethyl)-3-hydropyrimidin- 4-one.
[0326] A mixture of 6-hydroxy-2-methyl-3-(methylethyl)-3-hydropyrimidin-4-one (4.3g, 25.6 mmol), N-bromosuccinimide (5.1g, 28.67 mmol) in dichloromethane (50 mL) was stirred at 5 0C under nitrogen for 1 h and warmed up to room temperature overnight. The reaction mixture was concentrated under vacuum and the residue was purified by silica gel flash chromatography using 7% methanol in dichloromethane as eluant to give 5.0g (80%) of the desired product as a solid: 1H NMR (CD3OD/400MHz) δ 4.61 (br.lH), 2.60 (s, 3H), 1.60 (d, 6H, 7=6.4 Hz).
Step 3: Preparation of 2-{ r5-bromo-2-methyl-l-(methylethyl)-6-oxohvdropyrimidin-4- yloxylmethvπ-5-fluorobenzenecarbonitrile.
[0327] A mixture of 5-bromo-6-hydroxy-2-methyl-3-(methylethyl)-3-hydropyrimidin-4- one (2.55g,10.32 mmol), 2-cyano-4-fluorobenzyl bromide (2.32g, 10.836 mmol) and potassium carbonate (1.71g, 12.384 mmol) in DMF (30 mL) was stirred at room temperature for 18 h. The resulting mixture was diluted with ethyl acetate (25ml), washed with water (2 x 100 mL), brine, dried over sodium sulfate and concentrated under reduced pressure. The resulting material was purified by silica gel flash
chromatography using 40% EtOAc in hexanes as eluant to give 2.89g.(73.8%) of the desired compound as a white powder: 1H NMRCD3OD/400MHz) δ7.78 (m, IH), 7.60 (m, IH),
7.44 (m, IH), 5.60 (s, 2H), 4.62 (br, IH), 2.60 (s, 3H), 1.60 (d, 6H, 7=6.8 Hz); ES-MS m/z 382.07 & 380.10.
Step 4: Preparation of 6-{r2-(aminomethyl)-4-fluorophenyllmethoxy)-5-bromo- 2-methyl-3-(methylethyl)-3-hvdropyrirnidin-4-one.
[0328] BH3THF (IM solution, 11.2 ml, 11.2 mmol) was added dropwise to a solution of 2- { [5-bromo-2-methyl- 1 -(methylethyl)-6-oxohydropyrimidin-4-yloxy]methyl } -5- fluorobenzenecarbonitrile in THF(18 mL) at 0 0C under nitrogen. After stirring at this temperature for 30 min, the mixture was allowed to warm up to room temperature overnight. 5 mL of methanol was added to the mixture at 00C and its volume was reduced to 10 mL under reduced pressure. The resulting material was purified by silica gel flash chromatography using 10% methanol in dichloromethane as eluant to give 1.24g (57.5%) of the title compound as a white solid : 1H NMR (CD3OD/400MHz) δ 7.43 (m, IH), 7.20 (m, IH), 6.97(m, IH), 5.50 (s, 2H), 4.62 (br, IH), 3.86 (s, 2H), 2.60(s,3H), 1.60 (d, 6H, 7=6.8 Hz); ES-MS m/z 386.12, & 384.13.
Step 5: Preparation of N-r(2-| [5-bromo-2-methyl-l-(methylethyl)-6- oxohydropyrimidin-4-yloxy1methyl|-5-fluorophenyl)methyl1(ethylamino)carboxamide.
[0329] Ethylisocyanate(0.103 ml, 1.3 mmol) was added to a solution of 6-{[2- (aminomethyl)-4-fluorophenyl]methoxy}-5-bromo-2-methyl-3-(methylethyl)-3- hydropyrimidin-4-one (0.25g, 0.65 mmol) in dichloromethane (5 mL) at 0 0C under nitrogen. The reaction mixture was stirred at this temperature for 1.5 h, solvent was
removed in vacuo and the residue was purified by silica chromatography column using 10% methanol in dichloromethane as eluant to afford the title compound (0.29g, 98%) as a white solid: mp 79-800C; 1H NMR (CD3ODMOOMHZ) 67.75(m,lH),7.10 (m,lH), 6.89 (m, IH), 5.80 (s, 2H), 4.65 (br,lH), 4.44(s, 2H), 3.16 (q, 2H), 2.56 (s, 3H), 1.59 (d, 6H, 7 =6.8 Hz), 1.061(t, 3H, 7=6.8 Hz); Anal. Calcd for C19H24BrFN4O3: C, 50.12; H, 5.31; N,12.30. Found: C, 49.50; H, 5.37; N.11.75; ES-MS m/z 457.17& 455.18.
Example 123
N-[(2-{[5-chloro-2-methyl-l-(methylethyl)-6-oxohydropyrimidin-4-yloxy]methyl}-5- fluorophenyl)methyl](ethylamino)carboxamide
Step 1: Preparation of 5-chloro-6-hvdroxy-2-methyl-3-(methylethyl)-3-hvdropwimidin- 4-one.
[0330] A mixture of 6-hydroxy-2-methyl-3-(methylethyl)-3-hydropyrimidin-4-one (4.13g, 24.58 mmol), N-chlorosuccinimide (3.68g, 27.56 mmol) in dichloromethane (40 mL) was stirred at 5 0C under nitrogen for Ih and warmed up to room temperature overnight. The reaction mixture was concentrated under vacuum. The residue was purified by silica gel flash chromatography using 7% methanol in dichloromethane as eluant to give 4.83g (97%) of the desired product as a solid: 1H NMR (CD3OD/400MHz) 5 4.60 (br.lH), 2.60 (s,3H), 1.60 (d, 6H, 7=6.4 Hz).
Step 2: Preparation of 2-(r5-chloro-2-methyl-l-(methylethyl)-6-oxohvdropwimidin-4- yloxylmethyl ) -5-fluorobenzenecarbonitrile.
[0331] A mixture of 5-chloro-6-hydroxy-2-methyl-3-(methylethyl)-3-hydropyrimidin-4- one (4.65g, 22.96 mmol), 2-cyano-4-fluorobenzyl bromide (5.41g, 25.25 mmol) and potassium carbonate (4.75g, 34.44 mmol) in DMF (50 mL) was stirred at room temperature for 18 h. The resulting mixture was diluted with ethyl acetate (25mL), washed with water (2x100 mL), brine, dried over sodium sulfate and concentrated under reduced pressure. The resulting material was purified by silica gel flash chromatography using 40% EtOAc in hexanes as eluant to give 5.Og of the title compound as a white powder (64.9%): 1H NMR (CD3OD/400MHz) 57.77 (m, IH), 7.60 (m, IH), 7.43 (m, IH), 5.60 (s, 2H), 4.61 (br, IH), 2.60 (s,3H), 1.60 (d, 6H, 7=6.8 Hz); ES-MS m/z 338.10 & 336.12.
Step 3: Preparation of 6-{ r2-(aminomethviy4-fluorophenyl]rnethoxy)-5-chloro-2- methyl-3-(methylethyl)-3-hvdropyrimidin-4-one.
[0332] BH3THF(IM solution, 25.26mL, 25.26 mmol) was added dropwise to the solution of 2-{ [5-chloro-2-methyl-l-(methylethyl)-6-oxohydropyrimidin-4- yloxy]methyl}-5-fluorobenzenecarbonitrile(4.24g, 12.62 mmol) in THF(40 mL) at O 0C under nitrogen. After stirring at this temperature for 30 min, the mixture was allowed to warm up to room temperature overnight. 10 mL of Methanol was added to the mixture at 00C and its volume was reduced to 10 mL under vacuo . The resulting material was
purified by silica gel flash chromatography using 10% methanol in dichloromethane as eluant to give 2.65g (61.8%) of the title compound as a yellow solid: 1H NMR (CD3OD/400MHz) 5 7.43 (m, IH), 7.20 (m, IH), 6.98 (m, IH), 5.45 (s, 2H), 4.61 (br, IH), 3.86(s, 2H), 2.60(s, 3H),1.60(d, 6H, J = 6.8 Hz); ES-MS m/z 342.15 & 340.17.
Step 4: Preparation of N-[(2-{[5-chloro-2-methyl-l-(methylethyl)-6-oxohvdropyrimidin- 4-yloxy1methyl)-5-fluorophenyl)methyll(ethylamino)carboxamide.
[0333] Ethylisocyanate(0.119 mL, 1.5 mmol) was added to a solution of 6- { [2- (aminomethyl)-4-fluorophenyl]methoxy}-5-chloro-2-methyl-3-(methylethyl)-3- hydropyrimidin-4-one (0.255g, 0.75 mmol) in dichloromethane (5 mL) at 0 0C under nitrogen. The reaction mixture was stirred at this temperature for 1.5 h, solvent was removed under vacuo and the residue was purified by silica gel chromatography column using 10% methanol in dichloromethane as eluant to afford the title compound (0.265g, 86%) as a yellow solid: mpl60-161 0C; 1HNMR(CD3ODMOOMHz) δ 7.46 (m,lH),7.08 (m,lH), 6.98 (m, IH), 5.51 (s, 2H), 4.63 (br,lH), 4.45 (s, 2H), 3.16 (q, 2H), 2.62 (s, 3H), 1.58(d, 6H, J = 6.8 Hz), 1.10(t, 3H, 7 = 7.6 Hz); Anal. Calcd for C19H24C1FN4O3: C, 55.54; H, 5.89; N.13.64. Found: C, 55.41; H, 6.12; N.13.10. ES-MS m/z 413.28 & 411.29.
N-[(2-{[5-chloro-2-methyl-l-(methyIethyl)-6-oxohydropyrimidin-4-yloxy]methyI}-5- fluorophenyl)methyl][(methylethyl)amino]carboxamide
[0334] lsopropylisocyanate(0.11 mL, 1.125 mmol) was added to a solution of 6- { [2- (aminomethyl)-4-fluorophenyl]methoxy}-5-chloro-2-methyl-3-(methylethyl)-3- hydropyrimidin-4-one (0.255g, 0.75 mmol) in dichloromethane (5 mL) at 00C under nitrogen. After stirring for 2 h at room temperature, the reaction mixture was purified by silica gel chromatography using 10% methanol in dichloromethane to give 0.304g (95.4%) of the title compound as a yellow solid: mp 131-132 °C; 1NMR (CD3ODMOOMHZ) 67.46 (m, IH), 7.07 (m,lH), 6.97 (m, IH), 5.50 (s, 2H), 4.61(br,lH), 4.45 (s, 2H), 3.79 (m, IH), 2.62 (s, 3H), 1.58 (d, 6H, J = 6.8 Hz), 1.12 (d, 6H, 7 =6.4 Hz); Anal. Calcd for C20H26C1FN4O3: C, 56.54; H, 6.17; N, 13.19. Found: C, 56.39; H, 6.41; N.12.82. ES-MS m/z 427.31& 425.31.
Example 125
N-[(2-{[5-bromo-2-methyI-l-(methylethyl)-6-oxohydropyrimidin-4-yloxy]methyl}-5- fluorophenyl)methyl](cyclohexylamino)carboxamide
[0335] Cyclohexylisocyanate(0.124 mL, 0.975 mmol) was added to a solution of 6{ [2-(aminomethyl)-4-fluorophenyl]methoxy}-5-bromo-2-methyl-3-(methylethyl)-3- hydropyrimidin-4-one (0.25g, 0.65mmol) in dichloromethane (5mL) at 0 0C under nitrogen. After stirring for 2 h at room temperature, the reaction mixture was purified by silica gel chromatography using 10% methanol in dichloromethane as the eluant to give 0.325g (98%) of the title compound as a white solid: mp 109-110 0C; 1NMR (CD3OD/400MHz) δ 7.46 (m, IH), 7.07(m,lH), 6.98(m,lH), 5.49(s, 2H), 4.61(br,lH), 4.46 (s, 2H), 3.46 (m, IH), 2.61(s, 3H), 1.58 (d, 6H, 7 = 4.4 Hz), 1.15-1.89 (m, HH);
Anal. Calcd for C23H30BrFN4O3: C, 54.23; H, 5.94; N, 11.00. Found: C, 54.43; H, 6.00; N, 10.74. ES-MS m/z 511.29 & 509.30.
N-[(2-{[5-Chloro-2-methyl-l-(methylethyl)-6-oxohydropyrimidin-4-yloxy]methyl}- 5-fluorophenyl)methyl](cyclohexylamino)carboxamide
[0336] Cyclohexylisocyanate (0.142 mL, 1.125 mmol) was added to a solution of 6-{ [2-(aminomethyl)-4-fluorophenyl]methoxy}-5-chloro-2-methyl-3-(methylethyl)-3- hydropyrimidin-4-one (0.255g, 0.75mmol) in dichloromethane (5 mL) at 0 0C under nitrogen. After stirring for 2 h at room temperature, the product was purified by silica gel chromatography using 10% methanol in dichloromethane as the eluant to give 0.34 Ig (98%) the title compound as a yellow solid: mp 95-96 0C; 1H NMR (CD3OD/400MHz) δ 7.45 (m, IH), 7.05 (m,lH), 6.96(m,lH), 5.49 (s, 2H), 4.61(br,lH), 4.44(s, 2H), 3.45(m, IH), 2.61 (s, 3H), 1.57(d, 6H, 7 = 4.4 Hz), 1.10-1.88 (m, HH); Anal. Calcd for C23H30BrFN4O3: C, 59.41; H,6.50; N.12.05. Found: C, 59.11; H, 6.62; N, 11.31. ES-MS m/z 467.28 & 465.30.
Example 127
{[3-(tert-butyl)-l-(4-methylphenyl)pyrazol-5-yl]amino}-N-[(2-{[5-bromo-2-methyl- l-(methylethyl)-6-oxohydropyridin-4-yloxy]methyl}-5- fluorophenyl)methyl]carboxamide
[0337] To a solution of 6-{ [2-(aminomethyl)-4-fluorophenyl]methoxy}-5-bromo- 2-methyl-3-(methylethyl)-3-hydropyrimidin-4-one (0.384g, 1.0 mmol) in dichloromethane (15 mL) and saturated solution Of NaHCO3 (15 mL), phosgene (20% in toluene, 1.042 mL, 1.97mmol) was added. The mixture was stirred for 15 min, the organic layer was dried over Na2SO4 and concentrated in vacuo, and the residue was treated with a solution of 3-(tert-butyl)-l-(4-methylphenyl)pyrazole-5-ylamine (0.229g) in dichloromethane (10 mL). The resulting mixture was stirred for 17 h at room temperature. After removal of the volatiles in vacuo the residue was purified by flash chromatography using dichloromethane/hexanes/acetone (5:5:1) as elution to give the title compound (0.366g, 57.2%) as a white solid: mp 133-135 0C; 1H NMR (CD3OD/400MHz) δ 7.44 (m, 1H),7.28 (m,lH), 6.97 (m, IH), 6.29 (s, IH), 5.44 (s, 2H), 4.60 (br.lH), 4.45 (s,2H), 2.57 (s, 3H), 2.37 (s, 3H), 1.54 (d, 6H, J = 6.8 Hz), 1.30 (s, 9H); Anal. Calcd for C31H36BrFN6O3: C, 58.22; H, 5.67; N, 13.14. Found: C, 57.72; H, 5.24; N, 12.76. ES-MS m/z 641.39 & 639.39.
Example 128
{[3-(tert-butyI)-l-(4-methylphenyl)pyrazol-5-yl]amino}-N-[(2-{[5-chloro-2-methyl- l-(methylethyI)-6-oxohydropyridin-4-yloxy]methyl}-5- fluorophenyl)methyl]carboxamide
[0338] To a solution of 3-(tert-butyl)-l-(4-methylphenyl)pyrazole-5-ylamine (0.229g, lmmol) and saturated solution of NaHCO3 (15 mL) in dichloromethane (10 mL), phosgene (20% in toluene, 1.042 mL,1.97mmol) was added. The mixture was stirred for 15 min and the organic layer was dried over Na2SO4. After removal of the volatiles in vacuo, the residue was treated with a solution of 6-{ [2-(aminomethyl)-4- fluorophenyl]methoxy}-5-chloro-2-methyl-3-(methylethyl)-3-hydropyrimidin-4-one (0.34Og, 1.0 mmol) in dichloromethane (15 mL) and the mixture was stirred for 17 h at room temperature. The reaction mixture was concentrated in vacuo and the residue which was purified by flash chromatography using dichloromethane/hexanes/acetone (5:5:1) as eluant to give the title compound (0.5Og, 84.0%) as a white solid: mp 153-155 0C; 1H NMR (CD3OD/400MHz) δ 7.46 (m, 1H),7.29 (m,lH), 6.98 (m, IH), 6.29 (s, IH), 5.46 (s, 2H), 4.60 (br,lH), 4.44 (s,2H), 2.59 (s, 3H), 2.38(s, 3H), 1.56 (d, 6H, J = 6.8 Hz), 1.30 (s, 9H); Anal. Calcd for C31H36ClFN6O3: C, 62.57; H, 6.10; N, 14.12. Found: C, 62.98; H, 6.56; N, 13.61. ES-MS m/z 597.45 & 595.46.
Example 129
l-(2-((5-chloro-l,6-dihydro-l-isopropyl-2-(methylthio)-6-oxopyrimidin-4- yIoxy)methyl)-5-fluorobenzyI)-3-ethylurea
Stepl: Preparation of 5-chloro-6-hvdroxy-3-isopropyl-2-(methylthio)pyrimidin-4(3H)- one.
[0339] To a suspension of 6-hydroxy-3-isopropyl-2-(methylthio)pyrimidin-4-one (5.0 g, 25.0 mmol) in 50 mL of dichloromethane in a 3-necked round bottom flask equipped with a magnetic stirring bar, N-chlorosuccinimide (4.0 g, 30.0 mmol) was added in portions at 0-5 0C, The reaction mixture was allowed to warm up to room temperature over 48 h with stirring.. It was then poured into brine with vigorous stirring, the solid precipitate was collected by filtration and crystallized from a mixed solvent of acetone/hexane (1:5), dried in vacuo overnight to give 3.05g of the title compound as a white powder (52%): 1H NMR (DMSO-d6/400MHz) δ 12.08 (s, IH), 4.25 (m, IH), 2.58 (s, 3H), 1.43 (d, 6H); MS-ESI: m/z 235 (M+H).
Step 2: Preparation of 2-((5-chloro-1.6-dihvdro-l-isopropyl-2-(methylthioV6- oxopvrimidin-4-vloxv)methvl)-5-fluorobenzonitrile.
[0340] To a solution of 5-chloro-6-hydroxy-3-isopropyl-2-(methylthio)pyrimidin-4(3H)- one (3.0 g, 12.79 mmol) in DMF (25 mL), potassium carbonate (2.7 g, 19.18 mmol) was added and the mixture was stirred for 15 min, after which 2-cyano-4-fluorobenzyl bromide (3.1 g, 14.07 mmol) was added in portions. The resulting mixture was agitated at room temperature over night and then added brine at 0-5 0C with vigorously stirring. A light yellow precipitate was collected by filtration, washed with water, dried in vacuo to give 3.15 g (71%) of pink powder: 1H NMR (CDCl3, 400MHz) 5 7.63 (m, IH), 7.39 (m, 2H), 5.62 (s, 2H), 4.46 (m, IH), 2.58 (s, 3H), 1.61 (d, 6H); MS-ESI: m/z 368 (M+H).
Step 3: Preparation of 6-(2-(aminomethyl)-4-fluorobenzyloxy)-5-chloro-3-isopropyl-2- (methylthio) pyrimidin-4(3H)-one.
[0341] To a solution of 2-((5-chloro-l,6-dihydro-l-isopropyl-2-(methylthio)-6- oxopyrimidin-4-yloxy)methyl)-5-fluorobenzonitrile (22 g, 59.86 mmol) in dry THF (250 mL) BH3THF (IM, 120 mL) was added dropwise at -10 0C. The resulting mixture was stirred at -10 0C for 3 h, and then allowed to warm up to room temperature.16 h. The mixture was cooled to -0 0C and methanol (2OmL) was added slowly to remove excess of BH3. After the removal of the solvent in vacuo the crude syrup was purified by silica
gel column chromatography eluting with hexane/DCM/CH3OH (3:6:1) to give 12.1 g (^5%) of the title compound as a colorless syrup: 1H NMR (CDCl3, 400MHz) δ 7.38 (m, I1H), 1.17 (m, IH), 6.95 (m, IH), 5.46 (s, 2H), 4.52 (m, IH), 4.51 (s, 2H), 2.58 (s, 3H), % 16 (bs, 2H), 1.61 (d, 6H); MS-ESI: m/z 372 (M+H).
Step 4: Preparation of the title compound.
[0342] To a solution of 6-(2-(aminomethyl)-4-fluorobenzyloxy)-5-chloro-3- isopropyl-2-(methylthio) pyrimidin-4(3H)-one (0.35g, 0.943 mmol) in of dry dichloromethane (5 mL) was added ethyl isocyanate ( 0.1 mL, 1.886 mmol) dropwise at room temperature under nitrogen. The resulting mixture was stirred at room temperature for 8 h. After the removal of solvent in vacuo, the crude product was purified by silica gel chromatography using hexane/DCM (1:5) as the eluent to give 0.0 87 g of the title compound (21%): 1H NMR (CDCl3, 400MHz) δ 7.36 (m, IH), 7.22 (m, IH), 6.95 (m, IH), 5.42 (s, 2H), 4.48 (m, IH), 4.30 (s, 2H), 3.19 (q, 2H), 2.62 (s, 3H), 1.61 (d, 6H), 1.08 (t, 3H); MS-ESI: m/z 443 (M+H). Anal. Calcd for C19H24ClFN4O3S: C, 51.52; H, 5.46; N, 12.65. Found: C, 51.73; H, 5.53; N, 12.75.
Example 130
l-(2-((5-chloro-l,6-dihydro-l-isopropyl-2-(methylthio)-6-oxopyrimidin-4- yloxy)methyl)-5-fluorobenzyl)-3-isopropylurea
[0343] To a solution of 6-(2-(aminomethyl)-4-fluorobenzyloxy)-5-chloro-3- isopropyl-2-(methylthio) pyrimidin-4(3H)-one (0.5 g, 1.346 mmol) in dry
dichloromethane (5 mL), was added iospropyl isocyanate ( 0.41 g, 4.037 mmol) dropwise at room temperature under nitrogen. The resulting mixture was stirred at room temperature over night. After the removal of solvent in vacuo, the crude product was purified by silica gel chromatography eluting with hexane/dichloromethane (1:5 v/v) to afford 0. 204 g of the title compound as a white powder (35.0%): 1H NMR (CDCl3, 400MHz) 57.38 (m, IH), 7.21 (m, IH), 6.90 (m, IH), 5.45 (s, 2H), 4.58 (m, IH), 4.43 (s, 2H), 3.85 (m,lH), 2.61 (s, 3H), 1.61 (d, 6H), 1.15 (d, 6H); MS-ESI: m/z 457 (M+H0. Anal. Calcd for C20H26ClFN4O3S: C, 52.57; H, 5.73; N, 12.26. Found: C, 52.45; H, 5.80; N, 11.87.
Example 131
l-(2-((5-bromo-l,6-dihydro-l-isopropyl-2-(methylthio)-6-oxopyrimidin-4- yloxy)methyl)-5-fluorobenzyl)-3-ethylurea
Step 1: Preparation of 5-bromo-6-hvdroxy-3-isopropyl-2-(methylthio)pyrimidin-4(3H)- one.
[0344] A suspension of 6-hydroxy-3-isopropyl-2-(methylthio)pyrimidin-4-one (25.0 g, 125.0 mmol) in dichloromethane (250 mL) in a 3-necked round bottom flask equipped
with a magnetic stirring bar, added N-bromosuccinimide (25.0 g, 140.0 mmol) in portions and stirred at 0-5 0C under nitrogen. The reaction mixture was then allowed to warm to room temperature over a period of 48 h. The mixture was poured into brine with vigorously stirring, solid precipitate was collected by filtration, crystallized from a mixed solvent of acetone/DCM (6:1), and dried in vacuo overnight to afford 28.1 g of the title compound as a white powder (81.0%): 1H NMR (DMSO-d6/400MHz) δ 12.08 (s, IH), 4.57 (m, IH), 2.58 (s, 3H), 1.43 (d, 6H); MS-ESI: m/z 280 (M+H calcd for C8HnBrN2O2S 280).
Step 2: Preparation of 2-((5-bromo-l,6-dihydro-l-isopropyl-2-(methylthio)-6- oxopwimidin-4-yloxy)rnethyl)-5-fluorobenzonitrile.
[0345] To a solution of 5-bromo-6-hydroxy-3-isopropyl-2-(methylthio)pyrimidin- 4(3H)-one (2.0 g, 8.097 mmol) in DMF ( 25 mL), potassium carbonate (1.70 g, 12.15 mmol) was added and the mixture was stirred for 15 min, then added 2-cyano-4- fluorobenzyl bromide (1.90 g, 8.907 mmol) in portions. The resulting mixture was stirred at room temperature over night and poured into brine at 0-5 0C with vigorous stirring. A light pink precipitate was collected by filtration, washed with water, dried in vacuo to give 3.01 g (91%)of the title compound: 1H NMR (DMSO-d6, 400MHz) δ 7.98 (dd, IH), 7.70 (m, 2H), 5.62 (s, 2H), 4.53 (m, IH), 2.59 (s, 3H), 1.53(d, 6H); MS-ESI: m/z 412 ( M+H).
Step3: Preparation of 6-(2-(aminomethyl)-4-fluorobenzyloxy)-5-bromo-3-isopropyl-2- (methylthio) pyrimidin-4(3H)-one.
[0346] To a solution of 2-((5-bromo- 1 ,6-dihydro- 1 -isopropyl-2-(methylthio)-6- oxopyrimidin-4-yloxy)methyl)-5-fluorobenzonitrile (1.0 g, 2.427 mmol) in dry THF (250 mL) BH3.THF (IM, 5.0 mL) was added dropwise at -10 0C. The resulting mixture was stirred at -10 0C to 0 0C for 3 h, then allowed to warm up to room temperature overnight. It was heated to reflux for 30 min, cooled to 0 0C and added 3N NaOH solution to pH 12. The organic phase was isolated, washed with brine, dried over Na2SO4, filtered, and concentrated to dryness. The resulting syrup was purified by silica gel column chromatography, eluting with hexane/DCM/CH3OH (3:6:1) to give 830 mg (82%) of the title compound as a colorless syrup: 1H NMR (CDCl3, 400MHz) δ 7.36 (m, IH), 1.16 (m, IH), 6.93 (m, IH), 5.36 (s, 2H), 4.41 (m, IH), 4.38 (s, 2H), 2.58 (s, 3H), 2.35 (bs, 2H), 1.60 (d, 6H); MS-ESI: m/z 417 (M+H ).
Step 4: Preparation of the title compound.
[0347] To a solution of 6-(2-(aminomethyl)-4-fluorobenzyloxy)-5-bromo-3- isopropyl-2-(methylthio) pyrimidin-4(3H)-one (0.25g, 0.602 mmol) in dry dichloromethane (5 mL) was added ethyl isocyanate ( 0.065 g, 0.903 mmol) dropwise at room temperature and stirred under nitrogen for 16 h. After the removal of solvent in vacuo the crude product was purified by silica gel chromatography using hexane/dichloromethane (1:5) to give 0.175 g (60%) of the title compound as a white powder: 1H NMR (CDCl3, 400MHz) 57.38 (m, IH), 7.22 (m, IH), 6.97 (m, IH), 5.43 (s, 2H), 4.58 (m, IH), 4.45 (s, 2H), 3.21 (q, 2H), 2.62 (s, 3H), 1.63 (d, 6H), 1.11 (t, 3H); MS-ESI: m/z 488 (M+H). Anal. Calcd for C20H27BrFN4O5S: C, 46.82 %; H, 4.96 %; N, 11.50 %. Found: C, 47.18 %; H, 4.83 %; N, 11.27%.
Example 132
l-(2-((5-bromo-l,6-dihydro-l-isopropyl-2-(methylthio)-6-oxopyrimidin-4- yloxy)methyl)-5-fluorobenzyl)-3-isopropylurea
[0348] To a solution of 6-(2-(aminomethyl)-4-fluorobenzyloxy)-5-bromo-3- isopropyl-2-(methylthio) pyrimidin-4(3H)-one (0.50g, 1.202 mmol) in dry dichloromethane (8 mL) isopropyl isocyanate ( 0.2 mL, 1.715 mmol) was added dropwise at room temperature under nitrogen. The resulting mixture was stirred at room temperature for 16 h, concentrated in vacuo, and the residue was purified by silica gel chromatography eluting with hexane/ dichloromethane (l:5v/v) to afford 0.38 g (63%) of the title compound as a white powder: 1H NMR (CDCl3, 400MHz) δ 7.36 (m, IH), 7.19 (m, IH), 6.97 (m, IH), 5.45 (s, 2H), 4.55 (m, IH), 4.43 (s, 2H), 3.85 (m, IH), 2.62 (s, 3H), 1.62 (d, 6H), 1.12 (d, 6H); MS-ESI: m/z 501 (M+ ). Anal. Calcd for C20H26BrFN4O3S: C, 47.91 %; H, 5.23 %; N, 11.17 %. Found: C, 47.89 %; H, 5.58%; N, 10.74 %.
Example 133
2-[5-Bromo-2-(2-hydroxy-ethylamino)-l-isopropyl-6-oxo-l,6-dihydro-pyrimidin-4- yloxymethyl]-5-fluoro-benzonitrile
Stepl: Preparation of 2-((5-bromo-1.6-dihvdro-l-isopropyl-2-(methylsulfonyl)-6- oxopyrimidin-4-yloxy)methyl)-5-fluorobenzonitrile.
[0349] To a solution of 2-((5-bromo-l,6-dihydro-l-isopropyl-2-(methylthio)-6- oxopyrimidin-4-yloxy)methyl)-5-fluorobenzonitrile (10.0 g, 24.272 mmol) in THF (200 mL) containing de-ionized water (40 mL),added oxone (37.3 g, 60.679 mmol) in small portions. The resulting mixture was stirred at room temperature for 5 days. The reaction mixture was poured into ice-brine with vigorously stirring and the white precipitate was collected by filtration, washed with water, and air dried to afford 10.6 g (98.9 %)of the title compound as a white powder: 1H NMR (CDCl3, 400MHz) δ 7.69 (m, IH), 7.45 (m, 2H), 5.59 (s, 2H), 5.28 (m, IH), 3.46 (S, 3H), 1.68 (d, 6H); MS-ESI: m/z 444.03 (M+ ).
Step 2: Preparation of the title compound.
[0350] To a solution of 2-((5-bromo-l,6-dihydro-l-isopropyl-2-(methylsulfonyl)-6- oxopyrimidin-4-yloxy)methyl)-5-fluorobenzonitrile (0.5 g, 1.126 mmol) in dry DMF (5 mL) was added pyridine (0.445 g, 5.631 mmol), followed by the addition of ethanolamine (0.138 g, 2.252 mmol). The resulting mixture was stirred at room temperature for 2 h, cooled to -10 0C, added brine (3OmL) and the pH was adjusted to 3 with 0.5N HCl. The solid that precipitated was collected by filtration, washed with water, dried in vacuo to give 0.36 g (76%) of the title compound as a white powder: 1H NMR (DMSO-d6, 400MHz) δ 7.95 (m, IH), 7.68 (m, 2H), 7.40 (bs, IH), 5.48 (s, 2H), 4.70 (t, 2H), 3.51 (m, 2H), 3.38 (m, 2H), 1.42 (d, 6H); MS-ESI: m/z 426 (M+H). Anal. Calcd for
C17H18BrFN4O3: C, 48.02 %; H, 4.27 %; N, 13.17 %. Found: 47.85 %; H, 4.35%; N, 12.94 %.
Example 134
l-(2-((5-bromo-l,6-dihydro-l-isopropyl-2-(methylsulfonyl)-6-oxopyrimidin-4- yIoxy)methyl)-5-fluorobenzyl)-3-isopropylurea
[0351] To a solution of l-(2-((5-bromo-l,6-dihydro-l-isopropyl-2-(methylthio)-6- oxopyrimidin-4-yloxy)methyl)-5-fluorobenzyl)-3-isopropylurea (04-003-137a, 0.92 g, 1.836 mmol) in THF (10 mL) containing de-ionized water (1 mL), oxone (2.26 g, 3.673 mmol) was added. The resulting mixture was stirred at room temperature for 50 h. The reaction was cooled to -1O0C, added brine (50 mL), and stirred vigorously. The solid precipitate formed was collected by filtration, washed with water, dried in vacuo to afford 0.88 g (90%) of the title compound as a light yellow powder: 1H NMR (CDCl3, 400MHz) δ 7.39 (m, IH), 7.15 (m, IH), 7.02 (m, IH), 5.42 (s, 2H), 5.28 (m, IH), 4.43 (s, 2H), 3.83 (m, IH), 3.55 (s, 3H), 1.68 (d, 6H), 1.12(d, 6H); MS-ESI: m/z 533.24 (M+ ). Anal. Calcd for C20H28BrFN4O6S-H2O: C, 43.55 %; H, 5.08 %; N, 10.16 %. Found: C, 43.25 %; H, 4.63 %; N, 9.68 %.
l-(2-((2-(2-hydroxyethylamino)-5-bromo-l,6-dihydro-l-isopropyI-6-oxopyrimidin- 4-yloxy)methyl)-5-fluorobenzyl)-3-isopropylurea
[0352] To a solution of l-(2-((5-bromo-l,6-dihydro-l-isopropyl-2-(methylsulfonyl)- 6-oxopyrimidin-4-yloxy)methyl)-5-fluorobenzyl)-3-isopropylurea (04-003-165a, 0.25 g, 0.469 mmol) in dry DMF (3 mL), was added potassium carbonate (0.065 g, 0.469 mmol) and ethanolamine (0.039 mL, 0.516 mmol). The resulting mixture was stirred at room temperature for 2 h, then cooled to -10 0C and added brine (3OmL). The solid precipitate was collected by filtration, washed with water, and dried in vacuo to afford 0.136 g (58%) of the title compound as a white powder: 1H NMR (DMSOd6, 400MHz) δ 7.42 (m, IH), 7.38 (m, IH), 7.06 (m, 2H), 6.23(t, IH), 5.85 (d, IH), 5.38 (s, 2H), 4.78 (t, IH), 4.35 (d, 2H), 3.65 (m, IH), 3.55 (m, 2H), 3.42 (m, 2H), 1.42 (d, 6H), 1.05 (d, 6H); MS-ESI: m/z 514.25 (M+). Anal, calcd for C21H29BrFN5O4: C, 49.03 %; H, 5.68 %; N, 13.05 %. Found: C, 49.11 %; H, 5.89 %; N, 12.71 %.
l-(2-((2-(2-(dimethylamino)ethylamino)-5-bromo-l,6-dihydro-l-isopropyl-6- oxopyrimidin-4-yloxy)methyl)-5-fluorobenzyl)-3-isopropyIurea
[0353] To a solution of l-(2-((5-bromo-l,6-dihydro-l-isopropyl-2-(methylsulfonyl)- 6-oxopyrimidin-4-yloxy)methyl)-5-fluorobenzyl)-3-isopropylurea (0.25 g, 0.469 mmol) in dry DMF (2 mL) was added pyridine (0.076 mL, 0.938 mmol) and N,N- dimethylethylenediamine (0.057 mL, 0.515 mmol). The mixture was stirred at room temperature for 2 h, cooled to -100C and poured into brine (3OmL). The solid that precipitated was collected by filtration, washed with water, and dried in vacuo to give 0.196 g (78%) of the title compound as a white powder: 1H NMR (DMSO-d6, 400MHz) δ 7.42 (m, IH), 7.29 (m, IH), 7.08 (m, 2H), 6.23(t, IH), 5.85 (d, IH), 5.39 (s, 2H), 4.25 (d, 2H), 3.66 (m, IH), 3.45 (m, 2H), 2.42 (t, 2H), 2.18 (s, 6H), 1.41 (d, 6H), 1.03 (d, 6H); MS-ESI: m/z 539.37 (M-2H> Anal. Calcd for C23H34BrFN6O3: C, 50.69 %; H, 6.34 %; N, 15.41 %. Found: C, 50.68 %; H, 6.29 %; N, 14.92 %.
Example 137
l-(2-((5-chloro-l,6-dihydro-l-isopropyl-2-(methylthio)-6-oxopyrimidin-4- yloxy)methyl)-5-fluorobenzyl)-3-cyclohexylurea
[0354] To a solution of l-(2-((5-chloro-l,6-dihydro-l-isopropyl-2-(methylthio)-6- oxopyrimidin-4-yloxy)methyl)-5-fluorobenzyl)-3-cyclohexylurea (0.76 g, 1.53 mmol) in THF (6 mL) and H2O (0.6 mL), oxone ( 1.88 g, 3.061 mmol) was added and stirred at room temperature for 16 h. The reaction was quenched by the addition of brine (3OmL)
and stirred at 00C. The solid that precipitated was collected by filtration, washed with water, and dried in vacuo to afford 0.73 g (91%) of the title compound as a yellow powder: 1H NMR (CDCl3, 400MHz) δ 7.38 (m, IH), 7.17 (m, IH), 7.05 (m, IH), 5.41 (s, 2H), 5.26 (m, IH), 4.43 (s, 2H), 3.75 (m, IH), 3.53 (s, 3H), 3.43 (m, IH), 1.85 (m, 3H), 1.68 (d, 6H), 1.60 (m, 1 H), 1.30 (m, 2H), 1.05 (m, 2H); MS-ESI: m/z 529.31 (M+). Anal. Calcd for C23H30ClFN4O3S: C, 55.58 %; H, 6.08 %; N, 11.27 %. Found: C, 55.47 %; H, 6.21 %; N, 11.18 %.
Example 138
l-(2-((5-chloro-l,6-dihydro-l-isopropyl-2-(methylthio)-6-oxopyrimidin-4- yloxy)methyl)-5-fluorobenzyl)-3-cycIohexylurea
[0355] To a solution of l-(2-((5-chloro-l,6-dihydro-l-isopropyl-2-(methylthio)-6- oxopyrimidin-4-yloxy)methyl)-5-fluorobenzyl)-3-cyclohexylurea (0.76g, 1.531 mmol) in THF (10 mL) containing de-ionized water (1 mL), oxone (1.88 g, 3.061 mmol) was added. The resulting mixture was stirred at room temperature for 16 h. The mixture was cooled to -10 0C, brine (50 mL) was added with vigorous stirring. The solid that separated was collected by filtration, washed with water, and dried in vacuo to afford 0.73 g (90%) of the title compound as a light yellow powder: 1H NMR (CDCl3, 400MHz) δ 7.39 (m, IH), 7.15 (m, IH), 7.02 (m, IH), 5.42 (s, 2H), 5.28 (m, IH), 4.43 (s, 2H), 3.83 (m, IH), 3.55 (s, 3H), 1.68 (d, 6H), 1.12(d, 6H); MS-ESI: m/z 533.24 (M+). Anal. Calcd for C23H30ClFN4O5S: C, 52.22%; H, 5.72 %; N, 10.59 %. Found: C, 51.88 %; H, 5.88 %; N, 9.83 %.
l-(2-((5-chloro-l,6-dihydro-l-isopropyl-2-(methylthio)-6-oxopyrimidin-4- yloxy)methyl)-5-fluorobenzyl)-3-(3-tert-butyl-l-p-tolyl-lH-pyrazol-5-yl)urea
[0356] To a solution of 6-(2-(aminomethyl)-4-fluorobenzyloxy)-5-chloro-3- isopropyl-2-(methylthio) pyrimidin-4(3H)-one (0.4 g, 1.076 mmol) in dichloromethane(12 mL) was added a saturated solution of NaHCO
3 (12 mL). The mixture was cooled to 5
0C, and added dropwise a solution of phosgene in toluene (20%, 1.14 mL, 2.154 mmol) with vigorous stirring. The resulting mixture was stirred at 5
0C for 30 min, the organic phase was separated, dried over Na
2SO
4, filtered, concentrated to 6 mL and added l-(4-methyl)phenyl-3-t-butyl-pyrazolyl-5-amine (0.493 g, 2.152 mmol).. The resulting mixture was stirred at room temperature under nitrogen for 48 h and the product was purified by silica gel chromatography eluting with hexane/dichloromethane/acetone (5:5:1) to afford 186 mg (30%) of the title compound as a light yellowish powder:
1H NMR (CDCl
3, 400MHz) 57.30 (m, 3H), 7.20 (d, 2H), 7.05 (dd, IH), 6.95 (m, IH), 6.35 (s, IH), 6.31 (bs, IH), 5.55 (bs, IH), 5.40 (s, 2H), 4.45 (d, 2H), 2.60 (s, 3H), 2.36 (s, 3H), 1.58 (d, 6H), 1.15 (s, 9H); MS-ESI: m/z 627.37 ( M
+). Anal. Calcd for C
31H
36ClFN
6O
2S: C, 59.37 %; H, 5.79 %; N, 13.40 %. Found: C, 59.10 %; H, 6.00 %; N, 12.67 %.
6-[(2,4-difluorophenyl)methoxy]-3-cyclopropyl-2-methylthio-3-hydropyrimidin-4- one
Step 1 : Preparation of cyclopropyl isothiocyanate.
^NCS
[0357] To a stirred solution of cyclopropylamine (8.03 g, 0.141 mol) and triethylamine (15.65 g , 0.155 mol) in THF (100 mL), carbon disulfide (32.12 g, 0.422 mol) was added at 0-10 0C. Stirring was continued for 0.5 h, added dropwise a solution of hydrogen peroxide (30 wt %, 47.82 g, 0.422 mol) at room temperature. After 1 h, the reaction mixture was neutralized with diluted hydrochloric acid (1:1) to neutral pH and diluted with ether (50 mL). The organic layer was washed with brine, dried over Na2SO4, and evaporated under reduced pressure. The resulting residue was distilled to give 8.50g (61.0%) of the desired product as a colorless oil: mp 125-130 0C; 1H NMR (CDCl3/ 200 MHz) 52.94-2.82 (m, IH), 0.96-0.82 (m, 4H).
Step 2: Preparation of cyclopropylthiourea.
S >-NΛNH2
[0358] A methanolic solution of ammonia (7N, 23.5 mL) was added to a solution of cyclopropyl isothiocyanate (8.140 g, 82.2 mmol) in methanol (10 mL) at 0 0C. The resulting mixture was then stirred at room temperature for 5 h, cooled to -100C and filtered the white precipitate, it was washed with ether and dried to afford 10.251 g
(100%) of the title product as a white crystalline solid: 1H NMR (DMSO-(V 400 MHz) δ 7.95 (br, IH), 7.62 (br, IH), 7.15 (br, IH), 2.20 (br, IH), 0.68-0.62 (m, 2H), 0.50-0.42 (m, 2H); ES-MS m/z 117.07 (M+ H).
Step 3: Preparation of B-cyclopropyl-o-hydroxy^-methylthio-S-hydropyrimidin-'l-one.
[0359] A vigorously stirred mixture of cyclopropylthiourea (10.191 g, 87.8 mmol), diethylmalonate (14.060 g, 87.8 mmol), sodium methoxide (25-30% in methanol, 35.1 mL) was heated to reflux under nitrogen. After 4.5 h, the reaction mixture was cooled, and iodomethane (12.952 g, 91.3 mmol) was added while the temperature was maintained at 50 0C. The mixture was stirred for 30 min, then cooled to 10 0C, and treated with of acetic acid (21.1 mL). The product that precipitated was diluted with water (5OmL), filtered, washed with water, and air dried to afford 13.853 g (79.7%) of the desired product: 1H NMR (DMSO-do/ 400 MHz) 55.10 (s, 2H), 2.68-2.62 (m, IH), 2.50 (s, 3H), 1.18-1.06 (m, 2H), 0.88-0.82 (m, 2H); ES-MS m/z 198.09 (M+).
Step 4: Preparation of 6-[(2,4-difluorophenyl)methoxy1-3-cyclopropyl-2-methylthio-3- h vdrop yrimi din-4-one.
[0360] A mixture of 3-cyclopropyl-6-hydroxy-2-methylthio-3-hydropyrimidin-4-one (13.853 g, 66.9 mmol), 2,4 difluorobenzylbromide (14.760 g, 71.3 mol) and potassium carbonate (17.390 g, 125 mmol) in DMF (80 mL) was stirred at 0 0C under nitrogen.
After 30 min, it was stirred at room temperature for an additional 30 min and filtered and the filtrate was concentrated under vacuum. The residue was partitioned between dichloromethane (50 mL) and 5% hydrochloric acid (20 mL). The organic phase was washed with water, dried (Na2SO4), and concentrated to dryness under reduced pressure. The resulting material was purified by silica gel flash chromatography using 33% EtOAc in hexane as elution to give 11.144 g of the title compound as a white powder: mp 106.1- 106.8 0C; 1H NMR (CDCl3/ 400 MHz) δ 7.41-7.39 (m, IH), 6.91-6.81 (m, 2H), 5.56 (s, IH), 5.30 (s, 2H), 2.70-2.51 (m, IH), 2.51 (s, 3H), 1.29-1.24 (m, 2H), 1.02-0.98 (m, 2H); Anal. Calcd for C15Hi4N2O2SF2: C, 55.55; H, 4.38; N, 8.64. Found: C, 55.68; H, 4.44; N, 8.59; ES-MS m/z 325.
Example 141
6-[(2,4-difluorophenyl)methoxy]-2-{[2-(dimethyI-amino)ethyl]amino}-5-bromo-3- cyclopropyl-3-hydropyrimidin-4-one
Step 1: Preparation of 6-r(2,4-difluorophenyl)methoxyl-5-brorno-3-cvclopropyl-2- methylthio-3-hvdropyrimidin-4-one.
[0361] A mixture of 6-[(2,4-difluorophenyl)methoxy]-3-cyclopropyl-2-methylthio-3- hydropyrimidin-4-one (1.600 g, 4.93 mmol), N-bromosuccinimide (0.922 g, 5.18 mmol) in dichloromethane (15 mL) was stirred at room temperature under nitrogen for 1 h. The reaction mixture was concentrated under reduced pressure and the residue was purified by silica gel flash chromatography using 33% ethyl acetate in hexanes as elution to give
1.544 g (77.7%) of the title compound as a white powder: mp 121.0-122.4 0C. 1H NMR
(CDCl3/ 400 MHz) δ 7.52-7.44 (m, IH), 6.98-6.78 (m, 2H), 5.52 (s, 2H), 2.80-2.72 (m, IH), 2.52 (s, 3H), 1.32-1.24 (m, 2H), 1.08-1.02 (m, 2H); ES-MS m/z 403.05 & 405.04.
Step 2: Preparation of 6-r(2,4-difluorophenyl)methoxy1-5-bromo-3-cyclopropyl-2- (methylsulfonyl)-3-hvdropyrimidin-4-one.
[0362] A mixture of 6-[(2,4-difluorophenyl)methoxy]-5-bromo-3-cyclopropyl-2- methylthio-3-hydropyrimidin-4-one (0.758 g, 1.88 mmol), m-chloroperbenzoic acid (60%, 1.352 g, 4.70 mmol) in dichloromethane (40 mL) was stirred at room temperature for 16 h. The reaction mixture was filtered, the filtrate was concentrated under reduced pressure and the residue was purified by silica gel flash chromatography using 33% ethyl acetate in hexanes as elution to give 0.695 g (84.9%) of product as a white powder, mp 159.6-160.5 0C; 1H NMR (CDCl3/ 400 MHz) 8 7.48-7.41 (m, IH), 6.98-6.84 (m, 2H), 5.42 (s, 2H), 3.38-2.72 (m, IH), 2.52 (s, 3H), 1.32-1.24 (m, 2H), 1.08-1.02 (m, 2H); ES- MS m/z 434.97& 436.97.
Step 3: Preparation of 6-f(2,4-difluorophenyl)methoxyl-2-( [2-(dimethyl- amino)ethvHamino)-5-bromo-3-cvclopropyl-3-hvdropyrimidin-4-one.
[0363] A mixture of 6-[(2,4-difluorophenyl)methoxy]-5-bromo-3-cyclopropyl-2- (methylsulfonyl)-3-hydropyrimidin-4-one (0.378 g, 0.868 mmol), N,N- dimethylethylenediamine (0.230 g, 2.60 mmol), and potassium carbonate (0.144 g, 1.04 mmol) in DMF (4 mL) was stirred at room temperature for 12 h. The reaction mixture was concentrated under vacuum and the residue was purified by silica gel flash chromatography using 13% methanol in dichloromethane as elution to give 0.154 g (40.2%) of the title compound as a white powder: mp 125.8-127.2 °C;
1H NMR (CDCl
3/ 400 MHz) δ 7.52-7.46 (m, IH), 6.92-6.78 (m, 2H), 6.58 (br, IH), 5.41 (s, 2H), 3.48-3.44
(m, 2H), 2.64-2.55 (m, 3H), 2.31 (s, 6H), 1.28-1.24 (m, 2H), 0.92-0.88 (m, 2H); Anal. Calcd for C
I8H
21N
4O
2BrF
2: C, 48.77; H, 4.77; N, 12.64. Found: C, 48.58; H, 4.68; N,
Example 142
6-[(2,4-difluorophenyl)methoxy]-2-{[2-(dimethyl-amino)ethyl]amino}-5-chIoro-3- cycIopropyl-3-hydropyrimidin-4-one
Step 1: Preparation of 6-r(2,4-difluorophenyl)methoxy1-3-cvclopropyl-2- (methylsulfonyl)-3-hvdropyrimidin-4-one.
[0364] A mixture of 6-[(2,4-difluorophenyl)methoxy]-3-cyclopropyl-2-methylthio-3- hydropyrimidin-4-one (5.000 g, 15.4 mmol), m-chloroperbenzoic acid (60%, 13.306 g, 46.3 mmol) in dichloromethane (200 mL) was stirred at room temperature for 12 h.. The reaction mixture was filtered, the filtrate was concentrated under vacuum and the residue was purified by silica gel flash chromatography using 50% ethyl acetate in hexanes as elution to give 4.266 g (77.6%) of the title compound as a white powder, mp 113.5-114.2 0C; 1H NMR (CDCl3/ 400 MHz) 67.46-7.35 (m, IH), 6.98-6.84 (m, 2H), 5.82 (s, IH), 5.24 (s, 2H), 3.35 (s, 3H), 3.24-3.16 (m, IH), 1.35-1.32 (m, 2H), 1.18-1.12 (m, 2H); ES- MS m/z 357.10.
Step 2: Preparation of 6-r(2,4-difluorophenyl)methoxyl-2-f [2-
(dimethylamino)ethyl1amino}-3-cvclopropyl-3-hvdropyrirnidin-4-one.
[0365] A mixture of 6-[(2,4-difluorophenyl)methoxy]-3-cyclopropyl-2- (methylsulfonyl)-3-hydropyrimidin-4-one (1.000 g, 2.81 mmol), N,N- dimethylethylenedi amine (0.348 g, 3.93 mmol), and potassium carbonate (0.465 g, 3.37 mmol) in THF (10 mL) was stirred at room temperature for 6 h. The reaction mixture was concentrated under vacuum and the residue was purified by silica gel flash chromatography using 9% methanol in dichloromethane as elution to give 0.951 g (92.2%) of white powder as product: mp 103.5-104.0 0C; 1H NMR (CDCl3/ 400 MHz) δ 7.46-7.38 (m, IH), 6.92-6.68 (m, 2H), 6.52 (br, IH), 5.24 (s, 2H), 5.22 (s, IH), 3.52-3.42 (m, 2H), 2.58-2.52 (m, 3H), 2.52 (s, 3H), 1.28-1.22 (m, 2H), 0.90-0.84 (m, 2H); ES-MS m/z 365.27 (M+H).
Step 3: Preparation of 6-r(2,4-difluorophenyl)methoxy1-2-{ r2-(dimethylarnino)ethyll- aminol-S-chloro-S-cvclopropyl-S-hydropyrimidin^-one.
[0366] A mixture of 6-[(2,4-difluorophenyl)methoxy]-2- { [2- (dimethylamino)ethyl]amino}-3-cyclopropyl-3-hydropyrimidin-4-one (0.500 g, 1.37 mmol), N-chlorosuccinimide (0.192 g, 1.44 mmol) in dichloromethane (10 mL) was stirred at room temperature for 6 h. The reaction mixture was concentrated under reduced pressure and the residue was purified by silica gel flash chromatography using 10% methanol in dichloromethane as elution to give 0.329 g (60.2%) of the title compound as a white powder: mp 127.6-128.4 0C; 1H NMR (CDCl3/ 400 MHz) 5 7.51-7.47 (m, IH), 6.91-6.78 (m, 2H), 5.41 (s, 2H), 3.46-3.45 (m, 2H), 2.63-2.56 (m, 3H), 2.31 (s, 6H), 1.28-1.24 (m, 2H), 0.93-0.90 (m, 2H); Anal. Calcd for C,8H2iN4O2ClF2: C, 54.23; H, 5.31; N, 14.05. Found: C, 53.80; H, 5.23; N, 13.69; ES-MS m/z 399 (M+H).
6-[(2,4-difluorophenyl)methoxy]-5-chloro-3-(2-methyI- propyl)-2-methylthio-3-hydropyrimidin-4-one
Step 1: Preparation of isobutylthiourea.
[0367] A methanolic solution of ammonia (7N, 29.8 mL) was added to a solution of isobutyl isothiocyanate (12.000 g, 104 mmol) in methanol (15 mL) at 0 0C. The resulting mixture was then stirred at room temperature, the solid that separated was collected by filtration, washed with ether and dried to afford 12.950 g (94.2%) of the title com[pound as a white crystalline material: 1H NMR (DMSO-d6/ 400 MHz) δ 7.68 (br, IH), 7.58 (br, IH), 6.88 (s, IH), 3.18(br, IH), 1.80-1.76 (m, IH), 0.88 (br, 6H); ES-MS m/z 133.10 (M+H).
Step 2: Preparation of 6-hvdroxy-3-(2-methylpropyl)-2-methylthio-3-hvdropyrimidin-4- one.
[0368] A vigorously stirred mixture of isobutylthiourea (11.468 g, 86.9 mmol), diethylmalonate (13.900 g, 86.7 mmol), sodium methoxide (25-30% in methanol, 34.7 mL) was heated to reflux under nitrogen. After 4.5 h, the reaction mixture was cooled and added iodomethane (12.803 g, 90.2 mmol) while the temperature was maintained at
50 0C. The mixture was stirred at 50 0C for 1 h, then cooled to 0 °C, and treated with acetic acid (9.0 mL). The product that precipitated was diluted with water (100 mL), filtered, washed with water, and air dried to afford 17.010 g (91.6%) of the desired product: 1H NMR (DMSO-d6/ 400 MHz) δ 5.20 (s, IH), 3.76(d, IH, J = IA Hz), 3.56 (s, 3H), 2.18-2.10 (m, IH), 0.85 (d, 6H, J = 6.6 Hz); ES-MS m/z 215.14 (M+H).
Step 3: Preparation of 6-r(2,4-difIuorophenyl)methoxyl-3-(2-methylpropyl)-2- methylthio-3-hvdropyrimidin-4-one.
[0369] A mixture of 6-hydroxy-3-(2-methylpropyl)-2-methylthio-3-hydropyrimidin- 4-one (16.800 g, 78.4 mmol), 2,4 difluorobenzylbromide (21.098 g, 102 mol) and potassium carbonate (16.253 g, 118 mmol) in DMF (100 mL) was stirred at 0 0C under nitrogen. After 30 min, it was stirred at room temperature for an additional 30 min and filtered. The filtrate was concentrated under vacuum and the residue was partitioned between dichloromethane and 5% hydrochloric acid (20 mL). The organic phase was washed with water, dried (Na2SO4), and concentrated to dryness under reduced pressure. The resulting material was purified by silica gel flash chromatography using 20% ethyl acetate in hexanes as elution to give 15.680 g (58.8%) of the desired product as a white powder: mp 89.8-90.2 0C; 1H NMR (CDCl3/ 400 MHz) δ 7.44-7.38 (m, IH), 6.92-6.78 (m, 2H), 5.58 (s, IH), 5.30 (s, 2H), 3.88 (d, 2H, J = 7.8 Hz), 2.56 (s, 3H), 2.32-2.24 (m, IH), 0.96 (d, 6H, J = 6.3 Hz); Anal. Calcd for C16HnN2O2SClF2: C, 51.27; H, 4.57; N, 7.42. Found: C, 51.22; H, 4.65; N, 7.38; ES-MS m/z 341.20 (M+H).
Step 4: Preparation of 6-r(2,4-difluorophenyl)rnethoxy"|-5-chloro-3-(2-methyl- propy0-2-methylthio-3-hvdropyrimidin-4-one.
[0370] A mixture of 6-[(2,4-difluorophenyl)methoxy]-3-(2-methylpropyl)-2- methylthio-3-hydropyrimidin-4-one (1.400 g, 4.11 mmol), N-chlorosuccinimide (0.577 g, 4.32 mmol) in dichloromethane (15 mL) was stirred at room temperature for 5 h. The reaction mixture was concentrated under reduced pressure and the residue was purified by silica gel flash chromatography using 20% ethyl acetate in hexanes as elution to give i.457 g (94.6%) of the title compound as a white powder: mp. 58.0-58.8 0C; 1H NMR (CDCl3/ 400 MHz) δ 7.48-7.44 (m, IH), 6.92-6.82 (m, 2H), 5.51 (s, 2H), 3.90 (d, J = 7.6 Hz, 2H), 2.55 (s, 3H), 2.30-2.27 (m, IH), 0.95 (d, J = 6.4 Hz, 6H); Anal. Calcd for C16H17N2O2SClF2: C, 51.27; H, 4.57; N, 7.42. Found: C, 51.22; H, 4.65; N, 7.38; ES-MS m/z 375 (M+H).
Example 144
6-[(2,4-difluorophenyl)methoxy]-5-bromo-3-(2-methyl- propyl)-2-methylthio-3-hydropyrimidin-4-one
[0371] A mixture of 6-[(2,4-difluorophenyl)methoxy]-3-(2-methylpropyl)-2- methylthio-3-hydropyrimidin-4-one (1.400 g, 4.11 mmol), N-bromosuccinimide (0.769 g, 4.32 mmol) in dichloromethane (15 mL) was stirred at room temperature for 5 h. The reaction mixture was concentrated under reduced pressure and the residue was purified by silica gel flash chromatography using 20% ethyl acetate in hexanes as elution to give 1.672 g (97.0 %) of the title compound as a white powder: mp 70.1-70.8 0C; 1H NMR (CDCl3/ 400 MHz) δ 7.51-7.45 (m, IH), 6.94-6.81 (m, 2H), 5.50 (s, 2H), 3.91 (d, J = 7.2 Hz, 2H), 2.55 (s, 3H), 2.32-2.28 (m, IH), 0.95 (d, J = 6.8 Hz, 6H); Anal. Calcd for C16H17N2O2SBrF2: C, 45.83; H, 4.09; N, 6.68. Found: C, 46.07; H, 4.20; N, 6.63; ES-MS m/z 419 (M+H).
6-[(2,4-difluorophenyl)methoxy]-3-(2-methylpropyl)- 2-(methylsulfonyl)-3-hydropyrimidin-4-one
[0372] A mixture of 6-[(2,4-difluorophenyl)methoxy]-3-(2-methylpropyl)-2- methylthio-3-hydropyrimidin-4-one (3.000 g, 8.88 mmol), m-chloroperbenzoic acid (60%, 6.338 g, 22.0 mmol) in dichloromethane (150 mL) was stirred at room temperature for 16 h. The reaction mixture was filtered, the filtrate was concentrated under vacuum and the residue was purified by silica gel flash chromatography using 20% ethyl acetate in hexanes as elution to give 2.093 g (63.8%) of the title compound as a white powder: mp 110.5-110.9 0C; 1H NMR (CDCl3/ 400 MHz) 67.44-7.38 (m, IH), 6.96-6.85 (m, 2H), 5.88 (s, IH), 5.23 (s, 2H), 4.24 (d, J = 7.6 Hz, 2H), 3.40 (s, 3H), 2.41-2.37(m, IH), 0.95 (d, J = 6.8 Hz, 6H); Anal. Calcd for C16H18N2O4SF2: C, 51.61; H, 4.87; N, 7.52. Found: C, 51.71; H, 4.98; N, 7.33; ES-MS m/z 373 (MW: 372).
6-[(2,4-difluorophenyI)methoxy]-2-{[2-(dimethyl-amino)ethyl]amino}-3-(2- methylpropyl)-3-hydropyrimidin-4-one
[0373] A mixture of 6-[(2,4-difluorophenyl)methoxy]-3-(2-methylpropyl)- 2-(methylsulfonyl)-3-hydropyrimidin-4-one (1.49 g, 4.00 mmol), N,N- dimethylethylenediamine (0.529 g, 6.00 mmol), and potassium carbonate (0.744 g, 5.60 mmol) in DMF (10 mL) was stirred at room temperature for 5 h. The reaction mixture
was concentrated under vacuum and the residue was purified by silica gel flash chromatography using 10% methanol in dichloromethane as elution to give 1.156 g (76.0%) of the title compound as a colorless liquid oil: 1H NMR (CDCl3/ 400 MHz) δ 7.46-7.40 (m, IH), 6.90-6.79 (m, 2H), 5.94 (br, IH), 5.28 (s, IH), 5.22 (s, 2H), 3.72 (br, 2H), 3.45-3.41 (m, 2H), 2.55-2.52 (m, 2H), 2.29 (s, 6H), 2.07-2.03 (m, IH), 0.97 (d, J = 6.4 Hz, 6H); Anal. Calcd for C19H26N4O2F2: C, 59.99; H, 6.89; N, 14.73. Found: C, 59.64; H, 6.93; N, 14.82; ES-MS m/z 381 (M+H).
Example 147
6-[(2,4-difluorophenyl)methoxy]-2-{[2-(dimethyl-amino)ethyl]amino}-5-bromo-3-(2- methylpropyl)-3-hydropyrimidin-4-one
[0374] A mixture of 6-[(2,4-difluorophenyl)methoxy]-2- { [2-(dimethyl- amino)ethyl]amino}-3-(2-methylpropyl)-3-hydropyrimidin-4-one (0.400 g, 1.05 mmol), N-bromosuccinimide (0.197 g, 1.10 mmol) in dichloromethane (10 mL) was stirred at room temperature for 5 h. The reaction mixture was concentrated under vacuum and the residue was purified by silica gel flash chromatography using 9% methanol in dichloromethane as elution to give 0.410 g (85.1 %) of the title compound as a white powder: mp 133.8-135.6 0C; 1H NMR (CDCl3/ 400 MHz) δ 7.54-7.48 (m, IH), 6.92-6.79 (m, 2H), 5.99 (br, IH), 5.42 (s, 2H), 3.76 (br, 2H), 3.43-3.39 (m, 2H), 2.56-2.53 (m, 2H), 2.29 (s, 6H), 2.07-2.04 (m, IH), 0.97 (d, J = 6.8 Hz, 6H); Anal. Calcd for Ci9H25N4O2BrF2: C, 49.68; H, 5.48; N, 12.20. Found: C, 49.93; H, 5.55; N, 12.22; ES- MS m/z 459 (M+H).
Example 148
6-[(2,4-difluorophenyl)methoxy]-3-(2,2-dimethylpropyl)-2-methylthio-3- hydropyrimidin-4-one
Step 1: Preparation of 2,2-dimethylpropylamine.
To a suspension of lithium aluminum hydride (10.956 g, 0.289 mol) in diethyl ether (50 mL) was added trimethyl acetonitrile (20.00 g, 0.241 mol) in ether (100 mL) at 0 0C. The resulting mixture was stirred at room temperature for 2 h. Then 5% of sodium hydroxide solution was added to the reaction mixture till no gas was released. The mixture was filtered. The filtrate was dried over sodium sulfate. The resulting solution was used directly in the next step. Step 2: Preparation of 2,2-dimethylpropanisothiocvanate.
[0375] To a stirred solution of 2,2-dimethylpropylamine (8.03 g, 0.141 mol) was added triethylamine (26.228 g , 0.259 mol), and carbon disulfide (49.339 g, 0.648 mol) at 0-10 0C. Stirring was continued for 0.5 h, followed by dropwise addition of hydrogen peroxide (30 wt %, 73.46 g, 0.0.648 mol) at room temperature. After 1 h, the reaction mixture was neutralized with diluted hydrochloric acid (1:1) to neutral. The organic layer was washed with brine, dried over Na2SO4. The resulting solution was used directly in the following step.
Step 3: Preparation of aminor(2,2-dimethylpropyl)amino1methane-l-thione.
[0376] A methanolic solution of ammonia (7N, 55.0 ml) was added to a solution of 2,2-dimethylpropanisothiocyanate in ether at 0 0C. The resulting mixture was then stirred at room temperature for 4 h. After concentration, the yellowish residue was collected by filtration, washed with 30% ether in hexanes, and dried to afford 18.889 g (53.7% overall in 3 steps) of white solid as desired product: 1H NMR (CDCl3/ 400 MHz) 6 6.24 (br, IH), 5.80 (br, 2H), 2.92 (br, 2H), 0.98 (s, 9H).
Step 4: Preparation of 3-(2,2-dimethylpropyl)-6-hvdroxy-2-methylthio-3- hydropyrimidin-4-one.
[0377] A vigorously stirred mixture of amino[(2,2-dimethylpropyl)amino]methane- 1-thione (8.00 g, 54.7 mmol), diethylmalonate (8.767 g, 54.7 mmol), sodium methoxide (25-30% in methanol, 21.9 mL) was heated to reflux under nitrogen. After 5 h, the reaction mixture was cooled and added iodomethane (8.076 g, 56.9 mmol) while the temperature was maintained at 50 0C. The mixture was stirred at 50 °C for 1 h, then cooled to 10 °C, and treated with acetic acid (6.3 mL). The product that precipitated was diluted with water. The solid was filtered, washed with water, and air dried to afford 11.057 g (88.5%) of the desired product. 11.057 g (88.5%) of the desired product: 1H NMR (DMSO-d6/ 400 MHz) 5 5.22 (s, IH), 3.83 (br, 2H), 2.48 (s, 3H), 0.98 (s, 9H); ES-MS m/z 229.14 (M+H).
Step 5: Preparation of 6-r(2,4-difluorophenyl)methoxyl-3-(2,2-dimethylpropyl)-2- methylthio-3-hvdropyrimidin-4-one.
[0378] A mixture of 3-(2,2-dimethylpropyl)-6-hydroxy-2-methylthio-3- hydropyrimidin-4-one (11.057 g, 48.4 mmol), 2,4 difluorobenzylbromide (15.040 g, 72.6 mmol) and potassium carbonate (12.049 g, 87.2 mmol) in DMF (80 mL) was stirred at 0 °C under nitrogen. After 30 min, it was stirred at room temperature for an additional 30 min and filtered. The filtrate was concentrated under vacuum and the residue was partitioned between dichloromethane and 5% hydrochloric acid. The organic phase was washed with water, dried (Na2SO4), and concentrated to dryness under reduced pressure. The resulting material was purified by silica gel flash chromatography using 20% EtOAc in hexane as elution to give 9.366 g (54.6%) of the title compound as a white powder: 1H NMR (CDCl3/ 400 MHz) δ 7.42-7.39 (m, IH), 6.90-6.81 (m, 2H), 5.56 (s, IH), 5.31 (s, 2H), 3.95 (br, 2H), 2.51 (s, 3H), 1.04 (s, 9H); Anal. Calcd for C17H20N2O2SF2: C, 57.61; H, 5.69; N, 7.90. Found: C, 57.83; H, 5.81; N, 7.97; ES-MS m/z 355 (MW: 354).
Biological Evaluation
p38 Kinase Assay
Cloning of human p38a:
[0379] The coding region of the human p38a cDNA was obtained by PCR- amplification from RNA isolated from the human monocyte cell line THP.1. First strand CDNA was synthesized from total RNA as follows: 2 μg of RNA was annealed to 100 ng of random hexamer primers in a 10 μl reaction by heating to 70° C. for 10 minutes followed by 2 minutes on ice. cDNA was then synthesized by adding 1 μl of RNAsin (Promega, Madison Wis.), 2 μl of 50 mM dNTP's, 4 μl of 5X buffer, 2 μl of 100 mM DTT and 1 μl (200 U) of Superscript II™ AMV reverse transcriptase. Random primer, dNTP's and Superscript II™ reagents were all purchased from Life-Technologies, Gaithersburg, Mass. The reaction was incubated at 42° C. for 1 hour. Amplification of
p38 cDNA was performed by aliquoting 5 μl of the reverse transcriptase reaction into a 100 μl PCR reaction containing the following: 80 μl dH.sub.2 O, 2 . μl 50 mM dNTP's, 1 μl each of forward and reverse primers (50 pmol/μl), 10 μl of 1OX buffer and 1 μl Expand™ polymerase (Boehringer Mannheim). The PCR primers incorporated Bam HI sites onto the 5' and 3' end of the amplified fragment, and were purchased from Genosys.
The sequences of the forward and reverse primers were 5'- GATCGAGGATTCATGTCTCAGGAGAGGCCCA-3' and 5'GATCGAGGATTCTCAGGACTCCATCTCTTC-S' respectively. The PCR amplification was carried out in a DNA Thermal Cycler (Perkin Elmer) by repeating 30 cycles of 94° C. for 1 minute, 60° C. for 1 minute and 68° C. for 2 minutes. After amplification, excess primers and unincorporated dNTP's were removed from the amplified fragment with a Wizard™ PCR prep (Promega) and digested with Bam HI (New England Biolabs). The Bam HI digested fragment was ligated into BamHI digested pGEX 2T plasmid DNA (PharmaciaBiotech) using T-4 DNA ligase (New England Biolabs) as described by T. Maniatis, Molecular Cloning: A Laboratory Manual, 2nd ed. (1989). The ligation reaction was transformed into chemically competent E. coli DHlOB cells purchased from Life-Technologies following the manufacturer's instructions. Plasmid DNA was isolated from the resulting bacterial colonies using a Promega Wizard™ miniprep kit. Plasmids containing the appropriate Bam HI fragment were sequenced in a DNA Thermal Cycler (Perkin Elmer) with Prism™ (Applied Biosystems Inc.). cDNA clones were identified that coded for both human p38a isoforms (Lee et al. Nature 372, 739). One of the clones that contained the cDNA for p38a-2 (CSB-2) inserted in the cloning site of PGEX 2T, 3' of the GST coding region was designated pMON 35802. The sequence obtained for this clone is an exact match of the cDNA clone reported by Lee et al. This expression plasmid allows for the production of a GST-p38a fusion protein.
Expression of human p38a
[0380] GST/p38a fusion protein w as expressed from the plasmid pMON 35802 in E. coli, stain DHlOB (Life Technologies, Gibco-BRL). Overnight cultures were grown in Luria Broth (LB) containing 100 mg/ml ampicillin. The next day, 500 ml of fresh LB was inoculated with 10 ml of overnight culture, and grown in a 2 liter flask at 37° C.
with constant shaking until the culture reached an absorbance of 0.8 at 600 nm. Expression of the fusion protein was induced by addition of isopropyl b-D- thiogalactosidase (IPTG) to a final concentration of 0.05 mM. The cultures were shaken for three hours at room temperature, and the cells were harvested by centrifugation. The cell pellets were stored frozen until protein purification.
Purification of P38 Kinase-alpha
[0381] All chemicals were from Sigma Chemical Co. unless noted. Twenty grams of E. coli cell pellet collected from five 1 L shake flask fermentations was resuspended in a volume of PBS (140 mM NaCl, 2.7 mM KCl, 10 mM Na.sub.2 HPO.sub.4, 1.8 mM KH.sub.2 PO.sub.4, pH 7.3) up to 200 ml. The cell suspension was adjusted to 5 mM DTT with 2 M DTT and then split equally into five 50 ml Falcon conical tubes.1 The cells were sonnicated (Ultrasonics model W375) with a 1 cm probe for 3. times.1 minutes (pulsed) on ice. Lysed cell material was removed by centrifugation (12,000 x g, 15 minutes) and the clarified supernatant applied to glutathione-sepharose resin (Pharmacia).
Glutathione-Sepharose Affinity Chromatography
[0382] Twelve ml of a 50% glutathione sepharose-PBS suspension was added to 200 ml clarified supernatant and incubated batchwise for 30 minutes at room temperature. The resin was collected by centrifugation (600.times.g, 5 min) and washed with 2.times.l50 ml PBS/1% Triton X-100, followed by 4.times.40 ml PBS. To cleave the p38 kinase from the GST-p38 fusion protein, the glutathione-sepharose resin was resuspended in 6 ml PBS containing 250 units thrombin protease (Pharmacia, specific activity >7500 units/mg) and mixed gently for 4 hours at room temperature. The glutathione-sepharose resin was removed by centrifugation (όOO.times.g, 5 min) and washed 2.times.6 ml with PBS. The PBS wash fractions and digest supernatant containing p38 kinase protein were pooled and adjusted to 0.3 mM PMSF.
Mono Q Anion Exchange Chromatography
[0383] The thrombin-cleaved p38 kinase was further purified by FPLC-anion exchange chromatography. Thrombin-cleaved sample was diluted 2-fold with Buffer A (25 mM HEPES, pH 7.5, 25 mM beta-glycerophosphate, 2 mM DTT, 5% glycerol) and injected onto a Mono Q HR 10/10 (Pharmacia) anion exchange column equilibrated with Buffer A. The column was eluted with a 160 ml 0.1 M-0.6 M NaCl/Buffer A gradient (2 ml/minute flowrate). The p38 kinase peak eluting at 200 mM NaCl was collected and concentrated to 3-4 ml with a Filtron 10 concentrator (Filtron Corp.).
Sephacryl SlOO Gel Filtration Chromatography
[0384] The concentrated Mono Q- p38 kinase purified sample was purified by gel filtration chromatography (Pharmacia HiPrep 26/60 Sephacryl SlOO column equilibrated with Buffer B (50 mM HEPES, pH 7.5, 50 mM NaCl, 2 mM DTT, 5% glycerol)). Protein was eluted from the column with Buffer B at a 0.5 ml/minute flowrate and protein was detected by absorbance at 280 nm. Fractions containing p38 kinase (detected by SDS-polyacrylamide gel electrophoresis) were pooled and frozen at -80° C. Typical purified protein yields from 5 L E. coli shake flasks fermentations were 35 mg p38 kinase.
In Vitro Assay
[0385] The ability of compounds to inhibit human p38 kinase alpha was evaluated using two in vitro assay methods, hi the first method, activated human p38 kinase alpha phosphorylates a biotinylated substrate, PHAS-I (phosphorylated heat and acid stable protein-insulin inducible), in the presence of gamma 32P-ATP (32P-ATP). PHAS-I was biotinylated prior to the assay and provides a means of capturing the substrate, which is phosphorylated during the assay. p38 Kinase was activated by MKK6. Compounds were tested in 10 fold serial dilutions over the range of 100 μM to 0.001 μM using 1% DMSO. Each concentration of inhibitor was tested in triplicate.
[0386] All reactions were carried out in 96 well polypropylene plates. Each reaction well contained 25 mM HEPES pH 7.5, 10 mM magnesium acetate and 50 μM unlabeled ATP. Activation of p38 was required to achieve sufficient signal in the assay.
Biotinylated PHAS-I was used at 1-2 μg per 50 μl reaction volume, with a final concentration of 1.5 μM. Activated human p38 kinase alpha was used at 1 μg per 50 μl reaction volume representing a final concentration of 0.3 μM. Gamma 32P-ATP was used to follow the phosphorylation of PHAS-1. 32P-ATP has a specific activity of 3000 Ci/mmol and was used at 1.2 μ Ci per 50 μl reaction volume. The reaction proceeded either for one hour or overnight at 30° C.
[0387] Following incubation, 20 μl of reaction mixture was transferred to a high capacity streptavidin coated filter plate (SAM-streptavidin-matrix, Promega) prewetted with phosphate buffered saline. The transferred reaction mix was allowed to contact the streptavidin membrane of the Promega plate for 1-2 minutes. Following capture of biotinylated PHAS-I with P incorporated, each well was washed to remove unincorporated 32P-ATP three times with 2M NaCl, three washes of 2M NaCl with 1% phosphoric, three washes of distilled water and finally a single wash of 95% ethanol. Filter plates were air-dried and 20 μl of scintillant was added. The plates were sealed and counted.
[0388] A second assay format was also employed that is based on p38 kinase alpha induced phosphorylation of EGFRP (epidermal growth factor receptor peptide, a 21 mer) in the presence 33P-ATP. Compounds were tested in 10 fold serial dilutions over the range of 100 μM to 0.001 μM in 1% DMSO. Each concentration of inhibitor was tested in triplicate. Compounds were evaluated in 50 μl reaction volumes in the presence of 25 mM Hepes pH 7.5, 10 mM magnesium acetate, 4% glycerol, 0.4% bovine serum albumin, 0.4mM DTT, 50 μM unlabeled ATP, 25 μg EGFRP (200 μM), and 0.05 μCi 33P-ATP. Reactions were initiated by addition of 0.09 μg of activated, purified human GST-p38 kinase alpha. Activation was carried out using GST-MKK6 (5:l,p38:MKK6) for one hour at 30° C. in the presence of 50 μM ATP. Following incubation for 60 minutes at room temperature, the reaction was stopped by addition of 150 μl of AG l.times.8 resin in 900 mM sodium formate buffer, pH 3.0 (1 volume resin to 2 volumes buffer). The mixture was mixed three times with pipetting and the resin was allowed to settle. A total of 50 μl of clarified solution head volume was transferred from the reaction wells to Microlite-2 plates. 150 μl of Microscint 40 was then added to each well of the Microlite plate, and the plate was sealed, mixed, and counted.
[0389] The above protocol assays were used to determine the IC50 values for compounds in the above Examples. The results are shown in Table 1. ; Table 1
TNF Cell Assays
Method of Isolation of Human Peripheral Blood Mononuclear Cells:
[0390] Human whole blood was collected in Vacutainer tubes containing EDTA as an anticoagulant. A blood sample (7 ml) was carefully layered over 5 ml PMN Cell
Isolation Medium (Robbins Scientific) in a 15 ml round bottom centrifuge tube. The sample was centrifuged at 450-500.times.g for 30-35 minutes in a swing out rotor at room temperature. After centrifugation, the top band of cells were removed and washed
3 times with PBS w/o calcium or magnesium. The cells were centrifuged at 400 .times.g for 10 minutes at room temperature. The cells were resuspended in Macrophage Serum
Free Medium (Gibco BRL) at a concentration of 2 million cells/mi.
LPS Stimulation of Human PBMs
[0391] PBM cells (0.1 ml, 2 million/ ml) were co- incubated with 0.1 ml compound (10-0.41 μM, final concentration) for 1 hour in flat bottom 96 well microtiter plates. Compounds were dissolved in DMSO initially and diluted in TCM for a final concentration of 0.1% DMSO. LPS (Calbiochem, 20 ng/ml, final concentration) was then added at a volume of 0.010 ml. Cultures were incubated overnight at 37° C. Supernatants were then removed and tested by ELISA for TNF-a and ILl -b. Viability was analyzed using MTS. After 0.1 ml supernatant was collected, 0.020 ml MTS was added to remaining 0.1 ml cells. The cells were incubated at 37° C. for 2-4 hours, then the O.D. was measured at 490-650 nM.
Maintenance and Differentiation of the U937 Human Histiocytic Lymphoma Cell Line
[0392] U937 cells (ATCC) were propagated in RPMI 1640 containing 10% fetal bovine serum, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine (Gibco). Fifty million cells in 100 ml media were induced to terminal monocytic differentiation by 24 hour incubation with 20 ng/ml phorbol 12-myristate 13-acetate (Sigma). The cells were washed by centrifugation (200.times.g for 5 min) and resuspended in 100 ml fresh medium. After 24-48 hours, the cells were harvested, centrifuged, and resuspended in culture medium at 2 million cells/ml.
LPS Stimulation of TNF production by U937 Cells
[0393] U937 cells (0.1 ml, 2 million/ml) were incubated with 0.1 ml compound (0.004-50 μM, final concentration) for 1 hour in 96 well microtiter plates. Compounds were prepared as 10 mM stock solutions in DMSO and diluted in culture medium to yield a final DMSO concentration of 0.1% in the cell assay. LPS (E coli, 100 ng/ml final concentration) was then added at a volume of 0.02 ml. After 4 hour incubation at 37° C, the amount of TNF-. alpha, released in the culture medium was quantitated by ELISA. Inhibitory potency is expressed as IC50 (μM).
Rat Assay
[0394] The efficacy of the novel compounds in blocking the production of TNF also was evaluated using a model based on rats challenged with LPS. Male Harlen Lewis rats [Sprague Dawley Co.] were used in this model. Each rat weighed approximately 300 g and was fasted overnight prior to testing. Compound administration was typically by oral gavage (although intraperitoneal, subcutaneous and intravenous administration were also used in a few instances) 1 to 24 hours prior to the LPS challenge. Rats were administered 30 μg/kg LPS [salmonella typhosa, Sigma Co.] intravenously via the tail vein. Blood was collected via heart puncture 1 hour after the LPS challenge. Serum samples were stored at -20° C. until quantitative analysis of TNF-. alpha, by Enzyme Linked- Immuno¬ sorbent Assay ("ELISA") [Biosource]. Additional details of the assay are set forth in Perretti, M., et al., Br. J. Pharmacol. (1993), 110, 868-874, which is incorporated by reference in this application.
Mouse Assay
Mouse Model of LPS-Induced TNF Alpha Production
[0395] TNF alpha was induced in 10-12 week old BALB/c female mice by tail vein injection with 100 ng lipopolysaccharide (from S. Typhosa) in 0.2 ml saline. One hour later mice were bled from the retroorbital sinus and TNF concentrations in serum from clotted blood were quantified by ELISA. Typically, peak levels of serum TNF ranged from 2-6 ng/ml one hour after LPS injection.
[0396] The compounds tested were administered to fasted mice by oral gavage as a suspension in 0.2 ml of 0.5% methylcellulose and 0.025% Tween 20 in water at 1 hour or 6 hours prior to LPS injection. The 1 hour protocol allowed evaluation of compound potency at Cmax plasma levels whereas the 6 hour protocol allowed estimation of compound duration of action. Efficacy was determined at each time point as percent inhibition of serum TNF levels relative to LPS injected mice that received vehicle only.
Induction and Assessment of Collagen-Induced Arthritis in Mice
[0397] Arthritis was induced in mice according to the procedure set forth in J. M. Stuart, Collagen Autoimmune Arthritis, Annual Rev. Immunol. 2:199 (1984), which is
incorporated herein by reference. Specifically, arthritis was induced in 8-12 week old DBA/1 male mice by injection of 50 μg of chick type II collagen (CII) (provided by Dr. Marie Griffiths, Univ. of Utah, Salt Lake City, Utah) in complete Freund's adjuvant (Sigma) on day 0 at the base of the tail. Injection volume was 100 μl. Animals were boosted on day 21 with 50 μg of CII in incomplete Freund's adjuvant (100 μl volume). Animals were evaluated several times each week for signs of arthritis. Any animal with paw redness or swelling was counted as arthritic. Scoring of arthritic paws was conducted in accordance with the procedure set forth in Wooley et al., Genetic Control of Type II Collagen Induced Arthritis in Mice: Factors Influencing Disease Suspectibility and Evidence for Multiple MHC Associated Gene Control., Trans. Proc, 15:180 (1983). Scoring of severity was carried out using a score of 1-3 for each paw (maximal score of 12/mouse). Animals displaying any redness or swelling of digits or the paw were scored as 1. Gross swelling of the whole paw or deformity was scored as 2. Ankylosis of joints was scored as 3. Animals were evaluated for 8 weeks. 8-10 animals per group were used.
* * * * * * * * *
[0398] The above detailed description of embodiments is intended only to acquaint others skilled in the art with the invention, its principles, and its practical application so that others skilled in the art may adapt and apply the invention in its numerous forms, as they may be best suited to the requirements of a particular use. This invention, therefore, is not limited to the above embodiments, and may be variously modified.